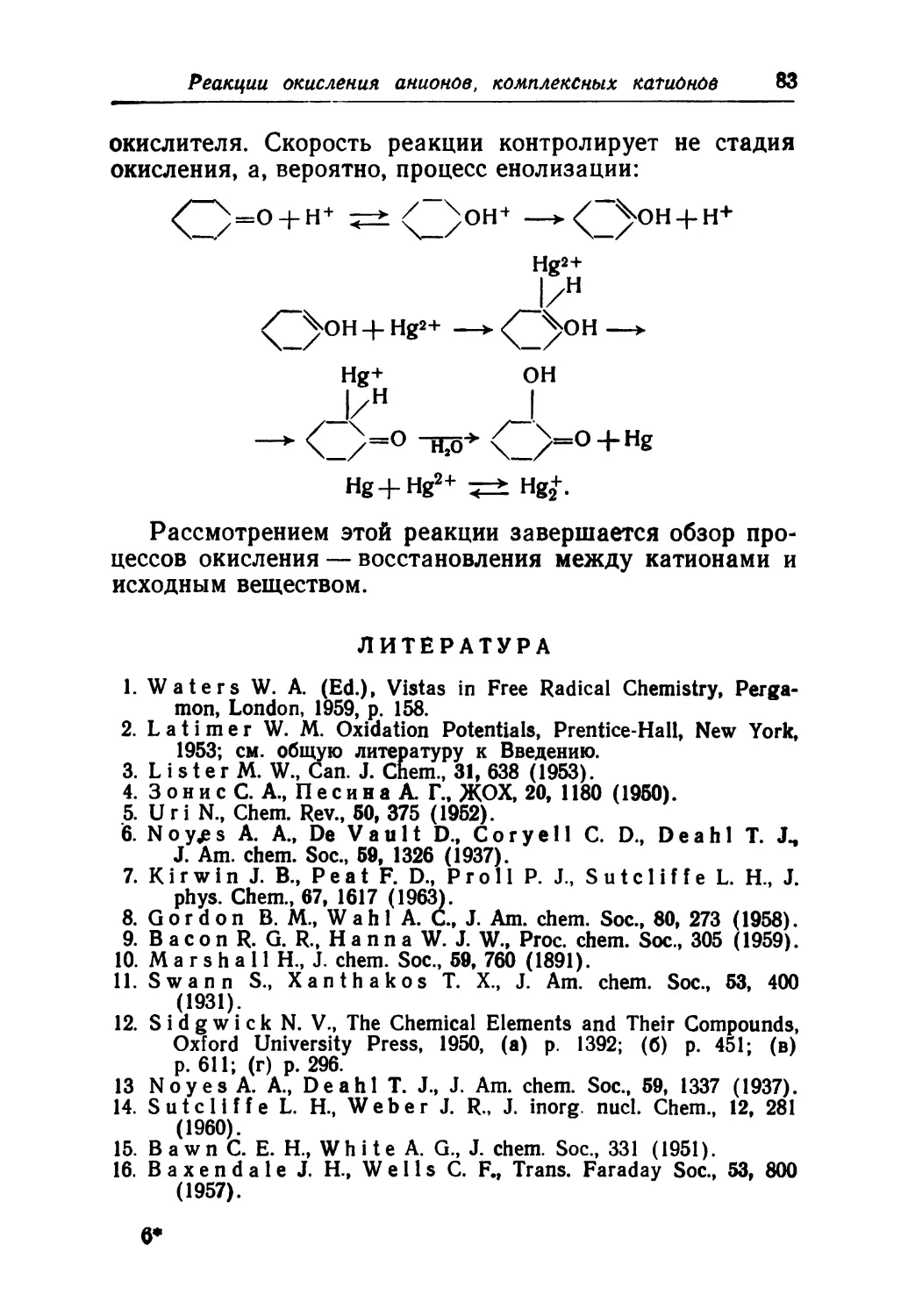
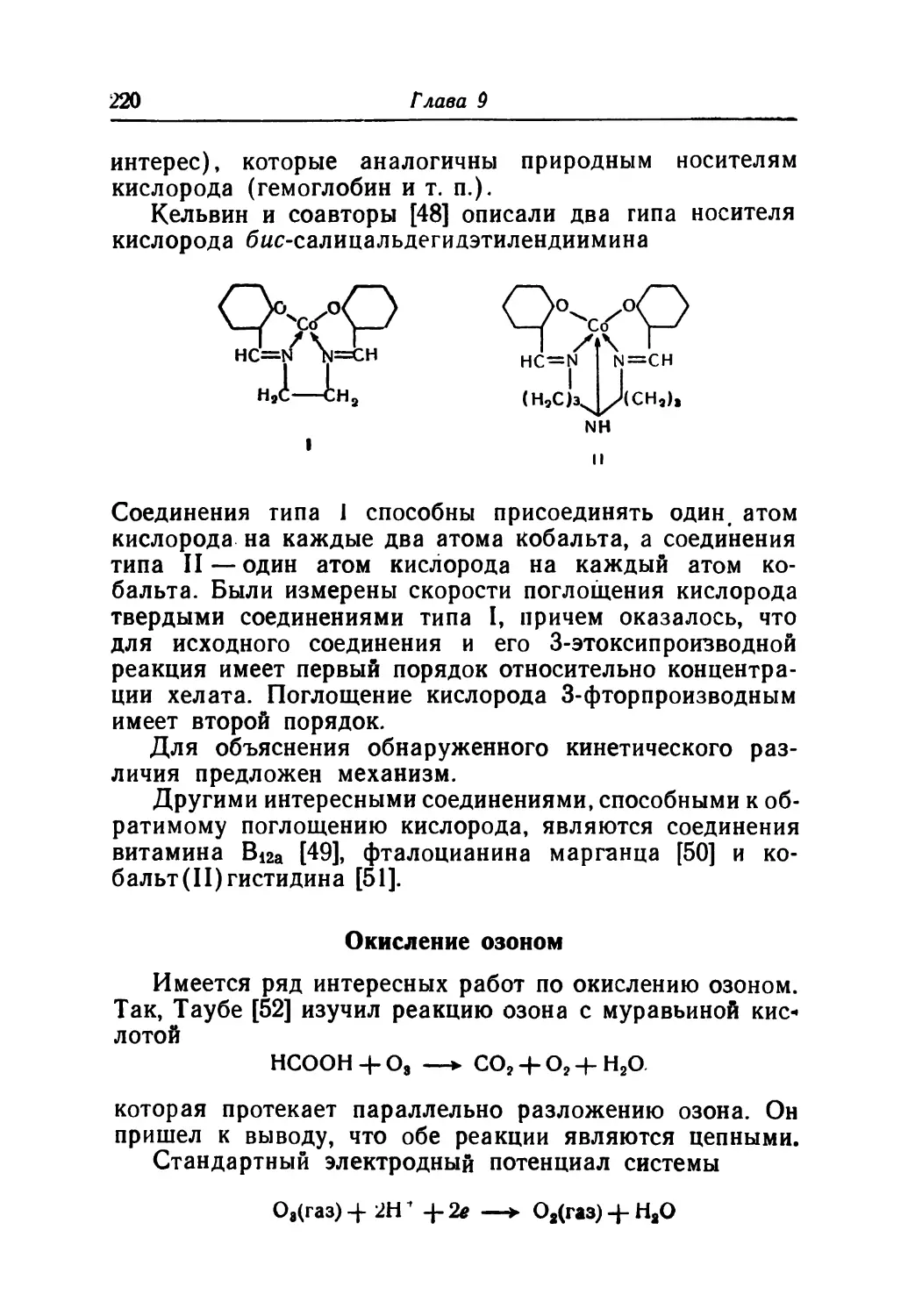
/



















































































































































































































































Text
МЕХАНИЗМЫ
РЕАКЦИЙ
ОКИСЛЕНИЯ-
ВОССТАНОВЛЕНИЯ
Больше химической литературы на
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
И ЗДАТЕЛЬСТВ О
«М И Р»
OXIDATION MECHANISMS
T. A. TURNEY, №. Sc. (N. Z.)
Senior Lecturer in Chemistry, University of Auckland,
Auckland, New Zealand
London
Butterworths
1965
Т. ТЕРНИ
МЕХАНИЗМЫ РЕАКЦИЙ
ОКИСЛЕНИЯ-
ВОССТАНОВЛЕНИЯ
Перевод с английского
канд. хим. наук К. Н. НИКИТИНА
Под редакцией
доктора хим. наук а. И. б У С в в а
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1968
УДК 543: 542.941+.943
В книге систематизированы данные по механизму
различных окислительно-восстановительных реакций,
имеющих значение для общей, неорганической, аналити-
ческой, препаративной химии и технологии. Способ из-
ложения, принятый автором, позволяет представить
сущность процесса в каждом конкретном случае. При-
нятая в книге классификация применяемых окислителей
и их реакций должна способствовать исследованию но-
вых окислителей и новых реакций окисления.
Книга представляет интерес для химиков-аналити-
ков, неоргаников, физикохимиков — работников научно-
исследовательских институтов и заводских лабораторий.
Редакция литературы по химии
Инд. 2-5-5
ПРЕДИСЛОВИЕ
Общеизвестно большое значение реакций окисления
в растворах для неорганической и аналитической химии,
а также химической технологии. Механизм таких реак-
ций интенсивно изучается последние 15—20 лет. Хотя
к настоящему времени накоплен большой материал, од-
нако очень многое остается неясным и весьма дискус-
сионным. Перед автором книги проф. Терни стояла труд-
ная задача попытаться систематизировать имеющиеся
нередко противоречивые и недостаточные сведения
относительно механизма ряда реакций окисления в рас-
творах. Автора можно было бы упрекнуть в некритиче-
ском отношении к имеющемуся материалу, но надо при-
знать, что вряд ли иное возможно при современном со-
стоянии знаний. Тем не менее в книге собран интересный
материал, который может быть полезен для широкого
круга химиков. Обзор аналитических методов, основан-
ных на реакциях окисления—восстановления, содержит-
ся в недавно изданной книге: Берка, Вултерин,
Зыка, Новые редокс-методы в аналитической химии М.,
«Химия», 1968.
Наименее изучен в настоящее время механизм мно-
гочисленных реакций окисления, на которых основаны
аналитические методы. Такие реакции, как правило,
протекают очень сложно.
При переводе сделаны отдельные небольшие сокра-
щения, главным образом материала, не имеющего пря-
мого отношения к теме книги или явно устаревшего.
В нужных случаях даны примечания.
Наряду с терминами «окисляемое вещество», «окис-
ляемая частица», «восстановитель» Терни часто приме-
няет термин «субстрат». Этот термин в нашей литера-
туре имеет другое значение, поэтому он переведен как
«окисляемое вещество» (ион, комплексная частица
и др.).
А. Бу се в
ИЗ ПРЕДИСЛОВИЯ АВТОРА
В настоящей книге автор сделал попытку избежать
традиционного деления в химии и рассмотреть все про-*
цессы окисления с единой позиции.
Не стремясь обсуждать механизм каждого процесса
окисления, автор сознательно выбрал такие примеры,
которые подтверждают общепринятые представления.
Возможно, подбор и размещение материала окажется
полезным читателю по следующим соображениям. Во-
первых, рассматриваемые реакции окисления с выбран-
ным окислителем могут быть использованы в качестве
основы для предположений о поведении его в других
процессах. Во-вторых, принятая в книге классификация
применяемых окислителей и их реакций может способ-
ствовать исследованию новых окислителей и новых реак-
ций окисления. В-третьих, эта книга может послужить
отправным пунктом для последующих исследований ме-
ханизма других реакций окисления.
За время подготовки рукописи вышли из печати две
книги иного содержания по данному вопросу: 1) Ross
Stewart, Oxidation Mechanisms, Applications to Orga-
nic Chemistry; 2) W. A. W a t e r s, Oxidation Mechanisms
in Organic Chemistry *.
Настоящая книга охватывает весь предмет с совер-
шенно иным подходом к процессам окисления.
* Есть русский перевод: Уотерс У., Механизм окисления ор-
ганических соединений, изд-во «Мир», М., 1966. — Прим. ред.
ВВЕДЕНИЕ
Химическая реакция осуществима в том случае, если
в результате ее протекания происходит уменьшение сво-
бодной энергии. Изменение стандартной свободной энер-
гии ДО0 реакции, протекающей в гальваническом эле-
менте, связано с разностью электродных потенциалов
ДЕ° известным уравнением
ДО0 = — Z • Д£о . F,
где Р — число Фарадея, Z — число электронов, уча-
ствующих в реакции. Это выражение не позволяет су-
дить, обратима ли реакция в гипотетическом гальвани-
ческом элементе, для которого рассчитано изменение
свободной энергии, и возможна ли она кинетически. Бо-
лее того, реакции, имеющие одинаковые электродные
потенциалы, могут протекать с совершенно различными
скоростями. Например, несмотря на близкие по вели-
чине электродные потенциалы реакций
£°
НСНО + Н2О —► НСООН + 2Н++2<? —0,06 в,
СН3ОН —► НСНО + 2Н++2<? —0,19 в,
первая из них может быть осуществлена в‘ присутствии
соединений серебра и меди, а вторая неосуществима.
Окислительно-восстановительные реакции органиче-
ских соединений вообще необратимы. Исключением из
этого правила являются системы с хинонами и такие,
которые обычно используются как редокс-индикатор.
В реакциях соединений последнего типа имеет место пе-
ренос протона либо к атомам кислорода, серы или азота,
либо от них. Если такой перенос сопровождается лег-
кой потерей или приобретением электронов, становится
понятной обратимость этих реакций.
8
Введение
Реакции неорганических соединений значительно
чаще обратимы, так как перенос электронов без пред-
шествующего разрыва связи более прост.
В настоящее время для составления уравнений реак-
ций окисления — восстановления принято пользоваться
ионно-электронным методом. Последний не дает сведе-
ний о механизме реакции, его ценность заключается
лишь в установлении стехиометрических соотношений
реагирующих веществ.
Так, реакция окисления альдегидов медью (II), со-
держащейся в растворе Фелинга, протекает по схеме
2Си2+4-2<? —> 2Си+,
—СНО + ЗОН" —> _С00“4-2Н20 4-2г.
Суммируя, получим
-CHO + 3OH- + 2Cu2+ —> — COO”H-2Cu++2H2O,
2OH“4-2Cu+ —> Cu2O + H2O.
Таким образом, 2 г-иона меди (II) эквивалентны 1 молю
альдегида.
Йодоформ образуется по схеме
2СН3СОСН3 + 312 —> 2CH3COCI34-6H++&?
+ 312 + 6г—>6Г
СН3СОСН3 + 312 —> СН3СОС1з+ЗН+4-ЗГ
СН3СОС13 + ОН" —► CH3COO4-CHI3
т. е. 3 моля иода эквивалентны 1 молю ацетона.
Более известны примеры из области неорганической
химии, например окисление ферро-ионов перманганатом:
5Fe2+ —► 5Fe34’+5^
MnO4- +8Н+ 4-5«? —► Mn2++4H2O
МпО4- Н-8Н+ +5Fe2+ —► Mn2+ +5Fe3+ +4Н2О
или окисление иодид-ионов йодатом:
6Г —► 3124-6й
1О^4-6Н+4-6е —► 1~4-ЗН2О
1О3 +бн+н-бг —> 3124-зн2о
Введение
9
Приведенные выше уравнения отражают теоретические
стехиометрические соотношения. В действительности мо-
гут протекать побочные реакции, изменяющие общее на-
правление процесса. Необходимо поэтому подчеркнуть
особую важность установления действительных стехио-
метрических соотношений перед началом исследования
механизма реакции. Общие принципы реакций окисле-
ния— восстановления хорошо изложены в монографии
Шарпа [1].
Механизм реакций
При рассмотрении механизма реакций необходимо
более детальное исследование, чем это требуется в по-
вседневной работе. Такое исследование может быть про-
ведено различными путями, в зависимости от поставлен-
ной цели.
Кинетический метод выяснения механизма реакции
сводится к сопоставлению наблюдаемой на опыте скоро-
сти реакции с теоретически рассчитанной на основе оп-
ределенных предположений о механизме реакции с учё-
том деталей электронных структур реагирующих частиц.
Согласно теории активного комплекса, константа ско-
рости реакции kCH описывается уравнением
kZK^kT/h-e^R-e-EfRT.
где kT/h — частотный фактор (эффективная скорость пе-
рехода активных комплексов через энергетический
барьер); AS* — так называемая энтропия активации;
Е — энергия активации. Для бимолекулярной реакции
величина e^s*iR эквивалентна отношению F*/FA • FB, где
F* — сумма по состояниям активного комплекса, a FA
и Fb — суммы по состояниям реагирующих частиц. Под-
становка соответствующих величин для сумм по состоя-
ниям приводит к следующему выражению для константы,
скорости:
^-(влйТ/Ю’АЧв-в'^..
которое совпадает с основным уравнением теории столк-
новений. Величина одв — сечение соударения частиц А
И В, р. — приведенная масса /Ид^вМа+^в-
10
Введение
Расчет Е более сложен. Для реакции X + YZ—>XY + Z
потенциальная энергия трехатомной системы может
быть выражена следующим образом:
£=Л + В + С-1[(а-₽Г + (₽-у)2 + (у-а)2]'/’.
где (Л + а), (В + р), (С+у)—полные потенциальные
энергии молекулярных пар XY, YZ и XZ. Первый член
в этих парах представляет собой энергию «кулонов-
ского» взаимодействия, а второй — «обменную» энергию.
Трудность расчетов заключается в распределении общей
энергии между этими двумя составляющими.
Откладывая значения энергии Е как функцию межъ-
ядерных расстояний XY и YZ, можно построить диа-
грамму пути реакции, наиболее высокая точка которого
дает величину энергии активации [2].
Интерес к подобного рода расчетам может быть вы-
зван чисто практической целью увеличения выхода про-
дукта данной реакции. При обычном лабораторном син-
тезе или расщеплении часто достаточно получить прием-
лемый выход на каждой стадии. Но если желательно
повысить выход, например при промышленном синтезе,
необходимо исследование механизма реакции. Напри-
мер, при нитровании фенола азотной кислотой в отсут-
ствие азотистой кислоты образуются о- и п-нитрофенолы
в отношении 7:3. В присутствии больших количеств
азотистой кислоты отношение этих продуктов соста-
вляет 1 :9. Поскольку такое же соотношение наблю-
дается при нитрозировании фенола азотистой кислотой,
вероятный механизм реакции в этом случае заклю-
чается, по-видимому, в быстром нитрозировании фенола
азотистой кислотой с последующим окислением азотной
кислотой образовавшегося нитрозосоединения до нитро-
соединения. Таким образом, выход n-нитрофенола мо-
жет быть увеличен путем нитрования азотной кислотой
в присутствии азотистой кислоты [3].
Изучение механизма реакции приносит также боль-
шую пользу при исследовании методов анализа. По-
скольку многие химические реакции, на которых осно-
ваны аналитические методы, не протекают абсолютно
количественно, знание их механизма в деталях помогает
Введение
11
повысить надежность аналитического метода. Например,
метилкетоны могут быть количественно определены по
реакции образования йодоформа, которая в случае аце-
тона протекает по уравнению
СН3СОСН3 -H3I2 + 4NaOH —► CHI3 + CH3COONa + 3NaI+3H2O
Одна из ошибок обычной аналитической методики воз-
никает вследствие протекания побочной реакции
310“ —► 10^4-21“.
Морган с соавторами [4], систематически варьируя кон-
центрации ацетона, иода и NaOH, экспериментально
обосновали условия количественного выхода йодоформа:
I) Ацетон и щелочь должны быть предварительно сме-
шаны, после чего медленно при помешивании до-
бавляется иод в умеренном избытке для уменьшения ско-
рости диспропорционирования. 2) Необходим большой
избыток щелочи, примерно в 20 раз больше стехиометри-
чески требующегося количества. Это способствует обра-
зованию иона енолята, замедляет побочные реакции,
ведущие к потере активных частиц иода, и катализирует
разрыв связи С—С.
Исследование механизмов реакций имеет и чисто
теоретический интерес. Подобные исследования в прош-
лом принесли большую пользу при объяснении резуль-
татов многих эмпирических и полуэмпирических наблю-
дений. В настоящее время их целесообразно продолжить
с целью попытаться установить корреляцию между
структурой молекул веществ и скоростью реакций.
Например, отношение скоростей реакций галогениро-
вания различных монозамещенных бензолов к скорости
реакции галогенирования бензола колеблется между 1018
для диметиланилина и 1,8 • 10-6 для нитробензола [5],
причем экстремальные значения отношения скоростей
определялись косвенно. Эмпирически была установлена
корреляция [5] между значениями скоростей реакций и
индукционным и таутомерным эффектами замещающих
групп.
При детальном исследовании механизма химиче-
ской реакции необходимо учитывать четыре фактора;
12
Введение
во-первых, природу исходного вещества; во-вторых,
влияние растворителя, в котором растворено это веще-
ство; в-третьих, реагент, приводящий к химическому из-
менению, и, наконец, возможное каталитическое влия-
ние на течение реакции.
Каждой молекуле обычно приписывается какая-либо
структура. Достоверность некоторых структур подтвер-
ждена экспериментально, в то время как для других
имеются недостаточные экспериментальные обоснования.
Одинарная ковалентная связь в молекуле А—В может
быть разорвана двояко. Если связь разрывается симме-
трично с образованием двух радикалов, каждый из ко-
торых имеет по одному электрону из электронной пары,
осуществлявшей химическую связь, т. е. А : В—>А • + • В,
процесс называется гомолитическим. Если же связь раз-
рывается несимметрично с образованием двух заряжен-
ных частиц, у одной из которых находятся оба элек-
трона, т. е. А В—>А++ : В, такой процесс носит назва-
ние гетеролитического. При первом типе разрыва связи
механизм реакции является «радикальным», при втдром
типе — «ионным».
Растворитель оказывает большое влияние на разрыв
связи. Для разрыва типа А : В—>А++ : В требуется более
высокая энергия, так как при этом возникают две раз-
ноименно заряженные частицы, для разделения которых
необходимо затратить работу, противодействующую
электростатическим силам притяжения. При разрыве
связи с образованием незаряженных частиц такая энер-
гия не затрачивается, поэтому в газовой фазе протекают
в основном гомолитические реакции Растворители с
высокой диэлектрической проницаемостью уменьшают
электростатическую работу разделения ионов, что в об-
щем благоприятствует гетеролитическому разрыву связи.
Растворители с низкой диэлектрической проницаемостью
способствуют гомолитическому разрыву связи. Четкого
разграничения здесь, однако, не существует.
В предлагаемой книге обсуждается механизм реак-
ций, протекающих в газовой фазе, в растворах воды и
некоторых других растворителях. Йз рассмотрения пол-
ностью исключены реакции в полярных растворителях
Введение
13
типа жидкого аммиака и в высокотемпературных систе-
мах (в расплавах солей).
Дальнейшее обсуждение отложим до тех пор, пока в
последующих главах не будут рассмотрены классифика-
ция окислительных агентов и каталитические эффекты.
Классификация реакций окисления
Процессы окисления часто встречаются в повседнев-
ной жизни. Старение резины, побурение яблока на срезе,
ржавление железа, высыхание масляных красок — все
это примеры окисления атмосферным кислородом.
В общей химии реакции окисления не менее обычны;
накоплено большое число эмпирических наблюдений и
выводов, характеризующих такие реакции. Цель настоя-
щей работы — систематически рассмотреть реакции
окисления, обобщив эмпирические наблюдения и вы-
воды.
Многие реакции окисления представляют собой не
что иное, как хорошо отработанные рецепты, поэтому
окислительные агенты в таких реакциях, как правило,
нами не рассматриваются.
При обсуждении механизма окисления обычно рас-»
сматривают реакции, включающие:
потерю веществом водорода,
присоединение кислорода к веществу,
потерю веществом электронов.
Эта классификация отражает общую схему изложения,
принятую в данной книге. По-видимому, нельзя дать
точное определение процесса окисления, но по ходу изло-
жения будет ясно, какие отдельные классы реакций рас-
сматриваются. Определение процесса окисления можно
сделать настолько широким, что оно станет бесполез-
ным. В то же время какие-либо ограничения приводят
к исключению некоторых реакций, обычно рассматри-
ваемых как реакции окисления.
При обсуждении будем исходить из того, что реак-
ции могут быть гомогенными или гетерогенными, гомо-
литическими или гетеролитическими. Таким образом, по-
лучается 4 класса реакций. Поэтому наиболее полезной
14
Введение
для наших целей можно считать следующую классифи-
кацию:
1) реакции, при которых электроны переходят от од-
ного катиона к другому (гл. 1);
2) реакции катионов с восстановителями, которыми
могут быть анионы, комплексные ионы или нейтральные
молекулы (гл. 2);
3) реакции, при которых из молекулы ХОН сначала
образуется катион Х+, который затем реагирует с осно-
ванием Льюиса, например
ХОН + Н+ Х++Н2О,
В -1- X + —► продукты (В — основание Льюиса);
такие реакции обсуждаются в гл. 3;
4) реакции окисления при взаимодействии анионов
друг с другом, анионов и комплексов или комплексов
между собой (гл. 4);
5) реакции окисления молекул и анионов различ-
ными простыми и комплексными анионами и комплекс-
ными катионами (гл. 5).
Реакции первых трех классов протекают преимуще-
ственно в кислой среде, так как при высоких значе-
ниях pH катионы Х+ осаждаются в виде гидроокисей.
Реакции последних двух классов протекают преиму-
щественно (но не исключительно) в щелочных раство-
рах. В самом деле, при низких значениях pH равновесие
диссоциации слабых кислот сдвигается в сторону обра-
зования недиссоциированных молекул, а комплексные
ионы имеют тенденцию распадаться с образованием ио-
нов металла.
Реакции окисления гликолей, проходящие через ста-
дию образования эфиров (например, окисление бихрома-
том, иодной кислотой), обсуждаются в гл. 6. В гл. 7 рас-
сматриваются гомолитические реакции в растворе, а в
гл. 8 — гомолитические реакции в газовой фазе.
При обсуждении процессов окисления в газовой
фазе умышленно исключены более сложные газовые ре-
акции, например протекающие в пламенах.
В гл. 9 рассматриваются гетерогенные реакции окис-
ления. Этот класс обычно включает некоторые биологи-
Введение
15
ческие реакции, геологические процессы, реакции элек-
тролитического окисления и процессы коррозии. Такие
более сложные реакции в данной книге подробно не об-
суждаются *.
ЛИТЕРАТУРА
Общая
В a solo F„ Pearson R. G., Mechanism of Inorganic Reactions,
Wiley, New York, 1958.
Clark W. M., Oxidation—Reduction Potentials of Organic Systems,
Williams and Wilkins. Baltimore, 1960.
Fieser L. F., Experiments in Organic Chemistry, Heath, Boston,
1958 (Chapter 50, section on reagents).
И н г о л ь д К. К., Механизм реакций и строение органических соеди-
нений, М., Издатинлит, 1959.
Латимер В. М., Окислительные состояния элементов и их потен-
циалы в водных растворах, М., Издатинлит, 1954.
Waters W. A., Oxidations Processes in Organic Chemistry ed. Gil-
man H., Vol. 4, Chapter 12, Wiley, New York, 1953.
Цитированная
1. Sharpe A. G., Principles of Oxidations and Reduction, R. Insti-
tute of Chemistry, Monographs for Teachers, 1961, № 2.
2. L a i d 1 e r K. J., Chemical Kinetics, McGraw-Hill, New York, 1950,
Chapter 13.
3. F i и a r I. L., Organic Chemistry, Longmans Green, London, 1954,
p. 533.
4. M о r g a п K. J., Bardwell J., C u 11 i s C. F., J. chem. Soc.,
3190 (1950).
5. Robertson P. W., de la Mare P. B. D., S w e d 1 u n d В. Е.»
J. chem. Soc., 782 (1953).
• О механизме реакций фотоокисления и фотовосстановления
неорганических ионов и органических красителей см. монографию
Теренина, Фотоника молекул красителей и родственных органи-
ческих соединений, Л., «Наука», 1967.
Глава 1
РЕАКЦИИ ОКИСЛЕНИЯ - ВОССТАНОВЛЕНИЯ
МЕЖДУ КАТИОНАМИ
В этой главе обсуждаются реакции, при которых
электроны переходят от одного катиона к другому,
с учетом состояния ионов в растворах и возможного рав-
новесия гидролиза. После общего рассмотрения кине-
тики и механизма таких реакций детально изучаются от-
дельные реакции.
Растворы электролитов
При обсуждении процесса окисления следует учиты-
вать состояние ионов в растворе.
Под термином «простой ион» подразумевается такой
ион, для которого не найдено экспериментально каких-
либо ассоциированных форм. Формула Х(Н2О){11, припи-
сываемая ионам в растворе, часто выводится из резуль-
татов рассмотрения структур кристаллических веществ.
Однако, несмотря на то что доказано существование
Be(H2O)Jr в кристаллах (тетраэдр), отсутствуют убеди-
тельные доказательства существования BefHkO)!1 в рас-
творе [1].
Результаты сравнения кривых радиального распре-
деления для водных растворов солей и чистой воды сви-
детельствуют, в частности, о том, что катионы К+ и Са2+
координируют по 6 молекул воды, при этом образуется
октаэдрическая конфигурация [2].
Изучение спектров комбинационного рассеяния света
показывает, что добавление солей изменяет частоту ва-
лентных колебаний гидроксила в жидкой воде, частота
деформационных колебаний при этом остается неизмен-
ной [3]. Для очень высоких концентраций солей экспери-
менты не проводились. Поэтому выводы из имеющихся
2 Зак. 1249
18
Глава 1
экспериментальных данных до некоторой степени неоп-
ределенны.
Таубе [4] показал, что путем сравнения спектров по-
глощения иона с известным стереохимическим окруже-
нием в кристаллах со спектром поглощения иона в вод-
ном растворе можно сделать заключение относительно
стереохимического окружения воды в растворе. Полу-
ченные данные позволяют предполагать существование
в водном растворе таких ионов, как Ni(H2O)^, Со(Н20)^
и Fe(H2O)H
Путем сравнения спектров поглощения ионов пере-
ходных металлов и некоторых их комплексов в водном
и тяжеловодном растворах было найдено, что при за-
мене обычной воды на тяжелую полосы поглощения гид-
ратированных ионов в видимой области спектра сме-
щаются в сторону более длинных волн. В случае окса-
латных комплексов такого смещения не наблюдается.
Это означает, что полосы поглощения смещаются вслед-
ствие замены молекул Н2О на D2O в первой координа-
ционной сфере [5].
Некоторые эмпирические соотношения были найдены
при изучении зависимости активности воды от моляль-
ности электролита [6]. Для электролитов типа 1—1 экс-
периментальные данные приводят к числу гидратации в
первичной сфере, равному 6. Это число,' по-видимому,
характеризует то минимальное число молекул воды, ас-
социированных с двумя ионами, которое будет сохранять
тетраэдрическую структуру воды в соответствии с мо-
делью, впервые предложенной Берналом и Фаулером [7].
Для электролитов типа 2—1 экспериментальные резуль-
таты дают число гидратации в первичной сфере, рав-
ное 10. Это согласуется с числом гидратации двухвалент-
ного иона, также равным 6.
Аналогичные выводы можно сделать при рассмотре-
нии функции кислотности Гамметта для концентриро-
ванных водных растворов кислот [8]. Быстрое увеличе-
ние функции кислотности с повышением концентрации
НС1, НВг, НС1О4 и H2SO4 (до 8 М) указывает на су«
шествование иона Н9О4+1Н+(Н2О)4]. Недавно опублико-
вана статья, содержащая всестороннюю оценку дру-
Реакции окисления — восстановления между катионами 19
гих фактов, подтверждающих существование такого
иона [9].
Робинсон и Стокс [10] также рассмотрели данные для
концентрированных электролитов и развили теорию Де-
бая — Хюккеля. Хотя их аргументы представляют зна-
чительный интерес, они не позволяют легко объяснить
значения чисел гидратации и поэтому не будут здесь бо-
лее подробно рассматриваться.
Результаты изучения скорости реакции обмена дают
основания предполагать, что с ионом хрома прочно свя-
зано 6 молекул воды. Опыты с использованием изотопов
18О показали, что обмен молекул воды между ионом
Cr(H2O)J11 и растворителем происходит достаточно мед-
ленно с полупериодом 7\2~40 час [11]. Изучение спектра
ЯМР 17О в водных растворах позволяет отличить моле-
кулы воды растворителя от молекул воды, находящихся
в сфере гидратации некоторых катионов, при условии,
если процесс обмена протекает сравнительно медленно
(Г|/2>10-4 сек) [12].
Полученные результаты согласуются с числом гидра-
тации 4 для иона бериллия и 6 — для иона алюминия.
Из сказанного видно, что пока еще существует неоп-
ределенность в вопросах точного числа, структуры и
прочности удерживания молекул растворителя, окру-
жающих «простой ион» в водном растворе. Такая неоп-
ределенность может быть полностью устранена с по-
мощью новейших физических методов изучения струк-
туры. Можно надеяться, что для расчета макротермоди-
намических свойств, таких, как активность воды, будут
разработаны методы статистической термодинамики.
Равновесие гидролиза и оксикатионы
Естественным развитием химии простых ионов яв-
ляется область гидролитических равновесий, весьма ин-
тересный обзор которых дан Силленом [13]. Речь идет
о равновесиях типа
ХП++Н2О Х<п-1)+ОН4-Н+
При гидролизе образуются оксикатионы, в которых атом
кислорода прочно связан с атомом металла.
20
Глава 1
По-видимому, наиболее достоверно существование
ионов трансурановых элементов ТиОг+ иТиОг' (Ти—обоб-
щенная формула трансурановых элементов), образова-
ние которых подтверждено различными физическими ме-
тодами [14]. Системы Tuv/Tuvi и Tuin/TuIV легко обра-
тимы, тогда как равновесие в системах Tuiv/Tuv и
TuIV/TuVI достигается медленно. Этот факт предста-
вляет интерес для понимания механизма реакции и до-
казывает сходство химических связей кислорода с Tuv
и TuVI в ионах и сходство состояния несвязанных ионов
Ти™ и Tuiv.
Доказательства существования ионов четырех- и пя-
тивалентного ванадия VO2+ и VO? получены при изме-
рении ЭДС гальванического элемента [15, 16]. Образова-
ние иона VO? подтверждается сравнением [17] энтропий
ионов VO* и NpOg > для которых эти величины оказа-
лись практически одинаковыми. Однако в более поздней
работе [18] высказывается сомнение относительно суще-
ствования простых ионов VO^.
В качестве примеров рассмотрим равновесия гидро-
лиза ионов Fe™, CeIV и UVI, которые обнаруживают
различную степень сложности поведения в растворе.
Для ферри-иона равновесие определяется уравне-
ниями [13]
Fe3+4-H2O 5=± Fe(OH)2+4-H+
Fe(OH)2+ -J- Н2О Fe(OH)+-|-Н+
2Fe«++2H2O Fe2(OH)£+ 4-2Н+
log Ki— 3.05,
log Ka — 3,26.
log p22 —2,91.
Церий (IV) в растворах хлорной кислоты находится
[19а] в виде смеси ионов Се4+, Се(ОН)3+ и димера
Се—О—Се6+, а в растворах серной кислоты [196] — в
виде смеси CeSO2+, Ce(SO4)2 и Се(8О4)з“
Гидролитическое равновесие для UVI в среде перхло-
рата отражают уравнения [20]
2UO|+ + Н2О (UO2)2OH3+ 4- Н+
И
2UO2+ 4-2Н2О (UO2)2(OH)|+ 4-2Н+,
Реакции окисления — восстановления между катионами 21
а результаты экспериментов при низкой кислотности
предположительно объясняются равновесием
3UO|+ +4Н2О 5Z± (UO2)3(OH)|+ + 4H+.
Гидролитические равновесия вносят дополнительные
усложнения при рассмотрении реакционной способности
ионов в растворе.
Обычно имеет место ассоциация катионов с анио-
нами, хотя не всегда известна ее степень. Считается, что
перхлораты обычно полностью диссоциированы, однако
отсутствие ассоциации нуждается в доказательстве.
Спектрофотометрические исследования свидетельствуют
в пользу ассоциации ферри-ионов с ионами перхлората
[21], а кинетические — подтверждают ассоциацию ионов
ртути с ионами перхлората [22]. Для установления воз-
можности образования комплексов было предпринято
изучение спектров комбинационного рассеяния раство-
ров перхлоратов различных металлов [23]. При этом, по-
мимо линий перхлорат-иона, было обнаружено не-
сколько дополнительных линий, что, однако, не является
достаточным доказательством существования ассоциа-
ций.
Кинетика и механизм
Скорости реакций можно выразить при помощи урав-
нения, выведенного из теории активного комплекса [24а]:
ЛСК = (ЛТ/А)^Д^.^^,
так что различия скоростей реакций сводятся к разли-
чиям в величинах энергии и энтропии активации.
В частности, в случае реакций между ионами в вод-
ных растворах энтропия активации AS* равна.— 10zaZb,
где zA и — заряды ионов [246]. На основании теории
соударений более вероятно протекание реакций с поло-
жительной энтропией активации, т. е. между противо-
положно заряженными ионами, и менее вероятно —
с отрицательной энтропией активации, т. е. между одно-
именно заряженными ионами.
В действительности энтропия активации в реакциях
с переносом электронов значительно меньше —10zazb*
22
Глава 1
Для реакции обмена между ферро- и ферри-иона-
ми [25] энтропия активации должна быть равной
—252 дж!молъ-град, в то время как экспериментально
найденная величина составляет —105 дж!моль* град.
Поскольку аналогичные результаты получены во всех
исследованных реакциях с переносом электронов, необ-
ходимо искать другие пути расчета скорости таких ре-
акций.
Абсолютные энтропии ряда активных комплексов для
реакций с участием ионов урана, нептуния и плутония
были рассчитаны из следующего соотношения [26]:
А*$эксп = акт. компл ^продуктов 5 реагентов*
Этим путем была обнаружена корреляция между энтро-
пией активного комплекса и его зарядом. Некоторый
разброс величин указывает на то, что, помимо заряда,
должны иметь значение также размеры и форма актив-
ного комплекса.
Очень несложные электростатические расчеты пока-
зывают, что введение отрицательно заряженного иона
между двумя положительно заряженными достаточно
для того, чтобы вместо очень большой энергии взаим-
ного отталкивания появилась энергия взаимного притя-
жения [27].
Легко представить протекание процесса переноса
электронов через следующие последовательные стадии*
Fe3+ + X" FeX21.
FeX2+-|-Fe24 7—> +2Fe« X-Fe2+ переходное состояние,
+2Fe-X-Fe2 + ^z± Fe2+ +FeX2+,
FeX2+ ^z± Fe3+ + X“.
Однако, если связи в активном комплексе слишком
прочны, он, по-видимому, не может распасться после пе-
реноса электрона, и, таким образом, скорость реакции
скорее понижается, чем повышается. Это соображение
подтверждается сравнением реакционной способности
иона T1CN2+, который уменьшает скорость обмена элек-
тронов между Т13+ и Т1+ и иона Tl(CN)4', который повьь
шает эту скорость [28].
Реакции окисления — восстановления между катионами 23
Хотя аналогичное соображение было высказано при
объяснении облегченного переноса электрона через ани-
онный мостик при обмене Fe3+—Fe2+, остается необъяс-
ненным тот факт, что обмен электронами может иметь
место даже в отсутствие какого-либо аниона, связываю-
щего ионы железа. Теория [29] предполагает первона-
чальную перестройку координационных сфер до наибо-
лее благоприятной конфигурации с последующим тун-
нельным переходом электрона от одного реактанта
к другому через барьер растворителя. Эта модель была
использована Лейдлером при развитии более общих
представлений [30]. Очень четко представляя процессы,
включающие перенос электрона от иона Fe2+ к иону Fe3+,
он вывел уравнение для общей константы скорости реак-
ции
. kT -Д^ифф/*’' -д4т/«г -Д<уннЛГ
Лек = * * ’ * • *
где ЛЛИфф — свободная энергия сближения двух ионов,
контролируемого процессом диффузии;
AFott — свободная энергия активации взаимного
электростатического отталкивания ионов;
АЛгунн — свободная энергия активации туннельного
перехода электрона по законам квантовой
механики.
В этой тщательно обоснованной работе, объединяющей
большое количество ранее полученных данных, исполь-
зованы достоверные предпосылки для расчета всех упо-
мянутых величин свободной энергии. Обнаружено, что
общая свободная энергия активации, представляющая
собой сумму факторов, определяющих диффузию, оттал-
кивание и туннельный эффект, проходит через минимум
при расстоянии между ионами 4 А. В последующей ра-
боте [31] эта модель признана слишком упрощенной.
Полная свободная энер1ия активации была разбита на
три слагаемых: 1) свободную энергию отталкивания в
рамках электростатической теории с допущением о ди-
электрическом насыщении; 2) энергию реорганизации
растворителя; 3) свободную энергию, соответствующую
квантовомеханическому туннельному переходу. Сумма
этих величин проходит через минимум при расстоянии
24
Глава 1
4,2 А, а рассчитанная величина свободной энергии акти-
вации 67 кдж1моль согласуется с найденной эксперимен-
тальной величиной 70,5 кдж!моль.
Возможно также, что обмен электронов связан с пе-
реносом атома водорода по схеме
Fe2a+ + H2O + Fe(OH)2+ ->
/ Н Н Н \4 +
I I I J
\Fe-O-H —> О—Н —► О-rfe/ —> Fe(OH)2+ + Fe2q+.
Таким образом, здесь атомы железа ассоциированы
с молекулами воды. Эта гипотеза была эксперименталь-
но проверена [32] при изучении скорости обмена элек-
трона между Fe2+ и Fe3+ в тяжелой воде. Удельная ско-
рость обмена в тяжелой воде понижалась примерно в
2 раза, что подтверждает участие атома водорода при
обмене электронами. К сожалению, этот вывод не со-
всем однозначен, так как не известно влияние тяжелой
воды на гидратацию.
Можно также представить, что некатализируемая ре-
акция между ионами Fe111 и Fe11 протекает через обра-
зование активного комплекса с обобществленной моле-
кулой воды:
Н
I
+2Fe_O_Fe3+
I
н
Очень высокий положительный заряд комплекса облег-
чает потерю протона, что приводит, к механизму обмена
через мостик. В случае плутония такому эффекту спо-
собствует более высокий заряд ионов, поэтому неката-
лизируемая реакция переноса электронов Ри1П—PuIV
имеет более низкую энергию активации (32,4 кдж/моль)
[33], чем для обмена Fe11—Fe111 (41,5 кдж/моль).
Скэтчард [34] исследовал влияние диэлектрической
постоянной среды на скорость ионных реакций и вывел
следующее уравнение:
d 1П k O/L.'T'l)
= — lkT
Реакции окисления — восстановления между катионами 25
где k'— константа скорости, D—диэлектрическая по-
стоянная, zA, — заряды ионов, е — заряд электрона,
k — константа Больцмана, Т—абсолютная температура,
г — расстояние между ионами.
За исключением нескольких реакций, при изучении
которых замена растворителя позволяла выяснить меха-
низм реакции в воде, отсутствуют данные для процессов
переноса электронов при реакциях в других растворите-
лях, кроме воды. Хотя протекание некоторых реакций ис-
следовалось и в других растворителях, накопленных экс-
периментальных данных еще недостаточно для каких-
либо широких обобщений.
Для ряда реакций наблюдается катализ металлами.
Так, общая скорость обмена Fe11—Feni увеличивается
в 4—5 раз в присутствии серебряной фольги [35]. Обмен
Т11—Т1Ш ускоряется платиновой чернью, а обмен
Ей11—Ей111 — в присутствии платиновой проволоки. Ин-
тересные взгляды на каталитическое действие метал-
лов высказал Спиро [36], который считает, что проте-
кание реакций облегчается переносом электронов через
металл.
Имеется превосходное общее описание эксперимен-
тальных методов исследования реакций с переносом
электронов [37].
Классификация реакций с переносом электронов
По числу электронов реакции этого типа можно клас-
сифицировать следующим образом:
I. Реакции с переносом одного электрона.
а) Реакции между простыми катионами одного и того
же металла, например
Pell 4-Fein 7~» Fe» -f-РеШ.
б) Реакции между оксикатионами одного и того же ме«
талла, например
NpO2+ +NpO^4 NpO+ -f-NpO£+.
26
Г лава 1
в) Реакции между простыми катионами различных ме-
таллов с образованием простых катионов, например
СеШ-)_СоШ —> Ceiv-I-Coii.
или с образованием оксикатионов, например
PuIV _(_VII1 + н2О —> Pu1114- VO,v + 2Н+
г) Реакции между оксикатионами различных металлов,
например
Puv+Amvl Puvi4-Amv
д) Реакции между простыми катионами и оксикатио-
нами одного и того же металла с изменением состоя-
ния окисления, например
Ри1П4-РиО^ —> PuIV + PuOV,
или сопровождающиеся изотопным обменом, напри-
мер
NpiV-f-NpV NpiV-t-NpV.
е) Реакции между простыми катионами и оксикатио-
нами различных металлов, например
Fen+Puvi —> Fem + Puv.
ж) Реакции диспропорционирования, например
2Pu,v4-6H2O —► Puin4-rPuO^4-4H3O+.
II. Реакции с переносом двух электронов.
а) Реакции между простыми катионами одного и того
же металла, например
Т11 -Н Т11П Т*1Ш + Т11.
б) Реакции между простыми катионами различных ме-
таллов с образованием простых катионов, например
Hg‘ + Tlin —► 2Hg" +Т11,
или с образованием оксикатионов, например
U1V+ Т11н4-2Н2О uO2v14-T1i4-4H\
Реакции окисления — восстановления между катионами
в) Реакции между простыми катионами и оксикатио-
нами одного и того же металла, например
й,у + иоу1 uIV + 0o^.
г) Реакции между простыми катионами и оксикатио-
нами различных металлов, например
UIV + PuOyI —> UO^ + Pu,v.
I. Реакции обмена с переносом одного электрона
Формально простейшей реакцией окисления является
перенос электрона между двумя различными катионами
одного и того же элемента в различном валентном со-
стоянии. Экспериментальное исследование таких реакций
возможно при соблюдении двух важных условий. Во-
первых, металл должен иметь изотоп с подходящим вре-
менем существования и удобной для измерений актив-
ностью, и, во-вторых, должна быть разработана методи-
ка разделения, обеспечивающая контроль за скоростью
реакции. При наличии требуемых условий можно иссле-
довать влияние кислотности и добавок анионов на про-
текание реакций. По результатам измерения темпера-
турного коэффициента, теплот и энтропий можно срав-
нивать различные реакции.
В первых же работах была показана возможность
обмена электронов и неожиданно обнаружено, что об-
мен часто происходит с очень высокой скоростью.
Для удобства изложения рассмотрим сначала обмен
электрона между одно- и двухзарядным катионами, за-
тем двух- и трехзарядным катионами и, наконец, трех-
и четырехзарядным катионами.
В 5,9 М хлорной кислоте скорость изотопного обмена
между Ag1 и Agn составляет /? = &[Agn]2. Обмен проте-
кает быстро. При 0° константа скорости равна 1,020±
±4 м2чал,олъ~х*секг\ а энергия активации составляет
52,5±5 кдж!моль [38]. Для объяснения резкого увеличе-
ния скорости обмена с понижением концентрации хлор-
ной кислоты предполагается следующий механизм этой
28
Г лава 1
реакции:
Затем
Agn + xH2O ±5: Ag(OH)(x2~x)+ + xH+.
быстрое установление равновесия
2Ag(OH)^ х)+ qr± [активный комплекс] у~> AgHi-|-Ag>.
стадия, определяющая скорость
Высокую скорость обмена, по-видимому, можно объяс-
нить ограниченной степенью гидратации ионов серебра
по сравнению с ионами других элементов более высокого
заряда.
Были приготовлены перхлораты Ag1 и Agn. Раствор
перхлората Ag11 готовился растворением AgO в 6 М
хлорной кислоте, причем AgO получали анодным окис-
лением нитрата серебра в растворе. Растворы перхлора-
тов Ag1 и Agn нагнетались в смесительную камеру, от-
куда реакционная смесь попадала в азотнокислый рас-
твор о-фенантролина. Последний немедленно прекращал
реакцию обмена вследствие осаждения комплекса Agn.
Обмен в системе Fe11—Feni изучен Сильверменом и
Додсоном [25].
Приготовленные растворы перхлоратов Fe11 и Fein
тщательно смешивались. Затем отбирали аликвотные ча-
сти смеси и прекращали в них реакцию путем добавления'
а, а'-дипиридила и ацетата натрия до pH около 5. Об-
разование стабильного красного комплекса железа (II)
с а, а'-дипиридилом прекращало реакции ферро-иона.
Ионы железа(III) затем отделяли осаждением в виде
Fe(OH)3 аммиаком. Осадок отфильтровывали, промы-
вали, высушивали и исследовали. Найденные таким пу-
тем скорости реакции отвечают эмпирическому уравне-
нию
In (У — Уа-ya3) = — R(a-\-b) tfab.
где i/o, У и t/oo представляют удельные активности соот-
ветственно вначале, в момент t и через бесконечно боль-
шое время, R — константа скорости обмена, а и b — кон-
центрации обоих реактантов.
В хлорной кислоте реакция обмена протекает по
первому порядку относительно каждого реактанта. Ско-
Реакции окисления — восстановления между катионами 29
рость возрастает с уменьшением концентрации ионов
водорода в соответствии с уравнением
Я/[Ге11] [Ге111] = А + в/[Н+1.
Общее кинетическое уравнение имеет следующий вид:
R=kt [Fe2+] [Fe3+| + к2 |Ре2+] [FeOH2+],
Если вместо [FeOH2+] поставить его выражение из реак-
ции равновесия
Fe3++OH_ 5z± Fe(OH)2+
то полученный результат будет находиться в согласии
с приведенным выше эмпирическим уравнением.
Скорость реакции можно выразить также как функ-
цию концентрации ионов хлора:
Я = A, [Fe2+ ] [Fe3+J + A, [Fe2+] [РеОН2+] -J-
+ а' [Fe2+] [FeCi+] + A" |Fe2+] [PeClf].
Некоторые значения констант скорости теплоты и
энтропии активации для реакций с ионом Fen представ-
лены в табл. 1.
Таблица 1
k, м9-кмоль-сек~ 0е С ДЯ* 10е дж!кмолъ Д$*, 10е дж/кмоль-град
Fe’+, kt 8,7-10"1 41,5 —105
Fe(OH)2+, k. 1.0-10® 31 -75,5
FeCl2+. k' 9,7 37 —101
FeCl2+. k" 1,5-10 40,6 —84
Обмен в системе Fen—Fe111 представляет типичный
пример реакций, на скорость которых влияют концен
трация ионов водорода и концентрация других коорди-
нируемых анионов.
Влияние анионов сложно. Константа скорости обмена
в реакции, протекающей с участием иона РеСгО?, со-
ставляет 700 ле3 • кмоль~1 • сект1, в то время как в реак-
ции с участием Fe^OOsT значение константы скорости
30
Глава 1
обмена 2500—4500 м3 • кмоль~1 • сек~{ [35]. Обнаружено,
что серная кислота значительно ускоряет обмен [39].
Банн с соавторами изучил реакцию обмена между Fe11
и азидом Fe111 в водном растворе [40]. Эксперименталь-
ные данные подтвердили механизм переноса электрона
через мостик Fe3+—N3 —Fe2+. При температуре ниже 13°
скорость реакции обмена определяется скоростью обра-
зования этого комплекса, тогда как при более высокой
температуре скорость реакции сравнима со скоростью
обмена электронами.
Обмен в системе Fe11—Fe111 в изопропиловом спирте
происходит в 108 медленнее, чем в воде [41]. Это согла-
суется с результатами, вытекающими из уравнения Скэт-
чарда [34]. Вычислено [41], что отношение &Спирт/&вода со-
ставляет приблизительно 10~9 для шестизарядных частиц
и 10~6 — для четырехзарядных, если принять расстояние
между реактантами в активном комплексе равным 6,8 А.
Скорость обмена в системе ферро — ферри исследова-
лась также при низких температурах, причем устано-
влено, что реакция протекает с измеримой скоростью
fe = 2,l • 10"3 м3 • кмоль'1 • сек~х даже при температуре
—78° [42].
Реакция обмена в системе Сг11—Сг1П в хлорной кис-
лоте протекает по первому порядку относительно ионов
хрома (II) и хрома (III) [43]. Скорость обмена выражает-
ся уравнением
/? = kx [Сг2+] [Сг3+] +*2 [Сг2+] [СгОН2+],
где &! = 1,93 • 10-5 и ^2 = 7,0*10-1 м3 • кмоль'1 • сек~х при
24,5° Если заменить концентрацию СгОН2+ на равно-
весную из следующего уравнения:
Сг3+ + ОН" Сг(ОН)24.
то скорость обмена можно выразить в форме аналогич-
ной скорости обмена в системе Fe11—Fe111
Я/[Сгн| [Сг111] = А + В/[Н+].
Скорость некатализируемой реакции значительно ниже
вследствие особой связи иона Сг3+ с водой. Большая ве-
личина k2 по сравнению с ki согласуется с механизмом
Реакции окисления — восстановления между катионами 31
переноса атома, но не может служить его доказатель-
ством. Катализ ионами хлора не наблюдается, поскольку
при обмене в системе Сг11—Сг1П невозможно переход-
ное состояние в виде активного комплекса [(Н2О)5Сг—
14+
из-за прочной связи воды в ионе
Cr(H2O)g+ и невозможности легкого соединения иона хлора
с ионом хрома. Таубе и Кинг показали, что в системе
Сг2+—СгС12+ происходит достаточно быстрый обмен элек-
тронов, доступный измерению [44]. Они пришли к заклю-
чению, что обмен происходит через мостик активного
комплекса с конфигурацией (Сг С1..Сг)4+, который
способствует переносу атома хлора. Такой механизм
справедлив также для обмена в присутствии других
анионов. Можно предположить, что образующий мостик
ион азида должен особенно сильно способствовать пере-
носу электрона [45]. Реакция обмена Ей11—Еиш в при-
сутствии хлорид-ионов протекает аналогично [46]. Ионы
водорода влияют на скорость обмена в этом случае
очень мало, так как концентрация продуктов гидролиза
невелика.
Для реакции обмена Риш—PuIV в растворе хлорной
кислоты, как и в указанных выше случаях [33], наблю-
дается отрицательный каталитический эффект ионов во-
дорода. Скорость реакции выражается уравнением
R = kx [Pu3+] [Pu4 f 1 + *2 [Pu3+] [Pu(OH)34 ],
где концентрация Ри(ОН)з+ определяется равновесием
гидролиза
Ри4+ +Н2О ^=± Ри(ОН)3+ +Н4
Экспериментально определенные величины теплоты и
энтропии активации реакции приведены в табл. 2. Эти
Таблица 2
Реакция дя*, 10е дж1кмоль д$* 10’ дж/кмоль-град
РиЗ-. . +- Ри44 32,4 -131
Ри8+ - 1- Ри (ОН)»4 11,8 —135
32
Глава 1
величины соответствуют скорости, в 100-4-1000 раз мень-
шей скорости обмена ферро — ферри. Предполагаемое
объяснение этого явления было дано при общем обсу-
ждении кинетики и механизма реакции обмена.
По сравнению с рассмотренными выше системами,
механизм обмена Сеш—CeIV значительно более сложен,
По данным, полученным несколькими исследователями
при различных условиях, скорость обмена для этой ре-
акции пропорциональна
1) [Се3+] [Се4+],
2) [Се3+] [47],
3) [Се3+] [Се4+]2 [48].
Первая зависимость обычна, тогда как третья может
иметь место только при образовании димера Се—О—Се6+
Для второго случая предполагается активация иона
Се3+ растворителем S [47]
л.___
Ce3++S Ce3++S,
«2
где Се3+ — активированный ион церия;
_________________________ к.
Се4+ + Се3+ обмен.
К4
Если предположить, что при образовании Се3+ наступает
стационарное состояние и что Л3[Се4+] fc^S], то ско
рость оказывается пропорциональной концентрации ионе
Се3+. Реакция обмена в системе Сеш—CeIV является
наиболее сложной из всех ранее рассмотренных намг
реакций вследствие сложности состояния иона церия(IV)
в растворе.
Следующие два примера иллюстрируют электронный
обмен между оксикатионами. В случае NpCV —NpO2"1
оба оксикатиона содержат одинаковое число атомов кис
лорода, а в случае VO24 —VO? — различное число ато
мов кислорода.
Результаты изучения реакций обмена в системе
NpC>2—NpOl+ приведены в серии статей [49—51]. В от
ношении каталитического влияния хлор-ионов эта реак
ция аналогична реакции обмена в системе ферро —
Реакции окисления — восстановления между катионами 33
Таблица 3
ДЯ*, дж/кмоль Д$*, дж/кмоль- • град ДЯ* дж/кмоль Д$* дж/кмоль- • град
NpOf/-NpO|+ 44,5-10» -53-10! Fe2 3+—Ре»+ 41,5-10» -105-10»
NpO^-NpO2Cl+ 69,5-10« 45,7.10» Fe2+-FeC12+ 37-10» -113-10»
NpO^-NpO2CI2 64,0-IO® 21-10» Ре»т-FeCl2 40,7-10» -84-10»
ферри (см. табл. 3). При 0° и ионной силе, равной 3, ско-
рость обмена выражается следующим уравнением [49в]:
Я= Ло [Npo+] [NpO2+] [NpO2+] [NpO2Cl+] +
4-ft2[NpO2+] [NpO2Cl2|.
Эти реакции отличаются друг от др^га в двух отно-
|цениях: скорость обмена NpO?—NpC>2+ в отличие от
юбмена Fe11—Fe111 не зависит от концентрации ионов во-
дорода. Энтропия активации в схемах с хлоридным ка-
тализом имеет положительную величину, в то время как
|йри обмене в системах с железом ее значения отрица-
тельны. Различия рассматриваемых реакций обмена
(олжны быть отнесены за счет несимметричной гидра-
тации иона нептуния. Было обнаружено [50], что изме-
нение величины диэлектрической постоянной раствори-
теля добавкой этиленгликоля или глюкозы не влияет на
скорость обмена. Отсюда был сделан вывод, что нека-
йлизируемая реакция протекает по механизму переноса
тома водорода. Это заключение подтверждается (но не
доказывается) опытами с заменой обычной воды на тя-
желую, уменьшающей скорость обмена [51].
Реакция обмена в кислых растворах, содержащих
Хлорид-ионы *, VIV—Vv, протекающая по схеме
tfo2++VO2+ VoJ+VO2+,
* Константа скорости реакции обмена в 6,5 н. кислоте (НС1 +
+НС1О4) составляет 1,5* 106 л2!моль2 • сек,
3 Зак. 1249
34
Глава 1
изучалась с помощью ЯМР. Было найдено, что скорость
этой реакции выражается в новой кинетической форме
следующим образом (52J:
я=л [v,v] [vv]2
Наши предварительные выводы можно закончить''
следующими общими соображениями. Характерными
особенностями переноса электрона от одного катиона
к другому являются, во-первых, зависимость пути пере-
носа электрона исключительно от растворителя; во-вто-
рых, легко наблюдаемый кислотный катализ, связанный
с образованием продуктов гидролиза при повышении
pH раствора; в-третьих, существование реакций обмена,
катализируемых анионами. Различные модификации
всех указанных особенностей не меняют общую харак-
теристику этого класса реакций.
Реакции между простыми катионами различных веществ
Вначале рассмотрим реакции, в результате которых
образуются простые ионы, а затем — реакции, одним из
продуктов которых является оксикатион, что приводит
к некоторым новым кинетическим эффектам.
Сатклиф и Вебер [53а] изучали скорость реакции ме-
жду ионами Сега—Сош в растворах хлорной кислоты
по изменению оптической плотности при длине волны
света 605 ммк (область поглощения иона Сога). По-
правки на поглощение света ионами церия(IV) и це-
рия (III) не нужны, но небольшое поглощение света
ионами Со11 следует учитывать. Было найдено, что ре-
акция протекает по кинетическому уравнению первого
порядка относительно ионов Со111 и Сеш, а ее скорость
уменьшается с повышением концентрации хлорной кис-
лоты и перхлорат-иона. При постоянной концентрации
перхлорат-иона константа скорости &'=а/[НСЮ4], а при
постоянной концентрации хлорной кислоты l/k' = b-^-
+(с/[сю4-])
k' = Я/[Се"1].
Поскольку скорость почти не зависит от концентра-
ции ионов водорода, можно предположить, что в стадии,
Реакции окисления — восстановления между катионами 35
определяющей скорость процесса, участвует перхлорат-
ный комплекс:
Се(С1О4)2+ + Со(ОН)24 —► Се (IV) + Со (II).
Те же авторы изучили влияние ионов нитрата и фторида
[536]. Фторид-ионы вызывают значительно более замет-
ное ускорение реакции обмена, чем нитрат-ионы, но ме-
ханизм их действия одинаков: оба иона участвуют в кон-
курирующих реакциях:
Со(ОН)2+ + Се(С1О4)2+ —> продукты,
Со(ОН)2+ -|- СеХ2+ —► продукты,
где X — ион F“ или NO3 •
Рассматриваемая реакция интересна как пример ка-
тализа перхлорат-ионами. Каталитический эффект ионов
нитрата и фторида известен.
Реакция обмена между простыми ионами PuIV и Fe11
в перхлоратной среде в присутствии хлор-ионов также
протекает с образованием простых ионов и особых от-
личий от ранее рассмотренной не имеет, за исключением
заметного каталитического эффекта серной кислоты [54].
Реакции обмена с образованием оксикатионов имеют
особые черты.
Гизенга и Мэгнуссон [55] для реакции
Np,v -h Fe111 +2Н2О NpO^ + Fe11 + 4H 4
*2
нашли следующее выражение скорости:
— d [NpivlIdt = k2 [Npvl [Fe11] [H + ] — kx [Np1 V1 [Feln]/[H+]3.
Константа скорости й2=7,8-10"2 м6 • кмоль~2 • сект' в 1 М
растворе хлорной кислоты прямо пропорциональна кон-
центрации ионов водорода. Константа скорости прямой
реакции составляет 5,7 м6 • кмоль~2 • се/r1, а энергия ак-
тивации 147 кдж!молъ. Эта величина значительно пре-
вышает аналогичные характеристики ранее встречав-
шихся реакций, что, вероятно, связано с образованием
прочных связей Np—О. Для прямой реакции посту-
лируется следующий механизм, согласующийся с
а*
36
Глава 1
экспериментально найденным кинетическим уравнением
Np(OH)4”* + Fe(OH)3”p —> NpO2++Fe2+4-H++Н2О,
где х+у = 3. Такая схема включает ряд неразличимых
равнозначных путей гидролиза и указывает на более вы-
сокий порядок обратно пропорциональной зависимости
скорости реакции от концентрации ионов водорода, чем
в предшествующем случае.
Прямая зависимость скорости обмена от первой сте-
пени концентрации ионов водорода соответствует реак-
ции диспропорционирования
Npni 4- Npv —.> 2Npiv,
протекающей со скоростью
— d [NpO2+]Idt = k [Np111] [NpO2+] [H+]
[56а].
Предполагается [56a, 57], что в этой реакции прояв-
ляется особый вид катализа вследствие наличия иона
водорода в активном комплексе, который непосред-
ственно приводит к образованию более стабильной фор-
мы иона NpIV, а именно NpOH3+, по схеме
Н
Np3+ + NpO^ + Н+ [О—Np—О—Np]6+
—> NpOH3+4-NpO2+
H+4-NpOH3+ —> Np4+ + H2O
2H+4-NpO2+ —► Np4+4-H2O
стадия, определяю-
щая скорость
быстрая стадия
быстрая стадия
быстрая стадия
Такое же объяснение приложимо к реакции окисления
ферро-иона.
Ванадий (III) окисляется по схеме
Vni + Npv _> VV + Np'V,
-d[Npv]/^ = Mvni][Npvl.
Скорость реакции [57] не зависит от концентрации ионов
водорода и определяется стадией
V3l4-NpO^ —> [О—Np—О—V]4+ —► VO2+ 4-NpO2+
Реакции окисления — восстановления между катионами 37
Присутствие ионов водорода не облегчает протекание
этой реакции из-за нестабильности иона VOH3+.
Для осуществления реакции
PuIV + v111 + Н2О —> Pu111 + VOIV + 2H +
имеются два различных пути [58]:
дж/кмоль
Д5*.
дж/кмоль*град
PuIV + V111 + Н2О —> (*)6+ 4- Н+ 72 • 10е 20.2 • 103
PuiV4-Vn,4-H2O —> (•)5+4-2н+ 90-106 87,5-103
где знаком (*) обозначен соответствующий активный
комплекс. Хотя при этом образуется связь V—О, вели-
чина энергии активации не слишком высока. Скорость
реакции не зависит от присутствия хлор-ионов, по-
скольку последние в данном случае не служат мостиком
для переноса электронов вследствие образования проч-
ной связи V—О [58].
Реакции между простыми катионами и оксикатионами
Реакции между оксикатионами различных металлов,
например РиО^ и АтО<^, по-видимому, не исследова-
лись. Рассмотрим реакции с переносом одного электрона
сначала между простыми ионами и оксикатионами од-
ного и того же металла, а затем — различных металлов,
после чего вкратце коснемся реакций диспропорциони-
рования.
Для скорости переноса электрона между V111 и VIV
в растворе хлорной кислоты Фермен и Гарнер [59] на-
шли следующее уравнение:
/?=[vInl[vIV] (л2+МнЧ).
которое согласуется с механизмом переноса через мо-
стик:
V3++OH" VOH2+.
voh2++vo2+ —► vo2++voh2+
В пределах экспериментальной ошибки константа k2 рав-
на нулю. Это неудивительно, так как некатализируемый
38
Глава 1
путь реакции
$3+_|_VO2+ -фО2++уЗ->
включает разрыв и образование связи металл—кислород
и может иметь высокую энергию активации.
Рабидо [60] измерил скорость следующей реакции:
PuO|+ + Vnl + Н2О —> PuO2++VO2+4-2Н+.
Реакция протекает по кинетическому уравнению второго
порядка, имея первый порядок относительно каждого
реагирующего вещества. Скорость реакции зависит от
[Н+]-1 или [Н+]"2. Это первый пример зависимости скоро-
сти реакции от концентрации водорода в степени минус
два для системы без конкурирующего равновесия гид-
ролиза.
Рассмотрим только один пример реакции диспропор-
ционирования [14, 61]. Реакция
Npiv + NpVi 2Npv
в растворе хлорной кислоты протекает в прямом напра-
влении следующим образом:
- d [NpIV]Idt = k} [NpIV] [NPVI]/[H+p.
Данных, характеризующих зависимость хода реакции от
присутствия ионов водорода, не имеется. Механизм ре-
акции для различных путей гидролиза остается неизмен-
ным. Итак, в результате рассмотрения реакций между
простыми ионами или между простыми ионами и окси-
катионами выявились новые характерные особенности,
заключающиеся в явлении катализа ионами перхлората,
наличии равноценных путей гидролиза, высоких значе-
ниях энергии активации и отсутствии катализа анио-
нами.
II. Реакции с переносом двух электронов
При изучении реакций с переносом двух электронов
прежде всего возникает вопрос: каким образом они про-
текают — в одну или в две раздельные стадии. В по-
следнем случае необходимо установить стадию, опреде-
ляющую скорость реакции. Если скорость лимитируется
Реакции окисления — восстановления между катионами 39
переносом первого электрона, кинетика реакции мало от-
личается от уже рассмотренной для реакций с перено-
сом одного электрона. Примером таких реакций яв-
ляется изотопный обмен
Эта реакция изучена более полно [62—69], чем какие-
либо другие реакции такого типа.
Экспериментальные данные для этой реакции сум-
мировал Росотти [63]. Установлено, что гомогенный об-
мен между одновалентным и трехвалентным таллием в
растворе хлорной кислоты протекает по кинетическому
уравнению первого порядка относительно общей концен-
трации таллия в каждом валентном состоянии. Данные
различных авторов согласуются между собой [62,64,65].
По мнению Росотти, независимо полученные данные по
гидролизу трехвалентного таллия позволяют выбрать
механизм реакции. Росотти пришел к выводу, что доля
продукта гидролиза Т1(ОН)2+, участвующего в обмене,
уменьшается от 40% в 1 М НС1О4 до 20% в 3 М НС1О4.
Общая скорость обмена описывается уравнением
kx [Т1+] |Т13+] +*2 [Т1 + ] [Т1ОН2+],
которое, учитывая равновесия гидролиза
Т13+ + ОН" ^=> Т1(ОН)2\
можно выразить в форме, аналогичной той, которая най-
дена для обмена в системе ферро — ферри:
Я/[тР] [Т1Ш1 = А-|-В/[нЧ
По-видимому, перенос двух электронов не вызывает зна-
чительных изменений в общем кинетическом уравнении.
Проверочное исследование реакции обмена в системе
Т11—Т1Ш при 25° позволило определить константы ско-
рости &i = 7,0’10“5 и /22 = 2,5’10-5 м3 • кмоль~х • секгх [66],
что указывает на недостаточную вероятность обмена че-
рез стадию образования Т1(ОН)^.
Скорость переноса электронов в среде 2,2 М серной
кислоты в 200 раз больше, чем в среде хлорной кислоты
при одинаковых ионной силе и концентрации ионов
40
Глава 1
водорода [67]. По-видимому, двухзарядпые сульфат-ионы
особенно эффективно влияют на перенос двух электро-
нов. В противоположность обмену Fe11—Feni скорость
реакции обмена в системе Т11—Т1Ш вначале уменьшает-
ся при добавлении хлорида, а затем после достижения
отношения [С1‘]: [Т1(Ш)] = 2,5 увеличивается в растворе
серной кислоты [68]. Джилке с сотрудниками [69] обна-
ружил, что это различие соответствует спектрофотоме-
трическим данным для соответствующих соединений Т11П.
Эти данные дают основание предполагать, что хлорид-
и бромид-ионы образуют комплексы (Т1С12+), которые
не участвуют в обмене электронов с таллием (I), а суль-
фат-ионы дают комплексы, принимающие участие в об-
мене электронов.
Вероятно, в реакции обмена Т11—Т11П имеет место
перенос двух электронов [70]. Если бы реакция между
Т11 и CeIV и обмен Т11—Т1га протекали через образова-
ние Т1П, тогда реакция с CeIV в присутствии Т11П не
могла бы протекать медленнее, чем обменная реакция,
поскольку последняя могла бы поставлять Т1п для реак-
ции с Ceiv. Но реакция обмена протекает значительно
быстрее реакции Т1п—C1IV, и поэтому образование Т1п
в указанных реакциях исключается. Очевидно, при об-
мене Т11—Т1Ш, или реакции TlI—CeIV, или в обоих слу-
чаях должен каким-то образом происходить перенос
двух электронов. Дорфман и Грайдер [71] считают, что
при взаимодействии Т11—CeIV происходят следующие
один за другим переносы одного электрона. Для реак-
ции в 6,2 н. азотной кислоте они предлагают следующую
схему:
Ceiv 4- ОН ^=> СеШ + .ОН,
ТР-НОН Т1п + бН,
ТВ +CeIV Т!п + Се1п,
T!n + CeIV ^=> Т11П + Се1П.
Кроме того, имеется дополнительная стадия, заключаю-
щаяся в объединении ионов Сеш и Cev. Поэтому обмен
в системе Т11—Т1Ш обычно считается одностадийным
переносом двух электронов.
Реакции окисления — восстановления между катионами 41
В заключение обзора реакции обмена Т11—Т1Ш сле-
дует отметить, что кинетическое уравнение данной реак-
ции, если исключить отрицательный каталитический эф-
фект однозарядных анионов, аналогично уравнению ре-
акции обмена в системе Fe11—Fe111. Вопрос о числе
переносимых за одну стадию электронов не может пока
считаться решенным.
Реакции между простыми катионами
различных веществ
Эксперименты показали, что главная реакция окис-
ления Т1+ при помощи Со3+ описывается кинетическим
уравнением первого порядка относительно Т1+ и Со3+;
скорость реакции не зависит от концентрации ионов во-
дорода [72]. Предлагаемый механизм сводится к двум
последовательным стадиям переноса одного электрона:
Со3+Н-Т1+ —> Со2 + + Т12+ стадия, определяющая скорость
реакции.
Со3+ + Т12+ —> Со2+ + Т13+ быстрая стадия.
Две равновероятные схемы, предполагающие образо-
вание перхлоратного комплекса, объясняют независи-
мость скорости реакции от концентрации ионов водо-
рода:
Я==£[ТЮН] [СоС1О^+] =
= k' [Т1+] [ОН"| [Со34"] [Н +] = k" [Т1+] [Со34 |
R = k [T1C1OJ [Со(ОН)2+].
Скорость реакции
Hg| + Tlln —> гН^+Т!1
в хлорной кислоте прямо пропорциональна первой сте^
пени концентрации ионов Hg£ и Т1ш и обратно пропор-
циональна первой степени концентрации Hgn [22]. Для
этой реакции предполагается механизм
Hg‘ HgH + Hg0,
Hg° + Tiln Hg« + Tl1.
42
Глава 1
Кинетическое уравнение для этой реакции имеет сле-
дующий вид:
-d [Hg^]/^ = [HgJ] [Tin,]/[Hgn|.
Экспериментально найденная константа скорости ре-
акции уменьшается с увеличением концентрации ионов
перхлората, которые, по-видимому, ассоциируют с ио-
нами Hg^. Добавление ионов галогенидов повышает ско-
рость, вероятно, вследствие образования комплексов с
ионами Hgn и смещения равновесия первой стадии реак-
ции вправо.
В случае реакции обмена Г!1—Т1Ш добавки неболь-
ших количеств галогенидов действуют в противополож-
ном направлении; следовательно, механизм влияния га-
логенидов в обоих случаях различен.
Механизм реакции
Hg|+4-2Mn3+ —> 2Hg2+4“2Mn2+
аналогичен. Реакция протекает по следующему кинети-
ческому закону [73]:
— d [N^+\!dt =
= k. [Mn3+] [Hgl+]/[Hg2+] +Л2 [Hg2+] [Мп3+]2/[Мп2+].
Член с константой ki описывает реакцию Мп3* с атомар-
ной ртутью, а член с константой Л2 — реакцию Мп4+
с Hg2+, в которой Мп4+ образуется по реакции диспро-
порционирования:
2Мпа+ ~7~~> Мп2+ + Мп4+.
При реакциях с участием иона UIV во всех случаях
образуется оксикатион UV1 [74—77]:
UIV4-2Fe’" — UVI + 2Fen,
U>v+2Pu,v UVI-f-2Pu,n,
UIV+2CeIV —> Uv, + 2Ce,n.
Реакции окисления — восстановления между катионами 43
Для первой реакции
Я=Л[Н+Гп[и4+] |Fe3+] [74].
Возможны два пути реакции с п=1 и п = 2 и значениями
энергии активации соответственно 94,5 и 101,5 кдж/моль.
Механизм предполагает две стадии:
U,v + Fenl —► Uv 4- Fe11 медленная стадия, определяющая ско-
рость реакции
Uv + Fe111 —► UVI + Fe11 быстрая стадия.
Стадию, определяющую скорость, можно интерпретиро-
вать, исходя из реакций с участием продуктов гидро*
лиза, которые могут протекать по-разному:
Fe(0H)3“x-f-U(OH)*“P —> Fen4-Uv для п = 1 и х±у « 1,
или
Fe(OH)3-' + U(OH)J“* —> Fen4-Uv для п = 2 и х + у = 2.
Аналогично протекает реакция между UIV и Т13+ [77];
кинетическое уравнение
я=*[нТл(и4Ч [Т13+],
где п может также быть равно 1 или 2. Величина энер-
гии активации составляет 103 кдж/молъ для первого
пути и 91 кдж!моль — для второго. Для реакции между
UIV и PuIV [75] преобладает схема с участием ионов
водорода. Скорость реакции обратно пропорциональна
квадрату концентрации ионов водорода, энергия актива-
ции составляет 102 кдж/моль. Небольшие количества
серной кислоты резко повышают скорость реакции.
Единственный путь реакции между UIV и CeIV [76]
определяет кинетическое уравнение, в которое входит
концентрация ионов водорода в степени —2. Интерес-
ная особенность этой реакции — малая величина теплоты
активации (58,8 кдж!моль) — объясняется, вероятно,
присутствием большей части ионов Сеш в виде Се (ОН)3+.
Реакция заметно катализируется ионами сульфата, ко-
торые должны легко образовывать удобный мостик для
переноса двух электронов.
44
Глава /
Реакции между оксикатионами различных веществ
Скорость реакции обмена
Qiv + uvi —yiv + (jvi
описывается сложным кинетическим уравнением [78]
[uiv|2[h+]-3
Эта экспериментально найденная скорость зависит от
квадрата концентрации Urv и обратно пропорциональна
третьей степени концентрации ионов водорода.
Для реакции постулируется следующий механизм:
и4* + Н2О UOH3+ + Н+
UOH3+ + UO|+ +2Н2О
/O=U—О—U—ОН\3+
I | | I 4-2Н+ быстро протекающая обрати*
\ ОН ОН / мая стадия
уЗ+
Y3+ -|- UOH3+ Z6+ стадия, определяющая скорость
активный
комплекс
—> (UOH)3+4-UO1+
Скорость реакции определяется как k [Y3+] [UOH3+];
после подстановки равновесной концентрации Y3+ полу-
чается следующее выражение:
R=kk' [UOH3+]2 [1Ю|+]/[н+]2,
U,v = U4+ 4- СЮН3\
откуда
/?=kk' [uivp [ио2+]/{( [H+I//Q+1}2[н+]2
где Кг — константа равновесия реакции гидролиза
и4+ 4- Н2О СЮН3+ 4- н+
Теория приводит таким образом к уравнению вто-
рого порядка относительно UIV и первого порядка от-
носительно UVI. Если концентрация ионов водорода вы-
сока, то относительно последних наблюдается порядок
Реакции окисления — восстановления между катионами 45
—4, а если низка, то —2. Исходя из экспериментального
значения Кг. теоретический расчет приводит к величине
—3 для порядка реакции, что соответствует эксперимен-
тально найденной величине Хотя в последней серии
примеров не продемонстрированы какие-либо новые важ-
ные принципы, они представляют интерес как новые
формы кинетических уравнений.
ЛИТЕРАТУРА
Общая
Amphlett С. В., Quart. Rev., 8, 219 (1954).
В a s о 1 о F., Pearson R. G., Mechanisms of Inorganic Reactions,
Wiley, New York, 1958 (Chapter 7).
Sykes K. W. (ed), The Kinetics and Mechanism of Inorganic Reac-
tions in Solution, Spec. Publ. № 1, Chemical Society. London,
1954.
Fraser R. T. M., Rev. pure appl. Chem. (Aust.), 11, 64 (1961).
H a 1 p e r n J., Quart. Rev., 15, 207 (1961).
Кац Дж. Дж., С и б о p г Г. Т., Химия актинидных элементов, М.,
Атомиздат, 1960.
Taube Н., Advances in Inorganic and Radio-Chemistry, Vol. 1, Chap-
ter 1, Academic Press, New York, 1959.
Цитированная
1. Wells A. F., Structural Inorganic Chemistry, Oxford University
Press, 1962 (p. 581).
2. E c k C. L. van P., Mendel H., В о о g W., Disc. Faraday Soc.,
24, 200 (1957).
3. Weston R. E., Sperctrochim. Acta, 18, 1257 (1962).
4. Taube H., Progress in Stereochemistry, Vol. 3, Chapter 3, But-
terworth, London, 1962.
5. Halpern J., Harkness A. C., J. chem. Phys.. 31, 1147 (1959).
6. C h r i s t i e L. D., Turney T. A., Disc. Faraday Soc., 24, 223
(1957).
7. Bernal J. D., Fowler R. H., J. chem. Phys., 1, 515 (1933).
8. В a s с о m b e K. N., Bell R. P., Disc. Faraday Soc., 24, 158
(1957).
9. С 1 e a v e r H. L., J. chem. Educ., 40. 637 (1963).
10. P о б и н с о н P. А., Стокс P. Г., Растворы электролитов, M.,
Издатинлит, 1963.
11. Н u n t J. P., T a u b e H., J. chem. Phys., 19, 602 (1951).
12. Jackson J. A., Lemons J. F., Taube H., J. chem. Phys., 32,
553 (1960).
13. Sillen L. G., Quart. Rev., 13, 146 (1959).
14. Katz J. J., S e a b о r g G. T, The Chemistry of the Actinide Ele-
ments, Methuen, London 1957; см. общую литературу.
46
Глава 1
15. Carpenter J. Е, J. Am. chem. Soc., 56, 1947 (1934).
16. R о s s о 11 i F. J. C, R о s s о 11 i H. S., Acta chem. scand, 9,
1177 (1955).
17. L a S a 11 e M. J, С о b b 1 e J. W, J. phys. Chem., 59, 519 (1955).
18. Mishra H. C, Symons M. C. R., J. chem. Soc., 4411 (1962).
19. Hardwick T. J., Robertson E., Can. J. Chem., 29, (a)
p. 818; (6) p. 828 (1951).
20. H i e t a n e n S, S i 11 e n L. G, Acta chem. scand., 13, 1828
(1959).
21. Sykes K. W., J. chem. Soc., 2473 (1959).
22. Armstrong A. M, Halpern J., Can. J. Chem., 35, 1020
(1957).
23. J о n e s M. M, Jones E. A., Harmon D. F., S e m m e r s R. T.,
J. Am. Chem. Soc.. 83, 2038 (1961).
24. L a i d 1 e r K. J., Chemical Kinetics, McGraw-Hill, New York and
London, 1950, (a) p. 75; (6) p. 134.
25. Silverman J., Dodson R. W„ J. phys. Chem., 56, 846 (1952).
26. N e w t о n T. W., R a b i d e a u S. W, J. phys. Chem., 63, 365
(1959).
27. Z w о 1 i n s к i B. J., Marcus R. J., E у r i n g H., Chem. Rev..
55, 157 (1955).
28. Penna-Franca E., Dodson R. W., J. Am. chem. Soc., 77,
2651 (1955).
29. Marcus R. J., Z w о 1 i n s к i B. J., E u r i n g H., J. phys. Chem.,
58, 432 (1954).
30. L a i d 1 e r K. J., Can. J. Chem., 37, 138 (1959).
31. S a c h e r E., L a i d 1 e r K. J., Trans. Faraday Soc., 59, 396
(1963).
32. Hu dis J., Dodson R. W., J. Am. chem. Soc., 78, 911 (1956).
33. Keenan T. K., J. phys. Chem., 61, 1117 (1957).
34. S c a t c h a г d G., Chem. Rev., 10, 229 (1932).
35. Horne R. A., J. phys. Chem., 64, 1512 (1960).
36. Spiro M, J. chem. Soc., 3678 (1960).
37. Современная химия координационных соединений, под ред.
Дж. Льюиса и П. Уилкинса, Издатинлит, 1963, гл. 2.
38. Gordon В. М., Wahl А. С.. J. Am. chem. Soc, 80, 273 (1958).
39. L i е s е г К. Н, Schroeder Н, J. inorg. nucl. Chem, 14, 98
(1960).
40. Bunn D, Da inton F. S, Duckworth S, Trans. Faraday
Soc, 57, 1131 (1961).
41. Sutin N, J. phys. Chem, 64, 1766 (1960).
42. Horne R. A, J. inorg. nucl. Chem.. 25, 1139 (1963).
43. A n d e r s о n A.. Bonner N. A, J. Am. chem. Soc, 76, 3826
(1954).
44. Taube H, King E. L, J. Am. chem. Soc, 76, 4053 (1954).
45. Ball D. L, King E. L, J. Am. chem. Soc, 80, 1091 (1958).
46 Meier D. J, Garner C. S, J. phys. Chem, 56, 853 (1952).
47 G г у d e r J. W„ Dodson R. W, J. Am. chem. Soc, 73, 2890
(1951).
48. D u k e F. R, P a r c h e n F. R., J. Am, Chem, Soc, 78, 1540
(1956).
Реакции окисления — восстановления между катионами 47
49. С о h е п D., Sullivan J. С., Hindman J. С, J. Am. chem.
Soc, (а) 76, 352 (1954); (б) 77, 4964 (1955).
50. Cohen D, Sullivan J. С, Amis Е. S., Hindman J. C.,
J. Am. chem. Soc., 78, 1543 (1956).
51. Sullivan J. C, Cohen D., Hindman J. C, J. Am. chem.
Soc, 79, 3672 (1957).
52. Giuliano C. R, M с С о n n e 1 H. M, J. inorg. nucl. Chem, 9,
171 (1959).
53. Sutcliffe L. H, Weber J. R, Trans. Faraday Soc, (a) 52,
1225 (1956); (6) 55, 1892 (1959).
54. Newton T. W, Cowan H. D, J. phys. Chem, 64, 244 (1960).
55. H u i z e n g a J. R, Magnusson L. B, J. Am. chem. Soc, 73,
3202 (1951).
56. H i n d m a n J. C, Sullivan J. C, Cohen D, J. Am. chem.
Soc, (a) 80, 1812 (1958); (b) 76, 3278 (1954).
57. A p p e 1 m a n E. H, Sullivan J. C, J. phys. Chem, 66, 442
(1962).
58. Rabi de a u S. W, Kline R. J, J. inorg. nucl. Chem, 14, 91
(1960).
59. F u r m a n S. C, Garner C. S, J. Am. chem. Soc, 74, 2333
(1952).
60. R a b i d e a u S. W, J. phys. Chem, 62, 414 (1958).
61. Hurst R. in The Kinetics and Mechanism of Inorganic Reacti-
ons in Solution, Spec. Publ. № 1, Chemical Society, London,
1954, p. 55.
62. P rest wood R. J, Wahl A. C, J. Am. chem. Soc.. 71, 3137
(1949).
63. Rossotti F. J, J. inorg. nucl. Chem, 1, 159 (1955).
64. Harbottle G, Dodson R. W, J. Am. chem. Soc, 73, 2442
(1951).
65. D о d s о n R. W, J. Am. chem. Soc, 75, 1795 (1953).
66. Ro ig E, Dodson R. W, J. phys. Chem, 65, 2175 (1961).
67. В rub acker С. H, Mickel J. P, J. inorg. nucl. Chem, 4, 55
(1957).
68. В r u b а с к e г С. H, G г о v e s К. О, M i с к e 1 J. P, К n о p С. P,
J. Am. chem. Soc, 79, 4641 (1957).
69. Gilks S, Rogers T. E, Wai nd G. M, Trans. Faraday Soc,
57, 1371 (1961).
70. Gryder J. W, Dorfman M. C, J. Am. chem. Soc, 83, 1253
(1961).
71. Dorfman M. C, Gryder J. W, Inorg. Chem, 1, 799 (1962).
72. A s h u r s t K. G, Higginson W. С. E, J. chem. Soc, 343
(1956).
73. R о s s e i n s к у D. R, J. chem. Soc, 1181 (1963).
74. Betts R. H, Can. J. Chem, 33, 1780 (1955).
75. Newton T. W, J. phys. Chem, 63, 1493 (1959).
76. Baker F. B, Newton T. W, Kahn M, J. phys. Chem, 64,
109 (1960).
77. H а г к n e s s A. C, Halpern J, J. Am. chem. Soc, 81, 3526
(1959).
76. Ron a E, J. Am. chem. Soc, 72, 4339 (1950).
Глава 2
РЕАКЦИИ ОКИСЛЕНИЯ АНИОНОВ,
КОМПЛЕКСНЫХ КАТИОНОВ
И МОЛЕКУЛ КАТИОНАМИ
В этой главе мы рассмотрим реакции окисления
анионов, комплексных катионов или нейтральных моле-
кул различными катионами. Характерной чертой этих
реакций является то, что они протекают в кислом рас-
творе, так как повышение pH вызывает осаждение гид-
роокисей металлов и тем самым прекращение реакций.
В то время как для реакций катионов с катионами
было затруднительно установить различие в истинном
механизме реакций с одноэлектронным и двухэлектрон-
ным переносом, в реакциях с участием окисляемого ор-
ганического вещества отделение одного или двух элек-
тронов от восстановителя обусловливает либо гомолити-
ческий, либо гетеролитический тип реакции, которые
можно четко разграничить. Имеется три основных типа
реакций с восстановителями, теряющими электрон:
-^CH4-R*+ - -► ^CH + R’-1, (1)
^CH+R. — ^C + RH, (2)
^СН + 'О—О’ - -> ^>С 4- но2. (3)
В этой главе мы обсудим реакции типа (1), реакции
типа (2) — в Главе 7 и реакции типа (3) — в главах
8 и 9.
Уотерс [1] считает, что легкость образования ради-
калов при атаке на органические молекулы опреде-
ляется величиной редокс-потенциала окислителя; наибо-
лее склонные к окислению органические молекулы имеют
цеспаренпые, легко отделяемые электроны.
Реакции окисления анионов, комплексных катионов
49
Вначале рассмотрим реакции окисления с участием
одноэлектронных систем [2]:
Е°, в
Agl — -> Ag"+e —1,98
Со" — -> Co"l-|-e —1,82
Се"1 — -> Ceiv-1-е —1,61
Мп" — -> Мп1"-Н —1,51
•/2 Н2О - -> н+ +•/„(),+<? —1,23
H2O4-VOiv - -> VO2v Н-2Н+ + е —1,00
Fen —> FeHi 4-^
—0,77
Названные системы разделяются на два класса в зави-
симости от того, существует или нет в данном случае
теоретически возможность окисления молекул воды.
Из двухэлектронных окислителей будут рассмотрены:
Е°, в
Pblv —> РЬп4-2г
Т11Н —Т!1-^
2Hgn —> Hg£ + 2*
—1,69
—1,25
—0,92
Кроме того, будут описаны реакции окисления ионов Сгп,
Мп11, Fe11, TiUI, V11, V™
В качестве окислителей могут быть использованы и
некоторые другие ионы, например Сип, Сиш, Nini и
MnIv. Последние три существуют, вероятно, только в
виде комплексов. Раствор ацетата меди в уксусной кис-
лоте представляет собой реактив Барфеда для опреде-
ления моносахаридов. Трехвалентный кобальт, соедине-
ния меди(III), никеля(III) и марганца(IV) получают
электролизом растворов. Реакции с Сога и Мпш хо-
рошо исследованы, реакции с Сиш и Niin, напротив, изу-
чены явно недостаточно. Получены [3] комплексы Сига
в твердом состоянииNa7Cu(IO6)2*16H2OиNa9Cu(TeO6)2•
• 20НгО. В литературе имеется упоминание об ацетате
MnIV как окислителе а-гликолей [4].
Другим типом рассматриваемых реакций является фо-
тохимическое окисление, на котором основан, например,
4 Зак. 1249
50
Глава 2
уранил-оксалатный актинометр. Фотохимические реак-
ции окисления, за несколькими исключениями, здесь не
рассматриваются. Интересующимся можно рекомендо-
вать обзор Ури [5] *.
В этом разделе книги при рассмотрении реакций мы
вначале будем обращать внимание на формы существо-
вания ионов в растворе, а затем последовательно обсу-
ждать стехиометрию и механизм реакции с анионом,
комплексом и молекулой восстановителя.
Окисление серебром (II)
Растворы, содержащие ион Ag11 (черного цвета),
можно приготовить действием озона на нитрат сереб-
ра [7] или исходя из окиси двухвалентного серебра, по-
лученного анодным окислением раствора нитрата се-
ребра в азотной кислоте [6]. Стандартный электродный
потенциал реакции Ag1 —*Agn4-e равен —1,98 в [8].
Ион Ag11 неустойчив и разлагает воду с выделением кис-
лорода
2Agn + Н2О —> 2Ag’ +2Н+ + i/2 О2.
Скорость реакции разложения Ag11 в среде НС1О4 [7]
прямо пропорциональна квадрату концентрации ионов
Ag11, обратно пропорциональна первой степени концен-
трации ионов Ag1, обратно пропорциональна квадрату
концентрации ионов Н+ и прямо пропорциональна кон-
центрации перхлората.
Изменения в спектре поглощения иона Ag11 в обла-
сти 350—750 нм в зависимости от концентрации аниона
С1ОГ свидетельствуют об образовании перхлоратного
комплекса. Предполагается двухстадийный механизм:
2Agi I у-* Agi -f- AgIH быстрая стадия,
Agni —► продукты стадия, определяющая скорость реакции.
Кинетическое уравнение:
-d[Ag«)/^-A[Ag«J2/|Ag>J |H+j2.
* См. примечание на стр. 15,
Реакции окисления анионов, комплексных катионов
51
Обратная зависимость скорости от квадрата концен-
трации ионов водорода, вероятно, объясняется последо-
вательно протекающими реакциями
Agnl-|-H2O 7—* AgO+-|-2H+ быстрая стадия,
AgO+ —► Ag+ 4- */г О2 стадия, определяющая скорость.
Было показано [10], что скорость переноса электрона
между ионами Ag1 и Agn определяется реакцией
2Agn ** Ag++Agni [8].
Двухвалентное серебро в виде комплекса с пиколи-
новой кислотой окисляет органические соединения [9].
Комплексное соединение получают при взаимодействии
AgNO3 в водном растворе пероксидисульфатами щелоч-
ных металлов и пиколиновой кислотой, при этом выде-
ляется оранжево-красный осадок пиколината Ag(II).
Это вещество используется как окислитель органических
соединений в водном растворе при температурах 0—90°
(при перемешивании). В ходе реакции малораствори-
мый оранжевый пиколинат Agn переходит в соединение
белого цвета Ag1.
Окисление кобальтом (III)
Ранее растворы сульфата Сош приготовляли элек-
тролитическим окислением сульфата Соп [10]. Мы при-
водим здесь усовершенствованную методику Свэнна и
Ксантакоса [11].
Анодное отделение электролизера представляет со-
бой пористый сосуд емкостью 800 мл, погруженный
в литровый стакан. Анодом служит тонкая платиновая
пластинка размером 80 см2, а катодом — медная пла-
стинка, помещенная между стенками пористого и стеклян-
ного стаканов. Анолитом и католитом является раствор
сульфата Со11 в 10 н. H2SO4 (растворимость сульфата
Со1” в таком растворе незначительна). Температуру
с помощью ледяной бани поддерживали ниже 10°.
Плотность тока варьировали в интервале 0,01—2 а/см2.
При более высокой плотности тока получали кристаллы
более мелких размеров, чем это требовалось. Ток не
должен быть более 5 а на 500 мл анолита, так как иначе
4*
52
Глава 2
невозможно поддерживать требуемую температуру.
Сульфат Со111 осаждали в виде зеленовато-голубых кри-
сталлов; максимальный выход достигался через 4 час.
Последующими исследователями этот метод был не-
сколько видоизменен.
Перхлорат Со111 готовили добавлением перхлората
бария к раствору Co2(SO4)3.
Соли Сош можно получить окислением солей Со11
такими сильными окислителями, как фтор, озон или
висмутат натрия [12а].
Нойес и Дил [13] нашли, что для реакции Со11—►
—>Сош + е редокс-потенциал равен —1,82 в. Поэтому
ион Со111 неустойчив также по отношению к воде
4СоП1 + 2Н2О —> 4Соп + 4Н+4-О2.
Поведение ионов кобальта(III) в водном растворе
хлорной кислоты изучалось спектрофотометрически [14].
Предполагается, что в водном растворе происходят сле-
дующие реакции:
Со3++Н2О Со(ОН)'2+-|-Н+ быстрая стадия
Со3++ Со(ОН)2+ —> Со—О—Со4+4-Н+
и (или)
Со(ОН)2+ +Со(ОН)2+ —> Со—О—СоОН3++Н+
медленная
стадия
Так как в водном растворе Со3+ обладает довольно вы-
сокой реакционной способностью, полученные экспери-
ментальные доказательства недостаточны.
Скорость реакции Со3+ с водой зависит от концен-
трации Со3+ следующим образом [15]:
— d [СоП1]/Л = k [Com] 4- V [Com]*
где k и k’ — константы скорости, сложным образом за-
висящие от кислотности.
Схема реакции предполагает две определяющие ско-
рость стадии
Со(ОН)2+ —► Со2+4--ОН
Со—О—СоОН3+4-Н+ —► 2Со(ОН)2+
—► Со24 +-ОН,
Реакции окисления анионов, комплексных катионов
53
устанавливающие сложную зависимость реакции от кон-
центрации ионов водорода. Бэксендейл и Уэллс [16],
проверяя эту зависимость, нашли, что скорость реакции
Сош с водой прямо пропорциональна концентрации
Сош в степени 3/2 и обратно пропорциональна квадрату
концентрации ионов водорода. На этом основании они
предложили уточненную схему реакции:
СоОН2+ + (НО-Со-О-СоОН)2+
—> ЗСо2+ Н-2НО • + НО, •.
2СоОН2+ (Со—О—Со)4+ + Н2О.
(Со—О—Со)4+Н-2Н2О (НОСо—О—СоОН)2+4-2Н+.
«2
Если допустить, что велико, a k2 мало, то
— d [Со1п[/<Я = k [Со(ОН)2+] [НОСо—О—СоОН]2+ =
= k' [(Co-О—Со)4+]’/г/[Н+12.
Если далее предположить, что Сош существует преиму-
щественно в виде (Со—О—Со)4+, то получающееся
уравнение
- Нсо'Ч/л = Исош1’7[н+12
соответствует найденному экспериментально кинетиче-
скому уравнению.
Начиная с 1951 г. опубликовано несколько статей
Боуна с сотрудниками [15, 17а, б] о реакциях между ио-
ном Сош и молекулами. Скорость стадии, определяю-
щей скорость реакции, описывалась уравнением
— d [Со1 Щ/dt = k [органическое вещество] [СоШ].
Вслед за этой стадией протекают реакции с участием
ранее образованных радикалов [15].
Муравьиная кислота окисляется ионами Сош в 2—
8 н. серной кислоте следующим образом [17а]:
2Со,п 4- НСООН —► 2Соп 4- 2Н+ 4- СО2.
Кинетика этой реакции описывается уравнением
— d [Colii]/<tt = Л, [НСООН] [Со111] 4-й2 [НСОб] [СоШ].
54
Глава 2
Скорость реакции окисления определяется стадией
атаки иона Сош на молекулу НСООН и на ион НСОО,
при этом в обоих случаях образуется радикал НСОг,
который затем быстро распадается различными возмож-
ными путями.
Скорость реакции окисления формальдегида в 5 и
10 н. серной кислоте [176] контролируется стадией взаи-»
модействия иона Со111 с НСНО
НСНО4-Со3+ —(НСНО-Со)3+.
Последующие стадии:
(НСНО-Со)3+ —* Со2+Н-Н+4-НСО-
НСО-4-Со3+ —* Со2+4-Н++СО,
НСО-Ч-Н2О НСНО + НО-,
нсо- + н2о —+ нсоон 4-н.,
НО • + нсно —-> Н2О + нсо •.
Н. + нсно —+ н2 4- нсо..
Скорость уменьшения концентрации ионов Со1П мо-
жно выразить уравнением
— d [СоПЦ/Л = &, [НСНО] (СоШ]4-Лэ[НСО-] [СоШ].
Если предположить существование стационарной кон-
центрации радикала НСОуравнение скорости реак-
ции приобретает вид:
— d (Col 11] tdt = (HCHO] [Co"i] +
+ ktk3 [Col 11]2 [HCHO]/(A3 (Col14 4- ks [H2O])
Порядок уравнения совпадает с экспериментально най-
денным порядком, реакции. Реакция протекает по пер-
вому или второму порядку [196] в зависимости от вели-
чины отношения Л3[СоП1]/Л5[Н2О] 1, причем сложность
кинетики увеличивается вследствие прохождения конку-
рирующей реакции радикала НСО • с органическим ве-
ществом и водой. Таким образом, мы здесь встречаемся
с более сложным случаем, чем приведенные в начале
этого раздела.
Реакции окисления анионов, комплексных катионов
55
Скорость окисления [176] метилового спирта
СН3ОН+2Сош —> НСНО 4-2Н+ Ч-2Соп
описывается кинетическим уравнением для стадии, опре-
деляющей скорость реакции:
— d [CoHi]/^ = kx [Coin] [СН3ОН].
За начальной медленной стадией следуют реакции, в ре-
зультате которых метиловый спирт превращается в
формальдегид. Такая же замедленная стадия наблю-
дается и при окислении этилового и пропилового спир-
тов.
Дальнейшие осложнения при обсуждении реакции
Со111 с формальдегидом [18а] возникают по причине при-
соединения протона к формальдегиду:
НСНО + Н2О Н2С(ОН)2
и
H2C(OH)2-f-H+ 7Z> H2COH-f-H2O.
Молекулы образующихся веществ затем подвергаются
атаке ионами кобальта(III), которые в серной кислоте
ассоциируются по схеме
Со3+ 4-2S01“ Co(SO4)-
и, возможно,
Co(SO4)2" + H+ HCo(SO4)2.
Реакция окисления ненасыщенных углеводородов в
разбавленной серноя кислоте протекает по кинетиче-
скому уравнению второго порядка [19а]. Это, по-види-
мому, объясняется непосредственной атакой двойной
связи ионом кобальта(Ш), сопровождающейся перено-
сом электрона:
RCH=CH2 Ч-СоШ —► RChCH2 + CoH
Ион карбония реагирует затем с водой, образуя ряд
продуктов, которые быстро окисляются ионами Сош.
Рассчитанная теплота реакции
[RCH=CH2J. aq + Coin . aq —► [RCHCHJ . aq + CqH . aq.
56
Глава 2
в пределах точности эксперимента приближается к ве-
личине энергии активации. Исходя из рассчитанных зна-
чений энергии активации, можно сделать вывод, что три-
алкилолефины реагируют в 6 раз быстрее, а диалкил-
олефииы — в 2,3 раза быстрее, чем моноалкилолефины.
Реакция окисления олефинов ионами кобальта (III)
в смеси уксусной и серной кислоты идет [196] по урав-
нению первого порядка относительно ионов Сош. Кон-
центрация олефинов не влияет на порядок реакции. Это
изменение кинетики, по-видимому, связано с образова-
нием стабильных сульфатацетатных комплексов кобаль-
та(Ш).
Реакция кобальта (III) с органическими гидроперок-
сидами [20] является бимолекулярной:
— d [Со3+] 'dt = k [Со3 Ч [RO • ОН].
Реакции с перекисью водорода [16] протекают по кине-
тическому уравнению второго порядка. В случае реак-
ции с Н2Ог константа скорости изменяется обратно про-
порционально концентрации ионов водорода вследствие
равновесия:
Со1114- Н2О2 ^=± Со111. но2" 4- Н+.
Результаты измерения относительной скорости окис-
ления циклогексанона и 2, 2, 6, 6-тетр а дейтероциклогек-
санона в воде и окиси дейтерия дают основания счи-
тать, что либо а-С—Н-связь не разрывается, либо, если
она и разрывается на первой стадии реакции, то это не
определяет скорости процесса. Можно прийти к заклю-
чению, что один эквивалент кобальта(III) преимуще-
ственно действует не на енольную, а на кетонную форму
циклогексанона, а медленной стадией может быть пере-
нос электрона [21]:
Coin -f-^>C=O —> Со"+^>С—6.
Другие исследователи [22] нашли, что скорость реак-
ции окисления циклогексанола ионами кобальта(III)
описывается уравнением
— d [Со11 []/dt = k [Со111] [циклогексанол],
Реакции окисления анионов, комплексных катионов
57
где k — константа скорости — изменяется обратно про-
порционально первой и второй степени концентрации
хлорной кислоты. Поскольку мы уже сталкивались
с явлением катализа перхлоратом при переносе элек-
трона (стр. 41), то влияние перхлората на величину
константы скорости не является неожиданным. Для
объяснения действия перхлората можно предложить
несколько реакций, представляющих вероятное взаимо-
действие реактантов с ионами перхлората или прото-
нами.
Все реакции окисления с участием ионов кобаль-
та (III) можно представить в общем виде кинетическим
уравнением
— d [СоШ]/^/ = k [Coin] [red],
где red — восстановитель. В этих реакциях после пере-
носа электрона от восстановителя последний подвер-
гается дальнейшим превращениям. Константа скорости
k может зависеть от концентрации ионов водорода, кон-
центрации анионов и от природы среды, в которой про-
исходит реакция.
Окисление церием (IV)
Редокс-потенциал для Cein-*CeIV равен —1,61 в [2]
и находится между потенциалами для Мпп-*Мпш и
Со11—>Сош. Исходя из термодинамических соображе-
ний можно предположить, что ион церия(IV) должен
быстро реагировать с водой, однако это не наблюдается,
вероятно, вследствие высокой склонности церия (IV) к
комплексообразованию [23]. Сульфат CeIV в твердом со-
стоянии имеет интенсивно желтый цвет, который при
растворении в серной кислоте дает красноватый рас-
твор [126] вследствие образования Н2[Ce(SO4)3]. Извест-
но несколько солей этой кислоты — цератов, например
соль аммония (МН4)2[Се(МОз)в]. Качественные опыты
показывают, что эта соль дает более интенсивную окра-
ску при взаимодействии с полиоксисоединениями, чем
сульфат CeIv.
Ион CeIV широко применяется в титриметриче-
ском анализе, поскольку многие реакции протекают
58
Глава 2
количественно [24]:
2Ce’v4-2N^ —► 3N24-2Ce111,
2Celv + Н3РО3 4- Н2О —► 2CeII, + H3PO44-2H+.
2Celv + H2O2 —► 2Сеп,4-2Н+ 4-O2,
2Ce,v + CH3COCOOH + H2O —► 2CeIIl+CH3COOH + 2H+-|-CO2.
Скорость реакции Celv с бромидом [25] описывается
уравнением
- d [Се^/л = [Ce'V]/[S01-] [Br"| 4- k2 [Br"]2}.
Этому уравнению соответствует следующий механизм:
Се(8О4)Г + Вг~ *=* Ce(SO4)2Br- + SO^-,
Ce(SO4)2Br- 4-Br- — Ce(SO4)2" + Br2-,
Ce(SO4)2Br- 4-aq —► Ce(SOj " 4-Braq,
Br2 4“ Br > Br2 4- Br .
Для окисления формальдегида'в хлорной кислоте [186]
HCHO4-H2O4-2CeIV —► НСООН4-2Н+4-2Сеш
предполагается протекание реакции в две стадии:
Ce,v 4-Н2С(ОН)2 —► СеП14-Н2С(ОН)О4-Н+.
CeIV 4-Н2С(ОН)6 —► Се1114-НСООН 4-Н+
В растворе серной кислоты схема этой реакции видоиз-
меняется, поскольку ион церия (IV) присутствует в виде
комплексного сульфата H[Ce(SO4)2].
Реакция перхлората CeIV с этиловым спиртом проте-
кает со скоростью [26]
— d [CeiV] ш [Ceiv] [C2HSOH]/{1 4- Л2 [C2H5OH]}.
Механизм, отвечающий этому кинетическому уравне-
нию, соответствует схеме
CetV4~C2H5OH (комплекс) —► продукты,
фотометрический метод Джоба дает соотношение 1 1
между ионом церия и этанолом в активном комплексе.
Реакции окисления анионов, комплексных катионов
59
Кинетика реакции окисления этилового спирта ана-
логична кинетике окисления циклогексанола [27]. Пред-
полагая, что скорость реакции определяется стадией
разрыва связи С—Н, нашли кинетический изотопный
эффект &h/&d равным 1,9. Однако малая величина эф-
фекта указывает на возможность существования конку-
рирующего механизма реакции (помимо переноса од-*
ного электрона), приводящего к образованию цикличе-
ского комплекса между спиртом и CeIV
При окислении пинакола сульфатом CeIV [28] урав-
нение константы скорости реакции имеет первый порядок
относительно каждого реагирующего вещества. Образо-
вания комплекса между церием (IV) и пинаколом не
обнаружено, поэтому предполагается следующая схема
реакции:
Се1¥ + (СНз)2С(ОН)С(ОН)(СН3)2 —>
—► (СН3)2С=О + (СН3)2СОН + Н+ + Сеш
и далее
CeIV + (CH3)2COH —> (СН3)2С=О + Н++Се1И.
На один ион церия (IV) образуется одна молекула аце-
тона, но в присутствии акриламида на образование
1 молекулы ацетона расходуется два иона церия (IV). В
последнем случае предполагается видоизмененная схема:
(СН3)2СОН4-М —► М’, где М — акриламид,
. м+м —► йя+1,
М„ 4“ Мт —► полимер,
Мя Се1П —► полимер 4- CeIV 4“ Н+
Эффективность захвата составляет около 100%.
При окислении миндальной, d,/-малоновой и молоч-
ной кислот сульфатом CeIV [29] образуется промежуточ-
ный комплекс, который затем распадается:
Ce(SO4)2 4-а-оксикислота ^"трая" (активный комплекс)
стадия
' медленнаяН+-|-Ce2(SO4), +свободный радикал
стадия
быстрая* продукты.
СТАДИЯ
60
Глава 2
В реакции, вероятно, участвуют ионы H2Ce(SO4)4~, ко-
торые образуются при взаимодействии Ce(SO4)2 и НЗОГ.
Предполагается следующая схема окисления ацетона
соединением нитратов аммония и церия [30]:
СН3СОСН3 + Н+ СН3С(ОН)=СН2 + н+
Се(ОН)3+4-СН3С(ОН)=СН2 —> CH3CO(iH2 4- Се111 + Н2О,
Се(ОН)3++ СН3СОСН2 —> СН3СОСН2ОН + Сеш,
очень быстрая стадия
СН3С(ОН)=СН2 —► СН3СОСН2-|-Н+.
Се(ОН)3+ -|- СН3СОСН2 —> СН3СОСН2-|-Сеш + ОН.
очень быстрая стадия
Как и при окислении пинакола, в этом случае также нет
указаний на образование комплекса с ионом церия (IV).
Вероятно, при этой реакции цмеет место отрыв элек^
трона от молекулы ацетона.
Новый аспект механизма реакций с участием иона
CeIV был предложен при изучении реакции окисления
глицерина. В среде серной кислоты механизм окисления
состоит, по-видимому, в непосредственном воздействии
CeIV на молекулу глицерина, но в среде хлорной кис-
лоты реакция протекает через образование промежуточ-
ного комплекса [31]. Аналогично окисляется метанол со-
лями CeIV [32].
Суммируя особенности реакции с участием солей
CeIV, можно сделать следующие выводы.
Вследствие большой склонности к комплексообразо-
ванию CeIV не выделяет кислород из воды, несмотря на
высокий редокс-потенциал. Соли CeIV количественно и
без образования побочных продуктов реагируют с раз-
личными веществами и, следовательно, пригодны в ка-
честве реагентов для титриметрических методов ана-
лиза. В тех случаях, где нет стерических препятствий,
окисляемое вещество с церием (IV) образует достаточно
стабильный промежуточный продукт. Тот факт, что суль-
фат и перхлорат CeIV реагируют неодинаково, можно
Реакции окисления анионов, комплексных катионов
61
объяснить конкуренцией между промежуточным соеди-
нением и сульфатом, который способен к комплексо-
образованию в большей степени, чем перхлорат. Если
отсутствует возможность образования циклического
промежуточного продукта, реакция может осуществ-
ляться прямым переносом электрона от окисляемого
вещества.
Окисление марганцем (III)
Тема этого раздела была предметом обзорной ра-
боты Уотерса [33]. Сульфат Мпш в растворе можно по-
лучить электролизом сульфата Мп11 аналогично мето-
дике приготовления сульфата Со111. Растворы Мпш в
концентрированной серной кислоте (>9 н.) имеют виш-
нево-красный цвет [34]. В водном растворе ион Мпш
подвергается диспропорционированию по реакции
2Мпш Mnn+MnIV, сопровождаемой осаждением дву-
окиси марганца. Путем комплексообразования с пиро-
фосфатом можно получить различные стабильные со-
единения от Mn(H3P2O7)3 до [Мп(Н2Р2О7)3]3Л в зависи-
мости от степени окисления. Растворы пирофосфатных
соединений марганца (III) стабильны до pH 6.
Для обратимой реакции
Mn(H2P2O7)2~ + Н4Р2О7 Mn(H2P2O7)f"4-2H++^
измеренный на платиновом электроде редокс-потенциал
составляет [36] 1,1—1,2 в (в зависимости от pH среды).
Величина потенциала показывает, что пирофосфат Мпш
представляет собой более сильный окислитель, чем соли
Fe111, но менее сильный, чем соли Ceiv.
Друммонд и Уотерс [37а] предлагают приготовлять
пирофосфатный раствор марганца (III) следующим об-
разом: 5 мл раствора сульфата Мп11 (114 г/л MnSO4-
•4Н2О) смешивают с раствором пирофосфата натрия
(70 г Na4P2O7-ЮН2О в 110 мл), доводят до pH 6 (по
стеклянному электроду) добавлением 4 н. серной кис-
лоты и оттитровывают раствором перманганата калия
до наступления скачка потенциала платинового индика*
торного электрода.
62
Глава 2
Ранее считалось, что окисление иона оксалата ионом
Мпш происходит по схеме [38]:
Mn3+-]-2С2О2' Мп(С2О4)” быстрая стадия
Mn3+ + C2Oj“ —> Мп2+ + СО2 4- СО' медленная стадия
Мп3+ 4- СО^ —► Мп2+ 4- СО2 быстрая стадия
Дальнейшие исследования [32, 40] показали, что в этой
реакции могут участвовать МпС2О4+ и Мп(С2О4)^.
Кинетическое уравнение реакции окисления пинако-
ла [376] ионами Мпш
— d [МпШ]Дй= k [пинакол]/{А' 4~[пинакол])
совпадает по форме с кинетическим уравнением реакции
окисления ионами церия(1У). Окисление, вероятно, про-
текает в три стадии:
1) быстрое и обратимое образование комплексов ме-
жду Мпш, пинаколом и пирофосфатом
[Мп (Pin) (Н2Р2О7)2]-;
2) медленный распад комплекса с образованием пи-
рофосфата Мп11, ацетона и радикала (СН3)2СОН;
3) быстрое окисление свободного радикала другой
молекулой пирофосфата Мпга.
Образование свободных радикалов удается установить
индуцированной полимеризацией.
Эксперименты показывают, что ион марганца (III)
является менее эффективным окислителем, чем ион ко-
бальта (III). Предполагается, что реакции с участием
марганца (III) имеют специфическую особенность, за-
ключающуюся в том, что окисляемое вещество должно
быть способно соединяться или образовывать хелатный
комплекс с ионом Мпш путем замещения пирофосфата
в координационной сфере. Этой точке зрения придава-
лось особое значение в работе [37в].
Реакция окисления малоновой кислоты пирофосфа-
том Мпш в атмосфере азота протекает количественно
следующим образом [37в]:
СН2(СООН)24-ЗО —► 2СО24-НСООН4-Н2О.
Вначале образуется комплекс, вероятно [Мп3+-
•СН2(СООН)2* (Н2Р2О7)гГ, который затем разлагается
Реакции окисления анионов, комплексных катионов
63
по обратимой реакции, определяющей скорость, на пиро-
фосфат Мп11 и радикал СН(СООН)2. Последний спо-
собен индуцировать винильную полимеризацию, окис-
лять спирты, эфиры и ионы Мп2+. В присутствии кисло-
рода малоновая кислота окисляется полностью по
схеме:
СН2(СООН)2 + 40 —> ЗСО2 + 2Н2О.
Интересно, что марганец(Ш) в виде пирофосфатного
комплекса не действует на одноатомные спирты в отсут-
ствие малоновой кислоты [376]. Аналогичный случай об-
ратимого образования промежуточного комплекса на-
блюдается при окислении а-оксикислот [41]. При окис-
лении кетонов [37в] комплекса, по-видимому, не
образуется, а стадией, определяющей скорость реакции,
вероятно, является енолизация кетона. Однако превра-
щение пировиноградной кислоты, которая количественно
окисляется до уксусной кислоты и С02, проходит через
стадию образования хелатного комплекса с Мпш, а не
стадию енолизации [37в]. Для акрилового альдегида [42]
начальная стадия, определяющая скорость реакции
окисления, катализируется кислотами; комплекс с окис-
лителем не образуется. Так как акриловый альдегид не
может образовать нормальную енольную форму, пред-
полагается, что реакция окисления протекает через
образование 1,4-конъюгированного енолгидрата. Пропио-
новый и масляный альдегиды окисляются по иному ме-
ханизму [37а]. Уравнение скорости реакции имеет нуле-
вой порядок относительно ионов марганца(III) и пер-
вый порядок относительно альдегида и ионов водорода.
Для механизма окисления альдегидов предполагается
следующая схема:
RCH2COR' + Н80+ RCH2CR'(OH) + Н20 быстрая стадия
В: +RCH2CR'(OH) ВН++ RCHCR'OH стадия с измери-
мой скоростью
RCH=CR'OH + Мп111 —► RCH=CR'O • + Мп11 + Н+
Хлоральгидрат, формальдегид и муравьиная кислота,
которые не могут образовать енольные формы, окис-
ляются с трудом.
64
Глава 2
На основании рассмотрения имеющихся данных для
реакций окисления марганцем (III) можно сделать сле-
дующие выводы. В тех случаях, когда это возможно,
окисляемое вещество вначале образует с марганцем(III)
достаточно стабильное промежуточное соединение цик-
лического строения, которое затем распадается с обра-
зованием продуктов окисления. Если образование таких
соединений невозможно, то реакция осуществляется
прямым переносом электрона от окисляемого вещества,
предположительно через стадии с участием ионов ено-
лята.
Окисление железом (III)
В противоположность ранее рассмотренным окисли-
телям ионы Fe111 существуют как стабильные частицы В
растворе при определенных условиях и подвергаются
гидролизу или комплексообразованию в очень малой
степени [43]. В качестве примера рассмотрим реакцию
Fe111 с иодидом.
Реакция имеет первый порядок относительно ионов
Fe111 и второй порядок относительно иодид ионов. Влия-
ние ионов железа (II) изучали путем добавления извест-
ных концентраций соли Fe11 перед началом реакции [44].
Кинетику реакции между Fe111 и Г можно описать
уравнением
[Fein] [Г]2/{1 +*2М/[Реш]}.
которое соответствует следующему механизму:
Fenl + I~ Fel'1
k2
Fel114-Г Fen + I^,
Feln + l2- Fen + I2.
В серии работ [45] описана реакция образования
FeS2O3, для осуществления которой был применен ме-
тод струй Промежуточный продукт темно-красного цве-
та быстро разлагается на ион железа(II) и тетратио-
Реакции окисления анионовL комплексных катионов
65
ната. Протекает следующая суммарная реакция:
2Fe3+ +2S2O|- —> 2Fe2++S4O|-
Экспериментально найденная скорость реакции описы-
вается уравнением
/?=*[FeS2O3+],/2[S2O|-],/‘1
которому соответствует следующая схема реакций:
Fe3++S2O23- ф Fe(S2O3)+,
Fe(S2O3)+ +S2O|- FeS2O? + S2O3,
Fe(S2O3)+ + S2O3~ Fe2+ + S4O2-,
FeS2Of + Fe2+ FeS2O3 + Fe2+,
Fe2+4-S2O2- 5=± FeS2O3.
Согласно этой схеме, по ходу реакции образуются два
промежуточных продукта, S2O3 и РеЗгОз, где а пред-
ставляет собой незаряженный тиосульфатный комплекс.
На начальных стадиях реакции (1—4) скорость обра-
зования ионов Fe2+ описывается уравнением
d [Fe2+ \!dt = k' [FeS2O3+]‘/2 [S2O|-]V*.
которое согласуется с результатами эксперимента.
Автор этих статей, комментируя усложнение наблю-
даемого общего кинетического уравнения, считает, что
оно возникает вследствие относительной медленности
и специфичности реакции между тиосульфатом Feni и од-
нозаряженным ионом тиосульфурила. Если бы эта реак-
ция протекала намного быстрее, ее кинетика была бы
аналогична поведению реакции между Fe111 и ионами I".
Для реакции Fein с сернистой кислотой [46]
H2SO3 + 2Feni 4- Н2О —> 2FeH + 4Н+ +SO^~
5 Зак. 1249
66
Г лава 2
предлагается следующий механизм:
H2SO3 H+ + HSO3",
Fe3+4-HSO7 Fe(HSO3)2+,
Fe(HSO3)2+ —► Fe2+ 4- HSO3 (свободный радикал),
HSO3 + Fe2+ —► Fe3+H-HSO3-,
HSO3 + Fe3+ + H2O —> SO2- + FeJ+ -|-3H+.
Реакция между железом (III) и H2SO3 ускоряется в при-
сутствии кислорода вследствие протекания дополнитель-
ной реакции:
HSO3 + O2 + H2O —> SO^--|-2H+4-HO2.
Интересные результаты получены при изучении про-
цесса окисления гидразина, который может распадаться
по двум конкурирующим реакциям [47]:
N2H4 —► '/2 N2 + NH34-H+ -f-е.
(1)
N2H4 —► Й24-4Н+4-4е.
(2)
В опытах с гидразином, обогащенным изотопом 15N,
было обнаружено, что в реакции (1) половина выде-
ляющихся молекул азота возникает при разложении
одной и той же молекулы гидразина, а другая поло-
вина — из двух различных молекул гидразина. В реак-
ции (2) с переносом двух электронов, которая проте-
кает со всеми окислителями в щелочных растворах, не
происходит разрыва имеющихся и образования новых
связей N—N. Реакция между Fe111 и гидразином прохо-
дит с переносом одного электрона [48]:
N2H44- Fe,n —> N2H3 • 4- Н+ 4- Fe“
N2H3.4-N2H,----► NH2NHNHNH2
—► 2NH34-N2
Аналогичные результаты получены другими исследова-
телями [49].
Примером реакции количественного окисления ио-
нами Fe111 служит окисление ацетоина (З-окси-2-бута-
нон) в водном растворе хлорной кислоты [50]. Оказа-
Реакции окисления анионов, комплексных катионов
лось, что два иона Feni расходуются для окисления од-
ной молекулы ацетоина. Скорость реакции описывается
уравнением
— d [FelIt]/dt = k [Feln] [CH3COCH (OH) CH3],
где константа скорости содержит член, не зависящий
от кислотности, и еще один член, в который входит кон-
центрация ионов водорода в степени минус единица. Эту
реакцию можно интерпретировать как перенос одного
электрона к иону Fe111 в две последовательные стадии:
Fen, + CH3COCHOHCH3 —> Fe11 +Н+ + СН3СОС(ОН)СН3,
Ре1п4-СН3СОС(ОН)СН3 —> Fen-[-H+ +СН3СОСОСН8.
При этом зависимость от концентрации ионов водорода
в степени минус единица объясняется переносом элек-
трона через ион Fe(OH)2+, образующийся по равновес-
ной реакции
Fe3+4-HjO ^z± Fe(OH)2+4-H+.
В органической химии часто применяют реакцию ио-
нов Fein с фенолами. Уэсп и Броде [51] отметили, что
окрашенные соединения некоторых фенолов с ионами
Fein медленно обесцвечиваются, вероятно, вследствие
их окисления с образованием хинонов. При окислении
гидрохинона ионами Fe111 образуется хингидрон. Изуча-
лась кинетика этой реакции; предполагается следующая
схема [52]:
Fe3+4-QH" 5=± Fe2+4-QH,
Fe3+4-QH2 Fe2++QH4-H+,
QH Q~ +H+,
Fe3+4-Q” «z* Fe3+ + Q.
Такая же схема предлагается для процесса окисления
толухинона и 2, 6-дихлорбензохинона [53].
Взаимодействие ионов Fe111 с перекисью водорода
изучалось неоднократно. Примером ее препаративного
применения служит получение d-арабинозы из глюко-
ната кальция [54]. В этом случае окисление происходит
главным образом по схеме >СНОН—►>€ О.
68
Глава 2
Описаны свойства ионной пары, образующейся в рас-
творе между Fe3+ и НОГ [55]. Сравнение числовых зна-
чений термодинамических констант (ДО0, ДЯ° и AS0)
для равновесия
Feni-|-HO2- Fe3+HO^
и для равновесия
Feln + HO~ Fe3+HO"
показывает, что энтропии ионов НОГ и НО' имеют
очень близкие значения. Предлагаемая схема разложе-
ния перекиси водорода имеет следующий вид:
Н2О2^± Н+ + НО2-,
Fe3+ + НО2" —► Fe2+ + НО2 •,
Н2О2 + НО2---► о2 + н2о + но.,
Fe2+ 4- НО---► Fe3+ 4- НО",
НО"4-Н+ —► Н2О.
Реакция разложения Н2О2 не является цепной, так как
каждый радикал НО2 реагирует с перекисью водорода,
образуя радикал НО', который затем окисляет ион желе-
за (II) [56].
Интересно приложение таких радикальных схей к
описанию реакции образования дифенила и фенола при
облучении водных растворов бензола, содержащих ионы
Fe111, ультрафиолетовым светом с длиной волны
313 нм [57]:
Fe3+OH" —> Fe2+-)-HO-
и затем
но • + с8н8н —> с8н8 • + н,о.
С,Н8 • + С8Н8. — (С8Н8)2,
Н2О + С8Н8. + Fe3+ — С*Н8ОНН+Fe3+,
Н2О 4- С8Н8 • 4- Fe2+ —> С8Н, 4- НО" 4- Fe2+
Та же реакция применяется для индуцирования поли-
меризации акриламида в водном растворе [58]. Выявле-
ны различные стадии реакции, включая стационарное
состояние. Квантовый выход фотохимически иницииро-
Реакции окисления анионов, комплексных катионов
69
ванной реакции уменьшается при увеличении общей
концентрации ионов Feni или концентрации ионов
Fe3+OH~ Результаты показывают, что при достаточно
высоких концентрациях ионов Fe111 стационарная ско-
рость полимеризации, /?р, пропорциональна интенсив-
ности поглощенного света и концентрации мономера и
обратно пропорциональна суммарному количеству ио-
нов железа(III) в обеих формах. Последовательность
происходящих при этом реакций представлена следую-
щей схемой:
Fe3+OH~ + hv —> Fe2+ 4-. ОН ]
# } зарождение цепи
• ОН + ml —► mj J
—> mj+i развитие цепи
m*y + Fe3+ —► P.OH4-H++Fe2+ ]
Л о л г обрыв цепи
m’+Fe3+OH —► P?OH-[-Fe2+ J
Окисление ванадием (V)
Ион VO? образуется при подкислении раствооа ва-
надата:
VO3 Ц-2Н+ VO?4-H2O.
Некоторые доказательства существования иона VO?
были приведены ранее (стр. 20). Ион VO? придает
раствору желтую окраску и является устойчивым в огра-
ниченном интервале значений pH [59]. Кинетические дан-
ные [62] позволяют предположить, что при повышении
концентрации НСЮ4 имеет место равновесие
VO? + H3O+ V(OH)2+.
Изменение цвета от желтого к красному при высокой
концентрации H2SO4 происходит, вероятно, вследствие
образования сульфатного комплекса. Интерпретация ки-
нетических данных осложняется отсутствием функций
кислотности, соответствующих данному равновесию.
70
Глава 2
При окислении пинакола по стехиометрическому
уравнению
(СН3)2СОН
| 2Vv —> 2(СН3)2СО + 2VIV + 2Н+
(СН3)2СОН
скорость реакции описывается следующим уравне-
нием [62а]:
— d [vV]ldt = kQ [пинакол] [Vv] (1 +f [H+] ).
Точная форма зависимости скорости реакции от кислот-
ности здесь не установлена. Добавление акрилонитрила
к полимеризации не приводит.
Для реакции окисления пинакола предлагается сле-
дующий механизм [60а]:
пинакол 4-VOJ -медленная^ пинакол (VO2)+ бы-СТрая->
стадия стадия
комплекс
—> (сн3)2Ся=о + (сн3)2сон4-н+4-уо2
VO24-H2O —> VO2+4-2OH“
(CH3)2COH4-VO2+ —> (СН3)2С=О + н+ + vo2.
Зависимость скорости реакции окисления от кислот-
ности, вероятно, объясняется образованием двухзаряд-
ного катиона V(OH)3+, который реагирует более актив-
но, чем однозарядный катион VO^. По мере протекания
реакции раствор становится голубым вследствие обра-
зования VO2+. Эта реакция аналогична окислению пина-
кола ионами церия(IV), исключая индуцированную по-
лимеризацию, которая в данном случае не наблюдается.
Реакция окисления циклогексанона до янтарной и
муравьиной кислот, а в растворе хлорной кислоты — до
адипиновой кислоты [606] также имеет первый порядок
как относительно ванадия (V), так и относительно цик-
логексанона. Предполагаемый механизм реакции вклю-
чает енолизацию циклогексанона и последующее мед-
ленное окисление. Отсутствие в уравнении скорости ре-
акции для этого более трудно протекающего процесса
окисления члена, не зависящего от кислотности, свиде-
Реакции окисления анионов, комплексных катионов
71
тельствует о том, что активным началом является более
электрофильный V(OH)g+
Скорость реакции окисления циклогексанола также
зависит от первой степени его концентрации и первой
степени концентрации окислителя — ванадия (V) [60в].
Оптические измерения дают возможность предполо-
жить, что в начале реакции образуется промежуточный
продукт [ROH, V(OH)3]2+, при разложении которого
почти количественно выделяется адипиновая кислота.
При этом на образование 1 молекулы адипиновой кис-
лоты расходуется 8 эквивалентов ванадия (V):
СбН12О + 8Н+ + 8VO2+ —> СбН10О4 + 5Н2О + 8VO2+.
При окислении 1-дейтероциклогексанола обнаружен
кинетический изотопный эффект [60в], указывающий на
то, что стадией реакции, определяющей скорость, слу-
жит разрыв связи С—Н. Зависимость скорости реакции
от сн3о* и отсутствие зависимости от fto доказывает
наличие в активном комплексе молекулы воды и участие
в реакции V(OH)3 При последующем исследовании [61]
было показано, что окисление монотретичных и дитре-
тичных гликолей, вероятно, происходит путем разрыва
связи, тогда как первичные и вторичные гликоли ведут
себя аналогично монозамещенным гликолям. Реакция
окисления 2-метилбутан-2,3-диола имеет кинетику про-
межуточного типа. Возможно, что электронодонорная
способность метильных групп, присоединенных к гли-
колю, в достаточной степени облегчает разрыв связи.
Окисление оксикислот (молочная, яблочная, мин-
дальная кислоты), согласно схеме [62]
RCH(OH)COOH 4- 2Vv —> RCHO-|-Н2О + СО2 + 2VIV,
происходит со скоростью
— d [Vv]/t/Z = k [оксикислота] [Vv] [1 4-аЛ0],
где hQ — соответствующая функция кислотности Гам-
мета.
Окисление изомасляного альдегида [63] протекает по
катализируемой кислотами реакции первого порядка
72
Глава 2
относительно ванадия (V). Показано, что скорость ее оп-
ределяется стадиейенолизации альдегида (тоже наблю-
дается при бромировании и иодировании). Молекулы
пропионового и я-масляного альдегидов енолизируются
значительно быстрее, чем окисляются. Скорость реак-
ции описывается следующим эмпирическим уравнением:
— d [Vv]/d/ = k [Vv] [альдегид]/{1 -\-k' [альдегид]}.
Происходит быстрое обратимое образование комплекса
1 : 1 и последующий его медленный распад:
rch2cho
RCH=CHOH + VO<t
RCH=CH—О—V=O + H+
ОН
RCHCHO и др. --XX---
। быстрая стадш
он
н+
7=^ RCH=CHOH
RCH=CHO—V=O
I
ОН
RCH=CH—О—V—ОН
I медленная qij
стадия
.CHRCHO4-Vlv
Окисление ионов хрома (II)
В качестве примера приведем результаты исследо-
вания кинетики реакции [64]
(NH3)5CoH2O3+ +Сг2+ Н-5Н+ —► Со2+ +Сг(Н2О)|+ + 5NH+.
Экспериментальные данные приводят к кинетическому
уравнению вида
— d [Cr11]Idt = kx [Cr11] [RН2ОШ] + k2 [Cr11] [RH2Oni]/[H+]
[здесь R представляет Co(NH3)5]. Это уравнение анало-
гично уравнениям, приведенным в гл. 1 для реакций
окисления катионами. Было показано [65], что при за-
мене легкой воды на тяжелую удельная скорость пер-
вого слагаемого в уравнении уменьшается в 3,8 раза,
а второго слагаемого — в 2,6 раза. Поскольку второй
Реакции окисления анионов, комплексных катионов
73
член в основном соответствует полному переносу кис-
лорода от окислителя (NH3)5CoH2O3+ к восстановителю
Сг2+, можно заключить, что первый член отвечает ско-
рости процесса, протекающего по аналогичному меха-
низму. Уменьшение скорости при этом свидетельствует
о наличии связи О—Н в активном комплексе.
В другой работе [66] при помощи изотопа 18О пока-
зано, что при реакции окисления кислород количествен-
но переходит от (NH3)5CoH2O3+ к Сг2+. Реакция окисле-
ния проводилась в достаточно кислой среде, при этом
оказалось, что по крайней мере на 50% реакция проте-
кает с участием воды, происходит образование промежу-
точного активного комплекса.
Таубе [67] изучил перенос электрона от ионов хро-
ма (И) к (NH3)5CoL2+, где L—SCN", N3*. РОГ, РзО?-’
СН3СОО", бутират, кротонат, оксалат, сукцинат, ма-
леинат. Малеиновая кислота в качестве лиганда уско-
ряет примерно в 100 раз перенос электрона по сравне-
нию с янтарной или уксусной кислотами. Последняя
действует не лучше уксусной кислоты. В случае приме-
нения малеиновой кислоты перенос электронов облег-
чается, по-видимому, наличием л-электронов двойной
связи. Поэтому представляет интерес изучить поведение
ацетилендикарбоновой кислоты в качестве мостика для
переноса электронов, поскольку поляризуемость л-элек-
тронов в этом случае меньше, чем в случае фумаровой
кислоты (см. [68]). Другие интересные случаи упомя-
нуты Дюком [69].
Если мостиком в реакции малонатопентаминко-
бальт(Ш)-аниона с ионом Сгп является малонат [70],
то в кинетическом уравнении реакции в добавление
к слагаемому, не зависящему от кислотности, и слагае-
мому, в знаменателе которого имеется величина концен-
трации ионов водорода, имеется еще слагаемое первого
порядка относительно ионов водорода. Член первого
порядка исчезает, если водород метиленовой группы за-
мещен этиленовой группой. Это дает основание пред-
полагагать, что член, прямо пропорциональный кислот-
ности, отражает диссоциацию метиленовой группы под
комбинированным действием кислоты и восстановителя
74
Глава 2
Возникающая при этом система делокализованных элек-
тронов обеспечивает перенос электрона от иона
хрома (II) к иону кобальта (III).
При исследовании реакции перхлората хрома (II) с
ионом азида [71]
2Сгп +2Н+ + HN3 —► 2Cr,n + NH3 + N2
среди ее продуктов обнаружен биядерный хром (III) ам-
мин:
(Н2О)5Сг—NH—Cr(H2O)j+ (выделен в виде сульфата)
Окисление ионов марганца (II)
Классической реакцией этого класса является реак-
ция Гайярда
ЗМп2+4-2МпС>4 +2Н2О 5=± 5МпО2 + 4Н+.
Она имеет большое значение в связи с использованием
перманганата в титриметрическрм анализе, так как для
предотвращения образования Двуокиси марганца необ-
ходимо создавать достаточную кислотность раствора.
Эта реакция была предметом многих исследований [72—
74], результаты которых суммированы Ледбери и Кал-
лисом [75].
Томпкинс [73] предложил для реакции Гайярда сле-
дующую схему:
Мпп4-МпО4-+Н2О Mnni + HMn<V Ч-ОН-, (1)
HMnO4~ + Мп" + ОН" меменвая> 2МпО2 + Н2О. (2)
стадия
2Мп,п —► Mnn + Mniv.
(3)
(замедляется образованием комплексов)
MnIV4-H2O —+ Мпш + ОН4-Н+,
Мп” + ОН —> МпП14-ОН“,
Mn,v4-OH~ —► Мп(ОН)3+ —> МпО2.
(4)
(5)
(6)
Основная часть двуокиси марганца образуется по реак-
ции (6), так как реакция (2) протекает медленно. При-
Реакции окисления анионов, комплексных катионов
75
соединение первого гидроксила по реакции (6), вероят-
но, является определяющей скорость стадией гидролиза
иона марганца(IV).
Адамсон [74] установил, что реакция в 3 н. НС1О4
между радиоактивным изотопом Мп2+ и перманганатом
происходит с малой, но измеримой скоростью. Кинети-
ческое уравнение реакции на стадии, определяющей ее
скорость, имеет вид
Л = k [Н+]4/> [Мп11]44 [МпО^’],/*.
Предполагается следующий механизм.
Быстрые обратимые равновесия:
6Н+ 4-ЗМп2+ 4- МпО4“ ^=± ЗМп3+ + МпО2+ +ЗН2О, (1)
Мп3+ + Н2О МпО+ + 2Н+. (2)
МпО2+ + 2Н+ Мп4+ + Н2О. (3)
Обмен, определяющий скорость:
МпО+ + MnO2+ МпО2+ 4- МпО+. (4)
Параллельные конкурирующие реакции:
МпО2+ 4- Н2О —> МпО2(8) 4-2Н+. (5)
2Н+ 4- Мп2+ 4- МпО2+ —► 2Мп3+ 4- Н2О, (6)
Мп»+4-Мп4+ 2Мп»+. (7)
Если предположить, что равновесие реакций (2) и
(3) очень мало сдвинуто вправо и что Мпш и MnIV в
основном присутствуют в форме ионов Мп3+ и МпО2+,
тогда, исходя из равновесия (1), отношение МпО2+/Мп3+=
= 1/3 и скорость реакции выражается следующим обра-
зом: k [МпО+] [МпО2+] или k' [н+] [Мп2+]/! [МпО?]'*, что
согласуется с результатами эксперимента.
Аналогично ионы марганца (II) окисляются перйода-
том до перманганата [76]. Предварительное исследова-
ние показало, что кинетика этой реакции очень сложна.
76
Глава 2
Окисление ионов железа (II)
В области реакций окисления ионов Fen имеются не-
которые интересные наблюдения. При исследовании ки-
нетики реакции окисления железа (II) с помощью различ-
ных комплексных ионов [77] было найдено, что между
константами скорости и редокс-потенциалами окисли-
теля имеется корреляция:
окислитель Eq
Ag (dipy)l+ 1,43
Fe (Men)*+ 1,06
Fe (dipy)3+ 0,97
Fe (tripy)f+ 0,93
Os (dipy)23+ 0,83
м1-кмоль~1-сек~1 (в 0,5AfH2SO4)
1,4-10*
3.06 • 105
2.2 • IO®
7,4 • 105
1,7.10s
Аналогичные результаты были получены Сатином и Гор-
доном [78] для реакции окисления иона Fe11 при помощи
Fe (р/геп)|+ При сравнении реакций окисления ионов
Fen ионами Fe (phen)3+ с другими реакциями окисления
Fe11 показано существование линейной зависимости ме-
жду свободной энергией активации и изменением стан-
дартной свободной энергии соответствующей реакции.
Энергия активации для реакции окисления — восстано-
вления между Fe11 и Fe(pfien.)f+ составляет только
3,36 • 106 дж/кмоль, а отрицательное значение энтропии
активации 1,56 • 105 дж/кмоль • град значительно превы-
шает соответствующие величины для реакций Fen—Fe™,
рассмотренных в гл. 1.
Представляет интерес классическая реакция окисле-
ния ионов железа(II) бихроматом:
Cr2O|" + 14H+ + 6Fe2+ —► 7H2O + 2Cr3++6Fe3+.
Кинетика этой реакции достаточно сложна [79]:
— d [Fe11]/^ = [Н+р [Fe»]2/[FeInl {*, [НСгО4“] + А2 [НСгО4 ]2}.
Реакции окисления анионов, комплексных катионов
77
Окисление некоторых катионов ионами перхлората
Эти реакции представляют интерес вследствие склон-
ности многих авторов приписывать свойство стабиль-
ности перхлоратам металлов. Для реакции окисления
Tiin перхлоратом
8Т11И + СЮ4 +8Н+ —> 8T1,V + Cl“ + 4Н2О
скорость описывается уравнением [80]
— d [Т1П1]/^ = [Т1Ш] [СЮ^] Aj [Т1С12+] [сю4 ] +
+*3 |Т1,П] [сю4-] [н+]+А4 [Т1С12+] [СЮ4-] [н+].
Этому сложному кинетическому уравнению соответ*
ствует следующий механизм реакции:
Т1,П + С1О4 (комплекс) —► Т1О2+4-С1Оз,
Tini -f. С1О4- + Н4 q=± (комплекс) —► ТЮН3++СЮз,
Т1С12+ -f- СЮ^ (комплекс) —► Т1О2+-|-С1О34-С1",
Т1С12+4-С1О4"4-Н4 (комплекс) —► ТЮН3+4-С1О34-С1".
При этом радикал С10з быстро восстанавливается до
хлор-иона.
Окисление V11 и V111 перхлоратом происходит по
уравнениям [81]:
8V3+ + С1О4" +4Н2О —> 8VO2+ -I-СГ + 8Н+,
8V2+4-C10^ + 8H+ —► 8V3+ 4- Cl" + 4Н2О.
Скорость уменьшения концентрации V11 выражается
следующим образом:
- d [Уп]/Л = kx [V11] [C1O4-] +2й2 [V1»] [C1O4-].
Для реакции между V111 и перхлоратом, не завися-
щей от кислотности среды, предлагается следующий
78
Глава 2
механизм [83]:
V34 4" CIO4 + Н20 —► VO^ 4-С1О34-2Н+ стадия, определяю-
щая скорость
V3+ 4“ СЮ3 —► продукты быстрая стадия
V3+ 4- VO^- —► 2VO2+ быстрая стадия
а для реакции между V11 и перхлоратом:
V2+ 4- С1О^ —► VO|+ 4- СЮ3 стадия, определяющая скорость
3V2+ 4- С1О^ —> 3VO2+ 4- СГ быстрая стадия
Для реакции, зависящей от концентрации ионов во-
дорода, предполагается существование равновесия дис-
социации:
Н+ 4“ С1О^" у-* НС1О4 быстро устанавливающееся равновесие
и затем
V3+ + НСЮ4 + Н2О —► VO2+ + СЮ3 +ЗН+ стадия, опреде-
ляющая скорость
Двухэлектронные окислители
Рассмотрим реакции окисления с участием окислите-
лей, присоединяющих два электрона.
Тетраацетат свинца
По методу, предложенному Криги [82], тетраацетат
свинца готовят растворением РЬ3О4 в горячей ледяной
уксусной кислоте до состояния насыщения, тетраацетат
выделяется при охлаждении [12в]. Тетраацетат свинца
можно также получить анодным окислением ацетата
свинца(II). По-видимому, таким путем нельзя получить
перхлорат четырехвалентного свинца. Стабилизация че-
тырехвалентного свинца достигается только ионами аце-
тата.
Стандартный электродный потенциал [2] для реакции
2Н2О 4~ PbSO4 —► PbO24-SO^"4-4H+4-2*
Реакции окисления анионов, комплексных катионов
79
составляет —1,69 в; следовательно, по термодинамиче-
ским данным свинец(IV) является сильным окислите-
лем.
Из простых реакций изучено взаимодействие ацетата
свинца (IV) с ацетатом Се1П [83] и ацетатом СоI 11 [84] в
уксусной кислоте. Реакции осложняются комплексооб-
разованием с ионами ацетата. Возможно участие РЬ1П
в реакциях образования промежуточного продукта.
Рассмотрим гетеролитическое окисление тетраацета-
том свинца. При повышенных температурах возможно
протекание гомолитических реакций.
Для окисления гликолей ранее предлагался следую-
щий механизм [85]:
I I
—С—ОН — С—ОРЬ(ОСН3СО)3
| +РЬ(ОСН3СО)4 —► I 4-СНзСООН
-С—ОН —С—он
I I
медленная бимолекулярная стадия
I I
—С—ОРЬ(ОСН8СО)8 —с—о ч
| | ЪРЬ(ОСН8СО), —*•
—С—ОН —С—о
I I
обратимое равновесие
I
-С—О
+ РЬ(ОСН8СО)2
—с=о
Белл с соавторами [86], исследуя реакцию взаимо-
действия этиленгликоля с тетраацетатом свинца в среде
уксусной кислоты при температурах 18, 25, 35 и 45°,
нашли, что реакция имеет первый порядок относительно
каждого реактанта и описывается уравнением Аррениу-
са со стерическим фактором, равным примерно единице.
Они предположили, что такой высокий стерический фак-
тор при реакции между двумя незаряженными моле-
кулами лучше объясняется участием свободных радика-
лов, а не ионов. В более позднем исследовании этой
80
Глава 2
реакции Кордиер и Позакер [87] предложили следую-
щий несколько видоизмененный механизм Криги:
С—ОН стадия 1
| +РЬ(ОСН3СО)4 / >
q q|_| быстрая
I
—С—ОРЬ(ОСН3СО)3
| +НОСН3СО ,
—с—он
I
| стадия 2, быстрая
I
_с=о
+ Pb(OCH3CO>;^S^-
—с—он
I
^стадия 4а, медленная
I
-С=О +РЬ(ОСН3СО)2 стадия4б1
^быстрая
—С=О +НОСН3СО
-С-0*
| + РЬ(ОСН3СО)3
—с—он
I
I стадия 36, медленная
—С—О*
| 4-РЬ(ОСН3СО)2 +
-с-о* + НОСНзСО
В более позднем сообщении Белл и соавторы [88]
указывают на наличие кислотного катализа при распаде
гликоля под действием тетраацетата свинца и на то, что
реакция в среде уксусной кислоты является гетероли-
тическим процессом. Подробно действие тетраацетата
свинца на сахара рассмотрено в обзоре Перлина [89].
Реакция окисления тетраацетатом свинца несколько
выпадает из общего плана этой главы, так как для ста-
билизации этого процесса необходимо применение ледя-
ной уксусной кислоты в качестве растворителя.
Ионы таллия (III)
Стандартный электродный потенциал для реакции
Т11 -* Т1ш4-2ё составляет —1,25 в, что свидетельствует
об относительно высокой окислительной способности ио-
нов Т1Ш.
В качестве примера рассмотрим перенос электронов
₽ системе Т11—Т1Ш, катализируемый ионами бромида.
Реакции окисления анионов, комплексных катионов
81
Скорость этого процесса выражается уравнением [90]
л = *о [Т1+1 [Т13+1 + *2 [Т1Вг2+] + л8 [TlBr3J +
+ Л4 [Т1+1 [Т1Вг4-] + Л6 [Т1Вг2“] [Т1Вг4-].
Каждый из более высоких членов уравнения отражает
окисление комплексными ионами таллия (III). Здесь мы
имеем предельный случай анионокатализируемого пере-
носа электронов, который обсуждался ранее в гл. 1.
Ирвин [91] измерил скорость реакции ионов ~Т11П с
Os (dipy)\+ При этом предполагался следующий меха-
низм:
Tlln+Os(rfZp^+ Т1П -р Os +,
Tl" + Os (dlpy)l+ т=± Tl’ + Os (dipyfr,
где dipy — молекула 2,2'-дипиридила.
В отличие от реакций, имеющих место при окислении
катионов катионами (глава 1), скорость данных реак-
ций с уменьшением концентрации ионов водорода уве-
личивается очень мало. Этот факт можно объяснить
предположением, что оба иона. Т13* и Т1(ОН)2+, могут
реагировать с комплексным ионом. Хлор-ионы, как обыч-
но, понижают скорость реакции.
Реакция взаимодействия ионов таллия(III) с му-
равьиной кислотой в водном растворе хлорной кислоты
[92] представляется уравнением
Т1П, +НСООН —> Т1’-|-2Н+4-СО2.
Реакция проходит через две стадии:
Т13+ + НСООН 5Z* Т1 • НСООН8+,
Т1 • НСООН3+ —► Т1+ -I-2H+ 4- со2.
Спектрофотометрические данные подтверждают суще-
ствование комплекса TI • НСООН3+.
В органической химии мало количественных иссле-
дований реакций с ионами таллия(Ш). Каббе [93] ка-
чественно исследовал окисление органических соедине-
ний ионами Т1т.
При окислении циклогексанона ионы таллия(Ш) ве-
дут себя аналогично ионам ртути(II) (см. ниже) и не
участвуют в стадии, определяющей скорость реакции [94].
6 Зек. 1249
82
Глава 2
Ионы ртути (II)
Стандартный электродный потенциал системы Hg1—
Hgn, равный —0,920 в [2], показывает, что ион ртути(II)
должен быть довольно сильным окислителем. Однако
окислительные свойства ртути(II) обычно понижаются
вследствие того, что она легко взаимодействует со мно-
гими органическими молекулами [12]. Во многих случаях
органические вещества могут подавить окислительные
свойства ртути(II).
Уравнение скорости реакции ионов ртути (II) и фор-
миата имеет первый порядок относительно каждого из
реактантов. Скорость реакции обратно пропорциональна
первой степени концентрации ионов водорода. Для этой
реакции выявлен значительный дейтериевый изотопный
эффект [95]. Предполагаемый механизм реакции сво-
дится к непосредственному переносу электрона от фор-
миат-иона к иону ртути(II) на стадии, определяющей
скорость процесса, и последующему быстрому разло-
жению.
Представляет интерес окисление окиси углерода
ионами ртути (II) [96]:
2Hgn + СО + Н2О —> Hg’4-CO2 + 2H+.
Скорость реакции окисления окиси углерода описывает-
ся уравнением
— d [СО]/Л = k [СО] [Hg11].
что соответствует следующему механизму:
О
II
Hg2+ + Н2О 4- СО —► [—Hg—С—ОН]+ + Н+ стадия, опреде-
ляющая скорость реакции
О
II
[—Hg—С—ОН]+ —► Hg-|-СО2 + Н+ быстрая стадия
Hg-|-Hg2+ —► Hg|+ быстрая стадия
При окислении циклогексанона [94] скорость реак-
ции имеет нулевой порядок относительно концентрации
Реакции окисления анионов, комплексных катионов
83
окислителя. Скорость реакции контролирует не стадия
окисления, а, вероятно, процесс енолизации:
Рассмотрением этой реакции завершается обзор про-
цессов окисления — восстановления между катионами и
исходным веществом.
ЛИТЕРАТУРА
1. Waters W. A. (Ed.), Vistas in Free Radical Chemistry, Perga-
mon, London, 1959, p. 158.
2. Latimer W. M. Oxidation Potentials, Prentice-Hall, New York,
1953; см. общую литературу к Введению.
3. L i s t е г М. W., Can. J. Chem., 31, 638 (1953).
4. Зонис С. А., Лесина А. Г., ЖОХ, 20, 1180 (1950).
5. Uri N., Chem. Rev., 50, 375 (1952).
6. N о y«e s A. A., De Vault D., Coryell C. D., D e a h 1 T. J-
J. Am. chem. Soc., 59, 1326 (1937).
7. Kirwin J. B., Peat F. D., Proll P. J., Sutcliffe L. H., J.
phys. Chem., 67, 1617 (1963).
8. G о r d о n В. M., W a h 1 A. C., J. Am. chem. Soc., 80, 273 (1958).
9. Bacon R. G. R., Hanna W. J. W., Proc. chem. Soc., 305 (1959).
10. M a r s h a 11 H., J. chem. Soc., 59, 760 (1891).
11. Swann S., Xanthakos T. X., J. Am. chem. Soc., 53, 400
(1931).
12. S i d g w i c k N. V., The Chemical Elements and Their Compounds,
Oxford University Press, 1950, (a) p. 1392; (6) p. 451; (в)
p. 611; (r) p. 296.
13 Noyes A. A., De ahi T. J., J. Am. chem. Soc., 59, 1337 (1937).
14. Sutcliffe L. H., Weber J. R., J. inorg. nucl. Chem., 12, 281
(1960).
15. Ba wn С. E. H., Whi te A. G., J. chem. Soc., 331 (1951).
16. В a x e n d a 1 e J. H., Wells C. F., Trans. Faraday Soc., 53, 800
(1957).
6*
84
Глава 2
17. В awn С. Е., White A. G., J. chem. Soc. (1951), (а) р. 339;
(б) р. 344.
18. Н а г g г е a v е s G., Sutcliffe L. Н., Trans. Faraday Soc., 51
(1955), (а) р. 786; (б) р. 1105.
19. В awn С. Е. Н, Sharp J. A., J. chem. Soc. (1957), (а) р. ,1854;
(б) р. 1866.
20. S h а г р J. A., J. chem. Soc., 2026 (1957).
21. L i 111 е г J. S., J. chem. Soc., 832 (1962).
22. Hoare D. G., Waters W. A., J. chem. Soc., 965 (1962).
23. Hardwick T. J., Robertson E., Can. J. Chem., 29, 818, 828
(1951).
24. Kolthoff I. M, Belcher R., Volumetric Analysis, Vol. 3,
Interscience, New York, p. 121.
25. King E. L, Pandow M. L., Л Am. chem. Soc., 75, 3063 (1953).
26. Ar don M, J. chem. Soc., 1811 (1957).
27. Litter J. S., J. chem. Soc., 4135 (1959).
28. MinoG., Kaizerman S., R a s m u s s e n E., J. Am. chem. Soc.,
81, 1494 (1959).
29. Krishna B., Tew ar i К. C., J. chem. Soc., 3097 (1961).
30. Shorter J., J. chem. Soc., 1868 (1962).
31. Guilbault G. G., McCurdy W. H., J. phys. Chem., 67, 283
(1963).
32. Muhammad S. S., Rao К. V., Bull. chem. Soc. Japan, 36, 943
(1963).
33. W a t e r s W. A., Quart. Rev., 12, 277 (1958).
34. Ubbelohde A. R. J. P., J. Chem. Soc., 1605 (1935).
35. С a r 11 e d g e G. H., Nichols P. M, J. Am. chem. Soc., 62,
3057 (1940).
36. Watters J. I., Kolthoff I. M, J. Am. chem. Soc., 70, 2455
(1948).
37. Drummond A. Y, Waters W. A., J. chem. Soc., (a) p. 440;
(6) p. 3119; (в) p. 3119 (1953); (r) ibid. (1954), p. 2456; (д)
ibid. (1955), p. 497.
38. Lin dwell О. M, Bell R. P., J. chem. Soc., 1303 (1935).
39. Duke F. R., J. Am. chem. Soc., 69, 2885 (1947).
40. Taube H., J. Am. chem. Soc., 70, 1216 (1948).
41. Levesley P., Waters W. A., J. chem. Soc., 217 (1955).
42. Land H., Waters W. A., J. chem. Soc, 4312 (1957).
43. S i 11 ё n L. G, Quart. Rev., 13, 146 (1959).
44. Fudge A. J, Sykes K. W, J. chem. Soc, 119 (1952).
45. Page F. M, Trans. Faraday Soc, 49, 635 (1953); 50, 120 (1954);
56, 398 (1960).
46. К а г г а к e r D. G, J. phys. Chem, 67, 871 (1963).
47. Higginson W. С. E, Sutton D, J. chem. Soc, 1402 (1953).
48. Higginson W. С. E, Wright P, J. chem. Soc, 1551 (1955),
49. Cann J. W, Powell R. E, J. Amer. chem. Soc, 76, 2568 (1954).
50. Thomas J. K., Trudel G, В у w a t e r S, J. phys. Chem, 64,
51 (1960).
51. Wesp E. F, В rode W. R, J. Am. chem. Soc, 56, 1037 (1934).
52. Baxendale J. H, Hardy H. R, Sutcliffe L. H, Trans.
Faraday Soc, 47, 963 (1951).
Реакции окисления анионов, комплексных катионов
85
53. BaxendaleJ. Н., Hardy Н. R., Trans. Faraday Soc., 50, 808
(1954).
54. Н о с к е 11 R. С., Hudson С. S., J. Am. chem. Soc., 56, 1632
(1934).
55. Evans M. G., George P., Uri N., Trans. Faraday Soc., 45,
230 (1949).
56. В a r b W. G., В a x e n d a 1 e J. H., George P., H a r g r e a-
ves K. R., Trans. Faraday Soc., 47, 591 (1951).
57. Baxendale J. H., Magee J., Trans. Faraday Soc., 51, 205
(1955).
58. D a i n t о n F. S., T о r d о f f M., Trans. Faraday Soc., 53, 666
(1957).
59. R о s s о 11 i F. J., R о s s о 11 i H., J. inorg. nucl. Chem., 2, 201
(1956).
60. L i 111 e r J. S., W a t e r s W. A., J. chem. Soc. (1959), (a) p. 1299;
(6) p. 3014; (в) p. 4046.
61. Littler J. S., Mallet A. I., Waters W. A., J. chem. Soc.,
2761 (1960).
62. J о n e s J. R., W a t e r s W. A., L i 111 e r J. S., J. chem. Soc., 630
(1961).
63. Jones J. R., Waters W. A., J. chem. Soc., 352 (1963).
64. M u r m a n R. K., Taube H., Posey F. A., J. Am. chem. Soc..
79 262 (1957).
65. Zwickel A., Taube H., J. Am. chem. Soc., 81, 1288 (1959).
66. Kruse W., Taube H., J. Am. chem. Soc., 82, 526 (1960).
67. Taube H., J. Am. chem. Soc., 77, 4481 (1955).
68. Robertson P. W., D a s e n t W. E., Milburn R. M., 01 i-
ver W. H., J. chem. Soc., 1628 (1950).
69. Duke F. R.. J. chem. Educ., 38, 161 (1961).
70. Svatos G., Taube H., J. Am. chem. Soc., 83, 4172 (1961)
71. Ard on M., Mayer В. E., J. chem. Soc., 2816 (1962).
72. P о 1 i s s а г M. J., J. phys. Chem., 39, 1057 (1935).
73. Tompkins F. C., Trans. Faraday Soc., 38, 131 (1942).
74. A d a m s 6 n A. W., J. phys. Chem., 55, 293 (1951).
75. Ladbury J. W., Cull is C. F., Chem. Rev., 58, 413 (1958).
76. W a t e r b u г у G. R., H a у e s A. M., M a r t i n D. S., J. Am. chem.
Soc., 74, 15 (1952).
77. G о r d о п В. M., Williams L. L., S u t i n N., J. Am. chem.
Soc., 83, 2061 (1961).
78. Sutin N., Gordon В. M., J. Am. chem. Soc., 83, 70 (1961).
79. E s p e n s о n J. H., King E. L., J. Am. chem. Soc., 85, 3328
80. Duke F. R., Quinney P. R., J. Am. chem. Soc., 76, 3800 (1954).
81. King W. R., Garner C. S., J. phys. Chem., 58, 29 (1954).
82. С r i e g e e R., Liebigs Annin, 481, 263 (1930).
83. В e n s о n D., Sutcliffe L. H., Trans. Faraday Soc., 56, 246
(1960).
84. Benson D., Proll P. J., Sutcliffe L. H., Walkley J.,
Disc. Faraday Soc., 29, 60 (1960).
85. Criegee R., Kraft L., Rank B., Liebigs Annin, 507, 159
(1933).
86
Глава 2
86. Bell R. P., Sut г rock J. G. R., Whitehead R. L. S., J.
chem. Soc., 82 (1940).
87. Cord n er J. P., Pa us acker К. H., J. chem. Soc., 102 (1953).
88. Bell R. P., R i v 1 i n V. G., Waters W. A., J. chem. Soc., 1696
(1958).
89. Perlin A. S., Rec. Adv. Carbonydrate Chem., 14, 9 (1959).
90. C ar p en t er L. G., Fо r d - S m i t h M. H., Bell R. P., Do d-
son R. W., Disc. Faraday Soc., 29, 92 (1960).
91. Irvine D. H., J. chem. Soc., 1941 (1957).
92. Halvorson H. N., Halpern J., J. Am. chem. Soc., 78, 5562
(1956).
93. К abb e H., Liebigs Annin, 656, 204 (1962).
94. Littler J. S., J. chem. Soc., 827 (1962).
95. Halpern J., Taylor S. M., Disc. Faraday Soc., 29, 174 (I960).
96. Harkness A. C., Halpern J., J. Am. chem. Soc., 83, 1258
(1961).
Глава 3
РЕАКЦИИ ОКИСЛЕНИЯ ОБРАЗУЮЩИМИСЯ КАТИОНАМИ
В данной главе рассматриваются реакции, которые
протекают при участии катионов, образующихся в кис-
лых растворах; повышение pH приводит к образованию
анионов и препятствует катионному пути реакции. Для
этих реакций принята следующая общая схема:
АВ А+4-В-,
л2
А + ।
А + основание —> продукты.
* Льюиса ¥
Скорость первой стадии определяется скоростью обра-
зования катионов А+. Относительная реакционная спо-
собность различных окислителей, устанавливаемая ки-
нетическим методом, является функцией константы ско-
рости ki. Во второй стадии скорость реакции зависит от
равновесной концентрации катионов А+, и их реакция
с основанием контролируется как термодинамически, так
и кинетически. Стадию k3 следует характеризовать пер-
вичным солевым эффектом, концентрация катионов А+
должна зависеть от вторичного солевого эффекта.
Другой возможной особенностью рассматриваемых
реакций является наличие равновесия молекул АВ с
ионами водорода по схеме
АВ+Н+ АВН+ ^=> А++НВ.
При этом может возникнуть такое положение, когда
необходимо или возможно различать реакции, проте-
кающие с участием катиона А+ или катиона АНВ+. Если
основанием В служит ион гидроксила, тогда следует
различать ионы А+ и Н2ОА+. Доказательства, которые
позволяют это сделать, обобщены в работе Де лд Мара
88
Глава 3
и Ридда [1а] следующим образом: (а) если реакция
имеет нулевой порядок относительно окисляемого ве-
щества, это соответствует образованию положительно
заряженного иона как стадии, определяющей скорость
реакции; (б) если известна скорость переноса 18О ме-
жду растворителем и НОА и если она меньше экспери-
ментально найденной скорости изучаемой реакции,
то последняя должна проходить через стадию об-
разования Н2ОА+; (в) если исходить из равновесия
Н+АОН АОН^, то концентрация иона AOHf опре-
деляется функцией кислотности Но; если же исходить из
равновесия Н++АОН^А++Н2О, то концентрация
ионов А+ определяется функцией кислотности Jo-
Мы рассмотрим реакции этих катионообразующих
веществ в следующем порядке: (а) положительно за-
ряженные ионы галогенов Х+ и источники их образо-
вания; (б) положительно заряженные кислородные
ионы галогенов ХО^+; (в) положительный ион гидрокси-
ла НО+; (г) ион нитрозония (NO+) и (д) ион нитрония
(no2+).
Положительные ионы галогенов Х+ и их источники
Ингольд [2а] предположил, что положительные ионы
галогенов и их носители по уменьшению электрофиль-
ности располагаются в ряд
Х+ XOHJ, X* ХВ, ХОН.
ХВ представляет вещества, в которых основание В по
силе занимает промежуточное положение между ионом
гидроксила и ионом галогена. Энбар и Гинзберг [3]
распространили эти идеи на органические носители га-
логенов (органические гипогалогениты) и рассмотрели
ряд
Х+, Н2ОХ+ ROHX+, сн3соонх +
Сначала рассмотрим теоретические доказательства
существования положительных ионов, а затем обсудим
свойства положительных ионов иода, брома и хлора и
их носителей-
Реакции окисления образующимися катионами
89
Белл и Гелле [4] рассчитали изменение свободной
энергии реакции
X2(aq) * X(aq) 4" X(aq)»
исходя из цикла
2 3
X2(g) -* 2X(g) -* X(g) + X(-g)
♦ / /
1 // //
/ /
X2(aq) > X(aq)+X(aq)
Затем они определили свободную энергию гидратиро-
ванных ионов Н2ОХ+, которые, по-видимому, предста-
вляют наиболее вероятную форму существования Х+ в
растворе. Расчет констант равновесия реакции
Х2 + Н2О т=± Н2ОХ+ + Х’
привел к значениям 10~10 в случае иода, 10~20 в случае
брома и 10-30 в случае хлора. На основании измерений
э. д. с. элеменга
Pt/I2, Ag+, Н+/солевой мостик/Н+, Iе, I2/Pt
они нашли экспериментальную величину константы рав-
новесия для иода, равную 1,2 • КН1.
Существование катионов галогенов доказывается пре-
паративным путем. В общепринятом методе получения
соединений, содержащих положительно заряженный га-
логен, используются соли серебра [5]. В обычной мето-
дике соли серебра вступают в реакцию с галогеном в
сухом слабополярном растворителе, например в хлоро-
форме, в присутствии основания, способного образовы-
вать координационное соединение, например пиридина
или хинолина.
Другие препаративные методы также доказывают
существование катионов галогенов. Серия статей [6,7]
посвящена исследованию свойств голубых растворов,
образующихся при растворении иода в олеуме. Измере-
ния магнитной восприимчивости показали, что эти рас-
творы содержат парамагнитное вещество, обязанное
90
Глава 3
образованию свободного катиона иода Г, существовав
ние которого подтверждается нелетучестью иода и устой-
чивостью растворов при высоких температурах. Реак-*
ция протекает по стехиометрическому уравнению
I2 + 2SO3 + H2SO4 —► 21+ 4- 2HSOJ" + SO^
Прямое иодирование бензола проводилось путем его
обработки иодом в присутствии раствора сульфата се-
ребра в концентрированной серной кислоте [8].
Бензол (23 г; 0,3 моля) интенсивно перемешивали с раствором
сульфата серебра (31 г; 0,1 моля) в смеси концентрированной сер-
ной кислоты (200 мл) и воды (20 мл) до получения эмульсии.
К последней в течение 2 час частями добавляли мелкодисперсный
иод (56 г; 0,22 моля). После часового перемешивания смесь разбав-
ляли водой (600 мл) и декантировали через фильтр; в остатке на-
ходились иодид серебра и свободный иод. Затем полученный продукт
обрабатывали сульфитом натрия, экстрагировали эфиром и очищали
фракционированной перегонкой под вакуумом. Выход иодбензола
с т. кип. 188° составлял 32 г (78% рассчитанного по расходу суль-
фата серебра).
Еще одно доказательство существования катионов
иода получено при исследовании кинетики иодирования
анилина [16]. Для этого процесса возможны две схемы,
описываемые уравнениями:
- d р2] /at=k [c6h5nh3+] [hoi]
ИЛИ
-d[[2]/dt = k' [C6h5nh2] [i+J.
По первой из схем замещение должно идти в пета-по-
ложение, в действительности оно происходит в орто- и
пара-положение. Это позволяет сделать вывод, что за-
мещение происходит путем взаимодействия нейтраль-
ной молекулы анилина с ионом иода 1+.
По процессам окисления иодом 12иего соединениями
имеется обширная литература [9]. Очевидно, молекула
иода так же легко распадается на атомы, как на ионы,
и поэтому параллельно с гомолитическим механизмом
реакции окисления может проявляться гетеролитический.
Кольтгоф [10] установил, что низкий редокс-потен-
циал для системы иод — иодид
12 + 2<? —> 21", £0 = — 0,54а
Реакции окисления образующимися катионами
91
с термодинамической точки зрения обеспечивает повы-
шенную избирательность иода по сравнению с другими
окислителями.
Для реакции между иодом и муравьиной кисло-
той [9]
f
НСООН 4-12 —► СО2 + 2Н+4-2Г
скорость описывается уравнением
Л = Л[НСОО~] [12]/(Г].
Это экспериментально найденное выражение соответ-
ствует двухстадийному механизму реакции:
k 7^ 1+ +1“.
t
I+ + НСОО —► СО2 + Н+ +1“ стадия, определяющая
скорость.
Реакция между 12 и Н2С2О4 протекает в две стадии.
Первая стадия идентична первой стадии окисления
Н2С2О4 бромом. Для второй стадии реакции кинетиче-
ское уравнение имеет вид
-4[1,)/Л«-М(С00-ШЧ'Л
В соответствии с этим вторая реакция протекает сле-
дующим образом:
(СОО-)2 + Ь —► (СОО)2- + Г,
(СОО-)2- 4-1. — 2СО24-Г.
Радикалы I • образуются при гомолитическом разло-
жении:
h 2Ь
Скорость реакции окисления титана (III) иодом [11]
- d [i8-] /at=[Ti1»] [i3]/[н+] [г] 4-
4-л2 [и»1] [i3-]/[н+] 4-л3 [Т1,п] [н+].
Основная доля значения константы скорости реакции
падает на первые два члена, выражающие взаимодей-
ствие между Ti(OH)2+ и Ь и la •
92
Глава 3
Растворы брома с высокой реакционной способно-
стью [12а] можно получить постепенным прибавлением
солей ртути(II) или серебра к раствору брома в воде
или уксусной кислоте в присутствии азотной или серной
кислот. Многие вещества, обычно с трудом присоеди-
няющие бром, легко бромируются в подобных растворах,
содержащих положительные ионы брома.
Бензол (22 лсл; 0,25 моля) и бром (6,5 мл\ 0,125 моля) переме-
шивали с 1 л 2 н. HNO3, после чего в смесь постепенно, в течение
1,5 400,добавляли раствор нитрата серебра (22 а; 0,125 моля в 100 мл
воды). Реакционная смесь находилась в темноте. После удаления
непрореагировавшего брома сульфитом натрия полученный бромбен-
зол экстрагировали эфиром, высушивали и очищали фракциониро-
ванной перегонкой. Выход бромбензола составлял 15 г (80%). Бы-
строе добавление нитрата серебра снижало выход* бромбензола за
счет образования дибромбензола. В отсутствие нитрата серебра ре-
акция не идет.
Исследование кинетики реакции бромирования то-
луол-со-сульфоната натрия [126] дает основание пред-
полагать, что активным бромирующим началом служит
Н2ОВг+.
Константа равновесия реакции образования Н2ОВг+
и реакция замещения выражаются следующим образом:
(Н2ОВг)+ + (C.H.CHjSO,)- —> H,O*+<C.H,Br-CHj-SO,)-.
Для этой реакции получены доказательства участия в
стадии, определяющей скорость, положительно заряжен-
ных частиц. Доказательства участия Н2ОВг+ в стадии,
определяющей скорость, были также получены путем
измерения скорости реакции бромирования бензойной
кислоты при высокой концентрации H2SO4 и HC1O*. Мо-
лекулы воды находятся в активном комплексе.
Рассмотрим реакции брома и бромноватистой кис-
лоты, которые могут (но не обязательно) протекать с
участием катионов брома. Символ Вг+ в наших уравне-
ниях в действительности может иметь форму Н2ВгО+.
Брей и Ливингстон [13] изучили окисление перекиси
водорода бромом по реакции
Н,О,+Br, —> 2Н + 4- 2Вг“ 4-
Реакции окисления образующимися катионами
93
авторы установили, что скорость уменьшения концен-
трации брома описывается уравнением
— d [Вг2]/Л = k [Н2О2] [Вг2]/|Н+] [Вг“],
которому соответствует следующий механизм реакции;
Н2О2 Н+ + НО2“,
Вг2 Т~** Вг+ + Вг”.
НО£"-|-Вг+ —► Н+ Н-Вг” Н-О2 стадия, определяющая скорость.
Под действием перекиси водорода происходит обратная
реакция окисления иона бромида до свободного брома.
Для реакции окисления щавелевой кислоты бромно-
ватистой кислотой [14] по уравнению
t
НС2О4- + НОВг —► 2СО2 + 2Н2О + Вг"
предполагается следующий механизм:
•Н+4-НВгО Вг+4-Н2О
СОО"
Вг+ + I —► Вг + 2СО2 стадия, определяющая скорость.
СОО’
Реакции между этиловым спиртом и бромом [15]
приписываются различные механйзмы. Во всех случаях
имеется стадия непосредственного взаимодействия бро-
ма со спиртом, определяющая скорость всей реакции:
1) СН3СН2ОН-|-Вг2 СН3СН2ОВг + НВг стадия, определяю-
щая скорость
СН3СН2ОВг —► СН3СНО + НВг
2) СН3СН2ОН 4- Вг2 —► СН3СНОН+ +НВг£" стадия, определяю-
щая скорость
СН3СНОН+ —► СН3СНО4-Н+
НВг^ —> 2Вг"+Н+
3) СН3СН2ОН 4-Вг2 —► СН3СНВгОН 4-НВг стадия, определяю-
щая скорость
СНзСНВгОН —► СН3СНО4-НВг
По экспериментальным данным невозможно было ука-
зать истинный механизм из трех возможных. Однако ис-
следование [16] водородного изотопного эффекта при
окислении этанола бромом показало, что скорость
94
Глава 3
окисления этанола-1-/ составляет всего 0,57 скорости
образования этилгипобромит.а и свидетельствует о том,
что реакция должна проходить через стадию, определяю-
щую скорость потери водорода метильной группой.
В аналогичных условиях ацетальдегид окисляется в
4 раза быстрее, чем ацетальдегид-\-D. Это дает ос-
нование предполагать, что окисление протекает через
образование гидрата СН3СН(ОН)2 по механизму, опи-
санному для этанола. При дальнейшем исследовании
дейтериевого изотопного эффекта при окислении альде-
гидов бромом [17] найдено, что изотопный эффект рас-
творителя в случае алифатических альдегидов
составляет 3,7, а в случае бензальдегида 1,3. Первая
реакция, очевидно, протекает через образование гидра-
та, а последняя через образование катиона бензоила
по стадиям:
СвН5СНО + Вг2 медлснн% свН6СО+ 4-Вг“ 4-НВг,
С8Н8СО++ Н2О 6ыстро » С,Н5СООН + Н+
Кинетика замещения водорода галогенами в арома-
тических соединениях в среде ледяной уксусной кислоты
во всех случаях лучше всего интерпретируется как пря-
мое воздействие нейтральных молекул галогенов на мо-
лекулу ароматического соединения [18]. Добавка элек-
тролитов влияет на скорость реакции в направлении,
противоположном ожидаемому действию положитель-
ных ионов.
Окисление диэтилфосфоната бромом происходит по
кинетическому уравнению реакции
где S — окисляемый диэтилфосфонат. Окисление, ката-
лизируемое кислотами, протекает по схеме [19]:
С2н8о о С2н8О ОН
)рС >р\ —>
С2НбО н С2НбО н
С2НбО ОН С2Н6О о
__\р/ быстрое \р^
/ •• окисление / \
С2НВО СцН.0 О“
Реакции окисления образующимися катионами
95
Затем реакция проходит по общей схеме, предложенной
для подобных реакций [20].
• Доказательства существования катионов хлора со-
держатся в работе Дербишайра и Уотерса [12в], кото-
рые исследовали реакцию хлорирования толуол-со-суль-
фоната натрия, используя в качестве катализаторов как
хлорную, так и серную кислоты. Активные катионы
хлора образуются из хлорноватистой кислоты в значи-
тельных количествах при более высокой кислотности,
чем катионы брома из бромноватистой кислоты. Разли-
чие хлорирующей способности ионов С1+ и Н2ОС1+ обна-
ружил Де ла Мар [1в], который показал, что реакция
с хлорноватистой кислотой таких реакционноспособных
соединений, как анизол и фенол, происходит по сле-
дующему кинетическому уравнению:
- d [СЮН]/dt = k [СЮН] + k' [СЮН] [Н+] + k" [СЮН] [Н+] [АгН],
где первый и второй члены описывают стадии, опре-
деляющие скорость и соответствующие распаду СЮН
и СЮН? соответственно, а последний член отражает
скорость взаимодействия органического соединения с
СЮН? при его равновесной концентрации.
Для окисления перекиси водорода хлором предла-
гается следующий механизм [21]:
Н2О2 + С12 7=± Н+4-СГ 4-HOOCL
Hooci —► о24-н+ + сг.
При концентрациях НС1 выше 1 М скорость разложе-
ния HOOCI определяет скорость всей реакции, а при
более низкой кислотности замедленной стадией являет-
ся взаимодействие перекиси водорода с хлором. Если
хлор полностью гидролизован до НС1О, то реакция про-
текает по схеме
нею —► Н+4-СЮ“,
С1СГ + Н2О2—> Н2О + О24-СГ,
где скорость реакции определяется скоростью стадии
взаимодействия перекиси водорода с ионом гипохло-
рита.
96
Глава 3
Ионы оксигалогенов и их источники
В этом разделе обсуждаются доказательства суще-
ствования ионов Ю+, Юг-, ВггО? и Юз- и данные по
равновесиям и кинетике реакций с их участием.
Мэссон и Хэнби [22] установили существование жел-
того сульфата (IO)2SO4, в котором, по-видимому, со-
держится иодозильный катион Ю+. При взаимодействии
этого вещества с нитробензолом в концентрированной
серной кислоте образуется л1-иодозонитробензол с выхо-
дом 50—60%, причём иодозогруппа непосредственно
вводится в ароматическое ядро.
Иодозонитробензол синтезировали следующим образом. Хорошо
растертые иод (4,0 миллимоля) и пятиокись иода (6,0 миллимоля)
механически встряхивали с 96%-ной серной кислотой (20 мл). Через
сутки количественно выделялся желтый твердый ЬОз’БОз, а рас-
твор над ним приобретал бледно-желтую окраску. К этой смеси до-
бавляли из бюретки при встряхивании чистый нитробензол (20 мил
лимолей). По исчезновении твердого вещества (примерно через
1 час) к гомогенной жидкости прибавляли чистый лед. Если полу-
ченный при этом раствор содержал серную кислоту концентрацией
не менее 7 н., выделялись кристаллы сульфата лс-иодозонитробензо-
ла. При дальнейшем разбавлении происходил гидролиз с образова-
нием объемистого осадка аморфного иодозосоединения. Нейтрализа-
ция раствора бикарбонатом натрия приводила к полному осаждению
твердого вещества, которое затем отфильтровывали.
В концентрированной серной кислоте желтый иодо-
зосульфат поглощает элементарный иод с образова-
нием темно-коричневого раствора. При действии этого
раствора на хлорбензол количественно образуется хлор-
трииодбензол, что доказывает присутствие в нем ак-
тивного иодирующего агента. Эти факты объясняются
следующей схемой реакции:
Ю++2Н+ 13+-|-Н2О,
13+ 4-12 31+
1+4-л12 <-21 11++2л»
где п=1 или 2, а катионы I+, I?, Is+ симметричны от-
рицательным ионам Г, 1з", 1Г- Изучение ИК-спектра
поглощения четырехокиси иода [23] показывает, что ее
структуру можно представить как 1О+ • Юз •
Реакции окисления образующимися катионами
97
Существование Ю2 доказывается результатами ки-
нетических исследований. Более ранние работы по это-
му вопросу рассмотрены Морганом [9]. Реакция
Юз + 51" + 6Н + —► 3124-ЗН2О
при высоких концентрациях иодида (>10‘4 Л1) проте-
кает по кинетическому уравнению [24]
- d [IO3-]/di = k [IO3-] [г]2 [Н+Р,
а при низких концентрациях иодида (<10-8 М)—по
уравнению
-d [io3-]/d< =л' [Юз ] [г| [н+|2.
Экспериментальные данные согласуются со схемой, со-
гласно которой Юг+. образующийся в равновесной
реакции
H+-|-IOf Ю2++ОН-,
принимает участие в определяющей скорость стадии,
которой является взаимодействие между ионом Юг и
иодидом. При высоких концентрациях иодида среднее
время жизни комплекса Юг • I- достаточно для того,
чтобы он успел прореагировать с ионами иодида. Поэто-
му скорость реакции описывается первым кинетическим
уравнением, а порядок реакции относительно иодида
приблизительно равен 2. При понижении концентрации
йодида вероятность распада комплекса увеличивается,
так что для реакции в растворах с очень низкой кон-
центрацией иодида преобладает ‘второе кинетическое
уравнение.
Полная схема реакции представляется следующим
образом [9]:
Ю3- + н+ Н1О3 (I)
HIO3+H+ Ю2+ + Н2о (2)
Ю2++2Г —>Г-|-21О' (3)
Ю2++Г — Ю+ + Ю- (4)
Ю~+Н+ —► НЮ (5)
НЮН-Н* —► Н2Ю+ (6)
Н21Оь + 1" —* к + НгО, (7)
7 Зак. 1249
98
Глава 3
где реакция (3) считается определяющей скорость ста-
дией.
Бромат взаимодействует с перекисью водорода по
реакции
ЗН2О2 “I” ВгОд > ЗН2О ЗО2 -1~ Вг
со скоростью [25]
R = k [Н+] [Н2О2] [ВгО3-].
Здесь определяющей скорость стадией может быть либо
реакция между НэО* и ВгОз\ либо реакция между
НО7 и ВгОз Чармиан О’Коннор (личное сообщение)
экспериментально с помощью 18О показала, что весь
выделяющийся при реакции кислород освобождается из
молекул перекиси водорода. Отсюда следует возможная
схема реакции:
t
ВгО^ + НО2 —► НВгО2Н-О2 стадия, определяющая скорость
НВгО24-Н+ —> ВгО+-|-Н2О
t
ВгО4+НО2“ —> НВгОН-О2
НВгОН-Н+ —► Вг++Н2О
Вг+ + НО2" —► Н+4-Вг“ + О2
Бантон и Левеллин [26] изучили разложение переки-
си водорода в присутствии Ag, MnO2, Fe111, CeIV и др.
в водных растворах, причем применялась вода, обога-
щенная изотопом 18О. Они показали, что атомарный кис-
лород во всех случаях выделяется из молекул пере-
киси водорода, что свидетельствует о прочности связи
—О—О— в молекуле перекиси водорода. Атом кисло-
рода между молекулами Н20 и Н2О2 не обменивается.
Представляет интерес и другая реакция бромата —
изотопный обмен с меченой водой. Кинетика этой реак-
ции выражается уравнением [27]
Я = й[Н + ]2 [ВгО3-]
Реакции окисления образующимися катионами
Ж
и соответствует следующему механизму обмена;
ВгО3" + Н+ НВгО3,
НВгО34-Н+ ВгО2+ 4- Н2О.
Реакция обмена с D2O происходит быстрее (&d/&h =
= 1,72).
Скорость выделения кислорода при взаимодействии
перекиси водорода и иодистой кислоты [28] в определен-
ных условиях периодически изменяется со временем.
Периодичность является результатом влияния двух
факторов: аутокаталитической природы реакции и фи-
зического удаления из сферы реакции образующегося
кислорода. Для этой реакции предлагается следующая
схема:
НЮ3 ю24 +ОН-,
н2о2+ю2+ —’•> н2о+О2+ю+.
Н2о2+Ю+ — -> Н2О + О2+1+,
H2o2+I+ —2Н * + О2 + 1“,
Н+ + ОН~ ---> Н2О,
Г +1- — > 1„
[ю2+] = К[н+|2[ю3-]/^,
[Ю+] = *2*[Н+]2 ik.Kv,
[1+| = *2Ин+|2[ю3]К*4.
Если в выражение скорости реакции
d \O^dt = 2*2 [Н2О2] [Ю2+] +2*3 [Н2О2] |Ю+1 + *< [Н2О2] fl+]
подставить значения концентраций, то получим
d [О2|/<« = *2К fH+12 [Ю3-] [Н202)/Кда.
Реакция имеет приблизительно второй порядок отно-
сительно ионов водорода и приблизительно первый по-
рядок относительно ионов йодата и перекиси водорода.
7*
100
Глава 3
Скорость реакции
(^[°2]МРедн = МН + |2 [Юз] [Н2О2].
Доказательства существования катионов Юз осно-
ваны на немногих данных, которые указывают на воз-
можность существования форм НгЮ^ или 1(ОН)+
Абель и Зибеншайн [30] нашли, что скорость реакции
между иодной кислотой и иодидом в кислом растворе
описывается уравнением
я=л[ю4-] [г][н+]2.
Эти данные можно интерпретировать [29] следующей
схемой реакции, приводящей к образованию йодата:
Ю4 +н+ ню4,
ню4н-н+ 5=* н2ю^,
н2ю4+ + г н2ю4+1,
н2ю4 —> Ю3+Н2О,
Г + Ю3 ю3- + 1.
Водные растворы перйодатов иодной кислоты сильно
поглощают свет в ультрафиолетовой области, а раствор
иодной кислоты в концентрированной серной кислоте не
обнаруживает такого сильного поглощения. Мишра и
Симонс [31] считают, что спектрофотометрические дан-
ные являются достаточными доказательствами суще*
ствования иона 1(ОН)^ в концентрированных растворах
серной кислоты.
Перекись водорода
Реакции окисления перекисью водорода привлекают
к себе внимание вследствие существования равновесий
Н2О2 + Н+ Н3О2+, (1)
н2о2+.н+ :р± но+ + н2о. (2)
Чармиан О’Коннор (частное сообщение) показала, что
обмен водорода перекиси с Н2,8О в 50%-ной H2SO4 при
Реакции окисления образующимися катионами
101
55,5° происходит всего лишь на 2% за 49 час. Это дает
основание считать, что реакции с перекисью водорода
в кислом растворе, которые происходят очень быстро,
например с иодидом, должны быть представлены сле-
дующим образом:
НЧНО ОН^ н3о+,
Г 4-—► Н2О1+ + ОН_ стадия, определяющая скорость
Н2ОГ 4-Г —> H2O-hI2.
Реакция с водой вместо иодида во второй стадии при-
водит к невероятному переходному состоянию. Обмен
поэтому не происходит. Ивенс и Ури [32] для реакции
Н3О4 + Н*О2 Н2О + Н3О+
определили порядок константы равновесия, который ока-
зался равным 10~3. Дербишайр и Уотерс [33] установили,
что в 10 н. кислоте конверсия перекиси водорода в НзО?
составляет около 1%. Ими показано, что при взаимодей-
ствии мезитилена с перекисью водорода в серной кис-
лоте основным продуктом является мезитол. Аналогич-
ные доказательства приводят к выводу о том, что пере-
кись водорода предотвращает образование RO+ при
гетеролитическом распаде органических перекисей [34].
Детальный обзор реакций органических перекисей опу-
бликован Дэвисом [34]. поэтому в этой книге они не
рассматриваются.
При взаимодействии перекиси водорода с С10Г, ЮГ,
МпОЛ Fe11, Fe111, CeIV, NO2 и NOi” происходит инду-
цированный изотопный обмен между перекисью водо-
рода и водой [35]. Изотопный обмен с водой облегчается
образованием перекисных комплексов типа ХООН по
схеме:
ХОН + НООН ХООН4-Н2О.
хоонч-н2д нодн+хон.
При реакции в азотной кислоте предполагается образо-
вание промежуточного продукта N2O5 [36]:
N2O64-HOOH —► HNO3+O2NOOH.
OBNOOH + над —► OeNdlI4-HOOH.
102
Глава 3
Наиболее полно изучено взаимодействие перекиси во-
дорода с иодидом. Вайс [29] на основании всестороннего
анализа этой реакции приписывает ей следующий меха-.
низм:
н2о2 + г — -> 1 + ОН“+ОН, (1)
Н3О2+ + Г - -> 1+Н2О + ОН, (2)
Г + ОН — -> I + OH" (3)
±1 1+ + г, (4)
НО2- + 1+ —> НО2—|—I. (5)
НО2 + Н2О2 —> О24-Н2О + ОН. (6)
Считается, что реакция (2) идет только в сильной кис-
лоте, тогда как реакция (6), вероятно, проходит через
образование иона OF. Кинетические уравнения имеют
вид
d [O2]/<tt = Л2Л3 [12] [НО2-]/*' [Г] + *3 [НО2-]
и
d [IJ/Л = [Н2О2] [Г] — '/2 d [O2]/dt.
В кислом растворе образуется только иод, выделения
кислорода не происходит; следовательно, пренебрегая
параллельно идущими реакциями с кислотным катали-
зом, уравнение можно представить в виде
d[l2]/dt = kt [Н2О2] [Г].
В нейтральном растворе протекает чисто каталитическая
реакция
d [l2)/<ft = 0,
d[O2]/dt=2kt [Н2О2] [Г].
Кинетическое уравнение
d[I2]/dt = kt [Н2о2] [Г]
соответствует следующей схеме реакции:
Н2О2-|-1~ —► Н20 + Ю" стадия, определяющая скорость
IO" +1- + 2Н+ —► 12-|-Н2О быстрая стадия.
Кинетика реакции перекиси водорода с иодидом при
концентрациях ионов водорода более 10~2 М описывается
Реакции окисления образующимися катионами
103
уравнением
<П12]/л = л' [Нэо7] [гЦн+].
Этому уравнению соответствует механизм реакции, рас-
смотренный выше.
Брей и Либхевский [37] дали ценное обобщение дан-
ных о реакции перекиси водорода с иодидом, иодом и
йодатом при различных pH.
Реакция окисления бромида, катализируемая кисло-
тами, выражается кинетическим уравнением [38]
— d [Н2О2]/^ = k [Н2О2] [Н Ч [Вг"].
Эта реакция аналогична рассмотренной выше реакции
с иодом.
Гальперин и Таубе [39] исследовали реакцию
H2SO3 + H2O2 —> H2so4 + H2o
при pH 1-5 и нашли, что образующийся сульфат содер-
жит два меченых атома кислорода из На этом
основании высказано предположение относительно су-
ществования промежуточного продукта пероксисерни-
* *
стой кислоты НО—S(O)—ООН, которая подвергается
внутримолекулярной перегруппировке с образованием
серной кислоты. При окислении щавелевой кислоты пе-
рекисью водорода [40] примерно 2/3 атомов кислорода
в образующемся СО2 поступает из окислителя. Скорость
реакции определяется, по-видимому, стадией замещения
группы —ОН на группу —ООН.
Азотистая кислота
Рассмотрение этих реакций послужило темой обзора
Терни и Райта [41а]. Азотистую кислоту можно рассма-
тривать как гидроксильный носитель иона нитрозония,
образующегося в кислом растворе по равновесной реак-
ции
HONO + Н+ NO+ + Н2О. (1)
Если обозначить как Х~ другое основание (С1~, Вг~,
NO3‘ и т. д.), равновесие приобретает форму
HONO + Н+ + X" NOX + Н2О. (2)
104
Глава 3
Особый интерес представляет случай, когда X' является
NOjT. Тогда равновесие реакции принимает вид
2HONO N2O3 + H2O. (3)
Величины некоторых констант равновесий определены
с достаточной степенью достоверности. Так, для равно-
весия (1) в среде хлорной кислоты при 25° имеет место
соотношение [416]
log С /С .=74- 8,20.
° hono/ no+
Константа равновесия (3) при 20° равна 0,20±0,05 [42].
Знание величины этих констант полезно в том отноше-
нии, что дает возможность рассчитать отношение
[N2O3]/[NO+J, важное для оценки реакционной способно-
сти азотистой кислоты.
Кинетические данные убедительно доказывают суще-
ствование иона нитрозония главным образом в форме
H2NO2 . Лидстон [43] считает, что ион H2NO? более ста-
билен по сравнению с ионом H2NO3» поскольку имеет
большее число резонансных форм.
Азотистая кислота легко вступает в реакцию с угле-
родным, азотным и кислородным центрами молекул, во-
дород замещается при этом нитрозогруппой так же, как
при окислении. Все реакции этого типа делятся на две
группы:
1) Реакции, скорость которых выражается уравне-
нием
R = k [окисляемое вещество] [HONO]2.
Реакции проходят две стадии:
2HONO N2O3+H2O,
окисляемое вещество + N2O3 —► продукт.
Если окисляемое вещество имеет достаточную реакцион-
ную способность, то N2O3 так же быстро расходуется во
второй стадии, как образуется в первой стадии. Тогда
уравнение скорости реакции принимает вид
R = k' [HONO]2.
2) Реакции, скорость которых описывается уравнением
R = k" [окисляемое вещество] [Н * j [HONO].
Реакции окисления образующимися катионами
105
Механизм реакции состоит из двух стадий:
HONOH-H+ h2no2+,
окисляемое вещество + H-jNO.^ —► продукт.
В этом случае оказалось невозможным определить за-
медленную стадию со скоростью
R = k"' [HONO] [н+],
которая имела бы место, если бы в качестве реакцион-
носпособного промежуточного продукта выступал ион
NO+.
Другие доказательства существования H2NO? как
промежуточного иона получены при изучении реакции
нитрозирования фенола [44]. Имеет место равновесие
HONO + Н+ # H2NO^ Ион представляет собой
нитрозирующий агент. Изучалось [45] взаимодействие
различных ароматических аминов и N2O3i При этом
было показано, что относительная реакционная способ-
ность N2O3 по сравнению с ионом H2NO^ повышается
с увеличением силы реагирующего основания (амина).
В работе Терни и Райта [41а] рассмотрено большое
число реакций окисления азотистой кислотой, например,
(SO3)2NO2', H2AsO3", sol’ S2O3- C2O42’, HCOO’,
SOeNHr, I’.ClOs’. Fe2+ При окислении иона (SOs^NO2-
обнаружена медленная стадия образования N2O3; ско-
рость реакции выражается как /? = fe[HONO]2. Скорость
окисления иона Н2АбОГ также ограничена величиной
равновесной концентрации N2O3. Окисление остальных
соединений описывается кинетическим уравнением:
R = Л' [red] [HONO] [Н+] (red — восстановитель),
что служит указанием на то, что действующим началом
является равновесная концентрация ионов TbNO^. Воз-
можно, при более полном исследовании всех этих реак-
ций удастся обнаружить обе формы кинетики. Приме-
ром может служить глубоко изученная реакция между
азотистой кислотой и аскорбиновой кислотой [46].
Эта реакция при pH 0—5 протекает по следующему
106
Глава 3
стехиометрическому уравнению:
аскорбиновая кислота H-2HONO —>
—> дигидроаскорбиновая кислота -f- 2NO + 2Н2О.
В среде 0,1—0,5 М НС1О4 окисление происходит со ско-
ростью
Я = А>3[Н + ] [HONO] [red0],
а в кислоте с концентрацией 0,5—2,0 М скорость реак-
ции следует функции кислотности Гамметта, HQ, что ука-
зывает на НгМ(Х как на активное начало. При pH 2
уравнение скорости реакции имеет форму
R = k'3 [HONO]2 [red0],
что свидетельствует о воздействии равновесной концен-
трации N2O3. При pH 4 скорость реакции не зависит от
концентрации аскорбиновой кислоты:
R = k2 [HONO]2.
Хорошо исследована реакция окисления азотистоводо-
родной кислоты азотистой кислотой [47, 48]:
HN34-HONO —► n2o + n2 + h2o.
При избытке хлорной кислоты при 0° указанная ре-
акция протекает через стадию нитрозилазида, который
может образовываться двумя путями: во-первых, нуклео-
фильным воздействием азида на N2O3
n3-+n2o3-^ non3+no2-
и, во-вторых, нуклеофильным воздействием азотистово-
дородной кислоты на ион H2NO^
HN3 + H2NO2+ —> NON3+H3O+
В обоих случаях образовавшийся нитрозилазид в даль-
нейшем распадается на закись азота и азот:
NON3 —> N2O + N2.
При большом избытке буферного раствора, состоящего
из азида натрия и азотистоводородной кислоты, ско-
Реакции окисления образующимися катионами
107
рость реакции определяется стадией
N- + H2NOj ;—> NON34-H2O.
Добавки хлорида, бромида и тиоцианата катализируют
другую реакцию:
N-4-NOX —> NON3+X“.
Опыты с меченной 18О водой показывают, что в водном
растворе нитрозилазид не гидролизуется, а полностью
распадается на азот и закись азота.
В ацетатной буферной смеси вначале идет реакция
CH3COO"4-H2NO2+ —► CH3COONO + H2O,
а затем или
CH3COONO + Nf —> СН3СОО~ + NONg,
или
CH3COONO 4- NO2- —> CH3COO“+N2O3.
Затем идет реакция
n2o3 + n3- —> non3+no2-.
Во всех случаях NON3 немедленно подвергается гидро*
лизу.
Азотная кислота
Азотная кислота — кислота средней силы. В раство-
рах умеренной концентрации содержится много недис-
социированных молекул HNO3. При нитровании пара-
финов, которое в основном происходит по гомолитиче-
скому пути
HONO2 —► НО + . NO2,
используется азотная кислота средней концентрации. На-
против, титрование ароматических соединений концен-
трированной азотной кислотой идет по гетеролитиче-
скому пути:
HONO2 —► НО“ +NOJ.
В то время как нитрование ароматических соединений
ионами нитрония исследовано подробно [26], работ по
108
Глава 3
гетеролитическому окислению азотной кислотой выпол-
нено значительно меньше.
Экспериментальные данные по процессам окисления
азотной кислотой укладываются в следующую схему
реакции:
2HNO3 H2NO^ + NO3" H2O+NO^4-NO3,
где главным реакционным началом служит ион нитро-
ния [1г]. Хотя прямые доказательства существования
иона H2NO3 отсутствуют, однако влияние нитрата на
скорость реакции нитрования свидетельствует о возмож-
ности существования иона H2NO3 Кроме того, скорость
реакции лучше следует функции кислотности /0, чем
функции Но [49—51], как это можно было бы ожидать
при нитровании ионом H2NO3
При определенных условиях реакция нитрования
имеет нулевой порядок, что соответствует определяю-
щей скорость стадии
H2NO3+ —► NO++до-
вода мало влияет на скорость этой реакции [2а]. В не-
которых работах окисление азотной кислотой осложня-
лось присутствием азотистой кислоты [52, 53]. Окисление
иона Сеш азотной кислотой протекает обратимо:
CeIH + Н3О + + HNO3 CeIVH-NO24-2H2O.
Кинетика окисления Сеш в 12—16 н. HNO3 при 90—110°
была изучена путем улавливания летучих окислов азота,
вытесняемых инертным газом; стадией, определяющей
скорость реакции [54], является
CeIH + NO2+ —> CeIV + NO2.
ЛИТЕРАТУРА
1. De la М а г е Р. В. D., R i d d J. H., Aromatic Substitution, Butter-
worth, London, 1959, (a) p. 35; (6) p. 120; (в) p. 116; (r) p. 58.
2. Ingold С. K-, Structure and Mechanism in Organic Chemistry,
Cornell University Press, 1953, (a) p. 288; (6) p 269; (в) p.
275; см. общую литературу к Введению.
3. Anbar М., Ginsburg D., Chem Rev., 54, 925 (1954).
4. В e 11 R. P., G e 11 s E., J. chem. Soc., 2734 (1951).
Реакции окисления образующимися катионами
109
5 Kleinberg J. (Ed.), Inorg. Synth., 7, 168 (1963).
6. Symons M. C. R., J. chem. Soc., 387, 2186 (1957).
7. А г о t s к у J., Mishra H. C., Symons M C. R., J. chem. Soc.,
12 (1961).
8. Barker I. R. L., Waters W. A., J. chem. Soc., 150 (1952).
9. Morgan K. 1. Quart. Rev., 8, 123 (1954)
10. Koi th off I. M., Belcher R., Volumetric Analysis, Vol. 3,
Interscience, New York, 1957, p. 199.
11. Johnson С. E., Win stein S., J. Am. chem. Soc., 73, 2601
(1951).
12. Derbyshire D. H., Waters W. A., J. chem. Soc., (a) p. 573,
(1950); (6) p. 564 (1950); (в) p. 73 (1951).
13. Bray W. C., Livingstone R S., J. Am. chem. Soc., 45, 1251
(1923).
14. Griffith R. O., McKeown A., Winn A. G., Trans. Faraday
Soc., 28, 107 (1932).
15. Farkas L., Perlmutter B., Schachter O., J. Am. chem.
Soc., 71, 2829 (1949).
16 К a p 1 a n L., J. Am. chem. Soc., 76, 4645 (1954).
17. McTigue P. T., Sime J. M., J. chem. Soc., 1303 (1963).
18. Robertson P. W, J. chem. Soc., 1267 (1954).
19. Silver B., Ljiz Z., J. Am. chem. Soc., 84, 1091 (1962).
20. Doak G. O., Freedman L. D., Chem. Rev., 61, 31 (1961).
21. Connick R. E., J. Am. chem. Soc., 69, 1509 (1947).
22 Masson I., Ha nd by W. E., J. chem. Soc., 1699 (1938).
23. W i s e J. H., Hannan H. H., J. inorg. nucl. Chem., 23, 31
(1961).
24. M о r g a n K. J., P e a r d M. G., C u 11 i s C. F., J. chem. Soc ,
1865 (1951).
25. Y о u n g H. A., J. Am. chem. Soc., 72, 3310 (1950).
26. Bunton C. A., Llewellyn D. R., Research (London), 5, 142
(1952).
27. H о e г i n g T. C., Butler R. C., McDonald H. O., J. Am.
chem. Soc., 78, 4829 (1956).
28. P e a r d M. G., Cull is C. F., Trans. Faraday Soc., 47, 616
(1951).
29. Weiss J., Rep. Progr. Chem., 44, 78 (1947).
30. Abel E., Sieben schein R., Z. phys. Chem., 130, 631 (1928).
31 Mishra H. C., S у m о n s M. C. R., J. chem. Soc., 1194 (1962).
32. Evans M. G., Uri N., Trans. Faraday Soc., 45, 224 (1949).
33. Derbyshire D. H., Waters W. A., Nature, Lond., 165, 401
(1950).
34. Davies A. G., Organic Peroxides, Butterworth, London, 1961,
p. 128.
35. Anbar M„ J. Am. chem. Soc., 83, 2031 (1961).
36. Anbar M., Guttman S., J. Am chem. Soc., 83, 2035 (1961)
37 Bray W C., L i e b h a f s к у H. A., J. Am. chem. Soc., 53, 38
(1931).
38 Livingstone R. S., J. Am. chem. Soc., 48, 53 (1926).
39. H a 1 p e г i n J., T a и b e H J. Am chem. Soc.. 74, 380 (1952)
40 Milburn R. M., T a u b e H., J. Am. chem. Soc., 81, 3515 (1959),
I 10
Глава 3
41. Turney 1 A., W r i g h t G. A. (a) Chem. Rev., 59, 497 (1959);
(6) J chem Soc., 2415 (1958)
42. T u г п e у T A., J. chem. Soc., 4263 (1960)
43 Lidstone A. G., Chem & Ind., 1316 (1959).
44. Morrison D. A.. Turney T. A., J chem. Soc., 4827 (1960).
45. S c h m i d H., E s s 1 e r C , Mh Chem., 91, 484 (1960).
46. Dahn H., Loewe L., Helv chim Acta, 43, 287 (1960).
47 Stedman G, J. chem. Soc 2943, 2949 (1959); 1702 (1960).
48 Bunton C. A., Stedman G., J. chem. Soc., 3466 (1959).
49. Westheimer F. H., Kharasch K. S., J. Am. chem. Soc., 68,
1871 (1946).
50. Lowen A. M., Murray M. A., Williams G., J. chem. Soc.,
3318 (1950).
51. Den о N. C., Stein R., J. Am. chem. Soc., 78, 578 (1956).
52. Longstaff J. V. L., Singer K., J. chem. Soc., 2610 (1954).
53. L о n g s t a f f J. V. L., J. chem. Soc., 3488 (1957).
54. Johnston R. W., Martin D. S., J. inorg. nucl. Chem., 10, 94
(1959).
Глава 4
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
МЕЖДУ АНИОНАМИ, АНИОНАМИ И КОМПЛЕКСАМИ,
МЕЖДУ АНИОННЫМИ КОМПЛЕКСАМИ
В этой главе рассматриваются реакции переноса
электронов с участием анионов и комплексов. Реакции,
разобранные в предыдущих главах, протекают только
в кислых растворах, так как повышение pH вызывает
осаждение катионов в виде гидроокисей и таким обра-
зом препятствует реакции. В случае слабых кислот по-
вышение pH приводит к появлению аниона кислоты, что
опять-таки исключает возможность катионного пути ре-
акции. Если, однако, исследуемый катион перевести в
прочный комплекс, то можно реакции с переносом элек-
тронов осуществлять в более щелочных растворах без
осаждения гидроокисей металлов. Аналогично в щелоч-
ных растворах может происходить перенос электронов
между анионами и между анионами и комплексами. Ре-
акции с переносом электронов между анионами или
комплексными ионами и нейтральными молекулами ис-
ходного окисляемого вещества будут рассмотрены в
гл. 5.
Реакции между комплексными ионами
Такие реакции интересны тем, что, несмотря на до-
статочно прочную с термодинамической точки зрения
связь металла с комплексообразующим агентом, ско-
рость обмена электронами может быть сравнительно вы-
сокой. Так, обмен электронами в реакции [1]
Fe(CN)64- 4- Fe(CN)3- —+ Fe(CN)|~ + Fe(CN)3~
происходит co скоростью 7,4-IO2 м3 • кмоль~х • сект* при
25°, т. е. более высокой, чем в случае обмена между
простыми ионами Fe2+—Fe3+, & = 4f0 м3 • кмоль~1 • сек~1
112
Глава 4
[2]. Этот результат особенно интересен, поскольку имеет
место также быстрый обмен простых ионов цианида
с ионами цианида в комплексах [3].
Возможные причины различной скорости обмена
электронами между «простыми» и комплексными ионами
рассмотрены в работе Либби [4]. В случае малых по раз-
меру ионов, имеющих в первичной координационной
сфере молекулы растворителя, для переноса электронов
сначала требуется значительная переориентировка рас-
творителя, что выражается в большой величине энерге-
тического барьера. В то же время структуры таких
комплексов, как цианидные комплексы железа (II) и же-
леза (III), настолько сходны между собой, что необхо-
дима лишь небольшая перестройка координационных
сфер, поэтому перенос электрона может происходить
очень быстро.
Более простое объяснение заключается в том, что,
когда молекула воды находится между двумя анионами,
лабильность гидроксила воды сильно возрастает и пере-
нос электрона легко осуществляется через мостик, ко-
торым служит ион водорода. Для простых ионов водо-
род воды исключается и в качестве мостика для пере-
носа электронов выступают ионы гидроксила. Поскольку
произведение зарядов ионов в случае ферро- и ферри-
цианидов значительно больше (zaZb=12), чем в случае
простых ферро- и ферри-ионов (zazb = 6), скорость пере-
носа электронов также значителньо выше в первом
случае.
Шеппард и Воль [5] показали, что обмен в системе
манганат — перманганат происходит со скоростью /? =
= £[МпОГ] [МпОМ, константа скорости реакции соста-
вляет ft = 710±30 м? • кмоль~х • секгх при 0°, а энтропия
активации AS* = —3,78 • 104 дж • к,моль~х • град~х. Ско-
рость реакции возрастает с увеличением концентрации
электролита, причем очень большое влияние оказывает
природа катионов и очень малое — природа анионов.
При постоянной концентрации электролита
^CsOH > ^KOH > ^NaOH = ^LlOH*
Специфическое явление катализа ионами цезия было
изучено более детально. Обнаружено, что зависимость
Реакции окисления — восстановления между анионами 113
скорости реакции от концентрации ионов цезия выра
жается уравнением [6]
Я= [MnOj"] [МпО4-] {*0 + *' |Cs+]],
где £о=71О±ЗО ж3 •/смоль-1 • сек-1, £'= 12000±3000 л«6Х
Хкмоль~2 • сек~'. Такой заметный катализ ионами цезия
указывает на возможность существования катионного
мостика. Дополнительные данные по механизму пере-
носа в этом интересном случае получены Волем [7]. Ско-
рость переноса электрона в системе ферроцианид — фер-
рицианид (10"4 М каждого) в 0,01 М растворе гидро-
окиси тетрафениларсония примерно в 10 раз меньше,
чем в 0,01 М растворе КОН. Уменьшение концентрации
последнего до 0,0011 М приводит к падению скорости
в 8—9 раз, тогда как природа аниона не влияет на ско-
рость реакции.
Другой вид переноса электрона через мостик обна-
ружен в реакции кобальт(II)этилендиаминтетраацетата
с феррицианидом [8]:
Co(EDTA)2" + Fe(CN)|" —> Co(EDTA)“ + Fe(CN)g".
Эта реакция, вероятно, протекает в две стадии по схеме:
Co(EDTA)2- -К Fe(CN)|" [Co(EDTA)Fe(CN)6]5-,
[Co(EDT A)Fe(CN)6]®- Co(EDTA)- + Fe(CN)<".
Для реакции переноса электрона
Fe(DMP)2+ + 1гС1|“ Fe(DMP)f+ + IrClg"
(DMP = 4, 7-диметил-1,10-фенантролин) найденные кон-
станты скорости составляют £=1,1*109 м3*кмолъ~^сек~{
и£ = 4«1013 е”6000/лт м3- кмоль~' -сек~'. Таким образом,
здесь наблюдается наибольшая скорость переноса элек-
тронов между комплексами металлов [9].
Реакции между простыми анионами и комплексными
анионами
Примерами реакций этого типа является изученная
Адамсоном [10] система
2Fe(CN)|" +21" —> 2Fe(CN4" +1,. 8
8 Зак. 1249
114
Глава 4
Реакция имеет приблизительно первый порядок относи-
тельно феррицианида и между первым и вторым — отно-
сительно иодида. Ее скорость увеличивается в кислых
растворах и уменьшается при добавлении ферроцианида.
Предполагается двухстадийный механизм процесса:
Fe(CN)|-+2r 5=± Fe(CN)g“ + 12",
12" + Fe(CN)|- FefCN^ + k
Реакция катализируется платиной, что, по-видимому,
объясняется переносом электронов через металл [11].
Реакции между анионами
Хотя теоретически возможны реакции между анио-
нами в чистом виде, однако зависимость таких реакций
от pH позволяет считать, что основным окислительным
началом служит слабая кислота или образующиеся из
нее частицы. В таких случаях реакции оказываются ана-
логичными тем, которые обсуждались в гл. 3.
Рассмотрим несколько примеров реакций окисления
между анионами. Хотя по таблице редокс-потенциалов
в щелочных растворах теоретически возможны многие
другие реакции, ограничимся рассмотрением следую-
щих:
1) реакции перманганата в щелочной среде;
2) реакции анионов кислородсодержащих галогенид-
ных кислот;
3) реакции анионов кислородсодержащих кислот
серы;
4) реакции перекиси водорода в щелочной среде.
Реакция перманганата в щелочной среде
Обмен в системе манганат — перманганат уже рас-
сматривался. Обстоятельно исследовалась [12] реакция
разложения перманганата в щелочном растворе
2 f
4МпО4" 4-4ОН- —> 4MnO^4-2H?O + Oj.
Реакции окисления — восстановления между анионами 115
На основании полученных кинетических результатов
можно считать, что вначале примерно 65% перманганата
разлагаются по следующей схеме:
МпО4-+ОН- ^=> МпО1" + 'он- (1)
• ОН + ОН" • О“+Н2О. (2)
МпО4-+ОН‘+ .0- МпО^- 4- НО<7, (3)
МпО^ + Н02- МпО|- + Н02 •, (4)
но2. +0Н" О2 + н2о, (5)
МпО^ + о2- —> t MnO^+Oj. (6)
Опыты с применением в качестве растворителя воды,
обогащенной 18О, показывают, что выделяющийся при
реакции кислород имеет тот же процент обогащения изо-
топом, что и вода. На этом основании был сделан вы-
вод, что весь выделяющийся кислород поступает из рас-
творителя, что соответствует указанной выше схеме.
Щелочные растворы перманганата применяются для
количественного определения оксикислот [13]. При окис-
лении перманганат переходит в манганат, причем даль-
нейшее окисление образовавшимся манганатом устра-
няют ионами бария (образуется малорастворимый
ВаМпО4).
Детально иссследована также реакция окисления
формиата
НО" + НСОО" +2МпО4- —► СО2+ Н2О + 2МпО^.
Хилл и Томкине [14] предложили следующую схему ре-
акции:
t
MnO4“ + НСОО" —> МпОз + ОН- + СО2
2МпОз + Н2О —> МпО2 + НМпО4- + ОН ’
НМпО4“+НСОО- —> MnO2 + СО2 4-2ОН"
медленная (ли-
митирующая)
стадия.
быстрая стадия.
быстрая стадия.
8*
116
Глава 4
Уиберг и Стюарт [15] показали, что скорость реакции не
зависит от pH среды, и обнаружили дейтериевый изо-
топный эффект, равный 7,4. Более третьей части кисло-
рода, входящего в состав продукта, поступает из окисли-
теля.
Предполагается, что реакция происходит по схеме
О О"
_Н _ I
о—С—Н 4- МпО4" О—С—Н —► НСО^ + МпОз .
ОМпО3
Вначале реакция имеет первый порядок относительно
каждого реактанта. Однако через некоторое время по-
рядок реакции по перманганату становится равным 2,
что объясняется, по-видимому, прямым взаимодействием
окисляемого вещества с перманганатом [16].
Цианид окисляется до цианата в щелочном растворе
количественно по уравнению [17]
2МпО4" + CN" + 2ОН" —► 2МпО4“ + CNCT + Н2О.
В области концентраций около 0,07 М кинетика реак-
ции описывается уравнением
— d [МпО4“]/^ = k [MnO4-] [CN“],
которое соответствует следующей стадии, определяющей
скорость реакции
CN" + МпО4" —► CNO" + МпО£\
Реакции кислородсодержащих галогенидных анионов
Реакция
С1О4" 4-8Вг“ 4-8Н+ —► СГ+4Вг24-4Н2О
катализируется солями рутения [18]. Скорость ее в пер-
вом приближении пропорциональна концентрациям пер-
хлората и иона Ruln и значительно повышается с уве-
личением концентрации бромида и кислоты. Выделяю-
щийся бром медленно переводит трехвалентный рутений
в четырехвалентный. Удаление свободного брома током
Реакции окисления — восстановления между анионами -117
С02 способствуем увеличению скорости реакции. Трех-
валентный рутений оказывает значительно большее влия-
ние на скорость реакции, чем четырехвалентный.
При pH 10—13 гипохлорит количественно окисляет
бромид до гипобромита [19]. Эта реакция имеет второй
порядок, причем константа скорости реакции приблизи-
тельно пропорциональна концентрации ионов водорода
в растворе. Лимитирующей стадией следует считать ну-
клеофильную атаку иона бромида на молекулу хлорно-
ватистой кислоты
Вг"+НОС1 —> НОВг + СГ
Скорость реакции гипохлорита с цианатом в щелоч-
ном растворе [20] пропорциональна концентрациям циа-
ната, гипохлорита и ионов водорода. Скорость этой ре-
акции предположительно определяется скоростью ста-
дии
CNO"+HOC1 —> HOCNO4-CF
после которой HOCNO распадается на карбонат и азот.
Такой же механизм имеет место при окислении фор-
миата гипохлоритом [21а]:
СЮ“ + НСОО" +НО” —> СГ +СО|" + Н2О.
Скорость этой реакции /? = &[НС1О][НСОО~]. Это кинети-
ческое уравнение соответствует стадии, определяющей
скорость:
НСОСГ + НСЮ —> СГ 4- Н2СО8.
Для быстро протекающих реакций гипохлорита с
сульфитом и иодидом [216] определяют скорость реакции
следующие наименее быстрые стадии:
cio"+so|- —> cr+so|-,
Г -J-HOCI —> НО1+СГ
При исследовании взаимодействия ионов гипохлорита
(или гипобромита) и нитрита [22] обнаружен почти пол-
ный перенос кислорода от гипогалогенита к нитриту
ХО“ + NO^- —> NO^ 4“ X где X — галоид.
Окисление нитрита гипохлоритом изучено детально
[21 в]. Фактически в реакции участвует хлорноватистая
118
Глава 4
кислота; замедленной стадией является реакция
NO^ + HOCl —> HONO2 + C1' —► H+4-NO3"+Cl“
Ионы йодата окисляются ионом гипохлорита
СЮ" +107 —► C14-IO7.
Реакция
Г + С10" —^Ю~+СГ
протекает по кинетическому уравнению [23]
d [\O~]ldt = k [Г] [C10"| [H+|.
Скорость реакции определяется замедленной стадией
Г + С10Н —► I0H+C1"
Представляет значительный интерес реакция разло-
жения гипоиодит-иона:
310' —► 10^ + 21"
Согласно Моргану [24], различными исследователями
для этой реакции предложено пять различных кинетиче-
ских уравнений:
— d [XO“]/tZ/= [Х“] [ХО"]2/[ОН~], (1)
= МНХОР[Х”]/[ОН-], (2)
= k3 [НХО]2 [Х0~], (3)
= МНХ0] [Х0“], (4)
= ks (Х0~]2. (5)
Из них только уравнение (2) вызывает сомнения.
Считается, что главное промежуточное соединение
1зОГ образуется по реакции
ЗНЮ + ОН' —► 13О2“ +2Н2О.
Райт [25] показал, что это соединение можно рассматри-
вать как комбинацию 10^ с двумя ионами иодида, тем
более что соответствующий фторид IO2F7 получен в
твердом состоянии. В качестве промежуточного продукта
предлагается также HO2I3. На этом основании в статье
Моргана разбираются различные схемы механизма ре-
акции, отличающиеся большой сложностью.
Реакции окисления — восстановления между анионами 119
При изотопном обмене брома между ионами гипо-
бромита и бромида в среде концентрированной щелочи
реакция протекает с участием в активном комплексе
бромноватистой кислоты и одного атома брома по схеме
[26]
НОВг + Вг" НОЙг ^=> Вг"4-НОВг
I
Вг
Для реакции между перйодатом и манганатом в ще-
лочном растворе [27] найдено сложное выражение ско-
рости, которому соответствует следующий механизм:
2МпО^“ МпО4~ + МпО?-,
«2
МпО4“ + Н3Ю|- МпО4" + IO£ +ЗОН-.
Добавление перманганата замедляет реакцию, сдвигая
равновесие первой стадии влево.
Скорость реакции окисления манганата до перманга-
ната гипохлоритом натрия пропорциональна квадрату
концентрации ионов манганата и первой степени кон-
центрации ионов гипохлорита и обратно пропорцио-
нальна концентрации ионов перманганата и квадрату
концентрации гидроксил-ионов [28].
Реакция, несомненно, протекает с участием проме-
жуточно образующихся ионов гипоманганата:
МпО^- + Н2О НМпО4"+ОН",
2НМпО4" Н2МпО4"+МпО4",
Н2МпО4" 4-СЮ“ —> МпО4‘ + СГ + Н2О.
При исследовании изотопного обмена кислорода ме-
жду ионами йодата и молекулами воды [29] обнаружено,
что при pH 4,5—10,5 реакция катализируется как кис-
лотами, так и основаниями. Скорость реакции обмена
R = 3,24 • 104 [Н+] [Юз] +1.35-103 [ОН”] [Ю3],
предполагается следующий механизм реакции:
Н2ю4‘ т=> Ю3‘ + Н2О,
онч-ню^" OH’-j-Hid^-.
120
Глава 4
Найтингейл [30] отметил, что кислородные анионы по
реакционной способности к обмену кислородом распола-
гаются в ряд
IO3 > ВгО^- > CIO3, СЮ^.
В такой же последовательности уменьшается длина кова-
лентной связи ХО. Понижение реакционной способности
связано с усилением л-связи. Усиление л-связи осуще-
ствляется р-орбиталями кислорода; • последние вслед-
ствие этого становятся менее доступными для образо-
вания межмолекулярных связей. Данные по электропро-
водности и вязкости свидетельствуют о том, что иодат
заметно гидратирован, тогда как бромат, хлорат, пер-
хлорат и перйодат гидратированы менее значительно
вследствие усиления внутримолекулярных связей.
Реакции серу- и кислородсодержащих анионов
Классическая реакция окисления иодида персульфа-
том явилась предметом недавно опубликованного крити-
ческого обзора [31].
21" + S2O|" —► 2SO4" + Lj.
Влияние добавляемых солей на скорость ионных ре-
акций в общих чертах было рассмотрено Ольсеном и
Симонсеном [32]. Кильпатрик [33] отметил, что, если на-
писать выражение для активного комплекса в реакции
А + В -> Продукты
в виде
сх = К • ск • св (fk •
концентрация активного комплекса будет определяться
величинами концентраций и коэффициентов активности
реагентов. Если для коэффициентов активности исполь-
зованы уточненные выражения, приведенное уравнение
даст правильное значение константы скорости. Труд-
ность заключается в том, что, за исключением очень раз-
бавленных растворов, когда применима теория Дебая—
Хюккеля, невозможно получить точное аналитическое*
выражение коэффициента активности. Некоторый про-
гресс в этом направлении достигнут при исследованиях
Реакции окисления — восстановления между анионами 121
растворов сильных кислот при помощи функций кислот-
ности.
Для нахождения функций кислотности желательно
разработать экспериментальную методику. Рассмотрим
диссоциацию слабой кислоты
НА Н+4-А".
Константу равновесия этой реакции можно выразить
следующим образом:
сн+ * са~ ^н+ * ^а~
СНА hik
Тогда для нахождения функций кислотности исполь-
зуется индикатор, в котором можно найти отношение
Са"/сна для раствора данной кислотности. Таким путем
определяется величина активности. Представляет инте-
рес варьировать при постоянной кислотности ионную
силу раствора и отсюда установить тип «функции кис-
лотности».
В реакции
C1O^4-3SO|“ —> cr+3so?-
происходит прямой перенос большей части кислорода от
хлората к сульфату [34а].
Далее была изучена реакция между сульфитом и ни-
тритом [35]. Механизм окисления сульфита нитритом за-
ключается в отнятии электронов от восстановителя.
К последнему присоединяется кислород, который посту-
пает от молекул растворителя. Происходящие в системе
изменения описываются схемой
2Н+ +NO2“ + 2SOf- + Н2О —> NH3OH+ +2SO$’, (1)
2Н+ + NO2“ + 3SO|- + H2O —> NH+ + 3SO^. (2)
Промежуточные соединения HON(SO3)2“ и N(SO3)2~
можно выделить из системы с хорошим выходом.
При исследовании реакции взаимодействия сульфита
с галогенатами [346] было найдено, что сульфат-ион, ко-
торый образуется при окислении сульфита хлоратом или
хлоритом, обогащенными изотопом 18О, содержит этот
122
Г лава 4
изотоп в более, чем обычном, соотношении. Во время
эксперимента обмен изотопом 18О между водой, с одной
стороны, и хлоратом и хлоритом, с другой, не происхо-
дит, и реакция должна протекать, по крайней мере ча-
стично, посредством переноса атома кислорода:
18ОСЮ2“ 4- SO^" —> СЮ2“ + 18OSO^“
Сульфат взаимодействует с хлоратом и броматом
при прямом переносе кислорода. В то же время сульфит
взаимодействует с нитритом по иному механизму, зави-
сящему от кислотности среды. Причина этого различия
в том, что азот является более электрофильным элемен-
том и образует NO+ гораздо легче, чем соответствующие
электрофильные соединения галогенов, например ВгОг •
В водном растворе ион цианида количественно реа-
гирует с тетратионатом с образованием сульфата, тио-
сульфата и тиоцианата [36]:
CN“ + S4O|“ + H2O —> SO^- + S2Oj“ 4" CNS”-|-2Н+.
Реакция описывается кинетическим уравнением
-d [S4O|-]/<tt = k2 [S4O|-] [CN-],
которое соответствует предполагаемой схеме
CN'4-~O3S—S—S—SO3 —► “O3SSCN4-S2O|-,
"O3SSCN4-2OH- —► so^"4-scn"4-h2o.
Реакции перекиси водорода в щелочной среде
Продуктами реакции между перекисью водорода и
тиоцианатом в интервале pH 4—12 являются сульфат,
цианат, аммиак и карбонат [37]. Реакция имеет первый
порядок относительно каждого реагирующего вещества
и не зависит от концентрации ионов водорода в иссле-
дованном интервале pH. Реакция осуществляется, оче-
видно, путем непосредственного нуклеофильного дей-
ствия тиоцианата на перекись водорода
SCN + Н2О2 —> HOSCN 4-ОН’ стадия, определяющая ско-
рость, с последующими быст-
рыми стадиями.
Реакции окисления — восстановления между анионами 123
Реакция окисления Сгш до CrVI в щелочных раство-
рах перекиси водорода [38]
2СгО2" + ЗНО2“ —> 2СгО^" + ОН" + Н2О
имеет первый порядок относительно как концентрации
Сгш, так и общей концентрации перекиси водорода. Ско-
рость реакции зависит также от квадратного корня из
величины эффективной активности ионов водорода, ко-
торая выражается видоизмененной функцией типа Гам-
мета:
Таким образом,
Я = МСгп,1 [НО2-] [Л-|*
Для реакции взаимодействия формальдегида и пере-
киси водорода в щелочном растворе [39] по уравнению
2НСНО 4-H2O2 + 2NaOH —> 2HCOONa+2Н2О +Н2
предполагается следующий механизм:
.ОН
НСНО + Н2О —> Н2С<
ХОН
/ОН _ /б
Н2С( + ОН —> Н2С< + Н2О
хон хон
/О /б
H2C< +HOOH —► НС< 4-бН4-Н2О
ХОН 4ОН
. /°" /О"
2нс< —> нс; + н2
ХОН X)
ЛИТЕРАТУРА
1. В a sol о F., Pearson R. G., Mechanisms of Inorganic Reacti-
ons, Wiley, New York, 1958, p. 318.
2. Silverman J., Dodson R. W., J. phys. Chem., 56, 846 (1952).
3. Thompson R. C, J. Am. chem. Soc., 70, 1045 (1948).
4. L i b b у W. F., J. phys, Chem., 56, 863 (1952).
124
Глава 4
5. Sheppard J. С., Wahl А. С., J. Am. chem. Soc., 81, 1572
(1959).
6. Gjertsen L., Wahl A. C., J. Am. chem. Soc., 81, 1572 (1959).
7. W a h 1 A. C., Z. Electrochem., 64, 90 (1960).
8 Adamson A. W., Gonick E., Inorg. Chem., 2, 129 (1963).
9. Halpern J., Legare R J., L u m г у R., J. Am. chem. Soc., 85,
680 (1963).
10. Adamson A. W., J. phys. Chem., 56, 858 (1952).
11. S p i г о M., J. chem. Soc., 3678 (1960).
12. Symons M. C. R., J. chem Soc., 3956 (1953); 3676 (1954).
13. Paul P. C., G a i n d V. S., M a 1 h u t a O. P.t N a hen P., Res.
Bull. E. Punjab Univ., 24, 123 (1953).
14. Hill L. M., Tomkins F. C., Trans. R. Soc. S. Afr., 29, 309
(1942).
15. W i b e r g К. B., S t e w a r t R., J. Am. chem. Soc., 78, 1214 (1956).
16. Lad bur у J. W., Cull is C. F., Chem. Rev., 58, 403 (1959).
17. Freund T., J. inorg. nucl. Chem., 15, 371 (1960).
18. Crowell W. R., Y о s t D. M., С a r t e r J. M., J. Am. chem. Soc.,
51, 786 (1929).
19. Farkas L., Lewin M., Bloch R.. J. Am. chem. Soc., 71,1988
(1949).
20. Lister M. W., Can. J. Chem., 34, 489 (1956).
21. Lister M. W., Rosenblum P., Can. J. Chem., 41, (a) 2727,
(6) 3013 (1963); (в) 39, 1645 (1961).
22. Anbar M., Taube H., J. Am. chem.'Soc., 80, 1073 (1958).
23. Chia Y. T., Con nick R. E., J. phys. Chem., 63, 1518 (1959).
24. Morgan K. J., Quart. Rev., 8, 132 (1954).
25. W r i g h t G. А., частное сообщение.
26. Anbar M., Rein R., J. Am. chem. Soc., 81, 1813 (1959).
27. Y о s h i n о Y., Can. J. Chem., 38, 2342 (1960).
28. Lister M. W., Yoshino Y., Can. J. Chem., 39, 96 (1961).
29. Anbar M., Guttmann S., J. Am. chem. Soc., 83, 781 (1961).
30. N i g h t i n g a 1 e E. R., J. phys. Chem., 64, 162 (1960).
31. I n d e 11 i A., P r u e J. E., J. chem. Soc., 107 (1959).
32. Olsen A. R., Simonsen T. R., J. chem. Phys., 17, 1167 (1949).
33. Kilpatrick M. A., Rev. phys. Chem., 2, 269 (1951).
34. H a 1 p e r i n J., Taube H., J. Am. chem. Soc., (a) 72, 3319
(1950); (6) 74, 375 (1952).
35. Rutenberg A. C., Halperin J., Taube H., J. Am. chem.
Soc., 73, 4487 (1951).
36. D a v i s R. E., J. phys. Chem., 62, 1599 (1958).
37. W i 1 s о n I. R., Harris G M., J. Am. chem. Soc., 82, 4515
(1960).
38. В a 1 о g a M. R., Earley J. E., J. Am. chem. Soc.. 83, 4806
(1961).
39. J ai Het J. B., Ou el let C., Can. J. Chem., 29, 1046 (1951).
Глава 5
РЕАКЦИИ ОКИСЛЕНИЯ МОЛЕКУЛ И АНИОНОВ
ПРОСТЫМИ И КОМПЛЕКСНЫМИ АНИОНАМИ,
КОМПЛЕКСНЫМИ КАТИОНАМИ
Эта глава посвящена рассмотрению реакций между
окислителями — анионами или комплексными ионами и
нейтральными молекулами восстановителей. Вначале мы
рассмотрим распространенный тип реакции между ве-
ществами, стабильными в щелочной среде. Затем перей-
дем к реакциям между веществами, которые достаточно
стабильны для того, чтобы реагировать также в кислой
среде. Наиболее важным примером реакций последней
группы служат реакции ионов перманганата.
При прохождении реакций в щелочных растворах
функция ионов гидроксила заключается в удалении про-
тона от молекулы окисляемого вещества, после чего про-
исходит быстрый отрыв одного или более электронов
молекулой окислителя. Наиболее вероятная схема про-
текания этих реакций может быть иллюстрирована на
примере окисления альдегидов:
ЯСНО + ОН RCH(OH)O быстрая стадия.
RCH(OH)O 4- ОН —► RC(OH)O + Н2О медленная стадия.
RC(OH)O окислитель —► продукт быстрая стадия.
Очевидно, что скорость реакции лимитируется процес-
сом отнятия водорода от альдегида, но не совсем ясно,
происходит ли это удаление непосредственно или путем
внутримолекулярного перемещения к некоторому дру-
гому месту молекулы с недостатком водорода. Тот факт,
что для успешного окисления необходимо наличие а-во-
дорода в молекуле, подтверждается тем, что реагент Фе-
линга и аммиачный раствор нитрата серебра с большим
трудом окисляют ароматические альдегиды по сравне-
нию с алифатическими [1].
126 Глава 5
Различные комплексы металлов и анионы могут быть
использованы в третьей стадии. Здесь мы обсудим под-
робно реакции комплексов меди, серебра и железа, а
также реакции перманганата и гипогалогенитов. Неко-
торые из реагентов этого типа представляют теперь
лишь исторический интерес. Так, щелочные растворы
цианидного комплекса ртути (II) и иодида ранее приме-
нялись для определения глюкозы. Щелочной раствор
тартрата висмута (реактив Нилендера) использовался
для обнаружения сахара в моче, причем при нагревании
выделялся осадок металлического висмута. Из других
щелочных растворов комплексов металлов, предложен-
ных для определения редуцирующих сахаров, отметим
щелочной тартрат, содержащий раствор соли никеля
или железа; первый раствор при нагревании образует
темно-красный осадок закиси никеля, второй при нагре-
вании дает осадок коричневого цвета [2].
Окисление комплексами серебра (I)
Наиболее чувствительным окислителем среди раство-
ров солей металлов является аммиачный раствор ни-
трата серебра, впервые примененный Толленсом [3] для
обнаружения редуцирующих сахаров.
Раствор аммиаката серебра готовят осторожным до-
бавлением 0,1 н. раствора аммиака к 0,1 н. раствору
нитрата серебра до исчезновения вначале образовавше-
гося осадка:
2Ag+4-2OH“ Ag2O4-H2O,
Ag2O + 4NH3 + H2O ^=± 2Ag(NH3)2++2OH’
Методика приготовления раствора аммиаката се-
ребра обеспечивает максимальную концентрацию ионов
Ag(NH3)^ при минимальной концентрации ионов гид-
роксила. Избыток аммиака, по-видимому, препятствует
реакции с редуцирующими сахарами, но добавление ще-
лочи облегчает ее протекание. Однако высокая концен-
трация щелочи нежелательна вследствие ее тенденции
отнимать протоны с последующей быстрой потерей элек-
тронов. Поэтому желательно поддерживать концентра-
Реакции окисления простыми анионами
127
цию щелочи на возможно низком уровне. Если опреде-
ляемые вещества нерастворимы в воде, то в качестве
удобного растворителя можно использовать пиридин.
Окисление комплексами меди (11)
Для обнаружения восстановителей применяются раз-
личные растворы соединений меди (II) с различной ве-
личиной pH и природой лигандов. Так, реактив Барфеда
представляет собой раствор ацетата меди (II) в уксус-
ной кислоте; реактив Бенедикта — раствор цитрата
меди (II) и карбоната натрия; реактив Фелинга — ще-
лочной раствор тартрата меди (II); реактив Пэви — ре-
актив Фелинга, содержащий много аммиака. Упомяну-
тые реактивы перечислены в порядке увеличения щелоч-
ности.
Во всех случаях в присутствии восстановителей оса-
ждается закись меди. В случае применения реактива
Пэви избыток аммиака препятствует осаждению закиси
меди и образуется бесцветный раствор аммиаката
меди (I). Момент окончания реакции устанавливают по
исчезновению голубой окраски [2].
Биуретные комплексы меди (II) похожи на содержа-
щиеся в реактиве Фелинга и аналогичны по реакцион-
ной способности тартратным комплексам меди (II).
Сахара окисляются медью (II), содержащейся в ре-
активе Фелинга, по сложному стехиометрическому урав-
нению. При изучении процесса окисления Л-арабинозы
Неф [4] обнаружил три различные ^одновременно проте-
кающие реакции:
1) 10—25% сахара окисляется до пентоновых кислот по
схеме
СбН10О5 + О —>- С5Н10Ов;
2) 35—45% сахара окисляется до муравьиной и триокси-
масляной кислот по схеме
С5Н10О5 + 2О —> НСООН + С4Н8О5;
3) 30—38% сахара образует муравьиную и гликолевую
кислоты по схеме
С5Н10Об + ЗО —> НСООН+ 2С2Н4О3.
Г28
Глава 5
Эти результаты заслуживают упоминания, поскольку в
ряде работ по окислению не анализировались образую-
щиеся продукты реакции. Реактив Фелинга широко ис-
пользуется для количественных определений, и во мно-
гих руководствах имеются эмпирически составленные
таблицы для перевода веса закиси меди в эквиваленты
восстановителей.
Холленд и Терни [5] проверили общий механизм этих
реакций на реакции между медным комплексом и глю-
козой, взятой в избытке, в буферном растворе. Наклон
прямой на графике концентрация меди — время зависит
от pH и концентрации сахара. Реакция протекает без
индукционного периода. Полученный результат согла-
суется с предложенным общим механизмом, так как,
если скорость не зависит от концентрации ионов
меди (II), то она должна быть пропорциональна произ-
ведению концентраций [RC (ОН) НО] [ОН]. Поскольку в*
буферном растворе концентрация гидроксила постоянна,
скорость должна быть пропорциональна концентрации
[RC(OH)HO]. Далее, если допустить, что быстро дости-
гаемое равновесие первой стадии сильно сдвинуто
вправо, то скорость должна быть пропорциональна
[RCHO]. Таким образом, при постоянном избытке вос-
становителя реакция должна иметь нулевой порядок,
что и наблюдается на опыте. Если реакцию проводить
в присутствии этилендиамина в качестве комплексообра-
зователя, ион меди стабилизируется в более низком ва-
лентном состоянии, закись меди не осаждается и ре-
акция становится гомогенной. В этом случае, однако,
возникают осложнения вследствие тенденции этиленди-
амина реагировать с карбонильной группой.
Синг с сотрудниками [6] обстоятельно изучали реак-
ции медных комплексов с сахарами. Они установили
следующие факты, характеризующие реакции окисления
глюкозы, галактозы и фруктозы' медью (II) в реактиве
Фелинга:
1) реакция имеет первый порядок относительно реду-
цирующего сахара;
2) реакция имеет нулевой порядок относительно ком-»
плекса меди (II), содержащегося в растворе;
Реакции окисления простыми анионами
129
3) скорость реакции не зависит от концентрации тар-
трата;
4) реакция катализируется ионами гидроксила, но ме-
жду ее скоростью и концентрацией гидроксила нет
прямой пропорциональности;
5) реакция имеет индукционный период;
6) предварительная обработка сахара щелочью повы-
шает скорость реакции в случае альдоз и замедляет в
случае кетоз;
7) относительная реакционная способность исследо-
ванных моноз изменяется следующим образом:
фруктоза > декстроза > глюкоза.
Скорость реакции, по-видимому, определяется ста-
дией взаимодействия щелочи и сахара с образованием
промежуточного продукта — ендиола
декстроза —м-е-ДЛеннБ^ ендиол, (1)
ендиол -|- медный комплекс —► Си2О -|- продукты. (2)
Первая стадия обусловливает первый порядок реак-
ции относительно концентрации сахара, а вторая стадия
объясняет нулевой порядок реакции относительно кон-
центрации медного комплекса. В этой работе и работе
Холленда и Терни авторы приходят к одному и тому же
выводу о нулевом порядке реакции относительно мед-
ного комплекса. Существенное различие состоит в том,
что Синг и сотрудники считают определяющей скорость
стадией присоединение гидроксила к молекуле сахара,
а не отрыв водорода от последней.
В более поздней работе Синг [7] изучал кинетику
окисления маннозы щелочным цитратным растворОхМ
меди (II). Эта реакция протекает медленнее, чем окисле-
ние глюкозы и галактозы, но сохраняет нулевой порядок
относительно медного комплекса и первый порядок отно-
сительно маннозы. Предварительная обработка маннозы
щелочью увеличивает скорость реакции, что объясняется
переходом альдегидной формы маннозы в более реак-
ционноспособную кетонную форму.
Окисление органических соединений солями меди (II)
изучалось еще одной группой авторов [8а]. Было показано,
9 Зак. 1249
130
Глава 5
что реакция окисления глюкозы и ацетоина щелоч-
ными растворами комплексных солей меди(II) характе-
ризуется индукционным периодом и имеет нулевой поря-
док относительно меди (II) и первый порядок относитель-
но органического вещества.
Ацетоин (0,222 г; 0,00252 моля) окисляли в течение 24 час при
40° избытком щелочного раствора цитрата меди (II) (0,01 моля
сульфата меди; 0,03 моля цитрата натрия и 0,15 моля карбоната
натрия). Через определенные интервалы времени отбирали по 5 мл
реакционной смеси и добавляли пробы к 5 мл 2 н. серной кислоты
в колбе с узким горлом. Затем немедленно добавляли избыток рас-
твора иодида калия и выделившийся свободный иод оттитровывали
стандартным 0,005 н. раствором тиосульфата в присутствии крах-
мала.
Реакция протекает по суммарному уравнению
СН3СН(ОН)СОСН3 4-2Си2+ + Н2О —►
—► СН8СОСОСН84-Си2О + 4Н+.
Кинетический анализ реакции показывает, что ацетоин
окисляется через ендиол, который, очевидно, образует
хелатный комплекс с медью (I). Комплекс затем легко
реагирует с ионами меди (II). Возможный механизм
реакции можно представить следующей схемой:
СН8СН(ОН)СОСН8 —> СН8С(ОН)=СОСН8 (1)
н3с—с—о.
СН8С(ОН)=х=СОСН84-Си+ —> II )iCu (2)
Н3С-С-О<ОН
н,с—с—с\ Н3С—со •
II >Cu 4-CU2+ —> || .Си4
Н8С—С—О\он н,с-со<он +Си4 (3)
(А)
А —► продукты (4)
После того как концентрация ионов меди (I) достигнет
величины, отвечающей произведению растворимости за-
киси меди, последняя начинает осаждаться. Скорость
окисления тогда становится постоянной и пропорциональ-
ной концентрации ендиола при условии, что реакция (2)
протекает медленнее реакции (1). Эти соображения очень
Реакции окисления простыми анионами
131
правдоподобны, так как концентрация меди (I) ограни-
чена очень малой растворимостью С112О в щелочном
растворе.
Для соединения А предполагается существование
двух резонансных структур:
Н3С—СО . Н3С—Сбч
II ZCu+ II >Си’+
н'с-со'он «•с-со\он
<0 <<0
Структура (/) представляет радикал комплекса меди (I),
структура (и)—енольную форму комплекса меди (II).
Считается, что ионы меди(II) соединяются с анионом
ендиола с образованием вещества А. Таким образом, пер-
вой стадией является реакция, приводящая к образова-
нию следов ионов меди (I), которые необходимы для на-
чала реакции (2).
В последующей работе [86] найдено, что бензоин окис-
ляется аналогично ацетоину. Результаты исследования в
этом случае подтверждают такой же механизм реакции,
как и при окислении ацетоина.
В случае окисления ацетатом меди (II) пропаргило-
вого спирта НС=ССН2ОН в пиридине при отсутствии
кислорода воздуха [9] необходимо присутствие меди (I).
Порядок реакции относительно меди (II) становится ну-
левым при высоких отношениях Си1: Си11. На основании
наблюдаемой зависимости скорости реакции от концен-
траций меди (I) и пропаргилового спирта возможен сле-
дующий механизм реакции, предполагающий образова-
ние в качестве промежуточного продукта соединения
меди (I) RCsCCu (R = CH2OH):
RC=CH 4- Си1 —> RteCCu + Н+
RCfeCCu + Си(СН3СОО)2 —> RteC. +2СиСН3СОО.
Окисление комплексами железа (III)
Комплексные соединения железа (III) взаимодей-
ствуют с участием одного электрона, при этом можно
было бы ожидать образования свободных радикалов
окисляемым веществом. Так, в реакциях окисления
9*
132
Глава 5
альдегидов, кетонов и нитропарафинов в щелочной среде
по схеме
red 4-Fe(CN)|- —► ox + Fe(CN)^
должны были бы образоваться свободные радикалы [10].
Однако в условиях эксперимента не наблюдается ката-
лиз винильной полимеризации, и, таким образом, пред-
положение об образовании свободных радикалов от-
падает.
Феррицианид широко применяется для количествен-
ного определения органических и неорганических соеди-
нений* Бриттон и Филипс [11] считают, что раствор
феррицианида более удобен для потенциометрического
титрования, чем реагент Фелинга. Раствор КзРе(СМ)6 в
присутствии Na2CO3 титруют потенциометрически с при-
менением платинового электрода при 100° анализируе-
мым раствором глюкозы.
При исследовании окисления альдегидов, кетонов и
нитропарафинов щелочными растворами феррицианида
[10] установлено, что, за исключением н-бутанола, началь-
ная скорость реакции их окисления пропорциональна
концентрации ионов ОН“, в большинстве случаев реак-
ции имеют первый порядок относительно органического
вещества. Однако порядок реакции относительно ферри-
цианида различен при окислении различных органиче-
ских веществ. Реакция имеет нулевой порядок в случае
изобутаналя, первый порядок при низких концентрациях
с тенденцией к переходу к нулевому порядку при высо-
ких концентрациях «-бутанола и диэтилкетона и первый
порядок в случае пропаналя, этилметилкетона, диизопро-
пилкетона и 2-нитропропана. В случае ацетона и нитро-
этана реакция имеет первый порядок при небольших
концентрациях феррицианида и становится реакцией вто-
рого порядка при повышенных концентрациях феррициа-
нида.
* Обзор аналитических методов имеется в монографии: Ber-
ka А., V и 11 е г i n J., Z у k a J., Newer Redox Titrants, Pergamon
Press, Oxford — London, 1965, p. 18 (есть русский перевод: Берка,
Вултерин, Зыка, Новые редокс-методы в аналитической химии,
М., «Химия», 1968). — Прим. ред.
Реакции окисления простыми анионами 133
Предлагается [10] следующий механизм реакции:
R—Н + ОН R 4-Н2О, (1)
об ратимая
енолизация
R + Fe(CN)|“ —> комплекс (X), (2)
X4-Fe(CN)g“ —> продукт 4-Fe(CN)g”, (3)
2Х —► димер4-2Ре(СМ)^-. (4)
Если обратимая енолизация (1) происходит много бы-
стрее, чем определяющая скорость стадия (2), тогда сум-
марная реакция имеет первый порядок относительно
окисляемого вещества, гидроксильных ионов и ферри-
цианида. Если же енолизация протекает медленнее ста-
дии (2), то в этом случае реакция окисления должна
иметь нулевой порядок относительно феррицианида.
Реакции (3) и (4), видимо, протекают быстрее реак-
ции (2).
Исследовано [12] окисление одноатомных фенолов
феррицианидом в щелочном растворе. Для реакции пред-
лагается схема
(ArO:)"4-Fe(CN)|- ArO. 4-Fe(CN)^,
ArO • 4- Fe(CN)|- АгО+ 4- Fe(CN)^,
Анализ кинетических данных приводит к следующему
выражению для скорости реакции
г .. .. d[ArO“]
-d [Fe(CN)^-]/^ =
kx [Fe(CN)3-]2 [ArO"]
*4Fe(CN)3-]+*3[Fe(CN)«-]'
В результате реакции окисления образуются димерные и
полимерные производные фенола.
Окисление галогенами в щелочной среде
Галогены обычно используют как окислители в ще-
лочном растворе. Хорошо известна реакция образования
галоформа по уравнению
RCOCH3+3NaXO->CHX3+ RCOONa +2NaOH.
134
Глава 5
На основе изучения реакции образования йодоформа
предлагается [13] следующий механизм:
RCOCH3+OH —> RCOCH2-|-H2O медленная стадия, опре-
деляющая скорость (1)
RCOCH24-H1O —► RCOCH2I + OH (2)
быстрая стадия
RCOCH2I —> RCOCIS (3)
быстрая стадия
RCOCI3 + 6H —► RCO6 + CHI3 (4)
При образовании йодоформа из спиртов RCH(OH)CH3
имеет место дополнительная реакция, заключающаяся в
окислении спирта до RCOCH3 по вероятной схеме:
CH3CH(OH)R + ОН CH3CHOR
CH3CHOR +12 —► CH3CHOIR +т
CH3CHOIR + 6H —► CH3COIR4-H2O
CH3COIR —> CH3COR + r
быстрая стадия
медленная стадия
быстрая стадия
быстрая стадия
(1)
(2)
(3)
(4)
Качественным подтверждением справедливости этой схе-
мы может служить относительная реакционная способ-
ность замещенных спиртов:
СН3ч СНЗЧ СНЗЧ
^СНОН> )СНОН> >СНОН
CH3Z CH3CH2Z Hz
Если бы реакция (1) была медленной, а реакция (2) бы-
строй, то такой порядок расположения спиртов по актив-
ности не имел бы места. В пользу указанной схемы сви-
детельствует также следующее наблюдение. Если вместо
иодноватистой кислоты использовать цианид иода, ско-
рость реакции резко понижается, поскольку значительно
уменьшается концентрация молекул свободного иода, не-
обходимая для осуществления второй стадии.
Исходя из рассмотрения равновесий и кинетики реак-
ций с участием иода и содержащих иод соединений в
водном растворе, можно найти оптимальные условия для
осуществления реакции получения йодоформа. Имеются
Реакции окисления простыми анионами
135
три важные равновесные реакции:
13- ^=> 12 + Г К = 1,4.10-3
НЮ Н+ + 10” К = 1.0. КГ11
12 4- Н2О Н+ + Г + НЮ К = 3.0 • 10“13
Максимальная концентрация иодноватистой кислоты на-
блюдается при pH 11,4, полностью образуется гипоиодит
при pH 13,0. Скорость реакции диспропорционирования
гипоиодита до йодата и иодида описывается уравнением
— d [IO"]/d/ = 2,9 • [Ю“]2 4- 104 [Ю”]2 [1”]/[ОН‘].
Отсюда для нахождения оптимальных условий получе-
ния йодоформа очень важно соотношение между ско-
ростью распада гипоиодита и скоростью иодирования [13].
Глюкоза взаимодействует с щелочным раствором
брома по уравнению [14]
НВгО4-СН2ОН(СНОН)4СНО —► СН2ОН(СНОН)4СООН4-НВг.
Эта реакция бимолекулярна, ее скорость достигает мак-
симума при pH 7,8. График зависимости константы ско-
рости этой реакции от pH почти идентичен графику за-
висимости концентрации бромноватистой кислоты в рас-
творе брома от pH. Это дает основания считать, что ак-
тивным окислителем в этих растворах служит бромнова-
тистая кислота.
Окисление перманганатом
Реакции окисления перманганатом в щелочных рас-
творах напоминают уже рассмотренные ранее примеры.
Уотерс отмечает [15], что окисление перманганатом
следовало бы по возможности проводить в нейтральной
среде, поскольку кислоты и щелочи могут вызвать раз-
личные осложнения реакции. Такие условия можно соз-
дать путем добавления нейтральных солей, например
сульфатов магния или цинка, произведение раствори-
мости гидроокисей которых определяет верхний предел
pH раствора.
В этом разделе обсуждаются равновесия, имеющие
место в растворах перманганата. Затем рассматривают-
ся реакции окисления ряда органических веществ. Цен-
ные обзоры реакций окисления перманганатом составле-
ны Ледбери и Каллисом [16] и Уотерсом [17].
136
Глава 5
Ниже перечислены состояния окисления марганца и
некоторые взаимные превращения его соединений.
Степень
окисления
+VII Мпб4 МпО4~ (сильно щелочная среда)
+VI МпО4~ МпО2 (слабо щелочная среда)
+V МпО4~ МпО4~ (очень сильно щелочная среда)
+IV МпО2 Мп2+ (кислая среда)
--III Мп3+—-> Мп2+ (кислая среда)
--II Мп2+
Реакции окисления ионов марганца(II) и марганца(III)
перманганатом рассмотрены вгл. 2, а реакции окисления
с участием анионов — в гл. 4. Гетерогенное окисление
марганцем (IV) в виде МпО2 рассматривается в главе 9.
Выделить марганцевую кислоту НМпО4 в чистом виде
нельзя; можно получить только ее растворы действием
серной кислоты на бариевую соль. Марганцевая кислота
является сильной кислотой, ее реакции идентичны реак-
циям иона перманганата [18].
Используя данные Латимера [19], можно вычислить
изменение свободной энергии реакции
4ОН~ 4-4МпО4- —> 4МпО1~+О2 + 2Н2О,
ДО = — 1.05 • 10е дж/кмоль.
При насыщении кислородом раствора NaOH с концен-
трацией выше 1 н. перманганат переходит в устойчивый
зеленый манганат. В растворах с концентрацией менее
1 н. NaOH манганат подвергается диспропорциониро-
ванию:
ЗМпО4~ +4Н+ —> 2МпО4~ + МпО2+ 2Н2О.
При сплавлении перманганата калия и NaOH при темпе»
ратуре красного каления можно получить слабораство-
римую соль К3МПО4, ион гипоманганата МпОд~ в раство-
ре имеет небесно-голубой цвет. Он устойчив только в
растворах с концентрацией щелочи выше 8н. [17]. Лотт
и Симонс [20] получили при растворении двуокиси мар-
ганца в концентрированном водном растворе КОН голу-
бые растворы, содержащие равные количества пятива-
лентного и трехвалентного марганца.
Реакции окисления простыми анионами
137
Исследования электропроводности и криоскопии рас-
творов перманганата в концентрированной серной кисло-
те показывают, что образование иона перманганила про-
исходит, по-видимому, по реакции [21а]
KMnO4 + 3H2SO4 —> K+ + H3O++MnO3+H-3HSO^
Изменение свободной энергии для реакции
2МпО4" Ч-2Н2ОЧ-ЗМп2+ 5МпО2 + 4Н+,
рассчитанное по данным Латимера [19], составляет
—2,57 • 108 дж)кмоль. Исходя из этой величины и значения
растворимости двуокиси марганца, можно показать, что
для предотвращения осаждения последней необходимо
присутствие ионов водорода с концентрацией не ниже
2н. Это согласуется с общепринятой практикой титриме-
трических определений с применением перманганата в
кислом растворе.
Свойства ионов МпО?- изучены Симонсом и его со-
трудниками [20, 21]. Ими показано, что окислительная
способность понижается в ряду
МпО4" > МпО4~ > МпО4“
Это естественно, поскольку при окислении происходит
присоединение электронов [17]. По данным Каррингтона
и Симонса [21], потенциал для системы манганат — пер-
манганат равен —0,588 в, а для системы манганат —
гипоманганат —0,285 в.
Приведем некоторые классические реакции окисле-
ния перманганатом:
ОН ОН
—cz —> Sc^ + o^/
Zl I4 7 Х
ОН он
/^^СНз —► <^—/СООН
—СН2ОН —► —соон
\снон —► \с=О
—сно —> —соон
R^CH —> R3COH
138
Глава 5
Обстоятельный обзор типов реакций окисления пер-
манганатом выполнен Ледбери и Каллисом [16]. Они раз-
личают четыре типа кинетического поведения реакций,
сопровождающихся уменьшением концентрации перман-
ганата во времени:
1) Реакции прямого взаимодействия перманганата с
окисляемым веществом, соответствующие кинетическому
уравнению второго порядка. Реакции окисления этого
типа рассматривались в главе 4.
2) Аутокаталитические реакции, в которых концен-
трация наиболее активных частиц окислителя вначале в
результате реакции увеличивается и только через неко-
торое время начинает уменьшаться. Наиболее известным
примером этого рода является реакция между перманга-
натом и оксалатом, рассматриваемая ниже.
3) Реакции, в которых быстрая начальная скорость
в последующий период падает почти до нуля, а затем
аутокаталитически увеличивается. В качестве примера
таких реакций можно привести окисление /г-нитрофенола
перманганатом.
4) Кинетические кривые реакций четвертого типа
имеют большой начальный наклон, который постепенно
уменьшается, давая под конец почти линейные участки.
Изменение исходной концентрации перманганата почти
не влияет на наклон линейного участка кривой. Это при-
водит к заключению, что скорость реакции контролирует-
ся, по-видимому, стадией енолизации окисляемого орга-
нического вещества. Этот тип реакции будет рассмотрен
ниже в данной главе.
Окисление ненасыщенных соединений
перманганатом калия
Реакция окисления разбавленным раствором перман-
ганата в щелочном растворе используется для качест-
венного определения некоторых органических веществ.
Растворяют испытуемое вещество в нескольких каплях разбав-
ленного раствора карбоната натрия, после чего прибавляют по кап-
лям разбавленный раствор перманганата калия. Если вещество не-
растворимо в водном растворе карбоната натрия, то его растворяют
в этанольно-водном растворе карбоната натрия и затем прибавляют
раствор перманганата.
Реакции окисления простыми анионами
139
Если вещество в указанных условиях мгновенно
обесцвечивает перманганат и если оно содержит только
С и Н, то можно сделать вывод о наличии в нем связей
^>С = С<^ или —С=С—. Спирты и другие способные к
окислению вещества в указанных условиях реагируют
медленно. Однако сохранение цвета раствора еще не до-
казывает отсутствия в нем веществ с ненасыщенными
связями. Так, например, дихлорэтилен реагирует с пер-
манганатом мгновенно, а трихлорэтилен и тетрахлор-
этилен медленно. Исследуемые растворы становятся ко-
ричневыми вследствие образования двуокиси марганца
по уравнению
3R + 2МпО4- + 4Н2О —► 3R(OH)2 + 2MnO2 + 2ОН".
При действии перманганата на двойную связь вна-
чале образуется гликоль. При определенных условиях
реакция окисления идет дальше, однако путем подбора
стехиометрического соотношения реагентов можно за-
держать реакцию на стадии образования двуокиси мар-
ганца. Поэтому стандартная методика приготовления
гликолей заключается в добавлении рассчитанного ко-
личества перманганата к раствору, охлажденному
льдом [15].
Бескен [22] показал, что при окислении пермангана-
том ряда алкенов в нейтральной или слабо щелочной
среде образуются ^uc-гликоли. При исследовании реак-
ции окисления олеиновой кислоты перманганатом, со-
держащим изотоп 18О в щелочном растворе, было об-
наружено, что образующаяся 9,10-диоксистеариновая
кислота содержит кислород из молекулы перманганата.
На этом основании предлагается механизм реакции, со-
гласно которому вначале образуется промежуточное со-
единение [23] циклического строения
। * । < I ±
Н—С Оч /О" Н—С—Оч /О' н—с—он
II + # >м< —► I )Мп< —► I
н—с о/ х° н—с—ХО н—с—дн
I I I
Однако детали механизма этой реакции не совсем ясны.
140
Глава 5
Окисление алкильных радикалов в ароматических
соединениях
Избыток перманганата в кислом или щелочной' рас-
творе окисляет алкильный радикал:
Аг(СН2)лСН3 —> АгСООН.
Установлено, что в щелочной среде реакция протекает
гетерогенным путем, однако детальных кинетических ис-
следований выполнено мало. Каллис и Ледбери [24] ис-
следовали реакции окисления серии ароматических
углеводородов, используя в качестве растворителя ле-
дяную уксусную кислоту или ее водные растворы, что
позволяет проводить реакцию в гомогенных условиях.
При изучении реакции окисления толуола [24а] установ-
лено, что главным местом атаки служит метильная
группа, толуол ступенчато окисляется и образуется бен-
зойная кислота. Реакции сопутствует частичный распад
ароматического кольца.
Начальная скорость реакции сложно зависит от кон-
центрации ионов водорода. Относительно перманганата
и толуола реакция окисления имеет первый порядок.
Результаты исследования влияния добавок комплексо-
образующих агентов — Mn(NO3)2 и других солей дают
основания считать, что толуол окисляется главным об-
разом образующимися ионами марганца (III), а не
ионами перманганата.
Каллис и Ледбери [246] исследовали скорость окисле-
ния различных производных толуола. Авторы пришли к
заключению, что введение нуклеофильных заместителей
понижает скорость окисления. Для реакции предлагает-
ся схема:
_2.i.
КМпО4 - промежуточный ион марганца (1)
«2
промежуточный ион марганца + органическое соединение —
—► продукт реакции. (2)
Если реакция (1) протекает быстрее реакции (2), то
/<нач = k2 [промежуточный ион марганца] • [органическое соединение]
= k2 [КМпО4] • [органическое соединение].
Реакции окисления простыми анионами
141
Это соответствует кинетическому уравнению второго по-
рядка. Влияние природы заместителя показывает, что
окислительными агентами являются катионы, так как
нуклеофильные и электрофильные заместители активи-
руются с образованием свободных радикалов.
При изучении процесса окисления этилбензола [24в]
в качестве первого промежуточного продукта был выде-
лен ацетофенон, который далее окисляется до бензойной
кислоты. Результаты исследования влияния добавок со-
лей показывают, что при окислении веществ с р-угле-
родом в боковой цепи ионы марганца (III), вероятно,
принимают меньшее участие.
Окисление ацетофенона предположительно протекает
по схеме
СвНбСОСН3 СвНбС(ОН)=СН2, (1)
*1
КМпО4 + СвН5СОСН3 —продукты, (2)
КМпО4+СвН5С(ОН)=СН2 продукты. (3)
Если k3>ki>k2, тогда начальная скорость окисления бу-
дет равна
k2 [C,H8COCH3J [KMnOJ + k3 [С8Н8С(ОН)=СН2] [KMnOJ,
но по мере протекания реакции концентрация енольной
формы уменьшается до тех пор, пока скорость стадии
(1) не станет ограничивать скорость стадии (3). По-
скольку ki>k2, то общая скорость реакции равна
k2 [С„Н8СОСН3] [KMnOJ + Л, [С3Н6СОСН3].
От концентрации перманганата скорость зависит в
очень малой степени. Экстраполяцией графика зависи-
мости скорости окисления от концентрации перманга-
ната до нулевого значения концентрации можно найти
слагаемое ^|[СвН5СОСНз], где ki — величина константы
скорости процесса енолизации (она согласуется с k, най-
денной для реакции иодирования).
Окисление спиртов
Избыток перманганата в щелочном или кислом рас-
творе окисляет первичные спирты до альдегидов, а вто-
ричные— до кетонов. В щелочном растворе происходит
142
Глава 5
гетерогенная реакция. Если необходимы гомогенные ус-
ловия, реакцию проводят в ледяной или умеренно раз-
бавленной водой уксусной кислоте.
Реакцию окисления бензгидрола (С6Н5)2СНОН ка-
тализируют ионы гидроксила, наблюдается положитель-
ный солевой эффект. На этом основании предполагает-
ся, что определяющей скорость стадией служит реакция
между ионами бензгидролата и перманганата [25].
В опытах с применением в качестве окислителя перман-
ганата, меченного изотопом 18О, не найдено 18О в про-
дуктах окисления. Этот факт и наличие изотопного эф-
фекта (6,6:1) при окислении бензгидрол-a-D показы-
вают, что реакция, вероятно, происходит путем переноса
иона водорода от иона бензгидролата к перманганату.
Механизм реакции можно описать целым рядом парал-
лельных реакций. Первой стадией должна быть иониза-
ция бензгидрола, за которой следует несколько опреде-
ляющих скорость стадий, например
(С6Н5)2СНб + МпО4- —> (С6Н5)2С(ОН)б + МпО3-.
Степень кислотности спирта (С6Н5)2СНОН такова, что
в условиях реакции он ионизируется очень слабо. По
этой причине были взяты фторпроизводные спирты,
которые проявляют достаточно ясно выраженные кис-
лотные свойства. Окисление фторированных спиртов
рассмотрено в статье Стюарта и Ван дер Линдена [26].
Окисление протекает по стехиометрическому урав-
нению
ArCHOHCF3 + 2MnO4-"4-2OH- —> ArCOCF3 + 2MnO^“ + 2Н2О.
Для этой реакции предлагается схема
ROH + OH” —► RO“ + H2O,
RO-4-MnO^ —► продукты.
Скорость реакции
— d [MnO4-]/d/ = k [RO~] [MnOj-].
Наблюдаемый большой дейтериевый изотопный эффект
(16:1) свидетельствует в пользу следующего меха-
Реакции окисления простыми анионами
143
низма:
О" “О
I I
Аг—С—Н -|- МпО4 —► Аг—С—Н—ОМпО3
CF, _ CFS
—► ArCOCFj + HMnO^’
Окисление альдегидов
Можно было бы ожидать, что катализируемые осно-
ваниями реакции протекают по механизму, аналогично-
му рассмотренному выше для окисления комплексов
металлов. Однако эти предположения не подтверждают-
ся. Уиберг и Стюарт [27] изучали окисление ряда аро-
матических альдегидов растворами перманганата при
pH 5—13, причем были обнаружены как кислотный, так
и основной каталитические пути реакции. Для послед-
него случая экспериментально установлено следующее:
1. Скорость /? —^[RCHO] [МпО?] [ОН-]7*.
2. Незначительный изотопный эффект при замещении
водорода альдегидной группы на дейтерий.
3. Главная часть присоединенного к альдегиду кис-
лорода поступает из молекул растворителя (воды).
Ледбери и Каллис [16а] отмечают, что реакция опи-
сывается кинетическим уравнением второго порядка от-
носительно перманганата. В определяющей скорость
стадии, вероятно, принимают участие две заряженные
частицы. Предлагаемая схема реакции:
МпО4 + НО“ — * МпО4“ + НО • зарожде-
ние цепи
НО - +RCHO RCH(OH)O-
RCH(OH)O • -|- МпО^" ——•* RCOOH -|- МпО^" -|- НО • развитие
цепи
2ОН . —Н2О2
4" МпО^" —О2 HMnO^- обрыв
цеои
144
Глава 5
Описанный механизм должен бы приводить к следую-
щему выражению для скорости реакции:
— d[MnO4-]/d/ = ЗЛ2[МпО4-][ОН~] + Л[КСНО][МпО^],/,[ОН_],/’
Поскольку степень разложения перманганата в щелоч-
ном растворе мала, величина первого члена уравнения
незначительна по сравнению со вторым.
Реакция, катализируемая кислотами, очевидно, про-
текает совершенно иначе. На это указывают следующие
экспериментально установленные факты: 1) скорость
реакции пропорциональна концентрациям альдегида и
перманганата; 2) определяющая скорость стадия заклю-
чается в разрыве связи С—Н, о чем свидетельствует
дейтериевый изотопный эффект; 3) кислород, присоеди-
няющийся к альдегиду, поступает главным образом из
окислителя, указывая тем самым, что между атомами
углерода, входящими в состав альдегида, и кислородом
окислителя на некоторое время возникает связь.
Для реакции предполагается механизм
RCHO + Н8О+ RCHOH + Н2О. (1)
ОН
RCHOH + MnO^- R—С—OMnO3(X), (2)
Н
х 4- В: —* RCOOH + НВ + МпО^, (3)
ЗМпОз + Н2О —► 2МпО2 4- MnO4 4-2ОН". (4)
В следующей статье [28] авторы рассмотрели процесс
окисления фторальгидрата растворами перманганата.
В интервале значений pH 2—8 скорость реакции очень
мала, но быстро увеличивается с ростом pH вследствие
образования анионов. Реакция окисления происходит по
суммарному уравнению
2MnO4 +CF?CH(OH)24-3OH —> 2MnO4~ 4-CF3COO- 4-ЗН?О.
Реакции окисления простыми анионами
145
а скорость реакции определяется стадией
0“ О"
I I
CF3—СН + М11О4 —► CF8—С - +НМпО4“
он он
CFjCOOH
Окисление соединений, содержащих водород
при третичном атоме углерода
Молекулы углеводорода с атомом водорода при тре-
тичном углероде окисляются перманганатом до третич-
ных спиртов. Кенион и Симонс [29] опубликовали инте-
ресные результаты по окислению карбоновых кислот с
атомом водорода при третичном углероде
R4 R4
>СН(СН2)„СООН —► >С(ОН)(СН2)„СООН
R'z R'z
В среде концентрированной щелочи эта реакция проте-
кает при м = 0 или п=2 и не происходит при п=1. При
реакции имеет место перенос одного электрона
МпО4 —> МпО^~.
Потеря оптической активности показывает вероятное
образование промежуточного соединения
^>С(СН2)„СОО-
В случае п=2 реакция идет в среде разбавленной
щелочи, причем имеет место перенос двух электронов
(MnOl- -> МпОг)- Оптическая активность сохраняется,
указывая на возможное образование промежуточного
соединения
R\ +
)С(СН2)„СОО~
R'z
|Q Зак. 1249
146
Глава 5
Окисление некоторых органических веществ
щелочными растворами перманганата,
манганата и гипоманганата
Друммонд и Уотерс [30] изучили реакции окисления
перманганатом в щелочных растворах, останавливая
реакцию на стадии образования манганата (МпОГ+г-*
-> MnCh-) путем добавления ионов бария для осаждения
BaMnOi. Избыток перманганата определяли по реакции
2Mn(V + CN“ + Н2О —► 2МпО1" + CN0” + 2Н+.
Реакция, вероятно, протекает по следующему пути:
МпО4“ + Н2О —+ МпО4“ + Н+ + НО •.
Экспериментально обнаружено, что такие вещества, как
эфир и диоксан, окисляются действием свободных ради-
калов гидроксила, а не щелочного раствора перманга-
ната. Однако ацетон, фумаровая и малоновая кислоты,
напротив, окисляются перманганатом, а не свободным
радикалом гидроксила.
Установлены некоторые общие правила:
1. Насыщенные карбоновые кислоты вообще не окис-
ляются, что объясняется прочностью связей С—Н и
С—С.
2. Некоторые реакции окисления, например метило-
вого спирта, бензилового спирта и бензальдегида, про-
текают количественно.
3. Алифатические кетоны инертны по отношению к
окислению в нейтральных, но разрушаются в сильноще-
лочных растворах перманганата.
4. В нейтральных растворах окисление по олефино-
вой связи останавливается на стадии образования цис-
гликолей, а в сильнощелочных растворах наблюдается
полный распад по месту двойной связи.
Ригби [31] отмечает, что tfuc-оксилирование двойных
связей можно осуществить также с помощью манганата
калия. Хотя эта методика имеет преимущество по срав-
нению с окислением перманганатом, но иногда манганат
способен окислять образующийся гликоль. Реакция про-
текает сложно и катализируется двуокисью марганца,
Реакции окисления простыми анионами
147
Реакции окисления органических соединений щелоч-
ными растворами манганата МпО4~ и гипоманганата
MnOi” рассмотрены в обзоре [32]. Растворы манганата,
не содержащие перманганата, можно приготовить при
концентрации щелочи от 1 до 10 н., тогда как типоман-
ганат достаточно устойчив только в холодных 8—10 н.
щелочных растворах. Реакции окисления органических
соединений щелочным раствором манганата столь же
неспецифичны, как и реакции окисления сильнощелоч-
ным раствором перманганата. Но ионы гипоманганата,
как более слабые окислители, проявляют повышенную
селективность.
Другие реакции окисления перманганатом
Рассмотрим некоторые не разбиравшиеся выше слу-
чаи окисления, а именно:
1) реакцию окисления Хукера;
2) реакцию между перманганатом и оксалатом;
3) реакцию окисления перманганатом формиата, на-
ходящегося во внутренней сфере комплекса.
При реакции окисления Хукера превращение
О О
Х°Н
о о
протекает в щелочном растворе перманганата калия,
являясь результатом одновременного действия двух
окислителей'—перманганата калия и кислорода. Меха-
низм этой интересной реакции подробно рассмотрен в
упомянутой статье [33].
Реакцию окисления оксалата перманганатом можно
представить суммарным уравнением
2МпО^ 4-5С2О^- 4- 16Н+ —► 2Мп2+ + 10СО2 + 8Н2О.
Непосредственно ионы МпО-Г и СгО^- взаимодейству-
ют очень медленно, но реакция между марганцем (IV)
10*
148
Глава 5
и СгО^Г и марганцем (III) и СгО^ протекает с измери-
мой скоростью. Лоунер [34] полагал, что важнейшим
промежуточным продуктом реакции является ион мар-
ганца (III), образующийся по уравнению
МпО4- + 4Мп2+ + 8Н+ —> 5Мп3+ + 4Н2О.
Однако позднее Лоунер и Иост [35] пришли к заключе-
нию, что ионы С2О4 окисляются как ионами Мп3+, так
и ионами Мп4+; последние образуются по уравнению
2МпО4- 4-ЗМп2+ + 16Н 'быстро -> 5Мп4+ +8Н2О. (1)
Относительная степень участия в окислении ионов Мп3+
и Мп4+ зависит от их концентраций; имеет место реак-
ция диспропорционирования
быстро
Мп4++Мп24 *• 2Мп3+. (2)
обратимо
Образовавшиеся ионы Мп3+ и Мп4+, вероятно, затем ре-
агируют с оксалатом по следующим стадиям:
Мп4+ + С2О4~ ЧХ™* Мп3+ + С°2 + С°2-> <3>
Mn4++COj 6ЫСТр°> Мп3+ +СО2, (4)
_ . о быстро
Мп3+ + 2С2О2- <==± Мп(С2О4)2-, (5)
Мп3+ + С2О2~ —> Мп2+ + СО24-СО7, (6)
Мп3++СО2- —► Мп2+-+-СО2. (7)
В присутствии ионов марганца (II) образующиеся
по уравнению (1) ионы марганца (IV) быстро перево-
дятся в ионы марганца (III) по уравнению (2), затем
ионы марганца (III) образуют комплекс с СгО!- по
уравнению (5). Последний медленно разлагается по
бимолекулярной реакции. Если не добавлять ионы Мп11,
то образующиеся ионы марганца (IV) быстро реаги-
руют преимущественно по уравнениям (3) и (4).
Если концентрация оксалата СгО-Г высока, то ре-
акции (5), (6) и (7) протекают относительно медленно.
Но при низкой концентрации СгО4“ создается по реак-
ции (6) относительно высокая концентрация ионов мар-
Реакции окисления простыми анионами
149
ганца (II), которые реагируют по уравнению (2) с об-
разованием Мпш. Хотя на основании результатов по-
следующих радиохимических исследований необходимо1
внести некоторые видоизменения, однако указанный
выше механизм реакции остается как основа для об-
суждения *.
Другим интересным случаем является окисление1
пентаминкобальт (III) формиата с помощью перманга-
ната [36]. Скорость реакции описывается кинетическим
уравнением
— d [комплекс]/^/ = k (комплекс] [МпО^“],
где константа скорости k не зависит от концентрации
ионов водорода. Общая стехиометрия реакции представ-
ляет нечто среднее между двумя уравнениями:
(NH3)5 • Со • О • СНО2+ + 7з МпО^ —► СО2 + Со2+ + 2/3 МпО2,
(NH3)6 • Со • О • СНО2+ + 2/3 МпО^ —►
—> СО2 + (NH3)5 • Со • Н2О3+ + 2/3 МпО2.
Замещение группы —CHOnaCDO—уменьшает скорость
реакции в 10,5 раза. На этом основании предполагается
механизм
|(NH3)5Coni(OCHO)]2+ + МпО4-—► [(NH3)5ConiCO^]2+ + НМпО4“
Со11 + СО2 |(NH3)5CoinH2Ol3+ + СО2
Высокое значение дейтериевого изотопного эффекта по-
казывает, что начальная реакция, по-видимому, пред-
ставляет собой отрыв атома водорода.
ЛИТЕРАТУРА
1. Waters W. A., Progress in Organic Chemistry, Vol. S, Buttery-
worth, London, 1961, p. 29.
2. Allans Commercial Organic Analysis, Vol. 1, Churchilft London,
1909.
* Обзор более поздних исследований см. в книге Л а йт и-
н е н Г А., Химический анализ, М., «Химия», 1966, стр. 399. —
Прим. ред.
150
Глава 5
3. То 11 ens В, Chem. Вег., 15, 1635 (1882); 16, 921 (1883).
4. Nef J. W., Liebigs Annin, 357, 214 (1907).
5. Holland R. V., Turney T. А., неопубликованная работа.
6. Singh M. P., Krishna B., Ghosh S., Z. phys. Chem., 205,
285 (1956).
7. S i n gh M. P., Z. phys. Chem., 208, 273 (1957).
8. Marshall B. A., Waters W. A., J chem. Soc., (a) 2392
(1960); (6) 1579 (1961).
9. Clifford A. A., Waters W. A., J. chem. Soc., 3056 (1963).
10. Speakman P. T., Waters W. A., J. chem. Soc., 40 (1955).
11. Britton H. T. S., Philipps L., Analyst, 65, 149 (1940).
12. Haynes C. G., Turner A. H., Waters W. A., J. chem. Soc.,
2823 (1956).
13. Seel ye R. N., Turney T. A., J. chem. Educ., 572 (1959).
14. Grover К. С., M e h г о t a R. C., Z. phys. Chem., 14, 345 (1958).
15. Waters W. A., Organic Chemistry (Ed. H. Gilman), Vol. 4,
Wiley, New York, 1953, Chap. 12.
16. Ladbury J. W., Cullis C. F., J. Soc. chem. Ind., 1010 (1959);
Chem. Rev., 58, 403 (1958).
17. Waters W. A., Quart. Rev., 12, 277 (1958).
18. S i d g w i c k N. V., The Chemical Elements and Their Compounds,
Clarendon Press, Oxford, 1950, p. 1267.
19. Latimer W. M., The Oxidation States of the Elements and
Their Potentials in Aqueous Solution, Prentice-Hall, New York,
1952; см. общую литературу к Введению.
20. Lott К. А. К., Symons М. С. R., J. chem. Soc., 829 (1959).
21. С а г г i n g t о n A., Symons M. C. R., J. chem. Soc., 3373
(1956).
21a. Royer D. J., J. inorg. nucl. Chem., 17, 159 (1961).
22. Boeskin J., Rec. trav. chim. Pays-Bas, 47, 683 (1928).
23. Wiberg К. B., Saegebarth K. A., J. Am. chem. Soc., 79,
2822 (1957).
24. C u 11 i s C. F.. Ladbury J. W., J. chem. Soc., (a) 555; (6)
1407; (в) 2850; (1955).
25. S t e w a r t R., J. Am. chem. Soc., 79, 3057 (1957).
26. Stewart R., van der Linden R., Disc. Faraday Soc., 29, 211
(1960).
27. Wiberg K. W., Stewart R., J. Am. chem. Soc., 77, 1786
(1955).
28. Stewart R., Мосек M. M., Can. J. Chem., 41, 1161 (1963).
29. Kenyon J., Symons M. C. R., J. chem. Soc., 3580 (1953).
30. Drummond A. Y., Waters W. A., J. chem. Soc., 435 (1953).
31. R i gb у W., J. chem. Soc., 2452 (1956).
32. Pode J. S. F., Waters W. A., J. chem. Soc., 717 (1956).
33. Shemyakin M. M., S c h i k i n a L. A., Quart. Rev. chem. Soc.,
Lond., 10, 276 (1956).
34. L a u n e r H. F., J. Am. chem. Soc., 54, 2597 (1932).
35. Launer H. F., Yost D. M., J. Am. chem. Soc., 56, 2571 (1934).
36. Candlin J. P., Halpern J., J. Am. chem. Soc., 85, 2518
(1963).
Глава 6
РЕАКЦИИ ОКИСЛЕНИЯ ЧЕРЕЗ СТАДИЮ ЭТЕРИФИКАЦИИ
В этой главе мы рассмотрим реакции окисления,
протекающие через стадию образования эфиров. Неко-
торые реакции, приближающиеся к такому типу, уже
обсуждались, в частности реакции ионов ванадила и
тетраацетата свинца. Типичными окислителями этой
группы являются неорганические кислоты: хромовая,
иодная, осмиевая и селенистая. Сюда примыкает также
реакция получения диэтилового эфира из этилового
спирта через образование промежуточного эфира сер-
ной кислоты. Среди других возможных реакций окисле-
ния можно отметить реакции с такими кислотами, как
молибденовая и вольфрамовая. Мы рассмотрим каждый
окислитель с точки зрения частиц, присутствующих в
растворе, и их реакционной способности с учетом суще-
ствующих равновесий.
Окисление хромовой кислотой
Бихромат широко применяется в качестве окислите-
ля. Хромовая кислота образуется в растворах при вза-
имодействии трехокиси хрома с водой:
СгО3Ч- Н2О Н+ + НСгО^.
Аналогичная реакция, очевидно, протекает с трет-бути-
ловым спиртом:
СгО3 + (СН3)3СОН 5=* (СН3)3С + НСгОГ-
Реагент для окисления готовят путем добавления
трехокиси хрома к избытку третичного бутилового спир-
та, охлаждения смеси и последующего растворения вы-
павших красновато-желтых кристаллов в неполярном
152
Глава 6
органическом растворителе. Раствор, содержащий избы-
ток бутанола, затем дегидратируют безводным сульфа-
том натрия [1].
В качестве других неводных растворителей трехоки-
си хрома используются ледяная уксусная кислота и пи-
ридин.
В последнем случае, вероятно, происходит образова-
ние комплекса с трехокисью хрома. Иногда применяют-
ся растворы Сг2О3 в ацетоне [2]. Поскольку уксусная
кислота, пиридин и вода смешиваются во всех отноше-
ниях, то было бы интересно спектрофотометрическим
методом изучить состояние трехокиси хрома в различ-
ных смесях этих растворителей.
В воде имеют место равновесия [3, 4]:
Н2СгО4 7Z* Н++НСгО4-
К
1,8. КГ1
Сг2°Г + н2° 2НСгО4"
2,3.10“2
НСгО4" ^=> Н+-|-СгО4-
3,0-10"7
Представляет также интерес исследование стехиомет-
рии реакции окисления хромовой кислотой в зависимо-
сти от условий применения последней. Так, реакция с
этиловым спиртом протекает по суммарному уравнению
Сг2О?- Ч-ЗСН3СН2ОН Ч-8Н+ —> 2С?++ЗСН3СНО+7Н2О,
т. е. при стехиометрическом соотношении реагентов:
1 моль бихромата на 3 моля спирта и 8 эквивалентов
кислоты.
Физер [б] перечислил все условия, в которых приме-
няется трехокись хрома как окислительный агент. По
его мнению, окислительная способность реагента возра-
стает с понижением содержания воды в растворителе.
Следует упомянуть также о некоторых интенсивно про-
текающих и потенциально опасных реакциях трехокиси
хрома [6].
Обзоры реакций окисления бихроматом опубликова-
ны Вестгеймером [7] и Уотерсом [8]. Реакции окисления
хромовой кислотой, по-видимому, лучше всего рассмат-
Реакции окисления через стадию этерификации
153
ривать по классам органических соединений, как это
предлагает Уотерс, а именно: окисление спиртов, окис-
ление альдегидов, окисление углеводородов.
Окисление спиртов
Вестгеймер и Новик [9] показали, что реакция окис-
ления изопропилового спирта хромовой кислотой имеет
первый порядок относительно ионов НСгОГ и второй
порядок относительно ионов водорода.
Для этой реакции авторы предложили следующий
механизм:
НСгО4“ 4- 2Н + 4- НО • С(СН3)2Н НС(СН3)2ОСгО2ОН2+ 4- Н2О
-медленно > НзО + + (СН3)2С=О + Н2СгО3-
В другой работе [10] было найдено, что 2-дейтеро-
пропанол-2 окисляется хромовой кислотой медленнее,
чем обычный изопропиловый спирт. Это показывает,
что определяющая скорость стадия заключается в от-
рыве атома водорода от вторичного атома углерода
спирта. Указывается [11] на возможное образование в
бензольном растворе нейтрального эфира хромовой кис-
лоты и изопропилового спирта. Этот эфир неустойчив,
его разложение ускоряется основаниями, например пи-
ридином. Свойства диизопропилхромата были детально
изучены [12]. В зависимости от условий это вещество
может подвергаться либо гидролизу, либо самоокисле-
нию— восстановлению. Изопропиловый спирт в 86,5Уо-
ном растворе уксусной кислоты окисляется трехокисью
хрома по реакции первого порядка относительно спир-
та, НСгОГ и. ионов водорода. Скорость реакции в этой
среде в 2500 раз выше, чем в воде [13]. Отношение ско-
ростей окисления (CH3)2CDOH и (СН3)2СНОН имеет
ту же величину, что и в водном растворе; что, по-види-
мому, указывает на сходный механизм реакций. Окис-
ление сильно замедляется в присутствии ионов хлори-
да, кинетические данные свидетельствуют о возможном
равновесии:
н+ 4- сг 4- нсгОг асгог 4- н2о.
154
Глава 6
Тритиевый изотопный эффект [14] при окислении
2-пропанол-2-Т имеет величину Лт/&н=0,04, которая со-
гласуется с величиной W&h, найденной Вестгеймером.
Цейсс и Мэтьюз [15] показали, что эфиры хромовой
кислоты и простых третичных спиртов можно выделить
методом лиофильной сушки. Ими описаны также новые
методы получения таких эфиров через хромилхлорид.
Факторы, определяющие переходное состояние при окис-
лении спиртов и других соединений хромовой кислотой,
рассмотрены в обзоре Куорта [16].
Окисление альдегидов
По данным Уиберга и Милла [17], бензальдегид в
91 %-ной уксусной кислоте окисляется хромовой кисло-
той со скоростью
/? = A[RCHO] [НСгО4] Ло.
Дейтериевый изотопный эффект составляет 4,3. Группы,
притягивающие электроны, увеличивают скорость окис-
ления. Реакция протекает по следующему суммарному
уравнению:
3RCHO + 2CrO3 + 6H+ —> 3RCOOH+2Сг3+ + ЗН2О.
Предлагаемый для этой реакции механизм:
НСгО4+Н+ Н2СгО4
ОН
I
CeH5CHO + H2CrO4 С4Н5СОСгО3Н
н
он
В + С3Н5СОСгО3Н —► C8H5COOH + CIV
I
н
где В — основание.
Скорость окисления бензальдегида в воде при 80° за-
висит от первой степени концентрации бензальдегида, но
является сложной функцией концентрации кислоты [18].
Реакции окисления через стадию этерификации
155
Предлагается следующий механизм реакции:
Н
I
С6Н5СНО 4- НСгО4 4-2Н+ C6H5COCrO3Hf (А)
ОН
А4-Н2О —► С«Н5СООН 4-Н3О++ Н2СгО3
CriV4-CrVi 2CrV
CrV4-CeH6CHO C.H5COOH + Crlll
Порядок реакции с формальдегидом [19] относительно
как хромовой кислоты, так и формальдегида равен еди-
нице. Относительно ионов водорода порядок реакции
равен двум при всех концентрациях ионов водорода.
Постулируется следующий механизм реакции:
HCrO4 4-СН2(ОН)2 —> CrIV4-HCOOH,
CriV4-CrVi —► 2CrV,
CrV 4- CH2(OH)2 —► Crin 4- НСООН,
2CrlV 4- CH2(OH)2 —► 2СгШ4-НСООН.
Окисление углеводородов
Первый пример показывает важность знания теоре-
тического стехиометрического соотношения реагентов
для практической методики синтеза. Циклогексен окис-
ляется трехокисью хрома до циклокетогексена с выхо-
дом 37% от теоретического [20]:
3^2^>4-ЗН2О —» 3<2^>=O4-6H+4-6*
2СгО,4-12Н+ 4-6<? —> 2Сг3+4-6Н2О
2СгО34-3/в=^>4-6Н+ —► 2Сг3+4*ЗН2О 4~3<^=у=О
Теоретическое соотношение таково, что 2 молекулы
трехокиси хрома расходуются на окисление 3 молекул
циклогексена.
Слек и Уотерс [21] исследовали реакцию окисления
дифенилметана трехокисью хрома в ледяной уксусной
кислоте, продуктом которой был бензофенон.
Найдено [21], что скорость реакции окисления понижа-
ется под действием ионов хрома (III). Это происходит,
156
Глава 6
вероятно, потому, что они соединяются со свободным
хроматом с образованием соли Сг(НСгО4) (ОСН3СО)2,
которая проявляет очень слабые окислительные свойст-
ва и мало диссоциирована. Реакция ускоряется при
добавлении 5%-ного раствора H2SO4-H2O и еще боль-
ше при добавлении безводной серной кислоты.
Другими исследователями при изучении реакции
окисления дифенилметана хромовой кислотой [22] най-
дено следующее выражение скорости реакции:
Л = k [(СвН5)2СН2] [СгО3] h0.
При 30° кинетический изотопный эффект составляет
£h/&d = 6,4, а нуклеофильные группы умеренно способ-
ствуют протеканию реакции. Поскольку этот эффект
действует в противоположном направлении по сравне-
нию с найденным для бензальдегида, механизм этих ре-
акций должен быть различен.
Механизм, предлагаемый для первой реакции, пред-
ставляет собой отрыв атома водорода и образование
радикала бензгидрила, который затем окисляется до
бензофенона. Скорость реакции окисления хромовой
кислоты, происходящей с отрывом атома водорода, за-
висит от концентрации CrVI. В то же время скорость
реакций, протекающих, по-видимому, по механизму эте-
рификации, зависит только от кислого хромата. Данное
отличие предлагается как метод различения этих меха-
низмов.
Окисление ( + )-3-метилгептана хромовой кислотой в
91 %-ном растворе уксусной кислоты дает ( + )-3-метил-
гептанол. При реакции на 70—85% сохраняется кон-
фигурация молекулы, что дает основание предполагать
следующий механизм [23]:
R3CH 4- CrVI —> R3C. + Н+ + Crv,
R3C. +CrVI —> R3C+,
R3C+ 4-H2O —► R3COH + олефин.
Интересные результаты получены при окислении хромо-
вой кислотой алкилбензолов [24]. В условиях Кюна—
Роша параллельно окислению кольца происходит окис-
ление боковой цепи. В случае третичного бутилбензола
Реакции окисления через стадию этерификации
157
реакция протекает исключительно посредством окисле-
ния ароматического кольца, имея первый порядок отно-
сительно концентрации углеводорода и показывая ли-
нейную зависимость logfc — HQ. Исследование ряда ме-
тилбензолов показывает, что окислению ароматического
кольца способствует увеличение числа замещающих
групп. Результаты дают основание считать, что окисле-
ние происходит посредством электрофильного замеще-
ния в ароматическом кольце:
НСгО4- + 2Н+ ^=> НСгОз" + Н2О
X—^4-HCrO3+ X—СгО3Н4-Н+
X—СгО3-4-Н2О —> X—</ ОН-|-Н2СгО3
Окисление перйодатом
Рассмотрим вначале состояние окислителя в рас-
творе, затем имеющиеся равновесия и, наконец, реакции
с восстановителем. Судя по спектрофотометрическим
данным, в разбавленном водном растворе хлорной кис-
лоты существуют три равновесия для иодной кислоты
Н5Ю6 Н+ + Н4Ю6-, Ki = 2,3 • КГ2,
Н4Ю6“ +± Н+ + Н3Ю|- К2 = 4,4 • КГ9,
Н3Ю3~ 5=* Н+ -|- H2IO|", Х3 = 1,1 • 1(Г16.
Наиболее устойчивой формой иодной кислоты является
HsIOe, в виде которой она кристаллизуется из водных
растворов [26]. Иодная кислота имеет формулу H5IO6
и не является гидратом НЮ4 • 2Н2О. Об этом свидетель-
ствует возможность получения многочисленных солей,
например Ba5(IOe)2, и отсутствие в ИК-спектре полос
поглощения воды [27].
В более позднем исследовании предполагается, что по-
ведение иодной кислоты в растворе лучше описывается
158
Глава 6
следующими равновесиями [28]:
Н5ю6 н+ + Н4Ю6-, К1а = 5,1 • 10-4.
Н41О6- 2Н2О 4-107, = *>»
Н4ю6- н+4-н3ю^-,
= 2,0 • 10~7.
Была сделана попытка [29] различить октаэдрическую
структуру Na4IO6 • Н20 и тетраэдрическую структуру
NaIO4*3H2O с помощью ИК-спектров поглощения три-
гидрата перйодата натрия в области 600—4000 см~1.
В спектрах обнаружены все особенности, характерные
для иона НДОв. а также полосы поглощения 1650 и
3400 см~1 кристаллизационной воды. Полученные спект-
ры очень похожи на спектры Na2H3IO6 и NasH2IOe и
резко отличаются от спектров NaIO4 и КЮ4, которые
характеризуются одной интенсивной полосой 840 слг1.
Данные о влиянии на иодную кислоту сильных кислот
[30] достаточно убедительно показывают существование
1(ОН)^. Описаны реакции [31], которые дают основания
считать, что фотохимическое разложение перйодата про-
исходит по механизму свободных радикалов
H4IOg + Av H3IO5- . 4- НО •, (1)
H3IOS- • —> Ю3- 4- H2o 4- HO.(2)
H4IO6- 4- HO. H3IO5-. 4- H2O2, (3)
H4IO6 4- H2O2 —> IO3- 4- 3H2O -P Oj. (4)
Исследуя окисление пинакола водным раствором пе-
рйодата, Прайс и Кролль [32] обнаружили, что реакция
протекает по простому бимолекулярному механизму.
Затем была изучена кинетика окисления этиленгликоля,
пинакола и цис- и гранс-циклогексенгликоля в широком
интервале pH [33]. Полученные результаты обсуждались
с точки зрения образования циклического эфира иодной
Реакции окисления через стадию этерификации
159
КИСЛОТЫ
I I
—С—ОН —с—о
| +Н6Ю.5Г> I \Ю4Н3 + 2Н2О
—С—ОН С—(У
I I А
А —► 2^>С=О-|-Н1О3+ Н2О
Реакциям окисления перйодатом посвящены обзоры
Джексона [34] и Боббита [35]. Последний высказал важ-
ное соображение о том, что наиболее «избирательные»
реакции окисления перйодатом протекают в гомогенной
смеси быстро и количественно.
При исследовании процесса окисления гликоля пе-
рйодатом Дюк [36] нашел, что скорость реакции описы-
вается уравнением
где А — общая концентрация перйодата, k'—константа
скорости, К — константа равновесия образования про-
межуточного комплекса, с — концентрация некоордини-
рованного гликоля. Форму кинетического уравнения
можно объяснить также, если предположить, что вна-
чале образуется комплекс между перйодатом и глико-
лем, который затем распадается. Эта реакция имеет
сходство с реакцией окисления гликоля ионами це-
рия (IV).
Более подробно исследовали эту реакцию Бантон с
сотрудниками. В предварительной заметке об окисле-
нии а-гликолей иодной кислотой [37] они отметили не-
которые особенности поведения комплексов между гли-
колем и перйодатом:
1) при смешении иодной кислоты с гликолем наблю-
дается повышение кислотности;
2) максимум светопоглощения л<ета-периодата при
длине волны 2225 А немедленно снижается при добав-
лении гликоля;
3) при окислении пинакола, где возможны стериче-
ские затруднения, нет доказательств образования про-
межуточного соединения.
I (И)
Глава 6
В последующих статьях авторами детально рассмот-
рены следствия из этих предварительных результатов и
предлагается схема протекания реакции:
и5ю6 ^=± Н4ю6- + н+ ^=± Н3ю|- 4- н+
/ СН2—О \
4-0 +°=1 / н-о
\ СН2—ох/
lb° IP' Ik
н3ою4 -—» Н2ою4- —-> HOIO*"
III
kt k{ k2
Определены [38] величины констант для всех стацио-
нарных равновесий и констант скорости.
Дюк и Бэлгрин [39] показали, что удельная скорость
окисления в гомологическом ряду гликолей, от этилен-
гликоля до пинакола, имеет разрыв непрерывности
между триметиленгликолем и пинаколом. Последний
медленно окисляется в катализируемой бимолекулярной
стадии, а триметиленгликоль — по некаталитической мо-
номолекулярной реакции.
Позднее было исследовано окисление а-гликолей
[40а]: 1,2-пропандиола, 2-метил-1,2-пропандиола, 2,3-
бутандиола и 2-метилбутан-2,3-диола. Общий эффект
замещения водорода метиловой группы заключается в
увеличении скорости распада а-гликолей. Константа
равновесия образования комплекса между гликолем и
перйодатом увеличивается при этом в 2 раза.
Освобождение электрона повышает, а стерические
затруднения понижают величину константы равновесия,
причем последний эффект зависит от стереохимического
строения молекулы гликоля. Обратимое образование
промежуточного соединения между иодной кислотой и
а-гликолем обычно происходит быстрее, чем его необ-
ратимый распад [406].
Реакции окисления через стадию этерификации
161
Однако в случае 2-метилбутан-2, 3-диола при pH 4,5
скорости образования и распада циклического проме-
жуточного соединения сравнимы [41].
Очень интересные результаты получены при иссле-
довании окисления перйодатом 1,2-диолов, дикетонов и
оксикетонов с использованием 18О [42]
Н6Ю6+Н18О Н5118О6 + Н2О
НО
R2COH I /О—CR2
H5I'»Oe+ I —> ‘»О=1< —> 2R2C=O.
r2coh I ХО—cr2
18Q
Оказалось, что кислородный атом в ацетоне при
окислении пинакола иодной кислотой поступает из мо-
лекулы 1,2-диола. Это соответствует предположению,
что промежуточное соединение образуется при электро-
фильной атаке перйодата. Напротив, один атом кисло-
рода уксусной кислоты, образующейся при окислении
биацетила перйодатом в щелочном растворе, поступает
из молекулы перйодата. Механизм реакции представ-
ляет собой нуклеофильное действие на углерод карбо-
нильной группы; в реакции, очевидно, не участвуют
гидраты кетона как промежуточные соединения.
Разработана новая методика окисления, в которой
применяется смесь перйодата и перманганата [43]. Ме-
ханизм такого окисления, очевидно, состоит в том, что
вначале происходит гидроксилирование двойной связи,
которая затем быстро разрывается под действием перйо-
дата. Образующиеся вещества могут затем последова-
тельно окисляться перманганатом.
Окисление ацетатом иодозобензола
Путем непосредственного хлорирования иодидов в
соответствующем растворителе можно получить хлориды
иода. При гидролизе последних возникают иодозосое-
динения, являющиеся сильными окислителями.
Иодозобензол ведет себя как двухкислотное ос-
нование. Ацетат иодозобензола представляет собой
П Зак. 1249
162
Глава 6
бесцветные кристаллы, хорошо растворимые в уксусной
кислоте, хлороформе и бензоле
Позакер [44а] рекомендует следующую методику по-
лучения ацетата иодозобензола: 70 мл 30%-ной пере-
киси водорода и 305 мл ангидрида уксусной кислоты
перемешивали при 40е в течение 4 час. К раствору при-
бавляли 50 г иодобензола, и смесь оставляли на ночь.
Выкристаллизовавшееся за ночь небольшое количество
фенилиодозоацетата отфильтровывали, а фильтрат кон-
центрировали под вакуумом (70 мм рт.ст.) и получали
вторую порцию кристаллов. Объединенные фракции
кристаллов промывали эфиром и высушивали в вакуу-
ме. Выход продукта с т.пл. 158° составлял 58 г.
Позакер с сотрудниками исследовал также реакции
окисления первичных ароматических аминов [446]. На-
пример, анилин окисляется по схеме
2CeH6I(OCH8CO)24-2ArNH2 2CeH6l(OCH3CO)NHAr-|-2CHaCOOH
I
2CeH5IOCHaCO • 4-ArNHNHAr 2ArNH . + 2CeH6IOCHaCO •
I
2СбНб1 + 2СН3СООН -|- ArN=NAr.
Предлагается интересный механизм [45] для реак-
ции окисления фенолов, содержащих электрофильные
заместители; происходит электрофильная атака арома-
тического кольца с последующей перестройкой моле-
кулы.
Окисление четырехокисыо осмия
Четырехокись осмия часто применяется в качестве
реагента для присоединения по двойным связям. В его
молекуле находится четыре атома кислорода, тетраэд-
рически расположенных вокруг осмия. Об этом свиде-
тельствуют ИК-спектры поглощения и спектры комби-
национного рассеяния четырехокиси осмия при 60° [46].
Стандартный потенциал для системы OsO2—OsO4 равен
—0,95 в [47].
ОвО2 (тв.) + 2Н2О —► OsO4 (раств.) + 4Н * 4" 4е.
Реакции окисления через стадию этерификации
163
Четырехокись осмия можно рассматривать как ангид-
рид гипотетической слабой осмиевой кислоты H2OsO5.
На основании спектрофотометрических измерений най-
дены кажущиеся константы диссоциации осмиевой кис-
лоты, Ki= 1,0 • 10-12 и К2 = 3,0 • 10-15, в растворе КОН с
ионной силой, равной 1, при 25° [48].
Методика применения четырехокиси осмия заклю-
чается, по Физеру [56], в следующем: четырехокись ос-
мия взаимодействует с эфиром или диоксаном при ком-
натной температуре, и через 2—4 дня выделяется
осмиевый эфир. Последний переводят в гликоль с по-
мощью водноспиртового раствора сульфида натрия, суль-
фита натрия совместно с цинковой пылью, щелочного
раствора формальдегида или цинковой пыли.
Четырехокись осмия присоединяется к олефинам с
образованием циклического соединения
НС—О
НС—о/
,0
°<
Если в качестве растворителя взять пиридин, то четы-
рехокись осмия присоединяет сразу две молекулы пири-
дина [49].
Препаративно исследовалась реакция различных кан-
церогенных углеводородов (3,4-бензпирен, 1,2-бензантра-
цен и их гомологи) с четырехокисью осмия [50] в бен-
зольно-пиридиновой среде. Кук и Шенталь применяли
следующую методику [50].
К раствору 0,0025 моля углеводорода в 15 мл бен-
зола прибавляли 0,0025 моля четырехокиси осмия в
0,005 моля очищенного пиридина. Раствор медленно тем-
неет и выпадают темно-коричневые микрокристаллики
комплекса.
Беджер [51] количественно исследовал реакции при-
соединения четырехокиси осмия по двойной связи боль-
шого числа канцерогенных углеводородов. В его работе
показано, что реакция имеет первый порядок относи-
тельно углеводорода и четырехокиси осмия. Замеще-
ние алкильными группами в «благоприятствующие»
11*
164
Глава 6
положения молекулы углеводорода повышает скорость
присоединения OsO4, в то время как группа циана в тех
же положениях понижает скорость присоединения.
Целиков и Тейлор [52] изучали кинетику окисления
фумаровой и малеиновой кислот реагентом, содержа-
щим четырехокись осмия и хлорат. Для этой реакции
ими предложен механизм
ноос. С—н
II +OsO4
ноос. с—н
/Н
ноос • с\
hooc.c<Os°2
\н
/Н
ноос.с<
ноос (.>0'0>+«=0 + ксю--ад*
nUUL •
\н
н
I
ноос-с—он
—> | +OsO4 + KCl
ноос. с—он
I
н
В ходе реакции четырехокись осмия регенерируется.
Реакция с четырехокисью осмия очень полезна при ис-
следовании структуры полициклических ароматических
углеводородов [53], поскольку этот реагент, очевидно,
всегда действует на наиболее реакционноспособную
связь. Теоретическое рассмотрение таких реакций дано
Брауном [54].
Опубликованы предварительные результаты приме-
нения для тех же целей четырехокиси рутения [55], кото-
рый менее токсичен по сравнению с четырехокисью ос-
мия. Четырехокись рутения легко реагирует с различ-
ными функциональными группами и ароматическими
соединениями, например с циклогексеном [56]
| || + RuO4—>
/\СНО
\/СНО
—h RuOg
Реакции окисления через стадию этерификации
165
Окисление двуокисью селена
Исследования по применению двуокиси селена опуб-
ликованы Райли с сотрудниками еще в 1932 г. [57].
Двуокись селена представляет собой ангидрид селе-
нистой кислоты, причем эти два соединения обычно реа-
гируют одинаково. По сообщению Дюка [58], ацетон ре-
агирует с селенистой кислотой по следующему стехио-
метрическому уравнению:
СН3СОСН3 + H2SeO3 —> CH3COCHO + 2H2O4-Se.
Скорость этой реакции описывается уравнением
— rf[H2SeO3]/^ = ^[CH3COCH3] [H2SeO3l [Н+].
Кинетика реакции селенистой кислоты с иодом [59]
4Н + + H2SeO3 4-41“ —> Se 4-ЗН2О 4-212
дает основание считать, что в процессе реакции возни-
кают два активных комплекса: SeIOH(H2O)n и
SeI3O(H2O)m. Известна также другая количественно
протекающая реакция между селенистой кислотой и
аскорбиновой кислотой [60]
2CeH8Oe4- H2SeO3 —> 2С6НбОб 4“ЗН2О 4-Ss.
В среде концентрированной серной кислоты двуокись
селена образует ярко-желтый раствор [61], вероятно,
вследствие ион из ации:
SeO24- H2SO4 —> SeO2H+ 4- HSO4"
Систематически исследовались процессы окисления ке-
тонов [62]. При окислении дезоксибензоина в 70%-ной
уксусной кислоте наблюдается каталитическое действие
как кислот, так и ионов ацетата:
ArCOCH2Ar'4-SeO2 —> АгСОСОАг'4-Se 4-Н2О.
В случае кислотного катализа найдено следующее вы
ражение для скорости реакции:
— d[SeO2]/^ = fc[SeO2] [СбН5СОСН2С6Н5] [Н+].
Кинетический изотопный эффект &h/&d=6.
166
Глава 6
В случае катализа основаниями скорость реакции
описывается уравнением
— d[SeO2]/^ = £ [SeO2] [CeH5COCH2CeH5] [СН3СОО~].
а кинетический эффект в этом случае &н/^п = 7,6. Для
случая кислотного катализа предлагается следующий
механизм:
OSeO2H
С6Н5СОСН2С6Н5 + H3SeOj <_медленно-> свН5С=СНСвНБ + Н8О+
^быстро
СвН5СОСОСвНб + Se + Н2О
При окислении 1,2-дибензоилэтана [63а] реакция ката-
лизируется как кислотами, так и основаниями
2СвНбСОСН2СН2СОСвН5 + SeO2 —► 2СбН5СОСН=СНСОСвНБ.
При замещении водорода а-метиленовой группы на дей-
терий наблюдается большая величина кинетического
изотопного эффекта, что соответствует определяющей
стадии образования эфира между енолом и селенитом.
После этой лимитирующей стадии следуют быстрые
стадии разложения эфира.
Измерены [636] скорости реакций окисления дезокси-
бензоина, n-нитробензилфенилкетона и и-метоксибензил-
фенилкетона в 80—99%-ных растворах уксусной кис-
лоты, содержащих ацетат натрия.
Для этих реакций обнаружены каталитическое дей-
ствие оснований и образование промежуточного эфира
селенистой кислоты.
Подводя итоги изложенному, можно отметить, что
хорошими примерами окисления через стадию этерифи-
кации служат реакции с бихроматом, перйодатом, че-
тырехокисью осмия и двуокисью селена.
ЛИТЕРАТУРА
i. Oppenauer R. V., Oberrauch Н. Chem. Abstr., 44, 3871
(1950).
2. В о w а г $ А., Н а 1 s а 11 Т. G., Jones Е. R. Н., Lemin A. J.,
J. chem. Soc., 254, 2548 (1953).
3. N е и s $ J. D., R е i m a n n W., J. Am chem. Soc., 56, 2238 (1934).
Реакции окисления через стадию этерификации
167
4. Н о w а г d J. R., N a i г V. S. К, NancollasG Н., Trans. Fa-
raday Soc., 54, 1034 (1958) .
5. Fieser L. F., Experiments in Organic Chemistry, Heath, London,
1955, (a) p. 309; (6) p. 331.
6. Tuey G. A. P, Chem. a Ind., 26, 766 (1948).
7. W e s t h e i m e r F. H., Chem. Rev., 45, 419 (1949).
8. Waters W. A., Quart. Rev. chem. Soc., Lond., 12, 277 (1958).
9. Westheim er F. H., Novick A., J. chem. Phys., 11, 506
(1943).
10. Westheimer F. H., Nicolaides N., J. Am. chem. Soc., 71,
25 (1949).
11. Holloway F., Cohen M., Westheimer F. H., J. Am. chem.
Soc., 73, 65 (1951).
12. L e о A., Westheimer F. H., J. Am. chem. Soc., 74, 4383
(1952).
13. С о h e n M., Westheimer F. H., J. Am. chem. Soc., 74, 4387
(1952).
14. Kaplan L., J. Am chem. Soc., 77, 5469 (1955).
15. Zeiss H. H., Matthews C. N., J. Am. chem. Soc., 78, 1694
(1956).
16. К wart H., Acta chem. fenn., 34, 173 (1961).
17. Wiberg К. B., Mill T., J. Am. Chem. Soc., 80, 3022 (1958).
18. Graham G. T. E., Westheimer F. H., Am. chem. Soc., 80,
3030 (1958).
19. C hatter ji A. C., Mukherjee S. K-, J. Am. chem. Soc., 80,
3600 (1958).
20. Whitmore F. С., P e d 1 о w G. W., J. Am. chem. Soc., 63, 758
(1941).
21. Slack R., Waters W. A., J. chem. Soc., 1666 (1948).
22. Wiberg К. B., Evans R. J., Tetrahedron, 8, 313 (I960).
23. Wiberg К. B., Foster G., J. Am. chem. Soc., 83, 423 (1961).
24. BrandenbergerS. G., MaasL. W., Dvoretzky I., J. Am.
chem. Soc., 83, 2146 (1961).
25. С г о u t h e m a 1 С. E., Meek H. V., Martin D. S., Banks
С. V., J. Am. chem. Soc., 71, 3031 (1949).
26. S i d g w i с к N. V., The Chemical Elements and Their Com-
pounds, Oxford University Press, 1951, p. 1236.
27. Van A г к e 1 A. E., F r i t z i u s С. P., Rec. Trav. chim Pays-Bas,
50, 1035 (1931).
28. С г о u t h e m a 1 С. E., Hayes A. M., МЪ r t i n D. S., J. Am.
chem. Soc., 82, 73 (1951).
29. Keen N., Symons M. C. R., Proc. chem. Soc., 383 (1960).
30. Mishra H. C., Symons M. C. R., J. chem. Soc., 1194 (1962).
31. Symons M. C. R., J. chem. Soc., 2794 (1955).
32. Price С. C., Kroll H., J. Am. chem. Soc., 60, 2726 (1938).
33. Price С. C., Knell M., J. Am. chem. Soc., 64, 552 (1942).
34. Jackson E. L., Org. React., 2, 341 (1944).
35. Bobbit J. M., Rec. Adv. Carbohydrate Chem., 11, 1 (1956).
36. Duke F. R., J: Am. chem. Soc., 69, 3054 (1947).
37. Buist G. J., Bunton C. A., Shiner V. J., Research, 645
(1953).
168
Глава 6
38. Bui st G. J., Bunton C. A., J. chem. Soc., 1406 (1954).
39. D u к e F. R., В u 1 g г i n V. C., J. Am. chem. Soc., 76, 3803 (1954).
40. В u i s t G. J., Bunton C. A., Miles J. H., J. chem. Soc., (a)
4567; (6) 4575 (1957).
41. Bui st G. J., Bunton C. A., J. chem. Soc., 4580 (1957).
42. Bunton C. A., Shiner V. J., J. chem. Soc., 1593 (1960).
43. Lemieux R. U., Rudi off E. von, Can. J. Chem., 33, 1701
(1955).
44. Pa us acker К. H., J. chem. Soc., (a) 107; (6) 1989 (1953).
45. Fox A. R., Pa us acker К. H., J. chem. Soc., 295 (1957).
46. Woodward L. A., Roberts H. L., Trans. Faraday Soc., 52,
615 (1956).
47. С a r 11 e d g e G. H., J. phys. Chem., 60, 1468 (1956).
48. S a u e r b r u n n R. D., S a n d a 11 E. B., J. Am. chem. Soc., 75,
4170 (1953).
49. С r i e g e e R., Marchand B., W a n n о w i u s H., Lieb’gs Ann.,
99, 550 (1942).
50. Cook J. W., Schoental R., J. chem. Soc., 170 (1948).
51. В a d g e r G. M., J. chem. Soc., 456 (1949).
52. Zelikoff M., Taylor H. A., J. Am. chem. Soc., 72, 5039
(1950).
53. Badger G. M., Quart. Rev. chem. Soc., London, 5160 (1951).
54. Brown R. D., Quart. Rev. chem. Soc., London, 6, 89 (1952).
55. Djerassi C., Eagle R. R., J. Am chem. Soc., 75, 3838 (1953).
56. В а г к о w i t z L. M., R у 1 a n d e r P. N., J. Am. chem. Soc., 80,
6682 (1958).
57. Riley H. L., et al., J. chem Soc., 1875, 2342 (1932); 391 (1933);
844 (1934); 801 (1935).
58. Duke F. R., J. Am. chem. Soc., 70, 419 (1948).
59. N e p t u n e J. A., King E. L., J. Am. chem. Soc., 75, 3069
(1953).
60. Deshmukh G. S., В a pat M. G., Chem. Ber., 88, 1121 (1955).
61. F1 о w e r s R. H., Gillespie R. J., Robinson E., J. inorg.
nucl. Chem., 9, 155 (1959).
62. Corey E. J., Schaefer J. P., J. Am. chem. Soc., 82, 918
(1960).
63. Schaefer J. P., J. Am. chem. Soc., 84, (a) 713; (6) 717 (1962).
Глава 7
ГОМОЛИТИЧЕСКОЕ ОКИСЛЕНИЕ В РАСТВОРЕ
В этой главе рассматриваются процессы окисления,
протекающие в растворе гомолитическими путями. Не-
сколько примеров реакций свободных радикалов, ини-
циированных ионами, уже обсуждалось в главе 2. Про-
цессы окисления с участием газообразного бирадикала
кислорода будут рассмотрены ниже в гл. 8 и 9. Для
протекания гомолитических процессов в растворе боль-
шое значение имеет природа растворителя. Здесь мы
ограничимся общим замечанием: чем более неполярным
является растворитель, тем выше вероятность протека-
ния реакции гомолитическим путем.
Из наиболее важных физических методов обнаруже-
ния и изучения свободных радикалов в растворе можно
отметить: измерение магнитной восприимчивости [1] и
электронный парамагнитный резонанс (ЭПР) [2]. По-
следний метод имеет особенно важное значение для
прогресса в этой области науки. При помощи ЭПР, на-
пример, доказано присутствие свободных радикалов в
таких средах, как биологические. Приведем в качестве
простейшего примера реакцию [3]
tihi4-h2o2 —> Tiiv-pHo. 4-но".
Если смешать кислые растворы, содержащие ионы ти-
тана и перекись водорода, и затем сразу же пропустить
смесь через резонатор спектрометра ЭПР, то возникает
синглет, который приписывается образованию радикала
гидроксила.
Обычный пример воздействия радикала на насыщен-
ные соединения описывается схемой [4]
R . + н-С^ —> rh + •
170
Глава 7
Действие радикала на водород в цепи насыщенного
углеводорода может быть заметно избирательным. Фак-
торами, определяющими направление этой реакции, осо-
бенно в отношении степени разрыва связи в переходном
состоянии, являются диссоциация связи С—Н и реак-
ционная способность исходных и возникающих при ре-
акции радикалов.
Обстоятельное описание реакций свободных радика-
лов приводится в книге Тротмен-Диккенсона [5]. Про-
блема определения места молекулы, подвергающейся
атаке свободными радикалами, рассматривалась теоре-
тически Фукуи и другими авторами [6]. Наиболее вос-
приимчивым к воздействию должно быть место, в кото-
ром наблюдалась бы повышенная плотность двух элек-
тронов, из которых один занимает наиболее высокую, а
другой-—наиболее низкую незанятую орбиту основного
состояния.
Маккинон и Уотерс [7] считают, что органические
свободные радикалы и находящиеся с ними в равнове-
сии катионы и анионы представляют пары термодина-
мически обратимых окислительно-восстановительных
систем:
Таким образом, каждый радикал характеризуется дву-
мя различными редокс-потенциалами Е(_е) и Е(+е). В ре-
акциях с неорганическими ионами радикал может вы-
ступать и как окислитель и как восстановитель:
R. + М (х) —R+ 4- М (х +1),
R. +2V(!/) —> R' 4"^(£/ —1).
где х и у — числа окисления для неорганических ионов.
Мерц и Уотерс [8] классифицировали органические
соединения (RH) на две группы по признаку: является
ли окисление данного вещества реагентом Фентона цеп-
ной или нецепной реакцией. Принадлежность соедине-
ния к одной из групп зависит от величины окислитель-
ного потенциала Е^ радикала в реакции
R. :p±R+-K
Гомолитическое окисление в растворе
171
При этом вещество, подвергающееся цепной реакции
окисления, имеет величину £(-е), значительно меньшую
Ео для реакции
Fe" Fe1" + е.
Дейнтон [9] рассматривает свободные радикалы как
кислоты и основания. Атомы водорода и радикалы гид-
роксила, по его мнению, способны принимать участие
в равновесиях
^2~(aq) ^(aq) + ’ ^(aq)»
’ H(aq)+ 0H(aq) H2°(aq) (T* e’ *aq)’
H2O(1q)^=> H+q)+.OH(aq),
•OHCaq) + OH(-aq) H2O + O(aq).
Продолжительность жизни радикалов H- и *ОН, по-ви-
димому, настолько мала, что нет возможности выполг
нить термодинамические измерения. Для различения от-
меченных равновесий необходимо использовать такие
реагенты, которые могут избирательно реагировать
только с одним видом радикалов. Так, например, с по-
мощью тяжелого изотопа кислорода можно отличить ча-
стицы ОН и О’, поскольку он участвует в изотопном
обмене с О’ и не реагирует с радикалом • ОН.
i«o" + i»ol8O х •> i«oi8O+ |8О“
' быстро
Если радикалы гидроксила возникают в воде, обога-
щенной изотопом 18О, то скорость изотопного обмена
резко увеличивается с повышением pH раствора, так
как параллельно другим реакциям с' участием радикала
"ОН начинает протекать следующая:
•OH(aq)+OH(;q) н2о+о(;ч).
Реакции свободных радикалов в растворе рассмат-
риваются здесь в следующей последовательности:
а) реакции радикала гидроксила (’ОН);
б) реакции замещенных радикалов гидроксила
(•OR);
в) реакции иона пероксидисульфата (БгОв );
г) некоторые другие реакции (тетраацетат свинца,
соль Фреми).
172
Глава 7
Радикал гидроксила и его реакции
Источником радикала гидроксила является перекись
водорода и перекисные кислоты. В молекуле перекиси
водорода связи атомов водорода на противоположных
концах цепочки —О—О— изогнуты наружу под углом
100° к линии цепочки. Угол между ними также состав-
ляет 100° [10]. Физические характеристики — дипольный
момент и ИК-спектр поглощения — подтверждают пра-
вильность этой модели. Перекисные кислоты, которые
также содержат группу —О—О—, можно разбить на
две категории: монокислоты (НООА) и дикислоты
(АООА) (см. обзор Эмеле и Андерсона [И]). Катали-
заторы способны повышать реакционную активность пу-
тем образования перекисных кислот.
Энергетику реакции разложения перекиси водорода
можно описать с помощью термодинамического цик-
ла [12]
Н2 -]- О2 qj > Н2О2
-dhJ | ~do2 |dho- - он
2H-J-2O 2ОН
Значение энергии диссоциации Оно «он тогда можно
найти из соотношения
Оно • . он = Qf + Он2 + О^ 2D0H
Она составляет 2,34 • 108 дж/кмоль. Радикал гидроксила
можно получить путем облучения перекиси водорода
ультрафиолетовым светом [13].
Наиболее простым способом получения радикала
гидроксила служит реакция перекиси водорода с катио-
нами X11, например с ферро-ионами
ХП+НООН —> Х1н + ОН'“+ - ОН
или интенсивное облучение воды
Н2о —► Н. + • ОН,
Гомолитическое окисление в растворе
173
при котором реакция протекает в следующей последо-
вательности стадий [14]
Н2О —> н2о+-н,
н2оь > н4 + • он,
Н2О+<? —> Н2О“,
н2о" —>н.4-он".
Фентон [15] открыл, что следы сульфата железа (II)
в охлажденной льдом перекиси водорода быстро окис-
ляют винную кислоту до диоксималеиновой кислоты.
В этих условиях окисляются также и другие а-оксикис-
лоты и гликоли [16, 17]. Эта реакция многократно ис-
следовалась. В первом систематическом исследовании
Габер и Вайс [18] предположили, что суммарная реак-
ция
2Feii + Н2О2 + 2Н+ —► 2FeHl4-2H2O
протекает через следующие стадии:
Fen + НООН —► Fe"! + • ОН + : ОН",
• ОН + НООН —> Н2О+ - ООН |
• оон + ноон —► о=о 4-н2о 4-. он J цепь*
FeH 4- .ОН —► Fein4-: ОН"
Редокс-потенциал системы ОН" -> *ОН+е равен
—2,0 в [19]. Для определения возможности протекания
данной реакции окисления с участием радикала гидро-
ксила необходимо рассчитать величину изменения сво-
бодной энергии реакции
Х+ .он —> Х+4-ОН".
Так, например, расчет показывает, что Соп можно окис-
лить до CoITI, a F" до F2 нельзя.
По мнению Бексендейла и сотрудников [20], полиме-
ризация, инициированная ферро-ионами, происходит по
схеме
Н2О24-Ре» —► ОН-4- • ОН + Fein,
• ОН 4-М —► —МОН,
—мон 4-м —► —(М)2он,
-(М)„ОН4-М —> -(М)я+1он.
174
Глава 7
Цепной механизм подтверждается очень высокой чув-
ствительностью этой реакции к кислороду. Мери и Уо-
терс [21] показали, что спирты и альдегиды окисляются
перекисью водорода в присутствии избытка солей Fe11
по следующему механизму:
Зарождение цепи Fe2+4 НО—ОН —> Fe31 -f- ОНОН)~,
Развитие цепи RCH2OH + • ОН RCHOH 4- Н—ОН,
RCHOH + H2O2 —> RCHO 4- -ОН4-Н2О,
Обрыв цепи (при низкой концентрации спирта)
Fe2++ ОН —> Fe3+ + (: ОН)"
Обрыв цепи (при высокой концентрации спирта)
2RCHOH — -> RCHO4-RCH2OH.
Те же авторы отмечают [22], что реакция окисления аро-
матических соединений перекисью водорода в присут-
ствии ионов Fen идет не по цепному механизму, не-
смотря на образование свободных арильных радикалов.
Свободный радикал фенила не реагирует с водой, а
частично димеризуется с образованием дифенила и ча-
стично соединяется с гидроксилом, образуя фенол. То-
луол в этих условиях дает дибензил, бензальдегид и
крезолы. При окислении бензойной кислоты, бензамида,
нитробензола и хлорбензола образуются ортозамещен-
ные фенолы и продукты дальнейшего окисления (только
не диарилы).
Исследуя механизм этой реакции, Барб с соавтора-
ми [23] пришли к заключению, что радикал НО2 не
взаимодействует с перекисью водорода, и предложили
другую модификацию механизма реакции:
Fen4-H2O2 ► Реш + -ОН + ОН' £0,
Fen-]- - ОН —► Fein + OH" kb
Н2О2+ - ОН —► Н2О + НО2 k2.
Feu + HO2- —► Fein + HO2
Feu + HO2. —>Fein4-H+ + O2 k4.
Для реакций, протекающих в системе Fe3+X", аро-
матическое соединение при облучении ультрафиолето-
вым светом, предлагается следующая схема [24]:
Гомолитическое окисление в растворе
175
1) Поглощение света
Fe3+X~ —Fe2+X
2) Первичная обратная, темновая реакция
Fe2+X —> Fe’+X"
3) Диссоциация возбужденного комплекса
Ре2+Х —> Fe2+4-X-
4) Вторичная обратная, темновая реакция
Fe2+ + X • —► Fe’+ -f- X-
5) Действие свободного радикала на окисляемое
вещество
RH + X- —». R. +НХ
6) Окисление радикала R-
R. 4-Fe3+X" —► RX-|-Fe2+
7) Обрыв цепи при низких концентрациях ионов
железа (III)
a) R. -f-х —> RX.
б) R. Ц-R. —► R,.
Продукт взаимодействия перекиси водорода с азо-
тистой кислотой (вероятно, пероксиазотистая кислота)
может инициировать реакции полимеризации метил-
акрилата, а также гидроксилирования и нитрования бен-
зола [25]. Реакция, по-видимому, протекает с участием
радикала гидроксила, образующегося по схеме
HONO4-HOOH НО, • ON 4- Н2О —» НО -|-NO2..
Для реакции радикалов гидроксила с водородом и
окисью углерода в водном растворе предлагается сле-
дующий механизм [26]:
1) Fe"4-H2O2 —► Fel"4-HO. 4-НО“
2) НО +Н2 —► Н2О4-Н. 1
3) НО. 4-Fell — НО-4-Fein ) ПРОИСХОДЯТ параллельно
4) Н- +О2 —► НО2- быстро и полно
5) НО2- H-Fe11 —► НО^ 4-Fe111 быстро и полно
6) НО?“ + Н+ —> Н3О3.
176
Глава 7
При использовании вместо водорода окиси углерода
стадия (2) заменяется реакцией
НО • +СО —> СО2+Н.
Реакции гидроксилирования бензола в водном рас-
творе в присутствии избытка ионов Feln приписывается
следующий механизм [27]:
1) АгН + НО. —> Аг - + Н2О,
2) FeHi + Н2О 4- Аг • —> АгОН 4-Ре" 4-Н+,
3) Fen + Н2О2 —> Fel 11 + HO • + HO"
Образование биарила происходит параллельно гидро-
ксилированию как преобразование молекулы окисляе-
мого органического соединения:
4) Аг - 4-Аг- —► Аг—Аг,
5) Аг - 4- Fe114- Н+ —► АгН 4- Fe111.
Эта реакция послужила предметом последующего де-
тального исследования [28].
Если добавлять по каплям перекись водорода к интенсивно пе-
ремешиваемой эмульсии бензола в разбавленной серной кислоте, со-
держащей сульфат железа(II), то фенол и дифенил образуются с
низким выходом. Их относительные количества зависят от скорости
добавления перекиси водорода. Доля дифенила повышается от 8,5
до 39% при увеличении скорости добавления перекиси в 60 раз. При
заданной скорости механического перемешивания и добавления пе-
рекиси водорода отношение продуктов реакции остается постоянным
и хорошо воспроизводится.
Исходя из величин выхода ионов железа (III) в вод-
ных растворах перхлората железа (II) и перекиси во-
дорода, подкисленных хлорной кислотой и содержащих
либо Н2, HD, либо D2 и О2, были найдены следующие
отношения констант скоростей реакций [29]:
-(1< ±
‘оН + 0,/‘<>Н + Ге„ "Об ±
Радикал гидроксила играет также важную роль при
радиолизе воды [30]. Были предприняты попытки рас-
считать отношение кинетических констант
*QH + s/AOH + HaO2 = roh, s
Гомолитическое окисление в растворе
177
для различных растворенных веществ S. Вследствие
большого значения перекиси водорода при радиолизе
воды как эталон была принята реакция
но. +Н2о2 —НО2. +Н2О.
Было исследовано действие у-облучения 60Со на водные
растворы бензола и измерены выходы фенола, дифенила
и Н2Ог как функции различных параметров. Реакция
в 0,8 н. растворе серной кислоты протекает по схеме
Н • Ц- СвНв —> СвН7 • —> полимер,
НО • + СвНв —> СвНб. + Н2О2
НО • + Н2О2 —► Н2О + НО2 •
Образующиеся по этим реакциям радикалы реагируют
в последующих стадиях.
В другой работе [31] изучалась реакционная способ-
ность радикалов гидроксила, образующихся под дейст-
вием ионизирующего излучения на водные растворы
ферро- и феррицианидов в интервале значений pH 2,5—
10,5. В присутствии добавок органических веществ можно
было определить отношение констант скоростей реакций
^ОН 1 феррицианид/^ОН-|-RH2*
Замещенные оксирадикалы (RO-)
Аналогично образованию гидроксильных радикалов
при взаимодействии перекиси водорода и ионов металла
из органических гидроперекисей могут быть получены
радикалы RO по реакции
МЛ++КООН —> Мл+1 + ОН-+ИО.
Радикалы ROO- образуются по реакции
мл+1+иоон —> мл+ + иоо.+н+.
Уотерс [32] отмечает, что радикалы ROO- могут быть
как слабыми окислителями, так и слабыми восстанови-
телями. Караш и сотрудники [33] нашли, что в водных
растворах, содержащих Fe11, гидроперекись а-кумила
разлагается на ацетофенон, метанол, этан и а, а'-диме-
тилбензиловый спирт. Эксперименты показывают, что
на первой стадии реакции, по-видимому, образуется ра-
дикал С6Н5С(СНз)2СО-, который затем может давать
различные продукты в результате либо восстановления,
12 Зак. 1249
178
Глава 7
либо диспропорционирования, либо под действием дру-
гой молекулы гидроперекиси а-кумила.
В развитие этих идей выполнено исследование реак-
ции между гидроперекисью кумола и комплексами
железа (II) с полиэтиленаминами общей формулы
H2N(C2H4NH)XH, где х = 2, 3, 4 и т. д. [34]. Реакция
имеет псевдопервый порядок относительно гидропере-
киси, а первой стадией, вероятно, является следующая:
ROOH + RNH2A —> RO. 4-H2O4-RNH. 4-а,
где А может быть Fe2+, Fe(OH)+ или Fe(OH)2.
В другой работе [35а] в качестве основных продуктов
реакции между ионами железа (II) и гидроперекисью
кумола обнаружены ионы железа (III), ацетофенон и
этан. Как в Н2О, так и в D2O реакция протекает со ско-
ростью /? = fc[Fen] [гидроперекись кумола]. Это дает ос-
нования предполагать, что реакция имеет следующий
механизм:
Feii+ROOH —Fem + RO - 4-НО",
RO- —> CeHeCOCH8 + CH,..
2СН,- —> С2Н„.
Реакция была видоизменена заменой ионов железа (II)
на комплексонат железа (II) и пирофосфат железа (II)
[356]. Стехиометрические соотношения, наблюдаемые в
присутствии и в отсутствие акрилонитрила, показывают,
что альтернативные пути реакции между свободными
радикалами полимера и комплексами железа (II), по-
видимому, могут происходить при низких концентрациях
реагирующих веществ. В присутствии мономера главными
продуктами реакции при низких концентрациях являются
комплекс Fera, ацетофенон и полимер. Схема реакции:
Fe(OH)mY-2-m4- ROOH ---*1—^ Pe(OH)mY-1-m+RO-+ОН~
1,
RO. -% СвНбСОСН8 + СН8.,
Зарождение цепи СН8. -|- М ,
Ч
Развитие цепи М, • М —-> М2 •,
м„_,- 4-м м„..
Fe(OH)mY“2-m 4- М„ - —> Fe(OH)/nY"1-'» 4- М;1,
m;4h+ —<► нм*.
Гомолитическое окисление в растворе 179
Стационарная концентрация кумилпероксирадикала
была определена Томасом [36] с помощью спектров ЭПР.
Пероксидисульфат
Реакции иона пероксидисульфата можно рассмат-
ривать как пример окисления пероксикислотой. Хаус
[37] следующим образом классифицирует реакции окис-
ления перокси дисульфатом:
1) Реакции, катализируемые ионами серебра. Их
скорость пропорциональна первой степени концентрации
ионов пероксидисульфата и концентрации ионов сере-
бра:
- d [S2O|-]/<tt = k2 [S2O|-] [Ag+|.
Считается, что скорость этих реакций определяется
стадией
Ag++S2O|- Ag^+USOt-
2) Некаталитические реакции окисления первого по-
рядка
S2of" 2SO^“ .;
их скорость описывается уравнением
-d[s2o|-]/d/ = *[s2o|-].
3) Реакции второго порядка, имеющие скорость
-rf[s2ol-]/<tt = *[s2o|-] 1г].
Последние реакции протекают без участия свободных
радикалов, как уже отмечено в гл. 2.
Наиболее простым примером реакции первого по-
рядка является разложение ионов пероксидисульфата,
которое изучалось в водно-спиртовых смесях с примене-
нием свободного радикала дифенилпикрилгидразила
(DPPH), связывающего другие радикалы [38]. Реакции
разложения соответствует механизм:
S2o£- —» 2SO4- •
SO^" DPPH —> конечный продукт.
12*
180
Глава 7
Реакция в водно-спиртовой смеси 1 : 1 по объему имеет
первый порядок и константу скорости £ = 3,7-Ю13’
. ^-1,19.ю-/₽т. Сек~\
В реакционном сосуде смешивали раствор DPPH известной кон-
центрации в этиловом спирте с водным раствором пероксидисульфа-
та; изменение концентрации DPPH во времени устанавливали фото-
метрическим методом.
Для эксперимента использовали химически чистые соли. Раствор
пероксидисульфата при хранении портится, поэтому необходимо еже-
дневно готовить свежий раствор. Спирт тщательно очищали кипяче-
нием с нитратом серебра и едким кали с использованием обратного
холодильника и с последующей перегонкой. Таким образом спирт
очищали от альдегида и других примесей, которые быстро реаги-
руют с DPPH; растворы DPPH в спирте разлагаются медленно, и
их достаточно возобновлять через несколько дней.
В присутствии малых количеств ионов серебра ско-
рость разложения ионов пероксидисульфата увеличи-
вается. Возможная схема реакции
Ag+ +S2O|- —> Ag2+ + SO4 • + SO2-,
Ag2+ + OH" —► Ag+ + • OH.
Ионы меди в этой реакции проявляют себя как более
слабые катализаторы по сравнению с ионами серебра.
Другим примером реакции первого порядка является
окисление меркаптанов пероксидисульфатом в гомоген-
ном растворе [39], для которого предполагается следую-
щий механизм:
S2O2" ^=Ё±25О4-.,
быстро
SO4~- 4-RSH . быстро > RS - +H+ + SO?-.
Для реакции окисления изопропилового спирта вначале
была предложена следующая схема [40]:
S2O|" so^+so4+-.
R2CHOH + SO^_ R2C—6—OSO3,
H
—► r2c=o + H+ + sol".
Гомолитическое окисление в растворе
181
Позже эту реакцию исследовал Уиберг [41], который
предложил видоизмененную схему реакции:
r2choh+s2o82- s2cho6so-+ SO2"
S2OI —> 2SO^" •,
SO4 • + Н2О — * НО • + HSO4“,
НО - 4-R2CHOH —-> H2O-t-R2tOH,
R2COH + S2o|" R2C=O + HSO4- + SO4- -,
R2COH + SO4“ • —> R2C=O 4- HSO4 .
Представляет большой интерес изучение образова-
ния радикалов НОг из Н2О и S2O|~ [42] по схеме
S2O|" —> 2SO4--,
so4 - +н2о —► HS07 + H0.,
Н2О2-|-НО. —>Н2О + НО2-
Если проследить дальнейшую судьбу радикала НОг •
при кислотности, достаточно высокой, чтобы можно
было пренебречь его ионизацией, оказывается, что он
реагирует двумя путями, не зависящими от pH:
НО2 - + s2o|- —► о2 + HSO4" + SO4" •,
НО2- +Н2о2 —► О2 + Н2О4-НО..
Цепь, развитием которой служит первая из реакций,
приводит к суммарному стехиометрическому уравнению
H2O2 + S2O|- —> O2+2HSO4-.
Аналогично вторая реакция приводит к другому суммар-
ному уравнению:
2Н2О2 —> 2Н2О + О2.
Изменение относительных концентраций БгОв” и Н2О2
позволяет выявить изменение относительного участия
каждой из двух последних реакций.
Реакция окисления фенола [43], вероятно, происхо-
дит по схеме
/О"
С6н5о- +s2o^- —> с6н4/ +so2- + h+
xoso3‘
182
Глава 7
Образующееся при этом промежуточное соединение гид-
ролизуется с образованием хинона.
Бекон и сотрудники [44] сообщают, что окисление
фенолов сильно катализируется ионами Ag1, Fe11 и
Fe111, тогда как другие ионы в этом случае неактивны.
Позднее было показано [45], что при окислении моноза-
мещеиных фенолов пероксидисульфатом нуклеофильные
группы облегчают, а электрофильные группы тормозят
реакцию.
Ирвин [46] установил интересную связь между ско-
ростями реакций окисления ряда комплексов и разно-
стью стандартных потенциалов пероксидисульфата и
комплексного иона:
log k0 = const 4- х ЬЕ° 70.
Тетраацетат свинца. Соль Фреми
Тетраацетат свинца способен реагировать по меха-
низму свободных радикалов. Примером может быть ре-
акция окисления некоторых метилфенолов [47], для ко-
торой предлагается следующая схема:
ОН
CHa + Pb(CHsCOO)4 —►
CHS
d
—* СНа + 15Ь(СН8СОО)э + СНзСООН
'cH, О 6 в j^-CH. —> 0-сн, - СН, СНз о II —> |Qj-CHs-|- .0СН3С0 сн8 0 в - Гксн’ - \/ OCH3CO СН, о II Q-CH* ]\сн3соо СНз
Гомолитическое окисление в растворе
183
Природа образующихся продуктов может изменяться в
зависимости от растворителя, относительной доли окис-
лителя в смеси, числа и положения метиловых групп.
Свободные радикалы, образующиеся при окислении
двуокисью свинца ряда замещенных фенолов в органи-
ческой среде [48], были идентифицированы по спектрам
ЭПР. Оказалось, что при этом образуются:
1) первичные радикалы путем отрыва атома водоро-
да от фенольной группы и
2) вторичные, более устойчивые радикалы; последние
образуются из первичных радикалов при реакции окис-
ления метиленовой группы в пара-положении к феноль-
ной группе.
Соль Фреми (SO3)2NO2‘ представляет собой инте-
ресное радикальное соединение, которое в кислом рас-
творе распадается по схеме [49]:
(SO,),NO2- • +Н3О+ (SO3)2NOH2"4-H2O+,
н2о+4-н2о 5=± hsof+ho.,
(SO3)2NO2- -[* НО • —промежуточный продукт,
—-> продукты
- d [(SO3)2NO2- • Vdt = [(SO3)2NO2- • I {Л, [H3O+] + k2 [HO . ])•
При окислении изомасляного альдегида солью Фреми
[50] реакция протекает путем отрыва электрона от анио-
на енольной формы
(СН3)2СНСНО + ОН > (СН3)2С=СНО + Н2О.
' равновесие
(СН3)2С=СНО + [ • ON (SO3K)J медленно *
—► [: ON(SO3K)2]“ + (СН,)20СНО>
Н2О + (СН3)2ЙСНО 4- • ON(SO3K)2 -gjjj—+
—► (CH3)2C(OH)CHO + HON(SO8K)2.
ЛИТЕРАТУРА
1. Уотерс У., Химия свободных радикалов., М., Издатинлит,
1948.
2. Инграм Д., Электронный парамагнитный резонанс в свобод-
ных радикалах, М., Издатинлит, 1961.
3. Dixon W, Т.» Norman R. О. C.f J. chem. Soc., 3119 (1963),
184
Глава 7
4. L е w i s E. S., S у m о n M. C R., Quart. Rev. chem. Soc., Lon-
don, 12, 320 (1958).
5. Trotman-Dickenson A. F., Free Radicals. An Introduction,
Methuen, London, 1959, p. 22.
6. Fukui K., Y о n e z a w a T., Nagata C., S h i n g u H., J. chem.
Phys., 22, 1433 (1954).
7. M с К i n n о n D. J., W a t e г s W. A., J. chem. Soc., 323 (1953).
8. M e г z J. H., W a t e г s W. A., J. chem. Soc., 15 (1949).
9. D a i n t о n F. S., Pure appl. Chem., 5, 325 (1961).
10. Wells A. F., Structural Inorganic Chemistry, Oxford University
Press, 1962, p. 407.
11. Emeleus H. J., Anderson J. S., Modern Aspects of Inorga-
nic Chemistry, Routledge and Kegan Paul, London, 1949, Ch. 10.
12. E v a n s M. G., Hush N. S., U r i N., Quart. Rev. chem. Soc.,
London. 6, 186 (1952).
13. Urey H. C., D a w s e у L. H., Rice F. O., J. Am. chem. Soc.,
51, 1371 (1929).
14. Waters W. A., Wistas in Free Radical Chemistry, Pergamon,
London, 1959, p. 157.
15. Fenton H. J. H., J. chem. Soc., 65, 899 (1894).
16. Fenton H. J. H., Jackson H., J. chem. Soc., 75, 1 (1899).
17. Fenton H. J. H., Jones H. L., J. chem. Soc., 77, 69 (1900).
18. Haber F., Weiss J., Proc. R. Soc., A147, 332 (1934).
19. Latimer W. M., Oxidation Potentials, Prentice-Hall, New York,
1952, p. 48; см. общую литературу к Введению.
20. В а х е n d а 1 е J. Н., Evans М. G., Park G. S., Trans. Fara-
day Soc., 42, 155 (1946).
21. Merz J. H., Waters W. A., Disc. Faraday Soc., 2, 179 (1947).
22. Merz J. H., Waters W. A., J. chem. Soc., 2427 (1949).
23. Barb W. G., Baxendale J. H., George P., Hargra-
ve K. R., Trans. Faraday Soc., 47, 462 (1951).
24. Bates H. G. C., Ur i N., J. Am. chem. Soc., 75, 2757 (1953).
25. Halfpenny E., Robinson R. L., J. chem. Soc., 939 (1952).
26. Dainton F. S., Hardwick T. J., Trans. Faraday Soc., 53,
333 (1957).
27. Dermer О. M., Edmison M. T., Chem. Rev., 57, 77 (1957).
28. L i n d s a у Smith J. R., Norman R. О. C., J. chem. Soc.,
2897 (1963).
29. Bunn D., Dainton F. S., Salmon G. A., Hardwick T. J.,
Trans. Faraday Soc., 55, 1760 (1959).
30. Ferradini C., Adv. inorg. Chem. Radiochem., 3, 181 (1961).
31. Rabani J., Stein G., Trans. Faraday Soc., 58, 2150 (1962).
32. Waters W. A., Progress in Organic Chemistry, Vol. 5, (Ed. by
J. W. Cook, W. Carruthers), Butterworth, London, 1961, p. 1.
33. К h а г a s c h M. S., F о n о A., N u d e n b e r g W., J. org. Chem.,
15, 763 (1950).
34. О г r R. J., W i 11 i a m s H. L., Disc. Faraday Soc., 14, 170 (1953).
35. Reynolds W. L., Kolthoff I. M., J. phys. Chem., 60, (a)
969; (6) 996 (1956).
36. Th о m a s J. R., J. Am. chem. Soc., 85, 591 (1963).
37. House D. M., Chem. Rev., 62, 185 (1962).
Гомолитическое окисление в растворе
185
38. В awn С. Е. Н., Magerison D., Trans. Faraday Soc., 51, 925
(1955).
39. Eager R. L., Winkler C. A., Can. J. Chem., 26, 527 (1948).
40. Levitt L. R., Malinowski E. R., J. Am. chem. Soc., 77, 4517
(1955).
41. Wiberg К. B., J. Am. chem. Soc., 81, 252 (1959).
42. Tsao М.-S., Wilma rth W. K., Am. chem. Soc. Monogr., 36,
113 (1962).
43. Sethna S. M., Chem. Rev., 49, 91 (1951).
44. Bacon R. G. R., Grime R., Munro D. J., J. chem. Soc., 2275
(1954).
45. Behrman E. I., J. Am. chem. Soc., 85, 3478 (1963).
46. I rvine D. H., J. chem. Soc., 2977 (1959).
47. C a v i 11 G. W. К., С о 1 e E. R., G i 1 h a m P. T., M с H u g h D. J.,
J. chem. Soc., 2785 (1954).
48. В e с с о n s a 11 J. L., С1 о u g h S., S с о 11 G., Trans. Faraday Soc.,
56, 459 (1960).
49. Murib J. H., Ritter D. M., J. Am. chem. Soc., 74, 3394
(1952).
50. Allen G. D., Waters W. A., J. chem. Soc., 1132 (1956).
Глава 8
ГОМОГЕННОЕ ОКИСЛЕНИЕ В ГАЗОВОЙ ФАЗЕ*
В этой главе рассматриваются гомогенные реакции
с участием кислорода в газовой фазе; в последующей
гл. 9 обсуждаются гетерогенные реакции с участием
кислорода. Следует отметить, что имеются и другие
пути введения кислорода в гомогенную систему. В виде
примера назовем газофазные реакции с окислами азота.
Здесь мы ограничимся рассмотрением реакций газооб-
разного кислорода и двуокиси азота.
Недостатки многих ранних работ были связаны с от-
сутствием хороших аналитических методов для контроля
продуктов реакций. Положение значительно улучшилось
в последнее время, когда стали доступными такие ана-
литические методы, как газовая хроматография, спект-
роскопия, спектроскопия ЯМР и масс-спектроскопия, ко-
торые обеспечены надежными приборами.
Молекулу кислорода можно считать нереакционно-
способным бирадикалом. Для начала реакции частотре-
буется затрата высокой энергии на активацию молеку-
лы кислорода; образующийся при этом радикал кисло-
рода обладает очень высокой реакционной способностью.
Классическим примером реакции этого типа служит
окисление водорода кислородом, которое мы вкратце
рассмотрим.
* Затрагиваемые в этой главе вопросы более полно и всесторон-
не рассмотрены в следующих книгах: Семенов Н. Н., О некото-
рых проблемах химической кинетики и реакционной способности
(свободные радикалы и цепные реакции), изд. 2-е, переработ. и до-
поли., М., Изд-во АН СССР, 1958, 686 стр.; Кондратьев В Н.,
Кинетика химических газовых реакций, М., Изд-во АН СССР, 1958,
стр. 688; Штерн В. Я., Механизм окисления углеводородов в газо-
вой фазе, М., Изд-во АН СССР, 1960, 496 стр. — Прим, ред.
Гомогенное окисление в газовой фазе
187
Очень высокая реакционная способность наблюдает-
ся для кислорода и радикалов, которые взаимодейству-
ют очень быстро. В виде примера рассмотрим реакцию
окиси азота с кислородом, которую можно считать двух-
стадийной реакцией между радикалами. Имеется ряд
исследований этой реакции, как классических, так и со-
временных. Она же послужила предметом детального
теоретического анализа.
Затем обсуждаются реакции кислорода с углеводо-
родами, у которых в реакцию могут вступать атомы во-
дорода, присоединенные к первичному, вторичному или
третичному атому углерода. Здесь следует учитывать
прочность и реакционную способность связей С—Н.
Наконец, рассмотрим реакцию кислорода с некото-
рыми другими классами соединений и реакции двуокиси
азота в газовой фазе. Область более сложных реакций,
как, например, воспламенение и взрыв, в данной книге
не затрагивается.
Одно из первых исследований реакций газофазного
окисления выполнено Боуном и Таунендом [1]. Обобще-
ние этих работ сделано Боуном [2].
В то время отдавали предпочтение теории, согласно
которой считалось, что углеводороды окисляются через
стадию гидроксилирования:
СН4 —СН3ОН —СН2(ОН)2
—> СН(ОН), —► CC^ + HjO + Hj.
Эта схема была распространена на горение топлива
(см. [3]):
углеродистое промежуточный ? инертные продукты
вещество 4~О2 —► продукт, М<^
* разветвление
I
активные центры цепи
Результаты экспериментальных исследований критиче-
ски обсуждались на основе представлений Семенова и
Гиншельвуда.
Историческое значение имеет книга Гиншельвуда и
Вильямсона [4]. Наиболее поздним обзором, охватывающим
188
Глава 8
все работы до настоящего времени, является книга
Минкова и Типпера [5]* (см. примечание на стр. 186).
Ценной является библиография по горению нефти [6].
В недавно опубликованном обзоре [7] рассматриваются
основные достижения и направления исследований га-
зофазного окисления.
Электронная структура кислорода рассматривалась
еще Милликеном [8], который приписал его молекуле
основное состояние 3S. Этим объясняется парамагне-
тизм кислорода. Рейд [9] отмечает, что известен ряд яв-
лений с участием кислорода и возбужденных молекул,
которые заставляют выделить реакции кислорода в осо-
бый класс.
Реакция между водородом и кислородом
Поскольку эта реакция очень подробно рассмотрена
в книге Минкова и Типпера [5]**, здесь отмечаются лишь
ее важнейшие особенности. Схема реакции между во-
дородом и кислородом предложена Гиншельвудом [10]:
1) Н2 —> 2Н,
2) Н + О2 —> НО + О.
3) О + Н2 —> НО + Н,
4) Н-|-О2 + М —> НО2 + М (М — какая-либо молекула).
5) НО2 —► удаление с поверхности,
6) НО24-Н2 —> Н2О4-НО,
7) НО4-Н2 —> н2о + н.
8) Н —► удаление с поверхности,
9) НО —► удаление с поверхности.
Существенной особенностью этой схемы является
удвоение числа активных радикалов на стадиях (1),(2),
и (3), что приводит к разветвлению цепи. Сложная ки-
нетика этой реакции, имеющей три предела взрыва,
• Здесь и далее в этой главе радикалы НО* и НО2* обозна-
чаются НО и НО2.
*• См. также монографию: Налбадян А. Б.» Воевод-
ский В. В., Механизм окисления и горения водорода, М.—Л.,
Изд-во АН СССР, 1949. — Прим. ред.
Гомогенное окисление в газовой фазе
189
возникает вследствие соревнования между разветвле-
нием и обрывом цепей.
Эта реакция и в настоящее время продолжает при-
влекать внимание исследователей. Так, Болдуин и Мей-
ер [11, 12] обнаружили ее новые кинетические характе-
ристики. Течение реакции они изучали в зависимости от
состава смеси, температуры, общего давления, добавок
инертных газов и диаметра сосуда. Для объяснения не-
обычных результатов авторами был предложен свой ме-
ханизм. В долго работавшем реакторе со стенками, по-
крытыми борной кислотой, сохраняющей НО2 и Н2О2,
выше второго предела взрыва протекают следующие ре-
акции:
но + Н2 —> Н2О + н,
н + о2 —> Н04-0,
О + Н2 —> НО + Н,
н4-о2 + м—> но2+м.
НО2 + НО2 —> Н2О2 + О2,
Н2О24-М' —> 2Н0 + М',
Обрыв цепи происходит, вероятно, двумя путями:
Н -Ь Н2О2 —> Н2О + но,
НО + Н2О2 —> Н2О + но2.
водородом и кислородом
фотосенсибилизированной
В другой работе [13] с помощью измерений выделяю-
щегося тепла реакция между
была изучена в присутствии
ртути. В этом случае реакция протекает по схеме
Hg -J- hv -> Hg 1 зарождение цепи
Hg* + H2 — > Hg + H2 J
Н + О2—’-»НС>2
но’ н+о2
НО2+М —но24-м
Н+НО2 —2НО
но + н2 —н2о + н
развитие цепи
НО2 -|- НО2 —•> Н2О2 4- О2 обрыв цепи
190
Глава 8
Другие реакции кислорода
Термин «простая» здесь означает простые стехио-
метрические отношения между реагирующими компонен-
тами. Классическая реакция
2NO + O2 —> 2NO2
была изучена Боденштейном [14]. Он показал, что ско-
рость образования двуокиси азота описывается урав-
нением
— d [NO2]/<ft = k [NO]2 [О2].
Наиболее вероятен двухстадийный механизм этой ре-
акции:
NO + O2 к -> ONO2
быстро
ONO2 + NO
ч
стадия, определяю-
щая скорость
-> О—О* —> 2N0o
I I
N N
I I
О О
На первой стадии быстро устанавливается равновесие.
Вторая, определяющая скорость стадия сводится к ата-
ке молекулы окиси азота. Вследствие квадрата концен-
трации окиси азота в кинетическом уравнении часто
считают, что первоначальное равновесие представляет
собой димеризацию окиси азота, Гершинович и Эйринг
[15] теоретически рассмотрели эту реакцию, их доводы
суммировал Лейдлер [16]. В указанных работах актив-
ному комплексу приписывается конфигурация, отмечен-
ная выше звездочкой (*}. Основываясь на теории актив-
ного комплекса, исследователи получили следующее вы-
ражение для константы скорости реакции:
k = ^T~^.e-E^T.
При условии, что Eq равна нулю или очень мала, кон-
станта скорости должна немного уменьшаться с ростом
температуры, что и подтверждено экспериментально. Ве-
личина Eq действительно близка к нулю, что можно
ожидать при рекомбинации радикалов.
Эта реакция продолжает привлекать внимание и по
сей день. Треси и Даниельс [17] измерили ее скорость
Гомогенное окисление в газовой фазе
191
при давлениях 1—20 мм рт.ст. и температурах 0—65°,
наблюдая за ходом реакции путем измерения поглоще-
ния света двуокисью азота. Результаты подтвердили ве-
личину порядка реакции, найденную Боденштейном. Ав-
торы предложили несколько иной механизм реакции:
1) N0 + 02 NO3,
2) NO3+ NO т=> NO3 • NO,
3) NO3 • NO + NO2 —► 2NO + O2 + NO2,
4) NO3 + NO2 N2O5,
5) NO3 + NO —> 2NO2,
6) NO3. NO + NO —> 2NO2 + NO.
Эшмор с соавторами [18] пришли к заключению, что
реакция протекает по схеме:
NO + O2 NO3,
NO3+NO —> 2NO2,
которая согласуется с простыми соображениями, при-
веденными в начале обсуждения.
Поскольку окислы азота играют большую роль в про-
мышленном производстве серной кислоты, детально ис-
следовалась система NO+SO2+O2 [19]. Предполагается,
чго в процессе окисления принимают участие радикалы
NO3, которые могут реагировать либо с NO, либо с SO2,
образуя NO2 по схеме:
NO + O2 ^=± NO3,
NO3+NO NO3 • NO,
NO3 • NO —► 2NO2,
NO3 • NO + N2(SO2) —► 2NO 4- O2 + N2(SO2),
NO3 + SO2 —> NO3 • SO2 —> NO2 4- SO„
no8-so24-n2 —► no34-so24-n2,
2NO2 —► 2NO4-O2,
so24-no2 —► SO3NO,
SO3NO 4- NO2 4- SO3 —> (SO8)2N2O8.
Число простых гомогенно протекающих реакций не-
велико. Дейнтон [20] довольно подробно изучил гомо-
генное окисление окиси углерода и фосфора. Россер и
192
Глава 8
Уайз [21] исследовали гомогенное окисление бромистого
водорода в газовой фазе
4НВг + О2 —> 2Н2О + 2Вг2.
Они нашли выражение для скорости реакции
d [Br2]ldt = 2k [НВг] [О2]
для случая [НВг]^>[02]
и предложили для нее следующий механизм:
НВг + О2 —► НО2 + Вг.,
НО24-НВг —> Н2О2Н-Вг-,
Н2о2 —> 2НО.
Если [02]^>[НВг], необходимо учитывать протекание па-
раллельно окислению следующих реакций:
НО2 —> н - +О2.,
НО2 —> распад на стенках.
Примером простой реакции газофазного окисления мо-
жет служить также фотохимическая реакция между хло-
ром и кислородом [22] с помощью флеш-фотолиза и
флеш-спектроскопии. Радикал CIO образуется легко,
концентрацию его определяют по светопоглощению
С1 —О2 —> С1ОО и С14-С1ОО —> 2С1О.
Окисление углеводородов кислородом
Превосходное общее описание реакций окисления ор-
ганических соединений составлено Каллисом [23].
На ранних стадиях окисление можно представить
так:
RH + X —> RH-XH
(X — свободный радикал)
R -|~ О2 —> RO2
RO2 —> продукт + X
первая реакция.
вторая реакция,
третья реакция.
По первой реакции могут возникать различные типы
радикалов, так как не все атомы водорода в молекуле
равноценны
ZR
RH->R'
Гомогенное окисление в газовой фазе
193
Вторая реакция также может протекать разными пу-
тями
— + окисление
—-> изомеризация
-_•> разложение
___-> рекомбинация
Однако наиболее быстрой является, вероятно, реакция
R + O2. Для нее мы имеем схему:
высокие ненасыщенное
соединение + пО2
Для третьей реакции имеется несколько возможностей:
RO24-RH —> ROOH-f-R.
или
RCHR' —> RCHO + R'O
I
оо-
или
RO2 + RO2- —> RO- +RO. +О2
Рассмотрим более подробно эту простую схему. Чет-
кую формулировку принципов окисления углеводородов
дал Уолш [24]. Он отметил, что в порядке убывания ре-
акционной способности связи С—Н можно расположить
следующим образом: третичная > вторичная >первич-
ная. Этот порядок возникает по двум причинам. Во-
первых, в результате уменьшения отрицательного заряда
связей С—Н и, во-вторых, вследствие повышения проч-
ности связи С—Н. При окислении парафинов кислород
атакует почти всегда связь третичного углерода с во-
дородом, если таковая имеется. Если такая связь от-
сутствует, тогда воздействие преимущественно сосредо-
точивается на вторичном водороде и частично затраги-
вается первичный.
Для сравнения было бы желательно исследовать ре-
акции окисления простых углеводородов в одинаковых
условиях. Однако Мэлкехи [25] нашел, что невозможно
определить скорость окисления более чем двух угле-
водородов с различным числом атомов углерода в цепи
13 Зак. 1249
194
Глава 8
при одной и той же температуре, поскольку слишком
мал температурный интервал между протеканием беско-
нечно медленной реакции и появлением холодного пла-
мени или взрыва. Холодные пламена появляются в слу-
чае окисления некоторых углеводородов в определенных
условиях температуры и давления [26].
Важнейшими стадиями окисления углеводородов яв-
ляются следующие [27]:
RH + O2 —► R4-HO2.
R-|-O2 —> R—О—О—.
R_О-О— + RH —► ROOR + H—,
ROOR —► 2R0.
Кинетика реакций газофазного окисления первичных
и некоторых вторичных хлорпарафинов аналогична най-
денной для соответствующих углеводородов. Скорость
реакции сильно зависит от концентрации хлорпарафина
и почти не зависит от давления кислорода. Первично за-
мещенный хлор увеличивает скорость окисления хлор-
парафина, во-первых, вследствие нарушения симметрии
группы метила и, во-вторых, вследствие прямого индук-
ционного эффекта. Противоположно направленное влия-
ние хлора и группы метила соответствует точке зрения,
что связи —О—О— перекисных соединений стабилизи-
руются электронодонорным действием и ослабляются
электроноакцепторным действием.
Мэлкехи [28] сравнил скорости реакций окисления не-
скольких веществ, содержащих три атома углерода в
молекуле, в одинаковых условиях. Различия в значении
скоростей реакции, наблюдаемые при низких темпера-
турах (<~300°), значительно уменьшаются при высо-
ких температурах (>~400°). Очевидно, различия воз-
никают вследствие того, что при высоких температурах
в реакции не участвуют перекиси, в то время как при
низких температурах влияние структур связано с при-
родой перекиси, принимающей участие в реакции.
При исследовании процесса окисления пропилена
[29] было найдено, что вблизи 200° реакция имеет ин-
дукционный период примерно 30 мин, в течение которого
малые, но заметные количества пропилена реагируют с
Гомогенное окисление в газовой фазе
1%
образованием формальдегида, высших альдегидов и
высших перекисей. Подобные индукционные периоды,
возникающие в результате прохождения побочных ре-
акций, довольно часто встречаются при гомогенном окис-
лении углеводородов и рассматриваются Минковым и
Типпером [5].
Некоторые интересные результаты получены при ис-
следовании параллельно протекающих реакций окисле-
ния. Так, при взаимодействии смесей этана и пропана
с кислородом [30] отношение скоростей уменьшения кон-
центраций двух газов составляет 0,44:1. Эта величина
почти не зависит от температуры в интервале 274—
495°, состава смеси и общего давления. Вероятно, в ре-
акциях окисления обоих веществ принимает участие
один и тот же радикал во всем интервале температур,
реакции с обоими углеводородами протекают с низкой
энергией активации.
Очень важным промышленным процессом окисления
является горение нефти. Молекулы третичных углеводо-
родов атакуются более легко, чем вторичных и первич-
ных. Разветвление цепных реакций усиливается в после-
довательности СН3>СН2>СН. По этой причине детони-
рующую способность жидкого топлива оценивают в
сравнении с детонирующей способностью изооктана
в стандартном двигателе.
Поскольку третичный углерод в молекуле углеводо-
рода окисляется наиболее легко, представляет интерес
рассмотрение реакции окисления простейшего третич-
ного углеводорода — изобутана. Процесс окисления по-
следнего был изучен Бозе [31]. Он обнаружил аномаль-
ный температурный коэффициент реакции, связанный с
увеличением диссоциации радикала RO2 , которая обры-
вает цепную реакцию. Автором предложены следующие
схемы механизма реакции при сравнительно низких и
при более высоких температурах. Механизм реакции
при температурах около 290° (общая схема)
RH —> R + H [R = (CH3)3C],
RH-pO2 —> R+HO2.
R -|- O2 > RO2,
RO2-HRH —► ROOR-f-H
13*
19£
Глава 8
или
RO2 + RH —> ROOH+R,
ROOR —> 2RO.
Затем протекают следующие реакции:
(СН3)з СО —> (СН3)2 СО + СН3.
сн3 + о2 —► НСНО, СН3ОН, СО, СО2, Н2,
(СН3)2СО + СН3 —> СН4 + СН2СОСН3,
СН3 + СН3 —> С2Н6.
Механизм реакции при более высоких температурах
(390°):
(СНз)зСН + О2 —> (СН3)3С. НО2,
(СН3)3С. +о2 —> (СНз)2С=СН2 + НО2,
(СН3)3С . —> (СН3)2С • +СН3,
—► СН3СН=СН2
или
(СН3)3СН + О2 —> (СН3)2СН=СН2+НО2,
(СН3)2СН=СН2 —► СН3СН=СН2 4- сн3,
CH34-RH —► CH44-R,
СН34-СН3 —> с2н6.
В более позднем исследовании реакции окисления
изобутана с применением изотопов [32] предлагается
схема, предусматривающая образование метилэтилке-
тона:
(СН3)3СН + О2 —> (СН3)3С. 4-НО2,
(СН3)3С • 4- О2 —> (СНз)гСОО •
(СНз)зСОО- —► (CH3)2tH2OOH,
—► СН3СОСН2СН3Н-но.
Окисление других органических веществ кислородом
Приведем только несколько примеров. Новым слу-
чаем окисления третичного атома водорода служит ре-
акция окисления изопропилового спирта, характеризую-
щаяся высоким выходом ацетона и перекиси водоро-
Гомогенное окисление в газовой фазе
197
да [33]:
СН(СН3)2ОН4-О2 —> СО (СН3)2 + Н2О2,
СН(СН3)2ОН 4- но2 —► С(СН3)2ОНЧ-Н2О2,
С(СН3)2ОН + о2 —► СО(СН3)2 + но2,
С(СН3)2ОН —> СН3СНО + СН3.,
При исследовании газофазного окисления диэтило-
вого эфира оказалось [34], что в области низких темпе-
ратур реакция имеет много общего с окислением угле-
водородов, за исключением того, что избыток кислорода
ингибирует скорость окисления. Для объяснения была
предложена следующая схема:
СН3СН2ОСН2СН3 + О2 —> СН3СНОСН2СН34-НО2,
сн3Сносн2сн3 —> СН3СНО + СН3СН2.»
СН3СН2- 4-О2 —> СН3СН2ОО ..
СН3СН2ОО. +RH —> CH:iCH2OOH + R.,
СН3СН2ООН —► СН3СН2О- 4-НО,
СН3СН2О . —> СН3. 4- нсно
или
—> н. + СН3СНО
В качестве примера ингибированной реакции можно
привести окисление альдегида, замедляемое углеводоро-
дами [35], что объясняется следующей схемой:
Инициирование цепи
СН3СНОЧ-Х —> СН3С=О4-ХН
(X — инициатор)
. /°°-
СН3С=О + О2 —► СНзС\чО
Развитие цепи
юо уоон
СН3С< 4-СН3СНО —► СН3С< 4-СН3С=О
^О X)
Ингибирование углеводородами заключается в замене
третьей стадии реакцией
/00 /000
СНзС<0 +RH ~* сн
IflB
Глава 8
В исследованном интервале температур радикалы R- не
могут производительно участвовать в развитии цепи
окислительных реакций.
Последним примером реакций медленного окисления
органических веществ (не углеводородов) является ре-
акция медленного окисления этилацетата [36], для ко-
торой предлагается схема
СН3СООС2Н5 + О2 —► сн3сооСнсн3+но2,
(R-)
R * -I- О2 —► RO2 •,
RO2- +RH —> ROOH + R..
Развитие цепи может происходить также посредством
реакции
ho2 + rh —> R- +н2о2..
Результаты показывают, что органическая перекись
разлагается различными возможными путями
, H2O-f-CH3CO-O COCH3
СН3СООСНСН3ООН^> СН3СНО 4- сн3. СО • ООН
сн3 • со • о • СНСН3О • + он
Реакции двуокиси азота в газовой фазе
Полезное обобщение реакций двуокиси азота состав-
лено Фишем и Уарисом[36]. В этой работе обсуждаются
разнообразные реакции, имеющие суммарный порядок
2. Величины предэкспоненциального множителя А во
всех случаях незначительны и изменяются не очень
сильно. Порядок изменения величины энергии актива-
ции соединений в точности соответствует порядку изме-
нения ионизационных потенциалов
СО > no2 > SO2 > СНО . сно > нсно > с2н4 >
> СН3СНО > СН3СН=СН2.
Главными продуктами реакции газофазного окисле-
ния ацетилена двуокисью азота являются глиоксаль,
окислы углерода и окись азота [38а]. Предлагается еле-
Гомогенное окисление в газовой фазе
199
дующий механизм реакции:
0N0
I
НС=СН + N0? СН=СН
(A)
ONO ONO
I I
(A) + NO2 —► HC-CH
ГНС—СНП
II II
о о _
+ 2NO
В другой статье [386] рассматривается окисление глиок-
саля двуокисью азота. Реакция протекает гомогенным
путем, и ее скорость приблизительно пропорциональна
концентрациям глиоксаля и двуокиси азота. В резуль-
тате реакции образуется окись азота, окись углерода,
двуокись углерода, вода и глиоксалевая кислота
СНО 1 сно 4-NO, - СО- -> I + hno2 сно
со. 1 сно 4* NO, - СОО. - 1 + NO сно
со 1 сно 4-NO, - -► 2СО 4- HNO2
соо • 1 сно 4* NO, - -> СО + со2 4- HNO<
соо• сно 1 +1 - сно сно СООН СО. -> 1 +1 СНО сно
2HNO2 —► Н2О + NO + NO2
Изучались реакции между хлористым водородом и
двуокисью азота (находящейся в равновесии с N2O4).
В интервале температур 25—55° реакция протекает гете-
рогенным путем; хлористый водород активно адсорби-
руется на поверхности стеклянного реакционного сосу-
да, и реакция протекает между адсорбированным НС1
и N2O4 [39]:
4НС1 + N2O4 —► 2Н2О 4- 2NOC1 4- Cl2.
200
Глава 8
Предлагается следующий механизм реакции:
Ы2О44-2НС1адс —► Н2Оадс+ NOC1 + NO2C1,
ЫОС1 + НС1адс —► C12+HNO,
HNO+N2O4 —► HNO2 + NO + NO2,
NO + NO2C1 —> NO2 + NOC1,
2NO + CI2 —► 2NOC1,
2HNO2 —► H2O4-NO + NO2.
При более высоких температурах реакция идет в гомо-
генной среде по схеме [40]
HC1 + NO, —> HNO2 + C1,
HNO2 + HC1 —> H2O+NOCI,
NOC1 + C1 —> NO4-C12,
NO + C1 + M —> NOC1 + M.
ЛИТЕРАТУРА
1. Bone W. A., Townend D. T. A., Flame and Combustion in
Gases, Longmans Green, London, 1927.
2. В о n e W. A, Proc. R. Soc., A137, 243 (1932).
3. Tipper С. H., Quart. Rev. chem. Soc., Lond., 11, 313 (1957).
4. Hinshelwood C. N., Williamson A. T., The Reaction bet-
ween Hydrogen and Oxygen, Oxford University Press, 1934.
5. M i n k о f f G. J., Tipper C. F. H., Chemistry of Combustion
Reactions, Butterworth, London, 1962.
6. American Chemical Society, «Literature of the Combustion of Pet-
roleum», Adv. Chem. Ser., № 20, 1958.
7. Khox J. H., Rep. Progr. Chem., 19 (1962).
8. M u 11 i k e n R. S., Rev. mod. Phys., 4, 1 (1932).
9. Rei d C., Quart. Rev., 12, 205 (1958).
10. Hinshelwood C. N., Proc. R. Soc., A188, 1 (1946).
11. Baldwin R. R., Mayor L., Trans. Faraday Soc., 56, 80; 103
(1960).
12. Baldwin R. R., Mayor L., Doran P., Trans. Faraday Soc.,
56, 93 (1960).
>3. Burgess R. H., Robb J. C., Trans. Faraday Soc., 54, 1008
(1958).
14. Bodenstein M., Z. phys. Chem., 100, 68 (1922).
15. G e г s h i n о w i t z H., E у ring H., J. Am. chem. Soc., 57, 985
(1935).
16. L a i d 1 e r K. J., Chemical Kinetics, McGraw-Hill, New York.
1950, p. 98.
17. T re асу J. C., Daniels F., J. Am chem. Soc., 77, 2033 (1955).
18. Ashmore P. G., Burnett M. G, Tyler B. J., Trans. Fara-
day Soc., 58, 685 (1962).
Гомогенное окисление в газовой фазе
201
19. Т i р р е г С. F. Н., W i 11 i a m s R. К., Trans. Faraday Soc., 57, 79
(1961).
20. Da in ton F. S., Chain Reactions, Methuen, London, 1956, p. 132.
21. Rosser W. A., Wise H., J. phys. Chem., 63, 1753 (1959).
22. Porter G., Wright F. J., Disc. Faraday Soc., 14, 23 (1953).
23. C u 11 i s C. F., Chern. Ind., 23 (1962).
24. W a 1 sh A. D., Trans. Faraday Soc., 42, 269 (1946).
25. Mulcahy M. F. R., Disc. Faraday Soc., 2, 128 (1947).
26. G a у d о n A. G., Quart. Rev., 4, 1 (1950).
27. Cull is C. F., Hinshelwood C. N., Mulcahy'M. F. R„
Proc. R. Soc., A196, 160 (1949).
28. Mulcahy M. F: R., Trans. Faraday Soc., 45, 537 (1949).
29. Mulcahy M. F. R., R i d g e M. J., Trans. Faraday Soc., 49, 906
(1953).
30. К n о x J. H., Smith R. F., T г q t m a n - D i с к e n s о n A F.,
Trans. Faraday Soc., 54, 1509 (1958).
31. Bose A. N., Trans. Faraday Soc., 55, 778 (1959).
32. C u 11 i s C. F., Fish A., T г i m m D. L., Proc. R. Soc., A273, 427
(1963).
33. Burgess A. R., C u 11 i s C. F., N e w i 11 F. J., J. chem. Soc.„
1884 (1961).
34. Waddington D. J., Proc. R. Soc., A252, 260 (1959).
35. Small N. J. H., Ubbelohde A. R., J. chem. Soc., 637 (1953).
36. F i s h A., W a r i s A., J. chem. Soc., 4513 (1962).
37. Rep. Progr. Chem., 91 (1954).
38. Thomas J. H., Trans. Faraday Soc., (a) 1142 (1952); (6) 630
(1953).
39. T a 1 b о t P. J., Thomas J. H., Trans. Faraday Soc., 55, 1884
(1959).
40. Gilbert J. R., Thomas J. H., Trans. Faraday Soc., 59, 1600
(1963).
Глава 9
ГЕТЕРОГЕННЫЕ РЕАКЦИИ ОКИСЛЕНИЯ
Гетерогенные реакции окисления газообразным кис-
лородом принадлежат к наиболее простым, но практи-
чески очень важным процессам. В этой главе рассмат-
риваются следующие вопросы:
1) аутоокисление различных органических веществ;
2) действие кислорода на поверхности металлов;
3) реакции кислорода на носителях;
4) реакции озона;
5) реакции двуокиси марганца.
Реакции аутоокисления протекают в растворах, при-
меняемых для поглощения кислорода из газовых смесей,
например в растворах пирогаллола, щелочного гипо-
сульфида и хлорида хрома (II). Хорошим примером ре-
акций второго класса служат коррозионные процессы.
Реакции гемоглобина в крови являются биологическим
прототипом реакций с искусственными носителями кис-
лорода, принадлежащих к третьему классу. Реакции
озона и двуокиси марганца, которые можно считать осо-
быми носителями кислорода, выделены в отдельную
группу.
Аутоокисление
В этом разделе кратко рассмотрены реакции окисле-
ния альдегидов, углеводородов, ненасыщенных соеди-
нений, фенолов, серусодержащих соединений, ионов ме-
таллов и некоторых других веществ.
Реакции аутоокисления в общем виде описаны в
книге Ландберга [1]. Следует отметить, что такие реак-
ции могут протекать в жидкой фазе или в растворе. Эту
тему так часто затрагивают в литературе, что трудно
найти к ней новый подход. Одним из наиболее четких
Гетерогенные реакции окисления
203
описаний реакций окисления молекулярным кислородом
в жидкой фазе является работа Твигга [2]. Такие реак-
ции протекают в три последовательные стадии:
1) Зарождение цепи
RH + M—► М—H-4-R-
где М — фактор, вызывающий зарождение цепи.
Зарождение цепи осуществляется различными путя-
ми: под действием радиации; в присутствии инициато-
ров, например перекиси бензоила; в результате термиче-
ского разложения гидроперекиси; при реакции металли-
ческих катализаторов с гидроперекисью.
На стадии зарождения цепи наблюдается индукцион-
ный период. Если углеводород имеет высокую чистоту,
может потребоваться значительное время для образова-
ния такого количества перекиси, которое достаточно
для поддержания цепной реакции.
2) Развитие цепи
r. _ро2 —► ROO •, Л2,
ROO • + RH —► ROOH + R .. k3.
Радикалы на этой стадии исчезают и затем регенери-
руются.
3) Обрыв цепи
2R • —► R—R, Л4,
R • -|- RO2 • —► ROOR, k3,
2RO2 • —► ROOR + O2, k3.
Применяя теорию стационарного состояния к концент-
рациям радикалов R- и ROs-, можно выразить умень-
шение концентрации кислорода при реакции окисления
следующим образом:
_ л Ю 1/Л = (<a W^)tRHKfe2-^)4O2]
2 k3 • л?» [R Н]+Л2. Л* I О2|+а* • # • Г, ’
где г< — скорость реакции зарождения цепи, а LRH] и
[OJ — концентрации углеводорода и кислорода. Если
длина цепи велика, то третьим членом знаменателя
можно пренебречь. Если давление кислорода велико,
204
Глава 9
формула приобретает вид
- d р2]/<« = (//«• л3/^«) • [RH]
и для аутоокисления, инициированного гидроперекисью,
-d [О2|/Л = (Мз/*^’) IROOH]*/’ [RH].
Аутоокисление альдегидов, кетонов, олефинов и па-
рафинов протекает легко. Ароматические амины и фе-
нолы функционируют как аутооксиданты с разрывом
цепи.
С помощью ЭПР показано, что ион [(CN)5Co3—О—
—О—Co4(CN)5]5~ образуется при окислении [Co2(CN5]3“
кислородом в водных растворах [3].
Окисление альдегидов
Реакциям окисления жидких альдегидов молекуляр-
ным кислородом посвящен квалифицированный обзор
Мак-Несби и Геллера [4]. Авторы отмечают, что полу-
чить воспроизводимые результаты очень трудно ввиду
возможного присутствия в альдегидах малых количеств
инициаторов и ингибиторов. Поэтому в опытах надо
применять либо очень чистые реагенты, либо вводить ин-
гибиторы и инициаторы в известных количествах.
Одной из обычных реакций аутоокисления альдегида
является окисление бензальдегида. Бекстром [5а] иссле-
довал термическое и фотохимическое окисление этого
соединения. В обоих случаях наблюдаются цепные ре-
акции. По цепному механизму протекает фотохимиче-
ское взаимодействие ацетальдегида с кислородом при
облучении УФ-радиацией.
Предложена цепная схема реакции окисления альде-
гидов [7, 8].
Каган и Любарский [9] изучили каталитическое влия-
ние соли марганца [Мп(ЫОз)2] на ход реакции между
ацетальдегидом и кислородом в среде уксусной кислоты.
В ходе этой реакции образуется ацетат марганца (III)
при действии пероксиуксусной кислоты на ионы марган-
ца (II). Ацетат марганца (III) сильно ускоряет указан-
ную реакцию окисления.
Гетероеенные реакции окисления
205
Боун и Вильямсон [10а] показали, что некатализируе-
мая и катализируемая следами металлов реакции окис-
ления ацетальдегида в растворе имеют две хорошо раз-
личимые стадии.
Вначале альдегид окисляется до пероксиуксусной
кислоты, и затем пероксиуксусная кислота взаимодей-
ствует с альдегидом с образованием перекиси. Сущест-
вование второй стадии подтверждается отдельным ис-
следованием реакции между ацетальдегидом и перокси-
уксусной кислотой.
Следующим этапом в обзоре реакций аутоокисления
является изучение их катализа солями кобальта [106].
Кинетику реакции в этом случае можно иллюстрировать
следующей схемой:
Зарождение цепи
Со111-|-СН3СОООН —► Сои4-Н+Н-СН3СООО.
и Сои + СН3СОООН. —► Coin + СН3СОО . + ОН"
СН3СООО- 4-СН3СНО —> СН3СОООН+СН3СО.
Развитие цепи
СН3СО • +О2 —> СН3СООО-
СНзСООО • + СН3СНО —► СН3СОООН 4- СН3СО .
Обрыв цепи
СН3СООО • +СН3СОООН —► неактивные продукты.
2СН3СООО • —► неактивные продукты.
Кинетика и механизм катализируемой и некатализируе-
мой реакций аналогичны. Это подтверждает предполо-
жение, что так называемое некаталитическое течение
реакции в действительности представляет собой реак-
цию, катализируемую следами металлов.
Купер и Мелвиль [Па] исследовали кинетику реак-
ции аутоокисления деканаля. Они нашли, что незави-
симо от способа инициирования реакции термического и
фотохимического окисления протекают по одному и то-
му же механизму. Экспериментальные результаты при-
водят к.следующим выражениям для скорости реакции:
Термическая реакция
R = [RCHOf* [О2]7’ (1)
206
Глава 9
Фотохимическая реакция
Я = Г [RCHO] [О2]° [/]'&
(2)
(/ — эффективная интенсивность света).
Если уравнение (1) представить в виде
Я = Л' [RCHO] [О2]° [RCHO]7’ [О,]7’
тогда, несмотря на наличие инициирования, выражения
для скоростей реакций термического и фотохимического
окисления становятся идентичными.
Предполагается следующий механизм реакции:
Термическое зарождение цепи
RCHO + O2—-> RCO- 4-НО2-
Фотохимическое зарождение цепи
RCHO4-ftv— * RCO- 4-Н-
Развитие цепи
RCO • + О2 RCOOO •.
RCOOO- 4-RCHO RCO • +RCOOOH.
Обрыв цепи
RCO - +RCO.
RCOOO• + RCO •
RCOOO - 4-RCOOO.
продукты
Указанная схема приводит к следующему выражению
скорости термической реакции:
/? = (*/. . Л3/Л7‘) [RCHO]7’ [O2J
и скорости фотохимической реакции
что согласуется с экспериментальными данными.
В следующей работе [116] теми же авторами иссле-
дован механизм торможения реакции аутоокисления де-
кана [11а] в присутствии гидрохинона. В этом случае
Гетерогенные реакции окисления
207
протекает еще одна реакция обрыва цепи
RCOOO • +h2q —► RCOQOH + H2Q“
Процесс аутоокисления бензальдегида в уксусной
кислоте, катализируемый соединениями кобальта [12],
можно объяснить следующей схемой реакций:
Со1114-СвНбСНО —► Со11 + СвНбСО • 4-Н+ зарождение цепи,
СвН5СО- 4-О2 —► СвН5СООО. |разви-
СвН6СООО.+СвН5СНО —> СвНБСОООН + СвНБСО• J цепи,
2СвНбСООО • —> нереакционноспособные обрыв цепи
продукты
Для реакции окисления ацетальдегида кислородом
в присутствии солей металлов в ледяной уксусной кис-
лоте найдено следующее кинетическое уравнение [13]:
— [О2]/^ = k' [ацетальдегид]*'2 [катализатор]*^.
Этому уравнению соответствует механизм реакции, ана-
логичный приведенному выше.
Металл удерживается в состоянии более высокой ва-
лентности в результате повторения цикла
RCOOOH+M11 —► RCOO • Ч-МП1 + ОН".
RCOOOH+M111 —> RCOOO + Мн + Н +
Реакции подобного типа в гомогенных условиях уже
были упомянуты в гл. 7 и среди других гомогенных ре-
акций ионов металлов в гл. 2.
Окисление углеводородов*
Одной из первых наиболее важных работ по ауто-
окислению углеводородов является исследование реак-
ции окисления тетралина, выполненное Робертсоном и
Уотерсом [14а]. В течение первых нескольких часов ауто-
окисление едва заметно, при этом поглощаются мизерные *
♦ Более подробно окисление тетралина, изопропилбензола (ку-
мола) и др. рассмотрено в книге: Эмануэль Н. М., Дени-
сов Е. Т., М а й з у с 3. К., Цепные реакции окисления углеводоро-
дов в жидкой фазе, М., «Наука», 1965, стр. 329. — Прим. ред.
208
Глава 9
количества кислорода. Содержание перекиси в жидкой
фазе постепенно увеличивается до тех пор, пока не
начинается совершенно явный аутокаталитический про-
цесс поглощения кислорода. В течение нескольких ча-
сов реакция ускоряется. После окисления примерно
5% тетралина реакция достигает стационарного состоя-
ния, которое сохраняется до тех пор, пока не окислится
30% тетралина. При окислении образуется вязкий ко-
ричневый продукт. В конце реакции скорость ее падает,
и окислить остающиеся 30% тетралина практически не-
возможно. В начальной стадии реакции образуется гид-
роперекись тетралина, в последующие стадии происхо-
дит ее распад.
В последующей работе [146] были изучены промежу-
точные продукты аутоокисления 'тетралина, представ-
ляющие собой главным образом а-тетралон и а-тетралол.
Позднее [14в] была количественно исследована реакция
разложения гидроперекиси тетралина. Была предпри-
нята попытка интерпретировать кинетику аутоокис-
ления с помощью метода стационарного состояния [14],
для чего рассматривалась следующая схема:
Зарождение цепи RCH2 -|- НО • —> RCH • + Н2О,
Развитие цепи RCH • -|-О2 —► RCHOO*,
RCHOO • +RCH2 —> RCHOOH+RCH.,
Обрыв цепи RCHOO • Н-НО- —> RCHOH-f-O2.
Возникающий свободный радикал гидроксила слу-
жит в одно и то же время инициатором зарождения и
инициатором обрыва цепи.
В случае фотохимического окисления изопропилбен-
зола (кумол), инициированного свободными радикала-
ми, которые образуются из азонитрилов [15], кинетика
реакции описывается уравнением
— d [O2]/dt = kl'1' • [С]’/а • [RH] [О2]°.
где I — интенсивность света, [С] — концентрация сенси-
билизатора, [RH] — концентрация углеводорода.
Гетерогенные реакции окисления
209
Этому кинетическому уравнению соответствует сле-
дующий механизм реакции:
Инициирование цепи R * или RO2 •,
Развитие цепи R • + О2 —> RO2 •,
RO2. 4-RH —> ROOH + R-
Обрыв цепи RO2 • + RO2 • —► продукты,
r. —свободный радикал, СбН5С(СН3)2.
Окисление ненасыщенных соединений
Обзор реакций окисления олефинов составлен Твиг-
гом [2] *. Этот автор предлагает следующую общую схе-
му: в начале реакции окисления возникают гидропере-
киси; образующиеся затем свободные радикалы атакуют
в a-положение метильную группу, атомы водорода ко-
торой активированы близостью двойной связи. В 1,4-ди-
олефинах группа —СНг активируется более сильно:
-СН=СН—СН2—СН=СН---------> — ей—CH—сй2—сн—ей-
Поскольку молекула кислорода может присоединиться
равновероятно к трем отмеченным местам, могут обра-
зоваться три перекиси.
Фармер с сотрудниками [16] отмечает, что в началь-
ной стадии аутоокисления ненасыщенных соединений,
таких, как каучук, происходит окисление а-метиленовой
группы. Болленд [17] показал, что реакции окисления
олефинов протекают по схеме, приведенной в начале
этой главы. Изучалась [18] эффективность радикальной
реакции
RO2- +RH —> ROOH4-R.
для 22 ненасыщенных углеводородов. При этом выясни-
лась легкость отрыва водорода а-метиленовой группы в
зависимости от природы замещающих групп. Показано,
что в олефине
СН3—СН-=СН?
а Ь с
* Подробный обзор исследований по окислению олефиновых уг-
леводородов см. в книге: Штерн В. Я., Механизм окисления угле-
водородов в газовой фазе, М., Изд-во АН СССР, 1960, стр. 367.
14 Зак. 1249
210
Глава 9
активность а-метиленовой группы не зависит от при-
роды алкильной группы в положении Ь, но увеличи-
вается при алкильном замещении в положении а и с.
Окисление фенолов
Представляют интерес реакции окисления фенолов
в щелочных растворах, поскольку такие растворы при-
меняются в качестве поглотителей молекулярного кис-
лорода. Джеймс и Вайсбергер [19] показали, что произ-
водные гидрохинона окисляются в щелочном’ растворе
молекулярным кислородом с количественным выходом
производных хинона и перекиси водорода
ОН
Н-2ОН
б
сн3/\сн3
сн311сн3
о
и
сн/\сн3
chJ^Jch, +°2
II
о
Скорость реакции окисления дурогидрохинона кис-
лородом пропорциональна квадрату концентрации ионов
гидроксила [19]. Это дает основание предполагать, что
на первой стадии образуется двухзарядный анион дуро-
гидрохинона, который затем сравнительно медленно ре-
агирует с молекулярным кислородом. Если одна из ме-
тильных групп дурогидрохинона замещена атомом водо-
рода, то протекает также следующая реакция [20]:
О О
CH’\/I\/H СН3 II он
I г -+-н2о2> Y I +Н2о
ch/Y^ch, ch/YXch3
о о
Гетерогенные реакции окисления
211
Образующийся при этом оксихинон полимеризуется с
образованием гуминовых кислот.
Представляет интерес самое обычное явление —
энзиматическое побурение фруктов на свежем срезе [21].
Скорость реакции окисления хлорогеновой кислоты
изучалась в различных условиях. Было найдено, что при
pH 7,49—8,74 начальная скорость окисления описыва-
ется уравнением
— [O2J/*# = [хлорогеновая кислота] • Роу[Н *|,
Скорость реакции окисления зависит от освещения и
наличия ионов меди.
Аутоокисление производных фенола было подверг-
нуто интересному теоретическому обсуждению [22].
Окисление серу содержащих соединений
Бекстром [56] показал, что аутоокисление сульфита в
растворе является цепной реакцией, но не высказал со-
ображений относительно механизма реакции. Уотерс
[23] дал схему реакции аутоокисления водных растворов
сульфитов, в которой ионы меди служат активными ка-
тализаторами:
Си11 + SO|“ —► Си1 + (. SO3)“,
(.SO8)“ + O2 —> (-OOSO3)
(. OOSO3)~ + (HSO3)" —> (HOOSO3)“ + (• SO3)"
Продукт реакции — это анион кислоты Каро (HSO3)",
которая является достаточно сильным окислителем для
превращения сульфитов в сульфаты через гидроксили-
рование
(HOOSO3)- +(SO3)2" —> (OSO3)2“+(HO.SO3)“
Доказательством участия радикала (‘SO3)“ в этом про-
цессе является то, что в отсутствие кислорода сульфат
меди и сульфит натрия реагируют с образованием за-
киси меди и дитионата натрия. Видимо, образуется ра-
дикал (-OOSO3)“, поскольку реакционная смесь может
индуцировать аутоокисление арсенитов, нитритов, аль-
дегидов и других органических соединений.
14*
212
Глава 9
Керакер [24], изучая реакцию взаимодействия серни-
стой кислоты и ионов Fe111, предложил следующую
схему влияния кислорода:
HSO34-O2 + H2O —► so^ -|-2Н+ + НО2-,
НО2- 4-Fe3+4-H2O —► Fe (ОН)2+4-Н2О2,
H2O24-Fe3+ —► Fe(OH)2+4-НО •.
HSO3+HO- —>2H++SO42"
Элеа и Бекстром [25] показали, что действие спир-
тов при инициировании реакции окисления растворов
сульфита должно сводиться к обрыву цепи как в тер-
мической, так и в фотохимической реакциях
Окисление органических моносульфидов было изу-
чено Бейтменом и Каннином [26] Формально насыщен-
ные сульфиды сами по себе нереакционноспособны. Ал-
лильные ациклические сульфиды вначале реагируют
значительно быстрее, чем соответствующие ненасыщен-
ные углеводороды, но аллиловые и виниловые цикличе-
ские сульфиды обладают более высокой реакционной
способностью. В интервале 45—75° для реакции пред-
лагается следующая схема:
rch=chch2sr(A) rch«-chChsr(B)>
в + о2--> ВО2,
ВО24-А----> ВО2Н-ЬВ.
ВО2Н-----во 4-он.
А 4-ОН----+ В4-Н2О.
ВО ----* CHR=CHCH04-RS.
2RS ----> r2s?.
В другой статье [27а] предлагается видоизмененный
механизм аутоокисления бут-2-енилметилсульфида, ме-
тил-1 -метилбут-2-енилсульфида и н-бутилметилсульфида.
Стадия 1
CH2RSCH3 —► CHRSCH, инициирование цепи,
CHRSCH, 4-О2 —» OOCHRSCH3 ]
OOCHRSCH, 4“ CH2RSCH3 —> | развитие цепи
— HOOCHRSCHj4- CHRSCH, I
Гетерогенные реакции окисления
213
Стадия 2
HOOCHRSCH8H-CH2RSCH3 —► HOCHRSCH3 + CH2RSOCHa.
Стадия 3
HOCHRSCH3 + CH2RSOCH8 —► (CH3)2S2 + H2O.
HOCHRSCH3 —► RCHO + CH3SH.
HOCHRSCH3 4-CH3SH —> CH3SCHR,CH=CHSCH3+H2O.
Далее было изучено [276] окисление тиоциклогекса-
на, 2-этил-2-метил-5-изопропилтиоциклопентана и тио-
циклогекс-3-ена.
Предлагается следующий механизм реакции;
Зарождение цепи RH —► R
Развитие цепи R • -)- О? —► RO2 •
RO2 +RH —► ROOH + R.
Образование сульфоокиси
ROOH + RH —► ROH 4- сульфоокись
Разложение ) ROH —► Н2О 4-полимеры
монотиоацеталя | ROOH —► НО • 4- RO
и гидроперекиси J НО • 4- RH —► Н2О 4- R
Окисление ионов металлов
Из ранних работ в области аутоокисления ионов ме-
таллов следует отметить исследования Френда и Прит-
чета [28], изучавших соли Fe11, и Фильсона и Уолтона
[29], работавших с солями Sn11. Поскольку стандартный
электродный потенциал реакции
4НЧ +О?4-4г —► 2Н2О
равен —1,23 в, можно составить большое число возмож-
ных редокс-систем с участием ионов металлов, напри-
мер со следующими ионами [30]:
Е,в
Till —> Tini-i-tf 0,37
uv UVI + * -0,10
Си» —► Cull +e —0,15
V’4 — vIV + ^ -0,36
Puin —► Puiv4-^ -0,97
214
Глава 9
Окисление иона ванадия (III) происходит по реакции
4Vni+O2-|-2H2O —> 4VO|++4H+,
кинетика которой описывается уравнением [31)
— d [V»4/^ = k [О2] [Vin|/[H+|.
Этому кинетическому уравнению соответствует следую-
щий механизм:
V3+ 4- Н2О 7~*• VOH2+ + Н+ быстро устанавливающееся равно-
весие.
VOH2+ -|- О2 —► продукты определяющая скорость стадия.
Реакция аутоокисления солей Т1ш [32] протекает со ско-
ростью
/?= [TiCl3] 4-0,94 [Т1С13]71.
Предлагается довольно сложная схема реакции:
Ti(OH)2+ 4- О2 4- Н2О —► Ti(OH)O3+ 4- он- 4- • он,
П(он)2+ 4-. он —► Т1(ОН)|+,
Ti(OH)O3+ 4-Ti(OH)2+ —► Т1(ОН)3+4-Т1(ОН)О2'
2Т1(ОН)О2+ —► 2Т1(ОН)2+ 4- О2,
Т1С12+4-2о2 —> TiCio|+4-o2,
TiClO^ 4- Н2О —> TiCIO2+ 4- 2НО.,
2Т1С1О2+ —> 2Т1С12+4-О,.
Кинетическое уравнение имеет вид
Я= Ло [Т1С13] [О2]/[НС1] +kb [TiCl3]7’ [O2J.
Легко заметить, что первый член уравнения имеет тот
же вид, что и для реакции окисления ванадия.
При окислении ионов Fe11 в среде концентрирован-
ной соляной кислоты [33] скорость реакции пропорцио-
нальна первой степени концентрации ионов Fe11 и кис-
лорода, но является более сложной функцией кон-
центрации ионов водорода. Предлагается следующий
Гетерогенные реакции окисления
215
механизм реакции:
H+4-Cl~4-Fen —► НРеС12+,
HFeCl2+ 4- О2 —> НО2- +Реш + СГ,
НО2. 4~Fen —► Fe,n 4-Н02“,
НО2~+Н+ — Н2О2,
H2O24-Fell —► НО - 4-HO-+FeHi,
НО - 4- Fell —► НО-4-Fein.
Для этой же реакции, но в среде хлорной кислоты най-
дено другое кинетическое уравнение [34]:
— d [Fell]/d/ = k [Fell]’ [O2],
причем величина константы третьего порядка слегка
увеличивается с понижением кислотности по эмпириче-
скому уравнению. Предлагается следующий механизм:
Fe’+4-O2 Fe2+• О2,
Fe2+-O24-H2O-Fe2+ —► FeO2H2+4-HOFe2+
Аутоокисление ионов меди (I) протекает со скоростью,
пропорциональной концентрациям кислорода и ионов
Си1.
В противоположность предыдущему случаю, скорость
реакции увеличивается с повышением концентрации ио-
нов водорода [35]. Реакция протекает по стехиометриче-
скому уравнению
4Си'4-4Н+4-О2 —► 4Сип4-2Н2О.
Для нее предлагается следующая схема:
Си+4-О2 —► CuO^,
CuO^4-H+ —* Cu2+4-HO2-
После этого последовательно протекают быстрые стадии
реакции, в результате которых каждой молекулой кис-
лорода окисляется дополнительно еще три иона Си1.
Реакция между Ри1п и кислородом в водном рас-
творе в присутствии ионов сульфата [36] имеет первый
порядок относительно О2, второй порядок относительно
Риш, нулевой порядок относительно PuIV и почти
216
Глава 9
нулевой порядок относительно ионов водорода. Скорость
реакции имеет сложную зависимость от концентрации
ионов сульфата. Исследовано влияние тяжелой воды
D2O на скорость этой реакции [37]. Найденные величины
отношения fen/^D= 1,17 — 1,30 оказались недостаточно
большими для того, чтобы подтвердить механизм реак-
ции через перенос атома водорода, а не через перенос
электрона.
Реакция UIV с кислородом в водном растворе хлор-
ной кислоты послужила предметом одного из наиболее
исчерпывающих исследований [38]. В большом интер-
вале условий результаты экспериментов можно описать
кинетическим уравнением
- d [U,v]/dZ = k [U,v] [О2]/[Н+].
Это уравнение имеет аналогичный вид с приведенным
ранее для кинетики окисления Tini и V111.
Реакция катализируется ионами меди (П) и инги-
бируется малыми количествами ионов серебра или хло-
ра. Реакция протекает по суммарному уравнению
2U4++O2-f-2H2O —> 2UO|++4H +
Предполагается следующий механизм:
и44 + н2о ион3+ + н+ первоначальное равновесие.
Зарождение цепи
ион3+ 4- О2 + Н2О —* UO2+ 4- НО2 4- 2Н+
Развитие цепи
ио2+ 4- о2 4- н2о —ио|+ 4- но2 4- но-,
ион3+ 4- НО2 4- Н2о ио2+ 4- 2Н+ 4- Н2О2.
Обрыв цепи
ио+ 4- НО2 4- Н2о ио|+ 4- Н2о24- НО-,
и44- 4- Н2О2 —*•> ио|+ 4- 2Н+
Применение метода стационарных состояний к проме-
жуточным продуктам данной схемы реакции приводит к
Гетерогенные реакции окисления
217
следующему выражению для ее скорости:
-d[U[V]/d/ = fe' [ulv] [О2)/[Н+].
где k' включает константы скорости указанных выше
последовательных реакций.
Каталитический эффект меди (II) объясняется воз-
никновением дополнительных стадий
Cu2++ ион3++ Н2О —► Cu+-|-UO2+-|-3H+,
Си+-|-О2 + Н+ —► Си2+-|-НО2.
а ингибирование реакции ионами серебра и хлора —
обрывом цепи:
UO2+ + Cl “ + 2Н2О —► и0Н3+4-С14-30Н~.
НО2 + С1-+Н2О —► Н2О2 + С14-ОН~,
иО2++С1 —► ио|++СГ,
НО2 + С1 —► О2 + СГ + Н+
При этом в последних двух реакциях ионы хлора реге-
нерируются. В присутствии ионов серебра возникают
реакции:
UO2+ + Ag+ —> UO2+ + Ag,
HO2 + Ag4 —> O24-H++Ag.
в результате которых серебро удаляется из раствора, в
связи с чем его ингибирующее действие сохраняется
лишь ограниченное время. Исследование реакций окис-
ления соединениями, содержащими изотоп кислорода
[39], показало, что только один атом кислорода в полу-
чаемом соединении UVI поступает из окислителя.
Другие реакции окисления
Известно много веществ, которые могут замедлять
реакцию окисления или приостанавливать ее [2]. Среди
таких соединений наиболее эффективны амины и фено-
лы, действие которых сводится к предотвращению раз-
вития цепи. Радикал RO$’ отнимает у молекулы фенола
218
Глава 9
(или амина) один атом водорода
ОН О
RO2- > ROOH+jQ
он он
Образующийся радикал не может присоединить кисло-
род и тем самым продолжить цепь, но способен реаги-
ровать с другим радикалом RO2-, теряя еще один атом
водорода и превращаясь в хинон. Обзор подобных реак-
ций ингибирования окисления в жидкой фазе составлен
Ингольдом [40].
Отклонения от обычного хода аутоокисления возни-
кают вследствие деструктивного аутоокисления некото-
рых хелатов металлов [41]. Оказывается, что цутоокис-
ление ацетилацетонатов металлов не протекает по
обычному цепному механизму. Скорость данной реакции
выражается уравнением
R = k [хелат]0,5 • [кислород]0,24
Этой кинетике соответствует механизм
Feiii(acac)3 R- + Feii(acac)2,
R- ^=± L-,
L • —|— O2 > LO2 • *
2LO2 • —> P,
LO2- +Fen(acac)2 —► Q.
FeH(acac)2 7""* Fe^S,
FeHS+ O2 —> S.
где P, Q и S представляют собой продукты, которые
могут подвергаться дальнейшим превращениям в ходе
реакции.
Если предположить, что радикал R-достаточно устой-
чив, чтобы избежать взаимодействия с кислородом, что
концентрации на ранних стадиях реакции обоих ради-
калов R- и L- могут достигать стационарных значений
и что на ранних стадиях R- и Fen(acac)2 присутствуют
в малых концентрациях, тогда описанная выше схема
реакции приводит к кинетическому уравнению, находя-
щемуся в согласии с экспериментальными данными.
Гетерогенные реакции окисления
219
Окисление поверхности металлов
В этом разделе дается очень краткий обзор мате-
риалов по данному предмету, так как полное описание
можно найти в книге Кубашевского и Гопкинса [42]. Для
зависимости увеличения веса образцов от времени на-
блюдается три вида кривых, которые описываются со-
ответственно логарифмическим, параболическим и ли-
нейным законами окисления. Краткий обзор этих зако-
нов дан Чилтоном [43].
Если скорость диффузии реагирующих веществ че-
рез окисную пленку представляет собой замедленную
стадию окисления и таким образом определяет скорость
реакции, тогда скорость окисления должна уменьшаться
с увеличением толщины окисной пленки. Если у —
толщина окисной пленки в момент времени t и она уве-
личивается до y+dy за время t+dt, тогда скорость ее
роста dyldt пропорциональна величине 1/у, что приво-
дит к параболическому закону окисления
y^k2 + 2k{t.
По параболическому закону окисляется кремний при
1200—1400° [44]. Логарифмический закон окисления
y = k3+k4 \gt
применим к металлам, которые вначале окисляются бы-
стро, а затем медленно, так что толщина пленки стано-
вится почти постоянной. Примером логарифмического
закона окисления служит поглощение кислорода герма-
нием [45].
В случае линейного закона
у = k3 4- k3t
окисление продолжается с не уменьшающейся во вре-
мени скоростью, несмотря на увеличение толщины окис-
ной пленки. Примером такого линейного закона явля-
ется окисление ниобия в интервале температур 450—
1050° [46].
Присоединение кислорода
В этом разделе также дано только краткое освещение
темы. Предметом недавно опубликованного обзора [47]
были такие соединения (представляющие значительный
220
Г лава 9
интерес), которые аналогичны природным носителям
кислорода (гемоглобин и т. п.).
Кельвин и соавторы [48] описали два типа носителя
кислорода быс-салицальдегидэтилендиимина
Соединения типа 1 способны присоединять один, атом
кислорода на каждые два атома кобальта, а соединения
типа II — один атом кислорода на каждый атом ко-
бальта. Были измерены скорости поглощения кислорода
твердыми соединениями типа I, причем оказалось, что
для исходного соединения и его 3-этоксипроизводной
реакция имеет первый порядок относительно концентра-
ции хелата. Поглощение кислорода 3-фторпроизводным
имеет второй порядок.
Для объяснения обнаруженного кинетического раз-
личия предложен механизм.
Другими интересными соединениями, способными к об-
ратимому поглощению кислорода, являются соединения
витамина Bi2a [49], фталоцианина марганца [50] и ко-
бальт(П)гистидина [51].
Окисление озоном
Имеется ряд интересных работ по окислению озоном.
Так, Таубе [52] изучил реакцию озона с муравьиной кис-
лотой
НСООН + О, —► СО, 4- 0,4- Н2О
которая протекает параллельно разложению озона. Он
пришел к выводу, что обе реакции являются цепными.
Стандартный электродный потенциал системы
О,(газ) 4- 2Н ’ 4- 2е —► Оа(газ) 4- Н4О
Гетерогенные реакции окисления
221
составляет —2,07 в. Эта величина показывает, что озон
принадлежит к сильнейшим окислителям.
Таубе и Брей [53] исследовали реакцию между озо-
ном и перекисью водорода и установили существование
двух суммарных реакций:
Н2О2 4- О3 — > Н2О 4“ 2О?.
2О3 —► ЗО2.
Для реакции перекиси водорода с озоном они предло-
жили следующий механизм:
Н2О2 4“ Оз > НО 4- НО2 • 4“ О2,
НО2 4-Оз —► НО. 4-2О2,
НО - 4-О3 —► НО2- 4-О2,
НО • 4- Н2О2 —► НО2 • + н2о.
При изучении распада озона в уксусной кислоте, ката-
лизируемого ионами кобальта (II), Хилл [54] пришел к
заключению, что реакция протекает по следующей
схеме:
Со11+О, + Н2О - -► Со(ОН)2+ 4- О2 -ь но •, (1)
Со(ОН)2+ 4-НСН3СО — -> Со(СН3СОО)2+ 4-Н2О. (2)
НО - +О3 - -> НО2- 4-0,. (3)
НО2. 4- Со(СНэСОО)2+ - -► Со2+ 4- 0,4- HCHjCO. (4)
Для реакции с ионами хлора [55]
2Н+ 4-2СГ 4-О3 —► O24-H2O4-CL
скорость образования хлора описывается уравнением
- d [Cl2] dt = kt |OS] |СГ | ч- Л21Н+1 |СГ I [Оз].
Этому кинетическому уравнению соответствует следую-
щий механизм:
О84-СГ —► О24“СЮ стадия, определяющая скорость
2Н+ 4- СП 4- CIO” —► С124“Н2О быстрая стадия.
222
Глава 9
При озонировании бензола в хлороформе было найдено,
что скорость реакции пропорциональна концентрациям
бензола и озона [56]. Дальнейшие исследования с заме-
щенными бензолами [57] показали, что озон действует
как электрофильный агент.
Реакции озонолиза послужили предметом обзора
Бейли [58]. При изучении озонолиза полициклических
ароматических углеводородов [59] найдено, что озон ата-
кует связь, имеющую самую низкую энергию локали-
зации.
Окисление двуокисью марганца
В нейтральных неводных растворах двуокись мар-
ганца может выступать как окислитель [60]. Было обна-
ружено, что при встряхивании концентрата витамина А,
растворенного в петролейном эфире, с подкисленным
раствором перманганата образуются продукты, спектр
поглощения которых указывает на присутствие ретине-
на. Очень высокий выход альдегида связан с образова-
нием гидратированной двуокиси марганца. Дальнейшие
эксперименты показали, что активным окислителем яв-
ляется двуокись марганца, а не перманганат. Бензило-
вый, цетиловый и изопропиловый спирты в сходных
условиях не окисляются; и замена окислителя хромовой
кислотой, двуокисью бария, двуокисью свинца или
окисью серебра остается безрезультатной.
Окисление можно проводить при комнатной темпера-
туре. Так, в случае применения а-, 0-этиленовых и аце-
тиленовых спиртов ненасыщенный спирт встряхивают
при комнатной температуре в нейтральном раствори-
теле с тонко диспергированной двуокисью марганца.
Можно также пропускать раствор через колонку, содер-
жащую двуокись марганца:
СН2==з=СНСН2ОН —► сн2==снсно
СН=С. СН«С(СН3)СН2ОН —► СН=С СН=С(СН3). сно.
Несмотря на избирательные действия и эффективность
этих реакций, применение двуокиси марганца ограни-
чено вследствие трудностей воспроизведения условий ее
приготовления.
Гетерогенные реакции окисления
223
ЛИТЕРАТУРА
1. Lundberg W. О., Autoxidation and Autioxidants, Vol. 1; 2;
Interscience, New York. 1961.
2. T w i g g G. H., Chem. Ind., 4 (1962).
3. В a у s t о n J. H., Looney F. D., Winfield M. E., Aust. J.
Chem., 16, 557 (1963).
4. Me Ne she у J. R., Heller C. A., Chem. Rev., 54, 325 (1954).
5. Backstrom H. L. J., J. Am. chem. Soc., 49, (a) 1460; (6) 1465
(1927).
6. В о w e n E. J., T i e t z E. L., J. chem. Soc., 234 (1930).
7. В о dens t ein M., Z. phys. Chem., B12, 151 (1931).
8. В acks trom H. L. J., Z. phys. Chem., B25, 99 (1934).
9. К a g a n M. J., Lubarsky G. D., J. phys. Chem., 39, 837
(1935).
10. В a w и С. E. H., Williamson J. B., Trans. Faraday Soc., 47,
(a) 721; (6) 735 (1951).
11. Cooper H. R., Melville H. W., J. chem. Soc., (a) 1984; (6)
1944 (1951).
12. Ba wn С. E. H., Jolley J. E., Proc. R. Soc., A237, 297 (1956).
13. В a w и С. E. H., Tobin T. P., Raphael L., Proc. R. Soc.,
A237, 313 (1956).
14. R о b e r t s о n A., Waters W. A., (a) Trans. Faraday Soc., 42,
201 (1946); (6) J. chem. Soc., 1574 (1948); (в) J. chem. Soc.,
1578 (1948); (r) J. chem. Soc., 1585 (1948).
15. Melville H. W., Richards S., J. chem. Soc., 944 (1954).
16. Farmer E. H., Bloomfield G. F., S u n d r a 1 i n g m a n A.,
Sutton D. A., Trans. .Faraday Soc., 38 348 (1942).
17. В о 11 a n d J. L., Quart. Rev., 3, 1 (1949).
18. В о 11 a n d J. L., Trans. Faraday Soc., 46, 358 (1950).
19. J a m e s T. H., W e i s s b e r g e r A., J. Am. chem. Soc., 60, 98
(1938).
20. James T. H., Snell J. M., W e i s s b e r g e r A., J. Am. chem.
Soc., 60, 2086 (1938).
21. I n g r a h a m L. L., С о r s e J., J. Am. chem. Soc., 73, 5550 (1951).
22. F u e и о T., R e e T., E у r i n g H., J. phys. Chem., 63, 1940
(1959).
23. Waters W. A., Rep. Progr. Chem., 152 (1945).
24. К а г г а к e r D. G., J. phys. Chem., 67, 871 (1963).
25. Al yea H. N., Backstrom H. L. J., J. Am. chem. Soc., 51,
90 (1929).
26. Bateman L., Cunneen J. I., J. chem. Soc., 1596 (1955).
27. Bateman L., Cunneen J. I., Ford J., J. chem. Soc., (a)
3056 (1956)*; (6) 1539 (1957).
28. F r i e n d J. A. N., Pritchett E. G. K-, J. chem. Soc., 3227
(1928).
29. Filson G. W., Walton J. H., J. phys. Chem., 36, 740 (1932).
30. Latimer W. M., Oxidation Potentials, Prentice-Hall, New York,
1952; см. общую литературу к Введению.
31. R a m s е у J. В., Sugimoto R., de V о г к i n H., J. Am. chem,
§oc., 63, 3480 (1941).
224
Глава 9
32. McKenzie Н. А. Е., Tompkins F. С., Trans. Faraday Soc.,
38, 465 (1942).
33. Р о s n е г А. М., Trans. Faraday Soc., 49, 382 (1953).
34. G е о г g е Р., J. chem. Soc., 4349 (1954).
35. Nord Н.. Acta chem. scand., 9, 430 (1955).
36. Newton T. W., Baker F. В., J. phys. Chem., 60, 1417 (1956).
37. Baker F. B., Newton T. W., J. phys. Chem., 61, 381 (1957).
38. Halpern J., Smith J. G., Can. J. Chem., 34, 1419 (1956).
39. Gordon G.t Taube H., Inorg. Chem., 1, 69 (1962).
40. I n go 1 d K. U., Chem. Rev., 61, 563 (1961).
41. Arnett E. M., Mendelsohn M. A., J. Am. chem. Soc., 84,
3824 (1962).
42. К у б а ш e в с к и й О., Гопкинс Б. Е., Окисление металлов и
сплавов, М., Издатинлит, 1955.
43. Chilton J. Р., Principles of Metallic Corrosion, R. Inst. Chem.
Monogr. Schools, 4, 5 (1961).
44. Evans J. W., Chatterji S. K-, J. phys. Chem., 62, 1064
(1958).
45. Green M., Lieberman A., J. Phys. Chem. Solids, 23, 1407
(1962).
46. McLintock С. H., Stringer J., J. less-common Metals, 5,
278 (1963).
47. V о g t L. H., F a i g e n b a u m H. M., W i b e r 1 e у S. E., Chem.
Rev., 63, 269 (1963).
48. С a 1 v i n M.. Bailes R. H., W i 1 m a r t h W. K., J. Am. chem.
Soc., 68, 2254 (1946).
49. Jaselskis B., Diehl H., J. Am. chem. Soc., 80, 2147 (1958).
50. Elvidge J. A., Lever А. В. P., Proc. chem. Soc.,- 195 (1959).
51. San о Y., Taube H., J. inorg. nucl. Chem., 25, II (1963).
52. Taube H., J. Am. chem. Soc.. 63, 2453 (1941).
53. T a u b e H., Bray W. C., J. Am. chem. Soc., 62, 3357 (1940).
54. Hi 11 G. R., J. Am. chem. Soc., 71, 2434 (1949).
55. Yea tt s L. R. B., Taube H., J. Am. chem. Soc., 71, 4100 (1949).
56. Sixma F. L. J., Boer H., Wibaut J. P., Rec. Trav. chim.
Pays-Bas, 70, 1005 (1951).
57. Wibaut J. P., Sixma F. L. J., Rec. Trav. chim. Pays-Bas, 71,
761 (1952).
58. В a i 1 ey P. S., Chem. Rev., 58, 925 (1958).
59 С о p e 1 a n d P. G., Dean R. E., McNeil D., J. chem. Soc.,
1232 (1961).
60. Evans R. M., Quart. Rev. Chem. Soc., London, 13, 61 (1959).
АВТОРСКИЙ УКАЗАТЕЛЬ
Абель (Abel Е.) 100
Адамсон (Adamson A. W.) 75,
113
Андерсон (Anderson J. S.) 172
Баксендейл (Baxendale J. Н.)
173
Банн (Bunn D.) 30
Бантон (Bunton С. А.) 98, 159
Барб (Barb W. GJ 174
Беджер (Badger G. М.) 163
Бекон (Bacon R. G. R.) 182
Бекстром (Backstrom H. L. J.)
204 211 212
Белл ’(Bell R. P.) 79, 80, 89
Берка (Berka A.) 5, 132
Бернал (Bernal J. D.) 18
Бескен (Boeskin J.) 139
Бейли (Bailey P. S.) 222
Бейтмен (Bateman L.) 212
Боббит (Bobbit J. M.) 159
Боденштейн (Bodenstein M.)
190, 191
Бозе (Bose A. N.) 195
Болдуин (Baldwin R. R.) 189
Болленд (Bolland J. LJ 209
Боун К. (Bawn С. E. H.) 53,205
Боун У. (Bone W. A.) 187
Браун (Brown R. D.) 164
Брей (Bray W. C.) 92, 103, 221
Бриттон (Britton H. T.) 132
Броде (Brode W. R.) 67
Бэксендейл (Baxendale J. H.) 53
Бэлгрин (Bulgrin V. C.) 160
Ван дер Линден (Van der Lin-
den R.) 142
Вайс (Weiss J.) 102, 173
15 Зак. 1249
Вайсбергер (Weissberger A.) 210
Вебер (Weber J. R.) 34
Вестгеймер (Westheimer F. H.)
153, 154
Вильямсон A. (Williamson A. T.)
187
Вильямсон Дж. (Williamson J. B.)
205
Воеводский В. В. 188
Воль (Wahl А. С.) 112, 113
Вултерин (Vulterin J.) 5, 132
Габер (Haber F.) 173
Гальперин (Halperin J.) 103
Гарнер (Garner С. S.) 37
Геллер (Heller С. А.) 204
Гелле (Cells Е.) 89
Гершинович (GershinQwitz Н.)
190
Гизенга (Huizenga J. R.) 35
Гинзберг (Ginsburg D.) 88
Гиншельвуд (Hinshelwood С. N.)
187, 188
Гопкинс (Hopkins В. Е.) 219
Гордон (Gordon В. М.) 76
Грайдер (Gryder J. W.) 40
Даниельс (Daniels F.) 190
Де ла Map (De la Mare Р. В. D.)
87, 95
Денисов Е. Т. 207
Дербишайр (Derbyshire D. Н.)
95, 101
Дейнтон (Dainton F. S.) 171,
191
Джексон (Jackson Е. L.) 159
Джеймс (James Т. Н.) 210
Джилке (Gilks S.) 4Q
226
Авторский указатель
Дил (Deahl Т. J.) 52
Додсон (Dodson R. W.) 28
Дорфман (Dorfman М. С.) 40
Друммонд (Drummond D. Y.) 61,
146
Дэвис (Davies A. G.) 101
Дюк (Duke F. R.) 73, 159, 160,
165
Либхевский (Liebhafsky Н. А.)
103
Ливингстон (Livingstone R. S.)
92
Лидстон (Lidstone A. G.) 104
Лотт (Lott К. А.) 136
Лоунер (Launer Н. F.) 148
Любарский (Lubarsky G. D.) 204
Зибеншайн (Siebenschein R.) 100
Зыка (Zyka J.) 5, 132
Ивеис (Evans М. G.) 101
Ингольд (Ingold С. К.) 88, 218
Иост (Yost D. М.) 148
Ирвин (Irvine D. Н.) 81, 182
Каббе (Kabbe Н.) 81
Каган (Kagan М. J.) 204
Каллис (Cullis С. F.) 74, 135,
138, 140, 143, 192
Каннин (Cunneen J. I.) 212
Караш (Kharasch М. S.J 177
Каррингтон (Carrington А.) 137
Кельвин (Calvin М.) 220
Кенион (Kenyon J.) 145
Керакер (Каггакег D. G.) 212
Кильпатрик (Kilpatrick М. А.) 120
Кинг (King Е. L.) 31
Кольтгоф (Kolthoff I. М.) 90
Кондратьев В. Н. 186
Корднер (Cordner J. Р.) 80
Криги (Criegee R.) 78, 80
Кролль (Kroll Н.) 158
Ксантакос (Xanthakos Т. X.) 51
Кубашевский (Kubaschewski О.)
219
Кук (Cook J. W.) 163
Куорт (Kwart Н.) 154
Купер (Cooper Н. R.) 205
Ландберг (Lundberg W. О.) 202
Латимер (Latimer W. М.) 136,137
Лайтинен Г. А. 149
Левеллин (Llewellyn D. R.) 98
Ледбери (Ladbury Y. W.) 74,
135, 138, 140, 143
Лейдлер (Laidler К. J.) 23, 190
Либби (Libby W. F.) 112
Маккинон (McKinnon D. J.) 170
Мак-Несби (McNesbey J. R.) 204
Майзус 3. К. 207
Мелвиль (Melville Н. W.) 205
Мерц (Merz J. Н.) 170, 174
Мейер (Mayor L.) 189
Милл (Mill Т.) 154
Милликен (Milliken R. S.) 188
Минков (Minkoff G. J.) 188, 195
Мишра (Mishra Н.) 100
Морган (Morgan К. J.) 11, 118
Мэгнуссон (Magnusson L. В.) 35
Мэлкехи (Mulcahy М. F. R.)
193, 194
Мэссон (Masson I.) 96
Мэтьюз (Matthews С. N.) 154
Налбандян'А. Б. 188
Найтингейл (Nightingale Е R.)
120
Неф (Nef J. W.) 127
Новик (Novick А.) 153
Нойес (Noyes А. А.) 52
О’Коннор (O’Connor С.) 98,100
Ольсен (Olsen A. R.) 120
Перлин (Perlin A. S.) 80
Позакер (Pausacker К. Н.) 80.
168
Прайс (Price С. С.) 158
Притчет (Pritchett Е. G. К.) 213
Рабидо (Rabideau S. W.) 38
Райли (Riley Н. L.) 165
Райт (Wright G. А.) 103, 105,
118
Рейд (Reid С.) 189
Авторский указатель
22?
Ригби (Rigby W.) 146
Ридд (Ridd J. К.) 88
Робертсон (Robertson А.) 207
Робинсон (Robinson R.) 19
Росотти (Rossotti F. J.) 39
Россер (Rosser W. А.) 191
Сатин (Sutin N.) 76
Сатклиф (Sutcliffe L. Н.) 34
Свэнн (Swann S.) 51
Семенов Н. Н. 186, 187
Силлен (Sillen L. G.) 19
Сильвермен (Silverman J.) 28
Симонс (Symons М. С. R.) 100,
136, 137, 145
Симонсен (Simonsen Т. R.) 120
Синг (Singh М. Р.) 128, 129
Скэтчард (Scatchard G.) 24, 30
Слек (Slack R.) 155
Спиро (Spiro М.) 25
Стокс (Stocks R.) 19
Стюарт (Stewart R.) 116, 142,
143
Таубе (Taube Н.) 18, 31, 73, 103,
220, 221
Тауненд (Townend D. Т. А.) 187
Твигг (Twigg С. Н.) 203, 209
Теренин А. Н. 15
Терни (Turney Т. А.) 103, 105,
128, 129
Тейлор (Taylor Н. А.) 164
Типпер (Tipper С. F. Н.) 188,
195
Толленс (Tollens В.) 126
Томас (Thomas J. R.) 179
Томпкинс (Tompkins F. С.) 74,
115
Треси (Treacy J. С.) 190
Тротмен-Диккенсон (Trotman-
Dickenson A. F.) 184
Уарис (Waris А.) 198
Уайз (Wise) 191
Уиберг (Wiberg К. W.) 116, 143,
154, 181
Уолтон (Walton J. Н.) 213
Уолш (Walsh A. D.) 193
Уотерс (Waters W. А.) 48, 61, 95,
101, 135, 146, 153, 155, 170, 174,
177, 207, 211
Ури (Uri N.) 50, 101
Уэллс (Wells С. F.) 53
Уэсп (Wesp Е. F.) 67
Фармер (Farmer Е. Н.) 209
Фаулер (Fowler R. Н.) 18
Фентон (Fenton Н. J. Н.) 173
Фермен (Furman S. С.) 37
Физер (Fieser L. F.) 152, 163
Филипс (Philipps L.) 132
Фильсон (Filson G. W.) 213
Фиш (Fish А.) 198
Френд (Friend J. A. N.) 213
Фукуи (Fukui К.) 170
Хаус (House О. М.) 179
Хилл Г. (Hill G. R.) 221
Хилл Л. (Hill L. М.) 115
Холленд (Holland R. V.) 128,
129
Хэнби (Hanby W. Е.) 96
Целиков (Zelikoff М.) 164
Цейсс (Zeiss Н. Н ) 154
Чилтон (Chilton J.*P.) 219
Шарп (Sharpe A. G.) 9
Шенталь (Schoental R.) 163
Шеппард (Sheppard J. С.) 112
Штерн В. Я. 186, 209
Элеа (Alyea Н. N.) 212
Эмануэль Н. М. 207
Эмеле (Emeleus Н. J.) 172
Энбар (Anbar М.) 88
Эшмор (Ashmore Р. G.) 191
Эйринг (Eyring Н.) 190
15*
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азота двуокись
как окислитель 198—200
реакции с ацетиленом 198, 199
— с глиоксалем 199
— с кислородом 187, 190, 191
— с хлористым водородом 199,
200
Азотистая кислота
как окислитель 103—107
------классификация реакций
104, 105
реакция с азотистоводородной
кислотой 106, 107
— с аскорбиновой кислотой
106
Азотистоводородная кислота,
окисление азотистой кислотой
106, 107
Азотная кислота
как окислитель 107, 108
реакции с ароматическими со-
единениями 107, 108
— с церием’(Ш) 108
Акриловый альдегид, окисление
марганцем (III) 63
Альдегиды
аутоокисление 204—207
окисление медью(II) 8
— перманганатом 143—145
— хромовой кислотой 154—157
Аммиакат серебра как окисли-
тель 126, 127
Анилин, окисление ацетатом
иодозобензола 162
L-Арабиноза, окисление
медью (II) 127, 128
Аскорбиновая кислота
окисление азотистой кислотой
106
Аскорбиновая кислота
окисление селенистой кислотой
165
Аутоокисление 202—217
механизм реакций 203, 204
Ацетальдегид
аутоокисление 204, 205
окисление кислородом 197, 198
Ацетил ацетонаты металлов,
аутоокисление 218
Ацетилен, окисление двуокисью
азота 198, 199
Ацетоин
окисление железом (III) 66,67
— комплексами меди(II) 130,
131
Ацетон
окисление селенистой кислотой
165
— церием(IV) 60
Ацетофенон, окисление перман-
ганатом 141
Барфеда реактив 49, 127
Бенедикта реактив 127
Бензальдегид
аутоокисление 204, 207
окисление хромовой кислотой
154, 155
Бензгидрол, окисление перман-
ганатом 142
Бензол
гидроксилирование 176
реакция с озоном 222
Бром
окисление перекиси водорода
92
реакция с диэтилфосфонатом 94
Предметный указатель
229
Бром
реакция с этиловым спиртом
93, 94
Бромистый водород, окисление
кислородом 192
Бромноватистая кислота, реак-
ция с щавелевой кислотой
93
Ванадий(П), реакция с перхло-
ратом 77, 78
Ванадий(Ш), реакция с перхло-
ратом 77, 78
Ванадий (III)-ионы
аутоокисление 213, 214
реакция с нептуний(V)-иона-
ми 36, 37
Ванадий (IV)-ионы, обмен с ва-
надий (V)-ионами 33
Ванадий (V)
как окислитель 69—72
реакция с изо масляным альде-
гидом 71, 72
— с а-оксикислотами 71
— с пинаколом 70
— с циклогексанолом 71
— с циклогексаноном 70
Винная кислота, окисление с уча-
стием радикала гидроксила 173
Гайярда реакция 74
Галогенидные кислородсодержа-
щие анионы, ряд реакционной
способности 120
Г алогены
доказательство существования
катионов 88—90
как окислители 133—135
Германий, окисление поверхно-
сти 219
Гетеролитический процесс, опре-
деление 12
Гидразин, окисление желе-
зом (III) 66
Гидроксила радикалы 172—177
источники 172, 173
и радиолиз воды 176, 177
реакция с водородом и окисью
углерода 175, 176
Гидрохинон окисление желе-
зом (III) 67
Гидрохинона производные, ауто-
окисление 210, 211
Гипоиодит-ион, разложение 118
Гипоманганат как окислитель
147
Гипоманганат-ион 136
Гипохлорит
реакция с иодидом 117
— с манганатом 119
— с нитритом 117, 118
— с сульфитом 117
— с формиатом 117
Гликоли, окисление перйодатом
159, 160
Глиоксаль, окисление двуокисью
азота 199
Глицерин, окисление церием (IV)
60
Гомолитический процесс, опре-
деление 12
Дезоксибензоин, окисление дву-
окисью селена 165, 166
Деканаль, аутоокисление 205,
206
1,2-Дибеизоилэтан, окисление
двуокисью селена 166
Диизопропилхромат 153
Дифенилметан, окисление хромо-
вой кислотой 155, 156
Диэлектрическая постоянная,
влияние на скорость ионных
реакций 24, 25
Диэтиловый эфир, окисление кис-
лородом 197
Диэтилфосфонат, окисление бро-
мом 94
Дурогидрохинон, аутоокисление
210, 211
Железо (II)-ионы
аутоокисление 212, 214, 215
обмен с железо (III)-ионами
22—25, 28—30
— с плутоний (IV)-ионами 35
окисление 76
— бихроматом 76
— перманганатом 8
230
Предметный указатель
Железо (III)
индуцирование полимеризации
акриламида 68, 69
как окислитель 64—69
реакция с ацетоином 66, 67
— с гидразином 66
— с иодидом 64
— с перекисью водорода 67,
68
— с тиосульфатом 65, 66
— с фенолами 67
Железо (III)-ионы
гидролитическое равновесие 20
обмен с нептуний (IV)-ионами
35, 36
реакция с сернистой кислотой
212
Железа(III) комплексы как окис-
лители 131—133
Изобутан, окисление кислородом
195, 196
Изомасляный альдегид
окисление ванадием (V) 71, 72
— солью Фреми 183
Изопропилбензол, фотохимиче-
ское окисление 208, 209
Изопропиловый спирт
окисление кислородом 196, 197
— пероксидисульфатом 180,
181
— - хромовой кислотой 153
Иод
окисление муравьиной кислоты
91
— титана(Ш) 91
Иодид
окисление персульфатом 120
реакция с иодной кислотой 100
— с перекисью водорода 101 —
103
Иодид-ионы
реакция с железом (III) 64
— с йодатом 8, 9
Йодистая кислота, реакция с пе-
рекисью водорода 99
Иодная кислота
равновесие в водном растворе
157, 158
реакция с иодидом 100
Иодозобензола ацетат
как окислитель 161, 162
получение 162
реакции с первичными арома-
тическими аминами 162
Иодозонитробензол, получение 96
Йодоформ
механизм реакции образования
134, 135
образование из ацетона 8, И
Ионный механизм реакции, оп-
ределение 12
Карбоновые кислоты с атомом
водорода при третичном угле-
роде, окисление перманганатом
145
Каро кислота 211
Каучук, окисление 209
Кислород
носители 219, 220
реакция с бромистым водоро-
дом 192
— с водородом 188, 189
— с окисью азота 187, 190,191
— с углеводородами 187,192—
198
— с хлором 192
структура молекулы 188
Кобальт (И) этилеидиаминтетра-
ацетат, реакция с феррициа-
нидом 113
Кобальт (III)
как окислитель 51—57
каталитическое действие при
аутоокислении 205, 207
реакция с водой 52, 53
— с метиловым спиртом 55
— с муравьиной кислотой 53.
54
— с олефинами 55, 56
— с перекисью водорода 56
— с формальдегидом 54, 55
— с циклогексанолом 56, 57
Малеиновая кислота, окисление
четырехокисью осмия 164
Малоновая кислота, окисление
марганцем (III) 62, 63
Предметный указатель
231
Манганат как окислитель 147
Марганец, степени окисления
136
Марганец(П)-ионы, окисление
74, 75
Марганец(Ш)
как окислитель 61—64
комплексообразование 61, 62
реакция диспропорционирова-
ния 61
— с акриловым альдегидом 63
— с малоновой кислотой 62,
63
— с оксалатом 62
— с а-оксикислотами 63
— с пинаколом 62
Марганца(III) ацетат, каталити-
ческое действие при аутоокис-
лении 204
Марганца двуокись как окисли-
тель 222
Марганцевая кислота как окис-
литель 136
Меди(II) комплексы
как окислители 127—131
реакция с ацетоином 130, 131
— с пропаргиловым спиртом
131
— с сахарами 127—129
Медь (I)-ионы, аутоокисление
213, 215
Меркаптаны, окисление перокси-
дисульфатом 180
Метилбензолы, окисление хромо-
вой кислотой 157
(+) -З-Метилгептен, окисление
хромовой кислотой 156
Метиловый спирт, окисление ко-
бальтом (III) 55
Моносульфиды органические, ау-
тоокисление 212, 213
Муравьиная кислота
окисление кобальтом (III) 53,
54
— озоном 220
— таллий (III)-ионами 81
реакция с иодом 91
Нептуний (III)-ионы, реакция с
нептуний (V)-ионами 36
Нептуний (IV)-ионы
обмен с железо(Ш)-ионами 35,
36
реакция с нептуний(VI)-иона-
ми 38
Нилендера реактив 126
Ниобий, окисление поверхности
219
Нитр ат-ионы, каталитическое
действие 35
Нитрозоний-ион, доказательства
существования 104, 105
Нитроний-ион, доказательства
существования 108
Нитрит
окисление гипохлоритом 117,
118
реакция с сульфитом 121, 122
Озон
как окислитель 220—222
реакции с ароматическими уг-
леводородами 222
— с бензолом 222
— с ионами хлора 221
— с муравьиной кислотой
220
— с перекисью водорода 221
— с уксусной кислотой 221
Окислители
двухэлектронные 49, 78—83
одноэлектронные 49—78
Окись углерода, окисление
ртуть (И)-ионами 82
Оксалат, окисление пермангана-
том 147, 148
Оксигалогенов ионы, доказатель-
ства существования 96—100
Оксикатионы и гидролитические
равновесия 19—21
Оксирадикалы замещенные 177—
179
Олефины, аутоокисление 209,
210
Осмиевая кислота 163
Осмия четырехокись
как окислитель 162—164
реакции с дикарбоновыми кис-
лотами 164
— с олефинами 163
232
Предметный указатель
Перекись водорода
изотопный обмен с водой 101
как окислитель 100—103, 172—
177
конверсия в Н3О2*Ю1
окисление хлором 95
реакция с ароматическими со-
единениями в присутствии
Fe2+ 174, 175
— с бромом 92, 93
— с иодидом 101—103
— с иодистой кислотой 99
— с озоном 221
— с тиоцианатом 122
— с формальдегидом 123
в щелочной среде 122, 123
энергетика разложения 172
Перенос электронов
двухэлектронный 38—45, 49,
78—83
— реакции между различными
ионами 41—45
-----обмена 39—41
классификация реакций 25—27
между комплексами металлов
111—113
одноэлектронный 25—27, 48,
49
— реакции между различными
ионами 34—38
----обмена 27—34
Перйодат
как окислитель 157—161
реакции с гликолями 159, 160
— с перманганатом 119
фотохимическое разложение
158
Перманганат
как окислитель 135—149
------типы реакций 137, 138
разложение в щелочной среде
114, 115
реакции с альдегидами 143
— с алкильными радикалами
в ароматических соединени-
ях 140, 141
— с ненасыщенными соедине-
ниями 138, 139
— с оксалатом 147, 148
— с пентаминкобальт(III)фор-
миатом 149
Перманганат
реакции со спиртами 141—143
— с формиатом 115, 116
— с цианидом 116
Перманганил-ион, образование
137
Пероксиазотистая кислота 175
Пероксисернистая кислота 103
Перхлорат как окислитель 77,
78
Пентаминкобальт (III) формиат,
окисление перманганатом 149
Перокоидисульфат
как окислитель 179—182
------классификация реакций
179
реакции с меркаптанами 180
— со спиртами 180, 181
— с фенолами 181, 182
Пинакол
окисление ванадием (V) 70
— марганцем(III) 62
— перйодатом 158, 159, 161
— церием (IV) 59
Плутоний (III) -ионы
аутоокисление 213, 215, 216
обмен с плутоний (IV)-ионами
31, 32
Плутоний (IV)-ионы, обмен с же-
лезо(II)-ионами 35
Пропаргиловый спирт, окисление
комплексами меди(II) 131
Пропилен, окисление кислородом
194, 195
Пэви реактив 127
Радикальный механизм реакции,
определение 12
Растворители, влияние на раз-
рыв связи 12
Ртуть (II)-ионы
как окислитель 82, 83
реакция с марганец(III)-иона-
ми 42
— с окисью углерода 82
— с формиатом 82
— с циклогексаноном 82, 83
Рутения четырехокись как окис-
литель 164
Предметный указатель
233
Сахара, окисление реактивом
Фелинга 128, 129
Свободные радикалы
обсуждение 169—171
реакции в растворе 171, 183
Свинца тетраацетат
как окислитель 182, 183
реакция с замещенными фено-
лами 182, 183
Селена двуокись
как окислитель 165,. 166
реакция с кетонами 165, 166
Селенистая кислота
реакция с ацетоном 165
— с иодом 165
Серебра(I) комплексы как окис-
лители 126, 127
Серебро(I)-ионы, обмен с сереб-
ро(П)-ионами 27, 28
Серебро (И)
как окислитель 50, 51
комплекс с пиколиновой кис-
лотой 51
Спирты
окисление перманганатом 141 —
143
— хромовой кислотой 153, 154
Сульфит
аутоокисление 211, 212
окисление галогенатами 121,
122
— нитритом 121, 122
Таллий(I)-ионы, обмен с тал-
лий (III)-ионами 39—42
реакция с кобальт(III)-ионами
41
— с церий(IV)-ионами 40
Таллий(1П)-ионы
как окислитель 80, 81
реакция с муравьиной кисло-
той 81
— с ртуть(1)-ионами 41, 42
Тетраацетат свинца
как окислитель 78—80
получение 78
реакции с гликолями 79
Тетралин, аутоокисление 207
Титан (II)-ионы, аутоокисление
213
Титан(Ш)
окисление иодом 91
реакция с перхлоратом 77
Титан (III)-ионы, аутоокисление
214
Углеводороды
аутоокисление 207—209
озонолиз 222
окисление кислородом 187.
102—198
Уксусная кислота, реакция сезо-
ном 221
Уран (IV)
реакция с железом(III) 42, 43
— с плутонием (IV) 42
— с таллием (III) 42, 43
— с ураном (VI) 44, 45
— с церием(IV) 42, 43
Уран(1У)-ионы
аутоокисление 216, 217
— ингибирование ионами се-
ребра и хлора 217
— каталитическое действие
Cu(II) 217
ypaH(V)-noHbi, аутоокисление
213
Уран(VI)-ионы, гидролитическое
равновесие 20, 21
Фелинга реактив 127, 128
реакция с L-арабинозой 127,
128
Фенолы
аутоокисление 210, 211
ингибирующее действие на ре-
акции окисления 217, 218
окисление пероксидисульфатом
181, 182
— феррицианидом 133
Феррицианид
как окислитель 132, 133
реакция с фенолами 133
Формальдегид
окисление кобальтом (III) 54,
55
— перекисью водорода 123
— хромовой кислотой 155
— церием (IV) 58
234
Предметный указатель
Формиат
окисление гипохлоритом 117
— перманганатом 115, 116
Фреми соль
реакция с изомасляным аль-
дегидом 183
схема распада в растворе 183
Фторальгидрат, окисление пер-
манганатом 144, 145
Фторид-ионы, каталитическое
действие 35
Фумаровая кислота, окисление
четырехокисью осмия 164
Хлор
реакция с перекисью водорода
95
фотохимическая реакция с кис-
лородом 192
Хлор-ионы, реакция с озоном 221
Хлористый водород, реакция с
двуокисью азота 199, 200
Хлорогеновая кислота, аутоокис-
ление 211
Хлорпарафины, окисление кисло-
родом 194
Хром (II) -ионы
обмен с хром(III)-ионами
30, 31
окисление 72—74
Хром (III)-ионы, окисление до
хром (IV)-ионов 123
Хромовая кислота
как окислитель 151—157
------ стехиометрия реакций
152
образование 151
реакции с альдегидами 154—
157
— со спиртами 153, 154
Хукера реакция 147
Цезий-ионы, каталитическое дей-
ствие 112, 113
Церий (III)-ионы
обмен с церий (IV)-ионами 32
окисление азотной кислотой 108
реакция с кобальт(П)-ионами
34. 35
Церий (IV)
как окислитель 57—61
реакции с ацетоном 60
— с бромидом 58
— с глицерином 60
— с а-оксикислотами 59, 60
— с пинаколом 59
— с этиловым спиртом 58, 59
Церий (IV) -ионы, гидролитичес-
кое равновесие 20
Цианид
окисление перманганатом 116
реакция с тетратионатом 122
Циклогексанол
окисление ванадием (V) 71
— кобальтом(III) 56, 57
Циклогексанон
окисление ванадием (V) 70
— ртуть(II)-ионами 82, 83
Циклогексан
окисление трехокисью хрома
155
— четырехокисью рутения 164
Щавелевая кислота, окисление
бромноватистой кислотой 93
Электродные потенциалы, связь
со свободной энергией 7
Электронный парамагнитный ре-
зонанс как метод изучения
свободных радикалов 169, 183
Энергия активации 23
Энтропия активации 21, 22
Этилацетат, окисление кислоро-
дом 198
Этилбензол, окисление перманга-
натом 141
Этиленгликоль, окисление тетра-
ацетатом свинца 79, 80
Этиловый спирт
окисление хромовой кислотой
152
— церием (IV) 58, 59
реакция с бромом 93, 94
Ядерный магнитный резонанс и
определение степени гидрата-
ции 19
СОДЕРЖАНИЕ
Предисловие 5
Из предисловия автора 6
Введение 7
Механизм реакций 9
Классификация реакций окисления 13
Литература 15
Глава 1. Реакции окисления — восстановления между
катионами 17
Растворы электролитов 17
Равновесие гидролиза и оксикатионы 19
Кинетика и механизм 21
Классификация реакций с переносом электронов 25
I. Реакции обмена с переносом одного электрона 27
Реакции между простыми катионами различных веществ 34
Реакции между простыми катионами оксикатионами 37
II. Реакции с переносом двух электронов 38
Реакции между простыми катионами различных веществ 41
Реакции между оксикатионами различных веществ 44
Л итерату р а 45
Глава 2. Реакции окисления анионов, комплексных
катионов и молекул катионами 48
Окисление серебром(II) 50
Окисление кобальтом(III) 51
Окисление церием (IV) 57
Окисление марганцем(III) 61
Окисление железом(III) 64
Окисление ванадием (V) 69
Окисление ионов хрома (II) 72
Окисление ионов марганца(II) 74
Окисление ионов железа(II) 76
236
Содержание
Окисление некоторых катионов ионами перхлората 77
Двухэлектронные окислители 78
Тетраацетат свинца 78
Ионы таллия (III) 80
Ионы ртути(II) 82
Литература 83
Глава 3. Реакции окисления образующимися катионами 87
Положительные ионы галогенов X* и их источники 88
Ионы оксигалогенов и их источники 96
Перекись водорода 100
Азотистая кислота 103
Азотная кислота 107
Литература 108
Глава 4. Реакции окйслеиия — восстановления между
анионами, анионами и комплексами, между
анионными комплексами 111
Реакции между комплексными ионами 111
Реакции между простыми анионами и комплексными анио-
нами 113
Реакции между анионами 114
Реакции перманганата в щелочной среде 114
Реакции кислородсодержащих галогенидных анионов 116
Реакции серу- и кислородсодержащих анионов 120
Реакции перекиси водорода в щелочной среде 122
Литература 123
Глава 5. Реакции окисления молекул и анионов простыми
и комплексными анионами, комплексными
катионами 125
Окисление комплексами серебра(I) 126
Окисление комплексами меди(II) 127
Окисление комплексами железа(Ш) 131
Содержание 237
Окисление галогенами в щелочной среде 133
Окисление перманганатом 135
Окисление ненасыщенных соединений перманганатом ка-
лия 138
Окисление алкильных радикалов в ароматических со-
единениях 140
Окисление спиртов 141
Окисление альдегидов 143
Окисление соединений, содержащих водород при тре-
тичном атоме углерода 145
Окисление некоторых органических веществ щелочными
растворами перманганата, манганата и гипоманганата 146
Другие реакции окисления перманганатом 147
Литература 149
Глава 6 Реакции окисления через стадию этерификации 151
Окисление хромовой кислотой 151
Окисление спиртов 153
Окисление альдегидов 154
Окисление углеводородов 155
Окисление перйодатом 157
Окисление ацетатом иодозобензола 161
Окисление четырехокисью осмия 162
Окисление двуокисью селена 165
Литература 166
Глава 7 Гомолитическое окисление в растворе 169
Радикал гидроксила и его реакции 172
Замещенные оксирадикалы (RO •) 177
Перокси дисульфат 179
Тетраацетат свинца. Соль Фреми 182
Литература 183
Глава 8. Гомогенное окисление в газовой фазе . 186
Реакция между водородом и кислородом 188
Другие реакции кислорода 190
Окисление углеводородов кислородом 192
238
Содержание
Окисление других органических веществ кислородом 196
Реакции двуокиси азота в газовой фазе 198
Литература . 200
Глава 9. Гетерогенные реакции окисления .202
Аутоокисление Окисление альдегидов Окисление углеводородов Окисление ненасыщенных соединений Окисление фенолов Окисление сер у содержащих соединений Окисление ионов металлов Другие реакции окисления Окисление поверхности металлов Присоединение кислорода Окисление озоном Окисление двуокисью марганца . 202 204 207 209 . 210 211 . 213 217 . 219 219 . 220 222
Литература Авторский указатель Предметный указатель 223 225 228
Т. ТЕРНИ
Механизмы реакций
окисления—восстановления
Редактор И. С. Чулкова
Художник К. П. Саратов
Художественный редактор Н. Фильчагина
Технический редактор Т. Мирошина
Корректор А. Рыбальченко
Сдано в производство 15/V 1968 г.
Подписано к печати 15/Х 1968 г.
Бумага глуб. печ. 84xl08’/32-3,75 бум. л.
12,6 усл. печ. л.
Уч-изд. л. 11,17. Изд. № 3/4443
Цена 98 коп. Зак. 1249
(Тем. план 1968 г. изд-ва «Мир», пор. № 131)
ИЗДАТЕЛЬСТВО .МИР*
Москва, 1-й Рижский пер., 2
Ленинградская типография № 2
имени Евгении Соколовой
Главполиграфпрома Комитета по печ
при Совете Министров СССР
Измайловский проспект, 29