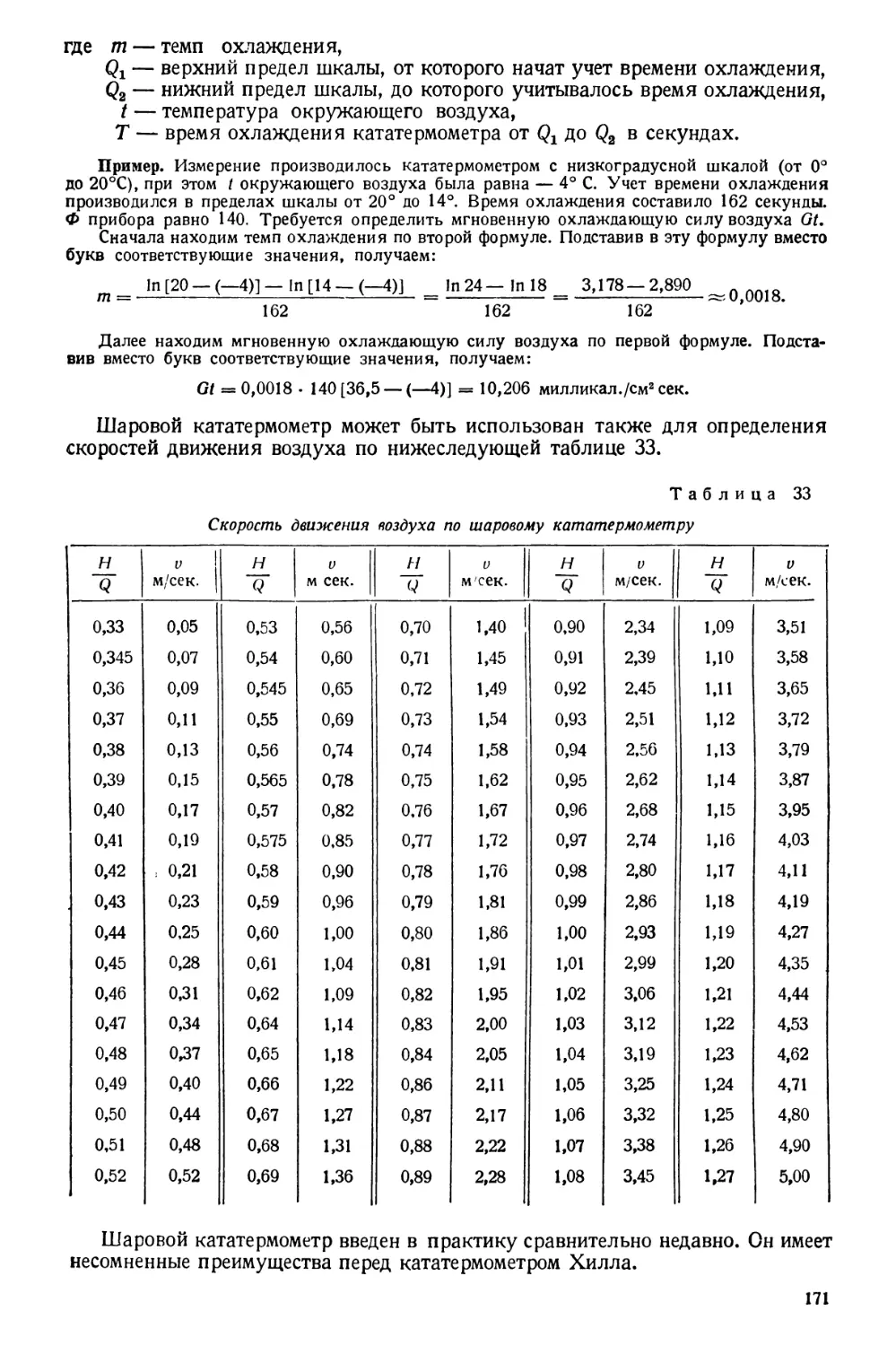
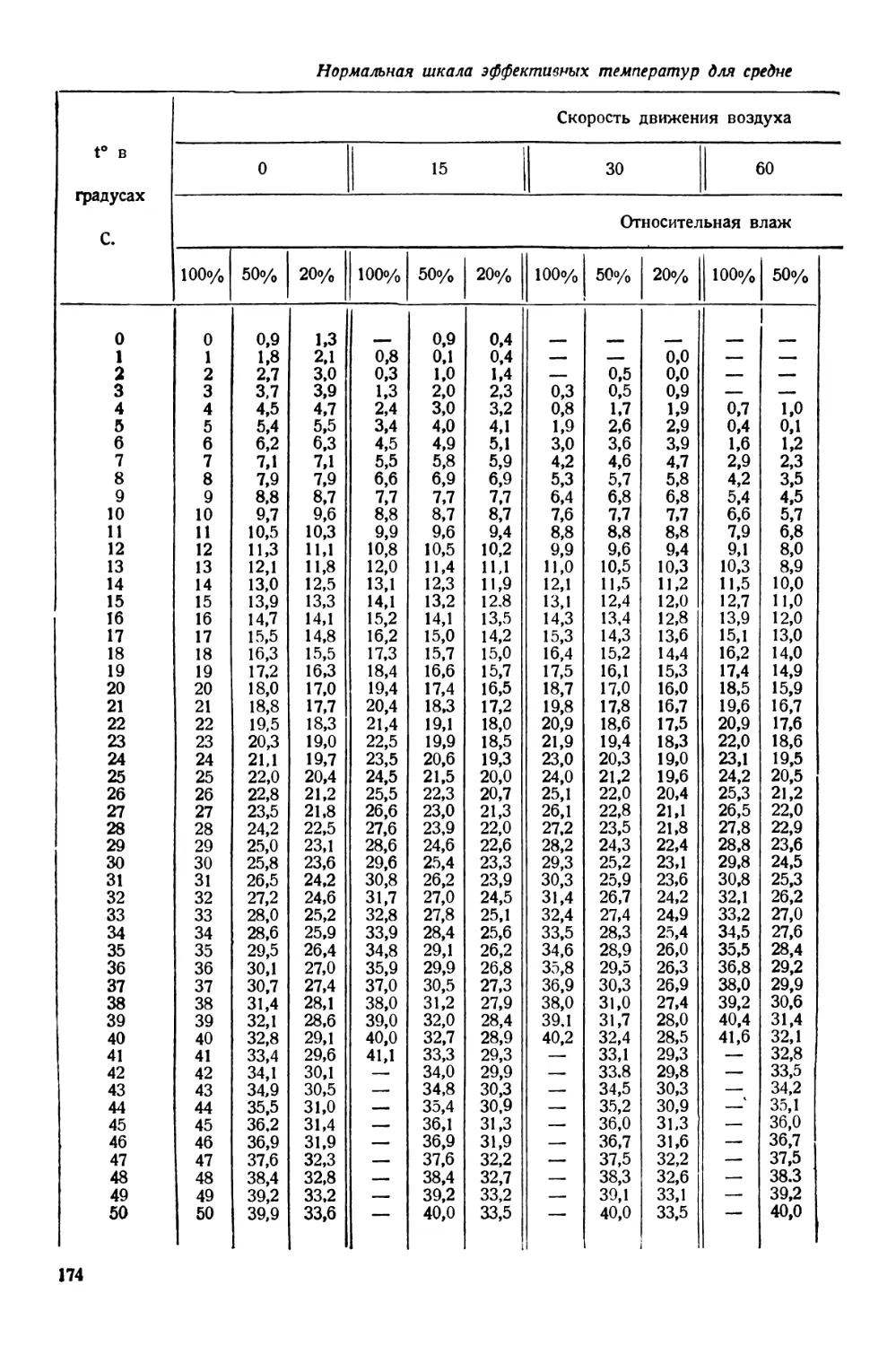
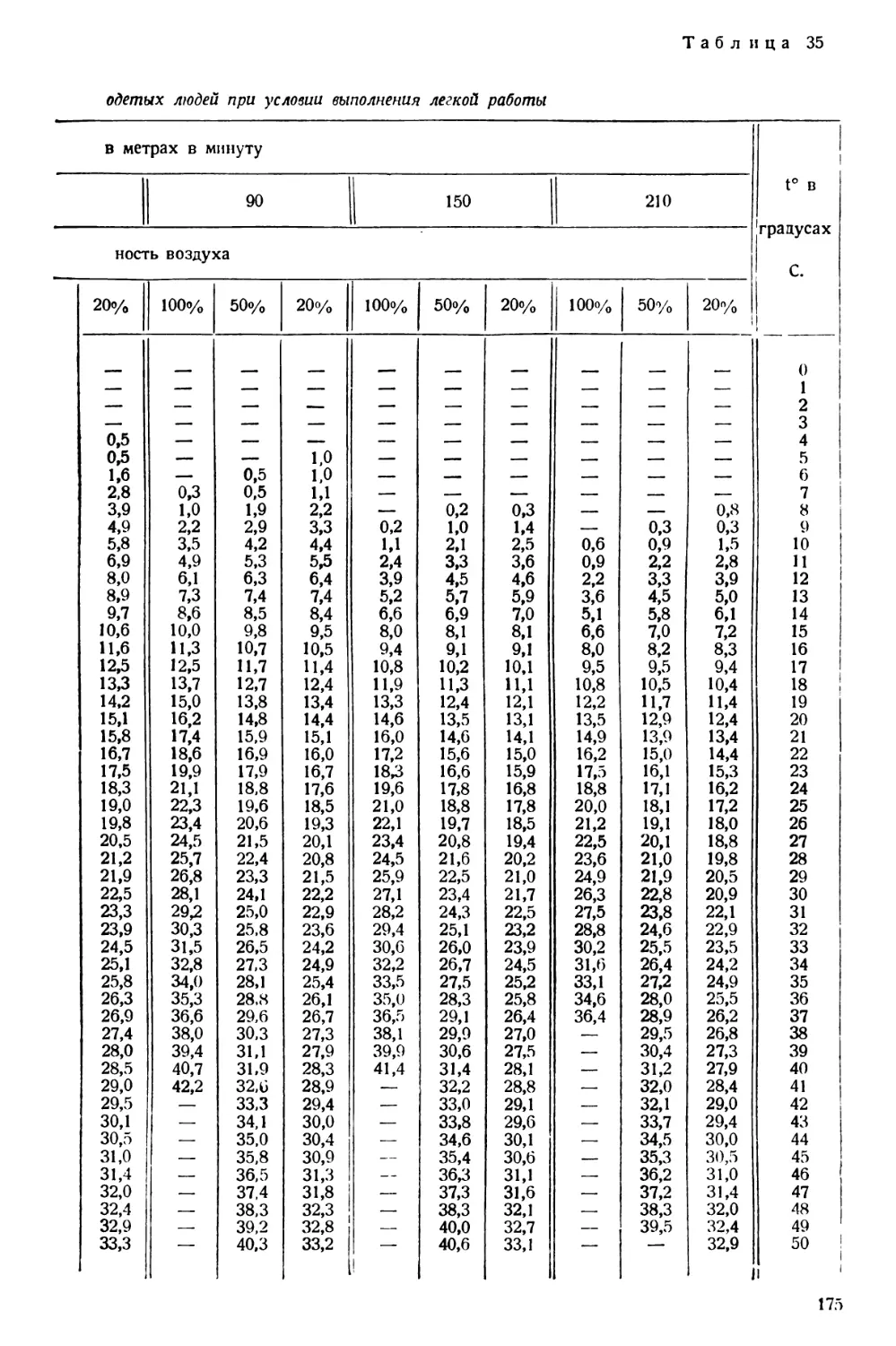
/











































































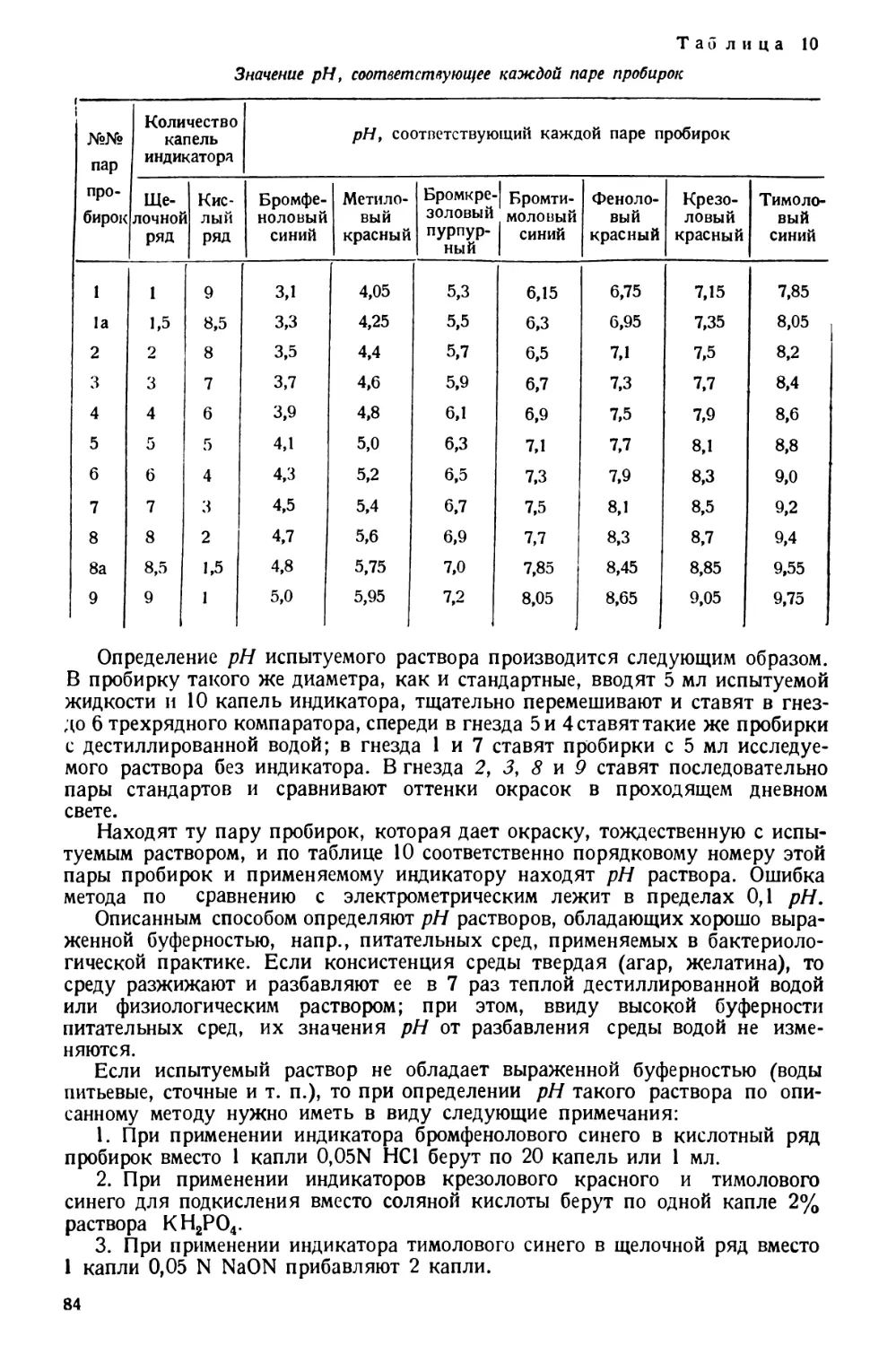









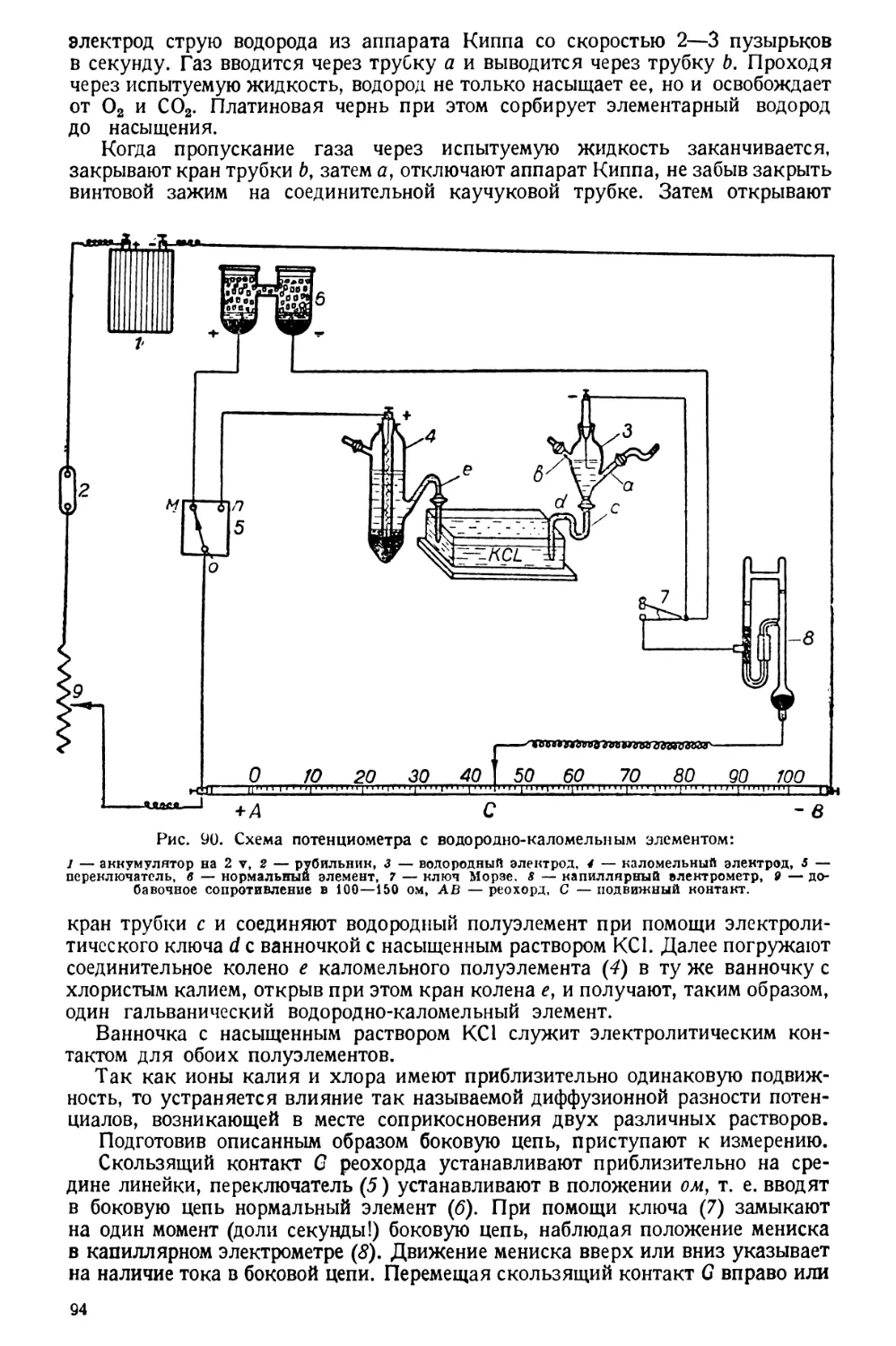


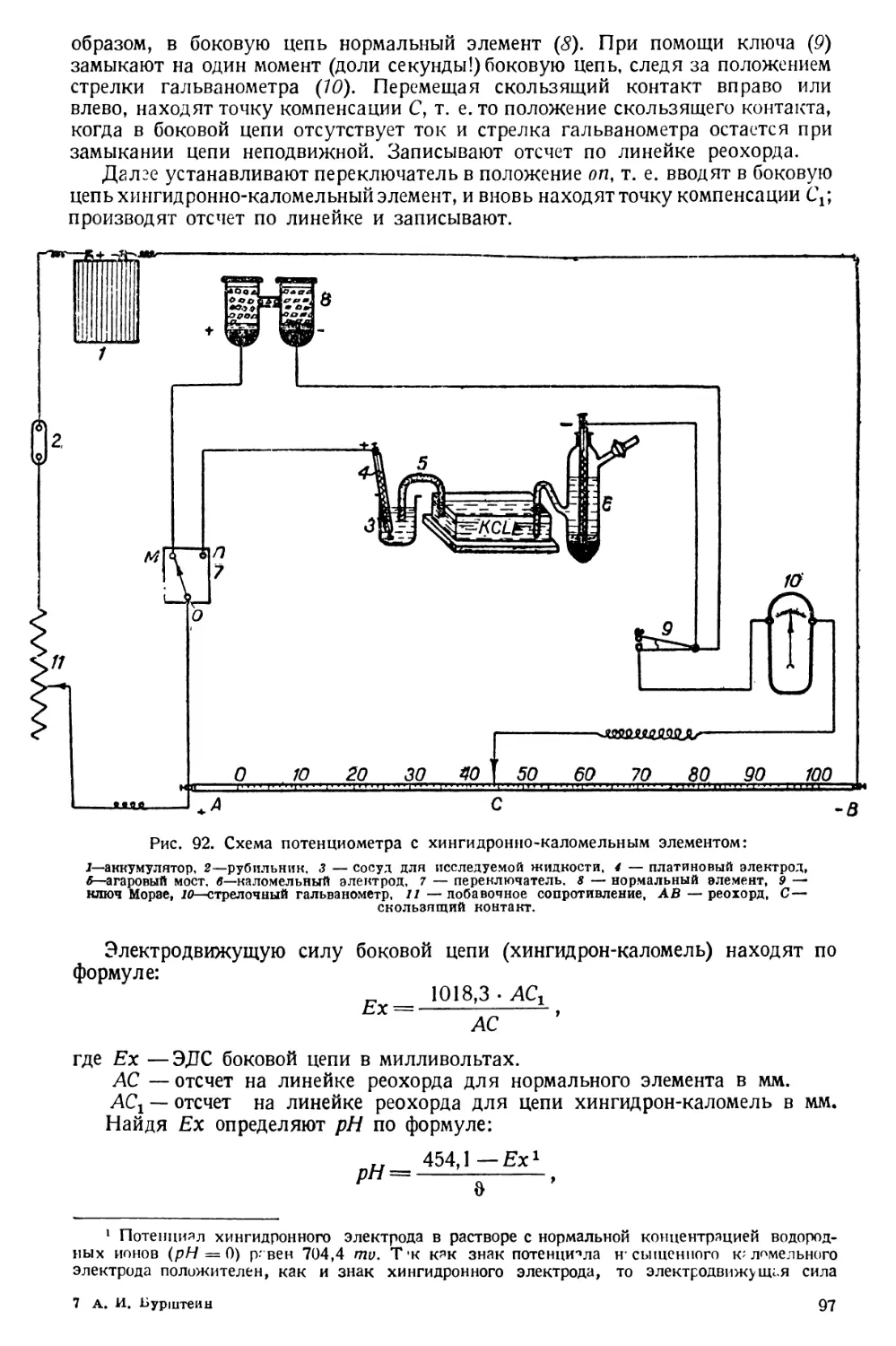



















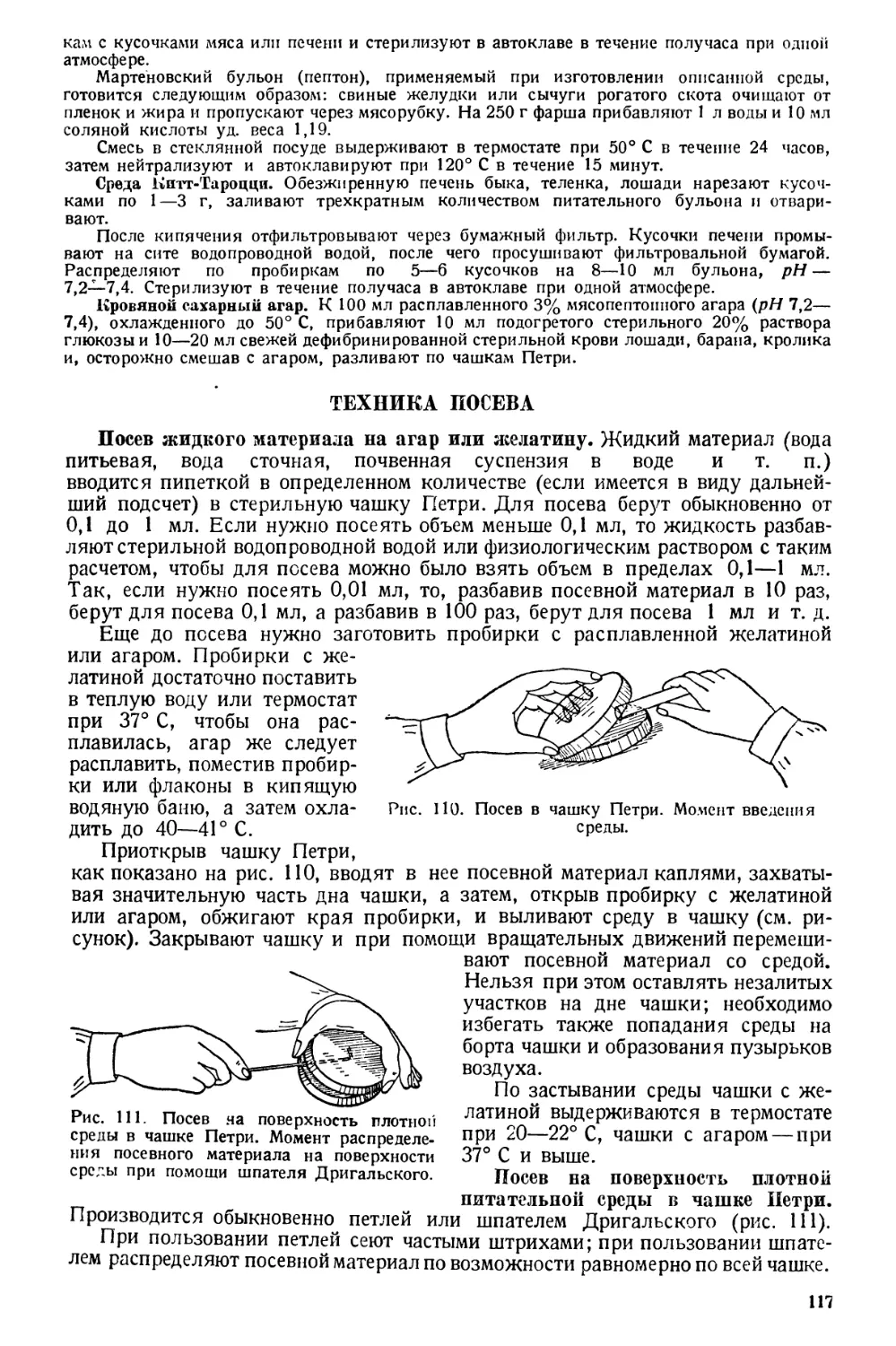








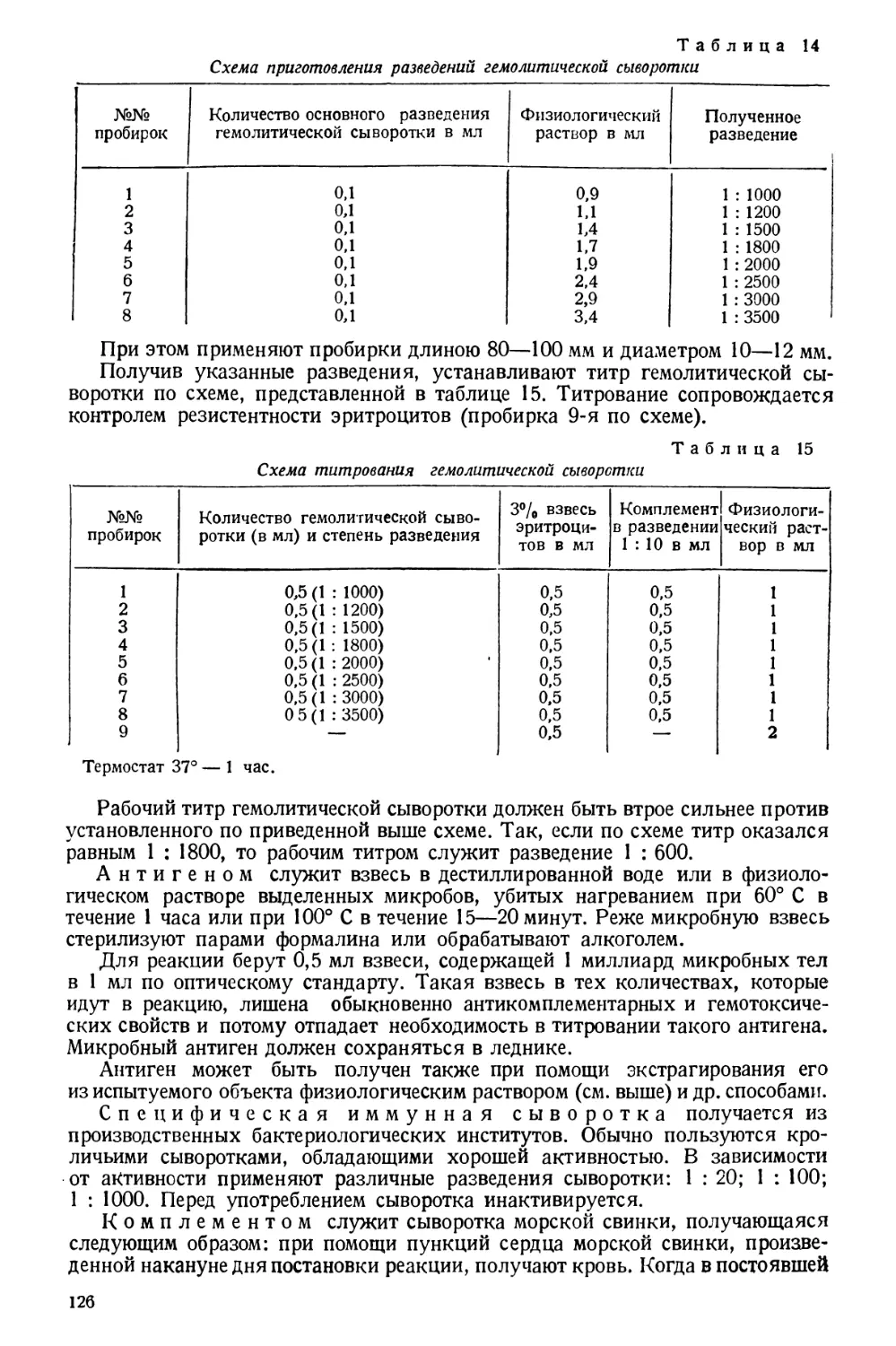
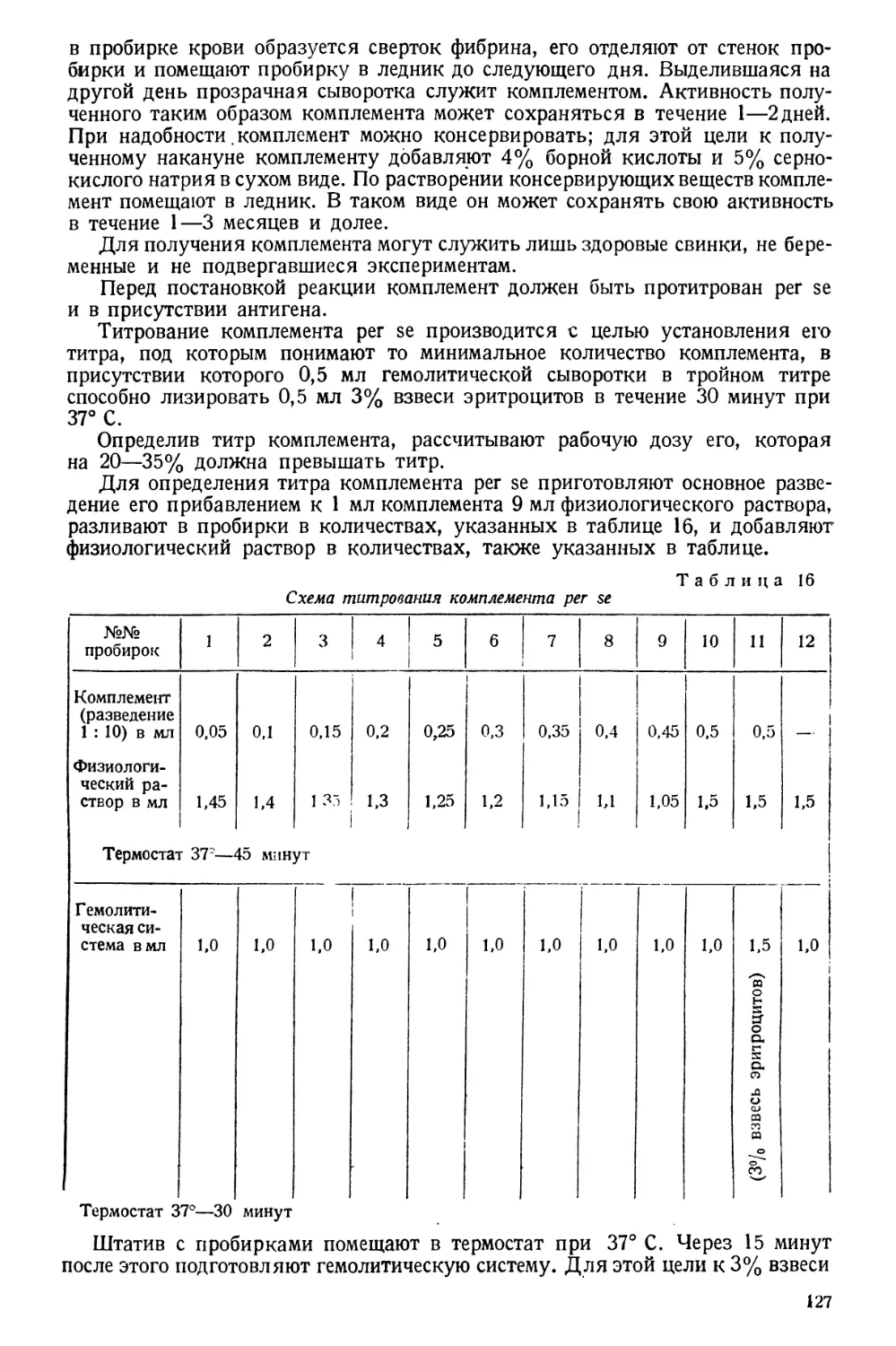

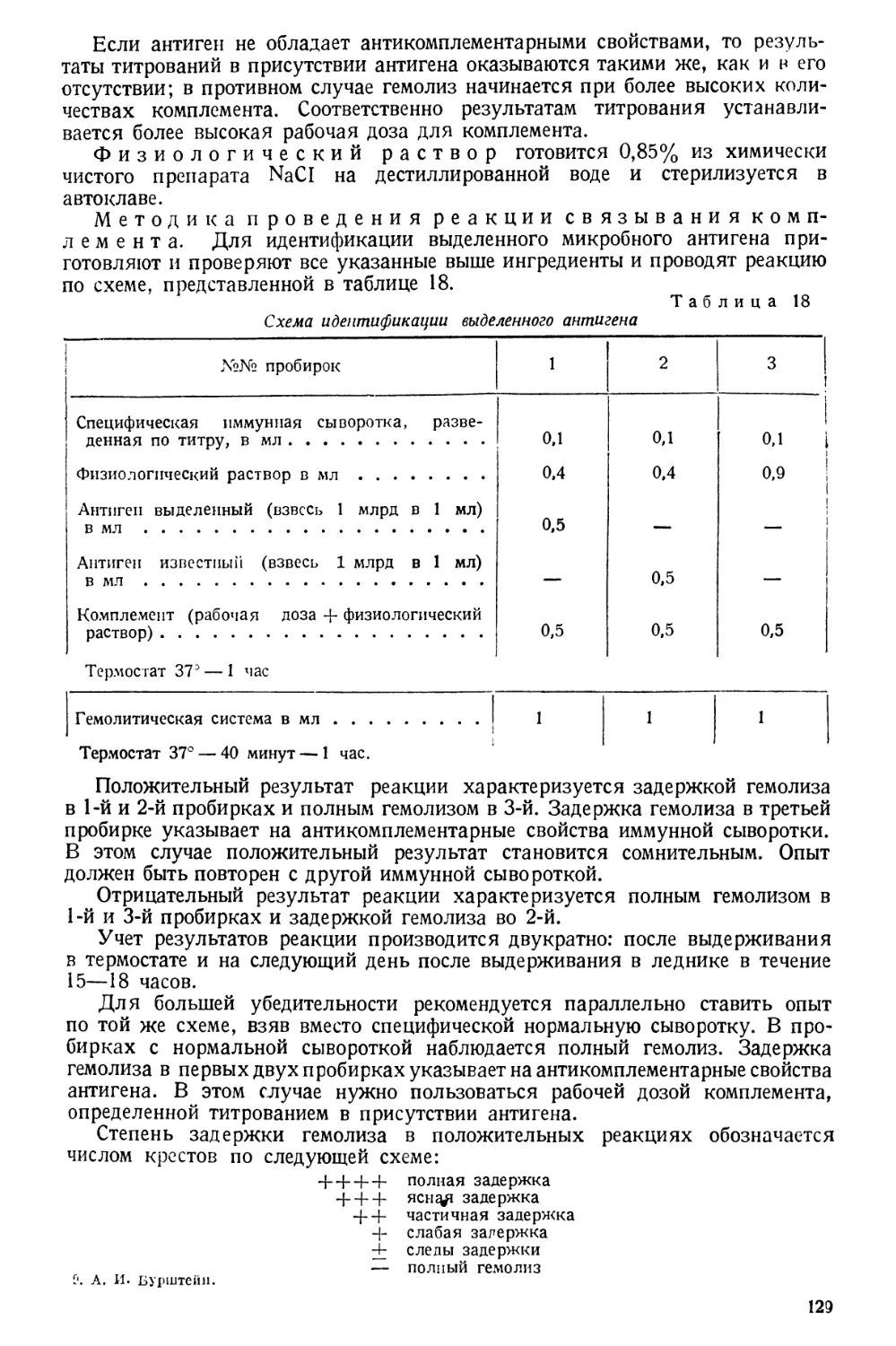

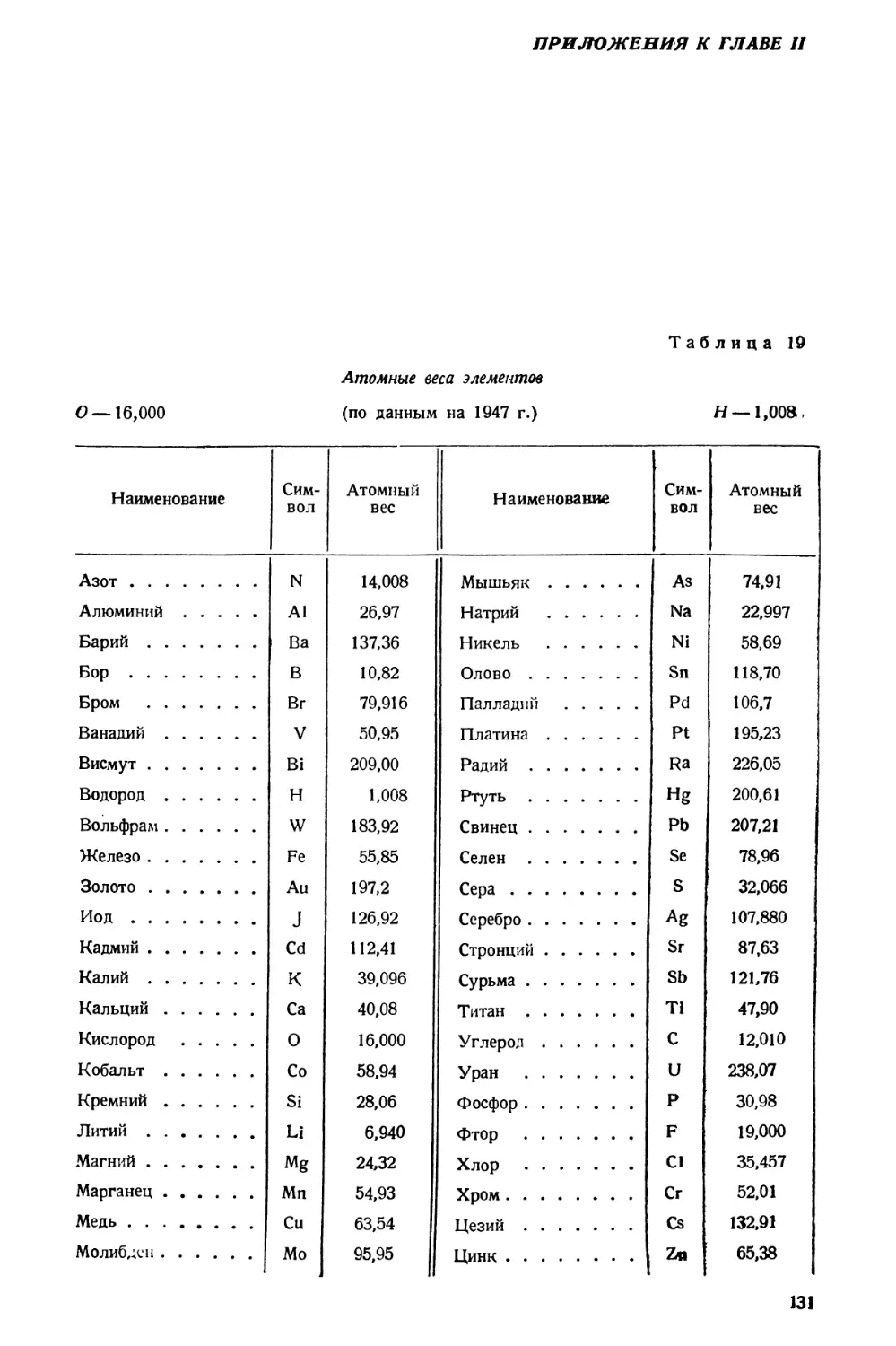












































































































































































































































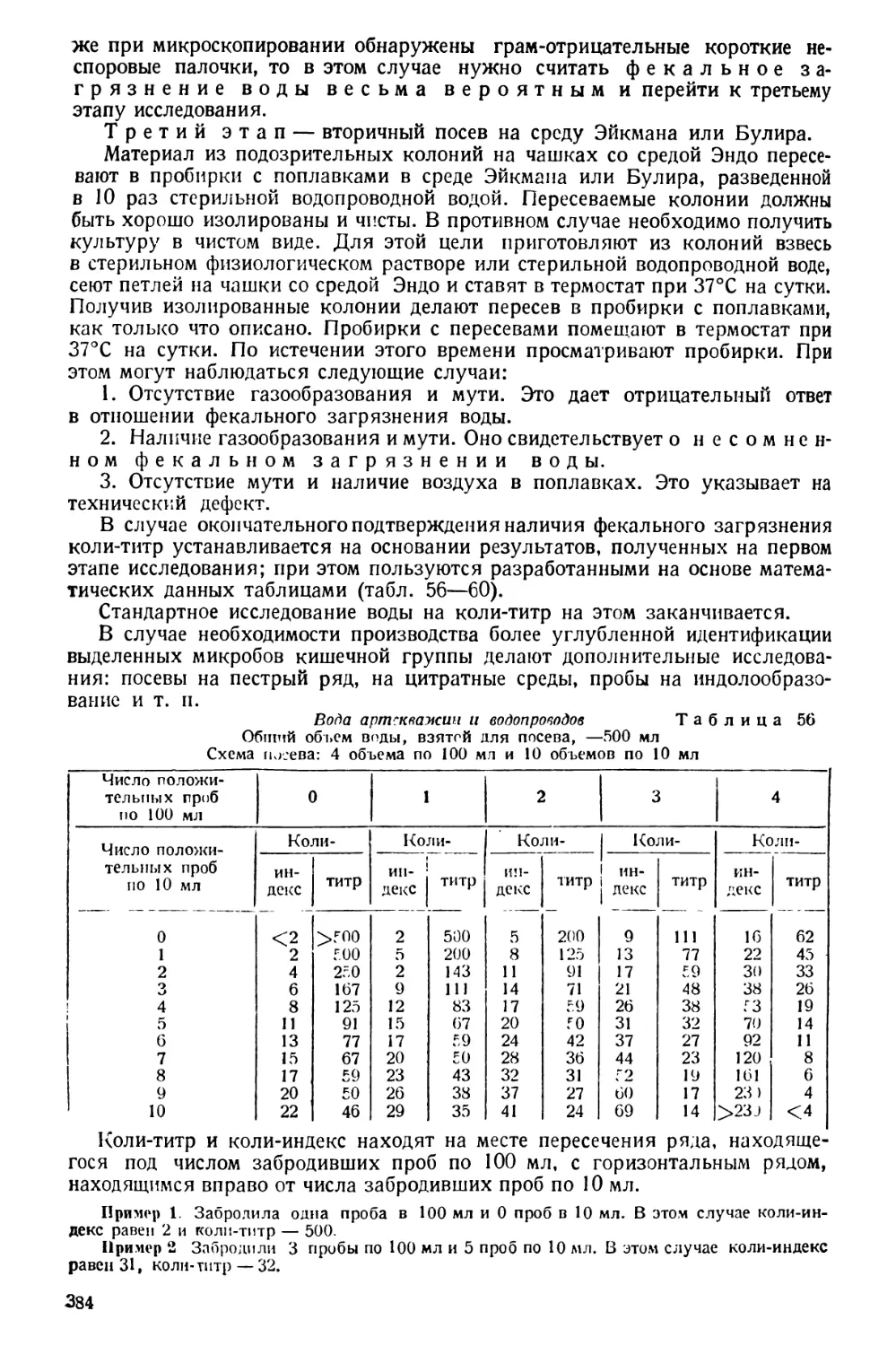
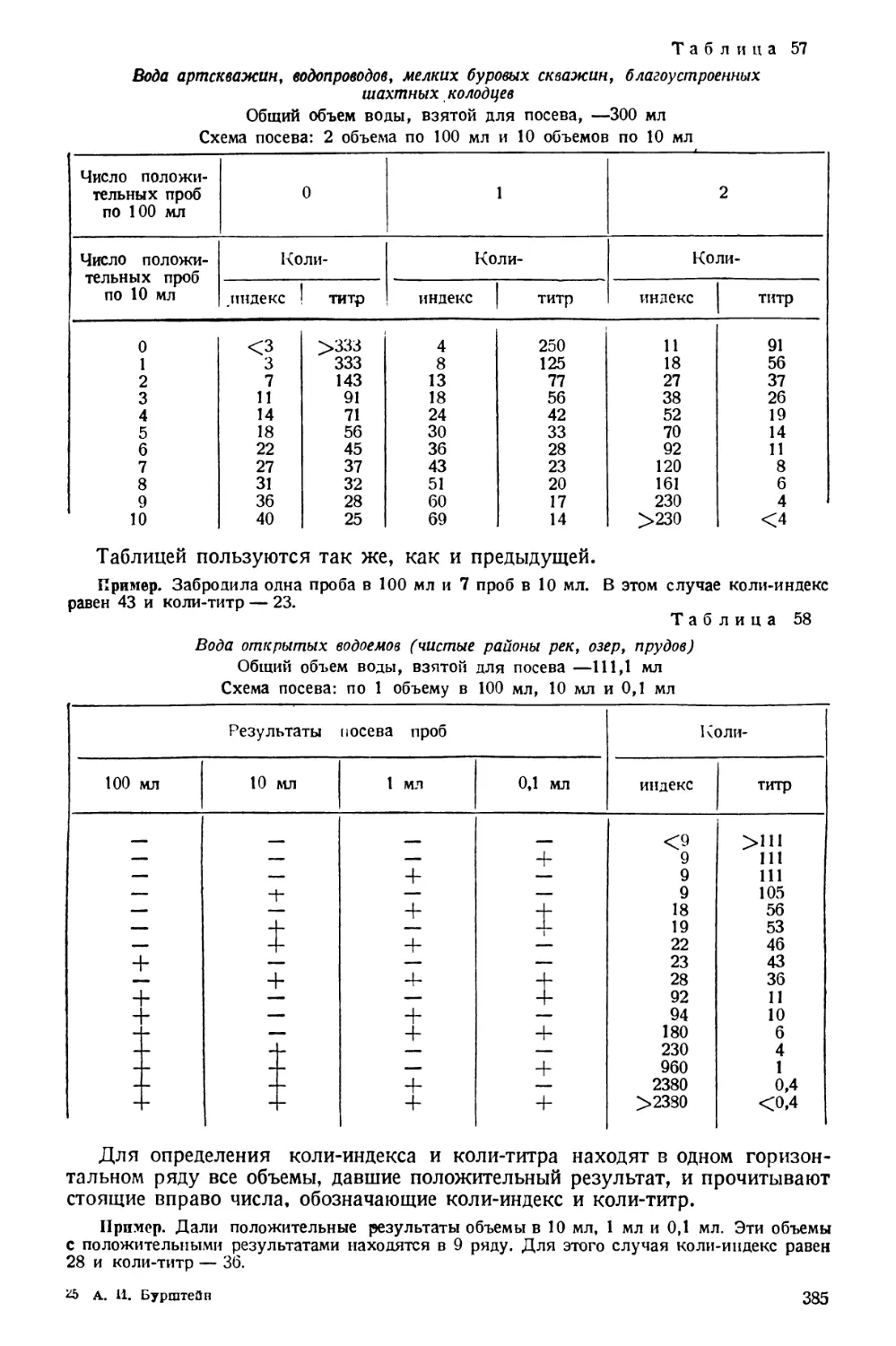
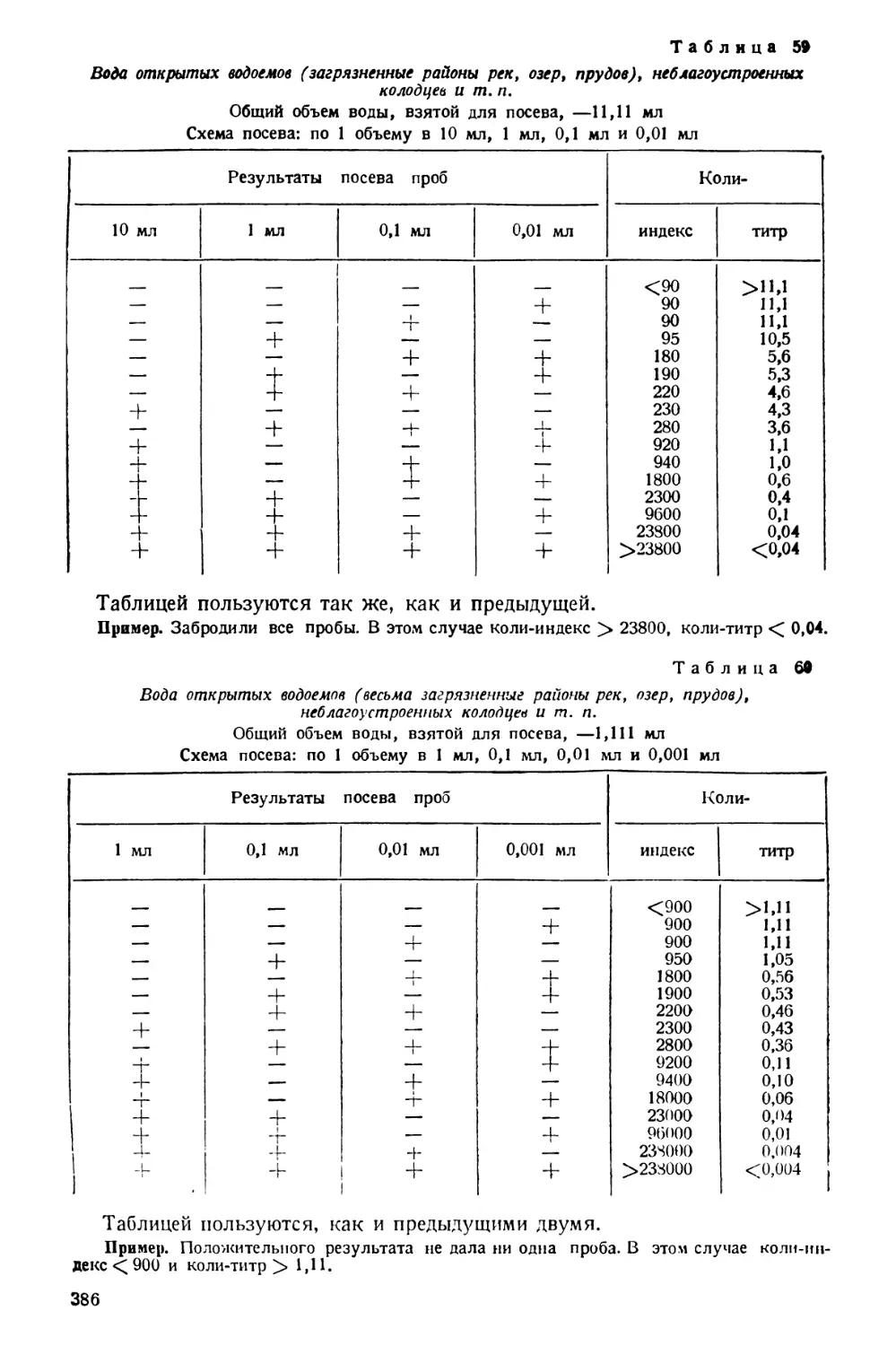






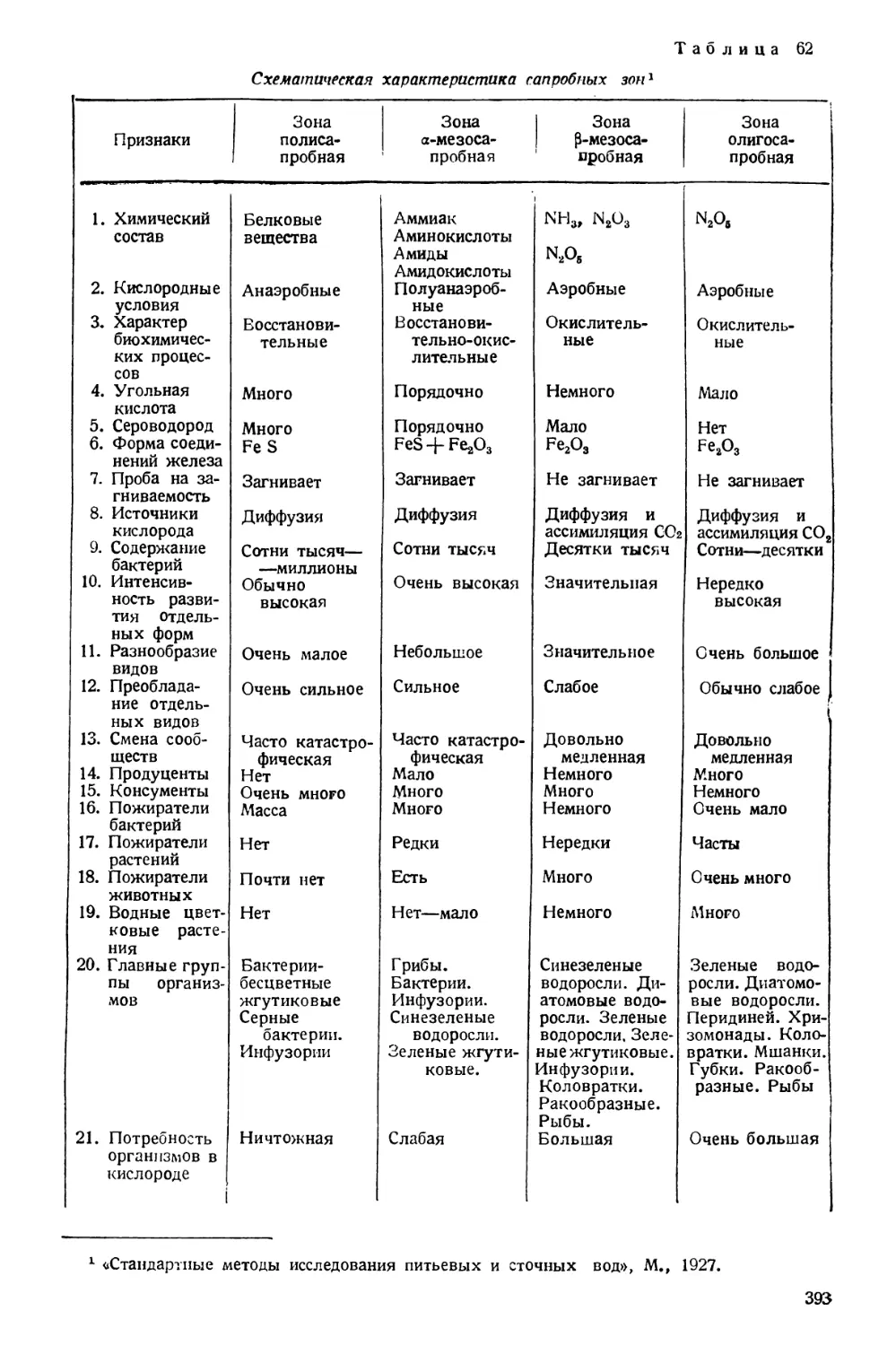



































































































































Text
Проф. А. И. БУРШТЕЙН
Заслуженный деятель науки
МЕТОДЫ
САНИТАРНО-ГИГИЕНИЧЕСКИХ
ИССЛЕДОВАНИЙ
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
ГОСУДАРСТВЕННОЕ
МЕДИЦИНСКОЕ ИЗДАТЕЛЬСТВО УССР
КИЕВ 1930
Книга является руководством по производству гигие-
нических анализов. Современная методика и техника
работы в лаборатории, оборудование, инструмента-
рий, реактивы описаны так, что книга должна стать
настоящим пособием для каждого работающего в
области гигиены. Руководство богато иллюстрировано.
ПРЕДИСЛОВИЕ
Настоящая книга предназначается в качестве руководства для санитарных
врачей и особенно лабораторных работников в их практической деятельности.
Нигде в мире гигиенические мероприятия не осуществляются в таком
масштабе и с такой последовательностью, как в Советском Союзе. В этом
большом и сложном деле лаборатория является одним из важных звеньев,
от состояния которого в высокой мере зависит успех всей работы в целом.
Вот почему мы вправе и должны предъявлять к лаборатории сугубо серьез-
ные требования в смысле надежности применяемых методов исследования
и точности их проведения. Это тем более возможно, что мы давно избавились
от необходимости пользоваться заграничной лабораторной аппаратурой,
лабораторным стеклом и химикалиями. В СССР налажено производство точной
оптической аппаратуры, метеорологических приборов, точных весов, лабо-
раторного стекла, химических реактивов и т. п., что создало благоприятные
условия для лабораторной работы.
В настоящей книге, поскольку разрешали размеры ее, автор стремился
максимально использовать большой и ценный методический материал, раз-
работанный в отечественных гигиенических институтах и лабораториях,
а также нормативные и методические указания ГОСТ-ов.
Все новое и ценное, получившее аппробацию в лабораторной практике,
автор старался отобразить в книге; напротив, старые, отжившие методы
отброшены.
Непосредственному изложению методического материала в книге предпо-
сланы две главы, в которых приводится классификация методов, применяемых
в санитарно-гигиенических исследованиях, и дается введение в лабораторную
технику. Остальные главы посвящены методике исследования воздуха,
питьевых и сточных вод, почвы, строительных материалов и помещений,
одежды, дезинфекционных и дезинсекционных средств. Это материал не охва-
тывает однако всех вопросов, которыми приходится заниматься санитарным
работникам в их повседневной работе. Остаются незатронутыми методика
исследования пищи и питания, определения энергетических затрат организма,
исследования реакций организма на воздействие внешней среды, методика
санитарно-статистических исследований и т. п.
Освещению всех этих вопросов должна быть посвящена отдельная книга.
В совокупности обе эти книги представят важнейший методический материал,
который поможет санитарному работнику в целеустремленном и едином
изучении среды и человека. Единство внешнего и внутреннего во всей
з
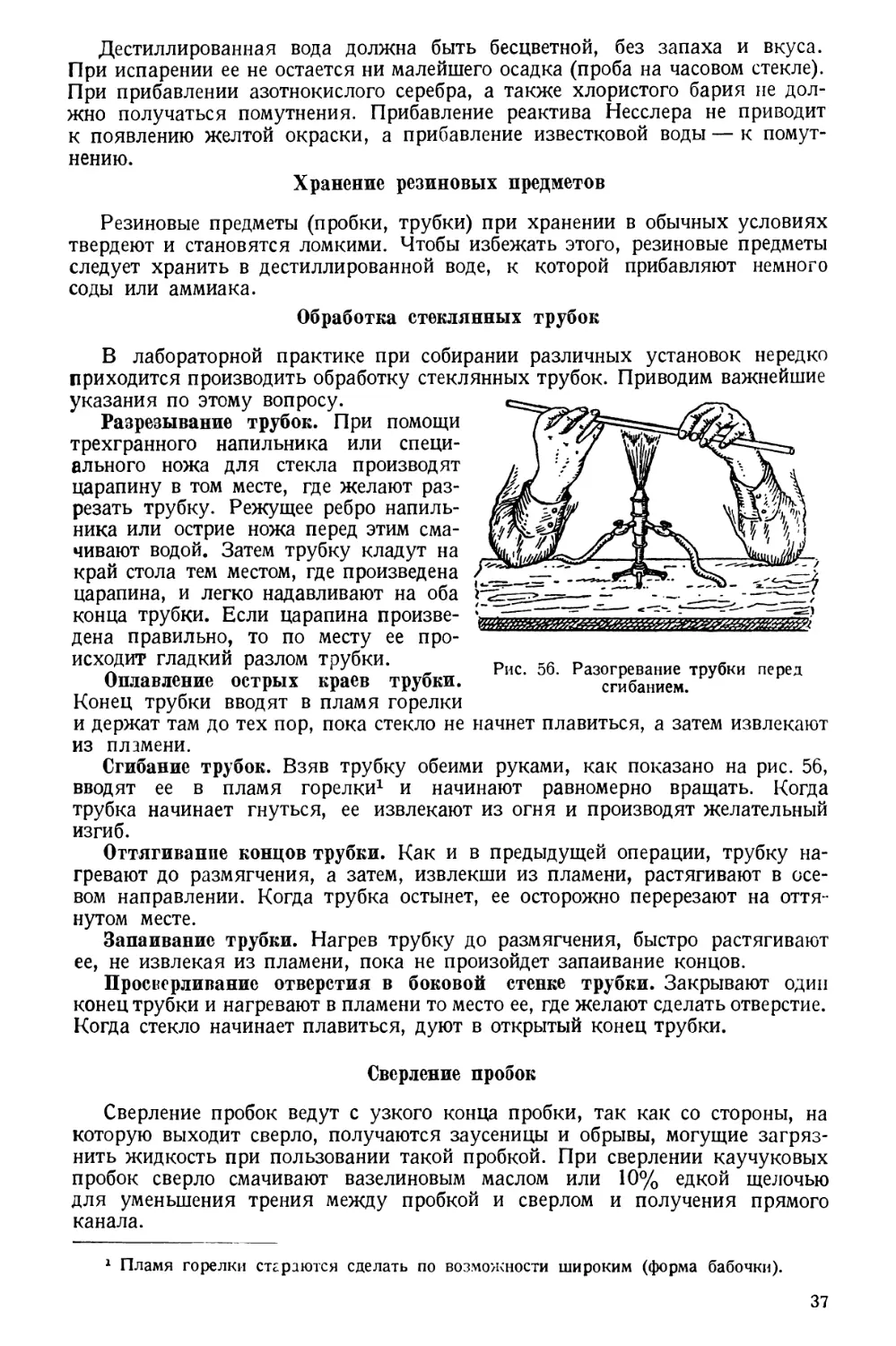
жизнедеятельности организма составляет, как известно, основу учения
И. П. Павлова — великого классика материалистического естествознания.
При составлении настоящего руководства автор имел возможность исполь-
зовать свой большой педагогический и практический опыт, тем не менее он
далек от мысли, что книга лишена дефектов. Напротив, он уверен, что при
изложении столь большого и разностороннего материала можно было до-
пустить некоторые неточности и промахи, и будет очень обязан всем читате-
лям, которые своими указаниями помогут устранить в дальнейшем допу-
щенные недочеты.
Одесса, октябрь 1950 г.
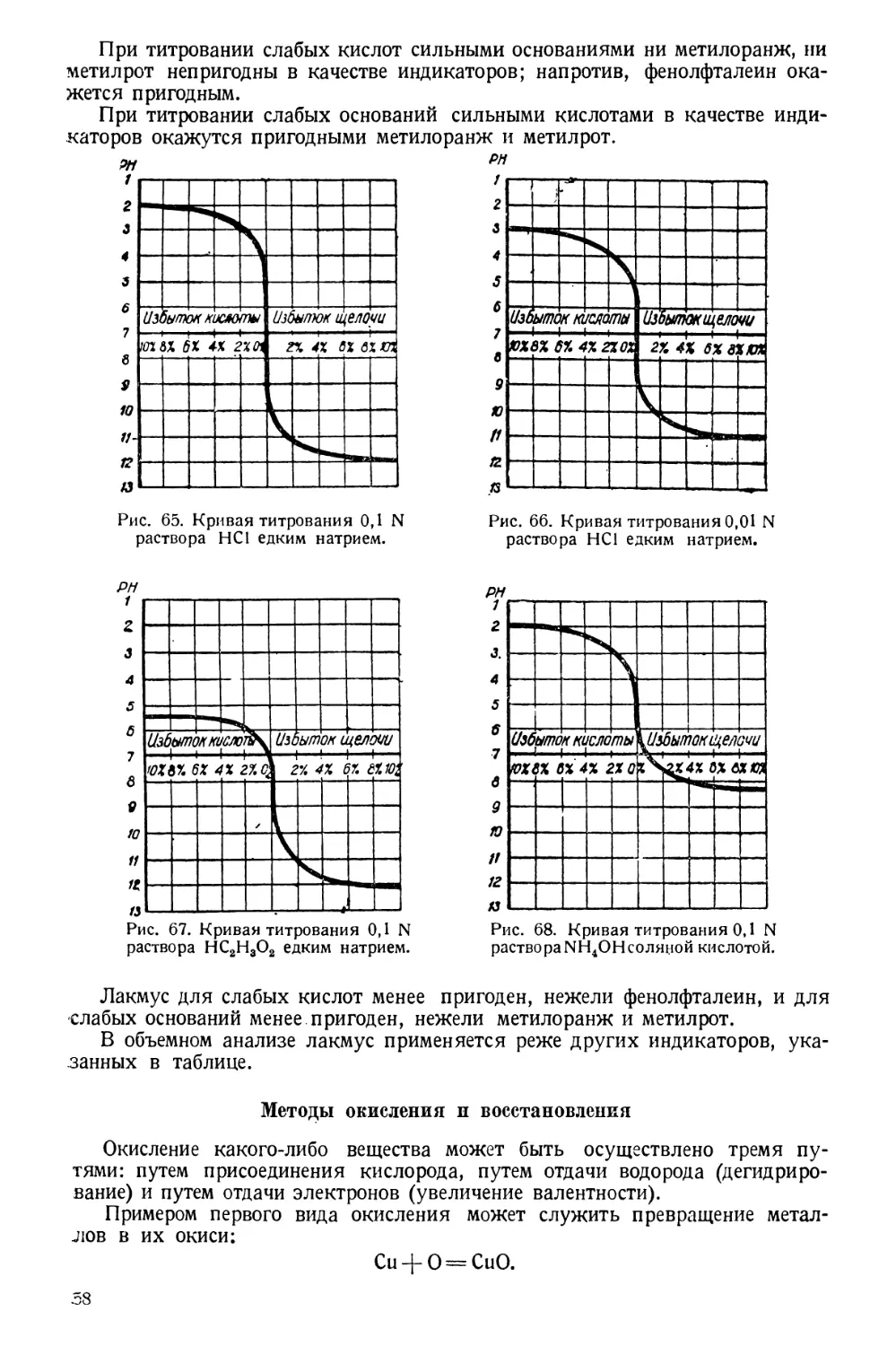
Автор.
ГЛАВА ПЕРВАЯ
МЕТОДЫ,
ПРИМЕНЯЕМЫЕ В САНИТАРНО-ГИГИЕНИЧЕСКИХ
ИССЛЕДОВАНИЯХ
Методы, применяемые в санитарно-гигиенических исследованиях, много-
численны и разнообразны. Важнейшие из них следующие: органолептические,
физические, физико-химические, химические, биохимические, микроскопи-
ческие, бактериологические, микологические, гельминтологические, сероло-
гические, биологические, физиологические и статистические.
Ниже приводится краткая характеристика каждого из этих методов и дается
оценка его с точки зрения значимости в санитарно-гигиенических исследо-
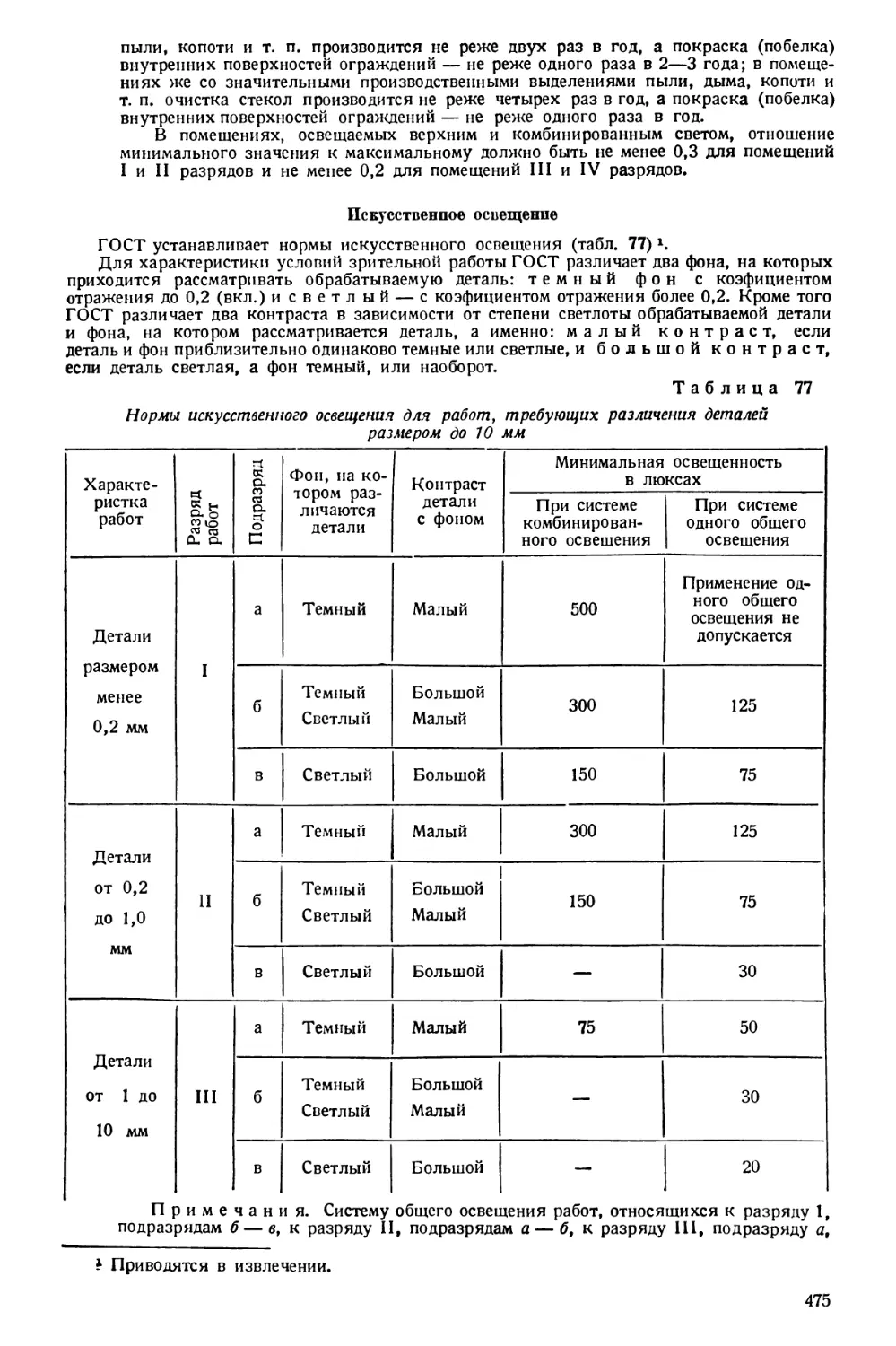
ваниях.
ОРГАНОЛЕПТИЧЕСКИЕ МЕТОДЫ
Органолептическими методами называются такие, при которых исследо-
вание осуществляется психофизиологической системой, состоящей из органа
чувств (экстрорецептора), нервных проводников и соответствуюших отделов
центральной нервной системы. Этим комплексным образованиям И. П. Павлов,
как известно, дал название анализаторов.
При органолептических методах исследования не применяются материаль-
ные приборы, исследуемый объект не подвергается воздействию каких-либо
химических агентов. Допускаются лишь воздействия физического порядка,
облегчающие восприятие экстрорецептором соответствующего раздражения,
например, придание большей поверхности испарения и легкое подогревание
испытуемой жидкости для лучшего ощущения запаха.
При помощи органолептических методов определяются запах, вкус, цвет,
консистенция, внешний вид (форма, рисунок) исследуемого объекта.
Органолептические методы отличаются большой чувствительностью. Так,
специфический запах метилмеркаптана ощущается уже при концентрации
0,000002 мг/м3, ванилина — при 0,0005 мг/м3 и т. п.; горький вкус хинина
ощущается уже от 0,02 мг вещества, сладкий вкус сахарина — от 0,05 мг
ит. п.; наименьшая мощность лучистой энергии, падающей на глаз и спо-
собной еще вызвать световое ощущение, составляет в среднем 1 х 1О~9 эрг.
сек. и т. д.
Необходимо однако отметить, что в зависимости от индивидуальных осо-
бенностей исследователя, состояния его здоровья, возраста и др. причин
органолептические определения могут оказаться неточными, а иногда совер-
шенно неверными. Так, страдающие цветовой слепотой не способны разли-
чать некоторые цветовые тона. Одни цветнослепые отождествляют светло-
красные цвета с темнозелеными, а фиолетовые и пурпурные с синими (даль-
тонизм); другие — отождествляют темнокрасные цвета с светлозелеными,
ь
а фиолетовые с голубыми. Всем известно резкое притупление обоняния
при катаральном состоянии слизистой оболочки носа (грипп, систематиче-
ское курение). Охлаждение языка до 0° С и нагревание его до 45° С парали-
зует вкусовое восприятие и т. п.
Наиболее надежные результаты при органолептических исследованиях
получаются тогда, когда они проводятся людьми с «нормальными» органами
чувств, т. е. не имеющими каких-либо недостатков. В сомнительных случаях
полезно для правильности суждения производить органолептическую оценку
не одному лицу, а нескольким (сотрудники лаборатории, эксперты произ-
водства). При участии нескольких лиц рекомендуется до окончания испытания
всем не выражать своего мнения ни словами, ни жестами, ни выражением
лица во избежание суггестии.
Значение органолептических методов в санитарном анализе довольно
велико и может оказаться решающим в ряде случаев при оценке качества
питьевой воды, пищевых продуктов и пр. Так, питьевая вода, получившая
по запаху оценку в пять баллов (очень сильный запах), признается непри-
годной для употребления.
ФИЗИЧЕСКИЕ МЕТОДЫ
Физическими методами называются такие, при проведении которых исполь-
зуется тот или иной физический принцип (физическое явление).
При помощи физических методов производятся самые разнообразные
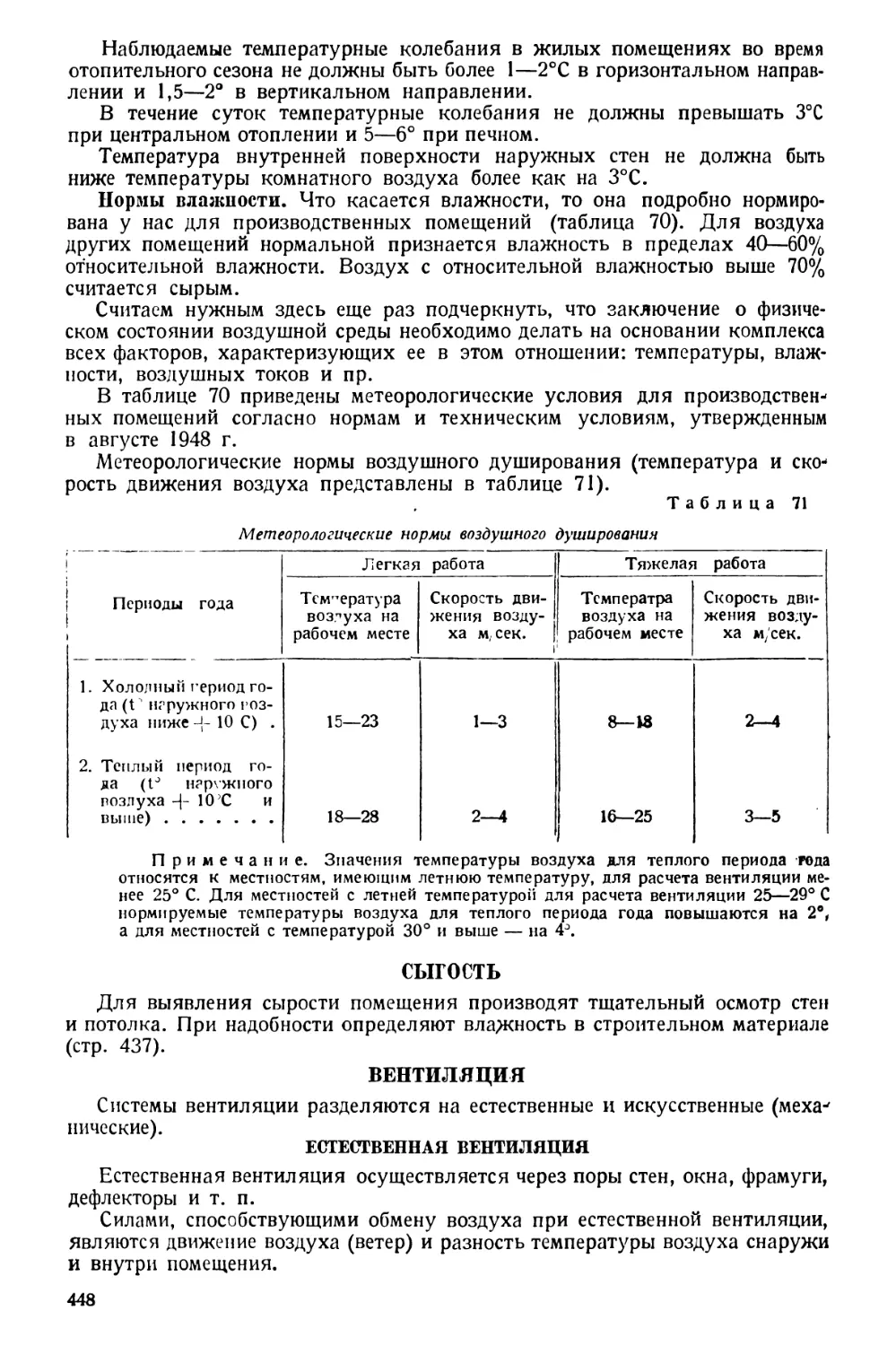
санитарно-гигиенические исследования: определение температуры, влажности,
движения, барометрического давления воздуха; измерения интегрального
потока лучистой энергии, ультрафиолетовой радиации, ионизации атмосферы,
теплопроводности тканей одежды, строительных материалов, освещенности
поверхностей, удельного веса жидкостей и плотных веществ, теплотворной
способности (калорийной ценности) топлива, пищевых продуктов и готовых
блюд.
Физические методы применяются не только для изучения физических
свойств и явлений, но и для химического анализа; так, спектрографический
анализ является тонким методом для обнаружения и идентификации ряда
веществ.
Физические методы исследования осуществляются при помощи измери-
тельных приборов, качество которых в смысле достоверности даваемых ими
показаний зависит от правильности, точности, чувствительности и постоян-
ства.
Под правильностью прибора понимают степень приближения
показания прибора к действительному значению измеряемой величины. От-
клонение показания прибора от действительного значения измеряемой вели-
чины называется погрешностью показания прибора. От
основной (инструментальной) погрешности приборов следует отличать
погрешности дополнительные, вызываемые воздействием внешних
условий на прибор (температура, атмосферное давление, магнитное поле и
пр.). Исключение погрешностей достигается иногда регулировкой прибора,
чаще же введением поправок.
Поправкой называется величина, которая должна быть алгебраи-
чески прибавлена к номинальному показанию прибора, чтобы получить дей-
ствительное значение измеряемой величины. Поправки к приборам указы-
ваются в их паспортах.
Под точностью измерительного прибора понимают степень досто-
верности результатов измерения, полученного с помощью данного измери-
тельного прибора. Точность тем больше, чем меньше по абсолютной величине
погрешность.
Под чувствительностью измерительного прибора понимают
отношение линейного или углового перемещения указателя прибора к изме-
6
нению значения измеряемой величины, вызвавшему это перемещение. Чув-
ствительность прибора выражается уравнением:
5 5Q’
где S — чувствительность,
?>а — угловое или линейное перемещение указателя,
SQ — изменение измеряемой величины.
Под постоянством измерительного прибора понимают степень
устойчивости показаний измерительного прибора при одних и тех же внеш-
них условиях его работы.
Значение физических методов в санитарно-гигиенических исследованиях
очень велико. Это видно хотя бы из приведенного выше перечня (далеко не-
полного) исследований, которые осуществляются при помощи физических
методов.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
Физико-химическими методами называются такие, при проведении кото-
рых используется тот или иной физико-химический принцип (физико-хими-
ческое явление).
При помощи указанных методов определяют различные физико-химиче-
ские показатели исследуемых объектов: вязкость, поверхностное натяжение,
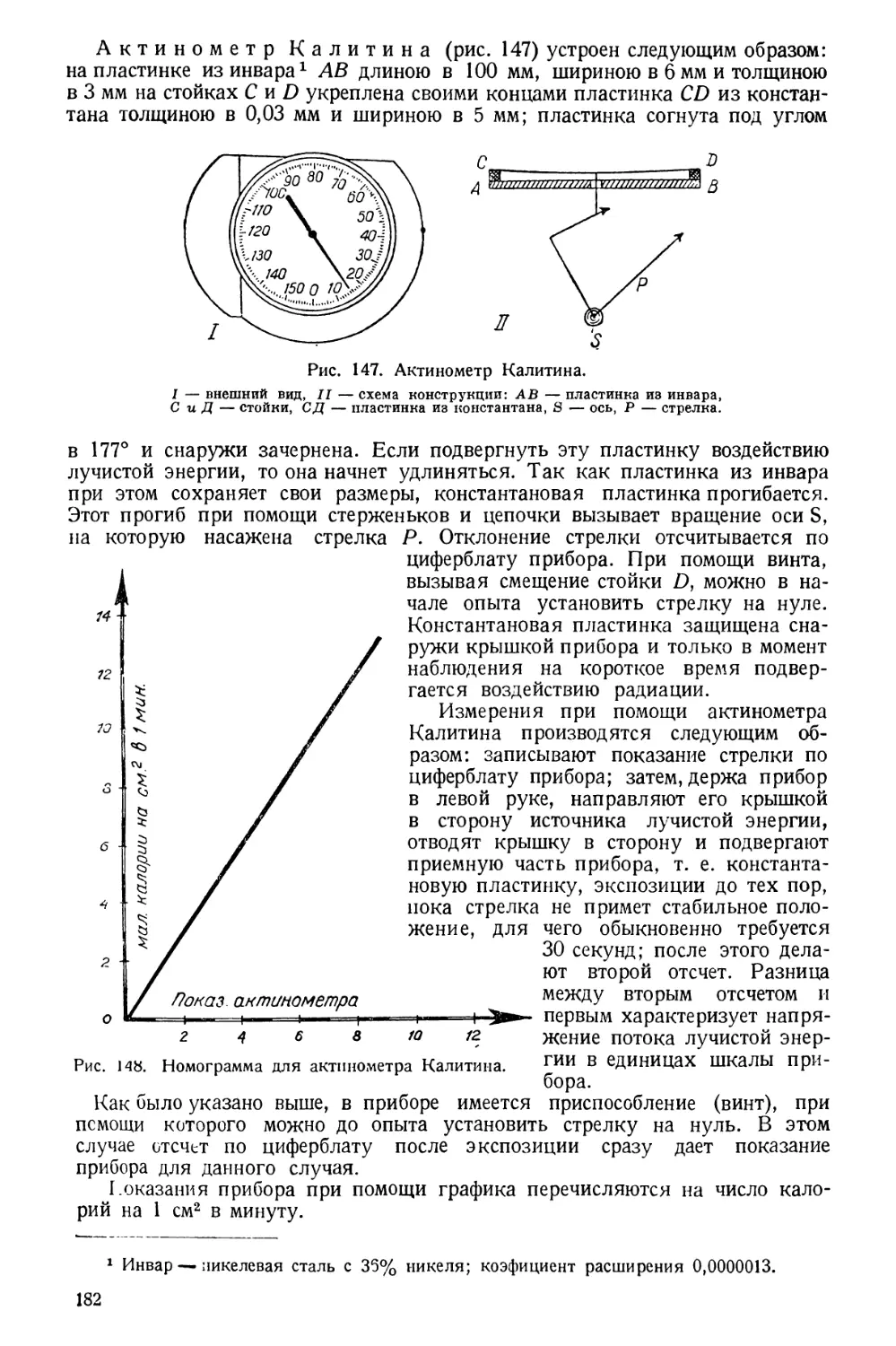
электропроводность, точки плавления, кипения и затвердевания и др.
Физико-химические методы применяются и с целью изучения химического
состава вещества; так, колориметрический и нефеломет-
рический методы исследования растворов дают возможность открывать
в них и количественно определять различные растворенные или взвешенные
вещества; поляриметрический метод исследования растворов дает
возможность открывать и количественно определять в них оптически деятель-
ные вещества; потенциометрический или колориметрический
метод определения pH растворов дает возможность оценивать активную
реакцию растворов и т. п.
Люминесцентный анализ, применяемый в основном при
исследовании пищевых продуктов, способствует выяснению качества их: све-
жести, натуральности.
Хроматографический метод, предложенный русским уче-
ным М. С. Цветом, нашел применение в исследовании витаминов (разделе-
ние пигментов).
Полярографический метод применяется в санитарно-гигиени-
ческих исследованиях для определения содержания тяжелых металлов в кон-
сервах, питьевых и сточных водах и т. п. То же можно сказать об анализе
при помощи электролиза.
Физико-химические методы осуществляются при помощи приборов, ка-
чество которых оценивается по тем же признакам, что и качество физических
приборов (см. выше).
Из физико-химических методов в санитарно-гигиенических исследованиях
особенно широко распространены методы определения pH растворов, а также
колориметрические и нефелометрические методы исследования растворов,
являющиеся одними из весьма распространенных методов количественного
анализа.
ХИМИЧЕСКИЕ МЕТОДЫ
Химическими методами называются такие, которые ставят своей задачей
исследование состава вещества. При этих методах исследуемое вещество по
ходу анализа подвергается различным химическим превращениям.
7
Одно лишь открытие составных частей вещества представляет качест-
венный анализ, количественное же определение составных частей
представляет количественный анализ.
Очень редко в санитарно-гигиенических исследованиях ограничиваются
одним лишь качественным анализом. Так, например, одно лишь качественное
открытие свинца в консервах служит основанием для их забракования и
потому количественного определения не требуется. В подавляющем же боль-
шинстве случаев качественное определение предшествует количественному и
определяет его целесообразность.
В санитарно-гигиенических исследованиях химические методы широко
представлены. Ими пользуются при исследовании воздуха, воды, почвы,
пищевых продуктов, дезинфекционных и дезинсекционных средств, реже
тканей одежды и пр.
Санитарные химико-аналитические методы отличаются довольно большой
чувствительностью. Так, при помощи современных методов можно открывать
и количественно определять в воздухе те или иные вещества еще в концентра-
циях 0,001—0,0001 мг/л. Методы для открытия и количественного опреде-
ления растворенных веществ в воде также отличаются высокой чувствитель-
ностью; так, соли аммония (выраженные через NH3) открываются еще в ко-
личестве 0,1 мг/л, нитриты (N2O3) — в количестве 0,001 мг/л, нитраты (N2O6)—
1,0 мг/л и т. д.
Значение химических методов в санитарно-гигиеническом анализе весьма
велико. В отношении воздуха они дают возможность ответить на вопрос,
не содержатся ли в нем те или иные вредные газообразные продукты в кон-
центрациях, превышающих допустимые санитарные нормы, а также на вопрос
о содержании в аэрозолях токсических веществ (свинца, ртути, мышьяка
и пр.). В отношении питьевой воды химический анализ дает ответ о соответ-
ствии состава воды требованиям ГОСТ’а и пригодности воды по химическим
показателям к употреблению. В отношении сточных вод химический анализ
дает ответ о составе их и возможности спуска по химическим показателям
в открытые водоемы. В отношении почвы химический анализ дает возможность
оценивать степень загрязнения ее отбросами человека и животных. В отно-
шении пищевых продуктов химический анализ отвечает на вопрос о соответ-
ствии продуктов по составу требованиям ГОСТ’ов, устанавливает свежесть
и натуральность их, наличие консервирующих и красящих веществ, тяжелых
металлов, мышьяка и пр. Определив содержание белков, жиров и углеводов
в пищевых продуктах и готовых блюдах, можно на основании полученных
результатов и калорических эквивалентов вычислить калорийную ценность
пищи. Исследуя состав дезинфекционных и дезинсекционных средств, можно,
учитывая необходимые концентрации, решить вопрос о количествах этих
веществ, какие нужно применить в том или ином случае, чтоб добиться необ-
ходимого эффекта.
Наряду с химическими методами в санитарно-гигиеническом анализе
иногда применяют и микрохимические методы, например, при анализе пыли
на присутствие ядовитых веществ и т. п.
БИОХИМИЧЕСКИЕ МЕТОДЫ
Биохимическими методами называются такие, которые применяются для
изучения химической природы и превращения веществ, входящих в состав
организма животных и растений.
В санитарно-гигиеническом анализе биохимические методы находят при-
менение при исследовании биологической полноценности пищевых продуктов
и готовых блюд и в первую очередь при определении в них витаминов. Реже
определяется солевой состав названных объектов и еще реже аминокислот-
ный состав белков.
в
Особое значение имеют исследования пищевых продуктов на содержание
в них тех или иных ферментов. Так, определение редуктазы (редуктазная
проба) и каталазы (каталазная проба) молока используется для оценки ка-
чества молока; определение пероксидазы (пероксидазная проба) — для выяс-
нения, подвергалось ли молоко нагреванию; определение пероксидазы (бен-
зидиновая проба) в мясе дает указание о свежести и доброкачественности
его и т. п.
Из сказанного следует, что биохимические методы, хотя и не охватывают
много вопросов, все же имеют большое значение в санитарно-гигиенических
исследованиях и в первую очередь в исследованиях пищевых продуктов.
Однако значение биохимических методов в санитарно-гигиенических ис-
следованиях должно значительно возрасти в связи с перестройкой, какая
происходит в настоящее время в гигиенической науке на базе учения
И. П. Павлова.
Такие исследования, как, например, определение некоторых профессио-
нальных ядов (Fb, Hg, As) в крови и моче, должны стать повседневным делом
санитарно-гигиенических лабораторий. Этим делом в настоящее время зани-
маются клинические либо судебно-медицинские лаборатории, когда налицо
имеются уже явления отравления в той или иной форме. Санитарно-гигие-
нические же лаборатории должны этими вопросами заниматься повседневно
с профилактической целью. То же примерно можно сказать об определении
витаминов в крови с целью выяснения витаминной насыщенности организма
и т. п.
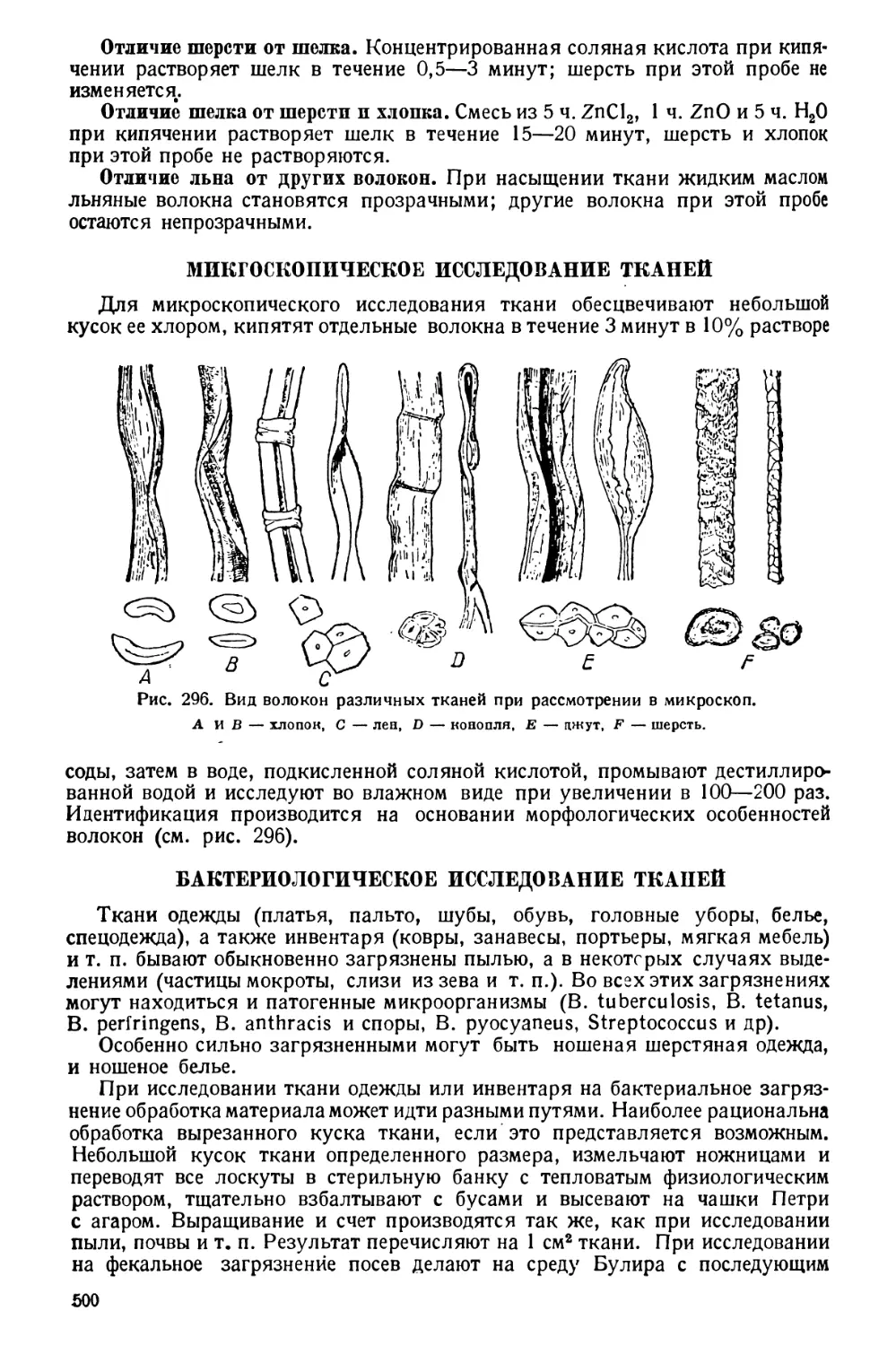
МИКРОСКОПИЧЕСКИЕ МЕТОДЫ
Микроскопическими методами называются такие, при которых изучение
морфологических особенностей (форма, размеры, строение) объектов, а ино-
гда и счет осуществляется при помощи микроскопа как основного прибора.
В качестве вспомогательных приборов служат счетные камеры, сетчатые
окулярные микрометры (для счета), окулярные и объективные микрометры,
винтовые микрометрические окуляры (для определения размеров). Иссле-
дуемые микроскопические объекты иногда зарисовываются без помощи при-
боров или с помощью рисовальных приборов различных конструкций, ино-
гда же фотографируются при помощи микрофотографических камер.
Микроскопические методы широко распространены в санитарно-гигиени-
ческих исследованиях. Они применяются при исследовании аэрозолей, гид-
ропланктона. пищевых продуктов растительного происхождения на наличие
посторонних примесей, тканей одежды с целью идентификации их и т. п.
Разрешающая сила обыкновенных микроскопов равна 0,2 ц, чего вполне
достаточно для обычных санитарно-гигиенических исследований.
В редких случаях, как, например, для изучения ультрамикроскопиче-
ских пылевых частиц (дымов), размеры которых лежат ниже разрешающей
силы обыкновенных микроскопов, приходится применять ультрамикроскопы,
а иногда электронные микроскопы, дающие увеличение в 20 — 30 тыс. раз,
которое фотографическим путем может быть повышено до 100—500 тыс. раз.
БАКТЕРИОЛОГИЧЕСКИЕ МЕТОДЫ
Бактериологическими методами называются такие, при помощи которых
устанавливают наличие бактерий в данном объекте (если они в нем имеются),
определяют число и природу их.
Бактериология в санитарно-гигиеническом анализе имеет столь большой
удельный вес, что диференцировалась в самостоятельную дисциплину —
санитарную бактериологию.
Бактериологические методы исследования приобретают первостепенное
значение при исследовании питьевой воды и пищевых продуктов, менее важ-
9
ное значение они имеют при исследовании воздуха, почвы, сточных вод, пред-
метов обихода, одежды и оборудования на предприятиях пищевой промыш-
ленности.
Особое значение в санитарной практике имеет обследование персонала
пищевых производств на носительство патогенных микробов.
Наиболее распространенным видом санитарно-бактериологического ана-
лиза является определение бактериального обсеменения объекта: воздуха,
воды, почвы, пищевых продуктов. Весьма важное значение имеет определение
коли-титра — показателя фекального загрязнения воды, пищевых продуктов,
почвы.
Наконец, санитарно-бактериологический анализ может быть направлен
на выделение из объекта и идентификацию патогенных и условнопатогенных
бактерий, если на то имеются соответствующие указания.
Основным прибором для бактериологических исследований служит микро-
скоп. Исследование под микроскопом морфологических особенностей бак-
терий, подвижности их и отношения к красителям носит название бакте-
риоскопии.
МИКОЛОГИЧЕСКИЕ МЕТОДЫ
Микологическими методами называются такие, при помощи которых
определяют наличие грибков в данном объекте.
Значение микологических методов исследования в санитарно-гигиениче-
ском анализе ограниченно и распространяется на небольшэй круг объектов:
обнаружение плесневых грибов в некоторых пищевых продуктах (хлеб, мяс-
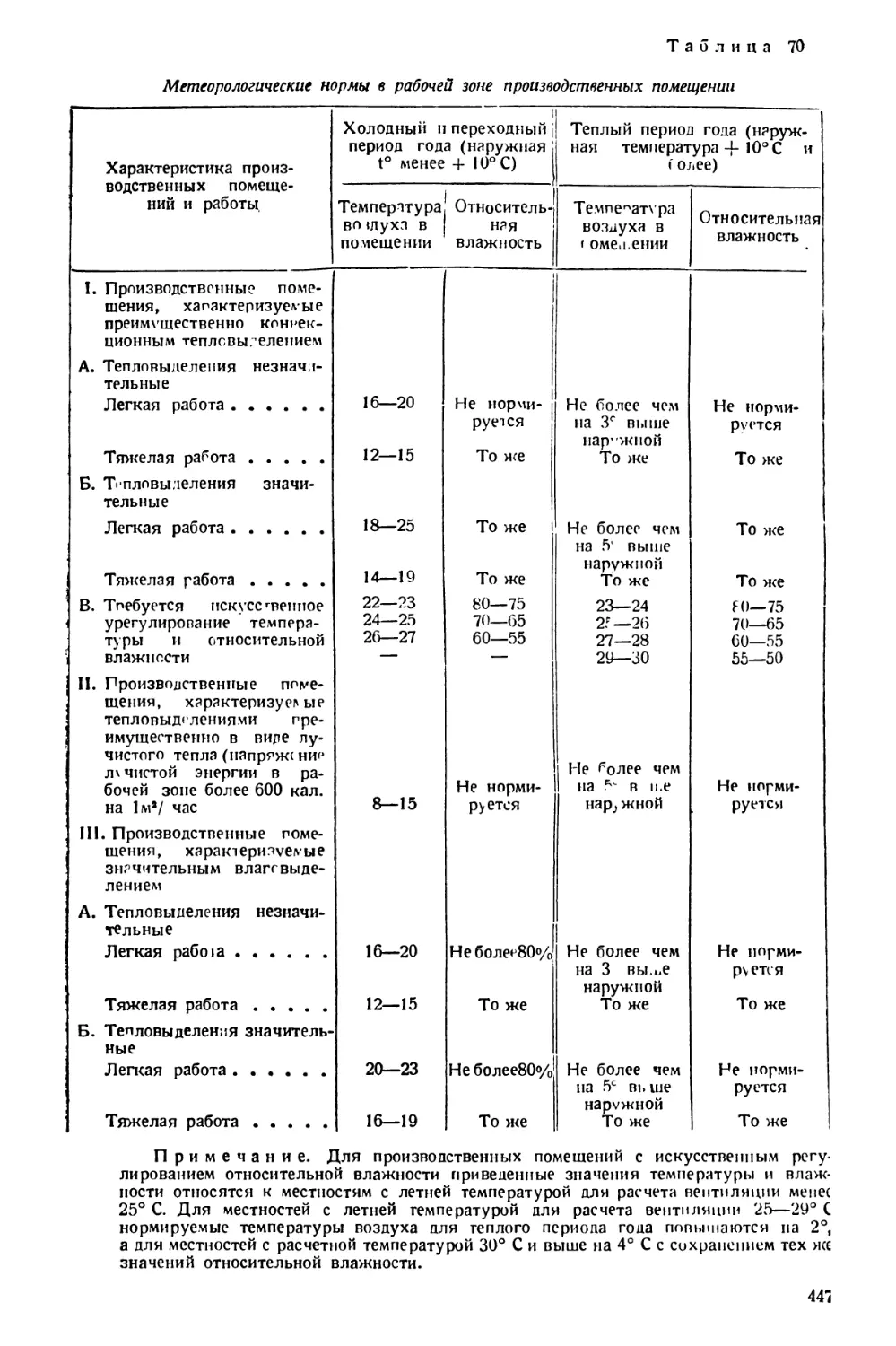
ные изделия) и домовых грибов в деревянных частях зданий.
Плесневые грибки определяются микроскопически на основании морфо-
логических особенностей. Домовые грибы определяются макро- и микро-
скопически на основании морфологических особенностей, а также путем
разведения чистых культур.
ГЕЛЬМИНТОЛОГИЧЕСКИЕ МЕТОДЫ
Гельминтологическими методами называются такие, при помощи которых
определяют наличие гельминтов (их яиц и личинок) в тех или иных объектах.
До недавнего времени гельминтологические исследования занимали в са-
нитарно-гигиеническом анализе очень скромное место и ограничивались
почти исключительно контролем мяса на трихинеллез и финноз.
В настоящее время, благодаря работам советских авторов (Васильковой,
Гнединой, Гефтер и др.), гельминтологические методы приобретают серьезное
значение в санитарно-гигиенических исследованиях воды, почвы, овощей
и пр. и, несомненно, явятся одним из важных звеньев в общей системе оздо-
ровления населения от гельминтов.
Яички и личинки гельминтов концентрируют на мембранных фильтрах
путем фильтрации или в осадках центрифугирования, а затем определяют
микроскопически по морфологическим особенностям.
Подробные вопросы гельминтологических исследований излагаются в спе-
циальных руководствах.
СЕРОЛОГИЧЕСКИЕ МЕТОДЫ
Серологическими методами (реакциями) называются такие, которые слу-
жат для идентификации антигена на основании его отношения к данной им-
мунной сыворотке. Серологические реакции применяются обыкновенно с
целью дополнительной проверки данных, полученных путем бактериологи-
ческих методов исследования.
10
Из серологических реакций в санитарно-гигиенических исследованиях
нашли применение реакции агглютинации, преципитации
и связывания комплемента.
Значение этих реакций как диагностического метода довольно велико.
Оно умаляется однако тем, что различные, но близкие по своей природе
антигены способны вступать в реакцию с одной и той же специфической сы-
вороткой. С другой стороны, возможное наличие в иммунной сыворотке кроме
специфического также и неспецифических антител приводит иногда к полу-
чению так называемых «групповых реакций», ввиду чего точная диагностика
становится невозможной (подробнее см. ниже).
БИОЛОГИЧЕСКИЕ МЕТОДЫ
Биологическими методами называются такие, при которых решение опре-
деленных санитарно-гигиенических вопросов достигается путем пробных
испытаний на животных (биопроб).
Сюда относятся опыты на животных по идентификации антигена в зара-
женном материале, по обнаружению токсических веществ, чаще всего бакте-
риального происхождения (токсины ботулинуса, тетануса и пр.), в исследуе-
мом материале, по определению наличия витаминов в различных продуктах
животного и растительного происхождения, по исследованию токсикологии
промышленных ядов («малая токсикология» по Н. С. Правдину) и т. п.
Испытуемый материал вводят тем или иным путем подопытным животным
и устанавливают за ними систематический надзор. При этом проверяют вес,
температуру, наблюдают поведение, походку, дыхание, реакции на внешние
раздражения, аппетит, характер выделений и т. п. Погибших животных под-
вергают патологоанатомическому и бактериологическому исследованиям. На
основании наблюдений при жизни животного и данных исследований после
смерти его делают соответствующие выводы.
При биологическом определении витаминов в продуктах требуется пред-
варительная подготовка животного: оно в течение определенного времени
выдерживается на диете, лишенной того или иного витамина; когда у жи-
вотного ясно обозначаются явления авитаминоза, к диете прибавляют испы-
туемый продукт, наблюдая воздействие измененной диеты на состоянии жи-
вотного и делая отсюда вывод о содержании соответствующего витамина
в испытуемом продукте.
Биологические методы, как видно из сказанного, имеют серьезное значе-
ние в санитарно-гигиенических исследованиях. Животные в этих исследо-
ваниях играют роль биологических индикаторов.
ФИЗИОЛОГИЧЕСКИЕ МЕТОДЫ
Физиологическими методами называются такие, при помощи которых
исследуется зависимость основных процессов, функций и проявлений орга-
низма от внутренних и внешних условий. В санитарно-гигиенических иссле-
дованиях изучаются реакции и сдвиги в организме под влиянием факторов
внешней среды.
В качестве примеров применения физиологических методов в санитарно-
гигиенических исследованиях могут служить исследования реакций организма
(сердечнососудистой системы, органов дыхания, обмена веществ и пр.) на
температуру, влажность и движение воздуха, лучистую энергию, ультра-
фиолетовую, рентгеновскую, радиоактивную радиации, барометрическое дав-
ление и пр.
Физиологические методы исследования проводятся на людях и животных.
Они имеют весьма важное значение, так как дают возможность установить
зависимость между внешней средой (биосферой) и организмом, которые,
по учению И. П. Павлова, рассматриваются как единое целое. При помощи
и
этих методов удается выяснить оптимальные условия среды для организма
человека; на основании их нормируются некоторые условия внешней среды,
например, метеорологический фактор помещений, освещенность рабочих
мест и т. п.
СТАТИСТИЧЕСКИЙ метод
Статистический метод применяется для изучения массовых явлений и их
сводных признаков. При помощи этого метода можно, группируя качественно
однородные и объективно связанные признаки в совокупности, делать обоб-
щения и устанавливать определяющие закономерности в изучаемых явлениях
и их связях.
Методом статистики пользуются все науки, в том числе и медицина, кото-
рая широко применяет его в научных исследованиях и практике здравоохра-
нения.
При помощи статистического метода в медицине изучаются:
а) санитарное состояние населения — его естественное движение (рож-
даемость, смертность, детская смертность), заболеваемость в ее многообразии,
физическое состояние и развитие отдельных групп населения;
б) лечебная работа клинических учреждений — в части выяснения этио-
логии и патогенеза, проведения диференциальной диагностики и эффектив-
ных лечебных и профилактических мероприятий;
в) санитарно-профилактическая работа органов здравоохранения — в ча-
сти проведения эффективных санитарно-гигиенических мероприятий;
г) состояние медицинских кадров (численность и состав), сети учрежде-
ний здравоохранения и деятельности ее с качественной и количественной
сторон.
Статистический метод широко применяется в научных экспериментальных
исследованиях для обобщений и установления закономерностей.
Для стандартизации и контроля статистической работы в практике здраво-
охранения существуют ведомственные статистические организации в системе
руководящих органов здравоохранения. Общий контроль и инструктаж
статистической работы в Союзе осуществляется Центральным Статистиче-
ским Управлением (ЦСУ).
Важное значение статистики для всех проявлений жизни страны четко
выражено в словах товарища И. В. Сталина: «Никакая строительная работа,
никакая государственная работа, никакая плановая работа немыслима без
правильного учета. А учет немыслим без статистики» (Отчет Центрального
Комитета XIII съезду ВКП(б), Стенографический отчет, стр. 130).
Изложенные выше методы, применяемые в санитарно-гигиенических ис-
следованиях, не всегда точно диференцируют по приведенной схеме: нередко
органолептические и физические методы объединяются под общим наимено-
ванием физических методов, физические и физико-химические — под назва-
нием физико-химических и т. д.
В настоящей книге приведена методика целого ряда органолептических,
физических, физико-химических, химических, микроскопических, бактерио-
логических, микологических, гельминтологических, серологических и биоло-
гических исследований.
Методика биохимических исследований и целого комплекса физиологиче-
ских исследований, применяемых для изучения влияния факторов внешней
среды на организм человека, а также основы статистического метода должны
быть изложены в другой книге, которая явится естественным продолжением
настоящей.
ГЛАВА ВТОРАЯ
ВВЕДЕНИЕ В ЛАБОРАТОРНУЮ ТЕХНИКУ
В настоящей главе приводятся основные сведения по оборудованию лабо-
раторий и лабораторной технике.
ПОМЕЩЕНИЕ ЛАБОРАТОРИИ
В зависимости от объема работы помещение лаборатории должно иметь
большие или меньшие размеры. Часть комнат отводится для химических
и физико-химических работ, другая — для микробиологических и гельмин-
тологических работ, третья — для биологических и физиологических иссле-
дований. Отдельные комнаты желательно иметь для взвешивания на анали-
тических весах (весовая), для термостатов (термостатная), для газоанали-
тических работ, для калориметрических определений и для заведующего
лабораторией.
В лаборатории необходимо выделить подготовительную комнату (препа-
раторскую), где готовятся реактивы, краски, питательные среды и т. д.; она
же обыкновенно служит и моечной. Для хранения всякого вида материалов
выделяют материальную комнату (кладовая); для работ с сероводородом
отводится отдельная комната (сероводородная), по возможности отдаленная
от центральной части лаборатории. Лаборатория должна иметь в своем
хозяйстве виварий.
Лабораторные комнаты должны быть сухими, светлыми и легко вентили-
руемыми. Нужно, чтобы все окна были снабжены большими, легко откры-
ваемыми форточками (ГОСТ-1507). Освещенность рабочего места должна быть
не ниже 100 люксов, коэфициент дневного света не ниже 1 : 5.
Необходимо защитить лабораторное помещение и в первую очередь весо-
вую, газоаналитическую и калориметрическую комнаты от прямого солнечного
света, который часто портит реактивы и аппаратуру и является помехой в ра-
боте; окна этих комнат должны быть обращены на север или в крайнем слу-
чае на восток.
Согласно постановлению НКТруда СССР от 3/VI 1929 г. на одно рабочее
место химика должно приходиться 10 м2 площади и на одно рабочее место
микробиолога 20 м2 площади при высоте помещения не ниже 4 м.
Стены лаборатории на высоте 1,8—2 м от пола покрываются масляной
краской светлого оттенка. Пол должен быть гладким паркетным либо хорошо
выкрашенным, либо покрытым линолеумом. Ввиду высокой ядовитости паров
ртути пол газоаналитической комнаты должен быть совершенно гладким,
без щелей и пазов, чтобы возможно было без остатка собрать случайно про-
литую ртуть. Материалом для такого пола может служить цемент либо сплош-
ной гладкий линолеум на деревянной основе.
Лабораторное помещение нужно обеспечить водопроводом с достаточным
количеством кранов и раковин, канализацией. Весьма важно иметь подводку
13
светильного или карбюраторного1 газа. Помещение лаборатории должно быть
электрифицировано.
Следует избегать для лаборатории таких помещений, где воздух может
загрязняться от соседства дымовых труб, выхлопных газов и т. п., а также
таких, где по тем или иным причинам наблюдается дрожание почвы.
Лабораторное помещение должно содержаться в совершенной чистоте.
ОБОРУДОВАНИЕ И ПРИБОРЫ
Важнейшее лабораторное оборудование состоит из следующих предметов.
Стол лабораторный
Лабораторный стол (рис. 1) должен иметь гладкий верх из тесно сбитых
сухих досок. Кафельные плитки менее пригодны, так как на таких столах
легко бьется лабораторная посуда. Крышку стола покрывают линолеумом,
который время от времени следует натирать смесью из 10 ч. воска, 20 ч. це-
резина, 9 ч. скипидара и 80 ч. бензина.
Рис. 1. Лабораторный стол.
Если стол не покрыт линолеумом, то для повышения стойкости дерева
в отношении разных химических реагентов свеже выстроганную крышку
стола обрабатывают следующим образом: раствор из 8 ч. хлористого аммония,
20 ч. хлористого анилина и 125 ч. воды наносят в горячем виде волосяной
щеткой на дерево; когда дерево подсохнет, наносят таким же образом дру-
гой раствор, состоящий из 10 ч. бертолетовой соли, 20 ч. сернокислой меди
и 125 ч. воды. Как только просохнет, вновь наносят первый раствор, а затем
второй и так 4 раза. Под конец дерево приобретает темнозеленый цвет. Тогда
его шлифуют стеклянной бумагой и покрывают несколько раз вареным мас-
лом, после чего окраска становится черной. Она отличается хорошей
кислотостойкостью и огнестойкостью (Созонов и Верховский).
Поверх стола во всю длину устанавливаются две деревянные полки: одна
на высоте 30 см от крышки стола, другая на высоте 50 см от нее. Толщина
полок 2—2,5 см, ширина 24—25 см. Через каждые 75 см погонной длины
ставят подпорку для придания полкам большей прочности.
1 Карбюраторный газ получается при помощи установки — карбюратора, дающего токи
воздуха, насыщенного парами углеводородов, чаще всего бензина.
14
Лабораторные столы оборудуются водопроводом, канализацией, подвод-
кой газа и электрического тока.
На одно рабочее место должно приходиться не менее 1,8 м погонной длины
стола при ширине его в 0,8—1 м и высоте в 0,9 м. Столы располагаются пер-
пендикулярно стене с окнами; расстояние между столами устанавливается
в 1,5—2 м. Свет от окон должен падать на рабочее место слева от работаю-
щего.
Для сидения у столов пользуются табуретами высотой в 60 см с ква-
дратным сидением 40 х 40 см.
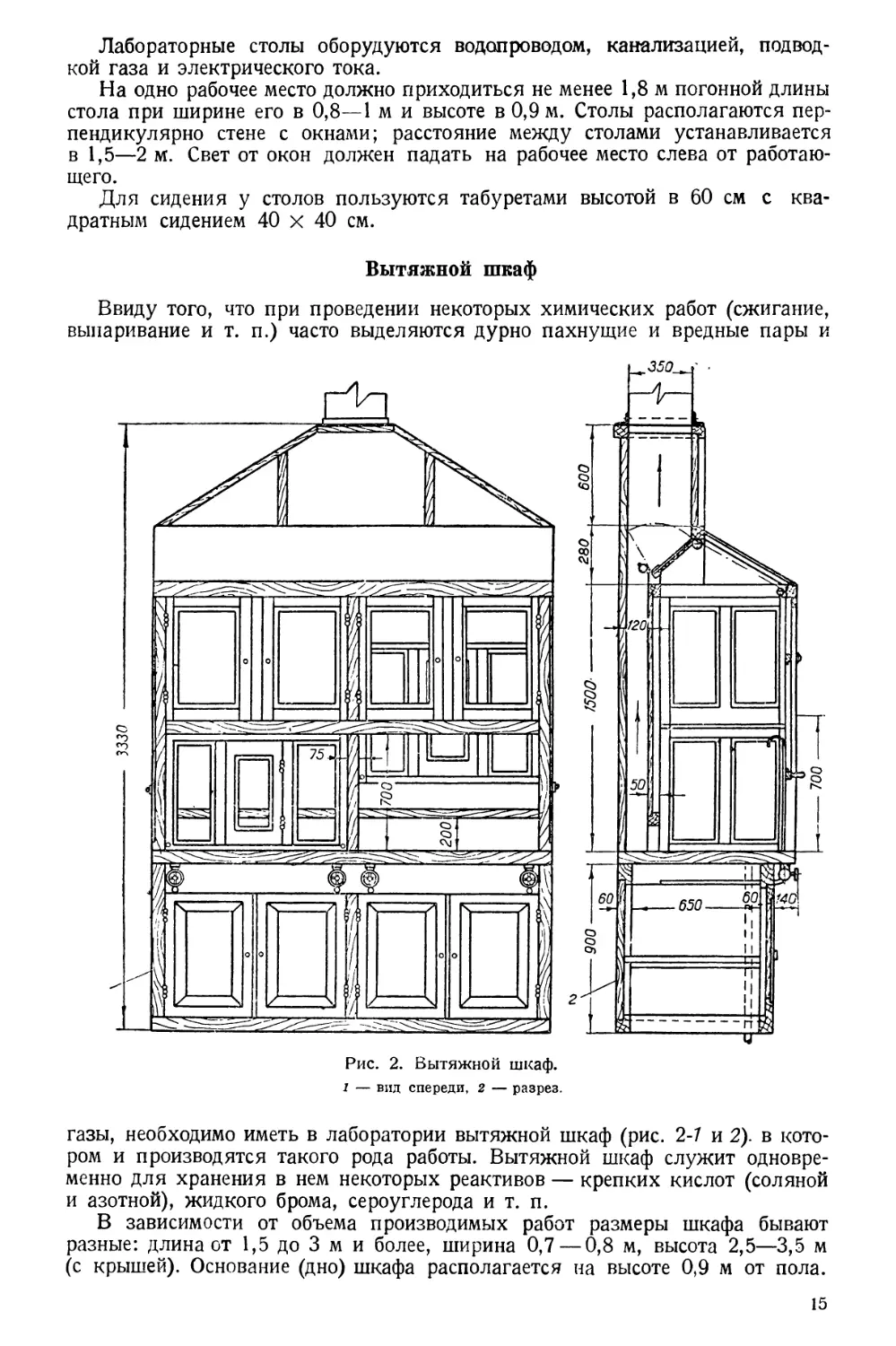
Вытяжной шкаф
Ввиду того, что при проведении некоторых химических работ (сжигание,
выпаривание и т. п.) часто выделяются дурно пахнущие и вредные пары и
газы, необходимо иметь в лаборатории вытяжной шкаф (рис. 2-7 и 2). в кото-
ром и производятся такого рода работы. Вытяжной шкаф служит одновре-
менно для хранения в нем некоторых реактивов — крепких кислот (соляной
и азотной), жидкого брома, сероуглерода и т. п.
В зависимости от объема производимых работ размеры шкафа бывают
разные: длина от 1,5 до 3 м и более, ширина 0,7 — 0,8 м, высота 2,5—3,5 м
(с крышей). Основание (дно) шкафа располагается на высоте 0,9 м от пола.
15
Внутренние стенки шкафа покрываются масляной краской либо кера-
миковыми плитками. Дно шкафа покрывается цементом или также керами-
ковыми плитками, или делается из дерева, которое обрабатывают, как и верх
лабораторного стола (см. выше). Стенки шкафа, как и крыша, должны быть
застеклены. В передней и боковых стенках шкафа устраивают подъемные
дверцы. Последние соединяют с помощью круглых ремней, перекинутых
через блоки, с противовесами. Это дает возможность остановить дверцу на
любой высоте.
Крыша шкафа делается наклонной под углом в 45° к вертикальной
стене.
Шкаф устанавливают близко от наружной стены, чтобы легче было обес-
печить вывод наружу газов, либо у стены, через которую проходит дымо-
ход; в последнем случае вентиляционная труба открывается в дымоход. Вы-
тяжка из шкафа осуществляется при помощи вентилятора большей или мень-
шей мощности, работающего от мотора, который располагается вне шкафа.
Отсос воздуха — комбинированный: снизу отсасывается 2 3, а сверху —
Чз воздуха.
Основной отсос воздуха снизу с верхней кромкой отверстия на 200—400 мм
от пола шкафа обеспечивает быстрое удаление газов в момент их образования;
дополнительный отсос сверху через воздуховод обеспечивает удаление легких
газов, неполностью захваченных нижним отсосом.
В больших вытяжных шкафах устанавливают внутри горизонтально
проходящую вентиляционную трубу; от нее отходит ряд патрубков,
входные отверстия которых открываются непосредственно над рабочими
местами.
Вентиляционные трубы готовятся из листового железа и покрываются
изнутри кислотоупорным лаком, предохраняющим от коррозии.
В правильно сконструированном шкафу скорость всасывания должна быть
не менее 0,3 м/сек., а в шкафах, предназначенных для работы с сильно ядови-
тыми газами, не менее 0,7—1,0 м/сек. При работе под дверкой следует оставлять
щель 50—100 мм для засоса воздуха. При плотном закрытии дверок цир-
куляция воздуха в шкафу недостаточна (Т. А. Фиалоковская). 1
В шкаф со стороны задней
стенки подводят водопровод-
ную, канализационную и га-
зопроводную трубы. Внутри
шкаф электрифицируется, при-
чем электрические провода,
проходящие внутри шкафа,
для предохранения «от разъ-
едания и возможного корот-
кого замыкания заключаются
в бермановские трубки. Под
собственно вытяжным шкафом
располагают часто обыкно-
венный шкаф.
Рис. 3. Технохимические весы.
технохимические весы
Технохимические весы
(рис. 3) пригодны для такого
взвешивания, которое не тре-
бует большой точности. Их
предельная чувствительность достигает обыкновенно 0,01 г. Как и аналити-
ческие весы, они имеют арретир.
1 Вытяжные зонты и шкафы, Стройиздат, 1947.
16
Аналитические весы
Аналитические весы (рис. 4) дают
до 0,1 мг. Коромысло аналитиче-
ских весов снабжено посредине
агатовой призмой, которая во
время взвешивания своим острым
ребром устанавливается на ага-
товой же пластинке, расположен-
ной наверху колонки весов. По
краям коромысла имеется еще по
одной агатовой призме. Обе дужки,
на которых подвешены чашки ве-
сов, снабжены агатовыми пластин-
ками, которые во время взвеши-
вания упираются в ребра агато-
вых призм, находящихся по краям
коромысла весов. Аналитические
весы всегда снабжаются арретиром
с целью предохранения ребер призм
от скорого изнашивания. При по-
вороте арретирного винта или ру-
коятки арретира вправо послед-
ний подымается и, подхватив ко-
ромысло весов, приподнимает его;
при этом и обе дужки, на кото-
рых подвешены чашки, также
приподнимаются; таким образом, в
арретированных весах ни одна
призма не касается своих опорных
пластинок. При повороте арретир-
ного винта налево арретир опус-
кается, сначала устанавливаются
на место дужки, а затем и коро-
мысло.
возможность определить вес с точностью
Рис. 4. Аналитические весы.
1 — коромысло, 2 — линейка для рейтера, з — сред-
няя призма, 4 — груз для смещения центра тяжести,
5 — груз для смещения нулевой точки, 6 — стрелка,
7 — шкала, 8 — стремечки, 9 — чашки, ю — руко-
ятка арретира, 11, 12 и 13— рычаги арретира, 14 —
чашечный тормоз, 15 — отвес, 16 — приспособление
для перемещения рейтера, 17 — крючок для захва-
тывания рейтера, 18 — ножки весов, 19 — подкладки
с выемками.
На верхнем ребре коромысла имеются зарубки для рейтера (см. ниже).
Каждые аналитические весы характеризуются предельной на-
грузкой, под которой понимают максимальный по весу груз, какой можно
О О о о О □ <» <з
500 200 Ю0 100 50 20 10 10
Рис. 5. Аналитический разновес.
взвешивать на данных весах без риска испортить
их. Обыкновенно аналитические весы рассчитаны
на максимальную нагрузку в 100 г. Реже встре-
чаются весы с максимальной нагрузкой в 50 и
200 г.
Большая или меньшая точность взвешивания
на аналитических весах зависит от чувстви-
тельности весов, т. е. от того, насколько
они легко отзываются на изменение веса
чашек.
О чувствительности весов судят по величине
угла отклонения стрелки, которое вызывается
прибавлением добавочного груза в 1 мг на какую-
либо из чашек весов, находящихся в равно-
весии.
Аналитические весы устанавливаются на дере-
вянной или мраморной подставке шириной в 0,7 м
и длиной в 1,5 м, приделанной к капитальной
стене на кронштейназ^ра высоте 0,8 м от пола.
2 А. II. Бурштейн.
17
Аналитический разновес и рейтер
Аналитические разновесы (рис. 5), представляющие собой комплекты
гирек в футлярах, тщательно проверяются перед выпуском. Каждая отдель-
ная гирька (разновеска) не должна отклоняться от принятого для нее веса
более чем на 0,00005 г. Гирьки весом в 1 г и более сделаны из латуни и позо-
лочены или платинированы. Меньшие гирьки готовятся из нейзильбера или
алюминия, реже из платины. Гирьки разновеса сохраняются в ящике, где
кроме них должны быть стеклянная линейка для прикрывания мелких гирек
Рис. 6. Рейтер.
а — форма рейтера, Ъ — положение на коромысле
(показывает 6,7 мг).
и пинцет, которым только и
разрешается брать гирьки и
рейтер.
При помощи гирек разно-
веса отвешивают граммы, а
также ’десятые и сотые доли
их.
Миллиграммы и десятые
доли их отвешивают при по-
мощи рейтера (рис. б).
Рейтер изготовляют из тонкой
золотой или платиновой проволоки; вес его обыкновенно равен 10 мг. Деле-
ния на коромысле показывают соответствующее изменение в весе при стоянии
рейтера на данном делении. Когда рейтер стоит на крайнем десятом делении,
то он показывает такое же изменение в весе, как если бы он находился на
чашке весов, т. е. изменение, соответствующее его собственному весу—10 мг.
При длительной работе гирьки могут несколько менять свой вес, ввиду
чего необходимо время от времени проверять их по точному разновесу.
В паспорте против веса каждой гирьки указывают соответствующую поправку.
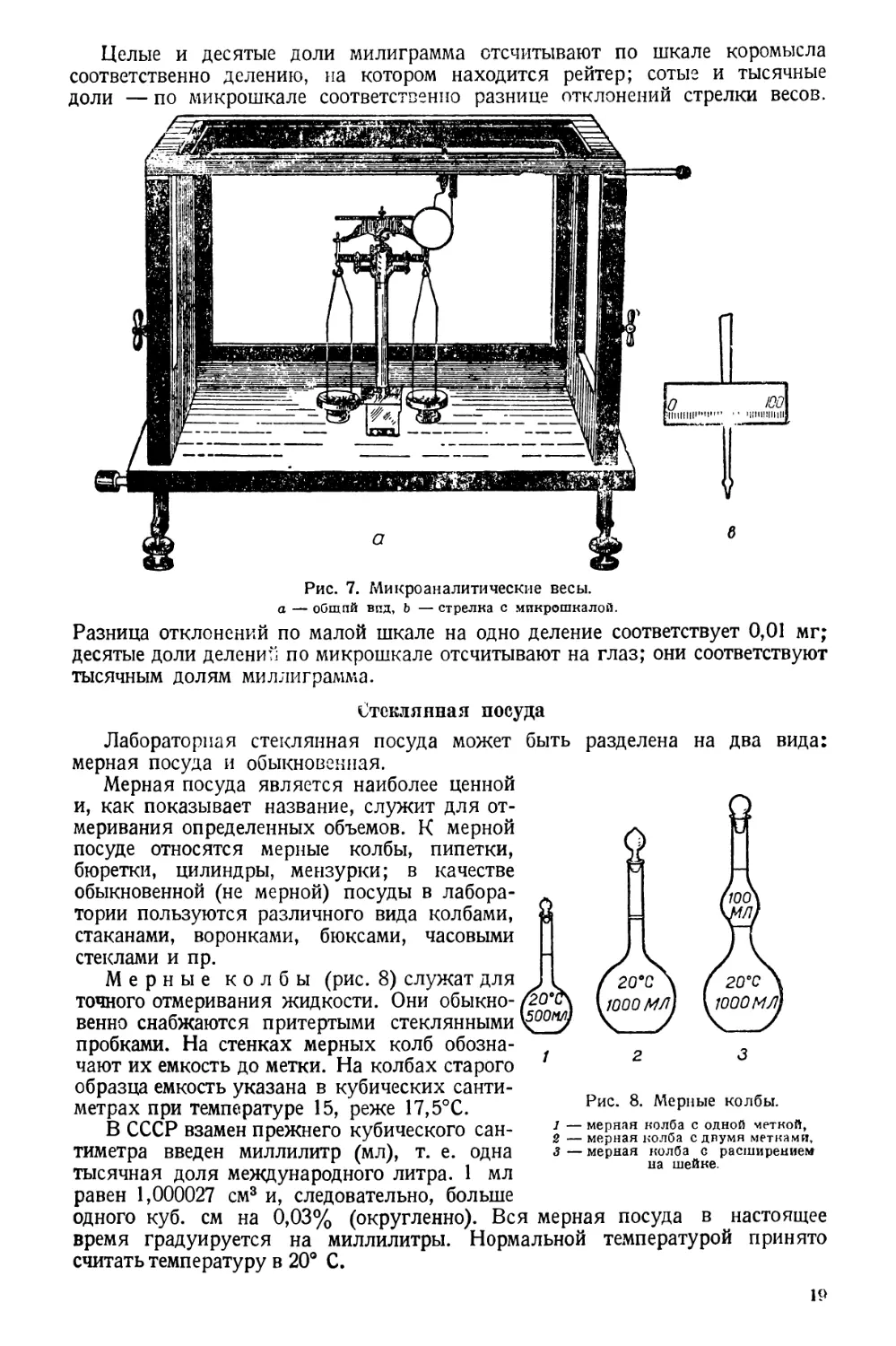
Микроаналитические весы
Для взвешивания с большой точностью малых количеств вещества при-
меняют микроаналитические весы (рис. 7).
Максимальная нагрузка их 20 г; чувствительность 0,001 мг или 1 рг1.
Минимальный груз для взвешивания 3—4 мг.
Изображенные здесь микроаналитические весы имеют в основном ту же
конструкцию, что и обыкновенные аналитические весы. Они отличаются
лишь меньшими размерами и некоторыми особенностями.
На верхнем ребре коромысла имеется 101 зарубка.
В микроаналитических весах, выпускаемых заводом «Техновес», крайняя
левая зарубка обозначена нулем, крайняя правая — числом 100. Средняя—
числом 50. Каждая зарубка соответствует 0,1 мг.
Вес рейтера 5 мг; не допускается применение деформированных рейтеров.
Во время взвешивания рейтер должен быть на коромысле весов. Ненагружен-
ные весы уравновешиваются при стоянии рейтера на первом делении слева;
при стоянии рейтера на последнем делении справа нагрузка на правой чашке,
обусловленная рейтером, равна 10 мг.
В весах, изготовляемых заводом «Метрон», средняя зарубка обозначена
нулем, а крайние левая и правая — числом 50.
Для ясности отсчетов (по шкале коромысла) к ручке, передвигающей
рейтер, прикреплена лупа.
К нижней части стрелки весов прикреплена микрошкала, градуированная
часть которой разделена на 100 частей. Шкалу наблюдают через зрительную
трубу (или микроскоп), проходящую через переднее стекло шкафа. Внутри
трубы натянута тонкая шелковая нить, которая при арретированных весах
должна совпадать со средними делениями микрошкалы.
1 Тысячная доля миллиграмма называется микрограммом и обозначается через
|лг или •/.
18
Целые и десятые доли милиграмма отсчитывают по шкале коромысла
соответственно делению, на котором находится рейтер; сотые и тысячные
разделена на два вида:
1 2 3
Рис. 7. Микроаналитические весы.
а — общий вид, Ь — стрелка с микрошкалой.
Разница отклонений по малой шкале на одно деление соответствует 0,01 мг;
десятые доли делений по микрошкале отсчитывают на глаз; они соответствуют
тысячным долям миллиграмма.
Стеклянная посуда
Лабораторная стеклянная посуда может
мерная посуда и обыкновенная.
Мерная посуда является наиболее ценной
и, как показывает название, служит для от-
меривания определенных объемов. К мерной
посуде относятся мерные колбы, пипетки,
бюретки, цилиндры, мензурки; в качестве
обыкновенной (не мерной) посуды в лабора-
тории пользуются различного вида колбами,
стаканами, воронками, бюксами, часовыми
стеклами и пр.
Мерные колбы (рис. 8) служат для
точного отмеривания жидкости. Они обыкно-
венно снабжаются притертыми стеклянными
пробками. На стенках мерных колб обозна-
чают их емкость до метки. На колбах старого
образца емкость указана в кубических санти-
метрах при температуре 15, реже 17,5°С.
В СССР взамен прежнего кубического сан-
тиметра введен миллилитр (мл), т. е. одна
тысячная доля международного литра. 1 мл
равен 1,000027 см3 и, следовательно, больше
одного куб. см на 0,03% (округленно). Вся мерная посуда в настоящее
время градуируется на миллилитры. Нормальной температурой принято
считать температуру в 20° С.
Рис. 8. Мерные колбы.
1 — мерная колба с одной меткой,
2 — мерная колба с двумя метками,
3 — мерная колба с расширением
на шейке.
19
Желая отмерить определенный объем жидкости мерной колбой, жидкость
наливают в колбу так, чтобы нижний край мениска касался метки: при этом
глаз наблюдателя должен быть в одной горизонтальной плоскости с чертою,
чтобы избегнуть ошибки на параллакс.
Мерные колбы с одной меткой на шейке рассчитаны на вливание, с двумя—
на вливание и выливание. Часть жидкости при выливании остается на стенках
посуды и равна по объему тому количеству жидкости, которое заключается
между двумя метками.
Объемы мерных колб необходимо проверить. Для этой цели определяют
вес тщательно вымытой и высушенной колбы; затем, наполнив ее дестиллиро-
ванной водой до метки, вновь взвешивают. По разности определяют вес воды
в колбе. Найденный практически вес сравнивают с истинным весом воды
соответствующего объема при данной температуре (табл. Л) и находят поправ-
ку для данной колбы.
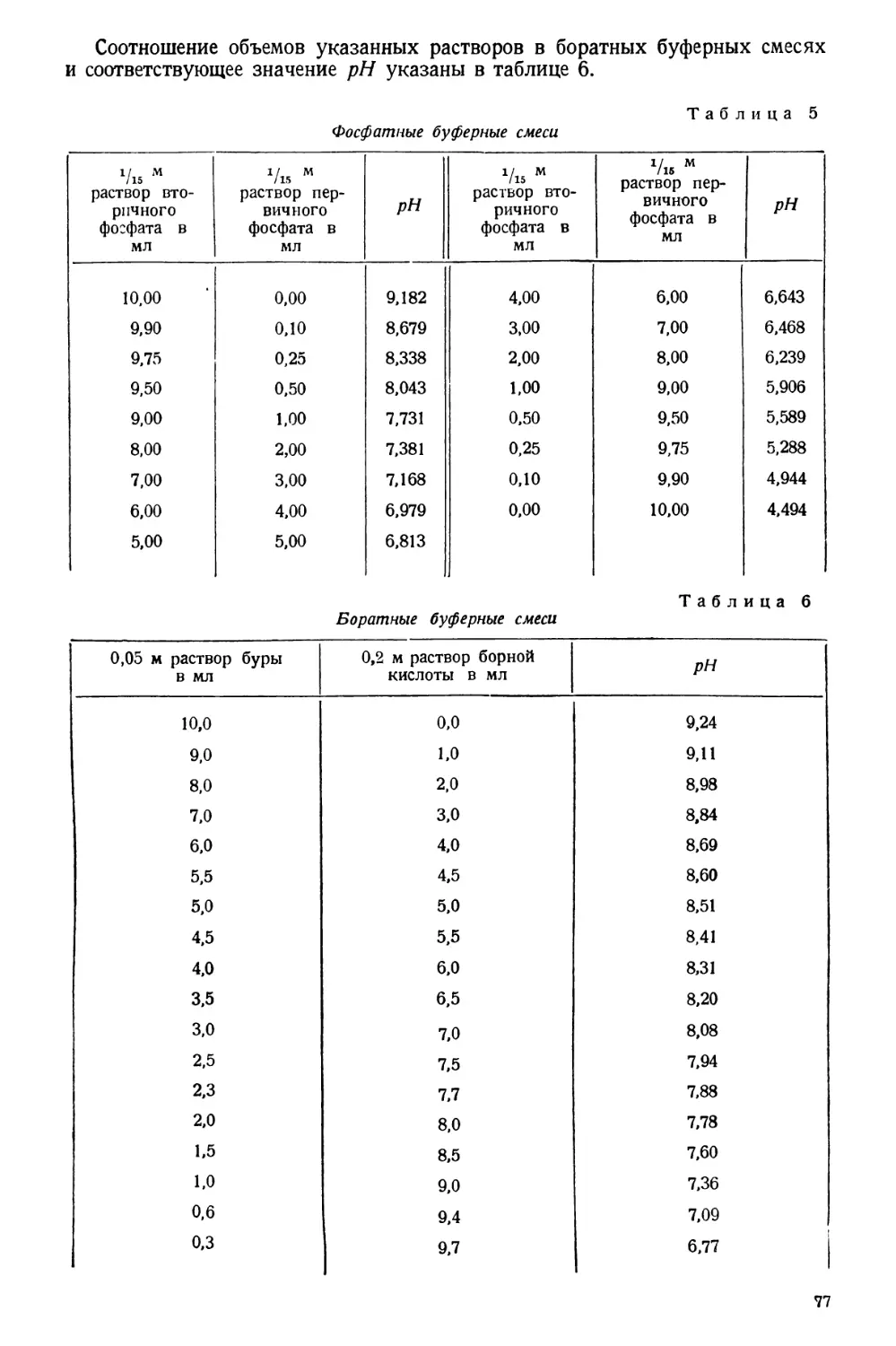
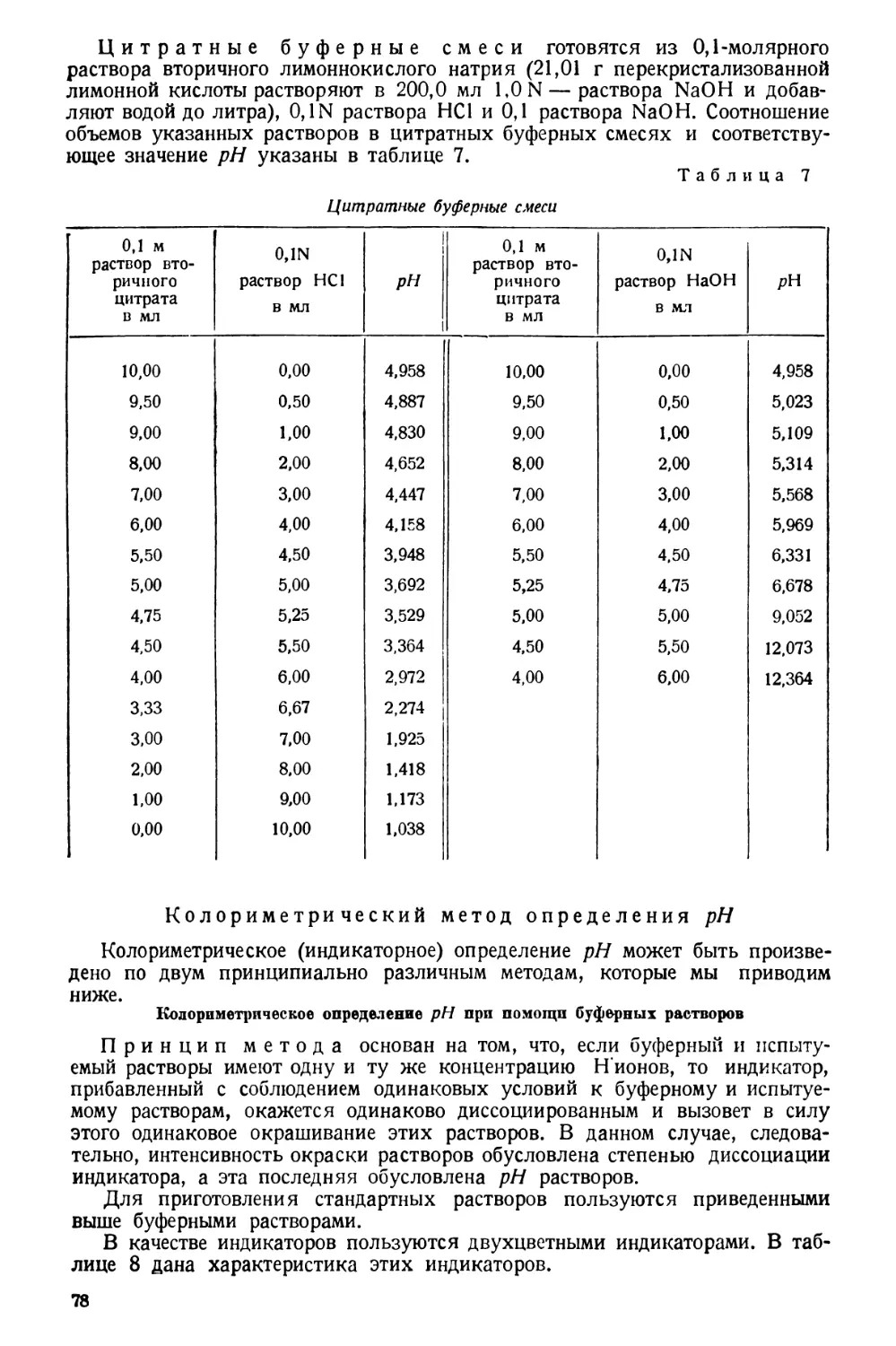
Таблица 1
Вес 1 л дестиллированной воды в граммах (приведенный к нормальной температуре 20°С),
взвешенный в воздухе латунными и бронзовыми разновесками при температуре 10—40°С
(плотность разновесок 8,4; коэфициент объемного расширения стекла 0,000026; плотность
воздуха 0,0012)
t° 0 1 2 3 4 5 6 7 8 9
10 998,41 998,41 998,40 998,40 998,39 998,38 998,37 998,36 998,36 998,35
и 998,34 998,34 998,33 998,32 998,31 998,30 998,30 998,29 998,28 998,27
12 998,26 998,25 998,24 998,23 998,22 998,22 998,21 998,20 998,19 998,18
и 998,17 998,16 998,14 998,14 998,12 998,11 998,10 998,09 998,08 998,07
14 998,06 998,05 998,04 998,02 998,01 998,00 997,99 997,98 997,96 997,95
15 997,94 997,93 997,92 997,90 997,89 997,88 997,88 997,85 997,84 997,82
16 997,81 997,80 997,78 997,77 997,76 997,74 997,73 997,71 997,70 997,68
17 997,67 997,66 997,64 997,62 997,61 997,59 997,58 997,56 997,55 997,53
18 997,52 997,50 997,48 997,47 997,45 997,44 997,42 997,40 997,39 997,37
19 997,35 997,34 997,32 997,30 997,28 997,27 997,25 997,23 997,22 997,20
20 997,18 997,16 997,14 997,12 997,10 997,09 997,07 997,05 997,03 997,01
21 996,99 996,98 996,96 996,94 996,92 996,90 996,88 996,86 996,84 996,82
22 996,80 996,78 996,76 996,74 996,72 996,70 996,68 996,66 996,64 996,61
23 996,59 996,57 996,55 996,53 996,51 996,49 996,47 996,44 996,42 996,40
24 996,38 996,36 996,33 996,31 996,29 996,27 996,24 996,22 996,20 996,18
25 996,15 996,13 996,11 996,08 996,06 996,04 996,01 995,99 995,97 995,94
26 995,92 995,89 995,87 995,84 995,82 995,80 995,77 995,75 995,72 995,70
27 995,67 995,65 995,62 995,60 995,57 995,55 995,52 995,50 995,47 995,44
28 995,42 995,39 995,37 995,34 995,31 995,29 995,26 995,24 995,21 995,18
29 995,16 995,13 995,10 995,08 995,05 995,02 994,99 994,97 994,94 994,91
30 994,88 994,86 994,83 994,80 994,77 994,74 994,72 994,69 994,66 994,63
31 994,60 994,58 994,55 994,52 994,49 994,46 994,43 994,40 994,37 994,34
32 994,31 994,28 994,26 994,23 994,20 994,17 994,14 994,11 994,08 994,05
33 994,02 993,99 993,96 993,93 993,90 993,86 993,83 993,80 993,77 993,74
34 993,71 993,68 993,65 993,62 993,59 993,56 993,52 993,49 993,46 993,43
35 993,40 993,37 993,33 993,30 993,27 993,24 993,21 993,17 993,14 993,11
36 993,08 993,94 993,01 992,98 992,94 992,91 992,88 992,85 992,81 992,78
37 992,75 992,72 992,68 992,65 992,61 992,58 992,55 992,51 992,48 992,44
38 992,41 992,38 992,34 992,31 992,27 992,24 992,21 992,17 992,14 992,10
39 992,07 992,03 992,00 991,96 991,93 991,89 991,86 991,82 991,79 991,75
40 991,72 — — — — — — — — —
Пример. Положим, что практически найденный вес воды проверяемой полулитровой
колбы оказался равным 499,14 г. Температура воды оказалась равной 21,4°С. При этой
температуре вес полулитра дестиллированой воды, согласно таблице 1, равен 996,92 : 2=
= 498,46 г. Поправка для данной колбы окажется равной 499,14 — 498,46 = 0,68 г.
Объем нашей колбы равен 500,0 + 0,68 = 500,68 мл.
20
I«11 [ । tun uiiiiiiiiiiiiniiii: жни itiiiiiniiiiinnii|ig I
Рис. 9. Бюретки.
1 — бюретка с носиком, 2 — бюретка
со стеклянным краном.
Бюретки (рис. 9) применяются для точного отмеривания определен-
ных объемов жидкостей. Чаще всего применяют бюретки на 25 и 50 мл с деле-
ниями на десятые доли миллилитра.
В лабораториях применяют бюретки, ко-
торые либо снабжены у основания стеклян-
ными кранами, либо заканчиваются суже-
нием, которое при помощи резиновой трубки
соединяется со стеклянным наконечником —
носиком. Во втором случае на резиновую
трубку между узким концом бюретки и сте-
клянным наконечником надевают винтовой
или обыкновенный металлический зажим
(рис. 10). Вместо крана или зажима можно
применять затвор, представляющий собой
стеклянный шарик, закрывающий каучуковую
трубку, надетую на бюретку (рис. 11). Ди-
аметр шарика должен быть несколько боль-
ше диаметра трубки. Вместо шарика пользу-
ются иногда стеклянной палочкой длиною в
5—б мм, оплавленной с обоих концов. При
надавливании пальцами на трубку и одновре-
менном оттягивании ее вбок около шарика
или палочки образуется узкий канал, через
который и вытекает жидкость. Нажимая на
каучук, нужно остерегаться выдавливать
жидкость ниже шарика (палочки), в против-
ном случае в носик проскочит пузырек воз-
духа, а жидкость, выжатая ниже шарика, не
войдет в отсчет бюретки.
Бюретки со стеклянными кранами применяют для таких жидкостей, ко-
торые действуют разъедающе на резину (раствор иода, перманганат, кислоты);
бюретки второго типа применяют для жидкостей, которые действуют разъе-
дающе на стекло и могут испортить кран (щелочи). Бюретки рассчитаны
для жидкостей при температуре 20°С.
Бюретки устанавливаются строго вертикально в шта-
тивах, снабженных лапками — держателями. Если лапки
штатива металлические, то на их концы нужно надеть ре-
зиновые трубки или сделать пробочные прокладки, чтобы
при зажимании не раздавить бюретки. Для защиты от
пыли бюретки закрывают сверху колпач- щ
ками.
Перед тем как заполнить бюретку дан-
ным раствором, ее тщательно промывают,
затем трижды ополаскивают изнутри дан-
ным раствором, чтобы не изменился его
титр при заполнении бюретки. Нужно сле-
дить за тем, чтобы после заполнения бю-
ретки раствором в нижней части крана или
наконечника не оставались пузырьки воз-
духа, что может привести к неправильному
отсчету. Чтобы удалить оставшийся воз-
дух, выпускают из бюретки быстрой струей
жидкость, а в бюретке со стеклянным наконечником при-
поднимают еще при этом наконечник кверху. Если указанные
мероприятия не приводят к цели, то на носик бюретки или стеклянного крана
надевают резиновую трубку, соединенную с воронкой, через которую вводят
жидкость.
Рис. 10. Металли-
ческие зажимы для
бюреток.
J — винтовой зажим,
2 — обыкновенный
зажим.
4
в
а
и
11. Стек-
Рис.
лянный затвор
для бюреток.
а — бюретка за-
крыта, Ь — резино-
вая трубка справа
оттянута, бюрет-
ка пропускает
жидкость.
21
Отсчет ведется по стоянию нижнего края мениска жидкости при условии,
что глаз
Рис. 12.
Бюретка с
линейкой
синего
наблюдателя находится на одном уровне с указанным краем мениска.
В непрозрачных растворах (KMnO4, J2 и т. п.) стояние мениска
лучше определять по верхнему краю его.
В лабораториях применяют иногда бюретки, задняя стенка
которых сделана из молочного стекла. По длине бюретки в заднюю
стенку вплавлена тонкая линейка из синего стекла. Мениск в
такой бюретке при правильном положении глаза имеет характер-
ный вид двух остриев, сходящихся в одной точке (рис. 12). Отсчет
ведется по перемещению этой точки.
Для проверки бюретки ее заполняют несколько раз дестилли-
рованной водой, выводя во взвешенные бюксы воду в интервалах
#0—5 мл, 0—10 мл, 0—15 мл, 0—20 мл и тЛд. до 0—50 мл. Каж-
дую порцию воды взвешивают на аналитических весах и делят вес
воды каждого интервала на вес 1 мл воды при данной температуре
(см табл. 1), определяя таким образом истинный объем интер-
вала бюретки. Найденными поправками пользуются в работе.
Согласно ГОСТ-6176 отклонения от номинальной вместимости бю-
реток объемом в 25, 50 и 100 мл не должны превышать соответ-
ственно + 0,05, + 0,06 и + 0,08 мл.
стекла. Микробюретки служат для точного титрования малых
объемов жидкости. Они рассчитаны на 1 или 2 мл, с делениями на
0,01 мл. На рис. 13 изображена микробюретка, представляющая один из
разнообразных видов микробюреток. Микробюретка снабжена двумя притер-
ными кранами и резервуаром для титрованного раствора. Один кран служит
Рис. 13.
Микробюретка
для перевода раствора из резер-
вуара в бюретку, другим кра-
ном пользуются для титрования.
Пипетки (рис. 14) упот-
ребляются для отмеривания оп-
ределенных объемов жидкости.
Применяют пипетки двух родов,
пипетки с расширением, рас-
считанные для отмеривания од-
ного определенного объема (от 1
до 100 мл), и пипетки цилинд-
рические, предназначенные для
отмеривания разных объемов
жидкости. Цилиндрические пи-
петки дают возможность отмери-
вать жидкость с точностью до
0,1 мл, а некоторые из них
(од по-двухмилли литровые) — с
точностью до 0,01 мл. Объемы
пипеток указаны на их стенках
и рассчитаны для температу-
ры 20°С. Проверка пипеток про-
изводится так же, как и про-
верка бюреток.
Пользуясь пипеткой, широ-
кий конец ее берут в рот и на-
Рис. 14. Пипетки.
1 — цилиндрическая пипетка,
2 — пипетка с расширением.
3 — пипетка с расширением и с
двумя шарообразными вздутия-
ми на шейке, / — штатив с
пипетками.
сасывают жидкость несколько выше метки.
Держа пипетку в руке так, чтобы указательный
палец был свободен, выпускают пипетку изо рта и быстрым движением указа-
тельного пальца закрывают широкое отверстие пипетки. Затем, держа пипетку
по возможности вертикально (рис. 15), так чтобы метка была на уровне глаз,
начинают постепенно выпускать жидкость. Это достигается посредством
22
легких движений пальца, закрывающего отверстие пипетки. Выпускание
жидкости продолжается до тех пор, пока нижний край мениска не подой-
дет до метки. В этот момент жидкость считается отмеренной, и ее переносят
в соответствующую посуду; при этом нужно прико-
снуться концом пипетки к стенке сосуда и тогда
лишь снять палец с широкого отверстия пипетки.
Жидкость по стенке стекает в сосуд. После того, как
вся жидкость из пипетки вытечет, ждут еще 15 се-
кунд, пока остатки жидкости со стенок пипетки не
стекут вниз; при этом обыкновенно из пипетки в сосуд
вытекает еще некоторое количество жидкости. Остав-
шуюся после этого жидкость у конца пипетки не сле-
дует вытряхивать, выдувать или вытеснять путем
нагревания широкой части пипетки теплотой рук,
так как пипетки рассчитаны на выливание.
Пользуясь цилиндрической пипеткой, дают жид-
кости вытекать, пока нижний край мениска не кос-
нется соответствующего деления
Пипетки устанавливают в штативе или в шкафу.
В первом случае для защиты пипеток от пыли их
Рис. 15. Правильное дер-
жание пипетки при выпу-
скании из нее жидкости.
закрывают колпачками.
Микропипетки на 0,01 и 0,02 мл имеют деления соответственно
тысячным долям миллилитра. Объемы указаны на стенках пипеток и рассчи-
таны для температуры 20°С. Микрспипетками пользуются так же, как и макро-
пипетками.
Мерные цилиндры и мензурки (рис. 16) изготовля-
ются разнообразной емкости. На стенках цилиндров и мензурок нанесены
деления, указывающие емкость в миллилитрах. В широких цилиндрах и
мензурках каждое деление соответствует не одному, а 5—10 мл. Указанная
емкость соответствует температуре 20°С. Для точного
2О°С
мл
0-500
0-1000
ЮО-±9СО
200^80.
300^700
400^600
500-+500
600^400
700^.300
800^200
90оЛ/00
отмеривания цилиндры и мензурки непригодны.
Колбы обыкновенные бывают различ-
ной формы и емкости (рис. 17). Они применяются
в лабораториях для разных целей—растворения веще-
ства, приготовления реактивов, нагревания, кипяче-
ния и т. п.
Колба с отводной трубкой (рис. 18),
толстостенная, имеет коническую форму и сбоку
отводную трубку. Такие колбы применяют для
Рис. 17. Колбы обыкновенные.
1 — колба круглая плоскодонная,
2 — колба коническая, 3 — колба
круглодонная.
Рис. 16. Мерные цилиндры
и мензурка.
1 п 2 — цилиндры, 3 — мензурка.
Рис. 18.
Колба кони-
ческая с от-
водной труб-
кой.
ускорения фильтрования. Отводную трубку сообщают с насосом. В шейку
колбы вставляют резиновую пробку с отверстием, а в отверстие — во-
ронку с фильтром, тигель с пористым дном и т. п. Благодаря действию насоса
воздух в колбе разрежается и фильтрование происходит быстрее.
23
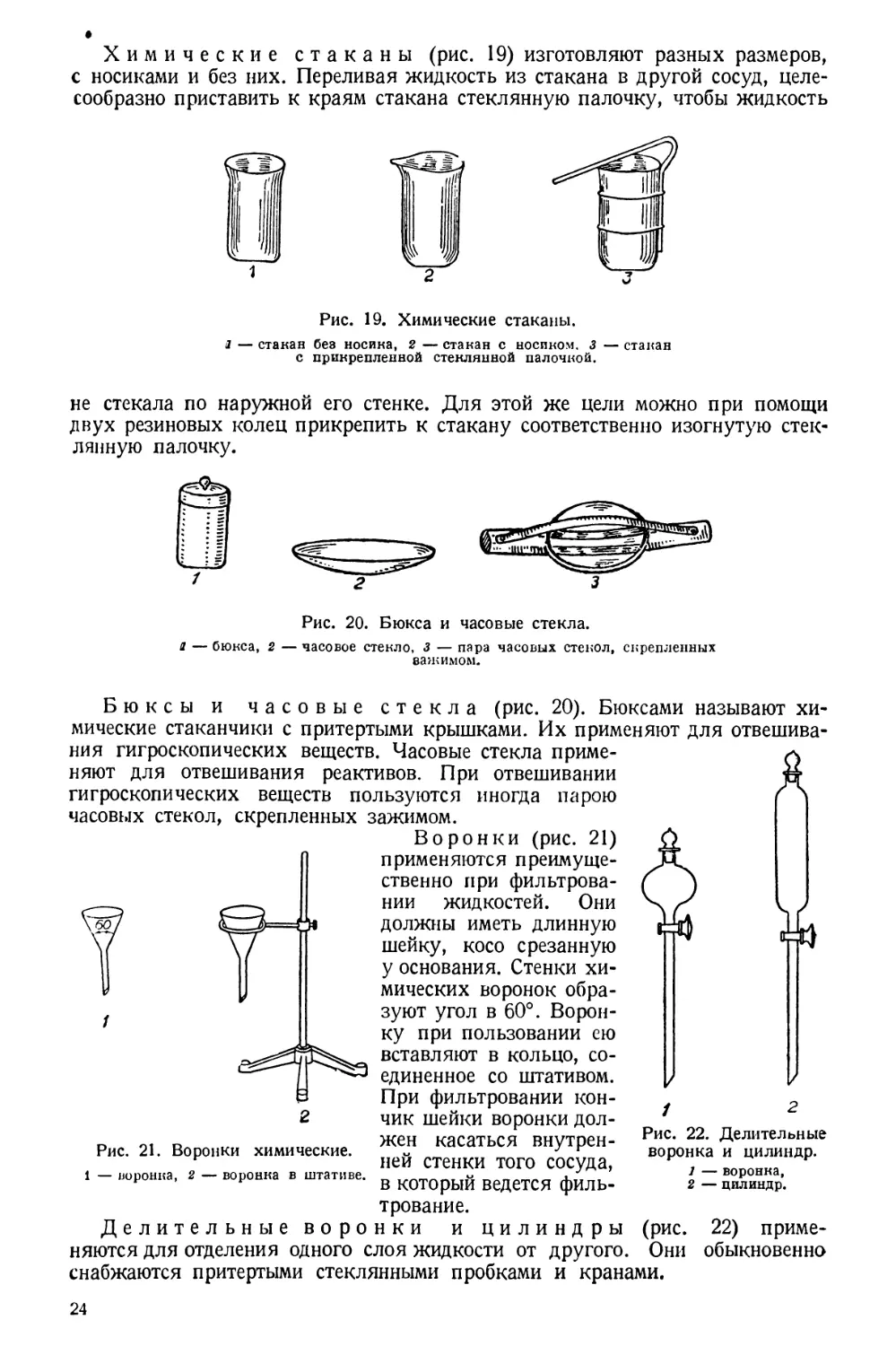
Химические стаканы (рис. 19) изготовляют разных размеров,
с носиками и без них. Переливая жидкость из стакана в другой сосуд, целе-
сообразно приставить к краям стакана стеклянную палочку, чтобы жидкость
Рис. 19. Химические стаканы.
1 — стакан без носика, 2 — стакан с носиком. 3 — стакан
с прикрепленной стеклянной палочкой.
не стекала по наружной его стенке. Для этой же цели можно при помощи
двух резиновых колец прикрепить к стакану соответственно изогнутую стек-
лянную палочку.
Рис. 20. Бюкса и часовые стекла.
1 — бюкса, 2 — часовое стекло, 3 — пара часовых стекол, скрепленных
зажимом.
Бюксы и часовые стекла (рис. 20). Бюксами называют хи-
мические стаканчики с
притертыми крышками. Их применяют для отвешива-
ния гигроскопических веществ. Часовые стекла приме-
няют для отвешивания реактивов. При отвешивании
гигроскопических веществ пользуются иногда парою
часовых стекол, скрепленных зажимом.
Воронки (рис. 21)
Рис. 21. Воронки химические.
1 — воронка, 2 — воронка в штативе.
применяются преимуще-
ственно при фильтрова-
нии жидкостей. Они
должны иметь длинную
шейку, косо срезанную
у основания. Стенки хи-
мических воронок обра-
зуют угол в 60°. Ворон-
ку при пользовании ею
вставляют в кольцо, со-
единенное со штативом.
При фильтровании кон-
чик шейки воронки дол-
V
1
жен касаться внутрен-
ней стенки того сосуда,
в который ведется филь-
Рис. 22. Делительные
воронка и цилиндр.
1 — воронка,
2 — цилиндр.
трование.
Делительные воронки и цилиндры (рис. 22) приме-
няются для отделения одного слоя жидкости от другого. Они обыкновенно
снабжаются притертыми стеклянными пробками и кранами.
24
Рис. 23. Промы-
ва лка для дестил-
лированной
воды.
Про мы валка или шприц (рис. 23) представляет собой обыкно-
венную колбу с резиновой пробкой, в которую вставлены две изогнутые труб-
ки; из них одна заканчивается сразу под пробкою, другая ж доходит почти
до дна. Промывалку заполняют дестиллированной водой и обыкновенно при-
меняют для промывания посуды, осадков и т. п. Воду из промывалки выли-
вают через короткую трубку. Если вдувать воздух в корот-
кую трубку, то вода будет вытекать тонкой струей из на-
ружного отверстия длинной трубки. Если при этом поднять
конец длинной трубки вверх, то и струя воды будет бить
вверх, что в некоторых случаях оказывается удобным (напр.
при вымывании осадка из опрокинутого стакана).
Экссикатор (рис. 24) представляет собой стеклян-
ный сосуд с пришлифованной стеклянной крышкой. Приме-
няется для охлаждения веществ, подвергавшихся прокали-
ванию или высушиванию. В некоторых случаях высуши-
вание ведется в самом экссикаторе. Нижнюю коническую
часть экссикатора заполняют гигроскопическим веществом
(СаС12, H2SO4, Р2О5). Пришлифованный край крышки эксси-
катора смазывают слегка вазелином, чтобы образовалось гер-
метически закрытое пространство. Над нижней конической
частью экссикатора устанавливается фарфоровая пластинка
с отверстиями для тиглей и чашек.
Для быстрого высушивания применяют экссикаторы, даю-
щие возможность
создать внутри вакуум (рис. 25). От крышки или боковой
стенки таких экссикаторов отходит трубка, снабжен-
ная краном. При желании получить вакуум, трубку
соединяют с насосом, открывают кран и выкачивают
воздух, после чего вновь закрывают кран.
Из гигроскопических веществ, применяемых в экс-
сикаторах для сушки, менее других пригоден хло-
ристый кальций, так как вследствие
- образования кристаллогидратов хло-
ристого кальция (СаС12 . Н2О; СаС12 .
. 2Н2О; СаС12 4Н2О и СаС12.6Н2О)
воздух в таких экссикаторах иногда
содержит влагу. Прокаленное веще-
ство, долго оставаясь в таком эксси-
каторе, может сорбировать некоторое
Справа показан
Рис. 24. Экссикатор.
вкладыш для экссикатора.
количество влаги.
Хорошими осушителями являются концентрирован-
ная серная кислота, пятиокись фосфора (Р2О5), а
также специальный осушитель, который готовится
следующим образом: зерна пемзы диаметром от 2 до
4 мм сушатся в течение 4 часов при 140°С и пропи-
тываются шестиводным хлористым кальцием — (СаС12.
6Н2О), расплавленным в сушильном шкафу при темпе-
ратуре не выше 60°С. Пропитка производится так:
зерна пемзы, залитые в колбе расплавленным хлори-
стым кальцием (температура 45—50°С), выдерживаются
в шкафу при 60°С в течение 3—4 часов до полного вы-
деления пузырьков воздуха. Избыток хлористого каль-
ция отсасывается быстро на бюхнеровской воронке,
после чего зерна пемзы сушатся на железном листе
4—5 часов при 140°С. Сохраняются зерна в гермети-
чески закрытом сосуде. Полученный таким образом
Рис. 25. Экссикатор
с краном.
осушитель очень хорошо сорбирует влагу и может
быть регенерирован нагреванием в течение 4—5 часов при 220—240°С*.
Фарфоровая и платиновая посуда
Фарфоровую и платиновую посуду применяют в виде чашек, тиглей, вс
ронок, конусов и т. п.
Фарфоровые и платиновые чашки (рис. 26) используют
для выпаривания жидкостей. Они бывают разных размеров и форм.
Рис. 26. Чашка
выпаривательная.
Рис. 27. Тигли.
1 —тигель фарфоровый обыкновенный,
2 — тигель с отверстием в крышке.
Фарфоровые и платиновые тигли (рис. 27) применяются
для прокаливания веществ. Они обыкновенно снабжаются крышками. Если
сжигание производится при помощи струи горящего водорода, то крышки
для тиглей применяются с отверстиями. В отверстие вставляют конец фар-
форовой трубки, служащей для привода
Рис. 28.
Воронка
фарфоровая
с ситчатым
дном.
Рис. 29 Тигель Гуча.
Показано соединение
тигля с колбой.
газа.
Фарфоровая воронка с
ситчатым дном (бюхнеровская
воронка—рис. 28) применяется для бы-
строй фильтрации жидкостей. Шейку
воронки вставляют в резиновую пробку,
а последнюю в горлышко колбы с от-
водной трубкой, которая присоединяется
к насосу. На дно воронки кладут не-
сколько кружочков фильтровальной бу-
маги, по диаметру соответствующих
диаметру воронки. Фильтр смачивается
дестиллированной водой и тщательно
прилаживается ко дну воронки, после
чего приступают к фильтрованию при
пониженном давлении.
Фарфоровый тигель с
ситчатым дном Гуча (рис. 29)
служит и для фильтрования жидкостей, и для прокаливания осадков. Каж-
дый такой тигель снабжается фарфоровой ситчатой пластинкой, которую
можно ввести до дна тигля. На дно тигля поме- п
щают слой крупноволокнистого асбеста, а поверх—
ситчатую пластинку, получая, таким образом, го-
___________ товый фильтр. Таким фильтром
пользуются в тех случаях, когда
\®° •* •___прокаливание осадка с бумажным
\**в *9 фильтром недопустимо (подробнее
S см. СТР- 41).
Фарфоровы е и плати-
п . и,,, новые конусы — шутны
Рис. 30. Шутц. . 7 7
J (рис. 30) имеют по стенкам множе-
ство отверстий. Если фильтрование
Рис. 31 Приборы с фильтру-
ющими пластинками:
1 — тигель, 2 — нутч,
3 — палочка
проводится ускоренно при пониженном давлении,
то такой конус помещают в воронку под бумажный фильтр. Благодаря
этому фильтр предохраняется от разрыва.
.26
Тигли, путчи и палочки с фильтрующим дном
(рис. 31) готовятся из стекла, фарфора и кварца. Стеклянные пористые пла-
стинки, представляющие дно в этих приборах, готовятся из слегка стоплен-
ного стеклянного порошка. Пластинки имеют разный диаметр пор в зависи-
мости от размера диаметра зерен, из которых они сплавляются.
Пористость стеклянных фильтров характеризуется условными обозначе-
ниями, представленными в таблице 2.
Тигли с фильтрующим дном служат
для той же цели, что и описанные выше
тигли с ситчатым дном, но удобнее в при-
менении. Нутчи весьма полезны при рабо-
тах по очистке разных веществ. Фильтру-
ющие палочки служат для отсасывания
раствора от выпавшего осадка, давая воз-
можность всю операцию отделения осадка
от раствора произвести в одном сосуде.
Пластинки №№ 1 и 2 применяют пре-
имущественно для изготовления газовых
фильтров и промывалок. Фильтры №№ 3
и 4 используют для количественных опре-
делений.
Таблица 2
Диаметр пор стеклянных фильтров
Условное обозна- чение диаметра пор Средний размер диаметра пор
1 100—120 (г
2 40—50 »
3 20—25 •>
4 4—10 »
Новые стеклянные фильтры перед упо-
треблением должны быть хорошо очищены кипячением в разбавленной соляной
кислоте. Через них нельзя фильтровать горячие растворы щелочей. Наилуч-
шие стеклянные фильтры выдерживают постепенное нагревание лишь до 600°С.
Фарфоровые пористые пластинки изготовляются из фарфоровой фритты;
поры их имеют в диаметре от 0,6—0,75р. . Как и стеклянные фильтры, они
непригодны для горячих щелочных растворов. При фильтровании раство-
ров, содержащих серную кислоту, фарфоровые фильтры заметно сорбируют
последнюю, и потому пластинки после фильтрования таких растворов сле-
дует тщательно промывать. Фарфоровые фильтры выдерживают нагревание
до 1100°С.
Кварцевые фильтры выдерживают фильтрование горячих, не слишком
концентрированных растворов и прокаливание до 1500°С.
Платиновые фильтровальные тигли и палочки с фильтром из губчатой
платины очень удобны для описанных выше работ, но весьма дороги.
Приборы для нагревания, прокаливания и высушивания
Спиртовая горелка (рис. 32) обыкновенно применяется для нагревания
небо л ыпи х коли честв
Рис. 32. Спиртовая
горелка.
Рис. 33. Бензиновая горелка.
жидкости (в пробирках).
Бензиновая го-
релка (рис. 33) служит
для получения высоких
температур. Еще до того
как разогреть горелку,
в резервуар при закры-
том кране накачивают
при помощи насоса воз-
дух. Затем разогревают
горелку, зажигая под
нею спирт. Когда спирт
сгорит, открывают кран,
бензин под давлением поступает в горелку, превращается в газ и сгорает.
Горелка Бунзена (рис. 34) имеет у основания трубку для подачи
газа; на трубку надевается резиновый шланг, второй конец которого соеди-
27
Рис. 34. Горелка
Бунзена.
1540*
/560°
520*
300*
Рис. 36. Пламя
горелки Бун-
зена. Показаг а
температура
пламени в раз-
личных точках.
няется с трубой магистрали, подающей светильный или карбюраторный газ.
Снизу горелки несколько выше трубки, подающей газ, имеются два отверстия
для поступления воздуха, который, сме-
шиваясь с газом, улучшает горение.
Для регулирования притока воздуха
горелка снабжается регулирующей гиль-
зой.
Горелка Теклю
(рис. 35) имеет некото-
рые преимущества перед
горелкой Бунзена: в ней
подача газа регулирует-
ся винтом а, а подача
воздуха — кольцом в.
Если кольцо подвинчи-
вать вверх, то оно по-
степенно закрывает ниж-
нюю расширенную часть
горелки, и доступ воз-
духа затрудняется. При
опускании кольца имеет
место обратное явление,
что в каждом пламени различают внеш-
оболочку и внутреннюю светящуюся
В первой оболочке происходят окислительные процессы
(окислительное пламя), в
Рис. 35. Горелка
Теклю.
Следует помнить,
нюю несветящуюся
оболочку (рис. 36).
Рис. 37. Тигельная
печь.
другой — восстановительные
процессы (восстанови-
тельное пламя).
Тигельная печь
(рис. 37) с нихромовой
спиралью нагревается
при помощи электротока
от сети до 800°С. Такая
же печь с платиновой
спиралью дает темпера-
туру до 1000°С. Тигель-
ные печи служат для
прокаливания (озоле-
ния) органического ма-
териала. Для регулиро-
вания температуры на-
грева печи в цепь вклю-
чают реостат.
печь (рис 38) работает так же, как и тигельная, от
Рис. 38. Муфельная печь.
Я
39. Водяная
баня.
Муфельна
электросети. Муфельные печи обыкновенно снабжаются реостатами, дающими
возможность регулировать высоту нагрева печи. Благо-
даря большим размерам муфельные печи дают возможность
вести одновременно прокаливание в нескольких тиглях.
Водяная баня (рис. 39) применяется для выпа-
ривания жидкостей. Она представляет собой медную или
алюминиевую круглодэнную чашку с крышкой, состоящей
из ряда отдельных концентрических колец. В зависимости
от размеров чашки, которая помещается на баню, снимают рис.
большее или меньшее число колец. Нагревание бани ве-
дется при помощи горелки или электроплитки.
При длительном выпаривании жидкостей применяют бани с
уровнем (рис. 40). Постоянный уровень обеспечивается специальным при-
ПОСТОЯННЫМ
28
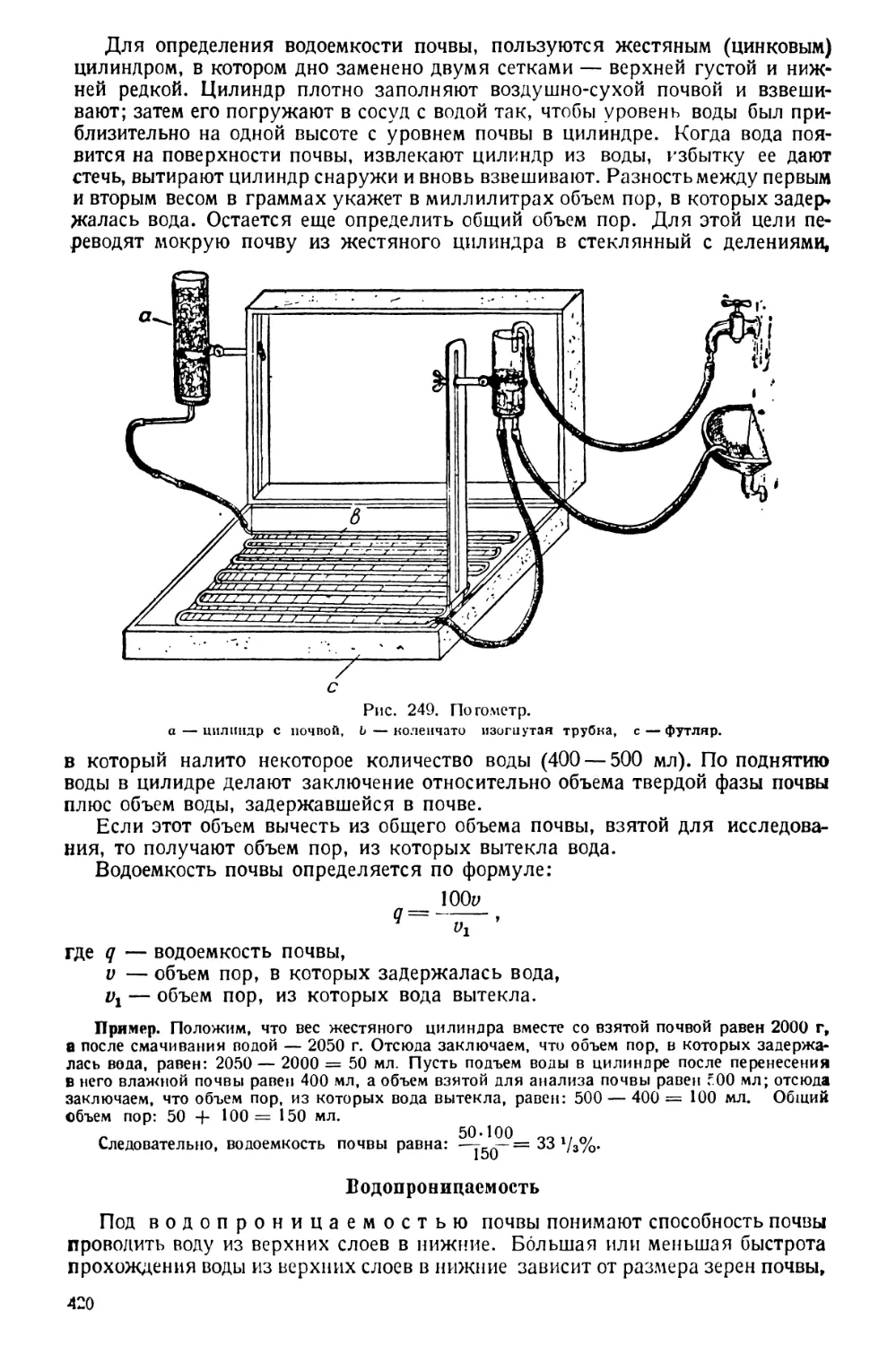
способлением, через которое в баню все время медленно поступает вода из
водопровода, а избыток отводится в отлив.
В лабораториях широко распространены удобные никелированные водя-
ные бани с электрическим нагревом и постоянным уровнем.
Воздушная баня устроена так же, как водяная,
объекта ведется в ней при помощи нагреваемого воз-
духа.
Песочная баня (рис. 41) представляет
электроплитку; спираль в ней скрыта под асбестом,
на который насыпают слой песка. Для получения
песочной бани можно использовать обыкновенную Д|||||| i | ।
баню, насыпав в нее песок и ведя подогревание горел- j| tsjg
кой или электроплиткой. Песочные бани примени-
ются для осторожного нагре-
вания до высокой температу-
ры (прокаливание соды, сжи-
гание по Кьельдалю и т. п.).
Нагревание
Рис. 41. Песочная баня. Рис. 42. Сушильный шкаф.
с постоянным уровнем.
Рис. 40. Водяная баня
Сушильный шкаф (рис. 42) применяется для высушивания раз-
личных веществ, а также влажной посуды. Изготовляется из листовой меди
или такого же железа. Наружные стенки шкафа покрываются асбестом.
В крышке шкафа имеются два отверстия: в одно вставляют термометр, другое
открывают, когда хотят снизить температуру внутри шкафа. В зависимости
от природы высушиваемого вещества температуру в шкафу поднимают до
той или иной высоты. Нагревание шкафа производится при помощи горелок
(газовых, бензинных), а также при помощи электричества.
Очень удобны шкафы, снабженные автоматическими тер-
морегуляторами.
Имеется много систем тер-
морегуляторов. На рис. 43
изображены терморегуляторы
для электрических сушильных
шкафов и термостатов. Принцип
их действия состоит в том, что
при достижении заданной темпе-
ратуры обогрев автоматически
выключается.
В терморегуляторе А это дости-
Рис. 43. Автоматические тер-
морегуляторы для электри-
ческого нагрева.
Рис. 44. Автомати-
ческий терморегу-
лятор для газо-
вого нагрева.
гается тем, что при повышении темпе-
ратуры гофрированная медная по-
душка а, заполненная частью эфиром,
от нагревания расширяется, при этом
стержень b перемещается вверх, под-
нимает рычаг е, вызывая разрыв контакта в точке d. При помощи винта с можно отрегу-
лировать терморегулятор так, чтобы момент разрыва контакта происходил при заданной
температуре.
В терморегуляторе В автоматическое выключение тока достигается при помощи биме-
таллической вилки а. При изменении температуры кривизна вилки меняется, а это приво-
29
дит к замыканию или размыканию тока в точке Ъ. Винт с служит для регулировки т<
регулятора на определенную температуру.
На рис. 44 изображен автоматический терморегулятор для термостатов или суш
ных шкафов, обогреваемых светильным или карбюраторным газом. Терморегулятор г
трубку А, заполненную внизу ртутью. Нижняя часть трубки помешается в обогревае
шкаф. От трубки А отходит сбоку отводная трубка О, в которую вставлен на шайбе г per
ровочный винт Р. Верхняя часть прибора устроена наподобие тройника В, который oi
коленом е вставлен в трубку А. Газ поступает в колено а, отсюда направляется вниз и ч
отвод d поступает в горелку. Как только температура поднимается выше нужного пред
выступившая ртуть закрывает поступление газа из тройника в отвод d, газ паправля
по трубке Ъ и через узкое отверстие стеклянного крана С поступает в горелку слабой стр
при этом нагрев шкафа уменьшается. Когда температура падает ниже желаемого пред
ртуть снова открывает доступ газу через колено е в отвод d.
Прочие лабораторные принадлежности
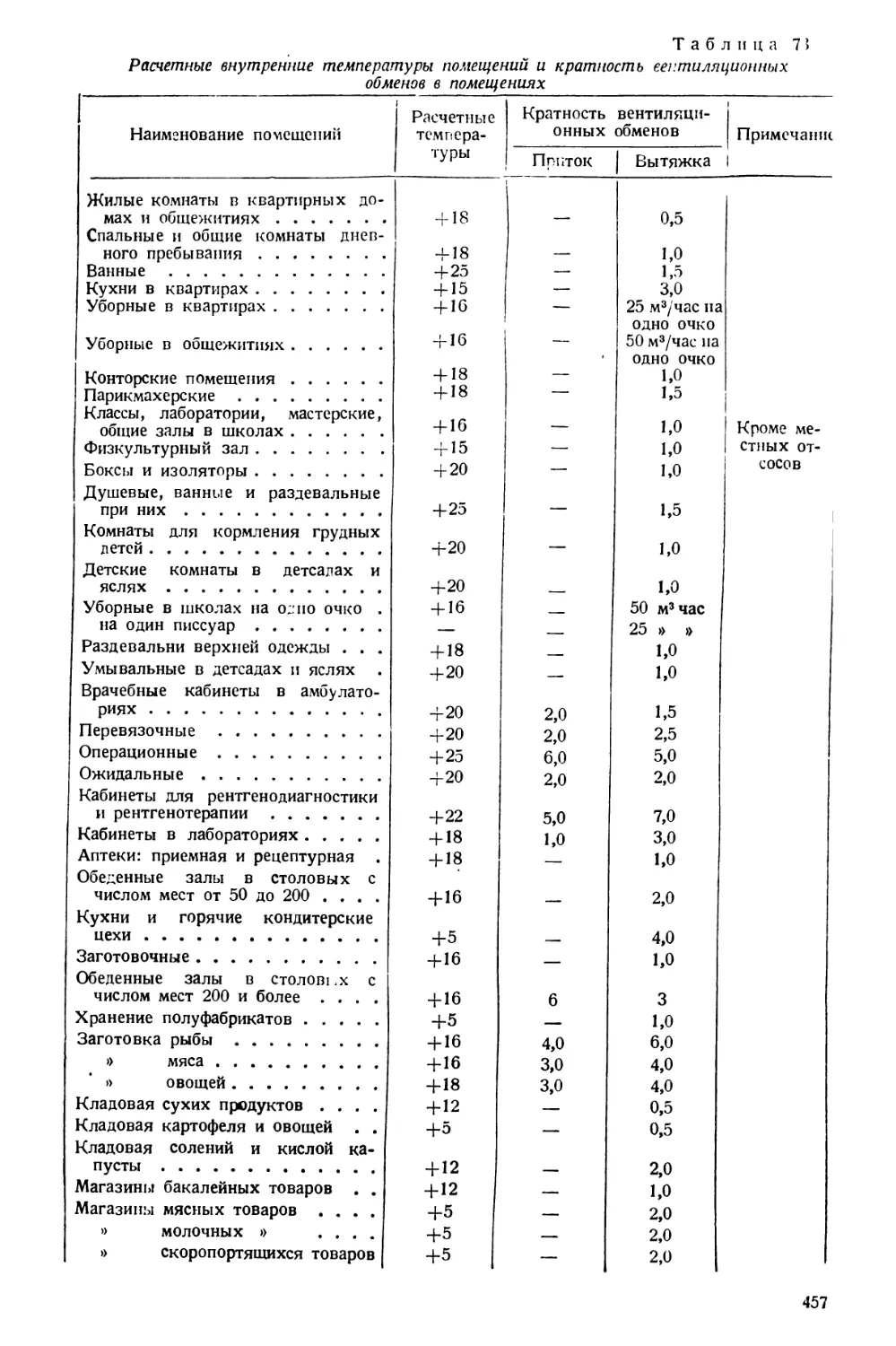
Тренога (рнс. 45) изготовляется из железа. На ней помещают предметы, под
жащие нагреванию.
Асбестовая сетка (рис. 46) представляет собой проволочную сетку, средь
часть которой покрыта асбестом. Сеткой пользуются в тех еле чаях, когда нагреваем
предметы имеют в виду защитить от действия «голого» пламени.
Рис. 45.
'Тренога.
Рис. 46. Асбесто-
вая сетка.
Рис. 47. Треуголь-
нике фарфоровыми
трубочками.
Треугольник (рис. 47) изготовляется из проволоки. Стороны его, образующи
непосредственно треугольник, заключены в фарфоровые, реже кварцевые трубочки. Приме
няются при нагревании и прокаливании тиглеи, которые в этом случае устанавливаются
треугольник.
Рис. 48. Тигель- Рис. 49. Сверла
ные щипцы. для пробок.
Рис. 50. Пробкомялка.
Щипцы тигельные никелевые (рис. 48) применяются для захватывания на-
гретой посуды; реже для этой же цели пользуются щипцами с платиновыми наконечниками.
Сверла для пробок (рис. 49) выпускаются в виде наборов с различными
диаметрами.
Пробкомялка (рис. 50) служит для обжима пробок.
ОСНОВЫ ЛАБОРАТОРНОЙ ТЕХНИКИ
Здесь приводятся общие указания по лабораторной технике и даны ос-
новы весового и объемного анализов.
30
мл техниче-
Рис. 51.
Прибор для
обработки
посуды
паром.
Правила обращения с химической посудой
Со стеклянной химической посудой необходимо обращаться бережно и
аккуратно.
При чистке посуды не следует применять вещества, способные поцарапать
стекло (песок, проволока). Загрязненную посуду моют горячей водой специ-
ально предназначенными для этой цели щетками (ерши). Иногда применяют
для чистки посуды горячий мыльный раствор, крепкую серную кислоту,
щелочной или кислый раствор КМПО4, хромовую смесь, которую готовят
следующим образом: 150—200 г технического К2Сг2О7 растворяют в воде
до общего объема в 600 мл и доливают осторожно по стенке 400
ской крепкой H2SO4. Все указанные растворы могут быть ис-
пользованы многократно и потому после употребления слива-
ются в склянку, где и сохраняются. Хромовая смесь при
многократном применении меняет свой темнооранжевый цвет на
темнозеленый, что служит указанием на ее дальнейшую непри-
годность. Хромовая смесь неприменима, если посуда загрязнена
нефтью или продуктами ее перегонки.
Чтобы удалить из посуды смолистые и другие нераствори-
мые в воде вещества, применяют серный и петролейный эфир,
ацетон, бензин, четыреххлористый углерод и другие органи-
ческие растворители. Выбор того или иного растворителя за-
висит от природы вещества, подлежащего растворению.
После применения перманганата на стенках посуды остается
нередко слабый налет бурой двуокиси марганца, который легко
удаляется соляной кислотой.
Удалив загрязняющее вещество, моют посуду теплой, затем
водопроводной водой и под конец хорошо ополаскивают де-
стиллированной. Хорошо вымытая посуда совершенно прозрач-
на; на стенках ее капельки воды не задерживаются, а стекают.
Высушивание посуды производится на колышках, сушильных
столах или в сушильном шкафу. Вытирать посуду изнутри
полотенцем или марлей не следует. Чтоб ускорить высушива-
ние посуды, можно промыть ее небольшим количеством спирта,
а затем эфира.
При нагревании жидкостей посуда помещается на асбесто-
вую сетку, а не на «голый» огонь, причем посуда должна быть
снаружи совершенно сухой. В лабораторной химической посуде
не следует хранить долго реактивы, способные действовать на
стекло: щелочи, кислоты.
Для уменьшения растворимости нового стекла рекомендуется обработать
его сильной струей пара (пропаривание). Обработка посуды паром произво-
дится при помощи прибора, изображенного на рис. 51. В колбу емкостью
в 2—3 л до половины наливают воду, для равномерного кипения опускают
несколько кусочков пемзы или несколько стеклянных бусинок. Шейку колбы
закрывают плотно пробкой, в которую вставляют воронку. В воронку также
вставляют пробку, а в нее стеклянную трубочку. Конец трубочки вводят
в сосуд, который подлежит пропариванию. Сосуд закрепляют при помощи
кольца или зажима в штативе (на рисунке не показано). Когда прибор собран,
кипятят воду в колбе на сильном огне. Пар вырывается через трубку струей,
а затем, конденсируясь на стенках пропариваемого сосуда, стекает в воронку.
Когда в воронке собирается много воды, ее сливают.
Пропаривание служит хорошим способом и для очистки химической посу-
ды. После пропаривания посуда должна быть тщательно вымыта водой.
Платиновая посуда также требует умелого обращения. Необходимо пом-
нить, что нельзя вести нагревание платиновых тиглей в светящемся пламени
горелки во избежание образования карбида платины, отчего посуда делается'
31
Рис. 52. Приспособление для пере-
кристаллизации.
хрупкой и ломкой. Нельзя раскаленный тигель приводить в соприкосновение
с железом (незащищенный треугольник, железные щипцы), так как раскален
ная платина сплавляется с железом и посуда теряет свою ценность. Нельз?
прокаливать в платиновом тигле вещества, содержащие железо, свинец
серебро, медь, кадмий, мышьяк, сурьму и олово: платина легко образует
сплавы с этими элементами; нельзя проводить реакций, при которых выде-
ляются хлор и бром.
Платиновую посуду чистят мелким круглозернистым песком с водою,
тальком, мокрым асбестом. Для удаления силикатов из платиновой посудь
в ней сплавляют безводную соду, а для удаления окисей металлов — ка-
лийгидросульфат (KHSOJ.
Правила обращения с реактивами, их очистка
Реактивы сохраняются в чистых банках, бутылках и т. п. сосудах, на
которых должны быть наклеены этикетки с обозначением названия реактива
и завода, выпустившего данный реактив.
Для чистых реактивов, годных для про-
изводства точных анализов, делается на
этикетке соответствующая отметка. На
наших препаратах, годных для точных
анализов, имеется обозначение: «х. ч.»
(«химически чистый»). На склянках с силь-
нодействующими ядами имеется еще допол-
нительная наклейка с изображением чело-
веческого черепа и скрещенных костей
(signum veneni). Посуда, в которой сохра-
няются реактивы, должна быть снабжена
притертой пробкой, и только для веществ,
сильно действующих на стекло, а также
очень гигроскопичных, предпочтение от-
дают корковым пробкам, которые заливают
парафином. Реактивы следует доставать из
склянок специальными реактивными фар-
форовыми или никелевыми ложечками. Такими же ложечками или стеклян-
ными лопаточками (палочками) нужно пользоваться при взвешивании реак-
тивов, ни в каком случае не применяя с этой целью пинцет для разновеса.
Перекристаллизация представляет собой операцию, приме-
няемую для очистки многих препаратов; нередко это касается препаратов,
идущих на изготовление титрованных растворов.
Подвергаемый очистке препарат растворяют в небольшом количестве го-
рячей дестиллированной воды до получения насыщенного раствора и филь-
труют через складчатый фильтр. Для получения складчатого фильтра листок
фильтровальной бумаги складывают вдвое и затем наподобие веера образуют
В—16 складок; свободные края фильтра обрезают так, чтоб при расправлении
листка получился правильный круг. Полученный фильтр вкладывают в во-
ронку без шейки; под воронку ставят фарфоровую чашку, которую помещают
на водяную баню с холодной водой. Горячий насыщенный раствор льют на
фильтр и одновременно помешивают стеклянной палочкой жидкость, которая
отфильтровывается в фарфоровую чашку (рис. 52). В результате быстрого
охлаждение и помешивания из маточного раствора выпадает мелкокристалли-
ческий препарат.
Фильтрование горячих насыщенных растворов иногда довольно затруд-
нительно, так как при их охлаждении начинают выпадать кристаллы, заку-
поривающие фильтр. С целью избежать этого затруднения применяют спе-
циальные воронки для горячего фильтрования (рис. 53). Эти воронки гото-
вятся из металла (меди); они имеют двойные стенки, пространство между
которыми заполняется водой; вода нагревается до кипения. В такую метал-
32
лическую воронку помещают стеклянную воронку, через которую ведется
фильтрование. Имеются приборы для горячего фильтрования и с электрическим
обогревом. Иногда насыщенный раствор после фильтрации и охлаждения
не кристаллизуется (пересыщенные растворы); в этом случае необходимо
в раствор ввести затравку — готовый кристаллик данного вещества, после
чего быстро наступает кристаллизация.
Мелкокристаллическую муку, полученную в результате
перекристаллизации, переносят в воронку с ситчатым
дном, в которую предварительно кладут 2—3 листка
фильтровальной бумаги, и отсасывают жидкость. Влажный
препарат размещают тонким слоем на фильтровальной & В ®
бумаге, сложенной в несколько слоев, покрывают такой же И
бумагой и оставляют на 12—24 часа. Чтобы убедиться, I
что препарат подсушен до постоянного веса, берут неболь- |
шое количество его на часовое стеклышко и взвешивают; И
затем, накрыв фильтровальной бумагой, оставляют еще I
несколько часов, после чего снова взвешивают. Если вес U
не изменился, то высушивание препарата считается за-
конченным; в противном случае оно продолжается, пока **
препарат не приобретет постоянный вес. Гигроскопические
вещества сушат под колоколом в разреженном простран- рис. 53 воронка
стве. Высушиванию в экссикаторе или в сушильном шкафу для горячего
можно подвергать лишь такие препараты, которые не содер- фильтрования,
жат кристаллизационной воды. Легко окисляющиеся веще-
ства можно сушить при высокой температуре лишь в атмосфере индиферент-
ного газа (СО2, N2). Полученный препарат испытывают на чистоту соответ-
ствующими реактивами. Иногда для очистки препарата его приходится пере-
кристаллизовывать несколько раз.
Приготовление различных растворов
Здесь описаны способы приготовления различных растворов за исключе-
нием титрованных, способы приготовления которых изложены отдельно
(см. ниже).
Растворы твердых веществ в жидком растворителе. Для приготовления
таких растворов указываются либо весовые отношения растворяемого веще-
ства и растворителя, либо, что бывает чаще, процентное содержание раство-
ряемого вещества в растворе. Приготовление растворов первого вида не тре-
бует разъяснений.
При изготовлении растворов второго вида поступают следующим обра-
зом. Отвешивают на технических весах с точностью до 0,01 г растворимое
вещество в количестве, равном требуемому проценту, и взятую навеску ра-
створяют в таком количестве воды, чтобы общий вес раствора был равен 100 г.
При этом нужно учесть следующие обстоятельства.
1. Растворимое вещество не должно содержать в заметном количестве
гигроскопической воды; если она имеется, то растворимое вещество либо
подсушивают, либо растворяют его с гигроскопической влагой, а затем вводят
необходимый корректив, как указано ниже (см. пример 2).
2. Если растворимое вещество содержит кристаллизационную воду, то
расчет ведут на вес вещества без кристаллизационной воды.
Пример 1. Требуется приготовить 100 мл 5% раствора хлористого натрия. Отвешивают
5 г сухой соли и растворяют в 95 мл дестиллированной воды. Получается нужный раствор.
Здесь объем воды принимается равным весу воды, что для обычных лабораторных условий
не совсем верно. Однако допущенной ошибкой можно пренебречь.
Пример 2. Требуется лригововить 100 мл 5% раствора едкого натрия. Ввиду высокой
гигроскопичности этого препарата и затруднений, с которыми связано высушивание его,
отвешивают 5 г на технических весах, не обращая внимания на влажность препарата, и раство-
ряют в 95 мл воды. Определяют при помощи ареометра удельный вес раствора Положим
з а. и. Бурштейн
33
что он оказался равным 1,052. Согласно таблице 22 (см. в конце главы) процентное содержа-
ние NaOH в таком растворе равно 4,64. Следовательно, на 100 г раствора нужно прибавить
еще 0,36 г. NaOH, чтобы получить требуемый раствор.
Пример 3. Требуется приготовить 100 мл 10% раствора хлористого бария. Учитывая,
что хлористый барий (ВаС12. 2Н2О) содержит две частицы кристаллизационной воды, нужно
для получения указанного раствора отвесить не 10 г, а во столько раз больше, во сколько
молекулярный вес ВаС12-2Н2О более молекулярного веса ВаС12. Находим отношение этих
244,306
молекулярных весов равное -208 274~ ~ 1Л73. Следовательно, для получения нужного ра-
створа отвешивают 10 . 1,173 = 11,73 г ВаС12 и добавляют дестиллированной воды 88,3 мл
(точнее 88,27 мл).
Растворы жидких веществ в жидких растворителях. Для приготовления
таких растворов указываются либо объемные отношения растворяемого ве-
щества и растворителя, либо процентное содержание (объемное или весовое)
растворяемого вещества в растворе, либо, наконец, приготовляется раствор
определенного удельного веса.
Пример 1. Требуется приготовить 200 мл раствора серной кислоты 1 : 3, т. е. такой
раствор, в котором на 1 объем серной кислоты приходилось бы 3 объема воды. Отбираем
цилиндром 50 мл практически безводной серной кислоты (уд. вес 1,84) и отдельно 150 мл
дестиллированной воды. Кислоту по стенке сосуда вводим осторожно в воду, а затем остатки
кислоты вымываем полученным раствором. Положим, что в нашем распоряжении нет без-
водной серной кислоти, а имеется серная кислота уд. веса 1,4. По таблице 20 (см. стр. 124)
находим, что содержание серной кислоты в таком растворе равно 50,11%. Следовательно,
100
приходится взять не 50 мл, а в jy- раз больше, т. е. приблизительно вдвое больше. Таким
образом, на 200 мл требуемого раствора (1 : 3) прийдется взять серной кислоти (уд. веса
1,4) 100 мл и 100 мл дестиллированной воды.
Пример 2. Требуется приготовить 100 мл раствора, содержащего 60 объемных процен-
тов абсолютного спирта. Для этой цели понадобилось бы к 60 мл абсолютного спирта приба-
вить дестиллированной воды до общего объема в 100 мл. Такие растворы готовятся обыкно-
венно не из абсолютного алкоголя. Положим, что в распоряжении имеется 95% ректификат.
60-100
Нужные нам 60 мл абсолютного спирта содержатся в —63,2 мл данного ректифи-
ката. Отмеряем в мерную колбу на 100 мл пипетками 63,2 мл 95% ректификата и доводим
до метки водой.
Пример 3. Требуется приготовить 100 мл соляной кислоты уд. веса 1,12. В распоря-
жении имеется соляная кислота уд. веса 1,19. По таблице 20 находим процентное содержание
хлористоводородной кислоты в имеющемся и требуемом растворах соляной кислоты; оно
равно соответственно: 23,82 и 37,23 весового процента. Для определения, сколько нужно
взять по весу имеющейся кислоты, чтобы это количество содержало 23,82 г хлористоводо-
родной кислоты, составляем пропорцию:
ЮО — 37,23
х —23,82.
Откуда:
100 • 23,82
х = 37,23 ~64 Г’
Таким образом, следует отвесить 64 г имеющейся в распоряжении соляной кислоты и
довести полученный объем до 100 мл водой.
Однако кислоту лучше отмерить по объему, нежели отвешивать. Чтобы перевести вес
на объем, нужно, как известно, вес разделить на уд. вес. В данном случае получим:
64
10 — 53,8 мл.
Таким образом, для получения требуемого раствора вводим в мерную колбу 53,8 мл
имеющейся соляной кислоты уд. веса 1,19 и доводим до метки водой.
Получаем 100 мл кислоты уд. веса 1,12.
Получение раствора определенной концентрации из двух растворов того
же вещества других концентраций. Иногда приходится получать растворы
определенной концентрации из имеющихся двух растворов других концен-
траций, причем один из имеющихся растворов крепче требуемого, другой
слабее. Чтобы быстро решить, какие объемы того и другого раствора нужно
смешать с целью получения требуемого раствора, прибегают к следующему
графическому способу.
34
Чертят два угла, обращенных вершинами один к другому. Между углами
следует написать требуемую концентрацию, напр. а. По концам сторон верх-
него угла пишут обе концентрации имеющихся растворов, напр. в и с:
b с
Пусть b > а и а > с. По концам сторон нижнего угла пишем разности а — с
и b — а. Эти разности указывают соответственно, какой объем раствора с
концентрацией b и с концентрацией с нужно взять для смешивания. Для
ясности нанесены стрелки.
В частном случае одна из смешиваемых жидкостей может являться чистым
растворителем (вода, эфир и т. п.); в этом случае на соответствующем месте
пишут О.
Пример 1. Нужно приготовить 60% спирт. Имеются в распоряжении 95 и 40%. Какие
количества того и другого растворов спрпта нужно смешать, чтобы получить требуемый?
Составив график, как указано выше, получаем:
Следовательно, нужно смешать 20 мл 95% спирта и 35 мл 40% спирта, чтобы получить
60%. В данном случае получится 55 мл 60% спирта. Если требуется больший объем, то для
смешивания берутся соответственно большие количества.
Пример 2. Требуется приготовить 80% спирт из 95% путем смешивания с водой. Соста-
вив график, получаем:
Следовательно, нужно взять 80 мл 95% спирта и 15 мл воды и смешать, чтобы получить
95 мл 80% спирта.
Хранение и очистка ртути
Ртуть должна храниться в закрытой посуде, ввиду того что установлены
ее способность испаряться при комнатной температуре и высокая токсич-
ность паров.
Так как во время работы ртуть часто загрязняется, ее приходится время
от времени очищать. Хорошая очистка ртути достигается следующим образом.
В толстостенную делительную воронку вводят ртуть, подлежащую очистке,
и приливают сюда же с избытком хромовую смесь (см. стр. 31), в которой
концентрация серной кислоты не должна превышать 15%. В течение несколь-
ких минут энергично взбалтывают ртуть с хромовой смесью, затем дают отсто-
яться. Ртуть выливают через кран, а хромовую смесь сливают через верх.
Тщательно промыв воронку, вводят в нее снова ртуть и промывают на этот
раз дестиллированной водой. Промывку дестиллированной водой повторяют
35
8—10 раз, причем каждый раз берут свежую порцию воды. Обычно такая
обработка ртути приводит к достаточной очистке. Однако для некоторых
целей (изготовление каломельного электрода и т. п.) необходимо очистку
дополнить еще промывкой ртути 5% раствором азотнокислой закиси ртути
(HgNO3), к которому прибавляют несколько капель 10% HNO3. Эта операция
приводит к освобождению ртути от посторонних металлов. После очистки
азотнокислой закисью ртути металлическую ртуть промывают еще несколько
раз дестиллированной водой, а затем для просушки пропускают через обыкно-
венный бумажный фильтр, помещенный в воронку. У кончика фильтра уко-
лом булавки делают небольшое отверстие. Такое фильтрование повторяют
2—3 раза до полной осушки ртути
Более совершенная очистка ртути достигается при помощи перегонки.
Получение и хранение дестиллированной воды
Дестиллированную воду в лабораториях получают при помощи перегон-
ных аппаратов. Устройство и применение их довольно просто и здесь не при-
водится. Для получения небольших количеств дестиллированной воды (3—5 л
в сутки) можно самому легко собрать перегонный аппарат (рис. 54).
Рис. 54. Прибор для получения небольших
количеств дестиллированной воды.
1 — колба для водопроводной (колодезной) воды,
2 — воронка для введения воды в колбу, 3 —
каплеуловитель, 4 — соединительная трубка, 5 —
холодильник, 6 — воронка, 7 — вата, 8 — колба
для дестиллированной воды.
Рис. 55. Бутыль с де-
стиллированной водой.
Дестиллированную воду со-
храняют в предназначенной для
этой цели бутыли, которую плот-
но закрывают каучуковой пробкой с двумя отверстиями. В одно отверстие
вводится сифон, в другое — трубка с хлопчатобумажной ватой для обеспы-
ливания воздуха, поступающего в бутыль. Для хранения дестиллированной
воды особенно пригодны бутыли, снабженные у основания трубкой и закры-
вающиеся сверху ватной пробкой, завернутой в марлю. Бутыль с дестилли-
рованной водой помещают на подставке, подвешенной к стене (рис. 55).
Чтобы получить особо чистую воду, перегоняют обыкновенную дестил-
лированную воду вторично, прибавив к ней несколько кристалликов КМПО4
и кусочек чистого NaOH для окисления органических веществ, которые могут
находиться в дестиллированной воде. Первые порции должны быть отброшены.
Получаемый затем дестиллят (aqua bis destillata) отличается большой чи-
стотой.
Зб
Дестиллированная вода должна быть бесцветной, без запаха и вкуса.
При испарении ее не остается ни малейшего осадка (проба на часовом стекле).
При прибавлении азотнокислого серебра, а также хлористого бария не дол-
жно получаться помутнения. Прибавление реактива Несслера не приводит
к появлению желтой окраски, а прибавление известковой воды — к помут-
нению.
Хранение резиновых предметов
Резиновые предметы (пробки, трубки) при хранении в обычных условиях
твердеют и становятся ломкими. Чтобы избежать этого, резиновые предметы
следует хранить в дестиллированной воде, к которой прибавляют немного
соды или аммиака.
Обработка стеклянных трубок
Рис. 56. Разогревание трубки перед
сгибанием.
В лабораторной практике при собирании различных установок нередко
приходится производить обработку стеклянных трубок. Приводим важнейшие
указания по этому вопросу.
Разрезывание трубок. При помощи
трехгранного напильника или специ-
ального ножа для стекла производят
царапину в том месте, где желают раз-
резать трубку. Режущее ребро напиль-
ника или острие ножа перед этим сма-
чивают водой. Затем трубку кладут на
край стола тем местом, где произведена
царапина, и легко надавливают на оба
конца трубки. Если царапина произве-
дена правильно, то по месту ее про-
исходит гладкий разлом трубки.
Оплавление острых краев трубки.
Конец трубки вводят в пламя горелки
и держат там до тех пор, пока стекло не начнет плавиться, а затем извлекают
из пламени.
Сгибание трубок. Взяв трубку обеими руками, как показано на рис. 56,
вводят ее в пламя горелки1 и начинают равномерно вращать. Когда
трубка начинает гнуться, ее извлекают из огня и производят желательный
изгиб.
Оттягивание концов трубки. Как и в предыдущей операции, трубку на-
гревают до размягчения, а затем, извлекши из пламени, растягивают в осе-
вом направлении. Когда трубка остынет, ее осторожно перерезают на оття-
нутом месте.
Запаивание трубки. Нагрев трубку до размягчения, быстро растягивают
ее, не извлекая из пламени, пока не произойдет запаивание концов.
Просверливание отверстия в боковой стенке трубки. Закрывают один
конец трубки и нагревают в пламени то место ее, где желают сделать отверстие.
Когда стекло начинает плавиться, дуют в открытый конец трубки.
Сверление пробок
Сверление пробок ведут с узкого конца пробки, так как со стороны, на
которую выходит сверло, получаются заусеницы и обрывы, могущие загряз-
нить жидкость при пользовании такой пробкой. При сверлении каучуковых
пробок сверло смачивают вазелиновым маслом или 10% едкой щелочью
для уменьшения трения между пробкой и сверлом и получения прямого
канала.
1 Пламя горелки стараются сделать по возможности широким (форма бабочки).
37
Некоторые рецепты
Здесь приводятся некоторые рецепты, полезные в лабораторной практике.
Замазки
Менделеевская замазка обыкновенно применяется для скрепления стекла с металлом.
Состав менделеевской замазки: 1000 ч. канифоли, 250 ч. желтого воска, 400 ч. безводной
окиси железа (Fe2O3, i .^аска «мумия») и от 3 до 10 ч. льняного масла. Сначала растапли-
вают в железной чашке i.а малом огне воск, затем прибавляют понемногу канифоль, помеши-
вая и продолжая нагревать, пока не спадет пена, которая при этом образуется. Далее при-
бавляют мумию и, наконец, масло.
Замазка из свинцового глета и глицерина. На одну весовую часть глицерина берут
В среднем до 10 весовых частей глета и замешивают в однородную густую массу. Поверхности,
подлежащие склеиванию, предварительно смазываит глицерином, а затем уже наносят слой
замазки.
Краска для надписей на стекле
12 ч. растворимого натриевого стекла (Вё30°), 15—18 ч. дестиллированной воды, 10 ч.
сернокислого бария и 1 ч. кремнекислоты смешивают и растирают до получения однородной
массы белого цвета. Для получения цветных красок добавляют в качестве красителей уль-
трамарин, сажу, сурик и т. п. Краска хорошо держится на стекле. Кремнекислота, применя-
ющаяся в данной краске, может быть получена путем обработки растворимого стекла соляной
кислотой (Na2SiO3 4- 2НС1 -> 2NaCl 4- Н2О 4- SiO2). Кремнекислота выпадает в коллоид-
ном состоянии. Ее отфильтровывают, промывают, просушивают и размельчают.
Карандаши для стекла
Карандаши для стекла имеются обыкновенно в продаже, однако иногда приходится
делать их самому по следующим рецептам.
Карандаш черный. В чашке расплавляют 40 ч. пчелиного воска и 10 ч. сала. В расплав-
ленную массу добавляют 10 частей ламповой сажи, все время помешивая. Полученную
массу в горячем виде выливают в бумажные цилиндрические формочки.
Карандаш красный. Готовится, как и черный, по следующему рецепту: воска пчелиного
25 частей, спермацета 100 ч., сала 15ч., сурика 150 ч.
Карандаш белый. Приготовляется из 20 ч. пчелиного воска, 30 ч. сала и 50 ч. окиси цинка.
Смазка для стеклянных кранов
12 ч. вазелина и 1 ч. парафина нагревают в выпаривательной чашке на песочной бане,
пока не образуется редкая масса, но без выделения дыма (температура смеси не выше 110°С).
В смесь небольшими порциями, при помешивании стеклянной палочкой, вносят 5 ч. чистого
невулканизированного каучука, причем каждая новая порция каучука вносится лишь после
того, как ранее внесенная порция целиком растворилась.
Полученная масса по охлаждении образует при растягивании между пальцами пучки
тончайших нитей. Если смесь получается очень вязкой, ее разбавляют вазелином, если же
опа очень жидка, то прибавляют еще каучука.
ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Весовой анализ (гравиметрия) предствляет собой метод
количественного определения тех или иных составных частей анализируемого
вещества при помощи взвешивания. Искомая составная часть выделяется из
общего вещества обыкновенно в виде нерастворимого осадка определенного
состава. Осадок собирается на фильтре с известным весом золы1, промывается,
высушивается вместе с фильтром, прокаливается в тигле до постоянного
веса, т. е. до такого момента, когда два последующие взвешивания не дадут
уже уменьшения в весе2, и затем взвешивается. Из полученного общего веса
вычитают вес тигля и золы фильтра и находят, таким образом, вес прокален-
ного осадка, а, зная его состав, определяют вес искомого вещества.
Из сказанного ясно, что основными моментами в весовом анализе явля-
ются: осаждение, фильтрование и промывание осадка, высушивание и про-
каливание, взвешивание.
1 Фильтры с известным весом золы, так называемые «беззольные фильтры», готовятся
у нас Моск, химико-техническим инет. им. Менделеева.
2 Допускаются колебания в весе не более чем в 0,2—0,3 мг.
38
Осаждение
Осаждение производится обыкновенной стаканах с носиками или в чашках;
для осаждения электролитическим способом применяется специальная пла-
тиновая посуда.
При операции осаждения необходимо строго придерживаться условий осаж-
дения данного вещества в отношении реакции, температуры, времени и т. п.
Полнота осаждения проверяется при помощи прибавления осаждающего
реактива к небольшой порции (одна-две капли) устоявшейся от осадка жид-
кости, перенесенной при помощи стеклянной палочки на часовое стекло.
Если при этой пробе на стекле появится муть, то осаждение не закончено,
в противном случае осаждение нужно считать законченным.
Если осаждение не закончено, то, смыв с часового стекла пробу в склянку,
продолжают прибавлять осаждающий реактив до полного осаждения соответ-
ствующего вещества.
Фильтрование и промывание осадка
Беззольный фильтр, представляющий собой кружок фильтровальной бу-
маги, складывают вдвое по диаметру; полученный полукруг вновь склады-
вают по радиусу. Затем отделяют один слой бумаги,
придав фильтру форму конуса, и вводят его в
воронку. Последняя должна быть такого размера,
чтобы края фильтра не доходили до ее краев.
Перед фильтрованием фильтр смачивают дестилли-
рованной водой и прилаживают к воронке так,
чтобы между ее стенками и фильтром не остава-
лось пузырьков воздуха. К фильтрованию жидко-
сти можно приступить лишь тогда, когда осадок
целиком осядет на дно. Не следует наливать филь-
труемую жидкость до краев фильтра; не следует
также прибавлять новую порцию жидкости на
фильтр, пока не отфильтруется вся предыдущая.
Перевод жидкости из стакана на фильтр рекомен-
дуется производить по стеклянной палочке, конец
которой упирается в фильтр (рис. 57).
Когда весь раствор отфильтрован, прибавляют
к осадку некоторое количество дестиллированной
воды, взбалтывают, дают осадку выпасть и снова
фильтруют. Такой способ промывания осадка назы-
вается декантацией. Не следует слишком
много промывать осадок, так как нет абсолютно не-
Рис. 57. Перевод фильтру-
емой жидкости из стакана
на фильтр.
растворимых осадков, и с каждой промывкой вода уносит хотя и незначитель-
ное количество самого осадка. Обыкновенно достаточно четырех-пятикрат-
ного промывания.
Полноту промывания осадка часто проверяют исследованием последней
порции фильтрата на наличие в нем реактива, которым велось осаждение.
Промывание считается законченным, когда этот реактив в промывных водах
больше не обнаруживается. Промыв осадок, переводят его на фильтр. Для
этой цели его вымывают из стакана струей воды, пущенной из шприца;
стеклянную палочку при этом кладут поперек стакана, придерживая ее
указательным пальцем левой руки, в которой держат стакан. Правой рукоь
направляют струю воды из шприца (рис. 58).
Остатки осадка в стакане тщательно собирают и перемещают на фильтр
при помощи каучуковой лопаточки либо такой же трубочки, насаженной
на стеклянную палочку, смывая эти остатки небольшим количеством воды.
Для трудно фильтрующихся жидкостей можно использовать колбу с отвод*
39
ной трубкой (см. рис. 18), соединенную с насосом; при этом под фильтр по-
мещают шутц (см. рис. 30).
Если имеют в виду произвести прокаливание осадка без бумажного филь-
тра1, то фильтрование производят при помощи тигля Гуча (см. рис. 29). На
дно тигля кладут небольшой слой
Рис. 58. Перевод осадка на фильтр.
крупноволокнистого, а затем мелковолок-
нистого асбеста, промытого горячей со-
ляной кислотой и водой. Над асбестом
помещают ситчатую пластинку и, со-
единив тигель с колбою, снабженной
отводной трубкой, а последнюю — с на-
сосом, промывают слой асбеста водою,
чтоб удалить мелкие волокна его, ко-
торые непрочно связаны с остальной
массой. Затем тигель вместе с фильтром
высушивают и определяют его посто-
янный вес. Через такой фильтр произ-
водят фильтрование так же, как через
бумажный фильтр, сначала слегка, а
затем сильно отсасывая фильтрат. Тут
же в фильтре производится промывание осадка. Один раз подготовленный
тигель с ситчатым дном может служить для целого ряда определений. Вместо
такого тигля можно с успехом использовать для той же цели тигель с фильтру-
ющим дном (см. рис. 31).
Рис. 59. Подготовка
фильтра для сжигания.
Высушивание и прокаливание
Прокаливание осадка производится после передварительного высушивания
его и отделения от фильтра или без этого. Предварительное высушивание
осадка должно производиться тогда, когда сжигаемый
фильтр может восстановить осадок. В таком случае
высушивают фильтр вместе с осадком в сушильном
шкафу при 100—105°С, осторожно отделяют осадок
от фильтра, собирая его на гладкой бумаге; отдельно
сжигают фильтр в платиновой спирали (рис. 59), а
затем, соединив в тигле золу фильтра вместе с осад-
ком, прокаливают до постоянного веса. Предвари-
тельно устанавливается постоянный вес одного тигля
после прокаливания и охлаждения его в экссикаторе.
Если фильтрование производилось в тигле Гуча,
то прокаливание осадка производится в нем же. При этом тигель устанавли-
вают в фарфоровый тигель большего размера, поместив между ними асбесто-
вое кольцо (рис. 60). Прокаливая наружный тигель, до-
стигают легкого прокаливания осадка.
Если не приходится опасаться восстанавливающего
действия на осадок сжигаемого фильтра, то можно про-
изводить прокаливание без предварительного отделения
осадка.
Фильтр высушивают сначала в сушильном шкафу при
100—105°С; высушивание заканчивают, когда фильтр
остается еще слегка влажным.
Затем фильтр вынимают из воронки и, не касаясь
руками низа фильтра, складывают его так, чтобы
осадок со всех сторон оказался окруженным бумагой.
Фильтр помещают в тигель, а последний в треугольник. Не закрывая
Рис. 60. Установка
тигля Гуча в дру-
гой тигель для про-
каливания.
1 Сжигаемый фильтр может в некоторых случаях восстанавливать осадок (галоидное
серебро, сульфат свинца и т._ д.).
40
тигля крышкой, приступают к прокаливанию на малом пламени горелки.
Когда фильтр обуглится, нагревание усиливают; при этом тигель помещают
в наклонном положении, неплотно прикрыв его крышкой, как показано на
рис. 61. Время от времени поварачивая тигель, нужно добиться полного
сгорания угля и побеления осадка. Когда прокаливание осадка закончится,
дают тиглю остыть, и еще теплым, но не горячим переносят в экссикатор-
и после охлаждения взвешивают. Полнота прокаливания проверяется повто-
рением той же операции до получения постоянного веса. Если из полученного
веса вычесть вес тигля и золы фильтра, то получится вес одного осадка.
В тех случаях, когда осадок при прокаливании изменяется независимо
от восстанавливающего действия сжигаемого фильтра (напр., K2PtCle), то огра-
ничиваются лишь высушиванием фильтра
вместе с осадком в сушильном шкафу и
последующим охлаждением в экссикаторе. От
веса фильтра с осадком отнимают вес одного
фильтра, предварительно высушенного при
той же температуре и взвешенного.
В этих случаях очень удобно пользоваться
тиглем Гуча вместо бумажного фильтра или
тиглем с фильтрующим дном.
Правила обращения с аналитическими
весами и способы взвешивания
В отношении весов необходимо твердо
придерживаться следующих правил:
1. При помощи винтов и ватерпаса или
отвеса, которые имеются при весах, послед-
ние должны быть правильно установлены на
неподвижной горизонтальной подставке. Раз
установленные, они не должны часто переме-
щаться.
2. Для уменьшения влажности воздуха
в шкаф весов ставят небольшую посуду с
концентрированной серной кислотой, пяти-
окисью фосфора или силикагелем.
3. В то время, когда весами не пользу-
Рис. 61. Положение тигля при
сжигании фильтра и прокаливании
осадка.
ются, они должны быть арретированы, т. е. находиться с приподнятым коро-
мыслом.
4. Взвешиваемое вещество ни в каком случае нельзя помещать непосред-
ственно на чашку весов, для этого специально предназначается посуда (бюк-
сы, часовые стекла и т. п.).
5. Взвешиваемое вещество в соответствующей посуде ставится на левую
чашку, гирьки — на правую (по возможности по средине чашки).
6. При взвешивании осторожным поворотом арретирного винта опускают
коромысло на подставку.
7. При каждом изменении груза на той или иной чашке, а также при пере-
мещении рейтера следует арретировать весы.
8. Гирьки подбирают по очереди, начиная с более крупных, а не наоборот.
При подборе разновесок не следует давать сильно качаться коромыслу: стрел-
ка весов не должна переходить за последнее деление шкалы.
9. Арретировать весы следует по возможности в момент прохождения
стрелки через среднее деление шкалы.
10. Посуду с веществом, которое взвешивают, нужно снимать с чашки
не руками, а соответствующими щипцами. Гирьки снимают пинцетом с ко-
стяными наконечниками, имеющимся при разновесе.
11. Класть или снимать груз на ту или иную чашку следует через боковые
41
ю 5 о 5 ю
Illi III! НН нн|
о 5 10 15 20
Ч......... -
Рис. 62. Шкала аналитических
весов.
дверцы. Передними дверцами для этой цели пользуются тогда, когда нет
боковых.
12. Во время взвешивания дверцы весов должны быть закрыты, чтобы
избежать влияния токов воздуха на качания коромысла с чашками.
13. Нельзя ставить на чашку весов предметы нагретые или сильно охлаж-
денные. Их следует предварительно поставить в экссикатор, чтоб придать
им комнатную температуру. Платиновая посуда при этом приобретает ком-
натную температуру за полчаса, стеклянная и фарфоровая — за час.
14. Нельзя взвешивать на аналитических весах груз, превышающий по
весу максимальную нагрузку для данных весов.
15. Регулировать весы нужно с большой осторожностью при помощи
специального груза, имеющегося на коромысле весов.
16. Весы следует сохранять в большой чистоте, защищать их от влияния
пыли, разъедающих газов и прямого солнечного света.
17. Гирьки и рейтер сохраняют в особом футляре, куда следует уклады-
вать их в соответствующие гнезда сейчас же после взвешивания.
Взвешивание на аналитических весах производится различно.
Взвешивание после предварительного отрегулирования весов. Наблюдают
качание стрелки ненагруженных весов. Если отклонения в правую и левую
стороны от среднего деления шкалы (рис. 62)
не одинаковы, то при помощи приспособле-
ния, имеющегося на коромысле весов (пере-
мещающийся груз), можно отрегулировать
весы, т. е. добиться одинакового отклонения
стрелки в правую и левую стороны от сред-
ней точки (состояние равновесия), а затем
приступить к взвешиванию. Взвешивание
считается законченным, когда стрелка нагру-
женных весов вновь дает одинаковые отклонения вправо и влево.
Взвешивание с поправкой. Рейтер помещают на коромысло весов так,
чтобы отклонения стрелки ненагруженных весов в правую и левую стороны
от средней точки были одинаковы, затем взвешивают груз, как и в преды-
дущем случае, и в найденный вес вводят соответствующую поправку.
Пример. Одинаковые отклонения стрелки ненагруженных весов вправо и влево наблю-
дались при стоянии рейтера на правом плече коромысла на делении 0,3. При взвешивании
тигля одинаковые отклонения стрелки вправо и влево получались при стоянии на правой
чашке весов груза в 8,35 г и положении рейтера на правом плече коромысла в точке 3,7.
Отсюда находим вес тигля равным 8,3537 — 0,0003 = 8,3534.
Взвешивание после нахождения нулевой точки. Под нулевой точкой весов
понимают то деление шкалы, которое находится в середине всей амплитуды
отклонений (колебаний) стрелки весов. Для определения нулевой точки
наблюдают качания ненагруженных весов. Первые два качания не записы-
вают, как могущие быть неправильными, вследствие сотрясения. Затем запи-
сывают результаты пяти качаний, из коих и выводится нулевая точка, как
среднее арифметическое отклонение стрелки.
Пример. Положим, что три последовательных качания влево, считая от крайнего пер-
вого деления слева, дали результаты 5,4, 5,6 и 5,8 (среднее арифметическое 5,6), а два кача-
ния вправо дали последовательно 14,9 и 14,7 (среднее арифметическое 14,8). На основании
5,6+ 14,8
этих данных находим нулевую точку равной --= Ю,2.
Нулевая точка весов не должна значительно отличаться от 10, находясь
в пределах между 9 и 11 делениями, в противном случае весы должны быть
отрегулированы.
Нулевая точка для неотрегулированных весов играет ту же роль, что
и средняя точка шкалы для отрегулированных весов: отвешивание считается
законченным, если отклонения стрелки вправо и влево от нулевой точки
будут одинаковы.
42
Само взвешивание производится следующим образом: при поднятом арре-
тире помещают взвешиваемый предмет на левую чашку весов, а гирьки раз-
новеса в приблизительно равном весе на правую чашку, после чего пробуют
очень медленно опустить арретир так, чтобы призмы мягко соприкоснулись
с пластинками, и наблюдают состояние стрелки весов. Всякое изменение
гирек можно производить лишь при арретированных весах. Гирьки помещают
с таким расчетом, чтобы постепенно от больших перейти к меньшим. Для
однозначного числа миллиграммов и десятых долей их пользуются обыкно-
венно рейтером. Вес груза определяют по гирькам разновеса и точке стояния
рейтера. Запись разновесов, положенных на чашку, делают сначала по пустым
гнездам, а затем проверяют при снимании с чашки весов и укладывании на
место.
Кроме взвешивания на весах того или иного груза нередко приходится
отвешивать на них определенные навески данного вещества, напр., при
изготовлении титрованных растворов.
Для отвешивания на весах определенной навески можно поступить так:
в соответствующую посуду (бюкса, часовое стекло и т. п.) насыпают данное
вещество, уравновешивают его, снимают часть разновесок соответственно
необходимой навеске, а затем из отвешенного вещества отсыпают в бюксу
или на часовое стекло столько, чтобы весы пришли вновь в состояние равно-
весия. Вес отсыпанного вещества соответствует требуемой навеске.
Можно поступить и так: сначала уравновесить (протарировать) посуду,
а затем, положив на правую чашку весов гирьки, соответственно необходимой
навеске насыпать в тарированную посуду столько вещества, чтобы весы вновь
пришли в состояние равновесия.
Если можно ограничиться взятием какой-либо точной навески, не задан-
ной наперед, то отвешивание часто производят по разнице. Отвешивают
некоторое количество препарата вместе с бюксой, затем отсыпают из бюксы
в склянку некоторое количество его и снова взвешивают бюксу с остатком
препарата. Разница между результатами первого и второго взвешивания
соответствует весу отсыпанного вещества.
Правила обращения с микроаналитическими весами
и способы взвешивания
Все правила обращения, указанные для обыкновенных аналитических
весов, приложимы и к микроаналитическим весам, в отношении которых
требуются еще большая осторожность и аккуратность.
Температура воздуха в весовой комнате должна быть в пределах 18—20°С.
Температурные колебания не должны превышать 1°С. Внутри шкафа весов
за время взвешивания температура не должна колебаться более чем на
0,1 — 0,2°С. Наблюдение за температурой производят по термометру, поме-
щенному внутри шкафа.
Чтобы уменьшить влияние на весы тепловых лучей, идущих от наблю-
дателя, рекомендуется уравновесить взвешиваемый предмет приблизительно
и отойти от весов на несколько минут, затем вновь подойти к весам и быстро
закончить взвешивание.
Нужно тщательно следить за чистотой весов, так как малейшее загряз-
нение (запыление) их может сделать неточным взвешивание.
Нулевая точка микроаналитических весов может меняться от многих
причин и потому ее следует проверять по микрошкале перед каждым взвеши-
ванием, что производится так же, как в обыкновенных аналитических весах.
Взвешивание на микроаналитических весах производится следующим
образом: сначала отвешивают данный груз при очень небольшой недогрузке
и вычисляют точку равновесия при недогрузке ве-
сов — nv производя нечетное число отсчетов отклонений стрелки колеблю-
щихся весов точно так, как при нахождении нулевой точки весов.
43
Затем таким же образом определяют точку равновесия при
перегрузке весов — л2.
п1 должна быть менее нулевой точки весов, а п2 — более нулевой точки.
Вес предмета находят по формуле:
Р = Pi + (Рг — А),
где р — искомый вес предмета,
Pi — вес разновесок вместе с показанием рейтера при недогрузке,
р2 — то же при перегрузке,
л0— нулевая точка,
— точка равновесия при недогрузке,
л2— то же при перегрузке.
Пример. Нулевая точка микроаналитических весов — равна 49,2. При взвешивании
груза с недогрузкой на правой чашке весов находилось 5,28 г; рейтер стоял на 46 зарубке,
т. е. вес с недогрузкой — рг = 5,2846.
При определении точки равновесия nY получены следующие отсчеты:
влево —64,5; 64,4; 64,3, отсюда среднее 64,4,
вправо —24,2; 24 ,2; отсюда среднее 24 ,2.
Следовательно,
64,44-24,2
п1 =----~2--— 44,3.
Передвигаем рейтер вправо на одно деление; при этом р2 = 5,2847. Определяя л2, полу-
чаем следующие отсчеты:
влево — 78,6; 78,5; 78,4; отсюда среднее 78,5,
вправо — 28,5; 28,5; отсюда среднее 28,5.
Следовательно,
78,5 +28,5
— 2 — Эо,5.
Подставляя в приведенную выше формулу соответствующие значения, получаем:
49,2 — 44,3
р = 5,2846 + 535 — 443 (5,2847 — 5,2846) = 5,284653 г.
ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Объемный анализ представляет собой один из видов количественного
анализа. Сущность его заключается в том, что раствор данного вещества,
подлежащего количественному определению, приводят в химическое взаимо-
действие с точно отмеренным объемом другого вещества известной концент-
рации (титра) и по количеству титрованного раствора, пошедшего в реакцию,
определяют искомое количество вещества на основании стехиометрических
расчетов. Методы, применяемые в объемном анализе, можно разделить на
три группы: метод нейтрализации, метод окисления и вос-
становления, метод осаждения (и комплексообразования).
Предварительные сведения
Прежде чем перейти к изложению основных методов объемного анализа,
приведем некоторые предварительные сведения.
Понятие о титрованных растворах
Титрованным раствором называется раствор строго определенной
концентрации.
Титром раствора называют количество вещества (в граммах) в 1 мл
раствора.
44
Различают нормальные, молярные и эмпирические
титрованные растворы.
Нормальным раствором называется такой, в литре которого
растворен один грамм-эквивалент данного вещества, т. е. такое
количество вещества в граммах, которое в данной реакции эквивалентно
одному грамм-атому водорода (Н= 1,008 г).
Тысячная доля грамм-эквивалента представляет миллиграмм-э к-
вивалент.
Грамм-эквивалент кислоты определяется путем деления грамм-молекулы кислоты на
число атомов водорода молекулы ее, которые принимают участие в реакции.
Пример:
НС1 + NaOH = NaCl + Н2О.
H2SO4 + 2NaOH = Na28O4 + 2H2O.
„ v HC1 36,46
Согласно первой реакции грамм-эквивалент для НС1 равен -у- = —j— = 36,46 г.
л H2SO4 98,086
Согласно второй реакции грамм-эквивалент для H2SO4 равен —— = —2—=49,043 г.
Грамм-эквивалент щелочи (едкой) определяется путем деления грамм-молекулы ще-
лочи на число гидроксильных групп (ОН) молекулы, участвующих в реакции.
Пример:
NaOH + HCI = NaCl + Н2О.
Ва(ОН)2 + H2SO4 = BaSO4 + 2Н2О.
„ л НаОН 40,0048
Согласно первой реакции грамм-эквивалент для NaOH равен —।।— =40,0048 г.
_ - г. Ва(ОН)2 171,3756
Согласно второй реакции грамм-эквивалент для Ва (ОН)2 равен —у— =---2--- =
= 85,6878 г.
Грамм-эквивалент соли в большинстве случаев определяется путем деления грамм-
молекулы соли на число атомов металла в молекуле ее и на его валентность.
NaCl 58,484
Пример: грамм-эквивалент для NaCl равен —j— = —j— = 58,454 г,
ВаС12 208,274
для ВаС12 равен —2— —----2— = 104,137 г,
A12(SO4)3 342,12
для A12(SO4)3 равен 2 . 3 — —§— = 57,02 г и т* д*
Грамм-эквивалент вещества, участвующего в реакции как окислитель, носит название
окислительного грамм-эквивалента. Окислительный грамм-эквивалент
данного вещества определяется по количеству граммов этого вещества, отдающему при
реакции окисления один грамм-эквивалент кислорода, т. е. 8 граммов кислорода.
Пример: 2КМпО4 + 3H2SO4 = K2SO4 + 2MnSO4 + ЗН2О + 5(0).
Из реакции видно, что 2 грамм-молекулы КМпО4, раскисляясь в кислой среде, отдают
Ю грамм-эквивалентов кислорода1. Следовательно, грамм-эквивалент КМпО4 как окисли-
2КМпО 2 • 158,03 Q1 _
теля согласно данной реакции равен —pg—=•---jg— =31,606 г.
Грамм-эквивалент вещества, участвующего в реакции как восстановитель, носит на-
звание восстановительного грамм-эквивалента.
Восстановительный грамм-эквивалент равен количеству граммов данного восстанови-
теля, потребляющему при реакции один грамм-эквивалент кислорода.
Пример: 5Н2С2О4 . 2Н2О + 5(0) = ЮСО2 + 15Н2О
Из реакции видно, что при окислении щавелевой кислоты 5 грамм-молекул ее потребляют
10 грамм-эквивалентов кислорода. Отсюда заключаем что восстановительный грамм-экви-
5 • Н2С2О4 • 2Н2О 5 • 126,0468
валент щавелевой кислоты равен: -----^д-----=------Jq---- = 63,U234 г.
Грамм-эквивалент не является постоянной величиной для одного и того же вещества,
а зависит от хода реакции, в которой оно принимает участие.
Пример 1:
Na2CO3 + НС1 = NaCl = NaHCO3,
Na2CO3 + 2HCI = 2NaCl + CO2 + H2O.
1 Один грамм-атом кислорода соответствует двум грамм-эквивалентам кислорода.
45
Если реакцию нейтрализации соды кислотой вести при индикаторе фенолфталеине, кото-
рый к NaHCO3 относится как к веществу нейтральному, то реакция заканчивается с обра-
зованием NaCl и NaHCO3 (см. первую реакцию). В этом случае грамм-эквивалент для Иа2СОз
Na2CO3 105,994
равен —j— = —j = 105,994 г. Если же реакцию нейтрализации соды кислотой вести
при индикаторе метилоранже, который к NaHCO3 относится как к щелочи, то реакция
нейтрализации заканчивается с образованием NaCl, СО2 и Н2О (см. вторую реакцию).
Na2CO3 105,994
В этом случае грамм-эквивалент для Na2CO3 равен —---= —2—=52,997 г.
Пример 2:
2КМпО4 = К2О + 2МпО + 5(0).
2КМпО4 = К2О 4- 2МпО2 + 3(0).
Если окисление какого-либо вещества вести перманганатом в кислой среде, то перманганат
раскисляется по схеме, изображенной первой реакцией. В этом случае его грамм-эквивалент
2КМпО4 2 . 158,03
равен —jo— =------pg--- = 31,606 г. Если же окисление вещества вести перманганатом
в щелочной среде, то оно протекает по схеме, изображенной второй реакцией. В этом случае
2КМпО4 2 . 158,03
грамм-эквивалент перманганата равен —g---=------g — = 52,6766 г.
Нормальные растворы для аналитических целей весьма крепки, ввиду чего готовят
более слабые растворы: в десять раз (децинормальные), в сто раз (сантинормальные), реже
еще слабее. Нормальные растворы обозначаются символами 1,ON либо 1,Оп, либо 1, ОН,
децинормальные — символами 0,1 N либо 0,1 и, либо 0,1 Н, сантинормальные — 0,01 N,
либо 0,01 и, либо 0,01 Н и т. д.
Молярным раствором называется такой, в литре которого растворена
одна грамм-молекула данного вещества. Молярные растворы реже приме-
няются в анализе, нежели нормальные.
Эмпирическим раствором называется такой, в котором навеска
растворенного вещества не связана правильным отношением с грамм-эквива-
лентом либо грамм-молекулой.
Понятие об индикаторах
Индикаторы представляют собой вещества, применяемые в объемном ана-
лизе с целью установления конца реакции между титруемым и титрованным
растворами, отсюда и само наименование этих веществ: «индикаторы», т. е.
«указатели» (конца реакции), от латинского indicare — указывать.
Конец реакции определяется по изменению цвета индикатора, которое
может возникать от самых разнообразных причин.
Простейший случай изменения окраски индикатора можно видеть на при-
мерах реакции осаждения: так, при титровании хлоридов в качестве инди-
катора берется хромовокислый калий (К2СГО4), имеющий в растворе лимонно-
желтый цвет. Конец реакции определяется по переходу этого цвета в кир-
пичнокрасный. Переход окраски возникает в силу образования к концу тит-
рования хромовокислого серебра (Ag2CrO4).
Здесь, следовательно, изменение цвета индикатора возникает в связи
с заменой его катиона другим катионом.
Аналогично объясняется изменение окраски железоаммиачных квасцов
[(NH4)2SO4Fe2(SO4)3 . 24Н2О], применяемых в качестве индикатора при тит-
ровании растворов солей серебра роданистым аммонием. Практически бес-
цветный раствор железоаммиачных квасцов при взаимодействии с роданистым
аммонием образует к концу титрования яркокрасного цвета родановое же-
лезо [Fe(CNS)3], окрашивающее раствор в красный цвет.
Здесь, следовательно, изменение окраски возникает в силу замены аниона
одного из компонентов индикатора другим анионом.
Применяемый в иодометрии индикатор крахмальный клейстер в присут-
ствии самых ничтожных количеств свободного иода дает синее окрашивание
в связи с образованием соединения иод-крахмал.
Таковы факты, лежащие в основе применения указанных выше индика-
торов.
46
Совершенно иное объяснение дается изменению цвета индикаторов, при-
меняемых в реакциях нейтрализации, а также при колориметрическом опре-
делении pH растворов.
Все эти индикаторы могут быть разделены на две группы: одноцветные
и двухцветные.
Одноцветные индикаторы, как, напр., фенолфталеин, нитрофенолы и др.,
при низких значениях pH не имеют окраски. При определенном значении
pH они приобретают окраску, которая усиливается с повышением pH, однако
до определенного предела, выше которого интенсивность окраски уже не
возрастает.
Двухцветные индикаторы, как, напр., метилоранж, метилрот, лак-
мус и др., в пределах определенного интервала значений pH спо-
собны менять свою окраску, причем каждый индикатор характеризуется
определенным интервалом значений pH, в пределах которого индикатор ме-
няет свою окраску. Ниже и выше определенного значения pH окраска инди-
катора не меняется.
Получение окраски одноцветным индикатором и изменение окраски двух-
цветным индикатором в зависимости от определенных значений pH объясняется
следующим образом.
Цветные индикаторы представляют собой либо слабые кислоты, либо
слабые основания, причем одноцветные индикаторы в недиссоциированном
(молекулярном) состоянии не окрашены, а в диссоциированном приобретают
окраску; двухцветные индикаторы в недиссоциированном (молекулярном)’
состоянии имеют одну окраску, а в диссоциированном—другую.
Изобразим диссоциацию индикаторов схематически равновесными уравне-
ниями:
Н Ind Ind'4-Н'
Молекулярная форма Ионная форма
индикатора —кислоты индикатора—кислоты
Ind ОН Ind’4-ОН'
Молекулярная форма Ионная форма
индикатора—основания индикатора—основания
Так как ни водородные, ни гидроксильные ионы, образующиеся при дис-
социации индикаторов, не имеют окраски, то приобретение одноцветным-
индикатором определенной окраски либо изменение окраски двухцветного
индикатора должно быть отнесено за счет образующихся при диссоциации
анионов или катионов индикатора: Ind'и Ind‘.
Очевидно, что степень диссоциации1 индикатора должна влиять на харак-
тер его окраски. Но степень диссоциации индикатора в свою очередь обуслов-
ливается pH той среды, в которой индикатор растворен. Следовательно, при-
обретение одноцветным индикатором окраски и изменение окраски двух-
цветного индикатора зависят от pH среды, в которой индикатор растворен.
Чем большую константу диссоциации имеет кислотный индикатор, тем
более высокая концентрация Н-ионов потребуется для того, чтобы подавить
его диссоциацию и вызвать переход окраски; то же можно сказать в отноше-
нии щелочных индикаторов и ОН-ионов.
Из сказанного ясно, что различные одноцветные индикаторы приобретают
и усиливают интенсивность окраски в различных пределах pH, которые мо-
гут довольно далеко отклоняться от нейтральной точки как в кислую, так
и в щелочную стерону. То же касается и двухцветных индикаторов, которые
в различных пределах значения pH меняют свою окраску. Так, из одноцветных
индикаторов фенолфталеин дает переход в пределах pH 8,3—10,0; а-дву-
нитрофенол — в пределах pH 2,8 — 4,4; из двухцветных индикаторов
тимоловый синий — в пределах pH 7,9 — 9,8, а метиловый красный — в
пределах pH 4,2 — 6,3.
1 Напомним, что под степенью диссоциации электролита а понимают отношение диссо-
цнированных молекул к общему числу растворенных молекул.
47
Значения pH, в пределах которых индикатор меняет свою окраску, со-
ставляют интервал или зону изменения окраски инди-
катора. Эти зоны могут лежать и в кислой и в щелочной среде, а иногда за-
хватывают и ту и другую.
Значения pH, при которых степень диссоциации индикатора равна поло-
вине (50%), носят название точки перехода индикатора.
При титровании эта точка носит название показателя титрова-
ния. Лишь немногие индикаторы имеют точку перехода при pH = 7 или
близко к этой величине; большинство же имеет точку перехода в кислой или
щелочной среде.
К сказанному необходимо добавить, что одна лишь диссоциация молекул
индикатора не в состоянии объяснить полностью наблюдаемые изменения
окраски. Имеются основания считать, что еще до диссоциации молекул ин-
дикатора они в связи с внутренней перегруппировкой атомов переходят из
одной таутомерной формы (неокрашенной) в другую (окрашенную), а эта
последняя, диссоциируя, дает окрашенные ионы. Так, напр., лактонная
форма бесцветных молекул фенолфталеина переходит сначала в хиноидную
форму окрашенных молекул, а эти последние дают окрашенные ионы:
Фенолфталеин
(лактонная форма)
Бесцветные молекулы
Фенолфталеин
(хиноидная форма)
Окрашенные молекулы
Фенолфталеин
(хиноидная форма)
Окрашенные аниионы
Весьма важно пользоваться в работе чувствительными индикаторами.
От чувствительного индикатора, применяемого в реакциях осаждения,
требуется, чтобы он давал отчетливый переход окраски раствора при самом
незначительном избытке титрованного (рабочего) раствора.
От чувствительного индикатора, применяемого в реакциях нейтрализации
и при колориметрическом определении pH, требуется, чтобы он давал отчет-
ливое изменение окраски в пределах определенного небольшого ин-
тервала кислотности или щелочности среды. Для реакций нейтрализации
важно еще, чтобы указанный интервал находился вблизи абсолютно ней-
тральной точки.
Способ изготовления и выбор индикаторов для того или иного случая
приведены далее.
Техника титрования
Титрование представляет собою операцию, при которой титруемый раст-
вор приводится во взаимодействие с титрованным (рабочим) раствором, при-
чем этого последнего берется столько, сколько нужно, чтобы реакция между
титруемым и титрованным растворами прошла до конца.
Имея точные бюретку и пипетку, а также соответствующие для данного
случая титрованный раствор и индикатор, можно произвести титрование.
Для этого отбирают пипеткой 25—50 мл титруемого раствора и вводят
в коническую колбу, прибавляют сюда же индикатор, наполняют бюретку
титрованным раствором и титруют из бюретки до эквивалентной
48
т о ч к и, т. до того момента, когда наступит конец реакции, что опреде-
ляется по изменению цвета индикатора, выпадению осадка и т. п.
Эквивалентная точка при титровании далеко не всегда совпадает с ней-
тральной точкой; так, при титровании слабой кислоты сильной щелочью в
силу гидролиза образующейся соли (щелочной реакции) эквивалентная точка
лежит в области щелочных растворов; напротив, при титровании слабых
щелочей сильными кислотами в силу гидролиза образующейся соли (кислой
реакции) эквивалентная точка лежит в области кислых растворов.
Количество прибавляемого при титровании индикатора должно соответ-
ствовать тому, которое указывается для каждого индикатора в отдельности
(см. далее). Избыточное прибавление индикатора ведет к уменьшению точ-
ности титрования.
При титровании рабочий раствор нужно приливать небольшими порциями,
взбалтывая осторожно жидкость в колбе. Скорость выхождения жидкости
из бюретки должна быть такова, чтобы мениск опускался на 1 см по длине
шкалы не быстрее, чем в 1—2 сек. Под конец титрования раствор прибавля-
ется отдельными каплями. На основании высоты стояния уровня жидкости
в бюретке в начале и к концу титрования определяют расход титрованного
раствора, а затем при помощи стехиометрических расчетов находят коли-
чество вещества в титруемом растворе (примеры стехиометрических расчетов
приведены далее).
Каждое титрование следует начинать с нулевого деления шкалы, благо-
даря чему уменьшается сшибка за счет неточности калибровки. Титрование
следует заканчивать по возможности содержимым одной бюретки. Титрова-
ние повторяют обыкновенно три раза и из результатов трех определений
берут среднее арифметическое.
Для точного определения момента эквивалентности (конца реакции) по
изменению цвета индикатора проводят так называемый холостой (слепой,
глухой) опыт, с целью выяснения количества титрованного раствора, которое
уходит на титрование самого индикатора. Для этого берут такое количество
дестиллированной воды, которое по объему равно сумме объемов взятого для
титрования раствора и расходуемого титрованного раствора, прибавляют
сюда столько же капель индикатора, сколько и в главном опыте, и титруют
до изменения цвета. Ушедшее при этом титровании количество титрованного
раствора вводят как корректив в главный опыт.
Пример. Допустим, что в главном опыте на 25 мл щелочи пошло 25 мл кислоты при
индикаторе метилоранже. Холостой опыт показал, что при 50 мл дестиллированной воды
с двумя каплями метилоранжа изменение цвета индикатора наступает от двух капель кис-
лоты. Учитывая, что объем одной капли равен 0,03 мл, вводим корректив в главный опыт
на основании результата холостого опыта в размере 0,03 х 2 = 0,06 мл, т. е. считаем,
что на нейтрализацию щелочи пошло не 25 мл кислоты, а 25 — 0,06 = 24,94 мл.
Холостой опыт полезно провести до главного опыта и титрование в глав-
ном опыте вести до той же окраски жидкости, как и в холостом опыте. Таким
образом, окрашенная жидкость холостого опыта служит, как выражаются,
«свидетелем» титрования в главном опыте.
Порядок титрования зависит в основном от того, какой индикатор приме-
няется при титровании. Напр., если индикатором служит метилоранж, то
лучше титровать щелочь кислотой или, как говорят, «титровать от щелочи
к кислоте», так как при этом легче проследить изменение окраски индика-
тора: человеческий глаз легче улавливает переход желтой окраски в розовую,
нежели наоборот.
Применяя в качестве индикатора фенолфталеин, лучше вести титрование
«от кислоты к щелочи», так как легче проследить появление розовой окраски
жидкости, нежели исчезновение этой окраски.
Наконец, следует заметить, что для получения точных результатов титро-
вания необходимо стремиться к тому, чтобы определяемый раствор по кон-
центрации был близок к титрованному раствору.
4 А. И, Бурштейн
49
Общие принципы приготовления титрованных растворов
Техника приготовления титрованных растворов сводится к следующему.
Приготовляют сначала так называемые основные или исходные тит-
рованные растворы, а по ним уже все другие. Таким основным раствором
в методе нейтрализации может служить раствор химически чистого углекислого
натрия (Na2CO3) или буры (Na2B4O7 . 10Н2О), в иодометрии — раствор хи-
мически чистого иодноватокислого калия (KJO3) и т. д.
Вещество, применяемое для приготовления исходного титрованного ра-
створа, должно удовлетворять следующим требованиям: 1. Оно должно быть
химически чистым. 2. Должно быть устойчивым на воздухе. 3. Должно хорошо
растворяться в воде и быть достаточно устойчивым в растворе, благодаря
чему раствор не меняет своего титра в течение более или менее продолжи-
тельного времени. 4. Оно должно иметь возможно больший эквивалентный
вес. Чем больше эквивалентный вес, тем большую навеску вещества прихо-
дится брать для установки титра и тем меньше относительная ошибка при
взвешивании. 5. Оно должно реагировать с раствором, титр которого уста-
навливается по строго определенному уравнению и с относительно большой
скоростью.
Имея исходные растворы, легко готовить другие. При этом, как правило,
поступают так. Приготовляют раствор несколько более крепкий и в несколько
большем количестве, нежели это требуется. Устанавливают его концентра-
цию по исходному раствору и вычисляют, сколько требуется прибавить воды
(алкоголя), чтобы получить заданный раствор. Для этой цели удобно пользо-
ваться колбой с расширением на шейке (см. стр. 19). Приготовив в ней до
другой черты 1100 мл раствора, расходуют 50—75 мл для установления тит-
ра. Отбирают пипеткой остаток раствора до первой черты, а затем прибав-
ляют из бюретки соответствующее количество воды и взбалтывают раствор.
Пример. Положим, что при изготовлении 0,1 N раствора HCI оказалось, что для центра-
лизации 20 мл 0,1 N раствора Na2CO3 уходит 18,5 мл кислоты. Следовательно, на каждые
18,5 мл кислоты нужно прибавить 1,5 мл дестиллированной воды, чтобы получить точно
1,5-1000 „
0,lN раствора. На литр это составит —j-g-g—^81,1 мл.
Отобрав пипеткой из колбы всю кислоту до первой черты, прибавляют к оставшемуся
литру раствора из бюретки точно 81,1 мл воды и взбалтывают, получая таким образом
раствор нужной концентрации.
Если в лаборатории не имеется колбы с расширением на шейке, то можно
приготовить раствор, как описано выше, воспользовавшись любой посудой
емкостью более литра; затем, установив титр приготовляемого раствора,
отбирают при помощи колбы на выливание точно один литр раствора, пере-
водят его в склянку с притертой пробкой, прибавляют из бюретки сюда же
точно необходимое количество дестиллированной воды, закрывают склянку
стеклянной пробкой и взбалтывают жидкость в склянке, получая таким об-
разом нужный раствор. Приготовляя титрованные растворы, пользуются
мерной посудой, рассчитанной для нормальной температуры 20°С. Если тем-
пература раствора выше или ниже нормальной, то необходимо ввести
поправку для получения необходимого раствора. Приведенная выше
таблица 1 (стр. 12) дает возможность рассчитать поправку.
Пример. Положим, что при изготовлении литра какого-либо 0,1 N раствора послед-
ний оказался температуры 25°С. Колба рассчитана для 20°С. Требуется найти поправку.
Согласно таблице вес литра дестиллированной воды 25°С, приведенный к нормальной тем-
пературе 20°С, равен 996,15 г, тогда как литр воды при 20°С имеет вес 997,18 г, т. е. на
997,18—096,15 — ] ,03 г больше. Отсюда заключаем, что наш раствор, доведенный до нормаль-
ной температуры (20°), будет на 1,03 мл меньше литра, следовательно, он станет крепче и,
чтобы выправить его, необходимо к нему прибавить 1,03 мл воды.
Взяв дестиллированную воду температуры близкой к нормальной (20°С),
можно пренебречь указанной поправкой.
В лабораториях очень часто пользуются растворами не точно нормаль-
ными, децинормальными, сантинормальными и т. д.; в таких случаях опре-
50
деляют коэфициент нормальности или к о э ф и ц и е н т
поправки раствора, т. е. отношение титра данного раствора к титру
раствора соответствующей точной нормальности. Эта величина обозначается
буквой К и должна быть указана на этикетке, имеющейся на бутыли, в ко-
торой хранится раствор1.
Коэфициент нормальности легко рассчитать на основании разультатов
титрования по формуле:
/< = —,
где К — коэфициент нормальности,
v — объем раствора точной нормальности,
v1—объем раствора, коэфициент нормальности которого определяется.
Пример 1. При проверке концентрации едкого натрия по 0,1 N кислоте оказалось,
что на 25 мл кислоты уходит 24,8 мл щелочи. Коэфициент нормальности едкого натрия
25
в данном случае окажется равным L00806. На этикетке обозначаем: 0,1 N NaOH К=
= 1,00806.
Пример 2. При проверке концентрации перманганата по 0,01 N щавелевой кислоте
оказалось, что на 25 мл щавелевой кислоты уходит 26,1 мл перманганата. Коэфициент нор-
25
мальности перманганата в данном случае окажется равным ~ 0,95785.
На этикетке обозначаем 0,01 N КМпО4К = 0,95785.
Для того чтобы пересчитать любой объем данного раствора с определен-
ным коэфициентом нормальности на соответствующий раствор точной нор-
мальности, нужно этот объем помножить на коэфициент нормальности.
Пример. При титровании некоторого количества кислоты 0,1 N щелочью с коэфициентом
нормальности, равным 0,99876, израсходовано 34,6 мл щелочи. Отсюда заключаем, что
при титровании этого же количества кислоты потребуется 34,6 мл X 0,99876 = 34,557 мл
0,1 N щелочи (приготовленной точно).
Вместо того, чтобы обозначать концентрацию не вполне точных растворов,
как деци, санти и т. д. нормальных с соответствующим коэфициентом нор-
мальности, можно указывать просто нормальность таких растворов,
т. е. число эквивалентов вещества или долей эквивалента в 1 л раствора.
Так в приведенном выше примере вместо того, чтобы титр едкого натрия
обозначать как 0,1 N-ный с коэфициентом нормальности 1,00806, можно ука-
зать нормальность этого раствора N = 0,1 • 1,00806 = 0,100806.
Тоже для перманганата в приведенном выше примере вместо того, чтобы
обозначать его титр как 0,01 N-ный с коэфициентом нормальности 0,95785,
можно указать нормальность этого раствора N = 0,01 • 0,95785 = 0,0095785.
Нормальность раствора должна быть выражена не менее как четырьмя
значащими цифрами. Нули, стоящие впереди десятичной дроби, не прини-
маются в расчет.
Капсулы «фиксанал». Для быстрого изготовления точных растворов можно
пользоваться имеющимися в продаже капсулами «фиксанал».
Капсула «фиксанал» представляет собой пробирку, в которой находится
точно десятая часть грамм-эквивалента данного вещества (реже грамм-экви-
валент, сантиграмм-эквивалент). Пробирка запаяна с обоих концов. На
одном из концов пробирки и сбоку у края другого конца имеются вдутия
внутрь пробирки, где стенка значительно тоньше, нежели в других местах
пробирки.
Для приготовления титрованного раствора данного вещества берут мерную
колбу емкостью в один литр, помещают в нее воронку 2, а в шейку воронки
х Коэфициент нормальности должен быть выражен до четвертого - пятого десятичного
знака.
2 Для этой цели рекомендуется пользоваться специальными воронками с расширенной
сверху шейкой.
51
нои у края, дают жидкости
Рис. 63. Вымывание содержи-
мого из пробирки «фиксанал».
вставляют специальную стеклянную вставочку, которая служит для проби-
вания капсулы.
На эту вставку опускают предварительно промытую снаружи дестилли-
рованной водой и просохшую пробирку «фиксанала» так, чтобы концевое
вдутие приходилось над вставкою. Легким надавливанием разбивают про-
бирку, жидкость при этом частью выливается в воронку, а отсюда в колбу;
если препарат твердый, то он высыпается в воронку; после этого пробивают
верхнее боковое вдутие пробирки при помощи стеклянной палочки, заострен-
или твердому веществу полностью выйти из про-
бирки и тщательно промывают капсулу изнутри,
вдувая в нее воду из шприца (рис. 63). Опола-
скивают пробирку снаружи, вынимают воронку
из колбы, доводят раствор до литра дестилли-
рованной водой и взбалтывают. Титрованный
раствор готов.
Хранение титрованных растворов
Титрованные растворы сохраняются в склян-
ках с хорошо притертыми стеклянными проб-
ками либо с каучуковыми пробками. На склянку
наклеивается этикетка с указанием названия
реактива, времени его изготовления и его титра.
Перед тем как применять титрованный раствор,
следует его взболтать, чтобы придать ему одно-
родный состав, так как во время стояния влага
конденсируется на свободной поверхности сосуда и постепенно стекает, от-
чего верхние слои раствора могут стать слабее по концентрации, нежели
нижние. Нередко склянки с титрованными растворами соединяются при по-
мощи сифона с бюреткой, что облегчает пользование раствором (см. ниже).
Не все титрованные растворы одинаково стойки. Слабые растворы вообще
мало стойки и меняют свой титр. Ввиду этого полезно время от времени про-
верять титр нестойких растворов и устанавливать коэфициент их нормаль-
ности, как указано выше.
Метод нейтрализации
Этод метод основан на реакции нейтрализации: кислотный Н-ион, сое-
диняясь с щелочным ОН’-ионом, дает весьма слабо диссоциированную воду.
Методом нейтрализации пользуются для количественного определения
щелочи при помощи титрованного раствора кислоты (алкалиметрия; alca’um—
щелочь) либо, наоборот, для количественного определения кислоты при по-
мощи титрованного раствора щелочи (ацидиметрия; aciduni — кислота).
Пример. Требуется определить содержание едкого натрия в растворе. Положим, что
при титровании 20 мл данного раствора 0,1 N раствором H2SO4 уходит (в среднем из трех
титрований) 12,5 мл раствора кислоты. Изобразив реакцию между едким натрием и серной
кислотой (2NaOH + H2SO, = Na2SO4 4- 2Н2О), находим, что на 80 весовых частей NaOH
(80 — удвоенный молекулярный вес NaOH) уходит 98 весовых частей H2SO4 (98 — молеку-
лярный вес H2SO4).
Титрование велось 0,1 N раствором H2SO4; каждый миллилитр этого раствора содержит
98
*2—= 4,9 мг H2SO4. На титрование ушло всего 12,5 мл рабочего раствора, т. е. было
затрачено 4,9 х 12,5= 61,25 мг H2SO4. Отсюда находим эквивалентное количество едкого
61,25-80
натрия из уравнения: X : 61,25= 80 : 98. Откуда х=-gg =50 мг NaOH. Следо-
вательно, в 20 мл определяемого раствора содержится 50 мг NaOH.
Расчеты можно упростить следующим рассуждением: каждый миллилитр 0,1 N H2SO4<
пошедший в реакцию, соответствует одной десятой миллиграмм-эквивалента NaOH, т. е.
4,0 мг; так как в реакцию пошло всего 12,5 мл 0,1 N кислоты, заключаем, что всего в растворе
4,0 х 12,5 = 50 мг NaOH.
52
Иногда определение щелочи или кислоты производится по методу так
называемого обратного титрования, сущность которого заклю-
чается в том, что к определяемому раствору прибавляют избыточное коли-
чество рабочего раствора, затем путем титрования определяют остаток непро-
реагировавшего рабочего раствора, откуда делают вывод о количестве про-
реагировавшего рабочего раствора и затем о концентрации определяемого
раствора.
Пример. Положим, что к 20 мл щелочи той же концентрации, что и в предыдущем при-
мере, прибавлено 20 мл 0,1 N H2SO4. Избыток H2SO4 оттитрован 0,1 N NaOH, при этом ушло
7,5 мл рабочего раствора; отсюда заключаем, что не прореагировавшей H2SO4 указанного
титра осталось также 7,5 мл. Следовательно, в реакцию с данным раствором щелочи пошло
20 — 7,5 = 12,5 мл 0,1 N H4SO4.
Дальнейшие рассуждения ведутся так же, как и в предыдущем примере.
Аналогично приведенным способам количественного определения щелочи
производят количественное определение кислоты.
Пример. Требуется определить процентное содержание уксусной кислоты в растворе;
известно, что на титрование 20 мл этого раствора уходит в среднем 36 мл 0,1 N NaOH.
Из реакции СН3СООН + NaOH = CH3COONa 4- Н2О находим, что 1 мл 0,1 N NaOH
соответствует б мг СН3СООН (6 мг coo BeTciBvei одной десятой миллиграм-эквива-
лента СН3СООН). Так как при титровании 20 мл раствора уксусной кислоты уходит
36 мл 0,1 N щелочи, заключаем, что в 20 мт испытуемого раствора находится б х 36 =
216 мг уксусной кислоты Следовательно, процентное содержание уксусной кислоты
216 • l00
в растворе равно [qqq = 1.68%.
Приводим описание способов изготовления важнейших растворов и инди-
каторов, применяемых в методе нейтрализации.
Титрованные растворы
Приготовление 0,lN раствора Na2(O3. Углекислый натрий может слу-
жить для приготовления основного раствора; при этом пользуются безводным
препаратом. Чистый безводный углекислый натрий представляет собой бе-
лый мелкозернистый аморфлый порошок; он должен без мути растворяться
в воде, не должен содержать сульфатов, хлоридов и др. солей. Получается
прокаливанием химически чистого двууглекислого натрия до постоянного
веса в платиновой чашке. Во избежание образования Na2O нагревание сле-
дует вести при температуре не выше 270—300°С при постоянном помешивании
платиновой проволочкой. М )жет быть п >лучен также из кристаллической
соды (Na2CO3 • 10Н2О) прокаливанием до постоянного веса.
Для получения из безводного углекислого натрия 0,1N раствора отвеши-
вают в закрытой бюксе точно на аналитических весах одну десятую грамм-
106
2 • 10
эквивалента этого препарата, т.
е.
= 5,3 г и растворяют полученную
навеску в стакане в небольшом количестве воды, тщательно смыв в стакан
при помощи шприца все остатки соды с часового стекла или из бюксы. Затем
все содержимое стакана переводят количественно в литровую колбу, пользу-
ясь при этом воронкой, многократно пршывая стакан и воронку свежими
порциями воды. Содержимое колбы доводят до метки водой и хорошо взбал-
тывают. Если вода взята соответствующей температуры (20°С), то раствор
получается точно 0,1N.
При изготовлении раствора соды следует пользоваться свеже прокипя-
ченной и охлажденной водой, не содержащей СО2. Охлаждение воды произ-
водится в закрытой склянке, в пробку которой вставляется хлор-кальциевая
трубка с натронной известью1 .
1 Натронная известь — белая пористая масса в зернах различной величины. Состав—
NaOH -f- СаО. Жадно поглощает влагу и СО2, переходя при этом в Na2CO3 и СаСО3. Полу-
чение: в железной чашке к крепкому раствору NaOH добавляют двойное количество не-
гашеной извести, перемешивают, выпаривают досуха и слегка прокаливают.
63
В качестве индикатора применяют метилоранж. Если индикатором служит
фенолфталеин, при не слишком большом разведении и охлаждении до нуля
происходит обесцвечивание, когда половина количества соды нейтрали-
зована (см. стр. 45).
Приготовление O,1N раствора тетраборнокислого натрия (буры
Na2B4 07- 10Н20). Бура в воде подвергается гидролизу согласно равновесной
реакции: Na2B4O7 + 7Н2О и 2NaOH -j-4B(OH)3, благодаря чему реагирует
с кислотами как щелочь и может служить для установки титра кислот.
Для получения чистой десятиводной буры имеющийся в продаже препарат
растворяют до насыщения в воде при температуре не выше 50—60°С. Насы-
щенный раствор фильтруют и оставляют, пока температура его не понизится
до 25—30°С. Затем колбу с раствором помещают в холодную воду и энергично
помешивают стеклянной палочкой; при этом выпадает мелкокристалличе-
ская десятиводная бура. Ее переносят на фильтр, промывают холодной во-
дой и сушат между листами фильтровальной бумаги до постоянного веса.
п , Na2B4O7 • 10Н2О 381,43
Дециграмм-эквивалент для буры равен 2 о щ ~—=— |П =
= 19,0715 г. Навеску в 19,0715 г перекристаллизованной буры переводят
количественно в колбу емкостью около 1 л, промывая многократно бюксу
(часовое стекло) теплой водой; прибавляют дестиллированной воды до 500—
600 мл и ставят на водяную баню, помешивая до растворения, затем пере-
водят количественно в литровую колбу. По охлаждении доводят раствор
до метки и тщательно взбалтывают. Как исходный раствор бура имеет неко-
торые преимущества перед содой: постоянство состава при хранении, малую
гигроскопичность и большой молекулярный вес. В качестве индикатора при-
меняют метилоранж или метилрот.
Приготовление 0,lN НС1. Имея 0,1 N раствор соды либо буры, легко при-
готовить 0,1 N раствор HCI, пользуясь тем, что нормальные, децинормаль-
ные и т. д. растворы кислот и оснований реагируют между собой в равных
объемах.
Грамм-эквивалент НС1 = 36,468 г, следовательно, для приготовления
литра 0,1 N раствора нужно взять 3,6468 г газообразной НС1. Практически
поступают так. Берут разбавленный раствор химически чистой соляной ки-
слоты, определяют посредством ареометра его удельный вес, находят по таб-
лице (см. стр. 132) процентное содержание газообразного НО во взятом раз-
бавленном растворе и на основании полученного результата определяют
сколько нужно взять данной соляной кислоты, чтобы получить нужный
раствор.
Пример. Положим, что удельный вес HCi — 1,1; процентное содержание в ней НС1,
согласно таблице, равно 20,01%. Отсюда легко рассчитать, сколько нужно взять данной
кислоты на литр, чтобы получить 0,1 N раствор:
х : 3,6468= 100 : 20,01,
3,6468 • 100
откуда х = —26761— = г’ Полученный результат необходимо рассматривать как
приблизительный, и титр приготовленной кислоты нужно установить по 0,1 N раствору
соды или буры.
Так как при установке титра расходуют около 100 мл раствора на титрование, приго-
18,2 • 1100 •
товляют не 1000 мл, а 1100. Очевидно, на 1100 мл понадобится j000——^6,02 г кислоты.
Раствор готовят умышленно несколько более крепким, чтобы затем разбавлением дестилли-
рованной водой довести его до надлежащего титра; в данном случае можно взять не 20,02,
а 20,5 г.
Кислоту легче отобрать в объемных единицах, нежели в весовых, и потому следует
перевести полученный вес на объем; это достигается делением веса в граммах на удельный
20,5
вес: -уу 18,6 мл.
Отмерив из бюретки или при помощи цилиндрической пипетки в колбу 18,6 мл данного
раствора НС1, доливают дестиллированной водой до 1100 мл и тщательно взбалтывают.
Затем определяют титр полученного раствора НС1 при помощи 0,1 N соды или буры. Для
этой цели наливают в одну бюретку полученный раствор НС1, а в другую — 0,1 N раствор
54
соды. Затем отмеривают в небольшую коническую колбу из бюретки 25 мл раствора соды,
прибавляют одну-две капли метилоранжа и титруют кислотой, пока желтый цвет раствора
не перейдет в розоватый. Титрование производят три раза и берут средний результат. Поло-
жим, что на нейтрализацию 25 мл соды уходит с учетом поправки на холостой опыт в сред-
нем 23,5 мл кислоты; следовательно, для получения соляной кислоты соответствующего
титра нужно на каждые 23,5 мл кислоты прибавить 1,5 мл дестиллированной воды, что на
1,5-1000 „„
литр составит —23-5— °3,8 мл.
Выяснив это, переводят при помощи мерной литровой колбы на выливание (с двумя
метками) литр приготовленного раствора кислоты в колбу (склянку) с притертой пробкой
и прибавляют сюда же при помощи бюретки или пипеток 63,8 мл дестиллированной воды,
после чего тщательно взбалтывают.
Если имеется колба с расширением па шейке, то исключается необходимость переливать
изготовляемый раствор из одного сосуда в другой; в этом случае, определив титр кислоты,
отбирают пипеткой излишек ее до первой метки и приливают сюда же из бюретки необхо-
димый объем воды. Титр полученного раствора вновь проверяют по соде и, если он не точно
децинормальный, устанавливают коэфициент нормальности. Так например, если на нейтра-
лизацию 25 мл 0,1 N раствора соды при первом титровании пошло 24,9 мл кислоты, при вто-
ром — 24,7 мл и при третьем — 24,8, то среднее из трех титрований составляет:
24,9 + 24,7 + 24,8 „ „
--------~--------= 24,8 мл. Отсюда делаем вывод, что коэфициент нормальности кислоты
25
равен 248’^ 1,00806.
Описанным способом могут быть приготовлены титрованные растворы
и других минеральных кислот.
Приготовление 0,1 N раствора NaOH. Литр 0,1N раствора содержит
4,0008 г NaOH. Приготовляют 1100 мл более крепкого раствора и из него
готовят раствор нужного титра. 4,5—5 г чистого NaOH ополаскивают дестил-
лированной водой, чтобы смыть карбонат натрия, который обыкновенно бы-
вает на поверхности, растворяют в 1100 мл дестиллированной воды и, когда
раствор приобретет комнатную температуру, титр его устанавливают по 0,1 N
соляной или серной кислоте при индикаторе метилоранже.
Пример. Положим, что среднее из трех титрований показало, что на 25 мл кислоты
идет 24,3 мл раствора едкого натрия. Отсюда делаем вывод, что на каждые 24,3 мл этого
раствора следует прибавить 0,7 мл дестиллированной воды, что при пересчете на литр дает
0,7 - 1000
—24~ч— — 28,8 мл. Отмерив колбой на выливание литр раствора, переводят его в колбу
большего размера или в склянку с притер-
той пробкой и прибавляют 28,8 мл воды, а
затем взбалтывают. Дестиллированная вода
должна быть свеже прокипяченной и свобод-
ной от СО2.
Если едкий натрий значительно
загрязнен углекислым натрием, то
NaOH может быть освобожден от
Na2CO3 следующим образом: раство-
ряют NaOH в равном количестве воды
в склянке с резиновой пробкой и
сильно встряхивают, отстаивают до
другого дня и прозрачный раствор
сливают с осевшего Na2CO3, который
в насыщенном NaOH нерастворим и
потому выпадает.
Из насыщенного раствора NaOH
приготовляют 0.1 N раствор, как
описано выше.
Приготовленный раствор едкого
Рис. 64. Установка для титрованных раст-
воров щелочи. Справа показано соединение
сифона с бюреткой при помощи тройника.
В точках а и b — стеклянные затворы.
натрия, как и всякой другой щелочи, сохраняют в склянке, закрытой рези-
новой пробкой, в которой имеются два отверстия: в одно из них вставляют
хлор-кальциевую трубку с натронной известью, а в другое — сифон, соеди-
ненный с бюреткой (рис. 64). Раствор нестоек и должен проверяться не реже
раза в две недели.
55
Индикаторы
Фенолфталеин — СбН4.СО2.С(Съ Н4ОН)2. Продукт уплотнения фено,
и фталевого ангидрида. Белый с желтым оттенком порошок, нерастворим!
в воде. Для получения индикатора растворяют один грамм фенолфталеи]
в 100 мл 70% этилового алкоголя. В нейтральной и кислой среде индик
тор бесцветен, в щелочной получает фиолетовокрасный цвет. Применяете
для титрования органических и неорганических кислот и сильных основани!
для титрования аммиака непригоден. Прибавляется к титруемому раствор
в количестве 2—3 капель. Угольная кислота, вследствие ее обесцвечивающе!
действия на фенолфталеин, должна удаляться из раствора кипячением. Ввид
этого титрование карбонатов кислотою при индикаторе фенолфталеине можн
производить лишь в кипящей среде.
Метилоранж 1 — диметиламиноазобензолсульфоновокислый натрий
NaSO3 • C6H4N : N • CGH4 • N(tH3)2. Порошкообразный препарат оранжевой
цвета. Для получения индикатора растворяют 0,1 г метилоранжа в 100 м.
горячей воды, охлаждают и фильтруют. В кислой среде метилоранж крас
ного цвета, в щелечной — желтого. К титруемой жидкости прибавляется
не больше одной-двух капель. Слабые кислоты, а следовательно, и СО2, в не
больших количествах не реагируют на метилоранж. Для титрования орга-
нических кислот метилоранж непригоден. Сильные и слабые щелочи титру-
ются при метилоранже очень точно.
Метилрот — р-диметоламиноазобензолкарбоновая кислота. Азокраска.
(СН3)2 • Nk 6H4N = N • (• СПОН Кристаллический препарат краснофио-
летового цвета. Как индикатор применяется в виде алкогольного раствора
(0,2 : 100). В кислых растворах имеет розовый цвет, в щелочных — желтый.
К титруемой жидкости прибавляется одна капля.
Лакмус. Красящее вещество растительного происхождения. Получается
из лишайников Rocella или Lecanora. Химичаская природа лакмуса еще не
выяснена. В продажу лакмус поступает в форме небольших кубиков и зерен
синего цвета. В состав его входят различные красители, из коих наиболее
важным является азолитмин; содержит также мел, гипс, известь и т. п. Как
индикатор применяется в виде лакмусовой настойки. Лакмусовая настойка
готовится следующим сбразсм. Кусе чки лакмуса экстрагируют спиртом и
экстракт сливают. Азолитмин, нерастворимый в спирте, в экстракт не пере-
ходит. Остаток после экстракции обрабатывают водой при взбалтывании до
получения раствора фиолетового цвета, затем фильтруют. Часть фильтрата,
представляющего насыщенную лакмусовую настойку, разбавляют дестил-
лированной водой и прибавляют сюда же по каплям раствор серной кислоты
(1—2 капли слабого раствора серной кислоты на 200 мл дестиллированной
воды), пока синее окрашивание не перейдет в цвет красного вина. Тогда в
этот раствор доливают концентрированный раствор лакмуса, пока раствор не
приобретет вновь синее окрашивание.
В кислой среде лакмус приобретает луковокрасный цвет, в слабо щелоч-
ной — краснофиолетовый, в концентрированных щелочных растворах — си-
ний; ввиду этого лакмус не может быть отнесен к очень чувствительным инди-
каторам. К титруемой жидкости прибавляется в количество 10—20 капель.
Так как СО2 действует на лакмус, титрование карбонатов при лакмусе воз-
можно лишь в условиях кипячения раствора.
Красные и синие лакмусовые бумажки часто применяются в лаборатор-
ной практике для определения реакции тех или иных веществ.
Ализарин — 1,2 — диоксиантрахинон, (6Н4 : (СО)2 : (6Н2(0П)2. Извле-
кался ранее из корней марены; в настоящее время получается синтетически
из антрацена. Оранжевокрасные иглы, в тонко измельченном состоянии бу-
роватожелтый порошок. Как индикатор применяется в */2 либо х/4% спиртовых
1 Как индикатор применяется и свободная сульфокислота диметиламиноазобензола.
56
(реже водных) растворах. В кислой среде окрашивается в желтый цвет, в
щелочной — в луковокрасный. Угольная кислота реагирует кисло, гидрокар-
бонаты — щелочно.
Розоловая кислота — С20Н1603. Получается окислением мышьяковой кис-
лотой смеси фенола с крезолом. Представляет собой зеленокрасные иглы или
призмы с металлическим блеском. Как индикатор применяется в виде одно-
двухпроцентного алкогольного раствора. В кислой среде окрашивается в
желтый цвет, в щелочной — в красный.
Конгорот — бензидинбисазонафтионовокислий натрии. Получается син-
тетически из бензидина действием на него азотистой кислоты и последующим
соединением полученного продукта с а-нафтиламиносульфокислотой. Тем-
нокрасный порошок. Как индикатор применяется в водном растворе 1 : 1000.
В кислой среде приобретает фиолетовое окрашивание, переходящее в синее
в концентрированных кислых растворах; в щелочной среде получает красное
окрашивание. СО2 действует на индикатор.
Описанные выше индикаторы изменяют свою окраску в пределах опре-
деленного значения pH.
То значение pH, при котором раствор принимает окраску, характерную
для конца титрования, называется показателем титрования
индикатора и обозначается через рТ.
В таблице 3 приведена краткая характеристика наиболее часто применяе-
мых индикаторов в нейтрализационном методе.
Таблица 3
Характеристика важнейших индикаторов
Наименование индикатора Изменение цвета от кислоты к щелочи Перехо । окраски в интервале pH Показатель титрования РТ
Фенолфталеин Бесцветный—красный 8,3—10,0 9,0
Метилоранж Розовы й—желты й 3,1— 4,4 4,0
Метилрот Красный—желтый 4,4— 6,3 5,5
Лакмус Красный—синий 5,0— 8,0 6,0 или 7,0 1
Выбор индикатора. При титровании сильных кислот сильными щелочами
(или обратно), а также при титровании слабых кислот сильными щелочами
и слабых щелочей сильными кислотами к концу титрования наблюдается
резкий скачок в величинах pH. Так при титровании 0,1N раствора HCi едким
натрием скачок в величинах pH охватывает интервал pH в пределах от 4,0
до 10,0 (рис. 65); при титровании более разведенной соляной кислоты, наир.
0,01N, скачок простирается от 5,0 до 9,0 pH (рис. 66); при титровании О,IN
уксусной кислоты едким натрием — в пределах 7,7 до 10,0 (рис. 67); при
титровании 0,1N раствора аммиака соляной кислотой — в пределах 4,0—6.25
(рис. 68).
Выбор индикатора должен быть произведен с таким расчетом, чтобы его
показатель титрования находился в пределах скачка pH для данного тит-
рования.
Отсюда можно заключить, что при титровании сильных кислот сильными
щелочами и обратно можно применить любой из индикаторов, представлен-
ных в таблице; однако при титровании очень разведенных сильных кислот
метилоранж окажется непригодным.
1 При рН 6,0 лакмус дает красную окраску с ясным .иним оттенком; при рН 7,0
окраска получается синяя с красным оттенком.
57
При титровании слабых кислот сильными основаниями ни метилоранж, ни
метил рот непригодны в качестве индикаторов; напротив, фенолфталеин ока-
жется пригодным.
При титровании слабых оснований сильными кислотами в качестве инди-
каторов окажутся пригодными метилоранж и метилрот.
Рис. 65. Кривая титрования 0,1 N
раствора НС1 едким натрием.
Рис. 66. Кривая титрования 0,01 N
раствора НС1 едким натрием.
Рис. 67. Кривая титрования 0,1 N
раствора НС2Н3О2 едким натрием.
Рис. 68. Кривая титрования 0,1 N
раствораГЧН4ОНсоляной кислотой.
Лакмус для слабых кислот менее пригоден, нежели фенолфталеин, и для
слабых оснований менее пригоден, нежели метилоранж и метилрот.
В объемном анализе лакмус применяется реже других индикаторов, ука-
занных в таблице.
Методы окисления и восстановления
Окисление какого-либо вещества может быть осуществлено тремя пу-
тями: путем присоединения кислорода, путем отдачи водорода (дегидриро-
вание) и путем отдачи электронов (увеличение валентности).
Примером первого вида окисления может служить превращение метал-
лов в их окиси:
Cu-]-O=CuO.
38
Примером окисления второго вида может служить превращение серово-
дорода в серу:
h2s + o==s + h2o.
Примером окисления третьего вида может служить переход окисленного
металла в высшую степень окисления:
Fe’* — (е)= Fe*".
Восстановление представляет собой процесс обратный окислению и может,
следовательно, осуществляться путем отнятия кислорода, присоединения во-
дорода (гидрирование) и присоединения электронов (уменьшение валентно-
сти).
Обобщая явления окисления и восстановления с точки зрения электрон-
ной теории, можно сказать, что всякое окисление сопровождается потерей
электронов, всякое восстановление — присоединением электронов.
Процессы окисления и восстановления протекают не изолированно, а
совместно, одновременно.
Методы окисления—восстановления объединяют различные объемноана-
литические определения: перманганатометрию, иодометрию, хроматометрию
и др. Здесь рассмотрены наичаще применяемые методы: перманганатометрия
и иодометрия.
Перманганатометрия
В перманганатометрии в качестве окислителя пользуются раствором пер-
манганата калия (КМпО4).
В кислой среде, в присутствии веществ, способных окисляться, восстанов-
ление перманганата калия при нагревании протекает по следующей реакции:
2КМпО4 + 3H2SO4 = K2SO4 + 2MnSO4 +3 Н2О + 5(0).
В результате раскисления КМпО4, как видно из приведенной реакции, об-
разуются две соли: K2SO4 и MnSO4. Первая из них в растворе бесцветна,
вторая в малых концентрациях также бесцветна. Вот почему при восстанов-
лении КМпО4 раствор обесцвечивается. Это обесцвечивание КМпО4 длится,
однако, до тех пор, пока в растворе присутствуют вещества, способные окис-
ляться. Как только окисление этих веществ закончится, то одна избыточная
капля приливаемого раствора КМпО4 окрашивает раствор в розовый цвет,
показывая конец реакции. В данном случае рабочий раствор служит одно-
временно и индикатором.
Из приведенной реакции видно, что 316,06 весовых частей КМпО4 (316,06—
удвоенный молекулярный вес КМпО4) при своем восстановлении выделяют
80 весовых частей кислорода in statu nascendi (80 — упятеренный атомный
о - гл 316,06
• вес О). Если приготовить нормальный раствор КМпО4, взяв на литр —— =
31,6062 г вещества, то весь литр при определенных условиях сможет выде-
лить 8 г кислорода, а каждый миллилитр—0,8 мг. В практике пользуются
более слабыми растворами — децинормальными и сантинормальными. Каж-
дый миллилитр слабого раствора выделит соответственно меньше кислорода.
В перманганатометрии в качестве исходного раствора, по которому уста-
навливают титр перманганата, пользуются щавелевой кислотой или ее со-
лями. Ниже приведены способы изготовления титрованных растворов щаве-
левой кислоты и перманганата.
Приготовление O,1N раствора щавелевой кислоты (С2Н2О4 • 2Н2О). Для
изготовления титрованного раствора щавелевой кислоты необходимо поль-
зоваться чистым препаратом. Растворенная в слабой азотной кислоте щаве-
левая кислота не должна давать реакции на хлор с азотнокислым серебром.
Навеска щавелевой кислоты в 2—5 г при прокаливании в платиновом тигле
не должна давать весомого остатка.
59
Если щавелевая кислота загрязнена х, то ее нужно подвергнуть очистке.
Эту операцию производят так: 100 г щавелевой кислоты растворяют в 125 мл
кипящей соляной кислоты (уд. веса 1,04); при этом щавелевая кислота, истер-
тая в порошок, вносится в соляную кислоту осторожно, небольшими порция-
ми, так как часто при этом жидкость вскипает. Горячий раствор фильтруют
через складчатый фильтр в чашку, помещенную на баню с холодной водой,
помешивая при этом фильтрат стеклянной палочкой (см. рис. 52). Когда мел-
кие кристаллы щавелевой кислоты выпадут, их переносят на фильтр, поме-
щенный в два-три слоя в бюхнеровскую воронку, отсасывают жидкость,
промывают ссадок на фильтре несколько раз небольшими порциями холодной
соляной кислоты и ледяной дестиллированной воды; затем заново растворяют
осадок в горячей дестиллированной воде и повторяют описанные операции.
Осадок, перенесенный на фильтр в воронку, промывают ледяной водой до
тех пор, пока промывная вода после подкисления азотной кислотой не пере-
станет мутиться от прибавления азотнокислого серебра (проба на хлориды).
Полученный кристаллический осадок сушат на воздухе в течение 24 часов
между двумя листами гладкой фильтровальной бумаги. Небольшую навеску
(5—7 г) испытывают на постоянство веса и чистоту. По получении удовлетво-
рительных результатов реактив считают годным к употреблению; в против-
ном случае его вновь перекристаллизовывают и сушат.
Для получения 0,1N раствора щавелевой кислоты берут тсчную навеску
в 6,3024 г, что соответствует 0,1 грамм-эквивалента = 6,30234^.
Взятую навеску растворяют в бюксе в небольшом количестве воды и
переводят в литровую колбу, пользуясь при этом воронкой. Бюксу и го-
ронку промывают многократно водой, затем содержимое колбы доводят де-
стиллированной водой до метки и тщательно взбалтывают. Воду для этой
цели следует брать дважды перегнанную.
При долгом стоянии (несколько месяцев) раствор щавелевой кислоты
несколько слабеет, ввиду чего титр должен время от времени проверяться
по щелочи (едкий натрий; индикатор — фенолфталеин).
Для придания большей стойкости раствору щавелевой кислоты к нему
можно прибавить во время приготовления некоторое количество крепкой
серной кислоты (50 мл на 1 л общего объема). В таком случае нельзя, конечно,
пользоваться щавелевой кислотой в алкалиметрии, но лишь в пермангано-
метрии. На этикетке склянки с таким раствором необходимо указать, что
он приготовлен с серной кислотой (cum acid, sulfuric.).
Вместо щавелевой кислоты в качестве исходных растворов в перманганэ-
метрии можно пользоваться химически чистыми солями щавелевой кислоты:
оксалатом калия (К2С2О4 • Н2О, грамм-эквивалент 92), оксалатем натрия
(Na2C2O4, грамм-эквивалент 67), оксалатом аммония f(NH4)2C2O4 • Н2О,
грамм-эквивалент 71]. Взятую навеску растворяют в разбавленной серной
Ki сл те, получая таким образом раствор щавелевой кислоты и соответствую-
щей соли серной кислоты.
Приготовление 0,lN раствора перманганата калия (КМпО4). Грамм-экви-
валент КМпО4 равен 31,606 г. Следовательно, для изготовления литра деци-
нормального раствора нужно взять 3,1606 г. Так как исходный препарат
обыкновенно содержит до 1% посторонних веществ (преимущественно серно-
кислые, хлористые и азотнокислые соли), навеску берут несколько большую,
чем это следует из теоретических соображений. На 1100 мл можно взять около
4 г КМпО4. Раствор тщательно взбалтывают и оставляют в закрытэй склянке
в теми ш месте на 7—10 иней. За это время происходит окисление способных
окисляться веществ, могущих присутствовать в воде да и в самом препарате.
По истечении указанного времени раствор фильтруют через асбест или через
нутч со стеклянной пористой пластинкой, после чего устанавливают титр.
1 Загрязнение часто обусловливается присутствием оксалатов калия и кальция.
60
Фильтрация раствора производится в основном для отделения выпавших
хлопьев перекиси марганца.
Установка титра раствора КМпО4 производится так: в коническую колбу
наливают 100 мл дважды перегнанной дестиллированной воды, 5 мл серной
кислоты (1 : 3), 5 мл испытуемого раствора перманганата калия (из бюретки
со стеклянным краном) и нагревают до начала кипения. К горячему раствору
приливают из бюретки 10 мл O,1N раствора щавелевой кислоты; при этом
раствор моментально обесцвечивается ввиду быстрого раскисления КМпО4
по реакции.
2КМпО4 + 5(С2Н2О4 • 2Н2О) + 3H2SO4 = K2SO4 + 2MnSO4 -J- ЮСО2 + 18H2O.
К обесцвеченному раствору прибавляют из бюретки испытуемый раствор
КМпО4, пока не появится легкое розовое окрашивание: последние порции
приливают по каплям. На основании полученного результата, разводят ра-
створ КМпО4 до нужного титра, либо устанавливают его коэфициент нор-
мальности.
Пример. Положим, что на окисление 10 мл 0,1 N раствора щавелевой кислоты пошло
всего 9,6 мл испытуемого раствора КМпО4. Следовательно, на каждые 9,6 мл раствора
0,4 • 1000
КМпО4 нужно прибавить 0,4 мл воды, что на литр составит-g~g-= 41,7 мл. Отмерив
колбой на выливание в склянку литр испытуемого раствора, приливают сюда же из бюретки
41,7 мл воды. Вместо разбавления раствора можно, как указано выше, установить коэфи-
10
циент его нормальности; в данном случае К = -g-g- 1,04166.
Раствор перманганата калия, изготовленный указанным образом, дол-
жен храниться в склянке из темного стекла с притертой пробкой. Раствор
довольно стоек, но все же ввиду медленного разложения КМпО4 несколько
слабеет: в течение нескольких месяцев титр уменьшится на 0,5%. Поэтому
титр КМпО4 следует проверять раз в 2—3 месяца.
Йодометрия
В основе иодометрии лежит обратимая реакция:
J2+H2O^2HJ(2H' + 2J') + O.
Свободный иод способен действовать как окислитель в отношении ряда
веществ: SO2, H2S, As2O3, Na2S2O3 и мн. др. При этом свободный иод перехо-
дит в ионное сс стояние — J'
С другой стороны ионы иода могут фигурировать в качестве восстано-
вителя в отношении ряда веществ: КМпО4 К2Сг2О7, МпО2 и мн. др. При этом
ионы—J' переходят в свободный иод — J2.
Эти окислительно-восстановительные процессы, связанные с превраще-
нием свободного J2 в ионы Jz и обратно, лежат в основе одного из точней-
ших аналитических методов — иодометрии.
В тех случаях, когда йодометрический метод используется как окисли-
тельный, необходимо иметь в своем распоряжении титрованный раствор
иода: в тех случаях, когда йодометрический метод используется как восста-
новительный, необходимо иметь иодистоводородную кислоту в качестве вос-
становителя и титрованный раствор тиосульфата натрия 1 (Na2S2O3 . 5Н2О)
для определения образовавшегося свободного иода Во всех случаях иодо-
метрических определений в качестве индикатора пользуются крахмальным
клейстером, который со свободным иодом дает синее окрашивание.
1 Наиболее часто применявшееся название для этого соединения было гипосульфит
натрия или просто гипосульфит Согласно предложению номенклатурной комиссии VI
Менделеевского съезда, Na2S2O8 • 5Н2О называют тиосульфатом натрия. Название же гипо-
сульфит натрия присвоено соединению Na2S2O, (ранее—гидросульфит натрия, подсерни-
стокислый натрий). Название гидросульфит натрия оставлено за NaHSO3.
61
Итак, для йодометрических определений нужно иметь следующие реак-
тивы: титрованные растворы тиосульфата натрия и иода и крахмальный
клейстер в качестве индикатора. Что же касается иодистоводородной кислоты,
то ее специально не заготовляют, а получают по ходу реакции путем под-
кисления ее соли, чаще всего KJ.
Ниже дается описание получения всех указанных растворов.
Приготовление O,1N раствора тиосульфата натрия (Na2S2O3.5Н2О). Име-
ющийся в продаже тиосульфат натрия, представляющий большие монокли-
нические призмы, перекристаллизовывают и высушивают между листами
фильтровальной бумаги.
Для изготовления литра децинормального раствора нужно взять десятую
часть грамм-эквивалента: —^"iTj ~ —10—~ 24,8148 г. Приготов-
ляют 1100 мл более крепкого раствора, взяв 28—28,5 г Na2S2O3. 5Н2О. Де-
стиллированную воду следует при этом брать свободную от СО2, т. е. свеже-
прокипяченную и охлажденную. Приготовленный раствор хранят в посуде
из темного стекла. В пробку вставляют хлоркальциевую трубку с натрон-
ной известью. Лишь по истечении 8—14 дней устанавливают титр тиосуль-
фата. За указанное время угольный ангидрид, попавший в раствор во время
его изготовления, а также из воздуха сосуда, войдет в обменное разложение
с тиосульфатом натрия, в результате чего возникают следующие реакции:
Na2S2O3 4- 2СО2 + 2Н2О = 2NaHCO3 + H2S2O3,
H2S2O,= H2SO3 +S.
Они приводят к изменению титра раствора. Помимо того, нужно указать на
действие микроорганизмов, могущее влиять на стойкость раствора.
С целью повысить стойкость раствора рекомендуется прибавлять неболь-
шие количества HgJ2 (10 мг на 1 л), либо Na2CO3 (200 мг на 1 л).
Проверка титра тиосульфата натрия может быть произведена одним из
следующих способов.
1. При помощи 0,1N раствора иодноватокислого калия (йодата калия,
К JO3), который может быть признан исходным в иодометрии.
При взаимодействии одной молекулы KJO3 с пятью молекулами KJ в
кислой среде освобождаются три молекулы свободного иода согласно реакции:
KJO3 + 5KJ + 6НС1 = 6КС1 + ЗН2О + 3J2.
Отсюда грамм-эквивалент KJO3 равен шестой части его грамм-молекулы.
Следовательно, для изготовления литра децинормального раствора KJO3
КJO3 214,02 Q
нужно взять = 3,567 г К JO3.
7 6-10 6-10 ' 3
Химически чистый реактив высушивают при 200°С, чтобы удалить гигро-
скопическую воду, охлаждают в закрытой бюксе, отвешивают точно 3,567 г
и переводят количественно в литровую колбу, долив до метки водой. Раствор
весьма стоек; хранить его следует в темной склянке.
Для проверки титра тиосульфата натрия по йодату калия поступают сле-
дующим образом: в небольшую коническую колбу с притертой пробкой на-
ливают 10 мл 10% свежеприготовленного раствора йодистого калия 1 (КJ),
5 мл соляной кислоты (1 : 5) и затем из бюретки приливают сюда же точно
25 мл 0,1N раствора йодата калия. Тотчас же происходит выделение свобод-
ного иода по приведенной выше реакции и раствор приобретает характерную
для иода окраску. Колбу закрывают пробкой и дают постоять минут 15—20.
Затем приливают сюда же 100 мл дестиллированной воды, смывая при этом
следы сублимированного иода с пробки и со стенок колбы. Свободного иода
1 Раствор KJ при подкислении не должен в течение 30 минут обнаруживать присутствие
свободного иода (проба с крахмальным клейстером).
62
в растворе будет точно столько, сколько его должно быть в 25 мл 0,1N раст-
вора иода, так как O,1N раствора йодата калия взято как раз 25 мл. Выделен-
ный иод титруют испытуемым раствором тиосульфата натрия по реакции:
J2 + 2Na2S2O3 = 2Na J + Na2S4O6.
При этом получаются две соли: иодистый натрий и тетратионовокислый на-
трий. И та и другая соли бесцветны, ввиду чего жидкость в колбе постепенно
обесцвечивается. Когда она приобретет бледножелтый цвет, прибавляют
2—3 мл 1% крахмального клейстера и посиневший раствор дотитровывают
до обесцвечивания.
Если оттитрованный раствор через некоторое время начинает медленно
синеть, что объясняется окисляющим действием воздуха на иодистый водород,
то это не принимается в расчет; если же посинение идет быстро и энергично,
то это указывает на то, что реакция не прошла количественно до конца; в
этом случае нужно повторить титрование.
Положим, что на 25 мл йодата калия пойдет 24,4 мл тиосульфата натрия.
Следовательно, на каждые 24,4 мл тиосульфата натрия нужно прибавить
0,6 мл дестиллированной воды, а на литр — ^24,6 мл воды, чтобы
получить 0,1N раствор. Вместо разбавления раствора можно установить
его коэфициент нормальности.
2. Проверка титра тиосульфата натрия может быть проведена также при
помощи бихромата калия (К2Сг2О7). Реакция выделения свободного иода
при этом протекает следующим образом:
K2Cr2O7 + 6KJ+ 14НС1 = 8КС1 4-CrCl3+7H2O + 3J2.
Бихромат калия перекристаллизовывают и высушивают при 130°С. Для при-
г К2Сг2О 294,23 л пП/1
готовления литра децинормального раствора берут £ j-g = 4,904 г
мелкокристаллического сухого препарата. Раствор стойкий и может служить
исходным в иодометрии.
Проверка титра тиосульфата натрия производится так, как описано выше.
В колбу наливают 10 мл свежеприготовленного 10% раствора KJ, приливают
5 мл соляной кислоты (1 : 5), а затем из бюретки 25 мл 0,1N раствора бихро-
мата калия. Свободный иод выделяется согласно приведенной выше реакции.
Раствор разбавляют дестиллированной водой (до 500 мл), а затем титруют
тиосульфатом натрия. Когда раствор становится светложелтым, прибавляют
крахмальный клейстер как индикатор и продолжают титрование до тех пор,
пока синий цвет не перейдет в яснозеленый, обусловленный присутствием
в растворе хлорного хрома (СгС13). Реакцию следует проводить при дневном
свете, чтобы легче уловить момент перехода одной окраски жидкости в другую.
Вместо соляной кислоты можно пользоваться серной. В этом случае ре-
акция протекает по следующему уравнению:
К2Сг2О7 4- 6К.J + 7H2SO4 = 4K2SO4 +Cr2(SO4)3 + 7Н2О + 3J2.
Титрование выделившегося иода при индикаторе крахмальном клейстере
идет в данном случае до обесцвечивания.
3. Наконец, установка титра тиосульфата натрия может быть произведена
при помощи 0,1N раствора перманганата калия; реакция выделения свобод-
ного иода при этом протекает следующим образом:
2KMnO4+ 10KJ+ 16НС1 = 12KCI + 2MnCl24-8H2O4-5J2.
Выделившийся иод оттитровывают тиосульфатом натрия при индикаторе
крахмальном клейстере до обесцвечивания.
63
Приготовление О,IN раствора иода Грамм-эквивалент иода 126,92. При-
готовляют более крепкий раствор, нежели 0,1N, взяв на 1100 мл около 14,5 г
кристаллического иода. Предварительно растворяют в небольшом количе-
стве дестиллированной воды 25—30 г чистого йодистого калия, отвешивают,
на технохимических весах в закрытой бюксе указанное количество иода и
переводят в раствор йодистого калия, тщательно взбалтывая. Иод, плохо
растворимый в воде, быстро растворяется в крепком растворе йодистого ка-
лия. По растворении иода раствор доливают водой до 1100 мл. Титр раствора
иода устанавливают по 0,1N раствору тиосульфата натрия.
Для этой цели наливают в коническую колбу из бюретки 25мл раствора
иода, прибавляют 100 мл дестиллированной волы и титруют 0,1N раствором
тиосульфата натрия до бледножелтого цвета; прибавляют 2—Змл 1% крах-
мального клейстера и посиневший раствор дотитровывают до обесцвечивания.
По расходу тиосульфата устанавливают титр раствора иода.
Раствор иода необходимо сохранять в темной склянке; он нестоек, ввиду
чего время от времени необходимо проверять его титр.
Приготовление 1% крахмального клейстера. Отвешивают на техни-
ческих весах 1 г пшеничного крахмала и растирают в ступке с небольшим
количеством холодной воды, пока не образуется равномерная жидкая
масса.
Вскипятив 100 мл дестиллированной воды, приливают сюда понемногу
полученный жидкий крахмал, после чего кипятят еще 1—2 минуты и филь-
труют через складчатый фильтр, смоченный горячей водой.
Крахмальный клейстер образует со свободным иодом иод-крахмал, при-
дающий раствору синее окрашивание еще в разведениях 1 : 1 500000. Реакция
протекает лишь в присутствии йодистого водорода или растворенных иодидов.
Крахмальный клейстер прибавляется к концу титрования, когда жидкость
приобретает слабожелтую окраску. При титровании следует брать 2—3 мл
крахмального клейстера, если объем жидкости в конце титрования равен
50 мл.
Крахмальный клейстер при стоянии портится вследствие развития в нем
плесени и уже через несколько дней обыкновенно становится непригодным
для употребления. Вместо синего окрашивания он дает с иодом бурое окра-
шивание, что указывает на порчу крахмального клейстера.
Чтобы уберечь крахмальный клейстер от порчи, его сейчас же по изго-
товлении разливают в маленькие бутылочки, стерилизуют и в таком виде
хранят. Рекомендуют также для сохранения стойкости крахмального клейстера
прибавлять к нему консервирующие вещества: на 100 мл клейстера 1 мл 2N
раствора HCI либо 0,125 г салициловой кислоты и т. п.
Особо прочный крахмальный клейстер, способный сохраняться до трех
лет, может быть получен следующим образом: 2 г крахмала растирают в ступ-
ке с 10 мг двуиодистой ртути и небольшим количеством холодной воды, пока
не образуется равномерная жидкая масса. Вскипятив около 100 мл дестилли-
рованной воды, вливают сюда понемногу полученную жидкую массу, после
чего кипятят еще 1—2 минуты и фильтруют через складчатый фильтр,
смоченный горячей водой. Фильтрат доводят до литра дестиллированной
водой.
При титровании берут 2—3 мл крахмального клейстера на каждые 50 мл
раствора из расчета общего объема титруемого и титрованного растворов.
В продаже встречается так называемый растворимый крахмал в порошке
или в виде пасты. Из таких препаратов предпочтительней готовить крахмаль-
ный клейстер, нежели из обыкновенного крахмала. Способ изготовления
тот же.
1 Еще не так давно раствор иода считался основным в иодометрии и по нему готовили
тиосульфат. Иод очищался возгонкой и бралась точная навеска. Теперь чаще иод готовят
по тиосульфату, который сам готовится по йодату калия или бихромату калия.
€4
Методы осаждения
В тех случаях, когда метод осаждения применяется для объемно-аналити-
ческих определений, речь идет о получении практически нерастворимых
осадков, возникающих в результате взаимодействия между определяемым
веществом и применяемым титрованным раствором. Конец реакции (точка
эквивалентности) определяется либо без индикатора наблюдением момента
прекращения образования осадка, либо чаще при помощи соответствующего
индикатора по окраске, которую приобретает раствор.
В важнейших реакциях осаждения в объемном анализе в качестве титро-
ванного раствора пользуются раствором нитрата серебра. Методы, основан-
ные на применении этого реактива, называются аргентометриче-
скими методами.
Типичным примером аргентометрического метода может служить опреде-
ление хлоридов (или других галоидных солей) при помощи титрованного
раствора AgNO3 по реакции:
Ag' + CI'-AgCl.
Определение точки эквивалентности при этом методе осуществляется
.различно:
1. Наблюдением момента прекращения образования осадка.
2. Титрованием с применением в качестве индикатора хромата калия —
(К2СгО4).
3. Обратным титрованием с последующим определением избытка AgNO3.
4. Применением адсорбционных индикаторов, преимущественно флуорес-
цеина и эозина.
Из всех указанных методов 1-й и 4-й применяются редко. Остальные ме-
тоды, часто применяемые, подробно рассмотрены далее. (Питьевая вода —
определение хлоридов).
Кроме определения хлор-иона методом осаждения в объемном анализе
определяются также ионы: Br', J', CNS', Ag, Zn”, Ва" и др.
К методу осаждения примыкает метод комплексообразования.
Примером титрования с образованием комплексного соединения может
служить определение ионов CN' ионами серебра:
2CN'4-Ag-^[Ag(CN)2]'.
Комплексный ион [Ag(CN)2]' остается в растворе. При незначительном из-
бытке Ag- образуется комплексное соединение Ag[Ag(CN)2], выпадающее
в осадок:
[Ag (CN)2]' + Ag' - Ag [Ag (CN)2].
Появление неисчезающей мути служит указанием конца реакции.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
Из физико-химических методов, часто применяемых в санитарно-гигиени-
ческом анализе, в настоящей главе рассмотрены колориметрия, нефеломет-
рия, определение концентрации водородных ионов, потенциометрическое тит-
рование и определение электропроводности.
Колориметрия
Колориметрия представляет ценный метод количественного анализа, осо-
бенно для определения малых количеств вещества. Этот метод применим
лишь тогда, когда определяемое вещество дает со специально подобранным
реактивом (индикаторный реагент) цветовую реакцию (color — цвет).
Колориметрический метод основан на следующих законах физики:
5 А. И. Бурштейн
65
1. Отношение интенсивности проходящего света к интенсивности пада-
ющего света — величина постоянная.
2. Слои одинаковой толщины одного и того же вещества поглощают оди-
наковую часть падающего света.
3. Поглощение света, проходящего через слой окрашенного растворенно-
го вещества, пропорционально концентрации растворенного вещества.
Из приведенных законов вытекает основной принцип колориметрии, ко-
торый сводится к следующему.
Если окрашенные одним и тем же красящим веществом два столба жид-
кости одинаковой высоты дают одинаковую интенсивность окраски при рас-
сматривании сверху вниз по высоте, то концентрация красящего вещества
в обоих растворах одинакова. При одинаковой же интенсивности окраски
двух столбов жидкости различной высоты концентрация красящего вещества
растворов обратно пропорциональна высотам столбов жидкостей, т. е.
Ci: С2 =
где Cj — концентрация одного раствора; Нг— высота его столба,
С2 — концентрация другого раствора; Н2— высота его столба.
Отсюда вытекает, что:
и с £1^1
Н2
Эти выводы приложимы, однако, не ко всем окрашенным растворам и
остаются точными лишь в определенных пределах концентраций, различных
для разных красителей и растворителей.
Сравнение интенсивности окраски может быть произведено при помощи
глаза наблюдателя (визуальная, субъективная колориметрия) и при помощи
фотоэлемента (фотоэлектрическая, объективная колориметрия).
Визуальная колориметрия
Визуальная колориметрия осуществляется по одному из следующих ме-
тодов: 1) метод стандартных серий или цветовой шкалы, 2) метод разбавления,
3) метод уравнивания (уравновешивания) окраски и 4) метод колориметри-
ческого титрования.
Метод стандартных серий (цветовой шкалы)
разлив в
:. 70. Штатив для пробирок.
100 ЧОО
ИЛ W
Рис. 69.
Подбирают ряд пробирок одинакового диаметра. Это проще всего сделать,
ряд пробирок из пипетки одинаковые объемы жидкости и выбрав
те пробирки, в которых высота
столбов жидкости окажется оди-
наковой.
Очень удобны для этой цели
пробирки одинакового диаметра с
плоским дном (рис. 69). Пробирки
нумеруют и устанавливают в шта-
тив (рис. 70). Нижние отверстия
штатива имеют бортики для удер-
живания пробирок. Под пробир-
ками на нижней части штатива
Пробирка для помещают под углом лист белой бумаги или молочное стекло.
рованияТпло- Таким образом, цвет, отражаемый бумагой (молочным стеклом),
ским дном, проходит через пробирки снизу вверх.
Приготовляют стандартный раствор, т. е. такой, в котором
имеется известное количество вещества, соответствующего определяемому,
и разливают при помощи точной цилиндрической пипетки или бюретки в
пробирки, начиная с малых количеств и переходя к большим по принципу
бб
геометрического ряда; это значит, что количество стандартного
раствора от одной пробирки к другой меняется в некоторое число раз, соот-
ветственно принятому показателю ряда.
Далее во все пробирки прибавляют дестиллированной воды в таком ко-
личестве, чтобы довести содержимое пробирок до одного и того же объема,
получая, таким образом, ряд стандартов (эталонов, шаблонов).
Затем в пробирку такого же диаметра (опытная пробирка) вводят раствор
определяемого вещества в количестве, соответствующем объему жидкости
в каждой из стандартных пробирок, после чего прибавляют во все пробирки
по одинаковому количеству индикаторного реагента и, тщательно переме-
шав содержимое в каждой пробирке, рассматривают столбы жидкостей сверху
вниз. Из ряда стандартов выбирают ту пару пробирок, между которыми
находится окраска, полученная в опытной пробирке.
Беря эти две пробирки в качестве крайних членов ряда, ставят между
ними новый ряд стандартов с меньшим показателем ряда и вновь колоримет-
рируют, находя в этом ряду то разведение, которое дает окраску соответственно
опытной пробирке. Концентрация искомого вещества в этой последней при-
нимается равной концентрации в соответствующем стандарте.
Пример. Приготовлен ряд стандартов из пробирок с показателем 1,25. Положим, что
в первую пробирку налит 1 мл раствора, содержащий 1 7 вещества; тогда ряд стандартов
будет содержать раствора (в мл) и вещества (в 7) соответственно: 1,0; 1,25; 1,56; 1,95;
2,44; 3,05; 3,81; 4,76; 5,95; 7,44. Положим, далее, что окраска раствора в опытной пробирке
оказалась между пятой и шестой стандартными пробирками. Тогда ставят новый ряд стан-
дартов с показателем 1,05 соответственно допускаемому в колориметрии пределу ошиб <и
в + 5%. Получаем новый ряд: 2,44; 2,56; 2,69; 2,82; 2,96; 3,05. Пусть окраска раствора
в опытной пробирке совпадает с окраской третьей пробирки второго ряда, тогда заключаем,
что во взятом для колориметрии объеме испытуемого раствора содержится 2,69 7 искомого
вещества.
Весьма часто метод стандартных серий проводится по принципу не геоме-
трического, а арифметического ряда, т. е. количество стандартного
раствора от одной пробирки к другой меняется на одну и ту же величину.
В некоторых случаях удается приготовить искусственные долговечные
стандарты при помощи растворов и смесей растворов некоторых окрашен-
ных соединений (хлористый кобальт, хлорное железо, медный купорос и т. п.).
Каждый такой искусственный раствор стандартизируется на известное коли-
чество определяемого вещества. Весьма важно, чтобы цвета стандартов точно
совпадали с цветами анализируемых растворов по оттенкам.
Долговечными реактивами можно пользоваться по крайней мере для
первого ориентировочного колориметрирования, что значительно облегчает
работу.
Метод разбавления
Подбирают две пробирки одинакового диаметра. В одну вводят опреде-
ленный объем стандартного раствора, в другую — такой же объем испытуемого
раствора. В обе пробирки вводят одно и то же количество индикаторного
раствора. Наблюдают растворы в горизонтальном направлении. В ту из
пробирок, где окраска раствора кажется более интенсивной, прибавляют из
бюретки небольшие порции воды, каждый раз взбалтывая раствор; так посту-
пают до тех пор, пока не будет достигнута одинаковая окраска растворов.
Количество вещества в каждом растворе прямо пропорционально объему
этого раствора.
Пример. В одну из пробирок налито 10 мл стандартного раствора, мл которого со-
держит 0,01 мг определяемого вещества; в другую пробирку налито 10 мл исследуемого
раствора. В обе пробирки налито по 1 мл индикаторного раствора, после чего для уравнения
окраски понадобилось прилить в стандартную пробирку 2,4 мл дестиллированной воды.
TaioiM образом, в стандартной пробирке оказалось всего 13,4 мл раствора, а в опытной—
Отсюда заключаем, что в одном мл исследуемого раствора содержится 0,0082 мг
13,4
определяемого вещества.
67
Метод уравнивания (уравновешивания) окраски
При этом методе для колориметрирования применяют специальные при-
боры, носящие название колориметров. Таких приборов предложено много,
однако в практической работе больше всего применяют простейший колори-
метр Генера и колориметр Дюбоско.
Колориметр Генера (рис. 71) представляет собою пару цилинд-
ров одинакового диаметра из бесцветного стекла. На стенках цилиндров
имеются деления, указывающие емкость их в миллилитрах
с точностью до 1 мл. Общая емкость цилиндров 105 мл.
У основания цилиндров имеются стеклянные краны.
В один из цилиндров (стандартный) вводят пипеткой опре-
деленный объем стандартного раствора (1 — 5 мл) и доли-
вают дестиллированной воды до общего объема в 100 мл;
в другой цилидр (опытный) наливают 100 мл исследуемого
раствора. Затем в оба цилиндра приливают одинаковые коли-
чества индикаторного реагента и тщательно перемешивают
жидкость в каждом цилиндре стеклянными палочками. Ци-
линдры после этого помещают рядом на лист белой бумаги
и рассматривают оба столба окрашенных растворов в верти-
кальном направлении (сверху вниз). Из того цилиндра, где
Рис. 71. окрашивание окажется более интенсивным, выливают через
Простейший кран столько жидкости, сколько нужно, чтобы интенсивность
колориметр, окрашивания растворов в обоих цилиндрах оказалась одина-
ковой. Содержание искомого вещества в определяемом
растворе находят на основании приведенного выше уравнения (стр. 66).
Пример. Положим, стандартный раствор приготовлен так, что в каждом миллилитре
его содержится 0,1 мг определяемого вещества.
В стандартный цилиндр налито 5 мл стандартного раствора и 95 мл дестиллированной
воды; следовательно, в стандартном цилиндре на 100 мл жидкости приходится 0,5 мг
определяемого вещества (0,1 • 5). В опытный цилиндр налито 100 мл исследуемого раствора.
Затем в оба цилиндра прилито по 5 мл индикаторного реактива.
Для получения одинаковой интенсивности окрашивания пришлось из опытного цилиндра
вылить 20 мл раствора. Нужно определить содержание искомого вещества в 100 мл иссле-
дуемого раствора.
Так как цилиндры имеют один и тот же диаметр, то вместо высот столбов жидкостей
можно для расчета взять объемы. Следовательно, можно написать:
= XV 2,
откуда
v-Ell1
Л -- 9
и2
где X — концентрация искомого вещества в испытуемом растворе в мг/100 мл,
q— концентрация искомого вещества в стандартном цилиндре в мг/100 мл,
vx — объем жидкости в стандартном цилиндре,
q — объем жидкости, оставшейся в опытном цилиндре после уравнивания окраски.
Подставив вместо q, q, и q их значения, получаем
0,5 • 105
X = ----g₽j--= 0,618 мг/100 мл,
или 6,18 мг/л.
Итак, для определения концентрации искомого вещества в исследуемом растворе, нужно
концентрацию раствора в стандартном цилиндре помножить на объем жидкости в стан-
дартном цилиндре и разделить на объем жидкости в опытном цилиндре.
Колориметр Дюбоско (рис. 72) является одним из наиболее
распространенных приборов этого рода. Он состоит из двух цилиндров А и
Ai, в новых образцах цилиндры сверху имеют расширения. В эти цилиндры
при помощи винтов погружают на большую или меньшую глубину призмы
В и Вр В новых образцах призмы неподвижны, а подвижными делаются
цилиндры. Боковые стенки затемнены или матированы. В цилиндры наливают
растворы — стандартный и испытуемый. Благодаря погружению призм в
68
растворы на большую или меньшую глубину высота рассматриваемых стол-
бов жидкости может быть доведена до желаемого размера; этим исключается
необходимость отливать жидкость через кран. Аппарат снабжен сверху оку-
ляром, снизу зеркалом либо пластинкой молочного стекла (D). Для защиты
цилиндров от постороннего света их закрывают вычерненной внутри короб-
кой. Отраженные от зеркала лучи света проходят через цилиндры, призмы,
а затем, благодаря стеклянным параллелепипедам (Е и Ег), претерпевают
полное внутреннее преломление,
сближаются, проходят через ди-
афрагму (К) и через окуляр (MN).
Поле зрения представляется в виде
кружка, разделенного на две по-
ловинки, разно окрашенные в за-
висимости от различной интенсив-
ности окраски жидкостей в ци-
линдрах, причем правая половина
поля зрения соответствует левому
цилиндру и наоборот (см. рис. 72).
В приборах нового образца, где
между окуляром и параллелепи-
педами помещена дополнительная
линза, правая половина поля зре-
ния соответствует правому цилин-
дру и левая — левому.
При колориметрировании, опу-
ская или поднимая призмы в том
Рис. 72. Колориметр Дюбоско.
I — Схема конструкции: А и — цилиндры (кюве-
ты), В и Bi — призмы, Е и Bi — параллелепипеды,
К — диафрагма, MN — окуляр, D — пластинка мо-
лочного стекла. II. Внешний вид колориметра.
или ином цилиндре, нужно до-
биться одинакового окрашивания обеих половин поля зрения, а затем при
помощи шкалы и двух нониусов, имеющихся на задней стенке прибора,
определить высоту столбов растворов в каждом цилиндре. Концентрация
испытуемого раствора определяется так же, как при пользовании парными
цилиндрами. Вместо объемов учитывается высота столбов жидкостей.
Для проверки правильности произведенного колориметрирования реко-
мендуется изменить положение одной из призм (цилиндров) и снова уста-
новить равенство окраски обеих половин поля зрения, соответственно пере-
мещая вторую призму. Если колориметрирование производится правильно,
то отношение высот столбов жидкости сохраняется во всех случаях полу-
чения одинакового окрашивания обеих половин поля зрения. Для этой же
цели рекомендуется поменять местами цилиндры и проверить вновь полу-
ченное отношение высот столбов растворов, сравнив его с ранее полу-
ченным.
Для получения возможно более точных результатов колориметрирования
необходимо строго придерживаться следующих правил: 1) стандартный и
испытуемый растворы должны быть близки по концентрации; в крайнем
случае разница не должна превышать 100%; 2) температуры обоих растворов
не должны отличаться более чем на 3°С; 3) освещение должно быть равномер-
ным и средним по силе.
В новых моделях при колориметре имеется и осветительный аппарат.
Для колориметрирования малых количеств растворов применяются микро-
колориметры, позволяющие обходиться 1 мл раствора для каждого цилиндра,
тогда как для обычных колориметров требуется 10—30 мл.
Метод колориметрического титрования
Колориметрическое титрование представляет колориметрический метод,
основанный также на уравнивании окраски растворов, однако этот метод
принципиально отличается от только что описанного.
69
Здесь высота столбов сравниваемых жидкостей остается одинаковой (вер-
нее, почти одинаковой), а изменение интенсивности окраски в одном из ци-
линдров (стандартном) достигается путем прибавления в него из микробю-
ретки или микропипетки такого количества стандартного раствора, чтобы
окраска в обоих цилиндрах оказалась одинаковой.
По израсходованному количеству стандартного раствора определяют ко-
личество искомого вещества в опытном цилиндре.
Пример. В стандартный цилиндр налито 100 мл дестиллированной воды и 5 мл
индикаторного реактива; в опытный цилиндр налито 100 мл исследуемого раствора и 5 мл
индикаторного раствора. Затем из микропипетки понадобилось прилить в стандартный ци-
линдр 1,12 мл стандартного раствора, чтобы добиться одинаковой интенсивности окраски
в обоих цилиндрах. Стандартный раствор приготовлен так, что в 1 мл его содержится
0,01 мл определяемого вещества.
Отсюда можно заключить, что в 100 мл исследуемого раствора содержится
0,01-1,12.106,12 Л „
-------105-----= 0,01132 мг искомого вещества или 0,1132 мг/л.
Фотоэлектрическая колориметрия
Ввиду того что точность визуальной колориметрии зависит в значительной
мере от чувствительности глаза наблюдателя, в последнее время в лаборатор-
ную практику вводятся объективные методы колориметрии, при которых
используются фотоэлементы, заменяющие в этих исследованиях глаз человека,
что дает возможность избежать субъективных ошибок.
Из различных известных фотоэлементов для указанной цели применяют
обыкновенно фотоэлементы с внешним фотоэффектом. Действие фотоэлемен-
тов этого типа основано на том, что, свет, падая на светочувствительную
поверхность, выбивает из нее электроны, которые летят к аноду, что обус-
ловливает появление в цепи фототока.
Рис. 73. Фотоэлемент.
С — окно, В — пучок света,
D — катод, А — анод.
Рис. 74. Схема включения фото-
элемента.
1 — фотоэлемент, 2 — аккумуляторная
батарея, 3 — сопротивление, 4 — галь-
ванометр.
Прототипом фотоэлементов указанного типа является фотоэлемент,
изображенный на рис. 73. Он представляет собой колбу, посеребренную вну-
три. Небольшая часть внутренней поверхности с остается свободной от сереб-
ряного слоя (окно) для прохождения в колбу пучка света В. Светочувстви-
тельным катодом фотоэлемента является слой щелочного металла (калий,
натрий, рубидий, цезий) Д, осажденный на части внутренней поверхности
колбы и находящийся в контакте с проводником, впаянным в стенку колбы.
Против катода находится кольцеобразный или сетчатый анод Д.
Схема включения фотоэлемента без усиления 1 показана на рис. 74. Здесь
7 — фотоэлемент, 2 — аккумуляторная батарея, 3 — сопротивление и 4 —
1 Значительное усиление фототока может быть достигнуто включением в цепь катод-
ной лампы.
70
гальванометр. Если светочувствительная поверхность фотоэлемента нс осве-
щена, то тока в цепи нет; если же фотоэлемент освещен, то в цепи появляется
ток, пропорциональный силе света.
Из фотоэлементов с внешним фотоэффектом наиболее пригодным для
фотометрии является селеновый фотоэлемент, так
мумом чувствительности к той же части спектра,
что и глаз человека.
Фотоэлектрическая колориметрия осуществля-
ется либо по схеме диференциальной с примене-
нием двух фотоэлементов, либо по схеме прямого
измерения с применением одного фотоэлемента.
Схема диференциальной фото-
колориметрии показана на рис. 75. С и
(?! — два фотоэлемента с одинаковыми характери-
стиками; В и В±— две кюветы, из коих одна
наполняется исследуемым раствором, другая —
стандартным; А — источник света; G — гальвано-
метр. Как видно из схемы, свет попадает на фото-
как он обладает макси-
Рис. 75. Схема диференци-
альной фото колориметрии.
С и С, — два фотоэлемента, В и
— две кюветы, А — источник
света, G — гальванометр.
элементы после прохождения через кюветы с
растворами. Так как оба фотоэлемента включены
навстречу друг другу, то при одинаковом освеще-
нии фотоэлементов тока в цепи не будет. Если же
свет пройдет через растворы разной концентрации,
то освещение обоих фотоэлементов окажется неодинаковым по интенсивности,
что вызовет отклонение стрелки гальванометра.
Установив зависимость между концентрациями растворов и отклонением
стрелки гальванометра для определенной серии колориметрических опре-
делений, можно, пользуясь полученными эмпирическими данными, по
отклонению стрелки делать заключение о концентрации испытуемого ра-
створа.
Для удобства рекомендуется вычертить кривую зависимости между кон-
центрациями данного раствора и показаниями стрелки гальванометра: так
на оси абсцисс можно отложить показания стрелки гальванометра, а на оси
ординат — соответствующие концен-
трации раствора или наоборот. Вы-
чертив кривую, можно пользоваться
ею, находя соответствующие концен-
трации на основании показаний стрел-
ки гальванометра. Понятно, что для
раствора каждого определяемого ве-
щества нужно вычертить отдельную
кривую по стандарту.
Схема прямой фотоко-
лориметрии показана на рис. 76.
Здесь А — фотоэлемент, G — гальва-
нометр, С — сопротивление, В — ба-
тарея, F — источник света, Вх — ба-
тарея, питающая лампу, —реостат
для поддержания режима накала
лампы, Е — светофильтр, К — кювета для растворов, D — ирис диафрагма.
При помощи описанной установки определяют отдельно отклонения стрел-
ки гальванометра при прохождении света через стандартный раствор, испы-
туемый раствор и через растворитель (вода). Искомая концентрация испы-
туемого раствора определяется по формуле:
Рис. 76. Схема прямой фотоколориметрии.
А — фотоэлемент, G — гальванометр, С — сопро-
тивление, В — батарея, F — источник света,
Bj — батарея, питающая лампу, Сг — реостат для
поддержания режима накала лампы, Е — свето-
фильтр, К — кювета для раствора, D — ирис-
диафрагма.
с = lg no~'gni . с
•g По — 1g п '
где С — искомая концентрация; п0— отсчет на гальванометре при испытаниг
растворителя; — то же при испытании определяемого раствора; п— тс
же при испытании стандарта; Со—концентрация эталона.
Фотоэлектрическая колориметрия дает лучшие результаты, нежели визу-
альная. Предел ошибки достигает + 0,2%1.
Нефелометрия
Нефелометрия представляет собой метод количественного анализа, весьма
близкий к описанному выше колориметрическому методу. Здесь при помощи
прибавления к раствору индикаторного реактива получают помутнение жид-
кости (nephelos — туман). Концентрацию испытуемого раствора определяют
сравнением его с полученным при тех же условиях обработки стан-
дартным раствором.
Как и колориметрия, нефелометрия проводится чаще либо по методу стан-
дартных серий, либо по методу уравновешивания.
Метод стандартных серий
Приготовляют ряд стандартных пробирок. В пробирку того же диаметра
наливают испытуемый раствор. Во все пробирки прибавляют одинаковое
количество индикаторного реактива и сравнивают муть, образовавшуюся
в опытной пробирке, с мутью в стандартных пробирках. Рассматривание
ведется сверху, пробирки же освещаются сбоку. В глаз наблюдателя не дол-
жен попадать посторонний свет.
Метод уравновешивания
Специальный прибор — нефелометр изображен на рис. 77. Для сравнения
степени помутнения растворов здесь использован феномен Тиндаля. Освети-
тельный прибор нефелометра построен так, что свет проходит через растворы
Рис. 77. Нефелометр.
I. Внешний вид. II. Схема конструкции; а и ах — кюветы, b и bt — призмы,
g и gi - металлические пластинки для регулирования высот освещенных слоев
взвесей, h — шкала для правой пластинки, Г и fг — призмы для преломления света,
с — окуляр.
перпендикулярно к продольной оси цилиндров; таким образом, в глаз на-
блюдателя попадает лишь свет, отраженный от частичек, обусловливающих
1 Подробно см. статью С. А. Стрелкова в книге «Новые методы технического
контроля в заводской лаборатории химической промышленности», 1935.
72
мутность растворов. В известных пределах между концентрацией взвешенных
в растворе частичек и интенсивностью отраженного света сохраняется зави-
симость, определяемая указанными на стр. 66 законами. Это дает возможность
определять концентрацию искомого вещества путем сравнения со стандар-
том. На рисунке прибор показан схематично сбоку. Две кюветы а и а± напол-
нены сравниваемыми мутными растворами. Свечение обоих конусов Тиндаля
рассматривается через призмы b и bv Высоты освещенных слоев взвеси регу-
лируются при помощи изменения положения металлических пластинок g и g±
и определяются по соответствующим шкалам (на рис. шкала Л для пластинки
g). При помощи призм / и /х отраженный от частичек свет преломляется и
попадает в глаз наблюдателя через окуляр С.
Способ применения нефелометра и производство расчетов те же, что и для
колориметра.
Для исследования необходима абсолютно чистая посуда. Растворы гото-
вятся при совершенно одинаковых условиях, ввиду чего размеры частичек,
обусловливающих явление Тиндаля, как в шаблонном, так и в испытуемом
растворах одинаковы. Рассматриваемые растворы должны быть совершенно
свободны от посторонних взвешенных частичек (волокна фильтровальной
бумаги) и пузырьков воздуха. Отношения концентраций сравниваемых ра-
створов не должны выходить за пределы 1 : 4.
Определение концентрации водородных ионов
Для определения концентрации водородных ионов предложены два ме-
тода: 1) индикаторный, или колориметрический, и 2}
электрометрический, или потенциометрический. Из
них наибольшей точностью отличается второй. Ниже рассматриваются оба
метода. Предварительно приводятся общие сведения по данному вопросу.
Общие сведения
Выше (стр. 44) было указано, как методами ацидиметрии и алкалиметрии
можно определить степень кислотности и щелочности растворов.
Эти методы, основанные на реакции нейтрализации, не дают, однако,
представления об истинной или активной реакции среды, обусловлен-
ной содержанием в данной среде свободных Н*- и ОН'-ионов.
Как известно, всякий электролит — кислота, основание или соль в раст-
воренном состоянии — распадается в большей или меньшей мере на ионы,
а именно: катионы, несущие положительный заряд, и анионы, несу-
щие отрицательный заряд.
Этот распад молекул на ионы называется электролитической
диссоциацией.
Согласно ионной теории такой, напр., электролит, как хлористый натрий,
находится в растворе в виде молекул недиссоциированного хлористого нат-
рия (NaCl), а также в виде катионов натрия, обозначаемых символом Na +
(либо Na’), и анионов хлора, обозначаемых символом С1“ (либо СГ). Процесс
электролитической диссоциации рассматривается, как обратимый. Следова-
тельно, для нашего примера можно написать:
NaCl Na —J- С1\
Для CuSO4, распадающегося на ионы, несущие двойной заряд, можно
написать:
CuSO^Cu’ + SOL
Чем выше степень разбавления данного электролита, тем выше и степень
его электролитической диссоциации. Для очень разбавленных растворов
электролитическая диссоциация электролита теоретически идет до конца.
При пропускании тока через раствор электролита электролитическая дис-
73
социация также идет до конца, причем катионы осаждаются на катоде, анио-
ны на аноде.
Во всех случаях диссоциации электролита сохраняется закон действу-
ющих масс. Согласно этому закону произведение из концентрации ионов,
отнесенное к концентрации недиссоциированного электролита, представляет
для данного электролита величину постоянную, носящую название кон-
станты диссоциации. Для вышеприведенных случаев, на основа-
нии закона действующих масс, можно написать следующие равенства:
JMd£± = K „ (с-1 [SO3 -
[Na Cl] [Си SOJ
Все физико-химические (и физиологические) свойства растворов электро-
литов зависят от находящихся в них (растворах) положительно и отрицатель-
но заряженных ионов.
Свойства кислот зависят от общего им водородного иона — Н’.
Большая или меньшая степень электролитической диссоциации кислоты,
т. е. большая или меньшая концентрация в ней водородных ионов, обуслов-
ливает большую или меньшую силу или активную реакцию кислоты. Кис-
лота тем сильнее, чем больше она диссоциирована.
Свойства щелочей зависят от общего им гидроксильного иона — ОН'.
Активная реакция щелочей обусловливается большей или меньшей концен-
трацией гидроксильных ионов. Как и кислота, щелочь тем силь-
нее, чем больше она диссоциирована.
При титровании кислоты щелочью равновесие между недиссоциирован-
ными молекулами и ионами диссоциированных молекул нарушается, так как
вместо связываемых прибавляемой щелочью Н-ионов тотчас же освобождаются
другие, образующиеся в результате диссоциации остающихся недиссоцииро-
ванных молекул кислоты. Точно такие же явления наблюдаются со щелочью
при титровании ее кислотой с тою лишь разницей, что вместо связанных ки-
.слотой гидроксильных ионов освобождаются другие в результате диссоциа-
ции недиссоциированных молекул щелочи. Вот почему титрование не может
дать представления об активной (истинной) реакции растворов.
Водородное число. Концентрация водородных ионов любого
раствора, выраженная в грамм-ионах на литр, называется его водородным
числом и обозначается одним из следующих символов [Н ], [Н+], Сн‘, Сн, сН.
Так, выражение: [Н’| раствора равно 10-6, означает, что в литре данного
1,008 1,008
раствора находится - ^в- = р^дд ggg г водородных ионов.
Химически чистая вода, реакцию которой считают нейтральной, также
несколько диссоциирована на водородные и гидроксильные ионы. В нейтраль-
ной воде ионная концентрация водородных ионов равна таковой же гидро-
ксильных ионов, т. е. [Н‘] = [ОН'].
При 22°С каждая из этих величин приобретает значение равное 10“1. Это
значит, что на литр химически чистой воды при указанной температуре при-
1,008 17,008
ХОДИТСЯ Ц)7 Г водородных ИОНОВ И —г гидроксильных ионов.
Произведение [Н‘] . [ОН'] носит название ионного произведе-
ния воды и обозначается символом Kaq. При 22°С оно равно 10~7 • 10-7,
т. е. 10-14. С повышением температуры значение для ионного произведения
воды возрастает.
Если к чистой воде прибавить кислоту, напр. НС1, то концентрация во-
дородных ионов в полученном растворе увеличится, так как часть молекул
кислоты окажется диссоциированной на ионы Н‘ и СГ. Подобное увеличение
концентрации водородных ионов подавляет диссоциацию воды, благодаря
чему концентрация гидроксильных ионов в полученном растворе окажется
74
меньше 10-7. Таким образом, во всех водных растворах, обладающих кислы-
ми свойствами, концентрация водородных ионов больше 10-7, а концентрация
гидроксильных ионов меньше 10~7.
Если к чистой воде прибавить щелочь, напр. NaOH, то в воде увеличится
число гидроксильных ионов, так как NaOH частью распадется на ионы Na’
и ОН'. Такое увеличение числа гидроксильных ионов подавляет диссоци-
ацию воды, благодаря чему концентрация водородных ионов в полученном
растворе окажется меньшей, нежели 10-7.
Следовательно, во всех водных растворах, имеющих щелочную реакцию,
концентрация водородных ионов меньше 10-7, а концентрация гидроксиль-
ных ионов больше 1СН7.
Из сказанного следует, что во всяком водном растворе концентрации Н"
и ОН'-ионов являются величинами сопряженными, обратно пропорциональ-
ными одна другой. Повышение концентрации Н’ вызывает равное понижение
концентрации ОН' и обратно. По концентрации любой из этих величин можно
судить о степени кислотности или щелочности раствора. Принято суждение
о степени кислотности или щелочности раствора делать по концентрации
Н‘ -ионов.
Если концентрация водородных ионов равна 10~7, то раствор имеет ней-
тральную реакцию. Кислые растворы имеют концентрацию водородных ионов
выше 10-7, и тем выше, чем кислее раствор. В щелочных растворах концентра-
ция водородных ионов ниже 10-7, и тем ниже, чем щелочность раствора
сильнее.
Показатель водородных ионов. Для выражения кон-
центрации водородных ионов введен символ Ph либо pH, называемый п о-
казателем водородных ионов (Potenz Hydrogenium).
Показатель водородных ионов представляет собой десятичный логарифм
абсолютной концентрации водородных ионов (водородного числа), взятый,
с обратным знаком, т. е. pH——log [Н‘].
Например, для нейтрального раствора с концентрацией водородных ио-
нов равной 10~7, показатель водородных ионов pH будет 7, так как логарифм
10-7 равен — 7, а взятый с обратным знаком равен 7.
Из сказанного следует, что для растворов кислой реакции pH меньше
7, для растворов щелочной реакции pH выше 7.
Рис. 78. Шкала значений pH.
Рис. 78 показывает шкалу значений pH. Середина шкалы соответствует
нейтральной реакции; справа от нее находится область оснований, слева —
кислот.
Для перевода [Н] на pH находят логарифм [Н”| и меняют в нем
знак на обратный. Так, например, если [Н ] равно 0,000447, то отыскав
логарифм этого числа, а именно — 3,3497, находят для pH значение
3,3497.
Для перевода pH на [Н ] меняют знак pH на обратный и находят число
для полученного логарифма. Так, в предыдущем примере, имея для pH зна-
чение 3,3497 и поменяв знак на обратный, получают логарифм искомого [Н ]
т. е. — 3,3497. По полученному логарифму находят значение для [Н], а
именно 0,000447.
Приведенная ниже таблица может служить для быстрого пересчета pH
на [Н ]. Так, напр., если pH = 4,45, то [Н ] = 3,55 . 10 и т. д.
75
Таблица 4
Пересчет pH на [Н-]
pH [Н-] рн 1Н-]
п.00 1 ,оо.ю“ п п.50 3,lG.10“(n+1)
п.05 8,91.10“(п+1) п.55 2,82.10“(П+1)
п.10 7,94.10“(п+1) п.60 2,51.10“(п+1)
п.15 7,18.ю“(п+1) п.65 2,24.10“(п+1)
п.20 6,31.10“(п+1) п.70 2,00.10“(п+1)
п.25 5,63.10“(п+1) п.75 1,78.10“(П+1)
п.30 5,02.10“(п+1) п.80 1,56.10“(П+1)
п.35 4,47.10“(П+1) п.85 1,41.10“(П+1)
п.40 3,98.10“(п+1) п.90 1,26.10“(11+1)
п.45 3,55.10“(п+1) п.95 1,12.10“(п+1)
Приводим примеры pH некоторых веществ:
pH обыкновенной дестиллированной воды, свободной от СО2, около 6,0
» свежей водопроводной воды.................................. 7,5—7,6
» питательного бульона (среда).............................. 7,0—7,5
» свежего молока............................................. 6,3—6,6
» муки....................................................... 5,5—5,95
» меда....................................................... 3,9
» винограда.................................................. 3,4—3,8
Буферы (регуляторы) представляют смеси слабых кислот с их щелоч-
ными солями, например, уксусной кислоты и уксуснокислого натрия, либо
смеси первичных и вторичных кислых солей, например, NaH2PO4 и Na2HPO4.
Такие смеси позволяют регулировать водородные числа растворов, так как
они связывают появляющиеся Н’-ионы и ОН’-ионы. Будучи прибавлены
к какому-либо раствору в количестве одной трети по объему и более, они
способствуют сохранению pH такого забуференного раствора.
Водородные числа буферных смесей оказываются в широких пределах
независимыми от разбавления водой, но буферная способность смесей от этого
слабеет.
Из сказанного следует, что посредством буферных растворов можно ре-
гулировать pH среды; кроме того, они имеют еще и то важное значение, что
дают возможность располагать растворами с любым значением pH.
Для изготовления буферных смесей можно пользоваться имеющимися
для этой цели готовыми таблицами. Ниже приведены таблицы для приго-
товления фосфатных, боратных и цитратных буферных смесей.
Фосфатные буферные смеси готовятся из 1 /15-молярного
первичного фосфата калия (9,078 г перекристаллизованного К Н2РО4 в литре
воды) и 1/15-мол яркого вторичного фосфата натрия (11,876 г перекристаллизо-
ванного Na2HPO4. 2Н2О в литре воды). Дестиллированная вода должна
быть освобождена кипячением от СО2. Соотношения объемов фосфатных ра-
створов в смесях и соответствующее значение pH указаны в таблице 5.
Боратные буферные смеси готовятся из 0,05-мол яркого
раствора буры (19,108 г кристаллической буры в 1 л раствора) и 0,2-моляр-
ного раствора борной кислоты (12,404 г кристаллической борной кислоты
и 2,925 г хлористого натрия в 1 л раствора).
76
Соотношение объемов указанных растворов в боратных буферных смесях
и соответствующее значение pH указаны в таблице 6.
Таблица 5
Фосфатные буферные смеси
i/15 м раствор вто- ричного фосфата в мл V1S М раствор пер- вичного фосфата в мл pH v15 м раствор вто- ричного фосфата в мл V15 М раствор пер- вичного фосфата в мл pH
10,00 0,00 9,182 4,00 6,00 6,643
9,90 0,10 8,679 3,00 7,00 6,468
9,75 0,25 8,338 2,00 8,00 6,239
9,50 0,50 8,043 1,00 9,00 5,906
9,00 1,00 7,731 0,50 9,50 5,589
8,00 2,00 7,381 0,25 9,75 5,288
7,00 3,00 7,168 0,10 9,90 4,944
6,00 4,00 6,979 0,00 10,00 4,494
5,00 5,00 6,813
Таблица б
Боратные буферные смеси
0,05 м раствор буры в мл 0,2 м раствор борной кислоты в мл pH
10,0 0,0 9,24
9,0 1,0 9,11
8,0 2,0 8,98
7,0 3,0 8,84
6,0 4,0 8,69
5,5 4,5 8,60
5,0 5,0 8,51
4,5 5,5 8,41
4,0 6,0 8,31
3,5 6,5 8,20
3,0 7,0 8,08
2,5 7,5 7,94
2,3 7,7 7,88
2,0 8,0 7,78
1,5 8,5 7,60
1,0 9,0 7,36
0,6 9,4 7,09
0,3 9,7 6,77 |
77
Цитратные буферные смеси готовятся из 0,1-молярного
раствора вторичного лимоннокислого натрия (21,01 г перекристализованной
лимонной кислоты растворяют в 200,0 мл 1,0 N — раствора NaOH и добав-
ляют водой до литра), 0,lN раствора НС1 и 0,1 раствора NaOH. Соотношение
объемов указанных растворов в цитратных буферных смесях и соответству-
ющее значение pH указаны в таблице 7.
Таблица 7
Цитратные буферные смеси
0,1 м раствор вто- ричного цитрата в мл 0,lN раствор НС1 в мл 1 pH 1 0,1 м раствор вто- ричного цитрата в мл 0,lN раствор НаОН В МЛ pH
10,00 0,00 4,958 10,00 0,00 4,958
9,50 0,50 4,887 9,50 0,50 5,023
9,00 1,00 4,830 9,00 1,00 5,109
8,00 2,00 4,652 8,00 2,00 5,314
7,00 3,00 4,447 7,00 3,00 5,568
6,00 4,00 4,158 6,00 4,00 5,969
5,50 4,50 3,948 5,50 4,50 6,331
5,00 5,00 3,692 5,25 4,75 6,678
4,75 5,25 3,529 5,00 5,00 9,052
4,50 5,50 3,364 4,50 5,50 12,073
4,00 6,00 2,972 4,00 6,00 12,364
3,33 6,67 2,274
3,00 7,00 1,925
2,00 8,00 1,418
1,00 9,00 1,173
0,00 10,00 1,038
Колориметрический метод определения pH
Колориметрическое (индикаторное) определение pH может быть произве-
дено по двум принципиально различным методам, которые мы приводим
ниже.
Колориметрическое определение pH при помощи буферных растворов
Принцип метода основан на том, что, если буферный и испыту-
емый растворы имеют одну и ту же концентрацию Н’ионов, то индикатор,
прибавленный с соблюдением одинаковых условий к буферному и испытуе-
мому растворам, окажется одинаково диссоциированным и вызовет в силу
этого одинаковое окрашивание этих растворов. В данном случае, следова-
тельно, интенсивность окраски растворов обусловлена степенью диссоциации
индикатора, а эта последняя обусловлена pH растворов.
Для приготовления стандартных растворов пользуются приведенными
выше буферными растворами.
В качестве индикаторов пользуются двухцветными индикаторами. В таб-
лице 8 дана характеристика этих индикаторов.
78
Таблица 8
Характеристика двухцветных индикаторов
Наименование индикатора Изменение цвета от кислоты к щелочи Промежуточная окраска Значение pH в интервале перехода окраски Значение pH для промежу- точной окрас- ки (точки пе- рехода)
Тимоловый синий Красный—жел- тый Желторозовая 1,2—2,8 2,6
Бромфеноловый синий Желтый—синий Зеленая 3,0—4,6 4,1
Метиловый крас- ный (метилрот) Малиновый— желтый Красная 4,2—6,3 5,0
Бромкрезолпур- пуровый Желтый—пур- пуровый Пурпурово— зеленая 5,2—6,8 6,3
Нейтральный крас- ный (нейтральрот) Малиновый— желтый Мясокрасная 6,8—8,0 7,0
Бромтимоловый синий Желты й—сини й Салатнозеле- ная 6,0—7,6 7,1
Феноловый красный Желтый—крас- ный Розовокрасная 6,8—8,4 7,7
Крезоловый красный Желтый—крас- ный Розовокрасная 7,2—8,8 8,1
Тимоловый синий Желтый—синий Синефиолето- вая 7,9—9,8 8,8
Для определения pH растворов, имеющих реакцию близкую к нейтраль-
ной, напр. бактериологических сред, пользуются чаще всего следующими
индикаторами: бромтимоловый синий, феноловый красный и крезоловый
красный, которые меняют окраску в интервале pH близком к 7.
Для приготовления двухцветных индикаторов поступают следующим об-
разом: 0,1 порошка индикатора растворяют в агатовой ступке с определен-
ными количествами 0,05 N раствора NaOH:
для бромфенолового синего............. 3 мл
» метилового красного................. 7,4 »
» бромкрезолового пурпурного.......... 3,7 »
» бромтимолового синего...............3,2 »
» фенолового красного................. 5,7 »
» крезолового красного ............... 5,3 »
» тимолового синего................... 4,3 »
Растворы готовятся водные или спиртовые, причем метиловый красный
и феноловый красный — в концентрации 0,02%, для чего растворенную на-
веску индикатора в едком натрии доводят водою или спиртом до 500 мл;
остальные растворы готовятся в концентрации 0,04%, для чего растворен-
ную навеску индикатора в едком натрии доводят водою или спиртом до 250 мл1.
Растворы индикаторов хранят в закрытых флаконах в прохладном месте.
Для колориметрического определения pH по буферному методу кроме
описанных выше индикаторов применяются также так называемые универ-
сальные индикаторы, пригодные для всего ряда изменений pH.
Универсальные индикаторы можно приготовить по следующим двум ре-
цептам:
1) 0,05 г метилоранжа; 0,15 г метилрота; 0,3 г бромтимолсинего; 0,35 г
фенолфталеина и 1 л 66% алкоголя;
1 В лабораторной практике обычно готовят растворы индикаторов в 10 раз крепче
(исходные) и по мере надобности разбавляют их в 10 раз (рабочие растворы).
79
2) 0,1 г метилоранжа; 0,04 г метилрота; 0,4 г бромтимолсинего; 0,32 г наф
толфталеина; 0,5 г фенолфталеина и 1,6 крезолфталеина в 100 мл 70% алкоголя.
Методика определения. При помощи буферных раствороЕ
(стр. 77—78) составляют ряд стандартов с известным значением pH с таким рас-
четом, чтобы значение pH испытуемого раствора находилось в интервале
значений pH стандартного ряда. При этом пользуются пробирками одинако-
вого диаметра. На пробирки наклеивают этикетки с обозначением соответ-
ствующего pH. В пробирку такого же диаметра наливают 10 мл испытуемого
раствора. Во все стандартные пробирки и в опытную пробирку прибавляют
одинаковое количество (напр., по 1 мл) соответствующего индикатора, выбор
которого зависит от приблизительного значения pH испытуемого раствора,
которое предполагается известным. Если окраска исследуемого раствора
совпадает с окраской
Рис. 79. Компаратор двухрядный,
а, Ъ и с — смотровые отверстия; справа номера отверстий
для пробирок.
одного из стандартных
растворов, то pH пер-
вого равно pH второго.
Если pH испытуемого
раствора неизвестно да-
же в приближении, то,
чтобы выяснить, с каким
индикатором следует ра-
ботать, можно устано-
вить pH раствора при-
близительно следующим
образом: к некоторому
количеству испытуемого
раствора прибавляют в
качестве индикатора та-
кой из них, изменение
окраски которого происходит в интервале pH, охватывающем и кислые и
щелочные растворы, напр., бромтимоловый синий. Прибавив к испытуемому
раствору такой индикатор, сразу видят, является ли реакция данного ра-
створа кислой, щелочной или она близка к нейтральной.
В зависимости от полученного результата подбирают затем соответству-
ющие индикаторы, пригодные для кислых или щелочных растворов, и по
окраске, которую они дают с испытуемым раствором, устанавливают приб-
лизительно pH этого раствора; затем следует точное определение pH раство-
ра, как описано выше.
Приблизительное значение pH раствора можно установить также при
помощи универсального индикатора. Для этой цели 3—5 капель испытуемого
раствора смешивают с 3—5 каплями универсального индикатора в маленькой
фарфоровой чашке (палетке) и сравнивают полученную окраску с цветной
шкалой, нанесенной на бумагу. Шкалу легко составить самому, зарисовывая
окраску, которая получается, если смешивать по 3—5 капель универсального
индикатора с 3—5 каплями буферного раствора с возрастающим и известным
значением pH.
Если исследуемая жидкость окрашена или мутна, то рекомендуется при-
менять для колориметрирования прибор, носящий название компаратор
(сошрагаге — сравнивать).
Компаратор двухрядный (рис. 79) представляет собой деревянную
колодку в форме призмы, в которой имеются гнезда 7, 2, 3, 4, 5 и 6 для про-
бирок и круглые отверстия а, b и с для наблюдения.
Применение компаратора производится следующим образом: в гнездо
4 помещают пробирку с исследуемым раствором + индикатор; в гнездо 3 ста-
вят пробирку с дестиллированной водой; в гнезда 7 и 5 — пробирки с испы-
туемым раствором без индикатора. В гнезда 2 и 6 помещают поочередно про-
бирки со стандартными растворами, к которым прибавлен такой же индика-
R0
тор, как и к испытуемому раствору. Стандартные растворы подбираются
такие, чтобы окраска испытуемого раствора по интенсивности совпала с ок-
раской одного из этих растворов или находилась между окраской того и дру-
гого раствора. Окраску сравнивают, смотря в отверстия а, b и с. Благодаря
такому способу влияние собственной окраски испытуемого раствора элими-
нируется.
Для лучшей ориентировки в нюансах окраски применяют матовый и си-
ний светофильтры, которые помещают позади смотровых отверстий.
Колориметрическое определение pH без буферных растворов
Колориметрическое определение pH без буферных растворов произво-
дится по одному из описанных ниже методов.
Метод колориметрического определения pH с одноцветными индикаторами.
Принцип метода основан на том, что применяемые одноцветные
индикаторы бесцветны в состоянии молекул и окрашены в диссоциированном
состоянии. Интенсивность окраски раствора индикатора в определенных
пределах зависит от концентрации ионов индикатора. Испытуемый раствор,
к которому прибавлен индикатор, сравнивают с рядом растворов щелочи,
к которым прибавлен тот же индикатор в постепенно повышающихся количе-
ствах. В этих щелочных стандартах индикатор полностью диссоциирован.
В данном случае, следовательно, разная интенсивность окраски индикаторов
объясняется не различной частичной диссоциацией взятых равных количеств
индикатора, а разными количествами индикатора при его полной диссоциации.
В этом и заключается принципиальное отличие данного метода индикаторно-
го определения pH от описанного выше.
На основании данных составления стандартов определяется их pH (см.
таблицу 9).
Отыскав среди стандартов такой, который имеет окраску, тождественную
с окраской испытуемого раствора, можно принять, что pH испытуемого рас-
створа равен pH стандарта.
Необходимые реактивы: 1) 0,05% водный раствор а—двунитрофе-
нола; 2) 0,025% водный раствор 7—двунитрофенола; 3) 0,1% водный раствор
паранитрофенола; 4) 0,3% водный раствор метанитрофенола; 5) децинор-
мальный раствор Na2CO3.
Для приготовления эталонов подбирают пробирки одинакового диаметра
и приготовляют с каждым индикатором по 8—9 смесей с разным значением
pH, получая таким образом 4 ряда эталонов. Для первого ряда берут 5 мл
раствора а—двунитрофенола и разводят до 50 мл 0,1N раствором соды;
для других рядов поступают так же с реактивами NN 2, 3 и 4. Затем смеши-
вают в указанных пропорциях (табл. 9) растворы индикаторов и 0,1N раствор
соды, получая нужные эталоны.
Таблица 9
Данные для приготовления эталонов с определенными pH
Ряд 1 Ряд 11 Ряд III Ряд I V
е' ео О п о • — е» о л о
£ pH Я* О й 0.0 И 1 о Я «6- И 0,1 N Na2 С' в мл pH д' о и S о. о 5 Н Я S Я О я •©• и Z.° 5 е» g — Г5 о Z со pH Пара- нитро фенот в мл o'Z со pH 2 о.® < я •©• я 0,1 N Na2 С в мл
1 2,8 0,51 6,49 4,0 0,74 6,26 5,4' 0,16 6,84 6,8 0,27 6,73
2 3,0 0,78 6,22 4,2 1,10 5,90 5,6 1 0,25 6,75 7,0 0,43 6,57
3 3,2 1,20 5,80 4,4 1,65 5,35 5,8 0,4 6,6 7,2 0,66 6,34
4 3,4 1,74 5,26 4,6 2,4 4,6 6,0 0,63 6,37 7,4 1,0 6,0
5 3,6 2,5 4,5 4,8 3,4 3,6 6,2 0,94 6,06 7,6 1,5 5,5
б 3,8 3,4 3,6 5,0 4,5 2,5 6,4 1,4 5,6 7,8 2,3 4,7
7 4,0 4,6 2,4 5,2 5,5 1,5 6,6 2,08 4,92 8,0 1 3,0 4,0
8 4,2 5,7 1,3 5,4 6,6 0,4 6,8 3,00 4,0 8,2 4,0 2,8
9 4,4| 6,7 0,3 — । — — 7,0[ 4,05 1 2,95 8,4 | 5,2 1,8
6 А. II. Бурштейн
81
Приготовив эталоны, герметически закрывают пробирки. Полученные
растворы могут сохраняться в темном месте довольно долго. На каждую про-
бирку наклеивают этикетку с указанием номера ряда и pH раствора.
Для определения pH раствора необходимо иметь, кроме описанных эта-
лонов, также индикаторные реактивы: 1, 2, 3 и 41.
Методика определения. Подбирают пробирку такого же ди-
аметра, как и в эталонах, вводят туда 6 мл испытуемой жидкости, прибав-
ляют 1 мл того индикатора, с которым данная жидкость дает окрашивание.
Если окрашивание получается с несколькими индикаторами, то берут послед-
ний из них. Затем из соответствующего ряда эталонов подбирают тот, который
по окраске ближе других подходит к окраске испытуемой жидкости, и про-
читывают pH на этикетке найденного эталона. Это и будет pH испытуемой
жидкости. Если окраска находится между двумя соседними эталонами, то
берут среднее значение этих двух эталонов.
Так поступают в том случае, если исследуемая жидкость не имеет своей
собственной окраски и не мутна; в противном случае применяют для срав-
нения компаратор.
В пробирку такого же диаметра, как и эталонные, вводят 2 мл испыту-
емой жидкости, 4 мл дестиллированной воды2 и 1 мл индикатора. Все тщатель-
но перемешивают и ставят пробирку в гнездо 4, а в гнездо 3 ставят такую
же пробирку с дестиллированной водой. В гнезда 1 и 5 помещают пробирки,
содержащие 2 мл испытуемой жидкости и 5 мл дестиллированной воды.
В гнезда 2 и 6 ставят по очереди эталонные пробирки с тем же индикатором,
какой был применен для испытуемой жидкости. Окраску сравнивают, смотря
в отверстия а, b и с. Для лучшей ориентировки в нюансах окраски приме-
няют светофильтры матовый и синий.
Точность индикаторного метода не превышает обычно 0,05 pH.
Присутствие в исследуемом растворе больших количеств солей, белков,
алкалоидов приводит к ошибкам при применении индикаторного метода опре-
деления pH. Это так называемые солевая, белковая и алка-
лоидные ошибки метода.
Метод колориметрического определения pH с двухцветными индикатора-
ми. Принцип метода основан на следующем: приготовляют два
раствора индикатора — кислый и щелочной. В кислом растворе все молекулы
индикатора находятся в недиссоциированном, в щелочном—в диссоциирован-
ном состоянии. Если рассматривать оба раствора в проходящем свете, то
суммарная окраска обоих растворов окажется такой, как если бы недиссоци-
ированные и диссоциированные молекулы индикатора находились вместе в
одном растворе.
Соотношения количеств индикатора в кислом и щелочном растворах под-
бираются так, чтобы суммарная окраска обоих растворов соответствовала
окраске, получающейся при определенной степени диссо-
циации данного индикатора и, следовательно, отвечала опреде-
ленному значению pH.
Так, если в кислый раствор ввести 9 капель раствора индикатора, а в
щелочной — 1 каплю того же раствора индикатора, то суммарная окраска
соответствует окраске при степени диссоциации индикатора, равной г/10
(или 10%); если в кислый раствор ввести 8 капель, а в щелочной 2, то сум-
марная окраска соответствует окраске при степени диссоциации индикатора,
равной 2/10 (или 20%) и т. д.
С такой суммарной окраской целого стандартного ряда из попарно состав-
ленных пробирок сравнивают окраску испытуемого раствора, к которому
прибавлен тот же индикатор.
1 Полный набор эталонов и индикаторных реактивов в портативном ящике вместе
с компаратором изготовляют у нас «Агроном» и «Гослаборснабжение».
* Если испытуемая жидкость содержит в растворе NaCl (биологические среды и пр.),
то вместо воды берут раствор NaCl соответствующей концентрации.
82
Найдя соответствующий стандарт (состоящий из пары пробирок), окра-
ска которого соответствует окраске исследуемого раствора, можно по pH
стандарта определить pH испытуемого раствора.
Реактивы и приборы. 1. Спиртовые растворы двухцветных
индикаторов (приготовление см. стр. 79).
2. 0.05N раствор NaOH (можно взять 0,2% раствор NaOH).
3. 0.05N раствор НС1 (можно взять 0,4% раствор НС1, исходя из HCI
уд. в. 1,19).
4. 2% раствор КН2РО4.
5. 20 пробирок одинакового диаметра (1,5 см), калибрированных на 5 мл,
и штатив к ним на два ряда.
6. Трехрядный компаратор (рис. 80) с девятью вертикальными гнездами
для пробирок и тремя смотровыми отверстиями. На задней стенке против
смотровых отверстий укреплен матовый светофильтр.
Рис. 80. Компаратор трехрядный;
а, Ъ и с — смотровые отверстия; справа номера отверстий
для пробирок.
Методика определения. Приготовляют стандартный ряд. Для
этого в штатив устанавливают 18 пробирок в два ряда. В 9 пробирок первого
ряда вносят по одной капле 0.05N раствора NaOH, в 9 пробирок второго
ряда— по одной капле 0,05N раствора НС1. В первую пробирку первого
ряда вводят одну каплю данного индикатора, во вторую — две, в третью
три и т. д. до 9 капель. В первую пробирку второго ряда вводят 9 капель
того же индикатора, во вторую — 8 капель, в третью — 7 и т. д. до одной
капли. Сумма капель индикатора в каждой паре пробирок передней и задней
составляет 10 капель. Содержимое каждой пробирки доливают водой до 5 мл.
Рекомендуется ввести еще дополнительно две пары пробирок 1-а и 8-а.
В пробирку 1-а щелочного ряда прибавляют 3 капли индикатора и две капли
щелочи, а в пробирку 1-а кислотного ряда—17 капель индикатора и
2 капли кислоты и доливают содержимое пробирок водой до 10 мл. В пробирку
8-а щелочного ряда прибавляют 17 капель индикатора и 2 капли щелочи,
а в пробирку 8-а кислотного ряда — 3 капли индикатора и 2 капли кислоты
и доливают водой до 10 мл.
Вместо капель лучше отмеривать индикатор пипеткой от 0,1 до 0,9 мл;
кислоты и щелочи при этом следует брать по 2 капли.
Пробирки закрывают парафинированными пробками и этикетируют. На
этикетках указывают соответствующий номер пары.
pH каждой пары пробирок для соответствующего индикатора указан в
таблице 10. Эти значения верны для температуры в пределах 25—30°С. При-
готовленный таким образом стандартный ряд может храниться несколько дней
83
Таблица 10
Значение pH, соответствующее каждой паре пробирок
1 — №№ пар про- бирок Количество капель индикатора pH, соответствующий каждой паре пробирок
Ще- лочной ряд Кис- лый ряд Бромфе- ноловый синий Метило- вый красный Бромкре-| Бромти- золовый моловый Феноло- вый красный Крезо- ловый красный Тимоло- вый синий
пурпур- ный | синий
1 1 9 3,1 4,05 5,3 6,15 6,75 7,15 7,85
1а 1,5 8,5 3,3 4,25 5,5 6,3 6,95 7,35 8,05
2 2 8 3,5 4,4 5,7 6,5 7,1 7,5 8,2
3 3 7 3,7 4,6 5,9 6,7 7,3 7,7 8,4
4 4 6 3,9 4,8 6,1 6,9 7,5 7,9 8,6
5 5 5 4,1 5,0 6,3 7,1 7,7 8,1 8,8
б 6 4 4,3 5,2 6,5 7,3 7,9 8,3 9,0
7 7 3 4,5 5,4 6,7 7,5 8,1 8,5 9,2
8 8 2 4,7 5,6 6,9 7,7 8,3 8,7 9,4
8а 8,5 1,5 4,8 5,75 7,0 7,85 8,45 8,85 9,55
9 9 1 5,0 5,95 7,2 8,05 8,65 9,05 9,75
Определение pH испытуемого раствора производится следующим образом.
В пробирку такого же диаметра, как и стандартные, вводят 5 мл испытуемой
жидкости и 10 капель индикатора, тщательно перемешивают и ставят в гнез-
до 6 трехрядного компаратора, спереди в гнезда 5 и 4 ставят такие же пробирки
с дестиллированной водой; в гнезда 1 и 7 ставят пробирки с 5 мл исследуе-
мого раствора без индикатора. В гнезда 2, 3, 8 и 9 ставят последовательно
пары стандартов и сравнивают оттенки окрасок в проходящем дневном
свете.
Находят ту пару пробирок, которая дает окраску, тождественную с испы-
туемым раствором, и по таблице 10 соответственно порядковому номеру этой
пары пробирок и применяемому индикатору находят pH раствора. Ошибка
метода по сравнению с электрометрическим лежит в пределах 0,1 pH.
Описанным способом определяют pH растворов, обладающих хорошо выра-
женной буферностью, напр., питательных сред, применяемых в бактериоло-
гической практике. Если консистенция среды твердая (агар, желатина), то
среду разжижают и разбавляют ее в 7 раз теплой дестиллированной водой
или физиологическим раствором; при этом, ввиду высокой буферности
питательных сред, их значения pH от разбавления среды водой не изме-
няются.
Если испытуемый раствор не обладает выраженной буферностью (воды
питьевые, сточные и т. п.), то при определении pH такого раствора по опи-
санному методу нужно иметь в виду следующие примечания:
1. При применении индикатора бромфенолового синего в кислотный ряд
пробирок вместо 1 капли 0,05N НС1 берут по 20 капель или 1 мл.
2. При применении индикаторов крезолового красного и тимолового
синего для подкисления вместо соляной кислоты берут по одной капле 2%
раствора КН2РО4.
3. При применении индикатора тимолового синего в щелочной ряд вместо
1 капли 0,05 N NaON прибавляют 2 капли.
84
В лаботаторной практике нередко приходится не только определять pH
растворов, но устанавливать наперед заданное для них pH, напр.,
при изготовлении питательных сред.
В этом случае поступают следующим образом: определяют pH среды од-
ним из описанных методов и, если оно не соответствует заданному, то прибав-
ляют во взятую пробу из микробюретки или микропипетки 0,05N раствор
НС1 или NaOH до получения нужного pH.
Расход кислоты или щелочи перечисляют затем на весь объем среды.
При этом нужно иметь в виду два обстоятельства:
1. pH сред, установленный при комнатной температуре (18—20°С), пони-
жается против установленных значений в условиях выдерживания в термо-
стате при 37°С.
2. После стерилизации pH сред несколько понижается, и только в том
случае, если усреднение среды было произведено содой (Na2CO3), pH после
стерилизации несколько возрастает. Пробной стерилизацией можно выяснить
изменения pH и учесть корректив при установлении pH среды.
Электрометрический (потенциометрический)
метод определения pH
В тех случаях, когда растворы заметно окрашены, мутны, а также и тогда,
когда они содержат большое количество белков, солей или алкалоидов, коло-
риметрическое определение pH дает неточные результаты, и предпочтительно
производит определение pH электрометрическим (потенциометрическим) ме-
тодом.
Этот метод является более точным, нежели колориметрический и потому
применяется еще и в тех случаях, когда требуется большая точность в изме-
рениях pH.
Принцип электрометрического определения pH основан на следую-
щем. При погружении металла в раствор его соли, между металлом и солью
возникает разность потенциалов, величина которой является функцией кон-
центрации металлических ионов в растворе.
Платиновый электрод, покрытый платиновой чернью и сорбировавший
на своей поверхности водород, ведет себя так, как водородный элект-
род. При погружении его в раствор между ним (электродом) и раствором
возникает разность потенциалов, величина которой представляет функцию
концентрации водородных ионов в данном растворе. Таким образом, по потен-
циалу водородного электрода можно судить о концентрации водородных
ионов в растворе, и следовательно, о pH данного раствора.
Очевидно, необходимо в каждом отдельном случае уметь измерить возни-
кающий потенциал на водородном электроде при погружении его в раствор,
чтобы иметь возможность определить pH этого раствора.
Измерение потенциала, возникающего на водородном электроде при погру-
жении его в испытуемый раствор, производится путем сравнения этого потен-
циала с каким-либо постоянным.
Постоянный потенциал можно получить посредством так называемого
нормального водородного электрода, т. е. водородного
электрода, погружаемого в нормальный раствор сильной кислоты.
На практике, однако, удобнее вместо нормального водородного электрода
пользоваться в качестве электрода сравнения каломельным электро-
дом, сохраняющим постоянство потенциала при данной температуре. Он пред-
ставляет собой ртутный электрод, соприкасающийся с насыщенным раствором
каломели в хлористом калии.
Оба полуэлемента — водородный и каломельный — соединяются в один
гальванический элемент. Если определить электродвижущую силу такого
элемента, то, зная потенциал одного из полуэлементов его (каломельного),
можно определить потенциал другого полуэлемента (водородного), а зная
85
& G
Рис. 81. Принципиальная схема электро-
метрического определения pH по компенса-
ционному принципу.
Еа — известная электродвижущая сила, Ех — не-
известная электродвижущая сила, АВ —реохорд,
G — гальванохметр, С — подвижный контакт.
потенциал водородного полуэлемента, как уже было указано, можно опреде-
лить pH раствора, в который погружен водородный электрод.
Электродвижущая сила водородно-каломельного элемента, ввиду ее малых
размеров, определяется по специальному так называемому компенса-
ционному методу, сущность которого легко уяснить себе из схемы,
представленной на рис. 81.
Здесь Еа известная електродвижущая сила (аккумулятор, сухой элемент и т. п.).
Клеммы аккумулятора соединены с концами тонкой констатановой1 проволоки сопротив-
ления АВ, лежащей на линейке, разделенной на сантиметры и миллиметры (реохорд). Об-
разованная таким образом цепь ЕаАВЕа
называется большой цепью.
Проволока сопротивления по всей длине
имеет строго одинаковое сечение и потому
падение потенциала происходит совершенно
равномерно от начала к концу измеритель-
ной линейки.
Ех представляет электродвижущую силу
элемента, которая должна быть определена.
От клемм этого элемента отходят две про-
волоки, из коих одна закреплена в точке А,
другая через гальванометр G соединена при
помощи скользящего контакта с проволокой
сопротивления, причем контакт может быть
создан в любой точке проволоки сопротив-
ления, на рисунке контакт установлен в
точке С. Цепь ExACGEx называется малой
(боковой) цепью. Отрезок АС яв-
ляется общим участком для обеих цепей.
Перемещая подвижный контакт, можно
добиться такого положения его, когда часть
напряжения элемента Еа, приходящаяся на
отрезок АС, окажется равной всему напря-
жению Ех. В этот момент гальванометр по-
кажет отсутствие тока — момент к о м-
при положении контакта в точке С, тогда,
проволок, как имеющих довольно большое
A
пенсации. Положим, что это произойдет
пренебрегая сопротивлением соединительных
сечение, можно написать:
откуда
Еа Ех
АВ = АС>
ЕаАС
Ех~ АВ ,
где Ех — искомая электродвижущая сила элемента,
Еа — известная электродвижущая сила аккумулятора,
АВ и АС — отрезки на реохорде.
По описанному (схематически) компенсационному методу определяется электродви-
жущая сила водородно-каломельного элемента, при помощи которого измеряется pH рас-
твора.
Определив электродвижущую силу водородно-каломельного элемента Ех и учтя по-
тенциал каломельного полуэлемента Ео, находят pH исследуемого раствора по следующей
формуле:
Ех— Ео
рН=—--------- .
где Ех — електродвижущая сила водородно-каломельного элемента,
Ео — потенциал каломельного электрода,
$ — абсолютная температура, при которой производится измерение, умноженная
на постоянный коэфициент 0,1983.
Приведенная формула выведена на основании следующих соображений. Если со-
ставить элемент из двух одинаковых металлических электродов, погруженных в растворы
соли разной концентрации этого же металла, то электродвижущая сила такого элемента
(концентрационной цепи) выразится следующим равенством:
RT In Ci
Ех =----- In —L ,
nF С2
1 Констатан — сплав, состоящий из 58% меди, 41% никеля и 1% марганца; коэфи-
циент расширения 0,000025.
66
где Ex — электродвижущая сила концентрационной цепи,
R— газовая постоянная, равная 8,313 джоуля или 8313 милли во льт-кулонам,
Т — абсолютная температура,
п — валентность ионов металла,
F — электрохимический эквивалент или число Фарадея, равное 96 500 кулонам
(округленно),
Cj — концентрация ионов металла в растворе одного электрода,
С2 — концентрация ионов металла в растворе другого электрода.
В частном случае, когда концентрационная цепь составлена из двух водородных электро-
дов, причем один из них (электрод сравнения) является нормальным, можно в приведен-
ное выше уравнение вместо л поставить единицу; вместо Сп т. е. концентрации водооодных
ионов в нормальном водородном элементе можно также поставить единицу. Если с2, т. е.
концентрацию водородных ионов в испытуемом элементе, обозначить через [Н+], то уравне-
ние примет следующий вид:
„ RT ,1 ,1 ExF . гтл+1
Ex =---- In----откуда In---------------- или —lnlH+J=--------
F |H + ] [Hb] RT RT
Переходя от натуральных логарифмов к десятичным, получаем:
— 2,3026 1g [ Н+] = или
RP
Вместо— lg [H+J можно написать pH; получим:
ExF
2,3026RT
ExF _ Ex
2,3026/?Т ~ 2,3026/?
F
Подставив вместо R и F их значения, получаем:
.. Ex Ех
pH =----------------= ----------.
2,3026 - 8313 т 0,1983Т
96 500
В каломельно-водородном элементе вместо нормального водородного электрода взят
каломельный электрод, потенциал которого по отношению к нормальному водородному
электроду известен. При вычислении значений pH следует значение потенциала каломель-
ного электрода по отношению к нормальному водородному элементу (Ео) вычитать из ЭДС
водородного электрода в исследуемой жидкости по отношению к каломельному электроду
(Ех). Таким образом, формула примет следующий вид:
Ех— Ео _ Ех — Ео
0,1983Т “ 0
Значения для Ео и 3 приведены в нижеследующих таблицах 11 и 12.
Таблица 11
Потенциал каломельного электрода (Ео) при разных температурах
t°C Напряжение в милливольтах t°c Напряжение в милливольтах t°c Напряжение в милливольтах
16 252,0 19 249.5 1 22 247,7
17 251,5 20 249,1 23 247,1
18 250,5 21 248,3 24 246,7
Таблица 12
Значения для 9 при разных температурах
t°C & t°C & !• 1 °
15 57,1 19 57,9 23 58,7
16 573 20 58,1 24 58,9
17 57,5 21 58,3 25 59,1
18 57,7 22 58,5 26 593
87
Описанная выше компенсационная схема лежит в основе большинства приборов, при-
меняемых для определения pH. В эту схему вносят некоторые дополнения.
Важнейшим дополнением является введение в схему нормального элемента Вестона,
электродвижущая сила которого постоянна. Введение в систему нормального элемента с по-
стоянной электродвижущей силой вызывается тем, что ЭДС аккумулятора непостоянна.
Нормальный элемент не заменяет в системе аккумулятора, а служит лишь для проверки
его электродвижущей силы. Другим дополнением является добавочное сопротивление, кото-
рое вводится в большую цепь с целью, если это вызывается необходимостью понизить напря-
жение элемента в главной цепи, напр., если оно выше 2 V и точка компенсации для нормаль-
ного элемента смещена значительно влево от середины линейки реохорда. Далее в систему
вводится ключ Морзе для одномоментного включения и выключения боковой цепи и пере-
ключатель для введения в боковую цепь нормального или исследуемого элемента.
Наконец, нужно указать еще на то, что нередко в установках для электрометрического
определения pH вместо водородного пользуются другими электродами.
Приводим описание различных установок для электрометрического определения pH.
Потенциометрическое определение pH с помощью водородно-каломельпого элемента
Для данного метода, принцип которого описан выше, необходимы следую-
щие приборы: 1) аккумулятор в 2V, 2) рубильник, 3) реостат в 130—150 ом,
Рпс. 82. Нормальный кадмиевый
элемент.
Н со впаянными в основание
4) реохорд, 5) нормальный кадмиевый эле-
мент, 6) водородный электрод, 7) кало-
мельный электрод, 8) «солевой мост» или
электролитический ключ, 9) капиллярный
электрометр или чувствительный гальвано-
метр, 10) переключатель, 11) ключ Морзе,
12) заряженный аппарат Киппа для полу-
чения водорода, 13) прибор для платини-
рования электродов.
Ниже дается описание всех указанных
частей установки за исключением извест-
ных из элементарных курсов физики.
Нормальный кадмиевый
элемент (рис. 82) представляет собой
стеклянный сосуд, имеющий форму буквы
платиновыми проволочками. В одно колено
сосуда наливают немного химически чистой ртути (положительный полюс
элемента) так, чтобы покрыть впаянную проволочку. В другое колено вводят
немного слегка подогретой кадмиевой амальгамы, которую готовят в фарфо-
ровом тигле из химически чистого кадмия и такой же ртути, взятых в отно-
шении 1 : 7 (отрицательный полюс элемента).
На поверхность ртути в первом колене наносят слой пасты, которую при-
готовляют, растерев в ступке несколько капель ртути и смесь из равных
частей сернокислой закиси ртути — Hg2SO4 и сульфата кадмия — CdSO4.
Сернокислая закись ртути предварительно промывается несколько раз насы-
щенным раствором CdSO4 и во влажном виде идет на изготовление пасты.
Слой пасты должен быть высотой около 0,5 см. Остальное пространство в
обоих коленах заполняется насыщенным раствором сульфата кадмия и кри-
сталлами последнего. Оба колена закрывают парафинированными пробками
так, чтобы под ними остался небольшой слой воздуха. Сверху пробки заливают
шелаком, парафином или менделеевской замазкой (см. стр. 38).
Схема описанного элемента может быть представлена в следующем виде:
+ Hg|Hg2SO4|CdSOJCd~.
Кадмий, обладающий большой электролитической упругостью растворения,
переходит в раствор и вытесняет ртуть из Hg2SO4. При этом кадмиевый элект- •
род заряжается отрицательно, а ртутный положительно.
Электродвижущая сила такого элемента равна 1,0183—l,0186v; она прак-
тически не зависит от температуры.
88
Рис. 83. Грушевидный водородный
электрод;
а — трубка длп ввода водорода, b — трубка
для выхода газа, с — трубка длп введения
исследуемого раствора в сосуд.
Нормальный элемент требует очень бережного отношения: необходимо
избегать толчков и сотрясений; резких смен температуры; длительного вклю-
чения в цепь и т. п. Хранение элемента рекомендуется при температурах не
ниже 4° С и не выше 40° С. Проверка элемента производится авторитетными
учреждениями.
Водородный электрод часто применяется в форме грушевид-
ного сосуда, изображенного на рис. 83. В стеклянную пришлифованную пробку
сосуда впаяна платиновая проволока, покрытая платиновой чернью (способ
платинирования описан ниже). Платиновая проволочка спаяна с медной про-
водящей ток проволочкой, а последняя — с клеммой электрода. От основной
грушевидной части прибора отходят три трубки, снабженные кранами. Через
трубку а в сосуд вводится струя водорода, через трубку b водород выводится
наружу. Трубка с служит для заполнения грушевидного сосуда испытуемой
жидкостью и для соединения водородного
полуэлемента с каломельным полуэлемен-
том в один элемент.
Введение испытуемой жидкости в гру-
шевидный сосуд производится следующим
образом: на трубку с одевают резиновую
трубку, конец которой погружают в испы-
туемую жидкость. Открывают краны в
трубках си би при закрытом кране трубки
а насасывают жидкость, высасывая воздух
из груши сосуда через трубку Ь. Наполнив
грушевидный сосуд до половины, закрывают
краны трубок с и Ь. Затем из аппарата
Киппа пропускают струю водорода через
испытуемую жидкость (подробнее см. ниже.)
Водород насыщает платиновую прово-
локу, нижняя часть которой погружена
в раствор, а верхняя находится в атмосфере
водорода. Насыщение электрода длится
5—8 минут, после чего пропускание водо-
рода через раствор прекращают. Дальнейшее насыщение платины водородом
поддерживается за счет водорода, оставшегося в электроде.
Сорбированные платиной молекулы водорода частью распадаются на
атомы:
Н2—> 2Н.
Часть атомов водорода, отщепляя по электрону, переходит в виде ионов в
раствор:
Н Н‘ + е.
С другой стороны ионы водорода из раствора выделяются на платине в виде
атомного водорода:
Н-}- е — Н.
Отщепленные водородными атомами электроны остаются на электроде, в
связи с чем водородный электрод приобретает отрицательный заряд. Вслед-
ствие электростатического взаимодействия выделенные электродом положи-
тельно заряженные ионы водорода располагаются у отрицательно заряженного
электрода, образуя двойной электрический слой.
Возникающие электростатические силы препятствуют как дальнейшему
переходу ионов в раствор, так и выделению их из раствора на электроде, благо-
даря чему концентрация водородных ионов исследуе-
мого раствора не меняется.
По окончании работы исследуемый раствор из сосуда удаляют. Сосуд про-
мывают через трубку с дестиллированной водой и заполняют таковой же
89
Рис. 84. U-образ-
ный водородный
электрод;
Е — пробна с плати-
нированным электро-
дом. V — стеклянная
пробка второго ко-
лена. R — отверстие
в стенне сосуда; ря-
дом второе отверстие
в стенке пробки.
Рис. 85. Каломельный
электрод;
а — трубочка со впаян-
ной платиновой прово-
локой в коние Ь— сосуд
каломельного электрода,
с — соединительное ко-
лено каломельного элек-
трода. d — стеклянная
пробка, переходящая в
трубку, р — кран.
амальгамирование
как катод, в 1 %
до следующего определения. Стеклянная пробка из сосуда не должна вы-
ниматься.
U-o бразный водородный электрод. В тех случаях, когда
исследуемый раствор содержит в большом количестве свободный угольный
ангидрид, пропускание через раствор водорода приводит к вытеснению СО2.
в связи с чем получаются повышенные результаты для pH.
Чтобы избежать этого, пользуются U-образным водо-
родным электродом (рис. 84). Испытуемый раствор вводят
в U-образную трубку так, чтобы колено Е оказалось все
заполненным; затем при помощи капиллярной трубки, рас-
ширенной с одного конца и присоединенной этим концом
к аппарату Киппа, вводят в колено и водород сначала, не
касаясь концом трубки жидкости, чтобы вытеснить из колена
воздух, а затем конец капилляра погружают в жидкость до
самого изгиба трубки и вводят очень маленькими пузырь-
ками водород, который собирается в колене Е, оттесняя жид-
кость от пробки. Нужно ввести столько газа, чтобы пла-
тиновая проволока была погружена на 1—2 мм в жидкость.
После этого колено v заполняют испытуемой жидкостью
до самого верха и закрывают пробкой. Излишек вытекает
через отверстие /?, после чего пробку поворачивают, чтобы
отверстие оказалось закрытым.
Водородный пузырек прогоняют 30—50 раз взад и впе-
ред через жидкость, наклоняя трубку в сторону то одного,
то другого колена. Жидкость оказывается насыщенной
водородом без потери СО2. Такой электрод называется «водородным
электродом с неподвижным пузырьком водорода».
«Солевой мост» («э л е кт ро л и ти че с к и й клю ч») пред-
ставляет собой небольшую трубочку, изогнутую в виде сифона и наполнен-
ную 3% раствором агара, к которому прибавлен хлористый калий из расчета
40 г на 100 мл агаровой жидкости. Приготовив указанную жидкость, ее в
горячем виде насасывают в трубочку, где и лают ей за-
стыть. Жидкость должна сплошным слоем заполнять
трубочку; нужно избегать попадания в нее пузырьков
воздуха, которые мешают проведению тока.
«Солевой мост» служит для включения полуэлементов
в цепь. На рис. 90 «солевой мост» в виде трубочки d
соединяет водородный электрод (3) с каломельным элек-
тродом (4) в один гальванический элемент. Это соедине-
ние осуществляется через ванночку с насыщенным рас-
твором КС1.
Вне работы «солевой мост» должен быть погружен
своими концами не менее как на 4—5 см в насыщенный
раствор КС1.
Каломельный электрод имеет форму,
изображенную на рис. 85. В трубочку а у нижнего конца
ее впаивается платиновая проволочка, служащая кон-
тактом. Платиновая проволочка, как и в водородном
электроде, спаяна с медной, проводящей ток проволоч-
кой, а последняя с клеммой электрода. Наружный конец
платиновой проволоки амальгамируется. Перед амаль-
гамированием его тщательно очищают серной кислотой;
проводится следующим образом: проволока вводится,
раствор азотнокислой закиси ртути HgNO3, к которой прибавлено несколько
капель азотной кислоты. Вместо 1 % раствора HgNO3 можно применять
2% раствор сулемы HgCl2. Анодом служит маленькая платиновая про-
волочка. Концы обеих проволочек соединяют с аккумулятором в 2v. Ток про-
90
ЧИСТЫМ
пускают в течение одной минуты. За это время платиновый контакт покры-
вается тонким слоем ртути; его промывают дестиллированной водой и
высушивают.
В сосуд Ь, предварительно промытый хромовой смесью и высушенный,
наливают немного химически чистой ртути так, чтобы в нее можно было
погрузить целиком платиновый контакт. Над ртутью помещают 2—3 г кало-
мели — Hg2CI2, сначала промытой декантацией в дестиллированной воде, а
затем в насыщенном растворе хлористого калия; над каломелью насыпают
8—10 г химически чистого хлористого калия в кристаллах, после чего в сосуд b
вводится трубочка а и насасывают через колено с в сосуд b насыщенный раст-
вор КС11; при этом в колено сив отверстие крана не должны попасть пузырьки
воздуха. Стеклянная пробка каломельного электрода а смаз1
вазелином, то же и кран е. Что касается крана колена с, то
он вазелином не смазывается, чтобы замыкание тока про-
исходило и при закрытом кране.
После работы, на соединительное колено с при помощи
резиновой трубочки одевается пробирка с насыщенным рас-
твором КС1.
Потенциал каломельного электрода представляет довольно
постоянную величину, незначительно изменяющуюся под вли-
янием температуры. Значения потенциала каломельного элек-
трода приведены в таблице 11 (стр. 87).
Каломельному электроду МОСКИП (Московский завод
контрольно-измерительных приборов) придает следующий вид
(рис. 86). Стеклянный сосуд Н имеет в нижней части шаро-
образное расширение Р, от которого отходит боковой отро-
сток с впаянной платиновой проволочкой и клеммой К.
В сосуд Н впаяна длинная трубочка AAV Сверху сосуд Н
имеет горлышко с пришлифованной пробкой. Сосуд должен
быть перед зарядкой промыт хромовой смесью или пропарен
и высушен.
Зарядка каломельного электрода производится следующим
образом.
Сначала заполняют агаровым студнем трубку AAV игра-
ющую роль «солевого моста» («электролитического ключа»).
Приготовляют агаровую массу как описано выше (стр. 90).
Чтобы насосать агаровый студень в трубку АА , поступают
так: вокруг той части трубки, которая проходит в сосуде Н, закла-
дывают тампон из ваты, чтобы захватить агар, вылившийся сверху трубочки
при насасывании в нее агарового студня. На горлышко сосуда Н одевают
каучуковую трубочку с зажимом. Нижний конец трубочки ААг погружают в
стакан с горячим еще агаровым студнем и, взяв каучуковую трубку в рот,
засасывают агар медленно до верха трубки ААг так, чтобы из трубки вышла
избыточная капля, попадающая на ватный тампон. Закрывают зажим и дают
массе застыть. Агаровая масса должна быть сплошной, так как сохранение
в ней пузырьков воздуха, как уже указывалось выше, затрудняет или делает
совершенно невозможным прохождение тока. После застывания агарового
студня трубку вынимают из стакана, снимают каучуковую трубку с горлышка
сосуда Н и осторожно извлекают ватный тампон из сосуда так, чтобы в пос-
леднем не остались частицы агаровой массы.
Далее следует амальгамирование платиновой проволоки, впаянной в отро-
сток шарообразной части сосуда.
Для амальгамирования, как уже указывалось выше, пользуются 2% раст-
вором сулемы или 1% раствором азотнокислой закиси ртути — HgNO3. В ша-
Рис. 86.
Каломельный
электрод
МОСКИП.
II — сосуд элек-
трода, Р — шаро-
образное расши-
рение сосуда длн
ртути, К—клемма
отростка сосуда,
AAt — трубочка
длн агарового
студня, п — слой
каломели, i — на-
сыщенный рас-
твор КС1.
1 Отсюда и сам каломельный электрод называется насыщенным в отличие от
нормального, наполняемого нормальным раствором КС1.
91
электрометр;
а — понровное стенлышно,
b — шарообразное расши-
рение электрометрической
трубки.
рообразную часть сосуда наливают раствор сулемы или закиси ртути; клемму
К соединяют с минусом двухвольтового аккумулятора, плюс которого сое-
динен с платиновой проволокой, погруженной в тот же раствор. Через 1—2 ми-
нуты амальгамирование прекращают. Сосуд освобождают от раствора, хорошо
промывают дестиллированной водой и дают ему высохнуть.
Далее следует зарядка сосуда. Через воронку с хорошо оттянутым капил-
ляром наливают в сосуд чистую сухую ртуть, заполняя ею шарик. При этом
надо следить, чтобы ртуть хорошо заполнила отросток, в который впаяна
платиновая проволочка, соединенная с клеммой, иначе не будет контакта и
каломельный электрод будет непригоден для работы.
После заполнения шарика ртутью, поверх нее наливают взвесь каломели
в насыщенном растворе хлористого калия. Каломель перед этим, как было
указано, промывают сначала дестиллированной водой,
а затем насыщенным раствором хлористого калия. Слой
каломели л, осевшей на поверхности ртути, должен иметь
в высоту около 3 мм. Поверх слоя коломели помещают
несколько кристалликов хлористого калия, взятых из
насыщенного раствора, и добавляют этого раствора в
таком количестве, чтобы слой его I покрыл трубочку
с агаром. Пробку сосуда смазывают вазелином и закры-
вают сосуд. Вне работы каломельный электрод своим
нижним концом должен быть погружен в сосуд S с на-
сыщенным раствором КС1.
Свежеприготовленный каломельный электрод должен
постоять не менее суток, пока не приобретет постоян-
ный потенциал.
Капиллярный электрометр (рис. 87)
служит в качестве «нуль-инструмента», дающего возмож-
ность обнаружить в цепи самые слабые токи и полное
исчезновение тока. Он состоит из электрометрической
трубки, микроскопа и штатива (микроскоп и штатив
на рисунке не показаны).
Часть электрометрической трубки сужена в капилляр
с просветом в 0,1 мм. К суженной части трубки снаружи
приклеено покровное стеклышко а. В трубку электро-
метра, предварительно промытую хромовой см?сью и
высушенную, наливают немного чистой ртути и, накло-
няя трубку в сторону шарообразного расширения Ь,
перемещают немного ртути из труики в это расширение
с таким расчетом, чтобы при вертикальном положении
трубки мениск приходился приблизительно на середине покровного стекла.
За положением мениска ртути в капиллярной трубке следят в микроскоп
(увеличение 50—60), в окуляре которого имеется шкала с делениями. Над
ртутью в оба колена наливают серную кислоту (1 : 5), уровень которой обо-
значен на рисунке.
Капиллярный электрометр представляет собой своеобразный гальвани-
ческий элемент: ртуть в нем служит электродами, серная кислота — электро-
литом. При пропускании тока через электрометр поверхностное натяжение
ртути в капилляре изменяется, вследствие изменения разности потенциалов
между ртутью и кислотой, в результате этого уровень ртути в капилляре то
поднимается, то опускается, в зависимости от направления тока. Покойное
положение мениска указывает на отсутствие тока в цепи.
Работая с капиллярным электрометром, следует избегать длительного про-
пускания через него тока, так как при этом возникает заметная поляризация
электродов. Включение электрометра в цепь должно производиться при помощи
контакта всего лишь на доли секунды. Если все же возникла заметная поля-
ризация электродов, то следует освежить ртутный мениск в капилляре. Для
92
пользоваться гальванометром
(рис. 88). Электрод,
концентрированной
Рис. 88. Сосуд для платиниро-
вания электродов;
а — сосуд для платинирующей
жидкости, Ъ — сосуд для серной
кислоты.
этой цели наклоняют электрометр так, чтобы несколько капель ртути перетекло
из капилляра в шарообразное расширение.
Вместо капиллярного электрометра лучше i
чувствительности не ниже 10“7Д.
Сосуд для платинирования
жащий платинированию, промывают сначала
кислотой, а затем дестиллированной водой. Про-
мытый таким образом электрод погружают в
сосуд а, куда наливают платинирующую жид-
кость (3% водный раствор хлорной плати-
ны PtCl4, к которой прибавлено небольшое
количество НС1. Электрод соединяют с катодом
аккумулятора, а платинирующую жидкость —
с анодом. Ток пропускают 5 минут, если элек-
трод платинируется в первый раз, и около
1—1,5 минут, если платинирование повторное.
Для удаления остатков платинирующей жидкости
с электрода, его промывают дестиллированной
водой, а затем погружают в сосуд Ь, в который
наливают раствор серной кислоты (1 :5). Теперь
ток пропускают счень короткое время (несколько
секунд) в обратном направлении, после чего
электрод промывается в дестиллированной воде. Платинирование повторяется,
подле-
серной
когда замечено, что платиновая чернь утратила свою равномерность.
Аппарат Киппа (рис. 89) в описываемом методе применяется для
получения водорода. Свободный от мышьяка чистый цинк обрабатывается
серной кислотой (1 объем H2SO4 уд. веса 1,84 на 8 объемов воды). Для уско-
рения реакции прибавляют несколько капель раствора CuSO4. До введения
водорода в водородный электрод газ для очистки пропускают последовательно
через три промывалки. В первую наливают 3% раствор КМпО4, под-
кисленный H2SO4, для окисления мышьяковистого водорода и сероводорода;
во вторую — щелочный раствор пирогаллола (5% раствор пирогаллола в
Рис. 89. Аппарат Киппа, соединенный
с тремя промывалками (на рис. склянки
Дрекселя).
22% растворе КОН) для освобожде-
ния водорода от О2 и СО2; в третью—
насыщенный раствор сулемы для за-
держания последних следов мышьяко-
вистого водорода. Полезно присоеди-
нить еще четвертую склянку с чистой
водой. Склянки должны быть герме-
тически закрыты и соединены между
собой при помощи резиновых трубок
на стык. На отводную трубку послед-
ней склянки одевается резиновая
трубка с винтовым зажимом.
Перед каждым измерением следует
выпускать из кипповского аппарата
газ приблизительно в течение 10 минут для удаления воздуха, если таковой
попал в аппарат. При свежей зарядке аппарата газ выпускается в течение
одного часа.
Методика определения. Собирают аппаратуру как указано
на рис. 90. При помощи рубильника (2) включают аккумулятор (7) в большую
цепь. Необходимо после этого подождать минут 10 пока ЭДС аккумулятора
станет стабильной.
За это время подготовляют водородно-каломельный элемент. В сосуд во-
дородного полуэлемента (5) вводят, как описано выше, исследуемую жидкость
в таком количестве, чтобы кончик платины был погружен в нее, после чего
закрывают кран трубки с и пропускают в течение 5—8 минут через водородный
93
электрод струю водорода из аппарата Киппа со скоростью 2—3 пузырьков
в секунду. Газ вводится через трубку а и выводится через трубку Ь, Проходя
через испытуемую жидкость, водород не только насыщает ее, но и освобождает
от О2 и СО2. Платиновая чернь при этом сорбирует элементарный водород
до насыщения.
Когда пропускание газа через испытуемую жидкость заканчивается,
закрывают кран трубки Ь, затем а, отключают аппарат Киппа, не забыв закрыть
винтовой зажим на соединительной каучуковой трубке. Затем открывают
Рис. 90. Схема потенциометра с водородно-каломельным элементом:
1 — аккумулятор на 2 v, 2 — рубильник, 3 — водородный электрод, 4 — каломельный электрод, 5 —
переключатель, в — нормальный элемент, 7 — ключ Морзе. 8 — капиллярный электрометр, 9 — до-
бавочное сопротивление в 100—150 ом, АВ — реохорд, С — подвижный контакт.
кран трубки с и соединяют водородный полуэлемент при помощи электроли-
тического ключа d с ванночкой с насыщенным раствором КС1. Далее погружают
соединительное колено е каломельного полуэлемента (4) в ту же ванночку с
хлористым калием, открыв при этом кран колена е, и получают, таким образом,
один гальванический водородно-каломельный элемент.
Ванночка с насыщенным раствором КС1 служит электролитическим кон-
тактом для обоих полуэлементов.
Так как ионы калия и хлора имеют приблизительно одинаковую подвиж-
ность, то устраняется влияние так называемой диффузионной разности потен-
циалов, возникающей в месте соприкосновения двух различных растворов.
Подготовив описанным образом боковую цепь, приступают к измерению.
Скользящий контакт С реохорда устанавливают приблизительно на сре-
дине линейки, переключатель (5) устанавливают в положении ом, т. е. вводят
в боковую цепь нормальный элемент (6). При помощи ключа (7) замыкают
на один момент (доли секунды!) боковую цепь, наблюдая положение мениска
в капиллярном электрометре (8). Движение мениска вверх или вниз указывает
на наличие тока в боковой цепи. Перемещая скользящий контакт 6 вправо или
94
влево по реохорду, находят точку компенсации, т. е. то положение сколь-
зящего контакта на реохорде, при котором мениск в капиллярном электро-
метре остается неподвижным при замыкании боковой цепи, что указывает
на отсутствие тока в этой цепи. Положим, что это произойдет при положении
подвижного контакта в точке С. Записав отсчет по линейке реохорда, устанав-
ливают переключатель в положении on, т. е. вводят в боковую цепь водородно-
каломельный элемент. Точно так, как и при включении нормального элемента,
находят точку компенсации. Положим, что этот момент наступит при поло-
жении подвижного контакта в точке С, (на рис. не показана). Производят
отсчет по линейке реохорда.
На основании полученных двух отсчетов АС и АСг находят электродви-
жущую силу водородно-каломельного элемента по формуле:
1018,3 • АСХ
АС
где Ех — ЭДС боковой цепи в милливольтах,
АС — отсчет на линейке реохорда для нормального элемента в мм,
ЛСХ — отсчет на линейке реохорда для элемента водород-каломель в мм.
Найдя электродвижущую силу водородно-каломельного элемента и зная
потенциал одного из полуэлементов его, а именно каломельного, находят pH
испытуемого раствора по формуле:
рН = Ех — Ео
и &
Ex —
В этой формуле Ех—найденная на основании вышеописанных измерений
величина, а Ео и v определяются из таблиц (см. стр. 87).
Пример. Отсчет по линейке в момент компенсации при включении нормального элемента,
т. е. АС = 560; отсчет при включении водородно-каломельного элемента, т. е. ACt = 420.
Измерение производилось при 17°С. Нужно найти pH испытуемого раствора.
Сначала находим Ех из равенства:
„ 1018,3 • 420, _
Ех =------------763,7 м. V.
560
Затем находим pH из равенства:
763,7 — 251,5
pH =
8,9.
57,5
Потенциометрическое определение pH с помощью хингпдронпо-каломе 1ьного элемента
Описанный выше метод определения pH имеет то важное преимущество
перед ниже описываемым хингидронно-каломельным методом, что позволяет
определять любые значения pH. Его отрицательной стороной является неко-
торая мешкотность и громоздкость.
Хингидронно-каломельный метод значительно проще, но он ограничен
определенным интервалом значений pH, в пределах коего он может быть
применим, а именно: хингидронно-каломельный метод может быть применим
при значениях pH не выше 8 — 8,3.
Учитывая однако, что многие биологические среды имеют значения, не
превышающие указанный предел, ценность этого метода становится очевидной.
Принцип в основном сохраняется тот же, что в предыдущем методе.
Вместо водородного применяется хингидронный полуэлемент.
Хингидрон — С6Н4О2 • С6Н4(ОН)2 представляет соединение, состоящее из
эквимолекулярных количеств гидрохинона — С6Н4(ОН)2 и хинона — С6Н4О2.
Он кристаллизуется в виде мелких темнозеленых игл. Хингидрон плохо
растворяется в воде, насыщенный раствор его содержит 0,005 моля в литре.
05
а 6 с
При растворении хингидрона в жидкости, он распадается на эквивалентные
количества гидрохинона и хинона. Гидрохинон подвергается диссоциации с
образованием хинона и водорода по реакции:
СвН4(ОН)2 = С6Н4О2+ н2.
Из сказанного ясно, что, если растворить некоторое количество хингидрона
в испытуемой жидкости, то операция пропускания через нее водорода, имею-
щая место при применении водородного электрода, окажется излишней.
Платинирование электрода также отпадает, так как окислительно-восстано-
вительная система гидрохинон-хинон при некотором избытке гидрохинона
обеспечивает стойкое наличие свободного водорода в растворе. Таким образом,
вся методика определения pH значительно упрощается.
Приборы те же, что и в предыдущем методе.
Хингидронный полуэлемент, который в данном методе заменяет водород-
ный полуэлемент, составляется весьма просто: в небольшой стакан или кол-
бочку наливают испытуемую жидкость, насыпают щепотку
хингидрона, взбалтывают и погружают платиновый электрод,
получая готовый хингидронный полуэлемент.
Платиновый электрод (рис. 91) представляет узень-
кую стеклянную трубочку, запаянную с одного конца; в этот
конец в трубочку впаяна платиновая проволочка, выходящая
несколько наружу; в противоположном конце к трубочке при-
делана клемма. Внутри трубочки проходит медная прово-
лочка, соединяющая клемму с платиновой проволочкой.
Вместо наружной, платиновой проволочки иногда при-
меняют маленькую платиновую пластинку.
Для удобства можно платиновый электрод заключить в
трубочку, открытую снизу и перекрывающую выходящую
наружу проволочку (пластинку); в этом случае предупре-
ждается изгибание проволочки и даже возможное отламы-
вание ее.
Вместо платинового электрода в данной установке можно
пользоваться точно так же устроенным золотым электродом.
Методика определения. На рис. 92 представ-
лена схема установки наичаще применяемой для потенцио-
метрического измерения pH при помощи хингидронно-кало-
мельного элемента. Эта схема от представленной выше отличается тем, что
вместо водородного взят хингидронный полуэлемент.
Так как в цепи с каломельным полуэлементом хингидронный полуэлемент
имеет более высокий потенциал, то каломельный полуэлемент включается
отрицательно по отношению к хингидронному полуэлементу, что и
видно из представленной схемы.
При помощи рубильника (2) включают аккумулятор (7) в большую цепь
и минут через 10, когда ЭДС аккумулятора станет стабильной, подготовляют
хингидронно-каломельный элемент.
Испытуемую жидкость наливают в небольшой химический стакан (3),
вводят сюда на кончике ножа щепотку хингидрона и тщательно перемеши-
вают стеклянной палочкой. В стакане должно остаться некоторое количество
перастворенного хингидрона. Затем в тот же стакан вводят платиновый
электрод (4), получая таким образом хингидронный полуэлемент. Его соеди-
няют с ванночкой с насыщенным раствором КС 1 при помощи электролитического
ключа (5). Соединительное колено каломельного полуэлемента (6) также вво-
дится в ванночку с КС1.
Получив таким образом хингидронно-каломельный элемент, приступают
к измерениям.
Скользящий контакт реохорда устанавливают приблизительно на средине
линейки и, установив переключатель (7) в положение ом, включают, таким
Рис. 91.
Платин о вые
электроды:
q — заканчиваю-
щийся проволо-
кой, Ь — заканчи-
вающийся пла-
стинкой, с—элек-
трод со стеклян-
ной муфтой сна-
ружи.
образом, в боковую цепь нормальный элемент (8). При помощи ключа (9)
замыкают на один момент (доли секунды!) боковую цепь, следя за положением
стрелки гальванометра (10). Перемещая скользящий контакт вправо или
влево, находят точку компенсации С, т. е. то положение скользящего контакта,
когда в боковой цепи отсутствует ток и стрелка гальванометра остается при
замыкании цепи неподвижной. Записывают отсчет по линейке реохорда.
Далзе устанавливают переключатель в положение on, т. е. вводят в боковую
цепь хингидронно-каломельный элемент, и вновь находят точку компенсации С\;
производят отсчет по линейке и записывают.
Рис. 92. Схема потенциометра с хингидронно-каломельным элементом:
I—аккумулятор, 2—рубильник, 3 — сосуд для исследуемой жидкости, 4 — платиновый электрод,
5—агаровый мост, в—каломельный электрод, 7 — переключатель. 8 — нормальный элемент, 9 —
ключ Морзе, 10—стрелочный гальванометр, 11 — добавочное сопротивление, АВ — реохорд, С—
скользящий контакт.
Электродвижущую силу боковой цепи (хингидрон-каломель) находят по
формуле:
1018,3 • АСГ
АС
где Ех —ЭД С боковой цепи в милливольтах.
АС —отсчет на линейке реохорда для нормального элемента в мм.
AC-l — отсчет на линейке реохорда для цепи хингидрон-каломель в мм.
Найдя Ех определяют pH по формуле:
„ 454,1—Ех1
рн=—5—'
1 Потенциал хингидронного электрода в растворе с нормальной концентрацией водород-
ных ионов (pH = 0) равен 704,4 mv. Т*к как знак потенциала н- сыщенного к; ламельного
электрода положителен, как и знак хингидронного электрода, то электродвижущая сила
7 А. И. Бурштейн
97
где Ex — найденная ЭДС боковой цепи,
— имеет значение соответственно таблице на стр. 87.
Пример. Ех оказалось равной 319,3 милливольтам. Измерение производилось при
18°С.
В этом случае:
РН = ^,1-319^
57,7
Нередко вместо приведенных формул пользуются формулами:
г 1,0183 • АС\ „ 0,4541—Ех
Ех — —---------1 и pH ---------------------,
АС 0,0577
что то же самое, так как в этих формулах те же компоненты, что и в приве-
денных выше, уменьшенные в 1000 раз. Для ft взято значение при 18° С также
уменьшенное в 1000 раз.
Потенциометр МОСКИП
В связи с тем, что потенциометр МОСКИП получил большое распростра-
нение, здесь приводятся описание прибора и методика применения его.
Все части прибора вмонтированы в портативный ящик. На рис. 93 показана
панель установки с обозначениями, соответствующими расположению частей
Рис. 93. Панель потенциометра МОСКИП
J5 — нлем*мы для включения батареи. ЭЛ—клеммы для включения влектролов с испытуемым раствором,
РД — ручка шкалы прибора для установки на десятки и единицы милливольт, S — окно и шкала при-
бора с делениями в милливольтах, R — ручка реостата настройки. RtR3 и Н3 — добавочные сопротив-
ления к реостату настройки, РК — ручка коммутатора для установки на согни милливольт, Г — галь-
ванометр нулевой стрелочный. К — корректор для установки стрелки гальванометра на нуль, Р —
ручка переключателя для установки в положении настройки (И) и в рабочем положении (Р), К, —
кнопка для регулировки прибора, — рабочая кнопка.
прибора. Аппаратура для хингидронно-каломельного и водородно-каломель-
ного элементов прилагается отдельно. К прибору приложена также инструк-
ция по применению его.
цепи хингидрон-каломель равна разности потенциалов обоих полуэлементов и определяется
из равенства:
Е = 704,4 — 250,3 = 454,1 mv.
При заполнении хингидронного электрода раствором с неизвестной концентрацией
водородных ионов,отличной от нормальной, электродвижущая сила цепи хингидрон-каломель
оказывается меньшей нежели 454,1 ти на величину, равную 0,1983 ТрН. Следовательно,
можно написать:
Ех = 454,1 — 0,1983 Тр/У,
откуда
454,1 — Ех 454,1 —Ех
pH -----------------------------.
0,1983 Г Ь
98
Для питания потенциометра пользуются аккумулятором, включая послед-
ний через реостат в 130—150 омов. Можно также пользоваться гальвани-
ческими элементами с ЭДС в 1,25—l,5v.
'Методика определения. К клеммам Б присоединяют источник
питания, соблюдая при этом правила знаков, а к клеммам ЭЛ присоединяют
хингидронно-каломельный или водородно-каломельный элемент.
Ручку переключателя PH устанавливают в положение Н (настройка).
Затем, подбирая сопротивление с помощью штепселей Rx, R2h R3 и вращая
ручку реостата Р, находят такое сопротивление, когда при одномоментном
нажатии кнопки регулировки прибора К2 стрелка гальванометра Г не откло-
няется от нулевого положения. Вслед за этим ручку переключателя PH уста-
навливают в положении Р (рабочее положение), включая этим самым в потен-
циометр боковую цепь. Нажимая кнопку /<2 и вращая ручку коммутатора
РК и ручку шкалы PD, находят положение, при котором стрелка гальвано-
метра не отклоняется от нулевого положения. Производят отсчет в сотнях
милливольт на коммутаторе РК по положению ползунка и в десятках и еди-
ницах милливольт на шкале S по нанесенной на стекле черте. Затем, поль-
зуясь таблицами, приложенными к прибору, или приведенными выше фор-
мулами, находят pH испытуемого раствора.
Пример. Произведено измерение pH при 22ЭС хингидронно-каломельным методом. Пол-
зунок коммутатора показывает 100, черта над шкалой — 45. Отсюда находим ЭДС элемента
равной 145. Соответственно этому отыскиваем по таблице:
для ЭДС 140................................ pH = 5,4430,
» » 5............................... pH = 0,0867,
» » 145, следовательно....................pH = 5,3563.
В полученный результат вводим поправку на температуру, которую находим также
по таблице. Для интервала pH 5 — 5V^ при 22°С поправка равна — 0,09. Следовательно,
pH раствора равна 5,3563—0,09 = 5,2663 или округленно 5,27.
Тот же результат получим н по формуле:
„ 0,4541 —0,001 Ех 0,4541—0,145
pH. =----------------=--------------~
0,001» 0,0585
Некоторые дополнительные замечания о потен биометрическом определении pH
Возможные ошибки. Ошибки при потенциометрическом опре-
делении pH могут возникать в следующих случаях:
1. При плохом платинировании водородного электрода, когда слой плати-
новой черни слишком тонкий или наоборот слишком толстый; в первом случае
потенциал электрода быстро достигает максимума, но затем быстро падает; во
втором случае наблюдается очень медленное нарастание потенциала, иногда же
он не достигает правильного значения. В обоих случаях измерение становится
невозможным.
Чтобы избежать этой ошибки, нужно платинирование электрода произво-
дить точно так, как описано выше.
2. При подсыхании и загрязнении водородного электрода, которые возни-
кают при небрежном пользовании электродом.
Чтобы избежать этого, следует после работы электрод промыть и держать
его в дестиллированной воде. Если загрязнение произошло, следует произ-
вести новое платинирование.
3. При загрязнении хингидрона в хингидронном методе.
Следует пользоваться химически чистым хингидроном и брать его чистой
ложечке й.
4. При загрязнении агара электролитического ключа испытуемым раство-
ром путем диффузии.
Чтобы избежать этого, следует агаровые трубки немедленно после работы
переносить в сосуд с насыщенным раствором хлористого калия.
99
5. При высыхании раствора хлористого калия в верхней части каломель-
ного электрода МОСКИП.
Необход! мо следить за стоянием уровня раствора, не допуская опуска-
ния его ниже верхнего конца агаровой трубки.
6. При попадании воздуха в соединительное колено каломельного элек-
трода, а также в агар электролитического ключа, что повышает сопротивление
току и делает измерение невозможным. Указанные дефекты должны быть
устранены.
Случаи, когда измерения невозможны. Потенциомет-
рические измерения pH становятся невозможными в следующих случаях:
1, Когда в испытуемой жидкости присутствуют вещества, способные вос-
станавливаться: соединения трехвалентного железа, двувалентной меди
и т. п. В этих случаях не удается добиться постоянства потенциала для водо-
родного электрода и определение становится невозможным.
2. Когда в испытуемой жидкости присутствуют вещества, способные «отрав-
лять» платиновый электрод, напр. мышьяковистый водород, сероводород,
синильная кислота, аммиак, свободный хлор, бром, иод и др.
Нередко отравление платинового электрода наблюдается при плохой
очистке водорода. В указанных случаях платиновый электрод становится не
чувствительным и выходит из строя.
3. В хингидронном методе при pH испытуемой жидкости выше 8,0—8,3.
4. В этом же методе, при высокой концентрации солей в испытуемой
жидкости, а также и в том случае, когда растворенные в этой жидкости
вещества вступают в реакцию с хингидроном.
Проверка установки. Каждая новая установка перед употреб-
лением должна быть проверена. Систематической проверке подлежат также
работающие установки. Безусловная проверка прибора должна быть произ-
ведена во всех случаях возможных сомнений в правильности получаемых ре-
зультатов.
Для проверки прибора можно воспользоваться любым буферным раствором:
фосфатным, боратным, цитратным и т. п. Предпочтительней брать растворы,
pH которых близки к 5.
Очень часто для указанной цели пользуются стандартным ацетат-
ным раствором pH = 4,62. Этот раствор готовится из равных объемов
0,1 N-ного раствора уксусной кислоты и 0,1 N-ного раствора ацетата натрия.
Раствор уксусной кислоты устанавливается по 0,1 N-ному раствору NaOH
при фенолфталеине. На литр 0,1 N-нсго раствора CH3COONa берут точно
8,202 г химически чистого препарата. Показания прибора должны соответ-
ствовать pH взятого для проверки буферного раствора.
Электрометрическое (потенциометрическое) титрование
Электрометрическое титрование может быть применено в отношении раз-
личных объемноаналитических определений; чаще, однако, оно применяется
в отношении определений, решаемых методом нейтрализации Большую услугу
оказывает электрометрическое титрование в тех случаях, когда определяемые
растворы окрашены или мутны и нет возможности применить обычное инди-
каторное титрование.
Из различных методов электрометрического титрования здесь описан
наиболее простой и доступный.
Принцип метода основан на том, что в испытуемый раствор погру-
жают платиновый электрод (индикаторный электрод) и соединяют его в один
гальванический элемент с электродом, погруженным в жидкость, pH которой
соответствует точке эквивалентности (электрод сравнения).
Испытуемую жидкость титруют до того момента, когда в цепи исчезает
ток, т. е. когда потенциал индикаторного электрода окажется равным потен-
циалу электрода сравнения. Исчезновение тока указывает конец титрования.
100
Так как описываемый метод хингидронный, то он пригоден для титрования
сильных и слабых кислот и оснований, pH которых не выше 8,3.
Прибор для электрометрического титрования показан на рис. 94. Он
состоит из конической колбы на 250—300 мл. В колбу вставлена пробка с
тремя отверстиями. В одно отверстие вставлена трубка Д, снабженная у
основания стеклянным краном. В трубку А при помощи резинового кольца
вставлен платиновый электрод В, Во второе отверстие пробки введен
платиновий электрод Сив третье — оттянутый конец бюреткп D. Оба
электрода при помощи звонковой проволки соединены с чувствительным
гальванометром Е. В цепь вводится ключ F.
Реактивы: 1. 0,1 N или 0,01 N раствор NaOH или кислоты (серной,
соляной).
2. Буферная смесь, pH которой соответствует точке эквивалентности \
3. Хингидрон.
Методика определения.
Трубочка А ополаскивается буферным
раствором; при этом необходимо про-
следить, чтобы шлиф крана и кончик
его были смочены этим же раствором:
такгм образом создается контакт меж-
ду буферным и испытуемым раство-
рами. Затем в трубочку А нали-
вают до половины ее высоты буферный
раствор и добавляют сюда же 10—20 г
хингидрона.
При помощи платинового электро-
да В перемешивают жидкость в трубке
А и устанавливают электрод на место.
В коническую колбу наливают
определенное количество (10—20 мл)
испытуемой жидкости, которую раз-
бавляют дестиллированной водой до
50—60мл. Бросают сюда же 10—20 мг
хингидрона и перемешивают жидкость
в колбе. Вставляют пробку в колбу,
Рис. 94. Схема прибора для потенциометри-
ческого титрования.
А — трубка для электрода сравнения, В — пла-
тиновый электрод, служащий электродом сравне-
ния, С — платиновый электрод, служащий в
качестве индикаторного электрода, 1) — бюретка,
Е — чувствительный гальванометр, F — ключ.
при этом концы электродов должны быть погружены в растворы.
Нажимая на ключ F, устанавливают наличие тока в цепи по отклонению
стрелки гальванометра. Затем приступают к титрованию, приливая небольшие
порции 0,1 N щелочи или кислоты и взбалтывают жидкость в колбе. Титрование
считается правильно проведенным, если после прибавления последней капли
щелочи или кислоты в цепи не обнаруживается тока.
Нажим на ключ должен быть коротким и быстрым.
После каждого определения колба, в которой ведется титрование, трубка А
и оба электрода ополаскиваются проточной и затем дестиллированной
водой.
Вне работы электроды сохраняются в дестиллированной воде. Время от
времени они очищаются хромовой смесью.
Расчет ведется, как при обычном титровании.
Измерение электропроводности растворов (кондуктометрия)
Под электропроводностью проводника понимают то коли-
чество электричества, которое проходит через поперечное сечение этого про-
водника в единицу времени при электродвижущей силе, равной единице.
1 Применяют также насыщенный раствор КС1.
101
Если обозначить электропроводность через L, то можно написать следующее
равенство:
R
где R — сопротивление, выраженное в омах.
Из приведенного равенства видно, что электропроводность представляет
величину, обратную сопротивлению, которое данный проводник оказывает
прохождению электрического тока.
Электропроводность принято выражать в обратных омах (мо).
Величина ее зависит от природы проводника; она прямо пропорциональна
площади поперечного сечения проводника и обратно пропорциональна длине его.
Электропроводность проводника, имеющего форму куба с длиной стороны
в 1 см, называется его удельной электропроводностью
и обозначается буквой х (каппа) или К.
Удельная электропроводность проводника представляет величину, обрат-
ную удельному сопротивлению. Напомним, что под удельным сопротивлением
проводника, понимают сопротивление проводника, имею-
щего форму куба с длиной стороны, равной 1 см. Эта
величина обозначается буквой р (ро). Из сказанного
вытекает следующее равенство:
Рис. 95.
Схема контакта в
индукционной
катушке.
Принцип метода определения электропровод-
ности растворов заключается в том, что измеряют
величину удельного сопротивления растворов и находят
обратную ей величину, которая и выражает удельную электропроводность,
выраженную в обратных омах.
Для определения удельной электропроводности растворов необходим при-
бор, состоящий из следующих частей:
1. Аккумулятор в 4v; 2. Индукционная катушка (зуммер); 3. Реостат:
4. Реохорд; 5. Телефон; 6. Сосудик для исследуемого раствора. Из перечислен-
ных приборов здесь рассмотрены лишь те, которые выше не описаны.
Индукционная катушка или зуммер должна быть малой с
небольшим числом витков. Пружина прерывателя должна производить воз-
можно быстрые колебания. Полезно контакт сделать пружинящим (рис. 95),
благодаря чему достигается беззвучный ход.
Реостатом для данной установки служит обыкновенный магазин
сопротивления (рис. 96) с набором в 5000, 2000, 1000, 500, 200, 100, 50 и 20 ом.
(Применяются и другие наборы сопротивлений).
Магазин сопротивления представляет собой ящик, на крышке которого
приделан ряд металлических пластинок из латуни; между каждой парой
пластинок оставлен зазор, имеющий в средней части форму круга. В полу-
чающиеся таким образом круглые отверстия вставляются штепсели, сделан-
ные из латуни и снабженные сверху эбонитовыми ручками. Каждая соседняя
пара пластинок при помощи проволоки соединяется с катушкой, на которой
намотана тонкая проволока, дающая то или иное сопротивление. Катушки
вмонтированы внутри ящика (рис. 97). Когда штепсель вставлен в гнездо, то
две соседние пластинки соединены через штепсель и потому проволочное
сопротивление, находящееся внутри ящика между этими пластинками будет
выключено; напротив, когда штепсель извлекается из гнезда, то этим самым
включается сопротивление соответствующей катушки, величина которого
Е
1 Приведенное равенство представляет частный случай известного равенства:
102
написана у гнезда. Штепсели, как и пластинки, должны содержаться в совер-
шенной чистоте. Они не должны быть смазаны во время работы салом или ва-
зелином, так как это создает добавочное сопротивление. По этой же причине
не следует штепсели
брать руками за метал-
лическую часть.
Т е л е ф о н. При-
меняется обыкновенная
слуховая телефонная
труб ка. Реком е нд у етс я
пользоваться трубками
с небольшим сопротив-
лением телефонной об-
мотки, которые приме-
няются в домовых уста-
новках. Можно приме-
нить также наушники.
Сосудики для
измерения сопро-
тивления готовятся
из стойкого стекла и
снабжаются платино-
Рис. 96. Магазин сопротивлений.
выми электродами. На
рис. 98 изображен один
из предложенных для
этой цели сосудов.
Электроды состоят из двух массивных круглых платиновых пластинок,
диаметром приблизительно в 2
Рис. 97. Расположение катушек
в магазине сопротивлений.
покрываются
смесью или
платиновой чернью. Для этой
98. Сосуд
измерения
Рис.
для
сопротивления
растворов.
см, соединенных с короткими платиновыми
проволоками. Последние впаяны в стеклян-
ные трубки, куда наливается ртуть служа-
щая для установления контакта со встав-
ленными сюда медными проволоками, вклю-
ченными в цепь.
Трубки устанавливаются параллельно
одна другой на расстоянии одного см, и в
таком положении закрепляются в эбони-
товой крышке прибора.
Платиновые пластинки
цели их промывают хромовой
серной кислотой, затем тщательно проточной и дестиллиро-
ванной водой, после чего спиртом и эфиром. После высуши-
вания электроды погружают в 3% раствор хлорной плати-
ны — PtCl4, к которому прибавлено 0,025% ацетата свинца.
Присоединив электроды при помощи проволоки к клеммам
4-вольтового аккумулятора, пропускают ток в течение 5—8
минут, ежеминутно меняя его направление. Затем элек-
роды погружают в дестиллированную воду, подкисленную
серной кислотой, и вновь пропускают ток 10—15 минут,
ежеминутно меняя его направление. Платинирование после
этого считается законченным. Электроды промывают дестил-
лированной водой и хранят в ней. После каждого измере-
ния сосудик и электроды следует тщательно ополаскивать
дестиллированной водой. В сосудик не следует наливать
сильных щелочей или кислот.
Сосудику придают цилиндрическую форму. Его размеры
должны быть таковы, чтобы электроды свободно в нем помещались. На ниж-
ней поверхности эбонитовой крышки имеется желобок, соответствующий верх-
юз
нему краю сосуда, что при цилиндрической форме последнего, обеспечивает
постоянное положение электродов в сосуде.
На рис. 99 представлена модель закрывающегося сосуда, который, как и
описанный выше, служит для плохо проводящих растворов. На рис. 100 изо-
бражен сосуд, применяющийся для хорошо проводящих раство-
ров.
Так как электропроводность сильно меняется с температу-
рой, то сосуд для измерения во время исследования помещают
в термостат или водяную ванну, где поддерживается необхо-
димая температура, чаще 20° С.
Объем жидкости в сосудике должен быть постоянным, так
как высота наполнения сосудика влияет на результаты измере-
ния.
Сопротивление, измеряемое при помощи описанных сосуди-
ков, только в том случае будет равно удельному, если отноше-
Рис. 99. ние расстояния между электродами к площади электродов равно
Сосуд для единице. Практически же оно часто отличается от единицы. Это
измерения
сопротивле-
ния плохо
проводящих
растворов.
отношение получило название емкости сопротивле-
ния сосудика и обозначается буквой С. Оно должно быть
установлено в отношении каждого сосудика в отдельности (см.
далее).
Емкость сопротивления сосудика представляет коэфициент,
на который нужно помножить найденную при помощи дан-
ного сосудика электропроводность любого раствора, чтобы
получить удельную электропроводность этого раствора.
O,1N раствор КС1, необходимый для установления емкости
сопротивления сосудиков, готовится из химически чистого
препарата. Предварительно слегка прокалив и охладив в
экссикаторе некоторое количество КС1, отвешивают точно
7,455 г и растворяют в литре дважды перегнанной, свобод-
?i
Рис. 100. Сосуд
для измерения
сопротивления
хорошо прово-
дящих раство-
ров.
ной от СО2 воды. Удельная электропроводность такого рас-
твора равна при 18° С — 0,01119 и при 20° С — 0,0167. Для
0,02 N раствора КС1 удельная электропроводность при 18° С
равна 0,002397 и при 20° С —0,002501.
Методика определения электропро-
водности растворов. Прибор собирают как пока-
зано на рис. 101. Здесь Ак — аккумулятор, J — индукционная катушка
(зуммер), R—реостат, г — сосудик для
исследуемого раствора, АС — реохорд,
D — скользящий контакт, Т — телефон.
Сначала калибрируется емкость со-
противления (С) данного сосудика.
В него наливают 0,1N раствор КС1 и
Рис. 101. Схема прибора для определе-
ния электропроводности растворов:
Ак — аккумулятор, J — зуммер, R — вводи-
мое сопротивление, г — испытуемое сопротив-
ление, АС — реохорд, BD — проводник («мо-
стик*), в который включен телефон T,D —
подвижный контакт.
R, в ВС — искомое сопротивление г.
доводят в водяннои ванне температуру
последнего до 20° С. Включают аккуму-
лятор; при этом приводится в действие
зуммер, трансформирующий постоянный
ток в переменный. Ток подается в
точки А и С. Разветляясь у точки Д,
ток идет с одной стороны по реохорду
АС и с другой стороны по ветви АВС.
В АВ включено известное сопротивление
Баластное сопротивление /? включают
таким образом, чтобы произошло максимальное заглушение звука, а затем,
вооружившись телефоном, перемещают скользящий контакт D по измеритель-
ной линейке, пока не находят такое положение его, при котором телефон
молчит. Индуктор накрывают при этом футляром, чтобы издаваемый им звук
104
не мешал измерению, т. е. воспринимался только через телефон. Опреде-
ляют положение подвижного контакта в момент исчезновения звука и нахо-
дят сопротивление раствора из равенства:
R : г = : DC,
откуда
R-DC
Т~ AD ’
где г — сопротивление раствора в омах,
R—введенное сопротивление реостата в омах,
AD и DC — отрезки на линейке реохорда.
Найдя г, определяют емкость сопротивления сосуда на основании следую-
щих соображений. Если г—найденное сопротивление взятого раствора КС1,
то —---его электропроводность. Чтобы наити удельную электропроводность,
1 г
нужно — помножить на емкость сопротивления сосуда, т. е. на С; получим
-р С. Учитывая, что удельная электропроводность 0,1 N раствора КС1 при
20° С равна 0,01167, можно написать:
— • С = 0,01167,
Г
откуда:
С = г • 0,01167.
Следовательно, чтобы найти емкость сопротивления сосудика, нужно най-
денное из опыта сопротивление взятого раствора КС1 помножить на удельную
электропроводность этого же раствора при той же температуре.
Так как при неосторожном обращении с сосудиком могут возникать незна-
чительные деформации электродов, что влияет на емкость сопротивления
сосудика, рекомендуется емкость сопротивления сосудика проверять перед
каждым определением электропроводности испытуемого раствора.
Определив емкость сопротивления сосудика, находят электропроводность
испытуемого раствора. Ее определяют описанным выше способом, т. е. находят
сопротивление раствора; отсюда определяют электропроводность его как
величину, обратную сопротивлению, и, наконец, для пересчета найденной
электропроводности на удельную умножают найденную электропроводность
на емкость сопротивления сосудика.
Таким образом, получаем для удельной электропроводности испытуемого
раствора следующее уравнение:
R • DC
где К — удельная электропроводность в обратных омах,
С — емкость сопротивления сосудика,
R — включенное сопротивление,
AD и DC — отрезки на линейке реохорда.
Пример 1. При определении емкости сопротивления сосуда посредством 0,01 N раствора
КС! при 20JC положение скользящего контакта при полном заглушении звука оказалось
на 600-м делении при введении баластного сопротивления в 120 ом.
Отсюда сопротивление раствора находится из равенства:
,-J20-4M-80.
600
Емкость сопротивления сосудика находим из равенства:
С = 0,01167.80 = 0,9336.
10S
Пример 2. В том же сосуде произведено определение удельной электропроводност
испытуемого раствора; при этом введенное спротивление оказалось равным 160 см. Скользи
щий контакт оказался на 320-м делении. Температура раствора 20°С. Отсюда удельную элект
ропроводность жидкости находим из равенства:
К =
0,9336 - 320
160 • 680
= 0,002746 = 27,46- Ю~4.
Прибор МОСКПП для измерения электропроводности
Прибор МОСКИП построен по тому же принципу, который описан выше.
Вся аппаратура вмонтирована в небольшой ящик. Аккумулятор в 4v, сосудик
для измерений и телефонные наушники прилагаются отдельно.
На рис. 102 показана панель ящика. R— переключатель сопротивлений,
г — клеммы для проводов от сосудика с испытуемой жидкостью, Ак — клеммы
Рис. 102. Верхняя панель
прибора МОСКИП для опре-
деления электропроводности
растворов:
А К— клеммы для проводов от
аккумулятора, г — клеммы для
электродов сосуда с испытуемым
раствором, J — зуммер, R — пере-
ключатель сопротивлений, Т —
клеммы для проводников теле-
фона, D — головка для перемеще-
ния подвижного контакта, F —
окно для отсчетов по шкале, В —
выключатель аккумулятора.
для проводов от аккумулятора, В — выключатель
питания, J — зуммер, D— головка для перемеще-
ния скользящего контакта по реохорду. В данном
приборе реохорд не прямой, а изогнутый по окру-
жности. При вращении головки D вместе с ней
двигается скрепленный с нею круг с делениями,
которые видны в окно F.
В окне отсчитывается деление, соответствующее
положению скользящего контакта. Круг содержит
деления от 350 до 650. Концы реохорда от Одо 350
и от 650 до 1000 выведены в виде катушек. Провода
телефона присоединяются к клеммам Т.
Измерения производят следующим образом:
присоединяют аккумулятор, сосуд с испытуемым
раствором и телефонные наушники. Последние на-
девают и поворотом выключателя В в правую сто-
рону включают аккумулятор в цепь. Регулируют
зуммер, пока не будет слышен ровный звук высо-
кого тона. Затем вращают рукоятку R и, поворачивая одновременно диск
с контактом при помощи головки D, добиваются максимального заглушения
звука в телефоне. После этого делают отсчет по шкале в окне F.
Как было указано, шкала градуирована лишь в пределах 350—650, так
как в этих пределах определения наиболее точны. Необходимо подобрать
сопротивление реостата, близкое по величине к определяемому сопротивлению
жидкости, и тогда удастся легко найти положение контакта в пределах указан-
ных делений реохорда. Вычисления ведутся как изложено выше.
Новая модель прибора МОСКИП для определения электропроводности
отличается лишь тем, что прибор присоединяют непосредственно к электросети
переменного тока в HOv. При помощи трансформатора ток переводится на
низкое напряжение. В связи с этим отпадает необходимость в аккумуляторе.
Зуммер и телефон здесь тоже отсутствуют. Момент компенсации определяется
по чувствительному О-гальванометру переменного тока — причем измерение
заканчивается, когда подобраны сопротивления, при которых гальванометр
дает минимальное отклонение стрелки.
ВАЖНЕЙШЕЕ оборудование для бактериологических работ
Микроскоп
Микроскоп (рис. 103) состоит из механической и оптической частей. Ме-
ханическая часть микроскопа слагается из штатива, предметного столика,
тубуса, макро-и микровинтов. Оптическая часть состоит из осветитель-
ного аппарата, объективов и окуляров.
106
Штатив состоит из тяжелой массивной ножки и колонки. Верхняя часть
колонки является держателем для тубуса. В верхней же части находятся
приспособления для перемещений тубуса — макро-и микровинт. Средняя
часть колонки служит ручкой для перенесения микроскопа. Колонка соединена
с ножкой при помощи шарнира, дающего возможность придавать микроскопу
наклонное положение.
Рис. 103. Микроскоп.
1 — разрез; А — рукоятка, Т — тубус. Н — вдвижная трубка тубуса, R — револьвер, S — винт
для грубой установки тубуса (макровинт), d — мпкровиит. Е — шарнир для наклонения верхней
части микроскопа. А — предметный столик. С — конденсор. 81 — винт для опускания осветительною
аппарата, ОЬ — объектив. D —окуляр. Sp — зеркало. // — ход лучей.
Предметный столик прикреплен к колонке; он имеет круглую или квадрат-
ную форму и делается неподвижным или подвижным. Подвижный круглый
столик вращается вокруг оси; кроме того, при помощи двух винтов можно
передвигать столик в двух взаимноперпендикулярных направлениях, что дает
возможность непрерывно и последовательно рассматривать препарат. Препарат
закрепляется на столике пружинящими зажимами.
Некоторые микроскопы снабжаются еще столиками с крестовым ходом,
дающим возможность передвигать препарат в двух взаимноперпендикулярных
направлениях. Такие столики имеют шкалу и нониус, благодаря которым
можно отмечать положение препарата.
107
Тубус, как было указано, укреплен в верхней части колонки. Он состоит
из двух вдвигающихся одна в другую трубок, благодаря чему длина тубуса
может быть увеличена или уменьшена. На наружной поверхности внутренней
трубки тубуса имеются деления в миллиметрах для установления «механи-
ческой длины» тубуса, при которой следует производить микроскопирование.
«Рабочая» длина тубуса равна обыкновенно 160—170 мм. Наружная трубка
тубуса снабжена зубчатой линейкой и может перемещаться при помощи зуб-
чатого колеса, находящегося в тубусодержателе. На оси зубчатого колеса
насажен макровинт, дающий возможность делать грубую установку микро-
скопа. Окончательная установка производится при помощи микровинта, по-
зволяющего передвигать тубус на очень незначительное расстояние (до I у.).
В нижней части наружной трубки имеется нарезка, в которую ввинчивается
револьвер. Револьвер имеет обыкновенно 3—4 гнезда с нарезками для ввинчи-
вания объективов. Поворачивая револьвер, подводят под тубус тот или
иной объектив.
Осветительный аппарат микроскопа состоит из зеркала, диафрагмы и кон-
денсора. Зеркало бывает обыкновенно двусторонним: с одной стороны плоским,
а с другой — вогнутым. При помощи особого приспособления зеркалу можно
придать различные полсжения. Свет, отражаемый зеркалом, проходит через
ирисдиафрагму, состоящую из ряда металлических пластинок, которые можно
при помощи движка плавно раздвигать или сближать, благодаря чему отвер-
стие для прохождения света, сохраняя круглую форму, может увеличиваться
или уменьшаться. Конденсор состоит из 2—3 сильных линз и служит для
концентрирования лучей света.
Объективы представляют самую существенную часть оптической системы.
Хорошие объективы дают увеличенное детальное изображение рассматривае-
мого объекта. По конструкции и качеству линз объективы делятся на апохро-
маты, ахроматы и флюориты.
Апохроматы являются наиболее ценными объективами. При пользовании
ими, благодаря устранению сферической и хроматической аберраций, полу-
чается наиболее четкое изображение объекта. Это весьма ценное качество
объектива достигается комбинацией линз различных систем и различного
состава.
Ахроматы представляют более простые объективы. Их способность устра-
нения сферической и хроматической аберраций выражена гораздо слабее,
чем у апохроматов, и поэтому изображение получается не столь четкое.
Флюориты по качеству занимают промежуточное место между названными
двумя видами объективов.
Объективы по способу применения делятся на сухие и иммерсионные.
Сухими объективами называются такие, при применении которых между рас-
сматриваемым объектом и объективом не требуется нахождение жидкости
(воды, кедрового масла и т. п.). Сухие объективы применяются обыкновенно
для увеличений до 700 раз.
Иммерсионными объективами называются такие, при применении которых
между фронтальной призмой объектива и рассматриваемым объектом поме-
щается какая-либо жидкость с коэфициентом преломления, близким к коэ-
фициенту преломления стекла. Такой жидкостью является кедровое масло
с коэфициентом преломления 1,515 (коэфициент преломления стекла равен
1,52).
Без применения преломляющей жидкости далеко не все лучи, идущие от
рассматриваемого предмета, попадают в объектив. Часть лучей, отражаясь
от верхней поверхности покровного стекла, претерпевает «полное внутреннее
отражение» и не попадает в объектив. Кроме того, часть лучей (крайних)
по выходе из покровного стекла может испытывать такое отклонение, что так-
же не попадает в объектив. Если же между фронтальной призмой и рассматри-
ваемым объектом поместить жидкость с коэфициентом преломления, близким к
коэфициенту преломления стекла, то получается как бы однородная среда и
108
лучи проходят, не преломляясь, что приводит к отчетливому изображению
рассматриваемого объекта.
Окуляры представляют менее сложную систему, нежели объективы. Они
служат для увеличения тех изображений, которые дают объективы. Обыкно-
венные окуляры, обозначаемые римскими цифрами, дают увеличение от 3
до 9 раз. Компенсационные, более ценные окуляры, обозначаемые арабскими
цифрами, дают увеличение от 3,5 до 25 раз.
Изображение, получаемое при помощи микроскопа, по отношению к рас-
сматриваемому объекту является мнимым, увеличенным и обратным.
Увеличение, которое дают обыкновенные оптические микроскопы, равно
1500—2000. «Разрешающая сила» их соответствует 0,0002 мм или 0,2р.. Это
значит, что объекты размерами менее 0,2 р. не могут быть различимы при
помощи обыкновенных оптических микроскопов.
При микроскопировании нужно придерживаться следующих правил. Для
грубой установки микроскопа пользуются макровинтом, пока не появляются
контуры рассматриваемого объекта, после чего окончательную установку
производят микровинтом. При установке тубуса следует избегать надавли-
вания объективом на препарат, так как можно повредить фронтальную линзу
объектива. При работе с иммерсионной системой на препарат наносят каплю
масла и в нее погружают объектив, после чего производят осторожно сначала
грубую, а затем окончательную установку.
Сильные окуляры поглощают часть света и уменьшают поле зрения, ввиду
чего к ним следует прибегать редко, когда получаемое изображение недоста-
точно четко.
При работе без конденсора для слабых увеличений пользуются плоским
зеркалом, а для средних и сильных — вогнутым. При работе с конденсором
при сильном увеличении применяют вогнутое зеркало, а при средних и слабых
увеличениях — плоское.
При искусственном освещении употребляют пластинку из синего стекла,
помещаемую на специальное кольцо под диафрагмой; зеркало в этом случае
применяют вогнутое.
Покровные стекла, которыми пользуются в работе, должны иметь толщину
в 0,17 мм. В апохроматах, а иногда и в других объективах имеются коррек-
ционные системы, рассчитанные на установку объектива в зависимости от
толщины покровного стекла. Для коррекции пользуются вращающимся коль-
цом в середине оправы; на нем нанесены деления от 0,10 до 0,20, соответствую-
щие толщине покровного стекла. Цифру, соответствующую толщине приме-
няемого покровного стекла, устанавливают против метки на неподвижной
части объектива. При отсутствии коррекши на объективах придерживаются
следующего правила: при тонких стеклах выдвигают внутреннюю трубку
тубуса, а при толстых — вдвигают.
Микроскоп требует самого бережного к себе отношения. В перерывах
между работой следует хранить микроскоп либо в специальном ящике, либо
под стеклянным колпаком. Нельзя подвергать микроскоп (его оптическую
часть) воздействию прямых солнечных лучей. Вытирать части микроскопа
от пыли следует мягкой кисточкой или мягкой полотняной тряпочкой. При
этом нельзя развинчивать объективы на их составные части.
После работы с масляной иммерсией следует немедленно стереть масло с
объектива мягкой тряпочкой, слегка смоченной бензином, а затем вытереть
насухо.
Термостат
Термостат (рис. 104) представляет собой шкаф, в котором поддер-
живается необходимая определенная температура. В завгсимости от объема
проводимой работы термостат может иметь различные размеры — вплоть до
размеров комнаты.
Различают два типа термостатов: водяные и воздушные.
109
Водяные термостаты — это металлические ящики с двойными
стенками, между которыми наливается вода; снаружи стенки обшиты изо-
ляционным материалом (линолеум, асбест).
Водяные термостаты снабжаются водомер-
ной трубкой. В крышке термостата име-
ются два отверстия: одно — для термо-
метра, другое —для терморегулятора.
Воздушные термостаты имеют
также двойные стенки. Для теплоизоляции
между стенками помещают изоляционный
материал (пробку, асбест и т. п.). Тепло-
устойчивость воздушных термостатов мень-
ше, нежели водяных.
Поддержание температуры в термостатах
достигается при помощи электрического
тока, газовых горелок, реже керосиновых
ламп.
Термостаты снабжаются автоматически-
ми терморегуляторами. (См. стр. 29).
Рис. 104. Термостат.
Приборы для стерилизации
Стерилизация (обеспложивание) различ-
ных объектов — питательных сред, посуды,
разных приборов — производится нагрева-
нием в сухих стерилизаторах, стерилиза-
торах Коха, автоклавах.
Сухим стерилизатором мо-
жет служить обыкновенный сушильный
шкаф с двойными стенками, описанный
выше (стр. 21). В таких шкафах стерилизуют обыкновенно посуду. Для
стерилизации пробирки закрывают ватными пробками и в проволочной кор-
зинке помещают в шкаф. Чашки Петри заворачивают в
бумагу и складывают в шкаф стопками по 5 чашек. Режим
стерилизации: 1—2 часа при 150—170° С.
Стерилизатор, изображенный на рис. 105, служит
для стерилизации текучим паром. Он представляет собой
луженый медный цилиндр с крышкой. Внутри на высоте
10—15 см от дна к стенке цилиндра приделаны лапки, на
которые кладется пластинка с отверстиями, служащая под-
ставкой для стерилизуемых предметов. На дно цилиндра
наливается небольшим слоем вода. Снаружи цилиндр
обшит асбестом или линолеумом. Внизу цилиндра сбоку
имеется небольшая водомерная трубка. Крышка цилиндра
снабжена отверстием для термометра. Пар свободно выхо-
дит через щели неплотно закрывающейся крышки. Нагре-
вание прибора производится примусом, газом или элек-
трическим током.
Обеспложивание в описанном аппарате осуществляется
по методу так называемой последовательной или дробной
стерилизации. Этот метод называется еще тиндализацией.
Сущность метода заключается в том, что стерилизуемый Рис. 105. Стерили-
объект нагревают в течение 0,75—1,5 часа в текучем паре затор Коха,
(т. е. при 100° С). При этом погибают вегетативные формы
бактерий, а споры остаются. Объект после этого оставляют на сутки при
температуре, благоприятной для прорастания бактериальных спор. Остав-
шиеся от первого нагревания споры прорастают, а развившиеся из них
по
Рис. 106. Автоклав.
вегетативные клетки не успевают образовать новых спор. По прошествии
первых суток вновь производят нагревание при 100° С в течение указан-
ного времени. Второе нагревание опять убивает развившиеся молодые веге-
тативные формы. Операцию повторяют еще через сутки, получая таким образом
стерильный материал.
Автоклав (рис. 105) представляет собою аппарат, в котором стерили-
зация производится насыщенным паром под давлением. Автоклав состоит из
вертикального толстостенного котла и толстостенной крышки. Между верхним
краем котла и крышкой помещается резиновая круговая прокладка. Крышка
скрепляется с котлом болтами с барашками. Благодаря резиновой прокладке и
прочному привинчиванию получается герметическое закрытие. В крышке
прибора имеется следующая арматура: а) манометр, отмечающий давление
внутри котла, б) кран, позволяющий выпускать из авто-
клава пар, и в) предохранительный клапан для выхода
излишка пара, устанавливаемый на определенное давле-
ние, выше которого клапан выпускает пар.
В автоклав до определенной метки наливают воду.
Над уровнем воды расположена решетка, на которую
помещают стерилизуемые предметы.
Загрузив котел автоклава, закрывают крышку, при-
винчивают плотно барашки — сначала руками, а затем
специальным ключом. После этого начинают нагревать
автоклав газом, примусом или электрическим током.
В начале нагревания автоклава кран для выпуска
пара должен быть открыт для выхода воздуха. Он за-
крывается лишь тогда, когда из него начинает выбивать
сильная струя пара. После закрытия крана давление в
автоклаве начинает подниматься. Когда давление до-
стигает необходимой высоты, регулируют нагрев так,
чтобы это давление оставалось на той же высоте нужное
времЯж Затем выпускают пар через кран или через пре-
дохранительный клапан. Выпуск пара нужно производить не поспешно, так
как понижение давления внутри сосудов не успевает за понижением давле-
ния в автоклаве, что приводит к выбрасыванию пробок. Открывать автоклав
можно лишь тогда, когда весь излишек пара вышел. Нужно открывать крышку
с осторожностью, чтобы не получить ожога от пара (открывающий становится
сзади автоклава и поднимает крышку «на себя»).
Режим стерилизации зависит от характера стерилизуемых объектов. При
двух ата (атмосферах), т. е. при одной дополнительной атмосфере по мано-
метру сверх нормального давления, темпера!ура внутри автоклава равна
120° С. Такая температура дает полную стерилизацию малых объектов (про-
бирки с жидкостью, колбочки со средами до 500 мл) в течение 20 минут.
Для других объектов устанавливается соответствующий режим. Таблица 13
Таблица 13
Упругость (давление) насыщенного пара и соответствующая ей температура
Давление в ата Температура в град. С j Давление в ата Температура в ! град. С j
1,1 102,68 1,7 115,54
1,2 105,17 1,8 117,30
1,3 107,50 1,9 118,99
1,4 109,68 2,0 120,60
1,5 111,74 3,0 133,91
1,6 113,69 4,0 144,00
111
показывает давление, которое нужно развить в автоклаве, чтобы получить
желаемую температуру. В таблице давление дано в виде суммы дополнитель-
ного давления, отсчитываемого по манометру +1 атмосфера (нормальное
барометрическое давление). Учтя это, легко определить, какое давление должно
быть по манометру, чтобы получилась желаемая температура.
Пример. Нужно получить температуру в 120°С. Этой температуре соответствует давле-
ние в 2 ата. Следовательно, когда манометр показывает 1 ата, температура в автоклаве
равна 120°С.
Сосуды для культур
Для выращивания культур применяют различные сосуды. Наиболее рас-
пространенные из них: пробирки, колбы, чашки Петри (рис. 107).
Пробирки применяют длиной в 16 см, диаметром в 1,6 см. Края про-
бирок не должны быть развернуты. Не бывшие в употреблении пробирки
Рис. 107. Сосуды для культур.
/ — пробирка (с косым агаром), 2 — колба
Виноградского. 3 — чашка Петри.
нужно предварительно прокипятить со
слабым раствором соляной кислоты или
обработать паром.
Питательную среду наливают в пробир-
ку в количестве, равном х/4—х/3 ее объема.
Пробирки закрываются ватными пробками.
Для получения ватных пробок берут пло-
ский лоскут ваты прямоугольной формы.
Длинные края складывают так, чтобы они
сошлись по средине и затем сворачивают
лоскут. При извлечении ватных пробок из
пробирок их следует вращать, чтобы ото-
рвать от стекла приставшие нити.
В пробирках делают посевы на жидкие и плотные среды. Для придания
плотным средам возможно большей поверхности им дают застывать при косом
положении пробирок.
Колб ы служат обыкновенно для посе-
вов на жидкие среды. Наичаще применяются
для этой цели колбы Виноградского кониче- 7
ской формы. Обработка новых колб произво- 4
дится тем же способом, что и обработка про- а
бирок. Ватные пробки для колб изготовля- А Г1
ются так же, как для пробирок. J
Чашки Петри широко применяются ।
для посевов на плотные среды (желатина, •
агар, среда Эндо и др.). Чашка имеет крышку i
несколько большего диаметра. Обычный раз- ! 1
мер чашек: диаметр 8—10 см, высота 1,5 см i г ! [
(с крышкой). При выращивании культур на !
чашках Петри последние помещаются в тер- ! !
мостат крышкой вниз. [ !
Приспособления для посева
4 5
Для посевов на питательные среды при-
меняются иглы, петли, шпатели, пипетки
(рис. 108).
Иглы готовят из платиновой или ни-
Рис. 108. Приспособления для
посева.
1 — игла. 2 — петли ? — шпатель ме-
таллический, 4 — шпатель стеклянный
Дригальского 5 — пипетка Пастера.
хромовой проволоки длиною в 8 см и диа-
метром около 0,4—0,5 мм. Держателями для игл служат металлические
(алюминиевые) или стеклянные палочки; в последнем случае один конец
иглы закрепляется в расплавленном конце стеклянной палочки. Иглы служат
для отвивок из колоний, или заражения уколом в плотную среду и т. п.
112
До взятия материала и после посева платиновые иглы подвергаются обжи-
ганию (фламбированию) на пламени спиртовой или газовой горелки; при этом
обжигают не только иглу, но и примыкающую к ней часть держателя. Это
необходимо производить обязательно, чтобы не внести в среду посторонние
микроорганизмы и не оставить на игле микробы после работы.
Петлю легко сделать из иглы, загибая конец ее завитком с диаметром в
3 мм. Петля служит для посевов. Обращение с петлей такое же, как и с иглой.
Шпатели готовят из толстой платиновой иглы путем расплющивания
ее конца. Назначение шпателя то же, что и петли. Для распределения посева
равномерным слоем по поверхности плотной питательной среды применяют
нередко шпатели из загнутых под прямым углом стеклянных палочек.
Пипетки применяют для заражения среды жидким материалом. Для
взятия малых количеств материала пользуются обыкновенно пипетка ми
Пастера, представляющими собой вытянутые в капилляр стеклянные
трубки. Конец капилляра запаян, а другая сторона трубки закрыта ватной
пробкой. В таком виде пипетки Пастера стерилизуются. Перед взятием мате-
риала конец пипетки обжигается в пламени и затем обламывается обожженным
пинцетом. Через капилляр засасывают посевную жидкость и затем выдувают
в среду.
Для посева относительно больших количеств жидкости служат обыкно-
венные пипетки.
Счетная камера
Счетная камера или пластинка (рис. 109) служит для
счета колоний на чашках Петри. Она состоит из квадратной стеклянной плас-
тинки и из квадратной деревян-
ной подставки для нее с зачер-
ненной поверхностью. Стеклян-
ная пластинка разделена на 144
квадрата в 1 см2 каждый. Квад-
раты, расположенные по диаго-
налям, в свою очередь разделены
на 9 малых квадратов каждый.
Исследуемую чашку со снятой
крышкой кладут кверху дном на
черную подставку и накрывают
стеклянной разграфленной пла- Рис- 109, Счетная камера,
стинкой.
С помощью лупы сосчитывают колонии не менее чем в 12 больших квадра-
тах; из них 4 в центре чашки и 8 в различных периферических участках. Если
рост густой, то при счете пользуются квадратами, разделенными на маленькие
квадратики.
Записывают отдельно число колоний в каждом подсчитанном квадрате и
сумму делят на общее число подсчитанных квадратов, определяя таким обра-
зом среднее содержание колоний на 1 см2 площади чашки. Для пересчета
на всю чашку умножают полученный результат на всю поверхность чашки,
выраженную в квадратных сантиметрах.
Пример. В 12 квадратах сосчитано 232 колонии. Диаметр чашки 10 см. Нужно определить
чиело колоний во всей чашке.
232
Находим среднее число колоний на поверхности в 1 см1. Оно равно "jy. Определяем
вою поверхность чашки:
Г. . 5^ 3,143.25^78,6 см1.
Общее число колоний на чашке равно
232 • 78,6
12
^1520.
• А. И. Бурпггейы.
Ш
ПРИГОТОВЛЕНИЕ ВАЖНЕЙШИХ ПИТАТЕЛЬНЫХ СРЕД
Мясопептонный бульон (МИБ)
Мясо коровы, лошади или другого животного освобождают от жира, сухожилий и
костей и пропускают через мясорубку. На каждые 500 г фарша приливают 1 л дестиллиро-
ванной воды (можно пользоваться и кипяченой водопроводной водой), тщательно перемеши-
вают и оставляют в прохладном месте на 12—24 часа.
По истечение этого срока жидкость фильтруют через марлю и прибавляют к фильтрату
воды до первоначального объема жидкости. К полученной мясной воде 1 прибавляют 1%
пептона в порошке и 0,5% поваренной соли. Кипятят до полного растворения пептона и
вновь доводят жидкость до начального объема.
При помощи 10% раствора соды (Na2CO3 или 0,1 N раствора NaOH доводят жидкость
до слабо щелочной реакции на лакмус, кипятят минут 20 и в горячем состоянии фильтруют
сначала через вату, затем через бумажный складчатый фильтр до полной прозрачности.
Устанавливают, как описано выше (стр. 77), реакцию в пределах pH 7,2—7,4. При этом
для подщелачивания пользуются раствором соды или едкого натрия, для подкисления —
раствором слабой соляной или фосфорной кислоты.
Полученную среду разливают в посуду и стерилизуют в автоклаве при 120°С в течение
15—20 минут.
Мясопептонный агар (МПА)
Сначала приготовляют мясопептонный бульон, как описано выше (до момента растворе-
ния пептона). Прибавляют затем 2% мелко изрезанного агара, кипятят до растворения агара
и доводят дестиллированной водой жидкость до начального объема. При помощи соды или
едкого натрия устанавливают слабощелочную реакцию на лакмус. Кипятят минут 20 и в
горячем состоянии фильтруют через вату, смоченную кипятком. Устанавливают реакцию
в пределах pH 7,0, разливают в посуду и стерилизуют в автоклаве при 120°С в течение
15—20 минут.
Под названием щелочного агара (для выращивания холерного вибриона) приме-
няют агар, pH которого равен 8,2—8,4.
Мясопептонная желатина (МПЖ)
Сначала приготовляют мясопептонный бульон, как описано выше (до момента раство-
рения пептона). Прибавляют зимой 10 процентов желатины, летом 15 процентов. Кипятят
до растворения и доводят дестиллированной водой до начального объема. Устанавливают
слабо щелочную реакцию на лакмус. Кипятят 10 минут, охлаждают до 5б°С. На литр жид-
кости прибавляют белок одного куриного яйца, взболтанный в 50 мл дестиллированной
воды, и смешивают. Кипятят и в горячем виде фильтруют через вату, смоченную кипят-
ком. Устанавливают реакцию в пределах pH 7,0. Стерилизуют три дня по часу при 100°С.
Среда Эйкмана
Состав: глюкозы 100 г, пептона 100 г, хлористого натрия 50 г, воды до 1000 мл. Раство-
ряют в воде при нагревании соль и пептон, доводят до кипения, отфильтровывают, добав-
ляют глюкозу и доводят до литра водой. Устанавливают pH в пределах 7,3—7,6, прибав-
ляя нужное количество 0,1 N раствора NaOH или 10% раствора Na2CO3. Разливают и сте-
рилизуют три дня по 30 минут при 100°С либо 15 минут при 120°С.
Из этого основного раствора готовят разведенный разбавлением в 8 раз, т. е. на 1 часть
основного раствора берут 7 частей воды. Стерилизуют, как основной раствор.
Среда Булира
Состав: мясопептонного бульона 1000 мл, маннита 12,5 г, 1% водного раствора краски
нейтральной красной (нейтральрот) б мл. Устанавливают pH среды в пределах б,8-—7,0.
Стерилизуют так же, как среду Эйкмана.
Из этого основного раствора готовят разведенный разбавлением в три раза, т. е. на 1 часть
основного раствора берут 2 части воды. Стерилизуют, как основной раствор.
При определении коли-титра среда Эйкмана и среда Булира являются равноценными,
так как санитарно-показательные Bact. coli разлагают при 43°С с образованием газа и глю-
козу (в среде Эйкмана) и маннит (в среде Булира).
Прибавление в среду Булира краски нейтральной красной имеет целью проследить,
кроме газообразования, еще и изменения окраски среды из красной в желтую вследствие
изменения pH среды. Этот признак не является, однако, обязательным, так как не все штаммы
типичных Bact. coli дают изменение окраски; поэтому можно пользоваться средой Булира
без краски.
1 Для получения мясной воды можно приготовлять 1% раствор мясного экстракта.
114
Среда Кесслера
1 л воды смешивают с 10 г пептона и 50 мл бычьей желчи. Смесь кипятят при помешива-
нии 20—30 минут. Фильтруют через вату. К фильтрату прибавляют 2,5 г лактозы и доводят
объем водой до 1 л. Устанавливают pH среды в пределах 7,4—7,6 и добавляют 2 мл 1%
водного раствора генцианвиолета. Среда разливается в пробирки с поплавками по 5 мл
и в колбочки по 50 и 100 мл и стерилизуется в автоклаве при 120° С в течение 15 минут.
Среда применяется для определения коли-титра таких продуктов, которые изобилуют
огромным количеством микрофлоры, сопутствующей Bact. coli (напр., почва, молоко и мо-
лочные продукты и т. п.) и способной подавлять рост Bact. coli.
Краска генцианвиолет прибавляется к среде как антисептик, подавляющий рост микро-
флоры, сопутствующей кишечной палочке. Желчь прибавляется в качестве защитного бу-
фера для Bact. coli. Таким образом, среда Кесслера оказывается элективной для Bact. coli.
Среда Эндо
К 1 л 3% мясопептонного расплавленного агара (pH 7,6) добавляют 10 г лактозы, рас-
творенной в 50 мл дестиллированной воды и простерилизованной текучим паром в течение
1 часа; затем прибавляют смесь из 5 мл насыщенного спиртового раствора основного фук-
сина и 2,5 г сернисто кис лого натрия (Na2SO3. 7Н2О), свеже растворенного в 25 мл дестил-
лированной воды и простерилизованного так же, как и лактоза. Эта смесь должна быть
бесцветной1, в противном случае прибавляют немного сульфита натрия. Прибавив смесь
к агару с лактозой, все тщательно перемешивают и разливают в чашки Петри. По застыва-
нии подсушивают в термостате 20—30 минут. Чашки хранят в темном месте, так как на свету
среда краснеет.
Среда Эндо служит диферснциальной для кишечной палочки. Последняя разлагает
лактозу, входящую в состав среды; при расщеплении лактозы образуются альдегиды, ко-
торые разрывают связь между фуксином и SO3, переводя лейкобазу фуксина в его исход-
ное состояние. Вот почему участки на среде Эндо, где развились колонии кишечной палочки,
приобретают яркокрасный цвет с металлическим оттенком, характерным для фуксина.
Висмут — сульфитная среда
По прописи проф. И. Е. Минкевича среда готовится следующим образом.
1. Расплавляют на водяной бане 100 мл 3% мясопептонного агара (pH 7,0) .одновременно
нагревают в той же бане 50 мл стерильной дестиллированной воды.
2. Пока агар расплавляется, приготовляют «предварительную смесь»: в стерильную
колбочку емкостью в 50 мл отвешивают 1 г глюкозы, 1 г сульфита натрия в порошке
(Na2SO3)n 1 г двуосповного кристаллического фосфорнокислого натрия (Na2HPO4 . 12Н2О).
Стерильной пипеткой вводят в колбочку 20 мл горячей (70—80°С) стерильной дестиллиро-
ванной воды и взбалтывают содержимое; при этом введенные в колбочку препараты раство-
ряются.
3. Параллельно приготовляют висмутовый раствор: в стерильную пробирку вводят
0,45 г цитрата висмутаммония, добавляют 5 мл горячей стерильной дестиллированной воды
и встряхивают до растворения. По растворении прибавляют 0,1 мл 10% раствора NaOH,
отчего раствор становится молочномутным.
Если лимоннокислого висмутаммония в распоряжении нет, то можно взять 0,45 г лимон-
нокислого висмута 2, добавить стерильной пипеткой 1 мл дестиллированной воды и 0,2 мл
раствора аммиака уд. веса 0,880 (или эквивалентный объем раствора аммиака другого уд.
веса).
Смесь подогревают слегка на водяной бане не долее 2 минут и прибавляют 4 мл стериль-
ной дестиллированной воды.
4. Приготовленный висмутовый раствор вводят в колбочку с «предварительной смесью»,
получая таким образом «окончательную смесь».
5. Приготовляют 5 мл 8% раствора сернокислой закиси железа (FeSO4): в стерильную
пробирку вводят 0,4 г железной соли и прибавляют 5 мл горячей стерильной дестиллиро-
ванной воды.
6. Приготовляют 1% раствор бриллиантовой зелени. Заготовленный впрок, он может
храниться очень долго.
7. Висмутсульфитная среда окончательно получается следующим образом: к 100 мл
расплавленного и охлажденного до 6О°С агара добавляют 1 мл раствора сернокислого же-
леза, хорошо перемешивают и добавляют всю подготовленную «окончательную смесь»;
1 Анион сульфита натрия (SO3"), соединяясь с фуксином, переводит его в бесцветное
соединение — лейкокраску.
2 Лимоннокислый висмут получают следующим образом: 10 ч. азотнокислого висмута,
7 ч. лимонной кислоты и 30—40 ч. дестиллированной воды нагревают несколько минут,
пока капля смеси с водным аммиаком не даст прозрачного раствора. Охлаждают. Выпав-
ший кристаллический осадок отфильтровывают, промывают и высушивают при умеренной
температуре.
115
вновь хорошо взбалтывают, прибавляют 0,4 мл 1% раствора бриллиантовой зелени и еще
раз тщательно взбалтывают.
Полученная среда имеет молочномутповатый цвет. Ее pH 7,8—8,1. Среда разливается в
чашки Петри.
Образующийся в процессе изготовления среды сульфит висмута, а также присутствую-
щий здесь же прибавленный в избытке сульфит натрия и прибавленный к среде раствор
бриллиантовой зелени подавляют рост Bact. coli. Если в посеве присутствуют Bact. typhi ab-
dominalis либо Bact. paratyphi, то они ферментируют присутствующую в среде глюкозу.
Продукты расщепления глюкозы восстанавливают сульфит натрия до сульфида (Na2S), а
последний с железом образует сернистое железо (FeS), отчего колонии становятся черными.
Среда электизпая для вибрионов азиатской холеры
100 мл дефибринированной бычьей крови смешивают со 100 мл нормального раствора
едкого калия (56 г КОН в 1 л воды) и стерилизуют текучим паром.
На каждые 3 части полученной жидкости берут 7 частей мясопептонного агара, смеши-
вают, разливают по чашкам Петри и подсушивают в термостате 30 минут при 60°С.
Пептонная вода
Основной раствор пептонной воды готовят по следующему рецепту:
Дестиллированной воды....................1 000,0 мл
Пептона...................................100,0 »
Хлористого натрия......................... 50,0 »
Калийной селитры (KNO.0.................... 1,0 »
Кристаллической соды (Na2CO3 . ЮН2О) .... 2,0 »
Жидкость фильтруют, разливают в колбы по 100 мл и стерилизуют в автоклаве при
120° в течение 20 минут.
Из основного раствора пептонной воды готовят разведенную пептонную воду разбавле-
нием в 10 раз.
Среды Барзикова
Готовят по следующему рецепту:
Дестиллированной воды. . . 100,0 мл
Нутрозы или пептона .... 1,0 »
Поваренной соли............ 0,5 »
Кипятят до полного растворения и фильтруют, после чего прибавляют один процент
углевода (глюкозы, лактозы, сахарозы и т. п.) или маннита и лакмусовой настойки до си-
неголубого цвета (в тонком слое). В случае надобности подщелачивают до нейтральной
реакции. Разливают по пробиркам с бродильными трубками и стерилизуют дробно три
дня по 20 минут при 100°С.
Рекомендуется углевод, маннит и лакмус стерилизовать отдельно и прибавлять сте-
рильно к остальному раствору.
Среды Барзикова применяют, чтобы выяснить отношение той или иной культуры к угле-
воду или манниту. Если культура вызывает гидролиз углевода или маннита, то в силу изме-
нения (уменьшения) pH среды меняется ее окраска — из синей становится красной. Эти
среды постоянно фигурируют в «пестрых рядах».
Молоко
Свежее снятое молоко разливают по пробиркам и стерилизуют три дня по 20 минут
при 100° С. До стерилизации можно молоко подкрасить лакмусом.
Эту среду применяют для выяснения отношения той или иной культуры к лактозе.
Если лактоза расщепляется культурой, то молоко свертывается. Присутствие лакмуса
указывает на изменение pH среды.
Желчь
Свежую бычью желчь фильтруют через бумагу, разливают по пробиркам или колбам
и стерилизуют а автоклаве 20 минут при 120° С.
Желчь применяют с целью развития и накопления некоторых бактерий, (напр,, Bast
typhi abdominal is).
Питательные среды для анаэробов
Печеночный бульон. Печень, изрезанную на мелкие кусочки, заливают водопроводной
водой в отношении 1 : 2, кипятят 30 минут и фильтруют.
Полученный фильтрат смешивают с мартеновским бульоном в равных объемах и добав-
ляют 1% поваренной соли. Устанавливают pH 7,6—7,8, фильтруют, разливают по пробнр-
116
кам с кусочками мяса или печени и стерилизуют в автоклаве в течение получаса при одной
атмосфере.
Мартеновский бульон (пептон), применяемый при изготовлении описанной среды,
готовится следующим образом: свиные желудки или сычуги рогатого скота очищают от
пленок и жира и пропускают через мясорубку. На 250 г фарша прибавляют 1 л воды и 10 мл
соляной кислоты уд. веса 1,19.
Смесь в стеклянной посуде выдерживают в термостате при 50° С в течение 24 часов,
затем нейтрализуют и автоклавируют при 120° С в течение 15 минут.
Среда Китт-Тароцци. Обезжиренную печень быка, теленка, лошади нарезают кусоч-
ками по 1—3 г, заливают трехкратным количеством питательного бульона и отвари-
вают.
После кипячения отфильтровывают через бумажный фильтр. Кусочки печени промы-
вают на сите водопроводной водой, после чего просушивают фильтровальной бумагой.
Распределяют по пробиркам по 5—б кусочков на 8—10 мл бульона, pH —
7,2—7,4. Стерилизуют в течение получаса в автоклаве при одной атмосфере.
Кровяной сахарный агар. К 100 мл расплавленного 3% мясопептоиного агара (pH 7,2—
7,4), охлажденного до 50° С, прибавляют 10 мл подогретого стерильного 20% раствора
глюкозы и 10—20 мл свежей дефибринированной стерильной крови лошади, барана, кролика
и, осторожно смешав с агаром, разливают по чашкам Петри.
ТЕХНИКА ПОСЕВА
Посев жидкого материала на агар или желатину. Жидкий материал (вода
питьевая, вода сточная, почвенная суспензия в воде и т. п.)
вводится пипеткой в определенном количестве (если имеется в виду дальней-
ший подсчет) в стерильную чашку Петри. Для посева берут обыкновенно от
0,1 до 1 мл. Если нужно посеять объем меньше 0,1 мл, то жидкость разбав-
ляют стерильной водопроводной водой или физиологическим раствором с таким
расчетом, чтобы для посева можно было взять объем в пределах 0,1—1 мл.
Так, если нужно посеять 0,01 мл, то, разбавив посевной материал в 10 раз,
берут для посева 0,1 мл, а разбавив в 100 раз, берут для посева 1 мл и т. д.
Еще до посева нужно заготовить пробирки с расплавленной желатиной
или агаром. Пробирки с же-
латиной достаточно поставить
в теплую воду или термостат
при 37° С, чтобы она рас- /
плавилась, агар же следует \ '
расплавить, поместив пробир-
ки или флаконы в кипящую ' '
ВОДЯнуЮ баню, а затем охла- Рис. ПО. Посев в чашку Петри. Момент введения
дить до 40—41° С. среды.
Приоткрыв чашку Петри,
как показано на рис. ПО, вводят в нее посевной материал каплями, захваты-
вая значительную часть дна чашки, а затем, открыв пробирку с желатиной
или агаром, обжигают края пробирки, и выливают среду в чашку (см. ри-
сунок). Закрывают чашку и при помощи вращательных движений перемеши-
вают посевной материал со средой.
Нельзя при этом оставлять незалитых
участков на дне чашки; необходимо
избегать также попадания среды на
борта чашки и образования пузырьков
воздуха.
По застывании среды чашки с же-
латиной выдерживаются в термостате
при 20—22° С, чашки с агаром — при
37° С и выше.
Посев на поверхность плотной
Рис. 111. Посев на поверхность плотно!!
среды в чашке Петри. Момент распределе-
ния посевного материала на поверхности
среды при помощи шпателя Дригальского.
питательной среды в чашке Петри.
Производится обыкновенно петлей или шпателем Дригальского (рис. 111).
При пользовании петлей сеют частыми штрихами; при пользовании шпате-
лем распределяют посевной материал по возможности равномерно по всей чашке.
117
Посев в жидкую среду. Может быть произведен в пробирку либо в колбу.
На рис. 112 показан посев в пробирку. Ватная пробка зажата между большим
Рис. 112. Посев в жидкую среду. Момент введения
посевного материала в среду при помощи петли.
и указательным паль-
цами. Петля введена в
жидкую среду пробирки.
После посева края про-
бирки и ватная пробка
обжигаются и пробирка
закрывается.
Пересев из одной
пробирки в другую
(рис. 113). Обе пробирки
зажимают между указа-
тельным и большим пальцами левой руки. Вынимают ватные пробки и за-
жимают одну между средним и безымянным
безымянным и мизинцем левой руки. Обжи-
гают края пробирки и, взяв стерильной пет-
лей часть материала из первой пробирки,
переносят его во вторую. Вновь обжигают
края пробирок и пробки и закрывают про-
бирки.
Если пересев производится на косой агар,
то по поверхности его проводят петлей черту
или змейку. Материал из подсохшей куль-
туры полезно сперва смочить в конденсаци-
онной воде посевной пробирки с агаром, а
пальцами, а другую — между
Рис. 113. Пересев из одной про-
бирки в другую.
затем уже вести посевную черту.
Посев уколом (рис. 114). Производится введением иглы с посевным матетриа-
лом в толщу питательной среды. Укол производится по оси пробирки на равном
расстоянии от краев и доходит почти до дна пробирки. Пробирку при этом
держат отверстием вниз, чтобы уменьшить возможность загрязнения из
Рис. 114. Посев уколом.
Момент введения иглы
с посевным материалом.
воздуха.
Посев уколом в желатину производится с целью
выяснения способности культуры выделять протео-
литические ферменты (разжижение желатины), а
также способность культуры разлагать среду с газо-
образованием.
Посевы анаэробов. Производятся в печеночный
бульон или среду Китт-Тароцци для первичного на-
копления анаэробов из исследуемого материала. Перед
посевом для удаления воздуха среда должна быть
«регенерирована» 10—15 минутным кипячением на
водяной бане.
Посевы производятся после быстрого охлаждения
среды и заливаются стерильным вазелиновым маслом
или расплавленным парафином.
Высевы из среды обогащения для получения по-
верхностных колоний производятся на кровяной са-
харный агар Цейслера.
Чашки для выращивания помещают в небольшой
герметически закрывающийся сосуд, напр., в обыкно-
венный экссикатор или прибор Аристовского. В сосуд помещают легко
окисляющееся вещество для поглощения кислорода, напр., подсернисто-
кислый натрий (Na2S2O4); препарат помещается в чашке Петри поверх соды
(на 20 г соды берут 3 г Na2S2O4, что достаточно для поглощения кислорода
из воздуха небольшого сосуда).
Вместо Na2S2O4 натрия можно применить пирогаллол — С6Н2(ОН)3Н.
118
ПРИГОТОВЛЕНИЕ МАЗКОВ И ИХ ОКРАСКА
Для приготовления мазков пользуются обезжиренными предметными стек-
лами толщиной в 1—1,2 мм. Обезжиривание стекол достигается погружением
их на сутки в крепкую серную кислоту или хромовую смесь. Обезжиренные
таким образом стекла хорошо промываются водой и помещаются в банку с
96° спиртом, где и хранятся до употребления.
Перед употреблением стекло насухо вытирают обезжиренной тряпочкой и
наносят на него каплю стерильной водопроводной воды, затем захватывают
прокаленной петлей исследуемый материал и, погрузив петлю в каплю воды
на стекле, тщательно размешивают материал, совершая при этом круговые
движения и расширяя поверхность, занятую каплей. Бульонную культуру
наносят непосредственно на стекло при помощи пастеровской пипетки.
После того как мазок высох, его фиксируют. Фиксация достигается наи-
чаще трехкратным медленным проведением предметного стекла мазком вверх
через пламя спиртовой или газовой горелки; длится фиксация обыкновенно
5—б секунд, из них 2 секунды препарат находится в пламени.
Описанный способ фиксации непригоден, если имеется в виду изучение
структуры клеток микроорганизмов. Для этих целей фиксируют метиловым
спиртом в течение 5 минут, этиловым спиртом — 20 минут или никифоров-
ской смесью, состоящей из равных объемов этилового спирта и эфира, —
20 минут и т. п.
Для окраски бактерий применяют почти исключительно основные краски:
фуксин, генциан, метиленовую синьку и др.
При окраске препарат кладут на стеклянную стоечку, которую легко
устроить, поместив в кювете поперек две стеклянных трубки, а вдоль кюветы
на стеклянных трубках — две стеклянные палочки.
Для окрасок, связанных с нагреванием, препарат зажимают в пинцет.
Приводим рецепты некоторых употребительных красок и методику окраски.
Щелочпый раствор метиленовой синьки
Насыщенный спиртовый раствор метиленовой синьки . . 30,0 мл
Дестиллированная вода................................ 100,0 »
1% водный раствор КОН.................................. 1,0 »
При окраске наливают раствор синьки на препарат и выдерживают 3—10
минут. Сливают краску, ополаскивают мазок дестиллированной водой и
сушат.
Карболовый фуксин
Основной фуксин.......................................... 1,0 г
Спирт 96°.............................................. 10,0 мл
Карболовая кислота (или Acid, carbol, liquefactum). . . . 5,0 г
Дестиллированная вода................................. 100,0 мл
Сначала растирают в ступке фуксин, прибавляя понемногу карболовую
кислоту и спирт, а затем небольшими порциями добавляют воду.
Применяется для окраски спор и кислотоустойчивых микроорганизмов.
Разбавленный раствор фуксина
Карболовый фуксин.................. 1,0 мл
Вода дестиллированная............. 10,0 »
Окрашивает быстро, и мазок не следует держать под краской долее 1 ми-
нуты ввиду возможности «перекрасить» препарат. По окончании окраски
краску сливают, ополаскивают мазок дестиллированной водой и сушат.
Применяется для простой окраски мазков, как и щелочная синька,
а также для дополнительной контрастной окраски в методе Грама.
119
Окраска по Граму
Для окраски по Граму нужны следующие краски и реактивы.
1. Кристаллическая фиолетовая по Граму
Кристаллическая фиолетовая...................... 2,0 г
Этиловый спирт 95°........................... 10,0 мл
1% водный раствор карболовой кислоты......... 100,0 »
Растирають краску в ступке, прибавляя спирт, а затем раствор карболовой
кислота.
2. Люголевский раствор в модификации Грама
Иод металлический....... 1,0 г
Иодистый калий........ 2,0 »
Вода дестиллированная . . . 300,0 мл.
Сохранять в прохладном месте.
3. Спирт 96
Более слабые концентрации мало пригодны.
4. Разбавленный фуксин
Приготовление указано выше. Вместо этой краски можно применять 1%
водный раствор краски нейтральная красная или 1% водный раствор эозина
и др.
Методика. На фиксированный жаром мазок кладут кусочек фильтро-
вальной бумаги, на которую наливают избыток краски (I). Окрашивание
продолжается 1—2 минуты.
Снимают с препарата фильтровальную бумагу и, не промывая его, нали-
вают на мазок раствор Люголя (2), который сливают через 1—2 минуты; не
промывая препарат, наливают на него немного 96° спирта (3) на 30 секунд.
Препарат после прибавления спирта покачивают в руке. Затем ополаскивают
мазок дестиллированной водой, после чего докрашивают одной из контрастных
красок (4) (фуксин, эозин, нейтральная красная и др.). Слив краску, промы-
вают водой и сушат.
При окраске по Граму комплекс краски и иода нацело извлекается спиртом
из грамоотрицательных бактерий и полностью удерживается грамположи-
тельными бактериями.
В виду этого грамположительные бактерии имеют темнофиолетовый цвет,
а граммотрицательные — цвет контрастной краски (красный).
Вместо кристаллической фиолетовой краски при окраске по Граму очень
часто пользуются генцианфиолетовой краской, которая готовится по следую-
щему рецепту:
Генциап фиолетовая краска
Генцианфиолетовая в Кристалах .... 0,1 г
Этиоловый спирт 95°.............. 10,0 мл
Фенол (кристаллический)........... 5,0 г
Растирать в ступке краску, прибавляя понемногу спирт и дестиллирован-
ную воду.
Окраска спор
Приготовляют на чистом покровном стеклышке мазок, который фиксируют
фламбированием. Переносят препарат на часовое стекло и обрабатывают в
течение 2 минут 5% раствором хромовой кислоты для разрушения оболочки
спор, что способствует затем лучшей их окраске.
120
Промывают препарат струей дестиллированной воды и переносят в раствор
карболового фуксина, налитый на часовое стекло. Часовое стекло
осторожно нагревают на небольшом пламени горелки до парообразования
(2—3 минуты). Затем покровное стекло извлекают, краску сливают и погру-
жают препарат секунд на пять в 30—32% этиловый спирт или 5% серную
кислоту. При этом происходит обесцвечивание сначала бактериального тела,
а затем споры. Если во-время извлечь препарат, то клетки окажутся бесцвет-
ными, а споры яркокрасными. Можно препарат окрасить дополнительно
щелочной синькой, тогда клетки окажутся синими, а споры красными.
Окраска жгутиков
Для окраски жгутиков бактерий предложено много способов. Приводим
один из наиболее простых.
Капля микробной суспензии наносится на край покровного стекла. Давая
капле стекать со стекла, получают тонкую полоску, которую высушивают
на воздухе. Затем стеклышко покрывают фильтровальной бумагой и поли-
вают краской следующего состава:
Таннин..................... 10,0 г
Хлористый цинк............. 10,0 »
Хлористый алюминий .... 18,0 »
Фуксин в порошке.......... 1,5 >>
Спирт 60%.................. 40,0 мл
Окраска препарата (без подогревания) длится 1%—2 минуты, после чего
смывают краску струей воды и докрашивают 1—2 минуты карболовым фукси-
ном.
СЕРОЛОГИЧЕСКИЕ РЕАКЦИИ
Из серологических реакций здесь рассмотрены лишь те, которые нашли
частое применение в санитарно-гигиенических исследованиях, а именно:
реакции агглютинации, преципитации и связывания комплемента.
Эти реакции применяются в санитарно-бактериологических исследованиях
для идентификации полученного антигена на основании его отношения к данной
иммунной сыворотке.
Специфичность серологических реакций, как уже указывалось выше,
довольно высока; их диагностическое значение, однако, умаляется тем, что
различные, но близкие по своей химической природе антигены способны всту-
пать в реакцию с одной и той же специфической сывороткой. С другой сто-
роны, в иммунной сыворотке кроме специфических могут содержаться и неспе-
цифические (групповые) антитела в результате чего получаются так называемые
«групповые реакции», делающие иногда невозможной точную диагностику.
Реакция агглютинации
Агглютинацией называется явление склеивания микробных клеток под
влиянием агглютининов специфической сыворотки.
Эта реакция проводится почти после каждого санитарно-бактериологи-
ческого исследования, в результате которого из данной среды (воздух, вода,
почва и т. п.) выделен соответствующий микроб. Природа микроба до реакции
агглютинации определяется различными другими методами: посев на электив-
ные среды, изучение культуральных, биохимических, морфологических и др.
особенностей. Реакция агглютинации, следовательно, призвана подтвердить
данные, полученные рядом других методов.
Взвесь выделенных микробных клеток смешивается со специфической
сывороткой, взятой в различных разведениях. В случае положительного ре-
зультата наблюдаются склеивание микробных клеток и образование больших
или меньших аггрегатов на предметном стекле (пробная агглютинация) или
осадка в пробирке.
121
В зависимости от характера антигенов и агглютининов реакция агглю-
тинации протекает различно.
Н — антигены (жгутиковые антигены), связанные с эктоплазмой микроб-
ной клетки и отличающиеся термолабильностью, вызывают образование Н —
агглютининов, устойчивых при нагревании до 70° С.
Н — антигены с соответствующими агглютининами дают крупные, рых-
лые осадки, появляющиеся при 37° С уже через 2 часа. При этом микробные
клетки склеиваются друг с другом жгутиками. Это так называемая «Н —
агглютинация».
О — антигены, содержащиеся в эндоплазме микробных клеток и отличаю-
щиеся термостабильностыо, вызывают образование О — агглютининов, раз-
рушающихся при нагревании уже при 40° С. О — антигены с соответствую-
щими агглютининами дают мелкие компактные зернистые осадки, выпадающие
на дно пробирки при 37° С в течение 24 часов. При этом микробные клетки
склеиваются своими телами. Это так называемая «О — агглютинация».
Для проведения реакции агглютинации необходимо иметь взвесь микроб-
ных клеток в физиологическом растворе и специфическую иммунную сыво-
ротку, из которой готовятся различные разведения.
Взвесь бактерий готовится из выделенных микробов; соответствующие
иммунные сыворотки получаются из производственных бактериологических
институтов, где они приготовляются путем иммунизации животных (чаще
всего кроликов), убитыми микробными культурами.
Агглютинирующая способность сывороток не одинакова. Максимальное
разведение сыворотки, при котором еще наблюдается явление агглютинации,
называется титром сыворотки.
Сыворотки сохраняются в лаборатории в запаянных ампулах в леднике.
Иногда сыворотки получаются в сухом виде. Для растворения сухой сыво-
ротки 1 часть ее заливается 10 частями физиологического раствора и на 1—2
часа помещается в термостат или водяную баню при 37° С.
Методика проведения реакции агглютинации.
Для ориентировочной идентификации выделенных микробов производят
так называемую пробную агглютинацию. На предметное стекло
наносят каплю разведенной 1 : 10— 1 : 100 агглютинирующей сыворотки и
вносят сюда немного микробной культуры, взятой петлей с косого агара или
из подозрительной колонии на чашке Петри. Все тщательно размешивают и
наблюдают, не появятся ли хлопья, особенно хорошо различимые на темном
фоне. Одновременно рекомендуется ставить контроль с тем же антигеном,
смешанным с физиологическим раствором или нормальной сывороткой.
Более обстоятельно реакция агглютинации проводится в агглютинацион-
ных пробирках длиной 70—80 мм и диаметром 6—8 мм.
В небольшой штатив помещают 6—10 таких пробирок. Число их колеб-
лется в зависимости от числа необходимых для реакции разведений сыворотки.
Начиная со второй, во все пробирки ряда наливают по 1 мл физиологического
раствора. В первую и вторую пробирки приливают по 1 мл разведения сыво-
ротки 1 : 100; из второй пробирки, перемешав жидкость, переносят 1 мл в
третью, из третьей в четвертую и т. д. Из предпоследней пробирки 1 мл выли-
вают. В последнюю пробирку сыворотку не добавляют; эта пробирка служит
контролем культуры.
Таким образом, во всех пробирках оказывается по 1 мл жидкости, причем
разведение сыворотки в первой пробирке равно 1 : 100, во второй 1 : 200,
в третьей 1 : 400 и т. д. Последняя пробирка содержит один лишь физиоло-
гический раствор.
В пробирки всего ряда добавляют по 1—2 капли взвеси испытуемых микро-
бов густотой приблизительно в 2 миллиарда микробных тел по оптическому
стандарту. Пробирки тщательно встряхивают до получения гомогенной взвеси
и помешают на 2 часа в термостат при 37° С, после чего оставляют еще на
18—20 часов при комнатной температуре. Испытуемая культура признается
122
соответствующей виду взятой сыворотки, если предельное разведение сыво-
ротки, при котором еще получается агглютинация, совпадает с титром взятой
сыворотки или равняется его половине.
Последняя пробирка, в которую сыворотка не прибавлена, служит конт-
ролем антигена.
Контроль имеет целью выяснить, не дает ли антиген самопроизвольного
образования и осаждения аггрегатов без воздействия агглютининов сыворотки.
Это явление, возникающее иногда в силу изменения коллоидно-химических
свойств антигена, носит название спонтанной агглютинации.
Такого рода измененный антиген не может служить для проведения реакции
агглютинации.
Иммунная сыворотка, как было уже указано, иногда дает групповые реак-
ции агглютинации, что исключает возможность постановки точного диаг-
ноза.
Пользуясь тем, что антиген, специфический по отношению к данной
сыворотке, дает реакцию агглютинации при более высоких разведениях
сыворотки, нежели другие антигены той же группы, можно путем повышения
разведения иммунной сыворотки уточнить диагноз.
Этот же результат достигается путем повышения концентрации хлористого
натрия, применяемого в реакции агглютинации. Вместо физиологического
раствора берут растворы хлористого натрия в 4—8—16 раз крепче. При этом
нужно учесть, что высокие концентрации соли могут вызвать выпадение гло-
булинов сыворотки; вот почему в этих случаях нужно ставить параллельно
контроль сыворотки, приготовленный разведением без добавления анти-
гена.
Реже для подавления побочных реакций агглютинации применяют метод
сорбции неспецифических агглютининов соответствующими антигенами.
Реакция преципитации
Реакция преципитации, как и реакция агглютинации, применяется в
практике санитарных лабораторий для идентификации антигена.
Применение реакции преципитации приобретает особо важное значение
для обнаружения антигена в таких материалах, бактериологическое исследо-
вание которых приводит часто к отрицательным результатам, как, например,
для обнаружения антигена сибиреязвенных бацилл в различных объектах
животного происхождения (кожа, кожаные изделия, овчина, меховые ворот-
ники, волос, волосяные изделия, шерстяные ткани, сукна, войлок, пыль
шерстяных фабрик), а также в почве пастбищ, фураже, иле из водоемов
и т. п.
В связи с термостабильностью антигена его извлекают экстрагированием
из подозрительного материала, применяя при этом нагревание при 100° С.
Экстрагирующей жидкостью служит 0,85% физиологический раствор, который
берется в количествах, в 5—10 раз превышающих взятый для исследования
материал.
Экстракция производится и на холоду при температуре не выше 15° С в
течение 6—24 часов. В этом случае к физиологическому раствору прибавляют
0,25—0,5% кристаллической карболовой кислоты с целью задержать рост
бактерий.
Получаемый экстракт для осветления подвергается центрифугированию
или чаще фильтрованию через прокаленную асбестовую вату, тальковый
фильтр и т. п.
Коллоидные растворы, применяемые в качестве антигенов в реакции пре-
ципитации, содержат ультрамикроскопические частицы порядка 0,001—0,1р..
Преципитины иммунной специфической сыворотки в присутствии элект-
ролитов (NaCl) вызывают аггрегацию и осаждение частиц антигена в виде
хлопьев или зерен.
123
Методика проведения реакции преципитации.
Для успешного проведения требуется соблюдение следующих условий: тем-
пература в пределах 20°—30° С, pH среды в пределах 7,0—7,4, полная про-
зрачность разведений антигена и специфической сыворотки.
Реакция проводится обыкновенно в форме кольцепреципитации: разве-
денный антиген наслаивается на специфическую сыворотку.
В положительных случаях на границе соприкосновения двух жидкостей
образуется белое кольцо, состоящее из преципитата.
Сыворотку, не разведенную или разведенную 1 : 2—1 : 5, разливают по
0,2 мл в ряд узких пробирок диаметром 3—4 мм. Приготовляют ряд разведе-
ний антигена 1 : 10— 1 : 1000 и выше.
Приготовленные разведения антигена наслаивают при помощи помеченной
пастеровской пипетки по 0,2 мл на разлитую в пробирки сыворотку. При этом
пробирки следует держать в наклонном положении и разведения антигена
медленно выдувать при помощи баллона на внутреннюю поверхность стенок
пробирок, не допуская перемешивания жидкостей.
Стекающий по стенке раствор антигена должен образовать над сывороткой
отдельный слой. Пробирки оставляются в штативе при комнатной темпе-
ратуре.
В положительных случаях уже через 5—10 минут на границе соприкоснове-
ния жидкостей появляется белое кольцо, размеры которого могут медленно
нарастать в течение 1—3 часов.
Реакцию необходимо сопровождать следующими контролями: 1) антиген +
нормальная сыворотка животного того же вида, которым пользовались
для получения иммунной сыворотки; 2) специфическая сыворотка + гомоло-
гический антиген, 3) специфическая сыворотка + физиологический раствор.
Реакция связывания комплемента
Реакция связывания комплемента, как и описанные выше реакции агглю-
тинации и преципитации, применяется в практике санитарно-гигиенических
лабораторий для идентификации антигена.
Реакция протекает в две фазы.
В первой фазе приводятся во взаимодействие полученный антиген, специ-
фическая сыворотка и комплемент в виде сыворотки морской свинки. Специ-
фическая сыворотка должна быть предварительно подвергнута, нагреванию
на водяной бане при 56° С в течение 30 минут. При этом комплемент специфи-
ческой сыворотки инактивируется.
В случаях, если между антигеном и специфической иммунной сывороткой
имеется специфическое сродство, происходит сорбция (связывание) компле-
мента, специально добавленного в систему в виде сыворотки морской свинки.
При этом образуется комплекс: антиген 4- специфические антитела сыворотки +
комплемент.
В случаях, когда отсутствует специфическое сродство между антигеном и
сывороткой, связывания комплемента не происходит: он остается свободным.
Во второй фазе решается вопрос, произошло ли связывание комплемента
или нет.
Для решения этого вопроса вводят в реакцию вторую систему, состоящую
из эритроцитов и специфической сыворотки, способной лизировать эти эри-
троциты.
Сыворотка и в этой системе должна быть также инактивирована нагре-
ванием.
Если связывание комплемента произошло в первой фазе, то лизирования
эритроцитов во второй фазе не произойдет, что подтвердит сделанное предпо-
ложение о природе антигена. Напротив, если связывания комплемента в пер-
вой фазе не произошло, то во второй фазе наблюдается лизирование эритро-
цитов, что опровергает сделанное предположение о природе антигена.
124
Для проведения реакции необходимы следующие ингредиенты: 1) эритро-
циты барана, 2) гемолитическая сыворотка, 3) антиген, 4) специфическая сы-
воротка, 5) комплемент, б) физиологический раствор.
Эритроциты барана получают от здорового животного возраста
не менее одного года, причем взятие крови не должно производиться чаще
двух раз в месяц. Через 1—1,5 года животное должно быть заменено
свежим.
Накануне постановки реакции производят пункцию яремной вены живот-
ного, собирая кровь через канюлю в банку, куда предварительно помещают
немного стеклянных бус. Путем встряхивания банки с содержимым в течение
10—15 минут производят дефибрирование крови, которую затем отфильтро-
вывают через нёсколько слоев марли. Эритроциты отмываются от сыворотки
путем смешивания с физиологическим раствором с последующим центрифуги-
рованием и декантацией промывной жидкости. Операция отмывания про-
изводится трижды; последняя порция промывной жидкости должна быть проз-
рачной. Из отмытых эритроцитов приготовляют 3% взвесь в физиологическом
растворе, для чего на каждый миллилитр массы эритроцитов прибавляют
33 мл физиологического раствора.
Все операции, связанные с приготовлением взвеси эритроцитов, прово-
дятся в асептических условиях.
Эритроциты могут сохраняться в леднике в течение 5—6 дней. Для консер-
вирования эритроцитов применяют раствор формалина, приготовленный раз-
ведением 2 мл 40% формалина в 100 мл физиологического раствора. На 10 мл
массы эритроцитов вводят 1 мл указанного раствора формалина и тщательно
взбалтывают. Консервированные эритроциты могут сохраняться до 2 недель.
Заметное потемнение эритроцитов указывает на их порчу.
Гемолитическая сыворотка. Для получения гемолити-
ческой сыворотки производят иммунизацию кроликов эритроцитами барана.
50% взвесь бараньих эритроцитов вводится животным внутривенно в коли-
честве 2—5 мл. Эта операция повторяется 3—4 раза, с промежутками в 2—3
дня. По окончании иммунизации берут у кролика немного крови из ушной
вены и испытывают ориентировочно титр сыворотки.
Под титром гемолитической сыворотки понимают максимальное разведение
предварительно инактивированной сыворотки, способное в объеме 0,5 мл ли-
зировать 0,5 мл взвеси эритроцитов в присутствии 0,5 мл комплемента (раз-
веденного 1 : 10) в течение одного часа при 37° С.
Если титр сыворотки оказывается не ниже 1 : 1200, то сыворотка призна-
ется пригодной для работы и производится кровопускание из уха кролика
путем поперечного разреза краевой ушной вены. Шерсть в месте намеченного
разреза предварительно сбривается. Кожа смачивается ксилолом с целью
вызвать гиперемию. Затем кожу обтирают ватой, смоченной физиологическим
раствором, и производят разрез. За один раз собирают 40—60 мл крови в
широкую пробирку, на дно которой для лучшего свертывания крови наливают
0,5—1 мл физиологического раствора.
Крови дают постоять. Когда образуется сверток фибрина, его отделяют
от стенок пробирки и помещают последнюю в ледник до следующего дня. На
второй день сыворотку отделяют от сгустка и подвергают инактивированию
нагреванием в водяной бане в течение 30 минут при 56° С.
Инактивированную таким образом сыворотку затем консервируют прибав-
лением 1% сухой борной кислоты.
Полученная сыворотка разливается в ампулы, которые запаиваются и хра-
нятся на льду.
Ввиду возможности колебания титра, сыворотка выдерживается 1—1,5
месяца, после чего устанавливается ее титр.
Для этой цели приготовляют ряд разведений от 1 : 1000 до 1 : 3500. Эти
разведения готовят из основного разведения 1: 100 (0,1 мл сыворотки +9,9 мл
физиологического раствора) по схеме, представленной в таблице 14.
125
Таблица 14
Схема приготовления разведений гемолитической сыворотки
№№ пробирок Количество основного разведения гемолитической сыворотки в мл Физиологический раствор в мл Полученное разведение
1 0,1 0,9 1 : 1000
2 0,1 1,1 1 : 1200
3 0,1 1,4 1 : 1500
4 0,1 1,7 1 :1800
5 0,1 1,9 1 : 2000
б 0,1 2,4 1 : 2500
7 0,1 2,9 1 : 3000
8 0,1 3,4 1 : 3500
При этом применяют пробирки длиною 80—100 мм и диаметром 10—12 мм.
Получив указанные разведения, устанавливают титр гемолитической сы-
воротки по схеме, представленной в таблице 15. Титрование сопровождается
контролем резистентности эритроцитов (пробирка 9-я по схеме).
Таблица 15
Схема титрования гемолитической сыворотки
№№ пробирок Количество гемолитической сыво- ротки (в мл) и степень разведения 3% взвесь эритроци- тов в мл Комплемент в разведении 1: 10 в мл Физиологи- ческий раст- вор в мл
1 0,5 (1 : 1000) 0,5 0,5 1
2 0,5(1 : 1200) 0,5 0,5 1
3 0,5(1 : 1500) 0,5 0,5 1
4 0,5(1 : 1800) 0,5 0,5 1
5 0,5(1:2000) 0,5 0,5 1
б 0,5(1 : 2500) 0,5 0,5 1
7 0,5(1 : 3000) 0,5 0,5 1
8 0 5(1 : 3500) 0,5 0,5 1
9 — 0,5 — 2
Термостат 37° — 1 час.
Рабочий титр гемолитической сыворотки должен быть втрое сильнее против
установленного по приведенной выше схеме. Так, если по схеме титр оказался
равным 1 : 1800, то рабочим титром служит разведение 1 : 600.
Антигеном служит взвесь в дестиллированной воде или в физиоло-
гическом растворе выделенных микробов, убитых нагреванием при 60° С в
течение 1 часа или при 100° С в течение 15-—20 минут. Реже микробную взвесь
стерилизуют парами формалина или обрабатывают алкоголем.
Для реакции берут 0,5 мл взвеси, содержащей 1 миллиард микробных тел
в 1 мл по оптическому стандарту. Такая взвесь в тех количествах, которые
идут в реакцию, лишена обыкновенно антикомплементарных и гемотоксиче-
ских свойств и потому отпадает необходимость в титровании такого антигена.
Микробный антиген должен сохраняться в леднике.
Антиген может быть получен также при помощи экстрагирования его
из испытуемого объекта физиологическим раствором (см. выше) и др. способами.
Специфическая иммунная сыворотка получается из
производственных бактериологических институтов. Обычно пользуются кро-
личьими сыворотками, обладающими хорошей активностью. В зависимости
от активности применяют различные разведения сыворотки: 1 : 20; 1 : 100;
1 : 1000. Перед употреблением сыворотка инактивируется.
Комплементом служит сыворотка морской свинки, получающаяся
следующим образом: при помощи пункций сердца морской свинки, произве-
денной накануне дня постановки реакции, получают кровь. Когда в постоявшей
126
в пробирке крови образуется сверток фибрина, его отделяют от стенок про-
бирки и помещают пробирку в ледник до следующего дня. Выделившаяся на
другой день прозрачная сыворотка служит комплементом. Активность полу-
ченного таким образом комплемента может сохраняться в течение 1—2 дней.
При надобности. комплемент можно консервировать; для этой цели к полу-
ченному накануне комплементу добавляют 4% борной кислоты и 5% серно-
кислого натрия в сухом виде. По растворении консервирующих веществ компле-
мент помещают в ледник. В таком виде он может сохранять свою активность
в течение 1—3 месяцев и долее.
Для получения комплемента могут служить лишь здоровые свинки, не бере-
менные и не подвергавшиеся экспериментам.
Перед постановкой реакции комплемент должен быть протитрован per se
и в присутствии антигена.
Титрование комплемента per se производится с целью установления его
титра, под которым понимают то минимальное количество комплемента, в
присутствии которого 0,5 мл гемолитической сыворотки в тройном титре
способно лизировать 0,5 мл 3% взвеси эритроцитов в течение 30 минут при
37° С.
Определив титр комплемента, рассчитывают рабочую дозу его, которая
на 20—35% должна превышать титр.
Для определения титра комплемента per se приготовляют основное разве-
дение его прибавлением к 1 мл комплемента 9 мл физиологического раствора,
разливают в пробирки в количествах, указанных в таблице 16, и добавляют
физиологический раствор в количествах, также указанных в таблице.
Таблица 16
Схема титрования комплемента per se
Штатив с пробирками помещают в термостат при 37° С. Через 15 минут
после этого подготовляют гемолитическую систему. Для этой цели к 3% взвеси
127
бараньих эритроцитов прибавляют равный объем гемолитической сыворотки,
взятой в тройном титре. Подготовленную таким образом гемолитическую си-
стему трижды переливают из одного сосуда в другой, затем помещают в термо-
стат при 37° С на 30 минут. По истечении этого срока, т. е. через 45 минут
после помещения комплемента в термостат, во все пробирки, в которых находятся
приготовленные разведения комплемента, прибавляют по 1 мл гемолити-
ческой смеси, которую предварительно тщательно размешивают повторным
всасыванием в пипетку и выдуванием. После прибавления гемолитической
смеси пробирки тщательно встряхивают и вновь помещают в термостат при
37° С на 30 минут.
Титрование сопровождают двумя контролями: 1) 0,5 мл основного разве-
дения комплемента + 1,5 мл физиологического раствора + 0,5 мл 3% взвеси
эритроцитов (см. пробирку 11-ю по схеме) и 2) 1,5 мл физиологического рас-
твора 4- 1,0 мл гемолитической смеси (см. пробирку 12-ю по схеме).
Смысл этих контролей ясен: первый из них имеет целью выяснить, не прои-
зойдет ли гемолиз эритроцитов в отсутствии гемолитической сыворотки, т. е.
проверяется участие гемолитической сыворотки в реакции; второй контроль
выясняет таким же образом участие антигена в реакции.
По истечении указанного времени, т. е. через 1 час 15 минут после помеще-
ния в термостат разведенного антигена, проверяют результаты реакции.
Практически за титр комплемента принимается то его количество, которое
было прилито в первую из пробирок, в которых наблюдается полный гемолиз.
Практически за рабочую дозу комплемента принимается количество его,
пошедшее в пробирку, следующую за той, по которой определяется титр ком-
племента.
Некоторый избыток комплемента в рабочей дозе против титра берется
из тех соображений, что антиген, а также гемолитическая сыворотка, участ-
вующие в реакции, могут проявлять антикомплементарные свойства и не-
сколько подавлять активность комплемента.
Хотя применяемые при описанной реакции полученные антигены не обла-
дают в применяемых дозах выраженным антикомплементарным свойством,
все же иногда представляется необходимость установить титр комплемента в
присутствии антигена, что производится по схеме, представленной в таблице 17.
Таблица 17
Схема титрования комплемента в присутствии антигена
№№ 1 пробирок 1 2 3 4 61 6 7 8 9 ' 10
Антиген (1 милли- ард микробных тел в 1 мл) 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5
Комплемент (разве- дение 1 : 10) в мл . 0,05 0,1 0,15 0,2 0,25 0,3 0,35 0,4 0,45 0,5
Физиологический . . раствор в мл . . . 0,95 0,9 0,85 0,8 0,75 0,7 0,65 0,6 0,55 0,5
>28
Если антиген не обладает антикомплемента рными свойствами, то резуль-
таты титрований в присутствии антигена оказываются такими же, как и в его
отсутствии; в противном случае гемолиз начинается при более высоких коли-
чествах комплемента. Соответственно результатам титрования устанавли-
вается более высокая рабочая доза для комплемента.
Физиологический раствор готовится 0,85% из химически
чистого препарата NaCl на дестиллированной воде и стерилизуется в
автоклаве.
Методика проведения реакции связывания комп-
лемента. Для идентификации выделенного микробного антигена при-
готовляют и проверяют все указанные выше ингредиенты и проводят реакцию
по схеме, представленной в таблице 18.
Таблица 18
Схема идентификации выделенного антигена
№№ пробирок 1 2 3
Специфическая иммунная сыворотка, разве- денная по титру, в мл 0,1 0,1 0,1 i 1
Физиологический раствор в мл 0,4 0,4 0,9 | 1
Антиген выделенный (взвесь 1 млрд в 1 мл) в мл 0,5 —
Антиген известный (взвесь 1 млрд в 1 мл) в мл — 0,5 —
Комплемент (рабочая доза 4- физиологический раствор) 0,5 0,5 0,5
Термостат 37э — 1 час
Гемолитическая система в мл............| 1 1 1
Термостат 37° — 40 минут—1 час.
Положительный результат реакции характеризуется задержкой гемолиза
в 1-й и 2-й пробирках и полным гемолизом в 3-й. Задержка гемолиза в третьей
пробирке указывает на антикомплементарные свойства иммунной сыворотки.
В этом случае положительный результат становится сомнительным. Опыт
должен быть повторен с другой иммунной сывороткой.
Отрицательный результат реакции характеризуется полным гемолизом в
1-й и 3-й пробирках и задержкой гемолиза во 2-й.
Учет результатов реакции производится двукратно: после выдерживания
в термостате и на следующий день после выдерживания в леднике в течение
15—18 часов.
Для большей убедительности рекомендуется параллельно ставить опыт
по той же схеме, взяв вместо специфической нормальную сыворотку. В про-
бирках с нормальной сывороткой наблюдается полный гемолиз. Задержка
гемолиза в первых двух пробирках указывает на антикомплементарные свойства
антигена. В этом случае нужно пользоваться рабочей дозой комплемента,
определенной титрованием в присутствии антигена.
Степень задержки гемолиза в положительных реакциях обозначается
числом крестов по следующей схеме:
4--I- + -I- полная задержка
+ + + ясн^1 задержка
+ + частичная задержка
слабая задержка
+ следы задержки
— полный гемолиз
9. А. И. Бурштейн.
129
Исследования на животных (биопробы)
Для идентификации антигена в зараженном материале, помимо описанн
выше бактериоскопических, бактериологических и серологических Иселер
ваний, применяют иногда и биопробы на животных.
Эти исследования являются заключительным звеном целой серии исследоь
ний инфекционного материала. Ими пользуются также для открытия токе
ческих веществ в зараженном материале (токсины ботулинуса, тетануса) и т.
В качестве подопытных животных используют обыкновенно белых мыше
крыс, морских свинок, кроликов, реже обезьян, кошек, хорьков и пр.
Животные, предназначенные для опыта, должны быть здоровы. Пер
опытом определяют их вес, температуру. При массовых опытах животне
отмечаются при помощи нумерованных бляшек.
Исследуемый материал вводят животным различными путями: подкожы
внутрикожно, внутривенно, внутримышечно, в брюшную полость; реже з<
раженный материал вводится per os.
Введение материала парентеральным путем производится в асептически
условиях. В месте введения материала шерсть сбривается, кожа дезинфицг
руется спиртом.
Подкожную инъекцию у мышей и крыс производят обыкновенно на спин
у корня хвоста, у свинок и кроликов — на спине или животе немного ниж<
подмышечной ямки.
При внутрикожном введении материал вводится тонкой иглой в эпидермис
При внутривенном заражении иглу вводят в вену по направлению токе
крови: при этом нужно следить, чтобы не проколоть противоположной стенкг
вены. При правильной инъекции не должно образовываться вздутия у места
инъекции. Кроликам внутривенная инъекция производится в краевую вену
уха, мышам и молодым крысам — в вены, расположенные по обеим сторонам
хвоста, морским свинкам и взрослым крысам — в яремную вену, которая
перед этим отсепаровывается.
При внутримышечной инъекции материал вводится в мышцу бедра.
При внутрибрюшном заражении материал вводится со стороны живота на
5—7 см выше симфиза; животное при этом держат головой вниз. Предвари-
тельно производят небольшой разрез кожи, вводят сюда иглу сначала в го-
ризонтальном направлении, а затем устанавливают вертикально и буравящими
движениями продвигают вперед. Когда достигают брюшной полости, сопро-
тивление сразу исчезает; в этот момент вводят материал.
Энтеральное введение материала производится либо простым скармли-
ванием, либо при помощи тонкого эластичного катетера, вводимого через нос
или рот.
Зараженные животные должны быть изолированы от других. За ними уста-
навливается систематический надзор. При этом проверяют вес, температуру,
наблюдают поведение, походку, аппетит животного, характер выделений и т. п.
Погибших животных вскрывают и подвергают патологоанатомическому
и бактериологическому исследованию. Делают посевы крови из сердца, экс-
судата из брюшной полости, если он обнаруживается, а также из селезенки,
печени и мезентериальных желез. Помимо посевов из указанных материалов
приготовляют мазки. При этом из крови и экссудата материал наносится на
предметное стекло пастеровской пипеткой; для получения мазков из органов
захватывают пинцетом кусочек органа и касаются им предметного стекла.
При вскрытии животных пользуются стерилизованным инструментарием,
чтобы не внести посторонней инфекции.
Трупы вскрытых животных подвергаются уничтожению (сжиганию).
ПРИЛОЖЕНИЯ К ГЛАВЕ И
Таблица 19
Атомные веса элементов
0 — 16,000
(по данным на 1947 г.)
Н —1,008-
Наименование Сим- вол Атомный вес Наименование Сим- вол Атомный вес
Азот N 14,008 Мышьяк As 74,91
Алюминий А1 26,97 Натрий Na 22,997
Барий Ва 137,36 Никель Ni 58,69
Бор В 10,82 Олово Sn 118,70
Бром Вг 79,916 Палладий Pd 106,7
Ванадий V 50,95 Платина Pt 195,23
Висмут Bi 209,00 Радий Ra 226,05
Водород Н 1,008 Ртуть Hg 200,61
Вольфрам W 183,92 Свинец Pb 207,21
Железо Fe 55,85 Селен Se 78,96
Золото . . Au 197,2 Сера S 32,066
Иод J 126,92 Серебро Ag 107,880
Кадмий Cd 112,41 Стронций Sr 87,63
Калий К 39,096 Сурьма Sb 121,76
Кальций Ca 40,08 Титан Ti 47,90
Кислород 0 16,000 Углерод c 12,010
Кобальт Co 58,94 Уран и 238,07
Кремний Si 28,06 Фосфор p 30,98
Литий Li 6,940 Фтор F 19,000
Магний Mg 24,32 Хлор Cl 35,457
Марганец Mn 54,93 Хром Cr 52,01
Медь Cu 63,54 Цезий Cs 132,91
Молибден Mo 95,95 Цинк Ze 65,38
131
Таблица 20
Удельный вес минеральных кислот
Уд. вес 15° при-jr (безвоздуш- ное про- странство) Весовые проценты Уд. вес 15° при-^г- (безвоздуш- ное про- странство) Весовые проценты
НС1 HNO3 H2SO4 HNO3 H2SO4 1
1,000 0,16 0,10 0,09 1,430 72,17 53,11
1,010 2,14 1,90 1,57 1,440 74,68 54,07
1,020 4,13 3,70 3,03 | 1,450 77,28 55,03
1,030 6,15 5,50 4,49 i 1,460 79,98 55,97
1,040 8,16 7,26 5,96 । 1,470 82,90 56,90
1,050 10,17 8,99 7,37 | 1,480 86,05 57,83
1,060 12,19 10,68 8,77 ! 1,490 89,60 58,74
1,070 14,17 12,33 10,19 1,500 94,09 59,70
1,080 16,15 13,95 11,60 1,510 98,10 60,65
1,090 18,11 15,53 12,99 1,520 99,67 61,59
1,100 20,01 17,11 14,35 1,530 — 62,53
1,110 21,92 18,67 15,71 1,540 — 63,43
1,120 23,82 20,23 17,01 1,550 — 64,26
1,130 25,75 21,77 18,31 1,560 — 65,08
1,140 27,66 23,31 19,61 । 1 1,570 — 65,90
1,150 29,57 24,84 20,91 j ! 1,580 — 66,71
1,160 31,52 26,36 22,19 ' 1,590 — 67,59
1,170 33,46 27,88 23,47 1,600 — 68,51
1,180 35,39 29,38 24,76 1 1.610 — 69,43
1,190 37,23 30,88 26,04 ' 1,620 — 70,32
1,200 39,11 32,36 27,32 1,630 — 71,16
1,210 33,82 28,58 1,640 — 71,99
1,220 35,28 29,84 1,650 — 72,82
1,230 । | — 36,78 31,11 1,660 — 73,64
1,240 38,29 32,28 1,670 — 74,51
1,250 39,82 33,43 1,680 — 75,42
1,260 41,34 34,57 1,690 — 76,30
1,270 42,87 35,71 1,700 — 77,17
1,280 ; 44,41 36,87 1,710 — 78,04
1,290 | 45,95 38,03 1,720 — 78,92
1,300 47,49 39,19 1,730 — 79,80
1,310 49,07 40,35 1,740 — 80,68
1,320 50,71 41,50 1,750 — 81,56
1,330 52,37 42,66 1,760 — 82,44
1,340 54,07 43,74 1,770 — 83,32
1,350 55,79 44,82 1,780 — 84,50
1,360 57,57 45,88 1,790 — 85,70
1,370 59,39 46,94 1,800 — 86,90
1,380 61,27 48,00 1,810 — 88,30
1,390 63,23 49,06 1,820 — 90,05
1,400 65,30 50,11 1,830 — 92,10
1,410 — 67,50 51,15 1,840 — 95,60
1,420 69,80 52,15 1,841 — 97,00
1,8485 99,95
।
[32
Таблица 21
Удельный вес водных растворов уксусной кислоты (при 15°С)
Удельный вес С2Н4О2 Весовые проценты Удельный вес С2Н4О2 Весовые проценты Удельный вес С2Н4О2 Весовые проценты Удельный вес С2Н4О2 Весовые проценты
1,0007 1 1,0298 21 1,0523 40 1,0679 59
1,0022 2 1,0311 22 1,0533 41 1,0685 60
1,0037 3 1,0324 23 1,0543 42 1,0691 61
1,0052 4 1,0337 24 1,0552 43 1,0697 62
1,0067 5 1,0350 25 1,0562 44 1,0702 63 1
1,0083 6 1,0363 26 1,0571 45 1,0707 64
1,0098 7 1,0375 27 1,0580 46 1,0712 65
1,0113 8 1,0388 28 1,0589 47 1,0717 66
1,0127 9 1,0400 29 1,0598 48 1,0721 67
1,0142 10 1,0412 30 1,0607 49 1,0725 68
1,0157 11 1,0424 31 1,0615 50 1,0729 69
1,0171 12 1,0436 32 1,0623 51 1,0733 70
1,0185 13 1,0447 33 1,0631 52 1,0737 71
1,0200 14 1,0459 34 1,0638 53 1,0740 72
1,0214 15 1,0470 35 1,0646 54 1,0742 73
1,0228 16 1,0481 36 1,0653 55 1,0744 74
1,0242 17 1,0492 37 1,0660 56 1,0746 75
1,0256 18 1,0502 38 1,0666 57 1,0747 76
1,0270 19 1,0513 39 1,0673 58 1,0748 77
1,0284 20
Таблица 22
Удельный вес водных растворов
КОН и NaOH (при 15°С)
Удельный вес Весовые проценты Удельный вес Весовые проценты
кон NaOH кон NaOH
1,007 0,9 0,61 1,220 24,2 19,58
1,014 1,7 1,20 1,231 25,1 20,59
1,022 2,6 2,00 1,241 26,1 21,42
1,029 3,5 2,71 1,252 27,0 22,64
1,037 4,5 3,35 1,263 28,0 23,67
1,045 5,6 4,00 1,274 28,9 24,81
1,052 6,4 4,64 1,285 29,8 25,80
1,060 7,4 5,29 1,297 30,7 26,83
1,067 8,2 5,87 1,308 31,8 27,80
1,075 9,2 6,55 1,320 32,7 28,83
1,083 10,1 7,31 1,332 33,7 29,93
1,091 10,9 8.00 1,345 34,9 31,22
1,100 12,0 8,68 1,357 35,9 32,47
1,108 12,9 9,42 1,370 36,9 33,69
1,116 13,8 10,06 1,383 37,8 34,96
1,125 14,8 10,97 1,397 38,9 36,25
1,134 15,7 11,84 1,410 39,9 37,47
1,142 16,5 12,64 1,424 40,9 38,80
1,152 17,6 13,55 1,438 42,1 39,99
1,162 18,6 14,37 1,453 43,4 41,41
1,171 19,5 15,13 1,468 44,6 42,83
1,180 20,5 15,91 1,483 45,8 44,83
1,190 21,4 16,77 1,498 47,1 46,15
1,200 22,4 17,64 1,514 48,3 47,60
1,210 23,3 18,58 1,530 49,4 49,02
133
Таблица 23
Удельный вес водных растворов аммиака (при 15° С)
Удельный вес Весовые проценты NH, Удельный вес Весовые проценты NH3
1,000 0,00 0,940 15,63
0,998 0,45 0,938 16,22
0,996 0,91 | 0,936 16,82
0,994 1,37 0,934 17,42
0,992 1,84 0,932 18,03
0,990 2,31 0,930 18,64
0,988 2,80 0,928 19,25
0,986 3,30 0,926 19,87
0,984 3,80 0,924 20,49
0,982 4,30 0,922 21,12
0,980 4,80 0,920 21,75
0,978 5,30 0,918 22,39
0,976 5,80 0,916 23,03
0,974 6,30 0,914 23,68
0,972 6,80 0,912 24,33
0,970 7,31 0,910 24,99
0,968 7,82 0,908 25,65
0,966 8,33 0,906 26,31
0,964 8,84 0,904 26,98
0,962 9,35 0,902 27,65
0,960 9,91 0,900 28,33
0,958 10,47 0,898 29,01
0,956 11,03 0,896 29,69
0,954 11,60 0,894 30,37
0,952 12,17 0,892 31,05
0,950 12,74 0,890 31,75
0,948 13,31 0,888 32,50
0,946 13,88 0,886 33,25
0,944 14,46 0,884 34,10
0,942 15,04 0,882 34,95
134
ЛИТЕРАТУРА
Оборудование лабораторий и лабораторная техника
1. Альянаки П., Жаворонков М., Леонов Б., Лабораторные весы и взвешивание, изд.
«Стандартизация и рационализация», 1935. а
2. Ангерер Е. Лабораторная техника, под ред. Яковлева К., ОНТИ, 1934.
3. Веселовский В. и LL1 маненков И., Нагревательные приборы в лабораторной практике,
1947.
4. Виноградов А. В., Организация заводских лабораторий в химической промышлен-
ности, Госхимиздат, 1948.
5. Винокуров П. Д., Техника санитарно-гигиенических исследований, Медгиз, 1939.
6. Воскресенский П. И., Техника лабораторных работ, Госхимиздат, 1941.
7. Иольсон Л. М., Заводские химические лаборатории, ОНТИ, 1937.
8. Кириллов В., Организация и оборудование лабораторий мясокомбинатов, журнал
«Мясная индустрия», №№ 1 и 2, 1935.
9. Рудо Н. М., Точное взвешивание, изд. ВНИИМ., Л., 1949.
10. Щеколдина Е., Прокофьева А. и Андреева Е., Организация лабораторий и прове-
дение химико-технического контроля на плодоовощных предприятиях, 1947.
Химический анализ
1. Алексеев В. Н., Курс аналитической химии, Госхимиздат, 1947.
2. Алексеевский Е., Гольц Р., Мусакин А., Количественный анализ, Госхимиздат, 1948.
3. Бер ль-Л у нее, Химико-технические методы исследований ОНТИ, 1935.
4. Гиллебранд В. и Лендель Г., Практическое руководство по неорганическому ана-
лизу, под ред. Лурье Ю., ОНТИ, 1935.
5. Кольтгоф И. М. и Сендэл Е. Б., Количественный анализ, под ред. Лурье Ю. Ю.,
изд. III, 1948.
б. Коренман И. М. Количественный микрохимический анализ, Госхимиздат, 1949
7. Петрашень В. И., Объемный анализ, Госхимиздат, 1946.
8. Тананасе Н. А., Объемный анализ, 1930.
9. Тананаев Н. А., Весовой анализ, Г НТ изд. Украины, 1934.
10. Тредвелл Ф. и Голл В., Курс аналитической химии, под ред. Комаровского, т.т.I
и II, ОНТИ, 1935.
11. Шилов Н. А., Объемный анализ, 1931.
12. Фаульз Г. Объемный анализ, под ред. Троицкого Е., ОНТИ, 1934.
Физико-химический анализ
1. Гилыпнер В., Практика потенциометрических анализов, под ред. Малярова К-9
ОНТИ, 1936.
2. Иоу Д., Колориметрия, под ред Успенского А., ОНТИ, 1935.
3. Иоу Д., Нефелометрия, под ред. Успенского А., ОНТИ, 1935.
4. Кольтгоф И. и Фурман Н., Потенциометрическое титрование, под ред. Николь-
ского Б. ОНТИ, 1935.
5. Мисловицер Е., Определение концентрации водородных ионов в жидкостях, под
ред. Беркенгейма Б., Госхимтехиздат, 1932.
6. Михаэлис Л., Практикум по физической химии, под ред. Кистяковского В., Гос-
издат, 1935.
7. Митрофанов П. П. и Северин Е. С. Учебник физической и коллоидной химии,
Медгиз, 1941.
8. Новые методы технического контроля в заводской лаборатории химической промыш-
ленности, под ред. Центрального совета заводских лабораторий тяжелой промышленности
и ВНИТО химиков, ОНТИ, 1935.
9. Оствальд-Лютер-Друкер, Физико-химические измерения, под ред. Рабиновича и др.,
ОНТИ, 1934—1935.
10. Рубинштейн Д. Л., Физическая химия, изд. Акад, наук СССР, 1940.
11. Современные физико-химические методы химического анализа, под ред. С. А. Щу-
ка рева, Лгнхимтехиздат, 1932.
12. Эйкен А., Физико-химический анализ в производстве, под ред. Вавилова С. и др.,
ОНТИ, 1936.
13. Щепкин С., Контрольно-измерительные и регулирующие приборы в химических
производствах, Госхимиздат, 1945.
Бактериологический анализ
1. Кальмет А. и др., Руководство по микробиологической и серологической технике,
под. ред. Бронштейна, 1938.
2. Кульчицкий Н., Учение о микроскопе и техника микроскопических исследований,
1907.
135
3. Макарова-Тарасевич Ю. Н., Основы анаэробной диагностики, Биомедгиз, 1934.
4. Миллер А. А., Санитарная бактериология, Госмедиздат, 1933.
5. Минкевич И. Е., Курс санитарной бактериологии, 1940.
О^мелянский В. Л., Практическое руководство по микробиологии, изд. Акад, наук
7. Синай Г. Я. и Биргер О. Г., Микробиологические методы исследования при инфек-
ционных заболеваниях, Медгиз, 1949.
8. Титов Л. Г., Микроскопы, их принадлежности и применение, 1934.
Биологические методы исследования
1. Ковалевский. К. Л., Разведение и содержание мелких лабораторных животных,
М., 1944.
2. Никитин В. Н., Атлас клеток крови сельскохозяйственных и лабораторных жи-
вотных, Госсельхозиздат, М., 1949.
3. Пауэлл M.f Способы обращения с мелкими животными в лабораториях, Лабор.
практика № 3, 1934.
4. Правдин Н. С., Методика малой токсикологии промышленных ядов, Медгиз, 1947.
5. Предтеченский В. Е., Боровская В. М. и Марголина Л. Т., Лабораторные методы
исследования, Медгиз, 1950.
6. Сахаров П. П., Лабораторные животные, Биомедгиз, 1937.
7. Синай Г. Я. и Баргер О. Г., Микробиологические методы исследования при
инфекционных заболеваниях, Медгиз, 1949.
Справочники
1. Карякин Ю., Чистые химические реактивы, ОНТИ, 1936.
2. Лейхман Л., Словарь названий химических реактивов, ГНТИ, 1932.
3. Лурье Ю., Расчетные и справочные таблицы для химика, Госхимиздат, 1947.
4. Палаузов В., Химические реактивы, их свойства, получение, методы испытаний и
применение, ОНТИ, 1935.
5. Перельман B.t Краткий справочник химика, Госхимиздат, 1947.
6. Справочник лабораторного оборудования, Союзлаборреактив, ОНТИ, 1936.
7. Спутник химика, тт. I, II и III, ОНТИ, 1932—1943.
8. Цион Р. А., Определитель микробов, Сельхозгиз., М., 1948.
ГЛАВА ТРЕТЬЯ
ВОЗДУХ
Газообразная оболочка, окружающая земной шар, называется атмос-
фере й. Она простирается на высоту в 1000 м.
Нижние слои атмосферы называются воздухом.
СОСТАВ И СВОЙСТВА АТМОСФЕРНОГО ВОЗДУХА
Атмосферный воздух имеет в среднем следующий состав в объемных про-
центах: азота 78%, кислорода 20,99%, угольного ангидрида 0,03%, аргона
0,94%, водяных паров 0,5—1,5%. Кроме этих газов в состав воздуха входят
еще в ничтожных количествах криптон, ксенон и неон, гелий и водород. Здесь
же непостоянно и в небольших количествах встречаются озон, перекись во-
дорода, аммиак, азотная и азотистая кислоты, сероводород и др. газы. В ка-
честве плотных примесей в воздухе встречаются пылевые частицы и микроорга-
низмы.
Вес одного литра воздуха при нормальных условиях (т. е.
при давлении 760 мм ртутного столба и при t 0°) равен 1,293 г; удельная
теплоемкость воздуха при постоянном давлении
(Ср) равна 0,237 м. кал/г, удельная теплоемкость воздуха
при постоянном объеме (Cv) равна 0,169 м. кал/г; тепло-
проводность воздуха равна 0,0000558.
САНИТАРНО-ГИГИЕНИЧЕСКИЕ ТРЕБОВАНИЯ К ВОЗДУХУ
Воздух по своим физическим качествам (температура, влаж-
ность, движение и лучистое тепло), суммарно действующим, должен произво-
дить приятное ощущение сохранения теплового равновесия организма. Баро-
метрическое давление не должно настолько отличаться от нормального (760 мм
Hg), чтобы вызывать расстройство здоровья.
По химическому составу воздух должен быть лишен вредных
для здоровья паров, газов и аэрозолейх.
Содержание СО2 в воздухе помещений не должно превышать 0,7—1,0%0.
Бактериальная флора воздуха не должна включать
патогенных микробов.
Органолептически воздух не должен обладать неприятными
запахами.
ОРГАНОЛЕПТИЧЕСКАЯ ОЦЕНКА ВОЗДУХА
Органолептическая оценка воздуха производится по запаху. Запах воздуха
определяется на основании субъективного ощущения. При помощи обоняния
можно открыть в воздухе присутствие лишь таких газов, которые отличаются
специфическим запахом (сероводород, аммиак и др.). Низкая температура,
сухость и неподвижность воздуха значительно ослабляют его запах.
1 Для производственных помещений устанавливаются предельно допустимые концент-
рации этих ингредиентов. Для атмосферного воздуха у нас также разрабатываются
соответствующие нормы, (см. далее).
137
ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ВОЗДУХА
Изучение физических свойств атмосферы и происходящих в ней физических
явлений составляет предмет специальной науки, носящей название метео-
рология. Ближайшие задачи метеорологии сводятся к изучению кли-
матов отдельных районов и погоды (прогноз погоды).
Главное Управление гидрометеорологической службы СССР при Совете
Министров СССР имеет в своем ведении ряд научно-исследовательских учреж-
дений и сеть метеорологических станций I, II и III разрядов.
Полные циклы основных наблюдений на метеорологических станциях
производятся 4 раза в сутки: в 1 час, в 7 час., в 13 час. и в 19 час. по мест-
ному времени.
Гигиеническая наука интересуется физическими свойствами и явлениями
не только атмосферного воздуха, но и воздуха помещений, ограниченных тер-
риториальных участков (санаторий, детдом, пляж и т. п.). В этом смысле в
число вопросов, которыми занимается гигиена, входят и макро- и микроклимат.
Физическое исследование воздуха сводится к определению его темпера-
туры, барометрического давления, влажности и скорости движения. Сюда же
можно отнести кататермометрию, фригориметрию, определение эффективных
температур, тепловой и ультрафиолетовой радиации, а также определение
качества и количества пыли, копоти и атмосферных осадков.
Температура
Температура воздуха определяется при помощи ртутных или спиртовых
термометров.
Для ртутных термометров пределы измерений: —39---------1-750° С; для
спиртовых: — 70---1-120° С.
К каждому точному термометру должен быть приложен его паспорт, в
котором указаны поправки, какие нужно вносить в наблюдаемые отсчеты.
Проверка термометров1 производится у нас Главной геофизической обсер-
ваторией в Ленинграде. Эталоном служит водородный газовый термометр.
В термометрах Реомюра (R) шкала разделена на 80 градусов (0° — точка
замерзания воды, 80° — точка кипения воды); в термометрах Цельсия (С)
шкала разделена на 100 градусов (0° — точка замерзания воды, 100° — точка
кипения воды;) в термометрах Фаренгейта (F) шкала разделена на 212 гра-
дусов (32° — точка замерзания воды, 212° — точка кипения воды). Для пе-
ревода градусов одной шкалы в градусы другой пользуются следующими
формулами:
5 /9 \
n°R = — п°С— — п-1-32 °F;
4 \ 4 /
Температурам выше нуля придают значение положительных
величин, а ниже — отрицательных.
Абсолютная шкала температур Кельвина обозначается буквой К:
t°K=t°CH-273.
Необходимо помнить, что термометр тогда лишь показывает истинную
температуру воздуха, когда на него не действует радиация солнца, небесного
свода или какого-либо иного источника. Ввиду этого для получения точных
1 Это касается и других метеорологических приборов.
138
температур воздуха следует пользоваться термометром, шарик которого за-
щищен от влияния радиации. Таким термометром может служить сухой тер-
мометр аспирационного психрометра (см. далее). Для этой же цели служит и
парный термометр. Опуская описание обыкновенных термометров, известное
из курсов физики, переходим к описанию максимальных и минимальных тер-
мометров, применяемых для определения максимумов и минимумов темпера-
туры за определенный период наблюдения.
Максимальные термометры изготовляются различно. В неко-
торых из них (рис. 115 А) со дна резервуара термометра поднимается стеклян-
ный стерженек, входящий узким концом в капиллярную трубку термометра;
ртуть при повышении температуры может подниматься, проходя пространство
между стержнем и стенками трубки; при снижении температуры ртуть не может
опускаться назад в резервуар, в виду сопротивления, возникающего в силу
трения ртути о стенку капиллярной трубки и стерженька. Благодаря этому
ртуть остается в трубке на прежней высоте, показывая максимальную темпера-
туру. Энергичными встряхиваниями удается перевести ртуть из трубки в ре-
зервуар. В других максимальных термометрах в капиллярной трубке термо-
метра над ртутью помещается стальной штифтик (указатель), снабженный по
краям головками. Ртуть, поднимаясь, проталкивает указатель вверх; когда же
Рис. 115. Термометры.
А — максимальный термометр, В — минимальный термометр. Справа разрез
основания максимального термометра.
ртуть, вследствие снижения температуры, опускается, указатель не идет за
ней, а остается на месте. Таким образом, по высоте стояния нижней границы
указателя можно видеть, каков был максимум температуры за данный период
наблюдения. Для снижения указателя применяют магнит.
Минимальный термометр (рис. 115 В) представляет собой спир-
товой термометр, в котором резервуару придана форма вилки. Внутри капил-
лярной трубки помещается подвижный стеклянный штифтик с головками по
концам. Своеобразная форма резервуара объясняется тем, что спирт значи-
тельно менее теплопроводен, нежели ртуть, и потому резервуару придают
форму, при которой получается большая поверхность соприкосновения
с воздухом. До наблюдения наклоняют термометр резервуаром вверх, держат
в таком положении, пока штифтик не прикоснется к поверхности спирта, и
оставляют термометр в горизонтальном положении. При повышении темпера-
туры спирт, перемещаясь по капиллярной трубке, обтекает штифтик, не увле-
кая его с собой; при снижении температуры и укорочении спиртового стол-
бика штифтик, упираясь в мениск спирта, увлекается последним и остается в
положении, соответствующем минимуму температуры за данный период на-
блюдения. Отсчет производится соответственно концу указателя, наиболее
удаленному от резервуара термометра.
Термограф (рис. 116) служит для автоматической регистрации дви-
жения температуры за определенный период наблюдения. Он состоит из плос-
кой металлической трубки А, наполненной алкоголем1. При температурных
1 Вместо такой трубки, наполненной алкоголем, применяют также биметаллические
пластинки.
139
колебаниях объем алкоголя изменяется, благодаря чему изменяется изогну-
тость трубки. Эти изменения кривизны трубки при помощи системы рычажков
приводят к подъему или опусканию рычажка а соответственно подъему или
снижению температуры. На конец рычажка а насажен писчик b (небольшой,
заканчивающийся пишущим краем челнок, заполняемый невысыхающими
чернилами). В приборе имеется барабан, приводимый во вращание посредством
часового механизма. На барабан надевается бумажная лента с нанесенной
на ней сеткой. По оси абсцисс отложено время, по оси ординат нанесены тем-
пературы.
В зависимости от быстроты вращения барабана можно получить запись
движения температуры за сутки, неделю, декаду и т. п.
Рис. 116. Термограф.
А — трубка, наполненная алкоголем, а — рычажок для писчика, Ь — писчик.
Парный термометр состоит из двух рядом помещенных термо-
метров: поверхность резервуара одного них (/J посеребрена и потому отра-
жает почти целиком падающий на нее поток лучистой энергии; поверхность
резервуара другого термометра (/2) почернена и потому поглощает почти це-
ликом падающий на нее поток лучистой энергии.
Температура воздуха определяется по формуле:
t~ G Л G2 ^i)>
где t — температура воздуха,
— показание посеребренного термометра,
/2 — показание почерненного термометра,
к— константа прибора, определяемая эмпирически и обозначенная в
паспорте прибора.
Барометрическое давление
Давление воздуха измеряется при помощи ртутных и металли-
ческих барометров. Ртутные барометры бывают двух видов: чашечные и
сифонные.
В чашечных барометрах (рис. 117 а) барометрическая трубка
своим открытым концом погружена в чашку со ртутью. Барометрическое
давление измеряют высотою ртутного столба от поверхности ртути в чашке
до горизонтальной плоскости, проведенной касательно вершины ртутного
мениска в барометрической трубке. При падении давления часть ртути перехо-
дит из трубки в чашку, благодаря чему уровень ртути в чашке повышается,
и наоборот. Так как нуль шкалы должен совпадать с поверхностью ртути
в чашке, делают подвижной либо шкалу, либо дно чашки.
140
Установив нуль шкалы на уровне поверхности ртути в чашке, определяют
стояние верхней границы ртути в трубке при помощи барометрической шкалы
и нониуса (рис. 117 Ь). Последний представляет собой небольшую металли-
ческую пластинку со шкалою в 9 мм, поделенной на 10 равных частей так,
что каждое деление шкалы нониуса равно
0,9 мм. При помощи микрометрического
винта можно перемещать нониус на ко-
роткие расстояния вверх и вниз. Нониус
устанавливают так, чтобы его нулевая точка
сливалась с вершиной ртутного мениска.
Глаз наблюдателя при этом должен нахо-
диться на одной горизонтальной плоскости
с этой вершиной. Затем определяют, какое
деление шкалы нониуса точно совпадает
с каким-либо делением шкалы барометра.
Это деление шкалы нониуса указывает
десятые доли миллиметра в том числе,
которое соответствует высоте ртутного стол-
ба; целое число миллиметров отсчитывают
по барометрической шкале непосредственно
под мениском ртути. На нашем рисунке по-
казание барометра соответствует 733,3 мм.
Сифонный барометр состоит
из барометрической трубки, нижний конец
которой сифонообразно загнут вверх
(рис. 117 с). Атмосферное давление в этих
барометрах определяется соответственно
Рис. 117. Барометры.
а — чашечный барометр, b — шкала баро-
метра — А и нониуса В, с — сифонный
барометр. При данном положении показа-
ние барометра соответствует 733,3 мм.
расстоянию между поверхностями ртути в коротком и длинном коленах.
Кроме давления воздуха на высоту стояния ртути в ртутных барометрах
влияет также и температура, ввиду чего показания барометра приводятся к
температуре 0° по формуле:
h0 = ht—ht 0,00016275/,
где Ло — показание барометра, приведенное к 0° С,
ht — показание барометра при данной температуре,
t — температура воздуха (ртути) во время наблюдения,
0,00016275 — коэфициент расширения ртути с поправкой на расширение
латунной шкалы.
В нижеприведенной таблице 24 даны вычисленные поправки для данных
температур и давления.
Кроме поправки на температуру вносят еще
поправку инструментальную, которая указывается
в аттестате прибора, и поправку на вес ртути,
которая образуется из поправки на широту места
и поправки на высоту барометра над уровнем моря.
Поправка на вес ртути вводится при очень точных
измерениях и здесь не приведена.
Металлический барометр или
анероид (рис. 118) состоит из изогнутой трубки,
внутри которой воздух разрежен. При повышении
давления концы изогнутой трубки приближаются
Рис. 118. Металлический
барометр (анероид). один к другому, при уменьшении давления удаля-
ются. Эти перемещения концов трубки передаются
при помощи рычажков стрелке, вращающейся по циферблату, на котором
нанесены деления, соответствующие миллиметрам ртутного столба.
Другой тип металлического барометра (рис. 119) представляет металли-
ческую герметически закрытую коробку А, из которой выкачан воздух до
141
Таблица 24
Поправки для приведения к 0° показаний ртутного барометра
Температура по Цельсию Показания барометра
730 мм 740 мм 750 мм 760 мм I 770 мм 1 780 мм
-10 м,2 -1,2 И,2 г-1,2 1-1,3 1-1,3
-8 -1,0 -1,0 -1,0 -1,0 -1,0 -1,о
- 6 . . . • -0,7 -0,7 -0,7 -0,7 -0,8 -0,8
-4 -0,5 -0,5 -0,5 -0,5 -0,5 -0,5
-2 -0,2 -0,2 -0,2 -0,2 -0,3 -0,3
0 0,0 0,0 0,0 0,0 0,0 0,0
+ 2 -0,2 -0,2 -0,2 -0,2 -0,3 -0,3
(-4 -0,5 -0,5 i । — -0,5 -0,5 -0,5 -0,5
-б -0,7 -0,7 -0,7 -0,7 -0,8 -0,8
-8 -1,0 -1,0 -1,0 -1,0 -1,0 -1,0
-10 -1,2 -1,2 -1,2 -1,2 -1,3 -1,3
-12 -1,4 -1,5 -1,5 -1,5 -1,5 -1,5
L14 -1,7 -1,7 -1,7 -1,7 -1,8 -1,8 !
Н1б -1,9 -1,9 -2,0 -2,0 -2,0 -2,о ;
-18 -2,1 -2,2 -2,2 -2,2 -2,2 -2,3 1
-20 . • -2,4 -2,4 -2,5 -2,5 -2,5 -2,5 i
-22 -2,6 -2,7 -2,7 -2,7 -2,8 -2,8 !
-24 -2,9 -2,9 -2,9 -3,0 -з,о -3,1 1
-26 -3,1 -3,1 ; -3,2 -3,2 -3,3 -з,з !
-28 -3,3 -3,4 -3,4 -3,5 -3,5 -3,6
-30 -3,6 -3,6 -3,7 -3,7 -3,8 -3,8
-32 -3,8 -3,9 -3,9 -4,0 -4,0 -4,1
-34 -4,1 -4,1 -4,2 -4,2 -4,3 -4,3
остаточного давления 50—60 мм Hg. Крышка
и дно для большей эластичности
гофрированы. Дно соединено с корпусом. Крышка коробки при помощи силь-
ной пружины Г удерживается от вдавливания атмосферным воздухом. Конец
пружины соединен с системой рычажков В, в свою очередь соединенных с
цепочкой Б. Последняя намотана на барабанчик со стрелкой Д, движущейся по
циферблату Е.
При увеличении атмосферного давле-
ния коробка сплющивается, при этом
передача от нее разматывает цепочку и
стрелка идет вправо. При уменьшении
давления коробка раздувается; при
этом под действием часового волоска,
надетого на ось стрелки, стрелка дви-
жется влево.
На шкале прибора деления соответ-
ствуют 0,5 мм Hg.
Рис. 119. Металлический барометр:
1 — внешний вид прибора, 2 — конструкция прибора, А — металлическая коробочка,
Г — пружина, В — система рычажков, Б — цепочка, Д — стрелка, Е — циферблат.
При помощи регулировочного винта можно точно устанавливать стрелку
соответственно показаниям ртутного барометра. Предел измерения анероида
142
от 600 до 800 мм Hg. Допустимая погрешность + 1 —2 мм Hg. На рис. 119-7
представлен внешний вид описанного барометра.
Барограф (рис. 120) построен по принципу анероида. Деформации
анероида при помощи системы рычажков передаются стрелке, на конце ко-
Рис 20. Барограф.
торой укреплен писчик. Последний отмечает соответствующие давления на
ленте, натянутой на барабане. Полный оборот барабан делает в сутки или в
неделю.
Применение барометра для определения высоты места
над уровнем моря
С подъемом над уровнем моря барометрическое давление падает. Зависи-
мость между барометрическим давлением и высотою места выражается форму-
лой:
Н =- 1600 g~g° [1 + 0,002 (/ — /0],
В + Bq
где Н — разность высот двух точек в метрах,
В — барометрическое давление в нижней точке,
BQ — то же в верхней точке,
t — температура воздуха в нижней точке,
— то же в верхней точке.
На циферблатах приборов альтиметров, представляющих собой
металлические барометры, непосредственно нанесена высота места над уровнети
моря.
ВЛАЖНОСТЬ
Различают абсолютную, максимальную и относительную влажность. Под
абсолютной влажностью понимают содержание водяных паров
в 1 м3 воздуха, выраженное в граммах, либо давление водяных паров воздуха,
выраженное в миллиметрах ртутного столба.
Под максимальной влажностью понимают абсолютную
влажность, при условии насыщения воздуха водяными парами при данной
температуре. Максимальная влажность для данной температуры представ-
ляет величину постоянную (табл. 25).
Под относительной влажностью либо гигроско-
пическим состоянием воздуха понимают процентное отношение
абсолютной влажности при данной температуре к максимальной влажности
при той же температуре.
143
Таблица 25
Напряжение и вес насыщающих воздух водяных паров
t°c Напряжение водяных паров в мм Hg Вес водяных паров насы- щающих воз- дух в г/м3 t°c Напряжение водяных паров в мм Hg Вес водяных паров насы- щающих воз- дух в г/м3
—15 1,400 1,571 20 17,391 ' 17,164
—10 2,093 2,300 21 18,495 18,204
—9 2,261 2,488 22 19,659 19,286
—8 2,456 2,674 23 20,888 20,450
—7 2,666 3,883 24 22,184 21,604
—б 2,890 3,111 25 23,550 22,867
—5 3,113 3,360 26 24,988 24,190
—4 3,387 3,614 27 . 26,505 25,582
—3 3,662 3,902 28 28,101 27,004
—2 3,955 4,194 29 29,782 28,529
—1 4,267 4,522 30 31,584 30,139
0 4,600 4,874 31 33,406 31,890
1 4,940 5,214 32 35,359 33,640
2 5,302 5,574 33 37,411 35,480
! 3 5,687 5,963 34 39,565 37,400
4 6,097 6,370 35 41,827 39,410
5 6,534 6.7Q: 36 44,201 41,510
б 6,998 7,260 37 46,691 43,710
7 7,492 7,734 38 49,302 46,000
8 8,017 8,252 39 52,039 48,400
9 8,574 8,793 40 54,906 50,910
10 9,165 9,372 41 57,910 53,520
11 9,792 9,976 42 61,055 56,260
12 10,457 10,617 43 64,346 59,090
13 11,162 11,284 44 67,790 62,050
14 11,908 12,018 45 71,391 65,140
15 12,699 12,763 46 75,158 68,360
16 13,536 13,552 47 79,093 71,730
17 14,421 14,391 48 83,204 75,220
18 15,357 15,329 49 87,499 78,860
19 16,364 16,203 50 91,982 82,630
Дефицитом насыщения называют разность между максималь-
ной и абсолютной влажностью Для данной температуры.
Для определения влажности воздуха применяют весовой метод, а также
психрометрический и гигрометрический методы.
144
Весовой метод
Принцип. Определенный объем обеспыленного воздуха просасывают
через U - образные трубки с гигроскопическим веществом и по прибыли в
весе трубок находят абсолютную влажность воздуха. Данный метод является
собственно не физическим, а физикохимическим.
Приборы и реактивы: три U - образные трубки, небольшая
стеклянная трубка, аспиратор хлористый кальций зернистый, крепкая
серная кислота, пятиокись фосфора (Р2О5), стеклянная вата, пемза.
Методика определения. Собирают прибор, как показано на
рис. 121. Впереди помещают трубку а, заполненную стеклянной ватой1 * для
обеспыливания воздуха, далее L’-образную трубку в с хлористым кальцием,
затем U - образную трубку с с зернами пемзы, смоченными крепкой серной
кислотой, и, наконец, U - образную
трубку d с пятиокисью фосфора. В обоих
коленах каждой U - образной трубки
поверх гигроскопического вещества по-
мещают небольшое количество стеклян-
ной ваты для предупреждения унесения
током воздуха мелких частичек данного
вещества. Последнюю U-образную труб-
ку соединяют с реометром е, а послед-
ний — с аспиратором (система бутылей)
или насосом.
Хлористый кальций берется прока-
ленный, с размерами зерен 3—7 мм.
Порошкообразный хлористый кальций
для работы не пригоден, так как
Рис. 121. Прибор для весового опреде-
ления абсолютной влажности воздуха.
а — трубка со стеклянной ватой. Ь, с и d —
U-ебразные трубки с влагопоглощающими
реактивами, е — реометр.
оказывает большое сопротивление при просасывании воздуха.
Через систему в течение часа просасывают 10—15 л воздуха. Трубки до и
после опыта при закрытых кранах выдерживают час-два в экссикаторе, чтобы
подсушить их с поверхности, и взвешивают на аналитических весах.
Если опыт проводится правильно, то вторая трубка либо вовсе не дает
привеса, либо привес ее составляет не более 10% привеса первой трубки.
Третья трубка, представляющая контрольную, совсем не должна давать
привеса.
Абсолютная влажность вычисляется по формуле:
л Ю00 , ,
А = р------ г/м3,
L
где А — абсолютная влажность,
р — найденный привес трубок в граммах,
L — число литров воздуха, пропущенного через трубки.
Психрометрия
Психрометрия (от греч. psychros — холодный и metron — мера)
представляет собой метод определения относительной влажности воздуха,
основанный на различной интенсивности испарения воды с данной поверх-
ности в зависимости от содержания в воздухе влаги и на связанной с этим
различной степенью охлаждения испаряющей поверхности.
Наичаще применяются для этой цели психрометры, обыкновенный (стан-
ционный) и аспирационный.
Психрометр станционный (рис. 122) состоит из двух
термометров — сухого а и влажного Ь, укрепленных на штативе. Резервуар
влажного термометра обернут батистом или марлей, конец которой погружен
в стаканчик с водой.
1 Хлопчатобумажную вату нельзя применять ввиду ее гигроскопичности.
1Q А. И. Бурштейн
145
Рис. 122. Психрометр
станционный.
а — сухой термометр,
Вода с поверхности марли постепенно испаряется, благодаря чему резер-
вуар термометра охлаждается. Испарение с поверхности марли и охлаждение
резервуара влажного термометра идет до тех пор, пока воздух, окружающий
резервуар влажного термометра, не окажется насыщенным влагой; в этот
момент дальнейшее снижение ртути в капилляре влажного термометра пре-
кращается. Тогда производят отсчет по сухому и влажному термометрам и по
приведенной ниже формуле рассчитывают абсолютную влажность.
Чтобы получить точные результаты по описанному психрометру, необходимо
придерживаться следующих условий: сухой термометр должен находиться на
одном уровне с влажным, на расстоянии от него в 4—5 см. Оба термометра
должны быть открыты так, чтобы воздух свободно омывал их. Резервуар
влажного термометра покрывают одним слоем обезжи-
ренного тонкого батиста или двумя слоями гигроскопи-
ческой марли. Батист или марлю перевязывают над
резервуаром и слабо под ним. Конец лоскута должен
свободно свисать в стаканчик с водой, который поме-
щают на 3—4 см ниже резервуара влажного термометра.
В стаканчик наливают дестиллированную или профиль-
трованную дождевую воду. В крайнем случае можно
пользоваться для этой цели профильтрованной кипяче-
ной речной водой, но не колодезной. Стаканчик с водой
должен быть покрыт крышкой с отверстием посредине
и узким прорезом до края для пропуска батиста (марли).
Психрометр подвешивают или устанавливают в дан-
ном месте так, чтобы на него не влияли случайные мо-
менты (радиация солнца или другого источника, случай-
ные токи воздуха и т. п.).
При низких температурах, когда влажный термо-
метр показывает ниже нуля, необходимо при работе
с психрометром придерживаться некоторых особых пре-
досторожностей: стаканчик с водой отставляют; свобод-
ный конец батиста (марли) обрезают под резервуаром
несколько ниже того места, где батист перевязан нит-
кой и подводят под резервуар влажного термометра
стаканчик с водой комнатной температуры. Выжидают,
пока батист хорошо увлажнится. Если на нем нахо-
дится слой льда от предыдущего смачивания, то, держа
все время резервуар в воде, дожидаются, пока лед совер-
шенно растает. Через полчаса после этого делают отсчеты показаний сухого и
влажного термометров; при этом нужно следить, чтобы на шарике сухого термо-
метра не было капелек воды, кусочков льда либо изморози. Батист влажного
термометра должен находиться в так называемом «нормальном» состоянии,
т. е. он либо должен быть пропитан переохлажденной водой ниже 0° С, нахо-
дящейся в жидком состоянии, либо вода, которая пропитывает батист должна
быть замерзшей, причем тонкий слой льда окружает со всех сторон резер-
вуар влажного термометра. В противном случае повторяют смачивание.
Вследствие того, что с поверхности резервуара влажного термометра испа-
ряется вода, он охлаждается, и показания влажного термометра оказываются
ниже показаний сухого термометра. Эта разница в показаниях сухого и влаж-
ного термометра тем больше, чем суше воздух, и наоборот. Абсолютная влаж-
ность вычисляется по формуле:
A = F —а(/ —ZJ./Y,
где А — абсолютная влажность в мм Hg,
F — максимальная влажность, соответствующая температуре влажного
термометра х,
Определяется по таблице 25 (стр. 144).
146
a — психрометрический коэфициент, который зависит от скорости движения
воздуха (равен 0,00128 в небольшой закрытой комнате, 0,001 в большой
закрытой комнате, 0,00077 в большой комнате при открытых окнах,
0,00074 на дворе при легком движении воздуха и 0,0000 на дворе
при отсутствии ветра),
t — температура сухого термометра,
— температура влажного термометра,
Н — барометрическое давление.
Согласно исследованиям Зворыкина установлена зависимость между ско-
ростью движения воздуха и значением психрометрического коэфициента
(табл. 26).
Описанный психрометр не дает вполне Таблица 26
точных результатов, ввиду того, что на
показания его термометров влияет ско-
рость движения воздуха, которая может
быть весьма разнообразна.
Более точным прибором является аспи-
рационный психрометр.
Аспирационный психро-
метр (рис. 123), как и станционный
психрометр, состоит из двух термомет-
ров: сухого а и влажного Ь. Резервуар
каждого термометра окружен двумя ме-
таллическими гильзами, которые защи-
щают его от влияния тепловой радиа-
ции. Гильзы переходят в общую трубку,
у свободного конца которой помещается
маленький аспирационный вентилятор.
Благодаря действию последнего через
гильзы во время наблюдения просасы-
вается воздух с постоянной скоростью,
равной 4 лг/сек. Вентилятор находится
под колпачком С и приводится в дви-
жение при помощи заводной пружины.
Пружина заводится ключом d. Батист
(марля) здесь не погружается в воду, а
смачивается при помощи пипетки е, снаб-
женной грушей. При смачивании батиста
наблюдают, чтобы не образовалась водя-
ная пробка, которая может затем мешать
свободному прохождению воздуха. Если
таковая образовалась, то излишек воды
удаляют при помощи фильтровальной
Значения для а в зависимости от скорости движения воздуха
Скорость движе- ния воздуха в м/сек Значение для a
0,13 0,00130
0,16 0,00120
0,20 0,00110
0,30 0,00100
0,40 0,00090
0,80 0,00080
2,30 0,00070
3,00 0,00069
1 4,00 1 0,00067
Относительная влажность опре-
деляется по формуле:
D А • 100
/V =-------.
F
где R — относительная влажность,
А — абсолютная влажность,
F — максимальная влажность,
соответствующа я темпе-
ратуре сухого термометра
(см. таблицу 25).
бумаги. Порибор снабжен приспособлением / для подвешивания.
Отсчет по термометрам производят летом через 4—5 минут, а зимою через
15 минут после начала действия вентилятора; в последнем случае вентилятор
приходится заводить дважды. За указанное время уровень ртути в обоих
термометрах успевает остановиться на определенной высоте.
В продаже имеются психрометры, в которых вентилятор приводится в дейст-
вие при помощи маленького мотора, работающего от электрической сети.
Абсолютная влажность вычисляется по формуле
A = F1 — 0,5 (^ — О
Н
755 ’
где А — абсолютная влажность в мм Hg,
Fv — максимальная влажность при температуре влажного термометра,
t — показание сухого термометра,
147
— показание влажного термометра,
Н — барометрическое давление.
Приведенную выше формулу можно написать и так:
О 5
7ээ
или
А = — 0,000662 (t — Q Н.
Как легко видеть,
Рис. 123. Аспирационный психрометр.
последнее выражение формулы ^1я аспирационного при-
бора соответствует формуле, приме-
няемой для обыкновенного психро-
метра, но с постоянным психроме-
трическим коэфициентом. Последний
приобретает постоянное значение
вследствие одинаковой скорости
движения воздуха у резервуаров
термометров в момент наблюдения
и равен 0,000662.
Главная геофизическая обсер-
ватория в Ленинграде издала пси-
хрометрические таблицы, при по-
мощи которых можно по пока-
заниям сухого и влажного тер-
мометров определить абсолютную
и относительную влажность.
Приводимые дальше таблицы 27,
28 и 29 также могут служить для
определения относительной влаж-
ности по данным сухого и влажного
термометров. В первом вертикаль-
ном столбце отыскивают число,
соответствующее показанию сухого
термометра, в первом горизонталь-
ном столбце — число, соответствую-
щее показанию влажного термо-
метра. Искомая относительная
влажность лежит на месте пере-
а — сухой термометр, Ъ — влажный термометр, с —
колпачок, в котором установлен вентилятор, d —
ключ для взвода пружины вентилятора, в — пипетка
для смачивания батиста влажного термометра, / —
приспособление для подвешивания.
сечения горизонтальной линии,
проходящей через первое число,
и вертикальной линии, проходя-
щей через второе число.
Пример 1. При скорости движения воздуха, близкой 0,8 м/сек., сухой термометр
обыкновенного психрометра показывает 15° С, влажный — 8°. Относительная влажность,
согласно показанию таблицы 28, равна 30%.
Пример 2. Сухой термометр аспирационного психрометра показывает 13,8, влажный —
6. По таблице 29 (после интерполяции) находим относительную влажность равной 26,2%.
Гигрометрия
Гигрометрия (от греч. hygros — влажный и metron — мера) пред-
ставляет собой метод определения влажности воздуха, основанный на способ-
ности гигроскопических тел (волос) поглощать из воздуха большее или
меньшее количество влаги в зависимости от относительной влажности воз-
духа и в связи с этим менять свою длину. На указанном принципе постро-
ены наиболее распространенные гигрометры.
148
о
Таблица 27
Относительная влажность воздуха по показаниям сухого и влажного термометров станционного психрометра при скорости движения в 0,2 м]сек,
Градусы Цельсия по сухому термометру Г р а I у' с ь j Цельсия по влажному термометру
12 5,3 5,7 6,0 6,4 6,8 7,2 7,6 8,0 8,4 8,7 9,1 9,5 9,9 10,3 10,7 11,0 11,3 • 11,7 12,0
13 5,9 6,4 6,8 7,2 7,6 8,0 8,4 8,8 9,2 9,6 10,0 10,4 10,8 11,1 11,5 11,8 12,2 12,6 13,0
14 6,6 7,1 7,5 8,0 8,4 8,8 9,2 9,7 10,1 10,5 10,9 11,3 11,7 12,1 12,5 12,8 13,2 13,6 14,0
15 7,3 7,8 8,2 8,7 9,2 9,6 10,0 10,5 10,9 11,4 11,8 12,2 12,6 13,0 13,4 13,8 14,2 14,6 15,0
16 8,0 8,5 9,0 9,4 9,9 10,3 10,8 11,3 11,8 12,2 12,6 13,1 13,5 14,0 14,4 14,8 15,2 15,6 16,0
17 8,6 9,1 9,7 10,2 10,7 Н,2 11,6 12,1 12,6 13,0 13,5 13,9 14,4 14,9 15,3 15,8 16,2 16,6 17,0
18 9,3 9,9 10,4 10,9 Н»4 11,9 12,4 12,9 13,4 13,9 14,4 14,8 15,3 15,7 16,2 16,6 17,1 17,5 18,0
19 10,0 10,6 11,1 11,7 12,2 12,7 13,2 13,8 14,3 14,8 15,3 15,7 16,2 16,7 17,2 17,6 18,1 18,5 19,0
20 10,6 11,2 11,8 12,4 12,9 13,4 14,0 14,5 15,1 15,6 16,1 16,6 17,1 17,6 18,1 18,5 19,0 19,5 20,0
21 11,2 11,9 12,6 13,1 13,6 14,2 14,8 15,3 15,9 16,5 17,1 17,5 18,0 18,6 19,1 19,5 20,0 20,5 21,0
22 11,8 12,5 13,2 13,8 14,4 15,0 15,6 16,1 16,7 17,3 17,9 18,4 18,9 19,5 20,0 20,5 21,0 21,5 22,0
23 12,5 13,1 13,8 14,4 15,1 15,7 16,4 17,0 17,6 18,2 18,8 19,3 19,8 20,4 20,9 21,5 22,0 22,5 23,0
i 24 13,1 13,8 14,5 15,2 15,9 16,5 17,1 17,8 18,4 19,0 19,6 20,1 20,7 21,3 21,9 22,4 23,0 23,5 24,0
25 13,7 14,5 15,2 15,9 16,6 17,2 17,9 18,5 19,2 19,8 20,5 21,1 21,7 22,2 22,8 23,3 23,9 24,4 25,0
Относ'тельная 1 влажность . . i 1 10»/о 15»/. 20% 25% 30% 35% 40% 45% 50»/. 55% 60»/. 65% 70»/. 75»/. 80»/. 1 85% 90»/. 95% 100о/о
Таблицд 28
Относительная влажность воздуха по показаниям сухого и влажного термометров станционного психрометра при скорости движения воздуха
около 0,8 м/сек.
Градусы Цельсия по влажному термометру
ео см 00 СО ио Ю ю Г-' 00 00 о О) о О 1 см _^©1 см ео СО ^с« Ю ю СО о* N
Градусы Цельсия по сухому термометру
Относительная влажность в процентах
8 22 28 34 40. 46 | 54 59 65 72 79 86 93’100 1 1 I । 8
8‘/, 17 23 29 35 141 48 54 60 66 73 79 87 93 100 ) i 8*/a
9 13 19 25 31 36 42 48 55 61 67 73 80 87 93 100 1 1 i । 9
Э‘/г 14 20 26 . 32 38 1 43 49 55 61 67 79 80 87 94 100 । 9’/,
10 11 16 22 28 33 39 45 50 56 62 68 74 80 87 94 100 10
ю»/, 12 18 ’ 23 29 34 40 45 51 57 63 69 74 81 88 91 100 । Ю*/,
н 14 19 25 30 36 41 46 52 58 63 69 74 81 87 94 100 11
11V. 15 21 26 31 36 41 47 53 58 64 70 76 82 88 95 100 Ш/,
12 13 18 22 28 33 38 43 48 54 59 65 71 76 83 89 95 100 12
121/, 13 18 23 28 33 39 44 49 54 60 65 71 77 83 (89 95 100 12*/,
13 11 16 20 25 30 35 40 45 50 55 61 66 72 77 83 89 95 100 1 i 1 13
13'/г 12 17 22 27 31 36 41 46 51 56 61 66 72 77 83 89 95 100 i 1 1 131/,
14 14 18 23 28 32 37 42 47 52 57 62 67 73 78 84 90 95 100 1 14
141/г 11 16 20 25 30 34 39 43 48 53 57 62 68 73 79 84 90 95 100 141Д
15 13 17 21 25 30 34 39 44 49 53 58 63 68 73 78 84 89 94 100 15
15*/, 15 18 22 27 31 36 40 45 49 54 59 63 68 74 79 84 89 94 100 15*/s
16 11 15 19 24 28 32 36 41 45 50 55 59 64 69 74 79 84 89 95 100 16
16*/, 12 16 21 25 29 33 38 40 47 51 56 60 65 69 74 79 84 90 95 100 161/,
17 14 18 22 26 30 34 38 43 47 52 56 61 65 70 75 80 85 90 95 100 17
17‘/г 1 11 16 19 .23 27 31 35 40 44 48 52 1 57 i 61 i 66 70 75 ”l 85 90 95100 IT/,
Градусы Цельсия по сужэму термометру
Таблица 28 (продолжение)
Относительная влажность воздуха по показаниям сухого и влажного термометров станционного психрометра
при скорости движения воздуха около 0,8 м/сек
| Градусы Цельсия по влажному термометру |
1 1 ^01 Cd ^е«1
00 00 о 1 О) О о ~ ~ CN CN 00 & ю ю Ю Ю £- 1 1'1
Относительная влажность в процентах
18 13 16 20 24 2832 36140 44 49 53 57 62|66|71 75 80 85 90 95 100 18
18‘/, 14 18 22 2629 33 37 42 46 50 54 58 62 67 71 76 80 85 90 95 100 187,
19 12 16 19 23 26 3034 38 42 46 50 54 5863 67 72 76 81 86 90 95 100 19
а 191/, 141721 24 28 31 35 39 43 47 51 55 5963 6872 77 81 86 90 95 100 197,
| 20 11 14 1821 25 28 32 36 40 43 47 52 55 5964 68 72 77 81 86 91 95100 20 и
| 201/, 12 1619 23 2630 33 37 41 45 48 52 55 60 64 69 73 77 82 86 91 95100 20’/, g
" 21 1417 20 23 27 3034 38 41 45 4953 57 61 65 69 74 78 82 87 91 95100 21 g
§ 211/а 12 14 18 21 25 28 32 3539 42 46 50 54 57 62 66 70 74 78 82 87 91 95100 21’/, Е?
* 22 О 12 1619 22 2529 32 363943 47 50 5458 62 66 70 74 78 82 87 91 95100 22 §
о 22’/, 14 17 20 23 27 3o|34 37 41 44 48 51 55 59 63 66 70 74 78 83 87 92 96100 22l/t §
<к 23 s 12151821 25 28 31 34 38 41 44 4852 55 59 63 66 79 74 78 83 88 91 96100 23
| 23’/, 13 1619 23 262932 35 3842 45 49 52 56 59 63 67 71 75 79 83 87 91 96100 23’/, g
5 24 11 14 172023 2629323539424650 53 57 60 63 67 72 75 79 83 87 91 96100 24
3 247, 121518 21 24 27 30 3336 40 43 47 50 53 56 60 64 68 72 76 80 83 88 92 96100 241/,
& 25 11 14 1619222528313437 4044 47 51 54 58 61 64 68 72 76 80 83 88 92 96100 25 g
£ 25’/, 13 151821 242629323539 42 45 48 51 55 58 62 65 69 72 76 80 84 88 92 96100 251/г
26 131618 2124 27 30 33 36 39 42 45 48 52 55 59 62 66 69 73 76 80 84 88 92 96100 26
267, 12,14 17120 22 25 28 31 |з4 37 40 43 46 49 53 56 59 63 66 69 73 77 80 84 88 92 96100 26’/,
27 1з[15182023262931|з4 37 40 43 46 49 53 56 60 63 66 69 73 77 80 84 88 92 96100 27
271/, 111141619 21 24 27.30'33 35 38 41 43 47 50 54 57 60 63 67 70 74 77 81 84 88 92 96100 27’/,
кр» СП 1 ! ! 11 1 II
Таблица 28 (продолжение)
Относительная влажность воздуха по показаниям сухого и влажного термометров станционного психрометра при скорости движения воздуха
около 0,8 м/сек.
Градусы Цельсия по влажному термометру
Относительная влажность в процентах
Градусы Цельсия по сухому термометру
28
2872
29
297,
30
301/2
31
зп/2
32
3272
33
3372
34
3472
35
351 /2
36
3672
37
3772
12 1547
13*16
12 14
12
20
18
16
22
20
19
11
14
13
11
17
16
14
13
12
11
25.27 30 33 36,39^42
24*26’28
24*26
22
21
19
18
16
21
19
15
13
12
17
16
24
23
21
19
31
29
27
25
24
22
12
14
18
16
21
19
12
18
11
15
14 16
13
12
15
13
12
11
33'3739
31 ;34[з7
29’32 35
28:
26:
25:
23:
21*24
20 22
21 :
19
16 18
14'17
13 15
12*14
30
29
27
25
19
17
33
31
29
27
26
24
23
21
20
19
18
16
13
12
11
15
14
Г 13
11214
44
43
40
37
36
34
32
30
28
27
25
24:
22
21f
19 21
18 20
17,19:
16^17
14116
15'17
47
45
43
40
38
36
34
32
30
29
27
26
24
51 5457 60 63
49 5255
46 49 52
43 -
41
39
37
35
33
31
30
23
46 49
44 47
42 45
40 42
3740
3738
34 36
32 34
28 30 33
27 29*31
25'27 29
24 26 28
22'24 27
20 23 25
19 21 23
18 20 22
192123
58 61
55'58
52 55
50 52
4749
45 48
43 46
41^43
3941
3739
3538
33 36
32 34
30 32
2931
27 29
2528
24*26
25 27
167
64
161
>58
55
53
50
48
46
44
42
40
38
37:
35
33:
31:
30:
28:
29:
70
67
64
61
59
55
53
51
49
47
44
42
41
39
37
35
33
32
30
31
73
70
67
64
62
59
56
54
52
49
47
45
43
41
40
38
77
74
71
67
65
62
59
57
54
52
50
36
34
32
33
81
77
74
70
68
65
62
60
57
55
53
50
48
46
42
41
85 89
81
78
74
71
68
65
63
60
58
55
53
45
49
46
45
43
41
47
45
44
42
40
38
36 38
35’3739
35*37 39
85
81
78
74
71
69
66
63
61
58
56
54
51
49
47
45
43
41
92 96
89
85
81
78
75
72
69
66
64
61
59
56
54
51
50
47
46
43
92
88
85
81
78
75
72
69
66
64
62
59
57
54
52
50
48
46
46
100
96
92
88
85
82
79
75
72
69
67
65
62
60
57
54
52
51
48
49
100
96
92
88
85
82
79
75
72
70
67
65
62
60
57
55
53
51
49
100
96
92
89
85
82
79
75
73
71
67
65
62
60
57
55
53
51
100
96
92
89
85
82
79
76
73
71
68
66
63
61
58
56
53
100
96
93
89
86
82
79
77
73
71
68
100
96
93
89
85
82
80
66
63
61
58
56
77
74
71
69
66
64
61
59
100
96
93
89
85
82
80
77
74
72
69
67
64
62
100
96
93
90
87
84
80
78
75
73
69
67
64
100
96
93
90
86
83
80
78
76
100
72
69
67
96 100
93 97 1 100
90 93 97 100 ।
86 90 93 97 100
84 86 90 93 97
81 84 87 90 93
78 81 84 87 91
7о| 78 81 84 87
72 75 78 81 84
70* 73 75 78 81
100
97
93
90
87
84
28
28i/2
29
291/2
30
ЗО'/2
31
зв/2
32
321/2
33
33V2
34
347'2
35
3572
36
3672
37
377,
Градусы Цельсия по сухому термометру
Таблица 28 (продолжение)
Относительная влажность воздуха по показаниям сухого и влажного термометров станционного психрометра
при скорости движения воздуха около 0,8 м!сек.
Градусы Цельсия по влажному термометру
м еч ei «Ч
20 /юг 04 /нг 22 04 04 23 231/ 24 25 26 Ю 04 04 \1г 28 5b 04 29 'тбг 30 301 я 311 32 04 00 й й 34 35
Градусы Цельсия по сухому термометру
Относительная влажность в процентах
38 12 14 16 18 19 21 23 25 27 29 31 33 36 38 40 42 44 46 49 51 54 56 59 61 64 67 70 72 75 79 81 38
38’;2 11 13 15 17 18 20 22 24 26 28 30 32 34 36 38 40 43 45 47 49 52 54 57 59 62 65 68 70 73 75 79 38%
39 12 14 16 17 19 21 23 24 26 29 31 33 35 37 39 41 43 45 47 50 52 55 57 59 62 65 68 70 73 76 39
39>/2 13 15 16 18 20 21 23 25 27 29 31 33 35 37 39 42 44 46 48 51 53 55 58 60 63 65 68 70 73 39%
40 12 14 15 16 18 20 22 23 25 28 30 32 34 36 38 40 42 44 46 48 51 53 56 58 60 63 65 68 70 40
4О‘/г 11 13 14 16 18 19 21 23 25 27 28 30 32 35 36 38 40 43 45 47 49 51 54 56 58 61 63 65 68 40%
41 12 13 15 17 18 20 22 23 25 27 29 31 33 35 37 39 41 43 45 47 50 52 54 56 59 61 64 66 41
41*/2 11 13 14 16 17 19 20 22 24 26 28 30 32 33 36 37 40 41 43 45 48 50 52 54 57 59 62 64 41%
42 И 12 13 15 16 18 19 21 23 24 26 28 30 32 34 36 38 40 41 44 46 48 50 52 55 57 59 61 42
42V, 11 12 14 15 17 19 20 22 23 25 27 29 30 32 34 36 38 40 41 44 46 48 50 52 55 57 59 42%
43 12 13 14 16 17 19 20 22 24 26 28 29 31 33 35 36 38 40 42 44 46 48 50 53 55 57 43
43>/г 12 14 15 17 18 20 21 23 24 26 28 30 31 34 35 37 39 41 43 45 47 49 51 54 56 43%
44 11 12 14 15 17 18 20 22 23 25 27 29 30 32 34 36 37 39 41 43 45 47 49 51 53 44
44>/г 12 13 15 16 18 19 21 23 24 26 28 29 31 33 35 36 38 40 41 44 46 48 50 52 44%
45 , । । i 1 i 1 11 12 14 15 17 18 । 20 22 23 1 25 26 28 30 31 33 35 । । 37 38 40 1 1 42 44 46 48 50 45
Градусы Цельсия по сухому термометру
S
Таблица 29
Относительная влажность воздуха по аспирационному психрометру
Градусы Цельсия по влажному те] эмометру
CN СО ‘/.£ ^©J ю ю со ^©< ю о ©9 1 00 1 X О) ея & о я о м CN Zhzi со СО ^©4 ю ^©1 ю со ^©1 ю
Градусы Цельсия по сухому термометру
Относительная влажность в процентах
8 29 34’ 40 45 51 57 63 69 75 81 87 94 100 ' । 1 1 8
8'/2 25 30 35 41 46 52 58 63 69 75 81 87 94 100 1 1 8i/2
9 21 26 31 39 42 47 53 58 64 70 76 82 88 94 100 । 9
9х/2 17 22 27 32 38 43 48 54 59 65 70 76 82 88 94 100 л ! 91/2
10 14 19 24 29 34 39 44 49 54 60 65 71 76 82 88 94 100 1 10
IO1/2 16 20 25 30 35 40 45 50 55 60 66 71 77 83 88 94 100 юу2
11 14 22 27 31 36 41 46 51 56 61 66 72 77 83 88 94 100 1 11
1Р/2 19 23 28 32 37 42 47 52 57 62 67 72 78 83 89 94 100 пу2
12 16 20 24 29 33 38 43 48 53 57 62 68 73 78 83 89 94 100 12
12Ч2 17 21 26 30 35 39 44 49 53 58 63 68 73 78 84 89 94 100 12i/a
13 14 18 23 30 31 36 40 45 49 54 59 64 69 74 79 84 89 95 100 13
131/2 16 20 28 28 32 37 41 46 50 55 60 G4 69 74 79 84 89 95 100 13*/2
14 17 24 25 29 33 38 42 46 51 56 60 65 70 74 79 84 90 95 ЮС 14
14i/2 14 21 22 26 30 35 39 43 47 52 56 61 65 70 75 80 85 90 65 00 141/г
15 18 20 23 27 32 36 40 44 48 52 58 61 66 71 75 80 85 । 90 1 05 100 15
1^/2 16 17 । 21 25 29 32 37 41 45 49 53 58 62 66 71 76 80 85 90 95 100 15у2
16 13 15 18 22 26 30 34 37 42 46 50 54 58 63 67 71 76 81 85 90 95 100 16
16i/2 12 16 20 23 27 31 34 38 42 46 50 58 59 63 67 72 76 81 86 90 95 100 1буа
17 * 14 17 21 24 2Я 32 36 39 43 47 51 55 59 64 68 72 77 81 85 90 95 100 17
Градусы Цельсия по сухому термометру
Таблица 29 (продолжение)
Относительная влажность воздуха по аспирационному психрометру
Градусы Цельсия по влажному термометру
17Vi
18
18V,
19
19V,
20
20V.
21
21V.
22
22V.
15
13
19
16
14
22|25|29|33 36 40 44 48 52 56 60
20 23 27 30....................
21|24 28 31 35 38 42
19 22--------------
17
15
34
37
41
45
17
15
13
20
18
16
14
29 32 36
23V.
24
24'/2
25
25V.
26
26i/2
27
27i/a
Относительная влажность в процентах
64 68 73 77 811 1 86 I91 95, 100
61 65 69 73 771 1 82 86 91 95 100
57, 61 । 65 69 73 I78 82 86 91 95 100
54 58 62 66 70 74 78 82 86 91 95 100
51 54 58 62 66 70 74 78 82 86 91 95 100
48 52 55 59 63 66 70 74 78 83 87 91 96 100
45 1 48 1 52 56 59 62 67 71 75 79 83 87 91 95 100
42 1 46 49 53 56 60 64 67 71 75 79 83 87 91 96 100
40 43 46 50 53 57 60 64 68 71 75 79 83 87 92 96
37 40 44 47 50 54 57 61 64 68 72 76 80 84 88 92
35 38 41 44 48 51 54 58 61 65 68 72 76 80 84 88
33* 1 36 39 42 45 48 51 55 58 62 65 69 72 76 80 84
30 33 36 39 42 46 49 52 55 59 62 66 69 72 76 80
28 1 31 34! । 37 40 43 46 49 53 56 59 63 66 70 73 77
27. 29 32 35 38 41 44 47 50 53 56 60 63 66 69 73
25 27 30 33 361 38 41 44 47 50 54 57 60 63 67 70
23 26, 28, .31 341 36 39 42 45 48 51 54 57 60 63 67
21' 24' |26 I29 32 '34 37 40 43 46 48 52 55 58 61 64
20 22 25 |27 30 32 35 38 40 43 46 49 52 55 58 61
18 21 23 25 1 28 ’30 ,33 36 38 41 44 47 50 52 55 58
17 19 21 I24 |26 |29 I31 36 39 42 44 47 50 53 56]
17 V,
18
181/.
19
19V,
20
20V.
21
100 1 1
96 100
92 96 100
88 92 96 100
84 88 92 96 100
80 84 88 92 96
77 81 84 88 92
74 77 81 84 88
70 74 1 77 81 85
67 71 74 77 81
64 68 71 74 78
62 65 68 71 75
. 59 ' 1 I62 65 68 72
49
46
43
40 43
37 41 44
35 38*41
32 36*39
30 33 36
I 1
25 28 31 34
I 1
23 26 2932
21^27*30
1922 25:28
1 I I
18 20 23 26
19*21*24
14 17 19 22
13 15*18*20
1416 19
13 15
14
12
39
25
23 26|30 33’36
24’27'30 34
122 25I28
20! 23*26
.18 21124
1619*22
14
13
53
49
46
56
53
50
47
21
19
17
15
13
31
29
27
17|20
16*18
14 17
12 15
13
16
17
16
14
100
96
92
88
85
81
78
75
100
96
92
88
85
81
78
100
96
92
89
85
82
100
96
92
89
85
100
96
92
89
100
96
92
100
96
100
21V.
22
22V2
23
231/.
24
24V2
25
25V.
26
26V.
27
27V.
Градусы Цельсия по сухому термомет]
о
Относительная влажность воздуха по аспирационному психрометру
Таблица 29 (продолжение)
Относительная влажность в процентах
Градуси Цельсия по сухому термометру
28 131 15 18 20 22 25 27 29 32 34 37 40 42 45 48 50 53 56 59
281/2 12 14 16 18 21 23 25 28 30 33 35 38 40 43 45 48 51 54 57
29 11 13 15 17 19 21 24 26 28 31 33 36 38 41 43 46 49 51 54
29i/2 10 12 14 16 18 20 22 24 27 29 31 34 36 39 41 44 46 49 52
30 11 13 15 17 19 21 23 25 27 30 32 34 37 39 42 44 47 50
30i/t 9 12 13 15 17 19 22 24 26 28 30 33 35 38 40 42 45 47
31 11 12 14 16 18 20 22 24 27 29 31 33 36 38 40 43 45
311/2 10 11 13 15 17 19 21 23 25 27 29 32 34 36 39 41 43
32 10 12 14 16 18 20 22 24 26 28 30 32 34 37 39 41
321/2 11 13 15 16 18 20 22 24 26 29 31 33 35 37 40
33 10 12 14 15 17 19 21 23 25 27 29 31 33 36 38
331/., 11 13 14 16 18 20 22 24 26 28 30 32 34 36
34 1 i 10 12 13 15 17 19 21 22 24 26 28 30 32 34
341/2 ! 1 11 12 14 16 18 19 21 23 25 27 29 31 33
35 1 | 10 11 13 15 16 18 20 22 24 26 27 29 31
35‘/2 1 1 10 12 14 15 17 19 21 22 24 26 28 30
36 1 1 i 11 11 13 14 16 18 20 21 23 25 27 29
36i/2 I 10 12 14 15 17 18 20 22 24 25 27
37 1 11 13 14 16 17 19 21 22 24 26
371/2 ! 1 1 1 l 10 12 13 15 16 18 20 21 23 25
38 1 i 11 13 14 16 17 19 20 22 24
381/2 1 1 I 10 12 13 15 16 18 19 21 22
39 1 11 12 14 15 17 18 20 21
391/о 1 i 12 13 14 16 17 19 20
40 ’ i 1 1 11 12 14 15 17 1 , 18 i 19 i 1
62
60
57
55
52
50
48
46
44
42
40
38
37
35
33
32
30
29
28
27
25
24
23
22
21
65
63
60
57
55
53
50
48
46
44
42
41
39
38
36
34
32
31
30
28
27
26
25
24
23
68
66
63
60
58
55
53
51
49
47
45
43
41
39
37
36
34
33
32
30
29
28
26
25
24
72
69
66
63
61
58
56
54
51
49
47
45
43
42
40
38
37
35
34
32
31
29
28
27
26
75
72
69
66
64
61
59
56
54
52
50
48
46
44
42
40
39
37
35
34
33
31
30
29
78
75
72
70
67
64
62
59
57
55
52
50
48
46
44
43
41
39
38
36
35
32
31
30
29
82
79
76
73
70
67
65
62
60
57
55
53
51
49
47
45
43
41
40
38
37
35
33
32
31
85
82
79
76
73
70
67
65
62
60
58
55
53
51
49
47
45
44
42
40
39
37
35
34
33
89
86
82
79
76
73
70
68
65
63
60
58
56
55
51
49
48
46
44
42
41
39
37
36
34
93
89
86
82
79
76
73
71
68
65
63
61
58
56
54
52
50
48
46
44
43
41
39
38
36
96
93
89
86
83
79
77
74
71
68
66
63
61
59
56
54
52
50
48
46
45
43
41
40
38
100
96 100
93 96 100
89
86
83
80
77
74
71
69
66
64
61
59
57
55
53
51
48
47
45
43
42
40
93
89
86
83
80
77
74
71
69
66
64
61
59
57
55
53
50
49
47
45
44
42
96
93
89
86
83
80
77
74
71
69
66
64
61
59
57
55
52
51
49
47
46
44
28
28i/2
29
29V2
30
301/2
31
ЗР/2
32
321/2
33
331/2
34
341/2
35
357,
36
361/2
37
37\'2
38
3872
39
3972
40
Градусы Цельсия по сухому термометру
Гигрометр, изображенный на рис. 124, состоит из обезжиренного
женского волоса, один конец которого прикреплен к металлической раме, а
другой, отягченный гирькой, обводится вокруг блока.
С блоком неподвижно соединена стрелка. Под влиянием
изменения влажности воздуха длина волоса увеличивает-
ся (при повышении влажности) или уменьшается (при
понижении влажности). Удлинение и укорочение волоса
вызывает поворот блока, а вместе с ним и стрелки,
которая показывает соответствующую относительную
влажность по шкале, где эмпирически нанесены деления
от 0 (слева) до 100 (справа)
Деление шкалы по величине не одинаковы: с увели-
чением относительной влажности деления становятся
все меньше и меньше, так как все меньше и меньше
увеличивается при этом длина волоса.
Гигрометр помещается в коробке, передняя стенка
которой стеклянная, а боковые и задняя металлические.
Передняя и задняя стенки подвижны и могут быть
удалены. Между задней стенкой и гигрометром поме-
щается марлевый экран. Желая проверить точность
показания гигрометра, смачивают марлю водою, благо-
даря чему в коробке воздух насыщается влагой; при
этом стрелка показывает 100. К прибору приложен
ключ, посредством которого можно регулировать по-
казания стрелки.
Рис. 124. Гигрометр.
При исследовании влажности воздуха удаляют пе-
реднюю и заднюю стенки прибора, а также
марлевый экран. Через 5—20 минут, когда по-
ложение стрелки становится стойким, производят
отсчет по шкале, определяя таким образом отно-
сительную влажность.
При приборе имеются два термометра (один
запасный) для одновременного определения тем-
пературы воздуха.
Зная температуру воздуха, можно по таблице
найти максимальную влажность, а зная макси-
мальную и относительную влажность, определить
абсолютную влажность по формуле:
А = ^,
100
где А — абсолютная влажность,
F — максимальная влажность,
/? — относительная влажность.
На метеорологических станциях применяют
гигрометры типа Соссюра (рис. 125). Такие
гигрометры, а также термометры (сухой и влаж-
ный, максимальный и минимальный) помеща-
ются в так называемые психрометрические
„ „ (термометрические) будки.
Рис. 125. Гигрометр Соссюра. Психрометрическая будка
(рис. 126) представляет собой деревянный
шкаф, одна из стенок которого, обращенная к северу, может откры-
ваться в виде дверцы наружу. Как эта стенка, так и три другие состоят из
рам, забранных внутри и снаружи планками, наклоненными к горизонту под
углом в 45° так, что каждая пара планок образует угол в 90°. Дно будки состоит
157
из трех досок, из коих средняя расположена
выше двух крайних. Крыша состоит из двух
сплошных настилов; нижний из них горизон-
тален, верхний же имеет скат к югу. Снаружи
будка покрывается белой эмалевой краской.
Высота будки берется с таким расчетом, чтобы
шарики термометров находились на расстоянии
2 м от земной поверхности. В психрометриче-
ской будке приборы защищены от солнечной
и рассеянной радиации. Наружный воздух сво-
бодно циркулирует в ней.
Гигрограф (рис. 127) служит для за-
писи относительной влажности за определен-
ный промежуток времени (сутки, неделя). При-
бор сконструирован на том же принципе, что
и описанный выше гигрометр. Вместо одного
волоса здесь взят пучок волос. Укорочение или
удлинение пучка волос передается при помощи
стерженьков писчику, который наносит кривую
влажности на бумажную ленту, натянутую на
вращающемся барабане.
Пучок волос в приборе защищен сеткою.
Рис. 126. Психрометрическая Прибор проверяют, насыщая при помощи смо-
булка. ченной марли воздух вокруг пучка волос вла-
гою. Показание пера на бумаге должно при
этом соответствовать 100, в противном случае прибор следует отрегулировать.
Рис. 127. Гигрограф.
Показание гигрографа должно контролироваться точным прибором, напр.,
аспирационным психрометром.
Движение воздуха
При изучении движения воздуха (ветра) определяют направление
движущегося потока, скорость движения, а иногда и оказываемое давле-
ние (напор).
Направление ветра
Направление ветра обозначается наименованием той части света, откуда
дует ветер. Например, северовосточный ветер — это ветер, дующий с северо-
востока, южный ветер — дующий с юга и т. д.
158
Различные направления ветра изображают в виде так называемой розы
ветров (рис. 128). Приведенные здесь буквенные обозначения показывают
соответствующие направления ветра (румб ы).
Приводим номенклатуру, соответствующую обозначенным на рисунке
буквам:
N — норд — север.
NNE — норд-норд-эст — северо-северо-восток.
NE — норд-эст — северо-восток.
ENE — эст-норд-эст — востоко-северо-восток.
Е — эст — восток.
ESE — эст-зюд-эст — востоко-юго-восток.
SE — зюд-эст — юго-восток.
SSE — зюд-зюд-эст — юго-юго-восток.
S — зюд — юг.
SSW — зюд-зюд-вест — юго-юго-запад.
SW — зюд-вест — юго-запад.
WSW — вест-зюд-вест — западо-юго-запад.
W — вест — запад.
Рис. 128. Роза ветров.
WNW — вест-норд-вест — западо-северо-запад.
NW — норд-вест — северо-запад.
NNW — норд-норд-вест — северо-северо-запад.
Определение направления ветра на метеороло-
гических станциях производится при помощи флюгера (рис. 129),
Прибор состоит из двух основных частей — нижней неподвижной, не-
сущей розу ветров в виде 8 горизонтальных стержней, и верхней подвижной,
представляющей собственно флюгер. Эта часть состоит из оси флюгера, на
Рис. 129. Флюгер.
Л — перо флюгера. С — ша-
рик, D — два крыла, В —
указатель скорости ветра,
Е — доска флюгера. К К —
ось, по которой происходит
отклонение доски флюгера.
N — буква па стержне розы
ветров, обращенном на
север.
которую насажены перо флюгера А и указатель ско-
рости ветра В, составляющие одно целое. Перо флю-
гера на одном конце имеет шарик С, на другом —
два крыла £>, образующих угол в 20°. При движении
воздуха перо флюгера устанавливается так, что ша-
рик направлен против ветра. Доска флюгера Е под-
вешена на оси Ж. Флюгером она устанавливается
нормально к направлению ветра.
По степени отклонения доски можно судить о ско-
рости ветра (см. далее).
Флюгер устанавливают на открытом месте на
высоте не менее 10 м над поверхностью земли; при
этом он должен быть ориентирован так, чтобы стер-
жень розы, несущий на своем конце букву N, был
обращен этим концом точно на север.
Желая определить направление ветра, наблюда-
тель становится по возможности ближе к флюгеру
так, чтобы роза была у него над головою. Затем он
определяет, в каком направлении в отношении розы
ветров проходит вертикальная площадь, проведенная
мысленно через пе™ флюгера, этим самым устанав-
ливая направление ветра.
Определяя направление ветра изо дня в день,
можно установить преобладющие ветры для
данного места, что имеет большое практическое зна-
чение при планировке городов, промышленных пред-
приятий, санаториев и т. д.
изображения распределения ветров в данном месте
Для графического
откладывают на восьми основных румбах отрезки, пропорциональные коли-
честву наблюдавшихся ветров данного направления, пользуясь при этом ка-
ким-либо условным масштабом. Концы отложенных прямых соединяют пря-
159
мыми линиями, получая, таким образом, розу повторяемости
ветров для данного места за определенный период времени.
На рис. 130 приведена роза повторяемости вет-
Рис. 130. Графическое изо-
бражение розы повторя-
е мости ветров.
ров, из которой видно, что преобладающими вет-
рами в данном месте являются западные и южные
(20%), затем по частоте следуют юговосточные
(17%), югозападные (15%) и т. д. Розы ветров
составляются за год, за период года, за месяц.
Определение направления дви-
жения воздуха в помещениях не
может быть произведено при помощи флюгера,
так как сила этих токов слишком ничтожна,
чтобы преодолеть сопротивление самого прибора.
В этих случаях направление воздушных токов
определяется по направлению дыма, получаемого
от сжигания какого-либо вещества. В последнее
Прибор состоит из двух склянок, снабжен-
ных резиновыми пробками. Через пробки про-
ходят по две стеклянные трубки, из коих одна
заканчивается у дна, другая у нижнего края
пробки. Наружные концы длинных трубок сооб-
щаются при помощи резиновых трубок и трой-
ника с резиновой грушей. Наружные концы
время стали применять для этой цели прибор, на-
зываемый фумигатор (рис. 131).
коротких трубок устанавливаются один возле Р|1С 13i фуМИГатор.
другого. В одну из склянок наливают нашатыр-
ный спирт, в другую — соляную кислоту. Сжимая слегка грушу, заставляют
одновременно выходить через короткие трубки из одной склянки пары NH3,
а из другой НС1. Тотчас же образуется густое облако NH4C1. Следя за его
движением, получают представление о направлении токов воздуха.
Скорость движения воздуха
Под скоростью движения воздуха понимают перемещение движущегося
воздуха в метрах в секунду.
В воздушных течениях, особенно быстрых, наблюдаются пульсации и по-
рывы, что создает турбулентное состояние атмосферы.
Приблизительную скорость ветра можно определить при помощи указа-
теля силы ветра, имеющегося на каждом флюгере (см. рис. 129). Указатель
силы ветра состоит из доски А 30 х 15 см весом в 200 г и собственно указателя
В, имеющего 8 штифтов; длинные из них соответствуют непарным, а короткие
парным номерам.
Сила ветра условно обозначается числом, показывающим, до какого штифта
ветер поднимает доску указателя либо между какими штифтами колеблется
доска указателя.
Между силою ветра, т. е. давлением, оказываемым им на единицу поверх-
ности, перпендикулярной направлению его движения, и скоростью движения
ветра существует функциональная зависимость; следовательно, можно по
величине угла отклонения доски флюгера судить о приблизительной скорости
движения воздуха. В таблице 30 показана эта зависимость.
Более точное определение скоростей движения воздуха производится при
помощи приборов, носящих название анемометров (от греческого — anemos —
ветер и metron — мера). В зависимости от конструкции приборов различают
динамические и статические анемометры.
Первые дают возможность определять скорость движения воздуха по
числу оборотов оси, на которой насажены стерженьки, снабженные крыль-
160
чатыми лопастями или полушариями; вторые — по отклонению крыльчатого
колеса или подвешенной вертикально пластинки.
Таблица 30
Скорость ветра по указателю флюгера
№ штифта Скорость ветра в м/сек. № штифта Скорость ветра в м/сек.
1 0 5 8
1—2 1 5—6 9
2 2 б 10
2—3 3 6—7 12
3 4 7 14
3—4 5 7—8 17
4 б
4—5 7 8 и > 8 20 и > 20
ц [Г
Динамический крыльчатый анемометр (рис. 132)
состоит из горизонтально поставленной оси, снабженной на одном конце че-
тырьмя стерженьками, на которых укреплены под
углом 40—45° к направлению потока слюдяные пла-
стинки а. На другом конце оси находится бесконеч-
ный винт (шнек) Ь, который по желанию может
быть приведен в контакт с системой зубчатых колес.
Прибор устанавливается так, чтобы направление дви-
жения воздуха было параллельно оси прибора.
Под влиянием давления движущегося воздуха
на лопасти прибора ось начинает вращаться; при
этом скорость вращательного движения оси оказы-
вается пропорциональной скорости воздушного по-
тока. При каждом обороте оси зубчатое колесо с
перемещается на один зубец перед указателем /. При
полном обороте колеса с колесо е перемещается стер-
женьком на один зубец d перед указателем g. Таким
образом, по колесу с можно определить единицы и
десятки числа оборотов оси вращения, а по колесу
е — сотни и тысячи. Установив прибор в отноше-
нии направления ветра, как указано выше, опре-
деляют его показания на основании счетчика прибора. Затем устанавли-
вают контакт между шнеком и системой зубчаток при помощи потягивания
за одну из тесемок прибора. Наблюдение продолжается несколько минут
(5 — 10). По окончании наблюдения потягиванием другой тесемочки отодви-
гают систему зубчаток от шнека. Вновь определяют показание счетчика при-
бора. По разнице между первым и вторым отсчетом определяют число оборо-
тов оси вращения, а затем вычисляют скорость движения воздуха при помощи
формулы:
v - а + Ьп
где v — искомая скорость,
а и b — постоянные коэфициенты для каждого прибора, обозначенные
в паспорте прибора \
п — найденное число оборотов в секунду.
Рис. 132. Динамический
анемометр крыльчатый.
а — слюдяная пластинка,
Ъ — бесконечный винт, с —
первая зубчатка, / — указа-
тель для отсчета по зубчат-
ке С, в — вторая зубчатка,
g — указатель для отсчета
по зубчатке в, d — стерже-
нек, перемещающий зуб-
чатку е.
1 а — наименьшая скорость движения воздуха, которая может вызвать вращение оси
прибора; b — сопротивление, обусловленное трением крыльев о воздух.
11 А. и. Бурштейн 161
Завод «Метприбор» выпускает динамические анемометры более прочные,
нежели описанный выше. В этих анемометрах (рис. 133) на оси вращения
сидят восемь стерженьков с алюминиевыми пластинками, огражденными ши-
роким металлическим кольцом. Перемещение стрелок по циферблатам соответ-
ствует всему пути, пройденному воздушным потоком за
время наблюдения и выраженному в метрах. Разделив
определенную по циферблатам длину пути воздушного
потока на время наблюдения, выраженное в секундах,
находят скорость движения воздушного потока.
Пример. В начале опыта показание стрелок на циферблатах
анемометра соответствовало 12 300; после опыта 13 560. Опыт
продолжался 10 минут. Отсюда определяют скорость движения
воздуха, равную
13 560— 12 300 ,
= 2,1 м/сек.
Рис. 133. Динамиче-
ский анемометр «Мет-
прибор».
ю. 60
Динамический
чашечный а немо-
метр (рис. 134) имеет вместо лопастей четыре полых
полушария из тонкой латуни, насаженных на концы
четырех стерженьков, образующих крест. Полушария
устанавливаются так, что все они своими выпуклостями
обращены в одну сторону. Крест с полушариями укреплен на оси, которая
может быть приведена в контакт с системой зубчаток.
Во время опыта ось устанавливается вертикально, и сидящая на ней кресто-
вина оказывается в горизонтальном положении. Принцип работы и способ
применения чашечного анемометра такие же, как и для крыльчатого.
Электрический анемометр системы
«Метприбор» (рис. 135) представляет собой ча-
шечный анемометр, у которого крестовина вращает
систему магнитов, расположенных над сердечниками
катушек. В результате вращения магнитов вырабаты-
Рис. 135. Электрический
анемометр «Метприбор».
а — система магнитов, b —
катушки, с — указатель ско-
рости ветра.
Рис. 134. Динамический чашечный анемометр.
Справа показано положение стрелок. Отсчет при
данном положении стрелок равен 6896.
вается электрический ток, напряжение которого пропорционально скорости
вращения электромагнитного якоря и, следовательно, скорости движения
ветра. Ток идет по проводам к указателю скорости ветра, построенному по
принципу вольтметра. Стрелка прибора все время показывает приблизитель-
ную скорость ветра в метрах в секунду.
Для определения средней скорости следят за показаниями стрелки в те-
чение 2 минут и берут среднее арифметическое из них.
162
Статический анемометр, изображенный на рис. 136, состоит
из оси, на которой насажены четыре стерженька, снабженные лопастями.
Один из концов оси соединен с часовой пружиной, что не дает возможности
оси свободно вращаться; она может лишь поворачиваться на больший или
меньший угол, в зависимости от силы ветра. Поворот оси определяют в гра-
дусах на циферблате по отклонению стрелки, сидящей на оси. Скорость дви-
жения воздуха определяют по формуле:
v = a^n.
Рис. 137. Статический
анемометр с подвешенной
доской.
где v — искомая скорость,
а — постоянный коэфициент, обозначенный в паспорте прибора,
п — угол отклонения стрелки в градусах.
Статический анемометр,
работающий по принципу
отклонения воздушным по-
током вертикально подве-
шенной доски, уже описан
выше вместе с флюгером
(стр. 160). Этот же прибор,
применяемый для более
точных определений ско-
ростей движения воздуха,
изображен на рис. 137.
Он состоит из пластин-
ки, подвешенной на раме,
которая устанавливается
вертикально по уровню.
В зависимости от давления,
образуемого движущимся
Рис. 136. Статический воздухом, пластинка от-
анемометр крыльчатый. клоняется на больший или
меньший угол, величину
которого определяют по дугообразной шкале. При
помощи эмпирически составленной таблицы определяют скорость движения
воздуха по углу отклонения доски прибора от начального положения.
Электроанемометры с охлаждаемым электропро-
водником построены по совершенно иному принципу, нежели описанные
выше. В основу работы этих анемометров положено охлаждение потоком воз-
духа электрического проводника (тонкой проволоки), нагреваемого электри-
ческим током.
В зависимости от скорости потока воздуха меняется степень охлаждения
проводника. По изменению температуры проводника определяют скорость
потока воздуха, применяя схему с измерительным мостиком сопротивления.
При этом прибегают к одному из следующих методов: 1) уравнивание сопротив-
ления; 2) уравнивание силы тока; 3) непосредственное измерение охлаждения
проволоки.
Ниже описан электроанемометр, работающий по приципу уравнивания
сопротивления.
Прибор состоит из головки (щупа), рукоятки и измерительной части.
Чувствительным элементом в головке является платиновая нить длиною от
нескольких до 140 мм, диаметром 0,025—0,15 мм.
На рис. 138-1 изображена головка анемометра а с рукояткой Ь. Нить с укреп-
лена при помощи спая золотом или серебром на двух константановых стер-
женьках d. От головки через рукоятку отходят провода е, присоединяемые к
измерительной части прибора (мостику сопротивления).
На рис. 138-11 дана схема соединения электроанемометра с мостиком сопро-
тивления для измерения по методу постоянного сопротивления. В одно из
163
плеч мостика включена чувствительная проволока анемометра а, нагреваемая
током от элемента Е до определенной температуры (обычно не выше 500° С).
Противоположное плечо сделано из константана.
Сопротивление всех ветвей и проволоки мостика подбирается так, чтобы
при нагреве
о
проволоки до желаемой температуры гальванометр показывал О
(отсутствие тока).
При измерениях проволоку а устанавливают перпендику-
лярно к направлению потока воздуха. Движущийся воздух
охлаждает проволоку, сопротивление ее изменяется и равнове-
сие мостика нарушается.
Действуя реостатом /?, изменяют силу тока и снова уравно-
вешивают мост. Сила тока измеряется либо амперметром, со-
единенным последовательно с нагретой проволокой, либо вольт-
метром высокого сопротивления, включенным параллельно про-
волоке. Зная силу тока или вольтаж, можно по градуировочной
кривой определить скорость движущегося воз-
духа.
Описанный прибор применяется для опре-
деления малых скоростей движения воздуха —
доли см/сек.
Кроме описанной конструкции электроане-
мометра существуют и другие — ЛИОТ (Ленин-
градский институт охраны труда), Рогозина,
Васильева и др.
Давление ветра
Напор или давление ветра в килограммах
на м2 зависит от скорости движения воздуха
и от величины угла, под которым он ударяется
о поверхность сопротивления. При перпендику-
лярном направлении ветра к поверхности со-
противления напор растет приблизительно про-
порционально квадрату скорости, при косом
направлении — пропорционально синусу угла падания. Напор ветра можно
определить при помощи описанных выше статических анемометров по формуле:
Рис. 138.
I — Электроанемометр: а — голов-
ка прибора, d — константановые
стерженьки, с — нить, Ъ — руко-
ятка прибора, е — провода.
II — схема соединения электроане-
мометра с мостиком сопротивления:
Е — элемент, R — реостат, а —
проволока анемометра, Г1 г2 г, —
сопротивления ветвей мостика,
и — вольтметр, g — гальванометр.
Pd=k—
S
где Pd — давление в килограммах на 1 м2 поверхности, нормальной к воздуш-
Рис. 139. Анемометр для определения дав-
ления воздуха.
а — пластинка, устанавливаемая нормально про-
тив движущегося воздуха, Ъ — спиральная пру-
жина. с — стерженек, перемещение которого
вызывает движение стрелки d, е — шкала.
ному потоку,
к — эмпирически определен-
ный коэфициент,
s — поверхность доски (м2),
v — скорость ветра (м/сек),
о — плотность воздуха.
Для этой же цели применяется
анемометр, изображенный на рис.
139. В нем движущийся воздух
давит на пластинку а, устанавли-
ваемую перпендикулярно к направ-
лению ветра. Позади пластинки
имеется спиральная пружина Ь,
к которой прикреплен стерженек с.
Перемещение пружины вместе со стерженьком передается стрелке d, указы-
вающей соответствующее давление на шкале е.
Для определения скорости движения воздуха, кроме анемометров, могут
служить кататермометры, а также пневмометрические трубки, описанные ниже.
164
Кататермометрия
Кататермометрия представляет собой метод определения охлаж-
дающей способности воздуха, зависящей от совокупного действия его темпе-
ратуры, влажности и движения. Влияние тепловой радиации при этом методе
не учитывается.
Прибор, применяющийся для данной цели, носит название кататер-
мометра (рис. 140), и представляет собой алкогольный термометр с цилинд-
рическим резервуаром поверхностью в 22,6 см и трубкою длиною
в 20 см. Шкала разделена на три градуса С (38 — верхнее и
35 — нижнее деление). Верхний конец капиллярной трубки при-
бора переходит в небольшой резервуар. Указанный температур-
ный интервал избран преднамеренно и дает возможность опреде-
лить охлаждающее действие внешней среды на прибор при тем-
пературах его, близких к температурам человеческого тела: среднее
арифметическое 38 и 35 равно 36,5, что соответствует средней
температуре человеческого тела.
При помощи кататермометра определяют потерю тепла в милли-
калориях1 на единицу поверхности (1 см2) его резервуара за
единицу времени (1 сек).
Прибор предназначен для определения охлаждающей способ-
ности внешней среды на тело человека, однако он может быть
рассматриваем лишь как отдаленный аналог кожи человече-
ского тела.
В санитарно-гигиенических исследованиях применяют сухой
и влажный кататермометры. Последний отличается от сухого
лишь тем, что на его резервуар надевается колпачок из хлопчато-
бумажной ткани, который во время исследования смачивается
(см. далее). В паспорте, а также на трубке каждого кататермо-
метра обозначен его фактор (F) — число, которое показывает,
сколько милликалорий тепла теряет каждый квадратный сантиметр
поверхности резервуара предварительно нагретого прибора при
охлаждении его с 38 до 35°. Фактор кататермометра представляет
собой величину постоянную для каждого прибора, зависящую от
его физических особенностей (теплопроводность алкоголя и стекла,
толщина стенки резервуара и т. п.).
Под временем охлаждения (Т) понимают число Р|1С 14о
секунд, которое в каждом отдельном случае протекает, пока ката- Кататер-
термометр не охладится от 38 до 35°. мометр.
Это время зависит от внешних факторов среды, где произво-
дится исследование, а именно от температуры, влажности и скорости дви-
жения воздуха.
Под величиной (силой) охлаждения (Н) понимают число
милликалорий тепла, которое теряет каждый квадратный сантиметр предва-
рительно нагретого прибора в одну секунду во время охлаждения его от 38 до
35°. Для влажного кататермометра принято величину охлаждения обозначать
через Hv
Проверка фактора. Фактор каждого кататермометра опреде-
ляется авторитетными учреждениями. При желании проверить фактор ката-
термометра пользуются формулой:
F = 0,27 . Q . Т,
где F — фактор,
Q — 36,5 — t° (t° — температура воздуха в месте наблюдения),
Т — время охлаждения кататермометра.
1 Милликалория — 0,001 малой калории.
165
Проверка фактора производится в термостате с двойными стенками. Объем
термостата должен быть не менее 30 л. Температура внутри термостата до-
пускается в пределах 10—20° С при совершенно спокойном состоянии воздуха.
Поместив в термостат точный термометр и предварительно нагретый ка-
татермометр, определяют по секундомеру время охлаждения кататермометра;
одновременно отмечают температуру внутри термостата. Наблюдения ве-
дутся через закрытую стеклянную дверь термостата.
Время охлаждения кататермометра определяется пять раз. Берут среднее
арифметическое Ч Температура внутри термостата должна оставаться во время
наблюдений постоянной; допускаются колебания не более 0,1° С.
На основании полученных данных находят фактор, пользуясь приведенной
выше формулой.
Пример. Положим, что t° термостата = 15° С, время охлаждения кататермометра
Т = ПО сек. Отсюда находим:
F = 0,27 X (36,5 — 15) X ПО = 639,55.
Методика определения Н и Hv Для определения силы охлаж-
дения кататермометра (Н) нагревают в стакане воду до 80° С и погружают
в нее резервуар кататермометра. Выжидают, пока поднимающийся вверх алко-
голь заполнит треть или половину расширения, в которое переходит сверху
капилляр. Затем кататермометр извлекают из воды, насухо обтирают резер-
вуар и, закрепив кататермометр в штативе, следят за падением алкоголя в
трубке. Штатив должен быть деревянным, а не железным, чтобы избежать
охлаждающего действия его на кататермометр. В тот момент, когда уровень
алкогольного столбика проходит через 38°, пускают в ход секундомер; когда
уровень достигает 35°, останавливают секундомер. Так повторяют пять раз,
записывая каждый раз отсчеты по секундомеру. Первый отсчет обыкновенно
отбрасывают как неточный. Среднее арифметическое из четырех отсчетов пред-
ставляет собой время охлаждения кататермометра. Определив Т,
находят величину охлаждения из равенства:
Н
где Н — величина охлаждения,
F — фактор кататермометра,
Т — время охлаждения.
Пример. Положим, что F кататермометра = 540; во время опыта выяснилось, что
Т = 108. Отсюда находим:
F
г ’
Для определения т. е. величины охлаждения влажного кататермометра,
надевают на резервуар прибора колпачок и поступают так же, как при работе
с сухим кататермометром, с той лишь разницей, что по извлечении резервуара
из горячей воды его не обтирают, а отжимают рукой избыток воды. Т и Нг
определяются так же, как при сухом кататермометре.
Сухим кататермометром определяют потерю тепла прибора, возникающую
вследствие теплоизлучения и теплопроведения; влажным же кататермометром
определяют теплопотерю, возникающую еще вследствие испарения влаги с
поверхности. Ввиду сказанного Нг >> Н. Разница между Нг и Н показавает
охлаждение, возникающее за счет испарения.
Определение скорости движения воздуха при
помощи сухого кататермометра. Сухой кататермометр может
служить хорошим прибором для определения скоростей движения воздуха в
определенных пределах.
1 Первое данное лучше отбросить, взяв среднее арифметическое из последующих че-
тырех.
166
Таблица 31
н Q Скорость движения в м/сек при н <0,6 и t° ч
10,0° 12,5° 15,0° 17,5° 20,5° 22,0° 25,0° 26,0°
0,27 — — — — 0,044 0,047 0,051 0,059
0,28 -— — — 0,049 0,051 0,061 0,070 0,074
0,29 0,041 0,050 0,051 0,060 0,067 0,076 0,085 0,089
0,30 0,051 0,060 0,065 0,073 0,082 0,091 0,101 0,104
о,31 0,061 0,070 0,079 0,088 0,098 0,107 0,116 0,119
0,32 0,076 0,085 0,094 0,104 0,113 0,124 0,136 0,140
0,33 0,091 0,101 0,110 0,119 0,128 0,140 0,153 0,159
0,34 0,107 0,115 0,129 0,139 0,148 0,160 0,174 0,179
0,35 0,127 0,136 0,145 0,154 0,167 0,180 0,196 0,203
0,36 0,142 0,151 0,165 0,179 0,192 0,206 0,220 0,225
0,37 0,163 0,172 0,185 0,198 0,212 0,226 0,240 0,245
0,38 0,183 0,197 0,210 0,222 0.239 0,249 0,266 0,273
0,39 0,208 0,222 0,232 0,244 0,257 0,274 0,293 0,301
0,40 0,229 0,242 0,256 0,269 0,287 0,305 0,323 0,330
0,41 0,254 0,267 0,282 0,299 0,314 0,330 0,349 0,36)4
0,42 0,280 0,293 0,311 0,325 0,343 0,361 0,379 0,386
0,43 0,310 0,324 0,342 0,356 0,373 0,392 0,410 0,417
0,44 0,340 0,354 0,368 0,385 0,401 0,417 0,445 0,449
0,45 0,365 0,381 0,398 0,412 0,429 0,449 0,471 0,478
0,46 0,396 0,415 0,429 0,446 0,465 0,483 0,501 0,508
0,47 0,427 0,445 0,464 0,482 0,500 0,518 0,537 0,544
0,48 0,468 0,481 0,499 0,513 0,531 0,551 0,572 0,579
0,49 0,503 0,516 0,535 0,556 0,571 0,590 0,608 0,615
0,50 0,539 0,557 0,571 0,589 0,604 0,622 0,640 0,651
0,51 0,574 0,593 0,607 0,628 0,648 0,666 0,684 0,691
0,52 0,615 0,633 0,644 0,665 0,683 0,701 0,720 0,727
0,53 0,656 0,674 0,688 0,705 0,724 0,742 0,760 0,768
0,54 0,696 0,715 0,729 0,746 0,764 0,783 0,801 0,808
0,55 0,737 0,755 0,770 0,790 0,807 0,807 0,844 0,851
0,56 0,788 0,801 0,815 0,833 0,851 0,867 0,884 0,894
0,57 0,834 0,852 0,867 0,882 0,898 0,915 0,933 0,940
0,58 0,879 0,898 0,912 0,929 0,911 0,959 0,972 0,977
0,59 0,930 0,943 0,957 0,971 0,985 1,001 1,018 1,023
0,60 0,981 0,994 1,008 1,022 1,033 1,044 1,056 1,060
167
Если скорость движения воздуха меньше метра в секунду, то пользуются
следующей формулой:
V =
— — 0,2
Q________
- 0,4
2
где v — искомая скорость,
Н — величина охлаждения кататермометра,
Q — 36,5 — t° (t° — температура помещения).
Приведенная формула для скоростей движения воздуха менее метра в
секунду требует поправок в зависимости от /° воздуха, так как при одном и
Н
том же значении-у скорость движения возрастает с повышением температуры
X
воздуха. В таблице 31 приведены скорости движения воздуха до 1 м/сек в зави-
Н
симости от значения с учетом поправок на температуру.
X
Для скоростей движения воздуха больше метра в секунду пользуются
следующей формулой.
V =
— — 0,13
Q________
0,47
2
где v, Н и Q сохраняют те же значения, что и в предыдущей формуле. Формула
применима для скоростей, не превышающих 17 м в секунду.
В таблице 32 приведены значения для v при скоростях более одного метра
в секунду.
Таблица 32
н
Скорость движения воздуха при “q">0,6
н Q Скорость в м/сек. Н Q Скорость в м/сек. н Q Скорость в м/'сек.
0,60 1,00 0,82 2,16 1,10 4,26
0,61 1,04 0,83 2,22 1,13 4,53
0,62 1,09 0,84 2,28 1,15 4,71
0,63 1,13 0,85 2,35 1,18 4,99
0,64 1,18 0,86 2,41 1,20 5,18
0,65 1,22 0,87 2,48 1,23 5,48
0,66 1,27 0,88 2,55 1,25 5,68
0,67 1,32 0,89 2,61 1,28 5,99
0,68 1,37 0,90 2,68 1,30 6,20
0,69 1,42 0,91 2,75 1,35 6,73
0,70 1,47 0,92 2,83 1,40 7,30
0,71 1,52 0,93 2,90 1,45 7,89
0,72 1,58 0,94 2,97 1,50 8,50
0,73 1,63 0,95 3,04 1,55 9,13
0,74 1,68 0,96 3,12 1,60 9,78
0,75 1,74 0,97 3,19 1,65 10,5
0,76 1,80 0,98 3,27 1,70 11,2
0,77 1,85 0,99 3,35 1,75 11,9
0,78 1,91 1,00 3,43 1,80 12,6
0,79 1,97 1,03 3,66 1,85 13,4
0,80 2,03 1,05 3,83 1,90 14,2
0,81 2,09 1,08 4,09 , 1,95 15,0
— — — 2,00 15,8
Для определения скоростей движения воздуха
общая формула:
2
предложена следующая
— — 0,14
0,49
168
Для определения скоростей движения воздуха при наличии источников
лучистой энергии пользуются кататермометром с посеребренным резервуаром.
Формула для указанной цели имеет следующий вид:
L 0,508 J
Определение скоростей движения воздуха по сухому кататермометру верно
лишь в том случае, если направление движущегося воздуха перпендикулярно
продольной оси прибора; в противном случае ошибки определений могут
достигнуть 50%. Учитывая это, необходимо при указанных исследованиях
придавать кататермометру такое положение, при котором направление потока
воздуха было бы перпендикулярно продольной оси прибора. Так, если воз-
дух имеет вертикальное направление (шахта), то кататермометру нужно
придать горизонтальное положение в момент, когда алкоголь охлаждаемого
прибора перейдет из верхнего расширения в капилляр.
В отношении применения кататермометра должны быть сделаны еще сле-
дующие указания.
При наличии лучистой энергии, кататермометр должен быть защищен от
ее влияния экраном, в противном случае показания кататермометра будут
неточны.
В помещениях с температурой между 35 и 38° С сухой кататермометр не
может быть применен вовсе, так как уровень алкоголя остановится между
35 и 38° и, следовательно, никаких расчетов нельзя будет делать.
В помещениях с температурой выше 38° С сухой кататермометр может
быть использован лишь для определения нагревающего действия воздуха.
Для этой цели кататермометр предварительно нагревают, как описано выше,
а затем охлаждают, погрузив резервуар в холодную воду, после чего ката-
термометр вытирают насухо и закрепляют в штативе, наблюдая время пока
уровень алкоголя с 35 делениями поднимется до 38.
Величину нагревания кататермометра определяют по формуле:
Большое увлечение кататермометром Хилла, как прибором, пригодным
для корреляции между величиной его охлаждения и теплоощущением чело-
века, прошло довольно быстро.
Ряд наблюдений, проведенных в СССР, показали, что указанная корреля-
ция имеет место лишь при средних температурах; при высоких и низких тем-
пературах корреляция отсутствует. Вот почему сухой кататермометр Хилла
в настоящее время применяют в основном для определения скоростей движе-
ния воздуха. Влажный же кататермометр почти оставлен.
Шаровой кататермометр
Физическая лаборатория Ленинградского института гигиены труда
и профзаболеваний разработала усовершенствованные типы кататермометра
(рис. 141).
Важнейшие особенности новых кататермометров сводятся к следующему:
1. Резервуар имеет не цилиндрическую, а шарообразную форму (диаметр
25—27 мм).
2. В целях возможного применения прибора при различных температурах
воздуха разработаны три шкалы: а) «нормальная» или «среднеградусная» с
пределами от 20° до 40° С для измерений при средних температурах воздуха;
б) «низкоградусная» с пределами от 0° до 20° С для измерений при темпера-
169
турах от 0 до 20°С и в) «высокоградусная» с пределами от 40° до 55° С для
измерений при температурах выше 40° С.
3. Деления шкалы (целые и половины градуса) нанесены непосредственно
на стекло (стержень) кататеруол:етра.
4. Фактор прибора указывает количество тепла в милликалориях, теряемое
1 см2 поверхности резервуара при охлаждении на 1° С. Фактор обозначается
буквой: «О»; его значение наносится на стержень прибора.
Методика определения охлаждающей силы воздуха та же, что и при приме-
нении обычного кататермометра. Разница лишь в температуре воды, применяе-
мой для нагревания прибора перед измерениями, а именно: для среднеградус-
ного кататермометра пользуются водой 65° — 70° С, высокогра-
дусного 75°—80° С и низкоградусного — 38° — 40° С.
Как и при работе с обычным кататермометром при каждом
определении охлаждающей силы воздуха производят четырехкрат-
ное измерение времени охлаждения, не считая первого, которое от-
брасывается; при чем время охлаждения можно учитывать между
любыми двумя течками шкалы прибора, т. е. не обязательно
учитывать время охлаждения от верхнего предела шкалы до
нижнего ее предела.
Охлаждающая сила воздуха вычисляется по формуле:
/у — ' (Qi Q2)
Т
где Н — охлаждающая сила воздуха,
Ф — фактор прибора,
QihQ2—верхний и нижний пределы отсчета по шкале при-
бора.
Т—время охлаждения в секундах.
Рис. 141.
Кататермо-
метр кон-
струкции
Ленинград-
ского инсти-
тута гигиены
труда.
Пример. При применении среднеградусного кататермометра (шкала от
20° до 40° С) Т оказалось равным 120 сек. Ф прибора 110. Отсчет времени
охлаждения произведен в пределах от 40 до 33 шкалы. Нужно опреде-
лить Н. Подставляя в приведенную выше формулу данные значения, по-
лучаем
„ 110(40—33)
Н =-------*--------6,42 милликал./См2 сек.
120
В тех случаях, когда вычисления охлаждающей силы требуется произ-
вести с большой точностью, а также и тогда, когда определения производятся
при высоких или низких температурах, или при средних температурах в
условиях резкого колебания их, рекомендуется определить так называемую
«мгновенную охлаждающую силу воздуха» Gt по следующей формуле «регу-
лярного режима»1.
Gt = m • Ф • (Q — 0,
где Gt — мгновенная охлаждающая сила воздуха при температуре охлаждения
тела, равной 36,5°, и при температуре окружающей среды, равной /° С.
Ф — фактор кататермометра,
Q — температура тела, принятая равной 36,5° С,
t — температура окружающей среды,
m — темп охлаждения, вычисляемый по нижеследующей формуле:
1п (б?! —/) — In (Q2— 0
т
1 См. Г. М. Кондратьев: «Испытания на теплопроводность по методам регуляр-
ного режима», Стандартгиз, 1936 г.
170
где т — темп охлаждения,
— верхний предел шкалы, от которого начат учет времени охлаждения,
Qa — нижний предел шкалы, до которого учитывалось время охлаждения,
t — температура окружающего воздуха,
Т — время охлаждения кататермометра от до Qa в секундах.
Пример. Измерение производилось кататермометром с низкоградусной шкалой (от 0°
до 20°С), при этом t окружающего воздуха была равна — 4° С. Учет времени охлаждения
производился в пределах шкалы от 20° до 14°. Время охлаждения составило 162 секунды.
Ф прибора равно 140. Требуется определить мгновенную охлаждающую силу воздуха Gt.
Сначала находим темп охлаждения по второй формуле. Подставив в эту формулу вместо
букв соответствующие значения, получаем:
In [20 — (—4)] — In [14 — (—4)] 1п24— In 18 3,178—2,890 _ ЛЛ1О
162 162 162
Далее находим мгновенную охлаждающую силу воздуха по первой формуле. Подста-
вив вместо букв соответствующие значения, получаем:
Gt = 0,0018 • 140 [36,5 — (—4)] = 10,206 ми л л икал./см2 сек.
Шаровой кататермометр может быть использован также для определения
скоростей движения воздуха по нижеследующей таблице 33.
Таблица 33
Скорость движения воздуха по шаровому кататермометру
н Q и м/сек. н Q м сек. Н Q и м'сек. н Q и м/сек. н Q и м/сек.
0,33 0,05 0.53 0,56 0,70 1,40 0,90 2,34 1,09 3,51
0,345 0,07 0,54 0,60 0,71 1,45 0,91 2,39 1,10 3,58
0,36 0,09 0,545 0,65 0,72 1,49 0,92 2,45 1,11 3,65
0,37 0,11 0,55 0,69 0,73 1,54 0,93 2,51 1,12 3,72
0,38 0,13 0,56 0,74 0,74 1,58 0,94 2,56 1,13 3,79
0,39 0,15 0,565 0,78 0,75 1,62 0,95 2,62 1,14 3,87
0,40 0,17 0,57 0,82 0,76 1,67 0,96 2,68 1,15 3,95
0,41 0,19 0,575 0,85 0,77 1,72 0,97 2,74 1,16 4,03
0,42 : 0,21 0,58 0,90 0,78 1,76 0,98 2,80 1,17 4,11
. 0,43 0,23 0,59 0,96 0,79 1,81 0,99 2,86 1,18 4,19
0,44 0,25 0,60 1,00 0,80 1,86 1,00 2,93 1,19 4,27
0,45 0,28 0,61 1,04 0,81 1,91 1,01 2,99 1,20 4,35
0,46 0,31 0,62 1,09 0,82 1,95 1,02 3,06 1,21 4,44
0,47 0,34 0,64 1,14 0,83 2,00 1,03 3,12 1,22 4,53
0,48 0,37 0,65 1,18 0,84 2,05 1,04 3,19 1,23 4,62
0,49 0,40 0,66 1,22 0,86 2,11 1,05 3,25 1,24 4,71
0,50 0,44 0,67 1,27 0,87 2,17 1,06 3,32 1,25 4,80
0,51 0,48 0,68 1,31 0,88 2,22 1,07 3,38 1,26 4,90
0,52 0,52 0,69 1,36 0,89 2,28 1,08 3,45 1,27 5,00
Шаровой кататермометр введен в практику сравнительно недавно. Он имеет
несомненные преимущества перед кататермометром Хилла.
171
Фригориметрия
Мы видели, что кататермометр не учитывает всех метеорологических фак-
торов, определяющих величину охлаждения внешней среды; так, например,
он не учитывает влияния тепловой радиации, атмосферных осадков.
Исходя из этих соображений, Г. X. Шахбазян предложил прибор, пред-
назначенный для определения величины охлаждения, обусловленной всеми
метеорологическими факторами, влияющими на размер этой величины. Этот
прибор назван фригориметром (от латинского frigor — холод и
греч. metron — мера). Он не нашел пока широкого применения в практиче-
ских и научных учреждениях, но ввиду большого интереса, который он пред-
ставляет, мы приводим здесь краткое описание устройства и принципа дей-
ствия прибора.
Рис. 142. Схема фригориметра Г. X. Шахбазяна.
Шг — штепсельные гнезда, Сч — счетчик, Ш — шар, О — нагревательная обмотка, Р — круговой
реостат, СР — ступенчатый реостат, ПР — приводной рычаг. К — контакт, р — реле, ТР—трансфор-
матор, В — вольтметр, П — переключатель, ТГ — трансформатор «Гном», Л — лампа сигнальная,
ч— часы, Вч — выключатель часов, ЭМ —электромагнит, А —аккумулятор, ВГ — выключатель
гальванометра, Г — гальванометр, АВ — гцупальцы. AxBi — контактные пластинки, Т — термометр,
М — мостик, 2, 2, 3, 4 — плечи мостика, РХ — реохорд.
Прибор, как видно из схематического чертежа (рис. 142), устроен следую-
щим образом: зачерненный снаружи медный шар Ш (поверхность 176,7 см2)
нагревается посредствэм электрического тока, проходящего от осветительной
сети через нижнее штепсельное гнездо ШГ, счетчик Сч, контакт К (если он
замкнут), ступенчатый реостат СР, нагревательную обмотку О, снова через
счетчик и верхнее штепсельное гнездо.
Во время работы прибора температура поверхности шара автоматически
поддерживается на определенной высоте. Автоматическая регулировка посто-
янства температуры поверхности шара достигается следующим образом:
внутри шара помещен термометр сопротивления Т, который включен в одно
из плеч равновесного мостика М. При помощи реохорда Рх, включенного
в другое плечо мостика, последний можно устанавливать так, что он будет
находиться в равновесии при любой заданной температуре, и тогда гальвано-
метр Г, включенный в вершину мостика, будет показывать отсутствие тока.
172
При изменении температуры шара (против заданной) равновесие мостика
нарушится и стрелка гальванометра отклонится от нуля вправо или влево.
Через каждые 15 секунд щупальцы контакта АВ, подвешенные под стрелкой
гальванометра, падают вниз, причем одно щупальце задерживается стрелкой
гальванометра, а второе, падая свободно, соприкасается с одной из контакт-
ных пластинок А1В1 и тем самым включает ток в одну из катушек перекид-
ного реле р, замыкая или размыкая контакт /{, благодаря чему осуществля-
ется нагрев шара, если его температура опустилась ниже заданной, либо
охлаждение, если температура поднялась выше заданной. Описанным спосо-
бом удается поддерживать температуру шара на определенной высоте (в опы-
тах автора Зб,5°С).
Пользование прибором весьма просто: включив его в осветительную сеть
и поставив указатель реохорда на желаемую температуру, нагревают шар
до заданной температуры и записывают время начала опыта и показание
счетчика. По окончании опыта снова отмечают время и показание счетчика,
а также показание часов прибора, учитывающих время прохождения тока
в приборе во время опыта.
На основании всех полученных данных определяют продолжительность
опыта и израсходованное за это время количество ватт-часов.
Охлаждающее действие среды определяют по формуле
W
q = 4861 —,
Т
где Q равно охлаждающему действию среды в милликалориях в 1 сек. с по-
верхности в 1 см2,
W — количество израсходованных ватт-часов,
Т — продолжительность опыта в секундах.
Пример. Во время опыта, длившегося 1 час 15 минут, израсходовано 4,2 ватт-часа
электроэнергии. Требуется определить охлаждающее действие внешней среды.
4,2
Оно равно 48G1 • ~ 4,54 милликалорий с 1 см2 в 1 сек.
Автор провел серию опытов с прибором, одновременно изучая влияние
той же среды на тепловое ощущение подопытных лиц, и вывел ряд данных,
сведенных в таблице 34.
Таблица 34
Тепловое самочувствие и показания фригориметра
Тепловое самочувствие Показания фригориметра
В неподвижном воздухе В движущемся воздухе
Жарко 1,2 2,3
Тепло 3,2 5,1
Хорошо 4,2 7,4
Прохладно 6,5 9,5
Автор считает свои данные ориентировочными, требующими дальнейших
наблюдений и экспериментов.
Эффективные и эквивалентно-эффективные температуры
Под эффективной температурой (ЭТ) понимают комп*
лексный индекс, при помощи которого можно учесть одновременное влияние
на тепловое ощущение человека двух компонентов метеорологического фак-
тора: температуры и влажности. Следовательно, об эффективных темпера-
турах может итти речь лишь для неподвижного воздуха.
173
Нормальная шкала эффективных температур для средне
Скорость движения воздуха
t° в
градусах 0 15 30 ( 50
Относительная влаж
С.
100% 50% 20% 100% 50% 20% 100% 50% 20% 100о/о 50%
0 0 0,9 1,3 0,9 0,4
1 1 1,8 2,1 0,8 0,1 0,4 —- — 0,0 — —.
2 2 2,7 3,0 0,3 1,0 1,4 — 0,5 0,0 —
3 3 3,7 3,9 1,3 2,0 2,3 0,3 0,5 0,9 —
4 4 4,5 4,7 2,4 3,0 3,2 0,8 1,7 1,9 0,7 1,0
5 5 5,4 5,5 3,4 4,0 4,1 1,9 2,6 2,9 0,4 0,1
б б 6,2 6,3 4,5 4,9 5,1 3,0 3,6 3,9 1,6 1,2
7 7 7,1 7,1 5,5 5,8 5,9 4,2 4,6 4,7 2,9 2,3
8 8 7,9 7,9 6,6 6,9 6,9 5,3 5,7 5,8 4,2 3,5
9 9 8,8 8,7 7,7 7,7 7,7 6,4 6,8 6,8 5,4 4,5
10 10 9,7 9,6 8,8 8,7 8,7 7,6 7,7 7,7 6,6 5,7
11 11 10,5 10,3 9,9 9,6 9,4 8,8 8,8 8,8 7,9 6,8
12 12 11,3 11,1 10,8 10,5 10,2 9,9 9,6 9,4 9,1 8,0
13 13 12,1 11,8 12,0 11,4 11,1 11,0 10,5 10,3 10,3 8,9
14 14 13,0 12,5 13,1 12,3 11,9 12,1 11,5 11,2 11,5 10,0
15 15 13,9 13,3 14,1 13,2 12,8 13,1 12,4 12,0 12,7 11,0
16 16 14,7 14,1 15,2 14,1 13,5 14,3 13,4 12,8 13,9 12,0
17 17 15,5 14,8 16,2 15,0 14,2 15,3 14,3 13,6 15,1 13,0
18 18 16,3 15,5 17,3 15,7 15,0 16,4 15,2 14,4 16,2 14,0
19 19 17,2 16,3 18,4 16,6 15,7 17,5 16,1 15,3 17,4 14,9
20 20 18,0 17,0 19,4 17,4 16,5 18,7 17,0 16,0 18,5 15,9
21 21 18,8 17,7 20,4 18,3 17,2 19,8 17,8 16,7 19,6 16,7
22 22 19,5 18,3 21,4 19,1 18,0 20,9 18,6 17,5 20,9 17,6
23 23 20,3 19,0 22,5 19,9 18,5 21,9 19,4 18,3 22,0 18,6
24 24 21,1 19,7 23,5 20,6 19,3 23,0 20,3 19,0 23,1 19,5
25 25 22,0 20,4 24,5 21,5 20,0 24,0 21,2 19,6 24,2 20,5
26 26 22,8 21,2 25,5 22,3 20,7 25,1 22,0 20,4 25,3 21,2
27 27 23,5 21,8 26,6 23,0 21,3 26,1 22,8 21,1 26,5 22,0
28 28 24,2 22,5 27,6 23,9 22,0 27,2 23,5 21,8 27,8 22,9
29 29 25,0 23,1 28,6 24,6 22,6 28,2 24,3 22,4 28,8 23,6
30 30 25,8 23,6 29,6 25,4 23,3 29,3 25,2 23,1 29,8 24,5
31 31 26,5 24,2 30,8 26,2 23,9 30,3 25,9 23,6 30,8 25,3
32 32 27,2 24,6 31,7 27,0 24,5 31,4 26,7 24,2 32,1 26,2
33 33 28,0 25,2 32,8 27,8 25,1 32,4 27,4 24,9 33,2 27,0
34 34 28,6 25,9 33,9 28,4 25,6 33,5 28,3 25,4 34,5 27,6
35 35 29,5 26,4 34,8 29,1 26,2 34,6 28,9 26,0 35,5 28,4
36 36 30,1 27,0 35,9 29,9 26,8 35,8 29,5 26,3 36,8 29,2
37 37 30,7 27,4 37,0 30,5 27,3 36,9 30,3 26,9 38,0 29,9
38 38 31,4 28,1 38,0 31,2 27,9 38,0 31,0 27,4 39,2 30,6
39 39 32,1 28,6 39,0 32,0 28,4 39,1 31,7 28,0 40,4 31,4
40 40 32,8 29,1 40,0 32,7 28,9 40,2 32,4 28,5 41,6 32,1
41 41 33,4 29,6 41,1 33,3 29,3 — 33,1 29,3 —— 32,8
42 42 34,1 30,1 — 34,0 29,9 — 33.8 29,8 — 33,5
43 43 34,9 30,5 —. 34,8 30,3 —. 34,5 30,3 —' 34,2
44 44 35,5 31,0 — 35,4 30,9 — 35,2 30,9 — 35,1
45 45 36,2 31,4 — 36,1 31,3 — 36,0 31,3 — 36,0
46 46 36,9 31,9 — 36,9 31,9 — 36,7 31,6 — 36,7
47 47 37,6 32,3 — 37,6 32,2 —. 37,5 32,2 — 37,5
48 48 38,4 32,8 38,4 32,7 — 38,3 32,6 — 38.3
49 49 39,2 33,2 —. 39,2 33,2 — 39,1 33,1 —> 39,2
50 50 39,9 33,6 — 40,0 33,5 — 40,0 33,5 — 40,0
174
Таблица 35
одетых людей при условии выполнения легкой работы
в метрах в минуту I t° в
90 150 210
ность воздуха градусах С.
20% 100о/о 50% 20% 100% 50% 20% 100% 50% 20%
0
— — — — — —— — — — — 1
— — — — — — — — — — 2
— — — — — — — — — — 3
0,5 — — — — . — — 4
0,5 — — 1,0 — — — — — 5
1,6 —• 0,5 1,0 — — — — — 6
2,8 0,3 0,5 1,1 — — — — — 7
3,9 1,0 1,9 2,2 — 0,2 0,3 0,8 8
4,9 2,2 2,9 3,3 0,2 1,0 1,4 — 0,3 0,3 9
5,8 3,5 4,2 4,4 1,1 2,1 2,5 0,6 0,9 1,5 10
6,9 4,9 5,3 5,5 2,4 3,3 3,6 0,9 2,2 2,8 11
8,0 6,1 6,3 6,4 3,9 4,5 4,6 2,2 3,3 3,9 12
8,9 7,3 7,4 7,4 5,2 5,7 5,9 3,6 4,5 5,0 13
9,7 8,6 8,5 8,4 6,6 6,9 7,0 5,1 5,8 6,1 14
10,6 10,0 9,8 9,5 8,0 8,1 8,1 6,6 7,0 7,2 15
11,6 11,3 10,7 10,5 9,4 9,1 9,1 8,0 8,2 8,3 16
12,5 12,5 11,7 И,4 10,8 10,2 10,1 9,5 9,5 9,4 17
13,3 13,7 12,7 12,4 11,9 11,3 И,1 10,8 10,5 10,4 18 !
14,2 15,0 13,8 13,4 13,3 12,4 12,1 12,2 П,7 11,4 19
15,1 16,2 14,8 14,4 14,6 13,5 13,1 13,5 12,9 12,4 20
15,8 17,4 15,9 15,1 16,0 14,6 14,1 14,9 13,9 13,4 21
16,7 18,6 16,9 16,0 17,2 15,6 15,0 16,2 15,0 14,4 22
17,5 19,9 17,9 16,7 183 16,6 15,9 17,5 16,1 15,3 23
18,3 21,1 18,8 17,6 19,6 17,8 16,8 18,8 17,1 16,2 24
19,0 22,3 19,6 18,5 21,0 18,8 17,8 20,0 18,1 17,2 25
19,8 23,4 20,6 19,3 22,1 19,7 18,5 21,2 19,1 18,0 26
20,5 24,5 21,5 20,1 23,4 20,8 19,4 22,5 20,1 18,8 27
21,2 25,7 22,4 20,8 24,5 21,6 20,2 23,6 21,0 19,8 28
21,9 26,8 23,3 21,5 25,9 22,5 21,0 24,9 21,9 20,5 29
22,5 28,1 24,1 22,2 27,1 23,4 21,7 26,3 22,8 20,9 30
23,3 29,2 25,0 22,9 28,2 24,3 22,5 27,5 23,8 22,1 31
23,9 30,3 25,8 23,6 29,4 25,1 23,2 28,8 24,6 22,9 32
24,5 31,5 26,5 24,2 30,6 26,0 23,9 30,2 25,5 23,5 33
25,1 32,8 27,3 24,9 32,2 26,7 24,5 31,6 26,4 24,2 34
25,8 34,0 28,1 25,4 33,5 27,5 25,2 33,1 27,2 24,9 35
26,3 35,3 28.8 26,1 35,0 28,3 25,8 34,6 28,0 25,5 36
26,9 36,6 29,6 26,7 36,5 29,1 26,4 36,4 28,9 26,2 37
27,4 38,0 30,3 27,3 38,1 29,9 27,0 — 29,5 26,8 38
28,0 39,4 31,1 27,9 39,9 30,6 27,5 — 30,4 27,3 39
28,5 40,7 31,9 28,3 41,4 31,4 28,1 — 31,2 27,9 40
29,0 42,2 32,6 28,9 — 32,2 28,8 — 32,0 28,4 41
29,5 — 33,3 29,4 — 33,0 29,1 — 32,1 29,0 42
30,1 — 34,1 30,0 — 33,8 29,6 — 33,7 29,4 43
30,5 — 35,0 30,4 — 34,6 30,1 — 34,5 30,0 44
31,0 — 35,8 30,9 — 35,4 30,6 — 35,3 30,5 45
31,4 — 36,5 31,3 — 363 31,1 — 36,2 31,0 46
32,0 — 37,4 31,8 — 37,3 31,6 — 37,2 31,4 47
32,4 — 38,3 32,3 — 38,3 32,1 — 38,3 32,0 48
32,9 — 39,2 32,8 — 40,0 32,7 — 39,5 32,4 49
33,3 — 40,3 33,2 1 40,6 33,1 — — 32,9 1 50 ! 1 1 1
175
Под 'эквивалентно-эффективнойтемпературой (ЭЭТ)
понимают комплексный индекс, при помощи которого можно учесть
одновременное влияние на тепловое ощущение человека трех компонентов
метеорологического фактора: температуры, влажности и скорости движения
воздуха. Следовательно, эквивалентно-эффективные температуры касаются
тех обычных условий внешней среды, когда воздух находится в движении.
Как эффективные, так и эквивалентно-эффективные температуры были
выведены на основании опытов, причем для выведения эффективных темпера-
тур подопытные лица ставились в условия различных комбинаций темпе-
ратуры и влажности в неподвижном воздухе, для выведения же эквивалентно-
эффективных температур подопытные лица подвергались воздействию раз-
личных комбинаций температуры, влажности и скоростей движения воздуха.
Те комбинации температур и влажности (для неподвижного воздуха) и
температур, влажности и скоростей движения воздуха (для воздуха, находя-
щегося в движении), при которых большинство испытуемых лиц давали одина-
ковую оценку своему тепловому ощущению («тепло», «очень тепло», «холодно»,
«очень холодно»), обозначались как одинаковые эффективные и эквивалентно-
эффективные температуры в условных градусах.
Лишь ЭТ при 100% относительной влажности воздуха обозначались чис-
лом, соответствующим показанию сухого термометра, и в этом случае
совпадают с температурами, определяемыми по сухому термометру, во всех
же прочих случаях ЭТ и ЭЭТ отличаются от показаний сухого термометра.
Так, напр., ЭТ в 25° при 100% относительной влажности соответствует тем-
пературе в 25°С по сухому термометру, но та же ЭТ в 25° при 50% относи-
тельной влажности соответствует уже температуре в 29°С по сухому термо-
метру, а ЭЭТ, очень близкая к 25°, именно, 25,1° при 50% относительной
влажности и движении воздуха в 150 м мин., соответствует уже 31°С по су-
хому термометру.
Таким образом, различные комбинации температуры, влажности и движе-
ния воздуха могут давать одинаковый тепловой эффект. Отсюда и само наиме-
нование эффективных температур.
ЭЭТ предназначаются для составления представления о комплексном воз-
действии на тепловое ощущение человека температуры, влажности и движе-
ния воздуха.
Результаты опытов по определению ЭТ и ЭЭТ для средне одетых и произ-
водящих легкую работу людей, сведены в таблицу 35. Таблицей пользуются
для определения ЭТ и ЭЭТ. При этом температура воздуха определяется
по сухому термометру аспирационного психрометра, относительная влаж-
ность — по этому же психрометру, а скорость движения воздуха — по ка-
татермометру.
Нахождение эффективной температуры по таблице производится следую-
щим образом: по первому или последнему вертикальному столбцу находят
соответствующую температуру, а по верхним двум горизонтальным столб-
цам — скорость движения воздуха и относительную влажность. Искомая
эффективная температура лежит на месте перекреста горизонтальной линии,
проходящей через первое данное, и вертикальной линии, проходящей через
два вторых данных. Если заданных условий в таблице нет, то для определения
эффективной температуры прибегают к интерполированию.
Пример. Нужно найти ЭЭТ при условии, что /° воздуха = 18,5° С, относительная
влажность / = 60% и скорость движения воздуха v — 20 м/м.
В приведенной таблице этих данных нет. Находим ЭЭТ для ближайших’меньших данных,
а именно: t° = 18° С, / = 50% и и = 15 м/мин. Для этих данных ЭЭТ равна 15,7.
0 9.5
Интерполируем в отношении /°: — = 0,45; 15,7 + 0,45 = 16,15;
» » » / : = 0,32; 15,7 + 0,32 = 16,02;
176
w —0,5.5
Интерполируем в отношении v:--—— ~—0,17;
Среднее арифметическое полученных трех значений:
15,7 —0,17= 15,53.
16,15 + 16,02 4- 15,53
3
представит ЭЭТ для заданных условий.
Рис. 143. Номограмма для определения ЭТ
и ЭЭТ нормальной шкалы.
По литературным данным, для людей средне одетых и выполняющих лег-
кую работу наиболее комфортное теплоощущение лежит в пределах 17,2—
— 21,7° ЭЭТ. Это и есть так называемая «комфортная зона» ЭЭТ.
Летом интервал для зоны комфорта находится в пределах 17,7—26°; зимою—
в пределах 15,5—23,2° ЭЭТ.
Приведенные выше зоны ком-
форта имеют лишь относитель-
ное значение. Необходимо уста-
навливать комфортные зоны для
разных географических широт,
для людей различно одетых, при
различных метеорологических
условиях с учетом и тепловой
радиации и при выполнении
профессионального труда раз-
личной тяжести.
Маршак установил, что ЭЭТ,
соответствующие зоне комфорта
для жителей средней части
СССР, средне одетых и выпол-
няющих легкую работу, лежат
в пределах 13,5—18,0° по нор-
мальной шкале эффективных
температур. Данные Маршака
должны быть проверены на
большем материале; помимо то-
го, желательно выяснение ком-
фортных зон для указанных
выше условий.
Для определения эффектив-
ных температур, кроме приведен-
нойтаблицы,можнопользоваться
еще соответствующими картами.
Из таких карт большой интерес представляет номограмма, составленная
В. А. Яковенко (рис. 143). Это так называемая термометрическая
карта человека. По ней можно найти значения ЭТ и ЭЭТ нормальной
шкалы по показаниям сухого и влажного термометров в пределах от 0 до
38°С и скоростям движения воздуха в пределах от 0 до 210 м/мин. На карте
имеются две шкалы: справа — для влажного и слева — для сухого термо-
метров. Длинные кривые, проведенные по диагонали карты, представляют
линии скоростей движения воздуха. Поперечные линии, перекрещивающие
кривые скоростей, представляют линии эффективных температур.
Как легко видеть из номограммы, кривые скоростей при повышении тем-
пературы воздуха постепенно приближаются одна к другой и при температуре
тела человека сходятся в одной точке. Эта точка называется нейтраль-
ной, так как в условиях, когда сухой и влажный термометры показывают
температуру человеческого тела, движение воздуха не производит на чело-
века никакого теплового эффекта; тело человека при этих условиях не те-
ряет тепла ни лучеиспусканием, ни теплопроведением, ни испарением влаги
с поверхности.
12 А. И. Бурштейн
177
Ниже нейтральной точки лежит зона охлаждения; движение
воздуха в этой зоне приводит к охлаждению тела.
Выше нейтральной точки линии скоростей движения воздуха располага-
ются в обратном порядке: движение воздуха в этих условиях производит
не охлаждение, а нагревание тела человека, ввиду чего зона эта называется з о-
н о й нагревания. Между шкалами сухого и влажного термометров
находится зона прямого действия влажного воздуха
на человека, т. е. в этих условиях чем выше влажность воздуха, тем
теплее при прочих равных условиях, и наоборот.
Слева от шкалы сухого термометра расположена зона обратного
действия влажного воздуха на человека; в этой зоне
чем выше влажность, тем холоднее, и наоборот.
Для определения по номограмме ЭТ соединяют прямой линией показания
сухого и влажного термометров и находят точку пересечения этой прямой
с кривой, соответствующей нулевой скорости, прочитывая тут ЭТ.
Для определения ЭЭТ точно так же соединяют прямой показания сухого
и влажного термометров и от точки пересечения этой прямой с соответствую-
щей кривой скорости движения воздуха проводят параллельную ближай-
шей поперечной линии, прочитывая ЭЭТ в точке пересечения проведенной
линии с кривой нулевой скорости.
Пример. На представленной карте при показаниях для сухого термометра 24° С и для
влажного 16° С в неподвижном воздухе ЭТ равна 21,3°, при движении же воздуха в 60 м/мин.
ЭЭТ равна 19,5° и т. д.
Кроме описанных выше методов, дающих возможность учесть комплекс-
ное воздействие на тепловое ощущение человека различных компонентов
метеорологического фактора, был предложен еще для этой же цели метод
радиационно-эффективных температур. Этот метод не
нашел однако у нас широкого применения в санитарно-гигиенической прак-
тике и здесь не описывается. Ознакомиться с этим методом можно по работам
проф. Яковенко и его сотрудников.
Лучистая энергия
Под лучистой энергией понимают электромагнитные колебания, распро-
страняющиеся в пространстве: радиоволны, инфракрасные лучи, видимые
лучи, ультрафиолетовые лучи, лучи Рентгена, д лучи и др.
Различные виды лучистой энергии отличаются между собой длиной
волны. Под длиной волны понимают наименьшее расстояние между двумя
точками луча, находящимися в одном состоянии возмущения Ч Длина волны
обозначается греческой буквой X (лямбда).
Под частотой — / понимают число колебаний электро-магнитных волн
в секунду. Частота выражается в герцах, сокращенно Hz.
Зависимость между длиной волны — X, частотой / и скоростью распро-
странения электромагнитной энергии v (ни) выражается следующей формулой:
Х = — .
/
Если учесть, что v в воздухе равно 3 • 108 м., то зависимость между Хи/
в воздушной среде выразится формулой:
. 3 • 108
А =-------.
/
1 Для измерений волн малой и очень малой длины применяют следующие единицы:
микрон (|i), ангстрем (А) и икс. 1 р. = 0,001 мм = 10 000 А = 10 000 000 икс.
178
Из различных видов лучистой энергии ниже рассмотрены лишь те. которые
входят в состав теплового спектра лучистой энергии, а именно: инфракрас-
ные, видимые и ультрафиолетовые лучи.
Инфракрасные лучи имеют длину волны от 343 ц и до 0.76 и- Так
как длинные инфракрасные лучи поглощаются водяными парами, углекислотой
и озоном, то практическое значение имеют лишь инфракрасные лучи с длиной
волны от 4 ц и до 0,76 у..
Видимые лучи имеют длину волны от 0,76 р. и до 0,4 р. Это те вол-
ны, которые вызывают зрительный эффект.
Ультрафиолетовые лучи имеют длину волны от 0,4 р и до
0,004 р. Однако, практическое значение имеют лишь ультрафиолетовые лучи
с длиной волны от 0,4 р и до 0,2 р.
Ниже приведены методы измерения лучистой энергии всего теплового
спектра и отдельно методы измерения ультрафиолетовой радиации.
Актинометрия
Под актинометрией понимают измерение лучистой энергии (от
греч. aktis — луч, metron—мера). В актинометрии принято выражать на-
пряженность потока лучистой энергии в малых калориях в одну минуту на
1 см2 поверхности перпендикулярной к направлению лучей.
Обыкновенно только лучи инфракрасной части спектра называют тепло-
выми. В действительности же все лучи обладают тепловым действием, кото-
рое уменьшается по направлению от инфракрасной к ультрафиолетовой части
спектра, т. е. с уменьшением длины волны.
Тепловое излучение тела является функцией его теплового состояния:
чем выше температура тела, тем больше излучение и наоборот.
Если повышать температуру тела, то количество излучаемой телом энергии
будет увеличиваться, при этом максимум излучения будет перемещаться
к коротким волнам.
При температурах источника ниже 650°С тело излучает одни инфракрас-
ные лучи; при более высоких температурах и до 1590°С вместе с инфракрас-
ными лучами излучаются и видимые лучи и, наконец, при температурах выше
1590°С наряду с инфракрасными и видимыми лучами излучаются и ультра-
фиолетовые лучи.
Лучистая энергия, попадая на какое-либо тело, может частью отразиться,
частью пройти насквозь, частью поглотиться и перейти в другие формы энергии.
Биологическое действие инфракрасных лучей сводится в основном к теп-
ловому воздействию на ткани. Видимые и ультрафиолетовые лучи дают тепло-
вой эффект, но он настолько незначителен, что не имеет существенного зна-
чения.
Ео данным Н. Ф. Галанина \ эффект действия лучистой энергии и вынос-
ливость к ней человека таковы (для радиации 1500°, К = 1,97 у.):
1. Зона 0,4—0,8 калорий — слабая радиация, переносимая неопределенно
долгое время.
2. Зона 0,8—1,5 калорий — умеренная радиация, переносимая 3—5 мин.
3. Зона 1,6—2,3 калорий — средняя радиация, переносимая 40—60 сек.
4. Зона 2,3—3 калорий — повышенная радиация, переносимая 20—30 сек.
5. Зона 3,0—4,0 калории — сильная радиация, переносимая 12—24 сек.
6. Зона 4,0—5,0 калорий — сильная радиация, переносимая 7—10 сек.
7. Зона больше 5,0 калорий — очень сильная радиация, переносимая
2—5 сек.
Принцип, применяемый в актинометрии для измерения напряжения лу-
чистой энергии, заключается в трансформировании лучистой энергии в тепло-
вую; по величине последней судят о напряженности потока лучистой энергии.
1 Н. Ф. Галанин. Труды научно-исследовательской секции охраны труда Ленин-
градского Гот’а т. 1, вып. 1 и 2, 1927 г. и Справочник для санитарного врача, Л. 1940.
179
Приемной частью каждого актинометрического прибора является поверх-
ность, зачерненная платиновой чернью или сажей и практически поглощаю-
щая всю падающую на нее лучистую энергию и трансформирующая ее в теп-
G
Рис. 144. Схема
компенсационного
актинометра.
1 и В — константа-
новые пластинки, 7
термоэлемент. D —
элемент. Е—перемен-
ное сопротивление.
Е— миллиамперметр,
G — гальванометр.
ловую.
В санитарно - гигиенической практике актинометрией приходится зани-
маться преимущественно при применении гелиотерапии и аэротерапии (соларии,
пляжи), а также на производствах, где приходится выяс-
нять напряженность потоков лучистой энергии, исходящей
от нагретых поверхностей (печи).
Ниже описаны наиболее распространенные актинометры.
Компенсационный актинометр (рис. 144)
состоит из двух тонких константановых пластинок А и В.
Одна сторона пластинок зачернена (покрыта слоем копоти).
На незачерненной стороне той и другой пластинки при-
клеены маленькие слюдяные пластинки, а к ним прикле-
ены спаи термоэлемента Т, концы которой- сообщаются
с гальванометром G.
Одна из пластинок (положим, А) подвергается воздей-
ствию лучистой энергии солнца или какого-либо другого
источника, другая (В) остается в тени. В результате не-
равномерного нагревания обеих пластинок в термоэлементе
возникает ток, вызывающий отклонение стрелки гальвано-
метра. Чтобы вернуть стрелку в исходное положение, на-
гревают пластинку В тс ком элемента D. Ток проходит
через переменное сопротивление Е и миллиамперметр F. Изменяя сопротивле-
ние, можно добиться того, что пластинка В окажется нагрета током в такой же
мере, как и пластинка А лучистой энергией. В этот момент стрелка гальвано-
метра будет стоять на нуле.
Напряженность лучистой энергии определяется по формуле:
Q - /л*2,
где Q — размер лучистой энергии в малых калориях на 1 см2 в минуту,
к — коэфициент прибора,
z — сила тока в миллиамперах.
' Пример. Коэфициент прибора 0,012.
Показание миллиамперметра при стоянии
стрелки гальванометра на нуле равно 24.
Отсюда находим:
Q = 0,012 X 2Г = 6,912.
Описанный актинометр принят в
качестве нормального.
Актинометр с биметал-
лической пластинкой
(рис. 145) построен на принципе
неодинакового расширения различных
металлов во время нагревания.
Прибор состоит из биметалличе-
ской пластинки, составленной из ме-
таллов с различным коэфициентом
расширения: меди И платины. Пла- Рис. 145. Актинометр с биметаллической
стинка, служащая приемной частью пластинкой,
прибора, зачернена платиновой чернью
или сажей й поставлена в центр массивного цилиндра из красной меди.
. Чер^з,сделанное в цилиндре окошко пластинка может быть подвергнута
воздействию лучистой энергии. Под влиянием последней один из листков
180-
(медный) биметаллической пластинки с большим коэфициентом расширения
удлиняется в бслыней мере, нежели другой (платиновый); так как биметал-
лическая пластинка одним концом своим закреплена неподвижно, другой
Конец при этом изгибается. К этому концу прикреплен кварцевый волосок.
За отклонением волоска следят в микроскоп с увеличением в 70 раз. В окуляре
микроскопа заключена шкала, по которой производят отсчет. Для пересче-
тов наблюдаемых отклонений в грамм-калории нужно установить сравнитель-
ный коэфициент, сопоставив показания данного актинометра с показаниями
абсолютного, напр., компенсационного актинометра.
Призер. Отсчет по шкале 62,4. Одно деление соответствует 0,0248 кал. Напряжение
лучистой энергии выразится: 0,0248 • 62,4 = 1,54752 кал. на 1 см2 в минуту.
Рис. 146. Актинометр
Араго-Деви-Калитина.
Прибор отличается быстрой восприимчивостью (15—20 сек.), точностью
и прост в применении.
Актинометр Араго-Деви-Кали-
тина (рис. 146) состоит из двух термометров,
установленных вертикально резервуарами кверху.
В ранее применявшихся актинометрах Араго-Деви
резервуары шарообразной формы, причем резервуар
одного из термометров покрыг сажей (черный тер-
мометр), другой остается непокрытым (блестящий
термометр). Оба термометра помещены в стеклян-
ные резервуары, из которых выкачан воздух.
Существенное изменение, внесенное проф.
Калитиным в конструкцию прибора, заклю-
чается в том, что резервуары термометров дела-
ются полушаровыми, причем плоская часть резер-
вуара одного термометра покрывается сажей, а
другого — окисью магния; полушаровые поверх-
ности резервуаров оставляются блестящими. В этой
модификации легче направить приемную поверх-
ность прибора в сторону изучаемого потока лу-
чистой энергии, что особенно :ажно там, где
одновременно имеются несколько источников лу-
чистой энергии (производство).
Направив приемную поверхность прибора в сторону источника лучистой
энергии, выжидают, пока показания термометров не окажутся стабильными,
на что уходит в среднем около 15 минут. Лучистая энергия, падая на поверх-
ность черного резервуара, почти полностью поглощается, тогда как с белой
поверхности значительно отражается. Разность показаний термометров про-
порциональна напряжению определяемой лучистой энергии.
Если сравнить показания данного прибора с абсолютным, определяющим
лучистую энергию в калориях, то можно установить коэфициент описывае-
мого прибора, т. е. найти, скольким калориям соответствует один градус
разности показаний термометров. Зная коэфициент прибора, определяют
напряженность лучистой энергии по формуле:
Q — к (^ /2)>
где Q — напряженность лучистой энергии, выраженная в малых калориях
на 1 см2 в минуту,
к — коэфициент прибора,
t и — температуры черного и белого термометров.
Пример. Коэфициент прибора равен 0,126. Показание черного термометра оказалось
равным 24,6° С, белого — 17,2° С. Отсюда находим:
Q = 0,126(24,6— 17,2) = 0,9324 кал/см2 в минуту.
181
Актинометр Калитина (рис. 147) устроен следующим образом:
на пластинке из инвара1 АВ длиною в 100 мм, шириною в 6 мм и толщиною
в 3 мм на стойках С и D укреплена своими концами пластинка CD из констан-
тана толщиною в 0,03 мм и шириною в 5 мм; пластинка согнута под углом
Рис. 147. Актинометр Калитина.
I — внешний вид, II — схема конструкции: АВ — пластинка из инвара,
С и Д — стойки, СД — пластинка из константана, 8 — ось, Р — стрелка.
в 177° и снаружи зачернена. Если подвергнуть эту пластинку воздействию
лучистой энергии, то она начнет удлиняться. Так как пластинка из инвара
при этом сохраняет свои размеры, константановая пластинка прогибается.
Этот прогиб при помощи стерженьков и цепочки вызывает вращение оси S,
на которую насажена стрелка Р. Отклонение стрелки отсчитывается по
циферблату прибора. При помощи винта,
вызывая смещение стойки £), можно в на-
чале опыта установить стрелку на нуле.
Константановая пластинка защищена сна-
ружи крышкой прибора и только в момент
наблюдения на короткое время подвер-
гается воздействию радиации.
Измерения при помощи актинометра
Калитина производятся следующим об-
разом: записывают показание стрелки по
циферблату прибора; затем, держа прибор
в левой руке, направляют его крышкой
в сторону источника лучистой энергии,
отводят крышку в сторону и подвергают
приемную часть прибора, т. е. константа-
новую пластинку, экспозиции до тех пор,
пока стрелка не примет стабильное поло-
жение, для чего обыкновенно требуется
30 секунд; после этого дела-
ют второй отсчет. Разница
между вторым отсчетом и
। । первым характеризует напря-
ю /2 жение потока лучистой энер-
Рис. 148. Номограмма для актинометра Калитина. гии в единицах шкалы при-
бора.
Как было указано выше, в приборе имеется приспособление (винт), при
псмощи которого можно до опыта установить стрелку на нуль. В этом
случае отсчет по циферблату после экспозиции сразу дает показание
прибора для данного случая.
[.оказания прибора при помощи графика перечисляются на число кало-
рий на 1 см2 в минуту.
1 Инвар — никелевая сталь с 35% никеля; коэфициент расширения 0,0000013.
182
На номограмме (рис. 148) по оси абсцисс отложены деления шкалы прибора,
по оси ординат малые калории. Номограммой пользуются следующим обра-
зом: на оси абсцисс отыскивают число делений шкалы, на которое передви-
нулась стрелка прибора, и из этой точки восстанавливают перпендикуляр
до пересечения с кривой. Из точки пересечения опускают перпендикуляр
на ось ординат и в точке пересечения определяют число малых калорий.
Прибор Калитина нашел у пас распространение в лабораториях промыш-
ленной гигиены благодаря его прочности, чувствительности, простоте для
применения.
Пирометрия
Интенсивность облучения человека нагретой поверхностью или из откры-
того отверстия печи можно определить не только по описанному выше ме-
тоду, но и при помощи следующих формул:
Случай Г.
0,78F
9i =--------
к. кал/м2 час.
R2
Случай 2:
к. кал/м2 час,
где Т — абсолютная температура излучающей поверхности или в печи в
градусах,
Л — излучающая поверхность или площадь излучающего отверстия в м2,
R — расстояние от рабочего до центра излучающей поверхности или
центра отверстия в м,
91 и 9г—интенсивность облучения рабочего в к. калм2 час.
Для решения приведенных формул необходимо кроме других данных
иметь температуру нагретой поверхности печи или температуру в самой печи.
Эти температуры определяются при помощи приборов, носящих название
пирометров.
Метод измерения температур при помощи пирометров носит название
пирометрия. Различают пирометры термоэлектрические и оптические.
Термоэлектрический пирометр состоит из термопары и измерительного
прибора (милливольтметр, чувствительный гальванометр).
Работа термопар основана на следующем явлении: если спаять концы двух
различных металлических проволок и один из спаев нагреть, оставляя дру-
гой холодным, то в цепи появляется электрический ток. Такая цепь носит
название термопары.
ЭДС, возникающая в цепи, зависит от разности температур не нагретого
(«холодного») и нагретого («горячего») спаев. Если температуру одного из
спаев — холодного — поддерживать на определенной высоте, напр. па 0°С, то
ЭДС термопары представит функцию температуры горячего спая. Следова-
тельно, поддерживая температуру одного из спаев термопары на определенной
высоте и, погрузив второй спай в какую-либо нагретую среду, можно по вели-
чине электродвижущей силы в цепи судить о температуре нагретой среды.
Каждую термопару нужно протарировать по милливольтметру и соста-
вить для нее график. На оси абсцисс откладывают температуру, па оси ор-
динат соответствующее число милливольт. Для тарировки* термопары се
холодный спай погружают в воду температуры 0°С (тающий лсд), а горячий
спай в расплавленные или кипящие сплавы химически чистых материалов,
определяя при этом температуру при помощи эталонных термопар. Для изме-
183
рения температур ниже 100—200°С термопары градуируют при помощи мас-
ляных ванн по нормальному термометру.
Нередко пользуются термопарами, снабженными своим измерительным
прибором (гальванометр, милливольтметр), на шкале которого нанесены гра-
дусы температуры.
При тарировке термопары, а также при пользовании ею термопара должна
быть соединена с измерительным прибором; таким образо;м в цепь вводится
третий проводник. Введение третьего проводника не изменяет ЭДС цепи
и, следовательно, не влияет на точность изме-
рения.
На рис. 149-/ представлена схема термо-
пары; здесь А и В—разнородные проводники,
tQ — холодный спай, / — горячий спай. На
том же рис. схемы: 2 и 3 показывают способы
введения в цепь третьего проводника (измери-
тельного прибора) С. Последний может быть
соединен с одной парой неспаянных концов
(схема 2), либо введен в одну из ветвей термо-
пары (схема 3). Воспринимающей частью в
1 —схема термопары, 2 и 3 —схемы ТерМОПаре ИЗ ТрСХ ПРОВОДНИКОВ ЯВЛЯЮТСЯ
введения в термопару третьего провод- КОНЦЫ обозначенные буквами /. Нулевую
температуру требуется обеспечить в точках,
обозначенных буквами /°. При схеме 3 в точках, обозначенных буквами tv
может Сыть любая, но одинаковая температура.
На рис. 150 показано присоединение термопары к измерительному прибору,
представляющему третий проводник. Все, сказанное относительно схем 2 и 3
рис. 149, приложимо и к схемам а и b рис. 150.
Наиболее пригодны в практике термопары: железо-константан для тем-
ператур до 6ОО°С и кратковременно до 800°С, медь-константан до 440°С и
мм и для микротермопар —
1
А
Рис. 150. Схемы присоединения тер-
мопары к измерительному прибору.
а — пулевая температура обеспечивается
в точках 1 и 3; воспринимающая часть в
точке 2; b — нулевая температура обеспе-
чивается в точке 7: в точках 3 и 4 обеспе-
чивается одинаковая(произвольная) темпе-
ратура; воспринимающая часть в точке?.
кратковременно до 500°С и серебро-константан до 600°С. Для более высоких
температур (до 1С00°С) применяются термопары платипа-платинородий. Про-
волока применяется диаметром в 1 мм и толще; в термопарах из драгоценных
металлов диаметр проволоки достигает 0,5—0,6
0,1—0,15 мм.
Проводники термопар (термоэлектроды)
изолируются один от другого при помощи
кварцевых капилляров, стеклянных или
фарфоровых бус, асбестовых обмоток и т. п.
Термоэлектроды помещаются в защитный
чехол, на верхнем конце которого укреп-
лена головка с клеммами для присоеди-
нения проводов.
При измерении термопарами в отсчеты
вводится ряд поправок: 1) на деления
шкалы гальванометра, 2) на сопротивле-
ния проводов и термопары и 3) на тем-
пературу холодного спая. Первые две по-
правки делаются обыкновенно в паспорте
прибора. Третья поправка находится по кривой градуировки термопары.
Здесь, как указано выше, на оси абсцисс отложена температура, на оси орди-
нат—ЭЛ С в m v. Откладывая на оси абсцисс значение температуры холодного
спая, имевшее место во время работы, находим соответствующую ЭДС.
К найденному значению ЭДС прибавляем значение ЭДС, полученное при
измерении искомой температуры и соответственно сумме этих двух величин
находим по оси абсцисс соответствующую температуру.
Иногда термопары со своим постоянным гальванометром тарируются
при той температуре холодного спая, при которой предполагается работать.
184
Описанный тип пирометра имеет большое применение в санитарно-гиги-
енической практике и применяется не только для описанной выше цели, но
и для определения кожной температуры, температуры стен и т. п.
Оптический пирометр применяется для измерения очень высоких тем-
ператур (выше 1500°С) раскаленных поверхностей и сред.
Принцип применения оптического пирометра основан на сравнении
яркости двух освещенных полей. Одно поле освещено раскаленным телом,
другое — источником света (лампой накаливания). Яркость освещения вто-
рого поля можно изменять, регулируя накал нити реостатом. Добившись
одинаковой яркости отсчитывают по шкале гальванометра температуру ис-
следуемой среды.
Прибор (рис. 151) состоит из оптической
нанометра.
Оптическая часть состоит из зрительной
трубы. Объектив трубы 4 сделан подвиж-
ным, чтобы можно было совместить дей-
ствительное изображение измеряемого объ-
екта с нитью лампочки накаливания 5.
Окуляр 7 сделан тоже подвижным для
возможности получения отчетливого изо-
бражения нити лампочки. В окуляр по-
мещен красный светофильтр 6. Накал лам-
почки регулируется реостатом 3. Ток в
лампочку поступает из аккумулятора 8.
Начальная установка гальванометра 7
производится при помощи переключателя
2, корректора на крышке прибора и ре-
остата 3,
Перед измерением температуры тела,
направляют трубу прибора при незажжен-
ной лампе на светлый экран и, передви-
гая окуляр, добиваются наиболее отчет-
ливого видения нити лампы. Затем на-
правляют зрительную трубу на тело, тем-
пература которого подлежит измерению.
Регулируя положение объектива, добива-
ются наибольшей ясности изображения
тела. При этом нить лампы на фоне раскаленного тела представляется
черной. Затем при полностью введенном реостате включают ток и, регули-
руя реостатом накал нити, добиваются такого момента, когда нить стано-
вится совершенно незаметной; в этот момент производят отсчет по шкале
гальванометра, градуированной в температурных градусах.
части, аккумулятора в 2 v и галь-
Рис. 151. Схема оптического пиро-
метра.
/ — гальванометр, ? — переключатель.
3 — реостат. 4 — объектив зрительной тру-
бы 5 —лампочка накаливания, в — крн-
пый светофильтр. 7 — окуляр, 6' —ак:.у-
м ул НТО р.
Ультрафиолетовая радиация
Ультрафиолетовые лучи входят в состав интегрального потока лучистой
энергии, посылаемой солнцем. В некоторых производствах (электросварка,
работа с ртутно-кварцевыми лампами и пр.) ультрафиолетовая радиация до-
стигает больших размеров.
По биологическому эффекту ультрафиолетовые лучи делят на три группы:
1. Лучи с длиной волн от 4000 до 3150 А, группа А;
2. » » » » » 3150 » 2800 » » В;
3. » » » » » короче 2800 » » С.
Лучи первой группы обладают слабым биологическим действием; лучи вто-
рой группы способы вызывать эритему и образование пигмента в коже. Они
обладают также антирахитическим действием; проявляют бактерицидный
185,
эффект. Лучи третьей группы обладают хорошо выраженным бактерицид
ным эффектом. о
Ультрафиолетовые лучи с длиной волны более 2000 А способны вызывал
не только эритему кожи с последующей пигментацией, но и дерматиты, г
также заболевание глаз (фотофтальмия).
По Галанину, за критерий предельно-допустимого количества ультрафио-
летовой радиации берется эритемная доза в 20—30 мгк/см2 мин. Время, не-
обходимое для получения такой дозы зависит от эритемного эквивалента
ультрафиолетового потока. Так при источнике радиации в виде угольной
вольтовой дуги (электросварка) за 90 сек. незащищенный глаз заболевает
фотофтальмией, тогда как при газовом пламени (автогенная сварка) фото-
фтальмия возникает лишь через 30 минут облучения (Галанин).
До сего времени не существует надежного и легко проводимого метода
измерения ультрафиолетовой радиации.
Предложенные методы могут быть разделены на три группы: фотохими-
ческие, фотоэлектрические и термоэлектрические.
Фотохимические методы измерения ультрафиолетовой
радиации основаны на способности ультрафиолетовых лучей разлагать не-
которые вещества. По степени их разложения можно судить об интенсив-
ности ультрафиолетовой радиации. В качестве примера такого метода может
служить разложение йодистого калия под влиянием ультрафиолетовых лучей.
Количество выделившегося свободного иода в единицу времени пропорцио-
нально ультрафиолетовой радиации. Для исследования берут смесь из равных
частей 1% KJ и 5% H2SO4. Облучение реакционной смеси производят
в камере с верхним кварцевым стеклом. Чтобы избежать окисляющего
действия озона, который образуется в атмосфере под влиянием ультрафиоле-
товых лучей, через камеру все время пропускают ток СО2. Выделившийся
иод оттитровывают 0,0025 N раствором тиосульфата натрия. Каждые 10 мл
такого раствора, пошедшие на титрование, принимают за единицу ультра-
фиолетовой радиации, которую обозначают через FL
К фотохимическим относится также литопонный метод, заключающийся
в том, что белая цинковая краска — литопон темнеет под влиянием ульт-
рафиолетовых лучей, вызывающих выделение из краски металлического
цинка.
Литопон состоит из ZnS и BaSO4 с примесью небольших количеств ZnO
и ZnCl2. При облучении ультрафиолетовыми лучами (X — 3500—2800 А)
из краски выделяется металлический Zn и краска темнеет. По степени потем-
нения краски определяют ультрафиолетовую радиацию в мг кал/см2 мин.
По Галанину, опыт проводится следующим образом. Берется навеска в
5—б грамм литопона и смешивается с 16—20 каплями насыщенного раствора
уксуснокислого свинца. Смесь тщательно растирается в ступке. Полученная
паста ровным слоем распределяется по стеклу; поверх пасты помещают квар-
цевую пластинку. Точно такое же количество пасты наносится на второе
стекло и поверх пасты кладут обыкновенное стекло. Второй препарат служит
контролем (обыкновенное стекло пе пропускает ультрафиолетовых лучей).
Перед облучением и после него определяют коэфициент отражения лито-
пона при помощи альбедометра и на основании уменьшения коэфициента
отражения определяют ультрафиолетовую радиацию.
Пример. Положим, что коэфициент отражения литопона до облучения составлял 72,5%,
а после облучения 45,3%. Опыт продолжался 2 минуты. Коэфициент для пересчета % потем-
нения литопона в мг кал/см2 мин. равен 0,6. Контроль не показал потемнения. Нужно опре-
делить размер ультрафиолетовой радиации в мг кал/см2 мин. Учитывая данные опыта, по-
лучаем:
(72,5 — 45,3) • 0,6
— —-----------=8,16 мг кал /см-мин.
2
Паста в данном методе должна готовиться ex tempore; длительность экспо-
зиции не более 5—10 минут.
186
Из фотохимических методов заслуживает также упоминания применение
светочувствительной (аристотопной) хлор-серебряной или бром-серебряной
бумаги, потемнение которой в силу выделения металлического серебра за-
висит на 95,6% от ультрафиолетовой радиации. Степень потемнения бумаги
сравнивают со шкалой потемнения.
Фотоэлектрические методы измерения ультрафиолетовой ра-
диации основаны на измерении фототока в фотоэлементе при облучении его
ультрафиолетовыми лучами. Сила возникающего фототока измеряется чув-
ствительным гальванометром.
Термоэлектрические методы определения ультрафиоле-
товой радиации ввиду сложности необходимой аппаратуры и малого приме-
нения в практике здесь не описаны.
Атмосферное электричество
Земля, как известно, заряжена отрицательно, а атмосфера положительно.
Вокруг земного шара наблюдается электрическое силовое поле, в котором
силовые линии имеют вертикальное направление.
Вопрос о значении атмосферного электричества для человека давно при-
влекает внимание физиологов и гигиенистов (Скворцов, Цунц, Минх и др).
Электрическое состояние атмосферы характеризуется: а) напряжением
электрических сил или так называемым электрическим потенциалом, б) спо-
собностью атмосферы рассеивать электричество и в) ионизацией атмосферы.
Вполне понятно, что между электрическим потенциалом атмосферы и
способностью ее рассеивать электричество существует тесная зависимость.
Определение электрического потенциала атмосферы может быть произве-
дено обыкновенным электрометром
и здесь не описывается.
Ниже приводится методика опре-
деления рассеивания электричества
в атмосфере и ионизации атмо-
сферы.
Рассеивание электричества в атмосфере
Чем электрический потенциал
воздуха выше, тем выше способ-
ность его к рассеиванию электриче-
ства. Электрическое напряжение
воздуха находится в прямой зависи-
мости от барометрического давления
и в обратной зависимости от влаж-
ности и температуры воздуха. Рас-
пыление воды и образование пыли
понижает потенциал. Приближение
циклона вызывает понижение по-
тенциала, а антициклона — повы-
шение.
В этом смысле состояние элек-
трического потенциала атмосферы
может давать основание для пред-
сказания погоды, как и состояние
барометрического давления атмо-
сферы.
Для определения степени рас-
Рис. 152. Прибор для определения степени
рассеивания электричества в воздухе.
А — рассеиватель электричества, В — электроскоп,
С — стержень длн крепления защитного цилиндра.
сеивания электричества в воздухе применяют прибор, изображенный на рис. 152.
Прибор состоит из электроскопа В и рассеивателя Д, который представ-
ляет собой цилиндр высотою в 10 см и шириною в 5 см. Электроскоп прока-
187
либрирован: деления его шкалы соответствуют вольтам и десятым долям во;
Для защиты системы от влияния электрического поля на стержне С ук
ляется открытый книзу цилиндр, более широкий, нежели рассеивателе
закрывающий этот последний (на рисунке не показан). Однако некотор
исследованиями установлено, что определение коэфициента рассеивания :
чительно обесценивается, если применять защитный цилиндр.
Определение способности воздуха рассеивать электричество производи
весьма просто. Рассеиватель заряжают положительно или отрицательно; i
этом листочки электроскопа удаляются один от другого на определен,
число делений шкалы. В результате рассеивания электричества заряд цилищ
уменьшается и листочки электроскопа постепенно сближаются. Наблюдег
производится в течение 15 минут. При этом определяют, на сколько делен
листочки приближаются один к другому. Если прибор заряжен положите;
но, то вывод делают о проводимости воздуха в отношении положительного эле
тричества (а+), и наоборот (а—). Отношение между этими величинами, пре
ставляющее коэфициент рассеивания электричества, обозначают символом (£
Ионизация атмосферы
В атмосферном воздухе постоянно присутствуют положительные и отри
нательные ионы, т. е. материальные частицы, несущие положительный ил.
отрицательный заряд.
Главными факторами, которые поддерживают атмосферу в ионизирован
ном состоянии, служат радиоактивные вещества, находящиеся в земной коре
а также космические атмосферные лучи. Меньшую роль в ионизации
атмосферы играют ультрафиолетовая радиация солнечного спектра, фото-
электрические вещества, которые при облучении выделяют электроны, несу-
щие отрицательный заряд.
Ионы атмосферы возникают в основном в силу распада газовых молекул
и атомов на отрицательно заряженные электроны и остатки, заряженные
таким же количеством положительного электричества.
Свободные электроны захватываются нейтральными молекулами, полу-
чающими благодаря этому отрицательный заряд. Оставшаяся часть молекул
остается положительно заряженной.
Носителями электричества могут быть не только газовые молекулы, но
и всякие материальные частицы воздуха, напр., частицы пыли (дыма).
Ионы атмосферы в зависимости от их заряда делят на положитель-
ные и отрицательные.
Применяется еще классификация ионов по их подвижности: легкие
ионы с подвижностью 1 см/сек. и более тяжелые с подвижностью менее
1 см'сек. Из группы тяжелых ионов выделяют иногда средние ионы
с подвижностью 0,1 —0,01 см сек.
Число легких ионов с положительным зарядом, содержащееся в 1 мл
воздуха, обозначают символом п + , для ионов с отрицательным зарядом при-
нято обозначение п_. Соответственно для тяжелых ионов: Nr и N_.
Сумма ионов одного знака в 1 мл воздуха, т. е. n , + N, и n_ + N_, со-
ставляет удельное число ионов данного знака.
п. +N
Отношение —1------называется коэфициентом у ни полярности
п— —р 7V—
и обозначается символом Q.
В нижних слоях атмосферного воздуха удельное число ионов каждого
знака достигает в среднем 500—700.
Отношение общего числа тяжелых ионов к общему числу легких ионов
в одном и том же объеме незагрязненного возд\ ха не превышает 50, т. е.
188
воздуха, влажностью и
Исследования ряда авторов показали, что ионизационный режим воздуха
оказывает прямое влияние на физиологические функции организма: дыхание,
сердечную деятельность и пр.
Весьма интересны исследования, показавшие зависимость между иониза-
цией атмосферы и горной болезнью, между ионизацией воздуха и его сани-
тарным состоянием.
Установлено изменение ионизации с высотой и, главным образом, преобла-
дание положительных ионов на высотах, где отмечается проявление горной
б< лезни (Ремизов)1.
Минх, изучая ионизационный режим в классных комнатах и больничных
палатах2, мог убедиться, что число легких ионов воздуха находится в обрат-
ном отношении с прозрачностью (запыленностью)
содержанием СО2, то1да как число тяжелых ионов
находится в прямом отношении с перечисленными
факторами. Общее число ионов улюньшается в мо-
мент ухудшения санитарных качеств воздуха.
Лесгафт, изучая ионизационный режим внешней
среды3, мог отметить, что по мере загрязнения воз-
духа количество легких ионов в нем уменьшается,
а количество тяжелых возрастает.
То же отметила Богословская Т. Н. для про-
изводственных помещений 4 *.
Ионизационный режим воздуха
определяется специальными при-
борами, носящими название счет-
чиков ионов. Мы приводим
описания наичаще применяемых
для указанной цели счетчиков ио-
нов Богоявленского и Эберта.
Счетчик ионов Богоявленского
выгодно отличается от нижеопи-
санного счетчика Эберта, позволяя
определять ионы различной подвиж-
ности.
Принцип, на котором по-
строен счетчик, сводится к следу-
ющему: через цилиндрический кон-
денсатор пропускают определенный
объем испытуемого воздуха. Ионы,
несущие положительный или отрицательный заряд, осаждаются на стержне,
присоединенном к нити струнного электрометра. По показанию электрометра
судят об общем заряде, который получил стержень от осевших на него
ионов. Если этот заряд, выраженный в ESE, разделить на элементарный
заряд б * *, который несет один ион, то можно определить общее число ионов
данного заряда.
Определение раздельно легких и тяжелых ионов достигается изменением
напряженности электрического силового поля и быстроты воздушного потока.
Конструкция счетчика. Существенной частью прибора яв-
ляется видоизмененный струнный электрометр (рис. 153-7). В нем металлизи-
Рис. 153. Схема счетчика ионов Богоявлен-
ского.
I. Электрометр; а — металлизированная кварцевая
пять, Ь и с — призмы (ножи). II. Счетчик; Л — вер-
тикальная труба. Б — труба меньшего диаметра,
Ь\ — дополнительная труба. В — эбонитовые кольца,
Г —стержень для передачи напряжения на трубу
Б (Б,). Д — батарея. Е — вентилятор. Ж — мптор,
3 — реостат, И — стержень. Щ —стержень Гоаьшего
диаметра, К — электрометр. Л — контакт, М — ма-
нометр, IV — трубка Вентури.
1 Труды Эльбрусской экспедиции Академии наук СССР и ВИЭМ, 1934—1935.
2 «Советский Врачебный Журнал», № 12,1937; «Гигиена и Санитария», № 2, 1938.
3 Сборник трудов Института коммунальной санитарии и гигиены, т. III, 1939.
4 Лучистая энергия на производстве, Сборник Ленингр. ин-та гигиены труда, 1938.
6 Элементарный заряд равен 4,77.10—10 ESE (абсолютных электростатических единиц).
Под абсолютной электростатической единицей заряда понимают таксе количество электри-
чества, которое действует в пустоте на равное количество электричества на расстоянии I см
с силой одной дины. Один кулон равл! 3 • Ю“9 ESE.
180
рованная кварцевая нить а натянута между двумя металлическими призма
(ножами) b и с. Нить присоединяется к телу, потенциал которого доля
быть измерен, и, находясь в электрическом поле ножей, изгибается в ту и
другую сторону, смотря по знаку сообщенного ей заряда.
Счетчик (рис. 153-//) состоит из вертикальной трубы Д, в которой помещу
более короткая и меньшого диаметра труба Б, изолированная при помои
двух эбонитовых колец В. При помощи стержня Г на внутреннюю тру(
Б (Бг) подается напряжение от батареи Д, другой полюс которой заземлс
через корпус прибора. Труба А снизу переходит в раструб, в котором поме
щается вентилятор Е, приводимый в движение 4—б-вольтовым мотором
скорость которого регулируется при помощи реостата 3. Мотор питается о
аккумулятора.
Внутри вертикальной трубы по длине ее располагается стержень И, изо
лированный при помощи янтарной пробки и присоединенный к описанном}
выше электрометру /(.
Стержень И может быть заменен другим — большего диаметра Иг. Первый
применяется при счете легких ионов, второй — средних и тяжелых. В послед-
нем случае вставляется еще дополнительная труба Bv
При помощи контакта Л каждый из электродов (И и может присоеди-
няться к заземленной наружной трубе А.
Объем воздуха, прошедшего через трубу, определяется по манометру М,
присоединенному к трубке Вентури W, вставляемой в верхнее отверстие
трубы А.
Методика определения. Для определения легкоподвижных
ионов в прибор вставляется тонкий электрод //, на трубуБ подается умерен-
ное напряжение от батареи, состоящей из ртутноцинковых элементов. Одно-
временно включают вентилятор на большую скорость. Легкие ионы, нахо-
дящиеся в воздухе, проходя по трубе, прилипают к стержню, сообщая ему свой
заряд; в то же время тяжелые ионы проходят через трубу наружу, не изменяя
своего направления, так как поле является недостаточно сильным, чтобы
изменить их направление.
Для определения тяжелых ионов вставляются толстый электрод и
дополнительная труба Бг На трубу Б подается напряжение порядка 300
вольт; одновременно включают вентилятор на малую скорость. Указанные
условия обеспечивают осаждение тяжелых ионов. На ножи электрометра
подается напряжение от двух батарей по 72 вольта.
Полярность ионов определяется знаком потенциала, поданного на трубу.
Во время наблюдения следят за быстротой отклонения нити в электро-
метре при помощи секундомера и микроскопа (с 80-кратным увеличением),
снабженного шкалой. По отклонению нити можно сучить о величине заряда,
полученного электродом И или а учтя величину заряда, время потребо-
вавшееся на отклонение нити и объем воздуха, прошедшего через трубу в
единицу времени, можно рассчитать число ионов по следующей формуле:
//ч.ч С Др
п (N) = — -
300Ewt
где N — число тяжелых ионов, п — число легких ионов,
С — электростатическая емкость электрода и электрометра,
Др — изменение потенциала на одно деление шкалы,
Е — элементраный заряд иона—4,77. 10 10 ESE,
W— объем воздуха,прошедшего через трубу, выраженный в см3/сек.
I — время в секундах, в течение которого наблюдается изменение по-
тенциала.
Для учета объема воздуха, прошедшего через трубу, пользуются формулой:
190
где Q — объем воздуха в см8;сек.,
а — коэфициент трубки Вентури,
F — отношение площадей сечений трубки Вентури (максимальной и
минимальной),
й — миллиметры водяного столба,
S — плотность воздуха.
Счетчик ионов Эберта. Принцип, на котором построен счетчик Эберта,
в общем совпадает с принципом, на котором построен счетчик Богоявлен-
ского. Здесь внутренний электрод конденсатора непосредственно соединя-
ется с нитью электрометра. На этот электрод наносят заряд, обратный тому,
который несут определяемые в данном опыте ионы, ввиду чего заряд электрода
по осаждении на него ионов уменьшается. По уменьшению заряда и по коли-
честву воздуха, пропущенного через конденсатор, определяют число ионов
в единице объема воздуха.
Рис. 154. Схема счетчика ионов Эберта.
7. Электрометр: НИ — металлическая коробка. N и N — платинированные кварцевые нити, а — груз,.
FF—футляр, В —янтарная пробна. Z — стержень, несущий нити электрометра. Ц. Счетчик: b —
электрометр, А — конденсатор, а — внутренний электрод конденсатора, И' — часовой механизм, Е —
вентилятор. G — анемометр.
Конструкция счетчика. Существенной частью прибора яв-
ляется электрометр Вульфа (рис. 154-/), состоящий из металли-
ческой коробки НН, в которую пропущены две платинированные кварцевые
нити N и N, соединяющиеся нижними концами, где к ним прикреплен малень-
кий груз а. Коробка помещена в футляр FF, от которого изолирована. Нити
N и N изолированы от коробки НН янтарной пробкой В. Через пробку про-
ходит стержень /., несущий нити электрометра.
В счетчике Эберта (рис. 154-//) электрометр Вульфа b соединен с конден-
сатором А; при этом внутренний электрод конденстатора а укреплен на стер-
жне, несущем нити электрометра. При помощи часового механизма W приво-
дится в движение вентилятор Е, просасывающий воздух через конденсатор.
Объем воздуха, прошедшего через конденсатор, регистрируется маленьким
анемометром G.
Методика определения числа ионов такова: заводят часовой
механизм и, не пуская еще в ход вентилятор, заряжают внутренний элект-
род конденсатора до потенциала порядка 200 вольт. В течение 5—10 минут
наблюдают натуральное рассеивание прибора, т. е. то разряжение, которое
возникает в результате ионизированного состояния воздуха, после чего
пускают в ход вентилятор.
Пустив секундомер и отмерив показания электрометра, включают ане-
мометр.
191.
Минут через 5—15, когда станет заметным спадение нитей электрометра
производят отсчет их положения; одновременно останавливают секундоме[
анемометр и, наконец, вентилятор. После этого вновь наблюдают натураль
ное рассеивание прибора. Число ионов определяется по формуле:
п_ С("о—"1)
300ЕН7 ’
где п — число конов,
с — емкость прибора,
v0—v1— падение потенциала с поправкой на натуральное рассеивание,
щ — объем воздуха в кубических сантиметрах, пропущенного через кон-
денсатор в 1 минуту,
t — число минут пропускания воздуха.
Е — элементарный заряд.
Пример. Пусть емкость прибора равна 25 см. Объем воздуха, пропущенного через
конденсатор в одну минуту, равен 162,5 л. Продолжительность опыта 15 минут. Падение
потенциала во время опыта с поправкой па натуральное рассеивание прибора — 9,8 вольта.
Отсюда находим:
4,47 • 1О~10 162500 • 15 • 300
Счетчик Эберта можно использовать для определения легких и тяжелых
конов, если применить дополнительный конденсатор и учесть поправку,
указанную В. И. Барановым. Это, однако, сложнее, чем в приборе Бого-
явленского.
пыль
Пыль представляет .собой твердое пли жидкое вещество в состоянии тон-
кого раздробления (измельчения). Находясь во взвешенном состоянии в газе
или смеси газов, в частности в воздухе, пыль образует аэрозоль. Пыль,
выпавшая из воздуха и осевшая на той или иной поверхности, образует а э-
р о г е л ь.
К о п о т ь представляет собой вид аэрозоля, в котором диспергированные
частицы состоят из сажи, часто с примесью смолистых веществ.
Исследование пыли сводится к определению размеров и форм пылевых
частиц, веса пыли в единице объема воздуха, числа пылевых частиц в единице
объема и удельного веса пыли. Иногда анализ пополняется еще химическим
исследованием пыли, определением ее взрывчатости и т. п.
Форма пылевых частиц
Для изучения форм и особенностей строения частичек пыли заготовляют
пылевые препараты.
На чистое предметное стекло в центре наносят каплю глицерина, которая
стеклянной палочкой равномерно распределяется тонким слоем по средней
трети предметного стекла, после чего последнее подвергается экспозиции
в течение 5—10 минут. Собрав пыль, покрывают препарат покровным стек-
лом и в таком виде рассматривают под микроскопом. Из различных слоев
воздуха для получения препарата пыли избирают обыкновенно тот, который
находится на высоте от пола, соответствующей росту человека (150—170 см),
чтобы иметь представление о пыли, вдыхаемой человеком. Забор проб про-
изводится и на других высотах, если интересуются распределением пыли
по вертикали.
Препараты пыли подвергаются микроскопическому исследованию сначала
при небольших (100—250), а затем при больших увеличениях (600—750);
при этом обращают внимание на форму пылевых частиц, строение углов,
граней и т. п. Препараты зарисовываются, реже с них снимают микрофото-
граммы.
192
Рис. 155. Шкалы объ-
ективного микрометра
(сверху) и окулярного
микрометра (снизу) в
поле зрения.
столик микроскопа.
Определение размеров пылевых частиц (микрометрия)
Пылевые частицы имеют различные размеры — от долей микрона до не-
скольких тысяч микронов.
В зависимости от размеров различают: собственно пыль, ча-
стицы которой имеют размеры больше 10 ц; такие частицы оседают в спокой-
ном воздухе с возрастающей скоростью; облако, состоящее из частиц
с размерами от 0,1 р. и до 10 р.; такие частицы оседают с постоянной скоростью;
наконец, дым, частицы которого имеют размеры меньше 0,1 р.; такие ча-
стицы пребывают в постоянном броуновском движении.
Частицы с размерами выше 50 у. задерживаются в верхних дыхательных
путях и санитарного значения не имеют; частицы с размерамы от 10 у. до
50 у., хотя и попадают в более глубокие дыхательные пути, но в ткань
легкого не заносятся; наконец, пылинки с размерами ниже 10 у. проникают в
глубочайшие отделы легких — альвеолы и бронхиолы —
и являются наиболее вредными для организма.
Для определения размеров пылевых частиц, как
вообще микроскопических объектов, применяют оку-
лярный и объективный микрометры.
На окулярном микрометре, представляющем собой сте-
клянный кружок, нанесена шкала с делениями (обыкно-
венно от 0 до 50); на объективном микрометре, имеющем
форму обыкновенного предметного стекла, также нане-
сена шкала с делениями на сотые доли мм (10 у.).
Окулярный микрометр помещают на диафрагму оку-
ляра, для чего необходимо отвинтить лупу окуляра
и, пометив окулярный микрометр на диафрагму, вновь
привинтить лупу.
Объективный микрометр помещают на предметный
На рис. 155 показаны шкалы окулярного (внизу) и объективного (сверху)
микрометров в поле зрения. Наложив одну шкалу на другую путем переме-
щения об’ективного микрометра, определяют, сколько целых делений шкалы
окулярного микрометра совпадает с целым числом делений шкалы объек-
тивного микрометра. Это дает возможность установить при данной оптической
системе размер делений шкалы окулярного микрометра, исходя из того,
что каждое деление шкалы объективного микрометра 10 р.
Пример. 12 делений шкалы окулярного микрометра совпадают с одним делением шкалы
объективного микрометра. Это значит, что 12 делений шкалы окулярного микрометра по
10
длине равны 10 р., следовательно, одно деление окулярного микрометра равно ~ 0,83 у.
Рис. 156. Винтовой
ми к ромет ри чески й
окуляр.
а — винт для передвиже-
ния нити, b — винт для
укрепления окуляра.
Установив размер одного деления окулярного микро-
метра, приступают к определению размеров пылинок.
С этой целью, сохраняя ту же оптическую систему
микроскопа, помещают на столик микроскопа препарат
пыли и определяют, сколько делений шкалы окулярного
микрометра укладывается по диаметру той или иной
пылевой частицы. Отсюда находят их размеры.
Пример. Требуется определить наибольший диаметр пылевой
частицы, который соответствует 3 делениям шкалы окулярного
микрометра.
Из предыдущего примера было выяснено, что одно деление
окулярного микрометра равно 0,83 у. Следовательно, наибольший
размер пылевой частицы равен: 0,83 х 3 = 2,49 у.
Для определения размеров микроскопических объектов применяют и дру-
гие приборы, напр. винтовой микрометрический окуляр
(рис. 156). Он представляет собой микрометрический окуляр с постоянной
шкалой; при помощи бокового винта а вправо и влево по шкале передвига-
13. А. И. Бурштейн.
193
стся тонкая нить параллельно черточкам, нанесенным на шкалу. Голов
винта снабжена шкалой со 100 делениями. При полном обороте винта ни
передвигается по окулярной шкале на одно деление, следовательно, од
деление по градуировке винта соответствует 0,01 размера одного делен)
окулярной шкалы. Винт b служит для укрепления окуляра тубуса микр
скопа.
Техника определения размера пылинки при помощи винтового микром
трического окуляра такова: полное число делений, занимаемых пылинко
отсчитывается обычным порядком; по части же деления, занятой пылинко
передвигается при помощи винта нить, при этом производится отсчет чис;
делений головки винта, на которое его пришлось повернуть при измереш
пылинки. Имея данные отсчетов по шкалам окуляра и винта и зная переводин
коэфициент микрометрического окуляра для данной оптической системь
который устанавливается как описано выше, легко определить размер
пылинок.
Пример. Пусть измеряемая пылинка занимает 2 полных деления окулярной шкал
и еще некоторую часть деления; для передвижения нити по этой части пришлось боково
винт повернуть на 23 деления. Положим, что переводный коэфициент для окулярной шкал
равен 2,5. Отсюда заключаем, что размер пылинки равен:
2,5 • 2 4- 0,025 • 23 = 5,575ч.
Винтовой микрометрический окуляр, дающий возможность определят)
размеры пылинок с точностью до тысячных долей микрона, представляе:
ценный прибор для точных микрометрических измерений.
Определение пылевых формул. На основании большого числа промеров
пылевых частиц (300—500) в различных полях зрения можно вывести таь
называемую пылевую формулу, т. е. вычислить процентное со-
держание пылевых частиц того или иного размера в данной пыли.
Пример. В данной пыли требуется определить процентное содержание пылинок с раз-
мерами до 10 р., от 10 до 50 |л и выше 50 ч. Произведено определение размеров 500 пылинок.
При этом оказалось, что пылинок с размерами до 10 ч было 226, с размерами от 10 р. до 50 р.—
148 и с размерами более 50 [i — 126.
Отсюда находим, что в исследуемой пыли:
1П 226-100 ,
пылинок с размерами до 10 pi ——-— = 4э,2%,
» » » от 10 до 50р. = 29,6о/о
126 100
» » » более 50 pi ---------= 25,2%.
500
Весовое определение пыли (гравиметрия)
Весовое определение пыли проводится различно: 1) фильтрацией воз-
духа через плотный или жидкий фильтр с последующим определением от-
фильтрованной пыли путем взвешивания; 2) электрическим осаждением пыли
из воздуха с последующим взвешиванием осажденной пыли; 3) фильтрацией
воздуха через плотный фильтр с последующим определением пыли колори-
метрическим путем и другими методами.
Весовое определение пыли по методу фильтрации с последующим взвешиванием
Фильтрация воздуха с последующим взвешиванием отфильтрованной пыли
представляет собой важнейший метод весового определения пыли. Метод
заключается в том, что определенное количество воздуха просасывают через
плотный фильтр или дестиллированную воду и по количеству пыли, задержан-
ной на фильтре или в воде, делают вывод о весовом содержании пыли в еди-
нице объема воздуха.
194
Рис. 157. Трубка для весового определения
пыли воздуха.
Рис. 158. Бутыльный аспи-
ратор.
Здесь нужно отдельно остановиться на применяемых фильтрах, способах
просасывания воздуха и измерении объемов воздуха, пропущенных через
тот или иной фильтр.
Фильтры. Для весового определения пыли обыкновенно применяют плот*
ные фильтры. В качестве таковых могут служить обыкновенная хлопчато-
бумажная вата, стеклянная ката, бумажные фильтры и т. п. Вату помещают
в тонкостенную стеклянную трубку, длиною в 8—10 см и поперечным сече-
нием в 1,5—2 см2. Длина самого
фильтра (ватного слоя) должна рав-
няться 3—4 см. На рис. 157 изобра-
жены форма трубки и детальные раз-
меры ее1. На границе перехода цилин-
дрической части трубки в коническую
имеются три вдавления для лучшего
удержания ватного фильтра во время
просасывания воздуха. Широкое от-
верстие имеет на наружной стороне шлиф и снабжено пришлифованной кры-
шечкой, чтобы во время транспортировки не потерять часть пыли.
На трубке и крышке обозначается номер фильтра.
Так как хлопчатобумажная вата обладает значительной гигроскопично-
стью, что затрудняет определение постоянного веса фильтра, лучше применять
нередко вместо хлопчатобумажной ваты стеклян-
ную. В этом случае со стороны, направленной к
аспиратору, ватный слой закрывают медной се-
точкой или тонким слоем хлопчатобумажной ваты,
чтобы отдельные волоски стеклянной ваты не уно-
сились током воздуха. Стеклянную трубку вместе
с фильтром высушивают в сушильном шкафу при
105°С до постоянного веса перед просасыванием
и после просасывания воздуха; по разнице в весе
делают вывод о количестве пыли, задержанной на
фильтре.
Пользуясь тем или иным фильтром, нужно про-
верить его задерживательную способность. Это
делается следующим образом: воздух, прошедший
через фильтр, пропускают еще через дестиллиро-
ванную воду, которая затем исследуется на плот-
ный остаток. Если задерживательная способность
фильтра полчая, то дестиллированная вода после
выпаривания не дает плотного остатка.
Приборы для просасывания воздуха. Для про-
сасывания воздуха через фильтр применяют раз-
личные приборы, которые здесь кратко описаны.
Бутыльный аспиратор (рис. 158)
состоит из двух тубулятных бутылей, сообщаю-
щихся, как показано на рисунке, при помощи
резиновой трубки, на которую надевают обыкно-
венный или винтовой зажим. В одну из бутылей
наливается вода, другая остается пустой. Наполненная бутыль помещается
на стол, закрывается пробкой со вставленной в нее стеклянной трубкой,
которая соединяется с фильтром при помощи резиновой трубки. Пустая
бутыль ставится на пол. Если пустить теперь воду из верхней бутыли
в нижнюю, то в верхней бутыли образуется вакуум, что вызывает засасывание
1 См. проект стандартизации методики определения количества пыли в воздухе промыш-
ленных предприятий и размера пылевых частиц, принятый на межинститутском совещании
в Москве 27—30 мая 1933 г.
193
воздуха. Когда верхняя бутыль опорожняется, ее ставят на место нижней
а нижнюю перемещают на стол; это повторяют желаемое число раз. Умножи:
емкость бутылей на число их опорожнений, получают общий объем воздуха
Рис, 159. Аспиратор из
двух баков.
пропущенного через фильтр.
Аспиратор из двух
баков (рис. 159) весьма удо-
бен для транспортировки. Он
состоит из двух металлических
баков с прямоугольными основа-
ниями. Каждый бак снабжен кра-
ном, водомерной трубкой, шка-
лой, указывающей объемы бака
на разных высотах, считая от
верхнего основания, трубкой
для присоединения бака к филь-
тру и закрывающимся крыш-
кой отверстием для вливания
и выливания воды.
Перекидной аспи-
ратор (рис. 160) представ-
ляет собой систему двух цилин-
дров А и А± одинакового объема,
установленных один на другом
и сообщающихся при помощи
трубки а, снабженной краном Ь.
Оба цилиндра имеют водомерные
Рис. 1G0. Перекидной
аспиратор.
А и — цилиндры,
а —трубка, сообщающая
п’.лпндры, Ъ — кран,
С и Сх — водомерные
трубки, е и ci — трубки
для прохождения воз-
духа, / — ось, по кото-
рой цилпн. ры могут
быть повернуты.
трубки С и С\. В оба цилиндра ввинчиваются трубки
г и cv открытые сверху и снизу. У основания трубки
несколько загнуты. По желанию систему цилиндров
можно повернуть по оси /, и тогда нижний цилиндр
оказывается сверху, и наоборот. В верхний цилиндр
Рис. 161. Ручной насос Петри.
А — цилиндр, а и Ъ — трубки для
всасывания и выдувания воздуха, с —
маховик, d — счетчик числа качаний
поршня.
наливают воду. Сообщают верхний конец
ввинчивающейся трубки с фильтром и откры-
вают кран. Вода переходит при этом из верх-
него цилиндра в нижний, а вместо нее в верх-
ний цилиндр поступает водзух, прошедший
через фильтр. Когда верхний цилиндр опорож-
нится, закрывают кран, отключают фильтр
от трубки, поворачивают систему и повто-
ряют все заново. Зная объем цилиндров и
число их опорожнений, определяют количе-
ство воздуха, прошедшего через фильтр.
Воздушные насосы ручные.
Насос Петри (рис. 161) состоит из цилиндра
А с поршнем. У основания цилг.кдра име-
ются приводящая (а) и отводящая (Ь) трубки,
снабженные клапанами. Приводящую трубку
сообщают с фильтром при помощи каучуко-
вой трубки. Движение поршня достигается
вращением маховика С при помощи рукоятки.
Насос снабжен счетчиком числа качаний d,
показывающим число перемещений поршня.
Вычтя из показания счетчика к концу опыта
показание в начале опыта, определяют число
сделанных накачиваний. Помножив это число на емкость цилиндра, получают
объем воздуха, пропущенного через фильтр. Ввиду частой порчи кожаных кла-
панов, необходимо насос Петри время от времени проверять по газовым часам.
106
в
насос системы
в
Рис. 163.
Водоструй-
ный насос.
А — муфта,
В — отводная
трубка, а —
трубка для
соединения о
водопровод-
ным краном,
Ъ — трубка
для выхода
воды.
Рис. 162. Ручной
автора.
Д — цилиндр, С и В — трубки для
всасывания и выдувания воздуха,
Е — шкала насоса, D — подвижная
муфта, F— остановка, К — счетчик.
засасывание воздуха через
Рис. 164. Воздушный ротационный
насос.
А — вращающийся цилиндр насоса,
С и D — отверстия для засасывания
и выдувания воздуха, G — непо юиж-
ный цилиндр насоса, S и St — пла-
стинки, прижимаемые пружиной к
стенкам цилиндра, V(Vt — полости
цилиндра G, изменяющиеся в объеме
при вращении цилиндра А.
Вместо насоса Петри можно пользоваться ручным насосом автора (рис. 162).
Насос состоит из цилиндра А, который у основания имеет два отверстия:
b — всасывающее и с — выбрасывающее. Стержень насоса соединен со шка-
лой Е, на которую надета подвижная муфта Д.
Муфту при помощи винта можно закрепить на
желаемого месте шкалы. Когда пор-
шень поднимают вверх, то муфта
упирается о неподвижную пластин-
ку F, благодаря чему положение
муфты регулирует высоту подня-
тия поршня и, следовательно, объем
вводимого и выводимого воздуха.
Насос снабжен счетчиком К. Кла-
паны насоса шариковые металли-
ческие.
Водоструйный насос
(рис. 163) готовится из стекла
или металла. Он состоит из муфты
А, имеющей приводную трубку
В. В муфту вделана трубка а,
суженная книзу; трубка эта соеди-
няется с водопроводным краном.
Снизу 4»- муфту вделана трубка Ь,
переходящая у основания в шаро-
образное расширение, от которого
отходит небольшая отводящая
трубка. На эту последнюю наде-
вается каучуковая трубка, свобод-
ный конец которой помещают в раковину отлива.
Если из крана пустить воду, то, проходя через
узкое отверстие трубки а под давлением в 2—3
атмосферы1, вода захватывает воздух, унося его из муфты наружу. В муфте
возникает некоторый вакуум, что вызывает
трубку В, которая соединяется с фильтром.
Воздушные насосы с элек-
трической тягой. На рис. 164 изоб-
ражен воздушный ротационный насос, приво-
димый в действие мотором, работающим от
сети. Неподвижный полый цилиндр G пересе-
кается эксцентрично осью вращающегося ци-
линдра А. Последний постоянно касается обра-
зующей цилиндра G. По диаметральной плос-
кости цилиндр А рассечен на две части, между
которыми помещены пластинки S и прижи-
маемые пружиной к стенкам цилиндра. В ци-
линдре G имеются два отверстия С и D, слу-
жащие для всасывания и выбрасывания воз-
духа. При вращении цилиндра А объем Vx
увеличивается, а объем Va уменьшается, бла-
годаря чему газ засасывается в С и выбра-
сывается в D.
Очень часто для просасывания воздуха
пользуются обыкновенными пылесосами,
приводится во вращение при помощи мотора.
Приборы для определения объема просасываемого воздуха. Выше было
где центробежный вентилятор
1 Обычное давление в водопроводной сети.
197
указано, как определяется объем просасываемого воздуха, если поль
ауются бутыльным или перекидным аспиратором либо насосами с оп
ределенной емкостью цилиндра, снабженными счетчиками. Если же поль
Рис. 165. Г азовыс часы (схема).
Л — цилиндр, В — барабан, а — отверстие
для входа воздуха, b — отверстие для выхода
воздуха.
зоваться воздушным насосом други:
типов, пылесосом или водоструйныд
насосом, то для определения объеме
пропущенного через фильтр воздухе
пользуются газовыми часами или рео-
метрами.
Газовые часы бывают двух
видов: сухие и гидравлические. Большей
точностью отличаются гидравлические
часы (рис. 165). Они состоят из метал-
лического цилиндра Д, заполняемого
несколько более чем на половину водой.
В цилиндре помещается металлический
барабан В, разделенный перегородками
на четыре камеры (секторы). Барабан
может вращаться вокруг горизонтальной оси. В каждой перегородке имеются
два отверстия: одно — вблизи оси для вхожде-
ния всасываемого воздуха, другое — у окруж-
ности барабана для выхождения воздуха в ци-
линдр, а отсюда наружу. На рис. 166 показан
внешний вид часов. Из рисунка видно, что в
цилиндре имеются отверстия для вхождения (а)
и выхождения (Ь) воздуха, вливания (на рис. не
показано) и выливания (К) воды. При вливании
воды в часы открывают отверстия для вливания
и выливания и заполняют часы водой, пока через
отверстие К не начнет вытекать вода. Газовые
часы снабжаются ватерпасом и винтами у осно-
вания В, чтобы можно было установить часы в
горизонтальном положении; иногда при часах
имеется и манометр с для определения давления,
Рис. 166. Газовые часы
(внешний вид).
а — трубка для поступления воз-
духа, Ь — трубка для выхода вов-
духа, с — манометр, d — винт для
регулирования скорости прохожде-
ния воздуха, С — циферблат, В —
основание часов.
при котором ведется проса-
сывание. Воздух поступает в
газовые часы по трубке, от-
крывающейся в барабан;
отсюда воздух направляется
в ближайший сектор, выте-
сняет отсюда воду, нарушает
этим равновесие барабана и
заставляет его вращаться.
Ось барабана соединена при
помощи бесконечного винта и
шестерни с системой стрелок,
вращающихся по своим ци-
ферблатам (С). На основании
показаний стрелок до опыта
и к концу его заключают о
Рис 167 Реометр количестве воздуха, прошедшего через часы за время опыта,
а и ь— Сш я Скорость прохождения воздуха можно регулировать вин-
в 1юленаГрТометрИа" том d. Время от времени газовые часы должны проверяться
е"—'откидная трубка*, П0 ГрЭДуирОВаННОМу ЭСПИраТОру.
с —диафрагма. Реометр (рис. 167) представляет собой U-образную
трубку с двумя расширениями а и Ь. Верхние концы обоих
колен сообщаются с горизонтальной трубкой, имеющей в средней части
сужение с, называемое диафрагмой. Отверстие d — приводное, отверстие
198
Рис. 168.
Реометр с тор-
мозящим капил-
ляром.
г — отводное. В реометр вводят небольшое количество окрашенной жидкости
(воды, керосина и т. п.). Для окрашивания керосина можно пользоваться
одной из следующих красок: Судан, шарлах красный, алькана и др. При про-
сасывании воздуха в правом колене возникает пониженное давление, в левом
же, наоборот, повышенное, благодаря чему жидкость в левом
колене опускается, а в правом подымается. Разность уровней
жидкости в обоих коленах тем больше, чем быстрее идет
просасывание воздуха. Калибровка реометра осуществляется
при помощи газовых часов и аспиратора. Цифры, нанесенные
на шкале реометра, указывают, какое количество воздуха
в литрах или миллилитрах проходит через реометр в минуту
при данном стоянии уровня жидкости в правом колене.
Реометр укреплен на штативе с тяжелым основанием.
В санитарной практике, кроме описанного реометра с
диафрагмой, применяют еще реометр без диафрагмы (рис. 168).
Он состоит из тормозящего капилляра, соединенного при
помощи каучуковых трубок с обыкновенным манометром,
нижняяпетля которого делаете я также из капиллярной трубки.
В этом реометре зависимость между быстротой просасывания
воздуха и разницей высот стояния уровней жидкости в обоих коленах выра-
жается прямой линией, в то время как в обыкновенном реометре с диафраг-
мой зависимость эта параболическая.
Методика весового определения пыли при помощи фильтрации
На рис. 169 изображена установка для определения весовой концентрации
пыли воздуха с ватным фильтром А, реометром В и пылесосом малого разме-
ра С.
Рис. 169. Установка для определения весовой концентрации
пыли.
А — ватный фильтр, В — реометр, С — пылесос.
На основании количества пыли, задержанной фильтром, и объема пропу-
щенного через фильтр воздуха делают вывод о количестве пыли в единице
объема воздуха.
Пример. При исследовании запыленности воздуха в угольной шахте через ватный фильтр
было пропущено 200 л воздуха. Постоянный вес трубки вместе с фильтром до опыта состав-
лял 12,458 г, после опыта — 12,482 г. Нужно определить весовую концентрацию пыли
в воздухе шахты.
Всего на фильтре задержано 12,482 — 12,458 = 0,024 г пыли, что соответствует
0,024 . 1000
----------= 0,08 г/м3 или 80 мг/м3.
199
Весовое определение пыли при помощи электрической преципитации
Принцип метода заключается в том, что воздух вместе с содер
щейся в нем пылью просасывают через трубку, в которой создается элею
ческое поле высокого потенциала. Пылевые частицы, получая электричек
заряд, выпадают на так называемом собирающем или пассивном электр
и могут быть взвешены (электрофильтрация).
Приборы. Здесь описаны электрофильтр или электрический преципита:
и электрофильтр Предводителева — Блинова.
Электрический преципитатор, представленный на рис. Г
состоит из стеклянной или кварцевой трубки а длиною в 26 см с двумя боь
выми тубусами: один из них d— для поступления, другой е — для выхо
Рис. 170. Электрический преципитатор пыли.
а — трубка, через которую просасывается воздух, b — преципитирующий электрод, с — металличе-
ская фольга, d — тубус для поступления воздуха, е — тубус для выхода воздуха, h — трансформатор,
g — фен для просасывания воздуха, / — реометр.
воздуха. Трубка снаружи обернута металлической (алюминиевой) фольгою
с и является собирающим или пассивным электродом. Этот электрод
заземляется. Концы трубки закрыты каучуковыми пробками. Через пробки
протянут тонкий металлический прут &, являющийся излучающим,
преципитирующим или активным электродом. Прут изо-
лирован от пробок посредством трубок из непроводящего материала. К пруту
подводится переменный ток, получаемый от четырех щелочных аккумуля-
торных батарей и проводимый через индукционный аппарат, либо от сети К
через трансформатор Л.
При прохождении тока достаточно высокого напряжения через излучаю-
щий электрод из него начинают выделяться электроны, которые ионизируют
проходящий слой воздуха. Так как ток применяется переменный, с излучаю-
щего электрода стекает то положительное, то отрицательное электричество.
Проходящие вместе с воздухом частицы пыли заряжаются попеременно
то положительными, то отрицательными зарядами. Разноименно заряженные
пылинки, взаимно притягиваясь, образуют более или менее крупные аггре-
гаты. Благодаря ионному давлению, которое частички испытывают со стороны
излучающего электрода, они относятся к собирающему электроду, где и осе-
дают.
В описанном методе воздух просасывается через преципитатор при по-
мощи фена g, приводимого в движение маленьким мотором. Последний пита-
200
ется током от тех же батарей. Количество пропущенного воздуха измеряется
реометром /.
Чтобы иметь возможность взвешивать и изучать под микроскопом соб-
ранную пыль, вводят внутрь стеклянной трубки целлулоидные цилин-
дрики, плотно пристающие к стенке трубки.
По окончании просасывания воздуха целлулоидная фольга извлекается
вместе с осевшей на ней пылью. Взвешивая фольгу до и после опыта, можно
определить количество выпавшей пыли. Если вырезать полоску фольги по
образующей, то можно посредством амилацетата монтировать ее на предмет-
ном стекле. Несколько капель амилацетата помещают на предметном стекле,
сверху кладут полоску целлулоида, расправляют ее и нагревают предметное
стекло на небольшом пламени. Полоска гладко пристает к стеклу. Пылинки
Рис. 171. Электрический фильтр Предводи-
телева—Блинова.
I. Фильтр; А и В — два цилиндра, С — отвер-
стие в крышке верхнего цилиндра, D — игла,
F — эбонитовый диск, Е — металлическая пла-
стинка, L — пружина, К — клемма, II. Приспо-
собление для взвешивания осевшей пыли; В и
Вх — диски из аллюминиевой фольги.
А — слюдяная пластинка.
можно измерять и сосчитывать.
Скорость просасывания воздуха
не должна превышать 9 л/мин. Если
использовать ток от сети и ввести в
цепь трансформатор, получая высо-
кое напряжение (от 5 до 20 тысяч
вольт), то скорость просасывания
воздуха может быть доведена до
50 л/мин.
Электрический фильтр
Предводителева — Блинов а
(рис. 171) состоит из двух цилиндров
А и В, надевающихся один на другой.
Стеклянный цилиндр А закрыт сверху
эбонитовой крышкой, в центре кото-
рой имеется круглое отверстие О диа-
метром в 1 мм. В отверстие вставлена
острая игла D толщиной приблизи-
тельно в 0,5 мм. Между острием иглы
и стенками отверстия имеется неболь-
шой кольцевидный промежуток. Ци-
линдр В сделан изэбонита; сверху в него вставлен эбонитовый диск F, диаметром
в 5 см. В диск вставлена металлическая пластинка Е диаметром в 1,6 см,
которая при помощи пружины L соединяется с клеммою /(. Для прохожде-
ния воздуха из верхнего цилиндра в нижний имеется ряд симметрично распо-
ложенных отверстий между стенками диска F и цилиндра В. На иглу D и
диск Е накладывается высокая разность потенциалов, для чего пользуются
током высокого напряжения (12 тысяч вольт). Тубус эбонитового цилиндра
В соединяют с аспиратором (водоструйный насос), поместив между аспира-
тором и тубусом прибор для определения скорости засасывания воздуха
(реометр).
При засасывании пыльного воздуха частички пыли, проходя через от-
верстие С, получают заряд у острия иглы и под влиянием электрического
поля относятся и осаждаются на диске F в виде колец или пятен. На диске
F пылинки могут быть подвергнуты измерению и сосчитыванию под микро-
скопом в отраженном свете.
Для взвешивания осевшей пыли система, на которой происходит осаждение
пыли, должна быть сделана более легкой (рис. 161-11). Ее можно приготовить
из очень тонкой слюдяной пластинки А и двух дисков В и из тончайшей
алюминевой фольги, соединенных между собой полоской из фольги.
Такую систему взвешивают до и после опыта на обыкновенных микровесах
или весах Предводителева и Витта1.
1 В. Блинов, «Гигиена, безопасность и патология труда», № 7, 1930; Предво-
ди т е л е в и Витт, «Прикладная физика», 1928.
201
Рис. 172. Держатель для
бумажных фильтров автора.
А — металлическая трубка,
В — гайка, С — втулка,
D — кольцевая пронладна.
Колориметрический метод определения весовой концентрации копоти
Принцип метода заключается в том, что определенный объем во
духа просасывают через бумажный фильтр и по степени потемнения фильт[
делают заключение о весовой концентрации копоти в воздухе, пользуяс
при этом таблицей эталонов.
Бумажные фильтры готовятся из фильтровальной бумаги
гладкой, а не шагреневой поверхностью. Для этой цели могут быть исполь
зованы беззольные фильтры химико-технического института им. Менделеева
Диаметр пор фильтровальной бумаги равен в среднем 1,5—3 ц; частичю
копоти имеют размер от 0,25 до 1 р, т. е. меньше диаметра пор. Однако, ввиду
небольшого отрицательного давления, развиваемого при просасывании воз-
духа, частички копоти не проходят через фильтр; помимо того, первые же
порции копоти забивают поры фильтра, уменьшая проходимость последнего
для твердых частиц.
Из фильтровальной бумаги вырезают кружки
соответственно применяемому держателю фильтров.
Нередко для этой цели пользуются охотничьими
высечками, при помощи которых из бумаги высе-
кают кружки соответствующего диаметра.
Держатели для фильтров должны
герметически зажимать фильтр. На рис. 172 изо-
бражен держатель, предложенный автором. Он со-
стоит из металлической трубки А длиною в 10 см
и с внутренним диаметром в 1 ,б см. Трубка имеет на
одном конце винтовую нарезку, на которую навин-
чивается гайка В. В гайку перед тем, как навинчи-
вать ее на трубку, вставляют втулку С, на которую
помещают вырезанное из тонкого картона колечко
D. Наружный диаметр кольцевой прокладки — 2 см, внутренний — 1,6 см,
ширина тела кольца — 2 мм. На кольцевую прокладку помещают бумажный
фильтр диаметром в 2 см. Поверх фильтра помещают вторую кольцевую про-
кладку, после чего фильтр присоединяют к трубке при помощи гайки.
Таблица эталонов готовится следующим образом. В маленькую
лампочку в качестве горючего наливают скипидар и зажигают фитиль. Опро-
кинув над коптящим пламенем фарфоровую чашку, получают быстро большое
количество копоти. Ее высушивают при 105°С до постоянного веса, а затем
приготовляют суспензию в хлороформе.
Для приготовления суспензии на определенный объем хлороформа берут
точную навеску сажи. Из полученной суспензии при помощи пипетки от-
бирают определенные объемы и наносят на кружочки ватманской бумаги,
стараясь при этом равномерно распределить взятую порцию по всему кружоч-
ку. На каждый следующий кружок наносят возрастающее количество
суспензии, получая, таким образом, шкалу стандартов. Приготовив ряд
эталонов, начиная с 0,0025 мг и до 0,0100 мг, с интервалами в 0,0025 мг,
а затем до 0,0500 с интервалом в 0,0050 мг, получают хорошую рабочую
шкалу, если диаметры кружочков равны в среднем 1,5 см.
Методика. Смонтировав фильтр в держатель, как описано выше,
просасывают через фильтр определенный объем воздуха, а затем сравнивают
полученный препарат со шкалою стандартов, подыскивая тот эталон, ко-
торый по окраске ближе других подходит к полученному препарату. Если
диаметры эталонных кружков одинаковы, то принимают, что количество
копоти на опытном кружке равно количеству копоти на соответствующем
эталоне. При разных диаметрах вносят в расчеты соответствующую поправку.
Пример. При исследовании задымленности воздуха в кузнечном цехе через фильтр
диаметром в 1,5 см пропущено 200 л воздуха. Полученная окраска фильтра соответствовала
эталону, на котором количество копоти равно 0,025 мг. Диаметр эталонного кружка, как
202
и опытного, равен 1,5 см. Отсюда можно сделать вывод, что концентрация копоти в воздухе
данного кузнечного цеха равна:
0,025 • 1000
200
= 0,125 мг/м3.
Определение числа пылевых частиц в воздухе (кониметрия)
Число пылевых частиц в единице объема воздуха определяют двояко:
1) из определенного объема воздуха осаждают взвешенные в нем пылевые
частицы, которые затем подсчитываются, и 2) в изолированном объеме воздуха
дают возможность пылевым частицам выпадать и подсчитывают число выпав-
ших пылевых частиц.
Счет пылевых частиц, взвешенных в воздухе
Принцип метода заключается в том, что определенный объем исследуе-
мого воздуха подвергают увлажнению, а затем адиабатическому расшире-
нию, вследствие чего воздух
охлаждается; влага конденси-
руется на пылевых частицах,
которые, благодаря удару,
прилипают к покровному
стеклышку, а затем подсчи-
тываются в микроскопе.
Прибор, применяемый для
указанной цели, носит назва-
ние счетчика взве-
шенной пыли Овенса
(рис. 173). Он состоит из трех
частей: корпуса А, увлажни-
тельной камеры В и насоса
С на 50 мл. Корпус прибора
в свою очередь слагается из
трех частей: крышки а, коль-
ца b и основания с. Все эти
части соединяются при по-
мощи винтовых нарезов и
А — корпус, В — увлажнительная камера, С — насос,
а, Ъ и с — крышка, кольцо и основание корпуса, d — щель
для прохождения воздуха, / — покровное стеклышко,
g — пружина, прижимающая покровное стеклышко. I —
желобовидные углубления, к — пружинящее колечко, в —
отверстие для привинчивания насоса, тип — нлапапы
насоса.
кожаных прокладок. Когда
части собраны, в центре кор-
пуса образуется небольшая
цилиндрическая камера (кон-
денсационная или охлажда-
ющая) диаметром в 10 мм
и высотой в 1 мм. Нижним основанием или дном камеры служат две металли-
ческие полулунные пластинки, между которыми имеется щель d длиной в 10мм
и шириной в 0.1 мм. Боковой стенкой камеры служит внутренняя поверхность
металлического колечка, укрепленного в части с; в колечке имеются два
желобовидных углубления /, расположенных перпендикулярно к щели (см.
деталь рис. 173). Верхним основанием камеры служит круглое покровное
стеклышко /, которое при помощи пружинки g, укрепленной у крышки а.
прижимается к верхней поверхности металлического колечка.
Увлажнительная трубка прибора представляет металлический рукав, вы-
ложенный по внутренней поверхности фильтровальной бумагой, которая
перед забором пробы увлажняется водой. Посредством пружинящего колеч-
ка к фильтровальная бумага прижимается к внутренней поверхности трубки.
С одной стороны трубка привинчивается к корпусу прибора, с другой — оста-
ется открытой.
203
Рис. 174.
Препарат
пыли, мон-
тированный
на предмет-
ном стекле.
Насос прибора емкостью в 50 мл ввинчивается в специальное отверс
е, имеющееся в части b корпуса прибора. Это отверстие находится на уро.
охлаждающей камеры. Насос снабжен двумя резиновыми клапанами т и
Методика. Отвинчивают от корпуса прибора увлажнительную тру(
и, не вынимая фильтровальной бумаги, смачивают ее водой посредством i
петки по возможности равномерно, но не слишком обильно; затем оп$
привинчивают увлажнительную трубку. Держа прибор одной рукой за нас
другой рукой производят быстро несколько качаний поршня, чтобы ввес
в увлажнительную камеру воздух, подлежащий исследованию. Затем быст;
отвинчивают крышку корпуса прибора и вкладывают в прибор предварител
но приготовленное, тщательно очищенное круглое покровное стеклыш!
так, чтобы оно легло на кольцо, ограничивающее с бою
охлаждающую камеру.
Привинтив крышку прибора, производят по возможност
быстро одно или несколько качаний поршня насоса до отказ;
При этом следует держать прибор за насос. Если держать ег
за увлажнительную камеру, то последняя нагревается, происхс
дит сильное испарение воды, вследствие чего препарат пыл
получается неясный. При полном качании поршня засасываете.
50 мл испытуемого воздуха. Последний, пройдя через увлажни
тельную камеру, насыщается здесь водой, а прорвавшись чере:
щель основания камеры и поступив в самую камеру, подвер
гается адиабатическому расширению и охлаждению. Влага
конденсируется на пылинках, и образовавшиеся ядра конден-
сации, встретив на своем пути экран — покровное стеклышко,
прилипают к нему. Обеспыленный воздух далее через желобо-
видные каналы поступает в насос, откуда при обратном ходе
поршня выходит через отверстие в основании поршня наружу.
Пылинки воздуха, прилипнув к покровному стеклышку,
по испарении влаги, дают сухой препарат пыли в виде пыле-
вой дорожки длиною приблизительно в 1 см, видной
и макроскопически. Готовый препарат пыли извлекается из
прибора следующим образом: отвинчивают осторожно крышку прибора
и, прикрыв образующееся отверстие ладонью одной руки, поворачивают
другой рукою прибор так, чтобы покровное стекло выпало на ладонь. Не
касаясь пальцами пылевой дорожки, которая обращена кверху, берут осто-
рожно покровное стеклышко, поворачивают и укрепляют его на картонном
колечке, наклеенном на предметное стекло, благодаря чему пылевая дорожка
не касается предметного стекла. Наружный диаметр кольца — 2 см, внутрен-
ний — несколько больше 1 см, ширина тела кольца — 2,5—3 мм. Описан-
ные колечки легко изготовляются из подходящего материала (ватманская
бумага, бристольский картон и т. п.) при помощи охотничьих высечек соответ-
ствующего размера. Покровное стеклышко осторожно прикрепляется к колеч-
ку при помощи клея так, чтобы пылевая дорожка шла параллельно попе-
речной оси предметного стекла. На предметное стекло наклеивается этикетка
с указанием даты, места забора пробы и количества воздуха, из которого
получен препарат. Монтированный таким образом препарат показан на
рис. 174.
Чтобы подсчитать пылинки на полученном препарате, пользуются микро-
скопом, в окуляр которого помечают сетчатый окулярный микрометр. По-
следний представляет собой круглую пластинку, на которой нанесена сеточка
площадью в один квадратный сантиметр, разделенный на 100 мм2.
Сначала находят дорожку при малом увеличении, а затем переходят на
иммерсионную систему, при которой и производится подсчет (вид поля зре-
ния показан на рис. 175). Подсчитывают число пылинок в нескольких попе-
речных полосках, взятых в разных местах по длине пылевой дорожки, и на-
ходят среднее арифметическое для поперечной полоски.
£04
Число пылинок в 1 мл воздуха определяется по формуле:
вде N — число пылинок в 1 мл воздуха,
п — среднее число пылинок в одной поперечной полоске по сетчатому
окулярному микрометру,
s — число поперечных полосок сетчатого окулярного микрометра, про-
ходящих по длине всей дорожки,
с — число миллилитров воздуха, пропущенных через прибор при взятии
пробы.
Рис. 175. Микроскопический вид препарата пыли.
а — при малом увеличении, Ъ — при большом увеличении.
Без большой погрешности можно считать, что длина дорожки представ-
ляет величину постоянную, равную 10 000 ц. Если рассмативание препарата
ведется с помощью иммерсионной системы при окуляре № 4 (т. е. при увели-
чении в 1000 раз), то ширина поперечной полоски в поле зрения оказывается
равной 15 р и потому s = -.— =667. Если к томуже принять количество воз-
1 э
духа, взятого для исследования, равным 50 мл, то у = yj- «= 13,3. Следо-
вательно, для определения числа пылинок в 1 мл воздуха нужно среднее
число пылинок одной поперечной полоски помножить на 13,3 (если препарат
рассматривать при увеличении в 1000 раз).
Пример. Среднее число пылинок в одной поперечной полоске оказалось равным 540.
Счет произведен при иммерсионной системе и окуляре № 4. Число миллилитров воздуха
равно 50. Отсюда заключаем, что число пылинок в 1 мл воздуха равно 540 х 13,3 = 7182.
Счет пылевых частиц, выпадающих из воздуха
Принцип метода сводится к тому, что из испытуемого воздуха изо-
лируется определенный объем; оседающие пылинки собираются на покров-
ных стеклах и здесь затем подсчитываются при помощи микроскопа и сет-
чатого окулярного микрометра. Зная размер поверхности покровного стекла,
на котором выпадали пылинки, и высоту воздушного слоя над ним, можно
сделать вывод относительно того объема воздуха, из какого выпали подсчи-
танные пылинки, а отсюда сделать вывод относительно числа пылинок, вы-
падающих из единицы объема воздуха.
Прибор Овенсадля счета оседающей пыл и (рис. 176)
состоит из тяжелой подставки А, на которую помещается цилиндрический
сосуд G, открытый с обоих концов. Чтобы придать устойчивость сосуду, в
подставке имеется циркулярное углубление N, в которое вставляется нижний
край цилиндра. Сверху цилиндр закрывается съемной крышкой Н. На ниж-
ней поверхности подставки находится углубление, в которое вставляется ба-
205
Рис. 176. Счетчик оседающей
пыли Овенса.
А — подставка, G — цилиндр, N —
циркулярное углубление в под-
ставке для цилиндра, Н — крышка
цилиндра, К — барабан, В — винт,
F — гайка, D — стерженьки для
перемещения барабана, С — отвер-
стие н барабане, Е — покровное
стекло, J — остановка.
крывают его крышкой так,
рабан К. Барабан насажен на винт В и поддерживается гайкой F. От барабана
по окружности его отходят на равном расстоянии один от другого шесть стер-
женьков D, при помощи которых можно перемещать барабан вокруг винта
В, как вокруг оси. Против каждого стерженька в барабане проделано круг-
лое отверстие С; над каждым отверстием по верхней поверхности барабана
сделано углубление несколько большего диаметра, нежели отверстие; в это
углубление закладывается покровное стекло Е. При вращении барабана
покровные стекла подводятся под круглое отверстие, сделанное в подставке
прибора и расположенное центрально по отношению к оси сосуда G (на ри-
сунке оно видно над покровным стеклом Е). Через это отверстие на покров-
ное стекло выпадает пыль из сосуда G.
В приборе имеется остановка у, ограничи-
вающая движение барабана. Она устроена так,
что при вращении барабана стерженьки цепля-
ются за остановку, что служит указанием на
то, что очередное отверстие барабана, вместе
с покровным стеклом над ним, стоит как раз
под отверстием подставки, в центре основания
сосуда.
На барабане имеется указатель, показываю-
щий номер покровного стекла, на которое в дан-
ный момент оседает пыль. По закраине крышки
сделаны несколько отверстий для свободного
выхода воздуха при помещении ее над цилин-
дром.
Методика. Забор проб пыли произво-
дится следующим образом: на верхней поверх-
ности барабана в соответствующие углубления
помещают покровные стекла, покрытые тонким
слоем канадского бальзама1. Барабан привинчи-
вают к подставке и последнюю устанавливают там,
где желательно взять для счета пыль. Затем
медленно опускают на подставку сосуд G и за-
чтобы отверстия в закраине крышки оказались
открытыми. Небольшим поворотом крышки закрывают эти отверстия и воз-
дух в цилиндре оказывается изолированным. Экспозиция данного покров-
ного стекла продолжается некоторое время, затем под отверстия основания
прибора подводится другое покровное стекло и т. д. По окончании забора
проб барабан вместе с покровными стеклами отвинчивается. Чтобы фиксиро-
вать пылинки на полученных препаратах, покрывают барабан колпаком
и кладут под него кусочек ваты, смоченной ксилолом. Через несколько се-
кунд под действием паров ксилола бальзам разжижается. Сняв колпак, дают
ксилолу испариться. Пылинки, благодаря описанной операции, оказываются
плотно фиксированными на препаратах.
Препарат монтируется затем на предметном стекле, при помощи бумаж-
ного кольца, как описано выше (стр. 204).
Счет пылинок ведетсяпри увеличении 100—150 на затемненном фоне. В оку-
ляр вставляется сетчатый микрометр, где площадь в квадратный миллиметр раз-
делена на 100 малых квадратиков. Сосчитывают пылинки в полной сетке
в различных местах препарата и затем находят среднее число пылинок для
одной сетки. Чтобы определить объем воздуха, из которого выпали сосчитан-
ные пылинки, определяют при помощи объективного микрометра ту повер-
хность в поле зрения, которой соответствует одна сетка, и умножают эту
1 Покровное стеклышко погружается в 2% раствор канадского бальзама в ксилоле.
По извлечении из раствора стеклышек ксилол через 1—1,5 мин. испаряется и остается тон-
кий слой бальзама.
206
поверхность на высоту столба воздуха в цилиндре. Число пылинок, выпавших
из 1 мл воздуха, определяется по формуле:
где N — число пылинок, осевших из 1 мл воздуха,
а — линейный размер, которому соответствует в поле зрения сторона
сетки,
п — среднее число пылевых частиц, сосчитанных в одной сетке,
Л — высота сосуда, в котором оседала пыль.
Для ббльшей точности рекомендуется от общего числа пылинок, подсчи-
танных в одной сетке, отнять среднее число пылинок, определенных на кон-
трольном препарате при холостом опыте.
Пример. Среднее число пылинок в одной сетке равно 450. Среднее число пылинок в од-
ной сетке при холостом опыте равно 6, линейный размер стороны сетки в поле зрения равен
0,15 см. Высота цилиндра равна 20 см. Отсюда находим, что из 1 мл воздуха на данный пре-
парат осело
450 — 6
----------= 987 пылинок.
[0,15]»• 20
Удельный вес пыли
Удельный вес пыли, как и всякого другого порошкообразного и порозного
вещества, может быть определен посредством пикнометра или волюмометра.
Рис. 177. Пикно-
метры,
образует в эксси-
Определение удельного веса пыли при помощи пикнометра
Пикнометр — маленькая колбочка определенной емкости (5—50мл) с при-
тертой пробкой. Иногда вместо пробки в шейку склянки вставляется притер-
тый термометр (рис. 177).
Некоторое количество пыли переводится в пикнометр
или обыкновенную мерную колбочку и взвешивается точно
на аналитических весах. От полученного веса пикнометра
вместе со взятой пылью вычитают вес одного пикнометра,
определяя таким образом вес взятого количества пыли.
Отмечают найденный вес пыли и температуру, при кото-
рой производилось взвешивание. Затем в пикнометр на-
ливают дестиллированной воды, так, чтобы она покрыла
взвешенную пыль, осторожно взбалтывают и помещают в
экссикатор или под стеклянный колпак на пришлифован-
ной подставке. Экссикатор или колпак должны быть снаб-
жены отводной трубкой. Последняя сообщается с водо-
струйным или воздушным насосом, посредством которого
каторе или колпаке вакуум.
Благодаря вакууму из смеси пыли с водой удаляются пузырьки воздуха,
которые задержались в пыли. После того, как прекращается выделение пу-
зырьков воздуха, пикнометр (колбочку) доливают до метки водой и поме-
щают в водяную баню или водяной термостат, где температура поддержи-
вается на определенной высоте, на которую рассчитан пикнометр (колбочка),
чаще всего 15—20°С.
По прошествии некоторого времени (20—30 мин.) проверяют уровень
воды в колбочке, доводя его посредством отсасывания фильтровальной бу-
магой или прикапыванием нескольких капель воды точно до метки. Затем,
извлекши пикнометр из термостата, тщательно обтирают его снаружи и
взвешивают вновь на аналитических весах.
207
На основании результатов взвешивания одного пикнометра и пикнометра
вместе с пылью и водой определяют удельный вес пыли по формуле:
Р+л + Рг— Рз
где d — удельный вес пыли,
р — вес пыли, взятой для исследования,
— вес пикнометра,
А — вес воды, вмещающейся в пикнометре до черты (обозначен на самом
пикнометре),
р8 — вес пикнометра вместе с пылью и водой после выдерживания в тер-
мостате.
Пример. Пусть вес
пикнометра равен 31 г, вес воды до метки 25 г, вес взятой навески
пыли 8,5 г, вес пикнометра с пылью и водой
после выдерживания в термостате 59,5 г. Отсюда
заключаем, что удельный вес пыли равен:
А
31 +25 + 8,5 — 59,5
Знаменатель показывает, какое количество
воды вытеснено навеской пыли, т. е. объем
последней.
Для тех сортов пыли, которые рас-
творимы в воде или подвергаются в
воде набуханию либо другим изменени-
ям, вместо воды при определении удель-
ного веса пользуются другой жидкостью
(бензол). Найденный удельный вес в от-
ношении к данной жидкости перечисля-
ют на истинный удельный вес умноже-
нием на удельный вес данной жидкости.
Рис. 178. Волюмометр Гуревича
и Вендта.
V — сосуд для пыли, В — пришлифованная
пробка. Е — кран, И7 — расширение капил-
лярной трубки, а и Ъ — метки на капиллярной
трубке. L — трубка с миллиметровыми деле-
ниями, Е — трехходовый кран. А — напорный
сосуд, Q — зажим, К — термометр, М — сосуд
с водой.
Пример. Положим, что удельный вес пыли
по отношению к бензолу равен 1,8. Удельный
вес бензола равен 0,9. Отсюда заключаем, что
удельный вес пыли равен 1,8 х 0,9 = 1,62.
Определение удельного веса пыли при помощи
водюмометра
На рис. 178 изображен волюмометр
Гуревича и Вендта х.
Волюмометр состоит из сосуда v ем-
костью примерно в 7 мл, который запи-
рается капиллярной трубкой с при-
шлифованной пробкой В; от последней
отходит капиллярная трубка, дважды
изогнутая под прямым углом. Неболь-
шая бранша капилляра снабжена кра-
ном Е. По ходу капиллярной трубки
имеется расширение W, ограниченное
метками а и Ь. Капиллярная трубка
переходит в широкую трубку L с мил-
лиметровыми делениями, образуя та-
ким образом LJ-образную трубку, ко-
торая может быть использована для
манометрических целей. У основания
U-образной трубки имеется трехходо-
* Гуревич В. и Вендт В., «Журнал Общей Химии», т. II, вып. 7, 1932.
208
вый кран F. От нижней части трубки L отходит тубус, соединенный по-
средством резиновой трубки с напорным сосудом А. На резиновую трубку
наложен зажим Q. В качестве напорной жидкости используют керосин. По-
следний предварительно перегоняется, причем первые порции отбрасываются.
Перегнанный керосин высушивается безводным хлористым кальцием и ок-
рашивается краской судан 111 в красный цвет.
Для значительного облегчения расчетов авторы предлагают составить
график, где по разности высот керосинового столба, выраженной в миллимет-
рах, можно было бы сразу определить объем взятой навески пыли. Такой
график, по предложению авторов, составляется следующим образом. В со-
суд V помещают различные навески ртути, удельный вес которой определя-
ется пикнометрически. По весу и удельному весу ртути определяются объемы
взятых навесок. В зависимости от обеъма взятых навесок ртути получаются
различные высоты керосинового столба.
На графике по оси абсцисс откладывают
объемы взятых навесок в миллилитрах, а
по оси ординат — разницы столбов керо-
сина между измерениями с пустым сосу-
дом V и с навесками ртути, налитыми в
этот сосуд. Сначала устанавливают «ну-
левую точку» прибора. Для этой цели при
открытом кране Е и закрытом кране F
надевают пустой сосуд V на пробку В
и помещают его в сосуд М с водой ком-
натной температуры. По термометру /(,
погруженному в воду, следят за постоян-
ством температуры. Установив кран F на
Рис. 179. График для нахождения
удельного веса пыли по данным во-
люмометра.
сообщении обоих колен U-об-
разной трубки и расслабляя зажим Q, пускают керосин в трубку L, пока уро-
вень его не дойдет до метки Ь. Если нужно снизить уровень, прибегают к
крану F. Затем закрыв кран Е, вводят керосин в трубку L из напорного со-
суда, пока мениск не дойдет до метки а. Высота стояния керосина в трубке
L при этом принимается за «нулевую точку». Найдя нулевую точку, вводят
в сосудУ ту или иную навеску ртути и таким же образом, как при определе-
нии «нулевой точки», определяют высоту стояния мениска в трубке L. Оче-
видно, чем больше объем навески, тем выше окажется мениск керосина в
трубке L при прочих равных условиях. Определяют разность стояния ме-
ниска при данной навеске и при «нулевой точке» и результаты наносят на
график, как указано выше (рис. 179).
Составив график, нетрудно определить объем любой навески пыли, поме-
щенной в сосуд V. По объему взятая навеска должна быть равна 113—14
сосуда V. Установив высоту стояния мениска керосина в трубке L при данной
навеске, отнимают от полученного результата высоту стояния керосина при
«нулевой точке», а затем по полученной разности, пользуясь графиком, опре-
деляют объем взятой навески пыли. Зная вес и объем взятой навески пыли,
устанавливают ее удельный вес.
При юр. Положим, что взято 1,534 г пыли. Нулевая точка прибора оказалась равной
540 мм. Высота столба керосина при навеске равна 620 мм. Следовательно, разница равна
620 — 540 = 8J мм Соответственно данной разнице находим по графику объем, равный
0,68 мл. Отсюда делаем вывод, что удельный вес пыли равен
1/34
0,68
2,256.
Атмосферная пыль
Выше описаны методы исследования пыли, при которых забор пробы
длится непродолжительное время и часто носит одномоментный характер.
Эти методы применимы при исследовании воздуха помещений, где запылен-
14 А. И. Бурштейн
209
ность имеет более или менее постоянную интенсивность или подвержена опре-
деленным закономерным колебаниям (производственные цехи, классные ком-
наты и т. п.).
Для исследования запыленности воздуха открытых пространств эти ме-
тоды мало пригодны ввиду резких и частых колебаний, которым подвержена
запыленность атмосферного воздуха из-за ветров, атмосферных осадков,
состояния дорог, интенсивности движения людей и транспорта, количеств
выбрасываемых в воздух аэрозолей дымовыми трубами и т. п.
Из многочисленных методов, предложенных для количественного опреде-
ления атмосферной пыли, здесь описан наиболее распространенный метод
собирания и взвешивания пыли, выпадающей из воздуха на единицу повер-
хности (1 м2, 1 км2) данного места за определенное время (месяц, год); при
этом не учитывается, из какого объема воздуха пыль выпала.
Известны три модификации собирания выпавшей пыли: а) в экспонируе-
мые сосуды, в) на марлевые экраны и с) на снежные покровы.
Метод весового определения атмосферной пыли,
выпавшей в экспонируемые сосуды, сводится к тому,
что приемник пыли (стеклянная или фаянсовая банка высотою в 25—30 см
и диаметром в 18—20 см) помещают в данном месте на уровне роста человека
и оставляют на определенный промежуток времени (10—15 дней). Для за-
щиты банки от попадания в нее пыли боковым заносом, а также от выдува-
ния из нее пыли банку помещают в деревянный ящик с ребром в 0,5 м (Баба-
янц). По истечении указанного времени пыль собирают из приемника, смы-
вая ее водой и тщательно собирая со стенок стеклянной палочкой с резино-
вым наконечником. Воду затем выпаривают, пыль доводят до постоянного
веса и взвешивают. На основании ряда исследований определяют количество
пыли, выпавшей в данном месте на 1 м2 в граммах за год, что количественно
соответствует числу тонн на 1 км2 за тот же промежуток времени.
Пример. Стеклянная банка цилиндрической формы с верхним отверстием, равным
200 см2, подвергалась экспонированию в течение года 20 раз, каждый раз на 15 дней. Сред-
нее количество пыли за одну экспозицию оказалось равным 0,125 г. Нужно определить
количество пыли в граммах, выпадающее на 1 м2 в данном месте за год. Оно равно:
<’-'2Э-ЗЯ 152., г-м..
15 • 200
Описанный метод прост, легко выполним и может дать представление
не только о средней запыленности воздуха в данном населенном пункте в
целом, но и о запыленности воздуха в отдельных районах и даже в отдельных
небольших зонах, напр., у заводских и фабричных зданий и т. п. Ввиду сказан-
ного метод широко применяется научно-исследовательскими учреждениями.
Метод весового определения атмосферной пыли,
выпавшей на марлевые экраны (экранирование),
разработан проф. П. Д. Винокуровым и сводится к взвешиванию пыли, вы-
павшей на марлевые экраны, которые укрепляются на переднем конце деревян-
ной флюгарки. Экран состоит из четырех слоев марли, натянутых на метал-
лическую рамку 10 х 13 см, которая привязывается за четыре угла к под-
весной рамке, укрепленной на флюгарке, марля смачивается минеральным
маслом (машинным или вазелиновым) так, чтобы просветы петель оставались
свободными (на один экран идет в среднем 8—10 мл масла), и хранится в пер-
гаментной бумаге. Флюгарки с экранами укрепляются на крышах зданий с
помощью деревянной рейки длиною около 1 м.
Для одновременного определения взвешенной и оседающей пыли приме-
няются комбинированные экраны, состоящие из горизонтальной и верти-
кальной рамок с натянутой марлей. Экраны сменяются каждые сутки. В лабо-
ратории в зависимости от степени запыленности экрана обрабатывается весь
экран или часть его. Обработка заключается в том, что марля разрезается
на лоскуты шириной в 1,5—2 см, которые затем промываются химически
210
чистым бензином последовательно в четырех заранее взвешенных центри-
фужных пробирках. Затем пробирки с содержимым подвергаются центрифу-
гированию в течение 5—10 минут. После этого раствор масла в бензине сли-
вают с осадка и для удаления остатка масла дважды промывают осадок бен-
зином и вновь центрифугируют. При каждой промывке пробирка заполняется
бензином, содержимое взмучивается и центрифугируется, а затем жидкость
сливается с осадка. После третьего центрифугирования и удаления жидкости
пробирки с пылью сушатся до постоянного веса и взвешиваются. Привес
показывает количество пыли, осевшей на обработанную часть экрана.
Метод, помимо сложности, имеет и ряд других недостатков: необхо-
димо устанавливать экран на большой высоте; оседание пыли на вертикаль-
ном экране происходит из движущихся потоков воздуха, получающих направ-
ление, перепендикулярное экрану, что не характеризует среднюю запылен-
ность атмосферы, и т. п. Ввиду сказанного метод не нашел большего распро-
странения.
Метод весового определения атмосферной пыли,
выпавшей вместе со снегом или на его поверхно-
сти (метод снеговых проб), заключается в том, что в данном
месте производят забор пробы снега при помощи снегового бура и опреде-
ляют пыль, содержащуюся во взятой пробе.
Снеговой бур представляет металлический цилиндр с острыми краями, снаб-
женный металлической ручкой. Диаметр цилиндра б—10 см, высота 15—20 см.
При заборе проб выбирают участки, где снеговой покров не поврежден.
При выемке бура нижний слой снега очищают от приставшей земли,
листьев и т. п. случайных примесей. Обыкновенно в данном месте берут
буром не одну, а несколько проб, располагая выемки по схеме конверта.
Все выемки считаются одной пробой для данной точки.
Подсчет ведут на 1 м2 поверхности, учитывая полученные данные исследо-
вания снега, площадь сечения бура и число выемок снега в данной точке.
Время считают с момента установившегося снегопада по данным метеостанций
до момента выемки проб.
Описанный метод весьма неточен и применим лишь в зимнее время при
наличии снегопада.
ОСАДКИ, ВЫПАДАЮЩИЕ ИЗ АТМОСФЕРЫ
На метеорологических станциях для измерения количества выпадающих
атмосферных осадков (дождь, снег, снежная крупа, град и др.) пользуются
прибором, носящим название до-
ждемер (рис. 180). Он состоит
из дождемерного ведра
с площадью поперечного сечения
500 см2. Ведро окружено усеченным
конусом В, расширяющимся кверху,
причем верхнее основание конуса на-
ходится вровень с верхним краем
дождемерного ведра. Усеченный ко-
нус не позволяет ветру вдувать или
выдувать из дождемерного ведра ка-
пельки и снежинки.
Внутри дождемерного ведра име-
ется перегородка аа в виде воронки
с несколькими отверстиями, через
которые осадки стекают в нижнее
отделение ведра. Благодаря перего-
Рис. 180. Дождемер.
— дождемернов ведро, В — усеченный коцуе
(защита Нифера), аа — перегородка, ь — отвод-
ная 1рубка, с — пробна.
родке задерживается испарение осадков, что особенно важно в теплое время
года.
211
100-
90—
80-
70—
60—
гоД
Рис. 181. Измеритель-
ный стакан для дожде-
мера.
Дождемер на столбе устанавливается на открытом месте на такой высоте,
чтобы верхний срез ведра возвышался на 2 м над поверхностью земли. Между
основанием защитного конуса и столбом, на котором он стоит, имеется про-
свет, через который осадки, попадающие между ведром
и защитным конусом, могут свободно стекать.
Измерение количества выпавших атмосферных осад-
ков на метеорологических станциях производится в 7
и в 19 час.
Воду выливают через отводную трубку Ь в измери-
тельный стакан (рис. 181), деления которого соответ-
ствуют слою осадков в ведре высотой в 0,1 мм* 1. Снег,
град предварительно переводят в жидкое состояние.
Количество выпадающих атмосферных осадков выра-
жают (в миллиметрах) высотой слоя воды, который они об-
разовали бы на горизонтальной поверхности, если бы вода
не просачивалась в почву, не стекала и не испарялась.
Количество осадков, выпавших за 1 мин., называют интенсив-
ностью осадков.
ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ВОЗДУХА
Химическое исследование воздуха сводится к качественному и количе-
ственному определению различных находящихся в нем газов.
Сюда же можно отнести и химический анализ воздушной пыли.
Общие сведения
Принципы качественного определения газов
воздуха. Качественное определение тех или иных газов воздуха про-
изводят очень часто при помощи реактивных бумажек, которые под влия-
нием искомого газа меняют свою окраску. Такие бумажки либо развешива-
ются в различных местах помещения, где подвергаются воздействию воздуха,
либо помещаются в трубочки, через которые просасывается испытуемый
воздух. Второй способ применения реактивных бумажек имеет некоторые
преимущества перед первым: при нем получается воздействие на бумажку
больших количеств воздуха и представляется возможность отделить из
воздуха те сопутствующие газы, которые могут давать такое же изменение
окраски реактивной бумажки, как искомый.
Для качественного определения того или иного газа в воздухе применяют
и другой метод: воздух просасывают через соответствующую поглощающую
жидкость (сорбент), а затем исследуют ее на присутствие того соединения,
которое должно образоваться в результате поглощения искомого газа.
Органолептическое определение того или иного газа в воздухе по запаху
имеет второстепенное значение и не всегда возможно.
Принципы количественного определения газов
в воздухе. Количественное определение газов воздуха во многих слу-
чаях производится по принт пу сорбции, в основе которой лежит хими-
ческое или физико-химическое присоединение.
Применяя метод сорбции, нужно для искомого газа подобрать соответ-
ствующий поглотитель (сорбент). Полнота поглощения газа зависит от при-
роды и концентрации искомого газа, природы сорбента, размеров поверхности
между газовой смесью и поглощающей средой, температуры реагирующих
веществ и, наконец, от длительности взаимодействия между сорбентом и га-
зовой смесью.
1 г 0,1-500
1 Каждое деление стакана равно в действительности---------—
сечение стакана в квадратных сантиметрах.
мм, где $ — поперечное
212
Определение количества поглощенного газа довольно часто производится
на основании уменьшения объема газовой смеси (газволюмометрия).
Чаще, однако, исследованию подвергают сам сорбент и по изменениям,
которые в нем произошли, делают вывод о количестве поглощенного газа.
Так, например, в основу выводов может быт’ положен факт изменения титра
раствора, которым велось поглощение; в других случаях полученное в
результате поглощения искомого газа вещество может быть определено
колориметрически либо весовым путем и на основании этого сделаны выводы
о количестве поглощенного газа; в третьих случаях о количестве поглощен-
ного газа можно судить по привесу, который приобрел поглотитель и т. д.
Из других принципов количественного определения газов следует упомя-
нуть о сжигании, применяемом для некоторых газов, с последующим исследо-
ванием тех изменений, которые произошли в газовой смеси после сжигания
искомого газа.
Место и время забора проб
воздуха для анализа. Воздух для
анализа берут в тех местах, где
предполагается изменение его со-
става.
Обыкновенно забор пробы про-
изводится на высоте, соответству-
ющей уровню верхних дыхатель-
ных путей человека, учитывая
необходимость выяснения действия
ганизм.
Рис. 182. Газовые пипетки;
— без кранов, ь — с кранами, bi — трехходовый
кран.
измененного состава воздуха на ор-
Забор проб может быть произведен в определенные моменты, если имеют
в виду выяснить динамику концентрации того или иного газа, либо в тече-
ние определенного длительного промежутка времени, если желательно вы-
яснить средние концентрации газа.
Взятие пробы воздуха. Очень часто для забора пробы воздуха пользуются
газовыми пипетками (рис. 182), которые изготовляются без кранов или с
fl
Рис. 183. Газовая
пипетка, соединенная
с уравнительным
сосудом.
А — момент вытеснения
ив пипетки воздуха,
В — момент образования
в пипетке вакуума.
Чтобы заполнить
кранами; в последнем случае полезно один из кранов
сделать трехходовым, что дает возможность точнее про-
изводить операции по заполнению пипетки испытуемым
газом и по выведению его из пипетки.
Газовые пипетки без кранов закрываются резино-
выми трубочками, в которые вставляются плотно сте-
клянные палочки (затычки). Такие пипетки менее при-
годны для работы, нежели пипетки со стеклянными
кранами.
Чтоб заполнить газовую пипетку испытуемым воз-
духом, поступают следующим образом. Через пипетку
при помощи резиновой груши или мехов многократно
прогоняют испытуемый воздух; при этом испытуемый
воздух вытесняет воздух, находившийся в пипетке, и
занимает его место.
Более точный метод заключается в том, что при no-
мощи ртути вытесняют из пипетки воздух, пользуясь
при этом уравнительным сосудом (рис. 183-Д). Когда
ртуть заполнит всю пипетку, дойдя до верхнего крана,
последний закрывают, удалив ртуть из пипетки опуска-
нием уравнительного сосуда (рис. 183-3), образуют
внутри пипетки вакуум и закрывают нижний край
пипетки. Такая вакуированная пипетка
при хорошо смазанных кранах не пропускает воздуха,
вакуированную пипетку испытуемым воздухом, откры-
вают кран пипетки; при этом воздух быстро заполняет пипетку. Если пипетка
213
снабжена трехходовым краном, то им пользуются для впуска воздуха, пред-
варительно продув при помощи груши входной канал испытуемым воздухом.
При манипуляциях с газовой пипеткой, связанных с забором про-
бы. нужно избегать согревания пипетки
связанных с забором про-
руках.
Близко к описанному здесь
методу вакуированных пипеток
стоит метод применения в а ку-
ированных склянок
(рис. 184). Склянка известного
объема А закрывается плотно
каучуковой пробкой В с отвер-
стием, в которое вставляется
стеклянная трубочка, снабжен-
ная краном С. Еще до забо-
ра пробы в склянку наливают
необходимое количество погло-
тителя и, плотно закрыв склян-
ку пробкой, присоединяют
трубку к водоструйному на-
сосу D, снабженному вакуум-
метром Е. Пустив насос, обра-
зуют внутри склянки некоторый
вакуум и закрывают стеклян-
ный кран. Чтобы исключить
возможность забрасывания воды
в склянку, помещают между нею и насосом обыкновенный клапан F.
Зная объем склянки и учитывая показания вакуумметра, можно определить
объем воздуха,
в
Рис. 184.
А — склянка, _ _______ .
стеклянным краном, D — водоструйный насос, Е — ва-
Приспособление для образования
вакуума в склянке.
В — каучуковая пробка, С — трубка се
куумметр, F — клапан.
удаленного
из склянки по формуле:
ир
Vx ~ 76 ’
из склянки воздуха,
удаленного
склянки,
где их — объем
v — объем
р — показание стрелки по циферблату вакуумметра.
Пример. Объем склянки до пробки без объема налитого в склянку реактива равен
1200 мл. Разрежение воздуха в склянке по показанию вакуумметра доведено до 30 см.
Отсюда заключаем, что из склянки удалено:
1200-30 /1Г7ОПО
----------------------------~ 473,68 мл воздуха.
Для определения объема удаленного из склянки воздуха можно присое-
динить к обыкновенному водоструйному насосу вакуумный манометр (рис. 185).
Объем удаленного из склянки воздуха определяется в этом случае по
формуле:
v(H — h)
vx = —--------
Н
где vx—объем удаленного из склянки воздуха,
и — объем склянки,
Н — барометрическое давление,
й — остаточное давление воздуха в склянке, определяемое по мано-
метру.
Пример. Объем склянки 2 л, барометрическое давление 762 мм, остаточное давление
в склянке 50 мм.
Отсюда делаем вывод, что из склянки удалено:
2000(762 — 50)
-------------1868,76 мл воздуха.
762
214
В некоторых ‘случаях забор пробы воздуха производится в бутыли ем-
костью в 5—6 л при помощи ручных мехов примерно такой же емкости. В
этом случае на металлическую трубку у входного отверстия мехов надевают
каучуковую трубку 1,— 1,5мдлиною. Конецтрубки вводят в сосуд до дна его
и производят выкачивание воздуха при помощи мехов; при этом воздух из
бутыли выходит и заменяется воздухом помещения. 40—50 качаний мехов
достаточно, чтобы можно было считать, что воздух бутыли целиком заменен
воздухом помещения. Забор воздуха в большие бутыли в некоторых случаях
производится еще и так: бутыль заполняется водой, которую выливают в
том месте, где желательно произвести взятие пробы; взамен выливаемой
Рис. 185. Склянка, соединенная с вакуумным манометром.
А — склянка вакуумный манометр, В — шкала манометра. При данном стоянии
менисков остаточное давление в склянке равно 80 мм.
воды в бутыль поступает испытуемый воздух. Бутыль, в которую взята проба
воздуха, плотно закрывается пробкой. Такой забор воздуха возможен в тех
случаях, когда оставшаяся на стенках бутыли вода не влияет на дальнейший
ход анализа.
Наконец, воздух забирается иногда в резиновые приемники — футболь-
ные камеры, резиновые мешки и т. п. Нужно, однако, иметь в виду, что в
этих случаях воздух не должен оставаться долгое время не проанализиро-
ваннным, так как через стенки такого мешка постепенно проходит СО2, что
вызывает изменение состава взятого воздуха.
Во всех описанных выше случаях анализ взятой пробы воздуха дает
представление о его составе в момент взятия пробы. Если же интересуются
изменением концентрации какого-либо газа воздуха в течение определенного
отрезка времени, т. е. динамикой содержания его в воздухе, то одномомент-
ного забора воздуха недостаточно и приходится производить последовательно
ряд одномоментных заборов, соблюдая большие или меньшие интервалы меж-
ду ними.
Если длительно (в течение нескольких часов) просасывать испытуемый
воздух через соответствующий сорбент, а затем определить концентрацию
того или иного газа в воздухе, то полученные цифры должны рассматрива-
ться как средние, не отображающие динамику искомого газа в воздухе.
Приведение объема воздуха к нормальным условиям. При каждом
заборе пробы воздуха отмечается температура его и барометрическое да-
вление, что дает возможность привести взятый объем воздуха к нормальным
215
условиям, т. е. к 0° температуры и давлению в 760 мм Hg. Приведение к
нормальным условиям производится по формуле:
игН
° (1+а/).760
где v0 — объем воздуха, приведенный к нормальным условиям,
ит — данный объем воздуха.
Н — барометрическое давление в мм Hg в момент взятия пробы,
а — коэфициент объемного расширения воздуха, равный 1/273 или
0,00367,
t — температура воздуха во время взятия пробы.
Подставив вместо a получают для той же формулы выражение:
273 • • Н
и0 =----------------.
(273 + 0 • 760
Если из общего давления водуха вычесть давление водяных паров при
данной температуре, то формула приобретает вид:
1)л ИЛИ с/л ,
° (14-а0 7бО (273 4-0-760
где все буквы сохраняют те же значения, что и в предыдущих формулах,
ай — соответствует давлению водяных паров.
Поправка на напряжение водяных паров не вносит значительных измене-
ний в результате подсчета и потому не всегда делается.
При помощи таблицы 36, в которой приведены значения для 1 + at при
уу
любом значении /, а также значения для при любом значении /7, можно
значительно упростить расчеты.
Поглотительные приборы представляют собой стеклянные сосуды,
в которые вводятся соответствующие поглощающие (сорбирующие) жид-
кости или твердые вещества, предназначенные для поглощения того или
иного газа. Газ может являться искомым или сопутствующим искомому и
подлежащим отделению от него. Для
тщательного соприкосновения исследу-
емого воздуха с сорбирующим веществом
воздух просасывается через поглотитель;
реже воздух вводится в поглотительный
прибор, где тщательно перемешивается
с сорбентом и оставляется здесь на не-
которое время, после чего переводится
в измерительный сосуд (волюмометри-
ческие определения).
Жидкий сорбент помещают в специ-
альные приборы — промывалку типа
Петри или ее видоизменения, склянкуТи-
щенко, фильтр Городецкого и ряд других.
Пром ы валка типа Петри (рис. 186) представляет собой стеклян-
ный тонкостенный цилиндр, в который впаяна центрально тонкая трубка,
доходящая почти до дна склянки и загнутая сверху под прямым углом.
У верхнего края цилиндра сбоку отходит трубка, также загнутая под пря-
мым углом.
Поглощающая жидкость вводится в промывалку через центральную труб-
ку при помощи пипетки или бюретки. Промывалку при этом держат на-
клонно. Удаление жидкости из промывалки осуществляется через боковую
трубку. Воздух просасывается в направлении, указанном стрелками.
216
Рис. 186. Промывалка Петри. Справа
штатив с тремя промывалками Петри.
Таблица 36
Значения для 1 + at и jjgg при различных значениях t и Н
t 1+<хГ Н Н 760
—10 0,9633 730 0,9605
—9 0,9670 731 0,9618
—8 0,9707 732 0,9632
—7 0,9743 733 0,9645
—б 0,9780 734 0,9658
—5 0,9817 735 0,9671
—4 0,9853 736 0,9684
—3 0,9890 737 0,9697
—2 0,9927 738 0,9710
—1 0,9963 739 0,9724
0 1,0000 740 0,9737
1 1,0037 741 0,9750
2 1,0073 742 0,9763
3 1,0110 743 0,9776
4 1,0147 744 0,9789
5 1,0183 745 0,9803
б 1,0220 746 0,9816
7 1,0257 747 0,9829
8 1,0293 748 0,9842
9 1,0330 749 0,9855
10 1,0367 750 0,9868
11 1,0403 751 0,9882
12 1,0440 752 0,9895
13 1,0476 753 0,9903
14 1,0513 754 0,9921
15 1,0550 755 0,9934
16 1,0586 756 0,9947
17 1,0623 757 0,9961
18 1,0660 758 0,9974
19 1,0696 759 0,9987
20 1,0733 760 1,0000
21 1,0770 761 1,0013
22 1,0806 762 1,0026
23 1,0843 763 1,0039
24 1,0880 764 1,0053 ;
25 1,0917 765 1,0066
26 1,0953 766 1,0079
27 1,0990 767 1,0092
28 1,1027 768 1,0105
29 1,1063 769 1,0118
30 1,1100 770 1,0132
217
На рис. 187 представлена промывалка, подобно описанной, снабженная
стеклянной пришлифованной пробкой, что облегчает введение в склянку
реактива.
На рис. 188 изображена та же промывалка, в которой длднная трубка
снабжена пористой стеклянной пластинкой, что создает лучшее перемеши-
вание воздуха с поглощающей жидкостью.
Промывалки с изоляцией пробы
о т СО2 воздуха: на рис. 189 изображена промы-
валка, предложенная Гуревичем, на рис. 190 — промы-
валка MOOT (Московского областного отдела труда),
на рис. 191—промывалка ЛИГТ и ТБ (Ленинградский
институт гигиены труда и техники безопасности).
Рис. 188. Склянка Дрекселя
с пористой пластинкой.
Рис. 189. Промы-
валка Гуревича.
Устройство промывалок понятно из рисунков. Эти промывалки приме-
няются, когда необходимо изолировать пробу от СО2 воздуха. В этом случае
титрование ведется в самом поглотительном приборе после отбора пробы;
носик бюретки при этом вводится в отверстие О по удалении из него пробки.
о Через длинную трубку во время титро-
Рис. 190.
Промывалка
MOOT
Рис. 191.
Промывалка
ЛИГТ.
Рис. 192.
Спиральная
промывалка.
вания вводят в поглотитель воздух,
освобожденный от СО2. Прохождение
воздуха через жидкость способствует
перемешиванию ее в склянке во время
титрования.
Спиральная промывалка
(рис. 192) состоит из цилиндра, в ко-
торый вставляется винтообразное полое
внутри тело, соединяющееся с пробкой
цилиндра. В нижнем конце винтообраз-
ного тела имеется небольшое отверстие.
Благодаря тому, что винтообразное тело
плотно соприкасается со стенками ци-
линдра, поступающий в поглощающую
жидкость воздух не может выделяться
непосредственно вверх, а вынужден про-
ходить по спирали, проделывая путь
длиною около метра; это способствует лучшему поглощению газа. Воздух
выходит через отводную трубку, отходящую от пробки цилиндра.
Промывалка Тищенко (рис. 193) представляет собой толсто-
стенную склянку на плотном стеклянном основании. Внутри склянка разде-
лена продольной стеклянной перегородкой на две одинаковые половины.
В перегородке у основания ее имеется отверстие, через которое обе половины
218
Рис. 194. Фильтр
Городецкого.
а и Ь — стеклянные
пористые пластинки.
Рис. 193. Промывалка
Тищенко.
склянки сообщаются. Сверху от каждой половины склянки отходят по од-
н^й боковой трубке. Реактив вводится в склянку через отверстие, которое
закрывается пришлифованной пробкой (на рис. не показано).
Фильтр Городецко-
го (рис. 194) представляет
U-образную трубку, одно из
колен которой кверху груше-
видно расширяется. У нижнего
края в это колено вставлена
стеклянная пористая пластин-
ка а. Поглощающая жидкость
наливается в таком количестве,
чтобы она несколоко покры-
вала пористую пластинку. Про-
сасываемый воздух, проходя
через пластинку, разбивается на
множество мелких струек, что
способствует энергичному перемешиванию его с поглощающей жидкостью.
Засасываемый воздух для обеспыливания пропускается через пористую стек-
лянную пластинку Ь. По полу-
чаемому эффекту в смысле пол-
ноты поглощения фильтр Горо-
децкого не уступает спиральной
промывалке, хотя и проще ее по
конструкции.
Десятишарикова я
трубка (рис. 195) состоит
из десяти соединенных между
собой шариков. От последнего
шарика отходит изогнутая под
углом в 60—70° трубка, закан-
чивающаяся баллоном. Баллон заканчивается шейкой, в которую встав-
ляется изогнутая под прямым углом трубка, имеющая недалеко от конца
аспиратору.
Рис. 195. Десятишариковая трубка.
шариковое раздутие. Конец этой трубки присоединяется
Змеевик (рис. 196) состоит из спиралеподобной
стеклянной трубки А. От нижнего конца спирали отходит
трубка В, которая впаяна в спираль узким
КОНЦОМ И
Рис. 196. Змеевик.
А — спиральная трубка,
В — вертикальная труб-
ка, С и D — шарообраз-
ные расширения.
Рис. 197. U-образная
трубка, снабженная
кранами.
Рис. 199.
Поглотитель-
ная колонка.
шариком С. Другой
Рис. 198.
Хлоркальци-
евая трубка.
конец спирали
заканчивается
сверху заканчивается
шариком D. 11оглощающая жидкость наливается не выше основания шарика D.
Приборы для твердых поглотителей. Для твердых поглотителей приме-
няют U-образные трубки без кранов или с кранами (рис. 197), хлоркальци-
евые трубки (рис. 198), поглотительные колонки (рис. 199) и др.
К
219
Рис. 200.
Прибор для
просасывания
через поглоти-
тель неболь-
ших объемов
воздуха.
А — трубка для
входа воздуха,
В — трубка с
отверстием С
для выхода
воздуха, D —
груша.
Все описанные приборы, предназначенные для жидких и твердых погло-
тителей, часто соединяют по два-три для более полного поглощения газов.
Приборы для просасывания воздуха. Если воздух, подлежащий исследо-
ванию, находится в бутыли, пипетке, бюретке и необходимо провести его
через тот или иной поглотитель, то для этой цели пользуются напорным со-
судом, наполненным водой, насыщенным раствором какой-либо соли (NaCl,
СаС12)либо ртутью. Напорный сосуд соединяют с сосудом, в котором нахо-
дится испытуемый воздух. Если поместить напорный сосуд выше сосуда с
испытуемым воздухом, то жидкость под давлением будет переходить в этот
последний и вытеснять оттуда воздух, который может быть проведен, таким
образом, через те пли иные поглотители.
Если же необходимо прососать через тот или иной поглотитель испытуемый
воздух, то для этой цели пользуются теми же приборами, какие применяют
при исследовании воздуха на запыленность (стр. 195).
Иногда просасывание воздуха производится при помощи ре-
зиновой груши. Один из таких приборов изобрг кен на рис. 200.
Он состоит из цилиндра, который закрывается сверху проб-
кой. В пробке имеются два отверстия: через одно из них
вводится трубка А служащая для привода воздуха и доходя-
щая нижним концом почти до дна цилиндра; во второе отвер-
стие вставлена трубочка В, на которую сверху надета груша D.
В этой трубочке имеется сбоку небольшое отверстие С. При
сжатии баллона отверстие С должно оставаться открытым,
благодаря чему воздух из баллона выходит наружу. При раз-
жатии баллона отверстие С закрывается пальцем, тогда в ци-
линдре образуется вакуум и наружный воздух через трубку А
поступает в цилиндр и проходит через поглощающую жидкость.
Зная объем баллона и число накачиваний воздуха, произведен-
ных при помощи его, можно определить общий объем воздуха,
пропущенного через поглотитель. Способ не может считаться
точным.
Приборы для определения объема воздуха. При просасы-
вании через поглотитель наружного воздуха объем последнего
определяется так же, как при исследовании воздуха на запы-
ленность.
Между аспиратором и поглотителем включают иногда мано-
метр, что дает возможность учесть под каким давлением шла
аспирация воздуха. Найденное давление учитывают при при-
ведении воздуха к нормальным условиям. Если отрицательное
давление, при котором ведется аспирация, невелико, то ука-
занную поправку на давление часто не вводят.
Выражание концентрации газа в воз-
духе. Содержание того или иного газа в воздухе выражают
в объемных процентах (об%, v%) либо в promille (°/00)1 и очень
часто в миллиграммах на литр (мг/л). Перечисляя объем газа на вес и на-
оборот, нужно помнить, что грамм-молекула любого газа, взятого при нор-
мальных условиях, имеет приблизительно один и тот же объем, равный в
среднем 22,4 л. Этот так называемый молярный объем для различных
газов лишь незначительно отличается от средней величины. Учитывая ука-
занное, легко объем любого газа, взятого при нормальных условиях, пере-
числить на вес и наоборот.
Пример. Концентрация угольного ангидрида (СО2) в воздухе оказалась равной 2 мг/л.
Требуется выразить эту концентрацию в объемных единицах. Так как грамм-молекула
СО2 равна 44 г, а молярный объем этого газа равен 22,26 л, отсюда заключаем, что 1 мг
1 Символом %о обозначают тысячную долю (promille); одна promille равна одной
десятой процента.
220
СО, имеет объем, равный
22,26 • 1000
44 • 1000
= 0,506 мл.
Следовательно, в литре данного воздуха обнаружено 0,506 x 2 = 1,012 мл или
1,012% СОа.
Кислород
Кислород (О2) — газ удельного веса 1,1053 по отношению к воздуху,
удельный вес которого принимается за единицу. Вес 1 л кислорода, взятого
при нормальных условиях, равен 1,429 г. В воздухе содержание кислорода
составляет 20,7% (объемных). В рудниках, угольных шахтах и т. п. содержа-
ние кислорода в воздухе может оказаться значительно ниже указанной кон-
центрации. При подъеме на возвышенности в связи с уменьшением атмосфер-
ного давления уменьшается и содержание кислорода. Падение концентрации
кислорода в атмосфере до 7—8% вызывает угрожающие жизни явления:
асфиксию, анурию, потерю сознания.
Качественное определение
1. Фильтровальная бумажка, смоченная раствором а — нафтиламина в
разведенной уксусной кислоте, синеет в присутствии кислорода.
2. Раствор индигокармина, восстановленный при помощи Na2S2O4, при-
обретает свою окраску при пропускании через него газовой смеси, содержащей
кислород.
Количественное определение
Из многочисленных методов, предложенных для количественного опреде-
ления кислорода, здесь описаны весьма совершенный йодометрический метод
Хлопина и волюмометрический метод Гемпеля. В санитарной практике метод
Хлопина нашел большее применение нежели другие.
Метод Хлопина
Принцип метода заключается в том, что в склянку, содержащую
испытуемый воздух, вводят раствор хлористого марганца, на который дей-
ствуют едким натрием, получая гидрат закиси марганца. Последний, погло-
щая кислород в присутствии воды, окисляется до гидрата окиси марганца.
Гидрат окиси марганца разлагают соляной кислотой; при этом выделяется
хлорный марганец. Последний разлагается с выделением свободного хлора,
который вытесняет из прилитого раствора йодистого калия свободный иод.
Иод оттитровывают тиосульфатом натрия и по израсходованному тиосуль-
фату делают вывод о количестве поглощенного кислорода.
Выражая сказанное уравнениями, получаем:
МпС12 + 2NaOH = 2NaCl + Mn(OH)2,
2 Mn(OH)2 + О + H2O = 2Mn(OH)3,
2 Мп(ОН)3 + 6 НС1 = 2 МпС13 + 6 Н2О,
2 МпС13 = 2 МпС12 + С12,
С12 + 2 KJ = 2 КС1 + J2,
J2 + 2 Na2S2O3 = 2 NaJ + Na2S4Oc>.
Приборы и реактивы. 1. Очень удобно для данного анализа
пользоЕ<аться специально предложенным для этой цели проф. Хлопиным
прибором (рис. 201). Прибор представляет собой склянку А на 150—180 мл
221
с тремя отверстиями сверху. В боковое отверстие а вставляется трубка b с
краном; она пришлифована к шейке склянки и доходит нижним концом
почти до дна склянки. На верхнюю часть трубки b одевается бюретка
с емкостью 20 мл, пришлифованная к коническому концу трубки. Над
другим боковым отверстием насажена трубка е, снабженная краном. В
среднее отверстие / вставляется термометр d со шкалой от 10 до 25°С с деле-
ниями на 0,2°. Термометр пришлифован к отверстию. Каждый прибор нужно
точно прокалибровать. Для этой цели взвешивают пустой прибор на техни-
ческих весах с точностью до 0,1 г, затем заполняют его дестиллированной
водой и вновь взвешивают; разница в граммах со-
ответствует объему склянки в миллилитрах.
2. Резиновый баллон.
3. Пипетка на 15 мл.
4. Раствор хлористого марганца, содержащий
на 100 мл 40 г соли; раствор приготовляют на
свежепрокипяченной и быстро охлажденной воде;
в соли не должно содержаться железо.
5. Раствор едкого натрия и йодистого калия,
содержащий на 100 мл 30 г KJ и 32 г NaOH;
воду берут свежепрокипяченную и остуженную.
6. Концентрированная соляная кислота без
свободного хлора.
7. 0,1 N тиосульфат натрия.
8. Однопроцентный крахмальный клейстер.
Методика определения. В прибор
вводят пипеткой 15 мл раствора хлористого мар-
ганца, после чего прибор помещают в воду комнат-
ной температуры. Через длинную трубку при по-
мощи груши вводят испытуемый воздух, много-
кратно продувая склянку при открытых кранах.
Отмечают температуру воздуха по термометру при-
бора и барометрическое давление. Затем, надев
на конец длиной трубки бюретку, вводят при
помощи ее в склянку 15 мл реактива, состоящего
из KJ и NaOH, закрывают оба крана склянки и,
сняв бюретку, сильно и часто встряхивают при-
бор, пока жидкость в склянке не приобретет цвет
толченого кирпича. Одна за другой протекают первая и вторая реакции,
указанные выше, приводящие к связыванию кислорода воздуха. Для пол-
ного поглощения кислорода требуется 10—12 часов, в течение которых
склянку время от времени встряхивают, а затем кладут на бок.
Далее через чистую бюретку приливают в склянку 20 мл концентрирован-
ной НС1, следя за тем, чтобы вместе с кислотой не попал в склянку снаружи
воздух. Закрыв кран, встряхивают жидкость до полного растворения осадка;
при этом протекают последовательно третья, четвертая и пятая реакции.
Выделившийся иод титруют 0,1 N раствором тиосульфата натрия при инди-
каторе крахмальном клейстере.
Объем воздуха, в котором определяется кислород, приводится к нормаль-
ным условиям по следующей формуле:
_(^i —30) (Н — 0,857й)
Ул-- ' у
(1Ц-а/)-760
где t>0 — искомый объем,
—объем склянки (от него, как видно из формулы, отнимают 30 мл
воздуха, вытесненных при приливании реактивов до поглощения
кислорода),
а — коэфициент объемного расширения газов (= 0,00367),
Рис. 201. Прибор Хлопина
для определения кислорода
воздуха.
А — склянка дли отбора пробы
воздуха, а, / и е — отверстия в
склянке, b — трубка для соеди-
нения склянки с бюреткой, с—
бюретка, d — термометр.
222
с уравнитель-
ным сосудом.
t — температура воздуха в склянке,
Н — барометрическое давление,
h — напряжение водяных паров, насыщающих воздух при данной тем-
пературе (см. стр. 144),
0,857 — коэфициент для пересчета этого напряжения на напряжение водя-
ных паров раствора хлористого марганца.
Найдя vQ и зная количество миллилитров тиосульфата натрия, пошедшего
на титрование выделившегося иода, находят содержание кислорода в испы-
туемом воздухе в объемных процентах по формуле:
0,5592 . и • 100
х =-----------------,
где х — искомое содержание кислорода в воздухе,
п — число миллилитров 0,1 N тиосульфата натрия, пошедшего в реак-
цию,
р0 —объем воздуха, приведенный к нормальным условиям,
0,55825—объем кислорода в миллилитрах при нормальных условиях, кото-
рому соответствует каждый миллилитр 0,1 N тиосульфата натрия,
пошедшего в реакцию.
Пример. В угольной шахте произведено исследование воздуха на содержание кислорода
по Хлопину. Объем склянки равен 182 мл, температура воздуха 18° С, барометрическое
давление 764 мм Hg; при титровании выделившегося иода израсходовано 42 мл 0,1 N тио-
сульфата натрия. Каково содержание кислорода в воздухе в объемных
Приведя исследованный воздух к нормальным условиям, получаем:
v _ (182-30). (764- 15,357.0,857) н0 9
° (1 +0,00367 • 18) • 760
Концентрация кислорода в исследуемом воздухе равна:
0,55825.42*100 _ г
-------------16,6 00 70.
140,9
Волюмометрическии метод
Принцип метода заключается в том, что определенный
объем воздуха, отмеренный бюреткой, приводится в соприкосновение
со щелочным раствором пирогаллола или с таким же раствором под-
сернисто-кислого натрия в пипетке, где происходит поглощение кисло-
рода по реакции:
2СиН2(ОН)аН + О = (ОН)3С6Н, — С6Н.,(ОН)3 + Н2О,
пирогаллол гексаоксидифенил
либо
Na2S2O4 + О2 + НХО = NaHSO4 + NaHSO3.
Из пипетки после поглощения кислорода газовая смесь переводится
назад в бюретку; по уменьшению объема делают заключение о концен-
трации кислорода в испытуемом воздухе (газволюмометрия).
Приборы и реактивы. 1. Бюретка на 50—100 мл, сооб-
щающаяся с уравнительным сосудом (рис. 202).
2. Пипетки для жидких (рис. 203) или твердых (рис. 204) поглоти-
телей.
3. Соединительная капиллярная трубка, изогнутая по концам под
прямым углом.
4. Деревянная подставка.
5. Щелочной раствор пирогаллола, приготовляемый смешением раствора из 5 г пиро-
галлола в 15 мл дестиллированной воды с раствором из 120 г едкого калия в 80 мл воды.
Вместо щелочного раствора пирогаллола можно пользоваться щелочным раствором под-
сернисто-кислого натрия, который готовится смешением раствора из 50 г Na2S2O4 в 250 мл
воды и 40 мл раствора едкого натрия (500 г NaOH растворяется в 700 мл воды).
Из твердых поглотителей применяют фосфор в палочках.
Методика определения. Сначала отбирают в пипетку 100 мл исследуемого
воздуха. Для этой цели наливают в уравнительный сосуд В (рис. 205) ртуть, пока он и бю-
223
ретка А не заполнятся приблизительно до половины; при этом оба крана бюретки оста-
ются открытыми. Затем поднимают уравнительную трубку до тех пор, пока ртуть не
покажется у верхнего края бюретки. Далее следует введение испытуемого воздуха в бю-
ретку.
Если поглощение кислорода имеется в виду вести щелочным раствором пирогаллола
или подсернистокислого натрия, то необходимо испытуемый воздух освободить от СО., в
противном случае сорбент поглотит не только кислород, но и СО2, что внесет ошибку в
результаты анализа.
Для указанной цели присоединяют к бюретке пару U-образных трубок с натронной
известью (СаО 4- NaOH), через которые еще до присоединения к бюретке просасывают не-
который объем испытуемого воздуха; затем опускают уравнительную трубку пока бюретка
не заполнится испытуемым воздухом несколько больше 100 мл. Закрывают верхний кран
бюретки и, несколько приподняв уравнительную трубку так, чтобы уровень жидкости в
бюретке точно совпал с сотым делением (глаз — на одной горизонтальной плоскости с
указанным делением), закрывают нижний кран бюретки. В бюретке, таким образом, ока-
зывается 100 мл испытуемого воздуха под давлением несколько
больше атмосферного. "
ний кран, то избыток
наружу, и в бюретке
сферпым давлением.
Рис. 205. Прибор для
определения газов по
Гемпелю.
Если на одно мгновение открыть верх-
воздуха со слабым свистом вырывается
оказывается 100 мл воздуха под атмо-
Далее следует заполнение пипетки С
Рис. 203. Пипетка для жидких Рис. 204. Пипетка для
поглотителей твердых поглотителей.
щелочным раствором пирогаллола. На свободный конец капил-
лярной трубки пипетки надевают каучуковую трубочку с
зажимом е. При расслабленном зажиме пипетка заполняется
А — бюретка, В — уравни-
тельный сосуд. С — пипетка,
тип — шарообразные рас-
ширения пипетки, р —на пил-
ляр пипетки, е — зажим,
К — соединительная трубоч-
ка, D — соединительная ка-
учуковая трубочка.
СО стороны шарика п раствором до тех пор, пока шарик т
не оказывается весь заполненным. Наклоняя пипетку вбок,
переводят жидкость из шарика в капиллярную трубку р, пока
жидкость не покажется у верхнего края каучуковой трубочки,
надетой на капиллярную; в этот момент зажимают зажим е.
Далее необходимо соединить при помощи капиллярной трубочки К бюретку А и пи-
петку С. Для этой цели на свободный копен бюретки одевают небольшую каучуковую тру-
бочку и затем при помощи обыкновенной пипетки заполняют эту соединительную каучуко-
вую трубочку, а также каучуковую трубочку, надетую на свободный конец пипетки, и,
наконец, соединительную стеклянную трубочку К ртутью, после чего концы трубочки К
вводят в концы каучуковых трубочек, следя за тем, чтобы воздух не попал в соедини-
тельные трубки.
Теперь необходимо переместить отмеренное количество воздуха из бюретки в пипетку.
Открыв нижний и верхний краны бюретки и расслабив зажим е, поднимают уравнительную
трубку В Ртуть, переходя из уравнительной трубки в бюретку, вытесняет отсюда воздух,
который переходит из бюретки в пипетку Уравнительную трубку В поднимают до тех пор,
пока ртуть не заполнит всю бюретку, а также соедин' тельные трубки В этом момент зажи-
мают зажим Воздух целиком оказывается переведенным в пипетку, где он должен оставаться
в соприкосновении с поглощающей жидкостью минут 15 Осторожными движениями ста-
раются перемешать жидкость с газом. Для более удобного перемешивания можно объеди-
нить пипетку от бюретки, предварительно закрыв кран бюретки
Поглощение кислорода в пипетке протекает по реакциям, приведенным выше.
По поглощении кислорода остаток газовой смеси переводят обратно из пипетки в бю-
ретку Для этой цели расслабляют зажим е, открывают кран бюретки и опускают уравни-
тельный сосуд В. Ртуть в бюретке начнет опускаться, вместо нее в бюретку будет поступать
газовая смесь из пипетки. Опускание уравнительной трубки ведут до тех пор, пока весь
224
газ не перейдет в бюретку, а жидкость из пипетки заполнит соединительные трубки и до-
йдет до верхнего крана бюретки. В этот момент верхний кран бюретки закрывают и, при-
близив уравнительную трубку к бюретке, устанавливают уровни ртути в них в одной и той же
горизонтальной плоскости (глаз — в той же плоскости). Отсчитывают деление, на котором
стоит жидкость в бюретке. Это деление показывает объем оставшейся газовой смеси. Приво-
дят объемы взятого для анализа воздуха и оставшейся газовой смеси к нормальным усло-
виям и из первого результата вычитают второй. Разность покажет объем поглощенного
кислорода, т. е. процентное содержание кислорода в испытуемом воздухе, освобожденном
от СО2.
Описанным методом (газволюмометрии) может быть определен целый ряд газов, для
которых известны соответствующие поглотители.
Озон
Озон (О3) — газ удельного веса 1,624 (воздух-1). В толстом слое за-
метно синего цвета. Обладает характерным запахом, слегка напоминающим
запах хлора. Встречается на производствах, где добывается для технических
целей, в лечебных кабинетах, где применяют ртутнокварцевые лампы, в рент-
генкабинетах; в небольшом количестве находится в воздухе открытых про-
странств, полей, лесов и т. п., особенно после грозы. Содержание озона в
воздухе открытых пространств колеблется от десятых долей до нескольких
миллиграммов на 100 м3 воздуха.
Производит ясное раздражение слизистых оболочек уже в концентра-
циях 0,001 мг/л воздуха. В концентрациях вдвое больших вызывает через
%—1 час кашель и сильную усталость. При хроническом отравлении в кон-
центрациях 0,0016—0,0035 мг/л вызывает плохое самочувствие, усталость,
боль в области лба.
Качественное определение
Иод-калий-крахмальная бумажка. Приготовляют крахмальный клейстер
из 1 г крахмала на 20 мл воды и прибавляют сюда 1 г KJ, не содержащего
KJO3. Фильтровальные бумажки смачивают полученным раствором и высу-
шивают в темном месте. Для реакции пригодны свежие бумажки, которые
перед употреблением увлажняются водой. В присутствии озона бумажка
синеет, так как из йодистого калия освобождается свободный иод, окраши-
вающий крахмал в синий цвет:
О3 + 2 К J + Н2О = О2 + 2 КОН + J2.
Бумажка синеет также от перекиси водорода, окислов азота, хлора и др,
окислителей.
Иод-калий-лакмусовая бумажка. Красная лакмусовая бумажка пропи-
тывается раствором йодистого калия, не содержащего KJO3. При действии
озона на бумажку последняя синеет, вследствие образования КОН (см. ре-
акцию). Вместо лакмуса можно брать в качестве индикатора фенол-
фталеин. К 15% водному раствору KJ прибавляют 1% спиртовый раствор
фенолфталеина до тех пор, пока не появится ясная опалесценция. Фильтро-
вальные бумажки, смоченные этим раствором, под влиянием озона краснеют.
Иод-калий-лакмусовая и иод-калий-фенолфталеиновая бумажки также
не специфичны для озона.
Урсоловая бумажка. Урсоловую краску «Д» или «Т», состоящую в основном
из р-фенилендиамина — CeH4(NH2)2, растворяют ex tempore в крепком или
абсолютном этиловом алкоголе и в полученный раствор погружают на не-
сколько минут полоски фильтровальной бумаги, которые затем высушивают.
Перед употреблением бумажки смачивают дестиллированной водой. Слабая
буроватая окраска бумажки под действием озона переходит сначала в фиоле-
товую, а затем в темносинюю.
По данным Хлопина, предложенная им урсоловая реактивная бумажка
сохраняет свою специфичность для озона: перекись водорода на нее не дей-
ствует; окислы азота, хлор и бром придают ей зеленоватую окраску, которая
быстро переходит в желтую.
t5 А. И. Бурштейн
225
Количественное определение
Здесь описан йодометрический метод количественного определения озоод
а также нитритный метод в видоизменении Городецкого.
Йодометрический метод
Принцип метода основан на взаимодействии озона с иодистым
калием, в результате чего выделяется свободный иод:
О3 + 2 KJ + Н2О = О2 + 2 КОН + J2. (1)
Выделившийся иод оттитровывают тиосульфатом натрия и по расходу
его заключают о количестве свободного иода и озона.
Приборы и реактивы. 1. 2—3 поглотительные склянки Ти-
щенко, либо 2 фильтра Городецкого.
2. Аспиратор.
3. Газовые часы или реометр.
4. 1—3% раствор KJ.
5. 0,01 N раствор Na2S2O3 . 5Н2О.
6. 1% крахмальный клейстер.
Методика определения. Воздух просасывают через 2—3 погло-
тительные склянки, в которые налито по 30—50 мл нейтрального 1—3% рас-
твора KJ.1 При наличии в воздухе 03 из йодистого калия выделяется
свободный иод. Раствор подкисляют серной кислотой и оттитровывают
0,01 N тиосульфатом натрия в первой склянке и во второй, если он здесь
обнаружен (проба с крахмальным клейстером). Реакция между иодом и
тиосульфатом натрия протекает, как известно, по уравнению:
J2 + 2 Na2S2O3 = 2 Na J + Na2S4Oe. (2)
Сравнивая первую и вторую реакции, легко видеть, что одна молекула
О3 приводит к вытеснению одной молекулы J2, а на одну молекулу J2 при
титровании уходят две молекулы Na2S2O3, т. е. 48 весовым частям озона со-
ответствует 2.248 весовых частей тиосульфата натрия или 24 весовым частям
О3 соответствует 248 весовых частей Na2S2O3. Если для реакции пользоваться
0,01 N раствором Na2S2O3, то каждый миллилитр его, пошедший в реакцию,
указывает на связывание 0,24 мг О3. Результат переводят на единицу объема
воздуха.
Пример. При определении озона воздуха кабинета, где производится лечение ртутно-
кварцевыми лампами, пропущено через раствор йодистого калия 500 л воздуха. На титро-
вание выделившегося иода затрачено 2 мл 0,01 N раствора тиосульфата натрия. Температура
воздуха в момент анализа 15° С, барометрическое давление 756 мм Hg. Какова концент-
рация, озона в испытуемом воздухе?
Каждый миллилитр тиосульфата натрия соответствует 0,24 мг О3. В реакцию пошло
2 мл тиосульфата, следовательно, всего из воздуха поглощено 0,24 • 2 = 0,48 мг О3.
Это количество озона приходится на 500 л воздуха или 463,5 л при нормальных условиях.
Следовательно, концентрация озона соответствует:
0’48 — 0,001 мг/л.
463,5
Основные недочеты описанного метода заключаются в следующем: 1) иод,
выделяющийся под действием озона, частью улетучивается при просасывании
воздуха; 2) из йодистого калия иод вытесняется не только озоном, но и дру-
гими окислителями, могущими одновременно присутствовать в воздухе;
1 Имеющиеся в литературе указания на необходимость подкисления до просасывания
воздуха раствора KJ неправильны, так как при этом освобождается Н2О2 и выделяется
из КJ свободный иод: 4О3 4- 10HJ + Н2О - 10J + ЗО2 + Н2О2 -f- 5Н2О.
226
3) иодистый калий разлагается под действием света, особенно ультрафиоле-
товых лучей.
Первая неточность может быть устранена тем, что вслед за поглотителем
с иодистым калием помещается поглотитель, содержащий на 20—30 мл воды
1 мл 0,01 N раствора Na2S2O3, для улавливания уносимого с воздухом иода.
По окончании просасывания воздуха раствор тиосульфата переводится в
склянку с поглотительной жидкостью, а затем иод дотитровывается из бю-
ретки. Расход тиосульфата исчисляется из суммы того количества, которое
находилось в поглотителе, и того, которое пошло на дотитровывание. Хотя
эта предосторожность уменьшает ошибку, но не устраняет ее совершенно,
так как иод частью задерживается на стенках соединительных трубок, осо-
бенно резиновых.
Вторая неточность метода может быть устранена тем, что окислители,
сопутствующие озону, задерживаются соответствующими поглотителями до
прохождения воздуха через раствор KJ. Так, перекись водорода может быть
задержана хромовым ангидридом (CrOJ, двуокись азота — подкисленным ра-
створом КМпО4.
Наконец, иодистый калий может быть заменен более стабильным к
свету калийкадмиййодидом (KCdJ3).
Нитритный метод в видоизменении Городецкого
Принцип метода основан на реакции взаимодействия озона с нит-
ритом натрия:
NaNO2 + О3 = NaNO3 + О2.
Озон определяется по уменьшению концентрации раствора нитрита на-
трия, через который велось просасывание воздуха.
Приборы и реактивы. 1. Бутыль емкостью 10 л.
2. U-образная трубка диаметром 15 мм с хромовым ангидридом; напол-
нение трубки хромовым ангидридом производится вперемешку со стеклянными
бусами и со стеклянной ватой; трубку с хромовым ангидридом вставляют
в кипящую водяную баню и пропускают чистый воздух в течение 30 минут.
3. Колориметр.
4. Стандартный раствор NaNO2; в литре свежеперегнанной воды раство-
ряют 0,1437 г сухого NaNO2; 5 мл данного раствора доводится водой до
литра; 1 мл последнего раствора соответствует 0,0005 мг 03.
5. 0,01 М раствор сернокислого марганца.
6. Реактив Грисса, дающий с нитритами розовое окрашивание. Ре-
актив готовится следующим образом: 0,1 г чистого а — нафтиламина по-
мещают в фарфоровую чашку, прибавляют 20 мл дестиллированной воды
и кипятят несколько минут. Прозрачный раствор сливают с грязносинего
осадка и растворяют в 150 мл 30% уксусной кислоты (уд. вес 1,0412). От-
дельно готовят раствор из 0,5 г сульфаниловой кислоты в 150 мл 30% ук-
сусной кислоты. Оба раствора хранят отдельно и по мере надобности сме-
шивают ex tempore в равных объемах.
Методика определения. Из сухой, чистой бутыли, закрываю-
щейся хорошо пришлифованной стеклянной пробкой, снабженной краном,
откачивают воздух насосом до 80 мм Hg. В месте забора пробы к бутыли
присоединяют U-образную трубку с хромовым ангидридом и впускают в бу-
тыль воздух, который, пройдя через хромовый ангидрид, освобождается
от перекиси водорода. В бутыль вводят 50 мл стандартного раствора
и 2 мл 0,01 М раствора сернокислого марганца. Закупоривают бутыль,
содержимое тщательно взбалтывают и оставляют до следующего дня.
Для анализа берут 20 мл из бутыли в мерную колбу на 25 мл, приливают
сюда же 3 мл реактива Грисса, помещают в водяную баню на 3—5
минут при температуре 70—80°С, охлаждают и доливают водой до метки
227
(опытная колба). Отдельно в сухую коническую колбу вливают 25 мл стан-
дартного раствора и 1 мл раствора MnSO4, взбалтывают, берут отсюда 20 мл,
переводят в мерную колбу на 25 мл и приливают Змл реактива Грисса. Затем
помещают в водяную баню на 3—5 минут, охлаждают, доливают водой до
метки (стандартная колба). Химизм реакций см. на стр. 255.
Растворы из обеих колб — опытной и стандартной — подвергают коло
риметрированию и по уменьшению концентрации нитрита натрия в опытной
колбе заключают о количестве поглощенного озона.
Городецкий, Кобозев, Темкин и ряд других авторов полагают, что одно
временное присутствие N02 в воздухе вместе с озоном невозможно, так как
NOj окисляется до N2O5 по уравнению:
2 NO2 + О8 = N2O5 4- О2.
Если в воздухе имеется NO2 в отсутствии озона, то при просасывании
воздуха через раствор нитрита натрия будет увеличиваться концентрация
ионов NO/ за счет образования азотистой кислоты:
2NO2 + Н2О = Н NO2 Н no8.
Таким образом, устраняется возможность определения озона вместо
окислов азота и наоборот, что может иметь место при йодометрическом
методе.
Угольный ангидрид
Угольный ангидрид (СО2) — бесцветный газ, без запаха, кисловатого
вкуса, удельного веса 1,5197 по отношению к воздуху. В атмосферном возду-
хе содержится в количестве 0,3—0,4 п100. В больших концентрациях наб-
людается в местах скопления людей при плохой вентиляции помещений, в
бродильных подвалах, шахтах и т, д. При концентрации в 4—6% вызывает
явления отравления: стеснение дыхания, цианоз, потерю сознания.
Доброкачественный воздух помещений не должен содержать более 0,7°/^
СО2. По мнению некоторых авторов крайним пределом содержания в воз-
духе СО2 можно считать l0/^. Большие концентрации СО2 свидетельствуют
о порче воздуха: неблагоприятном для здоровья изменении его физических
свойств и химического состава. Таким образом, СО2 является как бы инди-
катором чистоты воздуха.
Качественное определение
Просасывание воздуха, содержащего СО2, через прозрачный известковый
или баритовый раствор ведет к помутнению раствора ввиду образования
нерастворимого карбоната кальция или бария.
Количественное определение
Здесь описан весовой и два объемных метода (макро- и микрометоды).
Весовой метод
Принцип метода основан на том, что определенное количество
воздуха просасывают через поглотитель, по привесу которого судят о коли-
честве поглощенного СО2. Чаще всего в качестве поглотителя применяют
натронную известь (СаО + NaOH). Следовательно, в основе метода лежат
реакции:
СаО + СО2 = СаСО3,
1 4 О7
2 NaOH 4- СО2 = Na2CO8 4- Н2О.
Необходимые приборы и реактивы. 1. Пять U-образ*
ных трубок.
228
2. Газовые часы или реометр.
3. Аспиратор или насос.
4. Хлористый кальций безводный гранулированный.
5. Натронная известь в зернах величиной с горошину.
Методика определения. Собирают прибор следующим образом:
две U-образные трубки заполняют хлористым кальцием; третья такая же
трубка заполняется натронной известью, одно колено четвертой трубки—хло-
ристым кальцием, а второе—натронной известью; пятая U-образная трубка
(контрольная) заполняется натронной известью. Заполнение U-образных тру-
бок производится на две трети; поверх реактивов помещают кусочки стеклян-
ной ваты. Третья, четвертая и пятая трубки выдерживаются вэкссикаторе для
высушивания снаружи и взвешиваются на аналитических весах. В указанном
порядке трубки соединяются при помощи каучуковых трубок и подвешива-
ются при помощи проволочек на стержне, который закрепляется на штативах.
Контрольная трубка соединяется при помощи каучуковой трубки с газовыми
часами (реометром), а последние — с аспиратором (насосом). Проверяют,
«держит ли прибор», т. е. прочны ли соединения. Для этой цели закрывают
входное отверстие первой трубки и пробуют засасывать воздух. Если соеди-
нения прочны, то засасывание воздуха невозможно. Проверив прибор, от-
крывают входное отверстие первой трубки и просасывают через всю систему
100—200 л испытуемого воздуха со скоростью 40—50 л в час. В первых двух
трубках воздух освобождается от влаги, в третьей трубке идет связывание
СО2 по приведенным выше реакциям. Хлористый кальций в четвертой трубке
служит для связывания «конституционной» воды, которая освобождается
в третьей трубке (см. 2-ую реакцию); эта вода выделяется в результате хими-
ческой реакции и, следовательно, не должна быть потеряна при взвешива-
нии. Натронная известь в правом колене четвертой трубки служит для связы-
вания СО2, который мог «проскочить» через третью трубку. Третья и четвертая
трубки после опыта в закрытом виде (повернутые краны) выдерживаются
в экссикаторе и взвешиваются. Привес указывает количество поглощенного
СО2. Пятая трубка отдельно взвешивается и не должна показывать привеса
(контроль), в противном случае опыт нужно повторить с заменой реактивов
либо с присоединением дополнительной поглотительной трубки.
Для большей точности нужно проверить, целиком ли высушивается
воздух при прохождении через трубки с хлористым кальцием; поэтому по-
лезно включить третью осушительную трубку с хлористым кальцием либо
с зернами пемзы, смоченными крепкой серной кислотой, либо с пятиокисью
фосфора (Р2О5). Контрольная трубка на полноту поглощения влаги взвеши-
вается отдельно до и после опыта. Она не должна показывать привеса,
если воздух осушается полностью в первых двух трубках.
Объемный метод Субботина—Пагорского
Принцип метода основан на способности едкого барита жадно
присоединять СОа по реакции:
Ва(ОН)а + СОа = ВаСОя + НаО.
В герметически закрытой посуде приводят в соприкосновение определен-
ный объем испытуемого воздуха с определенным же объемом титрованного
раствора Ва(ОН)2 и по поглощении СО2 вновь определяют титр едкого барита.
По ослаблению титра делают заключение о концентрации СО2 в испытуемом
воздухе, учитывая при этом условия, при которых ведется определение.
Приборы и реактивы. 1. Бутыль А (рис. 206) емкостью в
5—6 л, прочно закрывающаяся резиновой пробкой В, в которой имеется
круглое отверстие; на горлышке бутыли сделана метка, до которой доходит
введенная пробка.
229
F
• Рис. 206. Прибор Субботина — Нагорского для опре-
деления угольного ангидрида воздуха.
А — бутыль для отбора пробы воздуха, В — каучуковая
пробка бутыли, а — склянка для раствора едкого бария,
Ъ — пробочка склянни, Е — бюретка, F — соединительная
трубка, D — хлоркальциевая трубка с натронной известью,
С — груша.
2. Длинногорлая склянка а емкостью 120—150 мл, прочно закрываю*
щаяся резиновой пробкой Ь, в которой имеется малое круглое отверстие,
закрывающееся стеклянной палочкой. Диаметр шейки склянки а должен со-
ответствовать диаметру отверстия пробки В. На шейке имеется метка, до
которой вставляется резиновая пробочка. Склянка, как и бутыль, должны
быть прокалибрированы: бутыль — посредством мерных колб на выливание
и бюретки, склянка — посредством взвешивания на технических весах в пу-
стом виде и при заполнении
дестиллированной водой.
3. Хлоркальциевая труб-
ка D, заполненная натронной
известью.
4. Резиновая груша С.
5. Бюретка Е со стеклян-
ным наконечником, закрыва-
ющаяся сверху пробкой, в
которую может быть встав-
лена соединительная трубоч-
ка Е, дважды изогнутая под
прямым углом.
6. Мехи.
7. Титрованный раствор
серной кислоты \ приготов-
ленный так, чтобы 1 мл со-
ответствовал 1 мг СО2. Литр
такой кислоты должен содер-
жать 98,086 : 44 = 2,2292
химически чистой H2SO4
(98.086 — молекулярный вес
H2SO4; 44—молекулярный
вес СО2).
Практически поступают
так. Определяют ареометром
удельный вес разведенной
серной кислоты. По таблице
(стр. 132) определяют кон-
центрацию и вычисляют,
сколько нужно взять на литр
данной кислоты, чтобы полу-
чить нужный раствор. Най-
денный вес перечисляют на
объем, поделив на удельный
вес. Отмеряют бюреткой или точной цилиндрической пипеткой найденный
объем кислоты, переводят в литровую колбу и доливают до метки водой.
Так, например, если удельный вес серной кислоты по ареометру равен 1,2,
то концентрация ее по таблице составляет 27,32%. Следовательно, для полу-
чения литра указанного эмпирического раствора кислоты нужно взять:
100.2,2299
27,32
8,1599 г
или, перечислив на объем:
8,1599
1,2
6,8 мл.
1 Можно пользоваться и другой кислотой: соляной, щавелевой.
230
Этот объем кислоты переводят в литровую колбу и доводят до литра водой.
Полученный таким образом раствор кислоты не может считаться точным.
Его нужно проверить по точному раствору щелочи, например по 0,1 N соде
(буре), учитывая, что 2,2 мл необходимого раствора кислоты эквивалентны
/44 \
1 мл 0,1 N кислоты ^20= 2,2 J. Следовательно, на 1 мл 0,1 N щелочи должно
уходить 2,2 мл приготовленного раствора серной кислоты; в противном случае
раствор нужно выправить или установить его коэфициент. Приготовленную
кислоту сохраняют в тщательно закрытой бутыли.
8. Титрованный раствор Ва(ОН)2, приготовленный так, чтобы 1 мл его
соответствовал I мл указанного выше эмпирического раствора H2SO4. Для
этой цели приготовляют насыщенный раствор едкого барита, растворив кри-
сталлический едкий барит в горячей дестиллированной воде. По охлаждении
и полном просветлении раствора отбирают пипеткой 10 мл и титруют приго-
товленным эмпирическим раствором кислоты при индикаторе фенолфталеине.
Положим, что на 10 мл крепкого раствора барита уходит 50 мл кислоты;
следовательно, раствор барита должен быть разбавлен в пять раз, чтобы
1 мл его соответствовал 1 мл кислоты. Отобрав некоторое количество креп-
кого барита посредством сифона, чтобы не взмутить осадка, разбавляют барит
водой и прибавляют на каждый литр раствора по 0,2 г хлористого бария.
Полученный раствор сохраняют в тщательно закупоренной бутыли; из нее
при надобности берут небольшие количества при помощи сифона. Чтобы
СО2 воздуха не изменил титр барита, вставляют в пробку бутыли хлор-
кальциевую трубку с натронной известью.
9. 1% спиртовый раствор фенолфталеина.
Методика определения. Анализ воздуха на СО2 по методу Суб-
ботина—Нагорского слагается из следующих моментов: взятие пробы воз-
духа для исследования; определение титра едкого барита по кислоте до
опыта; поглощение СО2 воздуха титрованным едким баритом; новое опре-
деление титра ослабевшего барита и расчеты.
Забор пробы воздуха производится в описанную выше бутыль посредством
мехов. На металлическую трубку у входного отверстия мехов одевают каучу-
ковую трубку длиною в 1—1 у2 м- Свободный конец трубки вводят до дна
бутыли и высасывают из нее воздух; вместо него в бутыль каждый раз посту-
пают новые порции испытуемого воздуха. При этом нужно стоять по возмож-
ности далее от бутыли, чтобы выдыхаемый воздух, богатый угольным анги-
дридом, не попадал в бутыль. 40—50 накачиваний мехами среднего размера
(на 5—6 л) достаточно для полного обмена воздуха в бутыли. В шейку бутыли
вставляют резиновую пробку без отверстия либо с отверстием; в последнем
случае в отверстие вставляют плотно маленькую пробочку, чтобы гермети-
чески закрыть бутыль. Отмечают температуру и барометрическое давление
и транспортируют взятую пробу воздуха в лабораторию.
Проверка титра едкого барита по кислоте производится следующим об-
разом: бюретка со стеклянным наконечником продувается воздухом, свобод-
ным от СО2, для чего пользуются грушей и хлоркальциевой трубкой, напол-
ненной натронной известью (см. рис. 206). Наконечник бюретки соединяется
посредством резиновой трубки с сифоном бутыли, в которой хранится едкий
барит. Бутыль помещается выше бюретки. Если теперь расслабить зажимы
сифона и бюретки, то раствор барита перейдет в бюретку. Наполнив бю-
ретку баритом, наливают в стаканчик 25 мл эмпирического раствора кислоты,
прибавляют сюда 2—3 капли фенолфталеина и, погрузив наконечник бюретки
в кислоту, титруют до появления фиолетоворозового окрашивания, не ис-
чезающего при взбалтывании жидкости. По числу миллилитров барита,
пошедшего в реакцию, определяют титр барита. Так, если на 25 мл кислоты
25
пойдет 26 мл барита, то титр барита выразится •
231
Далее следует введение барита в бутыль. Маленькую склянку (рис. 206)
продувают воздухом, свободным от СО2, для чего пользуются грушей и хлор-
кальциевой трубкой, наполненной натронной известью. Затем вводят рас-
твор барита в склянку доверху и закрывают ее пробочкой b с отверстием, в
которое вставляется стеклянная палочка. Большую бутыль помещают на
некоторое время в воду температуры, соответствующей температуре воздуха
при заборе пробы; затем извлекают из горлышка бутыли А пробку и заме-
няют ее пробкой с отверстием. Вынув пробочку из склянки а и наклонив бу-
тыль Д, вводят шейку склянки в отверстие пробки В, производя при этом
вращательные движения. Изолировав таким образом герметически воздух
бутыли от внешнего воздуха, взбалтывают барит минут 15—20, после чего
оставляют бутыль на 10—20 часов, время от времени взбалтывая жидкость.
За это время весь угольный ангидрид воздуха в бутыли окажется погло-
щенным баритом по реакции, приведенной выше.
Определение титра едкого барита после поглощения СО2 производят сле-
дующим образом. Повернув бутыль вверх дном, дают бариту стечь в склянку;
вытягивают горлышко скляеки из пробки и затыкают склянку пробкой со
стеклянной палочкой. Дают бариту отстояться до полного выпадения ВаСО3
при этом Ва(ОН)2 становится совершенно прозрачным. Известным уже об-
разом освобождают воздух бюретки от СО2, погружают наконечник бюретки
в склянку с отстоявшимся едким баритом, стараясь при этом не взмутить
осадка (ВаСО3), и насасывают в бюретку барит из склянки. Для этой цели
удаляют грушу с хлоркальциевой трубки и надевают на свободный конец
последней резиновую трубку, снабженную стеклянным наконечником. Нако-
нечник берут в рот и равномерным глубоким вдохом образуют вакуум в бю-
ретке, благодаря чему в нее поступает барит. Последний должен быть совер-
шенно прозрачным. Титр ослабевшего барита определяется, как описано
выше.
Определение концентрации СО2 в испытуемом воздухе производится на
основании объема воздуха, взятого для анализа, количества едкого барита,
введенного в бутыль, титра барита до опыта и после него, температуры
и барометрического давления в момент анализа.
Пример. В классной комнате к концу занятий взята проба воздуха в бутыль, емкостью
до метки в 5200 мл. Емкость склянки с баритом до метки — 140 мл. Поправка на пробку 1
склянки равна 1 мл. Эмпирический раствор серной кислоты приготовлен так, что каждый
миллилитр эквивалентен 1 мг СО2.
25 25
Титр раствора барита по данной кислоте равен до опыта 2g, после опыта 2g. Температура
воздуха 15° С, барометрическое давление 762 мм Hg. Нужно на основании указанных дан-
ных рассчитать содержание СО2 в воздухе в миллиграммах на литр и в объемных pro mille
(об°/01).
Объем воздуха в бутыли до метки с поправкой на пробку равен 5201 мл. Приведя этот
объем к нормальным условиям, получаем:
у,Н
(1 4-а/) 760
5201 . 762
(1 +0,00367 • 15) • 760
~ 4942,7 мл.
По разнице титров барита до опыта и после него можно сказать, что крепость каждого
,25 25\
миллилитра барита уменьшилась соответственно (^g— 29* мл серной кислоты; следова-
25 25
тельно, весь взятый барит ослабел соответственно 2g — 29' 140 мл серной кислоты. Это
значит, что барит поглотил столько же миллиграммов СО2. Отсюда находим, что концен-
трация СО2 в испытуемом воздухе в мг/л равна:
/25 _ 25\ 140 . юоо
\ 26 29 /
4942,7
- 2,82.
1 При извлечении пробочки из склянки в шейку ее над баритом попадает некоторое
количество воздуха; это количество воздуха нужно учесть для точности расчетов.
232
Для пересчета полученного весового количества на объемное, нужно исходить из того,
что грамм-молекула СО2, т. е. 44 г имеет объем 22,26 л при нормальных условиях; следо-
22,26
вательно, каждый мг СОа имеет объем в ~дд— = 0,506 мл.
Перечислив полученное число миллиграммов на миллилитры, получаем: 0,506 • 2,82 =
= 1,43 мл.
Это и соответствует содержанию СО2 в воздухе, выраженному в pro mi Не (°/оо)-
Объемный метод Реберга—Винокурова
Метод Реберга предложен сравнительно недавно и нашел довольно большое распростра-
нение в наших санитарно-гигиенических лабораториях. Из микрометрических методов он
отличается большей точностью. Здесь метод изложен с теми изменениями, какие внес в него
Винокуров.
Принцип метода заключается в том, что определен- д
ный объем воздуха (100—120 мл) просасывают через щелочной
раствор определенного титра и по уменьшению последнего делают
вывод о количестве поглощенного СО2; отсюда рассчитывают кон-
центрацию СО2 в испытуемом воздухе. В основе способа лежит, к.
следовательно, реакция:
Ва(ОН)2 4- СО» = ВаСО3 + Н2О. \\
Приборы и реактивы. Прибор для определения \\
СО2 состоит из эпруветок для щелочи, оригинальной микро-
бюретки, пипетки для щелочи, газовой пипетки, сообщающейся
с уравнительным сосудом, трех поглотительных колонок, не- Рис* 207. Эпруветка.
больших склянок для запаса щелочи и кислоты. В приборах
нового образца имеется еще и ртутный манометр для наблюдения за давлением, под ко-
торым ведется проталкивание воздуха через раствор щелочи. Из названных частей нужно
описать лишь эпруветки, микробюретку и пипетку для щелочи Устройство других частей
известно из предыдущего.
1. Эпруветка (рис. 207) представляет собой стеклянную трубку диаметром в 1 см
и длиною в 18—20 см. На одном конце трубки имеется шарообразное расширение, в другой
-----. конец трубки впаяна капиллярная стеклянная трубка такой же дли-
ны, как и первая Угол, образуемый обеими трубками, должен рав-
няться приблизительно 45°.
Рис 208. Микро-
бюретка Реберга;
а — внешний вид,
b — деталь, на кото-
рой показано соеди-
нение бюретки с ме-
таллическим пилин-
дром. снабженным
микровинтом.
Для анализа берут две эпруветки, которые соединяют между
собой так, что широкая трубка одной эпруветки сообщается с капил-
лярной трубкой другой. Для этой цели в свободный конец широкой
трубки одной эпруветки вставляют пришлифованную полую пробку,
сужающуюся кверху, либо обыкновенную резиновую пробку, в ко-
торую вставляют стеклянную трубочку Сообщение обеих эпруветок
осуществляется при помощи узкой резиновой трубки.
При больших концентрациях СО2 приходится брать три эпру-
ветки.
2. Микробюретка (рис. 208) представляет собой толстостенную
капиллярную трубку, загнутую у одного конца дважды под прямым
углом У другого конца трубка переходит в цилиндрическое расши-
рение. Это расширение снизу соединяется со стальной шайбой, в центре
которой ходит стальной винт В цилиндрическое расширение бюретки
вводят чистую ртуть. Если завинчивать винт в цилиндрическую
основу микробюретки, то ртуть вытесняется и заполняет капилляр.
Напротив, если винт вывинчивать, то ртуть переходит назад из
капилляра в цилиндр. Микробюретка укреплена на штативе, на ко-
тором сзади капиллярной трубки находится миллиметровая шкала.
Шкала дает возможность определять перемещение ртути в капил-
ляре. Объем каждого деления капилляра обозначается в паспорте
прибора и равен в среднем 0,0005 мл. Микробюретка, таким образом,
дает возможность титровать в среднем с точностью до 0,0005 мл.
Для заполнения микробюретки титрованным раствором кислоты
ввинчивают винт в цилиндр, вытесняя тем самым из него ртуть в капил-
ляр до момента появления маленькой капельки ртути у наружного от-
верстия капиллярной трубки, после чего подносят к свободному концу
капилляра сосуде раствором кислоты так, чтобы конец капилляра ока-
зался погруженным в жидкость. Если после этого начать вывинчи-
вать винт, то ртуть пойдет из капилляра в цилиндр, а вслед за ней, ввиду образования
вакуума, пойдет раствор из сосуда в капилляр.
Для выведения титрованного раствора из капилляра, напр. при титровании, завинчи-
вают винт r цилиндр; ртуть переходит из цилиндра в капилляр и вытесняет из него эквива-
лентное количество по объему титрованного раствора.
233
Рис. 210. Приспособ-
ление для отмерива-
ния раствора щелочи.
А — пипетка, В — резер-
вуар со щелочью, С и
С — хлоркальциевые
трубки с натронной из-
вестью.
Описанная микробюретка представляет наиболее дорогую часть прибора и иногда заме-
няется более дешевой микробюреткой Шилова, которую мы здесь опишем.
Пневматическая микробюретка Шилова (рис. 209) готовится из микропипетки с деле-
ниями на 0,001 мл. К нижнему концу микропипетки присоединяют при помощи резиновой
трубки стеклянный наконечник длиною в 5—6 см,
капиллярно суживающийся книзу. Верхний конец
микропипетки присоединяется
при помощи каучуковой проб-
ки к одному из колен U-об-
разной стеклянной трубки. Вто-
рое колено этой последней при
помощи каучуковой пробки со
вставленной в нее трубкой
присоединяется к толстостен-
ной капиллярной каучуковой
трубке, которая у свободно-
го конца сообщается с ма-
ленькой резиновой грушей.
Г рушу помещают между пла-
стинками винтового зажима,
закрепленного на подставке.
Для точного титрования необ-
ходимо в зажиме иметь тонкий
микрометрический винт. Для
заполнения пневматической
микробюретки жидкостью по-
гружают кончик микробюретки
в данную жидкость и, вывин-
чивая винт, создают в микро-
бюретке вакуум, в силу чего
жидкость входит в капилляр
микробюретки. Напротив, же-
лая вывести жидкость из
микробюретки (при титрова-
нии), завинчивают винт в за-
жим, сжимая тем самым грушу,
в силу чего жидкость выталки-
Рис. 209. Пневматическая
микробюретка Шилова.
вается из капилляра.
Пипетка для щелочи А (рис. 210) сообщается с резервуаром В, в который вводят
щелочь — раствор Ва(ОН2) либо NaOH, окрашенный фенолфталеином в фиолетовокрас-
ный цвет. Над расширением пипетки и под ним на стенке пипетки имеются метки.
Объем пипетки между обеими метками обозначен в паспорте прибора и равен в среднем
2—2,5 мл. И резервуар и пипетка защищены трубками с натронной известью.
4. Раствор НС1 или С2Н2О4 • 2Н2О концентрации 0,1—0,2 N.
5. Раствор Ва (ОН)2 такой крепости, чтобы г.
содержимое одной пипетки нейтрализовалось
содержимым одной микробюретки. Так как
объем микробюретки равен в среднем 0,1 мл то
в случае, если кислота взята 0,1 N и объем
пипетки равен 2,5 мл, титр щелочи должен быть
—2~5 ’ = 0,004 N. На литр раствора прибав-
ляют 10 г ВаС12. Щелочь подкрашивают фенол-
фталеином в красный цвет. Чем концентрация СО2
в воздухе выше, тем крепче готовятся растворы.
Методика определения. Собирают
прибор, как показано на рис. 211. Три погло-
тительные колонки С,С,С соединяются при по-
мощи стеклянных и каучуковых трубок. В пер-
вые две колонки насыпают зерна пемзы, смо-
ченные крепким раствором КОН для поглоще-
ния СО2 внешнего воздуха; в третью колонку
насыпают зерна пемзы, смоченные крепким
раствором H2SO4 для высушивания воздуха.
Третья колонка сообщается при помощи трой-
ника с двумя эпруветками D,D. Свободный ко-
нец тройника соединяют с газовой пипеткой А,
причем газовая пипетка при помощи винтовых
зажимов или стеклянных кранов в начале опыта
Рис. 211. Прибор Реберга — Виноку-
рова (показан в собранном виде)
ССС — поглотительные склянки, А — газовая
пипетка, В — уравнительный сосуд, DD —
эпруветки.
остается закрытой сверху и снизу. Газовую пипетку соединяют с уравнительным сосудом
В, в который наливают ртуть либо концентрированный раствор NaCl либо СаС1а.
234
Приводящая трубка первой колонки соединяется с газометром, из которого пускают
ток воздуха. Последний, проходя через поглотительные колонки, освобождается здесь от
СО, и Н2О и поступает в эпруветки. Таким образом, воздух эпруветок, содержащий СО2,
постепенно заменяется воздухом, свободным от этого газа.
Выждав некоторое время, отъединяют вторую эпруветку от первой, подводят послед-
нюю под носик пипетки со щелочью и при токе воздуха, свободного от СО2, наполняют
эпруветку точно отмеренным содержимым пипетки, т. е. тем количеством щелочи, которое
заполняет пипетку между верхней и нижней метками. Затем, присоединив вторую эпру-
ветку к первой и выждав некоторое время при беспрерывном токе воздуха, свободного от
СО,, вводят во вторую эпруветку такой же объем щелочи.
Далее, при продолжающемся токе воздуха, свободного от СО2, оттитровывают кисло-
той из микробюретки сначала щелочь второй эпруветки, а затем первой, погрузив при каж-
дом титровании кончик микробюретки непосредственно в щелочь и титруя до обесцвечи-
вания раствора. Так как титрование ведется при прохождении через щелочь воздуха, свобод-
ного от СО2,титр щелочи не меняется под влиянием внешнего воздуха. На титрование щелочи
в каждой из эп^^еток должно уходить одинаковое количество кислоты.
Так устанавливают титр щелочи.
Освободив эпруветки от содержимого и тщательно промыв их или взяв две другие чис-
тые эпруветки, присоединяют их к тройнику и так же, как и в первом случае, заполняют
щелочью. Расслабив зажимы, закрывающие газовую пипетку и приподымая осторожно
уравнительный сосуд, медленно вытесняют взятую пробу воздуха из газовой пипетки, при-
чем воздух последовательно проходит через первую, а затем вторую эпруветки со щелочью.
Скорость прохождения воздуха должна быть такова, чтобы образовалась беспрерывная
«цепочка» пузырьков воздуха. Полное вытеснение воздуха газовой пипетки заканчивается
в 30—40 минут. При поглощении СО2 баритом протекает реакция:
Ва(ОН)а + СО, = ВаСО3 + Н2О.
Для того чтобы образовавшийся карбонат бария не подвергался гидролизу по уравне-
нию:
ВаСО3 + 2Н,0 = Ва(ОН)2 4- Н,СО3,
что вызвало бы при титровании дополнительный расход кислоты и привело бы к неправиль-
ным выводам, в раствор едкого бария, как было указано, прибавляют хлористый барий
с целью подавления гидролиза.
По прохождении всего воздуха через эпруветки со щелочью оттитровывают избыток
ее при токе воздуха, свободного от СО2, и на основании результатов первого и второго титро-
ваний, учитывая взятый объем воздуха, делают вывод о концентрации СО2 в испытуемом
воздухе.
Пример. Положим, что объем воздуха, взятого для анализа, приведенный к нормаль-
ным условиям, составляет 120,5 мл. На титрование барита при определении его титра ухо-
дило по 180 делений 0,2 N НС1, а после поглощения СО2 на дотитровывание остатка щелочи
первой эпруветки ушло кислоты соответственно 135 делениям микробюретки, а второй —
170 делениям. Объем каждого деления микробюретки равен 0,00052 мл. Нужно на основании
этих данных определить концентрацию СО2 в испытуемом воздухе.
Из данных задачи заключаем, что после поглощения СО2 щелочь ослабела соответственно
180 4- 180 — 135 — 170 = 55 делениям кислоты, т. е. соответственно 0,00052-55 = 0,0286 мл
0,2 N НС1. Каждый миллилитр 0,2 N НС1 соответствует 4,4 мг СО2; следовательно, всего
поглощено 4,4.0,0286 мг СО,.
Отсюда заключаем, что в литре испытуемого воздуха находится:
4,4 • 0,0286 . 1000
—---------------- мг СО,
120,5
или
4,4 - 0,0286.1000.0,506 п „0,
---------—-------------«0,53о/оо СО,.
Описанный метод является довольно точным и дает ошибку в пределах 1—2%.
Окись углерода
Окись углерода (СО) — газ без цвета, запаха и вкуса. Удельный вес
0,96702 (воздух = 1). Горит синеватым пламенем. Смесь из двух объемов
СО и одного объема О при сжигании взрывает.
Выделяется при неполном сгорании продуктов, содержащих углерод.
Присутствует в каменноугольном светильном газе, водяном газе, генера-
торных, выхлопных и доменных газах.
235
Окись углерода — ядовитый газ. При вдыхании вытесняет кислород из
оксигемоглобина, образуя карбоксигемоглобин (карбонилгемоглобин), что
ведет к аноксемии и асфиксии при потере сознания и судорогах.
Уже в концентрации 0,25 мг л при длительном вдыхании производит
вредное действие на организм. Концентрации в 2,5—5,0 мг/л вызывают вскоре
явления отравления (ускорение сердечной деятельности и дыхания, тяжесть
в голове, ощущения слабости в ногах и т. п.). При длительном вдыхании
указанные концентрации вызывают смерть.
СО в малых концентрациях способна вызывать хроническое отравление.
Качественное определение
1. Бумажка, обработанная хлористым палладием (PdCl2), чернеет в при-
сутствии СО вследствие выделения металлического палладия:
PdCL + СО + Н,0 = Pd + 2 НС1 + СО,.
Реактивные бумажки изготовляют следующим образом: растворяют не-
которую навеску PdCl2 в соляной кислоте и доливают дестиллированной
воды до получения 1% раствора хлористого палладия. Полоски фильтро-
вальной бумаги смачивают в полученном растворе, сушат и сохраняют в тем-
ном месте.
Так как при применении описанных бумажек выделяющаяся соляная
кислота вновь переводит металлический палладий в PdC 12, реакция становится
мало показательной. Чтобы избежать этого, бумажки перед применением
смачивают в 5% растворе уксуснокислого натрия. При этом происходит ре-
акция:
CH3COONa + НС1 = СН3СООН + NaCl.
Ни СН3СООН, ни NaCl не действуют на металлический палладий.
Присутствие аммиака мешает реакции и потому его удаляют, пропуская
воздух через раствор кислоты (серной, соляной). Сероводород, как и окись
углерода, вызывает почернение бумажки вследствие образования сернистого
палладия, ввиду чего сероводород должен быть удален пропусканием воздуха
через раствор ацетата свинца — РЬ(С2Н3О.,)2. Ненасыщенные углеводороды
(ацетилен) могут также вызывать почернение хлорпалладиевой бумажки,
восстанавливая хлористый палладий до металлического. Их отделяют про-
пусканием воздуха через жидкий парафин, схлажденный до —8°С, или че-
рез свежий активированный уголь. Во всех перечисленных случаях реак-
тивная бумажка помещается в трубку, один конец которой соединяется с
поглотительным сосудом, другой — с аспиратором.
При изготовлении реактивных бумажек на СО вместо PdCl2 можно взять
натрий палладий хлорид — PdNa2Cl4.
2. Проба с гемоглобином основана на способности гемоглобина крови
давать с СО карбоксигемоглобин:
НЬ + СО = НЬСО.
Карбоксигемоглобин и оксигемоглобин при известной обработке крови
дают разные цветовые реакции. Применяют свежую дефибринированную
кровь, разведенную дестиллированной водой (1 : 4). В бутыль с испытуемым
воздухом вливают дефибринированную кровь и хорошо взбалтывают. Затем
отбирают из бутыли в пробирку 10 мл крови, прибавляют 15 мл 20% раствора
желтой кровяной соли — K4Fe(CN)6, 1 мл уксусной кислоты (1 : 2) и взбал-
тывают. При наличии карбоксигемоглобина проба дает красное окрашива-
ние; контрольная проба без карбоксигемоглобина дает серо- или чернобурое
окрашивание. Проба получается тотчас же и уже через несколько минут
разница в окрашивании исчезает.
Другая модификация той же пробы проводится следующим образом: к
5 мл крови после взбалтывания ее с воздухом прибавляют 15 мл однопроцент-
236
ного раствора таннина и взбалтывают. Через час (через 24 ч. яснее) при на-
личии карбоксигемоглобина получается красное окрашивание, тогда как
в контроле оно серобурое.
3. Спектроскопическая проба основана на способности гемоглобина давать
с СО карбоксигемоглобин, обладающий определенными спектральными свой-
ствами, а именно: он дает в желтозеленой части спектра между линиями D
и Е две темные полосы поглощения, как и оксигемоглобин. Разница, однако,
заключается в том, что, если прибавить к крови какой-либо восстановитель—
(NHJjS, Na2S2O4 и др., то две полосы поглощения, обусловленные присут-
ствием в крови карбоксигемоглобина, сохраняются, тогда как обусловленные
оксигемоглобином пропадают и заменяются одной широкой, нерезко огра-
ниченной полосой между теми же линиями D и Е.
Для данной пробы берут дефибринированную кровь, разбавленную де-
стиллированной водой 1 : 40, 1 : 100.
Количественное определение
Из количественных методов здесь описан метод окисления окиси углерода
в угольный ангидрид с последующим объемным определением последнего.
Метод окисления СО в СО2
Принцип заключается в том, что СО окисляется в СО2при помощи оки-
слителя, каким может служить желтая осадочная ртуть или пятиокись иода.
Реакции при этом протекают следующим образом:
СО + HgO = СО2 + Hg,
5 СО + J2O5 = 5 СО2 + J2.
Угольный ангидрид, образующийся при этих реакциях, улавливается
щелочью (едким баритом) и определяется титрометрически. Результат пере-
числяется на СО.
Для окисления СО в СО2 был предложен также платинированный нихром.
Метод, однако, не подвергся пока лабораторной проверке.
Приборы и реактивы. 1. Две тубулятные склянки, емкостью
в 8—10 л.
2. Три-четыре U-образные трубки.
3. Водяная баня.
4. Пара промывалок Гуревича либо MOOT, либо ЛИГТ.
5. Хлоркальциевая трубка.
6. 0,02 N раствор НС1.
7. 0,02 N раствор Ва(ОН)2 с прибавлением 10 г ВаС12 на 1 л раствора.
8. 1% спиртовый раствор фенолфталеина.
9. Я(елтая окись ртути в зернах диаметром в 2—3 мм или пятиокись иода
в зернах такой же величины.
Продажная окись ртути (HgO) не годится для работы, так как она обыкно-
венно бывает в порошке. Приходится готовить окись самому. В склянке тем-
ного стекла растворяют сулему до получения насыщенного раствора, охлаж-
даемого до —8°С на льду, к которому примешана поваренная соль. Отдельно
готовят раствор едкого калия или натрия с таким расчетом, чтобы две части
щелочи приходились на пять частей сулемы, и охлаждают раствор, после
чего приливают щелочь к раствору сулемы при помешивании, пока выпадаю-
щий осадок не примет яркожелтый цвет. Получившийся осадок желтой окиси
ртути промывают дестиллированной водой декантацией, пока промывная
вода не теряет щелочной характер (проба с фенолфталеином). Затем осадок
переносят в бюхнеровскую воронку, в которую помещают два-три
фильтра. Хорошо отсасывают жидкость. Чтобы достаточно просушить
препарат, его переносят на пористую фарфоровую кюветку и помещают в
237
экссикатор над крепкой серной кислотой на 7 — 8 дней. Полученную твер-
дую аморфную массу осторожно размельчают на зерна диаметром в 2—3 мм.
Мелкие частицы отсеивают через сито.
Пятиокисьиода (йодноватый ангидрид, J2O5) готовится следующим образом.
В колбу емкостью 150 мл наливают 70—75 мл дымящей азотной кислоты
(уд. вес 1,515—1,520), после чего колбу помещают в водяную баню и нагре-
вают до температуры 70—75°С (термометр внутри колбы). Небольшими пор-
циями вводят в колбу в течение 30—40 мин. 15 г дважды возогнанного иода
и повышают температуру до 80—85°С. Через час реакция образования пяти-
окиси иода заканчивается, бурые пары иода перестают выделяться и осадок
пятиокиси иода собирается на дне колбы. По выпадении осадка кислоту сли-
вают, прибавляют 50 мл дестиллированной воды и нагревают до кипения,
которое поддерживают до получения сиропообразной массы. Жидкость ох-
лаждают; при этом пятиокись иода выкристаллизовывается. Маточный раствор
Рис. 212. Прибор для определения окиси углерода воздуха.
А — напорный сосуд, В — сосуд с пробой воздуха, С, и С, — поглотительные tJ-образпые трубки,
D — водяная (масляная) баня, Е и — склянки Гуревича, F — хлоркальциевая трубка с натронной
известью.
сливают, осадок вновь растворяют в 10 мл дестиллированной воды, переносят
в малую фарфоровую чашечку и нагревают до 175—200°С при помешивании
до получения твердых комочков. Полученный реактив вносят в U-образную
трубку, которую помещают в масляную (глицериновую) баню при 180—200°С
и просасывают в течение 2—3 часов воздух, свободный от влаги и СО2. После
этого препарат готов к употреблению.
Методика определения. Пробу воздуха забирают в тубулятную
бутыль емкостью в 8—10 л (заполнение бутыли водой и последующее опо-
рожнение на месте взятия пробы). На трубку бутыли надевают маленькую
каучуковую трубку, которую плотно зажимают винтовым зажимом. Сверху
склянка закрывается каучуковой пробкой, в которую вставлена стеклянная
трубочка. На последнюю надевают маленькую каучуковую трубку, зажатую
зажимом, и в таком виде склянку транспортируют в лабораторию. В лабо-
ратории прибор собирают, как показано на рис. 212.
Три U-образные трубки С, Cj и С2, из которых первая наполнена зернами
пемзы, смоченными крепким раствором едкого калия, вторая — зернами
пемзы, смоченными крепкой серной кислотой, а третья —натронной известью,
соединяют в стык; третью трубку соединяют с U-образной трубкой, в которую
насыпают 40—50 г гранулированной HgO или J2O5. Эта трубка помещается
в водяную баню D (100°С для HgO) или масляную баню (145—150°С для J2O5).
Последняя трубка соединяется с двумя поглотительными склянками Гуре-
вича Е и Ev Склянка Е± соединяется с хлоркальциевой трубкой F, заполненной
натронной известью для задержания СО2 внешнего воздуха на случай обрат-
238
ного тока его. Еще до присоединения склянки с испытуемым воздухом присое-
диняют к хлоркальциевой трубке аспиратор и просасывают через всю си-
стему в течение 30—40 минут воздух с целью удалить из трубок и склянок
СО2. Затем в склянки Е и Ех при токе воздуха, свободного от СО2, вводят
из бюретки точно отмеренное количество Ва(ОН)2 определенного титра (при-
близительно 0,02 N), прибавляют по несколько капель фенолфталеина и за-
крывают склянки, после чего, удалив аспиратор, присоединяют к системе
склянку В с испытуемым воздухом. В свою очередь склянку В соединяют
со склянкой Д, наполненной водой и служащей напорным сосудом.
Доведя температуру в бане до необходимой высоты, пускают воду из
склянки А в склянку В. При этом испытуемый воздух из склянки В выте-
сняется и проходит последовательно через всю систему. В первой U-образной
трубке с едким калием он освобождается от СО2 и других кислых продуктов,
могущих присутствовать в воздухе; во второй U-образной трубке с крепкой
серной кислотой воздух освобождается от влаги, газообразных оснований
и непредельных углеводородов, если таковые имеются в нем; в третьей
U-образной трубке с натронной известью воздух окончательно освобождается
от СО2, после чего поступает в U-образную трубку с окислителем. Здесь СО
окисляется в СО2. Далее, при прохождении через барит СО2 вступает с ним
в реакцию по уравнению:
СО2 *4“ Ва(ОН)2 = ВаСО3 -|- Н2О,
понижая титр баритового раствора.
После вытеснения из склянки В всего испытуемого воздуха отъединяют
от системы склянку В и при помощи аспиратора, присоединенного к трубке F,
просасывают еще некоторое время воздух для того, чтобы провести через
всю систему те остатки испытуемого воздуха, которые застряли в самой си-
стеме. Просасывание все время должно вестить со скоростью не более 3—4 л
в час.
Если в качестве окислителя был взят J2O5, то выделяющийся при реакции
свободный иод должен быть уловлен, для чего между U-образной трубкой
с J2O5 и поглотителем с баритом помещают поглотительный сосуд с крепким
раствором К J (60 г в 100 мл раствора).
Если в воздухе присутствуют, кроме указанных выше, и другие органи-
ческие продукты, которые способны окислиться при прохождении через
систему, то они должны быть предварительно отделены соответствующим
поглотителем; так, для предельных углеводородов можно взять силика-
гель, для альдегидов и кетонов — бисульфит натрия (NaHSO3) и т. д. U-об-
разные трубки с соответствующими поглотителями вводятся в систему до
окислительной трубки.
После того как весь испытуемый воздух проведен через раствор барита,
продолжают просасывать воздух при помощи аспиратора и оттитровывают
барит кислотой до полного обесцвечивания. На основании уменьшения
титра раствора барита делают вывод о количестве поглощенного СО2, ко-
торое перечисляют на СО х.
Полезно до проведения опыта или после него поставить и холостой опыт
с целью выяснения поправки, какую нужно ввести в результат опыта в
смысле возможного изменения титра Ва(ОН)2 ввиду остатков СО2, недоста-
точно удаленных из системы.
Пример. При исследовании воздуха гаража на содержание СО для анализа взято 8,2 л
воздуха при температуре 12° С и давлении 764 мм Hg. Определение СО произведено ио ме-
тоду окисления его в СО2 с последующим поглощением СО2 баритом.
В две промывалки, в которых велось поглощение СО2, налито по 20 мл Ва(ОН)2. На
титрование барита каждой склянки до опыта уходит по 18 мл 0,02 N НС1. После опыта на
1 Гуревич и Горкин, Определение окиси углерода в воздухе, «Труды Укр.
ин-та патологии и гигиены труда», вып. VI, 1928.
239
титрование барита первой склянки ушло 12,8 мл 0,02 NHC1, второй — 18 мл. Нужно опре-
делить концентрацию СО в воздухе гаража в миллиграммах на литр.
На титрование барита обеих склянок после опыта ушло на 18 4- 18 —12,8 — 18 = 5,2 мл
кислоты меньше, нежели до опыта. Каждый миллилитр 0,02 N HCI эквивалентен 0,44 мг
СО2. Отсюда заключаем, что во время опыта поглощено 0,44 • 5,2 = 2,288 мг СО2, что соот-
ветствует
2,228 -28 ,
----------- = 1,456 мг СО.
По приведении взятого объема воздуха к нормальным условиям получаем:
________8,2 764
(1 + 0,00367 • 12) . 760
« 7,896 л.
Следовательно, концентрация СО в испытуемом воздухе соответствует:
1,456
7,896
0,184 мг/л.
Описанный метод в части определения полученного СО2 осуществляется
нередко при помощи прибора Реберга—Винокурова; в этом случае воздух
после окисления СО в СО2 проводится через эпруветки со щелочью, которая
тут же оттитровывается, как описано на стр. 235.
Определение СО2, О2и СО прибором ОРСА
Прибором Орса можно определить СО2, О2 и СО в газовой смеси, причем
СО2 и СО определяются при относительно больших концентрациях (топочный
газ).
Принцип метода волюмометрический: из определенного объема
отобранной газовой смеси поглощают соответствующими сорбентами последо-
вательно указанные газы и по уменьшению объема газовой смеси судят о
количестве поглощенных газов.
Поглощение СО2, О2 и СО протекает по следующим реакциям:
2КОН + СО2 = К2СО3 + Н2О;
2С,Н2(ОН)3Н + О = (ОН)3С6Н2 - С6Н2(ОН)3 + Н2О;
Си2С12 + СО + 2Н2О = СОСп2С12 • 2Н2О.
Приборы и реактивы. 1. Прибор Орса (рис. 213) состоит
из газовой бюретки а на 100 мл, снабженной сверху краном и сообщаю-
щейся при помощи каучуковой трубки с уравнительным сосудом Ь. Для
поддерживания во время опыта температуры газовой смеси на одной
и той же высоте бюретка погружена в термостат с, в который налита
вода комнатной температуры. Сверху от бюретки отходит капилляр d, от
которого в свою очередь отходят три капиллярные трубки, снабженные кра-
нами. Капиллярная трубка с тремя капиллярными отростками носит назва-
ние «гребенки». К каждому капиллярному отростку присоединяется при
помощи каучуковой трубки поглотительная пипетка.
Каждая из поглотительных пипеток (е, / и g) представляет собой цилиндр,
заполненный стеклянными трубками, для придания большей поверхности
соприкосновения между газом и той поглощающей жидкостью, которая на-
лита в цилиндр. Каждый цилиндр сверху заканчивается капилляром, на
котором имеется метка; снизу каждый цилиндр при помощи стеклянной дуго-
образно изогнутой трубки сообщается с другим таким же парным цилиндром.
Каждый из последних закрывается каучуковой пробкой, в которую встав-
ляется стеклянная трубочка. На стеклянную трубочку надевается каучуковая
трубочка, заканчивающаяся каучуковым же мешочком, назначение которого
сводится к защите реагентов от влияния атмосферного воздуха. (На рис. 213
виден полностью один лишь мешочек Л). Капилляр d заканчивается трехходо-
240
вым краном к. Один отросток крана соединяется с U-образной трубкой г,
наполненной ватой для обеспыливания газовой смеси. Другой отросток крана
соединяется с грушей т, при помощи которой нагнетают в U-образную
трубку испытуемую газовую смесь. Прибор монтирован в деревянном ящике.
2. Раствор КОН уд. веса 1,2—1,26.
3. Щелочной раствор пирогаллола (приготовление см. на стр. 223).
4. Аммиачный раствор однохлористой меди. Приготовление: растворяют
в склянке 250 г NH4C1 в 750 мл воды, прибавляют 200 г Си2С12 и, тщательно
закрыв склянку, взбалтывают до полного растворения Си2С12. В склянку
вводят медную спираль, доходящую до шейки, и закупоривают. Перед за-
полнением поглотительной пипетки три объема описанного раствора сме-
шивают с одним объемом NH„OH уд. веса 0,905.
Рис. 213. Прибор Орса:
а — газовая бюретка, Ъ — уравнительный сосуд, с — термостат, d — гребенка, i — кран, е, f, g — по-
глотительные пипетки, h — каучуковый мешочек, k — трехходовый кран, г — ^/-образная трубка с
ватой для обеспыливания газовой смеси, т — груша.
Вместо аммиачного раствора меди для поглощения СО применяют и кислый
раствор меди: 16—18 частей хлористой меди смешивают с 24—27 частями
концентрированной соляной кислоты и 55—60 частями воды с прибавлением
0,2—0,3 г хлористого олова (SnCl2 . 2Н2О).
Методика определения. Собирают прибор, как показано на ри-
сунке 213. В уравнительный сосуд b наливают насыщенный раствор NaCl (СаС1«)
либо ртуть,открывают кран бюретки и, установив кран к на сообщение бюретки
с наружным воздухом, поднятием уравнительного сосуда вытесняют из бюретки
весь воздух, после чего закрывают кран бюретки. Далее следует заполнение
поглотительных пипеток соответствующими реактивами. Пипетка е запол-
няется раствором „КОН. Для этой цели открывают кран этой пипетки; кран к
устанавливают на сообщение с наружным воздухом и, открыв парный сосуд
пипетки, а именно ег, наливают в него раствор КОН, пока пипетка и парный
сосуд не окажутся заполненными несколько больше половины. После этого
закрывают кран к и опусканием уравнительного сосуда доводят уровень
щелочи в пипетке точно до метки (в парном сосуде при этом уровень жидкости
опускается), после чего закрывают кран пипетки и парный сосуд пипетки.
Пипетка / точно таким же образом заполняется щелочным раствором ПИ po-
ll А. II. НурштеПп
241
галлола, а пипетка g—раствором хлористой меди. При открытом кране
бюретки и открытом кране к поднятием уравнительного сосуда вытесняют
воздух из бюретки, после чего закрывают ее кран. Прибор готов к работе.
Забор газовой смеси для анализа производится так: кран к устанавливают
на сообщение груши с U-образной трубкой г. Последняя свободным концом
должна быть направлена к источнику, из которого желательно произвести
забор газовой смеси; если нужно, то на свободный конец U-образной трубки
одевают еще каучуковую трубку, свободный конец которой выводят в про-
странство, из какого производится забор пробы газа. Несколькими накачива-
ниями груши заполняют U-образную трубку испытуемою газовою смесью.
Затем кран к устанавливают на сообщение с бюреткой, открывают кран по-
следней и опусканием уравнительного сосуда заполняют бюретку испытуе-
мою газовою смесью.
Так как в капиллярной трубке d находился воздух, который перешел
в бюретку, изменив несколько состав испытуемой газовой смеси, поднятием
и опусканием уравнительного сосуда несколько раз вытесняют из бюретки
газовую смесь, заменяя ее испытуемой. В бюретку забирают несколько больше
100 мл газовой смеси. Закрывают кран бюретки и поднятием уравнительного
сосуда устанавливают уровень жидкости в бюретке точно на сотом делении.
Если теперь зажать между пальцами каучуковую трубку уравнительного
сосуда и на одно мгновение открыть кран бюретки, то избыток газа выйдет
из бюретки, и в ней окажутся 100 мл его под атмосферным давлением.
Если прибор в порядке, то уровни жидкости в бюретке и в пипетках не
должны опускаться; в противном случае проверяют краны и каучуковые
соединения.
Далее следует определение газов: СО2, О2 и СО.
Закрывают кран к. Уравнительный сосуд поднимают вверх и открывают
кран пипетки е; при этом газовая смесь переходит из бюретки в пипетку,
вытесняя из нее щелочь в парный сосуд. Поднятие уравнительного сосуда
прэизводят до того момента, пока жидкость в бюретке не дойдет до крана
бюретки; тогда кран бюретки закрывают, оставляя на некоторое время (40—60
секунд) газ в пипетке. Затем, открыв кран бюретки, переводят в нее газ из
пипетки путем опускания уравнительного сосуда, следя при этом, чтобы жид-
кость в поглотительной пипетке не переходила выше метки. Так поступают
несколько раз до полного поглощения СО2. Полноту поглощения газа прове-
ряют следующим образом. Опускают уравнительный сосуд при открытых
кранах бюретки и пипетки до тех пор, пока жидкость в пипетке не подойдет
точно к метке, и, закрыв краны, измеряют объем оставшейся газовой смеси
в бюретке, установив уровни жидкостей в уравнительном сосуде и бюретке
в одной горизонтальной плоскости. После этого вновь переводят газ из бю-
ретки в пипетку и через некоторое время назад из пипетки в бюретку, измерив
описанным только что способом объем оставшейся газовой смеси. Если два
последовательных отсчета совпадают, то поглощение считается законченным.
По уменьшению объема газа в бюретке после поглощения СО2 делают
заключение о количестве СО2 в объемных процентах в испытуемом газе.
Определив СО2, переходят к определению О2, пользуясь второй поглоти-
тельной пипеткой, а затем СО, пользуясь третьей поглотительной пипеткой
и манипулируя точно так же, как при определении СО2.
На поглощение СО2 уходит 2—3 минуты, на поглощение О2— от 5 до 15
минут, на поглощение СО — от 5 до 10 минут. Все определение продолжается
в среднем 30—40 минут.
Поглощающие жидкости время от времени следует заменять свежими,
так как поглощающая способность их ограничена.
1 мл раствора КОН поглощает.......................
1 » раствора СвН2(ОН)3Н поглощает.................
1 » аммиачного раствора Си2С12 поглощает..........
1 л кислого раствора Си2С12 поглощает.............
до 40 мл СО2
» 8—9 » О2
» 6 » СО
» 1,8 » СО
242
В заключение необходимо отметить следующие факты: крепкие растворы
пирогаллола под влиянием О2 способны выделять СО, что может внести ошибку
в анализ; раствор хлористой меди, поглотивший большое количество СО,
начинает выделять его назад, что заставляет часто менять этот раствор. Ра-
створ хлористой меди поглощает не только СО, но и тяжелые углеводороды,
ввиду чего эти газы должны быть удалены при взятии пробы. Для этой цели
вслед за U-образной трубкой присоединяют поглотитель с силикагелем.
Описанный прибор Орса применяют преимущественно в техническом ана-
лизе.
Сернистый ангидрид
Сернистый ангидрид (SO2) — удушливый газ с удельным весом 2,2639
(воздух = 1). Легко растворим в воде. Выделяется при сжигании продуктов,
содержащих серу (уголь), при обжиге и плавлении сернистых руд, при вы-
плавке чугуна. Сернистым ангидридом пользуются для получения серной
кислоты (сернокислотные заводы). Он применяется также как белящее сред-
ство (целлюлозные, свечные, салотопенные и др. заводы), как консервант
(консервное дело), в холодильном деле, а также для целей дезинсекции и
дератизации. Действует раздражающе на слизистые оболочки дыхательных
путей и глаз, вызывает чиханье и кашель, при больших концентрациях —
спазм голосовой щели, одышку, расстройство сознания. 0,06 мг/л максималь-
ная концентрация, которую человек может перенести в течение получаса.
При хроническом воздействии наблюдается привыкание к газу.
Качественное определение
Сернистый ангидрид можно качественно определить: 1) по характерному
запаху, который ощущается при концентрациях, начиная с 0,008 мг/л; 2) при
помощи бумажки, смоченной азотнокислой закисью ртути (HgNO3); в при-
сутствии SO2 бумажка чернеет вследствие выделения ртути; от действия соляной
кислоты почерневшая бумажка не обесцвечивается (почернение бумажки на-
блюдается и при H2S и NH3); 3) фильтровальной бумажкой смоченной
крахмальным клейстером и окрашенной в синий цвет парами иода, которая
обесцвечивается от действия SO2: SO2 + J2 + 2 Н2О = H2SO4 + 2 HJ. Обес-
цвечивание бумажки наблюдается также и от действия других восстанови-
телей.
Количественное определение
Из методов количественного определения SO2 здесь приведены весовой,
объемный и нефелометрический.
Весовой метод
Принцип метода заключается в том, что воздух просасывают
через раствор иода в иодистом калии; при этом SO2 окисляется до H2SO4 по
приведенной выше реакции.
Образовавшуюся серную кислоту осаждают хлористым барием:
H2SO4 + ВаС12 = BaSO4 + 2НС1.
Полученный сульфат бария собирают на беззольном фильтре, который
затем просушивают и прокаливают вместе с осадком. По весу сульфата бария
определяют количество поглощенного из воздуха SO2.
Из приведенных двух реакций видно, что каждая частица SO2 приводит
к образованию одной частицы BaSO4. Выражая весовые отношения между
поглощенным из воздуха SO2 и найденным BaSO4, находим, что 64,06 ве-
совых частей SO2 соответствуют 233,42 весовым частям BaSO4. Следовательно,
переводный коэфициент для пересчета BaSO4 на SO2 равен:
-64’06 ~ 0,2744.
233,42
243
Приборы и реактивы. 1. Три поглотительные склянки типа
Петри или фильтр Городецкого.
2. Прибор для просасывания воздуха.
3. 0,01 N раствор иода в иодистом калии (см. стр. 64); при больших кон-
центрациях SO2 можно пользоваться 0,1 N раствором иода.
4. 10% раствор ВаС12.
5. 10% раствор НС1.
Методика определения. В три поглотительные склянки вводят
по 15—20 мл 0,01 N раствора иода в иодистом калии и просасывают 100 л
воздуха со скоростью 20—25 л в час
Содержимое всех склянок переводят в стакан, склянки тщательно про-
мывают дестиллированной водой и ополоски вводят в тот же стакан. Послед-
ний помещают на баню и нагревают до обесцвечивания. Обесцвеченный го-
рячий раствор подкисляют 10% соляной кислотой и тут же вводят по каплям
нагретый 10% раствор хлористого бария в избытке (1,5—2 мл). Стакан с со-
держимым оставляют на несколько часов до полного выпадения BaSO4, после
чего фильтруют через уплотненный беззольный фильтр. Осадок промывают
декантацией, затем переносят его на фильтр и промывают до исчезновения
в промывных водах ВаС12. Промывные воды не должны давать мути от при-
бавления AgNO3:
ВаС12 + 2 AgNO3 = Ba(NO3)2 + 2 AgCl.
Фильтр вместе с осадком переносят в платиновый тигель, высушивают
и прокаливают до постоянного веса.
От привеса вычитают вес золы фильтра; разность соответствует весу BaSO4.
Для пересчета на SO2 умножают, как было указано, на коэфициент 0,2744.
Пример. При исследовании воздуха кузнечного цеха на содержание SO2 пропущено
через раствор иода 100 л воздуха при температуре 18° С и барометрическом давлении 758 мм
Hg. Вес осажденного BaSO4 оказался равным 3,182 мг. Какова концентрация SO. в данном
цехе?
По приведении данного объема воздуха к нормальным условиям получаем 93,6 л. Пере-
числив сульфат бария па SO2 и поделив на 93,6, находим концентрацию SO2:
3,182 • 0,2744 n
------------= 0,0093 мг/л.
93,6
•
Вместо раствора иода для окисления SO2 в H2SO4 можно применить 5%
перекись водорода, свободную от серной кислоты. В этом случае реакция
протекает по уравнению:
SO2 + Н2О2 = H2SO4.
Нередко в качестве окислителя пользуются бертолетовой солью:
3SO2 + КС1О3 + ЗН2О = 3H2SO4 + КС1.
• Бертолетовую соль берут в 5% растворе. Соль должна быть чистой (дваж-
ды перекристаллизованной).
Методика весового определения при этих окислителях такая же, какая
описана для иода.
Объемный метод
В случаях, когда в воздухе имеется незначительная концентрация SO2 и
через поглотительный раствор просасываются небольшие объемы воздуха,
навеска BaSO4 получается ничтожная и весовой метод становится нена-
дежным.
В этих случаях лучше прибегать к одному из описанных ниже методов.
1 При применении фильтра Городецкого можно просасывание воздуха вести со ско-
ростью 80—100 л в час.
244
Предложен ряд методов объемного определения SO2. Здесь описан метод,
основанный на титрометрическом определении серной кислоты, образовав-
шейся при окислении SO2. Это проще всего сделать, оттитровывая серную
кислоту едким натрием при индикаторе метилоранже.
При этом в качестве окислителя берут 5% раствор перекиси водорода.
До просасывания воздуха раствор перекиси точно нейтрализуют слабым
раствором щелочи.
После отбора пробы образовавшуюся серную кислоту оттитрововывают
слабым раствором едкого натрия и по израсходованному количеству NaOH
делают вывод о количестве поглощенного SO2.
Так, из реакций:
SO2 + Н2О2 = H2SO4,
H2SO4 + 2NaOH = Na2SO4 + 2H2O
видно, что 1 мл 0,01 N раствора NaOH соответствует
SO2 64,06 n QOHQ СП
-----— =----------= 0,3203 мг ЬО2.
2 • 100 200
Пример. При исследовании воздуха литейного цеха на SO2 через нейтральный раствор
5% перекиси водорода было пропущено 80 л воздуха (температура 20° С, барометрическое
давление 762 мм Hg). При оттитровывании образовавшейся серной кислоты израсходовано
5,4 мл 0,01 N NaOH. Нужно определить концентрацию SO2 в воздухе обследован-
ного цеха.
Приведя воздух к нормальным условиям, получаем 74,72 л. Концентрация SO2 в воз-
духе равна:
0,3203 . 5,4 _ z
------------ж 0,023 мг/л.
74,72
Нефелометрический метод.
Принцип метода основан на том, что слабые растворы серной
кислоты при действии на них хлористого бария образуют муть, по интен-
сивности которой можно сделать вывод о количестве серной кислоты и отсюда
о количестве поглощенного SO2.
Приборы и реактивы. 1. Стандартный раствор K2SO4, 1 мл
которого соответствует 0,1 мг SO2. Для получения такого раствора раство-
ряют 0,272 г K.SO4 в 1 л воды ~ 0,272).
\ lu 002 /
2. 5% раствор бертолетовой соли.
3. 0,1 N раствор НС1.
4. 10% раствор ВаС12.
5. Набор пробирок одинакового диаметра либо нефелометр.
Методика определения. Забор пробы ведется, как и в преды-
дущем методе. Раствор из поглотительных приборов переводится в измери-
тельный цилиндр. Небольшими порциями воды, чтобы не разбавить сильно
раствор, промывают поглотительные склянки и переводят промывные воды
в тот же цилиндр.
Составляют шкалу из раствора K2SO4 соответственно от 0,02 до 0,2 мг
SO2 с интервалами в 0,02 мг. Для этой цели нужно в пробирки налить следу-
ющие количества стандартного раствора: 0,2; 0,4; 0,6; 0,8; 1,0; 1,2; 1,4; 1,6;
1,8; 2,0.
Во все пробирки прибавляют по 8 мл раствора бертолетовой соли и урав-
нивают водой объемы жидкости во всех пробирках, доводя их до 10 мл; затем
прибавляют по 1 мл 0,1 N раствора соляной кислоты и 1 мл хлористого бария.
Одновременно со стандартными пробирками приготовляют опытную про-
бирку, взяв 10 мл из поглотительной жидкости и прибавив сюда же по 1 мл
0,1 N НС1 и раствора ВаС12.
245
Через 10—15 минут производят сравнение на темном фоне. Муть держится
20—30 минут. На основании произведенного нефелометрирования делают
заключение относительно количеств поглощенного SO2.
Пример. Опытная пробирка дала такую же муть, как и в третьей пробирке. Отсюда
заключаем, что количество поглощенного SO2 в ней соответствует 0,06 мг. Положим, что
общее количество поглотительной жидкости (с промывными водами) было равно 48 мл.
Выводим, что общее количество поглощенного SO2 равно:
Нефелометрический метод применяется при малых концентрациях SO,
в воздухе.
При одновременном присутствии в воздухе кроме SO2 еще и H2SO4 в вы-
сокодисперсном состоянии (травление металлов, зарядка аккумуляторов)
необходимо перед поглотительными склянками поставить ватный или бу-
мажный фильтр; тогда H2SO4 при просасывании воздуха остается на
фильтре, a SO2 пройдет через фильтр и задержится в поглотителе. Если не
принять этой предосторожности, то в поглотителе задержится и H2SO4 и
SO2, что даст повышенные результаты определения.
При одновременном присутствии в воздухе SO2 и H2S нельзя в качестве
сорбента применять Н2О2, так как произойдет реакция:
H2S + 4 Н2О2 = H2SO4 + 4 Н2О,
т. е. окисленными окажутся и SO2 и H2S.
Сероводород
Сероводород (H2S) — бесцветный газ неприятного запаха с удельным
весом 1,1912 (воздух = 1). Ощущается явственно обонянием уже при кон-
центрациях 0,001—0,002 мг/л. В воздухе встречается при гниении органи-
ческих веществ (утильцехи, выгребные ямы), на некоторых химических про-
изводствах (получение сернистых красок, производство сероуглерода), при
получении искусственного шелка, на кожевенных заводах (отмочнозольные
цехи), на производствах клея и желатины, на костеобрабатывающих заводах
и т. п. В химических лабораториях сероводородом часто пользуются для
аналитических целей.
При концентрациях 0,5 мг/л вызывает раздражение слизистых оболочек
дыхательных путей, конъюнктивы. При концентрациях 0,7 мг/л воздействие
в течение 15—30 минут приводит, кроме указанных явлений, еще и к тош-
ноте, рвоте, выступлению холодного пота, потере сознания. Концентрации
1,8 мг/л и выше быстро приводят к смерти от остановки дыхания.
Качественное определение
1. Бумажка, смоченная щелочным раствором уксуснокислого свинца,
в присутствии H2S буреет или чернеет вследствие образования сернистого
свинца (PbS). При долгом лежании бумажка вновь белеет ввиду перехода
сернистого свинца в сернокислый свинец — PbSO4 (соль белого цвета).
Щелочный раствор уксуснокислого свинца получается прибавлением
крепкого раствора едкого натрия к 10% раствору уксуснокислого свинца до
растворения выпавшей гидроокиси свинца.
2. Бумажка, смоченная аммиачным раствором нитропруссида натрия —
Na3 [Fe(CN)5NO] — в присутствии H2S приобретает краснофиолетовый цвет.
3. Обоняние позволяет обнаружить H2S в концентрациях 0,00034 мг/л.
246
Количественное определение
Здесь описаны методы весовой, йодометрический и колориметрический.
Весовой метод
Принцип метода основан на окислении сероводорода до серной
кислоты и осаждении последней в виде сульфата бария:
H2S + J2 -= 2HJ + S; S + 3Br2 + 4H2O = H2SO4 + 6HBr;
H2SO4 + BaCl2 = BaSO4 + 2HC1.
Каждый миллиграмм осажденного BaSO4 эквивалентен
233Ж -°-l456 "r
H,S, соответственно .
BaSO4 ‘
Приборы и реактивы. 1. Две поглотительные склянки.
2. Аспиратор.
3. 0,01 N раствор иода в иодистом калии.
4. Бромная вода.
5. 10% раствор НС1.
6. 10% раствор ВаС12.
Методика определения. Воздух просасывают через две поглоти-
тельные склянки с раствором иода в иодистом калии со скоростью 20—25 л в час.
При этом сероводород окисляется до серы. Содержимое поглотительных склянок
переводят количественно в химический стакан, нагревают на водяной бане до
удаления избытка иода, подкисляют соляной кислотой и, прибавив бромной
воды, нагревают на водяной бане. Сера при этом окисляется до серной кис-
лоты. Нагревание ведется до полного обесцвечивания раствора. Далее сле-
дует осаждение серной кислоты посредством хлористого бария, выделение
сульфата бария и его взвешивание. Полученный вес перечисляют на H2S,
пользуясь коэфициентом 0,1456.
Вместо раствора иода для поглощения H2S можно пользоваться аммиач-
ным раствором перекиси водорода: к 94 мл 3% раствора Н2О2 прибавляют
6 мл 25% аммиака.
В этом случае H2S окисляется до H2SO4, и нет необходимости в приме-
нении брома:
H2S + 4 Н2О2 = H2SO4 + 4 Н,О.
Дальнейшие операции производятся, как описано выше (стр. 244).
Йодометрический метод Петровой и Яковцевской
Принцип метода заключается в том, что сероводород поглоща-
ется щелочью, из которой затем вытесняется кислотой и определяется
титрованным раствором иода. Ход реакции:
H2S + 2NaOH = Na2S + 2Н2О; Na2S + 2НС1 = 2NaCl + H2S;
H2S 4- J2 = 2HJ + s.
Из приведенных реакций видно, что 1 мл 0,01 N раствора иода соответ-
ствует 0,17 мг H2S.
Приборы и реактивы. 1. Две поглотительные склянки.
2. Аспиратор.
3. 0,01 N раствор NaOH (можно неточный).
4. 0,01 N раствор иода.
5. 0,01 N раствор тиосульфата натрия.
6. 1 N раствор НС1 или 2 N раствор СН3СООН (можно неточный).
7. 1% крахмальный клейстер.
247
Методика определения. Воздух просасывают через две по-
глотительные склянки с 15 мл 0,01 N раствора NaOH в каждой со скоростью
20—25 л в час.
Содержимое склянок переводится количественно в мерную колбу на 100 мл
и доводится до метки водой, лишенной свободного кислорода (свеже про-
кипяченной). Раствором наполняют бюретку на 50 мл.
В мерную колбу на 250 мл наливают 20мл 0,01 N раствора иода, прили-
вают 5 мл нормальной НС1 или 10 мл 2 NCH3COOH и прибавляют сюда из
бюретки понемногу 50 мл налитой туда жидкости, каждый раз сильно взбал-
тывая содержимое колбы. Через 10 минут избыток иода оттитровывают 0,01 N
раствором тиосульфата натрия.
Одновременно ставят холостой опыт: взяв 20 мл 0,01 N иода и подкислив
соляной или уксусной кислотой, прибавляют 50 мл NaOH такой же концен-
трации, как и в главном опыте. Иод оттитровывают через 10 минут тиосуль-
фатом натрия.
Разность между расходом тиосульфата в холостом и главном опытах
соответствует количеству 0,01 N раствора иода, которое пошло на связывание
H2S. На основании полученного результата делают расчет.
Пример. При определении H2S в воздухе отмочнозольного цеха кожевенного завода
через 0,1 N раствор NaOH пропущено 200 л воздуха (объем приведен к нормальным усло-
виям). Поглотительная жидкость переведена в колбу на 100 мл и доведена до метки водой.
В колбу на 250 мл налито 20 мл 0,01 N раствора иода, сюда прибавлено 5 мл нормальной
НС1 и затем из бюретки понемногу переведено сюда же 50 мл поглотительной жидкости.
Для оттитровывания избытка иода понадобилось 12 мл 0,01 N раствора тиосульфата натрия.
При оттитровывании иода в холостом опыте уходит 19,6 мл тиосульфата.
Отсюда находим, что на связывание H2S пошло 19,6— 12 = 7,6 мл 0,01 N раствора
иода, что соответствует 0,17 • 7,6 мг H2S. Так как из поглотительной жидкости для опыта
берется лишь 50 мл, то при пересчете на весь объем поглотительной жидкости получаем
0,17 • 7,6 • 2 мг H2S, что на литр испытуемого воздуха дает
~ 0,01292 мг H..S.
200
Колориметрический метод Сендерихиной—Арончук
Принцип метода основан на том, что диметилпарафенилендиамин
в присутствии хлорного железа и H2S образует метиленовую синь по реакции:
2[(CH3)2NC6H4NH2H2SO4] + 6FeCl3+ H2S =
N
(CH3)2NCeH3 \ / CeH8: N(CH3)2C1 + 2H2SO4 + NH4C1 + 6FeCI2 + 4HC1.
S
Заметная окраска раствора наступает уже при ничтожных количествах H2S.
Приборы и реактивы. 1. Две поглотительные склянки.
2. Аспиратор.
3. Диметилпарафенилендиамин.
4. НС1 уд. веса 1,19.
5. Хлорное железо.
6. Индикаторный реагент: 1 г диметилпарафенилендиамина растворяют
в 300 мл НС1 уд. веса 1,19 и прибавляют сюда раствор 1 г хлорного железа в
100 мл Н2О. Реактив сохраняется в темной склянке.
7. 0,01 N раствор NaOH.
Методика определения. Воздух просасывают через две про-
мывалки с 10 мл 0,01 N раствора NaOH в каждой со скоростью 20—25 мл
в час. При этом происходит реакция:
H2S + 2NaOH = Na2S + 2НаО
248
После просасывания воздуха во избежание окисления образовавшегося
Na2S тут же прибавляют 1 мл индикаторного реагента. Посинение появляется
через 5—30 минут. Чувствительность метода: 0,0002 мг H2S в 10 мл жидкости.
Для сравнения пользуются шкалой, приготовленной ex tempore из Na2S
и соответствующей от 0,0002 до 0,002 мг H2S.
Если в литре воды растворить 0,229 г химически чистого Na2S, соответ-
ственно -гг^т/г» то 1 мл раствора соответствуете,! мг H2S. Раствор разбав-
Г12о *10
ляют в тысячу раз, 1 мл разбавленного раствора соответствует 0,0001 мг
H2S. Из такого раствора готовят шкалу.
Ввиду того что Na2S окисляется на воздухе, необходимо раствор Na2S прове-
рить по раствору иода. 1мл 0,01 N раствора иода соответствует 0,39027 мг NaaS
согласно уравнению:
Na2S + J2 = 2NaJ + S.
Определение SO2 и H2S при совместном присутствии их в воздухе
При совместном присутствии SO2 и H2S в воздухе эти два газа могут быть
определены раздельно следующим образом.
1. Воздух просасывают через раствор иода в иодистом калии. При этом
протекают, как уже было указано, следующие реакции:
SO2 + J2 + 2Н2О = H2SO4 + 2HJ,
H2S + J2 = 2HJ + S.
Отогнав избыток иода, отфильтровывают поглотительную жидкость и
промывают фильтр. При этом в фильтрат переходит серная кислота, образо-
вавшаяся за счет SO2, а сера, образовавшаяся за счет H2S, задерживается
на фильтре.
Серную кислоту, перешедшую в фильтрат, осаждают хлористым барием
и по выпавшему BaSO4 определяют SO2 (см. стр. 243).
Фильтр вместе с серой переводят в стакан, подкисляют соляной кисло-
той и при помощи бромной воды окисляют серу в серную кислоту, которую
затем определяют осаждением хлористым барием.
2. Другой метод раздельного определения SO2 и H2S при совместном при-
сутствии их в воздухе основан на том, что нейтральный раствор КС1О3 пол-
ностью окисляет SO2 в серную кислоту; сероводород же окисляется ней-
тральным и слабосернокислым раствором бертолетовой соли всего лишь в
количестве от 3 до 4%. Если взять два поглотителя — один с нейтральным
раствором КС1О3, а другой с аммиачным раствором перекиси водорода —
и просасывать через них воздух, то в первом поглотителе задержится весь
SO2, окисленный в серную кислоту, а во втором — весь H2S, окисленный
в серную кислоту и переведенный затем в сульфат аммония:
3SO2 + КС1О3 + ЗН2О = 3H2SO4 + КС1,
H2S + 4Н2О2 = H2SO4 + 4Н2О,
H2SO4 + 2NH3 = (NH4)2SO4.
В качестве поглотительных сосудов берут фильтры Городецкого. В пер-
вый сосуд наливают 50 мл 5% раствора КС1О3, во второй — 47 мл 3% Н2О2
и 3 мл концентрированного аммиака. Максимальная скорость просасывания
1,5 л/мин. Для осаждения серной кислоты в виде BaSO4 раствор нагревается
до кипения и подкисляется соляной кислотой, после чего к нему прибавляется
кипящий раствор хлористого бария.
Содержимое второго сосуда предварительно кипятится полчаса для раз-
рушения избытка Н2О2 (Гуревич).
249
Серная кислота (H^SOJ и серный ангидрид (S08)
Серная кислота (H2SO4) представляет собой маслянистую жидкость, в
чистом виде бесцветную и не обладающую запахом.
Различают следующие сорта серной кислоты: камерную (62—65% HgSOJ,
гловерную (78% H2SO4), купоросное масло (92—96%
моногидрат (98—100% H2SO4); «олеум», или дымящая серная ки-
слота, представляет 12—14% раствор SO3 в концентрированной серной ки-
слоте.
Серная кислота широко применяется в промышленности (обработка ме-
таллов, приготовление соды, квасцов, взрывчатых веществ, искусственного
удобрения, красок и т. п.).
В воздух рабочих помещений серная кислота может попадать в виде мель-
чайшей пыли или тумана (аккумуляторные заводы, травление металлов).
При изготовлении серной кислоты по контактному способу в рабочие по-
мещения могут проникать пары SO3, которые с парами воды дают серную
кислоту.
В концентрациях 0,0005—0,002 мг/л переносится без видимого вреда.
Концентрации 0,006—0,008 мг/л вызывают раздражение слизистых оболочек
дыхательных путей.
Качественное определение
Воздух просасывают через раствор хлористого бария, подкисленный соля-
ной кислотой. При наличии в воздухе H2SO4 или SO3 получается муть, обус-
ловленная образованием сульфата бария по реакции:
H2SO4 + ВаС12 = BaSO4 + 2НС1.
Количественное определение
1. Воздух просасывают через ватный фильтр, смоченный водой. По окон-
чании просасывания воздуха ватный фильтр промывают дестиллированной
водой до тех пор, пока промывные воды не перестанут давать реакцию на
серную кислоту (проба с ВаС12). Серная кислота в промывных водах оттитро-
вывается едким натрием при индикаторе метилоранже по реакции:
H.,SO4 + 2NaOH = Na2SO4 + 2Н2О.
Если при титровании пользоваться 0,01 N раствором NaOH, то каждый
миллилитр его, пошедший в реакцию, соответствует 0,49 мг H2SO4 или 0,4 мг
SO3.
2. Из других объемных методов рекомендуется прибавление к промывным
водам раствора KJO3 и кристаллика KJ и оттитровывание выделившегося
яода тиосульфатом натрия (Гродзовский):
KJO3 + 5KJ + 3H2SO4 = 3K2SO4 + ЗН2О + б J
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
1 мл 0,01 N раствора тиосульфата натрия, пошедший в реакцию, соответ-
ствует 0,49 мг H2SO4 или 0,4 мг SO3.
Определение можно вести и весовым методом, осадив H2SO4 хлористым
барием. Полученный вес BaSO4 перечисляют на H2SO4 или на SO3. Коэфи-
циенты 0,4201 соответственно или 0,343 соответственно —
BabO4 BaSO4.
Сероуглерод
Сероуглерод (CS2) представляет собой бесцветную жидкость с удельным
весом 1,261. Запах чистого сероуглерода напоминает хлороформ; техниче-
ский сероуглерод имеет неприятный запах редьки. Сероуглерод легко испа-
250
ряется, причем пары его, будучи в два с половиной раза тяжелее воздуха,
стелются по земле. Один литр газообразного CS2 при нормальных условиях
весит 3,4009 г. Смеси паров CS2 и воздуха взрывчаты при содержании от
6,4 до 32°/0 CS2.
Применяется как растворитель серы, фосфора, жиров и пр., в резиновой
промышленности при холодной вулканизации каучука, для целей дезинсекции
и т. п.
Концентрация 1—1,2 мг/л причиняет головную боль, 6—10 мг/л вызывает
наркоз. Действует преимущественно как хронический яд, вызывая катар
дыхательных путей, расстройства функций пищеварительных органов, мало-
кровие, головные боли, параличи нервной системы, расстройства зрения,
психики.
Качественное определение
1. Обонянием по характерному запаху.
2. Воздух высушивают хлористым кальцием и просасывают через раствор
едкого калия в абсолютном спирте. При этом получается ксантогеновокислый
калий:
/ОС2Н5
CS2 -|- КОН + С2Н6ОН - CS С + Н2О.
XSK
Раствор подкисляют уксусной кислотой и прибавляют к нему некоторое
количество раствора сернокислой меди; при этом выпадает желтый осадок
ксантогеновокислой закиси меди. Если вместо раствора сернокислой меди
прибавить несколько капель раствора молибденовокислого аммония, то по-
лучается сначала красная, затем фиолетовая, и, наконец, синяя окраска.
3. Если высушенный газ пропустить через эфирный раствор триэтилфос-
фина, то образуется светлокрасный раствор или кристаллический осадок:
Р(С2Н5)3 + CS2 = Р(С2Н5)3 . CS2.
4. Бумажка, пропитанная раствором сернокислой меди, в избытке диме-
тиламина (CHa)2NH от действия сероуглерода окрашивается в коричневый цвет
вследствие образования диметилдитиокарбамата меди [(CN)2NCSS]2Cu (Куз-
нецов).
Количественное определение
Принцип метода. Высушенный воздух пропускают через 20%
раствор едкого калия в 96° спирте. При этом образуется ксантогеновокислый
калий по реакции, указанной выше.
Образовавшийся ксантогенат оттитровывают иодом:
/ОС2Н6
2CSf
XSK
S-CS—О—С2Н5
I +2KJ.
S—CS—о—С2Н5
По израсходованному иоду делают заключение о количестве поглощен-
ного CS2. Из сопоставления реакций видно, что 1 мл 0,01 N раствора иода
соответствует 0,7612 мг CS2.
Приборы и реактивы. 1.Три поглотительные склянки.
2. Две U-образные трубки, наполненные хлористым кальцием.
3. Аспиратор.
4. 5% спиртовый раствор едкого калия.
5. Разведенная уксусная кислота (1 : 3 по объему).
6. 1% спиртовый раствор фенолфталеина.
7. 0,01 N раствор иода.
8. 1% крахмальный клейстер.
Методика определения. Воздух просасывают через две U-об-
разные трубки с хлористым кальцием, за которыми помещают три поглоти-
251
тельные склянки с 5% спиртовым раствором едкого калия по 15—20 мл в
каждой. Скорость просасывания воздуха 30—40 л в час.
Поглотительную жидкость переводят в мерный цилиндр, ополаскивают
склянки спиртом, переводя ополоски туда же. Половину полученной жид-
кости оттитровывают уксусной кислотой при фенолфталеине. Другую поло-
вину нейтрализуют уксусной кислотой в отсутствии фенолфталеина на осно-
вании результатов первого титрования.
К нейтрализованному раствору прибавляют 2—3 мл крахмального клей-
стера и титруют иодом до получения стойкого синего окрашивания. Результат
титрования умножают на два и перечисляют на CS2.
Пример. При обследовании цеха холодной вулканизации каучука на наличие сероугле-
рода в воздухе было пропущено 100 л воздуха через 60 мл 5% спиртового раствора КОН.
После нейтрализации половины объема уксусной кислотой на оттитровывание ксантогената
пошло 25,5 мл 0,01 раствора иода. Нужно определить концентрацию сероуглерода в воздухе
обследованного цеха, учитывая, что в момент взятия пробы температура воздуха была
18° С, а атмосферное давление 758 мм Hg. По приведении воздуха к нормальным условиям
получено 93,6 л.
На оттитровывание половинного объема поглотительной жидкости ушло 25,5 мл 0,01 N
раствора иода, следовательно, на весь объем потребовалось бы 51 мл 0,01 N раствора иода.
Каждый миллилитр израсходованного иода соответствует 0,7612 мг CS±; следовательно,
во время опыта сорбировано всего 0,7612 • 51 мг CS2, что дает:
0,7612-51 .
----------~ 0,062 мг/л.
93,6
Аммиак
Аммиак (NH3) — бесцветный газ с резким специфическим запахом. Удель-
ный вес — 0,597 (воздух = 1). Легко растворим, в воде: 1 объем воды при
20°С растворяет 702 объема NH3 при давлении в 760 мм.
Встречается в воздухе на газовых заводах как побочный продукт; на про-
изводствах при получении соды по Сольвею; на кожевенных заводах при
обработке кожи; в холодильном деле; в зеркальном и шелковом производ-
ствах и т. д.
В концентрации 0,1 мг/л действует на слизистые оболочки раздражающе;
концентрации 2—4 мг/л опасны для здоровья (отек легких и голосовой щели,
легочные кровотечения).
Смеси воздуха и аммиака при концентрации последнего 16—29% взрыво-
опасны.
Качественное определение
1. Обонянием по характерному запаху.
2. Бумажка, смоченная азотнокислой закисью ртути (HgNO3), в присут-
ствии аммиака чернеет вследствие выделения ртути. Под действием НС1
черная окраска исчезает, так как образуется Hg2Cl2.
3. Влажная куркумовая1 бумажка от действия аммиака буреет; при
высыхании бурая окраска исчезает.
4. Влажная лакмусовая бумажка в присутствии аммиака синеет.
5. При просасывании воздуха через слабый раствор кислоты, с последу-
ющей нейтрализацией едкой щелочью и прибавлением реактива Несслера,
жидкость приобретает желтую окраску, если в растворе имеются малые кон-
центрации аммиака, и бурую, если концентрации аммиака велики. Это свя-
зано с образованием йодистого меркураммония:
zHg\
NH.OH + 2(HgJ2,2KJ) + 3KOH = )NH2J + 7KJ + ЗН2О.
Реактив Несслера . d
Йодистый мер-
кураммоний
Реактив способен обнаруживать еще 0,0001 мг NH3 в 1 мл раствора.
1 Куркума — травянистое растение; корневище куркумы содержит красящий пигмент
куркумин.
252
Количественное определение
Здесь изложены два метода: объемный и колориметрический.
Объемный метод
Принцип метода: испытуемый воздух просасывают через титро-
ванный раствор H2SO4. Аммиак связывается с кислотой по реакции:
2NH3 4- H2SO4 = N(H4)2SO4.
Остаток кислоты оттитровывают щелочью и отсюда заключают о коли-
честве кислоты, связавшейся с HN3. На основании этого рассчитывают ко-
личество поглощенного аммиака.
Приборы и реактивы. 1. Две промывалки.
2. Аспиратор.
3. 0,01 N раствор H2SO4.
4. 0,01 N раствор NaOH.
5. Метилоранж (1% водный раствор).
Методика определения. Через две промывалки, содержа-
щие по 10 мл 0,01 N H2SO4, просасывают испытуемый воздух (можно с боль-
шой скоростью ввиду быстрого взаимодействия между NH3 и H2SO4).
Содержимое промывалок переводят количественно в коническую колбу,
прибавляют 1—2 капли метилоранжа и оттитровывают остаток кислоты.
Отсюда делают заключение о количестве кислоты, пошедшей на связывание
с аммиаком, и о количестве последнего.
Пример. В зольном цехе кожевенного завода проведено исследование воздуха на аммиак.
Через поглотительные склянки пропущено 54,8 л воздуха (объем указан при нормальных
условиях). На оттитровывание остатка кислоты ушло 14,4 мл 0,01 N щелочи. Нужно опре-
делить концентрацию аммиака в обследованном цехе.
Из данных задачи видно, что на связывание с NH3 ушло 20— 14,4= 5,6 мл H2SO4.
Приведенная выше реакция показывает, что миллилитр 0,01 N кислоты, связавшейся с амми-
аком, соответствует 0,17 мг NH3. Следовательно, всего было поглощено 0,17 • 5,6 = 0,952 мг
NH3. При перечислении на литр получаем:
-^^-^0,0174 мг/л.
5,48
Колорите грический метод
Принцип метода. По поглощении NH3 кислотой усредняют
раствор и определяют количество поглощенного аммиака по интенсивности
окраски раствора с реактивом Несслера, сравнивая окраску со шкалой или
пользуясь колориметром.
Приборы и реактивы. 1. Две поглотительные склянки.
2. Аспиратор.
3. 10 пробирок одинакового диаметра.
4. Реактив Несслера: растворяют 50 г KJ в 50 мл горячей воды и прили-
вают сюда понемногу насыщенный горячий раствор сулемы до появления
красного осадка, не исчезающего при взбалтывании. Раствор фильтруют,
приливают к фильтрату раствор 150 г КОН в 300 мл воды, доливают водой
до литра, прибавляют еще 5 мл насыщенного раствора сулемы и оставляют
на несколько дней в темной посуде. Прозрачный раствор затем сливают для
пользования. Можно отфильтровать через асбестовый фильтр.
5. Стандартный раствор хлористого аммония, содержащий в 1 мл 0,01 мг
аммиака. Одна грамм-молекула нашатыря т. е. 53,5 г NH4C1 содержит
53 5
17 г NH3. Следовательно, если в литре воды растворить 3,147 г NH4C1,
то весь литр будет содержать 1 г аммиака, а каждый миллилитр такого ра-
створа — 1 мг NH3. Этот раствор перед употреблением разбавляют в сто
раз; 1 мл такого слабого раствора соответствует 0,01 мг NH3.
253
6. 0,01 N раствор H2SO4.
7. 0,1 N раствор NaOH.
Методика определения. Забор пробы производится, как и
в предыдущем методе. Составляют шкалу, взяв слабый раствор нашатыря
от 0,1 до 1 мл с интервалом в 0,1 мл. В пробирках получается содержание
аммиака в миллиграммах соответственно: 0,001, 0,002; 0,003; 0,004; 0,005;
0,006; 0,007; 0,008; 0,009: 0,01. Объем жидкости доводят до 10 мл. В такую
же пробирку отбирают 10 мл пробы. Во все пробирки приливают по 1 мл
0,1 N раствора NaOH, чтобы усреднить растворы, и затем приливают по
5 капель реактива Несслера.
Находят стандартную пробирку, которая по окраске соответствует опыт-
ной, и отсюда рассчитывают содержание аммиака в пробе.
Пример. Опытная пробирка по окраске соответствует 8-й стандартной. Следовательно,
в 10 п'п поглотительной жидкости находится 0,008 мг NH3. Положим, что всей поглотительной
жидкости вместе с промывными водами было 30 мл. Отсюда заключаем, что всего поглощено:
0,008 • 30
10
= 0,024 мг NH3.
Азотная кислота
Азотная кислота (HNO3) — бесцветная жидкость с едким запахом. В кон-
центрированном виде (уд. вес 1,52) содержит около 100% HNO3. Сильно
дымит на воздухе, поглощая влагу. Бурая дымящая кислота содержит окислы
азота и азотистую кислоту.
Пары азотной кислоты обыкновенно вместе с окислами азота встречаются
на производствах, где азотная кислота является предметом получения, а так-
же образуется при синтезе некоторых других кислот (серной, мышьяковой,
азотистой.) Встречается и там, где азотная кислота применяется для техни-
ческих целей: травление металлов, нитрирование органических продуктов
и т. п.
При концентрации 0,03 мг/л переносится без видимого вреда, вызывая
лишь легкое раздражение слизистых оболочек. Большие концентрации вызы-
вают заметное раздражение слизистых оболочек. Концентрации в 0,5—0,7 мг/л
опасны для жизни (отек легких, сердечная слабость).
Качественное определение
1. Воздух просасывают через небольшое количество воды. Незначитель-
ное количество сорбента переносят на часовое стекло, подкисляют крепкой
серной кислотой, свободной от азотной, и вносят сюда немного бруцина
(C23H26N2O4). В присутствии HNO3 появляется красное окрашивание.
2. На часовом стекле растворяют в крепкой серной кислоте, свободной
от азотной, несколько кристалликов дифениламина (C6H5)2NH и прибав-
ляют несколько капель сорбента. В присутствии азотной кислоты появляется
синее окрашивание. Реакция менее чувствительна, нежели с бруцином. Обе
реакции получаются и с азотистой кислотой.
Количественное определение
Воздух просасывают через трубку с фильтром из обезжиренной и смочен-
ной водой ваты. Фильтр и трубку затем промывают до тех пор, пока промыв-
ная вода не перестанет давать кислую реакцию. Для более быстрой экстрак-
ции HNO3 из ватного фильтра рекомендуется прокипятить фильтр с водой.
Все промывные воды собирают вместе и титруют 0,01 N раствором едкого
натрия при индикаторе метилроте. Из реакции
HNO3+NaOH=NaNO3+ Н2О
видно, что 1 мл 0,01 N раствора NaOH соответствует 0,63 мг HNO3.
254
Так как промывные воды чистой ваты сами могут оказаться кислой реак-
ции, то необходимо в холостой пробе выяснить поправку.
Окислы азота
К окислам азота относятся: закись (N2O), окись (NO), азотистый ангид-
рид (N2O3), двуокись (NO2) и азотный ангидрид (N2O5).
Закись и окись азота образуются при восстановлении азотной кислоты
(травление металлов, действие органических веществ на HNO3 и т. п.).
Окись азота на воздухе окисляется до двуокиси:
2NO + О2 = 2NO2.
Оба последних окисла, т. е. NO и NO2, образуются при изготовлении
серной кислоты камерным способом (получение нитрозилсерной кислоты),
а также азотной кислоты, нитроглицерина, тринитритолуола и других взрыв-
чатых веществ и нитропроизводных.
Азотистый ангидрид образуется при разложении азотистой кислоты.
Двуокись азота, соединяясь на воздухе с влагой, дает эквимолекулярную
смесь азотистой и азотной кислот:
2NO2 + Н2О = HNO2 + HNO3.
Азотистая и азотная кислоты, повидимому, представляют важнейшие
составные части нитрогазов воздуха; здесь, может быть, присутствуют еще
малостойкие N2O3 и N2O5.
Вдыхание небольших количеств окислов азота (0,07—0,1 мг/л) вызывает
раздражение верхних дыхательных путей, чувство стеснения в груди. При
относительно больших концентрациях (0,2—0,3 мг/л) через 8—10 часов скры-
того периода развиваются тяжелые симптомы: одышка, кашель, кровавая
мокрота, рвота, цианоз и нередко смерть при явлениях отека легких и ослаб-
ления сердечой деятельности.
Качественное определение
1. Для обнаружения окислов азота пользуются реактивом Грисса (стр. 227).
Воздух просасывают через дестиллированную воду; при этом окислы
азота с водой дают HNO2. К раствору прибавляют несколько капель реактива
Грисса. В присутствии HNO2 получается розовое окрашивание ввиду
образования азокрасителя:
NH, N = N—ООССН3
c6hZ '+нко2|сн3соон — с6н4; +2н2о.
XSO3H XSO3H
Сульфаниловая
кислота
N=N—OOCCHS n=N—c10h6nh2 • СН3СООН.
С*н4( +с10н7н2-* с6н4(
SO3H о—нафтиламия SO3H
Азокраситель
Реакция специфична, дает возможность обнаружить еще 1 ч. HNO2 в
1 000 000 000 ч. воды.
Коренман рекомендует для обнаружения в воздухе окислов азота поль-
зоваться фильтровальными бумажками, пропитанными указанным реакти-
вом, и развешивать такие бумажки в помещении. При наличии в воздухе
окислов азота бумажки розовеют. Следует пользоваться свежеприготовлен-
ными бумажками; причем они должны быть слегка влажными. Этим способом
удается обнаружить 0,0001—0,0002 мг окислов азота в 1 л воздуха.
255
Способ приготовления жидкого реактива Грисса приведен выше (стр. 219).
Здесь укажем рецепты приготовления сухого реактива Грисса. 1 г а-нафти-
ламинэ, 10 г сульфаниловой кислоты и 89 г винной кислоты растирают в одно-
родный порошок. Сохраняют в склянке с притертой пробкой. Перед употреб-
лением 1 г порошка растворяют в 10 мл воды или прибавляют щепотку по-
рошка в исследуемую жидкость и взбалтывают до растворения. Другой
рецепт сухого реактива: 0,1 г а-нафтиламина, 1г сульфаниловой кислоты и
5 г щавелевой кислоты смешивают и растирают в однородный порошок. Хра-
нят в склянке с притертой пробкой.
2. Для обнаружения азотистой кислоты можно пользоваться также сле-
дующим реактивом: 0,085 г дифениламина взбалтывают с 190 мл H2SO4 (1 : 3),
затем добавляют H2SO4 уд. веса 1,84 до растворения и доводят такой же ки-
слотой до 500 мл, после чего добавляют 200 мл воды. Полученный реактив
взаимодействует лишь с HNO2 и ее солями, но не с HNO3. Окраска полу-
чается от голубой до синей в зависимости от концентрации HNO2.
Предел чувствительности реакции — 0.1 мг азотистой кислоты в 1 л воды.
Количественное определение
Окислы азота определяются совместно с азотистой кислотой.
Йодометрическое определение
Воздух отбирают в сухую бутыль, вводят сюда 30 мл 10% раствора KJ,
подкисленного 1—2 мл IN H2SO4 и сильно взбалтывают.
При этом выделяется свободный иод:
2KJ + H2SO4 = 2HJ + K2SO4,
2HJ + 2HNO2 = 2NO -L- 2H2O + J2.
Иод оттитровывают 0,01 N раствором тиосульфата натрия из микробю-
ретки. 1 мл 0,01 N раствора Na2S2O3 соответствует 0,47 мг HNO2.
Присутствие в воздухе хлора, озона и других окислителей мешает опре-
делению,’так как тоже ведет к выделению свободного иода.
Колориметрическое определение
Воздух отбирают в сухую бутыль, вводят сюда 30 мл дестиллированной
воды и сильно взбалтывают. Через 2—3 часа пипеткой берут 10 мл жидкости
и определяют азотистую кислоту, как в воде (см. стр. 339).
Хлор
Хлор (С12)—газ желтозеленого цвета удельного веса 2,4494 (воздух = 1),
неприятного запаха; сильно раздражает дыхательные пути. Атомный вес хлора
35,457. Литр хлора весит 3,214 г. При 0°С газообразный хлор переходит в жид-
кое состояние, если подвергнуть его давлению в 3,7 атмосферы. В воде раство-
ряется хорошо. При 15°С один объем воды поглощает 2,68 объема хлора.
Если хлорную воду охлаждать ниже 8°С, то выделяется кристаллический
гидрат хлора (С12 . 8Н2О).
Хлор химически весьма активен В производственных условиях может
встречаться при его изготовлении, при использовании для целей дезинфекции,
а также при изготовлении хлорной извести, отбеливающих жидкостей и т. п.
Раздражающее действие хлора на дыхательные пути объясняется тем,
что с водою он образует соляную и хлорноватистую кислоты:
С12 + Н2О = НС1 + нею
1 Хлор был первым боевым отравляющим веществом, которое немцы применили 22 ап-
реля 1915 г.
256
Уже в концентрации 1 : 200 000 хлор вызывает раздражение слизистых
оболочек, а в концентрации 1 : 1000 — смерть от удушья при явлениях воспа-
ления и отека легких и спазма голосовой щели.
Качественное определение
1. Обонянием по характерному запаху определяются концентрации от
0,003 мг/л.
2. При пропускании воздуха, содержащего хлср, через раствор йоди-
стого калия, к которому прибавлен крахмальный клейстер, жидкость синеет
вследствие выделения свободного иода и образования иодкрахмала:
2KJ + С12 = 2КС1 + J2
Та же реакция проводится с иод-калий-крахмальной бумажкой (изготов-
ление бумажки см. стр. 225). Реакция неспецифична: озон, окислы азота,
бром и др. окислители приводят также к выделению свободного иода и по-
синению жидкости или реактивной бумажки.
3. При пропускании воздуха, содержащего хлор, через раствор азотно-
кислого серебра, подкисленный азотной кислотой, появляется белая муть
вследствие выделения хлористого серебра. Муть исчезает при добавлении
аммиака.
4. Флуоресцеиновая бумажка изменяет свой желтый цвет на розовый
либо красный, в зависимости от концентрации хлора. Для получения бума-’
жек приготовляют раствор по следующему рецепту: 0,2 г флуоресцеина рас-
творяют в небольшом объеме 10% NaOH, прибавляют 2,0 г углекислого
калия и доводят водой до 50 мл. Отдельно растворяют 30,0 г бромистого калия
в 50 мл воды; оба раствора сливают вместе и прибавляют 10 г глицерина.
В этом растворе смачивают фильтровальные бумажки и высушивают. Изме-
нение желтого цвета бумажки на красный под действием хлора объясняется
тем, что хлор вытесняет бром из бромистого калия, а бром дает с флуорес-
цеином яркокрасный эозин:
2КВг + С12 = 2КС1 + Вг2
Х6Н4х /С6Н3(ОН)Ч /С6Н4Ч /С6НВг2(ОН) ч
со; ;с; ;o + 4Br2 = coz ;с; ;о+4нвг.
х О хС6Н3(ОН)z х О z xC6HBr2(OH)z
флуоресцеин эозин
По данным Тейс и Каплун, чувствительность реактивной бумажки повы-
шается, если из рецепта исключить глицерин.
Количественное определение хлора
Здесь описаны два объемных метода: с иодистым калием и мышьяковистой
кислотой.
Объемное определение хлора иодистым налпем
Принцип метода: воздух просасывают через раствор йодистого
калия и выделившийся иод оттитровывают тиосульфатом натрия:
2К J + С12 = 2КС1 + J2
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
Сопоставляя обе реакции, мы видим, что 1 мл 0,01 N раствора тиосульфата
натрия соответствует 0,3546 мг хлора.
Приборы и реактивы. 1.Три поглотительные склянки.
2. Аспиратор.
17 А. н. ъуршгеин 257
3. 5% раствор KJ (свежеприготовленный), не содержащий свободного
иода.
4. 1% крахмальный клейстер.
5. 0,01 N раствор тиосульфата натрия.
Методика определения. В три склянки типа Петри наливают
по 15 мл 5% йодистого калия и просасывают воздух со скоростью 30 л в час.
Пожелтение раствора допускается лишь в первых двух поглотительных
сосудах. Раствор в последнем (контрольном) сосуде должен оставаться бес-
цветным, в противном случае включают еще одну поглотительную склянку.
Жидкость из всех склянок переводят количественно в коническую колбу
и оттитровывают иод 0,01 N раствором тиосульфата натрия.
Количество миллилитров израсходованного тиосульфата натрия умно-
жают на 0,3546 и получают количество поглощенного хлора в миллиграммах.
Присутствие в воздухе брома, озона, перекиси водорода, окислов азота
мешает реакции, так как и они вытесняют иод из йодистого калия.
Объемное определение хлора мышьяковистой кислотой
Принцип метода: воздух просасывают через раствор мышья-
ковистой кислоты, которая окисляется хлором в мышьяковую:
As2O3 + 2С12 4- 2Н2О = As2O5 + 4НС1.
Остаток мышьяковистой кислоты оттитровывают иодом:
As2O3 + 2J2 + 2Н2О = As2O5 + 4HJ.
По разности между взятым количеством мышьяковистой кислоты
шимся делают заключение о количестве мышьяковистой кислоты,
шей в реакцию с хлором, и отсюда выводят количество поглощенного хлора.
35 46
1 мл 0,01 N мышьяковистой кислоты соответствует - q - = 0,3546 мг хлора.
и остав-
вступив-
Приборы и реактивы. 1. Приборы те же, что и в предыдущем
методе.
2. 0,01 N раствор мышьяковистой кислоты. Приготовляют сначала более
крепкий раствор (напр. 0,1 N). Для этой цели 4,947 г* 1 химически чистой As2O3
растворяют в 40 мл 1 N раствора едкого натрия, добавляют 40 мл 1 N соля-
ной или серной кислоты и доводят водой до литра. Раствор должен быть ней-
тральным на лакмус. Из данного раствора готовят более слабые. Титр про-
веряют по иоду.
3. 0,01 N раствор иода.
4. 1% крахмальный клейстер.
Методика определения. В две склянки типа Петри наливают
по 20—30 мл 0,01 N мышьяковистой кислоты и просасывают воздух со ско-
ростью 15 л в час. Жидкость количественно переводят в коническую колбу
и, прибавив крахмальный клейстер, титруют иодом до посинения.
Бром и иод
Бром и иод определяется в общем так же, как хлор. 1 мл 0,01 N Na2S2O3
или As2O3 соответствует 0,7996 мг брома и 1,2685 мг иода.
Хлористый водород
Хлористый водород (НС1) — бесцветный газ, на воздухе дымит, жадно
поглощая влагу. Удельный вес—1,2684 (воздух=1), молекулярный вес —
36,465. Раствор хлористого водорода в воде представляет хлористоводород-
As О
1 Г рамм-эквивалент As^ = —- == 49,47 р.
258
ную (соляную) кислоту. Концентрированная, или дымящая, соляная кислота
имеет удельный вес 1,19 и содержит 37, 23% газообразного НС1.
Попадает в воздух при получении соляной кислоты, а также как побочный
продукт при производстве соды, искусственных удобрений и т. п.
При концентрации 0,16 мг/л действует раздражающе на слизистые обо-
лочки. Концентрация 0,5 мг/л опасна для здоровья.
Качественное определение
1. При пропускании воздуха, содержащего НС1, через раствор азотно-
кислого серебра, подкисленный азотной кислотой, образуется муть вслед-
ствие выделения хлористого серебра; муть легко растворяется при прибав-
лении раствора аммиака. Так как хлор дает такую же реакцию, то его отде-
ляют, пропуская предварительно воздух через сухой тиосульфат натрия.
2. Влажная синяя лакмусовая бумажка от действия НС1 краснеет (реакция
не специфична).
Количественное определение
1. Некоторое количество воздуха просасывают через слабый (3—5%)
раствор едкого натрия; при этом происходит реакция:
HCI + NaOH -=- NaCl + Н2О.
Избыток едкого натрия нейтрализуют уксусной кислотой и определяют
полученный в поглотителе хлорид, титруя азотнокислым серебром при инди-
каторе хромовокислом калии (2—3 капли 10% раствора). Титрование ведется
до тех пор, пока лимонножелтый цвет раствора не переходит в кирпично-
красный.
Между NaCl и AgNO3 происходит реакция:
NaCl + AgNO3 — AgCl + NaNO3.
Из приведенных двух реакций легко видеть, что 1 мл 0,01 N раствора
AgNO3, пошедший в реакцию, соответствует 0,365 мг НС1.
2. В склянку с пришлифованным краном наливают 10 мл 0,2% раствора
KJO3. Производят вакуацию склянки (см. стр. 214) и вводят испытуемый
воздух. Встряхивают в течение 10 минут, затем содержимое склянки пере-
водят количественно в коническую колбочку с притертой пробкой и прибав-
ляют сюда несколько капель 5% раствора KJ; происходит реакция:
5К J 4- KJO3 + 6HCJ = 6КС14- ЗН2О + 3J2.
Выделившийся иод титруют 0,005 N раствором тиосульфата натрия из
микробюретки. В конце титрования прибавляют две-три капли 1% крахмаль-
ного клейстера и дотитровывают.
1 мл 0,005 N Na2S2O3, пошедший в реакцию, соответствует 0,1825 мг НС1
(Петрова и Яковцевская).
Мышьяковистый водород
Мышьяковистый водород (AsH3) — бесцветный газ чесночного запаха с
удельным весом 2,695 (воздух = 1). Может попадать в воздух при получении
водорода действием кислот на цинк (технические серная кислота и цинк со-
держат обыкновенно примесь мышьяка), при паянии и протраве металлов,
цинковании, лужении, освинцевании, при наполнении аккумуляторных бата-
рей, при доступе влаги к ферросилицию (наряду с фосфористым водородом),
при выделении ацетилена из содержащего мышьяк карбида, при производстве
анилиновых красок и т. п.
Мышьяковистый водород — сильный яд: концентрации 0,02 мг/л опасны
при воздействии в течение 0,5—1 часа. Тяжелые симптомы отравления про-
259
являются часто не сразу, а через несколько часов (скрытый период) после
поступления яда в организм (тошнота, рвота, боли в животе, слабость сер-
дечной деятельности). Позднее, через 16—24 часа, развиваются общая жел-
туха, гемоглобинурия вследствие распада эритроцитов, воспаление почек,
анурия. Часто смертельный исход.
Качественное определение
1. Обонянием по чесночному запаху.
2. Раствор азотнокислого серебра чернеет при пропускании через него
воздуха, содержащего AsH3, вследствие выделения металлического серебра
по реакции:
2AsH3 4- 12AgNO3 4- ЗНаО = AsaO3 + 12HNO3 4- 12Ag.
H2S и PH3 мешают реакции. Освобождение воздуха от H2S достигается
посредством просасывания через прокаленный медный купорос. Кусочки
пемзы смачиваются концентрированным раствором CuSO4, которые прока-
ливаются затем в тигле.
От фосфористого водорода воздух освобождается просасыванием через
солянокислый раствор хлористой меди (Си2С1а).
Если AsH3 действует на азотнокислое серебро в отсутствии воды или при
наличии малого количества воды, напр. на реактивные бумажки, обработан-
ные раствором AgNO3, или на кристаллик AgNO3, то реакция протекает в
две фазы:
6AgNO3 4- AsH3 = AsAg3 • 3AgNO3 4- 3HNO3
AsAg3 • 3AgNO3 4- 3H2 О - As(OH)3 + 3HNO3 4- 6Ag.
Комплексное соединение AsAg3 . 3AgNO3 дает зеленоватожелтое окра-
шивание (первая фаза); когда же это соединение распадается с образованием
металлического серебра, то появляется черное окрашивание (вторая фаза).
3. Бумажка, обработанная спиртовым раствором сулемы или бромной
ртути (HgBr2), окрашивается в желтый цвет при малых концентрациях AsH3
и в коричневокрасный—при больших. При этом протекают следующие реакции:
2HgCla 4- AsH3 = 2HCI4- AsH(HgCl)2,
3HgCla 4- AsH3 = 3HCI4- As(HgCI)3.
Бумажку помещают в узкую трубку, через которую просасывают воздух.
Необходимо произвести предварительно поглощение H2S и РН3 (см. выше).
4. Воздух просасывают через бромную воду; при этом AsH3 окисляется
до As^Os. Сорбент насыщают аммиаком, бром удаляют нагреванием, перево-
дят жидкость в аппарат Марша, при помощи которого можно обнаружить
малейшие количества мышьяка — до 0,0007 мг.
Способ Марша заключается в том, что соединения мышьяка переводятся
водородом, взятым in statu nascendi, в мышьяковистый водород, который
при нагревании до 230°С разлагается с выделением металлического мышья-
кового зеркала. При этом протекают следующие реакции: 1) Zn4~H2SO4 =
= 2H4-ZnSO4, 2) AsaO34-12H = 2AsH3 + 3HaO, 3) 2AsH3=2As4-3H2.
Для производства определения конструируют прибор (рис. 214). В колбу
А со стеклянной пробкой вставляется капельная воронка с краном. Отвод-
ная трубка колбы соединяется с трубкой В. В последнюю помещают гигро-
скопическое вещество для сушки AsH3, напр., хлористый кальций. Трубка В
соединяется с тугоплавкой трубкой С диаметром в 5 мм, оттянутой в капил-
ляр диаметром 2 мм. Тугоплавкая трубка нагревается в одном месте бунзе-
новской горелкой до температуры 230°С (слабо-красное каление). Другая
часть трубки охлаждается током воды из бутыли D. В колбу А насыпают
3—5 г гранулированного цинка, сюда же наливают серной кислоты (1 : 10)
260
и испытуемый раствор. Колбу помещают в ванночку с водой комнатной
температуры с целью предупреждения нагрева жидкости в колбе выше 30°С.
Выделяющийся AsH3 разлагается при нагревании с выделением металличе-
ского мышьяка, который отлагается в холодной части трубки в виде зер-
кала. Очень часто применяются трубки с 2—3 сужениями. В этих случаях
охлаждаются суженные места, где и отлагается металлический мышьяк.
Применяемые Zn и H2SO. должны быть свободны от As, в чем можно убе-
диться при помощи холостой пробы.
Цинк хорошо применять активированный. Активирование цинка дости-
гается следующим образом: металлический цинк расплавляют и к нему до-
бавляют 1—1,5% (по весу) безводного хлористого магния. Расплавленную
Рис. 214. Прибор для определения мышьяка.
А — склянка для испытуемого материала (в ней As восстанавливают до АзНа). В — трубна с хлористым
кальцием для осушки газа, С — трубка из тугоплавкого стекла, D— склянка с холодной водой.
массу тонкой струйкой вливают в холодную воду. Получающийся гранули-
рованный цинк обладает достаточной активностью. Активирование при по-
мощи CuSO4 нецелесообразно: такой цинк удерживает мышьяк.
Так как Sb и Se также дают металлическое зеркало, нужно иметь в виду
следующие особенности мышьякового зеркала: оно свободно передвигается
по трубке при нагревании бунзеновской горелкой; зеркало при Sb и Se тре-
бует для перемещения долгого нагревания. Зеркало As полностью раство-
ряется в растворе хлорноватистокислого натрия, не содержащего хлора х.
Зеркала Sb и Se в этом реактиве нерастворимы. Мышьяковое зеркало нера-
створимо в 15% растворе виннокаменной кислоты, сурьмяное в этом реак-
тиве растворимо. Зеркало Se растворяется безводной H2SO4 (несколько ка-
пель) с образованием зеленой окраски.
Количественное определение
Здесь приведены два метода: объемный и колориметрический.
1 Раствор хлорноватистокислого натрия (NaOC!) получается обработкой фильтрован-
ного раствора хлорной извести раствором соды: Са(ОС1)2 4- Na3CO3 = СаСО3 4- 2 NaOCl.
261
Объемный метод
Принцип метода. Воздух просасывают через бромную воду. При
этом AsH3 окисляется в As2O3 по реакции:
2AsH3 + ЗН2О 4- 6Вг2 = As2O3 + 12НВг.
Мышьяковистый ангидрид оттитровывают иодом:
As2O3 + 2J2 -I- 2Н2О = As2O5 4- 4HJ.
По израсходованному раствору иода делают заключение о количестве
поглощенного AsH3.
Приборы и реактивы. 1.4 промывалки типа Петри.
2. Аспиратор.
3. Бромная вода.
4. Аммиак.
5. 0,01 N раствор иода.
6. Раствор едкой щелочи.
7. Крахмальный клейстер 1%.
Методика определения. Воздух просасывают через три склян-
ки Петри, содержащие по 15 мл бромной воды каждая. В четвертую склянку
наливают 15 мл раствора едкого натрия, чтобы задержать уносимый бром,
который может загрязнить воздух помещения. Бромную воду из склянок
переводят количественно в фарфоровую чашку, насыщают раствор аммиа-
ком и отгоняют бром на водяной бане (под тягой). Остывший раствор пере-
водят количественно в коническую колбу и титруют 0,01 N раствором иода
при индикаторе крахмальном клейстере. По расходу иода делают заключе-
ние о количестве поглощенного AsH3. Из приведенных реакций видно, что
1 мл 0,01 N раствора иода соответствует 0,3897 мг AsH3.
Колориметрический метод
Принцип метода основан на возниконовении синего окрашивания
при воздействии реактива Дениже на раствор, содержащий мышьяковистую
кислоту.
Приборы и р е a i: т и в ы. 1.4 промывалки Петри.
2. Аспиратор.
3. Стандартный раствор из мышьяковокислого натрия (Na3AsO4 . 12Н2О).
В литре воды растворяют 0,544 г реактива. 1 мл раствора соответствует
0,1 мг AsH3: Нужная навеска получается из расчета
Na3AsO4 • 12Н2О = 424,15 М4
AsH3 • 10 77,98 -10 ’
Раствор разбавляют в 10 раз; 1 мл слабого раствора отвечает 0,01 мг AsH3.
4. Реактив Дениже; 2,5 г молибденовокислого аммония растворяют в
25 мл воды, прибавляют 25 мл серной кислоты удельного веса 1,84 и остав-
ляют до охлаждения. Затем вносят 0,25 г медных опилок (электролитическая
медь). Жидкость взбалтывают в течение 30 минут, доливают водой до 250 мл
и оставляют до следующего дня. Она имеет темнобурый цвет и должна хра-
ниться в темной склянке.
Методика определения. Отбор пробы производится, как и
в предыдущем методе. По удалении брома раствор переводят количественно
в мерную колбу на 50 мл и доводят водой до метки.
Приготовляют стандартный ряд пробирок, введя в пробирки последо-
вательно от 0,2 до 1,0 мл слабого раствора Na3 AsO4 . 12Н2О с интервалом
в 0,2 мл. Содержимое пробирок соответствует содержанию AsH3 последова-
тельно от 0,002 до 0,02 мг Поводят содержимое пробирж водой до 10 мл.
Из подготовленной пробы отбирают в пробирку одинакового диаметра со стан-
262
дартными 10 мл жидкости. Во все пробирки прибавляют по 0,5 мл реактива
Дениже, взбалтывают и через 30 минут колориметрируют.
Нагревать пробирки не следует.
Фосфор и фосфористый водород
Фосфор (Р4) — твердое вещество; белый или желтый фосфор ядовиты,
красный не ядовит. Белый фосфор имеет характерный запах, обусловленный
образованием в его присутствии в воздухе озона. Ввиду легкой воспламе-
няемости фосфор хранят под водой. Пары фэсфора в 4,3 раза тяжелее воздуха,
образуют в воздухе белый туман, в котором кроме фосфора содержатся и
продукты его окисления — Р2О3 и Р2О5.
Встречается при производстве фосфора и на некоторых химических про-
изводствах.
Весьма ядовито соединение фосфора — фосфористый водород (РН3), газ
удельного веса 1,1829 (воздух = 1). Встречается на ацетиленовых производ-
ствах, так как содержится в виде примеси в исходном продукте — карбиде
кальция. Выделяется также из ферросилиция при хранении и применении его.
Пары фосфора и его окислов действуют обыкновенно как хронический
яд, вызывая прогрессирующее малокровие, заболевание слизистой оболочки
рта, некроз челюстей.
Фосфористый водород дает чаще острое отравление. Опасными являются
уже концентрации в 0,01 мг/л; при концентрациях 0,14 мг/л быстро вызы-
вает затруднение дыхания, потерю сознания, судороги, учащение пульса,
иногда смерть. Внутренние органы претерпевают жировое перерождение.
Качественное определение
1. Фильтровальная бумажка, смоченная крепким раствором ляписа
(AgNO3), от паров фосфора чернеет, от фосфористого водорода окрашивается
в желтокоричневий цвет. AgNO3 восстанавливается при этом до металла
и до фосфористого серебра:
8AgNO3 + РН3 + 4Н2О - 8Ag + 8HNO3 + Н3РО4
РН3 + 3AgNO3 = Ag3P + 3HNO3.
Мышьяковистый водород и сероводород мешают реакции.
2. Фильтровальная бумажка, смоченная слегка подкисленным раство-
ром медного купороса, чернеет в присутствии фосфора или фосфористого
водорода. При этом образуются Си3Р и Си3Р2. Реакция менее чувствительна,
нежели первая.
Количественное определение
Забор пробы и определение ведутся так же, как и при AsH3.
Через бромную воду просасывают 100—200 л воздуха. При этом нужно
соединение поглотительных склянок производить в стык, так как под дейст-
вием паров брома каучук захватывает фосфор. Бром затем отгоняют кипя-
чением.
Обесцвеченную жидкость подвергают колориметрированию. Стандартный
раств ip готовят из двузамещенного фосфорнокислого калия (К2НРО4). Навеску
в 0,5116 г чистой кристаллической соли растворяют в литре свежедестиллиро-
ванной воды. 1 мл полученного раствора соответствует 0,1 мг РН3. Расчет
нужной навески:
_К.НРО._= 174,25
РН3- 10 34,06 . 10
Раствор разбавляют в десять раз. 1 мл слабого раствора соответствует
0,01 мг РН3.
Приготовление шкалы и колориметрирование ведется точно так же, как
при определении AsH3.
263
Пары ртути
Ртуть (Hg) — жидкий металл с удельным весом (по отношению к воде)
13,595. Уже при обыкновенной температуре постепенно испаряется. Пары
ртути тяжелее воздуха в 7 раз.
В воздухе встречается на производствах, где ртуть добывают из ее руд,
а также при применении ртути для технических целей: наводка ртутных
зеркал \ работа с ртутными насосами, изготовление различных физических
приборов, золочение и серебрение через огонь (приготовление амальгам)
и т. п.
Вдыхание паров ртути может вести Кострому отравлению. Симптомы: гин-
гивит, рвота, боли в области живота, поносы. Острые отравления редки, чаще
наблюдаются хронические. Вдыхание от 0,05 до 0.1 мг и более ртути ежедневно
в течение нескольких месяцев способно обусловить хроническое отравление,
причем поражается центральная нервная система (раздражимость, интен-
ционное дрожание верхних конечностей). Как при остром, так и при хрони-
ческом отравлении поражаются паранхиматозные органы и кровь (разрушение
эритроцитов).
Качественное определение
1. Воздух, освобожденный от пыли и от влаги, просасывают через трубочку,
в которую помещают тонкие золотые листочки. Если в воздухе при-
сутствует ртуть, то листочки покрываются белым налетом ртути (амаль-
гама).
2. В трубочку длиною 25 см и диаметром 2—3 мм, слегка согнутую, по-
мещают крупинку иода и просасывают обеспыленный высушенный воздух
со скоростью б—8 л в час. В присутствии паров ртути получается налет жел-
тых и красных кристаллов иодной ртути HgJ2.
3. Реактивные бумажки для обнаружения паров ртути предложены Поле-
жаевым. Они готовятся следующим образом: смешивают равные объемы
10% раствора CuSO4. 5Н2О и 10% раствора KJ. Дают осесть осадку, а буро-
окрашенный раствор сливают. Осадок отмывают от свободного иода дестил-
лированной водой методом декантации. Затем промывают осадок один раз
1% раствором йодистого калия и один раз 1% раствором сульфата натрия,
после чего промывают осадок дестиллированной водой. Фильтровальной
бумажкой отсасывают с осадка воду, переносят его в баночку, прибавляют
этилового спирта до получения кашицеобразной массы и на каждые 50 мл
кашицы прибавляют одну каплю 25% азотной кислоты. Полученную массу
наносят тонким слоем на фильтровальные бумажки при помощи ваты,
навернутой на палочку. Бумажки сушат и хранят в экссикаторе. Они окра-
шены в слабый желтый цвет. Если развесить такие бумажки в помещении,
где имеются пары ртути, то окраска бумажек становится розовой. В зависи-
мости от концентрации ртути розовая окраска наступает быстрее или медлен-
нее (от нескольких часов до нескольких дней). Розовый цвет зависит от
примеси к красному цвету комплектной соли—CuJHgJ2 белого цвета йоди-
стой меди—CuJ.
Количественное определение
Весовой метод
Вышеприведенная (качественная) реакция, в результате которой полу-
чается HgJ2, предложена и для количественного определения ртути. После
просасывания воздуха и образования налета иодной ртути избыток иода
осторожно отгоняют и взвешивают трубочку. Зная вес трубки без иодной
ртути и вместе с последней, определяют по разности количество образовав-
1 В СССР при изготовлении зеркал вместо ртути применяют AgNO3, что не вредит
здоровью.
264
шейся иодной ртути и отсюда делают заключение о количестве ртути, содер-
жащейся в воздухе. Переводный коэфициент 0,441:
™^L=o,44L
HgJ2 454,46
lie! олометричесвпй метод
После получения в трубке налета иодной ртути вводят сюда же несколько
кристаллов йодистого калия и вымывают содержимое трубки небольшим
количеством воды. Избыток иода восстанавливают при помощи SO2, затем
подщелачивают раствор несколькими каплями аммиака, прибавляют 5 мл
% % раствора желатины и немного свежеприготовленного и промытого серо-
водорода. HgJ2 при этом переходит в HgS:
HgJ2 + H2S = HgS + 2HJ.
Желатина, находящаяся в растворе, препятствует выпадению HgS. Темно-
окрашенную суспензию HgS сравнивают со стандартными суспензиями, при-
готовленными из растворов сулемы с добавлением желатины и сероводорода.
0,1354 г сулемы растворяют в 1 л воды. 1 мл этого раствора соответствует
0,1 мг ртути:
0,1354 • Hg = 0,1354 • 200,6 j
HgCl2 271,5
При надобности готовят более слабые растворы.
Колориметрический метод Полежаева
Поглотительный иодный раствор готовится так: 2,5 г иода (возогнанного}
и 30 г йодистого калия растворяют в небольшом объеме воды и доводят водою
до литра.
В поглотительный сосуд Городецкого наливают 15 мл указанного раствора
и просасывают воздух со скоростью 150 л в час. Затем отбирают из поглоти-
тельной жидкости 4 мл, переводят в колориметрическую пробирку и прибав-
ляют сюда 3 мл индикаторного раствора Полежаева, который готовится по
следующему рецепту. К одному объему 7% раствора двухлористой меди
(СиС12 . 2Н2О) прибавляют 2 объема приблизительно 0,25 N раствора серни-
стокислого натрия (Na2SO3 . 7Н2О), перемешивают палочкой до растворе-
ния выпавшего осадка и к прозрачному раствору приливают полтора объема
8% раствора двууглекислого натрия (NaHCO3) и все тщательно перемешивают.
Раствор должен быть свежеприготовленным. После прибавления в пробирку
указанного раствора содержимое пробирок взбалтывают и через 10 минут
колориметрируют.
Стандартный раствор готовится растворением 0,1354 г сулемы в небольшом
объеме поглотительного иодного раствора и им же доводится до литра. Этот
раствор разбавляется в 10 раз поглотительной жидкостью. 1 мл разбавлен-
ного раствора соответствует 0,01 мг Hg.
Налив определенное количество указанной стандартной жидкости в стан-
дартные пробирки, доводят содержимое пробирок иодной жидкостью до 4 мл,
прибавляют 3 мл раствора Полежаева и через 10 минут колориметрируют.
Стандартная шкала дает постепенно увеличивающуюся желтовато розовую
окраску густой взвеси, зависящую от примеси к белой взвеси иодистой меди
(CuJ) того или иного количества комплексной ртутной соли CuJ HgJ2, обла-
дающей красным цветом.
Шкалу и пробу нужно готовить одновременно и перед сравнением взбал-
тывать.
Метод дает возможность обнаружить до 0,001 мг Hg.
265
Пары тетраэтилсвинца
Тетраэтилсвинец — ТЭС, РЬ(С2Н5)4— жидкость, горящая оранжевым пла-
менем с бледнозеленой каймой. Удельный вес при 18°С—1,6591. Вес литра
пара—13,44 г. Применяется в технике как антидетонатор. Смеси бензина и
тетраэтилсвинца (свинцовый или этилированный бензин, этиловая жидкость)
получили широкое применение в воздушном и автомобильном транспорте.
Чрезвычайно опасный яд для организма человека, способный вызывать
острые и хронические отравления при вдыхании его паров или всасывании
через кожу. Поражает главным образом центральную нервную систему.
Приятный и нерезкий запах, отсутствие раздражающего действия ТЭС
заставляет относить его к коварным ядам.
За последние годы русскими авторами разработана методика определения
паров ТЭС в воздухе (Быховская, Гуревич и Карлсон, Перегуд).
Здесь приводится метод, предложенный Е. А. Перегуд.
Принцип метода основан на разрушении ТЭС водным или спирто-
водным раствором иода при нагревании, с последующим нефелометрическим
определением свинца при помощи бихромата калия в обесцвеченном тио-
сул ьфатом раство ре.
Реактивы. 1. Поглотительный раствор приготовляют смешением од-
ного объема 5% спиртового раствора иода с полутора объемами 15% раст-
вора йодистого калия. Спирт должен быть абсолютно чистым, иначе при
дальнейшем обесцвечивании раствора может появится муть, что вызывает
необходимость дополнительной фильтрации раствора.
2. 1,0 N раствор Na2S2O3 . 5Н2О.
3. Раствор CH3COONa 50%.
4. Раствор К2Сг2О7 10%.
5. Стандартный раствор, 1 мл которого соответствует 0,1 мг ТЭС. Раство-
ряют 0,1025 г Pb(NO3)2 в 100 мл воды; получают раствор, 1 мл которого
1 Pb(C2H5h • 102,5 п
отвечает 1 мг ТЭС соответственно • При разбавлении
РЬ(Г\О3)2 -100 г г
раствора в 10 раз получают раствор, 1 мл которого соответствует 0,1 мг ТЭС.
Методика определения. Воздух просасывают со скоростью
40 литров в час через две поглотительные склянки с 5 мл иодного раствора
в каждой. Исследование производится в каждой склянке отдельно.
Стандартная шкала готовится следующим образом: в 8 колориметриче-
ских пробирок наливают по 4 мл поглотительного раствора, обесцвечивают
раствор титрованием 1 N раствором тиосульфата натрия, добавляя одну лиш-
нюю каплю. Начиная со второй пробирки (первая — контроль), вносят от
0,1 до 0,7 мл стандартного раствора с интервалами в 0,1 мл. Растворы взбал-
тывают, а затем добавляют во все пробирки по 1 мл 50% раствора уксусно-
кислого натрия и по 2 капли 10% раствора бихромата калия. При этом:
2Pb(NO3)2 4- К2Сг2О7 4- Н2О = 2РЬСгО4 + 2KNO3 + 2HNO3.
Нефелометрирование можно производить через 15—20 минут после при-
готовления шкалы, так как в течение этого времени идет нарастание мути.
Само определение ТЭС после просасывания воздуха ведется следующим
образом: 4 мл исследуемого раствора переводят в колориметрическую про-
бирку и нагревают на водяной бане в течение 10 минут при 65°С. После охлаж-
дения раствор осторожно обесцвечивают 1 N раствором тиосульфата натрия,
добавляя при этом лишнюю каплю, и доводят объем дестиллированной водой
до 9 мл. Приливают при встряхивании 1 мл 50% раствора уксуснокислого
натрия и 2 капли 10% раствора бихромата калия. Через 20 минут производят
сравнение пробы со шкалой стандартов в проходящем свете от лампы на-
каливания в 96 w, помещенной в колпак молочного стекла конусообразной
формы.
266
Ацетилен
Ацетилен (С2Н2) — бесцветный газ удельного веса 0,9056 (воздух = 1).
С воздухом образует взрывчатую смесь. Особенно опасны смеси, содержащие
7—11% ацетилена по отношению к воздуху. Технический ацетилен обыкно-
венно загрязнен H2S, РН3, AsH3 и др. газами.
Встречается на производствах при получении и применении его (ацетиле-
новая сварка), в химической промышленности (синтез ацетальдегида, ацетона,
каучука и пр.).
Действует наркотически. Концентрации в 10% могут переноситься в те-
чение 0,5—1 часа; вдыхание 25% опасно. Примеси в виде РН3, AsH3 и H2S
представляют главную опасность отравления.
Качественное определение
1. Растворяют 50 г NH4C1, 25 г CuSO4 и 0,5 мл НС1 уд. вес 1,19 в воде и до-
водят раствор до 250 мл водой. 4—5 мл полученного раствора нагревают с
0,3—0,4 г медных стружек до обесцвечивания и прибавляют 1 мл воды. Бы-
стро охладив раствор, смачивают им фильтровальные бумажки. В присутст-
вии ацетилена в воздухе бумажки окрашиваются в интенсивно красный цвет.
2. Растворяют 25 г HgJ2 и 30 г К J в 100 мл воды. Перед опытом раствор
подщелачивают. При пропускании через указанный реактив воздуха, содер-
жащего ацетилен, выпадает белый осадок.
Количественное определение
Весовой метод с использование и AgNO3
Воздух просасывают через аммиачный раствор AgNO3. В присутствии
ацетилена происходит реакция:
С2Н2 + 2AgNO3 + 2NH4OH = C2Ag2 + 2NH4NO3 + 2H2O.
Выпавший осадок ацетиленистого серебра (C2Ag2) отфильтровывают,
промывают и переводят в AgCl, обработав соляной кислотой:
C2Ag2 + 2HCI = 2AgCl + С2Н2.
Количество AgCl определяют весовым способом и перечисляют на ацети-
лен. 1 мг AgCl соответствует 0,0908 мг ацетилена.
Весовой метод с приг.ененлем реактива Илосвая
Приготовляют реактив Илосвая: 1 г Cu(NO3)2 растворяют в малом коли-
честве воды, прибавляют 4 мл 20% NH3, 3 г солянокислого гидроксиламина
(NH2OH . НС1) и разбавляют водой до 50 мл. Если в раствор опустить мед-
ную проволоку, то он может сохраняться до года.
В три склянки Петри вводят по 15 мл реактива Илосвая и просасывают
испытуемый воздух со скоростью 10 л в час. При наличии в воздухе С2Н2
выпадает красный осадок ацетиленистой меди (Си2С2 . Н2О), что может слу-
жить качественной реакцией на ацетилен. Для количественного определения
осадок отфильтровывают, промывают слабым раствором гидрэксиламина,
обрабатывают концентрированной HNO3, сжигают и прокаливают. Взвеши-
вают остаток СиО и пересчитывают на ацетилен. 1 мг Си О соответствует
0,1635 мг ацетилена.
Колориметрический метод
При малых концентрациях ацетилена в реактиве Илосвая осадок не
выпадает, а раствор лишь приобретает розоволиловое окрашивание.
Для стойкости окраски к поглотительной жидкости прибавлял т предва-
рительно по 3 мл 2% раствора желатины на каждые 50 мл раствора.
267
Окраску сравнивают со стандартными растворами, которые готовятся
следующим образом: 0,1 г метилрота растворяют в 1 л спирта и 0,04 г метил-
виолета растворяют в 1 л воды. Из этих основных растворов готовят три стан-
дартных рабочих раствора: 1) 5 мл метилрота + 5 мл метилвиолета, 2) 5 мл
метилрота +8 мл метилвиолета и 3) 5 мл метилрота + 10 мл метил-
виолета. Тем или иным рабочим стандартным раствором пользуются в зави-
симости от оттенка окраски, которая получается в связи с различными по-
сторонними примесями к ацетилену в воздухе (Березова). 1 мл рабочего
стандарта соответствует 0,034 мг ацетилена. Из рабочих стандартов состав-
ляют шкалу с интервалами в 0,2 мл, что соответствует 0,0068 мг ацетилена.
Акролеин
Акролеин (алиловый альдегид, СН2 : СН . СОН) представляет собой бес-
цветную жидкость с острым неприятным запахом; удельный вес ее при 0°С
0,82205, молекулярный вес 56,03, точка кипения 52°С. В воде растворяется
до 50 объемов, в спирте, эфире и ледяной уксусной кислоте растворяется
в любых отношениях. Легко окисляется на воздухе в акриловую кислоту.
Встречается в салотопенных предприятиях, на фабриках свеч, при производ-
стве стеарина, олифы и др. Оказывает слезоточивое и удушающее действие.
Концентрации в 0,1 мг/л опасны при воздействии в течение 0,5—1 часа.
Качественное определение
1. Реакция Левина: воздух пропускают через этиловый спирт, поглоти-
тельную жидкость переводят затем в фарфоровую чашку, прибавляют одну
каплю пиперидина и одну каплю едва окрашенного раствора нитропруссида
натрия. При наличии следов акролеина возникает зеленое окрашивание, при
больших концентрациях окраска получается темнокрасная. Предел чувст-
вительности 0,024 мг акролеина.
2. 1% раствор бензидина в ледяной уксусной кислоте дает с акролеином
желтое окрашивание. Предел чувствительности 0,005 мг акролеина.
Количественное определение
Колориметрический метод
Количественное определение основано на второй из приведенных выше
качественных реакций. Воздух просасывают через три трубки Петри с 15 мл
ледяной уксусной кислоты в каждой. Скорость просасывания 20 л в час.
Составляют шкалу с содержанием от 0,005 до 0,1 мг акролеина в ледяной
уксусной кислоте с интервалами в 0,005 мг.
Доводят содержимое пробирок до 10 мл, доливая уксусной кислоты. В про-
бирку такого же диаметра вливают 10 мл поглотительной жидкости и затем
во все пробирки прибавляют по 1 мл 1% раствора основного бензидина в
ледяной уксусной кислоте. Все пробирки ставят в кипящую баню на 15 ми-
нут, после чего колориметрируют.
Пары формальдегида, скипидара и фурфурола мешают реакции, так как
дают с бензидином аналогичное окрашивание.
Акролеин, необходимый для проведения реакции, может быть получен
из глицерина. НО г безводного глицерина нагревают в круглодонной колбе
и в горячую жидкость прибавляют 250 г кислого сернокислого калия. Смесь
оставляют на сутки, прибавляют еще 25 г KHSO4 и отгоняют акролеин на
масляной бане при 160—180°С, собирая акролеин в приемник, охлаждаемый
льдом. Полученную жидкость вновь отгоняют и собирают фракцию от 52—
53°С. Ее хранят в склянке с притертой пробкой. Для стабилизации при-
бавляют 0,1—0,2 г гидрохинона.
2G8
Ацетон
Ацетон (СН3 . СО . СН3) — жидкость удельного веса 0,797 с характерным
приятным запахом. Пары ацетона в два раза тяжелее воздуха. Хорошо ра-
створяет жиры, каучук, воск и т. п. Воздух, содержащий от 2,5 до 9,6% аце-
тона по объему, способен взрывать.
Применяется как растворитель в лаковой промышленности; для извле-
чения смол и масел. Нашел применение также при выработке бездымного
пороха, искусственного шелка и пр.
Случаи легкого острого отравления сопровождаются головными болями,
головокружением, легким опьянением, слезотечением, раздражением носо-
глотки. При концентрациях 0,6 мг/л способен вызывать хроническое отрав-
ление.
Качественное определение
1. Воздух просасывают через небольшое количество воды. К полученному
водному раствору ацетона прибавляют раствор иода в иодистом калии и по
каплям едкую щелочь до получения слабой желтой окраски. Образуется
желтоватобелый осадок йодоформа по реакции:
СН3 СО.СН3 + 4КОН + 3J2 = СН3СООК + 3KJ + ЗН2О + CHJ3.
Наилучшие условия выполнения реакции следующие: к водному раствору
ацетона прибавляют равные объемы 9N раствора КОН и 0,1 N раствора
иода, приготовленного с помощью минимально необходимого для растворе-
ния йодистого калия (Коренман).
Реакция не специфична и получается с рядом веществ: этиловым спиртом,
уксусным альдегидом и др.
2. К раствору ацетона прибавляют несколько капель свежеприготовлен-
ного 10% водного раствора нитропруссида натрия Na3Fe(CN)5NO и затем
около 1 мл 15% раствора КОН. Жидкость окрашивается в красножелтый
цвет. При больших концентрациях ацетона проба дает красный или красно-
бурый цвет. Реакция получается и при наличии уксусного альдегида и ке-
тонов с группой СН2 . СО.
3. К водному раствору ацетона прибавляют реактив Несслера в избытке.
Выпадает лимонножелтый кристаллический осадок.
Количественное определение
Йодометрический метод
В основе метода лежит приведенная выше реакция.
Воздух просасывают через поглотительный сосуд, содержащий 25 мл
0,1 N раствора иода и 5 мл 20% NaOH. Подкислив затем жидкость 2 N со-
ляной кислотой, оттитровывают избыток иода 0,1 N раствором Na2S2O3. По
разности определяют количество иода, пошедшего с ацето ом в реакцию.
Каждый миллилитр 0,1 N раств зра иода, пошедшего в реакцию с ацетоном,
согласно приведенному выше уравнению, соответствует:
— 0,96747 мл СН3СОСН3.
6-10 3 3
Бензин
Бензил — один из продуктов перегонки нефти (фракция между 52 и 120°С),
жидкость удельного веса 0,730—0,750. По химическому составу представляет
по преимуществу смесь различных углеводородов: метановых, ароматиче-
ских и нафтенов. Легко испаряется на воздухе. Пары бензина с воздухом
дают ' зрывчатые смеси. Взрывоопасные концентрации для бензина удель-
2G9
кого веса 0,722 лежат в пределах 2,9—8,1% (объемных), для обыкновенного
бензина — в пределах 2,9—4,8%.
Бензин — весьма распространенный продукт: применяется как горючее
в разных двигателях, как растворитель каучука в резиновом производстве,
для извлечения жира в утильцехах и т. д.
В настоящее время большие количества бензина получаются при помощи
крекинг-процесса, сущность которого заключается в том, что тяжелые моле-
кулы высококипящих составных частей нефти подвергаются термическому
разложению с образованием более простых веществ с низшей температурой
кипения.
При содержании бензина в воздухе в количестве 5—10 мг/л могут возни-
кать острые отравления, выражающиеся в головной боли, головокружении,
сердцебиении, психическом возбуждении и т. п. Большие концентрации
(35—40 мг/л) являются опасными для жизни, вызывая потерю сознания,
параличи. Хроническое отравление ведет к анемии и функциональным
нервным расстройствам.
Качественное определение
Специальной реакции нет.
Количественное определение
Поглощение жидким пара рипом
Воздух высушивают, освобождают от СО2 и пропускают через поглоти-
тельные трубки с жидким парафином со скоростью 10—20 л в час. Так как
осушительные трубки, а также трубки для поглощения СО2 могут сорбиро-
вать некоторое количество паров бензина, нужно пропустить через эти
трубки воздух еще до приключения поглотительных трубок с парафином.
Достаточно в течение получаса просасывать воздух, чтобы осушительные
трубки и трубки с натронной известью оказались насыщенными парами бен-
зина. После этого к ним приключают трубки с парафином.
Прибор собирают так: впереди 2—3 U-образные трубки для задержания
влаги (СаС12, Р2О5), затем 2 такие же трубки с натронной известью и, наконец,
2 U-образные трубки с зернами пемзы, насыщенными жидким парафином.
Трубки с парафином помещают во время опыта в охлаждающую смесь при тем-
пературе— 8—12°С (лед и поваренная соль). Парафиновое масло следует брать
уд. веса 0,88 — 0,89 с температурой кипения около 360°С, не выделяющее при
температурахО—2°С твердых веществ. При пропускании сухого воздуха через
такое масло оно не должно изменяться в весе. Если констатируется умень-
шение в весе, то для удаления из парафинового масла летучих веществ ре-
комендуется нагревать такое масло в течение нескольких часов на кипящей
водяной бане, просасывая через масло сухой воздух.
U-образные трубки до и после опыта снаружи высушиваются в экссика-
торе и взвешиваются. Привес указывает количество поглощенных паров
бензина. Третья трубка (контрольная) не должна показывать привеса; в
противном случае опыт повторяют, взяв четвертую трубку (Грибанов, Яков-
цевская, Гольденберг и др.).
Бензол, скипидар, амил и метилацетат также сорбируются парафиновым
маслом; эти соединения, однако, в значительной части задерживаются хло-
ристым кальцием.
Поглощение силикагелем
Силикагель представляет собою гидрат кремневой кислоты. Получается
действием НС1 на силикат натрия — Na2SiO3 (подробно получение см. у Ока-
това г). Был применен Пигулевским и Яковцевской для сорбции паров
бензина.
1 Журнал прикладной химии, 2, 21, 1928.
W0
В лаборатории силикагель освобождается от углекислоты и влаги сна-
чала нагреванием до 140°С в течение 2 часов. Затем препарат вводят в U-об-
разные трубки, через которые просасывают в течение часа 40—50 л воздуха,
предварительно высушенного и освобожденного от СО2. Трубки взвешивают
и вновь пропускают воздух, поступая так до установления постоян-
ного веса.
Доведенные до постоянного веса трубки применяются на производстве.
Воздух и здесь пропускается предварительно через U-образные трубки с хло-
ристым кальцием и пятиокисью фосфора для освобождения от влаги, а также
через натронную известь для освобождения от СО2. Просасывание воздуха
производится со скоростью 20—40 л в час. По привесу трубок судят о коли-
честве поглощенного бензина.
После двукратного применения силикагель должен быть регенерирован.
Для этой цели трубки с использованным препаратом нагреваются в течение
2 часов при 140°С и одновременном просасывании воздуха, освобожденного
от Н2О и СО2.
Поглощение активированным углем
Активированный уголь хорошо сорбирует пары бензина и других углево-
дородов. Предварительно уголь высушивается просасыванием сухого
воздуха и одновременным нагреванием при 200°С. Уголь путем отсеивания
очищается от пыли (Ключаров, Яковцевская).
Для определения в воздухе паров бензина и других нефтепродуктов
может быть использован и оптический метод, основанный на повышении пре-
ломляющей способности воздуха вследствие примеси указанных продуктов
(Давыдов).
Для испытания смеси воздуха и паров бензина или других углеводородов
на взрываемость может быть использован прибор Якобсона и Мирского, со-
стоящий из взрывной трубки, закрывающейся пробкой, в которую вставлены
два стерженька, соединенные посредством проводов с электролампой и вилкой.
Приключив прибор к электросети, трением стерженьков один о другой
вызывают искрение и наблюдают, произойдет ли в трубке вспышка. Так же
могут быть испытаны и другие взрывоопасные смеси.
Бензол
Бензол (С6Н6) — представитель ненасыщенных углеводородов ряда СпН2П_^
(ароматической группы). Бесцветная, легкоподвижная жидкость характерного
запаха. Удельный вес 0,879 (при 0°). Легко испаряется при обыкновенной
температуре.
Технический бензол содержит различные примеси: толуол, ксилол, серо-
углерод и др.
Применяется как растворитель в ряде производств: каучуковом, клееноч-
ном, линолеумном, лаковом и др.
При концентрации 10—15 мг/л вызывает легкое оглушение. При концен-
трации 20—30 мг/л ведет к потере сознания и судорогам. Концентрации
64 мг/л смертельны при воздействии в течение 5—10 минут. При хрониче-
ском поступлении в организм вызывает прогрессирующее малокровие, жиро-
вое перерождение органов.
Качественное определение
Воздух просасывают через небольшой поглотитель со спиртом. К 5 мл
спиртового раствора бензола добавляют 5 мл смеси равных объемов 3% Н2О2
и 4% раствора NaNO2, а затем 1—2 мл 2 N H.,SO4 и кипятят в течение 2 ми-
нут По охлаждении добавляют несколько капель концентрированного ра-
створа NaOH. При наличии бензола появляется красная окраска (Трифонов).
271
Количественное определение
Для количественного определения бензола применимы те же методы,
которые были описаны для весового определения бензина. Помимо этого
здесь приведены еще два метода, представляющие практический интерес.
Весовой метод
Воздух просасывают со скоростью 1—5 л в час через смесь равных объе-
мов концентрированной серной и дымящей азотной кислоты. Бензол при
этом переводится в твердое кристаллическое тело динитробензол по реакциям:
С6Н6 HNO3 = C6H5NO2 4- Н2О
C6H5NO2 4- HNO3 = C6H4(NO2)2 4- H2O.
Смесь нейтрализуют крепким едким натрием и переводят в делительную
воронку, в которой извлекают динитробензол эфиром. Эфирную вытяжку
динитробензола отделяют от всей остальной жидкости, переводя эту вытяжку
в предварительно взвешенную колбочку. Эфир отгоняют (в отсутствии огня!)
и взвешивают колбочку вместе с динитробензолом. По разности определяют
вес одного динитробензола и перечисляют его на бензол путем умножения на
коэфициент 0,4643 соответственно отношению: •
CcH6(NO2)2
Колориметрический метод Степанова
Воздух просасывают со скоростью 8—10 л в час через 20% раствор сухого
(высушенного при 80 С) NH4NO3 в H2SO4 удельного веса не менее 1.84.
Бензол при этом превращается в динитробензол C6H4(NO2)2. Поглотитель-
ную жидкость переводят затем в 50—60 мл охлажденной дестиллирован-
ной воды. Сосуд с жидкостью ставят в толченый лед. Бросают в жидкость
кусочек лакмусовой бумажки и нейтрализуют крепким аммиаком.
Затем жидкость переводят в делительную воронку и извлекают динитро-
бензол эфиром, прибавляя три раза по 10 мл эфира и каждый раз извлекая
и сливая через верхнюю шейку воронки.
Эфир отгоняют и оставшийся динитробензол растворяют в 10 мл ацетона.
Если к раствору прибавить 4—5 капель 5% NaOH, то появится сине-
фиолетовое окрашивание (реакция Яновского).
Предварительно готовят стандартные растворы динитробензола в ацетоне
с содержанием от 0,01 до 0,1 мг бензола с интервалами в 0,01 мг в 10 мл
ацетона.Затем в стандартные и опытную пробирки одновременно прибавляют
по 4—5 капель 5% NaOH и колориметрируют.
Если в литре ацетона растворить 0,2154 мг динитробензола, 1 мл рас-
твора соответствует 0.1 мг бензола.
Стандартный раствор динитробензола лучше готовить нитрованием бензола
точно так, как это выполняется в отношении пробы.
Лнилпп
Анилин (C6H5NH2) — бесцветная маслянистая жидкость характерного
ароматического запаха, удельного веса 1,025 (при 15°С). На воздухе быстро
желтеет. Пары анилина встречаются на производстве при получении его,
а также при изготовлении анилиновых красок, на текстильных фабриках
(красильные цехи), в каучуковой промышленности и т. п. Пары анилина
стойко пропитывают окружающие предметы. Отравление анилином может
протекать остро и хронически.
Концентрации 0,15—0,2 мг/л могут переноситься без последствий в те-
чение 6 часов, 0,4—0,6 мг/л — в течение 0,5 — 1 часа. Концентрация в 2 мг/л
опасна.
272
Качественное определение
1. Водный раствор анилина от. прибавления раствора хлорной извести
(СаОС12) окрашивается в фиолетовый цвет. Для лучшей сорбции анилина
как основания вместо воды можно взять раствор кислоты, который после
поглощения анилина до прибавления хлорной извести нейтрализуется ще-
лочью.
2. Бихромат калия (К2Сг2О7) в кислом растворе в присутствии анилина
окрашивается в темнозеленый до черного цвет.
3. Раствор из 0,5 мл азотной кислоты удельного веса 1,4 и 0,2 г бертолетовой
соли окрашивается от анилина в синий цвет, переходящий затем в желтова-
токрасный.
4. К 100 мл сиропообразного раствора хлората алюминия — А1(С1О3)3,
содержащего 17,5% хлора, прибавляют 4—5 капель хлористого ванадия
в качестве катализатора. Полученным раствором смачивают полоски батиста
и высушивают. Под действием анилина реактивные полоски ткани синеют.
Синяя окраска постепенно переходит в черную. Реакция весьма чувствительна
и дает возможность обнаружить 0,0005 мг анилина в 1 л воздуха уже через
15—30 минут.
Чем выше температура воздуха и чем ниже его влажность, тем быстрее
наступает окрашивание (Гродзовский).
Количественное определение
Бромометрический мегод
Бромометрический метод основан на реакции образования трибромани-
лина при действии брома на анилин:
CeH5NH, + ЗВГо = CeH,Br3NH, + ЗНВг.
Воздух просасывают со скоростью 100—200 л в час через 10% раствор
H2SO4. Нейтрализуют частично поглотительную жидкость и прибавляют
с избытком раствор гипобрэмита натрия. Для получения гипобромита натрия
растворяют 3,0—4,0 г брома в 1 л воды и приливают раствор едкой щелочи
до обесцвечивания:
Вг2 4- 2NaOH = NaOBr + NaBr 4- Н2О.
Из гипобромита натрия под действием кислоты освобождается бром, ко-
торый связывается анилином по приведенной выше реакции. Затем прили-
вают небольшое количество раствора йодистого калия; при этом избыток
брома, не вступивший в реакцию с анилином, вытесняет из йодистого калия
свободный иод, который оттитровывается 0,1 N раствором тиосульфата нат-
рия. Ход реакций:
NaOBr 4- NaBr 4- H2SO. = Na2SO4 4- H2O 4 Br2
2KJ 4- Br2 = 2KBr + J2
J2 4* 2Na2S,O3 = 2NaJ 4- Na2S4O6.
Отдельно определяется титр гипобромита натрия. Для этой цели опреде-
ленный объем его подкисляют H2SO4 и прибавляют раствор 1 г KJ в неболь-
шом количестве воды. Выделившийся иод титруют тиосульфатом натрия.
Ход реакции тот же. Зная титр гипобромита натрия, определяют количество
брома, вступившего в реакцию с анилином, а отсюда делают вывод о количе-
стве анилина, поглощенного из воздуха.
Из приведенной выше реакции видно, что 93 весовых части анилина
(93—молекулярный вес анилина) связываются с 6 . 79,92 весовыми частями
брома (79,92— атомный вес брома). После сокращения на 6 получаем, что
15.5 весовых частей анилина связываются с 79,92 весовыми частями брома.
Отсюда находим, что 1 мл 0,1 N брома связывает 1,55 мг анилина.
18 А. И. Бурштейн
273
Пример. Исследуя воздух на анилин в красильном отделении текстильной фабрики,
прососали через 10% раствор H2SO4 196 л воздуха (объем приведен к нормальным условиям).
К поглотительной жидкости после частичной нейтрализации H.,SO4 прибавлено 15 мл ги-
побромита натрия и некоторое количество раствора KJ. На титрование выделившегося
пода затрачено 12 мл 0,1 N тиосульфата натрия. При определении титра гипобромита нат-
рия на 20 мл его уходит 22 мл 0,1 N Na2S2O.v Нужно определить на основании приведенных
данных концентрацию анилина в воздухе обследованного цеха.
При определении титра гипобромита натрия найдено, что каждый миллилитр его соответ-
22
ствуетэд мл 0,1 N тиосульфата натрия. Так как в реакцию взято 15 мл гипобромита, то весь
22 . 15
объем его соответствует — ,jq-- — 16,5 мл 0,1 N тиосульфата.
На оттитровывание остатка гипобромита пошло 12 мл 0,1 N тиосульфата; следовательно,
количество гипобромита, пошедшего в реакцию с анилином, соответствует 16,5—12 =
= 4,5 мл 0,1 N раствора тиосульфата. Каждый миллилитр 0,1 N тиосульфата натрия экви-
валентен 1 мл 0,1 N иода, а каждый миллилитр последнего соответствует одному мл 0,1 N
брома. Следовательно, количество брома, вступившего в реакцию с анилином, соответ-
ствует 4,5 мл 0,1 N брома. Как было указано, каждый миллилитр 0,1 N брома может связать
1,55 мг анилина. Следовательно, во время анализа всего связано 1,55 • 4,5 мг анилина, что
на литр воздуха составит
1,55 • 4,5 Л .
--------~ 0,03.ээ мг/л.
196
Метод диазотирования
Метод диазотирования заключается в том, что полученную кислую соль
анилина после поглощения паров его переводят при помощи титрованного
раствора нитрита в диазосоединение и по количеству израсходованного
нитрита делают вывод о количестве поглощенного из воздуха анилина.
Реакция между кислой солью анилина и нитритом идет по уравнению:
Л
C6H5NH2HC1 + HNOo=CgH5 . Nf 4-2Н2О.
4 Cl
Титрование считается законченным, когда капля титруемого раствора,
нанесенная на иодкалийкрахмальную бумажку, вызывает моментальное по-
синение ее (выделение свободного иода из KJ). Реакция диазотирования
протекает медленно и требует затраты более часа времени. Титр раствора
нитрита устанавливается при помощи КМпО4 в кислой среде:
2КМпО4 + 5NaNO2 + 3H2SO4 = K2SO4 + 2Mn SO4 + 3H2O + 5NaNO3.
Из первой реакции видно, что 1 мл 0,01 N раствора нитрита соответствует
0,93 мг анилина.
Гродзовский рекомендует перед титрованием упаривать поглотительный
раствор на песочной бане до появления паров SO3. В 1 мл титруемого раствора
не должно содержаться более 5 мг анилина.
Колориметрический метод
Воздух поглощают слабым раствором серной кислоты. 5 мл поглотитель-
ной жидкости нейтрализуют едким натрием, разбавляют водой до 20 мл,
добавляют 1 мл раствора хлорной извести, содержащей 0,1% активного хлора,
и через 2 минуты подщелачивают, прибавив 1 мл 1 N раствора NaOH. При-
готовляют ряд стандартов с определенным содержанием анилина.
Для этой цели 1 г чистого свежеперегнанного анилина растворяют в воде
до общего объема в 1 л. 1 мл раствора содержит 1 мг анилина.
В 7 одинаковых цилиндров вводят от 0,1 до 0,7 мл стандартного раствора
анилина с интервалом в 0,1 мл, разбавляют водой до 200 мл и взбалтывают.
К 20 мл каждого из этих растворов, содержащих от 0,01 до 0,07 мг анилина
с интервалом в 0,01 мг, добавляют те же реактивы, как и к испытуемому ра-
274
створу, и колориметрируют. Получающееся желтое окрашивание довольно
устойчиво.
Другие методы колориметрического определения анилина см. у Алексе-
евой Ч
Никотин
Никотин (C10H14N2)—бесцветная маслянистая жидкость удельного веса 1.0097
при 20сС. На воздухе буреет вследствие поглощения кислорода. Хорошо
растворяется в воде, спирте, эфире, хлороформе и пр. При острых отравле-
ниях вызывает головную боль, тошноту, рвоту, сердцебиение, затрудненное
дыхание. При хроническом отравлении — атрофическое состояние слизистых
носа, глотки и гортани, потерю аппетита, нервные расстройства. Встречается
в воздухе табачных производств как при сушке, так и при ферментации и
обработке табака.
Качественное определение
Воздух просасывают через 1% (по объему) раствор серной кислоты со
скоростью 100 — 200 л в час. Так как одновременно с никотином в воздухе
табачных производств могут присутствовать и другие осно ания (пиридин,
аммиак), их следует удалить. Это достигается следующим образом: к погло-
тительной жидкости прибавляют в избытке известковое молоко и оставляют
на 3 дня в экссикаторе над крепкой серной кислотой. Аммиак и пиридин
при этом улетучиваются, никотин задерживается. Затем фильтруют, про-
мывают осадок, на фильтре подкисляют фильтрат слабой серной лсислотой
и упаривают на водяной бане до небольшого объема. Далее снова фильтруют,
промыв фильтр слабой серной кислотой. С фильтратом для обнаружения
никотина может быть проведена одна из следующих проб.
1. 5% раствор фосфоровольфрамовой кислоты дает с никотином белую
муть еще в разведениях 1 : 1 000 000. Подогревание пробы с последующим
охлаждением ускоряет появление мути. При больших концентрациях нико-
тина выпадает осадок.
2. 2% раствор фэсфэромолибденовой кислоты дает с никотином светло-
желтую муть еще в разведениях 1 : 200 000.
3. K(Bi J4) дает с никотином красный или коричневый осадок. Предел
чувствительности реакции 1 : 40 000. Для получения иодвисм уткали я ра-
створяют 10 г основного азотнокислого висмута в возможно малом количестве
крепкой азотной кислоты и полученный раствор вводят при взбалтывании
в насыщенный раствор 35 г KJ в воде. Хранить в темном месте.
4. Раствор р-диметиламинобензальдегида в дымящей НС1 дает с никоти-
ном фиолетовую окраску.
Количественное определение
Нефелометрический метод
По отделении пиридина и аммиака (см. выше) количественное содержание
никотина определяется нефелометрически: к раствору прибавляют 5% фосфор-
новольфрамовую кислоту и появляющуюся муть сравнивают со стандартными
растворами, приготовленными с содержанием никотина от 0,01 до 0,1 мг ни-
котина в 10 мл с интервалом в 0,01 мг (Бурштейн).
Колориметрический метод
К 10 мл раствора по удалении пиридина прибавляют 2—3 капли уксусной
кислоты, 5 мл щелочного раствора сульфанилата аммония и 5 мг водного
раствора BrCN. При наличии никотина появляется желтая окраска, приобре-
тающая затем золотистокрасный оттенок.
1 Журнал прикладной химии, № 1, 1931.
275
Стандарты готовят как в предыдущем методе. Сульфанилат аммония при-
готовляется следующим образом: 0,02 г сульфаниловой кислоты растворяют
в 300 мл воды и прибавляют 6 мг 5% NH4OH, свободного от пиридина.
Раствор цианбромида готовится прибавлением 10% KCN к насыщенной
бромной воде до исчезновения желтой окраски.
ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ АТМОСФЕРНОГО ВОЗДУХА
Атмосферный воздух населенных пунктов и промышленных центров может
загрязняться не только аэрозолями, но и вредными газами промышленного
и коммунального происхождения (сернистый ангидрид, хлор, хлористово-
дородный газ, окислы азота, окись углерода, аммиак, сероводород и т. п.).
По мнению проф. Р. А. Бабаянца, сернистый ангидрид может служить
хорошим показателем загрязнения атмосферного воздуха промышленных
пунктов, подобно тому как угольный ангидрид служит хорошим индикато-
ром загрязнения воздуха жилых и общественных помещений. Эту мысль
автор подкрепляет соображением, согласно которому количество сернистого
ангидрида, выбрасываемого в воздушный бассейн промышленными предпри-
ятиями, прямо пропорционально количеству выбрасываемой ими золы, сажи и
пыли \
Автор предлагает для воздуха промышленных городов считать в ка-
честве предельно допустимых величин сернистого ангидрида: в кварта-
лах промышленных районов — максимально 0,3 мг/м3, в жилых районах —
0,05 мг/м3.
Что касается методов исследования атмосферного воздуха, то они совпа-
дают с соответствующими методами, применяемыми для жилых и промышлен-
ных помещений.
Забор проб воздуха для анализа производится путем аспирации
(Берюшов, Бухалов, Могилевчик, Угрюмова-Сапожникова и мн. др.).
В связи с малой концентрацией вредных газов, какая наблюдается обы-
кновенно в атмосферном воздухе, приходится просасывать через поглоти-
тельную жидкость большие количества воздуха, что создает известные труд-
ности.
Некоторые авторы для устранения этих трудностей предлагают пользо-
ваться специальными аспираторами. Так Рязанов сконструировал три моди-
фикации таких аспираторов: стационарный аспиратор, батарейный, или сек-
ционный аспиратор и автоматический аспиратор. Угрюмова-Сапожникова
сконструировала аспиратор для круглосуточной аспирации воздуха. Все эти
аспираторы имеют, к сожалению, ряд дефектов — сложны, громоздки и т. п.
Ввиду этого вопрос о конструкции простого и портативного прибора для про-
сасывания больших количеств атмосферного воздуха еще ждет разрешения.
ХИМИЧЕСКИЙ АНАЛИЗ ПЫЛИ
. Химический анализ пыли сводится к определению органических и мине-
ральных веществ (золы) и кремнезема. Нередко пыль подвергается еще
специальному исследованию на присутствие каких-либо ингредиентов:
металла (свинец, цинк, медь и пр.), алкалоида (никотин, кофеин и т. п.).
Для анализа пыль собирается по возможности из разных мест данного
помещения. Для этой цели рекомендуется разостлать в разных местах листы
гладкой бумаги, оставить их на некоторое время и затем собрать осевшую
на них пыль. Еще лучше пользоваться для этой цели пылью, задержанной
на фильтрах при просасывании через них воздуха. Таким образом, анализу
подвергается пыль, которая носится в воздухе и, следовательно, вдыхается
человеком.
1 Р. А. Бабаянц, Загрязнение городского воздуха, М., 1948.
276
Чтобы собрать значительные количества пыли, достаточные для анализа,
приходится просасывать большие количества воздуха (от одного до несколь-
ких кубических метров), развивая большую скорость просасывания (0,5—
1 м3/час и более).
Приведем методику определения некоторых составных частей пыли.
Зола и органические вещества
Навеску пыли прокаливают в предварительно высушенном и взвешенном
платиновом или фарфоровом тигле до постоянного веса, охлаждают и взве-
шивают. Остаток после прокаливания представляет золу, а разница между
весом высушенной пыли и золой соответствует количеству органических
веществ, точнее потере при прокаливании.
Для определения зольности пыли и потери при прокаливании определен-
ную навеску высушенной пыли помещают в тигель и нагревают сначала на
малом огне, держа пламя горелки поодаль от тигля, а затем все больше и
больше приближая окислительное пламя к тиглю. Если сжигание происхо-
дит полностью, то зола приобретает однообразный цвет без примеси кусочков
угля.
Цвет золы обыкновенно серый, но может иметь и другой оттенок в за-
висимости от состава золы: бурокрасный от примеси окиси железа, черный
от окиси меди и т. д.
Если сжигание органических частей идет плохо, то, дав остыть тиглю,
смачивают содержимое дестиллированной водой или 3% перекисью водорода
и вновь прокаливают до получения однородной массы. Закончив прокаливание,
дают тиглю остыть сначала на воздухе, а затем в экссикаторе и взвешивают
на аналитических весах.
Зная вес тигля, можно по привесу сделать заключение о количестве золы
во взятой навеске пыли. Разность между взятой навеской и найденной золой
соответствует потере при прокаливании.
Кремнезем
Кремнезем представляет окись кремня (SiO2). Входит в состав многих
горных пород.
При вдыхании больших количеств кремнезема и его соединений возни-
кает силикоз легких.
Для количественного определения кремнезема золу пыли обрабатывают
концентрированной соляной кислотой, выпаривают, вновь обрабатывают та-
кой же кислотой, опять выпаривают и нагревают до 200°С. Затем прибавляют
дестиллированной воды, взбалтывают и фильтруют. На фильтре задержи-
вается кремнезем. Фильтр с осадком прокаливают в тигле. По привесу опре-
деляют количество кремнезема.
С гигиенической точки зрения большой интерес представляет вопрос
о содержании свободного кремнезема в пыли, так как собственно он вызывает
силикоз легких. Определение кремнезема в пыли производится так: исследуе-
мую пыль размешивают в жидкости с удельным весом, равным удельному
весу кремнезема (2,66). Полученную взвесь подвергают центрифугированию.
Тяжелые частицы при этом оседают, частицы кремнезема и более легкие
остаются во взвеси. Слив центрифугат, немного понижают удельный вес
жидкости, напр. до 2,64, и вновь центрифугируют. В осадок выпадают части-
цы кремнезема. Осадок собирают, высушивают и взвешивают (Е. В. Хухрина).
Свинец
Свинец (РЬ) — металл серого цвета с точкой плавления 327°С и точкой
кипения 1540°С.
277
Как промышленный яд встречается при добывании и выплавке из
свинцовых руд, при наводке глазури в гончарном производстве, изготовлении
свинцовых красок, в типографском деле и т. п.
Является протоплазматическим ядом. Поражает кровь, костный мозг,
нервную систему. Опасен уже в количестве 1 мг/м2 3 воздуха.
Для подготовки пробы пыли к анализу просасывают через ватный фильтр
1—3 м3 воздуха со скоростью 0,5—0,6 м3 час.
Ватный фильтр рекомендуется предварительно прокипятить в 3% уксус-
ной кислоте, промыть горячей водой, отжать и высушить при 60°С.
После забора пробы свинец экстрагируют из пыли. Для этой цели ватный
фильтр вместе с пылью переводят в стакан, приливают 3% уксусной кислоты
и кипятят 15 минут. Жидкость сливают, ватный фильтр переносят количе-
ственно на бюхнеровскую воронку и промывают 2—3 раза 3% уксусной
кислотой, а затем горячей водой до тех пор, пока промывные воды не пере-
стают показывать кислую реакцию.
Промывные воды присоединяют к слитому ранее раствору и полученный
раствор, в котором свинец находится в виде уксуснокислого свинца—
РЬ(СН3СОО)2, выпаривают на водяной бане досуха. Осадок растворяют в не-
большом количестве дестиллированной воды.
Свинец можно экстрагировать и 10% азотной кислотой; в этом случае
получают раствор нитрата свинца — Pb(NO3)2. Подготовив таким образом
пробу, приступают к качественному и количественному определению свинца.
Качественное определение
С раствором свинцовой соли может быть проведен ряд пробирочных ре-
акций, а также ряд микрохимических кристаллоскопических реакций.
Так как содержание свинца в пыли обыкновенно мало, рекомендуется
пользоваться микрохимическими реакциями.
1. Каплю испытуемого раствора переносят на предметное стекло, прибав-
ляют каплю уксуснокислой меди и выпаривают; к остатку прибавляют каплю
Рис. 215. Виды осадков, полученных при микрохими-
ческих реакциях на свинец.
а — трппелънитрнт свинка. налил в мели — K,PbCu(NOt\,
b — пи чистый свинец— I b.Tt, С — хлористый свинец — I’bClj,
слева — выпавший из концентрированного раствора,
справа — из разбавленного раствора.
концентрированного раство-
ра уксуснокислого аммония
(CH3COONH4) и KNO2 в 50%
уксусной кислоте. В присут-
ствии свинца выпадают чер-
ные кристаллы трипельнитри-
та калия, свинца и меди —
K2PbCu(NO2)6. От прибавле-
ния аммиака кристаллы обес-
цвечиваются; черная окраска
восстанавливается при при-
бавлении уксусной кислоты
(рис. 215 а). Предел чувстви-
тельности реакции 0,03 7 РЬ.
2. Каплю испытуемого раствора переносят на предметное стекло, под-
кисляют уксусной кислотой и вносят маленький кристаллик KJ; в присут-
ствии свинца выпадает иодистый свинец (PbJ2) в форме треугольных и шести-
угольных пластинок желтого цвета (рис. 215b). Предел чувствительности
реакции 0,2 7 РЬ. Избыток йодистого калия вредит реакции, так как раство-
ряет PbJ2 с образованием растворимой комплексной соли K(PbJ3).
3. К капле раствора, перенесенной на предметное стекло, прибавляют
каплю разведенной НС1. Из концентрированных растворов свинца выпадают
кристаллы хлористого свинца (РЬС12) в виде дендритов (рис. 215 с левая по-
ловина); из разбавленных растворов — в виде призм и шестиугольников
(рис. 215с правая половина)
Предел чувствительности 0,3 7 РЬ.
278
Количественное определение
Количественное определение свинца в пыли проводят обыкновенно объем-
ным или нефелометрическим методом.
Из объемных методов здесь описаны йодометрические: хроматный и йодат-
ный.
Хроматный метод
Хроматный метод основан на осаждении свинца бихроматом калия и по-
следующем йодометрическом определении избытка последнего. Реакция проте-
кает по следующим уравнениям:
2Pb(NO3)2 + К2Сг2О7+ Н2О — 2PbCrO4+ 2KNO3 Ц- 2HNO3
K2Cr2O7 + 6KJ + 7H2SO4 = 4K2SO4 + Cr2(SO4)3 7Н2О + 3J2
J2 + 2Na2S2O3 = 2NaJ + Na2 S4O6.
Раствор свинцовой соли, полученный, как описано на стр. 277, вносят в
малую коническую колбочку, нейтрализуют 0,1 N NaOH1 и при нагревании
на водяной бане осаждают небольшим избытком 0,01 N бихромата калия:
0,4904 г К2Сг2О7 вносят в литровую колбу, прибавляют 8,2 г уксуснокислого
натрия, предварительно растворенного в 100 мл воды и 25 мл 2N (12%)
уксусной кислоты и доводят объем водою до метки.
Введение бихромата в жидкость производят из микробюретки или из
микропипетки до тех пор, пока не перестанет образовываться осадок, и затем
добавляют еще небольшой избыток жидкости.
Колбочку нагревают на водяной бане 10—15 минут при постоянном взбал-
тывании; при этом выпадает кристаллический красножелтый осадок хро-
мата свинца (РЬСгО4).
Содержимое конической колбочки переводят в малую мерную колбочку
(10—25 мл) и доводят водою до метки. Взбалтывают и дают отстояться. От-
бирают пипеткой 10—15 мл и переносят в колбочку с притертой пробкой,
прибавляют 0.5 г KJ и 1 мл H2SO4 (1 : 3). Через 5 минут титруют выделив-
шийся иод 0,01 N Na2S2O3 при индикаторе крахмальном клейстере и таким
образом определяют количество непрореагировавшего бихромата во взятсм
объеме. Результат перечисляют на весь объем жидкости и вычитают из общего
объема взятого бихромата. Таким образом находят то количество милли-
литров 0,01 N К2Сг2О7, которое вступило в реакцию с солью свинца. Резуль-
тат умножают на 0,6907 и получают количество свинца во взятой навеске.
Переводный коэфициент 0,6907 находят из равенства:
2РЬ
6 • 100
2- 207,21
6 • 100
= 0,6907 мг.
Пример. Для анализа прососали через фильтр 2,5 м3 воздуха. Все количество пыли
вместе с ватой подвергнуто анализу. 0,01 N раствора К2Сг2О7 прибавлено всего 1,4 мл.
На оттитровывание иода, выделенного из К J, избытком бихромата ушло 0,8 мл 0,01 N тио-
сульфата. Отсюда заключаем, что в реакцию с солью свинца вступило:
1,4— 0,8 = 0,6 мл 0,01N К2Сг2О7.
Следовательно, в 1 м3 испытуемого воздуха находится:
0,6907 • 0,6 п
—---------— ж 0,1658 мг РЬ.
2,5
Йодатный метод
Йодатный метод основан на осаждении свинца йодатом калия (KJO3) и
последующем йодометрическом определении избытка последнего. Реакции
протекают по следующим уравнениям:
1 Индикатором служит фенолфталеиновая бумажка.
279
Pb(NO3)2 + 2KJO3 = Pb(JO3)2 4- 2KNO3
К J03 + 5K J + 3H2SO4 = 3K2SO4 4- 3H2O 4- 3J2
J2 + 2Na2S2O3 — 2NaJ + Na2S4O6.
Раствор свинцовой соли, полученный так, как описано выше, подкисляют
слабой уксусной кислотой (берут 2—3 капли 5% уксусной кислоты), прили-
вают из микрэбюретки или микропипетки точно отмеренное количество 0,01 N
KJO3 и оставляют на ночь. Свинец выпадает в виде белого кристаллического
осадка йодата свинца — Pb(JO3)2. В дальнейшем определение ведется так
же, как и при хроматном методе.
1 мл 0,01 N раствора KJO3, связавшийся со свинцовой солью, соответствует
207,21
2-6-100
= 0,1727 мг РЬ.
Нефелометрический метод
Нефелометрический метод основан на образовании мути в растворе свин-
цовой соли при добавлении H2S:
Pb(NO3)2 4- H2S = Pb 4- 2HNO3.
Приготовляют стандартный раствор свинцовой соли. Для этого 0,1599 г
Pb(NO3)2 растворяют в литре воды. 1 мл такого раствора соответствует 0,1 мг.
Расчет навески:
ззг22б 599 г
РЬ 10 207,21 • 10
Составляют шкалу с содержанием свинца от 0,01 до 0,1 мг. Доливают
содержимое пробирок до 10 мл. Пробу, подготовленную так, как описано
выше, берут в пробирку того же диаметра, что и остальные, и во все пробирки
приливают до 1 мл свежеприготовленной сероводородной воды. Через не-
сколько минут нефелометрируют на белом фоне.
Вместо сероводородной воды в каждую пробирку можно добавить по од-
ной капле 3% уксусной кислоты и по одной капле 10% бихромата калия,
через несколько минут нефелометрируют на темном фоне.
Мышьяк
Мышьяк (As) — металлоид серостального цвета с атомным весом 74,91.
На воздухе горит, превращаясь в мышьяковистый ангидрид (As2O3).
В воздушную пыль соединения мышьяка могут попадать при обжиге и
выплавке мышьяксодержащих руд, при изготовлении и применении некото-
рых красок (аурипигмент, реальгар, парижская зелень и др.). Мышьяк-
содержащая пыль может попадать в воздух из различных инсектисидов и
фунгисидов (белый мышьяк, арсенит натрия, арсенит кальция, арсенат каль-
ция и др.). Поэтому у нас при работе с указанными препаратами принимают
соответствующие меры предосторожности.
Если соединения мышьяка попадают с пылью в организм, то они дают
преимущественно хронические отравления, при которых на первый плач
выступают поражения кожи, слизистых, желудочнокишечного тракта и нерв-
ной системы.
Забор пробы пыли ведется так же, как и при анализе на свинец (стр. 277).
Для подготовки пробы пыли к анализу ватный фильтр вместе с задержан-
ной пылью подвергают влажному озолению. Для этой цели фильтр вместе
с пылью переносят количественно в коническую колбу на 250 мл, прибавляют
25 мл концентрированной соляной кислоты и небольшими порциями бертс -
летовую соль. Колбу закрывают пробкой с обратным холодильником и на-
гревают на водяной бане. По растворении всего содержимого в колбе и обе-
сцвечивании жидкости последнюю переносят количественно в фарфоровую
280
чашку и нагревают, прибавляя небольшие порции воды, до полного исчезно-
вения запаха хлора. Полученный раствор при надобности упаривают. Ок
может быть использован для качественных и количественных определений.
Качественное определение
Для качественного обнаружения мышьяка предложено много пробироч-
ных и кристаллоскопических реакций. Здесь приведены наиболее чувстви-
тельные кристаллоскопические реакции.
1. На предметное стекла помещают каплю испытуемого раствора и сюда
же прибавляют каплю магнезиальной смеси. В присутствии мышьяковой
кислоты выпадает NH4MgAsO4 . 6Н2О; при этом из слабых растворов выпа-
дают кристаллы в виде призм (рис. 216 а — левая половина), из концентри-
рованных же растворов получается осадок в виде дендритов (рис. 216а —
правая половина).
Так как соли фосфорной кислоты дают изоморфные осадки, для отличия
можно воспользоваться прибавлением раствора азотнокислого серебра; при
этом соль мышьяковой кислоты окрашивается в бурый цвет, соль фосфор-
ной кислоты — в желтый.
Магнезиальная смесь, применяемая при этой реакции, готовится растворе-
нием 55 г кристаллического хлористого магния и 70 г хлористого аммония
в 650 мл воды; раствор доливают до литра крепким аммиаком удельного
веса 0,96.
Чувствительность реакции 0,05 -( As.
2. Неско; ь'<о капель испытуемого раствора медленно выпаривают на пред-
метном стекле и после охлаждения вводят в центр остатка каплю реактива,
который готовится прибавле-
нием к 3% раствору AgNO3
одной пятой (по объему) ча-
сти аммиака крепостью пе
ниже 7—8 N. В присутствии
солей As получаются гра-
натового цвета кристаллы
As(OAg)3 гексаэдрическойили
формы (рис.
3. К капле нейтрального
или азотнокислого испыту-
емого раствора прибавляют
кристаллик Hg2(NO3)2. Выпадает сначала аморфный осадок Hg3A3O4, пере-
ходящий вскоре в светложелтые иглы, расположенные в виде звезд и
дендритов (рис. 216 с).
Предел чувствительности 0,3 у As.
Количественное определение
Количественное определение мышьяка в жидкости, полученной после
влажного озоления, производится колориметрически — так же, как и опре-
деление мышьяковистого водорода (стр. 262).
При больших количествах мышьяка возможно и весовое определение.
В этом случае жидкость, полученную после влажного озоления, переводят
количественно в колбу, прибавляют два объема концентрированной НС1,
помещают колбу в мелко истолченный лед и пропускают струю промытого
сероводорода. Оставляют на ночь. Выпавший осадок пятисернистого мышья-
ка — (As2S5) отфильтровывают через тигель Гуча, промывают водой и затем
спиртом, высушивают и взвешивают. Привес соответствует количеству Аз28й.
Для пересчета на As умножают на 0,4832 соответственно:
Asa = >49,82 ~ 0 4832.
As2S5 310,12
28?
Рис. 216. Виды осадков, полученных при микро-
химических реакциях на мышьяк.
а — кристаллы XIl4MgAsO4. 6HtO, слева — выпавшие из
слабых растворов, справа — из концентрированных, Ь —
кристаллы As(OAg)3, с — дендриты Hg3AsO4.
ромбической
216b).
ХИМИЧЕСКИЙ АНАЛИЗ АТМОСФЕРНОЙ ПЫЛИ
Химический анализ атмосферной пыли сводится к определению: 1) ко-
личества золы и потерь при прокаливании, 2) смолистых веществ, 3) хлори-
дов, 4) сульфатов и др.
Зола и потери при прокаливании
Под зольностью пыли, как указывалось выше, понимают сумму мине-
ральных веществ пыли, которые остаются после сжигания органической
части ее.
Зола пыли состоит в основном из минеральных веществ почвы и золы,
выбрасываемой из топок в атмосферный воздух. Потеря при прокаливании
пыли происходит преимущественно за счет углеродистых частей пыли (сажи),
а также веществ растительного и животного происхождения. Незначитель-
ные потери могут происходить за счет разложения во время прокаливания
некоторых минеральных веществ: улетучивание СО2 из карбонатов, S из
сульфидов и т. д. Определение см. на стр. 277.
Смолистые вещества
Смолистые вещества пыли попадают в нее вместе с дымом и золой, выбра-
сываемыми из топок в воздух.
Для определения смолистых веществ определенную навеску сухой пыли
вводят в колбочку, приливают немного сероуглерода и оставляют стоять,
время от времени взбалтывая; смолистые вещества при этом растворяются
в сероуглероде. Сероуглерод вместе с растворенными смолистыми веществами
сливают и повторяют экстракцию. По удалении смолистых веществ пыль
высушивают в отсутствии огня (взрывоопасно!) и взвешивают. Потеря в весе
соответствует количеству смолистых веществ. Экстракцию можно вести в ап-
парате Сокслета.
Сульфаты
Определенную навеску пыли (1—2 г) переводят в фарфоровую чашку,
приливают сюда 10—15 мл крепкой соляной кислоты и нагревают до кипения,
которое поддерживают 15 минут. Затем чашку переносят на водяную баню,
где испаряют жидкость досуха. К остатку прибавляют 100 мл горячей воды,
взбалтывают и, дав осадку выпасть, отфильтровывают, не взмучивая осадка.
Последний промывают декантацией еще два-три раза горячей водой, каждый
раз отфильтровывая жидкость в ранее полученный фильтрат.
Осадок переносят на фильтр и здесь продолжают промывание его неболь-
шими порциями горячей воды, пока промывные воды не перестанут показы-
вать реакцию на H2SO4 (проба с ВаС12).
Весь полученный фильтрат вновь переносят в фарфоровую чашку и вы-
паривают досуха. К сухому остатку прибавляют несколько капель НС1 и
разбавляют горячей водой; при этом выпадает осадок (SiO2). В чашку при-
бавляют 10% раствор аммиака и дают жидкости постоять до выпадения ги-
дроокисей алюминия и железа. Жидкую часть отфильтровывают и промывают
осадок на фильтре до полного исчезновения реакции на хлор в промывных
водах (проба с AgNO3). Доводят объем фильтрата до 300—350 мл, прибав-
ляют 1 мл НС1 удельного веса 1,19 и определяют сульфаты весовым путем,
как описано выше (стр. 243. См. также стр. 334).
Для пересчета на S умножают вес BaSO4 на 0,137 соответственно:
_^ = 2^„о,137.
BaS04 233,32
282
Хлориды
Хлориды экстрагируют из пыли водой и определяют в водной вытяжке,
как описано на стр. 259. См. также стр. 330.
Ниже приводится таблица предельно допустимых концентраций вредных
газов, паров и пыли согласно ГОСТ 1324-41 (табл. 37а).
Табл и ц а 37а
Предельно допустимые концентрации газов, паров и пыли в воздухе производственных
помещений
Наименование вещества Акролеин Аммиак Анилин, толуидин, ксилидин Ацетон Бензидин, дианизидин, а- и нафтилами ны Бензин, уайт-спирт, лигроин, керосин Бензол, толуол, ксилол, соль- вентнафта 1 и 11 Декалин, тетралин Марганец и его соединения . Мышьяк и его соединения в пересчете на мышьяк . . . Нитробензол, нитротолуол . . Ди- и тринитросоединения (динитробензол, тринитрото- луол и др.) Динитрохлорбензол Непредельные спирты жир- ного ряда (аллиловый и др.) Окислы азота Окись углерода1 2 Окись цинка Окись этилена Ртуть металлическая .... Свинец и его неорганические соединения j Скипидар 1 Сернистый газ* 1 Предельно j допустимые ( концентра- ции (мг/л) 1 0,002 0,02 ; 0,005 0,2 | 0,001 0,3 0,1 0,1 0,0003 0,0003 0,005 0,002 0,001 । 0,002 : 0,005 0,02 I 0,005 0,001 0,00001 0,00001 ! 0,3 0,02 ' Наименование вещества Сероводород Сероуглерод Серная кислота и серный ан- гидрид ! Спирт метиловый | Спирт этиловый 1 | Спирт бутиловый Сулема Табачная и чайная пыль . . Фенол . Формальдегид Фосген Фтористый водород Фтора соли Хлор Хлористый водород Хлорированные углеводоро- ды (четыреххлористый угле- род, трихлорэтилен, дихлор- этилен) Хлорнафталип Хлордифенил Хромовый ангидрид, хро- 1 маты, бихроматы । Цианистый водород ; Эфиры уксусной кислоты (этилацетат, пропилацетат, бутилацетат) Эфиры уксусной кислоты (амилацетат, метилацетат). . Этиловый (диэтиловый) эфир Предельно допустимые концентра- ции (мг/л) 0,01 0,01 0,002 0,05 1,0 0,2 0,0001 0,003 0,005 0,005 0,0005 0,001 0,001 0,001 0,01 0,05 0,001 0,001 0,0001 0,0003 0,3 0,1 0,3
Ни в одной стране в мире, кроме СССР, не соблюдаются столь строгие
нормы в отношении воздуха производственных помещений. Эти нормы в отно-
шении воздуха производственных помещений указывают на заботу Партии
и Правительства о здоровье трудящихся.
1 В доменных и мартеновских цехах, в газогенераторных, а также в производствах,
где окись углерода является сырьем (синтез метанола), предельно допустимая концен-
тр; ция для окиси углерода устанавливается в 0,03 мг/л.
2 При плавке и обжиге материалов, содержащих серу, допустимая концентрация
0,04 мг/л.
283
Для нетоксических сортов пыли приняты следующие нормы.
1. Предельно допустимые концентрации нетоксической пыли в воздухе
рабочей зоны производственных помещений не должны превышать:
а) 2 мг/м3 для видов пыли, содержащих кварц в количестве свыше 50%
(пыль кварца, песка, кварцита и др.)
б) до 10 мг/м3 для всех остальных видов пыли.
2. Предельно допустимые концентрации пыли по отдельным отраслям
промышленности, в зависимости от характера пыли и особенностей произ-
водственного процесса, в пределах норм, указанных в п. 1 устанавливаются
соответствующим министерством по согласованию со Всесоюзной Государ-
ственной Санитарной Инспекцией.
3. В отдельных случаях, при доказанной невозможности достигнуть при-
веденных концентраций, с разрешения министра соответствующего ми-
нистерства по согласованию со ВГСИ допускаются отступления от норм ука-
занных в п. 1.
Для атмосферного воздуха допустимые концентрации газов и пыли еще
точно не установлены.
Проф. Бабаянц Р. А. считает, что для аэрозолей, выпадающих из атмо-
сферного воздуха, можно принять (на ближайшие 10—15 лет) следующие
допустимые нормы: 50 т/км2 в год для мест с чистым воздухом и 200 т/км2
для воздуха промышленных городов. Эти цифры несколько выше указанных
другими авторами (Яковенко, Эттингер).
Согласно распоряжению Министерства Здравоохранения СССР от 30/VII
1948 г. Институт общей и коммунальной гигиены АМН СССР и Центральный
санитарный институт им. Эрисмана совместно с другими научно-исследова-
тельскими учреждениями разработали и представили во ВГСИ как ориенти-
ровочные «нормы предельно допустимых концентраций вредных веществ
в атмосферном воздухе».
В связи с постановлением Совета Министров СССР от 29/V 1949 г.
«О мерах борьбы с загрязнением атмосферного воздуха и об улучшении са-
нитарно-гигиенических условий населенных мест» значительно возрос интерес
со стороны научно-исследовательских учреждений к этим важнейшим сани-
тарным вопросам.
На Всесоюзной планово-тематической конференции по коммунальной ги-
гиене, протекавшей в Москве (31/1—4/IJ 50 г.), были приняты следующие
нормы в отношении 10 главнейших вредных ингредиентов, выделяемых различ-
ными промышленными предприятиями в атмосферный воздух (таблица 376).
Таблица 376
Предельно допустимые концентрации газов, паров и пыли в атмосферном воздухе
Наименование вещества г. '1 Предельно допустимая концентрация ! в мг/м3 в
одноразовой пробе среднесуточной пробе
Сернистый ангидрид I 0,75 0,25
Сероводород • . . . . 0,05 i ’ 0,015
Сероуглерод 0,5 0,15
Хлор 0,1 0,03
Окислы азота (при пересчете на N2O5) 0,5 0,15
Окись углерода 6,0 2,0
Свинец — 0,0007
Ртуть — О.ОнОЗ
Пыль нстоксичсская 0,5 0,15
Сажа 0,15 1 1 0,05 1
Приведенные нормы пока не узаконены и являются ориентировочными
для ВГСИ.
284
БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ВОЗДУХА
Живые и мертвые микроорганизмы (бактерии, дрожжевые клетки, пле-
сени и др.), находящиеся в воздушной среде, составляют аэропланктон.
(По аналогии с гидропланктоном в водоемах).
Подавляющее большинство микроорганизмов воздуха — сапрофиты: Ь.
subtilis, b. mesentericus vulgatus, b. megatherium, b. mycoides, b. prodigio-
sum, sarcina lutea, sarcina rosea, torula alba, torula lutea, penicillium, asper-
gillus, mucor и др. Однако в воздухе помещений неоднократно были нахо-
димы и патогенные микроорганизмы: b. tuberculosis, b. dyphtcriae, staphylo-
coccus pyogenes, streptococcus pyogenes, споры b. anthracis, b. tetani и др.
Микроорганизмы не встречаются в воздухе в свободном состоянии: они
оказываются обыкновенно фиксированными в капельках жидкости или на
пылевых частицах.
Некоторые авторы (С. С. Речменский)1 различают в бактериальном аэро-
золе три фазы: 1) крупноядерную, состоящую из крупных капель
диаметром от 0,1 до 1 мм, такие капли очень быстро оседают из воздуха,
дистанция рассеивания их до 1 м; 2) м е л к о я д е р н у ю, состоящую из
мелких капель диаметром менее 0,1 мм; такие капли остаются в воздухе до
двух суток, рассеиваются на большие пространства и обнаруживают способ-
ность глубоко проникать при аспирации в органы дыхания; 3) ф а з у бак-
териальной пыли, образующейся при подсыхании крупных и мел-
ких капель.
Наиболее опасным в смысле аэрогенных инфекций является аэропланктон
в виде мелкокапельной фазы и фазы бактериальной пыли.
При изучении бактериального загрязнения воздуха необходимо кроме
общего подсчета бактерий иметь и качественную характеристику микрофлоры.
Миронов рекомендует для этой цели выяснить содержание в воздухе споро-
носных микробов, b. coli, анаэробов группы perfringens, b. proteus и стрепто-
кокков. Спороносные микробы, относясь к почвенной микрофлоре, указы-
вают на загрязнение воздуха почвенной пылью. Присутствие в большом ко-
личестве b. coli в воздухе указывает на загрязнение его пылью, содержащей
фекалии. Анаэробы группы perlringens также указывают на фекальное про-
исхождение воздушной пыли. Нахождение в большом количестве b. proteus
указывает на загрязнение воздуха продуктами распада гнилостного харак-
тера. Наконец, присутствие большого количества стрептококков свидетель-
ствует о загрязнении воздуха выделениями из верхних дыхательных путей.
Шафир и др. предлагают в качестве биологического показателя бакте-
риального загрязнения воздуха считать зеленеющий и гемолитический стреп-
тококки, которые являются банальными обитателями человеческой носо-
глотки. По мнению авторов, эти стрептококки могут служить таким же инди-
катором загрязнения воздуха, как титр бактерий группы кишечной палочки
служит индикатором загрязнения воды.
В некоторых случаях, например, в больничных палатах, при изучении
бактериального загрязнения воздуха производят исследования и на наличие
тех или иных патогенных микробов посевом на специальные среды, привив-
кой животным и т. п.
Определение общего числа бактерий в воздухе
Для определения общего числа бактерий в воздухе применяются различные
методы: осаждение, фильтрация, центрифугирование и электропреципитация.
Метод осаждения
Наиболее простой способ определения общего числа бактерий в воздухе
заключается в том, что чашки Петри с агаром или желатиной размещают
в разных пунктах помещения и оставляют открытыми в течение 5—10 минут
1 Вопросы санитарной бактериологии, изд. Акад. мед. наук, стр. 93, 1948.
285
затем чашки закрывают и выдерживают в термостате в течение 5 дней при
22°С или двое суток при 37°C, после чего подсчитывают число выросших
колоний, применяя в случае надобности счетную камеру.
Описанный метод очень прост; его недочет заключается в том, что он не
дает представления о числе бактерий в единице объема воздуха, ввиду чего
может служить лишь для ориентировочного исследования воздуха.
Омел янский считает, что на площадь в 100 см2 в течение 5 минут оседает
столько микробов, сколько их содержится в 10 л (0,01 м3) воздуха. Для ра-
счета определяют поверхность чашки и перечисляют найденное количество
бактерий на единицу объема воздуха.
Пример. На чашке Петри, которая была подвергнута экспозиции в течение 5 минут,
выросло 125 колоний. Радиус чашки 5 см. Если принять расчетные данные Омелянского,
то в 1 м3 воздуха число бактерий окажется равным
125.100.100 125.100-100
= 1о 910.
кг2---------------3,143-25
Метод фильтрации
Метод фильтрации заключается в том, что некоторый объем воздуха про-
сасывают через плотный фильтр или жидкую среду и затем производят посев
фильтрующей массы на питательную среду. По числу выросших колоний
определяют количество задержанных из воздуха бактерий; результат пере-
числяют на единицу объема воздуха.
Из многочисленных методов фильтрации опишем методы Миронова, Ми-
лявской, Микеля, Дьяконова и фильтр Штрауса с жидкой средой.
Фильтр Миронова готовится из фильтровальной бумаги. Фильтр
в форме конуса вставляется в алонж диаметром в 3 см. Края фильтра заги-
бают за край алонжа и зажимают резиновым кольцом. Воздух просасывают
со скоростью 500—700 л в час. После просасывания воздуха фильтр помещают
в стерильную банку со стеклянными бусами, прибавляют сюда 50 мл стериль-
ной воды и взбалтывают. Полученная суспензия представляет собой материал
для определения бактерий посевом на питательную среду.
Фильтр Милявской. Автор применяет мембранные нитроцелюлозные
фильтры № 3 (подробно о мембранных фильтрах см. далее: «Полевой метод
определения фекального загрязнения воды», стр. 387).
Мембранный фильтр для стерилизации предварительно кипятится в воде,
высушивается и помещается в прибор Зейтца зеркальной поверхностью к сетке.
Воздух просасывается через фильтр со скоростью 5 л в минуту в продол-
жении 10 минут, т. е. всего 50 л.
Для учета скорости просасывания воздуха между прибором Зейтца и
насосом включают реометр.
Для выяснения вопроса, имеет ли место во время эксперимента проскок
бактерий, в шейку прибора Зейтца помещают ватную пробку. Прибор Зейтца
до опыта стерилизуется. После опыта ватная пробка засевается в бульон.
Если проскока бактерий не было, то роста не будет.
После фильтрации мембранный фильтр укладывается зеркальной стороной
на 1°/о агаР в чашки Петри. Агар до посева фильтра нужно настолько
остудить, что почти не образуется конденсата отделяющимися парами влаги.
Чашки помещают в термостат на 2 суток.
Мембранные фильтры по данным автора обеспечивают развитие различ-
ных сапрофитных видов микроорганизмов из воздуха, способных пропзро-
стать на мясо-пептонной среде, в том числе спороносных и пигментообра-
зующих.
Если фильтры засевать в специальные среды соответственно да!шму
виду микробов, то можно обеспечить развитие и пашенных микрсц 1аьиз-
мов, отфильтрованных из воздуха.
286
/4
Рис. 217. Фильтр с порошкообразным
материалом.
А и В — ватные пробки.
Рис. 218. Фильтр
Дьяконова.
Таким образом, мембранные фильтры дают возможность получить не только
количественную, но и качественную характеристику микрофлоры воздуха.
Так как на отдельных местах фильтра могут оседать группы бактерий,
дающие при выращивании единичные колонии, то число выросших колоний
далеко не всегда соответствует числу осевших микроорганизмов.
Вот почему для полноценного учета автор рекомендует производить отмы-
вание фильтра 15 мл физиологического раствора. Отмытый фильтр нужно
засевать отдельно, а отмытую жид-
кость фильтровать через несколько
мембран, которые также задевают.
Результаты подсчетов выросших
колоний на всех фильтрах сумми-
руются.
Фильтр Микеля с порош-
кообразной фильтрующей
средой отличается тем, что здесь в качестве фильтрующей массы применяется
пор. шкообразное вещество (сульфат натрия, сахарит, п.). Сульфат натрия вы-
сушивают при 200°С, размельчают в ступке и просеивают через сито. Затем
соль всыпают в стеклянную трубочку с перехватом, поместив между солью
и перехватом кусочек ваты (рис. 217). Трубку с обоих концов закрывают
ватными пробками и стерилизуют сухим жаром.
При исследовании воздуха ватную пробку у конца А извлекают, другую
же, у конца В, оставляют. Пропускают 100 л воздуха. После этого растворяют
соль в 50—100 мл стерильной водопроводной воды, засевают в расплавлен-
ный агар (42°С) или желатину (37°С) в чашках Петри, которые выдерживают
в термостате при 22°С. Через 5 дней подсчи-
тывают число колоний.
Ватная пробка у конца В может служить
в качестве контроля. Ее вводят в пробирку
с расплавленной желатиной, взбалтывают и
выливают желатину в чашку Петри. Если
желатина остается стерильной, либо выра-
стают единичные колонии, то опыт считается
удачным.
Фильтр Дьяконова (рис. 218)
состоит из цилиндра, в который насыпают
стеклянные бусы и наливают 30—50 мл воды.
Цилиндр закрывается пробкой, через которую
проходят две трубки: приводная, доходящая
почти до дна, и отводная, заканчивающаяся
под пробкою. Наружные отверстия трубок
заткнуты ватными пробками. Перед употреблением прибор
стерилизуется. Техника применения та же, что и для прибора
Миронова.
Фильтр Штрауса с жидкой средой (рис. 219)
представляет собою цилиндр а, суженный книзу и имею-
щий боковой отросток Ь, В цилиндре вставляется пришли-
фованная пипетка с.
II
Ь-
Рис. 219.
Фильтр
Штрауса
с жидкой
средой.
а — цилиндр,
суженный
книзу. Ь — бо-
ковой отро-
сток. с — при-
шлифованная
пипетка.
Для опыта наливают в цилиндр стерильный бульон, физиологический
раствор или расплавленную желатину. В последнем случае цилиндр во время
опыта погружают в стакан с теплой водой (30—35°С). Для просасывания
воздуха трубка b присоединяется к насосу.
Если в качестве фильтрующего материала взят физиологический раствор
или бульон, то из определенного объема делают посев на чашки Петри с ага-
ром. Если же была взята желатина, то по окончании опыта она распределяется
равномерно по стенкам цилиндра и в таком виде цилиндр помещается в тер-
мостат.
287
Метод центрифугирования
Принцип метода основан на том, что испытуемый воздух, поступая в быст-
ро вращающийся стеклянный цилиндр, вследствие трения о внутреннюю
поверхность его, сам получает вращательное движение. Под действием воз-
никающей центробежной силы взвешенные в воздухе частицы, в том числе
Рис. 220. Прибор Шафира для микробиологического исследования воздуха.
/ — влектродвигатель, 2 — аммортпзатор, 3 — металлический стакан. 4 — стеклянный цилиндр,
$—съемный металлический кожух, 6—металлическая воздухопроводящая трубка, 7—гайка, 8—
упорное кольцо, 9 — уплотняющее кольцо, 10 — опорное кольцо кожуха, 11 — винты, укрепляющие
шкалу микроманометра, 12 — установочные винты микроманометра. 13 — уровни, 14 — кольцевой
шаблон с квадратными окошечками для счета бактерий, 15 — микроманометр, 16 — задвижки для регу-
лировки поступления воздуха.
микроорганизмы, отбрасываются на внутреннюю поверхность цилиндра, по-
крытую липкой питательной средой, и здесь фиксируются. Цилиндр вместе
с осевшими бактериями выдерживается в термостате. Выросшее колонии
подвергаются подсчету и, если необходимо, дифзренциальному испытанию.
Прибор для микробиологического исследования воздуха по центрифуж-
ному методу, сконструированный проф. Шафи р А. И., изображен на рис.
220. Он состоит из вертикально установленного электродвигателя (7),
поддерживаемого с трех сторон резиновыми аммортизаторами (2). К оси дви-
283
гателя прикреплен металлический стакан (5). В стакан помещается сте-
рильный стеклянный цилиндр (4). Под основанием металлического стакана
находится улитка центробежного вентилятора. Над металлическим стаканом
с цилиндром помещается съемный металлический кожух (5).
К прибору прилагается металлическая воздухопроводящая трубка 1 (6),
суженная у верхушки и расширенная у основания. Через расширенный конец
на трубку надеваются гайка (7), упорное кольцо (8) и уплотняющее кольцо
(9). Трубка с надетыми перечисленными деталями вставляется в опорное
кольцо кожуха (10) и затягивается гайкой. У верхнего конца осевой воздухо-
проводящей трубки имеется штуцер, который при помощи резиновой трубки
сообщается с микроманометром (75).
На корпусе микроманометра при помощи винтов (77) укрепляется шкала,
соответствующая номеру трубки, применяемой в данном опыте. На шкале
указан расход воздуха в метрах в минуту. Установка микроманометра в гори-
зонтальное положение осуществляется путем вращения установочных винтов
(72). Проверка горизонтального положения микроманометра производится при
помощи двух уровней (73). В качестве индикаторной жидкости для микро-
манометра применяется керосин, окрашенный алканной в желтый цвет. Инди-
каторная жидкость наливается в колбочку микроманометра в таком количе-
стве, чтобы мениск жидкости в трубке манометра совпадал с нулевой чертой
шкалы. Регулировка количества вводимого в цилиндр воздуха производится
с помощью приспособлений (16) типа задвижек, имеющихся на крышке кожуха.
Чем больше открыты задвижки, тем меньше засасывается воздуха через
о:.вую трубку, и наоборот. Прибор допускает регулирование просасывания
воздуха в пределах от 12 до 40 л в минуту.
Тарировка микроманометра может быть осуществлена при помои и рео-
метра, присоединенного к наружному отверстию осевой воздухопроводящей
трубки.
Проведение опыта при помощи описанного прибора производится следую-
щим образом. Кожух вместе с укрепленной в нем воздухопроводящей трубкой
снимается и в металлический стакан вставляется стерильный стеклянный
цилиндо. В цилиндр вводится 20 — 25 мл предварительно расплавленной и
несколько охлажденной питательной среды. Включается мотор, пссле чего
кожух устанавливается на место. Жидкая питательная среда в силу вращения
цилиндра поднимается по его стенке, распределяясь слоем толщиной около
1,5 мм.
Пр должительность работы мотора в течение одного опыта равна в среднем
10 минутам. Бактерии из всасываемого в прибор воздуха, как было указано,
отбрасываются на липкую поверхность питательной среды и здесь фиксиру-
ются.
По окончании опыта стеклянный цилиндр вынимается из прибора и поме-
щается в термостат дном кверху, где подсушивается. Затем цилиндр закры-
вается стерильной ватной пробкой и помещается в шкаф для выращивания
колоний. Для количественного учета микробов воздуха в качестве питатель-
ной среды применяется мясопептонный агар. При качественном анализе ми-
крофлоры воздуха применяются соответствующие среды: для обнаружения
зеленеющего и гемолитического стрептококков — кровяной агар, для b. coli—
среда Эндо и т. п. Отдельные колонии могут быть извлечены петлей из цилин-
дра и высеяны на соответствующую среду.
Подсчет выросших в цилиндре колоний производится полностью, если
их немного (200—300). При обильном росте пользуются для подсчета при-
ложенным к прибору кольцевым шаблоном (14), в котором имеются квадрат-
ные окошечки. Подсчитывают число колоний в 15 — 20 квадратах и затсл!
находят среднее содержание числа колоний в одном квадрате.
1 Для особо точных работ прилагается еще стеклянная трубка, применяемая после
стсрилизац и.
19 А. И. Бурштейн 289
Зная размеры поверхности квадрата шаблона и внутренней поверхности
цилиндра и учтя пропущенный через прибор объем воздуха, можно опреде-
лить число микробов в 1 м3.
Пример. Через прибор пропущено 250 л воздуха. Среднее число колоний в одном квад-
рате шаблона равно 28. Поверхность квадрата шаблона равна 4 см2, а внутренняя поверх-
ность цилиндра — 195 см2 (высота цилиндра 170 мм и диаметр 50 мм). Число микробов
в 1 м3 испытуемого воздуха равно:
28.195-1000 г_
------------- = 5460.
4 • 250
Метод электронрецшштации
Прибор для определения числа микробов воздуха по методу электропре-
ципитации построен по тому же принципу, что и прибор, описанный на стр. 200.
Преципитация бактерий из воздуха происходит на поверхности среды,
которая в чашках непосредственно помещается на электродах.
Прибор улавливает все фазы бактериального аэропланктона. Он пока не
нашел распространения в лабораториях.
БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПЫЛИ, ОСЕВШЕЙ ИЗ ВОЗДУХА
Для бактериологического исследования пыли, осевшей из воздуха, неко-
торое количество ее (0,5—1 г) собирают сухим ватным стерильным тампоном
в предварительно взвешенную стерильную бюксу. Собранную пыль взвеши-
вают, после чего в бюксу вводят 50—100 мл стерильной воды и тщательно все
взбалтывают. Определенное количество взвеси засевают на агар или же-
латину в чашках Петри, которые затем выдерживаются в термостате при 22
или 37°С. Результат подсчета перечисляют на 1 г пыли.
Если требуется исследовать пыль на наличие патогенных бактерий, то
производят соответствующие испытания (посевы на специальные среды, при-
вивки животным, серологические реакции и т. п.).
ЛИТЕРАТУРА
Общие курсы, руководства, труды и сборники
1. Бабаянц Р. А., Загрязнение городского воздуха, изд. Акад. мед. наук СССР, 1948.
2. Гродзовский М. К. Анализ воздуха в промышленных предприятиях, ГСЭИ,1931.
3. Игнатов Н. К., Практическое руководство по методике санитарно-гигиенических
исследований, Биомедгиз, 1935.
4. Каплун С. И. (ред.), Методический практикум по гигиене труда, Госмедиздат, 1933.
5. Койранский Б. Б. (ред)., Справочник для санитарно-промышленных врачей, 1940.
6. Левицкий В. А. (ред.), Гигиена труда, Биомедгиз 1936.
7. Лейтис, Марцинковский и Хоцянов, Учебник по промышленной гигиене и профес-
сиональным заболеваниям, Медгиз, 1938.
8. Летавет А. А. (ред.), Курс гигиены труда, Медгиз, 1946.
9. Леман К., Краткий учебник рабочей и профессиональной гигиены, Госиздат, 1923.
Ю. Марзеев А. Н., Сысин А. И. и Яковенко В. А., Основы коммунальной гигиены, т. II,
1938.
11. Методы исследования загрязнений атмосферного воздуха, изд. Всесоюзного инсти-
тута коммунальной санитарии и гигиены, 1940.
12. Смоленский И. О., Простейшие способы исследования и оценки доброкачественности
съестных припасов, напитков, воздуха, воды и пр., 1909.
13. Сборник трудов Всесоюзного института коммунальной гигиены и санитарии, т. III,
1939.
14. Сборник работ по санитарно-промышленной химии института гигиены труда Лспип-
гр i некого здравотдела, 1940.
15. Сборник работ и материалов по санитарно-химическим методикам Горьковского
института гигиены труда и профзаболеваний, 1941.
16. Сборник работ Санитарной и технической инспекции на Украине, 1923—1940.
17. Труды и материалы Московского института охраны труда, т. т. IV и V, 1930—1931.
18. Труды и материалы Московского санитарного института им. Эрисмана Ф. Ф.
19. Труды Ленинградского института организации экономики и охраны труда, т. VII,
1934.
2Н
20. Труды научно-исследовательской секции охраны труда Ленинградского ГОТ,
т. I, 1927.
21. Труды Ленинградского института гигиены труда и техники безопасности, тт. I—III,
1927—1930.
22. Труды и материалы Украинского института патологии и гигиены труда, вып. 5—
10, 1926—1930.
23. Хлопин Г. В., Основы гигиены, т. 1, вып. I, изд. НКЗ, 1921.
24. Он же, Курс общей гигиены, Госиздат, 1930.
Метеорологический фактор
1. Бенуа К. М., Метеорология, Военмориздат, 1941.
2. Берг Л. С., Основы климатологии, Учпедгиз, 1938.
3. Бернштейн Н. Р. и Брюкман В., Введение в метеорологию, ОНТИ, 1935.
4. Брент Д., Физическая и динамическая метеорология, Гидрометиздат, 1939.
5. Вегенер Д., Термодинамика атмосферы, ОНТИ, 1935.
6. Гемфрис В., Физика воздуха, ОНТИ, 1936.
7. Извеков Б. И. и Кочин А, Н., Динамическая метеорология, Гидрометиздат, чч. I
и II, 1935—1937.
8. Кайгородов Д., Практическая метеорология, 1927.
9. Калитин Н. Н., Основы физики атмосферы в применении к медицине,Биомедгиз, 1935.
10. Он же, Актинометрия на курортах, Биомедгиз, 1937.
11. Кедролеванский В. Н., Метеорологические приборы, Гидрометиздат, 1937.
12. Клоссовский А. В., Основы метеорологии, 1910.
13. Колобков Н. В., Метеорологические приборы, 1932.
14. Кошмидер Г., Динамическая метеорология, ГОНТИ, 1938.
15. Лондис А. П. и Ханявский В. Д., Начальная метеорология, Гидрометиздат, 1939.
16. Молчанов П. Д., Аэрология, Гидрометиздат, 1938.
17. Оболенский В. Н., Основы метеорологии, Сельхозгиз, 1935.
18. Он же, Метеорология, чч. 1 и II, Гидрометиздат, 1938—1939.
19. Основы курортологии, ч. I, Госмедиздат, 1932.
20. Ремизов Н. Д., Учебник медицинской метеорологии и климатологии, Биомедгиз,
1934.
21. Шипчинский Л. В., Основы метеорологии и климатологии, Сельхозгиз, 1933.
22. Шталъ, Справочник метеоролога, Воениздат, 1939.
Электрическое состояние атмосферы
1. Бендорф Г., Атмосферное электричество, 1934.
2. Гесс В., Ионнизация атмосферы и ее причины, 1930.
3. Дессауер Ф., Ионизированный воздух и его физиологическое действие 1932.
4. Кауфман С., Неклюдов В., Божевольнов Д., Униполярно заряженный воздух по
методу Дессауэра и его биологическое действие, 1935.
5. Корф С., Счетчики электронов и ядерных частиц, 1947.
6. Скворцов Н. П., Основы гигиологии и гигиены, М., 1900.
7. Яицкий Я., Ионизированный воздух и его физиологическое действие, 1932.
Аэрозоли
1. Бурштейн Д. И., Методы исследования аэрозолей, Госмедиздат УССР, 1934.
2. Гиббс В., Аэрозоли, Химтехиздат, 1929.
3. Джиббс В., Пыль и ее опасность в промышленности, Химтехиздат, 1930.
4. Мельдау Р., Пыль в производстве и способы ее удаления, Гостехиздат, 1931.
5. Рязанов В., Методика изучения пылевого фактора в производстве, 1934.
6. Уайтлоу-Грей и Паттерсон, Дым. Исследование в области аэродисперсных систем,
ГХТИ., 1934.
7. Углов В. Д., Борьба с пылью, дымом и газами населенных пунктов, Медгиз, 1934.
Г а з ы и пары
1. Берлъ-Лунге, Химико-технические методы исследования, т. II, ч. 1, вып. 1,ОНТИ,
1936.
2. Гендерсон и Хаггард, Вредные газы в промышленности, Гострудиздат, 1930.
3. Гуревич В., Определение вредных веществ в воздухе промышленных предприя-
тий, 1937.
4. Деннис Л. и Николс М., Газовый анализ, ОНТИ, 1934.
5. Житкова А. С., Методика определения вредных газов и паров в воздухе, Оборои-
гиз, 1939.
6. Комарь Н. П., Руководство по определению промышленных ядов в воздухе, ОНТИ,
1934.
7. Коренман И. М., Органические вещества в воздухе промышленных предприятий,
ОНТИ, 1935.
291
8. Коренман И, М., Анализ воздуха промышленных предприятий, вып. 1, 2, 3, 4 и 5
Госхимиздат, 1947—1949.
9. Лидов А., Анализ газов, 1928.
10. Лазарев Н., Бензин как промышленный яд, 1931.
11. Лазарев Н. и Астраханцев П., Химические вредные вещества в промышленности,
чч. I и II, ОНТИ, 1933—1935.
12. Памфилов А. В. (ред.), Промышленно-санитарная химия, 1931.
13. Сабаличка Ф., Химическое открытие ядов, 1926.
14. Соколов В., Методы исследования природных газов, ОНТИ, 1932.
15. Степанов А. В., Судебная химия, Медгиз, 1947.
16. Тредвел Ф. и Гол В., Курс аналитической химии, т. П ч. 2, ОНТИ, 1935.
17. Флюри Ф. и Церник Ф.9 Вредные газы, ГОНТИ, 1938.
18. Францен Г. Газовый анализ, 1923.
19. Шуфтан П., Анализ газов в технике, 1933.
Аэропланктон
1. Батенин В. А., Курс общей эпидемиологии, Биомедгиз, 1936.
2. Вопросы санитарной бактериологии, под ред. Сысина А. Н. и Мац Д. И., изд. Амая,
наук СССР, 1948.
3. Лазарев Н. В., Общие основы промышленной токсикологии, М. Л., 1938.
4. Миллер А. А., Санитарная бактериология, Госмедиздат, 1933.
5. Минкевич И. Е., Курс санитарной бактериологии, 1940.
6. Правдин Н. С., Методика малой токсикологии промышленных ядов, М., 1947.
7. Смелянский В. Л., Практическое руководство по микробиологии, изд. Акад, наук
СССР, 1940.
8. Синай Г. Я. и Биргер О. Г, (ред.), Микробиологические методы исследования при
инфекционных заболеваниях, Медгиз, 1940.
9. Шафир А. И., Микробиологический метод гигиенического исследования воздуха,
Изд. Военно-мед. акад. 1945.
ГЛАВА ЧЕТВЕРТАЯ
ВОДА (ПИТЬЕВАЯ)
Всесторонняя санитарно-гигиеническая оценка питьевой водын водоисточ-
ника может быть дана на основании: а) подробного, систематически повторя-
емого в разные периоды года лабораторного анализа воды и б) детального
обследования источника водоснабжения.
Лабораторное исследование воды слагается из органолептического, фи-
зического, химического, бактериологического и биологического анализа ее.
Обследование водоисточника сводится к биологическому изучению
его, а также к геологическому, гидрологическому, гидрографическому,
топографическому, метеорологическому и общесанитарному изучению мест-
ных условий, характеризующих данный водоем.
СВОЙСТВА ДОБРОКАЧЕСТВЕННОЙ ПИТЬЕВОЙ ВОДЫ
Доброкачественная питьевая вода должна быть чистой, прозрачной и
бесцветной; не иметь неприятного вкуса и запаха; обладать освежающей
температурой (5—15°С), небольшой жесткостью; быть свободной от вредных
для организма соединений; не содержать паразиты и патогенные бактерии
(число других бактерий должно быть ограничено).
Некоторые гигиенисты и гигиенические комиссии установили для различ-
ных ингредиентов воды определенные количественные нормы. Так, например,
для почвенных вод России Эрисман Ф. Ф. в свое время предложил следующие
количественные нормы:
1. Плотного остатка после выпаривания . . . 500—G00 мг/л
2. Окиси кальция (СаО) и магния (MgO) сов-
местно.................................... 180—200 »
3. Окиси магния (MgO)..................... 40—50 »
4. Хлора (СГ) солевого.................... 20—30 »
5. Серной кислоты (SO3)....................... 80 »
6. Азотной кислоты (N2O5)................. 30—40 »
7. Азотистой кислоты (N2O3)................. следы
8. Аммиака.................................. следы
9. Окисляемость (в мг кислорода).......... 2—3 »
10. Общая жесткость (в градусах)........... 18—20 »
Современная гигиеническая наука считает, что при санитарной оценке
питьевой воды не следует пользоваться количественными нормами, уни-
версальными, так как разнообразные местные природные и санитарные
условия, определяющие режим водоисточника, могут в значительной мере
влиять на состав воды и в силу этого не позволяют применять шаблонные
требования к составу и качеству воды, о чем писал уже Ф. Ф. Эрисман.
Однако для питьевой воды, подвергшейся специальной обработке: очистке,
обеззараживанию, обезжелезиванию, умягчению и т. п., могут быть установ-
лены определенные требования в отношении некоторых количе-
ственных норм.
293
Так, для питьевой воды, подаваемой постоянными централизованнымя
хозяйственно-питьевыми водопроводами и техническими водопроводами, в
случае организованного использования их воды для питьевых и хозяйствен-
ных нужд, ГОСТ 2874-45 устанавливает следующие количественные нормы;
А. Общие для всех водопроводов
1. Запах и привкус при температуре 20° С........ не более 2 баллов
2. Общее число бактерий при посеве 1 мл неразбав-
ленной воды, определяемое числом колоний после
24-часового выращивания при 37° С ............... не более 100
3. Количество кишечных палочек в 1 л воды, опреде-
ляемое числом колоний на среде Эндо с примене-
нием концентрации бактерий на мембранных фильт-
рах ............................................. не более 3
4. Содержание свинца (РЬ)........................ не более 0,1 мг/л
5. » мышьяка (As).........................не более 0,05 »
6. » фтора (F)........................... не более 1,0 »
7. » меди (Си)........................... не более 3,0 »
8. » цинка (Zn).......................... не более 15, 0 »
9. Фсиолсодержащих соединений, считая на фенол . . не более 0,001 »
Примечание к п.п. 4—9. Вода не должна содержать следов других
ядовитых веществ (ртути, шестивалентного хрома, бария и др.), учитываемых
стандартными методами исследования.
Б. Для водопроводов, имеющих устройства для осветления, обезжелезивания
или умягчения воды
10. Мутность (по мутномеру Бейлиса):
в среднем за год ..............................
в отдельных определениях.......................
11. Цветность:
в среднем за год ..............................
в отдельных определениях ......................
12. Содержание остаточного активного хлора в наи-
более удаленных точках водоразбора (наружного
и внутреннего) ...................................
13. Содержание железа (Fe) и марганца (Мп):
в сумме .......................................
в том числе закисного железа...................
14. Общая жесткость................................
15. Активная реакция (pH) в пределах...............
16. Вода при хранении ее в стеклянном сосуде в ко-
личестве одного литра в течение 24 часов не должна
давать отлежки, т. е. мути на дне сосуда, замет-
ной при легком взбалтывании.
не более 1 мг/л
не более 2 »
не более 15 градусов
не более 35 градусов
не менее 0,1 мг/л
не более 0,3 *
не более 0,2 »
не более 40 градусов 1
6,5—9,5
Примечание к п.п. 10—16: нормы мутности, цветности, а также
предельное содержание остаточного активного хлора, железа и марганца, ой-
щей жесткости и активной реакции для водопроводов, подающих воду без
ее обработки, устанавливаются Главной ГСП СССР особо для каждого отдель-
ного случая.
Весьма важно отметить, что согласно последнему ГОСТ 2874-45 (раз-
работанному А. Н. Сысиным, С. Н. Черкинским и В. Т. Турчаниновым) «под
качеством воды, подаваемой потребителям, подразумевается совокупность ее
свойств, в точках разбора воды: в наружных водоразборах и в кра-
нах внутренних домовых водопроводов».
Согласно ранее изданному ГОСТ 2761-44, определенным количественным
нормам должна удовлетворять вода источников, предназначаемых для
централизованного хозяйственно-питьевого водоснабжения, а именно: плот-
ный остаток такой воды не должен превышать 1000 мг/л, общая жесткость
не должна превышать 40°; количество кишечных палочек в 1 л не должно
превышать 1000 для источников, намечаемых к использованию при условии
одного только хлорирования, и 10000 для источноков, намечаемых к исполь-
1 1° жесткости соответствует содержанию в воде солей жесткости в количестве, экви-
валентном 10 мг/л. СаО.
294
зованию при условии полной очистки и хлорирования воды. Оценка вкуса
и запаха воды не должна превышать 2 баллов в среднем и 3 баллов для
отдельных анализов.
Предельное содержание солей тяжелых металлов и других вредных ве-
ществ в воде источников устанавливается в каждом отдельном случае особо
органами ГСИ СССР.
В случае использования для водоснабжения источника без очистки или
без обеззараживания воды (артезианские колодцы) качество последней дол-
жно удовлетворять требованиям стандарта на питьевую воду (см. выше).
ОТБОР ПРОБ БОДЫ ДЛЯ АНАЛИЗА
При отборе проб воды нужно твердо придерживаться основного положе-
ния, согласно которому пробы отбираются при средних условиях,
характеризующих режим данного источ-
ника; для этой цели необходимо избегать отбирать
пробы при случайном загрязнении или взмучивании, за-
стаивании воды и т. п., за исключением тех случаев,
когда преследуются специальные задачи.
Вода должна отбираться из того места данного источ-
ника, где производится забор воды для пользования.
Воду набирают в чистые бутылки, предварительно
промытые два-три раза испытуемой же водой. Хорошо
для этой цели пользоваться бутылями из устойчивого
прозрачного стекла с притертыми пробками. При отсут-
ствии бутылей с притертыми пробками можно поль-
зоваться бутылями с корковыми пробками, применив
прокладку из
Рис. 222. Бато-
метр Виногра-
дова.
пергаментной бумаги. Закупорив склянку,
срезают пробку в уровень с шейкой бу-
тыли и заливают сургучом или парафином.
При исследовании воды открытых водо-
емов пробу берут не с поверхности, а из
глубины соответственно уровню забора
воды. Для этой цели пользуются еспе-
циальными приборами, носящими название
батометров.
На рис. 221 изоображен батометр по
Хлопину. Он состоит из медного гнезда
А с тяжелым основанием В из свинца. В
гнездо вставляется бутыль С. От гнезда
Рис. 221. Батометр
Хлопина.
А — меткое гнездо,
В — основание (из
свинца), С — бутыль.
D — бечевка для опу-
скания и извлечения
батометра, Е — бе-
чевка для извлечения
пробки из бутыли,
F — пружина, уста-
навливающая пробку
в бутыль.
отходит медная зацепка, к которой прикреплена бечевка D
для опускания и извлечения батометра. На бечевке делают
ряд узлов на определенном расстоянии один от другого, что
даст возможность легко определить глубину, на которую
погружается прибор в воду.
К пробке бутыли прикреплена бечевка Е для извлечения
пробки, когда батометр опущен на нужную глубину. Пру-
жина F возвращает пробку на место, если ослабить натяже-
ние бечевки Е.
На рис. 222 изображен батометр Виноградова.
При взятии пробы из водопроводных кранов, из колодцев с насосами обжи-
гают кран пламенем горелки, воду выпускают или отсасывают в продолжении
10—15 минут и тогда отбирают пробу. Фламбирование крана производится
с целью уничтожения случайной бактериальной флоры, спуск или отсос во-
ды — с целью устранения застоявшихся порций воды.
Бутыли с водой, предназначенные для пересылки на большое расстояние
в теплое время года, следует сохранять на льду с целью задержать развитие
295
микрофлоры и возможное изменение состава воды; зимою во избежание за-
мерзания пробу накрывают каучуковой подушкой с теплой водой.
Транспортируемые на дальние расстояния в теплое время года пробы
иногда подвергают консервированию. В качестве консервирующих веществ
на 1 л воды прибавляют 2 мл 25% серной
Рис. 223. Пробирки с оттянутыми
кончиками для отбора небольших
порций воды на бактериологиче-
ское исследование.
кислоты, если проба предназначена для опре-
деления окисляемости и аммиака; к пробам,
предназначенным для других видов исследо-
вания (определение плотного остатка, взве-
шенных веществ, потери при прокал! вании,
азотной и азотистой кислот), на каждый литр
воды прибавляют 2 мл хлороформа.
Одновременно с консервированной водой
следует брать и пробу натуральной воды.
В зависимости от вида анализа требуется
следующие количества воды: для общего хи-
мического анализа — 4 л; для определения
ВПК (биохимического потребления кислоро-
да)— 1,5 л; для определения растворенного
кислорода — две пробы по 0,25 л, причем для связывания
кислорода прибавляют на месте реактивы: хлористый марга-
нец и едкую щелочь (см. далее).
Пробы для бактериологического исследования берут в сте-
рильную стеклянную посуду (склянки с притертыми проб-
ками, пробирки с ватными пробками и т. п.), придерживаясь
при этом предосторожностей, требуемых бактериологической
техникой.
Для этой же цели в случае отбора небольших количеств
воды пользуются стерильными пробирками (рис. 223) с от-
тянутыми кончиками; внутри этих пробирок воздух должен
быть разрежен. Такие пробирки опускают в воду с помощью
прибора Склаво-Чаплевского (рис. 224) и потягиванием бе-
чевки отламывают кончик. Заполнившуюся водой пробирку
извлекают из воды и кончик запаивают.
Количество воды для бактериологического анализа должно
быть не менее 500 мл; только для явно загрязненной воды
можно отбирать меньшие количества. Для исследования воды
на присутствие патогенных бактерий отбирают не менее 3 л.
Для правильной оценки режима водоисточника
отбор проб и анализ их производится повторно и в разные
периоды года.
Из открытых водоемов пробы отбирают в весеннее по-
ловодье, летом в низкую воду, при осеннем подъеме и в
средине зимы (январь—февраль).
Из озер и больших водохранилищ берут дополнительно
пробы после длительного волнения, а из устьев рек, впада-
ющих в море, во время нагона воды с моря.
Для выяснения влияния атмосферных осадков пробы
берут до и после продолжительного выпадения осадков.
Из рытых колодцев и родников пробы отбирают два раза
в день: один раз — до разбора воды (рано утром), другой—
к концу расходования (конец дня).
Из подземных источников (скважин, колодцев) при отсут-
ствии постоянного излива воды пробы отбираются после две-
Рис. 224. При-
бор Склаво-
Чапле! ского
для взятия ма-
лых количеств
волы с целью
бактериологи-
ческого иссле-
дования.
надцатичасовой откачки воды и не ранее, чем будет получено одинаковое
содержание хлоридов и железа в трех пробах, взятых во время откачки
с промежутком не менее одного часа.
293
Химический анализ воды должен быть произведен не позднее чем через
72 часа после отбора проб незагрязненной воды, не позднее чем через 48 ча-
сов при отборе проб достаточно чистой воды и не позднее 12 часов при от-
боре проб загрязненной воды. Что касается бактериологического исследо-
вания, то весьма желательно производство посева взятых образцов на мес-
те. При невозможности выполнения этого условия посевы должны быть про-
изведены не позднее 1—3 часов после отбора проб.
В тех случаях, когда вода хлорирована и не имеется возможности про-
извести пссев воды на месте отбора пробы, пробу воды нужно дехлори-
ровать. Для этой цели в скля! ку, предназначенную для забора воды, вводят
по 2 мл 1,5% тиосульфата натрия Na2S2O3 . 5НО на каждые 500 мл
воды и стерилизуют, как обычно в автоклаве. Указанное количество тио-
сульфата натрия способно связать активный хлор в количестве до 2 мг/л
в объеме пробы в 500 мл. Можно, соблюдая стерильные условия, внести
указанное количество стерильного тиосульфата в стерильную посуду. В склян-
ку, подготовленную таким образом, отбирают пробу из данного источника.
В сопроводительном акте к отобранной пробе необходимо указать:
1. Наименование источника;
2. Место и точки взятия пробы (для открытых водоемов — расстояние
от берега и глубину, с которой взята проба, считая ст поверхности и со дна);
3. Дату взятия пробы (год, месяц, число и час);
4. Цель взятия пробы;
5. По чьему заданию и кем взята проба;
б. Способ взятия пробы;
7. Цвет, вкус, запах, прозрачность, мутность, осадок, опалесценцию,
температуру воды в момент взятия пробы;
8. Метеорологические данные в момент взятия пробы и за последние 10
дней перед взятием пробы; сюда относятся температура воздуха, движение
его (ветер) и атмосферные осадки;
9. Способ консервирования пробы в том случае, если оно было применено;
10. Подпись лица, производившего отбор пробы.
На склянку, в которой находится отобранная проба, наклеивается эти-
кетка с соответствующей надписью.
СХЕМА ПОЛНОГО САНИТАРНОГО АНАЛИЗА ВОДЫ
Физико-химические исследования1
Температура (на месте)
Запах
Вкус
Цветность
Прозрачность
Мутность и осадок
Реакция (pH)
Щелочность или кислотность
(титрирная)
Взвешенные вещества
Плотный остаток
Плотный остаток после прокаливания
Окисляемость
Жесткость: а) общая, б) карбонатная,
в) устранимая*, г) постоянная *
Кальций
Магний
Натрий*
Калин*
Хлориды
Сульфаты
Фосфаты*
Силикаты (кремнекислота) *
Азот аммонийный
» альбуминоидный
» нитритов
» нитратов
» органический
» общий
Алюминий*
Марганец*
Железо (общее, закисное* и окисное*)
Свинец,*, медь*, цинк* и олово*
Мышьяк*
Кислород, растворенный в воде*
Углекислота (свободная, гидрокарбонатная,
карбонатная, общая, агрессивная*)
Сероводород*
Остаточный хлор*
Фтор*
Число бактерий в 1 мл
Кили-титр и коли-пндеке
Бактериологические исследования
Патогенные бактерии*
Определения, отмеченные звездочкой, производятся по мере надобности.
297
СХЕМА САНИТАРНОГО АНАЛИЗА ВОДЫ Физико-химические исследования
Температура Цвет Запах Вкус Прозрачность Муть и осадок Реакция Жесткость: а) общая, б) карбонатная Железо Хлориды Азот нитратов » нитритов » аммонийный Окисляемость Бактериологии ескне исследокапля
Число бактерий в 1 мл Коли-титр
ОРГАНОЛЕПТИЧЕСКАЯ ОЦЕНКА ВОДЫ
Органолептическая оценка воды (запах, вкус и привкус) производится
с нефильтрованной водой по возможности у места отбора проб.
Запах
Различают запахи естественного и искусственного
происхождения. Первые обусловливаются естественными условиями: планкто-
ном, почвой, берегами и т. п., вторые возникают в силу загрязнения воды
промышленными сточными водами, обработки воды хлором с целью обез-
зараживания и т. п.
Для определения запаха испытуемую воду, доведенную до температуры
15—20°С, наливают в широкогорлую колбу емкостью 150—200 мл до 2/3 ее
объема; закрывают колбу часовым стеклом и встряхивают, производя враща-
тельные движения, затем открывают и испытывают обонянием, сильно втя-
гивая носом воздух из колбы.
Запахи естественного происхождения классифицируют по характеру: аро-
матический, болотный, гнилостный, древесный, землистый, плесневый, рыб-
ный, сероводородный, травянистый, неопределенный.
Запахи искусственные — фенольный, бензинный, хлорный и т. п. —
определяют так же, как и естественные. Запахи оценивают по пятибальной
системе следующим образом:
Балл Балл
Никакого запаха ... 0
Очень слабый ....... 1
Слабый ............. 2
Заметный............. 3
Отчетливый........... 4
Очень сильный........ 5
При оценке в 3 балла со стороны потребителя может быть дан неодобри-
тельный отзыв о воде, при оценке в 4 балла вода становится неприятной, при
оценке в 5 баллов вода непригодна для питья.
Запах хлорированной воды на месте хлорирования определяется через
30 минут после введения хлора.
Вкус п привкус
Вкус и привкус воды оцениваются также по пятибалльной системе.
Для определения вкуса и привкуса набирают в рот около столовой ложки
воды и держат несколько секунд.
Различают четыре вида вкуса: соленый, горький, сладкий, кислый.
Привкусы бывают различные: хлорный, рыбный, металлический и пр.
В зависимости от интенсивности вкуса и привкуса дают бальную оценку.
Привкус хлорированной воды на месте хлорирования определяют через
30 минут после введения хлора.
298
Вкус и привкус воды открытых водоемов, сомнительных в санитарном
отношении, определяется после кипячения и охлаждения до комнатной
температуры, что отмечается в протоколе анализа.
ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ВОДЫ
Физическое исследование воды сводится к определению ее температуры,
прозрачности, мутности и цветности. Указанные определения
производятся с нефильтрованной водой по возможности у места
отбора проб.
Недавно введенный ГОСТ 3351-45 уточняет эти определения.
Температура
Температура воды определяется либо в отобранной пробе, либо
непосредственно в самом водоеме.
В первом случае отбирают не менее литра воды в сосуд, тем-
пература которого доведена до температуры воды.
Во втором случае пользуются термометром, снабженным чер-
паком (рис. 225).
В таких приборах шарик термометра помещается в сосуд,
открытый сверху; когда прибор погружают в воду, сосуд наполня-
ется водой и термометр приобретает температуру зачерпнутой воды.
Рис. 225.
Термометр
с черпа-
ком.
и сравни-
Прозрачность
Прозрачность воды определяется качественно сравнением ее
с дестиллированной водой. Испытуемую воду наливают в стек-
лянный цилиндр, который помещают на хорошо освещенный
белый фон; также поступают и с дестиллированной водой
вают прозрачность испытуемой воды с прозрачностью дестиллированной
воды.
Степень прозрачности воды определяют словами: прозрачная, едва мутная,
мутная, опалесцирующая и т. п. Количественное определение прозрачности
производится по методу Снеллена либо «по кресту». По-
следний метод применяется при контроле очистных со-
оружений и при определении качества воды в водопровод-
ной сети.
Определение прозрачности по методу One члена
Прибор Снеллена (рис. 226) представляет собой сте-
клянный цилиндр с отъемным плоским дном, удержива-
емым через резиновую прокладку металлическими застеж-
ками. Цилиндр градуирован на сантиметры, начиная от
дна. Высота градуированной части равна 30 см, точность
отсчета — 0,5 см. Дно цилиндра находится на высоте 4 см
от края ножек, на которых цилиндр утвержден. У осно-
Рис. 226. Цилиндр вания цилиндра находится тубус, на который надевается
Снеллена. ’ резиновая трубка с зажимом.
Определение производят в хорошо освещенной ком-
нате не на прямом свету в отдалении 1 м от окна. Цилиндр наполняют
водой и помещают под цилиндр страницу шрифта Снеллена № 1 (см.
стр. 300). Высоту столба воды в цилиндре нужно отрегулировать так,
чтобы через этот столб еще возможно было чтение шрифта. По высоте
столба воды определяют ее прозрачность. Например, выражение: «про-
зрачность воды равна 15 см» обозначает, что наибольшая высота столба
воды, через который возможно еще чтение шрифта, равна 15 см.
200
Образец шрифта Снеллена № 1
для определения прозрачности воды.
Санитарно-гигиеническое исследование воды слагается из
физического, химического, бактериологического и биологи-
ческого анализа ее, а также из детального обследования
источника водоснабжения и из) чения местных условий, в
которых источник находится.
5381276409
Санитарно-гигиеническое исследование воды слагается из
физического, химического, бактериологического и биологи-
ческого анализа ее, а также из детального обследования
источника водоснабжения и изучения местных условий, в
которых источник находится.
5381276409
Санитарно-гигиеническое исследование воды слагается из
физического, химического, бактериологического и биологи-
ческого анализа ее, а также из детального обследования
источника водоснабжения и изучения местных условий, в
которых источник находится.
5381276409
Санитарно-гигиеническое исследование воды слагается из
физического, химического, бактериологического и биологи-
ческого анализа ее, а также из детального обследования
источника водоснабжения и изучения местных условий, в
которых источник находится.
5381 276409
Санитарно-гигиеническое исследование воды слагается из
физического, химического, бактериологического и биологи-
ческого анализа ее, а также из детального обследования
источника водоснабжения и изучения местных условий, в
которых источник находится.
5381276409
зоо
Определение прозрачности «по кресту»
Прибор (рис. 227) состоит из стеклянной трубы
диаметром в 3 см и высотой в 350 см, градуиро-
ванной на сантиметры. Нижний конец трубы за-
крыт резиновой пробкой, снабженной спускным
отверстием с зажимом. На пробке помещается
белый фарфоровый диск с черными расположен-
ными крестом линиями шириной в 1 мм и с че-
тырьмя черными точками диаметром в 1 мм по
одной в середине каждой четверти диска (см. де-
таль рис. 227).
Определение производится при искусственном
освещении с помощью лампы в 300 ватт, помещен-
ной у основания трубы в ящик с окошечком.
Глаз наблюдателя располагается на высоте около
5 см над верхним концом трубы. Труба напол-
няется испытуемой водой до полного исчезновения
креста. После выделения пузырьков воздуха воду
постепенно спускают до появления в поле зре-
ния отчетливо видимых точек. По высоте столба
оставшейся в трубе воды определяют ее прозрач-
ность.
Мутность
Определение мутности воды производится при
помощи прибора, носящего название мутномер
(рис. 228). Он представляет собой закрытый ящик
длиной 80 см и шириной 21 см; высота ящика у одно-
го конца 80 см, а у другого — 30 см. Крышка ящика
в высокой части горизонтальна на протяжении
20 см, а далее представляет собою покатую доску
на петлях. В горизонтальной части крыши ящика
имеются два круглых отверстия, отстоящих одно
от другого на 3 см. В отверстия вставляются стек-
лянные цилиндры с внутренним диаметром в 2 см
и высотой в 75 см. Цилиндры упираются в нижней
части ящика в края круглых отверстий деревянной
полки, расположенной на 8 см от дна и закрытой
спереди темносиним стеклом. Перед синим стеклом
у стенки низкой части ящика помещается электро-
лампа в 300 ватт. Свет от лампы освещает боко-
вые стенки цилиндров, а также их дно, проходя
через синее стекло. Стенки ящика изнутри около
1
Деталь 9
Рис. 227. Прибор для опре-
деления прозрачности «по
кресту».
1 — стеклянная трубка длиною
3500 мм. диаметром 30 мм, 2 —
лоска с делениями, к которой
крепится стеклянная трубка.
3 — трос от хомутика с грузом
4— хомутик с грузом для регу-
лирования спуска волы. 5 —
резиновая пробка с вырезами,
6 — белая фарфоровая пластин-
ка с крестом и четырьмя точ-
ками 7 — ящик с электрической
лампочкой в 300 w, 8— отводная
трубочка для спуска воды.
лампы обиты листовым железом и вы-
крашены в белый цвет.
Эталонный раствор приготовляют из
инфузорной земли или каолина, пред-
варительно просеянного через шелковое
сито (диаметр отверстий 0,1 мм). Раст-
вор должен содержать 100 мг взвеси
в 1 л дестиллированной воды.
Для приготовления эталона берут
навеску в 25—30 г инфузорной земли
или каолина и хорошо взбалтывают в
после чего оставляют жидкость на 24 часа.
Рис. 228. Мутномер.
1 — разрез, 2 — внешний вид.
3—4 л дестиллированной воды,
Через 24 часа отбирают сифоном, не взмучивая осадка, среднюю иеосвет-
ленную часть жидкости. К оставшейся части вновь приливают воду,
301
сильно взбалтывают и снова оставляют на 24 часа, после чего отбирают
опять среднюю неосветленную часть жидкости. Так повторяют до тех пор,
пока не удается извлечь по возможности полностью не оседающую в течение
24 часов муть. Собранную таким образом жидкость сливают вместе, тщательно
взбалтывают и оставляют в сосуде на 3 суток, после чего жидкость над
осадком сливают, а из оставшегося осадка приготовляют эталонный раствор.
Для этой цели переносят осадок в широкий сосуд, приливают понемногу
дестиллированную воду и помешивают, давая затем жидкости успокоиться.
Так поступают до тех пор, пока блестящая платиновая проволока диаметром
в 1 мм и длиной в 30 мм, погруженная горизонтально в постепенно разбавляе-
мый раствор, не начнет исчезать из зрения, находясь точно на глубине в 100 мм.
Платиновая проволока укрепляется перпендикулярно на нижнем конце де-
ревянного стержня длиною 1,22 м, на верхнем конце которого на расстоянии
1,2 м от платиновой проволоки имеется кольцо, через которое ведется наблю-
дение. Концентрация приготовленной таким образом суспензии прове-
ряется весовым методом путем фильтрации через беззольный фильтр с по-
следующим прокаливанием фильтра до постоянного веса. Если оказалось,
что суспензия получилась более концентрированной нежели 100 мг/л, то
ее разбавляют соответствующим количеством воды; если же она получилась
слабее 100 мг/л, то ее отстаивают в течение 3 суток и отбирают пипеткой
из верхних слоев, избыток воды.
Полученную суспензию консервируют сулемой из расчета 1 мл насыщен-
ного раствора HgCl2 на 1 л суспензии. В таком виде она сохраняет годность
в течение нескольких месяцев. Из полученной суспензии готовят более слабые
рабочие суспензии с концентрацией 0,05—0,08—0,1—0,2—0,4 и т. д. до 2 мг/л,
которые также консервируют сулемой. Эталоны с содержанием взвеси менее
0,2 мг/л ввиду их неустойчивости меняют каждую декаду.
Методика определения мутности воды весьма проста: в один
из цилиндров прибора наливают в уровень с краями испытуемую воду, в дру-
гой цилиндр—эталонный раствор, подбирая такой из них, который по мут-
ности окажется эквивалентным испытуемой воде. Как эталонный раствор,
так и испытуемую воду перед сравниванием тщательно взбалтывают. Сравне-
ние мутности начинают через 5—10 минут по заполнении цилиндров,
когда успевают выделиться пузырьки воздуха.
Мутность воды выражают в мг/л соответственно подобранному эталону.
Ниже приведена таблица 38 для пересчета прозрачности на мутностьи наоборот.
Для мутных вод прозрачностью 20 см и менее (мутность 45,5 мг/л и более)
дают характеристику выпадающего осадка и степени осветления воды при
спокойном ее стоянии. Для этого испытуемую воду наливают в мерный ци-
линдр высотою в 30см и оставляют в спокойном месте. Характеристика дает-
ся через один час для воды открытых водоемов и через 24 часа для воды из
подземных источников.
Осадок характеризуют: по количеству (незначительный, большой;
при очень большом указывают толщину в мм); по качеству (хлопье-
видный, илистый, песчаный); по цвету (серый, бурый, черный и т. п.).
Осветление характеризуется по степени интенсивности словами: осветле-
ние незаметное, слабое, сильное, вода прозрачна.
Цветность
Для качественного определения цветности воды поступают сле-
дующим образом. Берут два одинаковых цилиндра. В один наливают дестилли-
рованную воду, в другой — исследуемую воду. Высота каждого столба воды
должна быть не менее 40 см. Оба цилиндра помещают на белый фон и смотрят
сверху вниз, сравнивая окраску. Боковое освещение при этом лучше исклю-
чить. Если степень окраски изменяется под влиянием присутствия в испы-
туемой воде взвешенных веществ (глина, гидрат окиси железа и др.), то воду
302
до определения цветности следует профильтровать через бумажный фильтр
или отцентрифугировать. Цветность воды определяется словами: желтоватая,
желтая, бурая и т. п.
Для количественного определения цветности воды ее сравни-
вают со шкалой эталонных растворов и выражают окраску в условных гра-
дусах, соответственно интенсивности окраски эталона.
Табл и ц а 38
Таблица для перевода прозрачности па мутность_________
Проз- рачность см 1 Л &.О 5- S = S Проз- рачность ?м Мут- ность мг/л Проз- рачность см н £ «=: S х S Проз- рачность см Мут- ность мг/л Проз- рачность см Мут- ность мг/л
3,5 270 21 43,3 50 18,2 79 11,6 130 7,15
4 235 22 41,4 51 17,9 80 11,45 132 7,05
4,5 205 23 39,6 52 17,6 81 11,3 134 6,9
5 185 24 38,0 53 17,3 82 11,05 130 6,8
5,5 170 25 36,5 54 17,0 83 11,0 138 6,7
6 155 26 35,1 55 16,7 84 10,85 140 6,6
6,5 142 27 33,8 56 16,4 85 10,7 145 6,3
7 130 28 32,6 57 16,1 86 10,35 150 6,1
7,5 122 29 31,5 58 15,8 89 10,3 155 5,9
8 114 30 30,5 59 15,5 90 10,1 160 5,75
8,5 107 31 29,5 60 15,2 92 9,9 165 5,6
9 102 32 28,6 61 15 94 9,7 170 5,45
9,5 97 33 27,7 62 14,8 96 9,5 175 5,3
10 92 34 26,9 63 14,6 98 93 180 5,15
10,5 87 35 26,1 64 14,4 100 9,1 185 5,0
11 83 36 25,4 65 14,2 102 8,9 190 4,85
11,5 79 37 24,8 66 14,0 104 8,7 195 4,75
12 76 38 24,2 67 13,8 106 8,5 200 4,6
12,5 73 39 23,6 68 13,6 108 8,3 210 4,4
13 70 40 23,0 69 13,4 ПО 8,2 220 4,2
13,5 67,5 41 22,4 70 13,2 112 8,1 230 4,0
14 65,0 42 21,8 71 13,0 114 8,0 240 3,85
14,5 63,0 43 21,2 72 12,8 116 7,9 250 3,7
15 61,0 44 20,7 73 12,6 118 7,75 260 3,55
16 56,4 45 20,2 74 12,4 120 7,65 270 3,45
17 53,1 46 19,7 75 12,2 122 7,55 280 3,3
18 50,4 47 19,3 76 12,05 124 7,45 290 3,2
19 48,0 48 18,9 77 11,9 126 7,35 300 3,1
20 45,5 49 18,5 78 11,75 128 7,25 310 3,0
При* леняют j 1ва вида эталон! ов: платиново-кобальтовый и хромово -кобаль-
Приготовление платииово-кооальтовой шкалы. 1,245 г хл°Р0”ла™“аJ? калия (K2PtCle) и 1,01 г кристаллического хлористого кобальта (СоС1 Ьн2О)
растворяют в дестиллированной воде. Раствор переводят кол _ литровую колбу, приливают 100 мл концентрированной НС1 и'Доводят до литра дестиллированной водой. Полученный Раств°Р^вп^^о^Н°ВвН“еМ„ой
приготовления платиново-кобальтовои шкалы; он довольно стоек посуде может храниться до года, не меняя интенсивность окра к . Ц
такого раствора принимают условно равной 500 . K»nvT пяп Из основного раствора готовят шкалу следующим образом. беру р д мерных цилиндров из бесцветного стекла емкостью в 100 мл МЖШИ. ° “ линдры наливают основного раствора и дестиллированной воды соответс
303
но количествам указанным в таблице 39, получая таким образом эталоны
разной интенсивности. Эталоны, закрытые пробками, хранят в темном месте.
Через 2—3 месяца шкала должна быть возобновлена.
Таблица 39
Приготовление платиново-кобальтовой шкалы
Количество стандартного
раствора в мл............
Дестиллированная вода
в мл.....................
Градусы цветности . . .
О
100
0
1
99
5
98
10
3
97
15
96 95
20 25
94
30
35
8
92
40
10 12 14 16
90 88 86 84
50 60 70 80
4 5 6
7
При отсутствии химически чистого хлороплатината калия основной раствор
готовят следующим образом: 0,5 г платины растворяот в царской вэдке,
удаляют азотную кислоту повторным выпариванием досуха с прибавлени >м
избытка соляной кислоты. По удалении азотной кислоты растворяот полу-
ченный сухой остаток в вэде, прибавляют 1,01 г хлористого кобальта и дово-
дят раствор до литра водою.
Цветность такого эталона также равна 500°.
Нрпготэшэяиг хромово-коэальтово i шкалы. Готовят два раствора. Ра-
створ первый: 0,0375 г двухромовокислого калия (К2Сг.,О7) и 2 г сернокис-
лого кобальта (CoSO4) растворяют отдельно в дестиллированной вэде, затем
смешивают оба раствора, прибавляют 1 мл химически чистой серной кис-
лоты удельного веса 1,84 и доводят дестиллированной водой до литра. Получен-
ный раствор является основным и отвечает цветности в 500°. Раствор второй:
1 мл химически чистой серной кислоты удельного веса 1,84доводят дестиллиро-
ванной водой до 1 л. Из этих двух растворов готовят шкалу, беря количества,
указанные в таблице 40.
Таблица 40
П риготовление хромово-кобальтугой шкалы
Раствор № 1 в мл 0 1 1 2 3 4 5 6 8 10 12 14 16
Раствор № 2 в мл 100 99 98' 97 96 95 94 92 90 88 86 84
| Градусы цветности 1 1 0 5 10; 15 20 25 30 40 50 60 70 80
Полученная шкала должна храниться в темноте (цилиндры закрывают
пробкой). Через 2—3 месяца шкалу возобновляют.
Методика определения цветности. В цилиндр, одно-
типный с теми, в которых приготовлена шкала, наливают 100 мл испытуемой
воды и подбирают эталон, по окраске сходный с испытуемой водой. Цветность
воды выражается в градусах соответственно подобранному эталону.
Мутная вода (прозрачность ниже 20, мутность выше 45,5 мг/л) перед опре-
делением должна быть отцентрифугирована. Если испытуемая вэда имеет
цветность выше 80°, то до определения цветности ее разбавляют дестиллиро-
ванной водой. Цветность в этом случае получается умножением результата
определения на кратность разбавления.
Описанный метод дает возможность определить цветность от 1 до 50э с
точностью до 2°, от 51 до 100° — с точностью до 5°, от 101 до 250°—с точностью
до 10° и от 251 до 500°— с точностью до 20°.
Электропроводность
Электропроводность воды изменяется с изменением солевого состава ее;
оэтому по электропроводности воды можно сделать заключение об изменении
солевого состава, возникающем в связи с попаданием в водоисточник больших
количеств талой воды, сточных вод и т. п.
304
Определение электропроводности производится по методу, описанному
выше (стр. 101).
Были предложены также методы определения сухого остатка воды на осно-
вании ее электропроводности (Дорошевский и Дворжанчик и др.). Эти методы
не нашли, однако, практического применения в лабораторной практике.
ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ВОДЫ
Различают полный санитарно-химический анализ и санитарно-химиче-
ский анализ воды.
Полный санитарно-химический анализ воды про-
водится в тех случаях, когда имеется в виду дать подробную характеристику
водоисточника, намечаемого или уже применяемого для широкого водополь-
зования (река, озеро, водопровод). Такой анализ дает представление о всех
показателях и ингредиентах, могущих представить интерес для санитарной
оценки воды.
Сюда относится определение реакции, щелочности или кислотности, взве-
шенных веществ, плотного остатка, окисляемости, жесткости, кальция, маг-
ния, натрия, калия, хлоридов, азотсодержащих соединений, сульфатов,
фосфатов, кремнекислоты, железа, марганца, алюминия, свинца, меди, цинка,
мышьяка, фтора, фенолсодержащих веществ, кислорода, растворенного в
воде, углекислоты, сероводорода, БПК (биохимического потребления кисло-
рода), остаточного хлора и др.
Однако далеко не всегда полный санитарно-химический анализ охваты-
вает все перечисленные определения. Некоторые из них не проводятся, если
нет специальных указаний. Это может касаться, например, определения мар-
ганца, алюминия, свинца, меди, цинка, мышьяка, сероводорода, фенола и др.
Санитарно-химический анализ воды проводится в тех
случаях, когда требуется дать санитарную характеристику небольших водо-
источников, служащих для ограниченного водопользования (грунтовые ко-
лодцы). Такой анализ дает представление о тех данных, которые могут слу-
жить показателями чистоты или загрязнения воды, а также о жесткости воды и
содержании железа.
Сюда относится определение реакции, плотного остатка, жесткости, окис-
ляемости, хлоридов, азотсодержащих веществ, железа.
Как было уже указано, состав воды при продолжительном стоянии проб
может изменяться, и потому ряд химических исследований нужно произво-
дить на месте или вскоре после отбора проб. Сюда относится определение
реакции, нитратов, нитритов, солевого аммиака, окисляемости.
Для указанной цели необходимо иметь портативную походную лаборато-
рию, в которой должны содержаться необходимые пробы и реактивы.
Последовательность производства отдельных определений
санитарно-химического анализа
Производство полного санитарного-химического анализа воды требует мно-
го времени —до 3—4 дней и более; поэтому определения необходимо распре-
делить с расчетом максимальной экономии времени.
Только немногие определения производят в нефильтрованной воде (реак-
цию, взвешенные вещества и аммиак), другие же — в фильтрованной; поэтому
значительную часть воды отфильтровывают через складчатый фильтр.
0,5—1 л воды фильтруют через тигель Гуча для определения взвешенных
веществ. Для определения плотного остатка нужно обеспечить выпарива-
ние не менее 1 л воды. Для весового определения кальция, магния и серно-
кислых соединений отдельную пробу воды или раздельно две пробы подкис-
ляют и помещают на водяную баню для упаривания до определенного объема
с целью концентрирования.
20 А. И. Бурштейн
305
Обеспечив указанные операции — фильтрацию, выпаривание, концентри-
рование, проводят ряд качественных проб на нитраты, нитриты, аммиак,
тяжелые металлы и пр., что дает указания на необходимость дальнейшего
количественного определения тех или иных компонентов.
В первый же день и по возможности вскоре после доставки проб в лабо-
раторию определяют реакцию воды, pH и количественно: окисляемость,
нитраты, нитриты, аммиак, если эти компоненты не были определены на
месте отбора проб воды.
Если представляется возможность, то в первый же день в одной порции
упаренной воды, по отделении окиси железа и глинозема, осаждают окись
кальция, а в другой — сульфаты.
На второй день заканчивают определение плотного остатка и окиси каль-
ция, а также осаждают магнезиальные соединения.
На третий день в плотном остатке воды после его прокаливания опреде-
ляют окись железа, глинозема и марганца и заканчивают определение магне-
зиальных соединений.
На четвертый день и далее заканчивают все другие определения. Конечно,
в указанную схему распределения работы могут быть внесены те или иные
изменения.
Способы выражения результатов анализа
Результаты большинства определений выражают условно: так, жесткость
воды выражают в градусах; окисляемость — в миллиграммах кислорода,
идущего на окисление органических веществ 1 л воды в определенных усло-
виях проведения опыта; кальций в виде окиси (СаО) — в мг/л либо в виде
концентрации ионов кальция (Са") в мг/л; нитраты в виде азотного ангид-
рида (N2O5) — в мг/л либо в виде концентрации ионов (NO3') в мг/л; нитриты
в виде азотистого ангидрида (N2O3) — в мг,л либо в виде концентрации ио-
нов (N0'2) в мг/л и т. д.
Рациональным способом записи результатов анализа является выраже-
ние компонентов воды в виде концентрации отдельных ионов в мг/л. Однако
концентрации металлов и металлоидов выражают довольно часто в виде
окислов, а вместо анионов кислот в анализе обозначают ангидриды
этих кислот.
Ввиду сказанного необходимо четко указывать в анализе, в каком
виде представлены результаты, иначе могут возникнуть большие недо-
разумения.
Реакция
Реакцию воды следует определять в самом водоеме или на месте сразу
после взятия пробы, так как во время перевозки пробы реакция может изме-
ниться (выделение СО2, выпадение осадка и т. п.).
Качественное определение реакции производится при по-
мощи синих и красных лакмусовых бумажек, погружаемых на несколько
минут в исследуемую воду. Вынув из воды бумажки, сравнивают их с такими
же бумажками, смоченными дестиллированной водой.
Активная реакция (pH) воды согласно ГОСТ 2874-45 должна
быть в пределах 6,5—9,5. Она определяется электрометрически по хингид-
ронному методу, если pH воды не превышает 8,5.
Что касается колориметрического определения pH питьевых вод, то для
этой цели пользуются либо изложенным уже выше методом (стр. 82)
либо приводимым здесь методом.
Принцип метода основан на том, что применяемые одноцветные
индикаторы бесцветны в состоянии молекул и окрашены в диссоциированном
состоянии.
В зависимости от pH воды приходится прибавить к ней большее или мень-
шее количество индикатора, чтобы получить такую же окраску, которая
306
устанавливается от прибавления определенного количества этого же инди-
катора к стандартному раствору, где этот индикатор диссоциирован до конца.
Зная количество индикатора, прибавленного к стандартному раствору, и
количество индикатора, прибавленного к испытуемой воде, нахоядт pH испы-
туемой воды.
Приборы и реактивы. 1. Стеклянные цилиндры высотою 25 сл|
и диаметром 1,5 см (8—10 шт.).
2. Набор индикаторов (см. таблицу 41).
3. Стандартный раствор 0,02 N NaOH.
Методика определения. Располагают цилиндры в ряд. В один
цилиндр наливают определенное количество стандартного раствора и точно
отмеренное количество индикатора (для пресных и морских вод, pH которых
колеблется в пределах 6,7—8,4, хорошим индикатором является метанитро-
фенол). В другие цилиндры наливают испытуемую воду в убывающих коли-
чествах и тот же индикатор в возрастающих количествах с таким расчетом,
чтобы общий объем жидкости равнялся объему жидкости в стандартном ци-
линдре, напр.:
в цилиндр № 1 39,4 мл 0,02 N NaOH и 0,6 мл 0,03% раствора метанитрофенола
» » № 2 39,0 » испытуемой воды и 1,0 мл » »
» » № 3 38,5 » » » 1,5» » »
» » № 4 38,0 » » » 2,0 » » »
» >> № 5 37,0 » » » 3,0 » » >
» » № 6 36,0 » » » 4,0 » » »
» » № 7 35,0 » » » 5,0 » » »
Содержимое цилиндров тщательно взбалтывают и подбирают тот из ци-
линдров с испытуемой водой, в котором окраска оказалась одинаковой с
окраской стандартного раствора.
На основании количества индикатора, прибавленного в эти два цилиндра,
вычисляют степень диссоциации индикатора в воде по формуле:
где F — степень диссоциации индикатора (его цветность),
А — количество индикатора, прибавленного к стандартному раствору,
В — количество индикатора, прибавленного к воде.
Найдя величину F, определяют по таблице 42 логарифмическую функ-
/ F \
цию I ф = log 7---- , а затем — pH воды по формуле:
pH = pK + ^> + S+^
где pH — искомая концентрация водородных ионов,
рК — константа полной диссоциации примененного индикатора (табл. 41),
ср — логарифимическая функция найденного значения F (табл. 42),
S — поправка на содержание NaCl (табл. 43),
& — поправка на температуру испытуемой воды (табл. 44).
Пример. Для исследования взята вода t 20°С. Содержание NaCl в ней равно 0,1%.
При сравнении окраски оказалось, что стандартный раствор с содержанием 0,6 мл метани-
трофенола по интенсивности окраски соответствует исследуемой воде, к которой прилито
4 мл метанитрофенола. Нужно найти pH воды.
0,6
На основании данных определяем F; она равна -д- = 0,15.
По таблице 42 ср для найденного значение F равно—0,75; для данных задачи рК по
табли е 41 равно 8,33; S по таблице 43 равно 0,0; $ по таблице 44 равно—0,02. Подста-
вив все эти значения в формулу, получаем для испытуемой воды:
pH = 8,33 + (— 0,75) + 0,0 + (— 0,02) = 7,56.
307
Таблица 41
Индикаторы для определения pH воды, их рК
Наименование индика юра Приготовление индикатора pH рК при 18ЭС
а - динитрофенол Насыщенный водный раствор .... 2,2—4,0 3,69
р-динитрофенол 0,1 гв 200 мл воды 2,8—4,5 4,06
7 - динитрофенол 0,1 г » 400 » » 4,0—5,5 5,15
паранитрофепол 0,1 г » 100 » » 5,2—7,0 7,18
метанитрофенол 0,3 г » 100 » » G,7—8,4 8,33
фенолфталеин 0,04 г » 70 » 90° спирта 4~30 мл воды 8,5—10,5 9,73
Таблица 42
Логарифмическая функция (Ф) для разных величин F
F ф F 9 F ф
0,01 — 2,00 0,12 — 0,87 j 0,23 — 0,52
0,02 — 1,70 0,13 — 0,83 | 0,24 — 0,40
0,03 — 1,51 0,14 — 0,79 ! 0,25 — 0,48
0,04 — 1,38 0,15 1 — 0,75 | 0,26 — 0,45
0,05 — 1,28 0,16 — 0,72 0,27 — 0,43
0,06 — 1,20 0,17 — 0,69 0,28 — 0,41
0,07 — 1,12 0,18 — 0,66 0,29 — 0,39
0,08 — 1,06 0,19 — 0,63 0,30 — 0,37
0,09 — 1,00 0,20 — 0,60 0,35 — 0,27
0,10 — 0,95 0,21 — 0,56 0,40 — 0,18
0,11 — 0,91 0,22 — 0,55 0,50 — 0,00
Таблица 43
Поправки на содержание (NaCl) в воде (S)
Процентное содержание NaCl в воде $
От 0,0 до 0,6 «... 0,0
» 0,6 до 3,0 — 0,05
» 3,0 до 6,0 — 0,10
» 17,0 до 35,0 — 0,16
308
Таблица 44
Поправки на температуру (Я)
Температура воды а-динитро- фенол р-динитро- фенол 7-динитро- фенол Паранитро- фенол Метанитро- фенол
0,0 + 0,10 + 0,10 + 0,09 + 0,21 + 0,14
5,0 + 0,07 + 0,07 + 0,06 + 0,15 + 0,10
10,0 + 0,05 + 0,05 + 0,03 + 0,09 + 0,06
15,0 + 0,02 + 0,02 + 0,01 + 0,04 + 0,02
17,5 । 0,00 0,00 0,00 0,00 0,00
20,0 — 0,01 — 0,01 — 0,01 — 0,02 — 0,02
25,0 — 0,04 — 0,04 — 0,04 — 0,08 — 0,06
30,0 — 0,07 — 0,07 — 0,06 — 0,14 — 0,11
35,0 — 0,10 — 0,10 — 0,03 — 0,20 — 0,15
40,0 — 0,13 — 0,13 — 0,09 — 0,25 — 0,18
Пр имечание: Температура воды не должна
индикатора более чем на 2—3 градуса.
отличаться от температуры
Щелочность
Под общей щелочностью воды понимают сумму содержащихся в воде би-
карбонатов, карбонатов, гидратов и солей других слабых кислот, вступающих
в реакцию с соляной кислотой с образованием хлористых солей щелочных
и щелочно-земельных металлов.
Щелочность воды выражают обыкновенно числом миллилитров 1 N ки-
слоты, расходуемой на разложение указанных соединений, находящихся в
1 л воды. Это число соответствует мг-эквивалентам щелочей в 1 л воды.
Щелочность воды выражают также в градусах. Под градусом щелочности
понимают щелочность, эквивалентную 10 мг/л окиси кальция (СаО).
Так как 1 мл 1 N НС1 соответствует 28 мг СаО, то 1° щелочности = — ~
28
« 0,357 мг/экв и 1 мг/экв = — = 2,8° щелочности.
Для определения щелочности воды 100 мл ее титруют 0,1 N соляной ки-
слотой при индикаторе метилоранже. Расход кислоты в миллилитрах соответ-
ствует щелочности воды в мг-эквивалентах, так как такое же количество
миллилитров 1 N кислоты потребовалось бы при титровании литра воды.
Новейший стандарт рекомендует следующую методику определения об-
щей щелочности воды и раздельно бикарбонатной, карбонатной и гидратной.
Принцип метода основан на последовательном титровании воды
соляной кислотой сначала в присутствии индикатора фенолфталеина, даю-
щего переход окраски при pH 8,3, а затем — в присутствии смешанного
индикатора (метилоранж + индигокармин), дающего переход окраски при
pH 3,7.
При титровании в присутствии фенолфталеина имеют место следующие
реакции:
он- + Н+— Н2О
СОГ“ + Н+— НСОГ
309
При дальнейшем титровании в присутствии смешанного индикатора про-
текает реакция:
Т
нсог + н+ - со2 + н2о.
На основании титрования в присутствии фенолфталеина и смешанного
индикатора можно рассчитать содержание в воде бикарбонатов и карбона-
тов при pH воды менее 9,5, если удвоенный расход кислоты, пошедшей на
титрование с фенолфталеином, менее общего количества кислоты, израсхо-
дованной от начала определения до перехода окраски смешанного индикатора.
В остальных случаях определение бикарбонатов, карбонатов и гидратов
следует производить по номограмме (стр. 311) на основе данных об общей
щелочности воды и pH,
Реактивы. 1. 0,1 N НС1 и N.— НС1.
2. 1% спиртовый раствор фенолфталеина.
3. Смешанный индикатор: в 1 л дестиллированной воды растворяют 1 г
метилоранжа и 2,5 г индигокармина.
Методика определения. К 100 мл испытуемой воды добавляют
3 капли фенолфталеина. Если появится розовая окраска, то воду титруют
0,1 N раствором НС1 до обесцвечивания. Затем в туже пробу добавляют
5 капель смешанного индикатора и продолжают титрование той же кислотой
до перехода окраски из зеленой в фиолетовую (через серую).
Если при титровании расход 0,1 N НС1 оказался менее 0,4 мл, то титрова-
ние повторяют 1/28 N НС1 (при водах с общей щелочностью менее 1°).
Общую щелочность воды при титровании 0,1 N НС1 вычисляют по форму-
лам:
ОЩ — ка (в мг/экв)
0Щ=ка • 2,8 (в градусах),
где 01П—общая щелочность,
к — коэфициент нормальности,
а — количество мл 0,1 N НС1, израсходованной на титрование 100 мл
воды.
Если титрование ведется 1,28 N раствором НС1, то общая щелочность вы-
ражается формулами:
ОЩ = (в мг/экв)
ОЩ = кгаг (в градусах),
где ОЩ — общая щелочность,
/q — коэфициент нормальности,
— количество мл 1/28 N НС1, израсходованной на титрование 100 мл
воды.
Для расчета бикарбонатной, карбонатной и гидратной щелочности нужно
вычислить отдельно щелочность по фенолфталеину в градусах, как указано
выше. Обозначим полученный результат через — ф, При этом могут пред-
ставиться три случая:
1. ф = 0, т. е. при прибавлении фенолфталеина окрашивание воды не
наступило.
В этом случае общая щелочность воды обусловливается только бикар-
бонатной щелочностью, определяемой вышеприведенными формулами, на
основе титрования при смепрнном индикаторе.
2. 2ф < ОЩ при pH < 9,5.
В этом случае общая щелочность воды обусловливается в основном бикар-
бонатной и карбонатоной формами щелочности, определяемыми по формулам:
БЩ = 0Щ — 2ф
КЩ = 2ф,
310
где БЩ — бикарбонатная щелочность в градусах,
КЩ — карбонатная щелочность в градусах,
3. 2ф ОЩ.
В этом случае общая щелочность воды обусловливается бикарбонатной,
карбонатной и гидратной щелочностью, определяемыми по номограмме
(рис. 229) на основе данных о величине общей щелочности и pH.
Рис. 229. Номограмма для определения трех форм щелочности воды (Н СО3 , СО3 и ОН ).
Пример пользования номограммой. Дано: РН = 9,9; общая щелочность 5,2 Определить НСО—
СО““ и ОН— Решение IICQ— = 2,64; СО““ = 2.32; ОН— = 0,24 ’
Определение rtCOj'
Данные, полученные по номограмме, верны при общем солесодержании
не выше 20 мг/л, в противном случае в результаты, полученные по номограм-
ме, должны быть внесены поправки согласно таблицам. Окончательные зна-
чения щелочности бикарбнатной, карбонатной и гидратной находят по фор-
мулам:
БЩ — ^>(БЩ),
КЩ= 7 (КЩ),
ГЩ = 8 (ГЩ),
где БЩ, КЩ и ГЩ — искомые бикарбонатная, карбонатная и гидратная
щелочности исследуемой воды,
(БЩ), (КЩ) и (ГЩ)—значения бикарбонатной, карбонатной и гидратной
щелочности, определяемые по номограмме,
р, 7, и 8 — поправочные коэфициенты (см. таблицы 45,46 и 47).
Таблица 45
Значения коэфициента р
pH Плотный остаток мг/л 50 100 200 300 400 500
8 1,00 1,00 1,00 1,00 1,00 1,00
9 0,99 0,99 0,98 0,97 0,97 0,96
10 0,98 0,95 0,90 0,87 0,85 0,83
Н 0,96 0,91 0,84 0,80 0,76 | 0,73
311
Таблица 46
Значения коэфициента 7
^^^^Плотный^ остаток мг/л pH 50 100 200 300 400 500
8 1,04 1,11 1,25 1,24 1,33 1,39
9 1,04 1,09 1,18 1,24 1,30 134
10 1,02 1,05 1,09 1,12 1,14 1,16
11 1,00 1,00 1,01 1,02 1,02 1,02
Значения коэфициента & Табл и ц а 47
Плотный остаток мг/л 50 100 200 300 400 500
о 1,03 1,06 1,10 1,13 1,16 1,19
Примечания. 1. Анализ воды на щелочность следует производить, по воз-
можности, немедленно.
2 При прозрачности воды ниже 20 см, по Снеллену, исследуемая вода отстаи-
вается в закрытой посуде до прозрачности 20 см.
3. Изложенные методы определения щелочности бикарбонатной, карбонатной и
гидратной неприменимы к водам, обладающим цветностью более 40°.
Кислотность
Кислотность воды обусловливается свободной угольной кислотой, а также
слабыми органическими кислотами и случайно попавшими в воду сильными
минеральными кислотами (промышленные сточные воды). Она выражается
в миллилитрах 1,0 N щелочи, расходуемой на нейтрализацию кислот в одном
литре воды.
В зависимости от природы кислот, обусловливающих кислую реакцию,
определение кислотности ведется различно. Здесь рассмотрены отдельно два
случая:
1. Вода показывает кислую реакцию на метилоранж, что указывает на
наличие сильных минеральных кислот. В этом случае 100 мл воды титруют
при метилоранже 0,1 N раствором NaOH. Расход щелочи в миллилитрах
соответствует кислотности воды.
2. Вода при pH < 7 не показывает кислую реакцию на метилоранж.
В этом случае 200 мл титруют 0.1 N раствором NaOH при фенолфталеине
до совпадения окраски со стандартным раствором (см. ниже). Расход щелочи
в миллилитрах, поделенный на два. соответствует кислотности воды по фе-
нолфталеину (делят на два, так как титруют 200 мл воды).
Стандартный раствор готовится следующим образом: 2 мл
1% спиртового раствора фенолфталеина разбавляют дестиллированной водой
до 100 мл. В склянку такого же образца и стекла, как та, в которой ведут
титрование испытуемой воды, наливают 200 мл дестиллированной воды, при-
бавляют сюда 0,5 мл 10% раствора NaOH и 0,2 мл разбавленного раствора
фенолфталеина. Полученный раствор имеет яснорозовую окраску и отвечает
pH 8,4.
312
Взвешенные вещества
Нефелометрическое определение взвешенных веществ, обусловливающих
мутность воды, было изложено выше (стр. 301).
Количественное определение взвешенных веществ производят
также весовым путем.
Точно отмеренный объем воды (1—2 л) фильтруют через тигель Гуча,
постоянный вес которого предварительно определен. Затем тигель сушат
до постоянного веса при 105°С, охлаждают в экссикаторе и взвешивают.
Количество взвешенных веществ определяют по формуле:
х 1000 (g! — g)
V ’
где х — количество взвешенных веществ в мг/л,
gi — вес тигля со взвешенными веществами в граммах,
g — вес тигля до опыта в граммах,
v — количество взятой воды в литрах.
Потеря при прокаливании взвешенных веществ,
возникающая в основном за счет сгорания органических веществ, определяется
следующим образом.
Тигель со взвешенными веществами осторожно прокаливают до постоян-
ного веса, как описано выше (стр. 40), охлаждают в экссикаторе и взвеши-
вают. Потеря при прокаливании определяется по формуле:
I0(Wi-£2)
* и ’
где у —потеря при прокаливании в мг/л,
g2 — вес тигля со взвешенными веществами в граммах,
g2—вес тигля после прокаливания в граммах.
v — количество профильтрованной воды в литрах.
Определение взвешенных веществ и потерю при прокаливании можно
произвести и при помощи плотных беззольных фильтров диаметром в 9 см.
Для этой цели берут два фильтра, высушивают их раздельно в бюксах
с притертыми крышками до постоянного веса (в шкафу крышки должны быть
сняты) и по охлаждении в экссикаторе взвешивают.
Через один фильтр отфильтровывают определенный объем исследуемой
воды, через другой — такой же объем свежеперегнанной дестиллированной
воды. Оба фильтра помещают затем снова в бюксы, высушивают до постоян-
ного веса, охлаждают и взвешивают. Количество взвешенных веществ опре-
деляют по формуле:
х IOOO(gi — g — Р)
V 9
где х — количество взвешенных веществ в мг/л,
gj — вес бюксы с фильтром, через который фильтровалась исследуемая
вода, в граммах,
g — вес бюксы с тем же фильтром до фильтрования через него иссле-
дуемой воды в граммах,
р — поправка, равная разнице весов в граммах бюксы с фильтром до
и после фильтрования через него дестиллированной воды,
v — количество исследуемой воды, профильтрованной через фильтр,
в литрах.
Для определения потери при прокаливании взвешенных веществ, в случае
фильтрования воды через бумажный фильтр, поступают следующим образом:
фильтр после определения взвешенных веществ помещают в тигель, постоян-
ный вес которого был предварительно определен, и осторожно прокаливают
313
до постоянного веса. Для точности следует после сжигания органических
веществ прибавить в тигель немного карбоната аммония — (NH4)2CO3— для
превращения разложившихся при прокаливании углекислых солей снова
в карбонаты, после чего осторожно нагревают для удаления избытка добав-
ленной соли.
Потеря при прокаливании определяется по следующей формуле:
1000 • (& —g3 —?)
V *
где z — потеря при прокаливании в мг/л,
</4 — вес тигля с золой в граммах,
g3 — вес тигля без золы в граммах,
q — вес золы фильтра.
v — количество профильтрованной воды в литрах.
Рис. 230. Прибор для опре-
деления плотного остатка
воды.
Плотный остаток
Величина плотного остатка воды зависит в основном от растворенных неор-
ганических веществ воды, в меньшей степени — от растворенных органиче-
ских веществ.
Для определения плотного остатка воды наливают в платиновую чашку,
предварительно доведенную до постоянного веса, определенный объем воды
и нагревают на водяной бане; к концу выпаривания
добавляют новую порцию воды и вновь выпари-
вают и т. д., пока не выпарится 500—1000 мл
воды. Затем чашку вместе с остатком высушивают
при 105°С до постоянного веса, охлаждают в
экссикаторе и взвешивают. Разница между полу-
ченным весом чашки вместе с осадком и началь-
ным весом чашки показывает вес плотного остатка
общего объема выпаренной воды. Результат пере-
числяют на литр.
Для того, чтобы не приходилось приливать воду
в чашку несколько раз, пользуются приспособле-
нием, изображенным на рис. 230. В круглодонную
колбу вводят определенное количество исследу-
емой воды. В шейку колбы вставляют пробку,
а в пробку трубочку, наружный конец которой
скошен. Колбу переворачивают над чашкой. Бла-
годаря скошенному концу трубочки вода вытекает
небольшими порциями по мере испарения ее из
чашки. Так чашка автоматически наполняется,
пока не вытечет вся вода из колбы.
Если нужно определить вес органических и неорганических частей плот-
ного остатка, то сжигают органические вещества, как описано выше (см.
Взвешенные вещества).
Плотный остаток в литре хорошей воды не превышает обыкновенно 500—
600 мг. Потеря при прокаливании не превышает 10—15% веса плотного
остатка.
Окисляемость (органические вещества)
Окислясмость воды обусловлена наличием в ней многообразных органи-
ческих веществ растительного и животного происхождения (гуминовые ве-
щества, органические кислоты и др.). Говоря об окисляемости воды, имеют
в виду окисляемость именно этих веществ.
Окисляемость воды определяют путем окисления органических веществ
марганцевокислым калием в кислой или щелочной среде при кипячении,
314
так как на холоду большинство органических веществ воды плохо или вовсе
не окисляется.
Присутствующие нередко в воде неорганические восстановители (закис-
ные соли железа, нитриты, сульфиды, сульфиты и др.) способны окисляться
марганцевокислым калием на холоду, и потому расход марганцевокислого
калия на окисление этих веществ определяется отдельно, а в случае замет-
ного количества их учитывается при определении окисляемости воды.
Окисляемость воды выражают либо в миллиграммах КМпО4, необходимого
для окисления органических веществ, находящихся в 1 л воды, либо в милли-
граммах кислорода, расходуемого на это окисление.
Так как каждые две молекулы КМпО4 в кгслой среде выделяют 5 атомов
кислорода, весовые отношения между затраченным на окисление перманга-
натом калия и выделившимся при этом кислородом выражаются так:
2 . 158 . КМпО4~ 5.16(0), следовательно: ~КМпО4« 4КМпО4 ~ 1(0).
oU
Отсюда ясно, что для пересчета окисляемости, выраженной в миллиграм-
мах КМпО4, на окисляемость, выраженную в миллиграммах кислорода,
переводным коэфициентом является 0,25; для обратного пересчета коэфи-
циентом служит число 4.
По количеству кислорода, расходуемого на окисление органических ве-
ществ воды, можно сделать вывод о приближенном количестве органических
веществ в воде. Для этой цели расход кислорода умножают на эмпирически
установленный коэфициент 20 (Г. В. Хлопин).
Количество кислорода, расходуемого на окисление органических веществ
хорошей воды, обыкновенно не превышает 2—4 мг/л, что соответствует 8—16 мг
КМпО4. В такой воде содержание органических веществ приближенно соответ-
ствует 40—80 мг л.
Однако высокая окисляемость воды еще не говорит о непригодности ее
в качестве питьевой. Так, грунтовые воды, богатые гуминовыми веществами
(торфяные местности), при высокой окисляемости часто оказываются совер-
шенно безвредными в гигиеническом отношении. Таким образом, при вы-
сокой окисляемости воды для правильного заключения о ее пригодности
необходимо установить природу органических веществ.
Окисляемость воды необходмо определять не позднее, чем через 2 часа
после взятия пробы; если это невозможно, то воду следует подкислить серной
кислотой (25 мл серной кислоты — 1 : 3 по объему — на 0,5 л воды).
Согласно стандарту, определение окисляемости воды производится в
кислой среде при содержании в воде хлоридов 300 мг/л и менее. При
большем содержании хлоридов окисляемость определяется в щелочной
среде; в противном случае заметные количества соляной кислоты, полу-
чающейся при взаимодействии хлоридов и серной кислоты, могут привести
к разложению марганцевокислого калия и расходу повышенных количеств
его, что видно из следующих реакции:
2NaCl + H2SO4 = Na2SO4 + 2НС1
2KMnO4 + 16НС1 = 2КС1 + 2МпС12 + 5С12 + 8Н2О.
Определение окисляемости воды в кислой среде
Принцип метода заключается в том, что марганцевокислый калий
как сильный окислитель окисляет в кислой среде при кипячении органиче-
ские вещества; при этом каждые две молекулы КМпО4 выделяют 5 (0) по
реакции:
2КМпО4 + 3H2SO4 = K2SO4 + 2MnSO4 + ЗН2О + 5(0).
Реактивы. 1. 0,01 N раствор КМпО4 (приготовление — см. стр. 60).
2. 0,01 N раствор щавелевой кислоты (приготовление — см. стр. 59).
3. Серная кислота 1 : 3 (по объему).
315
Методика определения. Сначала проверяют титр КМпО4.
К 100 мл дестиллированной воды добавляют 5 мл раствора серной кислоты
(1 : 3), 1 мл 0,01 N раствора щавелевой кислоты, нагревают до кипения и
титруют 0,01 N раствором КМпО4 до появления очень слабой розовой окраски;
при этом окисляются органические вещества, которые находятся в дестилли-
рованной воде, а также и прибавленная щавелевая кислота. К полученному
раствору, свободному от органических веществ, прибавляют точно 10 мл
0,01 N раствора щавелевой кислоты и вновь титруют 0,01 N раствором КМпО4
-До появления бледнорозового окрашивания.
Коэфициент нормальности раствора КМпО4 вычисляется по формуле:
где К — коэфициент нормальности,
v — количество миллилитров раствора марганцевокислого калия,
пошедшего на титрование 10 мл 0,01 N раствора щавелевой кислоты.
Установив титр перманганата, определяют окисляемость воды.
В коническую колбу емкостью 250 мл вводят 100 мл исследуемой воды,
добавляют 5 мл серной кислоты 1: 3 и точно 10 мл 0,01 N раствора КМпО4.
Накрыв колбу часовым стеклсм, нагревают жидкость до кипения. Для равно-
мерного кипения рекомендуется опустить в колбу несколько капилляров
или кусочков пемзы. Кипячение поддерживают 10 минут, считая с момента
закипания, после чего вводят в колбу точно 10 мл 0 01 N раствора щавелевой
кислоты и перемешивают содержимое колбы взбалтыванием при помощи
кругообразных движений. При этом избыток взятого перманганата калия
восстанавливается по реакции:
2КМпО4 + 5(С2Н2О4 • 2Н2О) Н 3H2SO4 = K2SO4 + 2MnSO4 + 18Н.О+ 10С02.
Обесцвеченный горячий раствор титруют 0,01 N раствором КМпО4 до
появления слабого розового окрашивания.
Окисляемость воды определяется в мг/л кислорода по формуле:
= (pt —р,)К • 0,08 • 1000
— v ’
либо в мг/л КМпО4 по формуле:
(t>i —1>2)/< • 0 32 - 1000
где Vi — общее количество 0,01 N раствора КМпО4, израсходованного при
определении окисляемости воды, в мл,
г>2— количество 0,01 N раствора КМпО4, расходуемое на окисление
10 мл 0,01 N раствора щавелевой кислоты, в мл,
К — коэфициент нормальности КМпО4.
Число 0,08 соответствует количеству мг кислорода, которое выделяет
1 мл 0,01 N раствора КМпО4.
Число 0,32 соответствует (округленно) количеству мг КМпО4, которое
содержится в 1 мл 0,01 N раствора КМпО4.
Примечание 1. Если проба воды была консервирована серной кислотой,
то при определении окисляемости добавлять серную кислоту не следует.
Примечание 2. При прозрачности волы по Снеллену 15 см и менее допол-
нительно определяется окисляемость воды, предварительно профильтрованной через
плотный бумажный фильтр.
Примечание 3. Если при титровании жидкости после введения в иссле-
дуемую воду раствора щавелевой кислоты расход 0,01 N раствора КМпО4 оказывается
более 5 мл, то определение следует повторить с предварительным разбавлением i сле-
дуемой воды при помощи дестиллированной воды. Отдельным определением уста-
навливают расход перманганата на окисление дестиллированной воды и учи-
тывают этот расход при определении окисляемости воды. В расчетах учитывают
также кратность разбавления воды.
316
Определение окисляемости воды на холоду производится точно так, как
описано выше, но вместо кипячения проба после добавления в нее марган-
цевокислого калия взбалтывается и выдерживается при температуре
+ 15 + 20°С в течение 10 минут.
Марганцевокислый калий в этом случае почти полностью идет на окисле-
ние неорганических восстановителей. Расход его должен быть вычтен из
общего расхода марганцевокислого калия при определении с нагреванием,
если имеется в виду учесть окисляемость одних органических веществ.
Пример 1. Для анализа взято 100 мл. воды. При определении окисляемости (с нагре-
ванием) общий расход 0,01 N раствора КМпО4 равен 16,2 мл. На окисление 10 мл 0,01 N
раствора С2Н2О4-бН2О уходит 11,2 10 мл перманганата. Нужно определить окисляемость
воды в мг кислорода и перманганата. Она равна:
10
(16,2 — 11,4) т-ру • 0,08 • 10С0
----------Lbr___________3,43 мг (0)
100_____________________V ’
10
(16,2—11,4)775- • 0,32 • 1000
-----------13,72 мг КМпО4.
100 4
Пример 2. При определении окисляемости той же вэты на холоду окисляемость вы-
разилась 0,32 мг (0) и 1,28 мг КМпО4. В этом случае окисляемость, обусловленная орга-
ническими веществами, выразится через 3,43 — 0,32 = 3,11 мг (0) и 13,72— 1,28 =
= 12,44 мг КМпО4.
Определение окисляв мости воды в щелочной среде
Принцип метода сводится к восстановлению в щелочной среде
марганцевокислого калия с выделением из каждых 2 молекул 3 атомов ки-
слорода.
2КМпО4 + 2КОН + Н2О = 2МпО2 + 4КОН + 3/0/.
Реактивы. Те же, что и в предыдущем методе; дополнительно
раствор КОН или NaOH (50 г щелочи растворяют в 100 мл воды).
Методика определения. В коническую колбу емкостью в
250 мл вводят 100 мл испытуемой воды, добавляют 0,5 мл раствора едкой
щелочи и точно 10 мл 0,01 N раствора КМпО4; нагрев содержимое колбы
до кипения, поддерживают кипение в течение 10 минут, затем, охладив со-
держимое колбы до 50—60°С, приливают в нее 5 мл раствора серной кислоты
и точно 10 мл 0,01 N раствора щавелевой кислоты. При этом происходит
восстановление избытка КМпО4 и образовавшейся в результате приведен-
ной выше реакции двуокиси марганца. Обесцвеченный раствор титруют
марганцевокислым калием до появления слаборозовой окраски, устойчивой
в течение 5 минут.
Расчеты ведутся так же, как в описанном выше способе.
Примечание. Если испытуемая вода подвергалась консервированию
серной кислотой, то в отдельной пробе нужно выяснить количество щелочи, которое
требуется на нейтрализацию воды (по метилоранжу), и определенное титрованием
количество щелочи прибавить в исследуемую пробу сверх 0,5 мл едкого калия
или едкого 'натрия.
Жесткость (щелочно-земельные металлы)
Жесткость воды обусловлена растворенными в ней солями щелочно-зе-
мельных металлов: Са и М". В основном в воде присутствуют бикарбонаты
и сульфаты Са и Mg с преобладанием известковых солей над магнезиальными.
317
Жесткость воды выражается в градусах. Г жесткости соответствует со-
держанию вводе солей жесткости в количестве,эквивалентном 10,0 мг/л СаО.
Различают жесткость общую, карбонатную, устранимую и постоянную.
Под общей жесткостью понимают жесткость, обусловленную
всеми солями Са и Mg в сырой воде.
Под карбонатной жесткостью понимают жесткость, обуслов-
ленную гидрокарбонатами и карбонатами Са и Mg, растворенными в
сырой воде.
Под устранимой жесткостью понимают жесткость, которую
удается устранить при кипячении воды. В результате кипячения бикарбо-
наты щелочно-земельных металлов, растворенные вводе, превращаются в кар-
бонаты и выпадают1:
Са(НСО3)2 — СаСО3 + СО2 + Н2О.
Однако выпадение углекислых солей щелочно-земельных металлов не про-
исходит полностью: в литре воды в отсутствие свободной СО2 при комнатной
температуре может остаться в растворенном состоянии около 16 мг СаСО3
и до 1000 мг Mg СО3.
Таким образом, понятие «карбонатная жесткость» не идентично понятию
«устранимая жесткость». Устранимая жесткость меньше карбонатной.
Под постоянной жесткостью понимают жесткость кипя-
ченой воды. Постоянная жесткость равна общей жесткости за вычетом
устранимой.
Питьевая вода с жесткостью до 10° считается мягкой, от 10° и до 20°—
умеренно жесткой, выше 20°—жесткой.
Вода с высокой жесткостью мало пригодна для бытовых нужд: она обу-
словливает большую накипь на стенках посуды, плохое разваривание овощей,
мяса, непроизводительную трату мыла при стирке белья на связывание со-
лей щелочно-земельных металлов.
Вода с высокой жесткостью непригодна и для фабрично-заводских нужд,
особенно для питания котлов, ввиду выпадения больших осадков на стенках
и непроизводительной траты топлива для нагревания таких котлов.
Согласно ГОСТ 2874-45 жесткость питьевой воды должна быть не бо-
лее 40°.
Для санитарной характеристики воды большое значение имеет динамика
жесткости.
Наблюдающееся иногда заметное повышение жесткости может указывать
на загрязнение воды: повышенная минерализация органических веществ,
сопровождающаяся обогащением воды СО2, в конечном результате приводит
к лучшему выщелачиванию пород, обусловливающих жесткость воды.
Количественное определение жесткости воды производится объемным либо
весовым методом, чаще применяется первый.
Объемное определение жесткости воды
Объемное определение жесткости воды можно производить двумя спосо-
бами: 1) разложением солей щелочно-земельных металлов при помощи мыль-
ных растворов и 2) разложением тех же солей при помощи щелочных (не
мыльных) растворов.
Определение жесткости воды олеатным методом (олеатом калия).
Принцип метода основан на том, что прибавляемое к воде щелоч-
ное мыло (олеат калия) при взаимодействии с солями щелочно-земельных
металлов переводится в мало растворимые в воде кальциевые и магниевые
1 Такое выпадение карбонатов наблюдается и без нагревания воды при высокой кон-
центрации гидро карбонатов.
318
соли олеиновой кислоты — олеаты кальция и магния, выпадающие в осадок
и не дающие при взбалтывании пены:
2С17Н33СОО“+ Са ++— [С17Н33СОО]2Са |
2С17Н33СОО- + Mg++—» [C17H33COO]2Mg 1
Конец реакции совпадает с переводом в нерастворимое состояние всех
солей щелочно-земельных металлов, после чего избыток олеата калия дает
при встряхивании пену. Появление пены служит, таким образом, индика-
тором конца реакции.
Пена должна получиться мелкопузырчатой, обильной и стойкой, не исче-
зающей в течение 3 минут в положенной на бок склянке после 8 встряхи-
ваний; это так называемая «живая пена» в отличие от крупнопузырчатой
и нестойкой пены, появляющейся вначале.
Получение «живой пены» характеризуется еще тем, что при встряхивании
жидкости в склянке слышится мягкий звук.
Реактивы. 1. Спиртовый раствор олеата калия, 1 мл которого со-
ответствует 1 мг СаО.
Если для анализа брать 100 мл исследуемой воды, то каждый миллилитр
мыла, пошедшего в реакцию, указывает на присутствие вл воды 10 мг СаО.
т. е. соответствует одному градусу жесткости.
Для приготовления спиртового раствора олеата калия растворяют 15 г
чистой олеиновой кислоты (acidum oleinicum purum) в 600 мл нагретого до
60°С 96% этилового ректификованного спирта, добавляют 400 мл дестилли-
рованной воды, в которой растворено 4 г чистого КОН, и оставляют стоять
3—4 дня. Потом раствор фильтруют, прибавляют несколько капель 1 % спир-
тового раствора фенолфталеина и нейтрализуют раствор мыла 0,1 N раство-
ром NaOH или 0,1 N раствором НС1 до слаборозового окрашивания.
Титр мыла устанавливают так, как описано ниже.
2. Аммиачная смесь: растворяют 10 г хлористого аммония в небольшом
количестве дестиллированной воды, добавляют 100 мл 10% раствора аммиака
и доводят объем дестиллированной водой до 500 мл.
Этот раствор добавляют в небольшом количестве к исследуемой воде для
того, чтобы титрование мылом проводить в щелочной среде и тем самым умень-
шить гидролиз мыла водой, а также способствовать тому, чтобы реакция
между мылом и щелочно-земельными металлами протекала равномерно.
3. Растворяют 6 г чистого едкого калия в дестиллированной воде, добав-
ляют 100 г сегнетовой соли (KNaC4H4O6) и по растворении доводят объем
дестиллированною водою до 500 мл.
Этот реактив добавляется к исследуемой воде, чтобы воспрепятствовать
солям магния вступить в реакцию с олеиновой кислотой, благодаря чему
можно определить жесткость, обусловленную лишь солями кальция.
4. Раствор азотнокислого бария — Ba(NO3)3, 1 мл которого соответствует
1 мг СаО.
Молекулярный вес Ba(NO3)2 равен 261, молекулярный вес СаО равен
56; отсюда 261 г Ba(NO3)2 соответствует 56 г СаО; одному же грамму окиси
26 Г
кальция соответствует —— - 4,668 г азотнокислого бария. Растворив в литре
56
дестиллированной воды 4,668 г азотнокислого бария, получают раствор,
каждый миллилитр которого соответствует 1 мг СаО. Жесткость такого
раствора соответствует 100 градусам.
Методика определения. Сначала устанавливают титр мыль-
ного раствора. В склянку с притертой пробкой вливают 10 мл раствора азотно-
кислого бария указанного выше титра и доливают 90 мл дестиллированной
воды. Получают раствор 10° жесткости. К нему приливают 5 мл аммиачной
смеси (раствор № 2) и титруют раствором мыла, добавляя его понемногу и
319
каждый раз встряхивая жидкость. Мыло приливают до тех пор, пока не по-
явится стойкая пена.
Мыло считается надлежащей крепости, если в реакцию пойдет 10,2 мл
его, т. е. каждый миллилитр соответствует 1 мг СаО; 0,2 мл мыла идет на
образование пены.
Если мыло получается не надлежащей крепости, то его нужно выправить.
Так, если во время титрования пошло, например, 8 мл раствора мыла, то его
нужно считать крепким и к нему на каждые 8 мл нужно прилить 2,2 мл
56—58° спирта.
Для определения жесткости воды, зависящей от солей кальция, берут
100 мл исследуемой воды, приливают к ней 5 мл щелочного раствора сегне-
товой соли, (раствор № 3), после чего титруют мылом, приливая понемногу
(0,2 мл), каждый раз энергично встряхивают, пока не появится стойкая пена.
По числу миллилитров мыла, пошедшего в реакцию, определяют градусы
жесткости, зависящей от солей кальция.
Для определения жесткости, зависящей от солей кальция и магния,
берут 100 мл исследуемой воды, приливают к ней для лучшего хода реакции
100 мл дестиллированной воды, затем добавляют 5 мл аммиачной смеси (ра-
створ № 2) и титруют мылом. По числу миллилитров израсходованного мыла
определяют градусы жесткости, обусловленной солями кальция и магния.
Отняв от результата второго титрования результат первого, определяют
градусы жесткости, обусловленной солями магния.
Принимая во внимание, что жесткость выражают обыкновенно по солям
Са, нужно число градусов жесткости, зависящей от Mg, перечислить на гра-
дусы жесткости по Са, для чего множат это число на 3/4, так как количества
мыльного раствора, идущие на растворы солей кальция и магния одинаковой
жесткости, относятся одно к другому, как 3 : 4.
Результат умножения прибавляют к результату первого титрования.
Сумма покажет общую жесткость, перечисленную на соли кальция.
Пример. Результат первого титрования равен 8°, второго — 12°. Отсюда жесткость,
4-3
обусловленная солями магния, равна 12—8 = 4°, а это равно -д- = 3° жесткости от солей
кальция. Таким образом, общая жесткость, перечисленная только на соли кальция, равна
8+3 = 11°.
На основании результатов определения жесткости воды по описанному
способу легко определяется количественное содержание известковых солей,
перечисленных на окись кальция (СаО), и магниевых солей, перечисленных
на окись магния (MgO), а именно: каждый градус жесткости, обусловленной
солями кальция, указывает на присутствие в литре исследуемой воды 10 мг
кальциевых солей, перечисленных на окись кальция, а каждый градус жест-
кости, обусловленной солями магния, указывает на присутствие в литре
воды 7,2 мг магниевых солей, перечисленных на окись магния.
Для кальциевых солей число 10 выводят на основании определения гра-
дуса жесткости. Для магниевых солей число 10 нужно помножить на коэфи-
40,32 MgO 10-40,32
циент соответственно получим---------------— = 7,2.
5о СаО ' 56
Так, в нашем примере известковых солей, перечисленных на окись каль-
ция, содержится 80 мг (8х 10) и магниевых солей, перечисленных на окись
магния, 21,6 мг (7,2x3) в литре воды.
По описанному способу можно определить не только общую, но и постоян-
ную жесткость воды. Для этой цели определенный объем ее кипятят, затем
фильтруют; фильтрат доливают дестиллированной водой до первоначального
объема и определяют жесткость, как описано выше.
Стандарт рекомендует титр мыльного раствора устанавливать по смеси
растворов хлористого кальция (СаС12 • 6Н2О) и сернокислого магния
320
(MgSO4 • 7H2O), взятых в таких отношениях, чтобы кальциевая жесткость
составляла 75% общей жесткости смеси, а магниевая — 25% (наичаще встре-
чаемые соотношения в питьевых водах).
Для получения эталонного раствора солей жесткости растворяют 7,84 г
кристаллического хлористого кальция в дестиллированной воде и доводят
объем до литра водою. Содержание кальция в растворе соответствует 1428 мг.
Отдельно растворяют 1,1 г сернокислого магния в 300 мл воды; раствор
содержит 108 мг магния. Затем отмеривают такое количество раствора хло-
ристого кальция, которое содержит 537 мг кальция, смешивают в литровой
колбе отмеренный объем раствора хлористого кальция с приготовленным
раствором сернокислого магния и доводят объем раствора до литра водою.
Полученный раствор имеет 100° жесткости; из них 75% составляет каль-
537 \ 168
циевая жесткость ( — » 75) и 25% — магниевая жесткость (- ~ 25 ).
\/,1т / J
Здесь 7,14 — коэфициент пересчета Са++в градусы жестокости
/ 7,2 • 24 32\
4,33—коэфициент пересчета Mg++B градусы жесткости —).
Из полученного основного раствора готовят шкалу стандартных раство-
ров жесткостью: 0,1; 0.2; 0.3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1,0, 1,5 и 2,0 градуса.
Приготовленные стандартные растворы титруют приготовленным олеатом
калия и результат заносят в тарировсчную таблицу.
По расходу олеата калия на титрование 100 мл испытуемой воды опреде-
ляют соли жесткости в градусах во взятой порции воды; результат перечи-
сляют на литр.
Стандарт рекомендует также испытуемую воду перед титрованием нейтра-
лизовать (по фенолфталеину или метилоранжу).
Определение общей жесткости воды пальмитатом к\тич и карбонат-
ной жесткости сол Iной кисл )той. Принцип метода основан на
том, что бикарбонаты (карбонаты) разлагаются соляной кислотой: а щелочно-
земельные металлы хлоридов и сульфатов связываются пальмитатом калия:
таким образом, в основе метода лежат следующие реакции.
Са(НСО3)2 + 2НС1 = СаС12 + 2СО2 + 2Н2О,
2С15Н31СОО~+Са++—♦ [С15Н31СОО]2Са j
2C15H31COO“+Mg ++ - [С15Нз1С00;2 /Vg j
Реактивы. 1. 0,1 N раствор НС1.
2. 0,1 N раствор хлористого бария — ЕаС12 . 2Н2О либо 0,1 N раствор
азотнокислого бария Ba(NO3)2.
3. 0,1 N раствор пальмитата калия.
4. 1% спиртовый раствор фенолфталеина.
5. 1% водный раствор метилоранжа.
б. 10% спиртовой раствор КОН.
7. 0,1 N раствор NaOH.
Приготовление 0,1 N раствора пальмитата калия. Отвешивают 25,6 г
химически чистой пальмитиновой кислоты, что соответствует дециграмм-
эквиваленту ее (грамм-эквивалент С1бН31 СООН=256). Навеску переводят
количественно в литровую колбу, в которую предварительно наливают 300 мл
95% этилового ректификованного спирта и 400 мл нейтрального глицерина,
и нагревают смесь на водяной бане до полного растворения пальмитиновой
кислоты (температура плавления 60°С). К горячему раствору прибавляют
2—3 капли фенолфталеина и приливают из бюретки 10% спиртовый раствор
едкого калия до появления розового окрашивания. Оставляют смесь на 24
часа и по истечении этого времени к обесцветившемуся раствору приливают
21 А. И. Бурштейн
321
снова раствор КОН до появления розовой окраски. Смесь переводят коли-
чественно в литровую колбу и доливают 95° этиловым спиртом до литра.
Раствор пальмитата калия может быть приготовлен и из чистого пальми-
тиновокислого калия. В этом случае 29,4 г чистого пальмитиновокислого
калия растворяют в литровой колбе при нагревании на водяной бане в 400—
500 мл 96° этилового спирта, добавляют 400 мл глицерина, охлаждают до
15°С, переводят количественно в литровую колбу и доводят до метки спиртом.
Установка титра пальмитата калия производится по 0,1 N раствору хло-
ристого бария — 12,22 г ВаС12 . 2Н,0 на литр воды — или по 0,1 N раствору
азотнокислого бария— 13,07 г Ba(NO3)2 на литр воды.
Для этой цели 25 мл 0,1 N раствора хлористого бария вводят в склянку
с притертой пробкой емкостью 150 мл, доливают дестиллированной воды до
объема в 100 мл, затем через раствор в течение 3 минут продувают воздух
при помощи стеклянной трубки, соединенной с каучуковой грушей, для
удаления из раствора угольного ангидрида; остаток угольного ангидрида
нейтрализуют прибавкой 0,1 N едкого натрия при индикаторе фенолфталеине
до устойчивого слаборозового окрашивания.
Подготовленный таким образом раствор хлористого бария титруют паль-
митатом калия до интенсивного розовокрасного окрашивания, взбалтывая
энергично раствор в закрытой пробкой склянке. По израсходованному коли-
честву пальмитата калия устанавливают его коэфициент нормальности.
Методика определения жесткости воды. Сначала
определяют карбонатную жесткость, а затем общую.
В склянку с притертой пробкой емкостью на 150 мл наливают 100 мл испы-
туемой воды1 *, прибавляют 2 капли метилоранжа и титруют 0,1 N соляной
кислотой до розового окрашивания.
Каждый миллилитр израсходованной на титрование 0,1 N НС1 соответ-
ствует 2,8 СаО во взятой пробе (см. первую реакцию), что на литр соответст-
28
вует 28 мг или — 2,8° жесткости. Таким образом, для определения кар-
бонатной жесткости в испытуемой воде израсходованное на титрование число
миллилитров 0,1 N НС1 умножают на 2,8.
Определение общей жесткости производится после определения карбо-
натной жесткости в той же пробе. Через раствор продувают воздух, как
было указано выше, для удаления угольного ангидрида. Остаток свободного
угольного ангидрида нейтрализуется 0,1 N едким натрием при индикаторе
фенолфталеине. После этого приступают к титрованию пальмитатом калия,
сильно взбалтывая время от времени раствор. Титрование ведется до интен-
сивно розовокрасного окрашивания, которое возникает от фенолфталеина в
силу гидролиза первой избыточной капли пальмитата калия.
Каждый миллилтр израсходованного на титрование 0,1 N пальмитата
калия соответствует 2,8 мг СаО во взятой пробе (см. вторую реакцию), что
28
на литр соответствует 28 мг или = 2,8* жесткости.
Таким образом, для определения общей жесткости в испытуемой воде
израсходованное на титрование число миллилитров 0,1 N пальмитата калия
умножают на 2,8.
Примрр. При определении карбонатной жесткости израсходовано 3,6 мл 0,1 N НС1;
при определении обшей жесткости израсходовано 4,8 пальмитата калия. При установке
титра пальмитата калия на 25 мл 0,1 N хлористого бария ушло 26 мл раствора мыла. От-
сюда заключаем:
карбонатная жесткость равна 2,8.3,6 = 10,08°,
25
общая жесткость равна 2,8 5 4,8 • — = 12,92°.
1 Если жесткость исследуемой воды выше 30°, то ее разбавляют соответственно дестил-
лированной водой.
322
Определение жесткости воды при помощи щелочного раствора (соды и
едкого натрия). Принцип метода основан на том, что карбонатная
жесткость воды определяется (как и в предыдущем методе) титрованием со-
ляной кислотой, а общая жесткость — титрованием щелочным раствором
соды и едкого натрия.
В основе определения лежат следующие реакции:
Са(НСО3)2 + 2НС1 = СаС12 + 2СО2 + 2Н2О
СаС12 -}- Na2CO3 = СаСО3 -}- 2Nacl
MgCl2 + 2NaOH = Mg(OH)2 + 2NaCl.
Реактивы. 1. Щелочная смесь, состоящая из равных объемов 0,1 N
раствора соды и 0,1 N раствора едкого натрия.
2. 0,1 N раствор соляной кислоты.
3- I %о раствор метилоранжа.
Методика определения жесткости воды. Сначала
определяется карбонатная жесткость, а затем общая.
Определение ч карбонатной жесткости производят, как и в предыдущем
методе, титрованием 0,1 N НС1 при индикаторе метилоранже, причем титро-
вание в данном случае можно вести не в склянке с притертой пробкой, а в
обыкновенной конической колбе.
Определение общей жесткости производится в той же пробе после опре-
деления карбонатной жесткости. К раствору прибавляют из бюретки или
при помощи пипетки щелочную смесь; при этом для воды с жесткостью до 10°
достаточно 20 мл смеси. Такое количество смеси берется с расчетом, чтобы
она оказалась в избытке. Добавив смесь, жидкость кипятят несколько ми-
нут, затем охлаждают колбу под струей воды, переводят количественно со-
держимое в мерную колбу на 200 мл и доводят жидкость до метки дестилли-
рованной ведой, хорошо взбалтывают и фильтруют через складчатый фильтр.
143 фильтрата отбирают в коническую колбу 100 мл, прибавляют одну каплю
метилоранжа и титруют 0,1 N соляной кислотой до покраснения. Число милли-
литров израсходованной при обратном титровании соляной кислоты умножают
на два. Результат показывает, сколько миллилитров щелочного раствора
не вступило в реакцию с солями щелочно-земельных металлов. Полученный
результат отнимают от общего объема щелочного раствора. Разность показы-
вает, сколько миллилитров щелочного раствора вступило в реакцию с солями
щелочно-земельных металлов. Так как 1 мл щелочного раствора соответст-
вует 2,8 мг СаО, то, умножив полученный после вычисления результат на
2,8, определяют общую жесткость в градусах. Сказанное можно выразить
формулой:
А = (а — 2Ь) • 2,8,
где А — общая жесткость,
а — число миллилитров прибавленного щелочного раствора,
b — израсходованное число миллилитров 0,1 N НС1 при обратном титро-
вании.
Пример. При определении жесткости по описанному методу на 100 мл воды затрачено
при первом титровании 1,6 мл. 0,1 N соляной кислоты. При втором титровании израсходо-
вано 7 миллилитров 0,1 N соляной кислоты. Отсюда заключаем:
устранимая жесткость равна 1,6.2,8= 4,48°,
общая жесткость равна (20,0 — 2 . 7). 2,8 = 16,8°.
По описанному способу с некоторыми изменениям можно определить
отдельно жесткость, обусловленную солями магния и солями кальция. Для
этого после первого титрования приливают 0,1 N раствор NaOH, осаждающий
только соли магния, согласно приведенной выше реакции, если он не взят
в большом избытке, в противном случае могут выпасть и соли кальция в виде
323
Са(ОН)2. Дальше исследование проводится, как описано выше. Число милли-
литров 0,1 N едкого натрия, пошедшего на связывание магнезиальных солей,
MgO 40 32
умножают на 2,016 соответственно: ~= = 2,016.
Отняв полученный результат от общей жесткости, определяют жесткость,
обусловленную солями кальция.
Приведенные выше два метода определения карбонатной жесткости могут
давать иногда неправильные результаты, а именно, когда в воде кроме ги-
дрокарбонатов щелочно-земельных металлов присутствуют также гидрокар-
бонаты щелочных металлов — К и Na, а также гидрокарбонаты Fe и Мп.
В этих случаях при титровании соляной кислотой часть ее уходит на разло-
жение этих соединений, что при пересчете затраченной при титровании со-
ляной кислоты на СаО дает повышенные результаты для карбонатной жест-
кости.
В дальнейшем при определении общей жесткости при помощи мыльного
раствора или щелочного раствора хлориды натрия и калия не принимают
участия в обменном разложении с мылом или щелочью и, таким образом,
определенная общая жесткость может оказаться равной или даже меньше
карбонатной жесткости.
Это надо иметь в виду, и в таких случаях правильнее рассматривать ре-
зультат титрования соляной кислотой, пересчитанный на СаО, не как карбо-
натную жесткость, а как «общую щелэчнэсть, выэажшную в градусах жест-
кости» (термин, предлагаемыйТ. М. Слободской); тогда можно будет избежать
недоразумений.
Определение устранимой и постоянной жесткости. Устранимая жесткость
определяется как разность между карбонатной жесткостью сырой воды и
воды прокипяченнсй.
Определение карбонатной жесткости сырой воды было приведено выше.
Определение карбонатной жесткости прокипяченной воды производится так
же, как и сырой, после того как сырая вода подвергалась кипячению.
Около 500 мл воды кипятят в колбе с обратным холодильником в течение
часа. Затем колбу с водой быстро охлаждают под током холодной воды и филь-
труют через складчатый фильтр. Первую порцию фильтрата отбрасывают,
а затем отбирают 200 мл фильтрата, прибавляют метилоранж и титруют 0,1 N
соляной кислотой. Карбонатная жесткость прокипяченной воды выразится
формулой:
2,8 • п
А = —2------
где А — карбонатная жесткость прокипяченной воды,
п — число миллилитров 0,1 N соляной кислоты, пошедшей на титрование.
Разность между карбонатной жесткостью сырой воды и прокипяченной
покажет устранимую жесткость.
Отняв от общей жесткости сырой воды устранимую жесткость, получают
постоянную жесткость.
Веоовсе определение жесткости воды
Определение жесткости воды весовым способом сводится к весовому опре-
делению солей кальция, а затем солей магния в одной и той же пориции воды.
На основании полученных данных вычисляют жесткость воды.
Весовое определение солей кальция. Принцип метода основан
на том, что кальций, связываясь с оксалатом аммония, выпадает в виде окса-
лата кальция согласно реакции:
СаС12 4- (NH4)2C2O4 = СаС204 + 2NH4C1
324
Благодаря наличию избытка хлористого аммония соли магния остаются
в растворе.
Реактивы. 1. Соляная кислота.
2. Аммиак.
3. Хлористый аммоний.
4. Концентрированный раствор щавелевокислого аммония—(NH4)2C2O4.
5. Азотная кислота.
6. Раствор AgNO3.
Методика определения. 500 мл испытуемой воды подкисляют
соляной кислотой и выпаривают на водяной бане до 150 мл. Добавляют в
избытке аммиак и хлористый аммоний. Если в растворе есть соли железа,
алюминия и кремневой кислоты, то выпадает осадок, который нужно отфиль-
тровать. Осадок на фильтре промывают горячей водой и к горячему фильтрату,
имеющему запах аммиака, добавляют при помешивании концентрированный
раствор щавелевокислого аммония до тех пор, пока идет образование белого
осадка щавелевою слого кальция. Охлаждают и добавляют дестиллирован-
ной воды до общего объема в 250 мл. Оставляют на 12—15 часов в закрытой
посуде.
По истечении указанного времени фильтруют через фильтр с определен-
ным весом золы. Еще до промывания осадка отбирают 200 мл фильтрата для
определения солей магния. Осадок промывают декантацией горячей водой,
а потом промывают на фильтре до исчезновения реакции на хлориды1.
Фильтр вместе с осадком высушивают, переводят во взвешенный платиновый
тигель и прокаливают до постоянного веса. При этом щавелевокислый каль-
ций переходит в окись кальция. Привес тигля соответствует весу СаО и весу
золы фильтра. Зная вес золы фильтра, определяют вес СаО.
В(совое определение солей магния. Принцип метода основан
на том, что в фильтрате, полученном от предыдущего анализа, связывают
магний при помощи двузамещенного фосфата натрия—Na2HPO4 . 12Н2О.
При этом магний выпадает в виде фэсфэрноаммониевомагнезиальной соли —
Mg(NH4)PO4. Реакция идет по следующему уравнению:
MgCl2 + NH3 + Na2HPO4 = Mg(NH4)PO4 + 2NaCl.
Реактивы: 1. Соляная кислота.
2. Раствор двузамещенного фосфата натрия или аммония.
3. 10% раствор аммиака.
4. 2,5% раствор аммиака.
5. Азотная кислота.
6. Раствор AgNO3.
Методика определения. 200 мл фильтрата, полученного от
предыдущего анализа, подкисляют соляной кислотой, добавляет в избытке
раствор фосфата натрия (двузамещенного) или фосфата аммония, нагревают до
кипения, после чего доливают с^азу 10% раствор аммиака в количестве,
равном одной трети объема жидкости.
Дают отстояться, после охлаждения отфильтровывают через уплотненный
фильтр и промывают 2,5% раствором аммиака до исчезновения реакции на
хлориды. Фильтр с осадком высушивают, затем осадок перемещают в пла-
тиновый тигель, а фильтр с остатком осадка сжигают в платиновой спирали.
Золу стряхивают в тигель с осадком и прокаливают сначала на малом, а потом
на сильном пламени до постоянного веса. При этом фосфэрноаммониевомаг-
незиальная соль превращается в пирофосфорномагнезиальную соль:
2Mg(NH4)PO4 = Mg2P2O7 + 2NH3 + Н2О.
1 Проба с азотнокислым серебром после предварительного подкисления фильтрата
азотной кислотой.
325
Для перечисления на MgO полученный вес соли умножают на 0,3623 со-
ответственно: -дд^р ® 0,3623.
Mg2P2O7 222,6
Полученный вес окиси магния для перечисления на окись кальция умно-
. . СаО 56,08 , л
жают на 1,4 соответственно -ттгл-т? ~ 1,4.
MgO 40,32
Пример. Для анализа взято 500 мл воды. Окиси кальция оказалось 55 мг, что на литр
соответствует ПО мг. В 200 мл фильтрата оказалось 36 мг MgO; отсюда в 250 мл жидкости
36 . 250 л
количество окиси магния равно: —200—=45 мг»а эт0 на литр воды соответствует 90 мг. При
перечислении на окись кальция получаем 90 . 1,4 = 126 мг. Добавляя к весу окиси кальция,
получаем 110 4- 126 = 236, что отвечает 23,6° жесткости.
Вычисление общей жесткости на основе определения кальция и магния
может быть произведено также по формуле:
Ж0=Са^ + ^2,
7,14 4,33
где — общая жесткость воды в градусах,
Ся++ — содержание кальция в воде в мг/л,
— содержание магния в воде в мг/л,
7,14 — коэфициент пересчета Са++ в градусы жесткости,
4,33 — коэфициент пересчета Mg++ в градусы жесткости.
Щелочные металлы (Na п К)
Щелочные металлы выражаются через окись натрия (Na2O) и окись калия
(К2О) или через соответствующие ионы Na* и К*.
Качественное определение не производится ввиду постоянного
присутствия солей Na и К в питьевых водах.
Количественное определение щелочных металлов в воде произво-
дится только в полных санитарно-химических анализах.
Весовой метод определения натрия и калия.
Принцип метода основан на сжигании органических веществ
плотного остатка воды и последовательном отделении всех минеральных
веществ, сопровождающих щелочные металлы.
Реактивы. 1. Соляная кислота разбавленная.
2. Хлористый барий 10%.
3. Раствор едкого барита (насыщенный).
4. Щавелевокислый аммоний.
Методика определения. Выпаривают в платиновой чашке от
1 до 5 л испытуемой воды. Сжигают органические вещества. Остаток раство-
ряют в небольшом количестве дестиллированной воды, подкисляют соляной
кислотой и выпаривают досуха. Осадок смачивают разбавленной соляной
кислотой и снова выпаривают досуха. Прибавляют несколько капель раз-
бавленной соляной кислоты, а затем горячей воды и фильтруют. На фильтре
остается SiO2. Осадок на фильтре промывают несколько раз горячей водой.
К собранному фильтрату прибавляют 15—20 мл насыщенного раствора едкого
барита и 5 мл 10% хлористого бария и нагревают. В осадок выпадают в виде
окислов соединения глинозема, железа, кальция, магния и марганца; соеди-
нения серной, фосфорной и угольной кислоты выпадают в виде баритовых
солей Жидкость вместе с осадком переводят в мерную колбу на 250 мл, по охла-
ждении доливают до метки дестиллированной водой и фильтруют. 200мл филь-
трата переводят в платиновую чашку и для удаления избытка едкого барита
прибавляют по каплям раствор углекислого аммония при нагревании, пока не
326
перестанет выпадать осадок углекислого бария. Фильтруют в колбу на 250 мл,
осадок на фильтре промывают дестиллированной водой и доводят фильтрат
до метки водой. Переводят из колбы в платиновую чашку 200 мл фильтрата
и прибавляют сюда несколько капель раствора оксалата аммония для осажде-
ния оставшихся следов кальция и бария, выпаривают, а затем высушивают
при Ю0°С и осторожно прокаливают для удаления избытка оксалата аммония.
Остаток растворяют в небольшом объеме горячей дестиллированной воды
и фильтруют. На фильтре остаются соли кальция и бария. Осадок на фильтре
промывают горячей водой, собранный фильтрат высушивают в платиновой
чашке, прибавляя несколько капель соляной кислоты для перевода угле-
кислых солей Na и К в хлористые. Выпаривают и осторожно прокаливают,
охлаждают в экссикаторе и взвешивают. Из общего веса вычитают вес чашки
25 ,
и полученную разность умножают на (так два раза по ходу анализа
из 250 мл отбирали по 200 мл). Произведение соответствует суммарному ко-
личеству хлоридов Na и К.
Для отделения Na от К растворяют хлориды в небольшом количестве
дестиллированной воды, прибавляют сюда несколько капель разбавленной
серной кислоты (1 : 3) и по 1 мл 10% хлорной платины (PtCl4) на каждые
30 мг смеси хлоридов. Затем упаривают до густоты сиропа на водяной бане.
Сняв чашку с бани, дают содержимому высохнуть при комнатной температуре.
Остаток обрабатывают 80% винным спиртом и фильтруют. Осадок на фильтре
промывают 80% спиртом, пока фильтрат не сделается совершенно бесцветным.
Осадок вместе с фильтром сушат и затем растворяют осадок в горячей воде.
Раствор выпаривают досуха в платиновой чашке и взвешивают. Разность
между полученным весом и весом чашки соответствует хлорплатинату калия
(K2PtCl6).
Для пересчета полученного веса хлорплатината калия на окись калия
К о
его умножают на 0,1937 соответственно: ‘
Для пересчета на калий умножают на 0,1609 соответственно:
2КС1
Для пересчета на хлорид калия умножают на 0,3067 соответственно:
Из общей суммы хлоридов натрия и калия вычитают вес хлорида калия
и получают вес хлорида натрия. Для пересчета хлорида натрия на окись
гч гоно Na2O
умножают на 0,5303 соответственно: ттгу-угг-
Для пересчета хлорида натрия на натрий умножают на 0,3934 соответ-
Na
СТВеНН0: NaCl •
Согласно стандартной методике определение Na и К в воде проводится
следующим образом
Объем пробы воды для определения содержания натрия и калия должен
быть не менее 1500 мл. Если определение щелочей должно начаться более
чем через три дня после взятия пробы, то последняя должна быть взята в
склянку, изнутри покрытую парафином, илисклянку из безнатриевого стекла.
Весовой метод определения натрия
Принцип метода основан на осаждении натрия цинкуранил-
апетатом в виде натрцинкуранилацетата — NaZn(UO2)3(C2H3O2)9. 6Н2О9. Вы-
сушенный осадок взвешивают и пересчитывают на натрий.
Реактивы. 1. Уранил уксуснокислый — UO2(C2H3O2)2 . 2Н2О.
2. Цинк уксуснокислый — Zn(C2H3O2)2 . 2Н2О.
3. Кислота уксусная — СН3СООН(30% раствор).
327
4. Спирт этиловый ректификованный.
5. Эфир этиловый. ’
б. Раствор цинкуранилацетата: растворяют 10 г уксуснокислого уранила
в 20 мл дестиллированной воды и 50 мл 30% уксусной кислоты, нагревая
смесь до полного растворения уксуснокислого уранила. Отдельно растворяют
при нагревании 30 г уксуснокислого цинка в 10 мл 30% уксусной кислоты
и 50 мл дестиллированной воды. Первый и второй растворы в горячем состоя-
нии смешивают и оставляют стоять на сутки. Если выпадет осадок, то раствор
сливают с осадка, профильтровывают и хранят в парафинированной изнутри
склянке. Если при хранении выпадает осадок, то раствор перед употребле-
нием профильтровывают.
7. Насыщенный спиртовый раствор натрциикуранилацстата, применяе-
мый для промывания осадка. Для получения раствора берут 5—10 мл раствора
цинкуранилацетата, полученого так, как описано выше, и добавляют в него
по каплям при перемешивании насыщенный раствор поваренной соли. Выпав-
ший осадок отфильтровывают и отмывают от хлор-иона несколькими порциями
этилового спирта, после чего осадок переносят в склянку на 100 мл заливают
90% этиловым спиртом и оставляют стоять б часов, время от времени взбал-
тывая. Если раствор насыщен, то на дне склянки должен оставаться осадок
нерастворившегося натрциикуранилацстата.
Методика определения. В зависимости от содержания на-
трия в воде берут для исследования от 100 до 1000 мл. Взятую пробу ней-
трализуют раствором едкого калия по метилоранжу и упаривают в фарфо-
ровой чашке на водяной бане до 15—30 мл. Затем переносят в платиновую
чашку и упаривают до объема 1—Змл, после чего прибавляют в чашку 16—
30 мл раствора цинкуранилацетата, перемешивают тщательно стеклянной
палочкой и оставляют стоять на 2 часа при комнатной температуре.
Выпавший осадок натрцинкуранилацетата отфильтровывают при помощи
высушенного при 50°С и взвешенного фильтровального тигля с пористой
стеклянной пластинкой №4, применяя при этом отсасывание при пониженном
давлении. Фильтратом пользуются для смывания осадка с платиновой чашки.
Перенесенный на тигель осадок промывают насыщенным спиртовым раство-
ром натрцинкуранилацетата, отсасывая промывной раствор. На все промы-
вание расходуют малыми пропорциями 30—50 мл спиртового раствора, каж-
дый раз отсасывая полностью прибавленную порцию раствора. Затем таким
же образом промывают осадок на фильтре эфиром и, наконец, для удаления
остатков эфира просасывают через фильтр воздух в течение 10—15 минут.
Тигель после этого помещают на 2 часа в экссикатор и взвешивают. Привес
соответствует натрцинкуранилацетату. Для пересчета на Na умножают по-
лученный привес на 0,01495 соответственно: -кт „/1тА ч
NaZn (UO2)3 • (С2Н3О2)9 • 6Н2О
и перечисляют на литр воды.
Описанный метод применим при следующих условиях:
1. Отношение содержания калия к натрию не должно быть более 25.
2. В водах, содержащих более 300 мг/л сульфат-иона, последний следует
удалить осаждением хлористым барием (еще до упаривания взятой пробы
воды), при этом хлористый барий должен быть взят без избытка.
3. При высоком содержании органических веществ (цветность более 50°)
исследуемую воду упаривают досуха, осторожно прокаливают осадок до
обугливания органических веществ и, растворив его в небольшом количестве
горячей дестиллированной воды, отфильтровывают от частичек угля, а также
нерастворившихся карбонатов и окисей щелочно-земельных металлов.
Объемный метод определения калия
Принцип метода основан на осаждении калия в виде нитроко-
бальтиата калия-натрия и определении количества этой соли в осадке по
расходу марганцевокислого калия на ее окисление.
323
Реактивы. 1. Натрий азотистокислый — NaNO2 (10% раствор).
2. Кобальт хлористый—Со’С12 . 6Н2О (10% раствор).
3. Хлористый натрий (насыщенный раствор).
4. Калий марганцевокислый — КМпО4 (0.05 N раствор).
5. Кислота щавелевая — С2Н?О4 . 2Н2О (0,05 N раствор).
6. Кислота уксусная СН3 С ООН; 1 объем уксусной кислоты смешивают
с 9 объемами воды.
7. Кислота серная удельного веса 1,84; 1 объем серной кислоты смешивают
с 2 объемами воды.
8. Кислота серная (1,0 N раствор).
9. Спирт этиловый ректификованный; 1 объем спирта и 1 объем воды.
Методика определения. 100—500 мл профильтрованной воды
вносят в фарфоровую чашку, подкисляют серной кислотой (раствор N7) до
кислой реакции по метилоранжу и добавляют еще 1 мл той же кислоты.
Подкисленную воду упаривают в фарфоровой чашке на водяной бане,
переносят в платиновую чашку, выпаривают досуха, после чего осторожно
прскаливают до побеления осадка.
По остывании растворяют осадок в горячей воде и фильтруют через про-
мытый беззольный плотный фильтр, тщательно промывая фильтр водой.
Фильтрат и промывные воды переносят в платиновую чашку и упаривают
на водяной бане досуха.
Берут вторую такую же чашку для контроля и промывают ее дестиллиро-
ванной водой.
Затем в каждую из этих чашек вводят последовательно по 10 мл насыщен-
ного раствора хлористого натрия, 10 мл 10% раствора хлористого кобальта
и 15 мл 10% раствора азотистокислого натрия.
Содержимое чашек упаривают на водяной бане до кашицеобразного со-
стояния, перемешивая стеклянными палочками. Оставив палочки в чашках,
охлаждают последние до комнатной температуры, добавляют по 10 мл уксус-
ной кислоты (раствор N б), перемешивают содержимое чашек и через полчаса
добавляют при перемешивании по 10 мл дестиллированной воды. Оставляют
на 12—16 часов. После этого содержимое чашек медленно фильтруют через
фильтровальные тигли со стеклянными пористыми пластинками № 4, пе-
ренося на фильтры весь осадок из чашек. Остатки осадка из чашек смывают
на фильтры разбавленным раствором спирта (раствор N 9), подкисленным
уксусной кислотой (раствор N6). Для подкисления 100 мл спирта берут
4—5 мл уксусной кислоты. Осадки на фильтрах промывают тем же раство-
ром, после чего фильтры с осадками высушивают в течение 2 часов при
80°С.
В два чистых стакана помещают по 50 мл 0,05 N раствора КМпО4, закры-
вают часовыми стеклами, нагревают быстро до кипения, вливают по 5 мл
серной кислоты (раствор N 8) и вновь нагревают до кипения. Снимают ста-
каны с нагревательного прибора и опускают в горячий раствор фильтры
с осадками. Наклоняя и поворачивая стаканы, стараются тщательно промыть
дно и стенки фильтров. Через минуты две вливают в каждый стакан по 50 мл
0,05 N раствора щавелевой кислоты и вновь нагревают до 70—80°С, добавив
в контрольную пробу еще 10 мл 0,05 N раствора щавелевой кислоты. Сняв
стаканы с нагревательного прибора, оставляют их на 40—60 минут до полного
обесцвечивания фильтрующих пластинок. Нагревают стаканы до 70—80°С
и титруют 0,05 N раствором марганцевокислого калия до слаборозовой ок-
раски.
На основании расхода марганцевокислого калия на титрование опытного
й контрольного растворов определяют количество калия в воде по следующей
формуле:
[Р1_ (р2_ р3)]К • 0,344 • 1000
л. ——--------------------------,
32g
где X — количество калия в мг/л,
иг — количество миллилитров 0,05 N раствора КМпО4, израсходован-
ное на титрование пробы исследуемой воды,
v2 — то же на титрование контрольной пробы,
и3—количество миллилитров 0,05 N раствора КМпО4, расходуемое на
титрование 10 мл 0,05 N раствора щавелевой кислоты (опреде-
ляется отдельным титрованием),
К — коэфициент нормальности 0,05 N раствора КМпО4,
щ — объем испытуемой воды в миллилитрах,
0,344— коэфициент пересчета 1 мл 0,05 N раствора КМпО4 на1 мг калия.
Хлористоводородная кислота (хлориды)
Хлориды находятся в воде преимущественно в виде хлористого натрия,
меньше—в виде других хлористых солей (КС1, СаС12, MgCl2). Выражаются
в виде иона хлора — СГ.
Хлориды, находящиеся в воде, могут быть минерального и животного
происхождения. Хлориды минерального происхождения (солончаковый грунт)
не имеют санитарного значения, если их концентрация не приводит к порче
вкуса воды.
Хлориды животного происхождения указывают на загрязнение воды;
в этом случае налицо и другие показатели загрязнения воды: высокая окис-
ляемость, аммиак, нитриты и т. п.
Качественное определение
В пробирку наливают 10 мл исследуемой воды, подкисляют разведенной
азотной кислотой, свободной от СГ, и добавляют несколько капель раствора
азотнокислого серебра. В присутствии хлоридов в воде образуется хлористое
серебро (AgCl), которое дает белую муть.
Углекислые и фосфорнокислые соли дают также с азотнокислым серебром
белые осадки; однако они растворимы в азотной кислоте и при проведении
пробы, как указано выше, не мешают реакции.
Количественное определение
Количественное определение хлоридов в воде можно производить объемным
и весовым путем.
Объемное определение хлоридов
Объемное определение хлоридов в воде можно производить по методу
Мора или Фольгарда.
Метод Мора. Принцип метода основан на следующих реакциях:
NaCl + AgNO3 = AgCl -j- NaNO3
K2CrO4 + 2AgNO3 = Ag2CrO4 + 2KNO3.
Азотнокислое серебро в нейтральном водном растворе, содержащем смесь
хлористых и хромовых солей, сначала осаждает хлориды с образованием
хлорида серебра (первая реакция), после чего начинается образование хромо-
вокислого серебра (вторая реакция), придающего жидкости кирпичнокрасный
цвет, что указывает на конец осаждения хлоридов. По израсходованному
титрованному раствору азотнокислого серебра делают заключение о коли-
честве хлоридов в воде.
Реактивы. 1. Титрованный раствор азотнокислого серебра, 1 мл кото-
рого соответствует 1 мг хлора. Навеску азотнокислого серебра рассчитывают
на основании приведенной первой реакции. Из этой реакции видно, что одна
молекула азотнокислого серебра соответствует одному атому хлора, т. е.
330
169,888 весовых частей азотнокислого серебра (169,888—молекулярный вес
AgN03) соответствует 35,457 весовым частям хлора (35,457 — атомный вес
х 1 - 169,888 Л -70!
хлора); отсюда: 1 весовой части хлора соответствует _ ~ 4,791 весовой
□ 0 I
части азотнокислого серебра. Если на литр раствора взять 4,79 г азотнокислого
серебра, то весь литр будет соответствовать 1 г хлора, а 1 мл раствора — 1 мг
хлора. Раствор AgNO3 сохраняют в темной склянке.
Перед употреблением нужно проверять титр AgNO3 ввиду нестойкости
раствора. Для этой цели пользуются раствором хлористого натрия (см. ниже).
2. Раствор хлористого натрия, 1 мл которого соответствует 1 мг хлора. Для
получения такого раствора нужно в литре воды растворить 1,649 г химически
NaCl 58,454 ,
чистого высушенного хлористого натрия соответственно: ~с — = б>~Ге7~1>Ь49.
1 О J,4’J I
3. 10% нейтральный раствор К2СгО4, не содержащий хлоридов.
Методика определения. Сначала проверяют титр AgNO3 для
чего берут 10 мл раствора хлористого натрия, изготовленного вышеописан-
ным способом, добавляют 90 мл дестиллированной воды и получают 100 мл
раствора, отвечающего 10 мг хлора. Раствор титруют азотнокислым серебром
при индикаторе 10% растворе монохромата калия — К2СгО4 (несколько
капель). Реакция считается законченной, когда появляется стойкое кирпично-
красное окрашивание. Предположим, что при титровании пошло 11 мл ра-
створа. Следовательно, 1 мл AgNO3 отвечает мг хлора. Определив титр
раствора AgNO3, приступают к определению хлоридов в воде.
К 100 мл испытуемой воды добавляют несколько капель 10% раствора
монохромата калия (индикатор), после чего титруют азотнокислым серебром
до появления стойкого кирпичнокрасного окрашивания. По затраченному
количеству AgNO3 делают заключение о количестве хлоридов в воде, выра-
женных в хлор-ионе.
Пример. При титровании 100 мл воды ушло 5 мл раствора AgNO3, каждый милли-
литр которого соответствует 1? мг хлора.
Отсюда заключаем, что в литре исследуемой воды содержится:
10 • 5 • 10 . г- к
— 11-------- 45,5 мг хлора.
Вода, которая исследуется по описанному методу, должна быть нейтраль-
ной реакции. Если проба кислая, ее нейтрализуют содой; при наличии едких
щелочей прибавляют разбавленной серной кислоты до исчезновения на хо-
лоду окраски от фенолфталеина. При интенсивной цветности воду обесцве-
чивают продолжительным встряхиванием с промытой гидроокисью алюминия
(0.5 г на 500 мл пробы) и дают осадку осесть. Для определения берут освет-
ленную пробу.
Для получения гидроокиси алюминия растворяют 125 г калийных или
аммонийных квасцов в литре дестиллированной воды и осаждают алюминий
осторожным прибавлением аммиака. Гидроокиси дают выпасть, сливают воду
и промывают осадок по способу декантации дестиллированной водой до
освобождения от аммиака, хнорилов и нитритов.
Если концентрация хлоридов велика, то вода перед исследованием раз-
водится дестиллированной водой до концентрации 30—40 мг хлора на литр.
Необходимая степень разведения устанавливается предварительным титро-
ванием. Наоборот, если концентрация хлоридов очень мала, то воду упа-
ривают до 1/2—1/з первоначального объема. В результат анализа вводят
соответствующую поправку (при разведении результат множат на степень
разведения, при упаривании результат относят к первоначальному объему).
331
Метод Фольгарда. Принцип метода основан на том, что хлориды
осаждают титрованным раствором азотнокислого серебра, который берется
в избытке. Избыток затем оттитровывают роданистым аммонием в присутствии
железоаммиачных квасцов как индикатора. Определив избыток, находят,
сколько азотнокислого серебра связалось с хлоридами, и на основании най-
денного результата определяют количество хлоридов в воде. Реакции про-
текают по следующим уравнениям:
NaCl + AgNO3 = AgC! + NaNO3
AgNO3 + NHjCNS = AgCNS NH4NO3
(NH4)2SOiFe2(SO4)3 • 24H2O + 6NH4CNS = 2Fe(CNS)3 + ^NH^SO, 24H2O.
Из приведенных реакций видно, что роданистый аммоний связывает из-
быток азотнокислого серебра (реакция вторая). Когда этот избыток окажется
связанным, то избыточная капля роданистого аммония образует с железоам-'
миачными квасцами родановое железо — Fe(CNS)3 (реакция третья), прида-
ющее раствору красное окрашивание, что указывает на конец реакции. По
затраченному количеству азотнокислого серебра на связывание хлоридов
делают заключение о количестве их в воде. Исследование ведется в кислой
среде.
Реактивы. 1. 0,1 N раствор азотнокислого серебра: берут на литр
17 г AgNO3. Титр AgNO3 устанавливают по 0,1 N раствору хлористого натрия.
2. 0,1 N раствор хлористого натрия: 5,8454 г химически чистой высушен-
ной соли переводят в литровую колбу и приливают воды до метки, взбалты-
вая раствор.
3. Раствор железоаммиачных квасцов: готовят насыщенный на холоду
раствор железоаммиачных квасцов. Раствор приобретает бурое окрашивание
в силу гидролиза Fe2(SO4)3 по реакции:
Fe2(SO4)3 + 6Н2О 2Fe(OH)3 + 3H2SO4.
Чтобы уменьшить гидролиз, подкисляют раствор крепкой азотной кислотой
(уд. веса 1,4) до исчезновения бурой окраски. Большого избытка HNO3 сле-
дует избегать.
4. 0,1 N раствор роданистого аммония — NH4CNS: растворяют 8 г этой
соли в литре воды и проверяют титр при помощи 0,1 N раствора азотнокислого
серебра.
Методика определения. Сначала проверяют титр AgNO3, как
описано выше. По титрованному раствору AgNO3 устанавливают титр
роданистого аммония следующим образом: к 25 мл раствора AgNO3 при-
бавляют около 50 мл дестиллированной воды и 1мл насыщенного раствора
аммиачных квасцов и титруют раствором роданистого аммония до слабскрас-
ного окрашивания. Отдельно ставят холостой опыт и на основании обоих
титрований устанавливают титр NH4CNS.
Установив титры обоих растворов, приступают к определению хлори-
дов в воде.
100 мл исследуемой воды вливают в мерную колбу на 200 мл, добавляют
избыток азотнокислого серебра, подкисляют немного азотной кислотой, до-
водят до метки дестиллированной водой и, закрыв колбу, энергично взбал-
тывают. Потом отстаивают до тех пор, пока хлорид серебра не соберется на
дне, а раствор над ним не станет прозрачным, и фильтруют через сухой фильтр.
Первые 10 мл отбрасывают, берут 50 мл фильтрата, доливают 1 мл раствора
квасцов и титруют раствором роданистого аммония до стойкого розового
окрашивания.
На основании расхода роданистого аммония можно судить о том, сколько
осталось в избытке азотнокислого серебра, а отсюда сделать заключение
о количестве азотнокислого серебра, пошедшего на связывание хлоридов
и о количестве последних в воде.
332
Пример. Взято 100 мл воды, добавлено 20 мл раствора AgNO3. По осаждении хлоридов
и доведении объема до 200 мл жидкость отфильтрована. На связывание избытка AgNO3
в 50 мл фильтрата израсходовано 3,5 мл раствора NH4CNS.
При установке титра AgNO3 на 25 мл 0,1 N NaCl уходит 25,25 мл AgNO3.
При установке титра NH4CNS на 25 мл AgNO3 уходит 24,5 мл NH4CNS.
Нужно определить содержание хлоридов в исследуемой воде.
25
Определяем коэфициент нормальности раствора AgNO3. Он равен 25 25 • Следовательно,
20.25
к воде прибавлено 25 25 ^ *8,18 мл 0,1 N раствора AgNO3.
25 • 25
Определяем коэфициент нормальности раствора NH4CNS. Он равен 25~25^245* Следо-
3,5-25-25
вательно, к фильтрату прибавлено 2^25.24“^" ~ 3,53 мл 0,1 N раствора NH4CNS.
Отсюда заключаем, что на связывание избытка AgNO3 во всей пробе потребуется 3,53 .
.4 = 14,12 мл 0,1 N раствора NH4CNS. Следовательно, на связывание с хлоридами во
всей взятой пробе пошло бы 18,18— 14,12 = 4,06 мл 0,1 N раствора AgNO3. 1 мл 0,1 N
раствора AgNO3 отвечает 3,5457 мг СГ. Следовательно, в литре воды количество хлоридов,
перечисленное на СГ, равно 3,5457 . 4,06 . 10 = 143,96 мг.
Весовое определен.ie хлоридов
Принцип метода основан на том, что хлориды осаждаются при
помощи азотнокислого серебра. Выпавший хлорид серебра отделяется, вы-
сушивается, прокаливается и взвешивается.
Реактивы. 1. Азотная кислота, свободная от хлора.
2. Раствор азотнокислого серебра.
3. Раствор хлористого натрия.
4. Соляная кислота.
Методика определения. 500 мл исследуемой воды подкисляют
азотной кислотой, не содержащей хлора, и выпаривают до объема 100 мл.
К нагретому раствору добавляют в избытке раствор азотнокислого серебра,
помешивая стеклянной палочкой. Дают выпасть осадку и проверяют раствор
на полноту осаждения: на часовое стекло наносят каплю раствора AgNO3 и
сюда же добавляют стеклянной палочкой каплю исследуемого раствора (после
его осветления). Если осаждение произошло полностью, то проба на стекле
не дает мути; в противном случае ее смывают в раствор и продолжают осажде-
ние. Когда осаждение закончено, раствор оставляют на несколько часов
в темном месте, затем отфильтровывают через смоченный беззольный
фильтр. Осадок на фильтре промывают горячей дестиллированной водой,
подкисленной азотной кислотой. Промывание ведется до тех пор, пока фильт-
рат не перестанет давать муть с раствором хлористого натрия, что указывает
на полное удаление избытка азотнокислого серебра. Фильтр с осадком высуши-
вают, осадок отделяют, фильтр сжигают отдельно в платиновой спирали.
Золу собирают в фарфоровый (не платиновый) тигель, добавляют несколько
капель азотной кислоты и 1—2 капли соляной кислоты, выпаривают на во-
дяной бане. В тигель добавляют осадок, ранее отделенный и собранный на
листке глянцевитой бумаги, и прокаливают. Затем охлаждают в экссикаторе
и взвешивают. От полученного веса отнимают вес тигля и золы фильтра,
получая вес хлористого серебра. Результат перечисляют на хлор, умножая
С1
па 0,2474 соответственно:
AgCl
Серная кислота (сульфаты)
Сульфаты встречаются в воде в виде солей щелочно-земельных и щелоч-
ных металлов. Как и хлориды, сульфаты в воде могут быть минерального
и органического происхождения. Резкие колебания концентраций сульфатов
в воде свидетельствуют об их органическом происходжении.
Сульфаты выражаются в виде серного ангидрида (SO3) либо в виде
иона SO4".
ззз
Качественное определение
В пробирку наливают несколько миллилитров исследуемой воды, подкис-
ляют слабой соляной кислотой, добавляют раствор хлористого бария и на-
гревают до кипения. В присутствии солей серной кислоты появляется белая
муть, обусловленная образовавшимся сульфатом бария. Соляную кислоту
нужно добавлять для растворения других солей, которые могут выпасть
одновременно в осадок (карбонаты, фосфаты).
Количественное определение
Количественное определение сульфатов в воде производится объемным
и весовым методом.
Объемный метод
Принцип объемного метода, предложенного Комаровским,
заключается в том, что сульфаты воды связывают хромовокислым барием;
при этом освобождается эквивалентное количество хромовокислого калия
(натрия и пр.). Последний в кислой среде вытесняет из йодистого калия экви-
валентное количество иода, которое определяется титрованием тиосульфатом
натрия. По израсходованному на реакцию количеству тиосульфата натрия
делают вывод о количестве сульфатов в воде, перечисленных на SO3 или SO/'.
Ход реакции:
K0SO4 ВаСгО4 = BaSO4 -|- К2СгО4
2К2СгО4 + 16НС1 + 6KJ = 10КС1 + 2CrCl3 + 8Н2О + 3J2
J2 + 2Na2S2O3 = 2NaJ + Na2S4Ott.
Из приведенных реакций видно, что 1 мл 0,1 N раствора тиосульфата
натрия соответствует 2,667 мг SO3 или 3,2 мг SO4".
Реактивы. 1. 10% НС1.
2. Сухой хромовокислый барий (ВаСгО4); его готовят, осаждая из горя-
чего раствора хлористого бария хромовокислым калием. Осадок тщательно
промывают водой, подкисленной уксусной кислотой, затем чистой водой,
после чего высушивают.
3. Раствор NH4OH.
4. 20% раствор KJ.
5. Концентрированная НС1.
6. 0,1 N раствор Na2S2O3 . 5Н2О.
Методика определения. К 200 мл исследуемой воды прибав-
ляют 1 мл 10% НС1 и 0,2—0,5 г сухого хромовокислого бария. Нагревают
до слабого кипения, которое поддерживают 3 минуты, охлаждают на льду
или в токе водопроводной воды и подщелачивают аммиаком до слабощелочной
реакции на лакмус. При испытании реакции среды на лакмус продувают
воздух над жидкостью при помощи груши для удаления паров NH3. Пере-
водят жидкость в колбу на 250 мл , доводят до метки водой и фильтруют. 100 мл
фильтрата переводят в коническую колбу, в которую предварительно нали-
вают 5 мл 20% К J и 5 мл концентрированной НС1. Колбу ставят в холодную
воду на 15 минут и выделившийся иод оттитровывают 0,1 N раствором тио-
сульфата натрия. Результат титрования умножают на 12,5 и на соответствую-
щий коэфициент 2,667 или 3,2 соответственно:
Весовой метод
Принцип метода основан на осаждении сернокислых солей при
помощи хлористого бария по реакции:
Na2SO4 + ВаС12 = BaSO4 + 2NaCI
334
Выпавший сульфат отфильтровывают, прокаливают и взвешивают. Ре-
зультат перечисляют на SO3 или SO4"
Реактивы. 1. Разбавленная соляная кислота.
2. Разбавленная серная кислота.
3. 3—5% раствор хлористого бария.
4. Раствор AgNO3.
Методика определения. 200—300 мл исследуемой воды под-
кисляют слабой соляной кислотой, упаривают до объема в 100 мл и к горячей
жидкости добавляют по каплям из пипетки теплый 3—5% раствор хлористого
бария. Это добавление производят до тех пор, пока происходит осаждение
сернокислого бария. Когда образование мути от прибавления хлористого ба-
рия прекращается, производят пробу на полноту осаждения. На часовое
стекло переносят стеклянной палочкой 1—2 капли отстоявшейся жидкости
и добавляют сюда столько же раствора хлористого бария. Отсутствие мути
показывает, что в растворе нет сернокислых солей. Можно также к 2—3 ка-
плям отстоявшегося раствора добавить немного серной кислоты. Если осажде-
ние закончено и в жидкости имеется избыток хлористого бария, то при второй
пробе должна появиться муть (контроль).
По окончании осаждения жидкость кипятят 15—20 минут. Осадку дают
выпасть, на что требуется несколько часов спокойного стояния; после этого
жидкость фильтруют через уплотненный спиртом беззольный фильтр, декан-
тируя осадок горячей водой и потом промывая его на фильтре такой же водой,
пока промывная вода не перестанет давать реакцию на хлор (проба с AgNO3)
или на барий (проба с H2SO4). Фильтр вместе с осадком высушивают. Сухой
остаток переносят в платиновый тигель; фильтр сжигают отдельно в платино-
вой спирали; золу добавляют в тигель к осадку и прокаливают тигель до
тех пор, пока зола не станет белой. Затем тигель охлаждают и взвешивают.
Отняв от полученного результата вес тигля и золы фильтра, определяют
вес сульфата бария. Результат для перечисления на SO3 умножают на 0,343
SO
соответственно: а для перечисления на SO4" умножают на 0,4115 со-
ьаьо4
so4
ответственно: •„ *
BaSO4
Пример. Для анализа взято 200 мл воды. По осаждении сульфатов вес сульфата бария
оказался равным 30 мр.
Отсюда заключаем, что на литр воды приходится:
30 • 0,343 • 1000 t t 30 • 0,4115 . 1000 в
------2оо------= 51,45 mp«SO3 или -------200-------= 61,725 SO/*.
АЗОТСОДЕРЖАЩИЕ ВЕЩЕСТВА
Азотсодержащие вещества воды образуются преимущественно за счет
разложения белковых веществ, которые попадают в водоисточник с бытовыми
и промышленными сточными водами. Одним из продуктов распада белковых
веществ является аммиак, который в присутствии кислорода подвергается
окислению в азотистую кислоту, а последняя — в азотную кислоту.
Наличие в воде аммиака при отсутствии нитритов и нитратов указывает
на недавнее загрязнение воды; совместное присутствие аммиака, нитритов
и нитратов указывает на то, что аммиачные соединения находятся в стадии
минерализации; наконец, наличие в воде одних нитратов указывает на то,
что аммиачные соединения успели подвергнуться уже полной минерализации.
Необходимо иметь в виду, что азотсодержащие вещества воды могут быть
и чисто минерального происхождения: нитриты и нитраты, заносимые атмо-
сферными водами с поверхностных слоев почвы в глубинные слои воды, могут
подвергаться здесь восстановлению до аммиака преимущественно сероводо-
родом, образующимся из сульфидов (колчеданов) при воздействии на них
СО2. Таким образом, наличие одних азотсодержащих веществ при отсутствии
335
других показателей загрязнения не может еще свидетельствовать о недобро-
качественности воды особенно, когда речь идет о глубинных водах (артезиан-
ские колодцы).
При анализе питьевых вод на азотсодержащие соединения определяют:
азот аммиака и аммонийных солей (свободный и солевой аммиак); азот альбу-
миноидный (альбуминоидный аммиак); азот нитритов (азотистая кислота);
азот нитратов (азотная кислота); азот органический и азот общий.
Азот аммиака п аммонийных солей (свободный и солевой аммиак)
Выражается в виде (NH3).
Качественное определение
К 10—15 мл воды прибавляют 3—5 капель реактива Несслера. В присут-
ствии свободного или солевого аммиака появляется желтое окрашивание от
образования йодистого меркураммония: NH2Hg2OJ (см. стр. 252).
Предел чувствительности реакции 0,1 мг NH3 в 1 л воды.
Если вода окрашена, то ее предварительно обесцвечивают, для чего на
100 мл исследуемой воды прибавляют 0,1 г гидроокиси аллюминия (приго-
товление см. стр. 331), взбалтывают и отстаивают в узком цилиндре в течение
2 часов. Для пробы отбирают пипеткой 10—15 мл.
Количественное определение
Количественное определение в воде аммиака или солей аммония произ-
водят колориметрически при помощи реактива Несслера.
Принцип метода. Тот же, что и при качественном определении.
Реактивы. 1. Стандартный раствор NH4C1, 1,0 мл которого соответст-
вует 0,1 мг NH3 (приготовление см. стр. 253).
2. Реактив Несслера (приготовление см. стр. 253).
3. Раствор сегнетовой соли — KNaC4H4OG. 4Н2О. Растворяют 50 г соли
в 100 мл горячей воды. Фильтруют через стеклянную вату, добавляют 5 мл
реактива Несслера. Полученный раствор хранят в склянке темного стекла.
Если нужно фильтруют вновь через стеклянную вату.
4. Раствор едкого натрия и карбоната натрия. 50 г NaOH и 100 г Na2CO8
растворяют в 300 мл дестиллированной воды, свободной от аммиака. Раствор
кипятят 15 минут и фильтруют через стеклянную вату.
5. Раствор уксуснокислого цинка — Zn(CH3COO)2 — 30%.
Методика определения. В один из двух парных цилиндров
колориметра наливают 100 мл исследуемой воды, в друю i — 1 мл стандарт-
ного раствора NH4C1 (реактив 1), второй цилиндр доливают до 100 мл безам-
миачной дестиллированной водой. В оба цилиндра прибавляют по 3 мл рас-
твора сегнетовой соли и затем по 2 мл реактива Несслера. Содержимое цилин-
дров перемешивают стеклянными палочками. Помещают цилиндры на белый
фон и уравновешивают окраску. Из цилиндра, в котором столб жидкости
окрашен более интенсивно, выпускают понемногу жидкость через кран, пока
не будет достигнута одинаковая окраска.
Пример Положим, что для получения окрашивания одинаковой интенсивности при-
шлось из стандартного цилиндра вылить 45 мл и, таким образом, осталось 60 мл жидкости.
Отсюда делаем вывод, что в 100 мл исследуемой воды содержится мг NH3, а в лигре
0,1 • 60 • 10 п ~ 10э
—-------~0,э7 мг NH,.
Юз 3
Если для получения одинаковой интенсивности окраски приходится вылить, напр.,
30 мл из цилиндра с исследуемой водой, то значит, в 100 мл исследуемой воды содержится
0,1 • 105 1,0 -105-10 1 л NIU1
------мг, а в литре —----- «= 1,4 мг NH,.
75 75
Примечание 1. Если по ходу анализа окажется, что для выравнивания
окраски слой исследуемой воды должен быть уменьшен до величины меньшей чем
336
’ v3 слоя стандартного раствора, то определение нужно повторить, приготовив более
концентрированный стандартный раствор.
Примечание 2. При содержании азота аммиака и аммонийных солей
более 5 мг/л исследуемую воду предварительно соответственно разбавляют безам-
миачной дестиллированной водой.
Примечание 3. Цветность, жесткость, наличие в воде железа, марганца,
алюминия, сульфидов и свободной углекислоты влияют на точность определения,
и потому воду нужно в этих случаях подвергнуть соответствующей обработке. Цвет-
ность устраняют так же, как и при качественном определении. При жесткости более
10° воду умягчают, прибавляя на каждые 100 мл ее 2 мл раствора едкого натрия и
соды (реактив 4). Взбалтывают, отстаивают и прозрачную жидкость сливают или
сифонируют для анализа. Одновременно с умягчением устраняются соли железа,
марганца и алюминия, свободная углекислота. При жесткости меньше 10° к исследуе-
мой воде прибавляют раствор сегнетовой соли по ходу анализа, как было указано
выше. При наличии в воде сульфидов на каждые 100 мл воды прибавляют по 10 ка-
пель 30% раствора уксуснокислого цинка (реактив 5); после этого воду отстаивают
в течение 2 часов и прозрачную часть сливают для исследования.
Для быстрого колориметрирования в лабораториях пользуются иногда
колориметром, изображенным на рис. 231. Он представляет собой шестигран-
ную призму, грани которой оклеены бумажными
полосками, окрашенными в цвета от желтого до
Желтокрасного. Тут же обозначено содержание
аммиака в 1 л воды, которое дает соответствующее
Окрашивание. Цилиндр с исследуемой водой ставят
на специальную подставку сбоку призмы. Добавив
раствор сегнетовой соли и реактив Несслера и
хорошо перемешав воду, сравнивают полученный
цвет с цветом граней призмы, поворачивая по-
следнюю вокруг вертикальной оси. Найдя грань,
окрашенную в соответствующий жидкости цвет,
прочитывают концентрацию, обозначенную на ней1.
В тех случаях, когда вода сильно загрязнена
или не поддается обесцвечиванию, или общее коли-
чество солей в ней выше 3000 мг/л, определение
аммиака или солей аммония производится после
предварительной отгонки воды.
Собирают перегонный аппарат, как изображено на рисунке 232. В колбу
А емкостью в 800—1000 мл наливают 200 мл дестиллированной воды, прибав-
ляют 10 мл фосфатной буферной смеси, приготовленной растворением 14,3 г
однозамещенного фосфата-калия (КН2РО4) и 90,15 г двузамещенного фосфата-
калия (К2НРО4 ЗН2О) в 1 л воды. Вводят в колбу несколько кусочков про-
каленной пемзы или несколько капилляров для равномерного кипения и
отгоняют воду через холодильник В в колбу С. Испытывают дестиллят на
присутствие аммиака’. Первые порции дестиллята обыкновенно содержат
следы аммиака, находившиеся в дестиллированной воде или в воздухе прибора.
Когда убеждаются, что пошел дестиллят, свободный от аммиака, то, не удаляя
содержимого перегонной колбы, вводят в нее 300—500 мл испытуемой воды
и, продолжая нагревание, отгоняют 200 мл в чистую колбу, причем ранее
полученный дестиллят отбрасывают.
Весь аммиак, содержавшийся во взятой пробе, перейдет в погон. Для
контроля одну каплю дестиллята подхватывают лакмусовой бумажкой и
наблюдают окраску.
Дестиллят подвергают исследованию на аммиак точно так, как описано
выше.
Описанный метод определения аммиака в дестилляте применяется и тогда,
когда вслед за определением солевого аммиака имеется в виду произвести
определение альбуминоидного аммиака.
Рис. 231. Колориметр в виде
призмы с эталонами на гра-
1 Подобные колориметры имеются и для определения нитритов, нитратов, железа и пр.
22 А. и. Бурштейн Э37
Азот альбуминоидный (альбуминоидный аммиак)
Выражается в виде (NH3).
Принцип метода. После отгона солевого NH3 в оставшейся жид-
кости разрушают азотсодержащие органические вещества белкового про-
исходжения, превращая их азот в NH3, который подвергают отгонке и опре-
деляют в дестилляте, как описано выше.
Реактивы. Те же, что и для описанного выше метода, а также щелоч-
ной раствор КМпО4 для разрушения альбуминоидных веществ. Этот раствор
готовится следующим образом: 200 г КОН и 8 г КМпО4 растворяют
в 1 л дестиллированной воды. Раствор упаривают в колбе до половины
объема.
Методика определения. К жидкости, оставшейся после отгона
солевого аммиака, прибавляют 100 мл дестиллированной воды, свободной
от аммиака, и 10 мл щелочного раствора КМпО4 и продолжают дестилляцию.
Собирают 100 мл дестиллята в цилиндре и определяют аммиак колориме-
трически при помощи реактива Несслера.
Азот нитритов (азотистая кислота)
Выражается в виде азотистого ангидрида (N2O3) либо в виде иона (NO2).
Качественное определение
1. С реактивом Грисса (приготовление см. на стр. 227). В пробирку к
исследуемой воде добавляют несколько капель реактива. В присутствии азо-
тистой кислоты появляется розовое окрашивание различной интенсивности
от слабого до яркорозового в зависимости от концентрации нитритов в воде.
Для увеличения чувствительности пробу подогревают на водяной бане в те-
чение нескольких минут при температуре 70—80°С. Проба специфична для
нитритов. Предел чувствительности 0,01 мг N2O3 в литре воды. Получаемая
окраска неустойчива.
2. С сульфаниловой кислотой и фенолом. В 100 мл насыщенного раствора
NH4C1 растворяют при нагревании 1 г сульфаниловой кислоты (C6H4NH2 .
. SO3H), после чего добавляют 1,5 г фенола (С6Н5ОН) и 100 мл
2N НС1.
Пробу производят следующим образом: к 50 мл исследуемой воды при-
бавляют 1 мл указанного реактива и через 15 минут добавляют 10% раствор
аммиака до щелочной реакции. В присутствии нитритов появляется стойкое
желтое окрашивание. Предел чувствительности реакции 0,1 мг N2O3 в литре
воды.
Этот метод, хотя и мало применяется у нас в лабораториях, заслуживает
внимания особенно для количественных определений нитритов, так как в от-
личие от реактива Грисса указанный реактив дает стойкое окрашивание,
которое можно воспроизвести при помощи К2Сг2О7, получив устойчивую
шкалу.
3. С дифениламином (приготовление см. на стр. 256). Реактив, изготовлен-
ный по указанному рецепту, специфичен только для нитритов. Прибавление
реактива к воде, содержащей нитриты, вызывает появление голубой или
синей окраски. Предел чувствительности реактива 0,1 мг азотистой кислоты
в литре воды.
4. С бруцином. Реактив готовится растворением 0,25 г бруцина (C23H26N2O4)
в 100 мл H2SO4 удельного веса 1,84.
1 мл исследуемой воды смешивают с 2 мл реактива. В присутствии нитри-
тов появляется розовая или красная окраска, быстро переходящая в желтую.
Реакция не специфична для нитритов: такое же окрашивание получается
и с нитратами.
338
Количественное определение
Количественное определение нитритов в воде производится чаще других
методов при помощи реактива Грисса.
Принцип метода тот же, что и при качественной реакции.
Реактивы. 1. Реактив Грисса.
2 . Стандартный раствор NaNO2. Растворяют 1,816 г химически чистого
/ 2NaNO \
NaNO2 ^соответственно —j в литре воды. 1 мл такого раствора соответ-
ствует 1 мг No03. Если расчет желательно вести на NO2'-hoh, то в литре воды
“ / NaNO \
растворяют 1,4998 г NaNO21 соответственно 2j. 1 мл такого раствора
соответствует 1 мг NO2'. Раствор консервируют прибавлением 1 мл хлоро-
форма на литр. Ввиду малых концентраций нитритов в воде указанный ра-
створ перед употреблением разбавляется в 100 раз. 1 мл слабого раствора
соответствует 0,01 мг N2O3 или 0,01 мг NO2' в зависимости от приготовления
исходного раствора.
Методика определения. Определение ведется по методу стан-
дартных серий или по методу уравнивания окраски.
Метод стандартных серий: составляется стандартный ряд из 10 одинако-
вых пробирок. В первую вводят 0,1 мл стандартного раствора, во вторую
0,2 мл и т. д. до 1 мл. Содержимое каждой пробирки доводят до 10 мл дестил-
лированной водой. В такую же пробирку вводят 10 мл испытуемой воды.
Во все пробирки прибавляют по 1 мл реактива Грисса и, если нужно, на-
гревают в течение нескольких минут в водяной бане до 70—80°С. Подбирают
стандартную пробирку, где жидкость окрашена так же, как в опытной про-
бирке. На основании содержания стандартного раствора в подобранной про-
бирке делают заключение о содержании нитритов в воде.
Пример. Положим, что одинаковую окраску с опытной пробиркой дала третья стан-
дартная пробирка. Отсюда заключаем, что в 10 мл испытуемой воды содержание нитритов
соответствует 0,003 мг N2O3 (или NO/). Следовательно, содержание нитритов в литре воды
соответствует 0,003. 100 = 0,3 мг N2O3 (или NO/).
Определение можно вести и в цилиндрах.
Метод уравнивания окраски: положим, что исследование ведется при помощи
колориметра Дюбоска. В одну колбочку наливают 50 мл испытуемой воды,
в другую — 1 мл стандартного раствора и 49 мл дестиллированной воды.
В обе колбочки вводят по 1 мл реактива Грисса, взбалтывают, если нужно,
нагревают и быстро переводят содержимое той и другой колбочек соответст-
венно в кюветы колориметра, после чего приступают к колориметрированию.
Допустим, что уравнивание окраски достигнуто при высоте слоя жидкости в
стандартной кювете 40 мм и в опытной кювете — 50 мм. Отсюда делаем за-
0,01-40.1000 П1с
ключение, что в литре испытуемой воды содержится ------------= 0,16 мг
5U* 5U
N2O3 или NO/ в зависимости от того, каким стандартным раствором пользо-
вались при определении.
Азот нитратов (азотная кислота)
Выражается в виде азотного ангидрида (N2O6) или иона (NO3').
Качественное определение
1. С дифениламином. В фарфоровой чашке растворяют несколько кри-
сталликов дифениламина в крепкой серной кислоте, свободной от нитратов,
и добавляют 1—2 капли исследуемой воды. В присутствии азотной кислоты
появляется синее окрашивание.
339
Реакция проводится также при помощи реактива, который готовится
следующим образом: 0,085 г дифениламина — (C6H6)2NH2 — взбалтывают
с 1Фо мл H2SO4 (1 : 3), добавляют H2SO4 уд. веса 1,84 до растворения и раз-
бавляют такой же кислотой до 500 мл. Окрашивание то же.
Присутствие азотистой кислоты и солей окиси железа вызывает появление
такой же окраски. Азотистая кислота может быть разрушена добавлением
к воде, подкисленной серной кислотой, щепотки мочевины. Разрушение азо-
тистой кислоты идет по реакции: 2HNO2 + CON2H4 = 2N + СО2 + ЗН2О.
За 10—12 часов вода освобождается от азотистой кислоты. Если в воде при-
сутствуют и соли окиси железа, то они осаждаются раствором едкого натрия.
Предел чувствительности реакции 5 мг N2O5 в литре воды.
2. С бруцином. К 3 мл чистой серной кислоты в фарфоровой чашке до-
бавляют по каплям 1 мл исследуемой воды. Когда смесь охладится, добавляют
немного бруцина. В присутствии азотной кислоты появляется розовое или
красное окрашивание, переходящее затем в желтое.
Реакцию можно провести прибавлением в пробирке к испытуемой воде
0,25% раствора бруцина в крепкой серной кислоте. Окрашивание то же.
Эта реакция наиболее чувствительна на азотную кислоту. Предел чувстви-
тельности 1 мг N2O5 в литре воды. Азотистая кислота и соли окиси железа
мешают реакции и должны быть удалены, как и при дифениламине.
3. С реактивом Грисса. Пробу проводят после восстановления нитратов
в нитриты. К исследуемой воде, подкисленной серной кислотой, добавляют
немного порошкообразного цинка. Выделяющийся водород восстанавливает
азотную кислоту до азотистой, которая может быть определена реактивом
Грисса. Если в исследуемой воде одновременно с нитратами присутствуют
и нитриты, то они должны быть предварительно разрушены.
Количественное определение
Количественное определение нитратов производится колориметрически при
помощи бруцина или по пикратному методу.
Онределеняе нитратов при помощи бруцина
Принцип метода тот же, что и при качественном определении
нитратов.
Реактивы. 1. Раствор бруцина: 0,25 г бруцина растворяют в 100 мл
серной кислоты уд. веса 1,84. Готовится ex tempore.
2. Стандартный раствор азотнонатриевой соли (NaNO3): растворяют 1,574 г
хт хтгч 2NaNO3 ,
NaNO3 в литре воды соответственно — • 1 мл такого раствора соответ-
ствует 1 мг N2O5. Если расчет желательно вести на NO3-hoh, то в литре воды
растворяют 1,37 г NaNO3 соответственно J • 1 мл такого раствора соот-
ветствует 1 мг NO3 х.
Раствор перед употреблением разбавляют в 10 раз. 1 мл разбавленного
раствора соответствует 0,1 мг N2O5 (или 0,1 мг NO3).
Методика определения. Берут две колбочки по 50 мл. В одну
из них наливают 10 мл исследуемой воды, а в другую — 1 мл разведенного
стандартного раствора (1 мл раствора соответствует 0,1 мг N2O5 или NO3)
и 9 мл дестиллированной воды. Потом в каждую колбочку приливают по
20 мл раствора бруцина в серной кислоте; приливать раствор бруцина сле-
дует одновременно из двух маленьких колбочек, емкостью каждая по 20 мл.
Смесь взбалтывают и окрашенную жидкость из колб переводят одновременно
порознь в два мерных цилиндра Генера емкостью каждый по 100 мл.
1 Если вместо NaNO3 пользоваться KNO3, то берут соответственно 1,872 р или 1,6305 г.
340
Предварительно в цилиндры наливают по 73 мл дестиллированной воды.
В цилиндрах получается не по 103 мл смеси, а только по 100, так как
около 3 мл жидкости остается в колбочках во время переливания растворов
из одной в другую, а затем в цилиндры. Устанавливают в цилиндрах одина-
ковую окраску слоев жидкости, глядя сверху вниз, и на основании получен-
ных данных производят расчет.
Пример. Положим, что для уравнения окраски растворов пришлось из стандартного
цилиндра отлить 30 мл. Следовательно, в 10 мл исследуемой воды содержится 9а1 * 7^, а в
0 1 • 70 • 100 ИЮ
литре г?— -------= 7,0 мг N2O5 или NO' в зависимости от того, каким стандартным рас-
твором пользовались при определении.
Описанный метод дает хорошие результаты при концентрации нитратов
в пределах от 1 до 50 мг/л при пересчете па N2O5.
При больших концентрациях исследуемую воду разбавляют дестиллирр-
ванной водой; при меньших концентрациях воду упаривают в несколько
раз, повышая таким образом концентрацию нитратов.
Определение нитратов по пикратному методу
Принцип метода заключается в том, что при помощи сульфо-
фенолового реактива образуют с нитратами, находящимися в исследуемой
воде, пикриновую кислоту по реакции:
СсН5ОН + 3HNO3 = C6H2(NO3)2 ОН + ЗН2О.
Полученную пикриновую кислоту переводят затем при помощи аммиака
в пикрат аммония, обладающий яркожелтым цветом, что дает возможность
применить колориметрию: C6H2(NO3)2 + NH3 = C6H2(NO2)3ONH4.
Реактивы. 1. Сульфофеноловый раствор: 3 г кристаллической бесцвет-
ной карболовой кислоты растворяют в 37 г серной кислоты уд. веса 1,84, не
содержащей азотной кислоты.
2. Стандартный раствор NaNO3 (см. стр. 340).
3. 10% раствор NH4OH (уд. вес 0,96)
Методика определения. 10—100 мл исследуемой воды вы-
паривают в фарфоровой чашке на водяной бане досуха. К остатку прибавляют
1 мл сульфофенолового раствора, тщательно перемешивают стеклянной палоч-
кой и через 5 минут приливают 10—50 мл дестиллированной воды и 10 мл
10% раствора NH4OH.
Колориметрия проводится по методу стандартных серий или уравнивания
окраски, причем стандартные растворы подвергаются такой же обработке,
как и исследуемая вода.
Для получения стойких стандартных растворов можно воспользоваться
уксуснокислым ураном, дающим окраску, идентичную с той, которую дает
пикрат аммония. Растворяют 35 г уксуснокислого урана — (UrО2) (С2Н3О2)2—
в литре 1% борной кислоты; 1 мл такого раствора в 100мл 1% борной кислоты
соответствует приблизительно окраске пикрата аммония от 0,01 мг нитратного
азота. Из полученного раствора приготовляют стойкие эталоны, проверив их
по стандартным растворам, приготовленным из NaNO3 или KNO3.
Если вода мутна, то она до исследования коагулируется при помощи креп-
кого раствора сернокислого глинозема — A12(SO4)3 • 18Н2О. Для коагуляции
берется 0,2—1,0 мл раствора, приготовленного растворением 500 г серно-
кислого глинозема в литре воды.
Определение нитратов при помощи салициловой кислоты
(ускоренный метод)
Принцип основан на переводе салициловой кислотой нитратного азота воды в нитро-
производные фенола, образующие со щелочью соединения желтого цвета.
Реактивы. 1. 10% спиртовый раствор х. ч. салициловой кислоты.
341
2. Кислота серная уд. веса 1,84, свободная от азотной.
3. Раствор едкого натрия 30%.
4. Кобальт хлористый — СоС12 1% водный раствор.
5. 0,01 N раствор двухромовокислого калия —К2Сг2О7 (приготовление см. стр. 63).
Методика определения. 1мл исследуемой воды выпаривают досуха в фар-
форовой чашке, не допуская прокаливания. По охлаждении к сухому остатку прибавляют
3 капли раствора салициловой кислоты (реактив 1) и 0,5 мл. крепкой серной кислоты (реак-
тив 2). Сухой остаток тщательно перемешивают стеклянной палочкой с прибавленными
реактивами.
По истечении пяти минут прибавляют 5 мл дестиллированной воды и 3 мл раствора
едкого натрия (реактив 3). Содержимое чашки хорошо перемешивают и переводят в пробирку
с меткой на 10 мл и доливают до метки дестиллированной водой; вторично тщательно пере-
мешивают и переливают в пробирку с плоским дном и меткой на высоте 5 см. Полученную
окраску сравнивают с эталонами жидкой шкалы, рассматривая растворы сверху вниз.
Эталоны готовят следующим образом: в мерную колбу емкостью в 50 мл вносят в указанных
в таблице 48 количествах растворы СоС12 и К2Сг2О7 после чего доливают до метки водой.
Полученные растворы разливают в пробирки с плоским дном до метки на высоте 5 см и закры-
вают резиновыми пробками.
Эталоны пригодны в течение двух месяцев. Содержание нитратов в испытуемой воде
определяется по последней графе таблицы в соответствии с интенсивностью окраски про-
бирки.
Примечание 1. Вода окрашенная, а также с высокой жесткостью и содер-
жащая железо, марганец, сульфиды, обрабатывается как указано на стр. 337.
Примечание 2. При pH воды ниже 7, ее следует подщелочить до pH
7,5—8,4.
Таблица 48
Количество К2Сг2О7 и СоС1.? для изготовления эталонной шкалы
№№ п/п Вносится мл Соответствует количеству азота нитратов в мг/л
0,01 раствора двухро- мовокислого калия 1°/„ раствора хлористого кобальта
1 0,87 0,25 0,2
2 1,75 0,50 0,4
3 2,75 0,50 0,6
4 3,00 0,50 0,8
5 3,75 0,75 1,0
б 6,0 0,75 2,0
7 9,0 1,0 3,0
8 11,0 1,о 4,0
9 16,5 — о,о
10 20,0 — 8,0
11 24,0 — 10,0
Азот органический
Под органическим азотом понимают азот всех органических соединений —
от сложных протеинов до аминокислот включительно.
Органический азот определяют по методу Кьельдаля и выражают в виде (N).
Так как по данному методу вместе с азотом органических веществ опре-
деляется и азот аммонийных солей, нужно либо предварительно отогнать
аммиак аммонийных солей (см. стр. 337), либо определить в отдельной пробе
азот аммонийных солей и вычесть его из суммы азота органических соеди-
нений и аммонийных солей. Чаще применяется отгонка. Что касается азота
нитратов и нитритов, то они удаляются следующим образом: нитраты восста-
навливают в нитриты, а нитриты разрушают и удаляют в виде азота (см. ниже).
342
Принцип метода определения органического азота основан на
том, что органические азотсодержащие вещества подвергаются разрушению
с помощью крепкой серной кислоты при подогревании. При этом углерод
и водород органических соединений полностью окисляются до СО2 и Н2О
за счет кислорода, получаемого от разложения серного ангидрида. Азот орга-
нических веществ освобождается при этом в виде аммиака, который с серной
кислотой образует сульфат аммония.
При помощи крепкой щелочи разлагают затем сульфат аммония и осво-
бодившийся аммиак отгоняют и улавливают в титрованный раствор серной
кислоты. Обратным титрованием определяют, какое количество кислоты не
вступило в реакцию с аммиаком, а отсюда делают вывод о количестве кис-
лоты, связавшейся с аммиаком, и о количестве органического азота.
Сказанное можно представит в виде следующих реакций:
NH2CH2CH2COOH -I- H2SO4 = NH2CH2CH2OH 4- СО2 + SO2 4- Н2О (1)
NH2CH,CH2OH 4- 2H2SO4 = NH2CH2COOH 4- 2SO2 4- 3H2O (2)
nh2ch2cooh 4- h2so4 = nh2ch2oh 4- co2 4- so2 4- н2о (З)
NH2CH2OH 4- 2H2SO4 = NH3 4- CO2 4- 2SO2 4- 3H2O (4)
2NH3 4- H2SO3 = (NH4)2SO4 (5)
(NHJjjSO, 4- 2NaOH = Na2SO4 4- 2NH3 4- 2H2O (6)
2NH3 4- H2SO4 = (NH4)2SO4. (7)
Первые 4 реакции показывают схематически, как идет разрушение азотсо-
держащего вещества (аланина); реакция 5 показывает связывание освобож-
дающегося аммиака серной кислотой; реакция 6 показывает разложение
сульфата аммония щелочью и вытеснение аммиака, а реакция 7 — связывание
отгоняемого аммиака серной кислотой (титрованной). Из последней реакции
легко увидеть, что по количеству титрованного раствора серной кислоты,
пошедшего на связывание аммиака, можно сделать заключение о количестве
азота разрушенных органических соединений. Так, если для анализа была
применена 0,05 N H2SO4, то каждый миллилитр ее, пошедший на связывание
аммиака, соответствует 0,7004 мг азота (N . 0,05 = 14,008.0,05 = 0,7004).
Реактивы. 1. Серная кислота концентрированная (уд. вес 1,84).
2. 0,05 N серная кислота.
3. 0,05 N едкий натрий.
4. Концентрированный раствор едкого натрия (1 : 2) либо едкого калия
(1 : О-
5. Метилоранж 1%0 водный.
6. Лакмусовые бумажки или 1°/0 спиртовый раствор фенолфталеина.
7. Медь металлическая в виде стружек либо сернокислая медь в порошке.
8. Парафин в форме стружек либо тальк, либо крупнозернистая пемза,
либо стеклянные капилляры.
Методика определения. Определенный объем исследуемой
воды (500—1000 мл) подкисляют серной кислотой и упаривают в фарфоровой
чашке на водяной бане до небольшого объема (50—100 мл). При этом азот
аммонийных солей остается в растворе в виде сульфата аммония. Аммиак
отгоняют тут же в фарфоровой чашке, усреднив жидкость при помощи извест-
ковой воды или MgO. Конец отгонки аммиака определяется при помощи смо-
ченной красной лакмусовой бумажки, которую держат над чашкой. Если
бумажка не синеет, то аммиак считается отогнанным. Испытывают упарен-
ный раствор на присутствие нитратов. Если они обнаруживаются, то их вое-
становливают при помощи сернистой кислоты в нитриты, а последние удаляют
в виде окиси азота, прибавив хлорную закись железа и продолжая некоторое
время нагревание.
343
Рис. 232. Перегонный аппарат.
А — нолба для испытуемой воды, В — холодиль-
ник, С — приемная колба, D — алошк.
Если нитратов и нитритов в воде мало, то эти операции излишни, так
как под действием серной кислоты и нагревания указанные соединения пе-
реходят в соответствующие кислоты, которые в значительной мере улетучи-
ваются.
По отгонке аммиака содержимое фарфоровой чашки переносят в окис-
лительную колбу из устойчивого стекла с длинным горлом, прибавляют сюда
10—20 мл концентрированной серной кислоты и небольшое количество ме-
таллической меди в виде стружек либо 0,1—0,2 г сернокислой меди. Роль
меди или другого металла (ртути) сводится к каталитическому ускорению
сжигания и основана на способности этих металлов легко окисляться и раски-
сляться, в связи с чем они служат хорошими передатчиками кислорода от
серной кислоты к органическому ве-
ществу по схеме:
Си + H2SO4 = СиО + Н2О + SO2
2СиО = Си2О + О
Си2О H2SO4 = CuSO4 Си -J- Н2О.
Иногда вместо металла или в ком-
бинации с ним к жидкости прибав-
ляют еще какой-либо соли: K2SO4,
Na2So4, КС1. Эти соли способствуют
повышению температуры кипения
смеси, которая для чистой серной
кислоты равна 330°С. Повышение на-
грева серной кислоты также способ-
ствует более быстрому сжиганию ор-
ганических веществ.
Когда сжигание органических ве-
ществ заканчивается, жидкость ста-
новится прозрачной; от присутствия
CuSO4 она окрашена слегка в зеленоватый цвет.
По охлаждении содержимое из окислительной колбы переводят количест-
венно в перегонную колбу А (рис. 232), причем общее количество жидкости
вместе со смывными водами не должно превышать 300—400 мл.
Следует помнить, что в тех случаях, когда в качестве катализатора при-
меняется ртуть, при сжигании образуются ртутноамидные соединения, спо-
собные удерживать в осадке некоторые количества азота. Чтобы избежать
этого, добавляют в приемную колбу щепотку цинковой пыли, которая при
взаимодействии с серной кислотой начинает выделять водород; выделение
водорода продолжается и при последующем прибавлении щелочи, при этом
водород восстанавливает ртутноамидные соединения.
Zn + 2NaOH = Zn(ONa)2 + Н2
(NH2Hg)2SO4 + 2Н2 = (NH4)2SO4 + 2Hg.
Чтобы избежать толчков при кипячении жидкости, в колбу вводят 1—2 г
талька или стружек из парафина, или немного стеклянных капилляров, или
немного крупнозернистой пемзы. Наконец, чтобы иметь возможность просле-
дить конец нейтрализации жидкости в колбе, туда бросают кусочек красной
лакмусовой бумажки или приливают несколько капель фенолфталеина.
Перегонную колбу закрывают каучуковой пробкой, через которую прохо-
дит шейка делительного цилиндра и трубка насадки. Другой конец насадки
при помощи каучуковой трубки присоединяется к холодильнику В, а по-
следний при помощи каучуковой пробки соединяется с алонжем £), конец
которого опущен в колбу С>с титрованным раствором серной кислоты.
344
Через делительный цилиндр вводят в перегонную колбу раствор креп-
кого едкого натрия или калия до полной нейтрализации жидкости в колбе
с очень небольшим избытком щелочи (посинение лакмусовой бумажки или
порозовение жидкости от фенолфталеина).
На 20 мл крепкой серной кислоты уходит в среднем 60—70 мл едкого нат-
рия (1 : 2) или 100 мл едкого калия (1 : 1). Большого избытка щелочи сле-
дует избегать, так как это ведет к неравномерному кипению жидкости и
образованию толчков. Насадка, соединяющая перегонную колбу с холо-
дильником, служит не только для перевода паров из колбы в холодильник,
но и благодаря особому устройству для предупреждения забрасывания жид-
кости из перегонной колбы в холодильник, а затем в приемную колбу,
что может привести к полному извращению результатов анализа.
Когда жидкость в перегонной колбе нейтрализована, приступают к на-
греванию колбы, помещенной на сетку. Нагревание ведется до тех пор, пока
дестиллят не перестанет показывать щелочную реакцию; в этом убеждаются,
подхватив каплю дестиллята на красную лакмусовую бумажку. Отсутствие
посинения бумажки указывает на то, что отгон можно считать законченным.
Алонж извлекают из приемной колбы, дестиллированной водой обмывают
его конец, который был погружен в кислоту, причем промывная вода должна
попасть в приемную колбу, прибавляют сюда же 1—2 капли метилоранжа
и оттитровывают остаток серной кислоты 0,05 N раствором NaOH. По коли-
честву серной кислоты, связавшейся с аммиаком, делают заключение о коли-
честве органического азота.
Пример. В приемную колбу налито 25 мл 0,05 N H2SO4. При обратном титровании из-
расходовано б мл 0,05 N раствора NaOH. При отдельном определении выяснилось, что в
литре воды находится 1,2 мг аммонийного азота. Отсюда заключаем, что в литре иссле-
дуемой воды находится 0,7004. (25 — 6) — 1,2 = 12,1076 мг азота органического.
Азот общий
Под общим азотом воды понимают суммарное количество азота органи-
ческих и неорганических соединений. Он определяется только в загрязненных
водах и выражается в виде (N).
Принцип метода основан на том, что воду обрабатывают смесью
из крепкой серной кислоты и фенола в присутствии цинковой пыли. При
этом нитраты и нитриты переходят в нитрофенол, который восстанавливается
в амидофенол, а этот последний при кипячении с серной кислотой выделяет
аммиак, задерживаемый, как и азот других соединений, в виде сульфата
аммония. Дальнейшее вытеснение аммиака, отгон его и определение азота
основаны на тех же реакциях, что и определение органического азота (см.
выше).
Реактивы. 1. Смесь из 100 г серной кислоты уд. веса 1,84 и 5 г фенола.
2. Цинковая пыль.
3. 0,05 N серная кислота.
4. 0,05 N едкий натрий.
6. Концентрированный раствор едкого натрия (1:2) либо едкого калия
(1 1).
6. Метилоранж 1%0 водный.
7. Лакмусовые бумажки или 1% раствор фенолфталеина.
8. Сернокислая медь в порошке.
9. Крупнозернистая пемза.
Методика определения. Литр (или более) исследуемой воды
подкисляют серной кислотой и упаривают в фарфоровой чашке на водяной
бане до 20—25 мл. Переводят количественно в окислительную колбу, при-
меняя небольшие количества дестиллированной воды для промывания посуды.
Прибавляют в колбу 25 мл фенолосерной смеси, 2,5 г цинковой пыли и 0,2 г
сернокислой меди и немного пемзы. Смесь кипятят до появления слабозеле-
345
ной окраски. Переводят жидкость количественно в перегонную колбу, причем
общее количество раствора в колбе не должно превышать 200—250 мл. В даль-
нейшем определение ведется так же, как и определение органического азота.
Фосфорная кислота (фосфаты)
Фосфорная кислота обычно в питьевых водах отсутствует, так как энер-
гично поглощается почвой.
Присутствие фосфатов в воде может обусловливаться сильным загрязне-
нием почвы отбросами животного происхождения либо удобрением почвы
фосфорсодержащими соединениями (суперфосфаты).
Выражается в виде фосфорного ангидрида Р2О5 либо иона РО4"', реже в
виде Р.
Качественное определение
Качественное определение фосфорной кислоты производится при помощи
реактива Дениже, который дает в ее присутствии синее окрашивание.
Приготовление реактива см. на стр. 262.
Стандартная методика рекомендует определение фосфорной кислоты про-
водить по молибденово-оловянному методу, для чего нужно иметь следующие
два реактива:
1. 100 мл 10% молибденовокислого аммония (NH4)6Mo7O24 смешивают
с 300 мл 50% (по объему) раствора серной кислоты.
2. 0,1 г измельченного олова растворяют в 2 мл концентрированной со-
ляной кислоты, прибавляют 1 каплю 3—4% медного купороса и разбавляют
водой до 10мл. Раствор готовится ex tempore.
Для измельчения олова его нагревают до 250°С и раздавливают пестиком
в фарфоровой нагретой ступке.
Определение фосфорной кислоты при помощи указанных двух растворов
производится следующим образом: к 100 мл исследуемой воды прибавляют
2 мл раствора молибдата аммония (реактив № 1) и одну каплю раствора
хлористого олова (реактив № 2). При наличии фосфорной кислоты появ-
ляется синее окрашивание.
Предел чувствительности пробы 0,001 мг Р2О5.
Количественное определение
Количественное определение фосфорной кислоты производится колориме-
трически по методу стандартных серий или уравнивания окраски. В основе
количественного определения лежит тот же принцип, что и в основе качествен-
ного.
Реактивы. Те же, что и при качественном определении, и стандартный
раствор. Последний готовится из двузамещенного фосфорнокислого калия.
О i/unr. 2К2НРО4 348,5
Растворяют 0,2455 г К2НРО4 в литре воды соответственно "pg ~[q = ]4jg“g ~
0,2455. 1 мл такого раствора соответствует 0,1 мг Р2О5.
Если расчет ведут на ион РО4'", то в литре воды растворяют 0,18346 г соли
соответственно —• 1 мл такого раствора соответствует 0,1 мг РО/".
r(J4
Указанные основные стандартные растворы по мере надобности разбав-
ляются в 10 и 100 раз.
Кремнекислота (силикаты)
Кремнекислота находится в воде преимущественно в неионизированном
состоянии и в анализах выражается в виде SiO2. Лишь при pH > 8 кремне-
кислота может присутствовать в воде в ионизированном состоянии. В этом
случае ее можно выразить в виде иона Si Од .
346
Качественное определение
Качественное определение кремневой кислоты практического значения
не имеет.
Количественное определение
Количественное определение кремневой кислоты в воде производится
чаще весовым методом, реже колориметрически.
Весовой метод
500—1000 мл воды подкисляют чистой соляной кислотой и выпаривают
в платиновой чашке на водяной бане насухо. Остаток смачивают концентри-
рованной соляной кислотой и выпаривают, повторяя эту оперецию дважды.
При этом кремневая кислота переходит в нерастворимое состояние: гидрат
кремневой кислоты. Добавив к остатку дестиллированной воды, фильтруют,
промывают осадок на фильтре кипящей водой до исчезновения в промывных
водах реакции на хлор (проба с азотнокислым серебром). Фильтр с осадком
высушивают при 110°С, переносят осадок в платиновый тигель, сжигают
фильтр отдельно в платиновой спирали, пепел переносят в тигель вместе с
осадком и прокаливают сначала слегка, а потом сильнее; при этом гидрат
кремневой кислоты переводится в кремневый ангидрид—SiO2. Охлаждают
в экссикаторе и взвешивают. От полученного веса отнимают вес тигля и золы
фильтра, получают вес SiO2. Взвешивание нужно производить быстро, так
как SiO2 обладает большой гигроскопичностью.
Колориметрический метод
Колориметрически определяется лишь растворенная кремнекислота.
Принцип метода. Растворенная кремнекислота с молибденовой
кислотой образует кремнемолибденовую кислоту — H8[Si(Mo2O7)e]. 28Н2О,
которая в водных растворах имеет интенсивную желтую окраску, что и по-
ложено в основу колориметрического определения кремнекислоты.
Реактивы: 1. 20% раствор молибденовокислого аммония —
(ЫН4)6Мо7О24.
2. 10% раствор НС1.
3. Стандартный раствор хромата калия, содержащий 0,530 г К2СГО4 в
100мл раствора. 1 мл такого раствора, прибавленный к 100 мл дестиллирован-
ной воды, дает такую же окраску, какая получается при прибавлении молиб-
дата аммония и соляной кислоты (реактивы №№ 1 и 2) к воде, содержащей
10 мг SiO2 или 12,67 мг SiO3" в литре воды.
Методика определения. В один из парных цилиндров коло-
риметра наливают 100 мл испытуемой воды, прибавляют 5 мл 20% раствора
молибдата аммония и 5 мл 10% раствора соляной кислоты. Образовавшаяся
от взаимодействия первых двух реактивов молибденовая кислота дает с крем-
некислотой кремнемолибденовую кислоту, которая окрашивает жидкость
в желтый цвет, причем максимальная окраска наступает через 10 минут.
В другой цилиндр наливают 1—2 мл стандартного раствора и добавляют
испытуемой воды до 110 мл. Стеклянными палочками перемешивают жидкости
в том и другом цилиндрах и колориметрируют.
Колориметрирование можно провести и по методу стандартных серий.
Присутствие фосфатов и железа мешает реакции, ввиду чего их удаляют
из воды: к 100 мл исследуемой воды прибавляют 2 мл 8—10% уксусной ки-
слоты и 3 мл раствора фосфата натрия, содержащего 5 мг Р2О5 в 1 мл. Если
вода содержит железа более 10 мг в литре, то фосфата натрия прибавляют
соответственно больше. Смесь нагревают до кипения, фильтруют и охлаждают.
По охлаждении прибавляют 2 мл раствора хлористого кальция, содержащего
10 г безводной хлористой соли в 90 мл воды и 2 мл 5% аммиака, взбалтывают,
отстаивают и фильтруют.
С фильтратом производят определения, как описано выше.
347
Алюминий (глинозем)
Выражается в виде А12О3.
Качественное определение
Воду освобождают от кремнезема (стр. 347) и осаждают в ней соли железа
и алюминия (см. весовое определение солей кальция). Осадок отфильтро-
вывают и растворяют в соляной кислоте. К раствору прибавляют до сильно-
щелочной реакции раствор NaOH или КОН, свободный от алюминия1. При
этом железо выпадает, алюминий же остается в растворе. Вновь отфильтро-
вывают, фильтрат подкисляют соляной кислотой и осаждают алюминий ам-
миаком:
А1С13 + 3NH4OH = А1(0Н)3 + 3NH4C1.
Количественное определение
Отделив от воды кремнезем (см. стр. 347), подкисляют фильтрат азотной
кислотой для перевода солей закиси железа в соли окиси. Нагревают жид-
кость до кипения, добавляют хлористого аммония и раствор аммиака до ще-
лочной реакции. Продолжают нагрев до исчезновения запаха аммиака. Алюми-
ний и железо выпадают в виде гидратов окисей. Осадок переносят на фильтр,
промывают горячей водой и высушивают фильтр вместе с осадком при 110°С.
Осадок отделяют и сжигают фильтр в платиновой спирали; затем осадок
и золу фильтра прокаливают в платиновом тигле, охлаждают в экссикаторе
и взвешивают. Из полученного веса вычитают вес тигля и золы и получают
вес окиси железа Fe2O3 плюс вес окиси алюминия А12О3. Отняв отсюда вес
окиси железа, определяют вес одной только окиси алюминия. Вес окиси же-
леза можно определить, если результат анализа на железо (см. далее) помно-
1 л г* Fе0Оо
жить на 1,46 соответственно —.
Ре2
Для количественного определения алюминия в воле были предложены также колори-
метрические методы с алюминоном (аммониевая соль трикарбоксиловой кислоты аурина),
гематоксилином, ализарином и др. Из колориметрических методов, по данным Е. И. Бату-
риной, наиболее точные результаты дает метод с алюминоном.
Железо
Железо находится в воде чаще в виде бикарбоната закиси —Fe(HCO3)2.
Частично подвергаясь гидролизу, бикарбонат переходит в гидрат закиси,
который, окисляясь кислородом, растворенным в воде, переходит в коллои-
дальный гидрат окиси, а этот последний — в водную окись:
Fe(HCO3)2 + 2Н2О Fe(OH)2 + 2Н2О + 2СО3
4Fe(OH)2 + 2Н2О + О2 = 4Fe(OH)3
2Fe(OH)3 = Fe2O3 . ЗН2О.
Вода с большим содержанием железа (0,5 мг/л и более) выделяет при сто-
янии осадок, неприятна на вкус (чернильный привкус), непригодна для це-
лого ряда производств, в ней нередко развиваются железистые бактерии
(Crenotrix polispora, Chlamydotrix ochracea и др.), загрязняющие воду.
Общее содержание железа выражается через Fe, закись железа — через
Fe", окись железа —через Fe"\
Согласно ГОСТ 2874-45 суммарное содержаниее Fe и Мп в водопроводной
воде не должно превышать 0,3 мг/л, в том числе закиси железа не более 0,2 мг/л.
1 К слабому раствору щелочи прибавляют NH4C1 и кипятят до исчезновения запаха
аммиака; при этом не должна выпадать гидроокись алюминия.
348
Качественное определение
Соли закиси железа переводят в соли окиси и производят одну из харак-
терных для окиси железа реакций.
Для перевода солей закиси железа в соли окиси применяют один из сле-
дующих способов: а) к 100 мл воды прибавляют 4 мл крепкой соляной ки-
слоты уд. веса 1,12 и 2 мл насыщенного раствора хлората калия (КС1ОЭ),
взбалтывают и нагревают на кипящей бане 15 минут; в) 100 мл воды подкисл-
ляют крепкой серной или соляной кислотой, прибавляют 10 капель 3% не-
разложившейся перекиси водорода и взбалтывают. Окисление происходит
моментально на холоду.
Для перевода закисных солей в окисные можно применить также азотную
кислоту, марганцевокислый калий и другие окислители. Все применяемые
реактивы должны быть свободны от железа.
После перевода солей закиси железа в окись производят одну из следую-
щих качественных реакций на железо:
1. Добавляют 10% раствор роданистого аммония. В присутствии железа
жидкость окрашивается в красный цвет, который обусловливается образо-
ванием роданового железа по реакции:
Fe2Cle + 6NH4CNS = 2Fe(CNS)3 + 6NH4C1.
2. Добавляют 5% свежеприготовленный раствор железистосинеродистого
калия K4Fe(CN)6. В присутствии солей окиси железа образуется синяя ок-
раска в результате получения берлинской лазури по реакции:
2Fe2Cle + 3K4Fe(CN)e = Fe4[Fe(CN)6]3 + 12КС1.
Если содержание железа в воде незначительно, то определенный объем
исследуемой воды перед испытанием упаривают в 2—3 раза, предварительно
подкислив соляной кислотой.
Количественное определение
Количественное определение всего железа в воде производят колоримет-
рически после перевода солей закиси железа в соли окиси.
Принцип метода тот же, что и при качественном определении
железа.
Реактивы. 1. Раствор железных квасцов—Fe2(SO4)3K2SO4. 24Н2О. На
_ плаас Fe2(SO4),K2SO4-24H2O
литр раствора берут 0,9006 г квасцов соответственно —“—2 10Fe----- •
Перед взвешиванием квасцы сушат между листами фильтровальной бумаги
для удаления гигроскопической воды.
Вместо железных квасцов можно применить железоамиачные квасцы —
(NH4)8SO4Fe8(SO4)3. 24Н2О. На литр раствора берут 0,863 г квасцов со-
(NH1)2SO4Fe2(SO4)324H2O
ответственно * ——10 Fe ~ ’ Раств0Ряют в некотором объеме воды,
подкисляют 10 мл крепкой серной кислоты и доводят до литра водой. 1 мл
каждого из указанных двух растворов соответствует 0,1 мг Fe.
2. Раствор роданистого аммония (NH4CNS) или роданистого калия
(KCNS). Получается растворением 225 г роданида в 125 мл воды.
3. Соляная кислота уд. веса 1,12.
Методика определения. В один из парных цилиндров коло-
риметра наливают 100 мл исследуемой воды (после перевода солей закиси
железа в соли окиси); объем воды доводят до 105 мл дестиллирован-
ной водой. В другой цилиндр наливают 100 мл дестиллированной воды,
4 мл соляной кислоты и 1 мл стандартного раствора. В оба цилиндра
прибавляют по 1 мл роданида аммония или калия, взбалтывают содержимое
стеклянными палочками и колориметрируют.
349
По стандартной методике рекомендуется произвести колориметри-
ческое титрование: в этом случае в стандартный цилиндр наливают
100 мл дестиллированной воды, 4 мл соляной кислоты, 1 мл роданида и за-
тем из пипетки с делениями на 0,01 мл приливают по каплям стандартный
раствор, перемешивая при этом стеклянной палочкой. Прибавление стандарт-
ного реактива ведется до получения одинаковой окраски с опытным цилин-
дром. По прилитому количеству стандартного раствора делают заключение
о количестве железа в испытуемой воде.
Пример. Положим, что одинаковая окраска получилась, когда в стандартный цилиндр
было прилито 1,68 мл стандартного раствора квасцов, 1 мл которого соответствует 0,1 мр
Fe. Для колориметрирования было взято 100 мл воды. В этом случае содержание всего же-
леза в воде равно:
0,1 . 1,68 • 1000
----10Q----= 1,6 мг Fe.
Если имеют в виду произвести раздельно определение закисного железа
и окисного, то кроме определения суммарного содержания железа в отдель-
ной пробе воды определяют соли окиси железа точно так, как описано выше,
без предварительного перевода солей закиси железа в соли окиси. Из сум-
марного количества железа вычитают количество окиси железа и получают
количество закиси.
Марганец
В естественных условиях марганец обыкновенно встречается вместе с же-
лезом, сопровождая его. Содержится в воде преимущественно в виде бикар-
боната. В количестве 0,5 мг/л и выше сообщает воде плохие качества (непри-
ятный вид и вкус, выпадение окисей и т. п.). Выражается в виде Мп.
Качественное определение
Исследование нужно производить сейчас же после взятия пробы.
1. К 25 мл воды добавляют 10 мл концентрированной азотной кислоты,
нагревают до кипения и осторожно, избегая закипания, добавляют 0,5 г пе-
рекиси свинца (РЬО2), снова нагревают до кипения, после чего жидкость
отстаивают. Если в исследуемой воде был марганец, то жидкость приобретает
фиолетовокрасную окраску от образовавшейся во время реакции марганце-
вой кислоты = НМпО4. Реакции протекают следующим образом:
2Мп(НСО3)2 4- 4HNO3 = 2Mn(NO3)2 + 4СОа + 4Н2О
2Mn(NO3)2 + 5РЬО2 + 6HNO3 = 2НМпО4 4~ 5Pb(NO3)2 + 2Н2О.
Предел чувствительности 0,1 мг Мп в литре воды.
2. В коническую колбу вводят 100 мл исследуемой воды, добавляют 10 мл
10% раствора персульфата аммония [надсернокислый аммоний —(NH4)2S2O8],
1—2 капли разведенной азотной кислоты и 1—2 капли раствора азотнокис-
лого серебра (избытка избегать). Жидкость нагревают почти до кипения.
В присутствии марганца она окрашивается в розовый цвет от образующейся
марганцевой кислоты:
2Мп(НСО3)2 + 4HNO3 = 2Mn(NO3)2 + 4СО2 4- 4Н2О
2Mn(NO3)2 4- 5(NH4)2S2O8 4- 8Н2О = 5(NH4)2SO4 4- 5H2SO4 + 4HNO3+ 2HMnO4.
Предел чувствительности реакции 0,05—0,1 мг Мп в литре воды.
Присутствие большого количества солей железа и хлоридов мешает реак-
ции. Для удаления солей железа воду подкисляют азотной кислотой, упари-
вают до малого объема, усредняют содой и осаждают окись железа уксусно-
кислым натрием при кипячении. Осадок отфильтровывают и промывают.
Хлориды удаляют осаждением азотнокислым серебром.
850
Количественное определение
Количественное определение марганца в воде производится колориметри-
чески.
Принцип метода основан, как и при качественном определении,
на окислении солей марганца в марганцевую кислоту, которая окрашивает
воду в розовый цвет.
Реактивы. 1. Крепкая азотная кислота, свободная от окислов азота.
2. 10% раствор AgNO3.
3. Насыщенный на холоду раствор персульфата аммония —(NH4)2 S2O8
(58 г в литре воды) или персульфата калия —K2S2O8 (17 г в литре воды).
4. 0,01 N раствор KMnO4. 1 мл раствора соответствует 0,1098 мг Мп со-
0,316Мп
ответственно .
КМпО4
Методика определения. 100 мл исследуемой воды подкисляют
5 мл крепкой HNO3 и прибавляют 1 мл 10% AgNO3 в качестве катализатора.
Нагревают до кипения и, легко поддерживая его, прибавляют небольшими
порциями 5 мл насыщенного раствора персульфата аммония или калия. Упа-
ривают до 100 мл и переводят в один из парных цилиндров колориметра.
В другой цилиндр наливают 100 мл дестиллированной воды, подкисленной
крепкой HNO3 и прибавляют из пипетки с делениями на 0,01 мл 0,01 N ра-
створ КМпО4 до совпадения окраски с окраской исследуемой воды. На осно-
вании прибавленного количества марганцевокислого калия к дестиллирован-
ной воде делают заключение о содержании марганца в испытуемой воде.
Пример. В стандартный цилиндр для уравнения окраски прибавлено 0,12 мл 0,01 N
раствора КМпО4. Для исследования взято 100 мл воды. Отсюда заключаем, что в литре
испытуемой воды находится: • 0Л 2 • 1000 13175 мг доп.
Свинец, медь, цинк и олово
Эти металлы попадают в воду обыкновенно из водопроводных труб, из
сосудов, в которых вода хранится, а также с промышленными сточными во-
дами.
Согласно ГОСТ 2874-45 в водопроводной воде допускается РЬ не более
0,1 мг/л, Си —не более 3 мг/л, Zn —не более 15 мг/л. Содержание олова,
ввиду его малой ядовитости, стандартом не нормируется.
Качественное определение
Качественное определение всех указанных металлов удобно производить
последовательно в одной и той же пробе воды.
1—5 л воды подкисляют соляной кислотой и упаривают до 100—150 мл.
Если образуется осадок, то его отделяют на фильтре и отбрасывают, а филь-
трат обрабатывают током хорошо промытого сероводорода при подогревании;
при этом свинец, медь и олово выпадают в виде бурого или черного осадка
сульфидов —PbS, CuS и SnS, а цинк остается в растворе. Фильтруют. Полу-
ченный фильтрат А сохраняют для определения цинка, а осадок на фильтре
промывают сероводородной водой, после чего его вместе с фильтром поме-
щают в фарфоровую чашку, прибавляют азотной кислоты уд. веса 1,2 и кипя-
тят; при этом сульфиды свинца и меди переходят в раствор в виде Pb(NO3)2
и Cu(NO3)2, а сернистое олово, окисляясь азотной кислотой в метаоловянную
кислоту (H2SnO3), остается в нерастворенном состоянии. Фильтруют. Фильтр
вместе с осадком метаоловянной кислоты сохраняют для дальнейшего опре-
деления олова, а фильтрат В служит для определения свинца и меди.
Определение свинца. Фильтрат В выпаривают досуха на водяной бане
для удаления избытка HNO3. Оставшиеся азотнокислые соли свинца и меди
растворяют в горячей воде и фильтруют. Фильтрат упаривают до 10—15 мл,
351
прибавляют 5 мл крепкой H2SO4 и нагревают до прекращения выделения
белых паров серного ангидрида, прибавляют 150 мл 50% этилового алкоголя
и оставляют стоять на несколько часов (обычно на ночь). Если в фильтрате В
присутствовал свинец, то выпадает белый осадок сульфата свинца (PbSO4).
Его отфильтровывают и промывают на фильтре 50% алкоголем. Фильтр с
осадком идет для количественного определения свинца, а фильтрат С при-
меняется для открытия меди.
Качественное обнаружение свинца можно произвести также, если в про-
бирку налить немного фильтрата В, подкислить уксусной кислотой и при-
бавить кристаллик KJ. Образуется желтый кристаллический осадок PbJ2,
растворимый при нагревании и выпадающий при охлаждении на стенках
пробирки.
Определение меди. 1. Фильтрат С упаривают до удаления спирта (на за-
пах), прибавляют в избытке аммиак. В присутствии солей меди появляется
сначала синий осадок, переходящий затем в раствор от избытка аммиака.
При этом образуется аммиачномедная соль —[Cu(NH3)4] (SO4)H2O
2CuSO4 + 2NH4OH = (NH4)2SO4 + Cu2(OH)2SO4
Cu2(OH)2SO4 + (NH4)2SO4 + 6NH3= 2[Cu(NH3)4](SO4) • H2O.
Описанное качественное определение меди может быть положено в основу
количественного колориметрического определения ее.
2. К фильтрату С, подкисленному уксусной кислотой, прибавляют не-
сколько капель 5% раствора железистосинеродистого калия — K4Fe(CN)e.
При наличии меди получается краснобурое окрашивание или такого же цвета
осадок в результате образования железистосинеродистой меди —Cu2[Fe(CN)„]:
K4Fe(CN)6 + 2CuSO4 = 2K2SO4 + CuJFe(CN)6].
Определение цинка. 1. К фильтрату А добавляют в избытке уксуснокислый
натрий для перевода солянокислого раствора в уксуснокислый и затем про-
пускают ток сероводорода. В присутствии цинка выпадает белый осадок
сульфида цинка —ZnS.
2. Фильтрат А нагревают и прибавляют избыток аммиака, а затем избыток
фосфорнокислого аммония. При наличии цинка получается сначала аморфный,
а затем кристаллизующийся осадок фосфорнокислого цинк-аммония —
ZnNH4PO4. Этот метод может быть применен и для количественного опре-
деления цинка.
Определение олова. Фильтр с осадком метаоловянной кислоты высуши-
вают, отделяют осадок. Фильтр сжигают отдельно, золу присоединяют к
осадку и прокаливают. Метаоловянная кислота переходит в окись олова
(SnO2), которая может быть взвешена.
Количественное определение
Производится в той же пробе воды после качественного обнаружения
того или иного металла.
Определение свинца. Если осадок PbSO4 получился значительный, то он
может быть использован для весового определения свинца. Фильтр с осадком
высушивают, отделяют осадок. Фильтр сжигают отдельно, золу присоеди-
няют к осадку и прокаливают в фарфоровом тигле. Прокаленный осадок
смачивают 1—2 каплями азотной кислоты, выпаривают на водяной бане,
смачивают серной кислотой, вновь слабо прокаливают до полного удаления
паров серного ангидрида и по охлаждении взвешивают. Вес PbSO4 для пере-
РЬ
счета на РЬ умножают на 0,68325 соответственно pegQ~ -
Если осадок PbSO4 получился незначительный, то его растворяют ки-
пячением в фарфоровой чашке с 5% раствором уксуснокислого аммония
352
Фильтруют и в фильтрате определяют свинец объемным или нефелометриче-
ским методом, как описано выше (стр. 278 и далее).
Определение меди. Может быть приозведено колориметрически, для чего
используется фильтрат С. Принцип тот же, на каком основано качественное
определение.
В один из цилиндров колориметра наливают фильтрат С и крепкий раствор
аммиака, доводя затем объем дестиллированной водой до 100 мл. В другой
цилиндр наливают 1 мл стандартного раствора, содержащего 1 мг меди, при-
бавляют в избытке NH4OH и доливают до 100 мл дестиллированой водой,
после чего колориметрируют.
Стандартный раствор готовится растворением 3,927 г химически чистого
Г on С ГЛ 1 - CuSO4 • 5Но0
CuSO4-5H2O в 1 л дестиллированной воды соответственно -------.
1 мл раствора соответствует 1 мг меди.
Определение цинка. 1. В тех случаях, когда цинк содержится в воде в
относительно больших количествах и при качественном определении его
получается заметной осадок фосфата цинк-аммония (ZnNH4PO4), можно опре-
деление цинка вести весовым путем.
По выпадении осадка (см. качественное определение) продолжают нагре-
вание еще 15 минут, после чего дают жидкости постоять некоторое время
и фильтруют через тигель Гуча. Осадок на фильтре промывают 1% раствором
фосфата аммония до тех пор, пока промывные воды не дают более реакции
на посторонние примеси (хлорилы), затем заканчивают промывание холодной
водой, сушат 2 часа при 100—105°С и взвешивают. Привес, соответствующий
фосфату цинк-аммония, для пересчета на цинк умножают на 0,3664 соответ-
Zn
ственно мн рл • Если прокалить осадок при 900—1000°С, то фосфат цинк-
ZjIl ГМ ri4r vJ4
аммония перейдет в пирофосфат цинка (Zn2P2O7), и тогда привес для пере-
счета на цинк нужно умножить на 0,429 соответственно ——.
^^2* 2^7
2 . Если содержание цйнка в воде незначительно—0,02—0,20 мг/л, что
при упаривании 5 л даст от 0,1 до 1 мг Zn, —то может быть применен нефе-
лометрический метод, основанный на том, что при взаимодействии между
солями цинка и железистосинеродистым калием образуется нерастворимый
железистосинеродистый цинк-калий —K2Zn3[Fe(CN)6]2, обусловливающий по-
мутнение жидкости:
2K4Fe(CN)6 + 3ZnSO4 = K2Zn3[Fe(CN)6]2 + 3K2SO4.
Для нефелометрического определения цинка используют фильтрат А.
Прежде чем приступить к определению Zn, необходимо удалить из фильтрата
железо, которое с железистосинеродистым калием дает синее окрашивание,
что мешает реакции. Железо окисляют добавлением к фильтрату бромной
воды; затем бром отгоняют нагреванием в течение нескольких минут на во-
дяной бане и прибавляют раствор фосфата натрия (Na2HPO4). Охладив рас-
твор, нейтрализуют его при индикаторе метилоранже уксуснокислым на-
трием или аммонием. При этом появляется ясная муть, обусловленная выпа-
дением фосфата железа — (FePO4). Осадок отфильтровывают и фильтрат
используют для нефелометрирования.
Приготовляют стандартный раствор сернокислого цинка (ZnSO4 • 7Н2О)
с таким расчетом, чтобы 1 мл соответствовал 1 мг Zn. Для этой цели на 1 л ра-
створа берут 4,3981 г химически чистого препарата соответственно-—-—
По мере надобности приготовляют из основного стандатного раствора слабый
раствор, разбавляя основной в 10 раз. 1 мл слабого раствора соответ-
ствует 0,1 мг Zn.
23 А,. И. Бурштейн
353
Слабый стандартный раствор разливают в цилиндры одинакового диаметра
с таким расчетом, чтобы содержание цинка повышалось от 0,1 до 1 мг, и
доливают дестиллированной водой до 100 мл. В цилиндр такого же диаметра
наливают фильтрат Л, освобожденный от железа, доводя объем также до 100 мл.
Во все цилиндры наливают по 1 мл 0,1 N H2SO4 и 1 мл насыщенного раствора
сернистой кислоты для восстановления следов окиси железа и по 1 мл свеже-
приготовленного 2% раствора K4Fe(CN)6.
Жидкость в цилиндрах взбалтывают при помощи стеклянных палочек,
помещают в темном месте на 20 минут, после чего образовавшуюся муть,
представляющую собой суспензию железистоцианистого цинк-калия, сравни-
вают, держа цилиндр против темного фона.
Подыскивают стандартный раствор, в котором образовавшаяся муть была бы
по интенсивности равна мути в цилиндре с испытуемой жидкостью. Таким
образом заключают о количестве цинка в этой жидкости.
Определение олова. Фильтр с осадком метаоловянной кислоты (H2SnO3)
прокаливают и, как было указано (см. качественное определение), взвеши-
вают полученную окись олова. Для перечисления на Sn умножают на 0,7877
Sn
соответственно .
SnO2
Наряду с изложенной выше методикой определения РЬ, Си и Zn считаем
необходимым привести и новейшую стандартную методику.
Определение свинца. Метод основан на получении бурой коллоидной
взвеси сульфида свинца при введении в воду, содержащую свинец, сульфид-
иона:
Pb++ + S—-»PbS j
Окраску исследуемой воды после введения в нее сульфид-иона сравнивают
с окраской раствора с известным содержанием свинца после введения в него
того же количества сульфид-исна.
Реактивы 1. 50% раствор сегнетовой соли.
2. 27% раствор едкого натрия.
3. 10% раствор тиосульфата натрия.
4. Водно-глицериновый раствор сульфида натрия. 5 г кристаллического
сульфида натрия растворяют в смеси 30 г глицерина с 10 мл дестиллирован-
ной воды. Отстаивают 5 суток в герметически закрытой склянке в темном
шкафу. Декантируют в предварительно замоченный в течение 2 часов в де-
стиллированной воде фильтровальный тигель и фильтруют с отсосом. Фильт-
рат хранят в плотно закрывающейся склянке.
5. Азотная кислота 1 : 5.
б. Стандартный раствор азотнокислого свинца. 0,16 г химически чистого
сухого Pb(NO3)2 растворяют в небольшом количестве дестиллированной воды»
подкисляют 1 мл азотной кислоты (1 : 5) и доводят до 1 л водой. 1 мл раствора
содержит 0,1 мл РЬ.
Методика определения. 100—1000 мл исследуемой воды под-
кисляют азотной кислотой (2 мл раствора азотной кислоты 1 : 5 на 100 мл
исследуемой воды) и упаривают в платиновой или фарфоровой чашке до 50 мл,
нейтрализуют 27% раствором NaOH по лакмусовой бумажке и переносят
количественно в цилиндр Генера; доводят объем раствора до 100 мл дестил-
лированной водой, после чего добавляют 5 мл 50% раствора сегнетовой соли,
чтобы парализовать вредное влияние железа, затем Змл 27% раствора едкого
натрия, чтобы парализовать влияние алюминия и цинка, и 5 мл 10 70 раствора
тиосульфата натрия, чтобы парализовать влияние меди. После введения
каждого раствора содержимое цилиндра хорошо перемешивают.
Одновременно в такой же цилиндр Генера вводят 100 мл дестиллированной
воды и 0,5 мл стандартного раствора азотнокислого свинца. Добавляют сюда же
364
в такой же последовательности, как в опытный цилиндр, растворы сегнето-
вой соли, едкого натрия и тиосульфата натрия. Затем в оба цилиндра вводят
по 0,2 мл водно-глицеринового раствора сульфида натрия и через 2 минуты
нефелометрируют. Если при этом окажется, что высота столба жидкости
в одном из цилиндров в три раза с лишним меньше, чем во втором цилиндре,
то определение следует повторить, изменив количество взятой для опре-
деления исследуемой воды либо количество раствора азотнокислого свинца,
вводимого в стандартный цилиндр.
Примечание. Количество свинца во взятом для определения объеме иссле-
дуемой воды не должно быть менее 0,1 мг и более 5 мг.
Определение меди. Метод основан на сравнении окраски исследуемой воды
при прибавлении к ней раствора диэтилдитиокарбамата натрия с окраской
стандартных растворов с известным содержанием меди в тех же условиях.
Реактивы. 1. 1 г диэтилдитиокарбамата натрия [N(C2H4)2CS2Na]
растворяют в 1000 мл дестиллированной воды. Сохраняют в склянке темного
стекла с притертой пробкой.
2. Продажный 30% раствор аммиака разводят в 5 раз дестиллированной
водой.
3. Стандартный раствор CuSO4 • 5Н2О. Растворяют 0,393 г препарата
в 1000 мл дестиллированной воды. 10 мл раствора доводят водой до 1 л 1 мл
полученного раствора содержит 0,001 мг меди.
4. 0,25 г растворимого крахмала растворяют в 100 мл дестиллированной
воды. Раствор должен быть свежеприготовленным.
5. 50% водный раствор сегнетовой соли.
6. 5% раствор надсернокислого аммония — персульфат аммония,
(NH4)2S2O8.
Методика определения. Приготовляют стандартную шкалу.
Для этой цели в 10 ирсСпрок бесцветного стекла одинакового диаметра с мет-
кой на 10 мл вводят 0,2; 0,5; 1,0; 2,0; 3,0; 5,0; 7,0; 8,0; 9,0; 10,0 мл слабого
стандартного раствора сернокислой меди и доводят до метки водой.
Содержание меди в мг/л будет соответственно равно: 0,02; 0,05; 0,1;
0,2; 0,3; 0,5; 0,7; 0,8; 0,9; 1,0. В каждую из пробирок вводят по 0,1 мл рас-
твора сегнетовой соли, 1 мл раствора аммиака, 0,5 мл раствора крахмала и
1,0 мл раствора диэтилдитиокарбамата натрия. Содержимое пробирок пере-
мешивают. Стандартная шкала должна быть приготовлена не ранее, чем за
30 минут до проведения определения.
В пробирку такую, как и для стандартных растворов, отмеривают 10 мл
испытуемой воды и затем вводят последовательно те же реактивы, что и в
стандартные растворы. Содержимое пробирки перемешивают и через 10 минут
сравнивают окраску раствора в пробирке с окраской растворов стандартных
пробирок.
Примечание 1. Содержание меди в испытуемой воде должно быть не более
1 мг/л, в противном случае воду нужно соответственно разбавить дестиллированной
водой.
2. Если исследуемая вода имеет цветность более 20°, то ее предварительно сле-
дует обесцветить, добавив на 150 мл воды 10 мл 5 % риствора персульфата аммония,
и прокипятить воду в течение 10 минут.
Определение цинка. Сущность метода заключается в том, что цинк осаж-
дается оксихинолином в виде оксихинолята цинка:
Zn++ + 2C9H7ON 4- 2ОН——> Zn(C9HeON)24~ 2НаО.
Оксихинолят цинка разрушается затем соляной кислотой:
Zn(C9HBON)2 + 2Н + -»Zn++ -J- 2C9O,ON.
Выделившийся свободный оксихинолин, эквивалентный количеству цинка,
в аммиачной среде с диазотированной сульфаниловой кислотой образует
355
азокраску. Концентрацию азокраски определяют колориметрически при по-
мощи шкалы, приготовленной на чистых растворах цинка и обработанной
таким же образом, как и испытуемые растворы.
Реактивы. 1. Раствор уксуснокислого цинка—Zn(C2H3O2)2, 1 мл
которого содержит 0,01 мг цинка. Для получения раствора нужно в 1 л
дестиллированной воды растворить 36,4 мг химически чистого уксусноки-
слого цинка.
2. Спиртовый раствор оксихинолина. 1 г оксихинолина растворяют в
100 мл ректификованного этилового спирта,
3. Раствор сульфаниловой кислоты. 0,1 г сульфаниловой кислоты раство-
ряют в 50 мл ректификованного этилового спирта, добавляют 10 мл креп-
кой соляной кислоты и разбавляют в 100 мл дестиллированной воды.
4. 1% водный раствор азотнокислого натрия (NaNO2).
5. 25% водный аммиак.
6. Кислота лимонная или винная.
7. 10% водный раствор едкого натрия.
8. 0,1% спиртовый раствор фенолфталеина.
9. 1% спиртовый раствор метилрота.
10. Раствор НС1 1 : 5.
И. Уксуснокислый натрий кристаллический (CH3COONa • ЗН2О).
Методика определения. Для приготовления шкалы вводят
в центрифужные пробирки: 0,2; 0,5; 1,0; 1,5; 2,0; 2,5; 3,0 мл раствора уксус-
нокислого цинка и доводят объем жидкости в них дестиллированной водой
до 10 мл. В каждую пробирку прибавляют около 50 мг сухого уксуснокис-
лого натрия и по 0,3 мл раствора оксихинолина. При этом происходит осаж-
дение цинка по приведенной выше реакции. Образны помешают в водяную
баню и, доводя баню до кипения, оставляют в ней пробирки на 15 минут, после
чего центрифугируют. При этом на дне прсбирск собирается выпавший сксихи-
нолят цинка. Жидкость над ним сифонируют и производят промывание осадка
от избытка сксихинолина холодной водой с последующим центрифугирова-
нием до отрицательной диазореакции в промывных водах. Испытание на
полноту промывания производят добавлением к промывным водам 10 капель
раствора сульфаниловой кислоты, 10 капель раствора азотистокислого натрия
и 5 капель раствора аммиака. Получение светлосоломенной окраски пробы
в пробирке над осадком указывает на полноту промывания.
Промытый осадок растворяют при легком нагревании в 0,5 мл раствора
сульфаниловой кислоты с добавлением 0,5 мл раствора азотистокислого
натрия и 0,3 мл раствора аммиака.
Полученные растворы азокрасок переносят в градуированные плоскодон-
ные пробирки одинакового диаметра и доводят объемы растворов до 10 мл.
При концентрациях цинка менее 0,5 мг л шкала имеет светложелтую
окраску, при больших концентрациях — темножелтую и светлокоричнеьую.
Сравнение окрасок производят, рассматривая пробирки сверху вниз. Шкала
устойчива в течение одних суток.
По изготовлении шкалы приступают к обработке исследуемой воды. Так
как исследуемая вода кроме цинка может содержать железо, алюминий,
магний и медь, которые также осаждаются оксихинолином, поступают сле-
дующим образом.
Для связывания железа и алюминия в комплекс в исследуемую воду до-
бавляют винной или лимонной кислоты в количестве 100 мг на пробу, после
чего в пробу добавляют по каплям раствор едкого натрия до щелочной реакции
но фенолфталеину и 0,3—0,5 мл раствора оксихинолина; при этом выпадает
лишь осадок оксихинолята цинка, магния и меди. Раствор с выпавшим осад-
ком оксихинолята цинка, магния и меди нагревают на водяной бане и кипя-
тят 15 минут. Осадок центрифугируют, промывают холодной водой от избытка
оксихинолина, как при изготовлении шкалы. Осадок растворяют в 0,3 мл
раствора соляной кислоты в присутствии метилрота.
356
В образовавшийся раствор оксихинолятов цинка, магния и меди добав-
ляют кристаллического уксуснокислого натрия до перехода розовой окраски
раствора в желтую. Принтом выпадает осадок оксихинолята цинка и меди,
а магний остается в растворе.
Пробирку с осадком помещают на водяную баню, кипятят 15—20 минут,
центрифугируют, промывают осадок холодной водой, после чего его раство-
ряют при легком нагревании в 0,5 мл раствора сульфаниловой кислоты с до-
бавлением 0,5 мл раствора азотнокислого натрия и 0,3 мл раствора аммиака.
Полученный окрашенный раствор сравнивают с ранее приготовленной шкалой
азокрасок.
Предельные концентрации цинка, определяемые указанным методом,—
от 0,3 до 3 мг/л.
При м е ч а н и е. При содержании в исследуемой воде меди более 0,3 мг/л
следует в приготовленные стандартные растворы вводить медь в виде ее сернокислой
соли в количествах, соответствующих содержанию меди в исследуемой воде, или
из результатов определения цинка, выраженных в мг/л, вычесть количество меди
в мг/л, содержащейся в исследуемой воде.
Мышьяе
Мышьяксодержащие вещества могут попадать в водоисточник вместе со
сточными водами преимущественно металлообрабатывающих и химических
заводов.
Качественное и количественное определение мышьяка в воздухе и в воз-
душной пыли было изложено выше.
Согласно стандарту содержание мышьяка в питьевой воде не должно
превышать 0,05 мг/л.
По стандарту применяется следующий метод количественного определе-
ния мышьяка.
Объем пробы воды для определения мышьяка должен быть не менее 500мл.
Если анализ предполагается произвести позднее чем через 4 часа после взятия
пробы, то в нее следует ввести столько 1% раствора хлорной извести, чтобы к
моменту проведения определения в воде было не менее 1 мг/л свободного
хлора.
Принцип метода основан на восстановлении мышьяка до мышья-
ковистого водорода (ASH3), окрашивающего в коричневый цвет бумажки,
пропитанные хлорной ртутью. (Соответствующие реакции см. в главе о воз-
духе — определение AsH3).
Интенсивность окраски и длину окрашенной части бумажки, полу-
чившейся при исследовании воды, сравнивают с интенсивностью окраски
таких же бумажек, подвергнутых воздействию AsH3, выделившегося при
восстановлении мышьяксодержащих соединений в растворах с известным
содержанием мышьяка. Подыскав соответствующий эталон, делают заклю-
чение о содержании As в испытуемой воде. Это так называемая линейная
колориметрия.
Реактивы. 1. Раствор сулемы 5%.
2. Раствор KJ 10%.
3. Раствор КОН 30% водный.
4. Цинк металлический (свободный от мышьяка).
5. Кизлота серная 25% (свободная от мышьяка).
6. СЕинец уксуснокислый 5% водный раствор.
7. Двухлористое олово.
8. Стандартный раствор трехокиси мышьяка. Его готовят следующим
образом: растворяют в стакане 0,0264 г очищенной возгонкой трехокиси
мышьяка в смеси 1 мл 30% раствора КОН и 5 мл дестиллированной воды.
Полученный раствор переводят количественно в литровую колбу и доводят
до метки водой.
357
26,4 . As2
1 мл такого раствора содержит 0,02 мг As соответственно iqqq "д^о"*
Из полученного основного раствора трехокиси мышьяка готовят более
слабые растворы разбавлением в 20, 10 и т. д. раз. 1 мл слабых растворов
содержит соответственно 0,001 мл, 0,002 мл As и т. д.
9. Сулемовые бумажки. Получаются погружением плотной фильтроваль-
ной бумаги на 15 минут в 5% раствор сулемы (HgC 12). Вынутая из раствора
сулемы бумага просушивается в темноте при 20°С, разрезается на полоски
длиною 150 мм и шириной 5 мм и хранится в банке темного стекла с притер-
той пробкой.
10. Раствор хлорной извести 1%.
Методика определения. Собирают прибор, как показано на
рис. 233. Склянка а емкостью в 150—200 мл плотно закрывается пробкой,
Рис 233 При-
бор для опреде-
ления мышьяка
в воде.
а — склянка лля
испьи уемой волы,
Ъ — стеклянная
гофрированная
трубка с — стек-
лянная трубочка,
в — пробка к ней.
/ — вага, d — су-
лемевяя бумажна.
через которую пропущена гофрированная стеклянная трубка
b диаметром 12—15 мм и длиной в 150 мм. Гофрированная
трубка закрывается сверху пробкой, через которую пропу-
щена стеклянная трубочка с диаметром в 8 мм и длиной в
120 мм. Эта трубочка сверху закрывается резиновой проб-
кой е со слегка срезанной гранью.
Перед определением гофрированная трубка заполняется
до половины ватой /, пропитанной 5% раствором уксусно-
кислого свинца, для поглощения H2S, могущего выделиться
при наличии в воде сульфидов. В стеклянную трубочку с
вставляется полоска сулемовой бумажки t/так, чтобы конец
полоски проходил между срезанной гранью пробки и стенкой
трубки.
Собрав прибор, приготовляют шкалу. Для этой цели по-
мещают в склянку 1 мл слабого раствора трехокиси мышьяка,
содержащий 0,001 мл As, вносят сюда же 2мл 30% раствора
едкого калия и оставляют рас г вор на 5 минут. Затем поме-
щают в склянку 0,5 г двухлористого олова, 5 г цинка и 20 мл
25% раствора серной кислоты.
Склянку быстро закрывают пробкой с гофрированной
трубкой, несущей маленькую трубочку с сулемовой бумаж-
кой, и помещают прибор на 30 минут в темную комнату.
Затем извлекают из прибора сулемовую бумажку и по-
гружают ее в 10% раствор КJ. Через 5 минут бумажку
извлекают, промывают дестиллированной водой и сушат
между листами фильтровальной бумаги. Получившаяся на
бумажке от образования соединений AsH(HgCl)2 и As (HgCl)3
окрашенная полоска принимается за эталон, соответствующий 0,001 мг As.
Вымыв склянку, повторяют опыт, взяв 1 мл более крепкого стандартного
раствора с содержанием 0,002 мг As. Полученная на этот раз бумажка пред-
ставляет эталон в 0,002 мг As. Подобным образом приготовляют всю шкалу.
Шкала устойчива в течение суток. Она должна храниться в закрытой папке
из черной бумаги.
Имея шкалу, приступают к анализу испытуемой воды.
К 500—1000 мл воды добавляют столько 1% раствора хлорной извести,
сколько нужно для того, чтобы в воде через 2 часа осталось не менее 5 мг/л
свободного хлора. Подкисляют воду серной кислотой до слабокислой реак-
ции и упаривают на водяной бане в фарфоровой чашке до объема в 2—3 мл.
Полученный остаток вносят в склянку о прибора, смывая фарфоровую
чашку 30% раствором КОН (2—3 мл). Через 5 минут фарфоровую чашку
смывают 20 мл 25% раствора H2SO4 и смыв переносят также в склянку
а; прибавляют сюда же 0,5 г двухлористого олова и 5 г цинка.
Горлышко склянки быстро закрывают пробкой с гофрированной трубкой,
несущей маленькую трубочку с сулемовой бумажкой, и оставляют прибор
358
в темноте на 30 минут. По истечении указанного времени сулемовую бумажку
извлекают и обрабатывают точно так, как эталонные бумажки, после чего
сравнивают с подготовленной шкалой. Находят ту эталонную бумажку, на
которой длина и интенсивность окраски полоски совпадает с длиной и интен-
сивностью окраски полоски на реактивной бумажке, полученной при исследо-
вании воды; отсюда делают вывод о содержании As в испытуемой воде. Наи-
меньшее количество мышьяка, определяемое по этому методу, равно 0,001 мг.
Вместо HgCl2 лучше применять HgBr2. Вместо полосок лучше применять
небольшие кр'окки фильтровальной бумаги, зажимая их в трубке, снабжен-
ной зажимом наподобие изображенного на рис. 172. Эталоны хорошо сохра-
няются, если покрыть их тонким слоем парафина.
Кислород, растворенный в воде
Определение кислорода, растворенного в воде, дает в некоторых случаях
возможность сделать косвенное заключение о степени загрязнения воды.
Вода, бедная кислородом, обыкновенно богата органическими веществами,
которые в процессе минерализации потребляют растворенный в воде кисло-
род. Однако глубокие грунтовые воды, часто безупречные в санитарнсм отно-
шении, могут содержать очень мало свободного кислорода.
Высокое содержание свободного кислорода в воде способствует переведу
закисного двууглекислого железа в гидрат окиси, выпадающий из воды,
что вызывает уменьшение концентрации ионов железа в воде и может вести
к новому растворению железа. Таким сбразом, высокое содержание кисло-
рода в воде вызывает агрессивное поведение воды по отношению к железным
сооружениям (водопроводы, железные котлы и т. п.).
Хорошая питьевая вода температуры 5—15°С при среднем атмосферном
давлении содержит 6—7 мл кислорода в литре. В воде, очень бедной кисло-
родом (менее 1 мл на литр), не могут жить рыбы.
Для определения кислорода, растворенного в воде, предложено много
методов. Из них здесь описан наиболее применимый.
Принцип метода тот же, что и при определении кислорода воз-
духа. Реакции те же (см. стр. 221).
Из хода реакций видно, что (О) отвечает J2, а на J2 идет ?Na2S2O3. Следо-
вательно. 16 весовым частям ки.лорода соответствуют 2.243 bccoi ых частей
тиосульфата натрия. Если титрование производить 0,01 N раствором тио-
сульфата натрия, то каждый миллилитр раствора соответствует 0,08 мг (О)
или 0,055825 мл (9) при нормальных условиях.
Реактивы. 1. Раствор свободного от железа хлористого марганца
(МпС12) в дестиллированной воде (80 г соли на 100 мл воды).
2. Смесь химически чистого едкого натрия с иодистым калием (32 г
NaOH и 10 г KJ растворяют в 100 мл воды). Прибавление крахмального
клейстера к раствору не должно вызывать посинения.
3. Химически чистая соляная кислота уд. веса 1,19, не содержащая сво-
бодного хлора.
4. 0,01 N раствор тиосульфата натрия.
5. 1% крахмальный клейстер.
Методика определения. При взятии пробы воды для анализа
на свободный кислород нужно следить за тем, чтобы не изменилось содержа-
ние этого газа в воде.
Склянку в 250—300 мл с притертой пробкой наполняют исследуемой во-
дой, при этом обеспечивают по крайней мере трехкратный обмен воды в
склянке, иначе воздух, находящийся в склянке, растворяясь в воде, может
изменить количественное содержание в ней кислорода. Воду набирают в склян-
ку до края горлышка. После этого вводят пробку, следя, чтобы под ней не
оказалось пузырьков воздуха. Для этой цели хорошо пользоваться пробкой,
скошенной с одной стороны (рис. 234).
359
Рис. 234. Склянка со
скошенной пробкой.
При взятии пробы воды из крана к нему присоединяют резиновую трубку,
свободный конец которой пропускают через горлышко до дна склянки, и
дают воде несколько минут переливаться из горлышка склянки.
Для забора пробы воды в водоеме очень удобны батометры со съемными
крышками. К нижней крышке батометра присоединяют каучуковую трубку
с зажимом. Кислородную склянку сперва споласкивают дважды водой из
батометра, а потом трубку от батометра опускают на дно склянки и пускают
воду в таком количестве, чтобы произошел несколько раз обмен ее в склянке;
затем склянку закрывают пробкой.
Сейчас же после отбора пробы нужно ввести в воду реактивы за исклю-
чением тиосульфата натрия. Немедленное прибавление реактивов имеет
целью фиксацию кислорода на месте. Остальные операции можно произвести
в лаборатории.
Прибавление реактивов ведется следующим образом:
с помощью пипетки с длинным носиком вводят сначала
3 мл раствора хлористого марганца \ причем конец
носика пипетки доводят почти до дна склянки; потом
другой такой же пипеткой вводят 3 мл смеси едкого
натрия с иодистым калием; затыкают склянку пробкой,
перемешивают содержимое склянки, поворачивая по-
следнюю несколько раз дном вверх, и дают отстояться.
Извлекают пробку и приливают 5 мл соляной кислоты уд.
веса 1,192. Заткнув пробкой склянку, опять переме-
шивают содержимое, пока раствор не просветлится и
от выделившегося иода окрасится в желтый цвет. Ти-
трование можно произвести в лаборатории.
Перелив количественно содержимое склянки в колбу, приступают к ти-
трованию 0,01 N раствором тиосульфата натрия. Когда раствор приобретает
бледножелтый цвет, добавляют крахмальный клейстер и дотитровывают по-
синевший раствор. По количеству миллилитров израсходованного тиосуль-
фата рассчитывают количество кислорода в воде. При этом нужно учесть,
что при добавлении реактивов вытесняется эквивалентное количество воды
из склянки.
Пример. Вместимость склянки до края пробки — 325 мл; к взятой пробе прилито 11 мл
реактивов. На титрование израсходовано 32 мл тиосульфата натрия. Нужно вычислить
содержание кислорода в литре воды.
Каждый миллилитр 0,01 N тиосульфата соответствует 0,055825 мл кислорода. В нашем
случае израсходовано 32 мл тиосульфата натрия; отсюда заключаем, что кислорода в воде
было 0,055825 . 32.
Такое количество кислорода приходится на 314 мл волы, так как из взятой порции воды в
325 мл прилитыми реактивами вытеснено 11 мл. Таким образом, в литре исследуемой воды
свободного кислорода
0,055825 • 32 • 1000 ₽. 7Я мг1
—--------------Э,7о мл.
314
Полученный после исследования результат сравнивают с числами, при-
веденными в таблице 49. Эти числа показывают растворимость кислорода
в воде при температуре от 0 до 30°С и 760 мм атмосферного давления.
Разница между количеством кислорода, указанным в таблице и найден-
ным непосредственно из опыта, представляет дефицит содержания кислорода
в воде. Так, например, если температура воды в момент взятия пробы в преды-
дущем примере равна 6°С, а барометрическое давление 760 мм, то дефицит
содержания кислорода в этом случае равен 8,68—5,78 = 2,9 мл.
Если вода загрязнена и в ней имеются нитриты, то йодометрический спо-
соб, описанный выше, дает преувеличенные числа, так как нитриты освобож-
дают из йодистого калия свободный иод:
2KJ + 2KNO2 + 4НС1 = 2NO + 4КС1 + 2Н2О -j- J2.
1 Вместо этого раствора можно ввести 1 мл раствора MnSO4 (48 г соли в 100 мл раствора)
2 Вместо НС1 можно ввести 1 мл концентрированной H2SO4 (уд. веса 1,84).
360
Таблица 49
Растворимость кислорода воздуха в воде при нормальном барометрическом давлении
Температура Кислород мл/л Кислород мг/л Температура Кислород мл/л Кислород МГ/Л
0 10,19 14,56 16 6,89 9,85
1 9,91 14,16 17 6,75 9,65
2 9,64 13,78 18 6,61 9,45
3 9,39 13,42 19 6,48 9,26
4 9,14 13,06 20 6,36 9,09
5 8,91 12,78 21 6,23 8,90
6 8,68 12,41 22 6,11 8,73
7 8,47 12,11 23 6,00 8,58
8 8,26 11,81 24 5,89 8,42
9 8,06 11,52 25 5,78 8,26
10 7,87 11,25 26 5,67 8,11
11 7,69 10,99 27 5,56 7,96
12 7,52 10,75 28 5,46 7,82
13 7,35 10,50 29 5,36 7,68
14 7,19 10,28 30 5,25 7,54
15 7,04 10,06 I 1 — —
Если вода содержит много органических веществ, они могут связать
часть иода; в этом случае получаются малые цифры для свободного кисло-
рода в воде.
Ввиду сказанного, стандартная методика рекомендует для определения
свободного кислорода в воде модификацию описанного выше метода, устра-
няющую возможность подобных ошибок, а именно: до связывания кислорода
нитриты и органические вещества, способные окисляться на холоду, подверга-
ются окислению; во всем остальном исследование ведется, как описано выше.
В склянку после взятия пробы прибавляют 0,7 мл крепкой серной кис-
лоты (уд. веса 1,84), затем 1 мл 0,2 N раствора КМпО4 (6,32 г КМпО4 в литре
воды). Закрывают пробкой и несколько раз перемешивают содержимое склян-
ки, переворачивая ее. Если после 20 минут стояния раствор совершенно обес-
цветится, то прибавляют еще 1 мл перманганата и оставляют склянку еще
на 20 минут. Необходимо добиться, чтобы раствор после 20 минут стояния
был окрашен в розовый цвет, что указывает на избыток КМпО4 и, следова-
тельно, на закончившееся окисление нитритов и органических веществ. Из-
быток перманганата восстанавливают прибавлением 1 мл 2% оксалата ка-
лия (К2С2О4). В остальном опыт проводится, как описано выше. При вычи-
слении необходимо учесть дополнительное количество вытесненной воды.
Углекислота
Различают углекислоту свободную, гидрокарбонатную, карбонатную и
общую.
Свободная углекислота находится в воде в виде растворен-
ного газа — СО2.
361
Гидрокарбонатная углекислота входит в состав гидро-
карбонатов и находится в воде в виде гидрокарбонатных ионов — НСО8':
Са(НСО3)<2 Са" + 2НСОз-
Карбонатная углекислота входит в состав карбонатов и
находится в воде в виде карбонатных ионов — СО':
СаСО.^Са'+СОз-
Общая углекислота представляет суммарное количество всей
углекислоты в воде.
Свободная углекислота может присутствовать в воде в виде одной лишь
так называемой инактивной углекислоты или в виде инактивной и агрессив-
ной углекислоты совместно.
Ин активная углекислота представляет ту часть свободной
углекислоты, которая находится в равновесном состоянии с диссоциирующим
гидрокарбонатом кальция и не способна растворять новое количество среднего
карбоната кальция (СаСО3).
Агрессивная углекислота представляет ту часть свободной
углекислоты, которая находится в избытке (над инактивной) и способна вы-
звать диссоциацию среднего карбоната кальция:
СаСОв + Н2СО3 Са" + 2НСОз.
Присутствием агрессивной углекислоты объясняется разрушение бетон-
ных частей водопроводов и других сооружений, содержащих карбонат кальция.
Наряду с приведенной современной терминологией в литературе приме-
няются еще довольно часто устаревшие термины: «нолусвязанная» и «связан-
ная» углекислота.
Полусвязанная углекислота представляет ту часть гидро-
карбонатной кислоты, которую можно устранить кипячением воды; при этом,
как уже указывалось выше, гидрокарбонаты переходят в карбонаты по ре-
акции:
Са(НСО3)2 == СаСО3 -J- Н2О -ф- СО2.
Как видно из реакции, полусвязанная углекислота по количеству равна
половине гидрокарбонатной кислоты.
Связанная углекислота представляет ту часть гидрокарбо-
натной кислоты, которая при кипячении не удаляется, и остается связанной
с кальцием. Она по величине равна полусвязанной углекислоте и также со-
ответствует половине гидрокарбонатной.
Зная pH воды, можно судить о том, в каких формах углекислота присут-
ствует в ней. Если pH воды равен точно 8,37 1 (округленно 8,4). то в ней присут-
ствует одна лишь гидрокарбонатная кислота. Если pH воды менее 8,4, то в
воде, помимо гидрокарбонатной углекислоты, присутствует еще свободная
углекислота. Если pH воды выше 8,4, то в воде, кроме гидрокарбонатной,
присутствует и карбонатная углекислота.
Определение свободной, гидрокарбонатной и общей углекислоты
Если при добавлении фенолфталеина2 вода остается бесцветной или окра-
шивается слабее, чем стандартный раствор с активной реакцией, отвечающей
гидрокарбонатпому пункту {pH 8,4), то это свидетельствует о том, что угле-
кислота находится в воде в свободном состоянии и в виде гидрокарбонатной
углекислоты, которые и можно определить отдельно.
Необходимый для исследования стандартный раствор, pH которого равен
8,4, готовится, как указано выше (стр. 312).
1 pH гидрокарбонатного раствора равен 8,37.
2 Переход окраски фенолфталеина происходит в интервале pH 8,3—10.
362
пределение свободной углекислоты. В склянку емкостью 230 — 250 мл
штертой пробкой наливают осторожно, избегая взбалтывания, 200 мл
едуемой воды, добавляют 0,2 мл 1% раствора фенолфталеина и титруют
Я раствором Na2CO3 до появления окраски, соответствующей окраске
дарта; при этом свободная углекислота связывается с содой, образуя
юкарбонат по реакции:
СО2 + Na2CO3 + Н2О = 2NaHCO3.
Из реакции видно, что каждый миллилитр 0,1 N раствора соды, пошедший
титрование, отвечает 2,2 мг свободной углекислоты. Следовательно, если
титрование ушло t мл 0,1 N раствора соды, то в данной порции воды на-
,ится:
2,2-t свободной СО2 (в миллиграммах).
Результат перечисляют на литр.
Определение гидрокарбонатной углекислоты производится в той же самой
эбе после оттитровывания свободной СО2. Добавляют 3 капли 1% метило-
1Жа в ту же пробу и титруют 0,1 N соляной кислотой до слаборозового
зашивания. Гидрокарбонаты переходят при этом в хлориды по следующим
акциям:
Са(НСО3)2 + 2НС1 = СаС12 + 2СО2 + 2Н2О
NaHCO3 + HCI = NaCl + СО, + Н2О.
Из реакций видно, что каждый миллилитр 0,1 N соляной кислоты, пошед-
ей на титрование, соответствует 4,4 мг гидрокарбонатной углекислоты,
ак как при титровании часть соляной кислоты уходит на взаимодействие
теми гидрокарбонатами, которые образовались из свободной углекислоты
ри первом титровании (см. вторую реакцию), то для того, чтобы определить
олько ту гидрокарбонатную кислоту, которая была в исследуемой воде,
ужно от количества затраченной соляной кислоты отнять количество милли-
штров израсходованной соды и результат умножить на 4,4. Таким образом,
ели при первом титровании пошло t мл 0,lNa2CO3, а при втором Т мл
>, 1NHC1, то количество гидрокарбонатной кислоты в данной пробе равно:
4,4 (Т — t)CO2 (в миллиграммах).
Результат перечисляют на литр.
Определение общей углекислоты сводится к суммированию свободной и
гидрокарбонатной углекислоты. В данном случае оно равно:
2,2t + 4,4 (Т — t) = 2,2(2Т — t) СО2 (в миллиграммах).
Результат перечисляют на литр.
Определение кар Зон атной, гидрокарбонатной и общей углекислоты
Если от добавления фенолфталеина вода окрасится в цвет, более интен-
сивный, чем цвет стандарта, то это свидетельствует о присутствии в ней кар-
бонатной и гидрокарбонатной углекислоты.
Определение карбонатной углекислоты 200 мл исследуемой воды после
добавления 0,2 мл 1% раствора фенолфталеина титруют 0,1 N соляной кисло-
той до цвета, соответствующего стандарту; карбонаты при этом переходят
в гидрокарбонаты по реакции:
2СаСО3 + 2НС1 = Са(НСО3)2 + СаС12.
Из реакции видно, что каждый миллилитр 0,1 N соляной кислоты, пошедшей
на титрование, соответствует 4,4 мг карбонатной углекислоты. Таким образом,
если на титрование ушло всего t, мл соляной кислоты, то количество карбо-
натной углекислоты во взятой пробе будет равно:
4,4^002 (в миллиграммах).
зез
Определение гидрокарбонатной углекислоты производится в той же пробе
после первого титрования. Добавляют 3 капли 1% метилоранжа и титруют
до слаборозового окрашивания. При этом гидрокарбонаты переходят в хло-
риды по реакции:
Са(НСО3)2 + 2НС1 = СаС12 + 2СО2 + 2Н2О.
Так как при втором титровании часть соляной кислоты уходит на взаимо-
действие с теми гидрокарбонатами, которые образовались из карбонатов при
первом титровании, то из расхода соляной кислоты при втором титровании
вычитают расход соляной кислоты при первом титровании и результат умно-
жают на 4,4.
Таким образом, если при первом титровании пошло мл 0,1 N НС1, а при
втором — мл, то количество гидрокарбонатной кислоты во взятой порции
воды окажется равным:
4,4(Tj — tJCOg (в миллиграммах).
Определение общего количества углекислоты в данном случае сводится
к суммированию карбонатной и гидрокарбонатной углекислоты:
4,4tj + 4,4(Ti — tj = 4,4ТСО2 ( в миллиграммах).
Определение агрессивной углекислоты
Агрессивная углекислота определяется при помощи таблиц или титро-
метрически. Более надежным является титрометрический метод, который
здесь и приводится.
Принцип метода заключается в том, что к взятой пробе воды
прибавляют немного истолченного мраморного порошка (СаСО3). Если в воде
присутствует агрессивная углекислота, то она переведет часть карбоната
кальция в гидрокарбонатное состояние по реакции:
СаСО3 + Н2СО3 Са- + 2НССХ.
Таким образом, общее количество свободной углекислоты уменьшится,
а содержание в воде гидрокарбонатной углекислоты возрастает. Сопоставляя
содержание в воде гидрокарбонатной кислоты до прибавления мрамора и
после прибавления его, легко рассчитать количество агрессивной углеки-
слоты: оно равно половине разности этих двух величин, так как агрессивная
углекислота составляет лишь половину прибавки гидрокарбонатной угле-
кислоты, что легко видеть из приведенной выше реакции.
Реактивы. 1. 0,1 N раствор Na2CO3.
2. 0,1 N раствор НС1.
3. 1% фенолфталеин.
4. 1%о метилоранж.
5. Истолченный в порошок мрамор.
Методика определения. Всыпают в склянку емкостью 400—500
мл 3—5 г мелко истолченного мраморного порошка и с помощью сифона, ста-
раясь избежать потерь СО2, наливают сюда столько воды, чтобы под пробкой
осталось небольшое воздушное пространство. Оставляют пробу на 3—4 дня,
время от времени энергично взбалтывая. Дав отстояться жидкости, отбирают
пипеткой 200 мл и определяют количество гидрокарбонатной углекислоты в
данной пробе. Отдельно определяют количество гидрокарбонатной углекислоты
в 200 мл исследуемой воды без прибавления мрамора. Если содержание гидро-
карбонатной углекислоты во взятой пробе с прибавкой мрамора обозначить
через Р, а содержание той же углекислоты в пробе без обработки мрамором
обозначить через р, то содержание агрессивной углекислоты во взятом объеме
Р—п
воды выразится формулой: —СО2 (в миллиграммах).
Результат перечисляют на литр.
364
Пример 1. При добавлении фенолфталеина к воде окраска получилась слабее стандарт-
ной; следовательно, вода содержит свободную и гидрокарбонатную углекислоту.
При первом титровании взятой пробы (200 мл) затрачено 4,5 мл 0,1 N раствора Na2COr
При втором титровании затрачено 12,8 мл 0,1 N раствора НС1. Следовательно:
2,2 • 4,5 . 1000
свободной СО2 в литре воды содержится ---------------= 49,5 мг,
гидрокарбонатной СО2 » » »
общей СО2 » » »
полусвязанной и связанной СО2 »
4,4(12,8—4,5) • 1000
----------200------= 182,6 МГ’
2,2(2 . 12,8—4,5) • 1000
> ----------200------------' 232,1 МГ|
4,4(12,8—4,5) • 1000
» по---------200 72-------91,3мг.
Пример 2. При добавлении фенолфталеина вода окрасилась интенсивнее стандартного
раствора; следовательно, опа содержит карбонатную и гидрокарбонатную углекислоту.
При первом титровании взятой пробы (200 мл) затрачено 1,2 мл 0,1 N НС1. При втором титро-
вании — 16,6 мл 0,1 N НС1. Следовательно:
4,4-1,2.1000
карбонатной углекислоты в литре воды содержится ------------— 26,4 мг,
4,4(16,6— 1,2) . 1000 о ,
гидрокарбонатной » » » » » ---------2oq---------- 338,8 мг,
4,4 • 16,6 • 1000
оощеи » » » » » -----2Q0------ = 365,2 мг.
Пример 3. При определении агрессивной углекислоты взято для анализа 200 мл воды.
Количество гидрокарбонатной углекислоты без прибавления мрамора оказалось равным
38,2 мг; в другой пробе, по объему также 200 мл после прибавления мрамора содержание
гидрокарбонатной углекислоты оказалось равным 42,5 мг. Отсюда заключаем, что количе-
ство агрессивной углекислоти в 1 л воды равно:
(42,5 — 38,2) . 1000
200~2
= 10,75 мг.
Сероводород
Сероводород, открываемый иногда в воде, может быть минерального и
органического происхождения.
Минерального происхождения H2S может образовываться в грунтовой
воде за счет сульфидных соединений (колчеданов):
FeS 4- 2СО2 4- 2Н2О = Fe(HCO3)2 4- H2S.
В водах болотного происхождения присутствие H2S может обусловливаться
восстановлением сернокислых солей металлов до сероводорода присутствую-
щими в воде гуминовыми веществами.
Органического происхождения сероводород образуется за счет разложе-
ния белковых веществ. В этом случае вода оказывается сильно загрязненной.
Выражается в мг или мл H2S.
Качественное определение
1. По запаху.
2. С помощью уксуснокислого свинца. К 10 мл исследуемой воды при-
бавляют 1 мл 20% раствора сегнетовой соли и несколько капель щелочного
раствора свинца (приготовление см. на стр. 367). В присутствии сероводорода
жидкость вследствие выпадения сульфида свинца приобретает окраску от
желтобурой до черной:
Pb(C2H3O2)2 + H2S = PbS 4- 2С2Н4О2.
365
Эта же реакция получается, если между пробкой и горлышком склянки,
в которую налита испытуемая вода, зажать бумажку, обработанную щелоч-
ным раствором свинцовой соли, и оставить на несколько часов.
3. При помощи раствора диметилпарафенилендиамина и хлорного железа
в соляной кислоте (приготовление см. на стр. 248). К 10 мл воды прибавляют
3 мл указанного реактива и взбалтывают. При малых количествах сероводо-
рода жидкость приобретает зеленую окраску, при больших — синюю (обра-
зование метиленовой синьки). Одновременно проводят контрольную пробу
с дестиллированной водой. Метод неприменим при наличии в воде значитель-
ных количеств нитритов.
Количественное определение
При наличии в воде больших количеств H2S определение его можно вести
иодометрически, при малых количествах — колориметрически.
Йодометрическое определение
Принцип метода. Сероводород окисляют иодом, получаемым в
подкисленной воде из КJ при воздействии КМпО4. По количеству иода, по-
шедшего на окисление H2S, делают заключение о количестве последнего:
H2S + J2 = 2HJ + S.
1 мл 0,01 N раствора иода соответствует 0,17 мг H2S.
Реактивы. 1. H2SO4 (1 : 3).
2. 10% раствор KJ.
3. 0,01 N раствор КМпО4.
4. 0,01 N раствор Na2S2Os . 5Н2О.
5. 1% крахмальный клейстер.
Методика определения. В колбу вводят 100 мл исследу-
емой воды, подкисляют серной кислотой, прибавляют 1 мл 10% KJ, а затем
при взбалтывании содержимого колбы добавляют из бюретки 0,01 N раствор
КМпО4 до появления ясного желтого цвета. При этом протекают следующие
реакции:
2КМпО4 4- ЮК J 4- 8H2SO4 = 6K2SO4 4- 2MnSO4 4- 8НаО 4- 5Ja
Has4-J2=2HJ4-S.
Появление желтой окраски указывает на то, что весь сероводород окислен
выделившимся иодом и имеется избыток последнего. Этот избыток оттит-
ровывают 0,01 N раствором тиосульфата натрия:
J2 4- 2NaaS2O2 = 2NaJ + Na2S4Oe.
Разность между числом миллилитров прибавленного 0,01 N раствора
КМпО4 и числом миллилитров израсходованного на титрование 0,01 N ра-
створа тиосульфата натрия соответствует числу миллилитров 0,01 N раствора
иода, пошедшего на окисление H2S. Это число умножают на 0,17 и опреде-
ляют, таким образом, количество миллиграммов H2S в 100 мл исследуемой
воды. Результат перечисляют на литр. Полученное число миллиграммов H2S
л яс 22,48
для пересчета на миллилитры умножают на О,бб соответственно •
34, Uo
Колориметрическое определение
Колориметрическое определение с уксуснокислым свинцом. Принцип
метода. К исследуемой воде добавляют щелочпый раствор свинца (см.
ниже) и полученное окрашивание, обусловленное образованием PbS,
сравнивают с окраской, возникающей при постепенном введении стандарт-
ного раствора трехсернистого мышьяка в щелочный раствор свинцовой соли.
Сравнение ведется по методу колориметрического титрования.
Эбб
Реактивы. 1. 37 мг сухого As2S3 растворяют в нескольких каплях
аммиака и доводят дестиллированной водой до 100 мл. Каждый миллилитр
л с 102,27-0,37
раствора содержит 0,37 мг As2S3, что соответствует —246~13— мг
(102,27 — утроенный молекулярный вес сероводорода; 246,13 — молекуляр-
ный вес AsoS3). Перечислив полученное весовое количество H2S на объемное,
102 27*0 37 -22 48
получаем: —246 li 34 03— ~ мл H2S при нормальных условиях.
Следовательно, 1,0 мл стандарта соответствует 0,1 мл H2S.
Трехсернистый мышьяк (As2S3), необходимый для приготовления указан-
ного стандарта, получается следующим образом: раствор мышьяковистой
кислоты (As2O3) подкисляют серной кислотой и обрабатывают током серово-
дорода. Выпавший As2S3 отфильтровывают, промывают и сушат при 100°С.
2. Щелочной раствор свинца: 25 г сегнетовой соли, 1 г уксуснокислого
свинца и 5 г едкого натрия растворяют в воде и доводят объем до 100 мл.
Методика определения. В склянку из бесцветного стекла
емкостью в 150 мл вливают 5 мл щелочного раствора свинца, затем добавляют
100 мл исследуемой воды и взбалтывают. В другую такую же склянку вли-
вают также 5 мл щелочного раствора свинца, 100 мл дестиллированной воды,
взбалтывают и прибавляют из бюретки приготовленный, как указано выше,
стандартный раствор до тех пор, пока жидкость в склянке не примет такой
же цвет, как и исследуемая вода. Число миллилитров ушедшего на титрование
стандарта соответствует количеству миллилитров H2S в литре исследуемой
воды.
Колориметрическое определение с димстплпарафенилендиамипом. Принцип
«методика те же, что и при определении сероводорода в воздухе (см. стр. 248).
Остаточный активный хлор
Остаточный активный хлор (свободный хлор) определяют в воде, которая
была подвергнута хлорированию.
Согласно ГОСТ 2874-45 содержание остаточного активного хлора в во-
допроводной воде даже в наиболее удаленных точках водоразбора (наружного
и внутреннего) должно быть не менее 0,1 мг/л.
Качественное определение
1. Органолептически по запаху и вкусу.
2. С KJ в кислом растворе в присутствии крахмального клейстера.
50—100 мл воды подкисляют уксусной (соляной, серной) кислотой, при-
бавляют 0,2—1 мл 10% KJ и 1—2 мл крахмального клейстера (1%). При на-
личии свободного хлора из йодистого калия выделяется эквивалентное ко-
личество иода по реакции:
Cl2 + 2KJ = 2KCI + J2.
Выделившийся иод с крахмальным клейстером дает синее окрашивание.
Реакция дает возможность обнаружить еще 0,13 мг свободного хлора в литре
воды. Однако, если смотреть через высокий столб воды (20—25 см) в цилиндре
из бесцветного стекла, помещенном на белом фоне, то окрашивание заметно
еще при содержании 0,0002 мг С12 в литре воды.
Другие окислители — окись железа, нитриты и т. п. — дают в кислом
растворе идентичную реакцию.
Так, при наличии окиси железа при подкислении воды и прибавлении KJ,
протекают следующие реакции:
KJ + СН3СООН = СН3СООК = HJ
Fe2(SO4)3 + 2HJ = 2FeSO4 + H2SO4 + J*
зет
В случае наличия нитритов при тех же условиях протекают следующие
реакции:
KNO2 4- СН3СООН = СН3СО(Ж + HNO2
2HNO2 = N2O3 + Н,0
KJ + СН3СООН = СН3СООК + HJ
N2O3 + 2HJ = Н2О + 2NO + J2.
При наличии в воде большого количества органических веществ часть
иода может оказатся связанной, ввиду чего чувствительность реакции по-
нижается.
3. С ортолидином (диметил-бензидином) — [CH3C6H8NH2|2. По Георгиев-
скому, 1 г ортолидина растворяют в 800 мл дестиллированной воды, прибав-
ляют по частям 100 мл концентрированной соляной кислоты при постоянном
взбалтывании и доводят объем раствора дестиллированной водой до 1 л. По-
лучается почти совершенно бесцветный, хорошо устойчивый при условии
хранения в темном месте реактив.
1 мл раствора прибавляют к 100 мл испытуемой воды. В присутствии
свободного хлора получается синее окрашивание.
Количественное определение
При относительно больших количествах свободного хлора в воде опре-
деление ведут иодометрически; при малых количествах применяют крахмал-
иодо-колориметрическое определение.
В хлорированной питьевой воде, ввиду малого содержания в ней свобод-
ного хлора, его чаще определяют крахмал-иодо-колориметрически.
Крахмал-иодо-колориметрический метод
Принцип метода тот же, что и при качественном определении
свободного хлора в кислом растворе при помощи KJ и крахмального клей-
стера.
Реактивы. 1. Буферная смесь (pH 4,6): смешивают в литровой мер-
ной колбе 102 мл 1 N раствора уксусной кислоты с 98 мл 1 N раствора кри-
сталлического уксуснокислого натрия (NaC2H3O2. ЗН2О) и доливают раствор
водой до метки.
1 N раствор уксусной кислоты получают растворением 75,03 г 80% уксус-
ной кислоты в дестиллированной воде в литровой мерной колбе; раствор
затем доливают водой до метки.
1 N раствор уксуснокислого натрия приготовляют растворением 137,46 г
химически чистого уксуснокислого натрия в воде в литровой мерной колбе
с последующим добавлением воды до метки.
2. 10% KJ.
3. 1% крахмальный клейстер.
4. Стандартный раствор иода, 1 мл которого соответствует 0,1 мг актив-
ного хлора.
К 5 мл 10% йодистого калия прибавляют 10 мл буферной смеси и 10 мл
0,01 N марганцевокислого калия и доводят водой до 35,5 мл. В полученном
растворе общее количество выделившегося иода эквивалентно 0,355. 10 мг
. 0,355-10 П1 _
хлора, что на 1 мл дает: —— — 0,1 мг свободного хлора.
оэ,э
Составляют стандартный ряд из цилиндров одинакового диаметра; в ци-
линдры вводят стандартный раствор, начиная от 0,1 и до 1,0 мл, что соответст-
вует от 0,01 и до 0,1 мг хлора. В каждый стандартный цилиндр доливают
дестиллированной воды до общего объема 101,0 мл. В цилиндр такого же
диаметра наливают 100 мл исследуемой воды, прибавляют сюда 1 мл 10%
368
йодистого калия. Далее приливают во все цилиндры по 10 мл буферного
раствора, если щелочность исследуемой воды не выше 7 мг-эквивалентов;
если же щелочность исследуемой воды выше 7 мг-эквивалентов, то количе-
ство миллилитров буферного раствора, прибавляемого во все цилиндры,
должно быть в полтора раза бзлыне щелочности исследуемой воды. Наконец,
во все цилиндры прибавляют по 1 мл 1% крахмального клейстера и колори-
метрируют.
Иодометрнчееклй хетод
Принцип метода. Свободный хлор в подкисленной воде вытес-
няет из прибавленного йодистого калия эквивалентное количество иода,
которое оттитровывают тиосульфатом натрия.
Реактивы. 1. Буферная смесь (см. стр. 3 8).
2. Химически чистый иодистый калий кристаллический.
3. 0,005 N раствор тиосульфата натрия.
4. 1% крахмальный клейстер.
Методика определения. Вводят в коническую колбу 0,5 г KJ,
приливают 2 мл дестиллирсваннсй воды, перемешивают содержимое до ра-
створения KJ, приливают 10 мл буферного раствора, если щелочность иссле-
дуемой воды не выше 7 мг-экв! валснтсв. Если щелочность исследуемой
воды выше 7 мг-эквивалентов, то количество буферного раствора должно быть
в полтора раза более щелгчнссти исследуемой воды. Доливают 100 мл
исследуемой воды, все взбалтывают и выделившийся иод титруют 0,005 N
раствором тиосульфата натрия до появления бледнолжелтой окраски. Прили-
вают 1 мл крахмального клейстера и дотитровывают до исчезновения синей
окраски.
Каждый миллилитр израсходованного тиосульфата натрия отвечает
С1
0,1775 мг свободного хлора соответственно ^оо”’
Для определения свободного хлора в испытуемой воде пользуются форму-
лой:
где X — активный хлор в мг'л,
п — количество миллилчтоэв тиосульфата, израсходованное на титро-
вание ЮЭмл исследуемой воды.
Показате 1ь хторир емости поды
Под показателем хлорирузмости воды понимают вели-
чину той дозы хлора (в мг л), при введении которой в исследуемую воду в по-
следней после ЗО минутнсго к. нтакта остается 0.5 мг/л остаточного хлора.
Показатель хлгрирусмости тем выше, чем выше хлоропоглощае-
мость воды. Хлоре псглс щасмгсть же зависит от наличия в воде легко
окисляющихся веществ: в их присутствии происходит быстрый гидролиз
хлора и возрастает хлорспоглсшасм^сть воды:
Cl —С14-Н —ОН ^НС1 + НСЮ
НС1 О “* идет иа окисление.
1
24 А. И. Бурштейн
ci изымается
щелочами,
белками и т. п.
Ниже приводится метод определения показателя хлорируемости воды.
Принцип метода. В равные количества исследуемой воды прибав-
ляют возрастающие количества активного хлора. Через 30 минут опре-
деляют количества остаточного хлора во взятых пробах воды и находят ту
369
пробу, в которой остаточного хлора оказалось 0,5 мг/л (или близко к этому).
Количество активного хлора, прибавленного в эту пробу воды, соответствует
показателю хлорируемости воды.
Реактивы. 1. Хлорная вода, содержащая 100 мг С12 в литре.
Дестиллированную воду насыщают газообразным хлором. Определяют
в ней концентрацию хлора титрованием 0,005 N раствором тиосульфата натрия
и разбавляют водой до концентрации 100 мг/л.
Положим, что при титровании 50 мл хлорной воды (после прибавления
5 мл 10% KJ, 5 мл 5% СН3СООН и 1 мл крахмального клейстера) уходит
40 мл 0,005 N тиосульфата натрия, что соответствует 0,1775. 40 мг хлора
в 50 мл или 0,1775 . 40 . 20 = 142 мг С12 в литре. Следовательно, на каждые
100 мл хлорной воды нужно прибавить 42 мл дестиллированной воды, чтобы
получить нужную концентрацию.
2. 10% KJ.
3. Буферная смесь (pH 4,6; приготовление см. на стр. 368).
4. 1% крахмальный клейстер.
5. 0,005 N раствор тиосульфата натрия.
Методика определения. В 10 конических колб каждая ем-
костью в 250 мл с хорошо подогнанными резиновыми пробками вливают по
100 мл исследуемой воды. Затем вливают в первую колбу 0,5 мл приготовлен-
ной, как указано выше, хлорной воды, а в каждую последующую — на 0,25 мл
больше. В каждой колбе налитое количество хлорной воды соответствует
дозе хлора (в мг/л). Тщательно перемешав содержимое каждой колбы много-
кратным опрокидыванием закрытой колбы вверх дном, оставляют колбы на
30 минут для контакта. Затем прибавляют в каждую колбу по 5 мл раствора
KJ, далее приливают по 10 мл буферного раствора, если щелочность иссле-
дуемой воды не выше 7 мг-эквивалентов, если же щелочность исследуемой
воды выше 7, то количество миллилитров буферного раствора должно быть
в полтора раза больше щелочности исследуемой воды. Затем титруют
тиосульфатом до появления бледножелтой окраски, прибавляют крахмаль-
ный клейстер и дотитровывают. По расходу тиосульфата определяют оста-
точный хлор. Найдя колбу, где остаточный хлор соответствует 0,5 мг/л (или
близко к этому числу), устанавливают по дозе хлора (в мг/л), введенной в
эту колбу, показатель хлорируемости воды.
Пример. На титрование остаточного хлора в пятой колбе затрачено 0,28 мл 0,005 N
тиосульфата натрия, следовательно, остаточного хлора здесь 0,1755. 0,28, что на литр состав-
ляет 0,1775 . 0,28 . 10 = 0,497мг или округленно 0,5 мг/л. Показатель хлорируемости ис-
следуемой воды равен 1,5 мг, так как в пятую колбу с хлорной водой введено 1,5 мг С1г.
Фтор
Вопрос о содержании фтора в воде в настоящее время разрешен в том
смысле, что большие количества фтора ведут к заболеванию флюорозом,
а малые (менее 0,5 мг) — к кариесу зубов.
Согласно ГОСТ 2874-45 содержание фтора в воде не должно превышать
1 мг в литре воды.
Качественное определение
Воду подщелачивают в избытке известковым молоком и выпаривают в пла-
тиновой чашке до получения плотного остатка. Последний высушивают и
прокаливают. С золой проводят следующие реакции.
1. Помещают золу в платиновый тигель, смачивают несколькими каплями
дестиллированной воды, прибавляют 1 мл концентрированной серной кислоты
и быстро накрывают часовым стеклом; выпуклая сторона стекла, обращен-
ная к тиглю, покрыта воском (парафином), на котором нацарапана надпись
или рисунок (до стекла). Тигель, накрытый часовым стеклом, помещают на
асбестовую сетку и осторожно нагревают. На часовое стекло для охлаждения
370
помещают кусочек льда. Если в золе присутствуют фтористые соединения,
то выделяющаяся фтористоводородная кислота (HF) выщелачивает из сили-
ката стекла кремний (SiO2), который с HF дает летучий фтористый кремний
(SiF4). Часовое стекло в местах, обнаженных от воска (парафина), оказывается
изъеденным:
CaF, + H2SO4 = CaSO4 + 2HF
4HF + SiO2 = SiF4 + 2H2O.
2. Золу помещают в пробирку и приливают концентрированной серной
кислоты. В отверстие пробирки вставляют стеклянную палочку с каплей
воды на конце. Выделяющийся фтористый кремний, растворяясь в воде,
вызывает ее помутнение вследствие образования кремневой кислоты:
3SiF4 + ЗН2О - H2SiO3 + 3HoSiF6.
Количественное определение
Согласно стандарту количественное определение фтора в воде про-
изводится колориметрически с ализаринциркониевой смесью.
Принцип метода основан на способности фторидов изменять
в кислой среде окраску ализаринциркониевой смеси от розовой до желтой.
Чем больше фторидов прибавлено к ализаринциркониевой смеси, тем слабее
становится ее розовая окраска. Сравнивая окраску подкисленной исследуе-
мой воды после введения в нее ализаринциркониевой смеси с окраской раство-
ров фторида натрия с известным содержанием фтор-иона в тех же условиях,
определяют содержание фтор-иона в исследуемой воде.
Определение ведется с отгоном или без отгона воды (в зависимости от
солевого состава воды).
Определение с отгоном
При большом содержании SOJ, С1', РО4', Fe, Al, мешающих реакции,
определение ведется с отгоном.
Реактивы. 1. Раствор 0,5 г оксихлорида циркония (ZrOCl2.8Н2О)
в 100 мл дестиллированной воды.
2. Раствор 0,1 г ализаринмоносульфата натрия в 100мл дестиллированной
воды. Хранить в темном прохладном месте.
3. Ализаринциркониевый раствор: к 50 мл раствора оксихлорида цирко-
ния (раствор 1) прибавляют по каплям при непрерывном помешивании 50 мл
раствора ализаринмоносульфата натрия (раствор 2). Если раствор получается
мутным, то ему дают осветлиться в течение 8—10 часов в темном прохладном
месте, после чего разводят 200 мл дестиллированной воды. Раствор сохраняют
в темном прохладном месте. Пригоден он в течение 2 недель.
4. Соляная кислота 5 N раствор.
5. Раствор фтористого натрия (NaF), 1 мл которого соответствует 0,01 мг
фтор-иона. Растворяют 0,221 г химически чистого NaF в 50 мл воды и дово-
дят раствор до 100 мл водой. 1 мл такого раствора содержит 1 мг фтор-иона
221 . F
соответственно .
10 мл полученного раствора разбавляют до литра водой. 1 мл разбавлен-
ного раствора содержит 0,01 мг фтор-иона.
6. Кислота серная уд. веса 1,84
7. Кислота кремневая в порошке.
8. 5% раствор NaOH.
9. Фенолфталеин (спиртовый 0,1% раствор).
Все перечисленные реактивы, за исключением раствора фтористого натрия,
должны быть свободны от фтора.
371
для подачи пара в колбу
Методика определения. Собирают прибор, как показано на
рис. 235.
Колба 1 емкостью в 250 мл соединена при помощи отводной трубки с хо-
лодильником и при помощи трубки, дважды изогнутой под прямым углом,
с колбой 2, служащей в качестве паровичка.
В колбе 7 имеется сбоку шейка, в которую вставляют пробку, а через
нее пропускают термометр со шкалой до 200°С.
При отсутствии такой колбы можно использовать обыкновенную кругло-
донную широкогорлую колбу, закрывающуюся пробкой с тремя отверстиями:
в одно отверстие вводится шейка термометра, в другое — трубка от паро-
вичка, в третье — трубка для соединения с холодильником.
Через пробку, закрывающую колбу 2, вводятся две трубки: одна служит
7, а вторая, на которую надета резиновая трубка
с винтовым зажимом, служит для выпуска пара в
атмосферу и регулирования подачи пара в колбу 7.
Собрав, как описано, прибор, приступают к по-
лучению отгона из испытуемой воды.
Для этой цели наливают в стакан 250 мл иссле-
дуемой воды, подщелачивают ее 5% раствором
NaOH до щелочной реакции по фенолфталеину и
упаривают на водяной бане до объема 50—60 мл;
охлаждают и прибавляют по каплям 1 мл серной
кислоты, не допуская при этом разогревания жид-
кости.
Содержимое стакана переносят количественно
в колбу 7. Одновременно в ту же колбу помещают
10 стеклянных бус и 0,5 г кремневой кислоты в по-
рошке. Затем, поместив колбу в сосуд с холодной
водой, вносят в колбу по каплям 10мл серной кисло-
ты, не давая жидкости разогреваться. Извлекши
колбу из сосуда с холодной водой, насухо обтирают
стенки колбы и соединяют ее с холодильником и
колбой 2, как показано на рисунке, после чего
приступают к постепенному нагреванию колбы 7 до 100°С. После отгона пер-
вых 5—10 мл нагревание усиливают так, чтобы в минуту отгонялось 20—30
капель. Одновременно нагревают колбу 2, выпуская сначала пар в атмосферу,
когда же температура жидкости в колбе достигает 135—137°С, ее нагревание
уменьшают и, уменьшая выход пара в атмосферу, начинают подавать его
в кипящую жидкость в колбе 7, следя за тем, чтобы температура была в пре-
делах 138—140°С и отгон шел со скоростью 2—3 мл в минуту до тех пор,
пока объем отгона не составит 100 мл; тогда отгон прекращают. Фтор
находится в отгоне в виде четырехфтористсго кремния (SiF4) и кремне-
фтористоводородной кислоты (H2SiF6). Отгон используется для колориметри-
рования.
Стандартный ряд готовят следующим образом. В 10 цилиндров бесцвет-
ного стекла с меткой на 100 мл наливают по 100 мл дестиллированной воды
и вносят в первый цилиндр 0,5 мл стандартного раствора фтористого натрия,
во второй цилиндр — 1 мл, в третий — 2 мл, в четвертый — 3 мл и т. д. До-
бавляют в каждый цилиндр по 5 мл 5 N раствора НС1 и после перемешива-
ния содержимого вносят в каждый цилиндр по 5 мл ализаринииркониевого
раствора. Снова перемешивают и оставляют стоять 2 часа. Шкала пригодна
в течение суток.
Через час после приготовления шкалы в такой же цилиндр, как те, в ко-
торых приготовлена шкала, наливают 100 мл полученного отгона, добавляют
5 мл 5N НС1 и после перемешивания — 5 мл ализаринциркониевого раство-
ра. После этого пробу оставляют в течение одного часа рядом с цилиндрами
шкалы, чтобы температура исследуемой пробы сравнялась с температурой
Рис. 235. Прибор для опре-
деления фтора в воде.
1 — иолба для испытуемой иолы,
S— колба, служащая в качестве
паровичка, 3 — трубка с зажи-
мом.
372
раствора шкалы, после чего приступают к колориметрированию. При этом
раствор в цилиндре рассматривают сверху вниз на белом фоне.
Пример. Окраска отгона испытуемой воды оказалась равной окраске раствора в 4-л.
цилиндре шкалы. Для исследования взято 250 мл испытуемой воды. Нужно найти содержа-
ние фтора в литре воды.
0,03 • 1000
Оно равно:--250---= °’12 мг/л.
При содержании в исследуемой пробе более 0,1 мг фтора окраска полу-
чается слабее, чем в десятом цилиндре шкалы, где содержание фтора достигает
лишь 0,09 мг. В этом случае нужно повторить определение, разбавив испы-
туемую воду дестиллированной водой в несколько раз, или взять меньший
объем воды для анализа.
Определение без отгопа
При общем содержании солей не более 500 мг/л, в том числе сульфатов
(SO/) не более 200 мг л, хлоридов (СГ) не более 300 мг'л, железа (Fe) не более
2 мг/л, алюминия (А1) не более 0,5 мг л, фосфатов (РО4"') не более 1 мг/л и
цветности не более 25° платиново-кобальтовой шкалы, определение содержа-
ния фтора можно производить без отгона.
Принцип метода тот же, что и при определении с отгоном.
Реактивы: Раствор сернокислого натрия (Na2SO4): 14,8 г сернокис-
лого натрия (безводного) растворяют в дестиллированной воде и доводят
объем раствора до литра. Каждый миллилитр раствора содержит 10 мг суль-
14с<00 - SO4 _
фат-иона соответственно Остальные реактивы те же, что и
т 1000 • Na2SO4 г
для предыдущего метода.
Методика определения. Приготовляют стандартный ряд.
В 10 цилиндров бесцветного стекла с меткой на 100 мл вносят такое количество
миллилитров раствора сернокислого натрия, которое нужно для того, чтобы
при разведении его до 100 мл дестиллированной водой концентрация сульфат-
иона соответствовала концентрации сульфат-иона в испытуемой воде, и дово-
дят раствор до метки дестиллированной водой. В дальнейшем шкала гото-
вится так же, как в предыдущем методе. Шкала пригодна в течение суток.
Через час после приготовления шкалы берут 100 мл испытуемой воды
и поступают так же, как и при определении с отгоном воды. Расчет такой же.
ПОЛЕВЫЕ МЕТОДЫ ФИЗИКО-ХИМИЧЕСКОГО ИССЛЕДОВАНИЯ
ПИТЬЕВОЙ ВОДЫ
Стандарт приводит простейшие полевые методы физико-химического исследования
питьевой воды. Эти методы применимы, когда требуется на месте (срочно) произвести
анализ воды и в распоряжении нет полностью оборудованной лаборатории. В этих случаях
пользуются портативной переносной лабораторией. Сами методы являются упрощенными
и не могут претендовать па большую точность. Тем не менее они должны давать вполне
приемлемые результаты, не резко отличающиеся от точных.
ОТБОР ПРОБ
При отборе проб воды необходимо придерживаться тех указаний, которые были даны
выше (стр. 295). И здесь нельзя делать каких-либо отступлений.
Для анализа требуется около одного литра воды.
ФИЗИЧЕСКИЕ СВОЙСТВА ВОДЫ
Температура. Термометр погружают на 15 минут в водоем на ту же глубину, с которой
берется проба воды для исследования.
Из колодцев, буровых скважин и т. п. источников, доступ к которым затруднен, отби-
рают ведро воды и сразу замеряют в нем температуру.
Прозрачп кит. определяется в нефильтрованной воде при помощи чтения шрифта Снел-
лена № 1, подложенного под цилиндр, в который наливается исследуемая вода (стр. 299).
373
Цветность определяется только качественно. Если вода мутна, ее фильтруют. В колбу
белого стекла наливают около 100 мл воды и определяют окраску, рассматривая колбу
с водой на белом фоне. Цветность воды характеризуют словами: бесцветная, светложелтая,
желтая, интенсивно желтая и т. п.
Запах воды определяется при температуре 20° С и 40° С, как описано выше (стр. 298).
Вкус определяется после кипячения воды и охлаждения се до 25° С В случае подозре-
ния на бактериологическую или химическую зараженность воды проба на вкус не произво-
дится.
ХИМИЧЕСКИЙ СОСТАВ ВОДЫ
Щеючпость воды определяется, как описано выше (стр. 309), с той лишь разницей, что
вместо раствора метилоранжа применяют метилоранжевые бумажки, которые готовят следую-
щим образом: нарезают фильтровальную бумагу полосками 1 х 5 см, смачивают их раство-
ром 1% метилоранжа, высушивают и повторно смачивают, пока высушенные бумажки не
приобретут темноораижевую окраску. Полоски сшиваются в книжечку с обложкой из про-
стой бумаги. На определение берется 1 см2, такой реактивной бумажки, которая опускается
в колбу с испытуемой водой. Вода взбалтывается до равномерного распределения окраски.
Карбонатная жесткость определяется путем умножения щелочности воды, выраженной
в миллилитрах, на 2,8 (см. стр. 322).
В тех случаях, когда вода содержит бикарбонаты щелочных металлов, указанное опре-
деление карбонатной жесткости неприменимо.
Сульфаты определяются при помощи эмпирически со-
ставленной таблицы по интенсивности мути, получаемой от
добавления к испытуемой воде хлористого бария (в из-
бытке).
Реактивы. 1. 30% соляная кислота.
2. Сухой хлористый барий, истолченный в порошок.
Методика определения. Берут мутномер-
ную пробирку с плоским дном. На дне пробирки с на-
ружной стороны наклеивают кружок бумаги с начерченным
черным крестом (толщина линий — 1 мм). На стенке про-
бирки по высоте от дна наносят сантиметровые деления.
В пробирку наливают 20 мл испытуемой воды прибавляют?
капли соляной кислоты и всыпают при помощи стеклянной
лопаточки немного порошкообразного хлористого бария.
Взбалтывают воду в проб рке до равномерного распреде-
ления мути. Через 0,5 минуты, держа в обхват верхнюю
часть пробирки над белой бумагой, отмечают видимость
креста через столб мути. Если крест виден, то сульфатов в
воде м.нее 20 мг/л. Если же крест через муть не виден от-
бирают пипеткой часть содержимого пробирки с таким рас-
четом, чтобы крест стал виден, и затем вводят обратно из
пипетки жидкость в пробирку до тех пор, пока не будет
достигнут предел видимости креста. Определяют в этот мо-
мент высоту столба жидкости в пробирке и по приведенной
ниже’эмпирически составленной таблице 50 определяют содержание сульфатов в воде.
Если по прибавлении реактивов получается сильная муть, то испытуемую воду разбав-
ляют дестиллированной водой в несколько раз (от 2 до 10). Определяют содержание сульфа-
тов, как описано выше, и полученный результат умножают на кратность разведения.
Рис. 236. Компаратор для
колориметрирования в поле-
вых условиях.
а и b — смотровые отверстия,
с и d — пазы для планшетки
ири рассматривании жидкостей
н вертикальном направлении,
си/ — пазы для планшетки при
рассматривании жидкостей в
горизонтальном направлении,
-к и I — отверстия для отсчета
показаний, нанесенных на ребре
планшетки; справа—планшетка.
Содержание сульфатов в воде
Таблица 50
Высота столба мутной жидкости в мм Содержание сульфатов в мг/л Высота столба мутной жидкости в мм Содержание сульфатов В МГ/Л Высота столба мутной жидкости в мм Содержание сульфатов в мг/л
30 90 37 65 65 40
31 85 41 60 75 35
32 80 45 55 85 30
33 75 50 50 95 27,5'
35 70 57 45 102 25
374
Хлориды (см. стр. 330).
Аммиак (Принцип определения и реактивы см. на стр. 336).
Методика определения. Наливают в пробирку 10 мл исследуемой воды,
прибавляют 0,3 мл раствора сегнетовой соли и 0,3 мл реактива Несслера. Дают постоять
10 минут. Определяют приближенно количество азота аммиака в исследуемой воде, поль-
зуясь таблицей 51.
Таблица 51
Сооержачие аммиачного азота в воде
Окрашивание при рассматрива- нии сбоку Окрашивание гри рассматрива- нии сверху вниз Содержание азота аммиачного (N) в мг/л
Нет Нет Меньше 0,04
Нет Чрезвычайно слабожелтоватое « 0,08
Чрезвычайно слабожелтоватое Слабожелтоватое I » 0,2
Очень слабожелтоватое Желтоватое » 0,4
Слабожелто ватое Свет.ожелтоватое » 0,8
Светложелтоватое Интенсивное желтобуроватое » 2,0
Желтое Бурое, раствор мутный » 4,0
Мутноватое, резкожелтое То же » 8,0
Интенсивное бурое, раствор мутный » 20,0
Определение аммиака можно вести также с помощью стеклянных светофильтров —
эталонов, приготовленных по точным растворам.
Методика определения. Наливают в пробирку 5 мл испытуемой воды,
прибавляют 2—4 капли сегнетовой соли и 1 мл реактива Несслера. Перемешивают
воду с реактивом, вращая пробирку между ладонями: через 5 минут переливают воду из
пробирки в специальную пробирку компаратора (рис. 236). В другую пробирку наливают
6 мл испытуемой воды без реактивов и под пробирку пускают планшетку с эталонами.
Находят эталон, дающий окраску, сходную с окраской испытуемой волы, к которой
прибавлены реактивы, и в соответствии со значением эталона определяют аммиачный
азот в испытуемой воде.
Азот нитритов. (Принцип и реактивы см. на стр. 338).
Методика определения. В пробирку наливают 10 мл исследуемой воды,
прибавляют 0,5 мл реактива Грисса. Нагревают пробирку на водяной бане при 70° С
в течение 5 минут. Определяют приближенно содержание азота нитритов, пользуясь для
сравнения эмпирической таблицей 52.
Таблица 52
Содержание азота нитритов в воде
Окрашивание при рассмат- ривании пробирки сбоку | Окрашивание при рассмат- ривании пробирки сверху вниз । Содержание азота нитритов в мг/л
Нет Нет Меньше 0,001
Едва заметное розовое Чрезвычайно слабо розовое » 0,002
Очень слабое розовое Слаборозовое » 0,004
Слаборозовое Светлорозовое » 0,02
Светлорозовое Розовое » 0,04
Розовое Сильнорозовое » 0,07
Силыюрозовое Красное » 0,2
Красное Яркокрасное » 0,4
Определение азота нитритов можно вести также с помощью стеклянных светофильтров-
эталонов, приготовленных по точным растворам.
Методика определения. Проводят пробу, как описано выше. Содержимое
пробирки переливают в специальную пробирку компаратора до метки 5. В другую пробирку
компаратора наливают 5 мл испытуемой воды без реактивов и под пробирку пропускают
планшетку с эталонами. Находят эталон, дающий окраску, сходную с окраской испытуемой
воды, к которой прибавлены реактивы, и в соответствии со значением эталона определяют
азот нитритов в испытуемой воде.
Же<ею. (Принцип см. на стр. 348).
Реактивы. 1. 50% раствор роданистого аммония или роданистого калия; 2. Пер-
сульфат аммония кристаллический или перекись водорода 3%; 3. Соляная кислота уд.
веса 1,19. Все реактивы должны быть свободны от железа.
Методика определения. Наливают в пробирку 10 мл исследуемой воды»
приливают 0.2 мл соляной кислоты, вносят на кончике ножа несколько крупинок персуль-
фата аммония или 2 капли перекиси водорода. Взбалтывают, приливают 0,2 мл раствора
роданистого аммония или роданистого калия. Содержание железа определяют прибли-
женно, пользуясь таблицей 53.
Таблица 53
Содержание железа в воде
Окрашивание при рассматривании пробирки сбоку Окрашивание при рассматривании пробирки сверху вниз Содержание железа В МГ/Л
Окрашивания нет Окрашивания пет Меньше 0,05
Едва заметное желтоваторозовос Чрезвычайно слабое желтоваторо- зовое । »> 0,1
Очень слабое желтоваторозэвое Слабо желтоваторозовое » 03
Слабо желтоваторозовое Слабо желтоваторозовое » 0,5
Светло желтоваторозовое Желтоваторсзовое » 1,0
Сильно желтоваторозовое Желтоватокрасное » 2,0
Светло желтоватокрасное Яркокрасное » 5,0
Определение железа можно вести также с помощью стеклянных светофильтров—эта-
лонов, приготовленных по точным растворам.
Методика определения. Пробу проводят, как описано выше. По истече-
нии 3 минут жидкость переливают в специальную пробирку компаратора до метки 5. В дру-
гую пробирку наливают 5 мл испытуемой воды без реактивов и под пробирку пропускают
планшетку с эталонами. Находят эталон, дающий окраску, сходную с окраской испытуемой
воды, к которой прибавлены реактивы, и в соответствии со значением эталонов определяют
содержание железа в воде.
Окисляем ость определяется только на холоду.
Реактивы. 1 • 0,01 N раствор перманганата калия. 2 • 0,01 N раствор соли Мора —
3,916 г (NH4)2SO4FeSO4-6H2O в литре. 3. Раствор серной кислоты 1 : 3.
Методика определения. В коническую колбу наливают 100 мл испытуе-
мой воды из пипетки, прибавляют 5 мл серной кислоты и 10 мл 0,01 N раствора перманга-
ната калия. Оставляют пробу на 15 минут. По истечении этого времени приливают Ю мл
0,01 N раствора соли Мора и титруют обесцветившийся раствор перманганатом калия
до появления розовой окраски; вливают в ту же колбу еще 10 мл раствора соли Мора и вто-
рично титруют перманганатом калия до появления розовой окраски.
Окисляемость воды в миллиграммах кислорода на литр воды определяется по формуле:
Х = [(А1 +Д2) —10] • 0.8К,
где Д1 — первый расход перманганата калия (в мл),
Д2 — второй расход перманганата калия (в мл),
Л — коэфициент нормальности перманганата калия.
В случае необходимости перевода окисляемости на холоду в окисляемость при кипяче-
нии умножают первую на коэфициент пересчета, который, в зависимости от температуры
воды во время определения, приобретает следующие значения:
Температ ра 0—5° С—10’ 11—15° 16—20° 21—25° 26—30°
Коэфициент 1 5,0 4,3 3,9 3,25 3,0 ,
376
Приведенные коэфициенты пересчета должны рассматриваться как сугубо ориентиро-
вочные ввиду различного характера органических веществ в разных водах.
Серэвэдэрод определяется только качественно (см. стр. 357).
Помимо приведенных выше качественных проб рекомендуется и такая: в колбу наливают
немного испытуемой воды с запахом, похожим на сероводород, бросают туда кристаллик
сернокислой меди и взбалтывают. Запах сероводорода после этого должен исчезнуть; если
запах сохраняется, то он не вызван сероводородом.
Активный хлор. (Качественное определение см. стр. 359).
Приближенно количественное определение производится при помощи иодокрахмалыюй
бумажки, которая готовится следую ним образом: фильтровальную бумагу пропитывают
смесью равных объемов 5% водного раствора К J и 0,5% крахмального клейстера и высуши-
вают на воздухе, свободном от хлора. Хранить в темной склянке с притертой пробкой.
Если хлор нужно определить в присутствии нитритов, то реактивная бумажка полу-
чается пропитыванием фильтровальной бумаги следующим раствором: 5 г KJ и 5 г уксус-
нокислого натрия растворяют в 50 мл воды и смешивают с 50 мл 0,5% крахмального клей-
стера.
Методика определения. Иодокрахмальную бумажку опускают в испытуе-
мую воду и наблюдают характер и быстроту окрашивания бумажки.
Пользуясь таблицей 54, делают заключение о приближенном количественном содержании
активного хлора в воде.
Таблица 54
Содержание активного хлора в воде
Концентрация В МГ/Л Окраска Время наступления окраски Субъективное ощущение
1,43 Синяя Моментально Явный запах хлора
0,143 » » Слабый запах
0,0143 » 3—5 сек. Очень слабый запах |
0,00143 Голубая 10 сек. Запах отсутствует |
Количественное определение активного хлора (см. объемный метод на стр. 369).
Хлоропог.кнцаемость — понятие, идентичное показателю хлорируемости. Под хлоро-
поглощаемостью (хлоропотреблением) воды понимают то количество активного хлора, ко-
торое нужно прибавить к одному литру воды, подлежащей хлорированию, чтобы добиться
надежной дезинфекции. Принято считать, что надежная дезинфекция воды достигается,
если количество активного избыточного хлора после 30-минутного контакта воды с препа-
ратом хлора достигает 0,2—0,4 мг/л.
Реактивы. 1% раствор хлорной извести, который готовится следующим образом:
отвешивают 1 г хлорной извести, отмеряют в стакан 100 мл дестиллированной воды и
растирают навеску хлорной извести в фарфоровой чашке с малыми порциями отмеренной
воды, сливая раствор в тот же стакан. Сливание и растирание навески производят до пол-
ного удаления ее из чашки. Раствор в стакане перемешивают и отфильтровывают, слив из
стакана первые 10 мл раствора.
2. Раствор тиосульфата натрия, содержащий 6,707 г Na2S2O3 • 5Н2О в одном литре
воды.
3. Йодистый калии кристаллический.
4. Серная кислота 10% (вместо буферного раствора).
5. 1% крахмальный клейстер.
Методика определения. В 4 стакана наливают по 200 мл испытуемой
воды, добавляют 1% раствора хлорной извести в первый стакан 2 капли, во второй — 3 капли,
в третий —4 и в четвертый — 5. Хорошо перемешивают содержимое каждого стакана стек-
лянной палочкой и оставляют стоять в покое 30 минут. Через полчаса определяют количество
свободного хлора, как указано на стр. 361. Выбирают такой стакан, где количество свобод-
ного хлора находится между 0,2 и 0,4 мг/л, т. е. где на титрование идет 2 капли раствора
тиосульфата. Расчет ведут по формуле:
Х = 2А
где X — хлоропотребление хлорной извести (в мг) на литр воды,
А — число капель 1% раствора хлорной извести в выбранном стакане.
Зная процентное содержание активного хлора в хлорной извести, можно вычислить
хлоро потребление в миллиграммах активного хлора на литр воды.
Определение процентного содержания активного хлора и хлорной извести. Реактивы
те же, что и при определении хлоропоглощаемости.
Методика определения. В колбу наливают 100 мл дестиллированной воды,
добавляют 5 мл сорили кис поты и 10 капель 1% раствора хлорной извести. Прибавляют
377
20—30 кристалликов йодистого калия, перемешивают и титруют тиосульфатом, приливая
его по каплям до слабожелтого окрашивания. Прибавляют 1 мл крахмального клейстера и
дотптровывают до исчезновения синей окраски.
Процентное содержание активного хлора в хлорной извести равно количеству капель
тиосульфата, пошед нему па титрование.
Указанные соотношения становятся понятными, если учесть, что раствор тиосуль-
фата приготовляется таким образом, что 1 мл его соответствует 1 мг активного хлора (или
пПГ х С 1-6,707
точнее 0,96 мг) соответственно —.
Na2S2O3 • 5Н2О
Если учесть, что 1 мл раствора при титровании дает 19—20 капель (это следует прове-
рить для данной пипетки), то каждая капля тиосульфата соответствует ~ =0,05 мг ак-
тивного хлора (в среднем).
При определении процентного содержания хлора в хлорной извести для анализа берут
10 капель 1% раствора хлорной извести, что соответствует 5 мг хлорной извести. Каждая
капля израсходованного тиосульфата, как мы видели, соответствует 0,05 мг активного хлора,
т. е. как раз 1% по отношению к взятому для анализа количеству хлорной извести.
Вот почему процентное содержание активного хлора в хлорной извести равняется коли-
честву капель тиосульфата, пошедшего на титрование.
ОГЛИЧИЕ НЕКИПЯЧЕНОЙ ВОДЫ ОГ КИПЯЧЕНОЙ
Из многих методов, предлагавшихся для отличия некипяченой воды от
кипяченой, здесь описаны два метода: 1) по растворенным газам и 2) по со-
левому составу.
Методика определения по растворенным газам.
Всыпают в пробирку 0,5—1 г хлористого натрия и прибавляют 10—15 мл
испытуемой воды. Если вода кипяченая и бедна растворенными газами, то
она поглощает воздух, находящийся между частицами хлористого натрия и
на его поверхности. В этом случае выделения пузырьков газа из воды не на-
блюдается. Если же вода сырая и содержит в достаточном количестве раств: -
репные газы, то наблюдается выделение газа в виде мельчайших пузырьков.
Метод пригоден для кипяченой воды, простоявшей после кипячения не более
4—5 часов.
Методика определения по солевому составу. В
пробирку наливают 3 мл испытуемой воды и прибавляют 5 капель настойки
кошенили (2 г кошенили настаивают в течение 2 дней в 100 мл дестиллирован-
ной воды).
Если вода при этой пробе сохраняет розовокрасную окраску, то это пока-
зывает, что вода кипяченая. Если же в течение нескольких минут розово-
красная окраска исчезает и переходит в серую, причем появляются хлопья,
то вода сырая: бикарбонаты щелочно-земельных металлов сырой воды спо-
собствуют быстрому выпадению краски.
К мягким водам и водам с высокой постоянной жесткостью реакция не-
применима: в мягких водах мало солей щелочно-земельных металлов, в водах
с высокой постоянной жесткостью выпадение краски вызывается хлоридами,
сульфатами и другими солями Са и Mg, которых в такой воде много.
Рекомендуется при проведении первой и второй проб ставить испытание
и с исходной водой (контроль).
БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ВОДЫ
Микрофлора воды весьма разнообразна. Из воды выделено свыше 500
культур.
Микробное население воды в значительной мере отражает микрофлору
почвы, с которой вода соприкасается. Во время весенних паводков и осенних
дождей микробное население открытых водоемов (реки, озера, пруды) зна-
чительно возрастает в связи с загрязнениями, попадающими в воду вместе
с водными потоками.
Степень обогащения воды микробной флорой зависит, однако, не только
от метеорологических условий, но и от качества и количества сточных вод,
378
попадающих в водоемы, и от характера самого водоема (объем, скорость тече-
ния воды и т. п.).
Наряду с процессами загрязнения постоянно протекают и сложные про-
цессы самоочищения водоемов от микробной флоры благодаря отмиранию
бактерий в воде, осаждению их вместе с взвешенными частицами на дно во-
доема, а также в связи с поеданием бактерий простейшими (Flagellata и др.)
и литическим действием на бактерии бактериофага.
Наибольшей чистотой в отношении бактериального обсеменения отли-
чаются глубинные грунтовые воды артезианских горизонтов, хотя известны
случаи загрязнения глубинных вод через трещины, скважины, шахты и т. п.
Важнейшее эпидемиологическое значение имеет заражение воды возбу-
дителями желудочнокишечных заболеваний — брюшного тифа, паратифов,
дизентерии, холеры. Возможно инфицирование воды бациллами сибирской
язвы, сапа, туляремии, бруцеллами, а также возбудителями глистных инва-
зий и др.
Санитарно-бактериологический анализ воды слагается из:
1. Определения числа бактерий, содержащихся в 1 мл воды, как показа-
теля общего микробного обсеменения воды;
2. Определения коли-титра (коли-индекса) как показателя фекального
загрязнения воды;
3. Обнаружения патогенных бактерий.
Микробное число воды
Под микробным числом воды понимают число микроорганизмов
в 1 мл воды, определяемое путем посева микробов на плотную питательную
среду (агар, желатина) и подсчета выросших колоний. Такой способ опреде-
ления не дает точного представления о количестве бактерий в единице объема
воды, так как бактерии различных физиологических групп неодинаково раз-
виваются при одних и тех же условиях. Однако микробное число все же яв-
ляется хорошим показателем общего микробного обсеменения воды.
Значение этого показателя для санитарной оценки воды в высокой мере
зависит от характера водоисточника. Так, вода обыкновенного шахтного
колодца может считаться еще чистой при содержании до 1000 колоний в 1 мл.
Вода глубинного артезианского колодца дает лишь единичные колонии
в 1 мл.
Для воды, подвергшейся очистке и обеззараживанию, микробное число
приобретает первостепенное значение. Так, для воды, профильтрованной
через песочный фильтр, важнейшим показателем эффективности такого фильт-
ра является именно микробное число, которое при надлежащей работе фильтра
не должно быть свыше 100.
Временный стандарт качества водопроводной воды (изд. НКЗ и НК Ком.
хоз. РСФСР, 1937) предъявляет к воде требование, чтобы число колоний
(при посеве 1 мл воды на агар или желатину), развивающихся через 48 часов
при 20—22°С, в среднем за год не превышало 30. Среднее число колоний за
отдельные месяцы не должно превышать 50, а для отдельных опреде-
лений 75.
Методика определения. В зависимости от предполагаемого
обилия бактерий берут для засева на чашку Петри то или иное количество
воды. Воду из водопроводов и артезианских скважин засевают неразведенной
в количестве 0,5—1 мл. Воду из открытых водоемов перед засевом разбавляют
стерильной водопроводной водой 1 : 10 и 1 : 100. Из каждой пробы воды
должно быть использовано для посева не менее двух различных разведений.
Для каждого разведения берут отдельную стерильную градуированную
пипетку. При недостаточном количестве пипеток посев можно производить
одной пипеткой. В этом случае необходимо начинать с больших разведений
и переходить к меньшим.
375
Стерильную градуированную пипетку на 10 мл над огнем спиртовой (га-
зовой) горелки вводят в бутылку с испытуемой водой, предварительно хорошо
перемешанной. Легким продуванием через пипетку вода еще раз перемеши-
вается перед самым отбором пробы. Затем набирают в пипетку нужное ко-
личество воды и переносят в стерильную пробирку.
Из пробирки воду засевают неразведенной или разводят стерильной во-
допроводной водой в 10 и в 100 раз. При разведении в 10 раз 1 мл неразведен-
ной воды переводится в пробирку, куда предварительно налито 9 мл стериль-
ной водопроводной воды, и тщательно взбалтывается; при разведении в 100
раз 1 мл разведенной в 10 раз воды переводится в пробирку, куда предва-
рительно налито 9 мл стерильной водопроводной воды. Для каждого разбав-
ления пользуются отдельной пипеткой. Ватная пробка и край пробирки при
разведениях фламбируются.
Самый посев на чашки производится следующим образом: 0,5—1 мл воды
переносят над пламенем спиртовой (газовой) горелки в стерильную чашку
Петри, слегка приподняв крышку ее. После этого в чашку наливают 10—12 мл
растопленного и охлажденного до 42—44°С мясопептонного агара. Ватная
пробка и края пробирки или бутылки с агаром предварительно фламбиру-
ются. Вращательными движениями закрытой чашки агар тщательно пере-
мешивается с водой. Нельзя при этом оставлять незалитых участков на дне
чашки; необходимо избегать также попадания среды на борты чашки и обра-
зования пузырьков воздуха. После засева чашку оставляют на столе в гори-
зонтальном положении.
На чашках делают карандашом для стекла надпись с указанием номера
или названия пробы, разведения и даты посева. После этого чашку ставят
крышкой вниз в термостат при 37°С на сутки. Через 24 часа подсчитывают
число колоний при помощи счетной пластинки и лупы.
Если число колоний менее 300, то подсчитывают все колонии. Если число
колоний более 300, то можно подсчитать не все колонии, однако площадь,
на которой подсчет произведен, не должна быть менее одной трети всей пло-
щади чашки. При диаметре в 10 см площадь чашки может быть принята рав-
ной 80 см2 (округленно). Следовательно, нужно подсчитать не менее 25—27
квадратиков по 1 см2 в различных секторах чашки.
Результат подсчета, если нужно округляют. Если на чашке выросло
много плесневых колоний, занимающих половину или две трети чашки, то
подсчет неплесневых колоний затрудняется, что и должно быть отмечено
в протоколе анализа.
Определение коли-титра и колп-индекса воды
Титром кишечной палочки или coli-титром воды
условно называют тот наименьший объем воды, в котором обнаруживается
еще присутствие кишечной палочки (хотя бы одной особи).
Под coli-индексом подразумевается число кишечных палочек
в одном литре воды.
Для перевода коли-титра на коли-индекс нужно 1000 разделить на коли-
титр.
Для перевода коли-индекса на коли-титр нужно 1000 разделить на коли-
индекс.
Пример. При коли-титре, равном 250, коли-индекс равен 1000 : 250 = 4.
При коли-индексе, равном 4, коли-титр равен 1000 : 4 = 250.
Исследование воды на коли-титр лежит в основе санитарно-бакте-
риологического анализа воды, так как кишечная палочка, находясь в огром-
ных количествах в экскрементах людей и животных, своим присутствием
в воде указывает на фекальные загрязнения воды.
Будучи родственна тифо-паратифозным и дизентерийным бактериям, ки-
шечиая палочка выживает во внешней среде практически так же долго, как
380
ее патогенные сородичи; вот почему находка в воде большого количества
кишечной палочки (малый коли-титр и большой коли-индекс) может служить
прямым указанием на опасность пользования такой водой в эпидемиологи-
ческом отношении.
Наименование «кишечная палочка, Bact. coli» охватывает целый ряд
микробных разновидностей, близких по своим морфологическим и культу-
рально-биохимическим особенностям. Из них наиболее важными являются:
Bact coli commune (Escherichia coli), Bact. coli citrovcrum (Citrcbacter) u
Bact. coli aeregenes (Aerobacter).
Типичной Bact. coli признается микроб: 1. Морфологически представля-
ющий небольшую грам-стрицательную палочку (размеры 0,5—0,8 х 1,5—3 у.)
со слегка закругленными концами; 2. Обладающий слабой подвижностью *
(последняя может и отсутствовать); 3. С хорошо выраженным аэробным ро-
стом; 4. Не образующий спор; 5. Не разжижающий желатину в течение 48
часов при 37°С (разжижение определяется отсутствием способности засты-
вать при охлаждении пробирки с желатиной в струе холодной воды):
б. Свертывающий молоко в течение 48 часов при 37°С (если свертывание не на-
ступает, то подогревают пробирку с молоком и определяют, получается ли
свертывание при подогревании); 7. Вызывающий кислотообразэвание и газо-
образование в средах с глюкозой, маннитом, лактозой и мальтозой; 8. Нс
образующий ни газа, ни кислоты с сахарозой (некоторые штаммы разлагают
сахарозу); 9. Дающий характерный рост на среде Эндо— крупные плоские
колонии яркокрасного цвета с металлическим зеленоватым блеском (типы
Bact. coli commune u Citrobacter) или без блеска (тип Aerobacter); 10. Обра-
зующий индол через 72 часа (признак не постоянный).
Преф. И. Е. Минкевич считает, что из признаков, характеризующих при-
надлежность бактерий к группе коли и происхождение их от теплокровных,
основными и обязательными являются лишь следующие:
1. Морфология (небольшая грам-стрицательная неспоровая палочка); 2. Ука
занный характерный рост на среде Эндо; 3. Образование газа в среде с глю-
козой или маннитом при 43°С в течение 24 часов; 4. Отсутствие разжижения
желатины при 37°С в течение 24 часов.
Близко к названным выше представителям группы коли стоят Bact. ра-
racoli А и В, появляющиеся в изобилии в экскрементах кишечноинфекцион-
ных больных, особенно в период реконвалесцениции. Эти бактерии от описан-
ных выше отличаются в основном тем, что не расщепляют лактозу и потому
молока не свертывают (Bact. paracoli А иногда вызывает позднее свертыва-
ние). На среде Эндо они дают бесцветные колонии, более мелкие по раз-
мерам, нежели колонии Bact. coli.
В отношении норм для титра кишечной палочки питьевой воды можно
сказать следующее. Наш ГОСТ 2761-44 требует, чтобы вода источи и-
к о в, которые могут быть использованы для хозяйственно-питьевого водо-
снабжения, имела коли-титр не ниже 1 и коли-индекс не выше 1000, если
источники намечаются к использованию только с хлорированием воды, и
коли-титр не ниже 0,1, а коли-индекс не выше 10000 для источников, наме-
таемых к использованию с полной счисткой и с хлорированием воды.
Для природной воды, не подвергающейся очистке,
принято считать, что коли-титр, равный 10, и коли-индекс, равный 100, яв-
ляются еще удовлетворительными показателями санитарного состояния
воды: лишь при коли-титре 1 и коли-индексе 1000 вода считается сомни-
тельной.
Что касается питьевых вод, подвергающихся очи-
стке и обеззараживанию, то к ним предъявляются гораздо
более строгие требования. Так, согласно временному стандарту качества
водопроводной воды (изд. НКЗ и НК Ком. хоз. РСФСР, 1937) к воде предъяв-
Подвижные расы снабжены 2—б жгутиками, расположенными псритрихиально.
381
ляются требования, чтобы кишечная палочка отсутствовала при посевах
333 мл, а для Москвы и Ленинграда — при посеве 500 мл воды.
Для воды, пригодной к пользованию, необходимо придерживаться сле-
дующих норм: 1) воды артезианских скважин должны давать не более 100 ко-
лоний в 1 мл и коли-титр выше 500 мл, 2) грунтовые колодцы, родники и
т. п. — до 1000 колоний в 1 мл и коли-титр не ниже 250 мл.
В основе стандартного метода определения коли-титра воды лежит ряд
важнейших особенностей кишечной палочки: 1) способность ее развиваться
при относительно высокой температуре (43°С). При этой температуре разви-
тие сапрофитной флоры воды и кишечной палочки холоднокровных живот-
ных (рыб), не имеющей санитарного значения, задерживается, что создает
условия для избирательного выращивания кишечной палочки; 2) способность
кишечной палочки сбраживать глюкозу и маннит с образованием газа; 3) спо-
собность кишечной палочки давать характерные колонии на среде Эндо.
Для определения коли-титра воды необходимы следующие среды: среда
Эйкмана или Булира и среда Эндо (приготовление см. на стр. 114).
Методика определения. Исследование воды на коли-титр сла-
гается из трех этапов.
Первый этап — посев на среду Эйкмана или Булира.
Посев производится по определенным схемам в зависимости от ожидаемой
степени фекального загрязнения воды (см. схемы в приведенных ниже таб-
лицах). Количество воды, которое берут для посева в одну посуду (пробирка,
колба, флакон), варьирует от десятых долей миллилитров до 100 мл. Одно-
временно засевают несколько проб одной и той же воды, причем берутся ко-
личества, уменьшающиеся в 10 раз: 100—10—1 мл — 0,1—0,01—0,001—
0,0001 мл.
Для посева десятичных долей миллилитра воду разбавляют соответствен-
но в 10—100—1000 и 10 000 раз и для посева берут 1 мл разведенной воды.
Так, чтобы посеять 0,01 мл, следует испытуемую воду разбавить стерильной
водопроводной водой в 100 раз и взять для посева 1 мл этой разбавленной
воды. Посевы производятся стерильными пипетками, причем для посевов
десятых долей миллилитра можно пользоваться одной пипеткой, идя от боль-
ших разведений к меньшим.
Соотношения между количеством среды и объемом засеваемой воды при-
ведены в таблице 55.
Таблица 55
Соотношения количеств засеваемой воды и питательной среды
Количество засе- ваемой воды 100 мл 10 мл 1 мл 0,1 и менее
Количество среды Эйкмана 14 мл 2,5 мл 7 мл стерильной водо- проводной воды 0,9 мл -|- 6,1 мл | стерильной водо- приводной воды 0,9 мл 4-6,1 мл стерильной водо- проводной воды
Количество среды Булира 50 мл 5 мл 3 мл 4 5 мл сте- рильной водопро- водной воды 2,5 мл 4- 5 мл стерильной водо- проводной воды
Среда в соответствующих количествах заготовляется в пробирках и кол-
бах (флаконах), в которые вводятся поплавки дном вверх (рис. 237 и 238).
Поплавки представляют собой маленькие пробирки длиною 25—30 мм диа-
метром 5 — 6 мм. При стерилизации и дальнейшем охлаждении пробирок и
колб с находящимися в них поплавками воздух из поплавков удаляется и
они нацело заполняются средой. В таком виде посуда и используется для
посева.
3S2
Рис. 237. Рис. 238. Колба
Пробирка с с поплавком,
поплавком.
Пссуда с поплавками, в которых остаются пузырьки возду?<а, должна
подвергаться повторному нагреванию и охлаждению, пока поплавки не за-
полнятся целиком средой.
Посев производится стерильно над пламенем спиртовой (газовой) горелки.
После посева пробирки и колбы помещаются в термостат при температуре
43°С. (Перегрев термостата выше 46°С праводит к гибели посевов!). Через
24 часа производится регистрация результатов, которые заносятся в журнал.
При этом могут представиться следующие случаи:
1. Отсутствие газообразования и мути во всех пробирках и колбах. По-
плавки при этом остаются заполненными жидкостью, а среда не мутнеет.
Такое состояние проб свидетельствует об отсутствии кишечной палочки во
взятом для посева объеме воды. Если объем посеянной воды обозначить че-
рез и, то коли-титр в этом случае обозначается так: коли-титр >и, а коли-
1000
индекс < ——.
2. Наличие газообразования и мути в про-
бирках и колбах (не обязательно во всех).
При этом в той посуде, где обнаружено газо-
образование, жидкость из поплавков выте-
снена и заменена газом. Если посев был
сделан на среду Булира, то отмечается и из-
менение цвета среды из красного в желтый.
Однако изменение цвета среды не является
обязательным. Жидкость в колбах и пробир-
ках, где имело место газообразование, оказы-
вается мутной. Такое состояние проб указы-
вает на возможное фекальное
загрязнение воды, ив этом случае
требуется дальнейшая проверка результатов
(см. этап второй).
3. Отсутствие газообразования и наличие
мути дает отрицательный ответ. Однако, если анализ касается вод, посту-
пающих без очистки для питья и хозяйственных целей, а также оконча-
тельно очищенной воды, необходимо перейти ко второму этапу исследования.
4. Наличие газа в поплавках без появления мути указывает на техни-
ческий дефект, в результате которого воздух попал в поплавки. В этом случае
требуется повторение анализа.
Второй этап — пересев на среду Эндо.
Материал из забродивших и помутневших проб пересевают при помощи
платиновой петли отдельными штрихами на чашки Петри со средой Эндо
так, чтобы получить изолированные колонии. Чашки с посевами помещают
крышками вниз в термостат при 37°С на 24 часа. По истечении этого времени
производят просмотр результатов роста. Они могут оказаться различными:
1. Рост отсутствует, либо получился рост нетипичных колоний. В этом
случае нужно считать, что газообразование и помутнение среды вызвано
не кишечной палочкой1. Ответ дают такой, как при отсутствии газообразова-
ния на первом этапе.
2. Рост получился типичный для группы Bact. coli. В этом случае необ-
ходимо из материала типичных колоний приготовить мазки, окрасить их
по Граму и обследовать микроскопически. Технику окраски см. на стр. 120.
Если в мазках, приготовленных, как описано выше, не обнаруживаются
грам-отрицательные короткие неспоровые палочки, то это указывает на то,
что рост колоний, из которых сделан посев, вызван не кишечной палочкой,
и в этом случае ответ дается, как при отсутствии газообразования. Если
1 Газообразование в среде Булира могут давать: В. proteus vulgaris, В. cloacae, В. vis-
cosus ochraceiis u ;ip.
383
же при микроскопировании обнаружены грам-отрицательные короткие не-
споровые палочки, то в этом случае нужно считать фекальное за-
грязнение воды весьма вероятным и перейти к третьему
этапу исследования.
Третий этап — вторичный посев на среду Эйкмана или Булира.
Материал из подозрительных колоний на чашках со средой Эндо пересе-
вают в пробирки с поплавками в среде Эйкмана или Булира, разведенной
в 10 раз стерильной водопроводной водой. Пересеваемые колонии должны
быть хорошо изолированы и чисты. В противном случае необходимо получить
культуру в чистом виде. Для этой цели приготовляют из колоний взвесь
в стерильном физиологическом растворе или стерильной водопроводной воде,
сеют петлей на чашки со средой Эндо и ставят в термостат при 37°С на сутки.
Получив изолированные колонии делают пересев в пробирки с поплавками,
как только что описано. Пробирки с пересевами помещают в термостат при
37°С на сутки. По истечении этого времени просматривают пробирки. При
этом могут наблюдаться следующие случаи:
1. Отсутствие газообразования и мути. Это дает отрицательный ответ
в отношении фекального загрязнения воды.
2. Наличие газообразования и мути. Оно свидетельствует о несомнен-
ном фекальном загрязнении воды.
3. Отсутствие мути и наличие воздуха в поплавках. Это указывает на
технический дефект.
В случае окончательного подтверждения наличия фекального загрязнения
коли-титр устанавливается на основании результатов, полученных на первом
этапе исследования; при этом пользуются разработанными на основе матема-
тических данных таблицами (табл. 56—60).
Стандартное исследование воды на коли-титр на этом заканчивается.
В случае необходимости производства более углубленной идентификации
выделенных микробов кишечной группы делают дополнительные исследова-
ния: посевы на пестрый ряд, на цитратные среды, пробы на индолообразо-
вание и т. и.
Вода артекважин и водопроводов Таблица 56
Общий объем воды, взятой для посева, —500 мл
Схема посева: 4 объема по 100 мл и 10 объемов по 10 мл
Число положи- тельных гроб по 100 мл 0 1 2 3 4
Число положи- тельных проб Коли- Коли- Коли- 1 Коли- Коли-
ин- ин- • ин- 1 ! ин- ин- титр
по 10 мл декс титр деке титр декс титр 1 деке титр декс
0 <2 >500 2 500 5 200 9 111 16 62
1 2 500 5 200 8 125 13 77 22 45
2 4 250 2 143 11 91 17 59 30 33
о 6 167 9 111 14 71 21 48 38 26
! 4 8 125 12 83 17 59 26 38 S3 19
5 11 91 15 67 20 50 31 32 70 14
б 13 77 17 59 24 42 37 27 92 11
7 15 67 20 50 28 36 44 23 120 8
8 17 59 23 43 32 31 52 19 161 6
9 20 50 26 38 37 27 60 17 23) 4
10 22 46 29 35 41 24 69 14 >23J <4
Коли-титр и коли-индекс находят на месте пересечения ряда, находяще-
гося под числом забродивших проб по 100 мл, с горизонтальным рядом,
находящимся вправо от числа забродивших проб по 10 мл.
Пример 1. Забродила одна проба в 100 мл и 0 проб в 10 мл. В этом случае коли-ин-
декс равен 2 и коли-титр — 500.
Пример 2 Забродили 3 пробы по 100 мл и 5 проб по 10 мл. В этом случае коли-индекс
равен 31, коли-титр — 32.
384
Таблица 57
Вода артскважин, водопроводов, мелких буровых скважин, благоустроенных
шахтных колодцев
Общий объем воды, взятой для посева, —300 мл
Схема посева: 2 объема по 100 мл и 10 объемов по 10 мл
Число положи- тельных проб по 100 мл 0 1 2
Число положи- тельных проб по 10 мл Коли- Коли- Коли-
.индекс I титр индекс | титр индекс титр
0 1 2 3 4 5 б 7 8 9 10 <3 3 7 И 14 18 22 27 31 36 40 >333 333 143 91 71 56 45 37 32 28 25 4 8 13 18 24 30 36 43 51 60 69 250 125 77 56 42 33 28 23 20 17 14 11 18 27 38 52 70 92 120 161 230 >230 91 56 37 26 19 14 11 8 6 4 <4
Таблицей пользуются так же, как и предыдущей.
Пример. Забродила одна проба в 100 мл и 7 проб в 10 мл. В этом случае коли-индекс
равен 43 и коли-титр — 23.
Таблица 58
Вода открытых водоемов (чистые районы рек, озер, прудов)
Общий объем воды, взятой для посева —111,1 мл
Схема посева: по 1 объему в 100 мл, 10 мл и 0,1 мл
Результаты посева проб Коли-
100 мл 10 мл 1 мл 0,1 мл индекс титр
— — <9 >111
— — + 9 111
— + 9 111
— — 9 105
+ + 18 56
+ ± 19 53
+ 22 46
4- 23 43
+ + 28 36
— 4- 92 11
— + 94 10
— + + 180 б
— — — 230 4
— — + 960 1
— + 2380 0,4
-j- + + >2380 <0,4
Для определения коли-индекса и коли-титра находят в одном горизон-
тальном ряду все объемы, давшие положительный результат, и прочитывают
стоящие вправо числа, обозначающие коли-индекс и коли-титр.
Пример. Дали положительные результаты объемы в 10 мл, 1 мл и 0,1 мл. Эти объемы
с положительными результатами находятся в 9 ряду. Для этого случая коли-индекс равен
28 и коли-титр — 36.
^5 А. И. Бурштейн
385
Таблица 59
Вода открытых водоемов (загрязненные районы рек, озер, прудов), неблагоустроенных
колодцев и т.п.
Общий объем воды, взятой для посева, —11,11 мл
Схема посева: по 1 объему в 10 мл, 1 мл, 0,1 мл и 0,01 мл
Результаты посева проб Коли-
10 мл 1 мл 0,1 мл 0,01 мл индекс титр
— — <90 >11,1
— — — 4- 90 11,1
— — — 90 11,1
— + — — 95 10,5
— + 4- 180 5,6
— 4- 4- 190 5,3
— 4- + 220 4,6
4- — — 230 4,3
+ -4- 280 3,6
+ — + 920 1,1
4- — 4- — 940 1,0
4- — 4- 4- 1800 0,6
+ — 2300 0,4
4- — — 4- 9600 0,1
4- — 4- 23800 0,04
4- 4- 4- >23800 <0,04
Таблицей пользуются так же, как и предыдущей.
Пример. Забродили все пробы. В этом случае коли-индекс > 23800, коли-титр < 0,04.
Таблица 60
Вода открытых водоемов (весьма загрязненные районы рек, озер, прудов),
неблагоустроенных колодцев и т. п.
Общий объем воды, взятой для посева, —1,111 мл
Схема посева: по 1 объему в 1 мл, 0,1 мл, 0,01 мл и 0,001 мл
Результаты посева проб Коли-
1 мл 0,1 мл 0,01 мл 0,001 мл индекс титр
<900 >1,11
— — — 4- 900 1,11
—. — 4- 900 1,11
— + — — 950 1,05
— 4- 1800 0,56
— + — 4- 1900 0,53
— 4- + 2200 0,46
+ — 2300 0,43
+ 4- 4- 2800 0,36
-г 4- 9200 0,11
+ — 4- — 9400 0,10
—у— — 4- + 18000 0,06
+ 4- — 23000 0,04
4- — + 96000 0,01
-Г 4- — 238000 0,004
1 + 1 -И 1 + 1 4- >238000 <0,004
Таблицей пользуются, как и предыдущими двумя.
Пример. Положительного результата не дала ни одна проба. В этом случае коли-ин-
декс < 900 и коли-титр > 1,11.
386
ПОЛЕВОЙ МЕТОД
ОПРЕДЕЛЕНИЯ ФЕКАЛЬНОГО ЗАГРЯЗНЕНИЯ ВОДЫ
Описанный выше метод определения фекального загрязнения воды, при котором ответ
получается лишь к концу третьих суток, в ряде случаев (санитарная разведка, экспедиция и
т. п.) должен быть заменен экспресс-методом, при котором ответ может быть получен в тече-
ние одних суток и даже 8—12 часов.
Экспресс-метод от обычного отличается двумя особенностями:
1. Вместо коли-титра определяют бродильный титр в о д ы, т. е. тот наи-
меньший объем воды, который дал положительным результат (брожение) при первом же
посеве на среду Эйкмана или Булира по методу, описанному выше. Такой авторитетный
микробиолог, как И. Е. Минкевич, считает, что в 99% случаев имеется совпадение результа-
тов бродильной пробы с действительным присутствием в посеянных порциях воды сани-
тарно-показательных разновидностей Bact. coli.
2. Прямым определением Bact. coli на мембранных фильтрах; точность этого метода
соответствует результатам бродильного титра.
Принцип использования мембранных фильтров для прямого учета задержанных на них
бактерий был выдвинут еше в 1932 г. Диановой и Ворошиловой. Методика разработана
лабораторией Рублевской водопроводной станции и Украинским центральным институтом
коммунальной гигиены.
Сущность применения мембранных фильтров (ультрафильтров) заключается в двух
свойственных им особенностях: а) путем фильтрации можно задержать на их поверхности
всю бактериальную форму из профильтрованного объема воды, что особенно важно для тех
случаев, когда искомые бактерии находятся в единичных экземплярах в больших объемах
воды; в) возможно выращивать на их поверхности соответствующие бактериальные колонии,
которые в зависимости от питательной среды, на какую помещаются фильтры, дают ту
или иную типичную для данного микроба картину.
Мембранные фильтры (ультрафильтры), выпускаемые Рублевской водопроводной стан-
цией, представляют собой нитроцеллюлозные пленки, имеющие форму кружков диаметром
в 35 мм и толщиною около 0,1 мм. По внешнему виду они напоминают белую тонкую бумагу.
Если пропитать мембранный фильтр жидкостью с коэфициентом рефракции приблизительно
равным 1,5 (кедровое масло, касторовое масло), то мембранный фильтр становится совершенно
прозрачным. Одна сторона фильтра имеет матовую поверхность. Это так называемая «воз-
душная» поверхность фильтра. На ней нанесена метка карандашом в виде точки или номера
фильтра. Воздушную поверхность обращают кверху (к воздуху) при подготовке фильтра
для фильтрации воды и при последующем наложении его на среду. Другая сторона фильтра
имеет поверхность глянцево-блестящую. Это так называемая «зеркальная» поверхность
мембранного фильтра, которая обращена вниз при фильтрации воды и наложении фильтра
на среду.
Мембранные фильтры выпускаются пачками по 60—70 экземпляров под пятью номерами.
Размер пор повышается от меньших номеров к большим. Фильтрующая способность мембран-
ных фильтров тем выше, чем больше размеры пор.
В таблице 61 приведена характеристика мембранных фильтров: номер, размер пор и
средняя продолжительность фильтрации полулитра дестиллированной воды при разности
давления в 0,5 атмосферы.
Таблица 61
Характеристика мембранных фильтров
| №№ Средняя продолжитель- ность фильтрации <*> Интервал продолжитель- ности фильтрации Средний диаметр пор В р.
1 9 мин. 6—12 мин. 0,35
2 4,5 мин. 3—6 мин. 0,5
3 2,25 мин. 1,5—3 мин. 0,7
4 70 сек. 49—90 сек. 0,9
5 35 сек. 25—45 сек. 1,2
Фильтры хранятся в банках с 20% раствором этилового спирта, завернутые в фильтро-
альную бумагу и два слоя марли. При хранении свыше года в сухом состоянии фильтры
ановятся хрупкими, в спирту же сохраняются значительно дольше.
Кроме перечисленных 5 номеров ультрафильтров выпускаются еще так называемые
редварительпые фильтры» или «префильтры», быстро фильтрующие. Назначение их —
истить воду от грубо взвешенных в ней частиц (планктон, окись железа и т. п.). В тех
учаях, когда это требуется, вода предварительно пропускается через префильтр, а затем
рез соответствующий номерной мембранный фильтр. Префильтры в таких случаях исноль-
ются для дальнейшего анализа так же, как и номерные фильтры, т. е. тоже помещаются
среду и выдерживаются в термостате.
Методика определения коли-титра воды при помощи мем-
энных фильтров. Необходимо предварительно подсчитать объем воды, подле-
387
жащнй фильтрации. Учитывая незначительный размер мембранного фильтра, нужно по-
добрать такой объем воды, чтобы число выросших на фильтре колоний было не более 50.
Очевидно, что чем вода более загрязнена, тем меньший объем ее нужно пропускать через
фильтр. Ориентировочно можно исходить из предполагаемого коли-титра. Так, если предпо-
лагаемый коли-титр 0,01, то коли-индекс для такой воды предположительно 100 000, т. е.
в литре воды должно содержаться предположительно 100 000 кишечных палочек. Следова-
1000-50
тельно для получения 50 колонии нужно посеять: 'jqqqqq = 0,5 мл. Короче: для расчета
нужно предполагаемый коли-титр помножить на 50; в данном случае: 0,01 . 50 = 0,5.
При высоких коли-титрах получаются слишком большие объемы; в этих случаях можно
фильтровать меньшие из расчета получения менее 50 колоний. Так, если предполагаемый
коли-титр 500, то для получения 50 колоний нужно
фильтровать 25 л воды, что очень много. Можно в этом слу-
чае профильтровать от 1 до 5 литров из расчета получения от
2 до 10 колоний и т. д.
Определив объем воды, подлежащий фильтрованию,
приступают к подготовке мембранного фильтра и фильтрую-
щего прибора.
Мембранный фильтр перед употреблением стерилизуют.
Для этой цели фильтр, вынутый из спирта, слегка подсуши-
вают на воздухе, а затем бросают плашмя в кипящую
дестиллировапную воду. Стерилизация продолжается 15—
20 минут на малом огне.
Простерилизованный фильтр помещается в один из филь-
тровальных аппаратов, выпускаемых Рублевской водопро-
водной станцией (рис. 239). Аппарат А состоит из стек-
лянной градуированной части, имеющей форму широкогор-
лой воронки (емкость 600 мл) и металлического фильтро-
вального столика. Обе части могут быть плотно присоединены
одна к другой путем навинчивания первой части на вто-
рую. От фильтровального столика отходит отводная труб-
ка, ее вставляют плотно в отверстие резиновой пробки, кото-
рая в свою очередь плотно вставляется в горло конической
колбы, снабженной отводной трубкой. На последнюю наде-
вают резиновую трубку, свободный конец которой присое-
Рис. 239. Приборы для филь-
трации через мембранные
фильтры.
диняют к насосу.
В качестве фильтровального аппарата может служить прибор Зейтца (см. рис. 239 В).
Аппарат В построен по тому же типу; верхняя часть его в отличие от прибора А состоит
из металлического цилиндра.
Перед употреблением фильтровальный прибор должен быть простерилизован. Отъеди-
нив стеклянную часть от металлической в приборе А, протирают обе части тампоном, смо-
ченным чистым спиртом, а затем фламбируют. В приборе В также протирается и флам-
бируется верхняя часть.
Простерилизованный, как описано выше, мембранный фильтр захватывают стериль-
ным пинцетом, переносят зеркальной поверхностью книзу на фильтровальный столик,
тщательно прилаживают и, создав в колбе небольшое отрицательное давление насосом
(или ртом), присасывают фильтр к столику. Присоединяют стеклянную воронку к столику
и фильтруют необходимый объем воды. Затем захватывают пинцетом фильтр, если на зеркаль-
ной поверхности задержались капли воды, их стряхивают и переносят фильтр на чашку
со средой Эндо. Нужно следить за тем, чтобы фильтр плотно пристал к среде и чтобы между
ним и средой не остались пузырьки воздуха.
Проращивание ведется в термостате при 37° С. Составные части среды, диффундируя
через поры фильтра, поддерживают рост бактерий, развивающихся в колонии. Последние
сохраняют на фильтре свой'типичный вид. Подсчитывают лишь типичные для группы Bact.
coli колонии; все другие колонии в учет не берут.
На основании результата подсчета определяют коли-индекс и коли-титр.
Пример. Профильтровано 100 мл воды. Типичных колоний оказалось 20. Следовательно,
20-1000 1000
коли-индекс = —— = 200, а коли-титр = "20б~= 5’
Для получения на мембранных фильтрах ускоренного роста применяют по прописи
лаборатории Рублевской водопроводной станции улучшенную среду Эндо. В ней обычный
мясопептопный агар заменяется агаром, который готовится по одному из следующих двух
рецептов:
1. Мясного лизата (Москов-
ского мясокомбината) 30 г
Пептона................. 10 »
Агара..................... 10 »
Воды дестиллированной. 1000мл
Настоя ячменных ростков 1—2 мл
2. Дрожжевого лизата ... 50 г
Мясного лизата........... 10 »
Пептона ................. 10 »
Агара.................... 10 »
Воды дестиллированной . 1000мл
Настоя ячменных ростков 1—2 мл.
pH сред = 7,0.
Настой из ростков ячменя готовится следующим образом: 5 г ячменных ростков осы
пают в 100 мл дестиллированной воды, перемешивают и нагревают на кипящей водяной
бане 15 минут (хранить не более 2 дней).
Среда в целом готовится так: 1,5 г лактозы и 0,04 г соды растворяют в пробирке в 4—5 мл
стерильной воды при кипячении. Охладив, добавляют к раствору 0,4 г безводного сульфита
натрия (Na2SO3) или 0,8 г кристаллического сульфита (Na2SO3-7 Н2О) и 0,8 мл насыщенного
спиртового раствора основного фуксина. Содержимое пробирки перемешивают и переливают
в 100 мл предварительно расплавленного питательного агара. Перемешивают и разливают
по чашкам Петри.
На этой среде рост бактерий группы coli получается уже через 8—12 часов.
ОБНАРУЖЕНИЕ ПАТОГЕННЫХ БАКТЕРИЙ В ВОДЕ
Обычный санитарный анализ воды не включает исследований на присут-
ствие патогенных бактерий. В некоторых случаях, однако, могут понадо-
биться подобные исследования (водные вспышки кишечных инфекций, по-
дозрение на возможное инфицирование воды и т. п.).
Исследование воды на тифозно-паратифозную группу
Согласно новейшей систематике возбудители тифозно-паратифозных ин-
фекций отнесены к группе сальмонелл.
Бактерии тифозно-паратифозной группы представляют небольшие (раз-
меры 0,5—0,8 х 1,5—3 р.) грам-отрицательные палочки с закругленными
концами, весьма похожие на Bact. coli. Спор и капсул не образуют. Снабжены
большим числом перитрихиальных жгутиков. Подвижность хорошо выра-
жена.
Развиваются в аэробных условиях.
На агаре Bact. typhi abdominalis и paratyphi А дают колонии в виде слегка
выпуклых влажных дисков, почти прозрачных в проходящем свете. Колонии
Bact. paratyphi В несколько более грубы.
После 24-часового роста при 37°С и дальнейшем хранении при комнат-
ной температуре по периферии колоний Bact. paratyphi В образуются при-
поднятые слизистые валики.
На желатине Bact. typhi abdominalis образуют характерные колонии,
напоминающие форму виноградного листа.
На среде Эндо вся тифозно-паратифозная группа дает бесцветные
колонии.
Тифозные палочки разлагают глюкозу, мальтозу и маннит с образованием
только кислоты (газа не образуют); паратифозные палочки разлагают их
с образованием кислоты и газа.
Лактозу и сахарозу вся тифозно-паратифозная группа не разлагает, мо-
лока не свертывает, желатину не разжижает. Bact. typhi abdominalis u Bact.
paratyphi А вызывают на лакмусовом молоке слабое кислотообразование.
Bact. paratyphi В показывают на этом же молоке щелочение.
Тифозные и паратифозные бактерии редко обнаруживались в питьевой
воде, возможно, из-за несовершенства методики или запоздалого анализа
воды, когда микробы, вызвавшие инфекцию, в силу отмирания уже в воде
исчезли.
Для обнаружения в воде бактерий тифозно-паратифозной группы было
предложено множество методов. В настоящее время лучшей методикой счи-
тается следующая.
Воду в большом количестве (8— 12 л) и Солее в зависимости от степени
загрязнения) пропускают через мембранные фильтры.
Для выращивания микробов часть фильтров помещают в желчь, часть —
на чашки с висмут-сульфитной средой.
1. Выращивание в желчи. Бычью желчь фильтруют, разливают в колбы
и стерилизуют. Три-четыре мембранных фильтра с задержанной на них
389
микрофлорой помещают в колбу с 80—100 мл стерильной бычьей желчи
и выдерживают в термостате при 37°С до 10 дней.
Через одни, двое, пять суток и на 10-й день делают высевы из желчи на
среду Эндо.
На этой среде колонии тифозных и паратифозных микробов бес-
цветны.
2. Выращивание на висмут-сульфитной среде (приготовление см. на стр. 115).
На чашках с висмут-сульфитной средой помещают несколько мембранных
фильтров с задержанной на них микробной флорой. Вместо этого можно по-
сеять на чашках с указанной выше средой при помощи шпаделя смыв, полу-
ченный с мембранных фильтров. Для получения смыва 3—4 мембранных
фильтра помещают в колбу с небольшим количеством (15—20 мл) стериль-
ного физиологического раствора и взбалтывают. Смыв сливают в про-
бирку, декантируют от крупных частиц и, если нужно, разбавляют
стерильным физиологическим раствором до миллиардной густоты по
стандарту.
Брюшнотифозные палочки через 24 часа дают на висмут-сульфитной среде
плоские колонии, которые при рассматривании на белом фоне или в прохо-
дящем свете имеют темный, грязнодымчатый цвет. К концу вторых суток ко-
лонии увеличиваются в размере и приобретают темную зеленоватую окраску;
на периферии окраска может оказаться более светлой.
Паратифозные микробы дают слегка выпуклые колонии черного цвета
с темными ореолами. Сохранившиеся единичные кишечные палочки разви-
ваются в колонии, имеющие коричневый оттенок. Протей дает зеленые ко-
лонии.
Если на диференциальных средах (Эндо, висмутсульфитная) получились
типичные или подозрительные колонии, то окончательная диагностика под-
крепляется следующими пробами:
1) пестрий ряд (укороченный),
2) реакция агглютинации и
3) реакция отклонения комплемента.
Пестрый ряд. Из развившихся на среде Эндо бесцветных колоний
отвивают чистую культуру на косой агар и идентифицируют ее по пестрому
ряду с бульонной средой, (на индолообразование), с жидкой цветной средой,
с маннитом (кислотообразование и газообразование), с лакмусовым молоком
(кислото- и щелочеобразование, свертывание). При этом учитывают следу-
ющие данные, отмеченные уже выше:
Bact. typhi abdominalis не образуют индола, образуют кислоту на манните
и показывают слабое кислотообразование на лакмусовом молоке.
Bact. paratyphi А не образуют индола, показывают кислотообразование
и газообразование на манните и слабое кислотообразование на лакмусовом
молоке.
Bact. paratyphi В не образуют индола, показывают кислотообразование
и газообразование на манните и щелочение на лакмусовом молоке.
Реакция на индолообразование производится следующим обра-
зом: к бульонной культуре прибавляют равное по объему количество эфира,
смесь взбалтывают в течение 3—5 минут, закрывая при этом пробирку кор-
ковой пробкой; затем дают эфиру отстояться, причем в случае плохого отстаи-
вания прибавляют несколько капель спирта. Эфирную вытяжку сливают
в отдельную пробирку, добавляют 0,25 объема 4% диметиламидобензальде-
гида на спирту и взбалтывают тонкой стеклянной палочкой или пастеров-
ской пипеткой. На дно пробирки наливают около 0,5 мл крепкой соляной
кислоты, вводя при этом пипетку до дна пробирки и осторожно выпуская
кислоту так, чтобы жидкости переслоились. Появление красного кольца на
границе обоих слоев указывает на присутствие индола. Фиолетовый оттенок
кольца указывает на присутствие и скатола.. Если в течение 5 минут кольцо
не появляется, реакция считается отрицательной.
390
Проба с маннитом проводится как описано на стр. 383.
Реакцию агглютинации проводят на предметном стекле
с тифозной и паратифозными сыворотками в разведениях 1 : 10, 1 : 20, 1 : 100
и более. Методику проведения см. на стр. 121.
Для реакции связывания комплемента служит смыв
с мембранных фильтров миллиардной густоты по стандарту. Смыв предва-
рительно прогревают при 6О°С в течение часа и затем охлаждают до комнатной
температуры. В опыт берут по 0,5 мл смыва. Методику проведения реакции
см. стр. 124.
Результат реакции отклонения комплемента в 2 креста следует считать
подозрительным, результат в 3 и 4 креста считается несомненным.
Исследование воды на возбудителя бациллярной дизентерии
В настоящее время известны б биологически близких между собой видов
бактерий, составляющих дизентерийную группу (Schigella) и относящихся
к семейству coli-typhus.
Морфологически они представляют короткие палочки с закругленными
концами, как и другие бактерии этого семейства. Жгутики у них отсутствуют.
Развиваются бактерии в аэробных условиях. На среде Эндо дают бесцветные
колонии.
Из дизентерийных палочек чаще других видов этой группы могут
встречаться Bact. dysenteriae Flexner u Bact. dysenteriae Hiss-Russel.
Свежевыделенные штаммы этих бактерий на пестром ряду показывают
почти одни и те же свойства: разлагают с кислотообразованием глюкозу и
маннит, не разлагают лактозу и сахарозу, не свертывают молока, индол обра-
зуют не постоянно, по отношению к мальтозе ведут себя неодинаково—ба-
циллы Флекснера разлагают мальтозу с кислотообразованием, бациллы
Гисс—Русселя не разлагают мальтозу.
Исследование воды на наличие дизентерийных бацилл ведется вообще так
же, как и на присутствие тифозно-паратифозной группы.
Фильтруют воду через мембранные фильтры. Приготовляют с них смыв,
как описано выше, и засевают шпаделем на среду Эндо. Из развившихся
бесцветных колоний отвивают чистую культуру на косой агар и идентифици-
руют ее по пестрому ряду, реакции агглютинации и реакции связывания
комплемента.
Исследование воды на присутствие вибрионов азиатской холеры
Холерный вибрион — грам-отрицательная, изогнутая в виде запятой па-
лочка (длина 2—3 р, ширина 0,5 р). Спор и капсул не образует. Снабжен
одним жгутиком (монотрих), весьма подвижен. Отличается полиморфизмом.
Разжижает желатину, образует индол.
Методика исследования воды такова:
К 900 мл воды прибавляют 100 мл 10% пептона (pH 8,0—9,0) и ставят в
термостат при 37° на 8—10 часов с целью накопления.
При наличии холерных вибрионов в воде на поверхности образуется неж-
ная пленка. Из нее берут материал для бактериоскопии, а также для посева
на щелочные среды — щелочный агар, щелочный кровяной агар.
На последнем холерные вибрионы образуют через 24 часа круглые,
с гладкими краями, крупные колонии; в проходящем свете колонии про-
зрачны, в падающем — голубоватосерого цвета.
Подозрительные колонии подвергаются подробному исследованию ввиду
возможности присутствия в воде холероподобных вибрионов.
Материал из колоний подвергается бактериоскопии, реакции агглютинации
со специфической агглютинирующей сывороткой, реакции связывания ком-
племента, испытывается на способность разжижать желатину (посев уколом)
и на индолообразование.
391
БИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ВОДЫ И ВОДОЕМОВ
Биологическое исследование водоемов сводится к изучению гидрофлоры
и гидрофауны микро- и макронаселения водоема в целом.
На основании совокупности данных об организмах, населяющих воду,
дно, берег и т. п., можно сделать вывод о степени загрязнения водоема и о рас-
пределении в нем загрязнения.
Наиболее характерные данные получаются при исследованиях, произве-
денных во второй половине лета, осенью и отчасти зимой.
Организмы, жизнь и развитие которых находится в зависимости от за-
грязнения воды и по обнаружению которых можно, следовательно, судить
о степени чистоты или загрязнения воды, называются показатель-
ными или индикаторными организмами.
Таких организмов в настоящее время насчитывают около тысячи.
Все индикаторные организмы подразделяются на п о л и с а п р о б н ы е,
живущие в сильно загрязненных водах, мезо сапробные, жи-
вущие в водах средней степени загрязненности, и олигосапробные,
живущие в относительно чистых водах.
Мезосапробные организмы в свою очередь подразделяются на две групы:
а-мезосапробные и ^-мезосапробные. а - мезосапроб-
ные характеризуют биологически слабо очищенные сточные воды, р - мезо-
сапробные населяют биологически очищенные воды.
Таблица 62 дает схематическую характеристику различных сапробных зон.
Гидрофлору и гидрофауну подразделяют на бентос и планктон.
Бентос охватывает все формы растений и животных, заселяющих
преимущественно дно водоемов или поверхность различных подводных пред-
метов.
Планктон включает все формы растений и животных, проводящих
свою жизнь в толще самой воды, причем растительные организмы планктона
составляют фитопланктон, а животные — зоопланктон.
По размерам организмов, входящих в состав планктона, различают мезо-
планктон (большие организмы), микропланктон (мелкие орга-
низмы) и наннопланктон (весьма мелкие организмы).
Частицы мертвого характера, взвешенные в воде, образуют триптон
(абиосестон, детрит).
Под сестоном понимают все живые организмы и мертвые частицы,
взвешенные в воде, которые могут быть «отсеяны» из нее тем или иным путем
(планктическая сеть).
Под нейстоном понимают организмы, развивающиеся в слое по-
верхностной пленки воды.
Под псевдопланктоном понимают посторонние водоему тела,
попадающие в него извне: волокна хлопка, льна, шерсти, зерна крахмала,
мышечные волокна и т. п.
Главная масса планктона и бентоса источников питьевых вод принад-
лежит обыкновенно к олигосапробам. К ним в большей или меньшей мере,
в зависимости от степени загрязнения воды, примешаны мезо- и полисапроб-
ные формы. При этом дно водоема обыкновенно на одну ступень грязнее воды
того же водоема. Так, если в воде преобладают представители олигосапробов,
то на дне и в иле преобладают мезосапробы и т. п.
Для биологической оценки воды имеет значение не только нахождение
определенных организмов, но и их количественное соотношение.
Техника собирания материала
Для улавливания планктона и собирания бентоса пользуются различными
приборами: планктическими сетками, черпаками, илососами, дночерпате-
лями и т. п.
392
Таблица 62
Схематическая характеристика сапробных зон1
Признаки Зона полиса- пробная Зона а-мезоса- ' пробная I Зона 1 р-мезоса- иробная Зона олигоса- пробная
1. Химический состав Белковые вещества Аммиак Аминокислоты Амиды Амидокислоты NH3, N2O3 n2o5 NA
2. Кислородные условия Анаэробные Полуанаэроб- ные Аэробные Аэробные
3. Характер Восстанови- Восстанови- Окислитель- Окислитель-
биохимичес- ких процес- тельные тельно-окис- лительные ные ные
сов
4. Угольная Много Порядочно Немного Мало
кислота
5. Сероводород Много Порядочно Мало Нет
6. Форма соеди- Fe S FeS - j- Fe2O3 Fe2O3 FeaO3
нений железа
7. Проба на за- Загнивает Загнивает Не загнивает Не загнивает
гниваемость
8. Источники кислорода Диффузия Диффузия Диффузия и ассимиляция СОг Диффузия и ассимиляция СО2
9. Содержание бактерий Сотни тысяч— —миллионы Сотни тысяч Десятки тысяч Сотни—десятки
10. Интенсив- ность разви- Обычно высокая Очень высокая Значительная Нередко высокая
тия отдель- ных форм
11. Разнообразие Очень малое Небольшое Значительное Очень большое
ВИДОВ
12. Преоблада- ние отдель- Очень сильное Сильное Слабое Обычно слабое ।
ных видов
13. Смена сооб- Часто катастро- Часто катастро- Довольно Довольно
ществ фическая фическая медленная медленная
14. Продуценты Нет Мало Немного Много
15. Консументы Очень много Много Много Немного
16. Пожиратели Масса Много Немного Очень мало
бактерий
17. Пожиратели Нет Редки Нередки Часты
растений
18. Пожиратели Почти нет Есть Много Очень много
животных
19. Водные цвет- Нет Нет—мало Немного Много
ковые расте- ния
20. Главные груп- Бактерии- Грибы. Синезеленые Зеленые водо-
пы организ- бесцветные Бактерии. водоросли. Ди- росли. Диатомо-
мов жгутиковые Серные бактерии. Инфузории Инфузории. Синезеленые водоросли. Зеленые жгути- ковые. атомовые водо- росли. Зеленые водоросли. Зеле- ные жгутиковые. Инфузории. Коловратки. Ракообразные. Рыбы. вые водоросли. Перидиней. Хри- зомонады. Коло- вратки. Мшанки. Губки. Ракооб- разные. Рыбы
21. Потребность Ничтожная Слабая Большая Очень большая
организмов в кислороде
1 «Стандартные методы исследования питьевых и сточных
вод», М.» 1927.
393
Планктическая сетка (рис. 240) представляет сачок, изготов-
ленный из шелкового газа № 20. Сверху сачок прикреплен к медному кольцу,
& а книзу переходит в медный цилиндр, заканчивающийся рези-
? новой трубкой с зажимом.
А Планктон улавливают обыкновенно с лодки; при этом сачок,
/ \ прикрепленный к бечевке, опускается в воду до полного погру-
/1\ жения и тянется за кормой. Когда соберется достаточно мате-
риала, сачок извлекают из воды. Чтобы смыть планктон, осевший
gtilgg на внутренней поверхности в узкую часть сачка, погружают
шйМг несколько раз сачок в воду, оставляяверхнее кольцо над поверх-
ностью воды. Собранный в низу сачка планктон переводят затем
в широкогорлую банку емкостью в 50 мл с корковой пробкой.
Под пробкой необходимо оставить воздух для поддержания
мН жизни планктона. На банку наклеивают этикетку с номером
W пробы. В журнале под соответствующим номером отмечают
А? место и время забора пробы, метеорологические данные и т. п.
/ft Ввиду того что ячейки планктических сетей имеют размеры
в 70—80р. и более, очень мелкие формы организмов проходят
Рис. 240. через них, и для улавливания наннопланктона приходится при-
Плапктичес- бегать к отбору проб воды без фильтрации.
кая сетка. Черпак (рис. 241) служит для собирания бентоса с под-
водных предметов и зачерпывания ила. Применение его не
нуждается в пояснении.
И л о с о с Перфильева (рис. 242) состоит из широкогорлой склян-
ки а, закрывающейся каучуковой пробкой. В пробку вставляется один конец
латунной коленчатой трубки Ь; на другом конце прикрепляется груз с. Внутри
короткого колена впаяна
узкая латунная трубка d,
выходящая одним концом
наружу. На наружный ко-
нец ее надевается каучу-
ковая трубка е, свободный
конец которой находится
в руках наблюдателя. При
опускании илососа в воду,
свободный конец каучуко-
вой трубки должен быть
зажат. Когда илосос до-
стигает дна водоема, от-
крывают зажим. Под дав-
лением столба воды ил
заполняет через широкую
латунную трубку склянку,
Рис. 241. Черпак. Рис. 242. Илосос
Перфильева.
а — склянка, Ъ — латунная
коленчатая трубка, с—груа,
d —отводная латунная труб-
ка, е — каучуковая трубка.
а затем по узкой трубке подымается вверх, вытес-
няя отсюда воздух. Прибор пригоден для работы
на глубине не более б—7 м.
Дночерпатели построены и работают по
принципу экскаваторов. В зависимости от размеров они облавливают пло-
щадь в 0,1 м2 и 0,025 м2.
Отбор проб планктона и бентоса производится в заранее намеченных пунк-
тах и в разное время года.
Особое внимание уделяют местам с быстрым течением: у плотин, шлюзов,
перекатов и т. д.
Методика исследовапия планктона и бентоса
Исследование планктона. Собранный материал рекомен-
дуется исследовать живым, так как фиксация может значительно деформи-
394
ровать отдельные организмы. При необходимости отложить исследование
фиксация производится прибавлением 5% (по объему) 40% формалина.
Макропланктон исследуется невооруженным глазом или при помощи
лупы, микропланктон — под микроскопом.
Приготовленный на предметном стекле под покровным стеклом препарат
рассматривается под микроскопом с сухими объективами.
Определение рода и вида организмов осуществляется при помощи опреде-
лителей и таблиц. Особи, вызывающие сомнение, лучше не учитывать вовсе.
Частота находок обозначается терминами: много (преобладают); очень
часто; часто (не менее 10 в препарате); нередко (менее 10 в препарате); редко
(3—4 в препарате) и очень редко (1—2 в препарате).
С целью изучения наннопланктона, как было указано, вода берется не-
посредственно каким-либо сосудом.
Для приготовления препаратов вода из этих сосудов подвергается центри-
фугированию. Полученный осадок из центрифужных пробирок переносится
пипеткой на предметные стекла и исследуется под
микроскопом.
Для количественного учета планктона, т. е.
определения числа организмов в единице объема
воды, пользуются счетными камерами (рис. 243)
на 1 мл и на 20 мл.
Подсчет ведется по возможности тотчас же
после взятия пробы. Предварительно взболтав
исследуемую воду, наливают ее доверху в камеру
и накрывают покровным стеклом. Подсчет ведется
при увеличении в 20—40, а затем в 100—120 раз.
Увеличение в 20—40 раз дает возможность
подсчитать относительно крупные микроорганизмы
(водоросли, инфузории) во всей камере.
При увеличении в 100—120 подсчитывают более
мелкие микроорганизмы. При этом поступают
Рис. 243. Счетная камера.
следующим образом: устанавливают на фокус поверхность воды и под-
считывают видимые организмы в поле зрения; затем опускают мед-
ленно объектив и подсчитывают организмы в толще воды и на дне камеры.
Сумма подсчитанных микроорганизмов соответствует числу их в одном поле
зрения по высоте всей камеры. Так подсчитывают микроорганизмы в 10—15
полях зрения и находят среднее арифметическое соответственно одному полю
зрения во всей толще воды данной камеры.
Для определения числа организмов в целой камере умножают полученный
R2
результат на коэфициент, равный — , где/? — радиус камеры, аг — радиус
поля зрения, определенный по масштабу, помещенному под объективом.
Если в пробе много подвижных организмов, мешающих подсчету, то смачи-
вают внутреннюю поверхность крышки водным раствором сулемы или вводят
при помощи иглы маленькую каплю 1 % осьмиевой кислоты, или подогревают
пробу до 35—40°С.
Исследование бентоса. Отмечают характер грунта, цвет,
запах, примеси.
Промывают материал через систему сит:
нижнее.............0,5 мм,
среднее............ 1,0 мм,
верхнее............2,0—3,0 мм.
Промыв пробу, достают пинцетом из сит выловленные вместе с грунтом
организмы и переносят в банку для дальнейшего изучения. Отбирают также
пробы для микроскопического исследования, как описано выше. Все это
дает представление о качественном составе бентоса.
395
С количественной стороны бентонс не поддается точному учету, и в этом
отношении ограничиваются лишь представлениями об интенсивности разви-
тия тех или иных организмов.
На основании всестороннего и повторного изучения материала делают
заключение о характере сообщества флоры и фауны в данном водоеме и
отсюда — о степени чистоты водоема.
САНИТАРНОЕ ИЗУЧЕНИЕ ИСТОЧНИКОВ ВОДОСНАБЖЕНИЯ
Для полной санитарной характеристики водоснабжения необходимо, по-
мимо физико-химического, бактериологического и биологического анализа
воды, произвести также всестороннее санитарное обследование источника
водоснабжения. Особого внимания заслуживает та часть бассейна, которая
может влиять на качество воды в водозаборе.
Важное значение указанного метода изучения источников водоснабжения
заключается в том, что он может дать прямые указания на источники загряз-
нения водоема.
Обследование открытых водоемов
При изучении открытых водоемов (река, озеро, пруд) выясняют следующее:
а) особенности самого водоема — его расположение и размеры, режим по-
верхностного стока в различные сезоны, расход воды, скорость течения и
уровень воды в месте водозабора, средние сроки ледостава и вскрытия,
подъем воды, разливы, размер забора воды и т. п.; б) характер берегов — почва,
растительность, наличие лесов, возделываемых земель, населенных пунктов
ит. п.; в) отношение водоема к населенным пунктам — наличие на том или
ином берегу фабрик, заводов, мельниц, боен, бань, прачечных, скотных дво-
ров, отхожих мест, помойных, навозных ям и т. п., т. е. всех источников,
которые могут вести к загрязнению водоема; г) качественную и количествен-
ную характеристику сточных вод (бытовых и промышленных), спускаемых в
водоем, расстояние места спуска стоков от водозабора, состояние очистных
сооружений; д) степень защищенности водоисточника от проникновения
других загрязнений извне и т. п.
Обследование грунтовых источников
При обследовании грунтовых источников выясняют: а) местонахождение
источника — улица, площадь, двор, сад, огород, пустырь и т. п.; б) место-
расположение источника — возвышенное, низкое, на уклоне и т. п.; в) основ-
ные гидротехнические данные — тип источника (колодец шахтный, труб-
чатый, смешанный, ключ, родник и т. п.); время сооружения; для шахтного
колодца выясняют конструкцию, размеры шахты, материал стенок, высоту
стенки над поверхностью земли, наличие перекрытия; водоподъемные соору-
жения ит. п.; для трубчатого колодца выясняют тип его (буровой, вбивной),
диаметр труб, водоподъемные сооружения, наличие водоразбора у колодца,
водопровода от колодца и т. п.; г) основные гидрогеологические сведения—
характер пройденных скважиной пород, их последовательность и мощность,
характер водоносного горизонта и того слоя, который составляет дно колодца,
глубину источника до дна, глубину источника до поверхности, высоту над
уровнем моря и т. п.; д) содержание источника и его санитарное состояние—
прочность и состояние стенок, перекрытия и водоподъемных сооружений;
состояние почвы около колодца, наличие загрязнений у колодца (водопой
скота, стирка белья и т. п.); наличие вблизи колодца жилых домов, скотных
дворов, помойных, мусорных и навозных ям, отхожих мест и т. п.; е) водо-
пользование— для каких целей пользуются водоисточником; число домов
и жителей, пользующихся источником; когда пользуются водоисточником
396
(круглый год или в определенные сезоны), средний расход воды в сутки;
ж) хлорируется ли вода колодца и как часто.
Если возникает предположение о связи между колодцем и источником
загрязнения, то производят экспериментальную проверку такой возможности.
Для этой цели вливают 2% раствор флуоресцеина в буровую скважину
или в воронку, через которую стекают в почву поверхностные воды. Из источ-
ника (колодца), в котором ожидают появление краски, берут каждый час
воду для исследования на присутствие флуоресцеина. Если расстояние между
колодцем и предполагаемым источником загрязнения не превышает 5 км,
то достаточно ввести в почву 100—300 г краски. Присутствие флуоресцеина
в воде колодца можно обнаружить по зеленой флуоресценции уже при кон-
центрации 1 : 10 000 000, а с помощью флуороскопа —в разведении
1 : 1 000 000 000.
Флуороскоп состоит из ряда стеклянных трубок высотой 95 см, нижний
конец которых закрывается черной каучуковой пробкой. Трубки помеща-
ются в штатив.
Вместо флуоресцеина для данной же пробы пользуются и сапролом, ко-
торый при концентрации 1 : 1 000 000 придает воде вкус нафталина. Можно,
наконец, ввести в буровую скважину раствор поваренной соли и следить
за концентрацией хлоридов в обследуемом колодце и т. п.
Подробные схемы обследования различных водоисточников можно найти
в книгах: «Стандартные методы исследования питьевых и сточных вод», М.,
1927; проф. В. А. Углов, «Санитарное изучение источников водоснабжения
на месте и их характеристика», Госмедиздат УССР, 1931. Примерная про-
грамма обследования источников водоснабжения приведена также в ГОСТ
2761-44.
На основании всестороннего исследования воды и изучения источника
водоснабжения делают заключения о качестве водоисточника и воды, ука-
зывают мероприятия, которые необходимо осуществить для ограждения
источника от загрязнений: зоны санитарной охраны для открытых водоемов,
улучшение оборудования и содержания шахтных колодцев, устройство кап-
тажа для ключей и т. п.
Если качество воды требует улучшения, то указывают, какие мероприятия
необходимо осуществить в каждом отдельном случае: осветление, обеззара-
живание, обезжелезивание и пр.
ГЛАВА ПЯТАЯ
СТОЧНЫЕ ВОДЫ
САНИТАРНЫЕ ТРЕБОВАНИЯ К СТОЧНЫМ ВОДАМ
Санитарные требования, предъявляемые к сточным водам, спускаемым
в водоемы, имеют целью предупредить загрязнение водоемов. Эти требования
сформулированы в ГОСТ 1324-47. Они тем строже, чем более важную роль
в санитарном и рыбохозяйственном отношении играют общественные водоемы.
Последние по указанным признакам разделены на три категории:
1. Участки водоемов, используемые для централизованного водоснабже-
ния, находящиеся в пределах второго пояса зоны санитарной охраны водо-
проводов или граничащие с государственными рыбными заповедниками.
2. Участки водоемов, используемые для неорганизованного хозяйственно-
питьевого водоснабжения и водоснабжения предприятий пищевой промыш-
ленности, а также участки с местами массового нереста промысловых рыб.
3. Участки водоемов внутри населенных мест, не используемые для питье-
вого водоснабжения, но используемые для массового купания или имеющие
архитектурно-декоративное значение, а также используемые для организован-
ного рыбного хозяйства или находящиеся на пути прохода рыб к нерести-
лищам.
Показатели загрязнения
1. Взвешенные вещества. После спуска сточных вод в водоем и смешения
допускается увеличение содержания взвешенных веществ на 0,25 мг/л для
водоемов первой категории, 0,75 мг/л — второй категории и 1,5 мг/л—третьей
категории.
Для водоемов, содержащих в межень более 30 мг/л природных взвешен-
ных веществ, а также при периодическом спуске сточных вод, в период па-
водков и в тех случаях, когда согласно указанным нормам требуемая степень
осветления сточных вод не может быть достигнута существующими методами
очистки, условия спуска устанавливаются органами Государственной сани-
тарной инспецкии.
2. Плавающие примеси. Сточные воды не должны содержать масел, жиров,
нефтепродуктов и других плавающих веществ в таких количествах, которые
способны вызвать в водоеме массовое образование сплошных плавающих
пленок.
3. Запахп п привкусы. После разбавления сточных вод в водоеме вода
последнего не должна приобретать непосредственно или при последующем
хлорировании никаких специфических запахов и привкусов за счет сточ-
ных вод.
4. Окраска. Смесь сточной жидкости с дестиллированной водой в про-
порции, соответствующей расчетному разбавлению в водоеме, не должна
иметь ясно выраженной окраски в столбике высотой в 20 см для водоемов
398
первой категории, 10 см — второй категории и 5 см — третьей категории.
5. Реакция. Сточные воды не должны изменять активной реакции воды
в водоеме по pH ниже 6,5 и выше 8,5.
6. Растворенный кислород. Сточные воды после смешения с водой водоема
не должны снижать в последнем содержание растворенного кислорода ниже
5 мг/л, считая по среднему суточному содержанию кислорода в летнее время,
а для водоемов рыбохозяйственного значения — по суточному минимуму
в тот же период.
7. Биохимическая потребность в кислороде (ВПК). После смешения сточ-
ной жидкости с водой водоема пятисуточная потребность в кислороде (при
20®С) воды не должна превышать 2 мг/л для водоемов первой категории и
4 мг/л — второй категории. Для водоемов третьей категории этот показа-
тель не нормируется.
8. Ядовитые вещества. Сты лые воды ни в растворе, ни в взвешенном
состоянии не должны содержать ядовитых веществ, которые могли бы после
их разбавления в водоеме оказать прямо или косвенно вредное действие на
человека, животных или рыб. Предельно допустимые концентрации ядови-
тых веществ промышленных сточных вод, спускаемых в водоем, устанавли-
ваются Государственной санитарной инспекцией.
9. Возбудители заболеваний. Сточные воды, в которых возможно при-
сутствие возбудителей заразных заболеваний людей и животных (сточные
воды боен, кожевенных заводов, шерстомоек, биофабрик и т. п. или отдель-
ных цехов этих предприятий) к спуску запрещаются (без соответствующей
обработки). До спуска их в водоем после предварительного механического
осветления они должны подвергаться обеззараживанию.
Примечания. 1. Правила спуска сточных вод в водоемы, не предусмот-
ренные указанными выше тремя категориями, устанавливаются органами Государ-
ственной санитарной инспекции.
2. Зоны санитарной охраны водопроводов устанавливаются в порядке, преду-
смотренном постановлением ЦИК и СНК СССР от 17.5. 1937 г. за № 96/834.
3. Водоемы рыбохозяйственного значения определены постановлением СНК
СССР от 25.9.1935 г. за № 2157.
4. Условия спуска сточных вод в водоемы определяются с учетом существующего
санитарного состояния водоема у мест водопользования. Места водопользования1
в зависимости от его назначения устанавливаются органами Государственной сани-
тарной инспекции.
СХЕМА ПОЛНОГО САНИТАРНОГО АНАЛИЗА СТОЧНЫХ ВОД
Физико-химические исследования
Температура
Запах
Цветность
Прозрачность
Мутность
Осадок
Пленка
Реакция (pH)
Щелочность или кислотность (титрир-
ная)
Взвешенные вещества
Плотный остаток
Окисляемость
Хлориды
Сульфаты
Фосфаты
Азот аммонийный
Азот альбуминоидный
Азот нитритов
Азот нитратов
Азот органический
Азот общий
Углерод органический
Биохимическая потребность в кислорода
(БПК)
Проба на загниваемость
Проба на стабильность
Сероводород
Свободный хлор
Тяжелые металлы: свинец, медь,
цинк, ртуть
Мышьяк
Другие ядовитые вещества: фенол
и крезол, хром, цианистые и рода-
нистые соединения, нефть.
Бактериологические исследования
Коли-титр
Патогенные бактерии.
СХЕМА КРАТКОГО САНИТАРНОГО АНАЛИЗА СТОЧНЫХ ВОД
Температура Взвешенные вещества (по объему)
Запах Окисляемость
Цветность Хлориды
Прозрачность Азот аммонийный
Мутность Азот нитритов
Осадок Азот нитратов
Пленка Биохимическая потребность в кисло-
Реакция (pH) роде (БПК)
Щелочность или кислотность (титрир- Проба на загниваемость
на я) Сероводород.
ОТБОР ПРОБ
Ввиду значительной изменчивости состава сточных вод прибегают к так
называемым составным пробам: через определенные промежутки вре-
мени отбираются индивидуальные пробы, которые затем смешиваются, давая
среднюю пробу. Если дебет стока во времени постоянен, то для получения
средней пробы смешивают одинаковые объемы индивидуальных проб; в
противном случае смешивают объемы, пропорциональные кривой дебета.
Общий объем воды, взятой для анализа, должен достигать 3—4 л.
Если взятая проба не может быть подвергнута тотчас же анализу, то ее
консервируют: на литр воды прибавляют 5—б мл хлороформа и помещают
пробу на лед. Вместо хлороформа как консервант может быть применена сер-
ная кислота в количестве 1 г на литр. Пробы, предназначенные для опреде-
ления стабильности и биохимической потребности в кислороде, не подверга-
ются консервированию.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ СТОЧНЫХ ВОД
Сточные воды в общем исследуются теми же методами, какими исследуются
питьевые воды. Только в отношении некоторых проб выработана особая ме-
тодика, которая здесь приводится.
Температура, цвет, запах, муть, осадок и активная реакция определяются
на месте. Амиак, нитриты, нитраты и окисляемость должны быть опреде-
лены в первый же день по доставлении пробы в лабораторию. Для опреде-
ления органического азота, аммиака, окисляемости и органического угле-
рода вода должна быть профильтрована; другие испытания производят с не-
фильтрованной водой.
Температура, запах, цветность, прозрачность
Исследование производится на месте взятия пробы так же, как и при анализе
питьевой воды. При этом нужно учесть следующее: запах, кроме обычного
способа, определяется еще после подкисления для установления наличия
связанного сероводорода; цветность определяется обыкновенно только каче-
ственно с указанием, окрашена ли вода сильно, незначительно или бесцветна.
Муть и осадок, пленка
Мутность воды выражают описательно. Различают муть грубую, хлопье-
видную, волокнистую, коллоидальную и пр. Помимо характера указывают
еще цвет и интенсивность мути.
Осадок наблюдают, налив воду в стеклянный цилиндр, после предвари-
тельного взбалтывания пробы. Осадок определяют словами: илистый, хлопье-
видный, комковатый, зернистый, слизистый и пр. К осадку причисляют и те
скопления плотных веществ, которые остаются во взвешенном состоянии
в виде аггрегатов различной формы.
Особо отмечают наличие на поверхности воды пленок маслянистых, неф-
тяных и пр.
Реакция (pH), щелочность и кислотность (титрирные)
Определение производится, как и в питьевой воде.
Вследствие невысокой буферности сточных вод и невозможности ввиду
этого разбавления их для колориметрического титрования, предпочтительно
электрометрическое определение активной реакции.
Взвешенные вещества и плотный остаток
Взвешенные вещества и плотный остаток при 105°С и потеря при прока-
ливании их определяются, как в питьевой воде.
Взвешенные вещества, кроме обычного метода, определяются еще объемным
способом, что особенно важно при проектировании очистных сооружений.
Определение ведется в цилиндре, который конически суживается книзу,
переходя в градуированную трубку на 10—20 мл с делениями на 0,1 мл. Труб-
ка книзу снабжена краном.
После предварительного взбалтывания пробы, вода наливается в цилиндр
и отстаивается. Через час цилиндр приводится во вращательное движе-
ние между ладонями рук с целью оторвать частицы, приставшие к стенке,
и способствовать выпадению их вниз. Цилиндр после этого оставляют в по-
кое и через 55 минут повторяют вновь вращение цилиндра. Через пять
минут после этого делают отсчет.
ОкПСЛЯСМОСТЬ
При значительном содержании органических веществ в сточной воде для
определения окисляемости берется не более 5— 10 мл. Взятая проба
разбавляется ‘дестиллированной водой, освобожденной от органических
веществ перегонкой над щелочью и перманганатом, либо отдельным исследо-
ванием определяют расход перманганата на окисление органических веществ
дестиллированной воды и этот расход вычитают из общего объема потреблен-
ного перманганата при исследовании пробы.
Определение окисляемости производится после отстаивания пробы в те-
чение 2-х часов.
Методика и расчеты см. стр. 314 и далее.
Хлориды, сульфаты, фосфаты
Хлориды в сточной воде определяют так же, как в питьевой воде (стр. 330).
При этом нужно иметь в виду следующее: при применении метода Мора вода
до исследования подвергается усреднению и затем фильтруется. При кислой
реакции воды для усреднения применяют соду (аммиаком усреднять нельзя,
так как он растворяет AgCl); при щелочной реакции усреднение ведется серной
кислотой.
Сульфаты и фосфаты определяются так же, как в питьевой воде (стр. 333
и 346).
Азотсодержащие вещества
Азот аммонийный. Стандартная методика рекомендует прямую несслери-
зацию при условии, что испытуемая вода от прибавления реактива Несслера
не дает помутнения. Предварительно сточная вода подвергается осветлению
(если она мутна). Для этой цели к 100 мл сточной воды прибавляют 1 мл раст-
вора сульфата меди (100 г CuSO4 на 1 л дестиллированной воды, свободной
от аммиака) и тщательно взбалтывают; затем прибавляют сюда же 1 мл ра-
створа NaOH (500 г NaOH в 1 л воды) и вновь тщательно взбалтывают. Обра-
зующаяся гидроокись меди по реакции: CuSO4 + 2NaOH = Na2SO4 + Cu(OH)2
выпадая, увлекает взвешенные вещества и осветляет жидкость.
26 А. И. Бурштейн
401
Если проба содержит сероводород, то его удаляют следующим образом:
на 100 мл воды прибавляют 1 мл насыщенного раствора сернокислого цинка
либо 1 мл уксуснокислого свинца (100 г ацетата свинца в 1 л воды) и 1 мл
раствора едкого натрия, приготовленного, как указано выше.
5 мл (или меньше) осветленной и освобожденной от сероводорода сточной
воды отбирают пипеткой, разбавляют в цилиндре дестиллированной водой
до 50—100 мл и исследуют на аммонийный азот так же, как питьевую воду
(стр. 336).
Азот альбуминоидный определяют, как в питьевой воде (стр. 338).
Азот нитритов и нитратов определяют так же, как в питьевой воде
(стр. 338 и 339).
Вода предварительно обесцвечивается (если нужно) при помощи свеже-
приготовленной гидроокиси алюминия (приготовление см. на стр. 331).
Азот органический и общий определяют так же, как
в питьевой воде (стр. 342 и 345).
Углерод органический
Определение органического углерода сточной воды дает представление
о содержании органических веществ в воде.
Определяют отдельно углерод органических веществ, растворенных в воде,
и органических веществ, взвешенных в воде, для чего взвешенные вещества
отфильтровываются через тигель Гуча. Фильтрат и осадок анализируют от-
дельно.
Принцип метода. Органические вещества окисляются марган-
цевокислым калием в присутствии серной кислоты и сернокислой окиси
ртути. При этом углерод органических веществ окисляется до СО2. Образо-
вавшийся угольный ангидрид улавливают натронной известью в U-образных
трубках и определяют по привесу последних. Угольный ангидрид для пере-
счета на углерод умножают на 0,2727 соответственно-^-
Ре а кт и вы: 1. Серная кислота разбавленная (1 : 3).
2. Серная кислота концентрированная.
3. Марганцевокислый калий в кристаллах.
4. 20% раствор сернокислой окиси ртути (HgSO4).
5. Натронная известь.
6. Хлористый кальций в зернах.
7. Хромовая кислота.
Методика, определения. Вводят в колбу 250—500 мл филь-
трата, прибавляют 10 мл разбавленной серной кислоты, закрывают колбу
пробкой, в которую введен конец обратного холодильника. Кипятят в
течение получаса для удаления СО2 карбонатов, а также СО2 воздуха колбы.
По охлаждении прибавляют 3 г КМпО4 в кристаллах, 10 мл 20% раствора
HgSO4 и устанавливают прибор, как показано на рис. 244. Закрывают
колбу пробкой, в которую введены конец делительной воронки (а), дохо-
дящей почти до дна колбы, и конец холодильника (Ь), заканчиваю-
щийся на небольшом расстоянии от пробки. На свободный конец холо-
дильника надевается каучуковая трубка, присоединяющаяся к системе U-об-
разных трубок. Из них трубка с наполнена кусочками пемзы, смоченными
крепкой серной кислотой, трубки d и е заполняются хлористым кальцием,
насыщенным перед опытом угольной кислотой х, трубка g заполняется натрон-
ной известью, левые колена трубок / и h заполняются также натронной из-
х Насыщение хлористого кальция угольной кислотой имеет целью перевести окись
кальция (СаО), имеющуюся в продажном хлористом кальции, в карбонат кальция (СаО -|-
4- СО2 = СаСО3) и этим избежать возможности сорбции СО2 хлористым кальцием во время
опыта. Для указанной цели углекислый газ из аппарата Киппа проводится 2 минуты через
трубки с хлористым кальцием, после чего на протяжений 20 минут через них просасывается
воздух, не содержащий СО2.
402
вестью, а правые — хлористым кальцием. Через воронку а вводят в колбу
40 мл разбавленной серной кислоты. Закрыв кран воронки, постепенно на-
гревают содержимое колбы, пустив одновременно струю воды в холодиль-
ник (направление тока показано стрелками). Выделяющиеся во время опьпа
пары воды частично задерживаются в холодильнике и стекают назад в колбу:
остатки водяного пара задерживаются в первых трех трубках. Уголь-
ная кислота задерживается почти целиком в трубке g с натронной известью
и частично в трубке /. Хлористый кальций правого колена трубки / задержи-
вает воду, образующуюся от взаимодействия углекислого газа с едким натрием,
входящим в состав натронной извести, по реакции:
2NaOH + СО2 = Na2CO. + Н?О.
1 и Л О *
Что касается трубки й, то ее присоединяют к аппарату на случай образо-
вания обратного тока воздуха; при этом влага и СО2 будут задержаны в пре-
дохранительной трубке, и содержимое трубки / не изменится в весе.
Рис. 244. Прибор для определения органического углерода'в воде.
а — воронка с насаженной хлоркальциевой трубкой, b — холодильник,
с, d, е, g, /, h — поглотительные трубки, к — хлоркальциевая трубка
с натронной известью.
Газы пропускают через систему трубок медленно, что достигается умерен-
ным нагреванием колбы. Когда выделение СО2 прекращется, устраняют огонь,
присоединяют к воронке а хлоркальциевую трубку к с натронной известью,
соединяют свободный конец U-образной трубки h с аспиратором, открывают
кран делительной воронки а, медленно просасывают некоторое время через
всю систему наружный воздух, чтобы уловить остатки СО2 из колбы и
соединительных трубок; при этом углекислый газ внешнего воздуха задер-
живается натронной известью в хлоркальциевой трубке к.
Вес трубок g и / перед опытом и после него определяют на аналитических
весах и по увеличению в весе судят о количестве угольного ангидрида, задер-
жанного в трубках. Это количество соответствует угольной кислоте во взятой
пробе исследуемой воды. Результат перечисляют на литр и выражают в виде
углерода, умножив на коэфициент 0,2727.
Определение органического углерода взвешенных веществ сточной воды
проводится по тому же принципу, что и определение органических
веществ воды. Осадок после фильтрования воды в тигле Гуча вносят
вместе с асбестом в колбу. Прибавляют 10 мл 20% раствора сернокислой
окиси ртути, 5 г хромовой кислоты, вставляют в колбу пробку с делительной
воронкой и холодильником и вводят медленно 50 мл концентрированной
серной кислоты; в это время усиливают ток воды в холодильнике. Во всем
остальном исследование ведется, как описано выше. Предварительное удале-
ние СО2 карбонатов здесь не производится.
40i
Биохимическая потребность в кислороде (ВПК)
Под биохимической потребностью в кислороде или
биохимическим потреблением кислорода (ВПК) сточ-
ной воды понимают то количество растворенного в воде кислорода в милли-
граммах на литр, которое поглощают нестойкие органические вещества,
находящиеся в сточной воде. При этом должны быть учтены следующие усло-
вия: 1) сточная вода берется в том разбавлении с водой водоема, которое
имеет фактически место при спуске воды в водоем; 2) разбавленная вода на-
сыщается в начале опыта кислородом; 3) вода хранится в течение 5 суток
при 20°С.
Чем ВПК сточной воды выше, тем вода более загрязнена и тем больше
сна спссобствует задержке аэробных процессов самоочищения водоема.
Нормы ВПК для сточной воды, спускаемой в водоемы различной кате-
гории, указаны выше (стр. 399).
Принцип метода. Проба сточной воды после соответствующего
разбавления водою водоема и насыщения кислородом воздуха исследуется
на количественное содержание растворенного кислорода. Другая такая же
проба исследуется после пятисуточного хранения при 20°С. Арифметическая
разность получаемых двух данных, выраженная в миллиграммах на литр,
равна ВПК исследуемой воды.
Реактивы. Те же, что и при определении кислорода, растворенного
в воде (стр. 359).
Методика определения. Отбор пробы производится либо из
стока, и тогда сточная вода разбавляется водой водоема в такой степени,
чтобы это разведение соответствовало разведению в самом водоеме, либо проба
воды берется в водоеме после спуска сточной воды и естественного разбавле-
ния ее.
Склянка емкостью в 1,5 л заполняется пробой доверху и пересылается
в лабораторию летом на льду, а зимой согреваемая наложением на бутыль
пузыря с теплой водой.
В лаборатории воду переводят в большую колбу и нагревают на водяной
бане до 20°С. Закрыв колбу пробкой, взбалтывают воду в течение одной ми-
нуты для насыщения воздухом. Затем при помощи воронки с длинной шей-
кой заполняют исследуемой водой две склянки емкостью в 250—300 мл каж-
дая с притертыми пробками.
В одной из них определяют растворенный кислород тотчас же (см. стр. 351);
другую склянку помещают в термостат при температуре 20°С и выдерживают
здесь в течение 5 суток, после чего определяют оставшийся в ней растворен-
ный кислород. Разность между этими двумя определениями, выраженная
в миллиграммах, пересчитывается на литр.
Если сточная вода очень загрязнена, то наличие свободного кислорода
может оказаться недостаточным для покрытия потребности в кислороде;
в этом случае пробу разбавляют в 10—100 раз дестиллированной водой и
результат анализа умножают на степень разбавления.
Проба на загниваемость
Загниваемость сточной воды определяют по одному из признаков гние-
ния— появлению свободного сероводорода.
Наполняют литровую склянку почти доверху нефильтрованной исследуе-
мой водой, закрывают пробкой и выдерживают при 20°С в течение 7 дней
в темном месте. По истечении этого времени пробу исследуют на наличие
свободного сероводорода (см. качественные пробы на H2S, стр. 365).
Проба на загниваемость считается положительной, если обнаружен сво-
бодный сероводород. Проба производится с цельной сточной водой и с пор-
цией, разбавленной в 2—5 раз дестиллированной водой.
404
Проба на стабильность
Относительная стабильность сточных вод определяется по быстроте обес-
цвечивания прибавленной к воде метиленовой синьки. Чем больше в воде
органических веществ, способных окисляться при средней комнатной темпе-
ратуре, тем быстрее расходуется растворенный в воде кислород и наступает
восстановление метиленовой синьки — переход ее в лейкобазу.
В склянку емкостью в 150 мл вводят доверху испытуемую сточную воду,
соблюдая те же предосторожности, что и при отборе проб на кислород, раство-
ренный в воде (стр. 359). Прибавляют пипеткой 0,4 мл раствора метиленовой
синьки (0,5 г краски в литре дестиллированной воды), плотно закрывают
склянку притертой пробкой так, чтобы под пробкой не остались пузырьки
воздуха, и ставят в термостат при 20° С. Выдерживают пробу до обесцвечи-
вания; при этом наблюдение ведется два раза в сутки.
Относительную стабильность воды выражают процентным отношением на-
личного кислорода в виде растворенного нитритного и нитратного ко всему
кислороду, необходимому для покрытия всей биохимической потребности
в кислороде данной пробы. Это соотношение может быть вычислено по
формуле:
S = 100 (1—0,749*),
где S — стабильность в процентах,
t — время в днях, потребовавшееся для обесцвечивания раствора.
Вместо формулы можно пользоваться таблицей 63, где приводятся числа
относительной стабильности в зависимости от времени, потребовавшегося на
обесцвечивание.
Таблица 63
Числа относительной стабильности сточных вод *
Время, потребовав- шееся на обесцвечи- вание синьки, в днях Относительная стабильность воды в процентах Время, потребовав- шееся на обесцвечи- вание синьки, в днях Относительная стабильность воды в процентах
0,5 11 8,0 84
1,0 21 9,0 87
1,5 30 10,0 90
2,0 37 11,0 92
2,5 44 12,0 94
3,0 50 13,0 95
4,0 60 14,0 96
5,0 68 16,0 97
6,0 75 18,0 98
7,0 80 20,0 99
Если обесцвечивание наступает не ранее 7 суток, то относительная ста-
бильность такой воды считается достаточной, и эта вода допускается к спуску
в водоем.
Сероводород
Сероводород в сточной воде определяется так же, как в питьевой (стр. 357).
Для определения в воде сульфидов ее предварительно подкисляют.
405
Свободный хлор
Свободный хлор определяют в сточной воде так же, как в питьевой воде
(стр. 367).
Тяжелые металлы, мышьяк
Тяжелые металлы и мышьяк в сточной воде определяют так же, как в
питьевой воде (стр. 348 и далее).
Фенолы и крезолы
Фенолы и крезолы встречаются в сточных водах газовых, коксовых,
анилиновых и других заводов.
Качественное определение
К 100 мл испытуемой воды прибавляют 5 мл концентрированной серной
кислоты и отгоняют 20 мл. В дестилляте присутствие фенолов и крезолов
открывается одним из следующих способов:
1. Обонянием;
2. К 10 мл отгона прибавляют бромной воды; при наличии фенолов и
крезолов образуется желтоватобелый осадок или муть трибромфенола:
С6Н5ОН + ЗВг2 = С6Н2Вг3ОН + ЗНВг;
3. К 10 мл отгона прибавляют 0,2 мл реактива Миллона (см. ниже) и 0,1 мл
концентрированной азотной кислоты. Нагревают до кипения на голом огне.
При наличии фенолов или крезолов получается красное окрашивание, более
или менее интенсивное в зависимости от концентрации фенолов или крезолов.
Для получения реактива Миллона 1 г ртути растворяют в 1 ч. дымящейся
азотной кислоты при нагревании на водяной бане и разбавляют 2 ч. воды.
При употреблении прозрачная жидкость сливается с выделившихся кри-
сталлов.
Количественное определение
1— 3 л (или более) испытуемой воды подщелачивают едким калием и упа-
ривают до 50 мл. Полученную жидкость сильно подкисляют (при охлаждении)
серной кислотой, прибавляют воды до 150 мл и отгоняют 100 мл. Отгон мо-
жет быть использован для количественного определения фенолов и крезолов.
1. Колориметрическое определение с реактивом Миллона:
к 100 мл отгона прибавляют 2 мл реактива Миллона и 1 мл концентрирован-
ной азотной кислоты, нагревают до кипения и переводят в мерный цилиндр.
Стандартный раствор получают такой же обработкой дестиллированной воды
с определенным содержанием фенола.
2. Объемное определение: дестиллят подщелачивают двуугле-
кислым натрием и извлекают эфиром. Эфирную вытяжку выдерживают при
комнатной температуре для отгона эфира. Остаток растворяют в небольшом
количестве дестиллированной воды и прибавляют из пипетки с делениями
на 0,01 мл бромной воды до желтого окрашивания, указывающего на избы-
ток брома. Объем израсходованной бромной воды точно отмечают.
Спустя 15 минут прибавляют 3—5 мл 10% KJ. Избыток брома вытесняет
эквивалентное количество иода, которое затем определяют титрованием 0,01 N
раствором тиосульфата натрия.
Отдельно проводят холостую пробу с дестиллированной водой, взяв
то же количество бромной воды и К J. Разность между расходом тиосульфата
натрия в холостой пробе и в опытной пробе соответствует числу миллилитров
406
0,01 N раствора брома, вступившего в реакцию с фенолом. Реакции проте-
кают следующим образом:
С6Н5ОН + ЗВг2= С6Н2Вг3ОН = ЗНВг
Br2 + 2KJ = 2KBr-|-J2
J2 + 2Na2S2O3 = 2Na J + Na2S4O6.
1 мл 0,01 N раствора брома, вступившего в реакцию с фенолами и кре-
золами, соответствует 0,15685 мг фенола соответственно ~ ~5|qq~>
Соединения хрома
Соединения хрома встречаются в сточных водах текстильных, кожевенных
и химических заводов.
Качественное определение
Выпаривают 500—1000 мл воды досуха, остаток высушивают и осторожно
прокаливают. Золу сплавляют с несколькими граммами смеси, состоящей
из 1 г углекислой соды и 2 г калийной селитры. Сплав растворяют в горячей
воде, подкисляют уксусной кислотой и фильтруют. Для открытия хрома
фильтрат делят на несколько порций, с которыми проводят следующие
реакции:
1. Усреднив фильтрат, прибавляют раствор AgNO3; в присутствии хрома
получается красное окрашивание (осадок) хромовокислого серебра (Ag2CrO4),
растворимый в азотной кислоте и аммиаке.
2. Приливают к фильтрату раствор уксуснокислого свинца; в присутствии
хрома образуется желтый осадок хромовокислого свинца (РЬСгО4), нераство-
римый в уксусной кислоте и растворимый в азотной.
3. К фильтрату прибавляют раствор хлористого бария; в присутствии
хрома образуется желтый осадок хромовокислого бария (ВаСгО4).
4. 2—4 мл перекиси водорода подкисляют серной кислотой, прибавляют
2 мл эфира, вносят несколько капель фильтрата и взбалтывают; при наличии
хрома эфирный слой окрашивается в синий цвет вследствие растворения
в нем образующейся надхромовой кислоты (Н2Сг2О8).
5. К 10 мл фильтрата прибавляют 0,25 мл 2N серной кислоты, перемеши-
вают и приливают 0,2 мл 0,1% спиртового раствора дифенилкарбазида —
NH . NHC6H5
0 —.В присутствии шестивалентного хрома жидкость
xNH-NHC6H5
окрашивается в фиолетовый цвет.
Количественное определение
1. При относительно больших количествах хрома можно применить
йодометрический метод. К фильтрату (см. выше) прибавляют раствор хло-
ристого бария. Хром выпадает в виде ВаСгО4. Осадок отфильтровывают в
тигле Гуча либо в тигле с фильтрующей пористой пластинкой, промывают
и обрабатывают горячим раствором соды; при этом осадок перейдет в раствор.
Раствор собирают в колбу с притертой пробкой, прибавляют 10—20 мл
10% раствора KJ и подкисляют разведенной НС1. Спустя 15 минут выделив-
шийся иод титруют 0,01 N раствором тиосульфата натрия. Реакция идет по
уравнению (схематично):
2CrO3 + 6HJ = Сг2О3 + ЗН2О + 3J2.
1 мл 0,01 N раствора тиосульфата натрия отвечает 0,1737 мг Сг соответст-
2Сг
венно -7Г-.
о
Ю7
2. При малых концентрациях хрома в воде (доли мг/л) йодометрический
метод оказывается непригодным; определение ведут колориметрически с по*
мощью дифенилкарбазида.
Приготовляют стандартный раствор бихромата калия, растворяя 2,8284 г
К2Сг2О7 в 1 л дестиллированной воды. 1 мл такого раствора содержит 1 мг Сг.
1 мл этого основного раствора доводят дестиллированной водой до 100 мл.
1 мл слабого раствора содержит 0,01 мг Сг. Из слабого раствора бихромата
калия приготовляют стандартный ряд, вводя в пробирки для колориметри-
рования 0,1; ,0,2; 0,3; 0,4; 0,5 мл и т. д. этого раствора. Содержимое пробирок
доводят дестиллированной водой до 10 мл. Содержание хрома в побирках
соответствует последовательно 0,1; 0,2; 0,3; 0,4; 0,5 мг/л и т. д. В пробирку
такого же диаметра вводят 10 мл испытуемой воды. Затем во все пробирки
прибавляют по 0,25 мл 2N серной кислоты (9,8 г серной кислоты удельного
веса 1,84 растворяют в 100 мл дестиллированной воды), после чего содержимое
пробирок перемешивают и прибавляют в каждую из них по 0,2 мл 0,1 % спир-
тового раствора дифенилкарбазида. Растворы окрашиваются в фиолетовый
цвет. Колориметрирование производят не ранее 20 минут и не позднее 4 ча-
сов после прибавления реактивов. Колориметрирование можно вести и в
цилиндрах Генера малого размера (25—50 мл).
Концентрация хрома в исследуемой воде не должна превышать 2 мг/л,
в противном случае воду нужно соответственно разбавить.
При наличии в воде трехвалентного хрома он переводится в шестивалент-
ный, после чего определяется описанным выше образом.
Для перевода трехвалентного хрома в шестивалентный поступают сле-
дующим образом. Определенный объем испытуемой воды (напр., 50 мл) вводят
в химический стакан и прибавляют сюда 0,1 г тонко измельченной окиси
магния, после чего кипятят на слабом пламени 20—25 минут. При этом соль
трехвалентного хрома переводится в гидроокись:
2KCr(SO4)2 + 3MgO + ЗН2О = 2Сг(ОН)3 + 3MgSO4 + K2SO4.
Осадок окиси магния с сорбированной гидроокисью хрома — Сг(ОН)3 —
отфильтровывают через беззольный фильтр и тщательно промывают горячей
дестиллированной водой. Фильтр с осадком переносят в тигель, высушивают
и сжигают. По охлаждении в тигель вводят 2—3 мл 2N раствора серной ки-
слоты и нагревают до кипения. Содержимое тигля переносят количественно
в стаканчик и нагревают до растворения осадка. По охлаждении содержимое
стаканчика нейтрализуют едким натрием, применяя вначале нормальный
раствор, а подконец децинормальный. В качестве индикатора пользуются
при этом лакмусовой бумажкой. В полученный раствор прибавляют 2—3
капли двунормальной серной кислоты и 5 мл свежеприготовленного 0,1 — 0,2%
персульфата аммония — (NH4)2S2O8,— после чего стаканчик доливают дестил-
лированной водой приблизительно до 100 мл и выпаривают на слабом огне
до объема 20—30 мл. При этом трехвалентный хром окисляется до шестива-
лентного по реакции (схематично):
2Cr + 3S2084-7H20 = Cr2074-6S044- 14Н.
Избыток персульфата аммония при выпаривании жидкости разрушается.
По охлаждении раствора его переносят количественно в мерную колбочку на
50 мл, доливают дестиллированной водой до метки и перемешивают.
Для колориметрирования отбирают в пробирку 10 мл раствора. Отоб-
ранную пробу нейтрализуют едким натрием. Чтобы не вводит фенолфталеин
в раствор, производят отдельно титрование 10 мл такого же раствора в про-
бирке. Количество миллилитров едкого натрия, израсходованное на титро-
вание, прибавляют в первую пробирку, после чего приступают к колориме-
трированию, как описано выше.1
1 Подробнее см. «Санитарная охрана водоемов от загрязнения промышленными сточ-
ными водами», статья Ф. И. Гинзбурга, Медгиз, 1949.
408
Чувствительность метода 0,1 мг хрома в 1 л при условии взятия для ана-
лиза 10 мл испытуемой воды. При взятии больших объемов могут быть обна-
ружены соответственно меньшие концентрации.
Цианистые соединения
Цианистые соединения встречаются в сточных водах газовых и некоторых
химических заводов, гальванопластических мастерских, коксовальных пе-
чей и пр.
Качественное определение
К 50 мл воды прибавляют 1 мл 10% раствора FeSO4 и 0,5 мл 10% раствора
NaOH. Оставляют на 5 минут и подкисляют серной кислотой. Появление
синей окраски, обусловленной образованием берлинской лазури —
Fe4[Fe(CN)6i3,— указывает на присутствие цианистых соединений. Реакция
основана на том, что цианистые соединения с солями закиси железа дают
железистосинеродистый натрий. Одновременно образовавшийся от взаимо-
действия между железным купоросом и едким натрием гидрат закиси железа
переходит в щелочном растворе в гидрат окиси железа. После подкисления
получается сернокислая окись железа, которая с железистосинеродистым
натрием дает берлинскую лазурь:
HCN -j- NaOH = NaCN + Н2О
2NaCN -4- FeSO4 = Fe(CN)2 + Na2SO4
Fe(CN)2 + 4NaCN = Na4[Fe(CN)6]
FeSO4 + 2NaOH = Fe(OH)2 + Na2SO4
2Fe(OH)2 + H2O + О = 2Fe(OH)3
2Fe(OH)3 + 3H2SO4 = Fe2(SO4)3 + 6H2O
3Na4[Fe(CN)6] + 2Fe2(SO4)3 = Fe4[Fe(CN)6]3 + 6Na2SO4.
В зависимости от количества HCN появляется синий осадок или синее
окрашивание; при очень малых количествах окраска получается синезеленая
или зеленая; при этом появление окраски наблюдается лишь через 24—48
часов.
Количественное определение
Объемный метод
HCN отгоняют и улавливают в титрованном растворе AgNO3.
По количеству AgNO3, вступившего в реакцию с HCN, делают заключение
о количестве последнего.
К 500 мл сточной воды прибавляют 50 г бикарбоната натрия и отго-
няют 100 мл в коническую колбу с 10 мл 0,1 N раствора AgNO3, к кото-
рому прибавлено 10 мл разбавленной HNO3. Образовавшееся цианистое се-
ребро (AgCN) отфильтровывают и в фильтрате определяют количество не-
прореагировавшего AgNO3 (см. стр. 332). Отняв полученное число от 10,
находят число миллилитров 0,1 N AgNO3, вступившего в реакцию с HCN.
1 мл 0,1 N раствора AgNO3, вступивший в реакцию с HCN, отвечает
2HCN 2CN
5,4052 мг HCN соответственно —или 5,2036 CN соответственно
Колориметрический метод
Принцип основан на том, что в слабощелочном растворе пикрата в
присутствии HCN образуется соль изопурпуровой кислоты красного цвета.
Как и в предыдущем методе, воду подвергают отгонке. Дестиллят улавли-
вают в 10 мл 10% раствора Na2CO3. Всего отгоняют 90 мл, т. е. до общего
объема с раствором соды в 100 мл.
409
Для анализа берут 5 мл в колориметрическую пробирку, прибавляют
1 мл насыщенного на холоду раствора пикриновой кислоты и ставят на во-
дяную баню 70—80°С.
Стандартный ряд пробирок должен быть заготовлен одновременно из
раствора KCN или NaCN, содержащего 0,01 мг HCN в 1 мл. Для получения
такого раствора берут на литр 0,02409 г KCN или 0,01813 NaCN. В пробирки
наливают от 0,2 до 1 мл стандартного раствора с интервалом в 0,1 мл, что
соответствует от 0,002 до 0,01 мг HCN. Затем наливают в стандартные про-
бирки 1% раствор Na2CO3 в таком количестве, чтобы общий объем жидкости
в каждой пробирке соответствовал 5 мл, после чего в каждую пробирку на-
ливают по 1 мл насыщенного раствора пикриновой кислоты. Стандартные
пробирки ставят в водяную баню одновременно с опытной пробиркой. Че-
рез 5 минут пробирки вынимают из бани и по охлаждении колориметрируют.
Роданистые соединения
Роданистые соединения встречаются преимущественно в сточных водах
газовых заводов.
Качественное определение
Испытуемую воду подкисляют соляной кислотой и прибавляют раствор
FeCl3. В присутствии роданидов вода окрашивается в красный цвет. Красное
окрашивание возникает в связи с образованием роданового железа Fe(CNS)3.
Количественное определение
Основано на том же принципе, что и качественное. Воду предварительно
осветляют: к 200 мл воды прибавляют концентрированный раствор хлори-
стого цинка (ZnCl2) и отфильтровывают от осадка. К 100 мл фильтрата в ци-
линдре Генера прибавляют 1 мл НС1 и 1 мл FeCl3. Во второй цилиндр Генера
вливают 100 мл дестиллированной воды, прибавляют те же количества НС1
и FeCl3 и из микробюретки прибавляют 0,01 N раствор роданистого аммония,
до получения такой же окраски, как в первом цилиндре.
По расходу роданистого аммония делают заключение о количестве рода-
нидов в исследуемой воде.
Нефть
Нефть находит применение в самых разнообразных производствах в ка-
честве топлива; она служит и для обработки руды в качестве флотационного
средства, а также является исходным материалом для получения различных
нефтепродуктов и генераторного газа. Вот почему нефть со сточными водами
может попадать в водоемы из многих производств.
Существуют различные методы определения нефти в сточных водах: объем-
ный, весовой и колориметрический.
С. Д. Замыслова в Институте им. Эрисмана проверила и улучшила при-
меняемые методы определения нефти и разработала свой колориметрический
метод Ч
Здесь приводятся лишь два метода — объемный и весовой с извлечением
нефти из водного слоя, как наиболее простые и вместе с тем достаточно точные.
Определение нефти по объему
Сущность метода заключается в том, что вода с примесью нефти отстаи-
вается в течение часа и отделившийся на поверхности слой нефти измеряется
по объему.
1 Санитарная охрана водоемов от загрязнения промышленными сточными водами,
Медгиз, статья Замысловой С. Д., 1949.
С. Д. Замыслова сконструировала прибор, дающий возможность опреде-
лить слой нефти в количестве до 0,25 мг/л. Прибор представляет собой цилиндр
емкостью в 1 л, суживающийся к концам и закрывающийся сверху притер-
той пробкой, а снизу краном. Узкая часть цилиндра градуирована с точностью
до 0,05 мл. Переходная часть цилиндра калибрируется от 1 до 10 мл. Прибор
укреплен в штативе.
По данным Замысловой, отстаивание в течение 1 часа дает небольшие
отклонения в определениях и должно считаться достаточным по времени.
Прибавление к испытуемой воде 0,5% хлористого натрия улучшает отделе-
ние нефти.
Для большей точности прибор может быть предварительно проверен при
помощи воды, к которой прибавлены заведомо известные количества нефти;
на основании полученных результатов составляется диаграмма: на оси абсцисс
откладываются найденные количества нефти, а на оси ординат — прибав-
ленные.
Определение нефти весовым методом
Испытуемую воду вводят в литровую делительную воронку и к воде прибав-
ляют 20 мл петролейного эфира (температура кипения 50°С). Эфир взбалты-
вают несколько раз с водой и отстаивают около 20—30 минут. Затем вода
переливается в другую делительную воронку, а эфирная вытяжка — в кол-
бочку. Таким путем нефть извлекается из воды 4—5 раз. В эфирную вытяжку
добавляется около 5 г свежепрокаленного хлористого кальция и проба остав-
ляется на 12 часов.
Обезвоженную вытяжку фильтруют через обезжиренный фильтр во взве-
шенную колбочку объемом 50 мл. Эфир отгоняется через холодильник, а
нефть подсушивается в течение часа над хлористым кальцием и взвешивается.
Чувствительность данного метода достигает 1 мг/л. Потери при подсуши-
вании над хлористым кальцием в течение часа равны .0,6—4%.
ЛИТЕРАТУРА
Общие курсы, руководства, труды и сборники
1. Берлъ-Лунге, Методы технико-химического анализа, т. П,ч. I, ОНТИ, 1936.
2. Бруевич С. В., Коршун С. В., Озеров С. А. и Хецров И. Р., Исследование питье-
вых вод, изд. Мосздравотдела, 1925.
3. Бруевич С. В. и Хецров И. Р., Краткое руководство для санитарно-химических
исследований воды при помощи походных лабораторий, М., 1926.
4. Бутырин П., Полевой количественный химический гидроанализ, 1931.
5. Волжин В. А., Анализ воды, 1912.
6. Евланова А. и Шатуновская Л., Технический и санитарный анализ воды в условиях
экспедиции, 1934.
7. Дост К. и Гилъгерман Р., Практическое руководство к исследованию питьевых
и сточных вод, 1912.
8. Игнатов Н. К., Практическое руководство по методике санитарно-гигиенических
исследований, Биомедгиз, 1935.
9. Лапшин М. И. и Строганов С. Н., Химия и микробиология питьевых и сточных
вод, Госстройиздат, 1938.
10. Марзеев А. Н., Сысин А. Н. и Яковенко В. А., Основы коммунальной гигиены,
т. 1, Биомедгиз, 1936.
11. Смоленский П. О., Простейшие способы исследования и оценки доброкачествен-
ности съестных припасов, напитков, воздуха, воды и проч., 1909.
12. Стандартные методы исследования питьевых и сточных вод, изд. Постоянного бюро,
М., 1927.
13. Стандартные методы химического и бактериологического исследования воды, изд.
Санитарного ин-та им. Ф. Ф. Эрисмана, 1940.
14. Строганов С. Н. (ред.), Анализ городских сточных вод, Т945.
15. Сысин А. Н. (ред.), Учебник гигиены, Медгиз, 1938.
16. Углов В. А., Санитарное изучение источников водоснабжения на месте и их характе-
ристика, Госмедиздат, 1931.
17. Хлопин Г. В., Основы гигиены, т. I, изд. вып. 2, НКЗ, 1922.
18. Хлопин Г. В., Методы санитарных исследований, т. I, Анализ питьевых, сточных
и минеральных вод, вып. 1 и 2, Химтехиздат, 1930—1932.
<11
Г и дро би о логи я
1. Арнолъди В. M.t Введение в изучение низших организмов, М., 1925.
2. Артари А. 17., Руководящие принципы оценки воды по ее флоре, 1913.
3. Вислоух С. М., Биологический анализ воды. Учение о микроорганизмах, под ред*
Златогорова С., ч. II, 1916.
4. Воронков Н. В., Планктон пресных вод., 1913.
5. Геншель Э., Жизнь пресных вод, М., 1914.
6. Ламперт К., Жизнь пресных вод, СПБ., 1900.
7. Лимин А. Н., Пресные воды и их жизнь, М., 1926.
8. Рылов В. М., Жизнь пресных вод. Планктон, Л., 1924.
9. Рылов В. М., Краткое руководство к исследованию пресноводного планктона,
Саратов, 1926.
Бактериология воды
1. Вопросы санитарной бактериологии, под ред. Сысина А. Н. и Мац Л. И., изд. Акад,
мед. наук СССР, 1948.
2. Миллер А. А., Санитарная бактериология, Госмедиздат, 1933.
3. Минкевич И. Е., Курс санитарной бактериологии, 1940.
4. Минкевич И. Е., Бактерии группы кишечной палочки, как санитарно показатель-
ные микроорганизмы, Медгиз, 1949.
5. Омелянский В. Л., Практическое руководство по микробиологии, изд. Акад, наук
СССР, 1940.
6. Сборник инструкций по микробиологическим методам исследования, Медгиз, 1944.
ГЛАВА ШЕСТАЯ
ПОЧВА
Санитарно-гигиеническое исследование почвы слагается из физического,
химического, бактериологического и гельминтологического анализа ее, а также
санитарного изучения характера данной местности.
САНИТАРНЫЕ ТРЕБОВАНИЯ К ПОЧВЕ
При санитарной оценке почвы большое значение имеет вопрос о том, для
какой цели имеется в виду использовать данный участок. Так, например,
для строения жилищ лучше использовать более или менее возвышенные мест-
ности с плотной геологической породой, с малой силой капиллярного под-
нятия воды, не загрязненные органическими отбросами, с хорошим природ-
ным дренажем; для полей орошения хороши участки с значительной водо- и
воздухопроницаемостью, со средней водоемкостью, с хорошим дренажем
и достаточным стоком для дренажных вод; для кладбищ необходимы участки
с большой пористостью, с значительной водо- и воздухопроницаемостью,
с низким стоянием почвенных вод и т. п.
ОТБОР ПРОБ
При отборе проб для исследования необходимо избегать пользоваться
теми участками, где качества почвы могли измениться вследствие случайных
условий.
Пробы отбираются в объеме 1—2 л в чистые банки с притертыми пробками,
либо в жестяные коробки с крышками.
Для изучения физических свойств почвы пробы должны отбираться при
условии ненарушенной структуры, для чего предложены
приборы, представляющие собой цилиндры с режущими краями, вводимые
в почву. Таковы, напр., прибор Качинского и др. Для бактериологического
исследования почвы пробы берут по способу, описанному на стр. 425.
ФИЗИКО-МЕХАНИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПОЧВЫ
Физико-механическое исследование почвы слагается из определения тем-
пературы ее, размеров зерен, общего объема пор, воздухопроницаемости,
капиллярного поднятия воды в ней (волосности), водоемкости и водопро-
ницаемости.
Температура
Для определения температуры почвы на небольшой глубине (до 1 м) поль-
зуются геотермометрами в металлической оправе, представляющей собой сверло
(рис. 245). Вводя сверло в почву на желаемую глубину, оставляют его здесь
ча некоторое время и отсчитывают температуру по шкале, видимой над по-
верхностью почвы. Для определения температуры почвы на большой глубине
413
вводят в почву трубу из плохого проводника тепла (дерево, эбонит). В стен-
ках трубы сделаны отверстия для сообщения с почвой. Термометр укрепляют
на деревянном стержне и опускают в трубу на желаемую глубину. Сверху
труба закрывается крышкой. Время от времени стержень вместе с термомет-
ром извлекают и отсчитывают температуру. Чтобы во время под-
нятия термометра его показания не изменились, шарик термо-
т метра покрывают веществом, плохо проводящим тепло: парафи-
# ном, каучукоми т. п.
I Величина зерен почвы
I Под зернами почвы понимают отдельные частицы ее. От раз-
меров частиц почвы и их взаимного расположения зависит' пори-
стость почвы, ее воздухопроницаемость и отношение к воде.
Количественное взаимоотношение зерен почвы разных размс-
Ров определяется по принципу отсеивания и отму-
Jij чивания.
' I Отсеивание почвы
1| Из приборов, служащих для отсеивания почвы, можно ука-
I зать прибор, состоящий из 5 сит, расположенных одно над другим
j и имеющих отверстия разных размеров (рис. 246). Каждое сито
I образует дно жестяного цилиндра высотою 8—10 см.
и Первое сито имеет отверстия диаметром в 7 мм, второе — 5 мм,
W третье—2 мм, четвертое — 1 мм и пятое — 0,3 мм. Под пятым
ситом помещается цилиндр со сплошным дном. На первом сите
245 задерживаются части диаметром > 7 мм, на втором — от 4 до
Геотер- ? мм, тРетьем — от 2 до 4 мм, четвертом — от 1 до 2 мм, пятом—
мометр. ОТ 0,3 ДО 1 ММ.
В нижний цилиндр попадают частицы размерами менее 0,3 мм.
Зерна, задерживающиеся на первом сите, называются крупным хря-
щом или крупным гравием; на втором сите — средним
хрящом или средним гравием; на третьем —
мелким хрящом или мелким гравием; на
четвертом — крупным песком; на пятом — сред-
ним песком; зерна, попадающие в нижний цилиндр,
образуют мелкий песок (пыль).
Частицы диаметром более 1 мм называют почвенным «ске-
летом», частицы диаметром менее 1 мм объединяют под
общим названием «мелкозем».
Разделение взятой пробы почвы на отдельные фракции
по размерам при помощи описанного прибора производят
следующим образом: навеску почвы (50—100 г) сначала
высушивают при комнатной температуре, т. е. получают
воздушно-сухую пробу, осторожно измельчают в ступке
деревянным пестиком для нарушения связи между отдель-
ными зернами, затем просеивают всю навеску последова- Рис 24б Прибор
тельно через все сита, начиная с верхнего, имеющего наи- дл/ отсеивания
большие размеры. почвы.
Отдельные фракции почвы, задержавшиеся на каждом
сите, и последнюю фракцию, достигшую нижнего цилиндра, взвешивают и
определяют процентное отношение ко всей взятой пробе.
Вместо описанных выше сит для просеивания почвы применяют также
набор сит с отверстиями в 5, 4, 3, 2 ,2, 1 и 0,5 мм.
Отмучивание почвы
Определение размеров зерен почвы по принципу отмучивания производят
в текучей или неподвижной воде.
<414
Отмучппание в текучей воде
Для определения размеров зерен почвы применяют прибор, изображенный
на рис. 247. Он построен на принципе, по которому при равномерном течении
воды отдельные частицы почвы (зерна) относятся тем легче, чем меньше их
размеры. При этом допускается, что разница в удельном весе и конфигурации
отдельных зерен незначительна.
Прибор Небеля состоит из четырех грушевидных воронок, общая емкость
которых равна 4 л, а отношение объемов отдельных воронок равно 1 : 8 : 27 : 64,
т. е. I3 : 23 : З3 : 43. От верхнего отверстия каждой воронки отходит трубка
к нижнему отверстию следующей воронки. Наименьшая воронка при помощи
трубки сообщается со склянкой1, объемом несколько более 9 л, расположен-
ной выше воронки. От наибольшей воронки отходит трубка в стакан (бутыль)
объемом несколько более 5 л. Воронки утверждены на деревянном штативе.
Почва, подлежащая исследованию, высушива-
ется на воздухе и просеивается через сито с
отверстиями диаметром в 3 мм.
30 г такой пробы кипятят при помешивании
в течение нескольких часов в воде, для возможно
более полного отделения одних зерен от других,
дают пробе отстояться в течение нескольких ми-
нут, после чего мутную жидкость сливают в
воронку № 2, а остаток смывают в воронку № 1,
т. е. самую малую. В склянку, помещенную над
нею, наливают 9 л воды и открывают кран, пуская
воду через всю систему. Вся вода должна вытечь из
склянки в течение 20 минут. При этом наибольшие
частички — хрящевой песок — задержи-
Рис. 247. Прибор Небеля
для отмучивания почвы.
ваются в воронке № 1, более мелкие частицы — грубый песок —
попадают в воронку №2, мелкий песок — в воронку № 3, гли-
нистый песок — в воронку №4 и глина — в стакан. Фракцию
из каждой воронки переносят на предварительно высушенный и взвешен-
ный фильтр, сушат до постоянного веса при 125°С, взвешивают и опреде-
ляют постоянный вес отдельных фракций почвы.
Содержимое стакана трудно поддается фильтрованию. Вот почему реко-
мендуется, хорошо взболтав все содержимое стакана, отобрать пипеткой
некоторое количество, перевести в чашку и высушить до постоянного веса.
Вес полученного плотного остатка пересчитывают на все содержимое стакана.
Общий вес всех полученных фракций окажется меньше веса взятой для
анализа пробы почвы, так как навеска почвы берется при воздушно-сухом со-
стоянии ее, т. е. с содержанием гигроскопической воды, тогда как каждая
фракция высушивается до постоянного веса. Полученный вес каждой фрак-
ции выражается в процентах к общему весу всех фракций.
Отмучивание в стоячей воде
Метод осуществляется при помощи двойного отмучивания в стоячей воде.
Этим методом, предложенным Сабаниным, разделяют частички с размером
менее 0,25 мм. Прибор Сабанина (рис. 248) состоит из двух цилиндров —
малого а и большого &. Большой цилиндр высотою около 17 см имеет внут-
ренний диаметр 5,5 — 5,9 см. На стенке цилиндра нанесены деления на
расстоянии 2, 4, 8, 10 и 12 см от дна. Цилиндры помещают на уравнительный
столик с, снабженный тремя винтами у основания и уровнем, при помощи
которых столик устанавливают в горизонтальном положении. Для сливания
жидкости служит сифон rf, а для взмучивания — стеклянная палочка е, на
конце которой насажена каучуковая лопаточка. Предварительное отмучива-
ние проводят в фарфоровых чашках.
1 Для этой цели берут обыкновенно сосуд Мариотта.
415
Около 4 г мелкозема, т. е. зерен, прошедших через сито с отверстиями
в 1 мм в диаметре, смачивают в чашке водой до консистенции редкой ка-
шицы и растирают пальцем в течение 2—5 минут. Переносят содержимое
чашки в колбочку на 125 мл, доливают водой до половины объема колбы
и, присоединив к колбочке обратный холодильник, кипятят в течение од-
ного часа.
По охлаждении фильтруют через сито с отверстиями диаметром в 0,25 мм.
Для этой цели помещают сито в большую фарфоровую чашку, налив столько
Рис. 248. Прибор Сабанина для отмучивания
почвы.
а — малый цилиндр, Ъ — большой цилиндр, с — урав-
нительный столик, d — сифон, е — стеклянная палоч-
ка с каучуковой лопаточкой.
воды, чтобы дно сита оказалось
погруженным в нее. Переведя со-
держимое колбочки в сито, проти-
рают почву на сите пальцем под
водой, приподниМая и снова по-
гружая сито в воду. Наконец, при-
подняв сито, промывают на нем
остаток пробы водой, пока через
сито пойдет прозрачная вода.
Оставшиеся на сите частицы с раз-
мерами от 0,25 до 1 мм в диаметре
переводят в маленькую фарфоро-
вую чашку, отстаивают, сливают
избыток воды и помещают чашку
на водяную баню для испарения
остатка воды. После этого чашку
выдерживают, пока почва в ней
не придет в воздушно-сухое со-
стояние, и взвешивают. Привес
соответствует фракции с размерами
от 0,25 до 1,0 мм. По разнице в
весе между начальной навеской и
полученной фракцией определяют
вес частичек с размерами менее
0,25 мм, прошедших через сито.
Все эти частицы после отфиль-
тровывания их через сито вводят
в большую фарфоровую чашку;
здесь всю почвенную массу взму-
чивают при помощи каучуковой
лопаточки и дают отстояться в течение 20 секунд, после чего не
осевшую муть сливают по палочке в малую чашку. Через 45—50 секунд
мутную жидкость из малой чашки переводят по палочке лопатки в большой
цилиндр прибора до уровня 4 см. Цилиндр должен стоять на уравнительном
столике, установленном в горизонтальном положении. Еще до перенесения
в цилиндр почвенной массы в него вводят сифон, наполненный водой; ниж-
ний край сифона должен быть на уровне 2 см. Лопаточкой тщательно взму-
чивают в цилиндре почвенную массу и оставляют на 100 секунд, после чего
сливают при помощи сифона жидкость от 4 до 2 см в большой стакан емкостью
в 2—3 л; при этом вместе с водой окажутся слитыми частицы диаметром
менее 0,01 мм, так как эти частицы падают со скоростью 0,2 мм/сек. и, сле-
довательно, за 100 секунд пройдут путь в 20 мм или 2 см. Подобное отмучи-
вание и сливание повторяют 10—15 раз. Затем почвенную массу, оставшуюся
в фарфоровых чашках, старательно растирают пальцем, вновь взмучивают
и сливают, пока жидкость в чашках и в верхней чашке цилиндра совершенно
просветлится, что укажет на полное отмучивание частичек с диаметром менее
0,01 мм. Далее следует отделение частичек с размером от 0,05 до 0,01 мм в
диаметре. С этой целью наливают в цилиндр дестиллированной воды до уров-
ня 12 см, подставляют под сифон другой стакан, взмучивают жидкость и,
410
дав ей устояться в течение 30 секунд, сливают жидкость до уровня б см. При
этом окажутся слитыми частицы диаметром 0,05—0,01 см, так как частицы,
падающие со скоростью 2 мм/сек., пройдут путь в 60 мм, т. е. 6 см. Эту опе-
рацию взмучивания и сливания повторяют до просветления верхнего слоя
жидкости в цилиндре.
Почвенную массу, оставшуюся в цилиндре, переносят количественно
в чашку. Тут собрана фракция частичек с диаметром 0,25—0,05 мм.
Содержимому двух стаканов и чашки дают отстояться, пока жидкость
над почвой не станет совершенно прозрачной. Ее сливают осторожно сифо-
ном, стараясь не взмутить осадков. Последние переносят в предварительно
взвешенные небольшие чашки, выпаривают воду, а затем, оставив чашки
открытыми, пока почва в них не станет воздушно-сухой, взвешивают чашки
с почвенными фракциями и определяют вес этих фракций.
Пример. Положим, что для исследования взято 500 г воздушио-сухой почвы. Вес почвы,
прошедшей через сито с отверстиями диаметром в I мм, составлял 420 г. Из этой почвы взята
навеска в 4 г и пропущена через сито с отверстиями диаметром в 0,25 мм. При этом прошло
3,2 г почвы, разделенной затем на фракции диаметром:
менее 0,01 мм . ... 1,8 г,
от 0,05 до 0,01 мм . . 0,8 г,
от 0,25 до 0,05 мм . . 0,5 г.
Отсюда делаем вывод, что на 420 г приходится частичек с диаметром менее 0,01 мм:
1,8-420
—о 9 - = 286,25 г,
0,8-420
от 0,05 до 0,01 мм: —“ = Ю5 г,
Л 0,6-420
от 0,25 до 0,05 мм: —^2~~ = 78,75 г.
Относительно начальной навески почвы (500 г) полученные фракции составляют после-
довательно:
236,25 -100 105-100 78,75 • 100
500 = 47,25%; 500 — 21°/о и pqq = 15,75%.
Общий объем пор
Под общим объемом пор или порозностью понимают сумму объемов сво-
бодных промежутков почвы. Порозность выражают в процентах к кажу-
щемуся объему почвы. Величина порозности почвы зависит от расположения,
формы и диаметра частиц почвы, а также от «климата» почвы, т. е. влажности
и температуры ее.
Порозность определяет целый ряд важных свойств почвы, как, напр.,
водопроницаемость, влагоемкость, капиллярное поднятие воды и др.
Общий объем пор у гравия равен в среднем 38,4—40,1%; у песка — 35,5—
40,8%; у суглшка 36,2—42,5%; в смеси разных частей гравия и песка —
23,1—28,9% и т. д.
Общий объем пор почвы может быть определен по методу погружения
в воду и косвенно по разности между кажущимся и истинным объемом почвы.
Метод погружения в воду
Жестяной цилиндр высотою в 20 см и диаметром 10 см заполняют иссле-
дуемой воздушно-сухой почвой, которую затем переводят в литровый цилиндр,
налив в него предварительно 400—500 мл воды, взбалтывают и отсчитывают
полученный общий объем почвы и воды. Он менее суммы взятого объема почвы
и налитой в цилиндр воды, так как часть воды заполняет поры почвы. По-
ристость почвы вычисляется по формуле:
__ (а + & — с) 100
Р а
Z1 А, И. Еурппсйн
417
яле р — пористость, выраженная в процентах,
а — объем взятой почвы,
b — объем воды в цилиндре,
с — общий объем смеси почвы и воды в цилиндре.
Пример. Положим, что объем взятой для исследования почвы равен 400 мл, в цилиндр
налито 500 мл воды, смесь воды и почвы имеет объем в 700 мл. Следовательно, пористость
(400 + 500 — 700) 100
очвы равна: ----------------= 50%.
Вышеуказанный метод применим для рыхлых сортов почвы и не отли-
чается большой точностью.
Косвенный метод по удельному весу
Косвенный метод определения порозности почвы по удельному весу заклю-
чается в том, что от кажущегося объема почвы, т. е. объема твердой
фазы почвы вместе с порами, отнимают истинный объем почвы, т. е.
объем одной лишь твердой фазы, и находят процентное отношение полученной
разности к кажущемуся объему почвы.
Объем твердой фазы почвы определяется делением веса взятой почвы на
удельный вес.
Удельный вес почвы определяется так же, как удельный вес пыли (стр. 207).
Порозность по данному методу вычисляется по формуле:
\а----— ) 100 1АЛ/ ч
ще р — пористость почвы в процентах,
а — объем взятой для исследования почвы,
q — вес взятой почвы,
qx— удельный вес почвы.
Пример. Для анализа взято 500 мл почвы; вес взятой почвы равен 800 г, удельный
„ (500-2,6 — 800) 100
вес — 2,6. Отсюда находим пористость равной: -500>2 6---= 38,4%.
Объем пор в каменистой почве определяют так. Кусок камня данной почвы
высушивают и взвешивают. Затем его помещают в чашку с водой так, чтобы
он был погружен целиком в нее, и кипятят до полного удаления всего воздуха
из пор. По охлаждении воды камень извлекают, обтирают фильтровальной
бумагой и вновь взвешивают. Разность между первым результатом взвешива-
ния и вторым соответствует весу воды, заполнившей поры. Этот вес соответ-
ствует объему пор данного камня.
Для определения порозности необходимо знать еще объем взятого камня.
Для этой цели камень укрепляют на штативе посредством проволоки и по-
гружают во взвешенный предварительно стакан с водой, оставленный на
чашке весов. Чашка при этом понижается. Число граммов, которые прихо-
дится положить на противоположную чашку, чтобы уравновесить стакан,
соответствует объему камня в миллилитрах.
Зная объем пор камня и объем самого камня, определяют порозность по
формуле:
100 л
P = ~b~’
те р — порозность почвы в процентах,
а — объем пор камня,
b — объем камня.
448
Пример. Положим, что вес взятого для исследования камня равен 115 г, а по заполне-
нии его пор водою— 125 г. Следовательно, объем пор равен: 125— 115= 10 мл. Пусть
объем камня равен 30 мл; отсюда находим порозность камня: — *1??~ 33,3%.
Воздухопроницаемость почвы
Под воздухопроницаемостью почвы понимают способность почвы про-
пускать через свою толщу воздух. Это свойство почвы зависит от величины
частиц почвы и общего объема пор.
Для определения воздухопроницаемости почвы плотно заполняют ею
жестяной цилиндр длиною в 25—50 см и диаметром 5—10 см. Оба отверстия
цилиндра закрываются густыми сеточками. У одного конца поверх сеточки
надевается цилиндрический плоский колпачок с двумя отходящими от него
трубочками: одна из них сообщается с газовыми часами, другая — с мано-
метром. У другого конца поверх сеточки надевается также колпачок с одной
отходящей трубочкой, сообщающейся с газометром. Из газометра пускают
воздух, наблюдая по газовым часам, какой объем его пройдет через всютолщу
почвы; одновременно отмечают давление по манометру и время по секун-
домеру.
Результат выражается в объеме воздуха, прошедшем через цилиндр при
данном давлении в единицу времени.
В таблице 64 указана воздухопроницаемость различных видов почвы при
давлении в 20 мм водяного столба.
Таблица 64
Воздухопроницаемость различных видов почвы при давлении в 20 мм водяного столба
Сорт почвы Диаметр частичек в мм Объем пор в % Объем воздуха, прошедшего в 1 минуту, в м3
Абсолютные цифры Относительные цифры
Мелкий песок . . <0,3 55,5 0,00133 1
Средний » . . 1 — 0,3 53,5 0,112 84
Крупный » . . 2—1 37,9 1,28 961
Мелкий хрящ. . 4 — 2 37,9 6,91 5195
Средний » . . 7 — 4 37,9 15,54 11684
Воздухопроницаемость почвы может быть определена также при по-
мощи прибора, носящего название потометра (рис. 249).
Цилиндр а с почвой присоединяется к длинной градуированной трубке Ь,.
коленчато изогнутой и помещенной в плоский ящик с. С другого конца
в трубку под постоянным давлением пускают воду и при помощи непосред-
ственного отсчета наблюдают скорость движения ее в трубке. Воздухо-
проницаемость по данному методу выражают в процентах к свободному дви-
жению воздуха в той же трубке. Это последнее определяется заранее и ука-
зывается в паспорте прибора.
Водоемкость
Под водоемкостью или влагоемкостью почвы понимают
способность почвы удерживать в себе то или иное количество воды.
Водоемкость выражается процентным отношением между объемом тех пор,
в которых при свободном стоке удерживается вода, и общим объемом пор.
Иногда водоемкость выражается процентным отношением между весом воды,
задержавшейся в порах, к общему весу воздушно-сухой почвы.
419
Для определения водоемкости почвы, пользуются жестяным (цинковым)
цилиндром, в котором дно заменено двумя сетками — верхней густой и ниж-
ней редкой. Цилиндр плотно заполняют воздушно-сухой почвой и взвеши-
вают; затем его погружают в сосуд с водой так, чтобы уровень воды был при-
близительно на одной высоте с уровнем почвы в цилиндре. Когда вода поя-
вится на поверхности почвы, извлекают цилиндр из воды, избытку ее дают
стечь, вытирают цилиндр снаружи и вновь взвешивают. Разность между первым
и вторым весом в граммах укажет в миллилитрах объем пор, в которых задер»
Жалась вода. Остается еще определить общий объем пор. Для этой цели пе-
реводят мокрую почву из жестяного цилиндра в стеклянный с делениями,
в который налито некоторое количество воды (400 — 500 мл). По поднятию
воды в цилидре делают заключение относительно объема твердой фазы почвы
плюс объем воды, задержавшейся в почве.
Если этот объем вычесть из общего объема почвы, взятой для исследова-
ния, то получают объем пор, из которых вытекла вода.
Водоемкость почвы определяется по формуле:
100у
д V1 ’
где q — водоемкость почвы,
v — объем пор, в которых задержалась вода,
— объем пор, из которых вода вытекла.
Пример. Положим, что вес жестяного цилиндра вместе со взятой почвой равен 2000 г,
а после смачивания водой — 2050 г. Отсюда заключаем, что объем пор, в которых задержа-
лась вода, равен: 2050 — 2000 = 50 мл. Пусть подъем воды в цилиндре после перенесения
в него влажной почвы равен 400 мл, а объем взятой для анализа почвы равен с 00 мл; отсюда
заключаем, что объем пор, из которых вода вытекла, равен: 500 — 400 = 100 мл. Общий
объем пор: 50 + 100 = 150 мл.
г 50.100 ОО1/О/
Следовательно, водоемкость почвы равна: —= 33 1/3%.
Водопроницаемость
Под водопроницаемостью почвы понимают способность почвы
проводить воду из верхних слоев в нижние. Большая или меньшая быстрота
прохождения воды из верхних слоев в нижние зависит от размера зерен почвы,
420
наличия большего или меньшего количества коллоидных частиц и т. n.t .
а также от высоты слоя воды над почвой.
О водопроницаемости почвы обыкновенно судят по тому времени, которое
необходимо для просачивания воды через слой почвы толщиною в 200 мм при
высоте слоя воды над почвой в 40 мм.
Для определения водопроницаемости почвы берут стеклянную трубку,
высотой в 35 см и диаметром в 3—4 см, подвязывают ее кусочком полотна
и отмечают на стекле трубки высоту в 20 и 24 см. Затем наполняют трубку
почвой до высоты 20 см, равномерно распределяя почву в трубке при легком
постукивании о ее стенку. Трубку помещают в воронку, закрепленную в
штативе, и наливают на почву слой воды высотою в 40 мм, поддерживая уро-
вень воды постепенным приливанием новых порций по мере просачивания
воды в почву. Следят за появлением первой капли воды, прошедшей через
весь слой почвы. Это время и определяет водопроницаемость данной почвы.
Капиллярность
Капиллярность или волосность почвы представляет собой
способность ее поднимать по своим капиллярам воду из нижних горизонтов
в верхние. Капиллярность почвы зависит от механического строения почвы,
влажности ее, наличия солей и т. п.
В почвах крупнозернистых вода вначале поднимается скорее, чем в мел-
козернистых, но затем это поднятие замедляется. Во влажных почвах вода
поднимается быстрее, чем в сухих. Растворимые соли, особенно соли натрия,
замедляют поднятие воды.
Для определения капиллярности почвы берут высокую стеклянную трубку
диаметром около 2—3 см, подвязывают ее снизу полотном и наполняют
почвой доверху. Укрепив трубку в штативе, погружают нижнйй конец ее
в воду на 0,5 см и следят за быстротой поднятия воды по изменению окраски
почвы, возникающему вследствие смачивания последней.
ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПОЧВЫ
При химическом анализе почвы определяют: влажность (гигроскопиче-
скую воду), потерю при прокаливании, азотсодержащие вещества, хлориды,
органический углерод и др. ингредиенты. В некоторых случаях приходится
производить исследование почвы на присутствие экскрементов и мочи.
Подготовка почвы для исследования
Исследования производятся в отношении: 1) почвы без предварительной
подготовки («первоначальная» почва), 2) воздушно-сухой почвы, 3) водной
вытяжки и 4) солянокислой вытяжки.
Для получения водной вытяжки почвы вводят в склянку с притертой
пробкой 100 г воздушно-сухой почвы, приливают литр воды и настаивают
в течение двух дней, часто взбалтывая смесь. Затем фильтруют и доливают
фильтрат до литра дестиллированной водой. 1 мл водной вытяжки соответ-
ствует 0,1 г воздушно-сухой почвы.
Для получения солянокислой вытяжки почвы берут навеску в 50 г воз-
душно-сухой почвы, переводят ее в литровую коническую колбу, приливают
500 мл соляной кислоты удельного веса 1,049, перемешивают и прибавляют
крепкой соляной кислоты удельного веса 1,19 для полного разложения
карбонатных солей. Нагревают на водяной бане в течение 10 часов, часто
перемешивая. Затем вытяжку отфильтровывают через беззольный фильтр
диаметром в 15 см. Нерастворившийся остаток почвы промывают, деканти-
руя горячей водой, подкисленной соляной кислотой, затем переводят на
фильтр и тут продолжают промывать, пока промывные воды перестают давать
421
реакцию на железо (проба с роданистым аммонием). Фильтрат переводя!
в фарфоровую чашку и выпаривают до объема в 200 мл, прибавляют 25 мл
азотной кислоты удельного веса 1,4, выпаривают на водяной бане почти до-
суха, прибавляют немного царской водки, накрывают стеклом и продолжают
нагревать до окончания бурной реакции окисления, а затем высушивают до-
суха. Повторяют операцию окисления царской водкой до полного разрушения
органических веществ. Нагревают остаток в сушильном шкафу при 120°С.
При этом кремневая кислота переходит в нерастворимое состояние. Остаток
вновь смачивают царской водкой, выпаривают, смачивают повторно соляной
кислотой и полностью высушивают. Затем прибавляют 100 мл воды и подкис-
ляют соляной кислотой. Нагревают на водяной бане, фильтруют через беззоль-
ный фильтр диаметром в 7 см в колбу емкостью 500 мл. Промывают кремне-
вую кислоту на фильтре горячей 1°/0 соляной кислотой и по охлаждении
доводят фильтрат до 500 мл. 1 мл солянокислой вытяжки соответствует 0,1 г
воздушно-сухой почвы.
Влажность
5 г «первоначальной» или воздушно-сухой пробы отвешивают в бюксе
и сушат в шкафу при 105°С в течение 5 часов, после чего взвешивают. Опре-
деляют потерю в весе и выражают ее в процентах.
На основании полученного результата, вычисляют коэфициент для пере-
счета любого веса «первоначальной» или воздушно-сухой пробы на вес сухой
почвы. Коэфициент вычисляется по формуле:
100
К==100 —Д’
где А — гигроскопическая вода в процентах от «первоначальной» или
воздушно-сухой почвы.
Потеря при прокаливании
Высушенную при 105°С почву прокаливают сначала на малом, а затем
на большом пламени, часто помешивая платиновой иголкой или шпаделем.
Под конец смачивают золу крепким раствором углекислого аммония и снова
прокаливают, охлаждают в экссикаторе, взвешивают и определяют потерю
в весе; результат выражают в процентах абсолютно сухой почвы.
Растворимые в воде вещества
250 мл водной вытяжки выпаривают в платиновой чашке, высушивают
при 110°С и взвешивают. Вес плотного остатка соответствует количеству
растворимых в воде веществ почвы. Результат выражают в процентах абсо-
лютно-сухой почвы.
Хлориды
Для определения хлоридов в почве пользуются водной вытяжкой. К 250 мл
ее прибавляют немного соды и выпаривают до г/6 начального объема, прибав-
ляют раствор КМпО4 до розовой окраски и кипятят; при этом органические
вещества разрушаются. Фильтруют, подкисляют фильтрат азотной кислотой
и определяют хлориды так же, как в воде. Результат перечисляют в проценты
абсолютно-сухой почвы.
Азотсодержащие вещества
Минеральный (солевой) аммиак, органический азот и общее количество
азота в почве определяют, как и в воде (стр. 330, 342 и 345). Для анализа
берут небольшую навеску воздушно-сухой почвы.
Для качественного и количественного определения нитритов и нитратов
пользуются водной вытяжкой.
Результаты выражают в процентах абсолютно-сухой почвы.
422
Органический углерод
Для определения органического углерода вводят в колбу 3—5 г просеян-
ной почвы, приливают 30 мл воды и осторожно 30 мл концентрированной
H2SO4 для разложения карбонатов. Выделяющийся при этом угольный
ангидрид удаляется при нагревании. Чтобы удалить остатки СО2, пропускают
через содержимое колбы воздух, предварительно освобожденный от СО2 при-
сасыванием через натронную известь. Затем прибавляют в колбу 10 мл сер-
ной кислоты и 8 г бихромата калия (К2Сг2О7).
При этом выделяется кислород in statu nascendi:
K2Cr2O7 + H2SO4 = K2SO4 + H2Cr2O7
H2Cr2O7 = 2CrO3 + H2O
2CrO3 = Cr2O3 4-3(0).
Углерод органических веществ окисляется до СО2, который поглощают
в трубки с натронной известью, так же как при определении органического
углерода в сточных водах (стр. 402).
Перед трубками с натронной известью помещают трубки с тонкой желез-
ной проволокой для поглощения хлористоводородной и фосфористоводо-
родной кислот, трубки с хлористым кальцием и концентрированной серной
кислотой для поглощения влаги, трубку с зернами пемзы, обработанными
сульфатом меди, для поглощения сероводорода. Найденное количество уголь-
ной кислоты для пересчета на углерод умножают на коэфициент 0,2727 соот-
С
ветственно
Для пересчета на гумус, т. е. остатки органических веществ почвы, най-
денное количество СО2 умножают на коэфициент 0,471, так так в гумусе со-
держится приблизительно 58% углерода.
Серная кислота (сульфаты)
200 мл солянокислой вытяжки почвы переводят в склянку, добавляют
крепкой соляной кислоты до сильнокислой реакции, нагревают до кипения,
прибавляют горячий 5% раствор ВаС12 до полного осаждения сульфатов,
продолжают кипятить еще 15 минут и оставляют на сутки в теплом месте.
Проверяют полноту осаждения сульфатов. Затем отфильтровывают осадок
BaSO4, промывают горячей водой до исчезновения реакции на хлориды, вы-
сушивают, прокаливают и взвешивают. Отняв вес золы фильтра, находят вес
BaSO4, который для пересчета на SO3 умножают на 0,343 и для пересчета
на SO4 умножают на 0,4115. Результат выражают в процентах абсолютно-
сухой почвы.
Фосфорная кислота (фосфаты)
100 мл солянокислой вытяжки выпаривают в стакане на водяной бане
досуха, к остатку прибавляют слабой азотной кислоты и продолжают нагре-
вать до его растворения. Нейтрализуют аммиаком, слегка подкисляют азотной
кислотой и прибавляют 50 мл кипящей молибденовой жидкости г. Фос-
форная кислота выпадает в виде фосфорномолибденовокислого аммония:
Н3РО44- 12(NH4)2Mo04 4- 21HNO3 =
= (NH4)3PO4 • 12MoO34-21(NH4)NO34- 12H2O.
Осадку дают выпасть, сливают осторожно жидкость и промывают осадок
декантацией 5% раствором азотнокислого аммония. Промытый осадок раст-
1 Растворяют 150 г молибденовокислого аммония в 1 л воды. Раствор тонкой струей
вливают в литр азотной кислоты уд. веса 1,2. Через два дня раствор готов для пользования.
423
воряют в аммиаке и осаждают магнезиальной смесью1. Получают белый
осадок фосфорнокислой магний-аммониевой соли:
(NHJaPOj. 12Mo034-24NH/)H =
= (NHJ3PO4+ 12(NH4)2Mo04+ 12Н2О
(NH4)3PO4 4- (NH ,CI 4- MgClo) + NHj(OH) =
= A:gNH4PO4 4- 3NH4C1 4- nh4oh.
Полученный осадок отфильтровывают, промывают аммиачной водой, вы-
сушивают и прокаливают. При этом двойная соль ортофосфэрной кислоты
переходит в соль пирофосфорной кислоты:
2MgNH1PO4 = Mg,P2O7 4- 2NH3 Jr Н2О.
Тигель вместе с осадком охлаждают и взвешивают. Полученный вес пиро-
фосфэрной соли для пересчета на Р2О5 умножают на 0,638 соответственно:
Р2О5 142 г ч
р 0=222 64’ РезУльтат выражают в процентах абсолютно-сухой
почвы.
Кроме весового определения фосфатов, применим и колориметрический
метод (см. определение фосфатов в воде стр. 346).
Экскременты и моча
Присутствие экскрементов в почве можно обнаружить следующим обра-
зом. К 250 мл водной вытяжки прибавляют 0,2 — 0,3 г виннокаменной кис-
лоты— (СНОН)2 (СООН)2 — и выпаривают досуха. Из сухого остатка гото-
вят спиртовую вытяжку. Последнюю выпаривают досуха, к сухому остатку
прибавляют немного раствора КОН. Появление специфического запаха сви-
детельствует о наличии в почве экскрементов. Желательно при данном ме-
тоде ставить контроль с почвой, заведшо загрязненной экскрементами.
Присутствие в почве мочи определяется легко мурексидной реакцией.
ПО—150 мл водной вытяжки выпаривают насухо, сухой остаток нагревают
с небольшим количеством соды, прибавляют воды и, смешав, отфильтровывают.
Фильтрат выпаривают в фарфоровой чашке, прибавляют азотной кислоты
и вновь выпаривают насухо.
Если в водной вытяжке присутствовала мочевая кислота, то остаток после
выпаривания приобретает желтокрасный цвет, переходящий от аммиака
в багряный, а от едкого натрия — в фиолетовый.
ИССЛЕДОВАНИЕ ПОЧВЕННОГО ВОЗДУХА
Для исследования почвенного воздуха применяют те же методы, что и при
исследовании воздуха атмосферного. В почву вбивают железную трубку соот-
ветствующей длины. Трубка снабжена у основания острым стальным нако-
нечником. В стенках трубки у основания имеется несколько отверстий для
прохождения воздуха. Воздух просасывают при помощи аспиратора через
те или иные поглотители, в зависимости от искомого вещества.
БАКТЕГИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПОЧВЫ
Поверхностные слои почвы весьма богаты бактериями. В одном грамме
могут быть миллиарды их. Максимум числа бактерий обнаруживается на
глубине 10—20 см.
Растворяют 100 г хлористого магния и 210 г нашатыря в 1,3 л воды, к раствору прибав-
ляют 700 мл 24% аммиака.
424
В направлении от поверхности в глубину бактериальная флора становится
все беднее.
На глубине 4—5 м и более почва оказывается стерильной.
Подавляющее большинство бактерий почвы — сапроф ты. Из патогенных
бактерий в местах, заселенных человеком, довольна часто встречаются в
почве: Вас. perfringens, Вас. oedematiens, Vibrio septicus (общепризнанные
возбудители «газовой гангрены»), Вас. tetani (возбудитель столбняка), Вас.
botulinus (возбудитель ботулизма). Далее, в почвах, особенно унавоженных
(пастбища), нередко открывали споры Вас. anthracis, которые могут сохра-
няться здесь в течение многих лет.
Возбудители кишечных инфекций, попадая с фекалиями в почву, могут
сохраняться здесь различные сроки. Так, В. typhi и В. paratyphi во влаж-
ной почве могут сохраняться до года, реже долее. Дизентерийные микробы
менее резистентны и выживают во влажной почве до 10—15 дней. Холерный
вибрион также сравнительно мало стоек и может сохра-
няться во влажной почве до 48 дней.
Из патогенных микробов в почве открывали также тубер-
кулезную палочку, бруцеллы, чумную палочку, гноеродные
кокки и др.
Ввиду сказанного бактериологическое исследование почвы
представляет в отдельных случаях несомненный эпидемиоло-
гический интерес.
Отбор пробы почвы для бактериологического исследова-
ния с поверхностных слоев и с небольшой глубины произво-
дится при помощи обеззараженных лопаты и ложечки в сте-
рильную банку. Для выемки пробы почвы из глубоких слоев
пользуются прибором, представляющим собой металлический
стержень, заканчивающийся сверлом (рис. 250). Над сверлом
в стержне имеется небольшой желоб, закрывающийся вра-
щающейся гильзой. Гильза может повернуться лишь на
полоборота, так как дальнейшему вращению ее препятствует
выступ на стержне. При поворачивании сверла вправо желоб
закрывается, при поворачивании влево — открывается
и в него попадает почва. Для получения пробы желоб
обеззараживают фламбированием и закрывают. В таком
Рис 250. Бурав
для забора проб
почвы (показа-
на лишь ниж-
няя часть).
виде сверло
вводят в почву, поворачивая его все время вправо, благодаря чему
желоб остается закрытым. Когда сверло дойдет до желаемой глубины, его
поворачивают влево, и тогда в желоб попадает почва. Таких поворотов влево
делают несколько. Затем поворотом вправо закрывают желоб и извлекают
сверло из почвы. Небольшой обеззараженной платиновой ложечкой достают
из желоба почву для исследования.
Бактериологическое исследование почвы сводится к определению: 1) об-
щего числа микробов в 1 г почвы, 2) коли-титра и 3) наличия патогенных
микробов.
Определение общего числа микробов в 1 г почвы. Почву просеивают через
стерильное сито (сторона ячеек — 2 мм). 1 г просеянной почвы взбалтывают
со 100 мл стерильной воды. Чтобы получить равномерную суспензию бакте-
рий в воде, взбалтывают смесь с бусами в течение 10 минут. Общее число
встряхиваний должно быть не менее двухсот. Из отстоявшейся жидкости
приготовляют разведения 1 : 1000; 1 : 100 000 и 1 : 10 000 000. Из получен-
ных разведений делают посевы на мясопептонный агар так же, как при ис-
следовании воды. Выращивание ведется при 22°С. Подсчет производят при
помощи счетной пластинки не ранее чем через 48 часов. Результат пересчиты-
вают на 1 г почвы.
Определение коли-титра. Коли-титром почвы называют то наименьшее
количество почвы, выраженное в граммах и миллиграммах, в котором обна-
руживается присутствие хотя бы одной особи кишечной палочки.
425
Горовиц-Власова считает, что при коли-титре более 1 г почву можно счи-
тать достаточно чистой, при 50 мг — 1 г — умеренно загрязненной и при
1—2 мг — сильно загрязненной.
Для определения коли-титра почвы среды Эйкмана и Булира оказываются
непригодными, так как обильная сопутствующая микрофлора почвы заглушит
рост кишечной палочки. Вот почему при определении коли-титра почвы
пользуются средой Кесслера (приготовление см. на стр. 115).
Посев производят из цельной почвы и почвенной болтушки. Исходная
почвенная болтушка получается взбалтыванием 1 г почвы в 10 мл дестилли-
рованной воды. Из исходной болтушки приготовляют десятикратные разве-
дения.
Посев производится следующим образом:
на 1 мл исходной болтушки и ее десятикратные разведения берут 5 мл
среды Кесслера;
на 1 г почвы берут 50 мл среды Кесслера;
на 10 г почвы берут 100 мл среды Кесслера.
Пробы выдерживают в течение 24—48 часов при 43°С. Появление газа
в поплавках свидетельствует о весьма вероятном присутствии в пробе В. coli.
Из проб, давших газообразование, производят контрольные высевы на чашки
со средой Эндо, а из этих последних — на пестрый ряд. Из типичных коло-
ний на чашках со средой Эндо готовят препараты, которые красят по Граму
и бактериоскопируют.
Патогенные микроорганизмы. Выделение и идентификация патогенных
микроорганизмов почвы представляет редкую задачу
практике. Эти исследования производятся по общим
правилам бактериологической техники: посевы, высевы,
отвивки на соответствующие среды, заражение экспери-
ментальных животных и пр.
лабораторной
в
Рис. 252. Установка для система-
тического наблюдения над ск я-
нием уроння почвенных вод.
А — поплавок, b — шнур, с — проти-
вовес, D — стрелка-указатель.
Е — шкала.
САНИТАРНО-ТОПОГРАФИЧЕСКАЯ
И ГИДРОГЕОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА
МЕСТНОСТИ
Для всесторонней санитарной оценки почвы, кроме исследова-
ний, приведенных выше, необходимо ознакомиться также с сани-
I
$
тарно-топографическими и гид-
ро-геологическими особенностя-
ми данной местности. При этом
обращают внимание на поверх-
ностный рельеф местности, вы-
соту положения ее над уровнем
моря, покров почвы, гидрогра-
фию, геологическое строение,
природный дренаж, колебание
почвенных вод и т. п.
Для определения стояния
уровня почвенной воды, поль-
зуются чашечковым прибором
Петтенкофера (рис. 251). При-
бор состоит из измерительной
ленты, к свободному концу
которой прикреплен медный
стержень длиной в 30 см. По
окружности стержня располо-
жены 30 маленьких круглых
чашечек на расстоянии 1 см
одна от другой. Ленту опускают
в колодец или шахту до того
момента, пока некоторые чашеч-
ки не погрузятся в воду, после
чего извлекают из воды ленту, отметив перед тем
то ее деление, которое соответствует поверх-
ности почвы. К этому отсчету остается прибавить столько сантиметров, сколько осталось
Рис. 251.
шечковый
Ча-
при-
бор Петтенко-
фера.
426
сухих чашечек. Чашечный прибор снабжается иногда свистком, который издает свист,
когда прибор касается воды.
Для систематических наблюдений над стоянием уровня почвенных вод может служить
прибор, изображенный на рис. 252. Над водой помещается поплавок А. Шнур Ь, отходящий
от поплавка, перекинут через блок и снабжен у края противовесом С и стрелкой-указате-
лем D. При перемещении уровня воды в вертикальном направлении в таком же направ-
лении перемещается и поплавок, что дает возможность по шкале Е сделать соответствую-
щие отсчеты. Для данной цели применяют и приборы, автоматически отмечающие уровень
воды (самописцы типа ВГС, «Валдай» и др.).
Для определения движения почвенной воды в горизонтальных направлениях поль-
зуются методами, изложенными выше (стр. 397).
ЛИТЕРАТУРА
Общие курсы, руководства и труды
1. Игнатов Н. К., Практическое руководство по методике санитарно-гигиенических
исследований, Биомедгиз, 1935.
2. Моисеев С. В., Учебник общей гигиены, Медгиз, 1947.
3. Никитин А. Ф., Способы и приемы практических наблюдений и исследований яо
гигиене, 1911.
4. Смоленский П. О., Простейшие способы исследования и оценки доброкачественности
съестных припасов, напитков, воздуха, воды и проч., IV изд. 1909.
5. Сысин А. Н. (ред)., Учебник гигиены, Медгиз, 1938.
6. Труды Санитарного института им Ф. Ф. Эрисмана, вып. 4, М., 1929.
7. Хлопин Г. В., Основы гигиены, т. 1, 1918.
8. Эрисман Ф. Ф., Гигиена, т. 1, 1901.
Почвоведение. Физико-механический и химический
анализ почвы
1. Антипов-Каратаев И. Н., Физико-химические исследования почвы, 1933.
2. Ахромейко А., Структура почвы, 1930.
3. Виноградов В. И., Химический анализ почвы, 1930.
4. Гедройц К., Химический анализ почвы, 1932.
5. Глинка К. Почвоведение, 1927.
6. Домрачева Е. А. Физико-механический и химический анализ почвы, 1931.
7. Захаров С. А., Курс почвоведения, 1927.
8. Качинский Н. А., Изучение физических свойств почвы и корневых систем, 1930.
9. Коссович П. С., Краткий курс почвоведения, 1916.
10. Кравков С. /7., Почвоведение, 1935.
11. Принц И., Гидрогеология, 1932.
12. Ремезов Н. П.,Физико-химические методы исследования почвы, 1931.
13. О' Э., Геология, под ред. Милановского, 1935.
14. Ферсман А., Геохимия, 1934.
Бактериология почвы
1. Миллер А. А., Санитарная бактериология, Биомедгиз, 1935.
2. Минкевич И. Е., Курс санитарной бактериологии, 1940.
3. Минкевич И. Е., Бактерии группы кишечной палочки как санитарный показатель
микрофлоры, Медгиз, 1949.
4. Омелянский В. Л., Курс общей и почвенной микробиологии, 1929.
5. Омелянский В. Л,, Основы микробиологии, Биомедгиз, 1936.
427
ГЛАВА СЕДЬМАЯ
СТРОИТЕЛЬНЫЕ МАТЕРИАЛЫ
Исследования строительных материалов касаются их механи-
ческих, физических и химических свойств. В некоторых
случаях исследуют поражение материала грибами и другими вредителями
(исследование дерева).
Под механическими свойствами строительных материалов понимают их
способность сопротивляться воздействию внешних сил. Главными механи-
ческими свойствами являются прочность, упругость и твер-
дость.
При исследовании физических свойств определяют: вес единицы объема
материала, пористость, воздухопроницаемость, водопроницаемость, водо-
поглощаемость, теплопроводность, теплоемкость, звукопроводность и др.
При химическом анализе определяют состав строительных мате-
риалов.
Санитарно-гигиеническое исследование строительных ма-
териалов касается по преимуществу определения их физических свойств;
из других исследований проводят определение влажности, а для дерева, как
было указано, приходится иногда решать вопрос о поражении его грибами
или другими вредителями.
САНИТАРНЫЕ ТРЕБОВАНИЯ
Строительные материалы должны обладать хорошей воздухопроницае-
мостью, малой теплопроводностью при относительно большой теплоемкости,
сухостью, малой водопроницаемостью и плохой звукопроводностью.
Вес единицы объема материала
Вес единицы строительного материала характеризуют, определяя либо
удельный вес, либо объемный вес, либо насыпной вес,
В строительной практике под удельным весом понимают вес
единицы объема данного вещества, занимающего сплошь весь этот объем,
без каких-либо пор и промежутков.
Под объемным весом понимают вес единицы объема вещества
с присущими этому веществу порами. Под насыпным весом по-
нимают вес единицы объема сыпучих веществ с присущими этим веще-
ствам промежутками между отдельными зернами и порами в отдельных
зернах.
Определение удельного веса непористых материалов производится посред-
ством взвешивания и измерения объема данного образца. Частное от деления
428
веса на объем и дает удельный вес и выражается в г/см3 или кг/м3, реже т/м3.
Следовательно, для удельного веса можно написать формулу:
ъ р ’
где — удельный вес материала,
g — вес образца,
v — объем образца.
Если непористое тело имеет неправильную форму, то объем его опреде-
ляют взвешиванием при погружении в воду (стр. 418).
Для определения удельного веса пористого материала его сначала исти-
рают в тонкий порошок, из полученного порошка берут некоторую навеску,
определяют объем ее так, как описано для пыли (стр. 207), а затем делят
вес на объем.
Определение объемного веса непористых материалов, обозначаемого сим-
волом 70, производится так же, как и определение удельного веса, так
как для таких материалов удельный и объемный вес совпадает.
Определение объемного веса пористых материалов производится делением
веса данного образца на его объем; при этом объем образца правильной формы
определяется измерением, а образца неправильной формы — путем погруже-
ния в воду после предварительного заполнения всех пор водой, как описано
на стр. 418. Если данный образец в воде растворяется, то его с поверхности
покрывают тонким слоем парафина.
Определение насыпного веса для сыпучих материалов производится по-
средством наполнения мерного сосуда и взвешивания того количества, кото-
рое поместилось в этот сосуд; при этом мерный сосуд должен быть взят тем
большего объема, чем больше размер зерен сыпучего материала. Различают
насыпной вес материала в рыхлом и уплотненном состоянии.
В таблице 65 приведены величины веса единицы объема для некоторых
строительных материалов.
Таблица 65
Вес единицы объема строительных материалов
Наименование материала Удельный вес г, см* Объемный вес кг/м3 Насыпной вес кг/м3
Железо 7,85
Гранит 2,7 2700 —
Кирпич обыкновенный . . . 2,6 2800 —
Бетон — 2200
Портланд-цемент 3,05 —. 1300
Гравий 2,65 —- 1700
Песок 2,65 — 1500
Пористость
Под пористостью строительного материала понимают процентное отношение
объема пор данного материала к общему его объему. От пористости мате-
риала зависят многие другие его свойства: воздухопроницаемость, теплопро-
водность, водопоглощаемость и др. Пористость строительного материала
определяется так же, как пористость почвы (стр. 417).
Если известны 70 и то пористость определяется по формуле:
vn=( 1 —— 100,
\ Ъ/
где vn — пористость,
— объемный вес пробы,
— удельный вес пробы.
42У
В таблице бб приведены величины пористости некоторых строительных
материалов.
Таблица бб
Пористость строительных материалов
Наименование материалов Пористость в процентах
Кирпич обыкновенный, слабо обожженный * » сильно » Известковый туф Известка обыкновенная Гипс Песчаник разных сортов Мрамор Гранит 20—30 0—20 32 26 42 9,5—21,5 0,1 менее 0,1
Воздухопроницаемость
Воздухопроницаемость строительного материала представляет свойство,
от которого отчасти зависит размер естественной вентиляции помещений.
Пористость материала повышает его воздухопроницаемость; однако пря-
мой зависимости между этими свойствами не существует, так как среди пор
могут быть многие, не сообщающиеся, а замкнутые. С увеличением влажности
материала воздухопроницаемость его уменьшается. Воздухопроницаемость
стен зависит не только от характера материала, но в значительной степени
от воздухопроницаемости швов. Оштукатурка стен и окраска их клеевой кра-
ской в значительной степени понижает их воздухопроницаемость. Окраска
масляной краской и облицовка глазурью делают стены совершенно непро-
ницаемыми для воздуха.
Для определения размера воздухопроницаемости строительных материа-
лов и стен введено понятие: коэфициент воздухопроницае-
мости. Под коэфициентом воздухопроницаемости
понимают количество воздуха в литрах, проходящее в течение одного часа
через данный материал или стену толщиною в 1 м и площадью в 1 м2 (1 см2)
при разности давления в 1 мм водяного столба.
Коэфициент воздухопроницаемости строительного материала определяется
экспериментально, причем для сыпучего материала можно воспользоваться
такой же установкой, как для почвы (стр. 419). Если материал не сыпучий,
то из него вытачивают кусок правильной формы, чаще всего в виде цилиндра;
боковые поверхности его покрывают слоем менделеевской замазки. На один
из концов цилиндра насаживают герметически металлический колпачок,
снабженный двумя трубками — центральной и боковой. Центральную трубку
соединяют с газовыми часами, а последние — с газометром. Боковую трубку
соединяют с манометром и пропускают через систему сухой воздух. Из опыта
определяют время, в течение которого воздух пропускался через материал,
давление, при котором воздух был пропущен, и объем пропущенного воздуха.
Коэфициент воздухопроницаемости определяется из формулы:
s(p — pjt ’
где К — коэфициент воздухопроницаемости строительного материала,
v — количество воздуха в литрах, прошедшее через материал в течение
опыта,
430
I — длина взятого куска материала в метрик,
s — площадь сечения куска материала в м2 (см2),
р—Pi — разность давлений в миллиметрах водяного столба,
/ — время в часах.
Для определения воздухопроницаемости строительных материалов у нас
в лабораториях пользуются прибором, предложенным Галаниным1. Прибор
Галанина (рис. 253) состоит из газометра А, откуда воздух поступает в кол-
пак В с квадратным сечением. В колпак вмазывается образец испытуемого
материала.
Газометр состоит из двух цилиндров: верхнего С и более широкого ниж-
него Сг Верхний цилиндр движется по двум направляющим роликам и по-
гружается в нижний цилиндр, который заполняется маслом. Через нижний
цилиндр в верхний проходит газопроводная
трубка, снабженная внизу краном. Она служит
для ввода в газометр воздуха и для выпуска его.
При помощи резиновой толстостенной трубки
она соединяется с колпаком. Внутренний ци-
линдр соединен с манометром Мх, а колпак —
с манометром М2. При помощи указателя g
отсчитывают по зеркальной шкале 3 с точностно
Рис. 253. Прибор Г аланина для
воз ду хоп рони цаемости строите л ьн ых
материалов.
А — баллон с воздухом, В — газовые часы,
С —трубка, сообщающая газовые часы с ко-
жухом, D — стена, покрытая кожухом,
Е — микроманометры.
определения воздухопроницае-
мости строительных материалов.
А— газометр, В — колпак для
вмазывания образцов материала, С
и Ci — цилиндры газометра,
и Mt микроманометры, g — указа-
тель, 3 — шкала.
до 0,5 мм высоту стояния дна верхнего цилиндра до опыта и после опыта и
отсюда делают вывод относительно объема воздуха, выпущенного из цилинд-
ра. Сверху на дно верхнего цилиндра помещают тот или иной груз,
изменяя таким образом давление, под которым воздух находится в
цилиндре.
Испытуемый образец вмазывают в цилиндр при помощи замазки, состоящей
из 60% канифоли и 40% воска. Канифоль сначала расплавляют на слабом
огне, а затем добавляют воск. Замазку сначала наносят на образец с четырех
сторон в два-три приема; затем на образец надевают колпак и тщательно
заделывают той же замазкой стык между образцом и колпаком до получения
полной герметичности.
Для определения воздухопроницаемости строительных материалов боль-
ших размеров (части стен) применяют установку, схема которой изо-
бражена на рис. 254.
В таблице 67 приведены коэфициенты воздухопроницаемости для раз-
личных строительных материалов.
1 Журнал: «Строительные материалы» № 4, 1931.
431
Таблица 67
Коофициечт воздухопроницаемости строительных материалов
Название материалов Коэфициент воз-
духопроницаемости
Известковый туф j 7,980
Известка обыкновенная । 0,907
Бетон 0,258
Цемент портландск' й 0,137
Кирпич ручной, хорошо обожженный . 0,203
» машинный, •> » 0,132
i> » слабо » 0,087
Песчаник 0,118—0,130
Дубовое дерево (попер чный разрез) 0,007
Водопоглощенпе
Под водопоглощением понимают способность строительного
материала (камня) впитывають воду при обычном давлении.
Очищенные от рыхлых частиц и пыли образцы высушивают до постоян-
ного веса и помещают в стеклянный или фарфоровый сосуд. Заливают дестил-
лированной водой до !/4 высоты образца; через 2 часа доливают воды до
высоты и через 3 часа — до 3/4 высоты образца, после чего оставляют в покое
на сутки. Через сутки образец заливают полностью водой и вновь оставляют
на сутки. Затем образец вынимают, обтирают влажной полотняной тряпоч-
кой, взвешивают и снова опускают в воду на сутки. Так поступают до тех
пор пока образец не приобретет постоянного веса.
Водопоглощенпе в процентах по весу определяют по формуле:
wb = . юо,
gi
где IVb—водопоглощение в процентах по весу,
g2—постоянный вес образца после поглощения воды,
gt—постоянный вес образца до поглощения воды.
Водопоглощение в процентах по объему определяется по формуле:
IV 0 = То . юо.
£1
гце Wo — водопоглощение в процентах по объему,
7о — объемный вес образца,
#1И#2 — имеют такое же значение, как в предыдущей формуле.
Исследование проводят для трех образцов и находят среднее арифмети-
ческое.
Водояасыщепие
Под водонасыщением понимают способность строительного ма-
териала (камня) впитывать воду при давлении ниже атмосферного или при
к шячении. Белее простым способом является определение водонасыщения
при кипячении.
Образцы, высушенные до постоянного веса, помещают в водяную баню,
наливают дестиллированной воды до половины высоты образцов и оставляют
432
в покое на 2 часа. Затем доливают воды столько, чтобы образцы оказались
погруженными в нее полностью, и кипятят в течение 3 часов, поддерживая
постоянный уровень воды. Затем баню охлаждают, образцы извлекают, обти-
рают слегка влажной полотняной тряпкой и взвешивают.
Водонасыщение в процентах по весу и по объему вычисляют, пользуясь
теми же формулами, что и при определении водопоглощения.
Водоотдача
Под водоотдачей понимают скорость просыхания насыщенного
водой камня. С точки зрения гигиены быстрая водоотдача является поло-
жительным свойством стройматериала.
Высушенные до постоянного веса образцы насыщают водой до постоян-
ного веса точно так, как при определении водопоглощения. Взвешенные
образцы помещают в экссикатор над крепкой серной кислотой, отметив
время начала высушивания. По истечении суток образцы вынимают из
экссикатора, взвешивают и вновь помещают в экссикатор. Через каждые
4 часа производят взвешивание до получения постоянного веса.
Время, потребовавшееся для высушивания образца до постоянного веса,
носит название показателя водоотдачи.
Теплопроводность и теплоемкость
Теплозащитные свойства ограждающих конструкций обусловливаются пре-
имущественно их теплопроводностью и теплоемкостью.
Теплопроводность строительных материалов характеризуется к о э ф и ц и-
ентом внутренней теплопроводности.
Коэфициентом внутренней теплопроводности на-
зывают абсолютное количество тепла, которое проходит в течение одного
часа через 1 ма стенки из испытуемого материала при толщине ее в один
линейный метр, при разности температур на противоположных поверхно-
стях в 1°С, при перпендикулярном к поверхности стенки тепловом потоке
и при установившемся тепловом состоянии.
Коэфициент внутренней теплопроводности определяется по формуле:
? _ QI
s(«i-e2)/ ’
где X — коэфициент внутренней теплопроводности,
Q — количество тепла, выраженное в больших калориях, прошед-
шее через стенку,
I — толщина стенки в линейных метрах,
s — поверхность стенки, выраженная в м2,
0, — 0. — разность температур на противоположных поверхностях стенки,
/ — время (часы).
Величина, обратная коэфициенту внутренней теплопроводности (-т-), на-
А
зывается термическим сопротивлением материала или
удельным термическим сопротивлением и обозначается через г.
Коэфициент внутренней теплопроводности зависит не только от характера
вещества материала, но в высокой степени и от его пористости: ч?м пори-
стость больше, тем коэфициент теплопроводности меньше. Между объемным
весом материала и его коэфициентом теплопроводности существует прямая
зависимость.
Влажность материала также влияет на коэфициент теплопроводности:
с увеличением влажности повышается коэфициент теплопроводности. Послед-
ний до некоторой степени зависит и ст температуры материала.
28 А. И. Бурштейн
433
Для определения коэфициента внутренней теплопроводности можно вос-
пользоваться способом проф. Скобельцина. Схема установки представлена
на рис. 255 и заключается в следующем: между плоской электрической грел-
кой А и металлической пластинкой В, служащей холодильником, зажаты
две пластинки — одна (7) из материала, коэфициент теплопроводности кото-
рой (KJ требуется определить, другая (2) с известным коэфициентом тепло-
проводности (\>)- С боковых сторон обе пластинки покрываются хорошо
изолирующим материалом 5, для защиты от утечки тепла. Толщина плиток,
7—I и 2—12 определяется при помощи штангенциркуля. После наступле-
ния температурного равновесия определяют при помощи термопары разности
температур по ту и другую сторону плиток — и 02.
Если через Q обозначить количество тепла, переходящее от грелки к
нижней металлической пластинке в единицу времени через единицу пло-
щади, то можно написать следующее уравнение:
Q = 21 0= 210.
к h '
Решая уравнение в отношении получаем:
Коэфициент теплопроводности определяют еще следующим образом: уста-
навливают величину объемного веса материала, как описано выше; находят
по таблице материал с таким же объемным весом и коэфициент теплопровод-
Рис. 255. Схема при-
бора Скобельцина для
определения геплопро-
водности строитель-
ных материалов.
А — плоская электриче-
ская грелка В — метал-
лическая пластинка / —
пластинка из исследуе-
мого материала 2 —
пластинка с известными
коэф и 11 ие н та м и те пло 11 ро-
водности .? — изолирую-
щий материал.
ности этого материала принимают равным коэфициенту
теплопроводности испытуемого материала.
Наконец, для определения коэфициента теплопровод-
ности пользуются еще следующей формулой:
X = — 0,14+ 1/ 0,01196+ 0,00017 f-7» V
Г \ 28 /
Теплоемкость строительных материалов харак-
теризуют удельной теплоемкостью, т. е.
количеством тепла в малых калориях, необходимом
для нагревания одного грамма вещества на 1°С, или
количеством тепла в больших калориях, необходимым
для нагревания одного килограмма вещества на 1°С.
Теплоемкость определяется по формуле:
с= Q ,
g(02-«i)
где С — теплоемкость удельная,
Q — количество тепла, затраченное на нагревание тела,
# — вес тела,
— температура нагрева.
Удельная теплоемкость строительных материалов варьирует в небольших
пределах от 0,11 для железа до 0,65 для дерева.
С повышением влажности строительных материалов удельная теплоем-
кость их повышается.
В таблице 68 приведены значения теплопроводности и теплоемкости для
некоторых строительных материалов.
434
Таблица 68
Значения для теплопроводности и теплоемкости строительных материалов
Название материала Теплопровод- ность Теплоемкость
Железо 50 0,11
Из! естняк 1,08—1,14 0,21
П( счаник 1,33—1,53 0,22
Мрамор 2,5 0,21
Песок 0,27 —
Кирпич ручной выделки 0,33 0,2
» машинный 0,44 —
• сил катный 0,80 —
» саманный 0,35 —
Стекло 0,65 0,20
Бетон трамбованный 1,1 0,20
Сосна вдоль волокон 0,30 0,65
» поперек » 0,15 0,65
Дуб вдоль волокон 0,35 0,57
»> поперек » 0.20 0,57
Линолеум 0,16 —
Звукопроводность
Звуковая волна, доходя до перегородки, частью отражается, частью про-
ходит через перегородку. Передача звуковой энергии через перегородку
осуществляется разными путями: частью через поры перегородки, если
таковые имеются, частью благодаря колебаниям материальных частиц пере-
городки, частью вследствие упругих колебаний всей перегородки в целом.
Из сказанного ясно, что звукопроводность перегородки зависит от харак-
тера строительного материала, из которого перегородка построена. Так как
с гигиенической точки зрения звукопроводность стенки имеет определенное
значение (передача шума из одного помещения в другое), то и возникает не-
обходимость определять звукопроводность строительного материала.
Для этой цели предложено много способов; здесь описаны лишь два.
Олреде юн ле звукопроводности при помощи динамометра
В фокусе параболического зеркала (рис. 256) помещают источник звука А;
параллельный пучок звуковых лучей направляют на перегородку В. С по-
мощью динамометра С (шайба Релея) определяют энергию пучка лучей, про-
шедшего через перегородку. Полученный результат можно выразить в про-
центах по отношению к энергии падающего на перегородку пучка лучей.
Можно также выразить звукопроводность данного материала в процентах
по отношению к звукопроводности открытого просвета (окно), принимаемой
за единицу.
Шайба Релея представляет собой маленький диск с приклеенным зеркаль-
цем, подвешенный на кварцевой или стеклянной нити. Его устанавливают
под некоторым углом к направлению звука. Диск под влиянием звука откло-
няется от начального положения; эти отклонения тем больше, чем интен-
сивней звук. За отклонением диска следят по перемещению зайчика, отбра-
сываемого зеркальцем.
435
Определение звукопроводности при помощи сонометра
Прибор (рис. 257) состоит из двух камертонов, настроенных в унисон.
Камертоны поставлены на ящиках — резонаторах. Для приведения в звуча-
ние одного из камертонов служит электромагнит. Другой камертон — соно-
метр — отвечает на звучание первого
Камертона резонирующим звуком. Силу
Звука определяют оптически, проецируя
световой луч от колеблющейся ветви
Рис. 257. Сонометр.
А — камертон, В — ящик-резонатор. С — мо-
лоточек, D — клемма для включения элек-
трического тока.
Рис. 256. Прибор для определения зву-
копроводности строительных материалов,
стен, потолков, полов и т. п.
А — источник эвука, В — перегородка,
С — шайба Релея.
камертона на матовую пластинку. Для определения звукопроводности ма-
териала помещают сначала оба камертона рядом, и, приведя в звучание элек-
тромагнитный камертон, отмечают длину световой линии от колеблющейся
ветви камертона-сонометра на матовой пластинке. То же повторяют, поме-
стив между обоими камертонами испытуемый материал (перегородку). Зву-
копроводность определяется из отношения квадратов полученных двух
величин.
В таблице 69 приведена относительная звукопроводность различных стро-
ительных материалов.
Таблица 69
Относительная звукопроводность строительных материалов
Материал Толщина в мм Вес в кг От юс чтельная звукопровод- ность
Воздух — — 100
Пробка (прессованная) . . . 15 1,29 77,5
Войлок 15 1,8 70,0
Дерево 15 2,83 57,0
Бетон 15 14,6 15,0
Кровельное железо 5 16,75 13,2
Свинец (листовой) 2 25,2 9,4
436
Влажность
Влажностью строительного материала называют процентное отно
шсние веса гигроскопической воды материала к его общему весу.
Содержание гигроскопической воды в стенах зданий допускается не
выше 2%. Стены, содержащие 4—5% воды, должны признаваться сырыми
(Эрисман). На сырых стенах часто можно констатировать плесень, желтые
пятна от влаги и прилипшей пыли; на каменных (кирпичных) неоштукатурен-
ных стенах от сырости появляются нередко белые налеты так называемой
«стенной селитры», состоящей преимущественно из глауберовой соли. Сырые
стены отличаются большей теплопроводностью, нежели сухие, и кажутся
более холодными. В сырых помещениях ощущается неприятный запах пле-
сени и затхлости. Влажность воздуха в них повышена.
В санитарной практике в отношении влажности стен приходится решать
вопросы следующего порядка: а) находится ли содержание гигроскопической
воды в стенах в пределах, допустимых с санитарной точки зрения, т. е. должны
ли быть стены признаны достаточно сухими или сырыми, и б) если стены ока-
зываются сырыми, то каковы причины сырости, т. е. зависит ли сырость от
почвенных условий, характера строительного материала, климатических и
т. п. моментов, либо обусловлена небрежной эксплоатацией помещения (обиль-
ное продуцирование водяных паров вследствие варки пищи, стирки и сушки
белья и т. п.).
Определение влаги в строительном материале
Взятие пробы материала для определения влаги производится не в одном,
а обычно в 4—5 подозрительных местах стены. В качестве материала для
анализа берется не только штукатурка, но и другие части стены,
по возможности всей толщи. Для каждой пробы берется навеска в среднем
в размерах 30 г. Для взятия навески пользуются обычно пробойником. На-
вески помещаются в бюксы с хорошо пришлифованными крышками.
Для определения влаги в строительных материалах предложено много
способов, из которых ниже приведены лишь наиболее надежные.
Определение влажности высушиванием нагретым воздухом. Этот метод
является наиболее применимым. Если материал не содержит извести
(например, кирпич), то высушивание может быть произведено в обыкновен-
ном сушильном шкафу при 110°С до постоянного веса. Взятый материал
предварительно измельчается в агатовой ступке до величины зерен крупного
песка; из него отсыпают в платиновую или никелевую чашку навеску в 1—2 г,
которая и подвергается высушиванию до постоянного веса.
Влажность материала вычисляется по формуле:
х_ юо‘gi
S ’
где х — влажность в процентах,
— вес гигроскопической воды во взятой навеске,
? — первоначальная навеска.
Иногда влажность выражают в процентах по отношению к сухому веще-
ству материала.
Сухая навеска выражается формулой:
g„ (100-х)
100
где ge — сухая навеска,
?н — первоначальная навеска,
х — процентное содержание гигроскопической воды.
437
Если строительный материал, например, гипс, содержит кристаллизацион-
ную воду, то сушить его следует при температуре не выше 6О°С; в противном
случае происходит потеря кристаллизационной воды.
Если материал, например, штукатурка, содержит известь, то его нельзя
высушивать в обыкновенных условиях, так как при этом будет итти погло-
щение СО2 и одновременная потеря гидратной воды:
Са(ОН)2 4- СО2 = СаСО3 + Н2О.
В таких случаях высушивание ведется в атмосфере, свободной от СО,
(см. ниже).
Определение влажности высушиванием без подогревания. Навеску ма-
териала помещают в небольшом экссикаторе над пятиокисыо фосфора или
крепкой серной кислотой. В течение двух суток проба отдает гигроскопи-
ческую влагу. По прошествии указанного времени повторным взвешиванием
через каждые 4 часа нужно убедиться в постоянном весе пробы.
Рис. 258. Прибор Илькевича для определения влажности стро-
ительных материалов.
А — двустенный водяной шкаф, В — обратный холодильник, С — тер-
мометр D —экссикатор, а иЪ—трубки, проходящие через пробку
экссикатора, е — троиник, / — бюретка, с, d, к, I — поглотительные
склянки.
Материал, содержащий известь, не может подвергаться высушиванию
в указанных условиях, так как воздух в экссикаторе содержит СО2.
Определение влажности в среде, свободной от < ()2. Предложено много
методов для указанной цели. Здесь описан метод К. Я. Илькевича.
Высушивание по этому методу ведется в специальном приборе в токе
воздуха, свободном от СО2 и Н2О.
Прибор (рис. 258) состоит из двустенного водяного шкафа А с тремя от-
верстиями в верхней стенке.
В одно отверстие, сообщающееся с полостью шкафа, вводится конец обрат-
ного холодильника В, благодаря чему пары воды, образующиеся при нагре-
вании шкафа, конденсируются и попадают обратно в шкаф, и уровень воды
в шкафу остается постоянным. Для наблюдения за температурой служит
термометр С.
В шкаф помещается небольшой экссикатор D, куда вносят бюксы с испы-
туемым материалом. В крышке экссикатора имеется отверстие, в которое
вставляют каучуковую пробку; в последнюю вставлены две трубки: одна
из них (а) доходит почти до дна экссикатора, другая (Ь) — заканчивается
под пробкой. Наружный конец длинной трубки при помощи каучуковой
трубки соединяется через отверстие в крышке шкафа с системой сосудов,
предназначенной для поглощения влаги и СО2. В качестве таковых может
быть применена система U-образных трубок, наполненных натронной изве-
стью и хлористым кальцием. Илькевич поглощает СО2 60% раствором
438
NaOH. Полнота поглощения СО2 контролируется при помощи склянки с про-
зрачным раствором Ва(ОН)2. Влага поглощается концентрированным раство-
ром H2SO4.
Короткая стеклянная трубка при помощи каучуковой трубки соединяется
через отверстие в крышке шкафа с одним концом тройника е. К нижнему
концу тройника присоединяется небольшая бюретка /. Третий конец трой-
ника соединяется с двугорлой склянкой к с концентрированной серной ки-
слотой, а последняя — с такой же пустой склянкой I, присоединяемой на
случай забрасывания воды из водоструйного насоса в систему. Вторая склянка
присоединяется к водоструйному насосу.
Анализ ведется следующим образом: нагрев шкаф до 98—99°С, пускают
в ход водоструйный насос и медленно просасывают через экссикатор сухой
воздух, лишенный СО2 и Н2О. Воздух увлекает па-
ры воды, выделяющиеся при сушке пробы. Последние
конденсируются в тройнике и стекают в бюретку,
где потом определяются простым отсчетом. Анализ
продолжается 4—6 часов.
Если проба содержит кристаллизационную воду,
то высушивание ведется при температуре не выше
60°С.
Для определения гидратной воды в штука-
турке пользуются пробой, освобожденной от гигроско-
пической воды, как описано выше. Через нее пропус-
кают ток сухого угольного ангидрида. При этом
протекает следующая реакция:
Са(ОН)2 + СО2 = СаСО3 + Н2О.
Как легко видеть из приведенной реакции, каж-
дая молекула Са(ОН)2 переходит в молекулу СаСО3,
ввиду чего молекулярный вес повышается на 26.
Такому повышению веса соответствует 18 весовых
частей воды. Следовательно, на основании повыше-
ния веса исследуемой пробы можно сделать вывод
о количестве гидратной воды: привес нужно помно-
жить на 161№.
Определение влажности ацетиленовым методом.
Принцип ацетиленового метода, заключается в том,
навеску данного материала действуют измельченным карбидом кальция; при
этом между карбидом кальция и водой протекает следующая реакция:
Рис. 259 Прибор Яко-
венко для определения
влажности ацетиленовым
методом.
а — бюретка. Ъ — уравни-
тельный сосуд, с — тройник,
d — стаканчик с испытуемым
материалом, е — стаканчик
с карбидом кальцин.
что на определенную
СаСО2 + 2Н2О = Са(ОН)2 + С2Н2.
По выделенному ацетилену делают заключение о количестве гигроско-
пической воды в материале. Из приведенной реакции видно, что две молекулы
воды соответствуют одной молекуле ацетилена, т. е. 36 весовых частей воды
соответствуют 26 весовым частям ацетилена. Так как 1 мг С2 Н2 при нормаль-
ных условиях имеет объем в 0,861 мл, заключаем, что 1 мл выделенного ацс-
36
тилена соответствует —26^= 1,61 мг ^2О- На основании указанного
отношения легко вычислить содержание воды в испытуемом материале.
На рис. 259 изображен прибор, предложенный для данного исследования
В. А. Яковенко. Обыкновенная бюретка а соединяется при помощи толсто-
стенной каучуковой трубки с уравнительным сосудом Ь. Сверху бюретка
закрыта пробкой, в которую вставлена стеклянная трубка, на конец которой
надета каучуковая трубка. Последняя соединяется с одним концом тройника
с. К противоположному концу тройника при помощи каучуковой пробки при-
соединен стаканчик d, в который насыпают 1—2 г измельченного материала.
439
Третий конец тройника соединен со стаканчиком е, в который насыпают мелко
истолченный карбид кальция. Установив ртуть в бюретке и уравнительном
сосуде в одной горизонтальной плоскости и отметив стояние ртути в бюретке,
несколько опускают уравнительный сосуд. Затем переводят карбид кальция
из стаканчика е в стаканчик d так, чтобы карбид покрыл испытуемое вещество
толстым слоем, после чего помещают стаканчик d на песочную баню при 100—
105°С.
Когда реакция двойного обмена между карбидом кальция и водою ма-
териала закончится, на что уходит в среднем 10—15 минут, удаляют баню
и дают стаканчику d вместе с содержимым охладиться до комнатной темпе-
ратуры. Далее, вновь устанавливают уровень ртути в уравнительном сосуде
и в бюретке в одной горизонтальной плоскости и производят отсчет высоты
стояния ртути в бюретке. Разница между высотой стояния ртути в бюретке
в начале и в конце опыта указывает количество выделенного С2Н2 в милли-
литрах.
Процентное содержание воды в материале определяется по формуле:
_ W - V • Н • 100 _ W -V • Н
Х~ g- 1000(1 +&0 - 760 ~~ g- 10 (1 + af) . 760 ’
где х — искомое число,
IV— 1,61,
V — найденный объем С2Н2 в мл,
Н — барометрическое давление,
g — вес взятого продукта в г,
а — 0,00367,
t — температура воздуха помещения.
Так как технический карбид кальция при действии на воду дает несколько
меньший выход ацетилена, нежели можно ожидать на основании теорети-
ческих расчетов, в каждом отдельном случае нужно установить коэфициент
для данного карбида. Это проводится обыкновенно при помощи оксалата
аммония — (NH4)2C2O4 . Н2О. 1 г этого вещества содержит ——мг Н2О.
Взяв некоторую навеску вещества, вычисляют теоретически количество аце-
тилена, которое должно выделиться при действии на эту навеску карбида
кальция. На основании опыта находят то фактическое количество С2Н2, ко-
торое выделяется, и, сопоставляя оба числа, выводят коэфициент для данного
карбида кальция. На этот коэфициент и нужно помножить результат анализа,
чтобы получить истинное содержание воды в испытуемом материале.
Пример. Теоретическое количество С2Н2, которое должно выделиться при данной на-
веске оксалата аммония, равно 8,2008 мл . В опыте выделилось лишь 8,04 мл. Отсюда на-
8 9008
ходим фактор для карбида кальция: = 1,02. Положим, далее, что при этом коэфпци-
o,U4
енте карбида кальция и 1,2 г навески данного вещества выделилось 32 мл С2Н2; барометри-
ческое давление во время исследования 762 мм Hg, а температура 16° С. Нужно определить
содержание воды в данном материале.
Пользуясь приведенной выше формулой и найденным коэфициентом, получаем:
v 1,61 • 32 • 762 . 1,02 . ,
X = ---------------------------4,1э°/0.
1,2 • 10(1 -(-0,00367 • 16) • 760
П. М. Оржеховский и К. Б. Хайт показали, что и сам карбид кальция,
нагреваясь, выделяет ацетилен, количество которого достигает иногда больших
размеров, ввиду чего возникают большие ошибки при пользовании описанным
выше методом. Исходя изэтого авторы приходят к такому выводу: необходимо
либо установить холостой пробой количество ацетилена, выделяющееся на
единицу веса данного карбида кальция,»и затем вычесть соответствующее
количество газа как при установке коэфициента карбида кальция, так и при
440
определении влажности материала, учитывая вес взятого карбида кальция,
либо, и это проще, применять высушенный предварительно карбид кальция.
Метод Яковенко для определения влаги в строительном материале не-
применим, если материал содержит кристаллизационную воду или известь.
ВЫЯСНЕНИЕ ПРИЧИН СЫРОСТИ СТРОИТЕЛЬНОГО МАТЕРИАЛА
Выяснение причин сырости строительного материала производится в ряде
случаев довольно легко и скоро. Так, например, без большого труда можно
установить причины сырости отдельных участков стен, связанные с дефек-
тами канализационных или водопроводных сооружений, нарушением целости
крыш. Сырость стен второго и более высоких этажей обыкновенно обуслов-
лена небрежной эксплоатацией помещения, если можно исключить указанные
сейчас моменты. Напротив, сырость стен подвальных помещений, а также
первого этажа на высоте 1—1% м, проявляющаяся отдельными, часто резко
ограниченными участками, связана обыкновенно с высоким стоянием почвен-
ных вод и плохой изоляцией стен от грунта.
В других случаях выяснение причин сырости требует тщательного учета
почвенных, климатических и других моментов, анализа строительного мате-
риала и т. п.
ГРИБЫ, НАСЕКОМЫЕ И ЖИВОТНЫЕ, ПОВРЕЖДАЮЩИЕ ДЕРЕВО
Наиболее сильное повреждение древесины (гниение) вызывается целым
рядом грибов. ГОСТ 2618 насчитывает 29 различных видов гнили и грибов;
в действительности число их значительно больше.
Под домовыми грибами разумеют грибы, растущие на деревян-
ных частях жилых строений и вызывающие разрушение древесины. Неко-
торые авторы причисляют к домовым грибам и такие, которые не вызывают
разрушения древесины, но растут в домах (Илькевич). В большинстве слу-
чаев тело грибов состоит из тонких бесцветных или окрашенных нитей —
гифов, которые, сплетаясь, образуют мицелий. Размножение грибов
происходит при помощи спор. В зависимости от способа питания грибы
разделяются на паразиты, живущие на живых растениях, и сапро-
фит ы, поселяющихся на отмерших частях растений. Кроме указанных
двух основных групп различают еще группу полупаразитов, которые
поселяются на живых и отмерших частях растений. Большинство грибов,
вызывающих разрушение древесины, относится к группе полупаразитов.
Заражение древесины происходит чаще всего спорами, которые переносятся
с пораженного дерева на здоровое ветрами, насекомыми (жуками) и живот-
ными (грызунами). Реже заражение происходит через грибницу при контакте
зараженной древесины со здоровой. Домовые грибы лучше всего развиваются
в неподвижном сыром воздухе. Оптимальная температура для их развития
лежат в пределах 18—Зб°С, однако многие домовые грибы способны выдер-
живать и более высокие и низкие температуры; то же относится и к их спо-
рам. Из множества домовых грибов ниже описаны лишь те, которые являются
наиболее опасными как разрушители деревянных частей здания.
Гриб домовой белый (Poria vaporaria) встречается
на всех древесных породах, но особенно часто на сосне и ели. Мицелий этого
гриба в виде белого пуха покрывает пораженное дерево. Древесина становится
бурой, в ней образуется много поперечных и продольных трещин. Под конец
гниения древесина распадается на сухие призматические части, легко расти-
рающиеся в порошок. Споры гриба яйцевидной формы, длина их 4,5 — 6р.
ширина 2,5—3,5р.
Гриб домовой настоящий, плачущий гриб (Mcrulius
lacrymans) является еще более опасным, нежели гриб домовой белый. Гриб-
ница его имеет вид белой пушистой ваты с легким розовым или канареечно-
желтым оттенком. Грибница обычно выделяет капли желтоватой жидкости,
441
откуда и название «плачущий гриб». Этот гриб приносит большие разрушения:
пораженная им древесина приобретает желтокоричневый цвет. Характер раз-
рушений такой же, как и при poria vaporaria. Считавшееся одно время харак-
терным для слезоточивого гриба образование пряжек по ходу гифов, как
показали исследования, наблюдается и у других видов грибов (poria vaporaria,
coniofhora cerebella и т. д.). Пряжки эти видны под микроскопом и представ-
ляют короткие, полукруглые выросты гифов. Споры плачущего гриба яйце-
видной формы; длина их 9—11ц, ширина 4,5—6,5-»].
Домовой пленчатый гриб, погребной гриб (С oni-
op h о г a cerebella) особенно часто встречается в подвалах, поражает
также лесоматериалы на складах, шпалы и т. п. На поверхности поражен-
ного дерева образуются легко отделяющиеся мясистые пленки оливковоко-
Рпс. 260. Насекомые, повреждающие дерево.
А — жук короед, В — ?кук превоточеи-часовщик, С — жук усач, D — домовой точиль-
щик. Е — гребкеусый точильщик.
ричневого цвета с беловатыми волокнистыми краями, представляющие собой
плодовые тела грибов. Споры яйцевидны, размеры их: длина 10—14|i,
ширина б—7 р.
Ввиду того что домовых грибов встречается большое количество, для
диференцирования их предложены определители (Янчевский и др.),
к которым нужно обращаться в неясных случаях. Кроме того, для целей
диагностики рекомендуется разводка чистых культур. Макроскопический и
микроскопический вид чистых культур способствует решению вопроса. Хо-
рошей средой для роста домовых грибов может служить мальц-экстрактовый
агар состава: 25 ч. агара, 25 ч. мальц-экстракта, 1000 мл воды и 0.1 ч. лимон-
ной кислоты. Для этой же цели пользуются средой, в состав которой входят
древесные опилки: 100 ч опилок, 400 ч. воды и 2 ч. мальц-экстракта и т. п.
Борьба с домовыми грибами должна вестись в направлении отбора для
построек заведомо здорового дерева, предупреждения сырости в помещениях
(изоляция от почвы, вентиляция), а также применения различных пропиток
дерева (3% NaF, 3% ZnCl2 и др.).
Н а с е к о м ы е, повреждающие дерево (рис. 260), не столь многочислен-
ны, как грибы — разрушители дерева, и не столь опасны в смысле разрушений.
К этим насекомым относятся: жук короед, жук древоточец-часовщик, жук
усач, домовый точильщик, гребнеусый точильщик.
Из ж и в о т н ы х, портящих древесину, можно назвать свайного червя,
грызунов.
442
ЛИТЕРАТУРА
Общие курсы и руководства
1. Игнатов Н. К., Практическое руководство по методике санитарно-гигиенических
исследований, Биомедгиз, 1935. *
2. Марзеев А. Н., Сысин А. Н., Яковенко В. А., Основы коммунальной гигиены, т. II,
Медгиз, 1938.
3. Моисеев С. В., Учебник общей гигиены, Медгиз, 1947.
4. Хлопин Г. В,, Основы гигиены, т. II, 1923.
Специальная литература по строительным материалам
1. Алмазов С. А. и Коршун Л. Г., Новые строительные материалы, М., 1933.
2. Андреевский В. А., Строительные материалы из торфа, Госстройиздат, 1933.
3. Гогин Ф. А., Соломит и камы пит, ГНТИ, М., 1931.
4. Головина Е. П., Определение влажности строительных материалов, 1930.
5. Григорьев П. Н., Химический анализ строительных материалов, 1932.
6. Кинд Н. А. и Окороков С. Д., Строительные материалы, 1934.
7. Копелянский Г. Д., Новые строительные материалы, Госстройиздат, 1933.
8. Копейкин С., Справочник строительных материалов, 1928.
9. Лившиц С. Я., Акустика зданий и их изоляция от шума и сотрясений, 1931.
10. Лоренц В. Ф., Руководство по испытанию и приемке строительных матери-
алов, 1929.
11. Поморцев В. П., Строительная гигиена и санитария, Госстройиздат, М., 1934.
12. Философов /7. С., Строительные материалы, 1931.
13. Он же, Глзвнейшие строительные материалы, 1931.
14. Фокин К. Ф., Строительная теплотехника ограждающих частей зданий, Госстрой-
издат, 1933.
15. Эвальд В. В., Строительные материалы, 1933.
16. ГОСТ’ы
Грибы и другие вредители древесины
1. Ванин С. И., Домовые грибы, их биология, диагностика, и меры борьбы 1931.
2. Голдин М. М., Руководство по борьбе с домовым грибом, 1934.
3. Илькевич К. Я., Грибы—разрушители деревянных частей строений, 1911.
4. Инструкция по борьбе с домовым грибом—разрушителем древесины (Постановление
№ 45 Совета Народных Комиссаров УССР от 13 января 1935 г.
5. Инструкция Наркомхоза РСФСР о мерах борьбы с домовыми грибами — разруши-
телями древесины, 1935.
6. Макринов А. И., Домовой гриб (Merulius lacrymans), его распространение и меры
борьбы, 1920.
7. Миллер А. А., Санитарная бактериология, Госмедиздат, 1933.
8. Наумов Н. Н., Методы микробиологических и фитопатологических исследова-
ний, 1937.
9. Плавильщиков Н. Н., Жуки дровосеки — вредители древесины, 1932.
10. Флеров Б. К., Домовые грибы и меры борьбы с ними, 1935.
11. Ячевский А. А., Определение грибов, тт. 1 и II, 1913—1927,
ГЛАВА ВОСЬМАЯ
ЖИЛЫЕ, ФАБРИЧНО-ЗАВОДСКИЕ, ШКОЛЬНЫЕ
И ДРУГИЕ ПОМЕЩЕНИЯ
Санитарно-гигиеническое обследование помещения имеет целью выяснить:
размеры площадей и объемов; состояние стен, потолков, полов, окон и дверей;
качество воздуха, устройство и эффективность вентиляции естественной и
искусственной; состояние естественного и искусственного освещения; характер
и эффективность отопительных систем; состояние сооружений водоснабже-
ния и канализации; санитарный уход за помещением.
Подробное обследование должно включать еще данные о санитарном со-
стоянии всего здания в целом, дворовой усадьбы и ближайшей местности.
На основании всех данных обследования делается заключение о пригод-
ности помещения для предназначенной цели.
Имеется ряд постановлений, которых нужно придерживаться при даче
заключения о соответствии между данными обследования помещения и су-
ществующими нормативами.
Так, требования к составлению и утверждению проектов планировки и
социалистической реконструкции городов и других населенных мест Союза
ССР изложены в Общесоюзном стандарте ОСТ (ВСКХ-7028/8), Санитарных
правилах НКЗ РСФСР по постройке жилых зданий № Б-247/МВ, изданных
26.VH—1929 г.
Санитарные правила устройства, оборудования и содержания рабочих
общежитий изложены в постановлении ВГСИ от 28 февраля 1946 г.
Нормы естественного освещения промышленных зданий изложены в
ГОСТ 3291-46. Нормы искусственного освещения в ГОСТ 3825-47 и т. д.
Работа по обследованию помещений может быть разделена на две основных
части: санитарно-описательную и лабораторно-экспериментальную. Обсле-
дование ведется по так называемым санитарным картам. В качестве примеров
составления таких карт здесь приводится содержание карт по санитарному
обследованию жилого помещения и производственного цеха.
КАРТА САНИТАРНОГО ОБСЛЕДОВАНИЯ ЖИЛОГО ПОМЕЩЕНИЯ
1. Местонахождение.
2. В чьем ведении владение (жилищное управление, жилищное товарищество, застрой-
щик и т. п.).
3. Этаж.
4. Число комнат.
5. Стены (материал, отделка, состояние, наличие сырости, причины); потолки (мате-
риал, отделка, состояние); полы (материал, состояние).
6. Внутренние переборки (материал, состояние).
7. Общая площадь в м-, кубатура в мч.
8. Отдельно для каждой комнаты: длина, высота, площадь, кубатура.
444
9. Количестко живущих в комнатах (мужчин, женщин, детей); площадь и кубатура
на одного человека.
10. Окна (количество, высота, ширина, общая световая поверхность, в какую сторону
света обращены, прегражден ли доступ света через окна). Световой коэфициент. Коэфп-
циент дневного света.
11. Двери и выходы, лестницы и лестничные клетки.
Т2. Искусственное освещение, его характеристика.
13. Отопительные приборы. Система, число точек, действуют или не действуют, если не
действуют, то почему.
14. Вентиляция. Наличие форточек, число, размер. Прочие виды вентиляции.
15. Наличие подсобных помещений (кухня, ванная, коридоры). Как используются,
в каком состоянии.
16. Наличие водопровода: есть, отсутствует, действует, нет. Вода: берется из уличной
колонки, из дворового колодца, где и как хранится.
17. Канализация: общегородская, местная домовая, действует, нет. Ванная: есть,
нет, как используется. Уборная: есть, нет, как содержится.
18 Как содержится помещение в целом. Наличие насекомых, грызунов. Производится
ли стирка и сушка белья в квартире. Уборка.
19. Прочие замечания.
КАРТА САНИТАРНОГО ОБСЛЕДОВАНИЯ ПРОИЗВОДСТВЕННОГО ЦЕХА
1. Местонахождение.
2. Наименование производства.
3. Наименование цеха, данные о числе рабочих, их половом составе и пр.
4. Общее описание производственных работ. Характеристика •сновных типов обору-
дования.
5. Размеры помещения: длина, ширина, высота, площадь, кубатура (общая и на одного
рабочего).
6. Стены, полы, потолки (материал, отделка, состояние).
7. Двери и выходы, лестницы, количество дверей и выходов. Материал и устройство
лестниц, лестничные клетки.
8 Внутрицеховой транспорт, его организация.
9. Освещение естественное. Верхнее или боковое. Для верхнего: величина горизонталь-
ной проекции осветленной поверхности, состояние остекленной поверхности, световой коэ-
фициент Для бокового: одностороннее или двустороннее, число окон, их общая поверх-
ность, состояние окон, в какую сторону света обращены, прегражден ли доступ света через
окна. Световой коэфициент. Освещенность рабочих мест. Коэфициент естественной освещен-
ности в соответствии с характером производимых работ.
10. Искусственное освещение. Общее, местное или комбинированное, прямое, отражен-
ное и т. д Типы светильников, высота подвеса, ориентировка по отношению к рабочим
местам. Количество и мощность ламп; освещенность рабочих мест в люксах, освещенность
проходов Степень неравномерности освещения.
11. Отопление. Система, тип нагревательных приборов, их расположение, состояние.
Общая оценка отопления.
12. Вентиляция Естественная вентиляция. Количество форточек, фрамуг, дефлекторов,
их размеры, расположение и устройство. Режим работы естественной вентиляции в разные
времена гола Эффективность, общая оценка. Искусственная вентиляция. Назначение,
характер: общеобменная, местная. Система: приточная, вытяжная, приточно-вытяжная.
Способы очистки, подогрева и увлажнения приточного воздуха. Очистка выбрасываемого
воздуха. Применяется ли рециркуляция воздуха, процент вводимого рециркуляционного
и свежего воздуха. Кратность обмена в час. Конструкция и расположение воздуховодов
и патрубков. Режим работы вентиляции в разные времена года. Ее эффективность. Обслу-
живание Общая оценка.
13. Метеорологические условия рабочих помещений. Температура, влажность, дви-
жение воздуха. Эффективная температура в различных местах цеха в разные часы дня и в
различные времена года. Наличие лучистой энергии, ультрафиолетовой радиации, лучей
Рентгена и пр. Меры устранения ненормальных метеорологических условий.
14. Пыль, пары и газы. Качественная и количественная характеристика. Мероприя-
тия общего и индивидуального характера по защите рабочих от вредного воздействия.
Эффективность этих мероприятий.
15. Шум и сотрясение. Качественная и количественная характеристика. Меры борьбы
с шумом и сотрясением. Эффективность.
16. Опасность травматических повреждений. Состояние предохранительных мероприя-
тий: ограждение станков, трансмиссий, защитные очки и пр.
17. Наличие в цехе питьевой волы, как хранится. Наличие умывальников, расположе-
ние и устройство. Место и способы хранения одежды. Наличие уборных, расположение и
санитарная характеристика.
18. Общее содержание рабочих помещений, загромождения, уход за чистотой.
445
ПЛОЩАДЬ ПОЛА И КУБАТУРА ПОМЕЩЕНИЯ
Размеры помещений (длина, ширина, высота) определяются при помощи
измерительной ленты-рулетки, которую прикладывают к самым стенам либо
держат параллельно им. При этом лента должна быть натянута в строго го-
ризонтальной или вертикальной плоскости. Измерения проводят вдвоем,
причем один делает записи.
Длиной комнаты считается расстояние от окна до противоположной стены,
шириной — расстояние между двумя другими стенами. Высота жилых комнат
свыше 3,5 м в расчет не принимается, так как считают, что слой воздуха,
лежащий выше 3,5 м, принимает лишь незначительное участие в общем воз-
духообмене. На основании полученных данных измерения вычисляют квад-
ратуру и кубатуру помещений.
При вычислении так называемых полезных площадей и объемов
помещения нужно от полученных при измерении данных отнять размеры
площадей и объемов, занимаемые крупными предметами (большие шкафы,
печи и т. п.). Полученные результаты представляют квадратуру и кубатуру
нетто данного помещения.
Площадь пола в м2 на одного человека в жилых квартирах должна
быть не менее 9, в спальнях — 6, в общежитиях — 4,5, во временных жилых
бараках — 3,5 (при высоте не менее 2,5 м), в столовых, читальнях, школах
и других помещениях недолговременного пребывания 1,5—2,5.
Кубатура в м3 на одного человека должна быть в жилых комнатах
нс менее 25—30, в спальнях (квартир)—17—25, общежитиях—10—15,
классах для детей до 10 лет — 5—7, классах для детей свыше
10 лет — 7—10, мастерских — 25—75, палатах больниц —
30—50.
Высота помещений должна быть не менее 3—3,5 м.
ВОЗДУХ ПОМЕЩЕНИЙ
Санитарная оценка воздуха помещений производится на
основании данных, полученных в результате всесторонних
исследований, описанных в главе о воздухе.
Измерения температуры следует производить одновременно
в разных местах комнаты по горизонтальному и вертикаль-
ному направлениям. Если ограничиваются одним термометром,
то измерения производятся на высоте 1,5 м от
пола и на расстоянии 1,5—2 м от наружных
, стен и печей.
Рис. 261. Термометр для
определения температуры
стен.
Для измерения температуры внутренней по-
верхности наружных стен применяются термометры
с плоскими извитыми резервуарами (рис. 261).
Резервуар стенного терме метра защищают от
действия комнатного воздуха при помещи тонкого
слоя извести или алебастра. Температура стен
измеряется на высоте 1,5—2 м от пола и в нижних
холодных углах. Для измерения температуры стен
применяют и термопары (стр. 183).
Температурные нормы. Для жилых квартир —
18—20°С., спален, аудиторий, театров, учреж-
дений, контор, классов —16—18°; коридоров, лестничных клеток —
12—14°, кино, вокзалов — 12—15°; гимнастических залов, музеев —
10—15°; больничных палат, врачебных кабинетов, изоляторов, рент-
генкабинетов, умывальных — 20°; родильных, операционных, ванных
комнат — 25°; перевязочных, предоперационных, процедурных — 22—25°;
бань, раздевальных — 22—25°; мыльных — 30—40°, парильных — 40—50°.
44С
Таблица 70
Метеорологические нормы в рабочей зоне производственных помещении
Характеристика произ- водственных помеще- ний и работы. । Холодный и переходный j период года (наружная ! t° менее + 10° С) Теплый период года (наруж- ная температура-}-10° С и (oj.ee)
Температура, вп <духа в помещении , Относитель- ная влажность Темпепат\’ра воздуха в f омед.ении Относительная влажность
I. Производственные поме- щения, характеризуемые преимущественно конвек- ционным тепловыделением А. Тепловыделения незначи- тельные Легкая работа 16—20 Не норми- Не более чем Не норми-
Тяжелая работа 12—15 руется То же на 3е выше наружной То же руется То же
Б. Тепловыделения значи- тельные Легкая работа 18—25 То же Не более чем То же
Тяжелая работа 14—19 То же на 51 выше наружной То же То же
В. Тпебуется искусственное 22—23 80—75 23—24 80—75
урегулирование темпера- 24—25 70—65 25—26 70—65
туры и относительной 26—27 60—55 27—28 60—55
влажности — — 29—30 55—50
II. Производственные поме- щения, характеризуем* ые тепловыделениями пре- имущественно в вире лу- чистого тепла (напряжс нир л\ чистой энергии в ра- бочей зоне более 600 кал. Не норми- Не более чем на б- в п.е Не норми-
на 1 ма/ час 8-15 руется нар? жной руется
III. Производственные поме- щения, характеризуемые значительным влаге выде- лением А. Тепловыделения незначи- тельные Легкая pa6oia 16—20 Неболер80о/о Не более чем Не норми-
Тяжелая работа 12—15 То же на 3 вы.ие наружной То же руется То же
Б. Тепловыделения значитель- ные Легкая работа 20—23 Не более80<>/о Не более чем Не норми-
Тяжелая работа 16—19 То же на 5е вь ше наружной То же руется То же
Примечание. Для производственных помещений с искусственным регу-
лированием относительной влажности приведенные значения температуры и влаж-
ности относятся к местностям с летней температурой для расчета вентиляции менее
25° С. Для местностей с летней температурой для расчета вентиляции 25—29° (
нормируемые температуры воздуха для теплого периода года повышаются па 2°,
а для местностей с расчетной температурой 30° С и выше на 4° С с сохранением тех же
значений относительной влажности.
441
Наблюдаемые температурные колебания в жилых помещениях во время
отопительного сезона не должны быть более 1—2°С в горизонтальном направ-
лении и 1,5—2° в вертикальном направлении.
В течение суток температурные колебания не должны превышать 3°С
при центральном отоплении и 5—6° при печном.
Температура внутренней поверхности наружных стен не должна быть
ниже температуры комнатного воздуха более как на 3°С.
Нормы влажности. Что касается влажности, то она подробно нормиро-
вана у нас для производственных помещений (таблица 70). Для воздуха
других помещений нормальной признается влажность в пределах 40—60%
относительной влажности. Воздух с относительной влажностью выше 70%
считается сырым.
Считаем нужным здесь еще раз подчеркнуть, что заключение о физиче-
ском состоянии воздушной среды необходимо делать на основании комплекса
всех факторов, характеризующих ее в этом отношении: температуры, влаж-
ности, воздушных токов и пр.
В таблице 70 приведены метеорологические условия для производствен-
ных помещений согласно нормам и техническим условиям, утвержденным
в августе 1948 г.
Метеорологические нормы воздушного душирования (температура и ско-
рость движения воздуха представлены в таблице 71).
Таблица 71
Метеорологические нормы воздушного душирования
1 | Периоды года 1 । Легкая работа Тяжелая работа
Температура воздуха на рабочем месте Скорость дви- жения возду- ха М/Сек. Температра воздуха на рабочем месте Скорость дви- жения возду- ха м/сек.
1. Холодный период го- да (Г наружного роз- духа ниже 10 С) . 15—23 1—3 8—18 2—4
2. Теплый период го- да (tJ наружного воздуха + Ю’С и выше) 18—28 2—4 16—25 3—5
Примечание. Значения температуры воздуха для теплого периода года
относятся к местностям, имеющим летнюю температуру, для расчета вентиляции ме-
нее 25° С. Для местностей с летней температурой для расчета вентиляции 25—29° С
нормируемые температуры воздуха для теплого периода года повышаются на
а для местностей с температурой 30° и выше — на 4°.
сыгость
Для выявления сырости помещения производят тщательный осмотр стен
и потолка. При надобности определяют влажность в строительном материале
(стр. 437).
ВЕНТИЛЯЦИЯ
Системы вентиляции разделяются на естественные и искусственные (меха-'
нические).
ЕСТЕСТВЕННАЯ ВЕНТИЛЯЦИЯ
Естественная вентиляция осуществляется через поры стен, окна, фрамуги,
дефлекторы и т. п.
Силами, способствующими обмену воздуха при естественной вентиляции,
являются движение воздуха (ветер) и разность температуры воздуха снаружи
и внутри помещения.
448
Определение объема воздуха, поступающего в помещения
через поры стен
Для определения объема воздуха, поступающего через поры стен в поме-
щение, еще Петтен кофе ром был предложен метод, называемый антропомет-
рическим. В случае обнаружения значительного воздухообмена здания
через поры метод применяется и в настоящее время.
Сущность метода заключается в том, что, определяя уменьшение концен-
трации СО2 в помещении при закрытых окнах и дверях за час, находят по
формуле объем наружного воздуха, поступившего в помещение за это время.
Методика определения. Закрывают окна, двери, печные от-
душины и т. п., повышают искусственно концентрацию СО2 в помещении
зажиганием свечей (1—2 свечки на 20 м3 воздуха) или иным способом и, пе-
ремешивая воздух при помощи платков или картонных листов, с целью со-
здания равномерности распространения СО2 по помещению, производят отбор
проб воздуха в бутыли в разных местах данного помещения. Затем оставляют
закрытым помещение на час. Через час вновь берут пробы воздуха в тех же
местах. Исследуют воздух, взятый вначале и в конце опыта, на содержание
СО2. Объем воздуха, поступивший в помещение за час, находят по формуле:
С = 2,30257/n 1g Р1~а ,
р2 — а
где С — объем воздуха в м3, поступившего в помещение через поры стен
в течение часа,
т — объем помещения в м3 (если в помещении много предметов, то берут
кубатуру нетто),
рг— содержание СО2 в воздухе помещения до опыта (в м3 на 1 м3 воздуха),
р2— содержание СО2 в воздухе помещения после опыта в тех же еди-
ницах,
а — содержание СО.? в 1 м3 наружного воздуха, принимаемое равным
0,0005 м3 (0,5%0).
В помещениях для временного пребывания большого числа людей описан-
ное выше исследование можно производить без предварительного повышения
концентрации СО2: к моменту, когда помещение освобождается, закрывают
окна и двери и производят отбор проб воздуха; через час после оставления
помещения людьми вновь производят отбор проб воздуха в тех же местах и,
пользуясь приведенной выше формулой, определяют объем воздуха, посту-
пающего в помещение через поры стен.
Пример. В помещении с кубатурой в 300 м8 содержание СО2 в воздухе в начале опыта
было равно б,?0/^; через час после того, как помещение было закрыто, оказалось, что со-
держание СО2 равно Р/оо- Нужно определить воздухообмен в данном помещении.
Подставив данные значения в формулу, получаем:
С = 2,30257 • 300 • log 0,0062 ~ °’000- = 690,77 • 1,05690 — 730 м’.
0,001—0,0005
На основании полученного результата делаем вывод, что вентиляционный коэфициент,
т. е. отношение объема воздуха, поступившего в помещение в течение часа, к кубатуре поме-
щения равен:
Следовательно, в течение часа воздух обменивается в данном помещении почти в два
с половиной раза.
Антрокометрический метод дает удовлетворительные результаты при усло-
вии, что воздух поступает в помещение равномерно; в противном случае метод
может дать совершенно неприемлемые цифры.
29 А. И. Бурштейн 4^9
Определение объема воздуха, поступающего в помещение через
вентиляционные отверстия
Объем воздуха, поступающего в помещение через данное отверстие (окно,
форточка, фрамуга) в м3/час, вычисляется по формуле:
Q = 3600 vs,
где Q — объем воздуха, поступившего через данное отверстие, в м3/час,
v — скорость движения воздуха в м/сек.,
s — площадь отверстия в м2.
При определении скорости поступления воздуха в помещение через дан-
ное вентиляционное отверстие пользуются обыкновенно анемометром. Если
отверстие большого размера, то нужно сделать промер в различных местах
его и взять среднюю величину.
Пример. Воздух поступает в помещение через раскрытое окно размером в 2,5 м2. Из
5 исследований по углам и в центре окна найдено, что воздух поступает в помещение со ско-
ростью 1,5 м/сек. Нужно определить объем воздуха, поступившего в помещение в течение
часа.
Подставляя указанные данные в приведенную выше формулу, получаем:
Q = 2,5 - 1,5 - 3600 = 13 500 м3.
ИСКУССТВЕННАЯ ВЕНТИЛЯЦИЯ
Под искусственной (механической) вентиляцией понимают такую, при
которой желаемое изменение в воздухе достигается при помощи механических
средств.
В зависимости от подачи или отсоса воздуха различают приточную, вы-
тяжную и приточно-вытяжную вентиляцию.
В зависимости от объема действия различают общеобменную и местную
вентиляцию.
Вентиляционные установки имеют целью: 1) удаление пыли, 2) удаление
избытка влаги и обестуманивание, 3) удаление вредных газов и дыма, 4) сни-
жение высокой температуры, 5) в некоторых случаях вентиляционные уста-
новки имеют целью дать комбинированный эффект.
Схема обследования вентиляцонной установки
Санитарно-техническое обследование вентиляционных установок должно
проводиться по определенной схеме.
Здесь приведена в сокращенном виде карта обследования вентиляционных
установок, разработанная Ленинградским институтом гигиены труда и тех-
ники безопасности. Карта состоит из 5 разделов.
1. Общие сведения о мастерской, в которой произведено исследование
вентиляционной установки, с указанием: размеров помещения, дверей и
окон, организации работы, отопления, оборудования, производственного
процесса, характера профессиональных вредностей и источников загрязнения
воздуха, состояния оборудования и особенностей производственного процесса
в данной мастерской, влияющих на санитарно-гигиенические условия труда.
2. Подробное описание вентиляционной установки с указанием всех за-
меченных неисправностей.
3. Техническое обследование вентиляционной установки.
4. Исследование физико-химического состояния воздушной среды во
время действия и бездействия вентиляционной установки.
5. Заключение, в котором приводится эффективность вентиляционной
установки, оценка конструктивного выполнения ее, данные о соответствии
воздухообменов проектным предположениям, расхождения с проектными
данными, влияющие на эффективность вентиляционной установки, и указы-
45©
ваются мероприятиями для повышения эффекта вентиляционной установки,
как-то:
а) санитарно-техническая рационализация производства,
б) изменение принципов вентиляции,
в) конструктивные изменения,
г) дополнительные устройства,
д) соображения эксплоатационного характера,
е) изменения мощности вентиляционной установки.
Определение воздухообмена
Для определения объема воздуха, подаваемого или отсасываемого через
отверстие вентиляционной установки, находят площадь отверстия и умно-
жают ее на скорость движения воздуха при прохождении через данное отвер-
стие и на время в секундах (стр. 450). Точно так поступают, когда нужно
определить объем воздуха, проходящий через воздуховод; в этом случае пло-
щадь сечения воздуховода умножают на скорость движения и на время.
Если требуется определить суммарную производительность всей венти-
ляционной установки, то измерения производят в воздуховоде, через который
проходит все количество подаваемого или отсасываемого воздуха. Так, при
определении всего количества подаваемого воздуха правильно измерения
произвести в трубе, идущей вслед за калориферной камерой и вентилятором;
при этом будет учтено то количество воздуха, которое засасывается через
наружный воздуховод, а также и то количество, которое подсасывается через
неплотности калориферной камеры, т. е. будет определено суммарное коли-
чество воздуха, подаваемого во все ветви установки.
Площадь сечения воздуховода определяется на основании геометрической
формы площади (круг, квадрат, прямоугольник).
Измерение скорости движения воздуха в воздуховоде производится с по-
мощью так называемых пневмометрических трубок, сооб-
щающихся с манометром. Непригодность обычных анемометров для указанной
цели объясняется тем, что при больших скоростях движения воздуха, кото-
рые имеют место в воздуховодах, показания обычных анемометров не соот-
ветствуют действительным скоростям.
Принцип применения пневмометрических трубок основан на
том, что они дают возможность определить так называемое динамиче-
ское давление, производимое движущимся потоком воздуха. Опре-
делив динамическое давление, находят по формуле скорость движения
воздуха.
Прежде чем перейти к описанию устройства пневмометрических трубок
и способа применения их, необходимо рассмотреть те сопротивления, которые
встречает движущийся по воздуховоду поток воздуха.
Различают динамическое давление (скоростной на-
пор) — йу, обусловленное сопротивлением, оказываемым воздушной средой
потоку движущегося воздуха; статическое давление — As, обу-
словленное трением воздуха о стенки воздуховода, и общее или суммар-
ное давление — ftg, равное алгебраической сумме динамического и
статического давлений: = hv + hs.
Динамическое давление зависит от скорости движения воздуха и его удель-
ного веса и выражается формулой:
где hv — динамическое давление,
v — скорость движения воздуха в м/сек.,
g — ускорение силы тяжести, равное 9,8,
7 — вес воздуха в кг/м3 при данных температуре, влажности и давлении.
451
На основании предыдущего равенства можно написать:
v
(2)
Так как в правой половине последнего равенства £ и 7 известны, то найдя
hv, т. е. скоростной напор, можно определить и, т. е. скорость движения
воздуха.
Итак, все сводится к определению скоростного напора. Пневмометриче-
ская трубка, сообщенная с манометром, и дает возможность определить ско-
ростной напор.
Пнев мом етричес-
кая трубка (рис. 262)
применяется в различных
модификациях. В ней раз-
личают короткую часть —
головку и длинную —
державку. Обе части
располагаются одна по
отношению к другой под
прямым углом. В пневмо-
метрической трубке, изоб-
раженной на рисунке, го-
ловка состоит из двух
трубок, из которых одна
вставлена в другую. Вну-
тренняя трубка сообщается
с наружным воздухом че-
рез отверстие а, а наруж-
ная — путем кольцевой ще-
ли Ь. В державке обе труб-
ки продолжаются в виде
двух параллельно распо-
ложенных трубок. У осно-
вания обе трубки заканчи-
ваются тубусами, причем
тубус трубки, служащей
продолжением внутренней
трубки головки, обознача-
ется знаком +, а тубус
трубки, являющейся про-
должением наружной трубки головки, — знаком —.
Если головку трубки направить против движущегося потока воздуха,
то через внутреннюю трубку будет передаваться суммарное давление дви-
жущегося воздуха, а через наружную — статическое. Следовательно, если
при помощи каучуковых трубок оба тубуса пневмометрической трубки сооб-
щить соответственно с двумя браншами манометра, то показания такого мано-
метра будут соответствовать динамическому давлению или скоростному на-
пору. В качестве манометра применяют часто тягомер.
Т я г о м е р (рис. 263) представляет собой манометр, состоящий из ре-
зервуара d и сообщающейся с ним капиллярной трубки Ь, поставленной под
небольшим углом к горизонтальной плоскости; чем этот угол меньше, тем
показания прибора точнее; наичаще угол равен 15о1.
Так как объем резервуара значительно более объема капиллярной трубки,
при определении давления можно не учитывать снижения уровня жидкости
1 В диференциональных манометрах угол можно менять по желанию.
452
в резервуаре, а делать отсчет лишь по перемещению жидкости в капиллярной
трубке. В качестве жидкости пользуются спиртом удельного веса 0,8, окра-
шенным фуксином в красный цвет. Шкала прибора указывает давление в мил-
лиметрах водяного столба, фактический размер каждого деления шкалы — более
1 мм. Если перемещение жидкости в тягомере при некотором давлении
обозначить через /, то при том же давлении в обыкновенном U-образном ма-
нометре разница уровней воды в обоих коленах выразится через /sin 15°. 0,8.
При работе тягомер должен быть подвешен или установлен в строго гори-
зонтальном положении, для чего в приборе имеется ватерпас. В этом положении
прибор устанавливается точно на нулевой точке шкалы, что достигается
передвижением шкалы и ее последующей фиксацией, а также подливанием
или отливанием в резервуар спирта.
d —резервуар, Ъ — капиллярная трубна, с и а —тубусы для каучуковых трубой, е — уровень.
Методика определения скорости движения воз-
духа в воздуховоде. Для определения скоростного напора вводят
головку пневмометрической трубки в небольшое отверстие, сделанное в стенке
воздуховода, при этом открытое отверстие головки должно быть направлено
против движущегося воздуха, а продольная ось головки должна быть парал-
лельна такой же оси воздуховода. При помощи каучуковых трубок соеди-
няют тубус, обозначенный знаком + с резервуаром тягомера, а тубус, обозна-
ченный знаком —, с концом капиллярной трубки прибора. При этих условиях
в резервуар тягомера будет передаваться суммарное давление движущегося
воздуха, а в капиллярную трубку — одно лишь статическое давление; таким
образом, показание тягомера будет соответствовать разности этих давлений,
т. е. динамическому давлению или скоростному напору.
Таких измерений делают не менее 5 в круглых воздуховодах и не менее 10
в прямоугольных, с продолжительностью отсчета колебаний в течение 15 сек.
Среднее значение для скоростного напора (Л„) находят по формуле:
[lv 1 ---------------------- I ,
\ п /
где й,,„ й„,... hv- — отсчеты по манометру,
п — число отсчетов.
Найдя среднее значение для hv, подставляют его в формулу (стр. 452) и, заме-
нив g и f соответствующими значениями, находят скорость движения возду-
ха (и). На основании полученного значения для v находят затем объем
воздуха, проходящего через данное сечение воздуховода.
Пример. Положим, что среднее значение для скоростного напора оказалось равным 13,
вес 1 м3 воздуха при данных условиях равен 1,29 кг, площадь сечения воздуховода равна
0,1 м2. Нужно определить объем воздуха, поступающего в помещение через воздуховод
в течение часа.
453
Подставив в формулу (стр. 452) вместо g, hv и ; соответствующие значения, находим
скорость движения воздуха:
/2 • 9,81 • 13 .
---------~ 14 м/сек.
1,29
Отсюда выводим, что за час через воздуховод поступает 0,1 • 14.3600 = 5040 мв воз-
духа.
Расчет вентиляционного воздуха
Количество воздуха, которое необходимо подать в каждом отдельном
случае в помещение, зависит от качества вводимого воздуха, тех норм, какие
желательно установить в помещении в отношении воздуха, и тех фактиче-
ских данных, какие имеются или каких можно ожидать, если подача воздуха
не будет иметь места.
В зависимости от того, на какой фактор воздушной среды (температурный,
влажности, газовый) имеется в виду оказать воздействие, расчет вентиляци-
онного воздуха будет различный. Рассмотрим отдельно указанные случаи.
Расчет по температуре
Количество вентиляционного воздуха при расчетах по температуре вычи-
сляется по формуле:
у_ УИ(1+а/)
0,306 (f — Q ’
где V — объем вводимого воздуха в м3,
W — избыточное количество тепла, подлежащее удалению и погло-
щению,
а — 0,00367,
t — санитарно допустимая норма температуры в помещении,
0,306 — теплоемкость для 1 м3 сухого воздуха,
t — температура наружного воздуха.
Из всех указанных данных, какие необходимо иметь для расчета, только W
требует отдельного расчета.
Здесь нужно иметь в виду следующее:
а) Количество конвекционного тепла, выделяемое нагретой поверхностью
(печи, котлы, машины и т. п.); вычисляется по формуле Ньютона:
Wk = Kk(t2 — t) . S кг-кал/час,
где Wk — конвекционное тепло,
Kjc — коэфициент конвекции, равный при температурах нагретой поверх-
ности 50—100°С — 6—7,5, а при температурах от 100 и выше—
7,5—8,0,
t2 — температура нагретой поверхности,
t — температура помещения,
s — нагретая поверхность в м2.
б) Количество лучистого тепла, выделяемого нагретой поверхностью,
вычисляется по формуле Ньютона:
ггл = кл (t2 — f) • s кг-кал/час,
где — лучистое тепло,
кл — коэфициент лучеиспускания, равный в среднем 3,6 х,
f2, t и s — имеют те же значения, что и в предыдущей формуле.
в) Количество тепла, выделяемого с поверхностей, температура которых
постепенно падает, вычисляется по формуле:
1VO = Р • С • (/3— кг-кал/час,
1 Значение для различных поверхностей см. в справочниках.
454
где Wo — количество выделяемого остывающим предметом тепла в кг-кал/час,
Р — вес остывающего предмета в кг,
G — теплоемкость,
t3 — начальная температура тела,
/4— конечная температура тела.
г) При расходе механической или электрической энергии:
1 л. с. дает 637 кг-кал/час,
1 квт » 860 »
д) Количество тепла, получаемое помещением через 1 м2 оконных пере-
крытий в солнечный ясный день в средних широтах, равно 150 кг-кал/час
для южных окон и 100 кг-кал/час для югозападных и юговосточных окон.
Для кирпичных стен при тех же условиях количество тепла, получаемое
помещением, равно 7,5—10 кг-кал/м2 час и для плоских перекрытий 15 —
20 кг-кал/м2 час. Для окон и стен, обращенных на восток и запад, количество
тепла, получаемое помещением, равно 2/3 указанных значений.
В зимнее время наружное охлаждение стен ведет к теплопотере зданий,
вычисляемой по формуле:
W0=*/(/-f1),
где Wo — теплопотеря внешними ограждениями в кг-кал/час,
/ — площадь поверхности в м2,
t — требуемая температура в помещении,
— температура наружного воздуха,
к — коэфициент общей теплопередачи внешнего ограждения (стр. 459).
е) Количество тепла, отдаваемого поверхностью тела человека всеми
путями, равное 100—НО кг-кал/час.
ж) Если вводимый в помещение воздух подвергается увлажнению, то
необходимо учесть и теплоту испарения воды, равную для килограмма воды
при 20°С 590 кг/кал.
Учтя все источники тепла и рассчитав количество выделяемого ими тепла,
находят суммарное количество продуцируемого тепла в данном помещении,
подставляют эту величину в основную формулу (стр. 454) как значение для W
и решают формулу.
Расчет по влажности
Количество вентиляционного воздуха при расчетах по влажности опре-
деляют по формуле:
V = —£—,
а —b
где* v — объем подаваемого воздуха в м3,
g — влажность, выделяемая в помещении, в г,
b — содержание влаги в г/м3 в приточном воздухе,
а — санитарно допустимая влажность в г/м3 в помещении.
Для вычисления g нужно иметь в виду следующее:
а) Количество влаги, испаряющейся с поверхности; вычисляется по фор-
муле:
tJ 45,6.FK(S1 —Sa)
В
где Н — вес испаряющейся влаги в кг/час,
F — поверхность испаряющейся жидкости в м2,
<$! — напряжение водяного пара в мм Hg при температуре испаряющейся
воды,
S, — напряжение водяного пара в мм Hg в окружающем воздухе,
В — барометрическое давление,
455
К — коэфициент, зависящий от скорости движения воздуха над поверх-
ностью испаряющейся жидкости. Он равен 0,55 для спокойного
воздуха, 0,71 при умеренном движении и 0,86 при сильном движении.
б) Количество влаги, выделяемое человеком при выполнении умеренного
физического труда; при благоприятных метеорологических условиях в сред-
нем оно равно 45 г;час.
Расчет по газовым выделениям
Количество вентиляционного воздуха при расчетах по газовым выделе-
ниям находят по формуле:
v= — ,
^2
где v — количество подаваемого воздуха в м3 час,
кг — количество газовых выделений, поступающих в воздух помещения
в мг/час,
к2 — допускаемая нормами концентрация газа в мг/м3 (для СО2 —
1000 мг/м3, для других газов — см. табл, на стр. 283).
Для вычисления кр нужно иметь в виду следующее:
а) Количество СО2, выделяемое при дыхании взрослым человеком в час,
равно в среднем 45 г; внешний воздух содержит СО2 0,6—0,8 г/м3.
б) Количество газовых выделений, поступающих в помещение; можно
определить по формуле:
а — т(ип — у),
где а — количество газовых выделений, поступающих в помещение в мг/час,
т — средняя концентрация газовых выделений в помещении, найден-
ная опытным путем в мг/м3,
ип — объем подаваемого воздуха в помещение в м3 час,
v — кубатура помещения в м3.
Для расчетов вентиляционного воздуха могут оказаться необходимыми
данные табл. 72.
Таблица 72
Выделение людьми углекислоты, тепла и влаги
Характер работы СО2 в л/час Тепловыделение в к-кал/час Вл аго вы- деление в г/час
явное | скрытое
Взрослые : а) При физической работе легкой . 25 80 100 160
» » » тяжелой 40 80 125 200
б) При спокойной работе (учрежде- ния, учебные заведения) .... 15 70 45 70 в
в) в спокойном состоянии (театры, кино и т. II.) 15 70 30 45
Дети до 12 лет 12 35 15 23
ВЕНТИЛЯЦИОННЫЕ НОРМЫ
Количество вентиляционного воздуха в час на одного человека в кубометрах должно
соответствовать следующим нормам: производственные помещения при отсутствии фак-
торов, вызывающих значительную порчу воздуха (пылеобразование, газообразование,
влаговыделепия, тепловыделения)—70; при наличии факторов, вызывающих значитель-
ную порчу воздуха, — соответственно расчетам; жилые квартиры — 40—75; аудитории,
театры — 20—30; учреждения, конторы — 20—42; классы для детей до 10 лет— 12—20;
классы для детей старше 10 лет — 20—30; палаты больничные для заразных больных —
100; палаты больничные для незаразных больных — 60—75; палаты больничные для детей —
35; кухни на один очаг — 200—300; уборные — 60—100.
В таблице 73 приводятся кратность вентиляционных обменов и расчетные внутренние
температуры жилых и гражданских помещений согласно нормам и техническим условиям,
утвержденным в августе 1948 г. (в извлечении).
456
Таблица 7 >
Расчетные внутренние температуры помещений и кратность вентиляционных
обменов в помещениях
Наименование помещений Расчетные темпера- туры Кратность вентиляци- онных обменов Примечание 1
Приток | Вытяжка
Жилые комнаты в квартирных до- мах и общежитиях Спальные и общие комнаты днев- 4-18 — 0,5
ного пребывания 4-18 — 1,0
Ванные 4“ 25 — 1,5
Кухни в квартирах + 15 — 3,0
Уборные в квартирах + 16 — 25 м3/час на
1 одно очко
Уборные в общежитиях 4-16 — 50 м3/час на
+ 18 одно очко
Конторские помещения — 1,0
Парикмахерские 4-18 — 1,5
Классы, лаборатории, мастерские, общие залы в школах +16 — 1,0 Кроме ме-
Физкультурный зал 4-15 — 1,0 стных от-
Боксы и изоляторы Душевые, ванные и раздевальные + 20 +25 — 1,0 1,5 сосов
при них Комнаты для кормления грудных
4-20 1,0
детей Детские комнаты в детсадах и
яслях 4-20 — 1,0
Уборные в школах на одно очко . +16 — 50 м3час
на один писсуар — 25 » »
Раздевальни верхней одежды . . . 4-18 — 1,0
Умывальные в детсадах и яслях Врачебные кабинеты в амбулато- 4-20 — 1,0
риях 4-20 2,0 1,5
Перевязочные 4-20 2,0 2,5
Операционные +2э 6,0 5,0
Ожидальные Кабинеты для рентгенодиагностики +20 2,0 2,0
и рентгенотерапии +22 5,0 7,0
Кабинеты в лабораториях + 18 1,0 3,0
Аптеки: приемная и рецептурная Обеденные залы в столовых с + 18 — 1,0
числом мест от 50 до 200 .... Кухни и горячие кондитерские +16 — 2,0
цехи +5 — 4,0
Заготовочные Обеденные залы в столовых с +16 — 1,0
числом мест 200 и более .... + 16 6 3
Хранение полуфабрикатов +5 — 1,0
Заготовка рыбы + 16 4,0 6,0
» мяса +16 3,0 4,0
» овощей +18 3,0 4,0
Кладовая сухих продуктов .... + 12 — 0,5
Кладовая картофеля и овощей . . Кладовая солений и кислой ка- +5 — 0,5
пусты +12 — 2,0
Магазины бакалейных товаров . . +12 — 1,0
Магазины мясных товаров .... +5 — 2,0
» молочных » .... +5 — 2,0
» скоропортящихся товаров +5 — 2,0
457
ОТОПЛЕНИЕ
Отопление жилищ имеет целью обеспечить постоянство соответствующей
температуры помещения в течение всего отопительного сезона, вне зависи-
мости от колебания температуры наружного воздуха.
Необходимый температурный режим см. табл. 73.
Кроме указанного основного условия к отоплению предъявляются сле-
дующие санитарно-гигиенические требования:
1. Продукты горения (СО2? СО, SO2 и др.) должны полностью выводиться
наружу, не проникая в помещение.
2. Подача тепла должна протекать равномерно и в такой степени, чтобы
поверхности отопительных приборов не нагревались выше 100°С, так как
при более высокой температуре может возникать возгонка и пригорание
пыли.
3. Влажность воздуха не должна падать ниже 30—35%.
4. Загрузка топлива и удаление золы не должны вызывать запыления
и загрязнения жилых помещений.
При обследовании местного отопления помещения выясняют состояние
отопительных приборов, их размеры, температуру нагрева, исправность
действия.
При обследовании воздушного отопления необходимо ознакомиться с со-
стоянием следующих сооружений: калориферной камеры (размеры, темпе-
ратура нагрева калориферов, температура воздуха); каналов, подающих
наружный воздух (расположение, размеры, защита от внешних загрязнений);
каналов, подающих нагретый воздух в помещение; каналов, выводящих
комнатный воздух. Кроме того, необходимо выяснить температуру подаваемого
воздуха в различные часы дня, его влажность, а в некоторых случаях и
химический состав.
При обследовании водяного и парового отопления выясняют вид данной
системы (давление, разводка воды или пара), состояние котельного поме-
щения (размеры, освещение, состояние котла), вид, размеры, расположе-
ние и состояние радиаторов (поверхность, температура нагрева, запыле-
ние и т. п.)
Во всех случаях при санитарно-гигиенических обследованиях отопления
выясняют температурный режим, обеспечиваемый отоплением, влажность
воздуха, наличие топочных газов и продуктов пригорания пыли.
Полезно рассчитать теплопотребность помещения и количество тепла,
подаваемого отопительной системой, сопоставляя полученные данные.
Расчет теплопотребности здания
Теплопотребность здания определяется его теплопотерями. Теплопотеря
здания вычисляется по формуле:
И'= к/(/„-/„).
где W — потеря тепла внешними ограждениями в кг-кал в 1 час,
/ — площадь теплоотдающей поверхности в м2,
t„ — требуемая температура воздуха внутри помещения,
tn — минимальная расчетная температура наружного воздуха,
К — коэфициент общей теплопередачи внешнего ограждения.
Из всех указанных компонентов формулы необходимо остановиться
на tn и К..
tn можно узнать на метеорологических станциях данной местности либо вы-
числить по формуле проф. В. М. Чаплина:
tn ---- tep 0,6 (tfn tcp'jt
458
где tn — минимальная расчетная температура наружного воздуха,
/ср — средняя температура самого холодного месяца,
tm — минимальная температура, когда-либо наблюдавшаяся в данной
местности.
Что касается К, то он может быть рассчитан по формуле либо взят из
готовых таблиц (см. напр., табл. 74)
Таблица 74
Коэфициент общей теплопередачи через ограждения
Материал и толщина ограждений
К
Деревянная рубленая стена без обшивки и штукатурки из бревен
толщиною 0,178 м ......................................... 0,44
Деревянная рубленая стена, внутри оштукатуренная по войлоку,
снаружи обшитая тесом 0,025 м............................. 0,32
Рубленая стена без обшивки и штукатурки из бревен толщиною
0,223 .................................................... 0,35
То же со штукатуркой внутри и обшивкой снаружи............ 0,26
Деревянный каркас из брусков 10 х 10, обшитый внутри и сна-
ружи тесом в 2,5 см по бумаге; засыпка опилками........... 0,66
Кирпичная степа в \у2 кирпича; толщина — 0,38 м; без штука-
турки .................................................... 1,3
То же, оштукатурена с обеих сторон....................... 1,2
Кирпичная стена в 2 кирпича; толщина — 0,51; без штукатурки 1,1
То же, оштукатурена с обеих сторон........................ 1,0
Стена в 2V3 кирпича; толщина — 0,64; без штукатурки....... 0,9
То же, оштукатурена с обеих сторон........................ 0,8
Стена из силикатного кирпича в 2 кирпича; толщина — 0,51; без
штукатурки . . .•................• •..................... 1,3
То же, оштукатурена с обеих сторон........................ 1,2
Стена из силикатного кирпича в 2у2 кирпича; толщина — 0,64; без
штукатурки .............................................. 1,1
То же, оштукатурена............................................ 1,0
Кирпичная стена с воздушной прослойкой.................... 0,6—0,75
Стена из трамбованного бетона; толщина—0,40........• . . . 1,9
То же, толщина — 0,50......................................... 1,7
То же, толщина — 0,60......................................... 1,6
То же с воздушной прослойкой в 8 см; толщина бетона — 0,40 . 1,3
То же, толщина бетона — 0,50.................................. 1,2
То же, толщина бетона — 0,60.................................. 1,1
Стена из шлаковых камней без штукатурки; толщина — 0,5' .... 0,6
Стена из камней легкого бетона (пемза), сложенная на извести,
оштукатуренная с обеих сторон; общая толщина — 27 см . . 0,44
Бетонная стена из шлаковых камней 25 х 25 х 50 см с двумя воз-
душными прослойками; оштукатурена с обеих сторон; общая
толщина — 26 см........................................... 0,55
Пол деревянный с накатом, засыпанный землей;толщина досок —
5 см, воздушная прослойка между полом и землей — 15 см,
слой земли — 14 см и накат из пластин — 1 м............... 0,34
Перекрытие по деревянным балкам; между балками подшивка
с заполнением, снизу оштукатуренная подшивка:..........
для потолка............................................. 0,5
для пола................................................ 0,3
459
Расчет печей
Количество тепла, которое должна получить печь в те-
чение часа топки, вычисляется по формуле:
w = ^-,
т
где IV — количество тепла в кг-кал, которое должна получить печь в тече-
ние часа,
Q— теплопотеря помещения за 1 час в кг-кал,
т — продолжительность топки в часах.
Размер теплоотдающей поверхности печи определяют по
формуле:
„ 0,66 • Q
F ---------,
Q
где F — теплоотдающая поверхность печи в м2,
Q — теплопотребность помещения при низшей расчетной температуре
наружного воздуха.
q—средняя расчетная часовая теплоотдача 1 м2 поверхности печи
в кг-кал.
Печи большой теплоемкости, облицованные изразцами, при одной двух-
часовой топке в сутки дровами отдают в течение одного часа с 1 м2 поверхности
в среднем 155 кг-кал, при топке углем — 175 кг-кал. Такие же печи, оштука-
туренные или в железном футляре, отдают соответственно 220 и 250 кг-кал.
Количество топлива, которое должно быть сожжено, вычисляется
по формуле:
где Р — количество топлива в кг,
IV — количество тепла в кг-кал., которое должна получить печь в течение
часа (см. выше).
у — коэфициент полезного действия печи, равный в среднем 0,7,
А — теплопроизводительность топлива (для дров — 3400, каменного
угля до 8000, кокса — 7000, мазута — 10 000, светильного газа—
10 600 кг-кал на 1 кг топлива).
ОСВЕЩЕНИЕ
Различают освещение естественное (дневное) и искусственное.
Световые понятия и единицы
Световым потоком называется та часть потока лучистой энергии,
которая воспринимается нашим глазом как световое ощущение.
Если в вершине телесного угла в один стерадиан 1 поместить источник
света силою в одну международную свечу (см. ниже), то световой поток внутри
стерадиана представит единицу светового потока, называемую люме-
ном (лм).
Сила света представляет собой частное от деления светового по-
тока на величину телесного угла при условии, что световой поток, заклю-
ченный в телесном углу, равномерно в нем распределен. Итак:
F
1 Стерадиан (единичный телесный угол) вырезает из сферической поверхности радиу-
сом в 1 м площадь в 1 м2.
460
где / — сила света,
F — световой поток,
со — телесный угол.
В 1909 г. за единицу силы света принята международная свеча
(св), представляющая собой приблизительно х/20 свечи В иол я1. Под свечей
Виоля, как известно, понимают силу света, испускаемого с 1 см2 поверхности
расплавленной платины в момент ее затвердения.
Эталоны международной свечи осуществляются
при помощи электрических ламп. С 1925 т. поста-
новлением ВСНХ СССР у нас введена для свето-
технических измерений международная свеча как
обязательная единица силы света.
Освещенность представляет собой по-
верхностную плотность падающего светового по-
тока при условии равномерного его распределе-
ния по поверхности. Освещенность определяется
уравнением:
где Е — освещенность.
F — световой поток,
S — поверхность.
За единицу освещенности принимают люкс
(лк), равный освещенности поверхности, на каж-
дый м2 которой падает и равномерно распреде-
Рис. 264. Схема, показы-
вающая зависимость между
международной свечой, лю-
меном и люксом.
Е — международная свеча,
О — стерадиан; световой поток,
заключенный внутри стерадиа-
на, равен люмену (лм), ABCD—
сферичная поверхность в один
квадратный метр; ее освещен-
ность равна одному люксу (лк).
ляется один люмен.
Между освещенностью и силой света существует зависимость, выражаю-
щаяся уравнением:
Е = — cos а,
R2
где Е — освещенность,
/ — сила света,
R — расстояние, между освещенной поверхностью и источником света,
а — угол между нормалью и направлением светового потока.
На рис. 264 дана схема, показывающая зависимость между международной
свечей, люменом и люксом.
Под яркостью освещения понимают отношение силы света к видимой
светящейся поверхности данного источника света, выраженной в см2.
За единицу яркости принимают стильб (сб), равный яркости поверхно-
сти, излучающей в перпендикулярном направлении с каждого см2 одну между-
народную свечу (1 св/см2).
Зависимость между яркостью, светящейся поверхностью, силой света
и направлением светового потока выражается уравнением:
В = — cos а,
S
где В — яркость освещения,
/ — сила света,
S — поверхность,
а — угол между нормалью и направлением светового потока.
1 Точность до 2%.
4С1
ЕСТЕСТВЕННОЕ ОСВЕЩЕНИЕ
При обследовании естественного освещения выясняют:
1. Размеры световой поверхности окон, фонарей, их форму, расположение,
состояние стекол в отношении чистоты (прозрачности).
2. На какую сторону света обращены окна и на каком этаже находится
данное помещение.
3. На каком расстоянии от окон находятся соседние строения, деревья
и т. п.
4. Видно ли с данной рабочей поверхности небо и попадают ли солнеч-
ные лучи в помещение.
5. Коэфициент отражения поверхностей соседних строений, а также
стен и потолка данного помещения.
6. Относительную площадь остекления или световой коэфициент.
7. Размеры углов падения и отверстия.
8. Глубину заложения данного помещения.
9. Размер пространственного угла.
10. Освещенность рабочих мест в люксах.
И. Равномерность освещения.
12. Коэфициент естественной освещенности.
Определения, указанные в пунктах 1, 2,3, 4 и 5, не нуждаются в пояснении.
Переходим к описанию других определений.
Коэфициент отражения
Коэфициент отражения данной поверхности представляет
процентное отношение светового потока, отраженного от поверхности, к све-
товому потоку, падающему на эту поверхность. Коэфициент отражения опре-
деляет, таким образом, способность поверхности отражать падающий на нее
свет.
Размер косфициента зависит в основном от цвета данной поверхности
и равен:
для белой эмали и молочного стекла....... 65—80%
» чисто выбеленной стены................. 75 »
» светложелтой или светлозеленой мело-
вой окраски................................ 66 »
» коричневой окраски..................... 40 »
» желтых обоев........................... 40 >
» голубых и серых обоев.................. 25 »
» темнокоричневых обоев.................. 13 »
> чернобурой окраски...................... 4 »
» черной окраски........................ 1,2 »
Методика определения коэфициента отражения приведена ниже (стр. 470).
Относительная площадь остекления (световой коэфициент)
Под относительной площадью остекления или
световым коэфициентом понимают отношение остекленной по-
верхности к площади пола данного помещения. При определении остеклен-
ной поверхности измеряют одни лишь стекла; поверхность оконных перепле-
тов не должна входить в остекленную поверхность. Измеряют лишь ту осте-
кленную поверхность, которая приходится выше рабочей поверхности. Нормы
светового коэфициента см. ниже.
Световой коэфициент дает лишь очень приближенное представление о сте*
пени освещенности данного места.
462
Углы падения и отверстия
Под углом падения для данной точки понимают угол, образо-
ванный горизонтальной линией, проходящей через наблюдаемую точку,
к окну и другой линией, идущей от данной же точки касательно к верхнему
краю окна. Этот угол для рабочей поверхности не должен быть менее 27°.
Под углом отверстия
понимают угол, образованный двумя
прямыми, из которых одна идет от
данной точки касательно к наивысшей
точке противоположного строения
(дерева и т. п.) и другая — касатель-
но к наивысшей точке окна. Угол
отверстия для рабочей поверхности
должен быть не менее 4°.
Рис. 266. Угломер.
Рис. 265. Углы падения и отверстия.
а — угол падения, аг — угол отверстия.
Углы падения и отверстия могут быть вычислены математически на осно-
вании несложных измерений или определены инструментально при помощи
угломера.
Из чертежа 265 видно, что tg угла падения а может быть определен
из уравнения:
. тп
tga =-----.
пр
По tg а находят а.
Что касается угла отверстия аи то он определяется из равенства:
аг — а — Ь;
tg угла b находят из уравнения:
По tg b находят Ь.
Для инструментального измерения углов падения и отверстия может
служить угломер, изображенный на рис. 266.
Глубина заложения
Под глубиной заложения понимают отношение где В — расстояние
между наружной поверхностью стены и наиболее удаленной от окна точкой
помещения, а Н — высота от пола до верхнего края окна. В целях сохране-
ния надлежащего естественного освещения глубина заложения помеще-
ния должна быть не свыше 2,5, (С. И. Ветошкин).
46.3
Пространственный угол
Для гигиенической оценки дневного освещения определяют иногда
размер того участка небесного свода, который посылает свои лучи к дан-
ному месту.
Небесный свод представляют себе разделенным на равные квадраты, сто-
рона которых равна длине дуги большого круга в 1°; эти квадраты и называ-
ются квадратны ми градусами. Длина стороны квадратного
2кг 1 / 2кг |2 р
градуса равна: , а площадь квадратного градуса равна: „-^-1 . Сле-
довательно, число квадратных градусов во всей небесной сфере равно:
/ 2~г \2 4т?г2 • 3602
4кг2 . / = 273.
\ 360 / 4к2 • г2
Рис. 267. Пространственный угломер
А — подставка, В — экран, С — угломер, U — лупа.
Если от данной точки провести линии, а через них плоскости к четырем
углам квадратного градуса, то получится пирамида, представляющая собой
пространственный угол
в 1°. Вершина пирамиды — ра-
бочее место, основание — квадра-
ный градус небосвода.
Размер пространственного
угла данного рабочего места
определяют при помощи прибо-
ра, называемого простран-
ственным угломером
(рис. 267). Прибор состоит из
деревянной подставки Д, снаб-
женной ватерпасом и двумя
винтами, при помощи которых
подставка устанавливается стро-
го горизонтально. На подставке установлен экран В, вращающийся на
горизонтальной оси; угол вращения отсчитывается по шкале С. На экран
помещают кружок бумаги, разлинеенный на квадратики, сторона которых
равна 2 мм. Перед экраном на металлическом стержне, прикрепленном к
нему, находится двояковыпуклая лупа D, которая может перемещаться по
стержню. На стержне имеются деления. Лупа устанавливается на расстоянии
11,459 см от экрана. При таком расстоянии лупы от экрана каждый квадрат-
ный градус небосвода в проекции на экране равен одному малому квадратику
со стороной в 2 мм. Поставив прибор на рабочем месте, направляют лупу
в сторону окна, стараясь получить на разлинеенной бумаге совершенно ясное
изображение неба. Если нужно, поворачивают при этом экран по оси. Полу-
чив ясное изображение небесного свода на экране, обводят это изображение
карандашом и подсчитывают число квадратиков; оно и соответствует числу
градусов пространственного угла, если экран стоит вертикально; если же
пришлось экран повернуть на некоторый угол, то результат подотчета умно-
жают на синус угла отклонения экрана от вертикали. Хорошо освещенное
рабочее место должно иметь не менее 50 редуцированных квадратных градусов.
Для мест, значительно удаленных от окна, пространственный угол не
может служить хорошим показателем освещенности.
Освещенность
Освещенность рабочих мест в люксах как при естественном, так и при
искусственном освещении измеряется при помощи приборов, носящих назва-
ние фотометров (люксметров). Из предложенного большого числа фотометров
здесь описаны лишь те, которые наичаще применимы.
1 г — здесь радиус большого круга
464
Рис. 268. Фотометр упрощенной
конструкции.
А — винт для регулировки высоты пламе-
ни, В — окошечко в ящике фотометра,
С — кнопка для поворачивания внутрен-
него экрана, L) — шкала прибора, Е —
наружный экран, F — зрительная трубка
с окуляром, G — пластинка с затем ците-
лями, К — пластинка с цветными свето-
фильтрами.
Фотометр упрощенной конструкции
Фотометр (рис. 268) представляет собой небольшой четырехугольный
ящик, окрашенный снаружи и внутри черной матовой краской и закрытый
со всех сторон. Внутри прибора установлена маленькая бензиновая лампочка,
зажигаемая во время исследования. Высота пламени должна равняться 20 мм,
что достигается регулированием при помощи винта Д. В стенке фотометра,
недалеко от лампы, имеется окошечко В с нанесенной на нем поперечной
чертой. Если пламя имеет надлежащую длину, то кончик его должен совпа-
дать с указанной чертой. Внутри прибора имеется экран из белого картона.
При помощи кнопки С можно поворачивать экран по горизонтальной оси,
благодаря чему он оказывается то более, то менее освещенным лампой при-
бора. Освещенность экрана определяется по шкале D и колеблется в пределах
от 10 до 50 люксов. Снаружи фотометра имеется еще один экран Е из белого кар-
тона. По освещенности этого экрана судят об освещенности данной поверхности.
Зрительная труба F устроена так, что,
смотря в нее, можно видеть одновременно
оба экрана. Поле зрения представляется
в виде круга, разделенного на два полу-
круга. Если смотреть в трубу со стороны
прибора, где находится его шкала, то
правая половина круга соответствует
внутреннему экрану, левая — наружному.
Установив фотометр в данном месте, смот-
рят в зрительную трубу и поворачивают
при помощи кнопки внутренний экран,
стараясь добиться одинакового освещения
обеих половин поля зрения. Если такое
фотометрическое равновесие получено, то
освещенность рабочего места соответствует
освещенности внутреннего экрана и отсчи-
тывается непосредственно по шкале при-
бора D.
Если освещенность рабочего места вы-
ше 50 люксов, то не удается получить
одинаковое освещение наружного и внут-
реннего экранов. В этом случае для изме-
рения освещенности данного места прибе-
гают к затемнителям (светофильтрам),
вправленным в подвижную пластинку G. Затемнители прибора представляют
собой дымчатые стекла, которые, в зависимости от интенсивности окраски
стекла, пропускают х/2, х/5 или х/10 падающего на них светового потока. Они
обозначаются коэфициентом 2, 5 и 10.
В зависимости от того, какой затемнитель пришлось подвести под зритель-
ную трубу для получения фотометрического равновесия, умножают результат
отсчета по шкале прибора на коэфициент данного затемнитиля.
При исследовании дневного или электрического освещения не удается
описанным способом получить фотометрическое равновесие ввиду того, что
наружный экран дает белое освещение, в то время как освещение внутрен-
него экрана имеет желтоватый оттенок.
В этом случае для измерения освещенности прибегают еще к цветным
светофильтрам — зеленому и красному, вправленным в подвижную пла-
стинку К. Определяют освещенность при зеленом и красном светофильтре.
Результат измерения, полученный при зеленом светофильтре, делят на ре-
зультат измерения при красном светофильтре.
Для полученного частного находят по таблице 75 соответствующий мно-
житель М, на который нужно помножить результат измерения при красном
светофильтре, чтобы получить освещенность данного места в люксах.
30 А. И. Бурштейн.
465
Таблица 75
Значения для множителя М
Зелен. Красн. М Зелен. Красн. м | Зелен. Краев. М 1 Зелен. Краев. М Зелев. Красн. М
0,1 0,23 1,2 1,15 2,3 1,75 3,4 2,15 4,5 2,47
0,2 0,38 1,3 1,22 2,4 1,80 3,5 2,18 4,6 2,49
0,3 0,50 1,4 1,28 2,5 1,84 3,6 2,20 4,7 2,52
0,4 0,56 1,5 1,34 2,6 1,88 3,7 2,24 4,8 2,55
0,5 0,64 1,6 1,40 2,7 1,92 3,8 2,27 4,9 2,57
0,6 0,72 1,7 1,46 2,8 1,96 3,9 2,30 5,0 2,60
0,7 0,80 1,8 1,50 2,9 1,99 4,0 2,33 5,1 2,62
0,8 0,87 1,9 1,55 3,0 2,02 4,1 2,36 5,2 2,64
0,9 0,94 2,0 1,60 3,1 2,05 4,2 2,39 5,3 2,67
1,0 1,00 2,1 1,65 3,2 2,08 4,3 2,41 5,4 2,69
1,1 1,08 2,2 1,70 3,3 2,11 4,4 2,44 5,5 2,71
Пример 1. При измерении освещенности рабочей поверхности фотометрическое равно-
весие получено при показании по шкале 25 и применении затемнителя с коэфициентом 10.
Так как освещение в данном случае было керосиновое, то цветных светофильтров при фото-
метрии на пришлось применить.
В этом случае освещенность рабочей поверхности равна 25 х 10 = 250 люксам.
Пример 2. При измерении освещенности рабочей поверхности при зеленом светофильтре
результат оказался равным 45 люксам и при красном — 18 люксам. Нужно найти освещен-
ность данной поверхности.
к зелен. 45 _ _ „ КЛ
Находим отношение:-------= -77; = 2,5. Соответственно данному отношению М по
красн. 18 J
таблице равно 1,84. Следовательно, освещенность данного места равна 18 х 1,84 = 33,32
люксам.
Фотометр для точных измерений
Фотометр (рис. 269 и 270) состоит из металлического стержня S, который
ввинчивается в крышку деревянного ящика, трубы А, неподвижно при-
крепленной к стержню, и трубы В, присоединенной перпендикулярно к трубе А
и вращающейся по ее оси в вертикальной плоскости. По угломеру s можно
судить об отклонении трубы В от горизонтальной плоскости. На свободном
конце трубы А имеется фонарь, в котором помещена бензиновая лампа h
в форме свечи. Высота пламени лампы регулируется при помощи винта g.
Внутри неподвижной трубы имеется экран из молочного стекла, перемеща-
ющийся вдоль трубы при помощи винта Z. Расстояние между экраном и лам-
пой определяется по шкале е. Внутри подвижной трубы В против трубы А
вставлена оптическая система, изображенная на рис. 271.
Если смотреть в окуляр О трубы В, то поле зрения представляется поде-
ленным на два концентрических круга: средний круг получает освещение
от исследуемого места, перифирический — от бензиновой лампы (рис. 272).
Вблизи окуляра к нему приделана призма к с полным внутренним отражением,
при помощи которой можно видеть поле зрения при всяком положении трубы В,
если смотреть на призму сверху вниз, что облегчает наблюдение. Позади
окуляра помещается пластинка / с цветными светофильтрами — зеленым и
красным. Противоположный конец трубы В снабжен камерой (кассетой) d,
в которую по мере надобности вставляют пластинки (светофильтры) молоч-
ного или матового стекла. Коэфициент светопоглощения каждой пластинки
устанавливается учреждением, изготовляющим приборы, и указывается
466
в приложении-инструкции к каждому прибору. К фотометру прилагается
экран из матовобелого картона размером 20 х 20 см, закрепленный
на штативе.
Рис. 269. Фотометр для точных измерений.
S —стержень, А —труба неподвижная, В —труба подвижная, s — угломер, h — бензиновая лампа,
g — винт для регулирования высоты пламени, I — винт для перемещения молочного экрана, е — шкала.
о — окуляр, к — призма, / — пластинка с цветными светофильтрами, d — камсра-кассета, С — кар-
тонный экран.
При помощи описанного фотометра можно производить измерения осве-
щенности данной поверхности, а также определять силу света того или иного
точкообразного источника света. .
Былов внес существенные усовер-
шенствования в описанный прибор: бен-
зиновая лампа заменена четырехвольто-
вой электрической лампочкой, соеди-
ненной с точным амперметром или вольт-
метром и питаемой аккумуляторами;
в конце подвижной трубы помещено
молочное стекло, под которым располо-
жен горизонтальный диск с разного раз-
мера диафрагмами; вращением диска
можно уменьшить размер светового по-
тока, попадающего в фотометр, что
избавляет от необходимости пользовать-
ся матовыми и молочными светофильт-
рами.
Измерение освещенности поверхнос-
тен. Устанавливают картонный экран
в том месте, освещенность которого нуж-
но измерить, придав экрану желаемое
положение. Зрительную трубу устанав-
ливают на центр экрана. При этом,
Рис. 270. Фотометр для точных измере-
ний (схема).
еСЛИ ОСВещенНОСТЬ данного места на- F— оптическая система; остальные обозначе-
столько интенсивна, что приходится при- нпя те ше’ что и ” ₽|,с‘ 269'
менять при фотометрировании затемнители, то эти последние не должны
467
отстоять от картонного экрана более чем на 24 см. Продольная ось зритель-
ной трубы должна иметь направление, перпендикулярное к плоскости кар-
тонного экрана или близкое к нему. Пламя свечи должно быть длиною в 20 мм,
в чем убеждаются, смотря в окошечко фонаря. При надобности длину пламени
можно отрегулировать при помощи винта g. При этих условиях установки
фотометра, производят наблюдение, смотря в зрительную трубу. Перемещая
при помощи винта / экран из молочного стекла внутри трубы В, стараются
добиться исчезновения границ между центральным
и периферическими кругами поля зрения. Если это
не удается, то прибегают к затемнителям.
Освещенность данного места определяется по фор-
s' муле:
Рис. 271. Схема оптичес-
кой системы прибора.
где Е — освещенность данного места в люксах,
С — константа, имеющая разное значение в за-
висимости от того, производилось ли изме-
рение с затемнителями или без них; зна-
чение констант имеется при каждом фото-
метре,
г — число делений по шкале фотометра.
Пример. Положим, что значение для С при данном затемнителе равно 0,2346;
г — 8. При этих условиях:
Е = 0,2346 -1-0-02- = 36,656 люкса.
82
В том случае, когда освещение картонного экрана отличается по своему
оттенку от освещения бензиновой лампы, применяют цветные светофильтры.
Последние подводят позади окуляра при помощи передвижной пластинки /.
Производят измерение при зеленом и красном светофильтре. Находят отно-
шение полученных двух результатов измерения, а затем по таблице 75 находят
соответствующий множитель М. На него умножают
результат, полученный при измерении красным свето-
фильтром, и получают освещенность данного места в
Пример. Положим, что константа для данного затемнителя
равна 0,4826; г при измерении с зеленым светофильтром равно 8,
а при измерении с красным светофильтром— 10. Нужно на осно-
вапии этих данных найти освещенность исследуемой поверхности.
Подставив приведенные данные в указанную выше формулу,
получаем для зеленого стекла:
0,4826 . — = 75,40625,
Д-’1Я красного: Рис. 272 Вид поля
0,4826------= 48,26. зрения в фотометре.
102
Отношение первого результата ко второму равно
75,40625
48,26
По приведенной
выше таблице для 1,5 М равно 1,34, а для 1,6 М равно 1,4. Интерполируя, находим
для 1,56 М равным 1,376. Помножив результат, полученный при измерении красным
светофильтром, т. е. 48,26 на 1,376, находим освещенность данного места равной 66,4 люкса.
Измерение силы света источника освещения. Исключив посторонний свет,
устанавливают зрительную трубу по направлению к источнику света. Поме-
щают в камеру затемнитель молочного стекла № 3 и обеспечивают соответст-
вующий размер пламени бензиновой лампы. Перемещением экрана в трубе
468
стараются добиться одинакового освещения центрального и периферического
кругов поля зрения. Если нужно, помещают в камеру дополнительно другие
затемнители. Силу света испытуемого источника определяют по формуле:
где / — сила света в свечах,
С3 — коэфициент затемнителя,
R — расстояние от молочного стекла до источника света; для простоты
расчетов это расстояние устанавливают равным 100 см,
г — показание шкалы фотометра.
Пример. Положим, что С3 = 0,46, R = 100 см, а г = 10 см. Отсюда силу света дан-
ного источника находим равной:
0,46 • 29 — = 46 свечам.
102
Ж
Люксметр ГОИ
Люксметр ГОИ (Государственного оптического института), построенный
проф. Майзелем, (рис. 273 и 274) состоит из двух основных частей: собственно
фотометра А и ящика В, в ко-
тором находятся два никель-
кадмиевых аккумулятора со
щелочным электролитом, вольт-
метр V и реостат г. Часть А по
окончании исследования укла-
дывается в ящик В в специаль-
ное гнездо.
Собственно фотометр А пред-
ставляет собой небольшую ко-
робку, снабженную с одной
стороны окуляром а, а с дру-
гой—выдвижной металлической
пластинкой, на которой укреп-
ляется белый гипсовый экран о.
Внутри ящика находится дру-
гой экран к, который при по-
мощи кнопки может быть установлен в различное положение относительно
небольшой электрической лампочки п, помещающейся в коробке и зажигае-
мой во время исследования. Лампочка питается током от указанных выше
аккумуляторов. Ее зажигание производится посредством нажатия кнопки /.
Внутри коробки имеются еще два экрана: t и s, назначение которых —
передача освещения с экрана к в глаз наблюдателя. В коробке имеется око-
шечко /, против которого внутри прибора могут быть установлены дымчатые
фильтры х и z с коэфициентом 1000 и 100. При приборе имеются еще два
дымчатых светофильтра в виде насадок, надевающихся на окошечко сна-
ружи, с коэфициентом 5 и 10.
Между лампой и экраном к могут быть установлены светофильтры: серый d
или голубой е. Серый светофильтр d устанавливается в тех случаях, когда
освещенность внешнего экрана менее освещенности внутреннего. Голубой
светофильтр е устанавливается в тех случаях, когда внешний и внутренний
экраны освещаются неодинаковым по оттенку светом, например, при измере-
нии естественного освещения. Снаружи коробки имеется шкала F с деле-
ниями, соответствующими люксам.
При помощи люксметра ГОИ можно определить освещенность, коэфи-
циент отражения данной поверхности и яркость источников света.
469
Измерение освещенности поверхностей. Поместив люксметр в исследуемом
месте, включают лампу и при помощи реостата устанавливают по вольтметру
то напряжение тока, при котором нужно пользоваться данным прибором.
Это напряжение указано в паспорте прибора.
Если смотреть в зрительную трубу, то поле зрения представляется поде-
ленным на два полукруга: верхний полукруг соответствует внутреннему
экрану, нижний — наружному. Поворачивая кнопку, стремятся добиться
фотометрического равновесия. При этом, как правило, рекомендуется полу-
чать отсчеты по шкале в пределах 10—20 лк, вводя фильтры с возможно
Рис. 274. Схема люксметра ГОИ.
а — окуляр, о — гипсовый экран, к — внутренний экран, п — электри-
ческая лампочка, / — кнопка для зажигания лампочки. In s —дополни-
тельные экраны, I — окошечко, х и z — дымчатые фильтры, d — серый
светофильтр, е — голубой светофильтр.
малым поглощением. Если при фотометрировании вводят дополнительно
голубой светофильтр, то учитывают и его коэфициент.
Освещенность вычисляется по формуле:
Е = А • к • кг • fc2,
где Е — освещенность,
А — отсчет по шкале,
к, кги к2 — коэфициенты введенных светофильтров.
Пример 1. Положим, что при измерении освещенности были введены фильтры: дымчатый
с коэфициентом 100, и голубой, с коэфициентом 0,25. Отсчет по шкале оказался равным
20 лк. В этом случае освещенность данного места:
Е = 20 . 100 • 0,25 = 500 лк.
Пример 2. При измерении освещенности введен серый светофильтр с коэфициентом
0,1. Отсчет по шкале — 20. Освещенность:
Е = 20 . ОД = 2,0 лк.
Область измерений освещенности при помощи люксметра ГОИ лежит
в пределах от 0,1 до 600 000 лк. Точность измерений практически достаточна:
предел ошибки 8—10%.
Измерения коэфициента отражения поверхности. Производят два изме-
рения освещенности данного места: одно при условии удаления наружного
экрана, а другое—при наличии наружного экрана, установленного парал-
лельно испытуемой поверхности. Коэфициент отражения вычисляется по
формуле:
Е • л 100
, '—г~'
470
где г — коэфициент отражения данной поверхности,
Е — освещенность данной поверхности при отсутствии экрана,
Ег — освещенность данной поверхности при наличии экрана,
гг — коэфициент'отражения экрана, указанный в свидетельстве прибора.
Пример. Показание без экрана равно 7,6, при наличии экрана—102. Коэфициент
отражения экрана, согласно свидетельства, равен 0,86. Отсюда коэфициент отражения дан-
ной поверхности равен:
7,6 • 0,86 -100 „ . ,
-------------= 6,4о/о.
102
Измерение яркости. Не надевая наружный экран, наводят люксметр на
испытуемую поверхность под тем углом, под которым желательно измерить
яркость, и известным уже образом получают фотометрическое равновесие.
Яркость определяется по формуле:
В = А • • к2 • С,
где В — яркость,
А — отсчет по шкале,
и Аг2—коэфициенты введенных фильтров,
С — коэфициент для измерения яркости, указанный в паспорте прибора.
Пример. При измерении яркости колпака арматуры «Универсаль» отсчет по шкале
оказался равным 14,2; коэфициент дымчатого светофильтра—1000; коэфициент для определе-
ния яркости, согласно свидетельству, — 21 . 10—в. Какова яркость в стильбах?
Подставляя указанные значения в формулу, получаем:
В = 14,2 • 1000* 21 • 10-6 = 0,298 св /см2 или стильба.
После окончания работы головку реостата поворачивают до возвращения
стрелки вольтметра к нулю. По истощении аккумуляторов1 они заряжаются
вновь постоянным током силой 2,5 А в течение б часов. Окончив зарядку, дают
газам выделяться из электролита в течение 12 часов, после чего завинчивают
запорные винты. Электролит должен меняться раз в год. Пластинки аккуму-
лятора должны быть покрыты слоем электролита высотою в 5—12 мм. При
порче лампы люксметр должен быть направлен в ГОИ для установки новой
лампы и проверки прибора. Для пересылки прибора аккумуляторы разряжают,
выливают электролит, отверстия плотно закрывают резиновыми пробками
и тщательно смазывают крышку густым вазелином.
Фотоэлектрический люксметр
Рис. 275. Фотоэлект-
рический фотометр.
Прибор (рис. 275) состоит из двух соединенных между собой четырех-
угольных коробок. В верхней коробке заключен фотоэле-
мент, представляющий собой существенную часть при-
бора: в нижней коробке помещается чувствительный
гальванометр.
Под влиянием освещения фотоэлемент вырабатывает
электрический ток, сила которого пропорциональна *
интенсивности освещения. Фотоэлемент соединен про-
водами с гальванометром. Под действием вырабатывае-
мого электрического тока происходит большее или
меньшее отклонение стрелки гальванометра по шкале,
которая градуирована в люксах.
Освещенность интенсивностью До 250 люксов опреде-
ляют по нижней шкале. При помощи переключателя
можно расширить предел фотометрических измерений
от 1000 до 5000 (верхняя шкала).
Методика применения прибора весьма проста. Его берут в руки, как по-
казано на рис. 275, и, поместив на исследуемое место, отбрасывают щиток,
1 Зарядку следует производить, когда напряжение падает до 1,8 вольта.
471
закрывающий окно фотоэлемента. Стрелка люксметра немедленно откло-
няется, показывая освещение в люксах.
Достоинством прибора является объективность и быстрота измерений;
недостатком его является неодинаковая чувствительность к свету различной
длины волны.
Равномерность освещения
При исследовании равномерности освещения определяют освещенность
в различных точках данного помещения и находят число точек с достаточным
и недостаточным освещением. Результат выражают в процентах по отношению
к общему количеству исследованных мест.
Для выполнения исследования рекомендуется составить картограмму, на
которой площадь помещения изображена разделенной на квадратные метры
(при определенном масштабе). На каждом квадрате указывают силу освещения
соответствующего места.
Коэфициент естественной освещенности
Под коэфициентом естественной освещенности (КЕО) по-
нимают процентное отношение освещенности естественным светом данной
рабочей точки внутри помещения к горизонтальной освещенности, измерен-
ной в то же самое время под открытым небом (поверхность защищают от
прямых солнечных лучей).
Коэфициент естественного освещения может дать представление об осве-
щенности данной рабочей точки по часам дня в течение года, если знать го-
ризонтальную освещенность под открытым небом в то же время года.
Среднюю горизонтальную освещенность по часам дня в течение всего
года (по месяцам) называют световым климатом данной точки
земной поверхности. У нас в ряде городов ведется изучение светового кли-
мата путем непрерывной регистрации диффузной освещенности посредством
гальванографов.
Зная световой климат данного места и коэфициент естественного осве-
щения данной рабочей поверхности, можно предусмотреть, будет ли освещен-
ность данной рабочей поверхности достаточной в те или иные часы на протя-
жении года \
Пример. По характеру работы освещенность данной рабочей поверхности должна
равняться 100 лк. КЕО этой поверхности равен 2%. Нужно определить, будет ли освещен-
ность в полдень в январе достаточной для данной рабочей поверхности, если, согласно гра-
фику светового климата, горизонтальная освещенность под открытым небом в указанное
время равна 3200 лк.
На основании приведенных данных находим, что в указанное время освещенность рабо-
3200 • 2
чего места окажется равной - .— = 64 лк. Следовательно, одного естественного осве-
щения окажется недостаточно и понадобится еще искусственное освещение, обеспечиваю-
щее дополнительно 100— 64 = 36 лк.
ИСКУССТВЕННОЕ ОСВЕЩЕНИЕ
При обследовании искусственного освещения выясняют:
1. Количество световых источников (ламп), их силу, распределение, высоту
подвеса. 2. Характеристику осветительной арматуры. 3. Освещенность.
4. Равномерность освещения. 5. Яркость.
Из всех указанных пунктов нужно остановиться лишь на характеристике
осветительной арматуры, так как все другие вопросы рассмотрены уже выше.
1 В порядке предварительного надзора соответствие КЕО нормам может быть установ-
лено графическим методом по Р. Н. Данилюку (Труды 2-й Всесоюзной светотехнической
конференции, т. Ill, 1931).
472
Осветительная арматура
Под осветительной арматурой понимают приспособления, которыми снаб-
жается голая лампа, для защиты глаз работающих от блесткости, защиты
лампы от механических повреждений и загрязнений и для соответствующего
направления светового потока. Голая лампа вместе с арматурой носит название
светильника.
Основные гигиенические требования к осветительной арматуре могут быть
сведены к следующим: осветительная арматура должна обладать достаточным
защитным углом, хорошим коэфициентом полезного действия и давать соответ-
ствующее перераспределение светового потока.
Защитимы угол арматуры
Под защитным углом арматуры понимают угол между горизон-
тальной линией, лежащей в плоскости светящейся нити, и линией, проведенной от
края колпака до противоположного края нити лампы. В пределах защит-
ного угла светящаяся нить закрыта от глаз непрозрачным или малопро-
зрачным краем арматуры и глаз защищен от слепящего действия источ-
ника света. На рис. 276 защитный угол показан
в виде угла АВС (а). Д
Из чертежа видно, что: /д\
АС _ BD _ BD
АВ — CD ~ CE^-ED '
Обозначив BD через h, СЕ через 7? и ED через г,
получаем:
Рис. 276. Защитный угол
арматуры.
где h — расстояние от светящейся нити до плоскости, проходящей через
края колпака,
R — радиус колпака,
г — радиус тела накаливания (светящаяся нить).
Коэфициент полезного действия арматуры
Коэфициент полезного действия арматуры представляет отношение свето-
вого потока, вышедшего из арматуры, к световому потоку, излучаемому лам-
пой. Коэфициент полезного действия арматуры менее единицы и выражается
либо в виде правильной дроби, либо в процентах.
Обычный коэфициент полезного действия арматуры лежит в пределах
50—80%.
Кривая светораспре деления
Характер светораспределения светильника изображают графически в виде
кривой в полярной системе координат. От некоторой точки, представляющей
начало координат и совпадающей со световым центром светильника, откла-
дывают по радиусам-векторам в определенном масштабе силу света светиль-
ника в данном направлении. Соединив концы отрезков непрерывной линией,
получают кривую светораспределения светильника в вертикальной плоскости.
Для удобства отсчета силы света в различных направлениях проводят из
начала координат, как из центра, ряд окружностей на определенном рассто-
янии одна от другой, соответствующем в масштабе определенному количеству
свечей.
Кривая светораспределения строится обыкновенно по одну сторону от
вертикальной оси светильника, так как у большинства светильников обе
половины кривой светораспределения тождественны.
473
На рис. 277 дана кривая светораспределения для светильника Люцета.
Рис. 277. Кривая светораспределения для арматуры «Люцета».
1 — осветительная арматура <Люцета». II — кривая светораспределения.
НОРМЫ ОСВЕЩЕННОСТИ
Естественное освещение
ГОСТ устанавливает нормы естественного освещения промышленных зданий.
В таблице 76 приведены нормы1 в виде коэфициента естественной осве-
щенности (КЕО).
Таблица 76
Нормы естественного освещения
3S Нормы освещенности
*3 к 5 Р Наименование помещений При верхнем или комбини- рованном свете При боковом свете
£ Е КЕО в процентах
I. II. III. IV. Производственные помещения, в которых производится точная работа, требующая различения деталей раз- мерами менее 0,5 мм (в частности, с применением точ- ных измерительных инструментов), а также поме- щения для лабораторий Производственные помещения, в которых производится работа средней точности, требующая различения де- талей размерами 0,5—10,0 мм, а также администра- тивно-конторские помещения, помещения для общест- венных оргнизаций, комнаты для приема и приго- товления пищи и т. п Производственные и складские помещения, в которых производится грубая работа, требующая различения предметов или деталей размерами более 10 мм, а также помещения гардеробных, умывальных, душе- вых, уборных, лестниц и т. п Складские помещения для хранения крупных предметов, материалов в крупной таре и сыпучих материалов, а так- же проходы в производственных помещениях и ко- ридоры 5,0 з,о 2 1 1,5 1,0 0,5 0,3
Примечания: Приведенные нормы установлены, исходя из условий, что
очистка стекол в помещениях с незначительными производственными выделениями
Приводится в извлечении.
474
пыли, копоти и т. п. производится не реже двух раз в год, а покраска (побелка)
внутренних поверхностей ограждений — не реже одного раза в 2—3 года; в помеще-
ниях же со значительными производственными выделениями пыли, дыма, копоти и
т. п. очистка стекол производится не реже четырех раз в год, а покраска (побелка)
внутренних поверхностей ограждений — не реже одного раза в год.
В помещениях, освещаемых верхним и комбинированным светом, отношение
минимального значения к максимальному должно быть не менее 0,3 для помещений
I и II разрядов и не менее 0,2 для помещений III и IV разрядов.
Искусственное освещение
ГОСТ устанавливает нормы искусственного освещения (табл. 77) х.
Для характеристики условий зрительной работы ГОСТ различает два фона, на которых
приходится рассматривать обрабатываемую деталь: темный фон с коэфициентом
отражения до 0,2 (вкл.) и светлый — с коэфициентом отражения более 0,2. Кроме того
ГОСТ различает два контраста в зависимости от степени светлоты обрабатываемой детали
и фона, на котором рассматривается деталь, а именно: малый контраст, если
деталь и фон приблизительно одинаково темные или светлые, и большой контраст,
если деталь светлая, а фон темный, или наоборот.
Таблица 77
Нормы искусственного освещения для работ, требующих различения деталей
размером до 10 мм
Характе- ристка работ Разряд работ Подразряд Фон, на ко- тором раз- личаются детали Контраст детали с фоном Минимальная освещенность в люксах
При системе комбинирован- ного освещения При системе одного общего освещения
Детали размером менее 0,2 мм I а Темный Малый 500 Применение од- ного общего освещения не допускается
б Темный Светлый Большой Малый 300 125
в Светлый Большой 150 75
Детали от 0,2 до 1,0 мм 11 а Темный Малый 300 125
б Темный Светлый 1 Большой Малый 1 150 75
в Светлый Большой — 30
Детали от 1 до 10 мм III а Темный Малый 75 50
б Темный Светлый Большой Малый — 30
в Светлый Большой — 20
Примечания. Систему общего освещения работ, относящихся к разряду 1,
подразрядам б — в, к разряду II, подразрядам а — б, к разряду Ill, подразряду а,
* Приводятся в извлечении.
475
разрешается применять только в тех случаях, когда по технологическим усло-
виям неосуществимо применение комбинированного освещения.
Если напряженная зрительная работа в рабочем процессе, характеризуемая раз-
рядом 1, подразрядами б — в, разрядом II, подразрядами а — в и разрядом III,
подразрядом а, имеет место в течение рабочего времени непрерывно или почти непре-
рывно, то при системе освещения указанные в таблице нормы освещенности увеличи-
ваются вдвое, причем применение в этом случае следующего примечания не допус-
кается.
Приведенные в таблице нормы освещенности для работ II и III разрядов отно-
сятся к случаям, когда расстояние от глаза до рассматриваемого объекта менее 500 мм.
Если это расстояние больше 500 мм, то освещенность должна выбираться по сосед-
нему более высокому разряду.
При системе комбинированного освещения освещенность на уровне рабочей
поверхности от светильника общего освещения должна составлять не менее 10%
норм, приведенных в таблице, но должна быть не ниже 10 лк; применение освещен-
ности свыше 30 лк не обязательно.
Таблица 78
Нормы искусственного освещения
для работ с самосветящимися предметами или материалами, для грубых работ,
требующих различения деталей размерами более 10 мм, и складских помещений
Характеристика работ Разряд работ Минимальная освещенность при системе общего осве- щения в люксах
С самосветящимися предметами или материалами (напр., в кузницах, литейных, в мартеновских цехах) IV 50
Требующие различения предметов или деталей размером от 10 мм (вкл.) до 100 мм V 20
Требующие различения предметов или деталей размером свы- ше 100 мм, а также работа, требующая общего наблю- дения за ходом производственного процесса VI 10
Работа в складах громоздких предметов и сыпучих тел . . . VII 5
Примечание. Нормы освещенности, приведенные в таблице 77 (при си-
стеме одного общего освещения) для работ, относящихся к разряду II, подразряду в,
разряду 111, подразрядам б — в, и в таблице 78 для работ, относящихся к разрядам
V и VI, увеличиваются в два раза, если работы производятся при наличии в зоне
рабочего места элементов оборудования, прикасание к которым сопряжено с повы-
шенной опасностью травматизма. Однако увеличение освещенности свыше 50 лк
не обязательно.
Для жилых и общественных зданий нормы естественного освещения не уточнены так,
как для промышленных зданий (т. е. нет соответствующих ГОСТ’ов).
Считается, что световой коэфициент должен быть:
для жилых комнат в пределах....................... 1 : 8—1 : 10
» палат лечебных учреждений...................... 1 : б—1 : 8
»> служебно-вспомогательных помещений обществен-
ных зданий......................................... 1 : 10—1 : 12
» классов........................................ 1 : 4—1 : б
Коэфициент естественной освещенности (КЕО):
для операционных должен соответствовать............ 1,5%
» классных комнат................................. 1,25—1,0%
» жилых комнат..................................... 1,0 —0,5%
» вспомогательных помещений, коридоров и т. п. . . . 0,5 —0,3%
Нормы искусственного освещения для жилых и общественных зданий также не уточ-
нены.
Рекомендуется мощность в 10—15 ватт на 1 м2. Высота подвеса — не ниже 2,8 м. Защит-
ный угол — не менее 30°. Яркость видимых частей светильника не должна превышать 0,2 сб.
Нормирование освещения жилых и общественных зданий (в виде проекта) приводится
в работе Н. М. Данцига (см. литературный указатель).
476
ШУМ
Звуковые явления по характеру создаваемых ими ощущений делятся
на «звуки» и «шумы». Звук вызывается правильными (периодическими) ко-
лебательными движениями; шум обусловливается неправильными (апериоди-
ческими) колебательными движениями. Длительное воздействие сильных
шумов вызывает заболевание, проявляющееся в понижении слуховой спо-
собности—тугоухости, реже—к полной глухоте. (Малютин, Попов, Ермо-
лаев, Захер и др.).
В виду сказанного, измерение шумов представляет интерес с гигиени-
ческой точки зрения.
Объективно интенсивность звука измеряют количеством звуковой энергии
в эргах, протекающей через 1 см2 в секунду. Единицей для такого измерения
служит эрг/см2 сек.
Объективное измерение звука производят также по тому колебанию воз-
душного давления, которое возникает в звуковой волне. В этом случае еди-
ницей измерения служит бар, соответствующий дине/см2. Бар равен прибли-
зительно одной миллионной до-
ле атмосферного давления. Что-
бы получить представление об
этой величине, достаточно ска-
зать, что речь обыкновенной
громкости создает давление при-
мерно в 1 бар.
Зависимость между указан-
ными двумя единицами может
быть выражена следующей фор-
мулой: __
P = 6,4|J,
где Р — интенсивность звука
в барах,
J — интенсивность звука
в эрг/см2 сек.
Так как ухо человека улав-
ливает не абсолютный прирост
силы звука, а относительный,
Рис. 278. Область слухового восприятия, доступ-
ная уху человека. Нижняя кривая — порог
слышимости, верхняя кривая — порог «осязания».
Штриховкой показана область частоты и силы
звука речи.
то в качестве основной акустической единицы принимается децибел,
соответствующий увеличению звуковой энергии в 1,26 раза или увели-
чению звукового давления в 1,12 раза.
Предельную силу звука при данной частоте, едва слышимую ухом, назы-
вают порогом слышимости.
При 1000 гц1 звуковое давление порядка 2 . 10~4 дн/см2 ощущается нор-
мальным ухом как едва слышимый звук (порог слышимости).
Величина порога слышимости зависит в высокой степени от частоты; она
меньше всего для частот от 2000 до 3000 гц. Это значит, что ухо наиболее чув-
ствительно в этой области. В обе стороны от этой области порог слышимости
быстро растет.
Очень интенсивные звуки, вызывающие давление, начиная с 200 дн/сма
ощущаются как «осязание» — порог осязания звука. При
1000 дн/см2 звуки вызывают болезненное ощущение — порог болевого
ощущения.
На рис. 278 представлена область слухового восприятия, доступная уху
человека. Масштаб ординат на этом рисунке логарифмический. Нулевой
уровень для силы звука здесь взят условно и соответствует
10~9 эрг/см2 сек. Кривые линии, изображенные на рис., представляют кривые
1 Символ гц обозначает число колебаний в сек.
равной громкости. Все тоны, лежащие на одной и той же кривой громкости
имеют один и тот же уровень громкости. Уровни громкости выражены в фонах,
что равнозначно децибелам. Тон в 1000 гц принят для сравнения громкостей
всех других звуков. Кривая с уровнем громкости О представляет кривую порога
слышимости; кривая с уровнем громкости 120 предствляет порог осязания.
Из рисунка видно, что тоны различной частоты оказываются равногромкими
не при одинаковой силе звука. Область частоты и силы звука, где лежат звуки
речи, показана на рисунке штриховкой.
Зависимость между субъективной громкостью звука и объективной оцен-
кой его может быть выражена следующей формулой:
5аб=10 1g-/-,
Jo
где Soo — уровень интенсивности звука над порогом слышимости («уро-
вень ощущения»), выраженный в децибелах.
J — интенсивность звука в эрг/см2 сек.
Jn — интенсивность на пороге слышимости в тех же единицах.
Указанная зависимость может быть выражена и другой формулой:
Sa6 = 201g^-,
*0
где Sd6 — сохраняет то же значение, что и в предыдущей формуле,
Р — звуковое давление в барах,
Ро — звуковое давление на пороге слышимости.
Чтобы иметь представление о субъективной оценке шумов можно сказать,
что шумы порядка 100—130 децибел воспринимаются как оглушительные,
порядка 80—90 децибел — как неприятные, и порядка 50—70 децибел — как
умеренные.
Для измерения шумов предложено много приборов. Здесь описан шу-
момер и фонометр.
Шумомер Павяжского
Шумомер Навяжского (рис. 279) представляет объективный фонометр
Как и другие приборы подобного типа, он построен на следующем принципе:
Рис. 279. Схема шумомсра Навяжского.
М —микрофон, Тр —трансформаторы, Г —гальванометр, УБ, СБ —электронные лампы.
звуковая енергия при помощи микрофона превращается в электрические
колебания, которые усиливаются ламповым усилителем; затем ток выпрям-
ляется и измеряется гальванометром.
478
По аналогии с тем, как это делается при измерении уровня громкости,
тон в 1000 герц принят автором прибора в качестве тона сравнения, таким
образом показания прибора эквивалентны стандартным тонам. Главными
частями прибора являются микрофон, усилитель и гальванометр с медноза-
кисным детектором. Усилитель оформлен вместе с гальванометром. Особен-
ностью усилителя является его частотная характеристика. Чувствительность
усилителя увеличивается линейно с частотой в пределах 50—8000 герц. Уси-
литель имеет три каскада усиления: первый и третий каскад — на лампе
УБ-110, а второй на лампе СБ-154. В цепь анода этой лампы включен дрос-
сель. Изменение сопротивления последнего вместе с частотой дает наклон-
ную характеристику усилителя. В сетку второго каскада включен градуиро-
ванный в децибелах потенциометр, который дает возможность уменьшать
чувствительность прибора на 60 децибел (ступенями по одному децибелу).
Шкала гальванометра градуирована в децибелах для частоты 1000 герц.
Питание собрано в отдельном ящике.
Фонометр
Фонометр, схема которого дана на рис. 280, построен на следующем прин-
ципе: одно ухо исследователя воспринимает испытуемый шум, в то время
Рис. 280. Схема фонометра.
а — аккумулятор, d — индукционная капнша. г — реостат,
t —телефон, /—неоновая лампочка.
как к другому уху при помощи телефонной трубки подводится звук, вызы-
ваемый индукционной катушкой. Увеличивая или ослабляя этот звук,
добиваются одинаковой интенсивности звуковых ощущений и определяют
силу звука по шкале прибора.
Важнейшие части прибора: а — аккумулятор, d — индукционная катушка,
t — телефон, /—неоновая лампочка для регулирования напряжения, ко-
торое подается на клеммы телефона, иг — реостат.
Включив ток, подносят трубку телефона к уху; затем пользуясь реоста-
том, стремятся добиться одинаковой интенсивности слуховых ощущений
в обоих ушах, после чего производят отсчет интенсивности шума по шкале
реостата. Применение фонометра предполагает одинаковую чувствительность
обоих ушей, поскольку оба уха участвуют в измерениях.
При установлении баланса громкости рекомендуется сначала устанавли-
вать тон сравнения слабее, а затем сильнее измеряемого шума, после чего
путем ряда проб удается установить равногромкость обоих звуков.
Во время измерений индукционная катушка должна быть хорошо изоли-
рована, чтобы звук, создаваемый ею, не проникал в ухо помимо телефона.
СОТРЯСЕНИЕ
Влияние сотрясений на организм человека изучено недостаточно, однако,
известно, что длительные и сильные сотрясения вредно влияют на человека
(поражение нервного аппарата верхнего завитка улитки, невралгии и пр.).
Сопутствуя нередко шумам, вибрации усиливают их вредное действие на
организм (Попов).
479
Нижний предел, когда сотрясения начинают улавливаться человеком,
соответствуют скорости в 0,1 см/сек и ускорению в 10—15 см,сек2. Верхний
предел, когда сотрясения начинают беспокоить, соответствуют скорости
в 0,4 см/сек и ускорению в 30—50 см/сек2.
Ниже описаны два прибора, служащие для измерения сотрясений: вибро-
метр Голицына—Бончковского и виброграф конструкции Ленинградского
института гигиены труда и профессиональных заболеваний.
Рис. 281. Виброметр Голицина—Бончковского.
А — металлический параллелепипед, В — стальная линей-
ка, D — перо, С — груз.
Виброметр Голицы па—Бон ч ко вс кого
Прибор (рис. 281) представляет собой тяжелый железный параллелепи-
пед А (154 х 153 х 193 мм). К верхней стороне параллелепипеда прикреплена
стальная линейка В, на свободном конце которой насажена муфта с пером О.
D На верхней стороне линейки
б ----имеется миллиметровая шка-
------......................ГТ ла. ГРУ3 можно перемещать
вдоль линейки и закреплять
на желаемом месте ее. Ниж-
няя и одна из боковых сто-
рон параллелепипеда совер-
шенно гладкие и по возмож-
ности теснее должны сопри-
касаться с вибрирующим
телом.
Если поставить прибор на нижнюю сторону, то он действует как верти-
кальный маятник; если же поставить его на боковую сторону, то он действует
как горизонтальный маятник.
Колебания записываются пером на закопченной ленте, натянутой на
барабане.
Для измерения сотрясений в помещении, ставят прибор на пол и, пере-
мещая груз по линейке, находят такой период ее собственного колебания,
при котором достигается синхронность колебаний собственных и вызванных,
что выражается периодической кривой. Тогда производят запись.
Виброграф конструкции Ленинградского института гигиены труда
Ленинградский институт гигиены труда и профзаболеваний сконструиро-
вал два прибора для измерения сотрясений: виброграф с оптической реги-
страцией и виброграф с механической регистрацией. Здесь описан второй
из них, как наиболее простой и удобный для применения.
Рис. 282. Виброграф конструкции
Ленинградского института гигиены
труда.
А — вертикальный маятник, В — гори-
зонтальный маятник.
Прибор (рис. 282) имеет два маятника: горизонтальный —Ви вертикаль-
ный А для записи смещений в двух плоскостях. Виброграф устроен так, что
имеется возможность не только устанавливать его в горизонтальной плос-
480
кости, но и крепить к вертикальной. На конце каждого маятника закреплен
писчик, производящий запись. Регистрирующий прибор (на рисунке не по-
казан) смонтирован на общем основании с вибрографом. Увеличение прибора
в пределах от 20 до 50 раз. Кривые (виброграммы), получающиеся при записи,
дают возможность определить амплитуду колебательных движений и их
период.
Положим, что на ленте получилась виброграмма, изображенная на
рис. 283. Измерив расстояние в мм между точками а—b и b — с, складывают
полученные значения и сумму делят на 4, что дает амплитуду, отнесенную
к увеличению, которое дает виброграф. Разделив полученный результат на
увеличение, получаем амплитуду.
Для определения периода делят длину ленты, соответствующую 1 секунде,
на расстояние между двумя смежными точками (а — с, аг — q и т. д.).
Пример. Расстояние ab = 36 мм; Ьс = 38 мм; ас = 8,0 мм. За 5 секунд смоталось
200 мм ленты. Увеличение прибора = 40. Отсюда заключаем, что амплитуда равна
36 + 38 200
-4—4Q- 0,46 мм. Число колебаний в одну секунду равно = 5. Величина периодам
= = 0,2 сек.
ОСВЕТИТЕЛЬНЫЕ И ТОПЛИВНЫЕ МАТЕРИАЛЫ
Из осветительных материалов здесь рассмотрен керосин, из топливных — уголь.
Исследование керосина
Доброкачественный керосин должен быть прозрачным, бесцветным, без резкого запаха,
не содержать примесей смолы и серной кислоты, применяющейся для его очистки. Удель-
ный вес керосина должен быть в пределах 0,809—
0,8265. Минимальная температура вспышки паров
керосина, принятая у нас, равна 28° С.
Для определения степени очистки керосина при-
бавляют к небольшой порции его немного крепкой
серной кислоты. Хорошо очищенный керосин при
этом не изменяет своего цвета, плохо очищенный
буреет. Примесь смолы выявляют прибавлением
раствора AgNO3 (побурение пробы). Присутствие
серной кислоты обнаруживают прибавлением раст-
вора ВаС12 (белая муть сульфата бария).
Удельный вес определяется ареометром при
15° С.
Температуру вспышки паров керосина опреде-
ляют при помощи прибора, изображенного на рис.
284. Прибор состоит из медной двустенной водяной
бани а. В верхней стенке бани имеется углубление
цилиндрической формы Ь. В это углубление встав-
ляется такой же формы медный сосуд с. На внутрен-
ней стенке сосуда укреплен указатель в виде обращен-
ного вверх острия; он указывает уровень, до кото-
рого нужно наливать керосин в сосуд при исследо-
вании. Сосуд закрывается съемной крышкой, в кото-
рой имеется прямоугольное отверстие. Это отверстие
закрыто пластинкой, которая, при нажатии на пру-
жину d, отходит вбок, оставляя отверстие открытым
на 2 секунды, после чего вновь закрывает его. В тот
Рис. 284. Прибор для определе-
ния температуры вспышки керо-
сина.
момент, когда отверстие открывается, к нему авто-
матически наклоняется горящее пламя фитиля ма-
ленькой масляной светильни е, закрепленной на
крышке сосуда.
Исследование температуры вспышки произво-
дится следующим образом. В баню и сосуд для
а — водяная баня, b — углубление в
бане, с — медный сосуд для испытуе-
мого материала, d — пружина, е —
масляная светильня, t и G — термо*
метры, е — воронка, / — сифон,
g — лампа.
керосина вставляют термометры t и В сосуд наливают керосин до уровня указателя.
Через воронку е наливают в баню воду, нагретую до 58°С, при этом излишек ее вытекает
через сифон /. Воду несколько охлаждают и при помощи лампы g поддерживают ее темпе-
ратуру на высоте 55° С. Керосин нагревается. Когда температура его поднимется до 15,5 °Q
при барометрическом давлении в 715—725 мм либо 16,5° при давлении 745—755 мм, нажа-
31 И. А. Бурштейн
481
тием на пружину d открывают отверстие крышки. Зажженная светильня при этом накло-
няется к отверстию. Если пары керосина нагреты уже до температуры вспышки, они зажи-
гаются голубым пламенем. Если вспышка не произошла, то продолжают производить такие
же испытания через каждые 0,5° поднятия ртути в термометре до того момента, когда про-
изойдет вспышка.
Температуру вспышки определяют по показанию термометра и пересчитывают на нор-
мальное давление по приложенной к прибору таблице (см. табл. 79). Барометрическое
давление в момент исследования можно определить с помощью барометра-анероида, имею-
щегося при приборе.
Таблица 79
Для перерасчета температуры вспышки керосина на такую же при нормальном давлении
Показание барометра 710 720 730 740 750 760 770 780
Градусы Цельсия 17,3 17,6 18,0 18,3 18,7 19,0 19,4 19,7
» » 18,3 18,6 19,0 19,3 19,7 20,0 20,4 20,7
> » 19,3 19,6 20,0 20,3 20,7 21,0 21,4 21,7
Найдя в таблице температуру вспышки, полученную при данном барометрическом
давлении, прочитывают в соответствующей колонке температуру вспышки при нормальном
давлении.
Пример. Положим температура равна 18,6° при давлении в 720 мм. Находим это число
в колонке второй. Против этого числа в колонке шестой находим температуру вспышки при
давлении в 760: она равна 20°.
Исследование угля
С гигиенической точки зрения представляет интерес определение серы в угле, так как
сернистый ангидрид, выделяющийся во время горения угля, портит дымоходы и загряз-
няет воздух. Не меньший интерес представляет определение зольности угля.
Сера присутствует в угле в виде сульфатных соединений (нелетучая сера) и в виде оки-
сляемых сернистых соединений (летучая сера). Нелетучей серы значительно меньше, нежели
летучей. При анализах угля на серу обыкновенно определяют общую серу. Исследование
ведется с суховоздушпой пробой; результат перечисляют на сухое вещество.
Как стандартный принят следующий метод (Бюро теплотехнических съездов).
0,5—1 г топко измельченного угля смешивают в тигле с 2 г препарата, состоящего из
2 ч. жженой магнезии и 1 ч. соды и полученную смесь покрывают 1 г того же препарата.
Открытый тигель помещают наклонно в фарфоровый треугольник и осторожно нагре-
вают, следя за тем, чтобы содержимое тигля не распылялось.
Через час усиливают нагревание и, время от времени перемешивая содержимое тигля,
продолжают прокаливание, пока смесь не приобретет белый, желтоватый или красноватый
цвет. Затем смесь обрабатывают горячей водой и оставляют на час на водяной бане, после
чего фильтруют и промывают осадок методом декантации, а затем на фильтре два-три раза.
К фильтрату прибавляют бромной воды из расчета 1 мл на 100 мл жидкости, подкис-
ляют соляной кислотой, затем кипятят, пока не исчезнет желтое окрашивание от брома.
Сера присутствует теперь в растворе в виде сульфатов. К кипящему раствору прибавляют
50 мл кипящего раствора ВаС12 • 2Н2О (18 г. ВаС12 • 2Н2О на 500 мл Н2О), дают отстояться
не менее 12 часов для выпадения BaSO4, после чего фильтруют через уплотненный беззоль-
ный фильтр, промывают горячей водой; фильтр с осадком прокаливают в тигле и определяют
вес BaSO4. Результат, умноженный на 0,1373, соответствует количеству общей серы во
взятой пробе.
Зольность угля определяется так же как зольность пыли (стр. 282). Озоление
навески производится медленно без появления пламени.
ЛИТЕРАТУРА
Общие курсы и руководства
1. Игнатов Н. К., Практическое руководство по методике санитарно-гигиенических
исследований, Биомедгиз, 1935.
2. Левицкий В. А. (ред.), Гигиена труда, Биомедгиз 1936.
3. Летавет А. А. (ред.), Курс гигиены труда, Медгиз, 1946.
4. Марзеев А. Н., Сысин А. Н.. Яковенко В. А., Основы коммунальной гигиены,
т. II, Медгиз, 1938.
5. Моисеев С. В., Учебник общей гигиены, Медгиз, 1947.
6. Никитин А. Ф., Способы и приемы практических наблюдений и исследований
по гигиене, 1911.
482
7. Смоленский П. О., Простейшие способы исследования и оценки доброкачествен-
ности съестных припасов, напитков, воздуха, воды, почвы, жилищ и пр., изд. IV, 1909.
8. Справочник санитарного врача, под ред. Сысина А. Н., Биомедгиз, 1935.
9. Справочник для санитарно-промышленных врачей, под ред. Койранского Б. Б.,Л.,
1940.
10. Сысин А. Н. (ред.), Учебник гигиены, Медгиз, 1938.
11. Хлопин Г. В., Основы гигиены, т. II, Госиздат, 1923.
12. Эрисман Ф. Ф., Курс гигиены, т. III, 1901.
Гигиена жилых, фабрично-заводских, школьных и других
помещений
1. Ветошкин С., Запорожец В., Иванов А., Опытные дома, М., 1927.
2. Гофман В. А., Фабрично-заводская архитектура, чч. I—Ill, изд. КУ БУЧ, 1932.
3. Дробинский К., Справочник по гигиене и санитарии детских учреждений, 1926.
4. Поморцев В. П., Строительная гигиена и санитария, ОНТИ, 1934.
5. Сельское жилище (сборник), НКЗ, М., 1928.
6. Серк Л. А., Промышленная архитектура, тт. I—II, ОНТИ, 1934—1945.
7. Справочник по жилищному строительству, М., 1925.
8. Строительные правила, М., 1934.
9. Цветаев В. Д., Современная фабрично-заводская архитектура, Госстройиздат, 1933.
10. Френкель 3. Г., Основы общего городского благоустройства, Л., 1926.
11. Шахнер Р., Сайитарная техника в жилищном строительстве, ГТИ, 1930.
Отопление и вентиляция
1. Аше Б. М., Отопление и вентиляция, тт. I и II, Госстройиздат, 1934—1946.
2. Вентиляция промышленных предприятий, сборник под ред. Рафеса и Синева, Гос-
трудиздат, 1930.
3. Гартман К., Вентиляция промышленных заведений, 1926.
4. Г ришечко-Климов С. М., Отопление и вентиляция промышленных предприятий,
Госстройиздат, 1933.
5. Казанцев А. 77., Справочная книга по отоплению и вентиляции, НТИ., 1932.
6. Каплинский И. Л., Карачаров Г. С. и Куче рук В. В., Приемка, испытание и сдача
в эксплоатацию вентиляционных систем, ОНТИ, 1935.
7. К методологии технического обследования вентиляционных установок, Труды и
материалы Ин-та охраны труда, т. VII, вып. 1, Гострудиздат, 1930.
8. Максимов Г. А. и Орлов А. И. Отопление и вентиляця, I и II тт., Стройиздат,
1948—9 (второй том — Максимов Г. А.).
9. Нормы и технические усповия проектирования отопления и вентиляции промыш-
ленных зданий, Стройиздат, 1949.
10. Нормы и технические условия проектирования отопления и вентиляции жилых
и гражданских зданий поселков при промышленных предприятиях, Стройиздат, 1948.
11. Павловский А. К., Курс отопления и вентиляции, тт. I и II, 1923.
12. Ритчель Г. и Браббе К., Руководство по отоплению и вентиляции, тт. I и II, 1928.
13. Хренов Л. К., Отопление и вентиляция, ОНТИ, 1936.
14. Чаплин В. М., Курс отопления и вентиляции, вып. 1—2, 1923.
Освещение
I. Вавилов С. И., Известия Академии наук СССР, Серия физическая, т. IX, 4—5,
1945.
2. Гусев Н. М., Расчет и проектирование естественного освещения промышленных
зданий, Госстройиздат, 1933.
3. Данциг Н. М., Нормы и правила искусственного освещения школ. Освещение
школьных зданий, изд. Академии наук СССР, М.—Л., 1939.
4. Данциг Н. М., Гигиеническое нормирование освещения жилых и общественных
зданий, изд. Академии медицинских наук СССР, М., 1948.
5. Зеленцов М. Е., Световая техника, изд. КУ БУЧ, 1925.
6. Корольков А., Курс электрического освещения, ГНТИ, 1931.
7. Майзель С. О., Свет и зрение, ГНТИ, 1932.
8. Майзель С. О., Основы рационального освещения, 1929.
9. Мешков В. и Смелянский 3., Гигиена освещения, Биомедгиз, 1936.
10. Муравьев А. Е., Электрическое освещение, изд. КУ БУЧ, 1929.
11. Никитин А. Ф., Исследование освещения и санитарные нормы, 1922.
12. Тиходеев П. М., Световые измерения в светотехнике, ОНТИ, 1936.
13. Труды Ленинградского института гигиены труда и техники безопасности, 193(1
14. Труды Всесоюзных светотехнических конференций, тт. I—II и 111.
15. Френкель С. и Надеждин П., Методы светотехнических расчетов, ОНТИ, 1932.
16. Фрюлинг Г., Освещение помещений естественным светом, Госстройиздат, 1933.
483
Шум и сотрясение
1. Беляев Н., Курс архитектурной акустики, Стройиздат, 1933.
2. Кнудсен В. О., Архитектурная акустика, ОНТИ, 1936.
3. Лифшиц С. Я., Акустика зданий и их изоляция от шума и сотрясений, ГНТИ, 1931.
4. Лифшиц С. Я., Курс архитектурной акустики, ОНТИ, 1937.
5. Методы исследования шумов, сборник статей под ред. Ржевкина С. Н., Гостехиз-
дат, 1933.
6. Навяжский Г. Л., Производственный шум, методы его исследования и борьба
с ним, изд. Ленинградского института организации экономики и охраны труда, 1934.
7. Навяжский Г. Я., Учение о шуме, Медгиз 1948.
8. Труды Ленинградского научно-исследовательского ин-та гигиены труда и профес-
сиональних заболеваний, т. X, ч. 2, «Вибрации на производстве», Л., 1947.
Осветительные и топливные материалы
1. Зикеев Т. А. и Корелин А. И,, Анализ энергетического топлива, Госэнергиздат,
М.-Л. 1948.
2. Кусаков М. М., Методы определения физико-химических характеристик нефтяных
продуктов, ОНТИ, 1936.
3. Методы исследования товаров, под ред. Августинина и Воскресенского, разд. II.
Топливо, ст. Бородулина М. В., Снабтехиздат, 1932.
4. Моргулиусов-Турковский А., Справочник по топливу, осветительным продуктам
и смазочным материалам, 1926.
5. Надеждин А., Основные виды топлива России и их характеристика, 1925.
6. Окнов М. Г., Топливо и его сжигание, ОНТИ, 1934.
7. Орлов H.t Очерки по химии угля. Общая химия угля, 1934.
8. Топливо. Исследование, обработка, использование, Сборник под ред. Рубина П. Г,,
Госиздат Украины, 1925.
9. Чернобаев Д., Топливо, его горение, Госиздат Украины, 1931.
ГЛАВА ДЕВЯТАЯ
ТКАНИ ОДЕЖДЫ
САНИТАРНО-ГИГИЕНИЧЕСКИЕ ТРЕБОВАНИЯ
Гигиенические требования к тканям одежды не могут быть сформулированы
в виде определенных норм независимо от того, в каких условиях метеороло-
гического фактора и состояния организма применяется данная одежда.
Такие существенные гигиенические качества тканей, как теплопровод-
ность, воздухопроницаемость, гигроскопичность и т. п., должны иметь раз-
личное количественное выражение в зависимости от указанных выше условий.
Это с полной убедительстью показал П. Е. Калмыков.
При всех условиях одежда должна создавать вокруг тела наиболее благо-
приятный (комфортный) климат, но именно поэтому ткани, идущие на изго-
товление одежды, должны обладать неодинаково выраженными теплопровод-
ностью, воздухопроницаемостью, гигроскопичностью и др. гигиеническими
показателями.
Так, теплопроводность ткани должна быть возможно малой, если одежда
призвана защитить тело от охлаждения (низкая температура воздуха, холод-
ный ветер), но это требование к ткани отпадает в условиях относительно
высоких внешних температур (не выше температуры тела), особенно при
выполнении тяжелого физического труда. Воздухопроницаемость ткани одежды
должна быть хорошо выраженной при высоких внешних температурах и
слабо выраженной в условиях низких температур, особенно при холодных
ветрах. Высокая гигроскопичность является положительным показателем
для тканей, идущих на изготовление белья, внутренней обуви, но она ока-
жется излишней для тканей, идущих для верхнего платья, особенно в условиях
повышенной влажности воздуха и т. п.
Лишь немногие требования могут быть сформулированы в отношении
всех тканей одежды независимо от метеорологического фактора и состояния
организма: легкость, мягкость, прочность и носкость, отсутствие вредных или
ядовитых веществ в краске, аппретуре и т. п., малая способность сорбиро-
вать из окружающей среды пахучие, вредные и ядовитые газообразные и
парообразные вещества. Ткани ношеной одежды помимо указанных качеств
не должны содержать пыли, грязи, насекомых, патогенных бактерий.
Все другие гигиенические качества тканей (теплопроводность, воздухо-
проницаемость, гигроскопичность и проч.) должны определяться конкретными
назначениями той одежды, на изготовление которой идет данная ткань.
Одежда в целом не должна нарушать нормальных физиологических функций
организма — дыхания, кровообращения. Она должна соответствовать кли-
матическим особенностям местности, времени года, возрасту, образу жизни
и условиям профессионального труда.
Весьма важно, чтобы ткань сохраняла свои положительные качества
при носке одежды и не утрачивала их при смачивании, стирке, глажке и т. п.
воздействиях.
485
Как указал известный русский гигиенист Ф. Г. Кроткое, для более полного
и всестороннего представления о гигиенических качествах тканей необходимо
повторное обследование их в процессе носки, когда они применяются в виде
«комбинации тканей, а не изолированных элементов».
ФИЗИКО-МЕХАНИЧЕГКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
ТКАНЕЙ ОДЕЖДЫ
Крепость и растяжимость ткани, счет нитей, просвечиваемость,
усадка, сваливасмость и аппретура
Крепость и растяжимость ткани определяется при по-
мощи разрывных машин—динамометров.
Крепость ткани оценивается по максимальному сопротивлению на разрыв
при испытании по основе и утку и выражается в кг/см. Растяжимость опре-
деляется по максимальному растяжению ткани перед разрывом.
Счет нитей по основе и утку производится в квадратном окошечке
с длиной стороны в 1 см, сделанном в металлической дощечке, при увеличении
в 4—12 раз.
Просвечиваемостьткани исследуют, рассматривая ткань про-
тив света. Наблюдатель находится при этом в затемненной комнате. Обращают
внимание на томные пятна, полосы, «завалы» в основе, пропуски в утке и т. п.
Усадка. Кусок ткани измеряют в длину, ширину и толщину. Затем
его смачивают или пропаривают, высушивают и вновь измеряют. Уменьшение
размеров представляет усадку.
Сваливасмость ткани представляет уплотнение ее, возникающее
под влиянием носки; при этом уменьшаются пористость и толщина, увели-
чивается удельный вес. Ткань становится менее воздухопроницаемой и более
теплопроводной.
По П. Е. Калмыкову1, сваливасмость определяется прибором, приме-
няемым обыкновенно для исследования истираемости тканей. Из ткани вы-
резается кружок, который помещается на выпуклый цилиндр прибора; сверху
кружок вырезанной ткани покрывается прочной хлопчатобумажной тканью,
закрепляемой металлическим кольцом. Поверх ткани опускается массивный
диск с переменной нагрузкой.
Поверхность диска, обращенная к ткани, рифленая. Когда аппарат при-
водится в действие, цилиндр вращается, делая при этом и качательные дви-
жения, благодаря чему ткань подвергается сжатию и сминанию по всей по-
верхности. Опыт продолжается 24 часа. Сваливаемость оценивается по умень-
шению толщины взятого кружка ткани и выражается в процентах по формуле:
(Д-В)ЮО
/\-------------
А
где К— коэфициент сваливаем ости,
А — начальная толщина,
В — толщина после опыта.
Аппретуру ткани определяют кипячением в течение часа в 1%
растворе мыла и соды. После кипячения ткань промывают сначала в теплой,
затем в холодной воде и высушивают. Убыль в весе соответствует количеству
аппретуры (шлихты).
Толщина ткани
Толщина ткани имеет значение для целого ряда свойств: воздухопрони-
цаемости, водопроницаемости, водоемкости и др. Знание толщины ткани
необходимо для определения удельного веса и объема пор ткани. Ниже опи-
саны приборы, применяемые для определения толщины ткани.
1 «Гигиена и Санитария», № 9, 1947.
486
Сферометр Рубнера
Сферометр (рис. 285) состоит из штатива а, от которого отходит горизонтальный стер-
жень гл; у сводобного конца последнего отходит вниз гильза /. У основания гильзы, в на-
резке ее движется микрометрический винт, у нижнего конца которого укреплен диск Ь.
Диск по краю разделен на 100 частей. Полный оборот диска перемещает винт вверх или
вниз на 0,5 мм, следовательно, перемещение диска на одно деление соответствует перемеще-
нию винта на 0,005 мм. Отсчет полных оборотов диска производят по шкале g, а части
оборота отсчитывают по диску. В гильзу t сверху введена ножка столика с, имеющего раз-
мер в 5 сма. Ножка столика касается верхушки винта. При передвижении винта вверх или
вниз в том же направлении перемещается и столик. Над столиком с помещается другой сто-
лик d, ножка которого свободно перемещается вверх и вниз в канале стержня и. Конец ножки
столика в виде острия упирается в рычаг е на расстоянии 4 мм от оси рычага. Отклонение
рычага отмечается стрелкой и, двигающейся по циферблату /. На левое плечо рычага
насажен подвижной груз к для уравновешивания более длинного правого плеча. При опре-
делении сжимаемости ткани в правое плечо рычага ввинчивается стержень h с делениями,
на который насаживается подвижной груз I. К прибору
прилагается небольшой пробойник, с помощью которого
можно из данной ткани высекать кружочки диаметром
в 5 см.
Перед измерением толщины ткани устанавливают
нулевое деление диска против нулевого деления шкалы;
при этом столики с и d соприкасаются между собой.
Грузу к придают такое положение, чтобы левое плечо
и правое уравновешивали одно другое; при этом
стрелка v должна показывать среднее деление шка-
лы /. Коромыслу придают очень небольшой перевес
вправо. Затем поворотом микрометрического винта
опускают несколько диск вниз, причем оба столика
опускаются вниз, а коромысло наклоняется вправо.
Осторожно раздвинув оба столика, помещают между
ними кружок из данной ткани и поворотами диска в
обратную сторону устанавливают коромысло в прежнее
положение, т. е. чтобы стрелка v показывала среднее
деление шкалы /.
Теперь по положению диска отсчитывают на шкале
g число полных оборотов микрометрического винта, а
на самом диске части оборота, и отсюда делают заключе-
ние о толщине данной ткани.
Рис. 285. Сферометр для опре-
деления толщины ткани.
а — штатив, т — стержень, t —
гильза, b — диск микрометричес-
кого винта, а — шкала для отсчета
полных оборотов микрометричес-
кого винта, с — нижний столик,
d — верхний столик, е — рычаг,
v — стрелка, / — циферблат, h —
подвижной груз, h — стержень с
делениями, i — подвижной груз.
Пример. Положим, что при измерении толщины
ткани диск опустился на 2,35 полных оборота. Отсюда
делаем вывод, что толщина ткани равна 0,5 • 2,35 =
= 1,175 мм.
Так как толщина ткани, вследствие неровностей ее,
не везде одинакова, рекомендуется делать измерение не
одной, а нескольких проб и из полученных данных
взять среднее.
При помощи описанного сферометра можно производить определение толщины ткани,
находящейся под некоторым давлением. Для этого в правое плечо рычага ввинчивается стер-
жень h, на который насажен подвижный груз Z. Если известен вес груза и расстояние
его от оси стержня, то можно рассчитать давление на столик, а отсюда давление на каждый
см2 ткани.
Пример. На стержень h насажен груз весом в 10 г на расстоянии 80 мм от оси рычага.
Учитывая, что основание столика d находится от той же оси на расстоянии 4 мм, можно
написать пропорцию: X : 10 = 80 :4. Отсюда
Х =
10 - 80
4
= 200 г.
Итак, груз в 200 г распределяется на 5 см2; следовательно, на! см2 давление равно—- = 40 г.
Сферометр автора
Сферометр, предложенный автором1 (рис. 286), состоит из штатива А, на котором при
помощи винта В закреплен микрометре с полумиллиметровым ходом. На шпинделе D укреп-
лен столик Е, поверхность которого равна 5 см2. На барабане F закреплен диск G, разде-
ленный по краю на 500 равных частей. Полный оборот диска вызывает перемещение сто-
лика на 0,5 мм; поворот диска на одно деление соответствует перемещению столика на 0,001 мм
т. е. на 1 |х. В нижнем углу дуги микрометра закреплена шкала К с делениями на миллиметры
1 «Гигиена и Санитария», № 7—8, 1946.
487
Сферо-
и полумиллиметры. Следовательно, полный оборот диска вызывает перемещение его по шкале
на одно малое деление (0,5 мм).
В верхнем крае дуги микрометра неподвижно закреплен полый металлический конус L,
основание которого затянуто эластической резиной. Она и служит дном конуса. Диаметр
основания конуса равен 3,5 см, т. е. приблизительно на 1 см более диаметра столика, что
дает возможность столику, при соприкосновении его с основанием конуса, касаться лишь
эластического дна его.
В верхушке конуса вставлена узкая стеклянная трубка М высотою в 12 см с делениями
на 0,01 см. Сбоку конуса L к нему приделана металлическая трубочка N, сообщающаяся
с полостью конуса. Сверху трубочка N закрывается винтом R.
Измерение производится при постоянном давлении на ткань,
равном 10 г на 1 сма. Сначала устанавливают нулевую точку, для
чего столик Е приводят в соприкосновение с дном конуса, и про-
должают поворачивать барабан, пока жидкость в капиллярной трубке
не поднимется до метки на капиллярной трубке, отстоящей от дна
конуса на 10 см. Записывают показание прибора, т. е. показание
шкалы и диска. Затем при помощи барабана перемещают столик Е
вниз и кладут на него кружок ткани в 5 сма, вырезанный пробойником,
и вновь перемещают столик вверх, пока жидкость в капилляре не
поднимется на ту же высоту, как и ранее. При этом условии давле-
ние на 1 см2 равно 10 г. Записывают вновь показание прибора.
Разность между обоими показаниями соответствует толщине измеряе-
мой ткани.
Пример. Первое показание прибора соответствует 2,345 мм,
второе 3,879. Следовательно, толщина ткани равна
3,879 — 2,345 = 1,534 мм.
Прибор дает возможность измерять толщину ткани при любом
другом давлении. Для этой цели нужно оба отсчета вести при соот-
ветствующей высоте столба жидкости в трубке. Давление рассчи-
тывается умножением площади основания столика на высоту столба
жидкости, стоящего над ним. Высота столба жидкости равна вы-
соте конуса плюс высота жидкости в трубке.
Пример. Высота конуса равна 3 см, высота жидкости в
трубке — 6 см. Отсюда заключаем, что давление столба жидкости
на весь столик равно
5 X 9 = 45 г, а на 1 см2 = 9 г.
Толщемер
Толщемер, изображенный на рис. 287, дает возможность изме-
рять толщину ткани при постоянном давлении на ткань, равном
70 г на 1 см2. Устройство прибора не сложно. Он состоит из
1жней неподвижной и верхней
подвижной, накладываемой на нижнюю. Подвижная
пластинка посредством системы стерженьков соединяется
со стрелкой, вращающейся по циферблату. На цифер-
блате при помощи шкалы и нониуса определяют рас-
стояние между пластинками с точностью до 0,005 мм.
Если между пластинками помещена ткань, то отсчет
соответствует толщине ткани.
Толщина ткани зависит, главным образом,
от способа изготовления ее и колеблется в
широких пределах от десятых долей миллиметра
до нескольких миллиметров. Так, гладкая хлоп-
чатобумажная ткань имеет толщину 0,15 —
0,2 мм, хлопчатобумажное полотно — 0,7 —
0,9 мм, шерстяная фланель — 2 мм, сукно —
1 — 2,6 мм и более.
Удельный вес и объем пор
Удельный вес ткани, или вес 1 см3 ее, опре-
деляют следующим образом: взвешивают несколько проб данной ткани
определенного размера и отсюда делают вывод относительно веса 1 см2
Рис. 286.
метр автора.
А — штатив, В — винт
для укрепления ми-
крометра, С — микро-
метр. D — шпиндель,
Е — столик, F — ба-
рабан G — диск
с делениями, К —
шкала L — конус
для жидкости, М —
стеклянная трубка
с делениями, W—
металлическая трубо-
чка, R — винтовая
пробка.
пластинок:
Рис. 287. Толщемер.
488
ткани. Разделив полученный вес на толщину ткани, находят вес 1 см3, т. е.
удельный вес.
Пример. 100 см2 ткани толщиной в 2,4 мм имеют вес равный 6 г. Нужно определить
удельный вес ткани. 1 см2 данной ткани весит—г; разделив найденный вес на толщину
ткани, получаем:
--------= 0,25.
100 • 0,24
Удельный вес ткани зависит от способа изготовления ткани и от свойств
основного материала.
Приводим удельный вес некоторых тканей: шерстяная фланель — 0,1,
шерстяное трико — 0,18; хлопчатобумажное трико — 0,2 и т. п.
Общий объем пор ткани определяется следующим образом.
Удельный вес данной ткани делят на удельный вес волокон той же ткани,
получая таким образом объем плотного вещества ткани в 1 см3 данной ткани.
Разность между 1 см3 и полученным результатом соответствует объему пор
в 1 см3 ткани. Разность перечисляют на 100 см3, получая таким образом объем
пор в процентах.
Пример. Пусть удельный вес ткани равен 0,26, удельный вес волокон ткани 1,3. Тре-
буется определить объем пор ткани.
Разделив 0,26 на 1,3, находим, что объем плотного вещества в 1 см3 ткани равен
= 0,2 см3. Следовательно, объем пор в 1 см3 ткани равен: 1 — 0,2 = 0,8 см3,
т. е. 80%.
Что касается удельного веса волокон ткани, то он часто при расчетах
принимается равным 1,3. Точное определение удельного веса волокон в
каждом отдельном случае лучше определить отдельно, применив волю-
мометр Гуревича и Вендта (стр. 208).
Приводим объем пор некоторых видов тканей: объем пор шерстяной
фланели равен 92,3%, льняного трико—73,3%, гладкой льняной ткани—48,9%.
Теплопроводность
Под теплопроводностью ткани понимают количество тепла в малых кало-
риях, проходящее в 1 секунду через 1 см2 ткани при толщине ее в 1 см и
температурной разнице на противоположных поверхностях 1°С.
Здесь приведена методика измерения теплопроводности ткани при помощи
калориметра (диатермометра).
Определение теплопроводности ткани посредством калориметра (диатермометра)
Калориметр, или диатермометр (рис. 288), состоит из двух латунных цилиндров А
и вставляющихся один в другой. В верхней части цилиндра А ввинчена небольшая ме-
таллическая трубочка а, в которую вставлен конец толстостенной стеклянной трубочки В;
при помощи менделеевской замазки этот конец плотно приделан к трубочке а. Трубка В,
как это показано на рисунке, дважды изогнута под прямым углом: ее внутренний диаметр
равен 2 мм. Свободный конец трубки погружают в стаканчик С с глицерином удельного
веса 1,22, окрашенным в синий цвет метиленовой синькой. Длинное колено трубки снабжено
шкалой D. Высоту жидкости в трубке В устанавливают так, чтобы уровень ее был несколько
выше уровня глицерина в стакане С. Цилиндр А вместе с трубкой В представляет собой
воздушный термометр. Высота стояния жидкости в трубке колеблется в зависимости
от изменений температуры воздуха в цилиндре, а также от барометрического давления.
Над цилиндром А помещается крышка цилиндра Ап обозначенная на рисунке буквой Cv
К этой крышке припаян сверху цилиндрик с, в него плотно вставлена пробка, через которую
плотно же проходит правое колено трубки В. Между обоими цилиндрами помещают ткань,
подлежащую исследованию. Ее вырезают так, чтобы можно было равномерно обшить внут-
ренний цилиндр со всех сторон. Для этой цели вырезают три лоскута: один прямоугольной
формы для боковой стороны и два круглой формы для оснований. Взятую для исследования
ткань взвешивают: определяют толщину и удельный вес ткани. Внутренний цилиндр, об-
шитый тканью, помещают во внешний, на который плотно надевается его крышка. Внеш-
ний цилиндр после этого погружают в стакан с водой температуры 20° С и выжидают, пока
уровень жидкости в трубке остановится на некоторой высоте. Затем наружный цилиндр
погружают в стакан со смесью воды с толченым льдом. Охлаждение наружного цилиндра
489
вызывает охлаждение внутреннего цилиндра вместе с содержащимся в нем воздухом, благо-
даря чему глицерин в длинном колене трубки начинает подниматься. Чем теплопроводность
ткани выше, тем быстрее идет охлаждение воздуха во внутреннем цилиндре и тем, следо-
вательно, быстрее совершается подъем жидкости в трубке. За поднятием жидкости следят
по секундомеру, отсчитывая время поднятия на равные отрезки (2—3 см). Сделав несколько
отсч.тов, оставляют калориметр на некоторое время и отмечают максимальное поднятие
жидкости в трубке. В начале и в конце опыта отмечают также барометрическое давление.
Если за время опыта произошло изменение барометрического
давления, то в число, выражающее подъем жидкости вводят
соответствующий корректив.
Теплопроводность ткани определяется по следующей
формуле:
где К — абсолютная теплопроводность в малых калориях,
Р — вес внутреннего цилиндра в г,
С — удельная теплоемкость металла калориметра,
Д — расстояние между внешней поверхностью внут-
реннего цилиндра и внутренней поверхностью внеш-
него цилиндра в см2,
F — среднее арифметическое внешней поверхности
внутреннего цилиндра и внутренней поверхности
внешнего цилиндра в см,
е— = 2,7183, 1g е = 0,4343
W — водяной эквивалент исследуемого материала,
pig е — находят по формуле:
Рис. 288. Калориметр для
определения теплопровод-
где
Р — число, пропорциональное теплопроводности вещест-
ва, заполняющего пространство между цилиндрами,
a Ige имеет такое же значение, как и в предыду-
щей формуле, /?0 /1' и /ij — высоты стояния жидкости
в трубке в см в разные моменты наблюдения и в
конце опыта, /' и /0 — моменты отдельных отсче-
тов в секундах.
ности ткани.
zi п Aj — латунные цилин-
дры, а — металлическая
трубочка, В — стеклянная
трубка, Сг — крышка верх-
него цилиндра, с — метал-
лический цилиндрик, С —
стакан, D — шкала.
Выражение представляет для данного калориметра постоянный фактор, выводимый
на основании представленых выше значений для Р, С, Д, F и Ige. Для каждого калори-
метра это число означает количество тепла в малых калориях, проходящее через 1 см1 2 поверх-
ности в 1 секунду, если толщина слоя 1 равна 1 см, а температурная разница на противопо-
ложных поверхностях равна 1°С.
Результат, получаемый при проведении опыта описанным сейчас образом, зависит
в некоторой степени от количества ткани, помещенной в калориметр, и представляет
выражение для теплопроводности смеси воздуха и ткани, помещенной в калориметр.
Исходя из этих соображений, нужно перечислить найденную величину К на вес ткани,
равный 6 г. При этих условиях результат получается для одного и того же удельного
веса ткани, равного 6, поделенным на объем межцилиндрового пространства в см3. Это
так называемый «опытный» удельный вес, а редуцированная, таким образом, тепло-
проводность называется типической в отличие от истинной теплопроводности,
которую имеет ткань при ее естественном удельном весе.
Если, наконец, учесть толщину ткани и определить количество тепла в малых кало-
риях, которое проходит за 1 секунду через площадь в 1 см2 при разнице температуры в 1°С
ограничивающих плоскостей и при соответствующей толщине данной ткани, то получают
абсолютное количество тепла, которое проводит данная ткань в единицу времени.
Пример. Фактор калориметра — = 0,1018. Для исследования взято 5,8 г шерстяной
Flge
ткани. Удельный вес ткани 0,21, толщина 2,1 мм. Среднее значение для £lge из трех опре-
делений оказалось равным 0,00072. Подставляя эти значения в формулу для К, получаем:
К = 0,1018 • 0,00072 (1 + —— ’j.
\ 4PCI
Для вычисления W нужно вес материала помножить на удельную теплоемкость его; так как
удельная теплоемкость шерсти равна 0,552, а взято для опыта 5,8 г, то W окажется равным
1 Под толщиной слоя здесь понимают расстояние между ограничивающими поверхно-
стями.
3 Удельная теплоемкость для хлопка — 0,48, для льна — 0,46.
490
0,55 . 5,8; вместо Р и С нужно также поставить их значения. Пусть Р, т. е. вес внутреннего
цилиндра равен 172,5 г, а Сх, т. е. удельную теплоемкость латуни примем равной 0,094,
тогда получаем:
К = 0,1018 • 0,00072 (14-9’55_5’8----'l ~ 0,0000769.
4 • 172,5 • 0,094 /
Если теплопроводность воздуха принять равной 100, то теплопроводность исследуемой
ткани определится из пропорции:
X ; 100 = 0,0000769 : 0,0000558 * *.
Отсюда X = 137,8, т. е. относительная теплопроводность испытуемой ткани выше тепло-
проводности воздуха на 37,8%.
При перечислении на 6 г ткани эта разница повышается до 49,4, что видно из пропор-
ции:
Y : 37,8 = 6 : 5,8.
Следовательно, относительная теплопроводность исследуемой ткани выразится числом
Обозначив типическую теплопроводность через Z, находим ее из пропорции:
Z : 0,0000558 = 149,4 : 0,271, т. е. Z = 0,00008337.
Чтобы получить истинную теплопроводность, нужно учесть следующее: соответственно
объему межцилиндрового пространства, который в данном случае положим равным 22,1 см3,
и весу ткани в б г получаем «опытный» удельный вес, равный 0,271 (6 : 22,1), тогда как истин-
ный удельный вес для данной ткани, согласно условию задачи, равен 0,21; следовательно,
чтобы иметь в калориметре ткань в условиях естественного удельного веса, нужно было бы
взять не б г, а 4,65, что следует из пропорции:
X : б = 0,21 : 0,271.
При этих условиях относительная теплопроводность исследуемой ткани оказалась бы выше
теплопроводности воздуха не на 49, 4%, а на 38,28%, что находим из пропорции:
X : 49,4 = 4,65 : 6.
т. е. относительная теплопроводность исследуемой ткани при ее естественном удельном
весе равна 138,28.
В абсолютных цифрах это дает для истинной теплопроводности данной ткани 0,0000772
грам-калорий из пропорции:
X : 0,0000558 = 138,28 : 100.
Абсолютную теплопроводность для данной ткани при ее натуральной толщине, равной
2,1 мм, находим из пропорции:
X : 0,0000772 = 10 : 2,1, т. е. X = 0,0003676.
Приводим абсолютную теплопроводность некоторых тканей: шерстяное
трико—0,000635, хлопчатобумажное трико — 0,000994, хлопчатобумажное
полотно — 0,0018783, гладкая льняная ткань— 0,005795.
С момента появления кататермометра некоторые авторы (Скавронский,
Шальнов и др.) пытались применить этот прибор для определения теплопро-
водности ткани. Г. М. Кондратьев2 показал ошибочность такого рода опытов.
Сконструированный Г. М. Кондратьевым шаровой бикалориметр
для исследования теплозащитных свойств тканей пока не нашел применения
в наших лабораториях и здесь не описан.
Воздухопроницаемость ткани
Воздухопроницаемость представляет важное свойство ткани, от которого
зависит интенсивность обмена между воздухом подплатяным и наружным
(вентиляция подплатяных пространств).
Воздухопроницаемость ткани зависит от природы ткани, ее структуры,
аппретуры и степени увлажнения. В основном воздухопроницаемость опре-
деляется размерами отдельных пор, а также замкнутостью их в ткани.
1 0,0000558 — теплопроводность воздуха в мал. кал.
* См. Сборник ЛИГТ и Г13: «Лучистая энергия на производстве», Л. 1938.
491
По Рубнеру, коэфициент воздухопроницаемости ткани выражается числом
секунд, в течение которых через 1 см2 поверхности ткани при толщине ее
в 1 см проходит 1 см3 воздуха при давлении в 0,42 мм водяного столба; такое
давление испытывает тело человека при кажущемся полном отсутствии ветра.
Г. В. Хлопин и П. Е. Калмыков внесли два изменения в указанное опреде-
ление и тем самым дали коэфициенту воздухопроницаемости более рациональ-
ное выражение.
Хлопин предложил принимать за коэфициент воздухопроницаемости
ткани число куб. сантиметров воздуха, проходящего через 1 см2 поверхности
ткани при толщине ее в 1 см и давлении в 0,42 мм водяного столба; таким
образом, между значением коэфициента и проходимостью ткани для воздуха
устанавливается прямая, а не обратная зависимость, как это следует из опре-
деления Рубнера.
Рис. 289. Прибор для определения воздухопроницаемости тканей.
А — цилинд, D — закраина, Е — насадка, С — микроманометр, В — газовые часы, F — трехгорлая
склянка, g — винтовой зажим, Н — трубка для хлористого кальция, L — газометр.
Калмыков, а также Королев и Елюшина предлагают определять воз-
духопроницаемость ткани при ее естественной толщине, так как между тол-
щиной ткани и ее проходимостью для воздуха не существует закономерной
зависимости. Таким образом, учитывая указанные коррективы, под коэфи-
циентом воздухопроницаемости ткани следует понимать число куб. санти-
метров воздуха, проходящего через 1 см2 ткани при естественной ее толщине
в 1 секунду при давлении в 0,42 мм водяного столба.
Для определения воздухопроницаемости можно пользоваться прибором,
изображенным на рис. 289.
Прибор состоит из медного цилиндра А диаметром в 10 см и такой же при-
близительно высоты. От цилиндра отходят две трубки: одна присоединяется
к газовым часам В, другая — к диферценциональному манометру С. К верх-
нему краю цилиндра припаяна кольцеобразная закраина D, на которую
помещается медная насадка Е. К этой насадке сверху прикрепляется испы-
туемая ткань. Рубнер применял насадки с просветом в 109,3 см2 и 16,6 см2.
Перед газовыми часами помещается трехгорлая склянка F. Одно из горлы-
шек склянки соединяется с газовыми часами В, другое с газометром L, в третье
горлышко вставлена пробка со стеклянной трубкой, на которую надета кау-
чуковая трубка. На эту последнюю накладывают винтовой зажим g. При
помощи зажима увеличивают или уменьшают отток воздуха наружу, что
дает возможность регулировать струю воздуха, поступающего в газовые
часы, и поддерживать определенное давление в цилиндре А, регистрируемое
микроманометром С. Воздух из газометра перед пуском в колбу следует не-
сколько подсушить, пропустив его через U-образную трубку с хлористым
кальцием, иначе ткань, сорбируя влагу, уменьшает свою воздухопроницае-
мость.
492
Пустив из газометра струю воздуха и установив в системе соответствующее
давление, следят за показанием газовых часов и одновременно отсчитывают
время по секундомеру.
Калмыков предлагает для определения воздухопроницаемости пользо-
ваться установкой, изображенной на рис. 290. Здесь А — цилиндр для закреп-
ления испытуемой ткани диаметром в 5 см; в — реометр для измерения объема
проходящего воздуха; благодаря меняющейся диафрагме можно измерять
различные объемы воздуха — от десятков миллилитров до десятков литров
в минуту; С — колба, служащая для выравнивания давления в системе:
в пробку колбы вставлена трубка, сообщающаяся с реометром, и тройник,
Рис. 290. Прибор Калмыкова для определения воздухопро-
ницаемости тканей.
А — цилиндр для закрепления ткани, Б — реометр, С — колба,
Е — винтовой зажим, D — воздуходувка, G — трехходовый кран,
Fx, F, и FB — манометры.
одно колено которого
сообщается с воздухо-
дувкой D. На другое
колено тройника наде-
та короткая резиновая
трубка, на которую на-
ложен винтовой зажим
Е. Зажимом регули-
руют количество воз-
духа, просасываемое че-
рез ткань. Fx — обыч-
ный водяной манометр
для средних отрица-
тельных давлений, F2 —
ртутный манометр для
больших отрицательных
давлений, F3 — диферен-
циальный манометр для
малых отрицательных
давлений, g — треххо-
довый кран, благодаря которому в систему можно включить любой из мано-
метров, в зависимости от того, какое отрицательное давление нужно развить
при данном опыте. Очевидно, чем проходимость ткани для воздуха менее,
тем большее отрицательное давление приходится развить в данном опыте.
Установка Калмыкова отличается универсальностью и рекомендована
автором на основании длительного испытания ее.
Формула для определения коэфициента воздухопроницаемости имеет
следующий вид:
к v • 0,42
S-Р-Т ’
где К — коэфициент воздухопроницаемости,
v — объем воздуха (в см3), прошедшего через ткань за время опыта,
S — поверхность ткани (в см2),
Р — давление в мм водяного столба,
Т — продолжительность опыта в секундах.
Пример. В течение 10 минут через ткань поверхностью 19,6 см* прошло б л воздуха при
давлении 1,2 мм водяного столба. Нужно определить коэфициент воздухопроницаемости
ткани.
Подставив в приведенную выше формулу соответствующие значения получаем:
К-----6000 • °’42 ^0,18-
19,6 • 1,2.600
Водопроницаемость
Водопроницаемость тканей, в общем, зависит от тех же свойств, от которых
зависит и воздухопроницаемость их. Методы, применяемые для определения
водопроницаемости тканей, могут быть подразделены на два вида: либо
493
ткань подвергают давлению определенной массы воды, приведенной в непо-
средственное соприкосновение с тканью, и определяют время, необходимое
для прохождения воды через толщу ткани, либо орошают ткань водою,
которая падает на нее дождем, и определяют то же время.
Из методов первого вида здесь описан метод Виноградова—Волжинского,
из методов второго вида — метод Котельникова.
Метод Виноградова—Волжинского
Прибор Виноградова—Волжинского (рис. 291) состоит из капсулы а и
бюретки g. соединенных между собой каучуковой трубкой. Каждое деление
бюретки соответствует 0,1 мл. Кран
бюретки двухходовый, благодаря чему
бюретка может сообщаться с полостью
капсулы либо с наружным воздухом.
Сверху бюретка плотно закрывается кау-
чуковой пробкой с двумя отверстиями;
в одно из них вставлена шейка малень-
кой делительной воронки, в другое —
конец дважды загнутой под прямым
углом стеклянной трубки, на которую
надета каучуковая трубка е со стеклян-
ным наконечником. Капсула прибора,
как видно из рисунка, составляется из
трех частей: полого цилиндра d, имею-
щего внутри круговой выступ, на кото-
рый помещается кружок испытуемой
ткани диаметром в 26 мм; цилиндричес-
кой трубки с, плотно входящей в верхнее
расширенное отверстие цилиндра; коль-
ца навинчивающегося на цилиндр d
после того, как в него положен кружок
ткани и вставлена трубка с.
Методика исследования следующая:
сообщив бюретку с капсулой, на-
ливают в бюретку некоторое количест-
во воды и устанавливают капсулу так,
чтобы нижнее деление бюретки нахо-
дилось на одном уровне с поверхностью
выступа цилиндра, на который поме-
щают испытуемую ткань. Закрепив кап-
Рис. 291. Прибор Виноградова—Волжин-
ского для определения водопроницае-
мости тканей.
а — капсула, g — бюретка, е — каучуковая
трубка. Справа — деталь показывающая стро-
ение капсулы, d — полый цилиндр, с — цилин-
дрическая трубка, b — кольцо навинчиваю-
щееся на цилиндр d и скрепляющее части due.
сулу в этом положении, отсасывают
ртом при помощи трубки е из бюретки
воздух, пока уровень воды в бюретке
поднимется соответственно 5 мл, после
чего, повернув кран, разобщают бюрет-
ку с капсулой. Обтерев края выступа
цилиндра а фильтровальной бумагой,
помещают на этот выступ кружок иссле-
дуемой ткани, вводят в цилиндр трубку с и навинчивают кольцо b на цилиндр.
Поверх ткани помещают кружок фильтровальной бумаги, смачивание кото-
рого водой указывает момент проникновения воды через толщу ткани. Далее
вводят в бюретку через делительную воронку такое количество воды, чтобы
она соответствовала столбу высотой в 60 мм, и снова сообщают бюретку с кап-
сулой. Как только вода опустится в бюретке на 5 мл, т. е. уровень воды в кап-
суле поднимется до поверхности ткани, обращенной внутрь цилиндра, пускают
в ход секундомер и наблюдают момент, когда фильтровальная бумажка
494
окажется смоченной водой. Прохождение воды через ткань, как ясно из ска-
занного, совершается под давлением водяного столба высотою в 60 мм. Водо-
проницаемость ткани выражается в секундах, в течение ксторых происходит
промокание ткани.
В других приборах подобного типа водопроницаемость выражается макси-
мальным давлением, потребным для того, чтобы ткань промокла тотчас же,
как только придет в соприкосновение с водой (прибор Шоппера).
Метод Котельникова
Динамопенетрометр ВЦИООТ1 (рис. 292), представляющий собой видо-
изменение прибора Котельникова, состоит из бака С, наполняющегося водой
лишь до определенного уровня, благодаря
клапану Е. Уровень воды в баке контроли-
руется при помощи водомерной трубки D. От
бака отходит дождевая воронка, в отверстия
которой впаяны небольшие медные направ-
ляющие трубочки, благодаря которым вода
вытекает вертикальными струйками. Другая
часть прибора состоит из бака, в центре кото-
рого помещается диск В, где фиксируется
ткань. Диск представляет собой один полюс
электрической цепи, питающейся током от
небольшой батареи либо аккумулятора. По-
явление тока в цепи определяется при помощи
миллиамперметра К. Другой полюс цепи в
виде небольшой медной пластинки Д, при-
паянной к проводу, можно по желанию при-
близить или удалить от диска В. Установка
обеспечивает давление воды, равное 70 динам
на 1 см2 ткани.
Методика исследования заключается в
Рис. 292. Динамопенетрометр
ВЦИООТ.
С — бак, Е — клапан бака. D — водо-
мерная трубка. В — диск для фикси-
рования ткани, А — медная пластинка.
К — миллиамперметр.
следующем: кружок испытуемой ткани фик-
сируется на диске В, после чего на ткань
пускают из бака дождь. Время от времени
пластинку накладывают на смачиваемую
ткань, наблюдая, появится ли ток в цепи. До тех пор, пока ткань не про-
мокла насквозь, она не проводит тока, но лишь только она промокнет, в
цепи появляется ток, о чем можно судить по отклонению стрелки мил-
лиамперметра.
Водопроницаемость или водоустойчивость ткани оценивается по времени,
которое проходит с момента, когда пущен дождь, до момента появления
тока в цепи, т. е. по времени, потребном для промокания ткани.
Водоемкость
Различают максимальную и минимальную водоемкость.
Под максимальной водоемкостью ткани понимают то
количество воды, которое находится в ткани при условии заполнения всех
пор ее водою так, что избыток воды свободно стекает.
Под минимальной водоемкостью понимают то количество
воды, которое остается в мельчайших порах после максимального выжимания
руками воды из смоченной ткани. Сжимающая сила руки принимается равной
25 кг. Такой метод определения минимальной водоемкости ткани не может
считаться точным.
1 ВЦИООТ—Всесоюзный Центральный Институт Организации и Оздоровления Труда.
495
ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ТКАНИ
Химическое исследование ткани наичаще касается определения гигро-
скопичности ее, влажности, наличия ядовитых красок (мышьяк), способности
ткани к сорбции тех или иных паров и газов или определения сорбированных
уже газов и, наконец, некоторых проб с целью идентификации ткани.
Гигроскопичность
Различают гигроскопическую воду, т. е. ту, которая связана
с самим веществом ткани, ипромежуточную воду, т. е. ту, которая
в силу капиллярности содержится в порах между отдельными волокнами
ткани. Гигроскопичность ткани определяется по максимальному количеству
гигроскопической воды, которая .содержится в единице веса данной
ткани.
Для определения гигроскопичности кусочки ткани (20 х 20 см) высуши-
ваются в течение суток под колоколом, где помещают одновременно погло-
титель для влаги, затем кусочки ткани взвешиваются (в закрытой бюксе),
после чего выдерживаются в течение суток под колоколом над водой и вновь
взвешиваются. Прибыль в весе относят к 1 кг, либо выражают в процентах.
Для различных тканей способность сорбировать влагу колеблется в пределах
от 8,4 до 20% веса ткани, а по некоторым авторам и выше.
Влажность
Для определения влажности ткани взвешивают некоторое количество
ткани, высушивают при 105°С до постоянного веса и находят потерю в весе.
Влажность выражают в процентах к начальному весу или к весу сухого
вещества.
Определение мышьяка
Испытуемую ткань в количестве 20—30 г разрезают на куски, обливают
50—100 мл 25% серной кислоты и нагревают при 50—60°С втечение 18—24 43-
сов. Мышьяковые соединения при этом переходят в раствор.
Раствор фильтруют, доводят фильтрат до 200 мл и затем переносят частью
или целиком в аппарат Марша.
Для количественного определения необходимо органические вещества
пробы подвергнуть разрушению при помощи соляной кислоты и бертолетовой
соли, как описано выше (см. определение мышьяка в пыли). После этого
количественное определение может быть произведено одним из описанных
выше методов (см. стр. 262 и 357).
Сорбция газов
Способность тканей сорбировать газы (пары) из воздуха зависит от природы
ткани, ее структуры, окраски, аппретуры, а также от природы данного газа,
концентрации его; важное значение в данном вопросе имеют степень влажности
ткани, температура среды и продолжительность .^прикосновения ткани и
газа (экспозиция).
Вопросом поглощения газов тканями занимались многие авторы (Углов,
Васильева и др.). Здесь описаны некоторые из предложенных методов.
Метод Васильевой
По Васильевой, сорбционная способность ткани в отношении данного газа
определяется следующим образом: вырезаются куски ткани определенного
размера (для хлопчатобумажной ткани в 100 см2, а для шерстяной в 60 см2);
496
длительным кипячением и промыванием эти куски ткани освобождаются от
аппретуры (см. стр. 486). Исследования производятся в отношении ткани
в воздушно-сухом состоянии ее и в отношении ткани, высушен-
ной при 100°С. В последнем случае последние два часа высу-
шивание ведется в маленьких стеклянных трубках, состоящих
из двух частей, соединяющихся посредством шлифа и снабжен-
ных стеклянными кранами (рис. 293). Трубки взвешиваются
вместе с лоскутами ткани, после чего через них пропускается
ток высушенного газа до полного насыщения ткани газом, о
чем судят по прекращению увеличения веса трубки с тканью.
После этого в течение получаса пропускают через трубку с
тканью ток чистого воздуха для удаления избытка газовой
смеси и взвешивают трубку с тканью. Привес соответствует
количеству поглощенного газа.
На рис. 294 дана схема определения сорбции газов тканя-
ми одежды, по методу Васильевой.
Газ из баллона А проходит через склянку В, где он подвергается
осушке, затем через реометр D направляется в стеклянную бутыль Е
емкостью в 15 л, служащую для смешивания газа с воздухом и получения
нужной концентрации. Избыток газа через трубку С может быть выведен
наружу (в тягу). Перед входом в смеситель Е газ смешисается с током
воздуха, нагнетаемого в прибор насосом. Воздух из трубки/7 проходит через
газовые часы К, отсюда через осушительную колонку L и затем уже на-
правляется в бутыль Е, смешиваясь по пути с газом в трубке а. Из 6уты-
Рис. 293.
Трубки для
испытания
ли Е смесь газа с воздухом направляется в трубку Af, а затем в трубку N,
где находится испытуемая ткань. Из трубки N смесь воздуха и газа через
трубку R уходит в тягу.
При желании проверить правильность показаний реометра D газ из
трубки М направляется в склянку 5 с раствором, способным поглощать
данный газ. Просасывание газовой смеси через поглотитель, находящийся
осуществляется при помощи водяного аспиратора О. Проанализировав жидкость в склянке
тканей на
сорбцию
газов, по
Васильевой.
в склянке S,
Рис. 294. Установка Васильевой для определения сорбции газов тканями,
А — баллон для газа, В — склянка для сушки газа, D — реометр. Е — стеклянная бутыль для сме-
шивания газа с воздухом. С — трубка для отвода избытка газа, F — трубка для ввода возтуха. К — газо-
вые часы, L — осушительная колонка, а — трубка для смешивания воздуха и газа, М — трубна для
подвода газа к трубке N с испытуемой тканью, R — трубна для отвода газа в тягу, 8 — поглотитель-
ная склянка для газа. О — аспиратор.
S, можно учесть количество поглощенного газа и отсюда сделать заключение о правиль-
ности показаний реометра. При надобности вместо одной берут 2—3 поглотительные
склянки.
После пропускания газа трубка N отъединяется от прибора и, как уже было указано,
через нее пропускают ток чистого воздуха для удаления избытка газовой смеси, после чего
трубка с тканью, сорбировавшей газ, взвешивается.
32. А. И. Ьурштейн.
497
Метод автора
Прибор для волюмометрического определения сорбции газов твердыми телами и, в част-
ности, тканями одежды (рис. 295) состоит из бюретки (2) емкостью в 100 мл. Узкая часть
бюретки емкостью в 10 мл имеет деления соответственно 0,01 мл. Бюретка соединена при
помощи резиновой трубки с уравнительным сосудом (7). В бюретку и уравнительный сосуд
Рис. 295 Прибор автора для определения
сорбции газов тканями.
/ — уравнительный сосуд, 2 — бюретка, 3 — термо-
стат, 4 — термометр б — кран для сообщения урав-
нительного сосуда и бюретки, в — винтовой зажим,
7 — сосуд для испытуемой ткани, 8 — термостат,
9 — термометр, 10 — трехходовый кран. 11 — сосуд
для газообраэующей жидкости. 12 — термостат, 13 —
термометр, 14 — воронка 15 и 16 — трехходовые
краны, 17 — штатив прибора.
Капилляр, служащий продолжением пробки
наливают ртуть. Бюретка помещена в
термостат (5), в котором налита вода.
В термостате имеется термометр. Для
сообщения уравнительного сосуда и бю-
ретки служит кран (5). На чертеже он
показан на соединение Винтовой зажим
(6) служит для точной регулировки вы-
соты стояния ртути в бюретке. Испытуе-
мую ткань помещает в сосуд (7) емкостью
в 10—20 мл. В сбсуд вводят пришлифо-
ванную полую пробку, продолжением
которой служит капилляр, соединенный
с одной стороны (справа) с бюреткой и
с другой стороны (слева) дающий гори-
зонтальное ответвление, заканчивающееся
трехходовым краном (16) От горизон-
тального ответвления капиллярной труб-
ки вертикально вниз идет капилляр,
заканчивающийся полой пробкой, на
которую надевается пришлифованная
шейка сосуда (7 7). В сосуд через ворон-
ку (14) вводят газообразующую жид-
кость (хлорпикрин, акролеин и т. п.).
Капилляр, служащий продолжением по-
лой пробки сосуда (7), снабжен треххо-
довым краном (10), при помощи кото-
рого можно соединить этот сосуд со всей
остальной системой, как показано на
рисунке, а при надобности — с наруж-
ным воздухом. При помощи этого же
крана сосуд может быть одновременно
изолирован и от внешней среды и от всей
остальной системы. Сосуд (7) помещен в
термостат (8), в который наливается
вода. В термостате имеется термометр (9),
сосуда (7 7), снабжен трехходовым краном
(75), при помощи которого этот сосуд может быть соединен со всей системой, как это пока-
зано на рисунке, либо изолирован от нее. При помощи этого же крана сосуд может быть
сообщен с внешней средой. Сосуд (7 7) помещен в термостат (72), в котором имеется термометр
(73). Весь прибор смонтирован на штативе (77).
Определение сорбции газа (пара) тканью при помощи описанного прибора произво-
дится следующим образом. Сосуд (11) при помощи крана (75) изолируют от всей системы
и от наружного воздуха. Испытуемую ткань помещают в сосуд (7) и определяют получив-
шуюся навеску, после чего сосуд плотно надевают на его пробку. Установив кран (16) на
соединение с наружным воздухом, опускают ртуть в бюретке при помощи уравнительного
сосуда до узкой части, напр. до 91—92 деления; кран (10) при этом должен быть уста-
) новлен на сообщении сосуда (7) с системой.
В термостаты (3 и 8) наливают воду такой температуры, при которой желательно вести
наблюдение, и, подождав некоторое время, пока воздух в бюретке примет температуру
воды, изолируют при помощи крана (16) систему от наружного воздуха. Установив точно
при помощи винтового зажима (6) стояние ртути в бюретке при данном барометрическом
давлении и записав найденный отсчет, подвешивают уравнительный сосуд так, чтобы
внутри системы сохранилось давление, близкое к данному барометрическому, и оставляют
прибор, записав атмосферное барометрическое давление и температуру воды в термостате.
Теперь необходимо выжидать, пока между испытуемой тканью и воздухом системы
не установится гигроскопическое равновесие. Чтобы убедиться, что такой момент наступил,
приводят объем воздуха всей системы (объем свободной части бюретки -f- объем свободной
части сосуда (7) 4- объем капилляров системы) к нормальным условиям сейчас после за-
грузки ткани, а затем, установив через некоторое время (сутки) высоту стояния ртути
в бюретке при новых условиях температуры и барометрического давления, приводят опять
объем к нормальным условиям и сравнивают найденный при втором измерении объем с
полученным при первом измерении. Если результаты получились одинаковые, то считают,
что гигроскопическое равновесие наступило; в противном случае продолжают выжидать,
пока два последовательных измерения объема воздуха в системе не дадут одинаковые
результаты.
498
Когда гигроскопическое равновесие установилось, отключают при помощи крана (76>
сосуд (7) от всей системы и вводят в систему испытуемый газ. Эта операция производится
различно, в зависимости от того, какой газ (пар) желательно ввести в систему.
Положим, что в систему желательно ввести пары хлорпикрина; в этом случае в сосуд
(77) наливают жидкий хлорпикрин до крана (75), установив этот кран на сообщение сосуда
(7 7) с наружной средой, а затем, переводя кран в положение, указанное на чертеже, при
котором сосуд (7 7) оказывается сообщенным с системой, выжидают, пока в воздухе системы
не получится та концентрация хлорпикрина, которая интересует исследователя.
Ускорение испарения хлорпикрина, как и другой жидкости, может быть достигнуто
двояко: в термостат (72) можно налить теплую (горячую) воду либо можно создать в си-
стеме пониженное давление, установив в уравнительном сосуде ртуть ниже, нежели в бюретке.
Получение в системе необходимой концентрации паров хлорпикрина устанавливается
на основании измерении объема воздуха + пары хлорпикрина.
Если испытуемый газ вводится из газовой пипетки или бюретки, напр., хлор, сер-
нистый ангидрид и т. п., то, присоединив пипетку или бюретку к наружному открытому
концу горизонтальной трубки системы, устанавливают трехходовый кран так, как показано
на чертеже, промывают газом соединительные трубки и затем, переведя кран на соединение
с системой, вводят в систему необходимый объем газа, после чего кран (76) закрывают.
Получив в системе необходимый объем газа, перемешивают его с воздухом, что дости-
гается многократным поднятием и опусканием уравнительного сосуда.
После этого при помощи крана (76) устанавливают сосуд (7) на сообщение с системой
и наблюдают за изменением объема воздуха 4- газ в системе. Уменьшение объема соответ-
ствует объему сорбированного газа испытуемой тканью. Результат переводят на вес
газа и выражают сорбцию в мг/г испытуемой ткани.
Так как стекло внутри системы само сорбирует газ, то до основного опыта производят
холостой опыт.
Удаление того или иного газа из системы достигается многократным поднятием и опуска-
нием уравнительного сосуда при открытом кране (76).
Для тех случаев, когда речь идет об определении неизвестного количе-
ства газа, сорбированного тканью, извлекают газ из ткани при помощи
соответствующего поглотителя и исследуют этот последний.
Так, например, для определения хлора ткань обрабатывают раствором
йодистого калия и оттитровывают выделившийся иод; для определения фос-
гена, ткань обрабатывают спиртовым раствором едкого натрия или калия
и исследуют раствор на хлор-ион; аммиак определяется обработкой ткани
слабым раствором серной кислоты с последующим усреднением и несслереза-
цией и т. д. Во всех случаях, конечно, раствор должен быть слит в чистую
колбу, а ткань повторно промыта, причем промывные воды сливаются в ту
же колбу.
Некоторые химические пробы для идентификации ткани
Отличие животных волокон от растительных. 10% раствор NaOH или КОН
растворяет волокна животного происхождения; растительные волокна при
этой пробе лишь набухают.
Азотная кислота удельного веса 1,2—1,3 окрашивает белые волокна
животного происхождения в желтый цвет (ксантопротеиновая реакция);
растительные волокна при этой пробе не изменяют своего цвета.
Слабый раствор пикриновой кислоты окрашивает волокна животного
происхождения в желтый цвет и не окрашивает растительные волокна.
Тимол с концентрированной серной кислотой (реактив Молиша) окра-
шивает животные волокна в желтый цвет, а растительные — в карминово-
красный.
При нагревании испытуемой ткани с раствором розанилина и последую-
щем промывании ткани в холодной воде ткань окрашивается в красный
цвет, если она животного происхождения, и не приобретает окраски, если
она растительного происхождения.
Г1ри кипячении в растворе уксуснокислого свинца животные волокна
чернеют от образования сернистого свинца; растительные волокна не изменяют
своего цвета.
При сжигании животные волокна издают запах жженого рога, а расти-
тельные волокна этого запаха не издают.
Отличие шерсти от шелка. Концентрированная соляная кислота при кипя-
чении растворяет шелк в течение 0,5—3 минут; шерсть при этой пробе не
изменяется,.
Отличие шелка от шерсти п хлопка. Смесь из 5 ч. ZnCl2, 1 ч. ZnO и 5 ч. Н20
при кипячении растворяет шелк в течение 15—20 минут, шерсть и хлопок
при этой пробе не растворяются.
Отличие льна от других волокон. При насыщении ткани жидким маслом
льняные волокна становятся прозрачными; другие волокна при этой пробе
остаются непрозрачными.
МИКРОСКОПИЧЕСКОЕ ИССЛЕДОВАНИЕ ТКАНЕЙ
Для микроскопического исследования ткани обесцвечивают небольшой
кусок ее хлором, кипятят отдельные волокна в течение 3 минут в 10% растворе
Рис. 296. Вид волокон различных тканей при рассмотрении в микроскоп.
АИВ — хлопок, С — лен, D — конопля, Е — джут, F — шерсть.
соды, затем в воде, подкисленной соляной кислотой, промывают дестиллиро-
ванной водой и исследуют во влажном виде при увеличении в 100—200 раз.
Идентификация производится на основании морфологических особенностей
волокон (см. рис. 296).
БАКТЕРИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ТКАНЕЙ
Ткани одежды (платья, пальто, шубы, обувь, головные уборы, белье,
спецодежда), а также инвентаря (ковры, занавесы, портьеры, мягкая мебель)
и т. п. бывают обыкновенно загрязнены пылью, а в некотсрых случаях выде-
лениями (частицы мокроты, слизи из зева и т. п.). Во всех этих загрязнениях
могут находиться и патогенные микроорганизмы (В. tuberculosis, В. tetanus,
В. perfringens, В. anthracis и споры, В. pyocyaneus, Streptococcus и др).
Особенно сильно загрязненными могут быть ношеная шерстяная одежда,
и ношеное белье.
При исследовании ткани одежды или инвентаря на бактериальное загряз-
нение обработка материала может идти разными путями. Наиболее рациональна
обработка вырезанного куска ткани, если это представляется возможным.
Небольшой кусок ткани определенного размера, измельчают ножницами и
переводят все лоскуты в стерильную банку с тепловатым физиологическим
раствором, тщательно взбалтывают с бусами и высевают на чашки Петри
с агаром. Выращивание и счет производятся так же, как при исследовании
пыли, почвы и т. п. Результат перечисляют на 1 см2 ткани. При исследовании
на фекальное загрязнение посев делают на среду Булира с последующим
500
пересевом на среду Эндо и т. д. При исследовании на патогенные микробы
физиологический раствор со взвесью, полученной из ткани, подвергается
центрифугированию. Осадок высевают на соответствующие среды и произво-
дят идентификацию по обычным методам (мазки, пестрый ряд, серологические
реакции, опыты на животных).
Если вырезать кусок ткани не представляется возможным, то поступают
следующим образом. Когда подлежащий исследованию предмет невелик,
его целиком замачивают в теплом физиологическом растворе и сильно встря-
хивают в течение длительного времени; когда же размеры предмета значи-
тельны, то с намеченного участка ткани делают смыв при помощи стерильного
ватного или марлевого тампона, захваченного стерильным пинцетом. Смыв
с одного и того же места повторяют многократно, меняя тампоны, которые
бросают в банку с физиологическим раствором. При возможности делают
еще соскребывание данной поверхности при помощи стерильного шпаделя
или скальпеля. Собранный материал тщательно встряхивают с бусами, и с по-
лученной взвесью поступают так, как описано выше. В этом случае результаты
получаются менее точными, нежели при обработке кусочка вырезанной ткани.
Исследование кожи и меха (овчины) на присутствие сибиреязвенных па-
лочек или спор производится при помощи реакции кольцепреципитации.
Материал может быть предварительно простерилизован (20—30 минут
при 120°С), так как положительная реакция получается и в том случае, когда
палочки и споры сибирской язвы убиты. Кусок кожи 5 х 10 см измельчают
ножницами и заливают физиологическим раствором в банке с бусами. Если
материал не был простерилизован, то операция взятия материала и его измель-
чения производится с предосторожностями (взятие пинцетом и ножницами,
измельчение под тягой в вытяжном шкафу).
Методику проведения реакции см. стр. 124.
Нужно иметь в виду, что кожа скота, вакцинированного против сибирской
язвы, часто дает положительный результат на реакцию преципитации и в тех
случаях, когда животное не болело сибирской язвой.
ЛИТЕРАТУРА
1. Архангельский А. Г., Волокна, пряжа, ткани, 1914.
2. Васильева П. И., О поглощении хлора, фосгена и сернистого газа тканями, диссер-
тация, Библиотека Военно-мед. академии, 1922.
3. Калмыков П. Е., Статьи в журн. «Гигиена и Санитария», 1936, 1938, 1947.
4. Керш Г. Г., Материалы по вопросу об исследовании обуви, дисс., СПБ, 1913.
5. Кондратьев Г. М., Сборн. Ленинградского института гигиены труда и профзабо-
леваний, «Лучистая энергия на производстве» Л., 1938.
б. Костямин Н. Н., Способы исследования тканей одежды с точки зрения гигиены,
дисс., 1909.
7. Кузьмин А. М, Хлопкопрядение, 1930.
8. Лбов А. Г., Прядение льна, 1927.
9. Лейтес Л. и Елисеева А., Шерстяное дело, № 4, 1931.
10. Лукашевский В. А., Текстильные товары, Методы исследования товаров, под ред.
Августинина А. И. и Воскресенского А. А., 1932.
11. Монахов А. Д., Общий курс технологии волокнистых веществ, 1924.
12. Насекин Н., Фридбург Е., Беленкова П., Микроскопические и физико-механические
исследования волокнистых материалов, Гизлегпром, 1934.
13. Никитин А. Ф., Способы и приемы практических наблюдений и исследований
по гигиене, 1911.
14. Николаев А. И., Шерстоведение, 1930.
15. Санков Е. А., Конспективное руководство по лабораторным работам над прядиль-
но-волокнистыми материалами, 1936.
16. Хлопин Г. В., Основы гигиены, т. II, 1923.
17. Шапошников В. Г., Общая технология волокнистых и красящих веществ 1923.
18. Шафранова А. С., Сигалов П, Я., Спецодежда, НКТП, 1932.
19. Эрисман Ф. Ф., Гигиена, т. II, 1887.
501
ГЛАВА ДЕСЯТАЯ
ДЕЗИНФЕКЦИОННЫЕ И ДЕЗИНСЕКЦИОННЫЕ
СРЕДСТВА
ДЕЗИНФЕКЦИОННЫЕ СРЕДСТВА
Формалин
Формалин представляет собой раствор формальдегида в воде. Продажный
формалин содержит в среднем 35—40% формальдегида, однако нередко встре-
чается формалин со значительно меньшим содержанием формальдегида. Кроме
формальдегида формалин содержит еще муравьиную кислоту и метиловый
спирт.
Формальдегид (НСОН) представляет газ специфического резкого запаха.
Вес литра 1,25 г. Сильно раздражает слизистые оболочки. В концентрации
0,025 мг/л и выше вызывает раздражение носа и глаз.
При производстве дезинфекции применяют на 1 м3 помещения от 5 до
40 г и более формальдегида; при дезинфекции предметов обихода и обста-
новки в тех случаях, где неприменима камерная дезинфекция, пользуются
увлажнением, обтиранием 2—4% раствором формалина. Во всех указанных
случаях необходимо проверять содержание формальдегида в формалине.
Качественное определение
I. По специфическому запаху.
2. К 5 мл серной кислоты прибавляют небольшое количество раствора
салициловой кислоты и 2—3 капли испытуемого раствора, после чего нагре-
вают на водяной бане. При наличии в испытуемом растворе формальдегида
получается красное окрашивание.
3. 0,2 г фуксина (нейтрального или основного) растворяют в 120 мл го-
рячей воды. По охлаждении прибавляют 20 мл 10% раствора сернистокис-
лого натрия (Na2SO3) и 2 мл концентрированной соляной кислоты; при этом
фуксин переходит в лейкобазу и раствор обесцвечивается. Через 2 часа после
обесцвечивания раствор готов к употреблению.
Если к 2—3 мл полученного реактива прибавить 2—3 капли испытуемого
раствора, содержащего формальдегид, то получается красное окрашивание
вследствие перехода лейкобазы вновь в краску. Описанный реактив пригоден
для обнаружения и других альдегидов.
4. К 3 мл 1% раствора азотнокислого серебра в пробирке прибавляют
небольшое количество аммонийной соли и аммиака до растворения выпада-
ющего осадка, после чего добавляют 2—3 капли испытуемого раствора. При
наличии в нем формальдегида на стенках пробирки образуется зеркало вслед-
ствие выпадения металлического серебра.
5. При выпаривании на водяной бане раствора, содержащего формаль-
дегид, часть формальдегида по испарении воды остается в виде белого твердого
полимера формальдегида-пароформа —(СНгО)2 и (СН2О)3. При дальнейшем
нагревании пароформ улетучивается в виде формальдегида.
502
Количественное определение
Мы описываем два метода определения формальдегида:
1) по окислению его в муравьиную кислоту и 2) по переводу его в гекса-
метилентетрамин (уротропин).
Определение формальдегида по методу окисления его в муравьиную кислоту
Принцип метода заключается в том, что при помощи иода в при-
сутствии щелочи формальдегид окисляется в муравьиную кислоту. Если
к определенному количеству раствора формальдегида прибавить определен-
ное же количество титрованного раствора иода, то по остатку иода, не всту-
пившего в реакцию, можно определить количество иода, пошедшего на оки-
сление формальдегида, и отсюда сделать заключение о количестве формаль-
дегида в растворе.
Реактивы. I. 0,1 N раствор иода.
2. 0,1 N раствор тиосульфата натрия.
3. 1% раствор крахмального клейстера.
4. 5% раствор едкого натрия.
5. Разведенная соляная кислота (1 ч. НС1 уд. веса 1,12 на 2 ч. воды).
Методика определения. 5 мл испытуемого формалина вводят
в мерную колбу на 250 мл, добавляют дестиллированной воды до метки и
тщательно взбалтывают. Из полученного раствора берут 5 мл в коническую
колбочку с притертой пробкой, прибавляют 10 мл 5% раствора NaOH и 40 мл
0,1 N раствора иода; последний раствор прибавляют из бюретки небольшими
порциями, все время взбалтывая жидкость в колбочке, закрываемой пробкой.
Происходящие при этом реакции можно представить схематически в следую-
щем виде:
НСОН + J2 + 3NaOH = HCOONa + 2NaJ + 2H2O.
Так как иод прибавляется в избытке, то часть его остается неизрасхо-
дованной. Через 20—30 минут после прибавления иода, приливают в колбу
30 мл разведенной соляной кислоты и избыток иода оттитровывают 0,1 N раст-
вором тиосульфата натрия:
J2 J- 2Na2S2O3 = 2NaJ J- Na2S4Oe.
Разность между числом миллилитров прибавленного раствора иода и
неизрасходованного иода указывает, какое число миллилитров 0,1 N раствора
иода пошло на окисление формальдегида в муравьиную кислоту. Из первой
реакции видно, что 1 мл 0,1N раствора иода, пошедший в реакцию, отвечает
НСОН
1,5008 мг НСОН соответственноследовательно, по количеству иода,
вступившего в реакцию, можно судить о концентрации формальдегида в испы-
туемом формалине.
Пример При проведении анализа, как описано выше, израсходовано на титрование
избытка иода 15,6 мл 0,1 N раствора тиосульфата натрия, следовательно, в реакцию всту-
пили 40 — 15,6 = 24,4 мл 0,1 N иода, что соответствует 1,5008 24,4 мг формальдегида.
Отсюда заключаем, что содержание формальдегида в испытуемом формалине равно:
1,5008 24,4.250-100 Л ,
------------------36,6%.
1000 -5-5
Определение формальдегида по методу перевода его в гексаметилентетрамин
Принцип метода заключается в том, что формальдегид при дей-
ствии на него аммиака переводится ь гексаметилентетрамин. Если к опреде-
ленному количеству раствора формальдегида прибавить определенное же
количество титрованного раствора аммиака, то по количеству аммиака, остав-
шегося свободным, можно сделать заключение о количестве аммиака, всту-
503
пившего в реакцию с формальдегидом, а отсюда и о количестве формальде-
гида в растворе.
Реактивы. 1. 1N раствор аммиака (17,024 г NH3 в 1 л раствора).
2. 1N раствор H2SO4.
3. 0,1% раствор метилоранжа в воде.
Методика определения. В коническую колбу емкостью в
150—200 мл с пришлифованной пробкой вводят точно 5 мл испытуемого раст-
вора, приливают сюда точно 50 мл 1N раствора аммиака, закрывают плотно
пробкой и оставляют в покое на 1 час в водяной бане или в шкафу при 36—
40°С. Между формальдегидом и аммиаком происходит следующая реакция:
6НСОН + 4NH3 = (CH2)eN4 + 6Н2О.
По прошествии часа прибавляют в колбу 2—3 капли раствора метилоранжа
и титруют избыток аммиака 1N серной кислотой:
2NH3 + H2SO4 = (NH4 )2SO4.
По расходу кислоты делают заключение об избытке аммиака, а отсюда
о количестве аммиака, вступившего в реакцию с формальдегидом. Из первой
реакции видно, что 1 мл 1N раствора аммиака отвечает 45,04 мл формальде-
6.НСОН _
гида соответственно----------. Следовательно, по числу миллилитров аммиака,
вступившего в реакцию с формальдегидом, можно сделать заключение о кон-
центрации формальдегида в испытуемом растворе.
Пример. При проведении анализа, как описано выше, израсходовано на связывание
остатка аммиака 20,2 мл 1 N H2SO4, следовательно, в реакцию с формальдегидом вступило
50 — 20,2 = 29,8 мл 1 N раствора аммиака, что соответствует 45,04. 29,8 мг формальдегида.
Отсюда заключаем, что содержание формальдегида в испытуемом формалине равно:
45,04 • 29,8 • 100
100-5
= 26,82<>/o.
Сулема
Сулема (HgCl2) — белый кристаллический порошок, плавится при 265°С,
кипит при 307°С. При 20°С в 100 частях воды растворяется 7,4 частей сулемы,
при 100°С —34 части. Применяется в виде растворов 1 : 1000 или 1 : 500
как антисептическое средство.
Продажные сулемовые таблетки готовятся обычно из равных весовых
частей сулемы и хлористого натрия. Последний ускоряет растворение
сулемы. Для окрашивания таблеток с целью опознавания применяется эозин
или другая каменоугольная краска. Таблетки содержат 1 или 0,5 г сулемы.
Качественное определение
1. При прибавлении в пробирке к раствору сулемы 2—3 капель раствора
KJ выпадает яркокрасный осадок иодной ртути (HgJ2); с избытком йодистого
калия HgJ2 образует растворимую бесцветную комплексную соль K2HgJ4.
2. При прибавлении в пробирке к раствору сулемы 1—2 капель раствора
AgNO3 образуется белый творожистый осадок AgCl.
3. Капля раствора сулемы, нанесенная на блестящую медную пластинку,
оставляет на ней серое пятно (Hg). После просушки и трения пятно приобре-
тает серебристый блеск.
Количественное определение
Принцип метода. Сулему при помощи KJ переводят в иодную
ртуть. Действуя на последнюю щелочью, получают окись ртути. При помощи
формальдегида восстанавливают окись ртути до металлической ртути, кото-
504
рую удерживают в растворе при помощи гуммиарабика. Прибавляя к раствору
определенное количество иода, получают вновь иодную ртуть. Избыток иода
оттитровывают тиосульфатом натрия и по расходу иода на связывание ртути
делают заключение о количестве сулемы.
Реактивы. 1 КJ кристаллический.
2. 15% раствор КОН.
3. 33% раствор гуммиарабика.
4. Раствор формалина, разбавленный (3 : 10) дестиллированной водой.
5. 30% раствор уксусной килоты.
6. 0,1N раствор иода.
7. 0,1N раствор тиосульфата натрия.
8. 1% раствор крахмального клейстера.
Методика определения. Сулемовую таблетку растворяют
в 100 мл дестиллированной воды и 20 мл полученного раствора переводят
в коническую колбочку с притертой пробкой. Прибавляют сюда 1 г кристал-
лического KJ, Ю мл 15% КОН и несколько капель 33% раствора гуммиара-
бика. При осторожном встряхивании колбы прибавляют в нее небольшими
порциями 3 мл разбавленного формалина. Через минуту добавляют 25 мл 30%
уксусной кислоты и 20 мл 0,1 N раствора иода. Взбалтывают и по растворении
ртути избыток иода оттитровывают 0,1N раствором тиосульфата натрия при
индикаторе крахмальном клейстере. Происходящие реакции можно пред-
ставить в следующем виде:
HgCl2 + 2KJ = HgJ2+2HCl.
HgJ2 + 2КОН = HgO -J- 2KJ + Н2О.
HgO 4- НСОН = Hg 4- НСООН.
Hg + J2 = HgJ2.
J2 2Na2S2O3 = 2NaJ 4- Na2S40g.
Из реакций видно, что одна молекула иода соответствует одной молекуле
сулемы. В весовых отношениях имеем: 2-127 весовых частей иода соответ-
ствуют 271,6 весовым частям сулемы. По сокращении на 2 получаем: 127 весо-
вых частей иода соответствуют 135,8 весовым частям сулемы. Следовательно,
1 мл 0,1N раствора иода соответствует 13,58 мг сулемы. На основании этих
данных и производят вычисления содержания сулемы в испытуемой таблетке.
Пример. При производстве определения, как описано выше, на титрование избытка
иода пошло 4,6 мл 0,1 N тиосульфата натрия, следовательно, на связывание ртути пошло
20—4,6 = 15,4 мл 0,1 N раствора иода, что соответствует 13,58 . 15,4 мг сулемы. Так как
на исследование была взята лишь одна пятая раствора, полученного от одной таблетки,
то во всей таблетке имелось:
13,58 • 15,4 -5 , _
------------~ 1,046 г сулемы.
Для количественного определения сулемы в препаратах, не содержащих NaCl, фарма-
копея рекомендует следующий метод: точную навеску (около 0,5 г) растворяют в 25 мл воды
в конической колбе на 250 мл, прибавляют 2 г цинковой пыли и нагревают на сетке с обрат-
ным холодильником в течение 1 часа так, чтобы смесь слабо кипела. По охлаждении раствор
фильтруют, осадок промывают 60 мл горячей воды, присоединяя промывные воды к полу-
ченному ранее фильтрату. К фильтрату прибавляют 50 мл 0,1 N раствора AgNO3H 5 мл HNO3.
Избыток азотнокислого серебра оттитровывают 0,1 N раствором роданида аммония (NH4CNS)
при индикаторе железоаммонийных квасцах.
Протекающие реакции можно представить так;
HgCl2 + Zn = ZnCl2 + Hg.
ZnCl2 + 2AgNO3 = 2AgCl + Zn(NO3)2.
AgNO3 + NH4CNS = AgCNS + NH4NO3
(NH4)2SO4Fe2(SO4)3.24HO2 + 6NH4CNS = 2Fe(CNS)3 + 2(NH4)2SO4 + 24H2O
505
От общего объема прибагленного раствора 0,1 N AgNO3 отнимают остаток, не вошедший
в реакцию с ZnCl2 (количественно этот остаток равен расходу при титровании 0,1 N раствора
NH4CNS). Определив число миллилитров 0,1 N раствора AgNO3, вступившего в реакцию
с ZnCl2, умножают это число на 0,01358 соответственно -—— , получая таким обра-
зом вес сулемы в граммах во взятой навеске. Отсюда делают заключение о процентном со-
держании сулемы в препарате
Пример. При проведении исследования, как описано выше, взята навеска в 0,52 г.
Расход 0,1 N роданида аммония составлял 12,8 мл. Отсюда заключаем, что содержание
сулемы в препарате:
0^145^-1^100 = 97] ,
0,52
Карболовая кислота
Карболовая кислота, фенол (С6Н5ОН) представляет собою по сути не кис-
лоту, а ароматический спирт. Это нейтральные на лакмус бесцветные иглы,
храктерного запаха, окисляющиеся при стоянии на воздухе, окрашиваясь
при этом в красный цвет. Температура плавления 42—43°С, температура
кипения 182—183°С. Применяется как дезинфицирующее средство в 3—5%
растворах.
Качественное определение
1. По специфическому запаху.
2. Раствор (1%) фенола дает с каплей раствора хлорного железа сине-
фиолетовое окрашивание.
3. Раствор (1%) фенола при прибавлении бромной воды даст кристалличе-
ский осадок трибромфенола.
Количественное определение
Принцип метода. При взаимодействии иода с фенолом образуется
дииодфенолиод (C6H3J2 . OJ)—осадок красного цвета. Если к определен-
ному объему раствора фенола прибавить определенное же количество титро-
ванного раствора иода, взятого в избытке, то по остатку непрореагировав-
шего иода можно судить о количестве иода, вступившего в реакцию с фенолом
и, следовательно, о количестве последнего.
Реактивы. 1. 15% раствор NaOH.
2. 0,IN раствор иода.
3. Раствор серной кислоты 1 : 5.
4. 0,1N раствор тиосульфата натрия.
5. 1% крахмальный клейстер.
Методика определения. 2г испытуемой карболовой кислоты
растворяют в 40 мл 15% раствора NaOH и добавляют дестиллированной
воды до 250 мл.
10 мл полученного раствора, содержащего 0,08 г испытуемой карболовой
кислоты, вводят в коническую колбу емкостью в 250 мл, подогревают до
60—65°С и приливают из бюретки 0,1N раствор иода до окрашивания жид-
кости в темнобурый цвет. При этом иод взаимодействует с фенолом по реакции:
С6Н5ОН + 3J2 = C6H3J2OJ +3HJ.
По охлаждении жидкости ее подкисляют серной кислотой, прибавляют
дестиллированной воды до объема 250 мл и фильтруют. Из фильтрата отби-
рают в отдельную колбочку 100мл иоттитровывают избыток иода 0,lN раство-
ром тиосульфата натрия при индикаторе крахмальном клейстере. Число
миллилитров раствора тиосульфата, пошедшее на титрование, умножают на
2,5, определяя таким образом весь избыток 0,1N раствора иода, не вступив-
506
шего в реакцию с фенолом. Отнимают от общего объема прибавленного
раствора иода непрореагировавший избыток и определяют таким образом число
миллилитров иода, вступившего в реакцию с фенолом.
Из приведенной выше реакции видно, что каждый миллилитр 0,1N раствора
иода, вступившего в реакцию с фенолом, отвечает 0,001567 ч. фенола соответ-
С н ОН
ственно 6|q5 |QQ- Помножив на 0,001567 число миллилитров 0,lN раствора
иода, вступившего в реакцию с фенолом, определяют количество фенола во
взятой пробе.
Пример. При производстве определения, как описано выше, прибавлено 62 мл 0,1 N
раствора иода. При титровании 100 мл фильтрата израсходовано 4,8 мл 0,1 N раствора
тиосульфата натрия. Отсюда делаем вывод, что не прореагировало с фенолом 4,8 х 2,5 =
= 12 мл 0,1 N раствора иода Следовательно, в реакцию вступило 62 — 12 = 50 мл 0,1 N
раствора иода, что соответствует 0,001567 х 50 = 0,07835 г. В процентном выражении
получаем:
о.ота.ю»
0,08
Количественное определение фенола по фармакопее производится следующим образом:
около 0,5 г препарата (точная навеска) растворяют в воде; раствор вводят в мерную колбу
на 250 мл, доводят водой до метки и тщательно перемешивают
Из полученного раствора отбирают 25 мл и переводят в колбу с притертой пробкой,
приливают сюда же 50 мл 0,1 N раствора бромата калия, 1 г бромида калия и 10 мл 50 %
серной кислоты. Все хорошо перемешивают и оставляют на 15 минут К смеси добавляют
2 г иодида калия, смесь сильно взбалтывают, ставят ее на 10 минут в темном месте, после чего
выделившийся иод оттитровывают 0,1 N раствором тиосульфата натрия при индикаторе
крахмальном клейстере. Происходящие при этом реакции можно выразить следующим
образом:
КВгО3 + 5КВг + 3H2SO4 = 3K2SO4 4- ЗН2О + ЗВг2
CeH5OH -J- ЗВг2 = СвН3Вг2ОВг 4- ЗНВг
2KJ4-Br2 = 2KBr + J2
J., 4- 2Na2S.,O3 = 2NaJ + Na2S4Oe.
Параллельно ставят контрольный опыт. 1 мл 0,1 N раствора тиосульфата натрия отве-
С,Н5ОН
чает 0,001567 г чистого фенола соответственно g'. j q. f qqq >
Пример. Взятая навеска фенола равна 0,45 г. При производстве определения, как опи-
сано выше, на опытную пробу израсходовано 22,5 мл 0,1 N тиосульфата, на холостую пробу
49,6 мл того же раствора
Отсюда заключаем, что в испытуемом препарате содержится фенола
0,001567(49,6 — 22,5) • 100
—----------—------—-------94,4%.
0,045
Карболовая кислота (неочищенная)
Неочищенная карболовая кислота (черная карболка) представляет смесь
фенолов, крезолов (СвН4-СН3ОН) и др. продуктов перегонки каменного
угля. Это маслянитстая жидкость буроваточерного цвета, остающаяся после
выделения кристаллического фенола из каменноугольного дегтя. С водою
образует мутную смесь. Применяется для дезинфекции уборных, выгребных
ям и пр.
Содержание крезолов (орто-, мета- и пара-) в хороших сортах черной
карболки достигает 90—95%; в худших сортах —45—50%. Содержание
фенолов невелико, количество воды не должно превышать 5%.
Пробу для анализа берут после тщательного перемешивания жидкости.
Если тщательное перемешивание невозможно, то пробу берут в 3 разных
местах по высоте сосуда: у дна, посредине и сверху. Отобранные пробы в ко-
личестве 2 л сливают вместе, хорошо перемешивают и помещают в склянки
с притертыми пробками.
507
Качественное определение
Качественные реакции те же, что и для очищенной карболовой кислоты.
Количественные определения
Определение количества воды. В колбу на 250 мл с отводной трубкой
вводят 100 мл исследуемого продукта, соединяют с холодильником и отгоняют,
постепенно доводя нагрев содержимого колбы до 170°С. Погон собирают
в измерительный цилиндр емкостью в 25 мл. Отсчет водяного слоя производят
после отделения его от маслянистого слоя.
Определение фенолокрезолов. В мерный цилиндр с притертой пробкой
вводят 80 мл 10% раствора едкого натрия, приливают 10 мл испытуемой
черной карболки и 10 мл петролейного эфира. Закрыв цилиндр пробкой,
тщательно взбалтывают содержимое и оставляют в покое на 1 час. Феноло-
крезолы при этом переходят в раствор, образуя со щелочью фенолаты и кре-
золаты, а другие примеси всплывают на поверхность в виде маслянистого
слоя.
По истечении часа отбирают пипеткой от отстоявшейся нижней части
50 мл жидкости и переносят в небольшой градуированный цилиндр, под-
кисляют с избытком соляной кислотой, сильно взбалтывают и оставляют
в покое на сутки. По истечении указанного времени фенолокрезолы соби-
раются на дне цилиндра в виде заметно отграниченного слоя. Отсчитывают
объем этого слоя и умножают на 20, получая таким образом содержание фе-
нолокрезолов в процентах.
Хлорная известь
Белый или слегка сероватый порошок с сильным запахом хлора. Частично
растворяется в воде. Состоит из смеси ряда продуктов: Са(0С1)2, СаС 12, Са(ОН)2,
СаС1(0С1). Применяется для обеззараживания воды, почвы, а также отхожих
мест и т. п.
Дезинфицирующее действие хлорной извести основано на ее способности
выделять активный хлор. Количество активного хлора в хлорной извести
не должно быть ниже 25%.
Пробу для анализа достают из бочек при помощи железного щупа, который
вводится до 3/4 глубины бочки по вертикальной оси. Если партия большая,
то пробу берут не менее как из 10% всех бочек. Взятые из разных бочек пробы
тщательно перемешивают и помещают в банку с притертой пробкой. Общий
вес взятой пробы должен быть около 1 кг.
Качественное определение
1. По запаху хлора.
2. Если к подкисленному (серной, соляной, уксусной кислотой) раствору
препарата прибавить раствор йодистого калия, то выделяется свободный
иод, который можно обнаружить, прибавив 1% раствор крахмального клей-
стера.
3. Прокипяченный до полного удаления хлора раствор препарата в раз-
веденной уксусной кислоте дает с раствором оксалата аммония белый осадок
оксалата кальция, растворимый в минеральных кислотах.
Количественное определение активного хлора
Принцип метода. Активный хлор вытесняет из подкисленного
раствора иодида калия эквивалентное количество иода, которое оттитровывают
тиосульфатом натрия, и по расходу последнего делают заключение о коли-
честве активного хлора в хлорной извести.
508
Реактивы. 1. Раствор йодистого калия (0,3%).
2. Соляная кислота уд. веса 1,19.
3. Тиосульфат натрия (0,01N).
4. Крахмальный клейстер (1%).
Методика определения. Отвешивают 3,55 г хлорной извести,
растирают в фарфоровой ступке с небольшим количеством воды. Получен-
ную смесь переводят количественно в литровую колбу и доливают до метки
дестиллированной водой.
Перемешав тщательно жидкость, отбирают пипеткой 10 мл и переносят
в коническую колбочку с притертой пробкой, приливают 50 мл 0,3% раствора
KJ и 5 капель соляной кислоты удельного веса 1,19. Выделившийся иод
оттитровывают 0,01 N раствором тиосульфата натрия.
При этом протекают следующие реакции:
Са(ОС1)2 + 4KJ + 4НС1 = СаС12 + 4КС1 + 2Н2О + 2J2,
CaO(OCl) + 2KJ + 2НС1 = СаС12 + 2КС1 + Н2О + J2,
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
Из приведенных реакций видно, что одному атому активного хлора соот-
ветствует одна молекула тиосульфата натрия. Следовательно, 1 мл 0,01 N
раствора тиосульфата натрия соответствует 0,00035 г активного хлора.
Так как для анализа берется 0,0355 г хлорной извести, то каждый мил-
лилитр 0,01 N раствора тиосульфата натрия, израсходованного на титрование,
соответствует 1% активного хл^ра в хлорной извести (0,000355 г составляет
1% от 0,0355 г).
Сл д в 1тельно, затраченное на титрование число миллилитров 0,01 N
тиосульфата натрия точно соответствует процентному содержанию активного
хлора в хлорной извести.
ДЕЗИНСЕКЦИОННЫЕ СРЕДСТВА (ИНСЕКТИСИДЫ)
Дезинсекционных cpeaciв предложено множество. Здесь рассмотрены лишь важнейшие.
Сера
Сера — элементарное вещество кристаллической или аморфной структуры. Цвет светло-
желтый или серожелтый. Удельный вес колеблется от 1,92 до 2,07. Точка плавления 112—
120° С, точка кипения 445° С. Сера почти нерастворима в воде, хорошо растворяется в серо-
углероде, хуже в бензине, бензоле, толуоле.
Известны следующие препараты серы: 1) серный цвет, сублимированная природная
сера, почти чистый продукт, содержащий 95—99% S;2) сернистый концентрат — молотая
природная сера с 50—75% S; 3) комовая сера, полученная выплавкой природной; 4) черен-
ковая сера, полученная возгонкой и затем расплавленная и отлитая в форме палочек, и др.
Для целей дезинсекции применяется путем сжигания и окуривания помещений в коли-
честве 100 г/м3, что создает концентрацию в 7% SO2.
Сера и ее препараты находят также применение в борбе с вредителями сельскохозяй-
ственных растений.
Качественное определение
Сера горит синим пламенем, распространяя специфический запах сернистого ангидрида
Количественное определение
Здесь описаны два метода: 1) сжигание серы и окисление SO2 до H2SO4 с последующим
весовым или обьемным определением серы и 2) растворение серы при помощи сульфита
натрия с последующим йодометрическим определением серы.
Метод сжигания
Точную навеску (около 0,1 г) измельченной серы сжигают в печи, применяемой для
элементарного анализа. Продукты сжигания просасывают через две промывалки с 5%
раствором перекиси водорода:
SOa + Н2О2= H2SO4
509
Полученную серную кислоту определяют осаждением хлористым барием либо титро-
метрически раствором едкого натрия при метилоранже.
В первом случае вес BaSO, пересчитывают на вес серы, умноженный на 0,13737 соответ-
S
стве,"’° BaSO, •
Во втором случае число миллилитров израсходованного на титрование 0,1 N NaOH
умножают на 0,0032066, получая содержание серы во взятой навеске в граммах.
Йодометрический метод
Фармакопея рекомендует метод определения серы, основанный на растворимости серы
в сульфитах щелочных металлов с образованием тиосульфата:
Na2SO3 + S = Na2S2O3
Образовавшийся тиосульфат оттитровывают раствором иода и по расходу последнего
определяют содержание серы в препарате.
Исследование ведется так: точную навеску (около 0,1 г) измельченной серы высуши-
вают в экссикаторах над серной кислотой до постоянного веса, переводят в колбу на 250—
300 мл и приливают сюда 10 мл горячего 30% раствора сульфита натрия. Раствор льют
по стенкам, чтобы смыть серу на дно колбы. Колбу закрывают пробкой со вставленной в нее
стеклянной трубкой высотой около 75 см (обратный холодильник). Кипятят Р/2—2 часа
на маленьком огне, время от времени взбалтывая содержимое колбы. Трубку и горлышко
колбы ополаскивают водой, присоединяя промывные волы к основному раствору. Дают
несколько остыть и фильтруют в колбу с притертой пробкой, промывая колбу, в которой
велось нагревание, и фильтр 3 раза водой (по 10 мл). К охлажденному раствору прибавляют
30—35 мл раствора формалина. По истечении 15 минут приливают около 50 мл разведенной
уксусной кислоты до кислой реакции и титруют 0,1 N раствором иода до остающегося в те-
чение 30 секунд синего окрашивания при индикаторе крахмальном клейстере.
1 мл 0,1 N раствора иода соответствует 0,0032066 г серы.
Сернистый ангидрид (сжиженный)
Сжиженный сернистый ангидрид SO2 хранится в стальных баллонах.
Сернистый газ легко сгущается в жидкость, кипящую уже при 10° С. 1 л жидкого сер-
нистого газа весит 1450 г. и при испарении образует 529 л газа (при 15° С). 1 л сернистого
газа при 15° С гесит 2,74 г, следовательно, SO2 в среднем в два раза тяжелее воздуха.
Для исследования отбирают из баллона пробу в газовую пипетку. Анализ ведется так же,
как и при определении SO2 в воздухе.
Сероуглерод
Сероуглерод — ценный инсектисид. Отрицательная сторона его — взрывоопасность,
ввиду чего применяется редко. При дезинсекции зернохранилищ применяемые дозы —
80— 100 г/м3 помещения, при дезинсекции зерна дозы—100 — 250 г/м3 помещения. Свойства
и методы определения см. стр. 250.
Хлорпикрип
Хлорпикрин (трихлорнитрометан, CC13NO2) представляет собою бесцветную масля-
нистую жидкость с резким раздражающим запахом Удельный вес 1,605. Температура ки-
пения 113° С, температура плавления — 69,2° С При обычной температуре легко испаряется,
но несколько медленнее воды. Вес 1 л пара 6,84 г
Применяется для дезинсекции складских помещений, элеваторов, мельниц и т. п. Сред-
няя концентрация 20 г/м3. Является сильным ядом, уже в концентрации 1 : 200 000 000,
вызывает раздражение слизистых оболочек глаз и дыхательных путей; в концентрации
1 : 1 000 000 вызывает слезотечение. Смерть от удушья наступает при концентрации 0,2 мг/л.
Качественное определение
Реакция Некрасова и Мельникова. В пробирку с несколькими миллилитрами дести лли-
рованной воды вводят очень небольшое количество испытуемого продукта, подщелачивают
раствором КОН и приливают несколько капель раствора тиофенола (CeH5SH); при этом
получается белая муть, переходящая при больших концентрациях хлорпикрина в осадок.
Реакция основана на взаимодействии хлорпикрина с тиофенолом в щелочной среде
с образованием дифенилдисульфида, дающего муть:
ЗС6 H-SH 4- CCI3NO2 4- ЭКОН = ЗКС! + (C6H5S) 3CNO2 4- зн2о
2 (CGH- S)3 CNO2 = 3 C6H5 — s — S — CtiH5 + 2 CO2 + 2 NO
Дпфенплдпсульфпд
510
Реакция положительна при наличии не менее 0,01 мг хлорпикрина в 1 мл раствора-.
Реакция Алексеевского. К водному раствору хлорпикрина в пробирке прибавляют
0,3—0,5 г металлического кальция в стружках. Как только прекращается выделение водо-
рода, пробу подкисляют уксусной кислотой и прибавляют около 1 мл реактива Грисса.
В зависимости от концентрации хлорпикрина сразу или через минуту появляется розовое
окрашивание различной интенсивности.
Реакция основана на том, что среди продуктов восстановления хлорпикрина находится
и азотистая кислота, обнаруживаемая реактивом Грисса:
Са + 2Н2О = Са (ОН)2 + Н2.
CC13NO2 + 4 Н2 = HNO2 + 3 НО + СН4.
Реакция положительна при наличии не менее 0,002 мг хлорпикрина в 1 мл раствора.
Количественное определение
В коническую колбу на 250 мл вводят точную навеску (0,3—0,5 г) испытуемого продукта
и приливают 100 мл 4% спиртового раствора сернистокислого натрия. При этом образуется
нитрометандисульфокислота, гидросульфат натрия и хлорид натрия по реакции:
CC13NO2 + 3Na2SO8 + Н2О = CHNO2 (SO3Na)2 + NaHSO4 + 3 NaCl.
Образовавшийся хлорид натрия определяют затем титрованным раствором AgNO3^
который берется в избытке с последующим определением избытка роданистым аммонием
(индикатор железоаммонийные квасцы)
Из приведенной реакции видно, что 164,5 весовым частям хлорпикрина соответствуют
106,5 весовых частей хлора. Следовательно, 1 весовой части хлора соответствует 1,5446
весовых частей хлорпикрина.
1 мл 0,01 N раствора AgNO3 соответствует 1,5446. 0,355 = 0,548 мг CC13NO2
Для количественного определения хлорпикрина может быть применена реакция со
спиртовым раствором иодида калия
2 СС13 N02 + 6KJ = 3J2 + 2 СО2Ч-6 KC1 + N,
По окончании реакции пробу разбавляют водой, добавляют крахмальный клейстер
и титруют выделившийся иод тиосульфатом натрия.
1 мл 0,01 раствора Na2S2O3 соответствует 0,548 мг CCl3NOa
Цианистые препараты
Из цианистых препаратов в качестве инсектисидов применяют цианистые натрий и ка-
лий, цианплав и циклоны. Жидкую синильную кислоту в баллонах как илсектисид в СССР
не применяют.
Цианплав получается путем перевода цианамида кальция при помощи углерода
(графита) в цианид:
CaCN2 + С Ca(CN)2
Реакция протекает при температуре 1200—1500° С в расплавленном хлористом натрии.
Препарат содержит значительное количество примесей в виде NaCN, графита, извести,
кремнезема, серы и др.
Применяется в различных видах: а) рядовой — в виде темносерых чешуек, б) п о-
роховидный — в виде зерен величиною от 1 до 3 мм, в) пылевидный — серый
порошок с величиной зерен до 1 мм.
Содержание цианидов в цианплаве колеблется от 25 до 50%.
Циклон А (жидкий препарат), применявшийся ранее, в настоящее время вытеснен
более безопасными порошковидными циклонами В и С. Эти препараты состоят из
синильной кислоты и поглотителя (диатомовая земля, бумажная масса ит. п.); к препарату
примешивают иногда стабилизатор (метиловый эфир хлоругольной или бромуксусной ки-
слоты), предохраняющий синильную кислоту от разложения и полимеризации; кроме того,
в препарат вводят иногда сигнализатор в виде слезоточивого вещества (хлорпикрин) с целью
предупредить об опасности, если кто-либо случайно попадет в зараженную зону.
Применение NaCN и KCN основано на выделении паров HCN при воздействии H2SOt
на эти соли; применение цианплава и циклонов основано на способности этих препаратов
выделять пары HCN при соприкосновении с воздухом.
Синильная (цианистоводородная) кислота (HCN) представ-
ляет собою бесцветную жидкость. Точка плавления — 14,8°С, точка кипения 26,6° С. Удель-
ный вес 0,697, удельный вес паров 0,93 (воздух=1). На открытом воздухе легко испаряется.
Слабые концентрации HCN в воздухе имеют запах горького миндаля. Смеси паров синиль-
ной кислоты с воздухом взрывчаты при содержании от 5,6 до 12,8% HCN.
HCN — сильнейший яд для человека и уже в концентрациях 0,06—0,07 мг/л является
опасным. Смегть наступает от паралича дыхательного центра почти мгновенно.
511
Качественное определение
Исследуемый продукт смешивают с водой, подщелачивают едкой щелочью и нагревают
с небольшим объемом сульфата железа, фильтруют, добавляют несколько капель раствора
хлорида железа, охлаждают и подкисляют разбавленной серной кислотой (проба на лак-
мус). В присутствии синильной кислоты появляется синезеленое окрашивание или синий
осадок берлинской лазури:
FeSO4 + 2 КОН = Fe(OH)2 + K2S04
2 KCN + Fe(OH)2 = Fe(CN)2 + 2 KOH
Fe(CN)2 + 4 KCN = K4 [Fe(CN)e]
3 K, [Fe(CN)e] + 4 FeCl3 = Fe4 [Fe(CN)e]8 + 12 KC1.
Берлинская лазурь
Пары синильной кислоты, выделяемые самой кислотой и препаратами ее, могут быть
открыты:
1. По характерному запаху горьких мин дал ей, который ощущается уже при концен-
трации 0,01 мл паров HCN в 1 м3 воздуха. Однако прибегать к открытию синильной
кислоты обонянием не следует в виду опасности отравления (!).
2. Бензидиновой бумажкой, которую готовят следующим образом: 0,3 г ацетата меда
растворяют в 100 мл воды; отдельно приготовляют насыщенный раствор бензидина в 5°/е
уксусной кислоте и разбавляют этот । аствор равным об’емом воды. Смесь равных об’емов
ацетата меди и бензидина служит для пропитывания полосок фильтровальной бумаги. Они
бесцветны и применяются свежеприготовленными во влажном состоянии. Под действием
синильной кислоты бумажки окрашиваются в синий цвет, интенсивность которого зависит
от концентрации HCN. Окраска обусловлена продуктом окисления бензидина — бензиди-
новой синью
NH2CeH4 • CtiH4NH2+ О = NHC6H4 • CeH4NH + Н2О.
Бензидин Бензидиновая синь
Обнаружение HCN бензидиновой бумажкой возможно при концентрации не менее
0,007 мг/л. Свободные галоиды также вызыв; ют посинение бумажки.
3. Пикриновой бумажкой. Полоски фильтровальной бумаги смачивают раствором из
1 г пикриновой кислоты и 10 г соды в 100 мл воды. Бумажки имеют золотистожелтый
цвет. Под действием паров HCN они окрашиваются в красный цвет ввиду образования
соли изопурпуровой кислоты:
CeH2 (NO2)3 ONa + 3 HCN = CeH2 (NO2)2 N (CN)2ONa + H2O + HCNO.
Пикрат натрия Изопурнурат натрия
Количественные определения
Из количественных определений при испытании препаратов синильной кислоты
самым важным является определение циана, которое в различных препаратах произво-
дится следующим образом:
Определение CN в NaCN и KCN. Образец дробят под тягой в ступке на небольшие кусочки
(растирать в порошок не следует). Быстро отвешивают в бюксе около 0,5 г и смывгют в мерную
колбу на 500 мл, в которую налито 200 мл воды. Прибавляют небольшое количество РЬСО8
для осаждения сульфидов, которые могут присутствовать в испытуемом материале, и дово-
дят водой до метки. Тщательно перемешивают и фильтруют через сухой фильтр Первые
порции фильтрата отбрасывают. Затем отбирают 50 мл фильтрата и переводят в стакан ем-
костью в 400 мл. (Из осторожности отмеривание объема не следует производить пипеткой).
Прибавляют 200 мл воды, 5 мл раствора едкого натрия (100 г на 1 л воды), 10 капель насы-
щенного раствора или несколько кристалликов йодистого калия в качестве индикатора и
титруют 0,1 N раствором AgNO3 до появления слабой опалесценции, возникающей от обра-
зования AgJ. Стакан держат на черном фоне. При проведении испытания протекают следую-
щие реакции:
2NaCN + AgNO3 = NaCN . AgCN 4- NaNO3,
KJ + AgNO3 = AgJ + KNO3
Из приведенной первой реакции видно, что каждый миллилитр израсходованного 0,1 N
раствора AgNO3 соответствует 0,005204 г CN. На основании израсходованного при титро-
вании раствора AgNO3 делают заключение о содержании CN в препарате.
Пример. Взята навеска в 0,425 г NaCN. При производстве определения, как описано
выше, израсходовано 3,84 мл 0,1 N раствора AgNO3. Отсюда заключаем, что процентное
содержание CN в препарате равно:
0,005204 3,84.101 = 49 34о/о.
0,0425
512
Примечание. Для осаждения сульфидов рекомендуется пользоваться
свежеосажденным углекислым свинцом, который готовится следующим образом:
растворяют 20 г уксуснокислого свинца, объем доводят до литра и прибавляют 200 г
свободного от хлора углекислого натрия. Для осаждения сульфидов в реакцию идет
25 мл или более.
Определение CN в цианплаве. Принцип тот же, что и в предыдущем методе.
Отвешивают под тягой 20 г препарата из отобранной средней пробы и быстро переносят
в 2 %-литровую колбу, содержащую 1 у2 л воды. Не закрывая склянки1, часто взбалтывают
ее содержимое в течение 2 минут и оставляют па 1 час для выщелачивания, время от времени
взбалтывая. Затем полученный раствор сливают в 2-литровую мерную колбу, а нераство-
римый остаток смывают в ступку, растирают и переносят в ту же мерную колбу. Разлагают
сульфиды свежеосажденным углекислым свинцом, как описано выше, прибавляя 25 мл
или более реактива (см. примечание в предыдущем определении) и доводят водой до метки.
Тщательно взбалтывают, отбирают 50 мл, что соответствует 0,5 г взятой навески, переносят
в коническую колбу на 500 мл, разводят до 300 мл водой и титруют 0,1 N раствором AgNO3
при индикаторе иодистом калии.
При исследовании пылевидного препарата можно взять меньшую навеску (1—1,5 г).
Растворение в этолТ случае производится в мерной колбе на 500 мл.
Дихлордифепилтрихлорэтан (ДДТ)
Дихлордифепилтрихлорэтан — СС13СН(СсН4С1)2 — является весьма ценным инсекти-
сидным средством. Получается синтетически хлорированием этилового спирта с последую-
щей конденсацией полученного хлорала с хлорбензолом в при-
сутствии крепкой серной кислоты. Схематически:
С2Н6ОН + 4С12 = СС13СНО + 5НС1
СС13СНО + 2С6Н5С1 = СС13СН (С6Н4С1)2 + Н2О.
Чистый дихлордифенилтрихлорэтан представляет собой белое
кристаллическое вещество с легким ароматическим запахом. Темпера-
тура плавления в пределах 108,5—109° С. Практически нерастворим
в воде, умеренно растворяется в минеральных и растительных маслах
и хорошо—в обычных органических растворителях.
Технический продукт представляет аморфное светлосерожелто-
ватое вещество. Наряду с формой рр ДДТ в нем содержится и форма
ор ДДТ, а также форма, содержащая на 1 атом хлора менее, ряд эфи-
ров и других соединений.
ДДТ применяется в виде дустов, суспензий и эмульсий. Дусты
готовятся путем смешивания препарата ДДТ с каким-либо инерт-
ным наполнителем: тальком, пирофиллитом, глинами и даже мукой
с последующей добавкой 2% масла для улучшения прилипаемости.
Содержание ДДТ в дустах в среднем равно 5—6%.
Тонина помола имеет важное значение для эффективности пре-
парата. Согласно ВТУ МХП тонина помола должна быть таковой,
чтобы остаток на сите 175 меш (4900 отверстий на 1 см2) не превышал
3%. Влаги допускается не более 1,5%.
Определение влаги. Определение влаги производится по методу дести л л яци и в специаль-
ном приборе, изображенном на рис. 297.
Прибор состоит из приемника колбы а и холодильника с. Приемник представляет
собой коническую градуированную пробирку. Нижняя часть от 0 до 1 мл имеет деления
в 0,05 мл; выше — от 1 и до 10 мл — деления соответствуют 0,2 мл.
К верхней части пробирки на расстоянии 40—50 мм от верхнего края припаяна отвод-
ная трубка, отогнутая вниз параллельно пробирке. Конец отводной трубки косо срезан.
Колба емкостью в 500 мл (круглодонная или плоскодонная) делается из меняла или стекла.
Обратный холодильник стеклянный; размеры указаны на чертеже; конец трубки
косо срезан.
В колбу помещается навеска в 25 г препарата, взятая на технических весах с точностью
до 0,2 г; туда же вводится 100 мл безводного (совершенно прозрачного) бензола. Прибор
собирают, как указано на рис. 297. Нагревают бензол до кипения и кипятят до тех пор,
пока слой воды в приемнике не перестанет увеличиваться (но не менее одного часа)
Так как вместе с влагой отгоняется и часть бензола, в приемнике получаются два слоя:
нижний — водяной и верхний—бензольный. Для четкого отграничения этих слоев рекомен-
дуется до начала отгона поместить в приемник 1—2 мг красителя, растворимого в воде
и нерастворимого в бензоле (напр., метиленовую синьку).
Если бензол в приемнике после перегонки мутный, то приемник опускают на несколько
минут в воду 40—45° С и затем вновь охлаждают.
1 Присутствующий в пробе СаС2 при взаимодействии с водой выделяет газообразный
ацетилен (С2Н2), который может выбросить пробку.
33. А. И. Бурштейн.
513
Количество миллилитров собравшейся в приемнике воды, умноженное на 4, соответ-
ствует процентному содержанию влаги в препарате.
Количественное определение ДДТ в дустах. Принцип метода. При помощи
едкого калия разрушают определенную навеску ДДТ и определяют обшее количество титро-
ванного раствора AgNO3, затрачиваемого на связывание хлора. Из этого количества вы-
читают тот объем раствора AgNO3, который расходуется на связывание ионизированного
хлора. Разность соответствует раствору AgNO3, затрачиваемому на связывание хлора, вхо-
дившего в состав ДДТ. По этой разности делаю г заключение о процентном содержании
ДДТ в препарате.
Реактивы. 1. Спирт этиловый.
2. 0,5 N спиртовый раствор КОН.
3. 0,05 N раствор AgNO3.
4. 0,05 N раствор NH4CNS.
5. Раствор железоаммонийных квасцов (насыщенный).
б. Азотная кислота.
Методика определения. Навеску в 20 г дуста помещают в колбу емкостью
в 250 мл, прибавляют 80 мл 96° спирта, закрывают колбу пробкой, снабженной обратным
холодильником, и нагревают до кипения, которое поддерживают 30 минут. Охлаждают и
фильтруют раствор в мерную колбу на 250 мл. Остаток на фильтре промывают пять раз
горячим спиртом, беря для каждого промывания по 25 мл спирта. Полученный раствор
охлаждают до комнатной температуры и доводят до метки спиртом. Тщательно перемеши-
вают и отбирают пипеткой 50 мл раствора, переносят в коническую колбу на 500 мл, добав-
ляют 30 мл 0,5 N спиртового раствора едкого калия. Полученную смесь кипятят в течение
30 минут на водяной бане, закрыв колбу пробкой, снабженной обратным холодильником.
Охлаждают и разбавляют 75 мл дестиллированной воды, подкисляют азотной кислотой,
прибавляют 20 мл 0,05 N раствора AgNO3, избыток которого оттитровывают 0,05 N раство-
ром NH4CNS при индикаторе железоаммонийных квасцах.
Для определения ионизированного хлора отбирают из мерной колбы, как и ранее, 50 мл
спиртового раствора ДДТ и переносят в коническую колбу на 300 мл, прибавляют 50 мл
воды, подкисляют азотной кислотой, прибавляют 10 мл 0,05 N раствора азотнокислого
серебра и оттитровывают избыток его 0,05 N раствором роданистого аммония.
Число миллилитров раствора азотнокислого серебра, пошедшее на связывание ионизи-
рованного хлора, вычитают из числа миллилитров раствора азотнокислого серебра, пошед-
ших на связывание хлора после разрушения ДДТ едким калием. Разность соответствует
числу миллилитров раствора азотнокислого серебра, пошедших на связывание хлора, ко-
торый входил в состав ДДТ.
Расчет содержания ДДТ производится по формуле:
(100 — 5а — 5Ь)
%%ДДТ = ---------20------- -0,01773- 100
или
(20 — а — Ь). 0,44325,
где а — число миллилитров 0,05 N раствора роданистого аммония, пошедших на обратное
титрование раствора серебра,
b — число миллилитров 0,05 N раствора азотнокислого серебра, пошедших на связыва-
ние ионизированного хлора.
Пример. Взята навеска в 20 г дуста. При производстве определения, как описано выше,
на обратное титрование после разрушения ДДТ пошло 5,2 мл 0,05 N NH4CNS. На свя-
зывание ионизированного хлора затрачено 0,8 мл 0,05 N AgNO3.
Отсюда % ДДТ = (20 — 5,2 — 0,8) - 0,44325 = 6,2
Гексахлорциклогексап (ГХЦГ)
Гексахлорциклогексан (гексохлоран, гексид, гаммексан, CGHeCle) — синтетическое хлор-
органическое соединение; весьма сильное инсектицидное средство.
Получается при пропусканиия хлора через кипящий бензол на прямом солнечном свету
или под действием лучей ртутнокварцевой лампы.
Представляет собой желтоватобелый кристаллический порошок неприятного запаха.
Образует маслянистые на ощупь комки. В воде почти нерастворим, хорошо растворяется
в органических растворителях.
Технический продукт является смесью четырех изомерных форм ГХЦГ (а, 3, 7 и S);
кроме того, в состав его входят пентахлорциклогексан в двух изомерных формах и ряд других
соединений.
По характеру действия на насекомых, как и ДДТ, является нервным ядом. Применя-
ется в виде дустов, суспензий и эмульсий. Содержание ГХЦГ в дустах колеблется от
7 до 20%.
ГХЦГ выпускается в двух марках А и В. Содержание ГХЦГ, считая на сухое вещество,
согласно ВТУ МХП, должно быть не менее 95% для марки А и 92% для марки В. Влаги
должно быть соответственно не более 5 и 8%; свободной кислотности в пересчете на НС) —
не более 0,04% и 0,1%.
514
Определение влаги производится так же, как и в ДДТ.
Определение свободной кислотности. Навеска в Юг, взятая на технохимических веса^
с точностью до 0,2 г, растирается в фарфоровой ступке с 30 мл воды в течение 10 минут.
Полученная масса смывается на фильтр небольшим количеством холодной воды Осадок
на фильтре промывается два раза холодной водой. Общее количество воды, затраченное
на растирание и промывание, должно быть около 100 мл. Фильтрат собирается в коническую
колбу емкостью в 250 мл К нему прибавляют 2—3 капли метилоранжа и титруют 0,01 N
раствором NaOH. Одновременно, проводят холостой опыт с таким же количеством дестил-
лированной воды.
Процентное содержание свободной кислоты в пересчете на НС1 вычисляется по формуле:
%% НС1 = (а — Ь) к . 0,00365,
где а — число миллилитров 0,01 N щелочи, пошедших на титрование водной вытяжки,
b — число миллилитров 0,01 N щелочи, пошедших на титрование холостого опыта,
к — коэфициент нормальности щелочи.
Количественное определение ГХЦГ. Около 10 г препарата растирают в ступке в круп-
ный порошок и сушат в открытой бюксе 2 часа при 70° С. Из высушенного продукта бе-
рется 1—2 г и растирается в тонкий порошок.
Навеска тонко растертого вещества в 0,4—0,5 г, взятая на аналитических весах, помеща-
ется в коническую колбу, емкостью в 150—200 мл. Приливают30 мл этилового спирта и слегка
нагревают для растворения препарата. Затем приливают 25 мл 0,5 N раствора NaOH. Колбу
закрывают пробкой, в которую вставлена изогнутая под углом стеклянная трубочка, соеди-
ненная с хлоркальциевой трубкой, наполненной натронной известью. Раствор в колбочке
нагревают на электрической плитке до кипения и кипятят два часа. Затем охлаждают до
30—40° С. Трубку холодильника и горлышко колбы смывают водой в колбу и оттитровы-
вают избыток щелочи 0,5 N раствором НС1 при индикаторе фенолфталеине. Одновременно
в тех же условиях проводят холостой опыт без препарата.
Процентное содержание ГХЦГ вычисляют по следующей формуле:
»/0о/„ ГХЦГ = К-0.0485. 100 _ т>
п
где а — количество мл кислоты, пошедшее на титрование в холостом опыте,
b — количество мл кислоты, пошедшее на титрование в опыте с ГХЦГ,
К — коэфициент нормальности кислоты,
п — навеска в граммах,
т — кислотность ГХЦГ, найденная предыдущим определением, в пересчете на ГХЦГ
(т = %НС1 . 2,66).
Пример. Взята навеска в 0,42 г. При производстве определения, как описано выше,
на опыт с ГХЦГ затрачено на обратное титрование 7,2 мл 0,5 N кислоты, на то же титрова-
ние в холостом опыте затрачено 24,6 мл 0,5 N кислоты; К = 0,9986. Свободная кислот-
ность оказалась равной 0,03%.
_ . (24.6 - 7,2) . O.W86 -0,0485 UOO . . ,
0,42
Определение ГХЦГ в дустах. Около 3 г дуста взвешивают на аналитических весах,
переводят в коническую колбу емкостью в 250 мл, прибавляют 25 мл этилового спирта,
закрывают пробкой с обратным холодильником и кипятят в течение 10 минут. Затем охлаж-
дают до комнатной температуры, прибавляют 20 мл 0,2 N раствора NaOH и кипятят с обрат-
ным холодильником в течение 2 часов для разрушения ГХЦГ. Отфильтровывают с отсасы-
ванием, ополаскивают колбу дестиллированной водой и выливают промывную воду на тот же
фильтр. Осадок на фильтре промывают 3 раза водой, беря каждый раз по 15 мл. К фильтрату
прибавляют 5 мл концентрированной азотной кислоты и 40 мл 0,1 N раствора AgNO,
для связывания хлора Избыток серебра определяют обратным титрованием 0,1 N раствором
NH4CNS при индикаторе железоаммонийных квасцах. Отдельно проводят холостое титро-
вание с таким же количеством дестиллированной воды.
Процентное содержание ГХЦГ в дусте вычисляется по формуле:
(а —Ь — с). 0,0097 . 100
% о/оГХЦГ = *--------,
где а — количество миллилитров 0,1 N раствора AgNO3, прибавленных в главном опыте,
b—количество миллилитров 0,1 N раствора NH4CNS, прибавленных в главном опыте,
с — разность между числом затраченных миллилитров AgNO3 и NH4CNS в холостом
опыте,
п — навеска.
Мыла
Мыла представляют собой соли жирных кислот Различают твердые и жидкие
мыла. Твердые мыла получаются действием натриевой щелочи на твердые жиры (свиное
и говяжье сало, кокосовое масло и т. п.) Жидкие мыла получаются действием калийной
щелочи на жидкие жиры (конопляное, льняное и др. масла).
515
При взаимодействии между жирными кислотами и щелочью получаются глицерин и
соли жирных кислот (мыла):
C3H6(OCOR)3 4- 3 NaOH = С3Нб(ОН)3 + 3 RCOONa.
К жирозаменяющим или суррогатам жиров в мыловарении относят: канифоль (гар-
пиус), нафтеновые кислоты (асидол), синтетические кислоты и др.
Мыла способны гидролизироваться:
RCOONa + Н2О = RCOOH + NaOH.
Моющая и эмульгирующая способность мыл, повидимому, обусловливается нейтраль-
ными и диссоциированными мицеллами, образующимися из анионов жирных кислот.
Инсектисидные свойства мыл зависят в большей мере от природы жирных кислот, обра-
зующихся при гидролизе, нежели от недиссоциированных молекул мыла и свободных
щелочей. ;
Калиевое (жидкое, зеленое) мыло по внешнему виду представляет темнобурую или зеле-
новатую массу, растворяющуюся в 2 ч. горячей воды и 4 ч. воды комнатной температуры.
Растворы имеют щелочную реакцию, при взбалтывании сильно пенятся.
Натриевое мыло представляет однообразную твердую на ощупь, сухую массу. На поверх-
ности его не должно быть налетов и выделений. Пенистость мыла должна проявляться ясно
в воде при температуре 25° С и жесткости 12 градусов (и ниже).
Огбор пробы. Проба жидкого мыла берется после тщательного перемешивания образца.
Отбор пробы твердого мыла производится следующим образом. При обычных прямоуголь-
ных формах куска его режут на 4 части по двум взаимноперпендикулярным направлениям
и от всех свежесрезанных поверхностей равномерно настругивают достаточное для анализа
количество мыла, которое сохраняют в банке с притертой пробкой.
Опре i.e ten не влаги. При исследовании жидкого мыла 2—3 г его отвешивают в чашку,
прибавляют 10—15 мл этилового спирта, тщательно перемешивают, после чего на водяной
бане выпаривают спирт и высушивают остаток в шкафу при 100—105° С до постоянного
веса. При определении по описанному способу получают не только содержание воды, но и
всех могущих находиться в мыле летучих веществ.
Более точным является дестилляционный метод, описанный выше (стр. 513). При этом
методе отходящие с водой летучие вещества растворяются в органическом растворителе и не
изменяют объема воды. Для устранения образования устойчивой пены в колбу, из которой
производится отгонка, прибавляют двукратное (по отношению к мылу) количество сухой и
чистой олеиновой кислоты.
При исследовании твердого мыла навеску в 5—8 г, состоящую из маленьких кусочков
или стружки, помещают в бюксу и сушат в шкафу сначала при 65—70° С во избежании сплав-
ления, а затем при 100—105° С до постоянного веса.
Дестилляционный метод, как и при исследовании жидкого мыла, дает более точные
результаты.
Оиреде 1бние минеральных и других наполнителей. 5—6 г испытуемого мыла вводят
в колбочку, снабженную обратным холодильником, прибавляют 25 мл абсолютного алко-
голя и кипятят до максимального растворения. Фильтруют через фильтр, постоянный вес
которого предварительно определен. Фильтр с осадком высушивают до постоянного веса
и взвешивают. Разность между полученным весом и весом одного фильтра соответствует
посторонним примесям (глина, поваренная соль, сода и др.). Если хотят определить от-
дельно количество минеральных примесей, то полученный осадок озоляют и после охлаж-
дения взвешивают.
Определение жирных кислот. Навеску в 3—5 г мыла растворяют в 100 мл горячей воды,
прибавляют несколько капель метилоранжа, а затем приливают разведенной серной кислоты,
пока раствор не порозовеет, после чего приливают еще 5—10 мл кислоты и нагревают при
частом помешивании на водяной бане. При этом происходит разложение мыла:
2RCOONa 4- H2SO4 = 2RCOOH 4- Na2SO4.
Жирные кислоты в виде прозрачного слоя всплывают на поверхность раствора. После
охлаждения смесь переводят в делительную воронку. Остатки смывают туда же этиловым
эфиром. Экстрагируют жирные кислоты сначала 30 мл эфира, а затем еще три раза, беря
каждый раз по 20 мл. После каждого экстрагирования отделяют эфирный слой от водного.
Соединенные эфирные вытяжки собирают в делительной воронке.
Для осушки эфирного раствора прибавляют в воронку прокаленный сульфат натрия и
встряхивают содержимое. Отфильтровывают эфирный раствор в предварительно высушен-
ную и взвешенную колбу на 200 мл, а оставшийся в воронке сульфат натрия промывают
свежими порциями эфира, высушенного также сульфатом натрия. Эти порции эфира присое-
диняют к основному эфирному раствору. Эфир отгоняют, остаток в колбе высушивают при
температуре не выше 75° С, охлаждают в экссикаторе и взвешивают. Привес соответствует
содержанию жирных кислот во взятой пробе.
Если в состав мыла входят летучие кислоты, то сушить следует при температуре не выше
60° С. В присутствии легко окисляемых жирных кислот следует производить сушку в
струе ипдеферентного газа (СО2 и др.).
516
Если в мыле содержится большое количество наполнителей, то мыло отделяют от них пу-
тем кипячения со спиртом. Затем спирт отгоняют и с мылом поступают, как изложено выше.
Выделенные жирные кислоты могут быть подвергнуты полному или частичному жирово-
му анализу (иодное число, кислотное число, титр и ,пр.).
Определение смоляных кислот. Смоляные кислоты в виде канифоли находят применение
в мыловарении. Они представляют смесь изомерных кислот, объединяемых под общим наз-
ванием абиетиновой кислоты (C^H^OJ. Кроме смоляных кислот в состав канифоли входит
небольшое количество неомыляемых веществ. Канифоль получается из живицы хвойных
деревьев.
Качественное определение канифоли в мыле производится следую-
щим образом: в пробирку вводят небольшое количество жирных кислот, выделенных описан-
ным выше образом, прибавляют 1 мл уксусного ангидрида, слегка нагревают и по охлажде-
нии прибавляют 1 каплю серной кислоты удельного веса 1,53, опуская ее по стенке пробирки.
В присутствии канифоли появляется краснофиолетовое окрашивание, переходящее
в коричневожелтое, а затем зеленое флуоресцирующее.
Количественное определение смоляных кислот основано на
том, что жирные кислоты переводятся при помощи метилового спирта в сложные эфиры,
а смоляные кислоты остаются свободными.
2—5 г общей смеси жирных кислот растворяют в 10—20 мл метилового спирта, при-
бавляют к раствору 5—10 мл смеси, состоящей из одного объема концентрированной
серной кислоты и четырех объемов метилового спирта, в течение 2 минут кипятят с обратным
холодильником. Переводят затем все в делительную воронку, добавляют 5—10-кратное
количество 10% раствора чистой поваренной соли, извлекают эфиром, после чего водный
слой экстрагируют эфиром еще 2—3 раза.
Соединенные вместе эфирные вытяжки промывают в присутствии метилоранжа 10%
раствором поваренной соли для удаления серной кислоты.
Отмытые от минеральной кислоты соединенные эфирные вытяжки после прибавления
30 мл спирта титруют 0,5 N раствором КОН при фенолфталеине. Процентное содержание
смоляных кислот вычисляется по формуле:
%% смоляных кислот = 17,76-?.— 1,5,
п
где а — количество миллилитров 0,5 N щелочи, израсходованной на титрование,
п — навеска кислот.
Пересчет на канифоль достигается умножением на коэфициент 1,07, так как канифоль
содержит около 7°/0 неомыляемых веществ.
Определенно поомыленного нейтрального жира и неомыляемых веществ. К неомыляе-
мым относятся те вещества, входящие в состав жиров, которые не реагируют с едкими ще-
лочами в условиях, при каких производится омыление; при этом имеются в виду лишь
те из инертных к действию щелочей вещества, которые не растворяются в воде; высоко-
молекулярные алкоголи восков, стерины, углеводороды, некоторые красящие вещества и др.
Так как в мыле может присутствовать неомыленный нейтральный жир, следует его опреде-
ление и определение неомыляемых веществ производить отдельно.
20 г измельченного мыла растворяют в смеси 80 мл спирта и 70 мл волы. В воде пред-
варительно растворяют 2 г NaHCO3 для нейтрализации могущих присутствовать в мыле
в свободном состоянии щелочи или жирных кислот. Смесь нагревают до растворения мыла,
а затем охлаждают. Переводят в делительную воронку. Остатки смывают туда же 50%
спиртовым раствором. Далее производят троекратное извлечение неомыленного нейтраль-
ного жира и неомыляемых веществ, беря при каждом извлечении 50 мл петролейного эфира.
Если при встряхивании образуется эмульсия, то ее разрушают, прибавляя небольшое коли-
чество спирта. Соединенные петро лейно-эфирные вытяжки промывают три раза 50% спир-
том и фильтруют в предварительно высушенную и взвешенную колбу. Эфир отгоняют и высу-
шивают до постоянного веса. Привес соответствует смеси неомыленного нейтрального жира
и неомыляемых веществ.
Для определения отдельно нейтрального жира и неомыляемых веществ полученный
остаток омыляют при нагревании спиртовым раствором КОН и производят экстракцию
петролейным эфиром, как описано выше. При этом экстрагированными окажутся лишь
неомыляемые вещества. Их определяют после отгонки петролейного эфира. Разность между
первым количеством перешедших в петролейный эфир веществ и вторым соответствует нео-
мыленному нейтральному жиру.
Определение свободной щелочи. Под свободной щелочью в мыле понимают свободные
NaOH и КОН.
Качественное определение. Кусочек мыла в 1,0—1,5 г растворяют
в нейтральном спирте и полученный раствор испытывают фенолфталеином. В случае нали-
чия свободной щелочи раствор окрашивается в красный цвет
Количественное определение. 5 г мыла растворяют в 100 мл 60%
нейтрального спитра. В полученный раствор медленно при помешивании вводят небольшой
избыток 10% нейтрального раствора хлористого бария. При этом бариевые мыла выпадают
и выделяется свободная шелочь:
2RCOONa 4- Ва(ОН)а = (RCOO)2Ba 4- 2NaOH.
517
Выпавшие бариевые мыла отфильтровывают и осадок на фильтре промывают до нейтраль-
ной реакции промывных вод. Соединенные фильтрат и промывные воды титруют 0,1 N
раствором соляной кислоты в присутствии фенолфталеина.
Каждый миллилитр израсходованной 0,1 N НС1 соответствует 0,004 г NaOH или 0,0056р
ЛИТЕРАТУРА
1. Августинник А. И. и Воскресенский А. А., Методы исследования товаров (статья
проф. Надененко В. Н., Мыла), Снабтехиздат, Л.-М., 1932.
2. Безобразов Ю. Н. Молчанов А. В.. Никифоров А М. Гексахлоран и его примене-
ние для борьбы с вредителями сельскохозяйственных культур, Сельхозгиз, 1948.
3 Бей-Биенко Г. А., Богданов-Катьков и др., Сельскохозяйственная энтомология,
Сельхозгиз, 1949.
4. Бертель Р. Г. и Набоков В. А. Учебник медицинской энтомологии, под ред»
проф. Беклемишева В. Н., ч. 11, Медгиз, 1949.
5. Болдырев Т. и Окуневский Я., Практическое руководство по войсковой дезинфек-
ции, 1934
6 Бьюкенен Д. Г., Цианистые соединения и их анализ, Ленхимтехиздат, 1933.
7 Головянко 3. С., Руднев Д. Ф., Стражеско Д. Н., ДДТ в борьбе с вредными насеко-
мыми, изд. У АН, 1947.
8 Государственная фармакопея Союза ССР 8 изд., Медгиз, 1946.
9. Гродзовский М., Анализ воздуха в промышленных предприятиях, Соцэкгиз, 1931.
10 Жиры, их получение и переработка, Справочное руководство, том II, Пи щеп ром-
издат, 1938.
11 Зиновьев А. А., Химия жиров, Пищепромиздат, 1939.
12. Житкова А. С., Методика определения вредных газов и паров в воздухе, Оборон-
гиз, 1939.
13. Коренман И. М. Анализ воздуха промышленных предприятий, Госхимиздат, 1949.
14. Мартин Д., Мыловарение, М -Л., 1927.
15. Мельников Н. Н. ДДТ. Химические свойства и применение, Госхимиздат, 1950.
16 Окуневский Я , Практическое руководство по дезинфекции, чч. 1—111, 1926—1933.
17 . Петерс Г., Цианистые соединения и их применение в борьбе с вредителями и пара-
зитами, ОНТИ, Л., 1935.
18 Петров П. и Церевитинов Ф., Товароведение, т. 1, М.-Л., 1927.
19 Сысин А., Чистяков Г., и Левинсон Я., Курс дезинфекции, дезинсекции и дера-
тизации, 1932.
20 Флюри Ф. и Церник Ф., Вредные газы, ГОНТИ, 1938.
21 Фрир Д., Химия инсектисидов и фунгисидов, Гос. изд. иностр, лит., 1948.
22 Шмалько В. С., О применении цианплава для дезинсекции зерна, зернохранилищ
и мельниц, Госторгизлат, 1937.
23 Шорохов П. И., Шорохов С. И.9 Вредители запасов зерна и зерно продуктов, Сель-
хозгиз, 1938.
ПРЕДМЕТНЫЙ указатель
A
Автоклав 114
Агар мясопептонный (МПА) 114
— кровяной щелочной 117
— кровяной сахарный 117
Азот аммиака и а мм. солей
в воле 336
— альбуминоидный 338
— нитратов 339
— нитритов 375
— общий 345
— органический 342
Актинометр Араго-Деви-Каля-
тина 181
-----Катитина 182
— — компенсационный 180
— — с биметаллической пла-
стинкой 180
Актинометрия 179
Акролеин в воздухе 268
Ализарин 56
Алкалиметрия 52
Алюминий (глинозем) в воде 348
Аммиак в воздухе 352
Анализ весовой 38
— объемный 44
Ангидрид сернистый в воздухе
244
— серный в воздухе 250
— угольный в воздухе 228
Анемометр динамический 161,
162
-----для определения цавле*
ния воздуха 164
— — статический 163
— — электрический 162
Анилин в воздухе 272
Антиген 122, 126
Аппарат Киппа 93
— — перегонный 36
Аппретура ткани 486
Арматура осветительная 473
Аспираторы 195. 196
Ацетилен в воздухе 267
Апетон в воздухе 269
Апи.дометрия 52
Аэрогель 192
Аэрозоль 192
Аэропланктон 285
Б
Бактерии воздуха 285
— иолы 378
— тканей одежды 500
— почвы 424
— пыли 290
Баня водяная 28
— песочная 29
Бар 477
Барограф 143
Барометр металлический 141,
142
— сифонный 141
— чашечный 140
Батометр Хлопина 295
— Виноградова 291
Бензин в воздухе 269
Бензол в воздухе 271
Бентос 392
Биопробы 130
Бром в воздухе 258
Будка психрометрическая 158
Б ульон мясопептонный (МПБ)
114
— печеночный 116
Бурав для бактериологического
исследования почвы 425
Буферы боратные 77
— фосфатные 77
— цитратные 78
Бюксы 24
Бюретка макро 21
— микро 22
— Гем пел я 223
— Реберга 233
— Шилова 234
В
Вентиляция естественная 448
— искусственная 450
Вес атомный элементов(т?.бл.) 131
—^насыпной стройматериалов
Вес^объемный стройматериалов
— одного литра дест. воды
(табл.) 20
— удельный водных растворов
аммиака (табл.) 134
----растворов КОН и NaOH
(табл.) 133
— — растворов минеральных
кислот (табл ) 132
— — растворов уксусной кис-
лоты (табл.) 133
— — квросина 481
— — пыли 207
— — стройматериалов 428
— — тканей одежды 489
Вещества азотсодержащие в
почве 422
— — — сточных волах 401
— ряиешенные в питьевой воде
313
— — — сточных водах 401
Взвешивание (техника) 41
Весы аналитические 17. 43
— микроаналитические 18, 43
— технохимичсские 16
Виброграмма 480
Виброграф Л И ГТ 480
Виброметр Голицына-Бончков-
ского 480
Вкус волы, опенка 298
Влажность воздуха абсолютная
143
---- максимальная 143
----относительная 143
— — — определение по
табл. 149—156
— мыла 516
— почвы 422
— стройматериалов 437
— тканей одежды 496
Вола дважды лестиллирован. 36
— дестиллироьанная 36
— кипяченая (отличие от неки-
пяченой) 378
— пептонная (среда) 116
— питьевая 293
— сточная 398
Водоемкость почвы 419
— тканей 495
Водонасыщение стройматериа-
лов 432
Водоотдача стройматериалов 433
Водопоглощаемость 432
Водопроницаемость тканей 493
Водород мышьяковистый 259
— фосфористый 263
Водородное число 74
Воздух 137
Воздухопроницаемость стройма-
териалов 430 ,
— почвы 419
— тканей 491
Волюмометр Гуревича и Вендта
208
Воронка бюхнеровская 26
— делительная 24
— для горячего фильтрования32
— химическая 24
Г
Газ карбюраторный 14
Газволюмометрин 213
ГеЛахлорциклогексан (ГХЦП
514
(гексохлоран, гексид, гаммек-
сан) 514
Геотермометр 414
Гигрограф 158
Гигрометр 157
— Соссюра 157
Гигроскопичность тканей одеж-
ды 496
Горелка бензиновая 27
— Бунзена 28
— спиртовая 27
— Теклю 28
Гравиметрия (см. анализ весо-
вой)
— пыли 194
Грамм-эквивалент 45
Гриб домовой белый 441
----плачущий 441
----пленчатый 442
Д
Давление барометрическое 140
Движение воздуха 158
Держатель для фильтров 202
Дефицит насыщения 142
Дизентерийные бациллы (обна-
ружение в воле) 391
Динамопенетрометр 495
Диссоциация электролитическая
Дихлордифенилтрихлорэтан
(ДДТ) 513
Дночерпатели для улавливания
бентоса 394
Дождемер 211
Е
Единица электростатическая
(ESE)
Емкость сопротивления сосуда
для кондуктометрии 104
Ж
Желатина мясопептонная
(МШК) 114
519
Железо в воде 348, 376
Желчь 116
Жесткость воды общая 317
— — постоянная 324
----- устранимая 324
3
Зажимы для бюреток 21
Замазка менделеевская 38
— из РЬО и глицерина 38
Запах воды (опенка) 298
— воздуха (оценка) 137
Затвор стеклянный для бюре-
ток 21
Звукопроводность строймате-
риалов 435
Зерна почвы, определение раз-
меров 414
Змеевик (промывалка) 219
Зона комфорта ЭЭТ 177
Зоны сапробные, характеристи-
ка (табл ) 393
Зола (минеральные вещества)
плотного остатка воды 314
-----------пыли 277
И
Игла для бактериологических
посевов 112
Известь натронная 53
— хлорная, определение актив-
ного хлора 377, 508
Илосос Перфильева 394
Индикаторы, выбор 57
— общие сведения 46
Иод в воздухе 258
Иодометрия 61
Ионное произведение воды 74
Ионы атмосферы 188
К
Калориметр (диатермометр) 489
Калий в воле 326 •
Камера счетная для бактерий
ИЗ
— — — планктона 395
Капиллярность почвы ''21
Капсулы «фиксанал» 51
Карандаши аля стекла 38
Карта санитарного обследова-
ния жилого помещения 444
— санитарного обследования
производственного цеха 445
Кататермометр 165
— шаровой 169
Кататермометрия 165
Кислород воздуха 221
— растворенный в воде 359
Кислорода биохимическое по-
треблю ие (ВПК) сточных вод
404
Кислота азотистая в воде см.
азот нитритов в воде
— азотистая в воздухе см. окис-
лы азота в воздухе
— азотная в воде см. азот ни-
тратов в воде
— азотная в воздухе 254
— карболовая 506
— — неочищенная 507
— кремневая в воде см. кремне-
кислота в воле
— розоловая 57
— серная (сульфаты) в воде
333, 37':
----- в воздухе 250
-----(сульфаты) в почве 423
— — — в сточной воде 401
— угольная в воде см. угле-
кислота в воле
— угольная в воздухе см. ан-
гидрид угольный в воздухе
— фосфорная (Фосфаты) в воде
питьевой 346
—------в воле сточной 401
—------в почге 423
— хлористоводородная (хлори-
ды) в воде питьевой 430
— хлористоводородная (хлори-
ды) в воде сточной 401
— хлористоводородная (хлори-
ды) в почве 422
Кислота хлористоводородная в
воздухе 258
— щавелевая 59
Кислотность воды 312
Кислоты жирные в мыле 517
— смоляные в мыле 517
Клейстер крахмальный 64
— калии-иод-крахмальный 225
Колба Виноградского 112
Колбы мерные 19
— обыкновенные 23
Коли-индекс волы 380
— титр воды 380
Колонка поглотительная для
газов 219
Колориметр Генера 68
— Дюбоско 68
— в виде призмы 337
Колориметрия визуальная 65
— фотоэлектрическая 70
Компаратор двухрядный 80
— трехрядный 81
— для колориметрирования
в полевых условиях 375
Комплемент 126
Конгорот 57
Кониметрия пыли 203
Константа диссоциации 74
Концентрация водородных ио-
нов 73
Копоть в воздухе 192
Коэфициент дневного света (све-
товой коэфициент) z62
— естественного освещения
(КЕО) 472
— нормальности титрованных
растворов 51
— общей теплопроводности ог-
раждений 459
— отражения света 462
— полезного действия армату-
ры 473
— полезного действия печей 46(>
— теплопроводности строймате-
риалов 433
Краска для надписей на стекле
38
— генпианвполет по Граму 120
— карболовый фуксин по Цилю
119
— крпсталлвиолет по Граму 120
— метиленовая щелочная 119
— фуксиновая 119
Крахмал растворимый 61
Крезолы в сточной готе z06
Кремнезем в пыли 277
Кремнекислота в воде 346
Кривая с вето рас пределения 473
Кривые титрования 58
Кубатура помещений 446
Л
Лакмус 56
Лучи видимые 179
— инфракрасные 179
— ультрафиолетовые 179
Люкс 461
Люксметр ГОИ 469
— фотоэлектрический 471
Люмен 461
М
Магазин сопротивлений 103
Мазки, приготовление 119
Манометр вакуумный 214
Марганец в воде 350
Материалы строительные 428
Мезосапробы воды 392
Медь в воде 351
Мензурки 23
Метеорология 138
Метилоранж 56
Метилрот 56
Микробное число воды 379
Микробюретки см. бюретки
микро
Микрометр объективный 193
— окулярный 193
— винтовой 193
Микрометрия пыли 193
Микропипетки 23
Микроскоп 106
Микроскопия тканей одежды 50S
Миллилитр 19
Молоко (среда) 116
Мутномеры 301
Мутность воды 301
Мышьяк в воле 357
— в воздухе 259
— в пыли 280
— в тканях одежды 494
Мыла 514
Н
Напряжение водяных паров, на-
сыщающих воздух (табл.) 144
Насекомые, повреждающие де-
рево 442
Насос водоструйный 197
Насосы воздушные 197
НатрЛй в воде 326
Нейстон воды 392
Нейтрализация (метод) 52
Нефелометрия 72
Нефть в сточных волах 410
Никотин в воздухе 275
Номограмма для актинометра
Калитина 182
-----волюмометра Гуревича и
Вендта 209
-----определения щелочности
воды 311
-----определения ЭТ и ЭЭТ 177
Нормальность растворов 51
Нормы вентиляции промышлен-
ных помещений (табл.) 456
— естественного освещения
промышленных помещений
(табл.) 474
— искусственного освещения
для работ (табл.) 475, 476
— температурные помещений
446
— метеорологические в рабочей
воне (табл ) 4^7
— метеорологические дутпиро-
вания (табл.) 448
— предельные для газов, паров
и пыли (табл.) 283 и 284
Пуль гесов 42
Путч 26
О
Озон 225
Окисление-восстановление (ме-
тол) 58
О кислы азота в воздухе 255
Окисляемость волы питьевой 314
— — сточной 401
Окись углерода в воздухе 235
Окраска жгутиков бактерий 121
— мазков 119
— по Граму 120
— спор 120
Олигосапробы воды 392
Олово в воде 351
Осадки атмосферные 211
Осадок сточных вод 401
Осаждение (метод) 65
Освещение естественное ^62
— искусственное 472
— помещений ^60
Остаток плотный питьевой воды
314
— — сточной волы 401
Отопление помещений 458
Очистка ртути 35
П
Палочки фильтровальные 26
Перекристаллизация 32
Перманганометрпя 59
Петля для бактериологических
посевов 112
Печь муфельная 28
— тигельная 28
Пикнометры 207
Пипетки 22
— газовые 213, 224
— Пастера 113
Пирометр термоэлектрический
183
— оптический 185
Пирохметрия 183
620
Иламя (структура) 28
ланитой воды 392
Пленка сточных вод 400
Площадь помещений 446
Показатель водородных ионов
(общие сведения) 75
— водородных ионов (колори-
метрическое определение) 78
— водородных ионов (электро-
метрическое определение) 85
— титрования (рТ) 48
Полиса пробы воды 392
Помещение лаборатории 13
Помещения жилые и другие 444
Пористость почвы 417
— стройматериалов 429
— тканей одежды 488
Порог болевого ощущения эвука
477
— осязания звука 477
— слышимости 477
Посев бактериологический (тех-
ника) 117
Посуда лабораторная 19, 31
— стеклянная 19
— платиновая 26
— фарфоровая 26
Потометр 420
Потенциал каломельного элек-
трода (табл.) 87
Потенциометр с водородно-кало-
мельным элементом (схема)
94
Потенциометр с хингидронно-
каломельным элементом (схе-
ма) 97
— МОСКИП 101
Почва 413
Препараты цианистые 511
Преципитатор пыли электриче-
ский 200
Прибор Галанина 431
—для обработки посуды паро*м 31
-----определения электропро-
водности растворов (схема)
104
----- определения электропро-
водности МОСКИП 106
-----определения влаги де-
стилляцией 513
— — определения газов по Тем-
пе лю 224
-----определения звукопровод-
ности стройматериалов 436
-----определения окиси угле-
рода 238
-----плотного остатка воды 314
—------прозрачности воды 299
— — — прозрачности воды по
кресту 301
----- — рассеивания электри-
чества в воздухе 187
------- фтора в воде 272
Приборы просасывания воздуха
220
— стерилизации 110
Прибор Илькевича 438
— Марша 261
— Небеля 415
— Орса 240
— Реберга-Винокурова 233
— Сабанпна 415
— Склаво-Чаплевского 296
— Скобельцина (схема) 434
— Субботина—Нагорского 230
— Хлопина 222
— Чашечковый Петтенкофсра 426
— Шэфира 288
— Яковенко 439.
Проба на загниваемость сточной
воды 404
----- стабильность сточной
воды 405
Пробирки для колориметриро-
вания 66
— с оттянутыми наконечниками
для взятия проб воды 296
— с поплавками 383
Пробкомялка 30
Прокаливание осадка 40
Промывалка (фильтр) Городец-
кого 219
-----Гуревича 218
-----Дрекселя 218
-----ЛИГТ 218
Промывалка . — MOOT 218
— Петри 216
— спиральная 218
— Тищенко 219
Промывание осадка 39
Псевдопланктон 392
Психрометр аспирационный 147
— станционный 145
Психрометрия 145
Пыль помещений 192
— атмосферная 209
Р
Радиация ультрафиолетовая 185
Разновес аналитический 17
Размеры пылевых частиц (см.
микрометрия пыли)
Раствор 0,1 N бихромата калия
----буры 54
---- едкого натрия 55
---- иода 64
----ирдата калия 62
------- соляной 54
----щавелевой 59
---- перманганата калия 60
— соды 53
---- тиосульфата натрия 62
Растворы, приготовление 33
— нормальные 45
— молярные 46
— титрованные 44, 50, 52
— эмпирические 46
Расчет вентиляционного воздуха
454
— теплопроводности здания 459
— печей ^60
Реактив Грисса 227
— Дениже 262
— Молит а 499
— Несслера 253
— Полежаева 265
Реактивы, обращение и очистка
32
Реакция агглютинации 121
— воды, определение 306
— преципитации 123
— связывания комплемента 124
Рейтер 18
Реометр 198
Роданиды в воде 410
Роза ветров 159
Ртуть, хранение и очистка 35
Ртуть (пары) в воздухе 264
Румбы 159
С
Сгаливаемссть тканей одежды
486
Срррла для пробок 30
Свеча международна я 61
— Вполя 461
Свидетели титрования 49
Свинеп в воле 351
— в пыли 277
Сера 509
Сероводород в воздухе 246
---- роде 365
— — сточных волах 405
Сероуглерод в воздухе 250
— (ивсектисид) 510
Сестон 392
Сетка асбестовая 39
— планктичсская 394
Сита для отсеивания почвы 414
Склянки вакуированные 214
Смазка для стеклянных кранов
38
Сонометр 436
Сорбция газов тканями одежды
496
Сосуды для измерения электро-
проводности растворов 103,
104
---- бактериальных культур
112
Сосуд для платинирования элек-
тродов 93
Среда Булира 114
— висмут-сульфитная 115
— Кесслера 115
— Кпгт-Тароцци 117
— Эйкмана 114
Среда Эндо 115
Средства дезинсекционные (ин-
сектисиды) 509
— дезинфекционные 502
Среды Барэикова-Гетча 116
Стакан измерительный длп дож-
демера 212
Стаканы химические 24
Стекля часовые 24
Стерилизатор сухой 110
— Коха НО
Стильб 461
Стол лабораторный 14
Сферометр системы автора 488
— Рубнера 487
Схема санитарного анализа
питьевой воды 297, 298
--------сточной воды 399
Счетчик ионов Богоявленского
189
----- Эберта 191
— взвешенной пыли Овенса 203
— оседающей пыли Овенса 205
Сулема 504
Сыворотка гемолитическая 125
— специфическая (иммунная) 126
Т
Температуры эффективно-экви-
валентные 173
Теплоемкость стройматериалов
Теплопроводность стройматериа-
лов 433
— тканей одежды 489
Термограф 139
Термометр максимальный 139
— минимальный 139
— парный 140
— почвенный см. геотермометр
— с черпаком для воды 299
Термопары 183
Терморегуляторы автоматиче-
ские 29
Термостаты 109
Тетраэтилсвинец 266
Тигель Гуча 26
Тигли платиновые 26
— фарфоровые 26
Титр растворов 44
Тигр-коли см. коли-титр
Титрование (техника) 48
— потенциометрическое 100
Ткани одежды 485
Толщемер 488
Точка эквивалентная титрова-
ния 48
Тренога 30
Треугольник с фарфоровыми
трубками 30
Триптон 392
Трубка десятишариковая 219
— пнеймометрическая 451
— U-образная 219
— хлоркальциевая 219
Трубки стеклянные (обработка)
37
Тягомер 453
У
Углекислота агрессивная в воде
362, 364
— гидрокарбонатная в воде 362,
363, 364
— ппактивная в годе 362
— карбонатная в воде 362
— общая в воде 362, 363, 364
— полусвязанная в роде 362
365
— свободная в воде 361, 362
— связанная в воде 362, 36.'»
Углерод органический почвы
423
-----сточных вод 402
Угломер 463
— пространственный 464
Угол защитный арматуры 473
— отверстия 463
— падения 463
— пространственный 464
Уголь (определение серы) 482
521
У’пр у гость насыщенного пара и
соответствующая температу-
ря (габл.) 144
Усадка тканей одежды 486
Установка для определения ве-
совой концентрации пыли
воздуха 199
— для определения весовой
концентрации влаги воздуха
145
— для систематического наблю-
дения за стоянием уровня
почвенных вод 426
Ф
Фактор кататермометра 165
Фенолфталеин 56
Фильтрование 38
Фильтр Городецкого см. про-
мывалка Городецкого
— Предволителева-Блинова 201
Фильтры беззольные 38
— бумажные складчатые 32
— для бактериологического ис-
следования воздуха Дьяко-
нова 287
— для бактериологического ис-
следования воздуха Микеля
287
— для бактериологического ис-
следования воздуха Миляв-
ской 286
— для бактериологического ис-
следования воздуха Миро-
нова 286
— для бактериологического ис-
следования воздуха Штрауса
с жилкой средой 287
— мембранные для бактериоло-
гического исследования воды
387
Фенолы в сточных водах 406
Флуороскоп 397
Флюгер 159
Фонометр 479
Форма пылевых частип 192
Формалин 502
Формулы пылевых частиц 193
Фосфаты в питьевой воде, сточ-
ной вопе и почве см. кислота
фосфорная в тех же объектах
Фосфор 263
Фотометр упрощенной конструк-
ции 465
— для точных измерений 466
Фригориметр Шахбазяна 172
Фригоряметрия 172
Фтор в воде 370
Фуксин карболовый 119
Фумигатор 160
X
Хлор в воздухе 256
Хлор активный в воде 367, 377
Хлорилы в питьевой воде, в
сточной воде и в почве см.
кислота хлористоводородная
в тех же объектах
Хлорпикрин 512
Холерный вибрион в воде, обна-
ружение 392
Хранение дестиллированной во-
ды 36
— растворов титрованных 52
— реактивов 32
— резиновых предметов 37
— ртути 35
Хром в сточных водах 407
Ц
Цветность воды 302
Цианиды в сточных водах 409
Цилиндры Генера 68
— делительные 24
— мерные 23
— Снеллена 299
Цинк в воде 351
Я
Часы газовые 198
Чашки выпаривательные 26
— Петри 112
Черпак для улавливания бентоса
394
Число водородное 74
— удельное ионов атмосферы
Ш
Шкалы температурные 138
Шкаф вытяжной 15
— сушильный 29
Шпатель Дригальского 112
— металлический для бактерио-
логических посевов 113
Шприц (промывалка) 25
Шрифт Снеллена К» 1 (табл.) 300
Штатив для колориметрических
пробирок 66
Шум 477
Шумомер Навяжского 478
Шутц ,26
Щ
Щелочность воды 309
Щипцы тигельные 39
Э
Экссикаторы 25
Электро анемометры 163
Электрод водородный груше-
видный 89
-----СГ-образный 90
-----каломельный 90
— — каломельный МОСКИП
91
— — платиновый 96
Электриче<*гво атмосферное 18J
Электрометр капиллярный 92
Электропроводность воды 304
— растворов 101
— удельная 102
Элемент нормальный Вестона 88
Энергия лучистая 178
Эпруветка 233
Эритроциты барана 125
Я
Яркость освещения 461
ОГЛАВЛЕНИЕ
Предисловие..................................................................... 3
Глава первая. Методы, применяемые в санитарно-гигиенических исследованиях
Органолептические методы...................................................... 5
Физические методы............................................................... 6
Физико-химические методы........................................................ 7
Химические методы .............................................................. —
Биохимические методы............................................................ 8
Микроскопические методы ........................................................ 9
Бактериологические методы....................................................... —
Микологические методы ......................................................... 10
Гельминтологические методы .................................................... —
Серологические методы .......................................................... —
Биологические методы............................................................ И
Физиологические методы.......................................................... —
Статистические методы ......................................................... 12
Глава вторая. Введение в лабораторную технику
Помещение лаборатории.......................................................... 13
Оборудование и приборы........................................................ 14
Стол лабораторный 14. Вытяжной шкаф 15. Технические весы 16. Аналитиче-
ские весы 17 Аналитические разновесы и рейтер 18 Микроаналитические
весы 18 Стеклянная посуда 19 Фарфоровая и платиновая посуда 26 При-
боры для нагревания, прокаливания и высушивания 27. Прочие лабораторные
принадлежности 30.
Основы лабораторной техники . ........................................... 30
Правила обращения с химической посудой 31. Правила обращения с
реактивами, их очистка 32. Приготовление различных растворов 33.
Хранение и очистка ртути 35 Получение и хранение дестиллированной воды 36.
Хранение резиновых предметов 37 Обработка стеклянных трубок 37. Свер-
ление пробок 37 Некоторые рецепты 38.
Основы весового анализа . . .................................. 38
Осаждение 39. Фильтрование и промывание осадка 39. Высушивание и про-
каливание 40 Правила обращения с аналитическими весами и способы взве-
шивания 41. Правила обращения с микроаналитическими весами и способы
взвешивания 43.
Основы объемного анализа.................................................... 44
Предварительные сведения 44 Метод нейтрализации 52. Методы окисления
и восстановления 58 Методы осаждения 65.
Физико-химические методы ... ............................. 65
Колориметрия 65. Нефелометрия 72. Определение концентрации водородных
ионов 73. Электрометрическое (потенциометрическое) титрование 100. Измерение
электропроводности растворов (кондуктометрия) 1 01.
Важнейшее оборудование для бактериологических работ........................... 106
Микроскоп 106. Термостат 109. Приборы для стерилизации ПО. Сосуды для
культур 112. Приспособления для посева 112. Счетная камера ИЗ.
Приготовление важнейших питательных сред...................................... 114
Мясопептонный бульон (МПБ) 114. Мясопептонный агар (МПА) 114 Мясопеп-
тонная желатина (МПЖ) 114. Среда Эйкмана 114. Среда Булира 114 Среда Кес-
лера 115. Среда Эндо 115 Висмут-сульфитная среда 115 Среда элективная для
вибрионов азиатской холеры 116. Пептонная вода 116 Среда Барзикова 116.
Молоко 116. Желчь 116. Питательные среды для анаэробов 116.
Техника посева.............................................................. 117
Приготовление мазков и их окраска............................................. 119
Щелочной раствор метиленовой синьки 119. Карболовый фуксин 119. Разбав-
523
ленный раствор фуксина 119. Окраска по Граму 120. Окраска спор 120. Окраска
жгутиков 121.
Серологические реакции.................................................... 121
Реакция агглютинации 121. Реакция преципитации 123. Реакция связывания
комплемента 123. Исследования на животных (биопробы) 130.
Приложения к главе второй ................................................. 131
Атомные веса элементов 131. Удельный вес минеральных кислот 132. Удель-
ный вес водных растворов уксусной кислоты 133. Удельный вес водных растворов
КОН и NaOH 133. Удельный вес водных растворов аммиака 134.
Литература................................................................. 135
Глава третья. Воздух
Состав и свойства атмосферного воздуха..................................... 137
Санитарно-гигиенические требования к воздуху................................. —
Органолептическая оценка воздуха............................................. —
Физические методы исследования воздуха..................................... 138
Температура 138. Барометрическое давление 140. Влажности 143. Движение
воздуха 158. Кататермометрия 165. Фригориметрия 172. Эффективные и эквива-
лентно-эффективные температуры 173. Лучистая энергия 178. Атмосферное
электричество 187.
Пыль.................................................................. 192
Осадки, выпадающие из атмосферы ........................................ 211
Химические методы исследования воздуха......................................212
Общие сведения 212. Кислород 221. Озон 225. Угольный ангидрид 228. Окись
углерода 235. Сернистый ангидрид 243. Сероводород 246. Серная кислота и сер-
ный ангидрид 250. Сероуглерод 250. Аммиак 252. Азотная кислота 254. Окислы
азота 255 Хлор 256. Бром и иод 258. Хлористый водород 258. Мышьяковистый
водород 259. Фосфор и фосфористый водород 263. Пары ртути 264. Пары тетра-
этилсвинца 2(56. Ацетилен 267. Акролеин 268. Ацетон 269. Бензин 269. Бен-
зол 271 Анилин 272. Никотин 275.
Химическое исследование атмосферного воздуха.............................276
Химический анализ пыли.................................................... —
Зола и органические вещества 277. Кремнезем 277. Свинец 277. Мышьяк 28Э.
Химический анализ атмосферной пыли........................ ... ... 282
Зола и потери при прокаливании 282. Смолистые вещества 282. Сульфаты 282.
Хлориды 283.
Бактериологическое исследование воздуха ..................................... 285
Определение общего числа бактерий в воздухе 285 .........................
Бактериологическое исследование пыли, осевшей из воздуха..................290
Литература .................................................................. —
Глава четвертая. Вода (питьевая)
Свойства доброкачественной питьевой воды......................................293
Отбор проб воды для анализа.................................................• 295
Схема полного санитарного анализа воды ................................... 297
Схема санитарного анализа воды.......................,.....................298
Орга ноле пти чес кая оценка воды ..........................................г 298
Запах 298. Вкус и привкус 298.
Физические методы исследование воды.......................................... 299
Температура 299. Прозрачность 299. Мутность 301. Цветность 302. Электро-
проводность 304.
Химические методы исследования воды ....................................... . 305
Последовательность производства отдельных определений санитарно-химиче-
ского анализа 305. Способы выражения результатов анализа 306 Реакция 306.
Щелочность 309. Кислотность 312. Взвешенные вещества 313 Плотный оста-
ток 314. Окисляемость (органические вещества) 314. Жесткость (щелочно-земель-
ные металлы) 317. Щелочные металлы (Na и К) 326. Хлористоводородная ки-
слота (хлориды) 330. Серная кислота (сульфаты) 333. Азотсодержащие ве-
щества 335. Фосфорная кислота (фосфаты) 346. Кремнекислота (силикаты) 346.
Алюминий (глинозем) 348. Железо 348. Марганец 350. Свинец, медь, цинк,
олово 351. Мышьяк 357. Кислород, растворенный в воде 359. Углекислота 361.
Сероводород 365. Остаточный активный хлор 367 Фтор 370.
Полевые методы физико-химического исследования питьевой воды.................373
Отбор проб................................................................373
Физические свойства воды..................................................373
Химический состав воды 374.
Щелочность 374. Карбонатная жесткость 374. Сульфаты 374. Хлориды 375.
Аммиак 375. Азот нитритов 375. Железо 376. Окисляемость. 376. Сероводород 377.
Активный хлор 377. Хлоро поглощаемость 377. Определение процентного со-
держания активного хлора в хлорной извести 377.
524
Отличие некипяченой воды от кипяченой........................................378
Микробное число воды 379. Определение коли-титра и коли-индекса воды 380.
Полевой метод определения фекального загрязнения воды........................387
Обнаружение патогенных бактерий в воде.......................................389
Исследование воды на тифозно-паратифозную группу 389. Исследование воды
на возбудителя бациллярной дизентерии 391. Исследование воды на присут-
ствие вибрионов азиатской холеры 391.
Биологическое исследование воды ............................................ 392
Техника собирания материала 393. Методика исследования планктона и бен-
тоса 394.
Санитарное изучение источников водоснабжения ............................... 396
Обследование открытых водоемов 396. Обследование грунтовых источников 396.
Глава пятая. Сточные воды
Санитарные требования к сточным водам...................................... 398
Показатели загрязнения...................................................... —
Схема полного санитарного анализа сточных вод............................. 399
Схема краткого санитарного анализа сточных вод............................ 400
Отбор проб .................................................................... —
Физико-химические методы исследования сточных вод..........................
Температура, запах, цветность, прозрачность 400. Муть и осадок, пленка 400.
Реакция (pH), щелочность и кислотность (титрирные) 401. Взвешивание ве-
щества и плотный остаток 401. Окисляемость 401. Хлориды, сульфаты, фос-
фаты 401. Азотсодержащие вещества 401. Углерод органический 402. Биохими-
ческая потребность в кислороде (БПК)404. Проба на загниваемость 404. Проба
на стабильность 405. Сероводород 405. Свободный хлор 406. Тяжелые металлы,
мышьяк 406. Фенолы и крезолы 406. Соединения хрома 407. Цианистые соеди-
нения 409. Роданистые соединения 410. Нефть 410.
Литература........................................................... 411
Глава шестая. Почва
Санитарные требования к почве........................................ 413
Отбор проб............................................................. —
Физико-механические методы исследования почвы................................. —
Температура 413. Величина зерен почвы 414. Отсеивание почвы 414. Отмучивание
почвы 414. Общий объем пор 417. Воздухопроницаемость почвы 419. Водоем-
кость 419. Водопроницаемость 420. Капиллярность 421.
Химические методы исследования почвы........................................ 421
Подготовка почвы для исследования 421. Влажность 422. Потеря при прокали-
вании 422. Растворимые в воде вещества 422. Хлориды 422. Азотсодержащие
вещества 422. Органический углерод 423. Серная кислота 423. Фосфорная кис-
лота 423. Экскременты и моча 424.
Исследование почвенного воздуха ............................................. 424
Бактериологическое исследование почвы......................................... —
Санитарно-топографическая и гидрогеологическая характеристика местности .... 426
Литература................................................................... 427
Глава седьмая. Строительные материалы
Санитарные требования........................................................ 428
Вес единицы объема материала 428. Пористость 429. Воздухопроницаемость 430.
Водопоглощение 432. Водонасыщение 432. Водоотдача 433. Теплопроводность
и теплоемкость 433. Звукопроводность 435. Влажность 437.
Выяснение причин сырости строительного материала............................. 441
Грибы, насекомые и животные, повреждающие-дерево............................... —
Литература.................................;................................. 443
Глава восьмая. Жилые, фабрично-заводские, школьные
и другие помещения
Карта санитарного обследования жилого помещения..........................444
Карта санитарного обследования производственного цеха...................... 445
Площадь пола и кубатура помещения.............................................. 446
Воздух помещений............................................................... —
Сырость...................................................................... 448
Вентиляция .................................................................... —
Естественная вентиляция.................................................... —
Искусственная вентиляция................................................... 4з0
Расчет вентиляционного воздуха 454.
525
Вентиляционные нормы......................................................456
Отопление....................................................................458
Расчет теплопотребности здания 458. Расчет печей 460.
Освещение....................................................................460
Световые понятия и единицы 460.
Естественное освещение................................................... 462
Коэфициент отражения 462. Относительная площадь остекления (световой коэ-
фициент) 462. Углы падения и отверстия 463. Пространственный угол 464. Осве-
щенность 464. Равномерность освещения 472. Коэфициент естественной освещен-
ности 472.
Искусственное освещение ................................................. 472
Осветительная арматура 473. Кривая светораспределения 473.
Нормы освещенности....................................................... 474
Шум..........................................................................477
Сотрясение.................................................................. 479
Осветительные и топливные материалы........................................ 481
Исследование керосина 481. Иследование угля 482.
Литература.................................................................. 482
Глава девятая. Ткани одежды
Санитарно-гигиенические требования.......................................... 485
Физико-механические методы исследования тканей одежды...................... 486
Крепость и растяжимость ткани, счет нитей, просвечиваемость, усадка, свали-
ваемость и аппретура 486. Толщина тканей 486. Удельный вес и объем пор 488.
Теплопроводность 489. Воздухопроницаемость ткани 491. Водопроницаемость
493. Водоемкость 495.
Химические методы исследования ткани........................................ 496
Гигроскопичность 496. Влажность 496. Определение мышьяка 496. Сорбция газов —
Некоторые химические пробы для идентификации ткани 499.
Микроскопическое исследование тканей.......................................... 500
Бактериологическое исследование тканей........................................ —
Литература.................................................................... 501
Глава десятая. Дезинфекционные и дезинсекционные средства
Дезинфекционные средства...................................................... 502
Формалин 502. Сулема 504. Карболовая кислота 506. Карбаловая кислота
(i еочишенная) 507. Хлорная известь. 508.
Дезинсекционные средства (инсектисиды)...................................... 509
Сера 509. Сернистый анторид (сжиженный) 510. Сероуглерод 510. Хлорпикрин 510.
Цианистые препараты 511. Дихлордифепилтрихлорэтан (ДДТ) 513. Гекса-
хлорциклогексан (ГХЦГ) 514. Мыла 515.
Литература ................................................................. 518
Предметный указатель................................................., , , • * 519
Редакторы П. Е. Бейлин п М. И, Снежин.
Техредактор Е. Н. Роаенцвейг
Корректор А. И. Делецкоя
Обложка худ. А. Е. Миткевича
БФ 04401. Зак. № 381. Тираж 10 000. Подписано
к печати 24/XI 1950 г. Учетно-изд. листов 54,15.
Бумага 70 х 10S1/16= 16,5 бумажных—45,2 печати.
лист. Цена 40 руб. 70 коп. Переплет 3 руб. 50 коп.
Книжно-журнальная фабрика Укрполиграфиздата
при Совете Министров УССР.
Киев, ул. Воровского, 24.
МЕТОДЫ
cskmwiw УНГИЕНИЧГСК?!;
ЖСЛТЕДОМШЙ >