/































































































































































































Author: Джексон Р.А.
Tags: органическая химия химия переводная литература химические реакции издательство химия
Year: 1978




Text
p. v.t;i;i:i;t он
Введение
в изучение
механизма
органических
реакций
Oxford Chemistry Series
General Editors:
P. W. ATKINS, J. S. E. HOLKER, A. K. HOLLIDAY
RICHARD A. JACKSON
Mechanism:
an introduction to the study of
organic reactions
Clarendon Press Oxford 1972
© OXFORD UNIVERSITY PRESS-1972
Р.А.ДЖЕКСОН
Введение
в изучение
механизма
органических
реакций
ПЕРЕВОД С АНГЛИЙСКОГО
канд. хим. наук Н. М. ЛОЙМА
ПОД РЕДАКЦИЕЙ
докт. хим. наук 3. Н. ПАРНЕС
Москва
Издательство «Химия»
1978
УДК 547
Джексон Р. А.
Введение в изучение механизма органических
реакций.—Пер. с англ./Под ред. 3. Н. Парнес—
М.: Химия, 1978.— 192 с, ил.,,
192 с, 7 рис., 8 табл., список литературы 62 ссылки.
Книга является руководством к изучению механизмов
органических реакций. В ней даны рекомендации, как изучать механизм
органических реакций, каким образом и какую информацию о
механизме можно почерпнуть при исследовании реакции тем или другим
методом, показаны возможности этих методов и их ограничения.
Достоинством книги является включение в нее большого числа
задач, которые помогут усвоению материала.
Книга предназначена для широкого круга читателей как
специалистов-химиков, так и начинающих работников, исследующих
органические реакции. Она может быть полезна преподавателям,
студентам старших курсов и аспирантам химических специальностей.
20504-028 28-?8
050(01)-78
Редактор Технический редактор
Слуцкий О. И. Скитина В. М.
Художественный редактор Корректоры Козловская Н. А.
Су мнительный Б. А. Волкова Л. А.
Художник
Сумнительный Е. А
ИБ № 664
Сдано в наб. 15.03.78. Подп. в печ. 28.08.78. Формат бумаги 84ХЮв'/з*.
Бумага тип. № 2. Литературная гарнитура. Печать высокая.
УсЛ. Печ. Л. 10,08. Уч.-изд. л. 9,54. Тираж 9700 экз. Зак. 1044.
Цена 65 к. Изд. № 1510.
*•
Издательство «Химия» 107076, Москва, ул. Стромынка* 13
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой
«Союзполиграфпрома> при Государственном комитете
Совета Министров СССР по делам издательств,
полиграфии и книжной торговли.
198052, Ленинград, Л-52, Измайловский проспект, 29.
©Перевод на русский язык. Издательство «Химия», 1978 г.
Содержание
Предисловие редактора английского издания 7
Предисловие автора 8
Предисловие редактора русского перевода 10
Применяемая символика 12
Глава 1. Что такое механизм реакций и зачем надо его
изучать 13
Что такое механизм реакций? Зачем изучают
механизмы реакций? Каким условиям должен удовлетворять
предлагаемый механизм реакции? Установление
механизма органических реакций. Задачи.
Глава 2. Продукты реакций 29
Определение строения и выхода продуктов. Какие
связи образуются и разрываются в ходе реакции?
Перемещались ли группы или атомы от молекулы к
молекуле в процессе реакции? Доказательства,
основанные на образовании побочных продуктов. Резюме.
Задачи
Глава 3. Кинетика 46
Экспериментальные методы. Связь между механизмом
и кинетикой. Первичный кинетический изотопный
эффект. Резюме. Задачи.
Глава 4. Интермедцаты 83
Карбониевые ионы. Карбанионы. Радикалы. Карбены.
Производные дегидробензола.
Тетраэдрические интермедиаты. Кислотно-основной
катализ. Стабильные молекулярные интермедиаты. За»
ключение. Задачи,
Глава 5. Стереохимия
120
Замещение при тетраэдрическом центре и оптическая
изомерия. Реакции с инверсией у атома углерода:'
Замещение с сохранением конфигурации. Реакции,
включающие- промежуточное образование ионов кар-
бония, радикалов или карбанионов. Присоединение к
кратным связям и элиминирование. Согласованные
молекулярные реакции. Интерпретация данных о
новых реакциях присоединения и элиминирования.
Задачи.
Глава 6. Другие методы изучения механизма реакций 150
Стерическое ускорение и замедление. Эффекты
заместителей. Электронные эффекты заместителей:
уравнение Гаммета. Эффект растворителей. Постулат
Хэммонда. Задачи.
Заключение 173
Задачи.
Указание к решению задач 181
Ответы на задачи 192
Литература 188
Предметный указатель 191
ПРЕДИСЛОВИЕ РЕДАКТОРА
АНГЛИЙСКОГО ИЗДАНИЯ
Уже стало довольно обычным описывать реакции
органической химии при помощи ряда формул с
«изогнутыми стрелками» в подходящих направлениях.
Часто это делается вне зависимости от того, изучен
или не изучен механизм реакции. Как ни странно, но
часто многие студенты и научные работники считают
описание с помощью изогнутых стрелок
удовлетворительным объяснением механизма и, по-видимому, не
расположены к его изучению. Данная монография
показывает необходимость установления, механизма
реакций и дает представление о некоторых методах,
пригодных для этой цели. Реакции, выбранные для
иллюстрации использования отдельных методов,
охватывают существенную область типов органических
реакций.
Содержание книги доступно для понимания
студентов старших курсов, однако для глубокого
проникновения в сущность вопроса необходимо изучить
также другие монографии этой серии. Так, детальное
представление об использовании методов протонного
магнитного и электронного парамагнитного резонан-
сов как инструмента в изучении механизма требует
фундаментальных знаний принципов, лежащих в их
основе; с этим можно познакомиться в томе,
посвященном магнитному резонансу. Более полную картину
о применении стереохимических методов можно
получить в книге о стереохимии и механизме, а
последующая монография серии о быстрых реакциях
иллюстрирует специальную технику, необходимую для
исследования в этой важной области.
Дж. С. Э. Холкер
ПРЕДИСЛОВИЕ АВТОРА
Химию часто представляют как дауку, которая по
сути является фактологической и в которой почти
нет места для работы мысли. И хотя верно то, что
содержание химии в большой степени представляют
факты, однако теоретические исследования должны
привести к обобщению и созданию принципов,
которые позволят объединить и объяснить отдельные
факты и на основании этого привести к постановке
новых экспериментов для проверки этих принципов.
Примером такого подхода является изучение
механизмов органических реакций, проиллюстрированное
в этой книге рядом специально подобранных реакций.
При этом не ставилась задача осветить механизм
каждой важной реакции, в связи с чем многие
значительные области химии, такие как фотохимия и
гетерогенные реакции, оказались нерассмотренными.
Методы исследования в данной области органической
химии представлены в настоящей монографии с
указанием их возможностей и ограничений. В текст
книги включен ряд задач, иллюстрирующих
рассматриваемый материал, и автор надеется, что это
поможет читателям использовать приведенные в книге
методы для изучения других реакций, с которыми они
встретятся в своей работе. Можно надеяться, что
знакомство с книгой приведет читателя, чьи научные
интересы лежат в других областях химии, к мысли
использовать рассмотренные методы для изучения
неорганических, полимерных, гетерогенных и
биохимических систем и сравнить их со специфическими
приемами, применяемыми в этих областях.
Развитие химии зависит как от экспериментальных,
так и от теоретических исследований. При этом
развиваемые представления иногда способствуют
прогрессу, а иногда тормозят его. На успех проводимых
8
исследований влияют вопросы стоимости и
доступности аппаратуры (и человеческих усилий). Эту
взаимосвязь, хотя она и не образует основное содержание
книги, нельзя полностью упускать из вида, так как
эта монография посвящена как методам и идеям, так
и экспериментальным результатам.
Я. Л. Джексон
БРАЙТОН
Ноябрь, 1971 г.
ПРЕДИСЛОВИЕ РЕДАКТОРА
РУССКОГО ПЕРЕВОДА
Предлагаемая вниманию читателей монография
Р. А. Джексона посвящена фундаментальному
разделу органической химии — механизму реакций. В
последние годы в отечественной и зарубежной
литературе появилось немало книг и пособий (в том числе
учебных), в которых рассмотрены различные аспекты
этой области теоретической органической химии. Тем
не менее, настоящая монография является
уникальной, поскольку основана на оригинальном подходе
автора к проблеме. Традиционный путь состоит в
систематизации материала по типам реакций,
природе реагентов, реакционного центра, молекулярности
и т. п., при такой постановке авторы стараются
ответить на вопрос «как протекает реакция?». Р. А.
Джексон основное внимание уделяет ответу на вопрос «как
изучать механизм реакции?». Отвечая на
поставленный вопрос, автор после формулировки определения
механизма реакций переходит к строго логическому
изложению путей, которые могут привести к
выяснению механизма. При этом Джексон четко
формулирует, какие сведения о ходе реакции могут быть
получены при изучении основных и побочных продуктов
реакции, при кинетическом изучении, какие пути
ведут к определению строения и характера интермедиа-
тов (промежуточных соединений) и установлению
структуры переходного состояния, какую информацию
можно получить из знания стереохимии процессов,
эффектов растворителей и влияния заместителей и т. д.
Автор на частных примерах, охватывающих многие
типы органических реакций, демонстрирует
возможности и ограничения экспериментальных методов
исследования механизма реакций,
10
Весьма ценным для закрепления материала
является включение в книгу большого числа интересных
задач и подробных ответов на них.
Монография написана по строгому, логически
оправданному плану и может быть сравнена с
путеводителем по запутанному лабиринту выяснения
механизма органических реакций. Излагаемые автором
принципы и подходы к исследованию механизмов
выходят за рамки понимаемой в классическом смысле
органической химии и с успехом могут быть
использованы в неорганической, полимерной, биологической
и других областях химии.
Я уверена, что монография Р. А. Джексона окажет
существенную помощь широкому кругу научных
работников и, в первую очередь, начинающим
исследователям в освоении принципов и методов изучения
механизма органических реакций.
Перевод книги выполлен канд. хим. наук
Н. М. Лоймом.
Июнь, 1977 г.
Докт хим. наук
3. Н. Парнес
ПРИМЕНЯЕМАЯ СИМВОЛИКА
Реакция не идет.
Специальное указание на то, что
обсуждаемая реакция включает более чем
одну стадию.
*=± Обратимая реакция.
<="-* Обратимая реакция, равновесие которой
сдвинуто в сторону образования тех или
иных продуктов.
—т* Реакция с инверсией (обращением
конфигурации).
^\ Графическое изображение движения
пары электронов в ходе реакции из
положения, указанного хвостом стрелки, в
положение, указываемое головой
(концом) стрелки.
*-+ Обозначение, используемое для
соотношения между двумя или большим
числом структур, являющихся
«резонансными» или «каноническими» формами,
которые вносят вклад в стабилизацию
молекулы, но не существуют в
индивидуальном виде.
Ковалентная связь.
* В этой книге почти всегда используется
для обозначения асимметрического
атома, например атома углерода с четырьмя
различными заместителями. Этот знак
использован также для'указания изотопа
14С в ароматическом кольце. Во всех
остальных случаях изотопы обозначаются
обычным образом: D, Т, 14С, 180 и т. д.
Моим родителям
Глава 1
ЧТО ТАКОЕ МЕХАНИЗМ РЕАКЦИИ
И ЗАЧЕМ ЕГО НАДО ИЗУЧАТЬ
Что такое механизм реакций?
Молекулы состоят из атомов, занимающих
определенные положения относительно друг друга. Это
расположение атомов является, как правило, положением
стабильного равновесия: небольшие смещения
любого из атомов приводят к появлению сил,
возвращающих их в равновесные положения. Спектральные
и диффракционные методы часто позволяют
определять расстояния между атомами в молекулах (длины
связей) с точностью до ±1 нм, а значения углов
между связями — в пределах ±0,5°
Если смещения одного или нескольких атомов в
одной или нескольких молекулах сравнительно
велики, исходная структура молекул может не
сохраниться: вместо этого атомы займут новые стабильные
конфигурации в новых молекулах, т. е. произойдет
химическая реакция. Для осуществления достаточно
больших смещений и, следовательно, химической
реакции необходима энергия, превышающая среднюю
энергию, которой обладают молекулы при данной
температуре. При любой определенной температуре
столкновения между молекулами будут приводить к
молекулам с различной энергией. При этом некоторые
молекулы могут приобретать энергию, достаточную
для реакции. Чем выше температура, тем выше
средняя энергия молекул и тем большее число молекул
будет иметь энергию, большую критической,
необходимую для протекания реакции. Молекулы
могут приобретать энергию и другими путями,
наиболее важный источник энергий — видимый или
13
ультрафиолетовый свет, поглощение которого
приводит к возбуждению электронных уровней молекулы.
Для того чтобы полностью понять механизм
органической реакции, необходимо знать точное
положение атомов в реагирующих молекулах как функцию
от времени в процессе их превращения в продукты
реакции через любые возможные интермедиаты. Это —
цель, которая никогда не может быть достигнута в
полном объеме. Спектральные и диффракционные
методы, с помощью которых определяется химическая
структура молекулы, мало приспособлены для
наблюдения за изменениями структуры, которые
происходят в течение химических реакций за время 10"13 —
Ю-14 с, так как это время сравнимо со временем,
необходимым для молекулярного колебания или
столкновения молекул. Следовательно, наше знание
механизма реакций должно основываться на
косвенных данных.
Можно получить данные, которые опровергнут тот
или иной предлагаемый механизм реакции, однако
редко имеющиеся данные полностью доказывают
правильность предложенного механизма. Учитывая это,
механизм реакции считается установленным, когда
широкий ряд данных свидетельствует в его пользу.
Однако с течением времени могут появиться новые
факты, которые заставят видоизменить или отбросить
этот механизм.
Зачем изучают механизмы реакций?
Использование химической .реакции не зависит от
нашего понимания ее природы. Многие химические
процессы, такие как горение, выделение металлов из
руд, производство мыла, осуществлялись задолго до
возникновения концепции механизма реакций или
даже самой химии. Тем не менее существуют четыре
основные причины, требующие изучения механизмов
реакций.
(1) Оптимизация
Для получения наиболее высоких выходов продуктов
органических реакций часто (если это вообще
делается) пользуются так называемым методом проб и
14
ошибок. Однако знание механизма и скоростей
индивидуальных реакций в ряде случ-аев может помочь
выявить лучшие условия для оптимизации выхода
определенного продукта.
Например, алкены-1 могут присоединить
бромистый водород в результате двух различных процессов
с образованием 1-бром- или 2-бромалканов. Многое
известно об этих процессах; один из них протекает
через образование свободных радикалов, другой
включает промежуточное образование ионных частиц.
На основании этих сведений можно указать условия,
в которых будут почти количественно образовываться
каждый из двух возможных продуктов.
R—СН=СН2
+ НВг —
очень чистый олефин,
* R—СНВг—СНз (1)
отсутствие пероксидных '
соединений
следы пероксидных
-> R—СН2—СН2~Вг (2)
соединений или другого
источника радикалов
Детально эти реакции рассматриваются в гл. 4.
В качестве примера оптимизации промышленного
процесса на основании знания механизма реакции
можно сослаться на опыт в области хлорирования
углеводородов. Большие суммы, которые были
затрачены на изучение кинетики этой реакции (10 тыс.
фунтов стерлингов), привели в конечном счете к
уменьшению образования побочных продуктов и
увеличению производительности на 40%. Увеличенная
мощность позволила избежать капитальных вложений
в сумме 400 тыс. фунтов стерлингов, необходимых на
строительство нового завода.
(2) Систематизация
Исследование механизмов органических реакций
нскрывает сходство между различными реакциями.
Например, первичные и вторичные алкилгалогениды
реагируют с гидроксильными ионами с образованием
спирта и галогенид-ионов. Многочисленные факты,
описанные в этой книге, наводят на мысль, что в
определенных условиях эта реакция бимолекулярна
15
(т. е. в реакции участвуют — одна молекула алкилгало-
генида и один гидроксил-ион) и включает
одновременное образование связи С—ОН и разрыв связи С—Hal.
Н Н Н
+ **А-в" '
НО" + °С— Hal —► НО-С Hal —> НО—С + Hal (3)
/V /V - /V
R R' R R' R R'
Структурной особенностью гидроксильного иона,
которая позволяет ему реагировать таким образом,
является наличие нёподеленной пары валентных
электронов, которые могут использоваться для
образования ковалентной связи (реагенты такого типа
называются нуклеофилами). Многие нуклеофилы
отрицательно заряжены, однако ряд нейтральных
молекул, таких как Н20 и NH3, также имеют нуклеофиль-
ный характер. Установлено, что в реакциях
замещения галогенов аналогично ОН~ ведут себя другие
нуклеофилы, такие как HS~, NH3, CN~. Более того,
в таких реакциях могут замещаться не только атомы
галогенов, но и другие группы. Многие реакции алкил-
галогенидов могут быть разумно объяснены с позиций
бимолекулярного нуклеофильного замещения
(обозначаемого обычно SN2). Это помогает нам
классифицировать реакции алкилгалогенидов, а также
предположить некоторые новые реайции и понять
невозможность других.
Известно ограниченное число типов органических
реакций и многие тысячи самих реакций.
Большинство из них можно сгруппировать по этим типам,
внося таким образом порядок в данную область.
(3) Предсказание
Знание механизмов органических реакций делает
возможным его использование для предсказаний. Если
известен тип реакции, ее механизм, то на основании
данных, полученных из других реакций, может быть
предсказано качественно (ускорение или замедление)
или количественно влияние изменений условий
реакции (например, заместителей, растворителя или
температуры и т. д.) на ее протекание.
16
Такой подход оказался особенно успешным в
случае реакций ароматических соединений; некоторые
примеры рассмотрены в гл. 6. Известно, что
присоединение бромистого водорода и других молекул к оле-
финам приводит к насыщенным соединениям.
Выяснение механизма такого присоединения позволило
предложить экспериментальные условия для
осуществления более сложных процессов присоединения,
таких как присоединение уксусной кислоты коктену-1:
(СН3)зС-0О-С(СН3)з
СНз-СООН + СаН13-СН=СНг ^^ *
(30 моль) (1 моль)
—> СбН13(СН2)3СООН (4)
(69% из расчета
на олефии)
Интересный пример предсказания на основе
знания механизмов реакций дает область разработки
ракетных двигателей. Развиваемая при определенных
условиях тяга может быть оценена, исходя из
теплоты, выделяемой в процессе горения. Например:
2Н2 + 02 —> 2Н20
СН4 + 202 —> С02 + 2Н20
Однако эти расчеты дают завышенное значение
тяги. Лучшие значения были получены при
рассмотрении отдельных стадий процесса горения, т. е. на
основании знания механизма реакций. Расчеты,
проведенные с учетом скорости этих отдельных стадий,
показывают, что в реальных условиях не происходит
полного сгорания и, таким образом, развиваемая тяга
меньше, чем дает термодинамическая оценка,
основанная на постулировании полного сгорания.
Могут быть предсказаны и совершенно новые
реакции. Наши представления о реакциях S#2-THna
(см. предыдущий раздел), основанные на большом
числе примеров, позволяют предсказать с
определенной степенью вероятности новые реакции этого типа.
Трудно оценить относительное значение
предсказания (на основе уже известных сведений) в сравне-
нии со случайным наблюдением в химическом
исследовании. Безусловно, оба направления играют
17
важную роль, однако в публикуемых статьях
существует тенденция переоценивать элемент логического
предвидения.
(4) Любознательность
Постадийное выяснение химических реакций на
молекулярном уровне, в основном на основе косвенных
данных, дает значительное интеллектуальное
удовлетворение. Многие химики рассматривают эту
проблему как сущность предмета химии и считают,
что она заслуживает особенно интенсивного изучения
вне зависимости от практического значения
исследования.
Каким условиям должен удовлетворять
предлагаемый механизм реакций?
Как я уже отмечал, не существует абсолютного
доказательства истинности механизма реакции. Недавно
найдено, что казалось бы твердо установленный
механизм димеризации трифенилметильных радикалов
оказался ошибочным (смотри следующую главу). Можно
привести еще много менее эффектных примеров, когда
пришлось отказаться от принятых механизмов
реакций или сильно их модифицировать. Во всех этих
случаях новые представления о механизме явились
следствием новых данных.
Что позволяет нам принять или отвергнуть
механизм реакции? Основные принципы следующие.
(1) Предлагаемый механизм должен быть
по возможности простым, но в то же время
объясняющим экспериментальные факты
Реакция гидроксильных ионов с первичными алкилга-
логенидами имеет кинетически первый порядок по
каждому из реагентов. Это заставляет нас прежде
всего предположить, что реакция бимолекулярна и
что гидроксильный ион непосредственно реагирует а
молекулой алкилгалогенида, в результате чего
происходит замещение и образуется спирт. Это самое
простое объяснение, а альтернативные, более сложные
IS
механизмы следует рассматривать только в том
случае, когда простейший механизм не может объяснить
все известные факты. В качестве примера,
показывающего необходимость привлечения более сложного
представления о механизме, можно привести реакцию
иодбензола с амидом калия в жидком аммиаке,
продуктом которой является анилин. Простейшим
механизмом этой реакции было бы прямое замещение:
Нч NH2
Однако эксперимент с использованием 14С-мечен-
ного соединения показывает, что такое простое
описание процесса неверно, так как замещающая
группа NH2 входит в молекулу как в положение,
меченное 14С, так и в соседнее положение с
соотношением 1 : 1 (с точностью эксперимента). Для
объяснения этого результата был предложен двустадийный
механизм, включающий промежуточное образование
дегидробензола 2.
1 2 47% 53%
Из сказанного следует, что реакция может быть
простой или элементарной, т, е. проходить в одну
стадию без образования интермедиатов, или сложной,
состоящей из нескольких элементарных реакций.
(2) Предполагаемый механизм должен
по возможности указывать пути его
экспериментальной проверки
В приведенном выше примере постулирование
образования промежуточного реакционноспособного
дегидробензола 2 предполагает, что он может быть
19
обнаружен либо спектроскопически, либо в результате
взаимодействия с различными соединениями. В гл. 2—
6 описаны другие методы проверки предлагаемого
механизма реакций.
(3) Индивидуальные стадии (элементарные реакции)
предлагаемого механизма должны быть либо моно-у
либо бимолекулярны
Мономолекулярные реакции представляют собой
результат реорганизации связей внутри молекулы,
проходящей с разрывом молекулы на части или без разрыва.
Требуемая для этих процессов энергия возникает при
столкновении молекул друг с другом (термическая
реакция) или в результате захвата фотона
(фотохимическая реакция). Следующие реакции служат
примерами термических мономолекулярных
элементарных реакций:
(6)
СН3—СОО—СН2СН3 —> СН3—СООН + С2Н4 (7)
•*
т-рег-BuCl —> трет'Ви+ + СГ (8)
PhN£ —> Ph+ + N2 (9)
3 4
СНз
I
СНз—С—СН2+ —> СНз—С—СН2СН3 (10)
I I
СНз СНз
5 6
Из этих реакций только реакции (6) и (7)
полностью представлены написанными уравнениями.
Реакция (8) сопровождается дальнейшими
превращениями трет-бутилкатиона. В реакциях (9) и (10)
необходимо предварительное образование катионов 3
и 5 и последующее связывание катионов 4 и 6.
20
Бимолекулярные реакции протекают при
столкновении двух молекул (одинаковых или различных).
При этом между атомами обеих молекул образуется
по крайней мере одна новая ковалентная связь; кроме
того, могут разрываться или образовываться другие
связи.
2CF2=CF2
О
II
СО
II
О
СНз • + С2Н5.
НО" + СН3С1
Щ + свн,
—
—
—»•
->
—>
CF2—CFj
1 1
CF2—CF2
4
О
СзНа
СН3ОН + С1"
C,H5-N02 + H+
(П)
(12)
(13)
(И)
(15)
Тримолекулярные реакции должны включать
столкновения трех молекул (с ориентацией,
необходимой для взаимодействия). Такие события, однако,
маловероятны и фактически возможность тримолеку-
лярных реакций ничтожна (известно несколько
примеров газофазных неорганических реакций,
которые, возможно, являются тримолекулярными).
Следовательно, предлагаемые механизмы, в которых для
образования наблюдаемых продуктов предполагается
взаимодействие трех или более реагентов в одной
стадии, должны быть разбиты на стадии,
включающие не более чем две реагирующие частицы.
(4) Предлагаемый механизм не должен нарушать
принципа микроскопической обратимости
Этот принцип устанавливает, что в случае
элементарной реакции обратная ей реакция будет проходить
тем же путем, но в обратном направлении. Интуитивно
21
этот принцип кажется разумным, но он может быть
и подтвержден фактами, основанными на
временной обратимости. Значение принципа
микроскопической обратимости в изучении механизмов
заключается в том, что любые данные о механизме
элементарной реакции могут быть применены к ее обратной
реакции. Так как реакции, молекулярность которых
выше трех, не встречаются, этот принцип приводит к
важному качественному заключению, что реакции, в
которых на индивидуальной стадии образуется более
трех частиц, являются соответственно невероятными.
Например, тетраэтилсвинец разлагается при
нагревании с образованием свинца и этильных радикалов.
Однако механизм, вероятно, не должен включать в
качестве элементарного процесса реакцию (16), так как
обратная реакция в таком случае должна быть пен-
тамолекулярной.
PbEt4 —> Pb + 4Et. <16)
Следует отметить, что практически необратимые
реакции, которые приводят к образованию трех
частиц (молекул), довольно обычны для органической
химии, хотя реакции с надежно установленным
механизмом, включающие взаимодействие трех молекул,
редки.
В гл. 3 рассматриваются условия, при которых
этот принцип может быть использован для сложных
реакций.
(5) Индивидуальные реакции должны
согласовываться с существующими химическими
представлениями
Интермедиаты, постулируемые механизмом реакции,
не должны нарушать правила валентности, в
частности элементы первого ряда большого периода
Периодической системы для образования связей могут
использовать только 2s- и 2р-орбитали. Перемещение
атомов, вовлеченных в реакцию, должно происходить
возможными путями. Присоединение брома к двойной
связи олефинов имеет транс-характер. Это наблюде-
22
ние фактически исключает согласованный четырех-
цснтровый молекулярный механизм (17а), в котором
обе связи углерод—бром образуются одновременно,
так как такой процесс должен приводить к цис-при-
соединению. граяс-Присоединение (176) требует,
чтобы атомы брома подходили к молекуле олефина с
противоположных сторон (как в 7). Такой характер
приближения, естественно, несовместим с атакой не-
диссоциированной молекулы брома. В соответствии с
этим следует рассмотреть альтернативные механизмы
этой реакции.
н.
.вг
ноос
Вг ^с^
к?
н
С^ Вг
ноос н
7
,соон
Вг
Н;
Bl
I
ноос н
,соон
н
•соон
(17а)
„соон
Вг
А^А
(176)
ноос^ хн
наблюдаемый
продукт реакции
фумаровой кислоты
Для катализируемой основаниями конденсации
бензальдегида с ацетофеноном было первоначально
предложено два «механизма» (18) и (19) образования
спирта 10 (который при этих условиях реагирует и
дальше, давая ненасыщенный арилсодержащий кетон
PhCOCH = CHPh.
PhCOCH2H + "OEt
PhCOCH; + рьсно
PhCOCH; + ЕЮН
8
(18)
O"
PhCOCH2-CHPh
9
OH
EtOH
+ PhCOCH2—CHPh
10
23
О"
I
Ph—CHO + ~OEt «=* Ph—CH—OEt
и
0"
I
PhCO—CHa-CHPh
I I
H OEt
12
l
O" OH j
I EtOH I
EtOH+PhCOCHa—CHPh * PhCOCH2— CHPh
9 10 J
В первом механизме (18) этилат-анион отщепляет
протон от молекулы ацетофенона с образованием
аниона 8, который, в свою очередь, реагирует с бен-
зальдегидом по углеродному атому карбонильной
группы, приводя к 9. Наконец, последний отрывает
протон от молекулы этанола, завершая реакцию и
регенерируя этилат-анион. Все эти процессы можно
мысленно представить, учитывая движения атомов,
образование к разрыв связей в элементарных
реакциях, которые аналогичны известным процессам.
В альтернативной схеме (19) первая стадия также
вполне вероятна. Она заключается в присоединении
этоксильного иона к молекуле бензальдегида с
образованием 11. Однако трудно понять на основании
сведений об известных процессах образования и разрыва
связи, как интермедиат 11 может взаимодействовать с
молекулой ацетофенона с образованием 9. Так как
при этом должна образоваться новая
углерод-углеродная связь, следовало бы предположить переходное
состояние, аналогичное 12. Но такие четырехцентро-
вые переходные состояния, как известно для других
случаев, являются невыгодными и требуют очень
высоких энергий активации. Согласно этому, первый
путь (18), по-видимому, более приемлем в качестве
возможного механизма, в то время как механизм (19)
(которому авторы исследования отдавали
предпочтение) кажется неправдоподобным.
} (19)
24
(6) Индивидуальные реакции должны быть
энергетически приемлемыми
Можно оценить эндотермичность и, следовательно,
минимальную энергию активации процессов, особенно
в случае свободнорадикальных и молекулярных реак*
ций. Эти оценки энергии активации в сочетании с
оценками значений предэкспоненциального множите-»
ля в уравнении Аррениуса (см. гл. 3) могут дать
верхний предел скорости рассматриваемой реакции
при данной температуре. Если наблюдаемая скорость
оказывается значительно выше, чем ее
«теоретическая» величина, то мы получаем надежное
доказательство ложности постулированного механизма и
должны искать другой путь реакции.
Это положение иллюстрируют следующие два при*
мера. Хлорирование алканов молекулярным хлором
протекает чрезвычайно быстрр, и, как. полагают, оно
представляет собой цепной радикальный процесс,
включающий реакции (20) и (21) как стадии
передачи цепи. Величины Д# в уравнениях даны для
R = Me.
С12 —> 2CN
R—H + C1. —V R. + H—C1 АЯ = + 2 кДж• моль-1 (20)
R. + Cl—C1 —► R—C1 + CN ДЯ = -106кДж.моль-1(21)
Альтернативный цепной механизм включает реакции
(22) и (23).
CN + R-H —> CI—R + H. АЯ = +85 кДж-моль-1 (22)
Н. + С1—С1 —> Н—С1 + С1- АЯ = -189кДж-моль-1 (23)
Однако реакция (22) второй схемы высоко эндотер-
мична и требует очень высокой энергии активации
(не меньше, чем величина теплового эффекта
эндотермической реакции: 85 кДж-моль-1). Используя
разумные значения Л-фактора и других переменных
величин уравнения Аррениуса, можно рассчитать,
что реакция (22) будет приводить к образованию хлор-
алкана со скоростью около 10~12 моль • л-1 • с1;
другими словами, время полупревращения для этой
реакции должно равняться приблизительно 3000 лет.
Поскольку смеси алканов с хлором взрываются при
их облучении светом (реакция завершается за доли
секунды), мы должны сделать вывод, что реакции
25
(22) и (23) не могут соответствовать процессу
хлорирования.
Реакция фтора с алканами, составляющая
отдельную проблему фторирования, протекает в темноте
даже при —80 °С (и, по-видимому, также имеет
радикально-цепной механизм).
При этом возникает вопрос: откуда берутся
радикалы? Реакция F2 —► 2F- является
эндотермической (Д# =-[-158 кДж-моль-1), и можно
рассчитать, что при —80 °С продуцирует очень мало
радикалов. Следовательно, первоначальный радикал
должен возникать в другой реакции; наиболее
вероятно, что таким процессом является
бимолекулярная реакция (24), эндотермический эффект которой
составляет только 24 кДж-моль-1.
СНз—H + F—F —> CH3. + H-F + F.
Atf = + 24 кДж • моль-1 (24^
(7) Механизм должен обычно согласовываться с тем,
что известно для аналогичных реакций
Если при определенном наборе условий проведено
тщательное изучение механизма, например
гидролиза я-пропилацетата, естественно предположить, что
я-бутилацетат будет реагировать в тех же
условиях по аналогичному механизму (и приблизительно
с той же скоростью). Действительно, необходимо
иметь веское доказательство (вместе с
рациональным объяснением) для того, чтобы убедить химиков,
что эти реакции различны. Однако грет-бутилъное
соединение может реагировать по иному механизму, чем
я-бутильное соединение, и это не вызовет удивления,
так как имеется много аналогий в других известных
реакциях с грет-бутил-, я-бутил- и я-пропилпроизвод-
ными. В более общем смысле следует с подозрением
относиться к механизмам реакции, которые нарушают
хорошо установленные принципы органической химии,
такие как правило Бредта (в мостиковых системах с
небольшой величиной кольца двойная связь в голове
моста не образуется). Однако рабски доверять
аналогиям опасно, так как истинно новые типы
соединений и реакции открываются случайно. Так, открытое
в 1900 г, Гомбергом образование трифенилметильного
26
радикала при взаимодействии трифенилметилхлорида
с металлами не получило немедленного общего
признания, так как в то время углерод в органических
соединениях считали однозначно четырехвалентным.
Установление механизма органических реакций
Могут использоваться различные типы доказательств
для выяснения механизма органической реакции;
именно они и составляют содержание остальных глав
этой книги. Нет необходимости применять для
каждой реакции все методы. Кроме того, может меняться
последовательность исследования. Гипотеза о
механизме реакции может возникнуть на различных
этапах. Иногда она возникает перед первоначальным
экспериментом, иногда сразу после него и перед
дальнейшей серией экспериментов. Однако довольно часто
новый факт может заставить пересмотреть механизм и
предложить новые пути его проверки.
Задачи
(При решении этих задач, а также задач к другим главам см.
указания на с. 181.)
1.1. Прокомментируйте схемы следующих реакций на основе
соображений, высказанных на с. 18—27. (Каждый из процессов,
обозначенных стрелкой (не считая переносов на другую строку),
следует рассматривать как возможную элементарную реакцию.)
Ni(CN)2 |, „ , ч
►■ (а)
нс=сн
н н
с с
III III
с с
н н
нс=сн
(СН3)4РЬ —> РЬ + 4СН3- (б)
н н
I I .
С4Н9СН2СН2СН2СНО. —> С4Н9СН2СН2СНСНОН —>
н
I .
с4н9сн2снснсн2он —> с4н9снсн2сн2сн2он —*
Продукты реакции (почти исключительно из последнего
радикала) (в)
27
+l/°
о
H—Si(CH3)3
НаС—feC—Н
Нз(Х
4Si(CH3)3
(г)
(Д)
(в CH3COOD)
CH3COOD
H+ + L *
Г 1+СНзСОО"
■f нагревание •
(CH3)3NCH2CH2—Н * (CH3)3NCH2CH2-H
+ он~ + *он
—> (CH3)3N + СН2 = СН2 + Н20
(е)
(ж)
Глава 2
ПРОДУКТЫ РЕАКЦИИ
Определение строения и выхода продуктов
Как это ни кажется банальным, но первым этапом
исследования механизма реакции должно быть
качественное установление продуктов реакции и
определение их выходов. Без определения выхода нельзя
быть уверенным в том, что данное соединение
является единственным или даже основным продуктом
реакции. К тому же идентификация побочных
продуктов может явиться ключом к установлению природы
основной реакции.
В ряде случаев проводили изучение механизмов
реакций, продукты которых, как выяснилось
впоследствии, были определены неверно. Трифенилметиль-
ный радикал, полученный в результате
взаимодействия трифенилметилхлорида с цинком или другими
металлами, находится в растворе в равновесии с диме-
ром. Постепенно стало общепринятым (смотри почти
любой учебник по физической органической химии,
опубликованный до 1968 г.), что димером является гек-
сафенилэтан и, основываясь на этом представлении,
было выполнено большое число определений
константы равновесия и эффектов заместителей. Однако
некоторые из результатов, например эффект мета- и пара-
метильных групп, выглядели неожиданными (табл. 1).
Замена одной фенильной группы в дифенилтолильном
радикале на толильную мало сказывается на степени
диссоциации димера, но замена третьей фенильной
группы на толильную сказывается значительно.
Использование представлений о резонансных эффектах
метильной группы не может объяснить подобный
29
скачок, а также не может объяснить более резко
выраженного эффекта жега-замещения по сравнению с
пара-замещением.
Таблица 1. Кажущаяся степень диссоциации некоторых
метилзамещенных «гексафенилэтана»
0,1 М растворы в бензоле (То1=СНзСвН4)
мета-То\
napa-Tol
Степень диссоциации, %
для
[Ph2(ToI)C-l2
6,5
5
для
[Ph(Tol)2C-]2
для
[Tol3c-12
7
5,5
40
16
В 1968 г. было убедительно показано, что димер
трифенилметила имеет строение, описываемое
формулой 14, но не 13. В спектре ПМР гексафенилэтана
следовало ожидать наличие только ароматических
протонов, тогда как в случае 14 должны проявляться,
кроме того, олефиновые протоны, что и наблюдалось.
Таким образом, димеризация включает в себя атаку
а-углеродного атома одного трифенилметильного
радикала в /гара-положение другого трифенилметильного
радикала.
Ph3C-Cl^ Ph3C-^X=^ Ph3C-CPh3
*4Л PbscXXph «»
* на 14
Из новой формулы димера 14 следует, что мета-
(X в 14а) и пара- (Y в 14а) заместители оказывают
стерические препятствия димеризации триарилме-
тильного радикала. Однако при введении метильной
группы в одно или два бензольных кольца триарил-
метильного радикала атака в /гара-положение
незамещенной фенильной группы при димеризации не
встречает пространственных препятствий. При димериза-
30
ции тритолилметильного радикала это препятствие
возникает.
Другим примером использования ошибочных
данных по строению продуктов реакции является работа
Терпе, Ингольда и др., выполненная в 1920 г. Авторы
выяснили условия, благоприятствующие замыканию
циклопропанового кольца, и предположили, что
объемные заместители у р-углеродного атома в
соединениях, содержащих фрагмент 15, будут
отталкиваться друг от друга, увеличивая тем самым угол
между связями X—С—X и уменьшая угол С—С—С
(0), что приводит к более легкому образованию
циклопропанового кольца.
хч ,0* X С
15
Одной из изученных ими реакций была реакция
замыкания кольца (27), которая постулировалась как
обратимый катализируемый сильным основанием
процесс.
R4 /COCOOH otr r
r/ \:н2соон r/\
1вГ 17
он
соон
—соон
(27)
Результаты экспериментов с использованием
различных заместителей подтвердили приведенную выше
гипотезу. В случае R = Me, равновесие почти
исключительно сдвинуто влево, однако когда две группы R
были заменены на спироциклогексильную группу,
существование циклопропанового соединения 18 в
равновесии оказалось предпочтительным по сравнению с
формой с открытой цепью. В то время полагали, что
циклогексановое кольцо является плоским с углом
между связями 120°, который по этой реакции
должен вызывать? сокращение угла 0 фрагмента
31
С—С—С. При R = Et — случай, промежуточный
между циклогексилиденовым и диметильным
соединениями,— устанавливалось равновесие, в котором
соединение с открытой цепью 16 и производное цикло
пропана 17 присутствовали в соотношении 38 62.
о
ОН
J—соон
\'—соон
18
Ek.Et
НООСч X /Н
19 20
ноос ^ ^соон w yf ncooh
В 1959 г. Виберг и Холмквист провели новое
исследование соединений 16 и 17 (R=Et), используя
методы ИК- и ЯМР-спектроскопии, и обнаружили,
что структуры 16 и 17 были неверными. Соединение,
формулированное как 17, не содержит гидроксильной
группы (по данным ИК-спектроскопии), а 16 не
является кетоном (отрицательная реакция с 2,4-ди-
нитрофенилгидразином). По данным ЯМР, истинное
строение этих соединений соответствует формулам
19 и 20. Следует напомнить, что в 1920 г.
инфракрасная спектроскопия еще не была введена в
практику и не было установлено, что 2г4-динитрофенилгид-
разин является реагентом для определения кетонов.
Уже первые исследователи перегруппировки окси-
мов в амиды (перегруппировка Бекмана) столкнулись
С проблемами стереохимии. Несимметричные кетоны
RR'CO образуют два геометрически изомерных окси-
ма RR'C=NOH 21 и 22, один из которых при
действии пятихлористого фосфора или серной кислоты
перегруппировывается в амид RCONHR', а другой —
в R'CONHR. Однако как определить геометрию исход-
ного оксима? Ганч и Вернер предположили, что
мигрировать должна ближайшая к гидроксйлу группа, и
32
на этом основании сделали отнесение структур
изомерных оксимов. Однако они ошиблись.
R\ /ОН
>-\
он
21
22
R'
\ /0Н
r/
C=N-
.С—N(
r/
Еще до проведения рентгеноструктурного анализа
строение оксимов было установлено химическими
методами. Несмотря на сложность, Мейзенхеймеру и
другим удалось получить ряд данных в пользу гране-
миграции группы R в перегруппировке Бекмана. В
частности, могут быть получены оксимы 2-бром-5-нитр-
ацетофенона. Однако только один из них легко
превращается в гетероциклическое соединение 24 за счет
замыкания цикла. На этом основании ему
естественно приписать структуру 23:
OoN
02N
^_C^N/OH (зо) >
kA
Br
25
02N
26
He циклизующийся изомер 25 дает в результате
перегруппировки Бекмана амид 26. Следовательно,
перегруппировка 25 включает граяс-миграцию фе-
нильной группы. Этот вывод был подтвержден
данными ряда других работ,
2 Зак, 1044
33
Какие связи образуются и разрываются
в ходе реакции?
Вслед за установлением строения исходных
соединений и продуктов необходимо выяснить, какие связи
образуются и какие разрываются в процессе реакции.
Это часто кажется очевидным, однако можно
привести много примеров, когда очевидный ответ
является ошибочным. Так, в случае нуклеофильного
замещения можно предположить, что в результате
замещения уходящей группы вступающий нуклеофил
присоединяется к тому же атому углерода, с которым
первоначально была связана уходящая группа.
НО'^+СН^Нг-О^! —► СН3СН2~ОН + СГ (31)
но не
Н
НО^ОТ^СН2-Ол —►•НО-СН2СН2-Н + СГ (зг)
Это предположение, по-видимому, является
оправданным для большинства простых случаев реакции
подобного типа, однако в более сложных случаях,
которые на первый взгляд кажутся аналогичными, это
предположение оказывается неверным. Например, на
основе опытов с меченным 14С иодбензолом считают,
что реакция ароматических галогенпроизводных с
амидом калия в жидком аммиаке является двух-
стадийным процессом (33), включающим
промежуточное образование дегидробензола 27 (см. также
гл. 1, с. 19).
27 47% 53%
Применение меченых соединений для определения
того, какие связи рвутся и какие образуются в
химической реакции, является наилучшим. Результаты,
которые получают при использовании этого метода,
обычно оказываются достаточно определенными и часто
84
дают информацию, которую нельзя получить другими
методами. Однако он дорог и требует затраты
большого времени. Например, при гидролизе сложного
эфира возникает вопрос: атом кислорода из воды
оказывается в кислоте или спирте? Другими словами,
происходит ли при гидролизе сложных эфиров
разрыв связи алкил — кислород [реакция (34)] или связи
ацил — кислород [реакция (35)]?
,8Q-fc2H5 он
НоО _ /
/
С2Н5-С^ —2_> С2Н5-С 4 СгНзОН (34)
О Ъ
26
£о~-с2н5 он
С2Н5-С^ -^^> QjHg-c/ +C2H518OH (35)
о чо
28
Решение этого вопроса было получено при
использовании меченного по кислороду (0,7% обогащения
180) этилпропионата 28. Если протекает
расщепление связи алкил—кислород [реакция (34)], то весь
180 должен оставаться в кислоте, в то время как при
разрыве связи ацил—кислород [реакция (35)] метка
180 целиком окажется в молекуле спирта. В
действительности 180 полностью оказывается в спирте,
демонстрируя тем самым, что в выбранных
экспериментальных условиях происходит разрыв^ связи ацил —
кислород [реакция (35)].
Перемещались ли группы или атомы от молекулы
к молекуле в процессе реакции?
Многие реакции включают миграцию атомов или
групп от одной части мЬлекулы к другой. Становится
ли в ходе реакции мигрирующая группа в
действительности свободной или на всех стадиях процесса
остается связанной с исходной молекулой? Например,
2*
35
аллилфениловый эфир 29 перегруппировывается при
нагревании в фенол 30.
О
/
СНгСН^СНСгНб
НзС\^'ч/СН£
нагревание
KJ
29
ОН
XX
(36)
СН2СН=СНС2Нб
30
Становится ли группа — СН2СН = СНСгН5,
свободной от остальной части молекулы в процессе
реакции? Ответ на этот вопрос можно получить из
«перекрестного» («кросс») эксперимента, для
проведения которого синтезируют молекулу, имеющую
небольшие различия в строении как «мигрирующей»,
так и «стационарной» частей. Такой молекулой может
быть соединение 31.
На С
о
/
СН2СН=СНСН3
XX
СООСНз
ОН
Н3(Х X ^СООСНз
31
(3?)
СН2СН=СНСНз
32
Установив, что новое соединение 31
перегруппировывается в 32 при тех же условиях и с
приблизительно той же скоростью, как и в случае эфира 29,
осуществляют перегруппировку смеси двух эфиров, 29 и 31,
36
и анализируют образующуюся смесь продуктов на
содержание в ней перекрестных продуктов 33 или 34.
НзС^
ОН
|
if'V^"3
V
СН2СН=СНСНз
33
НзСч^
он
1
1/СООСН,
и
1
1
СН2СН=СНС2Н5
34
В рассматриваемом примере не было обнаружено
даже следов 33 или 34. Поэтому можно с уверенностью
сделать вывод, что мигрирующая группа не является
свободной от остальной части молекулы. Реакции
такого типа, которые протекают «внутри» молекулы,
называются внутримолекулярными в противоположность
межмолекулярным реакциям, в которых группы
переходят от одной молекулы к другой.
Литиевые соли бензиловых эфиров типа 35 быстро
перегруппировываются в инертных растворителях с
образованием алкоголятов 36. Является ли эта
реакция меж- или внутримолекулярной? Для ответа на
этот вопрос нагревали смесь двух литиевых солей 39
и 40, которые перегруппировываются с близкими
скоростями. Анализ продуктов реакции показал, что хотя
реакция преимущественно внутримолекулярна, на
~7% она протекает за счет перехода групп между
молекулами. Этот результат можно рассматривать
как доказательство образования в ходе реакции
«тесной» ионной пары типа 37. При этом два иона
удерживаются вместе в клетке молекул растворителя и
реагируют друг с другом с образованием продукта
внутримолекулярной реакции. Небольшой процент
ионов может диффундировать из клетки и реагиро*
вать межмолекулярно. Механизм, включающий
образование радикальной пары вместо ионной, также
будет согласовываться с этими результатами. Вместе с
тем приведенные факты не согласуются ни с полной
диссоциацией эфира на ионы или на радикалы, ни
С прямой внутримолекулярной перегруппировкой,
37
в которой новая углерод-углеродная связь образуется,
когда разрывается связь углерод—кислород.
Ph-CH-O
to
35
ИЛИ
Ph-CH=0-Li
37
["Ph-CH-O-Lil
38
Ph-CH-O-Li (38)
R
36
n-DC6H4CH(Li)-0-CHMeEt C6H5CH(U)-0-CHMe,
39
.40
Перекрестные эксперименты позволяют сделать
определенные выводы только тогда, когда скорости
реакций двух соединений примерно равны. Если же,
например, одно соединение перегруппировывается в
сто раз быстрее, чем другое, тогда его
перегруппировка фактически должна закончиться прежде, чем
начнется реакция другого, и следовательно, даже если
реакция была межмолекулярной, образование
перекрестных продуктов в заметных количествах не
может произойти.
Близость скоростей перегруппировки можно весьма
легко достигнуть при использований изотопной метки,
однако меченые соединения типа, иллюстрированного
реакциями (36) и (37), получить легче (и дешевле),
и часто их анализ является более простой задачей.
Обмен с растворителем
В реакциях, включающих перемещение водорода от
одной молекулы к другой, возможен обмен этого
водорода с атомами водорода растворителя. При
выдвижении механизма реакции необходимо учитывать,
происходит ли такой обмен на самом деле или нет.
В качестве примера рассмотрим две реакции,
протекающие в воде в присутствии оснований:
автоокисление-восстановление формальдегида [реакция 39)]
38
и рацемизацию втор-бутилфенилкетона [реакций
(41)]. При проведении первой из этих реакций в
D2O, в метильной группе метанола дейтерий не
обнаруживается. Из этого следует, что образование
метанола из формальдегида осуществляется за счет атома
водорода другой молекулы формальдегида. Исходя из
этого, можно не рассматривать механизм (40),
включающий гидридный переход водорода гидрок-
сильной группы, а постулировать механизм (39), в
котором одна молекула формальдегида
непосредственно атакует другую молекулу с миграцией
водорода, связанного с углеродным атомом."
он- НЧ/Н но н
2 * p^VrCHs-O —^ с + СНа-СГ (39)
"О Н cfk
он:
Нч РТН . НОН
2 * /\ СН2Ж° ► С + СН3-0~ (40)
-О Н
но
С другой стороны, рацемизация оптически
активного вгор-бутилфенилкетона 41 в водном диоксане в
присутствии основания сопровождается изотопным
обменом водорода, что было показано с
использованием D2O. В действительности скорость рацемизации
равна скорости внедрения дейтерия, что
подтверждает механизм, представленный уравнением (41).
* OD~ - D20
PhCOCHEt >- PhCOCEt ► PhCOCDEt (41)
I I I
Me Me Me
41
оптически рацемат
активный
Доказательства, основанные на образовании
побочных продуктов
В результате реакций редко получается
стопроцентный выход одного продукта; обычно с большим или
меньшим выходом образуются и другие продукты. По-
39
бочные продукты могут образоваться за счет
конкурирующей, но более медленной реакции, однако
часто их образование свидетельствует о том, что
промежуточные частицы могут реагировать по нескольким
направлениям.
При фторировании метана молекулярным фтором
образуется смесь CH3F, CH2F2, CHF3 и CF4.
Возможны два механизма фторирования: молекулярный
процесс (42) или свободно-радикальный (43).
СНз-Н СНз Н CH2F-H CH2F H
! ! —* I + I I !—>1 + I
F F F F F F F F
CHF2—H CHF2 H CF3-H CF3 H
! j —> I + I ! ! —> I + I
F F F F F F F F
Реакция инициирования, приводящая к F»
F2
F. + СШ —> HF + .СНз —► CH3F + F.
F2
F. + CH3F —> HF + »CH2F —► CH2F2 + F.
F2
F • + CH2F2 —► HF + • CHF2 *—> CHF3 + F •
F2
F. + CHF3 —> HF + .CF3 —> CF4 + F.
(42)
} (43)
При реакции в качестве побочного продукта
всегда образуется C2F6. Этот результат естественен
при радикальной схеме реакции: если в реакции
промежуточно образуются радикалы «CF3, то они могут
димеризоваться с образованием СгНв так же как
взаимодействовать с F2 с образованием CF4.
Следовательно, наличие C2F6 среди продуктов является
доказательством в пользу свободно-радикального (43),
а не молекулярного механизма (42) (см. также
с. 26).
Предполагаемый механизм реакции должен
обязательно объяснять образование всех продуктов или
40
отсутствие ожидаемых продуктов реакции. Гидролиз
замещенного хлористого аллила Ме2С=СНСН2С1
действием влажного оксида серебра приводит к смеси
первичного спирта Ме2С=СНСН2ОН (15%) и его
изомера Ме2С(ОН)СН=СН2 (85%).
Ме2С=СН—СН2С1
H20/Ag20
+
■> Ме2С = СН-СН2 ч-
42
Ме2С—СН=СН2
Ме2С—СН=СН2
Ме2С=СН—СН2ОН
- н2о
(44)
Ме2С—СН=СН2
I
ОН
Та же смесь продуктов и почти точно с тем же
соотношением образуется, если проводить гидролиз
Ме2С(С1)СН=СН2. Поскольку эта смесь не
является термодинамически равновесной смесью двух
изомеров (последняя должна содержать около 90%
первичного спирта), наблюдаемые результаты
наводят на мысль, что оба галогенида реагируют с
образованием общего интермедиата, который затем в
результате атаки по двум различным положениям
приводит к образованию смеси изомерных спиртов.
Наиболее вероятным интермедиатом в этой реакции
является мезомерный катион 42. В аналогичных
условиях бромид Ме2С=СНСН2Вг приводит к той же
смеси продуктов, что подтверждает сделанное
заключение.
В реакциях ароматических соединений часто
образуется смесь двух или более изомеров. N-Хлорацетани-
лид при действии НС1 в хлороформе
перегруппировывается с образованием смеси о- и я-хлорацетанилидов
41
(69,8:30,2). При непосредственном хлорировании
ацетанилида в том же растворителе возникает та же
смесь изомеров (с точностью ошибки опыта) (68,8
31,2). Эти данные могут быть объяснены двухстадий-
ной реакцией: на первой стадии происходит
образование хлора, вторая представляет собой обычное
ароматическое замещение ацетанилида. (Этот механизм
можно проверить другими способами: например, с помощью
перекрестных опытов или улавливанием промежуточ'-
но образующегося хлора.)
(45)
В противоположность приведенному, нагревание с
серной кислотой близкого по строению N-нитроанилина
приводит главным образом к о-нитроанилину
(соотношение о-: м-: п- = 93 : 0 : 7). Если бы при реакции
в качестве интермедиатов образовывались свободные
ионы нитрония или другие нитрующие частицы,
следовало ожидать возникновения смеси, состоящей
главным образом из мета- и пара-изомеров (при
нитровании анилина азотной кислотой в аналогичных
условиях наблюдаемое соотношение о-, м- и /г-/гродук-
тов равно 6 34 59). Полученные данные являются
веским доказательством в пользу того, что такие
интермедиа™ в ходе перегруппировки не образуются.
Этот вывод (с некоторыми оговорками) подтвержден
и другими экспериментами.
«
NHNOa
85X H2SO4
HNO3
85% H2SO4
6%
340/o
590/0
(46)
(47)
Резюме
Рассмотренные примеры показывают, какое
важное значение имеет установление строения
продуктов реакции и определение выхода каждого из них.
Наличие побочных и второстепенных продуктов, а
также отсутствие ожидаемых продуктов часто
позволяет предположить или отвергнуть тот или иной
механизм. Далее следует определить, какие связи
разрываются и образуются, является ли реакция
меж- или внутримолекулярной, происходит ли в
реакционной смеси обмен атомов с растворителем.
На этой стадии оказывается возможным (и
полезным) предложить один или несколько
альтернативных механизмов реакции, справедливость
которых может быть установлена методами,
описанными в следующих главах.
Задачи
2.1. Как показать, что простое нуклеофильное замещение
должно действительно протекать по механизму (31), а не (32J
(два различных эксперимента)?
2.2. Можно ли объяснить результаты реакции (5а) любой
из следующих схем?
43
ci.
:NHo
H
&"'
NH<
+ Г
I (протекают
совместно)
(а)
(б)
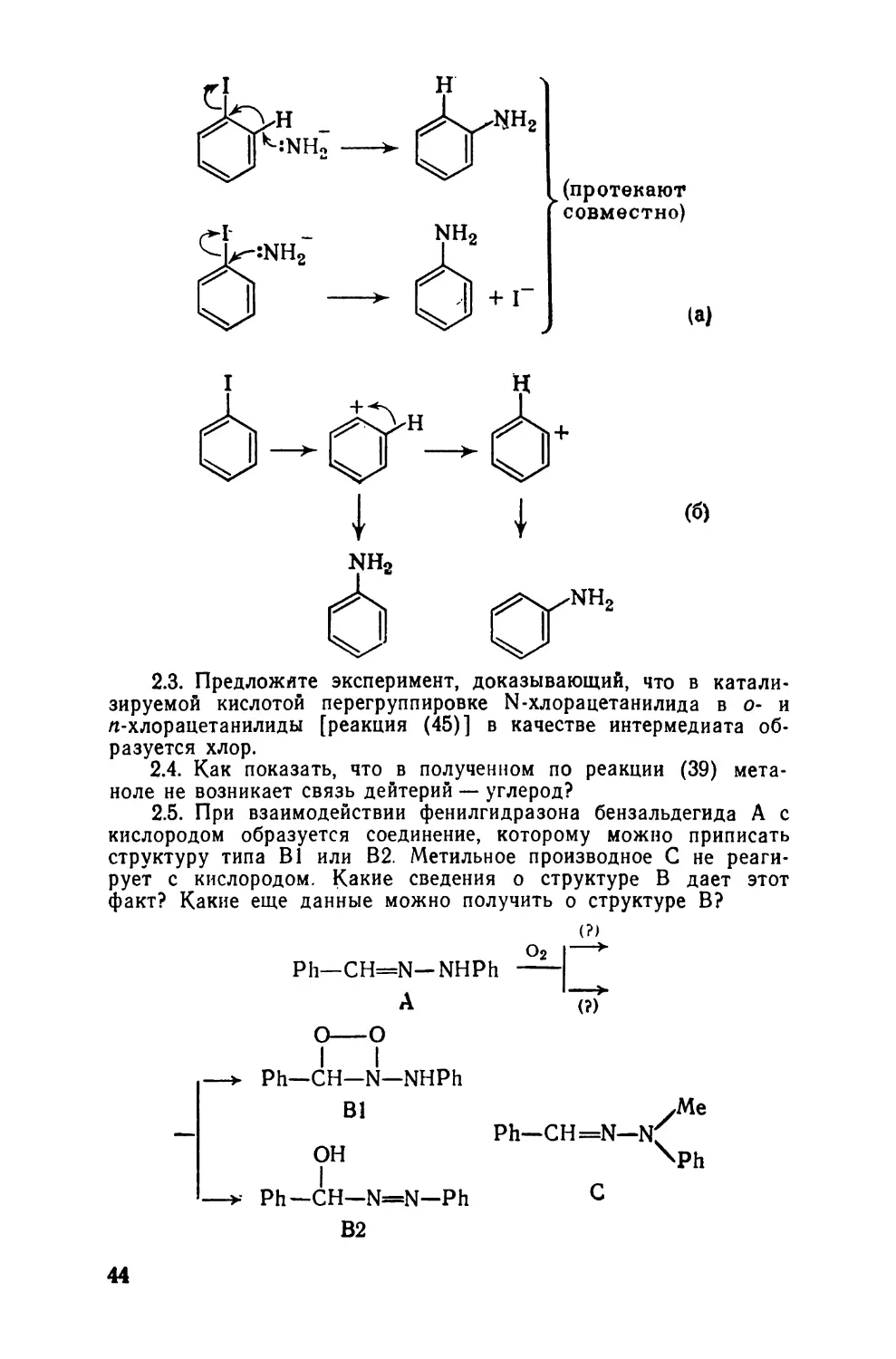
2.3. Предложите эксперимент, доказывающий, что в
катализируемой кислотой перегруппировке N-хлорацетанилида в о- и
л-хлорацетанилиды [реакция (45)] в качестве интермедиата
образуется хлор.
2.4. Как показать, что в полученном по реакции (39)
метаноле не возникает связь дейтерий — углерод?
2.5. При взаимодействии фенилгидразона бензальдегида А с
кислородом образуется соединение, которому можно приписать
структуру типа В1 или В2. Метильное производное С не
реагирует с кислородом. Какие сведения о структуре В дает этот
факт? Какие еще данные можно получить о структуре В?
Ph_CH=N— NHPh
О О
I I
Ph—CH—N—NHPh
о2
(?)
(?)
В1
ОН
I
Ph—CH—N=N-Ph
В2
Ph—CH=N—N;
/
Me
^Ph
44
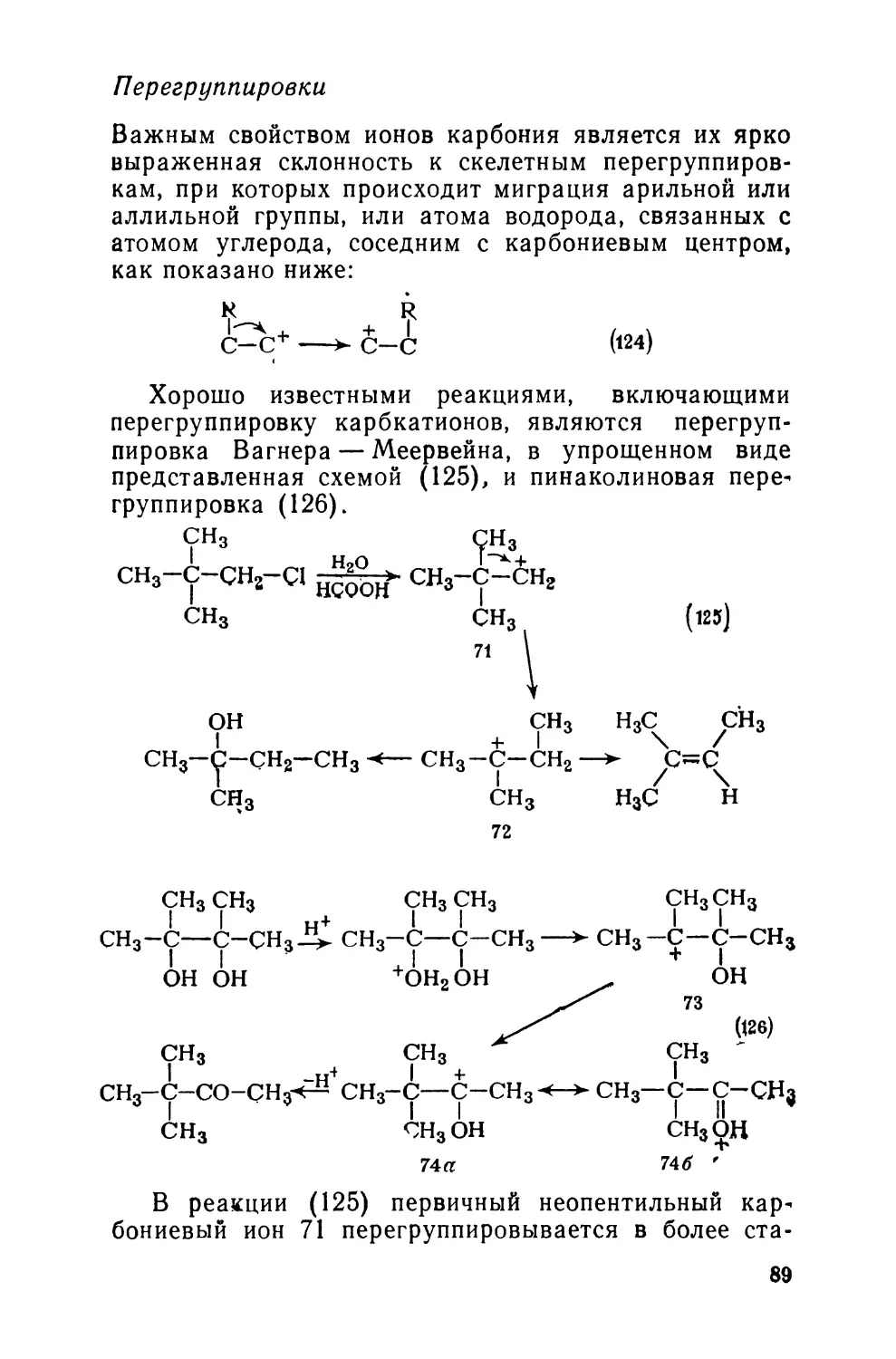
2.6. При обработке R3C—CH2C1 основанием образуется оле-
фин R2C=CHR. Перегруппировка, которая при R = Ph протекает
гораздо быстрее, чем при R=CH3, по-видимому, должна
проходить по механизму:
R3C-CH2-C1—KR2C-CH2->R2C-CH2RTnT^R2C = CHR
Если перекрестный эксперимент со смесью Ph3C—CH2C1 и
Ме3С—СН2С1 не. приводит к образованию ни Me2C = CHPh, ни
Ph2C = CHMe, будет ли это хорошим доказательством того, что
в ходе реакции группа R не становится свободной?
Гл ава 3
КИНЕТИКА
После установления строения продуктов реакции
наиболее важной характеристикой реакции является,
по-видимому, определение ее скорости. Данная глава
посвящена проникновению в механизм реакции с
помощью изучения ее скорости.
Экспериментальные методы
В идеале для определения скорости реакции надо знать
концентрацию реагентов и продуктов (при
определенных условиях) как функцию от времени после
смешения реагентов. Для этой цели может быть использован
любой химический или физический метод анализа,
приемлемый для данного реагента или продукта. Методы
анализа делятся на две группы: в первую входят
методы, использование которых для определения
скорости процесса требует отбора аликвотных частей
реакционной смеси через определенные отрезки
времени в течение всей реакции (либо за счет отбора
проб из реакционной смеси, либо запаиванием
реакционной смеси в ряд ампул, которые время от
времени вскрывают и анализируют). С помощью методов
второй группы проводят непрерывное наблюдение
какой-либо физической характеристики системы, не
прерывая реакции.
Первая группа методов включает титрование (для
кислот, оснований, окислительно-восстановительных
агентов, галогенид-ионов и т. п.), определение массы
осадка, разделение продуктов с помощью газовой
хроматографии. Это — прямые методы, они могут
быть очень точными и специфичными для некоторых
соединений и делают маловероятным ошибочное
толкование результатов. Часто, однако, процесс отбора
46
проб неудобен или требует большой затраты
времени. Эти трудности можно избежать, используя
аналитические методы второй группы, которые
представляют собой физические методы, позволяющие
следить за ходом реакции непосредственно в
реакционном сосуде. К ним относятся дилатометрия
(измерение изменений объема реакционной смеси),
измерение электропроводности и электродного потенциала,
различные типы спектроскопии, в особенности
ультрафиолетовая и, в меньшей степени, инфракрасная и
ЯМР. Эти методы часто оказываются удобными и
позволяют, сделать большое число измерений за время
протекания реакции. Однако результаты в большей
степени зависят от интерпретации регистрируемых
величин. Если, например, за реакцией следят по
увеличению абсорбции при определенной длине волны, при
которой должен поглощать один из продуктов,
необходимо установить, что наблюдаемое поглощение не
связано с присутствием небольшого количества другого
соединения, случайно обладающего большим
коэффициентом экстинкции при той #е длине волны.
В целях получения достоверных данных о скорости
процесса идеальным является использование
нескольких различных методов.
Быстрые реакции
Обычные методы химического анализа пригодны для
реакций со временем полупревращения более ~5 мин.
Физические методы анализа позволяют снизить это
время приблизительно до десяти секунд, не используя
при этом какой-либо специально разработанной
аппаратуры. В случае реакций, протекающих еще быстрее,
можно использовать струевую, импульсную или
релаксационную технику; эти методы позволяют в
благоприятных случаях изучать реакции со временем
полупревращения вплоть до ~10~9 с.
В какой области концентраций и за какой период
времени следует изучать реакции?
Обычно глубина протекания реакций должна быть
как можно более высокой, по крайней мере 80—95%.
Когда для наблюдения за ходом реакции используют
М
физический метод, например абсорбционную УФ-
спектроскопию, как правило, не возникает
затруднений в проведении большого числа измерений за время
реакции. В противоположность этому, при анализе
методом аликвотных проб, в частности, когда
реакционная смесь запаяна в ряд ампул, можно получить
только ограниченное число данных о ходе реакции
через определенные промежутки времени. В типичном
эксперименте такого рода, как правило, используют
только десять ампул. Обычно правильнее проводить
наблюдение за ходом реакции через равные
промежутки времени в течение двух или трех периодов
полупревращения, оставляя, если это необходимо, две
ампулы для анализа реакционной смеси через
«бесконечное» время реакции.
Мы должны стремиться охватить в различных
экспериментах максимально возможную область
концентраций исходных реагентов (часто, однако,
возникают ограничения, связанные с растворимостью,
временем реакции и т. п.). При изучении
температурной зависимости хода реакции необходимо
использовать, по возможности, наиболее широкий интервал
температур. Несомненно, «ito в результате таких
экспериментов будет накапливаться более полезная
информация, чем при проведении очень большого числа
повторных экспериментов при одинаковых условиях.
Однако двукратное (по крайней мере) повторение
эксперимента должно быть непреложным правилом.
Предостережение
Скорости многих реакций существенно изменяются в
присутствии в реакционной системе воздуха или под
воздействием света. Влияние воздуха можно
исключить, проводя реакции в атмосфере аргона или азота
или, что еще более строго, запаивая реакционные смеси
в стеклянные ампулы, из которых предварительно был
удален воздух. Действие света легко исключить,
закрашивая наружную поверхность реакционных сосудов
в черный цвет (или обертывая их металлической
фольгой) или проводя эксперименты в темноте.
Само собой разумеется, что чистота используемых
реагентов и растворителей должна быть по возмож-
48
ности максимальной для исключения
неопределенности результатов из-за наличия примесей.
Классическим примером трудностей в понимании природы
процесса, вызываемых наличием примесей, а также
присутствием или отсутствием воздуха или света,
является присоединение бромистого водорода к
терминальным олефинам, кратко упомянутое в гл. 1 и
детально рассмотренное в гл. 4. В этой реакции
образуется смесь соответствующих 1- и 2-бромалканов,
соотношение которых различно в разных
экспериментах. Однако если реагенты тщательно очистить и
реакцию проводить в отсутствие кислорода и света,
то продуктом реакции почти исключительно является
2-бромалкан. В этих условиях можно изучить
реакцию, приводящую к 2-бромалкану, изолированно от
другой реакции, которая дает 1-бромалкан.
Некоторые реакции протекают на поверхности
сосуда, а не в объеме жидкости*или газа; для
газофазных реакций это характернее, чем для жидкофазных
реакций. Важность поверхностных реакций можно
оценить, сильно увеличивая (скажем, в десять раз)
поверхность сосуда путем заполнения стеклянного
сосуда стеклянными стержнями, бусами или ватой
(шерстью). Если при этих условиях не наблюдается
изменения скорости реакции, то можно сделать вывод,
что реакция является гомогенной и не протекает на
поверхности. Реакцию, протекающую на поверхности,
можно подавить, изменяя материал сосуда или
покрывая его неактивным материалом.
Скорость реакции
Обычно измеряют концентрацию одного или
нескольких компонентов реакционной смеси через различные
промежутки времени. Более подходящей величиной с
точки зрения интерпретации механизма является
скорость реакции, т. е. dc/dt для данного исходного
вещества или продукта реакции. Скорость реакции в
любой данный момент времени обычно получают
построением графика зависимости концентрация —
время, проводя кривую через экспериментальные точки, а
затем касательную в различных точках этой кривой.
49
Этот процесс довольно утомителен и не свободен
от ошибок. В некоторых случаях необходимость
определения скорости таким путем можно избежать,
делая допущение 6 порядке реакции (см. ниже).
Порядок реакции
Для очень большого числа реакций установлено, что
скорость реакции меняется с изменением
концентрации одного или нескольких реагентов согласно
выражению типа:
--^=/с[А]'[ВГ[СГ (48)
где А, В, С, .. — молекулы реагентов. Степени /, т,
п, могут быть как целыми числами, так и
дробными. Если это выражение выполняется, говорят, что
реакция имеет общий (/ + т -{- п + .. ) порядок,
/-тый порядок по А, m-тый по В, n-тый по С и т. д.
На практике редко в такое выражение входят более
чем два реагента и довольно часто только один.
Константа к представляет собой константу скорости
реакции (/ + т -f n -f .. •) -ного порядка.
Для того чтобы оценить порядки реакции по
отношению к каждому компоненту, следует измерить
скорости реакции в условиях, когда все компоненты за
исключением одного находятся в большом избытке,
так что их концентрацию можно считать постоянной.
Затем строят график зависимости логарифма
скорости реакции (определенной, как описано выше) от
логарифма концентрации остающегося реагента А
([А]):
log (Скорость) = log (к') + I log [A]
Наклон прямой этого графика дает значение /
для порядка реакции по реагенту А, а отрезок
координаты, отсекаемый прямой, дает к' (которая
включает в себя концентрации реагентов В, С и т. д.).
Затем для определения порядка реакции по В
повторяют аналогичный эксперимент, в котором А и С
берутся в большом избытке, и т. д. Таким путем
можно определить порядок реакции по каждому
компоненту, а также константу скорости /с.
6Q
Этому способу определения порядка, связанному с
необходимостью получать значения скоростей, исходя
из графических данных, часто предпочитают
следующий метод. Предполагают кинетический закон для
реакции (часто по аналогии с другими реакциями или
исходя из предполагаемого механизма) и
интегрированием этого выражения получают зависимость
концентрации реагентов от времени. Например, в случае
реакции, включающей только один компонент, для
которой предполагается кинетика второго порядка,
имеем —d[A]dt = /c2[A]2. Это выражение может быть
непосредственно проинтегрировано, что приводит к
уравнению
1 ! 4
Следовательно, для того чтобы проверить
предположение о втором порядке реакции, необходимо
построить график зависимости обратной концентрации
реагента А от времени. Если график представляет
собой прямую линию, это является хорошим
доказательством второго порядка реакции при условии, что
реакцию осуществляли, по крайней мере, в течение
двух периодов полупревращения. Аналогичным
образом, график log[A] — t может быть использован (в
случае его прямолинейности) как тест реакции
первого порядка. Графики подобного типа можно
построить и для реакций других порядков, включая
дро.бные.
Преимущество описанного выше метода проверки
различных порядков реакции заключается в
отсутствие необходимости проведения касательных к
кривой концентрация — время. Однако его
недостаток в том, что с его помощью обычно не удается
выявить небольшие отклонения от (предполагаемого)
суммарного порядка, особенно если изучение
проводят в узкой области концентраций.
Определение порядка прямой реакции по данным
для обратной реакции
Часто константа равновесия реакции бывает
настолько мала, что трудно измерить скорость прямой
реакции. Если реакция элементарна (т. е. проходит без
51
образования интермедиатов), становится
применимым закон действующих масс и выполняется
уравнение
*ПРЯМ0Й = К (49)
^обратной
даже тогда, когда реакция не достигает равновесного
состояния. Поэтому, если известна константа
равновесия (измеренная или рассчитанная) и определены
кинетический порядок и констаута скорости обратной
реакции, из (49) могут быть рассчитаны порядок и
константа скорости прямой реакции. Если реакция не
элементарна, то соотношение (49) будет справедливо и
будет давать кинетическую информацию о прямой
реакции (в той же области концентраций, которую
использовали для обратной реакции) при следующих
условиях:
(а) реакция не является цепной (см. с. 66);
(б) концентрация любых интермедиатов низка;
(в)-стационарное состояние (см. с. 65)
устанавливается быстро.
Наглядным примером использования этого метода
является катализируемая основанием димеризация
ацетона в диацетоновый спирт ДАС [реакция (50)],
механизм которой рассмотрен ниже в настоящей
главе. Константа равновесия этой реакции
свидетельствует о предпочтительности обратной реакции (т. е.
образуется ацетон), скорость которой, по данным
дилатометрического изучения, равна /с_5о [ДАС] [ОН~!
Зон ОН
2СН8СОСНз «=^- СН8СОСН2С(СН8)2 (50)
ДАС
tfso = Wk-50 - [ДАС]/[СН3СОСН3]2 (50а)
Так как константу равновесия можно определить
экспериментально, то кинетический порядок прямой
реакции можно найти из уравнения скорости,
записанного в общем виде
Скорость = /с50 [СНзСОСНз]' [ДАС]* [ОРТ]'
и приравнивая это выражение в условиях равновесия
к скорости обратной реакции ас_50 [ДАС] [ОН~].
Из уравнений (506) и (50а) получают х = 2, у = 0 и
52
z = 1, что дает кинетический порядок прямой
реакции, не поддающийся измерению:
к ,к [ДАС] [ОРТ]
*50/'С-50 - [СН3СОСНз]-ЧДАС]НОНТ К )
Связь между механизмом и кинетикой
Предполагаемый механизм обусловливает
определенные кинетические закономерности, однако обратное
положение — неверно. Механизм реакции нельзя
непосредственно дедуцировать (вывести) из
кинетических данных. Тем не менее кинетические данные дают
возможность не рассматривать определенные
механизмы или позволяют сделать выбор между
альтернативными механизмами, которые представляются
правдоподобными для данной реакции.
Реакции целочисленного порядка
Теория скоростей реакций устанавливает
кинетический характер элементарной реакции: порядок по
каждому компоненту равен числу молекул этого
компонента, принимающих участие в реакции.
Кинетически важными являются только три основных типа
элементарных реакций (табл. 2).
Таблица 2. Кинетически важные тФгы реакций *
Общий
порядок
Порядок
по А
Порядок
по В
А-^Продукты (51) 1 1 О
А + А -> Продукты (52) 2 2 0
А + В -> Продукты (53) 2 1 1
* В данном обсуждении тримолекулярные реакции не рассматриваются.
Молекулярность элементарной реакции обычно
определяют как число молекул, являющихся
реагентами. Таким образом, реакция (51) — мономолеку-
лярна, тогда как (52) и (53)—бимолекулярны. Для
элементарных реакций молекулярность равна
кинетическому порядку. Следует подчеркнуть, что
53
кинетический порядок является экспериментальйой
характеристикой реакции, в то время как молекуляр-
ность — характеристикой индивидуальных
элементарных стадий предполагаемого механизма.
Если реакция имеет любой из трех указанных в
табл. 2 кинетических порядков, то ее поведение
согласуется с элементарной реакцией подходящего типа:
(51), (52) и (53). Это — простейшее объяснение
результатов, однако дополнительные данные могут
показать, что оно не состоятельно. Примерами
элементарных реакций указанных трех типов являются
следующие реакции:
Q-a— О+»с'
(54)
(55)
И
В качестве примера использования кинетики для
проведения различия между возможными
альтернативными механизмами рассмотрим нуклеофильное
замещение органических галогенидов. Два из наиболее
вероятных механизмов показаны на схемах (57) и
(58). (Nu:~— отрицательно заряженный нуклеофил)
(см, с. 16).
V
V
Nu;~ + R—Hal —■> Nu-R + НаГ (57)
(58а)
медленно
R—Hal < * Hal + R+
(-58а) (58)
1 Nu: Nu_R
(586) быстро
Механизм (57) должен приводить суммарно к
кинетике второго порядка (первый порядок по отноше-
54
нию к каждому компоненту), что было найдено для
ряда реакционных систем. Механизм (58) является
сложным и состоит из двух стадий. Если
предположить (и это вероятно), что стадия (58а) является
медленной, а стадия (586) быстрой по отношение к
реакции, обратной (58а) (это не всегда верно; см.
ниже), тогда скорость реакции будет целиком зависеть
от стадии (58а), поскольку образующийся R+ будет
быстро реагировать с нуклеофилом, образуя Nu—R.
Наблюдаемая кинетика должна, таким образом, быть
первого порядка по алкилгалогениду и нулевого —
по нуклеофилу (не зависеть от его концентрации).
Реакция (58) является примером процессов,
включающих скорость, определяющую стадию, так как ее
кинетика зависит только от стадии (58а). Реакция
подобного рода не дает кинетической информации о
стадиях процесса, протекающих после стадии,
определяющей скорость превращения. В реакции (58)
скорость стадии (586) может в значительной степени
увеличиваться или уменьшаться, не вызывая
изменения общей скорости расщепления R—Hal.
Следовательно, механизмы реакций нуклеофиль-
ного замещения (57) (бимолекулярной, SN2) и (58)
(мономолекулярной, SN\) можно разграничить
экспериментально. Схема (58) для реакций SN\ может
быть дополнительно проверена разнообразными
способами. Простая кинетика первого порядка будет
наблюдаться только в том случае, если реакция (58а)
очень медленна. Однако если скорости реакций
(—58а) и (586) сравнимы, суммарная скорость
реакции будет падать во времени быстрее, чем
предсказывается уравнением скорости первого порядка,
поскольку, когда накопится значительная концентрация
галогенид-иона, алкилгалогенид будет снова
образовываться по реакции (—58а). Падение скорости
должно также наблюдаться при добавлении к
реакционной смеси в начале реакции соответствующего
иона галогена. Это падение скорости часто объясняют
эффектом общего иона или законом действующих
масс. Реакционная схема (58) предполагает также,
что использование в реакции вместо одного нукле-
офильного реагента смеси нуклеофилов должно
привести к получению смеси продуктов, хотя скорость за-
59
мещения не должна изменяться. Если к раствору
п,я'-диметилбензгидрилхлорида 43 в водном ацетоне
прибавить азид натрия, наблюдается незначительное
изменение скорости (по-видимому, за счет солевого
эффекта*), однако продуктом реакции становится
смесь спирта 44 и азида 45. Отношение концентраций,
[45]Д44] линейно растет с увеличением концентрации
азида. Этот результат следовало ожидать при
конкуренции между реакциями (59а) и (596), так как
концентрация воды фактически постоянна.
н2о
(/г-СН3С6Н4)2СНС1 —> (А1-СНзС6Н4)2СН —I *
NaN3
>-
-* (ai-CH3C6H4)2CHOH (59а)
44
-> (ai-CH3C6H4)2CHN3 (596)
45
Различные реакции, протекающие с одинаковой
скоростью
В ряде случаев найдено, что несколько реакций
одного и того же соединения проходят с одной и той же
скоростью. Маловероятно, что это результат
случайного совпадения, особенно если близость скоростей
реакций сохраняется п*ри нескольких температурах.
Наиболее вероятным объяснением является то, что
все эти реакции имеют общий механизм вплоть до
скорости, определяющей стадию, и что реагенты,
приводящие к различным продуктам, реагируют только на
последующих стадиях. Примеры этого типа реакций
включают катализируемое основаниями хлорирование,
бромирование и иодирование ацетона, а также
сходные реакции вгор-бутилфенилкетона. В этих
процессах все реакции, включая галогенирование, дейтеро-
* Скорости реакций, в которых из нейтральных молекул
образуются ионы или в которых реагируют между собой ионы
одного знака, увеличиваются в присутствии солей (т.е. ионов).
Наоборот, скорости реакции между ионами противоположного
знака уменьшаются в-присутствии солей. Солевой эффект может
использоваться в качестве метода диагностики механизма; в
прошлом этот метод применялся при изучении главным образом
неорганических, но не органических реакций.
56
обмен и рацемизацию оптически активного кетона,
протекают с той же самой скоростью. В каждой из
этих двух групп реакций наблюдается первый порядок
реакции по отношению к основанию и кетону, из чего
следует, что скорость определяющей стадией является
отрыв протона от кетона, за ней следуют быстрые
реакции аниона.
медленно - Н20
НО" + PhCOCHEt >- PhCOCEt —► PhCOCHEt
I I I
Me Me Me
(рацемизация)
D2o
—> PhCOCDEt
Me *
Br2
—> PhCOCBrEt
I
Me
Реакции более высокого порядка, чем второй
Поскольку обычно элементарные реакции моно- или
бимолекулярны, то реакции, показывающие более
высокий порядок, чем второй, не являются
элементарными. Тем не менее, кинетически многие реакции
имеют третий или даже более высокий порядки.
Причины такого кинетического поведения часто связаны
с механизмом реакции, который включает ряд стадий,
в том числе обратимую реакцию, протекающую перед
стадией, определяющей скорость процесса.
Следующая схема является типичной для такого механизма:
А + В ^— с (61)
C + D —> Е (62)
Если (а) реакция (62) медленнее, чем прямая и
обратная реакции уравнения (61), и (б) равновесие
реакции (61) значительно сдвинуто влево, то
равновесие устанавливается быстро, а концентрация С
определяется уравнением:
[C] = (k61/k-6iHA][B] (63)
Скорость образования Е и, следовательно, общая
скорость процесса задается уравнением (64). Исходя
67
(60)
из уравнения, должна наблюдаться кинетика третьего
порядка.
d[E] _ d[A] _ d[B) [П\п\-
= (/c6i/C62//c-6i)[A][B][Dr (64)
Реакции, протекающие таким путем, довольно
обычны и включают большое число процессов,
катализируемых кислотами или основаниями. Примером
реакций кислотного катализа служит этерифика-
ция (65):
о он R,0H он
|| (65а) | медленно |
Н+ + R—С—ОН *=± R—С—ОН к ■» R—С—ОН
+ быстро |
46 0+
/\
Н R'
t
быстро
(65)
медленно
+он2
H+ + R—C=0 ?=± R—С—ОН < ■» R—С—ОН
| быстро I
OR' OR' OR'
Эта схема объясняет наблюдаемую зависимость
скорости реакции от [Н+] [RCOOH] [R'OH].
К реакциям, катализируемым основаниями и
имеющим кинетику третьего порядка, относится широко
используемая альдольная конденсация альдегидов и
кетонов, представленная на примере ацетона
уравнениями (66) и (67). [Для этой частной реакции,
равновесие в которой значительно сдвинуто влево,
большинство кинетических исследований выполнено на
обратной реакции; из этих результатов можно дедуцировать
кинетические данные для прямой реакции (см. с. 52).]
быстро
СНзСОСНз + ОН" *=* Н20 + СНзСОСН2" (66)
47
медленно
СН8СОСН; + СН,СОСН3 > СН3СОСН2С(СНз)2 (67)
О"
58
Почему так много реакций, катализируемых
кислотами и основаниями, подчиняются кинетике третьего
порядка? Обычно реакции переноса протона
протекают быстро, но равновесие большого числа реакций
этого типа значительно сдвинуто влево, поскольку
большинство протонированных органических
соединение (например, 46) являются более сильными
кислотами, чем Н30+, а большинство органических
оснований, образующихся при отрыве протона от связи
С—Н (например, 47), являются более сильными
основаниями, чем ОН-, OEt~" и т. д. Поэтому
равновесия реакций (65а) и (66) и многих других близких
им реакций значительно сдвинуты влево и вместе с
последующей реакцией органических интермедиатов
могут приводить к суммарной кинетике третьего
порядка.
Сложные реакции, которые случайно имеют
простую кинетику
Обычно первое представление, которое можно
высказать о механизме, наиболее простое. Для реакций с
общей стехиометрией А + В —► С + D,
описывающихся кинетикой второго порядка, первого
порядка по А и В, простейшим является представление о
бимолекулярности реакции, которая протекает между
молекулами А и В, по одной' от каждой. Ряд фактов
подтверждает мнение, что, например, реакции нуклео-
фильного замещения второго кинетического порядка
(57) действительно являются простыми
(элементарными) бимолекулярными реакциями.
Однако простое кинетическое поведение
наблюдается иногда и для реакций со сложным
механизмом. Пример подобного процесса из области
неорганики — реакция между водородом и иодом. Эта
обратимая реакция отвечает кинетике второго порядка:
первого по водороду и по иоду. «Очевидный»
механизм этого взаимодействия, простая бимолекулярная
реакция (68), был общепринятым в течение
шестидесяти лет.
н—н н н
—> I I (68)
I-I I I
59
И в самом деле, было бы более чем странным
предположить, без дополнительных фактов, что
реакция должна проходить по какому-либо иному
механизму. Но этот факт был, наконец, обнаружен в 1967 г.
Сэлливеном, который установил, что в реакции
участвуют атомы иода, находящиеся в термическом
равновесии с молекулярным иодом.
Рассмотрим схему альтернативного механизма
взаимодействия водород — иод [реакции (69) и (70)],
в которой вслед за установлением равновесия между
молекулами и атомами иода [реакция (69)] протекает
медленная реакция атомов иода с молекулярным
водородом [для простоты реакция (70) записана в виде
одностадийного процесса, хотя она может протекать
и в две стадии]:
быстро
h *=* 21. (69)
быстро
21 • + Н2 ^г* 2HI (в одну или две стадии) (70)
Поскольку равновесная концентрация атомов иода
равна
[I •] = ([Is] кв0/к-в9),/а
скорость образования HI на ранней стадии
реакции, когда можно пренебречь обратной
реакцией (2HI -> Н2 + Ь), будет иметь вид:
d ГНИ 2квэ*70
-=-У/ ^2 [I #1 [Hal K" - ^— [На] [1*] (71>
1X1 • К—69
Следовательно, на основании кинетического
изучения термической реакции нельзя провести
разграничение между простым бимолекулярным механизмом
(68) и сложным механизмом (69) и (70). Выбор
между этими механизмами был сделан с помощью
фотохимической генерации атомов иода в смеси иода и
водорода при достаточно низких температурах, что
позволяет пренебречь скоростью термической
реакции. Из скорости образования йодистого водорода
можно найти величину /с7о. Отношение констант
скоростей aWk-69 представляет собой известную
константу равновесия диссоциации молекулярного иода
на атомы при данной температуре. Исходя из этого,
Сэлливен рассчитал по уравнению (71) скорость, с
которой должен получаться йодистый водород в тер-
60
мической системе по механизму (69) и (70). С
точностью эксперимента оказалось, что эта скорость
равна скорости, которая в действительности наблюдается
для термической реакции, и, следовательно, вклад
непосредственной бимолекулярной реакции (68) в
суммарный процесс незначителен.
Реакции нецелочисленного порядка
Не все известные реакции имеют целочисленный
порядок по одному или большему числу компонентов
реакции. Мы уже видели, что даже в случае реакций
целочисленного порядка они необязательно должны
быть элементарными. Реакции, которые показывают
более сложных кинетический характер, не могут быть
элементарными. Ниже рассмотрены два обычно
встречающихся типа реакций нецелрчисленного порядка с
обсуждением того, какая информация о механизме
может быть при этом получена.
Реакции смешанного порядка
Многие реакции имеют смешанный кинетический
порядок. Их скорость может быть выражена суммой двух
или более членов, например:
Скорость = кх [А] + к2 [А] [В] (72)
Скорость = /с, [А] [В] + к2 [А] [В]2 (73)
Часто экспериментально трудно провести различие
между кинетической зависимостью в этих реакциях
и реакциями дробного кинетического порядка,
например
Скорость = к [А] [В]0,5 (74)
Скорость = к [А] [В] Ь5 (75)
если нельзя изучить кинетику реакции в достаточно
широком интервале концентрации В.^ Обычный способ
проверки реакций, обладающих смешанным
порядком, можно показать на примере, для которого
ожидают кинетическую зависимость типа (72). Если обе
части уравнения (72) разделить на [А], получаем
61
выражение (76) для «кажущейся» константы скорости
первого порядка (СкоростьДА]):
^Кажущаяся" константа скорости первого порядка =
= Скорость/[А] = к, + к2 [В] (76)
На рис. 1 приведен график зависимости
кажущейся константы скорости первого порядка от [В].
Если реакция действительно подчиняется
кинетическому поведению типа (72), этот график будет иметь
вид прямой линии, тангенс угла наклона которой
равен /с2, с отрезком, отсекаемым от оси ординат,
равным к\. Аналогичные графики могут быть
непосредственно построены для других типов кинетической
зависимости смешанного порядка.
I
Рис. 1. Кинетика смешанного
(первого и второго) порядка.
График зависимости
скорости [А] от [В] для
реакции, описываемой
уравнением (72).
Обычная причина кинетики смешанного порядка
связана с тем, что один из компонентов
реакционной смеси реагирует в двух различных направлениях.
Примером такого процесса является гидролиз изо-
пропилбромида при действии едкого натра в водном
этаноле, для которого наблюдаемая зависимость
скорости от концентрации реагентов имеет вид;
г d [(СНзЬСНВг]
Скорость = — jr. — =
= «| [(СНзЬСНВг] + к2 [(СНзЬСНВг] [ОН"] (77)
Sft 1-составляющая 5дг2-составляющая
Это интерпретируют в терминах конкурентного
гидролиза, протекающего по механизмам SNl и
SN2, обсуждавшимся ранее *. Поскольку при этих
* В настоящее время принято, что подобные процессы
протекают по одному механизму: через образование пар. — Прим.
ред.
Тангенс уела
наклона ра$енкг
W
S2
условиях реакции бромистый этил реагирует
исключительно по механизму S#2, а грвг-бутилбромид — по
механизму 5^1, нет ничего неожиданного в том, что
оба эти пути гидролиза одновременно возможны в
случае промежуточного (с точки зрения строения)
бромистого изопропила (о причине различного
поведения алкилбромидов см. гл. 6).
Многие реакции, катализируемые кислотами и
основаниями, описываются кинетикой смешанного
порядка. Например, ортоэтилацетат 48 гидролизуется с
образованием этанола и уксусной кислоты [реакция
(78)] в водных буферных системах ж-нитрофенол
(слабая кислота) — ж-нитрофенолят натрия. Показано, что
при постоянной ионной силе раствора скорость
расщепления ортоэтилацетата 48 подчиняется
кинетическому уравнению (79).
НА быстро
CH3C(OEt)3 >■ CM3C(OEt)2
медленно i несколько
(78а) HOEt стадий
+
48 49
—> СНзСООН + ЗЕЮН (78)
- ЛШ = Кц [48] + кп [НзО+] [48] + кп [*-HOCeH4NQ»] [48] (79)
Эта кинетическая зависимость с членами,
содержащими [Н30+] и [^-HOC6H4N02], предполагает,
что стадия, определяющая скорость всего процесса,
включает переход протона от молекулы кислоты к
ортоэтилацетату [реакция (78а)]. Донором протона
может служить НзО+ (Kh ^> /с„), однако если
раствор не слишком кислый, то [HOCeH4N02] ^>
>• [Н30+] и член с кп может внести значительный
вклад в скорость реакции. Аналогично, появление
члена с киу по-видимому, обусловливается наличием
в системе воды, действующей как кислота, т. е. этот
член в уравнении (79) следует заменить на /си[Н20]
[48].
Примером реакции, катализируемой основанием,
является реакция самоокисления — восстановления
альдегидов (реакция Канниццаро). В гл. 2 было
приведено доказательство того, что водород
63
непосредственно переходит от группы СНО одной
молекулы альдегида к другой [реакция (31)]. Найдено,
что при низкой концентрации основания реакция
имеет второй порядок по альдегиду и первый по гид-
роксильному иону. При более высокой концентрации
гидроксильных ионов кинетическая зависимость
приобретает вид:
d [RCHO]
dt
= /ci [RCHO]2 [ОН"] + к2 [RCHO]2 [ОН']2 (80)
Эти результаты поясняются схемой (81).
Направление реакции (81а) реализуется при низкой
концентрации основания. Поскольку между RCHO и
анионом 50 существует равновесие, вообще говоря, следует
ожидать уравнения скорости третьего порядка [ср.
реакции (66) и (67) в предыдущем разделе]. Однако
при более высокой концентрации основания реакция
50 с ОН- может привести к 51, открывая новый путь с
суммарным четвертым порядком [реакция (816)].
Реакции, катализируемые кислотами и основаниями,
будут рассмотрены далее в гл.. 4, при этом особое
внимание будет уделено интермедиатам типа 49 и 50.
RCHO + ОН" ^=±
О" О
I RCHO ||
±=+ R—С—Н * R—С—ОН + RCH20" (81а)
50
н2о||оьг
0" О
I RCHO ||
R-C-H ► R—С—О" + RCH20~ (816)
А-
51
Реакции дробного порядка
Реакции, показывающие кинетику истинно дробного
порядка (т. е. не кинетику смешанного поряда,
рассмотренную в предыдущем разделе), часто включают
64
образование свободно-радикальных интермедиатов.
Способы установления присутствия
свободно-радикальных интермедиатов описаны в следующей главе,
но с точки зрения кинетики наиболее важной чертой
большинства радикалов является их высокая
реакционная способность. В отсутствие подходящих
молекул, с которыми радикалы могут реагировать, они
изаимодействуют друг с другом и гибнут в
результате реакций рекомбинации и диспропорционирования
типа (82) и (83), скорость протекания которых
приближается к частоте столкновений. Вследствие
крайне высокой скорости рекомбинации и
диспропорционирования, концентрация радикалов обычно не
превышает величины Ю-8 М, и поэтому для того, чтобы
другие реакции могли конкурировать с
рекомбинацией и диспропорционированием, они должны быть
быстрыми:
2СН3СН2. —
• —> СНзСН2СН2СНз рекомбинация (82)
I—> СН3СНз + СН2=СН2 диспропорционирование (83)
Метод стационарного состояния
При использовании этого подхода упрощается
кинетическая обработка систем, включающих высокореак-
ционноспособные интермедиаты, такие как свободные
радикалы. Если концентрация интермедиата не
становится сравнимой с концентрацией реагентов и
продуктов, то после короткого периода времени с начала
реакций скорости образования и расхода
интермедиата станут практически равными, что приводит к так
называемому «стационарному состоянию».
Предположение о стационарности не означает, что
концентрация промежуточного соединения не изменяется в ходе
реакции (на самом деле после определенного времени
для большинства реакций концентрация интермедиата
будет снижаться до нуля), но имеется в виду, что в
любое данное время скорости образования и расхода
интермедиата можно считать равными.
В качестве простого примера использования
принципа стационарного состояния рассмотрим разложение
3 Зак. 1044
65
азобисизобутиронитрила 52 в бензольном растворе.
При этом по реакции (84) образуются 2-цианопро-
пильные-2 радикалы 53, которые, будем считать для
простоты, гибнут только по реакции (85).
(CH3)2C-N=N-C(CH3)2 —> 2(CH3)2C + N2 (84)
I I 1
CN CN CN
52 53
2(CH3)C —> (CH3),C—C(CH3)2 <85)
CN CN CN
Применение принципа стационарного состояния дает!
-^L - 2лс84 [52] - 2к85 [53]2 _ О (86)
ИЛИ
[53] = (к84[52]/к85)1/2 (87)
Из уравнения (87) следует важный вывод: в уело*
виях стационарного состояния концентрация
радикалов обычно пропорциональна квадратному корню
скорости их образования.
Следует подчеркнуть, что принцип стационарного
состояния может применяться также в случае
реакций, в которых образуются не радикальные интерме-
диаты, а например ионы карбония, карбанионы и
карбены.
Цепные реакции
Большинство свободнорадикальных реакций, с
которыми обычно приходится сталкиваться,
представляют собой цепные процессы. Возникающий в
цепной реакции радикал реагирует с молекулой с
образованием нового радикала, который в свою очередь
способен взаимодействовать с другой молекулой и
так далее. В конце концов радикал исчезает в
результате рекомбинации или диспропорционирования,
однако прежде чем это произойдет, прореагирует
очень-большое число молекул (обычно ~1000). Роль
первоначального (или инициирующего) радикала
аналогична роли молекулы кислоты или основания в
реакциях кислотного или основного катализа.
Типичными свободнорадикальными цепными реакциями мо-
66
гут служить реакции замещения и присоединения
молекул к ненасыщенным соединениям.
Ниже приведена общая схема довольно простой
цепной реакции. Эта схема пригодна, в частности,
для описания хлорирования алканов (X = Hal;
А= Н; B = R).
нагревание или hv
X—X > 2Х« инициирование (88)
v i л -п ^Y A,n ч развитие цепи; суммарный
A»-t-A—о —► л л-\-а» 1 эффект этих двух реакций— (89)
+ Х. J
В. + X—X —> В—X + X • J Х. + АВ + Х2 —>
т J —* АХ + ВХ + Х- (90)
Х. + Х. —► Х-Хч (91)
Х. + В» —► X—В \ обрыв цепи (92)
В. + В. —> В—В J (93)
Реакция, в которой (говорят) НВг присоединяется
к олефину, в общем протекает по аналогичной
схеме, однако одна из стадий развития цепи должна
включать присоединение Bv к олефину [реакция
(94)], сопровождаемое реакцией перемещения
водорода [реакция (95)].
Вг • + СНзСН=СН2 —* СН3СНСН2Вг (94)
СН3СНСН2Вг + НВг —> СН3СН2СН2Вг + Вг • (95)
Кинетический анализ цепных реакций до сих пор
представляет собой сложную задачу, даже если
используется приближение стационарного состояния.
Однако, как правило, в цепных реакциях одна из
стадий развития цепи [(89) или (90)] проходит
значительно быстрее другой либо вследствие различия
в константах скоростей, либо (если они близки) из-
за избытка X—X или А—В. Если реакция (89) много
быстрее реакции (90), то основным радикалом,
присутствующим в системе, будет радикал В». При этих
условиях обрыв цепи будет проходить в основном за
счет реакции (93) и метод стационарного состояния
приведет к выражению (96). Наоборот, если реакция
(90) много быстрее реакции (89). [X • ]»[В • ],
обрыв будет проходить главным образом за счет
стадии (91), и кинетическое уравнение приобретает вид
3*
67
(97). Эти альтернативные механизмы можно легко
различить.
<*[ВХ] к&&
dt к*
[X2]1/s при [В.]>[Х.] (96)
d [BX] Кьскк и
,-i°» [Х,]*[АВ] при [Х.]>[В.] (97)
dt кЦ
Кинетический порядок, которому подчиняется
данная реакция, показывает, какая из реакций развития
цепи является быстрой стадией; а из составной
константы скорости можно получить информацию о
скорости определяющей стадии развития цепи.
Например, реакция брома с хлороформом,
приводящая к образованию бромистого водорода и бром-
трихлорметана, в газовой фазе при 150—180 °С
подчиняется кинетическому уравнению:
- d [Br2]/dt = к„абл [СНС13] [Br2]v' (98)
Такая зависимость предполагает, что реакция
представляет собой радикальный цепной процесс,
включающий две стадии развития цепи: (99) и
(100).
Вг. + СНСЬ —> НВг + *СС13 (99)
.СС13 + Вг2 —> ВгСС13 + Вг* (100)
Так-как выражение (98) имеет тот же вид, что
и уравнение (97), но не (96), то реакция (100)
является быстрой, а стадия (99) более медленной,
т. е. скорость определяющей стадией, и практически
единственным радикалом, присутствующим в системе,
является бром.
Используя известные величины константы
равновесия диссоциации брома при данной температуре
(kWkqi; Х=Вг), можно из общей (суммарной)
скорости рассчитать константу скорости реакции (99).
О реакции (100) можно сказать только, что она
должна иметь константу скорости, значительно
большую, чем к99. Для реакций, в которых /с8э и
/Сдо — величины одного порядка, лимитирующие
скорость выражения (96) и (97) можно получить при
использовании большого избытка одного или другого
68
реагента. До тех пор, пока этого сделать нельзя,
кинетические данные будут относиться только к более
медленной из двух стадий развития цепи и
единственный вывод, который можно сделать
относительно другой реакции, заключается в том, что она
протекает быстрее.
Реакции с еще более сложной зависимостью
скорости от концентрации
Не все реакции можно описать кинетическими
зависимостями целочисленного, дробного или смешанного
порядка. Более сложное (кинетическое) поведение
обусловлено двумя простыми причинами.
(а) Накопление интермедиата в реакционной смеси
В этих случаях скорость образования конечных
продуктов не будет равна скорости исчезновения
реагентов и будет наблюдаться сложная кинетическая
зависимость скорости образования продуктов от
концентрации реагентов. В реакциях этого типа можно
измерить концентрацию интермедиата. Это дает
возможность разложить суммарное кинетическое
выражение реакции на два или более членов,
соответствующие отдельным последовательным стадиям
данного процесса. Простыми примерами реакций, для
которых было установлено поведение этого общего
типа, являются реакции гидролиза дигалогенангидри-
дов и эфиров дикарбоновых кислот.
(б) Ингибирование реакции одним из ее продуктов
В любой обратимой реакции, в которой после
достижения равновесия наблюдается заметная
концентрация реагентов и продуктов уже на ранних стадиях
прямой реакции, важное значение могут приобрести
обратные процессы. Кроме того, реакция может инги-
бироваться (замедляться) при добавлении в
реакционную смесь одного или нескольких продуктов
реакции. В качестве примера ингибирования можно
рассмотреть бромирование хлороформа [реакции
(99) и (100)]. В этой реакции обратный процесс
69
[реакция (—99)] обладает заметной константой
скорости, так что наличие в системе НВг,
образовавшейся в ходе реакции или добавленной специально,
будет снижать скорость расхода брома и приводить
к кинетическому выражению типа:
d [Br2] АСнабл [СНС1э] [Br]Va
dt 1 + /с-99 [НВг]/ас100 [Br2] U '
Это выражение при нулевой концентрации
бромистого водорода переходит в выражение (99).
Кинетические выражения типа (101), содержащие
в знаменателе концентрацию продукта, показывают,
что реакция ингибируется продуктом.
Экспериментально обычно выгоднее упростить ситуацию:
измерять начальные скорости (т. е. проводить реакцию
лишь на несколько процентов); в этом случае можно
пренебречь членом, учитывающим ингибирование, и
получить'более простое уравнение типа (98).
Зависимость скорости реакции от температуры
Скорость почти всех реакций увеличивается с ростом,
температуры. Найдено, что изменение скорости
элементарных реакций (и некоторых сложных) в
зависимости от температуры обычно описывается (с
точностью эксперимента) выражением типа (102),
известным под названием уравнение Аррениуса:
K = Aexp(—E/RT) (102)
В рамках теории столкновении в теории
скоростей реакций энергия активации Е представляет
собой критическую энергию, которая необходима
молекуле для протекания мономолекулярной реакции.
В случае бимолекулярной реакции Е — суммарная
энергия, которой должны обладать две
молекулы в момент столкновения, чтобы осуществилось
их взаимодействие. Предэкспоненциальный
множитель уравнения Аррениуса А для бимолекулярных
реакций рассматривают как скорость, с которой
проходят эффективные столкновения между молекулами,
и часто составляют из двух членов: A =PZt где Z —
частота столкновений, а Р — стеричёский фактор.
Поскольку обычно А меньше Z, можно предположить,
70
что не все столкновения являются эффективными и
что реакция может произойти только в случае
определенным образом ориентированных в момент
столкновения молекул. В случае мономолекулярных
реакций интерпретация предэкспоненциального
множителя является несколько более сложной. Однако в
последнее время для описания как моно-, так и
бимолекулярных реакций в большей степени
используют теорию переходного состояния.
Рис. 2. Вид зависимости
энергии от координаты
реакции для эндотермических
реакций.
" Координата реакции
Согласно этой теории считают, что, реагенты
находятся в псевдоравновесии с переходным
состоянием, представляющим собой высшую точку на
диаграмме потенциальной энергии (рис. 2),
изображающей переход реагентов в продукты. Координату
реакции на рис. 2 можно рассматривать как
расстояние (иЛи подходящую произвольную функцию от
него) между любыми двумя атомами в реагирующей
системе, между которыми завязывается или рвется
ковалентная связь в ходе реакции. Все другие длины
и углы связей следует рассматривать выбранными
так, что это приводит к минимуму энергии в любой
точке кривой. Поэтому переходное " состояние на
диаграмме потенциальная энергия — координата
реакции следует представлять не как максимум пика,
а как точку перегиба седла.
Используя это приближение, можно получить
следующее уравнение для константы скорости
химической реакции:
(kT/h) exp (AS*//?) exp (- AH*/RT) (103)
В этом уравнении k и h — константы Больц-
мана и Планка соответственно, AS* и АЯ* —
5
71
разность между энтропией и энтальпией
переходного состояния и реагентов (например,
AS^=^ о переход, сост. — 25Реагентов). В случае некоторых
реакций существует возможность априорной оценки
величин AS** и АНФ> которые затем можно сравнить
с результатами кинетических экспериментов,
поскольку для реакций в растворе справедливы
соотношения (104) и (105) (для газофазных реакций эти
формулы нуждаются в небольших изменениях).
AH^^E-RT (104)
А = (kT/h) ехр [1 + (AS^lR)] (105)
Использование энергии активации при изучении
механизмов
Обычно трудно провести априорную оценку энергии
активации реакции. Однако для эндотермических
реакции энергия активации должна быть по крайней
мере такого же порядка, что и тепловой эффект
реакции (см. рис. 2). Действительно, в случае
эндотермических реакций с большим поглощением тепла
(таких, которые часто включают образование
нестабильных интермедиатов) тепловой эффект и энергия
активации часто оказываются приблизительно равными.
Тепловой эффект эндотермических реакций (который
зависит только от теплот образования реагентов и
продуктов) можно часто рассчитать. Такой подход
позволяет указать минимальную величину энергии
активации эндотермических реакций. Если
экспериментальная величина оказывается значительно ниже
расчетной, тогда данный механизм реакции следует
исключить из рассмотрения.
В качестве наглядного примера подобного
подхода рассмотрим термическое разложение тетраэтил-
свинца, при котором образуется свинец и ряд
органических продуктов, возникающих из этильных
радикалов:
(106)
PbEt4 *■ Pb + 4Et • —> Этан, этилен, бутан
(l^7) I i одна или
▼ I несколько (106, 107)
Et. + PbEts. ' стадий
72
На схеме (106,107) показаны два возможных пути
образования наблюдаемых продуктов. Реакция (106)'
нключает одновременный разрыв всех Четырех
связей свинец—углерод. Реакция (107) включает
первоначально разрыв только одной из этих связей с
последующим разрывом остальных связей.
Протекающие вслед за этим реакции этильных радикалов с
образованием этана, этилена и бутана, как
известно, имеют фактически нулевую энергию активации и
должны быть очень быстрыми. Поэтому реакция
(1Q6) или (107) или любая другая постулируемая
первая стадия термолиза должна определять
скорость всего процесса.
Исходя из известных значений теплот
образования тетраэтилсвинца, свинца и этильного радикала,
можно оценить тепловой эффект реакции (106) в
515 кДж-моль-1. Эта величина значительно больше
наблюдаемой энергии активации (155 кДж-моль-1).
Исходя из этого, должен быть сделан вывод, что
реакция (106) не имеет места и что стадия,
определяющая скорость процесса разложения, представляет
собой реакцию типа (107).
В гл. 1 (см. с. 25) приведен пример того, как на
основании оценки величины энергии активации был
исключен один из двух возможных радикальноцеп-
ных механизмов хлорирования алканов.
Выбор механизма на основании такого метода
обычно делается с учетом уже накопленных данных.
Тем не менее автор готов рискнуть использовать
этот метод для следующего недостаточно широко
изученного примера. Органогидриды металлов IVB
группы Периодической системы R3MH (М = SI, Ge,
Sn, Pb) могут присоединяться к алкенам. При
М = Si, Ge или Sn в подходящих условиях реакция
представляет <:обой радикально-цепной процесс.
R3M . + CH2=CHR' —> R3M—CH2—CHR' (108)
R3M-CH2-CHR' + HMR3 —> R3M-CH2—CH2R' + R3M . (109)
Органогидриды свинца также присоединяются к
олефинам, однако в этом случае, используя величины
энергии диссоциации связи, можно рассчитать тепло-*
иой эффект реакции (108) (~88 кДж-моль-1). Если
73
принять разумную величину для Л-множителя и
считать тепловой эффект реакции равным минимальной
величине энергии активации этого процесса, то
скорость реакции (108) окажется слишком мала для
того, чтобы конкурировать с процессами
рекомбинации и диспропорционирования радикалов.
Следовательно, для этой реакции должен быть предложен
другой механизм, например четырехцентровый
молекулярный механизм (НО).
R3M Н
! l —> R3M-CH2—CH2R' (НО)
CH2=CHR'
Предэкспоненциальный множитель А
Можно предположить, что предэкспоненциальные
множители Л (Л-факторы) уравнения Аррениуса в
пределах данного класса реакций будут иметь
близкие значения. С точки зрения теории столкновений
это следует из того, что стерические требования не
должны слишком сильно различаться внутри одного
класса соединений. С другой стороны, в рамках
теории переходного состояния разность энтропии
реагентов и переходного состояния должна оставаться
одинаковой внутри одного типа реакций. Это положение
согласуется с экспериментом: очень большое число
элементарных реакций имеют А-факторы, лежащие
в интервалах, приведенных в табл. 3.
Наиболее просто различие между отдельными
классами реакций можно провести с точки зрения
энтропийного фактора. Для мономолекулярных
реакций гомолитическое расщепление на два (или
более) фрагмента приводит к значительно большей
свободе движения (колебательного и вращательного) в
переходном состоянии, чем в основном состоянии
молекулы. Следовательно, энтропия активации, будет
положительна и Л-фактор велик. С другой стороны,
четьгрехцентровые процессы включают частичное
образование двух новых связей в переходном состоянии,
приводящее к «жесткому» переходному состоянию с
меньшей энтропией и таким образом к более низким
А-факторам, чем при мономолекулярном гомолизе.
Для бимолекулярных реакций, например
рекомбинации двух радикалов, Л-фактор велик, что согла-
74
Таблица 3. Пред экспоненциальные Л-факторы уравнения
Аррениуса
Моле-
куляр-
ность
реакции
Тип реакции
Пример
lg A
1 Гомолиз С2Нб -> 2СН3- 15—17
1 Четырех- С2Н5Вг -> СН2=СН2+НВг 12,5-14
центровый
процесс
2 Рекомбина- 2СН3» -> С2Нб 9—10,5
ция
радикалов
2 Радикаль- СНз^+СгНв -> СН44-С2Н5« \ 7__Q
ный перенос OV+CH^CHa -> СН3СН2СН2. J ' *
или
присоединение
к двойной
связи
гент—органический
радикал)
2 Тежереак- Ci-fCH4 -> CH^+HCl 10-11
ции, но с
одновалентным атомом
в качестве
реагента
суется с переходным состоянием, в котором между
двумя радикалами завязывается слабая связь. В
случае реакций присоединения и переноса радикалов
больший Л-фактор наблюдается, когда в качестве
реагента выступают одновалентные атомы, у которых
отсутствует энтропийный член, связанный с
вращением, который, следовательно, не может уменьшаться
при достижении переходного состояния.
По-видимому, в ближайшем будущем в нашем
распоряжении появится достаточное количество
примеров реакций других классов, которые смогут
расширить данные табл. 3. Следует обратить внимание
на отсутствие данных для ионных реакций. Ионные
реакции часто сопровождаются очень большими
изменениями энтропии, связанными с перестройкой
75
сольватных оболочек (или с их разрушением) в ход^
реакции. По этой причине сделать обобщения
относительно Л-факторов для ионных реакций
оказывается более сложным, чем в случае свободноради-
кальных и молекулярных реакций, для которых
взаимодействие с растворителем (если оно имеет место)
является гораздо более слабым.
Использование Л-факторов уже не раз проливало
свет на механизм реакций. Пиролиз циклобутана
представляет собой реакцию первого порядка,
приводящую к образованию двух молекул этилена.
Первоначально считалось, что этот процесс является че-
тырехцентровой молекулярной реакций (111), так
как попытки ингибировать эту реакцию окисью азота
или пропеном (которые должны реагировать с
радикалами, если они образуются в процессе) оказались
безуспешными. Однако наблюдаемая беличина А-фак-
тора (1015'6) в соответствии с данными табл. 3
более характерна для процессов гомолитического
расщепления, чем для истинной четырехцентровой
реакции. Детальные расчеты энтропии активации
подтверждают эту^ точку зрения, и теперь считают,
что более вероятной является схема (112). Для
объяснения того факта, что пропен и оксид азота не
оказывают влияния на реакцию, полагают, что
вторая стадия, в которой бирадикал 54 расщепляется на
две молекулы этилена, протекает очень быстро.
<?**£-<№* ^ 9н2=<гН2 СН^СЩ . Y
I I —>- ! ! —►• (in
сн2—сн2 сн2=сн2 сн2=сн2
т
сн2— сн2- быстт)0 сн2=сн2
I :-> (112
сн2— сн2« сн^сн,,
54
В прошлом использование Л-фактора для выбора
возможного механизма практиковалось
сравнительно редко. Однако по мере накопления данных об
элементарных реакциях становится возможным оценить
величину (или область значений) Л-фактора для все
большего числа типов химических реакций. Таким
76
образом, появляется возможность более широкого
применения этого критерия для выяснения механизма
реакций.
Первичный кинетический изотопный эффект
Особым случаем использования кинетики для
определения механизма является применение изотопов
(почти всегда дейтерия или трития) путём введения их
в положения, по которым осуществляется реакция.
Силы, обусловливающие притяжение и
отталкивание атомов в молекуле, имеющие
электростатическую природу, не будут изменяться, если атом
водорода в молекуле заменить на атом дейтерия, так как
заряды ядер двух изотопов равны. Однако большая
масса атома дейтерия приведет к меньшей величине
нулевой энергии для дейтерированного соединения по
сравнению с протиевым (водородным) соединением.
Таким образом, энергия диссоциации связи в дей-
терированном соединении будет примерно на
5 кДж-моль-1 выше, чем в случае водородного
соединения. В химических реакциях, в которых, связь
С—D или С—Н по крайней мере частично
разрывается, следует ожидать более высокой энергии
активации для дейтерированного соединения (рис. 3),
хотя любая остаточная нулевая энергия в переходном
состоянии будет уменьшать разность между ED и
Ен. Если в реакции скорость определяющая стадия
включает ослабление связи X—Н, то замена этого
водорода на дейтерий будет замедлять реакцию.
Отношение констант скорости реакций недейтерированной
и дейтерированной молекул, /ch/kd, называют
первичным изотопным эффектом изучаемой реакции.
Максимальная величина первичного изотопного
эффекта зависит от разности нулевых энергий; для
реакций, проходящих по связям С—Н, Kh/kd не
может быть больше, чем ~7 при 25 °С, а Кн//Ст не
больше, чем ~17, если не действуют другие
факторы. Несколько большие величины возможны для
связей N—Н и О—Н. Величина первичного изотопного
эффекта, близкая максимальному значению,
показывает, что данная связь почти Полностью
разрывается в переходном состоянии. Меньшие величины
могут свидетельствовать в пользу частичного разрыва
77
связи в переходном состоянии,.в то время как
близость изотопного эффекта к единице подразумевает,
что связь либо не разрывается, либо это происходит
в незначительной степени. --
Использование этого метода подтвердило
представление, что механизм электрофильного
ароматического замещения [например, реакция нитрования
Расстояние между Ru H Координата, реакции
п В
Рис. 3. Первичный изотопный эффект при замене водорода (протия) на
дейтерий:
(а) — гомолитическая диссоциация алкана. Разность между энергиями
диссоциации связей R— Н и R—D обусловлена разностью нулевых энергий
(преувеличена на рисунке); (б) —реакция, в которой нулевая энергия в
переходном состоянии частично сохраняется. Ер — £н—меньше, чем
D(R-D)-ZMR-H).
(113)] включает присоединение иона нитрония к
ароматическому ядру с образованием интермедиата
Уэланда 55, а не является альтернативным
синхронным процессом, проходящим через переходное
состояние 56;
56
переходное сдртоянив
78
В механизме (а) на медленной стадии, ведущей к
образованию интермедиата Уэланда 55, не
происходит значительного растяжения связи С—Н, тогда как
по альтернативному механизму (б) связь С — Н в
переходном состоянии 56 приблизительно наполовину
разорвана. Согласно этому, для реакции,
протекающей по механизму (б), в противоположность (а),
можно ожидать заметного изотопного эффекта.
Экспериментально бензол, содержащий очень небольшое
количество трития, был превращен в моно- и динитро-
бензолы. Обогащенных тритием продуктов
нитрования с точностью эксперимента не обнаружено; из
этого следует, что реакции проходят одинаково
быстро как по положениям С—Т, так и по С—Н. Эти
данные согласуются с механизмом (а) и не
согласуются с механизмом (б).
Примером реакции, в которой наблюдали
изотопный эффект, является окисление спиртов под
действием Cr(VI). В разбавленном кислотном
растворе изопропанол окисляется ионом НСгО~ до
ацетона со скоростью, пропорциональной произведению
[Спирт] [HCrOJ"][H+]. Если заменить изопропанол на
дейтерированное соединение (CbhhCDOH, то
скорость окисления уменьшается в семь раз.
Следовательно, связь углерод — дейтерий в дейтерирован-
ном изопропаноле должна быть в переходном
состоянии частично разорвана. Наряду с другими данными
этот результат подтверждает механизм (114),
включающий равновесие между спиртом и хроматом 57.
Вслед за этим протекает определяющий скорость
отрыв водорода (или дейтерия) от 67 под действием
основания с одновременным образованием двойной
связи С=0 и расщеплением связи хром—кислород,
как показано в (114) [роль основания в уравнении
(114) играет вода, однако им может быть при
некоторых условиях один из атомов кислорода хромат-
иона].
О.
Н3С H ОНз Н3С
(CKj)j,CHOH ^=fc Д) —*■ ^=0 + Нз6+НСЮ1
Н3С O-^-OjH Н3С
67 <"4)
79
Аналогичный изотопный эффект (около семи)
наблюдался в случае катализируемого основанием
бромирования ацетона, в котором, как полагают,
скорость определяющей стадией является отрыв
протона основанием [реакция (115)]:
ОН- Вг2
медленно
* СН3СОСН- £^£ СН3СОСН2Вг (115)
СНзСОСНз
(CH3COCD3)
Введение дейтерия в метальные группы изопро-
пилбромида не вызывает заметного изменения
скорости его 5^2-реакции (116) с этоксильным ионом,
однако конкурентный процесс элиминирования
[реакция (117)] в случае дейтерированного соединения
замедляется в 6,7 раза.
ЕЮ~(СН3)2^СН-^Вг —^EtO-CH(CH3)2 + Br*
[(CD3)2CH-Br]
(«в)
сн3
EtO~^-^CH^C-43r ► EtOH + СД2=СНСН3 + Br"
Н
CD3
D-CD2—С—Br
H
(117)
Использование изотопов других элементов
В принципе для изучения кинетического изотопного
эффекта могут использоваться изотопы и других
элементов помимо водорода. Из числа обычных
элементов, входящих в состав органических соединений,
можно применять изотопы 13С, 14С, 15N и 180.
Однако максимальный кинетический изотопный
эффект, который можно наблюдать в случае любого из
этих изотопов, составляет лишь 10% или около того
(в противоложность 700%, часто наблюдаемым для
дейтериевого изотопного эффекта). Все дело в том,
что замена изотопов в соединении (например, 12С на
80
,3С) приводит к гораздо меньшему в процентном
отношении изменению массы и нулевого уровня
энергии по сравнению с заменой *Н на 2D. Из этого
следует, что при определении изотопных эффектов для
элементов, отличных от водорода, требуются крайне
точные кинетические измерения, поэтому до сих пор
опубликовано относительно мало примеров таких
изотопных эффектов.
Резюме
После того как измерена скорость химической
реакции, проводят анализ ее зависимости от
концентрации реагентов. Выводы о механизме можно
получить в зависимости от того, является ли
порядок реакции по реагентам целочисленным,
дробным или смешанным или наблюдается более
сложный характер зависимости. Если можно
определить константы скорости элементарных реакций
при нескольких температурах, становится
возможным получение параметров уравнения Аррениуса,
которые сами по себе могут дать информацию о
механизме реакции или сделать выбор между
альтернативными механизмами. Наконец, изучение
кинетического изотопного эффекта может
показать, разрывается или нет данная связь на стадии,
определяющей скорость всего процесса.
Задачи
3.1. Как измерить скорость следующих реакций?
(а) С2Н5Вг + ОН" —► С2Н5ОН + Вг"
газовая
(б) СНзСООСНСНз — * СНзСООН + PhCH=CH2
| фаза
Ph
н+
(в) СНзСОСНз + Вг2 > СН3СОСН2Вг + НВг
3.2. Напишите схемы реакций, которые согласуются со
следующими наблюдениями:
а) разложение дибензилртути в инертных растворителях
приводит к образованию ртути и дибензила и показывает кинетику
первого порядка;
В)
б) разложение (Me3Si)2Hg в инертных растворителях дает
Hg + Me3SiSiMe3 и показывает кинетику второго порядка.
3.3. Скорость нитрования бензола смесью азотной и
уксусной кислот не зависит от концентрации бензола. Какой вывод
можно сделать из этого? „^
3.4. Ниже представлена упрощенная схема реакции
автоокисления углеводородов кислородом:
Инициатор —► 2R • (1)
быстро
R* + 02 > R—00. (2)
медленно
R-00 • + RH >• R . + ROOH (3)
Обрыв цепи происходит главным образом за счет реакции (4)
2R—00* —> R—00—R + 02 (4)
Можно предположить, что кислород присутствует в избытке.
Используйте метод стационарного состояния для описания
ожидаемой кинетики реакции. Почему стадия обрыва (4), которая
включает в себя два радикала R—00», по-видимому, является
гораздо более важной, чем альтернативные реакции обрыва цепи?
3.5. Предложите механизм или механизмы бензидиновой
перегруппировки (А->В), которые согласуются со следующими
фактами: (1) реакция имеет первый порядок по А и второй по
[Н+]; (2) реакция строго внутримолекулярна и (3) изотопный
эффект при замещении Н-атомов в пара-положении на дейтерий
не наблюдается.
0_NH_NH-0 J^ H2N-Q_0-NHa
А В
3.6. Аррениусский Л-фактор для газофазного пиролиза
первого порядка этилацетата равен 1012-5. Какой из двух
постулированных механизмов лучше согласуется с этим фактом?-
о--* он
Н3С-С СН2 ► СНз-С + СН2
о—сн2 о сн2
медленно J быстро
сн3соос2н5 v сЦ,соо+ сгн5--—*• сн3соон + с2н4
Глава 4
ИНТЕРМЕДИАТЫ
Только незначительное число наблюдаемых
химических реакций являются элементарными, т. е. такими,
в которых молекулы реагентов (не более двух или в
крайнем случае трех) взаимодействуют в одну
стадию с образованием молекул продуктов. Чаще
реакция представляет собой сложный процесс,
включающий две или более элементарные реакции с
образованием одного или более интермедиатов. Рассмотренные
в предыдущей главе кинетические характеристики
реакций часто указывают или даже доказывают,
что в реакции образуются некие интермедиаты.
Более того, иногда кинетические данные
позволяют предположить тип образующихся
интермедиатов.
В этой главе мы более детально рассмотрим
методы установления истинной природы интермедиатов.
Существуют два класса таких методов:
спектроскопические методы, в частности ЯМР-, ЭПР-, УФ-спект-
роскопия и масс-спектрометрия; с другой стороны,
интермедиаты могут быть идентифицированы либо
путем выделения самого интермедиата, либо —
более обычно — путем образования аддуктов при
добавлении к реагирующей системе соединения, которое
может взаимодействовать с постулируемым интерме-
диатом. В органических реакциях встречаются с
различными типами интермедиатов: ионы карбония, кар-
банионы, радикалы, карбены, бензины (производные
дегидробензола), тетраэдрические интермедиаты и
стабильные молекулы (перечисленные не в порядке
их важности). Рассмотрим каждый тип
интермедиатов в отдельности.
83
Карбониевые ионы, радикалы(с радикальным центром
на углероде) и карбанионы
Все эти три типа интермедиатов содержат три
группы, связанные с центральным атомом углерода.
Четвертая несвязывающая орбиталь содержит 0, 1 или
2 электрона в соответствии с типом интермедиата.
Стереохимические и полярные следствия образования
этих интермедиатов будут детально рассмотрены в
гл. 5 и 6 соответственно, здесь достаточно лишь
упомянуть, что образование любого из перечисленных
интермедиатов при асимметрическом центре может
привести к потере оптической активности.
Следовательно, потеря оптической активности в ходе
реакции является указанием на то, что процесс
возможно протекает через один из этих типов
интермедиатов. Однако этого еще недостаточно, чтобы можно
было сделать выбор между ними.
Все три типа интермедиатов будут вызывать
полимеризацию олефинов. Например, при добавлении к
реакционной смеси, в которой образуются радикалы,
олефина протекает цепная реакция, заключающаяся
в последовательном присоединении молекул
олефина к радикалу-инициатору. В конце концов это
приведет к образованию полимерной молекулы:
CH2=CHR'
R. + CH2=CHR' —> R—СН2—СН >•
—> R—СН2—СН—СН2—СН —> —► Полимер (118)
Я' R'
Полимер либо осаждается в ходе реакции, либо
его удается осадить метанолом после окончания
реакции, и поэтому его можно легко идентифицировать.
Однако не все олефины (мономеры) в одинаковой
степени полимеризуются под действием катионов,
радикалов и анионов. Например, радикалы и анионы
вызывают полимеризацию метилметакрилата, тогда
как. катионы не полимеризуют его. В
противоположность этому, стирол легко образует полимеры под
действием радикалов и катионов, но стабилен к
действию анионов. Предпочтительное (по сравнению со
стиролом) взаимодействие метилметакрилата с анио-
84
нами, вероятно, связано с тем, что в результате
атаки по СН2-группе метилметакрилата образуется
анион 58, стабилизованный за счет делокализации
отрицательного заряда на атоме кислорода (59).
Стабилизация этого типа сохранится и в ходе
дальнейшего присоединения аниона 58 к другой молекуле
метилметакрилата. При атаке анионом молекулы
стирола такая стибилизация будет отсутствовать.
О О
IT + СН2=С-С-ОСН3 —> R-CH2-C-C—OCH3 «->-
I I
СН3 СН3
58
О"
ч-> R—СН2— С=С-ОСН3 (119)
СНз
59
Различная реакционная способность различных
олефинов к действию указанных инициаторов
использована для создания метода определения типа ре-
акционноспособного интермедиата, образующегося в
реакции. К реакционной системе добавляют экви-
мольную смесь стирола и метилметакрилата. После
окончания реакции полимер осаждают метанолом и
анализируют сожжением для определения его
состава. Анионы реагируют с образованием полимера,
состоящего из фрагментов метилметакрилата; катионы
дают только полимер, включающий звенья стирола,
Радикалы приводят к сополимеру, который
содержит приблизительно равное число звеньев стирола и
метилметакрилата. Данные табл. 4 показывают, что
состав полимера и, следовательно, природу
инициатора можно без труда определить из результатов
элементного анализа, обычно с точностью ±0,2%.
Примером использования этого метода является
исследование природы' реакционных частиц,
образующихся при облучении олефинов у-лУчами. Показано,
что при использовании совершенно сухих олефинов
проходит катионная полимеризация. Однако в
присутствии даже следов воды за процесс
инициирования ответственны радикалы. Эти результаты означают,
85
Таблица 4. Сополимеризация метилметакрилата
и стирола в присутствии различных интермедиатов
Строение полимера
Содержание
углерода,
%
Содер
жание
водорода,
%
Инициатор
Полистирол 92,3 7,7 Катион
Поли(метилметакрилат) 71,4 9,6 Анион
Сополимер стирола и метилмет- 82,9 8,6 Радикал
акрилата (1:1)
что при облучении образуются и катионы, и
радикалы (и анионы). Но катионы являются гораздо
более реакционноспособными инициаторами
полимеризации. В присутствии следов воды катионы, в
отличие от радикалов, гибнут, так что полимеризация
начинает протекать по более медленному радикальному
механизму.
Перейдем теперь к рассмотрению методов
обнаружения и доказательства существования отдельных
типов интермедиатов.
Карбониевые ионы
Во многих распространенных органических
реакциях в качестве интермедиатов образуются ионы кар-
бония. Их образование уже отмечалось в реакциях
Sn\ при непосредственной ионизации органических
галогенидов в полярных растворителях. Соли
металлов, такие как хлориды ртути и железа, способствуют
ионизации за счет комплексообразования с галоге-
нид-ионом. К реакциям, в которых генерируются
карбониевые ионы, относятся также электрофильное
присоединение к олефинам, кислотная дегидратация
спиртов и реакция аминов с азотистой кислотой.
Я MP-спектроскопия
При растворении алифатических фторидов в пяти-
фтористой сурьме протекает обратимая реакция,
ведущая к образованию ионов карбония в
концентрации, достаточно высокой для их регистрации
методом ЯМР.
R-F + SbF6 z<==± R++SbF6" (120)
86
Спектры однозначно относятся к карбониевому
иону: все резонансные линии сдвинуты в сторону
слабых полей по сравнению с алифатическими галогени-
дами, как это и следует ожидать для мало
экранированных ионов карбония. В спектре трет-бутилкатио-
на присутствует синглет при б 4,35 млн-1, тогда как
спектр изопропилкатиона содержит дублет при
б 5,03 и септет при 6 13,5 млн-1, относящиеся к
протонам метильных и С—Н-групп соответственно.
Данные ЯМР подтверждают следующее: (а)
стабильность карбониевых ионов увеличивается в ряду:
первичные < вторичные < третичные; (б) карбоние-
вьге ионы легко перегруппировываются; так,
например, при растворении любых изомерных бутилфтори-
дов (1-, 2-, изо- и трет-бутил-) в пентафторсурьме
образуется исключительно трет-бутилкатион.
Метод химической фиксации (химической ловушки)
Карбониевые ионы легко взаимодействуют с нуклео-
филами, будь то растворитель (например, этанол)
или специально прибавленный в реакционную смесь
нуклеофил (такие как галогенид- или азид-ион).
Присоединение брома к этилену (в отсутствие
радикальных инициаторов) протекает, как полагают, по
механизму (121). Доказательство промежуточного
образования карбониевого иона 60 получено с
помощью метода химической ловушки в результате
прибавления к реакционной смеси хлор- и нитрат-ионов*
При этом наряду с нормальным продуктом, 1,2-диб-
ромэтаном, получаются соединения 61 и 62 *.
СН2=СН2
вР-вг >- сн2-сн2 5^вгсн2сн2вг
Вг
60 п- / v
^> ВгСН2СН:С1 (121)
61
^С BrCH2CH2ON02
62
* О структуре катиона 60 см, также с. 139.
87
Для сольволиза диэфира 63 постулируется
механизм, включающий образование промежуточного кар-
бониевого иона 64, что было доказано его фиксацией
этанолом с образованием орго-эфира 65.
/OS02C6H4CH3 /ч^.
^ОСОСНз ^^
63
Н20/АсОН |
\
Уж Лу
^ОСОСНз ^^
;с-сн3
о
64
EtOH
г
у0\/снз
^/ \oEt
(122)
65
В предыдущей главе отмечалось снижение
выхода спирта (без изменения скорости процесса) в
ходе сольволиза алкилгалогенидов в результате
добавления других нуклеофилов [реакция (59)]. Другое
логическое следствие образования карбкатиона
заключается в том, что, если два конечных продукта
образуются из одного и того же иона карбония,
отношение концентраций этих продуктов не должно
изменяться в зависимости от способа получения карбо-
ниевого иона. Так, хотя расщепление бензгидрилбро-
мида 66 в 90%-ном водном ацетоне при 50 °С в
присутствии 0,101 М азида натрия протекает в 335 раз
быстрее, чем в случае соответствующего хлорида 67,
соотношение спирта 68 и азида 69 не изменяется (с
точностью эксперимента), что указывает на
образование общего интермедиата — иона карбония 70 —
в обоих процессах сольволиза.
(123)
Ph2CHBr -
66
Ph2CHCl -
67
—> Ph2CH+ -
70
—► Ph2CHOH (66о/о)
68
—> Ph2CHN3 (34%)
69
88
Перегруппировки
Важным свойством ионов карбония является их ярко
выраженная склонность к скелетным
перегруппировкам, при которых происходит миграция арильной или
аллильной группы, или атома водорода, связанных с
атомом углерода, соседним с карбониевым центром,
как показано ниже:
К R
сРс+ —V £-i (124)
Хорошо известными реакциями, включающими
перегруппировку карбкатионов, являются
перегруппировка Вагнера — Меервейна, в упрощенном виде
представленная схемой (125), и пинаколиновая
перегруппировка (126).
СН3 <рнз
сн3-с-снг-с1 ;|gU сн3-с-сн2
СН3 СН3 (125)
71 \
ОН СН3 Н3С СН3
I + I \ _ /
СН3 С—СНд-^СНз "^ СН3 С—СН2 —► С*—0
сн3 сн3 н3с н
72
сн3сн3 сн3сн3 сн3сн3
сн3-с—c-CH3i£ сн3-с—с-сн3—► сн3-с—с-сн3
II' . I I + I T
он он +он2он ^ он
73
(t26)
CH3 CH3 CH3
СНз-С-СО-СНз-^ СН3-С—С-СНз-^^ СН3—С—С-СН3
сн3 <:н3он сн3он
74а 746 '
В реакции (125) первичный неопентильный кар-
бониевый ион 71 перегруппировывается в более ста-
89
бильный третичный ион 72. В реакции (126)
первоначально образующийся карбкатион 73 — третичный,
но образовавшийся в результате перегруппировки
катион 74 обладает дополнительной стабилизацией за
счет делокализации заряда. Изомеризация я-пропил-
катиона в изопропильный при растворении я-пропил-
фторида в пятифтористой сурьме (см. предыдущий
раздел), очевидно, связана с миграцией гидрид-
иона:
и
сн3 -сРсНо —>. сн3 -с-снз (т)
и н
Еще легче, чем алкильные группы и водород,
мигрируют арильные группы.
В случае'реакций, включающих промежуточное
образование карбанионов или радикалов,
перегруппировки являются чрезвычайно необычным явлением.
Поэтому если в ходе реакции имеет место скелетная
перегруппировка, это доказывает (хотя и не
окончательно) промежуточное образование иона карбония.
С другой стороны, если для реакции постулируется
промежуточное образование иона карбония,
склонного к перегруппировке (т. е. соседние группы могут
мигрировать с образованием катиона равной или
большей стабильности), и вместе с тем продукт
перегруппировки отсутствует, то в таком случае
свободные ионы карбония,- по-видимому, в реакции не
образуются, так как перегруппировки карбониевых
ионов происходят довольно легко.
Карбанионы
Карбанионы довольно часто встречаются в качестве
интермедиатов в реакциях, где сильное основание
может отрывать протон от связи С—Н органической
молекулы. Углеводороды инертны к такой атаке*, одна-
* Автор ошибается, см., например, Э. Стрейтвизер мл.,
Дж. Хеммонс. Новые проблемы физической органической химии.
Пер. с англ. Под ред. И. П. Белецкой. М., «Мир», 1969, с. 7—
35. — Прим. ред.
90
ко введение в молекулу таких групп, как СО или
—COOEt, активирует соседние связи С—Н и
облегчает атаку основанием. Активирующий эффект кетон-
ной и эфирной группы рассматривают как результат
стабилизации образующегося карбаниона за счет де«
локализации- заряда (например, 75). Две кетонные
или эфирные группы оказываются более
эффективными, чем одна: тогда как равновесие (128)
сдвинуто влево, равновесие (129) в значительной степени
сдвинуто вправо и 76 фактически можно получить в
виде его натриевой соли.
О
CH3COOEt + EtO~ ч=* ЕЮН + ~СН2—С—OEt ч->
75а
от
I
ч-> СН2=С—OEt (128)
756
CH3COCH2COOEt + ЕЮ" *=*
О О
II - II
*=fc ЕЮН + СНз—С—СН-С—OEt *->
76
0 0" О" О
II I I II
ч-> СНз—С—СН=С—OEt «-► СН3—0=СН—С—OEt (129)
Винильная группа также может стабилизировать
карбанионный центр, хотя и не в такой степени, как
кетогруппа. Если циклогексадиены-1,3 или -1,4
нагревать при 95 °С в смеси трет-СьНпОК и трет-
C5H11OD в течение времени, достаточного лишь для
достижения малой степени превращения [реакция
(130)], то образуется смесь монодейтерированных
соединений 77 и 78 с одним и тем же соотношением
4 : 1 в обоих случаях. Поскольку это соотношение не
отвечает равновесной смеси, это указывает на
образование общего интермедиата из обоих исходных
91
веществ, которым, почти наверняка, является карб-
анион 79.
6
7 7 (4 части) (13о)
О.
78 "
(1 часть)
Карбанионы, в которых отрицательный заряд
находится на sp-гибридизованном атоме углерода,
также стабилизованы, причем лучше, чем анионы с
sp2- и 5р3-гибридизованным атомом углерода, так
как sp-гибридизованный атом углерода более
электроотрицателен, чем sp2- или 5р3-атомы углерода, и
поэтому может лучше удерживать отрицательный
заряд. Благодаря этому терминальные ацетилены легко
дают натриевые соли. Карбанионы образуются также
при реакциях металлоорганических соединений.
Основной способ химической фиксации карбанио-
нов состоит в их присоединении к поляризованным
кратным связям. Так, карбанионы легче инициируют
полимеризацию метилметакрилата, чем стирола (см.
вводную часть настоящей главы, с. 84). Карбанионы
легко присоединяются также к положительно
поляризованному атому углерода карбонильных групп.
"На этом основании считается, что альдольная
конденсация включает присоединение карбаниона к молеку-»
ле альдегида или кетона:
он- - rcho н2о
R—СН2СНО *=* R—СН ► R—СН—СН—О" <=*
1 I I
СНО СНО R
3=* RCH-CH-OH (131)
I I
CHOR
Кажется маловероятным, что в процессе декарб-
оксилирования а-карбоновых кислот пиридина и хи-
mpem-C5HnOD
^ трет-СъИиОК
е
.рет-СъНпОТ) 7y
трет- C5HltOK
92
нолина (например, пиколиновой кислоты 80) может
возникнуть карбанион. Однако значительно более
легкое по сравнению с Р- и у-карбоновыми
кислотами декарбоксилирование а-кислот позволяет
предположить, что атом азота гетероциклического ядра
может отщеплять протон от соседней карбоксильной
группы с образованием цвиттер-иона 81. Этот ион,
вероятно, может терять С02 с образованием карбанио-
иа 82, который за счет протонного переноса должен
быстро таутомеризоваться в пиридин 83.
Промежуточное образование карбаниона 82 было изящно
показано путем проведения реакции в присутствии
ацетофенона, что привело к образованию аддукта 84
за счет присоединения карбаниона к атому углерода
карбонильной группы.
а
i
н—о
80
Радикалы
Свободнорадикальные интермедиаты часто
наблюдаются в реакциях, проводимых при высоких
температурах, особенно в газовой фазе или в неполярных
растворителях. Радикалы могут возникать в
реакционной системе за счет фотолиза или пиролиза
подходящих молекул, как было обсуждено в предыдущей
главе. Кинетические зависимости, свидетельствующие
в пользу промежуточного образования свободных
радикалов, часто имеют дробный порядок по
концентрации реагентов.
93
Спектроскопия
Радикалы очень быстро гибнут в результате
рекомбинации, поэтому концентрация этих интермедиатов
редко достигает величины выше примерно 10~8 М.
Это обстоятельство в большинстве случаев исключает
использование ультрафиолетовой, инфракрасной и
ЯМР-спектроскопии для их обнаружения. Однако
спектроскопия электронного парамагнитного
резонанса, которая очень чувствительна к
парамагнитным частицам и позволяет регистрировать
концентрации радикалов вплоть до ~ 10~9 М, представляет
собой чрезвычайно полезный метод обнаружения
частиц с неспаренным электроном (молекулы, в которых
отсутствуют неспаренные электроны, не
регистрируются в этом виде спектроскопии). Сигнал
электронного парамагнитного резонанса является
убедительным доказательством присутствия в системе
свободного радикала, хотя и ничего не говорит об
источнике образования радикала (каким путем, в какой
-реакции он возник). В случае простых органических
радикалов однозначное структурное отнесение часто
можно провести на основе анализа сверхтонкого
расщепления или путем сравнения спектра с известным
ЭПР-спектром радикала, который был получен
другим путем.
В настоящее время считают, что автоокисление
углеводородов атмосферным кислородом является
радикально-цепным процессом, в котором в качестве
первого стабильного продукта образуется гидропер-
оксид. Проиллюстрируем эту реакцию на примере
изопропилбензола (кумола) 85. Радикалы в системе,
по-видимому, образуются за счет реакции (133), а
цепной процесс [реакции (134) и (135)]
обусловливает превращение углеводорода в его гидропероксид.
PhCH(Me)2 + 0=0 —^ PhC(Me)2 • + • ООН (133)
85 86
PhC(Me)2 • + 0=0 —> PhC(Me)2—00 • (134)
87
PhC(Me)2-00. + PhCH(Me)2 —>
—> PhC(Me)2—ООН + PhC(Me)2 • (135)
88
94
Когда эту реакцию осуществили в ячейке ЭПР-
спектрометра, то обнаружили синглетный сигнал,
характерный для радикала 87. Поскольку радикал 86
должен иметь очень сложный спектр, полученный
результат одновременно подтверждает
радикально-цепной механизм автоокисления и означает, что реакция
(134) протекает быстрее по сравнению с реакцией
(135), так как в противном случае наряду со
спектром 87 или даже вместо него должен наблюдаться
спектр радикала 86.
ХДПЯ (химически индуцированная динамическая
поляризация ядер)
Обычно свободные радикалы нельзя зафиксировать
с помощью ЯМР из-за их низкой концентрации и
парамагнетизма, который приводит к очень широким
резонансным линиям. Однако, можно ожидать, что
продукты, образующиеся по радикальной реакции,
обладают характеристическими спектрами ЯМР.
Поскольку неспаренный электрон за счет магнитного
момента может поляризовать ядерные спины
протонов в радикале, продукты радикальной реакции
могут иметь распределение по уровням ядерной
энергии, заметно отличающееся от нормального больц-
мановского распределения. Это обстоятельство
приводит к значительным различиям в
интенсивности сигналов наблюдаемого ЯМР-спектра. Если
перезаселенными оказываются низшие
энергетические уровни, то происходит увеличение поглощения
радиочастотной энергии, в то время как при
перезаселенности высших уровней имеет место эмиссия
энергии и в ЯМР-спектре появляются отрицательные
пики.
В период написания этой книги метод ХДПЯ все
еще находился на стадии разработки, в связи с чем
этот метод в большинстве случаев применялся для
реакций, в которых промежуточное образование
радикалов было доказано другими путями. Однако
существует один пример — термическая
перегруппировка соединения 89 в более стабильную незаряженную
:труктуру 90 — для которого на основании эффекта
ХДПЯ был предложен радикальный механизм вместо
95
первоначально принятого ионного. До того как было
проведено изучение методом ХДПЯ, считали, что
перегруппировка протекает либо по согласованному
механизму, либо через «тесную» ионную пару
[реакция (136)]. Однако изучение методом ХДПЯ
показывает сильный положительный эффект для бензиль-
ных протонов в продукте 90, (помечены звездочкой),
наводя на мысль, что перегруппировка включает го-
молиз связи азот—углерод с последующей клеточной
рекомбинацией радикалов, как показано на схеме
(137).
Me2N—N-СОСНз
+СН2
(136)
Me2N—NCOCH
I
сн2
Me2N—N-СОСНз
(♦) I (*)
н—с—н
Me2N—N—СОСНз ■<-► Me2N—N—СОСН3
сн2
(137)
ХДПЯ кажется многообещающим методом для
регистрации и изучения радикальных интермедиатов,
96
Радикальные ловушки и родственные методы
На образование в системе свободных радикалов
часто указывает присутствие среди продуктов
реакции небольших количеств димеров. Образование в
качестве побочного продукта фторирования метана
C2F6 уже упоминалось как доказательство
промежуточного образования в реакции радикалов *С¥г.
С другой стороны, отсутствие димера указывает, что
соответствующий радикал может не присутствовать
(хотя следует помнить, что в цепных реакциях
количество образующихся продуктов рекомбинации может
быть чрезвычайно мало). Например, фенильные
радикалы, возникающие при нагревании пероксида бен-
зоила, взаимодействуют с ароматическими
соединениями с образованием фенилированных продуктов 91.
Отсутствие среди продуктов биарильного соединения
Аг—Аг отвергает схему (138),, которая включает в
себя радикалы Аг», и делает предпочтительной
альтернативную схему реакции (139). Другим
доказательством схемы (139) является факт
выделения в ряде случаев димера промежуточных
радикалов 92.
(PhCOO)2 j, PhH + Аг» -^V Ph-Ar (138)
]нагре-
увание
Ph*+ArH
Ph—Ar + R—H (139)
Ph y=\ /=\ Ph
■ О 1Q<&
ArH
R-любой радикал
Свободные радикалы можно зафиксировать
также добавлением к реакционной системе соединений,
охотно реагирующих с ними: например, добавлением
брома или оксида азота. Так, при нагревании азобис-
изобутиронитрила 93 [R = (CH.o)2C(CN)] в
растворе в присутствии оксида азота образуется
4 За к, 1044
97
производное гидроксиламина 94, вероятно в
результате присоединения трех 2-цианопропильных-2
радикалов к молекуле оксида азота.
ц—N=N—R —► R- + N2 + R-
93
R (НО)
NO + 3R. —► —> ;N-OR
W
94
Однако такие аддукты не всегда легко выделить
из реакционной смеси. Метод спиновых ловушек
позволяет избежать выделения продукта. Этот метод
заключается в добавлении к реакционной системе
молекул, взаимодействие которых с реакционноспо-
собным свободным радикалом будет приводить к
образованию более стабильного радикала. Такой
молекулой является 2-метил-2-нитрозопропан 95,
присоединение которого к нестабильным интермедиатам
R • дает относительно стабильный N-оксидный
радикал 96, для которого можно зарегистрировать
ЭПР-спектр. Исходя из наблюдаемой сверхтонкой
структуры спектра, можно получить информацию
о строении R*.
rper-BiK
трет-Ви—N=0 + R • —► ^N—0- (141)
W
95 96
Выше в этой главе (см. с. 84) был рассмотрен
еще один важный метод определения присутствия
радикалов в системе, основанный на введении в
реакционную смесь способного к полимеризации оле-
фина с последующим анализом образующегося
полимера.
Ингибирование
В радикально-цепных реакциях, например в
реакциях (99) и (100), на стадиях развития цепи
происходит превращение молекул реагентов в молекулы
продуктов с одновременной регенерацией радикала-
инициатора. Если в реакционной системе
присутствует соединение,.способное к взаимодействию с этим
№
радикалом с образованием либо нейтральной
молекулы [например, реакция (142)], либо радикала,
который слишком стабилен для того, чтобы
продолжать цепной процесс [например, 97 в реакции
(143)], то скорость цепной реакции уменьшится или
она вообще остановится. Соединения, которые
действуют таким образом, называются ингибиторами; к
ним относятся оксид азота, олефины, фенолы,
ароматические амины и нитросоединения.
R. + N=0 —► R—N=0 (142)
ОН 0.0
R-H + P I ч-> || | и т. д. (143)
ОН ОН ОН
97
стабилизованный
нереакционноспо-
собиый раликал
Примером реакции, чувствительной к ингибирова*
нию, является пиролиз насыщенных углеводородов.
Скорость пиролиза может снижаться (часто на
пять порядков или около того), если реакции
проводить в присутствии оксида азота. Считают, что
пиролиз углеводородов инициируется за счет разрыва
углерод-углеродных связей. Образующиеся при этом
радикалы реагируют в различных направлениях,
включая атаку молекул углеводородов, что ведет
к цепной реакции. Такой процесс приведен ниже на
примере пиролиза этана (реакции обрыва цепи
опущены). Выведение из сферы реакции алкильных
радикалов с помощью оксида азота уменьшает общую
концентрацию радикалов в системе и, следовательно,
снижает скорость расщепления молекул алкана в
реакциях, таких как (147).
В
ва
СНз—СНз
СНз • + СНз—СНз
С2Н5 •
Н • + СНз—СНз
противоположность
НИИ
—►
—>
—►
—►
2СНз«
сн4 + с2н5.
СгН4 + Н •
Н2 + C2H5 •
пиролизу алканов
(144)
(145)
(146)
(147)
при на-
этил- и более высших алкилацетатов
4*
99
протекает молекулярная реакция, приводящая к
уксусной кислоте и соответствующему алкену.
Кинетически эти реакции имеют первый порядок и не ингиби-
руются при добавлении циклогексена.
.О а Н
СН3-О ^ (cHY —►■ СН3СООН + CHX-CHY
о^-снх
(148)
Аргументы, основанные на ингибировании
реакций, следует применять с осторожностью, так как
ингибиторы одной реакции могут служить
промоторами или инициаторами для других реакций.
Например, оксид азота ингибирует пиролиз алканов, но
вызывает пиролиз ацетальдегида. В качестве примера
ошибочного вывода, полученного из результатов
экспериментов по ингибированию, приведем
автоокисление триалкилборанов. Было найдено, что этот
процесс не ингибируется производными гидрохинона. На
этом основании было высказано предположение, что
автоокисление боранов имеет молекулярный
механизм (149), но не является цепным процессом
[реакции (150) и (151)], аналогично автоокислению
углеводородов [реакции (134) и (135)].
R О R—О
I + II —> I (Н9)
R2B О R2B—О
R* + 02 —> R—00* (150)
R-OO. + R3B —> R-00-BR2 + R. (151)
трет-Ви трет-Ви
СН—/ \—0. + R
трет-Ви
97а
трет-Ви
трет-Ви трет-Ви
OR (152)
100
Однако в действительности более мощные
ингибиторы, такие как гальвинойсил 97а, ингибируют
автоокисление триалкилборанов. Кроме того, тот факт,
что оптически активные органобораны рацемизуются
в процессе автоокисления (сравни следующую главу,
с. 134), также указывает, что радикальный механизм
более вероятен, чем молекулярный. Тот факт, что
автоокисление триалкилборанов не ингибируется
гидрохиноном, можно связать (с учетом других данных)
с реакционной способностью радикала 98. Так как
этот радикал с трудом отрывает водород от алканов,
то соответствующий гидрохинон должен быть
хорошим ингибитором автоокисления алканов. Однако
радикал 98 обладает реакционной способностью,
достаточной для замещения алкильного радикала в
молекуле триалкилборана [реакция (153)]. Этот
процесс снова приведет к цепной реакции и,
следовательно, производные гидрохинона не будут
действовать в качестве ингибиторов автоокисления
триалкилборанов.
НО—\\—О • + R3B —► НО—(~Л—OBR2 + R . (153)
Инициирование
Скорость свободно-радикальных реакций ~часто
можно увеличить при введении в реакционную смесь
соединений, называемых инициаторами или
промоторами, которые дают при нагревании или фотолизе
радикалы. В качестве термических инициаторов
часто используют азосоединения и пероксидные
соединения; кетоны и ряд других соединений образуют
радикалы при фотолизе. Как уже отмечалось ранее,
оксид азота ингибирует пиролиз алканов, но
инициирует пиролиз альдегидов (вероятно, за счет отрыва
относительно слабо связанного атома водорода
группы СНО):
СНзСНО+NO —► СНзСО. + HNO (154)
СНзСНО
СН3СО* —> СО+.СНз ► СН4 + СН3СО. (155)
Увеличение скорости в результате добавления к
реакционной смеси инициатора радикалов не может
101
рассматриваться как однозначный критерий
радикального механизма реакции. Реакция может быть
ионной или молекулярной, а возрастание ее скорости
при добавлении радикального инициатора может
быть связано g совершенно иной реакцией, которая
не протекает в его отсутствие. В качестве примера
рассмотрим присоединение бромистого водорода
к чистым алкенам, приводящее при исключении света
или радикальных катализаторов к алкилбромидам.
Скорость реакции не снижается в присутствии
ингибиторов. Этот факт можно с уверенностью
использовать в качестве доказательства того, что реакция
не протекает по свободно-радикальному механизму.
Действительно, считается, что реакция включает
промежуточное образование иона карбония:
R—СН=СН2 + НВг —> RCH—СНз + Вг- —>
—► RCH-СНз (156)
Вг 99
Скорость расхода олефина значительно
возрастает в присутствии свободно-радикальных инициаторов
(кислород, пероксидные соединения и т. д.). Однако
это связано не с тем, что реакция (156) имеет
радикальный механизм, а с протеканием совершенно иной
радикально-цепной реакции (157).
р НВг
Br. + RCH=CH2 —► RCH—CH2Br *
—> RCH2CH2Br + Br • (157)
10'J
В случае этилена и симметричных внутренних
олефинов эти реакции нельзя различить на
основании их продуктов. Однако если используется
терминальный олефин, реакции (156) и (157) можно
различить по их продуктам: вторичный бромид 99
образуется по карбониево-ионной реакции, а
первичный бромид 100 — по радикально-цепному процессу
(см. также с. 153, 154).
Карбены
Карбены представляют собой нейтральные реакцион-
носпособные частицы, в которых центральный атом
углерода ковалентно связан лишь с двумя группами,
102
а два несвязанных электрона сохраняют
электронейтральность. Эти два электрона могут быть
спаренными или неспаренными, что вызывает некоторые
различия в их реакционной способности. Карбены
можно генерировать несколькими путями: например,
фотолизом диазоалканов или взаимодействием гало-
формов с сильным основанием.
+ - hv
R—CH=N=N >■ R—CH: (158)
трет-ВиО' + НСС13 +=* трет-ВиОН + СС1J —>
—у :СС12 + С1" (159)
Карбены — чрезвычайно реакционноспособные
частицы и по многим свойствам похожи на радикалы.
Наиболее распространенные методы обнаружения
карбенов основаны на их способности образовывать
две новые ковалентные связи. Карбены :CR2 внедря-
ются по —С—Н-связям органических молекул с
образованием продуктов типа —С—CR2 —H. К
сожалению, эта реакция не обладает селективностью по
отношению к различным связям С—Н в
органической молекуле, что ограничивает ее диагностическое
использование, так как обычно образуется сложная
смесь продуктов. Однако карбены внедряются и по
связям Si — Н, благодаря чему такое соединение, как
триэтилсилан, представляет собой хорошую ловушку
для карбенов, поскольку в этом случае образуется
только один продукт.
Et3Si—H + R2C: —> Et3Si—CR2—H (160)
Триэтилсилан не удается использовать в качестве
ловушки карбенов, если реакцию проводят в
присутствии основания, так как при этом силан легко гид-
ролизуется до силанола HOSiEt3 и водорода. В таких
случаях лучше использовать другой метод,
основанный на способности карбенов образовывать
производные циклопропана за счет присоединения по углерод-
углеродной двойной связи. Часто (в этом методе) в
качестве ловушки карбенов применяют стирол, цик-
логексен и октагидронафталин 101. Реакцию можно
103
использовать как удобный метод синтеза циклопро
Пановых соединений.
101
CHCls + rper-BuO" —> СС12: ► | , |>< (161)
102
Другое доказательство, что, по крайней мере,
в некоторых случаях образуются истинные свободные
карбены и реакции не обусловлены их
предшественниками, было получено в случае дихлоркарбена,
который генерировали различными путями, включая
реакцию (159) и пиролиз хлороформа, приводящий
к :СС12 и хлористому водороду. Дихлоркарбен
вводили в конкурентное взаимодействие со смесью оле-
финов. В случае нескольких различных пар олефи-
нов соотношение продуктов оказалось одинаковым,
независимо от способа получения дихлоркарбена.
Этот результат является хорошим доказательством
того, что дихлоркарбен действительно представляет
"собой свободную частицу. И верно, если бы в
реакциях участвовали его предшественники, различные
методы получения привели бы к различным
соотношениям продуктов.
Производные дегидробензола
Эти реакционноспособные интермедиаты
представляют собой дегидроароматические соединения. Сам де-
гидробензол (бензин) 103 может быть представлен
структурой с формальной тройной связью, хотя она,
вероятно, вносит очень небольшой вклад в
стабилизацию молекулы.
(162)
Доказательство промежуточного образования
производных дегидробензола, основанное на необычном
104
характере ароматического замещения, уже
упоминалось. Другим доводом в пользу существования
производных дегидробензола является факт
обнаружения с помощью масс-спектрометрии короткоживущего
иона с т/е 76, образующегося при фотолизе
соединения 108, а также результаты экспериментов по
химической фиксации (ловушкам). По способности
образовывать две новые связи с молекулами реагентов
производные дегидробензола до некоторой степени
аналогичны карбенам. Хорошим примером является
реакция производных дегидробензола с 1,3-диенами,
приводящая к образованию шестичленных
циклических соединений. Эта реакция аналогична реакции
Дильса — Альдера, в которой роль активированного
олефина играет дегидробензол. Так, дегидробензол,
полученный при действии NH~ на РЫ (или другим
путем), присоединяется по положениям 9, 10
антрацена с образованием триптиЦена 104. В качестве
ловушки (фиксатора) производных дегидробензола
наиболее широко применяется соединение 105,
которое с элиминированием оксида углерода дает
производные нафталина..
105
Промежуточное образование «свободных»
бензинов (но не их предшественников, которые реагируют
аналогичным образом) было установлено в случае
самого дегидробензола при его конкурентном
взаимодействии со смесью фурана и циклогексадиена-1,3
[реакции (165) и (166)]. Дегидробензол генерировался
четырьмя различными путями при пиролизе
соединений 108—111. Во всех случаях отношение /CiesMee
оказалось почти идентичным (21,5 ± 0,9). Это
указывает на то, что во всех реакциях присутствовал один
и тот же интермедиат — свободный дегидробензол.
n: ^ J<
'2
Ч/Чсоо- ^^Чо, ^ ^и
108 109 110
^
MgBr
ill
Тетраэдрические интермедиа™
Считается, что ряд важных реакций карбонильных
соединений (альдегидов, кетонов, карбоновых кислот
и их производных) протекает через присоединение
реагента к атому углерода карбонильной группы
с образованием тетраэдрического интермедиата
(общая схема, заряды опущены):
;с=о + х —> X <167)
/ / \х
112 113
Если 112 — кетон или (в особенности) альдегид,
то интермедиа™ типа 113 относительно стабиль-
106
ны; эти интермедиаты или полученные из них
насыщенные продукты даже могут быть выделены. Если
112 — карбоновая кислота, сложный эфир, амид
и т. п., интермедиаты типа ИЗ более реакционно-
способны и их идентифицировать (и зафиксировать)
труднее. Однако даже в случае реакций производных
карбоновых кислот имеются неопровержимые
доказательства образования структур типа 113 как
истинных интермедиатов, а не как переходных
состояний.
180 180-
// но- i
R-C ^=± R-C-OH
OR' OR'
114
115 \ 109
\\ R-C-OH + OR (168)
О 18ОН «»
// ^ I
R-C +± R-C-O
\ 18 _ i
OR'"H °" OR'
116 117
О ОН ОН ОН ОН
R-C + H+^R-C(+ ^±rR-C-OH2^R-C-OH —► R-C(+
OR' OR OR HOR у ОН
+
^ #
oh2 ,o
(169)
R-C-OH R-C +H
I , \
OR OH
119
Наиболее веским доказательством
промежуточного образования тетраэдрических соединений 115
в ходе гидролиза сложных эфиров [реакция (168)]
является тот факт, что если реакцию обмена эфира,
меченного 180 по карбонильной группе, прервать до
ее завершения, то остаточный эфир будет содержать
меньше 186, чем исходный эфир, за счет обмена
180 карбонильной группы эфира с растворителем.
107
Такой обмен не мог бы произойти, есяи бы 115 был
только переходным состоянием между 114 и 118
и в соответствии с простейшим объяснением 115
является истинным интермедиатом, который в
результате перемещения протона может переходить
в 117. Последний, в свою очередь, может терять
18ОН~, превращаясь в немеченый эфир 116.
Аналогичные аргументы справедливы и в случае
катализируемого кислотами гидролиза [реакция (169)], для
которого также можно показать потерю 180
карбонильной группой в ходе реакции.
Интермедиаты типа 115 или 119 не были с
определенностью идентифицированы или
зафиксированы в простых реакциях этерификации или
гидролиза эфиров. Тем не менее дополнительные доводы
их промежуточного образования были получены из
результатов родственных реакций. Так, с помощью
ИК-спектроскопии по уменьшению интенсивности
полосы карбонильной группы было показано
образование 120 в обратимом процессе (170), который
протекает при обработке этилтрифторацетата этила-
том натрия в ди-«-бутиловом эфире:
л О" Na+
CF3-C^ + ЕЮ" Na+ ^=± CF3—С—OEt (170)
^0Et Jvr,
OEt
120
До сих пор не удалось зафиксировать тетраэдри-
ческие интермедиаты при гидролизе сложных эфиров
или в родственных реакциях .за счет присоединения
к интермедиату других молекул. Однако реакцион-
носпособные группы, находящиеся внутри той же
молекулы, могут осуществлять такое взаимодействие
и тем самым доказывать образование подобного ин-
термедиата. Так, доказательством образования ин-
термедиата 122 в ходе аммонолиза эфира 121 до
амида 123 (который в условиях реакции
подвергается циклизации в 124) служит нахождение среди
продуктов реакции соединения 125, образующегося за
счет нуклеофильного замещения атома брома в 122
группой —О",
109
с
о
NH3
Br [CHgCN]
/
Ph CH2—CH2
121
Ph CH2—CH2
122
(171)
Ph CONH2
123
Ph CONH2
124
Наконец, существует много кинетических
доказательств в пользу образования тетраэдрических интер-
медиатов в этих реакциях. Рассмотрим только один
пример. Скорость катализируемого основаниями
гидролиза N-метилацетанилида ~в воде при 25 °С
описывается уравнением, в которое входят члены с
первым и вторым порядком по концентрации основания
(ср. с автоокислением — восстановлением
альдегидов, с. 64). Крайне трудно понять присутствие в
кинетическом выражении члена с квадратом
концентрации основания, если не предположить схему
реакции (172), включающую интермедиат 127, который
в условиях очень высокой основности может терять
протон с образованием двухзарядного аниона 128.
СНз—С
\
он-
СН
О"
,-£-
I
ОН
127
NMePh
NMePh
ОН'
t
О"
I
СНз-C-NMePh
L
О
СНзСОО" + PhNHMe (172)
128
109
Применение принципа стационарного состояния к
схеме (172) приводит к уравнению скорости
гидролиза в виде:
- d [126]ЛИ = кх [126] [ОН"] + к2 [126] [ОН"]2
Легче получить доказательство образования тет-
раэдрических интермедиатов в реакциях альдегидов
и кетонов, так как в некоторых случаях образуются
стабильные продукты присоединения, такие как
хлоральгидрат СС1зСН(ОН)2, и цианогидрины
RCH(OH)CN. Однако во многих реакциях
образующиеся тетраэдрические интермедиа™ из кетонов
и альдегидов быстро теряют воду; типичным
примером является конденсация гидроксиламина с
ацетоном:
СНзЧ
V=0 + NH2OH
ОН
О" -1
I +
СНз— C-NH2OH
13
129
СНз'
\
+=t СНз—C—NHOH >• ;C=NOH (173)
I -н2о сн/
L.ri3
130
При этом скорость исчезновения ацетона,
оцененная по его УФ-спектру, выше, чем скорость
появления оксима 130. Следовательно, в реакции должен
образовываться интермедиат, которым, по-видимому,
является 129. Дополнительное доказательство
образования интермедиата было получено при изучении
влияния рН среды на скорость реакции. Найдено,
что максимальное значение скорости достигается при
рН 4,5; при переходе как к более кислым, так и к
более основным средам скорость падает. Такое
поведение характерно для реакции, протекающей по
крайней мере через две последовательные стадии и,
следовательно, включающей образование
интермедиата, поскольку если бы реакция была простым
процессом, катализируемым кислотами, то ее
скорость непрерывно увеличивалась бы с ростом
кислотности среды. Наблюдаемая зависимость согласуется
НО
с механизмом (173). В нейтральных или основных
средах равновесие между исходными соединениями
и 129 устанавливается быстро и скорость
определяющей стадией является катализируемая кислотой
дегидратация с образованием оксима 130. В кислых
растворах NH2OH находится в протонированной
форме [NH3OH]+, которая не может
взаимодействовать с кетоном. Поэтому при достаточной
кислотности раствора определяющей скорость стадией
становится присоединение оставшегося непротонированного
NH2OH, за которой следует быстрая стадия
катализируемой кислотой дегидратации. Следовательно,
в кислом растворе скорость превращения должна
быть ниже максимальной из-за низкой концентрации
свободного гидроксиламина. С другой стороны,
в сильноосновной среде скорость образования оксима
будет снижаться вследствие низкой концентрации
протонов в растворе.
Кислотно-Ьсновной катализ
Большое число реакций катализируется кислотами
или основаниями. В типичных примерах, таких
как катализируемый кислотами гидролиз сложных
эфиров [реакция (169)], реакция протекает через
ряд последовательных стадий. Многие из них
включают только переход протона от одной молекулы
к другой или миграцию протона внутри молекулы,
т. е. процессы, которые не требуют большей энергии
активации (если они не очень эндотермичны). В
итоге суммарная реакция, протекающая через
несколько стадий, часто оказывается более быстрой, чем
возможные конкурентные некаталитичэские
процессы. Например, для гидролиза сложных эфиров
можно предложить два некаталитических
направления:
СНз-С^ 5F=fc СНзСГ >
X)Et "OEt
—► СН3СООН + ЕЮН (174)
111
СН3-(Г —> СНз-Cf +ЕЮН (175)
/ \ Х0Н
ног ;oEt
\ /
н
Прямой гетеролиз [реакция (174)] должен
проходить с громадной энергией активации, так как
образующиеся ионы не обладают специфической
стабилизацией. Процесс (175) запрещен с точки зрения
орбитальной симметрии (см. гл. 5, с. 143), и для него
также можно ожидать высокой энергии активации.
Поэтому катализируемая кислотой реакция (169),
которая не включает стадий, характеризующихся
особенно высокой энергией активации *, является
более вероятной, чем оба приведенных более прямых
процесса.
Некоторые реакции (в воде) специфически
катализируются Н30+. Иные реакции катализируются,
кроме того, другими кислотами, присутствующими
в системе. Эти два типа взаимодействия называются
специфическим и общим кислотным катализом.
Аналогично, возможен специфический и общий основной
катализ. Наличие специфического или общего
катализа проливает свет на детали механизма. Например,
ряд катализируемых кислотами реакций включает
присоединение протона к субстрату, за которым
следуют дальнейшие реакции аддукта.
(а) . (б)
Н+ + Х ч=ь НХ+ ► Продукты (176)
(—а)-
Н+ + Х +=± НХ+ 1
* Продукты (177)
НА + Х *=>: А" + НХ+ '
Если между субстратом и его протонированной
формой НХ+ устанавливается равновесие [реакция
(176)] [что происходит тогда, когда оба процесса
* Переход протонов к молекуле эфира и обратный процесс
следует в действительности рассматривать как перемещение
протона от Н30+ к молекуле эфира и обратный переход Н+ к
молекуле воды. Следовательно, одна связь рвется и одна
образуется, что делает переход приблизительно термонейтральным.
112
(а) и (—а) много быстрее по сравнению с (б)], то
концентрация НХ+ будет зависеть только от
кислотности (рН) раствора. Следовательно, наблюдается
специфический кислотный катализ. Если скорость
стадии (б) сравнима со скоростью стадии (—а), то
НХ+ не будет достигать ее равновесной величины.
В этом случае начинает играть роль скорость, с
которой протоны присоединяются к X. Донором
протонов может быть либо Н30+, либо другие
присутствующие в системе кислоты. Следовательно, в
кинетическом уравнении будут присутствовать члены,
включающие концентрацию всех кислот данной
системы, т. е. имеет место общий кислотный катализ.
Например, гидролиз ортоэтилацетата 48 [см. гл. 3,
реакция (78)] представляет собой случай общего
кислотного катализа, тогда как обе довольно
близкие реакции гидролиза ортоэтилформиата 131 и ди-
этилацеталя 133 [реакции (Ь78) и (179)
соответственно] специфически катализируются Н30+.
Возможное объяснение такого различного поведения
состоит в том, что этанол легче отрывается от
промежуточного катиона 49, чем от 132 или от 134, так
как образующийся катион 135 более стабилизован,
чем 136 (метильный заместитель стабилизует
катион) и 137 (в котором только одна ЕЮ-группа
может стабилизовать положительный заряд). Исходя
из этого, кажется обоснованным, что в этих
реакциях только в случае ортоэтилацетата стадия (б)
оказывается сравнимой по скорости со стадиями (а) и
(—а), что приводит к общему кислотному катализу.
Следует напомнить, что для рассмотренных
соединений бтношения К(б)/К(а) и к(б)/К(-а) могут принимать
любые значения. Таким образом, если реакции,
подверженные специфическому катализу, проводить в
буферном растворе с очень высокой концентрацией
кислоты, то они могут вести себя как реакции
общего кислотного катализа.
OEt OEt _
I (a) I +/Et
H+ + CH3-C-OEt z*=t CH3—C-O,
I ("a) I \h
OEt OEt
48 49
113
49
I (б)
OEt +OEt OEt
l+ И I
CH3—С •<—** CH3—С •<—*■ CH3—С —>- —>► Продукты
OEt OEt +OEt (/б'
135
OEt OEt
H+ + H—C-OEt q=> H—C-O^
OEt OEt
131 132
I
OEt +OEt OEt
l+ II I
H—C+ +-> H—С ч-> H—С —-> —> Продукты (178)
OEt OEt +OEt
136
OEt
/0Et | Et
H++CH3—CH( ч=± СНз-С—O^
X)Et I \h
H
133 134
I
OEt +OEt
I. II
СНз—С+ ч-> СНз—С —> —► Продукты (179)
H H
137
Стабильные молекулярные интермедиа,™
В ряде реакций в качестве интермедиата может
образовываться относительно стабильная молекула.
Присутствие такого интермедиата можно показать
114
с помощью кинетических исследований, если
начальная скорость исчезновения исходных веществ
больше, чем скорость образования продуктов. Если
реакция изучается с помощью спектрофотометрии, то на
присутствие или отсутствие стабильных интермедиа-
тов часто указывают изменения в спектрах в ходе
реакции (рис. 4). Предположим, что соединение X
Длина волны, Л
Рис. 4. Изобестическая точка. Кривые поглощения в УФ-области спектра,
характерные для реакции X -> Y.
Кривая 1 соответствует чистому X; кривые 2—4 — смесям X и Y в
соотношении 75 : 25, 50 : 50; 25 : 75; кривая 5 отвечает чистому Y.
(УФ-спектр- которого представлен кривой 1)
превращается в Y (УФ-спектр — кривая 5) и что в системе
отсутствуют другие вещества, поглощающие в этой
области спектра. Если X и Y имеют достаточно
различные спектры, то будут существовать одна или
более точек, в которых их спектры пересекаются.
В этих точках, называемых изобестическими, два
соединения будут иметь одинаковые коэффициенты
экстинкции и таким образом при соответствующих
этим точкам длинах волн оптическая плотность
раствора будет всегда постоянна в* ходе реакции.
Следовательно, в промежуточные моменты реакции
будут наблюдаться спектры типа кривых 2, 3 и 4, но
каждый из них будет проходить через изобестиче-
скую точку. Показанный на рис. 4 тип спектральной
зависимости является хорошим свидетельством
против образования в реакции долгоживущего интерме-
диата, тогда как спектры, которые не содержат
115
изобестические точки, представляют собой хорошее
доказательство присутствия такого интермедиата.
Стабильный интермедиат можно выделить или
зафиксировать, используя методы, которые зависят
от конкретного случая. Катализируемая соляной
кислотой перегруппировка N-хлорацетанилида (см.
гл. 2) дает ту же смесь о- и n-хлорацетанилидов, что
и прямое хлорирование ацетанилида. Это наводит
на мысль, что в качестве интермедиата в реакции
(180) образуется свободный хлор. Такое
предположение было подтверждено двумя способами. Когда
Ы,2,4-трихлорацетанилид нагревали с соляной
кислотой в присутствии аминов, таких как м-броманилин,
то в заметных количествах образовывался 4-бром-
2,6-дихлоранилин. Этого следовало ожидать, так как
/г-броманилин хлорируется гораздо легче, чем 2,4-ди-
хлорацетанилид. NHj
(180)
Еще более прямое и убедительное доказательство
было получено, когда через реакционную систему
продували азот. При этом из раствора уносился хлор,
что непосредственно доказало его образование в ходе
реакции.
В ряде случаев относительно небольшое
изменение условий (обычно понижение температуры)
позволяет выделить интермедиат, который при
нормальных условиях реагирует слишком быстро для
того, чтобы его можно было обнаружить. Реактивы
116
Гриньяра взаимодействуют с кислородом по хорошо
известной реакции с образованием спиртов. Это
окисление трудно правдоподобно представить как
одностадийную реакцию. Кажется очевидным, что ин-
термедиатом в этом процессе является пероксид 138.
Такие интермедиаты были выделены при
пропускании кислорода в реактивы Гриньяра при — 70 °С.
RMgX Н20
R MgX > 2R0—MgX ——*
R-MgX —> -
+ I I
0=0 0-0
138
—► 2R0H + 2MgX0H (181)
Разнообразные эксперименты с мечеными
соединениями в случае /гара-пере^руппировки Кляйзена
аллиловых эфиров фенолов указывают, что алкиль-
ная группа остается связанной с исходной
молекулой, что подразумевает существование интермедиата
типа 139, который представляет собой диен и как
таковой должен вступать в реакцию Дильса — Аль-
дера с подходящим олефином. Действительно,
введение в реакционную смесь малеинового ангидрида
приводит к аддукту 140, что еще более увеличивает
уверенность в предполагаемом механизме.
сна-сн=сн2
Н3Сч^Лч^СН3
о
НзС^А|г^СНз
н сн2-он=сн2
dea)
сн2-сн
сн2
117
Заключение
Присутствие в системе интермедиата
определенного типа обычно постулируется на основании
продуктов реакций или результатов изучения
кинетики. Интермедиат может быть затем
идентифицирован или зафиксирован, как описано выше.
При этом, однако, существуют две опасности,
которых следует избегать.
(1) Некоторые методы оказываются чрезвычайно
чувствительными, это в особенности относится
к ЭПР, ХДПЯ и инициируемой полимеризации,
поэтому следует всегда иметь в виду возможность
гого, что детектируемый с помощью такого метода
«интермедиат» является продуктом параллельной
второстепенной реакции и не характеризует
рассматриваемую основную реакцию. Эта
возможность менее вероятна при использовании метода
химической фиксации (химической ловушки),
в частности если фиксирующий агент превращает
весь или большую часть интермедиата в
соединение, отличное от обычного продукта реакции.
(2) Химические фиксаторы (агенты
улавливания) или ингибиторы могут обладать
недостаточной реакционной способностью по отношению
к интермедиату для того, чтобы заставить его
реагировать в направлении, отличном от
нормального течения реакции. Эти же агенты могут
реагировать также с образованием другого
интермедиата, как было показано в рассмотренном примере
автоокисления боранов. Такие результаты могут
ошибочно привести к заключению об отсутствии
в системе определенного интермедиата или
интермедиата определенного типа.
Задачи
4.1. Соединение PhCHMe—CHMeNH2 реагирует с азотистой
кислотой в уксусной кислоте, образуя смесь: PhCHMe—СНМеОАс
(44%), PhCMe(OAc)—СН2Ме (24%) и PhCH(OAc)—СНМе2
(32%). Можно предположить, что в качестве интермедиата
образуются структуры PhCHMe—CHMe+, PhCHMe—CHMe- или
PhCHMe—СНМел Какой интермедиат является более вероятным?
116
4.2. Дибензилртуть (раствор в октане) термически
разлагается с количественным образованием ртути и дибензила как
основного органического продукта. Однако получаются также
следы 1,2,3-трифенилпропана и толуола. Предложите механизм,
согласующийся. с этими данными, и дополнительные
эксперименты, которые могут подтвердить ваш механизм.
4.3. Реактивы Гриньяра [органомагнийгалогениды, структура
б- б+
которых для простоты может быть записана R—Mg—Hal
(А) или R" +Mg—Hal (В)] не разлагают диэтиловый эфир,
тогда как метилнатрий реагирует с диэтиловым эфиром с
образованием этилена, метана и этилата натрия. Предложите механизм
этой реакции. Проливает ли какой-либо свет на структуру (ко-
валентную А или карбанионную В) реактивов Гриньяра разница
в реакционной способности между метилнатрием и реактивами
Гриньяра?
4.4. Первичные алифатические спирты окисляются
соединениями Cr(VI) в кислом растворе с образованием альдегидов и
Сг(Ш). Если к раствору добавлять соли Мп(П), то осаждается
Мп02. Однако Мп(Н) не окисляется спиртами, альдегидами,
Cr(VI) или Сг(Ш). Предложите (в общих чертах) объяснение.
4.5. Известно, что соединение PhCHN2 образует при
фотолизе PhCHS; в присутствии бутена-1 образуется эквимольная
смесь цис- и граяс-1-этил-2-фенилциклопропанов. Те же два
продукта в соотношении 2: 1 образуются при обработке PhCHBr2
литием в присутствии бутена-1. Можно предположить, что
PhCHBr2 реагирует с литием, образуя PhCH(Br)Li, который либо
непосредственно реагирует с бутеном-1 с получением
наблюдаемых продуктов, либо разлагается с образованием бромистого
лития и карбена PhCHS, который, в свою очередь, взаимодействует
с бутеном-1, приводя к циклопропановым соединениям. Какой
механизм более, вероятен?
Глава 5
СТЕРЕОХИМИЯ
Каждый этап исследования механизма реакции
приводит к более детальному пониманию природы
изучаемого процесса, хотя, как уже отмечалось в гл. 1,
получение полной картины химической реакции
•является невыполнимой задачей. Стереохимические
исследования реакции позволяют глубже проникнуть
в механизм процесса, так как приводят к
установлению направления, в котором реагент подходит к
молекуле, или направления перемещения групп во
время реакции.
Как ни странно, в действительности оказывается,
что многие представления в области стереохимии
органических реакций возникли на основании наших
знаний о механизме, как это будет показано ниже,
В основном существуют два типа стереоизомерии:
оптическая и геометрическая. Информация, полезная
для изучения механизма реакции, может быть
получена при использовании молекул, обладающих как
одной, так и другой формой стереоизомерии.
Замещение при тетраэдрическом центре
и оптическая изомерия
Органические соединения, в которых при одном
атоме углерода имеются четыре различных группы,
существуют в двух изомерных формах. Такие
оптические изомеры обладают одинаковой химической
реакционной способностью *, но' различаются по их
оптическим свойствам. В частности, они имеют
одинаковое по величине, но противоположное по знаку
* Кроме реакций с ассиметрическими реагентами.
120
оптическое вращение (растворы равной
концентрации вращают луч плоско-поляризованного света в
одинаковой степени, но в противоположных
направлениях).
Для реакции замещения, в результате которой
группа, связанная с центральным атомом углерода,
заменяется на новую группу, существуют две стерео-
химические возможности. Либо новая группа
занимает то же положение относительно других
заместителей, что и уходящая группа [реакция
(183)—процесс, который включает сохранение конфигурации],
либо образуется противоположный (зеркальный)
оптический изомер [реакция (184)]. На данном этапе
мы не будем рассматривать пути, по которым это
может происходить, но следует отметить, что при
этом положение по крайней мере одной из нереаги-
рующих групп относительно других групп должно
изменяться. Этот процесс называется инверсией
(обращением) конфигурации по причинам, которые- бу«
дут детально рассмотрены ниже. Образующееся
соединение 143 будет обладать противоположным
оптическим вращением по сравнению с 142, которое
образуется при замещении с сохранением
конфигурации. Реакции (183) и (184) иллюстрируют эти две
возможности. В макроскопическом масштабе
реакция может осуществляться либо по одному из
указанных направлений [(183) или (184)], либо может
иметь место конкуренция обеих реакций, в
результате чего наблюдается полная или частичная потеря
оптической активности или рацемизации.
1183)
Ц84)
121
Доказательство сохранения или обращения
конфигурации в процессе реакции не является
тривиальной задачей, так как (по крайней мере до самого
последнего времени) не было возможности связать
знак вращения оптического изомера с его
абсолютной конфигурацией (т. е. отвечает ли ему структура
142 или 143?). Это связано с тем обстоятельством,
что соединения с одной и той же относительной
конфигурацией (т. е. конфигурации, которые связаны
друг с другом так же, как 141 с 142) не обязаны
иметь одинаковый знак оптического вращения. Это
можно продемонстрировать на примере трех
реакций (185), в которых одновременно меняется только
одна группа и всегда с сохранением относительной
стереохимии. Однако в результате этих реакций
образуется оптический изомер исходного соединения.
Ясно, что либо одна, либо все три реакции должны
приводить к изменению знака вращения.
(Ю5)
X X х х
Для того чтобы определить стереохимический
путь реакции замещения, необходимо знать
относительные конфигурации как исходных соединений,
так и продуктов. В следующих трех разделах
описаны различные методы определения относительной
конфигурации.
Вальденовское обращение
В 1896 г. Вальден осуществил превращения,
показанные на верхней схеме с. 123. Очевидно, что
полученные результаты, когда молекула в две стадии
превращается в ее зеркальное отображение, можно
объяснить либо тем, что реакции, показанные на схеме
вертикально, включают сохранение, а горизонтальные
реакции — обращение конфигурации, либо наоборот.
Но как различить эти две возможности?
122
„AgOH"
нооссн2снсоон * нооссн2снсоон
I I
CI OH
(Ч-)-хлорянтарная
кислота
PCls
КОН,
н2о
(^+)-яблочная кислота
PCls (186)
кон,
н2о
„AgOH"
НООССН2СНСООН ■< НООССН2СЦСООН
ОН
( —Ьяблочная
кислота
С1
( — )-хлорянтарная
кислота
В 1923 г. Филлипс получил (+)- и (—)-формы
этилового эфира метилбензилкарбинола из (+)-ме-
тилбензилкарбинола 144 при помощи следующих
двух схем реакции. Процесс включает четыре
индивидуальные реакции. Объяснить стереохимический
результат можно, предполагая, что либо одна, либо
три из них проходят с обращением конфигурации
(инверсией), а остальные с сохранением
конфигурации.
PhCH2
СНз/ ^Н
PhCH24 /OK
(+)
РЬСН2ч /ОН
С\\/ \\
( + 33,0)
144
SO2C6H4CH3
i
CH3CeH4S02Cl
реакция (187)
реакция (189)
К2СО3/С2Н5ОН РЬСН2ч уН
реакция (188) у~\
сн3/ х
(- 19,9)
с2н5вг РЬСН2ч /ОС2Н5
OC2Hfi
реакция (190) / \
Сп3 х
Н
(+ 23,5)
Три реакции [(187), (189), (190)] не включают
атаку по асимметрическому центру, и почти
несомненно, что конфигурация оптически активного атома
углерода в этих реакциях не изменяется. Из этого
123
следует, что реакция (188), представляющая собой
бимолекулярное нуклеофильное замещение (Sw2)
молекулой этанола у асимметрического атома
углерода, должна сопровождаться фактически 100%
инверсией. Дальнейшими работами в этой области
установлено, что и в ряде других примеров нуклео-
фильного замещения у атома углерода также
наблюдается инверсия *.
Еще более прочная связь между бимолекулярным
нуклеофильным замещением и инверсией
установлена Хьюзом и сотрудниками в 1935 г. Они изучили
параллельно рацемизацию 2-иодоктана при
действии иодид-ионов и накопление в 2-иодоктане
радиоактивного иода в результате взаимодействия с
радиоактивными иодид-ионами. Обе эти реакции
оказались второго порядка; первого порядка по каждому
реагенту (2-иодоктану и иодид-иону). В
экспериментах с радиоактивным иодид-анионом была
установлена скорость, с которой протекает реакция
замещения:
128P + R—I —> 1281—R + Г (191)
Стереохимические последствия замещения были
показаны при помощи экспериментов по рацемизации.
Если каждый индивидуальный акт обмена проходит
с сохранением конфигурации, то никакой потери
оптической активности наблюдаться не будет. Если
же в реакции замещения стереохимическая
предпочтительность отсутствует, то скорость обмена иода
должна быть той же, что и скорость пот.ери
оптической активности. Однако если при каждом акте
замещения имеет место инверсия, скорость
рацемизации должна быть в два раза выше скорости
накопления радиоактивного иода, так как каждый акт
замещения в молекуле (+)-иодида должен дать
одну молекулу (—)-иодида, которая будет
компенсировать вращение, обусловленное другой молекулой
(+)-иодида.
128P + R—I —> ,28I-R + P (192)
(+) (-)
* Для обозначения реакции, которая протекает с
обращением конфигурации, используют символ ---**.
124
С точностью эксперимента было установлено, что
константа скорости рацемизации в два раза больше
константы скорости обмена радиоактивного иода. Из
этого следует, что акт замещения приводит к
инверсии.
Аналогичные результаты были получены для
реакции бромид-иона с оптически активным фенил-
этилбромидом. Таким образом, для бимолекулярных
нуклеофильных реакций, в которых входящая и
уходящая группы одинаковые (примеры с 2-иодоктаном
и 1-фенилэтилбромидом) или включают в себя тот
же элемент (кислород), который связан с
асимметрическим углеродом [реакция (188)], всегда
наблюдается инверсия. Исходя из этих результатов, было
сно соон „_ соон
| ^й2^ Т <HN°2 |
I'
CH2NH2
н он н он н он
D-(-f)- глицериновый
х ' альдегид NODr
соон
н он
ка/нд
СООН
ДхСНз
Н ОН
ft
он
СООН , СООН СООН
< H2/Pt | < NaN, .
сн3 х^ЧГснз х^Ч"снз
Н NH2 H N3 Br H
D-И-аланин (193)
125
высказано предположение, что обращение
конфигурации будет происходить даже в тех случаях, когда
нуклеофил и уходящая группа различны. На
основании этого предположения было проведено очень
много структурных корреляций для оптически активных
соединений углерода и ни в одном из случаев, когда
было доказано, что реакция нуклеофильного
замещения бимолекулярна, не было получено
противоречивых результатов. Рассмотрим следующий пример
структурной корреляции, осуществленной таким
способом. Было показано, что Ь-глицериновый альдегид
имеет относительную конфигурацию,
противоположную природной аминокислоте (+)-аланину. Так как
все обычно встречающиеся в природе а-аминокислоты
имеют одинаковую конфигурацию, то все они
относятся к соединениям L-ряда (см. схему на с. 125).
Метод псевдорацематов*
Несмотря на то, что многие физические свойства
(в частности, температура плавления) оптических
изомеров одинаковы, эти изомеры, являясь на самом
деле различными соединениями, вызывают
депрессию температуры плавления смешанной пробы по
сравнению с индивидуальными изомерами.
Эквимольная смесь двух изомеров может либо
представлять собой эвтектическую " (рацемическую)
смесь, либо образовывать рацемическое соединение.
Рассмотрим пример из химии кремнийорганических
соединений. Представим себе два различных
оптически активных соединения, такие как Ph(doH7)MeSiH
и Ph(Ci0H7)MeSiF (СюН7 — а-нафтил), которые
содержат относительно небольшие структурные
различия. Два соединения с одинаковой конформацией
часто образуют твердый раствор, в котором молекулы
обоих изомеров беспорядочно занимают узлы
решетки в зависимости от их относительного содержания.
Если два соединения имеют противоположные
конфигурации, то обычно твердый раствор
невозможен: образуется или простая эвтектическая смесь,
* По мнению Илиела, в случаях, аналогичных описанным
ниже, следует использовать термин «квазирацематы». — Прим.
пер.
126
или «псевдорацемическое» соединение. Используя
диаграммы плавления или, чаще, диффракцию
рентгеновских лучей, можно различить эти варианты. С
помощью описанного метода установлено, например,
что ( + )-Ph(Ci0H7)MeSiCl, (—)-Ph(Ci0H7)MeSiF
и (—)-Ph(CioH7)MeSiH имеют одинаковую
конфигурацию, что дает возможность определить стереохими-
ческое течение различных взаимопревращений
оптически активных соединений кремния, таких как
доказаны на схеме (194). Становится очевидным, что
стереохимия замещения при атоме кремния не столь
проста, как в случае углерода, и в частности
бимолекулярное нуклеофильное замещение при атоме
кремния иногда проходит с обращением
конфигурации [схема (194)], а иногда с ее сохранением [как
в (195)]. [Если бы эта реакция проходила с
инверсией, то в каждом отдельном акте возникали бы
одна молекула (+)-и одна (—^-продукта, давая в
результате оптически неактивный (рацемический) продукт.]
СЬ/ССЦ ЫА1Н4
Ph(C10H7)MeSiH - >- Ph(C10H7)MeSiCl —о—>
(а> (б)
{ + 32) сохранение (— 6,3) иверсия
конфигурации
—> Ph(C10H7)MeSiH (194)
(-32)
КОН, ксилол
Ph(CI0H7)MeSi-O-SiMe(CI0H7)Ph >■
( + 9,8)
—>■ 2Ph(CI0H7)MeSiOK (195)
(-70)
Другие методы
Другим важным методом определения относительной
конфигурации является дисперсия оптического
вращения (изменение оптического вращения в
зависимости от используемой длины волны света). Как уже
отмечалось, знак вращения оптически активного
соединения — ненадежный ключ к установлению его
стереохимии. Если измерять оптическое вращение как
функцию длины волны, получается кривая одного из
типов, показанных на рис. 5. Если соединение имеет
полосу поглощения в видимой или ультрафиолетовой
области спектра за счет хромофора, находящегося
вблизи асимметрического центра, то наблюдается
кривая типа а. Обычно с уменьшением длины
127
волны оптическое вращение изменяется сначала
медленно, а затем быстрее, вплоть до максимального
значения (положительного или отрицательного),
после чего быстро падает и пересекает нулевую линию
при длине волны, приблизительно соответствующей
Ятах для хромофора. Этот тип зависимости
называется эффектом Коттона и для двух соединений,
приведенных на рисунке, определяется как
положительный (отрицательным эффектом Коттона было бы
зеркальное отображение кривых (а) по отношению
Рис. 5. Дисперсия оптического вращения (ДОВ):
fl —кривые ДОВ для двух соединений А и В. Обе кривые показывают
положительный эффект Коттона, но оптическое вращение этих соединений имеет
противоположный знак при D-линии натрия; б—плавные кривые, т. е.
кривые без эффекта Коттона в доступной области УФ-света.
к горизонтальной оси). Если максимум поглощения
лежит в области слишком коротких длин волн, то
наблюдается плавная кривая (как показано на
рис. 56). Можно предположить, что если бы удалось
измерить оптическое вращение при более коротких
волнах, то такие кривые обладали бы
положительным эффектом Коттона. Знак эффекта
Коттона является гораздо более надежным критерием
относительной стереохимии, чем оптическое вращение
при любой одной данной длине волны. Например,
все природные а-аминокислоты RCH(NH2)COOH в
том случае, если они не содержат никакого другого
хромофора кроме карбонильной группы,
характеризуются положительным эффектом Коттона и,
следовательно, по-видимому, обладают одинаковой
относительной конфигурацией, несмотря на то, что их
128
оптическое вращение, в частности при D-линии
натрия, может иметь противоположные знаки. С
помощью этого метода было проведено очень большое
число корреляций относительной конфигурации
разнообразных природных продуктов, например
стероидов.
Относительно недавно в результате модификации
обычной техники рентгеноструктурного анализа
удалось определить абсолютные конфигурации ряда
соединений. Это сделано для ограниченного числа
соединений, однако таким образом удалось проверить
относительные конфигурации различных классов
веществ, установленные ранее другими методами.
Реакции с инверсией у атома углерода
Обращение конфигурации, которым сопровождаются
реакции 5^2, означает атаку "нуклеофила с
противоположной стороны молекулы, по отношению к
уходящей группе, что приводит к переходному состоянию
146, и затем к продукту 147, с конфигурацией,
противоположной 145. При атаке по такому
направлению силы отталкивания между вступающей и
уходящей группами (часто объемистыми) минимальны, что
позволяет образовываться новой связи, в то время
как старая разрывается. В примере, показанном на
схеме (196), обе группы, вступающая и уходящая,
имеют отрицательный заряд.
СИ,
но—е.—Вг
/ \
С2 H5 H
146
сн,
*- НО < + Вг~
Н С2И5
147
СхЩ
5 Зак. 1044
129
Однако электростатическое отталкивание между
одноименными зарядами не может являться
причиной наблюдаемого направления атаки, так как когда
положительно заряженная сульфониевая соль
атакуется гидроксильным ионом, все же наблюдается
инверсия конфигурации, хотя вступающая и
уходящая группы обладают противоположными зарядами:
C6H13CHSMea + ОН" -о~» СбН13СНМе + Me2S (197)
I I
Me ОН
Механизм 5^2-замещения подтверждается тем
фактом, что если структура молекулы не допускает
атаку молекулы с противоположной стороны, как в
случае бициклического соединения 148, то нуклео-
фильное замещение проходит с трудом. трет-Бутиль-
ный заместитель в неопентилбромиде 149 также
достаточно объемен для того, чтобы затруднять атаку
нуклеофила по СН2-группе.
Н3С
Вг НзС—С-СДг-Вг
Н3С
148 149
В общем случае первичные соединения вступают
в реакции замещения S^2-Tnna легче, чем
вторичные, а 5^2-реакции третичных соединений
протекают с большим трудом, если вообще имеют место.
В переходном состоянии 5^2-реакций 146
центральный атом углерода увеличивает свое
координационное число от четырех до пяти и пространственное
взаимодействие между заместителями должно быть
более сильным, чем в. основном состоянии. Алкиль-
ные группы занимают большее пространство, чем
атомы водорода, следовательно, такое стерическое
отталкивание должно быть более сильным в ряду:
третичные > вторичные > первичные соединения.
Замещение с сохранением конфигурации
Такие реакции для углеродных систем встречаются
крайне редко. Атаке с фронтальной стороны
молекулы благоприятствуют два обстоятельства.
о-
130
(1) При атаке электрофилом (т. е. электроноде-
фицитной частицей) отталкивание между
вступающей и уходящей группами может оказаться
небольшим, поскольку обычно электрофилы имеют
свободную орбиталь,.. которая может перекрываться со
связью С—X в 150, образуя трехцентровую орбиталь.
R\ X
R-q
r/ V+
R\ A
r-c; ;b
*/ v
150 151
(2) Сохранение конфигурации следует ожидать
в%тех случаях, когда замещение имеет характер 4-цен-
трового молекулярного взаимодействия с
переходным состоянием 151._
Различие между возможностями (1) и (2) часто
провести нелегко.
К примерам реакций, протекающих с сохранением
конфигурации, относится ряд реакций металлоорга-
нических соединений, такие как реакция (198).
Интересно, что для элементов, начиная со второго
большого периода Периодической таблицы, более общим
случаем являются реакции, включающие сохранение
конфигурации. Ранее в этой главе был уже приведен
пример процесса,- протекающего с сохранением
конфигурации у атома кремния [реакция (194)].
С,1 ОМе
Я1—Hfi,
(198)
н ОМе
PhCMe2CH2-Hg
—>- PhCMe2CH2— HgCl +
Cl-^Hg-
н
5*
131
Реакции, включающие промежуточное образование
ионов карбония, радикалов или карбанионов
Ионы карбония
На основании кинетических и ряда других данных
считается, что реакции SN\ алкилгалогенидов
протекают с промежуточным образованием карбониевых
ионов. Если оптически активный алкилгалогенид
реагирует по этому пути, то продукт оказывается
более или менее рацемизованным, из чего следует,
что свободный ион карбония плоский или почти
плоский. Дополнительное подтверждение плоскостного
строения карбониевых ионов дают исследования
реакционной способности мостиковых циклических
соединений, для которых возможный промежуточный
ион карбония не может стать плоским из-за
напряжения кольца. Например, соединение 153 гидроли-
зуется в водном этаноле при 25 °С со скоростью
примерно в миллион раз меньшей, чем в случае
гидролиза 154. Соединение 152, для которого
образование плоского карбониевого иона должно быть еще
более трудным, реагирует примерно в десять
миллионов раз медленнее, чем 153.
нзс.
Br ^V8' Н3С—С—Вг
^^ Н3С
152 153 154
В действительности реакции, включающие
промежуточное образование ионов карбония, не всегда
приводят к полностью рацемическим продуктам.
Когда в результате реакции наблюдается частичная
инверсия, полагают, что хотя свободный ион
карбония и образуется, но уходящая группа (X в 155)
может частично экранировать (защищать) эту
сторону молекулы от атаки. В результате вступающая
группа Y предпочтительно атакует молекулу с про-
о-
132
тивоположнои
инверсии.
стороны, что приведет к частичной
Вступающая группа
предпочтительно
атакует с этой стороны
®
Уводящая группа
С+ (X) экранирует фронтальную
/ \ сторону карбониевого иона
155
Некоторые реакции нуклеофильного замещения у
атома углерода протекают с частичным сохранением
конфигурации: например взаимодействие хлорянтар-
ной кислоты с гидроксидом серебра [реакция (186)].
В родственной реакции гидролиза 156 при
использовании концентрированной щелочи протекает
бимолекулярная (S)v2) реакция и наблюдается
обращение конфигурации. При использовании более
разбавленных растворов щелочи реакция становится
мономолекулярной и приводит преимущественно к
продукту с сохранением конфигурации. Полагают, что
в промежуточном ионе карбония 157 существует
взаимодействие между карбоксильной группой и карбо-
ниевым центром, что одновременно стабилизует ион
и препятствует подходу атакующего нуклеофила с
этой стороны молекулы. В результате приближение
нуклеофила преимущественно осуществляется со
стороны уходящей группы и вызывает преобладающее
сохранение конфигурации.
(+)-Ме—СН-СОСГ -
Вг
конц.
щелочь
разбавл.
щелочь
г
156
1—> (—)-Ме—СН—COO" S^2, инверсия
ОН
1 . Ь'еУ 1
(199)
/ \
Me H
157
(+)-Ме—СН—COO" S„l, (200)
I
Ли сохранение конфиг.
133
Радикалы
Когда замещение у асимметрического углеродного
центра осуществляется в ходе свободнорадикальной
реакции, продукт оказывается оптически
-неактивным. Так, хлорирование оптически активного
1-хлор-2-метилбутана 158 по оптически активному
центру приводит к неактивному продукту 160.
СН3 СМ3 СН3
С8Н5 СН2С1 С2Н5 С1 С2Н5 СН2С1
159 160
(201)
CU СН3 СН,
^Н5 СН2С1 -Jc^ I
(so^ ^Цс^с! илй ^a + *
с2н5 а с2н5 ch2ci
161 162
Из этого следует, что промежуточно
образующийся радикал 159 практически плоский, что и
приводит к рацемическому продукту. Если бы
замещение проходило непосредственно за счет атома хлора
[реакция (202)], то следовало бы ожидать стереохи-
мического предпочтения либо для сохранения
(продукт 162), либо для обращения конфигурации
(продукт 161). Предпочтительная плоская структура
алифатических радикалов подтверждается спектром
ЭПР радикала 13СН3в и ИК-спектром метильного
радикала. Хотя простые алкильные радикалы имеют
преимущественно плоскую структуру, галогенирован-
ные радикалы, такие как CF3e оказываются
неплоскими. Неплоскими являются также циклопропильные.
радикалы. В этих случаях возможно протекание
радикальных реакций без полной рацемизации. Хотя
алкильные радикалы стремятся принять плоскую
134
конфигурацию, однако это стремление невелико.
В противоположность карбониевым ионам,
невыгодность образования которых в голове моста би-
циклических соединений затрудняет их S#l-реакции,
радикальное замещение у атомов углерода в голове
моста или получение радикалов по этим положениям
любым другим способом — довольно обычное
явление.
Карбанионы
Считается, что кетоны реагируют с основаниями
с образованием карбанионов, которые могут
вступать в другие реакции или отрывать протон от
растворителя, вновь образуя рацемический кетон [см.
реакцию (41)]. Поскольку скорость рацемизации
кетона равна скорости различных катализируемых
основаниями реакций кетона, отсюда следует, что
либо карбанион является плоским, либо он
пирамидален, но в нем легко проходит инверсия. Однако,
как и в случае карбониевых ионов, некоторые
реакции, включающие, как считают, образование
карбанионов, протекают с преобладающим сохранением
конфигурации или с некоторым преобладанием
инверсии. Оптически активный флуорен 163' в реакции
с аммиаком в относительно неполярных
растворителях приводит к преимущественному сохранению
конфигурации [реакция (203)]. Полагают, что в
реакции образуется ионная пара, в которой значительное
взаимодействие между положительными и
отрицательными ионами делает более вероятным обратный
переход ионной пары в 163 по сравнению с
диссоциацией на полностью свободные, ионы. Однако
вращение иона аммония внутри ионной пары может
привести к тому, что генерируемый флуорен 166
будет содержать водород вместо дейтерия, но тем не
менее будет наблюдаться преобладающее сохранение
конфигурации. С другой стороны, в более полярных
ионизующих растворителях, таких как метанол,
молекула протонированного основания будет
обладать большей тенденцией дальше отойти от аниона
Л 67.
135
н н
X
р&г Н D CHj
NH3, "I
Vr* 1
1 И
Н D
V/
N
/\
Н Ц СН3
-1-
н сн3
-*. I
I
163
164
165.
166
Ь*
(203)
D СН3
СН<
<$
CQW^ Pr3NDLHOM0
молекула, проекция которой
из точки наблюдения
представлена на схеме
Н3С Н
167
168
(2Q4)
Протон от молекулы растворителя с большей
вероятностью присоединится к 167 со стороны,
противоположной уходящему иону Pr3NH+, образуя
таким образом продукт 168 с преимущественной
инверсией [реакция (204)].
Перегруппировки ионов карбония
Одной из характерных черт, которые отличают кар-
бониевые ионы от радикалов и карбанионов,
является их склонность к перегруппировке за счет сдвига
группы от соседнего атома к катионному центру.
Если между мигрирующей группой и а-углерод-
ным атомом может возникать некоторое
взаимодействие при отходе группы X в виде Х~, то эту реак*
136
цию можно рассматривать как внутримолекулярную
реакцию «5^2-типа» по Са, и конфигурация у
Садолжна стать противоположной (первоначальной).
Взаимодействие, показанное в 170, возможно
только в том случае, если четыре атома R—С—С—X
лежат приблизительно в одной плоскости с указанной
зигзагообразной (анти) конформацией: практически
легко мигрируют только те группы, которые могут
занять примерно такую конформацию. Например, ди-
ол 171 перегруппировывается в кетон 172
приблизительно в шесть раз быстрее, чем 173. В 171 после
отрыва протонированной тидроксильной группы
р-фенильная группа, расположенная в транс-положе-
нии к уходящей группе, мигрирует к а-углеродному
атому. В 173 ни одна из фенильных групп не
находится в благоприятном граяс-положении к уходящей
гидроксильной группе; перегруппировка
осуществляется гораздо медленнее Ъ, возможно,
наблюдаемая медленная перегруппировка на самом деле
является результатом предварительной
изомеризации 173 в 171.
/JQpj быСТрО
ОН
172
173
(206)
Если мигрирующая группа при перегруппировке
иона карбония [R в реакции (205)] имеет
асимметрический атом углерода, непосредственно связанный
с р-положением в 169, то его конфигурация в ходе
реакции сохраняется. По отношению к R эту
реакцию можно рассматривать как электрофильное
замещение у Ср под действием Са. «Вступающая» и
«уходящая» группы находятся с одной стороны
асимметрического атома углерода, в результате чего его
конфигурация сохраняется.
137
Присоединение к кратным связям и элиминирование
При изучении реакций, включающих присоединение
к кратным связям [реакция (207)], или обратного
процесса, который называется элиминированием,
возникают два вопроса.
\с=с/ _ -U
X—Y + ,C=C —* -С—С— (207)
/ \ II
X Y
(1) Происходит ли образование обеих новых
связей одновременно?
(2) В каком направлении реагент атакует
кратную связь?
Используя подходящие олефины, можно
определить, является ли направление присоединения X—Y
цис- (т. е. обе новые связи образуются" в результате
ата^и с одной стороны молекулы), транс- (одна
новая связь с одной стороны, другая с
противоположной) или беспорядочным. Рассмотрим эти
возможности отдельно и ту информацию, которую они дают
о механизме.
Транс- (и беспорядочное) присоединение
Когда наблюдается транс- или беспорядочное
присоединение, ответ на поставленный выше вопрос (1)
относительно того, одновременно ли образуются две
новые связи, будет отрицательным. Переходное
состояние для ту?а«с-присоединения должно включать
атаку X с одной стороны л-связи и атаку Y — с
другой. Однако при таких условиях X и Y не могут в
значительной степени сохранять связь друг с другом.
Таким образом, для реакций с ту?а«с-присоединением
следует исключить молекулярный четырехцентровый
механизм:
\ / I' I
V=C; —► —С—С— (208)
/\ !\ II
X—Y X Y
Известно очень большое число реакций транс-прк-
соединения и элиминирования. Например, _присоеди-
нение брома к малеиновой кислоте 174 дает (±)-ди-
138
бромкислоту 175, которую можно -разделить на
антиподы, в то время как присоединение брома к фу-
маровой кислоте 176 приводит к нерасщепляемой ме-
зо-кислоте 177.
Н СООН
н соон н соон
малеиновая (±)- расщепляется
кислота *
(2Q9)
174
175
тоЯ/ ноосч ^
С Br V:^
А
Вг "С
н' чсоон н^ чсоон
фумаровая мезо-у не расщепляется
кислота /__
л™ 177
176
граяс-Стерёохимия отмечена также для многих
реакций присоединения к ненасыщенным
циклическим соединениям и к ацетиленам. г/?а«с-Присоедрне-
ние часто наблюдается в реакциях, которые (как
предполагают на основе данных об образующихся
интермедиатах) включают первоначальное
присоединение электрофила или радикала к одному концу
двойной связи. При этом возникает другое
затруднение. Если в качестве интермедиата при
присоединении, например брома к малеиновой кислоте [реакция
(209)], образуется ион карбония 178, то можно
ожидать не ту?а«с-присоединения," а смешанный
механизм, так как в 178 вращение вокруг ординарной
углерод-углеродной связи (а) должно быть быстрым,
что делает равновероятным присоединение Вг~ в
любом направлении. Эту трудность можно
преодолеть, если предположить, что между атомом брома
и карбониевым центром имеется взаимодействие
либо в виде частичного связывания, как представлено
в 179, либо за счет образования симметричного
интермедиата 180. В любом случае свободное вращение
139
вокруг связи С—С замедлится или прекратится
вовсе и катион будет способен присоединить Вг~
с противоположной стороны. Такое направление
реакции обусловлено как стереохимией, так и (по
аналогии с 5л^2-реакциями) тем, что при этом связь
между бромом и карбониевым центром может
сохраняться (и поэтому энергия переходного состояния
будет достаточно низкой).
VA-COOH IH^C-COOH /^Сх. н
14 hr j^C COOH
ъу "соон н' "соон н соон
178 179 180
Реакции элиминирования обычно проводят в
условиях, весьма отличающихся от условий
соответствующих реакций присоединения. Элиминирование га-
логеноводородов из алкилгалогенидов под действием
сильных оснований [реакция (211)] и отщепление
брома от дибромидов, промотируемое иодид-ионом
[реакция (212)], являются характерными
реакциями этого типа.
н Y: V
(Г Hal ** ЕЮН + с + НаГ (211)
4 \
«от Ь у^
р^1>, *■ EtOH + ||
***** Л
I-O Rx /R R\ /R
Br )C \/
С Br* *~ IBr + Д + ВГ" (212)
На основании изучения стереохимии реагентов и
продуктов установлено, что два уходящих атома от-»
щепляются, как показано, из сшти-положений
относительно друг друга. Отсутствие заметных количеств
продуктов tjuc-элиминирования означает, что
центральная связь в переходном состоянии обладает
значительным характером двоесвязанности.
140
цис-Присоединение и элиминирование
Рассмотренные в предыдущем разделе реакции
граис-элиминирования и присоединения включают
промежуточное образование радикалов или ионных
частиц. Установлено, что образование связей,
изображенных в 179 и 180, способствует протеканию
реакции, при которой пространственные препятствия
минимальны. Тем не менее в ряде случаев
присоединение по двойной связи, а также элиминирование
являются цис-реакциями. Широко известные
примеры этих процессов — присоединение ВН3 к олефи-
нам (гидроборирование) и газофазное
элиминирование галогеноводородов из алкилгалогенидов.
Элиминирование галогеноводородов из
алкилгалогенидов под действием основания проходит как
граяс-отщепление. Наиболее вероятное объяснение
этого факта с позиции ионного механизма приведено
ранее. В газовой фазе отщепление галогеноводорода
протекает по первому порядку и, по-видимому,
является простой мономолекулярной реакцией. В 1952 г.
Бартон, Хэд и Вильяме показали, что
элиминирование является цис-процессом. При
нагревании (—)-ментилхлорида 181 они получили (-f)-neH-
тен-2 182 и (+)-ментен-3 183 наряду с хлористым
водородом:
(СНзЬСн/^Н
(СН3)2СН
182(25%) 133(75%)
Образование 183 с выходом 75% означает, что по
крайней мере на 75% происходит ^ис-элиминирова-
ние, так как в случае траяс-элиминирования
образовывался бы только 182. Поскольку в ходе реакции
не образуются ни атомы водорода, ни атомы хлора,
можно сделать вывод, что реакция представляет
собой четырехцентровый процесс, в котором
одновременно происходит образование новых связей Н—С1
и я-С=С и разрыв связей С—Н и С—С1.
141
Гидроборирование, реакция алкенов с дибораном
с образованием ди- или триалкилборанов, часто
используется в синтетических целях. Присоединение
носит электрофильный характер, но является цис-сте-
реоспецифичным, что указывает на молекулярный
механизм реакции.
н-в' >СНЖ
&
(214)
Согласованные молекулярные реакции
Ряд реакций присоединения, элиминирования и
перегруппировок, по-видимому, имеет чисто
молекулярную природу, поскольку нет данных об образовании
в них ионных интермедиатов.
Реакции присоединения данной категории обычно
имеют цнс-характер; исходя из этого, разумно
предположить, что обе новые связи образуются
одновременно. Среди указанных процессов особое значение
имеет реакция Дильса — Альдера, в которой диен
реагирует с (активированным) олефином с
образованием производного циклогексена [реакция (215)].
Ряд примеров молекулярных реакций показан на
схемах.
(215)
а
цмс-Присоединение, проходит легко; используется в
препаративных целях.
*-*-
| | (216)
Эта реакция вообще не идет. Энергия активации должна
быть, вероятно, не менее 182 кДж/мюль.
т — т
R;
R/Xl R^^R CI
(217).
142
Реакция ^wc-элиминирования; требует очень высокой
энергии активации.
ь
BR2 ^у-^ВКз
-| > Т (218)
н У^н
^^-Присоединение осуществляется при комнатной
температуре или при небольшом нагревании.
Н2СЦ;
(219)
Протекает при нагревании довольно легко, если иметь в виду,
что в интермедиате теряется энергия стабилизации
ароматического кольца.
к чи —►
/\V\
0 СН? СН3
н ~-
о \
II О
С + I —)
Н2С СНз
Легко протекает при на
сн3—с=о
-
Протекает только выше
1гревании.
ц II
1 + С
8 о
500 °С.
^-о-н4^ 1 А-н
1
(220)
/ \
HgC CH3
(2J1>
(222)
Почти для всех органических функциональных групп эта
реакция не идет.
Как видно, некоторые из этих реакций протекают
очень легко, в то время как другие — очень медленно
или вообще не наблюдаются. Этот факт легко
объяснить, предположив, что в ходе приведенных реакций
при синхронном разрыве и образовании новых
связей связывание между атомами сохраняется. Образо-
143
вание и разрыв одинакового числа связей должны
приводить к переходным состояниям с не слишком
высокими значениями энергии. Однако эта простая
картина не вполне оправдывается. Наиболее
очевидная черта этих реакций заключается, по-видимому,
в том, что благоприятным условием для их
протекания является одновременный разрыв и образование
трех связей. В противоположность этому, реакции,
в которых должны разрываться и образовываться
только'две связи, либо не осуществляются, либо
протекают с различной скоростью, в том числе и
быстро. Важное значение для протекания
молекулярных реакций играют не отдельные связи и орбитали,
а все орбитали, которые принимают участие в
реакции, так как в переходном состоянии приобретают
значение молекулярные орбитали всей реагирующей
системы. Для систем, которые включают три
разрывающиеся и три образующиеся связи, т. е. шесть ор-
биталей, в переходном состоянии будет наблюдаться
частичное перекрывание всех этих орбиталей с их
ближайшими соседями в шестичленном кольце. В
результате возникает ситуация (шесть
перекрывающихся орбиталей, шесть электронов), которая
напоминает картину в бензоле, кс^гда существуют три
низколежащие молекулярные орбитали, которые
могут принимать шесть электронов. В итоге переходное
состояние стабилизовано за счет такого «бензолопо-
добного» резонанса и потому будет энергетически
относительно доступно для реагентов.
Аналогичное плоское переходное состояние для
четырехэлектронных систем должно приводить
к переходному состоянию циклобутадиенового типа.
Так как циклобутадиен не является резонансноста-
билизированной молекулой, энергия таких
переходных состояний будет гораздо выше, чем для
соответствующих молекул реагентов, и эти реакции будут
осуществляться чрезвычайно трудно, если вообще
будут иметь место. Поэтому реакции типа (216),
(217), (221), (222) протекают с трудом или не
осуществляются вовсе.
С этой точки зрения интересна реакция (223). По
Хюккелю, циклические л-системы с 4м -J- 2 электро-
144
нами являются ароматическими, в то время как
системы с 4п электронами — аяп/-ароматическими.
Однако если можно было бы создать я-систему,
и которой бы имелось отрицательное перекрывание
между одной парой * соседних орбиталей и
положительное перекрывание между всеми остальными, то
ситуация будет обратной: предпочтительным числом
п таком случае было бы 4я электронов. Трудно
создать карбоциклические соединения этого типа
(которые иногда называют системами Мёбиуса), однако
ситуацию такого рода можно наблюдать для более
сложных систем, и она может часто достигаться
в переходных состояниях. В реакциях (223) цикло-
бутановое кольцо может раскрываться в двух
направлениях: две орбитали, образующие рвущуюся
о-связь, могут вращаться или в одном и том же
направлении (конротаторное раскрытие цикла), или
в противоположных направлениях (диеротаторное).
Способ раскрытия цикла можно установить, исходя
из их продуктов. Если имеет место диеротаторное
раскрытие кольца, то образуется переходное
состояние 185 хюккелевского типа, тогда как
конротаторное направление приводит к переходному состоянию
184 мёбиусского типа, в котором одна пара орбита-
лей имеет отрицательное перекрывание. Поскольку
система включает четыре электрона, мёбиусское
переходное состояние будет обладать более низкой
энергией, в результате чего наблюдается
конротаторное раскрытие кольца.
П—Л конрота- диерота- п—л
Н3СН Н Н ' Г
сн2
н н
(223)
орбитали
* Или между нечетным числом пар.
И5
Гидроборирование
На первый взгляд реакция гидроборирования (218)
кажется аномальной, так как, по-видимому,
является четырехцентровой реакцией в смысле Хюккеля, но
обладает низкой энергией активации. Однако в
отличие от большинства органических соединений
соединения бора содержат свободную валентную орби-
таль. Наличие этой дополнительной орбитали делает
возможным согласованное протекание
гидроборирования (т. е. обе новые связи образуются
одновременно), хотя реакция не включает истинного "
циклического переходного состояния, так как новая связь
атома бора может использовать орбиталь бора,
отличную от орбитали, которая участвует в
образовании связи В—Н (186).
Таким образом согласованная реакция может
осуществляться по пути, не включающему
высокоэнергетический циклический интермедиат циклобутадие-
нового типа. Молекулы типа НС1 не имеют
вакантной валентной орбитали. Следовательно, как в
146
реагентах, так и в продуктах должны
использоваться одни и те же орбитали, и поэтому молекулярное
присоединение должно проходить через переходное
состояние циклобутадиенового типа с высокой
энергией.
Бирадикальные интермедиа™
Энергия активации, необходимая для протекания
некоторых «запрещенных» четырехцентровых
реакций, приближается или даже превышает энергию,
которая требуется для разрыва одной из ковалент-
ных связей системы. В таком случае может
оказаться возможным альтернативный путь реакции через
бирадикальный интермедиат. Имеются данные в
пользу того, что раскрытие кольца циклобутана с
образованием двух молекул этилена может проходить
через бирадикальный интермедиат 187, а не в
результате непосредственной четырехцентровой
реакции (225).
!_► СН2=СН2 (224)
187
,—> СН2=СН2 (225)
Доказательство образования бирадикального ин-
термедиата в реакции этого типа, проведенной в
обратном направлении, было получено Бартлеттом
с сотрудниками в 1964 г. Они нагревали смесь
1,1-дихлор-2,2-дифторэтилена 188 с транс,транс- или
цис,транс-гексалиеном-2у4 (189 и 190). Промежуточно
образующийся в этих реакциях радикал
стабилизован за счет сопряжения с двойной связью, что
приводит к аллильному радикалу. Оказалось, что
вращение в интермедиатном радикале конкурирует с
замыканием цикла. Это следует из того, что хотя
и 189, и 190 в большей степени образуют 193, чем
194, соотношение содержания 193/194 зависит от
конфигурации исходного диена и оказывается выше,
если исходят из 189 (5,1 1), чем в случае 190 (3,2: 1)
147
(в случае 190 имеет место также атака и по другой
двойной связи молекулы).
CF2=CCi2 188
н3с + н
н сн3
189
CF2=CCI2
Н
CF2-CC12-
Н3С | Н
н сн.
191
вращение
HiC)=H
н н
190
РН<Г
F2C-CGI2-
H3C4j
Н''
н
192
СН,
Н
F C1
H3C-J
CI
■н н
н сн,
193
F *Г Я1 CI
(226)
H3~cnJ
н сн,
н н
194
Н
Интерпретация данных о новых реакциях
присоединения и элиминирования
Реакции, которые приводят к траяс-присоедине-
нию по двойной связи, обычно включают
образование интермедиата (возможно радикального или
катионного). Если наблюдается цмс-присоедине-
ние, следует предположить молекулярную
реакцию.
Если предполагаемый механизм включает
циклическое переходное состояние, то следует
выяснить, является ли оно' «ароматическим». Если
нет, то должна наблюдаться высокая энергия
активации (порядка величины энергии разрыва
самой слабой связи системы).
Задачи
5.1. При действии гидроксида калия в ксилоле на
(+)-Ph (C10H7)MeSiOCOCH3 образуется (—) -Ph(C;0H7)MeSiOK,
который, в свою очередь, под действием хлористого ацетила мо-
148
жет переходить в (—)-Ph(CioH7)MeSiOCOCH3. Что можно
сказать о стереохимии этих двух реакций?
5.2. 5^2-Реакции вторичных алкилгалогенидов проходят с
инверсией. Как вы могли бы показать, что инверсия имеет место
также и в ходе 5^2-реакций первичных галогенидов?
5.3. Чистые триалкилбораны перегруппировываются при
нагревании с образованием смеси изомеров, в которой преобладают
соединения с бором, связанным с первичными атомами углерода.
Например: (3-С6Н13)зВ—► (1-С6Н13)зВ. Предложите механизм,
который объясняет перемещение атома бора вдоль углеродной
цепи. При этом примите во внимание факт, что этот процесс не
наблюдается, если углеродная цепь содержит четвертичный атом
углерода, например группировку С—СМе2—С.
5.4. Объясните, почему ментилхлорид 181 при обработке
спиртовым раствором КОН будет давать исключительно мен-
тен-2 182.
5.5. При нагревании в течение пяти часов при 260 °С
(+)-PhMeEtCNC перегруппировывается в оптически неактивный
изомер PhMeEtCCN. Предложите правдоподобный механизм, а
также возможный контрольный эксперимент.
Глава 6
ДРУГИЕ МЕТОДЫ ИЗУЧЕНИЯ
МЕХАНИЗМА РЕАКЦИЙ
В гл. 2—5 описаны основные методы, используемые
для установления механизма реакций. Существует,
кроме того, ряд других методов, которые обычно
в меньшей степени используются, однако часто дают
важные дополнительные сведения или в пользу
предполагаемого механизма и приводят к более
детальному пониманию природы процесса, или против
первоначальных представлений.
Стерическое ускорение и замедление
Многие реакции включают изменение числа групп,
координированных с определенным атомом, при
образовании интермедиата или переходного состояния.
Например, в реакциях 5^2 первоначально
четырехвалентный атом углерода приобретает
дополнительную группу в переходном состоянии, тогда как в
реакциях SnI интермедиатом является ион карбо-
ния, содержащий только три связанные с углеродом
группы. Исходя из этих соображений, при прочих
равных условиях, реакции SNl должны быть
предпочтительны, если заместители R представляют
собой объемистые группы (т. е. не водород).
ОН"
R3C-Hal -
> HO-CR3 Hal6'
1
->- Har + R3C+ OH_
R3C-OH + НаГ (227)
В соответствии с этим метил- и этилбромиды почти
исключительно подвергаются реакциям SN2. Изопро-
150
пилбромид гидролизуется по обоим направлениям,
а грег-бутилбромид реагирует главным ч>бразом по
направлению SN\.
Катализируемый кислотами гидролиз сложных
эфиров обычно проходит по следующей схеме:
1 /=\ ^°
?-0Н I? \J-C\0H (228)
+ ROH ОН
Когда молекула воды атакует промежуточно
образующийся ион карбония 195, координационное
число центрального атома углерода увеличивается
от трех до четырех. В концентрированной серной
кислоте более затрудненный эфир 196 гидролизуется
по альтернативному направлению (229), в котором
из-за промежуточного образования ацильного иона
197 лишь с дважды координированным атомом
углерода стерические препятствия для подхода воды
оказываются минимальными.
Ч_/ ~C\6r
197
(229)
Ш
Стерические эффекты редко можно с
определенностью отделить от других возможных эффектов
заместителей (см. следующий раздел).
Эффекты заместителей
Переходные состояния или интермедиа™ часто
обладают зарядом или имеют радикальный характер.
В таком случае подходящие заместители у
реакционного центра могут стабилизовать или дестабилизо-
вать переходное состояние или интермедиат.
Стабилизация катионов
Экспериментально найдено, что образование иона
карбония облегчается при замещении атомов
водорода у карбониевого центра на алкильные группы.
Так, при растворении я-пропилфторида в пятифтори-
стой сурьме первоначально образующийся я-пропил-
катион перегруппировывается в более стабильный
изопропильный катион. Еще более стабильными
являются третичные катионы: трет-бутилбромид
растворяется в жидком S02 с образованием растворов,
проводящих электрический ток, которые содержат
грег-бутилкатион; первичные и вторичные галоге-
ниды ионизуются труднее. Такие группы как CF3,
дестабилизуют ионы карбония. Стабилизующий
эффект заместителей (алкил > Н > CF3) отчасти
объясняется с позиций индуктивного эффекта: некоторые
группы (СН3) могут более эффективно предоставлять
свои а-электроны для стабилизации катиона, чем
другие (CF3); в последнем случае поляризация
электронов осуществляется по направлению к CF3-rpynne,
что вызывает дестабилизацию карбониевого иона.
Стабилизация под действием заместителей
является экспериментальным фактом, и для многих
задач не имеет значения то, как можно эту
стабилизацию распределить между различными возможными
причинами (индуктивный, стерический эффекты или
сверхсопряжение, изменение прочности связи с
изменением гибридизации и т. д.). Если алкильные
группы при реакционном центре ускоряют реакцию, в то
время как группы CF3 замедляют ее, то это
является хорошим доводом в пользу интермедиата с
некоторым характером иона карбония.
152
Например, считается, что присоединение галогено-
водородов к олефинам, если оно проводится с
чистыми реагентами, включает промежуточное
образование ионов карбония типа 198, 199 и 200. Найдено,
что пропен реагирует с галогеноводородами быстрее,
чем этилен, и дает изопропильные соединения
[реакция (231)], а не я-пропильные производные. С другой
стороны, 3,3,3-трифторпропен реагирует медленнее,
чем этилен, и приводит к первичному бромиду 202.
Относительные скорости и ориентация в этих
реакциях согласуются с образованием промежуточного
иона карбония. В реакции (231) изопропильный
катион 199 более стабилен, чем я-пропильный, что
объясняет предпочтительное направление присоединения;
изопропильный катион 199 также более
стабилизован, чем этильный катион 198, что объясняет, почему
присоединение к пропену проходит быстрее, чем к
этилену. В реакции (232) наличие CF3-rpynnbi де-
стабилизует любой из возможных катионов 200 или
201, поэтому эта реакция протекает медленнее, чем
(231). Однако в первичном катионе 200 дестабили-
зующая CF3-rpynna дальше удалена от карбоние-
вого центра, чем в альтернативном катионе 201,
поэтому в качестве продукта реакции образуется
первичный бромид 202.
сн2=сн2-^^ сн3сн2 —^с2н5вг
198 (230)
СН3-СН=СН2 iffi^ CH3-CH-CH3 —> СН3-СНВг-СН3
199
сн3-сн2-сн2 (231)
менее стабилен
CF3TCH=CH2 -ЫЙ^ CF3-CH2-CH2 ► CF3-CH2-CH2Br
200 202
CF.?-CH-CH3
201 (232)
менее стабилен
153
Свободные радикалы
Алкильные заместители стабилизуют радикальные
центры, однако эффекты выражены слабее, чем в
случае карбониевых ионов, так как в радикалах
отсутствует требующий стабилизации формальный
положительный заряд. Экспериментально показано, что
третичные радикалы более стабильны, чем
вторичные, которые, в свою очередь, более стабильны, чем
первичные. Например, энергия диссоциации
связей D(R—Н) растет в ряду: трет-Ви—Н 380,
изо-Рт—Н 345, Et—H 410 и Me—H 437 кДж/моль.
Поскольку имеются убедительные данные в пользу
того, что, например
D (Et—Н) « D («-Рг—Н)
ясно, что вторичные радикалы лучше стабилизованы,
чем первичные, тогда как третичные радикалы
являются еще более стабильными. Эти различия в
стабильности радикалов наряду с другими факторами
углубляют наше понимание цепных свободнорадикаль-
ных реакций бромистого водорода с олефинами, в
которых найдейо, что пропен реагирует быстрее, чем
этилен, и образует в качестве продукта 1-бромпро-
пан [реакция (233)]. При присоединении атома
брома к пропену предпочтительным оказывается
образование более стабильного (по сравнению с
первичным радикалом 204) радикала 203, что и определяет
направление присоединения; радикал 203
является также более стабилизованным, чем радикал
• СН2СН2Вг, так что присоединение к пропену
осуществляется быстрее, чем к этилену.
СЯ^Сй=СНз +Вг—* Шз-СН-СН2ВгН^СНзСН2СН2Вг+Вг-
^^с 203
^^"^ (233)
^ CH3-CHBr-C*V
2Q4
154
Сопряжение
Введение ненасыщенных группировок, таких как
С = С, С = 0 или Ph, у реакционного центра
облегчает реакции, проходящие через ионы карбония, карб-
пнионы или свободные радикалы (благодаря этому
ускоряются, например, реакции 5^2). Принимая во
внимание такую потерю избирательности, этот метод
не может быть очень надежным диагностическим
приемом проверки определенного типа механизма.
Однако такой способ может иметь отрицательное
предсказательное значение: если постулируется, что
реакция протекает через промежуточный ион карбония,
то она не должна ускоряться при наличии фениль-
ного заместителя у предполагаемого карбониевого
центра; в таком случае этот механизм является
сомнительным. Действие способной к сопряжению
группы заключается в распределении заряда или
плотности свободного электрона в молекуле; делокализо-
ванная структура является более стабильной, чем
(незамещенная) локализованная структура.
СН2 ч-> + ( \=СН2 и т. д. (234)
Электронные эффекты заместителей:
уравнение Гаммета
Было предпринято несколько попыток
количественной оценки эффектов заместителей. Одна из
наиболее удачных таких схем получена из сравнения
кислотности ж- и n-замещенных бензойных кислот,
которая может быть измерена с высокой точностью.
м- или я-ХСбН4СООН —> ХСбН4СОСГ + Н+ (235)
Константа заместителя а для каждого
заместителя была определена по уравнению:
. Кв (ХС6Н4СООН) _
10g /Са (С6Н5СООН)
= ptfa (СбН5СООН) - р/Са (ХСбН4СООН) = <х (236)
Поскольку электроноакцепторные заместители
стабилизуют анион кислоты, они будут увеличивать
о-
155
Таблица 5. Значения констант Гам мет а для ряда
заместителей *
Заместитель
NMe2
СНз
СН30
F
С1
Вг
I
COOEt
CF3
CN
N02
мета-
a
-0,21
-0,07
0,11
0,34
0,37
0,39
0,35
0,37
0,42
0,56
0,71
о
-0,83
-0,17
-0,27
0,06
0,23
0,23
0,18
0,45
0,54
0,66
0,78
пара-
о+
-1,70
-0,31
-0,78
-0,07
0,11
0,15
0,13
0,48
0,61
0,66
0,79
о"
0,68
0,87
1,24
* Для более полной сводки значений констант Гаммета см., например,
К. Д. Риче% У. Ф. Сэджер. В кн. Современные проблемы физической
органической химии. Пер. с англ. Под ред. М. Е. Вольпина. *Мир*, М., 1967, с.
498.
силу соответствующей кислоты и обладать, таким
образом, положительными значениями а. В
противоположность этому электронодонорные заместители будут
иметь отрицательные значения а. Примеры некоторых
констант даны в табл. 5.
Как можно использовать эти константы
'заместителей для других реакций ароматических
соединений? Для очень большого числа различных реакций
замещенных соединений АгХ, так же как и для
незамещенного АгН, найдено, что график зависимости
log (katx/kath) против а представляет собой прямую
линию. Это означает, что справедливо соотношение
типа:
Это выражение известно как уравнение Гаммета.
Наклон прямой р является характеристикой данной
реакции и представляет собой меру чувствительности
переходного состояния к электронным
взаимодействиям. Сила бензойных кислот увеличивается в
присутствии электроноакцепторных заместителей и,
поскольку р для кислотной диссоциации бензойных
кислот равно единице по определению, положительное
156
значение р для реакции означает, что в переходном
состоянии аккумулируется отрицательный заряд или
что положительный заряд исходной молекулы
уменьшается в переходном состоянии. Наоборот, реакции,
в которых положительный заряд аккумулируется на
реакционном центре в переходном состоянии, имеют
отрицательное значение р. Некоторые примеры
реакций и соответствующие им значения р приведены
в табл. 6.
Константы о+ и о"
Стабилизация или дестабилизация замещенного бен-
зоат-иона 205, которая ответственна за различие
в кислотности бензойных кислот, осуществляется
главным образом за счет индуктивного эффекта
(поляризация электронов вдоль а-связей), так как
отрицательный заряд бензоат-иона не сопряжен с
ароматическим кольцом (приемлемые резонансные структуры
с отрицательным зарядом в кольце изобразить не
удается). Однако в некоторых реакциях могут
возникать ионные частицы или переходные состояния,
заряд в которых способен находиться в сопряжении
с ароматическим радикалом. Для таких реакций
становится существенной делокализация как по кольцу,
так и на подходящих /zapa-заместителях. Такие
группы, как МеО и NMe2, эффективно стабилизуют
положительный заряд, тогда как такие группы, как
СООМе, CN и N02, эффективно стабилизуют
отрицательный заряд. Типичные резонансные структуры
показаны ниже на примере замещенного бензильного
катиона 206 и феноксильного аниона 207.
Заместители в жега-положениях не обладают подобным
резонансным эффектом.
СНо СНг
J I'
ОМе +ОМе
206
157
207
Таблица 6. Значения р-фактора Гаммета для ряда реакций
Среда
Вода 1,00
Этанол 1,96
Вода 0,46
Вода 2,77
SOa -3,97
Ацетон/вода —4,54
Уксусный ангидрид —7,29
Хлорная кислота/диок- —6,2
спи/вода
Реакция
(235) АгСООН ^± АгСОО" + Н+
(245) АгСООН ^± АгСОО" + Нн
(246) АгСН2СООН ^± АгСН2СОО~ + W
(247) ArNH* ^± ArNH2 + Н*
(248) Аг3СС1 ^± Аг3С+ + СГ
(249) АгСМе2С1 + Н20 -> ArCMe2OH + HCI
(250) АгН + НШз -> ArN02 + Н20
(251) АгН + НОВг -> ArBr + H2Q
(З&с) АгН + Вг2 -* АгВг + НВг
(253) ArCl * + МеСГ -> АгОМе + СГ
(254) АгСГ + СН2—СН2 -> АгОСН2СН20"
О
(255) АгСНГ2. -> АгСН* + е"
(256) АгСНСНз -> АгСН=СН2 + НС1
С1
(257) АгСНз + С1. -> ArCH2.+ HC1
(258) ArCOCl + «-Bu3Sn -> АгСО + /i-Bu3SnCl
* ArCl-
N02
Уксусная кнслота/вода —12,1
Метанол +3,9
Этанол —1,12
Газ —20
Газ —1,36
СС14 -0,66
м -Ксилол
+2,6
Для определения констант заместителей,
пригодных для описания ситуаций, в которых существенное
значение имеют резонансные эффекты, были
предложены две стандартные реакции, рассмотренные ниже.
Это кислотная диссоциация замещенных фенолов
[равновесие (238)] и сольволиз замещенных кумил-
хлоридов в водном ацетоне [реакция (239]. Величины
р для этих двух реакций были установлены в
результате изучения лишь жета-замещенных соединений
и использованы для определения серий а"- и
(^-величин на основании скоростей (или констант
равновесия) реакций яара-замещенных соединений. Эти
константы должны быть более пригодными (чем
а-значения) для описания других реакционных
систем, в которых положительный или отрицательный
заряд в ходе реакции находится в сопряжении с
ароматическим кольцом.
(239)
Уравнение Гаммета и механизм реакций
Уравнение Гаммета может быть использовано для
получения информации о механизме реакции.
Представляет интерес как знак и величина р, так и то,
какая из трех констант заместителей а, а+ или о~
дает лучшее соотношение Гаммета. Информация
о переходном состоянии, которую можно получить
из этих параметров, суммирована в табл. 7. Иногда
категория реакции (в смысле соотношения
параметров Гаммета), приведенная в табл. 7, позволяет
сделать выбор между двумя возможными механизмами,
однако чаще категория подтверждает уже постули-
160
рованный механизм, а величина р дает более
количественную информацию о заряде, возникающем в
переходном состоянии.
Таблица 7. Определение типа переходного состояния
по знаку р и природе наиболее подходящей константы
а, а+ и от в уравнении Гаммета *
Положительное р
Отрицательное р
а- {
[ Реакционный центр **
сопряжен с групппой
Аг — в переходном
состоянии на реакционном
центре возникает
отрицательный заряд. Пример:
[ реакция (238)
Реакционный центр
не сопряжен с группой
Аг — переходное
состояние в меньшей степени
нуждается в электронном
взаимодействии, чем
реагент. Пример: гидролиз
ароматических сложных
эфиров под действием
оснований
Реакционный центр
сопряжен с группой Аг
и стабилизован электро-
нодонорными
заместителями — сопряжение
менее важно в
переходном состоянии. Примеры
редки
Реагент стабилизован за счет
сопряжения, уводящего
электроны к группе Аг — это
сопряжение играет меньшую роль
в переходном состоянии.
Пример: реакция (254)
Реакционный центр не
сопряжен с группой Аг — переходное
состояние более чувствительно
к электронам, чем реагент.
Пример:
Ar2C=N=N+PhCOOH ->
-> Ar2CHCOOPh+N2
Реакционный центр сопряжен
с группой Аг — переходное
состояние более чувствительно
к электронам, чем реагент.
Пример: реакция (241)
* Значения а используются тогда, когда значения а или а"
непригодны.
** Под реакционным центром в этой таблице подразумевается
положение, на котором в ходе реакции возникает или исчезает заряд.
В качестве примера использования уравнения
Гаммета для выбора между альтернативными
механизмами реакции рассмотрим катализируемую кислотой
перегруппировку Ы-нитро-Ы-метиланилинов в
соответствующие о-нитро-М-метиланилины, два возможных
6 За к. 1044
161
механизма которой показаны на схемах (240) и
(241):
N Me-+N-H N
н+
(а)
+ N0* (240)
• N +
+ »N02
N
X^N°2
IIJ <241>
Если скорость реакции определяется стадией j(a),
то следует ожидать, что р будет положительным и
должна наблюдаться корреляция с <т, так как элек-
тронодонорные заместители в большей степени
(индуктивно) должны стабилизовать 208, чем
незаряженный метиланилин 209. С другой стороны, если
реализуется путь (б), сопряжение положительного
заряда с кольцом в 210 будет вызывать большую
стабилизацию, чем индуктивная стабилизация в реагенте
208. Следовательно, для этого направления логично
ожидать отрицательного значения р и корреляции
с от1". Действительно, экспериментально р найдена
равной — 3,7, с лучшей корреляцией для а+, что
служит веским подтверждением направления (241),
а не (240).
Иногда зависимость Гаммета проливает свет на
природу интермедиата реакции. Свободнорадикаль-
ное бромирование замещенных толуола
молекулярным бромом представляет собой цепной процесс (242).
Сходное бромирование можно провести при
использовании вместо брома N-бромсукцинимида 211. Пер-
162
воначально считалось, что механизм этой реакции
описывается схемой (243). Однако недавно
высказано предположение, что функция N-бромсукцинимида
заключается в генерировании небольших количеств
молекулярного брома, которое затем поддерживается
за счет реакции (244) (как известно, быстрой
реакции), так что бромирование фактически
осуществляет молекулярный бром [реакция (242)]. Значения
р, полученные при изучении реакций замещенных
толуола с использованием в качестве бромирующих
реагентов соединений 211, 212, 213 и молекулярного
брома, подтверждают такой механизм. С точностью
эксперимента величины р идентичны, хотя 213,
содержащий в качестве заместителей четыре атома
фтора, и 212, имеющий четыре метильных
заместителя, и -бром — совершенно различные соединения.
Весьма маловероятно, что радикалы, образующиеся
из четырех бромирующих агентов, будут атаковывать
замещенные толуола с одинаковой легкостью.
Единственно возможным объяснением этому является то,
что во всех случаях образуется один и тот же ин-
термедиат, атом брома, а механизм бромирования
описывается схемами (244) и (242), но не (243).
сна
Вг
медленно
+ НВгз£%о^
СН2Вг
СН2Вг
(243)
6*
163
N-Br + HBr 5^ I N-H + Br2
(244)
1
Г>-Вг
Ъ
211
/5=-l,46±0,07
„?\Л
н3с b
212
-1,45 ±0,07
Br
fM!
pT N-Br Br2
F Ъ
213
-1,36 ±0,05 -1,36 ±0,5
Значение р-фактора является количественной
мерой заряда, сосредоточенного на реакционном центре
в переходном состоянии. Самые высокие значения
наблюдаются для процессов ионизации, проходящих
в газовой фазе в масс-спектрометре [реакция (255) J,
где отсутствует растворитель, стабилизующий ионы,
и поэтому стабилизующий эффект заместителей
максимален. Меньшие значения р факторов найдены:
(а) в растворах полярных растворителей — р для
диссоциации бензойной кислоты в этаноле выше, чем
в воде;
(б) если ароматическое кольцо удалено от центра,
несущего заряд, — р для фенилуксусной кислоты
приблизительно в два раза меньше, чем для бензойной
кислоты;
(в) если образование заряда в переходном
состоянии наблюдается лишь в небольшой степени, как,
например, в радикальных реакциях (257) и (258)
(ск. табл. 6).
Для оценки величины заряда, возникающего в
ходе реакции, полезно рассмотреть полученные
значения р в сравнении с величинами р, найденными для
реакций в .аналогичном растворителе, которые
проходят с полным разделением или исчезновением
заряда и которые (реакции), таким образом, должны
давать максимальное значение р в рассматриваемом
растворителе. Примерами таких реакций являются
сольволиз кумилхлорида [реакция (249)] и
ионизация бензильного радикала [реакция (255)]. Для
164
оценки степени разделения зарядов в переходном
состоянии реакции (256) следует сравнить величину
р, равную—1,36 при 335 °С, что соответствует
значению — 2,6 при 25 °С, с величиной р —20,
полученной для реакции (255).
Аг -СН-СН2
I I
С1 Н
Аг-СН=--СН2
£"
-Н
Аг-СН=СН2
Ci—Н
(256)
Сравнение показывает, что разделение зарядов
в (256) хотя и значительно, но не достигает полной
ионизации.
Эффект растворителей
Органические кислоты, такие как бензойная кислота,
в водном или спиртовом растворе ионизованы в
большей степени, чем, скажем, в растворе ацетона или
гексана. Вода и спирт стабилизуют анионы и
катионы за счет сольватации, что благоприятствует
диссоциации нейтральной органической кислоты на ионы.
Эти растворители можно назвать ионизующими в
противоположность неионизующим растворителям,
таким как ацетон и гексан (заметим, что хорошие
ионизующие растворители обладают высокими ди-
польными моментами, а также способны к
образованию водородных связей).
НА 5F=± H+ А' (259)
природа растворителя
играет меньшую роль
для недиссоциированной
кислоты, чем для ионов
стабилизованы
ионизующими
растворителями
Хьюз и Ингольд показали, что эффект того же
типа действует и в химических реакциях и что в
общем случае, если заряд в переходном состоянии
увеличивается по отношению к исходным реагентам,
скорость реакции будет существенно возрастать при
проведении реакции в «более высокоионизующем
165
растворителе. Наоборот, если заряд исчезает в
переходном состоянии, то скорость реакции будет падать
при использовании высокоионизующего
растворителя. В некоторых реакциях заряд не возникает и не
исчезает, а делокализуется в переходном состоянии.
Например, в реакциях нуклеофильного замещения,
таких как вторая реакция в табл. 8, заряд
распределен между вступающей и уходящей группами.
Протеканию таких реакций способствует использование
неионизующих растворителей, так как в
ионизующей среде исходные ионные реагенты будут
стабилизированы в большей степени, чем переходное
состояние. Однако в этих случаях эффект
растворителя будет меньше, чем для реакций, в которых
происходит образование или исчезновение заряда.
Этот принцип может быть использован для
широкого круга реакций, включающих частицы
различного заряда. Некоторые примеры приведены в табл. 8.
Из сказанного следует, что варьирование
растворителя дает удобный способ проверки
постулированного механизма. Для эффективного использования
этого способа необходимо изменять ионизующую
силу растворителя лишь в небольшой степени,
например изменяя соотношение компонентов системы
ацетон/вода от 80 : 20 до 70 : 30, так как при этом
опасность изменения механизма мала. Более резкое
изменение растворителя (например, переход от
ацетона к воде) может привести к изменению
механизма реакции; например от механизма 5^2 можно
перейти к реакции 5И.
Постулат Хэммонда
В заключение этой главы рассмотрим концепцию,
которая позволяет проникнуть в природу изменения
реакционной способности в большей степени, чем
большие различия в механизме. Качественно
экзотермические, термонейтральные и эндотермические
реакции можно представить, как показано на рис. 6,
в виде графика зависимости энергии реакционной
системы от произвольной координации реакции,
Постулат Хэммонда устанавливает, что для
высоко экзотермичных реакций переходное состояние
166
Таблица 8. Влияние растворителя на скорость реакций
Реагенты
Переходное состояние
Продукты
Тип
Влияние увеличения
полярности
растворителя
на скорость реакции
(СНз)зС—Вг -+ (СН3)зС+ Вг"
н2о
(СН3)зС—ОН2 + Вг" Возникновение Большое ускоре-
заряда ние
НО" + СНз—Вг
б" б+
НО •■•• СНз—* Вг
НО^-СНз + Вг*
Делокализация Небольшое сниже-
заряда ние
о+ б+
Н20 -f +S(C2H5)3 -> Н20 С2Н5 S(C2H5)2 -> Н2ОС2Н5 + S(C2H5)2 Делокализация Небольшое сниже-
заряда ние
б" б+
HO~ + +S(C2H5)3 -> НО-С2Н5 S(C2H5)2 -* HOC2H5+S(C2H5)2 Уничтожение
заряда
Большое снижение
близко напоминает исходные соединения [рис. 6(a)],
тогда как для сильно эндотермичных реакций
переходное состояние похоже на продукты реакций
[рис. 6 (в)]. В случае приблизительно
термонейтральных реакций переходное состояние будет
представлять собой нечто среднее между исходными
соединениями и продуктами, в котором образование
Рис. в. Постулат Хеммонда:
(а) —экзотермические реакции; (б) —
термонейтральные реакции;
(в)—эндотермические реакции.
Координата реакции
(б)
Координата, оеанции
и разрыв связей происходит примерно наполовину.
Эта концепция может быть с успехом использована
для широкого ряда органических реакций. Если
реакции включают образование интермедиатов, то
постулат применим лишь к индивидуальным реакциям, а
не ко всему процессу в целом.
Для реакций, которые примерно термонейтральны,
этот постулат означает, что любая частичная стаби-
168
лизация или дестабилизация продукта по отношению
к исходному соединению будет примерно наполовину
эффекта отражаться в переходном состоянии. В
качестве примера рассмотрим процесс переноса
водорода в реакции метильного радикала с метаном
и этаном. Термонейтральная реакция метильного
радикала с метаном имеет энергию активации
61 кДж/моль. Аналогичная реакция между метиль-
ным радикалом и этаном включает атаку по связи
С—Н, которая приблизительно на 28 кДж/моль
слабее, чем связь С—Н в метане.
СН3* + Н—СН3 —>
Л//, Е,
кДж/моль кДж/моль
—► СНз—Н+СНз- 0 61 (260)
СН3 • + Н-СН2СН3 —*
—* СНз—Н + СНзСНз» —28 49 (261)
В переходном состоянии связь С2Н5—Н будет
разорвана примерно наполовину и, следовательно,
разница в энергии двух переходных состояний для
реакций (260) и (261) должна быть примерно
равной 14 кДж/моль, что хорошо согласуется с
экспериментальной разницей в 12 кДж/моль.
В случае эндотермических реакций переходное
состояние в большей степени напоминает продукты,
и любые причины, вызывающие стабилизацию
молекул продуктов, будут сильно отражаться на
переходном состоянии. Так, реакции атомов брома с метаном
и этаном [(262) и (263)] являются достаточно
эндотермическими. Для этой пары реакций разница в
энергии активации равна 21 кДж/моль, что
составляет 75% разницы энтальпий двух реакций.
ЛЯ £,
кДж/моль кДж/моль
Вг« + Н-СНз —*
—> Вг—Н+СНз» +72 76 (262)
Вг. + Н—СН2СН3 —*
—> Вг—Н + СН3СН2. +44 65 (263)
169
В сильно эндотермичных реакциях, таких как
термический, гомолиз ковалентных связей или стадия
ионизации в реакции S^l, переходное состояние и
продукты почти идентичны и таким образом любые
эффекты стабилизации (делокализация и т. п.),
которые имеют место в продуктах, будут почти в той
же степени присутствовать в переходном состоянии
и, следовательно, будут оказывать большое влияние
на скорость реакции.
В реакциях, в которых молекула может
подвергаться атаке реагентов в более чем одно положение
или если реагент реагирует с двумя близкими
соединениями, обычно в различных условиях продукты
образуются в различном соотношении. Для таких
реакций было установлено общее правило, согласно
которому чем выше реакционная способность
реагента, тем он менее селективен при атаке одной из
молекул (или одного из положений в молекуле).
Постулат Хэммонда дает объяснение этим различиям в
селективности.
Бромирование толуола бромноватистой кислотой*
в водном диоксане дает смесь п- и ж-бромтолуолов
в отношении 10:1. Бромирования можно достичь,
хотя и медленнее, действием брома в уксусной
кислоте; в этом случае отношение пара-1мета-
возрастает до 200:1. Существуют убедительные данные
в пользу того, что бромирующим агентом в первом
примере служит катион Вг+ или его гидратирован-
ная форма; такой реагент представляет собой реак-
ционноспособную, обладающую высокой энергией,
частицу. Таким образом, реакция (264) должна быть
гораздо более экзотермичной, чем реакция (265), в
которой используется молекулярный бром. Исходя
из этого, переходное состояние реакции (264) будет
ближе к исходным соединениям, и электронное
влияние метильной группы будет относительно невелико.
С другой стороны, бромирование молекулярным
бромом (реакция 265) — эндотермичный процесс и
переходное состояние должно близко напоминать интер-
медиат Уэланда 214. Так как стабилизующее влияние
метильной группы несомненно больше при атаке в
дшра-положение, чем в жега-положение, отношение
17Q
пара-/мета- оказывается гораздо выше, чем для
реакции (264).
СИ и
переходное состояние
ближе к исходным (264)
СН3 соединениям
СН3 Вг Н
^ _ переходное состояние
Вг —Вг + Г ll -> fl+JL + Br ближе к интермедиату
°Н3 214 (265)
Такое положение имеет достаточно общее
значение: реакционноспособные реагенты
характеризуются относительно низкой селективностью, в то
время как менее активные реагенты имеют
тенденцию к большей селективности действия. Если
оказывается, что при изменении условий реакции
наблюдается существенное изменение селективности, то это
может означать, что в новых условиях присутствует
иная реагирующая частица (например Вг+, а не Вгг).
Однако могут встречаться и более тонкие изменения;
например, растворители, образующие комплексы с
реагентом, часто понижают его реакционную
способность и увеличивают его селективность.
Задачи
6.1. СН3Вг, С2Н5Вг, изо-С3Н7Вг. и трег-С4Н9Вг реагируют с
иодид-ионом в ацетоне с относительными значениями констант
скоростей второго порядка 256 000: 1667 13: 1. Предложите
объяснение этих различий.
6.2. Константа скорости бимолекулярной реакции изо-СзН7Вг
с ОН" в водном спирте уменьшается в 1,6 раза при увеличении
содержания воды от 20 до 40%. Когда аналогичное изменение
растворителя было проведено для бимолекулярной реакции
ызо-СзН7Вг с водой, скорость возросла в 2,8 раза. Объясните
эти различия.
6.3. Предложите ряд /шра-заместителей, для которых (а) а+
значительно отличается от а и (б) а* — значительно отличаются
от а.
6.4. 1-Арилэтилацетаты разлагаются при нагревании в
газовой фазе с образованием уксусной кислоты и арилэтиленов; эти
171
превращения подчиняются кинетике первого порядка. Для
заместителей в ароматическом кольце наблюдается гамметовская
корреляция при использовании о+ и р = —0,7. О чем, с точки
зрения механизма разложения, свидетельствуют эти данные?
6.5. Установлено, что как цис-, так и т/?анс-2-арилциклопен-
тилтолуолсульфонаты АгСбНвОБОгСб^СНз элиминируют толуол-
сульфокислоту при взаимодействии с грег-бутилатом калия,
давая арилциклопентены. Величины р уравнения Гаммета для цис-
соединений равны +1,48, для г/?а«с-соединений +2,76. Объясните
эти результаты с позиции вероятного переходного состояния.
6.6. Предложите реакции (иные, чем описанные в этой главе),
которые будут иметь (а) положительные и (б) отрицательные
значения р и для которых лучшая корреляция достигается с
(1) а, ("2) а+ и (3) а-, т.е. в общей сложности шесть категорий.
При поиске примеров для некоторых из этих категорий могут
возникнуть затруднения.
ЗАКЛЮЧЕНИЕ
В предыдущих главах были рассмотрены некоторые
методы изучения механизмов органических реакций.
Выбор общей методики исследования зависит от кон-
кретного случая; на рис. 7 изображена типичная
последовательность изучения механизма реакции.
Обычно первая стадия исследования включает установление
продуктов реакции. Затем можно перейти к
кинетическому изучению, в результате которого
постулируется механизм (или несколько возможных
механизмов) реакции. Помимо этого, представления о
возможном механизме могут возникнуть и на стадии
изучения продуктов реакции. Следующий этап
должен включать проверку постулированного механизма
с помощью максимально возможного,, в идеальном
случае, числа различных методов. При этом можно
руководствоваться следующим общим принципом: чем
больше постулированный механизм отличается от
механизмов, установленных для близких реакций, тем
более строго следует его проверять. Напротив, если
реакция лишь незначительно отличается от серии
реакций с известным механизмом, можно довольно
уверенно предположить, что будет иметь место тот
же механизм без какого-либо его изучения.
Например (как уже отмечалось в гл. 1), если гидролиз
и-пропилацетата был тщательно изучен при
определенных условиях и был установлен его механизм,
разумно предположить, что в тех же условиях
гидролиз и-бутилацетата должен проходить таким же
образом. Однако, с другой стороны, было бы
опрометчиво предложить тот же механизм для трет-бу*
тилацетата, в котором заместители находятся близко
к реакционному центру. В этой книге описан ряд
частных методов проверки механизма. Этот перечень
173
Изучаемая
реакция
Идентификация
продуктов
.Установление
Кинетическое
изучение
разрывающихся и (или,
образующихся связей
Постулирование
механизма*^
}
Побочные
продукты ?
j
/^Проверка механизма
iвсеми) максимально воз-у^
можными путями ^
((((((( \ a
Стерео- Нине- Ннтер- Изотопное Здлрект Залрект Другие Новые методы,
химия тика медиа ты изучение замести- раство- методы предназначен-
телеа рителей \ мыс для
частного сличая
к к к к к
Объясняет
другие
результаты
Результаты согласуются
с предполагаемым
механизмом
Результаты не сог-\
ласу юте я с пред по
лаеаемым
механизмом
По-видимому
„истинный"
механизм •
Рис. 7 Схема исследования механизма реакции.
нельзя считать исчерпывающим; следует учитывать,
что новые методы проверки механизма изобретаются
постоянно. Результаты контрольных экспериментов
могут согласовываться с предлагаемым механизмом;
если же они противоречат ему, следует провести
усовершенствование данного механизма или выбрать
новый. Механизм, согласующийся со всеми
известными фактами, можно считать приемлемым для данной
реакции.
Далее, уверенность в механизме будет возрастать,
если механизм согласуется с общепринятыми
химическими представлениями, и в еще большей степени,
если он связывает отдельные химические факты в
более общую картину. Наконец, «истинный»
механизм может часто привести к разработке иных
реакций и идей, которые, в свою очередь, можно
проверить, в то время как механизм, который содержит
объяснение лишь частных результатов, редко
позволяет сделать такие выводы.
«Открытие» пилтдаунского человека
Представляет интерес рассмотреть в
ретроспективном плане некоторые реакции, о механизме которых
были первоначально выдвинуты неверные
представления. Мы разберем два уже упомянутых выше
примера: димеризацию триарилметильных радикалов и
равновесие между открытой и циклической формами
некоторых циклопропановых соединений. Каждый
случай характеризуется различной степенью
неправдоподобности.
Равновесие триарилметил/гексаарилэтан никогда
не позволяло удовлетворительно объяснить, почему
на константу равновесия столь большой эффект
оказывает наличие трех пара-заместителей по
сравнению с ди-яара-замещенными. Когда было
установлено, что димеризация осуществляется за счет
образования а-яара-связывания, стало ясно, что причина
пара-эффекта имеет стерическую природу.
Представление о димере трифенилметила как о гекса-
фенилэтане привело также к другому затруднению.
Скорость образования пероксида трифенилметила
при взаимодействии димера с кислородом воздуха
175
оказывается выше, чем скорость диссоциации димера
на свободные радикалы. Для того чтобы объяснить
этот факт, было высказано предположение, что
образование пероксйда должно протекать по цепной
реакции (266). Однако достоверные примеры
реакций типа, представленного последней стадией
схемы (266) — прямое замещение одного радикала
другим у ациклического углерода, неизвестны. Кроме
того, в этой реакции атом углерода, по которому
должна проходить атака, экранирован объемистыми
заместителями. Новая структура димера в виде 215
позволяет дать более вероятную схему механизма
[(267) и (268)]. Первая стадия последовательности
реакций (268) представляет собой присоединение
радикала по двойной связи, вторая — процесс,
обратный присоединению, встречающемуся в реакциях
ароматического замещения. Известно много реакций,
аналогичных обоим этим процессам.
медленно. Л.~. _, быстроv -, Л*ЛЛ« V
Ph3C-CPh3 —~ + 2Ph30 У Ph3COO* —*
Ph3C-cph3> ph3C-.0-0-CPh3+ Ph30 (266)
V>/h ^=V 2Ph3C ^> PbCOO
Ph3c \=/ )>h
215
(267)
Ph
Y~Vc{ .OOCPh,-d >Qb?-0OCPh:
_ Ph
—>- Ph3C- + ^>-i^oOCPh3 (268)
Ph
Идея эффекта Торпе — Ингольда возникла на
основании наблюдения, что в реакциях типа (269),
когда хотят получить циклопропановый цикл,
замене
стители X, увеличивающие угол Ч* (между
связями X—С—X), сокращают угол в (между связями
С—С—С) и облегчают образование кольца. Так,
циклизация производного циклогексана 218 (угол Ч**
в кольце циклогексана принимался в то время
равным 120°) должна осуществляться, по-видимому,
гораздо легче, чем в случае диметильного
соединения 216 (Х = СН3).
X СОСООН
.п
СН2СООН
210
\ / г
(269)
/—Ч СОСООН
(в !$=> < XI ? (270)
СН2СООЯ ^^ ^СООН
218
Это объяснение оставалось в силе даже после
того, когда было установлено, что кольцо
циклогексана имеет складчатую структуру, а угол между
связями С—С—С является нормальным тетраэдри-
ческим. Позже было показано, что циклопропанолы
крайне чувствительны к нагреванию, что делает
образование таких соединений в условиях реакций
(269) и (270) удивительным. Таким образом, был
сделан шаг в направлении окончательного
обнаружения, что первоначально исследованные соединения
не имели никакого отношения к циклопропанолам.
Ситуацию, аналогичную описанной выше, можно
привести и из другой области науки, палеонтологии.
В 1912 г. в Пилтдауне (Суссекс, Англия) были
обнаружены остатки, по-видимому, древнего черепа.
Реконструкция этого черепа показала, что он
принадлежит человеку — представителю недостающего
звена в эволюционной цепи. В свете дальнейших
открытий некоторые аспекты, увязанные с этим чере*
пом, становились все более и более загадочными.
Наконец, в 1959 г. определение возраста черепа с
помощью радиоуглеродного метода показало, что
177
имела место фальсификация. Первоначальному
доверию к находке способствовал тот факт, что этот череп
в первом приближении соответствовал тому, что
ожидали. Последующие открытия с очевидностью
показывали большую аномалию пилтдаунского черепа, и,
когда была обнаружена подделка, эта область
палеонтологии человека значительно прояснилась. Так
и с механизмами реакций, которые были выдвинуты
в рамках имевшихся к тому времени сведений.
Прямая фальсификация результатов встречается редко
(хотя некоторые подобные случаи известны многим
химикам, проводящим практические исследования).
Более частный случай — пренебрежение
существенными данными о реакции, рассмотрение их как
аномальных. По мере накопления «аномалий»
первоначальные представления видоизменяют, приводят их
в соответствие с новыми фактами. В конце концов,
появление новых результатов может привести к тому,
что доводы в пользу предложенного ранее
механизма исчезают как дым. Несмотря на все усилия,
которые были затрачены на изучение равновесия «гек-
саарилэтан»/триарилметил, и факт принятия
большинством . химиков структуры замещенного ' этана
для димера, все это цельное здание было разрушено
одним единственным коротким сообщением без
экспериментальной части. Теоретические представления
имели слишком умозрительный характер, пока не
была предложена новая структура димера.
Аг С у— Аг
Аг3С-САг3 Z&&: 2Ar3C-+=t: 3 )Г==\=С (271)
\
АГ
Тенденциозность
В первой части этой главы была проведена мысль
о том, что большее доверие к механизму может воз*
никнуть, если результаты в общем согласуются с
общепринятыми химическими представлениями. Однако
такая постановка вопроса имеет отрицательную
сторону. Идеи, которые выходят за рамки
укоренившейся схемы, часто наталкиваются на препятствия
на пути их признания. Основополагающее открытие
!78
Гомберга трифенилметила встретило значительное
недоверие, так как было «известно», что углерод
всегда четырехвалентен. Позже, когда радикалы уже
были «признаны», замечательная работа Кёлыиа,
который получил радикал 219, была признана
неприемлемой для публикации (хотя в конце концов она
была опубликована спустя двадцать пять лет) на
том основании, что радикалы, «как известно»,
реагируют с кислородом, а 219 оказался стабильным к
действию воздуха.
219
Очевидно, в действительности новые
представления о механизме реакции требуют более детального
и однозначного подтверждения, чем идеи, которые
являются лишь незначительным развитием уже
существующих. Во всяком случае, так это обычно
и происходит.
Вывод
В этой книге описаны различные методы
исследования механизмов органических реакций. При отборе
материала книги многие интересные методы и
концепции остались вне поля зрения. Проведенное
рассмотрение было ограничено гомогенными
органическими реакциями, что также заставило
обойти многие важные и интересные реакции-. Тем не
менее можно надеяться, что использованный в
этой книге подход окажется полезным и для тех,
кто стремится к более осмысленному изучению
механизма реакций и в других областях химии; не
только для «чисто органических», но также для
реакций полимеров, реакций проходящих на
179
поверхности, неорганических и биохимических
реакций. Каждая из этих областей характеризуется
своими методами исследования, которые дополняют
(или заменяют) методы, описанные в этой книге.
Создавая новые представления, следует помнить
до сих пор справедливое утверждение философа
шестнадцатого* столетия Вильяма из Оккама:
«Сущности (концепции) не следует умножать без
необходимости». Время от времени появляются
различные новые концепции. Например,
неклассические ионы карбония и различного типа ионные
пары в органической химии, гель-эффекты в
процессах полимеризации, представления о
взаимодействии фермент-субстрат в биохимии. Прежде чем
признать необходимость введения какой-нибудь
новой концепции, следует спросить: какие
существуют доказательства в пользу этого? Можно ли
объяснить факты более простым способом без
использования новой концепции? Признать
справедливость и необходимость новой концепции следует
лишь в том случае, если на оба этих вопроса
можно дать удовлетворительные ответы.
Задачи
7.1. Хлорид фенилдиазония реагирует с азид-ионом с
образованием фенилазида и азота:
Ph-rWj + N=N=N —> Ph-N=N=N + N=N
(1) (2) (3) (4) (3) (а) (б) (в) (г) (г)
(а) Предложите механизм этой реакции.
(б) Найдено, что если атом (1) в соли диазония пометить
16N, весь 15N оказывается в положении (а) фенилазида.
Переработайте или измените (если необходимо) первый
постулированный вами механизм в свете этого факта.
(в) Если ввести метку в положение (2) фенилдиазонийхло-
рида, то 83% 15N оказывается в положении (б) фенилазида,
остальная метка присутствует в выделившемся азоте. В
положениях (а) или (в) метка lgN не найдена. Снова скорректируйте
ваше представление о механизме, исходя из этих дополнительных
данных (по-видимому, необходимо рассмотреть две конкурентные
реакции). Какие дополнительные эксперименты можно
предложить для проверки вашего механизма?
* Вильям из Оккама жил в четырнадцатом веке н. э.. (ок.
1285—1349 гг.). — Прим. переводи.
180
7.2. (Для студентов, интересующихся вопросами механизма
реакций в других областях химии: например, неорганической
химии, биохимии, химии полимеров или поверхностных процессов.)
Все ли описанные в этой книге методы применимы в других
областях? Какие дополнительные методы имеются в распоряжении
или частично пригодны для изучения механизмов реакций в
выбранной вами области?
7.3. Соберите как можно больше данных (используя эту
книгу или другие источники) о механизме какой-либо реакции
(например, перегруппировке Кляйзена). Кажутся ли
убедительными доводы, рассмотренные в целом? Какие другие
эксперименты, если они вообще возможны, вы можете предложить сами?
УКАЗАНИЯ К РЕШЕНИЮ ЗАДАЧ
1.1. Цифры указывают разделы, приведенные на с. 18—26:
(а) 1,3; (б) 3,4; (в) 1; (г) все верно; (д) 5; (е) 6; (ж) 5.
2.1. Изотопы.
2.3. Как вы докажете присутствие хлора в любой
реакционной смеси?
2.6. Фенильные производные реагируют гораздо быстрее ме-
тильных производных.
3.2. Рассмотрите моно- и бимолекулярные реакции ртутных
производных как скорость определяющие процессы.
3.3. Два реагента, присутствующие в реакционной смеси,
образуют интермедиат, который .затем быстро атакует оставшееся
соединение.
3.4. См. показанную на с. 67 обработку реакции, которая до
некоторой степени аналогична данной.
3.6. См. табл. 3, с. 75.
4.1. В одном из продуктов происходит перегруппировка
углеродного скелета.
4.2. Обдумайте пути, по которым из дибензила может
образоваться 1,2,3-трифенилпропан.
4.3. Натрий является значительно более
электроположительным, чем магний.
4.4. Мп02 должен образовываться за счет окислителя,
который отсутствует в исходном растворе (т. е. за счет интермедиа-
та). Предложите один или несколько вероятных интермедиатов,
которые могут окислять Мп(Н).
4.5. Такое соотношение продуктов обычно дает общий
интермедиат.
5.1. Одна реакция должна включить сохранение, а другая —
обращение конфигурации (инверсию).
5.2. Дейтерий.
5.3. Рассмотрите реакцию гидроборирования.
5.4. Обычные реакции «ионного» элиминирования протекают
с граяс-стереохимией.
5.5. Диссоциация на радикалы или молекулярная реакция,
включающая «перевертывание» группы NC, которая сохраняет
связь с молекулой (т.е. несвободна)?
6.1. Рассмотрите влияние стерических факторов на
переходное состояние в реакции SN2.
181
6.2. См. табл. 8, с. 167.
6.3. Рассмотрите заместители, которые способны
стабилизовать положительный и отрицательный заряд.
6.4. Величина р и корреляция с а+ должны подсказать
величину и тип заряда, возникающего в положении 1 в ходе
реакции.
6.5. В положении 2 гракс-соединения возникает больший
отрицательный заряд.
7.1. Возможен следующий ряд реакций:
(а) прямое замещение N2 в фенилдиазонийхлориде на NJ;
(б) атака азид-иона N" по атому азота (2) с образованием
+
в качестве интермедиата Ph—N=N—N=N=N (A);
(в) образование (А), который затем циклизуется в
N
Ph—N4 I (В); (А) и (В) могут потом разлагаться, приводя
N
к продуктам.
Путь (а) исключается на основании одного из фактов,
указанных в вопросе (какого?). Попытайтесь объяснить все
экспериментальные данные, основываясь на реакциях (б) и (в).
Предлагая другие возможные пути доказательства
механизма, помните, что А и В постулируются как интермедиаты.
7.3. Для ответа на этот вопрос обратитесь к предметному
указателю и библиографии.
ОТВЕТЫ НА ЗАДАЧИ
1.1. (а) Реакция, как она записана, является тетрамолеку-
лярной. Кроме того, этот механизм не объясняет роль Ni(CN)2.
(б) Реакция, обратная этой, должна быть пентамолекуляр-
ной.
Следовательно, исходя из принципа микроскопической
обратимости, прямая реакция будет также проходить по другому
направлению.
(в) Приведенная схема реакции, по-видимому, довольно
сложна; кроме того, она не объясняет, почему продукты
образуются почти исключительно из последнего радикала данной
схемы, а не, например, из предыдущего или из радикала в
начале цепи. Альтернативный механизм
СН2 СН2
Н2СМ). Н2С/ М)
Н2С\ /Н Н2СХ Н
сн сн.
I I
C4H9 С4ГК
проще и объясняет наблюдаемую специфику реакции,
(г) Все правильно.
182
(д) Молекулярное присоединение указанного типа с
одновременным образованием обеих связей должно приводить к
возникновению двух связей с одной и той же стороны молекулы
ацетилена. В результате будет получаться противоположный
геометрический изомер.
(е) Ионизация бензола в растворе уксусной кислоты
является энергетически крайне неблагоприятным процессом.
(ж) Постулированный промежуточный азотный радикал
содержит девять электронов в валентной оболочке азота.
2.1. CD3CH2C1+ OH" —► CD3CH2OH + Cr
I4CH3CH2C1 + ОН" —* иСН3СН2ОН + СГ
2.2. (а) Вероятное объяснение. Однако факт образования
приблизительно 50% каждого из двух продуктов при проведении
реакции в различных условиях решительно свидетельствует в
пользу того, что такой результат не может быть обусловлен
лишь простым совпадением.
(б) Если реакция проходит через ион карбония с
последующим гидридным перемещением, как показано на схеме, следует
ожидать во всяком случае некоторое количество продукта(тов),
обусловленного дальнейшими гидридными переходами, которые
должны осуществляться так же легко.
Кроме того, образование ионов карбония в основной среде,
такой как жидкий аммиак, маловероятно.
2.3. Два способа, которые были использованы для этой цели,
описаны в гл. 4, на с. 116. Возможны и многие другие методы.
2.4. Например, выделите метанол и ищите полосу валентных
колебаний связи С—D в ИК-спектре. Помимо этого, обработайте
метанол химическим путем с образованием производного, в
котором атом водорода (дейтерия), связанный с кислородом,
отсутствует, например метилацетат. Затем сравните ИК- и масс-
спектры этого образца с заведомым недейтерированным
соединением.
2.5. Тот факт, что А окисляется, тогда как С нет, наводит
на мысль, что при окислении А затрагивается водород,
связанный с атомом азота. Следовательно, более вероятно, что продукт
имеет строение В2. Кроме того, соединение В окрашено в
желтый цвет, что указывает на наличие в В2 сопряженной
группировки N = N—Ph. B1 не содержит группы, которая могла бы
сообщить молекуле желтую окраску. В1 имеет N—Н группу, а
В2 — О—Н, которые можно различить с помощью ИК- и ЯМР-
спектров.
2.6. Нет. Фенильное соединение должно практически
полностью прореагировать за период времени, в течение которого
разложится лишь незначительное количество метильного
соединения. Следовательно, не должно быть почти никаких изменений
при проведении перекрестных реакций.
3.1. (а) Титрование невошедшего в реакцию ОН- или
образующегося Вг~. (б) Измерение изменения давления (или
изменения объема при постоянном давлении) (в) Спектрофотометри-
ческое определение количества оставшегося брома.
!83
3.2. (a) (PhCH2)2Hg —► PhCH2CH2Ph + Hg
или —* PhCH2 • + . HgCH2Ph —>>
—> Hg + .CH2Ph
или —► 2PhCH2. + Hg
с последующей реакцией: 2PhCH2 • —>- PhCH2CH2Ph
(б)
Me3Si—Hg—SiMe3 Me3Si Hg—SiMe3 Hg SiMe3
i i —> i + | —> +
Me3Si—Hg—SiMe3 Me3Si Hg— SiMe3 Hg SiMe3
Me3Si «HgSiMes
или —► | + —> 2Hg+2Me3Si»
Me3Si «HgSiMes
с последующими реакциями:
Me3Si. + Hg(SiMe3)2 —* Me3Si—SiMe3 + Hg + • SiMe3
2Me3Si« —> Me3Si—SiMe3
Для того чтобы показать, что вторая схема также приводит
к кинетике второго порядка, можно применить принцип
стационарного состояния.
3.3. На медленной стадии, включающей азотную и,
по-видимому, уксусную кислоты, должен образовываться интермедиат.
Этот реакционноспособный интермедиат (вероятно, нитроний-ка*
тион) исчезает за счет быстрой^ реакции с бензолом.
Следовательно, скорость нитрования равна скорости образования
промежуточного иона нитрония, а концентрация бензола
несущественна.
3.4. Скорость = (ki/ka) ,/2 кг [Инициатор] ч>2 [RH]. R02»
присутствует в значительно большей концентрации, чем R-.
C6H6-NH-NH-C6H5 + 2Н+ +
H2N-<\ Л-\ /bNH2 ^
Н-Г/ NT TJ
184
Отсутствие дейтериевого изотопного эффекта исключает
любой механизм, в котором разрыв связей С—Н в Ашра-положении
происходит на кинетически важной стадии. Данный механизм
достаточно хорошо согласуется с фактами, указанными в вопросе,
однако к моменту написания книги детали механизма этой
реакции все еще остаются невыясненными.
3.6. Реакция (а) является молекулярной. Радикальное
разложение (б) должно иметь значительно более высокую величину
Л-фактора.
hno2 +
4.1. PhCHMe—CHMeNH2 >• PhCHMe—CHMe.
Миграция метильной группы или гидрид-иона и последующее
взаимодействие с уксусной кислотой дает два продукта; третий
продукт образуется без миграции или за счет миграции фенила.
Если реакция проходит через радикал или карбанион, этой
перегруппировки не было бы.
1 или 2
4.2. (PhCH2)2Hg >• Hg + 2PhCH2. —> PhCH2CH2Ph
стадии
PhCH2 •
PhCH2CH2Ph + • CH2Ph —> PhCH3 + PhCH—CH2Ph ►■
PhCH2CHCH2Ph
Ph
Введите в реакционную смесь дибензил; это должно
увеличить количество образующегося трифенилпропана. Добавление
соединения, которое реагирует с бензильными радикалами
(например, нитробензола), должно уменьшать количество
образующегося дибензила.
3. СН3 Н-СИ2^СН2-ОС2Н5 ► СН4 + СН2=СН2 +~OC2Hij
Поскольку реактив Гриньяра не участвует в такой реакции,
он должен обладать меньшим ионным характером, чем натрий-
органические соединения. Следовательно, связь С—Mg в
реактиве Гриньяра должна иметь значительный ковалентный
характер.
4.4. В реакции должен образовываться интермедиат, который
является более сильным окислителем, чем Cr(VI) или альдегид.
Предполагается Cr(IV).
4.5. Поскольку отношение продуктов в двух реакциях
различно, образование общего интермедиата маловероятно.
Поскольку PhCH2N2 дает PhCH:, другая реакционная система, по-
видимому, его не образует, и, вероятно, имеет место прямое
взаимодействие PhCHBrLi с бутеном-1.
5.1. Вторая реакция протекает не по атому Si и поэтому
осуществляется с сохранением конфигурации. Следовательно,
инверсия происходит в первой реакции. См. также, с. 127.
183
5.2. Для этой цели следует использовать оптически активное
дейтерированное соединение CH3CHDBr.
5.3. С3Н7СН^=СНСН3 —>■ С3Н7СН=*СНСН3—>•
(сен13)2в—н * (свн13)2в-н
^" CaHfCHoCHCH^j
I и т.д
Двойная связь не может образовываться у четвертичного
атома углерода. Следовательно, атом бора не может пройти этот
атом углерода в ходе перегруппировки.
5.4. Элиминирование галогеноводородов под действием
основания осуществляется довольно легко, если атомы водорода и
галогена находятся в граяс-положении по отношению друг к
другу. Для того чтобы в результате реакции образовывался 183,
должно проходить цнс-элиминирование.
5.5. Протекает термическая диссоциация на PhMeEtC- и CN-
с последующей рекомбинацией радикалов в PhMeEtC—CN.
Поскольку промежуточно образуется радикал (плоский), можно
ожидать рацемизацию. Если же группа NssC просто
«перевертывается» в ходе молекулярной реакции, то конфигурация
должна сохраняться. Оптически активный PhMeEtC—CN не рацеми-
зуется в условиях приведенного эксперимента, что исключает,
таким образом, изомеризацию с последующей рацемизацией.
6.1. В переходном состоянии для 5#2-реакции у центрального
атома углерода координируются пять групп, тогда как в
исходном галогениде — четыре. Следовательно, в переходном
состоянии стерические эффекты имеют большее значение, и чем больше
алкильных групп связано с центральным атомом, тем этот
эффект будет больше.
6.2. В переходном состоянии первой реакции происходит
рассредоточение заряда, а во второй — его образование. Поэтому
с увеличением ионизующей силы растворителя скорость первой
реакции будет снижаться, второй — возрастать (в значительной
степени).
6.3. or+: NMe2 NH2 ОМе SMe ОН CI
а": СНО СОМе СООМе CN N02
6.4. В положении 1 возникает положительный заряд, однако
малая величина р указывает на то, что молекула не ионизуется.
Вполне вероятно следующее молекулярное переходное состояние:
&- 8+
О—СНАг
HsC С СН2
V-h'
186
6.5. Согласованное элиминирование толуолсульфокислоты
может протекать довольно легко, если группы Н и OS02CeH4CH3
находятся в граяс-положении друг к другу (т.е. в цнс-соедине-
нии). В согласованном процессе положение 2 обладает
относительно небольшим карбанионным характером. В граяс-соединении
группы занимают не столь благоприятное положение для
согласованного элиминирования, и переходное состояние обладает
ббльшим карбанионным характером.
7.1. Прямое замещение N2 на N3 исключается результатом
эксперимента с меченым в положение (1) атомом азота.
Распределение ,5N, первоначально находившегося в положении (2),
объясняется следующей схемой:
Ph -N=I5N
+
N=N=N
быстро
►
+ 34%
Ph—N=,5N—N=N=N —>
Ph—N:
i5N
N
66 %
(a)
17*
{6)
Ph—N=,ffN=N + N=N Ph—N=N=N + ,5N=N
суммарно 83 % 17%
Если реакцию проводить при — 50 СС, то примерно 75% Na
выделяется по реакции первого порядка (обогащение l6N в
выделившемся Na не наблюдается). При нагревании реакционной
смеси до 0СС выделяется остальное количество Na (снова по
реакции первого порядка), обогащенного 16N в соответствии со
схемой.
Л итератуpa
Все монографии, приведенные в разделе «Общая
литература», кроме книги Вайсбергера, рассматривают механизмы
реакций с точки зрения типа реакции. Они приведены в порядке
возрастающей «сложности». В большинстве из них приведены
ссылки на обзоры и оригинальные научные работы. Литература,
данная в специальных разделах, не является исчерпывающей и
не всегда отражает ключевые работы в данной области. Однако
.эти работы позволяют найти более ранние статьи по затронутым
в данной книге темам.
Общая литература
П. Сайке. Механизмы реакций в органической химии. Изд. 3-е.
Пер. с англ. Под ред. Я. М. Варшавского. М., «Химия», 1977.
Е. S. Gould. Mechanism and Structure in Organic Chemistry,
Holt, N. Y., 1959.
/. Hine. Physical organic chemistry, McGraw-Hill, N. Y., 1962.
/(. К Ингольд. Теоретические основы органической химии.
Пер. с англ. Под ред. И. П. Белецкой. М., «Мир», 1973.
/. March. Advanced organic chemistry: reaction, mechanisms,
and structure, McGraw-Hill, N. Y., 1968.
R. W. Alder, R. Baker and J. M. Brown. Mechanism in organic
chemistry, Wiley, N. Y., 1971.
E. M. Kosower. Introduction to physical organic chemistry.
Wiley, N. Y., 1968.
A. Weissberger (ed.). Techniques of organic chemistry,
Vol. VIII, Investigation of rates and mechanisms of reactions, In-
terscience, N. Y., 1961.
Глава 1
Оптимизация. /. D. Rose. Chem. Br., 7, 421 (1971).
Ракетное топливо. R. M. Lawrence, W. H. Bowman. J. chem.
Educ, 48, 458 (1971).
Реакция бензальдегида с ацетофеноном. Е. Coombs,
D. P. Evans. J. Chem. Soc, 1295 (1940).
Бензин и радикальные реакции — см. гл. 4.
Глава 2
Проблема димера трифенилметила. Н. Lankamp, W. Th. Nau-
ta, С. Maclean. Tetr. Letters, 249 (1968); W. Theilacker, Wessel-
Ewald Af.-L, Ann., 594, 214 (1955).
Эффект Торпе-Ингольда. К. В. Wiberg, H. W. Holmquist.
J. Org. Chem., 24, 578 (1959).
Перегруппировка Бекмана. W. V. Sidgwick. The Organic
Chemistry of Nitrogen. Clarendon Press, Oxford (1969).
Гидролиз сложных эфиров — см. гл. 4, тетраэдрические ин-
термедиаты.
Реакции, включающие образование карбониевых ионов, кар-
банионов, радикалов и производных дегидробензола — см. гл. 4.
188
Глава 3-
A. A. Frost and R. G. Pearson. Kinetics and Mechanism,
Wiley, N. Y., 1961.
G. L. Pratt. Gas kinetics, Wiley, London, 1969.
E. Колдин. Быстрые реакции в растворе. Пер. с англ. Под
ред. Н. М. Эмануэля. М., «Мир», 1966.
R. Huisgen. Кинетическое доказательство образования в
реакциях интермедиатов. Angew. Chem. Int. Ed., 9, 751 (1970).
Реакции SN\ и Sn2 — см. монографию К. К. Ингольда.
Реакция иода с водородом. /. Н. Sullivan. J. Chem. Phys., 46,
73 (1967); R. Hoffmann. J. Chem. Phys., 49, 3739 (1968).
Бромирование хлороформа. /. H. Sullivan, N. Davidson.
J. Chem. Phys., 19, 143 (1951).
Расщепление циклобутана. С. Г. Denaux, F. Kern, W. D.
Walters. J. Amer. Chem. Soc, 75, 6196 (1953); S. W. Benson. J. Chem.
Phys., 34, 521 (1961).
Изотопные эффекты. R. P. Bell, The proton in chemistry, Met- -
huen, London, 1959; в ароматическом замещении. L. Melander,
Nature, Lond., 163, 599 (1949).
Окисление хроматами. F. H. Westheimer. Chem. Rev., 45, 419
(1949); F. Holloway, M. Cohen, F. H.. Westheimer. J. Am. Chem.
Soc, 73, 65 (1951).
Глава 4
Сополимеризация стирола и метилметакрилата. С. Walling,
Е. R. Briggs, W. Cummings, F. R. Mayo. J. Am. Chem. Soc, 72,
48 (1950).
Карбониевые ионы. R. H. DeWolfe, W. G. Young. Chem. Rev.,
56, 753 (1956); Д. Бетл, В. Голд. Карбониевые ионы, Пер. с англ.
Под ред. И. П. Белецкой. М., «Мир», 1970.
P. de Mayo (ed.), Molecular rearrangements, Vol. I, Inter-
science, N. Y., 1963.
Радикалы. W. A. Pyror. Free radicals, McGraw-Hill, N. Y.,
1968; W. A. Pyror. Introduction to free radical chemistry, Prentice-
Hall, New Jersey, 1966.
Фторирование метана. E. H. Hadley, L. A. Bigelow. J. Am.
Chem. Soc, 62, 3302 (1940).
Автоокисление кумола. /. R. Thomas. J. Am. Chem. Soc, 85,
591 (1963).
Автоокисление борорганических соединений. A. G. Davies,
K. U. Ingold, B. P. Roberts, R. Tudor, J. Chem. Soc, B, 698 (1971).
Применение ХДПЯ. R. W. Jemison, D. G. Morris. Chem. Com-
mun., 1226 (1969).
Карбанионы. Д. Крам. Основы химии карбанионов. Пер. с
англ. Под ред. И. П. Белецкой. М., «Мир», 1967.
Декарбоксилирование пиридин-1-карбоновой кислоты.
М. R. F. Ashworth, R. P. Daffern, D. LI Hammick, J., Chem. Soc,
809 (1939).
Перегруппировка литиевых солей сложных эфиров. U. Scholl-
kopf, D. Nalter, Angew. Chem., 73, 545 (1961).
Карбены и производные дегидробензола. Т. L. Gilchrist,
С. W. Rees, Carbenes, Nitrenes, and Arynes, Nelson, Lond., 1969.
189
14С-Дегидробензол. /. D. Roberts, D. A. Semenov, H E. Simmons
Jr., L. A. Carlsmith J. Am. Chem. Soc, 78, 601 (1956).
Конкурентная реакция фурана и циклогексадиена с дегидро-
бензолом. R. Huisgen, R. Knorr. Tetr. Letters, 1017 (1963).
Тетраэдрические интермедиаты. 180 в гидролизе сложных
эфиров. Д. Н. Курсанов, Р В. Кудрявцев, ЖОХ, 26, 1183 (1956);
М. L. Bender, Chem. Rev., 60, 53 (1960).
Циклизация тетраэдрических интермедиатов. Н. Е. Zaugg,
V. Papendick, R. J. Michaels. J. Am. Chem. Soc, 86, 1399(1964).
Глава 5
Э. Илиел Стереохимия соединений углерода. Пер. с англ.
Под ред. В. М. Потапова. М., «Мир», 1965.
G. Hallas. Organic stereochemistry. McGraw-Hill, London,
1965.
Стереохимия соединений кремния. L. N. Sommer e. a.t J. Am.
Chem. Soc, 83, 2210 (1961).
Дыс-элиминирование. D. H. R Barton, A. J. Head, R. J.
Williams. J. Chem. Soc, 453 (1952).
Гидроборирование. H. С. Brown. Hydroboration, Benjamin,
N. Y., 1962.
Бирадикальные интермедиаты. L. К. Moutgomery, К. Schuel-
ler, P. D. Bartlett. J. Amer. Chem. Soc, 86, 622 (1964).
Орбитальная симметрия. R. В. Woodward, R. Hoffmann. The
conservation of orbital symmetry, Verlag, Weinheim, 1970;
H. E. Zimmerman. Acct. Chem. Research, 4, 272 (1971).
Глава в
Уравнение Гаммета. P. R. Wells. Chem. Rev., 63, 171 (1963).
a+ H. С Brown, Y. Okamoto, J. Am. Chem. Soc, 80, 4979 (1958).
Потенциалы ионизации бензильных радикалов A. G.
Harrison, P. Kebarle, F. P. Lossing. J. Am. Chem. Soc, 83, 777 (1961).
N-Нитроамины. W. H. White, J. R. Klink. J. Org. Chem., 35,
965 (1970).
N-Бромсукцинимид. R. E. Pearson, /. C. Martin. J. Am. Chem.
Soc, 85,354 (1963).
Электрофильное ароматическое замещение. R. О. С. Norman,
R. Taylor, Electrophilic substitution in benzenoid compounds,
Elsevier, Amsterdam, 1964. ,
Пиролиз алкилгалогенидов. А. Массой. Chem. Rev., 69, 33
(1969).
Пиролиз арилэтилацетатов. R. Taylor, О. О. Smith.
Tetrahedron, 19, 937 (1963).
Эффекты растворителей — см. монографии Ингольда и Koso-
wer в разделе «Общая литература».
Глава 7
Реакция азид-иона с хлористым диазонием (задача). /. Ugi,
R. Huisgen, К. Clusins, M. Veechi. Angew. Chem., 68, 705, 753
(1956).
190
Предметный указатель
Автоокисление
триалкилборанов 100, 101
углеводородов 94, 100
Азобисизобутиронитрил 66, 97
Активации энергия 70, 72
Альдольная конденсация 58, 92
Аммонолиз
флуорена 135
эфиров 108
Ароматичность и антиароматичность
144. 145
Аррениуса уравнения 25, 70, 74—76
Ассиметрические атомы 12
Бекмана перегруппировка 32, 33
Бензидиновая перегруппировка 82
Бензильный радикал 165
Бензины см. Дегидробензол и его
производные
Бимолекулярные реакции 15, 16, 21
Бредта правило 26
Бромирование
ацетона 56. 80
малеиновой кислоты 138, 139
олефинов 22, 23, 87
толу о лов 162, 170
фумаровой кислоты 139
хлороформа 68. 69
грег-Бутилкатион 87, 152
Вагнера — Меервейна
перегруппировка 89
Вальденовское обращение 122. 123
Гальвиноксил 101
Гаммета уравнение 155 ел.
Гексафенилэтан 29. 30
Гидроборирование 141, 142, 146, 147
Гидробромирование олефинов 15, 49,
67. 102, 154
Гидролиз
алкилгалогенидов 41, 62, 132, 171
дигалогенангидридов 69
N-метилацетанилида 109
ортоэфиров 63
сложных эфиров 35. 69. 107. 108,
111. 112. 151. 173
Дегидратация спиртов 86
Дегидробензол 19. 34, 104—106
производные 104—106
Дильса — Альдера реакция 142
Дихлоркарбен 104
Заместителей эффекты 152 ел.
Изобестическая точка 115
Изопропилкатион 87. 90, 152. 153
Изотопные эффекты 77—81
Инверсия (обращение
конфигурации) 121 ел.
Ингибиторы и ингибирование
реакций 98-101
Инициаторы и инициирование
реакций 101
Интермедиаты 10, 83 ел.
бирадикальные 147
стабильные 114—117
Иодирование
ацетона 56
водорода 59. 60
Ионов карбония перегруппировка
136, 137
Канницаро реакции 63, 64
Канонические структуры см.
Резонансные структуры
Карбанионы 84—86. 90—93. 135
Карбены 102—104
Карбониевые (Карбония) ионы 84—
90. 132. 133. 152
Квазирацематы 126
Кинетика реакции 46 ел.
Кинетический изотоппый эффект
77-81
Кислотно-основной катализ 111—114
Кляйзена перегруппировка 117
Коттона эффект 128
Кросс (перекрестные) эксперименты
36
Ловушки
карбенов 103
радикальные 97, 98
спиновые 98
химические 87, 88, 103, 105
Метильный радикал 169
Механизм реакции определение 13,
14
Мёбиуса системы 145
Миграция атомов и групп 35
Молекула, определение 13, 14
Молекулярность реакции 53
Мономолекулярные реакции 20
Неопентильный катион 90
Нитрование аренов 78, 79, 82
Нуклеофильное замещение
бимолекулярное 16, 55, 129, 130,
133
мономолекулярное 55, 132
Обратимости микроскопической
принцип 21. 22
Обращение конфигурации
(Инверсия) 121 ел.
Общего иона эффект 55
Окисление
реактивов Гриньяра 117
спиртов 119
углеводородов 82. 94, 100
триалкилборанов 100. 101
Органогидриды металлов 73
191
Перегруппировки см. по названиям
соединений и именам
"первооткрывателей
Перекрестные (Кросс) эксперименты
36
Пилтдаунский человек 175, 177, 178
Пинаколиновая перегруппировка 89
Пиролиз
углеводородов 99
хлороформа 104
Порядок реакций 50 ел.
Присоединения реакции 138—141
Промежуточные соединения см. Ин-
термедиаты
Промоторы радикальные 101
Пропильные катионы 87, 90, 152, 153
Псевдорацематов метод 126, 127
Радикальные реакции 66—70. 84. 85
93—102, 134. 135, 154
Растворителей эффекты 38, 39, 165,
166
Реакции химические
бимолекулярные 15, 16, 21
быстрые 47
выход 29. 39. 40
заместителей эффекты 152 ел.
именные см. по именам
исследователей
индивидуальные стадии 20
кинетика 46 ел.
молекулярность 53
молекулярные 142, 143
мономолекулярные 20
необратимые 22
нуклеофильного замещения 16;
55. 129 ел.
обратимые 21, 22
определение 13
оптимизация 14. 15
побочные 39. 40
порядок 50 ел.
предсказание механизма 16—18
присоединения 138—143
продукты 29 ел.
радикальные 66—70, 84. 85. 93—
102. 134. 135, 154
растворителей влияние 38, 39.
165. 166
систематизация 15, 16
скорость 46. 49, 50
согласованные 142, 143
стадии 20
стереохимия 120 ел.
стерическое ускорение и
замедление 150—152
Реакции химические
температуры влияние 70—72
тримолекулярные 21
цепные 66—69
электрофильного замещения 78,
79. 131
элементарные стадии 20
элиминирования 138—143
энергетика 25
энергия активации 70, 72
Резонансные структуры 12
Скорость определяющая стадия 55
Скорость реакций 46, 49, 50
Согласованные реакции 142, 143
Солевой эф-фект 56
Сольволиз
алкилгалогенидов 56. 88
кумилхлоридов 160, 164
Стационарное состояние 65, 66
Стереохимия реакций 120 ел.
Стерическое ускорение и
замедление 150—152
Тесные ионные пары 37
Тетраэдрические интермедиаты 106—
111
Триптицен 105
Торпе — Ингольда эффект 176
Трифенилметильные радикалы 16,
26 ел., 175 ел.
Фенильные радикалы 97
Фиксация химическая интермедиа*
тов 87. 88
Фотолиз диазоалканов 103
Фторирование алканов 26, 40, 97
ХДПЯ метод 95, %
Хлорирование
алканов 25. 67, 134
ацетанилида 42
ацетона 56
углеводородов 15
Хэммонда постулат 166 ел.
Цепные реакции 66—69
Электрофильное замещение 78, 79,
131
Элиминирования реакции 138, 143
Энергия активации 70, 72
ДЖЕКСОН Р. А.
Введение в изучение
механизма органических реакций