/



































































































































Author: Гурвич М.Ю.
Tags: экономическая геология месторождения полезных ископаемых минералы горные породы горное дело минералогия минеральные ресурсы
Year: 2009




Text
М.Ю. Гурвич
СОВРЕМЕННЫЕ МЕТОДЫ
ИССЛЕДОВАНИЯ МИНЕРАЛОВ,
ГОРНЫХ ПОРОД И РУД
Учебное пособие
Москва 2009
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ
РОССИЙСКОЙ ФЕДЕРАЦИИ
Российский государственный
геологоразведочный университет
М. К). Гурвич
СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ МИНЕРАЛОВ,
ГОРНЫХ ПОРОД И РУД
Учебное пособие
Допущено УМО по образованию в области прикладной геологии в качестве учебного
пособия для студентов высших учебных заведений, обучающихся по специальности
130301 «Геологическая съемка, поиски и разведка месторождений полезных ископае-
мых» и 130306 «Прикладная геохимия, петрология, минералогия» направления 130300
«Прикладная геология»
МОСКВА 2009
УДК 553.08
Г 95
Аннотация
К учебному пособию Гурвича М.Ю. «Современные методы исследования
минералов, горных пород и руд»
В учебном пособии рассмотрены физические методы определения
содержания тех или иных элементов в природных и техногенных объектах -
это оптический спектральный метод (эмиссионный и атомно-
абсорбционный), рентгеноспектральный ( рентгенофлуоресцентный,
микрорентгеноспектральный и рентгенорадиометрический), активационный
анализ. Рассмотрены метрологические параметры этих методов.
Для исследования форм нахождения элементов в объекте освещены
методы рентгенографии, электронной микроскопии и радиографии.
143 стр. ил. 49.
ВВЕДЕНИЕ
На современном этапе геолог практически любой специализации сталкивается с
задачей исследования вещественного состава природных или техногенных объектов.
Для успешного решения этих вопросов требуется применение разнообразных анали-
тических методов, каждый их которых обладает своими метрологическими парамет-
рами.
В то же время, как показывает опыт преподавания в геологоразведочном универ-
ситете, многие студенты- выпускники, даже геологоразведочного факультета, плохо
ориентируются в вопросах выбора необходимого круга аналитических методов и ме-
тодик для решения конкретных геологических задач, что зачастую приводит лишь к
потере сил и времени.
В данном пособии рассмотрены методы рентгенографии, методы элементного
анализа и электронной микроскопии, т.е. тех методов, которые позволяют ответить на
вопросы содержания тех или иных элементов и формы их нахождения в исследуемых
объектах. Развитие этих методов вместе с ростом технического прогресса обусловило
появление справочной литературы, рассчитанной на узкий круг специалистов - анали-
тиков. В связи с этим возникла необходимость написания учебного пособия, где в
доступной форме были бы изложены основы и возможности различных методов ана-
лиза вещества.
Данное учебное пособие написано на основе курса лекций, читаемого студентам
геологоразведочного факультета РГГРУ на протяжении последних 10 лет. В нем рас-
смотрены вопросы теоретических основ методов, во многих случаях
конкретная аппаратура, рассмотрены вопросы метрологических параметров методов
аппаратуры (порог чувствительности, воспроизводимость результатов, требуемая н:
веска и т.д.).
Рекомендованный список литературы включает только основные источник
публикаций по рассматриваемым вопросам.
ДИФРАКЦИОННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
Кристаллография зародилась еще в древности в связи с наблюдением и опи-
санием внешних форм кристаллов, закономерностей их огранения. Это так назы-
ваемая геометрическая кристаллография. На ее основе возникла гипотеза (конец
XIX века), что в кристалле атомы и молекулы образуют трехмерную периодиче-
скую решетку. Периоды решеток кристаллов (исходя из известной к тому времени
постоянной Авагадро) должны были составлять от единиц А до и 10 2 - и '103 А
(А=10-,нм = Ю^мкм = 10'7мм= 10'8см = 10~’°м).
Возможность непосредственного исследования внутреннего строения
кристаллов появилась после открытия в 1895г. немецким физиком Рентгеном X-
лучей (впоследствии названных его именем - рентгеновские лучи).
Рентгеновские лучи - электромагнитные волны с длиной волны 103- п КГ4 А
Переходя к понятию дифракции, отметим, что дифракционные явления мы
наблюдаем в природе часто. Говорим о дифракции волн на воде, о дифракции света
и звука. Согласно теории дифракции в любой произвольной точке волновое поле
складывается из вторичных волн, испускаемых фиктивными источниками. Так,
имея непрозрачный для света экран с несколькими отверствиями, размеры которых
соизмеримы с длиной волны света, мы за экраном в области тени получим интерфе-
ренционную картину, состоящую из светлых и темных зон. Светлые зоны соответ-
ствуют вторичным волнам, пришедшим в фазе, а темные - в противофазе.
ГЛАВНОЕ:
Дифракционная картина наблюдается тогда, когда размер фиктивного источ-
ника совпадает с длиной волны. Так, дифракция на воде наблюдается при размерах
фиктивного источника, равного « п 102см;
• дифракция звука возникает при размерах фиктивного источника,
равного ~п 10 см;
• дифракция света в воздухе образуется при размерах фиктивного источника,
равного 10'4 — 10'5 см.
Если теперь сравнить известные данные на сегоднешний день значения иара-
мегров элементарной ячейки некоторых минералов (табл.1.) со средней длиной вол-
ны рентгеновского излучения (п ’ 10° А), то можно предположить, что при облучении
кристаллической решетки рентгеновским излучением будет происходить дифракция
лучей.
Таблица 1
Параметры элементарной ячейки некоторых минералов
МИНЕРАЛ А НАЗВАНИЕ сингония
Аи 4,06 ЗОЛОТО кубическая
PbS 5,92 галенит 44 _
CusFeSf 10,93 борнит
Ag2PbsSb4Sl5 а=22,8 b=27,3 с= 8,9 овихиит ромбическая
Впервые дифракцию рентгеновских лучей на кристаллической решетке наблю-
дал в 1912г. Лауэ (с помощниками). Он пропустил пучок рентгеновских лучей че-
рез кристалл медного купороса (CuSCV 5Н2О) и зафиксировал его на фотопластин-
ке. На последней были получены правильно расположенные точки (дифракционная
картина), что можно было объяснить только тем, что:
1) рентгеновские лучи представляют собой волны;
2) частицы в кристалле медного купороса располагаются периодично.
Позже Лауз дал обоснование возникновению дифракции в своем опыте.
Как происходит дифракция рентгеновских лучей?
При воздействии на вещество рентгеновских лучей (обладающих определен-
ной энергией, равной Е= chA) они возбуждают электроны на оболочках атомов.
Возбужденные электроны отдают эту энергию в виде вторичных рентгеновских лу-
чей, распространяющихся по всей сфере. Из множества вторичных рентгеновских
лучей, рассеянных двумя (как минимум) или большим числом атомов, найдутся та-
кие, которые в определенной точке пространства придут в фазе, г.е. эти лучи на пе-
риодической решетке кристалла дифрагируют. Поставив в точке, где лучи приходят
в фазе, приемник рентгеновского излучения, мы получим рефлекс. В качестве при-
емника рентгеновского излучения применяют фотоматериалы или счетчики
рентгеновского излучения (ионизационные камеры, пропорциональные
счетчики, сцинцилляционные, полупроводниковые и т.д.).
Для получения мощных пучков рентгеновского излучения применяют рентге-
новские трубки. Это электровакуумный прибор, в котором вакуум составляет окало
КУ4 мм.рт.ст. Имеется нить накала из тугоплавкого материала (вольфрам). В качест-
5
ве анода применяются различные “чистые” материалы (медь, железо, кобальт и дру-
гие металлы). Между нитью накала и анодом прикладывается разность потен-
циалов, равная десяткам киловольт.
Не рассматривая вывод формулы, запишем уравнение дифракции, выведенное
Бреггами и Вульфом:
nA, = 2d sinO,
где d - межплоскостное расстояние между атомными сетками, дав-
шими “отражение”, “рефлекс”;
6 - угол, под которым рентгеновский пучок попадает на образец;
• X - длина волны рентгеновского излучения;
п - порядок отражения — 1, 2, 3 и т.д.
Из формулы видно, что при d = const дифракцию (рефлекс) можно получить ли-
бо меняя X, либо 6.
Помимо рентгеновских лучей дифракцию на кристаллическом веществе можно
получить от потока электронов, нейтронов и др. частиц. Ничего в этом странного
нет, если вспомнить, что по квантомеханическим представлениям движущуюся
микрочастицу можно рассматривать как волну (корпускулярно-волновой дуализм).
Согласно квантовой механике, движение частицы массой “т” со скоростью “о“
(энергией Е) можно представить как плоскую монохроматическую волну (волну де
Бройля), которая равна
h
V2mE
где h - постоянная Планка, равная 6,6'10'27эрг с; и« “с” (скорость света)
Впервые экспериментально дифракцию электронов наблюдали в 1927 году
Дэвиссон и Джермер. Они направили пучок электронов, ускоренных электростати-
ческим полем, на кристалл Ni , который вращался. Регистрация “отраженных”
электронов осуществлялась с помощью гальванометра, расположенного под неко-
торым углом к первоначальному направлению пучка электронов.
Расположение “max” на полученной кривой зависимости отраженных элек-
тронов от угла поворота кристалла вокруг оси соответствовало той же формуле
дифракции:
nl = 2d sin 0
Так, впервые было экспериментально подтверждено, что электроны имеют и
волновые свойства, т.е. обладают явлением дуализма.
Вскоре была обнаружена дифракция и на “просвет” вещества. Так экспери-
ментаторы получили еще один “инструмент” (метод) для исследования внутренней
структуры вещества.
Сегодня для получения картины дифракции на электронах (электронограмм)
используют электронографы или электронные микроскопы, в которых получают уз-
кий направленный пучок электронов. Их источником является раскаленная
(~ 2700°С) вольфрамовая нить. Электроны разгоняются высоким напряжением
(30 кВ и выше) и фокусируются с помощью диафрагм и электромагнитных линз. На
оси острофокусного моноэнергетического пучка электронов размещается исследуе-
7
мый образец. Рассеянные электроны попадают на фотопластинку и создают ди-
фракционную картину (электронограмму). В электронографах для получения луч-
шей дифракционной картины имеются устройства для нагревания образца, охлаж-
дения, испарения, вращения его.
В электронографах и электронных микроскопах создается вакуум до
lO^-lO'6 мм. рт.ст.
Дифракционная картина тонкого монокристаллического вещества представ-
ляет собой правильно расположенные точки (рефлексы), а от поликристаллического
- равномерно зачерненные окружности (рис. I).
Мсжплоскостные расстояния определяются по формуле
d = LX/г,
где L - расстояние от образца до фотопластинки,
к - длина волны,
г - расстояние от рефлекса до центрального пятна.
Рис.]. Электронограммы от монокристаллического (а), текстуриро-
ванного (в) и поликристаллического (с) вещества
Сильное взаимодействие электронов с веществом ограничивает размеры ис-
следуемых образцов. При просвечивании толщина образцов ограничивается деся-
тыми долями мкм. (1 О^см).
Впервые дифракцию нейтронов наблюдали в 1936г. на кристалле MgO.
Метод дифракции нейтронов на кристаллическом веществе получил толчок своего
развития в связи с созданием ядерных реакторов. Ядерный реактор является мощ-
ным источником нейтронов (“п”). Используются тепловые нейтроны, т.е. нейтро-
ны с Е= 0,5 - 5 ’Юэв (длина волны « 2А). Уравнение дифракции нейтронов на кри-
сталлическом веществе имеет такую же формулу как уравнение дифракции рентге-
новских лучей и электронов.
Природа взаимодействия различных частиц и рентгеновских лучей с вещест-
вом различна. Так, электроны рассеиваются в результате взаимодействия электри-
ческого заряда электрона с электростатическим потенциалом атома, который скла-
дывается из потенциала положительно заряженного ядра и потенциала электронной
оболочки.
Взаимодействие рентгеновского излучения осуществляется с электронами
атома, а нейтроны взаимодействуют с ядром, а также происходит взаимодействие
магнитного момента атомов исследуемого вещества с магнитным моментом ней-
трона.
Наличие магнитного упорядочения в исследуемом веществе ведет к появле-
нию на нейтронограммах наряду с ядерным рассеиванием дополнительных рефлек-
9
сов за счет магнитного рассеяния. Для разделения рефлексов от различной природы
рассеивания требуются дополнительные меры. По расшифровке дифракционных
максимумов от магнитных центров рассеяния можно установить магнитную струк-
туру кристалла
Рассеивающую способность атомов вещества характеризуют атомной ам-
плитудой рассеяния (f). Для электронов f зависит от порядкового номера вещества
(Z) и с увеличением его увеличивается как Z2H. Для рентгеновских лучей f увели-
чивается с увеличением Z как Z2. Для нейтронов атомная амплитуда рассеяния не
зависит от порядкового номера вещества.
Интенсивность рассеяных атомами электронных волн гораздо больше, чем
интенсивность рассеяния рентгеновских лучей. Так, подсчет показал, что при Z>30
атом рассеивает в 106 раз больше электронов, чем рентгеновские лучи. Сильная рас-
сеивающая способность электронных волн атомами является характерной чертой
электронографии. Поэтому для получения электронограмм требуются малые коли-
чества вещества и небольшая экспозиция. Электроны проникают на глубины до
1000А.
Рассматривая зависимость рассеивания рентгеновских лучен, электронов и
нейтронов от Z, видим, что для получения соизмеримого рассеивания от легких и
тяжелых атомов всего лучше использовать нейтроны, затем электроны.
Сведем изложенные сведения о методах в таблицу 2.
ю
Таблица 2
ДИФРАКЦИОННЫЕ МЕТОДЫ СТРУКТУРНОГО АНАЛИЗА
Рентгенография Электронография Нейтронография
О бшая формула дифракции: ПХ — 2d SillG
Год открытия 1912 1927 1936
Источник излуче- ния Рентгеновская трубка Электронная пушка Ядерный реактор
Характер взаимо- действия С электронами ато- мов С электростатиче- ским потенциалом от электронной оболоч- ки и ядра С ядром и магнит- ным моментом атома
Достоинства и не- достатки Большая проникаю- щая способность, легко получить рент- геновское излучение Малая проникающая способность, можно исследовать объекты с толщиной не бо- лее 1000А, с другой стороны - это досто- инство; источник элекгронов помеща- ют в вакуум Большая проникаю- щаяспособность, можно хорошо ана- лизировать положе- ние легких атомов в соединениях; воз- можность магнитно- го рассеяния; источ- ник нейтронов мало- доступен
Наиболее широко из дифракционных методов применяется рентгенография.
Существуют несколько методов получения рентгенограмм:
1. Метод Лауз
для монокристаллов; используется сплошной спектр (белый); 0 = const
2. Метод Вейсенберга
для монокристаллов; метод вращающегося кристалла; X = const
3. Метод Дебая - Шеррера
для порошковых объектов; образец вращается; X = const
В геологии чаще используются два метода структурного анализа: рентгено-
графия и электронография. В рентгенографии наибольшее распространение полу-
чили порошковые методы.
РЕНТГЕНОГРА ФИ Я
Рентгеновское излучение - электромагнитное ионизирующее излучение, за-
нимающее спектральную область между гамма- и УФ излучением в пределах длин
волн от КГ4 до 103 А (от 10'12 см до 10 '5см)_
Если рассмотреть шкалу электромагнитных волн, то рентгеноское излучение
занимает довольно узкую область (рис.2).
Родиобояны
ЮООЛМ /кн 1мм (ма 1ц*
1С,6А Й,3Л lO’A 10 Л ЮА Я А 1(Г Л
Рис. 2. Шкала электромагнитных волн
Электромагнитные волны - это электромагнитные колебания, распростра-
няющиеся в пространстве со скоростью 3 1010 см/сек. (в вакууме) Существование
электромагнитных волн было предсказано анг. физиком Фарадеем в 1832г. В опы-
тах Герна (нем.) в 1888г. было подтверждено, что электромагнитные волны ни что
иное, как электромагнитные колебания.
Радиоволны, свет ( ИК-, видимый, УФ ) , рентгеновские лучи и у-излучение
- это все электромагнитные волны с различной длиной волны (X). Причем, между
12
соседними диапазонами шкалы электромагнитных волн нет резких границ. Длина
волны связана с другими физическими величинами следующими соотношениями:
X = c/v ; Е = hv ; X = ch/E , где h - пост. Планка, с - скорость света, Е - энер-
гия у - квантов, v = частота излучения.
Электромагнитные волны различной длины характеризуются различными
способами возбуждения и регистрации, они по-разному взаимодействуют с вещест-
вом.
Процессы излучения и поглощения электромагнитных волн от самых длин-
ных до ИК-излучения достаточно полно описываются законами электродинамики.
На более коротких волнах доминируют процессы, имеющие существенно кванто-
вую природу, а в диапазоне рентгеновского и у-излучения описываются только на
основе представлений о дискретности этих процессов . Во многих случаях электро-
магнитное излучение ведет себя не как набор монохроматических волн с частотой v
и волновым вектором “к”, а как поток квазичастиц - фотонов с энергией hv и им-
пульсом р- htfc.
Волновые свойства проявляются в явлениях дифракции и интерференции, а
корпускулярные - в явлениях фотоэффекта и Комптон эффекта.
РЕНТГЕНОВСКИЕ СПЕКТРЫ
Рентгеновские спектры - это спектры испускания и поглощения излучения в
области Х= ПУ4 до 103 А. Рентгеновское излучение с Х< 2А называется условно
“жестким”, а с Х>2А - мягким.
13
Для регистрации и исследования рентгеновских спектров применяют:
1) спектрометры с диспергирующим элементом (кристалл-анализаторы - слюда,
кварц, LiF и т.д. или дифракционная решетка) и детектором рентгеновского из-
лучения;
2) бездифракциоиную аппаратуру, состоящую из детектора рентгеновского излу-
чения (сцинтилляционные, газопропорциональные или полупроводниковые
счетчики) и амплитудного анализатора импульсов;
3) фотоматериаллы.
Применение обычных призм для изучения рентгеновских спектров, как это
делается в области видимого спектра, здесь невозможно, так как коэффициент пре-
ломления рентгеновских лучей близок к 1, в то время как для видимого света он
равен приблизительно 0,5.
Рентгеновские спектры испускания - это спектры тормозного и характери-
стического излучения.
Спектр тормозного излучения
Основные закономерности распространения рентгеновского излучения опи-
сываются классической электродинамикой. Электромагнитная волна может соз-
даться ускоренно-движущимися заряженными частицами. Изменяющееся во време-
ни электрическое поле (Е) создает переменное магнитное поле (Н), которое, в свою
очередь, создает вихревое электрическое поле. Обе компоненты “Е” и “Н”, непре-
рывно изменяясь, возбуждают друг друга и распространяются в пространстве с ко-
14
печной скоростью, удаляясь от источника. Источник может исчезнуть, а поле его
распространяться.
Это все классическая теория излучения (заложена Фарадеем и Максвеллом),
объясняющая характерные черты процессов излучения, но она не смогла объяснить
спектры рентгеновского излучения. Это удалось решить только с помощью кванто-
вой теории, разработанной Планком, Эйнштейном, Бором, Бройлем и др.
Простейшим источником электромагнитного излучения является движущий-
ся ускоренно точечный заряд. Если он покоится или движется равномерно, то излу-
чения нет. В зависимости от природы ускорения заряженной частицы излучение
имеет определенные названия. Так, излучение, возни-
кающее при торможении частицы в веществе в результате
взаимодействия с кулоновскими полями ядер и электро-
нов атомов, называется тормозным излучением. Излуче-
ние, возникающее в результате воздействия на заряжен-
ную частицу магнитных полей, называется синхронным
излучением.
Интенсивность тормозного излучения пропорцио-
нальна квадрату ускорения заряженной частицы “а”, а
так как “а” обратно пропорционально, при прочих рав-
Рис.З. Зависимость
спектральной плотности
для W - анода
ных условиях, массе частицы, то интенсивность излуче-
15
ния при торможении электрона будет в миллионы раз больше, чем протона
( ГПр/Ше ~ 1800).
Рентгеновский тормозной спектр (рис.З) получают в рентгеновских
трубках. Его основные характеристики:
1)он сплошной, непрерывно распределен по всем длинам вплоть до УФ све-
та ; его коротковолновая граница (1о) равна - Хо = he / eV = 12,8 / U , где U -
анодное напряжение на рентгеновской трубке в Кв, а X - в А;
2)Хта> « 1,5 Amin ( Х„) ;
3)1,шт.= i U2Z, где i и U - ток и напряжение на рентгеновской трубке, a Z- поряд-
ковый номер в таблице Менделеева атомов мишени.
Интенсивность тормозного рентгеновского спектра с возрастанием энергии
электронов возрастает и смещается в сторону коротких длин волн. Интенсивность
возрастает с увеличением Z мишени, поэтому для получения интенсивных тормоз-
ных рентгеновских спектров берут мишени с большим Z.
Тормозной рентгеновский спектр имеет еще названия - “сплошной” и “бе-
лый”.
Характеристические рентгеновские спектры
Характеристические (или дискретные) рентгеновские спектры испускаются
атомами мишени (анод рентгеновской трубки) при столкновении с заряженной час-
тицей высокой энергии (первичные рентгеновские спектры) или рентгеновским фо-
16
тоном (флуоресцентные). Природа этих спектров одинакова. Прежде чем рассмат-
ривать природу этих спектров, вспомним основы квантовой теории твердых тел.
Энергетическое состояние электронов в атоме определяется четырьмя кван-
товыми числами; n, I, т,, и т8.
1) и - главное квантовое число. Может принимать только целое значение: 1,2,3 и
т.д.. Оно определяет энергию электрона в атоме. Совокупность электронов с одина-
ковым “и” образует слой. Наибольшее число электронов в слое равно 2п2. Слои
имеют свои обозначения: К-слой, L-слой, М-слой, N-слой и т.д. Количество элек-
тронов по слоям равно соответственно: 2, 8, 18, 32 и т.д.
2)1 - побочное квантовое число (орбитальное или азимутальное). Принимает
также только целое значение, равное 0, 1, 2, .... (и - 1). Совокупность электронов с
одинаковым “п” и “I” образует оболочку.
3) Магнитное квантовое число - mi = ±0, ±1, ±2 .... ±1.
4) Спиновое квантовое число - ms = + S.
По той же квантовой механике возможные значения внутренней энергии ато-
мов могут принимать только определенные дискретные значения, соответствующие
устойчивым состояниям атомов. Совокупность этих устойчивых значений внутрен-
ней энергии атомов образует ее энергетический спектр. Самый нижний уровень этой
энергии называется основным, все остальные - возбужденными. Энергия атомов мо-
жет изменяться только скачкообразно, путем квантового перехода атома из одного
„ I Учебная
библиотека
стационарного состояния в другое. Для перехода атома в возбужденное состояние
ему необходимо сообщить дополнительную энергию извне.
Эту энергию атом и получает при облучении его электронами или рентге-
новскими лучами. В результате этого облучения с одной из внутренних оболочек
атома (К-, L-, М- ...) выбивается электрон. Состояние атома с вакансией на внут-
ренней оболочке неустойчиво, и спустя некоторое время (время жизни на возбуж-
денном уровне энергии - » n 1СГ8 с) электрон с одной из внешних оболочек может
заполнить эту вакансию. Атом переходит при этом в более устойчивое энергетиче-
ское состояние с меныпей энергией, испуская избыток энергии в виде фотона ха-
рактеристического излучения (рис. 4).
Поскольку энергия начального состояния и конечного квантованы (Е| и ЕД
возникает линия рентгеновского спектра с v=(E| - Ei) / h Все возможные кванто-
вые переходы из начального К - состояния образуют наиболее жесткую (коротко-
Рис.4. Схема образования характеристического спектра
18
волновую) К-серию. Однако переходы возможны не между любыми энергетиче-
скими состояниями атома; возможность перехода определяется правилами отбора.
Одно из основных правил отбора - вероятность перехода очень мала, если кванто-
вое число I не меняется при этом на 1 единицу.
Вот почему нет перехода с уровня Li на К уровень, так как Li ( п=2,1=0,
Ш|=0, ms=±l/2), а К (п=1,1=0, mj=0, ms=±l/2) и Л1=0
Переход К —>L3 - называется линией Kat
Переход К ->L2 - называется линией
В табл.З приведены некоторые характеристические линии, начальные и ко-
нечные орбиты переходов электронов.
Исходя из рассмотрения всех энергетических состояний, можно объяснить
закономерности спектров, которые были найдены экспериментально:
Таблица 3
Линия Начальное состояние Конечное состояние
Kai К L3
Ко2 К U
K₽t К М3
Кр2 к N23
Крз к мг
Lal L3 м5
м4
И Т.Д.
19
1) лучи каждой серии возникают тогда, когда для возбуждения затрачивается
энергия выше критической энергии (или потенциала возбуждения Ц для данной се-
рии), необходимой для выбивания электрона с внутренней орбиты; при дальнейшем
повышении энергии (анодного напряжения U) интенсивность линий этого спектра
растет пропорционально (U-Uj)2; затем рост интенсивности замедляется и при
U » 11 Uj начинает падать;
2) так как возбуждается одновременно громадное количество атомов, то од-
новременно появляются все линии серии;
3) К- серия, по сравнению с другими сериями, имеет волны с меньшими 1;
4) спектр характеристического излучения состо-
*.А
Рис.5. Кривые распре-
деления интенсивности
характеристического и
тормозного спектров по
длинам волн
ит, как правило, из нескольких серий; при наличии К-
серии всегда есть и другие серии - L, М и так далее се-
рий, так как потенциалы возбуждения этих серий ниже,
чем для К-серии;
5) несмотря на го, что спектры различных эле-
ментов содержат одни и те же линии, длина волны
линии однозначно характеризует элемент, ис-
пускающий эту линию; связь между длиной волны
и Z элемента впервые была обнаружена Мозли
v = 2,48 105(Z - I)2;
6) интенсивность линий любой серии определяется
20
заселенностью уровней электронами и вероятностью перескоков их с уровняна на
уровень; так соотношение интенсивностей в К- серии
I<xl: 1а2: 1рр 1р2 = 100 : 50 : 23 :1 ;
7) при наличии характеристического спектра всегда имеется сплошной
(рис.5); обратное положение неверно, так как для возникновения характеристиче-
ского спектра необходимо напряжение на трубке выше икрм^ческого; в рентгеновских
трубках при их эксплуатации возникают, чаще всего, оба спектра.
Спектры поглощения
Данные спектры получаются в результате пропускания рентгеновского спек-
тра того или иного вида через слой вещества - поглотителя (фильтр). При этом рас-
пределение интенсивности относительно первоначального спектра изменяется, т.е.
возникает новый спектр - спектр поглощения.
Начальная нтенсивность (1О) уменьшается за счет поглощения и рассеяния
до величины
I = 1<,е , где
ц - коэффициент ослабления, зависящий от 2. падающего излучения;
х - толщина поглотителя.
Эта зависимость используется для фильтрации 0-излучения от рентгенов-
ских трубок. Дело в том, что характеристическое излучение, испускаемое анодом
рентгеновской трубки, состоит из нескольких серий. Рассмотрим только К-серию,
21
так как другие имеют значительно меньшую интенсивность. Но и у К-серии есть а-
и P-излучение. От P-излучения легко освободится с помощью селективнопогло-
щающих фильтров, что позволяет получать практически моноэнергетическую ли-
нию при работе рентгеновской трубки.
Рентгеновские Р - лучи поглощаются сильно, когда край полосы поглощения
фильтра лежит между длинами волн Х^ и Хр (рис.6). Например, чтобы отфильтро-
вать Р-лучи К-серии, полученные от трубки с Fe-анодом, необходимо взять фильтр,
изготовленный из марганпа. так как край полосы поглощения марганца при X =
1,869 А между длинами волн Ха=1,938А иХр=1,75бА.
Рис. 6. Селективное поглощение фильтром Хр - излучения
Таким образом, использование селективнопоглощающих фильтров (табл.4)
позволяет отфильтровывать не только p-излучение, но и почти польностью избавить-
ся от “белого” излучения, если анодное напряжение на рентгеновской трубке не бу-
дет превышать потенпиал возбуждения той или иной серии в 1,5 раза.
22
Таблица 4
Некоторые характеристики селективных фильтров
Материал Эмиссионная линия Материал К-край Ослабление р - линии
в 500 раз
анода Z А фильтра Z А толщина ЦК процент потерь Ка
Сг(24) Ка, 2,2896 Ко2 2,2935 К₽ 2,0848 V(23) 2,269 17 51
Fe(26) К.,! 1,9360 Ка2 1,9399 К₽ 1,7565 Мп(25) 1,869 18 53
Устройство рентгеновских трубок
В зависимости от области применения рентгеновские трубки различаются
по конструкции, способу получения и фокусировки электронов, вакуумированию,
охлаждению анода, размеру и форме фокуса, по веществу анода.
В рентгенографии наиболее широко применяют стеклянные отпаенные рент-
геновские трубки с вакуумом Ю^-Ю^мм-рт.ст., с термоэмиссионным катодом и во-
дяным охлаждением. Катод представляет собой вольфрамовую нить, накаливаемую
до « 2700°С электрическим током. Между катодом и анодом прикладывается уско-
ряющее напряжение до ~ 50Кв. Мощность трубок для рентгенографии достигает 1,5
- 2,0 квт. Если учесть, что только » 1% бомбардирующих анод электронов превра-
щается в рентгеновское излучение, а остальная энергия - в тепло, то требуется его
охлаждение. Для охлаждения анода применяется проточная вода. Для выхода рент-
геновского излучения имеются 1-4 окна, закрытых бериллиевой (как самого легкого
металла) фольгой.
Для рентгенографии применяют трубки типа БСВ с анодами из Сг, Fe, Со,
Ni, Си и т.д. Кроме потребляемой мощности и материала анода трубки различают-
ся между собой размерами фокусного пятна, которые варьируют по ширине от 0,4
до 2,0мм и по длине от 8 до 10мм.
Методы рентгенографии
После открытия Лауэ явления дифракции рентгеновских лучей на кристал-
лической решетке появился мощный инструмент (метод) для исследования кри-
сталлов.
Вся информация о кристаллическом строении вещества содержится в наборе
“d” и интенсивности рефлексов (I). Исходя из формулы дифракции рентгеновского
излучения (пХ = 2 d sinO) для получения информации о “d”, можно:
I) не меняя положения образца, менять X или использовать белый (сплошной)
рентгеновский спектр, где есть набор всех длин волн, начиная с Хо;
2) использовать моноэнергетическое излучение (X=const) и вращать образец.
24
Для создания условий дифракции и регистрации дифрат ированного излуче-
ния слу'Жат рентгеновские камеры, рентгеновские дифрактометры и рентге-
новские гониометры. Регистрация излучения осуществляется с помощью фотопле-
нок или детекторов ядерных излучений, в частности, детекторов у-излучения.
Метод Лауз
11олучение рентгенограмм от монокристалла, когда он неподвижен, а используемое
рентгеновское излучение имеет непрерывный спектр. Расположение дифракцион-
ных пятен на лауэграммах (рис.7) зависит от симметрии кристаллов и их ориента-
ции относительно падающего луча. Метод позволяет установить принадлежность
кристалла к одной из 11 лауэвских групп симметрии и определить направление
его кристаллографических осей с точностью до нескольких угловых минут.
Рис.7. Лауэграмма образца
Метод применяется в основном для ориентировки монокристаллов. Образен
при исследовании закрепляется на гониометрической головке, позволяющей пово-
25
рачивать кристалл в любом направлении. Путем съемки нескольких лауэграмм на-
ходят положение, которое дает симметричную картину дифракционных пятен на
рентгеновской пленке. Это положение соответствует тому, что кристалл располо-
жен по направлению пучка с каким-то элементом симметрии. После этого кристалл
на этой же гониометрической головке готов к более тщательному рентгенографиче-
скому анализу.
Метод качания и вращения образца
Метод используется для определения периодов повторяемости (постоянной
решетки) вдоль кристаллографического направления в монокристалле. В данном
исследовании используется монохроматическое излучение (1= const), а образец
вращается или приводят в колебательное движение вокруг оси, совпадающей с кри-
сталлографическим направлением. Пятна на рентгенограммах, полученных в ци-
линдрических кассетах, располагаются на семействе параллельных линий (рис.8).
Зная расстояние между линиями (измерив), длину' волны (зная анод рентге-
новской трубки) и диаметр кассеты, можно вычислить параметры кристалла.
Метод исследования поликристаллов (метод Дебая-LIleppepa)
Метод “порошка” основан на исследовании поликристаллических образцов
монохроматическим излучением. Образец исследуется в виде порошковой пробы,
истертой до 200-300 меш. (до пудры). Дифрагируемые лучи регистрируются либо
26
Рис. 8. Рентгенограмма «качания»
на пленку (фотометод) в специальных камерах, либо с помощью счетчиков
у - излучения на дифрактометрах.
Фотометод
При исследовании фотометодом получается интерференционная картина в
виде системы конусов с углом 46. Ось конусов совпадает с направлением первич-
ного пучка, а пересечение конусов дифрагированных лучей с фотопленкой образует
интерференционные линии. Каждая линия представляет собой результат дифракции
от определенной серии параллельных атомных плоскостей, расположенных относи-
тельно друг от друга на расстоянии d. Дифракционная картина, получения данным
методом, называется дэбаеграмма (рис. 9).
Основное достоинство фотометода — это возможность получения дифракци-
онной картины с пробы весом ~ 10 мкг. Если рассматривать порошковую пробу, как
поликристалл, то в дифракции участвуют множество кристалликов. При
27
Рис. 9. Дебаеграмма образца
размере исследуемого образца в фотометоде около 0,7-0,5мм в диаметре, количест-
во кристалликов равно около ~106. Образец в камере вращается.
Дебаевские рентгеновские камеры имеют определенные размеры, которые
удобны для расчета d. Чаще всего используются камеры РКД-57,3; РКУ-86; РКУ-
114; где номер камеры обозначает ее диаметр в мм.
Для каждого образца, каждого кристаллического вещества, к которым отно-
сятся и минералы, имеется своя интерференционная картина. Нахождение неиз-
вестной фазы осуществляется путем сравнения с эталонными дебаеграммами или
путем ее расчета и сравнения полученных межплоскостных расстояний (d) и их ин-
тенсивностей с эталонными, приведенными в специальных рентгеновских опреде-
лителях.
Дифрактометрический метод исследования
Рентгеновский дифрактометр - прибор для измерения интенсивности и на-
правления дифрагированных на кристаллическом объекте рентгеновских пучков.
Он позволяет измерять интенсивность дифрагированного излучения с точностью до
десятых долей процента и угол дифракции с точностью до десятых долей минут.
28
Дифрактометр включает в себя источник излучения (рентгеновская трубка), рентге-
новский гониометр (устройство, в которое помещают исследуемую порошковую
пробу), детектор излучения (сцинтилляционные, пропорциональные, полупровод-
никовые счетчики) и электронное измерительно-регистрирующее устройство. Го-
ниометр позволяет вращать под рентгеновским пучком образец и перемещать де-
тектор для регистрации в каждой точке интенсивности дифрагированного излуче-
ния за определенный промежуток времени.
По сравнению с рентгеновскими камерами дифрактометр позволяет произ-
водить угловые измерения дифрагированных пучков с гораздо большей точностью,
обладает большей экспрессностью. В дифрактометрах процесс получения информа-
ции может быть полностью автоматизирован (последние модели дифрактометров
даже снабжены управляющими ЭВМ); дифракционная картина (дифрактограмма)
выводится на перфоленту или может передаваться с помощью интерфейса во внеш-
нюю ПЭВМ, где с помощью определенных программ осуществляется обработка
дифрагированного рентгеновского спектра и вывод его на дисплей или распечаты-
вающее устройство. С помощью этих же программ осуществляется поиск иссле-
дуемой минеральной фазы путем сравнения “d” и интенсивностей спектров образца
с эталонами, банк которых может составлять несколько десятков тысяч кристалли-
ческих веществ. На рис. 10 приведена дифрактограмма исследованного образца,
полученная на дифрактометре и обработанная с помощью программы РФА (рентге-
иофазовый анализ); исследованный образец является малахитом: мономинераль-
ная фаза.
29
Рис.10. Дифрактограмма малахита
По сравнению с фотометодом исследования с помощью дифрактометров тре-
буют несколько большего количества материала - не менее 100 мг. С другой сторо-
ны исследования на дифрактометре требуют гораздо меньшего времени, имеют
значительно лучше разрешение, чем исследования с помощью рентгеновских камер
и возможность компьютерной обработки полученных результатов.
В зависимости от задачи исследования поликристаллов (метод порошка) не-
обходимо выбирать условия съемки. Так, например:
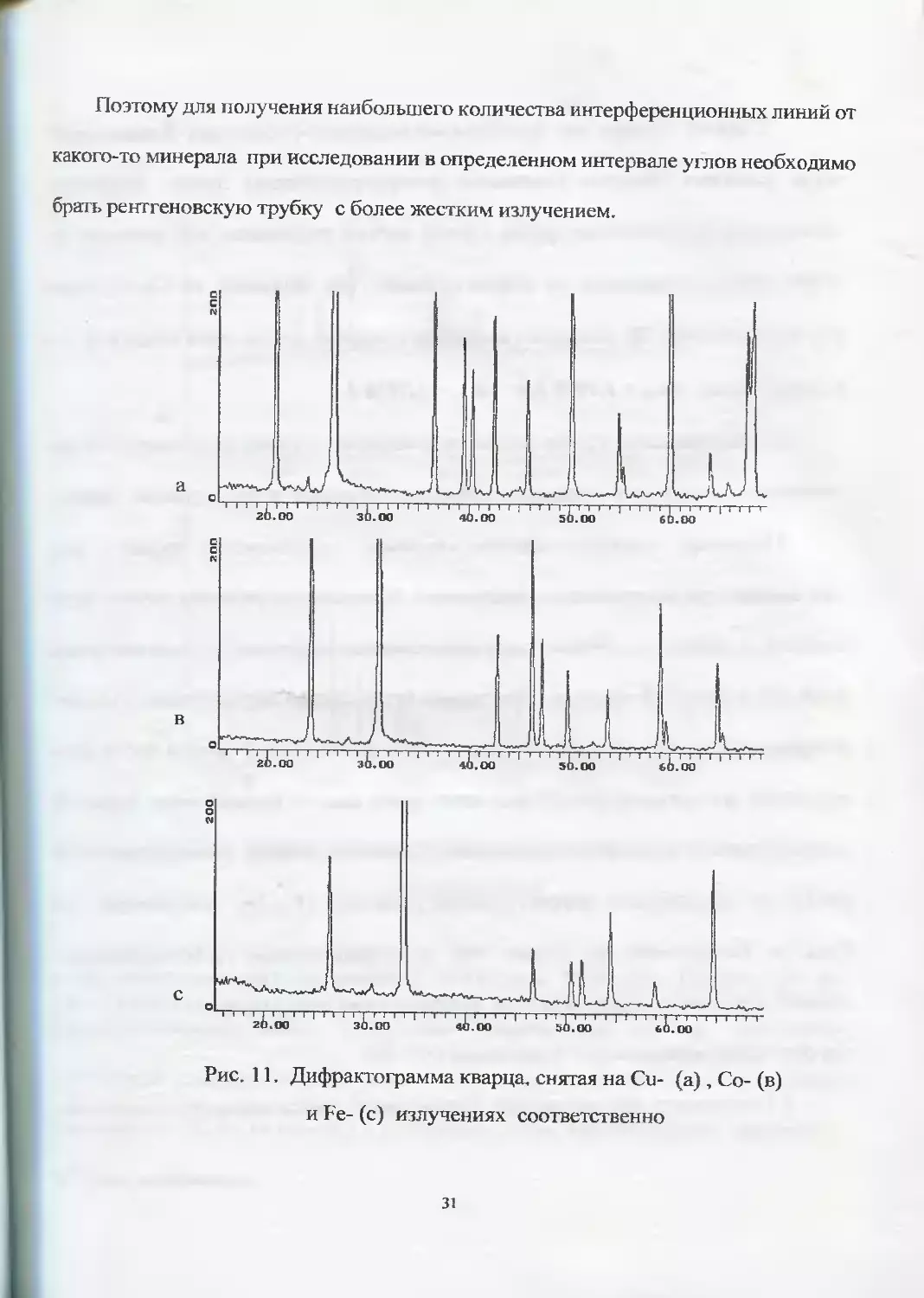
I. Использование рентгеновских трубок с различным анодом будет приводить
к растяжению (сжатию) дифрактограмм по горизонтали. Чем более мягкое излуче-
ние имеет рентгеновская трубка, тем более растянута рентгенограмма по оси углов
(рис. 11).
зо
Поэтому для получения наибольшего количества интерференционных линий от
какого-то минерала при исследовании в определенном интервале углов необходимо
брать рентгеновскую трубку с более жестким излучением.
Рис. 11. Дифрактограмма кварца, снятая на Си- (а), Со- (в)
и Fe- (с) излучениях соответственно
31
С другой стороны, при исследовании минералов с более низкой симметрией,
когда возникает большое количество интерференционных линий, желательно
использовать рентгеновские трубки с более мягким излучением, что позволяет по-
лучить лучшее разрешение на дифрактоурамме. Так, например, на Со-излучении
уже на углах 50-60 °20 возникают интерференционные линии даже от двух (К а( и
Кц2) длин волн: ХКа1 = 1,7889 А и = 1,7928 А.
2. Рентгеновскую трубку необходимо выбирать с таким излучением, которое
не вызывает вторичного характеристического излучения в исследуемом образце.
Например, характеристическое излучение рентгеновской трубки с мед-
ным анодом при исследовании минералов с большим содержанием железа будет
вызывать в образце вторичное характеристическое излучение, и рентгенограмма
будет иметь большой «фон», а дебаеграмма будет сильно завуалирована. Для пре-
дотвращения этого необходимо выбирать анод рентгеновской трубки таким обра-
зом, чтобы его материал (его Z) был ниже, равен или, по крайней мере, только на
единицу выше Z исследуемого вещества. Сказанное хорошо иллюстрируется на
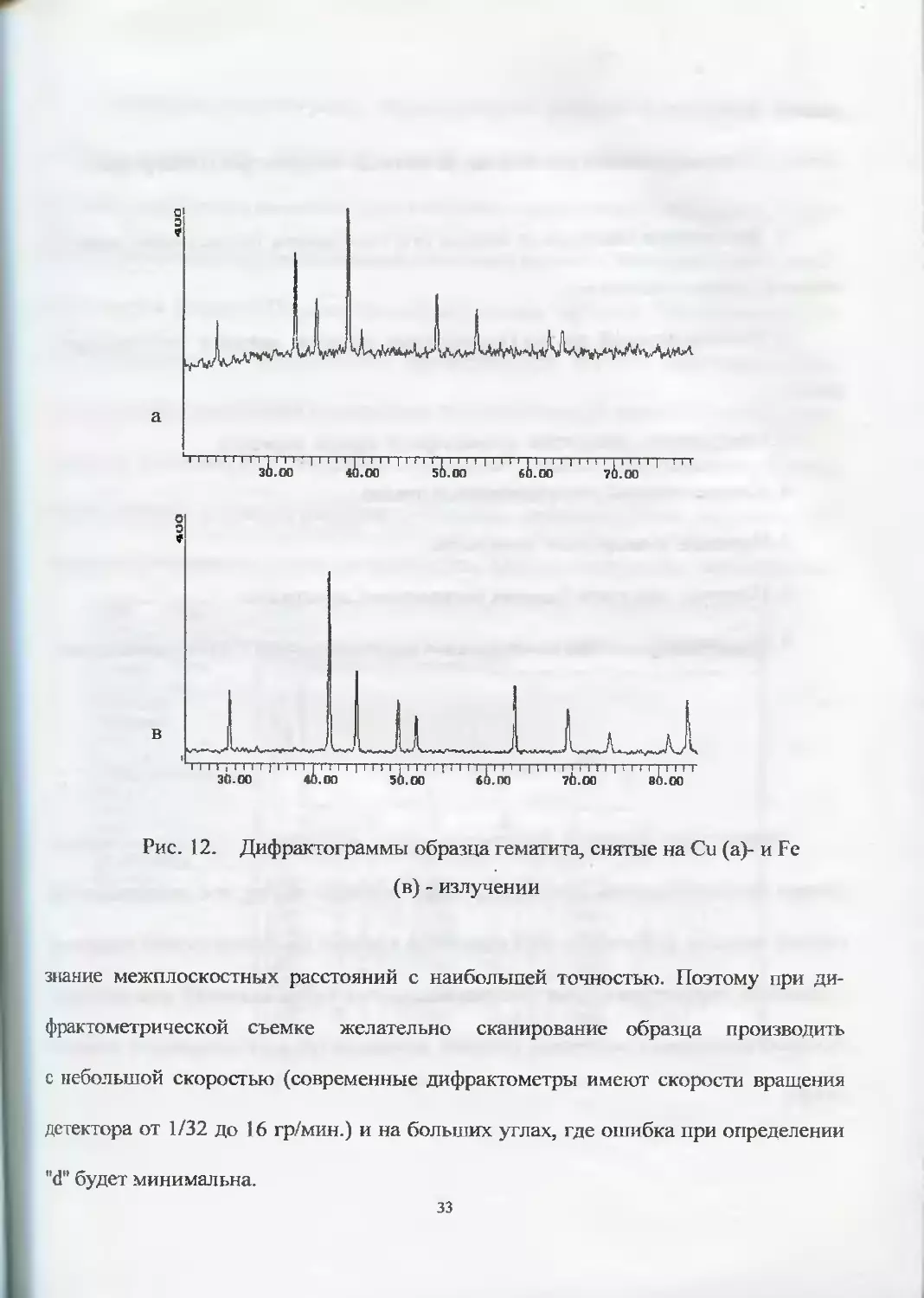
рис. 12, где представлены дифрактограммы гематита (Ре20з), полученные на
Си-(а) и Fe-излучении (в). Видно, что на дифрактограмме (а) большой «фон»,
который объясняется возникновением харкт ерист ического излучения от Fe (Z = 26)
при облучении минерала Си - излучением (Z = 29).
3. При определении параметров элементарной ячейки минералов необходимо
32
Рис. 12. Дифрактограммы образца гематита, снятые на Си (а)- и Fe
(в) - излучении
знание межплоскостных расстояний с наибольшей точностью. Поэтому при ди-
фрактометрической съемке желательно сканирование образца производить
с небольшой скоростью (современные дифрактометры имеют скорости вращения
детектора от 1/32 до 16 гр/мин.) и на больших углах, где ошибка при определении
"d" будет минимальна.
зз
Минералогические задачи, решаемые методом рент! енографии
1. Диагностика минерала по данным рентгенограммы (исследуемое вещество
является мономинеральным).
2. Рентгенофазовый анализ (исследуемое вещество является полиминераль-
ным).
3. Определение параметров элементарной ячейки минерала.
4. Количественный рентгенофазовый анализ.
5. Изучение изоморфизма минералов.
6. Изучение характера фазовых превращений минералов.
7 Определение степени совершенства кристаллической структуры вещества.
Диагностика минерала
Диагностика минерала (кристаллического вещества) по данным рентгено-
граммы (дифрактограммы или дебаеграммы) основана на том, что дифракционная
картина является диагностическим признаком каждого кристаллического вещества.
Основными параметрами рентгенограмм являются набор значений межплоскост-
ных расстояний кристаллической решетки минералов (d) и интенсивности отраже-
ний (I).
34
Сравнивая рентгенограмму исследованного минерала с эталонной, можно
однозначно определить минерал или группу минералов. Данные эталонных рентге-
нстрамм содержатся в различных рентгеновских определителях минералов, публи-
куются в различных научных изданиях, систематизируются Международной комис-
сией, которой создана “Порошковая дифракционная картотека Объединенного ко-
митета порошковых дифракционных стандаргов (PDF JCPDS). Картотека содер-
жит на сегодня около 60000 порошковых рентгенограмм. В качестве примера ниже
приведена эталонная рентгенограмма пирита, где указаны: номер карточки образца,
условия съемки, ссылка на источник публикации, межплоскостные расстояния, ин-
тенсивность отражений и индексы плоскостей. Иногда приводятся параметры эле-
42-1340 Quality “
CAS Number. 1303-36-0
^Molecular Weight 11997
Wolume|CO| 159.04
Рк 5.OT0 Dm.______________
jSG.. Pa3{205j
Ce! Parameters.
.a 5.417 b c
£_________P_______1_______
-SS/FOM: F27*98( 0095,23)
l/lcot 16
,Rad CuKal
‘Lambda, 1540598
Fta Gteph
-d-sp: dlftaclometa_______
Mineral Name-
ipyifte
FeS2
Iron SUfcie
Ret Nodland. D, Syrinski. W. McCarthy G.. "Bajfcs. P„ North Dakota State Univ., Fargo,
North Dakota. USA, ICDD Grant in-A'id. 4 989]
d|A) mt-v h k I
3.1280 23 1 1 1
2.7055 87 2 0 0
24203 52 2 1 0
22107 43 2 1 1
1.9160 44 2 2 0
1.8061 1 2 2 1
1.6333 100 3 1 1
15639 17 2 2 2
1.5023 20 0 2 3
d(A| mt-v h k I
1.4479 26 3 2 1
1.3141 2410
1 2770 <2411
1 2429 11 3 3 1
12115 14 4 2 0
1 1822 14 1 2 4
1.1552 6 3 3 2
1 1059 15 4 2 2
1 0834 <2 4 3 0
d[A] mt v h k I
10625 <2 4 3 1
10427 45 3 3 3
10061 16 2 3 4
98918 10 1 2 5
95770 30 4 4 0
94320 <3 4 4 1
.91581 5 5 3 1
90302 16 4 4 2
.89072 2 6 1 0
Рис. 13. Эталонная дифрактограмма пирита
35
ментарной ячейки, химический состав минерала, величина его плотности и показа-
тели преломления (рис. 13).
Помимо “ручной” диагностики исследуемого минерала по данным рентгено-
грамм имеются различные компьютерные программы, помогающие значи-
тельно облегчить эту задачу.
Определение параметров элементарной ячейки минерала
Определение параметров элементарной ячейки минерала производится на
основе квадратичных уравнений, связывающих межплоскостные расстояния (d),
индексы кристаллографических плоскостей (hkl) и размеры ячейки. Ниже приведе-
ны квадратичные уравнения для некоторых сингоний (табл.5).
Наиболее просто определяются параметры элементарной ячейки для известных ми-
нералов, т.е. минералов с известной сингонией и известными hkl. Расчет параметров
на сегоднешний день производится, как правило, по несложным программам на
ЭВМ и занимает несколько минут. Точность определения параметров элементарной
ячейки минерала, как следует из квадратичных формул, зависит от ошибки изме-
рения межплоскостных расстояний. Для уменьшения этой ошибки необхо-
димо использовать линии с большими углами отражения, использовать «внутрен-
ний» эталон, который позволяет вводить угловые поправки и устраняет практиче-
ски все систематические погрешности измерений, производить съемку рентгено-
граммы с небольшой скоростью (0,25-0,5 гр/мин.).
36
Квадратичные формулы для определения параметров
элементарной ячейки минералов
Таблица 5
Сингония Квадратичная формула
Кубическая 1 ft2+*2+/2 dh
Гексагональная и тригональная 1 4 к2 +Ы +к2 t I2 dlu 3X a2 + сг
Тетрагональная 1 h2+k2 dlu~ “ 'c~
Ромбическая 1 Л2 к2 I2 d2^ ьг+г
Моноклинная 1 h2 k2 2/rfcos/? d^u a2 sin2/? b2 c2sin2/? acsin2/?
В качестве "внутреннего" эталона чаще всего используют порошок металли-
ческого кремния или германия высокой чистоты. В настоящее время для метроло-
гического обеспечения дифракционных исследований применяют некоторые отрас-
левые стандартные образцы (СО), и разработан первый отечественный государст-
венный стандартный образец (ГСО) - параметр решетки на основе силицида вана-
дия (V3Si). Применение СО и ГСО позволяет производить определение параметров
кристаллической решетки с погрешностью ~ 0,0005 А.
37
Таким образом, имея экспериментальную рентгенограмму какого-либо об-
разца, определяют точно (введением поправки на эталон) межплоскостные рас-
стояния и интенсивности отражений, что позволяет определить минерал, знать его
сингонию и индексы плоскостей. Выбрав соответствующую квадратичную форму-
лу, вручную или по соответствующим программам, определяют параметры элемен-
тарной ячейки.
В качествые примера ниже приведены экспериментально определенные па-
раметры гранатов (табл. 6)
Таблица 6
Параметры элементарной ячейки граната трех различных
рудопроявлений
Рудопроявление Количество определений а, А
Камчатка 10 12,064+0,0015
Полярный Урал 12 12,059+0,0015
Средний Урал 10 12,059+0,0015
Рентгенофазовый анализ
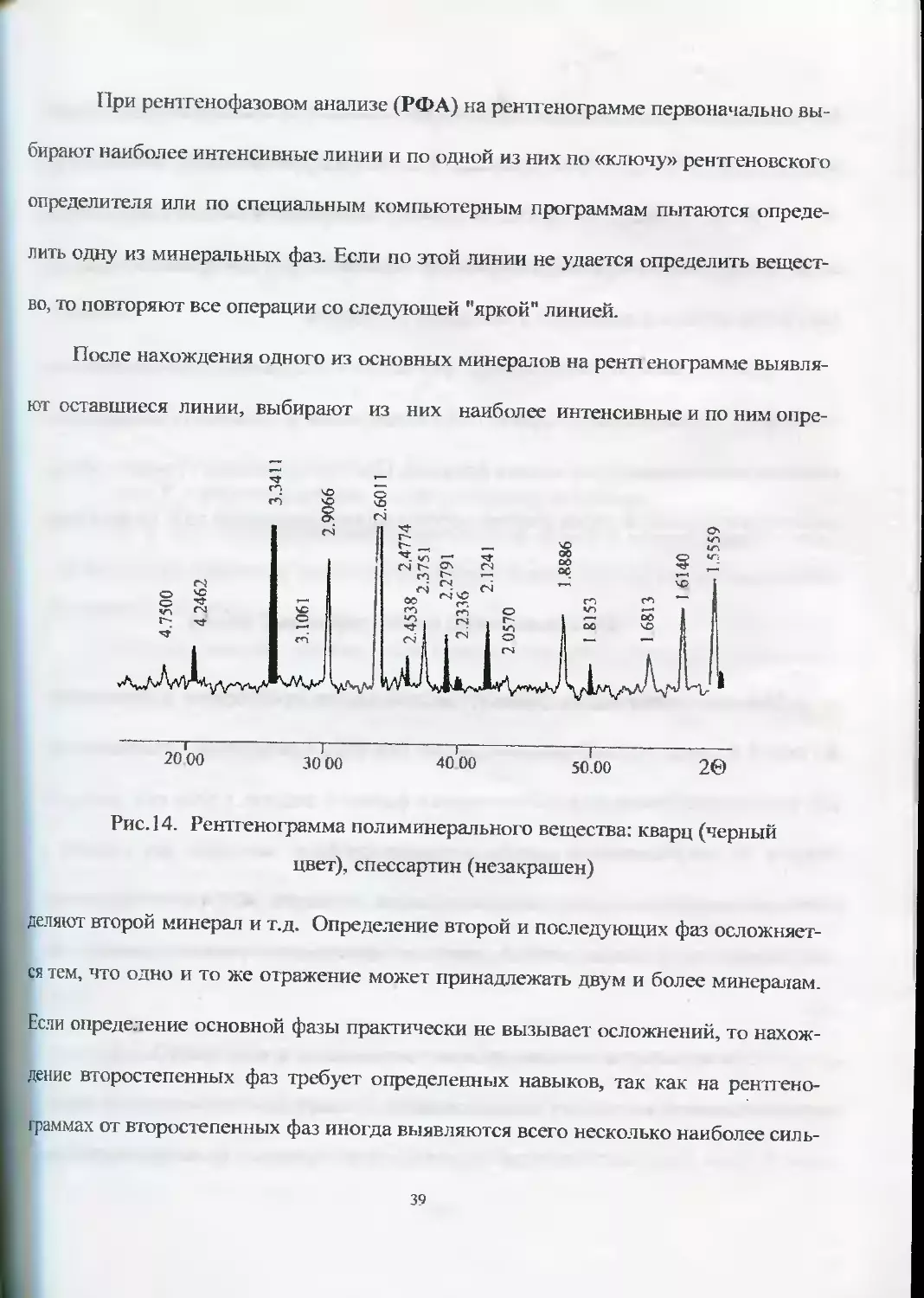
Рентгенография позволяет производить качественный (рис. 14) и количест-
венный фазовый анализ гетерогенных смесей. Каждая фаза (минерал) дает на рент-
генограмме свои дифракционные картины, что позволяет диагностировать минера-
лы, входящие в смесь, а интенсивность дифракционных отражений каждой фазы
находится в прямо пропорциональной зависимости от ее содержания в смеси.
38
При рентгенофазовом анализе (РФА) на рентгенограмме первоначально вы-
бирают наиболее интенсивные линии и по одной из них по «ключу» рентгеновского
определителя или по специальным компьютерным программам пытаются опреде-
лить одну из минеральных фаз. Если по этой линии не удается определить вещест-
во, то повторяют все операции со следующей "яркой" линией.
После нахождения одного из основных минералов на ренп енограмме выявля-
ют оставшиеся линии, выбирают из них наиболее интенсивные и по ним опре-
20 00 30 00 40 00 50.00 2©
Рис. 14. Рентгенограмма полиминерального вещества: кварц (черный
цвет), спессартин (незакрашен)
деляют
второй
минерал и т.д.
Определение второй и последующих фаз осложняет-
ся тем, что одно и то же отражение может принадлежать двум и более минералам.
Если определение основной фазы практически не вызывает осложнений, то нахож-
дение второстепенных фаз требует определенных навыков, так как на рентгено-
аммах от второстепенных фаз иногда выявляются всего несколько наиболее силь-
39
ных отражений. Порог обнаружения минерала в многокомпонентной смеси изменя-
ется в зависимости от состава, кристаллической структуры минерала, от общего фа-
зового состава исследуемой пробы, от условий эксперимента (длины волны исполь-
зуемого излучения, скорости сканирования образна и т.д.). Как правило, порох об-
наружения минерала находится в интервале 1-3 отн.%.
Для обнаружения минеральных фаз низкого содержания целесообразно ис-
пользовать предварительное фракционирование пробы и произвести дифрактомет-
рическое исследование полученных фракций. При благоприятном стечении обстоя-
тельств в гетерогенной смеси (горной породе) можно определить до 8-10 фаз (мине-
ралов).
Количественный рентгенофазовый анализ
Помимо качественного фазового анализа можно производить и рентгеногра-
фический количественный фазовый анализ (РКФА). Существует несколько мето-
дик рентгенографического количественного фазового анализа. Среди них: методика
анализа по искусственным смесям, методика добавок, методики внутреннего и
внешнего стандарта и др. Необходимо заметить, что перед РКФА для определения
всех минералов, входящих в смесь, проводят качественный рентгенофазовый ана-
лиз.
Одна из методик - метод добавок, заключается в том, что в исследуемую
многокомпонентную систему вводят известное количество определяемой мине-
ральной фазы. Далее получают две дифракционные картины - от исследуемой пре-
до
бы и от той же пробы после добавления в нее известного количества анализируемо-
го минерала. Интенсивность отражений анализируемой фазы на второй дифракто-
грамме при этом увеличивается.
Концентрация определяемого минерала (С, вес.%) в смеси определяется по
формуле
С, =Р1
к2-к}
где Р( - величина добавки в г на 1г исследуемой смеси;
К । и К2 - сотношение интенсивностей на первой и второй дифракгограм-
мах одного из рефлексов анализируемой фазы к рефлексу другой какой-то фазы,
находящейся в смеси.
Методика хорошо работает для определения концентрации минеральных
фаз, величина которых в смеси небольшая. Относительная погрешность анализа не
более Зотн.%. Оптимальная величина добавок - 104-20%.
Изучение изоморфизма минералов
Для минералов природных и синтетических характерно содержание в них, в
тех или иных количествах, элементов-примесей. Эти примеси в минерале-хозяине
могут:
1. Образовывать в минерале-хозяине минеральные включения, и тогда мы
говорим о механической примеси (например: рутил в кварце, флюорит в топазе,
магнетит в гранатах, самородные Au и Pt в сульфидах и т.д.).
41
2. Либо замещать какой-либо элемент (или несколько элементов) с обра-
зованием кристаллов переменного состава (твердые растворы замещения), и тогда
мы говорим об изоморфной примеси или изоморфном замещении; при этом не ме-
няется атомно-кристаллическое строение и внешняя форма кристаллов, остается
неизменным и тип химической связи между атомами.
В первом случае механическая примесь в минерале-хозяине, при ее доста-
точности, дает на рентгенограмме вторую минеральную фазу, т.е. это задача рент-
генофазового анализа.
При наличии серии твердых растворов с закономерно и постепенно
изменяющимся элементным составом между крайними членами этого ряда проис-
ходит непрерывное изменение параметров элементарной ячейки (как метрических,
так и угловых) от одного крайнего члена до другого. Это так называемый непре-
рывный изоморфный ряд или полный изоморфизм.
Изучение изоморфизма минералов методом рентгенографии заключается в
обнаружении наличия твердых растворов между изоструктурными минеральными
видами посредством выявления характера зависимости параметров элементарной
ячейки от количества изоморфной примеси.
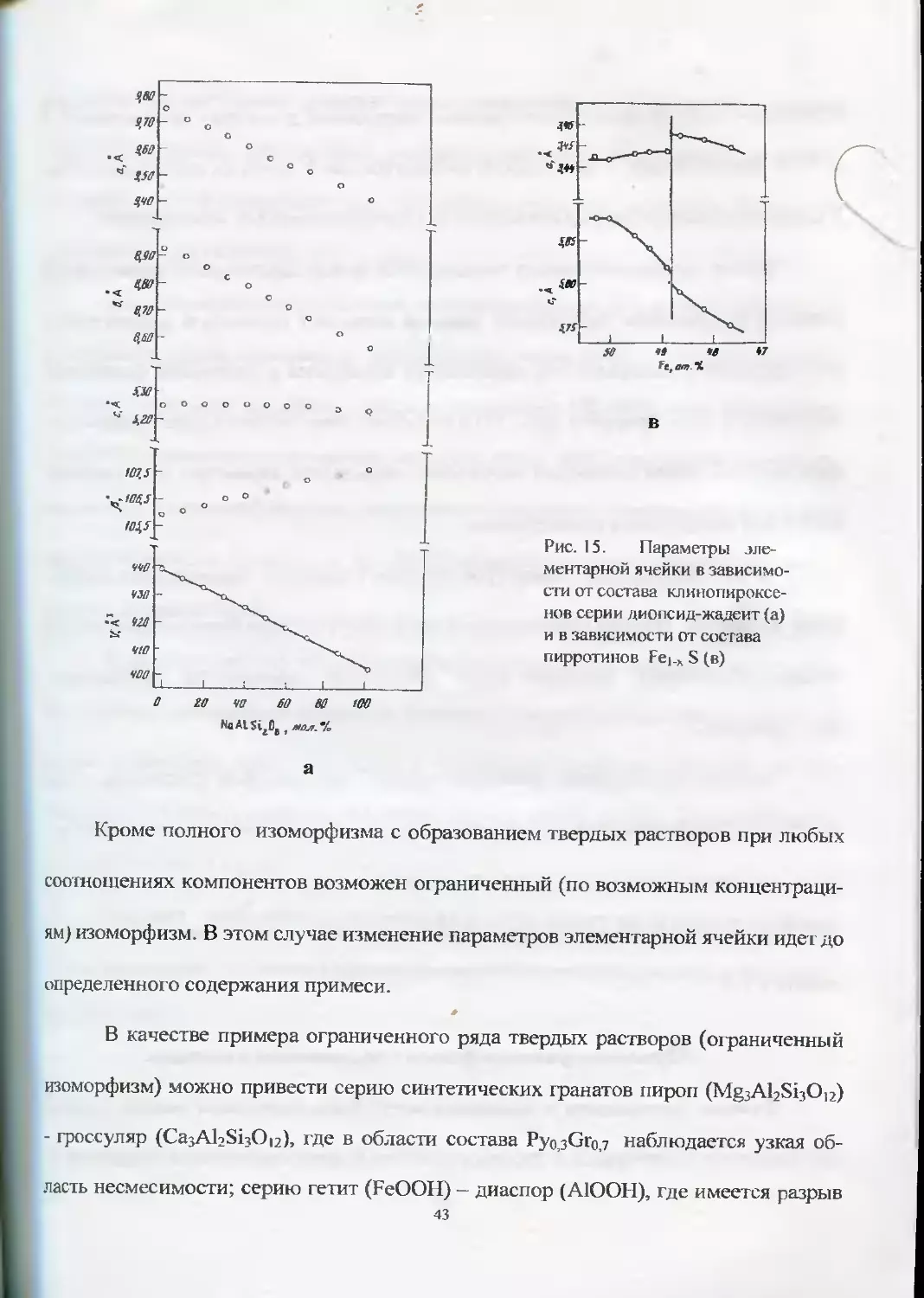
Примерами полного изоморфизма могут служить следующие серии изо-
структурных минеральных видов: уранинит (ПО2) - торианит (ThO2); андрадит
(Ca3Fe2Si3Oi2) — гроссуляр (CajAbSijOn); пироксены (рис. 15а) серии диопсид
(CaMgSi2O6) - жадеит (NaAlSi2O6) и т.д.
42
Рис. 15. Параметры эле-
ментарной ячейки в зависимо-
сти от состава клинопироксе-
нов серии диопсид-жадеит (а)
и в зависимости от состава
пирротинов Fei.x S (в)
Кроме полного изоморфизма с образованием твердых растворов при любых
соотношениях компонентов возможен ограниченный (по возможным концентраци-
ям) изоморфизм. В этом случае изменение параметров элементарной ячейки идет до
определенного содержания примеси.
В качестве примера ограниченного ряда твердых растворов (ограниченный
изоморфизм) можно привести серию синтетических гранатов пироп (MgjAbSbOp)
- гроссуляр (CasAKSijOu), где в области состава PyopGroj наблюдается узкая об-
ласть несмесимости; серию гетит (FeOOH) - диаспор (А1ООН), где имеется разрыв
43
в области 45-65% Fe; серию искусственных пирротинов, у которых при составе 48,8
ат.% Fe имеется разрыв в зависимости параметров «а» и «с» от их состава (рис.15в).
В последнем примере разрыв связывают со сверхструктурными изменениями.
Знание экспериментальных зависимостей между параметрами элементарной
ячейки и содержанием изоморфной примеси позволяет определять концентрации
этих примесей в минерале. Эти зависимости приводятся в различной справочной
литературе в виде графиков (рис. 15) или таблиц. Чем меньше концентрация при-
меси, тем с большей точностью необходимо определять параметры элементарной
ячейки для обнаружения изоморфизма.
Точное определение параметров позволяет выявлять типоморфные особен-
ности минералов. Именно микропримеси часто могут быть индикаторами процесса
минералообразования: скорости роста кристаллов, температуры кристаллиза-
ции, давления.
Изучение изоморфизма позволило создать синтетические кристаллы, когда
введение небольших количеств добавок меняет многие их свойства: изменение ок-
раски ювелирных камней (искусственные гранаты, корунды, фианиты): изменение
типа электрической проводимости в полупроводниках, изменение механических
свойств и т.д.
Изучение характера фазовых превращений минералов
Фазовые превращения в минералах могут быть следствием выноса отдель-
ных элементов из минералов в процессе различных метасоматических процессов, в
44
результате их окисления, действия воды, температуры и т.д. В качестве примера
применения метода для изучения фазовых превращений можно привести рентге-
нографические исследования при нагревании уранового минерала болтвудита
((К, Na)UO2 [SiO3OH]' Н2О ).
Исследования осуществлялись по порошковой пробе с использованием вы-
сокотемпературной приставки к дифрактометру типа ДРОН. Было установлено,
что: при нагревании минерала в пределах температур 1 ЗО-ЗОО°С идет потеря воды;
при 35О°С начинается аморфизация вещества, и при 540°С образец становится пол-
ностью рентгеноаморфным; при дальнейшем повышении температуры начинается
раскристаллизация вещества, и при 900°С образуются новые минеральные фазы -
уранаты натрия и калия.
В качестве другого примера можно привести хорошо известные в минерало-
гии фазовые превращения минералов, имеющих одинаковый химический состав, но
разное количество воды, входящей в кристаллохимическую формулу эпсомит
(Mg [SO4]. 7Н2О) - гексагидрит (Mg [SO4] .6Н2О); гипс (Са [SO4] ' 2Н2О) - бассанит
(Са [SO4]' 0,5Н2О) и т.д.)
Эпсомит через небольшое время на воздухе теряет часть воды и превраща-
ется в гексагидрит, а бассанит образуется при обезвоживании гипса при температу-
рах 120—130°С.
45
Аналогичный результат получается при обезвоживании сульфата никеля, ко-
гда он из семиводного превращается в шестиводный (мореназит NiSO4'7H2O -> рет-
герсит МБОд'бНоО).
Другим примером фазовых превращений могут служить явления окисления
магнетита (Fe3O4) до гематита (Fe2O,) и т.д.
Все эти исследования сводятся к простой диагностике минерала или фазово-
му анализу, если в пробе присутствует более одного минерала.
Определение степени совершенства кристаллической структуры вещества
Структура минерала, как кристаллического вещества, часто бывает несовер-
шенна. Причинами этого мсгут служить не только изоморфные замещения атомов,
но и нарушения в периодичности, вызванные смещением атомов; смещением атом-
ных плоскостей или их «искривлением». Все эти нарушения являются результатом
условий образования минерала или последствием его изменения в процессе гидра-
тации или механического воздействия. Так или иначе, изменения степени кристал-
личности может служить типоморфным признаком для какого-либо минерального
вида, образовавшегося при конкретных физико-химических условиях.
Следствием несовершенной кристаллической структуры является изменение
межплоскостного расстояния каждого из отражений в некоторых пределах, что вы-
ражается в расширении дифракционных пиков. Однако далеко не всегда удается
связат ь расширение отражений с каким-либо дефектом в структуре, поскольку при-
46
чиной расширения отражений может быть и высокая степень дисперсности минера-
ла.
В различной литературе приводятся описания методик определения степени
кристаллического совершенства для различных минералов. В основном они приме-
няются для глинистых минералов, имеющих слоистое или смешанно-слойное
строение. Но существуют методики и для других минералов.
Методика определения степени кристалличности кварца (СКС) была опи-
сана в журнале Геология и разведка, № 6 за 1980г. За показатель СКС может быть
выбрано отношение ширины анализируемой линии к ширине аналогичной линии
эталона. В качестве эталона предлагается жильный кварц Кожимского месторожде-
ния. Коэффициент кристалличности, рассчитанный по этой методике, оценивается в
процентах. Иногда при расчете полученные результаты могут иметь коэффициент
более 100%. Это объясняется тем, что не очень удачно выбран эталон.
Аналогичные методики предлагаются для определения степени упорядо-
ченности каолинита, позволяющие чувствовать смещение слоев относительно друг
друга вдоль осей “Ь“ и “а“ и к разворотам отдельных слоев (~ 120°), а также и к дру-
гим нарушениям. Согласно методу Хинкли (одного из методов расчета), показате-
лем совершенства структуры является отношение высот пиков (11 0) и (11Т), изме-
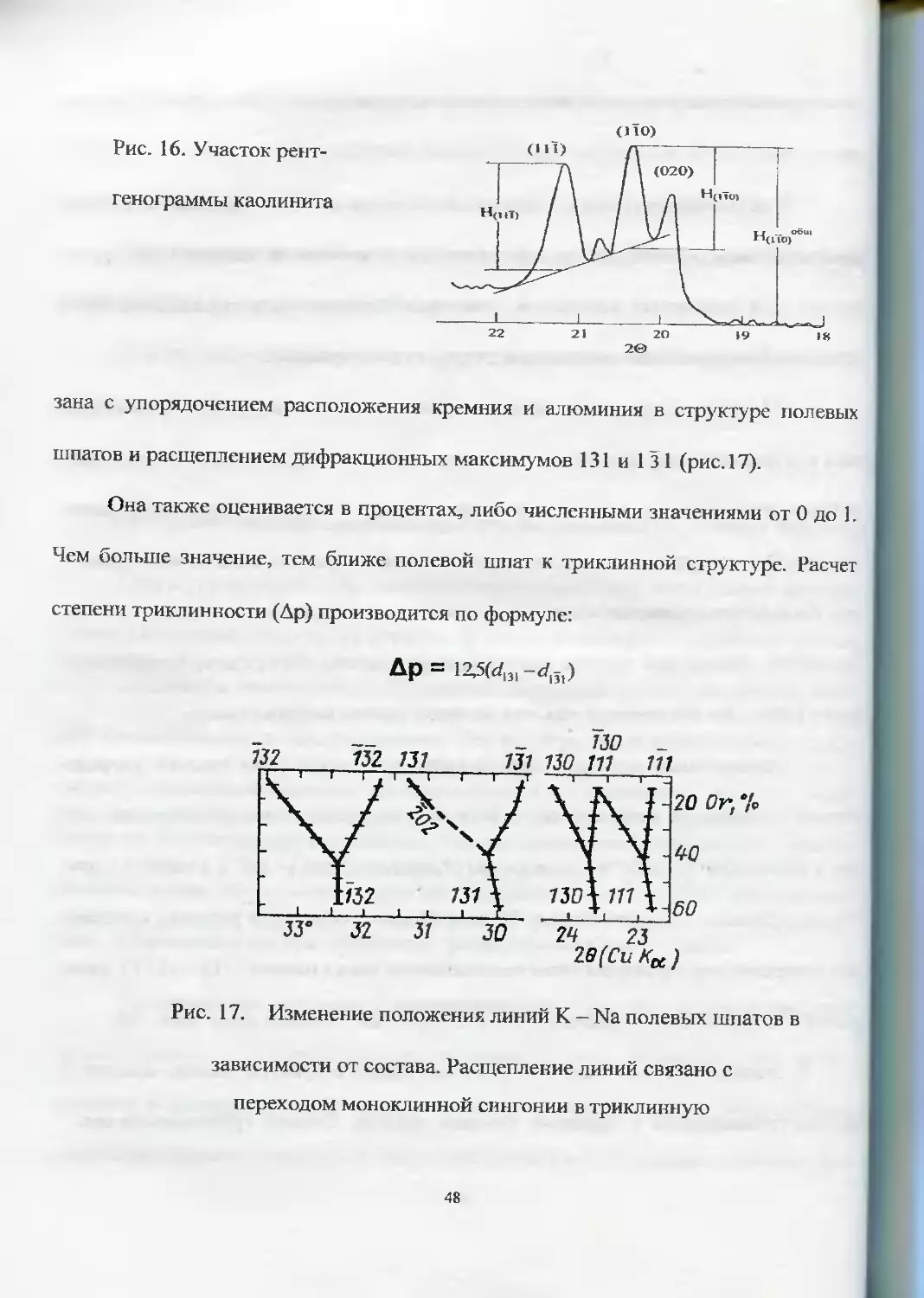
ренных от их основания, к высоте (11 0), измеренной от линии фона (рис. 16).
К степени упорядоченности кристаллической структуры можно отнести и
степень триклинности в калиевых полевых шпатах. Степень триклинности свя-
47
Рис. 16. Участок рент-
генограммы каолинита
зана с упорядочением расположения кремния и алюминия в структуре полевых
шпатов и расщеплением дифракционных максимумов 131 и 131 (рис. 17).
Она также оценивается в процентах, либо численными значениями от 0 до 1.
Чем больше значение, тем ближе полевой шпат к триклинной структуре. Расчет
степени триклинности (Др) производится по формуле:
Др = 12,5Ц5,-</151)
Рис. 17. Изменение положения линий К — Na полевых шпатов в
зависимости от состава. Расщепление линий связано с
переходом моноклинной сингонии в триклинную
48
Проводя исследования степени кристалличности минералов по разрезам
одного месторождения, можно сделать выводы об условиях их образования или
дальнейших генетических изменениях.
ЛИТЕРАТУРА
1 Васильев Е.К., Васильева Н.П. Рентгенографический определитель карбонатов.
Изд-во "Наука", Новосибирск, 1960.
2 Васильев Е.К., Васильева Н.П. Рентгенографический определитель оловосо-
держащих минералов. Изд-во "Наука", Новосибирск, 1977.
3 Васильев Е.К., Кашаева Г.М. и др. Рентгенометрический определить минералов
( класс фосфатов). Изд-во "Недра", М,. 1974.
4 Завьялов Е.Н. Рентгенографический определитель минералов. Ред.-изд. отдел
MITA, М., 1996.
5 Кондратьева В.В. Рентгенометрический определитель боратов. Изд-во "Недра",
Л., 1969.
6 Косолапов Г.Ф. Рентгенография. Изд-во "Высшая школа", М., 1962
7 Михеев В.И. Рентгенометрический определитель минералов. Изд-во "Госгео-
лтехиздат", М., 1967.
8 Пушаровский Д.Ю. Рентгенография минералов. Изд-во ЗАО "Геоинформмарк",
М., 2000
9 Рентгеновские методы изучения и структура глинистых минералов. Под ред.
Г.Брауна. Изд-во "Мир", М., 1965.
10 Рентгенография основных типов породообразующих минералов. Под ред.
В.А.Франк-Каменецкого. Изд-во "Недра", Л., 1983.
11 Руководство по рентгеновскому исследованию минералов. Под.ред. В.А.
Франк-Каменецкого. Изд-во "Недра", Л., 1975.
12 Сидоренко Г.А. Рентгенографический определитель минералов урана. Изд-во
"Энергоиздат", М., 1981.
13 Чичагов А.В, Сипавина Л.В. Рентгенометрические параметры твердых раство-
ров. Изд-во "Наука", М., 1982.
14 JCPDS- International Centre for Diffraction Data. All rights reserved PCPDFWIN.
1997.
49
МЕТОДЫ ЭЛЕМЕНТНОГО АНАЛИЗА
При определении элементного состава образца перед аналитиком стоит задача
выбора наиболее подходящего метода, что требует от него ответа на следующие во-
просы:
1. Каковы ожидаемые концентрации определяемого элемента в образце?
2. Что представляет собой матрица вещества, в котором определяется тот
или иной элемент?
3. Какова навеска исследуемого образца?
4. Какие требования к воспроизводимости результатов анализа?
5. Предполагаемое’количество проб, которое необходимо проанализировать.
б. Рассмогреть экономические вопросы.
Для ответа на эти вопросы требуется рассмотреть метрологические параметры
методов элементного анализа.
МЕТРОЛОГИЧЕСКИЕ ПАРАМЕТРЫ МЕТОДОВ
ЭЛЕМЕНТНОГО АНАЛИЗА
Все методы элементного анализа делятся на валовые и локальные. В валовых
- результаты анализа относятся ко всему объему анализируемого вещества, а в ло-
кальных - к точке исследования.
Кроме такого деления методы делятся на разрушающие и неразрушающие,
когда вещество в процессе исследования не исчезает и может использоваться для
повторных анализов или для других исследований.
50
Основными метрологическими параметрами методов элементного анализа яв-
ляются
следующие.
1. ПОРОГ ЧУВСТВИТЕЛЬНОСТИ МЕТОДА (II) - это минимальное ко-
личество
определяемого элемента, которое можно проанализировать с погрешно-
стью
не хуже
33%
отн.
Порог чувствительности метода измеряется в г\т;
г\г;
вес%.
г\т= 10’6г\г= 10’4вес. %.
2. ВОСПРОИЗВОДИМОСТЬ МЕТОДА (а)
Характеризуется рассеянием многочисленных результатов анализа относи-
тельно их
среднеарифметического значения
(х)и
оценивается среднеквадратичным
отклонением.
Различают внутрилабораторную воспроизводимость (внутренний контроль) и
межлабораторную (внешний контроль). Единицы измерения те же самые, что и по-
рог чувствительности.
По воспроизводимости методы элементного анализа делятся на:
количественные - когда о < 33% отн;
полуколичественные — когда о > 33% отн;
качественные — когда метод позволяет только ответить на вопрос присутствия
или
отсутствия
анализируемого элемента.
51
Более подробно о достигнутом уровне воспроизводимости аналитических оп-
ределений различных компонентов в минеральном сырье в зависимости от изме-
ряемых содержаний, классификация лабораторных методов анализа в зависимости
от их воспроизводимости можно найти в / 9 /.
3. ПРАВИЛЬНОСТЬ МЕТОДА
Характеризует отклонение среднеарифметического значения большого коли-
чества измерений от « истинного » содержания компонента в исследуемой пробе.
Это отклонение может быть обусловлено изменением по тем или иным причинам
содержания определяемого элемента в эталоне, применяемого при анализе.
4. ЛОКАЛЬНОСТЬ МЕТОДА
Помимо трех основных метрологических параметров для локальных мето-
дов существует еще один - локальность. Это минимальный размер выделения, ко-
торый можно проанализировать с погрешностью не хуже 33% отн.
Вспомогательными параметрами методов элементного анализа являются:
стоимость анализа, необходимая навеска, диапазон определяемых элементов, диа-
пазон определяемых концентраций, производительность.
Методы определения элементного состава проб подразделяются на следуею-
щие виды анализа: химический, рентгеноспектральный, спектрально-оптический,
ядерно-физический, масс-спектрометрический.
52
РЕНТГЕНОСПЕКТРАЛЬНЫЕ МЕТОДЫ
Методы рентгеноспектрального анализа основаны на изучении характери-
стических рентгеновских спектров, возникающих в результате облучения иссле-
дуемого вещества так называемым первичным излучением. В качестве первично! о
излучения применяются рентгеновское излучение, электронные пучки либо жест-
кое у-излучение от ампульных ядерных источников. Длина волны характеристиче-
ской линии (или энергия) однозначно характеризует элемент ее испускающий, а ин-
тенсивность этой линии пропорциональна концентрации этого элемента.
В зависимости от первичного возбуждающего излучения все рентгеноспек-
тральные методы разделяются на три группы:
Рентгенофлуоресцентный анализ — рентгеновское излучение;
Микрорентгеноспектральный анализ (микрозондовый) - электроны;
Рентгенорадиометрический - жесткое ядерное у -излучение.
РЕНТГЕНОСПЕКТРАЛЬНЫЙ ФЛУОРЕСЦЕНТНЫЙ АНАЛИЗ
Выделение нужной характеристической линии осуществляется либо дифрак-
ционным методом ( кристалл-дифракционный способ; используются кристалл-
анализаторы) с последующей регистрацией счетчиками ядерного излучения, либо за
счет регистрации такими счетчиками у-кваитов характеристического рентгеновско-
го излучения с последующим разделением импульсов по энергиям (энергодиспер-
сионный способ; используются амплитудные анализаторы). В первом случае ис-
53
пользуется дисперсия длин волн, а во втором - дисперсия энергии характеристиче-
ских у-квантов. На рис. 18 приведена блок-схема рентгеноспектрального кристалл-
дифракционного флуоресцентного прибора.
Рис. 18. 1 -диафрагма: 2-рентгеновская трубка: 3-детектор
рентгеновского излучения: 4,6-коллиматры: 5-кристалл-анализатор:
7- образец
В качестве кристалл-анализаторов используются кварц (отражающие плоско-
сти ЮТО или 10 1 1), LiF (220 или 200), слюда (002) или искусственные кристаллы.
Если в рентгенофлуоресцентной спектральной дифракционной аппаратуре ре-
гистрируются 1-2 линии спектра, прибор называют рентгеновским спектромет-
ром. Если одновременно большое количество линий (до 24-х) — квантометром. В
таком приборе в каждом спектрометрическом канале имеется свой счетчик рентге-
новских у-квантов, т.е. анализ идет одновременно на все элементы (количество оп-
ределяемых элементов соответствует количеству спектрометрических каналов).
В качестве примеров квантометров можно указать такие отечественные при-
боры как КРФ-18 и КРФ-24, в которых 12 спектрометрических каналов или СРМ-1
с 16 каналами. Все эти приборы выпускаются ЕШО «Буревестник».
54
В некоторых рент генофлуоресцентных приборах с кристалл-дифракционным
способом выделения характеристических линий применяются подвижные узкие
приемные щели, настроенные на определенные элементы. В качетве примера можно
указать отечественные флуоресцентные рентгеновские анализаторы ФРА-4 и
АРФ-6.
Другой вариант флуоресцентных рентгеновских приборов — это когда счет-
чик характеристического рентгеновского излучения подвижен и настраивается на
нужную линию, т.е. определяемый элемент. Это такие приборы как VRA-20 и
VRA-30 (производство - Германия). В этих приборах за одно измерение анализиру-
ется один элемент.
Флуоресцентный анализ осуществляется по порошковым пробам (валовый
анализ) и является неразрушающим, т.е. вещество не уничтожается и может исполь-
зоваться для повторного или других видов анализа.
Порог чувствительности данного элементного анализа от 10’2 до 10"4 вес.% в
зависимости от определяемого элемента.
В геммологии для элементного анализа могут использоваться приборы -
«МЕКА 10-44», который позволяет установить элементный состав закрепленных в
изделии драгаценных камней или рентгенофлуоресцентный прибор
«Портаспек-2501», определяющий элементы в диапазоне от Ti до Ag и использую-
щийся для диагностики имитаций бриллиантов.
В последнее время рентгенофлуоресцентные приборы снабжаются ЭВМ, что
позволяет автоматизировать процесс анализа и резко повысить их производитель-
55
ность. Так, производительность квантометров может достигать до 300 тыс. проб в
год, что позволяет сократить срок окупаемости приборов до 2-3 лет
В энергодисперсионных спектрометрах регистрируется одновременно весь
спектр характеристических у- квантов от всех элементов, входящих в пробу, причем
могут регистрироваться не только линии К-серии но и L-серии от одних и тех же
элементов.
Основное преимущество энергодисперсионного способа выделения нужной
спектральной линии - это гораздо больший сбор возникающего характеристичекого
излучения за счет расположения детектора вплотную к образцу (в кристалл-
дифракционном способе между детектором и образцом расположен кристалл-
йнализатор). Если в кристалл-дифракционных спектрометрах сбор информации
идет с угла 0,015-0,04 стерадиана (т.е. используется лишь несколько тысячных из
общего потока в 4тг излучения), то во втором случае почти с угла 2тг стерадиан. Та-
ким образом, за счет чисто геометрических особенностей сбор информации идет
в гораздо большем телесном угле и число у-квантов попадающих на детектор
увеличивается на 1-2 порядка. Это обстоятельство позволяет в рентгенофлюорес-
центных приборах с энергодисперсионным спектрометром применять миниатюр-
ные рентгеновские трубки, где ток анода не превосходит нескольких мА. Если
сравнивать разрешение кристалл-дифракционных (КДС) и энергодисперсионных
(ЭДС) спектрометров, то в КДС оно в 5-10 раз больше.
56
Среди энергодисперсионных флуоресцентных рентгеновских приборов
можно привести аппаратуру таких фирм как Philips (Голландия), Siemens (ФРГ) или
Японской фирмы JEOL (элементные анализаторы JXS-3211, JXS-3201, JXS-3220).
Эти приборы снабжены Si(Li) полупроводниковыми детекторами, процесс анализа
автоматизирован, определяются элементы от Na до U, диапазон определяемых кон-
центраций от 0,01% до 100%.
Среди отечественных энергодисперсионных флуоресцентных приборов необ-
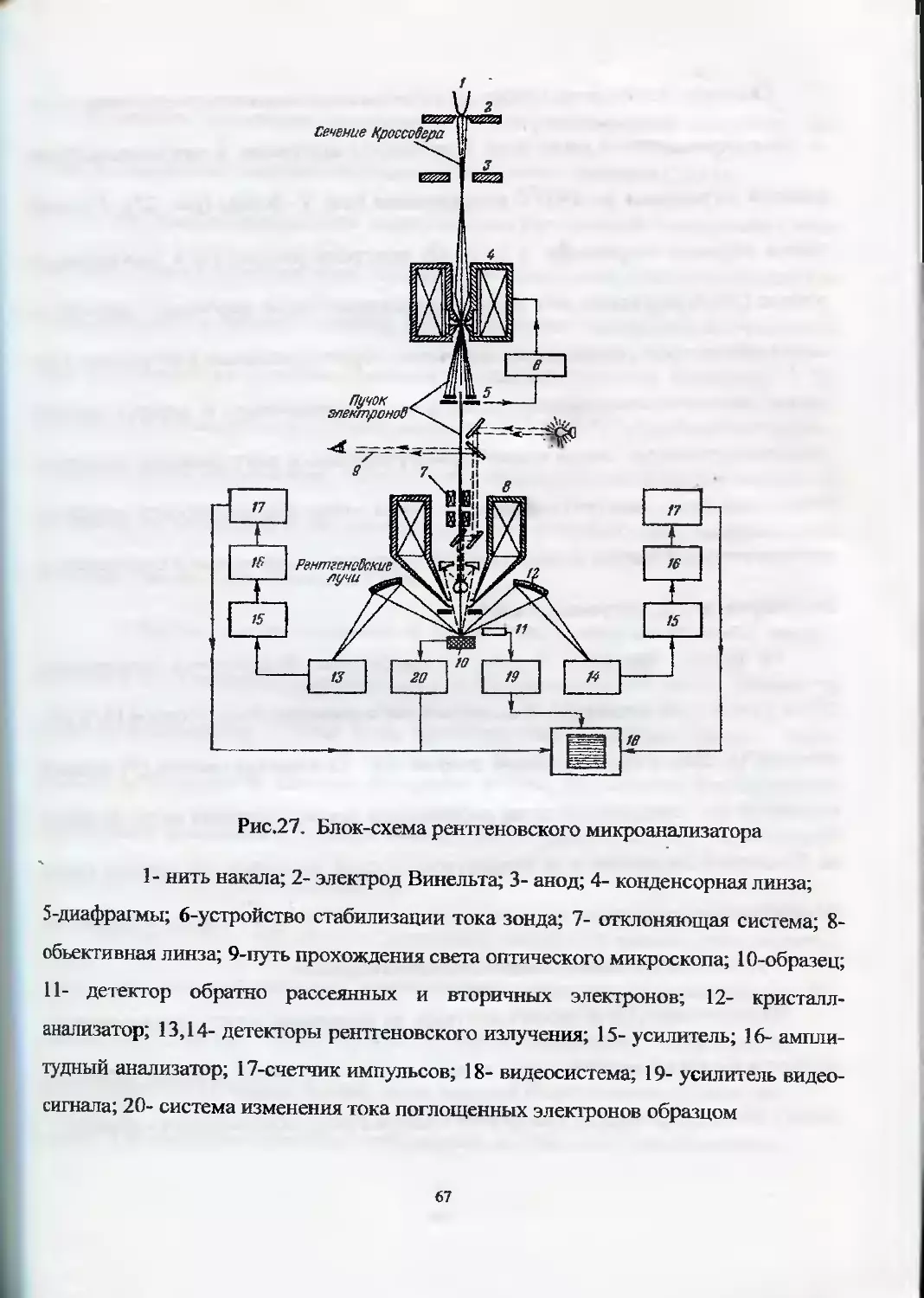
ходимо отметить рентгеновский спектрометр «РеСПЕКТ» (рис. 19). В приборе преду-
смотрена полная автоматизация процесса анализа, одновременно определяется до 50
элементов (от Na до U), диапазон определяемых концентраций от 10’4 % до 100% в
зависимости от анализируемого элемента, энергетическое разрешение ~ 150 эВ, в
приборе имеются две миниатюрные рентгеновские трубки (с Ti и Ag анодами). Бла-
годаря большой светосиле прибора удается анализировать образцы очень мелких
размеров, до 0,1 мм.
Прибор выпускается с вакуумированием камеры для образцов и без. Во вто-
ром случае на рентгеновской трубке с Ag-анодом определяются элементы от С1 до
U. Имеется съемная карусель на 16 кювет. Система водяного охлаждения рентге-
новских трубок - замкнутая.
Прибор позволяет анализировать порошковые пробы, ограненные кристал-
лы различной формы и размеров без разрушения образцов, жидкие растворы по
«методу высушенной капли». При навеске порошковой пробы 0,9 г, растертой до
57
Рис. 19. Рентгенофлюоресцентный прибор’ «РеСПЕКГ» с энерго-
дисперсионным спектрометром
пудры, возможен количественный анализ. Анализ жидких растворов осуществляет-
ся с использованием внутреннего стандарта.
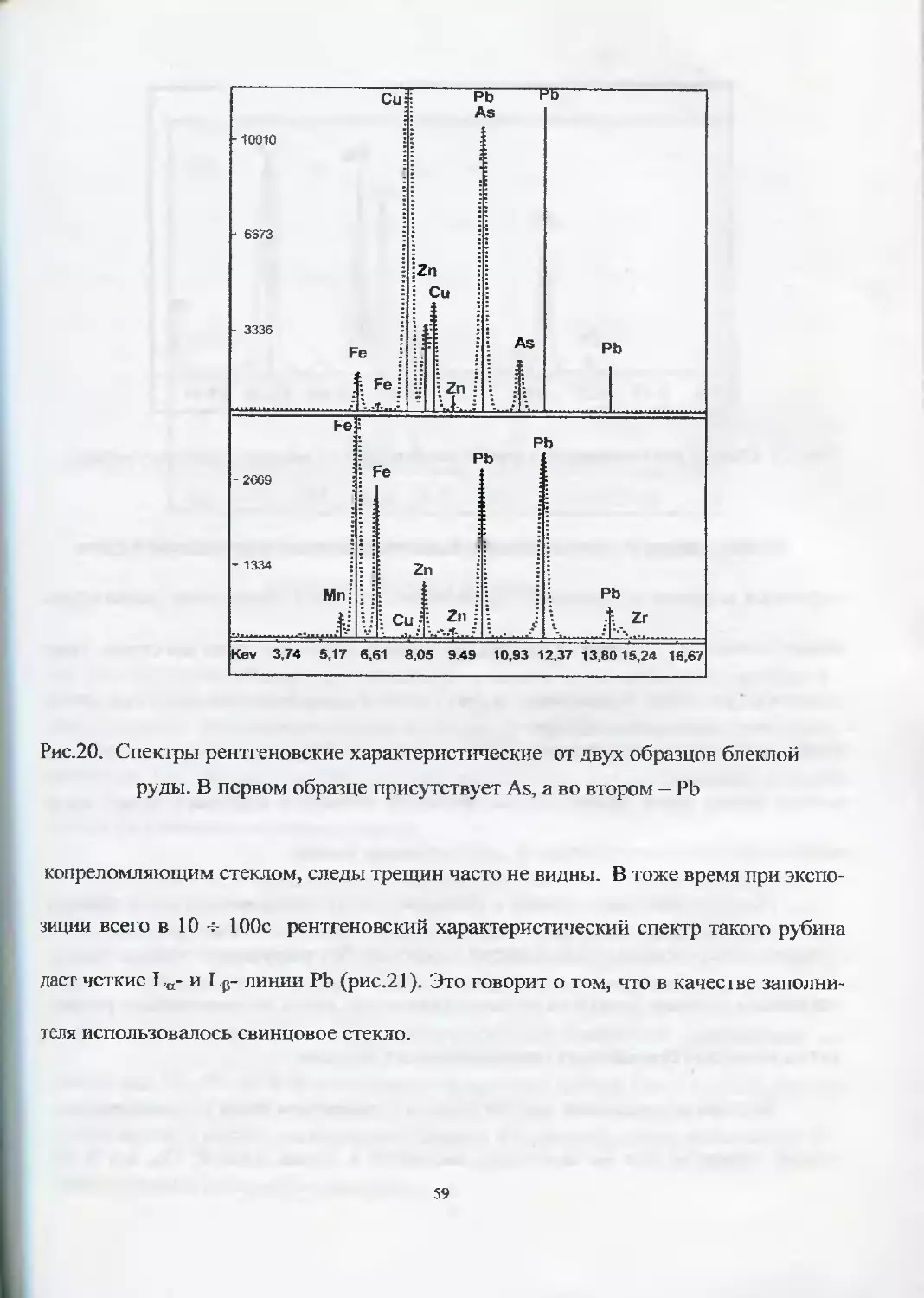
Иногда характеристические линии от двух элементов в приборах с ЭДС
могут накладываться друг на друга. Так, на рис.20 представлены результаты иссле-
дования двух образцов блеклой руды. Видно, что характеристическая линия Кд от
As и La от РЬ совпадают. Для однозначного ответа о присутствии одного или
другого элемента необходимо использовать p-линии от этих элементов. Так, на
первой спектрограмме видно, что характеристическая линия Lp от РЬ отсутствует,
что говорит о наличии в образце только As. На второй спектрограмме видно при-
сутствие только РЬ.
Широкое применение нашел прибор при решении различных геммологиче-
ских задач.
Так, например, в рубинах, облагороженных путем заполнения трещин высо-
58
Рис.20. Спектры рентгеновские характеристические от двух образцов блеклой
руды. В первом образце присутствует As, а во втором — РЬ
копреломлякмцим стеклом, следы трещин часто не видны. В тоже время при экспо-
зиции всего в 10 -5- 100с рентгеновский характеристический спектр такого рубина
дает четкие Ln- и Lp- линии РЬ (рис.21). Это говорит о том, что в качестве заполни-
теля использовалось свинцовое стекло.
59
Рис. 21. Спектр рентгеновский характеристический от облагороженного рубина
Можно привести другой пример. В последнее время для изменения цвета
природных корундов и топазов на 1рани вставок наносят тонкие слои (пленки) раз-
личных металлов. Толщина покрытия составляет несколько сотен ангстрем. Такая
технология усиливает блеск камня за счет сильной интерференции света при прохо-
ждении слоев с различной оптической плотностью. Обработанные таким образом
вставки имеют очень разнообразные цветовые оттенки и благодаря этому могут
имитировать многие популярные и дорогостоящие камни.
Исследования таких камней с помощью pern генофлюоресцентного прибора
с энергодисперсионным спектрометром позволяет без разрушения образца быстро
установить наличие пленки на вставках даже тогда, когда это невозможно устано-
вить с помощью стандартных геммологических методов.
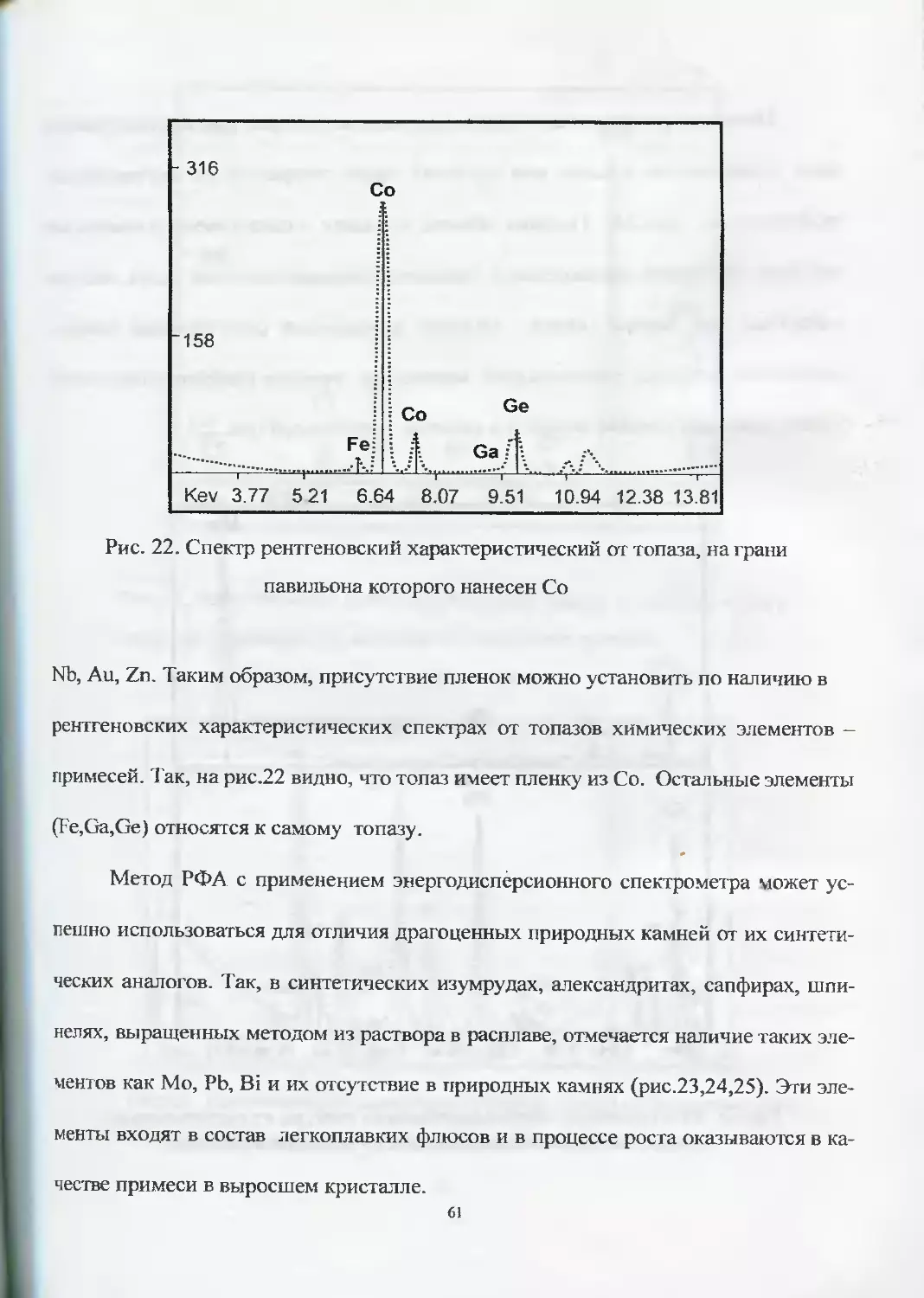
Так, при исследовании партии топазов с покрытием были установлены сле-
дующие элементы или их сочетания, входящие в состав пленок: Со, Cr, Ti, Fe,
60
Рис. 22. Спектр рентгеновский характеристический от топаза, на грани
павильона которого нанесен Со
Nb, Au, Zn. Таким образом, присутствие пленок можно установить по наличию в
рентгеновских характеристических спектрах от топазов химических элементов -
примесей. Так, на рис.22 видно, что топаз имеет пленку из Со. Остальные элементы
(Fe,Ga,Ge) относятся к самому топазу.
Метод РФА с применением энергодисперсионного спектрометра может ус-
пешно использоваться для отличия драгоценных природных камней от их синтети-
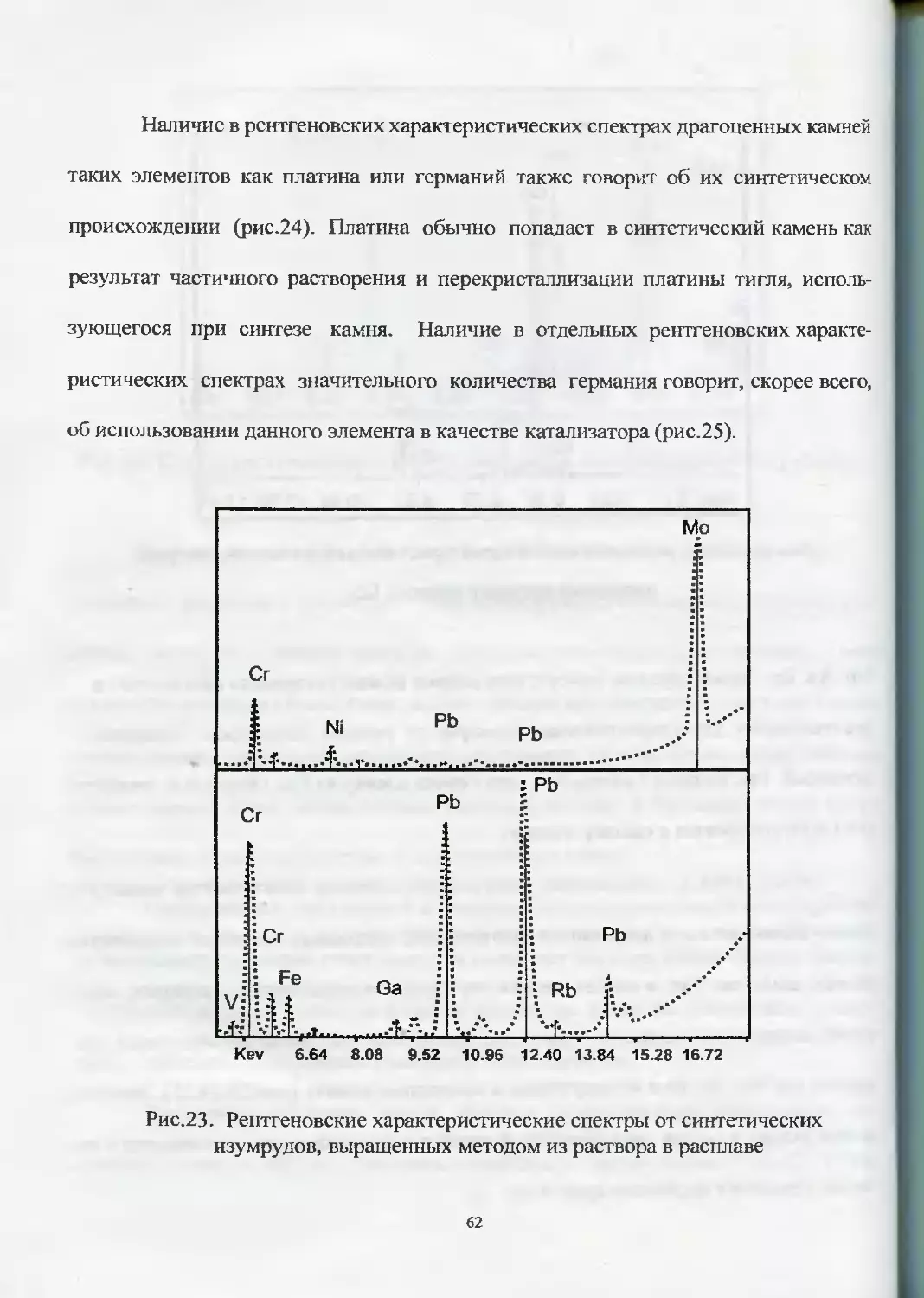
ческих аналогов. Так, в синтетических изумрудах, александритах, сапфирах, шпи-
нелях, выращенных методом из раствора в расплаве, отмечается наличие таких эле-
ментов как Mo, Pb, Bi и их отсутствие в природных камнях (рис.23,24,25). Эти эле-
менты входят в состав легкоплавких флюсов и в процессе роста оказываются в ка-
честве примеси в выросшем кристалле.
61
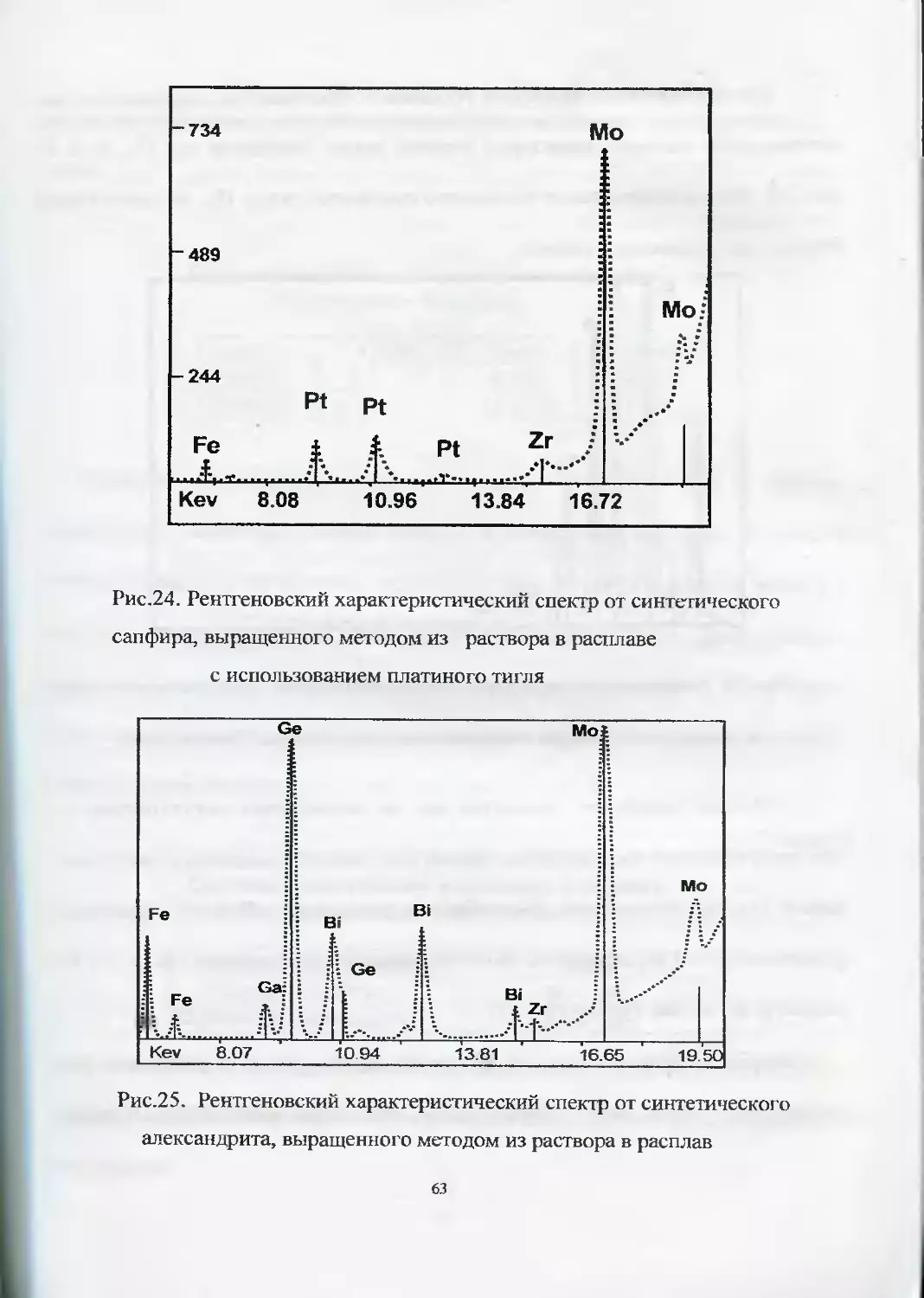
Наличие в рентгеновских характеристических спектрах драгоценных камней
таких элементов как платина или германий также говорит об их синтетическом
происхождении (рис.24). Платина обычно попадает в синтетический камень как
результат частичного растворения и перекристаллизации платины тигля, исполь-
зующегося при синтезе камня. Наличие в отдельных рентгеновских характе-
ристических спектрах значительного количества германия говорит, скорее всего,
об использовании данного элемента в качестве катализатора (рис.25).
Рис.23. Рентгеновские характеристические спектры от синтетических
изумрудов, выращенных методом из раствора в расплаве
62
Рис.24. Рентгеновский характеристический спектр от синтетического
сапфира, выращенного методом из раствора в расплаве
с использованием платиного тигля
Рис.25. Рентгеновский характеристический спектр от синтетического
александрита, выращенного методом из раствора в расплав
63
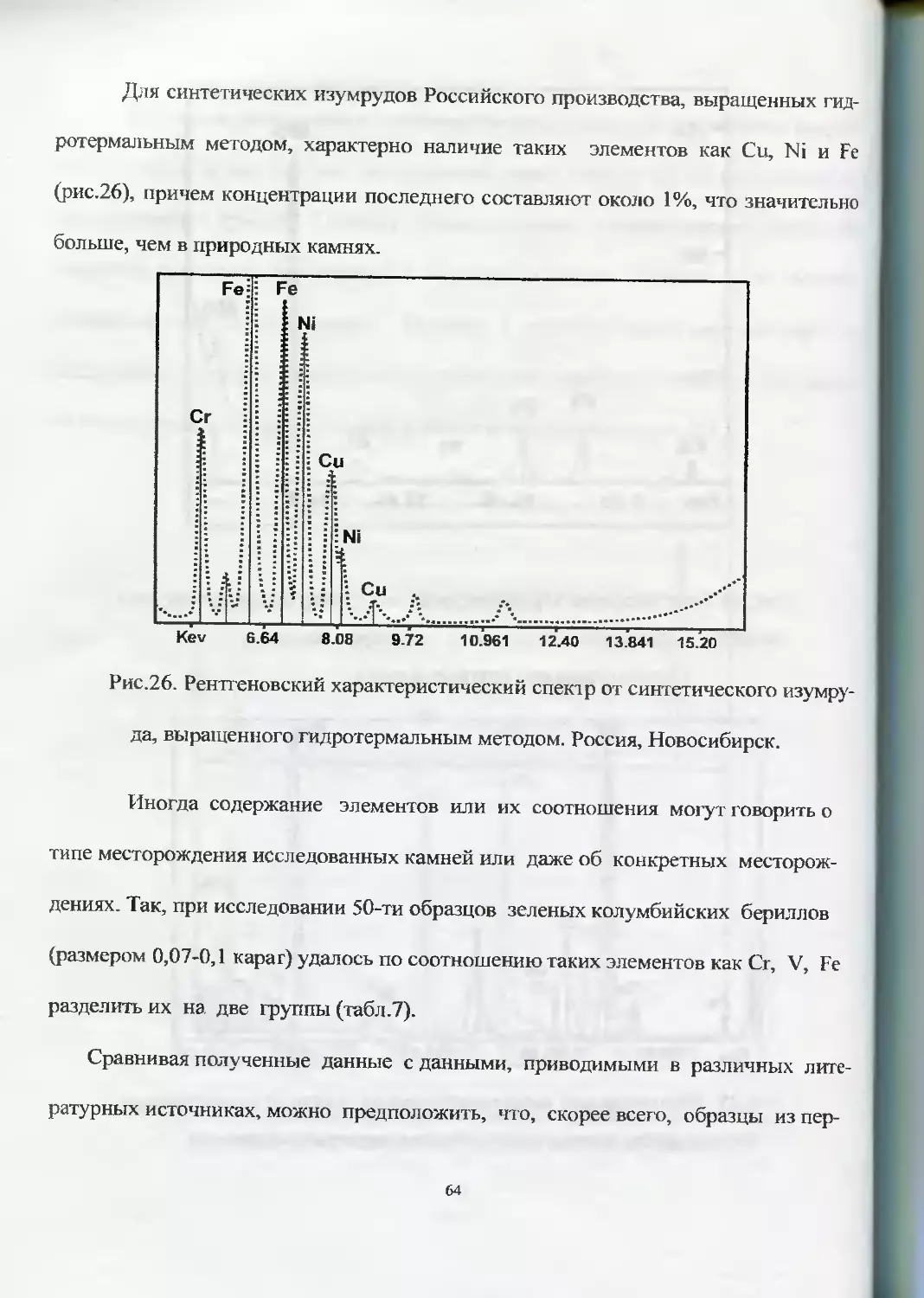
Для синтетических изумрудов Российского производства, выращенных гид-
ротермальным методом, характерно наличие таких элементов как Си, Ni и Fe
(рис.26), причем концентрации последнего составляют около 1%, что значительно
больше, чем в природных камнях.
С Fejj F Г : : : е Ni Н Си = Ё i :Ni < И i Си v V VK.A Л •-
Kev 6.64 8.08 9.72 10.961 12.40 13.841 15.20
Рис.26. Рентгеновский характеристический спектр от синтетического изумру-
да, выращенного гидротермальным методом. Россия, Новосибирск.
Иногда содержание элементов или их соотношения могут говорить о
типе месторождения исследованных камней или даже об конкретных месторож-
дениях. Так, при исследовании 50-ти образцов зеленых колумбийских бериллов
(размером 0,07-0,1 караг) удалось по соотношению таких элементов как Cr, V, Fe
разделить их на две группы (табл.7).
Сравнивая полученные данные с данными, приводимыми в различных лите-
ратурных источниках, можно предположить, что, скорее всего, образцы из пер-
ед
вой руппы относятся к месторождению Коскуэс, а из второй — к месторождению
Музо.
Таблица 7
Соотношения некорых элементов в зеленых
колумбийских бериллах
Г руппа Cr/Fe Сг/V Fe/V
Группа 1 ' 0,1 - 0,2 4—27 44-65
Группа? 3-н5 0,6- 1,6 0,1- 0,31
Сравнение соотношений содержания определенных элементов в образцах
бывает также полезно при отличии морского жемчуга от пресноводного. Различный
солевой состав и их концентрация приводят к тому, что в пресноводном жемчуге в
качестве элементов-примесей присутствуют стронций и марганец. В жемчужинах,
выросших в морских условиях, марганца очень мало и в основном присутствует
только стронций. Поэтому отношения содержания стронция к марганцу в морском
и пресноводном жемчуге будут сильно отличаться (табл.8).
Таблица 8
Соотношения стронция и марганца в морских
и пресноводных жемчужинах
Объект исследования Sr/Mn
Морской жемчуг 50 - 140
Пресноводный жемчуг 0,2 - 1,6
Эти примеры наглядно говорят о преимуществе рентгенофлюоресцентного
прибора с энергодисперсионным спектрометром перед другими методами элемент-
ного анализа.
65
Основными преимуществами такого прибора являются: неразрушающий
метод элементного анализа; возможность исследования мелких образцов;
высокая скорость анализа; небольшой размер прибора; высокий порог чувст-
вительности (до 105%) на отдельные элемент ы.
МИКРОРЕНТГЕНОСПЕКТРАЛЬНЫЙ АНАЛИЗ
В качестве первичного излучения в микрорентгеноспектральном анализе ис-
пользуются электронные пучки от электронной пушки. Основное достоинство ме-
тода - возможность локального определения химического состава. Локальность
метода — 1-2 мк. Порог чувствительности метода составляет 10' - 10'2вес.%.
Регистрация возникающих характеристических спектров осуществляется с
помощью кристалл-дифракционных спектрометров, либо энергодисперсионных. В
современных электронно-зондовых приборах чаще всего применяются оба способа
регистрации возникающих рентгеновских характеристических спектров; количество
спектрометров достигает 5-6, причем один из них является энергодисперсионным.
66
Сечение Кроссовера
Рис.27 Блок-схема рентгеновского микроанализатора
1- нить накала; 2- электрод Винельта; 3- анод; 4- конденсорная линза;
5-диафрагмы; 6-устройство стабилизации тока зонда; 7- отклоняющая система; 8-
обьективная линза; 9-путь прохождения света оптического микроскопа; 10-образец;
11- детектор обратно рассеянных и вторичных электронов; 12- кристалл-
анализатор; 13,14- детекторы рентгеновского излучения; 15- усилитель; 16- ампли-
тудный анализатор; 17-счетчик импульсов; 18- видеосистема; 19- усилитель видео-
сигнала; 20- система изменения тока поглощенных электронов образцом
67
Основное достоинство метода достигается использованием электронного пуч-
ка сфокусированного в узкий зонд. Источником электронов в микроанализаторах
является нагреваемая до~2900пС вольфрамовая нить V- формы (рис. 27). Под дей-
ствием анодного напряжегйя в 10-50 кВ электроны ускоряются и бомбардируют
образец (10), в результате чего возникает несколько видов излучений: тормозное и
характеристическое рентгеновское излучение, обратно рассеяные и вторичные элек-
троны, катодорентгенолюминесценция и др. В микрозонде, в первую очередь,
представляет интерес возникающее характеристическое рентгеновское излучение.
Именно анализ его спектра (определение длины волны и интенсивности каждой ха-
рактеристической линии) позволяет индентифицировать элементы и определять их
концентрации в исследуемом объекте.
На рис.27 приведена блок-схема микрозонда. Фокусировка электронного
пучка в узкий зонд диаметра 1-2мк достигается с помощью конденсорной (4) и объ-
ективной (8) лннз, а также системой диафрагм(5). Оптическая система (9) позволя-
ет вывести под электронный пучок необходимое для исследования место на образ-
це. Последний закрепляется на предметном столике, имеющим две степени свобо-
ды. Увеличение оптической системы составляет 400-500 крат. Столик образцов по-
зволяет устанавливать одновременно несколько образцов.
Видеосистема (18) позволяет получить во вторичных электронах картину по-
верхности объекта и распределение элементов по поверхности образца. Это дости-
гается с помощью развертки электронного пучка в растр но поверхности образца.
68
В качестве детекторов характеристического рентгеновского излучения чаще
всего используются газопроточные пропорциональные счетчики (13,14).
В кристалл-дифракционных спектрометрах (брэгговский спектрометр), изме-
няя угол падения рентгеновского характеристического излучения к плоскости кри-
сталл-анализатора (вращая кристалл), можно настроить спектрометр на нужную ли-
нию интересующего элемента. Диапазон изменения этого угла составляет 12-75°,
что позволяет фиксировать диапазон длин волн 0,4- l,9d ("d" - межплоскостное рас-
стояние кристалл-анализатора). Поэтому для определения как можно большего чис-
ла элементов таблицы Д.И.Меделеева применяют несколько спектрометров с раз-
личными кристалл-анализаторами (табл. 9).
Образцы для исследования на микрозонде готовят в виде полированных
аншлифов или прозрачно полированных шлифов на эпоксидной основе. Мелкие об-
разцы запрессовывают в сплав Вуда, протокрил или эпоксидные смолы с после-
дующей их полировкой. Качество полировки должно обеспечивать безрельефность
исследуемой поверхности. Размеры исследуемых образцов зависят от конструкций
держателя образцов микроанализатора.
Полированные, прозрачно полированные шлифы или мелкие зерна, запрессо-
ванные в тот или иной материал, должны хорошо проводить электрический ток. В
противном случае при бомбардировке образцов электронами в точке исследо-
вания скапливается отрицательный заряд, который будет отклонять пучок элек-
тронов, что приведет к искажению результатов анализа. Для устранения этого
69
Таблица 9
Используемые кристалл-анализаторы в рентгеноспектральном анализе
Кристалл Отражаю- щая пл. d, А” Диапазон атомных номеров анализируемых элементов
К -серия L-серия
Кварц 1011 3,443 15(Р) — 46 (Pd) >40(Zr)
Кварц 1010 4,25 14(Si)- 41 (Nb) >37 (Rb)
LiF 220 1,424 23(V)- 68 (Er) >58 (Ce)
LiF 200 2,014 19(K)- 58 (Ce) >49 (In)
Слюда 002 9,92 9(F)- 27 (Co) >26 (Fe)
КАР (кислый фталат калия) ЮТО 13,316 8(0)- 23 (V) >23(V)
LS (стеарат свинца) - 50 5(B)- 12 (Mg) >20 (Ca)
эффекта на исследуемую поверхность наносят электропроводящий слой, в ка-
честве которого используются тонкие слои углерода (чаще всего), золота, се-
ребра или алюминия. Напыление осуществляется на ВУПах (вакуумный уни-
версальный пост).
В качестве примера в табл. 10 приведены результаты исследования на микро-
зонде золотин (размером ~ 0,1мм), запрессованных в протакрил. Видно, что образец
2 представляет собой техногенный материал (латунь), попавший в протолочку, ви-
димо, от буровой коронки. Исследовать такие мелкие выделения никаким другим
методом не представляется возможным.
70
Таблица 10
Результаты микрорентгеноспектрального анализа золота (вес.%)
Образец 1 Au Ад Си Zn Z
Зерно 1 центр 97,4 2,4 - 99,8
Зерно 1 край 99,7 0,1 - 99,8
Зерно 2 центр 96,0 3,6 - 99,6
Зерно 2 край 98,4 1,4 - 99,8
Зерно 3 центр 99,5 - - 99,5
Зерно 3 край 99,8 - - 99,8
Зерно 4 центр 97,3 1,8 - 99,1
Зерно 4 край 95,4 4,3 - 99,7
Зерно 5 центр 99,0 0,6 - 99,6
Зерно 5 край 95,2 4,1 - 99,3
Образец 2
Зерно 1 ~60 -40
Зерно 2 ~60 -40
Зерно 3 ~60 -40
В качестве другого примера можно привести результаты исследования оди-
ночного зерна минерала размером около 2 мм. В результате исследования (табл. 11)
оказалось, что данный минерал относится к гранату гроссуляр-уваровитового ряда.
Помимо элементного анализа в точке (1-2 мк) микрозондовый анализ позво-
ляет получить распределение элементов по площади образца (рис.28).
71
в
г
Рис. 28. А. Повышенные содержания Au и As приурочены к центральной части об-
разца. Б.Выделение серебросодержащего золота (1) в гелените (2) с продуктами
замещения (3) В.Выделенис самородного золота (Au-79,1 %; Ag-20,9%) в арсенопи-
рите (Ars) с кварцем. Г.Выделение селенсодержащего теллурида серебра (1) в сра-
стании с галенитом (2) в халькопирите (3)
72
Таблица 11
Химический состав зеленого граната
SiO2 TiO2 Ai2O3 Cr2O3 FeO MnO MgO CaO V2O5 Сумма
Край зерна 40,50 0,62 16,68 4,86 2,13 1,37 0,16 33,69 0,04 100,04
Центр зерна 39,73 0,67 16,21 7,11 2,42 1,36 0,16 32,41 0,04 100,11
. Таким образом, микрозонд является уникальным прибором для ло-
кального химического анализа. Локальность составляет 1-2 мк. Порог чув-
ствительности метода составляет ПГ'-Ю’2 вес.%, что вполне достаточно
для определения основных элементов, входящих в состав минералов. По-
мимо этого данный вид анализа позволяет получить пространственное
распределение элементов по площади образпа.
РЕНТГЕНОРАДИОМЕТРИЧЕСКИЙ АНАЛИЗ
В качестве первичного излучения используется ядерное у-
излучение маломощных ампульных радионуклидных источников. Определение эле-
ментного состава основано на регистрации и изучении характеристического рентге-
новского излучения атомов элементов, входящих в исследуемое вещество. Регист-
рация рентгеновского излучения осуществляется энергодисперсионным спектро-
метром. В качествые примера можно привести переносные анализаторы элементно-
го состава материалов ПРИМ-1 и ПРИМ-2. Эти анализаторы способны определять
73
до 50 элементов - от Al (Z=13) до U (Z=92). Для обработки спектров, их регистра-
ции, идентифицирования элементов применяют малогабаритные компьютеры клас-
са Notebook.
Прибор является портативным, размещается в металлизированном кейсе (для
защиты от излучения), масса приборов составляет 8,5 кг.
Приборы используются в качестве штатного средства таможенного контроля
для исследования состава экспортируемой продукции (металлы, руда, шихта и дру-
гие материалы). При таможенном контроле прибор используется для проверки пра-
вильности сведений по качественному и количественному составу декларируемых
материалов. Время анализа составляет от 10 до 300с. Диапазон измеряемых концен-
траций от 0,01 до 100%.
Другой пример рентгенорадиометрического лабораторного прибора - РАЛ-
М-102. Данный прибор предназначен для определения в порошковых пробах содер-
жания химических элементов с атомными номерами от Z=19(калий) до Z=83 (вис-
мут). В качестве источника первичного излучения применяется кадмий -119. Порог
чувствительности достигает 10’1 — 10'3вес.%.
ОПТИЧЕСКИЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
По современным представлениям о строении атома, он может находиться в
различных энергетических состояниях, но эти энергетические состояния строго
74
определенные (дискретные) для каждого атомам: Е Е2, Е3 и т.д. Промежуточные
значения атом принимать не может.
Чаше всего атом находится в основном (стабильном) состоянии, наиболее ус-
тойчивом и обладает при этом минимальной энергией. Все другие состояния явля-
ются возбужденными и менее устойчивыми. Время жизни атома в возбужденном
состоянии —10 Л. Поглощение или излучение энергии атомом в виде у-кванта
света возникает в результате переходов между дискретными энергетическими уров-
нями атомов. Количество энергии, поглощаемое или излучаемое при этих перехо-
дах, связано с длиной волны X следующим соотношением
Х= ch / (Е2 -Е]) , где
h = 6,6 ‘ 10'27 эрг' с - постоянная Планка;
с = 3 ’ 108 м/ с — скорость света.
Таким образом, набор длин волн спектральных линий атома определяется на-
бором энергетических состояний, в которых он может находиться.
Чем сложнее строение атома химического элемента, тем больше в спектре
элемента спектральных линий. Например, атом водорода имеет около 30 линий, Со
- 600 линий, а атом Mo —1 000 линий. Длины волн этих линий лежат в ПК, видимой
и УФ областях света. Поэтому эти атомные спектры называются оптическими и об-
разуются они при испускании или поглощении электромагнитного излучения сво-
бодными или связанными атомами (в газах или парах).
75
Изучение атомных оптических спектров и лежит в основе оптического спек-
трального анализа элементного состава образца. Он подразделяется на эмиссионный
спектральный анализ (по спектрам испускания атомов) и атомно-абсорбционный (по
спектрам поглощения анализируемых элементов).
ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Атомные спектры испускания (эмиссионные) получают при переходе атома из
возбужденного состояния в такое же возбужденное, но с меньшей энергией или пе-
реходе в основное состояние. Источником возбуждения (атомизатор) являются
вольтова дуга переменного или постоянного тока, электрическая дуга или ацетиле-
нокислородное пламя.
Для максимальной чувствительности в эмиссионном спектральном анализе
отношение количества возбужденных атомов к количеству атомов, находящихся в
основном состоянии, должно быть возможно большим.
При вольтовой дуге напряжение подается на электроды, между которыми соз-
дается электрическая дуга. Температура на концах графитовых электродов (при
переменном токе в 10 А) достигает приблизительно 2500°С. При дуге постоянного
тока - 3000-3800°С.
Но даже эти температуры недостаточны для полного испарения труднолету-
чих элементов, что сказывается на пороге чувствительности и воспроизводимости
метода. При увеличении силы тока эти недостатки частично уменьшаются.
76
Приборы для эмиссионного спектрального анализа называются спектрогра-
фами, например ДФС-8 или ДФС-13. Основной деталью этих приборов являются
призма или дифракционная решетка (600 - 1000 штрихов на 1 см), которые разлага-
ют излучение света возбужденных атомов в пространстве по длинам волн.
Регистрация оптических спектров производится либо на фотоматериалы (в
’’оптическом” приборе), либо фотоэлектрическим способом (приборы с прямой ре-
гистрацией). Для этого в современных спектрометрах применяют счетчики фото-
нов для УФ области, термоэлементы и болометры для 14К области и т.д.
Спектрограммы (регистрация на фотоматериаллы) изучают на спектропроек-
торах или микрофотометрах (например, МФ-2). Изучается при этом 10-15 мм спек-
трограмм. В качестве реперных линий применяют спектральные линии от Fe. Ин-
тенсивность спектральных линий, в принципе, пропорциональна концентрации ато-
мов в исследуемом веществе. Образец для анализа представляет собой порошковую
пробу массой несколько мг.
На рис.29 в качестве примера приведен участок спектрограммы образца гор-
ной породы, полученный на спектрографе ДФС 13, блок-схема которого представ-
лена на рис.30. В табл. 12 приведены пороги чувствительности эмиссионно-
спектрального анализа при определении некоторых элементов.
77
Рис.29. Участок спектрограммы образца горной породы
Таблица 12
Пороги чувствительности определения некоторых элементов эмиссионно-
спектральным анализом
Элементы П, вес.% Элементы П, вес.% Элементы П, вес.%
Си 1 IO"4 Mo 1 10’3 V 2'10’3
РЬ 2 Ю’3 Zr 5' 10’3 Cr 1 IO’3
Zn 5" 10’3 Ti 1' 10’3 Mn 5 ’ IO-4
Sn 2'10’3 Sr 5" 10’3 Co 2' 10’3
Ag Г IO"4 Ba 5 IO"4 Ni 210’3
Au <5- KT* Al 1 10'3 Fe 5' 10"4
78
Рис.30. Блок схема спектрографа
1 - угольные электроды; 2 — пламя электрической дуги; 3 — оптические линзы;
4 - щель; 5 - сферическое зеркало; 6 - призма; 7 - объектив; 8 - фотопластинка
Основные достоинства метода.
1. Довольно высокий порог чувствительности (от I0'2 до 10 5 вес. % на отдель-
ные элементы);
2. Имеются методики, позволяющие вести одновременный анализ на 40 эле-
ментов;
3. Малая навеска порошковой пробы ~ несколько мг;
4. Большая производительность;
5. Небольшая стоимость анализа.
Но! Существенный недостаток этого метода - плохая воспроизводимость
данной методики. Повторный анализ может отличаться от первичного в 2 - 3
раза, что объясняется нестабильностью пламени атомизатора.
Этот недостаток устраняется при применении в качестве атомизатора индук-
тивно-связанной плазмы (ИСП). Температура "горения" плазмы достигает 6000-
10000” К. При таких температурах достигается практически полная атомизация ве-
щества, и за счет этого воспроизводимость метода резко повышается и достигает 1-
3%. При анализе исследуемая проба переводится в раствор и в виде аэрозоля пере-
носится потоком газа (Аг) в "пламя" плазмы.
В установках с ИСП применяют спектрометры с фотоумножителями, преобра-
зующими энергию у - квантов в электрические сигналы. Спектрометры могут быть
полихроматическими и сканирующими.
В полихроматических имеются много фотоумножителей, настроенных на оп-
ределенные длины волн (т.е. на определенные элементы), и все линии тогда изме-
ряются одновременно. В таких установках одновременно можно определять десятки
элементов за - 30с при расходе пробы 0,5 мл.
В сканирующем спектрометре (один спетрометрический канал; один детектор
у- квантов) выбор определяемых элементов неограничен (кроме Аг), но измерение
их идет последовательно. Поэтому расход пробы и время исследования несколько
увеличиваются.
Как правило, установки с ИСП связаны с ЭВМ, что позволяет ускорить про-
цесс расчета концентраций исследуемых элементов.
Основными достоинствами эмиссионного спектрального анализа с использо-
ванием ИСП являются.
80
1. Широкий диапазон определяемых элементов периодической системы (прак-
тически все, кроме Аг);
2 Высокая воспроизводимость метода, удовлетворяющая требованиям анали-
тической службы геологии на данном этапе;
3. Небольшая навеска для анализа ( ~10 мг), но она должна быть престави-
тельной относительно всей массы пробы.
Среди ограничений метода можно отметить.
1. Необходимость перевода пробы в раствор;
2. Высокую стоимость анализа за счет дорогостоящей аппаратуры.
АТОМНО-АБСОРБЦИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Абсорбционный спектральный анализ осуществляется путем пропускания
света от внешнего дискретного источника света через пары атомов анализируемых
элементов. Перевод пробы в парообразное состояние производится в атомизаторе
(пламя различных горючих газов: воздух-пропан; воздух-ацетилен; закись азота-
ацетилен и др.). Выбор газовой смеси определяется той необходимой температурой,
при которой наиболее полно атомизируются те или иные элементы. Так, окислы
алюминия, кремния, титана, стронция, вольфрама и некоторых других элементов
требуют для полной атомизации более высоких температур. В этом случае применя-
ется смесь закиси азота - ацетилен.
81
1 емпература пламени этих смесей составляет <3000° С. При этих условиях от-
ношение числа атомов, находящихся в возбужденном состоянии, к числу атомов,
находящихся в основном (стабильном) состоянии, составляет от ~10’4 до 10‘9 для от-
дельных элементов. Таким образом, степень поглощения излучения, пропускаемого
через исследуемую пробу, будет определяться практически только атомами, нахо-
дящимися в стабильном состоянии. В качестве внешнего источника применяют га-
зоразрядные лампы (300-400В, 3-30мА) с полым катодом, который сделан из того
металла, спектр которого мы желаем получить. Корпус лампы изготовляют из про-
зрачного для УФ-излучения материала (кварца). Лампы заполняются инертным га-
зом (неон, аргон) под небольшим давлением (~1 Омм.рт.ст.). Разряд возникает внутри
полого катода, и ионы газа сталкиваются с атомами металла, что приводит к излуче-
нию характеристического дискретного спектра металла.
Степень ослабления интенсивностей спектральных линий внешнего источника
с длинами волн, соответствующих набору длин волн спектральных линий атомов
определяемых элементов, является показателем их концентрации в пробе. Интен-
сивность прошедшего (неадсорбированного) через пары атомов исследуемых эле-
ментов излучения фиксируется детектором.
В качестве детектора применяются фотоэлектронные умножители (ФЭУ), элек-
трический сигнал с которых подается к усилителю переменного тока и затем к из-
мерительному прибору. Основные узлы атомно-абсорбционного прибора приведены
на рис. 31.
82
Для количественного анализа используют калибровочную кривую, построен-
ную по результатам измерения образцов с известным содержанием элемента
Для выделения нужной абсорбционной линии используются монохроматоры
(дифракционные решетки), работающие в диапазоне длин волн 190-850 нм.
Рис.31. Основные узлы атомно-абсорбционного прибора
1 - источник света - лампа с полым катодом; 2- линзы; 3- распылитель - горелка с
атомизированной пробой; 4-монохроматор; 5-фотоэлектронный умножитель;
б.усилитель и регистрирующее устройство.
Основными достоинствами атомно-абсорбционного метода элементного ана-
лиза являются:
1. Применим для анализа более чем на 60 элементов;
2. Порог определения элементов в растворе ниже, чем 1 мкг/мл;
3. Очень хорошая воспроизводимость (1-2 отн %);
4. высокая скорость анализа.
Среди ограничений метода можно отметить.
1 Необходимость перевода пробы в раствор;
2 . Одновременно определяется лишь один элемент.
83
Наиболее перспективно применение метода атомной абсорбции при анализе
большого количества проб на отдельные элементы, содержащиеся в ’’следовых” ко-
личествах, а также при анализе пресных и морских вод, рассолов.
ЯДЕРНО - ФИЗИЧЕСКИЕ МЕТОДЫ
Ядерно-физические методы элементного анализа основаны на регистрации
ядер и изучении вторичных излучений, возникающих в результате облучения иссле-
дуемых элементов элементарными частицами или у - квантами. При этом протекает
ядерная реакция, которую можно записать в следующем виде:
А (а, веб) В*, где
А - ядро исследуемого элемента;
а - бомбардирующие частицы или у-квант;
B,c,d - частицы или у-квант, испускаемые при ядерной реакции;
В*- образующийся радионуклид.
При регистрации излучения радионуклида (В’) ядерно-физические методы на-
зываются активационными. В тоже время существует много ядерно-физических ме-
тодов, когда для элементного анализа используются продукты ядерных реакций, ре-
гистрация которых идет одновременно с облучением исследуемого образца. Среди
них можно указать на радиографические методы анализа с помощью регистрации
продуктов ядерных реакций на твердотельные детекторы (см. раздел Радиография).
84
АКТИВАЦИОННЫЙ АНАЛИЗ
Активационный анализ - наиболее распространенный ядерно-физический ме-
тод определения элементного состава вещества
Он заключается в активации атомов исследуемого образца путем облучения
его пот оками нейтронов (нейтронный активационный анализ), у-квантов (фотоак-
тивационный), заряженных частиц (анализ с помощью заряженных частиц) и изме-
рении наведенной радиоактивности: интенсивности и энергетического спектра вто-
ричного излучения, сопровождающего распад образовавшихся радиоактивных нук-
лидов, а также периодов полураспада (T1Z2).
Наиболее широкое распространение получил нейтронно-активационный
анализ, что обусловлено его высокой чувствительностью, связанной с большой ве-
роятностью реакции захвата нейтронов (наиболее используемая ядерная реакция в
данном анализе) и наличием мощных источников нейтронов (ядерных реакторов).
В качестве источников нейтронов в нейтронно-активационном анализе при-
меняют научно-исследовательские ядерные реакторы, нейтронные генераторы,
ампульные источники нейтронов и т.д. Наиболее мощным источником нейтронов
являются ядерные реакторы, в которых в отдельных экспериментальных верти-
кальных каналах (ВЭКах) потоки нейтронов достигают 1О10 - 1013 н/см2'с, т.е. за
реальное время можно набрать плотность потока нейтронов до 1016-1017 н/см2.
Среди этих каналов имеются специальные (тепловые) каналы, где основное количе-
85
ство нейтронов являются тепловыми, т.е. нейтроны, на которых реакция захвата
нейтронов идет с большей вероятностью.
Среди ампульных источников нейтронов наиболее широкое распространение
получили калифорниевые и сурьмяно-бериллиевые источники.
Для получения мощных потоков у- излучения в фотоактивационном анализе
применяют различного типа линейные ускорители электронов (ЛЭУ), бетатроны и
микротроны, дающие тормозные спектры у - квантов, ампульные источники,
у-контуры на ядерных реакторах. На линейных ускорителях электронов пучок
электронов с большой энергией (9-25Мэв) направляют на тяжелый металл (ми-
шень). Возникает тормозное у-излучение. Эффективность преобразования кинетиче-
ской энергии электронов в тормозное у - излучение весьма велика — около 2/3 (на-
пример, в рентгеновских трубках всего 1%). Мощность дозы тормозного излучения
в таких ускорителях достигает на расстоянии 1 метра от мишени до 1,5 105 р/мин.
Для активационного анализа на заряженных частицах используют ускорители
электронов и прогонов, ампульные источники сс-частиц.
Горные породы и минералы имеют сложный элементный состав, и при актива-
ции образуется множество радионуклидов. Селективность (избирательность) на тот
или иной элемент достигается выбором излучения, временем активации, временем
"остывания", периодом полураспада образовавшихся изотопов, энергией регистри-
руемого излучения этих изотопов.
86
Измерение наведенной радиоактивности производится с помощью детекторов
ядерного излучения. Чаще всего используются у - спектрометры высокого разреше-
ния (~150-170 эВ) с использованием полупроводниковых детекторов, что позволяет
разделить основное число изотопов по энергии у - квантов (рис.32 ).
Рис.32. Гамма-спектр от активированного золотосодержащего образца
При выборе оптимального временного режима, т.е. выбора наиболее выгодных
значений времени облучения (активации) и времени "остывания", учитывается
желание исследователя определить максимальное число элементов в образце.
Время активации обычно ограничивают тремя периодами полураспада обра-
зующихся изотопов, при этом накапливается до 90% атомов от числа, соответст-
вующего насыщению активации.
87
Используя различное время "остывания", мы в процессе измерения можем вы-
делить короткоживущие, среднеживущие и долгоживущие радионуклиды. Так, на-
пример, в табл. 13 показаны группировки некоторых нуклидов по периодам полу-
распада.
Таблица 13
Периоды полураспада некоторых нуклидов
Нуклид Т1/2. мин Нуклид Т|/2, дни Нуклид T|/2, ГОД
28al 2,26 sbRb 18,8 ,j4Cs 2,06
5,1 ’’’Ст 27,7 wlCo 5,27
1 'Mg 9,46 ' *5Sr—~ 63,9 T5Jeu 12,7
Из таблицы видно, что для короткоживущих нуклидов требуется неболь-
шое время "остывания" (часто используется пневмопочта для доставки образцов из
зоны облучения до места измерения радиоактивности), а для определения долго-
живущих - соответственно максимальное время "остывания". Таким образом,
двух - или трехкратным измерением спектров у - лучей мы можем увеличить число
определяемых элементов при однократной активации образца.
Так, например, метод нейтронно-активационного анализа применялся для
оценки изменчивости геохимической обстановки на больших площадях в рамках
выявления антропогенных аномалий, связанных с загрязнением окружающей среды
предприятиями по выплавке и переработке металлов. С этой целью определялось
88
среднее содержание микроэлементов в волосах человека - конечного звена природо-
охранных мероприятий.
Использование двух различных времен облучения потоками нейтронов образ-
цов волос и двух времен "остывания" наведенной радиоактивности при этих облу-
чениях позволили определить содержания 24-х микроэлементов. Первое облучение
потоком нейтронов 5 ' К)'1 п/ см2с в течении 15с и измерение спектров активирован-
ных образцов волос через Юс (анализ Cl, I) и 1ч (Na, Си, Мп). При повторном облу-
чении (через неделю) в течение 15ч и измерении спектров вторичного излучения
через неделю (Sc, Br, As, Mo, Cd, La, W, Au, Np) и месяц после облучения (Sc, Cr,
Fe, Co Zn, Se. Ag, Sb, Cs, Ng).
Данные исследования показали, что происходит накопление в организме чело-
века некоторых элементов, специфичность которых отражает характер выплавки и
переработки металлов.
Активация у - квантами расширяет возможности активационного анализа. Ак-
тивация идет преимущественно по реакциям (у,п) и (у,р). Эти реакции являются по-
роговыми, т.е. идут с £-, > Епор Как правило, этот порог составляет 10-30 Мэв. Све-
дения о некоторых реакциях активации под действием у-кавантов приведены в
табл. 14.
Наиболее перспективной областью применения у-активационного анализа
является анализ рудообразующих минералов на такие элементы как Си, Zn, Ti, Zr,
НС Mo, W, Au, Ag и т.д.
89
Таблица 14
Активация у-квалтвми
Реакция Распростра- ненность изо- L Порог, Мм И Hi Период полу- распада 1 ъ 5 чЛ
СИ (у. п) С» 98,89 18,6 10,4 20,5 мин 0,96
О‘« (у, п) О11 99,76 15,6 14,1 2,03 мин 1,73
Si’» (у> п) Si.T 92,27 17,4 21,0 4,2 сек 3,85 0,84
Si” (у, р) А!” 4,68 12,8 31,0 2,3 мин 2,86 1,01 1,78
Si” (у, р) А1” 3.05 12,9 32,0 6,6 мин 2,5 1,4 2,43 1,28
N1* (у. п) № 99,63 10,5 2,8 10 мин □ ,19
А1« (у, п) А1'5т 100 12,8 8,С 6,5 сек 3,21 —_
Mg’* (у. n) Mg« 78,6 16,5 9,8 12 сек 3,0 0,44
Mgs* (у, р) Na’5 11,29 14,3 25,0 60 сек 3,8 2,8 1,61 0,98 0,58 0,40
S” (у, n) S’1 95,02 14,7 15,С 2,6 сек 4,42 1,27
Р” (у. п) Р” 100 12,4 17,С 2,55 мин 3,3
Са‘» (у, п) Са” 96,97 15,9 15,0 0,9 сек 5,5 2,5
К3» (у, п) К” 93,08 14,5 11,2 0,97 мин 7,7 мин 4,8 2,7 2,1
Fe5* (у, и) Fe5’ 5,84 13,8 67,0 9,0 мин 2,6 0,37
Высокая селективность анализа этих элементов на фоне элементов, слагаю-
щих породообразующие минералы, обусловлена низкими порогами активации.
Источником у-излучения в этих методиках является тормозное излучение бе-
татронов и микротронов.
Пороги определения этих элементов составляют 102- 10’3 вес.%. Навески
образцов составляют десятки грамм.
Помимо использования реакций (у,п) и (у,р) в фагоактивационном анализе
большое практическое применение нашел анализ, основанный на фотовозбужде-
90
нии изомерных состояний ядер некоторых элементов по реакции (у, у ')•
Небольшое количество элементов, на ядрах которых идет данная реакция,
невысокая энергия у-квантов, при которых она идет, широкий Диапазон периодов
полураспада и энергий регистрируемого излучения обуславливают отличную селек-
тивность на ряд элементов.
Наиболее ярким примером реализации данного вида фотоактивационного анали-
за является создание для определения Au и сопутствующих элементов на базе гор-
но-металлургического комбината экспрессного высокопроизводительного комплек-
са "Аура”.
Комплекс размещен в 3-х этажном здании на площади около 900 м2. В качест -
ве источника у - квантов используется тормозное излучение линейного ускорителя
электронов типа ЛЭУ-8-5А. Весь процесс анализа (взвешивание проб, транспорти-
ровка их к месту облучения и регистрации, измерения наведенной радиоактивности,
обработки и выдачи результатов) полностью автоматизирован.
Производительность комплекса на начальном этапе эксплуатации составляла
260 тыс. анализов в год. Порог определения Au составляет 0,2 ’ 10'6вес.%.
Масса анализируемой навески 0,5кг при крупности зерна в 1мм. Мешающие эле-
менты при производстве анализов практически отсутствуют.
Выводы:
1. Нейтронно-активационный анализ позволяет определять большинство
элементов с порогом чувствительности 10 s— 10 8вес%. Среди ограничений ме-
91
года следует отметить необходимость использования из-за самоэкранирования
нейтронов небольших навесок (мг), но тогда возникают вопросы представи-
тельности пробы; высокая активация элементов матрицы образца; труднодос-
тупность мощного источника излучения; возникновение наведенной радиоак-
тивности, при которой требуется соблюдение мер безопасности.
Основная область применения метода — это анализ содержания следовых
количеств редких и рассеянных элементов.
2. Гамма- активационный анализ имеет порог чувствительности на основ-
ное количество элементов 1(Г2- 10'7 вес.%. Среди достоинств метода необхо-
димо отметить высокую экспрессность, селективность, представительность
навески, слабаю активацию мат рицы.
Из ограничений этого метода можно отметить необходимость применения
из-за малых сечений взаимодействия у - квантов с ядрами определяемых эле-
ментов сильноточных ускорителей электронов.
92
МЕТОД
РАДИОГРАФИИ
Радиография (от латинского - radio-излучение, grapho-пишу) — метод ис-
следования содержания и распределения элементов в различных объектах
(сплавах, биологических тканях, породах и минералах), заключающийся в полу-
чении их изображения путем регистрации их собственного или наведенного ра-
диоактивного излучения, продуктов ядерных реакций, а также при просвечива-
нии излучением внешнего источника.
Регистрация излучения осуществляется на фотографические материалы или
твердотельные детекторы.
Эти методы элементного анализа относятся к локальным методам. Локаль-
ность их, в лучшем случае, около 50 мкм.
Существующие радиографические методы можно разделить на несколько
принципиально различающихся (по их физической сущности) между собой групп:
1. Собственно радиография (авторадиография);
2. Активационная (эмиссионная) радио! рафия;
3. Гамма-просвечивание;
4. Методы радиографии, где регистрируются продукты ядерных реакций
на твердотельные детекторы, в частности f - радиография и а - радио-
графия
АВТОРАДИОГРАФИЯ
Первый из рассматриваемых методов зародился, можно считать, в момент от-
крытия естественной радиоактивности, когда Беккерель зафиксировал почернение
93
завернутой в черную бумагу фотопластинки, на которую накладывались некоторые
соли урана.
Из курса общей физики вспомним, что большинство естественных радиоак-
тивных изотопов входят (образуют) в три семейства (цепочки, ряда) радиоактивных
нуклидов: ряд урана ( 92U238); ряд актиноурана ( 92U23S ); ряд тория (%Тй234).
Все начальные радионуклиды радиоактивных рядов являются а- излучателя-
ми, а конечными продуктами их распада - стабильные изотопы свинца. В каждой
цепочке помимо а- излучателей имеются и р - излучатели. Все эти превращения
сопровождаются еще у - излучением. В результате а - распада нуклид теряет в массе
4 единицы и заряд его уменьшается на две единицы, так как испускается а-частица
( 2Не4). Так ад U238 переходит в wTh234 , тот в свою очередь является р - активным и
переходит в p-активный протактиний 91 Ра234 и т.д.
Скорость этих превращений различна, поэтому вводится понятие периода
полураспада (Т1/2) - время, в течение которого первоначальное количество изотопа
уменьшается вдвое.
В каждой цепочке количество а и р-излучателей различно. Так, в цепочках
урана, актиноурана и тория количество а-излучателей 8, 7 и 6 соответственно. Ес-
ли учесть Т,,2( U238-4,51 109лет; U23s- 7,13108ne4;Th232-l,389 101влет), количество а-
излучателей в цепочках, изотопическое соотношение U238/ U235( 99,282 ) то при од-
ном и том же количестве U и Th количество а-частиц от превращений в ряду акти-
ноурана, тория и урана соотносятся как 0,03:0,24:1 .
94
Видим, что основное количество а-частиц (при Сь = С-п,) дает U238 и его це-
почка распада.
Для проведения анализа методом авторадиографии образец (в виде шлифа без
покровного стекла, аншлифа или просто каменного материала) прикладывается к
фотоматериалу (фотопластины, фотопленка или жидкие фотоэмульсии) на опреде-
ленное время (время экспозиции). После этого фотоматериал проявляется в фоторе-
активах и просматривается на оптических микроскопах.
Необходимо подчеркнуть, что во время экспозиции (т.е. регистрации излуче-
ния) все должно находится в темноте, чтобы не происходило засвечивание фотома-
териала. Степень “почернения” , интенсивность излучения (I) равна
I С(; гьХ Сэке, где:
С - концентрация урана и тория в образце;
t - время экспозиции.
Схема проведения анализа:
фотоматериал
образец
При реальном времени экспозиции (не более одного месяца) порог чувстви-
тельности метода (П) составляет не более 104 %. Для регистрации излучения при-
меняются фотопленки, ядерные пластинки и жидкие эмульсии. Эти фотоматериалы
%
фиксируют (после проявления) радиоактивное излучение, а а-частицы проявляются
в виде характерных следов (треков), возникающих в результате взаимодействия ра-
диоактивных часгиц со светочувствительным слоем. Метод позволяет по различной
95
длине пробега а - частиц (длине треков) в фотоматериале различать (за счет разной
энергии а-частиц) радиоактивность от урана и тория. Подсчет треков осуществля-
ется в оптическом микроскопе. Более подробно об этом методе радиографии можно
найти в / 6 /.
ВЫВОДЫ 1. До недавнего времени это единственный способ радиографии;
2. Порог чувствительности составляет не более 10'4%;
3. Во время экспозиции требуется защита от видимого
света, УФ, рентгеновского и у - излучения;
4. Это метод анализа содержания и распределения урана
(U238) и тория.
АКТИВАЦИОННАЯ РАДИОГРАФИЯ (эмиссионная)
Это метод выявления содержания и распределения исследуемых элементов
путем их активации и последующей регистрации излучения активированных изото-
пов этих элементов с помощью фотоматериалов. Из самого названия ясно, что ис-
следуемый элемент необходимо сначала активировать, а затем уже регистрировать
образовавшийся изотоп. Активация, чаще всего, осуществляется нейтронами или
у-квантами. В результате этого облучения идет ядерная реакция на атомах иссле-
дуемого элемента и образуется радиоактивный изотоп.
Схема реакции: А (а, в) В , где
96
A - исходное ядро;
а - ядерная частица, взаимодействующая с исходным ядром;
в - ядерные частицы, образующиеся в результате реакции;
В - радиоактивный изотоп, образующийся в результате реакции.
Регистрируя как и в первом методе с помощью фотоматериалов радиоактив-
ный изотоп В, мы определяем содержание и распределение исследуемого элемента
(А). Этот метод ничто иное, как разновидность активационного анализа.
Схема проведения анализа:
Обр. 1) активация определяемого элемента А
Излучение
Обр.
Фотоматериал 2) регистрация радиоактивного изотопа В*
Для наилучшей селективности (избирательности) на тот или другой элемент
подбирают излучение для активации, время активации (облучения), время “остыва-
ния” образца (время между концом облучения и началом регистрации наведенной
активности образца), время экспозиции.
Сегодня этой методикой пользуются для анализа содержания и распределе-
ния в шлифах золота, серебра, скандия, вольфрама, редких земель (самария, евро-
пия, лантана, церия) и т.д.
97
ВЫВОДЫ: 1. Метод появился сравнительно недавно, после того, как
появились мощные источники “л “ и у -квантов (реакторы, уско-
рители электронов и т.д.).
2. В связи с наведенной радиоактивностью исследуемого об-
разца метод требует специальных защитных действий (защита
временем, работа с манипуляторами, защитные экраны).
3. Необходимо применять (на втором этапе проведения ис-
следований) защитные меры для фотоматериалов от видимого
света, УФ света, рентгеновского излучения.
4. Большой набор выявляемых элементов.
РЕНТГЕНОГРАФИЯ или у - ПРОСВЕЧИВАНИЕ
Рентгенография или гамма-просвечивание основаны на зависимости эффекта
поглощения у-квантов от /(порядковый номер в таблице Менделеева) вещества. Ос-
новным эффектом взаимодействия у-излучения с веществом, в результате которого
у-квант исчезает, является фотоэффект. Вероятность его (Оф) зависит от Е, и Z ядра.
В общем случае <3ф» Z5/ Еу 7/2, при Er > I,, где Is - потенциал ионизации.
Сравним, как'соотносятся фотоэффекты на урановом минерале, находящем-
ся в матрице из гранита:
стц / °гр ~ (238)5/ (13,6)’ ~ 6 ’ 105 , где - 13,6 - атомная масса гранита.
Вот на этом принципе и основан эффект; т.е. контраст в изображении возни-
кает благодаря неодинаковому' поглощению излучения различными участками объ-
екта (рис.33). За объектом помещается фотоматериал, на котором и регистрируется
98
изображение. Так этот метод применялся для диагностики искусственных и естест-
венных жемчужин по наличию или отсутствию в них затравки 12 1.
Это явление заложено и в проекционном рентгеновском микроскопе, вклю-
чающем в себя рентгеновский источник со сверхмикрофоку сным пятном, камеру
для размещения исследуемого объекта и регистрирующее устройство. Такой рент-
геновский микроскоп может быть оснащен преобразователем рентгеновского изо-
бражения в видимое с последующей его передачей на телевизионный экран.
Схема проведения гамма-просвечивания:
ВЫВОДЫ. 1. Редко применяется в геологии.
2. Нашел широкое применение в технике и медицине.
РАДИОГРАФИЯ, ОСНОВАННАЯ НА ПРИМЕНЕНИИ
ТВЕРДОТЕЛЬНЫХ ДЕТЕКТОРОВ
На сегодняшний день, несмотря на свою молодость (в России метод начал
разрабатываться под руководством академика Т.Н. Флерова в 60-тые годы),
99
радиография получила огромное развитие. С ее помощью геологи вышли на совер-
шенно новую ступень понимания в области определения содержания и распределе-
ния урана, тория, бора, лития и бериллия в минералах и горных породах.
С чего все началось? В 1963 году в кристаллических веществах с помощью
электронных микроскопов удалось наблюдать область искажений в кристаллах, об-
разуемых осколками деления ядер урана.
Что такое реакция деления ядер? В результате спонтанного (самопроиз-
вольного) деления или в результате облучения (п, у, е) ядра урана и тория делятся
на два радиоактивных осколка со средними массами в районе 98 и 140 ед.. При
этом освобождается ~ 2,5 нейтрона и другие элементарные частицы. На один
акт деления освобождается около 200 Мэв энергии, которая распределяется сле-
дующем образом:
кинетическая энергия осколков - 168 Мэв
энергия нейтронов деления - 5
энергия мгновенных у - квантов - 7
энергия р - частиц продуктов
деления - 5
энергия у - квантов продуктов
деления - 5
энергия антинейтрино продуктов
деления - 10
200 Мэв
Видим, что осколки деления обладают огромной энергией и значительной
массой. Если этот осколок попадает в вещество, то на пути своего движения
юо
(до полной остановки) он оставляет область искажений (нарушений). Не рассмат-
ривая механизм образования этих нарушений, отметим, что если применить
избирательное химическое травление, то можно увеличить область этих нарушений
до размеров, видимых в обычный оптический микроскоп.
Вот как выглядят после химического травления эти следы (треки) в некоторых
материалах (рис. 33).
Рис.33. Следы от осколков деления в мусковите (а), в стекле (б) и в лавсане (в)
Режимы химического травления для некоторых материалов приведены ниже.
101
Стекло - 2,5% HF, Т-20°С . t=20 мин, е=-42 %
Слюда - 48% HF, Т=20°С, t=30 мин, е=96 %
Лавсан - 40% КОН, Т=60°С, Г -ЗОмин, £=72%
Нитрат целлюлозы - 40% NaOFT, Т=60°С, t=20-60 мин, е=40-70%,
где е - эффективность регистрации осколков.
После этого открытия сразу стали развиваться методы радиографии с ис-
пользованием этих материалов, названных твердотельными детекторами.
1, Классификация слюд по следам от осколков деления
Из рис.33 видно, что различные твердые тела имеют различную форму сле-
дов от осколков деления урана. Для кристаллических веществ форма фигур трав-
ления будет определяться структурой вещества. Работа с различными слюдами по-
казала, что для каждой из них характерна своя форма фигур травления. Удалось да-
же провести классификацию слюд по форме фигур травления, которая приведена
в специально выпущенном атласе / 4 /.
Интересны результаты работы, например, со слюдами подгруппы биотита,
где по форме фигур травления следов от осколков деления урана удалось располо-
жить слюды в ряд по возрастающему' общему коэффициенту железистости (флого-
пит, железистый флогопит, биотит, лепидомелан, сидерофиллит).
2. Возраст минерала
На чем основан этот метод определения возраста минералов?
В любом минерале содержится в том или ином количестве уран. За геологическое
время жизни минерала в нем в результате спонтанного (самопроизвольного) деле-
102
ния ядер урана, открытого в 1939г. Флеровым и Петржаком, накапливаются треки
(следы от осколков деления урана). Известно, что основной вклад в плотность сле-
дов от осколков спонтанного деления тяжелых элементов в минералах вносит уран-
238. Их количество равно:
Реп. = Си I238 NosRTX I2 ц, где
рсп - плотность следов от осколков спонтанного деления ядер урана, см'2;
Си - концентрация урана в минерале, г/г;
I238- изотопическая доля урана-238 в естественной смеси;
Мо- число Авогадро, моль1;
в - эффективность регистрации следов данным минералом;
R- эффективный пробег осколков деления в минерале, г/ см'2;
Т- геологическое время, год;
А, - постоянная спонтанного деления ядер урана-238, год.'1;
ц - атомный вес урана, г/ моль.
В данном уравнении I238, е, R, A., No, р. величины постоянные. Поэтому количе-
ство следов от осколков спонтанного деления ядер урана в минерале будет пропор-
ционально только концентрации урана в минерале и времени его жизни.
Если этот же образец облучить тепловыми нейтронами, то количество следов
от осколков вынужденного деления ядер урана (рвь,„.) будет равно:
Рвын = Cu 1235 No ейстФ / 2ц где
о - сечение вынужденного деления ядер урана-235, см2;
Ф - интегральный поток тепловых нейтронов , см'2;
юз
В данном уравнении, как и в предыдущем, I, е, R, о, NOi ц величины постоян-
ные. Поэтому количество следов от осколков вынужденного деления ядер урана в
минерале будет пропорционально концентрации урана в минерале и интегральному
потоку излучения.
Решая систему этих двух уравнений относительно возраста, получим:
Т = А ФрС1,/рвы„; А-коэфф. пропорциональности.
Подсчитав экспериментально количество следов от осколков вынужденного и
спонтанного деления ядер урана, и зная интегральный поток излучения, однозначно
определяется возраст минерала.
Этим методом определялся возраст слюд, кальцитов, апатитов, цирконов, тор-
бернитов и т.д.
3. Определение содержания и пространственного распределения
Урана
Основное количество работ по радиографии с применением твердотельных
детекторов относится к выявлению содержания и пространственного распределения
урана в минералах и горных породах.
Метод заключается в следующем. Если взять исследуемый образец в виде
прозрачного шлифа без покровного стекла и приложить к нему детектор, то при об-
лучении этого “сэндвича” нейтронами (у-квантами) в результате реакции деления
образуются осколки деления, которые вылетают из приповерхностного слоя толщи-
ной R и попадают в детектор (лавсан, стекло, слюда). Содержание и распределение
104
треков на детекторе, выявленное с помощью избирательного химического травле-
ния, однозначно отвечают содержанию и распределению урана в исследуемом об-
разце. Иногда этот метод называется f-радиография (по английскому названию ре-
акции деления - fissin ) или осколочная радиография.
Чаще всего в качестве детектора используется лавсан (толщиной 3-5мк), так
как наведенная в нем в результате облучения радиоактивное! ь самая маленькая по
сравнению с другими детекторами. Одновременно за одно облучение, например,
облучение нейтронами на атомном ректоре, можно исследовать до 30 шлифов. По-
сле спадения наведенной активности до допустимых величин детекторы отделяют
от шлифов и протравливают для выявления в них следов от осколков деления. В
дальнейшем детектор исследуют под оптическим микроскопом и сравнивают рас-
пределение треков на нем со шлифом.
Для определения концентрации урана в том или ином месте на шлифе подсчи-
тывают плотность треков на детекторе в этом же месте. Зная нейтронный поток и
определив плотность треков, подсчитывают концентрацию урана по формуле:
С = р2р / N„eoR<I>
Порог чувствительности определения урана этим методом составляет ~ 10'8 вес.
Мешающих элементов при определении урана практически нет. Только при содер-
жаниях тория в 1000 раз больше чем урана сказывается его влияние. Необходимо
отметить широкий диапазон определения содержания урана: 102 10"*вес.% Помимо
определения содержания урана метод позволяет выявлять его распределение. На ос-
105
нове анализа пространственного распределения урана в большом количестве мине-
ралов и горных пород (магматических, метаморфических и осадочных) можно кон-
статировать, что уран распределен в них как равномерно (при разрешающей спо-
собности данного метода в 50 цк), так и неравномерно (рис. 34).
Рис. 34. Равномерное распределение урана в глинистых бокситах (а) и
глинистых фосфоритах (б); приуроченность урана к акцессорным минералам: сфе-
нам в аляскитовых гранитах (в) и гранатам в нефелиновых сиенитах (г); к трещи-
нам (д), межзерновым швам (е), мельчайшим точечным урансодержащим вклю-
чениям в эпилейцитовых порфирах (ж)
106
Относительно равномерное распределение урана наблюдается в мелоподоб-
ных и глинистых фосфоритах (34 б), в глинистых бокситах (34 а), в обсидианах и
т.д. Однако чаще всего уран в горных породах распределен неравномерно.
При неравномерном распределении урана в породах он может быть
приурочен к различным урансодержащим минералам (рис.34в, г), трещинам
(рис.34 д) и межзерновым швам (рис.34 е), мельчайшим минеральным образовани-
ям (рис.34 ж), На рис.34 и последующих фотографиях слева приведены фотографии
шлифов, а справа — распределение исследуемого элемента; отображения — зеркаль-
ные.
В неизмененных урансодержащих акцессорных минералах (цирконах, цирто-
литах, сфенах, апатитах, ортитах, гранатах и др.) элемент распределен, как правило,
равномерно (рис.34 в, г). При этом величина концентрации урана в минералах в
значительной степени варьирует в зависимости от типа пород, содержащих эти ми-
нералы.
Кроме равномерного распределения урана в неизмененных минералах иногда
наблюдаются специфические формы неравномерного распределения, обусловлен-
ные условиями роста и формой кристаллов.
Так, на рис.35 распределение урана в кристаллах вавеллита отражает сферо-
литовую форму выросшего агрегата (радиально-лучистое распределение) и пульси-
рующее поступление элемента в раствор во время роста минерала (зонально-
концентрическое распределение).
107
Рис.35. Распределение урана в вавеллите (а) и гранате (в)
На рис.35в в неизмененных гранатах из щелочных сиенитов видны зоны
роста минерала, по которым наблюдается различное содержание урана с изменени-
ем концентрации в 2-5 раз.
В то же время неравномерное распределение урана по минералу говорит, как
правило, о том, что минерал подвергался изменениям. В этом случае повышенное
или пониженное содержание элемента, по сравнению со средним его содержанием в
минерале, наблюдается в некоторых, чаще в периферийных, участках кристаллов, в
трещинах и во включениях инородных минералов. Указанные изменения
108
содержания урана обусловлены замещением одних минералов другими
(рис.Збв); циркуляцией урансодержащих растворов по трещинам и плоскостям
спайности кристаллов, где рассматриваемый элемент либо сорбируется на стенках,
либо входит в виде примеси в решетки вновь образуемых минералов, либо образует
собственные урановые минералы (рис. 36с). При этом нередко трещинам во вме-
щающей породе, выполняющим роль подводящих к минералам каналов, соответст-
вуют повышенные содержания урана. На рис.36а приведены частично изменен-
ные минералы сфена из щелочных сиенитов, в процессе замещения которого лей-
коксеном происходило увеличение содержания урана.
Ms
Рис.36. Распределение урана в сфенах (а), в гранатах (в) и в поструд-
ных кальцитах с урановых месторождений (с)
109
На рис.Збв представлены гранаты из щелочных сиенитов, в которых отдель-
ные участки минерала содержат пониженные концентрации урана; эти участки кри-
сталлов замещены в результате метасоматических процессов карбонатом. На рис.37
видно увеличение содержания урана по периферии пирита, где он замещается тон-
кой каемкой гидроокислов железа, сорбирующих, очевидно, данный элемент.
400mk
kJ
Рис.37. Распределение урана в пирите
Интересные формы распределения урана в слюдах. Как правило, в неизме-
ненных минералах содержание урана низкое. Но при постмагматических процессах
биотит, например, замещается хлоритом, мелкими зернами сфена и лейкоксена, рас-
полагающимися цепочками по плоскостям спайности (рис.38). В указанных участ-
ках уран либо сорбируется, либо входит в решетки замещающих минералов.
Приведенные выше примеры показывают несомненное достоинство метода:
по
Рис. 38. Распределение урана в слюдах
возможность определять концентрацию равномерно распределенного урана в образ-
це и концентрацию, которая приходится на участки неравномерного распределения.
Проведенные многочисленные исследования по пространственному распре-
делению и анализу локальных содержаний урана в минералах подвели к вопросу
рассмотрения правомерности определения содержания данного элемента по моно-
минеральным фракциям методами, дающими “валовое” значение содержания урана
(например, радиохимический, люминесцентный, активационный и т.д.). Только в
редких случаях при исследовании неизмененных минералов (а при оптических ис-
следованиях эти изменения зачастую не видны) результаты анализа перечисленны-
ми выше методами будут отражать истинное содержание урана в исследуемых ми-
нералах.
Так, например, результаты исследования закономерностей распределения
урана в интрузивных породах Северного Казахстана, полученные с помощью мето-
111
да f-радиографии, показали, что даже частичное изменение породообразующих или
акцессорных минералов, выражающиеся в замещении их вторичными продуктами,
приводит к увеличению содержания урана в 3 - 6 раз.
Подчеркнем, что при облучении тепловыми нейтронами анализ идет по изо-
топу уран-235. Это обстоятельство позволяет, используя авторадиографию (регисг-
рация урана-238) и f-радиографию, выявлять миграцию урана и продуктов его рас-
пада .
После краткого рассмотрения возможностей метода f-радиографии для оп-
ределения содержания и пространственного распределения урана можно сделать
выводы.
ВЫВОДЫ. 1. Метод обладает большой чувствительностью; порог
определения составляет 10~8 вес %.
2. Практически нет мешающих элементов (до Cftl/C( =104).
4. Анализ содержания и пространственного распределения
тория,бора, лития и бериллия
Кроме определения содержания урана и выявления его пространственного
распределения метод позволяет анализировать радиоактивный элемент - торий.
Возможность его анализа обусловлена определенной вероятностью деления ядер
этого элемента при облучении теми же излучениями что и урана. Для большей се-
лективности выявления тория на фоне имеющегося в исследуемом образце урана
подбирают режим облучения нейтронами или вид излучения.
112
Так, если при облучении тепловыми нейтронами вероятность деления ядер
урана значительно превосходит деления ядер тория, то при облучении быстрыми
нейтронами вероятность деления ядер этих двух элементов соизмерима. Применяя
двукратное облучение исследуемого образца и сравнивая содержание и распределе-
ние следов от осколков деления на детекторах в этих двух случаях, можно одно-
значно определить содержания и пространственное распределение урана и тория.
Другой способ раздельного анализа этих двух элементов заключается также в
двукратном облучении: одно - тепловыми нейтронами, другое - у-квантами, исполь-
зуя для получения этого излучения ускоритель электронов.
На рис.39 приведены формы распределения урана и тория в пирохлорах из кар-
бонатитов. Сравнивая распределения осколков деления (треков) на двух детекторах,
можно однозначно говорить о различном распределении этих двух элементов: уча-
стки минералов, которым соответствует одинаковая плотность треков на двух де-
текторах, содержат преимущественно торий; участки же минерала, на которых
плотность треков резко различна, содержат преимущественно уран.
Так, на рис.39 уран приурочен к отдельным зонам роста кристаллов (рис.39а ), к
центральным его участкам - «урановые ядра» (рис.39в,д ). к периферийным участ-
кам -«кайма» (рис.39с,д ). Торий же приурочен к отдельным зонам роста кристаллов
(рис.39а,в) и распределен равномерно по минералу (рис.39с). Однако эта методика
радиографии на торий получила небольшое распространение.
из
300 мк
300 мк
Рис.39. Распределение урана и тория в пирохлорах из карбонатитов
1 - шлиф; 11 - детектор после первого облучения; 111 - дегектор по-
сле второго облучения
2 мм
2 ММ
Кроме методик на естественные радиоактивные элементы уран и торий ме-
тод радиографии с применением твердотельных детекторов позволяет оп-
ределять содержание и выявлять пространственное распределение таких легких
элементов как бор, литий и бериллий.
114
Методики анализа на эти элементы методом радиографии удалось разрабо-
тать после того, как обнаружилось, что нитрат и ацетат целлюлозы регистрируют
а-частицы. Так же как при выявлении осколков деления для наблюдения а-частиц в
оптическом микроскопе применяют избирательное химическое травление этих де-
текторов: 40% NaOH, Т=20°С, т = 20мин. Эффективность регистрации а-частиц
этими детекторами оказалась, в зависимости от марки целлюлозы, от 40 до 90%.
После того, как удалось подобрать режимы избирательного химического
травления для выявления а - частиц, возникли методики определения содержания и
выявления пространственного распределения бора и лития, основанные на исполь-
зовании ядерной реакции (п,а). Отсюда и названия методик - методики (п,а) - ра-
диографии.
Для выявления этих двух элементов прозрачные шлифы горных пород без
покровного стекла с прилегающими к ним вплотную твердотельными де-
текторами облучают тепловыми нейтронами на ядерном реакторе. В результате об-
лучения на ядрах этих элементов с большой вероятностью идут следующие реакции:
5 В10 + n -> 3Li7 + 2Не4 (а-частица)
з1л6 + п -> ]Н3 + 2Не4 (а-частица)
Возникающие в результате ядерных реакций а-частицы и ядра отдачи попа-
дают в детектор, и после химического травления последнего следы от этих частиц
видны в оптическом микроскопе.
115
Многочисленные исследования распределения бора и лития в минералах и
горных породах позволили выяснить, что формы их распределения те же самые, что
и урана. Так, на рис.40 представлены некоторые формы распределения бора в раз-
личных минералах, а на рис.41а - распределение лития в кристаллах кварца и отра-
жение процесса лепидолитизация мусковита (рис.41 в) и т.д.
Лоо»*,
Рис. 40. Зональное распределение бора в кристаллах салита (а),
приуроченность бора к многочисленным тончайшим вросткам данбурита в
кристаллах граната (б) и включениям гидротерм в кристаллах кварца (в)
При определении содержания и распределения этих двух элементов они яв-
ляются взаимомешающими; поэтому необходимо быть осторожным в оценке влия-
ния каждого из элементов на характер распределения треков на детекторах и приме-
нять дополнительные методы для безошибочного анализа.
,ЙСО«,
Рис. 41. Распределение лития по зонам роста кристаллов кварца (а) и
отражение процесса лепидолитизации мусковита (в)
И, наконец, определение содержания и выявление пространственного рас-
пределения бериллия. Для анализа этого элемента используется фотонейтронная ре-
акция на Be9 с порогом реакции, равным 1,78 Мэв. В результате этой реакции обра-
зуется радиоактивный изотоп Be8, который распадается с периодом полураспада
равным 0,61с на две а-частицы. Вот эта реакция:
4Ве9 + у -> 4Ве8 + п ; 4Ве8 -» 2 2Не4 (2 а-частицы)
Т |/2~0,61С
Эту методику можно отнести и к активационной радиографии, так как снача-
ла идет реакция активации, а потом уже и регистрация излучения продукта актива-
ции.
117
Однако использование для регистрации а-частиц твердотельного детектора
позволяет обособить эту методику и назвать ее (у, а)-радиографией.
Методика применялась для изучения фазового минералогического состава
полиминеральных тонкодисперсных бериллиевых руд и определения количествен-
ного соотношения между отдельными бериллийсодержащими минеральными фаза-
ми, отличающимися по содержанию этого элемента.
Кроме использования твердотельных детекторов а-частиц для разработки
методик анализа на бор, литий и бериллий они могут широко применятся в автора-
диографии, при этом картина распределения урана более четкая, чем при использо-
вании фотоматериалов. Это объясняется тем, что при использовании фотомате-
риалов почернение обусловлено у - квантами, электронами и а-частицами, а
твердотельные детекторы регистрируют только а-частицы, пробег которых в ве-
ществе значительно меньше, чем у - квантов.
Таким образом, использование твердотельных детекторов позволило расши-
рить круг выявляемых элементов, включив в него такие легкие элементы как бор,
литий, бериллий.
Порог чувствительности методик радиографии с применением твердотель-
ных детекторов составляет: уран -10’8%, торий - 10'5%,бор - КГ* %, литий - КГ5 %,
бериллий - 10’1 %.
118
ЛИТЕРАТУРА
Активационный анализ. Методология и применение. Изд-во "ФАН", Ташкент,
1990.
Б.Андерсен. “Определение драгоценных камней”, Изд-во “Мир камня” М.,
1996.
Афонин В.П., Гуничева Т.Н. Рентгеноспектральный флуоресцентный анализ
гор- ных пород и минералов . Изд-во "Наука", Новосибирск, 1977.
И.Г.Берзина, Д.П.Попенко “Диагностика слюд по фигурам травления на
следах от осколков деления”, Изд-во “ОНТИ ВНИИЯГГ”, М., 1970.
Бурмистенко Ю.Н. Фотоядерный анализ состава вещества. Энергоатомиздат.
М., 1986.
Е.И.Железнова, И.П.Шумилин, БЛ.Юфа. Радиометрические методы анализа
естественных радиоактивных элементов. Изд-во “Недра”, М., 1968.
А.Н. Зайдель Основы спектрального анализа. Изд-во "Наука", М.1965.
Кузнецов Р.А. Активационный анализ. Атомиздат, М., 1967.
Методические основы исследования химического состава горных пород, руд и
минералов. Под ред. Г.В.Остроумова, ''Недра1'', М, 1979.
Методы минералогических исследований. Справочник. Под ред. Гинзбурга
А.И. Изд-во "Недра", М.,1985.
Павлова Л.Ф., Белозерова Щ.Ю. и др. Рентгеноспектральный электронно-
зондовый микроанализ природных объектов. Изд-во "Наука", Новосибирск, 2000.
Плаксин И.Н., Старчик Л.П. Ядерно-физические методы контроля
вещественного состава. Изд-во "Наука", М., 1966.
Рентгенорадиометрический метод при поисках и разведке рудных месторож-
дений. Под ред. Очкура А.П. Изд-во "Недра", Л., 1985.
119
Ривес Р.Д., Брукс Р.Р. Анализ геологических материалов на следы элементов.
Изд-во "Недра", М. 1983.
Современнные методы исследования минералов, горных пород и руд. Под
ред. Гавриленко В.В. Изд-во Санкт-Петербургского горного ин-та. СПб. 1997.
Г.Н.Флеров, И.Г.Берзина. “Радиография минералов, горных пород и руд”,
Атомиздат, М., 1979.
Г.Юднг. Инструментальные методы химического анализа. Изд-во "Мир11, М.
1989.
ЭЛЕКТРОННАЯ МИКРОСКОПИЯ
Электронная микроскопия - метод исследования, который получил широкое
распространение в различных областях науки и техники и занял прочное место в
ряду современных методов исследования минералов.
Значение этого метода определяется тем, что электронная микроскопия,
благодаря своей высокой разрешающей способности и огромным увеличениям по-
зволяет наблюдать такие особенности и детали микроструктуры объектов, которые
другими методами выявлены быть не могут.
Основными параметрами оптических приборов и электронного микроскопа,
в частности, являются их увеличение и разрешающая способность, которая опреде-
ляется как минимальное расстояние между двумя точками объекта, при котором их
можно еще распознать как отдельные.
120
Электронная микроскопия позволяет получать увеличение до 106 раз. Раз-
решающая способность (d) определяется формулой
, _ o,6U
wsincz где>
X- длина волны применяемого излучения;
а- апертурный угол объектива;
п- показатель преломления среды.
Угол альфа стремится к 90°, т.е. sina к 1. Показатель преломления
изменяется от 1 до 2,5.
Из этой формулы видно, что разрешаемое расстояние зависит, главным обра-
зом, от длины волны применяемого излучения.
Разрешающая способность глаза - 0,2мм.
Разрешающая способность светового микроскопа (при длине волны види-
мого света =0,4-0,7 мкм) составляет примерно 0,2 мкм. Применение более корот-
кого ультрафиолетового излучения с длиной волны =0,25-0,3 мкм позволяет улуч-
шить разрешающую способность прибора только в два раза
В электронном микроскопе изображение формируется пучком электронов,
длина волны которых зависит от скорости летящих электронов, которая в свою
очередь зависит от ускоряющего напряжения.
При средних ускоряющих напряжениях длина волны электронов составляет
порядка 0,04 А
121
Применение в электронном микроскопе излучения с длиной волны меньше 1
А позволяет получать предельное разрешение 2-4 А.
ПРИНЦИПИАЛЬНАЯ СХЕМА ЭЛЕКТРОННОГО
МИКРОСКОПА
На рис.42 показаны оптические схемы светового и электронного микроско-
пов.
Основными узлами электронного микроскопа являются (рис.42):
1 . Источник освещения - электронная пушка;
2 .Система электромагнитных линз, с помощью которых формируется изо-
бражение;
3 .Флюоресцирующий экран.
Источником электронного пучка в микроскопе является электронная пушка,
которая состоит из катода, анода и фокусирующего электрода.
Катод - это источник электронов. Он представляет собой тонкую вольф-
рамовую нить, диаметром около 0,1мм, изогнутую в ^юрме буквы V.
При разогревании катода до- 2500-2900’С, происходи! процесс термоэлек-
тронной эмиссии, т.е. испускание электронов при высокой температуре.
Между катодом и анодом создается высокое напряжение, ускоряющее ис-
пускаемые катодом электроны, которые проходят сквозь отверстие в аноде и в виде
электронного пучка выходят из пушки. В электронном микроскопе (среднего клас-
са) используется ускоряющее напряжение от 15 до 200 кВ.
Система линз включает в себя;
а) конденсорную линзу;
б) объективную линзу;
в) проекционную линзу (см. рис.42).
122
Конечное изображение объекта выводится на флуоресцирующий экран, под
которым помещается кассета с фотопленкой для фиксации изображения.
Рис.42. Схема распространения лучей в оптическом и электрон-
ном микроскопах
Поскольку электроны сильно рассеиваются при прохождении через вещест-
во, в том числе и в воздухе, то для обеспечения большой величины свободного про-
бега электронов вся система находится в вакууме порядка НТ*- КУ6 мм.рт.ст. Ваку-
ум в электронном микроскопе поддерживается с помощью вакуумных насосов.
123
ТИПЫ ЭЛЕКТРОННЫХ МИКРОСКОПОВ
Различаются два основных типа электронных микроскопов:
а) просвечивающие электронные микроскопы. В этих приборах пучок элек-
тронов проходит сквозь исследуемый объект, как бы просвечивая его (отчего и на-
звание - просвечивающий электронный микроскоп);
б) растровые или сканирующие.
При формировании изображения в этих электронных микроскопах пучок
электронов падает под небольшим углом к поверхности ’’массивных” образцов и от-
ражается от него. Изображение формируется за счет неодинакового отражения
электронов от различных участков образца.
Растровый (сканирующий) электронный микроскоп имеет ряд характерных
особенностей:
а) возможность наблюдения объектов без специальной их подготовки (в
отличие от просвечивающих, о чем будет сказано в дальнейшем);
б) изображение в растровом электронном микроскопе характеризуется очень
большой глубиной резкости (0,6-0,8мм) и объемностью, что и является его главным
преимуществом по сравнению с просвечивающим. Однако разрешающая способ-
ность растровых микроскопов на порядок ниже, примерно 20-50 А по сравнению с
2-5 А - в просвечивающих;
124
в) большой диапазон постепенно возрастающих увеличений от светоопти-
ческих (20-1000) до электроннооптических (100000 - 500000 раз);
Применение того или иного типа электронного микроскопа определяется за-
дачами, поставленными перед исследователем, а также характером самого объекта.
Электронные микроскопы могут быть оснащены различными дополнитель-
ными приспособлениями, что позволяет значительно расширить возможности ис-
следования объектов.
Так, электронный микроскоп-микроанализатор представляет собой элек-
тронный микроскоп с встроенным в него энергодисперсионным ( как правило) рент-
геновским спектрометром. Приставка- микроанализатор позволяет определять
элементный состав исследуемого объекта. Принцип работы такой приставки за-
ключается в использовании возникающего характеристического рентгеновского из-
лучения при попадании пучка электронов на объект, т.е. приставка работает анало-
гично прибору микроанализатору.
Таким образом, такой прибор позволяет одновременно наблюдать как
микроструктуру поверхности объекта, так и определять его химический состав.
В некоторых электронных микроскопах предметный столик оснащается уст-
ройством поворота и наклона образца, что позволяет получить электронограммы от
разных сечений его кристаллической решетки. Эта позволяет однозначно иденти-
фицировать минерал.
125
ЗАДАЧИ, РЕШАЕМЫЕ С ПОМОЩЬЮ ЭЛЕКТРОННОЙ
МИКРОСКОПИИ
1. Изучение микроморфологических особенностей минеральных агрегатов и
индивидов:
а) исследование частиц тонкодисперсных минералов - установление фор-
мы, размера, относительной толщины и ориентировки отдельных минеральных ин-
дивидов, соотношение частиц различной формы и размера. Примером таких иссле-
дований могут быть глинистые минералы, относящиеся обычно к фракции менее
1 мкм. Морфология, размер частиц, их фазовый состав часто не могут быть опре-
делены другими методами, кроме электроннной микроскопии. Большинство глини-
стых минералов отличаются чисто морфологическими признаками. Например, гал-
луазит имеет удлиненную, нитевидную, трубчатую форму (рис.43,а), в то же время,
у каолинита хорошо образованные с четкими контурами кристаллы правильной
гексагональной формы (рис.43б); монтмориллонит имеет хлопьевидный облик агре-
гатов и нечеткие, размытые контуры частиц (рис.43в).
б) установление тонких структурно-текстурных особенностей минеральных
агрегатов: наблюдение зарождения, роста кристаллических зерен, их перекристал-
лизация и замещение, изучение микроскульптуры граней (фигур роста, растворе-
ния, характер двойникования и др. тонкие детали микроструктуры). Примером та-
ких исследований является изучение под электронным микроскопом минералов
группы кварца.
126
С помощью электронного микроскопа были выявлены различия в микро-
морфологии разлиных типов кварца; микроскульптурные особенности граней кри-
сталлов, двойниковое строение.
Рис.43. Формы выделения галлуазита (а), каолинита (б) и
монтмориллонита (в). Ув. 12000
Различные разновидности кварца характеризуются различными микромор-
фологическими особенностями. Для сколов крупнокристаллического кварца харак-
терен раковистый рельеф (рис.44а). Скрытокристаллические разновидности кварца
(халцедоны, хризопраз) при оптическом наблюдении не выявляют своего кри-
127
сталлического строения, тогда как под электронным микроскопом наблюдаются
мельчайшие кристалики размером менее 1 мк.
Обычный опал (неиризирующий) сложен кремнеземом глобулярного строе-
ния с разным размером глобулей и их беспорядочным расположением (рис.44).
Рис.44. Крупнокристаллический кварц (а) и неиризирующий опал (б). Ув. 12000
а У в. 12000 б У в. 24000
Рис.45. Благородный опал
128
Исследование благородного опала под электронным микроскопом позволило
дать объяснение их замечательному свойству - игры цвета. Под электронным
микроскопом было выявлено, что поверхность благородного опала сложена
мельчайшими шариками близкого размера в пределах от 100 до 4000 А. Упорядо-
ченное расположение этих шариковидных частиц по принципу плотнейшей упа-
ковки с образованием между ними пустот правильной формы(рис.45б) вызывает
дифракцию света, чем и обусловлен наблюдаемый эффект игры света. При этом
установлена определенная зависимость между размером частиц кремнезема с дли-
ной волны дифрагированного света, чем и объясняется определенный цвет благо-
родных опалов (розовый, зеленый).
2. Электронномикроскопический контроль степени однородности минералов.
а) установление фазовой неоднородности минералов - выявление микровк-
лючений, тонких структур распада твердых растворов, зональной неоднородности
кристаллов, определение продуктов изменения минералов. Примером установления
тонких структур распада твердых растворов могут быть исследования титаномагне-
тита. Исследования показали, что даже оптически гомогенные образцы титано-
магнетитов из различных месторождений не являются однородными, а представ-
ляют собой закономерное срастание нескольких минеральных фаз. Наиболее часто
встречаются тонкие структуры распада твердого раствора на магнетит (FeFe2O4) и
ульвошпинель (TiFejOj). Характерными для титаномагнетита являются топкоре-
шетчатые структуры распада твердого раствора, имеющие в зависимости от
129
ориентировки образца вид прямо - 0(a) и косоугольной сетки (б), составленной из
пластинчатых выделений ульвошпинели шириной обычно в десятые доли мкм сре-
ди основной массы магнетита (рис.46). Ульвошпинель в этих выделениях диагно-
стируется с помощью микродифракции и локального микроанализа.
Рис.46. Структуры распада гитаномагнетита на магнетит (основная фаза)
и ульвошпинель.Ув. 18000
б) расшифровка фазового состава полиминеральных агрегатов, диагностика
отдельных минеральных частиц, уточнение химического состава минералов. Для
исследования фазового состава микровключений применяется метод микродифрак-
цни, по возможности дополняемый методом локального микроанализа (если имеется
рентгеновская приставка).
Сущность метода микродифракции заключается в том, что электроны, так же
как рентгеновские лучи, при прохождении через кристаллическую решетку взаимо-
действуют с веществом и рассеиваются по определенным направлениям
130
Рассеянные электроны дают дифракционные максимумы, упорядоченное
расположение которых соответствуют расположению атомов в кристаллической
решетке вещества. Для каждого вещества характерна своя микродифракционная
картина, позволяющая рассчитывать межплоскостные расстояния, определять па-
раметры ячейки и, в результате, диагностировать микровключения. Метод микро-
дифракции в электронном микроскопе аналогичен методу рентгеноструктурпого
анализа, но имеет ряд особенностей. Ввиду того, что длина волны электронов на
два порядка меньше, чем у рентгеновского излучения, размер минимальной частицы
определяемою вещества составляет 1мкм. Поэтому, метод микродифракции приоб-
ретает особое значение для изучения рентгеноаморфных образцов, когда рентгено-
структурные исследования не обнаруживают кристаллического строения вещест-
ва, однако дающего рефлексы на электропограммах, что позволяет говорить о сте-
пени кристалличности вещества. Кроме того, если для определения какой-то фазы
методом рентгеноструктурного анализа содержание ее не должно быть меньше 3-
5%, то метод микродифракции позволяет нам определять единичные микровключе-
ния размером ~ 1 мкм.
Чтобы получить микродифракционную картину, необходимо сфокусирован-
ный пучок электронов направить на извлеченную частицу вещества. При взаимо-
действии электронов с веществом получаем микродифракционную картину, расчи-
тав которую можно определить состав микровключения.
Так, на рис.47а среди общей массы коффинита выделяются овальные
131
а б
Рис.47. Коффинит с циппеитом (а) и микро дифракционная кар-
тина от циппеита (б)
кристаллы циппеита, которые были определены с помощью микродифракции
(рис.47б).
Таким образом, с помощь электронной микроскопии, микродифракции и
локального микроанализа можно одновременно наблюдать микроструктуру объек-
та, диагностировать микровключения и определять их химический состав.
3. Изучение реального внутреннего строения минералов.
Установление структурной неоднородности минералов, выявление очень
тонких (вплоть до атомарного уровня) деталей поверхности минеральных зерен,
граней кристаллов, дефектов минералов и их связь с особенностями кристалличе-
ской структуры.
Для исследования очень тонких деталей поверности минеральных зерен, гра-
ней кристаллов, плоскостей спайности применяется метод декорирования. Сущ-
ность метода заключается в том, что нанесенные на поверхность образца зародыши
какого-либо вещества концентрируются в виде скоплений и цепочек на отдельных
132
деталях рельефа: ступеньках скола, микровключениях, местах выхода дислокаций,
тем самым подчеркивая (декорируя) неоднородность. Например, методом декориро-
вания были выявлены слои роста галита (рис.48), серии равномерно расположенных
ступеней элементарных каолинитовых слоев тощиной 7 А и др.
Рис.48. Слои роста галита. Ув.50000
4. Изучение метамиктных минералов
Очень хорошие результаты были получены при исследовании метамиктных
I минералов, являющихся обычно рентгеноаморфными. Для метамиктных минералов
I типичной является колломорфная микроструктура. При прокаливании этих образ-
I цов происходит раскристаллизация метамиктного вещества, в результате чего обра-
I зуются микрокристаллические агрегаты с различной морфологией слагающих его
индивидов. При этом в электронном микроскопе можно наблюдать все стадии рас-
I кристаллизации метамиктного вещества: начало кристаллизации, рост кристаллов,
I оформление граней кристаллов (рис.49).
вз
Рис.49. Раскрисгаллизация метамиктного бритолита при прокаливании.
Ув. 16000
Таким образом, применение электронной микроскопии в минералогии по-
зволяет решать широкий круг задач по, исследованию минеральных индивидов.
Правильное и наиболее полное решение этих задач зависит от:
а) применения различных методов исследования вещества (микродифракция,
локальный микроанализ);
б) выбора метода и технологии подготовки препарата для просмотра в элек-
тронном микроскопе.
Правильная подготовка объекта для исследования под электронным микро-
скопом составляет одну из главных задач для получения достоверной информации
об исследуемом объекте.
134
ОСНОВНЫЕ МЕТОДЫ ПРЕПАРИРОВАНИЯ ОБРАЗЦОВ ПРИ
ЭЛЕК! РОННОМИКРОСКОПИЧЕСКИХ ИССЛЕДОВАНИЯХ
Методика подготовки объекта для исследования определяется, прежде всего,
тем, на каком микроскопе будет он изучатся: просвечивающем или растровом.
Растровый электронный микроскоп позволяет непосредственно наблюдать
массивные объекты без их предварительной подготовки. Необходимым является
только напыление на образец угольной пленки.
При исследовании объекта на просвечивающем электронном микроскопе
главным требованием является прозрачность образца для электронного пучка,
т.е. толщина препарата, сквозь который проходит электронный луч.
Оптимальной является толщина 100 - ЗООА. Более толстые препараты погло-
щают поток электронов и разрушаются.
В настоящее время разработаны разнообразные методы приготовления пре-
паратов для исследования на просвечивающем электронном микроскопе. Эти ме-
тоды могут быть разделены на две основные группы:
1)прямые методы, при которых исследуется сам объект.
2)косвенные методы, с помощью которых изучается не сам объект, а отпеча-
ток с его поверхности, называемый репликой.
К прямым методам исследования относятся:
а) метод суспензий;
б) метод ультратонких срезов.
135
Метод суспензий применяется для изучения изолированных частиц,
главным образом глинистых и других тонкодисперсных минералов.
Сущность этого метода заключается в изучении морфологии тонкодиспсрс-
ных минералов, размер частиц менее 1 мкм, в основном глинистых минералов, ко-
торые легко разделяются по границам соприкосновения отдельных зерен, часто
имеют характерную форму кристаллитов, листоватое или волокнистое строение.
Для приготовления качественного препарата необходима предварительная
диспергация образца - разделение тончайших частиц друг от друга. Растирание
этих частиц проводить не рекомендуется, так как при растирании может разрушать-
ся или изменяться морфология частиц. Обычно для получения суспензии небольшое
количество вещества насыпают в пробирку и заливают дистиллированной водой.
Затем проводят простое энергичное встряхивание пробирки (5-10мин.) для отделе-
ния частиц друг от друга.
Лучшие результаты дезинтеграции частиц получаются с применением ульт-
развука.
На ультразвуковом диспергаторе подбирают оптимальный режим работы для
данного образца (частота колебаний, мощность излучателя, время) с таким расче-
том, чтобы твердый агрегат распадался по границам зерен или по спайности без
нарушения их формы. После диспергации суспензия может быть использована
после того, как осядут наиболее крупные частицы.
Хорошего качества считается почти прозрачная суспензия. Затем пипеткой
из верхней части суспензии набираем каплю раствора и капаем на стекло, слюду,
136
каменную соль, т.е. на абсолютно гладкую поверхность. После высыхания суспен-
зии поверх препарата напыляем угольную пленку. Угольная пленка выполняет
роль подложки, на которой удерживаются частицы исследуемого объекта. После
напыления стекло, соль или слюда осторожно помещаются в чашку с водой для от-
деления суспензии. При этом реплика с суспензией остается плавать на поверхно-
сти воды. Затем полученный препарат вылавливают на медную сеточку и помещают
для просмотра в электронный микроскоп. При исследовании суспензии наблюдают
непосредственно сами частицы вещества: их размер, морфологию, толщину,
однородность.
Метод ультратонких срезов.
Сущность метода заключается в получении тонких срезов исследуемого ве-
щества толщиной 100-500А, которые затем просматриваются на электронном мик-
роскопе. Однако, в минералогии вследствие специфики объектов, а также ввиду
своей сложности и трудоемкости, этот метод применяется ограниченно.
Поэтому основное применение этод метод имеет в металловедении (тон-
кие пленки), в биологии, медицине (срезы тканей). Резка мягких образцов произ-
водится с помощью стеклянных ножей, а для получения срезов с твердых мате-
риалов (в том числе и в минералогии) применяются алмазные.
Для получения среза образцы обычно затачивают в виде прямоугольной
пирамиды (с площадью сечения 0,1 мм). Процесс резки осуществляется путем пе-
ремещения образца навстречу режущей кромке ножа. Прибор, на котором получа-
ют ультратонкие срезы, называется ультрамикротомом. Недостатком метода ульт-
137
ратонких срезов является неизбежная деформация образца, искажающая реальную
картину микроморфологии объекта, вследствие чего этот метод в минералогии не
получил широкого распространения.
Косвенные методы исследования.
Эти методы применяются для исследования на просвечивающем электрон-
ном микроскопе поверхности массивных, непрозрачных для электронов объектов.
Сущность этих методов заключается в том, что в электронном микроскопе
для просмотра помещается не сам объект, а отпечаток с его поверхности, толщина
которого не должна превышать 100-500А, чтобы сквозь него мог проходить элек-
тронный луч. Этот отпечаток с поверхности называется репликой. Основное требо-
вание к реплике состоит в том, чтобы она была тонкой, обладала хорошей разре-
шающей способностью и максимально точно отражала микроструктуру поверхно-
сти образца. Кроме этих требований необходимо, чтобы реплика была механически
прочная, электронейтральная, вакуумостойкая, термостойкая. Всем этим требования
отвечает угольная реплика.
Для получения реплик - отпечатков на свежий скол поверхности образца на-
носится тонкая пленка (100-300А) .какого-либо вещества, на котором отпечатывает-
ся микрорельеф поверхности. Затем этот отпечаток тем или иным способом отде-
ляется от образца и просматривается на электронном микроскопе. Рельеф поверх-
ности не должен быть слишком грубым, чтобы не рвалась реплика, но и не абсо-
лютно гладким, иначе невозможно получить какую-либо информацию об объекте.
138
При приготовлении реплик с полированной поверхности (аншлифов) или с очень
гладкой поверхности объекта ее сначала протравливают, тщательно подбирая
режим травления, чтобы получить рельеф, соответствующий данному образцу.
В минералогии применяется, в основном, метод одноступенчатых угольных
реплик. Изготавливаются они следующим образом. Образец со свежим сколом по-
мещается в напылитель. Это прибор, в котором под вакуумным колпаком разме-
щаются угольные стержни, заточенные под определенным углом, чтобы создать
между ними плотный контакт в точке. С помощью насоса под колпаком создается
вакуум, а затем через соприкасающиеся графитовые стержни пропускается ток 60-
80 ампер. При сильном разогревании стержней происходит термическое испарение
угля в вакууме. При этом тонкая пленка испаряющегося угля покрывает поверх-
ность образна, повторяя все неровности микрорельефа поверхности. Таким обра-
зом, рельеф образца как бы отпечатывается на угольной пленке. Затем эта плен-
ка отделяется от образца и помещается для просмотра в электронный микроскоп.
Такие реплики называются одноступенчатыми угольными репликами. Они облада-
ют хорошей прозрачностью для электронов, химической инертностью по отноше-
нию к различным кислотам; прочностью, высокой разрешающей способностью.
Толщина такой пленки не должна превышать ЮО-ЗООА. При более толстых репли-
ках ухудшается один из главных параметров электронного микроскопа - его разре-
шающая способность.
Достаточно зрудоемкой является задача отделения этой угольной пленки
от образца. Это может быть сделано двумя способами:
139
а) химический способ - в этом случае образец помещается в соответствую-
щую кислоту для растворения. Образец растворяется, при этом угольная реплика,
которая, как говорилось выше, инертна к химическим кислотам, при растворении
образца всплывает на поверхность. Эту реплику вылавливают на сеточку, промыва-
ют и просматривают на электронном микроскопе. Этот способ не очень удачен тем,
что образец пропадает при растворении, и невозможно повторить его исследование
или передать его на изучение другими методами;
б) вторым, наиболее употребительным способом, является отделение уголь-
ной пленки (реплики) от образца с помощью желатина, который выполняет роль
клея. Горячий желатин (жидкий) капают на напыленную поверхность образца. При
охлаждении желатин застывает. Через 24 часа застывший желатин отделяют пинце-
том от образца. При этом реплика отделяется от образца вместе с желатином. За-
тем реплику с желатином опускают в теплую воду (обязательно желатином вниз).
При нагревании воды желатин растворяется, а реплика остается плавать на поверх-
ности. Полученную реплику вылавливают и исследуют в электронном микроскопе.
Эгот способ более прост, и, самое главное, сохраняет образец, что позволяет прово-
дить повторные исследования.
Существует множество других, дополнительных методов исследования
минералов на электронном микроскопе, которые применяются в зависимости от
поставленных задач.
140
ЛИТЕРАТУРА
1. Методы минералогических исследований. Под ред. А.И.Гинзбурга.
Изд-во "Недра", М., 1985.
2. Методы электронной микроскопии минералов. Грицаенко Г.С. и др.
Изд-во "Наука", М., 1969.
3. Н.Е.Сергеева. Введение в электронную микроскопию минералов.
Изд-во Моск, ун-та, 1977.
141
ОГЛАВЛЕНИЕ
Стр.
Введение..................................................1
Дифракционные методы исследования.........................3
Рентгенография...........................................12
Методы рентгенографии...................................24
Минералогические задачи, решаемые методом рентгенографии . . 34
диагностика минералов..................................34
определение парамеров элементарной ячейки минерала.....36
реитгенофазовый анализ.................................38
количественный рентгенофазовый анализ..................40
изучение изоморфизма минералов.........................41
изучение характера фазовых превращений минералов.......44
определение степени совершенства кристаллической структуры
вещества.................................................46
Литература...............................................49
Методы элементного анализа................................50
Метрологические параметры методов элементного анализа.50
Ретгеноспектральные методы.............................53
ретгеноспектральный флюоресцентный метод..............53
микрорентгеиоспектральиый анализ......................66
рентгенорадиометрический метод анализа.................73
142
Оптический спектральный анализ.........................74
эмиссионный спектральный анализ......................76
атомно-абсорбционный спектральный анализ.............81
Ядерно-физические методы...............................84
активационный анализ.................................85
метод радиографии....................................93
Литература.............................................119
Электронная микроскопия...............................120
Литература............................................141
Подписано в печать 14.04. 2009г. Объем 9,0 п.л.
Тираж 100 экз. Заказ № 9
Редакционно-издательский отдел РГГРУ
Москва, ул. Миклухо-Маклая, 23