/






































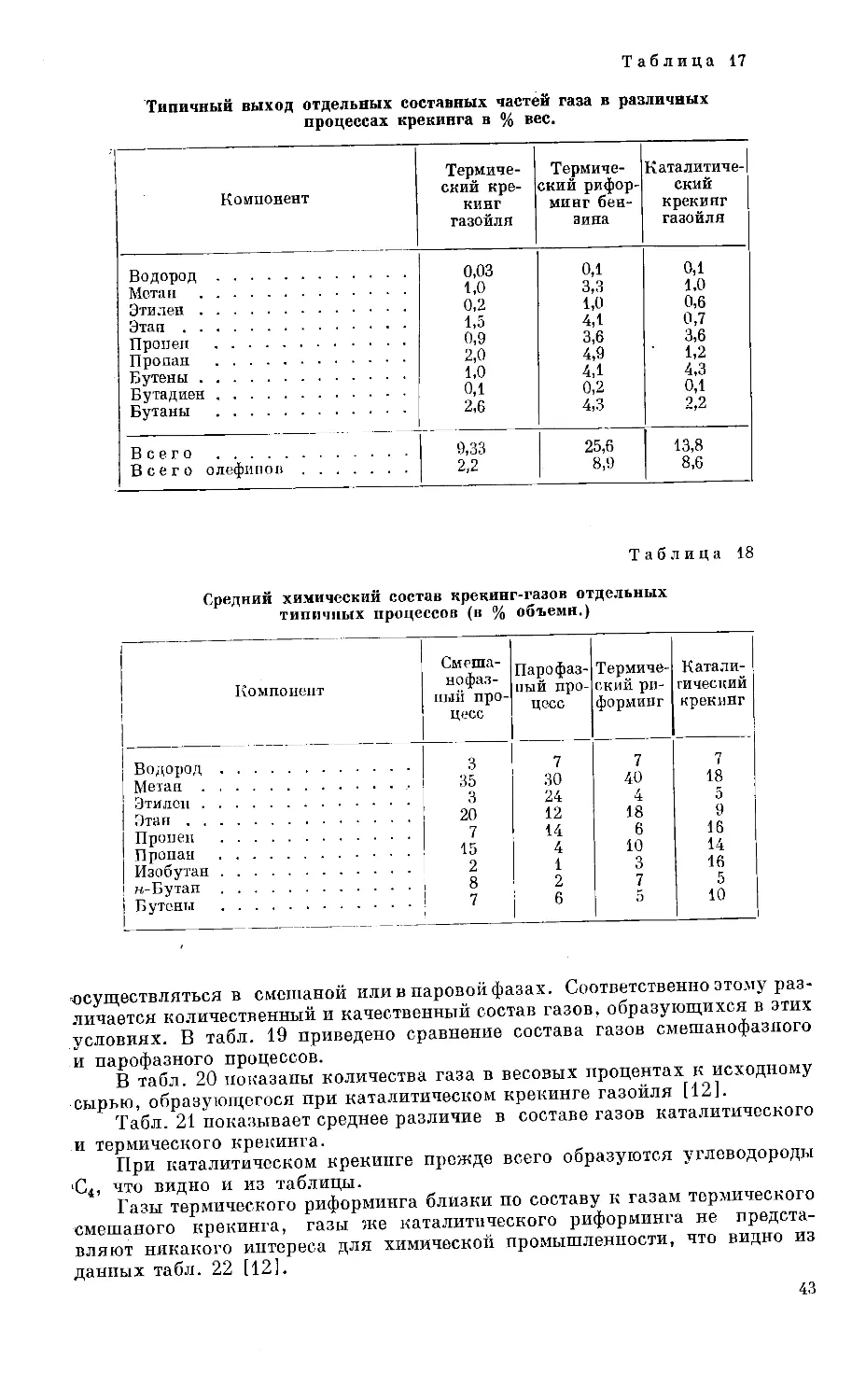
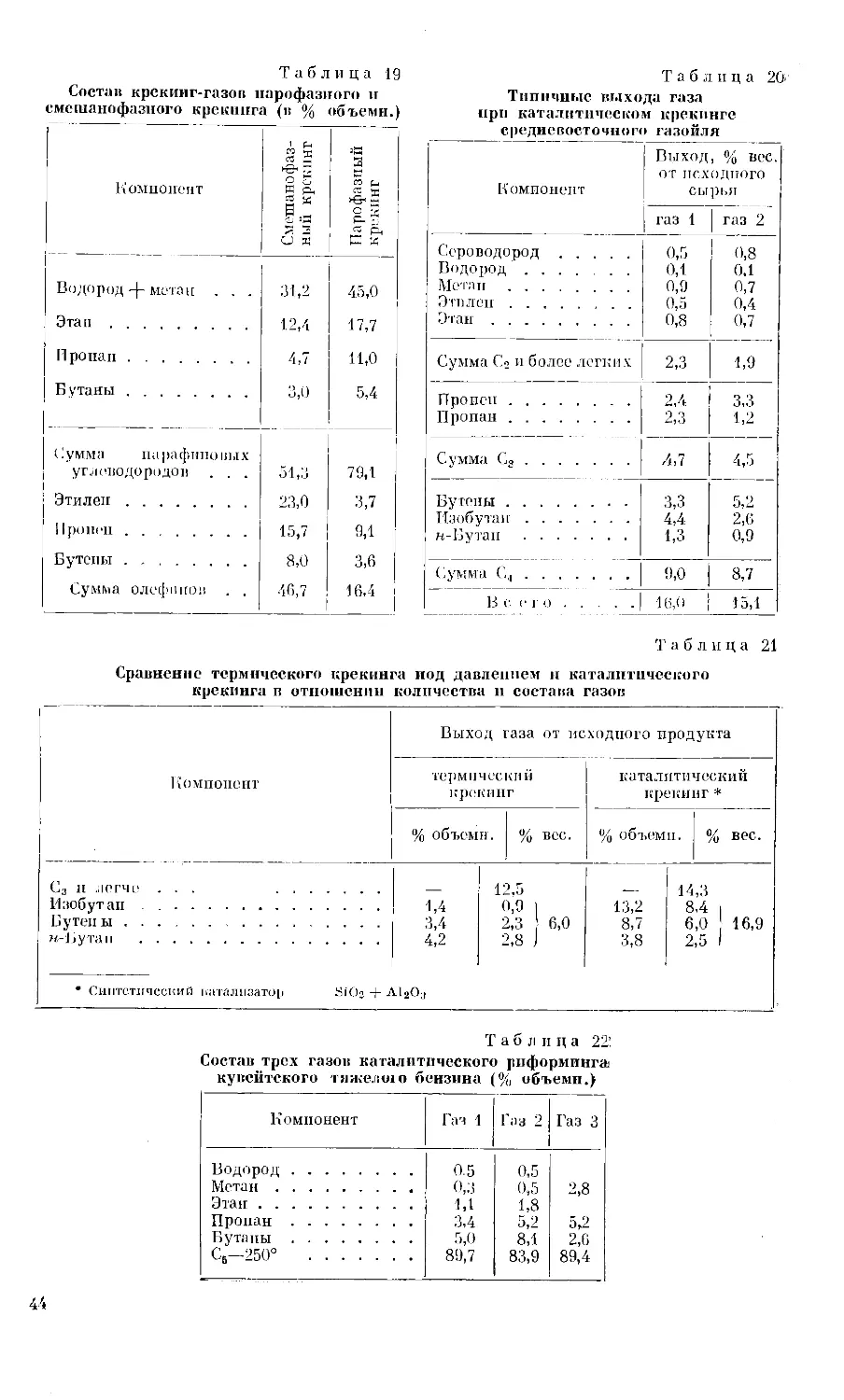






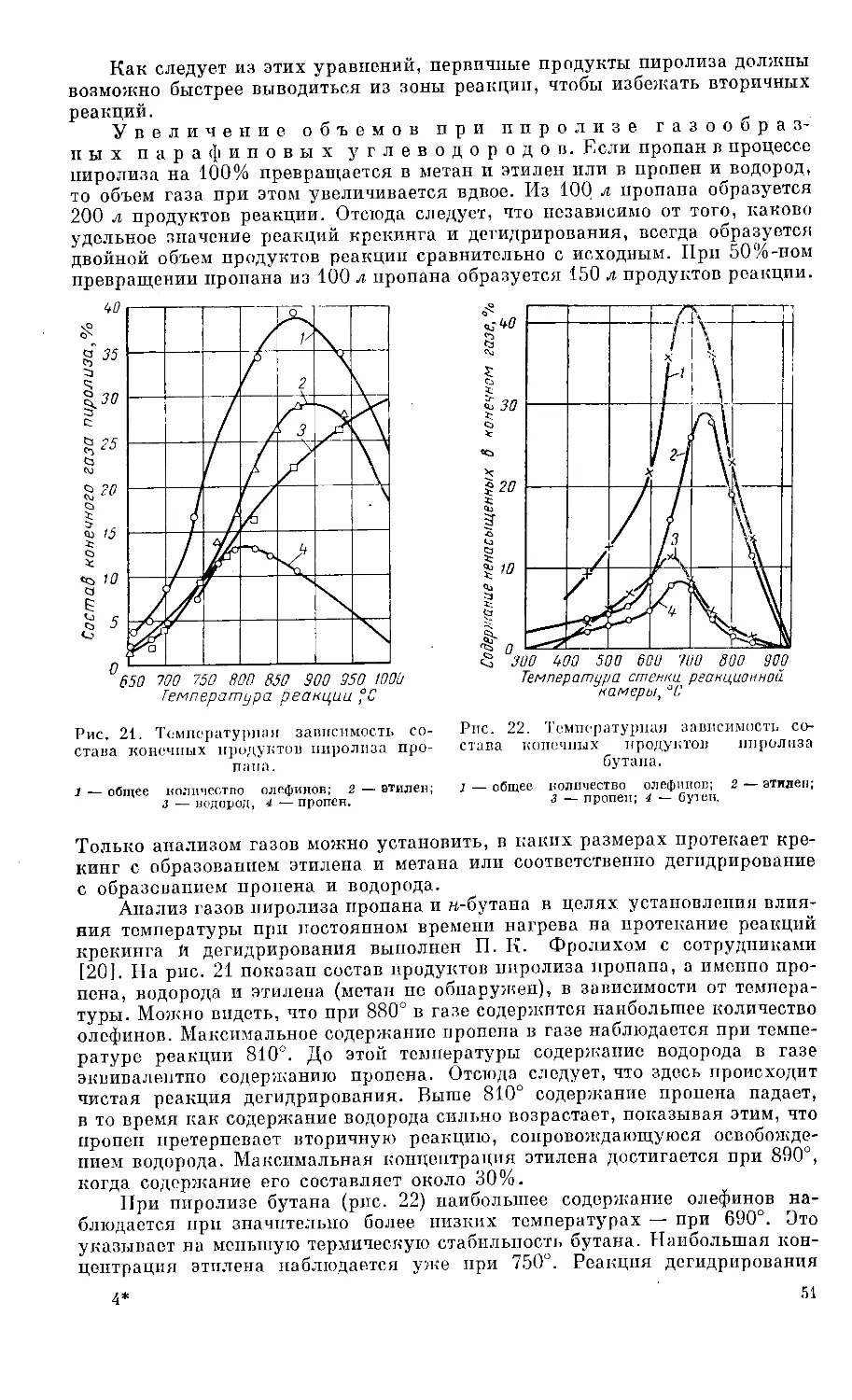
































































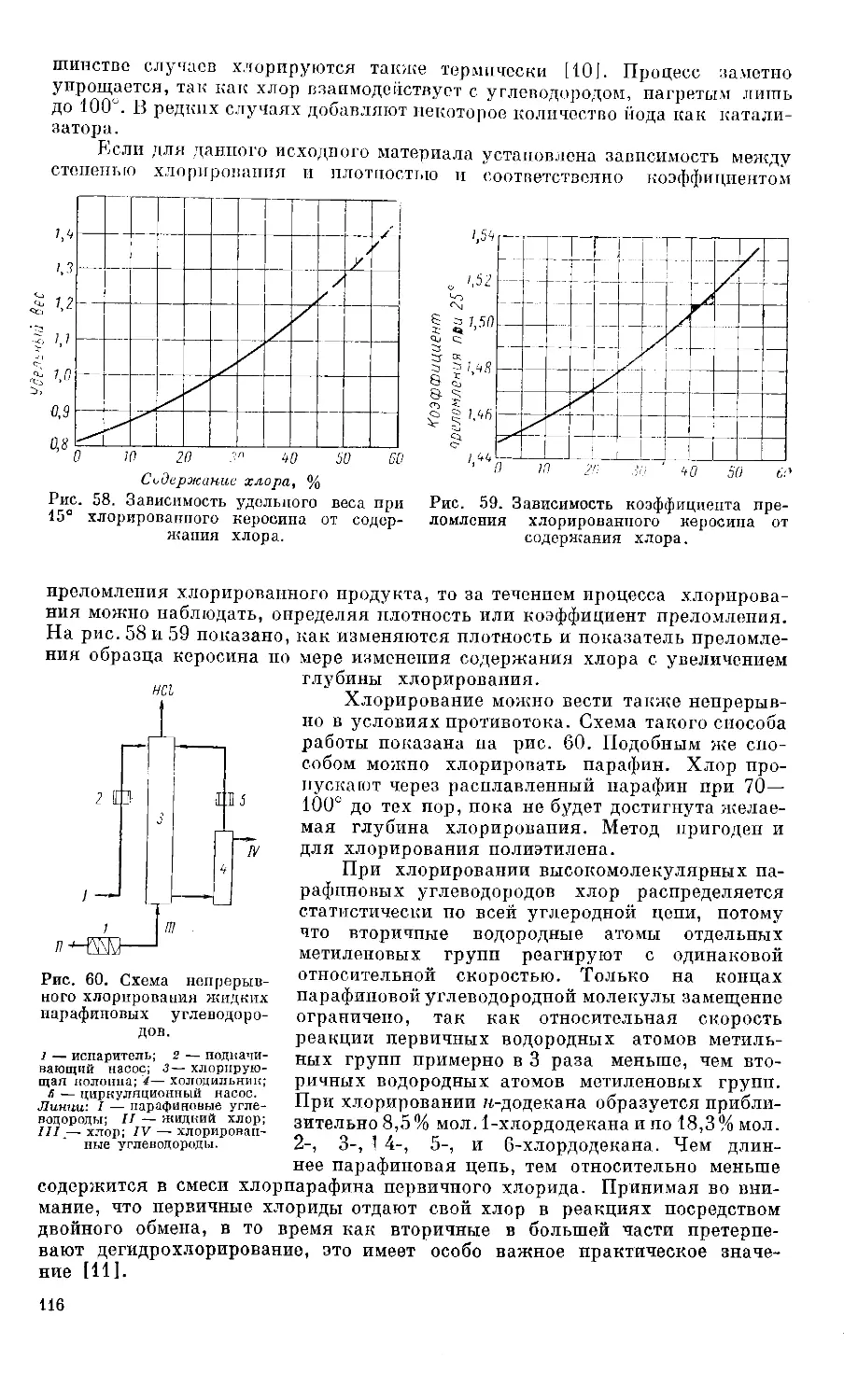



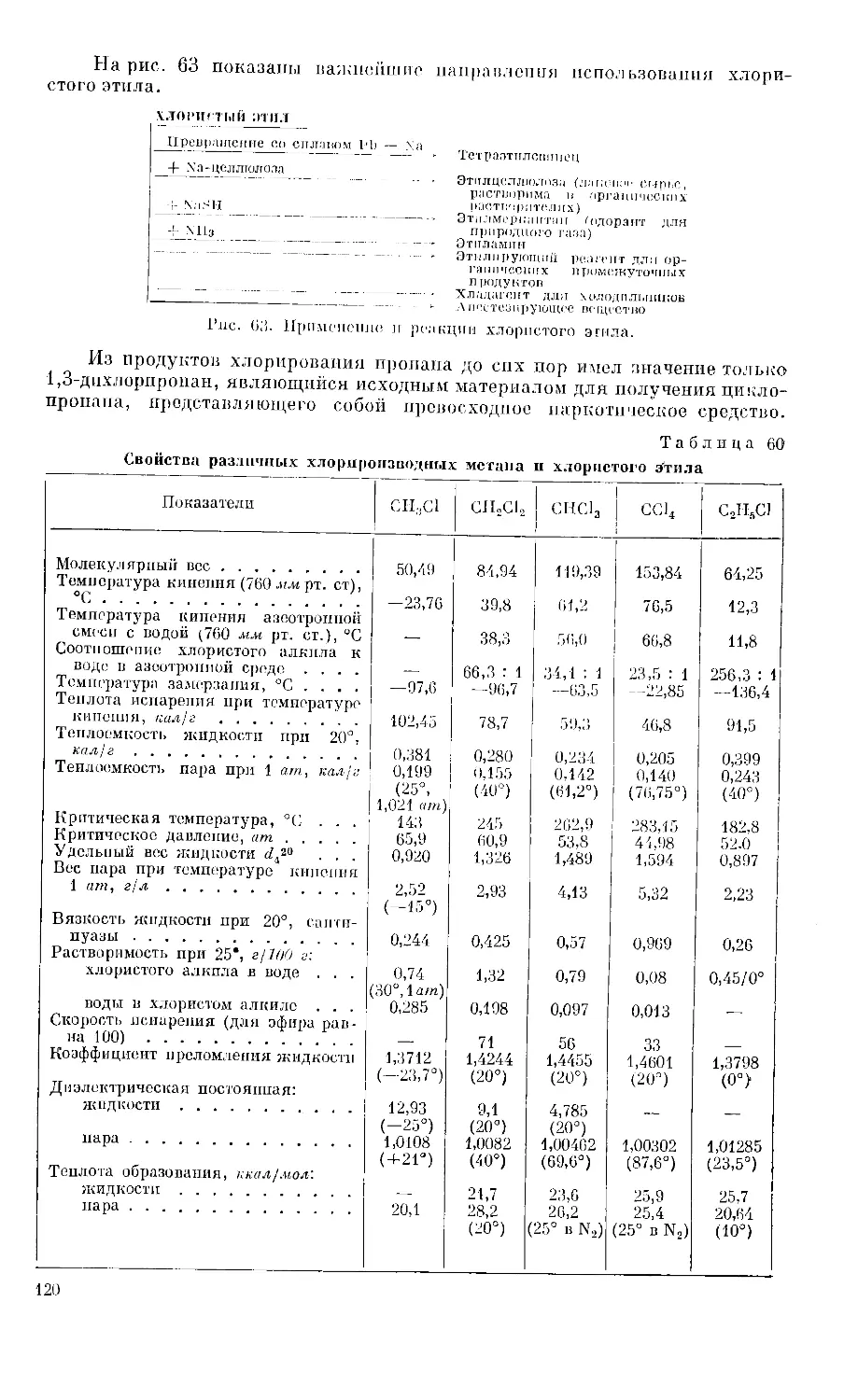





































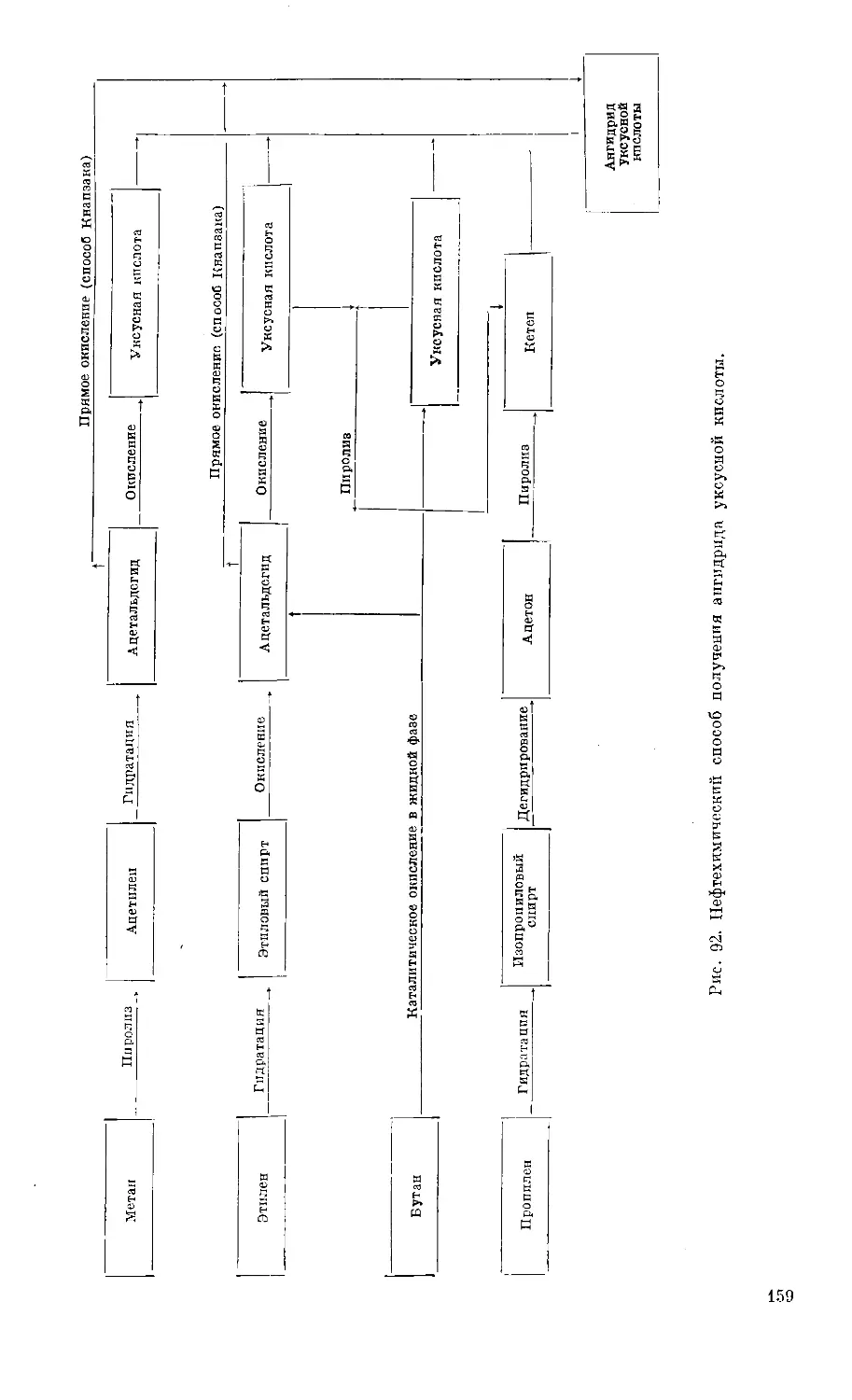


















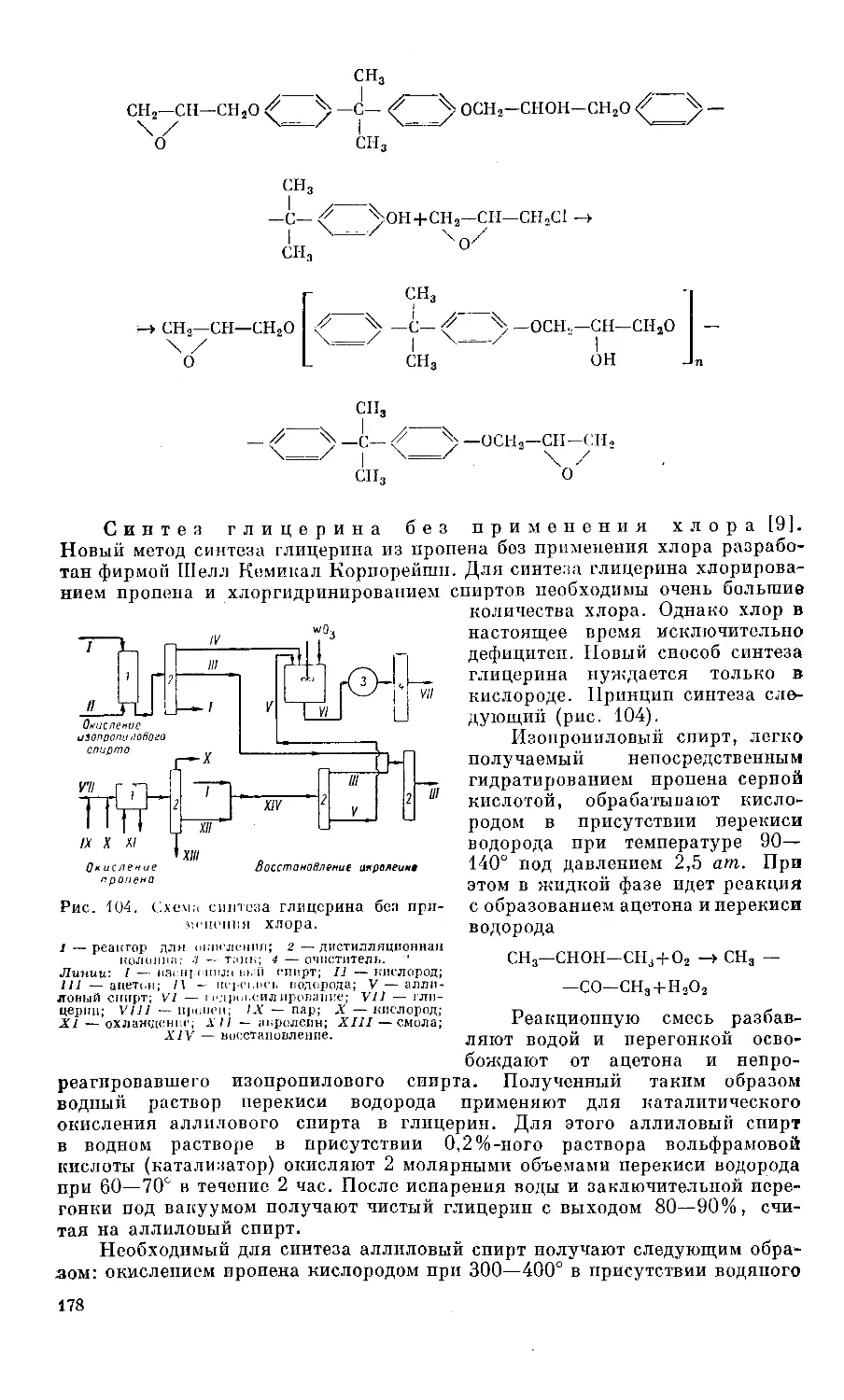



















































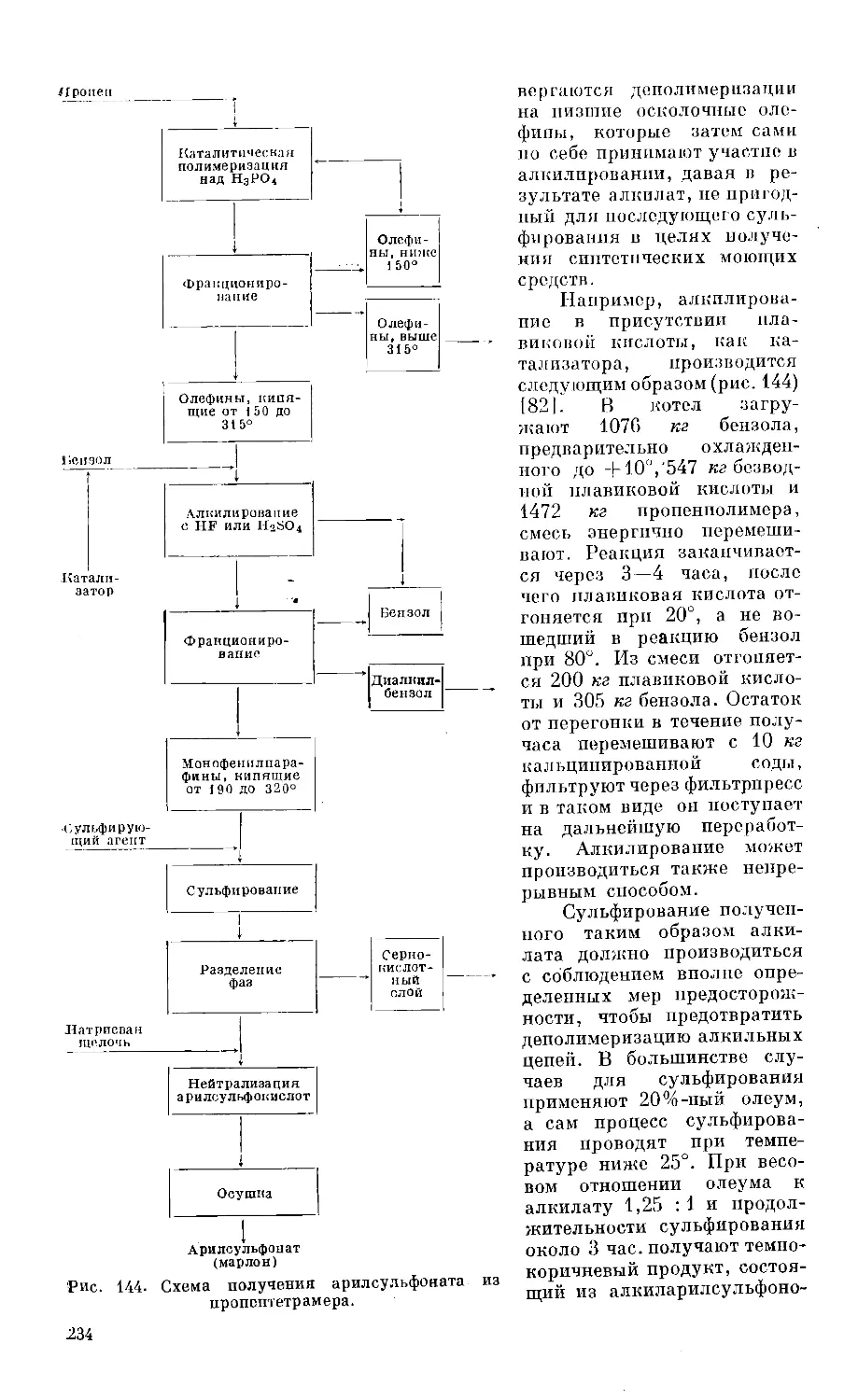

























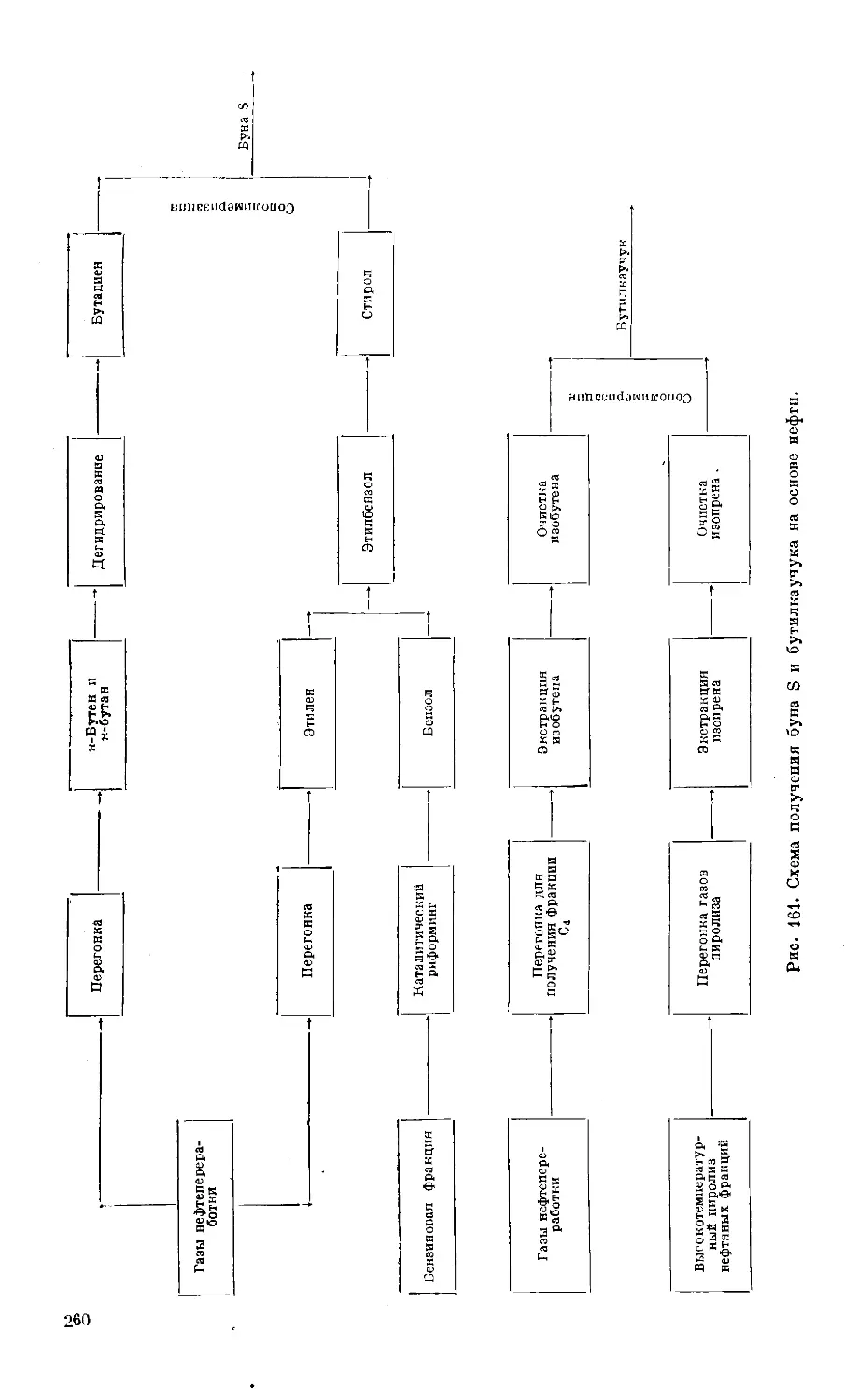
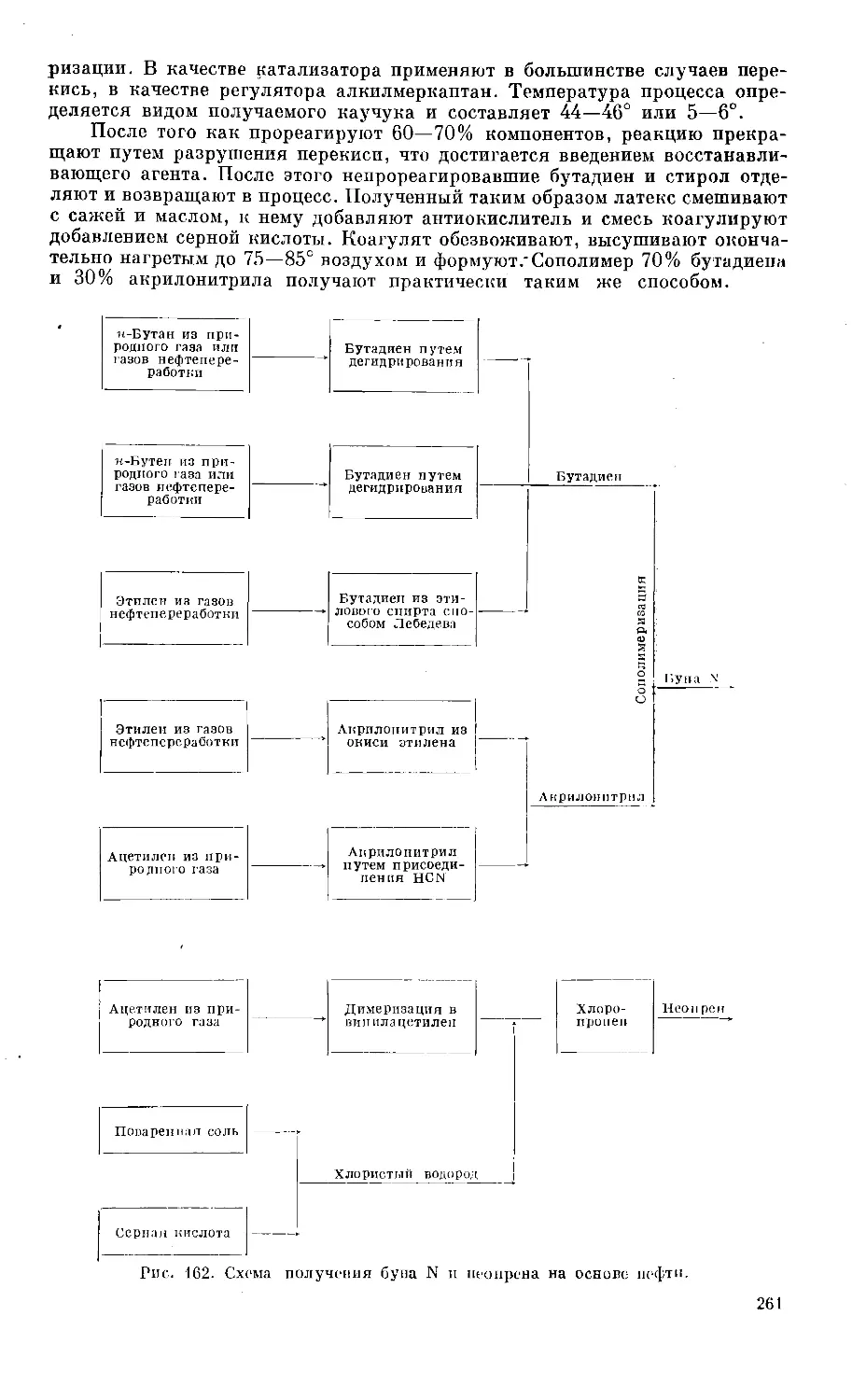







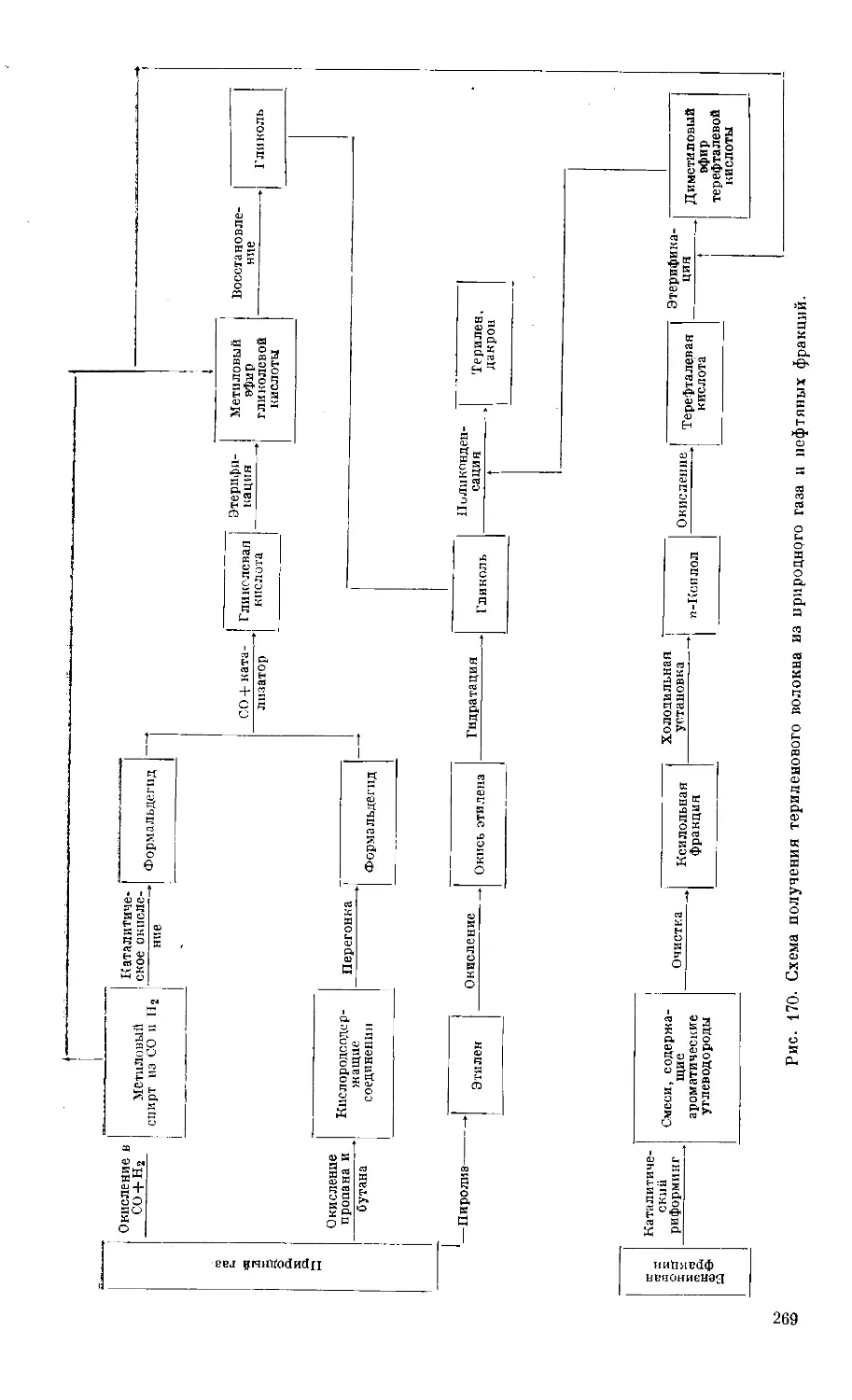






















Text
ВВЕДЕНИЕ В НЕФТЕХИМИЮ
Ф.АЗИНГЕР
EINFUHRUNG
IN DIE PETROLCHEMIE
von
Professor Dr. Ing habit. Friedrich Asinger
1959
AKADEMIE — VERLAG - BERLIN
Ф. АЗИНГЕР
ВВЕДЕНИЕ
В НЕФТЕХИМИЮ
Перевод с немецкого и редакция
д-ра техн, наук, проф. Б. В. ЛОСИКОВА
С предисловием действительного члена Академии наук Азерб. CCS
проф. М. А. ДАЛИНА
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
НЕФТЯНОЙ И ГОРНО-ТОПЛИВНОЙ ЛИТЕРАТУРЫ
Москва 1961
14-5-3
Л И II О Т А Ц II Я
В книге изложены достаточно полные и хорошо подоб-
ранные сведения по всем основным процессам химической'
переработки нефтяного сырья. В первой части книги опи-
сываются углеводороды как исходное сырье для нефтехи-
мической промышленности, во второй части приводятся
теоретические основы нефтехимии, принципиальные техно-
логические схемы промышленных установок технические
показатели процессов химической переработки нефтяного
сырья.
Книга рассчитана па широкие круги ппжеиерпо-техпи-
ческих работников, занимающихся химической переработкой
нефтяного сырья и газов, п может быть использована как
учебное пособие студентами вузов.
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Последние годы характеризуются бурным развитием нефтехимической
промышленности.
Достаточно сказать, что объем нефтехимикатов в общей продукции
химической промышленности США в 1937 г. составлял лишь 1%, а в 1957 г.
уже 26%. Предполагается, что к 1965 г. удельный вес нефтехимического
производства достигнет 40%.
В Советском Союзе нефтехимическая промышленность развивается
еще более быстрыми темпами. За семилетие 1959—1965 гг. при среднем
увеличении выпуска важнейших химических продуктов примерно в 3 раза,
производство синтетических и искусственных волокон возрастет в 4 раза,,
синтетических смол и пластмасс более чем в 7 раз.
Для выполнения этих грандиозных задач стране нужны многочислен-
ные кадры химиков и технологов, специализированных в области нефте-
химического производства. В связи с этим в настоящее время очень велика
потребность в учебных пособиях и руководствах по нефтехимии.
Изданные на русском языке монографии Ф. Азингера «Химия и техно-
логия парафиновых углеводородов» и «Химия и технология мопоолефипов» хо-
рошо известны советским читателям.
Нефтехимическим процессам, рассматриваемым во «Введении в нефтехи-
мию», посвящены многие опубликованные в Советском Союзе отечественные и
переводные монографии. К таковым относятся «Общая химическая техно-
логия» С. И. Вольфковича и др., «Синтетические каучуки» Н. И. Смирнова,
«Окись этилена» П. В. Зимакова, «Химическая переработка нефти» Р. Гольд-
штейна, «Технология синтетических пластических масс» Э. И. Барга, «Хими-
ческое использование нефтяных углеводородных газов» А. С. Некрасова
и Б. А. Кренделя, «Введение в химию и технологию полимеров» Ф. Биль-
мейера, «Основы синтеза алифатических спиртов из нефтяных углеводоро-
дов» Б. А. Кренделя, «Алкилирование бензола олефинами» М. А. Далина,
«Введение в нефтехимический синтез» Эстля, «Технология основного орга-
нического синтеза» И. И. Юкельсона, «Основы технологии нефтехимического
синтеза» под редакцией А. И. Дипцеса и Л. А. Потоловского и др.
Много фундаментальных статей по современному состоянию нефтехими-
ческой науки и производства напечатано в последние годы в журналах
«Химическая наука и промышленность», «Химическая промышленность»
и в других периодических изданиях, а также в выпущенных сборниках
трудов («Химическая переработка нефтяных углеводородов», «Труды
НИИСС» и др.).
Достоинством книги Ф. Азингера «Введение в нефтехимию» является
то, что автор в сравнительно небольшом объеме сумел осветить вопросы
химии и технологии основных нефтехимических процессов. Следует
также отметить, что эта книга снабжена богатым справочным матери-
алом в виде большого числа различных таблиц.
В первой части книги подробно описываются сырьевые ресурсы для
нефтехимической промышленности и способы получения индивидуальных
углеводородов.
В настоящее время постоянно возрастает роль природного и попутного
газов как сырья для нефтехимических производств. В связи с этим автор
останавливается на методах выделения бензиновых фракций из попутного
газа нефтедобычи.
Большое внимание уделяется моноолефинам — основному сырью
нефтехимии. Детально излагается основной способ их получения путем
пиролиза газообразных и жидких нефтяных углеводородов. Подробно рас-
сматривается проведение пиролиза с применением водяного пара. Осве-
щаются вопросы разделения газов, указаны способы выделения олефинов
из газовых смесей.
Рассматриваются процессы экстракции олефинов селективными раство-
рителями и экстрактивной ректификации смесей углеводородов с равным'
числом углеродных атомов.
При рассмотрении процессов получения ацетилена автор выделяет
два наиболее перспективных метода: окислительный пиролиз метана и вы-
сокотемпературный пиролиз легких и средних нефтяных дистиллятов.
Подробно излагаются каталитические риформинг-процессы, являю-
щиеся основным путем получения ароматических углеводородов.
Следует отметить, что некоторые вопросы освещены недостаточно полно
или требуют некоторого уточнения. В частности, при описании методов
разделения газовых смесей Ф. Азпнгер совершенно недостаточное внимание
уделяет наиболее современному методу глубокого охлаждения.
Кроме того, не отражены современные методы получения этилена и
пропилена высокой степени чистоты, что необходимо для осуществления
большинства синтезов на их основе. В частности, в процессах полимериза-
ции и сополимеризации этилена и пропилена к сырью предъявляются весьма
жесткие требования. По этому вопросу в советской и зарубежной печати
опубликован ряд работ.
Рассматривая методы дегидрирования бутана, автор указывает, что
основной трудностью процесса является необходимость быстрого подвода
большого количества тепла в зону реакции. Однако он не упоминает
о широко распространенном методе дегидрирования в «кипящем» слое ката-
лизатора, при котором эта проблема решается наиболее удачно.
Вторая часть книги посвящена химической переработке углеводо-
родов. Эта часть состоит из шести различных по объему глав, описыва-
ющих синтезы нефтехимических продуктов на основе предельных углеводо-
родов, олефинов, ароматических соединений, ацетилена и других со-
единений.
Автор подробно разбирает процессы хлорирования парафиновых угле-
водородов различными способами. Хорошо написаны также разделы нитро-
вания и сульфохлорирования.
Большое внимание уделено автором переработке моноолефинов и аце-
тилена, а также процессам алкилирования и окисления ароматических
углеводородов.
Автор описывает способы получения отдельных нефтехимических про-
дуктов, их свойства и направления дальнейшей переработки.
Некоторый интерес представляет приводимое в отдельных случаях
технико-экономическое сравнение различных методов получения одного
и того же продукта.
Вместе с тем следует отметить, что ряд нефтехимических процессов,
являющихся весьма важными и имеющих большое промышленное зна-
чение, описан автором недостаточно. Это в первую очередь относится
к производствам полиэтилена, алифатических спиртов и синтетического
6
каучука, на описание которых отводится по дв₽-три страницы. Некоторые
процессы, нашедшие в промышленности нефтехимического синтеза большое
развитие, вообще не упоминаются.
Нельзя не отметить, что автор почти не ссылается на известные работы
русских и советских ученых в области нефтехимии.
Но, несмотря на указанные недостатки, книга Ф. Азингера «Введение
в нефтехимию» найдет своего благодарного читателя и принесет ему извест-
ную пользу. Книга эта, несомненно, вызовет большой интерес у работников
заводов и научно-исследовательских институтов нефтехимической промыш-
ленности. Ее можно также рекомендовать в качестве учебного пособия для
студентов нефтехимических факультетов.
М. Далин
действительный член
Академии наук Азерб. ССР
ПРЕДИСЛОВИЕ
Химия на основе природного нефтяного газа и нефти раньше всего
получила развитие в США, где в настоящее время около 80% алифатических
продуктов производится нефтехимическим путем. Сырьем для этой промыш-
ленности служат в первую очередь алифатические углеводороды (парафины,
циклопарафины, моноолефины, диолефины и ацетилен). Значительную роль
играют также ароматические углеводороды, в прошлом типичный продукт
углехимпческой промышленности, теперь во все возрастающем количестве
они получаются из нефти и ее фракций.
Количество отраслей промышленности, которым нефтехимические
заводы поставляют свою продукцию, постоянно возрастает. В США произ-
водство ряда определенных химических продуктов базируется почти исклю-
чительно на основе нефтяных газов или нефти.
Другие высокоразвитые в промышленном отношении страны, как имею-
щие, так и не имеющие собственных источников нефти и газа, также, особенно
за последние 15 лет, вложили большие средства в развитие собственных
крупных центров нефтехимической промышленности. В Европе этой отрасли
промышленности уделяется большое внимание. Круг химиков, занятых
в нефтехимической промышленности, стал очень значительным.
Предлагаемая книга должна дать этим специалистам, а также тем,
кто хочет дальше совершенствоваться в этой области, краткие, но, насколько
это возможно, полные сведения как в отношении сырья, так и в части важ-
нейших процессов его переработки.
Этой цели служат многочисленные иллюстрации с упрощенными схе-
мами способов получения и сопоставлением возможностей получения и
использования важнейших продуктов нефтехимической промышленности.
В тексте имеются ссылки на изданные в 1956 и 1957 гг. издательством
Akademie Verlag книги автора «Химия и технология парафиновых угле-
водородов» и «Химия и технология моноолефинов», в которых часть веществ,
упоминаемых в настоящей книге, была рассмотрена значительно более
широко и подробно. Процессы, которые не могут рассматриваться как
нефтехимические, в особенности сортировка нефтей, получение карбюра-
торного горючего, а также производство высокооктановых бензинов мето-
дами алкилирования и полимеризации, рассматриваются в настоящей
книге лишь вкратце.
Автор будет очень признателен специалистам за советы, пожелания
и указание ошибок.
Дрезден, лето 1958 г. Ф. Азингер
ЧАСТЬ ПЕРВАЯ
УГЛЕВОДОРОДЫ КАК СЫРЬЕ
ДЛЯ НЕФТЕХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Углеводороды, служащие сырьем для нефтехимической промышлен-
ности, принадлежат к алифатическому, циклоалифатическому и ароматиче-
скому рядам. Алифатические углеводороды включают насыщенные или
парафиновые углеводороды, олефины, диолефины и ацетилен. Из цикло-
алифатических углеводородов в нефтях содержатся только производные
циклопентапов и циклогексанов; важнейшую роль играет циклогексан как
сырье для получения чистой адипиновой кислоты. Кроме того, он имеет
особое значение как промежуточный продукт в производстве ароматических
углеводородов методом каталитического риформинга.
К ароматическим углеводородам, получаемым и перерабатываемым
на нефтехимических заводах, относятся бензол, толуол и ксилол. Их полу-
чают каталитическим риформингом определенного нафтенового сырья.
В меньшем масштабе при помощи специальных процессов получают и другие
ароматические углеводороды — нафталин, его гомологи, а также ряд дру-
гих конденсированных ароматических углеводородов.
Глав а I
ПАРАФИНОВЫЕ УГЛЕВОДОРОДЫ
К парафиновым углеводородам, служащим исходным сырьем для нефте-
химической промышленности, относятся в первую очередь низкомолеку-
лярные, при нормальных условиях газообразные или жидкие низкоки-
пящие парафиновые углеводороды: метан, этан, пропан, бутаны и пен-
таны .
Парафиновые углеводороды с 6 —10 атомами С, кроме использования
их в качестве специальных растворителей, находят лишь ограниченное
применение в нефтехимической промышленности. Напротив, важную роль
играют высокомолекулярные углеводороды с 10—20 атомами С. Газообраз-
ные члены парафинового ряда, содержащиеся в природном нефтяном газе,
в газах, сопровождающих нефть при ее добыче, и в отходящих газах нефте-
перегонных установок вследствие большой разницы в температурах кипения
могут быть сравнительно простыми методами разделены па технически
чистые индивидуальные углеводороды. Для получения углеводородов, *
кипящих при более высоких температурах, чем бутан, сырьем может слу-
жить газовый бензин, ниже рассматриваемый подробно. Из пего методом
четкой ректификации можно получать пентан, гексан и гептан. Парафино-
вые углеводороды с 6—10 атомами С и парафиновые углеводороды с 10—
20 атомами С в настоящее время получают в чистом виде из нефтяных фрак-
ций посредством экстрактивной кристаллизации с мочевиной. Парафин,
являющийся смесью высокомолекулярных парафиновых углеводородов пре-
имущественно с прямой цепью, получают в больших количествах депара-
финизацией масляных фракций. Продукт этот является чрезвычайно цен-
ным сырьем.
Крекинг-газы, имеющие большое значение как источник получения
олефиновых углеводородов для нефтехимической промышленности, не
могут рассматриваться в качестве экономически выгодного сырья для полу-
чения парафиновых углеводородов. Для этого следовало бы подвергать
крекинг-газы каталитическому гидрированию с использованием водорода,
содержащегося в самом крекинг-газе. Так как олефины, однако, составляют
основную массу крекинг-газа, то такой способ работы является по существу
нецелесообразным.
Прежде чем изучать отдельные источники получения низко- и высоко-
молекулярных парафиновых углеводородов, остановимся на важнейших
физических константах этих углеводородов.
В табл. 1 даны константы газообразных при нормальных условиях
или низкокипящих углеводородов, а в табл. 2 — жидких или твердых
парафиновых углеводородов.
О
Таблица 1
Физические константы низкомолекулярных парафиновых углеводородов
Парафиновый углеводород Мол. вес Температура, °C Критиче- ское давле- ние, ат
критиче- ская кипения плавления
Метан 16,03 —82,5 — 161,5 —182,6 45,7
Этан 30,06 32,5 —88,6 —183,5 48,8
Пропан 44,06 97,0 —42,3 —188,0 45,0
и-Бутап 58,08 152,0 —0,5 —138,5 35,7
Изобутан 58,08 134,5 —12,0 —159,6 36,5
и-Пентан 72,09 197,2 36,1 —129,0 —
Изопентан 72,09 187,7 28,0 —159,0 —
Таблица 2
Важнейшие физические константы н-парафиновых углеводородов,
начиная с гексана
Число атомов углерода Температура, °C Показатель пре- ломления п^1 Плотность С?4°
кипения плавления
При 760 мм рт. ст.
6 68,7 —95,3 1,3749 0,6594
7 98,4 —90,6 1,3877 0,6838
8 125,7 —56,8 1,3979 0,7025
9 150,8 —53,5 1,4054 0,7176
10 174,1 —29,6 1,4119 0,7301
11 195,6 —25,6 1,4172 0,7402
12 216,3 —9,6 1,4217 0,7487
13 235,5 —5,3 1,4256 0,7563
14 253,6 + 6,2 1,4290 0,7629
15 270,0 9,9 1,4320 0,7685
16 286,5 18,2 1,4347 0,7737
17 302,5 21,8 1,4367 0,7776
18 317,3 28,0 1,4388 0,7815
19 331,5 31,4 — —
20 345,4 36,6 1,4346/40 0,7755/40
При 15 мм )Т. ст.
21 215 40,4 1,4352/48 0,778/40
22 224 44,1 1,43'58/45 0,779/44
234 47,0 1,4270/70 0,7786/47
24 243 -45 50,6 1,4283/70 0,7665/70
9 5 9 7) 53,3 -
26 262 55,8 1,4312/70 0,7586/90
27 270 59,1 — —
28 278 61,2 1,4327/70 0,7747/70
29 285—90 63,2 — —
30 304 65,7 — 0,7795/70
31 ,— 68,0 1,4278/90 0,7709/90
32 .— 69,5 1,4360/70 0,7840/70
33 — 72,0 — —
34 79,9 (?) 1,4296/90 0,7728/90
35 — 75,0 1,4301/90 0,7734/90
36 — 76,0 1,4347/80 0,7803/80
И
I. ПРИРОДНЫЙ НЕФТЯНОЙ ГАЗ
Л. ОБЩИЕ СНЕДЕНИЯ
Нефтяной газ выделяется из земных недр непосредственно пли вместе
с нефтью. Месторождения природного нефтяного газа распространены
иа земном шаре повсеместно. Наиболее крупные из них находятся в США
(добыча газа в 1955 г. составила около 300 млрд, nt3) и в Советском Союзе.
В Европе наибольшими месторождениями обладают Италия [1J
(4,5 млрд, м3) и Румыния. В Германии добыча газа относительно ограничена
(370 млн. nt3) и запасы его также малы. Значительными источниками природ-
ного газа обладает Австрия, где годовая добыча газа составляет в настоящее
время 900 млп. м3. Газовые месторождения имеются в прилегающих к Пи-
рипеям областях Франции, где годовая добыча газа, содержащего до
15% H2S, достигает 750 —1000 млн. nt3.
По составу природные газы подразделяются на две группы: сухие
и жирные. Сухой газ содержит, кроме метана, лишь небольшие количества
этана. Жирный газ содержит еще некоторое количество высокомолекуляр-
ных углеводородов, из которых при определенных условиях может быть
выделен так называемый’ ожиженный" газ или углеводороды, кипящие
в интервале температур кипения бензина. Разницу в составе этих газов
на основании их анализа можно видеть из табл. 3.
Таблица ,3
Состав сухого и жирного газов (% объемн.)
Газ Метая Этан Пропан Бутан Пентан
Сухой .... 84,7 9,6 3,0 1,1 —
Жирный . . . 36,8 32,6 21,1 5,8 3,7
Природный газ часто содержит большие количества углекислоты,
сероводорода и в редких случаях также гелия. Чаще всего газ находится
под повышенным давлением. Смесь газообразных углеводородов, выделяю-
щаяся из сырой нефти при ее нагреве, в противоположность природному
газу богата высокомолекулярными углеводородами, такими как пропан
и бутан. Количество ее, включая бутан, может составлять 1—2% вес. от
нефти. Средний состав ее (в % объемн.).
Метан.............5
Этап.................10
Процап...............30
Бутан................35
Пентан и выше .... 20
Жидкая часть природного газа, особенно жирного (ожиженный газ или
газовый бензин), представляет большой интерес для нефтехимической про-
мышленности. Под ожиженным газом понимается смесь газообразных при
нормальных условиях углеводородов, в основном состоящая из пропана,
бутанов, пропена и бутенов. Он может содержать еще и рядом стоящие угле-
водороды, способные сжижаться при нормальной температуре под давлением,
не превышающим 20 ат. Как показывает табл. 1, метан при нормальной '*
температуре не может быть превращен в жидкость, а этан может быть ожи-
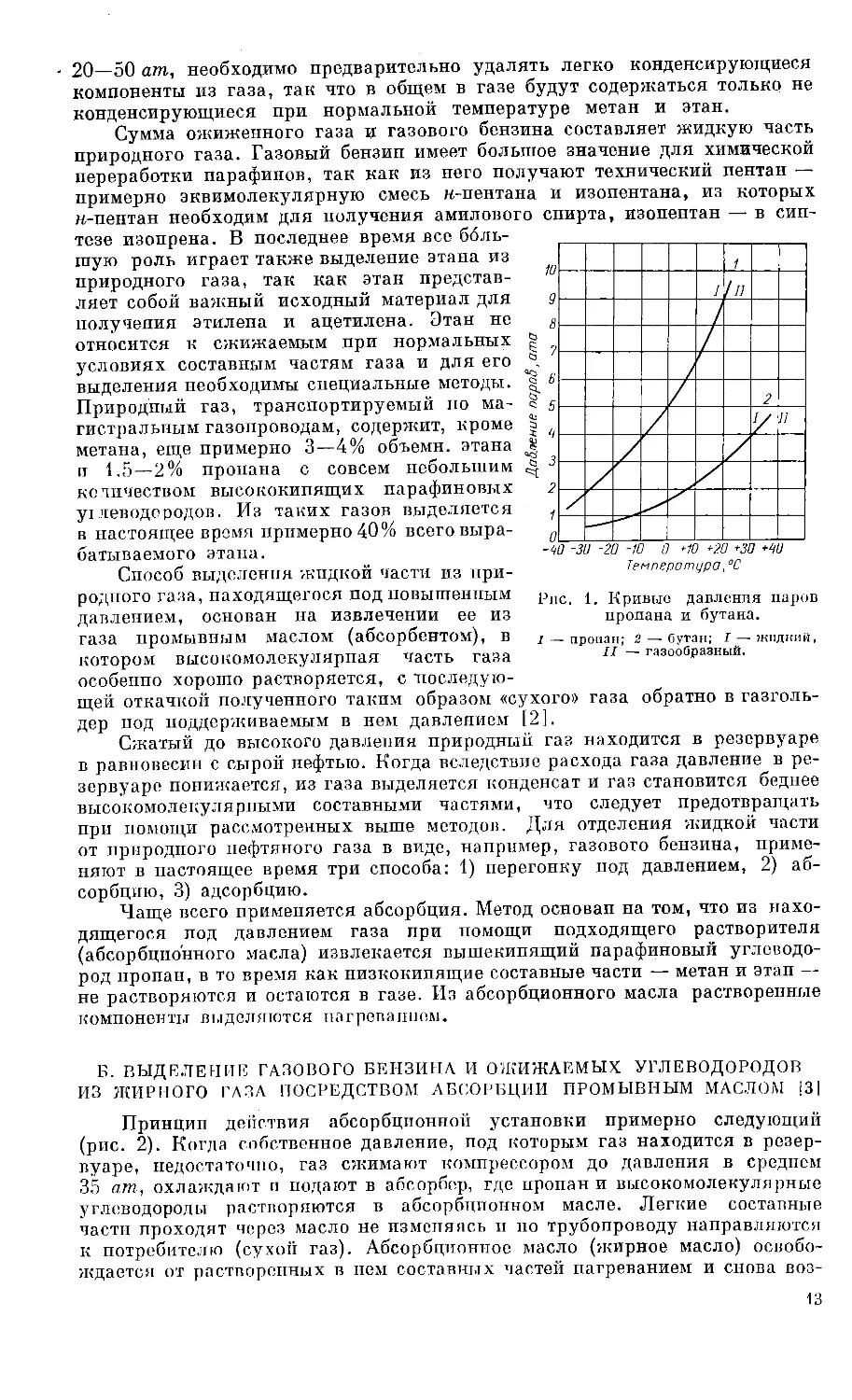
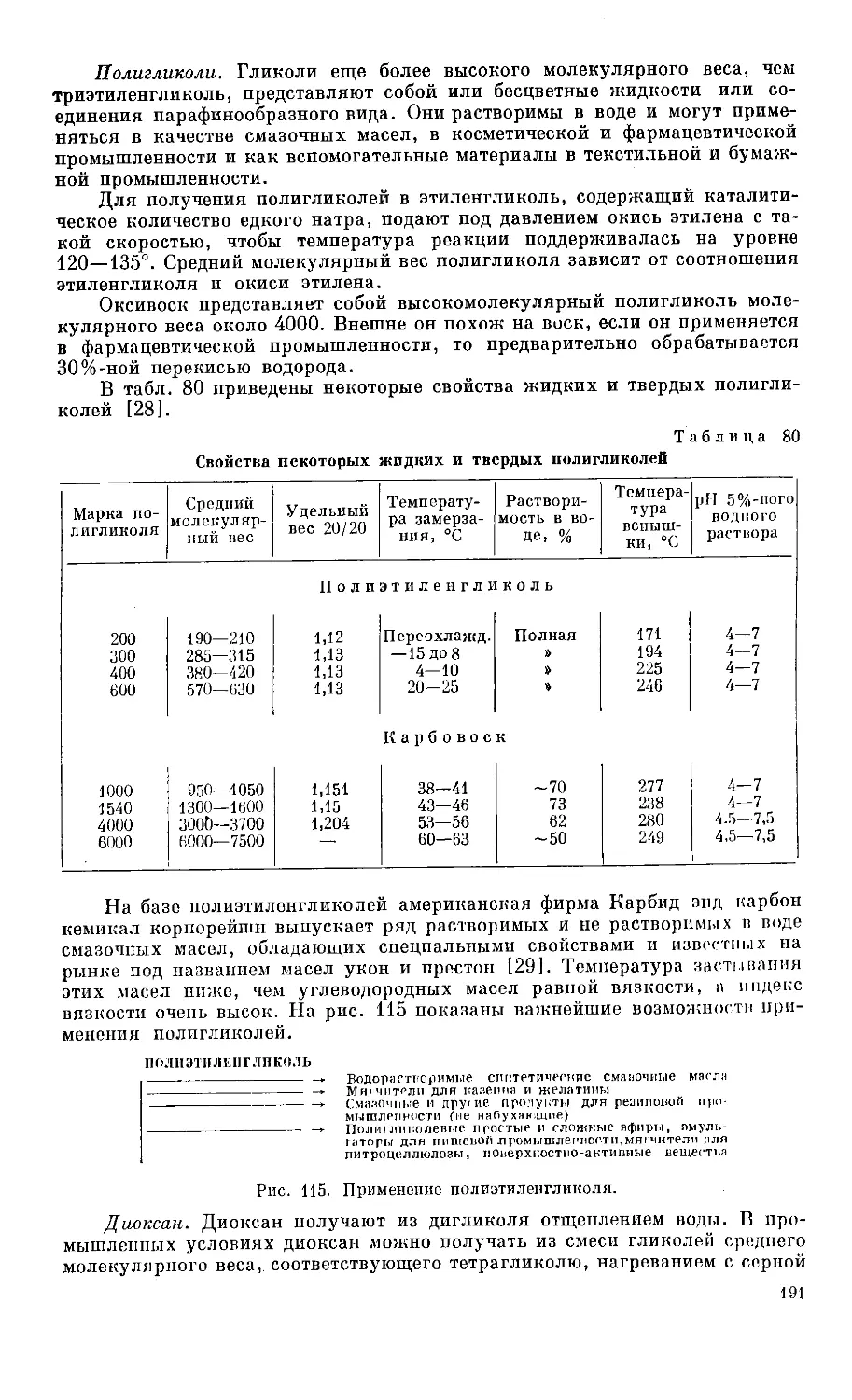
жен лишь при применении более высокого давления. На рис. 1 даны кривые
упругости паров пропана и бутана. Газовый бензин, составляющий около
17% от всего вырабатываемого в США бензина, выделяется из жирного
природного газа. т
Чтобы избежать конденсации части жирного газа при транспортировке
природного газа по трубопроводам, осуществляемой обычно под давлением
12
20—50 ат, необходимо предварительно удалять легко конденсирующиеся
компоненты из газа, так что в общем в газе будут содержаться только не
конденсирующиеся при нормальной температуре метан и этан.
Сумма ожиженного газа и газового бензина составляет жидкую часть
природного газа. Газовый бензин имеет большое значение для химической
переработки парафинов, так как из него получают технический пентан —
примерно эквимолекулярную смесь н-пентана
н-пентан необходим для получения амилово
тезе изопрена. В последнее время все боль-
шую роль играет также выделение этана из
природного газа, так как этан представ-
ляет собой важный исходный материал для
получения этилена и ацетилена. Этан не
относится к сжижаемым при нормальных
условиях составным частям газа и для его
выделения необходимы специальные методы.
Природный газ, транспортируемый по ма-
гистральным газопроводам, содержит, кроме
метана, еще примерно 3—4% объемн. этана
и 1,5 — 2% пропана с совсем небольшим
количеством высококипящих парафиновых
углеводородов. Из таких газов выделяется
в настоящее время примерно 40% всего выра-
батываемого этана.
Способ выделения жидкой части из при-
и изопентана, из которых
родного газа, находящегося под повышенным
давлением, основан на извлечении ее из
газа промывным маслом (абсорбентом), в
котором высокомолекулярная часть газа
особенно хорошо растворяется, с последую-
Рпс. 1. Кривые давления паров
пропана и бутана.
2 — пропан; 2 — бутан; I — жидкий,
II — газообразный.
щей откачкой полученного таким образом «сухого» газа обратно в газголь-
дер под поддерживаемым в нем давлением [2].
Сжатый до высокого давления природный газ находится в резервуаре
в равновесии с сырой нефтью. Когда вследствие расхода газа давление в ре-
зервуаре понижается, из газа выделяется конденсат и газ становится беднее
высокомолекулярными составными частями, что следует предотвращать
при помощи рассмотренных выше методов. Для отделения жидкой части
от природного нефтяного газа в виде, например, газового бензина, приме-
няют в настоящее время три способа: 1) перегонку под давлением, 2) аб-
сорбцию, 3) адсорбцию.
Чаще всего применяется абсорбция. Метод основан на том, что из нахо-
дящегося под давлением газа при помощи подходящего растворителя
(абсорбционного масла) извлекается вышекипящий парафиновый углеводо-
род пропан, в то время как низкокипящие составные части — метан и этан —
не растворяются и остаются в газе. Из абсорбционного масла растворенные
компоненты выделяются нагреванием.
Б. ВЫДЕЛЕНИЕ ГАЗОВОГО БЕНЗИНА И ОЖИЖАЕМЫХ УГЛЕВОДОРОДОВ
ИЗ ЖИРНОГО ГАЗА ПОСРЕДСТВОМ АБСОРБЦИИ ПРОМЫВНЫМ МАСЛОМ (3|
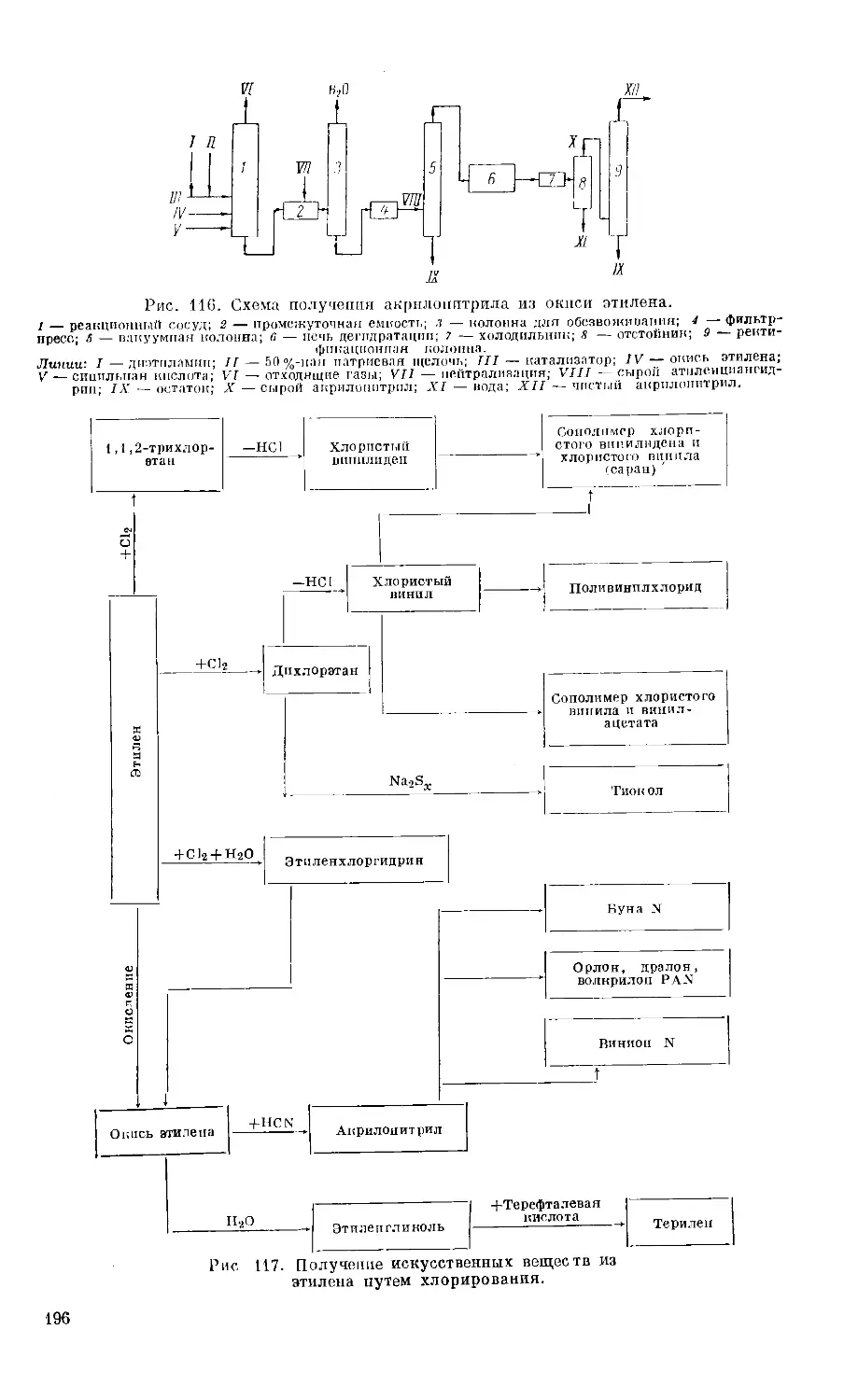
Принцип действия абсорбционной установки примерно следующий
(рис. 2). Когда собственное давление, под которым газ находится в резер-
вуаре, недостаточно, газ сжимают компрессором до давления в среднем
35 атп, охлаждают и подают в абсорбер, где пропан и высокомолекулярные
углеводороды растворяются в абсорбционном масле. Легкие составные
части проходят через масло не изменяясь и по трубопроводу направляются
к потребителю (сухой газ). Абсорбционное масло (жирное масло) освобо-
ждается от растворенных в нем составных частей нагреванием и снова воз-
13
вращается в установку (бедное или тощее масло). Для получения газового
бензина выделенные из масла парафиновые углеводороды конденсируются,
затем их перегоняют под давлением для освобождения от еще содержаще-
гося в них довольно большого количества пропана и бутана (стабилизация).
Стабилизационная колонна имеет большое число тарелок (40 или больше).
Выделяющаяся при стабилизации из верхней части колонны смесь
этана, пропана и бутанов разделяется перегонкой под давлением на отдель-
Рис. 2. Схема получения газового бензина из
сырого природного газа абсорбционным методом.
1 — абсорбер и холодильник; 2 — разделитель и на-
греватель; <3 — конденсатор; 4 — резервуар; 5 —
стабилизатор.
Линии: I — сырой природный газ; II — сухой газ; III —
жирное масло; IV — тощее масло; V — сырой бензин;
VI — газы стабилизации на переработку; VII — гото-
вый газовый бензин.
ные составные части: пропан,
н-бутан и изобутан. Процесс
ведут при таком соотношении
давлений, чтобы при данной тем-
пературе в верхней части ко-
лонны часть продуктов всегда
конденсировалась для ороше-
ния. Схема абсорбционной уста-
новки показана на рис. 3. Ко-
лонна 1, из которой еще выде-
ляются небольшие количества
метана и этана, работает при-
мерно при 17,5 ат и имеет около
30 тарелок. В колонне 2 угле-
водороды С3 и С4 отделяются
от пентанов и более высоко-
кипящих углеводородов. Ко-
лонна работает примерно при
9 ат. Температура верха ее 78°, низа 120 — 140°. В колонне 3 разде-
ляются углеводороды С3 и С4. Пропан уходит через верх колонны, а угле-
водороды С4 из низа колонны 3 переходят в колонну 4, где разделяются на
изо- и н-бутаны. Колонна 3 работает примерно при 17,5 ат и имеет 30 та-
релок. Температура верха колонны около 60°, низа 115°. Колонна 4 имеет
50 тарелок и работает при 8,7 ат; температура верха 70°, низа 85°.
Рис. 3. Получение пропана и бутана из газов стабили-
зации газового бензина путем перегонки.
В табл. 4 показан состав сырого газового бензина непосредственно после
выделения из абсорбционного масла, стабилизированного, а также пол-
ностью дебутанизированного в дебутанизаторе — колонне, работающей под
давлением, обеспечивающем отделение бутана.
Вследствие все возрастающего спроса на ожиженный газ полное выде-
ление пропана и бутанов из природного газа становится главной целью
абсорбционного процесса. Состав газового бензина при высоком содержании
пропана и бутанов следующий (в % мол.).
Метан....... —
Этан.....Следы
Пропан .... 38,7
Изобутан . . . 10,0
н-Бутан.............18,3
Пентаны.............20,0
Тяжелые углеводоро-
ды .................13,0
14
Содержание «-бутана в смеси бутанов из природного газа составляет
около 70%. Пентановая фракция природного газа представляет собой смесь
примерно равных частей н-пентана и изопентана.
Таблица 4
Состав сырого, стабилизированного и дебутанизированного
газовых бензинов
Показатели ; Сырой Стабилизи- рованный Дебутанизи- рованный
Удельный вес при 16° ... . 0,6309 0,6761 0,6869
Давление паров при 37,7°, ат Состав, % объемн.: 4,2 1,22 0,77
этан 1,5 — —
пропан 14,7 — —
изобутан 10,2 1,5 —
н-бутан 30,0 15,3 3,2
изопентан 4,8 7,2 8,5
и-пентан 15,0 21,0 24,5
тяжелые углеводороды 23,5 55,0 63,8
Выделение высокомолекулярных составных частей из сухих газов
адсорбционными методами (гиперсорбцией) рассматривается ниже.
Выделение этана из сухих газов, в которых, как указывалось выше,
содержится 3—4% этана, абсорбционным методом осуществляется следую-
щим путем.
Природный газ высушивается, охлаждается до —30° и в абсорбере
поглощается легким маслом. Неабсорбированный газ проходит через второй
абсорбер, в котором тяжелым маслом задерживается легкое масло, увле-
ченное из главного абсорбера остаточным газом. Легкое масло из главного
абсорбера направляется в деметанизатор, а затем в работающую под да-
влением колонну, в которой этан и малые количества пропана и бутана отго-
няются и отводятся через верх колонны. Этот дистиллят в следующей ко-
лонне разделяется на этан, небольшое количество сжижаемых газов и газо-
вый бензин. Принцип работы здесь в основном такой же, как показано на
схеме рис. 3.
Необходимо отметить, что природный нефтяной газ как сырье для хими-
ческой промышленности используется еще в очень небольшой степени.
В настоящее время он потребляется в первую очередь как тепло- и энерго-
носитель. Его теплота сгорания, также как и других технических и чистых
газов, применяемых в энергетике, дана ниже.
Газ Теплота сгорания, ккал/м* Газ Теплота сгорания, ккал/мг
Колошниковый .... Генераторный Даусоновский Водяной Коксовый Осветительный .... Природный или нефтя- ной 750 850 1200 2600 4000—5000 5200 9500 Крекинг-газ Водород Метан Этан Пропан Бутан 12500 3200 9525 1241X1 19900 25000
15
IL ВЫСОКОМОЛЕКУЛЯРНЫЕ, ЖИДКИЕ ПРИ НОРМАЛЬНОЙ ТЕМПЕРАТУРЕ
ПАРАФИНОВЫЕ УГЛЕВОДОРОДЫ ИЗ НЕФТИ (С7—С20)
Прежде чем рассматривать парафиновые углеводороды этого вида,
следует коротко остановиться на свойствах, составе и способах переработки
нефти.
А. СОСТАВ НЕФТИ
Важнейшим сырьем для нефтехимической промышленности наряду
с природным газом является нефть. Мировая добыча нефти (без газового
бензина и сжиженного газа) составила в 1956 г. около 840 млн. т. Мировые
запасы нефти, разведанные к настоящему времени, составляют около
32 млрд, т, из которых примерно 62% (около 20 млрд, т) находится на
Ближнем Востоке [4].
Нефть представляет собой сложную смесь различных углеводородов,
относящихся к парафиновому, нафтеновому и ароматическому гомологиче-
ским рядам. В зависимости от того, парафиновые или нафтеновые углево-
дороды преобладают в нефти, принято относить последнюю к нефтям пара-
финового или нафтенового основания.
Нефти парафинового основания содержат лишь относительно немного
ароматических углеводородов, нефти нафтенового основания богаче ими.
К нефтям парафинового основания в первую очередь относится пенсиль-
ванская нефть, к нафтеновым и ароматическим — многие румынские и со-
ветские нефти. Кроме этих видов нефтей имеются различные промежуточные,
здесь нс рассматриваемые.
В табл. 5 приведен состав нефтей нафтенового и парафинового основания
15]. Из таблицы видно, что в нефтях парафинового основания содержание
парафиновых углеводородов во фракциях понижается по мере повышения
их температуры кипения, а в нефтях нафтенового основания увеличивается
содержание ароматических углеводородов.
Таблица 5
Ссстав нефтей парафинового и нафтенового оснований
Фракция нефти Нефть парафинового основания Нефть нафтенового основания
пара- фины нафтены ароматиче- ские пара- фины нафтены ароматиче- ские
Бензин 65 30 5 35 55 10
Керосин 60 30 10 25 60 25
Газойль 35 55 15 — 65 35
Тяжелый дистиллят 20 65 15 — 55 45
Б. ПЕРЕРАБОТКА НЕФТИ [6]
(и е р е г о и к а, к р е к и пг, легкий крек и и г)
Основным процессом переработки нефти является ее разгонка на отдель-
ные фракции. Важнейшими фракциями являются: бензиновая, выкипаю-
щая в пределах 20—200“, керосиновая — в пределах 175—275°, газойль
разного рода, кипящий в интервале температур от 200 до 400° и смазочные
масла, выкипающие в пределах 300—500°. Отдельные фракции могут подвер-
гаться дальнейшему разделению в целях получения специальных продук-
тов — петролейного эфира, бензина-растворителя, медицинского бензина
и т. д.
Все эти фракции, выделяемые из нефти посредством простого нагрева-
ния, называются продуктами прямой перегонки. Сырая нефть содержит
16
также газы, выделяющиеся при нагревании нефти и состоящие исключительно
из парафиновых углеводородов (отходящие газы нефтеперегонки). Прин-
цип переработки нефти перегонкой дан на схеме рис. 4.
Перегонка нефти вначале проводится при нормальном давлении; по-
следней фракцией этой стадии процесса является газойль. Получающийся
остаток далее разгоняется под вакуумом. Первой фракцией разгонки под
вакуумом является газойль, последние фракции представляют собой сма-
зочные масла. Остаток от перегонки нефти может быть различным в зависи-
мости от природы нефти. Нефти нафтенового основания дают асфальтсодер-
жащий остаток; остаток нефти парафинового основания представляет собой
смесь высоковязких углеводородов, используемый для получения смазочных
масел (брайтстоков).
Диаграмма рис. 5 дает представление о фракциях, выделяемых из нефти
при атмосферной и соответственно при вакуумной перегонке.
Значительная часть газов,
выделяющихся при перегонке
нефти, растворяется в бензине.
Основное количество их должно
быть удалено из бензина. Эта
операция называется стабилиза-
цией бензина, а выделяющиеся
при стабилизации газы называют-
ся газами стабилизации. Они со-
стоят главным образом из этана,
пропана и бутана. Схема стабили-
зационной установки показана на
рис. 6. Такие установки очень
широко применяются в нефтяной
промышленности.
Нестабилизованный бензин,
предварительно подогретый паром
в специальном теплообменнике,
поступает в колонну, работаю-
щую под давлением 6—10 ат.
Рис. 4. Упрощенная схема переработки сы-
рой нефти путем перегонки.
I — трубчатая печь; 2 — фракционирующая колон-
на; 3—конденсатор — холодильник. Линии: I —
сырая нефть; II — газ; III — остаток; IV — смазоч-
ное масло; V — газойль; VI —керосин; VII — бен-
зин.
В нижней части стабилизационной колонны при помощи пара под-
держивается температура порядка 130—140°, чтобы бензин мог освобо-
диться от всех газообразных углеводородов, в присутствии которых зна-
чительно повышается упругость его паров при нормальных условиях.
Чаще всего колонна имеет 35—40 тарелок. Большая часть удаляемых через
верх колонны газов конденсируется и собирается в находящемся под давле-
нием сосуде, из верхней части которого отводятся неконденсирующиеся
газы, главным образом метан и этан. Жидкий продукт удаляется со дна
сосуда. Вторичной перегонкой под давлением этот жидкий продукт может
быть разделен на составляющие компоненты.
В прошлом нефть служила в основном для получения керосина, смазоч-
ных масел и котельного или печного (отопительного) топлива. С распростра-
нением двигателей внутреннего сгорания и с постоянно возрастающим спро-
сом на бензин перед нефтяной промышленностью была поставлена задача
получать из нефти больше бензина, чем его в ней первоначально содержится.
Эта задача была решена при помощи крекинг-процесса. Процессы расщепле-
ния под влиянием тепла (термический крекинг) или тепла и катализатора
(каталитический крекинг) позволяют получить из нефти не только больше
бензина, чем было первоначально в нефти, но и бензин лучшего качества.
Крекингу подвергают чаще всего высококипящие фракции, представляющие
собой остаток после отгона от нефти при нормальном давлении бензина
прямой перегонки, керосина и в отдельных случаях дизельного топлива.
Сырьем для каталитического крекинга служат только дистиллятные
продукты, так как содержащиеся в золе остатков ют перегонки тяжелые
2 Заказ 5 9li.
17
металлы, отлагаясь на катализаторе, делают его со временем не-
активным.
Термический крекинг проводится двумя способами. Первый способ за-
ключается в том, что сырье крекируют до образования жидкого крекинг-
остатка (крекинг-мазута), во втором способе конечным продуктом крекинга
является кокс. В первом случае высококипящие составные части продуктов
крекинга, кипящие выше температуры кипения бензина, удаляются и не
возвращаются на крекинг; во втором случае все фракции, кипящие выше
температуры кипения бензина, возвращаются в крекинг-установку и там
после нагревания в специальном сосуде остаются до образования кокса.
Рис. 5. Фракции нефти, получаемые пере-
гонкой при нормальном давлении и под
вакуумом.
1 — перегонка при нормальном давлении; 2 —
перегонка под вакуумом.
Линии: I — сырая нефть; II — отходящие газы
(парафиновые углеводороды); III — легкий бен-
зин; IV — средний бензин; V — тяжелый бензин
(бензин-растворитель, лаковый бензин); VI —
керосин; VII — дизельное топливо; VIII — лег-
кий газойль; IX — остаток от атмосферной
перегонки на перегонку под вакуумом; X —
отходящие пары вакуумной перегонки; XI —
тяжелый газойль; XII — веретенное масло;
XIII — дистилляты машинного масла (а —
легкий, б — средний, в — тяжелый); XIV —
цилиндровое масло; XV — остаток вакуумной
перегонки: асфальт из сильно ароматизирован-
ных нефтей, цилиндр — сток из парафинистых
нефтей.
11
Рис. 6. Схема установки для стабили-
зации бензина.
1 — колонна для фракцйонировки под давлением;
2 — сепаратор, работающий под давлением для
разделения газа от жидкости.
Линии: I — нестабилизированный бензин; И —
газообразные углеводороды с примесью жидких;
III — орошение; IV — отделение метана-этана;
V — ожиженный газ (фракция Сз и С<); V’l — во-
дяной пар; VII — стабилизированный бензин.
Образующиеся в процессе крекинга газы содержат олефины, которые
полимеризацией или алкилированием могут быть превращены в полимер-
бензин или алкилат, которые могут быть присоединены к крекинг-бензину.
Этот процесс, не относящийся к нефтехимическим, здесь не рассматривается.
В других случаях, например при значительном спросе на мазут, целесооб-
разно в качестве сырья для крекинга использовать прямогонные фракции,
выкипающие в пределах 200—400°, а остаток от прямой перегонки нефти
использовать как отопительный мазут. Такое топливо, однако обладает
чрезмерно высокой вязкостью. Его можно подвергать легкому крекингу,
при котором образуется лишь немного бензина, но заметно понижается вяз-
кость остатка. Это явление, называемое «разрушением вязкости», весьма
часто используется в технологии. Бензиновая фракция нефти, так называе-
мый прямогонный бензин, разделяется далее на две фракции: легкий и тя-
желый бензины. Тяжелая бензиновая фракция для улучшения моторных
свойств подвергается термическому или каталитическому риформингу,
заключающемуся в кратковременном нагреве при высоком давлении в при-
сутствии катализатора или без него, улучшающему антидетонационные
свойства бензина. Принципиальная схема современного метода переработки
нефти представлена на рис. 7 [7].
18
Бензив, тяжелый бензин, керосин, дизельное и легкое котельное топлива
Сырая нефть
Продукты прямой
перегонки
Перегонка
Газойль
Легкий
газойль
Каталитический
крекинг
Фракции смазочных
масел
Вакуумная |
перегонка |
Газ
Полимеризация
или
алкилирование
Бензин
Отопительный газ
Очистка
смазочных
масел
Смазочное масло
и парафин
I
Остаток от
перегонки
Термический
крекинг или
коксование
Бензин
Мазут или кокс
Циркулирующее масло
Рис. 7. Основные способы переработки нефти.
В. ВЫСОКОМОЛЕКУЛЯРНЫЕ С7-С20 ПАРАФИНОВЫЕ УГЛЕВОДОРОДЫ
11.3 НЕФТИ
Когда говорят, что парафиновые углеводороды и углеводороды, отно-
сящиеся к другим гомологическим рядам, существуют во многих изомерных
формах, значительное число которых содержится в нефтях различных ме-
сторождении, то кажется очевидным, что выделение отдельных высокомолеку-
лярных углеводородов путем перегонки невозможно. Представление о числе
структурных изомеров дают следующие цифры [8].
Число атомов С Число изомеров Число атомов С Число изомеров
4 2 16 10.359
5 3 18 60323
6 4 20 366319
7 9 25 36797585
8 18 30 4111846763
10 12 75 355 40 62491178805831
Кроме того, большие количества парафиновых углеводородов содер-
жатся лишь в нефтях определенного происхождения. Но даже и в этом слу-
чае, кроме парафиновых углеводородов, во фракциях этих нефтей имеется
большое число равнокипящих циклических углеводородов — нафтеновых
и ароматических.
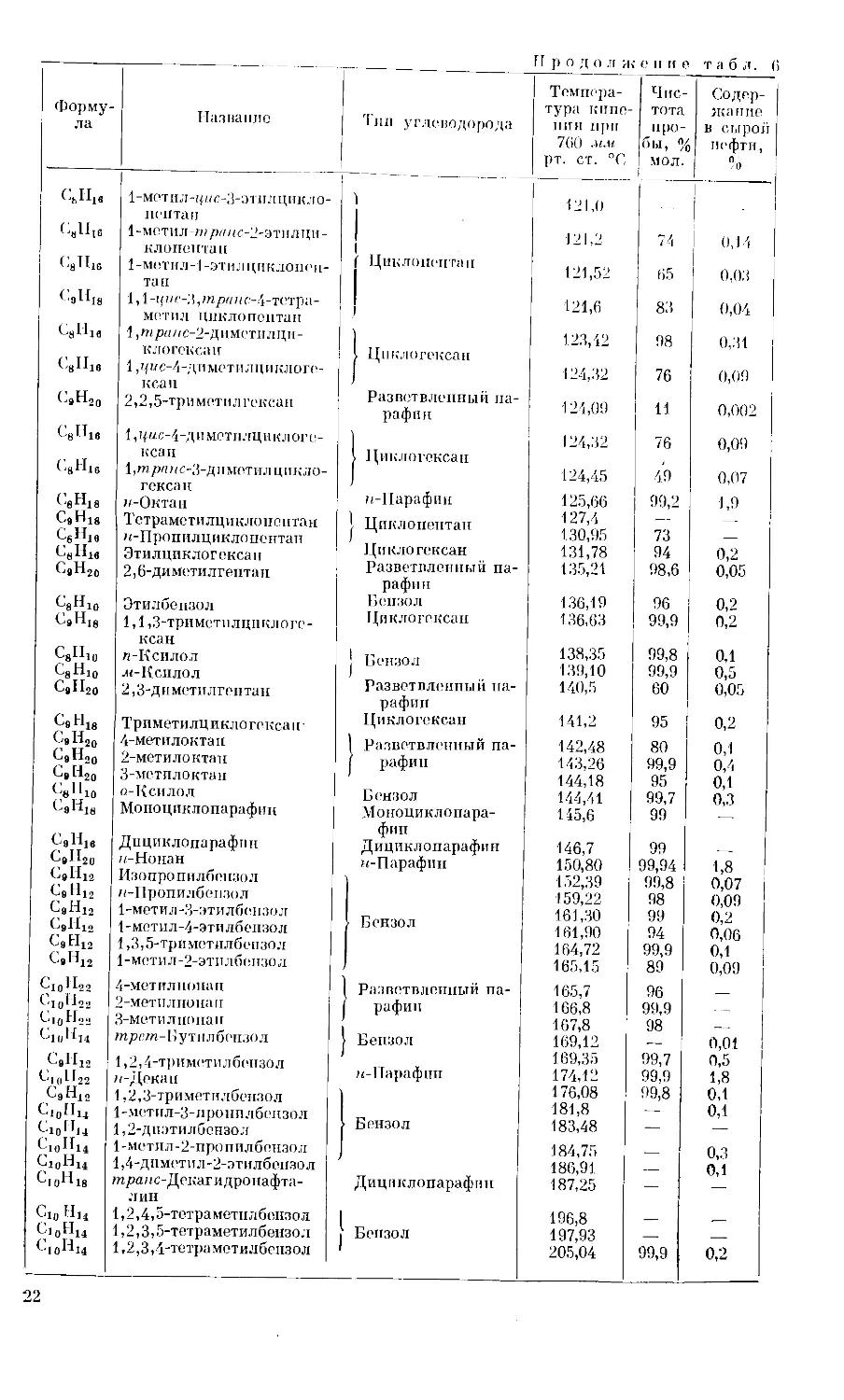
В табл. 6 дан перечень углеводородов, выделенных из нефти Попка
(Оклахома, США) [9]. Здесь представлены углеводороды только с 7 —10 ато-
мами С в молекуле. Из таблицы следует, что было бы бесполезно пытаться
выделять чистые парафиновые высокомолекулярные углеводороды из нефти
методом перегонки.
Г. ВЫДЕЛЕНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
ИЗ НЕФТЯНЫХ ФРАКЦИЙ ПОСРЕДСТВОМ ЭКСТРАКТИВНОЙ
КРИСТАЛЛИЗАЦИИ С МОЧЕВИНОЙ
В настоящее время парафиновые углеводороды с прямой цепью выде-
ляют из нефти и ее фракций при помощи мочевины. Как наблюдал впервые
в Германии Ф. Бенген [10], мочевина (карбамид) дает с н-парафинами
кристаллические аддукты, в то время как разветвленные парафиновые угле-
водороды, а также нафтеновые и ароматические этой способностью не обла-
дают. Эти аддукты могут быть отделены от жидкой фазы фильтрованием или
центрифугированием, промыты подходящим растворителем, а затем разру-
шены горячей водой. В результате отделяется маслообразная смесь парафино-
вых углеводородов нормального строения. Так как аддукты образуются
только с нормальными парафинами, а изопарафины, имеющие в общем
меньшее значение для дальнейшей химической переработки, одновременно
отделяются, то этот новый способ с точки зрения химической переработки
содержащихся в нефтях парафинов приобретает еще большее значение.
В настоящее время способ экстрактивной кристаллизации с мочевиной
применяется в первую очередь для удаления парафиновых углеводородов
из смазочных масел или топлив с целью улучшения их температуры засты-
вания [11]. Интересно, что тиомочевина образует аддукты с изопарафино-
выми углеводородами и не образует с н-парафинами.
Так как образование аддуктов с мочевиной представляет собой равно-
весную реакцию, необходимо чтобы мочевина присутствовала в реакции
в большом избытке. При депарафинизации масел, особенно богатых парафи-
20
Таблица 6
Углеводороды е 7—10 атомами С, выделенные из нефти Понка (Оклахома, США)
Форму- ла Название Тип угле- водорода Темпера- тура кипе- ния при 760 мм рт. ст., °C Чис- тота про- бы, % мол. Содер- жание в сырой нефти, %
с„н14 н-Гексан к- Парафин 68,74 99,9 1,8
СвНв Бензол Бензол 80,10 99,6 0,2
СвН12 Циклогексан Циклогексан 80,74 99,9 0,7
С7Н14 1,1-диметилцпклопептаи Цикло пентан 87,85 98,0 0,2
С7 н1в 2,3-диметилпептан 1 Разветвленный 89,78 58 0,1
C7Hje 2-метилгексан / парафин 90,05 97 0,7
С,Н14 1,траис-3-диметплцикло - 90,77 85 0,9
с,н14 пентан 1,1|мс-з-диметилцикло- | Циклопептан 91,72 51 0,2
пентан 1 Разветвленный
с,н1в З-метилгексаи 91,85 93 0,5
С7Н14 парафин
1 ,т/>аис-2-ДПметилци- Цпклонептап 91,87 93 0,5
клопентан
U7Hxe З-этилпентаи Разветвленный па- 93,47 98,7 0,05
рафии
с,н4. н-Гептан и-Парафин 98,43 99,9 2,3
С7Н]4 Метилциклогексан Циклогексан 100,93 99,8 1,6
с7н14 Этилцпклопентан 103,47 98 0,2
CgHj, 1,1,3-триметилциклопен- | Циклопептан 104,89 98,1 0,3
таи 1
QjHis 2,2-Диметилгексан ) Разветвленный па- 106,84 50 0,01
СаН]8 2,5-диметилгексан 1 рафии 109,10 55 0,06
1,траис-2,цис-4-трвме- Циклопептан 10,29 84 0,2
тилциклопептап
С8Н18 2,4-Д пмети лгексан Разветвленный па- 109,43 41 0,06
с8н18 2,2,3-триметилпентан рафии 109,84 1,0 0,004
С8Н1в 1,траис-2,ч«с-3-триме- Циклопептан 110,2 98,6 0,3
тилциклопептап
С,Н8 Толуол Бензол 110,62 98 0,5
СдНщ 3,3-диметилгексан Разветвленный па- 111,97 86 0,03
С8Н18 2,3,4-тр и метилпентан рафип
CgHie 1,1,2-триметилцикло- Циклопептан 113,73 98 0,06
пентан
csHls 2,3,3-трпметилпептан Разветвленный па- 114,76 10 0,006
C8HI8 2,3-диметил гексан 115,61 65 0,07
CgHi8 2-метил-З-этилпептан 115,65 51 0,06
C8Hie 1 ,цис-2,тр:1н с-4-трпмс- 116,73 84 0,01
C8Hie тилциклопептап {,ф7С-2,^рп7ге-3-триме- Циклопептан 117,5 90 0,07
тилциклопептап
CgHie 2-метилгептан Разветвленный па- 117,65 90 0,90
CfiHig 4-метилгептап рафии 117,71 34 0,02
C7H14 Циклогептап Циклогептап 117,7 90 0,01
cgH]8 3,4-диметилгсксан Разветвленный па- 117,72 40 0,13
З-метил-З-этплпсптап 11826 /6 0 20
CgH18 3-этилгсксап рафии 118,53 43 0 09
C8H18 З-метилгептап 118,92 98 0,30
C8Hie 1,траис-4-диметилци- 119,35 75 0,25
клогексан
CgHje C8H16 1,1-димет и л циклогексан Циклогексан 119,54 84 0,06
1 ,^ИС-3-ДИМСТИЛЦПКЛОГР- 120,09 84 0,63
ксап
C.8ITie 1-метил-шр jhc-3-этплцп- клопентан Циклопептан 120,8 57 0,12
21
IT p о д о л ж е н и е т а б л . 6
форму- ла Название Тип углеводорода Темпера- тура кипе- нии при 760 •»•« рт. ст. °C Чис- тота про- бы, % мол. Содер- яса п не в сырой нефти, %
СЬН14 1-МОТПЛ-ЧМС-3-ЭТ11ЛЦПК.ПО- 1 121,0 -
пентан
Са1116 1-метил-м/и/<с-2'Этилци- 121,2 74 0,14
клопептап 1 СЦиклопептаи
Q 1116 1-метил-1-эти.и циклопе! i- , г чти I См । 0,03
С0И18 тан 1
1,1-1{»г-3,т/аднс-4-т<’гра- 1 121,0 83 0X14
Qi 111 в метил циклопоптан
1 ,тр«/(с-2-Диметплци- 123,42 98 0,-31
клогексаи | Цикюгексан
Сй111б 1,1(ис-4-диметилциклого- 1‘>2Ц2. 76 (Ц)9
ксан Разветвле нный па- рафин
С9Н2о 2,2,5-тримет и л гексан 124,09 11 0,002
О И16 1, ц и с-4 -д и мет п л ц и к л ог е - 124.32 76 Q 09
ЦЛЬв ксаи | Циклогексан
1,тринс-3-д и метил цикло- 124/15 49 0,07
гексан п-Парафип
(’вН18 н-Октап 12566 | .992 1,9
С9Н18 Тетраметилциклопентан Циклопептан Циклогексан 127,4 130,95 131,78 73 94 —
^6111 в /i-Пропплциклопентан Этилциклогексаи 0,2
С9Н20 2,6-ди метилгептаи Разветвленный па- 135,21 98,6 0,05
рафии
^81110 Этилбензол Бензол 136,19 96 ОС’
СвН]в 1,1,3-триметилцпклоге- Циклогексан 136,63 99,9 0,2
ксан 138,35 139,10 99,8 99,9
CglllQ CgHiQ п-Ксилол .«-Ксилол } Бензол 0,1 0,5
C9H2g 2,3-дпметнлгептан Разветвленный па- рафин 14(15 60 0,05
Св Hie Тримети лц икло гексан- Циклогексан 141,2 95 02
СвН2п 4-метилоктан 1 Разветвленный па- 142 48 80 01
С9 Нод 2-метилоктап I рафии 143 26 99 9 0 4
С9 Н20 З-метплоктан 144 18 95 01
Q Ню о-Ксилол Бензол 144 41 99,7 0,3
СэНщ Мопоциклопарафиц Моноциклопара- 145,6 99 —
фип
С9111в Дцциклопарафип Дициклопарафин 146,7 99 —
С9Н20 и-Нонан «(-Парафин 150,80 99,94 13
С91112 Изопро пилбеизол 152,,39 99,8 0,07 i
С9 Н12 и-Пропилбепзол 159 22 98 009 ,1
с9 н12 1-метил-З-этилбепзол Бензол 161 ,30 99 02 1
С9Н12 1-метил-4-этилбеизол 161,90 94 0,06
С9Н12 1,3,5-триметилбснзол 164 Т2. 9Ц 9 01
С 9 Н1 о 1-метил-2-этнлбензол , 16,5,1.5 89 0,09
ОцДЦг 4-метплнопап | 1 Разветвленный па- 1657 96
СД-» 2-метпллонап | 1 рафии 166 8 99,9 - -
(_< 1 q Н о о З-метплпонан 167,8 98 —
СцДЦ-! трет -1 >у т 11 л бои.to л j Бензол 169,12 — 0,01
С9П12 СщЩо C9Hj2 1,2,4-триметилбепзол н-Декан 1,2,3-триметилбепзол /(-Парафин 169,35 174,12 176,08 181,8 183,48 99,7 99,9 99,8 0,5 1,8 0,1 0,1
С1ОПЦ ^10^14 1-метил-З-лроиплбе изол 1,2-днэтилбензол Бензол —
с10п14 1-метил-2-пропилбепзол 184,75 — 0,3
C1qH14 1,4-дпметил -2-этилбепзо л 186,91 — 0,1
CjgHig трапс-Декагидронафта- Дицпклопарафин 187,25 — —
ЛИН
Ci0 Н14 1,2,4,5-тетраметппбопзол 1 196,8 — —
Ci 0н14 1,2,3,5-тетраметилбензо л । Бепзол 197,93 — —
С|0Н14 1,2,3,4-тетраметилбепзол 1 205 J04 99,9 02
22
ном, необходимо добавление разбавителя такого, как метилизобутилкетон,
изооктан, или хлористый метилен. Мочевина может применяться в спирто-
вом или водном растворе.
Постоянная высокая концентрация мочевины во время образования
аддукта поддерживается или постоянным добавлением новых порций моче-
вины к реакционной массе или, что еще проще, работой с раствором моче-
вины, насыщенным при повышенной температуре, сравнительно с темпера-
турой образования аддукта. Благодаря охлаждению из насыщенного при по-
вышенной температуре раствора выделяется каждый раз столько мочевины
в свободном состоянии, сколько используется на образование аддукта.
Таким образом, раствор мочевины остается всегда концентрированным.
Соотношение между парафиновыми углеводородами и мочевиной в ком-
плексных соединениях таково, что на 4 атома С приходится примерно 4 мо-
лекулы мочевины, а на 10 атомов С примерно 8 молекул.
В табл. 7 даны соотношения компонентов в отдельных аддуктах [121.
Таблица 7
Молекулярное соотношение между органическим компонентом и мочевиной в аддукте
Органический компонент Молей мочеви- 1 ны на 1 моль органического | компонента Органический компонент Молей мочеви- ны на 1 моль органического компонента
название формула название формула
Гексан Гептан Октан Нонан Декан Ундекан Додекан Гексадекап Тетракозан Октакозан 1-хлороктан .... 1-хлортетрадекап . . 1-бромоктан .... 1-бромдекан .... Метилэтилкетоп . . свн14 с,н„ С8Н1Й С9Н20 С10Н22 СцН24 С12В26 С16Н34 С24И6О ^28 Ща С8Н1,С1 С14Н29С1 С8Н17Вг Сц)Н21Вг С4Н8О 5,5 6,1 7,0 7,7 8.3 9,7 12,2 18,0 21,4 7,7 10,0 7,2 8,9 4,0 Диэтилкетон .... Дипропилкетон . . . н-Масляная кислота Валериановая » Капроновая » Энантовая » Каприловая » Пеларгоновая » Каприновая » Ундекановая » Лауриновая » Миристиновая » Пальмитиновая » Стеариновая » nnnonnnnnnnccn ® ft rf». to — 0 м >7-1 Гр* HH >-ri i->- Д Д Д Д Д ДЯ3^ ti « Ю tO ti W ® Й О О U '-' U L-Q Oto » ю 0 » tO to tO tO to 4,8 6,0 4,0 4,6 5,4 6,0 6,7 7,6 8,2 8,9 10,0 11,6 12Д 14,2
Соотношение мочевины и парафина в комплексах, образованных с па-
рафином, имеющим от 7 до 18 атомов С в молекуле, составляет около 2,48 г
мочевины на 1 мл парафина.
В промышленности разработаны различные способы депарафинизации
экстрактивной кристаллизацией с мочевиной. Л. Хоппе разработал процесс,
в котором в качестве растворителя успешно применяется хлористый метилен.
Мочевина применяется в водном растворе [13]. Важное технологическое зна-
чение имеет тот факт, что вода, применяемая как растворитель для мочевины,
если ее не более 40%, считая на мочевину, удерживается аддуктом. При
последующем разделении образуется только жидкая фаза (раствор депарафи-
нированного масла) и твердая фаза — аддукт. Последний выделяется в круп-
нозернистой форме, легко отделяемой при помощи сита, когда количество
введенной вместе с мочевиной воды составляет 10—40%, считая на мочевину.
При этом способе депарафинизации масел не только отпадают расходы
на охлаждение, достигающие больших величин в старом способе, но уда-
ляются также и те парафины, которые при нормальном охлаждении из масла
не выделяются [14]. Схема процесса А. Хоппе представлена па рис. 8.
Равные объемы масла, хлористого метилена и насыщенного при 70е
водного раствора мочевины поступают при перемешивании в реакционный
сосуд, где мгновенно происходит образование кристаллов. Хлористый мети-
лен, испаряющийся за счет тепла, выделяющегося при экзотермическом
образовании аддукта, конден-
сируется в обратном холо-
дильнике и возвращается в
реакционный сосуд. При этом
в реакционном сосуде под-
держивается температура
35—45°. Кристаллизат под-
вергается фильтрации. Часть
реакционной массы приме-
няется для следующей за-
грузки как вызывающий на-
чало кристаллизации мате-
риал. Депарафинированное
масло с фильтра поступает
на установку регенерации
растворителя. Осадок на
фильтре промывается для
освобождения от захвачен-
ного им масла и поступает
в сосуд для разложения,
где при энергичном пере-
мешивании водяным паром
разлагается на парафин и
VII]
Рис. 8. Схема депарафинизации нефтяных фракций
при помощи мочевины.
1 — фильтр; 2 — приемный сосуд; 3 — аппарат для восста-
новления концентрации мочевины; 4 — аппарат для рас-
щепления аддукта; .5 —сепаратор; 6 — отстойник для отде-
ления раствора мочевины.
Линии: I — парафинистое масло; II — мочевина; III —
циркулирующий хлористый метилен; IV — орошенпе; V—
вода; VI — пар; VII — парафин; VIII — депарафиниро-
ванное масло.
водный раствор мочевины.
Разделение обоих слоев про-
исходит в отстойнике. Пара-
финовый слой поступает в
дистилляционную колонну
для удаления растворителя.
Разбавленный водный рас-
твор мочевины нагревают до
80° и выдерживают при этой
температуре столько вре-
мени, сколько необходимо, чтобы за счет испарения излишка воды сделать
раствор насыщенным при 70°. Раствор такой концентрации возвращается
в процесс. Таким путем можно получать парафиновые углеводороды 95%-ной
чистоты. Парафин, выделенный из мазута, содержавшего примерно 13% па-
рафина [11], имеет следующий состав.
Компонент Содержание, % объемы. | Компонент Содержание, % объемы.
Додекан 5 Октадекан .... 12
Тридекан 10 Нонадекан .... 6
Тетрадекан .... 15 Эйкозан 5
Гексадекан .... 16 Вышекипящие . . 7
Гептадекан .... 14
III. НЕФТЯНОЙ ПАРАФИН
А. ОБЩИЕ СВЕДЕНИЯ
Нефтяной парафин получают депарафинизацией фракций смазочных
масел. Смазочные масла должны обладать определенной температурой засты-
вания, т. е. при температурах ниже 0° из них не должен выделяться парафин.
24
Для этого масла должны быть освобождены от парафина. При этой операции
в качестве побочного продукта получают смесь парафиновых углеводородов,
которые в настоящее время приобрели столь большое промышленное зна-
чение, что на многих заводах депарафинизация масел ведется специально
в целях получения парафина. Депарафинированные масла, если они не
могут быть использованы как смазочные, служат сырьем для крекинга.
Мировое производство парафина достигает в настоящее время 500 000 т.
Пенсильванские масла содержат в среднем около 2,4% парафина, вы-
сокопарафинистые американские и советские масла могут содержать до 7 %
парафина.
Большая часть содержащихся в нефти парафинов кипит в тех же пре-
делах, что и масляные дистилляты, и поэтому перегонкой и ректификацией
парафин не может быть выделен из масляных фракций.
Масляные фракции могут содержать до 30% парафина и для достижения
необходимой температуры застывания должны быть освобождены от него,
так как уже 1% парафина в смазочном масле вызывает обращение масла
в гель при 10—20°.
Б. ВЫДЕЛЕНИЕ ПАРАФИНА ИЗ ФРАКЦИЙ СМАЗОЧНЫХ МАСЕЛ
Депарафинизация смазочных масел осуществляется в настоящее время
большей частью при помощи растворителей [15]. Принцип этого метода
заключается в том, что фракция смазочного масла растворяется в подходящем
растворителе и из этого раствора посредством охлаждения выкристаллизо-
вываются парафины, которые отделяются. После фильтрации раствор осво-
бождается от растворителя, последний возвращается в процесс. Остаток
перерабатывается на смазочные масла. Оставшийся на фильтре осадок —
парафин — подвергается дальнейшей очистке, заключающейся в обезмасли-
вании парафина при помощи растворителей. В большинстве случаев вспомо-
гательный растворитель, применяемый при депарафинизации, является
смесью метилэтилкетона и технического бензола. Применяется также смесь
ацетон-бензол. Превосходным растворителем для депарафинизации является
жидкий пропан, применение которого позволяет решить одновременно две
задачи [16]. С одной стороны, он служит растворителем, а с другой вслед-
ствие низкой температуры кипения является охлаждающим агентом. Так
как при этом имеет место внутреннее охлаждение кристаллизующейся массы,
то потери тепла за счет теплопередачи полностью отсутствуют. Содержащее
парафин смазочное масло и пропан совместно нагреваются под давлением
до температуры, необходимой для полного растворения масла в пропане.
Для нагревания берут 1—3 объема жидкого пропана на 1 объем масла.
Затем вследствие испарения пропана смесь постепенно охлаждается до
температуры около —35°, причем, как правило, температура охлаждения и
фильтрации должна лежать примерно на 20° ниже желаемой температуры
застывания масла. Выделившийся парафин фильтруют под давлением и оста-
ток иа фильтре промывают пропаном.
В. ОЧИСТКА ПАРАФИНА
Освобожденный от растворителя парафин содержит еще 30—35% масла
и имеет коричневую окраску. Последние следы масла можно удалить рас-
творением в горячем растворителе, например в хлористом этилене, с после-
дующим охлаждением раствора и отделением выделившегося парафина на
центрифуге [17].
Другим широко применяемым способом является так называемый
процесс выпотевания, состоящий в том, что содержащая масло парафиновая
лепешка помещается на сетчатую тарелку и нагревается до температуры,
при которой из парафиновой лепешки выделяется масло и часть низкоплав-
ких парафинов, тогда как высокомолекулярный парафин остается на сетке.
Зтот медленный и неудобный на практике процесс будет, несомненно, в бу-
дущем полностью заменен процессом обезмасливания при помощи раствори-
телей. Обезмасляный парафин подвергается далее очистке серной кислотой
и отбеливающей землей.
Г. ПУТИ ПЕРЕРАБОТКИ ПАРАФИНА
Нефтяной парафин представляет собой смесь нормальных парафиновых
углеводородов с длинной цепью. Мягкий парафин (температура плавления
40—42°) применяется в спичечной промышленности, в производстве импре-
гпироваппой бумаги, в кожевенной промышленности и др. Твердый парафин,
плавящийся при 50—52“, применяется в основном в свечном производстве.
Парафин применяется также для многих других целей — для консервации
фруктов, в косметике, в фармацевтическом производстве, для специальных
сортов бумаги, смазочных материалов, в фотографии и т. д.
Микрокристаллический парафин, который может быть выделен в пер-
вую очередь из остатков от перегонки нефтей парафинового основания,
представляет большую ценность, чем нормальный парафин. Конечно, вслед-
ствие разветвленной структуры он мало пригоден для дальнейшей химической
переработки. Получение такого парафина из обычного из-за плохой филь-
труемости и высокой вяйкости исходного продукта представляет большие
трудности. Микрокристаллический парафин вязок и пластичен. Он имеет
высокую температуру плавления 60—80° (сорт церезин). Церезин получают
в общем тем же способом. Возможности применения парафина показаны
на рис. 9.
ПАРАФИН
"Производство свечей
"Пропитка спичек
"Пропитка бумаги
"Консервирование фруктов
‘“"Косметика
"Текстильная и ледериновая промышленность, парафиновые
эмульсии, гидрофобирование
"Типографские краски
"Фармацевтическая промышленность
"Фотографическая промышленность
----"Смазочные материалы
----"Получение крскинг-олефинов (350°, водяной пар)
"Окисление парафина (спирты, жирные кислоты и др.)
----"Хлорпарафин (около 15% хлора). Получение депрессаторов
----"Хлорпарафин (40 — 70% хлора). Получение негорючих бумаги,
текстиля, древесины. Пластификаторы для поливинилхлорида.
Клей для борьбы с гусеницами.
----"Парафип-сульфонаты путем сульфохлорировапия.
Рис. 9. Возможности применения парафина (С22—С25).
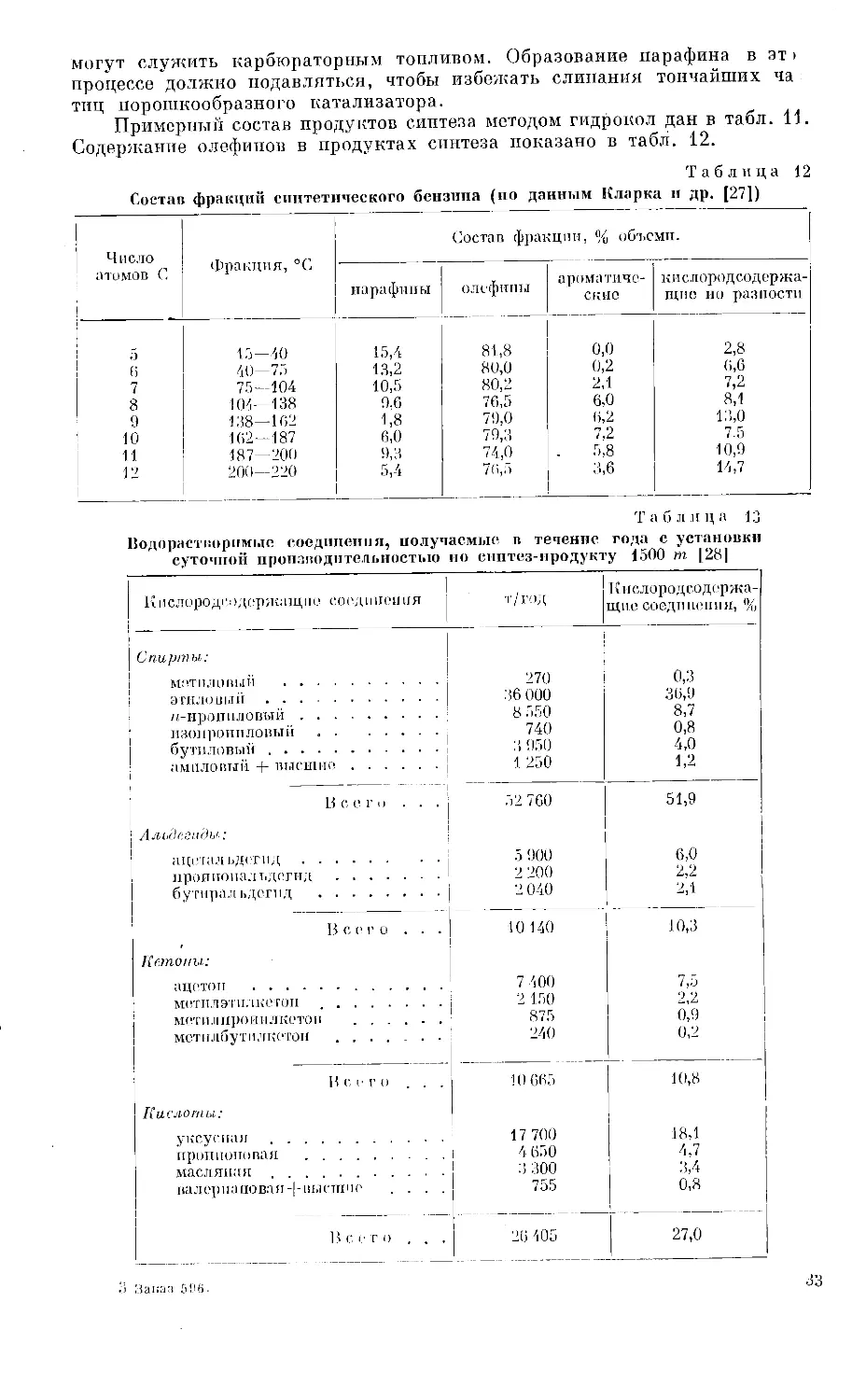
IV. ПОЛУЧЕНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ СИНТЕЗОМ
ФИШЕРА-ТРОПША
А. ОБЩИЕ СВЕДЕНИЯ
Под синтезом Фишера-Тропша понимается каталитическое гидрирование
окиси углерода в присутствии кобальтового или железного катализаторов
до образования олефиновых и парафиновых углеводородов с различным
числом атомов С. начиная от метана и кончая самыми высокомолекулярными
углеводородами. Первоначально для этого синтеза применялся кобальтовый
2в
катализатор. В настоящее время на новых установках работают только с ка-
тализатором на железной основе.
Синтез с кобальтовым катализатором протекает главным образом по
следующему уравнению:
CO-J-2I12 —> СН2+Н2О+39,4 ккнл/моль (Со=уравиепие).
Отсюда следует, что кислород окиси углерода выделяется в виде воды.
Формально синтез но Фишеру-Тропшу с кобальтовым катализатором
является синтезом олефинов, так как можно принять, что образующиеся
промежуточные метиленовые группы затем полимеризуются. Так как,
однако кобальт в условиях синтеза (200°, нормальное или низкое давление)
действует как активный катализатор гидрирования, то большая часть олефи-
нов насыщается до парафинов.
При синтезе с железным катализатором кислород окиси углерода выде-
ляется не в виде воды, а в виде двуокиси углерода. Реакция протекает по
следующему уравнению:
ДСО+Н2 —> СО2+СН.,-|-48,9 ккал/молъ (Ре=уравиеипс)
Так как железо для гидрирования в условиях синтеза (320 \ 25 ат) зна-
чительно менее активно, чем кобальт, то продукты синтеза Фишера-Тропша
с железным катализатором содержат значительно больше олефинов.
Большая трудность при проведении синтеза по Фишеру-Тропшу с ко-
бальтовым катализатором состоит в том, что на 1 л3 синтез-газа развивается
приблизительно 600—700 ккал тепла, которое должно быть отведено, по-
тому что температура катализатора должна поддерживаться с точностью
до 1°. Промышленный катализатор на кобальтовой основе содержит па
100 частей кобальта 5 частей окиси тория, 8 частей окиси магния и 200 ча-
стей кизельгура. Катализатор отличается чрезвычайно низкой теплопровод-
ностью и поэтому проблема отвода тепла становится особенно трудной.
Контактная камера установки Фишера-Тропша, вмещающая 10 м3 кобаль-
тового катализатора, может из-за плохого отвода тепла пропустить лишь
1000 л3 синтез-газа в час. Требуемая поверхность охлаждения для 1000 л3
синтез-газа составляет около 3000 л2. Из 1 л3 газа получают 165 —175 г
целевых углеводородов. В настоящее время современные установки синтеза
Фишера-Тропша работают только с железным катализатором, состоящим
практически только пз железа и обладающим значительно лучшей тепло-
проводностью.
На современных установках синтеза Фишера-Тропша могут осуще-
ствляться два различных процесса: с неподвижным и с подвижным железным
катализатором. Отвод тепла производится или как при работе с кобальтовым
катализатором по принципу парового котла или путем наружного охлажде-
ния печей синтеза.
После второй мировой войны в Германии получил развитие синтез
Фишера-Тропша с железным катализатором методом, разработанным Рур-
хеми-Луржп, и методом Кольбсля с сотрудниками. При этих методах отвод
реакционного тепла осуществляется при помощи охлаждающих поверх-
ностей, установленных внутри печей. В то время как по способу Рурхсми-
Луржи работают с неподвижным катализатором (большие заводы в Сасоль-
бурге, в Южной Африке), но способу Кольбеля работают с взмученным
в жидкой фазе контактом. В США, особенно для получения бензина, раз-
работан способ синтеза «с летучим катализатором» (способ псевдоожиженного
слоя), в котором отвод тепла реакции осуществляется при помощи располо-
Htennoro в печи охладителя.
Синтез Фишера-Тропша с кобальтовым катализатором в настоящее время
не имеет значения для нефтехимической промышленности и мы не будем
27
более па нем задерживаться. Здесь следует остановиться несколько подроб-
нее па железном катализаторе.
Чтобы процесс Фишера-Тропша мог быть отнесен к нефтехимической
промышленности, синтез-газ (смесь окиси углерода — водорода) в пашем спе-
циальном случае должен получаться нз природного газа.
Получение его основано па неполном сгорании метана в кислороде или
на термическом или каталитическом процессе превращения метана в кон-
такте с водяным паром.
Первый процесс идет согласно уравнению
CII4-f-1/2 О2 —> СО-|-2Н24-8,5 ккал/моль,
Второй — по уравнению
СН4-Г 112О —> СО-рЗИ2 — 30 ккал!молъ
Обе реакции могут комбинироваться, как, например, в процессе Фау-
зера, где смесь метана и кислорода (1 ж3 + 0,6 ж3) в присутствии водяного
пара при 950° превращается над катализатором в смесь СО и Ш (2,8 ж3).
Особенно значительную роль этот способ получения смеси окиси угле-
рода и водорода играет в гидрокол-процессе — американском способе осу-
ществления синтеза Фишера-Тропша на базе природного газа (18].
Кислород, нагретый примерно до 315°, и предварительно нагретый
до 650° природный газ под давлением 20 am (рабочее давление синтеза)
подаются в футерованную огнеупорным материалом камеру сгорания,
где температура достигает 1350°.
Газы сгорания отдают свое тепло в утилизационном котле, где давление
достигает 45 ат [19]. При синтезе освобождается значительное количество
энергии, часть которой может быть использована для получения кислорода.
Термическое взаимодействие метана с водяным паром происходит при
1200—1300°. В присутствии никелевого катализатора взаимодействие ста-
новится возможным при 700—800°. Каталитический способ, в котором
природный газ (в целях предотвращения отравления никелевого катали-
затора) должен предварительно освобождаться от сернистых соединений,
в промышленности уже давно разработан [20]. .Грубая очистка преду-
сматривает удаление неорганической серы, главным образом в виде серо-
водорода. Она происходит над так называемой люкс-массой (окись железа —
красный шлам бокситиых отходов) или над бурым железняком при обычной
температуре. Тонкая очистка, имеющая целью удаление органической
серы в виде сероуглерода или сернистого карбонила, осуществляется над
щелочной люкс-массой при температуре 250—300°.
При этих условиях сера органических сернистых соединений превра-
щается в сероводород, который одновременно удаляется с катализатора.
Новейший способ, очень хорошо зарекомендовавший себя на практике,
одновременно позволяет очистить газ не только от сернистых соединений,
но и от углекислоты, синильной кислоты, аммиака и смолистых загрязнений
(ректизол-способ); он заключается в промывке газа глубоко охлажденным
метиловым спиртом, растворяющим все перечисленные загрязнения [21].
Способ работы примерно следующий (рис. 10). Сырой газ при рабочем давле-
нии синтеза, равном примерно 20 ат, подается в нижнюю часть промывной
колонны 1, имеющую температуру —20°, где промывается метиловым спир-
том, поступающим в среднюю часть промывной башни с температурой
порядка —75°. Стекая вниз по колонне, метиловый спирт нагревается от
— 75 до —20° за счет теплоты абсорбции углекислоты. Газ на этой первой
ступени очистки очищается настолько, что в нем остается только 3% СО2.
98% сероводорода, большая часть органических сернистых соединений
и углеводороды при этом также удаляются. На второй ступени газ про-
мывается метиловым спиртом, охлажденным до —62°, и здесь практически
полностью освобождается от оставшихся загрязнений, так что уже может
быть использован для синтеза.
28
Обогащенный углекислотой спирт поступает затем в колонну 2 под
атмосферным давлением, понижаемым затем на 0,2 ат, освобождается там
от углекислоты и при этом температура его понижается до —75°. Этот спирт
возвращается в среднюю часть колонны 1. Отработанный метиловый спирт
со второй ступени освобождается от углекислоты в колонне 3 и после охла-
ждения в теплообменнике
возвращается в процесс.
В этой связи следует
остановиться на получении
из природного газа чистого
водорода — промышленном
процессе, применяемом в
широких масштабах, так
как водород потребляется
для получения аммиака и
его производных (мировое
производство аммиака со-
ставило в 1957 г. около
8,7 млн. т [22]). Этим про-
цессом нефтехимическая про-
мышленность объединяется
Чистый газ
Рис. 10. Схема ректизол-процесса по Герберту [22].
с большой промышленно-
стью неорганической химии (аммиак, азотная кислота, нитраты).
Способ получения водорода из углеводородов схематически представлен
на рис. 11. Пропан (который здесь взят в качестве примера) предварительно
подогревается до ~370° и приводится в контакт с бокситом; здесь содержа-
ние. И. Схема получения водорода из газообразных углеводородов.
/ — нагреватель; 2 — установка для обессеривания бокситом; 3 — холодильник/ 4 — колонна для
промывки щелочью; -5 — колонна для промывки водой; 6 — печь с никелевым контактом* 7— СО-кон-
вертор I; 3 — отделенно СОз; у — СО-нонвертор II; 10 —СО-конвертор III; 11 — регенератор.
Линии: I — пропап; II — натронная щелочь* Ш — подо* IV — дренаж; V — пар; VI — аминовый
раствор, насыщенный СОз; VII — аминовый раствор; VIII — водород 99,9%-ной чистоты; IX— сво-
бодный от СОг аминовый раствор обратно в процесс.
щиеся в газе сернистые соединения (меркаптаны и сульфиды) превращаются
в сероводород. На следующей стадии предварительно охлажденный газ
промывается щелочью для удаления сероводорода. После промывки водой
освобожденные от серы углеводороды при температуре 760—980° вместе
с водяным паром проходят над никелевым катализатором; нагрузка на ката-
лизатор равна примерно 600 (600 объемов газа на 1 объем катализатора
в час).
При этом имеет место следующая реакция:
СН3—СН2—СН3-ЬЗН:О
Катализатор
ЗСО+7Н.2
29
и далее через реакцию конверсии окись углерода превращается в угле-
кислоту и водород согласно уравнению
CO+1LO СО..+Щ+Ю «ш/шмъ
Углекислота из газа удаляется при помощи моноэтаполамипа по спо-
собу «Гирботол» (Girbotol) или методом алкацид-процссса (Alkazid) с амино-
кислотами, и, наконец, промывкой газа водой под давлением .
Конверсия осуществляется пропусканием смеси окиси углерода и паров
воды над окисью железа, активированной окисью хрома, при температуре
около 370°. Вследствие экзотермического характера реакции температура
повышается приблизительно до 430" . Контактная нагрузка в этом процессе
достигает 100 и выше.
На схеме показано, что газ последовательно проходит через три кон-
вертора, после каждого из которых подвергается промывке для удаления
углекислоты; в результате газ полностью освобождается от окиси углерода.
В настоящем примере газ отмывается от углекислоты примерно 20%-ным
водным раствором моноэтаноламина. Насыщенный углекислотой этано-
ламиновый раствор регенерируется продувкой острым паром. Количество
углекислоты составляет около 300 л3 на 1000 л3 водорода. Чистота водо-
рода 99,8%. Из 1 л3 этаноламинового раствора вымывается 2.5 л3 угле-
кислоты [23].
Полученный таким способом газ практически свободен от сернистых
соединений и может быть использован для синтеза. Синтез-газ из других
источников, например каменноугольный или буроугольный, должен под-
вергаться особой очистке, так как железный катализатор в синтезе Фишера-
Тропша также чувствителен к действию серы.
Г>. ПРАКТИЧЕСКОЕ ПРОВЕДЕНИЕ СИНТЕЗА ФИШЕ Р А.-ТРОПША
С ЖЕЛЕЗНЫМ КАТАЛИЗАТОРОМ
а) Кольбель-процесс [24]
В процессе Кольбеля катализатор в реакционной зоне находится
во взмученном состоянии. Тепло реакции отводится при помощи находя-
щегося внутри печи охладителя водой, прокачиваемой через него под давле-
нием. Температуру устанавливают путем регулирования давления пара
в охлаждающем элементе. Отвод реакционного тепла настолько хорош,
Рис. 12. Схема синтеза по способу Копьбеля.
1 — система охлаждения; 2 — печь; 3 — холодильник для
орошения; 4 — главный холодильник для масла и воды;
«5 — отделитель низших углеводородов.
Линии: I — парафин; II — свежий катализатор; III —
свежий сиптеэ-гаэ; IV — газы циркуляции; V — жидкие
продукты; VI — газообразные продукты реакции; VII —
отходящие газы.
чго нагрузка печи может
быть в 6—8 раз больше, чем
при работе с кобальтовым
катализатором. Синтез-газ
за одну ступень претерпевает
превращение на 90% . Состав
продуктов реакции может
изменяться в чрезвычайно
широких пределах. Они
могут на 80% состоять из
бензина и ожиженного газа
с содержанием 65—90%
олефинов, или из 50% га-
зойля с содержанием 25—
70% олефинов, или 65%
мягкого и твердого пара-
фина.
Продукты синтеза от-
водятся вместе с отходя-
щими газами и конденси-
30
руются. Нелетучий парафин удаляется отдельно (рис. 12). Катализатор
состоит из железного порошка с небольшим количеством меди или щелочи
в качестве промотора. Можно применять также порошкообразный катали-
затор синтеза аммиака. Давление около 10 ат, температура 200—320°.
Средний выход целевого продукта 160—175 г на 1 л3 СО/Нг. Состав конеч-
ных продуктов очень сильно зависит от температуры синтеза. При высоких
температурах образуется значительно больше низкомолекулярных соеди-
нений, чем при низкотемпературном процессе. Табл. 8 показывает возмож-
ности процесса Кольбеля в отношении изменения состава продуктов реак-
ции. Процесс Кольбеля настолько гибок, что при возвращении в процесс
высококипящих составных частей, когда режим направлен на получение
бензина, можно и эти высококипящие углеводороды превратить в пизко-
кипящие. Напротив, при использовании для синтеза низкокипящих угле-
Таблица 8
Возможности управления синтезом в жидкофазном процессе
Кольбеля в связи с составом продуктов реакции при
нормальных рабочих условиях (синтез-газ 1,5СО : 1Н2,
нагрузка 300)
Показатели
Условия синтеза на-
правлены па получение
бензина I парафина
•' I % I i %
Выход из 1 „и3 СО -: Н.,
Углеводороды:
С3-:-С4 .
Фракция
»
20—200°
200—300°
» выше 300°..........
Спирты С2 — Са...............
168
30
104
22
8
4
100 184 100
18
62
13
7
20
28
120
9
водородов, когда задачей является образование парафина, в нужном на-
правлении происходит перестройка и этих углеводородов, как показано
в табл. 9.
Таблица 9
Повышение выхода низко- или высококипящих продуктов
путем возврата части продуктов синтеза в печь
в жидкофазном процессе Кольбеля с железным катализатором
Показатели Условия синтеза на- правлены па получение
бензина парафина
С 1 % е | %
Выход из 1 .и3 СО-| 1b 168 100 185 100
Углеводороды: t-З + 1'4 27 16' 9
Фракция 211—200° 117 69 7 4
» 200-300° П 1 10 12 6
» выше 300 ’ .) 137 85
Спирты Со — Со 3 1 2 1
Содержание олефинов в продуктах синтеза с железным катализатором,
как выше указывалось, довольно значительно. Но в процессе Кольбеля
путем соответствующего регулирования температуры и состава газа оно
может быть увеличено еще более, как показано is табл. К).
б) Рурхеми-Луржи-процесс [25]
Работа этим способом про-
водится с неподвижным желез-
ным катализатором и с отводом
тепла реакции через вмонти-
рованный внутрь печи охла-
дитель. Поддержание необхо-
димой температуры регулирует-
ся давлением пара в охлаж-
дающем агрегате. Выход про-
дукта составляет 185 г на 1 .и3
смеси СО/Нг, включая фрак-
цию Сз. Это соответствует вы-
Таблица 10
Пределы содержания олефинов в продуктах
жидкофазного процесса по Кольбелю
с железным катализатором
Фракция Содержание олефинов, %
минимум максимум
Углеводороды С3 + С4 . . 74 92
20—200° 65 90
200—300° 25 70
> 300° 20 66
ходу около 90% от теоретического. Здесь также содержание олефинов
исключительно высокое и (что особенно важно при использовании их в
химическом направлении) олефины очень равномерно распределены по
всем фракциям. Их содержится около 75% во фракции С5 и 62% во
фракции С18. В среднем у 70% олефинов двойная связь находится у
конца молекулы. Степень разветвленности углеводородной смеси, кипя-
щей в интервале кипения среднего масла, составляет около 25%.
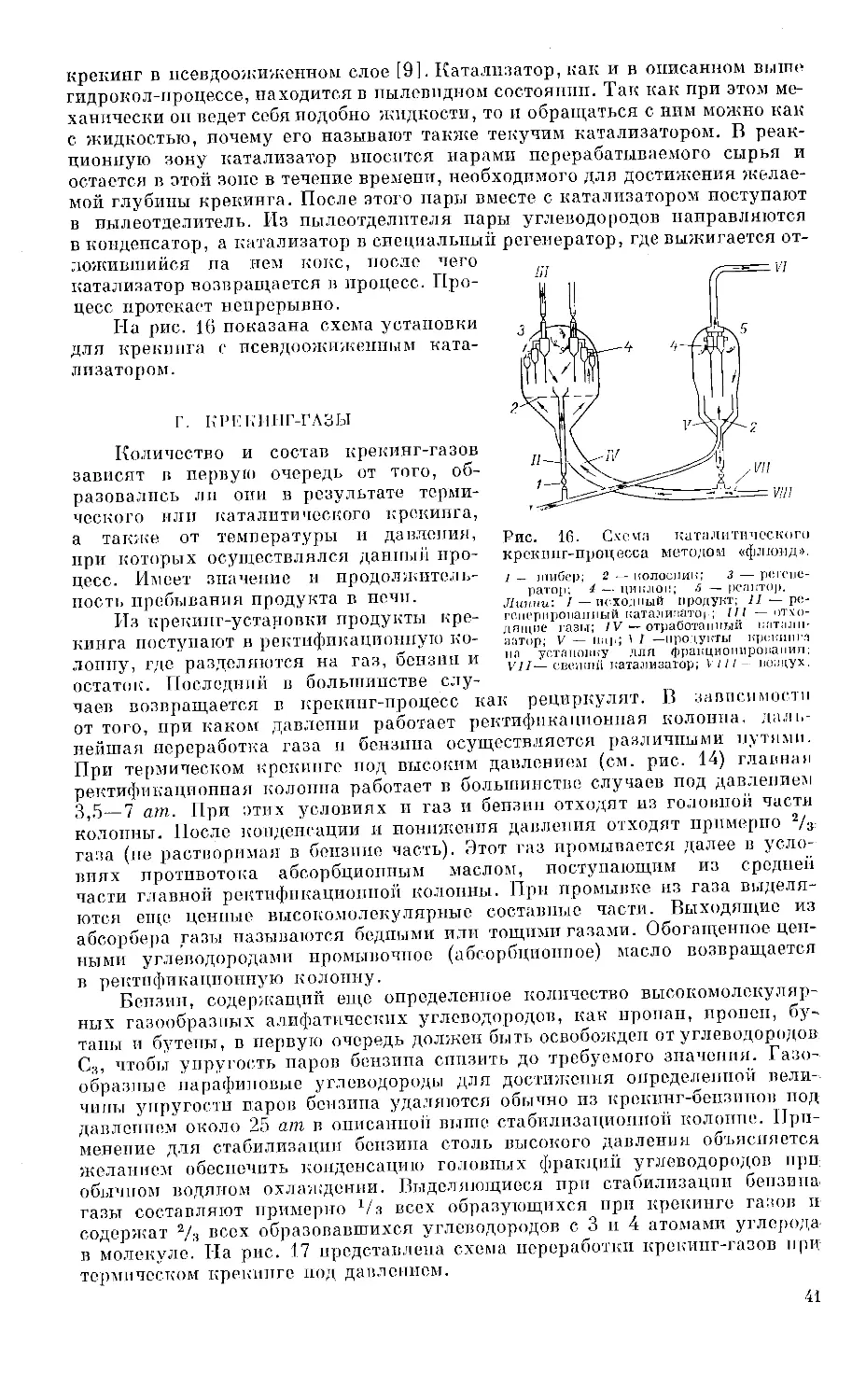
в) Способ псевдоожиженного слоя (гидрокол-процесс)
В гидрокол-процессе [26] в качестве катализатора применяется тонкая
железная пыль, поддерживаемая во взвешенном состоянии синтез-газом.
Температура регулируется при помощи подвесного теплообменника. Рабо-
Рис. 13. Схема гидрокол-процесса.
I — реактор для получения синтез-газа; .2 — газоочиститель; з —
печь для синтеза в условиях псевдоожиженного слоя; 4— охлаждаю-
щее устройство; 5 — разделительная установка.
Линии: I — природный газ; II — кислород; \Ш — циркулирую
щие газы; IV — продукты реакции.
чая температура составляет 350—460°, давление 30—45 ат. Катализатор
состоит из природного магнетита с 0,5% КгО. Может применяться также
катализатор синтеза аммиака. Предварительно катализатор восстанавли-
вается в потоке водорода по способу «псевдоожиженного слоя» при 350—
460°. Степень превращения га-
за составляет около 90%. Таблица 11
На рис. 13 приведена схе-
ма гидрокол-процесса. Синтез-
газ получают сжиганием при-
родного газа в кислороде. Про-
дукты синтеза, после выделения
из них кислородсодержащих
водорастворимых соединений
и перевода высших кислород-
содержащих составных частей,
особенно спиртов, путем де-
гидратирования, в олефины,
Средний состав продуктов синтеза
гидрокол-процесса
Фракция % вес. от обще- го выхо- да Содержа- ние оле- финов, % объемн.
Сь+С4 32 80
Легкий и тяжелый бензины 56 70
Дизельное топливо .... 8 65
Остаток 4 —
32
могут служить карбюраторным топливом. Образование парафина в эт >
процессе должно подавляться, чтобы избежать слипания тончайших ча
тиц порошкообразного катализатора.
Примерный состав продуктов синтеза методом гидрокол дан в табл. 11.
Содержание олефинов в продуктах синтеза показано в табл. 12.
Таблица 12
Состав фракций синтетического бензина (по данным Кларка и др. [27])
Число атомов С Фракция, °C Состав фракции, % объемн.
парафины олефины ароматиче- ские кислородсодержа- щие ио разности
5 15—40 15,4 81,8 0,0 2,8
6 40—75 13,2 80,0 0,2 6,6
7 75—104 1.0,5 80,2 2,1 7,2
8 104—138 9,6 76,5 6,0 8,1
9 138—162 1,8 79,0 6,2 13,0
10 162—187 6,0 79,3 7,2 7.5
11 187—200 9,3 74,0 5,8 10,9
12 200—220 5,4 7(i,5 3,6 14,7
Т а б л и ц а 13
Водорастворимые соединения, получаемые в течение года с установки
суточной производительностью по спптез-иродукту 1500 т |28]
Кислородсодержащие соединения | т/год Кислородсодержа- щие соединения ,%
Спирты:
метиловый 270 0,3
эгпловый 36 000 36,9
/ьирояшювый 8 550 8,7
изопропиловый 740 0,8
бутиловый 3 950 4,0
ампловы'й + высшие 1 250 1,2
В с его • . . 52 760 51,9
ЛлыС.гиды:
ацетил ьдегцц 5 900 6,0
пропиональдегид 2 200 2,2
бутиральдегид 2 040 2,1
Всего . . . 10 140 10,3
Кетоны:
ацетол 7 400 7,5
метплэтплкепш 2 150 2,2
мстилuponилкетон 1 875 0,9
мстплбутилкотон 240 0,2 }
В с е го ... 10 665 10,8
Кислоты:
уксусная 17 700 18,1
пропионовая 4 650 4,7
масляная 3 300 3,4
валериановая+выспгие .... 755 0,8
Всего , . . 26'0Ъ 27 0
Особое значение синтез Фишера-Тропша с железным катализатором
имеет потому, что в этом процессе всегда получается то или иное количество
кислородсодержащих соединений. В гидрокол-процессе образование кисло-
родсодержащих соединений имеет первостепенное значение. Табл. 13 дает
представление о количестве спиртов, альдегидов, кетонов и кислот, полу-
чаемых в течение года с установки гидрокол суточной производительностью
1500 т продуктов синтеза.
ЛИТЕРАТУРА
1. Pastonesi G. Chem. Ing. Techn., 27, 465, 1955.
2. Robertson W. T., Stampfer H. N. Am. Gas. J., 169, 6, 21, 23, 1948.
3. Oil a. Gas J., 46, 47, 108—188, 1948; P r a 11 A. W., Foskett N. L. Science
of Petroleum, 5, Teil 11, 109, 1955.
4. Brennstoffchem. Wirtschaftsteil, 38, 7, 8, 31, 1957.
5. V 1 ugt er J. C., Waterman H. I., van Westen H. A. J. Inst. Petrol.
Techn., 21, 661, 1935.
6. R u m p f К. K. Erdol und Kohle, 5, 745—747, 1952; Ault W. С. J. Inst.
Petr. Techn., 33, 598—607, 1947; Dietz W. А. с сотрудн. Ind. Engng. Chem., 44, 1818,
1952; Reisemann E. Erdol und Kohle, 5, 161, 1952.
7. К i rk R. E., О t h m e г D. F. Encyclopedia of chem. Techn. 10, 109, 1953.
8. H e n z e H. R., Blair С. M. J. Am. Chem. Soc., 53, 3045, 3084, 1931.
9. R о s s i n i F. D., Mair B. J. Progress in Petroleum Technology, 341, 1952:
Advances in Chem., Series 5, Amer. chem. Soc.
10. В e n g e n F., Schlenk W. Experimentia, 5, 200, 1949; Schlenk W.
Fortschr. chem. Forsch., 2, 92—145, 1951.
11. Bailey Wm. А. с сотрудн. Ind. Engng. Chem., 43, 2125, 1951.
12. S c h 1 e n k W. Fortschr. chem. Forsch., 2, 92—145, 1951.
13. Hoppe А., Пат. ФРГ. 928726, 1955, Edeleanti G. m. b. H., Zbl., 127, 315,
1956; Hoppe A., Franz H. Erdol und Kohle, 8, 411—413, 1955.
14. Chem. Engng., 63, 11, 115, 1956.
15. Oil a. Gas J., 46, 48, 158, 1948, Petrol. Refiner, 33, 9, 231—235, 1945.
16. Chamberlin N. F., Dinwiddie J. A., Franklin J.L. Ind.
Engng. Chem., 41, 556—570, 1949.
17. Y о u n g D. A. Oil a. Gas. J., 40, 11, 34—37, 1941; 42, 46, 98—101, 137—157,
1945; Ebner E. B., Mertens F. T. Petrol. Refiner, 23, 230—234, 1944.
18. Keith P. Oil a. Gas J., 45, 6, 102—112, 1946.
19. Latta J. E., Walker S. W. Chem. Engng. Progr., 44, 173, 1948; Sher-
wood P. W. Petrol. Process, 5, 1308, XII, 1950.
20. Schiller G. Chem. Fabrik, 11, 505, 1938.
21. Gerbert W. Erdol und Kohle, 9, 77—81, 1956.
22. Chem. Trade J. (London), 140, 1408, 1957; Oil a. Gas J., 55, 35, 119, 1957.
23. Sherwood P. W. Petrol., 18, 8, 296—300, 1955.
24. Third World Petroleum Congress the Hague, Proceedings 1951, Sect. IV, 4.
25. Tramm H. Chem. Ing. Techn., 24, 237—247, 1952; Erdol und Kohle, 5, 10—
17 1952.
26. Latta J. E., Walker S. W. Chem. Engng. Progr., 44, 173—176; 1948.
Third Petrol. Congress, 1951, Sect. IV, 42.
27. Clark. Ind. Engng. Chem., 41, 1527, 1947.
28. Chem. Engng., 54, 12, 19, 1947.
Глава II
МОНООЛЕФИНЫ
Физические константы газообразных, жидких и твердых представителей
ряда моноолефинов даны в табл. 14.
Важнейшими исходными материалами для нефтехимической промышлен-
ности являются газообразные при нормальных условиях или низкокипящие
олефины. К ним относятся: этилен, пропен, бутены (бутен-1, цис- и транс-
бутены-2), изобутен (2-метилпропеп) и пентены, такие как пентен-1, цис-
и транс-пентены-2 и 2-метилбутен-3, которые известны также под наз-
ванием амиленов. Спрос на эти олефины исключительно велик и будет воз-
растать еще более с развитием нефтехимической промышленности. Как
пример можно привести данные статистики США [1] о предполагаемом
росте спроса на олефины в ближайшие годы (табл. 15).
Эти олефины содержатся в большом количестве в крекинг-газах; нахо-
дятся они там в качестве побочного продукта. Первоначально эти газы
были относительно богаче этиленом. С совершенствованием крекинг-про-
цесса содержание этилена в продуктах крекинга уменьшается и вслед-
ствие этого затраты на его извлечение постоянно возрастают. Это выну-
ждает к поиску иных источников получения этилена и других газообразных
олефинов. Таким является прежде всего пиролиз природного газа, содер-
жащего пропан, который при этом расщепляется на этилен и метан. Затем
следует приобретающий первостепенное значение процесс пиролиза этана.
При нагреве до высокой температуры этан расщепляется на этилен и водород
(термическое дегидрирование).
Термическое дегидрирование высших парафиновых углеводородов,
как пропан или бутаны, с образованием олефинов, имеющих равное с исход-
ным углеводородом число атомов С, или вообще невозможно или протекает
с очень малыми выходами, так как сопровождается обычно крекингом.
Однако возможно дегидрирование каталитическим путем — пропусканием
сырья над смешанным катализатором (окись хрома — окись алюминия) при
температуре около 500°.
Кроме получения олефинов термическим дегидрированием этана и
крекингом, вернее каталитическим дегидрированием пропана и бутана,
возможен еще пиролиз высокомолекулярных углеводородов, таких как
тяжелый бензин и газойли. Этот пиролиз протекает со значительным образо-
ванием кокса. Чтобы уменьшить затруднения, связанные с образованием
кокса, имеются три пути:
1) пиролиз в присутствии водяного пара в определенном процентном
содержании, что заметно снижает коксообразование;
2) пиролиз в присутствии сильно нагретых каменных шариков, на кото-
рых отлагается слой кокса, выгорающий при последующем нагреве —
Пебблес — Хитер-процесс (Pebbles-Heatep-Proze|3);
3*
35
Табл и ц а 14
Физические константы важнейших моноолефииок
()леф'1 и Температура Mi п сипя np'j 7(j;l .и.и рт. ст. 3'м,'|иаи1К'т I Хцелъпш "г во с Ilpli 20'' 1 " 1.)
Отплеи -103,<3 — 169,5 1),5699 (—103,8°) 1,363 (-1()oi).
Пр шоп .... --47,7 — 185,3 0,6095 (-47,4°) 1,3540 ( —50°)
/<-1 >у теп-1 . -6,28 i —1 85,3 0,6.356 ( — 15,1°) 1,3791 (—25°)|
дис-Вутсн-З + 3, 72 —138,9 0,6447 (0°) 1,3783 (0°)
тринс-Буп'П-'Л + 0,88 —105,5 0,626!) (0°) 1,3695 (0°)
Изобутен -6,9 —140,35 0,6398 (—20°) 1,3776 (—20°)
Пентен-1 +29,97 — 167.2 0,6 3)5 1,3718
[еиген-2 . + 37,1 - 151,3 0,6554 1,3828
/при/ic-lleu ген-2 36,36 -140,2.3 0,6482 1,3798
2- метплбутен-1 31,16 -137,5 0,6504 1,3.778
3-метилбу ген-1 20,1 —168,5 0,6332 1,3640 ।
2-метилбутеи-2 38,6 —13.3,6 0,6625 1,3874 1
Гексон-1 63,5 —140,0 0,67:12 1,3878 1
((.ш;-Гсксеп-2 68,8 -1.41,8 0,6969 1,3976
тртис-Гоксои-2 67,5 —13.3,5 0,678(1 ' 1,39.15
ifuc-Гсксен-З 66,5 -137,8 0,6794 1.3951
тп/икс-Гексен-З 68,0 -113,4 0,6778 1,3937
Гептен-! . 9.3,8 —119,6 0,6393 1,3998
+ 7а.р<шс-Гептеп-2 . . . 98,2 - 0,7034 1,4041
(fnc-Гептеп-З ...... 95,8 - 0,70:10 I 1,4059
траис-Гептои-3 95,68 -1.36,3 0,6981 1,4043 1
Октон-1 121.3 — 102,7 0,711'0 1,4086
гцтс-Октеп-2 125,6 —100,5 0,7243 1,4146
траи.с-Октеп-2 12-1,5 —87,8 0,7184 1,4132
ifHC-Октои-З 122,7 -126 0,7223 1,4140
тря/.с-Октен 3 123,:', -100,0 0,7149 1,4129
1(ис-Онтс‘п-4 122,5 — 118,1 0,7212 1,4150
тр'«ис-Октен-4 +2+,:>> -9.3,8 0,71:16 1,4120
Ноиоп-1 146,8 - 81,6 0,7292 1,4157
Но поп-2 149,4 — 149,9 j - (1,7407 1,4201
Нопеп-З 148,2(165 .п.и) 0,7334 1,4191
Иопеп-4 148,0 — 0,7343 1,4200
Децеи-1 170,57 — 0,7313 1,4200
Децеп-З 173—1.73,5 — 0,7447 1,4221
Децен-4 170,6 — 0,7404 1,4243
'(•'"'-Доцоп-5 .... . . 169,5 (739 ,и,п) — 112 0,7445 1,4252
7»р,/«с-Децо1Г-5 । 170,2 —73 i 0,7412 1,42:15 I
Уидецон-1 192,67 — 49,1 0,750.33 1,4261
Уидецеп-5 192,2 0,7511 1,4289 1
Додецеи-1 213,3 —33,6 0,7583 1.4300
Додецон-6 212,9 (745 мм) — 0.7597 1,4328
Трпдецен-1 104 (10 мм) —22,2 0,7654 1,4335
Тетрадсцон-1 12.) (14 мм) — 0,7726 1,4367
Пента децеи-1 1.33,5 (10 мм) -3,8 0,7763 1,4389
Гексадецен-1 155 (15 мм) + 4,0 0,7811 1,4412
Геитадецеп-1 159 (10 мм) + 10,7 0,7860 1,44.36
Октадецен-1 176—78 (14 мм) + 18,0 0,7888 1,4450
Эйно.чеп-1 1.51 (1,5 мм) + 28,b — 1,4440 (30°)
Геиейкоиоп-1 134 (0,04 мм)- 0,7985 1,4510
Триакоитеи-1 213 (0,5 .нл) + 62—63 0,7620 (Ю0°) —
3) работа по способу катарол-процесс (Catarol-Prozef)), пиролиз высоко-
кипящих углеводородов (тяжелый бензин или газойль) в присутствии ката-
лизатора и при низкой температуре, прп которой коксообразование ые про-
является в такой степени. Он состоит в том, что фракция тяжелого бензина
пропускается над медной стружкой, причем пиролиз идет в сторону образо-
вания олефинов.
При всех процессах пиролиза газообразных или жидких углеводородов
образуется как побочный продукт жидкость, содержащая до 60% аромати-
36
-
ческих углеводородов. Выделение этих соединений является в большинстве
случаев дорогим процессом. Однако можно долю ароматических углеводо-
родов в продуктах пиролиза
значительно повысить, если
вести процесс при очень вы-
соких температурах, по это
связано с потерей газообраз-
ных олефинов. Катарол-про-
цесс позволяет наряду с газо-
образными олефинами получать
жидкую часть пиролиза, со-
стоящую па 95% из ароматп-
Таблица 15
Предполагаемый рост потребности в олефинах
в США в 1960 и 1975 гг. (в тыс. т)
Олефин 1955 г. I960 г. 1975 г
Этилен 1608 2356 4711
Пропел 707 925 1614
Бутоны 988 1121 2129
ческих углеводородов.
Таким образом, в настоящее время в нашем распоряжении имеется
олефиновое сырье из двух источников: 1) из крекинг-газов и 2) из газов
пиролиза газообразных парафиновых углеводородов и высококипящих
углеводородных смесей.
I. ОТХОДЯЩИЕ ГАЗЫ НЕФТЕПЕРЕГОННЫХ УСТАНОВОК
Под отходящими газами нефтеперегонных установок понимаются все
газы, выделяющиеся с установок прямой перегонки сырой нефти, крекинг-
установок и установок риформинга. В зависимости от типа и режима работы
установок состав отходящих газов меняется в чрезвычайно широких пре-
делах. Приблизительно 25—30% этих газов приходится па долю установок
прямой перегонки нефти. Остальное — крекинг- и риформииг-газы. Ниже
приведен типичный состав газа с установки прямой перегонки нефти [2].
Компоненты
% мол. j Компоненты
il .___
мол.
Инертные (N2, 0.2, СО) . .
Водород................
Метан..................
Этилен ...............
Этап ..................
Пропен ................
4,1
6,1
39,1
7,3
17,5
8,9
Пропан ...............
Путаны ...............
Пентаны...............
i Сероводород .........
Углекислый газ . . . .
9,4
2,6
1,4
3,0
0,6
Для нефтехимической промышленности в первую очередь представляет
интерес содержание олефинов в газах крекинга и риформинга. Ниже мы
коротко рассмотрим процессы, в которых эти газы образуются.
П. КРЕКИНГ-ПРОЦЕСС
А. ОБЩИЕ СВЕДЕНИЯ
Крекинг-процесс служит для превращения высококипящих (выше
температуры кипения бензина) составных частей нефти в смесь углеводоро-
дов, кипящих в интервале, типичном для бензиновых фракций. В прин-
ципе возможны два различные вида крекинга — термический и каталити-
ческий.
Термический крекинг также может быть двух видов: крекинг в паровой”
или в смешанной фазе. В газофазном, или, как более принято говорить,
парофазном крекинге, иысококипящие составные части нефти (чаще всего
фракция, выкипающая в пределах 200—400°, пли газойль) подвергаются
расщеплению при высокой температуре, низком давлении, в течение корот-
.37
кого времени. В крекинге в смешанной фазе процесс идет под давлением
в течение более длительного времени и при менее высокой температуре,
чем в парофазном процессе.
Так как при парофазпом процессе образуется очень много (до 30% вес.)
газообразных побочных продуктов, богатых олефинами, которые в прошлом
не находили применения, был разработан процесс, протекающий в смешан-
ной фазе, под давлением, способствующим тому, что олефины в большей
части претерпевают термическую полимеризацию, превращаясь в жидкие
продукты.
К термическому крекингу относится также так называемый риформинг.
Его назначение — улучшение аптидетонационных качеств (октанового
числа) тяжелой бензиновой фракции прямой перегонки путем нагрева под
давлением до 550—600°.
Каталитический крекинг, в котором пары нефтепродукта при темпера-
туре около 500° и выше пропускают над катализатором, состоящим в основ-
ном из алюмосиликатов, дает в результате примерно столько же бензина,
как и термический крекинг. Преимущество каталитического процесса в более
высоких антидетонационных свойствах получаемого бензина.
Б. ТЕРМИЧЕСКИЙ КРЕКИНГ
Крекинг-процесс в общем включает не только реакции расщепления,
в которых под влиянием теплового воздействия образуются смеси низко-
молекулярных углеводородов, но и реакции, приводящие к образованию
смесей углеводородов, кипящих при более высокой температуре, чем исход-
ный материал, и богатых ароматическими углеводородами. Таким образом,
суммарный эффект крекинга измеряется пе только образованием низко-
кипящих продуктов в результате расщепления исходного сырья, по также
и количеством вновь образовавшихся продуктов, кипящих при температурах
более высоких, чем исходное сырье и являющихся результатом реакций
конденсации.
Образование низкокипящих продуктов при крекинге можно рассма-
тривать как результат первичных реакций. Они протекают по следующей
схеме:
RCH=CH2-]-R1CH2—СН3
2R—СН2—СН2. . . СН:—СН2—Rx<^
RCH2—CJX.j-|-R1CEI=C[I2
Реакции эти идут тем быстрее, чем выше температура. При равных
температурах глубина разложения зависит от продолжительности теплового
воздействия.
Процесс, при котором образуются более высоко кипящие продукты,
чем исходное сырье, можно рассматривать как результат вторичных реакций
при крекинге. В результате этих вторичных реакций по большей части
и идет образование кокса. Образование кокса при крекинге в общем тем
больше, чем тяжелее исходное сырье. Это связано с повышенным содержа-
нием ароматических углеводородов в сырье и, следовательно, с его обедне-
нием водородом, что ведет к образованию высококонденсированных, не рас-
творимых в углеводородах веществ. Кокс не является чистым углеродом —
он содержит еще некоторое количество водорода и летучих соединений.
С другой стороны, крекинг идет тем труднее, чем ниже пределы выкипания
фракций. Поэтому, если очень широкая фракция подвергается крекингу
в условиях, обеспечивающих расщепление ее наиболее низкомолекулярной
части, то одновременно более высококипящая часть ее, расщепляясь, дает
много кокса. Чтобы этого избежать, необходимо крекинг-сырье предвари-
тельно разделять на фракции, кипящие в относительно узких пределах,
и каждую из фракций подвергать крекингу в наиболее подходящих для
нее условиях (селективный крекинг).
38
К селективному крекингу может быть отнесен и так называемый легкий
крекинг или висбрекинг, заключающийся в том, что высококипящий остаток
перегонки нефти, большая вязкость которого препятствует его исполь-
зованию в качестве котельного топлива, подвергается термическому раз-
ложению. Последнее осуществляется в условиях, исключающих образование
кокса; при этом образуется небольшое количество бензина, но вязкость
продукта значительно понижается.
Селективным крекингом является и названный выше риформинг, кото-
рый проводится при значительно более жестких условиях температуры
и давления, чем крекинг га-
зойля, потому что тяжелый
бензин кипит при значительно
более низкой температуре.
При использовании в каче-
стве сырья для крекинга га-
зойля, выкипающего в преде-
лах 200—400°, можно за один
проход сырья через крекинг-
установку получить около 30%
бензина без заметного образо-
вания кокса. Крекинг более
высококипящего сырья может
дать лишь около 20% бензина.
Выход бензина при висбрекин-
ге составляет всего 5—10%.
I
Рис. 14. Схема термического крекинга с рабо-
той на жидкий остаток.
Практически термический
крекинг осуществляется сле-
дующим образом: подлежащий
крекингу исходный материал
поступает в трубчатую печь,
2 — трубчатая печь; 2 — испарительная камера; 3 —
фракционирующая колонна; 4— газовый сепаратор.
Линии: I — исходное сырье; II — крекинг-гааы; III —
крекиш'-бензин на стабилизацию; IV — мазут (остаток);
V — продукты циркуляции обратно в крекинг-уста-
новку.
стальные трубы которой нагреваются непосредственно пламенем сжигаемого
в форсунках жидкого топлива, в печи продукт нагревается до необходимой
для крекинга температуры, приблизительно до 500—600° [3]. После нагрева
до указанной температуры продукт из печи поступает в реакционную ка-
меру, где он остается некоторое время, необходимое для реакции крекинга,
при той же температуре. Далее продукт поступает в испаритель, где в боль-
шей части испаряется, а легко коксующийся остаток удаляется из низа испа-
рителя (крекинг-мазут). В современных установках (рис. 14) крекинг пол-
ностью протекает уже в трубчатой печи, что делает реакционную камеру из-
лишней. В этих установках продукт из трубчатой печи поступает непосред-
ственно в испаритель. Отделившийся в нем остаток в количестве, примерно
равном количеству крекинг-бензина, применяется как котельное топливо.
Испаренные в испарителе продукты крекинга направляются в ректифика-
ционную колонну, работающую при том же давлении, что и испаритель.
Там они разделяются на газ, крекинг-бензин и высококипящую часть.
Последняя возвращается на крекинг (рециркулят). Этот вид термического
крекинга определяется как крекинг-процесс с работой на жидкий остаток.
В этом процессе кокса образуется очень немного и возможен длительный,
безостановочный пробег установки. После примерно трехмесячного пробега
установки требуются ее остановка и очистка от кокса трубчатой печи и
других элементов.
Во втором способе термического крекинга под давлением предусматри-
вается работа до образования кокса [4]. Способ состоит в том, что склонный
к образованию кокса остаток, включающий все кипящие выше температуры
кипения бензина составные части, из испарителя возвращается в крекинг-
процесс. Реакционная камера в этом случае выполняется так, чтобы обеспе-
чить возможность очистки ее от отлагающегося в ней кокса. Продуктами
крекинга являются здесь бензин, газ и кокс. В качестве исходного сырья
39
применяются продукты, особенно склонные к образованию кокса, перера-
ботка которых по другому варианту оказалась бы затруднительной. Выход
бензина и газа при этом способе работы выше, чем при крекинге до жидкого
остатка. На рис. 15 дана схема установки, работающей до коксового
остатка.
Интерес к крекингу с
образованием кокса значи-
тельно вырос за последнее
время. Некоторые остатки
от перегонки нефти, пред-
ставляющие интерес лишь
как котельное топливо, при
использовании в качестве
исходного сырья для этого
вида крекинга дают в ре-
зультате бензин и газойль
(исходный материал для ка-
талитического крекинга), а
Рис. 15. Схема термического крекинга с работой. также кокс .
ла коксовый остаток. В последнее время иро-
1— трубчатая печь; 2 — камера коксования; .3 — фрак- блема КОКСОВапНЯ решается
циопиругощая колонна; 4 — холодильник; 5 — газовый
сепаратор. также при помощи метода
«Линии: I — исходное сырье; II крекинг газы; III кокслрлтгтгя р пррвпллжтгжртт—
иренииг-бевзин стабилизацию-, IV —гоне. КОкСОВаиид в иссвдоижн/ксн
ном слое 151.
Парофазпый крекинг как способ получения бензина играет в настоя-
щее время очень ограниченную роль.
Для термического риформинга применяется в большинстве случаев
тяжелый бензин с началом кипения выше 100°, так как более низкокипя-
щие составные части бензина прямой перегонки сами по себе обладают
удовлетворительными антидетопацпоипымп свойствами. Процесс идет при
температуре 550—600°, давлении 17 — 70 ат, при продолжительности 40—
45 сек. [6].
В. КАТАЛИТПЧ Г; 1.11 (I КРЕКИНГ
При каталитическом крекинг-процессе, как п при термическом, обра-
зуются газ, бензин и высококпиящие продукты. Склонные к коксообразова-
пию, богатые ароматическими углеводородами продукты конденсации отла-
гаются па поверхности катализатора, который через определенные проме-
жутки времени регенерируется. Жидкий остаток (мазут) в этом процессе
не отделяется.
Каталитический крекинг отличается двумя важными особенностями .
Во-первых, получаемый этим способом бензин, как уже указывалось выше ,
по антидетонационпой стойко сти и по химическому составу значительно
лучше бензина термического крекинга при одном и том же исходном сырье .
Во-вторых, образующийся при каталитическом крекинге газ содержит зна-
чительно меньше метана и фракции Са и очень богат углеводородами с 3.
4 и 5 атомами углерода. Превращение за один проход через крекинг-печь,
здесь может быть значительно выше, чем при термическом крекинге, вслед-
ствие меньшего образования кокса.
В каталитическом крекинге применяются природные или синтети-
ческие катализаторы. В качестве природных катализаторов используется
отбеливающая земля типа монтмориллонита, активированная соляной кис-
лотой. Синтетический катализатор состоит примерно из 10% окиси алюми-
ния и 90% кремневой кислоты. Каталитический крекинг имеет еще и другие-
преимущества перед термическим. Процесс может идти или с неподвижным
(процесс Гудри) [7] или с подвижным катализатором. В последнем способе-
может применяться гранулированный или пылевидный катализатор [8].
Важнейшим способом каталитического крекинга является каталитический
40
т —— w
Рис. 16. Схема каталитического
крекинг-процесса методом «флюид».
1 — шибер; 2 — колосник; 3 — регене-
ратор; 4 — циклоп; -5 — реактор.
Лш(Нй’. I — исходный продукт; II — ре-
генерированный катализатор; III — отхо-
дящие газы; IV — отработан ный it.it.ijiи -
затор', V — пар; I I —продукты крешпп л
на установку для фракционировании;
VII—свежий катализатор; V 111 — воздух.
крекинг в псевдоожиженном слое [9]. Катализатор, как и в описанном выше
гидрокол-процессе, находится в пылевидном состоянии. Так как при этом ме-
ханически он ведет себя подобно жидкости, то и обращаться с ним можно как
с жидкостью, почему его называют также текучим катализатором. В реак-
ционную зону катализатор вносится парами перерабатываемого сырья и
остается в этой зоне в течение времени, необходимого для достижения желае-
мой глубины крекинга. После этого пары вместе с катализатором поступают
в пылеотделитель. Из пылеотделителя пары углеводородов направляются
в конденсатор, а катализатор в специальный регенератор, где выжигается от-
ложившийся па нем кокс, после чего
катализатор возвращается в процесс. Про-
цесс протекает непрерывно.
На рис. 16 показана схема установки
для крекинга с псевдоожиженным ката-
лизатором.
Г. КРЕКИНГ-ГАЗЫ
Количество и состав крекинг-газов
зависят в первую очередь от того, об-
разовались ли они в результате терми-
ческого или каталитического крекинга,
а также от температуры и давления,
при которых осуществлялся данный про-
цесс. Имеет значение и продолжитель-
ность пребывания продукта в печи.
Из крекинг-установки продукты кре-
кинга поступают в ректификационную ко-
лонну, где разделяются на газ, бензин и
остаток. Поел едини в большинстве слу-
чаев возвращается в крекинг-процесс как рециркулят. В зависимости
от того, при каком давлении работает ректификационная колонна, даль-
нейшая переработка газа и бензина осуществляется различными путями.
При термическом крекинге под высоким давлением (см. рис. 14) главная
ректификационная колонна работает в большинстве случаев под давлением
3,5—7 ат. При этих условиях и газ и бензин отходят из головной части
колонны. После конденсации и понижения давления отходят примерно 2/3
газа (не растворимая в бензине часть). Этот газ промывается далее в усло-
виях противотока абсорбционным маслом, поступающим из средней
части главной ректификационной колонны. При промывке из газа выделя-
ются еще ценные высокомолекулярные составные части. Выходящие из
абсорбера газы называются бедными или тощими газами. Обогащенное цен-
ными углеводородами промывочное (абсорбционное) масло возвращается
в ректификационную колонну.
Бензин, содержащий еще определенное количество высокомолекуляр-
ных газообразных алифатических углеводородов, как пропан, пропен, бу-
таны и бутены, в первую очередь должен быть освобожден от углеводородов
С3, чтобы упругость паров бензина снизить до требуемого значения. Газо-
образные парафиновые углеводороды для достижения определенной вели-
чины упругости паров бензина удаляются обычно из крекинг-бепзипов под
давлением около 25 ат в описанной выше стабилизационной колонне. При-
менение для стабилизации бензина столь высокого давления объясняется
желанием обеспечить конденсацию головных фракций углеводородов при
обычном водяном охлаждении. Выделяющиеся при стабилизации бензина
газы составляют примерно Дз всех образующихся при крекинге газов и
содержат 2/3 всех образовавшихся углеводородов с 3 и 4 атомами углерода
в молекуле. На рис. 17 представлена схема переработки крекинг-газов при
термическом крекинге иод давлением.
41
В табл. 16 дан средний состав бедного газа после абсорбера и газов
стабилизации.
Стабилизация бензина может проводиться также в двух колоннах.
В первой колонне — депропанизаторе — отделяется фракция С3, а во вто-
I! V
Рис. 17. Схема переработки продуктов тер-
мического крекинга под давлением.
1 — фракционирующая колонна; 2 — холодильник;
3 — газовый сепаратор; 4 — абсорбер.
Линии: I — крекпнг-продунты из сепаратора; II —
газ -|- бензин; III — абсорбент; IV — газ; V —
бедный газ из абсорбера; VI — крекинг-бензин па
•стабилизацию; VII — абсорбент, насыщенный га-
зообразными углеводородами; VIII — остаток обрат-
но на крекинг-установку.
рой—дебутанизаторе—фракции С4<
Депропанизатор работает при
25 ат, дебутанизатор — при 7 ат.
В условиях парофазного кре-
кинга, в частности каталитиче-
ского, обработка газа осущест-
вляется практически тем же спо-
собом {10]. Так как в главной
ректификационной колонне под-
держивается лишь совсем неболь-
шой избыток давления, отходя-
щие газы увлекают с собой боль-
шое количество бензина. Поэтому
необходимо особое компримирова-
ние газа, сопровождающееся ожи-
жением части его. Газ и жид-
кость затем охлаждаются и на-
правляются далее в разделитель,
где газ отделяется от жидкости.
Окончательно газ обрабатывается
уже описанным выше способом.
Таблица 16
Состав крекинг-газов термического крекинга газойля
% мол.
Компонент Бедный газ из абсор- бера Газ с установки стабилизации
из депропа- низатора из дебутани- затора
Метан Этилен Этан Пропен Пропан Бутены Бутаны Пентан -{- высшие 58,20 6,71 13,09 6,05 10,62 2,06 2,08 1,19 11,01 5,98 19,10 18,38 5,67 4,89 4,97 0,0 1,0 н-Бутен 32,6 Изобутен 18,4 48,0
Всего олефинов. . 14,82 29,95 51,0
а) Состав крекинг- и риформинг-газов
Средний выход газов, включая фракцию С4, при различных про-
мышленных способах крекинга показан ниже.
В табл. 17 дано сравнение
количеств отдельных составных
частей газа в весовых процентах,
отнесенных к исходному сырью,
при различных типичных про-
мышленных способах крекинга,
а в табл. 18 состав тех же газов
в объемных процентах [И].
Термический крекинг-про-
цесс, как уже указывалось, может
Крекинг Выход газа, % вес. от исходного сырья
В смешанной фазе . . . Парофазный Термический риформинг Каталитический .... 10—12 20—25 15—20 8-12
42
Таблица 17
Типичный выход отдельных составных частей газа в различных
процессах крекинга в % вес.
Компонент Термиче- ский кре- кинг газойля Термиче- ский рифор- минг бен- зина Каталитиче- ский крекинг газойля
Водород 0,03 0,1 о,1 1,0
Метан 1,0 3,3
Этилен 0,2 1,0 0,6
Этан 1,5 4,1 0,7
Пропей 0,9 3,6 3,6
Пропан 2,0 4,9 1,2
Бутены 1,0 4,1 4,3
Бутадиен 0,1 0,2 0,1
Бутаны 2,6 4,3 2,2
Всего 9,33 25,6 13,8
Всего олефинов 2,2 8,9 8,6
Таблица 18
Средний химический состав крекинг-газов отдельных
типичных процессов (в % объемн.)
Компонент Смеша- нофаз- иый про- цесс Парофаз- пый про- цесс Термиче- ский ри- форминг Катали- тический крекинг
Водород 3 7 7 7
Метан . 35 30 40 18
Этилен 3 24 4 5
Этан 20 12 18 9
Пропен 7 14 6 16
Пропан 15 4 10 14
Изобутан 2 1 3 16
и-Бутан 8 2 7 5
Бутены 7 6 5 10
осуществляться в смешаной или в паровой фазах. Соответственно этому раз-
личается количественный и качественный состав газов, образующихся в этих
условиях. В табл. 19 приведено сравнение состава газов смешанофазного
и парофазного процессов.
В табл. 20 показаны количества газа в весовых процентах к исходному
сырью, образующегося при каталитическом крекинге газойля [12].
Табл. 21 показывает среднее различие в составе газов каталитического
и термического крекинга.
При каталитическом крекинге прежде всего образуются углеводороды
'СЛ, что видно и из таблицы.
Газы термического риформинга близки по составу к газам термического
смешаного крекинга, газы же каталитического риформинга не предста-
вляют никакого интереса для химической промышленности, что видно из
данных табл. 22 [12].
43
Таблица 2С
Таблица 19
Состав крскинг-газов нарофазпого и
смсшанофазного крекинга (в % объемы.)
Компонент Смешанофаз- Нып крекинг Парофазпып Крекинг
Водород + метан . . . 31,2 45,0
Этап 1.2,4 17,7
Пропап 4,7 11,0
Б утаны 3,0 5,4
Сумма парафиновых углеводородов . . . 51,3 79,1.
Этилеп 23,0 3,7
Пропен 15,7 9,1
Бутены 8,0 3,6
Сумма олефинов . . 46,7 16,4 i
Типичные выхода газа
при каталитическом крекинге
средневосточного газойля
Компонент Выход, % вес. от исходного сы рья
| газ 1 I газ 2
Сероводород 0,5 0 8
Водород 0,1 0,1
Метан 0,9 0,7
Этилен 0,5 0,4
Этан 0,8 0,7
Сумма Со и более легких 2,3 1,9
Пропен 2,4 3,3
Пропан 2,3 1,2
Сумма С, 4,7 4,5
Бутены 3,3 5,2
Пзобутаи 4,4 2,6
н-Бутап 1,3 0,9
Сумма С4 9,0 8,7
В сего | 16,0 1 15,1
Табл и ц а 21
Сравнение термического крекинга под давлением и каталитического
крекинга в отношении количества и состава газов
Выход газа от исходного продукта
Компонент термический крек ннг
% объемы. % вес.
каталитический
крекинг *
% объемн. % вес.
С3 п легче ... 12,5 14,3
Изобут ап 1,4 0,9 I 13,2 8,4 ।
Бутен ы 3,4 2,3 6,0 8,7 6,0 16,9
н-Бутап 4,2 2,8 J 3,8 2,5 1
• Синтетический катализатор 81Ог + А12О3
Табл и ц а 22’
Состав трех газов каталитического риформинга!
кувейтского тяжелою бензина (% объемн.)
Компонент Гач 1 Газ 2 Газ 3
Водород 0.5 0,5
Метан 0,3 0,5 2,8
Этан 1,1 1,8
Пропан 3,4 5,2 5,2
Бутаны 5,0 8,1 2,6
С6—250° 89,7 83,9 89,4
44
б) Газ стабилизации
Как выше указывалось, газы стабилизации содержат большую часть
всех образующихся при крекинге углеводородов С3 и С4, а потому имеют
исключительно большое значение для нефтехимической промышленности как
сырье для получения пропена и бутенов. Средний состав газов стабилизации
смешанофазного и каталитического крекинга приведен в табл. 23.
Таблица 23
Средний состав газов стабилизации бензинов
счешанофазиого п каталитического крекинга (в % объемн.)
Смешано- | Каталитиче-
Компо пент фазный СК ИЙ
крекинг крекинг
Метан 10 8
Этплеп 2 9
Утан 15 6
Пропен 16 17
Пропан 26 14
Изобута п Л 29
//-Бутан 14 7
Бутопы 12 15
Высшие углеводороды 2 2
в) Бутан-бутеновая фракция (Б-Б-фракция)
Бутан-бутеповая фракция выделяется в дебутанизаторе. В зависимости
от вида процесса крекинга среднее количество фракции С4 изменяется сле-
дующим образом.
Из этих цифр следует, что
при каталитическом крекинге,
проводимом при очень высокой
температуре, преобладает фрак-
ция С4 [13].
Состав фракции С4, отходя-
щей из дебутанизатора при паро-
фазном крекинге, следующий (в %
объемн.).
Крекинг-процесс Количество С4, % вес.
Смешапофа.!иый 4—6
Термический риформинг . . . 4-6
Парофазпым 6-8
Каталитический 6-8
Каталитический крекинг при
повышенной температуре . . 20
Бутаны................10—12 | Бутадиен.................12—14
Изобутен.............. 20—24 I Углеводороды Cs и С5 2
/i.-Бутен .............50—55
Среднее распределение углеводороде]! С4 в Б-Б-фракцип каталитиче-
ского крекинга, дано ниже (в % объемн.).
Изобутан..............30—50 I Изобутен................ 10
/(-Бутан ...10—15 I /(-Бутен ...........................30—45
III. ВЫСОКОМ!).ПЕКУЛЯРНЫЕ ОЛЕФИНЫ В ПРОДУКТАХ КРЕКИНГА НЕФТИ
Продукты крекинга нефти не имеют значения как источник получения
высокомолекулярных олефинов. Эти олефины очень неодинаковы по своей
природе и едва ли могут быть использованы практически.
В табл. 24 дано содержание ароматических, олефиновых, нафтеновых и
/парафиновых углеводородов в различных крекппг-бонзинах [13, стр. 254].
45
Таблица 24
Химический состав различных крекинг-бензинов с концом кипения 204°
Крекинг-процесс Исходное сырье Состав, % объемн.
арома- тиче- ские оле- фины наф- тены пара- фины
Смешанофазный Парафиновый газойль . . 8 39 16 37
Смешанофазный Нафтеновый газойль . . . 15 30 25 30
Термический риформинг Смесь тяжелых бензинов . 15 20 30 35
Паро фазный Газойль 20 45 15 20
Каталитический, 454° Парафиновый газойль . . 15 16 23 46
Каталитический, 454° Нафтеновый газойль . . . 22 13 32 33
Каталитический, 525° Парафиновый газойль . . 40 5 19 36
Каталитический риформинг Смесь тяжелых бензинов. . 28 5 26 41
IV. ПРЯМОЕ ПОЛУЧЕНИЕ ОЛЕФИНОВ
А. ОБЩИЕ СВЕДЕНИЯ
В предыдущих разделах были рассмотрены газообразные и жидкие угле-
водороды, образующиеся при крекинг-процессе, и их состав. Теперь необ-
ходимо рассмотреть получение низко- и высокомолекулярных олефинов,
в процессах, где эти олефипы являются не сопутствующим, а целевым конеч-
ным продуктом. Крекинг-газы должны подвергаться химической перера-
ботке непосредственно на нефтеперегонном заводе или в крайнем случае на
близ расположенных химических заводах, так как их транспортировка обхо-
дится довольно дорого. С другой стороны, нефтехимическая промышленность,
стремится получать олефиновое сырье, и в первую очередь этилен, от пред-
приятий нефтяной промышленности. Способы, которые применяются для
получения олефинов, в технологическом отношении отличны от обычного,
крекинг-процесса, так как здесь уже не бензин, а газ является целевым,
продуктом.
В общем все процессы, позволяющие из низкомолекулярного или из.
высокомолекулярного исходного сырья получать газообразные олефины,,
основаны на процессах пиролиза, т. е. на процессах, при которых газ или
нефтяная фракция короткое время нагревается до высокой температуры,
предпочтительно в присутствии водяного пара.
Если углеводороды нагревают до 600° и выше, то говорят вообще-
о пиролизе. При нагреве до высокой температуры низкомолекулярных
парафиновых углеводородов, как пропан и бутан, идут две первичных
реакции.
1. Реакция крекинга, при которой парафиновые углеводороды превра-
щаются в парафин и олефин, причем сумма атомов С олефина и парафина-
равна числу атомов углерода в исходном углеводороде
r1ch2ch3-)-r2ch=ch2
2R1CH2CH2CH2CH2R2<^
RjCH=CH2+R2CH2CHs
2. Реакция дегидрирования, при которой от парафинового углеводо-
рода отщепляется водород с образованием двойной связи и сохранением
строения (остова) парафинового углеводорода:
СН3СН2СН2СН2СН3 -> сн3сн2сн=снсн3+н..
46
Удельное значение протекающих одновременно реакций крекинга в
дегидрирования зависит в первую очередь от числа атомов С в исходном
материале. В то время как этан при высоком нагреве превращается практик
чески только в этилен и водород и, следовательно, здесь в основном идет
реакция термического дегидрирования, при нагреве пропана уже большее
значение имеет реакция крекинга с образованием этилена и метана. При
нагреве бутана до высокой температуры образуется совсем немного бутена.
Бутан расщепляется главным образом на этилен и этан или, соответственно
на пропен и метан. Изобутан, напротив, примерно на 50% превращается
в изобутен.
Б. КАТАЛИТИЧЕСКОЕ ДЕГИДРИРОВАНИЕ
ГАЗООБРАЗНЫХ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ В ОЛЕФИНЫ
С РАВНЫМ ЧИСЛОМ АТОМОВ УГЛЕРОДА
Реакцию дегидрирования парафиновых углеводородов до гексана вклю.
чительно можно вести с хорошими выходами, если парафиновый углеводо-
род нагревать в присутствии соответствующего катализатора
СН3СН2СН2СНа —> СН3СН=СН—СН3-(-Н2—29,5 ккал]молъ
Таким катализатором является прежде всего смесь окиси хрома и окиси
алюминия (10% Сг20з, 90% А120з). Пропуская над подобным катализато-
ром, например, бутан, можно при однократном проходе получать более чем
90%-ный выход бутена и водо-
рода [14].
Реакция дегидрирования
сильно эндотермична. Для де-
гидрирования 1 моля бутана в
1 моль бутена и водород необ-
ходимо затратить 29—30 ккал.
В промышленных условиях
для полного превращения 1 кг
бутана требуется примерно
550 ккал. Подведение такого
большого количества тепла
представляет технически труд-
ную проблему. Для решения
ее имеется в принципе три
возможности. Во-первых, рас-
положение катализатора в труб-
ках, обогреваемых снаружи
газом (UOP-процесс) [15]. Во-
вторых, тепло, необходимое
Рис. 18. Схема каталитического дегидрироиа-.
ния я-бутапа.
г — трубчатый нагреватель; 2 — печь; 3 — компрессор;
4 — абсорбционная колонна; &—десорбционная нолоппа.
Линии: I — н-бутан; II — газы реактивации; III —
выход газов реактивация-, /V — водород 4- низшие угле-,
водороды; V — бутан-бутеновая фракция на экстрактив-
ную перегонку; V/—циркулирующее промывочное масло.
для дегидрирования, предварительно накапливается в реакторе таким
образом, что совместно с катализатором в зону дегидрирования вводится
некатализирующий материал, обладающий высокой теплоемкостью. Так
как катализатор для освобождения от коксовых частиц, делающих его
неактивным, время от времени подвергается регенерации путем выжига-
ния в струе воздуха, и при этом освобождается большое количество тепла,
то в дальнейшем тепло, приносимое катализатором в реактор, используется
для осуществления реакции дегидрирования. Но количество тепла, нако-
пленное при этом в катализаторе, вернее в теплоносителе, ограничено, по-
этому необходимо, чтобы процесс регенерации проходил за возможно корот-
кое время (7—15 мин.). В случае необходимости можно также в период
регенерации подводить к катализатору еще искусственное тепло (процесс
Гудри [16]).
В-третьих, одновременно с дегидрируемым углеводородом в реактор
подают высоко нагретый водяной пар, являющийся теплоносителем (про-
цесс Стандард-Ойл [17]). Этот процесс применяется также для дегидриро-
47"
нация этилбензола в стирол, а также н-бутаиа в бутадиен, с применением
устойчивого против действия водяного пара катализатора.
Продукт, получаемый при каталитическом дегидрировании бутана,
только в идеальном случае состоит из непрореагировавшего бутана, бутена
и водорода. Практически он всегда содержит некоторое количество низко-
молекулярных и высокомолекулярных углеводородов. Процесс идет так,
как показано на рис. 18: углеводороды абсорбцией маслом отделяются от
ле растворимого в масле водорода, вместе с которым отделяются и образо-
вавшиеся в результате крекинга низкомолекулярные осколочные, газообраз-
ные соединения. Выделенная затем из масла смесь бутена и бутана или при-
меняется как таковая или разделяется на парафиновую и олефиновую со-
ставные части. На процессе разделения методом экстрактивной перегонки
мы остановимся ниже.
11. ТЕРМИЧЕСКОЕ ДЕГИДРИРОВАНИЕ ЭТАНА В ЭТИЛЕН
а) Способ дегидрирования в трубчатой печи
Одним нагревом до высокой температуры (850—900°) этан расщепляется
па этилен и водород. Процесс проводится часто в трубчатых пёчах, выпол-
ненных из хромистой стали (25—30% Сг), содержащей некоторое количество
кремния. Этан пропускают через систему труб так, чтобы время его пребыва-
ния в системе составляло
1 сек. it ио выходе из си-
стемы он тотчас быстро ох-
лаждался, чтобы предотвра-
тить протекание вторичных
реакций (рис. 19). За один
проход через печь этан рас-
щепляется примерно на 50%,
так что в конечном продукте
этилена содержится пример-
но 39% объемн. [18]. Кроме
этилена образуется еще не-
большое количество ацети-
лена, который превращается
затем в этилен методом пар-
циального гидрирования над
хромо-пинелевым катализа-
тором (на схеме не показано).
Необходимый для гидрирова-
ния водород находится в
газовой смеси процесса де-
гидрирования. Этилен из
ние. I!). Схема получения этилена пиролизом этапа.
I — трубчатая печь; 2 — система быстрого охлаждения;
— компрессор; 4 — метапонам колонна; <5 — этиленовая
колошга; 6' — этаповая колонна.
Лшнш: I — свежий этап; И — водород и метан; III —
этилен; IV—-этан; V— жидкие продукты чромчтпчесгоп
природы; VI — цирк ул прующпй этан.
газов пиролиза выделяется, например, так, что газовая смесь подвергается
сжатию, а затем разделяется па установке Линде при таких температуре и
давлении, что водород и метан уходят через верх первой колонны. Затем
фракция С2 разделяется на этан и этилен. Этан возвращается на повторный
пиролиз. На разделении при помощи медно-щелочного раствора мы остано-
вимся ниже.
Технологически трудной проблемой является выбор материала змееви-
ков для установок дегидрирования. Здесь мы встречаемся с теми же трудно-
стями, как и при каталитическом дегидрировании, с той, однако, разницей,
что приходится работать с еще более высокими температурами. Материал
труб подвергается снаружи окисляющему, а изнутри восстанавливающему
действию и чрезвычайно напряжен. В связи с этим уже давно делались по-
пытки обойтись в процессе пиролиза без трубчатых печей. Некоторые опыты
в этом направлении рассматриваются позднее.
• 48
б) Способ автотермического дегидрирования этана в этилен [19]
Интересный способ, для осуществления которого требуется кислород,
состоит в том, что необходимое для дегидрирования тепло образуется за
счет сгорания части парафинового углеводорода в кислороде в самом реак-
торе. Реакционная печь для частичного сгорания этана выполняется из кера-
мического материала и относитель-
но проста по конструкции. Она
наполнена фарфоровыми тарами.
Способ этот, известный как автотер-
мическое дегидрирование, заклю-
чается в следующем (рис. 20).
100 объемных частей этана и
30 объемных частей кислорода после
прохождения через подогреватели
смешиваются при остаточном давле-
нии 410 мм, при этом происходит
Рис. 20. Схема автотермического деги-
дрирования этапа.
1 — подогреватель; 2 — печь; 3 — промывка га-
за; 4 — компрессор.
Линии: I — кислород; II — этан; III — на раз-
деление посредством перегонки; IV — вода; V —
пода и органические кислоты.
гз освобождается от небольших коли-
сгорание и температура повышается
приблизительно на 880°. Образуется
примерно 170 объемов газа, в кото-
ром содержится около 30 объемов
этилена. При последующей промывке
честв органических кислот. Сжатием до 17,5 ат газ освобождается от
высокомолекулярных углеводородов типа бензина и от двуокиси угле-
рода. Газовая смесь поступает затем на дальнейшую переработку перегон-
кой под давлением. Этилен получается 98—99 %-ной чистоты. Средний
состав газов автотермического дегидрирования (в % объемн.).
Этилон.......................30,5
Водород .....................26,0
Этап.........................16,5
Окись углерода..............11,1
Метан ........................7,5
Азот..........................4,3
Двуокись углерода.............1,1
Пропан......................0,9
Ацетилен ...................0,6
Пропел .....................0,5
Кислород....................0,5
Путай.......................0,2
Путей.......................0,2
Пелтаи и высшие углеводороды 0,1
Так как содержащийся в газе этан возвращается в реакционную печь,
общий выход этилена составляет около 67% от теоретического.
Аналогичный способ, при котором тепло, необходимое для эндотерми-
ческой реакции получения ацетилена из метана, образуется за счет сжигания
части газа в смеси с кислородом, так называемый Захсё-процесс, рассмотрен
ниже.
Г. ПОЛУЧЕНИЕ ГАЗООБРАЗНЫХ ОЛЕФИНОВ ПИРОЛИЗОМ
ГАЗООБРАЗНЫХ ИЛИ ЖИДКИХ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
а) Общие сведения
В предыдущих разделах были рассмотрены способы получения олефи-
нов дегидрированием парафиновых углеводородов без уменьшения числа
углеродных атомов в молекуле. Этан дегидрируется в этилен простым
нагреванием до высокой температуры, более высокомолекулярные углеводо-
роды, как пропан, бутан, пентан, дегидрируются каталитическим способом.
Высокомолекулярные парафиновые углеводороды — гексан, гептан и т. д. —
не могут быть превращены экономически приемлемым способом в олефины
с равным числом атомов С, так как в этом случае преобладают процессы
крекинга.
Получение этилена возможно технически сравнительно простым спо-
собом — крекингом при нормальных условиях таких газообразных пара-
финовых углеводородов, как пропан и бутан. Так, нагревая в течение корот-
4 Зана:: '>96. 49
кого времени до высокой температуры пропан, получают этилен и метан
с примесью пропена, из бутана получают этилен и этан с примесью пропена
и метана. Разделяя далее тем или иным способом продукты пиролиза, можно
выделить чистые этилен и пропен.
Низкомолекулярные парафиновые углеводороды, выделяемые в боль-
ших количествах из природного газа и отходящих газов нефтеперегонных
установок, были в течение длительного времени важнейшим исходным про-
дуктом для получения олефинов. По этой причине в США нефтехимическая
промышленность концентрируется в первую очередь в районах больших
газовых месторождений, например в Тексасе. В районах, где нет достаточ-
ного количества природного газа и газов крекинга, олефины можно полу-
чать пиролизом смесей жидких углеводородов нефти. Пиролиз жидких угле-
водородов можно проводить двумя способами: в одном способе процесс идет
в условиях, обеспечивающих максимальный выход олефинов и одновре-
менно высокоароматизированпой части, которая далее используется для
получения высокооктанового бензина. Ароматические углеводороды в чистом
виде в этом случае из продуктов пиролиза не выделяются. В другом способе
процесс направлен на получение жидких продуктов, практически целиком
состоящих из ароматических углеводородов. Последние легко выделяются
в чистом виде из продуктов пиролиза. Высококипящие нефтепродукты, на-
пример остатки прямой перегонки нефти, также могут подвергаться пиролизу
для получения олефинов в условиях, исключающих помехи, связанные с об-
разованием кокса.
б) Пиролиз газообразных парафиновых углеводородов (газовый крекинг)
В разделе рассматривается газовый крекинг или, иными словами, пиро-
лиз парафиновых углеводородов, газообразных при нормальных условиях.
Общие сведения. При высоком нагреве газообразных при нор-
мальных условиях парафиновых углеводородов температуры, вызывающие
те или иные изменения, могут быть различными в зависимости от числа ато-
мов С в молекуле. Метан, являющийся низшим парафиновым углеводородом,
устойчив до 500°. Разложение начинается примерно с 700°. Разложение
идет быстро при 1400° и выше с образованием радикалов. Отще-
плением водорода из метана образуются радикалы СН3—, СН2= и СП~, кото-
рые могут соединяться в этап, этилен и ацетилен. Чем выше температура,
тем больше получается радикалов СН= и тем значительнее образование аце-
тилена. Этот вопрос ниже рассматривается подробнее. Этан распадается при
кратковременном нагреве до высокой температуры на этилен и водород.
Этот процесс уже рассматривался нами выше. При нагреве пропана наряду
с реакцией термического дегидрирования происходит разрыв углеводород-
ной цепи с образованием метана и этилена:
СНа—Clio—СН3 —» CH2=CH2-|-CIIj—16,3 кксл/моль (крекинг)
СНЭ—СИ2—СН3 —> СН3—СН=СН2 -|-Нг—30,0 ккал/моль (дегидрирование)
Тогда как крекинг протекает по необратимой реакции, дегидрирование
представляет собой реакцию равновесную, которая в случае пропана тре-
бует для своего осуществления примерно вдвое больше тепла, чем крекинг.
Отсюда следует, что предпочтительно будет протекать реакция крекинга.
При высоком нагреве бутана могут протекать три различные первичные
реакции:
СН3—СН2—СН2—СН3 —> СН3—СН=СН2-)-СН4—17,6 ккал)молъ (крекинг)
СН3—СН.,—СП2—СП3 —> СН2=СП2-]-С113—СН3—17,0 ккал)моль (крекинг)
СН3—СН2—СН2—СП3 —> СМ3—СН2—СН=СН2-|-Н2—30,0 ккал/моль (дегидрирование)
50
Как следует из этих уравнений, первичные продукты пиролиза должны
возможно быстрее выводиться из зоны реакции, чтобы избежать вторичных
реакций.
Увеличение объемов при пиролизе газообраз-
ных парафиновых углеводородов. Если пропан в процессе
пиролиза на 100% превращается в метан и этилен или в пропен и водород,
то объем газа при этом увеличивается вдвое. Из 100 л пропана образуется
200 л продуктов реакции. Отсюда следует, что независимо от того, каково
удельное значение реакций крекинга и дегидрирования, всегда образуется
двойной объем продуктов реакции сравнительно с исходным. При 50 %-ном
превращении пропана из 100 л пропана образуется 150 л продуктов реакции.
Рис. 21. Температурная зависимость со-
става конечных продуктов пиролиза про-
пана.
J — общее количество олефинов; 2 — этилен;
3 — водород, 4 — пропен.
Рис. 22. Температурная зависимость со-
става конечных продуктов пиролиза
бутана.
I — общее количество олефинов; 2 — этилен;
з — пропен; 4 — бутен.
Только анализом газов можно установить, в каких размерах протекает кре-
кинг с образованием этилена и метана или соответственно дегидрирование
с образованием пропена и водорода.
Анализ газов пиролиза пропана и н-бутана в целях установления влия-
ния температуры при постоянном времени нагрева на протекание реакций
крекинга й дегидрирования выполнен П. К. Фролихом с сотрудниками
[20]. На рис. 21 показан состав продуктов ппролиза пропана, а именно про-
пена, водорода и этилена (метан не обнаружен), в зависимости от темпера-
туры. Можно видеть, что при 880° в газе содержится наибольшее количество
олефинов. Максимальное содержание пропела в газе наблюдается при темпе-
ратуре реакции 810°. До этой температуры содержание водорода в газе
эквивалентно содержанию пропена. Отсюда следует, что здесь происходит
чистая реакция дегидрирования. Выше 810° содержание пропена падает,
в то время как содержание водорода сильно возрастает, показывая этим, что
пропен претерпевает вторичную реакцию, сопровождающуюся освобожде-
нием водорода. Максимальная концентрация этилена достигается при 890°,
когда содержание его составляет около 30%.
При пиролизе бутана (рис. 22) наибольшее содержание олефинов на-
блюдается при значительно более низких температурах — при 690°. Это
указывает на меньшую термическую стабильность бутана. Наибольшая кон-
центрация этилена наблюдается уже при 750°. Реакция дегидрирования
4*
51
бутана с образованием бутена и водорода происходит в очень ограниченных
масштабах. Максимум дегидрирования наблюдается около 670°. Из практики
применения газового крекинга для получения олефинов следует, что пропан,
как исходное сырье более всего подходит для этой цели, так как фракция
С2 при этом практически нацело состоит из этилена, в то время как фракция
Сз представляет собой смесь пропена и пропана. Если хотят получать про-
стейшим способом чистый пропен, то используют бутан. В этом случае фрак-
ция Сз будет представлять собой смесь этана и этилена, а фракция Сз —
практически один пропен.
Промышленные способы газового крекинга [21 ].
В промышленности газовый крекинг, проводимый в основном для получения
этилена, осуществляется в большинстве случаев в трубчатых печах. Способ
работы в принципе такой же, как при термическом дегидрировании этана
(см. рис. 19). Сырье и газы циркуляции поступают в змеевик трубчатой печи,
обогреваемый снаружи за счет тепла от сгорания мазута или газа, сжигае-
мых в топке. Газовая смесь при этом быстро нагревается до температуры
пиролиза, а затем должна быстро охлаждаться, чтобы продолжительность
реакции была ограничена 1 сек. В большинстве случаев это достигается
прямым впрыском воды пли нефтяного масла. Далее газ компримируют,
обезвоживают, а затем подвергают фракционировке под давлением. Метан
и водород отходят как остаточные газы и могут быть использованы в качестве
топлива. Во второй колонне отделяется этилен. Газовая смесь из третьей
колонны, когда главной целью является получение этилена, направляется
обратно па крекинг в качестве газов циркуляции. Температура реакции лежит
менаду 780—800°. Время реакции 0,8—1,0 сек. Исходным сырьем для газо-
вого крекинга является, например, пропан, выделяемый из природного газа
как головной продукт депропанизации при стабилизации газолина (около
25% этана и 75% пропана). Также может быть использован содержащий
олефины газ с установок стабилизации крокинг-бепзипов.
В табл. 25 показаны условия, в которых в промышленности осуще-
ствляется крекинг этана и пропана. Там же показан состав продуктов.
Таблица 25
Пиролиз парафиновых газообразных углеводородов в трубчатой печи
Показатели Исходный продукт
этан этан пропан
Температура газов, выходящих из нагре- вателя, °C Давление, ат ................. 775 800 780
2,1 1,1 1,05
Продолжительность реакции, сок 1,05 1,5 —
Полнота превращения, % Состав продуктов, % объемн.: водород 38,5 71,1 —
26,0 40,8 9,4
метан 2,5 2,9 21,9
этилен 25,5 36,5 19,7
этап 45,2 17,2 0,5
пропен 0,2 0,6 24,5
пропан — 0,3 0,6
углеводороды С4 0,3 0,5 1,7
С5 + высшие углеводороды 0,1 0,7 10,5 J
Внутренняя поверхность змеевиков печи должна время от времени очи-
щаться от углистых отложений путем выжигания в смеси воздуха с водяным
паром.
52
Все здесь сказанное дает представление лишь о принципе работы. Можно
также отходящие газы нефтеперегонных установок перерабатывать непо-
средственно на этилен. При этом каким-либо способом выделяют метан и
водород, используемые затем как топливо. Оставшуюся высокомолекуляр-
ную часть разделяют на этан-этиленовую и пропан-пропеновую фракции.
Этан-этиленовую фракцию разделяют перегонкой на этан и этилен. Этан
подвергают пиролизу в специальной трубчатке, отдельно от фракции С3,
так как последняя для разложения требует несколько иных температурных
условий, чем этан. Переработка пиролиз-газа с обеих трубчатых установок
может производиться совместно с крекииг-газами.
Высокосортная сталь, являющаяся материалом для трубчатых нагрева-
телей печей, очень дорога и, кроме того, трубы требуют частой замены.
Стремление избежать применения трубчатых печей при крекинге привело
к широко применяемому в настоящее время способу нагрева в печах с твер-
дым (галечным) теплоносителем. Специально для газового крекинга разрабо-
тан процесс в печах с твердым теплоносителем фирмой Филлипс Петролеум
компани.
Газовый крекинг в печах с твердым теплоносителем [22]. Установка для
газового крекинга с твердым теплоносителем состоит из двух заполненных
огнеупорными шариками (камень, галька) цилиндров, расположенных один
над другим и соединенных огнестойкой трубой (рис. 23). Верхний цилиндр,
или подогреватель, служит для нагрева газами сгорания каменных шариков
диаметром около 1 см. Нижний цилиндр, называемый реактором, содержит
горячие шарики, поступающие сюда из подогревателя, и подвергаемый
пиролизу продукт. Они соприкасаются друг с другом в условиях противо-
тока. Пройдя реактор, шарики поступают в промежуточный бункер, откуда
они при помощи горячего воздуха транспортируются обратно в подогрева-
тель- Глубина крекинга контролируется температурой и удельным весом
крекипг-газов. Продукты пиролиза возможно быстрее охлаждаются и перера-
батываются далее, как указывалось выше.
В табл. 26 показаны результаты пиролиза бутана описанным способом
при температуре 943°. Из 100 кг бутана получают 44,1 кг этилена и 12,5 кг
пропена, когда степень превращения н-бутапа составляла 91%. При полном
превращении бутана путем повторного пиролиза выход этилена составил
48,4 кг и пропена — 13,3 кг.
Таблица 26
Состав газов пиролиза и-бутапа методом
Филлипс Петролеум
Компонент Исход- ный про- дукт, % мол- Газ пи- ролиза , % мол.
Водород 16,6
Метан — .30,5
Ацетилен — 1,2
Этилен .— 37,2,
Этап — 2,0
Пропен — 6,7 .
Пропап 0,3 0,1
Бутадиен — 0,6
Бутены .— 9,5
Изобута и 3,7 0,3
и-Бутап 93,6 3,5 |
Другие углеводороды . . . 0,4 0,7
Степень превращения бутана, . . . 91,0
Рис. 23. Схема парофазпого
крекинга с твердым теплоноси-
телем.
1 — предварительный подогреватель;
2 — подогреватель; з — реактор; 4 —
контейнер.
Линии: I — исходный продукт; И —то-
почные газы и воздух; III — продукты
пиролиза ти переработку; IV — горячий
воздух длт транспортировки тенлопо -
ептеля.
53
Термофор-пиролиз-процесс (ТPC-ProzeB). Этот метод, являющийся раз-
витием известного метода каталитического крекинга «термофор» и приме-
няемый для пиролиза этапа п пропана, детально рассматривается ниже
Таблица 27
Результаты опытов пиролиза этапа и пропана
по регенеративному способу
Исходный газ Этап Пропан
Целевой продукт Этилен Этилен
Температура, °C 954 1010
Состав реакционных газов, % объемн.: двуокись углерода окись углерода водород азот метан этан этилен ацетилен пропан пропен С4ЦС5 Глубина превращения, % . . . Выход этилена, % вес 0,5 0,3 42,5 1,4 10,8 8,9 30,0 2,6 0,3 83,9 50,7 0,3 0,3 25,8 1,8 30,6 1,9 25,6 1,7 5,8 4,2 2,0 87,8 34,3
вместе с процессами аро-
матизации.
Газовый крекинг реге-
неративным способом(Кор-
pers-Hasche-Wul jj-Verjah-
reri) [23]. Способ пиро-
лиза, основанный на ре-
генерационном принципе,
применяется как для про-
изводства этилена пироли-
зом этапа, так и для
получения ацетилена. Тех-
ническое совершенство
печей системы Копперс-
Хаше делает особенно вы-
годным применение прин-
ципа регенерации и обе-
спечивает максимально
возможное использование
тепла. Здесь могут быть
достигнуты значительно
более высокие темпера-
туры, чем при пиролизе
в трубчатых печах, в ре-
зультате чего может быть
сокращено время реакции.
В интервале температур 870—1110° пропан расщепляется на 85—90% с обра-
зованием 34% вес. этилена. Этан при 900—980° превращается на 75—85%,
давая до 52,5% этилена. Все выходы достигаются за однократный пропуск
сырья через печь и могут быть увеличены еще более при работе с циркуля-
цией, т. е. когда не подвергшаяся пиролизу часть парафиновых углеводоро-
дов возвращается обратно в процесс. Табл. 27 показывает результаты полу-
промышленного опыта пиролиза регенеративным способом.
Д. ПОЛУЧЕНИЕ ОЛЕФИНОВ ПИРОЛИЗОМ ЖИДКИХ УГЛЕВОДОРОДОВ,
В ЧАСТНОСТИ НЕФТИ И ЕЕ ФРАКЦИЙ
а) Общие сведения
Страны, не располагающие собственными источниками нефти и газа,
имеют в настоящее время возможность получать этилен, являющийся осно-
вой нефтехимической промышленности, из легкотранспортируемых продук-
тов, например из определенных фракций нефти. Эта задача решается в пер-
вую очередь пиролизом нефтяных фракций в присутствии водяного пара при
600 — 700°. Водяной пар служит одновременно разбавляющей средой и тепло-
носителем и уменьшает коксообразование. Процесс во многом подобен паро-
фазному крекинг-процессу. При этих процессах до 30% всего вводимого
сырья превращается в газообразные продукты, в большинстве с высоким
содержанием олефинов, которые в недавнем прошлом считались нежела-
тельными. Целевым продуктом являлся бензин. Процесс пиролиза, имеющий
целью получение олефинов, о котором здесь идет речь, должен проводиться
таким образом, чтобы' обеспечить максимальный выход олефинсодержащих
газов и минимальный — жидких продуктов, кипящих в интервале темпера-
туры кипения бензина. Выход последних может быть различным в зависимо-
сти от состава сырья и условий пиролиза.
54
Жидкие продукты пиролиза содержат часто до 60% и больше ароматиче-
ских углеводородов, выделение которых очень сложно. Можно, однако,
при определенных условиях ведения процесса пиролиза повысить долю
ароматических углеводородов в продуктах пиролиза до 95%, что, конечно,
связано с определенным уменьшением выхода олефинов. Из таких высоко-
ароматизированных фракций чистые ароматические углеводороды могут быть
выделены сравнительно легко.
Далее рассматривается процесс пиролиза, направленный исключи-
тельно на получение олефинов. Жидкая часть продуктов пиролиза при
этом не имеет особого значения. После надлежащей очистки она исполь-
зуется как компонент бензина.
б) Способ фирмы Шелл [24]
Способ Шелл (рис. 24) заключается в том, что легкая нефтяная фракция
подвергается пиролизу в присутствии водяного пара. Процесс идет следую-
щим образом.
Газойль в трубчатой печи нагревается до 400° и почти полностью испа-
ряется. Пары затем разделяются в колонне (на схеме не показана). Остаток
из колонны идет на топливо,
а головная фракция вместе
с водяным паром, перегретым
до 600—700°, поступает в
трубчатую печь. Водяной
пар уменьшает коксообра-
зование в пиролизной печи
и одновременно понижает
парциальное давление газа,
благодаря чему уменьшается
возможность протекания вто-
ричных реакций олефинов.
Выходящие из пиролизной
Рис. 24. Схема пиролиза способом Шелла.
7 —трубчатый нагреватель; 2 — система охлаждения; з —
фракционирующая колонна; / — водоотделитель.
Линии: I — перегретый водяной пар; II — фракция лег-
кого газойля; III— пар высокого давления; IV — мазут;
V —дренаж; VI —на установку для разделения и пере-
работку.
печи продукты пиролиза
быстро охлаждаются и по-
ступают далее во фракцио-
нирующую колонну, где по-
добно продуктам термиче-
ского крекинга разделяются па газ, бензин и остаток, используемый как
топливо. Газ разделяется далее обычным способом. Таким образом, без
труда могут получаться этилен и другие олефины.
Выход отдельных фракций при пиролизе способом Шелл в % вес.
С2 и легче...............29
Са-фракцпя ..............15
С4-фракция ..............10
Ароматизированный дистил-
лят . . .'..............20
Мазут..................20
в) Процесс Келлога [25]
Этот процесс, основанный на аналогичном принципе, осуществляется
следующим образом (рис. 25).
Тяжелый бензин или газойль в присутствии водяного пара нагревается
в трубчатой печи до 590—680°. При этом уже происходит частичное расще-
пление углеводородов. Продукты, выходящие из трубчатой печи, смеши-
ваются с водяным паром, перегретым в отдельном пароперегревателе до
930°, и короткое время остаются в адиабатических условиях в изолированной
трубе. Водяной пар отдает тепло на протекание эндотермической реакции
пиролиза и, понижая парциальное давление углеводородов, препятствует
возникновению вторичных реакций. Глубина пиролиза определяется отно-
шением перегретого пара к парам углеводородов. Затем газы снова быстро
охлаждаются, причем выделяющееся при охлаждении газов тепло ис-
55
пользуется для генерации водяного пара. Благодаря конструктив-
ным особенностям, обеспечивающим регенерацию тепла, этот требую-
щий очень много пара процесс становится технически приемлемым. Время
'______________________________ пребывания продукта в труб-
IV
Рис. 25. Схема процесса пиролиза при помощи
высокоперегретого пара по способу Келлога.
7 — подогреватель; 2 — изолированная труба, в которой
происходит процесс пиролиза; з — система охлаждения;
4 — фракционирующая колонна; .5 — водоотделитель.
Линии: I — перегретый водяной пар; II — газойль;
III — пар высокого давления; IV — мазут; V — жидкие
продукты пиролиза; VI—дренаж; VII— газ на раз-
делительную установку.
чатои печи составляет около
1 сек. Дальнейшая переработ-
ка газов и жидких продуктов
производится так, как было
описано выше. Табл. 28 по-
казывает результаты, полу-
ченные при переработке тяже-
лой бензиновой фракции при
различных условиях.
г) Процесс коксования [26]
(Der Hochsler Koker-Prozefl)
Процесс предназначается
для непрерывного пиролиза
нефти и нефтяных остатков.
Табл п ц а 28
Продукты пиролиза тяжелого бензина методом Келлога
Газообразные продукты Работа с низким коэф- фициентом превращения Работа с высоким коэф- фициентом превращения
выход па исходный продукт, % вес. содержание олефинов во фракции, % выход па исходный продукт, % вес. содержание олефинов во фракции, %
Водород 0,4 0,8 .—
Метан 6,2 — 10,9 —
Этилен 11,8 — 22,5 —-
Этан 3,0 80 2,4 90
Пропен 10,5 — 12,4 —
Пропан 0,4 96 0,4 97
Сумма . . 32,3 49,0
В качестве исходного сырья
для получения олефинов могут
использоваться такие мате-
риалы, которые не могут быть
переработаны другими методами
из-за высокой склонности к об-
разованию кокса. В принципе
этот метод подобен описанному
Рис. 26. Схема работы по способу
фирмы Хохстср.
1 — коксоуловптель; 2 — коксораздели-
тель; 3 — промежуточный сосуд; 4 —
подогреватель кокса; 5 — смесительная
зона; 6 — реактор; 7 — подъемник; 8 —
воздуходувка для горячего газа; 9 —
циклон; 10 — охладитель; 11 — фракцио-
нирующая колонна.
Линии: I — предварительно подогретое
сырье; II — пар; III — тяжелое масло
расщепления; IV — газ; V — газовый
бензин; VI — газойль; VII — легкое
котельное топливо.
56
выше методу газового крекинга с твердым теплоносителем. Каменные
шарики заменены здесь коксовыми шариками. Образующийся при пиро-
лизе кокс отлагается на коксовых шариках, а затем отделяется в осо-
бых разделителях.
Способ работы в основном следующий (рис. 26). Предварительно подо-
гретое сырье для пиролиза подается непосредственно на коксовые шарики,
подогретые в трубчатом подогревателе 4 до 650—750°, и подвергается раз-
ложению. Образование кокса полностью завершается в примыкающем
реакторе 6. Газы пиролиза идут далее в охладитель 10, где они быстро охла-
ждаются тяжелым маслом. Наконец в колонне 11 они разделяются па газ,
бензин, газойль и мазут. Газ идет далее на разделительную установку.
Кокс проходит испарительную зону и из нее в бункер подъемника 7, откуда
он горячим газом пневматически транспортируется в коксоулавливатель 1.
Отсюда коксовые шарики через разделитель 2, где они сортируются, напра-
вляются в промежуточный сосуд 3 и далее в коксонагреватель. Газы газ-
лифта очищаются от твердых частиц в циклоне 9 и горячей воздуходувкой 8
возвращаются в бункер газлифта. Результаты работы подобной установки
приведены в табл. 29.
Т а б л н Ц а 29
Результаты работы полупромышленной установки пиролиза
методом кокеообразовапия
Показатели Сырье
сырая нефть буроуголь- ная смола остаток головной перегонки вакуумный остаток
Плотность при 20° 0,855 0,996 0,940 0,980
Коксовое число, % 3,8 2,25 6,0 19,5
Содержание асфальта, % Элементарный состав, %: 1,31 — 2,27 61,0
углерод 85,70 82,64 86,02 80,01
водород 13,00 10,34 11,54 10,57
кислород . 0,43 4,68 1,16 —
вода 0,31 0,30 0,3 0,44
сора Фракционный состав по ASTM, %: 0,54 1,91 1,17 1,63
до 170° 16 0 0 0
170—340° 49 25 10 10
остаток Условия работы: 35 75 90 90
подача продукта, кг]час ... 193 80 112 140
температура в зоне смешения, °C 680 630 580 680
давление в реакторе, мм вод. ст. 400 250 200 400
циркуляция кокса, кг/час . . . Продукты реакции: газообразные углеводороды, 4000 2600 3500 4000
% вес. па сырье олефины, % вес. от выхода 53,3 55,0 14,0 40,6
газов жидкие продукты, % вес. па 58,8 49,0 58,4 48,6
сырье 38,2 42,0 75,0 11,0 34,2
кокс, % вес. иа сырье .... 6,5 3,0 25,2
Е. ПОЛУЧЕНИЕ ГАЗООБРАЗНЫХ ОЛЕФИНОВ ПИРОЛИЗОМ ЖИДКИХ
УГЛЕВОДОРОДНЫХ СМЕСЕЙ, В ЧАСТНОСТИ НЕФТЯНЫХ ФРАКЦИЙ,
В УСЛОВИЯХ ПРАКТИЧЕСКИ ПОЛНОЙ АРОМАТИЗАЦИИ
ЖИДКИХ ПРОДУКТОВ ПИРОЛИЗА
а) Общие сведения
При нагревании в течение определенного времени тяжелого бензина,
керосина или газойля до 700—1000° образуется до 50% газообразных про-
дуктов реакций, содержащих до 50% олефинов. Другие 50% от исходного
сырья приходятся на жидкие продукты реакции, почти нацело состоящие
из ароматических углеводородов. Последние сравнительно легко могут быть
выделены в чистом виде.
Такое совместное получение олефинов и ароматических углеводородов
интересно в том отношении, что при этом возможно одновременное алкили-
рование ароматических углеводородов олефинами. Таким путем получаются,
например, этилбензол, стирол, фенол, синтетические вещества — мягчи-
тели, моющие средства и т. д.
Следовательно, нефтехимическая промышленность в полном смысле
слова может быть построена на основе нефти и ее фракций. Прежде чем
рассматривать эти процессы более детально, необходимо сделать некоторые
основные замечания о процессах термической ароматизации парафиновых
углеводородов.
Определенная часть жидких и газообразных алифатических углеводоро-
дов при нагреве до высоких температур без давления и без катализатора
независимо от состава исходного материала превращается в смесь жидких
при нормальных условиях веществ, практически полностью состоящих из
^Таблица 30
Пиролиз парафинистого сураханского газойля
при различных температурах
Показатели Максимальная температура, °C
600 650 700 750 800 850
Выход смолы, % вес. на сырье . . 85,8 54,8 47,8 41,1 42,0 47,0
Выход кипящих до 205°, % вес. Ароматические бензольной фрак- ции, % Ароматические в толуольной фрак- 17,0 24,5 24,5 19,5 20,0 17,9
13,5 39,7 69,2 88,9 96,9 95,5
НИИ, % 12,5 43,6 69,4 88,9 96,9 95,5
Выход газа, % вег. 14,2 45,2 52,2 58,5 58,0 52,9
Состав газа, % объема.:
всего ненасыщенных 52,1 53,1 48,9 42,1 42,6 31,9
водород 8,3 9,1 10,1 10,6 13,2 24,3
метан 26,9 26,4 31,5 39,0 38,4 40,9
ацетилен — — — — — 0,8
этилен 29,4 29,1 28,6 29,6 32,6 27,7
этан 10,2 7,9 7,0 6,7 5,2 3,5
пропен 17,0 16,8 15,7 10,5 10,0 3,4
пропан 1,8 2,2 2,1 1,4 0,6 0,3
б у тепы 5,7 7,4 4,6 2,0 0,0 0,0
бутаны 0,7 1,0 0,4 0,2 0,0 0,0
Выход компонентов газа, % вес.:
водород 0,1 0,3 0,4 0,5 0,71 1,4
метан 2,2 6,7 10,2 15,9 16,5 19,6
ацетилен — — — — — 0,6
этилен 4,3 13,0 16,2 21,2 24,6 23,4
этап 1,6 3,8 4,3 5,1 4,2 3,2
пропен 3,7 11,0 13,4 11,3 11,3 4,3
прэпап 0,4 2,9 1,9 1,6 0,7 0,4 ,
бутены 1,7 6,4 5,3 2,9 0,0 0,0
бутаны 0,2 0,9 0,5 0,3 0,0 0,0
Всего углеводородов, % вес.: . . 14,1 44,9 51,8 48,3 57,3 51,5 ।
Всего ненасыщенных углеводоро-
дов, % вес 9,7 30,6 34,9 35,4 35,9 28,3
58
ароматических углеводородов. Качественный состав конденсата (называе-
мого смолой) не зависит от того, газообразные или жидкие углеводороды
применялись в качестве исходного материала. Природа сырья влияет только
на количество образующейся смолы. Существенное значение имеют темпера-
тура и продолжительность пиролиза. Например, если пропускать газойль
определенного состава через печь, нагретую до 550°, с временем пребывания
в ней 20 сек., то пары продукта выходят из печи почти не изменившимися;
при 600° уже наблюдается заметное разложение, сопровождающееся газо-
образованием. С повышением температуры газообразование еще более уве-
личивается. Жидкая часть, всегда богатая ароматическими углеводородами,
при 800° состоит почти только из них. Из табл. 30 видно влияние темпера-
туры на газообразование и содержание ароматических в жидких продуктах
реакции при одинаковом времени превращения [27]. Начиная от 700°, коли-
чество смолы остается приблизительно постоянным. В толуольной и соот-
ветственно бензольной фракциях содержится около 70% ароматических,
остаток же состоит из алифатических и циклоалифатических соединений.
При 800° содержание ароматических достигает 95%.
Табл. 31 показывает состав смол и газов, образующихся при пиролизе
некоторых высокомолекулярных углеводородных смесей при 800° в усло-
виях ароматизации. Из таблицы видно, что независимо от природы
исходного материала состав смолы всегда одинаков, откуда следует, что
процессы ароматизации во всех случаях лежат в основе механизма реакции.
Таблица 31
Состав смол и газов, образующихся при пиролизе высокомолекулярных
углеводородных смесей различной природы в условиях ароматизацип
Показатели Сырье пиролиза
углеводо- родный газ парафини- стый га- зойль ароматизи- рованный экстракт процесса Эделеапд
Температура, °C 800 800 800
Состав смолы, % вес.'.
олефины, кипящие до 75° 1,0 4,5 0,65
бензол 36,0 23,2 4,90
толуол 7,0 8,8 4,75
ксилолы и стирол 4,0 4,5 7,20
трпметилбспзол — 3,7 &70
высокомолекулярные производные бензола — 3,0 5,10 ]
Всего кицящих до 205°, % вес 48,0 47,7 31,3 J
нафталин 18,0 10,0 13/)
алкилнафталииы 3,0 6,9 2 13 ,
антрацен и фенантрен 10,0 6,0 5,7 .
алкнлаптрацены 12,0 26,3 23,0
тяжелый остаток смолы 9,0 26,3 27,0
Ароматических в бензольной фракции .... 93,3 94,2 82,7'
Ароматических в толуольной фракции .... 92,7 96,9 90,1
Состав газов, % объемп.: В среднем В среднем
водород 20,0 13,0 2>,0
метай 50—60 38,4 48,0
этан 0—5 40
этилен 20—25 32,6 20,0
пропоп 2-7 10,0 7,5
пропан — 06 1 0,.5
бутены — 0,0 0,0
бутаны — 0,0 1 —
Всего непредельных 20—30 42,6 27,5
59
В данный момент можно в общем принять, что образование ароматиче-
ских вызвано реакцией Дильса-Альдера между бутадиеном и низкомоле-
кулярными олефинами [28]. Как известно, бутадиен и этилен реагируют
друг с другом при высоких температурах с образованием циклогексена.
Так же, путем реакции бутадиена с пропаном, образуется метилциклогексеи.
Нафтены превращаются в ароматические углеводороды термическим дегидри-
рованием. Все эти ответвления классической реакции Дильса-Альдера дока-
заны прямыми опытами. Образование конденсированных ароматических
соединений можно при этом объяснить реакцией бензола и его гомологов
с бутадиеном. Необходимо, конечно, заметить, что при диеновом синтезе
одновременно протекают реакции изомеризации и дегидрирования.
б) К ат арол-процесс [29]
Промышленный процесс пиролиза нефтяных фракций, идущего с образо-
ванием только газообразных олефинов и высокомолекулярных ароматиче-
Т а б л н ц а 32
Изменения выходов продуктов при катарол-процееее е различным сырьем
Показатели Тексасская нафта Тяжелыц парафини- стый иран- ский бензин Парафини- стый иран- ский керо- син
Пределы кипения по Энглеру от 5 до 95%, °C 90—205 113—183 175—261
Удельный вес при 20° 0,799 0,756 0,796
Газообразные продукты, % вес. от общего вы- хода: водород . . 0,5 0,9 0,5
метан 18,3 24,0 13,7
отплои 7,4 11,6 11,6
этап 6,5 9,6 7,4
пропен 9,6 10,6 3,9
пропап 1,9 1,3 1,4
бутены 4,8 4,5 .3,9
бутаны 1,3 0,8 0,5
Всего газообразных алифатических углеводо- родов 49,8 62,4 49,4
Всего олефинов газообразных 21,8 26,7 26,4
Жидкие продукты, % вес. от общего выхода: кипящие ниже бензола 1,1 0,4 1,0
бензольная фракция 11,0 6,1 7,5
толуольная » 11,0 6,5 7,5
ксттлольная » 6,0 5,1 5,8
алкплбензольпая фракция 2,6 4,5 9,4
нафталиновая фракция 3,6 2,6 3,7
алкилпафталиновая фракция 4,0 2,7 3,8
антраценовая фракция 2,7 2,6 2,4
хризеновая » 2,5 2,6 1,6
остаток 5,5 3,9 7,3
Всего жидких продуктов 50,0 37,0 50,0
Чистые продукты, % вес. от общего выхода: нафталин 2,3 1,2 1,7
антрацен 0,2 0,1 0,15
фенантрен 80%-ный 0,5 0,3 0,4
хризен 0,3 0,2 0,2
пирен 0,2 0,1 0,15
60
ских продуктов, разработан в Англии К. Вейцманом с сотрудниками под
названием катарол-процесса. Катализатором в процессе служит медь; удается
достигнуть полной ароматизации, ведя процесс при сравнительно низких
температурах и с незначительным коксообразованием. Эксплуатируемая
фирмой Петрокемикел установка работает при температуре около 650°
и обеспечивает полную ароматизацию за время 0,5 —1,0 мин. Горячие пары
пропускают над медной стружкой, находящейся в обогреваемых снаружи
трубах. После трехсуточной работы необходимо освобождать катализатор
от коксовых отложений сжиганием кокса в струе воздуха.
Выходящий из печи продукт быстро охлаждается и далее перерабаты-
вается, как описано выше. Табл. 32 дает представление о выходах и составе
продуктов катарол-процесса.
в) Термофор-пиролиз-процесс [30]
Этим способом можно подвергать пиролизу как тяжелые нефтяные фрак-
ции, так и газообразные парафиновые углеводороды. Очень сильное коксо-
образование не имеет значения для этого процесса, так как корунд, приме-
няемый в качестве теплоносителя, освобождается от кокса прямым нагрева-
нием. Установка работает непрерывно. Труднейшей задачей в этом процессе
является подача шариков в подогреватель, так как здесь они подаются
не газлифтом, а при помощи элеватора.
Как уже указывалось, можно также крекировать пропан в этилен и
дегидрировать этан. Можно вести процесс при условиях, обеспечивающих
максимальный выход олефинов при только частичной ароматизации исход-
ного сырья, по можно также путем применения особо жестких условий
(высокая температура, продолжительное пребывание продукта в печи) осуще-
ствить полную ароматизацию жидких продуктов реакции.
г) Рургаз-процесс
Рургаз-процесс представляет собой процесс пиролиза, также предусма-
тривающий полную ароматизацию жидкой части продуктов [31]. При исполь-
зовании остатков перегонки нефти в качестве сырья получают около 40%
вес. олефинов от исходного продукта, из которых примерно 50% этилена.
Ж. ПОЛУЧЕНИЕ ВЫСОКОМОЛЕКУЛЯРНЫХ ОЛЕФИНОВ
а) Общие сведения
При крекинге нефти, осуществляемом в целях получения бензина,
образуется крекинг-бензин, содержащий олефины, которые пока еще не
находят применения вследствие их неоднородности и непостоянства состава.
Высокомолекулярные олефины, которые требуются нефтехимической про-
мышленности во все возрастающих количествах, могут быть получены при-
мерно четырьмя способами.
Во-первых, использованием продуктов синтеза Фишера-Тропша в при-
сутствии железного катализатора, содержащих очень много олефинов и
являющихся исключительно важным исходным материалом для их полу-
чения. Этот процесс был рассмотрен выше.
Во-вторых, получением высокомолекулярных относительно однородных
олефинов термическим крекингом парафина. Парафин из нефти, полученный
синтезом Фишера-Тропша или из бурого угля, разлагается при высоком
нагреве (пример 550°) в присутствии перегретого водяного пара. Образую-
щиеся при этом олефины смешаны с парафинами, так как при крекинге
парафиновых углеводородов образуются олефины и парафины, причем сумма
атомов С олефина и парафина равна числу атомов С исходного парафина.
61
Эти олефины, смешанные с парафином, представляют собой технически
исключительно ценный, но относительно дорогой исходный материал.
13-третьих, можно получать олефпны каталитическим дегидратирова-
нием спиртов, например этилового. Дегидратирование высокомолекулярных
спиртов, которые могут быть получены из природных продуктов , как коко-
совое и пальмовое масла и др., также иногда играет некоторую роль.
Четвертый важнейший способ получения высокомолекулярных олефинов
состоит в каталитической ди-, трп- и тетраморизацив газообразных при
нормальных условиях олефинов, как пропей и изобутен. Образующиеся при
этом олефины представляют собой важнейший исходный материал для алки-
лирования ароматических углеводородов, служащих для получения про-
межуточных продуктов.
б) Полимеризация газообразных при нормальных условиях олефинов
в высокомолекулярные олефпны
Общие сведения. Олефпны могут полимеризоваться двумя
путями: во-первых термически, путем нагревания до высокой температуры
под давлением и, во-вторых, нагреванием до умеренных температур в при-
сутствии подходящего катализатора.
При термической полимеризации легче всех реагирует этилен, труднее
пропен, а изобутен реагирует очень медленно. Совсем иначе реагируют они
при каталитической полимеризации: здесь легче всех реагирует изобутен,
а этилен (при наиболее благоприятных для изобутена условиях) практически
не полимеризуется.
Термическая полимеризация, как способ получения высокомолекуляр-
ных олефинов в качестве сырья для нефтехимической промышленности,
еще не играет никакой роли. Такие олефины получают исключительно ката-
литическим путем. Направленная полимеризация низкомолекулярных,
газообразных при нормальных условиях олефинов в высокомолекулярные
может происходить только при строго определенных условиях. Важнейшими
катализаторами для полимеризации олефинов являются серная и фосфор-
ная кислоты. При применении для полимеризации олефинов серной кислоты
несколько более высокой концентрации, чем требуется, возникают явления
так называемой «гидрополимернзации», при которой образовавшиеся поли-
меры олефинов смешаны с заметным количеством пеолефиновых продуктов.
То же имеет место при применении фосфорной кислоты, если температурные
условия выдерживаются недостаточно точно [32].
Количество насыщенных (неолефиновых) углеводородов, образующихся
при гидрополимернзации, тем больше, чем выше концентрация серной кис-
лоты. Так, например, в смеси пентенов с 98%-ной серной кислотой 70%
исходного продукта превращаются в полимеризат, выкипающий в пределах
90—350° п состоящий в большей части пз парафиновых углеводородов.
При этом растворимая в серной кислоте часть, выделяемая при разбавлении
ледяной водой, оказывается сильно ненасыщенной и обнаруживает до трех
и более двойных связей на молекулу. Реакция протекает по карбониум-
ионному механизму. В присутствии концентрированной серной кислоты
водород олефинов может переноситься из одной молекулы в другую, причем
одна молекула превращается в парафин, а другая в диолефин, который
еще раз может служить донором водорода, в то время как моноолефин
является акцептором.
Даже если полимеризация идет при самых благоприятных условиях,
все же наблюдается явление так называемой гетерополимеризации [33],
заключающееся в том, что наряду с ожидаемыми олефинами образуются
олефиновые продукты, не кратные по составу исходным олефинам. Так,
например, полимеризат, образующийся при полимеризации пропена в при-
сутствии фосфорной кислоты при 150—200°, хотя и состоит исключительно
из олефинов, однако кривая разгонки его показывает, что наряду с ожидае-
62
мыми ди-, три- и тетрамерами (изогексен, изононен, изододецен) полимери-
зат содержит также и олефины с промежуточным числом углеродных атомов.
Из этих небольших примеров следует, что при полимеризации газообраз-
ных при нормальных условиях олефинов, которые имеют большое значение
для нефтехимической промышленности как промежуточные продукты, усло-
вия проведения реакции должны строго выдерживаться, если главной целью
является не получение полимербензина, применяемого в качестве карбюра-
торного топлива.
Полимеризация в присутствии серной кис-
лоты. Действие серной кислоты как катализатора для полимеризации
олефинов -было установлено еще А. М. Бутлеровым, который нашел, что при
действии изобутена па 70%-ную серную кислоту образуются ди- и триизо-
бутены.
CH
СН,
I
CH3—C—CH=C—CH3
СН, СИ,
с=сп2 + ^>С=|СВ2
СН,' Сн/
2, 4, 4-триметилпеятеп-2
Ппмсризация
\н3х Y1’
CH3—C-CH2—C=CH2
сн/
2, 4. 4-триметилпептеп-1
Гидрирование
ClT3
C1L,
-> СИ3—С- СН2—СН—сн3
/ I
сн3 сн,
2, 2, 4-триметилпентан (изооктан)
Серная кислота применяется в настоящее время как катализатор при
получении диизобутена, который каталитическим гидрированием превра-
щается в изооктан (2,2,4-триметилпентан) — широкоизвестный эталонный
углеводород (октановое число равно 100), применяемый для определения
антидетонациониых свойств карбюраторных топлив.
Получение высокооктановых карбюраторных топлив или их компонентов
не относится к области нефтехимии. Все же здесь следует отметить, что
2,2,4-триметилпентан (изооктан) в смеси с другими высокоразветвлеппыми
изооктапамп как 2,2,3-, 2,3,4- п 2,3,3-триметилпентаны имеет октановое
число, также практически равное 100; смесь их может быть получена катали-
тическим алкилированием изобутана и-бутеном или изобутеном. В настоящее
время это единственный промышленный способ его получения. В качестве
катализаторов при алкилировании применяют концентрированную серную
кислоту, безводную фтористоводородную (плавиковую) кислоту и в некото-
рых случаях безводный хлористый алюминий. Реакция протекает фор-
мально согласно следующему уравнению, причем должен образоваться
2,2,3-триметилпептан с октановым числом, равным 105:
сп3 сп3 сн3
\ \ I
С.Л.,—С]1+С1Т„=СН—СН2—СН3 -> СИ—С—СП—СН,—СН,
/ “ I
СПз сн3
Практически этот изомер образуется в небольшом количестве. Вслед-
ствие структурной изомеризации, обусловлеппой карбониум-ионным механиз-
мом реакции, получается главным образом изооктан (2,2,4-триметилпентан)
одновременно с 2,3,4- и 2,3,3,-триметилпеитанами.
6
Димеризация с серной кислотой при низкой температуре. В промышлен-
ности димеризация изобутена при помощи серной кислоты в большинстве
случаев проводится при температуре, не превышающей 30° (рис. 27). Способ
заключается в том, что при температуре приблизительно 20—25° содержащая
изобутен фракция C-i кре-
Рис. 27. Схема холодной полимеризации изобутена
с серной кислотой.
кинг-газа или газов пиро-
лиза интенсивно переме-
шивается с 60—70%-иой
серной кислотой. При
этом изобутен растворяет-
ся в кислоте с образова-
нием mpem-бутилсуль-
фата, а другие олефины и
парафиновые углеводорды
не растворяются [34].
Абсорбция изобутена
осуществляется в про-
тивотоке, при котором
кислота, содержащая уже
относительно много трет-
бутплсульфата, приходит
1 — абсорбер I; 2 — абсорбер II; з — полпмсрпзацпонпая
колонна; 4 — сепаратор;
Линии: I — Франция С4, содержащая изобутен; II — свежая
кислота; III—газ, бедный изобутеном; IV—н-бутан и н-бутсп;
V — регенерированная серная кислота; VI — серная кис-
лота, насыщенная изобутеном; VII — полимеризат на про-
га ы вку и р е I гр и ф и к а ц ню.
в контакт с газом, содер-
жащим изобутен (колон-
на /), тогда как бедный
изобутеном газ смешивает-
ся со свежей серной кис-
лотой (колонна 2). Серная
времени нагревается до 100°
кислота после насыщения в течение короткого
под давлением в полимеризационной части установки (колонна 5), где про-
исходит ди-, три- и тетрамеризация. Полимеризат отделяется от кислоты,
промывается и ректифицируется. Смесь диизобутенов, состоящая примерно
из 80% 2,4,4-триметилпентена-1 и 20% 2,4,4-триметилпентена-2 отде-
ляется, а тетрамеры изобутена уходят в остаток. Выход диизобутенов соста-
вляет около 75%. Диизобутен может быть превращен в и-ксилол с помощью
процесса, разработанного в промышленных масштабах фирмой Хёхстер
Фарбверкен [35].
Физические константы обоих диизобутенов, получаемых каталитической
димеризацией изобутена, приведены в табл. 33.
Таблица 33
Физические константы чистых продуктов каталитической димеризации
изобутена [36]
Углеводород Температу- ра кипения при 760 мм рт. ст.; °C ^20 4 20 Темпера- тура замер- зания, °C
2,4,4-тримстилпентен-1 2,4,4-триметилпентен-2 101,2 104,5 0,7151 0,7211 1,4082 1,4158 —93,6 ±0,1 —106,5 ±0,1
Действительный состав диизобутеновой фракции, полученной полиме-
ризацией изобутена [37], следующий (в % объемн.):
2,4,4-триметилпептен-1.....................................80,7
2,4,4-триметилпентен-2..........................•.........17,6
3,4,4-триметилпентен-2 I
2,3,4-триметилпентен-2 I....................................1,1
64
2,3,3-триметилпентен-1 ]
2,3,4-триметилпентен-1 >..................................0,4
5,5-диметилгексен-2 ]
Спирты, кетоны и другие соединения........................0,2
Физические константы изоокта и а [39]
Температура кицения при 760 мм рт. ст., °C........99,27
Температура замерзания, °C .....................—107,4
Плотность ....................................... 0,6920
Коэффициент преломления nD20 .................... 1,3915
Полимеризация, при повышенной температуре [38]. По второму способу
полимеризация проводится с горячей кислотой. Этим способом, при котором,
кроме изобутенов присутствует также н-бутен (сополимеризация), получают
неоднородную смесь олефинов, используемую в дальнейшем для производ-
ства карбюраторного горючего. Применять метод для получения нефтехими-
ческих продуктов нецелесообразно.
Рис. 28. Схема получения тетрамера пропена.
* —[промывочная колонна; 2 — сборная емкость очищенной фракции Сз; 3 — компрессор; -1 — ката-
лнзаторная камера; Л — депропанизатор; 6 — фракционирующие колонны.
Линии: 1 — пропан-пропеновая фракция; II —[фракция Сз, промытая щелочью; Ill — фракция
Сз,(промытая водой; IV — щелочь; V — вода; VI — пропан для понижения содержания пропена в ис-
ходном продукте; VII — бедный пропеном газ из депропанизатора для поддержания температуры;
VIII — газ депропанизации; IX— пропан для получения ожиженного газа,- X — низшие полимеры
пропена, возвращаемые па полимеризацию-, XI — тетрамер пропена; XII — высокополимерпый оста-
ток.
Полимеризация с фосфорной кислотой как ка-
тализатором. Важнейший способ полимеризации газообразных оле-
финов состоит в пропускании их над обработанными ортофосфорной кисло-
той кизельгуром, активированным углем или асбестом при температуре
170—220° и давлении 15—40 ат. Наилучшим катализатором является смесь
75% ортофосфорной и 25% пирофосфорной кислот на кизельгуре. Содержа-
ние олефинов в полимеризате при применении этого катализатора составляет
около 90%. Катализатор находится в виде шихты в специальных камерах;
тепло полимеризации (около 16 ккал/моль при полимеризации пропена) сни-
мается добавлением холодного газа. Течение реакции полимеризации исход-
ного сырья, очень богатого олефинами, регулируется добавлением газа
депропанизации, в котором содержание олефинов сильно понижено поли-
меризацией.
Схема полимеризационной установки для получения тетрамера пропена
показана на рис. 28. Необходимая пропен-пропановая смесь поступает из
.депропанизатора установки для стабилизации крекинг-бензина. После
5 Заказ 596.
65
промывки щелочью и водой для очистки от сероводорода, аммиака и аминов
газ поступает в сборник, откуда компрессором пагнетается в печь. Там при
14—40 ат и 170—200е над фосфорнокислым катализатором происходит
полимеризация. Для регулирования температуры в печи в различные точки
ее реакционного пространства подают пропап из депропанизатора. Вслед-
ствие снижения содержания олефинов, вызываемого впуском пропана, наблю-
дается понижение температуры. Из печи продукты реакции идут в депро-
панизатор, где пропан отделяется. Часть его возвращается в печь для под-
держания температуры, другая часть удаляется из установки. Освобожден-
ные от пропана продукты полимеризации поступают в фракционирующую
колонну, где от них отделяются ди- и тримерпропены, часть которых воз-
вращается в цикл для дальнейшем полимеризации, а часть отводится из уста-
новки для использования в качестве добавки к карбюраторному топливу.
Тетрамер пропена отделяется от высокополимерных продуктов в особой
колонне.
Часть пропана из депропанизатора можно добавлять также непосред-
ственно к исходному сырью для понижения содержания в нем пропена.
В промышленных установках получаются, например, общие выходы,
приведенные в табл. 34 [40].
При применении олефинового полимеризата как промежуточного про-
дукта для нефтехимической промышленности и особенно как исходного
материала для алкилирования бензола или фенола необходимо, чтобы сырьем
для полимеризации служили олефины близкого состава. В первую очередь
для этого применяется пропен-пропановая фракция крекинга и установок
стабилизации бензинов. Сополимеризаты из пропена и и-бутена пли изобу-
тена мало пригодны как компоненты алкилирования, так как в условиях
Таблица 34
Выход отдельных продуктов при получении тетрамера
пропена полимеризацией фракции С3 над твердой
фосфорной кислотой [40]
Продукт м31 сутки % объемп.
Исходный :
пропан-пропсповая фракция . . 95,0 100,0
пропен, % — 49,3
пропап, % Продукт полимеризации: использовано исходного про- — 50,7
дукта полимеры, как добавка к беп- 51,5 45,6
зииам 0,3 0,3
тетрамер 27,6 29,2
высококипящио полимеры . . полнота превращения пропе- 1,3 1,1
па, % 92,3
алкилирования возможна их деполимеризация. Как компоненты алкилиро-
вания применяются в большинстве смеси тетра- и в меньшей степени пента-
меры пропена. В каких количествах они применяются, видно из того, что
в США в 1955 г. было получено 200 000 т тетрамера пропена [41].
в) Мюльхеймский способ димеризации олефинов
Если смешать, например, пропен с 1% вес. трипропилалюминия или
триэтилалюминия и нагревать его в автоклаве при 200°, пока давление не
упадет на 100 ат, то получается продукт димеризации, а именно 2-метил-
66
уже в условиях опыта проис-
пентен-1 с практически количественным выходом [42]. Реакция протекает
так, что молекулы пропена вклиниваются между алюминием и пропильными
группами, что приводит к образованию гексила алюминия, из которого
гексильные группы тотчас вытесняются пропильными группами с образо-
ванием изогексена. Катализатор не вызывает никаких изменений этого
димера. Принципиально так же протекает реакция с бутеном и другими
а-олефипами. Олефины с внутренними двойными связями (Р и у-олефины) так
не реагируют.
Так как бутен может существовать в двух изомерных формах, отличаю-
щихся положением двойной связи, и так как
ходит изомеризация связей, то при диме-
ризации бутепа-1 появляется также бутеп-2,
который непосредственно катализатором не
димеризуется. Однако он находится в
равновесии с бутеном-1, поэтому переходит
постепенно в димер бутена-1.
Высшие а-олефипы ведут себя также.
В начале превращения они быстро димери-
зуются. Наряду с этим часть мономеров,
еще не успевших димеризоваться, претерпе-
вает изомеризацию двойных связей. Из
находящихся в равновесии изомеров только
изомеры-1 превращаются в димерные про-
дукты, но эти олефины всегда имеются
только в ограниченном количестве, а уста-
новление равновесия требует времени. По-
этому процесс этот протекает медленно.
Димеризация пропена технически легко
осуществима. Пропен под давлением 100 ат
и трипропилалюминий в объемном соотно-
шении 3 : 1 подаются в нагретую до 200°
трубу (рис. 29), являющуюся реакционным
сосудом. Вытекающий с низа колонны димер
увлекает с собой катализатор, от которого
Димер пропена, который кипит при 62°, при этом освобождается от содер-
жащегося в нем непрореагировавшего пропепа. Трипропилалюминий после
отделения димера нагнетается обратно в реакционный сосуд. Время пре-
вращения составляет около 60 мин., полнота превращения 80—90%. На 1 л
реакционного объема в час образуется примерно 1 л димерного продукта.
Из бутена-1 при этой реакции получается смесь димеров.
Этилен ведет себя па первый взгляд так, как будто он принадлежит
к другому гомологическому ряду, чем а-олефипы с большим числом углерод-
ных атомов [43]. При действии этилена на триэтилалюминий при температуре
100—120° и давлении этилена около 100 ат молекулы этилена внедряются
между алюминием и этильными группами
zC,H5
Al (С2Н5)»+С2Н4 -> А1—С2Н5 и т. д.,
\с2[Т4—С2Н5
так, что образуются высокомолекулярные алкилаты алюминия. Но внедре-
ние молекул этилена происходит не бесконечно, так как этот процесс, кото-
рый может быть назван «реакцией роста», (Wachstumreaction) обрывается
реакцией вытеснения, при которой длинные, связанные с алюминием алкиль-
ные цепи вытесняются этиленом. Алкильные группы выделяются в виде
олефинов. Высокие температуры очень способствуют протеканию реакции
вытеснения
А! (С„Н2п+1)3+ЗС2Н4 -+ А1 (С2Н5)3+ЗСПН,П
5* 67
Рис. 29. Схема димеризации
пропена по Циглеру.
1 — реакционный сосуд; 2 -- компрес-
сор; 3 — депропанизатор; I — дистил-
ляционная JtOJJOJJJJJ.
Линии: I — сухой пропен под давле-
нием 100 am; II — нролилалюмииий
обратно в реактор*, III —пропен*,
IV — дпмеризат.
затем отделяется перегонкой.
Образование алкилатов алюминия из триэтилалюминия и этилена про-
текает не единообразно; когда еще не все этильные группы перешли в бутиль-
ные уже образуются гексильные группы, до образования всех гексильных
групп появляются уже октильные группы и т. д. Поэтому выделяющиеся
в результате реакции вытеснения олефины имеют не однородный состав и
представляют собой смесь.
Полученные описанным способом алкилаты алюминия можно также
подвергнуть разложению водой с образованием смесей парафиновых угле-
водородов, начиная от мягкого и твердого парафина и до углеводо-
родов, аналогичных полиэтилену, но не столь высокого молекулярного
веса.
Ровная и единообразная димеризация этилена, подобная димеризации
пропена и бутена-1, может быть достигнута в присутствии никеля, чрезвы-
чайно ускоряющего реакцию вытеснения. Тогда уже при 100—110° образо-
вавшийся в результате реакции роста бутилат алюминия в присутствии
этилена дает бутен-1 и триэтилалюминий.
Для синтеза высокомолекулярных олефинов из этилена необходимо
разделить реакции роста и вытеснения. Тогда на первой ступени можно
действовать этиленом под давлением на триэтилалюминий, причем обра-
зуется высокомолекулярный алкилат алюминия, а затем также под давле-
нием этилена проводить реакцию вытеснения в присутствии никеля.
г) Крекинг-олефины
Крекинг-олефины, называемые также вторичными олефинами, полу-
чаются при парофазном крекинге парафина в присутствии водяного пара.
Превращение парафина происходит лишь на 25—30% и неразложившийся
парафин возвращается на повторный крекинг. В табл. 35 показаны резуль-
таты крекинга парафина в паровой фазе [44] в условиях, когда парафин
путем повторного крекинга непревращенной части, был переработан пол-
ностью. Высокомолекулярная часть крекинг-олефинов применяется в первую
очередь для производства моющих средств, получаемых присоединением
серной кислоты по двойной связи (типоль).
Таблица 35
Содержание олефинов в крекинг-олефинах, выкипающих
в различных интервалах температур
Фракция Температура кипения при 760 мм рт. ст., °C Количе- ство, % Число уг- леродных атомов Содержа- ние олефи- нов в сред- нем, %
Головка: До 75 21,3 <7
1 75—100 4,9 7 85
2 100—126 4,9 8 80
3 126—152 5,75 9 80
4 152-175 5,75 10 79
5 175—197 10,53 11 82
6 197—216 9,05 12 80 -
7 • ' 216—236 7,24 13 78
8 236—255 8,31 14 72
9 255-273 7,24 15 58
10 273-290 8,84 16 50
11 290-305 4,48 17 40
Остаток Выше 305 1,7 >17 —
68
V. КОНЦЕНТРАЦИЯ И ВЫДЕЛЕНИЕ ОЛЕФИНОВ ИЗ СМЕСЕЙ
ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ
А. ОБЩИЕ СВЕДЕНИЯ
Концентрация и выделение чистых олефинов, например из крекинг-
газов, газов пиролиза, риформинг-газов и т. д., исключительно важны для
нефтехимической промышленности. В принципе эти процессы заключаются
в том, что смеси газообразных алифатических углеводородов разделяются
на этан-этиленовую, пропан-пропеновую
Каждую фракцию можно затем разделить
части. Обычно из таких га-
зовых смесей прежде всего
выделяют водород и метан.
От газов, содержащих
ацетилен, необходимо пред-
варительно его отделить;
чаще всего его отделяют
селективным гидрированием
в этилен. Таким путем аце-
тилен выделяется из газа
почти количественно. Метан
и водород можно отделять
промывкой газовой смеси
маслом, в котором раство-
и бутан-бутеновую фракции,
на олефиновую и парафиновую
/Г1*
Перегонка
Adcopbuut.
Рис. 30. Важнейшие пути промышленного раз-
деления газовых смесей.
1 — компрессор; 2 — осушитель; — 3 деметанизатор; 4—
деэтанизатор; 6 — масляный абсорбер; б — адсорберы с
активированным углем.
Линии: I — исходный газ: Нз 4- СН4 + Со -f- Сд и высшие
углеводороды; II — тогцее масло; III —уголь обратно в
адсорбер.
____________углеводороды
-^CHt ^Г{~Сг
Т_«. С3 и бывшие
____углеводороды
ряются углеводороды с дву-
мя и большим числом угле-
родных атомов, метан и во-
дород не абсорбируются
маслом и удаляются из
установки. Газообразные
углеводороды выделяются
затем из промывочного масла и отделяются друг от друга. Этот абсорб-
ционный метод п его значение подробно рассмотрены выше.
Другим способом отделения водорода и метана с одновременным раз-
делением высокомолекулярной части по числу атомов С является адсорб-
ционно-десорбционный, с использованием активированного угля и непре-
рывной промышленной газовой хроматографии (гиперсорбционный метод).
Наиболее часто применяемым способом полного разделения газовых
смесей на отдельные составные части является фракционировка. Газ сжи-
жают путем охлаждения до низких температур при относительно высоком
давлении или только лишь охлаждением до очень низкой температуры,
затем разделяют на составные части фракционировкой.
На рйс. 30 дана схема важнейших промышленных способов разделения
газовых смесей. В табл. 14 были приведены важнейшие физические кон-
станты моноолефинов, в табл. 36 сопоставлены температуры кипения низко-
молекулярных, газообразных при нормальных условиях парафинов и оле-
финов.
Разделение олефинов и парафинов с равным числом углеродных атомов
для фракции Сг проходит довольно легко и успешно, так как разница между
температурами кипения этана и этилена составляет около 15°. Пропан и
пропен, разница между температурами кипения которых составляет всего
5,5°, разделить значительно труднее. Для фракции С4, которая может вклю-
чать в себя уже шесть различных индивидуальных углеводородов, разделе-
ние фракционировкой невозможно. Здесь в лучшем случае удается изолиро-
вать две группы углеводородов, а именно изобутен, изобутан и н-бутен-1,
с одной стороны, и н-бутен-2 и н-бутан — с другой.
Отделение олефинов от парафинов с равным числом углеродных атомов
возможно абсорбционным методом. Так, например, этилен растворяется
69
Таблица 36
Сопоставление температур кипения важнейших низкомолекулярных,
газообразных при нормальных условиях или нпзкокипящих парафинов и олефинов
Парафины Температура кипения при 760 мм рт. ст., °C Олефины Температура кипения при 760 мм рт. ст., °C
Метан Этап Пропан —161,5 —88,6 —42,3 Этилен Пропен -103,8 —47,7
и-Бутап Изобутан —0,5 —11,7 Бутеи-1 г(ис-Бутеп-2 тра/1с-Бутсн-2 Бутадиен-1,3 Изобутен —6,26 + 3,72 -0,88 —4,4 —6,9
н-Пептан Изопентан (2-метплбу- тан) 36 28,0 Пентеп-1 Ц14с-Пептен-2 траПс-Пептеи-2 2-метилбутеп-1 2-метилбутеп-2 З-метнлбутен-1 29,97 , 37,1 36,36 31,16 38,6 20,1
в растворе медной соли определенного состава, этан же в пей не растворяется.
Из фракции С4 изобутен, например, можно извлечь 65%-ной серной кисло-
той с соблюдением условий, при которых другие компоненты по фракции
в реакцию не вступают.
Из серной кислоты изобутен может быть сравнительно легко выделен
в чистом виде. На этом основан относительно дешевый и простой метод полу-
чения изобутена.
Исключительно важным для разделения практически равнокипящих
олефинов и парафинов является способ экстрактивной фракционировки.
При этом газовая смесь приводится в контакт с движущейся ей навстречу
экстракционной средой, причем олефиновая составная часть поглощается
этой средой, парафины же не абсорбируются и удаляются из установки.
Этот процесс играет также большую роль в получении чистого бутадиена
дегидрированием бутана.
Б. РАЗДЕЛЕНИЕ ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ ФРАКЦИОНИРОВКОЙ
Этилен из газов крекинга и пиролиза часто выделяют фракционировкой.
Эти газы в большинстве случаев содержат водород и метан. Чтобы можно
было отделить фракцию С2 от водорода и метана без потерь перегонная
колонна должна иметь метановое орошение. Для достижения этого необхо-
Рис. 31. Способ разделения крскинг-газа
путем низкотемпературной перегонки под
давлением.
димы давление и низкая темпера-
тура.
На рис. 31 показано разделе-
ние крекинг-газа низкотемператур-
ной фракционировкой под давлением.
Крекинг-газ давлением 40 ат охла-
ждается, освобождается от воды при
помощи силикагеля и при темпера-
туре —18° поступает в метановую
колонну 1 и далее проходит через
все остальные, показанные на схеме
колонны, рабочие условия которых
70
даны в табл. 37. В той же таблице приведены и результаты разделе-
ния [45].
Таблица 37
Рабочие условия низкотемпературной перегонки крекинг-газа под высоким
давлением
Показатели Метановая колонна Этиленовая колонна Этановая колонна С3 колоппа
Давление, ат Температура верха, °C » низа, °C 40 —90 + 38 27 —18 + 43 24 + 4 + 65 15 + 38 + 93
Состав газа, % объемн. Исходный Верх Низ Верх Низ Верх Пиз Верх Низ
Водород Метан Этплеп Этан Пропен Пропан С4 + С5 11,5 16,5 19,4 17,7 89 24,0 2,0 40,0 56,0 4,0 0,5 25,6 24,9 12,5 33,7 2,8 ООО 11 1 1 0,9 32,6 17,0 45,7 3,8 2,8 95,0 2,0 1,4 24,3 68,6 5,7 1,5 25,7 72,2 0,6 5,0 95,0
Метан для орошения первой колонны ожижается при помощи этилена,
выходящего из второй колонны и дросселируемого со снижением давления
от 27 ат до нормального. В колоннах 2 и 3 орошение получают за счет
охлаждения этапа и этилена испаряющимся пропаном, тогда как колонна 4
работает с водяным охлаждением орошения.
В. СЕЛЕКТИВНОЕ ГИДРИРОВАНИЕ АЦЕТИЛЕНА В ЭТИЛЕН
В ГАЗАХ ПИРОЛИЗА
Прежде чем разделять газ фракционировкой на составляющие компо-
ненты необходимо освободить его от содержащегося в нем некоторого коли-
чества ацетилена. Это достигается селективным гидрированием ацетилена
в этилен.
С этой целью газ пропускают над хромо-
никелевым катализатором, состоящим приблизи-
тельно из 95% окиси хрома и 5% никеля. Ката-
лизатор получают растворением в воде хромовой
кислоты (Н2СтО4) и азотнокислого никеля, с по-
следующим нагревом раствора при перемешивании
до полного удаления воды и прекращения выделе-
ния двуокиси азота. Частичное гидрирование газов
пиролиза, богатых водородом, ведут при темпера-
туре около 200° и скорости подачи около 800 л газа
(в пересчете на нормальное давление) на 1 л ката-
лизатора в час. В газах, бедных водородом, скорость
подачи должна быть меньше, а температура выше.
Схема рис. 32 показывает, как проводится такое
селективное гидрирование. Сухой, находящийся под
давлением газ крекинга или пиролиза нагревается
в зависимости от содержания в нем водорода до
150—200° и поступает в один из двух, связанных
друг с другом ацетиленовых конверторов, заполнен-
ных катализатором, расположенным в виде многих
Рис. 32. Схема удаления
ацетилена путем пар-
циального каталитиче-
ского гидрирования.
1 — печь для гидряропяштн
1; 2 — печь для гидриро-
вания 11.
Линии: I — сухов алстилсп-
содергкащнй газ под давле-
нием; II— свободный от аце-
тилена газ на разделитель-
ную установку.
71
отдельных слоев. Постоянная температура в конверторах поддерживается под-
водом свободного от ацетилена газа в различные секции реакторов. Это очень
важно, потому что иначе не может быть обеспечена селективность гидриро-
вания. Катализатор должен быть вначале восстановлен чистым водородом
и во время работы в определенной степени должен регенерироваться подводи-
мым чистым водородом.
В табл. 38 показаны результаты частичного гидрирования над хромо-
никелевым катализатором газовой смеси, полученной автотермическим
дегидрированием этана.
Таблица 38
Результаты частичного гидрирования ацетилена
в газе автотермического дегидрирования этана
над хромо-никелевым катализатором
Компонент Исходный газ, % объемн. Газ после гидрирова- ния, % объемн.
Бутаны и бутены . . 3,46 3,50
Этилен 38,10 39,00
Водород 49,18 49,50
Окись углерода .... 8,00 8,00
Ацетилен 1,26 0,00
Г. РАЗДЕЛЕНИЕ ГАЗОВЫХ СМЕСЕЙ АБСОРБЦИОННО-КОМПРЕССИОННЫМ
СПОСОБОМ (АБСОРБЦИЯ МАСЛОМ ПОД ДАВЛЕНИЕМ)
Способ был уже подробно рассмотрен, когда речь шла о переработке
природного газа. В данном случае он применяется или для концентрации
жидкой составной части (С3 и С4 — углеводороды) крекинг-газа, или для
отделения водорода и метана. Этим очень сильно облегчается дальнейшее
разделение сконцентрированной таким образом углеводородной смеси. Прин-
цип разделения основан на том, что углеводородная смесь вступает в контакт
с промывочным маслом (абсорбентом) при таких условиях температуры и
давления, при которых метан и водород в нем не растворяются и удаляются
из установки. Свободный от метана и водорода газ, абсорбированный маслом,
выделяют из последнего нагревом и затем разделяют. Табл. 39 показывает
результат разделения пирогаза путем абсорбции при комнатной температуре
и давлении 20 ат.
Таблица 39
Концентрация пирогаза i *5 ( ь ри обычной
температуре и 23 »»
Компонент Исходный газ, % объемн. Неабсорби- ровавный газ, % объемн. Газ из абсорб- ционной ко- лонны, выде- ленный нагре- вом, %
Метан 31,4 63,0 6,4
Фракция С2 . . . . 22,9 36,4 13,0
* с3 . . . . 23,3 0,0 46,6
» с4 . . . . 17,7 0,0 31,4
72
Выделенная нагреванием из абсорбента газовая смесь состоит, как видно
из таблицы, главным образом из углеводородов С3 и С4. Эта смесь может
быть далее разделена фракционировкой под давлением.
Во многих случаях особое значение придается выделению фракции С2,
содержащей этилен, при условии возможно более полного освобождения ее
от метана и водорода. Чтобы повысить растворимость фракции С2 в абсор-
бенте применяют более высокое давление и охлаждение абсорбента (масла).
Этими методами удается также разделить абсорбированную смесь газов на
составляющие непосредственно из масла. Последнее должно иметь соответ-
ствующую высокую температуру кипения.
На рис. 33 схематично показан способ отделения низкомолекулярных
компонентов (метана и водорода) от высокомолекулярных, путем абсорбции,
Рис. 33. Схема получения чистого этилена
из газов пиролиза пропана и этана абсорб-
ционным способом с разделением угле-
водородов в отсутствии промывочного
масла.
1 — абсорбционная колонна; 2 — разделитель;
3 — деэтанизатор; 4 — этиленовая колонна.
Линии'. I — свободный от ацетилена и находящий-
ся под давлением газ пиролиза пропана и этана;
II— отходящие газы (С4 и Но), используемые как
топливо; III — свободное от газа промывочное
масло (тощее масло); IV—насыщаяое промы-
вочное масло (жирное масло); V — франция Сз
обратно на пиролиз; VI — этан обратно на пиро-
лиз; VII — фракция Со и Сз; VIII — фракция Со;
IX — этилен.
Рис. 34. Схема получения чистого этилена?,
из газов пиролиза пропана и этана с раз-
делением газовой смеси в присутствии
промывочного масла (абсорбента).
1 — абсорбционная колонна; 2 — деэтанизатор;
з — этиленовая колонна; 4 — депропанизатор.
Линии: I —свободный от ацетилена и находящий-
ся под давлением газ пиролиза; II — отходящие
газы (СН4 и Но); III — жирное масло; IV — этан
и этилат; V— этилен* V/— фракция Сз обратно
на установку пиролиза; VII — этап обратно на
установку пиролиза; VIII — тощее масло.
последних маслом. Высокомолекулярная часть затем выделяется из масла и
оно вновь возвращается в абсорбционную колонну. Выделенные из масла
углеводороды разделяются фракционировкой под давлением.
В настоящем примере при переработке газов пиролиза пропана и этана
применяется низкомолекулярное промывочное масло (абсорбент) молеку-
лярного веса 82 (бензол-78).
Абсорбционная колонна состоит из секций, отделенных днищами (та-
релками). Условия, при которых водород и метан не абсорбируются, следую-
щие: давление 31 am, температура верха колонны 21°, низа 120°. При весо-
вом соотношении абсорбента и газа 4,2:1 абсорбируется 98—99% этилена,
водород освобождается полностью, а метан на 96%.
Отгонная колонна работает при 30 am. В нижней части колонны поддер-
живается температура 230°. Уходящий через верх колонны продукт охла-
ждается водой и поступает в деэтанизатор. Отсюда через верх колонны отхо-
дит фракция С2, а снизу — фракция С3, возвращающаяся на пиролиз. Фрак-
ция С2 поступает далее в этиленовую колонну, где разделяется на этилен,
уходящий через верх колонны, и этап, отбираемый снизу и возвращаемый
на пиролиз.
На рис. 34 показана схема переработки пирогаза на аналогичной уста-
новке, по в присутствии абсорбента. Насыщенное углеводородами масло из
абсорбера под давлением 31,5 am поступает в деэтанизатор, где освобождается
73.
•от фракции С2, которая в колонне, работающей под давлением и при
глубоком охлаждении, разделяется на этилен (головной продукт) и этан,
отбираемый с низа колонны и возвращаемый па пиролиз. Абсорбент напра-
вляется далее в депропанизатор, где он освобождается от фракции С3 и воз-
вращается для повторного использования в абсорбционную колонну. Этан
и фракция С3 идут снова на пиролиз.
Д. ВЫДЕЛЕНИЕ ОЛЕФИНОВ ИЗ СМЕСЕЙ ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ
АБСОРБЦИЕЙ СЕЛЕКТИВНЫМИ РАСТВОРИТЕЛЯМИ
В специальных случаях выделение определенных олефинов из газовых
смесей может производиться при помощи селективных растворителей. Так,
из газа, богатого этиленом, последний можно выделить промывкой раство-
ром медной соли под давлением. Растворимость олефинов в этаноламиновом
растворе одпохлористой меди при 20° представлена в табл. 40.
Промывка медным раствором осо-
бенно пригодна для выделения этилена
из продуктов дегидрирования этана.
Растворимость олефинов в этаноламиновом
растворе однохлористой меди при 20° в л/кг
в зависимости от давления
(Количество газа дано в литрах при
760 мм рт. ст. и 0°)
Олефин Давление, ат.
1 5 10 20
Этилен .... 8,4 15,8 21,2 24,2
Пропен .... 1,1 4,3 6,1 —
и-Бутеи .... 1,0 — — —
Вутадпеп . . . 11,0 — — —
Таблица 40
Рис. 35. Очистка этилена медно-щелоч-
ным раствором.
1 — компрессор; 2 — абсорбер.
Линии'. I— исходный газ, содежащий этилен;
II — С2Н4 и менее растворимые газы; III —
газ, свободный от C0H4; IV—медный раствор,
свободный от газа; V — 96 %-ный этилен на
кислотную и щелочную промывку.
‘Сжатый до 15 ат газ подается в абсорбционную колонну, где на-
встречу ему движется медный раствор (рис. 35) следующего состава (в г/л).
Медь (1) ...............215 Моноэтаиоламин.................570
Медь (2) ................ 15 Аммиак ........................ 30
Азотнокислый аммоний . . . 350 Вода ........................220
Насыщенный этиленом медный раствор проходит затем обработку
в несколько ступеней. При понижении давления от ступени к ступени с 15
до трех, а затем до 1,4 ат раствор освобождается от части газов, направляю-
щихся затем обратно в абсорбционную колонну. Эти газы наряду с этиле-
ном содержат также водород и этан.
При дальнейшем понижении давления до 0,1 ат выделяющийся газ
уже примерно на 96% состоит из этилена. Он промывается сначала
разведенным раствором кислоты для удаления небольших количеств аммиака,
.а затем щелочью, после чего вполне пригоден для дальнейшей хими-
ческой переработки.
Е. АДСОРБЦИОННЫЕ МЕТОДЫ
а) Общие сведения
Углеводороды тем легче и полнее адсорбируются на активированном
угле, чем больше их молекулярный вес. Адсорбированные на активирован-
ном угле низкомолекулярные углеводороды могут быть вытеснены с него
74
'(десорбированы) углеводородами большего молекулярного веса. Наконец,
все адсорбированные активированным углем углеводороды независимо от
их молекулярного веса могут десорбироваться водяным паром. Если же акти-
вированным углем адсорбированы углеводороды разного молекулярного
веса, то под воздействием водяного пара в первую очередь будут десорбиро-
ваться углеводороды наименьшего молекулярного веса. На этом принципе
основан метод непрерывного разделения газовых углеводородных смесей,
известный под названием гиперсорбции. Этот способ называют также непре-
рывной промышленной газовой хроматографией.
б) Гпперсорбционнып процесс [46]
Особый интерес этот способ представляет для выделения этилена из
бедных этиленом газов. Он может применяться также для непрерывного
выделения фракций С3 и С4, а также этана из природного газа. Разделение
парафинов и олефинов с равным числом углеродных атомов проходит этим
методом недостаточно гладко. В промышленных
условиях принцип реализуется следующим образом.
Разделяемый газ идет навстречу непрерывно
движущемуся слою активированного угля и, в за-
висимости от условий работы и молекулярного веса
составляющих газа, в большей или меньшей степени
адсорбируется углем. Активированный уголь после
насыщения, двигаясь к низу колонны, в части ее,
расположенной ниже места ввода исходного газа,
приходит в соприкосновение с тяжелыми углеводо-
родами, испарившимися из угля в нижней части
колонны. Тяжелые углеводороды вытесняют из угля
адсорбированные им углеводороды меньшего моле-
кулярного веса и последние выводятся пз колонны
через специальный боковой газоотвод. При этом
происходит фракционирование и при соблюдении
необходимых рабочих условий возможно разделе-
ние, как и в обычных ректификационных колон-
нах. Схема гпперсорбционного процесса приведена
на рис. 36.
Колонна для гиперсорбционного процесса имеет
в верхней части холодильник для охлаждения по-
ступающего в процесс активированного угля.
В нижней части колонны имеется испарительное
устройство, в котором посредством водяного пара вы-
деляются из угля адсорбированные им тяжелые
углеводороды. Разделяемый газ поступает примерно
в верхнюю треть колонны, так называемую адсорб-
ционную ее часть. Неадсорбированные газы (абгаз)
I
Рис. 36. Упрощенная
схема гиперсорбционно-
го процесса.
1 — охлаждающая секция;
2 — адсорбционная часть;
3—ректификационная часть;
4 — испарительная секция.
Линии- I — исходный про-
дукт; II — возврат активи-
рованного угля; III—десор-
бированный активирован-
ный уголь обратно в гипер-
сорбер; IV — не абсорбиро-
ванные газы; V — боковой
газовый поток; VI — глав-
ная фракция (готовый про-
дукт); VII — пар.
ОТВОДЯТСЯ из колонны
п
ниже холодильника.
Насыщенный газом уголь попадает из адсорбционной части в ректифи-
кационную часть колонны, где соприкасается с обратным потоком тяжелых
углеводородов, которые были вытеснены водяным паром из угля в испари-
тельной секции. Эти тяжелые углеводороды вследствие их большего моле-
кулярного веса вытесняют из угля более легкие углеводороды и таким обра-
зом в испаритель попадает уголь, содержащий высокомолекулярные углево-
дороды, находившиеся в разделяемой газовой смеси.
Вытесненная в ректификационной части колонны из угля газовая смесь,
которая при соответствующем режиме работы содержит углеводороды с опре-
деленным числом углеродных атомов, удаляется через боковой отвод. В испа-
рителе уголь освобождается от высокомолекулярных углеводородов (глав-
ная фракция). Основная часть угля после прохождения через испарительную
75
часть колонны, с помощью лифта возвращается в ее верхнюю часть. Мень-
шая часть активированного угля подается на регенерационную установку,
где при высокой температуре обрабатывается водяным паром для освобо-
ждения от продуктов полимеризации, которые не удаляются в испарительной
секции и которые со временем понижают адсорбционную способность угля
(на схеме не показана).
В табл. 41—44 даны примеры применения гиперсорбционного процесса.
В табл. 41 показан результат разделения водорода и метана при очистке
газов гидроформинга.
Таблица 41
Освобождение от водорода газов гидроформинга
посредством гиперсорбции
Компонент Отходящие газы гидро- форминга , % объемн. Головной продукт адсорбц. КОЛОППЫ, % объемн. Адсорбат, вытеснен- ный ВОДЯ- j ным паром, % объемн.
Водород 52,2 100 Следы '
Метан 43,5 0 91,0
Фракция Сг 2,7 0 5,6
» са 1,6 0 3,4
Табл. 42 показывает концентрацию этилена, выделенного из бедного
газа. В табл. 43 дан анализ гиперсорбционного процесса при условиях отбора
бокового потока, в табл. 44 — результат выделения жидкой фракции из
газа, состоящего в основном из метана.
Таблица 42
Концентрация этилена, выделенного из бедного этиленом
газа посредством гиперсорбции
Компонент Исходный газ, % объемн. Испаренный газ3 % объемн.
Водород 37,2 0,0
Кислород 0,3 0,1
Азот 6,0 0,1
Окись углерода 1,0 0,0
Метан 49,2 0,0
Ацетилен 0,3 3,7
Этилен . 5,7 92.0
Этан 0,1 1,0
Двуокись углерода 0,2 3,1
Таблица 43
Разделение этиленсодержащего газа при отводе бокового потока
Компонент Исходный газ, % Боковой поток из ректифика- ционной части, % Продукт из низа колонны, % Продукт из головы ко- лонны, %
Метан 40,1 0,5 Следы 99,6
Этилен 18,9 32,8 0,3 0,3
Этан 37,9 65,9 1,2 0,1
Пропан 3,1 0,8 98,5 Следы
76
Таблица 44
Отделение фракций С3 и С4 из газа, содержащего
преимущественно метан посредством гиперсорбции
Компонент Исходный, % объемн. Донный продукт, % объемн. Головной продукт, % объемн.
Метан 92,2 1,2 95,2
Фракция С2 4,6 0,7 4,7
» С3 2,9 87,0 0,1
1 » С4 I 0,3 11,1 Следы
Ж. РАЗДЕЛЕНИЕ ОЛЕФИНОВ И ПАРАФИНОВ ЭКСТРАКТИВНОЙ
ПЕРЕГОНКОЙ [471
а) Общие сведения
Выше было показано, что ректификацией под давлением, абсорбцией
под давлением и гиперсорбцией парафины могут быть разделены на фракции
по числу атомов углерода в молекуле, а в отдельных случаях могут быть
разделены парафины и олефины с равным числом атомов углерода. Однако
имеются смеси олефинов и парафинов с равным числом атомов С, например
во фракции С4, которые уже не могут быть разделены перегонкой.
Представим себе, что температуры кипения и соответственно упругости
паров двух не разделимых перегонкой соединений очень близки друг к
другу. Добавляя к смеси третье соединение, жидкое при условиях, когда
два первых находятся в газообразном состоянии, и способное значительно
увеличить первоначально малую разницу в упругостях паров обоих компо-
нентов, мы получаем возможность разделить их благодаря тому, что один
из компонентов остается растворенным в этом третьем соединении, тогда как
второй отгоняется. Растворимый компонент тот, у которого упругость паров
относительно ниже. Практически разделение выполняется таким образом,
что разделяемая смесь испаряется и пары промываются веществом, сдвигаю-
щим величину упругости паров. Так как этот способ разделения подобен
экстракции, имеющей в нашем случае селективный характер, то принято
называть этот процесс экстрактивной перегонкой. В США этот процесс полу-
чил название дистекс-процесса (Distillation-Extraction-Process).
б) Экстрактивная перегонка для разделения фракции С4
Процесс служит не только для разделения узкокипящих парафинов и
олефинов, но также и для разделения жидких углеводородных смесей.
К этому вопросу мы вернемся позднее при рассмотрении способов получения
чистых ароматических углеводородов. Особое значение имеет дистекс-процесс
при получении чистого бутадиена методом ступенчатого дегидрирования
бутана.
Так как последний процесс является одним из важнейших в нефтехими-
ческой промышленности, необходимо здесь также остановиться на бута-
диене. Приводим температуры кипения важнейших углеводородов С4 при
760 мм рт. ст. в ° С.
Изобутап.................—11,7
Изобутен.................—6,9
Бутен-1 .................—6,24
Бутадиен-1,3.............—4,4
н-Бутан .................—0,5
траис-Б-утон-2...........+0,88
гщс-Бутен-2 ............ +3,72
Винилацетилен........... -(-5,0
Этилацетилен............ +8,1
Диацетилен.............. +10,3
Диметилацетилен......... +27,0
77
Если углеводороды фракции С4, за исключением ацетиленовых, располо-
жить по их летучести в сравнении с бутадиеном, причем летучесть опреде-
лить в виде частного:
упругость паров углеводорода
упругость паров бутадиена ’
то получим при 41° и 4,55 ат значения летучести [48], приведенные-
в табл. 45 [48].
Табл п ц а 45
Относительная летучесть некоторых углеводородов С4
в сравнении с бутадиеном при 41° и 4,45 пт
Компонент Летучесть в сравнении с бутадие- ном Температу- ра кипения при 760 мм рт. ст., °C
Изобутап 1,209 —11,7
Изобутен 1,070 —6,9
Бутен-1 1,040 —6,26
Бутадиен-1,3 1,000 —4,4
и-Бутан 0,871 —0,5
тданс-Бутсп-2 0,843 -4-0,88
гр<с-Бутен-2 0,776 -4-3,72
В качестве растворителя в дистекс-процессе для разделения олефинов и
парафинов фракции С4 применяется фурфурол, содержащий 4—6% воды.
При добавке этого полярного растворителя отношение летучестей для при-
веденных выше углеводородов изменяется таким образом, что становится
возможным разделить углеводороды, имеющие при нормальных условиях
практически равные температуры кипения.
Эффект, вызываемый добавкой фурфурола, виден из относительной
летучести некоторых углеводородов С4 по отношению к бутадиену в присут-
ствии фурфурола, содержащего 4% воды, при 54,5° и 4,55е ат.
Изобутап................. 2,600
п-Бутан.................. 2,020
Бутон-1 ................. 1,718
Изобутен................. 1,666
ягр<шс-Бутен-2 .............. 1,190
1{ис-Бутен-2................. 1,065
Бутадиен..................... 1,000
Выделение изобутена из Б-Б-фракции [49]. Прежде
чем подробно рассматривать разделение парафинов и олефинов, которые мо-
гут содержаться во фракции С4, следует коротко остановиться на выделении
изобутена экстракцией 65%-ной серной кислотой. Экстракция фракции С4
65 %-ной серной кислотой проводится под давлением, гарантирующим проте-
кание процесса в жидкой фазе. При этом образуются два слоя: нижний,
состоящий из шрет-бутилсерной кислоты и верхний — свободный от изобу-
тена. При поддержании определенной температуры, концентрации кислоты
и времени контакта можно практически количественно извлечь изобутен из
верхнего слоя. Из тпрет-бутилсерной кислоты большую часть изобутена
удается регенерировать разбавлением mpem-бутилсерной кислоты, примерно
до 45%-ной крепости, водой и последующей отдувкой водяным паром. Осво-
бождающийся при этом газ после промывки водой компримируется, конден-
сируется и подвергается ректификации под давлением.
Экстракция серной кислотой проводится примерно при 35—40°; при этой
температуре бутадиен еще не затрагивается и другие олефины также прак-
тически не растворимы в кислоте.
78
На рис. 37 показана упрощенная схема экстракции изобутена. Исход-
ный продукт — фракция С4, содержащая от 10 до 35% изобутена, экстраги-
руется в условиях противотока 65 %-ной серной кислотой. Свежая фракция
С4 поступает в колонну 2, где встреча-
ется с уже содержащей изобутен серной
кислотой; при этом часть изобутена аб-
сорбируется. Готовый экстракт в колон-
не 3 продувкой водяным паром освобо-
ждается от изобутена, который поступает
на очистную установку, где освобождается
от сернистого ангидрида, полимерных про-
дуктов и т. д., а затем перегоняется.
Оставшаяся от этой операции 45%-ная
кислота концентрируется до 65 %-ной и
подается в колонну 1, где встречается с
поступающей из колонны 2 фракцией, со-
держащей еще некоторое количество изобу-
тена. Отсюда кислота с растворенным в пей
изобутеном направляется в колонну 2, где
встречается со свежей фракцией С4; свобод-
ная от изобутена фракция удаляется через
верх колонны 1.
Дальнейшее разделение
свободной от изобутена смеси
углеводородов С4. Дальнейшее раз-
деление не содержащей изобутена газовой
Рис. 37. Упрощенная схема экст-
ракции изобутена серной кисло-
той из фракции С4 и последующей
регенерации.
1 — абсорбционная колонна I; 2 —
абсорбционная колонна II; 3 — колон-
на для регенерации.
Линии-. I — фракция С4, содержащая
изобутен; II — тощий экстракт; III—
регенернропанная кислота; IV — гото-
вый экстракт; V — из’ установки дли
концентрации кислоты-, VI — 45 %-кис-
лота; VII — пар; VIII — к установке
для очистки изобутена-, IX— свободная
от изобутена фракция С4; X— газ.
содержащий остатки изобутена.
углеводородной смеси С4, при условии, что
она не содержит также бутадиена, производится следующим образом (рис. 38).
Сначала посредством предварительной ректификации в соответствую-
щей колонне из смеси выделяют три первых углеводорода из указан-
Рис. 38. Схема разделения
бутанов и бутенов посред-
ством экстрактивной перетопки
с применением фурфурола в
качестве растворителя.
1 —ректификационная колонна;
2 — колонна экстрактивной пере-
гонки; 3 — разделитель.
Линии: I — свободная от изобутена
и бутадиена фракция С4; II — пзо-
бутан и бутен-I; III—н-бутан и
бутен-2; IV — пзобутан*, V— фур-
фурол; VI — бутен-f; VII— н-бу-
тав; VIII —бутен-2.
ных в табл. 4, имеющих низкие температуры
кипения, а именно: изобутан, изобутен и
бутен-1. Большая часть обоих бутенов-2 и н-бу-
тап остаются в колонне как остаток. Изобу-
тан и бутен-1 разделяются экстрактивной
перегонкой. Бутен-1 остается в фурфуроле,
а пзобутан отгоняется. Бутен-1 выделяется из
фурфурола нагреванием в особой колонне.
Смесь «.-бутана и бутенов-2 разделяется
также экстрактивной перегонкой, причем н-бу-
тан отгоняется, а оба бутена-2 остаются в фур-
фуроле. Они выделяются из него затем в осо-
бой колонне.
Таким образом, относительно сложная по
составу фракция С4 легко разделяется на
составные части.
Колонна для экстрактивной перегонки
представляет собой агрегат со 100 тарелками,
в большинстве случаев разделенный па две
секции по 50 тарелок в каждой.
3. РАЗДЕЛЕН!]Г, УГЛЕВОДОРОДОВ С4 ПРИ
ПОЛУЧЕНИИ БУТЛДИЕНА СТУПЕНЧАТЫМ
ДЕГИДРИРОВАНИЕМ и-БУТЛИА
а) Общие сведения
В принципе бутадиен из н-бутана может
получаться двумя методами дегидрированием
и-бутана в бутеп, который затем очищается и
79
на второй установке дегидрируется до бутадиена (способ Филлипс-Пет-
ролеум), или дегидрированием бутана в бутадиен за один проход, без
промежуточной бутеновой стадии (способ Гудри). Способы выделения
бутадиена при этом также различны. По способу Гудри бутадиен выде-
ляется из печных газов селективно раствором медной соли, а по способу
Филлипса для очистки бутена с первой ступени и бутадиена со второй
ступени применяется метод экстрактивной перегонки (дистекс-процесс).
б) Первая ступень дегидрирования
При способе Филлипса необходимо, чтобы как «-бутан — исходный мате-
риал для первой ступени, так и «-бутен — исходный материал для второй
ступени были очень чистыми. Однако реакция дегидрирования «-бутана про-
текает не настолько полно, чтобы в послереакционной смеси находились
лишь непрореагировавший «-бутан, водород и
н-бутен — в ней содержится еще некоторое коли-
чество бутадиена, изобутена и изобутана и одно-
временно немного углеводородов С3 и высокомо-
лекулярных, содержащих более четырех угле-
родных атомов в молекуле.
Для переработки газовой смеси, выходящей
с первой ступени дегидрирования, ее комприми-
руют, абсорбцией под давлением освобождают от
водорода и, наконец, стабилизируют, т. е. отде-
ляют от углеводородов, кипящих ниже темпера-
туры кипения углеводородов С4.
Полученная таким образом фракция С4 в
отдельной колонне подвергается тщательной
ректификации, в ходе которой бутен-1, кипящий
при—6,26°, отделяется от и-бутана (температура
кипения —0,5°) и от обоих бутенов-2, кипящих
при +0,88 и +3,72° (рис. 39).
Ректификационная колонна имеет 100 таре-
лок и разделена на две части. Бутадиен, содер-
жащийся в смеси углеводородов С4, поступающей
с первой ступени дегидрирования в виде азеот-
ропной смеси с «-бутаном, кипящей при —5°,
отделяется вместе с бутеном-1. Далее с бутеном-1
уходят содержащиеся в малых количествах изо-
бутан и изобутен, а также последние следы
углеводородов, кипящих ниже С4, которые не
были полностью отделены при стабилизации.
Полученная таким образом фракция бутена-1 поступает на стабилизацион-
ную установку (депропанизатор), где освобождается от всех низкокипя-
щих загрязнений, а оттуда направляется на вторую ступень дегидриро-
вания.
Остаток из колонны со 100 тарелками состоит из и-бутана, который
только в незначительной части, как азеотроп, уходит с бутадиеном, и обоих
бутенов-2, которых при каталитическом дегидрировании н-бутана получается
значительно больше, чем бутена-1. Этот остаток депентанизируется, а затем
подвергается экстрактивной перегонке для отделения бутана от бутенов.
Перегонка производится также в колонне со 100 тарелками, состоящей из
.двух секций. Отходящий в качестве дистиллята бутан, содержащий еще
3—4% олефинов, возвращается на первую ступень дегидрирования. Раство-
ренная в фурфуроле смесь обоих бутенов-2 идет в разделительную колонну,
•откуда после освобождения от фурфурола она направляется на вторую сту-
пень дегидрирования.
Рис. 39. Схема первой сту-
пени получения бутадиена
каталитическим дегидриро-
ванием «-бутана.
1 — ректификационная колон-
на; 2 — депентанизатор; 3 —
колонна экстрактивной пере-
гонки; 4 —колонна для выде-
ления бутена-2 из фурфурола;
6 — депропанизатор.
Линии: I — свободный от во-
дорода сжатый газ после де-
гидрирования н-бутана; II •—
н-бутан и бутен-2; III—бутен-1;
IV — н-бутан; V — фурфурол;
VI — Сэ и более легкие углево-
дороды; VII — Сб и высшие
углеводороды; VIII — бутен-1
на дегидрирование в бутадиен
(вторая ступень).
.80
в) Вторая ступень дегидрирования
На второй ступени дегидрирования смесь трех бутенов (бутена-1 и цис-
и транс-бутенов-2) дегидрируется в бутадиен. Здесь, как и на первой ступени
дегидрирования, степень превращения не превышает в среднем 22%. Выхо -
дящий из реакционной печи газ в основном состоит из смеси н-бутенов, бута-
диена и водорода. Вследствие крекинга, изомеризации и других побочных
реакций в газе содержатся также ограниченные количества изобутена, изо-
бутана, гомологов ацетилена, в частности диметилацетилен, и выше- и ниже-
кипящие составные части.
Разделение газа производится
примерно следующим образом
(рис. 40). После компримирования
и отделения водорода абсорбцион-
ным способом фракция С4 стабили-
зируется. При этом отгоняются
кипящие при —23° метилацетилен
и пропан, образующие азеотропную
смесь. Смесь углеводородов С4 затем
ректифицируется в колонне, имею-
щей 100 тарелок. Здесь отделяется
смесь из бутена-1 и бутадиена с
некоторым количеством изобутана,
изобутена и н-бутана (бутадиеновый
концентрат), причем н-бутан частич-
но уходит с дистиллятом, а частью
остается в остатке. В остатке оста-
ются оба бутена-2, часть н-бутана и
гомологи ацетилена (С4). В этой
связи интересно сопоставить темпе-
ратуры кипения отдельных изоме-
ров в нормальных условиях (см.
стр. 11 и 36) с летучестью в усло-
виях экстрактивной перегонки (см.
стр. 78). Остаток поступает в депента-
низатор, где от него отделяются выс-
шие углеводороды, а головной
продукт, состоящий из бутена-2,
возвращается па вторую ступень
Так называемый бутадиеновый концентрат поступает для дальнейшего
разделения в колонну экстрактивной перегонки, имеющую 100 тарелок.
Рис. 40. Схема получения бутадиена на
второй ступени дегидрирования при по-
мощи дистекс-процесса.
1 — ректификационная колонна; 2 — колонна
экстрактивной перегонки; з — разделительная
колонна для бутадиена; 4 — депентанизатор.
Линии'. I — фракция С4 со второй ступени, сво-
бодная от водорода и стабилизированная; II —
бутадиеновый концентрат (бутадиен, бутен-1 и
другие углеводороды); III— фурфурол, насыщен-
ный бутадиеном; IV — фурфурол, свободный от
бутадиена; V — бутен-1, н-бутан и другие угле-
водороды; VI — чистый бутадиен; VII — бутен-2;
VIII — бутен-2 обратно во вторую ступень;
IX —С5 и высшие углеводороды.
дегидрирования.
Таблица 46
Результат переработки бутен-бутадиеновой смеси после дегидрирования
н-бутена посредством экстрактивной перегонки
Компоненты Головной про- дукт обычной колонны со 100 тарелками, % объемн. Головной продукт из колонны экстрактивной пе- регонки со 100 та- релками, % объемн. Головной продукт из фурфуролотде- лительной колон- ны, % объемн.
Изобутан .... 5,8 8,3
Изобутен .... 6,8 9,8 —
Бутен-1 29,7 42,8 —
Бутадиен-1, 3 . . 26,2 1,2 83,8
н-Бутан 15,2 21,6 —
транс-Бутен-2 . . 13,0 14,6 9,2
^ис-Бутеп-2 . . . 3,3 1,7 7,0
6 Заказ 596.
81
Из этой колонны как головной продукт получают бутен-1, н-бутан, изобутан
и изобутен, тогда как бутадиен и часть бутена-2 остаются в растворе.
Табл. 46 дает представление о результатах экстрактивной перегонки
бутадиенового концентрата.
Полученный таким образом сырой бутадиен подвергается ректифика-
ции в колонне со 120 тарелками. Бутадиен со следами изобутена, бутена-1
и тп/ишс-бутена-2 отходят в качестве головного продукта; часть бутена-2
сохраняется в остатке. Анализ полученного описанным способом бутадиена
после перегонки сырого бутадиена в колонне со 120 тарелками дает следую-
щие результаты (в % вес.).
Бутадиен................. 98,71
транс-Бутеп-2 ...... 0,87
цис-Бутен-2 ................ 0,11
Бутен-1 ................... 0,11
Изобутен................... 0,07
Ацетилен................... 0,07
н-Бутан.................... 0,06
Изобутап................... 0,00
ЛИТЕРАТУРА
1. Chem. Engng. News, 30, 2983, 1952.
2. С и г t i s s E. O. Chem. a. Ind., 18—23, Aug., 1953.
3. Read D. Oil a. Gas J., 46, 44, 72—74, 92—93, 1948; К i mb a 1 1 T. B.,
Scott J. A. Petrol. Refiner, 27, 6, 326, 1948; S c h m e 1 i n g F., Erdol und Kohle,
5, 513, 1952; Willing E. Erdol und Kohle, 7, 561—565, 1954; Pollock A. W.
Petrol. Refiner, 34, 2, 127—128, 1955.
4. Petrol. Process., 5, 1058—1062, 1952; Petrol. Refiner, 33, 9, 181, 1954; Wal-
ther C. Erdol und Kohle, 3, 327—330, 1950.
5. M urph гее E. V. Chem. Engng. Progr., 49, 9, 28, 1953; Johnson F. C.,
Wood R. E. Petro], Refiner, 33, 11, 157—160, 1954; Barr F. T., J a h n i g [С. E.
Chem. Engng. Progr., 51, 167—173, 1955.
6. R e a d D. Petrol. Process, 3, 986, 1948; Petrol. Refiner, 33, 9, 1954.
7. Stadtherr J. Petrol. Refiner, 25, 2, 109, 1946; Carlsmith L. E.,
Johnson F. B. Ind. Engng. Chem., 37, 451, 1945.
8. Oil a. Gas J., 53, 46, 142—143, 1955; Simpson T. P., Bergstrom E. V.,
Eastwood S. C. Science of Petrol., V, 11, 239—252, 1953.
9. Hofmann H. Chem. Ing. Techn., 27, 194, 1955; Wickham H. P., К a m p-
tner H. Erdol und Kohle, 6, 624, 1953; Monrad С. C. Science of Petrol., V, II, 201,
1953; Murphree E. V. с сотрудн. Science of Petrol., V, 11, 253, 1953.
10. Gilmore F. E., Bauer R. D. Petrol. Refiner, 30, 9, 196, 1951.
11. Murphree E. V., Semaine C. R. Techn. de L’AFTP, 20—25, VI,
1949, 300.
12. W h a I 1 e у H. К. Chem. and Ind., 3—8, Aug., 1953.
13. S a c h a n e n A. N. Conversion of Petroleum, 2 aufl. Reinhold Puhi, Co. N. Y.,
1948, 543.
14. И п а т ь e в В. H. и др. Ind. Engng. Chem., 32, 268—272, 1940.
15. W a t s о n С. C., Newton F. с сотрудн. Trans. Am. Inst. Chem. Engr.,
40, 309-315, 1944; Dodd R. H., Watson К. M. Trans. Ibid., 42, 263—291, 1946.
16. H о r n a d a у G. F. Petrol. Refiner, 32, 9, 131—134, 1953.
17. R u s s e 1 R. P. с сотрудп. Trans. Am. Inst., chem. Engr., 42, 1—14, 1946.
18. S c h u t H. C. Chem. Engr. Progress, 43, 103—116, 1947.
19. Petrol. Process., 1, 135, 1946; Legutke G., Geiseler G. Chem. Techn..
10, 719—724, 1955.
20. F г о 1 i c h P. K., W i e z e v i c h P. J. Ind. Eng. Chem., 27, 1055—1062,
1935.
21. Schutt H. C. Chem. Engng. Progress., 43, 103—116, 1947; Cornell P. W.
с сотрудн. Petrol. Engr., 26, 12, C 34—41, 1954.
22. К i 1 p a t r i с k M. О. с сотрудн. Oil a. Gas J., 53, 1, 162—165, 1954- Petrol.
Refiner, 33,4, 171—174, 1954.
23. F a r n s w о г t h J. F. с сотрудн. Ind. Engng. Chem., 47, 1517—1522, 1955;
С о b e r 1 у C. W. с сотрудн. Petrol. Process., 8, 377—382, 1953.
24. E d g a r J. L. Ind. Chemist., 27, 345—349, 1951.
25. King С. C., Warburton J. Oil a. Gas J., 51, 31, 92—94, 1952.
26. Winnacker K. Chem. Ind. Techn., 27, 400, 1955.
27. Groll H. P. A. Ind. Engng. Chem., 25, 789, 793, 1933.
28. R о w 1 e у D., Steiner H. Discuss. Faraday Soc., 10, 198—213, 1951.
29. Weizmann с сотрудн. Ind. Engng. Chem., 43, 2312—2325, 1951; Bor-
rows E. T. с сотрудн. Third World Petrol. Congr. 1951, Proceedings, V, 159—171-
Brill E. G., Leiden, 1951.
30. E a s t w о о d S. С., P о t a s A. E. Petrol. Engr., 19, 12, 43—46, 1948.
31. W u s t г о w W., Kratz H. Erdol und Kohle, 8, 82—86, 1955.
82
32. Ипатьев В. Н. и П и н е с Г. Ж. орг. хим., I, 464—489, 1936; Н амет-
кин С. С. и А бак у мо век ая Л. Н. Ж. общ. хим., 15, 358—362, 1945.
33. L a n g 1 о i s G. Е. Sympos. on Catalisis in Hydrocarbon, Chem. Am. chem.
Soc. Meeting, Atlantik City Sept. 79, 1952.
34. M c A 1 1 i s t e r S. H. Oil a. Gas J., 36, 26, 139—142, 1937.
35. W i n n a с k e г К. Kunststoffe, 47, 401, 1957.
36. Tongberg С. O. J. Am. Chem. Soc., 54, 3706, 1932.
37. Pomerantz P. J. Research nat. Bureau of Standards, 48, 76—81, 1952.
38. Howard F. L. J. Research nat. Bureau of Standards, 38, 365—395, 1947.
39. McAllister S. H. Refiner and nat. Gasoline Manufacturer, 16, 493, 1937;
Ind. Chem., 518, Sept. 1946.
40. Oil a. Gas. J., 55, 35, 118, 1957.
41. Birch S. F. J. Inst. Petrol., 38, 69-87, 1952.
42. Ziegler К. с сотрудн. Brennstoffchem., 33, 153, 1952; Angew. Chem., 64,
323, 1952.
43. Ziegler К. исотрудн. Brennstoffchem., 35, 21/22, 321,1954.
44. G u i 1 1 e m i n А. и сотрудн. Third World Petrol. Congress 1951, Proceedings,
IV, 184—191, E. G. Brill, Leiden.
45. Goldstein F. R. The Petroleum Chemicals Industry, E. a. F. N. Spon Ltd,
London, 112, 1949.
46. Berg C. Trans. Amer. Inst. chem. Engr., 42, 655, 1946; Petrol. Refiner, 32,
11, 132, 1953.
47. Buell С. K., Boatright R. G. Ind. Engng. Chem., 39, 695, 1947; A t-
k i n s G. T., Boyer С. M. Chem. Engng. Progr., 45, 553, 1949.
48. Benedict M., Rubin L. C. Trans. Amer. Inst. chem. Engr., 41, 353,
1945; Happel J. и сотрудн. Trans. Amer. Inst. chem. Engr., 42, 189—214, 1946.
49. Baumann G. R., Smith M. R. Petrol. Refiner, 33, 5, 156—159, 1954.
6*
Глава III
ДИОЛЕФИНЫ
I. БУТАДИЕН
А. ОБЩИЕ СВЕДЕНИЯ
К важнейшим продуктам нефтехимической промышленности относится
бутадиен. При совместной полимеризации со стиролом бутадиен дает син-
тетический каучук Буна GR-S или S. Общее производство бутадиена со-
ставило, например, в США в 1956 г. 650 тыс. т.
Хотя природный каучук представляет собой полимер изопрена (2-метил-
бутадиен), однако бутадиен получается значительно проще и исключительно
легко полимеризуется; поэтому в настоящее время в качестве основы для
производства синтетического каучука применяют почти исключительно
бутадиен. Получение бутадиена из ацетилена через ацетальдегид-ацеталь-
доль и 1,3-бутиленгликоль по так называемому четырехступенчатому спо-
собу большого интереса не представляет. В данной книге не рассматривается
детально способе. В. Лебедева получения бутадиена из этилового спирта,
хотя этиловый спирт является исключительно важным и массовым про-
дуктом нефтехимической промышленности (гидратирование этилена,
см. стр. 200).
Получение бутадиена из этилового спирта разработано С. В. Лебедевым
[2] и осуществлено в Советском Союзе в больших масштабах. Пары спирта
пропускают над катализатором, представляющим собой комбинацию окиси
алюминия и окиси цинка, при 400° и пониженном давлении (0,25 ат). Ката-
лизатор обладает одновременно дегидрирующим и дегидратирующим дей-
ствием. Выход бутадиена составляет около 60% вес. от спирта. Может при-
меняться также катализатор окись магния — окись хрома или окись ко-
бальта — окись магния.
В США во время второй мировой войны был разработан фирмой Карбид
энд Карбон Кемикел компани двухступенчатый метод получения бутадиена
из этилового спирта. Спирт над медью при 400° дегидрируется в ацеталь-
дегид, который затем на второй ступени с трехмолярным избытком спирта
при 350° над катализатором из 2% пятиокиси тантала и 98% силикагеля
преобразуется в бутадиен [3].
Вторая ступень в синтезе бутадиена представляет альдольную конден-
сацию, влекущую за собой дегидратацию:
ОНИ О
// / / //
2СН3—С -> СН3—С—СН2—С -» СН3—СН=СН—С
ХН ^ОН \
84
Образующийся кротоновый альдегид тотчас же восстанавливается спир-
том в кротиловый спирт, который затем также быстро дегидратируется:
О О
Л Л
СН3СН=СН—С 4-СН3СН2ОН-> СН3СН=СН—СН2ОН+СН3-С
\i Хн
CHj—СН=СН—сн2он -> сщ=сн—сн=сн2+н2о
В настоящее время важнейшим способом получения бутадиена является
каталитическое дегидрирование н-бутана и соответственно н-бутена, полу-
чаемых из природного нефтяного газа и газов нефтеперерабатывающих за-
водов. Получение бутадиена из бутана возможно двумя способами: либо
сначала дегидрируют н-бутан в первой ступени, получая н-бутен, который
затем после выделения дегидрируют на второй ступени в бутадиен, либо
одноступенчатым способом, при котором за один проход через печь из н-бу-
тана получают бутадиен. Дегидрирование н-бутана в н-бутен и выделение
последнего в чистом виде были кратко рассмотрены выше.
Б. ДВУХСТУПЕНЧАТЫЙ СПОСОБ ДЕГИДРИРОВАНИЯ БУТАНА
В качестве сырья для двухступенчатого процесса может применяться
смесь газов, содержащая н-бутен, которая образуется при крекинге и исполь-
зуется для получения высокооктанового карбюраторного топлива. Этот кре-
кинг-газ, как указывалось выше при помощи 65—70%-ной серной кислоты
может быть освобожден от изобутена, а затем экстрактивной перегонкой
с фурфулом из него может быть выделена газовая смесь, примерно на 90%
состоящая из бутенов. Первую
ступень дегидрирования можно
проводить так, как это предусмо-
трено при получении бутадиена
методом Стандард Ойл.
Дегидрирование н-бутена в
бутадиен можно осуществить одним
из двух способов. По способу
Филлипса 14] н-бутеновый кон-
центрат пропускгиот над катали-
затором окись хрома — окись алю-
миния при температуре 670—680°.
Этот катализатор пригоден также
для дегидрирования н-бутана в
н-бутен. Тепло для дегидрирования
подводится извне путем нагрева
заполненных катализатором тру-
бок. В процессе Стандард Ойл
дегидрирование бутенового кон-
центрата происходит над специальным катализатором [5], устойчивым
против действия водяного пара (рис. 41). При этом уже не требуется
наружный обогрев. Теплоносителем является применяемый в большом
избытке водяной пар; преимуществом является то, что очень сильно пони-
жается парциальное давление бутена, а это благоприятствует протеканию
дегидрирования как равновесной реакции
Рис. 41. Схема получения бутадиена методом
Стандард Ойл.
1 — перегреватель; 2 — реактор с контактом; 3 —
котел, обогреваемый отходящим теплом; 4 —холо-
дильник; 5 — компрессор; 6 — масляный абсорбер;
7 — разделитель; 8 — стабилизационная колонна.
Линии-. I — н-бутен; II — водяной пар; III —
конденсат для быстрого охлаждения; IV — жирное
масло; V — тощее масло; VI — Се + высшие угле-
водороды; VII — С3 + более легкие углеводороды,
Но, СО, СОо; VIII —сырой бутадиен.
С„Н]0«=» Бутен-1 + Н2
C4H]0j± 1щс-Бутен-2 + Н2
С4И]0£2 транс-Бутен-2 + Н2
1щс-Бутен-2# Бутадиен-1,3 + Н2
—31,4 ккал/моль
—28,2 ккал/моль
—27,7 ккал/моль
—30,2 ккал/моль
85
Кроме того, вследствие большого избытка водяного пара уменьшается
отложение кокса на катализаторе. Катализатор, известный под № 1707,
имеет следующий состав: 72,4% MgO, 18,4% Ее20з, 4,6?4К2О, 4,6% GuO.
При избытке водяного пара порядка 10—15 молей на 1 моль бутена
последний дегидрируется примерно на 25%. Предварительно пар перегре-
вается до 700°, бутеновая смесь до 530°. Оба газа смешиваются и в течение
около 0,2 сек. пропускаются над катализатором, имеющим форму таблеток
и находящимся в трубках из легированной стали. Температура дегидриро-
вания на входе в печь около 670°. Разница между температурами на входе
и выходе равна примерно 25°, что объясняется эндотермическим характером
реакции. В некоторых установках, чтобы обеспечить возможность непрерыв-
ного ведения процесса, имеется два реактора, из которых в одном все
время происходит регенерация. Последнюю проводят прекращая подачу
бутена в реактор. Перегретый водяной пар реагирует с высокоактивным
коксом с образованием водяного газа.
После выхода из печи газы быстро охлаждаются примерно до 500°.
Затем они отдают тепло котлу-утилизатору, в котором образуется часть
необходимого для процесса пара. После сжатия и дальнейшего охла:кдения
газы промываются легким маслом при условиях, обеспечивающих растворе-
ние в масле углеводородов С4, в то время как углеводороды С3 и более легкие,
а также водород, окись углерода и двуокись углерода отделяются как го-
ловной продукт.
Углеводороды С$ выделяются из абсорбционного масла нагреванием,
а затем в отдельной колонне освобождаются от Сз и высокомолекулярных
углеводородов. В результате получается сырой бутадиен.
Условия работы реакторов дегидрирования в способе Стандард Ойл
компани [6] следующие
Температура, °C................................ 620—680
Объемное отношение водяной пар : бутен .... 14—18 : 1
Парциальное давление бутена на входе, мм рт. ст. 60—80
Давление в печи над катализатором, ат............... ~0,7
Давление в печи за катализатором, » ........ ~0,21
Скорость прохождения, объем бутена/объем ка-
тализатора в час ................................. 125—225
Превращение бутена при однократном проходе через печь составляет
20—30%. В бутадиен переходит 70—75% от всего превращенного бутена.
В бутадиеновом процессе Филлипса исходный материал — бутан — на
первой ступени дегидрируется в бутен, который на второй ступени превра-
щается в бутадиен. Вторая ступень работает практически так же, как первая,
т. е. с катализатором окись хрома — окись алюминия, который находится
в обогреваемых снаружи трубках. Дегидрирование на второй ступени идет
при температуре около 670°, т. е. примерно на 140° выше, чем на первой сту-
пени. Водяной пар подается в значительно меньшем количестве, чем в про-
цессе Стандард Ойл. Здесь он не является теплоносителем, а служит лишь
средством понижения парциального давления и уменьшения отложения
кокса на катализатор.
В. ОДНОСТУПЕНЧАТЫЙ СПОСОБ ПОЛУЧЕНИЯ БУТАДИЕНА
(ПРОЦЕСС ГУДРИ)
Этот процесс получения бутадиена является единственным до настоящего
времени одноступенчатым процессом, в котором бутан, минуя стадию бутена,
превращается непосредственно в бутадиен. Процесс осуществлен теперь
также и на химических заводах Хюльса в Марл (округ Реклинхаузен).
Поэтому необходимо рассмотреть его более подробно. В США также соору-
жаются новые установки, работающие по этому способу.
В процессе Гудри работают с катализатором окись хрома — окись
алюминия примерно при 600° и абсолютном давлении 100—150 мм рт. ст.
86
Фракция С4 после прохождения бутан-бутеновой смеси через печь содержит
8—12% бутадиена. В этом процессе представляет интерес решение вопроса
о подводе тепла, необходимого для эндотермической реакции дегидриро-
вания. Подвод тепла при помощи перегретого водяного пара здесь невозмо-
жен, так как устойчивый против действия водяного пара катализатор № 1707
не пригоден для прямого дегидрирования «-бутана в бутадиен и может приме-
няться только в двухступенчатом процессе.
При дегидрировании на катализаторе отлагается довольно много угле-
рода, понижающего его активность. Этот углерод должен удаляться сжига-
нием в струе воздуха. В процессе Гудри таблетированный катализатор сме-
шан с большим числом алундовых шариков, которые сами каталитическим
действием не обладают, но имеют большую
теплоемкость. Тепло, освобождающееся при
регенерации, воспринимается этим теплоноси-
телем и отдается им в процессе дегидриро-
вания. Теплоноситель препятствует также
чрезмерному повышению температуры при
регенерации, что чрезвычайно важно, так как
при нагревании до 700—750° активность ката-
лизатора быстро ухудшается.
Печи горизонтальные, с огнеупорной изо-
ляцией реакторов, изготовляемых из высоко-
хромистой стали и заполняемых катализатором
и теплоносителем. Установка состоит из ряда
печей, каждая из которых находится в работе
8—10 мин., а затем переключается на регене-
рацию. Температурные условия здесь более
жесткие, чем в установках, работающих по
способу Филлипса или Стандард Ойл. В про-
цессе работы важно, чтобы катализатор не за-
грязнялся железом, потому, что загрязнения
сильно повышают количество коксовых отло-
жений и способствуют образованию низко-
Рис. 42. Схема экстракции
бутадиена из сырого бута-
диена в газовой фазе.
1 — бутадиеновый абсорбер; 2 —
установка для концентрации бута-
диена; 3— бутадиеновый десорбер.
Линии: I — сырой бутадиен; II —
С4 + бутадиен; III — свободная от
бутадиена фракции С< и аммиак для
промывки; IV — медно-щелочной
раствор; V — десорбированный бу-
тадиен; VI—бутадиеновый раствор;
VII — бутадиен на промывку ам-
миаком и перегонку.
молекулярных газов.
В период регенерации эти печи сильно перегреваются. Высокая темпера-
тура в свою очередь ускоряет коксообразование и это приводит постепенно
к столь значительному повышению температуры, что катализатор дезакти-
вируется.
С другой стороны, какая-то часть катализатора может пметх, температуру
ниже необходимой для протекания реакции дегидрирования. В этом случае
на катализаторе будет отлагаться мало кокса, поэтому и количество тепла,
выделяющегося при регенерация, будет очень малым. Однако, как показы-
вают вычисления, это различно в температурах может в процессе регенера-
ции выравниваться, если воздух, применяемый для выжигания кокса, обла-
дает в массе теплоемкостью, составляющей одну четвертую часть теплоем-
кости катализатора.
Процесс протекает следующим образом . «-Бутан и н-бутен из газов цир-
куляции проходят над катализатором, дегидрирующим н-бутап в /(-бутен,
а «-бутен в бутадиен (рис. 42). После быстрого охлаждения газ комприми-
руется и, как обычно, путем абсорбции освобождается от водорода и низко-
молекулярных продуктов крекинга. Выделенная из абсорбента фракция C.i
для извлечения 8—12% бутадиена обрабатывается на экстракционной уста-
новке аммиачно-ацетатным раствором меди. Отделяющаяся смесь «-бутана
и н-бутена (газ циркуляции) вместе со свежим «-бутаном возвращается в ре-
актор для дегидрирования.
По окончании дегидрирования реактор эвакуируют (удаляют еще остав-
шиеся в нем углеводороды), а затем выжигают кокс с подачей в реактор
предварительно подогретого воздуха.
87
Условия работы в одноступенчатом бутадиен-процессе Гудри сле-
дующие.
Средняя температура, °C........................ 607
Абсолютное давление, мм рт. ст................. 100—150
Объемная скорость подачи, считая на жпдкую
фракцию С4, объем С4/па объем катализатора
в час........................................ 1,0—1,5
Продоля?ительиость рабочего периода, мин. . . . 8—10
В табл. 47 дай примерный состав газов — сырого, циркуляции, посту-
пающего в реактор и выходящего из реактора (печи). За один рабочий период
(однократный проход газа через печь) содержание бутадиена в газе достигает
11 —11,5%. Конечный выход бутадиена при многократной циркуляции
н-бутана и соответственно бутенов составляет около 52%.
Таблица 47
Состав газов в процессе Гудри (% вес.)
Компонент Сырой газ Газ цирку- ляции Газ, посту- пающий в реактор Газ, выхо- дящий из реактора Конечный выход па к-бутан
Водород 1,9
Метан — — — 1,0 ——
Са — — — 1,1 —
Са — — — 1,2 —
Изобутап 1,0 11,2 9,2 9,0 —
н-Бутены — 37,6 29,9 30,0 —
к-Бутан 98,5 50,2 60,1 40,0 —
Бутадиен — 0,7 0,5 11,0 51,7
Сб и выше 0,5 0,3 0,3 0,5 —
Кокс — — — 2,6
Инертные газы . . . — — — 1,7 —
Г. ДРУГИЕ НЕФТЕХИМИЧЕСКИЕ СПОСОБЫ ПОЛУЧЕНИЯ БУТАДИЕНА
Во время второй мировой войны, да и в настоящее время, только неболь-
шая часть бутадиена получается пиролизом тяжелых бензиновых, кероси-
новых и газойлевых фракций. Бутадиен в этих условиях является побочным
продуктом, главные же продукты — этилен и пропен.
Пиролиз проводится при 700—780° в регенеративных печах в присутствии
водяного пара. В трубчатых печах первостепенной проблемой является от-
ложение кокса. Трубы выполняются из высокохромистой стали. Пиролиз
ведут в присутствии большого количества водяного пара, чтобы уменьшить
коксообразование.
Состав газа сильно зависит от состава исходного сырья и условий ве-
дения процесса.
Средний состав газов пиролиза тяжелого бензина или керосина в % мол.
приведен ниже.
Водород .................... 8
Метан ......................20
Этилен......................20
Этан........................10
Фракция С3 ................30
» С4 ...................10
» С5 и выше ............ 2
Образующиеся при пиролизе жидкие углеводороды имеют в основном
ароматический характер. В составе газов основное место занимают фракции
С» и Сз, количество же фракции С*, содержащей бутадиен, невелико.
88
Содержание бутадиена во фракции Са составляет 20—50% в зависимости
от условий пиролиза, или не более 3—5% считая на исходное сырье. Выделе-
ние бутадиена из фракции С4 производится уже известным способом (стр. 81).
В США таким путем должно производиться около 40 тыс. т бутадиена.
Д. ОЧИСТКА СЫРОГО БУТАДИЕНА
Получение бутадиена путем ректификации фракции С4 невозможно.
Для выделения бутадиена применяют два процесса — экстрактивную пере-
гонку и экстракцию бутадиена аммиачно-ацетатным раствором меди.
а) Способ экстрактивной перегонки
О получении бутена и бутадиена способом экстрактивной перегонки,
а также о принципах, лежащих в основе этого метода, уже говорилось выше
(см. стр. 80).
б) Экстракция бутадиена аммиачно-ацетатным раствором
одновалентной меди
Аммиачно-ацетатный раствор одновалентной меди является наилучшим
средством выделения бутадиена из смеси других углеводородов фракции С4.
Способ экстрактивной перегонки особенно пригоден, если концентрация бу-
тадиена очень мала. Аммиачно-ацетатный раствор свободен от недостатков,
присущих аммиачному раствору хлористой меди, который обладает чрезмер-
ной коррозийной агрессивностью и муравьинокислому раствору, выделяю-
щему со временем карбонат
меди.
Аммиачно-ацетатный рас-
твор меди применяется при
экстрактивной перегонке ана-
логично фурфуролу. Экстрак-
ция бутадиена может произво-
диться из смеси, находящейся
в жидком или газообразном
состоянии. Чаще всего процесс
ведется в жидкой фазе спосо-
бом, разработанным Стандард
Ойл Компани. Преимущество
этого способа связано с повы-
шенной растворимостью бута-
диена при низкой температуре
Таблица 48
Примерный состав аммиачно-ацетатного
раствора меди (1)
Компонент Содержание
моли! л % вес.
Медь 3,3 17,5
Уксусная кислота . . 4,0 20,0
Аммиак 11,0 15,5
Вода 31,3 46,9
и с уменьшением количества тепла реакции, выделяющегося в процессе.
Медный раствор поглощает также бутадиен-1,2 и углеводороды Сз
и С4 ацетиленового ряда. Последние растворимы в нем лучше бутадиена-1,3,
накапливаются в растворителе и отходят вместе с бутадиеном, способность
к полимеризации которого они сильно понижают. С другой стороны, ацети-
лид меди легко детонирует и, кроме того, в результате реакций полимери-
зации образует соединения, действующие как эмульгаторы. Поэтому аце-
тилены должны быть удалены, что может быть сделано путем нагрева мед-
ного раствора после выделения из него бутадиена. При этом образуются
продукты полимеризации, которые в последующем удаляют фильтрацией или
промывкой.
Состав аммиачно-ацетатного раствора дан в табл. 48.
Растворитель проходит через ряд ступеней, каждая из которых включает
турбосмеситель и отстойник, где в условиях противотока приходит в кон-
такт с жидкой углеводородной смесью, абсорбируя бутадиен (рис. 43).
Нерастворившаяся часть промывается водным раствором аммиака и возвра-
89
щается на установку дегидрирования. Насыщенный бутадиеном раствори-
тель подается в концентрационную колонну, где он орошается чистым бута-
диеном из десорбера, причем выделяются еще растворенные в нем монооле-
фины Ct. Образовавшаяся газовая смесь добавляется к сырому бутадиену п
возвращается в абсорбционную колонну. Медный раствор поступает в де-
сорбционную колонну, где выделяется бутадиен. После промывки водой он
еще раз перегоняется и в результате получают 98,5%-ный бутадиен. Усло-
вия ведения процесса в жидкой фазе следующие.
Соотношение медного раствора и исходного продукта,
кг/кг ’...................................... 8,0
Температура медного раствора при экстракции, °C —13
Давление при экстракции, ат ............ 3,1
Температура выделения (десорбции), °C....... 77
Давление в десорбере, ат.................... 1,05
Качества исходного и конечного продуктов, а также выход последнего
показаны в табл. 49.
Таблица 49
Состав исходного и конечного продуктов при экстракции бутадиена
медным раствором
Компонент Исходный про- дукт, % объемн. Продукт, свобод- ный от бутадиена, % объемн. Бутадиен, % объемн.
Сз 0,2 0,3 0,3
Бутадиен l(i,7 0,9 98,5 (мин.)
Изобутен 4,9 5,7 0,2
и-Бутен 62,8 75,1 1,0
Бутаны 15,3 17,9 —
с5 0,1 0,1 —
Сумма ... 100,0 100,0 100,0
Для синтеза бутадиена по способу Гудри в Хюльсе [9] потребляется
около 70 000 т бутана, поставляемого частью в цистернах, частью по трубо-
Рис. 43 Л Схема получения бутадиена одноступен-
чатым дегидрированием и-бутапа по Гудри.
1 — предварительный подогреватель; 2 — реактор с ката-
лизатором; з — компрессор; I — абсорбер; 5 — испаритель.
Линии: I — н-бутан; II — тощее масло; III — Щ + наи-
более легкие углеводороды; IV — н-бутаи + н-бутен
(циркуляция); V—бутадиен; VI — жирное масло.
проводам.
Для получения 45 тыс. т
каучука (сырой резины), в
котором бутадиен составляет
примерно 77% углеводород-
ной части (остальное сти-
рол), необходимы 35 тыс. т
бутадиена, каковые при
54 % -ном выходе могут быть
получены из 70 тыс. т
бутана.
В США для отдельных
сортов каучука введены не-
давно новые обозначения, а
именно: BR — бутадиено-
вый каучук, 1R — изопре-
новый каучук, CR — хлоро-
преновый каучук, NR —
синтетический натуральный
каучук, ABR — акрилбута-
диеновый каучук, 11R — изо-
бутенизопреновый каучук
90
бутилкаучук), NBR — нитрилбутадиеновый каучук (пербунан — буна),
NCR — нитрилхлоропреновый каучук, PBR — пиридинбутадиеновый кау-
чук, SBR — стиролбутадиеновый каучук (буна S), SGR — стиролхлоро-
преновый каучук, SIR — стиролизопреновый каучук.
Эти сорта каучуков и эластомеров для обозначения химического состава
маркируются еще буквами: М — цепь полиметиленового типа, N — азот-
содержащий, О — кислородсодержащий, Р — фосфорсодержащий, R — не-
насыщенный, Т — серусодержащий и U — кислород- и азотсодержащий.
II. ИЗОПРЕН
Изопрен (2-метил-1,3-бутадиен) в последнее время вновь привлек к себе
внимание благодаря тому, что на его основе в присутствии 0,1% лития при
40—50° или мюльхеймского катализатора полимеризации (комбинация три-
алкилалюминия и хлористого титана) можно получить синтетический каучук,
вполне идентичный натуральному и благодаря чистоте даже превосходящий
•его в некоторых свойствах. В присутствии лития происходит 1,4-полимери-
зация с предпочтительной ^пс-конфигурацией двойной связи. Полиизопрено-
вый каучук, называемый в США корал-каучуком или америполом SN, не
получил еще широкого промышленного значения из-за отсутствия дешевого
способа получения изопрена.
Изопрен является также компонентом смешанной полимеризации для
получения бутилкаучука (см. стр. 224). Его можно получить крекингом при-
родного (натурального) каучука и терпентина. Главная составная часть
терпентинового масла (живичного скипидара) при пиролизе на 60% превра-
щается в изопрен. Фракция Cs продуктов пиролиза на 90—95% состоит из
изопрена. Стоимость сырья, однако, высокая.
Главным источником изопрена является фракция Cs высокотемпера-
турного пиролиза нефтяных фракций, подобно тому как это показано выше
для бутадиена. Фракция Сз чрезвычайно сложна по составу и в зависимости
от условий пиролиза (650—760°) содержит 15—25% изопрена. Другими
главными составными частями являются пиперилин (пентадиен-1,3,), цикло-
пентадиен и пентен-1. Чистый изопрен можно выделить экстрактивной пере-
гонкой — способом, описанным выше для бутадиена. В качестве экстракци-
онной среды здесь применяется, например, ацетон с 5% воды. Перед экстрак-
тивной перегонкой богатые изопреном фракции отделяют обычной перегон-
кой от «пипериленовой фракции». Температуры кипения отдельных предста-
вителей -фракции Сз следующие.
Компонент Температу- ра кипения при нор- мальных условиях, °C Компонент I 1 Температу- ра кипения при нор- мальных условпях, °C
Изопентан .... 27,9 ци<:-Пептсп-2 37,0
Пентеп-1 30,1 2-мстилбутен-2 . . . . 1 38,6
2-метилбутсп-1 . . . 31,0 Ппперплсн (смесь изо-1
Изопрен 34,5 меров) 42,0
и-Пентан 36,1 Циклопентадиен .... 42,0
траис-Пентен-2 . . 35,9 Циклопеитен 44,4
Циклопентан 49,3
Получаемый таким образом изопрен применялся главным образом для
получения бутилкаучука и для опытных работ в области полимеризации.
Изопрен может быть также получен дегидрированием изоамилена,
например 2-метилбутена-1 или триметилэтилена посредством процесса,
91
разработанного Стандард Ойл Компани для бутена. Амилены могут быть
выделены из крекинг-бензинов.
О возможности получения изопрена из ацетона и ацетилена следует
только упомянуть.
В течение некоторого времени в технике применялся способ получения
изопрена из изобутена и формальдегида. Формальдегид взаимодействует
с изобутеном в присутствии 20 %-ной серной или 40 %-ной фосфорной кислоты
при комнатной температуре с примерно 60%-ным выходом 4,4-диметилдиок-
сана-1,3, который затем при пропускании над фосфорнокислым катализато-
ром при 220° разлагается на изопрен, формальдегид и воду.
СН3 О
^>С=СН2+2НС<^
СЩ И
сн2=с—сн=сн2+н2о+нсон
I
сн3
H3PO4
пары НаО
ЛИТЕРАТУРА
1. Ruebensaal С. F. Petrol. Refiner, 36, 3, 144, 1957.
2. Л е б е д е в С. В. Англ. пат. 331482, Zbl, П, 4319, 1930; франц, пат. 665917
Zbl, I, 3484, 1930.
3. Toussaint W. J., Dunn J. T., Jackson D. R. Ind. Engng. Chem.,
39, 120, 1947.
4. H a n s 0 n G. H., Hays H. L. Chem. Engng. Progr., 44, 431—442, 1948.
5. R u s s e 1 R. P. и сотрудн. Trans. Amer. Inst. chem. Engr., 42, 1—14, 1946.
6. Petrol. Refiner, 33, 12, 173, 1955.
7. H or n ad ay G. F. Petrol. Refiner, 33, 12, 173, 1954.
8. Oil a. Gas J., 55, 35, 113, 1957.
9. Erdol und Kohle, 9, 9, 631—653, 1956; Chem. Ztg., 79, 62, 1955; Chem. Ztg., 80,
296, 1956; Brennstoffchem. (Wirtschaftsteil), 36, 84, 1955.
Глава IV
АЦЕТИЛЕН
I. АЦЕТИЛЕН ИЗ КАРБИДА
В настоящее время большая часть ацетилена еще получается из карбида
кальция воздействием на него воды. Получение карбида кальция, требую-
щее исключительно много энергии, более всего развито там, где имеется
дешевая водяная энергия, как в Норвегии, Канаде и т. д. В Германии источ-
ником энергии для получения карбида является уголь. Получение карбида
не нефтехимический процесс. Недавно карбид начали получать из нефтяного
кокса. Этот весьма реакционноспособный и почти беззольный кокс является
исключительно ценным сырьем для получения карбида. Только в этом
смысле производство карбида можно рассматривать в качестве нефтехими-
ческого процесса.
Ацетилен из карбида получают двумя путями. Так называемый «влаж-
ный процесс» заключается в том, что карбид в мешалке обрабатывают во-
дой и образующееся согласно уравнению
CaC2-f-H2O -> НС^СН-|-Са (ОН)2
известковое молоко отводится из мешалки,
В «сухом процессе» наряду с ацетиленом образуется твердая гидро-
окись кальция.
Измельченный карбид подается в цилиндрические барабаны с несколько
большим, чем требуется по расчету, количеством воды, при этом образуется
свободный ацетилен. Ацетилен выделяется в виде примерно 97 %-ного про-
дукта. При'разложении карбида образуется еще некоторое количество серо-
водорода и фосфористый водород (фосфин), от которых ацетилен перед ис-
пользованием должен быть освобожден. Это можно сделать промывкой газа
разбавленной хлорной водой, которая разрушает оба эти загрязнения.
В заключение ацетилен промывают концентрированной натронной щелочью
и просушивают.
Недавно процесс получения карбида кальция на базе кокса и извести
значительно улучшен. В одном еще находящемся в опытной стадии методе,
разработанном Баденскими содовыми и анилиновыми фабриками, известь
и кокс обжигают в атмосфере кислорода в футерованной графитом шахтной
печи (1). Образование СО по уравнению
С + 1/2О2 —-► СО + 29,2 ккал
сопровождается значительным повышением температуры. Высококонцентри-
рованный жидкий карбид кальция периодически отводится из печи.
93
При оценке экономичности этого способа должна быть принята во вни-
мание ценность окиси углерода, которая выходит из печи в почти чистом
виде. В основе этого способа лежит процесс газификации угля, при котором
карбид отделяется как высокоценный шлак [2]. Окись углерода можно,
кроме того, конвертировать и смесь окиси углерода с водородом применять
как синтез-газ.
И. ДУГОВОЙ ПРОЦЕСС ПИРОЛИЗА МЕТАНА [3J
Из таких углеводородов, как метан, этан и пропан, содержащихся
в отходящих газах гидрирования угля или в природном газе пиролизом
при очень высоких температурах можно получить ацетилен. Проблема
подвода большого количества тепла, необходимого для эндотермического
процесса пиролиза, может решаться различными способами. Превращение
метана согласно уравнению
2СП4—> НС = CH-f-3H.>—97 ккал/моль
требует исключительно большого количества энергии. В течение длительного
времени эта задача решалась пропусканием смеси углеводородов через воль-
тову дугу. На химических заводах в Хильсе таким путем производилось
около 80 000 m/год ацетилена. Чем выше число углеродных атомов в парафи-
новом углеводороде, тем меньше энергии необходимо затратить при пиролизе
для получения ацетилена. Когда исходным материалом служит метан, рас-
четная потребность в энергии на 1 м3 ацетилена составляет 11 —12 квт-ч.
Если среднее число углеродных атомов на молекулу составляет от 1,4
до 1,5, что имеется при наличии в газовой смеси некоторого количества этана
и пропана, то расход энергии на производство 1 м3 ацетилена составляет
уже 8—9 квт-ч. При получении ацетилена методом дугового крекинга
в качестве побочного продукта всегда образуется этилен в количестве
20% вес. от ацетилена. В табл. 50 приведен состав исходного материала и
продукта пиролиза дуговой установки для производства ацетилена в Хюльсе.
Таблица 50
Анализ газов, поступающих на дуговую ацетиленовую установку и
выходящих с нее
Газ, поступающий на установку Газ, выходящий с установки
компонент % вес. компонент % вес.
Метан Этилен Пропап Пропсп Этап Бутан Бутен Высшие углеводоро- ды 84 9 2 2 1,4 0,6 0,6 0,4 Водород Метан и высшие . . . Ацетилен Этилен Азот Окись углерода .... Кислород ....... 50 25 16,8 3,9 2,5 0,9 0,2
Тотчас по выходе из дуговой печи газ охлаждается до 150°, путем
впрыска воды, затем освобождается от сажи в циклонах или посредством
суконных фильтров. Смолообразные полимеры удаляются из газа промывкой
маслом, синильная кислота — водой, а сероводород — окисью железа.
Газ в четыре ступени сжимается до 18 ат и после удаления высших ацетиле-
нов абсорбцией маслом под давлением промывается водой для извлечения
ацетилена. Водород, этилен и этан при этом не растворяются и выводятся из
абсорбера. Над водным раствором ацетилена давление понижают до 2 ат,
94
а выделяющийся при этом газ возвращают на компрессионную установку.
При понижении давления до атмосферного получают 93%-ный ацетилен.
Остаток ацетилена удаляют эвакуацией. Образующийся при получении
ацетилена этилен, еще смешанный с метаном и этаном, перерабатывается
далее на дистилляционной установке. Метан и этан возвращаются на уста-
новку дугового пиролиза.
А. ПИРОЛИЗ МЕТАНА ПО СПОСОБУ ЗАХСЕ [4]
Подвод больших количеств тепла, необходимых для осуществления
эндотермической реакции пиролиза метана в ацетилен, возможен также пу-
тем сжигания части газа в чистом кислороде. При этом выделяется тепло
в количестве, достаточном для расщепления оставшейся части углеводородов
в ацетилен.
Этот процесс, разработанный Захсе для БАСФ, широко применяется
в настоящее время многими фирмами США и Италии. В США по этому спо-
собу работают Монсанто Кемикал Компани, Карбид энд Карбон Кемикал
Компани и Америкен Цианамид Корпорейшн.
Предполагается, что в 1975 г. в США выработка ацетилена должна со-
ставить 1,3 млн т, из которых 75% будут получены из природного газа по
способу Захсе.
Способ состоит в том, что метан и кислород раздельно подогревают
примерно до 500° в подогревателях с непосредственным огневым подогревом,
а затем в молярном отношении 1 : 0,65 сжигают в специальных горелках.
Для достижения оптимального выхода ацетилена газы должны быть идеально
смешаны, а продукты реакции мгновенно охлаждаться, чтобы предотвратить
или по крайней мере уменьшить дальнейшее расщепление, особенно с обра-
зованием сажи. Приблизительно 2/з метана при этом сгорает, а ’/з превра-
щается в ацетилен. Температура пламени 1500—1600°, время пребывания
газов в аппарате 0,001—0,01 сек. Давление поддерживается несколько выше
1 ат. Метан используется на 95%, кислород на 100%.
Процесс сходен с автотермическим пиролизом этана в этилен (см. стр. 49).
Газы, получаемые при автотермическом пиролизе метана в ацетилен, имеют
следующий состав (в % объемн.).
Ацетилен ......... 8,5 Двуокись углерода . . 3,5
Водород.......... 56,0 1 г ___ Метан................... 6,5
Окись углерода . . 25,0 / Высшие ацетилены . . 0,5
Из приведенных данных следует, что большая часть метана при сгораний
переходит в смесь окиси углерода и водорода, которая представляет собой
отличный синтез-газ, или может быть использована для получения водорода
конверсией окиси углерода. Все это должно учитываться при оценке эконо-
мической целесообразности процесса Захсе. Из 1000 кг метана получается
в среднем 230 кг ацетилена и 1160 кг синтез-газа [5].
На 1 т ацетилена из синтез-газа после конвертирования можно полу-
чить 4 т аммиака.
По расходу энергии процесс Захсе является наилучшим, так как при
получении ацетилена из карбида кальция коэффициент использования энер-
гии составляет примерно 50%, в дуговом процессе — 66%, а в способе Захсе
эта величина достигает 75%. Для получения 1 м3 ацетилена пз карбида
требуется 11 квт-ч электроэнергии, 2,6 кг кокса и 3,6 кг извести. Для полу-
чения того же 1 м3 ацетилена способом Захсе необходимы 6 мя метана и
3,5 м3 кислорода.
Процесс Захсе является в настоящее время простейшим промышленным
процессом производства ацетилена, основанным на переработке природного
газа. Для получения 1 кг ацетилена необходимы следующие исходные про-
дукты: 4,3 кг парафиновых углеводородов, 4,9 кг водяного пара и 1,2—
2,0 квт~ч электроэнергии, расходуемой для работы компрессоров.
95
На каждые 50 м3 газа образуется примерно 1 кг сажи. Очень целесооб-
разна комбинация производства ацетилена с получением аммиака или циан-
амидных продуктов. Необходимый для получения аммиака или цианамида
кальция чистый азот может получаться из жидкого воздуха. Освобождаю-
щийся при этом кислород может быть использован для получения ацетилена.
Выходящие из печи газы после охлаждения и очистки от сажи сжи-
примерно до 10,5 ат. При последующей абсорбционной очистке
выделяются высшие ацетилены и, наконец, абсорбцией диметил-
формамидом — ацетилен. От-
сюда
99 % -ный
процесса
переработку сырого
приведена на рис. 44. Для
очистки ацетилена может
также применяться с очень
хорошими результатами ги-
персорбциоиная очистка,
основанная на принципе не-
прерывной газохроматогра-
фии.
маются
из
газа
44.
Рис.
далее получают
ацетилен. Схема
Захсе, включая
газа,
Схема получения ацетилена способом
Захсе.
1 — предварительный подогреватель; 2 — камера сгора-
ния; 3 — холодильник; 4 — оросительный холодильник;
5 —сажеотделитель; 6 — компрессор; 7—масляный абсор-
бер; 8 —диметилформамидный абсорбер; 9 — колонна ста-
билизации от легколетучих составных частей; 10— колонна
для отделения ацетилена.
Линии: I — кислород; II — метан; III — вода; IV — ди-
метилформамид; V — 99,8%-ный ацетилен; VI — высшие
ацетилены; VII —диметилформамид обратно на абсорбцию.
Б. ПИРОЛИЗ МЕТАНА
МЕТОДОМ ВУЛЬФА [б]
Процесс Вульфа для по-
лучения ацетилена состоит
в пиролизе природного газа
или пропана при темпера-
туре 1200—1400° и низком
парциальном давлении в пе-
чах, работающих по регене-
ративному циклу с периодами пиролиза и нагрева. Процесс Вульфа наи-
более применим там, где имеется много дешевого углеводородного сырья,
а смесь окиси углерода и водорода, получающаяся при пиролизе по методу
Захсе, не нашла бы применения.
Метод основан на том, что отходящие газы, образовавшиеся при пиро-
лизе, сжигаются в смеси с воздухом для нагрева огнестойкого материала,
подготовляя таким образом печь для пиролиза. Чтобы обеспечить регуляр-
ный и непрерывный поток пирогаза, установка состоит из очень многих
печей. В каждый данный момент в одной половине печей идет пиролиз исход-
ного сырья (газа), в то время как другая половина печей нагревается за счет
сжигания отопительного (отходящего) газа. Оборот каждой печи 60 сек.
В качестве отопительного газа используется отходящий газ (абгаз), полу-
чающийся при переработке газов пиролиза на ацетилен. Продукты сгорания
выбрасываются в атмосферу.
Пирогаз, как и в ранее описанных процессах, быстро охлаждается,
а затем перерабатывается. Понижение парциального давления газов в печах
пиролиза достигается добавкой водяного пара. Время пребывания продукта
в печи составляет около 0,1 сек. При этом способе работы сажа не образуется.
После сжатия до атмосферного давления газ проходит через установку
Котрелля, далее сжимается до 10 ат и поступает на дальнейшую перера-
ботку практически таким же методом, как и в описанном ранее способе
Захсе. Состав газов, выходящих из печей пиролиза, при использова-
нии в качестве исходного сырья пропана и природного газа показан в
табл. 51.
Газ содержит около 50% На, который также может найти промышлен-
ное применение.
«6
В табл. 52 дан выход ацетилена, отнесенный к проценту углерода, содержавшегося в исходном сырье и перешедшего при пиролизе в ацетилен для случаев переработки природного Таблица 51 газа, этана и пропана. В той же таблице Состав пирогаза в процессе Вульфа приведен выход ацетилена в килограм- при использовании пропана и мах на jQQ кг ИСХодного сырья, природного газа в качестве исходного 1 сырья
Компонент Исходное сырье, % объемн. Выход ацетилена в % углерода при пиролизе природного газа, этапа и пропана способом
про- пан природ- ный газ Вульфа
Исходный про- дукт Содержание углерода в ис- ходном продук- те, полученно- го при перера- ботке в виде ацетилена, % Выход аце- тилена, кг на 100 кг исходного продукта
Ацетилен Этилен Метан Этан Высшие ацетилены . Высшие углеводоро- ды (не ацетилены) Водород Азот Окись углерода . . Двуокись углерода . 9,6 7,0 15,1 0,1 0,8 0,3 50,8 6,6 8,2 1,5 5,5 0,8 25,8 0,1 0,6 0,3 51,5 5,6 8,2 16
Природный газ (метан) . . Этан Пропан . . . 21,8 48,6 38,4 16,2 37,9 30,7
III. СПОСОБ ВЫСОКОТЕМПЕРАТУРНОГО ПИРОЛИЗА ПО МЕТОДУ
ФИРМЫ ХОХСТЕР
(Das Hochster hochtemperatur — Pyrolyse Verfahren)
Другой путь получения ацетилена и этилена, развившийся в самое
последнее время, состоит в высокотемпературном пиролизе легких и средних
нефтяных фракций, а также газообразных углеводородов, начиная
с этана [7]. Тепло для эндотермического процесса в этом способе получают
от сжигания отходящих при переработке продуктов пиролиза остаточных
газов в смеси с кислородом, т. е. получение тепла основано здесь на том же
принципе, как и в автотермических процессах получения этилена и ацети-
лена. Разница заключается в том, что для получения тепловой энергии
используется не исходное сырье, а отходящие газы процесса. Выход синтез-
газа в этом процессе (смесь СО/Нг) значительно меньше, чем в процессе
Захсе.
Газ сжигается в охлаждаемых водой металлических горелках. Про-
дукты сгорания (дымовые газы), имеющие очень высокую температуру,
примешиваются к подвергаемой пиролизу смеси жидких или газообразных
углеводородов и после короткого времени пребывания в зоне высокой тем-
пературы тотчас охлаждаются. Соотношение этилена и ацетилена в пирогазе
регулируется относительным количеством продуктов сгорания, добавляемых
к углеводородной смеси. Температура и время пребывания могут также
оказывать большое влияние на величину указанного соотношения. Здесь
также используется большая часть тепла, содержащегося в газах.
Процесс переработки газа включает отделение образовавшихся при
пиролизе ароматических углеводородов, очистку газа и абсорбцию ацетилена.
Из остаточных газов выделяются этилен и окись углерода. Остающаяся
часть используется как топливо.
Пирогаз содержит приблизительно 30% ацетилена и этилена. Выход
составляет 50—60% вес. на исходное сырье, а при применении в качестве
сырья одного этана может достигать 80% вес. Остаточный газ можно исполь-
зовать и как синтез-газ, в этом случае для получения необходимого тепла
используется в определенной части само исходное сырье. Схема установки
показана на рис. 45 [7].
7 Заказ 596.
97
Рис. 45. Схема получения ацетилена и этилена высокотемпературным пиролизом по
способу фирмы Хохстер.
1 — горелка; 2 — реакционная зона; 3 — зона быстрого охлаждения; 4 — маслниый абсорбер; 5 — ди-
стилляционная колонна; в — газовый холодильник; 7 — газометр; 3 — компрессор; 9 — отделитель
H»S и СО»; 10 — установка Линде; 11 — отделитель для ацетилена.
Линии: I — кислород; II — остаточный газ пиролиза; III — нефтяная фракция; IV — вода; V — масло;
VI — остаточный газ; VII — смола; VIII —тяжелые ароматические углеводороды; IX —легкие аро-
матические углеводороды; X — окись углерода; XI — чистый этилен; XII — чистый ацетилен.
IV. ПРОЦЕСС ВЫСОКОТЕМПЕРАТУРНОГО ПИРОЛИЗА ПО ИСТМАНУ [8]
Процесс Истмана основан на том же принципе, что и рассмотренный
выше метод высокотемпературного пиролиза. Пропан из натурального газа
или газолин, предварительно подогретые до 600°, смешиваются в камерной
печи с также подогретым до 600° кислородом или воздухом и сгорают. Коли-
чество кислорода составляет в обоих случаях около 95% от стехиометри-
ческого. Вычисленная температура пламени лежит около 2000°.
Горячие продукты сгорания соединяются в зоне смешения с идущим
на пиролиз сырьем, предварительно также подогретым до 600° в присут-
ствии водяного пара. В зоне смешения, которой заканчивается камера сго-
рания, сужением поперечного сечения достигаются очень высокие скорости
газового потока. Отсюда смесь поступает в реакционную зону, где пиролиз
заканчивается. После выхода газа из этой зоны они охлаждаются до темпе-
ратуры ниже 100° посредством впрыска воды, чтобы стабилизировать про-
дукты пиролиза.
Дальнейшая обработка производится описанными выше методами
(гиперсорбция и т. д.). Соотношение ацетилена и этилена в смеси зависит
в первую очередь от температуры пиролиза и может варьировать в широких
пределах. Оно не зависит от того, как происходит сгорание — в атмосфере
чистого кислорода или в воздухе. Выход ацетилен-этиленовой смеси соста-
вляет при пиролизе пропана или газолина в среднем 55% вес. или более,
считая на исходный продукт, независимо от того, каково соотношение аце-
тилена и этилена в смеси, которое может изменяться от 1 : 2 до 4 : 1.
Ведя процесс так, чтобы образовывался практически один ацетилен, и
используя в качестве исходного материала пропан, можно довести выход
ацетилена до 45% вес. от исходного сырья.
ЛИТЕРАТУРА
1. Chem. Trade J. Chem. Engr.,. 138, 1274, 1956.
2. Wurst er C. Chem. Ing. Techn., 28, 1, 1, 1956.
3. В a u m a n n P. Z. Angew. Chem., B. 20, 257—259, 1948.
4, Sachsse H. Dechema Monographien, 24, Nr. 283—292, 9—38, 1955.
5. W inn acker K. Kunststoffe, 47, 399, 1957.
6. Updegraff N. C., Reynolds J. C. Petrol. Refiner, 32, 9, 163, 1953;
Bixler H. G., С о b e r 1 у C. W. Ind. Engng. Chem., 45, 2596, 1953.
7. Winn acker K. Kunststoffe, 47, 402, 1957.
8. A k i n G. A., Reid T. F., Schrader R. J. Chem. Engng. Progress,
54, 1, 41-48, 1958.
Глава V
ЦИКЛОГЕКСАН
1. ОБЩИЕ СВЕДЕНИЯ
Циклогексан является важнейшим исходным материалом для получения
адипиновой кислоты окислением его воздухом. Для этой цели гидрируют
бензол и полученный таким образом циклогексан окисляют. В связи с тем,
что бензол в нефтехимической промышленности получают путем дегидриро-
вания циклогексана в различных процессах каталитического риформинга,
а затем снова в чистом виде его гидрируют в циклогексан, высказывались
сомнения в целесообразности этого процесса. Сомнения эти однако не осно-
вательны, и по следующим причинам. Во-первых, циклогексан в исходных
фракциях, выделенных из нефти перегонкой, содержится не только как
таковой, а в смеси со значительным количеством метилциклопентана, который
изомеризуется в циклогексан при каталитическом риформинге и тотчас же
дегидрируется в бензол. Во-вторых, к тому времени как вырос спрос на
циклогексан, в промышленности уже была создана серия установок для
получения бензола нефтехимическим путем.
Получение циклогексана из определенных нефтяных фракций бази-
руется на изомеризации содержащегося в этих фракциях вместе с цикло-
гексаном метилциклопентана в циклогексан. Процесс проводится в усло-
виях, при которых дегидрирование не имеет места, а именно с хлористым
алюминием в присутствии хлористого водорода как промотора. Количество
метилциклопептана и циклогексана во фракциях некоторых американских
нефтей показано в табл. 53, в которой дан состав углеводородных нефтяных
фракций, выкипающих в пределах 36 —118° [1].
Таблица 53
Состав углеводородных смесей Са и С, фракций 36 — 118° из различных американ-
ских сырых нефтей, % объемн.
Компоненты Coalinga Kalifomien Jenning Con- roe Hasdings Segno Old Ocean Tom O'Connor
св Метилциклопеитан . . 10,29 5,01 6,51 7,42 6,40 5,42 6,70
Бензол 2,22 3,61 3,27 0,16 1,82 2,28 0,58
Циклогексан 7,63 7,13 10,40 12,66 9,64 7,30 10,94
С, Этил- и диметилцикло- пентан 17,52 6,89 6,59 12,41 7,56 9,59 10,94
Метилцпклогексан . . . 14,55 18,07 22,00 32,39 25,58 17,20 23,02
Толуол 7,94 12,02 16,19 0,77 9,65 5,87 3,39
7*
99
11. ПОЛУЧЕНИЕ ЦИКЛОГЕКСАНА
А. ИЗ НЕФТЯНЫХ ФРАКЦИЙ
В процессе изомеризации метилциклопентана по способу фирмы Шелл
применяется очень узкая фракция, выкипающая в пределах 66—85°, состав
ее в % объемн. приведен ниже.
Метилпентан................ 2
и-Гексап.................. 30
Диметилпептан.............. 3
Метилциклопсптан .... 30
Циклогексан ................. 20
Бензол................... 7
Различные углеводороды . . 2
Такая смесь углеводородов подвергается обработке в присутствии хло-
ристого алюминия и хлористого водорода, при этом происходит изомери-
зация метилциклопентана в циклогексан (рис. 46). При температуре 80°
и времени пребывания в реакторе 20—
30 мин. совершается превращение
Равновесие между метилциклопентаном
и циклогексаном сильно сдвинуто в правую
сторону, что позволяет достигнуть довольно
полного превращения. После изомеризации
катализатор отделяется в отстойнике, а со-
держащийся в количестве 1 %, считая на ис-
Рис. 46. Схема процесса изоме-
ризации мстилциклопентана в
циклогексан методом Шелла.
1 — изомериаационная установка; 2—
отстойник; з— колонна для отделения
хлористого водорода; 4 — фракциони-
рующая колонна; <5 — колонпа для
промывки щелочью.
Линии: I — исходный продукт; II —
циркулирующий катализатор; III —
свежий катализатор; IV — отработан-
ный катализатор; V— циркулирующий
хлористый водород; VI— свежий хло-
ристый водород; VII — циркулирую-
щий метилциклопентан; VIII — сырой
циклогексан па очистку или дегидри-
рование в бензол.
ходный продукт, хлористый водород удаляет-
ся перегонкой. Углеводородная смесь после
щелочной промывки ректифицируется. При
этом отбирают фракцию, выкипающую в
пределах 74—85°. Головной погон, содер-
жащий главным образом непревращенный
метилциклопентан, возвращается на изоме-
ризацию. Фракция 74—85° имеет следующий
состав: диметилпентана 7%, циклогексана
88%, бензола 5%.
Бензольная составляющая может быть
удалена очисткой серной кислотой. Парафи-
< новый углеводород удаляется в виде продуктов крекинга, для чего
^фракцию подвергают пиролизу при 550—650° со временем пребывания в
реакторе 10—20 сек. После повторной перегонки и очистки серной кис-
лотой для удаления небольших количеств олефинов получают 96%-ный
; циклогексан. Очистка циклогексана может производиться также методами
экстрактивной, вернее азеотропной перегонки. О путях промышленного
решения этой задачи надежных данных нет.
! мерно 20 л:. раким
: ( Б. ИЗ БЕНЗОЛА
При, гидрировании бензола в циклогексан, если бензол не содержит сер”
нистых соединений, работают с никелевым катализатором. В противном слу-
чае применяют устойчивый против действия сернистых соединений комбини-
; рованный катализатор, состоящий из сульфида никеля и сульфида вольфрама.
! t В присутствии никелевого катализатора (например окись никеля,
’ нёнёсенЬгая нц окПсь алюминия) процесс ведут при давлении водорода 200 ат
5 и3температура! 270°. Процесс можно вести с 2,7-кратной нагрузкой на кон-
такт, т. е. на !1 л контакта может быть пропущено 2,7 л бензола в час. Одно-
; временно на |1 л контакта должно быть дано в час 4 л3 На (приведенного
' к<' нормальны^ условиям). Под давлением 200 ат это соответствует при-
получают полностью насыщенный циклогексан.
ЛИТЕРАТУРА
1. Science of Petrol., 1, 14, Oxford University Press, 1938.
Глава VJ
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ ИЗ НЕФТИ
I. ОБЩИЕ СВЕДЕНИЯ
Ароматические углеводороды прежде получали исключительно из
каменноугольной смолы. Уже во время первой мировой войны были про-
ведены опыты получения толуола из нефтяных фракций. Для этой цели
был применен пиролиз при 700—750° процесс, уже рассмотренный нами
коротко (см. стр. 57).
Первый патент на производство ароматических углеводородов из нефти
посредством пиролиза был получен русским химиком А. Н. Никифоровым
еще в 1910 г. Промышленный процесс пиролиза для получения ароматиче-
ских углеводородов, главным образом толуола, разработан Галлом в Англии
и Ритманом в США в годы первой мировой войны.
В процессе Галла тяжелая бензиновая фракция нагревается в трубчатой
печи до 750° при очень высокой скорости потока. При этом наблюдается
значительное газообразование. Жидкая составная часть продуктов реакции
содержит 17—18% толуола, 18% бензола и 6% ксилолов. В настоящее время
такой процесс в измененном виде и в условиях максимального ограничения
коксообразования применяется в первую очередь для получения газообраз-
ных олефинов. Ароматические углеводороды при этом в известных условиях
являются желательным побочным продуктом.
Для прямого получения ароматических из нефти используются узкие
фракции бензина прямой перегонки определенного происхождения, которые,
если они отобраны в интервале, близком к температуре кипения толуола,
содержат 25% и более толуола. Для обогащения такие фракции можно под-
вергать термическому крекингу, при котором ароматическая часть сохра-
няется, а неароматическая часть, как менее стабильная, в основном превра-
щается в нокс и газ. Дальнейшая обработка включает в себя кислотную
очистку и перегонку.
Разработка этого способа прекращена. В настоящее время ароматиче-
ские углеводороды из содержащих их фракций извлекаются при помощи
селективных растворителей, например жидким сернистым ангидридом,
по способу Эделеану и другими, которые ниже рассматриваются детальнее.
Во время второй мировой войны проблема получения толуола из нефти
стояла особенно остро. После этого, особенно в США, возник огромный
спрос на бензол, что явилось причиной постановки опытов получения его
из нефти. Этот чрезвычайный спрос на бензол был обусловлен постоянно
возрастающим объемом его переработки. Достаточно назвать лишь некото-
рые важнейшие продукты его переработки — стирол, арилсульфонаты,
фенол, найлон, ДДТ, гексахлорциклогексан, малеиновая кислота, промежу-
точные продукты в производстве красителей и т. д.
Производство каменноугольной смолы связано в первую очередь с про-
цессами коксования углей для нужд металлургии. Однако спрос на кокс
101
возрастает далеко не в такой мере, как потребность в ароматических угле-
водородах для химической промышленности. В табл. 54 показаны размеры
производства ароматических углеводоро-
Та блица 54 дов из угля и нефти в США в 1955 г. и
предположительное производство этих
продуктов в 1960 и 1975 гг. [1].
И. КАТАЛИТИЧЕСКИЕ РИФОРМИНГ-
ПРОЦЕССЫ
Современные способы получения бен-
зола, толуола и ксилолов из нефти осно-
ваны на том, что подходящая по составу
прямогонная бензиновая фракция, бога-
тая нафтеновыми углеводородами и уже
Производство бензола, толуола и
ксилола из угля и нефти в США
(млн. м3)
Год Из угля Из нефти
1955 269 286
19Г>0 281 437
1975 311 1060
содержащая некоторое количество ароматических, подвергается каталити-
ческому дегидрированию, при котором циклогексаны дегидрируются в аро-
матические углеводороды, а алкилциклопентаны изомеризуются в цикло-
гексаны, которые тотчас же дегидрируются в производные бензола. Как
можно видеть из табл. 8, бензин из нефти нафтенового основания содержит
до 55% нафтеновых углеводородов, которые в процессе риформинга пре-
вращаются в ароматические.
А. ГИДРОФОРМИНГ-ПРОЦЕСС
Первым промышленным процессом дегидрирования циклоалифатических
углеводородов был гидроформинг-процесс. Он был разработан в нефтяной
промышленности для повышения октанового числа бензинов посредством
ароматизации его нафтеновой части и мог быть очень быстро перестроен
для прямого получения ароматических углеводородов [2].
Гидроформинг-процесс представляет собой каталитический риформинг-
процесс для бензиновых фракций, проводимый в присутствии катализатора
(окись молибдена — окись алюминия) при температуре 480—540° и давле-
нии водорода 7—20 ат. В Германии этот процесс известен как процесс DHD.
Процесс гидроформинга сильно эндотермический, что можно видеть из сле-
дующих уравнений
. С6Н!2 —> СвНв+ЗН2—53 ккал/моль
—> С6Н5—CH3-f-3H2—52,7 ккал[моль
С8Н1в —4 С6Н4 (СНа)2-|—ЗГ12—52,9 ккал)моль.
Катализатор обладает не только дегидрирующим действием, но, что
очень важно и уже отмечалось выше, также и изомеризующим [3]. Так,
метилциклопентан превращается в циклогексан, этилциклопентан и диметил-
циклопентан соответственно в метилциклогексан, которые затем дегидри-
руются в бензол или толуол. Переход парафиновых углеводородов в арома-
тические лишь очень незначителен.
102
В зависимости от того, что хотят получить — бензол, толуол или кси-
лол — применяют исходный продукт, выкипающий в тех или иных пределах.
В табл. 55 показан состав продуктов гидроформинга в зависимости от пре-
делов выкипания исходного сырья.
Таблица 55
Распределение ароматических углеводородов в продуктах
гидроформинга в зависимости от пределов выкипания исход-
ного материала (% объемн.)
Ароматические из продукта гидроформинга Пределы выкипания сырья, °C
93 — 135° 121 — 177°
Бепзол 0,9 0,6
Толуол 29,1 3,6
Этилбензол и ксилолы 9,3 25,4
Ароматические С9 0,7 16,6
» С1о и высшие . . . 0,2 6,2
Ниже приведен состав типичного исходного сырья для гидроформинг-
процесса для получения толуола (в % объемн.).
Диметилциклопеитан ..................... И
Метилциклопоитан i .................... „г
Этилциклопентан I .....................
Толуол (уже содержащийся).............. 3,5
Схема гидроформинг-процесса дана на рис. 47.
Исходный продукт нагревается до температуры реакции в трубчатой
печи и отсюда поступает в реакционную печь, в которой имеется катализатор.
Одновременно с исходным продук-
том над контактом пропускают во-
дород, чтобы не допустить чрезмер-
ного образования кокса. Продукты
реакции конденсируются и посту-
пают в разделитель, где водород
отделяется и возвращается в про-
цесс. Жидкая часть из разделителя
поступает в стабилизационную ко-
лонну, где при нормальных усло-
виях выделяются газообразные угле-
водороды. Жидкий остаток из низа
стабилизационной колонны посту-
Я /V
Рве. 47. Схема процесса гидроформинга.
1 — печь; 2 — реакционная печь с катализатором;
3 — газо-жидкостный сепаратор; 4 — стабилиза-
тор; <5 — дистилляционная колонна.
Линии: I — исходный материал; II — циркули-
рующий водород; III — газ; IV — продукт гид-
роформинга; V — полимеры.
паст в дистилляционную колонну,
где продукты гидроформинга при
нормальном давлении отделяются от
высококипящих примесей (поли-
меров).
Дальнейшая переработка продукта гидроформинга рассматривается
позднее. В этом процессе получают примерно 70%-ный (остальное газ
и кокс) выход сырого продукта, в котором содержание толуола почти точно
соответствует названному выше значению. Отсюда путем перегонки можно
получить фракцию, содержащую 60% толуола.
Для получения ксилолов гидроформинг-процессом необходимо, как
видно из табл. 55, более высококипящее сырье. Интересен состав фрак-
ции С8 (ксилольиой фракции) и в первую очередь содержание п-ксилола
(табл. 56).
103
Таблица 56
Состав типичной ксилольной фракции гидроформпнг-процесса
Компонент Температура, °C Плотность при 20° Содержание, % вес. от фракции
кипения замерзания
Этилбензол 137,2 —95 0,8670 14,3
га-Ксилол 138,4 + 13,3 0,8610 18,4
ж-Ксилол 139,1 —47,8 0,8642 45,9
о-Ксилол ...... 144,4 —25,2 0,8802 21,4
Гидроформинг-процесс проводится сейчас в промышленности также
методом псевдоожиженного слоя. Хотя в процессе гидроформинга в резуль-
тате дегидрирования освобождается водород, и дегидрирование и гидриро-
вание представляют собой равновесный процесс, гидроформинг ведут под
давлением водорода. В присутствии водорода под давлением коксообразо-
вание значительно меньше, чем в отсутствие водорода, а благодаря высокой
температуре равновесие сильно сдвинуто в сторону дегидрирования. Регене-
рация катализатора при работе методом псевдоожиженного слоя происходит
непрерывно.
Б. ПЛАТФОРМИНГ-ПРОЦЕСС И ДРУГИЕ СПОСОБЫ
КАТАЛИТИЧЕСКОГО РИФОРМИНГА
а) Общие сведения
Дальнейшим большим достижением в области каталитического рифор-
минга явился процесс платформинга [51- В этом процессе применяется пла-
тиновый катализатор (платина на окиси алюминия). В основе этого процесса
лежит также дегидрирование нафтеновых углеводородов в ароматические и
изомеризация соответствующих циклопентанов в циклогексаны.
Платиновый катализатор, что для получения ароматических не очень
важно, но имеет большое значение для улучшения антидетонационных
свойств бензина, способствует изомеризации парафиновых углеводородов,
крекингу их и гидрированию ненасыщенных продуктов крекинга (гидро-
крекинг). Последние реакции представляют собой экзотермический процесс,
в ходе которого используется часть водорода, освобождающегося в про-
цессе дегидрирования.
К ранее формулированным реакциям дегидрирования и изомеризации
циклических углеводородов должны быть присоединены теперь следующие
реакции.
Изомеризация парафинов
СН3СН2СН2СН2СИ2СН2СН3 pi сн3сн2сн2сн2сн—сн3
н-Геитан ।
сн3
Изогептан
Гидрокрекинг парафинов
СНаСН2СН2СН2СН2СН2СП2СН2СН2СН3 р± СН3СН2СН2СН2СН3+СН3СН2СНСН3
н-Пентан |
сна
Изопентан
Гидрирование олефине
СНаСН2СН2СН=СН2 ——- СН3СН2СН2СН2СП3
Амилен Изомеры пентана
Платформинг-ироцесс получил дальнейшее развитие в ряде процессов
каталитического риформинга, которые все направлены на улучшение анти-
детонационных свойств бензиновых фракций и основаны в первую очередь
104
на реакциях ароматизации и изомеризации. Число предложенных катализа-
торов очень велико и среди них главное место занимают катализаторы на
платиновой основе.
Способы работы также часто различны. Как и в каталитическом кре-
кинге, здесь различают три вида установок: установки с неподвижным
катализатором, в которых контакт находится в виде таблеток, установки
с подвижным катализатором, в которых контакт, в большинстве случаев
имеющий форму шариков, непрерывно циркулирует через установку и реак-
тивируется (регенерируется) в особой печи и, наконец, установки, работаю-
щие по принципу псевдоожиженного слоя, в которых катализатор находится
в пылевидном состоянии и поддерживается парами бензина в постоянном
завихренном движении. Так как процесс эндотермический, то часть необхо-
димого тепла подводится за счет предварительного подогрева бензиновых
паров циркулирующим водородом, а другая часть катализатором, который
в процессе регенерации (выжигание кокса в струе воздуха) поглощает много
тепла.
Неподвижный катализатор с платиной в качестве действующей состав-
ной части применяется в процессах: платформинг, гудриформинг, каталити-
ческий риформинг Синклер-Беккера, изоплюс, рексформинг, соваформинг,
ультраформинг.
Подвижный катализатор на кобальто-молибденовой (или хром—окись
алюминия) основе применяется в процессах гиперформинг и каталити-
ческий термофор-риформинг.
По способу с псевдоожиженным (молибден — окись алюминия) катали-
затором работают в процессах флюид-гидроформинг и ортоформинг.
Все катализаторы должны быть «кислыми», т. е. быть в состоянии отда-
вать протоны, так как отдельные процессы протекают по карбониум-ионному
механизму. Это может быть достигнуто или применением окислов, входящих
в состав контакта, или добавкой определенных кислых веществ в исходное
сырье.
При использовании каталитических риформинг-процессов со специаль-
ной целью получения ароматических углеводородов лучше каждый раз
исходить из очень узких фракций. Условия риформинга, необходимые
для перевода углеводородов С6 в бензол, могут оказаться слишком жесткими
для фракции С8 и наоборот. Здесь имеются те же соотношения, что и при
крекинге нефтяных фракций для получения бензина. Следовательно, если
хотят получить бензол, то следует для риформинга применять в первую
очередь фракции, — содержащие циклогексан и метилциклопентан. Для
получения толуола применяют фракции, по составу отвечающие приведенной
на стр. 103. Для ксилолов справедливо то же самое. Выход аромати-
ческих тем выше, чем выше концентрация соответствующих нафте-
нов.
Получение ароматических углеводородов из нефти осуществляется,
следовательно, в три стадии: получение четкой ректификацией необходимых
нефтяных фракций, собственно каталитический риформинг этих фракций,
включающий с химической точки зрения два основных процесса — дегидри-
рование и изомеризацию нафтенов — и, наконец, переработка высокоарома-
тизированных продуктов риформинга для получения чистых индивидуаль-
ных углеводородов, как бензол, толуол и ксилольная фракция.
б) Промышленное осуществление платформинга [6]
В качестве типичного примера современного каталитического процесса-
для получения ароматических углеводородов из нефти рассмотрим несколько-
подробнее платформинг-процесс.
Катализатор состоит из окиси алюминия с 0,5—1,0% платины и некото-
рого определенного количества галоидов. Катализатор помещается в реак-
ционной камере в виде таблеток и не регенерируется. После отработки
105
'контакта содержащаяся в нем платина извлекается. Чтобы действенность
контакта была возможно продолжительнее и чтобы уменьшить отложение
кокса, работают с фракциями, имеющими конец кипения не выше 200е.
Способ работы представлен на схеме рис. 48. Тепло подводится к реак-
ционным печам от подогревателя, в котором пары углеводородов и водород
подогреваются до вступления в реакционную камеру. Исходный продукт
проходит последовательно три печи (камеры), в которых протекает желаемая
Рис. 48. Схема процесса платформинг.
J — печь; 2 — реактор; 3 — газо-жидкостный
сепаратор; 4 — стабилизатор; 5 — фракцио-
нирующая колонна.
Линии: 1 — бензиновая франция; II — цирку-
лирующий водород; III — газ (на отопле-
ние); IV — остаток*, V — продукт платфор-
минга.
реакция. В зависимости от исходного
сырья температура составляет 450—
540°. Давление водорода равно 14—
70 ат. Высокое давление водорода
здесь, как и в гидроформинг-процес-
се, позволяет практически полностью
предотвратить коксообразование и по-
лучить насыщенные продукты реакции.
Сера должна предварительно удалять-
ся из сырья каким-либо каталитиче-
ским способом, например гидроочист-
кой, так как катализатор чувствите-
лен к ее действию.
При стабилизации продукта плат-
форминга из него удаляются газо-
образные фракции, включая Сд. Стаби-
лизат идет на дальнейшую переработ-
ку для выделения чистых ароматиче-
ских углеводородов.
Важнейшее значение для нефтехимической промышленности имеет
большое количество водорода, получаемое при каталитическом риформинге.
Подсчитано в среднем, что при каталитическом риформинге 100 л прямогон-
ной бензиновой фракции освобождается около 8,2 л3 водорода. Так как
в будущем термический риформинг будет, вероятно, полностью вытеснен
каталитическими процессами, количество производимого таким способом
водорода окажется очень значительным. Чистота водорода составляет
70—90% и он находится под давлением 14—50 ат.
В США 11% производства аммиака базируется на таком водороде.
Предварительное выделение его в чистом виде легко осуществляется при
помощи гиперсорбции.
III. ПОЛУЧЕНИЕ ЧИСТЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ ПРОДУКТОВ
КАТАЛИТИЧЕСКОГО РИФОРМИНГА
А. СПОСОБ ЭДЕЛЕАНУ [7]
Освобождение высокоароматизированных концентратов от равнокипя-
щих алифатических углеводородов и получение таким образом чистых
индивидуальных углеводородов принципиально осуществимо различными
путями. Выделение ароматических углеводородов из ароматизированных
жидкостей возможно, например, путем экстракции. Для этого применяют
в большинстве случаев жидкую двуокись серы (сернистый ангидрид). Способ
был предложен для этой цели в 1907 г. Эделеану и первоначально приме-
нялся для очистки керосина [7]. Экстрагируемый исходный материал сме-
шивается с жидким сернистым ангидридом (рис. 49), который растворяет
ароматические углеводороды и как тяжелый слой оседает вниз (экстракт).
Вследствие растворяющего действия ароматических углеводородов вместе
с ними переходит в экстракт и определенная часть неароматических состав-
ных частей. Для удаления их экстракт промывают высококипящей парафи-
нистой фракцией, извлекающей эти неароматические углеводороды. Затем
из экстракта удаляют сернистый ангидрид, который возвращается на уста-
106
яовку. Экстракт очищают серной кислотой, промывают щелочью и перего-
няют. В результате получают чистые ароматические углеводороды.
В качестве верхнего слоя в экстракционной колонне собирается неаро-
матическая часть (рафинат), содержащая еще небольшое количество раство-
ренного сернистого ангидрида, от которого она освобождается в отделителе.
Парафинистое промывочное масло, ко-
торое было использовано для извлечения
неароматических из экстракта, сначала осво-
бождается от небольших количеств раство-
ренного в нем сернистого ангидрида, затем ,
перегонкой — от неароматических, извле-
ченных им из экстракта, и возвращается
® абсорбционную колонну.
Б. СПОСОБ УДЕКС ДЛЯ ЭКСТРАКЦИИ
АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ [8]
Новый процесс для экстракции арома-
тических был разработан фирмой Доу Кеми-
кал Компани и применен Юниверсал Ойл
Продектс Компани под названием «удекс-
процесс» (рис. 50).
Он основан на применении гликолево-
водной смеси (8—10% воды), обладающей
очень высокой селективностью по отноше-
нию к ароматическим углеводородам. По-
этому нет необходимости применять при
экстракции узкие фракции, но можно бен-
зол, толуол и ксилолы экстрагировать сов-
местно. Экстракция производится, как и в
методе Эделеану, в условиях противотока
в очень эффективной, специально для этого
Рис. 49. Схема экстракции аро-
матических углеводородов при
помощи жидкого сернистого ан-
гидрида по методу Эделеапу.
J — экстракционная колонна; 2 — ко-
лонна дли отделения SO2; 3 — промы-
вочная колонна; 4 —дистилляционная
колонна; .5 — колонна для отделения
растворителя; 6 — очиститель; 7 —
дистилляционная колонна.
Линии: I — исходный материал; II —
жидкий SOo; III — свободный от аро-
матических углеводородов рафинат;
IV — циркулируюпц 1й SO*; V— па-
рафинистое промыв очное масло; VI —
промывочное масло; VII — рафинат;
VIII — остаток-, IX —чистые арома-
тические углеводороды.
процесса разработанной ко-
лонне. Экстрагирующую среду (растворитель) подают в голову колонны,
экстракт отводится снизу. Экстрагируемое масло поступает в среднюю (по
высоте) часть колонны. Часть ароматических подается в низ колонны как
«орошение». Обогащенный ароматическими растворитель поступает в разде-
литель, где ароматические отделяются от растворителя, который возвра-
щается в экстракционную колонну.
Выходящие из разделителя пары после
охлаждения и конденсации разделяются
на две фазы. Водная фаза частью исполь-
зуется для освобождения рафината от
Таблица 57
Рис. 50. Схема процесса удекс для
экстракции ароматических из про-
дуктов каталитического риформинга.
1 — экстракционная колонна; 2— колонна
для промывки рафината водой; 3 — колон-
на для отделения ароматических угле-
водородов; 4 — водоотделитель.
Линии: I — фракция, содержащая арома-
тические углеводороды; II— рафинат;
III — циркулирующий гликоль; IV —
циркулирующие ароматические углево-
дороды; V — вода; VI — ароматические
углеводороды на перегонку.
Результаты экстракции ароматических
методом удекс
Продукт Плотность при 20° Содержание ароматиче- ских, %
Исходный 0,7715 51,3
Рафинат 0,6806 35
Бензол 0,885 99,9
Толуол 0,873 998
Смесь ксилолов . . 0,873 99,0
107
остатков гликоля, после чего она идет обратно в экстрактор, этим содер-
жание воды в гликоле доводят до желаемого уровня. Остаток возвра-
щается в разделительную колонну. Ароматическая фаза (слой) предста-
вляет собой исключительно чистую смесь ароматических углеводородов
и может быть легко разделена на отдельные индивидуальные углеводороды
перегонкой. Выходы очень хорошие.
Удекс-процесс в большинстве случаев комбинируется с платформингом.
Результаты применения экстракции методом удекс представлены в табл. 57.
Ксилольная фракция содержит 14,5% вес. этилбензола, 19% вес. п-
ксилола, 42% вес. .м-ксилола и 24,5% вес. <э-ксилола. Выход составляет:
бензола 99%, толуола 98% и ксилолов около 94%.
/I
В. СПОСОБЫ ЭКСТРАКТИВНОЙ ПЕРЕГОНКИ [9]
Способы основаны на том же принципе, что и разделение бутанов и бу-
тенов или, вернее, бутенов и бутадиена, посредством перегонки в присут-
ствии фурфурола (см. стр. 77), содержащего 4% воды.
В данном случае неароматическая часть очи-
щаемой смеси отгоняется в присутствии фенола.
Ароматическая часть остается растворенной в феноле
при условиях, в которых равнокипящие парафины
отгоняются. После этого ароматическая часть выде-
ляется из фенола в разделителе, а фенол возвра-
щается на циркуляцию. Принцип работы в виде
упрощенной схемы приведен на рис. 51.
В процессе, разработанном фирмой Шелл,
ароматические получают 98—99 %-ной чистоты. Про-
цесс применяется в больших масштабах для по-
лучения чистых бензола и толуола. В качестве
экстракционной среды применяют также крезол.
Для получения бензола сначала из определен-
ной бензиновой фракции методом четкой ректифи-
кации выделяют концентрат бензола, который дол-
жен содержать около 10% последнего, а затем
методом экстрактивной перегонки получают 98—
99%-ный бензол с 90%-ным выходом.
Г. МЕТОДЫ АЗЕОТРОПНОЙ ПЕРЕГОНКИ [10]
Процесс основан на том, что неароматическая
часть образует со смесью метанол — вода илиметил-
этилкетон — вода тройную азеотропную смесь,
от которой ароматические углеводороды могут
быть отделены перегонкой. На рис. 52 дана упрощен-
ная схема выделения чистого толуола из продуктов
гидроформинга. Из продуктов гидроформинга выделяется кипящая в узких
пределах толуольная фракция, которую подают в колонну вместе с азеотропо-
образователем, в данном случае с водным метилэтилкетоном. Азеотропная
смесь (метилэтилкетон — вода — неароматическая часть) отгоняется, а полу-
чающийся в виде остатка чистый толуол отбирают из низа колонны, и далее
очищают серной кислотой и промывают щелочью, водой и повторно перего-
няют.
Выходящая из головной части первой колонны азеотропная смесь по-
ступает во вторую колонну, где экстрагируется водой, неароматическая часть
отходит сверху, а водный раствор метилэтилкетона с низа колонны. В сле-
дующей колонне из водного раствора отгоняется азеотропная смесь, содержа-
щая 88,7% метилэтилкетона, которая возвращается в колонну азеотропной
перегонки. Необходимое количество освобожденной от метилэтилкетона
воды также возвращается в колонну азеотропной перегонки.
Рис. 51. Схема отделе-
ния ароматических от
равнокипящих неарома-
тических составных ча-
стей посредством экс-
трактивной перегонки с
фенолом.
/ — колонна экстрактивной
перегонки; 2 — колонна для
отделения ароматических
углеводородов.
Линии: I — смесь раввоки-
пящпх парафиновых и аро-
матических углеводородов;
II — неароматическая состав-
ная часть; III — фенольный
экстракт; IV — ароматиче-
ские углеводороды на кис-
лотную очистку; V— свежий
фенол; VI — циркулирую-
щий фенол.
108
Толуол можно получать также из фракций некоторых нефтей непосред-
ственно. Например, содержание толуола в восточнотексасской нефти соста-
вляет 0,4%, а в некоторых западнотексасских нефтях 0,5%. Из таких нефтей
четкой ректификацией на колонне с 50 тарелками можно выделить фракцию,
содержащую 23—25% толуола, из которой затем методом ступенчатой азео-
тропной перегонки можно выделить толуол 98%-ной чистоты. Из приведен-
ных на стр. 103 цифр можно видеть, однако, что значительно выгоднее, когда
наряду с ограниченным количеством толуола во фракции содержится относи-
тельно много нафтеновых углеводородов, которые могут быть превращены
в толуол посредством каталитических процессов.
Рис. 52. Схема выделения пеарома-
тпческих составных частей из смеси
с ароматическими посредством азео-
тропной перегонки (на примере очи-
стки толуола).
1 — колонна азеотропной перегонки; 2 —
колонна для экстракции водой; 3 — ко-
лонна регенерации метилэтилкетона; 4—
дистилляционная толуольная колопна; 5—
установка для обработки кислотой, ще-
лочью и водой.
Линии: I — исходный продукт; II —
метилэтплкетон и HiO; III — циркули-
рующий водный метилэтилкетон; IV —
.нсароматические; V— 99%-ный толуол;
VI — остаток; VII — чистый толуол.
Рис. 53. Схема аросорб-процесса для выделе-
ния ароматических углеводородов из углево-
дородных смесей.
1, 2t 3—колонны.
Линии: I — исходный продукт; II — возврат проме-
жуточной фракции; III—неароматичеснис; IV— аро-
матические-, V — неароматические продукты (рафинат);
VI — бензол; VII — толуол; VIII — десорбированная
часть.
Д. АДСОРБЦИОННЫЕ СПОСОБЫ (АРОСОРБ-ПРОЦЕСС) [11]
Процесс основан на том, что силикагель адсорбирует ароматические
углеводороды раньше олефинов и насыщенных углеводородов. Поэтому,
если пропускать углеводородную смесь, содержащую ароматические, через
камеру, заполненную гелем кремнекислоты, то они будут задерживаться си-
ликагелем, а насыщенные углеводороды и моноолефины пройдут через ка-
меру. Когда силикагель полностью насытится ароматическими (практически
применяют избыток силикагеля до г/з от всей загрузки), приступают к де-
сорбции. Для этого берут смесь высокомолекулярных ароматических угле-
водородов, которые вытесняют ранее адсорбированные ароматические угле-
водороды с силикагеля и выводят ее из адсорбера. Низкокипящие углево-
дороды можно затем легко выделить из смеси перегонкой.
При получении бензола и толуола для десорбции применяют ксилол.
Перед собственно десорбцией силикагель промывают, чтобы освободить
•его от неароматических составных частей. Это может быть сделано тем же
десорбентом, который затем направляют в колонну 1 (рис. 53), где неаромати-
ческие отделяются от десорбента. За этой фракцией идет промежуточная
фракция, которая в основном еще содержит неароматические углеводороды,
но одновременно и часть десорбированных ароматических. Эту фракцию
возвращают на повторную адсорбцию. И, наконец, следующая за этой фрак-
ция содержит уже только десорбированные ароматические углеводороды.
В колонне 2 ее фракционировкой освобождают от десорбента, а в колонне 3
разделяют бензол и толуол. Десорбент возвращают в цикл.
109
[Е. РАЗДЕЛЕНИЕ КСИЛОЛОВ [121
Изложенные выше принципы получения ароматических из нефти при-
менимы и для ксилольной фракции С8. Однако в этом случае описанные про-
цессы должны быть дополнены разделением изомеров.
Ароматическая фракция С8 состоит из трех ксилолов и этилбензола.
Важнейшие физические константы этих четырех углеводородов даны
в табл. 58.
Сначала отделяют о-ксилол, что может быть сделано посредством ректи-
фикации в колонне с 60 тарелками при отношении орошения 10 : 1, причем
как остаток получают 97%-ный
Таблица 58 о-ксилол.
Важнейшие константы компонентов ксилольной Выделение о-ксилола фрак- фракции ционированием в промышлен-
Компонент Температ плавле- ния ура, °C кипения Плотность при 20° ных масштабах еще не приме- няется. Как следует из табл. 58г о-ксилол является наиболее высококипящим из всех изоме- ров ксилола. Его применяют для получения фталевого ангид- рида. Процесс основан, как и окисление нафталина, на газо-
о-Ксилол jh-Ксилол . п-Ксилол . Этилбензол —25,2 —47,8 + 13,3 —95,0 144,4 139,1 138,4 137,2 0,880 0,864 0,861 0,867
фазном окислении над вана-
диевым контактом (оронит-процесс). Равным образом и n-ксилол пред-
ставляет большую ценность как исходный материал для получения те-
рефталевой кислоты, применяемой в производстве волокна (териленовое
волокно в Англии, декроновое в США, тревира в Германии). С этой целью
смесь м- и n-крезолов охлаждают до —60° и выкристаллизовавшийся п-
крезол отделяют центрифугированием. Выход w-ксилола ограничивается
образующейся эвтектикой, состоящей из 88% .и-ксилола и 12% ге-ксилола.
В 1960 г. в США предполагается произвести 50 тыс. т м-ксилола, более
90% которого должно быть получено из нефти путем каталитического рифор-
минга. Ниже коротко рассматривается работа установки Гумбл Ойл Рефай-
нипг Компани в Вайтоуне (рис. 54).
Сырая смесь, состоящая примерно из 15,8% ге-ксилола, 39,6% л-кси-
лола, 20,0% о-ксилола, 18,9% этилбензола, 3,5% толуола и 2,5% парафи-
новых и нафтеновых углеводородов, просушивается над окисью алюминия,
чтобы полностью освободиться от влаги. Далее смесь проходит в тепло-
обменник, где охлаждается до —32°; здесь уже начинается кристаллизация.
Холодильник снабжен скребковым устройством для устранения помех в те-
плопередаче. После этого предварительного охлаждения продукт поступает
в главный холодильник, где охлаждается до —70° и, наконец, центрифуги-
руется. В виде твердой фазы выделяется 80%-ный n-ксилол, составляющий
в совокупности 55—60% всего содержавшегося во фракции ге-ксилола.
Отжатые на центрифуге кристаллы га-ксилола поступают далее в специаль-
ную емкость, где подогреваются до +24° и при этом расплавляются, а затем
вновь подвергаются ступенчатой кристаллизации, охлаждаясь сначала
до +7°, а затем до —18°; при этой температуре они центрифугируются.
Чистота полученного таким образом n-ксилола составляет 95%. Фильтрат,
содержащий еще 40% n-ксилола, смешивается со свежим исходным про-
дуктом.
Разделение м- it re-ксилолов после выделения ректификацией о-ксилола
может производиться различными способами. Так, например, л-ксилол можно
выделить из смеси ксилолов, используя его способность образовывать с двой-
ным соединением трехфтористый бор—фтористоводородная кислота (BF3 • HF)1
стабильные комплексы, растворимые в этом двойном соединении. Экстрак-
ция производится под давлением, а выделенный комплекс затем разру-
110
шается перегонкой [13]. Представление об этом способе разделения дает-
схема рис. 55.
Из смеси ксилолов л-ксилол можно выделить также селективным суль-
фированием его в л-ксилолсульфокислоту, из которой ксилол может быть
затем регенерирован перегонкой с перегретым водяным паром. Можно также
сульфировать всю ксилольную фракцию, а затем выделить л-ксилол селек--
тивным гидролизом смеси сульфокислот [14].
Рис. 55. Схема выделения лг-ксилола-
из ксилольной фракции посредством
экстракции фтористым бором и фтори-
стоводородной кислотой.
1 — колонна для экстракции; 2 — конденса-
тор; 3 — разделительный сосуд; 4 — раздели-
тельная колонна.
Рис. 54. Схема выделения n-ксилола из фрак-
ции Св на заводе Гумбл Ойл Рефайнинг
Компани.
1 — осушитель; 2 — холодильная установка; 3 —
центрифуга; 4 — емкость для расплавления; 5 —за-
пасная емкость.
Линии: I — ароматическая фракция Cg; II — филь-
трат; III — п-ксилол.
Проводились также опыты селективного алкилирования ксилольной
смеси изобутеном, на их основе разработан соответствующий процесс, трет-
Бутил-л-ксилол может быть затем деалкилирован в л-ксилол, последний
получается 98 %-ной чистоты [15].
Возможно также получение и-ксилола из диизобутена (см. стр. 64).
Полученная димеризацией смесь, состоящая из 80% 2,4,4-триметилпентепа-1
и 20% 2,4,4-трпметилпентана-2 в присутствии водорода проходит над катали-
затором, состоящим из смеси окиси хрома и окиси алюминия активированной
окисью церия при 500°. При частичном превращении образуется ароматиче-
ская фракция Cs, состоящая из 97% га-ксилола и небольших количеств о-и
л-ксилолов [16].
ЛИТЕРАТУРА
1. S i t t i g M. Petrol. Process., II, 68, 1956.
2. Chem. Engng. News, 29, 3015, 1951.
3. Hettinger W. P. Ind. Engng. Chem., 47, 719—730, 1955.
4. Marshall С. H. Chem. Engng. Progr., 46, 313, 1950.
5. H a e'n s e 1 V. Petrol. Process., 5, 356, 1950; Petrol. Engr., 22, C (4), 9—14,
1950; H a e n s e 1 V., Donaldson G. R. Ind. Engng. Chem., 43, 2102, 1951.
6. H a e n s e 1 V., Donaldson G. R. Proceedings of the Third World Pet-
rol. Congr., IV, 225, 1951; Chem. Engng., 59, 5, 242, 1952; Oil a. Gas J., 53, 46, 149, 1955.
7. Petrol. Refiner, 32, II, 166, 1953; 33, 9, 230, 1954; Wilkinson W. F. и
сотрудн. Chem. Engng. Progr., 49, 257, 1953; Ratliff R. A., S t г о b e 1 W. B. Petrol.
Refiner, 33, 5, 151—155, 1954.
8. R e a d D., Petrol. Refiner, 31, 5, 97—103, 1952; 33, 9, 154, 1954; Oil a. Gas J.,
53, 46, 149, 1955; 55, 35, 117, 1957.
9. D u n n C. L., L i e d h о 1 m G. E. Oil a. Gas J., 51, 5, 68—70, 1952.
10. Petrol. Refiner, 34, 12, 194, 1955.
11. Chem. Engng. News, 29, 3015, 1951; Williams C. R., Sutherland 1!. E.
Oil a. Gas J., 50, 14, 84, 88, 101, 1951; Petrol. Refiner, 32, II, 162, 1953.
12. Haines H. W. и сотрудн. Ind. Engng. Chem., 47, 1096, 1955.
13. В г о о k e L. F. Am. пат. 2251444, 1950; Chem. Abs. 45, 649g, 1951.
14. McCauley D. А. и сотрудн. Ind. Engng. Chem., 42, 2103, 1950; К и ж-
п e p H. M., В e п д e л ь ш т e й H Г. ЖРФХО, 57, I, 1946.
15. S c h n e i d e r A. Am. пат. 2648713, 1953, Chem. Abs. 48, 8258, 1954.
16. S c h m e t t e r 1 i n g A., Krekeler H. Am. пат. 2785209, 2785210, Chem..
Abs., 51, 12971, 1957.
ЧАСТЬ ВТОРАЯ
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА УГЛЕВОДОРОДОВ
Глава I
ПЕРЕРАБОТКА ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Реакции хлорирования относятся к числу важнейших процессов нефте-
химической промышленности. Парафины и особенно олефины легко реаги-
руют с хлором, давая в результате продукты, являющиеся важнейшими
промежуточными и конечными продуктами современной промышленности
алифатической химии. Значение продуктов хлорирования метана, этана,
этилена, пропена, пентана, а также высокомолекулярных парафиновых угле-
водородов, получаемых из парафинистых нефтяных фракций или синтезом
Фишера-Тропша, в настоящее время очень велико.
1. ХЛОРИРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
А. ОБЩИЕ СВЕДЕНИЯ
Хлорирование парафиновых углеводородов может осуществляться тремя
способами: фотохимическим, каталитическим и термическим. Оно протекает
согласно реакции:
RH+C12 —> RCl-|-HCl+24 ккал/молъ
В промышленных расчетах процессов хлорирования считают, что на
каждый килограмм вошедшего в реакцию хлора выделяется 360 ккал тепла.
Б. ФОТОХИМИЧЕСКОЕ ХЛОРИРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Фотохимическое хлорирование может с успехом применяться для газо-
образных и жидких парафиновых углеводородов. При хлорировании жидких
углеводородов газообразный хлор подают при перемешивании и облучении
ультрафиолетовым светом непосредственно в углеводород. Для хлорирования
газообразных углеводородов целесообразно применять инертный к хлору
растворитель, например четыреххлористый углерод, в который при облучении
ультрафиолетовым светом одновременно вводят хлор и парафиновый угле-
водород. Фотохимическое хлорирование легко идет уже при низких темпе-
ратурах — важное преимущество перед рассматриваемым ниже термическим
хлорированием, позволяющее полностью избежать разложения, вызывае-
мого пиролизом, а также реакций перегруппировки.
Фотохимическое хлорирование является типичным радикально-цепным
процессом [1]. Подвод энергии в форме ультрафиолетового света вызывает
расщепление молекулы хлора на атомы:
С12 + fc v = 2СГ
112
Хлор-атом отрывает от углеводородной молекулы атом водорода,
причем образуется алкильный радикал и хлористый водород:
RH-j-Cl’ -> R'+HC1
Алкил-радикал тотчас вступает во взаимодействие с находящимися
в большом избытке молекулами хлора, образуя алкилхлорид и атом хлора:
R4-CL, -> RC1+C1-
Таким образом, в результате реакции всегда образуется новый атом
хлора, обеспечивающий возможность дальнейшего протекания цепной реак-
ции.
Прекращение реакции может наступить в результате обрыва цепи,
вызываемого прежде всего действием кислорода, который вступает в соеди-
нение с алкил-радикалом и с атомом хлора. Так как в технических газах
всегда содержится большее или меньшее количество кислорода, обрыв цепи
в промышленных условиях наступает относительно быстро. В то время как
при использовании химически чистых газов квантовый выход достигает
30 000—40 000, в технических процессах эта величина не превышает 2000.
Под квантовым выходом понимается число реакций, вызываемых одним све-
товым квантом до обрыва цепи.
В. КАТАЛИТИЧЕСКОЕ ХЛОРИРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Каталитическое хлорирование основано на применении переносчика
хлора, такого как йод [2], сера [3], фосфор, сурьма и другие, в виде соответ-
ствующих хлоридов, которые растворяются в хлорируемом углеводороде
или при хлорировании газообразных парафиновых углеводородов — в рас-
творителе. Применяются исключительно элементы, имеющие по крайней
мере два значения валентности. В качестве гомогенных катализаторов могут
также применяться вещества, образующие радикалы, как, например, диазо-
метан, тетраэтилсвинец и гексафенилэтан [4]. Они обладают способностью
разделять молекулу хлора на атомы, которые тотчас же вызывают возникно-
вение цепной реакции.
Гетерогенный катализ применяется главным образом при газофазном
хлорировании. В качестве катализаторов используют активированный уголь,
пемзу, отбеливающие земли и т. и., пропитанные металлическими солями,
особенно медными. В соответствии с теорией Тэйлора их действие основано
на способности их активных центров вызывать ионизацию хлора. Гетероген-
ное каталитическое хлорирование протекает по криптоионному механизму
и нечувствительно к обрыву цепи, особенно если он вызывается кислородом.
Благодаря этой нечувствительности к кислороду становится возможной
разработка такого процесса хлорирования, при котором хлор будет исполь-
зоваться целиком именно потому, что процесс будет проходить в присут-
ствии кислорода. При этом применяются такие контактные массы, которые
делают возможным превращение образовавшегося хлористого водорода под
воздействием кислорода в воду и хлор [5].
Г. ТЕРМИЧЕСКОЕ ХЛОРИРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Для хлорирования газообразных при нормальных условиях парафино-
вых углеводородов наибольшее значение имеет термический способ. Термиче-
ское хлорирование протекает в отсутствие воздуха и катализатора. Реакция
эта протекает также по цепному механизму, опа сильно тормозится кисло-
родом и другими соединениями, способными обрывать течение реакционных
цепей, например окислами азота.
Атом хлора, освобождающийся при фотохимическом хлорировании за
счет световой энергии, здесь образуется в результате термической диссоциа-
8 Заказ 596.
113
ции. Этим определяется величина энергии активации, составляющая при тер-
мическом хлорировании 20 ккал/моль, на 8 ккал больше, чем при каталити-
ческом.
а) Промышленные способы термического хлорирования
парафиновых углеводородов
Для термического хлорирования низкомолекулярных парафиновых
углеводородов предложены различные технические способы, выбор которых
определяется тем, какая степень хлорирования должна быть достигнута
в том или ином случае. Значительная трудность в осуществлении этих про-
цессов обусловливается тем, что парафиновый углеводород и хлор не дают
абсолютно гомогенной смеси. Этим вызывается опасность местного чрезмерно
глубокого хлорирования и связанного с этим образования сажи.
С другой стороны, парафиновый углеводород всегда необходимо приме-
нять в избытке, чтобы избежать взрывов. Например, процесс получения
четыреххлористого углерода из метана, разработанный Хассом с сотруд-
никами [6] (рис. 5G) состоит в том, что смесь хлора и метана в количествен-
ном соотношении, исключающем опасность взрыва, протекает через нагре-
тую трубу, по длине которой устано-
влены насадки для подачи хлора.
Через эти насадки по мере израсхо-
дования каждый раз подается хлор,
так что в конечном счете достигается
Рис. 56. Схема хлорирования по Хассу
и Mait lin.
1 — реакционный змеевик; 2 — вентиль; з —сме-
сительные насадки; 4 — нагревательная баня.
Линии'. I — хлор; II — метал; III — четырех-
хлористый углерод и хлористый водород.
Рис. 57. Схема хлорирования метана и
переработки продуктов хлорирования.
1 — печь для хлорирования; 2 — колонна для
промывки водой; 3 — колонна для промывки
щелочью; 4 — холодильник; «5 — хлорметиловая
колонна; 6 — хлорметиленовая колонна; 7 —
хлорсформовая колонна без давления.
Линии: I — углеводороды (метан); II — хлор;
III — циркулирующий метан; IV — вода; V —
щелочь; VI— соляная кислота; VII — дренаж;
VIII — хлористый метил; IX —хлористый мети-
лен; X — хлороформ; XI — четыреххлористый
углерод.
такое молярное соотношение между хлором и метаном, какое необхо-
димо для получения четыреххлористого углерода.
Промышленный способ хлорирования метана, как он был применен
впервые Хохстер Фарбеверкен, состоит в том, что метан и хлор в соотноше-
нии 5 ; 1 смешиваются и подаются в стальную, облицованную бетоном трубу,
обогреваемую газом [7] (рис. 57). При этом образуются одновременно все
продукты хлорирования метана и от молярного соотношения зависит, какие
продукты хлорирования будут преобладать. Образование отдельных продук-
тов хлорирования метана в зависимости от молярного соотношения хлора
к метану представлено данными табл. 59.
Переработка продуктов хлорирования сильно усложняется тем, что оно
идет с большим избытком углеводорода. Если целью процесса в первую
очередь является получение высших продуктов хлорирования метана, то
хлористый метил и хлористый метилен могут быть возвращены в процесс.
Если хотят получить хлористый метилен, то на повторное хлорирование воз-
вращают в первую очередь хлористый метил.
114
Хлорирование метана
Таблица 59
Температура реакции, °C Молярное соотно- шение СВ:СН4 СН3С1 СН2С12 СНС13 СС14
% мол.
440 1 : 2 62 30 7 1
440 1,1 : 1 37 41 19 3
440 1,68 : 1 19 43 33 4
440 1,98 : 1 И 35 45 9
440 2,28 .' 1 I) 29 52 14
440 3,02 : 1 3 15 53 29
440 3,31 : 1 — 6 43 51
460 3,88 : 1 — — 4 96
После успешного завершения реакции газы охлаждают и хлористый
водород отмывают водой, а затем щелочью. Далее газ охлаждают до —50°.
При этом оставшийся газообразный метан возвращают в установку для хло-
рирования. Из конденсата непрерывной перегонкой под давлением отделяют
хлористый метил и хлористый метилен. Остаток, состоящий из хлороформа
и четыреххлористого углерода, разделяют особо.
Большой интерес представляет способ термического хлорирования в при-
сутствии взвешенных веществ, как он был разработан в промышленности
Герольдом, Гриммом и Зексауером [8]. Уже упомянутые трудности, связан-
ные с образованием сажи и отложением угля и смолистых продуктов в трубо-
проводах и в других частях аппаратуры, в этом способе исключаются. Способ
заключается в том, что, например, угольные шарики из специального бун-
кера увлекаются потоком поступающего в печь газа и. в течение всего про-
цесса находятся в состоянии «кипящего» движения. Сажа и углистые ча-
стички, выделяющиеся в процессе хлорирования, непрерывно измельчаются
трущимися друг о друга угольными ядрами и с газовым потоком выносятся
из установки.
Этот способ имеет еще то достоинство, что благодаря непрерывному
«кипящему» движению взвешенного контакта достигается быстрее внутрен-
нее смешение газов и более действительный и полный теплообмен между по-
ступающим свежим газом и реакционным. Способ полностью оправдал себя,
особенно для хлорирования этана, пропана и др. Дальнейшая переработка
продуктов хлорирования после освобождения от хлористого водорода про-
мывкой водой и щелочью производится перегонкой под давлением.
Термическое хлорирование находит очень большое применение для по-
лучения хлористого амила [9] из технического пентана (см. ниже рис. 64).
Хлористый амил омыляют в амиловый спирт (пентазол), который сам по себе
или в виде ацетата является важнейшим растворителем для лаковой про-
мышленности. Пентан получают из газового бензина перегонкой, он предста-
вляет собой смесь примерно равных частей н-пентана и изопентана. С недав-
него времени стали использовать только н-пентан.
В промышленных условиях термическое хлорирование пентана проводят
следующим образом: на 15—20 частей пентана берут 1 часть хлора и смесь
пропускают через трубчатую печь при температуре около 260° с продол-
жительностью пребывания в печи примерно 2,5 сек. Незначительное время
превращения обусловливается исключительно высокой скоростью газа,,
при которой достигается хорошая гомогенизация смеси. На практике струю
хлора вдувают в поток пентана со скоростью 90 тыс. м/час. Дальнейшая
переработка производится перегонкой, что в данном случае (при жидких
продуктах реакции) осуществляется сравнительно просто. Непрореагиро-
вавший пентап возвращается в процесс.
Высокомолекулярные парафиновые углеводороды, когда они предста-
влены высокопарафпппстыми нефтяными фракциями или когазипом, в, боль-
8*
115,
шинстве случаев хлорируются также термически [10]. Процесс заметно
упрощается, так как хлор взаимодействует с углеводородом, нагретым лишь
до 100 . В редких случаях добавляют некоторое количество йода как катали-
затора.
Если для данного исходного материала установлена зависимость меящу
Рис. 58. Зависимость удельного веса при
15° хлорированного керосина от содер-
жапия хлора.
содержания хлора.
Рис. 59. Зависимость коэффициента пре-
ломления хлорированного керосина от
преломления хлорированного продукта, то за течением процесса хлорирова-
ния можно наблюдать, определяя плотность или коэффициент преломления.
На рис. 58 и 59 показано,
ния образца керосина по
как изменяются плотность и показатель преломле-
мере изменения содержания хлора с увеличением
глубины хлорирования.
Хлорирование можно вести также непрерыв-
но в условиях противотока. Схема такого способа
работы показана на рис. 60. Подобным же спо-
собом можно хлорировать парафин. Хлор про-
пускают через расплавленный парафин при 70—
100° до тех пор, пока не будет достигнута желае-
мая глубина хлорирования. Метод пригоден и
для хлорирования полиэтилена.
При хлорировании высокомолекулярных па-
рафиновых углеводородов хлор распределяется
статистически по всей углеродной цепи, потому
что вторичные водородные атомы отдельных
метиленовых групп реагируют с одинаковой
Рис. 60. Схема непрерыв-
ного хлорирования жидких
парафиновых углеводоро-
дов.
1 — испаритель; 2 — подначи-
вающий насос; 3— хлорирую-
щая колонна; 4— холодильник;
5 — циркуляционный насос.
Линии: I — парафиновые угле-
водороды; II — жидкий хлор;
III — хлор; IV — хлорирован-
ные углеводороды.
относительной скоростью. Только на концах
парафиновой углеводородной молекулы замещение
ограничено, так как относительная скорость
реакции первичных водородных атомов метиль-
ных групп примерно в 3 раза меньше, чем вто-
ричных водородных атомов метиленовых групп.
При хлорировании н-додекана образуется прибли-
зительно 8,5 % мол. 1-хлордодекана и по 18,3 % мол.
2-, 3-, ! 4-, 5-, и 6-хлордодекана. Чем длин-
нее парафиновая цепь, тем относительно меньше
содержится в смеси хлорпарафина первичного хлорида. Принимая во вни-
мание, что первичные хлориды отдают свой хлор в реакциях посредством
двойного обмена, в то время как вторичные в большей части претерпе-
вают дегидрохлорирование, это имеет особо важное практическое значе-
ние [11].
116
б) Хлоролиз
К термическим процессам хлорирования относятся также способы
хлорирования под высоким давлением и хлоролиз [12]. Высокохлорированные
парафиновые углеводороды при нагреве разлагаются; например октахлор-
пропан при нагреве выше 200° (лучше при 300°) разлагается на четырех-
хлористый углерод и тетрахлорэтилен.
С1 С1 С1 С1 С1
\ I / \ /
С1—с-с— с—С] -> СС14+ с.--Ж
/ I \ / \
С1 С1 С1 С1 С1
В присутствии небольшого количества безводного хлористого алюминия
октахлорпропан расщепляется уже при значительно более низкой темпера-
туре с хорошими выходами в течение непродолжительного времени. Если
вести этот процесс в присутствии хлора, то образуются почти исключительно
четыреххлористый углерод и гексахлорэтан. Последний может подвергаться
дальнейшему пиролизу с образованием четыреххлористого углерода и тетра-
хлорэтилена. Реакция идет, вероятно, таким образом, что сначала в резуль-
тате отщепления хлора от гексахлорэтапа образуется тетрахлорэтилен, а
освобождающийся при этом хлор реагирует с еще содержащимся в смеси
гексахлорэтаном с образованием четыреххлористого углерода. Четырех-
хлористый углерод и гексахлорэтан можно получить из хлорпропана, нагре-
вая последний под давлением в смеси с жидким хлором. Этот процесс назы-
вается хлоролизом.
Четыреххлористый углерод и тетрахлорэтилен могут получаться хлоро-
лизом высокохлорнрованного пропана с хорошими выходами без применения
давления. Равным образом из высокохлорнрованного пентана можно полу-
чать гексахлорциклопентадиен [13], применяемый как компонент для син-
теза «хлордана», являющегося отличным инсектисидом. Хлоролизом поли-
хлорбутана при нормальном давлении может также получаться гекса-
хлорбутадиен с выходом около 75%.
в) Фторирование парафиновых углеводородов
Непосредственное бромирование и йодирование парафиновых углево-
дородов не имеют значения, хотя хлорбромметан широко применяется как
огнегасящее средство. В данном случае, одттако, хлор заменяется бромом,
о чем позднее будет сказано особо. Фторированные парафины и олефины
приобретают в настоящее время все возрастающее значение. Фреон 12 (ди-
хлордифторметаи) является важнейшим хладагентом. Его также получают,
исходя из четыреххлористого углерода1.
Прямое фторирование парафиновых углеводородов протекает так бурно,
что сопровождается воспламенением. Поэтому для введения фтора приме-
няют непрямые способы, например пропускание паров парафинового угле-
водорода, разбавленных азотом, над трехфтористым кобальтом при 200—
300° [14]. При этом трехфтористый кобальт отдает один атом фтора парафи-
новому углеводороду, сам превращаясь в двухфтористый кобальт, который
затем реакцией с элементарным фтором при 200° снова фторируется в трех-
фтористый кобальт. Из выделяющегося при фторировании фтористого во-
дорода можно снова получить элементарны и фтор электролизом. Электро-
1 Номенклатура фреонов имеет цифровое обозначение. Производные метана обо-
значаются двухзначным числом, производные этана — трехзпачпым. 1F слсдияя цифра
показывает число атомов фтора. Предпоследняя цифра означает: 1 — пергалопдироваи-
иыи (не содержащий водорода), 2 — один атом водорода, 3 — два атома водорода, 4 —
три атома водорода в молекуле. Например: F 11— фторхлороформ (CFC13), F142 - ди-
фторхлорэтаи (F2C1C—СН3), F114 — тетрафтордпхлорэтап (F.,C1C—CF3C1).
117
лизу подвергают расплавленное двойное соединение, состоящее пз фтористого
водорода и фтористого калия (KF-HF) с добавкой 1,0—1,5% LiF.
Для получения перфторированных соединений фторированный угле-
водород должен пропускаться через фторирующий аппарат много раз.
Фторировать можно также, пропуская смесь углеводорода и фтора,
сильно разбавленную азотом, над обработанной фтористым серебром медной
стружкой при 150—300°. Считается, что реакция идет через перфторид се-
ребра, который превращается во фтористое серебро, последнее вновь фтори-
руется элементарным фтором. При этих условиях можно, например, пз
н-октана получить с ограниченным выходом перфтороктап.
Перфторпарафины можно также получать вытеснением фтором хлора
из высокохлорированпого углеводорода, воздействуя на него фтористым
водородом в присутствии фтористой сурьмы. Полученный таким образом
высокофторированпый парафиновый углеводород обрабатывают затем эле-
ментарным фтором в условиях большого разбавления до образования пер-
фторида [15].
Фторированные парафины исключительно устойчивы против действия
таких химических агентов, как азотная и серная кислоты, олеум, нитрую-
щая смесь и т. п. Они совершенно негорючи и по крайней мере до 500° вполне
стабильны.
Большое значение имеет фреон 22 (хлордифторметан), который в усло-
виях пиролиза при 650° дает тетрафторэтилеп и хлористый водород [16].
Хлордифторметан получают действием фтористого водорода на хлороформ
в присутствии фтористой сурьмы как катализатора. Тетрафторэтилеп можно
также получать действием цинковой пыли на си.иж-дихлортетрафторэтан.
Он представляет собой газ, кипящий при —76,3°, затвердевающий при
—142,5°. Полимеризацией его получают исключительно стойкое искусствен-
ное вещество (тефлон) [17].
г) Использование продуктов хлорирования парафиновых углеводородов
X л о р п р о и з в о д н ы е метана. Продукты хлорирования ме-
тана являются прежде всего растворителями. Хлористый метил часто при-
меняетсятак же, как метилирующий агент (метплцеллтолоза и др.). На рис. 61
показаны возможные направления использования хлористого метила.
ХЛОРИСТЫЙ МЕТИЛ
4-Na-целлюлоза
---------------* Мет ил целлюлоза (загустители, аппретура, на-
бивочные насты, косметика)
---------------- Дпметилдихлорснлан и т. п. (для получения
силикопов)
---------------*• Хладагенты для холодильных машин
--------------Растворитель, например при получении бутил-
каучука
---------------> Алкилирующий материал (органический проме-
жуточный продукт)
+NaSH
---------------—* Метилмеркаптап
+Na2S , О2
---------------► Диметилсульфид —диметилсульфоисид (рас-
творитель)
+Na2S2 к Н2О+С12
1--------------* Диметилдисульфид —-----> метаисульфохлорид
Рис. 61. Реакции и применение хлористого метила.
Хлористый метилен, получаемый прямым хлорированием метана, как
растворитель находит все более широкое применение и является в настоящее
время единственным из хлорметанов, получаемым хлорированием метана.
Для получения всех остальных хлорметанов применяют специальные методы.
Например, хлористый метил получают этерификацией метилового спирта
хлористым водородом, хлороформ — хлорированием спирта или ацетона в ще-
118
лочной среде, четыреххлористый углерод — из хлористой серы и сероугле-
рода.
Хлористый метилен является исключительным растворителем для жи-
ров, масел, смол и часто применяется как растворитель для удаления старой
краски и как экстрагирующая среда. Он все шире применяется как раство-
ритель для поливинилхлорида в производстве клеящих средств.
Хлороформ также является превосходным растворителем для жиров,
масел, смол и канифоли. В смеси с четыреххлористым углеродом он приме-
няется как морозостойкая огнегасящая жидкость, имеющая температуру
застывания около —50°. Он является исходным продуктом для получения
хладагента хлордифторметана (фреон 22) и тетрафторэтилена. Большое
количество хлороформа применяется (в качестве растворителя) в производ-
стве пенициллина.
Четыреххлористый углерод — наиболее широко применяемый в промыш-
ленности растворитель для самых различных органических продуктов.
Большое количество четыреххлористого углерода применяется как негорю-
чее очищающее средство в прачечных и в предприятиях химической чистки
(азордин). Он служит растворителем в различных процессах хлорирования.
Из него получают также смешанный хлорированно-бромированный метан,
являющийся исключительно эффективным огнегасящим средством.
При воздействии фтористого водорода на четыреххлористый углерод
в присутствии фтористой сурьмы как катализатора получают дихлордифтор-
метан, кипящий при —30°, не горючий и лишь мало ядовитый газ, обладаю-
щий исключительными свойствами как хладагент. Представление о возмож-
ных путях использования четыреххлористого углерода дает рис. 62.
ЧЕТЫРЕХ ХЛОРИСТЫЙ УГЛЕРОД
Fc-i-HsO, восстановление
Растворитель для смол, поливинил-
хлорида масел и т. и.
Получение фреона 12, дихлордифтор-
метана (хладагент в холодильных ма-
шинах, домашние холодильники и
т. и.)
Обезжиривающее средство (для метал-
лов и др.)
Средство для борьбы с вредителями в
сельском п лесном хозяйстве
Противопожарное средство (в смеси с
хлороформом)
Средство для «сухой» химической чист-
ки
Получение хлороформа
Рис. (,2. Применение и реакции четырсххлорпстого углерода.
Хлористый эти л. Хлористый этил применяется в качестве
хладагента и для этилирования целлюлозы. Реакция между щелочной
целлюлозой и хлористым этилом проводится в никелированном автоклаве-
мешалке при 200°. Степень этилирования может быть различной в зави-
симости от условий работы. Этплцеллюлоза в противоположность метил-
целлюлозе ужо не растворима в воде, по растворима в органических жид-
костях и служит важным компонентом в производстве лаков.
Хлористый этил применяется во все расширяющихся масштабах в про-
изводстве тетраэтилсвинца, который в составе этиловой жидкости доба-
вляется к карбюраторным топливам для повышения их октанового числа.
Тетраэтилсвинец получают действием хлористого этила иа евппцово-
иатриевый сплав в автоклавах |.18]:
4PbNa-HC..Ilr,Cl -4 Pb (C2II5)4T3Pb+4NaCl
Из продуктов реакции его изолируют перегонкой с водяным паром и
получают бесцветную, кипящую при 200°, очень ядовитую жидкость со
сладковатым запахом.
119
На рис. 63 показаны важнейшие направления использования хлори-
стого этила.
ХЛОРИСТЫЙ этил
Превращение си сплином. ГЬ —х &
-----------------------------— - Тетрэлтплсгшнсц
+ Na-цсллюлоза
------------------ - - --- -- - Эт1ллцсллюлг>за (лакеи'.. .м- сырье,
растворима в органических
.;. \ । > ' н IK1C т I;' ; t тел их)
---------------------------------- ------------------------------ эт НМ1 epj.-iiKTun ( одорант для
j_ хп,-------------------------------природного газа)
—-------------- ------- - -- — ЭГП’ПМИП
---- — ------ ----------- —- - - * Этилирующая реагент ч.дкщр
гаи п чсск и х промежуточных
л родуктоп
----— ----- — - --------------- Хладагент для холодильников
-------------------------------- ' Анестезирующее всгщсство
Рис. 6;’. Применение и реакции хлористого эглча .
Из продуктов хлорирования пропана до спх пор имел значение тонко
1,3-днхлорпронан, являющийся исходным материалом для получения цпхло-
пропапа, представляющего собой превосходное наркотическое средство .
Таблица 60
Свойства различных хлорпроизводных метана п хлористого этила
Показатели CIL.C1 СИ2С12 СЛС13 СС14 С2Н5С1
Молекулярный вес Температура кипения (760 мм рт. ст), °C Температура кипения азеотропной смеси с водой (760 мм рт. ст.), “С Соотношение хлористого алкила к воде в азеотропной среде . . . . 1 Температура замерзания, °C . . . , Теилота испарения при температуре 1 кипения, г,ал/г ......... Теплоемкость жидкости при 20°, кал!г . . . , . . . . . Теплоемкость пара при 1 ат, кал/г Критическая температура, °C . . . Критическое давление, нт Удельный вес жидкости . . . Вес пара при температуре кипения 1 ат, г/л Вязкость жидкости при 20°, санти- пуазы Растворимость при 25*, г/1()0 г: хлористого алкила в воде . . . воды в хлористом алкиле . . . Скорость испарения (для эфира рав- на 100) Коэффициент преломления жидкости Диэлектрическая постоянная: жидкости пара Теплота образования, г кал/.мол: жидкости вара 50,49 -23,76 —97 6 102,45 0,381 0,199 (25°, 1,021 ат) 143 65 9 Q 920 2,52 (-15°) 0,244 0 74 (30°, 1am) 0,285 1,3712 (-23,7°) 12,93 (-25°) 1,0108 ( + 21°) 20,1 84,94 39,8 38,3 66,3 г 1 —96 7 78,7 0,280 0,15э (40°) 245 609 1,32’6 2,93 0,425 132 0,198 71 1,4244 (20°) 9,1 (20°) 1,0082 (40°) 21,7 28,2 (20°) 119,39 61,2 56,0 ,34,1 : 1 —6+5 59,3 0,234 0,142 (61,2°) 262,9 53,8 1,189 4,13 0,57 0 79 0,097 56 1,4455 (20°) 4,785 (20°) 1,00462 (69,6°) 23,6 20,2 (25° в N.,) 153,84 76,5 66,8 23,5 •• 1 —2285 46,8 0,205 0,140 (76,75°) 283,15 44,98 1,594 о ,о2 0,969 0 08 0,013 33 1,4601 (20°) 1,00302 (87,6°) 25,9 25,4 (25° в NJ 64,25 12,3 11,8 256,3 : 1 - 4'16 ,'ч 91,5 0,399 0,243 (40°) 182,8 . 72.0 1 0,897 223 0,26 0/5/0° 1,3798 . (0°) 1,01285 (23,5°) 25,7 | 2064 (10°) '
120
Последний можно получить с 95 %-ним выходом, например действием цин-
ковой пыли в присутствии йодистого натрия на раствор дихлорпропана
в 75 %-ном этиловом спирте.
В табл. 60 даны важнейшие физические свойства различных хлорметанов
и хлористого этила.
Использование продуктов хлорирования пен-
танов [19]. Как уже указывалось, продукты хлорирования пентанов
применяются для получения амиловых спиртов. С этой целью смесь моно-
хлорпентанов, выкипающую в пределах 85—107°, подвергают гидролизу
в щелочной среде в присутствии олеата натрия. Процесс ведут при 170—
180° под давлением. Олеат натрия является диспергирующим агентом, улуч-
шающим контакт хлорида с омыляющей средой. Схема установки показана
на рис. 64.
Рис. 64. Схема получения амилового спирта из пентана.
I — печь для хлорирования; 2 — колонна для отделения хлористого водорода и пентана; .V— ректифи-
кационная колонна, разделение моно- и дихлоридов; 4 — реактор для омыления монохлорнда; 5 — раз-
делитель; 6 — колонна.
Линии: I — хлор; II — пентан; III — 11С1 и пентан, IV — монохлорид; V — олеат натрия; VI — ди-
хлоран; УН — раствор поваренной соли; VIII — амилен; IX — хлористый амил; X — веди; XI —
амиловый спирт; XII — диамнловый эфир.
По окончании гидролиза не растворимый в воде слои разделяется
в нескольких колоннах. В первой колонне отделяются пентены (амилены),
образовавшиеся в результате дегидрохлорирования. В следующей колонне
отделяется непрореагировавший хлористый амил. Наконец, после обезвожи-
вания получают смесь амиловых спиртов. В остатке остается диамнловый
эфир.
Смесь амиловых спиртов примерно на 60% состоит из первичных и на
40% из вторичных спиртов. В табл. 61 показан состав сырой смеси амиловых
спиртов, как она получается в результате гидролиза смеси амилхлоридов.
Пентены, образующиеся в результате дегидрохлорирования, при щелоч-
ном омылепйи смеси амилхлоридов состоят главным образом из пентена-2
и триметплэтилена. Последний используется для алкилирования фенола,
причем получается трет-амил фенол.
Техническая смесь амиловых спиртов, помимо использования ее в произ-
водстве лаков, применяется также в производстве флотационных реагентов.
Взаимодействием хлористого амила с гидросульфидом натрия получают
амилмеркантан, обладающий исключительно неприятным запахом, он при-
меняется в США в качестве одоранта природного газа (пенталарм). Ампл-
хлориды могут служить также для получения амиламинов, амилфенолов,
амилнафталипа и т. д. Конденсацией амилфенола с формальдегидом полу-
чают растворимые в маслах смолы. Амиламин дает с высокомолекулярными
жирными кислотами превосходные эмульгаторы.
На рис. 65 показаны возможные области применения хлористого амила,
а на рис. 66 соответственно амиловых спиртов.
Применение продуктов хлорирования высоко-
молекулярных углеводородов. Продукты хлорирования
121,
Табл и ц’а 61
Состав сырой смеси амиловых спиртов, полученной гидролизом смеси амплхлоридов
Продукт Температу- ра кипения, °C Содер;ка- мие, % все. Формула
А. Первичные, спирты
Пептапол-1 3-мстилбута пол-1 2-метилбута пол-1 138,0 132,0 129,5 25,5 11,9 16,8 сн,сп,сн,сн»сн.он (СН3),СИСН2СН2ОП СН3'СНгСНСНоРН
Б. Вторичные спирты Пептапол-2 Пентанол-З 3-мстплбута пол-2 119,3 115,6 112,0 25,1 9,4 2,2 СП-; СН3СНоСН„СНГОП)СП3 СН3СИ,СП(ОН)СП,СП3 СН3СНСН(ОН)СН3
В. Т ретпичные спирты 2-мстилбутапол-2 101,8 6,0 СН3 CT^CHjCHfOHJCHa 1 сп3
АМПЛХЛОРНДЫ
Омыление с NaOH и
олеатом Na
Алкилирование
фенола
_Ал пилирование
нафталина
H-NaSII
! +NH,______________________________
Амиловые спирты (пентазолы пептаацетаты)
Амилфенолы (маслорастворпмые. светоустойчивые про-
дукты конденсации с формальдегидом)
Смачивающие и моющие
вещества
Амилнафталин
Мягчители, теплоносители,
растворители
Амилмеркаптан (одорант для природного газа)
Амил а мины
Рис. 65. Реакции амилхлоридов.
АМИЛОВЫЕ СПИРТЫ
--------------------------------* Растворители для нитроцеллюлозных ла-
ков, мочевипо-формальдегидных смол и
т. п.
--------------------------------* Компоненты этерификации (амилацетат,
амилфталат как растворители и мягчите-
+CS2 + NaOlI ли)
---------------------------------* Амилксантогенаты (флотационные реаген-
ты)
---------------------------------* То рмо 311ые жидко ст и
Рис. 66. Применение и реакции амиловых спиртов.
высокомолекулярных парафиновых углеводородов, содержащихся в опре-
деленных, богатых парафином нефтяных фракциях или в когазине, при-
меняются, в основном, в производстве моющих средств, смазочных веществ,
депрсссаторов, гербойля и т. п.
Синтетическое смазочное масло пз продуктов хлорирования среднего
масла может получаться двумя путями: конденсацией хлорпарафина с аро-
матическими углеводородами, особенно с нафталином, в присутствии безвод-
ного хлористого алюминия методом Фриделя-Крафтса, пли действием безвод-
ного хлористого алюминия, или активированного алюминия на хлорпара-
фин как таковой. При этом, вероятно, происходит полимеризация олефинов,
образующихся в качестве промежуточных продуктов.
Конденсация хлорированных средних масел с ароматическими углево-
дородами изучалась Р. Кольбелем с сотрудниками, установившими основные
зависимости между качеством смазочных масел и степенью хлорирования
парафина, длиной углеводородной цепи парафинового компонента и т. д.
Ими же был разработан промышленный процесс, обеспечивающий получе-
122
иие исключительных по качеству смазочных масел. В качестве парафинового
компонента они применяли в первую очередь когазин [20] и нашли, что
с увеличением степени хлорированности когазина вязкость смазочного
масла растет, вязкостно-температурные свойства ухудшаются, коксовое
число увеличивается. Чем длиннее цепь парафинового компонента, тем
лучше вязкостно-температурные свойства и тем больше выход масла. Они
нашли далее, что выход масла тем больше, чем выше в реакции отношение
нафталина к хлорированному компоненту.
Получение депрессаторов (для понижения температуры застывания
масел) конденсацией алкилхлоридов с нафталином (парафлоу). В связи
с установленной способностью парафиновых углеводородов улучшать вяз-
костно-температурные свойства смазочных масел представляется выгодным
сохранить в масле максимально возможное количество парафинов. Однако
известно также, что это влечет за собой повышение температуры застывания
масла до более высоких значений, чем допускается эксплуатацией. Добавле-
ние к маслу продуктов конденсации нафталина с высокомолекулярными
алкилхлоридами значительно понижает температуру застывания содержащих
парафин масел.
Для получения такого депрессатора (присадки для понижения темпера-
туры застывания масел типа парафлоу) конденсируют твердый парафин,
хлорированный при температуре 80° до содержания хлора, равного 14%,
с нафталином в присутствии хлористого алюминия. В качестве разбавителя
применяют хлористый этилен. Конденсацию ведут при температуре 30—
35°, повышая ее перед концом реакции до 60°.
На 1000 кг твердого парафина требуется 150 кг нафталина, 70 кг безвод-
ного хлористого алюминия и 1000 кг хлористого этилена.
Добавка 1 % парафлоу к пенсильванскому смазочному маслу понижает
его температуру застывания с —1 до —20°. В отдельных случаях понижение
температуры застывания достигает 25°.
Керилбензол (арилсулъфонат). Важной областью применения высоко-
молекулярных алкилхлоридов в нефтехимической промышленности является
получение алкилбензолов, служащих промежуточным продуктом в про-
изводстве синтетических и моющих средств и вспомогательных материалов
в текстильной промышленности. Ниже па примере додекана показана по-
следовательность реакций, приводящих к получению желаемого продукта:
Сп11о0+С1о С„НЩС1+ПС1
С12Н.25С1+<~> С1211’-5 \ / 4 ПС1
С121Т25 / +H.2SO4 -> Cellos _J^ + If2O
S0.J1
tK-jU-s \ ^>+NaOII -> CI2IJ;:5 / ^> + ll2O
SO3II SO3Na
В качестве исходного материала для получения смеси алкилхлоридов
применяется, папример, фракция пенсильванской нефти, выкипающая в пре-
делах 200—300° (керосин), глубокоочпщеппая горячей концентрированной
серной кислотой. Это масло затем хлорируют в таких условиях, что па
1 моль углеводорода присоединяется 1 моль хлора. Таким образом получают
смесь алкилхлоридов, называемую керилхлоридом.
Керилхлорид смешивают затем с бензолом в соотношении 1 : 6 и в при-
сутствии безводного хлористого алюминия (как катализатора) получают
керилбензол [21].
123
Сырой продукт далее разделяют перетопкой на установке, состоящей
из трех колонн. В первой колонне отделяется избыток бензола, во второй —
непрореагировавший керосин, в третьей, работающей при пониженном да-
влении, керилбензол.
При сульфировании керилбензола олеумом при комнатной температуре
получают упомянутые сульфокислоты, которые при последующем разбавле-
нии смеси водой отделяются в виде масляного слоя. Известно, что высокомо-
лекулярные алифатические или ароматическо-алифатические сульфокислоты
сразу после образования плохо растворимы в воде и могут быть, кроме того,
легко вытеснены из их водных растворов минеральными кислотами. Сульфо-
кислоты нейтрализуют далее едким натром, отбеливают гипохлоритным
раствором и высушивают. Упрощенная схема установки для получения ке-
рилбензола и его сульфирования показала на рис. 67.
Рис. 07. Схема получения арилсульфопатов из парафиновых углеводородов путем хло-
рирования, алкилирования и сульфирования.
1 — емкость для углеводородов; 2 — промежуточная емкость (керилхлорид); 3 — освинцованная ме-
шалка для проведения хлорирования; 4 — мешалка для приведения алкилирования; 5 — промежу-
точная емкость (сырой керилбензол); G — абсорбер для соляной кислоты; 7— вакуумная колонна;
8 — отгонный куб; 9 — алкилатная емкость; ю —сульфатор; 11 — нейтрализатор; 12 — распылитель-
ная установка или вальцовая сушилка.
Линии: I — хлор; II — бензол; III — отходящие газы; IV — соляная кислота,- V — бензол обратно
на алкилирование; VI — углеводороды п хлорированные углеводороды; VII — несульфпрованные угле-
водороды; VIII — отработанная серная кислота; IX — сульфированные углеводороды; X — гипо-
хлорит; XI — сульфонатный раствор.
Полученный таким образом почти бесцветный арилсульфонат приме-
няется в настоящее время для производства большинства синтетических
моющих и очищающих средств. Варьируя длину алкильных остатков, можно
получать многочисленные различные специальные продукты.
Стабилизация продуктов хлорирования вы-
сокомолекулярных парафиновых углеводородов.
Продукты хлорирования среднего масла, которые, как, например, гербойль,
применяются для получения клея, должны стабилизироваться путем доба-
вления к ним определенных веществ, чтобы избежать выделения хдористого
водорода при длительном хранении. Стабильность продуктов хлорирования
является необходимым условием их практического применения. Для стаби-
лизации стремятся применять такие стабилизаторы, которые уже в ограни-
ченной концентрации обладают способностью количественно связывать
хлористый водород. Превосходным средством является феноксипропеноксид
(1,2-эпокси-З-феноксипропан), 1% которого полностью стабилизирует высо-
кохлорированное среднее масло. Когазин, хлорированный до содержания
40% хлора, с добавкой феноксипропеноксида хорошо эмульгируется и
остается вполне стабильным при его применении в качестве замасливающего
средства в текстильной (шерстяной) промышленности.
Хорошим заменителем ворвани является смесь 25% поливинилового'
эфира и 75% хлоркогазина, содержащего 40% хлора. При хлорировании
124
до содержания 60—65% хлора получают бальзамообразньгй продукт, обла-
дающий свойствами живицы.
Легко получается и продукт, содержащий до 75% хлора. Интересно
отметить, что этот продукт может быть таким же стабильным, как малохло-
рированный.
Хлорированные парафины. Парафин, как уже выше упо-
миналось, хлорируется при температуре приблизительно 100—110°. В про-
типа хлорированного парафина.
мышленности получают три главных
1. Хлорпарафин, содержащий
около 47% хлора, что соответствует
примерно 7 атомам хлора на парафи-
новую молекулу с 25 атомами угле-
рода. Это желтая, нелетучая жид-
кость, отщепляющая при нагреве до
135° хлористый водород. Железо
сильно катализирует отщепление хло-
ристого водорода, так что уже при
незначительном содержании железа
дегидрохлорирование начинается при
37°, т. е. на 100° ниже.
2. Хлорпарафин, содержащий
60% хлора (примерно 15 атомов
хлора на С25), с температурой плав-
ления около 50°.
3. Хлорпарафин, содержащий
70% хлора, что соответствует
Рис. 68. Зависимость температуры затвер-
девания хлорированного парафина от
содержания хлора.
22 атомам хлора на Саз.
Высокохлорированный парафин твердый и хрупкий начинает размяг-
чаться только при 90—100°.
В США такой высокохлорированный парафин применяется под назва-
нием хлорвакса [22]. Хлорвакс 70 содержит 70% хлора и химически исклю-
чительно инертен. Он не конденсируется и не полимеризуется, устойчив
против действия кислот и разбавленных щелочей. Последние вызывают сла-
бый гидролиз только после очень длительного воздействия. Он растворим
в сухих маслах и применяется главным образом для пропитывания и полу-
чения огнестойкой бумаги, тканей и других материалов, тогда как хлор-
вакс 40, содержащий 40% хлора, является пластификатором для винилполи-
меризата, для этилцеллюлозы и др. Применение хлорвакса как пластифика-
тора для поливиниловых соединений возможно при одновременной добавке
стабилизатора, известного под названием дифос п представляющего собой
двухосновный фосфит свинца [23].
При хлорировапии парафина сначала получают продукты, температура
плавления которых при повышении содержания хлора от 0 до 35% законо-
мерно понижается. При дальнейшем увеличении содержания хлора отме-
чается, как показано па рис. 68, повышение температуры плавления [24].
На рис. 69 показаны важнейшие направления в использовании продуктов
хлорирования высокомолекулярных парафиновых углеводородов.
Хлорированное среднее масло
Соединение хлорированного
среднего масла с нафталином
реакцией Фриделя-Крафтса
Высокохлорированный парафин
(4 0% хлора)
Высокохлорированный парафин
(70% хлора)
Низнохлориро ванный парафин
(14% хлора)
Керилхлорид -* Керилбензол -* Арилсуль-
фонат
Синтетические смазочные масла
Хлорвакс 40, пластификатор для поли-
винилхлорида. клей
Хлорвакс 70 для получения огнестойких
бумаги, тканей и древесины
Получение депрессатора (парафлоу) реак-
цией с нафталином по Фриделю-Крафтсу.
Рис. 69. Важнейшие реакции и применение продуктов хлорирования
высокомолекулярных парафиновых углеводородов.
125
II. НИТРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
А. ОБЩИЕ СВЕДЕНИЯ
Прямое нитрование парафиновых углеводородов путем нагрева углево»
дородов с разбавленной азотной кислотой было впервые успешно выполнено
М. И. Коноваловым в 1893 г. Предшествовавшие опыты нитрования парафи-
новых углеводородов потерпели неудачу вследствие применения чрезмерно
агрессивных нитрующих средств.
Еще в 1834 г. Митчерлих нитровал такие ароматические углеводороды
как бензол, толуол и т. д., нагревая пх со смесью концентрированных азот-
ной и серной кислот. В ранних опытах Ф. Бейльштейн, В. Курбатов и др.
для нитрования парафиновых углеводородов применяли высококонцентри-
рованную, часто дымящую азотную кислоту. Главным результатом этих
опытов было окисление. Проводя нитрование парафиновых углеводородов
нагреванием с 13 %-ной азотной кислотой в запаянных трубках при 130—
150°, М. Коновалов получил питропарафпны и с хорошим выходом.
После классических работ Коновалова [25] нитрование парафиновых
углеводородов не получило практически дальнейшего развития до 1930 г.,
когда Хасс с сотрудниками в университете Пурдю начали широкое изучение
газофазного нитрования низкомолекулярных парафиновых углеводородов
[26]. Например, они нагревали пропан с азотной кислотой в газовой фазе
при 410° и получили при этом нитрометан, нитроэтан и оба изомерных пп-
тропропапа. Метод применен в промышленных масштабах фирмой Ком-
мершиал Сольвенте Корпорейшн (Иллинойс, США). В настоящее время
в США и в Англии производится много тысяч тонн низкомолекулярных
питропарафинов.
Нитрование высокомолекулярных парафинов проводят в настоящее
время двумя способами. Способ, разработанный Грундмаиом [27], состоит
в том, что нагретый до 170—180° парафиновый углеводород взаимодействует
с перегретыми парами азотной кислоты. В этих условиях нитрование идет
исключительно быстро. Метод применим при условии, чтобы температура
начала кипения углеводородной смеси составляла 160—170°. Для углево-
дородов с 7—12 атомами С газофазное нитрование Хасса не может быть
применено из-за возможности пиролиза, способ Грундмана не пригоден
вследствие низкой температуры кипения этих углеводородов. Для таких
углеводородов Гейзелер разработал изящный способ нитрования в присут-
ствии четырехокиси азота под давлением при 160—170° [28].
а) Нитрование пропана
Промышленное нитрование пропана, имеющее в настоящее время перво-
степенное значение, осуществляется следующим образом. Пропан под давле-
нием 7 ат нагревается до температуры 430—450° и в изолированном реак-
торе приводится в соприкосновение с потоком тонко распыленной 75 %-ной
азотной кислоты. Азотная кислота подается через насадки (жиклеры) в раз-
личные точки потока пропан-газа (рис. 70).
Насадки расположены таким образом и
количество подаваемой через них кис-
лоты дозировано так, чтобы теплота ис-
парения кислоты полностью компенсиро-
вала теплоту реакции нитрования. В то-
Рис. 70. Схема нитрования газообразных пара-
финовых углеводородов.
1 — насадки для подачи азотной кислоты; 2— реактор;
3 — холодильник; 4 — отделитель.
Линии'. I — азотная кислота; II — циркулирующий
пропан; III — пропан; IV — нитропарафины на пере-
гонку; V — разбавленная азотная кислота.
126'
время как общее молярное отношение пропана к азотной кислоте соста-
вляет примерно 5:1, подача кислоты через насадки установлена такой,
что это соотношение на каждой ступени всякий раз равно 25 :1 [29]. Печь вы-
полнена из нержавеющей стали и имеет внутри облицовку из стекла пирекс.
Выходящие из реактора пары охлаждаются, непрореагировавший
углеводород возвращается в процесс, а продукты нитрования разделяют рек-
тификацией. Состав нитропарафинов, получаемых описанным выше спосо-
бом, примерно следующий: нитрометана 25%, нитроэтана 10%, 1-нитро-
пропана 25%, 2-нитропропана 40%.
Это соотношение не всегда отвечает потребности в тех или иных продук-
тах, что вынуждает при помощи специальных мер изменять его в требуемом,
направлении. Работами Бахмана и его учеников показано, что добавкой
кислорода, хлора или того и другого можно влиять па распределение раз-
личных нитропроизводных в продуктах реакции [30]. Определенное значе-
ние имеют также температура реакции, соотношение компонентов и время
пребывания реакционной смеси в реакторе.
Комбинируя различные условия, в настоящее время при нитровании
пропана в газовой фазе можно получить следующие выходы продуктов,
реакции (в %):
нитрометана .................9—49 1-нитропропапа ...........9—35
нитроэтапа ............. 6—36 2-питропропапа ..........5—45
б) Нитрование высокомолекулярных парафиновых углеводородов
При нитровании по Грундману, которое протекает без применения по-
вышенного давления, наивыгоднейшая температура реакции 160—180°.
Необходимые для реакции пары азотной кислоты получают пропусканием
азотной кислоты по стеклянной труб-
ке, расположенной в сосуде с углево-
дородом, нагретым до указанной тем-
пературы. При этом кислота нагре-
вается до 160—180° и полностью пере-
ходит в парообразное состояние. Пары
Рис. 71. Аппарат для нитрования высо-
комолекулярных парафиновых углеводо-
родов.
Рис. 72. Схема нитрования парафиновых
углеводородов двуокисью азота под давле-
нием.
1— реактор; 2 — холодильник; 3 — вентиль;
4 — отделитель.
Линии: I—подача Na под давлением; ТТ— н итропа-
рафины па переработку; III — азотная кислота.
1 — реакционный сосуд; 2 — нагревательная
баня; 3 — змеевик перегревателя; 4 — газорас-
пределительная пластинка нз пористого мате-
риала; <5 — н гааоотводу; 6' — капельная во-
ронка.
азотной кислоты поступают затем через газораспределительную пластинку
из пористого материала в горячий углеводород и быстро нитруют его (рис 71).
Нитрование в этих условиях является вполне безопасным процессом,
так как нет одновременного контакта углеводорода с большим количеством
такого сильного окислителя, как азотная кислота.
127
По способу Гейзелера жидкая четырехокись азота, способная смеши-
ваться с углеводородами в любом соотношении, вместе с углеводородом
проходит по трубке, нагретой электрической грелкой или теплоносителем
до 170—180° (рис. 72). При работе по методу Грундмапа вся сумма про-
дуктов нитрования до конца реакции, т. е. в течение нескольких часов,
остается при температуре 170—180°, в методе Гейзелера время превращения
составляет всего несколько минут, что исключительно благоприятно для
продуктов нитрования.
В ходе дальнейшей переработки продуктов нитрования используется
способность нптропарафинов растворяться в щелочи (образование аци-
формы). Таким путем удается отделить иитропарафины от непрореагиро-
вавшего углеводорода. Из водно-щелочного раствора продукты нитрования
выделяются затем путем продувания через раствор углекислоты. Образовав-
шиеся при нитровании в качестве побочных продуктов карбоновые кислоты
остаются в растворе в виде натриевых солей. Моно-, дп- и полинитропроиз-
водные разделяются далее перегонкой (рис. 73).
ИСХОДНЫЙ ПРОДУКТ
I
Нитрование
Смесь из нитропарафинов и углеводородов
Обработка спиртовой щелочью, разбавление
водой и смешение с пстролитным эфиром
В щелочи растворяются:
первичные и вторичные нитропарафины
и карбоновые кислоты
Обработкой углекислотой выделяются
первичные и вторичные иитропарафины
Подкислением минеральной кислотой
выделяются карбоновые и нитрокарбоно-
вые кислоты
В щелочи не растворяются, во раство-
ряются в петролейном эфире
третичные нитросоедипения, непрореаги-
ровавшие углеводороды (нейтральное
масло), а также нейтральные продукты
окисления (спирты, кетоны)
Рис. 73. Схема переработки продуктов нитрования высокомолеку-
лярных парафиновых углеводородов.
При нитровании высокомолекулярных парафинов, как и при хлори-
ровании (см. стр. 116), одинаково вероятно присоединение нитрогрупп к лю-
бому из метиленовых радикалов парафиновой цепи, в то время как первич-
ные нитропарафины образуются в очень ограниченных количествах.
Б. ФИЗИЧЕСКИЕ И ФАРМАКОЛОГИЧЕСКИЕ СВОЙСТВА
НИЗКОМОЛЕКУЛЯРНЫХ НИТРОПАРАФИНОВ
В настоящее время низкомолекулярные нитропарафины играют исклю-
чительную роль в промышленности. Они являются превосходными раствори-
телями, особенно в смеси со спиртами, для нитро- и ацетилцеллюлозы.
Эфиры целлюлозы, виниловые смолы, бутират и пропионат целлюлозы также
прекрасно растворяются в нитропарафинах. В табл. 62 приведены важней-
шие физические константы нитропарафинов, полученных нитрованием про-
пана [31].
При перегонке нитропарафинов небольшая добавка борной кислоты
(0,1 —1,0%) сильно тормозит разложение. Относительно ядовитости низко-
молекулярных парафинов мнения расходятся. Исследования показывают,
что иитропарафины ядовиты и обращение с ними требует большой осторож-
ности [32]. Нитрометан является сильным ядом для центральной нервной
системы. Ядовитость увеличивается с длиной молекулы.
Однако ядовитость нитропарафинов не выше, чем обычно применяемых
растворителей лаков, так что при некоторой осторожности они вполне могут
применяться в качестве растворителей.
128
Таблица 62
Важнейшие физические константы промышленных образцов — нитропарафинов,
полученных нитрованием пропана
Показатели Нитрометан Нитроэтан 1-нитро- пропан 2-нитро- пропан
Удельный вес 20°/20° 1,139 1,052 1,003 0,922
Температура плавления, °C . . —29 —90 —108 —93
Температура кипения при 760 мм рт. ст., °C 101,2 114,0 131,6 120,3
Упругость паров при 20°, мм рт. ст 27,8 15,6 7,5 12,9
Температура вспышки в откры- том тигле, °C Коэффициент преломления, 20° 45 41 49 40
1,3818 1,3916 1,4015 1,3941
pH в 0,01 мол. водном растворе при 20° 6,4 6,0 6,0 6,2
Испаряемость (и-бутплацетат-100) 180 145 100 124
Растворимость при 20°, мл! 100 мл: пптропарафппа в воде . . . 9,10 4,5 1,4 1,7
воды в нптропарафипе . . . 2,2 0,9 0,5 0,6
Температура кипения азеотроп- ной смеси с водой, °C .... 83,6 87,1 88,4 91,2
Содержание нитропарафипа в азео- тропной смеси, % вес 77,1 73,6 73,1 64,5
13. ХИМИЧЕСКИЕ РЕАКЦИИ НИТРОПАРАФИНОВ
а) Нитроспирты
При действии формальдегида па нитропарафины в присутствии таких
соединений, как едкие щелочи, щелочные карбонаты или амины, образуются
нитроспирты. Реакция протекает согласно уравнению
О
В—C1L—NOa-f-HC^ -> В—CH—NO2
\ I
н сн,он
Формальдегид реагирует с водородным атомом того углеродного атома,
с которым связана нитрогруппа. Нитрометан, имеющий три активных водо-
родных атома, реагирует с тремя молями формальдегида.
Подобно формальдегиду действуют и другие альдегиды, например,
ацетальдегид или бутиральдегид, при этом, однако, в реакции участвуют
только два'водородных атома нитрометана.
В табл. 63 приведены важнейшие физические свойства промышленных
образцов продуктов реакции иптропарафинов с формальдегидом [31, 33].
Нитроспирты с органическими кислотами дают эфиры. С уксусным
ангидридом, например, достигается практически 100%-ный выход. Получен-
ные таким способом эфиры представляют собой бесцветные жидкости, являю-
щиеся превосходными пластификаторами.
Сложные эфиры неорганических кислот и нитроспиртов, кроме нитро-
метана, не имеют особого значения. Нитрометан с формальдегидом в присут-
ствии 0,1%-ного раствора едкого натра очень легко дает три-(гидроксиме-
тил) нитрометан, превращающийся с азотной кислотой в присутствии концен-
трированной серной кислоты в трппитрат. Этот продукт, называемый также
трипитробутилглпцсрипом, обладает очень высокой взрывной силой (на
70% выше нитроглицерина) и может быть получен из воздуха, воды и метана
или другого низкомолекулярного парафинового углеводорода, следующим
путем.
9 Заказ 596.
129
Таблица 63
Физические свойства продуктов реакции четырех простейших
нитропарафинов с формальдегидом
Показатели 2-питро- бута- нол-1 2-нитро-2- метилпропа- пол-1 2-нитро-2- метилпро- пандпол-1,3 2-питро- 2-этилпро- пандиол-1,3 Три-(гидро- ксиметил) нитрометан
Молекулярный вес . . Температура плавления, °C Температура кипения при 100 мм рт. ст. pH 0,1 мол. водного рас- 119,12 119,12 135,12 145,15 151,12
—47 до —48 105 90—91 95 147—149 56—57 Разлагается 165—170
твора, 20° Растворимость в воде 4,5 5,1 5,4 5,5 5,6
при 20°, .ил/100 мл 20 350 80 400 220
Удельный вес . Коэффициент преломле- 1,1332 — — — —
ПИЯ 1,4420 — — — —
1. Воздух —> Азот-|-Кислород (установка Линде)
2. СН4+Н2О -Кат^3- СО+ЗН2
.. Катализ „ „ ,
3. Получение водорода конверсией для синтеза аммиака СО+Н2О----------- СО2 —|—
-|-Н2. Углекислота отмывается водой под давлением
4. СО+2Н2 —тал-- СН3ОН
„ . ,.тт Катализ+ 02
5. Получение формалт.дегида или из метилового спирта СН3ОН----------------
СПгО4-Н2О, или непосредственно из метана или других низкомолекулярных пара-
финовых углеводородов частичным окислением
Катализ „_,ттт , ,
6. N2-j-3H2-------► 2NH3 (синтез аммиака)
7. NH3+2O2 -Ката—3-> HNO3-I-H2O (окисление аммиака в азотную кислоту)
8. CH4+HNO3 —> CH3NO24-H2O (нитрование метана)
9. CH3NO2-|-3HCOH Щелочь-. no2—С (СН2ОН)3 (конденсация)
10. O2N-C (CH2OH)s+3HNO, -°"Цен28О4ВаННа^ N°a—с (CH20N0»)8+H20 (эстери-
фикация нитроспиртов)
Нитропарафины могут также реагировать по Манниху. Опять реагирует
водородный атом, связанный с тем же углеродным атомом, что и нитрогруппа
[34]. Так, например, при совместном воздействии формальдегида и изопро-
пиламина на 2-нитропропан после 5-дневного стояния образуется N-(2-
нитроизобутил)-изопропиламин:
СН3 О СН3 СН3 СН3
I II I I I
NO2-C—H-I-H—С—H+H2N—СП -» N02—С—СН2—NH—сн+н2о
I III
сн3 сн3 сн3 сн3
Реакцией 2 молей формальдегида и 2 молей изопропиламина с 1 молем
нитроэтана образуется с 70%-ным выходом 5-нитро-2,5,8-триметил-3,7-
диазононан: СН3—СН2—NO2+2HCOH-|-2CH3—СН—СН3 NH, сн3 no2 сн8 -* CH—NH—СН2—С—СН2—NH—СН4-2Н2О 1 1 1 си3 сн8 сн8
130
Такие диаминопроизводные могут далее конденсироваться с формальде-
гидом с образованием циклических соединений. Названный диазононан
в ходе дальнейшей конденсации с формальдегидом замыкается в пиримидино-
вое кольцо с образованием 5-нитро-5-метил-1,3-диизопропилгексагидропири-
мидина:
и с СН3 [Т 1
сн2 н сн \ - । /^п3 \ г гн N СН: ПС. Л Л?1
1 /Н Cll3 \
СНа—С—NO2+ О =с< -+ п2о+нас 1 I хсн3
ХН сн3 Н2С сн.
сн2 II N сн/ 3 \ /
хсн3 с
СН3 NO2
Нитроспирты легко восстанавливаются в аминоспирты. Эта реакция про-
водится преимущественно при нагревании с никелем Ренея в растворе мети-
Таблица 64
Важнейшие свойства некоторых аминоспиртов и аминогликолей
Продукт Формула Температу- ра кипения прп 10 мм рт. ст., °C Темпе- ратура плавле- ния, °C „20 nD
2-аминобутанол-1 СН3-СН2-СН-СН2ОН 1 NH, 80 —2 0,944 1,453
2-амипо-2-метилпропа- нол-1 СН3 1 СН3—с—СН2ОН 1 NH2 67 25—26 0,934 1,449
2-амино-2-метилпропап- диол-1,3 сн3 НОН2С—С—СН2ОН 1 nh2 151—152 109—111 —
2-амино-2-этилпроиан- диол-1,3 с2н6 1 НОН2С—С-СН2О1 NH2 152—153 37,5—38,5 1,099 1,490
2-амипо-2-пропилпропаи- диол-1,3 С3Н, 1 НОН2С—С—СН2ОН 1 NH3 — 58 — —
2-амино-2-изопропилпро- пандиол-1,3 i-C3H, 1 НОН2С—С—CIIsOH 1 nh2 — 74 — —
Три-(гидроксиметил) аминомстап уСНаОН NH«—С—СН2ОН /СН»ОН 219—220 171—172 — —
9*
131
левого спирта под давлением водорода. Восстановление должно проводиться
при возможно низкой температуре. В табл. G4 приведены важнейшие свой-
ства некоторых ампноспиртов.
б) Действие минеральных кислот на питропарафины
При действии минеральных кислот на первичные питропарафины, как
нитроэтаи и 1-нитропронан, образуются карбоновые кислоты и гидрокси-
ламин [35]. Например, смесь 1 моля 1-питропропана с 1 молем 85 %-ной
кислоты нагревают до 120е до начала бурно протекающей реакции, по оконча-
нии которой смесь еще нагревают ири 140° в течение 8 час. с обратным хо-
лодильником для завершения реакции. В результате получают около 92%
пропионовой кислоты и 88% гидроксиламипа. Нитроэтан дает уксус-
ную кислоту и гидроксилампи. При действии минеральной кислоты
на ациформу нитроиарафинов образуются альдегиды или кетоны (реакция
Нефа) [36]:
2Н—CII=N00Na-|-2II2S0.1 -> 2II- -С +2NaIISO1+N.,p+H2O
И\ 1{\
2 ''c=NOONa+2II,SO4 -> 2 C=O+2NaHSO4 + N2O + H,O
r/ r/
в) Дальнейшее нитрование парафиновых углеводородов
Дальнейшее, более углубленное нитрование моионитропарафинов до
дипитропарафинов должно производиться при более низкой температуре,
чем газофазное нитрование, чтобы предотвратить пиролиз. Нитрованием
2-нитропропана 70%-пой азотной кислотой при температуре 210—230°
и давлении 60—100 ат получается с хорошим выходом 2,2-дипитропропан
{37]. Такие динитросоединепия обладают способностью значительно повы-
шать цетановое число дизельных топлив.
Высоконитрированные алифатические соединения как тетрапитрометан
не могут быть получены нитрованием нитрометана. Их получают действием
концентрированной азотной кислоты на ангидрид уксусной кислоты или
ацетилен [38].
При обработке нитроиарафинов в щелочной среде окислителями пер-
вичные нитропарафины превращаются в альдегиды или карбоновые кислоты,
а вторичные питропарафины в кетоны [39].
Действием азотной кислотой на первичные нитросоедпнепия получают
нитронитрозосоединения, способные еще к образованию ациформы. Рас-
творы этих соединений, называемых нптролами, в щелочи имеют алую
окраску. Вторичные питросоединения, реагируя с азотной кислотой, дают
нитронитрозосоединения, уже не растворимые в щелочи, по растворимые
в хлороформе с зелено-синим окрашиванием (псевдонитролы [40]).
г) Галоидирование пптропарафииов
При действии хлора или брома на нитросоединенпя в щелочной среде
мгновенно образуются геминально-замещенные галоидонитросоединения.
Первичные нитросоединения могут присоединять два атома галоида, вто-
ричные — только один. Действием гипохлорита на нитрометан получают
хлорпикрин (NO2CG13). В первую мировую войну хлорпикрин применялся
как боевое отравляющее вещество, но в настоящее время он утратил это зна-
132
чение, так как полностью адсорбируется активированным углем. Сейчас
он применяется как инсектисид и для стерилизации почвы.
Другие хлориитропарафины, как, например, 1,1-дихлор-1-нитроэтан„
обладают практически таким же действием, как и хлорпикрин, но не вызы-
вают слезотечения.
В табл. 65 приведены физические свойства некоторых низкомолекуляр-
ных хлорнитропарафинов, а на рис. 74—76 — возможные пути превращения
нитрометана, питроэтапа и питропропапа.
Таблица 65
Физические свойства низкомолекулярных хлорнитропарафпнов
Показател и 1-хлор-1- питроэтап 1-хлор-1 - гштр о про- пан 2-хлор-2- яитро- пропап 1,1-дп- ХЛОР'1- нп тро- этан 1,1-дп хлор- 1-пптро- пропап
Молекулярный вес .... 109,52 123,54 123,54 143,97 157,99
Удельный вес 20°/20° . . Температура выкипания 90% при 760 -им рт. ст., 1,258 1,209 1,193 1,405 1,314
°C Температура вспышки 112—128,5 139—143,3 129—132 122-125 141—143,6
(открытый тигель), °C . . Коэффициент преломления 57 62 57,5 76 66
„20 nD Растворимость при 20°, лгл/100 мл'. хлорпнтропродуктов в 1,423 1,430 1,425 1,441 1,443
воде воды в хлорнптропро- 0,4 0,8 0,5 0,5 0,5
дуктс 0,5 0,4 0,5 0,5 0,5
д) Дальнейшая переработка высокомолекулярных нитроиарафинов
Дальнейшая переработка продуктов нитрования высокомолекулярных
парафиновых углеводородов может проводиться так же, как и низкомолеку-
лярных членов ряда. При восстановлении с никелем Ренея в спиртовом
растворе при 40—50° и под давлением 100 ат водорода они превращаются
в амины [41]. При окислении перманганатом калия в щелочной среде,
перекисью водорода или озоном образуются с очень хорошим выходом высо-
комолекулярные кетопы, которые каталитическим гидрированием могут
быть легко переведены во вторичные спирты, а эти последние при необходи-
мой длине алкильного остатка дают при сульфировании каппллярно-актпв-
пые вещества.
Взаимодействие с формальдегидом в щелочной среде ведет к образова-
нию питросвиртов, которые могут быть далее превращены в амппоснирты,
применяемые для самых различных целей.
Хлорированием высокомолекулярных нитроиарафинов равным обра-
зом получают гемптталыго-замещенпые хлорнитросоединения, в которых
хлор обладает очепь большой подвижностью.
Ш. СУЛЬФСХЛОРИРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
При действии сернистого ангидрида и хлора на парафиновые углево-
дороды в условиях ультрафиолетового облучения или в присутствии обра-
зующих радикалы веществ образуются алифатические сульфохлориды по
уравнению
RII4-SOH-CI2 -> BSO2C]-|-]IC1
133
HCHO 2HCHO знсно
НОН2С—CH2NO2 (HOH2C)2CHNO2 (HOH2C)SCNO2
Кислоты
Эфиры
CHgXOj
СвН6СНО
С12
_CH2CiNO2
CHC12NO2
Восстановление
CH3NHOH
chsnh2
(СНэ)2СО
NaOH
КОН
0
---CH2 = N<^
ONa
Кислоты
Эфиры
Кислоты
Эфиры
Восстановление Восстановление Восстановление
НОН2С—CH2NH2 (HOH2C)2CHNH2 (HOH2C)3CNH2
1
Высшие жирные кислоты
I
МЫЛА ОКСАМИНОВЫХ СОЕДИНЕНИЙ
Нагрев
I
АМИДЫ
Кислота,
гидролиз
*
CClaNO2
C2HaNa
I
(C2H5O)4C
HSN
HN = C (X'H2)2
Щелочные соли ни-
тропарафинов
CcH5CHOH—CH2NO2
Кислоты
Гидролиз
ДИНИТРОПАРАФПНЫ
СвН6СНОН—COOH
Рис. 74. Производные нитрометана.
NOH
О
CH-CH = N-^
OK
HgCl2
CH2KO2
(CHa)2C^
CH2NO2
(C = NO)2Hg
I
Альдегиды
и кетоны
ОКСИМЫ
Восстановление
АМИНЫ
2HCH0
C3H7CHO
CH2OH
-----------СН3-------
cno2
СН2ОН
Кислоты
Эфиры
Восстановление
I
СН2ОН
I
СНэ-CNH-2-
СН2ОН
CHs-CH2NO;
I
СбНзСНО
С6Н5СНОН—CH-NO2.CH3
I
С12
CH3CHC1NO2
СНзСС12КО2
NaOH
C3H7CHOH.CH.NO2.CH3
О
ch8.ch = n^
О Na
I
H2SO4 + H.O
I
CH3COOH
NH2OH.H2SO^
Кислоты
Эфиры
Дегидратация
C6H6CH = C.NO2-CH3
Восстановление
Восстановление
I
I C6H5.CH2.CH.NH2.CH3
CaH7CHOH.CH.NH2.CH3
Высшие жирные кислоты
I
I
МЫЛА
Нагрев
I
АМИДЫ
Натриевые соли
нитропарафина
Конденсация
и гидролиз
Альдегиды и
кетоны
ДИНИТРОПАРАФИН
ИЗОКСАЗОЛ
Восстановление
CH3.CH2.NH2
H2SO4
ОКСИМЫ
СНдСНО
Восстановле-
ние
Восстановление
СвНБСНОН.СН.КН2.СНз
АМИНЫ
Рис. 75. Производные питроэтана.
(1-нитропропан дает те же самые реакции, что и нитрсэтан, но образующиеся при этом продукты содержат еще одну группу СН2.)
Кислоты
Эфиры
CH3CH.NO2CH3
нсно
СНз
ХО2-С—СН2ОН
СНз
Восстановление
I
СНз
nh2c—СН2ОН
СНз
Высшие жирные
кислоты
J.
МЫЛА
Нагрев
ЛМПДЫ
С3Н7СНО
| С\3
С3Н,СНОН.СКОг
СНз
Восстановление
СНз
С3Н7СНОН.Сх\Н2
СНз
Кислоты
Эфиры
Cl2
С1
СНз-С-СНз
NO2
ДИНПТРОПЛРАФПН
Рис. 76. Производные 2-нптропропана.
NaOH
О
I /
(СН3 2 C=N
H2SO4
(СНз)2 СО
Восстановление
СНз
сн—nh2
/
СНз
Соединения с высокой реакционной способностью, относительно легко
превращаемые в различные промежуточные и целевые продукты современной
технологии соединений алифатического ряда.
Одновременно с собственно сульфохлорированием, как важнейшая по-
бочная реакция, протекает только одно хлорирование углеродной цепи без
одновременного присоединения двуокиси серы. При проведении сульфохло-
рирования в условиях рассеянного освещения, реакции сульфохлорирова-
ния и хлорирования углеродной цепи протекают с практически одинаковой
скоростью, так что в молекуле на каждый атом серы приходится примерно
двойное количество атомов хлора. Если реакция сульфохлорирования прово-
дится в условиях облучения ультрафиолетовым светом или в присутствии
образующих радикалы веществ, как перекиси, тетраэтилсвинец, диазометап
и т. п., хлорирование углеродной цепи приобретает второстепенное значение
и практически идет только сульфохлорировапие.
При сульфохлорировапии, как и при других реакциях замещения, на-
ступает момент, когда в реакционной смеси еще имеется известное количе-
ство пепропеагировавшего (незамещенного) углеводорода, по уже происхо-
дит образование дисульфохлорида, которое может принять такие размеры,
что сульфохлорированию подвергнется только часть углеводородов, тогда
как для остальной части пе хватит сульфохлорирующих реагентов.
Процесс сульфохлорировапня представляет собой типичную цепную
реакцию.
1. С]2+Л V -> С1--фС1-
2. RH-f-CI' -4 R'4-l-ICl
3. R'4-SO, -> R—SO»'
4. R—S(V+(X BSOoCl+CJ-
Считается, что в этой реакции сначала под действием ультрафиолетового-
света молекулы хлора расщепляются на атомы. Атом хлора отнимает от
углеводородной молекулы одни атом водорода, причем образуются хлори-
стый водород и алкильный радикал. Алкильный радикал соединяется с дву-
окисью серы с образованием алкилсульфонового радикала, который реаги-
рует с молекулой хлора, давая сульфохлорид и освобождая атом хлора.
Квантовый выход при технологическом сульфохлорировапии составляет
около 2000.
В присутствии веществ, образующих радикалы, атом хлора освобо-
ждается в результате реакции молекулы хлора с алкильными или ациль-
ными радикалами. Исключительно эффективной в этом отношении оказы-
вается перекись ацетилциклогексилсульфопила, распадающаяся на следую-
щие радикалы [42]:
CcHnSOaOOCOCH3 -> CeIInSO2O'+C]I3COO'
1
, CO2 + CU3
Количество сульфохлоридов, образующихся при сульфохлорировании,
можно определить титрованием спиртовым раствором метилата натрия,
дающим в присутствии фенолфталеина красное окрашивание. По уравнению
R—CIl—C]bSOXl + NaOCH3 -+ NaCl+R—СИ—CH2SO.,OCH3
1 I
Cl Cl
происходит мгновенная реакция с образованием сложного эфира алкилсуль-
фоновой кислоты, сопровождающееся обесцвечиванием мстапол-феполфта-
леинового раствора. Первая капля избыточного раствора метилата натрия
вызывает опять красное окрашивание.
Процентное содержание дисульфохлоридов в продуктах сульфохлори-
рования углеводородов может быть определено растворением сульфохлорпд-
ной смеси в большом количестве пентана с последующим охлаждением до
—60°. При этом дисульфохлориды выпадают в осадок.
137
А. ОТНОШЕНИЕ ОТДЕЛЬНЫХ ТИПОВ УГЛЕВОДОРОДОВ
К СУЛЬФОХЛОРИРОВАНИЮ
Лучше всего сульфохлорируются и-парафины. Разветвленные парафино-
вые углеводороды также легко реагируют, но при этом значительно увели-
чивается значение реакции хлорирования углеводородной цепи, так как
третичный углеродный атом не сульфохлорируется, а предпочтительно
хлорируется.
Циклопарафины (цпклопентан и циклогексан) сульфохлорируются очень
хорошо. Циклопарафииы с боковыми цепями ведут себя так же, как развет-
вленные парафиновые углеводороды с открытой цепью.
Ароматические углеводороды — бензол, нафталин и т. д. — не суль-
фохлорируются; при наличии у ароматических углеводородов боковых цепей
сульфохлорировапие этих алифатических боковых цепей возможно, но
в ограниченных размерах.
Нефть поддается сульфохлорированию только после основательной
очистки от азот-, кислород- и серусодержащих соединений, а также, видимо,
ароматических углеводородов. Путем гидрирования под высоким давлением
или сернокислотной очистки из нефти может быть выделена смесь углеводо-
родов, поддающихся сульфохлорированию. Получающиеся прй этом сульфо-
хлориды имеют темную окраску и содержат относительно много хлора в угле-
родных цепях.
Для технических целей наиболее подходящим исходным материалом
может служить гидрированный при высоком давлении когазин II синтеза
Фишера-Тропша с кобальтовым катализатором. Гидрирование проводится
примерно при 320° и 200 ат давления водорода над сульфидным никель-
вольфрамовым катализатором. При этом получают с 99%-ным выходом
смесь бесцветных вполне насыщенных углеводородов, очень мало развет-
вленных, так называемые мепазины. При сульфохлорировании получается
смесь всех теоретически возможных моносульфохлоридов. Если в качестве
исходного материала применяется смесь парафиновых углеводородов с пря-
мой цепью и четным числом углеродных атомов в цепи, то образуется равное
количество всех возможных вторичных сульфохлоридов, так как сульфо-
хлорирование любой из метиленовых групп одинаково вероятно. Первичных
сульфохлоридов получается очень мало, во-первых, потому, что реакционная
способность водородных атомов метильных групп меньше, чем водородных
атомов метиленовых групп, а во-вторых, потому, что с увеличением длины
молекулы парафиновых углеводородов число метиленовых групп значи-
тельно увеличивается.
Б. РЕАКЦИИ АЛИФАТИЧЕСКИХ СУЛЬФОХЛОРИДОВ
Реакции алифатических сульфохлоридов основаны на исключительной
подвижности связанного с серой хлора. Важнейшей реакцией является ще-
лочное омыление сульфохлоридов в сульфонаты:
RSO2Cl + 2NaOH -> RSO2ONa + NaCl-|-H2O
Сульфонаты, у которых парафиновая цепь состоит из 12—20 углерод-
ных атомов, обладают очень высокой капиллярной активностью.
Действием аммиака на сульфохлориды получают алифатические суль-
фамиды
RSO2C1 + 2NH3 -> RSO2NH2+NH2C1
в свою очередь способные ко многим дальнейшим превращениям.
С такими аминами, как анилин, диэтиламин и т. д., сульфохлориды
дают N-алкилированные сульфонамиды. Сульфохлориды реагируют также
легко с фенолятами, алкоголятами и тиоалкоголятами, давая соответствую-
щие алкилсульфокислые эфиры. Продукт превращения алкильного ради-
138
кала (в среднем 12—16 атомов С) с фенолятом натрия является превосход-
ным пластификатором для поливинилхлорида (мезамол), с успехом применяе-
мым вместо трикрезилфосфата.
Различный эффект может быть достигнут варьированием длины алкиль-
ного остатка, степени сульфохлорирования и заменой фенольных компонен-
тов. При кигДтчении алифатических сульфохлоридов с низкомолекулярными
спиртами, как метиловый пли этиловый, образуются не эфиры, а сульфо-
кислоты, в то время как спирт превращается в соответствующий галоидал-
кил:
RSO3Cl + ROH-> RSO2OH + RC1
Этим путем из сульфохлоридов алифатических углеводородов можно
легко получить в чистом виде соответствующие сульфокислоты.
При нагреве сульфохлориды отщепляют двуокись серы и превращаются
в ал кил хлориды. Эта реакция, называемая десульфированием, протекает
относительно легко, особенно с высокомолекулярными сульфохлоридами,
когда нет перегонки.
Сульфофториды, легко получаемые реакцией между сульфохлоридами
и фтористым калием, применяемым в виде концентрированного водного
раствора, обладают высокой термической устойчивостью. Они легко перего-
няются в вакууме.
В. ПРАКТИЧЕСКОЕ ПРОВЕДЕНИЕ СУЛЬФОХЛОРИРОВАНИЯ
Сульфохлорирование газообразных при нормальных условиях парафино-
вых углеводородов проводится пропусканием смеси парафинового углево-
дорода с двуокисью серы и хлором через инертный растворитель, например
четыреххлорпстый углерод, при одновременном облучении ультрафиолето-
Рис. 77. Лабораторный аппарат для сульфохлорирования жидких и газообразных
углеводородов.
/ — ртутная лампа; 2 — пористая пластинка; з — охладительный змеевик; 4 — реометр; <5 — гидра-
влический затвор.
Линии'. I —_вода; II — углеводородный газ из бомбы; III — газообразный SO2 из бомбы; IV — газо-
образный хлор из бомбы.
вым светом. Если хотят получить в основном моносульфохлорид, то приме-
няют избыток парафинового углеводорода. На рис. 77 показан аппарат для
сульфохлорирования газообразных и жидких углеводородов в лаборатор-
ных условиях. При соответствующем увеличении этот аппарат может быть
139
превращен в полупромышленную установку, па которой процесс может осу-
ществляться непрерывно.
При сульфохлорировании газообразных парафиновых углеводородов газ
(двуокись серы + парафиновый углеводород -4- хлор) подают в четырех-
хлорпстый углерод снизу, через пористый стеклянный фильтр. По окончании
реакции четыреххлористый углерод отгоняют. В остатке — смесь дип-
моносульфохлоридов. Если целью процесса является получение дпеульфо-
хлорида, то углеводорода в реакцию берется значительно меньше, чем тре-
буется стсхиометрически для получения мопосульфохлорида. Из приведен-
ных в табл. 66 данных можно видеть, какое влияние па выход мопосульфо-
хлоридов и дисульфохлоридов при сульфохлорпровапип пропана и бутана
оказывает избыток или недостаток углеводорода в реакционной смеси [43].
Таблица 66
Влияние различных объемных соотношений углеводорода, двуокиси серы
и хлора при сульфохлорнроканпи пропана п к-бутапа на образование моно-
п дпеульфохлоридов п хлорсульфохлорпдов (в %)
Объемное соотно- шение углеводо- родов : SO2: С12 Мопосульфохлорпд Дпсул! фохлорид Хлорсульфохлорпд
пропап м-бутап пропан /г-бутан пропан к-бутап
2,5 : 1,1 : 1 80 85 10 1.0—13 7 3—5
1,0 : 1,1 : 1 48 — 42 — 10 —-
0,55 :1,1 : 1 15 10 60 85 15 3-5
При сульфохлорированных жидких при нормальных условиях пара-
финовых углеводородов, как, например, очищенные нефтяные фракции или
мепазин, двуокись серы и хлор в соотношении 1,1:1 подаются непосред-
ственно в углеводород при одновременном облучении ультрафиолетовым
светом. Для отвода тепла реакции служит расположенный в реакционной
жидкости охлаждающий змеевик. Охлаждение возможно также путем про-
качки насосом реакционной смеси через холодильник.
Так как капиллярно-активное действие продуктов омыления высокомоле-
кулярных сульфохлоридов зависит от степени сульфохлорировапия, полу-
чают три различных типа продуктов сульфохлорировапия мепазина — так
называемых мерзолеп. Их состав приведен в табл. 67.
Таблица 67
Состав трех типичных промышленных образцов мерзолей
Марка Содержание в продукте, Содержание мопо- еульфохлорида в Содержание дпеуль- фохлорида в обез-
/и
морзоля сульфохло- рида нейтрального масла обезмасляной смеси, % вес. масляной смеси, % вес.
30 30 70 -94 ~6
н -45 — 55 -84 -16
D 80—82 18—20 ~60 -40
Капиллярная активность продуктов омыления высокомолекулярных
сульфохлоридов при равной длине алкильных остатков зависит от глубины
сульфохлорировапия, а именно с увеличением содержания гидролизуе-
мого хлора в молекуле сульфохлорида его капиллярная активность умень-
шается. Последнее связано с увеличением количества продуктов ди- и поли-
сульфохлорирования при повышении степени сульфохлорирования [44].
Получаемые в результате омыления ди- и полпсульфонаты обнаруживают
лишь весьма ограниченную капиллярную активность.
140
Мерзоль 30 получается, когда в мепазин вводится примерно 30% дву-
окиси серы и хлора от того количества, которое теоретически необходимо
для полного превращения углеводородов в моносульфохлориды. Мерзоль Н
соответствует 50%. Мерзоль D отвечает тому случаю, когда в мепазин
введены все необходимые 100% двуокиси серы и хлора.
F. ПРЕВРАЩЕНИЕ СУЛЬФО ХЛОРИДОВ
При омылении сульфохлоридов растворами щелочей образуются сульфо-
наты, растворимые в воде, которая удаляется выпариванием после отделе-
ния от раствора непрореагировавших парафиновых углеводородов. Эти
продукты называются мерзолятамп [45]. Во время войны фирма Дюпон
в США выпускала продукты сульфохлорирования нефтяных фракций под
названием дюпонолов. В Германии продукты сульфохлорирования мепазина
(сульфонаты) производятся под маркой витолата и майнолата.
Продукты реакции высокомолекулярных сульфохлорпдов типа мерзоля
D с фенольными, крезольнымп и ксиленольными смесями использовались
в Германии во время войны как пластификаторы, особенно для поливинилхло-
рида. С этой целью фенолы смешивали с мерзолем и через эту смесь при тем-
пературе около 40° продували аммиак. После отделения хлористого аммо-
ния и промывки 2%-ным раствором хлористого кальция полученный эфир
освобождали от избыточного парафинового углеводорода продувкой водя-
ным паром под вакуумом. В заключение продукт обрабатывался 2% тонсиля
(отбеливающая земля) и фильтровался [46].
Высокомолекулярные алифатические сульфамиды реакцией с хлори-
рованными жирными кислотами в присутствии щелочи превращаются в алкил-
сульфамидокарбоновые кислоты, являющиеся превосходными эмульгато-
рами для минеральных масел и обладающие исключительными антикорро-
зийными свойствами. Из хлоруксусноп кислоты таким путем получают
сульфампдоуксуспую кислоту, применяемую под названием эмульгатора
STH в маслах для сверления.
При реакции сульфамидов с формальдегидом и хлористым водородом
в растворе хлористого метилена образуется N-хлорметилсульфамид, в кото-
ром хлор благодаря своей высокой реакционной способности может вступать
далее в реакции с тиомочевпной, пиридином, триметиламином и другими со-
единениями . Таким образом получают вспомогательные продукты для тек-
стильном, бумажной и кожевенной промышленности:
RSO2NIIa+ClCH2COOH RSO2NIICHsCOOH4-NaCl + H2O
Алкиле ульфами дкарбо-
пован кислота
RSO2NII2+HC1 + IICOH RS0oNHCH2C1+H30
N-хлорметнл-
сульфампд
При действии на сульфамиды смеси формальдегида с бисульфитом обра-
зуются растворимые в воде соединения по реакции:
RSO2NH2+IICOH + NaIISO3 -> RS0ojNIICIT2S03Na+H,0
Эта реакция, известная как реакция сульфометилирования, гладко
проходит также с другими соединениями, как фенолы, амины, меркаптаны
И Т. д.
Сульфохлорированием определенных галоидалкилов получают хлор-
сульфохлориды, которые при омылении дают оксисульфокислоты, переходя-
щие при нагреве (сопровождающемся отщеплением воды) в сультоны:
С1СП2—СН2—СН2—CH3+SO2-|-C12—> ОСИ—СН2—СН—СН3 -Ха01Г
I
SO3Cl
-> НОСН2—СН3—СН—СН3 Иа'^% СИ2—СН3—СН—СН3
I I I
SO2OH о so3
141
Такие сультоны являются исключительно ценными вспомогательными
веществами для введения сульфогрупп в соединения, содержащие актив-
ный водород, которые благодаря этому приобретают способность раство-
ряться в воде:
СН2— СИ»—СП— CIIh4-ROII I’.OCIb—СИ»—СИ—СП3
II I
О--------SOs SO2OII
Интересное применение как в Германии, так и в США находят высоко-
молекулярные сульфохлорпды типа мерзоля D. Чаще всего они применяются
для дубления кож. Хорошие сорта кож получают прп применении продуктов
сульфохлорирования, называемых «иммерган» (Германия) и «скелт» (США).
Д. ПРОДУКТЫ СУЛЬФОХЛОРИРОВАНИЯ ПОЛИЭТИЛЕНОВ [47]
В последнее время особое значение приобретают продукты сульфо-
хлорирования полиэтиленов. При взаимодействии полиэтилена с хлором
и сернистым ангидридом получаются продукты, содержащие' около 2G—
29% хлора и от 1,3 до 1,7% серы. Отсюда можно подсчитать, что при моле-
кулярном весе полиэтилена, равном 20000, каждый седьмой атом С связан
с атомом хлора, а каждый девяностый атом с сульфохлоридной группой.
Такой продукт вулканизируется добавкой ароматических диаминов, как,
например, бензидина или диоксима, тиурамена и аналогичных соединений.
При этом получается цепное каучукообразное вещество (гипалон S2 фирмы
Дюпон). Возможности различных вариаций состава и свойств продуктов,
которые могут быть получены на основе полиэтиленов, как в связи с различ-
ной глубиной сульфохлорироваиия, так и путем применения полиэтиленов
различного молекулярного веса, очень велики.
IV. СУЛЬФООКИСЛЕНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
А. ОБЩИЕ СВЕДЕНИЯ
Под сульфоокислением понимается совместное воздействие сернистого
ангидрида и кислорода па парафиновые углеводороды, протекающее со-
гласно следующему уравнению:
RH4-SO2-H/2 О2 -22^ RS03H
Эта реакция, как установил Платц в 1940 г., протекает под воздействием
ультрафиолетового света или в присутствии перекисей, например перкислот
[48]. Реакция идет также в присутствии некоторого количества озона.
В настоящее время парафиновые углеводороды посредством сульфоокисле-
ния могут также легко превращаться в сульфокислоты, как и ароматические
углеводороды, реакцией с концентрированной серной кислотой.
Натриевые соли продуктов сульфоокисления высокомолекулярных па-
рафиновых углеводородов, таких как мепазины, применяются в качестве
моющих средств, пенообразователей, эмульгаторов, смачивающих веществ
и флотационных реагентов.
Углеводороды, используемые в качестве исходного материала для
сульфоокисления, делятся на две группы. К первой группе относятся со-
единения (циклогексан, гептан и др.), которые после инициирования облу-
чением или добавки перкислот продолжают реагировать и в отсутствие
этих инициаторов. Вторая группа требует облучения или добавок органиче-
142
ских катализаторов в течение всего времени, пока идет реакция. К послед-
ним относятся высокомолекулярные парафиновые углеводороды.
Течение реакции сульфоокисления было изучено Графом на примере
циклогексана. Граф показал, что образуется (пока еще точно не известно
каким образом) гексильный радикал [42], дающий с двуокисью серы алкил-
сульфоиильпый радикал. Последний, реагируя с кислородом, дает алкилсуль-
фоперекисный радикал, вступающий во взаимодействие с новой молеку-
лой циклогексана, с образованием сульфоперкислоты и нового радикала
циклогексила:
R-|-SO2 -> RSO2’ — - RSOoOO' — - R’+RSO2OOH
Благодаря распаду сульфоперкислоты, протекающему в соответствии
с уравнениями
RSO2OOII RSO2O4-OH-
rso2o+rh -> RSO2OH+R-
RH-|-OH’—R’+H2o
каждый раз образуются новые радикалы, так что реакция один раз начав-
шись, дальше продолжается самопроизвольно.
У высокомолекулярных углеводородов, как, например, содержащихся
в мепазине, реакция прекращается после удаления инициирующего реакцию
источника света.
По Графу поведение отдельных углеводородов в рассматриваемой реак-
ции определяется соотношением между числом обрывов цепей и числом вновь
образовавшихся радикалов. Даже при применении совершенно сухих
исходных материалов в продуктах реакции всегда обнаруживается некоторое
количество серной кислоты, что указывает на образование воды при сульфо-
окислении. Последнее по Графу является результатом дегидрирования
циклогексана в циклогексен перкислотой по уравнению:
CeII11SO2OOH+CeH12 —» СвНпЗО2ОН-ГСвН10+Н2О
Вода реагирует затем в присутствии сернистого ангидрида с сульфо-
перкислотой по уравнению
CeHuSp2OOH+H2O+SO2 -*CeHuS0201I+H2S04
с образованием серной кислоты и сульфокислоты.
Сульфоперкислота высокомолекулярных парафиновых углеводородов
может быть стабилизирована добавлением уксусного ангидрида, который
образует с ней смешанный ангидрид по уравнению
СН3СО
CeTTnSO2OOH+ 'о -> CeHnSO2OOCOCn3+CH3COOH
CHSCO
Этот ацетилалкилсульфонилпероксид значительно более стабилен, чем
персульфокислота, но также постепенно разделяется на свободные ради-
калы в степени, достаточной, чтобы вызвать начало реакции.
143
При сульфоокислспнп парафиновых углеводородов сульфогруипы рас-
полагаются равномерно по всем метиленовым группам цепи. Образование
сульфокислот с сульфогруппами у конечных углеродных атомов тем менее,
чем длиннее парафиновая цепь.
В практике для сульфоокислепия применяют два метода: во-первых,
султ.фоокпслепие при облучении ультрафиолетовым светом (фотохимический)
[491, во-вторых — сульфоокпслеппе в присутствии ацилирующих соедине-
ний.
Б. ФОТОХИМИИ ЕСКИЙ СПОСОБ С ВОДНОЙ ЭКСТРАКЦИЕЙ (рис. 78)
В облучаемую ультрафиолетовым светом реакционную трубку подают
мепазпп, а навстречу ему сернистый ангидрид п кислород. Контакт осу-
ществляется при температуре 25—30°. Одновременно в реакционную трубку
вводят некоторое количество воды для отвода образующихся сульфокис-
лот и серной кислоты. Для поддержания температуры в реакционной трубке
Рис. 78. Схема сульфоокисления мепазина.
1 — сульфоокислительная башня; 2 — лампа; <3 — колонна для отделения мепазина; .4 — обогревае-
мая колонна для отделения сульфокислот; 5 — нейтрализатор; 6 — испаритель.
Линии: I — свежий мепазпп; II — вода; III — кислород; IV — сернистый ангидрид; V — циркули-
рующий мепазнн; VI — мепазип, сульфокислота, вода и H2SO4; VII — сульфокислота, вода и H0SO4;
VIII — разбавленная серная кислота; IX — сульфокислота и небольшие количества RH; X — мспа-
зпн и вода; XI —сульфонат
па необходимом уровне, ее содержимое прокачивают через выносной холо-
дильник. Часть содержимого постоянно отбирают в колонну, где из реак-
ционной смеси выделяется углеводород, возвращаемый через верх колонны
па сульфоокпслеппе. Тяжелый нижний слой, состоящий из серной кислоты,
сульфокислот и гидротрогшо растворенного в них мепазина, разделяется
во второй обогреваемой колонне. Здесь с низа уходит разбавленная серная
кислота, а не растворимая в ней при повышенной температуре мепазин-
сульфокислота отводится через верх колонны. Затем она нейтрализуется и
в испарителе освобождается от воды и небольшого количества гпдротропно
растворенного мепазина.
В. СУЛЬФООКПСЛЕППЕ В ПРИСУТСТВИИ АЦИЛИРУЮЩИХ ВЕЩЕСТВ
Сульфоокисление проводится в две стадии. На первой стадии процесс
ведут в условиях, благоприятствующих образованию стабилизированной
ангидридом персульфоновой кислоты. На второй стадии смесь парафиновых
углеводородов реагирует с двуокисью серы и кислородом, причем перекис-
ные соединения служат возбудителем реакции.
144
Первая реакция — образования перекисного, соединения протекает
согласно уравнению
RH4-SO2+O2+(CHaCO)2 О-* RSO2O2COCH3+CH3COOH
Она инициируется ультрафиолетовым светом или добавкой некоторого
количества перекиси водорода. В дальнейшем реакция протекает самопроиз-
вольно при условии непрерывной подачи уксусного ангидрида. Темпера-
тура на этой ступени реакции около 40°. Готовый продукт непрерывно отво-
дится, а в реакцию вводится свежая смесь углеводородов и уксусного ан-
гидрида.
Реакционная смесь из первой колонны поступает затем во вторую,
куда также подают сернистый ангидрид и кислород, но сюда дают уже не
уксусный ангидрид, а разбавленный водный раствор уксусной кислоты,
который при температуре 55—60° растворяет образовавшиеся сульфокис-
лоты. Способ работы ясен из схемы рис. 79.
Рис. 79. Схема сульфоокпслепия парафиновых углеводородов по способу с уксусным
ангидридом.
1 — башля I; 2 — холодильник I; 3 — насос I; 4 — башпя II, 5 — холодильник II; б — насос II;
7 — очиститель дли циркулирующего мепазнна; 8 — отделитель.
Линии'. I— отходящие газы; II— мепазин; III — ангидрид; IV — разбавленная уксусная кислота;
V — циркулирующий мепазин; VI — сырые сульфокислоты.
V. РЕАКЦИЯ ФОСФОНИЛИРОВАНИЯ [50]
Интересной реакцией парафиновых углеводородов, не нашедшей пока
еще широкого применения в практике, является превращение углеводородов
при одновременном, совместном воздействии на них треххлористого фосфора
и кислорода.,Она открыта Графом (Хохстер-Фарбверке) в 1943 г.
Реакция идет без участия катализаторов, без индукционного периода,
в условиях низкой температуры согласно уравнению
О
RH-|-2PC].H-O2 -> R—Р-С14-НС1 + РОС|3
'ci
с образованием дихлорида алкилфосфоновой кислоты.
Так получают, например, дихлорид пропилфосфоновой кислоты, про-
пуская кислород через жидкую смесь пропана с треххлористым фосфором
при —40°. Хлор в таких соединениях отличается исключительной реакцион-
ной способностью. Эти соединения легко вступают в реакции с аминами,
алкоголятами, фенолятами и т. п.
10 Заказ 59С.
145
VI. КАРБОКСИЛИРОВАНИЕ [51]
При действии оксалилхлорида или фосгена на парафиновые углеводе-
роды, лучше на циклопарафины, под действием света или в присутствии
радикалобразующих веществ, как, например, органических перекисей,
протекает реакция образования хлорангидридов карбоновых кислот со-
гласно уравнению
С,Н1г+ (СОС1)2 -- CeIInCOCl-1-CO+HC
С циклогексаном и и-гептаном реакция проходит очень гладко. С вы-
сокомолекулярными парафиновыми углеводородами реакция идет с исклю-
чительно малыми выходами, так что с технической точки зрения интереса
не представляет.
VII. РЕАКЦИИ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ С СЕРОЙ
А. ОБЩИЕ СВЕДЕНИЯ
При нагревании паров серы и бутана до температуры около 570° с про-
должительностью пребывания паров в реакционной зоне около 2 сек. обра-
зуется тиофен [52]. Можно полагать, что реакция проходит через несколько
фаз. Сначала, вероятно, происходит дегидрирование бутана серой в бутадиен,
который затем реагирует с серой с образованием тиофена:
CH3CH2CH2CH3+4S->
сп-сн
| || +3H2S
сн сн
Выходящий из печи газ мгновенно охлаждается впрыском воды, чтобы
предотвратить протекание дальнейших реакций. После сжатия примерно
до 12 ат жидкая часть отделяется от газообразной, стабилизируется под
давлением и, наконец, подвергается перегонке. В результате получают
около 70% вес. тиофена 99%-ной чистоты. Из реакционной печи выходят
газы следующего состава (в % вес.).
Тиофен...................... 8
Бутадиен................... 3
Бутен....................., 6
Бутан.......................26
Сероводород..................37
Сероуглерод ................. 2
Легкие углеводороды .... 3
Остаток......................15
В остатке находятся тиофентиолы, как, например, 3-тиофентиол и
тиофен-3, 4-дитиол.
Тиофен может получаться из бутана или бутенов взаимодействием их
с двуокисью серы над катализатором окись молибдена — окись алюминия
или окись хрома — окись алюминия. Из высокомолекулярных парафино-
вых углеводородов наряду с тиофеном получаются алкилтиофены. В табл. 67
даны некоторые примеры этого. Тиофен и алкилтиофены могут получаться
при помощи названных выше катализаторов дегидрирования также из пара-
финовых углеводородов и сероводорода.
Тиофен является чрезвычайно реакционноспособным веществом и слу-
жит в технике исходным материалом для проведения многих важных реак-
ций. Он легко алкилируется и ацетилируется. В США ежегодно произво-
дится синтетическим путем около 300 т тиофена.
146
Таблица 67а
Выход серусодержащих продуктов превращения из 100 кг
исходного углеводорода
Углеводород Температу- ра реакции, °C Серусодержащий продукт Выход, кг
н-Бутан 595 Тиофен 46,0
Бутен-2 545 » 46,2
Бутадиен-1,2 500 » 50,2
Изопентан 540 3-метилтиофен .... 51,4
2,3-диметИлбутеп . . . 545 3,4-диметилтиофсн . . . 22,4
Б. ПОЛУЧЕНИЕ СЕРОУГЛЕРОДА ИЗ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
И СЕРЫ
При нагревании природного газа с серой или сероводородом до темпера-
туры порядка 1000° образуется сероуглерод. Отдельные реакции, которые
при этом могут быть, следующие [53].
CH4+2S2 =4 CS2-f-2H2S
CH4+S2 -> CS2+2H2
CH4+2H2S -> CS2+4H2
Последняя реакция равновесная, что видно из температурной зависи-
мости образования сероуглерода из метана и сероводорода.
Температура, °C.............. 500 600 700 800
Превращение в CS2, % .......... 5 10 17 35
Температура реакции метана с серой может быть значительно понижена
применением соответствующих катализаторов. Так, в присутствии неболь-
ших количеств окиси хрома, окиси марганца или пятиокиси ванадия серо-
углерод получается с 90%-ным выходом уже при 700° [54].
Промышленные способы получения сероуглерода из природного газа
(метана) и сероводорода разработаны в США фирмой Пур Ойл Компани.
Использование реакции сероводорода с метаном особенно целесообразно
в тех случаях, когда природный газ уже содержит достаточное количество
сероводорода, как, например, газ месторождения Лакк во Франции, где
содержание сероводорода достигает 15%.
VIII. КАТАЛИТИЧЕСКОЕ ПОЛУЧЕНИЕ СИНИЛЬНОЙ КИСЛОТЫ
РЕАКЦИЕЙ МЕТАНА С АММИАКОМ И ВОЗДУХОМ ПРИ ВЫСОКИХ ТЕМПЕРАТУРАХ
(Способ Андрусова [56])
При сжигании смесей низкомолекулярных углеводородов, например
природного газа, с воздухом в присутствии аммиака, над определенным ката-
лизатором, например применяемым для окисления аммиака в окислы азота»
образуется синильная кислота согласно следующему уравнению:
CH4-|-NH3 + 1,5 О2 —> HCN-f-3H2O-|-114,9 ккал/моль
10*
147
Температура реакции в этом процессе, разработанном Андрусовым,
составляет 1000° (рис. 80) [56 ]. Газ, поступающий в печь, состоит из
11 — 21%объемн. аммиака, 12—13% объемн. метана и 72—75% объемн. воз-
духа. Выходящий из печи газ содержит около 8% объемн. синильной кислоты
V!
Рис. 80. Схема получения синильной
кислоты по способу Андрусова.
I — печь для сжигания с платиновым контак-
том; 2 — холодильник; 3 — аммиачный абсор-
бер; 4 — абсорбер для синильной кислоты
водой при 5°; 5 — ректификационная ко-
лонна.
Линии: I — аммиак, метан и воздух; II —
дренаж; III — разбавленная серная кислота;
IV — к установке сульфата аммонии; V —
отходящие газы; VI — 100%-ная синильная
кислота.
вместе с небольшими количествами
метана, водорода, окиси углерода и
азота. Газ быстро охлаждают и осво-
бождают от избытка аммиака обработ-
кой сернокислым раствором суль-
фата аммония. Отсюда газ поступает
в скруббер, где синильная кислота
абсорбируется из газа водой при 5°.
При этом получается примерно 3%-ный
водный раствор синильной кислоты,
из которого затем перегонкой выде-
ляют 100%-ную синильную кислоту1.
IX. ПОЛУЧЕНИЕ САЖИ ИЗ ПРИРОДНОГО
ГАЗА И НЕФТЦ 158]
Важнейшим продуктом нефтехими-
ческой промышленности уже давно
является сажа. Мировое производство
сажи приближается к 1 млн. т!год.
Большие количества сажи приме-
няются в производстве синтетического
каучука (па 100 кг синтетического каучука идет около 40 кг сажи), в произ-
водстве типографских красок и т. п. Благодаря примеси сажи продолжи-
тельность жизни автомобильной покрышки повышается с 10 тыс. до
60 тыс. км. Таким образом нефть и природный газ являются сырьем не
только для получения карбюраторного топлива, но и являются исходными
материалами для производства автомобильных покрышек и камер в виде
бутадиена, стирола, сажи и изобутена.
Получение сажи основывается или на неполном сгорании углеводородов
в воздухе, или на термическом расщеплении углеводородов на элементы
углерод и водород.
Получение сажи неполным сгоранием природного газа осуществлено
в так называемом канальном процессе (рис. 81). Природный газ сжигают
в условиях недостатка воздуха при помощи множества маленьких горелок
из плавленного базальта (лавы). Коптящее пламя попадает на вертикально
расположенные, охлаждаемые железные желоба, находящиеся в состоянии
медленного возвратно-поступательного движения, с которых осаждающаяся
на них сажа снимается шабером. Температура пламени достигает 1000—
1200°. Технологическое оформление процесса очень сложно.
Печной процесс получения сажи основывается на том, что газ или лег-
кий газойль непрерывно сжигается в печи в условиях точно регулируе-
мого недостатка воздуха (рис. 82). Выделяющейся тепловой энергии доста-
точно для термического расщепления оставшихся углеводородов на угле-
род и водород. Полученная таким путем сажа отделяется от газообразных
продуктов на установках Котрелля и в циклонах.
Другой процесс получения сажи заключается в том, что углеводород-
ный материал подают на нагретый до высокой температуры камень, в ре-
зультате углеводороды распадаются на элементы по уравнению
СН4—> С-Ь2Нг—22 ккал/моль
1 Ф. Эндтер недавно описал промышленный способ синтеза синильной кислоты из
метана и аммиака без участия кислорода над платиновым катализатором с 80—90%-ным
выходом [57].
148
Примерно 50% всей образовавшейся сажи выносится водородом и от-
деляется затем от него фильтрами. Через некоторое время каменную кладку
снова нагревают до высокой температуры, оставшаяся на поверхности
кладки сажа выгорает, давая часть необходимого для процесса тепла. После:
Рис. 81. Упрощенная схема канального процесса полу-
чения сажи.
1 — каналы; 2 — базальт; з — горелка; 4 — каналы; 5 — воронка;
6 — камера сгорания; 7 — элеваторы; 8 — сажеприемник; 9 —
вагонетка.
Рис. 82. Схема печного процесса с установкой Котрелля.
1 — печь; 2 — холодильник; з — установка Котрелля; 4 — циклонный
сборник; <5 — сажеприемник.
Рис. 83. Схема термического способа получения сажи.
1 — холодильник; 2 — транспортер; 3 — фильтр; 4 — сажеприемник.
подогрева над кладкой снова пропускают углеводородный газ (регенератив-
ный принцип).
В настоящее время в США примерно 50% сажи получают канальным
способом и 50% — печным.
149
X. ОКИСЛЕНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
А. ОБЩИЕ СВЕДЕНИЯ
Окисление низкомолекулярных, газообразных при нормальных усло-
виях парафиновых углеводородов осуществлено на нескольких больших
установках США. Окисление относится к числу типичных нефтехимических
процессов. Целью его в настоящее время при использовании в качестве
исходного сырья пропана и бутана является получение формальдегида и
уксусной кислоты, вернее уксусного ангидрида; важнейшим промежуточным
продуктом в большинстве случаев является ацетальдегид.
Относительная реакционная способность типичных водородных атомов
понижается от третичных к вторичным и первичным. При 300° скорости их
окисления относятся как 10 : 2 : 1. Поэтому изобутан окисляется очень
легко. Метан и этан, содержащие только первичные водородные атомы, чрез-
вычайно устойчивы к окислению. Пропан и бутан, имеющие первичные и
вторичные водородные атомы, занимают среднее положение. В настоящее
время еще не известен промышленный способ окисления метана в метило-
вый спирт пли формальдегид.
Уже рассмотренные выше в различных главах этой кйиги процессы
окисления метана не ставили своей целью получение кислородсодержащих
продуктов (ацетилен по Захсе, получение синтез-газа, сажи и т. п.). Этан
также не применяется в промышленности как исходный материал для окис-
ления при получении кислородсодержащих соединений. Вместе с тем все
возрастает значение автотермического получения этилена, при котором часть
этана сжигается, чтобы получить энергию, необходимую для процесса.
Б. УСЛОВИЯ ОКИСЛЕНИЯ ГАЗООБРАЗНЫХ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
В научно-исследовательских лабораториях процессы окисления низко-
молекулярных углеводородов изучены весьма широко, тогда как о техноло-
гии этого процесса, осуществленного в промышленных масштабах фирмой
Силениз Корпорейшн в США и другими, до сих пор точных сведений нет.
Важнейшими переменными пвп окислении парафиновых углеводородов
для получения кислородсодержащих продуктов являются соотношение
между углеводородом и воздухом или кислородом, температура, давление
и продолжительность реакции (время превращения). Окисление углеводоро-
дов связано с большой энергией активации и поэтому даже незначительные
изменения температуры оказывают очень большое влияние на скорость
реакции.
На практике окисление парафиновых углеводородов ведут в условиях
избытка углеводорода. Реакция между углеводородом и воздухом или кис-
лородом протекает в большинстве случаев в газовой фазе, без применения
каких-либо катализаторов, под давлением 7—10 ат при 330—370°. По окон-
чании процесса продукты окисления быстро охлаждают путем впрыска
воды, кислородсодержащие соединения абсорбируют водой, а непрореаги-
ровавшие, находящиеся в определенном избытке углеводороды снова воз-
вращают в процесс.
Некоторое представление о влиянии соотношения между углеводородом
и воздухом на состав продуктов окисления пропана при 275° дают цифры
табл. 68.
Отсюда следует, что с увеличением избытка углеводорода выход про-
дуктов конденсации повышается. Однако количество углеводорода, окислен-
ного за один проход через печь, при этом уменьшается вследствие малого
содержания кислорода в газовой смеси, количество альдегидов, кислот и
кетонов изменяется лишь незначительно, выход спиртов сильно возрастает.
В практике концентрация кислорода в газе составляет 4—5%.
150
Таблица 68
Состав продуктов окисления пропана
Продукт окисления Соотношение пропан : воздух
1:20 1 :15 | 1 : 3,6 | 1 : 1,25 1 :0,15
Выход, % углерода окисленного пр опана
Альдегиды 12,5 8,8 12,0 16,1 16,7
Спирты 17,3 25,5 23,0 33,1 34,5
Изопропиловый спирт 2,7 6,9 5,2 5,2 14,4
Ацетон 1,2 1,4 1,3 0,3 7,4
Кислота 13,9 13,4 15,2 8,9 12,5
Всего продуктов конденсации 47,6 56,0 56,7 63,6 85,5
СО2 31,5 25,0 22,1 10,5 7,6
СО 20,9 19,0 4,3 25,9 8,0
Кислород вводится в исходный продукт в виде воздуха, его концентра-
ция регулируется или величиной избытка углеводорода, или добавлением
к реагирующей смеси водяного пара.
Понижение температуры увеличивает выход кислородсодержащих про-
дуктов, однако при этом сильно понижается скорость реакции, поэтому
практически нижняя температурная граница лежит около 250°. Если взята
чрезмерно высокая температура, то преобладает образование олефинов,
выход кислородсодержащих соединений понижается.
Увеличение давления повышает при прочих равных условиях скорость
реакции, а если в качестве исходного материала применяется пропан,, то
повышается выход изопропилового спирта.
Время превращения ограничивается 1,0—1,5 сек. При увеличении вре-
мени пребывания смеси в зоне реакции содержание кислородсодержащих
продуктов не увеличивается. Преобладающим процессом становится обра-
зование углекислоты.
Все сказанное выше о влиянии условий ведения процесса на выход
отдельных продуктов реакции справедливо для некаталитического окисле-
ния парафиновых углеводородов в газовой фазе. Но в то же время существует
процесс каталитического окисления бутана в жидкой фазе в присутствии
растворителя, например уксусной кислоты, и катализаторов, как ацетат ни-
келя, кобальта и марганца.
Каталитическое окисление в жидкой фазе имеет то преимущество перед
газофазным процессом, что позволяет более точно регулировать состав
конечных продуктов [60]. Так, при окислении н-бутана в жидкой фазе
образуется в первую очередь уксусная кислота при полном отсутствии фор-
мальдегида. При окислении же пропана в газовой фазе, напротив, обра-
зуются главным образом пропионовый альдегид, пропиловый спирт, ацетон,
уксусный альдегид, уксусная кислота, формальдегид, метиловый спирт,
окись пропилена, окись этилена. При окислении н-гексана теоретически
можно получить около 60 различных продуктов окисления, не считая вто-
ричных продуктов, образующихся за счет дальнейших реакций кислород-
содержащих компонентов. Метан и этан не только содержатся в значительно
больших количествах в природном газе, чем пропан или бутан, по они пред-
ставляют интерес и для применения в качестве исходного сырья, так как при
окислении дают продукты более простого состава. Именно сложный состав
продуктов газофазного окисления был причиной того, что внедрение этого
процесса в промышленную практику сильно задержалось.
151
Для разделения различных кислородсодержащих соединений применяют
различные способы. Трудность разделения подобных сложных смесей обу-
словливается двумя причинами. Во-первых, температуры кипения боль-
шинства соединений настолько близки друг к другу, что разделение их
посредством перегонки очень сложно. Во-вторых, большое значение имеет
то, что кислородсодержащие соединения дают большую группу двойных
и тройных азеотропных смесей. Число возможных азеотропных смесей со-
единений, образующихся при окислении бутанов, составляет около 100.
Так, например, метилал СН2 (ОСН3)2, имеющий температуру кипения 42,3°,
и метиловый спирт с температурой кипения 64,7° дают азеотропную смесь
с температурой кипения 41,8°, которая может быть разделена экстрактив-
ной перегонкой в присутствии воды. Выделение чистого н-иропилового спирта
из смеси с близко кипящим аллиловым спиртом также может быть осуще-
ствлено экстрактивной перегонкой в присутствии воды, причем н-пропиловый
спирт отходит в качестве головного продукта, а аллиловый спирт остается
в растворе.
Чистый метилэтилкетон из содержащей его фракции можно выделить
добавлением к фракции н-гексапа, дающего с метилэтилкетоном азеотроп-
ную смесь (см. стр. 108, где метилэтилкетон используется в качестве ком-
понента для образования азеотропной смеси с парафиновыми углеводоро-
дами в целях выделения последних из смеси с ароматическими). Отделяю-
щаяся в качестве головного продукта азеотропная смесь метилэтилкетона
и w-гексана разделяется затем при помощи воды, в которой метилэтилкетон
растворяется. Из водного раствора метилэтилкетон получают в виде азео-
тропной смеси с водой, из которой затем воду выделяют в форме азеотроп-
ной смеси с пентаном.
Выделение чистого формальдегида из смеси продуктов окисления бу-
тана возможно экстрактивной перегонкой с этиленгликолем в качестве рас-
творителя.
Применяются также адсорбционные методы, а также разделение при по-
мощи ионнообменных смол, однако детали этих процессов пока не известны.
Впервые промышленное окисление парафиновых углеводородов осу-
ществлено фирмой Сити Сервис-Ойл Компани (Таллант, Оклахома, США).
Исходным материалом является природный газ, окисляемый непосредственно
воздухом. Кислородные соединения экстрагируются водой, а остаточный
газ используется как топливо. Подобный же процесс применяется фирмой
для окисления бутана. Окисление природного нефтяного газа ведут при
430° и 20 ат над фосфатом алюминия в качестве катализатора. Абсорбат
состоит из 15% метилового спирта, 22% формальдегида, 3% ацетальдегида
и 60% воды с небольшой примесью других кислородсодержащих продуктов,
как этиловый спирт, уксусная и муравьиная кислоты и др. [61].
Условия газофазного некаталитического окисления пропана и бутана на
принадлежащих фирме Силениз Корпорейшн установках в Бишопе (Тексас,
США) и Эдмонтоне (Канада) приблизительно следующие: смесь, состоящая
примерно из 7 объемов газа циркуляции, 1 объема свежего газа и 2 объемов
воздуха под давлением 7 ат, проходит через нагретую до 370° печь, где
в результате экзотермической реакции температура повышается до 450°.
Горячие газы поступают затем в орошаемый водой абсорбер, где быстро
охлаждаются до 90°, причем образуется водный раствор формальдегида,
обогащаемый затем до концентрации порядка 12—14%. Выходящие из этого
абсорбера газы промываются водой вторично. Из газов извлекаются аце-
тальдегид, метиловый спирт, ацетон и т. д., а углеводороды и азот остаются
в газообразном состоянии. Приблизительно 75% отходящего газа как газ
циркуляции возвращается в печь, где он смешивается с исходным углеводо-
родным газом и воздухом и подвергается повторному окислению. Меньшая
часть (25%) выходящего из последнего абсорбера газа подается на специаль-
ную установку, где пропан и бутан отделяются от азота и низкокипящих
152
составных частей. Чистые углеводороды С3 и С4 подобным же образом воз-
вращаются на окисление. Таким образом часть содержавшегося в воздухе
азота постоянно удаляется из установки. Дальнейшая переработка абсорбата
производится перегонкой, азеотропной перегонкой и другими методами,
кратко рассмотренными выше.
Табл. 69 дает некоторое представление о примерных выходах отдельных
продуктов в описанном процессе (61].
Таблица 69
Выход кислородсодержащих продуктов при окислении
газообразных парафиновых углеводородов воздухом
Продукт Выход жидкого исходного продукта в г/л из
пропана н-бутапа изобутана
Ацетальдегид 165 180 92
Формальдегид .... 180 190 120
Метиловый спирт . . . 144 123 74
Смешанный растворитель 43,5 72 73
Ацетон 11,9 24 144
Общий выход 544,4 589 503
Следует отметить, что в настоящее время все более переходят к окисле-
нию парафиновых углеводородов чистым кислородом. Выход кислородсодер-
жащих продуктов при этом повышается на 30%, установки значительно
меньше по габаритам, соответ-
ственно уменьшению объема про-
ходящих через установку газов,
исключены потери углеводородов
вместе с азотом, отделяемым от
газов циркуляции. Часть устано-
вок фирмы Силениз Корпорейшп
в Бишопе, предназначенных для
окисления пропана и бутана, пе-
реведены уже на работу с чистым
кислородом.
Процесс газофазного окисле-
ния пропана чистым кислородом
вместо воздуха освоен также с
1954 г. на заводе Варрен Петро-
леум Компани в Конрое (Тексас,
США).
При окислении пропана или
бутана чистым кислородом содер-
жание последнего в смеси, должно
составлять 3—6%. Температура
предварительного подогрева 350—
370° , температура реакции око-
ло 430°. Давление поддерживает-
ся на уровне 7—10,5 ат. После
Рис. 84. Схема окисления пропана и бу-
тана кислородом.
1 — смеситель; 2— предварительный подогреватель;
з — реактор; 4 ~ формальдегидный абсорбер; 5 —
абсорбер для летучих продуктов окисления; 6 —
колонна для отделения летучих продуктов окисления.
Линии: I — исходные парафиновые углеводороды;
II — кислород; III — циркулирующий углеводо-
род; IV — раствор формальдегида на переработку;
V — водяное орошение; VI — углеводороды на
очистку; VII — летучие кислородсодержащие про-
дукты па переработку.
охлаждения в теплообменнике до 150°
реакционные газы при 60—70° обрабатываются водой, извлекающей из
них нелетучие кислородсодержащие составные части, как формальдегид
и кислоты. Летучие продукты промывают водой во второй колонне, при этом
Часть их растворяется, а газы возвращаются через верх абсорбера на уста-
новку для повторного окисления. Схема процесса окисления и последова-
тельность разделения продуктов окисления представлены на рис. 84 и 85.
153
Парафиновые углеводороды С3+С4
Окислительная
печь
Холодильник
Абсорбер
Разделение
растворитель Пропанол
Рис. 85. Выделение продуктов при окислении смеси пропапа-бутана.
В. ПЕРЕРАБОТКА ПРОДУКТОВ ОКИСЛЕНИЯ
а) Отходящие газы
При окислении пропана отходящие через верх абсорбционной колонны
газы имеют следующий состав: 69% пропана, 6% двуокиЬи углерода, 14%
окиси углерода, 5% азота и 6% метана, этана и этилена. Большая часть
газов (75%) возвращается на окислительную установку, меньшая — в уста-
новку для выделения углеводорода.
Рис. 86. Переработка пропана при
окислении.
На составные части газ может раз-
деляться промывкой маслом на уста-
новке, работающей под давлением, как
описано выше (см. стр. 72). Пропан
растворяется в масле, а низкомолеку-
лярные углеводороды, окись углерода и
азот удаляются из абсорбера. Другая
возможность разделения состоит в том,
что сжатием до 35 ат ожижается боль-
шая часть пропана и после промывки
водой возвращается на установку для
окисления. Оставшаяся газообразная
часть отделяется от инертных газов
фракционировкой под давлением
(рис. 86).
1 — компрессор; 2— газо-жидкостный сепара-
тор; з — колонна для перегонки под давле-
нием; 4 — колонна для промывки жидкого
пропана водой.
Линии: I — головной продукт из абсорбера;
II — СО, СО2, сн4| C2He> С2Н4, используемые
как топливо; III — вода; IV — пропан об-
ратно на окисление; V — вода в дренаж.
дегид, ацетали, метиловый спирт,
растворимые в воде оксидаты, как
б) Сырой формальдегид
Сырой формальдегидный раствор
содержит около 20—25% формальде-
гида, от 10 до 20% летучих кислород-
содержащих соединений, как ацеталь-
ацетон, а также высокомолекулярные,
гликоль, глиоксаль и т. п. От летучих
соединений раствор освобождается нагревом под давлением порядка 0,7 ат
(рис. 87). Остаток продувают паром под давлением около 3 ат, при этом
154
формальдегид отдувается, а в остатке концентрируются тяжелые, раствори-
мые в воде продукты окисления.
Формальдегидный конденсат содержит 34—40% формальдегида. Этот
концентрированный формальдегидный раствор экстрагируется, например,
хлористым метиленом или трихлорэтаном и, наконец, в установке для от-
деления растворителя и далее в вакуумной колонне концентрируется до
45%. Отделяющийся конденсат, содержащий около 4% формальдегида,
используется в качестве промывочной жидкости для абсорбции продуктов
окисления из газов. Для удаления из раствора муравьиной кислоты приме-
няются ионнообменные смолы [62].
Рис. 87. Схема переработки формальдегида.
1 — колонна для отделения летучих продуктов окисления; 2 — колонна для выделения формальдегида;
-'} — экстрактор для формальдегидного раетвора; 4 — колонна для перегонки растворителя; 5 — ем-
кость для растворителя; С — вакуумная перегонная колонна для концентрации формальдегида.
Линии: I — сырой формальдегид; II — летучие продукты на дальнейшую переработку; III — дренаж;
IV— водяной пар; V — экстрагированный формальдегид; V7 —хлорированный растворитель-, VII—
свежий растворитель; VIII — 4 %-пый формальдегидный раствор; IX — концентрированный 45 %-ный
раствор формальдегида.
Производство формальдегида в США в 1956 г. составило 625 тыс. т
и должно повыситься к 1960 г. до 1 млн. т [63]. Главным потребителем
является промышленность фенольных смол, потребляющая 25% всего выра-
батываемого формальдегида. Вторым крупным потребителем является про-
мышленность мочевино-альдегидных смол. Большие количества расходуются
на производство пентаэритрита, гексаметилентетраамина, меламиновых смол
и клеящих веществ. На рис. 88 показаны важнейшие направления исполь-
зования формальдегида,
ФОРМАЛЬДЕГИД
Конденсация с фенолом-----—•
Конденсация с мочевиной----*
Конденсация с меламином ----
Конденсация с аммиаком------
Конденсация с ацетальдегидом—*
Реакция с СО + ЩО----------*
Полимеризация с HCOI-I------*
Бакелит--------------«-Фенольные смолы
Мочевинные смолы----------------> Клеи
Меламиновые смолы
Гексаметилентетрамин
Пентаэритрит
Гликолевая кислота------«-Этиленгликоль
Параформальдегид
Рис. 88. Важнейшие направления использования формальдегида.
в) Летучие кислородсодержащие соединения
В состав летучих соединений входят ацетальдегид, ацетали, ацетон,
метиловый спирт, изо- и и-пропиловые спирты, пропионовый альдегид,
акролеин и 10% воды. Схема переработки летучих продуктов окисления
представлена па рис. 89.
155
В колонне 7, имеющей 70 тарелок, при 2,1—3,5 ат отделяется чистый
ацетальдегид. В колонну подают небольшое количество воды для гидролиза
окиси этилена, которая может содержаться в продуктах окисления.
Донный продукт из колонны 1 соединяется с летучими составными
частями, отходящими с установки для получения чистого формальдегида
(см. рис. 87), и поступает в колонну 2, где разгоняется под давлением 1,4 ат.
При этом отделяется щце некоторое количество ацетальдегида, который воз-
вращается в колонну 1. Свободный от ацетальдегида донный продукт ко-
лонны 2 поступает в колонну 3, где от него отгоняются все летучие соеди-
нения. Остаток (17%-ный раствор формальдегида) поступает на установку
для получения формальдегида.
Рис. 89. Схема переработки летучих продуктов окисления пропапа и бутана.
1 — ацетальдегидная колонна; 2 — колонна для отгонки ацетальдегида; 3 — колонна для отгонки
летучих кислородсодержащих соединений; 4 — колонна авеитропной перегонки метилового спирта;
6 — колонна для отделения ацетона; 6 — колонна чистого метилового спирта; 7 — колонна для обезво-
живания высших спиртов; 8 — печь гидрирования; 9 — смеситель; 10 — сепаратор.
Линии: I — летучие кислородсодержащие продукты окисления пропана или бутана; II — чистый
ацетальдегид; III —летучие соединения из установки очистки формальдегида; IV — водород; V —
па установку для очистки формальдегида; VI — гептан; VII—дренаж; VIII — чистый метиловый
спирт; IX — этиловый, изопропиловый и н-пропиловый спирты.
Выходящая из колонны 3 смесь кислородсодержащих соединений гидри-
руется далее при 100—150° и 28—56 ат давления водорода над никелевым
катализатором для получения спиртов.
Продукты гидрирования смешивают с гептаном и смесь подвергают
азеотропной перегонке в колонне 4. При перегонке отгоняются метиловый
спирт, гептан и вода, которые разделяются путем добавления небольшого
количества щелочи. Гептан возвращается в колонну 4, а метиловый спирт
поступает в колонну 3, где от него отгоняются ацеталь и ацетон, возвра-
щающиеся в колонну 3. Остаток из низа колонны 5 подают в колонну 6,
где отделяется чистый метиловый спирт. Остаток из этой колонны возвра-
щается в колонну 4. Высшие спирты, содержащие около 25% воды, из ниж-
ней части колонны 4 поступают в смеситель, где смешиваются с гептаном,
а бблыпая часть воды выделяется и удаляется из системы. Гептано-алко-
гольная смесь разгоняется затем в колонне 7, гептан и спирт отводятся
через верх колонны в разделитель, где разделяются на два слоя, а вода
дренируется из низа колонны 7. Находящийся в верхнем слое гептан возвра-
щается в колонну 7, а свободные от воды спирты могут ректифицироваться
или использоваться как присадки к карбюраторному топливу для умень-
шения образования льда в системе питания двигателей автомобилей в зимнее
время.
На рис. 90 показаны важнейшие возможности использования метилового
спирта.
В табл. 70 дан примерный выход отдельных продуктов в описанном
процессе [61].
156
Каталитическое окисление
)+~НС1____________________
+ N Нд = катализатор___
Этерификм ц ия
+ Изопропиловый спирт
МЕТИЛОВЫЙ СПИРТ
Формальдегид и его производные
Хлорметил
Моно-, ди- и триметиламины
Эфиры акриловой, терефталевой и фталевой
ияслот
Растворители, денатурирование алкоголей,
удаление лаков и красок
Рис. 90. Реакции и пути возможного применения метилового спирта.
Окисление бутана воздухом в жидкой фазе в присутствии кобальтовых
или марганцевых солей в качестве катализатора производится в уксуснокис-
лом растворе. Процесс проводят примерно в следующих условиях: на
1300 частей уксусной кислоты, содержащей около 0,3% вес. уксуснокислого
кобальта или уксуснокислого хрома, подают в минуту 3,5 части жидкого
бутана и 16 вес. частей воздуха. Температура реакции 165—170°, давление
60 ат. Отходящие при понижении давления пары конденсируются и обра-
зуется два слоя.
Таблица 70
Выход кислородсодержащих продуктов при окислении
газообразных парафиновых углеводородов кислородом
Продукт Выход жидкого исходного продукта, в г/л из
пропала я-бутапа пзобутаиа
Ацетальдегид 216 192 91
Формальдегид .... 192 240 144
Метиловый спирт . . . 168 120 24
Ацетон 18 24 168
Смешанный растворитель 120 180 192
Общий выход 714 756 619
Углеводородный слой промывают водой и вновь возвращают на уста-
новку для окисления. Водный слой очищают и он поступает на дальнейшую
переработку. Из 100 частей п-бутана таким путем получают около 79,2
(вес. частей) уксусной кислоты, 12,6 метилацетата, 7,2 этилацетата, 1,9
спирта и 6,6 метилэтилкетона. Высокий выход уксусной кислоты может быть
получен за счет других кислородсодержащих соединений, если образовав-
шийся в результате конденсации углеводородный слой возвратить в уста-
новку для окисления непосредственно, не промывая его предварительно
водой. Работая таким образом можно из 100 частей бутана получить 103,7
части уксусной кислоты, 9,3 смешанных эфиров, 4,5 метилэтилкетона и
1 часть спирта. Уксусная кислота и ацетальдегид, получаемые окислением
парафиновых углеводородов, используются в первую очередь для получе-
ния ангидрида уксусной кислоты, потребляемого в исключительно больших
количествах в производстве ацетилцеллюлозы.
Получение ангидрида уксусной кислоты возможно двумя путями:
из уксусной кислоты по способу Баккера, в котором часть уксусной кислоты
при 750—850" в присутствии триэтилфосфата как катализатора расще-
пляется па воду и кетен, а последний, реагируя с другой частью уксусной
кислоты, дает ангидрид уксусной кислоты:
СН3СООНт-^^г СН2=С=О+Н2О
СН3СОЧ
СН3СООН-|-СН2=С=-=О -> >0
СН3ССи
157
Другой способ, разработанный Кнапзаком, основан на окислении аце-
тальдегида непосредственно в уксуснокислый ангидрид с примерно 70—
75%-ным выходом. Одновременно образуется уксусная кислота.
С этой целью (рйс. 91) воздух под давлением 4—5 ат при 50—70°
подают в ацетальдегид, растворенный в каком-либо растворителе — уксус-
ном эфире, пропилацетате или бензоле. В качестве катализатора приме-
няется, например, медь-кобальтацетат, в молярном соотношении 10 : 1,
в количестве около 2%, считая на ацетальдегид. Марганцевый катализатор
способствует главным образом образованию уксусной кислоты. В зависи-
мости от температуры и природы катализатора процесс направляется в сто-
рону преимущественного образования уксусной кислоты пли ангидрида-
Рис. 91. Схема получения уксусного ангидрида из ацетальдегида (способ Кнапзака).
1 —' очиститель для разбавителя; 2 — реактор для окисления; з — колонна для промывки водой; /_
дистилляционная колонна; Л — альдегидная колонна; 6 — ректификационная колонна.
Линии-. 1 — ацетальдегид; II — катализатор; III — разбавитель; IV — вода; V — воздух; V/___вод-
ный ацетальдегид; VII — вода и разбавитель; VIII — азот; IX — ацетальдегид обратно на окисление-
X — уксусная кислота; XI — уксусный ангидрид; XII — катализатор обратно на установку ’
Выходящие из печи (где происходит окисление) газы освобождаются
промывкой водой от ацетальдегида и выбрасываются из установки. Оксидат
разделяется в ряде колонн. Сначала от оксидата отделяют избыточный
ацетальдегид, воду и этилацетат (растворитель), после чего остаток в дру-
гой колонне разделяют на уксусную кислоту, уксусный ангидрид и катали-
затор. Последний возвращается снова в аппарат, где происходит окисление.
Смесь, состоящую из ацетальдегида, этилацетата и воды, отделяют в специаль-
ной колонне от ацетальдегида, который также возвращается на окисление.
Этилацетат и воду далее также разделяют и первый вновь используют как
разбавитель и растворитель.
Способы получения уксусного ангидрида из метана, этилена, бутана
и пропена представлены схематически на рис. 92.
Ацетальдегид и формальдегид, получаемые окислением пропана или
бутана, являются сырьем для получения пентаэритрита, в 1956 г. его было
произведено в США 70 тыс. т [64]. Он применяется главным образом в про-
изводстве искусственных смол (алкидные смолы). Небольшое количество
его используется в производстве взрывчатых веществ. Интересное примене-
ние находит ацетальдегид в виде паральдегида для получения метилэтил-
пиридина, который каталитическим дегидрированием может быть превра-
щен в метилвинилпиридин [651. На рис. 93 представлены основные
пути использования ацетальдегида, а на рис. 94 — то же уксусной
кислоты.
158
Прямое окисление (способ Кнапэака)
Рис. 92. Нефтехимический способ получения ангидрида уксусной кислоты.
АЦЕТАЛЬДЕГИД
Окисление
Мп-катализатор
Окисление
Способ Кнапзака
Кетен—Уксусный ангидрид—Ацетил-
/ целлюлоза
Уксусная кислота
\Уксусный эфир
Уксусный ангидрид
Хлорирование
^ддт
Хлораль —Хлоральгидрат
\Хлороформ
Акрилонитрил
-ь нем
Альдегидциангидрин —Молочная кислота
хМ-метилаланин (алкацид М)
4- NHa
Конц. H2SO4 при
высокой т- -ре
Конц. H2SO4 при
низкой т-ре
^Никотиновая кислота
Метилэтнлниридин .
^Метилвинилпиридин
Паральдегид
Метальдегид (топливо)
(Бутиральдсгид—2-этилгексанол
Щелочи
Альдоли —Кротоновый альдегид—Бутиловый спирт
‘Бут иленгликоль—Бутадиен
Формальдегид
жидкая фаза
Формальдегид
газовая фаза
Ацетилен
^Искусственные смолы
Пентаэритрит4^
Взрывчатые вещества
Акролеин
Бутинол-3—Гексин-З-диол-2,5 — гександиол-2,5
Реакция
Тищенко
— Уксусный эфир—Ацетоуксусный эфир—Антипирин—Пирамидон
Рис. 93. Реакции ацетальдегида.
УКСУСНАЯ КИСЛОТА
Пиролиз
4- Глицерин
4- Ацетилен
Кетен-------------^Уксусный ангидрид
Компонент (растворитель) при этерификации
Триа цетин (мягчитель)
Вин илацетат
Окислы металлов
Хлорирован ие
+ SO С Ь
Поливиниловый
Гликолевые эфиры целлюлозы
Поливинилацетат ------
<Поливипилформаль
Поливинилбутираль
Ацетаты различных металлов
Монохлоруксусная кислота
Ацетилхлорид
Вспомогательные продукты в красочной, текстильной и других отраслях
промышленности
Рпс. 94. Реакции и применение уксусной кислоты.
Г. КАТАЛИТИЧЕСКОЕ ОКИСЛЕНИЕ ГАЗООБРАЗНЫХ ПАРАФИНОВЫХ
УГЛЕВОДОРОДОВ В ГОМОГЕННОЙ ФАЗЕ
Кроме уже описанного каталитического окисления в конденсированной
системе, возможно окисление парафиновых углеводородов в гомогенной
системе, в газовой фазе. Катализатором в этом случае служит бромисто-
водородная кислота. Руст и Вейган нашли, что парафиновые углеводороды,
160
имеющие третичные водородные атомы, как, например, изобутан, в присут-
ствии бромистоводородной кислоты хорошо окисляются уже при 160° с обра-
зованием трет-бутилгидроперекиси с выходом около 75%. Для окисления
вторичных водородных атомов необходима уже температура 190°, а первич-
ного 220° [66].
Из пропана в этих условиях получается ацетон с 75%-ным выходом,
из этана с таким же выходом — уксусная кислота. Промышленное значение
имеет ди-трет-бутилперекись, применяемая как катализатор полимериза-
ции и как присадка к дизельным топливам. Ди-трет-бутилперекись обра-
зуется в результате конденсации треш-бутилгидроперекиси с mpem-бутило-
вым спиртом в уксуснокислой среде:
CH3v ------------ СН3 СН3. /СНа
СН3ДС—00 I НД-ОН I —С-—СН3->СН3-С—О—О—с/сн3+н2о
СН3/ ----------- \сн3 сн3/ \сн3
Она может быть получена также прямым окислением изобутана по
уравнению
СН3. /СН3 СН3, ,СН3
СН3-^СН-|-НС^СН3+3/о О,--. СН3-2С—О—О—С2-СН3-|-НгО
сн3/ \сн3 сн3/ \сн3
Реакция каталитического окисления газообразных парафиновых угле-
водородов протекает по радикально-цепному механизму, схема которого
может быть представлена следующим образом:
НВг+О2->Вг-|-(НО2?)
1\ Вч
ВДсН-|-Вг -> В-С'+НВг
В/ В/
вч вч
вДсч-о2 -» ВДС—00-
в/ в/
R\ Вч
в22С—ОО' + НВг—> В^с—ООНД-Вг'
Выделяющийся в последней фазе атом брома может положить начало
новой цепи.
В табл. 71 приведен состав продуктов окисления пропана и бутана
в присутствии бромистого водорода.
Таблица 71
Продукты окисления пропана и бутана в присутствии бромистого
водорода
Окисление пропана при 189°
Выход, моли па 100 молей
Ацетон . . . 71,6
Бромацетон .... . . . 2,9
Уксусная кислота . . . 3,8
Пропионовая кислота . . 6,7
Бромистый алкпл . . . э.Ь
Этилен . . . 5,7
Пропен . . . 0,1
со2 . . . 2,3
СО . . . 5,5
Окисление бутана при 183°
превращенного углеводорода
Метилэтилкетон 42,2
Кислоты (уксусная) .... 26,5
Диацетил 9,6
Бромкетоп 7,2
лтор-Бромистый бутил . . . 6.0
Дпбромбутап 3,6
СО + СО2 14,5
И Заказ 596.
161
Особый технический интерес по-прежнему представляет окисление
метана в формальдегид. В этой области получили развитие два промышлен-
ных процесса [67]. Один из них — фирмы Гутенофнунгсхютте — основы-
вается на окислении метана при атмосферном давлении и высое<ой темпера-
туре в присутствии небольших количеств двуокиси азота как катализатора.
Мотан и воздух в соотношении 1 : 3,7 добавляются к циркулирующему
метану после его промывки водой под давлением. На 9 объемных частей
циркулирующего метана дают 1 объемную часть свежей метано-воздушной
смеси. К газовой смеси добавляют 0,03% окислов азота, полученных окис-
лением аммиака. Реакционную смесь нагревают до 600 , после чего из нее
промывкой водой извлекают формальдегид. Освобожденные от формальде-
гида газы из абсорбера возвращаются в процесс.
Водный раствор, содержащий около 10% формальдегида, нейтрализуют,
а затем перегоняют под давлением. Таким путем получают 34%-пый раствор
формальдегида, содержащий 3% метилового спирта. Выход 100%-ного фор-
мальдегида составляет около 10% от теоретического [68]. Процесс фирмы
Хиберния предусматривает окисление метана озонированным кислородом
в присутствии перекиси бария как катализатора. Температура реакции
110—120°, соотношение кислород : метан равно 2 : 3. Катализатор активи-
рован окисью серебра. Выход формальдегида составляет около 2б?4.
Д. ОКИСЛЕНИЕ ПАРАФИНА
Окисление парафина с целью получения жирных кислот получило боль-
шое развитие в Германии во время второй мировой войны. В качестве исход-
ного материала здесь применяют или очищенный нефтяной парафин, или
что дает более благоприятные результаты, буроугольпый парафин (ТТН-
процесс), или синтетический парафин, полученный процессом Фишера-
Тропша.
Нефтяной парафин должен предварительно очень хорошо очищаться,
чтобы удалить содержащиеся в нем природные ингибиторы окисления,
которые могут или полностью затормозить процесс окисления или сильно
его замедлить. Такими ингибиторами являются в первую очередь серусо-
держащие соединения и фенолы, которые можно удалить, например, очист-
кой разбавленной азотной кислотой или безводным хлористым алюминием.
Для окисления используют парафин молекулярного веса 250—420,
что соответствует 18—30 углеродным атомам в молекуле с температурой
плавления от 28 до 65°. Наиболее подходящей для этой цели является смесь
парафиновых углеводородов с 19—24 атомами С, с температурой плавления
32—52°, содержащихся в мягком парафине 40/42 и твердом парафине 50/52.
Так как при окислении парафина кислород распределяется по всем
метиленовым группам примерно равномерно, при окислении получаются
кислоты разного молекулярного веса, из которых перегонкой отделяют
кислоты, пригодные для мыловарения. Окисление проводят при возможно
низких температурах порядка 105—120° [69]. Образующиеся жирные кис-
лоты, особенно высокомолекулярные, окисляются далее, при этом обра-
зуются оксикислоты, кетокислоты и двухосновные жирные кислоты,
не растворимые в бензине. Чтобы свести к минимуму образование
этих нежелательных побочных продуктов, окисление ограничивают
30—50%-ным превращением всей окисляемой углеводородной смеси. В каче-
стве катализатора применяют в большинстве случаев перманганат калия
в количестве 0,3% вес. от всего парафина. Перманганат калия вводят при
перемешивании в нагретый до 150° парафин в виде концентрированного
водного раствора, вода испаряется, а перманганат восстанавливается орга-
ническим веществом до двуокиси марганца, которая распределяется в реак-
ционной смеси в исключительно тонко распыленном состоянии. Окисление
ведут без применения давления. Важно, чтобы применяемый для окисления
воздух поступал в парафин в возможно тонко распыленном состоянии.
162
В аппаратах большой емкости на 1 т парафина должно подаваться 40—60 ма
воздуха в час. Схема промышленной установки для окисления парафина
показана на рис. 95.
Чтобы ускорить начало реакции, парафин в таких установках смеши-
вают перед загрузкой с уже частично окисленным продуктом. Печь, в кото-
рой идет окисление, выполняется или из кислотоустойчивой стали или из
алюминия и имеет диаметр 1—1,25 м и высоту 8—12 м. Рабочий объем
для жидкого парафина составляет 30—60 м3.
Течение процесса контролируется определением в оксидате кислотного
числа и числа омыления. При применении хорошо очищенного парафина
длительность реакции составляет около 21—23 час. Процесс заканчивают,
когда кислотное число оксидата достигает 70—75 мг КОН на 1 г. Тепло
реакции (на 1 т парафина при 40 %-ном превращении выделяется около
500 000 ккал) отводится охлаждением реакционной колонны водой.
Рис. 95. Схема получения жирных кислот окислением парафина.
1 — окислительная колонна; 2 — конденсатор; 3 — реактор для омыления; 4 — отделитель для неомы-
ленных 1; 5 — колонна для экстракции неомылеяных II бензином; 6 — колонна для отгонки бензина;
7 — колонна для отгонки спирта; 8 — реактор для разложения мыл кислотой.
Линии: I — парафин; II — раствор катализатора; III — воздух; IV — отходящие газы; V— масля-
ный и водный конденсат; VI — скежий спирт; VII — несмыленныс I; VIII — мыльный раствор; IX —
свежий бензин; X — циркулирующий спирт; XI — бензин; XII — неомыленные II; XIII — минераль-
ная кислота; XIV — смесь сырых жирных кислот на перегонку.
Первой операцией в переработке сырого продукта окисления является
промывка оксидата водой, чтобы удалить низкомолекулярные кислые соеди-
нения, оставшиеся в процессе окисления воздухом. Затем оксидат омы-
ляют нагреванием до 150° с несколько меньшим, чем теоретически необхо-
димо количеством щелочи (3,5%-ный раствор натриевой щелочи), в авто-
клаве при перемешивании.
От омыленного продукта парафин отделяют следующим образом 170].
Продукт обрабатывают 45%-ным этиловым спиртом, при этом неомыляемые
отделяются в виде маслянистого слоя, мыла же растворяются в разбавлен-
ном спирте. Затем спиртовый раствор освобождают от остатков неомылен-
ных экстракцией бензином в условиях противотока. От мыльного раствора
отгоняют спирт и остаток подкисляют для выделения свободных жирных
кислот. Таким образом получают смесь практически всех теоретически воз-
можных кислот с 6 — 20 атомами С. Смесь перегоняют под вакуумом с водя-
ным паром и получают отдельные фракции:
1) головной погон жирных кислот, выкипающих до 140® при
2 Л1Л4 рт. ст.;
2) основной погон (мыльные жирные кислоты), выкипающий в преде-
лах 140—260° при 2 мм рт. ст.;
3) высшие жирные кислоты, выкипающие в пределах 260—320® при
2 мм рт. ст.
Результаты разгонки смеси сырых жирных кислот, полученных окисле-
нием парафинового гача, приведены в табл. 72.
163;
11*
Таблица 72
Результаты разгонки смеси сырых жирных кислот из парафинового гача
Показатели Головной погон Основной погон Высшие кислоты Остаток
Температура, °C 1 До 120 1 120—170 270—315 —
Выход, % 15—20 56—60 7—12 1—13
Молекулярный вес Св—t'io Сю—С20 ^20—^28 —
Кислотное число 350-460 245 155—160 80—90
Число омыления 350—4(10 255 175—180 I 110—120
Эфирное число .... 10 20 30
Средний выход отдельных фракций жирных кислот, образующихся
в результате окисления парафинового гача, и их важнейшие свойства пока-
заны в табл. 73.
Таблица 73
Средний выход отдельных фракций жирных кислот, полученных окислением
парафинового гача
Показатели Головной погон Основной ПОГО1Г | Высшие кислоты | Остаток
Число атомов С . , . . Выход, % Кислотное число . . . Число омыления . . . Карбоксильное число . Карбонильное » . . Йодное число Неомыляемых, % . . До 10 16 410—425 425—430 0-3 7—12 9-13 0,5-0,8 10-20 63 250—258 256—262 0—7 7—12 11—15 1,2—1,6 20—25 12 155—165 170—185 5—10 35-43 33—45 8—12 Высокомолекуляр- ные 9 Не перегоняющие- ся дикарбоновые окси- и кетокислоты и неомыляемые
Отходящая при окислении вместе с воздухом парообразная смесь лету-
чих продуктов после конденсации разделяется на два слоя. Нижний водяной
слой содержит главным образом водорастворимые низкомолекулярные
жирные кислоты, состав которых следующий (в %).
Муравьиная кислота............................
Уксусная кислота .............................
Пропионовая кислота...........................
Масляная кислота .............................
Кислоты выше С4, эфиры, лактоны, альдегиды,
спирты и т. д................................
Вода..........................................
8-10
11—14
1,3-1,5
1,5
~3,0
Остальное
до 100
Верхний маслянистый слой, составляющий около 3—5% от количества
участвующего в процессе парафина, на 40—50% состоит из неомыленных,
остаток же в большей своей части состоит из жирных кислот.
Головной погон жирных кислот, количество которого может составлять
6—16% от участвующего в процессе парафина, имеет средний состав, при-
веденный в табл. 74.
Головной погон жирных кислот или непосредственно или в виде эфиров
гидрируется в спирты, являющиеся исключительно ценным компонентом
этерификации для адипиновой или фталевой кислот в производстве мягчи-
телей. Нагреванием до 300° с железным порошком под давлением они могут
быть с хорошим выходом переведены в кетоны со средним расположением
кетонной группы. Последние могут применяться или в виде сульфатов
164
Таблица 74
Показатели двух головных погонов различного происхождения
Голо вной погон жирных кислот I1 2
Кислотное число 435 375
Число омыления 440 389
Гидроксильное число .... 3 2
Карбонильное » .... 6 4
Неомыляемых 0,3 0
С4 . . . 2 С4-1-5 • 5
С5 . . . 9 Се . . .15
Фракционный состав (раз- Св . . .20 С7 . . .20
гонкой), % С, . . .23 С8 . . .20
С8 . . .23 С9 . . .15
Со . . .18 С1о • • Ю
Выше С9 . . 5 Ctl + 12 + i3 • 15
1 Проба 1 отобрана при окислении парафинового гача , проба 2 —твердого
парафина.
в качестве смачивающих, пенообразующих и эмульгирующих веществ,
или в виде продуктов оксиэтилирования, как вспомогательные материалы
в текстильной промышленности. Смеси жирных кислот, содержащихся
в головном погоне, с высокомолекулярными жирными кислотами с большим
успехом применяют в производстве смазочных материалов. Вопрос о при-
менении определенных фракций жирных кислот для производства мыл
выходит за пределы собственно нефтехимии.
Этерификацией высокомолекулярных жирных кислот глицерином полу-
чают синтетические жиры. Схема синтеза жиров из угля, воды и воздуха
дана на рис. 96.
С + Н2О
-•СО + Но
2NaCl—
Шз + 2NaOH
(электролиз)
Синтез Фишера-Тропша
(парафиноиый гач)
Алкоголь-синтсз
((пропанол)
Окисление
воздухом
Пр пев или пролен
из синтеза Фишера-Тропша
Хлорирование
Жирные кислоты
Аллилхлорид
Хлоргидрирование
и омыление
Глицерин
Этерификация
Жиры
Рис. 96. Схема синтеза жиров из угля, воды, воздуха и поваренной соли.
Из кислот, предназначенных для получения жиров, должны быть пол-
ностью удалены кислоты разветвленного (изо) строения. Из кислот нормаль-
ного строения для получения жиров пригодны кислоты с нечетным числом
атомов С.
165
Окисление высокомолекулярных парафиновых углеводородов, не имею-
щее целью получение кислот для мыловаренной промышленности, осуще-
ствляется в США фирмой Алокс Корпорепшн. Оксидат используется для
получения импрегнирующих средств для пропитывания тканей в виде метал-
лических солей, в качестве мягчителей, антикоррозийных средств и т. д.
Сырой оксидат применяется для пропитывания пеньковых канатов, исполь-
зуемых в мореплавании и в рыболовном промысле. Средство обладает не
только хорошими смазочными свойствами, но и хорошо защищает канаты
от гниения. Окисление ведется воздухом при 95 —17(1° и 10—20 ат давле-
ния. Температура реакции устанавливается в зависимости от необходимых
свойств конечного продукта. Кислород воздуха используется практически
полностью. В качестве катализатора применяют металлические соли жир-
ных кислот. Переработка оксидата производится способом, уже описанным
выше.
ЛИТЕРАТУРА
1. S t a u f f J., Schumacher H. J. Zt. Electrochem., 48, 271, 1942; Schu-
macher II. J., Stauff J. Pie Chemie, 55, 341, 1942.
2. Франц, пат. 773679, 1934; Zbl., I, 3597, 1935.
3. F i e r z - P a v i d H. E. Naturwissensch., 17, 13, 1929.
4. Vaughan W. E., Bust F. F. J. Org. Chem., 5, 449, 1940.
5. M о issn er II. P., T h о d e E. F. Ind. Engng. Chem., 43, 129, 1951; С r a w-
ford R. M. Chem. Engng. Progr., 46, 483, 1950.
6. Hass H. B.,McBee E. T. Am. пат. 2004073, 1935; McBee E. T„
Hass H. B. Ind. Engng. Chem., 34, 296, 1942; 41, 799—803, 1949.
7. Nicodemus O. In Winnacker—Weingaertner, Org. Chem. Technol. I, 680,
Carl Hanser — Verlag — Munchen, 1952.
8. Herold P., Grimm II., Sexauer T. Герм. пат. 720079, 1942; Chem.
Abs., 37, 2553, 1943.
9. Kenyon R. L. и сотрудн. Ind. Engng. Chem., 42, 2388, 1950.
10. Nat. Petrol. News, 37, 45, 1945.
11. Asinger F. Chemie und Technologic der Paraffinkohlenwasserstoffe, 595—
626, Akademie — Verlag, 1956.
12. M с В e e E. T., H a s s H. В., P i e r s о n E. Ind. Eng. Chem., 33, 181,
1941.
13. M с В e e E. T., В a r a n a u c k a s C. F. Ind. Engng. Chem., 41, 806, 1949.
14. Barbour A. K., Barlow G. В., T a t 1 о w J. C. J. appl. Chem. (Lon-
don), 2, 127, 1952; Massingham W. E. и сотрудн., Idem, 2, 221, 1952.
15. W h a 1 e e у W. В. J. Soc. chem. Ind. (London), 66, 427, 1947.
16. Fergusson W. C. Chem. and Ind., 586—590, 1949.
17. P 1 u n k e t t R. Y. Am. пат. 2230654, 1941; Chem. Abs., 35, 3365, 1941.
18. Edgar G. Ind. Engng. Chem., 31, 1439, 1939; Zscharn A. Chem. Ztg., 64,
498, 1940; Chem. and Process. Engng., 35, 4, 115, 1954; Ind. Chemist, 30, 429—436, 1954.
19. К e n у о n R. L. и сотрудн. Ind. Engng. Chem., 42,'2388, 1950.
20. К б 1 b e 1 H. Erdol und Kohle, 1, 308—318,1948; К б 1 b e 1 H. и сотрудн. Brenn-
stoffchem., 30, 73, 300, 302, 1949.
21. S t u p e 1 H. Synthetische Wasch — und Reinigungsmittel, 104, Konradin —
Verlag Stuttgart, 1954.
22. Scheer W. E. Chem. Industries, 54, 203, 1944.
23. H e n d r i c k s J. G. и сотрудн. Ind. Engng. Chem., 42, 899, 1950.
24. T у м a p к и н a E. С. и др. Ж. прикл. хим., 23, 958, 1950.
25. К о н о в а л о в М. И. Вег., 26, R 878, 1893; ЖРФХО, 25, 472, 1893.
26. Hass Н. В., Hodge Е. В., Vanderbildt G. М., Ind. Engng.
Chem., 28, 339, 1936; Hass H. В., Patterson J. A. Ibid., 30, 67, 1938; H i b-
shman H. J., Pierson E. H., Hass H. B. Ibid, 32, 427, 1940,
27. Grund mann Ch. Die Chemie, 56, 159, 1943.
28. Geiseler G. Angew. Chem., 67, 270, 1955.
29. R e i d e I J. C. Oil. a. Gas J., 54, 36, 110—114, 1956; Sherwood P. W.
Chim. et Ind., 76, 1, 85, 86, 1956.
30. Bachman и сотрудн. J. org. Chem., 17, 935—954, 1952.
31. Bogin C., W a m p n e r H. L. Ind. Engng. Chem., 34, 1091, 1942.
32. Chem. Trade J. chem. Engr. 107, 310, 1942.
33. Gabriel C. L. Ind. Engng. Chem., 32, 887, 1940.
34. Senkus M. J. Am. Chem. Soc., 68, 10—12, 1946; Johnson H. G. Ibid,
68, 12—14, 1946.
35. L i p p i n с о 11 S. B., Hass H. B. Ind. Engng. Chem., 31, 118, 1939.
36. N ef J. U. Liebigs Ann. Chem., 280, 263, 1894.
37. Denton W. I. с сотрудн. Ind. Engng. Chem., 40, 381, 1948.
166
/
38. Hager К. F. Ind. Engng. Chem., 41, 2168, 1949.
39. Наметкин С. С. и др. ЖРФХО, 45, 1420, 1913.
40. S с о t t E. W., Treon J. F. Ind. Engng. Chem. Anal. Ed., 12, 189, 1940;
Tur ba F., Haul R. Angew. Chem., 61, 74—75, 1949.
41. J ohnson K., D e g e r i n g Ed. F. J. Am. Chem. Soc., 61, 3194, 1939.
42. Graf R. Liebigs Ann. Chem., 578, 50, 1952.
43. Asinger F. с сотрудн. Ber., 75, 42, 1942.
44. A s i n g e r F., Richter G, J. prakt. Chem. (4), 2, 228, 1955; A s i n g e r F.
с сотрудн. J. prakt. Chem. (4), 2, 203, 1955.
45. Asinger F. Герм. пат. 715747, 1942; Франц, пат. 853686, 1940; Вельг. пат.
434487, 1939.
46. Herold Р., S m е у k а 1 К., Asinger F., Horst II. D. Герм. пат.
715846, 1942.
47. Chem. Engng. News, 30, 1966, 1952; India Rubber World, 126, 86, 1952; 127,
791, 1953; 128, 54, 348, 1953; Ind. Engng. Chem., 44, 6, A1952; Gilbert E. E., J o-
n e s E. P. Ind. Engng. Chem., 45, 2041, 1953; Smook M. A., Pieski E. T.,
Hammer C. F. Ind. Engng. Chem., 45, 2731, 1953.
48. Platz C., S c h i m m e 1 s c m i d t К. Герм. пат. 735096, 1940; Chem.
Abs, 1249, 1944.
49. Orthner L. Angew. Chem., 62, 304, 1950.
50. Graf R. Ber. 85, 9—19, 1952.
51. К h a r a s c h M. S., Brown II. C. .1. Am. Chem. Soc., 62, 454, 1940; 64,
329, 1942; К h a r a s c h M. S., Kane S. S. Ibid., 64, 333, 1631, 1942.
52. Rasmussen H. E., Hansford R. C., S a c h a n e n A. N. Ind.
Engng. Chem., 38, 376, 1946.
53. S t u 1 1 D. R. Ind. Engng. Chem., 41, 1968—1973, 1949; Thacker С. M.,
Miller E. Ind. Engng. Chem., 36, 182, 1944.
54. F о 1 к i n s II. О. с сотрудн. Ind. Engng. Chem., 42, 2202—2207, 1950.
55. Kautter С. T., Leitenberger W. Chem. Ind. Techn., 25, 697, 1953.
56. Андрусов JI. Angew. Chem., 48, 593, 1935; Chem. Ing. Techn., 27, 469—472,
1955
57. Chem. Ing. Techn., 30, 305—310, 1958.
58. Cohan L. H. Science of Petroleum, V, 78—88, 1953; Cohan L. H., M a-
c к e у J. F. Ind. Engng. Chem., 35, 806, 1943; Petrol. Refiner, 32; II, 174, 1953; Oil a. Gas
J., 55, 35, 122, 1957.
59. Mitchell R. L. Petrol. Refiner, 35, 7, 179—182, 1956.
60. S h e г w о о d P. W. Chem. Age, 24, 21, 1363—1369, 1955.
61. Meyer R. E. Oil a. Gas J., 54, 7, 82—86, 1956.
62. Meyer R. E., Petrol. Refiner, 35, 12, 172, 1956.
63. Chem. Trade J., 139, 3620, 914, 1956.
64. S h e r w о о d P. W. Petrol. Refiner, 35, II, 171 —179, 1956.
65. Petrol. Refiner, 34, 12, 170, 1955.
66. Rust F. F., Vaughan W. E. Ind. Engng. Chem., 41, 2595, 1949.
67. FIAT. Final Rept., Nr. 1085.
68. FIAT. Final Rept., Nr. 608.
69. P r u с к n e r H. Chem. Techn., 4, 196, 1952.
70. W i e t z e 1 G. Z. Angew. Chem., 51, 531,1938; Fette und Seifen, 46, 24, 1939;
Mannes L. Chemie, 57, 6, 1944.
Г лава II
ПЕРЕРАБОТКА МОНООЛЕФИНОВ
I. ХЛОРИРОВАНИЕ ОЛЕФИНОВ
А. ОБЩИЕ СВЕДЕНИЯ
Хлорирование олефинов может проводиться двумя принципиально
различными способами — присоединением и замещением. При хлорирова-
нии присоединением насыщается двойная связь олефинового углеводорода,
при хлорировании замещением двойная связь сохраняется. Как пример
хлорирования присоединением хлора можно назвать получение хлористого
этилена (1,2-дихлорэтапа) из этилена и присоединение хлорноватистой
кислоты с образованием хлоралкоголей, так называемых хлоргидринов.
Последняя реакция имеет большое промышленное значение.
В качестве примера можно также привести реакцию гидрохлорирова-
ния, основывающуюся на присоединении хлористого водорода ио месту
двойной связи (хлористый этил из этилена).
Не меньшее значение имеют реакции хлорирования олефинов замеще-
нием. С олефиновыми углеводородами изостроения, у которых углерод
с двойной связью находится в боковой цепи, реакция хлорирования путем
замещения идет уже при комнатной и даже при значительно более низкой
температуре. Хлорирование перазвствленных углеводородов, в частности
пропена, для которого эта реакция играет большую роль, идет только при
очень высоких температурах (горячее хлорирование при 500 ).
1>. ХЛОРИРОВАНИЕ ЗАМЕЩЕНИЕМ
По поведению при хлорировании замещением низкомолекулярные
олефины можно разделить на две группы. К первой группе относятся оле-
фины с прямой цепью, как этилен, пропен, и-бутен и и-пентен, реагирующие
с хлором при окружающей температуре только с образованием продуктов
присоединения. Вторая группа включает в себя углеводороды, которые
при равных условиях реагируют исключительно путем замещения с сохра-
нением двойной связи в молекуле вновь образовавшегося хлорпроизводного.
К ним относятся олефины, у которых двойная связь находится в боковой
цепи, как, например, изобутен, триметилэтилен.
Однако олефины первой группы могут при определенных условиях,
а именно при высоких температурах (400—500е), также подвергаться хло-
рированию замещением. Эти закономерности были установлены Гроллем
с сотрудниками в исследованиях, проведенных ими для фирмы Шелл [1].
На примере пропена в табл. 75 показано, как при повышении темпера-
туры реакции присоединения постепенно уступают место реакциям заме-
щения.
При поверхностном рассмотрении хода реакции можно придти к выводу,
что первой фазой реакции горячего хлорирования является присоединение
168
хлора по месту двойной связи с образованием 1,2-дихлориропана, от кото-
рого затем в результате пиролитического процесса отщепляется хлористый
водород с образованием хлористого аллила. Но если хлорирование 1,2-ди-
хлорпропана проводить в условиях хлорирования пропена в нагретой
до 500е кварцевой трубке, то он отщепляет лишь очень незначительное коли-
чество хлористого водорода. Реакция дигидрохлорирования начинает про-
текать с достаточной скоростью примерно при 700°.
Таблица 75
Температурная зависимость присоединения и замещения при хлорировании пропена
Средняя температу- ра реакции, °C Молярное соотношение пропен : хлор Расход хлора, % Расход хлора, г на 100 мл реак- ционного объема
в реакции при- соединения в реакции замощения
210 .3,44: 1 74,7 25,3 0,081
320 6,03 : 1 22,5 77,5 0,200
400 6,43 : 1 3,8 96,2 0,550
510 6,32 : 1 1,3 98,7 4,060
590 6,60 : 1 0,3 99,7 10,900
В продуктах реакции, однако, содержится только 58% хлористого
аллила, наряду с 40% 1-хлорпропена-1 и 2% 2-хлорпропена-1, откуда
следует, что горячее хлорирование пропена в хлористый аллил следует
рассматривать как специальную реакцию.
а) Применение хлорирования замещением в промышленности
Хлорирование изобутена. Процесс хлорирования изо-
бутена в промышленных условиях весьма прост (схема рис. 97). Изобутен
и хлор в молярном соотношении 1,5 : 1 пропускают через нагретую до 150°
Рис. 97. Схема хлорирования
изобутена.
камера хлорирования; 2— промы-
вочная колонна; 3 — отделитель; 4 —
дистилляционная колонка.
Линии-. I — изобутен; II — хлор;
III—циркулирующий изобутен; IV—
вода; V — разбавленная соляная кис-
лота; VI — хлористый металлил;
VII — высокохлорврованный продукт.
Рис. 98. Схема горячего хлорирования пропена в
хлористый аллил.
I—предварительный подогреватель; 2 — реактор; з — ко-
лонна для промывки щелочью; 4 — колонна для промывки
водой; 5 — холодильник; в — ректификационная н<яояпа.
Линии: I — пропен, 5 молем; II — хлор, 1 моль; 111 —
циркулирующий пропен; IV— дрснинс V — 2-хлорпропен;
VI—дихлориропеп и дихл<рироп‘Н| ;VI1 —хлористый алчил .
керамическую или железную трубу, где и протекает реакция. Выходящие
из печи газы быстро охлаждаются прямым впрыском воды, образовавшийся
хлористый металлил конденсируется, а избыток газообразного изобутена
просушивается и возвращается в процесс. Хлористый металлил получается
с 85%-ным выходом. Он кипит при 72,2°, плотность его = 0,9257. Его
169
атом хлора исключительно реакционноспособен. Свойства его рассматри-
ваются далее (см. стр. 171).
Хлорирование пропена. Для горячего хлорирования пропена
(схема рис. 98) применяют обычные стальные трубки, в которых предварительно
подогретые от нормальной температуры до 400° пропен и хлор реагируют
друг с другом при 500—530°. Чтобы избежать избытка хлора и связанного
с ним псрехлорирования, смесь перед поступлением в печь тщательно пере-
мешивают при помощи специальных насадок. Оптимальным молярным соот-
ношением, при котором перехлорирование и связанное с ним сажеобразова-
ние сводится к минимуму для пропена и хлора будет 5:1. Необходимое
время пребывания газа в горячей зоне равно 2—3 сек. Давление составляет
около 1 ат. Использование хлора в реакции достигает 99%.
Продукты реакции охлаждают и в ректификационной колонне при тем-
пературе верха колонны —40° отделяют хлористый водород и пропен от
хлористого аллила и других хлорпроизводных углеводородов. Из смеси
пропена с хлористым водородом последний отмывают водой, получая в ре-
зультате 32%-ную соляную кислоту. После длительной промывки для уда-
ления следов хлористого водорода пропен возвращается в процесс.
Сырой хлористый аллил в специальной колонне освобождается от низ-
кокипящих составных частей, прежде всего от 2-хлорпропсна, а затем в сле-
дующей ректификационной колонне получается в чистом виде как головной
продукт. В нижней части колонны собираются дихлорпропан и дихлорпро-
пеп. При этих условиях легко достигается 80%-ный выход хлористого
.аллила.
Результаты горячего хлорирования пропена на полупромышленной
установке можно видеть из приводимых цифр [2].
Температура предварительного подогрева пропена, °C.......... 390—410
Реакционная температура в печи, °C.......................... 500—510
Молярное соотношение пропен : хлор . ........................ 5:1
Выход продуктов, считая па прореагировавший хлор, % вес.:
хлористый аллил........................................... 80
2-хлорпропеп-1 ........................................... 2,5
1-хлорпропен-1............................................ 0,5
смесь насыщенных и ненасыщенных дпхлоридов ............. 10,0
трихлориды и тяжелый остаток............................. 1,0
В качестве побочного продукта при горячем хлорировании пропена
получается смесь дихлорпропана и дихлорпропена, обозначаемая в технике
как смесь D — D. Эта смесь представляет собой превосходное средство для
окуривания почвы, особенно в борьбе с нематодами. На 10 тыс. м2 площади
почвы необходимы 250—500 кг смеси D — D.
В табл. 76 даны физические константы составных частей фракции моно-
хлоридов, получающихся при горячем хлорировании пропена.
Таблица 76
Физические константы составных частей фракции монохлоридов, образующихся
при горячем хлорировании пропена
Компонент Температура кипения при 760 мм рт. ст., °C Плотность при 20° Коэффици- ент прелом- ления при 20° Выход, % вес. от фракции монохлоридов
Хлористый аллил . . . 44,96 0,9382 1,415 96,0
цис-а-Хлоропропен . . 33—32,2 0,930 1,4055 | 1,0
транс-а-Хлоропропен 36,7 — 1,4054
Р-Хлоропропен .... 22,5 0,9093 1,3973 3,0
170
б) Дальнейшая переработка хлористого металлила
Вследствие очень высокой реакционной способности атома хлора хло-
ристый металлил легко омыляется в металлиловый спирт. При смешении
хлористого металлила с 10%-ным раствором едкого натра при 116°
полное омыление достигается за 15 мин. При 200° реакция протекает прак-
тически мгновенно, побочным продуктом является диметаллиловый эфир.
Металлиловый спирт — исключительно реакционноспособное соедине-
ние. При нагревании с 12%-ной серной кислотой он практически количе-
ственно превращается в изомасляный альдегид. Так как диметаллиловый
эфир реагирует точно так же, то при омылении хлористого металлила можно
получать спирто-эфирную смесь. Изомеризацию металлилового спирта
в изомасляный альдегид можно представить себе таким образом, что сна-
чала происходит гидратация с образованием гликоля, который затем дегид-
ратируется, давая в результате изомасляный альдегид [3]:
Н0СН2< н+ HOCIU уон _н 0
>С=СН24-Н2О —- >(2-СН2 — - СН3—С—СН3 —-
сн/ сн/ II
снон
СНз>сн-с^°
сн/ хн
Изомасляный альдегид легко окисляется кислородом воздуха в изо-
масляную кислоту с 95%-ным выходом. Таким путем из изобутена хлори-
рованием и омылением, изомеризацией и окислением можно получить изо-
масляную кислоту с 60%-ным выходом, считая на изобутен. Из 1000 кг
изобутена можно получить около 1000 кг изомасляной кислоты.
В присутствии водяного пара над серебряной сеткой как катализатором
металлиловый спирт окисляется кислородом в метакролеин с 90%-пым вы-
ходом. Метакролеин образуется также с количественным выходом при
нагреве метилглицерина с 12 %-ной серной кислотой.
Жидкий метакролеин под действием кислорода под давлением 14 ат,
при нормальной температуре и продолжительности реакции 5 час. в при-
сутствии катализатора медь — никель — ацетата дает метакриловую кис-
лоту с 96%-ным выходом.
При взаимодействии хлористого металлила с аммиаком в автоклаве
при 90° иод давлением быстро образуются металлиламины. В случае приме-
нения молярного соотношения хлористый металлил : аммиак, равного 1 : 10,
теоретически возможные металлиламины образуются в следующем
соотношении — первичный : вторичный : третичный : четвертичный -=
= 56 : 26 : 8<: 5.
Металлиламин в присутствии водяного пара и серебряного катализатора
при 450—600° может окисляться воздухом в метакрилонитрил с почти
90%-ным выходом [4]. Последний является важнейшим исходным материа-
лом для получения метакриловой кислоты. До сих пор метакрилонитрил
в промышленных условиях получался почти исключительно из ацетона
и синильной кислоты, через циангидрин:
СН3
C=CH2+NH3 ► H2N—СН2—С.= СН2 -°2. NC—с=сн2
/ I Ае I
С1СН2 сн3 сн3
он
СНЭ—СО—CHS+HCN —> CHS—С—СН8 ~Нг- -
I
CN
171
Необходимо отметить, что таким же путем получаемый из хлористого
аллила и аммиака аллилам ин может быть переведен в акрилонитрил.
Реакционноспособный хлор-атом хлористого металлила реагирует в при-
сутствии карбонила никеля, давая хлористый никель и диметаллил (2,5-ди-
метилгексадиеи-1,5) с температурой кипения 114,3° [51:
СН3
СН.2=с/
СН2С1
+ Ni(CO)
СП2С1
СН2=с/
СН,
сн2=с-сн3
I
сп2
---- 4-NiC]2+4CO
СН,
I
СП2=С—СН3
Взаимодействием с водой и хлором через промежуточное образование
хлорноватистой кислоты хлористый металлил может быть переведен в соот-
ветствующий хлоргидрин. Отщеплением хлористого водорода от дихлор-
гндрипа образуется р-метилэпихлоргидрин. Гидролизом в присутствии кис-
лот может быть получен Р-метилглицерин-а-мопохлоргидрин, а этот послед-
ний после отщепления хлористого водорода переходит в р-метилглицидол,
который при последующем гидролизе превращается в р-метилглицерин [6].
С1СН2Х С1СН2Ч /ОН Na0H С1СН,Ч .О,
>С=СН2-ЬН2О+С12----- >С-СН2С)---------> >CZ ХСН2
сн/ сн/ CH3Z
О\
jj_l С1СН2Х /ОН хаон । \ н+ НО(.Н2- .он
;с-сн2он “ сн,—С—СН2ОН >с<
Н2° сн/ I Н2° CH3Z \сн2он
СНз
Р-метилглицерин в тепловом отношении неустойчив. На рис. 99 дан
обзор возможных продуктов реакции хлористого металлила.
в) Дальнейшая переработка хлористого аллила
Общие сведения. Хлористый аллил является важнейшим проме-
жуточным продуктом нефтехимической промышленности. Он легко омы-
ляется в аллиловый спирт, являющийся исходным материалом для получе-
ния синтетического глицерина и многих эфиров, из которых важнейшими
являются эфиры фталевой, фосфорной и угольной кислот. Эфиры аллило-
вого спирта и низших жирных кислот, таких как уксусная, масляная или
капроновая, а также коричной и фенилуксусной кислот, имеют особое
значение для промышленности душистых веществ. Представляют интерес
также эфиры аллилового спирта и крахмала или сахаров. Их получают
взаимодействием спиртовых гидроксильных групп с хлористым аллилом.
На рис. 100 показаны важнейшие направления использования хлористого
аллила в нефтехимическом синтезе.
ХЛОРИСТЫЙ АЛЛИЛ
Гидролиз
+ NH3
+ НОС1
_ « NaSH г, . „
ВГ2^Дибромпропанол-----БАЛ
Аллиловый спирт-—-Глицерин
^Аллиловый эфир------Полимер
диаллилфталата
+ NaOH___________
+ аллиловый спирт
+ Mg_____________
+крахмал или сахар
+ KCNS
. Окисление .
Алли ламин----------Акрилонитрил
Дихлорпропанол (дихлоргидрин)^-Глицерин
^Эпихлоргидрин
Эпоксидные смоль»
_ „ Пиролиз ,
Диаллиловый эфир-----------Акролеин
------» Диаллил
------- Аллилкрахмал, аллилсахароза
—------ Аллилгорчичное масло
Рис. 100. Реакции хлористого аллила.
172
Хлоргпдрп-
рование
Реакция со
щелочью и метал-
лиловым спиртом
Гидрилиа
Хлористый металлил
Реакция со
щелочью п
спиртами
Реакция с
аммиаком
Гидратация
Изомеризация
с H2SO4
X л о рированп е
Реакция с Mg
Рис. 99- Реакция хлористого металлила.
Получение аллилового спирта. Омыление хлористого аллила
в аллиловый спирт в большинстве случаев производится разбавленным рас-
твором натриевой щелочи или соды [7].
Наилучшие результаты дает омыление содовым раствором, так как
в этом случае сводится к минимуму образование побочного продукта —
диаллилового эфира, которого получается тем больше, чем концентрирован-
ное омыляющий раствор щелочи. При применении соды в качестве омыляю-
щего раствора необходимо непрерывно удалять образующуюся углекис-
лоту. При этом имеют место значительные потери органического вещества.
Для избежания этого в реакционную смесь непрерывно добавляют натрие-
вую щелочь в количестве, необходимом для поддержания щелочности среды,
Рис. 101. Схема получения аллилового
спирта из хлористого аллила.
1—сосуд для омыления; 2 — дистилляционная
колонка; 3 — колонна для обезвоживания;
4 — отделитель; 5 — ректификационная ко-
лонна .
Линии: I —хлористый аллил; II — натрие-
вая щелочь; III — пар; IV — сырой алли-
ловый спирт; V— раствор поваренной соли;
VI — обезвоженный аллиловый спирт; VII—
разбавленный аллиловый спирт; VIII—чистый
аллиловый спирт; IX — остаток.
контролируемой величиной pH. Таким
образом можно длительно работать с
карбонатно-щелочной средой без выде-
ления угольной кислоты.
В непрерывном процессе для омы-
ления обычно применяют 5%-ный рас-
твор натриевой щелочи (рис. 101).
Гидролиз проводится при 150—160е
и 14—15 ат, продолжительность про-
цесса около 10—15 мин. Значение pH
равно 10—12. Из верха сосуда, в ко-
тором производится омыление, продук-
ты реакции поступают в дистилляцион-
ную колонну, где аллиловый спирт,
диаллиловый эфир и вода, поступаю-
щая в колонну в виде водяного пара,
образуют азеотропную смесь (сырой
аллиловый спирт), а раствор хлористо-
го натрия с небольшим количеством
аллилового спирта отходит из низа
колонны. Кипящая при 89° азеотропная смесь может непосредственно при-
меняться как исходный материал для синтеза глицерина.
Для получения 100%-ного аллилового спирта, сырой аллиловый спирт
обезвоживается при помощи азеотропной смеси, состоящей из аллилового
спирта, воды и диаллилового эфира. С этой целью сырой аллиловый спирт
подают в колонну для обезвоживания, где от него отгоняется тройная азео-
тропная смесь, состоящая из 9% аллилового спирта, 79% диаллилового
эфира и 12% воды. После охлаждения и конденсации смесь разделяется
на два слоя. Нижний слой, состоящий из 90% воды, 10% аллилового спирта
и следов диаллилового эфира, возвращается в дистилляционную колонну.
Верхний слой, содержащий 90% диаллилового эфира, 9% аллилового
спирта и 1% воды, возвращается в колонну, где происходит обезвоживание.
Из низа этой колонны отводится обезвоженный аллиловый спирт, посту-
пающий далее в ректификационную колонну, откуда отбирают 100%-ный
продукт.
При гидролизе хлористого аллила в аллиловый спирт в только что
описанных условиях в качестве побочного продукта образуется около 10%
диаллилового эфира. Последний является хорошим растворителем, кипя-
щим при 94,5°. Кроме того, посредством пиролиза при 520° этот эфир рас-
щепляется на пропен и акролеин с 90%-ным выходом;
СН2=СН—СН2ОСН,-СН=СН, СН3—СН=СН2+СН2=СН—С^°
ХН
Важнейшие физические свойства аллилового спирта приведены ниже.
Аллиловый спирт
Температура кипения при 760 мм рт. ст., °C....................... 96,9
........................................................ 0,8535
174
d20 ............................................... 0,8540
n2D° ...............................................1,4133
Температура вспышки (закрытый тигель), ЭС ........... 22,3
Упругость паров при_20°, мм рт. ст................ 17,3
Азеотропная смесь с водой
Температура кипения при 760 мм рт. ст., °C.......... 88,89
Содержание аллилового спирта, % вес. ................ 72,3
Простые и сложные эфиры аллилового спирта способны полимеризо-
ваться. Полимеры представляют интерес как искусственные вещества и по-
крытия. Полидиаллилфталат, например, применяется для внутренней обли-
цовки молочных бидонов. Диаллиловый эфир фосфорной кислоты полиме-
ризуется в прозрачную и огнестойкую смолу с высоким коэффициентом пре-
ломления.
Полимеры аллилкрахмала и аллилсахарозы в составе лаков обладают
такими же свойствами, как нитроцеллюлоза. Они представляют особый
интерес в составах для окраски мебели, как заменитель шеллака и для
других целей.
Аллиловый спирт может получаться также изомеризацией окиси про-
пена при температуре 230—270е над фосфатом лития в качестве катализа-
тора. При 20—30%-ном превращении окиси пропена за проход через печь
можно получить 70—80%-ный выход аллилового спирта:
сн2—сн—сп3 СН2ОП—сн=сн2
Аллиловый спирт может получаться также из ацетилена и формальде-
гида через пропаргиловый спирт методом Реппе.
Интересным специальным продуктом, получаемым из аллилового спирта,
является дитиоглицерин. Его получают из 1,2-дибромпропанола-З реакцией
с гидросульфидом натрия в щелочном растворе. Дибромпропапол получается
прямым бромированием аллилового спирта. Дитиоглицерии под названием
«БАЛ» (Бритиш Анти-Люизит) известен как средство против люизита и дру-
гих боевых отравляющих веществ. В настоящее время он применяется
в терапии.
Синтез глицерина. Синтез глицерина, осуществляемый в боль-
ших масштабах фирмой Шелл, проводится в соответствии со следующими
уравнениями:
СН8—СН=СН2 горячее хлорировани^ СИ2С1_СН=СП9 А"—А СП2ОП-СЛ=СН, ------
Хлоргидрированпе^ С112ОН_СНОН_СН2С1 АН—% СН2ОН-СНОН-СН2ОН
Процесс получения хлористого аллила и его гидролиз в аллиловый
спирт были уже рассмотрены выше. Глицерин-а-хлоргидрин получают ил
аллилового спирта продуванием хлора через 5%-ный водный раствор алли-
лового спирта (схема рис. 102). При этом получают около 94% монохлор-
гидрина. Для отвода тепла (хлоргидрирование должно проходить примерно
при 15°) реакционная масса непрерывно прокачивается через охладитель.
Для омыления монохлоргидрина применяют смесь из едкого натра и угле-
кислого натрия. Реакция идет при 150° в течение 30 мин.
Содержащий хлористый натрий водный раствор глицерина перерабаты-
вают методом, принятым в мыловаренной промышленности. В испарителе
удаляется большая часть воды. Выделившиеся кристаллы хлористого нат-
рия отделяют фильтрованием, а сырой глицерин подвергают перегонке.
Для удаления хлорсодержащих соединений дистиллят экстрагируют ксило-
лом, а затем подвергают ректификации в вакууме.
Синтетический глицерин можно также получать из хлористого аллила.
Проводить хлоргидринирование хлористого аллила при помощи хлора и
175.
воды нецелесообразно, так как хлористый аллил не растворим в воде. В этом
случае работают со свободной хлорноватистой кислотой, которую получают,
пропуская хлор через раствор гипохлорита, по уравнению
CJ.,4-H2O+NaOCl -> 2HOCl+NaCJ
Таким путем можно получить 92%-ный выход глицерппдихлоргидрппа.
Глицериндихлоргпдрин перерабатывается в глицерин таким образом,
что сначала при помощи гидроокиси кальция получают эпихлоргидрин,
который затем в присутствии щелочи легко омыляется в глицерин:
2СН,С1-СНС1-СН2ОН ——- 2СН2С1-СН-СН, — -
СН2С1-СНОН-СН2ОН ^521% СН2—СН-СН2ОН ^2- СН2ОН-СНОН-СН2ОН
о
Рис. 102. Схема синтеза глицерина из аллило-
вого спирта.
На рис. 103 показано по-
лучение глицерина хлорирова-
нием пропена.
Э пи х лоргидрин.
Эпихлоргидрин является важ-
нейшим промежуточным про-
дуктом нефтехимической про-
мышленности, что связано с
наличием в нем очень подвиж-
ного атома хлора и исключи-
тельно реакционноспособной
эпоксигруппы. Реакцией эпи-
хлоргидрина с аммиаком, ами-
нами, серной кислотой, мер-
каптанами и т. д. получают
большое число ценных проме-
1"— сосуд для хлоргидринирования; 2 — циркуляцион-
ный холодильник; 3 — сосуд для гидролиза; 4 — ис-
паритель; 5— фильтр; 6 — дистилляционная колонна;
7 — экстракционная колонна; 8 — вакуумная дистилля-
ционная колонна.
Линии: I — 5 %-ный этиловый спирт; II — хлор; III —
отходящие гааы; IV — щелочь; V — остаток; VI — кси-
лол на очистку; VII — ксилол; VIII — остаток; IX—
глицерин.
жуточных и специальных про-
дуктов. Взаимодействием с
фенолятом натрия получают
уже упоминавшийся выше фе-
ноксипропеноксид—важнейший
стабилизатор для хлоралкилов.
Особое значение эпихлоргидрин
имеет для получения так называемых эпоксидных смол. Их получают вза-
имодействием эпихлоргидрина с двухатомными фенолами, особенно с 4,4'-ди-
оксидифенилпропаном. Это соединение, известное под названием «диан», по-
лучают конденсацией ацетона и фенола в присутствии концентрированной
серной кислоты. В щелочной среде из диана и эпихлоргидрина образуется
диандиглицидный эфир, присоединяющий к себе еще молекулы диана с одно-
временным разрушением эпоксидного кольца. Таким путем получают поли-
эфир со степенью полимеризации, достигающей 15 [8]:
СН.
___ । 3
СН2—СН-СН2С1+НО<^ Ч-С— { Ч ОН+С1 • СН2-СН—СН2 21523
\ / \=z I \=/ \ /
о сн3 о
сн3
СН2—СН—СН2О / S -С- 4 Ч ОСН2—СН—СН2 -2^н-.
\ / \=/ I \=/ \ /
О СН3 о
Диандиглицпдовый эфир
176
I I
: Пропен
______
Горячее
хлориро-
вание
I:
Глицерин
Рпс. ЮЗ. Получение глицерина из пропена посредством хлорирования.
сн3
сн2—сн—сн2о —с— осн2—снон—СН2О
Q СН3
сн3
—С— { ^он+сн2—СН—СН,С1 ->
СН3
СН2—СН—СН2О { S —С— 4 S —осн„—сн—СН2О
\ / \.=/ I \—/ - I
о L сн3 он
Синтез глицерина без применения хлора [9].
Новый метод синтеза глицерина из пропена без применения хлора разрабо-
тан фирмой Шелл Кемикал Корпорейшн. Для синтеза глицерина хлорирова-
нием пропена и хлоргидринированием спиртов необходимы очень большие
количества хлора. Однако хлор в
Рис. 104. Схема синтеза глицерина без прп-
v.c пен и и хлора.
1 — реактор дли окисления; 2—дистилляционная
колонна; .7 — Т.Н'г,- 4— очистителе ’
Линии: I — нз< hi ( пол» oi. ii спирт; 11 — кислород;
111 — ацетон; 1\ — перекись водорода; V — алли-
ловый спирт; VI — ।•1'Др(;1.силир.овапие; VII — гли-
церин; VIII —пропен; /А — пар; X — кислород;
XI — охлаждение*, ХИ — акролеин*, XIII — смола*,
XIV — восстановление.
настоящее время исключительно
дефицитен. Новый способ синтеза
глицерина нуждается только в
кислороде. Принцип синтеза сле-
дующий (рис. 104).
Изопропиловый спирт, легко
получаемый непосредственным
гидратированием пропена серной
кислотой, обрабатывают кисло-
родом в присутствии перекиси
водорода при температуре 90—
140° под давлением 2,5 ат. При
этом в жидкой фазе идет реакция
с образованием ацетона и перекиси
водорода
СН) — СНОН—СН) +о2 -> сн3 —
—со-сн3+н2о2
Реакционную смесь разбав-
ляют водой и перегонкой осво-
бождают от ацетона и непро-
реагировавшего изопропилового спирта. Полученный таким образом
водный раствор перекиси водорода применяют для каталитического
окисления аллилового спирта в глицерин. Для этого аллиловый спирт
в водном растворе в присутствии 0,2%-ного раствора вольфрамовой
кислоты (катализатор) окисляют 2 молярными объемами перекиси водорода
при 60—70е в течение 2 час. После испарения воды и заключительной пере-
гонки под вакуумом получают чистый глицерин с выходом 80—90%, счи-
тая на аллиловый спирт.
Необходимый для синтеза аллиловый спирт получают следующим обра-
зом: окислением пропена кислородом при 300—400° в присутствии водяного
178
пара над карбидом кремния, обработанным окисью меди [10], получают
с 85%-ным выходом акролеин, который отделяют перегонкой:
О
II
сн3—сн=сн2+о2 -> н—с—сн=сн2+н2о
Пропуская' смесь парообразного акролеина с 2-х—3-х кратным моляр-
ным количеством изопропилового спирта над катализатором, состоящим
из окиси цинка в окиси магния, получают с 77%-ным выходом, считая на
акролеин, аллиловый спирт, являющийся исходным продуктом для окисления
в глицерин:
О
сн,—сн—с<^ +сн3—СНОН—СН3 ZnO+--sO- СН2ОН—сн=сн2+сн3—со—сн,
н
WOi
сн2=сн—сн2он+н2о2 —-- сн2он—СНОН—СН2ОН
Глицерин находит в практике исключительно широкое применение.
Так как он совершенно не токсичен, он применяется в кондитерском произ-
водстве, в косметической и фармацевтической промышленности. Большие
количества глицерина употребляются для поддержания влажности табака.
Он применяется далее как мягчитель для целлофана, особенно когда целло-
фан используется для обертки колбасы.
В промышленности глицерин применяется для производства динамита
и взрывчатой желатины, продуктов конденсации глицерина с фталевой
кислотой, эфирных смол, в бумажной промышленности, для получения
типографской краски, в производстве канцелярских принадлежностей
(копировальное оборудование, штемпельная краска), при получении кремов
для обуви, в производстве гидравлических и амортизационных жидкостей
и т. д.
Описанный способ получения акролеина является в настоящее время
наилучшим и наиболее дешевым. Ранее акролеин получали пиролизом диал-
лилового эфира, образующегося в качестве побочного продукта при полу-
чении аллилового спирта из хлористого аллила [11]:
О
СН,=СП-С112-О-СН2-СН=СН2 Рир-°^по3оп?Л СН2=СН-С^ +сн3-сн=сн3
н
АКРОЛЕИН
X л о р и р о в а н и с_____
П рисоединение хлористого
водорода
Присосди некие сероводорода
Присогдпневпе меркаптанов
Присоединение спирта________
При сое ди пение синильной
кислоты
Присоединение уксусной
кислоты
Присоединение N а НSОз
Присоединение воды
JSH3
Уксусный ангидрид
-R.xIIg "7
2-питропропан
Диены
Виниловый спирт
Восстановлен не
Восстановление
Окисление
Кетен
Акролеин ____
Акролеин (реакции Тищенко)
а, p-дихлорпропионовый альдегид
Р-хлорпропионовый альдегид
р, p'-тиодипропионовый альдегид
Р-алкилмернаптопропионовый альдегид
Р-алконейпропионовый альдегид
Акролеинциангидрин
р-ацетоксипропионовый альдегид
Na-соль р-сульфопропионового альдегида
Гидракрилальдегид
р-пиколин
Алли лиде п диацетат
1,3-пропендиамин
4-нитро-4-мети лпептапол
Тетрагидробенэплъдсгид
2-алкоксидигидропропая
Аллиловый спирт
Пропионовый альдегид
Акриловая кислота
Лактон З-гидрокси-4-пентеновой кислоты
Димер акролеина
Аллилаврилат
Рис. 105. Реакционные возможности акролеина.
12*
179
или взаимодействием формальдегида с уксусным альдегидом в газовой фазе
112]:
0 0 о
СИ3—i+IIC^ -> сн,=сп—с/
'н п \
Акролеин представляет собой исключительно реакционноспособное со-
единение. Учитывая его дешевизну, можно предсказать ему в будущем самое
широкое применение. Рис. 105 иллюстрирует сказанное [13].
ш
В. ХЛОРИРОВАНИЕ ОЛЕФИНОВ РЕАКЦИЕЙ ПРИСОЕДИНЕНИЯ
а) Хлористый этилен (дихлорэтан)
Хлорирование олефинов, основанное па реакции присоединения,
имеет особо большое значение для этилена. При действии газообразного
хлора на газообразный этилен образуется хлористый этилен (1,2-дихлор-
этан):
СН2=СН2+С12 -> СН,С1— СНоС1 + 41 кглл/молъ,
ь качестве побочного продукта получается значительное количество три-
хлорэтана. Образование трихлорэтана, наблюдаемое и в тех случаях, когда
имеется избыток этилена, обязано сопряженным реакциям, так как в слепом
опыте при обработке дихлорэтана газо-
образным хлором в тех же условиях обра-
зования трихлорэтана не наблюдается. Об-
разование трихлорэтана можно очень силь-
но уменьшить, если вести процесс в усло-
виях конденсированной системы, при
которых хлор и этилен вдуваются сов-
местно в охлажденный дихлорэтановый
раствор. Для дальнейшего уменьшения
замещения [14] в реакционную смесь це-
лесообразно добавить 0,05—0,25% безвод-
ного хлорного железа.
В промышленных условиях при полу-
чении хлористого этилена (рис. 106) хлор
и этилен продувают через охлаждаемый
хлористый этилен при температуре 20—
25°, при этом вновь образовавшийся хло-
ристый этилен выводится из реакционного
сосуда. Отходящие из реакционного со-
суда газы содержат хлористый этилен.
Для извлечения его газы промывают ох-
лажденным жидким хлористым этиленом.
После промывки водой и нейтрализации хлористый этилен перегоняют.
Его температура кипения 83,5° и плавления —35°. Хлористый этилен в за-
метном количестве получается как побочный продукт в производстве эти-
ленхлоргидрина.
Хлористый этилен является превосходным растворителем жиров, масел,
поливинилхлорида, льняного масла, пиретрума и др. Все расширяется
применение его в процессах депарафинизации смазочных масел, точнее для
обезмасливания парафина. Для химической промышленности хлористый
этилен имеет большое значение, как промежуточный продукт. Химическим
или каталитическим дегидрохлорированием из хлористого этилена получают
хлористый винил, полимеризуемый затем в важнейшие искусственные
вещества. Реакцией с аммиаком под давлением получают этилендиамин,
Рис. 106. Схема получения хлори-
стого этилена.
1—реакционный сосуд, заполненный ди-
хлорэтаном; 2 — щелочная промывка сы-
рого продукта; .3 —отстойник; 4 —ди-
стилляционная колонна.
Линии: I — хлор; II — этилен; III —
отходящие газы; IV — 6%-пая щелочь;
V —дренаж; VI — чистый дихлорэтан;
VII — высокохлорировакные продукты.
180
представляющий собой очень ценный промежуточный продукт для хими-
ческой промышленности.
Реакцией хлористого этилена с тетрасульфидом натрия получают тио-
кол, продукт конденсации которого содержит 82% серы и может вулкани-
зироваться нагреванием с окисью цинка примерно при 140°. При этом по-
лучают каучук, исключительно устойчивый против действия ароматических
углеводородов [15]. Тиокол вследствие высокой его устойчивости приме-
няется для производства масло- и бензостойких шлангов, для изготовления
резиновых вальцов печатных машин и т. д. Для производства покрышек
тиокол не пригоден.
Дихлорэтан применяют также для окуривания почвы. Хлоразол пред-
ставляет собой смесь из 75% дихлорэтана и 25% четыреххлористого угле-
рода. О важнейших возможностях применения дихлорэтана дает предста-
вление рис. 107.
сими-ДПХЛОРЭТЛН
Дегидрохлорирование
Хлорирование
Хлорирование
Винилхлорид (полимеры и сополимеры)
Трпхлорэтап -------> виннлиденхлорпд (полимеры и сопо-
лимеры)
Гексахлорэтап
о!
1,1,1,2-тетрахлорэтан
Монсхлоруксусная кислота
— 600° g
+ KCN
Гидролиз (сода)
+NH3
+Na2SO4
1 С1о „
Трихлорэтилен-----— Псптахлорэтан
I
Са(ОН)2
Псрхлорзтилсн
Янтарная кислота
Динптрил янтарной кислоты
Тетраметнлеидиамин
Этиленгликоль
Этилсядпамил (диэтилеитрпамин, триэтилентетрамин)
I
Трилоп В (этилепдпаминтетрауксусная кислота)
Тиокол (ненабухающая резина)
Добавка к этиловой жидкости (выноситель)
Растворитель для масел, жиров, смол, поливинилхлорида,
восков п т. и»
Средство для окуривания почвы.
Рис. 107. Реакции и пути использования сил/ж-дпхлорэтаиа (хлористого
этилена).
б) Хлористый винил из дихлорэтана
Дегидрохлорирование дихлорэтана может осуществляться при помощи
спиртовой щелочи, при этом с 95%-ным выходом получается очень чистый
хлорвинил. Термическое дегидрохлорирование идет при температуре 300—
350° в присутствии катализатора, например активированного угля, окиси
алюминия и т. д. Хлорвинил может получаться также присоединением хлори-
стого водорода к ацетилену. Он кипит при —13,8°, упругость его паров при
25° составляет 2,66 ат.
Присоединением хлора к хлористому винилу получают 1,1,2-трихлор-
этан, который может также получаться хлорированием хлористого этилена.
181
Равным образом при газофазном хлорировании этилена в присутствии метал-
лического железа или марганцевого контакта имеет место реакция
СП2=СН2+2С12 -> СНС12—СН,С1+НС1
с хорошим выходом трихлорэтана.
При взаимодействии с натриевой щелочью от трихлорэтана отщепляется
хлористый водород с образованием хлористого винилидена (СН2=СС]2).
Обработкой трихлорэтана известковым молоком при повышенной температуре
получают смесь асимметричного п симметричного дихлорэтиленов.
Хлористый винилиден кипит при 31,7°. Сополимеризацией хлорвини-
лидена с хлорвинилом получают искусственные материалы (саран). Другими
важными сополимерами хлорвинила являются сополимер с винилацетатом
(винион) и с акрилнитрилом (винион-N). На рис. 108 показаны важнейшие
направления использования хлористого винила.
ХЛОРИСТЫЙ винил
Полимеризация
Хлорирование и де-_____
гидрохлорирование
Смет энная полиме-_____
ризация с винилацетатом
Поливинилхлорид (игелит, абартен) ,
,, Полимеризация
Хлористый винилиден -------------—* саран
Винион (90% винилхлорида 4- 10% винилацетата)
Смешанная полиме-
ризация с акрилнитрилом
Сметанная т олим с р и за -
ция с эфиром акриловой
кислоты
Винион N, динель (60%
Астралон
винилхлорида + 40% акрил-
нитрила)
Рис. 108. Важнейшие продукты полимеризации хлорвинила.
При дальнейшем хлорировании трихлорэтана образуется 1,1,1,2-тетра-
хлорэтан, который при нагреве до 600° переходит в трихлорэтилен. В ка-
честве сырья для получения трихлорэтилена трихлорэтан конкурирует
с ацетиленом. Для получения трихлорэтилена на базе этилена требуется,
однако, на 1 моль хлора больше, чем на базе ацетилена (см. стр. 242).
в) Этилендиамин
При взаимодействии дихлорэтана с избыточным количеством аммиака
при 180° под давлением в автоклаве образуется этилендиамин. Наряду с этим
в результате реакции этилендиамина с дихлорэтаном образуются диэтилен-
триамин и триэтилентетрамин.
Этилендиамин является исходным продуктом для трилона В (этилен-
диаминтетрауксусная кислота), получаемого из этилендиамина, формаль-
дегида и синильной кислоты:
„ CH2CN ch2cn
U I I
NH2-CH2-CH,-H2N+4HC/ +4HCN -> N—CH2—CH2—N
H I I
H ch2cn ch2cn
'CH2COOH CH2COOH
^5. A-CH2-CH2-N
I I
CH2COOH CH2COOH
Трилон B
182
Г. ПРИСОЕДИНЕНИЕ ХЛОРА К ОЛЕФИНАМ ПРИ ПОМОЩИ
ХЛОРНОВАТИСТОЙ КИСЛОТЫ (ХЛОРГИДРИРОВАНИЕ)
При присоединении хлорноватистой кислоты к олефинам, например
к этилену, образуются хлоралкоголи — соединения, в которых атом хлора
и гидроксильная группа находятся у соседних углеродных атомов. Такие
соединения называют хлоргидринами. Реакцией хлоргидринов со щелочами,
сопровождающейся отщеплением хлористого водорода, очень легко обра-
зуются циклические эфиры, так называемые окисные соединения:
СН2=СН2+С12+Н2О -> СН2ОН—СН2С1+НС1
НОСН2—CH2Cl+NaOH -4 СН2—CH2+NaCl+H2O
Таким путем из этилена получают окись этилена — исключительно
важный промежуточный продукт для промышленности алифатического
синтеза. Реакцию этилена с хлорноватистой кислотой можно осуществлять
также в условиях образования последней,
пропуская одновременно хлор и этилен через
воду (процесс Гомберга [16]). По уравнению
С12+Н2О -> НС1+НОС1
в результате гидролиза хлора образуется хлор-
новатистая кислота, которая тотчас вступает в
реакцию с этиленом путем непосредственного
присоединения хлора к этилену. Процесс сле-
дует вести так, чтобы образование хлористого
этилена не препятствовало хлоргидрированию.
В промышленности процесс ведут таким обра-
зом, чтобы концентрация хлоргидрина в воде
не превышала 8—10% при концентрации соля-
ной кислоты 4—5%. Принимая количество
хлоргидрина за 100%, количество образую-
щегося при хлоргидрировании хлористого
этилена составляет в среднем 15%.
Для получения хлоргидрина (рис. 109)
хлор и этилен подают в реакционную камеру,
облицованную изнутри кислотостойким мате-
риалом. Хлор подают в колонну ниже места
ввода потока этилена, чтобы иметь необхо-
димое время для реакцип с водой и образо-
вания хлорноватистой кислоты. Этилен не весь
вступает в реакцию, часть его возвращается
Рис. 109. Схема хлоргидри-
нового процесса.
1 — насос для циркуляции газа;
2 — конденсатор; з — реакционная
колонна; 4 — газоотделитель.
Линии: I — этилен; II — отходя-
щие газы; III — вода; IV — цирку-
лирующий этилен; V — охлаждаю-
щая вода; VI — газ; VII — пере-
ток; VIII— хлор; IX — fO^-пый
хлоргидриновый раствор с хлори-
стым этиленом и хлористым водо-
родом.
на циркуляцию, осуществляемую газовым циркуляционным насосом.
В реакционной колонне при помощи водяного охлаждения поддержи-
вается температура 50—60°. Выходящий из верха реакционной колонны
раствор хлоргидрина и соляной кислоты сразу поступает на дальнейшую
переработку в окись этилена, так как выделение 100%-ного хлоргидрина
из такого раствора не целесообразно. Для получения окиси этилена выходя-
щий из хлоргидринирующей колонны раствор, содержащий приблизительно
10% хлоргидрина, вливается в 12%-ный раствор гидроокиси кальция (из-
вестковое молоко). При этом образуются окись этилена и хлористый каль-
ций. Окись этилена, кипящая при 12°, отгоняется, раствор хлористого каль-
ция дренируется.
Чистый 100%-ный хлоргидрин представляет собой бесцветную, хорошо
растворимую в воде жидкость, кипящую при 128,7° (760 мм рт. ст.). Про-
стейший способ его получения заключается в том, что газообразные окись
183
этилена и хлористый водород подают в охлажденный этиленхлоргидрин,
при этом сразу начинается реакция, сопровождающаяся заметным выделе-
нием тепла, количественно протекающая по уравнению
СН2—СН2+НС1 —>2СН2ОН—СН2С1+36 кпал]молъ
Так как выделение тепла составляет 36 ккал/молъ, необходимо исклю-
чительно интенсивное охлаждение.
ЭТИЛЕНХЛОРГИДРИН
+ Са(ОП)2
+ NaCN __ + анилин
+ 7 5% H0SO4
Ч- хлор + этилен
Окисление с 6 0%-ной
HNOg + Ka2S
+ Na-целлюлоза
Окись этилена и ее производные
Нитрил оксипропиопоиой кислоты (циангидрин)
I- Н2О
Акрилонитрил
Оксиатилаиилин, дпоксиэтплаиилип
Дихлордиэтиловып эфир (хлорекс)
Дихлорэтпловый эфир (хлорекс)
Момохлоруксуеная кислота
Тиодиглииоль
Гилроксиэтитовый эфир ’целлюлозы (водорастворимые
вспомогательные средства, эмульгаторы и т. п.)
Рис. 110. Реакции этиленхлоргидрииа.
Содовым раствором при 105° этиленхлоргидрин практически количе-
ственно омыляется в этиленгликоль. При нагревании с концентрированной
серной кислотой до 90—100° образуется (3, Р'-дихлордиэтиловый эфир (хло-
рекс), применяемый в качестве селективного растворителя в производстве
нефтяных смазочных масел. Небольшие количества хлорекса образуются
также как побочный продукт при хлоргидринироваиии этилена по реакциям
(С1С112—CIL,OH + C12 -> C1CIL—СН2ОС1+1-1С1
gjClCHj— CI]2OC1 + C»H4 -> С1СН2— СН2— О—СН2—СН2С1
Действием хлора на хлоргидрин получают гипохлорит, который затем,
реагируя с этиленом, дает хлорекс.
С фенолятами хлоргидрин дает этиленгликольмонофепиловый эфир,
применяемый в качестве фиксатора в производстве душистых веществ (на-
пример, арозоль). Окислением 60%-ной азотной кислотой хлоргидрин
превращается с 90%-ным выходом в монохлоруксусную кислоту [17].
Этиленхлоргидрин, который может также получаться продуванием
хлористого водорода через этиленгликоль, исключительно ядовит. Вдыха-
ние его паров опасно для жизни. Попадание на кожу менее 5 мл этиленхлор-
гидрина уже смертельно [18].
На рис. НО представлены важнейшие реакции этпленхлоргидрина.
Как уже выше указывалось, окись этилена получают действием извест-
кового молока на технический раствор хлоргидрина при 100°. При этой опе-
рации через реакционную смесь целесообразно продувать водяной пар, что
способствует отделению образующегося в качестве побочного продукта хло-
ристого этилена. В промышленных условиях переработка окиси этилена про-
водится в непрерывно действующих колоннах. Хранится она в охлаждаемых
до —10° стальных резервуарах под давлением азота [19].
184
Д. ПОЛУЧЕНИЕ ОКИСИ ЭТИЛЕНА КАТАЛИТИЧЕСКИМ ОКИСЛЕНИЕМ
ЭТИЛЕНА
а) Общие сведения
Окисление этилена идет исключительно легко до сгорания с образова-
нием углекислоты и воды:
2СН2=СИ2+6О2 —> 4СО2+4Н2О + 631 ккал/моль
Присоединение кислорода по двойной связи с образованием окиси
этилена идет со значительно меньшим выделением тепла:
2СН2=СН2+О3 —> 2СН2—СН2+56 ккал/молъ
Соответствующим подбором катализатора необходимо ограничить воз-
можность протекания реакции горения, выдвинув на первый план образо-
вание окиси этилена. В настоящее время в промышленности применяются
для этой цели серебряные катализаторы различных состава и способа при-
готовления.
Каталитическое окисление этилена в окись этилена представляет осо-
бый интерес вследствие того, что дальнейшее увеличение производства окиси
этилена за счет развития процесса хлоргидринирования уже более невоз-
можно из-за трудности удовлетворения огромного спроса на хлор. Получение
окиси этилена хлоргидриновым методом требует па 1 кг окиси этилена около
2 кг хлора, 0,9 кг этилена и 2 кг гидроокиси кальция. Одновременно полу-
чается от 0,20 до 0,22 кг хлористого этилена. Если исключить отсюда расход
хлора на образование хлористого этилена, то на 1 кг окиси этилена потре-
буется около 1,8 кг хлора и 0,74 кг этилена. Новейшие способы получения
окиси этилена путем окисления требуют около 1,04 кг этилена на 1 кг окиси
этилена. Таким образом, увеличение расхода этилена равно 0,3 кг. Применяя
хлоргидриновый процесс, мы сберегаем 0,3 кг этилена за счет расходования
2 кг хлора и 2 кг гидроокиси кальция или в пересчете на 1 кг сберегаемого
этилена 6 кг хлора и 6 кг гидроокиси кальция. Таким образом, с точки
зрения стоимости расходуемых реагентов хлоргидриновый процесс мог
бы быть признан равноценным окислительному только в том случае, если
бы стоимость этилена была по меньшей мере в 6 раз выше стоимости хлора,
что, однако, в настоящее время не имеет места.
б) Промышленные способы
О промышленных способах каталитического окисления этилена в окись
этилена точных данных нет. Процесс ведут над неподвижным катализатором
в присутствии воздуха или чистого кислорода как окисляющего агента.
В последнем случае требуется применение очень чистого этилена и очень
чистого кислорода.
Окись этилена получают также с катализатором, находящимся в псе-
вдоожиженном слое [20]. При использовании в качестве окисляющего
агента воздуха содержание этилена в газовой смеси должно быть ниже
2,9%. Температура реакции лежит между 260 и 290°, продолжительность
реакции 1—4 сек. Когда работают с чистым кислородом, температура реак-
ции может быть 230—240°.
Газы реакции, содержащие окись этилена, пропускают через активи-
рованный уголь, которым окись этилена адсорбируется и далее она выде-
ляется посредством испарения. Окись этилена может также абсорбироваться
подкислеппой водой с одновременным образованием этиленгликоля, кото-
рый далее перерабатывается способом, описанным ниже.
185
в) Переработка окиси этилена
Общие сведения. Окись этилена является одним из наиболее
реакционноспособных соединений в алифатической химии. Реакционная
способность ее связана с расщеплением окисного кольца. Ниже приво-
дится несколько примеров, иллюстрирующих возможности превращения
окиси этилена:
СН2—СН2+НС1 —> СН2ОН—СН2С1 (этиленхлоргидрип)
х^
СН2—СН2+Н2О —> НОСН2—СН2ОН (этиленгликоль)
Х^
СН2—СН2+НОН —> НОСН2—СН2ОЙ [(моноэфир гликоля) R-алкил, арил, цикло-
X'
алкил или аралкил
СН2—СН2+соединения Гриньяра —> НОСН2— СН2В (первичные спирты) В-алкил, арил,
х^
циклоалкил или аралкил
СН2— CH24-HCN —» НОСН2—CH2CN (этиленциангидрин, нитрил р-оксипропионовой
кислоты)
СН2—CH2+H2S —> НОСН2—CH2SH (тиогликоль)
Ху
2СН2—CH2+H2S —> НОСН2—СН2—S—СН2—СН2ОН (тиодигликоль)
Х^
СН2—GH2 + NH3 -> НОСН2—CH2NH2 (этаноламин)
х/
Производство окиси этилена составило в США в 1956 г. около 360 тыс. т
[21]. Этиленгликоля получено около 320 тыс. т, причем около 83% реак-
цией окиси этилена с водой.
Из всего количества выработанной окиси этилена 64% использовано
для получения этиленгликоля, 11% в производстве гликолевых и полигли-
колевых эфиров, 10% для получения моющих и вспомогательных продук-
тов, 8% для получения этаноламина, 5% в производстве акрилонитрила
и 2% для различных других целей.
Главная масса (80%) этиленгликоля расходуется в настоящее время
для получения антифриза, 6% в качестве мягчителя для целлофана (целло-
фан содержит 10—20% этиленгликоля для поддержания необходимой влаж-
ности), 5% этиленгликоля используется в производстве взрывчатых ве-
ществ, 4% для производства терилена и 5% для других целей.
Гидролиз окиси этилена в гликоль. Гидролиз
окиси этилена водой проводится или при 200е под давлением, или при 50—
100° в присутствии небольших количеств кислоты, которая значительно
ускоряет гидратацию. Эта кислота удаляется из продуктов реакции до их
разгонки при помощи основного ионнообменного поглотителя.
Реакция закапчивается примерно в полчаса. Реакция экзотермическая.
Количество выделяющегося при реакции тепла составляет 23 ккал!моль.
По окончании реакции вода отделяется перегонкой и возвращается в гидра-
тационную установку. Содержание гликоля в циркулирующей воде около
1%. Остаток от перегонки, содержащий 85% гликоля и 15% воды, разго-
няется затем в ряде колонн под пониженным давлением.
Из смеси 1,5 Л13 окиси этилена и 9 лр воды получают примерно 87,5%
моноэтиленгликоля, 10% диэтиленгликоля и 2,5% триэтиленгликоля. Со-
186
отношение количеств отдельных гликолей в продуктах реакции опре-
деляется начальным соотношением числа молей окиси этилена и воды
при гидролизе. Численные величины этой зависимости показаны в
табл. 77 [22].
Таблица 77
Распределение моно-, ди-, три- и полигликолей при омылении окиси этилена
в зависимости от количества воды (% мол.)
Моли воды на 1 моль окиси этилена Моногли- коль Дигликоль Тригликоль Тетрагли- коль Полигликоль
10,5 82,3 12,7
4,2 65,7 27,0 2,3 — .—
2,1 47,2 34,5 13,0 0,3 —
0,61 15,7 26,0 19,8 19,0 14,5
Таблица 78
Некоторые константы гликолей из табл. 77
Гликоль Температура кипения в °C при rf15 “4 45
760 мм рт. ст. 14 мм рт. ст.
Моногликоль 197,6 97 1,1170 1,4332
Дигликоль 244,5 133 1,1197 1,4488
Тригликоль 285,0 165 1,1274 1,4578
Тетрагликоль 328,0 198 1,1285 1,4609
Таблица 79
Температура замерзания различных смесей этиленгликоля с водой
Содержание этиленгли- коля Температура замерзания, °C Содержание этиленгли- коля Температура замерзания, °C
% вес. % объемн. % вес. % объемн.
0,0 0,0 0,0 30 28,0 —15,0
2 1,8 — 0,6 32 29,9 —17,0
4 , 3,6 — 1,3 34 31,9 —18.0
6 5,4 — 2,0 36 33,8 —20,0
8 7,2 — 2,7 38 35,8 —22,0
10 9,1 — 3,5 40 37,8 —24,0
12 10,9 — 4,4 42 39,8 —26,0
14 12,8 — 5,3 44 41,8 —28,0
16 14,6 — 6,3 46 43,8 —31,0
18 16,5 — 7,3 48 45,8 —33,0
20 18,4 — 8,0 50 47,8 —36,0
22 20,3 - 9,0 52 49,8 —38,0
24 22,2 —11,0 54 51,9 —41,0
26 24,1 —12,0 56 53,9 —44,0
28 26,0 —13,0 j 58 56,0 —48.0
Если целью процесса является получение дигликоля как исходного
материала для производства взрывчатых веществ, то процесс ведут в смеси,
содержащей, кроме окиси этилена и воды, уже готовый моногликоль. Так,
например, из смеси 1,5 nt3 окиси этилена, 4,5 л3 воды и 4,5 .и3 моногликоля
187
получается смесь, состоящая примерно из 80% дигликоля и 20% три гли-
коля .
В табл. 78 даны важнейшие физические константы некоторых этилен-
гликолей, в табл. 79 соотношение между содержанием этиленгликоля и
воды в смеси п температурой замерзания смеси.
Получение этиленгликоля из водяного газа.
Этиленгликоль может получаться не только из окиси этилена, но и другими
путями. Так, по способу фирмы Дюпон де Немур (Западная Вирджиния,
США) действием углекислоты и воды (2 части) на формальдегид (1 часть)
в присутствии гликолевой кислоты (2 части) как растворителя при темпера-
Рис. 111. Схема получения этиленгликоля из формальдегида и окиси углерода.
1 — смеситель; 2 — реактор; 3 — вакуумная колонна; 4 — установка для этерификации; 3 — уста-
новка гидрирования.
Линии: I — гликолевая кислота и вода; II — разбавленная серная кислота; III — формальдегид и
водяной лар из промесса окисления метилового спирта; IV — циркулирующая СО; V — свежая окись
углерода; VI — отходы; VII — циркулирующий водород; VIII — свежий водород; IX — алнплен-
глпколь.
туре 200° и давлении 700 ат и при использовании серной кислоты
(0,02 части) или фтористого бора как катализатора получают гликолевую кис-
лоту (рис. 111). Эфиры гликолевой кислоты восстанавливают над смесью
хромитов бария и меди (катализатора) при 200—225° и 20—40 ат водорода
и получают этиленгликоль и обратно спирт. Лучше для этой цели приме-
нять выходящий из печи продукт окисления метилового спирта, состоящий
из формальдегида и водяных паров, которые растворяются в подаваемой
в процесс гликолевой кислоте.
После завершения реакции в печи, на что требуется около 5 мин., про-
дукты реакции подвергают перегонке под вакуумом, в ходе которой в
остатке получается чистая, свободная от воды гликолевая кислота, а в погоне
смесь воды и гликолевой кислоты. Разбавленная гликолевая кислота снова
применяется для абсорбции смеси паров воды и формальдегида, а получен-
ная в виде остатка чистая гликолевая кислота этерифицируется и затем ги-
дрируется 123].
Способ получения этиленгликоля на основе водяного газа как исходного
материала можно представить себе следующими реакциями:
О
СО+ЗН2 -> СН3ОН -> Н—
II
О
н—+со+п2о сн,он—соон
н
СН2ОН—СООН+СИ3ОН -4 сп2он—соосн3+н2о
СН2ОН—СООСНз -каталИ2ааТ01Т СИ2ОН-СН2ОН+СН3ОН
188
Применение этиленгликоля. В качестве низкозамер-
зающей жидкости может применяться смесь этилен- и пропенгликолей,
как она получается при совместной гидратации окиси этилена и окиси про-
пена. Эта смесь окисей получается из хлоргидрина, являющегося продук-
том совместного действия хлора и воды на получаемую крекингом пропана
смесь этилена и пропена (процесс Виандотта). В малых масштабах гликоль
в настоящее время получают прямым окислением этилена перекисью
водорода.
Нитрогликоль применяется как заменитель нитроглицерина. Он имеет
то преимущество перед нитроглицерином, что существует не в двух формах,
как последний, что в случае нитроглицерина приводит к выделению тепла при
изомеризации.
Поликонденсацией гликоля с терефталевой кислотой получают очень
важное искусственное вещество — терилен (см. стр. 268).
Полигликоли, получаемые дальнейшим действием окиси этилена на мо-
ногликоль, находят все расширяющееся применение. Ниже описаны не-
которые возможности применения и дальнейшей переработки гликоля.
На рис. 112 даны важнейшие направления использования гликоля.
Глиоксаль [24]. Каталитическим окислением этиленгликоля над окисно-
медным катализатором при температуре около 270—280° и давлении 3,5 ат
в газовой фазе можно гликоль окислить в глиоксаль, получающийся в виде
водного раствора глиоксальгидрата. Возможности применения глиоксаля
в промышленности многочисленны и разнообразны. Он является исходным
материалом для получения пира.зин-2,3-дикарбоновой кислоты — витамина,
применяемого при лечении пеллагры.
ЭТИЛЕНГЛИКОЛЬ
Ката литическое________
окисление
Нитрование
Этерификация___________
Реакция с диметилтере-
фталатом
-4- HCI__________________
этерификация
Этерифипция с фталевой
кислотой + глицерин
яУпрочнителп для текстильной промыгплен-
Глиоксаль < пости
* Получение не растворимых в воде полио-
ксисоедннений
Гликольдинитрат взрывчатое вещество)
Гликольацетат, -стеарат, -фталат (мягчители)
Терилен» дакрон, тренира
H.NO3 монохлоруксусная кислота
Хлоргидрин
дихлордиэтиловый афир (хлоренс)
Алкидные смолы с ограниченной сетчатостыо
Низкозамерзающие жидкости (глизантии, церекс)
Увлажнители для целлофана, кожи, фибры
Тормозные жидкости для автомобилей
Рис. 112.
Реакции и возможности применения этиленгликоля.
Вследствие бифункциональной природы глиоксаль при соединении
с аминными или гидроксильными группами может образовывать сетчатые
молекулы. Эти свойства глиоксаля используются в процессах упрочения
волокна. В процессе «санфорсет» для пропитки применяется 4%-ный, считая
па волокно, раствор глиоксаля. После пропитки при 150—200° материал
становится значительно прочнее [25].
Крахмал, сахар и другие полиоксисоединения, как декстрин-поливини-
ловый спирт, после обработки глиоксалем становятся не растворимыми
в воде. Обработанные глиоксалем обои можно мыть. Протеины также обра-
зуют с глиоксалем сетчатые молекулы, практически почти совершенно устой-
чивые против действия воды. Глиоксаль применяется для защиты ковров
от моли, для бальзамирования трупов, для склеивания корковой пробки
и т. д.
189
Диэтиленгликоль. Диэтиленгликоль, называемый также дигликолем,
применяется как растворитель, как средство регулирования влажности
материала, в качестве пластификатора, смазочного средства, тормозной
жидкости, для получения взрывчатых веществ, клеев, типографской краски,
текстильных препаратов, как средств осушки газов и т.д. Он легко раство-
ряется в воде. С многоосновными кислотами образует полиэфирные смолы.
Особый интерес представляют продукты реакции дигликоля с малеино-
вой и фумаровой кислотами, могущие сополимеризоваться с винилацета-
том, стиролом и т. д. Важные продукты превращения, способные к много-
численным и разнообразным реакциям, образуются при взаимодействии
дигликоля с фосгеном. Бис-хлоруглекислый эфир дигликоля в присутствии
натриевой щелочи может вступать в реакцию с аллиловым спиртом. При по-
лимеризации диаллиловых эфиров получают прозрачные смолы (аллимер
СВ-39). На рис. 113 приведены важнейшие возможности использования
дигликоля.
ДИЭТПЛЕНГЛИКО.ТЬ
I Нитрование
Этерификация с малеиновой
кислотой
Этерификация с жирными
кислотами, например пелар-
гоновой
Взаимодействие с фосгеном
и аллиловым спиртом
•4-H2SO4
Этерификация с канифолью
Этерификация в смеси с гли-
церином и фталенсГ! кислотсй
Диглинольдипитрат (взрывчатое вещество)
Полимеризующиеся эфиры, сополимеры со стиролом
и винилацетатом
Мягчители для смол и каучука
Полимерный продукт аллимер CR-39
Диоксан
Увлажнители для табака, типографской краски,
целлофана, декстрина, штемпельной краски и т. д.
Осушка природного нефтяного и других газов
Растворители для химических производств
Смолы для лаковой промышленности
Алкидали с ограниченной сетчатостью
Тормозные жидкости
Рис. 113-
Возможности применения и реакции диэтиленгликоля.
Триэтиленгликолъ. Триэтиленгликоль представляет собой бесцветную,
легко растворимую в воде, вязкую жидкость. Он применяется в качестве
тормозной жидкости, для осушки газов, особенно природного нефтяного
газа [26], и служит для дезинфекции воздуха в больницах, театрах, концерт-
ных залах и т. и., так как уже в малых концентрациях обладает сильным
стерилизующим действием [27]. Эфиры триэтиленгликоля и монокарбоно-
вых кислот являются превосходными пластификаторами. Известны 2-этил-
масляный эфир триэтиленгликоля под названием флексол 3GH и смесь
эфиров триэтиленгликоля и смеси жирных кислот с 6—10 атомами С из
кокосового масла под названием пластификатора SG. На рис. 114 приведены
основные направления использования триэтиленгликоля.
ТРПЭТПЛЕНГЛИКОЛЬ
Эфиры жирных КИСЛОТ
Се — Сю
Эфир 2-втилмасля-
ной кислоты
Мягчители (пластификатор SC, пластомель KF, желотон.
моллит KF)
Мягчитель (флексол CGH)
Увлажняющие вещества для табака, клея, желатины
Упрочняющие вещества для волокон и тканей
Осушка природного taoa
Добавки к типографским краскам
Дезинфекция воздуха в больницах, конторах, театрах,
кино, концертных залах и пр.
Рис. 114. Реакции и применение триэтиленгликоля.
190
Полигликоли. Гликоли еще более высокого молекулярного веса, чем
триэтиленгликоль, представляют собой или бесцветные жидкости или со-
единения парафинообразного вида. Они растворимы в воде и могут приме-
няться в качестве смазочных масел, в косметической и фармацевтической
промышленности и как вспомогательные материалы в текстильной и бумаж-
ной промышленности.
Для получения полигликолей в этиленгликоль, содержащий каталити-
ческое количество едкого натра, подают под давлением окись этилена с та-
кой скоростью, чтобы температура реакции поддерживалась на уровне
120—135°. Средний молекулярный вес полигликоля зависит от соотношения
этиленгликоля и окиси этилена.
Оксивоск представляет собой высокомолекулярный полигликоль моле-
кулярного веса около 4000. Внешне он похож на воск, если он применяется
в фармацевтической промышленности, то предварительно обрабатывается
30 %-ной перекисью водорода.
В табл. 80 приведены некоторые свойства жидких и твердых полигли-
колей [28].
Таблица 80
Свойства некоторых жидких и твердых полигликолей
Марка по- лигликоля Средний молекуляр- ный нес Удельный вес 20/20 Температу- ра замерза- ния, °C Раствори- мость в во- де, % Темпера- тура вспыш- ки, °C pH 5%-пого водного раствора
Полиэтиленгликоль
200 190—210 1,12 Переохлажд. Полная 171 4—7
300 285—315 1,13 —15 до 8 194 4—7
400 380—420 1,13 4—10 » 225 4-7
600 570—630 1,13 20—25 246 4—7
Карбовоск
1000 950—1050 1,151 38—41 —70 277 4-7
1540 1300—1600 1,15 43—46 73 238 4—7
4000 3006—3700 1,204 53—56 62 280 4.5—7,5
6000 6000—7500 — 60—63 -50 249 4,5—7,5
На базе полиэтиленгликолей американская фирма Карбид энд карбон
кемикал корпорейшп выпускает ряд растворимых и не растворимых в воде
смазочных масел, обладающих специальными свойствами и известных на
рынке под названием масел укон и престоп [29]. Температура застывания
этих масел ниже, чем углеводородных масел равной вязкости, а индекс
вязкости очень высок. На рис. 115 показаны важнейшие возможности при-
менения полигликолей.
ПОЛПЭТИЛЕПГЛВКОЛЬ
------------------* Водорастворимые синтетические смазочные масла
------------------* М я । ч и тел и для казеина и желатины
-----------------Смазочные и другие продукты для резиновой про-
мышленности (не набухающие)
-------------------- Полиглш.'олевые простые и сложные эфиры, эмуль-
гаторы для пншевой лромышлег’ностп,мягчители лля
нитроцеллюлозы, поверхностно-активные вещества
Рис. 115. Применение полиэтиленгликоля.
Диоксан. Диоксан получают из дигликоля отщеплением воды. В про-
мышленных условиях диоксан можно получать из смеси гликолей среднего
молекулярного веса, соответствующего тетрагликолю, нагреванием с серной
191
кислотой до 150—160°. Выход диоксана составляет около 80%. Он является
превосходным растворителем для многих труднорастворимых или нераство-
римых соединений, однако ядовит.
О к си этилирование органических соединений
с реакционноспособным водородом в м о л е-
к у л е. Подобно реакции с водой, верное с гидроксильной группой, приводя-
щей к образованию этиленгликоля, окись этилена может вступать во взаи-
модействие с другими соединениями, содержащими реакционноспособный
водород, как, например, спиртами, аминами, меркаптанами, кислотами,
амидокислотами, сульфамидами и т. д.
Реакции окиси этилена с соединениями, содержащими гидроксильные
группы. При взаимодействии окиси этилена со спиртами образуются мопо-
алкиловые эфиры этиленгликоля, называемые целозольфами. Реакция
протекает по уравнению
С0Н5ОИ 1-СТП— СП, -> CjH6O—СП,—СП,ОП пли С2П5О—СП,—СИ,—ОСН2—СП2ОН
О
Собственно целозольфом называется моноэтиловый эфир этиленгли-
коля. Другие представители ряда целозольфов называются в соответствии
с названием спирта, образующего эфир с этиленгликолем, например, метил-
пропил-, бутилцелозольф.
Моноалкиловые эфиры диэтиленгликоля называются карбнтолями. Соб-
ственно карбитолем называется моноэтиловый эфир диэтиленглпколя. Дру-
гие карбитоли обозначаются в соответствии с названием спирта, использо-
ванного в реакции этерификации диэтиленгликоля: метил-, пропил-,
бутилкарбитоль.
В промышленности целозольф и карбитоль получают взаимодействием
окиси этилена с соответствующим абсолютным спиртом, соотношение коли-
честв целозольфа и карбитоля в продуктах реакции зависит от начального
соотношения между окисью этилена и спиртом. При 12-часовой реакции
этилового спирта с жидкой окисью этилена при 15%-ном избытке спирта
в автоклаве с начальной температурой 150° и давлением 17—18 ат полу-
чают смесь, содержащуго около 70% целозольфа и 30% карбитоля. Целозольф
кипит при 135°, карбитоль при 202°.
В табл. 81 даны важнейшие физические константы некоторых сортов
целозольфа.
Таблица 81
Физические свойства некоторых моноэфиров этиленгликоля
Метил- Этил- Бутил- Фенил- 2-этил- бутпп
эфиры моноэтиленглпколя
Температура кипения,°C:
при 790 мм рт. ст 124,5 135,1 171,2 244,7 196,8
» 50 » » » 55 64 94 157 116
» 10 » » » 27 35 61 118 84
Температура вспышки, °C .... 46 55 74 121 82
Температура замерзания, °C ... —85,1 — — 14 —90
Коэффициент преломления , 1,4021 1,4076 1,4193 1,5386 1,4304
Растворимость в воде при 20°, % Растворимость воды в целозольфе, % СО оо о? 2,1 1,2
со со со 10,8 10,0
Удельный вес 0,9663 0,9311 0,9019 1,1094 0,8954
Упругость паров при 20° 6,2 3,8 0,6 0,03 0,17
192
Целозольф является превосходным растворителем для нитроцеллюлозы,
но не растворяет ацетилцеллюлозы. Метилцелозольф прекрасно растворяет
также ацетилцеллюлозу. Карбитоль является хорошим растворителем
для нитроцеллюлозы, канифоли, шеллака, он широко применяется в кос-
метике, красильном деле, текстильной промышленности и т. д.
При действии окиси этилена на высшие спирты, например лауриловый,
стеариловый и другие, образуются так называемые оксиэтилированные
продукты — алкилполигликолевые эфиры — общей формулы
RO (СН2— СН2Ок СН2СН2ОН 1 |
растворимые в воде и обладающие хорошими поверхностно-активными свой-
ствами, что обеспечивает им широкое применение в ^кстильной промышлен-
ности.
Для проведения реакции оксиэтилировапия окись этилена пропускают
через нагретый до приблизительно 170° высокомолекулярный спирт
в присутствии этилата натрия или едкого натра как катализатора. В про-
мышленности оксиэтилирование ведут жидкой окисью этилена под давле-
нием около 3,5 ат и при температуре 165°. В зависимости от соотношения
между окисью этилена и спиртом полигликолевые эфиры содержат больше
или меньше оксиэтильных групп.
Олеиловый спирт с 15 молями окиси этилена образует растворимый
в воде полигликолевый эфир, 30%-ный раствор которого под названием «лео-
нил О» применяется для мойки шерсти. Продукт оксиэтилирования стеарило-
вого спирта 20 молями окиси этилена под названием «эмульфор С» приме-
няется для изготовления эмульсий для промывки шерстяной пряжи.
Также легко поддается оксиэтилированию окисью этилена касторовое
масло, содержащее глицериды оксикислот. Продукт взаимодействия 40
молей окиси этилена с касторовым маслом выпускается в продажу под на-
званием «эмульфор EL».
Подобно спиртам способны оксиэтилироваться и фенолы. Так как гидро-
ксильная группа фенолов обладает более сильно выраженными кислотными
свойствами, чем гидроксильные группы спиртов, то удается нагреванием
в автоклаве до 200° 1 моля фенола с 1 молем окиси этилена получить этилен-
гликольмонофенпловый эфир с 95%-ным выходом. Этот продукт, известный
под названием «арозол», уже упоминался ранее (см. стр. 184).
Для промышленности особый интерес представляют продукты окси-
этилирования додецил- и тетрадецилфенола, которые легко получают алкили-
рованием фенола тетрамерами или петамерами пропена (см. стр. 231).
В результате получается чисто синтетическое, превосходно действующее
и вследствие неионной природы совершенно устойчивое моющее средство,
широко используемое в настоящее время. Так, продукт оксиэтилирования
изододецилфенола 7 молями окиси этплена исключительно пригоден для
мойки шерсти, а с 9 молями окиси этплена получается продукт, дающий
особенно хорошие результаты при мытье хлопка. Возможности вариаций
здесь изменением характера алкильного остатка и степени оксиэтилирования
исключительно велики.
Карбоновые кислоты также могут реагировать с окисью этилена с обра-
зованием эфиров этих кислот и полигликоля:
ОН
R—с/ +СН2—СГГ2->1:—СОО—СН„—СН2ОН
н О
Продукты применяются в первую очередь в текстильной промышлен-
ности. Эмульфор А, например, представляет собой продукт оксиэтилиро-
вания олеиновой кислоты 6 молями окиси этилена.
Оксиэтилирование соединений, содержащих активный водород, не свя-
занный с кислородным атомом. Реакции с сероводородом
13 Заказ 596. 193
и меркаптанами. Окись этилена очень легко реагирует с сероводо-
родом, с образованием тиоэтиленгликоля или, соответственно, тиодигликоля
(0, Р'-диоксидиэтилсульфид).
Тиодигликоль легко получается при 90° в результате реакции газооб-
разной сухой окиси этилена с сухим сероводородом, в молярном соотноше-
нии 2:1, температура кипения его 168° при 14 мм рт. ст. Эта глицерино-
образная жидкость применяется в красильном деле и в производстве
красителей в качестве вспомогательного вещества.
Также легко реагируют с окисью этилена меркаптаны. Так, додецил-
меркаптан с 5 молями окиси этилена в присутствии метилата натрия обра-
зует растворимый в воде продукт, обладающий хорошими капиллярными
свойствами.
Окись этилена взаимодействует с гидросульфитом натрия, находя-
щимся в водном растворе в 30%-ной концентрации, с образованием изатио-
нокислого натрия, который при нагревании до 280° при 200 ат переходит
в N-метилтаурин. Реакцией изатионокислого натрия с хлорированной олеи-
новой кислотой получают игепон А, а аналогичной реакцией с N-метил-
таурином — игепон Т.
Реакции с аммиаком. Окись этилена легко вступает в реак-
ции с активным водородом, связанным с атомом азота. При действии окиси
этилена на аммиак в водном растворе образуется смесь моно-, ди- и триэта-
ноламинов:
сн2-ch2+nh3 НОСН2—СН2—nh2
V
сн.—ch2+nh2—СН2—СНаОН -> НОСН2—СН2—NH—сн.—-СН2ОП
СП 2—СН2ОН
сн2—сн2+носн2—ch2-nh—сн.-сн2он -> n-ch2—сп2он
C(I2-CH2OH
Соотношение количеств трех этаноламинов в продуктах реакции зави-
сит от молярного отношения окиси этилена к аммиаку [30]. При большом
избытке аммиака в продуктах реакции преобладает моноэтаноламин. Разде-
ление трех этаноламинов производится перегонкой и ректификацией. Все
они легко растворимы в воде. Их важнейшие физические свойства приве-
дены в табл. 82.
Таблица 82
Важнейшие свойства трех этаноламинов
Показатели Моноэта- ноламин Диэтанол- амин Триэтанол- амин
Температура кипения, °C: при 760 мм » 50 » ,* » 10 » .20 “20 Температура вспышки, °C . . . Температура замерзания, °C . . „20 пп pH 0,1 н. водного раствора . . 172,2 1,0220 93 10 1,4538 12,05 268 160 1,0920/30° 138 28 1,4776 11,0 360 244 208 1,1258 179 21 1,4852 10,5
194
В технике наиболее широко применяется триэтаноламин. В больших ко-
личествах он используется для извлечения углекислоты и сероводорода
из промышленных газов (гирботоль-процесс). Мыла жирных кислот и три-
этаноламина являются хорошими эмульгаторами и широко используются
для получения эмульсий минерального масла в текстильной промышлен-
ности, для сверления и резания и холодной обработки металла, моющих
эмульсий, косметики, дезинфекционных средств и в других областях.
Диэтаноламин при нагревании с 70%-ной серной кислотой дает морфо-
лин — циклический иминоэфир. Последний может получаться также дей-
ствием аммиака на р, Р'-дихлордиэтиловый эфир (хлорекс):
СН2—СН2ОН СН2—СН2
hn/ > hn/ \q+H2O
сн2—снаон сн2—си2
СН2—СНаС1 сн2—сн2
о/ +NH3 —► 0^ ^>NH + 2HCI
СН2—СН2С1 СН2—СН2
Этиленимин. Действием тионилхлорида на моноэтанолаадингид-
рохлорид при 80—90° или действием газообразного хлористого водорода
на моноэтаноламин при 160° [311 получают Р-хлорэтиламингидрохлорид,
который под действием водных щелочей при 90—100° переходит в этилен-
имин:
CH2OH-CH2NH2-HC1 + SOC12 80~90- CHaCl-CH2NH2-HCl + SOa+HCl
CH2OH-CH2NH2+HC1 1600 ► CH2Cl-CH2NH2-HCl + lI2O
СП2С1—CH2NHa+NaOH -► СН,—СН2
NHZ
Этот продукт, кипящий при 46° и по строению аналогичный окиси эти-
лена, исключительно ядовит. Применяется он для введения амипоэтильных
групп в органические соединения с реакционноспособным атомом водорода.
Реакции окиси этилена с синильной кисло-
той (акрилонитрилы) [32]. Исключительно большое значение
в использовании окиси этилена в промышленности синтетических и искус-
ственных веществ имеют реакции окиси этилена с синильной кислотой,
протекающие по уравнениям
СН2—CH2+HCN Датализат°Р. СН2ОН-СН2—CN
X. / Этиленцмангидрин
' О (Р-оксипропионитрил)
НОСНа—СН2—CN СН2=СН—CN
Акрилонитрил
с образованием p-оксипропионитрила, дегидратацией которого получают
акрилонитрил.
Акрилонитрил представляет собой бесцветное, как вода, соединение,
кипящее при 77,3°, легко полимеризующееся и исключительно реакционно-
способное, но оно может стабилизироваться посредством олеата меди или
диоксидифенила.
В промышленности присоединение синильной кислоты к окиси этилена
производится в водной среде в присутствии диэтиламина и натриевой щелочи
как катализатора (рис. 116). Реакция идет при температуре около 55°.
Реактор заполняют синильной кислотой и окисью этилена и оставляют в нем
в течение 6 час. при 60°. Отсюда содержимое перекачивают в промежуточный
13* 195
✓
Рис. 116. Схема получения акрилонитрила из окиси этилена.
1 — реакционный сосуд; 2 — промежуточная емкость; .3 — колонна для обезвоживания; V — фильтр-
пресс; .5 — вакуумная колонна; 6 — печь дегидратации; 7 — холодильник; 8 — отстойник; 9— ректи-
фикационная колонна.
Линии.4 I—дпотиламии,- 7I— 50 -паи натриевая щелочь; III — катализатор; IV — окись этилена;
V — синильная кислота; VI — отходящие газы; VII — нейтрализация; VIII — сырой атиленциансид-
рпп; IX. — остаток*, X —сырой акрилонитрил; XI — иода; XII — чистый акрилонитрил.
Рис 117. Получение искусственных веществ из
этилена путем хлорирования.
196
сосуд, где нейтрализуют уксусной кислотой, а затем в специальной
колонне при температуре 110° и пониженном давлении смесь освобождается
от воды. После отстаивания соли отделяют фильтрованием, а сырой этилен-
циангидрин поступает в вакуумную колонну, откуда он получается в чистом
виде с 90%-ным выходом. Дегидратирование может производиться в газо-
вой фазе над окисью алюминия как катализатором при 250—350°. Газы
конденсируются, вода отделяется от акрилонитрила в сепараторе и сырой
акрилонитрил очищается в реактификационной колонне.
Дегидратирование может производиться также в жидкой фазе перегон-
кой оксинитрила в присутствии боксита или окиси магния при 280°.
Другие пути получения акрилонитрила — присоединение синильной
кислоты к ацетилену (см. стр. 247), окисление аллиламина (см. стр. 172)
и реакция этиленхлоргидрина с цианистым натрием с последующей дегидра-
тацией полученного хлоргидрина.
Полимеризацией и сополимеризацией акрилонитрила получают много
технически ценных полимеров. Полимеризацией акрилонитрила полу-
чают исключительное по качеству волокно, поступающее в продажу под
названием «ПАН-волокно», «орлон», «волкрплон» и т. д.
Важнейшим продуктом смешанной полимеризации акрилонитрила
является каучук буна N. Продукт смешанной полимеризации акрилонитрила
ОКИСЬ ЭТИЛЕНА
Г ГТ д р О Л И 3______
Р е а к ц и я с гликоле м_
Полиме ризация________
Реакция с низшими
спиртами
Реакция с низшими_____
спиртами
Реа иди я с алкалицеллю-
лозой
Этиленгликоль
Ди-, три- и полиэтилен гликоли
Полиэтиленоксид
Этилснгликольмоиоэлкиловый эфир (целозольф)
Дпэтиленгликольмоноалкиловый эфир (нарбптол)
Гидроксиэтиловый эфир целлюлозы (цсллозив)
Реакция с NH3
Продукты конденсации с жир-
ными кислотами, вспомогатель-
ные средства для
текстильной промышленности
/
Моно-, ди- и триэтаноламин
: I
Этиленимин । \
Морфолин Промывка кислых газов
Реакция е HCN
+ H-.S _________ _
+алнилфенол
4-высшие спирты
4-угольная кислота
+ 1 моль фенола
Буна К
ПоО Л
Эткленциангидрин — Акрилонитрил^—«•ПАИ, орлон, волкрилон
Акриловый эфир, полиакри-
ловый эфир
Океиэтилмеркаптан 4- тиодигликоль
+НС1
+NaHSO3
•(-бензол + А1С1з___
Катализатор, высокие
температуры
4-анилин ____________
+РОС13
+KCNS_____________
4-Ацетоуксусный эфир
Гидрирован ие _
Алкилполигликолевый эфир (неиипое моющее средство)
Алкилполигликолевый эфир
Сложные эфиры алкилиолнгликолевых эфиров
Этплснгликольмоиофениловый эфир (арозол)
j----- Монохлоруксусная кислота
100%-ный этиленхлоргидрин
Lt гтс* Дихлордиэтиловый эфир
Н2Ы>4
__ - .. CH3NH2 ,,
Иэатионокислыи натрии —------*Метилтаурин
I
Хлорид олеиновой кислоты
Игелон А и Т
р-фепилэтиловый спирт
Ацетальдегид
Оксиэтнланилин, диоксиэтиланплин
РО (ОСН2СН2С1)з, три-(2-хлорэтил)-фосфат
Этпленсульфид
Ацетилпропанол (СН3СОСН2СН2СН2ОН)
Этиловый спирт
Т-газ (90% окиси этилена и 10% СОо)
Картоне, инсектисид (10% окиси этилена, 90% СО2)
Рис. 118. Реакции и применение окиси этилена.
197
(искусственное волокно под названием «динел») на 40% состоит из акрило-
нитрила и на 60% из хлорвинила. Винион N и акрилан представляют собой
смешанный полимеризат акрилонитрила и винилацетата. На рис. 117 пока-
заны возможности получения искусственных веществ па базе этилена хло-
рированием. Важнейшие реакции окиси этилена показаны на рис. 118,
а на рис. 119 известные виды искусственного волокна и источники их полу-
чения.
Полихлорвинил
Полиакрилонитрил
Полиамид
Полиуретан
Полиэфир
Полипропилен (Монтекатини)
Сополимер 60% хлорвинила и 40% акрило-
нитрила
Сополимер 85% акрилонитрила и 1 fjo/o винил-
а цетата
Сополимер 85% хлорвинилидена и 15% хлор-
винила
Сополимер хлорвинила и винилацетата
Сополимер стирола и изобутена
Ре Се
ПАН, орлон, волкрилон
Найлон 66, перлон (най-
лон 6)
Перлон U
Терилен, дакрон
Мопрен
Винион N, динель
Акрилан
Саран, велон
Винион
S-полимер
Рис. 119- Наиболее известные синтетические волокна.
Е. ГИДРОХЛОРИРОВАНИЕ ОЛЕФИНОВ
До сих пор процесс гидрохлорирования олефинов представлял большой
интерес только в связи с получением хлористого этила. Этот продукт, полу-
чаемый хлорированием этана, применяется в больших количествах как ком-
понент этиловой жидкости.
Хлористый водород присо-
единяется к этилену согласно
реакции
СН2=СН2 + НС1 -> СН2С1-
— СН3 + 13,4 ккал/моль
Для получения хлористого
этила в промышленных усло-
виях сухой этилен и сухой
хлористый водород в примерно
эквимолекулярных количест-
вах, при 35° и 2,5—3,0 ат
подают в реактор. Реакция
Рис. 120. Схема получения хлористого этила из
этилена и хлористого водорода.
1 — реактор; 2 — испаритель; 3 — катализатор; 4 —
колонна для промывки водой; 5 — дистилляционная
колонна.
Линии: I — этилен; II — хлористый водород; III —
катализатор, растворенный в хлористом этиле; IV —
свежий катализатор; V — отработанный катализатор;
V/ — вода; VII—разбавленная соляная кислота; VIII—
хлористый этил; IX — остаток.
идет в присутствии хлористого
алюминия, растворенного в
хлористом этиле (рис. 120).
Образовавшийся хлористый
этил испаряется [33].
Газ, состоящий из хлори-
стого этила, этилена, хлори-
стого водорода и инертных
газов (метан, этан и др.), промывается водой для удаления следов
хлористого водорода, а затем очищается перегонкой. В испарителе отде-
ляются катализатор и высококипящие полимерные продукты, которые
возвращаются в реактор. Часть хлористого алюминия выводится из про-
цесса и заменяется свежим.
198
Ж. ГИДРАТАЦИЯ ОЛЕФИНОВ В СПИРТЫ
Гидратация низкомолекулярных олефинов, особенно этилена, пропена
и бутенов, в спирты может производиться двумя способами. При непрямой
гидратации олефин обрабатывают серной кислотой, при этом образуется
алкилсульфат, который затем гидролизуется с образованием спирта и сер-
ной кислоты:
R—СН=СН, -Н2-РО-4- R—СН—СН3 R—СП—C1T3+H2SO4
I I
OSO2OH он
Так как для присоединения серной кислоты к олефину требуется более
концентрированная кислота, чем остающаяся после гидролиза, то кислоту
перед возвращением в процесс необходимо концентрировать. Это довольно
дорогое мероприятие вследствие высокой стоимости энергии.
ВтороГ! способ гидратации олефинов в спирты заключается в прямом
каталитическом присоединении воды по олефиновой двойной связи. В этом
процессе олефин (этилен) вместе с водяным паром при высоких темпе-
ратуре и давлении пропускается над соответствующим катализатором,
например фосфорной кислотой, нанесенной на кизельгур, активированный
уголь или асбест. Процесс прямой каталитической гидратации представляет
собой равновесный процесс, поэтому при однократном пропуске компонен-
тов реакции через печь только небольшой процент олефинов превращается
в спирты, так что требуется вести процесс с многократной циркуляцией
реагирующих веществ, требующей довольно значительных затрат энергии.
Несмотря на это процесс прямой гидратации все же дешевле.
а) Непрямая гидратация
При присоединении серной кислоты к олефинам, имеющим общую фор-
мулу Bj—СН = СН—В2, образуются алкилсульфаты, которые после ги-
дролиза дают вторичные спирты.
СП—СП,—В,
/ OSOoOII
в,—сн=сн—h,+h,so4
^Rj—СН2—СН—R,
I
OSO2OII
г, г гы и HgO . R1—СПОН—сп2—r2+h2so4
И £— /11— U1J о—
OSO2OH
Rj—C1I2—CH—R, Rj—CH2CIIOII—R2+H,SO4
1
OSOoOH
Из разветвленных олефинов получаются исключительно третичные
спирты
R R /OSOoOH R ОН
\c-=Clb.+II,SO4 -> /-С—СП3 - ^с/ +H,SOj
r' Ji R CII3
Описанным выше процессом только из этилена образуется первичный
спирт — этиловый.
Легкость, с которой протекает реакция присоединения серной кислоты
к олефинам, увеличивается от этилена к гексенам. Особенно гладко
199
проходит реакция с изоолефппамп, например с изобутеном. Тогда как для
присоединения серной кислоты к этилену требуется 98%-пая кислота,
высокие температуры, а желательно и повышенное давление, присоединение
серной кислоты к изобутену идет гладко уже с 65 %-ной кислотой при 0°.
Для каждого олефина должна быть точно установлена наиболее целе-
сообразная концентрация кислоты, так как применение слишком концентри-
рованной кислоты вызывает полимеризацию. Если, например, изобутен
обрабатывать серной кислотой при условиях, оптимальных для пропена или
особенно этилена, то это приведет к полимеризации п осмолению. Для про-
пена и //.-бутена наплучшей концентрацией серной кислоты следует считать
85%-пую.
Способ непрямой гидратации олефинов в спирты играет в настоящее
время еще очень большую роль в нефтехимической промышленности. Изо-
пропиловый спирт получают почти исключительно непрямой' гидратацией'
пропена через изопропплсульфат. Значительная часть общей продукции
этилового спирта из этилена получается способом непрямой гидратации.
Чтобы дать представление о количестве спиртов, производимых только
в США, укажем, что в 1955 г. там было получено 620 тыс. т этилового
спирта, причем около 80% из этилена, одна пятая часть этого количества
была получена прямой гидратацией [34]. Производство метилового и изо-
пропилового спиртов составило 630 тыс. т и 450 тыс. т соответственно.
Получение этилового спирта непрямой ги-
дратацией этилена [35]. Процесс протекает в четыре стадии:
1) абсорбция этилена серной кислотой с образованием этилсульфата;
2) гидролиз этилсульфата в спирт и серную кислоту;
3) выделение спирта из разбавленной серной кислоты;
4) восстановление концентрации разбавленной серной кислоты.
Абсорбция этилена производится 97—98%-ной кислотой при 80—90е и
давлении 35 ат. Для абсорбции применяют колонны из нержавеющей стали,
процесс ведут в течение времени, необходимого для образования смеси,
состоящей из 50% моноэтилсульфата и 50% диэтилсульфата, что соответ-
ствует расходу примерно 75% от всей введенной в процесс кислоты. Расход
кислоты па 10 л 100%-ного спирта составляет около 13 кг.
Теплота абсорбции при образовании диэтилсульфата равна 23 ккал/моль,
для моноэтилсульфата 35 ккал/моль. Тепло выделяется по высоте колонны
неравномерно, поэтому отвод тепла необходимо точно регулировать.
Колонна работает непрерывно (рис. 121). В верхнюю часть колонны
подается 98%-ная кислота, поступающая из регенерационной установки,
в основание колонны нагнетается этилен. Насыщенная кислота отводится
из низа колонны и поступает в гидролизер, где она разбавляется водой и
где под давлением происходит гидролиз. Отсюда омыленная смесь подается
в дистилляционную колонну, в нижнюю часть которой подают водяной пар
для отдувки органических продуктов от серной кислоты. Здесь они разде-
ляются и серная кислота, имеющая концентрацию 50—60%, поступает на ре-
генерационную установку.
В гидролизере происходит не только гидролиз с образованием спирта,
но также образование эфира в результате действия спирта на диэтилсуль-
фат по уравнению
(C2H5)2SO4+C2H5OH -4 C2H6HSO4+ С2Н6ОС2Н6
Образование эфира можно значительно снизить, если предварительно
отделить диэтилсульфат. Этого можно достигнуть разбавлением водой,
выходящей из абсорбционной колонны смеси, состоящей из концентрирован-
ной серной кислоты, моноэтилсульфата и диэтилсульфата. Последний вы-
деляется в нижиий слой и затем отдельно подвергается гидролизу 30%-ной
серной кислотой. После омыления оба слоя соединяются для дальней-
шей переработки. Эфира в этом случае образуется не более 2%.
200
Выходящие из верха колонны вместе с водяным паром пары спирта и-
эфира промывают разбавленной натриевой щелочью и водой, конденсируют,
а затем подвергают переработке в специальных колоннах для получения
чистых продуктов.
Непрямая гидратация пропена в изопропи-
ловый спирт. Одно из наиболее значительных мест в ряду алифати-
ческих спиртов занимает изопропиловый спирт, первый из спиртов, полу-
ченный синтетическим путем — гидратацией олефина (петрохол). Большая
часть изопропилового спирта применяется для получения ацетона. Важней-
шие возможности применения изопропилового спирта показаны на рис. 122.
Рис. 121. Схема получения этилового спирта из этилена непрямой гидратацией посред-
ством серной кислоты.
J — абсорбционная колонна; 2 — гидролизирующая установка; ,3 — разделительная колонна; —
колонна для промывки щелочью; 5 — установка для концентрации серкой кислоты; 6 — колонна для
отгонки эфира; 7 — колонна для отгонки спирта.
Линии: 1 — этилен и этан; II — серная кислота; III — в отопительный газ; IV — вода; V — пар;
VI — 50—60 %-ная серная кислота; VII — 98 %-ная серная кислота обратно в абсорбционную колонну;
VIII — натриевая щелочь; IX — отработанная щелочь; X — этиловый эфир; XI — этиловый кспирт,
96 %-ный.
ИЗОПРОПИЛОВЫЙ СПИРТ
Дегидрирование________
Этерификация
Этерификация
_4- CS2 4- NaOH ______
4- метиловый спирт
Ацетон
Растворители для нитроцеллюлозных лаков
Диизопропиловый эфир (антидетонатор)
Ксантогенаты (флотационные реагенты)
Антифриз
Присадка к бензинам для предотвращения обле-
денения карбюратора зимой
--------------------- растворитель для лаков, красок, нитроцеллю-
лозы, антисептик вместо этилового спирта и др.
Рис. 122. Реакции и возможности применения изопропилового
спирта.
Непрямое гидратирование пропена в изопропиловый спирт при помощи
серной кислоты может быть осуществлено двумя способами с помощью кон-
центрированной и разбавленной кислоты.
Гидратация концентрированной кислотой. 20—24%-ный
пропен, как он получается из газов стабилизации крекинг-установок, сжи-
мается до 8—10 ат и абсорбируется в условиях противотока при 20° 92 %-ной
серной кислотой. После окончания экстракции экстракт направляют в раз-
делитель, где в виде масла отделяется верхний слой. В отличие от процесса
абсорбции этилена в процессе для пропена на отдельных тарелках колонны,
кроме серной кислоты, применяют еще фракцию минерального масла, облег-
чающего абсорбцию олефинов серной кислотой. Часть этого масла вместе
с серной кислотой поступает из абсорбционной колонны в разделитель и
оттуда возвращается в колонну. Применение абсорбционного масла на от-
дельных тарелках колонны облегчает также переработку низкопроцентного
пропена.
201
Покидающий абсорбер экстракт содержит иа 1 моль серной кислоты
около 1,1—1,3 моля пропена. Для получения 10 л 100%-ного изопропилового
спирта расходуется около 12 кг серной кислоты.
Отвод тепла с отдельных тарелок должен быть так отрегулирован, чтобы
температура везде поддерживалась около 20° в целях избежания значитель-
ных потерь олефина и полимеризации его. Из отстойника экстракт поступает
в освинцованный гидролизер, снабженный турбосмесителем, куда добавляется
вода, чтобы образовался примерно 40%-ный раствор. После четырехчасо-
вого гидролиза при 50° спирт и образовавшийся эфир отгоняются в колонне
с водяным паром, после чего смесь паров воды, спирта и эфира промывается
разбавленной щелочью и конденсируется. Конденсат разбавляют таким ко-
личеством воды, чтобы образовался примерно 25%-ный раствор, который
оставляют отстаиваться 1 — 2 дня для отделе-
ние. 123. Схема получения
изопропилового спирта спо-
собом со слабой кислотой.
I — абсорбционная колонна,,
без тарелон; 2 — колонна для
омыления и последующего раз-
деления. Линии: I — пропан-
нропен; II —сырой 85 %-ный
спирт на переработку; III —
паровой нагрев (открытый);
IV — 70 %-нан серная кислота .
пня полимеризата. Затем его подвергают -ректи-
фикации. В качестве головного погона полу-
чают около 2% объемн. изопропилового эфира
и легколетучих полимеров. Спирт, отбираемый с
тарелок первой колонны, поступает во вторую
колонну с 50 тарелками, откуда отгоняется в
виде азеотропной смеси с водой, с выходом 85—
90% от теоретического, считая на израсходован-
ный пропен. Азеотропная смесь содержит 91%
изопропилового спирта и кипит при 80,4°. Усло-
вия работы установки аналогичны описанным
для получения этилового спирта (рис. 121).
Гидратация разбавленной кислотой [35а].
Пропен при температуре около 65° и давлении
25 ат абсорбируется 70%-ной серной кисло-
той (рис. 123). Выходящая из абсорбционной
колонны кислота поступает во вторую, работаю-
щую при пониженном давлении колонну, из ко-
торой после добавки некоторого количества воды
отгоняется изопропиловый спирт. Вследствие
добавки воды концентрация серной кислоты
поддерживается на уровне 70%. Свободная от спирта кислота снова отка-
чивается в первую колонну.
Абсорбционная колонна не имеет тарелок, высота ее на больших уста-
новках достигает 14 м. Колонна заполняется 70%-ной серной кислотой,
в которую через газораспределитель подается газ. В колонне серная кис-
лота насыщается до степени, соответствующей примерно 1 молю пропена на
1 моль серной кислоты. Из газа, содержащего 25% пропена, последний извле-
кается в такой степени, что в остатке сохраняется всего около 6% пропена.
Этот остаточный газ поступает в колонну с колпачковыми тарелками, в кото-
рой он в условиях противотока приводится в контакт со свежей серной кис-
лотой, выходящей из колонны, где происходит омыление (иа схеме не пока-
зана). Таким путем удается извлечь при помощи серной кислоты последние
остатки пропена (около 1%). В колпачковой колонне серная кислота насы-
щается до уровня, соответствующего 0,2 моля пропена на 1 моль серной
кислоты, и направляется в бестарельчатую колонну. Отсюда экстракт по-
ступает в колонну, работающую при остаточном давлении 0,2 ат, где про-
исходит отделение спирта. Собственно в этой колонне происходят омыление
и отгонка изопропилового спирта, который направляется далее в конденса-
тор, откуда выходит 85%-ной крепости. Отделившаяся от спирта вода воз-
вращается в разделительную колонну.
Гидратация бутена. Гидратация бутена производится таким
же способом, как и гидратация пропена. Применяются приблизительно
80%-ная серная кислота и давление немного выше атмосферного, температура
40-50°.
202
Для поддержания необходимой температуры отдельные тарелки абсорб-
ционной колонны снабжены охлаждающими змеевиками. Строгое соблюде-
ние температурного режима в данном случае особенно важно по причине
высокой склонности этого олефина к полимеризации.
В ходе дальнейшей переработки смесь алкилсульфата и серной кислоты
разбавляют на 30% водой. Спирт и эфир отгоняют с водяным паром. Вос-
становленную до прежней концентрации серную кислоту используют, как
уже было описано ранее.
Из н-бутана получается втор-бути л оный спирт, кипящий при 99,5°.
Он является исходным материалом для получения метилэтилкетона.
Если в распоряжении имеется только смесь бутенов в том виде, как полу-
чается с установок дебутанизации крекинг-заводов, в которой содержится
также изобутен, то в процесс переработки должны быть внесены изменения,
предусматривающие предварительное удаление изобутена, так как при тех
условиях, которые необходимы для гидратации бутена, неизбежна сильная
полимеризация изобутена.
Изобутен из Б—Б фракции выделяют обработкой ее 65%-ной серной
кислотой, которая не реагирует с н-бутеном, а с изобутеном при 0° дает
трет-бутилсульфат. Из оставшейся части углеводородов С4 при помощи
аммиачно-щелочного раствора хлористой меди выделяется бутадиен, содер-
жащийся в газах в небольшом количестве. В остатке получают газ, из кото-
рого можно получить emop-бутиловый спирт.
mpem-Бутиловый спирт получают путем разбавления водой смеси из
65 %-ной серной кислоты и mpem-бутилсульфата с последующей отдувкой
спирта водяным паром. Конденсат состоит из азеотропной смеси 78% mpem-
бутилового спирта и 22% воды. Азеотропная смесь кипит при 81°.
На схеме рис. 124 представлено получение обоих бутиловых спиртов
из Б — Б-фракции крекинг-газов. Из амиленовой фракции крекинг-бензина
соответствующий спирт можно получить аналогичным способом через амил-
сульфат. На рис. 125 показаны важнейшие возможности применения втор-
и mpem-бутиловых спиртов.
С4-фр,акция (В — 13-фракция)
60 — 65%-ная серная кислота
I I
Абсорбция изобутена при 0 — 5°
под давлением или при —6 4—7°
без давления
Разделение слоев отстаиванием
Нижний слой — трет-бутнлсернля
кислота
Вода
I
Гидролиз при
0°
Отстой-----------------
*
Слой полимеризата
I
Верхний слой — Б — В-фракция,
свободная от изобутена
I
I
Отделение бутадиена аммиачно-ще-
лочным раствором хлористой
меди (1)
Водный спой
Бутадиен
I
Абсорбция бутена
йО^Ы-ной серной
кислотой
| Вода
I
Гидролиз
Перегонка
с водяпым паром
Перс он на
водяным паром
Разбавленная
серная кислота
I
Разбавленный водный
раствор mprm-бути-
лового спирта
I
Ректификация 7 8%-ной
азеотропной смеси с во-
дой при 81°
1
Разбавленная
серная кислота
1
Ректификация
спирта в виде азеот-
ропной 72 ,7%,-пой
смеси при 87,5°
Рис. 124. Схема получения третичного и вторичного бутиловых спиртов
из бутенов Б—Б-фракции.
203
/ГГШ*-ПУТИЛОВЫ» СПИРТ
Дегидри роваппе
Эстерпфикация
Т1’1!Т-НУ 111ЛОВЫЙ СПИРТ
Д ег и др а тацпя_______
Метилэтилкетон
Растворители дли лаков
Компоненты для тормозных жидкостей и
растворителей
Изобутен
Компоненты алкилирования
Компоненты для растворителей, денату-
рирующие добавки
Рис. 125. Реакции и применение етор- и лпрел?г-бутиловых
спиртов.
Непрямая гидратация олефинов может осуществляться также периоди-
ческим способом. Так, например, по способу Рейнско-Прусского акционер-
ного общества в стационарных условиях получают изопропиловый и втор-
бутиловый спирты. Для этого смесь фракций С3 и С4 с общим содержанием
олефинов около 30% при температуре 40° смешивают в автоклаве с
75 %-ной серной кислотой. Молярное отношение кислоты к о'лефипам соста-
вляет 3 : 2. Продолжительность реакции 1 час. За это время превращение
пропена протекает практически на 100%, бутена на 29%. Около 30% бутенов
дают при этом полимерные продукты.
По окончании реакции верхний слой, содержащий пропан, бутан и
продукты полимеризации, отделяется. Сернокислотный слой, содержащий
алкилсульфаты, настолько разбавляется водой, чтобы в результате образо-
валась 30 %-пая серная кислота. Гидролиз и выделение спиртов производятся
непрерывным способом. Ректификацией на ряде колонн из конденсата выде-
ляют изопропиловый и emop-бутиловый спирты и соответствующие эфиры.
Важнейшие физические свойства чистого изопропилового спирта
Температура замерзания, “С............................. —85,8
Температура кипения при 760 мм рт. ст., °C............. 82,4
dt/dp, spadjMM ........................................ 0,033
Давление паров при 20°, мм рт. ст. .................... 32,4
Плотность при 20“...................................... 0,7887
Плотность при температуре кипения...................... 0,7283
Вязкость при 20°, пуазы................................ 0,02431
Температура вспышки (закрытый тигель), 'С.............. 11,7
Коэффициент проломления при 20°....................... 1,37757
» » » 15°.......................' 1,37925
Критическая температура, °C............................
Критическое давление, ат . . . ........................ 53,0
Скрытая теплота при температуре кипения, ««л/,1........ 160
Теплота сгорания, к,ал!г ................... 7970
Теплоемкость при 0°, кал/;: . ........... . 0,563
» » 30° • • . ... 0,677
» » 50° .................................. 0,740
Поверхностное натяжение при 20°, дн/см................. 21,7
Диэлектрическая постоянная при 20“..................... 26,0
Упругость паров при 100°, ат........... ............... 2,127
Азеотропная смесь изопропилового спирта с водой кипит при 80,4° и со-
держит 91% спирта. Во многих случаях эта азеотропная смесь может под-
вергаться дальнейшей переработке.
б) Прямая гидратация [36]
Реакция С2Н4 + HtO С2Н5ОН + 10,9 ккал./моль представляет собой
экзотермическую равновесную реакцию. Чтобы равновесие было сдвинуто
в желательную сторону, реакцию следует вести при возможно низкой тем-
пературе и высоком давлении. Практически, однако, температуру реакции
нельзя поддерживать на уровне, лежащем ниже некоторого определенного
значения, так как в противном случае скорость реакции и присоединение
204
воды будут столь малы, что не представят уже технического интереса.
С другой стороны, температура при соответствующем давлении не должна
быть слишком низкой, так как для реакции безусловно необходим водяной
пар. При низкой температуре и относительно низком давлении процесс
можно вести только с применением достаточно активного катализатора.
Первые практические результаты по прямой гидратации этилена были
получены в США фирмой Шелл Кемикал Корпорейшн [37]. Способ этой
фирмы состоит в том (рис. 126), что этилен и водяной пар в молярном отно-
шении 0,6 : 1 при 300° и 70 ат давления пропускают над катализатором,
состоящим из фосфорной кислоты, нанесенной на диатомовую землю. На
1 м3 катализатора в час может быть пропущено 1800 м3 газа (приведенного
к нормальному давлению). Этилен должен быть 97 %-ной чистоты. Превра-
щение этилена за один проход составляет около 4,5%, что вынуждает ра-
Таблица 83
Температура кипения и состав азеотропных смесей
этилового спирта с важнейшими органическими
растворителями __________________________________
Компонент азеотропной смеси Содержание в смеси, % вес. Температу- ра кипения, °C
Бензол 68,0 68,2
Сероуглерод 91,0 42,4
Четыреххлористый углерод 84,2 64,9
Хлороформ 93,0 59,4
Циклогексан 69,5 64,9
Этилацетат 69,0 71,8
н-Гептап 52,0 72,0
н-Гексан 79,0 58,6
Метилэтилкетон 66,0 74,8
Метилпроппонат 48,0 93,2
Перхлорэтилен 19,0 78,0
Толуол 32,0 76,6
Трихлорэтилен 73,0 70,9
Рис. 126. Схема получения
этилового спирта прямой ка-
талитической гидратацией эти-
лена.
1—предварительный подогреватель:
2 — реактор с фосфорнокислым ка-
тализатором; з— холодильник; 4—
промывочная колонна.
Линии: I —свежий этилен; II —
во'дпноп пар; III —циркулирую-
щий этилен; IV — разбавленный
спирт; V — пары спирта; VI — от-
ходящие газы; VII — разбавлен-
ный спирт па переработку.
ботать с многократной циркуляцией. Расход этилена возмещается подачей
в процесс свежего этилена. Когда вследствие постепенного обогащения
инертными составляющими содержание этилена в газах циркуляции дости-
гает 85%, часть этих газов выводится из системы. Отходящий 85%-ный эти-
лен перерабатывается на установке Линде в чистый этилен.
Выходящий из печи под давлением 70 ат продукт при охлаждении
выделяет часть жидкости, состоящей из разбавленного спирта. Остаток содер-
жащихся в газе паров спирта выделяют из газа промывкой водой. Конден-
сат и промывочные воды соединяют и от смеси отгоняют этиловый спирт.
Полученный таким образом сырой продукт не выдерживает еще перманга-
натной пробы, таг; как содержит некоторое количество ацетальдегида и кро-
тонового альдегида. Продукт подвергают гидрированию над никелевым ката-
лизатором, при котором легко восстанавливаемые соединения гидрируются.
Этиловый спирт представляет собой бесцветную жидкость с температу-
рой кипения 78,3°. Температура кипения 96%-ного спирта 78,2°. Этиловый
спирт образует азеотропные смеси со многими органическими растворителями.
В табл. 83 приведены некоторые такие смеси. Обезвоживание технических
спиртов может осуществляться азеотропной перегонкой. Для этой цели при-
меняют или бензол, который образует тройную азеотропную смесь из
18,5% вес. спирта, 74,1 % бензола и 7,4% воды, кипящую при 64,9°, или три-
хлорэтилен, дающий тройную азеотропную смесь, содержащую 64,9 объемн.
части трихлорэтилена, 6,8 объемн. части воды и 23,8 объемн. части этилового
-спирта и кипящую при 67,2°.
205
Этиловый спирт является одним из наиболее важных растворителей.
Значительная часть его перерабатывается в химической промышленности
в ацетальдегид, уксусную кислоту, уксусный ангидрид, хлористый этил,
этилацетат и др. На рис. 127 показаны основные направления использова-
ния этилового спирта.
этиловый спирт
Пропускание над
бокситом
Дегидрирование
+НС1_________
4С12
Д с ги дратация
Этерификация
Эстерифинация
Рис. 127. Реакции и
Бутадиен
Бутиловый спирт
Z
/ 2-этилгексиловый спирт
Ацетаatдеги'д и роиаводпые
\ Уксусный ангидрид
Уксусная кислота
Хлористый этил
Хлораль -----* DDT
Этилен
Диэтиловый эфир
унсусноэтиловый яфир и другие растворители
Растворители, антиобледнители и др.
пути использования этилового спирта.
в) Дегидрирование вторичных спиртов в кетоны
Общие сведения. Изопропиловый и втпор-бутиловый спирты
в основном применяют для получения каталитическим дегидрированием
соответствующих кетонов — ацетона и метилэтилкетона. Производство аце-
тона в США в 1956 г. составило 250 тыс. т, из которых 230 тыс. т
были получены из изопропилового спирта.
В последнее время все возрастающие количества ацетона получают
из кумола, также постоянно растет производство ацетона из продуктов
окисления газообразных парафиновых углеводородов.
Ацетон является исходным материалом для получения ряда продук-
тов, как, например, диацетопового спирта, являющегося превосходным рас-
творителем для ацетата целлюлозы. Окись мезитила, метилизобутилкетон
и др. являются растворителями для искусственных веществ и лаков. На
рис. 128 показано, какими возможностями располагает нефтехимическая
промышленность для получения важнейших растворителей, на рис. 129—
то же в отношении мягчителей и пластификаторов. На рис. 130 приведена
принципиальная схема получения растворителей и пластификаторов на ос-
нове нефти и природного нефтяного газа.
Ацетон является также сырьем для получения ангидрида уксусной кис-
лоты. При нагреве до 700—800° ацетон разлагается на кетен и метан:
СН3СОСН3 -+ СН4+СНг=С=О
Пропуская кетен через ледяную уксусную кислоту, получают уксусный
ангидрид. Последний потребляется в больших количествах в производстве
ацетилцеллюлозы. Кипящий при —41° кетен легко димеризуется в дикетен,
кипящий при 127° и реагирующий со спиртом с образованием ацетоуксус-
ного эфира, а с анилином дающий анилид ацетоуксусной кислоты.
Ацетон служит также исходным продуктом для получения эфиров
метакриловой кислоты, продукты полимеризации которых находят широчай-
шее применение.
206
ИСХОДНЫЙ МАТЕРИАЛ
Хлорирование
ПРИГОДНЫЙ ГАЗ Метан »
Превращение
в смеси СО и Н2 Нитрование —-
Пропан
Окисление
Окисление
Бутан | ~
Хлорирование
Пентан I
Диметилсульфид —» Диметилсульфоксид.
Хлористый метил, хлористый метилен, хлороформ,
четыреххлористый углерод
л. NHg „ СО -т _
Метиловый спирт —--* Диметиламин •-» Диметилфор-
мамид
Изобутиловое масло (н-ттропиловый, изобутиловый,
изоамиловый, изогексиловый, изогептиловый спирты)
Этиловый, н-проииловый и н-бутиловый спирты, аце-
тон, метилэтилкетон, ацетальдегид, уксусная кислота
Синтез Фишера-Тропша с железным катализатором
(гидрокол-процесс)
Нитрометан, нитроэтап, 1- и 2-нитропропаны и их
питросиирты и ацетаты
а) В газовой фазе-, спирты, кетоны, альдегиды, кислоты
б) В жидкой фазе: главным образом уксусная кислота
а) В газовой фазе: спирты, кетоны, альдегиды, кислоты
б) В жидкой фазе: главным образом уксусная кислота
Амилхлорид, амиловый спирт (пентазол) и его ацетаты
ГАЗЫ КРЕКИНГА И ПИРОЛИЗА
Гидратация
Хлорирование
Хлоргидринирова-
ние, окисление Гидроформилиро- вание Гидратация j
Этилен Пропен
Гидроформилиро-
вание Гидратация
Гидроформилиро-
вание Гидратация
Ацетилен Хлорирование |“*
> Реакция с NH3
Ацетальдегид—> Кротоновый альдегид-— >
। —* Г.утанал —* Бутиловый спирт
| 2-метилгексанол
Этиловый спирт, диэтиловый эфир
Хлористый этилен
I—► Дигликольмоноалкиловый эфир и его
ацетаты
Окись этилена—* Гликольмоноалкиловый эфир
I—> Глинольацетат
н-Проииловый спирт
t
I-----
Пропионовый альдегид—* Пропионовая кислота
_----- emop-Пропиловый спирт, диизопрониловый эфир.
Ацетон—* Диацетоповый спирт—> Окись мези-
тила —2-* Метилизобутилкстон — Метилизобутил-
карбивол
•н-, Изобутиловый спирт
н-, Изомасляный альдегид
и-, Изомасллная кислота
бтпор-Бутилопый спирт —Метилэтилкетон
Амиловый альдегид—> Амиловый спирт
Ацетальдегид —* см. выше
Тетрахлорэтан —* Трихлорэтилен
Пептахлорэтан —* Перхлорэтилсп
Ацетонитрил
СПЕЦИАЛЬНЫЕ НЕФТЯНЫЕ ФРАКЦИИ
Очищенная фракция прямой перегонки
Циклогексан из специальной нефтяной фракции
Лаковый бензин 135 — 200°
Петроле,йный эфир 40 — 60°
Эталонный бензин 130 — 200°
Специальный бензин 130 —180°
|->Циклогенсанол
Окисление
I -*1Динлогексанон
|Специальные нефтяные франции для получения арома-
тических посредством каталитического риформинг-
процесса
4-Пропеп
Бензол----------* Кумол
Окисление
Расщепление
—* Фепол + Ацетон
Циклен -сксамол
I
Циклогексанон
Рис. 128. Получение важнейших растворителей нефтехимическим способом.
207
ИСХОДНЫЙ МАТЕРИАЛ
Парафин
Хлорирование
Вы со ко клир к ронин Hi.tii парафин
Парафиновые углеводороды
среднего масла
Сульфохлорировакне
Превращение в мсрзоляты —- Мезамолл
Крезол Гиз продуктов кре- Реакция с POCI3
•кинга (крезиловые кислоты)
—* Трикреэилфосфат
Этилен
Хлор гидринированне
или окисление
Окись этилена---► АТоло-, ди- и триэти-
ленгликоли и их сложные и простые
эфиры
Изогептен
.Диизобутсн
Реа।«ция Ройлена____
Гидроформилирование
Октанол 1 Эфиры фталевой, адипиновой
Нонанол | и себациновой кислот
Окисление бензола
Ароматические от катали- тического риформинга Алкилирование бензола
Окисление
о-ксилола
Малеиновый ангидрид —- диэфиры
диацетоновый спи т
Кумол-----—> Фенол 4- Ацетон
диацетат
t
2-мети л-2,4 —
пеитадиол«—
I
Ц иклогексапол
Адипиновая кислота
Фталевая кислота------►Фталат
Бутадиен
Дибутадиен Na + СО-2
Иэосебациновая кислота
-----► Д и эф и р ы
Ацетилен
Гидратация
Ацетальдегид-------- Кротоновый
альдегид
I
БУтанал-------- 2-этилгексанол
I
Бутанол
Фталаты и адипаты
Циклогексан
Окисление
— Циклогексанон--------► Адипиновая
кислота
Рис. 129. Получение важнейших пластификаторов нефтехимическим путем.
208
Важнейшим продуктом конденсации ацетона с фенолом является
известный под названием «дйан», димётйл-п,п'-дйоксйдифенилметан (дифе-
нилолпропан)
СН3
СН3СОСН3+2^ Чон —ОН^ S-C-<f ^-ОН+Н4О
сн3
Реакцией этого продукта с эпихлоргидрином получают эпоксидные
смолы (см. стр. 176).
Недавно удалось простым способом конденсировать ацетон с аммиаком
с образованием производных тетрагидропиримидина — новое убедительное
доказательство исключительной реакционной способности ацетона [38].
Введением кетена в ацетон в присутствии небольших количеств серной
кислоты получают изопропенилацетат
СН,-СО—СН3^2 СН,—С=СН2 сн2=с = о сн3—с=сн,
I I
он ососн3
представляющий собой исключительный ацетилирующий агент, так как
в кислой среде он снова расщепляется на ацетон и кетен [39].
Недавно перхлорированием ацетона был получен гексахлорацетон,
который взаимодействием с эквимолекулярным количеством едкого натра
расщепляется на хлороформ и уксуснокислый натрий [40].
Важнейшие реакции и возможности превращения ацетона показаны
на рис. 131.
Каталитическое дегидрирование изоп р о п и-
лового спирта. Каталитическое дегидрирование изопропилового
спирта осуществляется пропусканием его паров в смеси с водородом при
400° над пемзой, обработанной окисью цинка или сульфидом цинка над
латунью или железно-медно-цинковым сплавом и т. д.
По уравнению
СН,—СНОП—CH3;KaTa4JI™T°S СН3-СО—СН3+Н2—15,9 ккал/мцль
образуются ацетон и свободный водород. Таким же образом идет дегидрирова-
ние вшор-бутилового спирта. Схема установки показана на рис. 132.
Изопропиловый спирт переводят в парообразное состояние при такой
температуре, что водород увлекает с собой примерно равный объем паров
спирта. Эту газовую смесь пропускают над катализатором. Дегидрирование
втор-бутилового спирта проводится при несколько более низкой темпера-
туре, примерно при 350°. Пучок заполненных катализатором трубок омы-
вается горячими газами, поддерживающими необходимую для дегидрирова-
ния температуру. Отходящие газы реакции проходят через холодильник,
где конденсируется около 50 % ацетона и 80% метилэтилкетона (на схеме
не показан). Водород в условиях противотока промывают водой и освобо-
ждают таким образом от последних следов кетонов. Когда содержание аце-
тона повышается до 20%, его отделяют перегонкой. Спирт, не вошедший
в реакцию, возвращается в процесс. Водород выходит из процесса 99%-ной
чистоты.
Катализатор должен время от времени регенерироваться выжиганием
отложений кокса в смеси азота с 2% кислорода.
Процесс каталитического дегидрирования сильно эндотермический.
Можно, однако, вести процесс дегидрирования также в присутствии воздуха
по уравнению
СН3—СНОН—СН34—02 —> СН3—СО—СН3 + Н2О + 43 ккал/молъ
14 Заказ 696.
2 09
Нефть
Рис* 130- Схема получения растворителей и пластифи
t t t
210
каторов на базе нефти и природного нефтяного газа.
14*
211
АЦЕТОН
Действие щелочей в
мягких условиях
Гексилен гликоль —- Метилпентадиен —- Диметил-
сульфолан
Диацетоновый спирт
Окпсь мезитила - * Метилизобутилкетоц —► Метил-
изобутилкарбинол
1,3,5-ксиленол
Дейст вие щелочей в
жестких условиях
Изофорон.
1,3,5-кси лидии
Форой
Мезитилен
Гидратация „ - Гидратация л
——--------► Диизобутилкетон----------► Диизобутилкарби-
НОЛ
Триацетопамин
1NH3
Диацетонамин, триацетопамин
Ацетонин (пентаметилтетрагидропиримидин)
HCN
Ацетонциангидрин QMbljeH~^r Метакриловая кислота
I
Метакрилаты
Хлорирование
Хлорацетон
Гексахлорацетон
Трихлоруксусная кислота
Щелочь /
Хлороформ
Пиролиз_______________
700-800°
Реакция с кетеном
Реакция с 2 молями______
формальдегида
Превращение с ацетиленом
Уксусный ангидрид —* Ацетилцеллюлоза
Кетен^
Дикетен —* Ацетоуксусный эфир—►Ацетоуксусный анилид
Изопропенилацетат (кетен-донатор)
2-дцетилпропандиол-1,3
Гидратация п * «
З-метилбутинол-З ——---------- З-метилбутенол-З
„ Дегидратация
Изопрен t-1--------1
Превращение с мерная та-
нами
Окисление „ .
Меркаптали---------► Сульфоны
Диметилбутадиен
Восстановление в
Мягких условиях
Пинакон
Пинаколин
Превращение с фенолом
Дифенилолпропан (диан), бисфенол А—►Эпоксидные смолы
Растворители для ацетилцеллюлозы, ацетилен, лаки, смолы.
Рис. 131. Реакции ацетона.
212
Таблица 84
Важнейшие физические свойства некоторых алифатических кетоиов
Показатели Ацетон Метил- этил- кетон Метил- пропил- кетон Метил- бутил- кетон Метил- амил- кетон
Молекулярный вес 58 72 86 100 114
Температура кипения при 760 мм рт. ст., °C Плотность при 20° 56,1 79,6 102 127,2 150,2
0,7915 0,8050 0,8089 0,8209 0,816
Коэффициент преломления при 20° 1,3587 1,3791 1,3895 1,4024 1,4110 ,
Давление паров при 20°, мм рт. ст. 184,8 77,5 30,0 10,0 3,6
Температура вспышки (закрытый тигель), °C —16,7 -7,2 7,2 22.8 41,1
Поверхностное натяжение, дн)см . 23,7 24,6 25,2 25,5 —
Температура кипения азеотропной смеси с водой, °C Нет азео- 73,4 83,3 86,4 —
Кетона в азеотропной смеси, % вес. тропа 88,7 80,5 84,3 —
Растворимость воды в кетоне, % вес. со 12,5 3,6 2.12 1,5
Растворимость кетона в воде, % вес. со 27,0 6,0 3,5 0,4
протекает сильно экзотермический
процесс, но выход ацетона полу-
чается очень небольшим. В табл. 84
приведены физические свойства не-
которых алифатических кетонов.
г) Присоединение серной кислоты
к высокомолекулярным олефинам
Присоединение серной кислоты
к высокомолекулярным олефинам с
образованием соответствующих суль-
фатов не ставит целью последую-
щее их омыление для получения
спиртов. Его целью является полу-
чение натриевых солей алкилсуль-
фатов, которые, если алкильный
остаток содержит от 12 до 18 угле-
водородных атомов, обладают хоро-
шими капиллярными свойствами и
могут применяться как вспомога-
тельные, моющие и эмульгирующие
средства. Особенно большое число
Рис. 132. Схема дегидрирования изопро-
пилового спирта в ацетон.
1 — испаритель; 2 — печь дегидрирования с ка-
тализатором; з — колонна для промывки водой;
4—ацетоновая колонна; 5—колонна изопропи-
лового спирта.
Линии; I — свежий водород; II — циркулирую-
щий водород; III — водород; IV— циркулирую-
щий пропиловый спирт; V — свежий пропиловый
спирт; VI — вода; VII — ацетон; VIII — изо-
пропиловый спирт.
синтетических моющих средств на
этой основе (тиноль) получают в Англии, Франции и Голландии [41].
Реакция присоединения серной кислоты к высокомолекулярным олефи-
нам проводится в так называемых сульфаторах при низкой температуре и
в условиях непродолжительного контакта (рис. 133). Концентрация серной
кислоты должна составлять минимум 98%. Применяется 15%-ный молярный
избыток кислоты.
По окончании продукт реакции тотчас нейтрализуют 20 %-ной натрие-
вой щелочью, причем образуется паста. Для разрушения диалкилсульфатов
эту пасту нагревают 1 час. Часть нейтрального масла гидротропно раство-
ренного в продуктах реакции осаждают добавкой пропилового спирта, остав-
шуюся часть удаляют извлечением низкокипящей бензиновой фракцией
213
в присутствии спирта. От нейтрального масла бензин отделяется перегонкой
и возвращается в экстракционную колонну.
Экстрагированный, свободный от нейтрального масла раствор алкил-
сульфата освобождается от пропилового спирта в специальной колонне и
иногда подвергается после этого еще
дополнительному отпариванию.
Типоль выпускается в большин-
стве случаев в виде пасты или в рас-
творе. Типоль 410 представляет со-
бой 20—22%-ный раствор, тпполь
710—40%-ный раствор. Сырьем слу-
жит фракция олефинов С12 — С16,
получаемая при термическом кре-
кинге парафина (см. стр. 68).
3. ГИДРОФОРМИЛИРОВАНИЕ
Рис. 133. Схема получения алкилсуль-
фатов (типоль) сульфированием высоко-
молекулярных олефинов.
7 — сульфатор; 2 — нейтрализатор; 3 — гидро-
лизер; 4 — отделитель для нейтрального масла;
& — экстракционная колонна; 6 — колонна для
отгонки бензина; 7—колонна для отгонки спирта.
Линии: I—серная кислота; II—олефины; III—
нейтральное масло; IV — растнор алкилсульфата
в иоде и спирт; V — циркулирующий пропило-
вый спирт; VI — возврат бензина; VII — ней-
тральное масло; VIII — паста типоль; IX —
пропиловый спирт.
ОЛЕФИНОВ (РЕАКЦИЯ РОЙЛЕНА [42])
При совместном действии окиси
углерода и водорода на олефины
под давлением в присутствии кар-
бопила кобальта как катализатора
и при температуре около 150° про-
исходит образование алифатических
альдегидов. Эта реакция, известная
под названием реакции гидрофор-
милирования или по имени ее автора
(Ройлена), имеет исключительно важное значение для нефтехимической
промышленности. Реакция протекает по уравнению
О
RCH2-CH2—С<^
2R—СН=СН2+2СО + 2Н2^ И
RCII—СН3
I °
1 z
Н
с образованием альдегидов, которые при известных условиях могут быть
восстановлены в первичные спирты.
Первичные спирты с различным числом атомов С в молекуле в современ-
ной промышленности алифатических соединений играют особую роль как
промежуточные и целевые продукты.
Для реакции применяют смесь окиси углерода и водорода в отношении
1 : 1. Наивыгоднейшее парциальное давление для каждого из компонентов
равно 100 ат, так что общее давление составляет 200 ат.
Гидрирование альдегидов в первичные спирты в известной мере может
протекать в сочетании с реакцией Ройлена. Оно идет как гомогенная ката-
литическая реакция само по себе и основано на том, что карбонилгидрид
кобальта при определенной температуре и определенном соотношении окиси
углерода и водорода может функционировать как восстанавливающий
агент [43].
Таким образом, в настоящее время, получение первичных спиртов,
исходя из альдегидов, возможно посредством их гидрирования тремя спо-
собами. Во-первых, гидрированием альдегидов в газовой фазе в присутствии
избытка водорода и, например, никелевого катализатора без давления
или под небольшим давлением гетерогенно-каталитической реакцией. Во-
вторых, в дополнение к реакции Ройлена можно по окончании образования
214
альдегидов вывести из зоны реакции смесь окиси углерода — водород и,
подняв давление водорода до 200 ат при 220°, провести гидрирование
альдегидов, являющееся также гетерогенно-каталитическим процессом. Под
действием чистого водорода при 220° из растворенного в продуктах реакции
карбонила-кобальта образуется металлический кобальт, служащий гете-
рогенным катализатором гидрирования.
Новейшим третьим способом является гомогенно-каталитическое гидри-
рование. Оно осуществляется таким образом, что по завершении реакции
гидроформилирования температура повышается до 220°, а понизившееся
в результате реакции давление выравнивается подачей в реактор водорода.
Катализатором гидрирования является растворенный в масле и, следо-
вательно, находящийся в гомогенной фазе карбонил гидрид кобальта. Он
термически диссоциирует на карбонил кобальта и водород. Последний
действует гидрирующе.
Присутствие окиси углерода под давлением 100 ат препятствует раз-
ложению карбонила-кобальта на кобальт и карбонил, а присутствие водо-
рода под давлением 100 ат способствует тому, что из карбонила-кобальта
вновь образуется карбонилгидрид кобальта. Таким образом в этом гомогенно-
каталитическом процессе расходуется только водород.
Исходным материалом для реакции Ройлена, поставляемым нефтехими-
ческой промышленностью, являются высокомолекулярные олефины, как
изогептен и диизобутен, получаемые полимеризацией низших олефинов.
Получаемые из них первичные спирты являются важными компонентами
этерификации алифатических и ароматических дикарбоновых кислот, в пер-
вую очередь адипиновой и себациновой, а также фталевой. Наряду с этим
для реакции Ройлена используются также низкомолекулярные олефины
как этилен, пропен и другие.
В германской промышленности для этой цели используются в первую
очередь содержащиеся в продуктах крекинга олефины от додецена до окта-
децена. Из этих олефинов получают спирты, необходимые в производстве
поверхностно-активных веществ. Очень хорошим материалом являются
также высокоолефиновые продукты синтеза Фишера-Тропша с железным
катализатором. В качестве катализатора может применяться любая кобаль-
товая соль или любое соединение кобальта, которое при температурах
~150° и суммарном давлении окиси углерода и водорода 200 ат спо-
собно образовать карбонил кобальта. В настоящее время работают
чаще всего с растворимыми в масле кобальтовыми солями органических
кислот.
В области механизма реакции гидроформилирования проведены
обстоятельные псследования. Основным катализатором реакции гидро-
формилирования, как установлено в настоящее время, является карбонил-
гидрид кобальта, как и было ранее предложено Ройленом [44]. Различные
теории процессов здесь не рассматриваются.
Карбонилгидрид кобальта, как показано исследованиями Реппе с со-
трудниками [45], представляет собой сильную кислоту, способную подобно
хлористому водороду присоединяться по месту двойной связи с образованием
аддуктов, способных расщепляться на альдегид и кобальткарбониловый
радикал. В ходе гидроформилирования всегда в определенном размере
происходит изомеризация двойных связей, так что даже если исходят из
строго определенных олефинов с двойной связью у конечного атома, альде-
гиды и соответственно спирты получаются со спиртовой группой, располо-
женной ближе к центру молекулы. В присутствии карбонилгидрида кобальта
направление изомеризации связей изменяется на обратное. Равным образом
при использовании олефинов с двойными связями, расположенными иа
некотором расстоянии от конца молекулы, получаются первичные спирты
с гидроксильной группой, стоящей у концевого углеродного атома, так
как двойные связи в течение реакции Ройлена передвигаются от центра
к периферии молекулы.
215
У олефинов, для которых изомеризация связи невозможна, как, напри-
мер, у пропена, образуются все же два спирта, потому что присоединение
формильной группы может происходить по двум направлениям.
Из бутена-1 и бутенов-2 получаются равные количества обоих теорети-
чески возможных амиловых спиртов. Различные метилбутены (изоамилены)
дают при гидроформилировании одинаковый выход каждого из гексиловых
спиртов, что снова подтверждает изомеризацию связей. Ниже дан процент-
ный выход спиртов при гидроформилировании различных олефинов и после-
дующем восстановлении альдегидов [46].
Исходный олефин
Спирты, получающиеся после
восстановления альдегидов
Пропен
Б у тен-1
Бутен-2
Изобутен
Пентен-1
2-метил бутен-1
2-метилбутси-2
З-метилбутеи-1
Гексен-1 . . .
2-метил nei 1 тен-3
/t-бутилового спирта
пзобутилового спирта
/t-амилового спирта
2-метил бу танола-1
/t-амилового спирта
2-метилбутанола-1
60»/0
40%
50%
50%
50%
50%
100% З-метилбутанола-1
50%
40%
10%
50%
45%
5% 2,3-ДИметилбутапола-1
н-гептилового спирта
2-метилгексанола-1 . .
2-этил пентанола-1
2,4-Димети л пентапола-1
5-метилгексанола-1
З-метилгексанола-1
н-гексилового спирта . .
2-метилпентанола-1
2-этилбутанола-1
4-метилпентапола-1
3-метилпентанола-1
50%
30%
20%
30%
40%
30%
После войны реакция гидроформилирования стала применяться и
в нефтехимической промышленности США, и в настоящее время там имеются
большие промышленные установки для получения первичных спиртов
указанным методом.
Так фирма Эссо Стандард Ойл Компани в Батон-Руж получает из изо-
гептена изооктиловый спирт непрерывным способом. Тоже на заводах
Стандарт Ойл оф Индиана. Спирт используется главным образом для полу-
чения диоктилфталата. Диизобутен таким же образом перерабатывается
в изононановый спирт, фталаты и адипаты которого применяются в качестве
пластификаторов.
Низкомолекулярные спирты и альдегиды, как пропионовый альдегид
и н-пропиловый спирт, изомасляный альдегид и соответствующие спирты,
получают гидроформилированием этилена и пропена.
Пропионовый альдегид переводят в пропионовую кислоту или кон-
денсируют с формальдегидом в триметилолэтан, который далее может
перерабатываться так же, как и пентаэритрит:
СН2ОН СН2ОН
О О , | О О | О
СН,—СН2—С^ +2НсГ-^1-’СН,-С-С^ +нс^-> сн,—с—сн2он+нс^
Н . н ) Н н | он
СН2ОН СНоОН
216
ПРОПИОНОВЫЙ АЛЬДЕГИД
Конденсация с
формальдегидом
Окисление
Триметилолатан
Пропионовая кислота-----Пропионат нальция
Реакция с нетоном Пропиоцеллюлоза
Смешанный ангидрид Перегонка , Пропионо-
ацетилпропионилоксид I вый ангидрид
] Уксусный
I ангидрид
I
I
Ацет илцеллюлоза
Рис. 134. Реакции пропионового альдегида.
Н-БУТИЛОВЫЙ СПИРТ
Эстерификация
Дегидрирование
Растворитель для нитроцеллюлозных лаков, смол,
денатурирующее средство
Бутилацетат, растворитель для нитроцеллюлозы
Фталаты и себанаты как пластификаторы
2-этилгексиловый спирт
н-Масляный альдегид
Масляная кислота ^ете?£» Ангидрид
I
I
Бутилцеллюлоа
Компонент для тормозных жидкостей
ИЗОБУТИЛОВЫЙ СПИРТ
Каталитическое
дегидратирование
Эстерификация
Изобутен
Растворитель для нитроцеллюлозных лаков
Растворитель
Компонент для тормозных жидкостей
Рис. 135. Реакции и возможности применения и- и изобутилового спирта.
На рис. 134 показаны важнейшие возможности применения пропионо-
вого альдегида, а на рис. 135 то же для н- и изобутилового спирта.
В промышленных условиях реакция гидроформилирования может
осуществляться двумя способами: или зумпфовым способом, когда катали-
затор находится в виде сусла, или
затором.
В первом случае процесс идет
непрерывно (рис. 136). Полученная
в специальной мешалке паста ката-
лизатора в масле нагнетается в
печь, где осуществляется процесс
гидроформилирования и куда при
орошением с неподвижным катали-
Рис. 136. Схема гидроформилирования
высокомолекулярных олефинов.
1 — заторный сосуд; 2 — реактор для гидрофор-
минирования; з — отделитель; 4 — печь для гид-
рирования; Д — фильтр.
Линии'. I — олефины; II — катализатор; III —
циркулирующий водяной газ; IV — свежий во-
дяной газ; V — циркулирующий водород; VI —
циркулирующий катализатор; VII — свежий
водород; VIII — сырой спирт на перегонку.
•температуре 150—160° и под давлением 200 ат поступают окись углерода,
водород и олефины. После прохождения через печь продукты реакции осво-
бождаются от смеси окиси углерода с водородом и подаются в так называе-
мую ступень гидрирования, где при температуре 200—220° и под давлением
200 ат водорода альдегиды гидрируются в первичные спирты. На обеих сту-
пенях реакции находящийся в виде суспензии катализатор поддерживается
во взвешенном состоянии или при помощи смеси окиси углерода и водо-
рода, или водородом. По выходе со ступени гидрирования катализатор
отделяется от спирта фильтрованием и возвращается в емкость, где гото-
вится паста.
Этот процесс применен в Германии для получения высокомолекулярных
спиртов. Для синтеза используют узкую фракцию, кипящую в интервале
15—20°. Крекинг-олефины (см. стр. 68) всегда смешаны с довольно значи-
тельным количеством кипящих в тех же пределах парафиновых углеводо-
родов.
В результате перевода олефиновой составной части в спирты разница
в температурах кипения спиртов и парафиновых углеводородов становится
настолько значительной, что последние уже могут быть выделены из сырой
смеси спиртов путем перегонки в виде головного погона. Если для реакции
гидроформилирования применяется чрезмерно широкая фракция, то тем-
пература кипения спиртов, полученных гидрированием низкокипящих
олефинов, будет совпадать или накладываться на температуру кипения
наиболее высококипящих из содержащихся в смеси парафинов.
Способ работы с неподвижным катализатором в том виде, как он получил
свое развитие в Германии (людвигсгафенский «способ орошения»), заклю-
чается в следующем: смесь олефинов подается сверху в реакционную печь,
где она в условиях противотока приходит в контакт с поступающей снизу
смесью окиси углерода и водорода при 200 ат и 160—180°. Катализатор
состоит из нанесенного на пемзу кобальта (1 — 2% Со) и получается про-
питкой пемзы раствором азотнокислого кобальта с последующим восста-
новлением в струе водорода.
Продукты реакции, выходящие из печи, где идет гидроформилирование,
и состоящие главным образом из альдегидов, после отделения смеси окиси
углерода и водорода подаются во вторую печь, заполненную только пемзой,
где при температуре 120° и давлении 180 ат водорода растворенный в про-
дуктах реакции кобальткарбонил осаждается на пемзе в виде тонко распы-
ленного кобальта. После накопления в этой печи примерно 10% металли-
ческого кобальта катализатор регенерируется. Потери кобальта в первой
печи пополняются добавкой маслорастворимых кобальтовых мыл. Угле-
водородно-альдегидная смесь после отделения кобальта гидрируется и
разделяется на составляющие ректификацией.
На базе этого метода построено в настоящее время получение изоок-
танола из изопентена. Отделение кобальта от продуктов гидроформилиро-
вания возможно простым нагреванием до 150—160° под давлением 7 —10 ат
водорода. Кобальт затем отфильтровывается в виде осадка. Для восста-
новления альдегидов в спирты можно, кроме никеля, использовать также
хромит меди или устойчивый против действия серы катализатор, состоящий
из сульфида никеля и сульфида вольфрама. В этом случае восстановление
(ведут при 200° и 200 ат давления водорода.
И. РАЗЛИЧНЫЕ РЕАКЦИИ ПРИСОЕДИНЕНИЯ ОЛЕФИНОВ
Присоединение сероводорода к олефинам постольку имеет промышлен-
ное значение, поскольку получающиеся при этом меркаптаны используются
в качестве регуляторов процессов эмульсионной полимеризации и в неко-
торых других областях. Особенно часто применяется присоединение серо-
водорода к олефинам изостроения, причем образуются третичные меркап-
218
таны. Такие изоолефины получаются предпочтительно полимеризацией
олефинов, газообразных при нормальных условиях.
Пропуская изододецен, изопентадецен или диизобутен при температуре
около 100° и давлении 70 ат с избытком сероводорода над катализатором,
состоящим из кизельгура и 1—5% окиси алюминия, получают соответ-
ствующий меркаптан с почти количественным выходом [47]. Такие меркап-
таны могут затем каталитическим путем окисляться в дисульфиды [48],
являющиеся присадками к маслам для работы в условиях высоких давлений,
к маслам для холодной обработки металлов, флотационными реагентами
и т. д. Меркаптаны в присутствии окислов азота как катализатора могут
также сравнительно легко окисляться через дисульфиды в алкилсульфоновые
кислоты. При оксиэтилировании меркаптаны дают полигликолевые эфиры,
которые могут применяться как неионогенные капиллярно-активные
вещества.
а) Реакции олефинов с окисью углерода и водяным паром
с образованием карбоновых кислот
Совместным действием на олефины окиси углерода и водяного пара
при очень жестких условиях в присутствии кислых катализаторов получают
карбоновые кислоты:
R—СП=СНо + С0+Н20 -> R—СН=СН3
I
соон
Так, например, из этилена (2% объемн.), окиси углерода (90% объемн.)
и водяного пара (8% объемн.) в присутствии фосфорной кислоты па активи-
рованном угле при 325° и 700 ат получают пропионовую кислоту [49].
Из высокомолекулярных олефинов получают карбоновые кислоты с развет-
вленным алкильным радикалом.
В качестве катализатора в этих реакциях может применяться также
фтористый бор. Таким образом, например, из тетраметилэтилена при 75°
и давлении окиси углерода 600 ат в присутствии тройного гидрата фтори-
стого бора получают 2,2,3-триметилмасляную кислоту [50].
По способу, разработанному Реппе, реакция олефинов с окисью угле-
рода и водой может легко проходить в присутствии карбопила никеля и
некоторых органических кислот при нагревании в автоклаве до 170 ° [51].
Течение реакции можно представить следующим уравнением:
4R—CH=CH,+4HnO+Ni(CO)4+2CH3COOH->4R—ClI-CII3+(CII3COO)sNi+H2
I
coon
В присутствии спиртов образуются соответствующие сложные эфиры.
Дальнейшее исследование этой реакции показало, что нет необходи-
мости работать со стехиометрическим количеством карбонила никеля и
что того же эффекта можно достичь с каталитическим количеством йоди-
стого никеля. В присутствии спиртов при температуре 180—220° и давлении
окиси углерода 100—200 ат можно, используя в качестве исходного мате-
риала олефины с прямой цепью из 4—18 углеродных атомов, получить
с 90%-ным выходом сложные эфиры.
Можно, наконец, получить также свободные кислоты из олефинов,
воды и каталитических количеств карбонила никеля без добавления йоди-
стого никеля. При 250—280° и 200 ат окиси углерода в присутствии карбо-
нила никеля из этилена получают пропионовую кислоту с 85%-ным выходом.
Если ввести в эту реакцию пропионовую кислоту, то в присутствии про-
пионата никеля образуется с 85%-ным выходом пропионовый ангидрид,
важный исходный материал для получения пропиоцеллюлозы. Подобным
же образом из олефинов, окиси углерода и первичных спиртов в присутствии
219
карбонила никеля, как катализатора, образуются эфиры. Из этилена,,
метилового спирта и окиси углерода получают метиловый эфир пропионовой
кислоты.
Присоединение окиси углерода и воды к олефинам в присутствии ката-
лизаторов, особенно концентрированной серной кислоты, с образованием-
карбоновых кислот разветвленного строения идет с исключительно хоро-
шими выходами при определенных условиях даже в отсутствие давления.
Целесообразно работать при температуре от 0 до 50° и при давлении окиси
углерода 50—100 ат в присутствии 96—97%-ной серной кислоты. В этих
условиях из пропена получают изомасляную кислоту, а из изобутена —
триметилуксусную кислоту [52]. Реакция идет в строгом соответствии
с правилом Марковникова:
СП3-СП=СГ12+СО+Н,О -> СП3 -СП -СПд
I
соои
Изомасляная кислота
сн3
3/С=СН2+СО+Н2О ->сн3 — с-соон
СНз СН-Ч
Триметилуксусная кислота
б) Действие окиси углерода и аминов па олефины [53]
При совместном действии окиси углерода и, например, анилина нэ
олефины при 180—220°, давлении 300—500 ат и отношении олефинов к ани-
лину в реакционной смеси 1 : 1 в присутствии кобальтового катализатора,,
образуются анилиды карбоновых кислот. Реакция протекает согласна
уравнению
Ri
сн—conh/ Ч
Ri / I
I / сн2
CH / I
2 11 / R2
CH+2CO+2C«H5NH2
I \ Ri
r2 \ I
\ CH3
4 сн—conh/ 4
I /
r2
Из анилина, окиси углерода и этилена при 220° и давлении 400 arm
получают с 80%-ным выходом анилид пропионовой кислоты. Вместо анилина
можно применять циклогексиламин, октадециламин и т. д. Равным образом
в этой реакции с успехом может применяться аммиак. При 250° и 500—
800 ат давления окиси углерода из этилена и аммиака в присутствии кар-
бонила кобальта получают амид пропионовой кислоты с выходом 70% от
теоретического [54].
в) Реакция гомологенизацпи
Реакции основаны на действии водяного газа на спирты при условиях:
гидроформилирования. Температура реакции здесь, однако, около 200°.
В результате реакции образуется следующий высший гомолог спирта. Из
mpem-бутилового спирта получают изоамиловый, из внгор-пропилового —
изобутиловый. Сущность реакции, вероятно, заключается в том, что в при-
сутствии карбонилгидрида кобальта, представляющего собой очень сильную»
кислоту, спирт дегидратируется с образованием олефина, который затеял
220
гидроформилируется, давая в результате следующий в гомологическом
ряду спирт. Важно при этом, что образующийся как промежуточный про-
дукт альдегид в результате гомогенно-каталитического действия карбонил-
гидрида кобальта восстанавливается в спирт. Это предположение подтвер-
ждается также тем фактом, что третичные спирты вступают в реакцию очень
легко, тогда как первичные значительно труднее, что соответствует изве-
стной различной способности этих спиртов к реакции дегидратирования.
Однако метиловый и бензиловый спирты способны к реакции гомологениза-
ции. Из метилового спирта образуется с 40%-ным выходом этиловый спирт,
л из бензилового с 30%-ным выходом (3-фенилэтиловый спирт [55].
г) Мюльхеймский способ синтеза спиртов [56]
Необходимо рассмотреть и мюльхеймский способ синтеза спиртов.
Юн также исходит из олефинов и, как и при реакции гидроформилирования,
этим способом получаются первичные спирты. Но в то время как при Гидро-
формилировании и последующем гидрировании образуются- спирты не
только с прямой цепью, но вследствие изомеризации двойных связей также
и разветвленные первичные спирты, по мюльхеймскому способу получаются
•спирты только с прямой цепью. Спирты получают, исходя из высокомоле-
кулярных олефинов с прямой цепью и двойной связью на конце молекулы.
Крекинг-олефины или чистые олефины с двойной связью, расположен-
чтой у конца молекулы, как они могут быть получены полимеризацией эти-
лена способом Циглера, совместным действием металлического алюминия
и водорода при температуре около 120° и давлении превращаются в триал-
жилалюминий [56]:
A1+3RCH=CH2 + 1,5H2 -4 Al (CH2CH2R)3
В то время как низкомолекулярный триалкилалюминий уже при про-
стом соприкосновении с воздухом воспламеняется, высокомолекулярный
алкилалюминий легко окисляется воздухом в алкоголят алюминия. Окисле-
ние проходит так гладко, что отходящие газы, если для окисления приме-
няется воздух, состоят практически из одного азота. Гидролизом алкоголята
лолучают затем спирты. Синтез протекает согласно следующим уравнениям:
AlR3-f-1,5О2 —> Al (OR)3
Al (OR),+3H2O ->3ROH+A1 (ОН),
д) Присоединение органических кислот к олефинам
Особый интерес, как уже выше указывалось, представляет прямое присо-
единение карбоновых кислот к олефинам, приводящее к образованию эфиров.
Так, присоединение уксусной кислоты к пропепу дает emo/j-пропилацетат,
который обычно получается этерификацией изопропилового спирта уксусной
кислотой. Так как, однако, изопропиловый спирт получают непрямой гидра-
тацией пропена с серной кислотой, то прямым присоединением уксусной
кислоты к пропену удается избежать операции гидратации. Имеется и еще
одно преимущество, заключающееся в том, что при использовании в реакции
с олефинами ледяной уксусной кислоты или другой не содержащей воды
кислоты не образуется воды, чем устраняется препятствие к установлению
равновесия между эфиром, спиртом и кислотой.
Исследованиями, проведенными в последнее время, установлено, что
смесь фтористого бора и фтористого водорода очень ускоряет реакции при-
соединения кислоты к олефинам. Растворяя в ледяной уксусной кислоте
при охлаждении фтористый бор (3% вес. считая на реагирующие компоненты:
олефин и кислоту) и также при охлаждении фтористый водород (3% вес.)
и подавая затем в автоклав при температуре 90—100° жидкий пропан,
221
получают после протекания реакции в течение 30 мин. изопропилацетат'
с 75—80%-ным выходом. Вместо уксусной кислоты можно применять также
фталевую и муравьиную кислоты и т. д. [57].
К. ПОЛИМЕРИЗАЦИЯ МОПООЛЕФИНОВ В ИСКУССТВЕННЫЕ ВЕЩЕСТВА
а) Общие сведения
Ди-, три- и тетрамеризация газообразных при нормальных условиях
олефиновых углеводородов, в частности пропена, для получения проме-
жуточных продуктов нефтехимической промышленности была уже вкратце
рассмотрена ранее. Получение полимербензина переработкой газов стаби-
лизации крекинг-бензинон в производстве карбюраторного горючего в насто-
ящей книге не рассматривается.
Соединение друг с другом большого числа олефиновых молекул в зави-
симости от степени полимеризации приводит к образованию маслообразных
или твердых полимеров. Маслообразные полимеры получают, например,
при обработке олефинов, особенно этилена, а также более высокомолекуляр-
ных олефинов, безводным хлористым алюминием. При этом получают
полимер, являющийся превосходным смазочным маслом. Этот процесс
также не относится к области нефтехимической промышленности и здесь
не рассматривается.
В плане настоящей книги интерес представляют лишь те процессы, в кото-
рых из низкомолекулярных олефинов получаются искусственные вещества,
такие как полиэтилен, полиизобутилен и бутилкаучук.
б) Получение полиэтилена
В настоящее время полиэтилен в промышленности получают тремя’
различными способами:
1) при высоком давлении; способ заключается в том, что этилен под.
давлением 1000 ат и более в присутствии небольших количеств кислорода
как катализатора превращается в полиэтилен;
2) при среднем давлении; в этом способе этилен полимеризуется в поли-
этилен под давлением 35 ат в присутствии растворителя, например ксилола,
и окиси хрома как катализатора;
3) при нормальном давлении; способом, разработанным в последние
годы Циглером и его сотрудниками в институте Макса Планка, предусматри-
вающим полимеризацию этилена в присутствии растворителя, например,
гидрированного когазина (мопазина) при помощи триалкилалюминия,.
активированного четыреххлористым титаном.
Полиэтилену предсказывают широчайшее применение в ближайшем
будущем. Достаточно сказать, что мировая продукция полиэтилена соста-
вила в 1956 г. 1,5 млн. т [58].
Получение полиэтилена при высоком давлении.
Полиэтилен впервые был получен при высоком давлении английской
фирмой Империал Кемикалс Индастри [59]. Способ получения заключается
примерно в том, что этилен при температуре 120—130° и давлении 1000—
20ОО ат полимеризуется в присутствии небольших количеств чистого
кислорода. Молекулярный вес полимеризата получается тем больше, чем
ниже температура полимеризации. Практически, однако, оптимальной
рабочей температурой признана 120—130°, потому что уже при этих усло-
виях температура плавления полимеризата составляет около 110°. Поли-
меризация проводится при полном отсутствии растворителя. Содержание
кислорода лежит практически в пределах 0,05—0,1%, считая на этилен.
Время пребывания этилена в установке составляет 2—6 мин. при
10—15%-ном превращении этилена за один проход через печь. Схема работы
при получении полиэтилена представлена на рис. 137.
222
Этилен, содержащий 0,05—0,1% кислорода, забирается компрессором
1, сжимается до 300 ат и подается в работающий под давлением приемник 2,
откуда газ поступает в компрессор 3, где давление газа доводится до 1500 ат.
Из приемника высокого давления 4 этилено-кислородная смесь через предо-
хранительную трубку 5 поступает в полимеризационную установку 6.
Полимеризационный змеевик омывается горячей водой и таким образом
температура поддерживается на требуемом уровне. В первой трубке поли-
меризация начинается при температуре 200—220°, из последнего сектора
змеевика продукт полимеризации выходит с температурой 130°. Продукты
полимеризации поступают далее через расширительный вентиль 7 в раз-
делитель высокого давления 8, в котором поддерживается давление 200 ат
и температура 130°, так что продукты реакции остаются жидкими.
Отделившийся в разделителе высокого давления этилен освобождается
в очистительной установке 9 от формальдегида и снова подается к компрес-
сору. Жидкий полиэтилен приводится к нормальному давлению и заливается
в формы, где он застывает в виде твердых плит.
Рис. 137. Схема получения полиэтилена.
Этот полимер, называемый в Англии полиэтиленом, а в Германии лупо-
леном Н, обладает исключительно высокими диэлектрическими свойствами
и находит широчайшее применение.
В Германии Хопф с сотрудниками разработали способ получения низ-
ших полимеров этилена [60]. В принципе способ заключается в том, что
этилен под давлением 200—300 ат в присутствии метилового спирта как
растворителя и перекиси бензоила как катализатора полимеризуется при
100—120°. В то время как луполен Н имеет молекулярный вес порядка
10 000, молекулярный вес получаемого таким образом луполена N равен
2000-3000.
Получение полиэтилена при среднем давле-
нии. Способ получения полиэтилена при средних давлениях разработан
в США фирмой Филлипс Петролеум Компани [61]. Процесс ведется при
температуре 180—250° и давлении 35—105 ат. Этилен, предварительно'
полностью освобожденный от сернистых соединений, кислорода, водяных
паров и углекислоты, растворяется под давлением при 20—30° в ксилольной
фракции в количестве 7—9% вес. и подвергается полимеризации в труб-
чатом автоклаве над катализатором из окисей хрома и молибдена, нане-
сенных на окись алюминия или алюмосиликат. Целесообразнее применять-
большой избыток растворителя, чтобы полиэтилен оставался в растворе,
а не отлагался на катализаторе, пассивируя его. Кроме того, при этом
223.
получается полимер большого молекулярного веса и улучшается отвод
тепла. Процесс ведется непрерывно.
Продукт реакции фильтруют в горячем состоянии, катализатор
в особой установке промывают ксилолом и затем регенерируют. Горячий
ксилольный раствор полиэтилена охлаждают до 25—60° и выделяющийся
в осадке полимер отделяют фильтрованием. Для дальнейшего выделения
полиэтилена к фильтрату добавляют жидкий пропан, бутан или спирт.
Затем от фильтрата перегонкой отделяют ксилол, возвращающийся на
полимеризационпую установку. В остатке остаются низшие полимеры эти-
лена и алкилированный ксилол. Полиэтилен освобождается от остатков
растворителя. Превращение взятого для полимеризации этилена составляет
около 98%.
Мюльхеймский способ полимеризации при нор-
мальном давлении [62]. Ранее упоминавшийся разработанный
Циглером с сотрудниками способ полимеризации при нормальном давлении
позволяет получать полимеры молекулярного веса от 10000 до 3000000. Ка-
тализатор полимеризации (около 1 % от количества получаемого полиэтилена)
состоит из триэтилалюминия и четыреххлористого титана. Наилучшая тем-
пература реакции около 70°.
Уже без применения давления этилен полностью, на 10(5%, полимери-
зуется с образованием высокополимерных продуктов. Молекулярный вес
полимера регулируется в заданных границах особыми мероприятиями.
Наибольший интерес в настоящее время представляют полимеры эти-
лена молекулярного веса 50000—100000. Полиэтилен, полученный при нор-
мальном давлении, обладает прямой (перазветвленной) структурой, что
сообщает ему исключительно ценные для применения в технике качества.
Катализатор, применяемый в мюльхеймском способе, может также
с успехом применяться для полимеризации пропена и бутена-1. При этом
получают два типа полимеров, обладающих совершенно неожиданными
свойствами (изотактическая полимеризация [63]). Фирма Монтекатини
получает из пропена так называемый мопрен, устойчивый против действия
растворителей, плавящийся при 160°, не чувствительный к действию воз-
духа, кислорода и атомного излучения. Волокно из него по величине сопро-
тивления разрыву равноценно найлоновому волокну [64].
в) Полимеризация изобутена (оппанол В, бутилкаучук)
При смешении жидкого изобутена при —80° с небольшим количеством
•фтористого бора, растворенного в жидком этилене, практически мгновенно
и почти количественно происходит полимеризация изобутена с образованием
каучукообразного вещества (оппанол В) [65]. В случае применения очень
чистого изобутена полимер имеет молекулярный вес около 200000, т. е.
в нем соединяется примерно 3500 молекул изобутена. При добавлении выс-
ших олефинов, например ди- и триизобутена, молекулярный вес полимера
снижается. Добавка же 0,015% диизобутена понижает молекулярный вес
на 50000 единиц. Поэтому для регулирования молекулярного веса получае-
мого полимера к изобутену добавляют большее или меньшее количество ди-
изобутена. Освобождающееся тепло реакции отводится за счет испарения эти-
лена, пары которого затем конденсируются и жидкий этилен возвращается
в процесс.
В промышленных условиях смесь жидких этилена и изобутена смеши-
вают на транспортерной ленте, выполненной из листового металла марки
V2A, с растворенным в жидком этилене фтористым бором, причем теп-
ло реакции отводится кипящим этиленом. Полимер снимается с транспор-
терной ленты шабером, а .затем вальцами ему придается форма плит.
Оппанол В очень устойчив против действия кислот, щелочей и других
-агрессивных веществ.
:224
При полимеризации изобутена в растворе метилового спирта с фтори-
стым бором получают масла различной вязкости.
Оппанол В не вулканизируется. Если, однако, добавить к изобутену
около 2% вес. диенов, как, например, изопрена или бутадиена, то в ре-
зультате полимеризации при •—80° в присутствии хлористого алюминия
получают легко вулканизируемый сополимер (бутилкаучук), производи-
мый в настоящее время в очень больших количествах вследствие его
некоторых исключительно ценных свойств. Он приблизительно в 10 раз
менее проницаем для воздуха, чем натуральный каучук, исключительно
устойчив против действия озона и значительно менее подвержен старению.
Широчайшее применение он находит в производстве автомобильных ка-
мер [66].
Его получение показано па схеме рис. 138 [67]. Жидкая смесь из хло-
ристого метила, изобутена и изопрена в весовом соотношении 146 . 53 : 1
смешивается в реакторе при температуре от —80 до —90° с 0,3%-ным раство-
ром безводного хлористого алюминия в хлористом метиле и полимеризуется.
Тепло реакции отводится непосредственно с испаряющимся этиленом.
Ряс. 138. Схема получения бутилкаучука.
1 — реактор с посредственным этиленовым охлаждением; 2 — газо-
отделнтель; 3 — разделитель; 4 —мешалка; <5 — аммиачный холо-
дильник; 6 — этиленовый холодильник'; 7 — фильтры; <$ — осуши-
тель; Р — фитильный (прутковый) пресс; 10 — каландр.
Линии: 1 — хлористый метил, *146 частей-, 11 —изобутен, 53 «псти-,
III — изопрен, 1 часть; IV — катализатор; V— циркулирующий
хлористый мстил; VI — стеарат цинка; VII — антиокислитель.
Продукты реакции поступают затем в газоотделитель, заполненный во-
дой при ,55е, где в условиях интенсивного перемешивания испаряются не
вошедшие в реакцию углеводороды и хлористый метил. В этот сосуд по-
даются стеарат цинка и антиокислитель. Содержимое фильтруют, высуши-
вают и подают на пресс, где оно проходит через калапдер, охладитель и
выдается как,готовый продукт в виде плиток.
Выходящий из газоотделнтеля газ перерабатывается на специальной
установке п в чистом виде возвращается в установку полимеризации.
В США бутилкаучук обозначается символом IIR (изобутенизопрен-
каучук).
г) Теломеризация
При полимеризации этилена в присутствии четыреххлористого углерода
или хлороформа под влиянием перекиси бепзопла образуется полимеризат,
включающий оба хлорметана. Факт, что при реакциях полимеризации неко-
торые хлорсодержащие соединения или соединения с активным водородом
могут принимать участие в реакции, был впервые установлен Бреитепбахом
[68 |. Этот процесс, известный под названием теломеризацип, обязан своим
протеканием радикально-цепной реакции.
Радикал растущей цепи реагирует, например, с четыреххлористым угле-
родом, причем атом хлора отщепляется и вступает в реакцию с алкил-ради-
15 Заказ 596. 225
калом, давая в остатке радикал СС13‘. Радикал трихлорметил вступает теперь
с этиленом в новую реакцию:
Rx + Cl—СС13-> RxCl+CCl3 (где R — этилен)
CClj+zR ->С13—CRi
Этот процесс повторяется:
CCl,|Ri+С1СС13 -> СС13RXC1 + СС13
так что в конечном счете протекает следующий процесс:
Cl3CCl+xR->C-Cl3RxCl
При присоединении четыроххлорпстого углерода к октепу-1 в присут-
ствии перекиси бензоила получают с 75—85%-пым выходом 1,1,1,3-те-
трахлорнонан [69]. Подобная реакция протекает также с четырехбромистым
углеродом.
Этилен реагирует с четыреххлорпстым углеродом при наличии перекиси
с выделением большого количества тепла, причем образуется смесь, состоя-
щая из тетрахлорпропана, тетрахлорпентана и тетрахлорнойапа. Реакция
протекает столь бурно, что необходимо добавлять воду в качестве разбави-
теля [70]. С четырехбромистым углеродом этилен превращается практически
полностью в 1,1,1, 3-тетрабромпропан.
Реакция теломеризации протекает также со спиртом и окисью угле-
рода. Так, Циглер нашел, что этилен теломеризируется с альдегидами, когда
реакция возбуждается радикалом, возникающим при разложении азосоеди-
нений Тиллс [71]. При этом образуется ряд гомологов метилкетона, которые
могут быть разделены перегонкой. Пропионовый п масляный альдегиды реа-
гируют подобным же образом. При полимеризации этилена в присутствии
ди-трепг-бутилперекиси при высоком давлении и температуре 120—190°
в присутствии спирта получают путем теломеризации смесь высокомолеку-
лярных спиртов [72].
Новый класс поликетонов получают теломеризацией этилена с окисью
углерода в присутствии ди-треш-бутилперекиси. Состав кетона зависит от
отношения этилена к окиси углерода, общего давления, температуры и рода
примененного растворителя. Бензол является очень хорошим растворителем
для предпочтительного вхождения окиси углерода в состав полимеризата.
Поликетоны посредством гидрирующего амидирования (обработки боль-
шим избытком аммиака в присутствии никелевого катализатора и водорода
при 150—200° и давлении 600 ат) превращаются в полиамины с очень ин-
тересными свойствами [73].
Л. АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ОЛЕФИНАМИ
а) Общие сведения
При действии хлористых алкилов или олефинов на ароматические
углеводороды или фенолы легко протекает реакция с образованием алкили-
рованных соединений. Реакция алкилирования должна проводиться в при-
сутствии различных катализаторов, к числу наиболее широко применяемых
относятся хлористый алюминий, безводная фтористоводородная кислота,
фтористый бор и серная кислота. При работе с хлористым алкилом как алки-
лирующим компонентом в качестве катализатора применяется хлористый
алюминий и в некоторых случаях безводное хромное железо (реакция
Фриделя-Крафтса).
Практическое применение ракции алкилирования в нефтехимической
промышленности исключительно велико. Большие количества этилбензола
получают алкилированием бензола этиленом и кумола — алкилированием бен-
226
зола пропеном. Большое значение имеет также алкилированный бензол
с высокомолекулярными алкильными цепями (изододецил- и изопентаде-
цилбензол), получаемый алкилированием бензола пропенполимеризатом
в присутствии серной кислоты, безводной плавиковой кислоты или хлори-
стого алюминия.
Большую роль играет также особенно легко протекающее алкилирова-
ние фенолов. Алкилфенолы с длинными боковыми цепями, например изодо-
децилфенол, представляют большую ценность как промежуточные продукты
для получения ряда синтетических неионогенных моющих средств.
Этилбензол сам по себе применяется мало, но каталитическим дегидри-
рованием он практически полностью превращается в стирол, являющийся,
как известно, важнейшим компонентом смешанной полимеризации с бута-
диеном для получения синтетического каучука (буна S, буна SS, GR—S-
буна). Кроме того, значительная часть стирола полимеризуется в полисти-
рол, широко применяемый в электротехнической промышленности.
Кумол, до 1942 г., изготовлявшийся в лабораториях в ничтожных коли-
чествах, с этого времени внезапно превратился в один из важнейших продук-
тов нефтехимии. Его получение вначале оправдывалось чисто военными це-
лями. Бензол, имеющий температуру замерзания +6°, мог добавляться
к авиационным бензинам лишь в очень ограниченном количестве. Кумол
с температурой замерзания —96° можно добавлять в значительно большем
количестве, не рискуя закупоркой бензопроводов при низких температурах.
Антидетонациопные свойства кумола при применении в двигателях внутрен-
него сгорания такие же, как и бензола.
В настоящее время кумол в первую очередь используется для получения
фенола и ацетона через гидроперекись кумола, которая легко может быть
получена окислением воздухом.
Многие продукты алкилирования ароматических имеют особое назна-
чение, например алкилфенолы. Гексил-, гептил- и октилфенолы обладают
исключительным бактерицидным действием. Гексилфенол очень эффективен
против бактерий тифа, гептил- и октилфенолы сильнее действуют на стафи-
лококки, против которых гексилфенол мало эффективен. Алкилированные
фенолы могут в общем применяться как стерилизующее средство. Они ли-
шены запаха и в отличие от фенола не раздражают кожу. Ниже приведены
сравнительные данные действенности различных алкилфенолов против
стафилококков [74].
Фенол (карболовая кислота) 1,0
mpem-Бутплфенол............ 87,5
Крезол ....................... 3,1
т рст-Бутилнрезол ........ 75,0
Ксиленол................... 5,6
т/^т-Бутилксилевол .... 150,0
Продукты алкилирования фенолов, например тгаретп-бутилфенол, трет-
амилфенол, имеют значение для получения растворимых в масле продуктов
формальдегидной конденсации. Другие алкилфенолы, как, например, 2,6-
дипзобутил-га-крезол, являются очень эффективными антиокислителями.
Представляют интерес флуоресцирующие вещества для смазочных
масел. Природные смазочные масла из нефти почти все обнаруживают спо-
собность к флуоресценции, а именно они имеют красный цвет в проходящем
свете и зеленовато-синий в отраженном. Характер флуоресценции нахо-
дится в определенной связи с качеством смазочного масла. Сейчас известно,
что многие полициклические ароматические углеводороды, алкилированные
высокомолекулярными олефинами в присутствии относительно большого
количества безводного хлористого алюминия, дают алкилат, добавление
которого в количестве 0,1 — 0,2% вес. к нефлуоресцирующим маслам сооб-
щает им зелеповато-сингою флуоресценцию в проходящем свете.
б) Получение этилбензола [75]
Промышленное получение этилбензола основывается на совместном
испарении бепзола и этплена в присутствии безводного хлористого алюминия.
Скорость, с какой этилен вступает в реакцию, исключительно велика и за-
15*
227
висит от скорости растворения этилена в бензоле, откуда следует, что ско-
рость реакции должна увеличиваться с интенсивностью перемешивания.
Реакция протекает по уравнению
\*СНг=С1 ч;—СП.,—СП;,+ 26 ккал!моль
В промышленных условиях реакция алкилирования проводится без
перемешивания. Поэтому реакция идет достаточно медленно, чтобы был
Отношение при роботе
в промышленных усло&иях
О 0,2 0,6 0,6 0,6 1,0 1,2
Отношение олеильных групп н
бензольным кольцом
Рис. 139. Влияние отноше-
ния между алкильными
группами и бензольными
кольцами в реакционной
смеси на выход продуктов
реакции.
1 — бензол; 2 — ятилбензол;
з — полиэтилбенаол.
хлористого алюминия и
возможен отвод довольно значительного количе-
ства тепла, выделяющегося при реакции, про-
стым наружным охлаждением реактора.
Так как скорости этилирования бензола и
ути образовавшегося этилбензола примерно
равны , то хотя еще в смеси имеется большое
количество не вступившего в реакцию бензола,
одновременно уже идет образование ди- и
триэтилбензола . Чтобы избежать преимуществен-
ного образования полизамещенных продуктов, в
реакции алкилирования ограничиваются лишь
частичным превращением, а затем отделяют друг
от друга непрореагировавший бензол, моноэтил-
бензол и полиэтилбензол посредством перегон-
ки и ректификации.
Бензол и полиэтилбензол (ди-, три- и тетра-
этилбензолы) возвращаются на алкилирование.
При этом всегда часть полиэтилбензола превра-
щается в этилбензол. Безводный хлористый алю-
миний в присутствии большого количества
бензола оказывает на полиэтиленбензол дис-
пропорциоиирующее действие, так что расщепле-
ние к примеру триэтилбензола в присутствии
большого избытка бензола идет согласно сле-
дующему уравнению:
хс2тт5
+ % + А1С13 —> 3^ ^€2н5 + (ж—2).^
С2Н6
При реакции 1 моля этилена с 1 молем бензола при 95° образуется в рав-
новесном состоянии около 51% мол. моноэтилбензола, в то время как
18% мол. бензола остаются не использованными. На рис. 139 показано со-
отношение бензола, этилбензола и полиэтилбензола при проведении процесса
алкилирования в промышленном масштабе в зависимости от отношения числа
этильных групп к числу бензольных колец в реакционной смеси.
Для поддержания скорости реакции в процессе алкилирования можно
также применять небольшие количества хлористого водорода как промотора
для хлористого алюминия, путем добавления к этилену небольшого количе-
ства хлористого этила.
Для промышленного этилирования бензола этиленом последний дол-
жен быть чистым. Он не должен содержать гомологов этилена, как пропен
или бутен, потому что образование даже небольших количеств изопропилбен-
зола может сильно мешать разделению бензола, моноэтилбензола и поли-
этилбензола из-за налегания друг на друга температур кипения компонентов
смеси. Этилен должен быть практически свободен также от кислорода и
окиси углерода, так как эти газы увеличивают расход безводного хлористого
алюминия.
228
Также чистым должен быть и применяемый для алкилирования бензол,
содержание серы должно быть менее 0,1% и, исходя из изложенных выше
соображений, он не должен содержать каких-либо гомологов. Температура
кипения бензола должна лежать в пределах 1°, температура замерзания
должна быть выше 5°.
Очистка бензола, который в Германии получается главным образом
из каменноугольной смолы, производится серной кислотой, перегонкой
с ректификацией и каталитическим гидрированием под давлением. Безвод-
ный хлористый алюминий должен быть 98 %-ной чистоты.
Схема промышленного способа получения этилбензола дана на рис. 140.
Алкилирование в большинстве случаев проводится без применения давления
и без перемешивания при 90°. Колонна, содержащая реакционную смесь,
Рис. 140. Схема этилирования бензола этиленом в присутствии хлористого алюминия.
1 — обратный холодильник; 2 — реакционная колонка с наружным водяным охлаждением; 3 — отде-
литель; 4 — колонна для промывки водой; 5 — колонна для промывки щелочью; в — бензольная ко-
лонна; 7 — этилбензольная колонна; 8 — полнзтилбензольная колонна; 9 — смолоотделитель.
Линии: I — бензол и полиэтилбензол; II — свежий AICI3; III — отходящие газы; IV — этилен; V —
возврат катализатора; VI — отработанный катализатор; VII — дренаж; VIII — щелочь; IX — бензол;
X — этилбензол; XI и XII — полиэтилбензол; XIII — смола; XIV — циркулирующий бензол и поли-
этилбензол.
имеет рубашку, через которую прокачивается охлаждающая вода. Внутри
колонны, в ее газовом объеме, расположены два охлаждающих змеевика.
Над колонной имеется обратный холодильник. Колонна снабжена также
штуцером, через который в колонну может подаваться безводный хлористый
алюминий. Этилен подается непосредственно через дно реакционного сосуда.
Колонна находится в работе непрерывно.
Продукты реакции отводятся из верхней части колонны, а снизу посту-
пают свежий бензол и циркулирующий бензол, содержащий еще ди- и поли-
этилбензол. Выходящие из верха колонны продукты реакции поступают
в сепаратор. Здесь отделяются тяжелые соединения хлористого алюминия,
которые возвращаются в реакционную колонну. Алкилат промывают водой,
щелочью и снова водой и подают на перегонку, процесс которой ясен из
схемы. Остаток из последней колонны собирают, еще раз разгоняют на
периодически действующей установке. Выделенные в процессе перегонки
остатки иолиэтилбензола возвращаются на установку для алкилирования.
Высокоалкилироваппые продукты можно так же, как это делает амери-
канская фирма Доу Корнинг Корпорейшп, подвергать деалкилированию
в специальном деалкплаторе при 200° в присутствии хлористого алю-
миния.
Расход хлористого алюминия в промышленных условиях составляет
около 2,5 кг на 100 кг моноэтилбензола, выход моноэтилбензола составляет,
считая на бензол, около 94%, считая па этилен 93%.
229
в) Получение кумола
Кумол может получаться так же, как и этилбензол, а именно реакцией
пропена с бензолом, смешанным с хлористым алюминием. Во время войны,
когда чистоте кумола придавалось не столь большое значение, как теперь,
когда кумол применяется в качестве исходного продукта для получения фе-
нола и ацетона, алкилирование бензола пропеном в присутствии фосфорно-
кислого катализатора под давлением проводилось в такой же аппаратуре,
в какой осуществлялась каталитическая полимеризация газов стабилизации
крекинг-установок (смесь пропена и бутенов) для получения полимер-
бензо ла.
Способ допускает использование разбавленного пропена в том виде,
в каком получается фракция С3 с установок депропанизации. В качестве
катализатора применяется так называемая «твердая» фосфорная кислота,
представляющая собой нанесенную на кизельгур ортофосфорную кислоту
Рпс. 142. Схема жидкофазного алкилирования бен-
зола в кумол с серной кислотой в качестве ката-
лизатора.
1 — циркуляционный насос; 2 — реактор; 3 — отстойник;
4 — нейтрализатор; <5 — депропанизатор; 6 — колонна длн
отгонки бензола; 7 — колонна для отгонки кумола.
Линии: I — свежая кислота; 11 — отработанная кислота;
III — отработанная гцелочь; IV— фракция Cg; V — свежий
бензол; VI — циркулирующий бензол; VII — остаток;
VIII — пропан; IX — бензол; X — кумол.
Рис. 141. Схема получения кумо-
ла газофазным алкилированием
бензола над «твердой» фосфорной
кислотой.
1 — смеситель; 2 — реакционная печь,
фосфорнокислый катализатор; 3 —
депропанизатор; 4 — колонки для от-
гонки бензола; 5 — колонна для от-
гонки кумола.
Линии: Г — свежий бензол; ZZ — Сд-
фракция; III — циркулирующий бен-
зол; IV — пропан; V — бензол; VI —
кумол; VII — остаток.
[76] . При давлении порядка 20 ат и при температуре 250° можно добиться
превращения 90—95% пропена. Бензол здесь также должен быть очень
чистым, а пропен не должен содержать гомологов. При алкилировании
в присутствии фосфорнокислого катализатора необходимо работать с значи-
тельным избытком бензола (в промышленности применяется отношение бен-
зола к пропену 5:1), чтобы в максимальной степени предотвратить образо-
вание полизамещенных продуктов. Обратное превращение высокоалкили-
рованных продуктов в этом процессе невозможно, так как фосфорная кислота
в условиях алкилирования не обладает диенропорционирующим действием
по отношению к полиизопропилбензолу в присутствии бензола.
Схема получения кумола газофазным алкилированием бензола про-
пеном в присутствии фосфорнокислого катализатора показана на рис. 141.
Смесь бензола и фракции С3 нагнетается в печь, заполненную фосфорноки-
слым катализатором, где при температуре 250° и 20 ат в газовой фазе проте-
кает реакция. Продукты реакции направляются в депропанизатор, где от-
деляется содержавшийся во фракции С3 пропан. В следующей колонне от-
гоняется бензол, возвращающийся затем на установку для алкилирования,
наконец в последней колонне изолируется кумол.
230
Другой процесс промышленного получения кумола основывается на
жидкофазном алкилировании бензола пропеном в присутствии серной ки-
слоты как катализатора [76]. Исходные материалы должны обладать такой
же чистотой, как и при газофазном алкилировании. Способ работы показан
на схеме рис. 142.
Тонкая эмульсия бензола в серной кислоте соединяется в смесительном
насосе с пропан-пропеновой смесью и подается в реакционный сосуд, где про-
исходит реакция между бензолом и пропеном. Смесь в реакционном сосуде
непрерывно перемешивается циркуляционным насосом, причем небольшая
часть алкилата и серной кислоты постоянно отбирается от циркулирующей
реакционной смеси и подается в отстойник, где в виде нижнего слоя отде-
ляется серная кислота, которая вновь возвращается на установку алкили-
рования. Часть серной кислоты из процесса выводится и заменяется свежей.
Верхний слой из отстойника нейтрализуют и подают в депропанизатор,
где при 170° и 14 ат давления пропан отгоняется. В депропанизаторе нахо-
дится некоторое количество щелочи для омыления следов эфиров серной
кислоты, которые могли образоваться в результате взаимодействия серной
кислоты с олефинами в процессе алкилирования. Затем во второй колонне
при давлении 1,7 ат отгоняется бензол.
Кумол от высокоалкилированных продуктов отделяется в колонне,
работающей при нормальном давлении. Само алкилирование проходит
при давлении 11,5 ат и температуре 30—40°, т. е. при условиях, обеспечи-
вающих протекание реакции в жидкой фазе. Молярное соотношение бензола
к пропену составляет 5:1, объемное соотношение серной кислоты к угле-
водородной смеси 1:1, время пребывания в реакционном сосуде 20—30 мин.
г) Алкилирование тиофена [77]
Тиофен, который в последние годы производится в промышленных
масштабах, также легко может алкилироваться каталитическим путем.
Алкилирование тиофена бутенами или пентенами, а также исключительно
пригодным для этого циклогексеном может осуществляться пропусканием
тиофена и олефинов или циклоолефинов над катализатором кремневая ки-
слота — окись алюминия при 200° или над «твердой» фосфорной кислотой,
как было выше описано для получения кумола, или также с серной ки-
слотой.
Алкилирование в присутствии алюмосиликатного катализатора пропеном
дает 72% изопропилтиофена, изобутеном—70% изобутилтиофена, циклогек-
сеном — 100% циклогексилтиофена.
д) Получение флуоресцирующих веществ
В большинстве случаев для этой цели нафталин алкилируют высоко-
молекулярными олефинами, содержащимися, например, в продуктах кре-
кинга мягкого парафина. Катализатором служит хлористый алюминий.
Количество ароматического компонента должно составлять от 20 до 70%
от количества олефинов, взятых в реакцию. В общем применяется, как пра-
вило, 50%. Хлористого алюминия берут от 20 до 60% (чаще 50%) от количе-
ства олефинов. Температуру реакции постепенно повышают до 180й.
После завершения процесса, как обычно принято для реакции Фри-
деля-Крафтса, от продуктов реакции отгоняют фракцию, кипящую ниже
150° при 2 мм рт. ст. Остающаяся после перегонки вязкая жидкость, будучи
добавлена к минеральному смазочному маслу в количестве 0,1—0,2%, со-
общает ему исключительно красивую флуоресценцию [78].
с) Алкилирование фенолов олефинами [79]
Как выше уже указывалось, алкилфенолы с низкомолекулярными и высо-
комолекулярными алкильными группами являются важными промежуточ-
ными продуктами нефтехимической промышленности. Продукты алкилиро-
231
вания фенола динзобутеном, тетра-пли пептапропепом получаются с очень
хорошими выходами при взаимодействий фенола с соответствующими олефи-
нами в присутствии следов концентрированной серной кислоты, при относи-
тельно низких температурах, около 100 . В качестве катализаторов алкилиро-
вания могут также с успехом применяться фтористоводородная кислота,
фтористый бор или безводный хлористый алюминий.
Продукты оксиэтилирования алкплфеполов с длинными алкильными
группами особенно широко применяются в производстве синтетических
моющих средств. Алкилфенолы являются антиокислителями для минераль-
ных масел. Они часто применяются в форме кальциевых, бариевых или цин-
ковых солей.
ж) Получение фенола и ацетона из кумола [80]
Получение фенола и ацетона из кумола основывается на расщеплении
гидроперекиси кумола в кислой среде. Ацетон и фенол образуются в со-
ответствии со следующим уравнением:
Гидроперекись кумола и ее расщепление на фенол и ацетон впервые
описаны Хоком и Ланге. Установленные ими закономерности легли в обнову
ряда промышленных способов получения гидроперекиси кумола. По одному
Рис. 143. Схема получения фенола и ацетона из
кумола.
1 — установка гидрирования; 2 — аппарат для окисления;
з — расщепительная установка; 4 —фильтр; б — газовый
сепаратор; 6 — насос; 7 — сепаратор; 8 — промывочная
башня; 9 — ацетоновая колонна; 10 —кумоловая колонна;
11 — фенольная колонна; 12 — кристаллизатор.
Линии'. I — свежий кумол; II—водород; III — воздух
(кислород); IV — разбавленная серная кислота; V — цирку-
лирующая кислота; VI— возврат кумола и а-метнлстирола;
VII — инертный газ; VIII — вода п кислота; IX —сырой
фенол; X — ацетон; XI — твердый фенол; XII — ацето-
фенон.
из методов кумол в виде
эмульсии с водой при зна-
чении pH, равном 8,5—10,5,
окисляется воздухом при тем-
пературе около 130е и дав-
лении 5 —10 ат. Наряду с
этим так называемым «влаж-
ным» методом разработан су-
хой способ, состоящий в
продувании воздуха или
кислорода через нагретый
до 120° кумол в присутствии
металлической меди как ка-
тализатора.
Схема работы по «влаж-
ному» способу представлена
на рис. 143. Способ заклю-
чается примерно в следую-
щем.
Свежий кумол и кумол
циркуляции смешиваются
в отношении 1:4 и гид-
рируются в автоклаве с
мешалкой при 100° в при-
сутствии никеля как ката-
лизатора. Целью этого гидрирования является перевод в кумол неболь-
ших количеств а-метилстирола, являющегося, подобно фенолу, сильным
ингибитором окисления. После гидрирования чистый кумол фильтруют через
фильтрпресс для освобождения от катализатора. Отсюда кумол поступает
в автоклавы, в которых он эмульгируется в присутствии эмульгатора с 2—
3-кратным объемом 1%-ного содового раствора. В эмульсию при постоянном
перемешивании нагнетают воздух при температуре 130° идавлении 5—10 ат
до тех пор, пока в реакции примет участие примерно 25% кумола. Затем,
232
прервав окисление, отделяют масляный слой от водного и обрабатывают
масляный слой в освинцованной емкости 10 %-ной серной кислотой при тем-
пературе около 120°. При этом гидроперекись расщепляется на фенол
и ацетон.
В качестве побочных продуктов образуются ацетофенон п метиловый
спирт, а также а-метилстирол:
а = Метилстирол
Полученное таким образом масло после отделения от кислоты разде-
ляется перегонкой. В колонне, работающей при нормальном давлении,
отделяют ацетон. Затем при пониженном давлении отгоняют кумол и а-
метилстирол, которые после описанной ранее очистки гидрированием воз-
вращаются в процесс. Остаток в кумоловой колонне состоит из фенола с при-
месью примерно 6% ацетофенона. Эта смесь разделяется затем перегонкой
в вакууме, фенол отгоняется, а ацетофенон остается в остатке.
При сухом способе окисление кумола производится в облицованной
внутри медью колонне, заполненной медными кольцами, при 120°. Медь
является катализатором процесса. Расщепление перекиси кумола произво-
дится практически так же, как было описано ранее.
з) Алкилирование ароматических в целях получения промежуточных
продуктов для производства синтетических моющих средств [811
Получение синтетических моющих средств типа арилсульфонатов алки-
лированием бёпзола хлорпарафином и последующим сульфированием в водо-
растворимые сульфонаты было уже рассмотрено выше в разделе о хлориро-
вании парафиновых углеводородов. Такие сульфонаты обладают часто не-
приятным запахом п окрашены в желтый или светло-коричневый цвет. По
этой причине от «ксрилбепзола» как промежуточного продукта отказа пись
и перешли к применению продуктов алкилирования бензола полимерами
пропена.
Алкилирование проводится главным образом в присутствии плавико-
вой кислоты или коицентрированной серной кислоты как катализатора .
Бензол применяется в значительном избытке, чтобы уменьшить образование
диалкилбепзола. В качестве алкилирующих компонентов более всего подхо-
дят пзододецеп и изопептадецеп, получаемые полимеризацией пропена
в присутствии фосфорнокислого катализатора (см. стр. 65)- Менее пригоден
для этой цели диизобутеп и совсем неудовлетворительные результаты дает
сополимер смеси алифатических газообразных олефинов , например пропена
и бутенов. Эти полимеры под действием катализаторов алкилирования под-
233
Пропен
Арилсульфонат
(марлон)
рис. 144. Схема получения арилсульфоната иа
пропентетрамера.
дергаются деполимеризации
на низшие осколочные оле-
фины, которые затем сами
по себе принимают участие в
алкилировании, давая в ре-
зультате алкилат, не пригод-
ный для последующего суль-
фирования в целях получе-
ния синтетических моющих
средств.
Например, алкилирова-
ние в присутствии пла-
виковой кислоты, как ка-
тализатора, производится
следующим образом (рис. 144)
[82]. В котел загру-
жают 107G кг бензола,
предварительно охлажден-
ного до 10°,'547 кг безвод-
ной плавиковой кислоты и
1472 кг пропенполимера,
смесь энергично перемеши-
вают. Реакция заканчивает-
ся через 3—4 часа, после
чего плавиковая кислота от-
гоняется при 20°, а не во-
шедший в реакцию бензол
при 80°. Из смеси отгоняет-
ся 200 кг плавиковой кисло-
ты и 305 кг бензола. Остаток
от перегонки в течение полу-
часа перемешивают с 10 кг
кальцинированной соды,
фильтруют через фильтрпресс
и в таком виде он поступает
на дальнейшую переработ-
ку. Алкилирование может
производиться также непре-
рывным способом.
Сульфирование получен-
ного таким образом алки-
лата должно производиться
с соблюдением вполне опре-
деленных мер предосторож-
ности, чтобы предотвратить
деполимеризацию алкильных
цепей. В большинстве слу-
чаев для сульфирования
применяют 20%-ный олеум,
а сам процесс сульфирова-
ния проводят при темпе-
ратуре ниже 25°. При весо-
вом отношении олеума к
алкилату 1,25 : 1 и продол-
жительности сульфирования
около 3 час. получают темно-
коричневый продукт, состоя-
щий из алкиларилсульфоно-
234
вой кислоты, избыточной серной кислоты и 1,5—2,0% не вошедшего
в реакцию алкилата. Продукты реакции разбавляют далее при охлаж-
дении водой так, чтобы температура не повышалась более чем до 70°,
и чтобы в результате образовалось примерно 70%-ная кислота, в которой
алкиларилсульфокислоты не растворяются. Последние нейтрализуют
и раствор натриевых солей упаривают.
Приводим условия сульфирования изододецилбензола 20%-ным олеу-
мом [83].
20%-лого олеума, кг на 1 кг алкилата........................ 1,25
Температура сульфирования, °C............................... 25—35
Продолжительность реакции, часы............................. 1—1,5
Время перемешивания при дополнительной реакции после введе-
ния олеума, часы............................................. *2
Температура при дополнительной реакции, С...................... 25
• » разбавлении водой, °C......................... 52—70
Продолжительность разбавления, часы ....................... 0,2—1,0
Температура в отстойнике, °C................................ 50—60
Продолжительность отстоя, часы ............................. 1—8
Концентрация отходящей серной кислоты, % ................... 63—78
Алкилирование бензола тетрамером пропена может проводиться
также в присутствии серной кислоты, лучше 100%-ной, при 10—20° непре-
рывным способом. Компоненты энергично перемешивают в смесителе в те-
чение 1 часа, а затем подают в разделитель, где кислота быстро отделяется
от углеводорода. Серная кислота возвращается в процесс, а углеводородный
•слой нейтрализуется и перегонкой освобождается от избыточного бензола.
Условия работы при алкилировании бензола тетрамером пропепа с серной
кислотой как катализатором следующие.
Температура, °C............................... 10—20
Давление................................... ... Нормальное
Концентрация серной кислоты, % 100
Расход кислоты, кг па 1 кг алкплата........... 1,15
Молярное соотношение бензол: тетрамер пропена 3,5:1
Время реакции, часы................................ 1
Соотношение кислота : углеводород.................. 1:1
Для алкилирования ароматических углеводородов могут применяться
также спирты. Процесс проводится преимущественно в присутствии серной
кислоты как катализатора. Реакцию можно представить себе таким обра-
зом, что собственно алкилирующим агентом является алкилсульфат, обра-
зующийся в результате действия серной кислоты на спирт. Подобная реак-
ция имеет место и при алкилировании олефинами в присутствии серной
кислоты. Здесь также продукт присоединения серной кислоты к олефину —
алкилсульфат — является промежуточным, алкилирующим продуктом.
В этом случае алкилирование может проходить одновременно с сульфирова-
нием. Типичным примером может служить получение продукта пекал. Он
относится к первым полностью синтетическим, капиллярно-активным со-
единениям, получавшимся в промышленных количествах. С этой целью
нафталин алкилировали при 50° в присутствии 96 %-пой серной кислоты
бутиловым или гексиловым спиртом или смесью пропилового и бутилового
спиртов, затем в тот же реакционный сосуд добавляли 25%-пый олеум п
начиналась реакция сульфирования.
Получение смачивающего вещества, известного под названием искал ВХ ,
можно иллюстрировать следующим примером. В котле емкостью 6000 л
тщательно перемешивают 1000 кг безводного бутилового спирта, 850 кг
нафталина и 1860 кг 96 %-ной серной кислоты. Котел снабжен мешалкой и
змеевиками для охлаждения и подогрева реакционной массы. Температура
реакции поддерживается на уровне 25°. После одного часа перемешивания
добавляют в котел 2400 кг 24%-ного олеума, перемешивают 30 мин. при 25°,
235
а затем поднимают температуру до 50° п перемешивают еще 4 часа. Затем
смесь оставляют па 8 час. отстаиваться, при этом происходит разделение па
два слоя. Пплший слон — серную кпсзоту — удаляют, верхний слой —бу,-
тплнафталипсульфокнслоту — разбавляют 2000 л воды, нейтрализуют
1700 иг 50%-ной натриевой щелочи .отбеливают 350 кг гипохлорита, филь-
труют и упаривают.
н) Получение стирола пз этилбензола
Как указывалось ранее, стирол является важнейшим промежуточш ям
продуктом для производства каучука. Кроме того, стирол сам по себе поли-
меризацией превращается в полистирол, представляющий' исключительно
ценный синтетический материал. Сополимеры стирола и изобутена, получае-
мые способом, аналогичным применяемому для получения бутилкаучука,
и известные под названием полимера S, обладают свойствами, совершенно
отличными от свойств полистирола или полиизобутена. Его важнейшая осо-
бенность — абсолютная влагонепроппцаемость, что делает его исключительно
ценным упаковочным материалом для гигроскопических товаров. Кроме
того, вследствие высоких диэлектрических свойств полимер S широко при-
меняется в электротехнике [84].
В настоящее время стирол получают исключительно каталитическим
дегидрированием этилбензола над определенными катализаторами. Ранее
для этой цели применялись и некоторые другие способы [85]. В одном из
таких способов этилбензол подвергался хлорированию, 1-хлор-2-фенил-
этан отделялся, омылялся в фенилэтиловый спирт, а последний дегидрати-
ровался в стирол:
СН2СН3+С12^-СН2СП2С1 -> V-CH2CH2OH ->
\-СП=СН2
В другом способе, также исходят пз этилбензола, каталитическим окис-
лением получали ацетофенон, который затем восстанавливали в метил-
фенилкарбинол, а последний каталитически дегидратировался в стирол:
^>-СН2СНз+О2-><^ S-C0CII3 + n,0
СОСНз-ч^ ^-СНОНСНз-к^' СН=СП2
Окисление этилбензола в ацетофенон протекает при 125° и 2 ат. Превра-
щение этилбензола за один цикл составляет 25—30%. Реакция экзотермиче-
ская. Сырые продукты реакции, состоящие примерно из 73% этилбензола,.
17% ацетофенона, 8% метилфенплкарбинола п 2% побочных продуктов,
разделяют разгонкой. Полученную таким образом смесь, состоящую из
68% ацетофенона и 32% метилфенолкарбинола, гидрируют при 14 ат водо-
рода и 130—170° над медно-хромо-железным катализатором. При гидрирова-
нии получается практически чистый метилфенилкарбинол. Дегидратация
его в стирол производится над нанесенной на боксит окисью титана, в отсут-
ствие давления при 250°.
По способу, разработанному И. Г. Фарбенпндустрн, пары этилбензола
в смеси с примерно равным количеством водяных паров пропускают через,
заполненную контактом трубчатую печь, обогреваемую снаружи до 600°.
При этом способе работы за один проход образуется примерно 40% стирола.
Катализатор состоит главным образом из окиси цинка с добавкой окисей
алюминия, кальция и магния, сульфата калия и хромовокислого калия.
Состав нескольких типичных катализаторов дегидрирования приведен в
236
Таблица 85
Состав некоторых катализаторов дегидрирования для получения стирола
из этилбензола
Компонент Состав катализаторов 1—Jt в % вес.
1 2 3 4
Окись цинка 85,6 76,0 86,0 77.4
» алюминия 2,3 8,0 5,0 7,6
» кальция 5,1 5,0 5,0 4,7
» магния 0,0 5,0 0,0 4,7
Сульфат калия 3,0 3,0 2,0 2,8
Хромовокислый калий 3,0 3,0 2,0 2,8
табл. 85. Состав сырого продукта дегидрирования этилбензола в весовых
процентах:
Этилбензол.................... 58,1
Стироль ...................... 40,0
Толуол........................... 0,8
Смолы............................ 0,7
Бензол........................... 0,4
Добавка водяного пара при дегидрировании по этому способу имеет
целью понижение парциального давления паров этилбензола и, таким обра-
зом, способствует протеканию реакции дегидрирования, являющейся равно-
весным ’процессом. Кроме того, введение водяного пара заметно уменьшает
отложения кокса на катализаторе.
Второй способ подвода тепла для обеспечения протекания эндотермиче-
ской реакции дегидрирования основан на введении в реакционную смесь
большого количества перегретого до высокой температуры водяного пара
[86]. По способу, применяемому фирмой Доу Кемикал Компани, работают
с весовым отношением водяного пара к этилбензолу 2,6 : 1. Водяной пар
предварительно перегревается до 710°, температура поступающих в процесс
паров этилбензола 520°. Над катализатором оба пара смешиваются, темпера-
тура смеси составляет около —625°. Газы остаются в печи ровно 0,5 сек.
и за это время (за один проход через печь) достигается превращение 37%
этилбензола. Применение метода стало возможно после разработки катали-
затора, устойчивого против действия перегретого водяного пара. Такие
катализаторы были разработаны фирмой Стандард Ойл Девелопмент и
.Шелл Девелопмент Компани под названием катализаторов 1707 и 105 [87].
Состав катализаторов 1707, %
MgO . . < ...................
Fo.,O3 ....
СиО..........
Ко() ....
72,4
18,4
4,6
4,6
Состав катализатора 105, %]
БгоОя...................... 90,0
СгоОч .......................... 4,0
К2Сб3......................... 6,0
Применение большого избытка водяного пара полностью предотвращает
отложение кокса на катализаторе, так что дегидрирование может проводиться
без периодической регенерации катализатора. Так как необходимое для
дегидрирования тепло подводится с перегретым водяным паром, то отпадает
необходимость в устройстве какого-либо обогрева реакционной печп, по-
этому конструкция ее, естественно, сильно упрощается. Способ работы пока-
зан на схеме рис. 145.
Дальнейшая переработка сырых продуктов дегидрирования связана
с перегонкой п ректификацией при пониженном давлении. Температуры
кипения этилбензола и стирола различаются на 9° (136° и 145,2° соответ-
ственно), поэтому необходима очень эффективная ректификационная колонна
с высоким коэффициентом орошения. Трудность заключается еще в том, что
стирол при тепловом воздействии сравнительно легко полимеризуется.
237
Ректификация производится преимущественно в оцинкованной железной
колонне, чтобы предотвратить влияние железа па полимеризацию сти-
рола.
До поступления в первую ректификационную колонну сырые продукты
дегидрирования проходят через сосуд, содержащий элементарную серу.
При этом в продукте растворяется некоторое количество серы, которая
затем при прохождении продукта через систему ректификационных колонн
образует иа поверхности металла сульфидную пленку, пассивирующую
каталитическое действие металла и предотвращающую таким образом поли-
меризацию.
В первой колонне, работающей при остаточном давлении 178 мм рт. ст.
и отношении орошения к сырью 12 : 1, отделяются бензол и толуол. Темпера-
тура верха колонны 57°, температура низа 96°. Выходящая из этой колонны
бензолыю-толуольпая смесь затем в специальной колонне с 40 тарелками,
работающей при нормальном давлении, освобождается от бензола, который
возвращается на алкилирование. Отбираемый из низа этой колонны сырой
толуол подвергается ректификации па колонне с 35 тарелками, откуда он
выходит совершенно чистым.
Рис. 145. Схема получения стирола каталитическим дегидрированием этилбензола с во-
дяным паром как теплоносителем.
z — перегреватель; 2 — реактор, температура 625°, катализатор контакт 1707 или 105, время реакции
0,5 сек.; з — конденсатор; 4 — смеситель; 5 — бензол-толуольная колонна; 6 — этилбепзольная ко-
лонна I; 7 — этилбенэольная колонна II; 8 — стирольная колонна; 9 — бензольная колонна; 10 — то-
луольная колонна.
Линии: I — водяной пар, 10%; II — этилбензол; III — водяной пар, 90%; IV — отходящие газы;
у — вода; VI — сырой стирол; VII — сера; VIII — этилбензол обратно на дегидрирование; IX — сти-
рол; -У — бензол; XI — толуол; XII — смола; XIII — остаток.
Остаток из первой (бензольно-толуольной колонны) поступает в первую-
этилбензольную колонну, где при остаточном давлении 35 мм отделяется
этилбензол (с примесью около 1% стирола), возвращаемый на установку
дегидрирования. Остаток первой этилбензольной колонны поступает на вто-
рую колонну, в которой от стирола отделяются последние остатки этилбен-
зола. Остаток из второй этилбензольной колонны поступает далее в периоди-
чески работающую при 35 мм колонну тонкой ректификации. Чистый стирол
отходит при температуре верха колонны 57°, температура низа колонны 74°.
В эту колонну сверху поступает стабилизирующий раствор в виде гидро-
хинона или ге-тре/ге-бутилпирокатехипа. Благодаря этому термическая
полимеризация стирола полностью предотвращается. Эти ингибиторы при-
меняются также для стабилизации стирола в условиях хранения. Необ-
ходимая концентрация составляет 10 частей ингибитора на 1 млн. частей
стирола.
Перед полимеризацией эти ингибиторы количественно удаляют из сти-
рола простой промывкой щелочью.
238
Ниже показан общий выход стирола при получении его из бензола
и этилена по способу Доу Корнинг.
Общий выход стирола при получении
его из бензола и этилена в % вес.
Состав чистого стирола
Бензол в этилбензол............ 95,8
Этилен в этилбензол............ 96,8
Этилбензол в стирол............ 90,1
Сырой стирол в чистый стирол 99,4
Бензол в стирол в совокупности 86,5
Этилен в стирол в совокупности 86,5
Стирол................. 99,7
Альдегиды ............ 0,010
Перекиси.............. 0,001
Сера ................. 0,003
Хлориды .............. 0,004
Полимеры.............. Следы
Этил- и изопропилбензол 0,03
n-znpcm-Бутилпирокате-
хин.................. 1/1000
Стирол, как ранее уже много раз указывалось, относительно легко,
полимеризуется под влиянием теплового воздействия [88]. Термическая
полимеризация стирола (блокполимеризация) проводится следующим
образом: в мешалке при 80" стирол полимеризуется до образования сиропо-
образной жидкости, содержащей примерно 33% полимера. Дальнейшая
полимеризация производится непрерывным способом в условиях ступенча-
того повышения температуры до 140—180е. Расплавленный стирол пропу-
скается затем через тонкие щели высотой 1 мм и шириной 30 леи на охлаждае-
мые стальные вальцы, при этом он затвердевает, а затем размалывается в.
порошок на мельничной установке.
Второй род полимеризации представлен так называемой эмульсионной
полимеризацией. Процесс полимеризации протекает в эмульсии, состоящей,
например, из 2 частей воды, 1 части мономерного стирола с 0,1% сульфата
калия как катализатора, 0,5% Na2HaP2O7, выполняющего функцию регуля-
тора, и 1% мыла как эмульгатора. Эмульсию затем разрушают при помощи
муравьиной кислоты, полимер отфильтровывают, промывают и сушат..
ЛИТЕРАТУРА
1. G г о I 1 II. Р. A., Hearne G. Ind. Engng. Chcm., 31, 1530, 1939.
2. Williams E. C. Trans. Am. Inst. Chem. Engr., 37, 157, 1941.
3. II e a r n c G. Ind. Engng. Chem., 33, 805, 1941.
4. Peters L. M. Ind. Engng. Chem., 40, 2046, 1948.
5. P e i n z e G., S t r i e g I e г А. Пат. ФРГ, Nr. 886907, 1954, Zbl. 4718, 1954..
6. H e a r n e G. и сотрудн. Ind. Eng. Chem., 33, 940, 1941.
7. Chubb L. W. Ind. Chemist, 30, 491, 1954; N e 1 s о n C. R., С о u r t e r M. L.
Chem. Engn. Progr., 50, 526, 1954.
8. J a h n II. Plaste und Kautschuk, 1, 50—56, 1954, W eg 1 e r R. Angew. Chem.,
67, 582—592, 1955.
9. Forbeth T. P., Gaffney В. J. Petrol. Refiner, 34, 12, 160, 1955.
10. 11 e t I i n g K. D., S k e i T. Am. пат. 2608585, 1952. Chem. Abs. 47, 810b
1953; Англ. пат. 668867, Chem. Abs. 47. 1729d, 1953; Hearne G. W., Adams M. L.
Am. пат. 2451485, 1948; Chem. Abs., 43, 2222f, 1949.
11. Watson F. G. Chem. Engng., 54, 12, 106—109, 1947.
12. Schulz II., Wagner H. Angew. Chem., 62, 105—118, 1950.
13. В a 1 1 a r d S. А. и сотрудн. Fourth World Petro]. Congr., Sect., IV/c, 2, 141,
1955.
14. G a 1 i t z e n s t e i n E. G-, Woolf С. J. Soc. Chem. Ind. (London), 69,
289, 19 50.
15. Fettcs E. M., Jorczak J. S. Ind. Engng. Chem., 42, 2217, 1950, Chem.
Abs., 43, 1795g, 1949.
16. Gomberg M. J. Am. Chem. Soc., 41, 1414, 1919.
17. Burrows L. A., Fuller M. F. Am. пат. 2455405, 1948.
18. Smyth II. F., Carpenter С. P. J. Ind. Hyg-Toxikll., 27, 93, 1945.
19. Skeen J. R. Chem. Engng., 57, 331, 1950; Sherwood P. W. Petrol.
Refiner, 28, 3, 129, 7, 10, 1949; Messing R. F. Chem. Ind. 67, 41 — 48, 148, 150, 1950;
Sherwood P. W. Petrol. Process., 9, 1592—1597, 1954.
20. Chem. Engng., 59, 11, 114, 1952; Corrigan T. E. Petrol. Refiner, 32, 2,
87, 1953.
21. К a t z e n R. Petrol. Refiner, 34, 12, 110, 1955.
239
22. M a t i g п о п и сотрудн. Bull. Soc. Chim. Franco, 5, 1, 1.308, 1934,- Ua visP. C.
л сотрудн. Chem, Engng. Progr. Symposium, 48, 4, 91, 1952, Chem. Abs., 8645a, 1953.
23. Chem. Ind., 62, 381, 1948; Loder D. J. Am. пат. 2152852, 1939, Chem. Abs.,
3.3, 5006, 1939; Larson A. P. Am. пат. 2153064, 1939, Chem. Abs., 33, 5006, 1939.
24. Boh in talk J. F. с сотрудн. Ind. Engng. Chem., 43, 786. 1951.
25. P f e f f e r E. С., Epclberg J. Am. пат. 2436076, 1948, Chem. Abs. 42.
1901, 1948.
26. Ц c i d L. S. Gas. 22, 6, 35, 1946; Oil a. Gas J., 50, 28, 218, 1952; Sher-
wood P. W. Petrol cum (London), 17, 96—97, 101, 1954.
27. Сосу S. C. Operating Eng., 12/VII 1948.
28. C u r tn e G. O. Jonsto n F. Glicols, 183, Reinhold Publ. Co., N.Y., 1952.
29. Millet W. II. Ind. Engng. Chem., 42, 2436, 1950; Russ J. M. Lubrica-
tion Eng., 2, 151, 1946.
30. Ferrero P. Bull. Soc. Chim. Belg., 56, 349—368, 1947.
31. К 5 n с с к e II. G., G a w a 1 e к G. Chem. Techn., 8, 734, 1956.
32. Messing R.F. James R. L. Chem. Inds. Week., 68, 2, 19, 1951.
33. Petrol. Refiner, 32, 11, 134, 1953.
34. В e n n i s t e г II. L. Manufacturing Chemist. 54—62, Fcbr. 1957.
35. Aries R. S. Chim. et Ind., 59, 231, 1948; Oil a. Gas J., 48, 46, 108, 1948; Mea-
m er С. B. Chem. Engng. Progr., 1, No. 3; Trans. Am. Inst. Chem. Engrs., 43, 92, 1947.
35a. Schneider II. G., Mistretta V. F. Am. пат. 2473224, 1949, Chem.
Abs., 43, 6645g, 1949.
36. Nelson C. R., Coorter M. L. Chem. Engng. Progr., 50, 526, 1954;
Sherwood P. W. Erdol und Kohle, 8, 465, 1955.
37. Johnson A. J., Nelson C. R. Chem. and Ind., 28—31, Aug. 1953.
38. Bradbnry R. В. и сотрудн. J. Chem. Soc. (London), 1394, 1947.
39. Gwynn В. II., D e g e r i n g. Ed F. J. Am. Chem. Soc., 64, 2216, 1942;
Hagenmeyer H. jr., Hull D. C. Ind. Engng. Chem., 41, 2920, 1949.
40. G i 1 b e r t E. E., Kelly D. II., Woolf С. Am. пат. 2695918, 1954;
Chem. Abs., 50, 399b, 1956; Chem. Engng., 62, 5, 234, 1955.
41. I n s к e e p G. С., M u s s a r a A. Ind. Engng. Chem., 47, 2, 1955.
42. R о e 1 e n O. Angew. Chem. Ausg. A., 60, 3, 213, 1948; Roelen О. в книге
К. Ziegler [45], стр. 157.
43. W e n d e г I. и сотрудн. J. Am. Chem. Soc., 72, 4842, 1950; 74, 4079, 1952;
Bachmann W. E. Cont rou 1 is J. Ibid, 73, 2656, 1951.
44. Roelen О. Сообщение 28/IX 1951 па собрании Общества химиков в Кельне.
45. Ziegler К. Praparative org. Chemie, 1, 149, 1948.
46. Ke ule mans A. I. M. и сотрудн. Rec. Trav. chim. Pays—Bas, 67, 298,
1948.
47. Schulze W. А. и сотрудп. Ind. Engng. Chem., 40, 2308, 1948.
48. P г о e 1 1 W. A. Am. пат. 2433396, 1947; Chern. Abs., 42, 2270, 1948; Ind. Engng.
Chem., 40, 3793, 1948.
49. H a rd у D. V. N., J. Chem. Soc. (London), 1936, 364.
50. Ford T. А. и сотрудн. .1. Am. Chem. Soc., 70, 3793, 1948.
51. H e c h t О., К г о p e r W. в книге К. Ziegler [45], стр. 126, Prapara-
tive org. Chemie, Toil I.
52. Koch H. Brennstoffchem., 36, 321, 1955.
53. Pino P., Magri R. Chimica e Industrie, 34 , 511, 1952.
54. Gresham Wrn. F. Англ. пат. No. 628659, 1949, Chem. Abs., 44, 8364g, 1950.
55. Wonder I. Science, 113, 206, 1951; J. Am. Chem. Soc., 71, 4160, 1949.
56. Ziegler К. и сотрудн. Angew. Chem., 67, 425, 1955.
57. Morin R. D., Bearse A. E. Ind. Engng. Chem., 43, 1596, 1951; В e-
a r s e A. E., Morin R. D. Am. пат. 2419999, 1947, Chem. Abs., 41, 3479d, 1947.
58. Prakt. Chemie, 8, 2, 60, 1957.
59. Allen P. C. Plastiks, 9, 69, 1948; Ind. Chemist, 31, 362, 131, 1955; К r a u s e A.
Chem. Ztg., 79, 657, 1955; Chem. Engng. News, 33, 1822, 1955.
60. H о p f f H., Kern R. Mod. Plastics, 23, 153, 1946; H о p f f H., Goe-
bel S. Ibid, 23, 141 — 145, 1946.
61. Petrol. Refiner, 34, 12, 181, 1955.
62. Ziegler К. и сотрудн. Angew. Chem., 67, 541 — 547, 1955; Ziegler K.,
G> 1 1 e r t H. G. Пат. ФРГ, 878560, 1950, Zbl. 662, 1954.
63. Angew. Chem., 67, 430, 1955.
64. Prakt. Chem., 8, 4, 124, 1957.
65. T h о m a s R. M. и сотрудн. J. Am. Chem. Soc., 62, 276, 1940.
66. T h о m a s R. M. и сотрудн. Ind. Eng. Chem., 32, 1283; Haworth J. P.,
Baldwin F. P. Ind. Engng. Chem., 34, 1301, 1942; Sparks W. J. Am. пат. 2241488,
1941; Zbl., I, 2356, 1942; Англ. пат. 585434, 1947, Chem. Abs. 41, 4006, 1947.
67. Petrol. Refiner, 32, II, 194, 1953.
68. Breitenbach J. W., M a s c h i n A. Z. phys. Chem., Abt. A., 187, 184,
1940.
69. Kharasoh M. S. и сотрудн. Science, 102, 128, 1945; J. Am. Chem. Soc.,
69, 1100, 1947.
240
70. I о у c e R. М. и сотрудн. J. Am. Chem. Soc., 70, 2529, 1948.
71. Ziegler К. Brennstoffchem., 30, 181, 1949.
72. G i 1 I a n d E. R., К a 1 1 a 1 R. J. Chem. Engng. Progr., 49, 647, 1953.
73. Coffman D. D. и сотрудн. J. Am. Chem. Soc., 76, 6394, 1954.
74. S t о с к e 1 b a c h F. E. Am. пат. 2081284, 1937, Chem. Abs., 31, 51107, 1937.
75. Mitchell J. E. J. Trans. Amer. Inst. Chem. Engr., 42, 293, 1946.
76. McAllister S. H. и сотрудн. Chem. Engng. Progr., 43, 4, 189, 1947.
77. К u t z W. M., Corson В. B. J. Am. Chem. Soc., 68, 1477, 1946;
Appleby W. G. и сотрудн. Ibid, 70, 1552, 1948.
78. de M e n t I. Petrol. Refiner, 22, 8, 83, 1943.
79. N a t e 1 s о n S. J. Am. Chem. Soc., 56, 1583, 1934.
80. Hock H., Lang S. Ber. 76, 1130, 1943; Sherwood P. W. Erdol und
Kohle, 7, 154, 362, 1954.
81. Sharrah M. L., F e i g h n e r G. C. Ind. Engng. Chem., 46, 248, 1954.
82. McAllister S. H. из книги В. T. В г о о Jc s, The Chemistry of Petroleum
Hydrocarbons, vol. 3, p. 601, Reinhold Publ. Co, New York, 1955.
83. Gilbert E. E. Там же, vol. 3, p. 633, Reinhold Publ. Co., New York, 1955.
84. Murphree E. V. Oil a. Gas J., 49, 12, 222, 1950.
85. Emerson W. S. Chem. Reviews, 45, 347, 1949.
86. D о w W. Ind. Engng. Chem., 34, 1267, 1942; Mitchell J. E. jr. Trans.
Am. Inst. Chem. Engrs., 42, 293, 1946.
87. Britton E. С.и сотрудн. Ind. Engng. Chem., 43, 2871, 1951.
88. Ward A. L., Roberts W. J. Styrene, Interscience Publ. Inc., New York,
1951.
16 заказ 596.
Глава Ш
РЕАКЦИИ АЦЕТИЛЕНА
Из большого числа возможных реакций ацетилена в настоящей книге
рассматриваются только важнейшие и имеющие в данный момент промышлен-
ное значение.
Большое число важнейших продуктов нефтехимической промышлен-
ности можно получать из этилена и из ацетилена. Выбор того или иного
исходного сырья зависит от его доступности и часто от исторических условий.
Так, хлористый винил, акрилонитрил, ацетальдегид, монохлоруксусную
кислоту и, наконец, бутадиен можно получать как из этилена, так и из
ацетилена (рис. 146).
В Германии ацетальдегид получают почти исключительно из ацетилена,
а в США большая его часть получается из этилена через этиловый спирт.
Акрилонитрил можно получать из обоих исходных веществ.
Различные реакции, к которым способен ацетилен, приведены па
рис. 147.
I. ТРИХЛОРЭТИЛЕН [1]
Получение трихлорэтилена, являющегося исключительно важным рас-
творителем и экстракционным агентом, основано главным образом на деги-
дрохлорировании гетрахлорэтапа. Тетрахлорэтан получают присоединением
хлора к ацетилену:
СП еСН+2С1., -> CJ2CH—СНС12
2С12СП- СПС)2+Са (ОП)2 -> 2С12С=СНС1+СаС!2+2Н2О
Ацетилен и хлор вводятся в тетрахлорэтан при температуре ~80° и
при исключительно хорошем перемешивании. Катализатором служит хлор-
ное железо или хлорная сурьма. В большинстве случаев тетрахлорэтан
переводится в трихлорэтан нагреванием тетрахлорэтана с 10%-ным извест-
ковым молоком до температуры кипения. Принцип работы можно видеть на
схеме рис. 148.
Хлор и ацетилен раздельно подаются в тетрахлорэтан, содержащий
0,1% вес. хлорной сурьмы, и приводятся в тесное соприкосновение в охла-
ждаемом снаружи смесителе. Объемное отношение ацетилена к хлору соста-
вляет примерно 2,00 : 2,05. Конструкция установок на разных заводах
различная.
Образовавшийся тетрахлорэтан перегонкой освобождается от катализа-
тора, который возвращается на установку. Далее тетрахлорэтан поступает
в обогреваемую колонну для дегидрохлорирования, снабженную тарелками
242
рис. 146. Сравнительны? возможности этилена и ацетилена как исходных материалов для нефтехимической промышленности.
АЦЕТИЛЕН
2С12 , Тртряу пиратап —* Трихлорэтилен —* МОПОХЛОРУКСУСНЯЯ
Хлор (в избытке) сшим-Дихлорэтилеи (СНС1-СНС1)
НС1 упористый пинии * Поливинилхлорид
Х-Гр гт'-> —* Акрилонитрил —* Полиакрилонитрил и сополимеры
1120 V.— Ацетальдегид H2SO4, HgSOi ^Пропаргиловый спирт Формальдегид /
^Бутиндиол-! ,4 Частичное восстановление w Этилен
Сополимеры с акрилонитрилом —► Акрилан СН3СООН - Винилацетат^
^Поливипилацетат 1Т ' , । Полипинилформаль Поливиниловый спирт/ ^Ноливппи лбу тира ль Полимеризация Бензол
Полимеризация Купрен
Полимеризация _ Циклооктатетраен
N1 (CN)2 Ацетальдегид _ вутиц.^ол-з
л ДкПИТТЛПЯЯ Т-НГЛЛОТЯ
Ni (СО)4 HCI v 1 ►Хлоропрен НС—СН ’ ( пипнля патиттр,я пивипи папетилен *
Cu2Cio + NH4Cl ьипилацетипеп, дивинила^ Неопрен, совиреп ^Ангидрид уксусной кислоты СО + СПзСООН /
^Ангидрид пропионовой КИСЛОТЫ
Н2О Апетон
Газовая фаза ROH г рпттмттппьлА пфиры —> Ппттипинм.ттОпыр, дфиры
RHS
А8С18 С.1СН СН АчС.Ы ('лИ)Ияпт1
Карбазол
а-Пирролидон л
CO + RSH .
СО 1 ROH
Пиролиз i— Ацетиленовая сажа
слота
Рис. 147. Реакции ацетилена.
244
или заполненную кольцами Рашига. В эту же колонну сверху подают в
10 %-ном избытке 10%-ный раствор известкового молока. Лучший контакт
поступающих в колонну паров тетрахлорэтана с известковым молоком дости-
гается подачей в нижнюю часть колонны водяного пара.
Рис. 148. Схема ^получения трихлорэтилена из
ацетилена.
Азеотропная смесь, со-
стоящая из 93% трихлор-
этилена и 7% воды, отгоняет-
S ся при 77°. У дна колонны
поддерживается температура
102°. Головной продукт этой
колонны затем освобождает-
ся от воды и перегоняется.
Чистый трихлорэтилен
необходим в первую очередь
для обезжиривания металли-
ческих поверхностей, для
экстракции жиров и масел,
как растворитель и для дру-
гих целей.
1—смеситель; 2— колонна для отгонки тетрахлорэтана;
3 — колонна дегидрохлорирования; 4 — отделитель; <5 —
колонна для отгонки трихлорэтилена.
Линии: I — хлор; II — тетрахлорэтан; III — ацетилен;
IV — возврат SbClg; V— известковое молоко; VI — раствор
CaCh; VII — иар; VIII — отходящая вода; IX — трихлор-
этилен; X — дренаж.
II. ХЛОРИСТЫЙ ВИНИЛ [2]
(см. стр. 181)
Хлористый винил может
получаться как дегидрохло-
рированием 1,2-дихлорэтана,
получаемого присоединением хлора к этилену, так и гидрохлорированием
ацетилена:
НС л СН+НС1 -> СН2=СНС1
Газообразный хлористый водород, содержащий менее 0,02% воды,
смешивается с сухим ацетиленом. Хлористый
избытке против стехиоме-
трически необходимого ко-
личества. Тщательный кон-
троль отсутствия воды не-
обходим, чтобы предотвра-
тить коррозию и не до-
пустить образование аце-
тальдегида.
Газовая смесь посту-
пает в реактор (рис. 149),
представляющий собой
трубчатую печь, в которой
находится катализатор —
активированный уголь,
пропитанный хлорной
ртутью. Перед началом ре-
акции температура в реак-
торе при помощи теплоно-
сителя доводится до 140°.
После начала экзотерми-
ческой реакции теплоно-
водород берется в 10%-ном
Рис. 149. Схема получения хлористого винила реак-
цией хлористого водорода с ацетиленом.
1 — смеситель; 2 — трубчатая печь с катализатором; 3 — ста-
билизационная колонна*, 4 —ректификационная колонна для
хлористого винила; <5 — колонна для промывки; 6 — приемная
емкость для хлористого винила; 7 — перегонное устройство.
Линии: I — хлористый водород, II — ацетилен; III — охла-
ждающая среда или теплоноситель; IV —дренаж; V — остаток;
VI — фенол (стабилизатор); VII—альдегид-, VIII —хлори-
стый этилен.
ситель служит уже охлаж-
дающим агентом. По мере старения катализатора температура постоянно
повышается и доводится до 200°. Продуктами, протекающих при этом
побочных реакций, являются ацетальдегид и дихлорэтилен (1, 1-дихлор-
этан) .
245
Выходящие из реактора газы охлаждаются и в первой колонне освобо-
ждаются от еще содержащихся в них ацетилена и хлористого водорода. Оста-
ток из этой колонны поступает во вторую колонну, где отделяется хлори-
стый винил, кипящий при —13,9°. Ацетальдегид и дихлорэтилен получаются
как остаток и могут затем подвергаться перегонке.
Хлористый винил промывают едким натром и водой и после добавки
некоторого количества фенола, как ингибитора полимеризации, он может
храниться. Выход составляет примерно 80—85%.
Ш. ВИНИЛАЦЕТАТ [3]
Винилацетат получают присоединением ледяной уксусной кислоты
к ацетилену:
НС = СН + СИ3СООН -> И.,С=СН—ОСОСПз
Ацетилен пропускают через нагретую до 70—80° уксусную кислоту,
поддерживая такие условия, чтобы отношение уксусной кислоты к ацети-
лену в смеси отвечало отношению 15 : 85 по объему. Далее эту смесь подогре-
Рис. 150. Схема получения винилацетата из ацетилена и уксусной кислоты.
1 — испаритель для уксусной нислоты; 2 — реактор с катализатором; з — сепаратор; 4 — ректифика-
ционные колонны.
Линии: I — ацетилен; II — ледяная уксусная кислота; III — циркулирующий ацетилен; IV — цир-
кулирующая уксусная кислота; V— ацетилен, ацетальдегид, «чцетоц- VI— винилацетат VII— ук-
сусная кислота; VIII — этилидендиацетат; IX — уксусный ангидрид.
вают до 170—180° и пропускают над активированным углем, обработанным
ацетатом цинка (20—30% вес. ацетата цинка, рис. 150). При малой актив-
ности катализатора температуру повышают до 200°. После трех месяцев
работы катализатор, как правило, заменяют. В час подается около 300 л
газовой смеси на 1 л катализатора. Теплота реакции, составляющая
24 ккал/моль винилацетата, отводится посредством циркуляции силиконо-
вого теплоносителя.
После охлаждения чистый кипящий при 76° винилацетат отделяют
перегонкой, а избыток ацетилена возвращают в реактор. В первой колонне
отделяются ацетилен, ацетальдегид и ацетон. Ацетилен идет обратно на
установку. Во второй колонне винилацетат отделяется от уксусной кислоты.
В третьей колонне уксусная кислота отгоняется от высококипящих состав-
ных частей, как этилидендиацетат, уксусный ангидрид и др. Выделившаяся
здесь уксусная кислота также возвращается в процесс.
IV. АКРИЛОНИТРИЛ [4]
Кроме ранее описанного способа получения акрилонитрила реакцией
окиси этилена с синильной кислотой и последующего дегидратирования
получаемого таким путем нитрила оксипропионовой кислоты, важнейшее
246
промышленное значение имеет получение акрилонитрила взаимодействием
синильной кислоты с ацетиленом:
НС = CH+HCN -> CH2=CHCN
Так как и синильная кислота и ацетилен могут получаться из метана,
то, очевидно, метан можно рассматривать как основное сырье для производ-
ства акрилонитрила.
Способ состоит в том, что ацетилен и синильную кислоту в молярном
соотношении 10 : 1 подают в насыщенный при 40° раствор хлористой меди
(одновалентной) и хлористого алюминия, подкисленный соляной кислотой
до pH = 3,5 (рис. 151). При этом в реакцию вступает около 10% ацетилена,
так что синильная кислота используется практически без остатка. Реакция
идет при температуре около 80°. На 1 л катализаторной жидкости в час
образуется 15—18 г нитрила. После одного-двух месяцев работы катализатор
должен регенерироваться. Давление в процессе немного выше атмосферного.
Рис. 151. Схема получения акрилонитрила реакцией синильной кислоты с ацетиленом.
1 — реактор с катализатором; 2 — водяной абсорбер; 3 — разделительная колонна; 4 — отделитель’
5 — промывочная колонна; 6 —дистилляционные колонны.
Линии: I — свежий ацетилен; II — синильная кислота; III — циркулирующий ацетилен; IV — ацети-
лен и высшие ацетилены; V — сырой акрилонитрил; VI — винилацетилен; VII — легкие составные
части, ацетальдегид, винилацетилен; VIII — чистый акрилонитрил; IX — загрязненный акрилони-
трил; X — остаток.
Выходящая из реактора газовая смесь (акрилонитрил кипит при 78°)
поступает в абсорбер, где обрабатывается водой, в которой акрилонитрил
растворяется. Растворимость акрилонитрила равна 6 г в 100 мл воды. Ацети-
лен и моновинилацетилен разделяются затем в специальной промывочной
колонне. Ацетилен возвращается в процесс.
Вода в абсорбере обогащается акрилонитрилом до содержания послед-
него около 2%. Далее водный раствор поступает в разделитель, где 80%-ный
сырой акрилонитрил отгоняется с водяным паром. Слой водного конденсата
возвращается в разделитель, а масляный слой подвергается дальнейшей
перегонке. В первой колонне отделяются низкокипящие загрязнения, состоя-
щие главным образом из ацетилена, моновинилацетилена и ацетальдегида.
В следующей колонне при остаточном давлении 140 мм рт. ст. отгоняется
акрилонитрил 99,5%-ной чистоты. Остаток из этой колонны, еще содержа-
щий акрилонитрил, разделяется на загрязненный акрилонитрил, возвра-
щающийся в первую колонну, и остаток. Выход акрилонитрила, считая
на синильную кислоту, составляет 85%, а считая на ацетилен 75%.
Некоторое представление о реакционной способности акрилонитрила дает
рис. 152.
247
АКРИЛОНИТРИЛ
Полимеризация_____
С12; Вг2_________
НО_________________2^
Н0С1_______________
HCN_______________
Н20; н+
RSH; 0Н~~____________
ROH_________________
NH3________________*
r-nh2
Сополимеризация
с хлористым винилом
Сополимеризация
с винилацетатом
Сополимеризация____
с бутадиеном
Рис. 152.
ПАИ-волокко, орлон, волкрилои, дралин
а,0-Дигалопропионитрил
0-Хлорпропионитрил
а-Хлор-р-гидроксипропионитрил
Динитрил янтарной кислоты
Акриловая кислота —»'Эфиры акриловсн
кислоты —► Полиакрилаты
Р-Алкилмернаптопропионитрил
Р-Алкоксипропионитрил
Р-Аминопропионитрил
Р-Алкиламииопроииопитрил
Винион N, динель
Акрилен
Буна N
Реакции акрилонитрила.
V. АЦЕТОН ИЗ АЦЕТИЛЕНА
Промышленное получение ацетона из ацетилена основано на пропуска-
нии смеси ацетилена и водяного пара над соответствующим катализатором.
Реакция протекает по уравнению
2НС -СН+ЗН2О СН3—СО—СН3 + СО2 + 2Н2 + 40 ккал/моль
Рис. 153. Схема получения ацетона из ацети-
лена.
Отдельные стадии процесса,
вероятно, могут быть пред-
ставлены следующими уравне-
ниями:
О
нс -= сн+н2о ->сн3—
хн
о
сн3-с^ +н2о->сн3-
н
—СООН+Н2
2СН3—соон ->сн3—со—
— сн3+со2+н2о
I — осадитель; 2 — центрифуга; з — шаровая мельни-
ца; 4 — реактор; 5 — абсорбционная колонна; 6 — ди-
стилляционная колонна.
Линии: I— раствор хлорного железа; II— водный
раствор аммиака; III — вода; IV — отходящая вода;
V — окись цинка; VI — катализатор; VII — водяной
пар; VIII — ацетилен; IX — отходящие газы; X—аце-
тон; XI — вода в дренаж.
В промышленных условиях
для этой цели может непосред-
ственно применяться газ про-
цесса Захсе (см. стр. 95), содер-
жащий 8—9% ацетилена. Силь-
но обводненный сырой ацети-
лен после освобождения от
сажи пропускают над цинковым катализатором при 400°.
В США для этой цели в большинстве случаев применяют технически
чистый ацетилен и катализатор из окиси железа и окиси цинка, находящийся
в виде шариков в трубчатой печи. Ацетилен и водяной пар смешивают в
объемном отношении 1 : 10 и пропускают над катализатором. Продукты
реакции промывают водой, а затем раствор подвергают перегонке. Незначи-
тельное количество ацетальдегида получают как побочный продукт.
Схема процесса приведена на рис. 153.
248
VI. АЦЕТОНИТРИЛ
При пропускании разбавленной водородом смеси ацетилена и аммиака-
в соотношении 1 :1 над осажденным на кизельгуре сульфатом цинка при 400°'
образуется ацетонитрил с 90%-ным выходом. Реакция идет без применения,
давления по уравнению
НСнСН+КНа-»СН3—cn+h2
Ацетонитрил кипит при 82°. Он ядовит. Продукт его трихлорирования,
применяется как инсектисид под названием «тритокс».
Реакцией аммиака с ацетиленом в жидкой фазе при 30—40° и 20 ат
в присутствии ацетилида меди как катализатора получают 2-метил-5-этил-
пиридин по уравнению
С8Н5\ СП^
с/ СН
4CH CH + NH3-> || I
СН С—СН8
N
VII. ВИНИЛИРОВАНИЕ АЦЕТИЛЕНОМ [5]
Реакцией ацетилена со спиртом в присутствии щелочи образуются вини-
ловые эфиры. Реакция идет в соответствии с уравнениями
C2HSOH4-КОН -> С2Н5ОК4-Н2О
C2H6OK+HCsCH -> С2Н5ОСН=СНК
С2Н6ОСН=СНК4-С2Н6ОН -> С2Н6ОСН=СН24-С2Н6ОК
Щелочи берут 1—2%, считая на спирт. Температура реакции для низ-
ших спиртов 120°, для высших спиртов 160—180°. Давление достигает 3—
10 ат.
Ацетилен дает с воздухом и осо-
бенно с кислородом очень взрывчатые
смеси. При сжатии чистый неразбав-
ленный ацетилен может распадаться
со взрывом и с образованием сажи.
Поэтому его нельзя сжимать более чем
до 2 ат. При работе с высокими дав-
лениями содержание ацетилена в газах
не должно превышать 70%. В качестве
разбавляющего агента обычно приме-
няют азот.
Схема работы при винилировании
представлена на рис. 154. Спирт и 1%
щелочи насосом подают в нагретый
до 150—180° реактор под давлением,
равным давлению в реакторе. Одновре-
Рис. 154. Схема получения виниловых,
эфиров из ацетилена и спиртов.
1 — компрессор; 2 — автоклав; 3 — установ-
ка глубокого охлаждения; 4 — ректифика-
ционная колонна.
Линии: I — спирт с 1 % КОН; II — циркули-
рующий ацетилен; III — свежий ацетилен и
Но; IV — сырой эфир; V — чистый винплал-
киловый эфир; VI — остаток.
менно в реактор поступает разбавлен-
ный азотом ацетилен. Выходящая из
верха реактора газовая смесь захваты-
вает с собой эфир, кипящий при зна-
чительно более низкой температуре, чем
спирт (этилвиниловый эфир кипит при 35°, метилвиниловый эфир при 8°).
Путем глубокого охлаждения газ освобождается от эфира и возвращается
в реактор. Эфир очищается перегонкой. Небольшая часть газов циркуляции
постоянно отводится из установки и заменяется свежим газом.
249-
Ллкилвиниловые эфиры легко полимеризуются. Такие продукты, как
лутопал, игевин и денсоидрип, являются полимерами различных алкилвини-
ловых эфиров.
Фенолы ведут себя подобно спиртам. С ацетиленом они дают винилфе-
ниловые эфиры. Иа рис. 155 представлены важнейшие реакции виниловых
эфиров.
ВИНИЛОВЫЕ ЭФИРЫ
Полимеризация
________HGN_________
НС1 '
NH34-CO—Ni
Карбоновые кислоты
Спирты
Поливиниловые эфиры (склеивающие ве-
щества, сырье, для получения лаков, ис-
кусственные вещества)
Но ,
।---—*Эфироамипы
Эфиронитрилы
1 ->Эфирониелоть1
ОН.
ц-Хлоралкиловые эфиры
Производные пиридина (из этплвипило-
вого эфира----*2-метил-5-этилпиридин)
Эфиры карбеновых кислот
Ацетали
Рис. 155. Реакции виниловых эфиров.
Азотсодержащие соединения, в которых азот связан с водородным ато-
мом, способным реагировать со щелочами, также способны винилироваться.
Из карбазола и едкого кали при 180° и 10—20 ат с ацетиленом (разбавлен-
ным азотом) при использовании в качестве растворителя метилциклогексана
получают винилкарбазол, дающий полимер, известный под названием «луви-
кап», применяемый в электропромышленности.
Пирролидон, получаемый из у-бутиролактона и аммиака, дает с ацети-
леном винилпирролидон, полимер которого перистон применяется как за-
менитель крови. у-Бутиролактон в свою очередь получается посредством
алкинольного синтеза из ацетилена и формальдегида через бутиндиол.
VIII. ЭТИНИЛИРОВАНИЕ АЦЕТИЛЕНОМ ПРОПАРГИЛОВЫЙ СПИРТ,
БУТИНДИОЛ, ТЕТРАГИДРОФУРАН [5]
Под этинилированием по Реппе понимается реакция ацетилена с альде-
гидами с образованием спиртов ряда ацетилена. С формальдегидом полу-
чают пропаргиловый спирт и далее бутиндиол; с ацетальдегидом — бутин-
З-ол-2
НС = СН + СН2О -> СП - с—СН2ОН
Пропаргиловый спирт
HCl.С-СП2ОН+СН2О -> НОСН2—С = С—СНоОН
1,4-бутпядиол
нс - сн+СИзСОН —> НС = с—снон—сн3
Бутин-З-Ол-2
Эти спирты являются исключительно реакционноспособными соедине-
ниями и могут претерпевать самые разнообразные превращения. Особую
техническую ценность имеет 1,4-бутиндиол — исходный продукт для полу-
чения бутадиена на базе ацетилена и смеси окиси углерода и водорода.
Этинилирование проводится в присутствии ацетиленида меди, осажденного
на носителе.
В оросительной колонне (рис. 156) 30%-ный раствор формальдегида
встречается над катализатором под давлением 3—5 ат и при температуре
90—100° с ацетиленом, разбавленным азотом. Продукты реакции, выходя-
щие из нижней части колонны, охлаждаются и поступают в разделитель.
В разделителе, работающем под давлением, отделяется непрореагировавший
ацетилен, который возвращается в процесс. Спирт из нижней части раз-
делителя поступает на ректификацию.
250
В первой колонне в качестве остатка получается примерно 35%-ный
водный раствор бутиндиола. Во второй колонне в остатке получается про-
паргиловый спирт, который может быть возвращен в процесс или исполь-
зован как таковой. Через верх этой колонны отходит метиловый спирт,
содержавшийся в сыром формальде-
гиде. Выход бутиндиола составляет
около 98% от теоретического.
Пропаргиловый спирт кипит при
113°, бутиндиол при 1 мм рт. ст. —
при 105—108°. Пропаргиловый спирт
частичным восстановлением может
быть переведен в аллиловый спирт,
последний дальнейшим восстановле-
нием — в пропиловый спирт. Рас-
твором хлорной меди (двухвалентной)
медная соль пропаргилового спирта
окисляется в гександиин-2,4-диол-1,6
согласно реакции
2CuCaC—CH2OH+2CuC12 -> 2Cu2C12 +
+ НОСН2—С = С—С - С—СН2ОН
который при 40—60° в присутствии
никеля может быть гидрирован водоро-
дом в гександиол-1,6, применяемый
как компонент этерификации полиэфи-
Рис. 156. Схема этинилирования альде-
гидов ацетиленом.
j — компрессор; 2 — оросительная колонна,
катализатор; 3 — отделитель, работающий под
давлением; 4—дистилляционная колонна.
Линии: I—ацетилен и N2; II—30%-пый
формальдегид; III — пропаргиловый спирт;
IV — циркулирующий ацетилен-, V — возврат
пропаргилового спирта; VI — метиловый
спирт; VII — бутиндиол 35 %-ный.
ров, а также для получения полиизоцианатов. Окислением гександиола
азотной кислотой получают адипиновую кислоту. На рис. 157 даны важней-
шие реакции пропаргилового спирта и возможности его использования.
При частичном восстановлении над железным катализатором бутин-
.диол переходит в бутендиол, который при каталитическом дегидратировании
над окисью алюминия при 250° легко переходит в 2,5-дигидрофуран:
HOCH, —С = С —СН.ОН НОСИ,—СН=СН—СИ,ОН ^°?
ПРОПАРГИЛОВЫЙ СПИРТ
Восстановление
“(Н2, Fej
Восстапоиление
(Н2> Си)
Дегилрир о вапи е
Окисление
(О,, Сп2С|7)
Окисление
Гидратации
H2SO4
Реакция с формальдегидом
Гидрохлори ропанне
SOCI2_
NaOCl
Рис. 157. Реакции
си2 сн2
o'
Аллиловый спирт
Пропиловый спирт
Гуа нидип
Пропаргиловый альдегид--------*
—- 2-аминопирпмидин
Гександиин-2 ,4-диол-1 ,6 —-* Гександиол
|окпсление
Лдипивовая кислота
CnClo TTZ4^
Пропаргиловая кислота------► НООС
НООС—С С—С С—С
Окисление (СНд—СО—СН2ОН)
Г.утиндиол-!, 4
2-хлораллпловнй спирт (СНо_-=СНС1 —
—СП2ОН)
З-хлорпропип-1 (НС-С—СН-эС1)
I -хлор-З-гидроксипропин-1 (С1Сг"С—
-СН2ОН)
пропаргилового спирта.
251.
Дигидрофуран можно каталитическим окислением над пятиокисью
ванадия перевести в ангидрид малеиновой кислоты:
СН = СН
I I
сн2 с
Катализатор ц \
----°*-----I /°
СН—СС)
Бутендиол можно далее с безводной синильной кислотой превратить
в динитрил дигидромуконовой кислоты, который затем можно гидрировать
в гексаметилендиамин.
Полное гидрирование бутиндиола дает бутандиол-1,4. Этот процесс
протекает исключительно легко при 180—200° и давлении водорода 200 ат
над кизельгуром, обработанным никель — медь — хромовым катализатором..
Из бутандиола получают омылением хлористым водородом 1,4-дихлорбутанг
который при взаимодействии с цианистым калием дает с 90%-ным выходом!
динитрил адипиновой кислоты, последний может быть гидрирован в гекса-
метилендиамин.
Бутандиол-1,4 при нагревании с водной фосфорной кислотой при 300°
и давлении 100 ат почти количественно отщепляет воду с образованием
тетрагидрофурана, кипящего при 64—65°:
НОСН2—СН2 СЕ12—СН2ОН2000_ юо ат | | +Н2О
СН2 СН2
Тетрагидрофуран реагирует под давлением с хлористым водородом и
образует 1,4-дихлорбутан. Промежуточным продуктом этой реакции является
4-хлорбутанол-1. При нагревании с аммиаком в автоклаве получается пир-
ролидин, с аминами — азотом замещенный пирролидин — соединение, могу-
щее найти разнообразное применение.
Тетрагидрофуран является последним продуктом бутадиенового синтеза
Реппе. Так как каталитическая дегидратация бутандиола в бутадиен про-
ходит недостаточно гладко и сопровождается образованием побочных про-
дуктов реакции — пропена и формальдегида, бутандиол превращают сна-
чала в тетрагидрофуран, который затем при 260—270° в присутствии водя-
ного пара легко дегидратируется в бутадиен.
Следующим важным производным бутандиола является бутиролактон.
Его получают с 90%-ным выходом, пропуская при 200° безводный бутандиол
в смеси с кислородом над силикагелем, обработанным медным катализатором:
г.. СН2 — СН2
НОСН2~СН2—СН2—СН2ОН-q2—| | +2н2о
сн2 с=о
Лактон кипит при 204,5°, он способен ко многим реакциям. При нагре-
вании с аммиаком до 200° под давлением он дает а-пирролидон:
СН2 — СН2 СН2 — СН2
| | 4-NH3—>| | +Н2О
сн2 с=о сн2 с=о
NH
252
Бутиролактон реагирует с хлористым водородом с образованием у-хлор-
ыасляной кислоты. С сульфитом натрия дает у-сульфомасляную кислоту.
<3 натриевой щелочью образует у, у'-дикарбоксидипропиловый эфир, с гидро-
сульфидом натрия у, у'-тиодимасляную кислоту. Обе последние кислоты
как дикарбоновые применяются для получения пластификаторов, полиами-
Т.УТИНДИОЛ
Гидрирование
< Дигидрофуран--------- Малеиновая кислота
Динитрил дигидромуконовой кислоты
Гексаметилендиамин
1,4-дихлорбутан-------► Адипонитрил ------------?
Гидрирование
Бутавдиол
Гексаметилендиамин
I л Пирролидин
------- Тетрагидрофуран
=* Бутадиен
Полимеризация
Окисление
Дегидратация
“► Бутиролактон
сн2он
носн2-.-^\-сн2он
HOCH2-^J-CH2OH
сн2он
Гексаметилолбензол
Окисление
СООН
НООС—СООН
HOOC-L "-СООН
СООН
Меллитовая кислота
Эстерификация с HC1
Гидратация
БУТИРОЛАКТОН
NH3 ч
HC1______
NaOH.
NaSH
Na2SO3>
NaCN
, Дегидрохлорирование „
1,4-дихлорбутпн--------------------► Диацетилен
2-кетобутандпол-1,4
(СН2ОН-СО—СН2-СН2ОН
I <----Восстановление
I
Бутантриол-1.2.4
(СН2ОН—СНОН—СН2—СН2ОН)
Рис. 158. Реакции бутипдиола.
а-Пирролидоп
у-Хлормасляпан кислота
V.V'-Дикарбоксипропиловый эфир
[О (СН2СН2СН»СООН) 2]
Тиодимасляная кислота
[S(CH2CH2CH2COOH)2]
V-Сульфомасляная кислота
Глутаровая кислота
Z
Цианомасляпан кислота
Пиперидон
Рис. 159. Реакции бутиролактона.
доп и искусственных смол. Из бутиролактона и цианистого натрия получают
натриевые соли у-цианомасляной кислоты, которая может быть омылена
в глутаровую кислоту или гидрирована и циклизирована' в пиперидон.
На рис. 158 и 159 показаны важнейшие реакции бутиндиола и бутиролак-
тона [6].
IX. КАРБОНИЛИРОВАНИЕ АЦЕТИЛЕНА
Под карбонилированием понимается открытая Реппе реакция присоеди-
нения окиси углерода и воды к ацетилену в присутствии карбонила никеля,
ведущая к образованию акриловой кислоты:
ПС - СН + СО +Н2о - СИ2=СН—СООН
Ока основана на воздействии карбонила никеля на ацетилен и идет
без применения давления в присутствии соляной кислоты. В присутствии
спиртов образуется соответствующий эфир акриловой кислота. Выход соста-
вляет до 95% от теоретического. Реакция может быть представлена следую-
щим образом:
НС - CH + ROII+ -г N* (С0)4+ -^-ПС1 —> СНг=С1 [—СООП + ~ NiCk+ П2
4 ~ 4 4
Добавлением аммиака к раствору хлористого никеля получают комплекс-
ное соединение вида [Ni (NH3)e] Ch, которое при обработке его окисью угле-
рода под давлением 50—100 ат при 80° позволяет количественно получить,
обратно карбопил никеля:
[Ni (NHa)e] С12+5СО+2И2О -> Ni (CO)4+2N1I4C1 + (NH4)2 COS+2NII3
Реакция проводится непрерывно следующим образом. Смесь ацетилена
и окиси углерода в соотношении 1:1с температурой 170° и под давлением
30 ат нагнетается в реакционную камеру, содержащую спирт с растворен-
ным в ном катализатором. Последний состоит из бромистого пийеля и бро-
мистого алкила в присутствии трифенилфосфита, который служит комплексо-
образователем. Алкильный состав бромида соответствует примененному
в реакции спирту.
Эфир акриловой кислоты отделяется от комплекса перегонкой, послед-
ний возвращается в процесс.
X. ПОЛИМЕРИЗАЦИЯ АЦЕТИЛЕНА
Давно известен под названием «купрен» рыхлый, мучнистый продукт
полимеризации ацетилена, получаемый вдуванием ацетилена в рорячее
масло в присутствии медного порошка. Соединением двух молекул ацетилена
образуется моновинилацетилеп (мова), три молекулы дают соответственно
дивипилацетилеи (дива):
2СН 7 СИ НС” С—СН = СИ., (мова)
ЗСН СИ -о П2С=СИ—С-С—CH - CH., (дива)
Эти соединения легко разлагаются со взрывом. Моновинилацетилен
получается пропусканием ацетилена через концентрированный раствор
хлористого аммония и хлористой меди (одновалентной), слегка подкислен-
ный соляной кислотой, при 60—70° и 2—3 ат. Продукты реакции пропу-
скают через холодильник, где большая часть воды отделяется от целевого
продукта, имеющего температуру кипения 5°. Затем сырой винилацетилен кон-
денсируется путем глубокого охлаждения и затем очищается ректифи-
кацией.
При действии хлористого водорода в присутствии солянокислого рас-
твора хлористой меди (одновалентной) при 40—45° образуется 2-хлорбута-
диен (хлоропрен). Это соединение кипящее при 60°, легко полимеризуется
в особо маслостойкий каучук (неопрен, совпрен).
Интересный новый вид полимеризации, соединенной с циклизацией,
представляет впервые осуществленная Реппе реакция получения цикло-
октатетраена. Циклооктатетраен получают, пропуская смесь ацетилена с
азотом под давлением 15—20 ат (давление азота около 6 ат) через суспен-
зию цианистого никеля в тетрагидрофуране при 60—70°.
Циклооктатетраен — чрезвычайно реакционноспособное соединение. Ка-
талитическим гидрированием из него получают циклооктан, который при оки-
слении переходит в пробковую кислоту. При действии гипохлорита цикло-
октатетраен дает терефталевый ангидрид и соответственно терефталевую
кислоту [7].
254
ЛИТЕРАТУРА
1. Petrol. Refiner, 34, 12, 185, 1955.
2. Petrol. Refiner, 34, 12, 189, 1955.
3. Petrol Refiner, 34, 12, 188, 1955.
4. Petrol. Refiner, 34, 12, 125, 1955; Chem. Engng., 62, 9, 288, 1955.
5. R e p p e W. Proceedings Fourth World Petrol. Congr. Sect., IV/C, Paper 14,
p. 291-325, 1955.
6. R e p p e W. Nene Entwicklungen auf dem Gebiet der Chemie des Acetylens und
des Kohlenoxids. Springer— Verlag, 1949, Berlin/Gottingen/Heidelberg; Reppe W.
Chemie und Technik der Acetylen — Druck — Reaktionen, 2 Aufl., Verlag Chemie, 1952,
Weinheim — Bergstrasse.
7. R e p p e W. (5) u Proceedings Fourth World Petrol. Congr. Sect. IV/C Paper 14,.
p. 291 — 325, 1955; Reppe W., Chemie und Tcchnik der Acetylen — Druck — Reaktio-
nen, Verlag Chemie, 2 Aufl., 1952, Weinheim — Bergstrasse.
Глава IV
ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ БУТАДИЕНА
I. ХЛОРИРОВАНИЕ
При присоединении хлора к бутадиену при низкой температуре
•образуются с хорошими выходами 1,4-дихлорбутен-2 и 3,4-дихлор-
•бутен-1. Оба соединения дают при нагревании с едкими щелочами хло-
ропрен.
Особый интерес представляет 1,4-дихлорбутен-2, обладающий двумя
исключительно подвижными атомами хлора. При взаимодействии с циани-
•стым натрием хлор легко замещается нитрильными группами, образуя
ненасыщенный динитрил, который гидрированием может быть переведен
в гексаметилендиамин. Кроме того, ненасыщенный динитрил может быть вос-
становлен в динитрил адипиновой кислоты, а этот последний омылен в ади-
пиновую кисл»ту. С этой точки зрения бутадиен можно рассматривать как
важнейший исходный материал для получения найлона.
На рис. 160 дана схема получения найлона из крекинг-газов.
Бутадиен может присоединять также 1 пли 2 моля хлорноватистой кис-
лоты с образованием хлоргидрина, который может быть переведен в эпоксид
бутадиенмонооксид и бутадиендиоксид).
II. ПРИСОЕДИНЕНИЕ SOa (СУЛЬФОЛАНЫ)
Бутадиен и другие 1,3-диены легко присоединяют SO2 с образованием
циклических сульфонов, из которых они могут регенерироваться нагре-
ванием:
СН2=СН—CH=CH2+SO
Высокая СН СН
Температура
сн2 сн2
Так, например, бутадиен можно регенерировать из такого соединения
с двуокисью серы нагреванием до 125°. Этот способ может быть использо-
ван для получения чистых диенов, когда они находятся в смеси с другими
близко кипящими моноолефинами или парафинами. В табл. 86 приведены
температуры плавления и разложения некоторых продуктов присоединения
двуокиси серы к диенам.
.256
17 Заказ 596-
Рис. 160. Получение найлона на основе крекинг-газов.
Ненасыщенные сульфоны гидрированием могут быть превращены в насы-
щенные соединения, растворимые в воде и являющиеся одновременно селек-
Таблица 8С Температуры плавления и разложения некоторых продуктов присоединения SO2 к диенам тивнымп растворителями для ароматических угле- водородов, обладающими резко выраженной изби-
Диен Температу- ра плавле- ния сульфо- на, °C Температу- ра разло- женпя, °C рательностью. Они совер- шенно инертны, не вызы- вают коррозии и с успе- хом применяются для
Бутадиеп-1,3 Пипсрилен Изопрен 1,3-диметилбутадиеп . . . 2,3-диметилбутадиен . . . 65 63 40 135 125 100 125 100 140 экстрактивной перегонки. Фирма Шелл выпускает эти продукты на рынок под названием сульфола- нов 11]. Из ацетона можно по-
лучить 2,4-диметилсуль-
фолан через следующие промежуточные реакции:
СН3 СП3
2СН3-СО-СП3 £”^2.\С-СН2-С0-СЦ. 5®= >С—CII2—CIIOII
СН3| СПз! I
on он СП;!
Детатация^ CHo=c_CH=CH _^н з
I
СНз
СП3—с=сн
I
П2С СП- сн3
so2
СНз-СН-СП,
Восстановление | |
Н,С СН—сп3
so2
2,3-диметилсульфолан имеет температуру замерзания —3е и кипения 281 °.
Ш. РЕАКЦИЯ ДИЛЬСА-АЛЬДЕРА
Бутадиен реагирует с моноолефинами по схеме с образованием про-
СН2
СН СН2
СП2=СН—СН=СН24-СН2=СН— Н___а. I I
СН СП—в
(JL
изводных циклогексена. Радикал R содержит обычно группу СО, как
она находится в карбоновых кислотах, ангидридах, хлоридах эфирокислот,
кетонах, альдегидах и т. д. Радикал R может означать также нитро-,
циан- и винтилгруппу или наконец водородный атом.
Одним из известных продуктов присоединения является ангидрид
тетрагидрофталевой кислоты, получаемый реакцией ангидрида малеиновой
кислоты с бутадиеном. Он может служить промежуточным продуктом для
получения синтетических смол. Диметиловый эфир соответствующей кислоты
является сильным инсектисидом.
258
Циклопентадиен также может реагировать с ангидридом малеиновой
кислоты. При этом получается ангидрид 3,6-эндометилен-1,2,3,6-тетра-
гидро-цнс-фталевой кислоты
СН
/\ со
нсГ
| СН2| о
НС сн Z
//ЧСО
сн
реагирующий подобно ангидриду тетрагидрофталевой кислоты с гликолями
и глицерином с образованием алкидных смол.
Реакция Дильса-Альдера возможна также между двумя молекулами
бутадиена с образованием винилциклогексена
СН2 СН2
Z /\
СН СН2 СН СН2
• I Ч —II
СН СН-СН=СН2 СН сн—сн=сн2
сн2 ''сн2
Эта реакция может осуществляться нагревом бутадиена до 425° под
давлением 14 ат в присутствии карбида кремния. Если работать при высоком
давлении в присутствии нафтената меди или хрома, то можно применять и
значительно более низкие температуры порядка 110—150°. При этом полу-
чают 60—85% винилциклогексена, остаток представляет собой линейный
полимер [2].
Возможны также реакции гидроформилирования бутадиена, при которых
путем насыщения двойных связей водородом получают изовалериановый
альдегид. При окислении бутадиена воздухом над пятиокисыо ванадия
получают малеиновую кислоту с выходом до 85% от теоретического [3].
При действии на бутадиен тонко измельченного металлического натрия
образуется соединение, состоящее из 8 углеродных атомов, с обоих концов
водород в нем замещен натрием. При обработке этого соединения углекис-
лым газом получают смесь дикарбоповых кислот, так называемую изосеба-
циновую кислоту, состоящую из 72—80% 2-этилсубероновой кислоты,
12—18% 2,5-диэтиладипиновой кислоты, 6—10% себациновой кислоты и
небольшого количества З-этилсубероиовой кислоты.
Эта смесь дикарбоповых кислот вырабатывается в США в количестве
до 5000 т. и применяется для получения смазочных масел [4] и пластифика-
торов, в производстве виниловых и алкидных смол.
IV. ПОЛИМЕРИЗАЦИЯ БУТАДИЕНА
Наибольшее промышленное значение имеет смешанная полимеризация
бутадиена со стиролом и акрилонитрилом. Полимеризация со стиролом в
большинстве случаев проводится при соотношении бутадиена к стиролу
70 : 30. При этом получают синтетический каучук, известный в Германии
как буна S, а в США под маркой GR-S.
Смешанная полимеризация бутадиена с акрилонитрилом дает каучук
бупа N.
Сополимеризация бутадиена со стиролом проводится следующим обра-
зом: бутадиен и стирол подают для полимеризации в облицованный изнутри
стеклом автоклав в соотношении 70 : 30. Процесс ведется при хорошем пере-
мешивании, в присутствии катализатора, эмульгатора и регулятора полиме-
17*
25!)
Сополимеризации
Газы нефтепере-
работки
Бутилкаучук
Рис. 161. Схема получения буна S и бутилкаучука на основе нефти.
ризации. В качестве катализатора применяют в большинстве случаев пере-
кись, в качестве регулятора алкилмеркаптан. Температура процесса опре-
деляется видом получаемого каучука и составляет 44—46° или 5—6°.
После того как прореагируют 60—70% компонентов, реакцию прекра-
щают путем разрушения перекиси, что достигается введением восстанавли-
вающего агента. После этого непрореагировавшие бутадиен и стирол отде-
ляют и возвращают в процесс. Полученный таким образом латекс смешивают
с сажей и маслом, к нему добавляют антиокислитель и смесь коагулируют
добавлением серной кислоты. Коагулят обезвоживают, высушивают оконча-
тельно нагретым до 75—85° воздухом и формуют."Сополимер 70% бутадиена
и 30% акрилонитрила получают практически таким же способом.
Рис. 162. Схема получения бупа N и неопрена на основе нефти.
261
На схемах рис. 161 и 162 показана последовательность операций при
получении различных сортов вулканизированного каучука из нефтехимиче-
ского сырья — нефти или природного газа.
Интересный новый вид полимеризации бутадиена при помощи катали-
затора Циглера, применяемого для полимеризации этилена в полиэтилен,
предложен Вильке [5]. Таким путем можно из бутадиена получить с 80—
90%-ным выходом оба стереоизомера циклододекатриена-1,5,9. Этот цикли-
ческий тример представляет особый промышленный интерес в связи с воз-
можностью получения из него додекандикислоты и соответственно со-амино-
додеканокислого лактама.
ЛИТЕРАТУРА
1. Staaterman И. G. Chem. Engng. Progr., 43, 4, Trans. Amer. Inst. chem.
Engr., 148, 1947.
2. S t a n 1 e у E. E. Am. пат. 2544808, 1951, Chem. Abs., 45, 4961a, 1951.
3. В г e t t о n II. H., Shen W n W a n, Dodge B. F. Ind. Engng., 44,
594, 1952.
4. Chem. Week, 60, 10/XII 1955.
5. Angew. Chem., 69, 397, 1957.
Глава V
ИСПОЛЬЗОВАНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
В НЕФТЕХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Нефтехимическая переработка бензола в этилбензол и стирол (стр. 227)
и в изододецил- и керилбензолы для получения арилсульфонатов (стр. 233),
а также в кумол, как исходного продукта для получения фенола и ацетона
(стр. 232) были уже рассмотрены выше и на рис. 163 дана сводка важнейших
способов получения ароматических промежуточных продуктов. На рис. 164—
168 показаны важнейшие реакции основных ароматических углеводородов.
В больших количествах ароматические углеводороды окисляются в
ангидриды и кислоты. Из бензола получают ангидрид малеиновой кислоты,
из о-ксилола — ангидрид фталевой кислоты, из лг-ксилола — изофталевую
кислоту и из «.-ксилола — терефталевую кислоту.
Для окисления в промышленности применяют в первую очередь воздух
и азотную кислоту,
[I. ОКИСЛЕНИЕ КСИЛОЛА ВОЗДУХОМ (ФТАЛЕВАЯ КИСЛОТА)
В присутствии ванадиевого катализатора о-ксилол окисляется воздухом
в ангидрид фталевой кислоты [1, 2]. Другие ксилолы превращаются в этих
условиях в бензойную кислоту, ангидрид малеиновой кислоты, окись угле-
рода и воду, т. е. подвергаются дальнейшему окислению.
В жидкой фазе ксилол легко окисляется воздухом в присутствии катали-
заторов в толуиловую кислоту. Однако дальнейшее окисление ее воздухом
протекает с большим трудом.
Окисление воздухом о-ксилола ведут над пятиокисью ванадия при
температуре 450—600° со временем превращения 0,01—0,1 сек. Выход
составляет около 50—70% от теоретического.
Пары о-ксилола из обогреваемого водяным паром испарителя поступают
в смеситель, где смешиваются с предварительно фильтрованным воздухом,
сжатым до необходимого давления и подогретым (рис. 169). Полученная
таким образом газовая смесь подается в реакционную печь. Катализатор
в печи находится в трубчатом коллекторе, окруженном соляной ванной
для отвода тепла. Соляной раствор непрерывно циркулирует через холо-
дильник. Выходящие из печи газы поступают в котел, где отдают свое тепло
для генерации водяного пара, а затем направляются в конденсатор, где
происходит полная конденсация их. Отсюда твердый продукт периодически
отбирают в плавильную установку, где он освобождается от влаги. В заклю-
чение продукт подвергают перегонке, отбирая в качестве главной фракции
фталевый ангидрид.
Превращение га-ксилола в терефталевую кислоту и л-ксилола в изофтале-
вую кислоту проводится в большинстве случаев в две стадии. На первой
263
to
о
Ксилолы Пропен Бензол Этилен
Эпоксидные смолы
Рис. 163. Получение промежуточных ароматических продуктов нефтехимическим путем.
Искусственные вещества, моющие средства
Искусственное волокно, терн леи, дакрсц
БЕНЗОЛ
+ Этилен
aTcG
Этилбензол
Стирол
Бутадиен
Хлорирование и
гидролиз
Сульфирование и
плавленный натрий
Окисление в присут-
ствии катализаторов
+ Пропен----------
Фенол
Фенол
Фенол
Кумол —
Фенол + Ацетон
Окис л.
Изобутен
Буна S
Полистирол
S-полимер
вещества, най-
Алкилирование с
керилхлоридОм
Алкилирование
тетрамером пропена
Керилбензол
Додецилбепзол
Хлорирование
Гексахлоргексая
Каталитическое
окисление
Нитрование
ксилолы
Дезинфекционные
лон, капролактам, фармацевти-
ческие препараты, бакелит, сред-
ства для борьбы с вредителями,
моющие средства, неионогенные
моющие средства
h2so4
—-—Арилсульфопат, марлоп
(гаммексап)
Хлорбензол —» ДДТ
Ангидрид малеиновой кислоты--------* Малеиновая
к ислота
(текстильные красители, капиллярно-активные вещества»
искусственные смолы)
Нитробензол и получаемые из пего продукты
Рис. 164. Реакции бензола.
ГОЛУО.1
Окисление
Окисление
Нитрование
Хлорсульфоновая кисло-
та + NH3
Бензальдегид (красители, душистые веще-
ства)
Бензойная кислота (консервирующие ве-
щества, лечебные средства и т. д.)
Тринитротолуол, флороглюцин
Сахарин ,хлорамин Т
Растворители для эмалей, лаков, красок,
жиров, масел, смол и т. д.
Рис. 165. Реакции и применение толуола.
о-Ксилол Окисление At-КСИ лол Фталевая кислота (искусственные вещества, смолы .пластификаторы}- Изофталевая кислота (искусственные вещества, смолы, пластификаторы)
п-Ксилол ' Терефталевая кислота (искусственное волокно: терилен, дакрон,, тревира)
Рис. 166. Реакции ксилолов.
ЭТИ.1 БЕНЗОЛ
Дегидрирование
/+Бутадиен-------*Буна S
Стирол —> /Полистирол
(+Изобутен------- S-полимер
--------------> Лаковые смолы
Окисление
Ацетофенон
Восстановление ,
--------------* Метилфенилкарбин ол
I -Н2р
Стирол
Хлорирование
---------------. ш-Хлорацетофспон
Рис. 167. Реакции этилбензола.
265
О'
□5
КУМОЛ
Фенол
Окисление
Расщепле-
Гидроперекись кумола---------
ние н+
Ацетон
—* Бакелит
— Найлон, перлон,
адипат
—► Дифенилолпропан
---* Диан
Эпоксидные смолы
Рис. 168. Реакции кумола.
Воздух
Рис. 169- Схема получения фталевого ангидрида каталитическим окислением о-ксилола воздухом.
стадии ксилол окисляется в толуиловую кислоту, которая затем в виде эфира
окисляется дальше. На этой стадии окисление и-ксилола, например, произ-
водится в присутствии 1 % растворимого в ксилоле нафтената кобальта при
температуре 100—200° и давлении от одной до нескольких атмосфер. При
этом окисляется от 30 до 50% исходного ксилола. Выход п-толуилевой
киолоты составляет 80—90% от теоретического. Условия работы следующие.
Исходного ксилола, г ........................
Чистота га-ксилола, %........................
Катализатор..................................
Вес катализатора, г..........................
Воздух, л)мин ...............................
Температура, °C..............................
Продолжительность реакции, часы..............
Давление, ат.................................
Превращение в толуиловую и терефталевую
кислоты, % вес...............................
Образование n-толуиловой кислоты, г/100 г кси-
лола в час ..................................
Выход гг-толуиловой кислоты, % от теории . .
Выход терефталевой кислоты, % от теории . .
Общий выход, % ...........................
1000
87
Кобальтовая соль
кислот С8 — С10
2
1,5
125
15
1
45
2,5
76,5
8,7
85,2
Дальнейшее окисление эфира толуиловой кислоты производится практи-
чески в тех же условиях. Выход составляет около 100%. Приводим в каче-
стве примера условия окисления метилового эфира га-толуиловой кислоты.
Количество эфира, г ..............
Катализатор.......................
Воздух, л/час.....................
Количество катализатора, г........
Температура, °C...................
Давление, ат......................
Продолжительность реакции, часы
Коэффициент превращения, % . .
Выход, % от теории ...............
1000
Кобальтовая соль
кислот С8
90,7
2
120
1
8
— 20
~ 100
П. ОКИСЛЕНИЕ л-КСИЛОЛА АЗОТНОЙ КИСЛОТОЙ
(ТЕРЕФТАЛЕВАЯ КИСЛОТА)
Для прямого окисления и-ксилола, проводимого при температуре
около 200°, применяется 30%-ная азотная кислота. Вместе с терефталевой
кислотой образуется также re-толуиловая кислота, которая отделяется и воз-
вращается в процесс. Выход приблизительно равен 85% от теоретического.
Азотнад кислота при окислении переходит частью в окись азота, частью
в закись азота и элементарный азот. Первая метильная группа и-ксилола
тоже окисляется значительно легче, чем вторая.
При разделении процесса на две стадии, что, однако, не типично для
промышленных условий, получают 85—90% толуиловой кислоты и 95—98%
терефталевой кислоты на второй ступени. Процесс на первой ступени проте-
кает при 150°, на второй — при 200°.
Состав газов, выделяющихся на отдельных ступенях процесса, также
различен: на первой ступени выделяются главным образом двуокись азота
и элементарный азот, на второй ступени — окись азота [2 ] .
Условия окисления n-ксилола азотной кислотой в терефталевую кислоту.
Температура, °C............................
Давление, ат...............................
Концентрация азотной кислоты, % ...........
Соотношение [весовое (HNOa) 100 %]:п-ксилол
Превращение ксилола, %.....................
Толуиловой кислоты в продуктах реакции, %
Выход, % от теории ........................
150—250
14,6—28,2
28-40
2—4:1
100
1—10
80-85
Для окисления ксилола может применяться также комбинация воздуха
и азотной кислоты. Так, например, и присутствии 0,3% нафтената кобальта’
n-ксилол окисляется воздухом под давлением 5 ат в толуиловую кислоту,,
а последняя с 20 %-ной азотной кислотой превращается в терефталевую'
кислоту.
Таким же путем лг-кснлол может превращаться в изофталевую кислоту.
При этом можно работать подобным же образом, т. е. совместно воздухом и
азотной кислотой [3].
Новейший способ окпслепия м- и и-ксилолов в соответствующие дикарбо-
новые кислоты состоит в нагревании ксилола с элементарной серой в при-
сутствии водного раствора сульфата аммония при температуре 200—700е
под давлением. При окислении получаются амиды пли соли аммония, кото-
рые легко могут быть переведены в соответствующие кислоты [4].
В настоящее время важнейшим направлением использования терефтале-
вой кислоты является конденсация ее с гликолем для получения искусствен-
ного волокна терилен. Схема получения волокна терилен из нефтяного
сырья представлена на рис. 170.
Ш. ОКИСЛЕНИЕ БЕНЗОЛА В АНГИДРИД МАЛЕИНОВОЙ КИСЛОТЫ
Ангидрид малеиновой кислоты получают окислением бензола воздухом
в паровой фазе над катализатором — пятиокисью ванадия, нанесенной на
окись алюминия:
НС-СО
j Ц-М'ЛОо-----' (I О+2СО2+2П2О
НС—СО х
Катализатор представляет собой модификацию контакта, применяемого
для получения ангидрида фталевой кислоты. Однако условия окисления
очень отличаются от условий окисления нафталина, так как бензол значи-
тельно устойчивее против окисления. Вследствие жестких условий процесса
снижается выход малеинового ангидрида, составляющий 50—60% от теорети-
ческого, тогда как выход фталевого ангидрида достигает 86—88%.
В промышленных условиях процесс ведут следующим образом (рис. 171)..
Воздух и бензол каждый в отдельности подогревают и смешивают в смеси-
теле. Воздуха — большой избыток, примерно 26—50 кг на 1 кг бензола.
Печь состоит из трубок с катализатором и снабжена соляной ванной для
обогрева. В большинстве случаев для ванны применяют нитрат-нитритную
смесь. В печи имеется приблизительно 3000 трубок, заполненных катализа-
тором в виде пилюль диаметром 5 мм. Реакция идет под давлением 2—3 ат
без циркуляции сырья, так как процесс окисления завершается за однократ-
ный проход реакционной смеси через печь.
Пятиокись ванадия наносится на окись алюминия или диатомовую
землю. Температура поддерживается 400—450°. Время пребывания реак-
ционной смеси в печи около 0,1 сек.
Вследствие большого разбавления реакционной смеси воздухом пере-
работка выходящих из печи газов сложна. Эти газы содержат около 1,0%
объемн. малеинового ангидрида, 8,6% кислорода, 5,0% углекислого газа и
77,4% азота. Газ охлаждают в холодильнике до 100—150° и затем промы-
вают водой в башне, заполненной кольцами Рашига. Промывка производится
циркулирующей водой, которая постепенно (в результате гидролиза малеи-
нового ангидрида) обогащается малеиновой кислотой. Если целью процесса!
является получение кислоты, то примерно 40%-ный раствор малеиновой
кислоты обесцвечивается затем при помощи угля, вода испаряется и в остатке'
получается кристаллическая малеиновая кислота.
Для получения ангидрида малеиновой кислоты кислый раствор напра-
вляется в колонну для обезвоживания, где получается сырой малеиновый
ангидрид, очищаемый затем перегонкой.
2(58
Рис. 170. Схема получения териленового волокна из природного газа и нефтяных фракций.
Малеиновый ангидрид может отделяться также другим способом. Выходя-
щие из иечи пары подвергают частичной конденсации в холодильнике, где-
поддерживается температура около 60°. Большая часть малеиновой кислоты
и малеинового ангидрида при атом конденсируется, а оставшиеся пары
поглощаются каким-либо неводным растворителем, например дибутилфтала-
том. Для перевода малеиновой кислоты в ангидрид конденсат обезвоживают
азеотропной перегонкой с применением ксилола. Обезвоживание открытым
нагревом не может применяться, так как малеиновая кислота при нагрева-
Рис. 171. Схема получения ангидрида малеиновой кислоты каталитическим окислением
бензола.
1 — предварительный подогреватель; 2 — трубчатая печь, катализатор; 3 — холодильник; 4 — смо-
лоотделитель; б — колонна для промывки водой; 6 — обезвоживающая колонна; 7 — дистилляционная
колонна; 8 — испарительная часть колонны.
Линии: I — воздух; II — бензол; III — отходящие газы; IV — вода; V — сырой ангидрцд; VI — ан-
гидрид малеиновой кислоты; VII — остаток.
нии выше температуры плавления легко превращается в фумаровую кислоту,
не образующую ангидрида.
Ароматическая монокарбоновая кислота — n-mpem-бутилбензойная кис-
лота — может получаться каталитическим окислением n-mpem-бутилтолуола.
Окисление ведут воздухом в присутствии нафтената кобальта при темпера-
туре около 170е [6]. Кислота может служить заменителем жирных кислот
в произсодстве алкидных смол. Применение ее сообщает смолам лучший
цвет и запах и большую стойкость. При этерификации ее ненасыщенными
спиртами получают легко полимеризующиеся эфиры, обладающие особыми
свойствами.
IV. ПОЛУЧЕНИЕ АДИПИНОВОЙ КИСЛОТЫ КАТАЛИТИЧЕСКИМ ОКИСЛЕНИЕМ
ЦИКЛОГЕКСАНА ВОЗДУХОМ
В присутствии катализатора циклогексан окисляется воздухом или
в смесь циклогексанола и циклогексанона или в адипиновую кислоту:
/СЩ—сн2— соон
(____________--- сн2 +н,о
7 \сн2—соон
В жидкой фазе в присутствии ацетата кобальта получается смесь
циклогексанола и циклогексанона с 90%-ным выходом.
Для прямого получения адипиновой кислоты процесс необходимо вести
в более жестких условиях. Циклогексан и циклогексанон в примерном соот-
ношении 40 : 1 в присутствии половинного объема ледяной уксусной кислоты
и 0,7% шестиводного . хлористого кобальта обрабатывают продуваемым
270
через реакционную смесь воздухом при 100—120° и 10,5 ат давления. Отхо-
дящие газы конденсируют и затем промывают этилбензолом для отделения
циклогексана. Продукты реакции перегонкой освобождают от непрореаги-
Рис. 172. Схема получения адипиновой кислоты каталитическим окислением цикло-
гексана воздухом.
1 — окислительная колонна; 2 — холодильник; з — промывочная колонна; 4 — дистилляционная ко-
лонна; б — перегонка с водяным паром; 6 — кристаллизатор; 7 — осушитель.
Линии: I — хлористый кобальт; II — уксусная кислота; III — циклогексанон; IV — циклогексан;
V — отходящие газы; VI — воздух; VII — этилбензол; VIII — циклогексан и уксусная кислота об-
ратно на окисление; IX — на переработку; X — циклогексанол-циклогексанон; XI — адипиновая
кислота.
ронявшего циклогексана и уксусной кислоты, которые возвращаются в про-
цесс (рис. 172). Остаток перегоняют с водяным паром для отделения цикло-
гексанола и циклогексанона. После этого продувку водяным паром повто-
ряют еще раз уже в присутствии небольших количеств серной кислоты для
Рис. 173. Схема получения iiaii/iona 66 п перлона (нчйаои 6) из циклогексана.
расщепления эфиров п лактонов. После удаления серной кислоты в виде
бариевой соли фильтрат подвергают кристаллизации. Выход адипиновой
кислоты составляет 50—70%.
Для получения циклогексанона образовавшийся при окислении цикло-
гексанол возвращается в процесс. Циклогексанон является исходным мате-
риалом для производства капролактама, превращаемого полимеризацией
в перлон. Таким образом, как найлон, так и перлон (найлон 6) получают
из циклогексана.
Схема получения обоих этих продуктов дана на рис. 173.
271
ЛИТЕРАТУРА
1. L е v i n e I. E. Chem. Engng. Progr., 43, 4, Trans. Am. Irist. Chem. Engr., 168,
1947; Toland G. and Nimer E. L. Proceedings Fourth World Petrol. Congr. Sect.
IV/B, Paper I, p. 39, 1955.
2. T о 1 a n d W. G. and N i m e r E. L. Proceedings Fourth World Petrol. Congr.
Sect. IV/B, Paper I, p. 39, 1955.
3. Chem. Engng., 63, 142, 144, 146, 1956.
4. T о 1 a n d W. G. Am. пат. 2722546, 2722547, 2722549, 1955; Aroyan H. J.
Am. пат. 2722548.
5. S h e r w о о d P. W. Petrol. Process., 82—89. Nov. 1956.
6. Cole II. M. Chem. Engng. Science Special Suppl., vol. 3, 67 — 77, 1954.
Глава VI
РАЗЛИЧНЫЕ ПРОДУКТЫ НЕФТЕХИМИЧЕСКОЙ
ПРОМЫШЛЕННОСТИ
I. АММИАК И ЕГО ПРОИЗВОДНЫЕ КАК НЕФТЕХИМИЧЕСКИЕ ПРОДУКТЫ
Получение водорода как важнейшего исходного продукта в производ-
стве аммиака уже рассматривалось ранее (см. рис. 11). Чтобы показать
связь между производством аммиака и нефтехимической промышленностью,
заметим, что из произведенных в США в 1956 г. 3,5 млн. т аммиака около
75% получено из природного газа [1].
Получение аммиака из водорода и дальнейшая переработка аммиака
в азотную кислоту и в минеральные удобрения на страницах этой книги не
рассматриваются.
Производство мочевины из аммиака и углекислоты, для осуществления
которого имеется большой выбор различных методов, относится к важней-
шим нефтехимическим процессам [2]. Мочевина требуется в больших коли-
чествах для производства удобрений, получения продуктов формальдегид-
ной конденсации и для других целей. С недавнего времени мочевина стала
применяться для экстрактивной кристаллизации в целях выделения нормаль-
ных парафиновых углеводородов из нефтяных фракций. Относительно не-
большие количества мочевины применяются в производстве вспомогательных
средств для текстильной промышленности, в производстве фармацевтических
препаратов и косметических средств.
АММИАК
+CO2______________________
Ацетальдегид +На_______
катализатор
+СН3ОН_________________
катализатор
Паральдегид 200—250°
+ацетат аммокия, 50 ат
Формальдегид
Бутиролактон
Окись этилена
Диэтиленглиноль
СН4 + О2
Pt—катализатор
Мочевина
Этиламин
Моно-, ди- и триметиламины
Никотиновая
Л кислота
2-метил-5-атилпиридин
Метилвинил-
пиридин
1
Полимеры
Гексаметилентетрамин
Пирролидон —- винилпирролидон
Моно-, ди- и триэтаноламины
Морфолин
Синильная кислота
Рид других соединений для органиче-
ского синтеза
Рис. 174. Применение аммиака в промышленности
органического синтеза.
18 Заказ 596.
273
Некоторые данные о применении аммиака в промышленности органиче-
ской химии приведены на рис. 174.
II. СЕРА ИЗ НЕФТИ И ПРИРОДНОГО ГАЗА
Важным нефтехимическим процессом является получение элементарной
серы и серной кислоты из сероводорода природного газа и отходящих газов
нефтеперерабатывающих заводов 13].
Сероводород выделяется во всех процессах крекинга и пиролиза и содер-
жится в природном нефтяном газе большинства месторождений, иногда
в очень значительном количестве. Так, например, газ нового большого
месторождения в Лакк (Пиринеи) содержит до 15% объемн. сероводорода.
Для получения из сероводорода элементарной серы содержащие серо-
водород газы сжигают в воздухе, количество последнего регулируют таким
образом, чтобы образовавшаяся в результате сгорания двуокись серы нахо-
дилась в молярном отношении к еще оставшемуся в смеси сероводороду, как
1:2. Тогда имеет место реакция:
2H2S+SO2->3S+H2O
Суммарная реакция будет тогда выглядеть так.
2H2S + O2 —> 2S+2H2O
При сжигании газа в печи температура пламени поддерживается около
1350°. Тепло отводится с водяным паром. При этом уже идет образование
элементарной серы. Для обеспечения полного превращения газ проходит
через несколько конверторов, в которых в присутствии боксита как катали-
затора происходит дальнейшее превращение в элементарную серу. Горячие
газы утилизируются для образования пара. Жидкая сера собирается. Выход
может быть доведен до 95%. Не вошедший в реакцию сероводород сжигается
в избытке воздуха в двуокись серы и через высокую трубу выбрасывается
в атмосферу.
Серная кислота получается известными способами. О получении серо-
углерода было сказано выше (стр. 147).
Находящиеся в продуктах крекинга органические сернистые соедине-
ния являются продуктами расщепления сложных сернистых соединений,
содержащихся в сырых нефтях часто в количестве 2,5—3,0%. В большин-
стве случаев в продуктах крекинга преобладают меркаптаны. Количество
их может быть довольно значительным. Для отделения и изоляции поль-
зуются их растворимостью в щелочах.
Метилмеркаптан применяется для получения метионина (реакцией
с акролеином) и как добавка к топливу для двигателей внутреннего сгора-
ния. Этилмеркаптан применяется как одорант в природном газе.
Интересными свойствами обладает дпметилсульфоксид. Он является
исключительно хорошим растворителем для многих органических продуктов,
кипит при 189°, растворим в воде, не имеет запаха и бесцветен. Он может
служить заменителем диметилформамида как растворителя в производстве
орлонового волокна.
Высокомолекулярные меркаптаны, применяемые в производстве син-
тетического каучука, могут быть получены каталитическим присоединением
сероводорода к третичным олефинам [4]. Они могут окисляться в дисуль-
фиды и сульфокислоты, которые в свою очередь находят разнообразное
применение.
III. ПРОДУКТЫ, ПОЛУЧАЕМЫЕ НЕПОСРЕДСТВЕННО ИЗ НЕФТИ
А. РАСТВОРИТЕЛИ
К продуктам, получаемым непосредственно из нефти простым выделе-
нием, относятся выкипающие в известных границах фракции, применяемые
как растворители или разбавляющие агенты в лаковой и красочной про-
274
мышленности. Они являются также важными вспомогательными материа-
лами для химической промышленности.
Эти растворители разделяются на две основные группы: одну, состоя-
щую почти исключительно из насыщенных алифатических углеводородов,
и вторую, богатую ароматическими соединениями и потому представляющую
особый интерес как растворитель для многих веществ.
Растворители первой группы практически не токсичны. Обычно при-
меняется фракция, выкипающая в пределах 90—150°, которая для очистки
может подвергаться вторичной перегонке. Такие углеводородные смеси
применяются, в частности, в производстве лаков, используемых для внутрен-
ней окраски. Эти растворители коррозийноустойчивы и вполне стабильны.
Для наружной окраски могут применяться краски и лаки, полученные
с использованием ароматизированных растворителей.
Растворители нефтяного происхождения применяются также в произ-
водстве синтетического каучука. Необходимые физические свойства раство-
рителя определяются его назначением [6].
Б. НАФТЕНОВЫЕ КИСЛОТЫ
В зависимости от происхождения нефти могут содержать различные
количества нафтеновых кислот, однако количества эти во всех случаях очень
незначительны. Из нефтей нафтеновые кислоты экстрагируются спиртовой
щелочью. Строение нафтеновых кислот во всех нефтях одно и то же и не
зависит от происхождения нефти.
Нафтеновые кислоты могут разделяться посредством ректификации
или как таковые или в виде эфиров. Исследованиями Брауна и Лохте с сотруд-
никами показано, что простейшими членами ряда нафтеновых кислот явля-
ются производные циклопентана с карбоксильной группой непосредственно
у кольца или соединенной с кольцом через углеродный атом:
СООН СН2СООН
0 и
Производных циклогексана содержится значительно меньше. Разница
молекулярного веса и температур кипения отдельных кислот определяется
различной длиной боковых цепей циклопентанового кольца. Молекулярный
вес может быть от 110 до 1000.
Кислоты, представляющие наибольший промышленный интерес, содер-
жатся в керосиновых и газойлевых фракциях нефтей. Молекулярный вес
их лежит в пределах 180—350. Их свинцовые, кобальтовые и марганцевые
соли хорошо растворимы в маслах и применяются в качестве агентов, уско-
ряющих сушку лаков. Нафтенат меди применяется для консервации древе-
сины, нафтенаты кальция и алюминия — в качестве присадок к смазочным
маслам. Нафтеновые кислоты с 14—30 атомами С в молекуле во многих
отношениях ведут себя подобно жирным кислотам с прямой цепью 17].
В. ФЕНОЛЫ
В нефти и ее фракциях, особенно в продуктах крекинга, содержатся
фенолы, в первую очередь крезол, а также ксиленол с небольшим количе-
ством высокомолекулярных фенолов. Из крекинг-продукта, кипящего в
интервале 150—230°, выделяются фенолы, состоящие из 45% крезола, 25%
ксиленола, 20% фенола и 10% загрязнений. Выделение производится экс-
тракцией щелочью с последующим отделением карбоновых кислот и нейтраль-
ной части. От сернистых соединений фенолы освобождаются путем продувки
воздуха через щелочной раствор.
18*
275
ЛИТЕРАТУРА
1. Chem. Trade J. (London), 140, 1408, 1957.
2. Тонн W. H. Chem. Engng., 62, 10, 186, 1955; Chem. Ing. Techn., 27, 2, 99,1955;
Gambon Th., Chim. et Ind., 74, 912—928, 1955.
3. Oil a. Gas J., 55, 35, 121, 1957; Hixon F. E. Chem. and Ind., 332, 1955.
4. S c h u 1 z e A. W. Ind Engng. Chem., 40, 2308, 1948.
5. Schulze A. W. Ind. Engng., Chem., 42, 916, 1950.
6. Frost II. N. Petrol, licfiner, 29, 2, 95—99; 3, 135—139; 4, 137—140, 1950.
7. J e z 1 J. L. Petrol. Process., 8, 89—90, 1953.
ОБЗОР ОСНОВНЫХ РЕАКЦИЙ ПАРАФИНОВ И ОЛЕФИНОВ
В заключение на рис. 175—187 даны основные реакции отдельных
наиболее важных представителей рядов парафинов и олефинов, а также их
современное и возможное промышленное значение.
МЕТАН (из природного газа и газов крекинга)
Термическое расщепление
—* Сажа и водород
Хлорирование
—► Хлористый метил, хлористым метилен, хлороформ
четыреххлористый углерод
Реакция с серой
• - Сероводород
Реакция с серой
- Сероуглерод
Частичное сжигание в 0? „„
— СО 4-Н2 4-ацетилен (процесс Захсе)
2 : 1 Неполное сжигание в воздухе
• Сажа (шинная промышленность)
Реакция с водяным паром над „ Конверсия „ Г*^0^1”'113
Ni-ката лизатором глхчи ' 1 cu-t-jri2 ^Азотная кислота
Пиролиз в алектричесной дуге
► Ацетилен 4-этилен
Пиролиз по способу Коперс-Хаше-
^уЛЬф ~ * Ацетилен 4- этилен
Сжигание в присутствии NH3 над „ ,
Pt-катализатором Синильная кислота (процесс Андрусова)
2 : 1 Метиловый спирт
Превращение с О, „ — /
> Синтез-газ СО 4-На'
синтез Фишсра-Тропша
— — * для повышения теплоты сгорания светильного газа
12 млн. ккал
4000—5000 jk3 водорода (в зависимости от способа)
' Из 1000 кг метана 3000—8000 кг продуктов хлорирования
230 кг ацетилена +1100 кг синтез-газа по способу
Захсе
Рис. 175. Основные реакции метана (Winnacker, Kunstoffe, 47, 8, 397, 1957).
ЭТАН
Хлорир ование
Дегидрирование
термичсские
Дегидрирование
автотермическое
Дуговой пиролиз
Пиролиз методом
Коппере-Хаше
Высокотемпературный
пиролиз во Хохстер
Превпатение с НоО
с а\ 1-катализатором
Хлористый этил
Этилен
Этилен
Ацетилен 4- этилен
Ацетилен 4- этилен
Ацетилен 4- этилен
СО 4- Н’_ (синтез-газ —-* Водород)
2 : 5
Рис, 176. Основные реакции этана.
277
ПРОПАН
Пиролиз
Каталитическое
дегидрирование
Окисление в газовой_____
фазе с до осз катализатора
Окисление в газовой фазе
с катализатором (НВг)
Окисление в жидкой
фазе каталитическое
Дуговой крекинг
Частичное сгорание
Пиролиз ио Копперс-Хатде
Превращение с П2О
над N i-катализатором
Нитрование в газовой фазе
Сульфохлорирование
Этилен, пропен
Пропен
Ацетальдегид, формальдегид, спирты,
кислоты, ацетали и т. д.
Ацетон
Уксусная кислота преимущественно
Ацетилен 4- этилен
Конверсия
СО 4- Н2 -----------*Водород
Ацетилеп 4- этилен
Конверсия
СО 4- П2 -----------► Водород
Нитрометан, нитроэтан, 1- и 2-нитропропан
Моно- и дисульфохлориды пропана
Рис. 177. Основные реакции пропана.
н-БУТАН
Каталитическое
дегидрирование
Пиролиз
Каталитическая _____
изомеризация
Превращение с серой или H2S
при высокой температуре
Окисление в газовой________
фазе
Каталитическое окисление
в жидкой фазе
н-Бутен, бутадиен
Этилен, пропен
Пзобутан
Тиофен
Ацетальдегид, формальдегид, уксус-
ная кислота, спирты, ацетали и т. д
Главным образом уксусная кислота
Рпс. 178. Основные реакции /(-бутана.
ИЗОБУТАН
Каталитическое ______
дегидрирование
Каталитическое алкилирование
с н-бутеном или изобутеном
Каталитическое окисление____
в газовой фазе (НВг)
Сульфохлорирование
Нитровали е
Хлорирование
Изобутен
Нзооктан
wpem-Бутилгидроперекись, ди-mpem-
бутилгидроперекись
2-метилпропансульфохлорид-1
2-нитропропан, нитрометан, 1- и 2-
нитроиэобутан
mpem-Хлористый бутил, хлористый
изобутил
Рис. 179. Основные реакции изобутана.
н-ПЕНТАН
Хлорирование
Нитрование
Каталитическая
изомеризация
Каталитическое
дегидрирование
Пиролиз
Хлористый амил------------ Амиловый
спирт
Нитрометан, нитроэтан, нитропропан,
нитробутан, 1-, 2- и З-нитропентая
Изопентан---------♦ Изопрен
н-Пептен, пиперилен
Этилен, пропен, бутен
Рис. 180- Основные реакции н-пентана.
278
ИЗОПЕНТАН
Каталитическое дегидрирование
Хлорирование________________
Алкилирование олефинами_____
Пиролиз_____________________
Изоамилен, изопрен
Хлористый изоамил
Высокооктановый бензин
Этилен, пропен
Рис, 181. Основные реакции изопентана.
УГЛЕВОДОРОДЫ С12 — C1S
Окисление
Хлорирование
Сульфохлорирование
Крекинг_______________
Сульфоокисление_______
Нитрование в жидкой фазе
Хлорфосфонилирование
Жирные кислоты с различной длиной цепей
Хлористые алкилы для получения арилсуль-
фонатов
Омыление в сульфонаты (дюпонол, меза-
пон и др.)
Олефины с различной длиной цепи
Гостапон
Нитропарафины
Хлориды алкилфосфоновых кислот
Рис. 182. Важнейшие реакции парафиновых углеводородов,
кипящих в интервале среднего масла (С12—С18).
ЭТИЛЕН
Каталитическое окисление Окись этилена
Хлоргидринирование Этиленхлоргидрин —* Окись этилена Этиловая жидкость
Хлорирование 1,2-дихлорэтан^* Винилхлорид Тиокол (пердурен)
Полимеризация над А1С1з „ Полимеризация по Циглеру, А1(С2Нз)з нормальное давление Полимеризация по Филлипс-Петролеум, пониженное давление, окись хрома или окись молибдена Полимеризация при высоком давлении, способ I. С. 1., следы О2 Смазочные масла синтетические Полиэтилен Полиэтилен Полиэтилен Буна S
Алкилирование бензола Этилбензол —- Стирол^
Термическое алкилирование изобутана Каталитическое алкилирование изобутана Гидратация через этилсульфат Прямая каталитическая гидратзция (Н3РО4) Полистирол Неогексан Диизопропил Этиловый спирт Этиловый спирт ^Триметилолэтан
Гидроформилирование Пропионовый альдегид^*Пропионовая кислста n-Пропиловый спирт
Присоединение НС1 Присоединение Вг Присоединение S2C12 Присоединение СО + Н2 Присоединение СО и пропионовой кислоты Каталитическая обработка Присоединение H2S Хлористый этил Бромистый этилен (этиловая жидкость) Дихлордиэтилсульфид Пропионовая кислота Ангидрид пропионовой кислоты Бутадиен Диэтилсульфид, этилмеркаптан
Рис. 183. Основные реакции этилена.
279
ПРОПЕН
Гидратация посредством H2SO4
Гидроформилирование
Присоединение СО4-Н2О
Алкилирование бензола
Полимеризация с катализатором Циглера
Каталитическое окисление над СиО
Хлорирование при нормальной________
температуре
Хлорирование при высокой____________
температуре
Хлоргидринирование
Полимеризация с НзРСЦ-катализатором
Превращение с изопропиловым спиртом
„ Дегидрирование
Изопропиловый спирт ----------------- Ацетон
—> Уксусный ангидрид
Гидрирование
н- и изомасляныи альдегид----------» н- и иэобу-
тиловый спирт —* 2-этилгексанол
Изомасллная кислота
Фенол
Кумол
^Ацетон
Полипропен (мопрен)
Акролеин (кроме того, из аллилового спирта путем
дегидрирования, дегидратацией глицерина, пиролизом
диаллилового эфира, из ацетальдегида и формальде-
гида), метионин
1,2-дихлорпропан
Хлористый аллил
.Моно-, ди- и трипропа-
/ нола^ин
„ Окись
Пропенхлоргидрин —> пропена
^Аллиловый спирт
Изододецеп, изопентадецен (тетра-и пентапропен) —*
—* Арилсульфонаты и неионогенные моющие ве-
щества
Диизопропиловый эфир (растворитель и экстракцион-
ный агент для масел, жиров, восков, этилцеллюлозы
и красящих веществ)
Рис. 184. Основные реакции пропена.
м-БУТЕН
Гидратация с H2SO4 „ .. Дегидрирование _ emop-Бутиловыи спирт *
Метилэтилкетон
Каталитическое . Бутадиен
дегидрирование
Гидроформилирование Амиловый альдегид >Амиловый спирт
Каталитическое алкилиро- . Изобутилфенол (маслорастворимые продукты
вание фенола формальдегидной конденсации)
Каталитическая полимери- ► Ди- и трибутены
зация над HsI’Oj
Рис. 185. Основные реакции н-бутена.
ИЗОБУТЕН
Гидратация с H2SO4
Хлорирование
Гидроформилирование
Димеризация CH2SO4 или Н3РО4
Полимеризация над А1С1з_______
или BFg при —80°
Алкилирование фенола
Сополимеризация с 2%
изопрена
Сополимеризация со стиролом
mpem-Бутиловый спирт---------*Тринитро-
mpem-бутилтолуол (искусственный мускус)
Металлилхлорид и его производные
Изоамиловый альдегид----------- Изоамиловый
спирт
Диизобутен
Полиизобутен (оппанол В, вистанекс)
mpem-Бутилфенол (маслорастворимые смолы с
формальдегидом)
Бутилкаучук
S-полимер
Рис. 186. Основные реакции изобутена.
280
ВЫСОКОМОЛЕКУЛЯРНЫЕ ОЛЕФИНЫ
Сульфирование Типоль
Гидроформилирование _ Сульфирование
— ----------------------► Высшие первичные спирты ---------- —
--------►Алнилсульфаты
Полимеризация, А1С1я л ,
-----------=!-----3----------► СмавочныеХмасла
Алкилирование бензола Алкилбеетзол Сульфирование Арялоульфонаты
Алкилирование Фенола . Онсиэтилирование
------- ---------------------- Алкилфенолы-----------------—•-*
-----------------------------------------------------------► Неионогенные моющие^оредства
Рис. 187. Важнейшие реакции высокомолекулярных олефинов.
t
СОДЕРЖАНИЕ
. Стр.
Предисловие к русскому изданию.......................................... 5
Предисловие ........................................................... 8
ЧАСТЬ ПЕРВАЯ
УГЛЕВОДОРОДЫ КАК СЫРЬЕ ДЛЯ НЕФТЕХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Глава I
Парафиновые углеводороды
I. Природный нефтяной газ.............................................. 12
А. Общие сведения .................................................. 12
Б. Выделение газового бензина и ожижаемых углеводородов из жирного газа
посредством абсорбции промывным маслом [3]......................... 13
II. Высокомолекулярные, жидкие при нормальной температуре парафиновые угле-
водороды из нефти (С,—С20).......................................... 16
А. Состав нефти ................................................... 16
Б. Переработка нефти [6] (перегонка, крекинг, легкий крекинг)....... 16
В. Высокомолекулярные С7—С90 парафиновые углеводороды из нефти .... 20
Г. Выделение парафиновых углеводородов из нефтяных фракций посредством
экстрактивной кристаллизации с мочевиной ...................... 20
III. Нефтяной парафин................................................. 24
А. Общие сведения ................................................. 24
Б. Выделение парафина из фракций смазочных масел.................... 25
В. Очистка парафина ............................................... 25
Г. Пути переработки парафина....................................... 26
IV. Получение парафиновых углеводородов синтезом Фишера-Трошиа......... 26
А. Общие сведения .................................................. 26
Б. Практическое проведение синтеза Фишера-Трошпа с железным катализато-
ром ............................................................... 30
Литература............................................................. 34
Глава II
Моноолефины
I. Отходящие газы нефтеперегонных установок............................ 37
II. Крекинг-процесс ................................................... 37
А. Общие сведения ................................................. 37
Б. Термический крекинг ............................................. 38
В. Каталитический крекинг ......................................... 40
Г. Крекинг-газы..................................................... 41
III. Высокомолекулярные олефины в продуктах крекинга нефти............. 45
IV. Прямое получение олефинов.......................................... 46
А. Общие сведения ................................................ 40
Б. Каталитическое дегидрирование газообразных парафиновых углеводородов
в олефины с равным числом атомов углерода.......................... 47
В. Термическое дегидрирование этана в этилен....................... 48
282
Стр.
Г. Получение газообразных олефинов пиролизом газообразных или жидких па-
рафиновых углеводородов ................................................ 49
Д. Получение олефинов пиролизом жидких углеводородов, в частности нефти
и ее фракций............................................................ 54
Е. Получение газообразных олефинов пиролизом жидких углеводородных сме-
сей, в частности нефтяных фракций, в условиях практически полной арома-
тизации жидких продуктов пиролиза....................................... 57
Ж. Получение высокомолекулярных олефинов................................ 61
V. Концентрация и выделение олефинов из смесей газообразных углеводородов 69
А. Общие сведения .................................................... 69
Б. Разделение газообразных углеводородов фракционировкой............... 70
В. Селективное гидрирование ацетилена в этилен в газах пиролиза........ 71
Г. Разделение газовых смесей абсорбционно-компрессионным способом (аб-
сорбция маслом под давлением) .......................................... 72
Д. Выделение олефинов из смесей газообразных углеводородов абсорбцией се-
лективными растворителями .............................................. 74
Е. Адсорбционные методы................................................. 74
Ж. Разделение олефинов и парафинов экстрактивной перегонкой [471 .... 77
3. Разделение углеводородов С4 при получении бутадиена ступенчатым деги-
дрированием н-бутана ................................................... 79
Литература ................................................................ 82
Глава 111
Диолефины
I. Бутадиен ............................................................... 84
А. Общие сведения ...................................................... 84
Б. Двухступенчатый способ дегидрирования бутана......................... 85
В. Одноступенчатый способ получения бутадиена (процесс Гудри) ...... 86
Г. Другие нефтехимические способы получения бутадиена................... 88
Д. Очистка сырого бутадиена............................................ 89
II. Изопрен ............................................................... 91
Литература ................................................................ 92
Г л а в а 1V
Ацетилен
I. Ацетилен из карбида..................................................... 93
II. Дуговой процесс пиролиза метана [3].................................... 94
А. Пиролиз метана по способу Захсе [4].................................. 95
Б. Пиролиз метана методом Вульфа [6].................................... 96
III. Способ высокотемпературного пиролиза по методу фирмы Хохстер.......... 97
IV. Процесс высокотемпературного пиролиза по Истману [8].................. 98
Литература ................................................................ 98
Г л а в а V
Циклогексан
I. Общие сведения ................................................... 99
И. Получение циклогексана ................................................. 100
А. Из нефтяных фракций............................................... 100
Б. Из бензола ................................................... 100
Литература ................................................................ 100
Г л а в а VI
Ароматические углеводороды из нефти
I. Общие сведения......................................................... 101
II. Каталитические риформинг-процессы .................................... 102
А. Гидроформинг-процесс ............................................... 102
Б. Платформинг-процесс и другие способы каталитического риформинга . . . 104
III. Получение чистых ароматических углеводородов из продуктов каталитиче-
ского риформинга ........................................................ 106
А. Способ Эделеану [7] ............................................... 106
Б. Способ удекс для экстракции ароматических углеводородов [8]......... 107
283
Стр.
В. Способы экстрактивной перегонки [9]................................ 108
Г. Методы азеотропной перегонки [10]................................ 108
Д. Адсорбционные способы (аросорб-процесс) [11]........................ 109
Е. Разделение ксилолов [12]............................................. НО
Литература................................................................ 111
ЧАСТЬ ВТОРАЯ
ХИМИЧЕСКАЯ ПЕРЕРАКПТКА УГЛЕВОДОРОДОВ
Глава 1
Переработка парафиновых углеводородов
I. Хлорирование парафиновых углеводородов................................. 112
А. Общие сведения .................................................... 112
Б. Фотохимическое хлорирование парафиновых углеводородов............... 112
В. Каталитическое хлорирование парафиновых углеводородов............... ИЗ
Г. Термическое хлорирование парафиновых углеводородов.................. 113
II. Нитрование парафиновых углеводородов.................................. 126
А. Общие сведения ..........................................'......... 126
Б. Физические и фармакологические свойства низкомолекулярных нитропа-
рафинов ............................................................ 128
В. Химические реакции нитропарафинов.................................. 129
III. Сульфохлорирование парафиновых углеводородов......................... 133
А. Отношение отдельных типов углеводородов к сульфохлорированию .... 138
Б. Реакции алифатических сульфохлоридов................................ 138
В. Практическое проведение сульфохлорпрования......................... 139
Г. Превращение сульфохлоридов.......................................... 141
Д. Продукты сульфохлорирования полиэтиленов [47].................... 142
IV. Сульфоокислейие парафиновых углеводородов............................ 142
А. Общие сведения .................................................... 142
Б. Фотохимический способ с водной экстракцией.......................... 144
В. Сульфоокисление в присутствии ацилирующих веществ.................. 144
V. Реакция фосфонилирования [50] 145
VI. Карбоксилирование [51] 146
VII. Реакции парафиновых углеводородов с серой............................ 146
А. Общие сведения ..................................................... 146
Б. Получение сероуглерода из парафиновых углеводородов и серы.......... 147
VIII. Каталитическое получение синильной кислоты реакцией метана с аммиа-
ком и воздухом при высоких температурах (способ Андрусова [5]) .... 147
IX. Получение сажи из природного газа и нефти [58]....................... 148
X. Окисление парафиновых углеводородов................................... 150
А. Общие сведения .................................................... 150
Б. Условия окисления газообразных парафиновых углеводородов............ 150
В. Переработка продуктов окисления.................................... 154
Г. Каталитическое окисление газообразных парафиновых углеводородов в го-
могенной фазе ...................................................... 160
Д. Окисление парафина.................................................. 162
Литература ............................................................... 166
Глава II
Переработка моноолефинов
I. Хлорирование олефинов .............................................. 168
А. Общие сведения ................................................. 168
Б. Хлорирование замещением ......................................... 168
В. Хлорирование олефинов реакцией присоединения..................... 180
Г. Присоединение хлора к олефинам при помощи хлорноватистой кислоты
(хлоргидрирование) ............................................... 183
Д. Получение окиси этилена каталитическим окислением этилена........ 185
Е. Гидрохлорирование олефинов....................................... 198
Ж. Гидратация олефинов в спирты..................................... 199
3. Гидроформилирование олефинов (реакция Ройлена [42]).............. 214
И. Различные реакции присоединения олефинов......................... 218
К. Полимеризация моноолефинов в. искусственные вещества............. 222
Л. Алкилирование ароматических соединений олефинами................. 226
Литература..................... . . . ............................ 239
284
Стр.
Глава III
Реакции ацетилена
I. Трихлорэтилен [1] 242
II. Хлористый винил [2]................................................ 245
III. Винилацетат [3]................................................... 246
IV. Акрилонитрил [4] 246
V. Ацетон из ацетилена................................................ 248
VI. Ацетонитрил ...................................................... 249
VII. Винилирование ацетиленом [5]...................................... 249
VIII. Этинилирование ацетиленом пропаргиловый спирт, бутиндиол, тетрагид-
рофуран [5] 250
IX. Карбонилирование ацетилена ....................................... 253
X. Полимеризация ацетилена............................................ 254
Литература ............................................................ 255
Глава IV
Химические превращения бутадиена
I. Хлорирование ....................................................... 256
II. Присоединение SO2 (сульфоланы) .................................... 256
III. Реакция Дильса-Альдера............................................ 258
IV. Полимеризация бутадиена........................................... 259
Литература............................................................. 262
Глава V
Использование ароматических углеводородов в нефтехимической промышленности
I. Окислейие ксилола воздухом (фталевая кислота)....................... 263
II. Окисление n-ксилола азотной кислотой (терефталевая кислота) ....... 267
III. Окисление бензола в ангидрид малеиновой кислоты .................. 268
IV. Получение адипиновой кислоты каталитическим окислением циклогексана
воздухом ............................................................ 270
Литература ........................................................... 272
Глава VI
Различные продукты Нефтехимической промышленности
I. Аммиак и его производные как нефтехимические продукты............... 273
II. Сера из нефти и природного газа..........’......................... 274
III. Продукты, получаемые непосредственно из пефти..................... 274
А. Растворители..................................................... 274
Б. Нафтеновые кислоты............................................... 275
В. Фенолы z......................................................... 275
Литература ............................................................ 276
Обзор основных реакций парафинов и олефинов........................... 277
Ф. Азиигер
ВВЕДЕНИЕ В НЕФТЕХИМИК)
Перевод с немецкого
Нод рода к цней
Бориса Витальевича Л о с и ко в а
Ведущий редактор Л. А. Львова
Корректор Е. Я. Гусецкая
Технический редактор Э. А. Мухина
Подписано к набору 23/VI 19G0 г.
Подписано к печати 23/IX 1960 г.
Формат 7о у. i08ll i<j. Физ. печ. я. 18.
Уел. нем. л. 24,66. Уч.-изд, л. 25,13.
Т-11093. Тираж 7000 экз. Зак. 596/822-
Цена 1 р. 91 к.
Гоетоптехиздат.
Москва, К-12, Третьяковский проезд, 1/19.
Типография «Красный Печатник».
Ленинград, Московский проспект, 91-
14 сверху
НЕФТЯНОЕ
ППДПИСЫВАИТЕСЬЭ^
I
на год 72 Р
НА 6 МЕС 36 р
Строка
го&>
6<-лС
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Напечатано
— -J__________Следует печатать
7 сверху
12 и 17 снизу
2» 23, 29 сверху
20 сверху
1/822.
гидрирование
СН»
+НС
гидрирования
[Я
£Л
хлоргидрирование
-* R—сн=сн8
СООН
+НС1
гипохлорирование
(хлоргидринирование)
- R—СН—СН3
СООН
а
1 IUIII г аиддп/А
ПериоЪигностпь журналов-12 номеров в гоЪ
ПОДПИСКА ПРИНИМАЕТСЯ БЕЗ ОГРАНИЧЕНИЯ
В ГОРОДСКИХ ОТДЕЛАХ СОЮЗПЕЧАТИ, КОНТОРАХ И
ОТДЕЛЕНИЯХ СВЯЗИ И ПУНКТАХ ПОДПИСКИ.