/


































































































































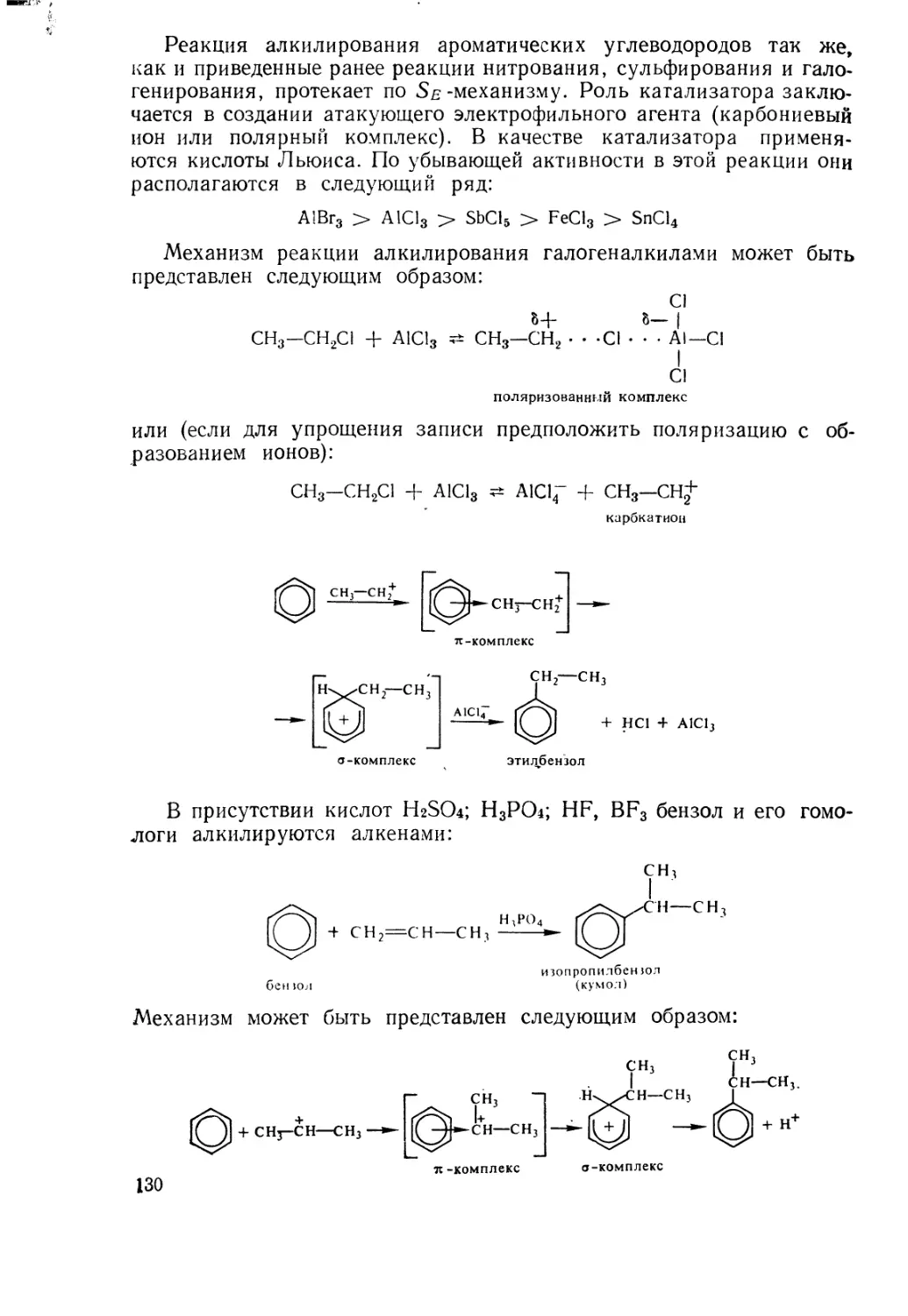












































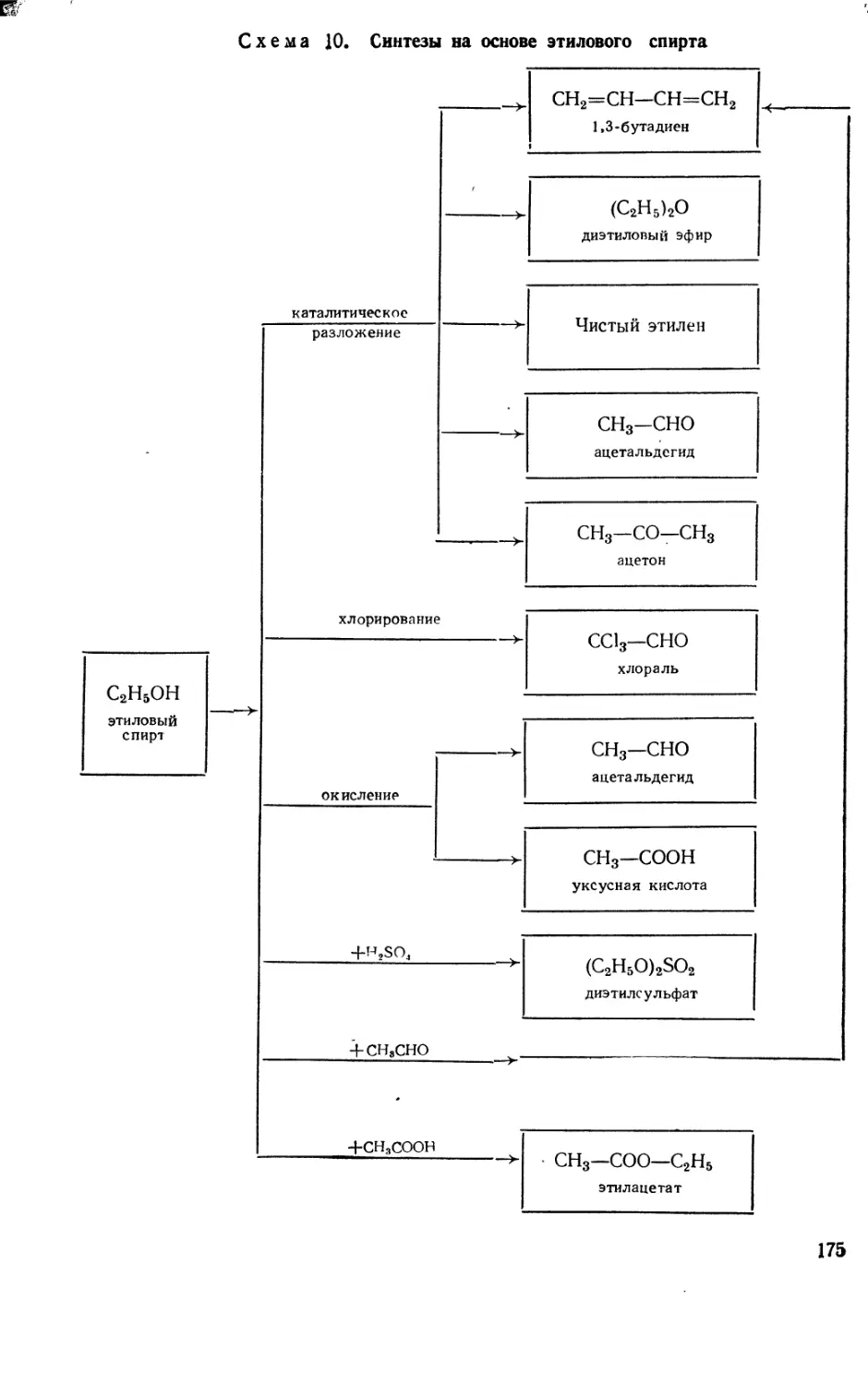






































































































































































































































































































Text
А. П. НЕЧАЕВ, Т. В. ЕРЕМЕНКО
Органическая
химия
-
для технологических
специальностей
пищевых вузов
А. П. НЕЧАЕВ, Т. В. ЕРЕМЕНКО
Органическая
химия
Допущено
Министерством высшего и среднего
специального образования СССР
в качестве учебника
для студентов,
обучающихся по специальностям
технологии продовольственных продуктов
МОСКВА ♦ВЫСШАЯ ШКОЛА» 1985
ББК 24.2
Н59
УДК 547
Рецензенты:
кафедра органической химии Киевского технологического института пище-
вой промышленности (зав. кафедрой прэф. А. А. Герасименко) и проф.
И. И. Грандберг (Московская сельскохозяйственная академия им. К-А. Ти-
мирязева)
Нечаев А. П., Еременко Т. В.
Н59 Органическая химия: Учебник для пищ. ин-тов. — М'.:
Высш, шк., 1985. — 463 с., ил.
В пор.: 1 р. 30 к.
В основу построения учебника положен принцип классификации органических
соединений по функциональным группам. В учебнике широко используются элек-
тронные представления, излагаются современные взгляды на механизмы важнейших
органических реакций.
Учебник состоит из двух частей. Ч. 1. Углеводороды. Функциональные произ-
водные углеводородов. Элементорганические соединения. Ч. 2. Азотсодержащие ор-
ганические соединения. Гетероциклические соединения. Биоорганические соединения
и некоторые соединения изопреноидного характера. Фактический материал дается
в соответствии с программой по курсу органической химии для студентов техноло-
гических специальностей пищевых вузов.
1803000000—215
001(01)—85
85—86—85
ББК 24.2
547
© Издательство «Высшая школа'», 1985
Предисловие
Предлагаемая книга предназначается в ка-
честве учебника по органической химии для учащихся высших учеб-
ных заведений, готовящих специалистов для системы заготовок,
пищевой и микробиологической отраслей промышленности. Совре-
менная технология хранения и переработки сельскохозяйственного
сырья, получение из него пищевых продуктов требуют глубоких зна-
ний химических и биохимических процессов, протекающих на всех
этапах производства, начиная от заготовок сырья, кончая получением
из него готовых продуктов и их хранением. Питательная ценность
последних определяется составом, количеством и соотношением со-
держащихся в них белков, углеводов, липидов, витаминов и других
соединений. Не меньшее значение имеет знание органической химии и
для инженеров-технологов микробиологической промышленности,
призванной обеспечить получение сложных органических соедине-
ний — белков, ферментов, липидов, витаминов, аминокислот и т. д.
Особое значение все эти вопросы приобретают в связи с реализа-
цией Продовольственной программы, призванной теснее сомкнуть
сельское хозяйство с отраслями, занимающимися хранением и пере-
работкой его продукции.
Курс органической химии излагается на основе современных тео-
ретических представлений в соответствии с утвержденной программой.
Авторы уделили особое внимание общим закономерностям, останав-
ливаясь в то же время на свойствах наиболее важных органических
соединений. Подробно рассматриваются те соединения, которые со-
ставляют основную массу органического вещества пищевого сырья
и готовой продукции (карбоновые и аминокислоты, углеводы, липиды,
белки).
В основу построения учебника положена классификация органи-
ческих соединений по функциональным (характеристическим) группам.
Большинство теоретических представлений излагается по мере опи-
сания основных классов органических соединений, что, по мнению
авторов, способствует их лучшему восприятию.
Учебник состоит из двух частей. В первой части излагаются све-
дения об углеводородах, их функциональных производных и элемент-
органических соединениях; вторая часть посвящена азотсодержащим
органическим соединениям, гетероциклическим соединениям, био-
органическим соединениям и некоторым соединениям изопреноид-
ного характера, здесь же даются сведения о методах выделения, очист-
ки и идентификации органических соединений.
Разделы «Введение», «Углеводороды», «Галогенпроизводные»,
«Спирты и фенолы», «Оксосоединения», «Стереохимия», «Карбоно-
вые кислоты и их производные», «Оксикислоты», «Липиды», «Белки»,
«Изопреноиды» написаны проф., д-ром техн, наук А. П. Нечаевым;
3
главы «Альдегидо- и кетокислоты», «Элементорганические соеди-
нения», «Азотсодержащие органические соединения», «Гетероцикли-
ческие соединения» и «Углеводы» — доц., канд. хим. наук Т. В. Ере-
менко; раздел книги, посвященный выделению и анализу органиче-
ских соединений, — доц., канд. хим. наук С. И. Петровым. Материал
учебника, посвященный природе ковалентной связи и квантовомеха-
ническим методам расчета в органической химии, взят, с согласия
автора, из учебника И. И. Грандберга «Органическая химия» (М.,
Высшая школа, 1980).
Авторы выражают глубокую признательность рецензентам ру-
кописи учебника проф., д-ру хим. наук И. И. Грандбергу, проф.
А. А. Герасименко, доц. Д. А. Бороде, а также проф., д-ру хим. наук
В. Г. Кульневичу, проф., д-ру техн, наук М. П. Попову, доц., канд.
хим. наук Н. В. Зотчик и сотрудникам кафедры органической хи-
мии Московского технологического института пищевой промышлен-
ности, просмотревшим рукопись, за ценные замечания и советы. За
большую помощь в работе над рукописью авторы сердечно благодарят
канд. техн, наук А. А. Кочеткову.
Авторы заранее признательны за все полезные критические за-
мечания и пожелания, которые позволят улучшить учебник.
Авторы
Часть
1
Н 2
н-с-с=с-н
ИНН
Углеводороды
Функциональные
производные
углеводородов
Введение
Органическая химия и причины выделения ее в
самостоятельную дисциплину. Органическая
химия изучает соединения углерода с дру-
гими элементами (органические соединения) и
законы, которым подчиняются превращения
этих веществ.
Чем вызвано, что изучение соединений толь-
ко одного элемента — углерода — составляет
содержание целой науки? Причины в много-
численности и разнообразии органических со-
единений, в их специфическом строении и свой-
ствах, особенностях протекания органических
реакций, а также в большом практическом
значении соединений углерода.
Число известных органических веществ со-
ставляет несколько миллионов. Некоторые про-
стые соединения углерода, такие, как оксиды
углерода, соли угольной и синильной кислот,
изучаются в курсе неорганической химии. Не-
органических веществ известно значительно
меньше (приблизительно 500 тысяч).
Многочисленность соединений углерода вы-
звана рядом причин: способностью его соеди-
няться с атомами большинства других элемен-
тов; образовывать углерод-углеродные цепи
различного строения с практически неограни-
ченным числом атомов; соединяться в кольца
или циклы:
I I I I I I I I I I
—С—С—С—С—С— — С—С—С—С—С—
1,111 1 ии 1
нормальная углер щ-угле- ~
родная цепь |
разветвленная углерод-
углеродная цепь
Элемент-
органические
соединения
циклическая углерод-уг-
леродная цель
5
Атомы углерода способны образовывать друг с другом не только
одинарные, но и двойные, и тройные связи (каждая черточка услов-
но обозначает одну связь):
одинарная связь
двойная связь
—с=с—
тройная связь
Органические соединения — это газообразные, жидкие или твер-
дые вещества. Температура плавления твердых органических ве-
ществ обычно не превышает 400°С. Многие органические вещества
горючи. Большинство же неорганических соединений — твердые, не-
горючие, высокоплавкие вещества.
Большая часть реакций, в которых участвуют органические со-
единения, протекает медленно, часто с невысоким выходом. Реакции
в водных растворах между неорганическими электролитами протека-
ют мгновенно, с количественным выходом.
Органические соединения сложнее неорганических. Органическая
химия имеет дело с более высокоорганизованной материей.
Соединения углерода имеют большое практическое значение: нефть,
природный газ, пластмассы, каучук, синтетические и искусственные
волокна, красители, органические соединения, применяемые в сель-
ском хозяйстве (инсектициды, фунгициды, гербициды, ростовые
вещества), медицинские препараты, витамины, ферменты и др.
Созданные в последние десятилетия искусственные полимерные
вещества (пластмассы, каучуки, волокна, пленки, полимерные по-
крытия) не только восполнили недостаток в традиционных материа-
лах, известных человеку с давних пор, но благодаря многим ценным
свойствам способствовали прогрессу в ряде отраслей промышлен-
ности. Применение органических соединений в сельском хозяйстве
привело к резкому росту урожайности, снижению себестоимости и
увеличению выпуска сельскохозяйственной продукции. Использо-
вание синтетических лекарственных препаратов в медицине избавило
человека от многих недугов и явилось одной из причин увеличения
средней продолжительности жизни. Органическая химия прочно во-
шла в жизнь человека. Сейчас невозможно представить его сущест-
вование без всех перечисленных соединений.
Все это и было причиной выделения органической химии в само-
стоятельную науку.
В то же время было бы неправильным считать, что между органи-
ческими и неорганическими соединениями существует четкая гра-
ница. «Подобно тому как одна форма движения развивается из дру-
гой, — пишет Ф. Энгельс, — так и отражение этих форм, различные
науки, должны с необходимостью вытекать одна из другой»*.
Краткий исторический очерк развития органической химии. Чело-
век знаком с органическими соединениями очень давно. Его пища,
одежда, топливо и другие предметы первой необходимости состоят
* Энгельс Ф. Диалектика природы. — Маркс К., Энгельс Ф. Соч. 2-е изд.,
т. 20, с. 565, М., 1°61.
6
из органических веществ. Постепенно человек приобретал большой
практический опыт по переработке и использованию природного орга-
нического сырья. Он научился делать пищу более вкусной, способ-
ной храниться продолжительное время, получать пьянящие напитки
(вино, пиво, брагу, напитки из меда), дубить кожу, варить мыло,
красить ткани, приготовлять целебное питье, получать мази. Это были
первые химические производства на Земле, созданные человеком.
Этот период развития химии получил название древнейшего и продол-
жался приблизительно до IV в. нашей эры. И хотя в это время был
накоплен большой практический опыт, химии как науки, по сущест-
ву, не было.
Последующий период (приблизительно до XVI в.) получил на-
звание периода алхимии. Основное внимание исследователи в это
время уделяли поискам «философского камня» — особого вещества,
с помощью которого можно было бы превратить неблагородные ме-
таллы в благородные, в первую очередь в золото; получению уни-
версального растворителя; эликсира долголетия^ Химические соеди-
нения разделяли в этот период на группы по их внешним свойствам.
Так, например, в группу масел (маслообразных жидкостей) включа-
лись серная кислота и оливковое масло. Названия некоторых соеди-
нений, данные им в тот период, сохранились и до наших дней: купо-
росное масло (концентрированная серная кислота), нашатырный
спирт — водный раствор аммиака.
Начиная с конца XVI в., темпы развития химии резко возраста-
ют, накапливается обширный материал о свойствах и элементном сос-
таве отдельных соединений (М. В. Ломоносов, Лавуазье). Значи-
тельные успехи были достигнуты и в изучении органических соедине-
ний. В начале XIX в. выдающийся шведский ученый И. Берцелиус
(1779—1848), отмечая ряд особенностей органических соединений,
выделил органическую химию в самостоятельную науку. Он, как
и его современники, считал, однако, что органические вещества мо-
гут быть получены только в живом организме, с помощью «жизненной
силы». Это идеалистическое представление об участии «души» или
«силы» в явлениях жизни, в том числе и в синтезе органических со-
единений, получила название витализма, от латинского «vitalis» —
жизненный. Но уже при жизни Берцелиуса успехи органической хи-
мии наносят сокрушительный удар этим представлениям. В 1828 г.
ученик Берцелиуса — Вёлер синтезировал из неорганических ве-
ществ (цианистого калия и сульфата аммония) органическое ве-
щество — мочевину. В 1842 г. наш соотечественник Н. Н. Зинин
получил из нитробензола анилин, который до этого выделяли из
природного красителя. Французский ученый Бертло (1854) синте-
зировал вещество, относящееся к классу жиров, а в 1861 г. знамени-
тый русский химик А. М. Бутлеров получил из параформальдегида
сахароподобное соединение. Стало ясно, что органические вещества
могут быть получены синтетическим путем.
Современный период развития органической химии начинается
с 60-х годов XIX в., когда была создана А. М. Бутлеровым теория
химического строения органических соединений. С этого времени
7
начинается ее бурное развитие. За последующие годы органическая
химия добилась выдающихся успехов.
т; На основе новейших достижений физики дальнейшее развитие по-
лучила теоретическая органическая химия. Применение электронных
представлений дало возможность по-новому подойти к вопросу о стро-
ении органических соединений, их поведении в химических реакциях,
понять механизм их превращений. В настоящее время все большее
значение для понимания строения и поведения органических соеди-
нений начинает приобретать применение методов квантовой механики.
Развитие теоретических представлений, разработка разнообразных
способов получения органических соединений, успехи в области ка-
тализа, внедрение физико-химических методов исследования, новых
методов выделения и очистки веществ привели к бурному развитию
органического синтеза, в том числе сложных природных соединений
(витаминов, алкалоидов, антибиотиков, липидов, углеводов). Син-
тезирован ген; человечество вплотную подошло к синтезу белка. В
промышленности в больших масштабах ведется синтез полимерных
материалов с разнообразными, в отдельных случаях не известными
ранее свойствами, красителей, поверхностно-активных веществ и
многих других соединений. Отдельные разделы органической химии
развиваются настолько интенсивно, что выросли в самостоятельные
разделы: биохимия, химия высокомолекулярных соединений, элемент-
органических соединений, красителей, белков, липидов, углеводов,
витаминов, ферментов и т. д.
Достижения органической химии открыли большие возможности
для совершенствования технологии и повышения эффективности ра-
боты в системе заготовок и пищевой промышленности. Понимание
химизма протекающих процессов, введение разнообразных химиче-
ских добавок способствовало повышению качества пищевых продук-
тов, позволило лучше их сохранить, снизить себестоимость. Было
организовано производство синтетического спирта, моющих средств,
синтетического клея, что помогло высвободить большие количества
пищевого сырья и продуктов, которые раньше расходовались на тех-
нические нужды. Создание новых полимерных материалов дало воз-
можность не только усовершенствовать ряд машин и технологических
процессов, но и создать принципиально новые виды дешевых упа-
ковочных материалов, повышающих сохранность пищевых продуктов.
Исключительное значение для получения органических соединений
(белка, ферментов, витаминов, антибиотиков и др.) сыграло развитие
микробиологической промышленности.
Своими успехами органическая химия во многом обязана трудам
таких выдающихся русских и советских химиков-органиков, как
Н. Н. Зинин, А. М. Бутлеров, Е. Е. Вагнер, В. В. Марковников,
Н. Д. Зелинский, А. Е. Фаворский, С. В. Лебедев, М. Г. Кучеров,
А. Н. Несмеянов и многих других.
Основными направлениями экономического и социального раз-
вития СССР на 1981—1985 гг. и на период до 1990 г., принятыми
на XXVI съезде КПСС, предусматривается дальнейшее развитие хи-
мической и нефтехимической промышленности. Должен быть значи-
8
тельно увеличен выпуск пластических масс и синтетических смол,
каучуков, химических волокон и нитей, консервантов и антисептиков,
биологически активных веществ для медицинских нужд и сельского
хозяйства, реактивов, различных пленок для расфасовки пищевых про-
дуктов, химических заменителей жиров и другого пищевого сырья,
расходуемого на технические цели, синтетического глицерина, кра-
сителей, лаков, вспомогательных веществ для улучшения качества тка-
ней, трикотажных изделий, обуви и искусственной кожи.
Элементный состав, источники и способы получения органиче-
ских соединений. Находясь в четвертой группе периодической таб-
лицы элементов Д. И. Менделеева, углерод может образовывать со-
единения практически со всеми элементами периодической системы.
Однако в состав большинства органических соединений входят атомы
небольшого числа элементов: углерода, водорода, кислорода, азота,
серы, фосфора, галогенов. Они получили название органогенов. Из
перечисленных элементов, кроме углерода, который, естественно,
входит в состав всех соединений, практически в каждом из них при-
сутствует водород, широко распространены кислород, азот.
Первые органические вещества были выделены человеком из рас-
тительных и животных организмов. Постепенно все большую роль в
их получении начинают играть продукты коксования каменного угля,
нефть и химический синтез. Перечень основных источников органи-
ческих веществ, расположенных по их значимости, выглядит следую-
щим образом. Природные источники: нефть, природные и попутные
газы, каменный уголь и сланцы, древесина, продукты сельского хо-
зяйства. Синтетические методы: химический синтез и микробиологи-
ческий синтез.
Появившийся в последние годы микробиологический синтез ста-
новится все более важным источником органических веществ. Способ-
ность микроорганизмов синтезировать сложнейшие органические со-
единения, многие из которых пока еще не удается создать химическим
путем, используется для получения белка, ферментов, липидов, ви-
таминов, антибиотиков, аминокислот.
Синтетическим путем можно не только воспроизвести сложнейшие
соединения, создаваемые природой, но получить новые, ранее не из-
вестные, с лучшими, чем природные, свойствами.
Способы получения органических соединений можно разделить
на: выделение из природных источников; утилизацию отходов произ-
водства; синтетические (промышленные и лабораторные) и т. д. Такое
деление весьма условно, так как на практике часто выделяют нуж-
ный компонент из природных источников, а затем «достраивают» его
химическим путем, превращая в необходимое соединение. Методы,
разработанные для получения органического вещества в лабораториях,
по мере роста потребностей народного хозяйства в этом соединении
применяются в промышленности. Однако каждый химик и технолог
должен четко представлять разницу в требованиях, предъявляемых
к промышленным и лабораторным методам получения.
Промышленные способы связаны с получением значительных ко-
личеств вещества, поэтому при их разработке большое внимание сле-
9
дует уделять наличию и доступности исходного сырья, его стоимос-
ти, возможности осуществления непрерывного и автоматизированного
производства, использованию побочных продуктов и отходов произ-
водства, безопасности труда.
Лабораторные методы обеспечивают получение чистого вещества
в небольших количествах с максимальной быстротой. Для каждого
класса органических соединений существуют свои характерные или,
как их иногда называют, «общие» способы получения. Теоретически
этими способами можно получить большинство из представителей
данного класса веществ. Однако на практике каждый из этих способов
удобен для получения только небольшой группы соединений, что свя-
зано с различиями в скоростях реакций, приводящих к их
образованию, незначительными выходами, трудностями выделения
нужного соединения из смеси образовавшихся продуктов реакции и
другими причинами. /
При описании методов получения органических соединений они
будут разделены на промышленные и лабораторные (учитывая ус-
ловность этого деления). В отдельных случаях будут приведены ме-
тоды получения, не имеющие большого практического значения,
но представляющие исторический интерес или способствующие раз-
витию у читателя химического мышления.
Краткие сведения о развитии теоретических представлений в ор-
ганической химии. Первой теорией строения химических соединений
была электрохимическая теория Берцелиуса, примененная к орга-
ническим соединениям в виде теории радикалов (Берцелиус, Либих,
Вёлер; первая треть XIX в.). Авторы ее обратили внимание на то,
что во многих превращениях органических веществ одна из частей мо-
лекулы, названная ими «радикал», не изменяется, а переходит из ис-
ходного соединения в продукт реакции. Молекула органических со-
единений, по мнению авторов этой теории, состоит из двух противо-
положно заряженных радикалов, связанных электростатическим вза-
имодействием. Однако постепенно накапливались экспериментальные
данные, которые не нашли объяснения с позиций «теории радикалов»,
и в 40-х годах XIX в. на смену ей приходит «теория типов», созданная
Жераром и Лораном. Они, в противоположность сторонникам теории
радикалов, основное внимание уделяли той части молекулы органи-
ческого соединения, которая изменяется в процессе реакции. Авторы
«теории типов» считали, что существует глубокое сходство в поведе-
нии органических и неорганических веществ. По их мнению, органи-
ческие вещества можно рассматривать как производные неорганиче-
ских, образованных замещением атомов в молекуле на радикалы,
которые они назвали «остатками». Полученные при этом органические
вещества вступают во все реакции, свойственные неорганическим
веществам. Они выделяли четыре основных типа: тип водорода, хло-
ристого водорода, воды, аммиака:
н
н)
Н
Н
Н 1
Cl J
Н'
Н N
н,
10
Например,
Hi
о
н J
вода
к представителям «типа воды»
ими были отнесены:
Н 1
О
СНз j
мети левый
спирт
СН3 1
О
CH3J
ди метиловый
эфир
С2Н3О1
0
Н J
уксусная
кислота
С2Н3О 1
°
с2н3о J
уксусный
анг идрид
Расположив органические соединения по типам, авторы не только
определили место для большинства известных в то время химических
соединений, но по аналогии указали и некоторые способы получения
еще неизвестных представителей отдельных групп. Однако сторонни-
ки этой теории стояли на идеалистических позициях. Так, они счита-
ли, что строение молекул органических соединений не может быть
познано в ходе химического эксперимента (агностицизм).
К 60-м годам прошлого века в органической химии накопился
большой экспериментальный материал, ставящий под сомнение мно-
гие положения теории типов. К этому же времени было сформулиро-
вано положение о четырехвалентности атома углерода (Кольбе, Ке-
куле), его способности соединяться с другими углеродными атомами
с образованием углерод-углеродных цепей (Кекуле, Купер). Шот-
ландский ученый Купер предложил систему изображения строения
химических соединений, в которой связи указываются с помощью
черточек.
Возникла острая необходимость в создании научной теории строе-
ния органических соединений. Автором такой теории стал выдающий-
ся русский химик А. М. Бутлеров (1828—1886).
Теория химического строения органических соединений А. М. Бут-
лерова. Теория химического строения органических соединений была
наиболее полно изложена А. М. Бутлеровым в его труде «Введение
к полному изучению органической химии». Ученик А. М. Бутлерова
В. В. Марковников писал впоследствии, что эта книга составила
«эпоху в развитии теоретических представлений, положенных в ос-
нову современной химии и... открывала обширный горизонт для со-
вершенно новых исследований». Ее возникновение именно в России
не было случайностью. В стране в это время происходил подъем рево-
люционно-демократического движения, бурно развивались естествен-
ные науки.
В своей теории А. М. Бутлеров выступает как ученый-материалист,
он признает реальность существования атомов и молекул и воз-
можность познания в ходе эксперимента внутренней структуры орга-
нического соединения. Сущность теории А. М. Бутлерова состоит в
следующем:
1. Свойства органических веществ определяются не только при-
родой и количеством входящих в состав молекулы атомов, но и по-
рядком их соединения, получившим название химического строения,
который заключается в последовательности соединения атомов и
характере соединяющих их связей.
Из понятия химического строения вытекает объяснение явления
изомерии, изученное А. М. Бутлеровым.
2. Химическая структура молекулы может быть представлена
и
только одной рациональной формулой, выражающей основные хи-
мические свойства данного вещества.
Это положение имеет большое практическое значение, оно дало
возможность на основании формулы органического соединения пред-
сказать его свойства и поведение в химических реакциях.
3. Атомы, входящие в молекулу органического соединения, ока-
зывают взаимное влияние друг на друга и на ее химическое поведение.
Вопросы взаимного влияния атомов в молекуле органического
соединения были подробно изучены учеником А. М. Бутлерова
В. В. Марковниковым.
Сформулированные А. М. Бутлеровым положения легли в основу
современной теоретической органической химии, которая по существу
и начинается с его работ. А. М. Бутлеров создал большую химическую
школу, представители которой, развивая дальше теорию химического
строения, создали новые,оригинальные научные направления.
Краткие сведения о химической связи. Теория химического строе-
ния органических соединений А. М. Бутлерова не касалась природы
сил, обеспечивающих связь между атомами в молекуле органических
соединений и свойств этой связи. Достижения науки дают возможность
ответить на эти вопросы. По современным представлениям химическая
связь возникает в результате взаимодействия атомов, сопровождаю-
щегося перестройкой их электронных оболочек.
При написании структурных формул органических соединений
связи между атомами обычно изображаются в виде черточек. Каждая
черточка символически изображает единицу валентности.
Химические связи делятся на типы по принципу их электронного
строения.
Гетерополярная (электровалентная, ионная ) связь. Большинство
неорганических соединений построены с помощью электровалентной,
или ионной, связи. При образовании гетерополярной связи происхо-
дит передача электрона от одного атома к другому, причем первый
превращается в положительно заряженный ион (катион), второй — в
отрицательный заряженный ион (анион). Схематично это можно пред-
ставить следующим образом:
А^+ШГ --А+ + ’.В
(точками условно изображены валентные электроны атомов). Две
разноименно заряженные частицы связываются силами электроста-
тического взаимодействия. Например:
Na* 4- :ci: -Na+ + СГ
Образовавшийся ион натрия обладает электронной конфигураци-
ей неона, а ион хлора — конфигурацией аргона, следовательно, оба
иона имеют устойчивый октет электронов на внешней оболочке. Ион-
ная связь характерна для атомов с резко различной электроотрица-
тельностью (т. е. с различной способностью притягивать электроны).
12
Ионная связь обладает рядом особенностей: значительной поляр-
ностью, не имеет определенного направления в пространстве, а молеку-
лы соединений с такой связью отличаются способностью к диссоциа-
ции на ионы и сольватацией последних в высокополярных раствори-
телях; высокой скоростью протекания реакций с их участием; раство-
римостью в полярных растворителях; электропроводимостью раство-
ров и расплавов; высокими температурами плавления и кипения.
В молекулах, построенных с помощью гетерополярной связи,
заряженные частицы (ионы) не находятся в жестком (фиксированном)
положении друг относительно друга, поэтому в последнее время часто
употребляют термин не «ионная связь», а «ионное взаимодействие».
Ковалентная (гомеополярная) связь. Большинство молекул орга-
нических соединений построены с помощью ковалентной связи, воз-
никающей за счет образования одной или нескольких электронных
пар, которые становятся общими для взаимодействующих атомов:
А’ 4- В‘ -> А: В
Ковалентную связь образуют атомы, обладающие близкой или
равной электроотрицательностью. Возникает единое электронное
облако, плотность которого между ядрами особенно велика, что обес-
печивает устойчивость молекулы. Кроме водорода, с помощью кова-
лентной связи построены молекулы хлора, азота, хлороводорода и
ряда других неорганических соединений, а также и большинство орга-
нических соединений.
Возникновение устойчивой электронной конфигурации инертного
газа для таких атомов происходит за счет обобществления двух, че-
тырех или шести электронов. Каждая из обобществленных пар элект-
ронов образует одну гомеополярную (ковалентную) связь:
Н* + Н- -> Н:Н
н—н
н
Н:С:Н
Н
Н Н
С: :С
Н Н
Н
I
Н—С—Н метан
I
Н
н н
С — С этилен
Н Н
Н:С:::С:Н Н—С = С—Н ацетилен
Атомы, связанные ковалентной связью, обладают полным окте-
том (в случае водорода — дублетом) электронов, так как электронная
пара ковалентной связи принадлежит в равной мере обоим связанным
ею атомам. Изображают ковалентную связь двумя точками (пара
электронов), либо черточкой. Имеющиеся в молекулах у отдельных
атомов электронные пары, не принимающие участия в образовании
ковалентной связи, называются свободными или неподеленными
электронными парами.
13
Во многих случаях образование ковалентной связи происходит
за счет обобщения электронов, ранее принадлежавших двум соеди-
няющимся атомам, но возможен и другой механизм. Она может обра-
зоваться за счет электронной пары только одной из реагирующих
частиц. Такая ковалентная связь называется координационной связью
или донорно-акцепторной. Атом или ион, отдающий свою электрон-
ную пару для образования связи, называется донором, обобщающий
чужую электронную пару — акцептором. Так, при взаимодействии
аммиака с соляной кислотой протон соединяется с аммиаком за счет
неподеленной пары электронов атома азота:
NH.3 + НС! -> NH4C1
Н Г Н Г
H:N: + Н+ -> H:N:H
Н L Н _
аммиак ион аммония
Заряд в этом случае находится не на присоединившемся протоне, а
на атоме азота иона аммония. Возникшая новая N—Н-связь ничем
не отличается от остальных ковалентных связей, которые имелись в
молекуле аммиака. Возможен случай присоединения не иона, а нейт-
рального атома; при этом на атоме, отдающем электронную пару, воз-
никает положительный заряд, а на присоединяющемся атоме — от-
рицательный. В результате два атома оказываются связанными как
бы двумя способами: ковалентной связью с помощью отданной элект-
ронной пары и электровалентной связью:
А: + В -> А+ : В“
Это характерно, например, для реакции трехфтористого бора с аммиа-
ком:
F Н F Н
F:B + ;N: н -> F:"B :N+;H
F Н F Н
молекула с не-
заполненным
электронны м
слоем (дефицит
электронов)
Иные способы изображения:
BF3-<-NH3 или BF3-Sh3
Такая координационная связь называется семиполярной связью (от
латинского «semi» — полу). Семиполярная связь влияет на свойства
соединений.
Ковалентная связь может быть охарактеризована длиной связи
(межъядерным расстоянием), направленностью в пространстве, энер-
гией образования, полярностью, поляризуемостью.
Длина связи может быть определена различными физическими
методами (по интерференции рентгеновских лучей, электронов или
14
нейтронов; на основании спектроскопических исследований в микро-
волновой и ПК областях, а также спектров комбинационного рассея-
ния света). Ниже приведены некоторые значения длин связей (нм):
С—Н (СН4)
С== С (этилен)
С= С (ацетилен)
0,154 С—F
0,133 С—С1
0,120 С—I
0,138
0,176
0,213
Одной из особенностей ковалентной
ная пространственная направленность,
теризована с помощью валентного
угла (угол между направлениями свя-
зей, образуемых многовалентным ато-
мом). Валентный угол между связями
атома углерода зависит от типа гиб-
ридизации (см. с. 26), вида атома* с
которым он связан. Определение ва-
лентных углов производят рентгено-
графическим, электронографическим
связи является ее определен-
которая может быть охарак-
модель
Рис. 1. Пространственная
молекулы метана
и спектральными методами. В моле-
куле метана СН4 ($р3-гибридизация)
угол между направляющими связей
109°28', т. е. атомы водорода распола-
гаются в углах правильного тетраэдра
ческом строении молекулы метана впервые была высказана более ста
(рис. 1). Гипотеза о тетраэдри-
лет назад французским ученым Ле-Белем и голландским исследователем
Вант-Гоффом и послужила основой для создания раздела органиче-
ской химии, называемого стереохимией. В молекуле этилена (sp2-
гибридизация) валентный угол между о-связями 120°. В молекуле
метиленхлорида CH2CI2 угол между с равен 111,8° и т. д.
\ci
В оценке химической связи большая роль принадлежит энергии
связи, которая определяет ее прочность и характеризуется энергией,
затрачиваемой на разрыв связи (кДж/моль).
Полярность связи обусловлена несовпадением центра отрицатель-
ных и положительных зарядов и количественно характеризуется
электрическим моментом диполя: р = 1е, где р — момент диполя,
Кл-м; е — элементарный заряд; I — расстояние между центрами
тяжести всех положительных и отрицательных зарядов. Являясь
векторной величиной, электрический момент диполя имеет простран-
ственную направленность. Его значение зависит от ряда факторов.
Решающая роль при этом принадлежит электроотрицательности ато-
мов, образующих связь. Чем больше отличаются атомы по своей
электроотрицательности, тем связь более полярна. По электроотри-
цательности атомы могут быть расположены в следующий ряд:
F>O>Cl>N>Br>C>H
Практически все ковалентные связи более или менее полярны.
Неполярны лишь связи в двухатомных молекулах, состоящих из оди-
наковых атомов:
15
Н —Н;
Cl —Cl
и в совершенно симметричных более сложных молекулах, в которых
центр симметрии совпадает с серединой рассматриваемой связи, на-
пример связь С—С в Н3С—СН3 и С13С—СС13.
В определенных условиях (например, под влиянием внешнего
электрического поля) ковалентные связи проявляют способность к по-
ляризации (поляризуемость связи); при этом происходит смещение
электронов по отношению к ядрам и самих ядер по отношению друг
к другу.
Водородная связь. Связь, возникающая между электроотрицатель-
ным атомом, содержащим по крайней мере одну свободную электрон-
ную пару (О, N, S) и ковалентно связанным атомом водорода, обла-
дающим повышенной подвижностью («протонизированный водород»),
называется водородной связью. По своему характеру она является
электростатической и обозначается пунктиром:
СН3 СН3 СН3 СН3
Г I I !
н—о • • • н— о • • • н—о • • • н—о
Специфическая роль протона в образовании водородных связей
заключается в том, что он способен близко подходить к
электроотрицательным атомам. Энергия водородной связи мала
(12,57—20,44 кДж/моль). Водородные связи влияют на физико-хими-
ческие свойства многих органических соединений (повышенная тем-
пература кипения спиртов, кислот и т. д.). Важна их роль во многих
биологических процессах.
Физическая природа ковалентной связи. Атомные орбитали.
Сформировавшаяся к 1913 г. планетарная теория строения атома
Бора уже через несколько лет оказалась неспособной объяснить не-
которые спектральные данные даже для атомов, содержащих только
два электрона, не говоря уже о более сложных атомах и тем более мо-
лекулах.
Усовершенствование теории стало необходимостью. Зоммерфельд
вводит второе квантовое число I — азимутальное квантовое число
и предполагает, что электроны могут находиться не только на круго-
вых, но и на эллиптических орбитах. При главном квантовом числе п
второе квантовое число / могло принимать значения от 0 до п—1. Каж-
дому значению / отвечала определенная форма электронной орбиты,
которая обозначалась как s (при I = 0), р (при I — 1), d (при / =
= 2) и т. д.
В 1924 г. де Бройль высказал предположение, что любую движу-
щуюся частицу можно отождествить с волной, длина X которой об-
ратно пропорциональна импульсу частицы р, равному mv:
X = h/(tnA-
Соотношение справедливо для любых частиц, но при больших т
длина волны X исчезающе мала и поддается реальному измерению
лишь для очень малых частиц, по массе близких к массе электрона.
16
В 1928 г. Томсон, изучая дифракцию электронов, экспериментально
обосновал соотношение де Бройля.
В 1927 г. Гейзенберг сформулировал свой принцип неопределен-
ности, в котором в применении к движению электрона утвержда-
лось, что обе характеристики движения электрона в атоме (коорди-
наты в пространстве и скорость движения в какой-либо момент вре-
мени) не могут быть одновременно найдены с такой точностью, как
этого требовала теория Бора. Из принципа неопределенности следо-
вало, что чем точнее вычисляется скорость электрона, тем большая
ошибка допускается в установлении его координат, и наоборот. Воз-
никла насущная необходимость в создании более удачного метода опи-
сания движения электронов в атомах.
Волновое уравнение Шредингера. Такой метод и соответствующий
математический аппарат был предложен в 1926 г. ШредингерОхМ на
базе начинавшей формироваться к этому времени квантовой механики,
исходными основными положениями которой были: 1) движение
электронов носит волновой характер; 2) наши знания об этом движе-
нии могут иметь лишь вероятностный (статистический) характер.
Поскольку движение электрона рассматривается как волновое
движение, то его описание возможно с помощью волнового уравнения.
По аналогии с уравнениями, описывающими упругие механические
звуковые и световые волны, уравнение движения электрона по ор-
битали получило название волнового уравнения Шредингера.
Что означает положение о вероятностном характере наших зна-
ний о движении электрона? Согласно принципам квантовой меха-
ники можно определить лишь вероятность нахождения электрона
в данной области пространства, окружающего точку с координатами
(х, у, z), но не его точные координаты. Обычно функция вероятности
обозначается через р(х, у, г), и тогда электрон с максимальной ве-
роятностью будет находиться в той области пространства, где р
максимальна.
Для любого уравнения волнового движения очень важную роль
играет квадрат амплитуды волны, который, например, для уравнения
колебания струны пропорционален ее энергии колебания; для энер-
гии электромагнитного поля плотность энергии пропорциональна
величине (Е2 + Я2), где Е — вектор электрический, а Н — магнит-
ная составляющая электромагнитного поля.
Если обозначить решение волнового уравнения Шредингера че-
рез гр(х, у, z) и назвать его волновой функцией, то гр2(х, у, г) оказыва-
ется пропорциональнььм р(х, у, z). Подобрав соответствующий посто-
янный числовой множитель, можно получить равенство
ф2 (*, У, z) = p(x, у, z),
при этом волновая функция останется решением уравнения Шредин-
гера.
При решении уравнения Шредингера оказывается, что в некото-
рых областях гр положительна, а в некоторых отрицательна. Посколь-
ку вероятность имеет смысл лишь в пределах положительных значе-
ний от 0 до 1, обычно пользуются величиной гр2, а не просто гр, когда
17
хотят связать волновую функцию с понятием плотности вероятности.
В тех же случаях, когда имеют дело с формами атомных или молеку-
лярных орбиталей, пользуются понятием самой волновой функции с
возможными разными знаками (+ и —) ее долей (см. рис. 3).
При решении конкретной задачи о движении электронов в атоме
необходимо: 1) по заданным условиям составить волновое уравнение;
2) решить это уравнение, т. е. найти волновую функцию ф(х, у, г);
3) определить плотность вероятности р = ф2(х, у, г).
Плотность зарядового облака. Часто применя-
ется несколько другая, более наглядная, хотя и менее точная интер-
претация ф. При движении электрона вокруг ядра можно представить
себе его как бы размазанным в пространстве в виде некоторого облака,
при этом плотность этого облака в каждой точке должна быть про-
порциональна гр2. Где [ф]2 максимальна, там облако наиболее плот-
но, и в этой части пространства сосредоточен максимальный отри-
цательный заряд.
Принципиальное отличие этой интерпретации от приведенной вы-
ше состоит в том, что здесь речь идет о реальной электронной плот-
ности в какой-то области пространства в отличие от ранее сформу-
лированной задачи о вероятности обнаружения электрона в этой об-
ласти.
Между обоими способами выражения существует прямая связь.
Представим себе, что все же в какой-то момент времени можно точно
определить положение электрона и отметить его точкой в простран-
стве. Если эту операцию провести достаточно большое число раз, то
совокупность точек будет иметь вид облака, в котором наиболее плот-
ными частями будут те, где вероятность обнаружения электрона наи-
большая. Следовательно, введенное понятие зарядового облака мож-
но с успехом применять вместо понятия вероятности нахождения элек-
трона в данной области пространства.
Интерпретация ф как зарядового облака оказалась очень нагляд-
ной при изображениях атомных и молекулярных орбиталей. В этих
случаях для каждой ф существует некая граничная поверхность, внут-
ри которой сосредоточено, например, 90 или 99% заряда. Формы этих
поверхностей являются важнейшими стереохимическими факторами
и определяют ход многих химических реакций.
Формы атомных орбиталей. Рассмотрим простей-
шую модель — атом водорода. В этом случае единственный электрон
вращается вокруг ядра, находящегося в начале координат. При этом
он может находиться на различных энергетических уровнях и соот-
ветственно этим уровням может существовать в нескольких энерге-
тических состояниях, причем основное состояние отвечает минимуму
энергии. Для атома водорода в основном состоянии полученные ре-
шения уравнения Шредингера имеют сферическую симметрию. Су-
ществует несколько способов их изображения. Мы рассмотрим наи-
более наглядные.
1. Можно изобразить сечение зарядового облака (рис. 2, а) плос-
костью, проходящей через начало координат (рис. 2, б). Этот способ
изображения удобен, когда необходимо составить представление о рас-
18
z
z
Рис. 2. Различные способы изображения s-орби-
тали
пределении заряда. В этом случае наиболее зачерненные части раз-
реза зарядового облака соответствуют максимальной плотности за-
ряда.
2. Можно изобразить сечение граничной поверхности (рис. 2, в),
внутри которой будет находиться основная доля (например, 90%)
полного электронного заряда, плоскостью, проходящей через начало
координат (рис. 2, г).
Хотя эта схема наиболее проста, она дает весьма наглядное пред-
ставление о форме волновой функции ф и используется наиболее ши-
роко. Не следует забывать о трехмерности движения электрона и что
изображение s-орбитали на рис. 2, г представляет собой лишь сечение
сферы соответствующей плоскостью. Хотя согласно принципу не-
определенности понятие «траектория электрона» не имеет явного смыс-
ла, все же вследствие того, что волновая функция ф прямо связана
с распределением электронной плотности, можно утверждать (правда,
с некоторой натяжкой), что волновая функция ф описывает орбиту
движения электрона вокруг ядра. В теории химической связи такие
волновые функции получили название атомных орбиталей, сокра-
щенно АО.
Итак, графически АО можно представить как разрез граничной
поверхности, соединяющей точки пространства с постоянным значе-
нием ф, т. е. область нахождения электрона, вне которой находится
лишь небольшая доля (около 10%) полного заряда электрона. Очень
важно иметь ясное представление о форме и симметрии наиболее часто
встречающихся атомных орбиталей.
АО, изображенная на рис. 2, является единственной для основ-
ного состояния атома водорода. Однако существуют и другие раз-
решенные значения энергии и соответствующие им атомные орбитали,
определяемые набором квантовых чисел (табл. 1).
19
Таблица 1. Связь типов атомных орбиталей с квантовыми числами
Наиболее важной является классификация орбиталей по типам
s; р\ d и т. д. Все АО s-типа (рис. 2) сферически симметричны, поэтому
распределение заряда зависит только от радиуса. Орбитали всех дру-
гих типов не имеют сферической симметрии. Например, имеются три
АО р-типа, граничные поверхности которых похожи на гантели
(рис. 3, а). Эти орбитали имеют ясно выраженную направленность,
поэтому они могут быть обозначены как рх\ ру\ pz, где х, у, z соот-
ветствуют трем осям координат, относительно которых симметрична
соответствующая /7-орбиталь. Все три /7-орбитали совершенно экви-
валентны, за исключением их направления, и все они линейно не-
зависимы. Упрощенная форма их изображения приведена на рис. 3, б.
Рис. 3. Зарядовые плотности р-орбиталей и разрез их граничных
поверхностен
20
Существует также пять АО d-типа с более сложной конфигурацией.
Орбитали других типов в органических соединениях почти не встре-
чаются.
Подразделение атомных (но не молекулярных) орбиталей на типы
s; р; d и т. д. довольно четкое; не существует промежуточных гибри-
дизованных орбиталей между s- и р-орбиталями. По этой причине
целесообразно ввести обозначение типов АО с помощью квантовых
чисел.
Благодаря квантовой механике была решена основная проблема
стационарных состояний электрона в атоме водорода. Набор стацио-
нарных состояний зависит от квантовых чисел: /г, /, m, s. От различ-
ных значений этих чисел зависят симметрия и ориентация волновой
функции ф и ее узловые свойства. В табл. 1 были приведены законо-
мерности в значениях квантовых чисел.
Главное квантовое число п определяет общий размер зарядового
облака. Это означает, что число п определяет общий уровень энергии
электрона.
Квантовое число I характеризует свойства симметрии АО и свя-
зано с моментом импульса движущегося электрона. Квантовое число
/ может иметь значения от 0 до (п—1), т. е. I = 0 при п = 1; I = 0, 1
при п = 2; I = 0, 1,2 при п = 3 и т. д., что соответствует типам ор-
биталей s, р, d и т. д. Для нас более важны геометрические характе-
ристики орбиталей, указываемые символами s, р, d, ..., чем цифровые
значения /, поэтому всегда целесообразнее пользоваться первыми.
Магнитное квантовое число т указывает на количество и направ-
ление в пространстве орбиталей данной формы. Оно имеет 21 + 1
значений: от —I до +/. Так, при / = 0, /и = О возможна только одна
s-орбиталь сферической симметрии. При / = 1, т = —1, 0, +1 и,
следовательно, возможны три р-орбитали, направленные по трем осям
координат (рх, ру, pz) (рис. 3).
Спиновое квантовое число s соответствует двум возможным ориен-
тациям магнитного момента электрона в магнитном поле: вдоль си-
ловых линий или против. Это записывается как —1/2 и или знач-
ками
Согласно принципу Паули в атоме не может быть двух электронов
с одинаковым значением всех четырех квантовых чисел. Это значит,
что одну и ту же АО могут занимать только два электрона с разными
значениями спина (спаренные электроны с антипараллельными спи-
нами).
В соответствии с возможными значениями квантовых чисел, по
мере возрастания заряда ядра электроны могут располагаться на АО
со все более высокой энергией, образуя нейтральные атомы периоди-
ческой системы (табл. 2). Последовательность энергий АО следующая:
ls<2s<2p<3s<3p<4s<3d ... . Три 2р-орбитали (рх, руу pz) имеют
одинаковую энергию и отличаются только направлением в простран-
стве, поэтому они называются трижды вырожденными.
При заселении вырожденных орбиталей электроны сначала рас-
полагаются на них по одному, а затем происходит окончательное за-
полнение вырожденных орбиталей двумя электронами (правило Гунда).
21
Таблица 2. Электронные конфигурации атомов элементов начала
периодической системы
п 1 2 Состояние электронов
1 0 0 1
т 0 0 -1 0 1
Тип орбиталей 1 5 2 s 2pv 2Pz
Н f Is1
Не If Is2
Li и ♦ 1
Be If If ls22s2
В If If f \s22s22px
С If It f f \s22s22pxlpy
N If If f f f \s22s22p^2py2p\
0 If If If f f \s22s2lpx 2py2p\
F If If If If f ls22s22px2py2pz
Ne If If If If If ls22s22p2 2p22p2
Молекулярные орбитали. Точное решение волнового уравнения
наталкивается на непреодолимые трудности даже при использовании
ЭВМ; эти трудности проявляются при нахождении гр для атомов,
содержащих несколько электронов. Но задача особенно усложняется
в случае молекул. Так, при попытке решить волновое уравнение для
этана (8 ядер и 18 электронов) мы будем иметь 26 X 3 = 78 неза-
висимых переменных. Решить дифференциальное уравнение с таким
числом переменных крайне сложно и поэтому всегда прибегают к
приближенным методам решения волновых уравнений.
Наиболее распространенным и простым приближенным методом
является модификация вариационного метода — метод линейных
комбинаций, получивший название «метод линейных комбинаций атом-
ных орбиталей», сокращенно ЛКАО.
Метод ЛКАО. Представим себе, что
два одинаковых атома с энергией Еа находят-
ся на расстоянии (точка рис. 4), исклю-
чающем какое-либо взаимодействие, и посте-
пенно сближаются. Если при этом может об-
разоваться устойчивая молекула (например,
Н Н-> Н2), то при некотором (достаточном)
сближении (точка R2, расстояние соизмеримо
с длиной связи) ядро каждого из атомов нач-
нет притягивать электрон, принадлежащий
другому атому. Это будет характеризоваться
Рис. 4. Кривая потенци-
альной энергии для
двухатомной молекулы
уменьшением значения потенциальной энергии системы. При даль-
нейшем сближении энергия будет уменьшаться до минимальной
величины Ем, которая представляет собой энергию молекулы при
R = г, где г — длина связи в устойчивой молекуле. Последующее
сближение приводит к быстрому росту энергии вследствие сильного
отталкивания ядер и возрастания кинетической энергии электронов.
Рис. 5. Схема образования связывающей молекулярной ls-орбитали
при сближении двух атомов водорода и пространственное изобра-
жение МО в виде заряда облака
Следовательно, в тот момент, когда R близко или равно г, вместо
атомных орбиталей грА и грв (ls-орбиталей, если сближались два ато-
ма водорода) возникает общая молекулярная орбиталь (МО) гр, ко-
торая в отличие от АО является двухцентровой орбиталью. Это объ-
ясняется тем, что, оказавшись в пространстве между атомами, элект-
рон атома А уже находится в поле и атома В; таким образом, он мо-
жет перейти на его орбиталь и наоборот. Наглядно образование такой
МО показано на рис. 5 для атомов водорода.
При интерпретации такой картины весьма существенно, что когда
электрон, находящийся на МО гр, движется вблизи ядра Л, МО гр
весьма сходна с АО грА, а когда электрон движется вблизи ядра В,
МОф весьма сходна с АО грв, т. е. гр попеременно сходна с грА и грв
и, следовательно, ее можно представить в виде линейной комбинации
гр = грА + грв.
Физический смысл знака «+» в этом равенстве означает, что плот-
ность электронов в пространстве между ядрами значительно выше,
чем в других частях МО, а это способствует связыванию ядер, и что
оба электрона в молекуле водорода охватывают оба ядра. МО такого
типа гр = грА + грв, образовавшиеся при сложении двух АО, назы-
ваются связывающими орбиталями, так как при этом процессе проис-
ходит соединение атомов в молекулу.
В общем случае образование МО из двух АО должно приводить обя-
зательно к двум МО. Дело в том, что на каждой АО могут иметься два
электрона и при образовании МО на них должно разместиться четыре
электрона. Так как, согласно принципу Паули, на одной орбитали не
может находиться более двух электронов, то должно образовываться
два МО. Из метода линейных комбинаций легко выводится, что вторая
МО гр* образуется при операции вычитания АО: гр* = грА — грв.
23
При образовании МО такого типа вероятность нахождения элект-
ронов между ядрами равна нулю.
Электроны на ф* = фА — фв находятся дальше от ядер, чем даже
на фА и фв, не говоря уже о ф = фА + Фв- Отсюда следует, что
нахождение электронов на такой МО энергетически менее выгодно,
чем их нахождение на ОА фА и фв, поэтому атомы стремятся разъ-
единиться, и МО такого типа называются разрыхляющими или, реже,
антисвязывающкми орбиталями. Для них принято обозначение ф*
(со звездочкой) или фа (рис. 6). Связывающие МО (фА + фв) обо-
значаются просто ф (без звездочки) или фь.
Рис. 6. Образование разрыхляющей молекулярной 1s*-
орбтали при сближении двух атомов водорода
Рис. 7. Образование молекулярной а-орбитали при ком-
бинации рхд- и рхв-атомных орбиталей
24
Электроны, находящиеся на любой связывающей МО гр, всегда
стремятся сблизить атомы, а электроны, находящиеся на любой раз-
рыхляющей МО гр*, стремятся разъединить их.
Молекула водорода, у которой оба электрона находятся на свя-
зывающей МО гр. обладает минимумом энергии и устойчива. При со-
общении молекуле достаточной энергии можно перевести один или
оба электрона, например, на гр*-орбиталь и система станет неустой-
чивой.
Форма молекулярных орбиталей, о- и л-Связи.
Рассмотренное выше образование МО происходило за счет линейной
комбинации АО s-типа. Образующиеся при этом орбитали гр и гр* ока-
зываются симметричными относительно поворота вокруг оси, соеди-
няющей ядра (см. рис. 5 и 6). Поэтому МО такого типа обозначаются
как о-орбитали; связывающая просто о,
дочкой). Отнесение МО к
типу о означает только,
что МО симметрична отно-
сительно оси связи.
Точно так же при обра-
зовании МО из двух АО р-
типа, ориентация которых \ у
соответствует оси связыва- Т /
ния (сближение атомов по (+J
оси х и выбор рх-орбита-
лей), образующаяся связы- *
вающая орбиталь тоже от-
носится к o'-типу, так как
она симметрична относи-
тельно оси х (рис. 7); со-
ответствующая разрыхля-
ющая орбиталь по той же
причине относится к
о*-типу.
Однако образование МО
из орбиталей p-типа может z
происходить и за счет ру- | у
или р2-орбиталей при обра- £ /
зовании связи по оси х
(рис. 8). МО этого типа (из- ш
за разных знаков долей р- *
орбиталей) антисимметрия-
ны относительно поворота /
на 180° вокруг оси АВ и ‘
обозначаются как л-связы-
вающие и л*-разрыхляю-
щие орбитали. Следует спе-
циально отметить принци-
а разрыхляющая о* (со звез-
изображение образо-
л*-орбиталей
пиальные различия л- и
а-орбиталей. Кроме уже
Рис. 8. Графическое
вания л- и
25
указанных свойств симметрии относительно поворота на 180° вокруг
оси связи, степень перекрывания для л-орбитали значительно
меньше, поэтому энергия ее образования также меньше, чем для
о-орбитали. Нельзя рассматривать части МО л-типа (рис. 8) со зна-
ками «+» и «—» как самостоятельные части, так как они составляют
единое целое. То же относится и к МО о-, о*- и л*-типа (рис. 7, 8).
Гибридизация орбиталей. Из табл. 2 видно, что в невозбужденном
атоме углерода (ls22s22pх2ру) на первом электронном уровне нахо-
дятся два спаренных электрона (1s2), а на втором электронном уровне—
два спаренных электрона на 25-орбитали (2s2) и по одному неспаренно-
му электрону на двух р-орбиталях (рх и ру), а одна р-орбиталь оста-
ется свободной (pz). В таком состоянии углерод должен бы быть двух-
валентным. Однако хорошо известно, что в подавляющем большинстве
соединений углерод четырехвалентен и что все четыре связи в такого
рода симметричных структурах (СХ4) одинаковы. Для объяснения
этого факта было введено и математически обосновано понятие о гиб-
ридизации орбиталей.
Смысл этого понятия (а не явления) заключается в том, что близ-
кие по энергии и имеющие общие элементы симметрии молекулярные
(но не атомные) орбитали могут взаимодействовать между собой, об-
разуя так называемые гибридные орбитали с более низкой энергией.
Так, при взаимодействии s-орбитали с рх-орбиталью (рис. 9) об-
разуются две sp-гибридизованные орбитали, имеющиеся, например,
у атомов углерода ацетилена и ориентированные по оси х. Такой тип
гибридизации называется s/2-гибридизацией (или, правильнее, ди-
гональной гибридизацией). Если взаимодействуют две р-орбитали
(рх и ру) и s-орбиталь, то три образовавшиеся $р2-орбитали (триго-
нальная, или 5/72-гибридизация) лежат в одной плоскости (рис. 10),
образуя между собой угол в 120°. При взаимодействии s-орбитали с
тремя /лорбиталями (рх; ру и pz) четыре образовавшиеся орбитали
Рис. 9. Образование sp-гибридизованных орбиталей из s-орби-
тали и рх-орбитали
26
(тетраэдрическая или $р3-гибридизация) направлены к вершинам
правильного тетраэдра и образуют между собой угол 109°28' (рис. 11)
Итак, при образовании ковалентных связей один из 2«-электрр-
нов невозбужденного атома углерода возбуждается (с затратой
419—628,5 кДж/моль) и переходит на свободную р-орбиталь. В воз-
Рис. 10. 5р2-Гибридизо-
ванная орбиталь
Рис. 11. $р3-Гибридизо-
ванная орбиталь
бужденном состоянии у атома углерода уже четыре неспаренных элект-
рона на 2s-, 2рх-, 2ру- и 2р2-орбиталях. Далее в зависимости от того,
в какой фрагмент молекулы войдет этот атом углерода, происходит
один из типов гибридизации (в СХ4—sp3-; этилене—sp2-, ацетилене —
sp-). Энергия образования четырех связей с избытком покрывает те
419—628,5 кДж/моль, которые требовались для возбуждения атома.
В случаях тригональной и дигональной гибридизации одна или соот-
ветственно две р-орбитали остаются негибридизованными и имеют вид
неискаженных восьмерок. Так, атом углерода в этилене имеет три
тригонально-гибридизованные орбитали и одну чистую р-орбиталь,
направленную перпендикулярно плоскости, в которой расположены
все три 5р2-орбитали (рис. 12).
Электроотрицательность атома углерода, равная 2,5 (по сравне-
нию с Li), характеризует атом углерода в степени гибридизации sp3.
Рис. 12. Орбитали атома углерода тригональной гибриди-
зации:
а — три $р2-орбитали; б — рг-орбиталь; в — атом углерода триго-
нальной гибридизации
27
При увеличении s-характера орбитали электроотрицательность уве-
личивается: для углерода в степени гибридизации sp2 она равна
2,8, а для ацетиленового атома углерода (sp) — даже 3,1.
Итак, при образовании ковалентных связей орбитали связываю-
щихся атомов могут взаимодействовать двояким образом. Например,
в молекуле водорода Н2 ls-орбитали двух атомов объединяются в одну
молекулярную орбиталь так, что область максимальной электронной
плотности, или область перекрывания атомных орбиталей, лежит на
линии, соединяющей центры атомов (рис. 13, а). Такой тип связи
называется о-связью. о-Связи образуют и гибридизованные орбитали.
Например, во фрагменте С—Н образуется о-связь между гибриди-
зованной орбиталью углерода и ls-орбиталью атома водорода
(рис. 13, б), а во фрагменте С—С — между двумя гибридизованными
орбиталями (рис. 13, в). о-Связь обладает большой прочностью, так
как основная масса электронной плот-
ности сосредоточена в небольшом про-
( ю ) странстве между ядрами атомов.
\ V / При взаимодействии двух р-орбита-
j '-Д'' лей соседних атомов (рис. 14) образу-
—/X—7х— —-------ется так называемая л-связь. В этом
/ \ / \ случае образуются две области макси-
/ X \ х........... мальной электронной плотности услов-
( ф ) но выше и ниже оси связи (рис. 14).
44—' Поэтому л-связь менее прочна, чем <т-
а связь, и ее электроны могут значитель-
Рис. 14. Перекрывание орбита-
лей при образовании л-связей
но легче смещаться в сторону одного
из атомов.
Между двумя атомами может быть
только одна о-связь, так как в против-
ном случае не будет соблюдаться правило, по которому на одной
орбитали не может находиться более двух электронов (следствие
принципа Паули). Таким образом, в предельных углеводородах, а так-
же в других соединениях, содержащих только простые связи, все связи
о-типа. Для таких атомов углерода иногда используют термин: атом
углерода в первом валентном состоянии.
В соединениях с двойными связями (например, в этиленовых
углеводородах ) одна из кратных связей является а-связью,
28
образованной гибридизованными орбиталями, а вторая — л-связью,
образованной негибридизованными р-орбиталями (рис. 15). В этих
соединениях атомы углерода, связанные кратными связями, нахо-
дятся в так называемом втором валентном состоянии. При этом все
три (т-связи каждого углеродного атома лежат в одной плоскости, а
л-связь располагается в перпендикулярной ей плоскости.
Рис. 15. Пространственное расположение
орбиталей в молекуле этилена
Рис. 16. Пространственное рас-
положение орбиталей в моле-
куле ацетилена
Аналогичное строение имеет и тройная связь (НС=СН). Углерод-
ные атомы здесь находятся в третьем валентном состоянии, т. е. все
о-связи лежат на одной линии, а четыре р-электронные облака об-
разуют две л-связи во взаимно перпендикулярных плоскостях
(рис. 16).
Как уже говорилось, л-связи менее прочны, чем о-связи, так как
область перекрывания электронных облаков лежит в стороне от оси
связи. Это приводит к тому, что соединения с кратными связями зна-
чительно более реакционноспособны, чем соответствующие алканы.
За счет разрыва л-связей и превращения их в более устойчивые о-свя-
зи идут реакции присоединения по кратным связям. В этом случае
атом углерода дигональной или тригональной гибридизации превра-
щается в атом углерода тетраэдрической гибридизации (с минимумом
свободной энергии).
Квантово-механические методы расчета в органической химии. Исследова-
ние природы химической связи является центральной проблемой всей теоре-
тической химии. Изучение строения и реакционной способности вещества дает
богатую информацию о характере взаимодействия между атомами в молекуле,
способствуя все более углубленному моделированию химических процессов.
Обобщение экспериментальных данных приводит на определенных этапах раз-
вития химии к теоретическим концепциям, которые наряду с чисто познаватель-
ным аспектом имеют и громадное практическое значение, так как позволяют
вести исследование более целенаправленно. Однако только с созданием аппарата
квантовой механики — науки о движении микрочастиц (атомов, ядер, электронов
и т. д.) — ранее существовавшие теории химической связи получили естест-
венное объяснение. Современная квантовая химия является частью квантовой
механики, в снове которой лежит представление о корпускулярно-волновом
дуализме микрочастиц. Если раньше электрон рассматривался как точечная
частица, положение и скорость которой в принципе можно было точно устано-
вить, то в дальнейшем было установлено, что электрон может обладать также и
29
волновыми свойствами (например, мы можем при определенных условиях наблю-
! дать дифракцию электронов).
* Рассматривая молекулу, можно только условно говорить об отдельных
электронах, связях и пр. С точки зрения квантовой химии правильнее говорить
о некоторой электронной плотности в данной точке, на данном атоме и ее распре-
делении по молекуле. Подобное распределение электронной плотности описыва-
ется математически некоторой функцией координат и времени ф (х, у, г, 6, полу-
пившей название волновой функции, причем [ф(х, у, z, /)]2 есть электронная
плотность в точках к, и, z в момент времени t. Если известно ф, мы можем опре-
делить практически любое интересующее нас свойство молекулы: электронную
энергию, электрические заряды на атомах, дипольный момент, поляризуемость
и т. д. Эти свойства во многих отношениях определяют особенности поведения
молекулы в химических реакциях, т. е. ее реакционную способность. Отсюда
ясно, какое громадное значение приобретает для химика знание волновой функ-
ции.
Чтобы найти ф, следует решить уравнение Шредингера: Нф = Еф, где ф—
искомая многоэлектронная волновая функция; Е — полная энергия молекулы;
Н — оператор полной энергии (так называемый оператор Гамильтона, или га-
мильтониан), равный сумме операторов кинетической и потенциальной энергии.
Вся сложность проблемы заключается в том, что мы не в состоянии точно
решить это уравнение даже для простых схем, например для молекулы водорода,
и должны прибегать к ряду весьма существенных упрощений. Предложение
рассматривать молекулярные волновые функции как линейную комбинацию
одноэлектронных атомных волновых функций легло в основу широко распрост-
раненных приближенных методов нахождения.
Математическая идея одного из таких методов физически соответствует сле-
дующей качественной модели. Каждый электрон в молекуле рассматривается
как находящийся на определенной молекулярной орбитали (подобно электрону в
атоме, находящемуся на определенной атомной орбитали), которая охватывает
всю совокупность атомов молекулы и характеризуется определенной энергией.
Общая энергия молекулы равна сумме энергий электронов на занятых МО.
Существуют полные теоретические расчеты молекул. Выполненные в на-
стоящее время для весьма небольшого числа простейших двухатомных и ряда
трехатомных молекул, подобные расчеты чрезвычайно трудоемки, и даже при
современной электронно-вычислительной технике их вряд ли можно будет про-
вести для систем, содержащих более чем 20 атомов.
Обычно конкретные расчеты молекулярных систем проводят на основе так
называемых чисто эмпирических или полуэмпирических методов, простейшим ва-
риантом из которых является метод Хюккеля, применяемый с наибольшим успе-
хом для расчета сопряженных систем, например полиенов, ароматических угле-
водородов и их производных. Специфика таких систем заключается в том, что их
можно рассматривать в так называемом ^-электронном приближении, т. е. опре-
делять молекулярные орбитали, их энергию, заряды на атомах и т. д. только для
я-электронов, не принимая во внимание а-электронные уровни.
Для многих органических молекул это приближение часто оказывается оп-
равданным, так как, во-первых, позволяет без большой погрешности пренеб-
речь взаимодействием между ст- и ^-электронами, а во-вторых, реакционная спо-
собность таких соединений в основном действительно определяется более высоко-
лежащими и более «подвижными» ^-электронными уровнями.
Для органической химии и биохимии ценность квантовомеханических рас-
четов определяется тем, что они не только помогают объяснить реакционную
способность молекулы, наблюдаемую в химических реакциях, но и во многих
случаях дают возможность предсказать ее. В этой связи большое значе-
ние приобретает знание так называемых реакционных индексов (заряды на ато-
мах, энергия делокализации, порядок связи, энергия локализации, граничная
электронная плотность и др.), которые позволяют выяснить способность мо-
лекулы к участию в тех или иных органических реакциях.
В настоящем разделе мы рассмотрим только некоторые наиболее простые
из реакционных индексов, которые представляют наиболее существенный
интерес.
30
Порядок связи р. Этот индекс позволяет количественно оценить сте-
пень ^-связывания между соседними атомами в молекуле. Если принять, что
порядок a-связи равен единице, то для этилена он равен двум условным едини-
цам:
2,0
Н2С =СН2
Путем несложных расчетов для бутадиена в рамках приближения Хюккеля мож-
но получить порядок С—С-связей:
1,8942 1,4473 1,8942
Н2С сн —^сн сн2
Из приведенных значений порядков связей видно, что в бутадиене связь
2—3 не является чистой ^-связью: она обладает частично я-характером (степень
я-связывания равна 0,4473). Это объясняется явлением, которое в органической
химии получило название эффекта сопряжения, оно приводит к эффективной де-
локализации электронов по молекуле.
Индекс свободной валентности/7. Этот индекс служит ме-
рой ненасыщенности атома — его неиспользованной способности к образованию
дополнительных связей. Согласно теоретическим расчетам максимальная степень
связывания для углерода в молекуле должна составлять 4,732 условных единиц.
Если теперь мы из этого значения вычтем сумму порядков связей, образуемых
атомом углерода с другими атомами в молекуле, то получим значение F для дан-
ного атома углерода. Так, для молекулы бутадиена F1= F±~ 0,8378, a F2 =
= F3 = 0,3905.
В радикальных реакциях наибольшую активность будут проявлять те ато-
мы углерода, которые имеют наибольшее значение F, Как показывает экспери-
мент, это положение подтверждается в большинстве случаев. Так, в частности,
концевые атомы углерода (Ft и F±) в бутадиене реагируют легче, чем централь-
ные. Однако возможен случай, когда атаке подвергаются одновременно два цент-
ра в одной и той же молекуле, например реакция Дильса — Альдера. Парамет-
ром, определяющим направление такой гомолитической реакции, может слу-
жить сумма индексов F двух атакуемых атомов. Нетрудно видеть, что для бута-
диена наиболее уязвимым местом для двойной гомолитической атаки являются
положения 1 и 4.
Электронная плотность и распределение заря-
до в q. Эти характеристики молекулы приобретают важное значение при интер-
претации результатов гетеролитических реакций, так как направление атаки
электрофильного или нуклеофильного агентов существенно связано с распреде-
лением условных зарядов атомов в молекуле. Наиболее четко эти характерис-
тики проявляются, если молекула содержит, например, гетероатом.
Сравним распределение F-электронной плотности в бензольном ядре анили-
на и нитробензола (в молекуле бензола электронная плотность на атомах угле-
рода равна 1):
Из приведенных молекулярных диаграмм можно сделать вывод, что при
электрофильном замещении в анилине атака будет в основном направлена на
о- и n-углеродные атомы, тогда как в нитробензоле следует преимущественно
ждать образования ^-изомера. В этих случаях эксперимент находится в полном
согласии с выводами теории.
31
Энергия делокализации ЭД. Согласно основной идее метода
МО электроны в молекуле располагаются на молекулярных орбиталях, охваты-
вающих всю совокупность атомов, т. е. являются делокализованными. Важней-
шим следствием делокализации электронов, как показывают соответствующие
квантовомеханические расчеты, является повышение стабильности молекулы,
что наиболее отчетливо проявляется в случае сопряженных и ароматических
структур. Величина, характеризующая с энергетической стороны глубину де-
локализации электронов в молекуле, получила название энергии делокализа-
ции — ЭД. Значение ЭД для многих сопряженных и ароматических молекул не
только может быть рассчитано, но и в ряде случаев допускает прямую экспери-
ментальную проверку. Например, для молекулы бензола как теоретическое,
так и экспериментальное значение ЭД= 149,94 кДж/моль. Это означает, что внут-
ренняя свободная энергия реальной молекулы бензола на 149,94 кДж/моль ни-
же, чем у гипотетической молекулы бензола с тремя локализованными двойными
связями.
Ниже приведены в качестве примера значения ЭД для трех ароматических
гетероциклов:
пиррол
(87,99 кДж/моль)
Если исходить из этих значений ЭД, то можно предположить, что ароматические
свойства (см. ч. 5) будут выражены наиболее ярко у тиофена. Это подтверждается
экспериментальными данными по реакционной способности и устойчивости при-
веденных гетероциклов.
Перечисленных основных реакционных индексов, однако, явно недостаточ-
но, чтобы охватить всю совокупность экспериментальных фактов. Особенно от-
четливо это проявляется в случае углеводородов с сопряженными связями, при-
мером которых может служить бутадиен или нафталин. Расчет по методу ЛКАО
МО дает эффективные заряды на всех атомах углерода в бутадиене равны нулю.
Поэтому могло бы показаться, что к электрофильной атаке будут одинаково
чувствительны все четыре углеродные атома молекулы. На самом же деле, как
это следует из опыта, наиболее подверженным действию электрофильных аген-
тов оказываются концевые атомы бутадиена.
На основании большого числа примеров было доказано, что в подобных слу-
чаях следует применять метод граничных электронов (МГЭ). При таком подхо-
де считается, что электрофильный агент атакует электроны, которые находятся
на высших заполненных молекулярных орбиталях в точках наибольшей элект-
ронной плотности, т. е. следует принимать во внимание не полную я-электрон-
ную плотность на атомах в молекуле, а только ту ее часть, которая связана с
высшей занятой молекулярной орбиталью. В таких случаях, если нас интересует
направление нуклеофильной атаки, то согласно МГЭ наиболее активным будет
то положение в молекуле, которое характеризуется максимальной электронной
плотностью на низшей незанятой (свободной) молекулярной орбитали.
Для понимания физических и химических свойств органических молекул
существенное значение имеет не только знание граничных электронных плотнос-
тей, соответствующих высшей занятой и низшей незанятой МО, но и их относи-
тельных энергий. Так, в частности, было доказано, что потенциалы ионизации
многих органических молекул находятся в хорошей корреляции с разностью
энергий низшей свободной и верхней занятой молекулярных орбиталей. Кроме
того, знание энергий этих ДЮ приносит большую помощь при интерпретации
электродонорных и электроноакцепторных свойств молекул, что особенно су-
щественно в случае образования комплексов с переносом заряда, когда элект-
рон, находящийся на высшей занятой МО одной молекулы (донор), переходит
на низшую незанятую МО другой молекулы (акцептор), причем образующийся
32
комплекс тем устойчивее, чем выше энергия МО донора и чем ниже энергия со-
ответствующей МО акцептора.
Рассмотрим несколько иллюстраций применения приведенных понятий и
методов расчета в одной из наиболее бурно развивающихся областей молекуляр-
ной биологии — в квантовой биохимии. Объектом исследования квантовой био-
химии являются электронная структура биомолекул, образующих живое ве-
щество, внутренняя природа взаимодействия между ними, специфика и уровень
их организации.
Успехи, достигнутые квантовой биохимией в последние годы, позволяют
сделать вывод, что в основе жизнедеятельности биологических организмов на мо-
лекулярном уровне лежат хотя и очень сложные, но в основном обычные физико-
химические процессы, изучение которых возможно методами квантовой ме-
ханики.
Известно, что все основные участвующие в фундаментальных функциях
живой материи биомолекулы построены из систем, полностью или частично со-
пряженных, богатых я-электронами (нуклеиновые кислоты, ферменты и пр.).
Существует широко распространенная точка зрения, что наличие далеко идущей
делокализации электронов в этих биомолекулах является тем фундаментальным
основанием, которое определяет специфику стабильности и функционирования
биологических объектов.
Весьма существенным в этой связи представляется недавно полученный ре-
зультат, показывающий, что из пяти оснований нуклеиновых кислот аденин,
имеющий наибольшее значение энергии делокализации, легче всего синтезиру-
ется электронным облучением смеси метана, аммиака и воды. Кроме того, было
обнаружено, что между резистентностью (сопротивляемостью) этих оснований
действию ионизирующих излучений и соответствующими ЭД существует глубо-
кая связь.
Совершенно ясно, что энергия делокализации приобретает важное значение
как фактор стабильности, или «выживаемости», биомолекул. Тот факт, что био-
молекулы содержат большое число сопряженных связей, является весьма благо-
приятным обстоятельством, так как в этом случае даже такой грубый метод,
как ЛКАО МО в приближении Хюккеля, может значительно облегчить задачу
установления электронной структуры и в связи с этим определение центров био-
химических процессов. В ряде случаев подобные расчеты даже позволяют ре-
шать чрезвычайно сложные биохимические проблемы, такие, как природа мута-
ций, проблема канцерогенности, изучение противоопухолевой активности пури-
новых антиметаболитов в химиотерапии рака и пр.
Рассмотрим с позиций квантовой химии проблему канцерогенности конден-
сированных ароматических углеводородов. Огромная трудность, препятствую-
щая всякой попытке установить связь между структурой и канцерогенной
активностью ароматических соединений, заключается в чрезвычайной чувстви-
тельности этого явления к изменениям структуры. Так, из более чем 50 исследо-
ванных в настоящее время конденсированных циклов только 9 оказались несом-
ненно канцерогенными. В частности, из шести приведенных ниже углеводородов
лишь 3,4-бензфенантрен обладает свойством вызывать злокачественные новооб-
разования:
нафтацен
1,2-бензантрацен
2—682
33
3,4- бензфен антрен
пирен
Изучение структуры большого числа конденсированных ароматических
ядер позволило сделать вывод, что в проявлении канцерогенных свойств сущест-
венную роль играет соотношение реакционной способности областей в молекуле
мезофенантренового (/(-область) и мезоантраценового (L-область) типов:
Сравнение реакционных индексов паралокализации (L-область) и ортоло-
кализации* (/(-область) приводит к двум важным заключениям, касающимся
канцерогенной активности ароматических углеводородов:
1) для того чтобы полициклический углеводород был канцерогеном, он дол-
жен обладать «активной» /(-областью; соответствующий параметр ортолокализа-
ции не должен превышать значения 3,31/7 (р — величина резонансного интегра-
ла; Р « 74,97 кДж/моль);
2) если молекула содержит также и L-область, то необходимо, чтобы она бы-
ла, наоборот, малоактивна, соответствующий индекс паралокализации должен
быть больше или равен 5,66.
Эти правила имеют лишь небольшое число исключений и в большинстве слу-
чаев могут быть успешно применены для решения вопроса — проявляет данный
конденсированный углеводород канцерогенные свойства или нет.
Таким образом, уже на основании приведенных примеров становится ясно,
в какой степени расчеты могут помочь исследованию не только при интерпрета-
ции и предсказаниях чисто химической реакционной способности молекул, но
также способствовать решению сложных биохимических проблем.
Однако не следует представлять себе дело таким образом, что метод молеку-
лярных орбиталей непогрешим и всегда приводит к верным результатам. Это
связано в основном с тем обстоятельством, что в полуэмпирических методах су-
ществует определенный произвол в выборе используемых в расчете кулоновских
и обменных интегралов, определявших типы связей в молекуле.
Так, для атома азота в пиридине значение а принималось разными авторами
от а -Ь 2,0)6 до а + 0,2)0, где а и Р — некоторые стандартные значения соответ-
ственно кулоновского и резонансного интегралов. Поэтому результаты расчетов
могут существенно отличаться или даже противоречить друг другу. В целом вы-
* Эти реакционные индексы характеризуют, подобно граничной электрон-
ной плотное)и, способности атомов данной области к взаимодействию с химичес-
кими реагентами.
34
бор а и Р часто определяется тем, в какой степени полученный результат отвечает
действительной картине, наблюдаемой* в эксперименте.
В последнее время, особенно в связи с совершенствованием техники элект-
ронно-вычислительных машин, с созданием новых расчетных программ, простой
метод Хюккеля практически вытеснен более совершенными полуэмпирическими
вариантами приближения, в которых непосредственно учитывается межэлектрон-
ное взаимодействие, что значительно повышает надежность получаемых резуль-
татов.
Общее представление о механизмах химических реакций. Некото-
рые вопросы реакционной способности органических соединений.
Органические соединения в определенных условиях (температура,
давление, наличие катализатора и т. д.) могут вступать во взаимо-
действие с другими органическими или неорганическими соединения-
ми или претерпевать разнообразные превращения.
Типы химических реакций. Реакции и превращения органических
веществ можно подразделить на четыре типа:
1. Реакция замещения (Substitution; индекс S)
нн нн
II II
R—С—С—R' + АВ -> R—С— С— R' + НВ
II II
НН АН
2. Реакции присоединения (Addition; индекс А или Ad)
\с=С^+ АВ ->\с—C<f
/ \ /| |\
А В
3. Реакции отщепления, или элиминирования (Elimination; ин-
декс Е)
R4 Л" R к ZR"
/С — С АВ 4- =
R'/ I I \R'" R'/ \R'"
4. Молекулярные перегруппировки
В ходе этих реакций взаимодействующая система молекула—ре-
агент, превращаясь в конечный продукт, проходит через ряд после-
довательных промежуточных состояний. Совокупность этих состоя-
ний получила название механизма реакции. При этом происходит
перераспределение валентных электронов между взаимодействующи-
ми молекулами или между реагирующей молекулой и реагентом. В за-
висимости от способа нарушения ковалентной связи в реагирующей
молекуле различают гомолитические (радикальные) А*4-|Б—>-А* +Б* и
гетеролитические (ионные) реакции: АйБ->А:+В+ При гомоли-
тических реакциях электронная пара ковалентной связи разъединя-
9*
35
ется с образованием свободных радикалов — нейтральных атомов или
групп атомов, имеющих неспаренные электроны. При написании сво-
бодных радикалов рядом с химическими символами (обычно справа)
ставят точку, условно обозначающую наличие неспаренного электро-
на. Гомолитические (свободнорадикальные) реакции характерны для
соединений, построенных из атомов с близкой электроотрицатель-
ностью. Они инициируются и успешно протекают под действием вы-
соких температур (паровая фаза), света с высокой энергией (видимый
свет, ультрафиолетовые лучи, у-излучение), в присутствии пере-
кисных соединений, легко распадающихся на свободные радикалы:
О-О—2R0.
перекисное свободный
соединение радикал
Свободнорадикальные реакции проходят в неполярных растворите-
лях; тормозятся веществами, легко реагирующими со свободными ра-
дикалами. Реакции замещения, протекающие по свободнорадикаль-
ному механизму, принято обозначать S в; присоединения — Ad^
(или Ar).
Гетеролитические, или ионные, реакции в зависимости от дей-
ствующего реагента могут быть нуклеофильными (S N) или электро-
фильными (SE). В первом случае реагент (В) предоставляет свою
электронную пару для вновь возникающей связи:
А*Б + : В- ->Б:В + А* (SN)
реагирующая нуклеофиль-
молекула ный реагент
во втором случае на образование новой ковалентной связи исполь-
зуется электронная пара, принадлежащая реагирующей молекуле
АБ:
А: Б + В+ -> Б : В + А+ (SE )
реагирующая электро-
молекула фильный
реагент
Нуклеофильными реагентами («имеющими сродство к ядру») обычно
бывают анионы или молекулы, содержащие атомы с неподеленными
электронными парами: ОН"; СГ; СН3СОО"; QHsO"; С2Н5ОН; Н26;
CN"; NH3. К электрофильным реагентам («имеющим сродство к элект-
ронам») относятся катионы: Н+; Cl+; R—С=0; N02; SO3H или моле-
кулы с незаполненными валентными орбиталями: А1С13; BF3; FeCl3;
S03 и др.
Ионные реакции катализируются кислотами или основаниями,
они редко проходят в паровой фазе и зависят от природы растворителя.
На них не оказывают влияние свет, свободные радикалы и их акцеп-
торы.
Отдельные превращения органических соединений проходят через
образование в качестве промежуточного этапа активированных комп-
36
лексов, при этом одновременно с разрывом старой связи возникает
новая. Эти превращения получили названия молекулярных реакций
и будут подробно разобраны в соответствующих разделах курса.
Взаимное влияние атомов в молекулах органических соединений.
Реакционная способность органических соединений во многом зави-
сит от постоянной поляризации связей, их поляризуемости и взаим-
ного влияния атомов в молекуле. Особое значение имеют два вида вза-
имного влияния атомов: индуктивный эффект и эффект сопряжения
(мезомерный эффект). Индуктивным эффектом (/-эффект) называют
эффект, связанный со смещением электронной плотности вдоль о-
связи, которое вызывается разностью электроотрицательностей ато-
мов, входящих в соединение. Индуктивный эффект атома водорода
равен нулю. Если вместо атома водорода ввести в молекулу какие-то
иные заместители, то может быть два варианта индуктивного влия-
ния:
г- г* 8* 8g
Х^-СН2^-СН2^-СН2—(СН2)П—СН3 -/-эффект
> *2 >
г+ 82 53
Y -> СН2 -> СН2 -> СН2 — (CH2)rt — СН3 + /-эффект
> Ь2 > Ь3
В 1-м случае (—/-эффект) происходит смещение электронной плотнос-
ти от атома углерода к заместителю X (электроноакцептор), во 2-м
(+/-эффект) — от заместителя Y (электронодонор) к углеродному
атому. Основные электроноакцепторные заместители: NO2; CN; ОН;
F; Cl; Вг; С6Н5; СН2=^СН—. Основные электронодонорные замести-
СНзх СН3
тел и: металлы; СН3—С—; СН — ; СН3—СН2—; СН3—,
СНз/ СН3
Особенность индуктивного эффекта — быстрое затухание по мере
удаления от атома или группы атомов, вызывающих этот эффект (прак-
тически четвертый атом уже не подвержен влиянию индуктивного
эффекта).
В органических соединениях возможны такие структуры, в кото-
рых через одну простую связь чередуются двойные связи (I), двойная
связь и атом, имеющий неподеленные электронные пары (II), арома-
тическое ядро и двойная связь (III), ароматическое ядро и атом, имею-
щий неподеленные электронные пары (IV).
Такие системы называются сопряженными:
1 сн2=сн—сн=сн, 1,3-бутадиен сн2=сн—сн=о акролеин 37
II CH2=CH—Cl
сн2=сн—о—С2Н5
хлористый винил
этил этиловый эфир
стирол
хлорбензол
Приведенные примеры не исчерпывают всех возможных вариантов
сопряженных систем, в которых наблюдается эффект сопряжения
(мезомерный эффект М), связанный с перераспределением электрон-
ной плотности в молекуле и образованием единого л-электронного об-
лака. При этом происходит деформация связей, понижение энергии
системы, изменение ее реакционной способности. Мезомерный эффект
положителен (+М), если в результате его повышается электронная
плотность в сопряженной системе, и отрицателен (—М), если она пони-
жается. Мезомерный эффект, действующий в нереагирующей моле-
куле, называется статическим, а в реагирующей — динамическим
эффектом сопряжения. Более подробно об эффекте сопряжения см.
с. 102.
Кинетика и энергетика химических реакций. Раздел химии, занимающийся
изучением скорости и механизмов химических реакций с учетом всех факто-
ров, влияющих на эти процессы, называют химической кинетикой, которая яв-
ляется одним из разделов физической химии. Знание кинетики химических ре-
акций позволяет установить общие закономерности протекания органических
реакций с участием различных продуктов, использовать эти знания для прове-
дения реакций в нужном направлении с максимальным выходом необходимых
веществ, правильно построить технологию получения или переработки веществ.
Превращение исходных веществ в конечные продукты реакции протекает
через ряд промежуточных стадий. Иногда промежуточные продукты, образовав-
шиеся в ходе химической реакции, можно выделить и исследовать или же обна-
ружить с помощью физических методов; в других случаях предположение об их
возникновении подтверждается только косвенными данными. Скорость хими-
ческих реакций определяется изменением концентрации любого из участников
реакции в единицу времени. Реакции могут протекать при постоянном давлении
(изобарные реакции), неизменном объеме (изохорные реакции) или температуре
(изотермические реакции). Гомогенные реакции в ходе всего процесса проходят
в одной фазе (обычно газообразной или жидкой), гетерогенные — в нескольких
фазах. Скорость химической реакции пропорциональна концентрации реагиру-
ющих веществ и может быть выражена следующими уравнениями:
dCA
А -> продукты реакции (мономолекулярная реакция) v = —-----= kCA
dCA
А + Б -> продукты реакции (бимолекулярная реакция) и=— —— = kCACb
где v — скорость химической реакции, Сл, Сь—концентрации реагирующих
веществ к моменту времени т; т — время, k — константа скорости реакции (ско-
рость реакции, отнесенная к концентрации, равной единице).
Важной характеристикой в химической кинетике является порядок реакции,
который равен сумме показателей степеней при концентрациях в уравнении ско-
38
рости реакции. Порядок реакции определяется экспериментально по скорости
расходования реагирующих веществ, а не по суммарному стехиометрическому
уравнению, так как последнее является суммарным выражением и не учитывает
особенностей механизма реакций.
Для протекания химической реакции А + Б -> В должно произойти столк-
новение (соударение) молекул А и Б (теория столкновений). Число столкновений
ход реакции
Рис. 17. Изменение энергии си-
стемы в ходе реакции А + Б->В
пропорционально концентрации реагирующих
веществ. Однако эффективными (приводящи-
ми к образованию В) будут только те соуда-'
рения, в которых участвуют активные моле-
кулы, избыточная энергия которых не ни-
же необходимой для этого энергии, получив-
шей название энергии активации £а. Толь-
ко в этом случае удается преодолеть взаим-
ное отталкивание электронных оболочек реа-
гирующих молекул, осуществить разрыв ста-
рых и создание новых химических связей.
Схематически изменение энергии системы
А + Б в систему В показано на рис. 17. Ре-
агирующая система А и Б, энергия которой
(энтальпия) Яр для перехода в конечное со-
стояние В, энтальпия которого Н2, должна
иметь энергию, равную £1а (энергия актива-
ции). Активация молекул происходит при нагревании, поглощении квантов
излучения, растворении вещества, а также за счет теплоты, выделяемой в
ходе реакции, и т. д. Химическая реакция может сопровождаться выделе-
нием (экзотермические) или поглощением (эндотермические) теплоты. Тепло-
вой эффект реакции А Я равен разности между начальной Я4 и конечной
Я2 энтальпией. Реакция А + Б -> В экзотермичная, следовательно, вслед-
ствие уменьшения энтальпии системы А Я < 0; для эндотермических АЯ>0.
Как следует из рис. 17, обратная реакция В -> А + Б будет эндотермична, энер-
гия активации в этом случае равна: £2а = Яр + А Я и, следовательно, больше
энергии активации экзотермической реакции на величину теплового эффекта.
Зависимость между константой скорости реакции и энергией активации выра-
~^акт
жается уравнением Аррениуса: k = koe , где k — константа скорости
реакции; R — газовая постоянная; Т — температура.
Превращения исходных веществ в продукты реакции может происходить
через состояние активированного комплекса или переходное состояние. Схема-
тически это можно представить следующим образом:
А + БВ А • • • Б • • • В -> АБ + В
начальный переходное сос- конечный
этап
тояние
Теория переходного состояния предполагает,
что при достаточном сближении молекул А
и ВБ будет возникать взаимодействие между
А и Б, в то время как связь между Б и В бу-
дет ослабевать. На определенном этапе воз-
никнет активированный комплекс А ... Б ...В,
в котором связь между БВ ослаблена,
между АБ сформировалась не до конца и час-
тица Б в равной степени связана с А и В. По
мере приближения А и Б взаимодействие
между ними будет возрастать, а связь БВ
ослабевать, что в конечном итоге приведет к
ее разрыву.
Для перехода исходных продуктов в
продукты реакции через состояние активи-
этап
Рис. 18. Изменение энергии
системы в теории переход-
ного состояния
39
рованного комплекса необходимо преодолеть энергетический барьер, равный
величине энергии активации (Еа). Переходное состояние — это предельно не-
устойчивое состояние взаимодействующих между собой молекул, которое не
является промежуточным соединением. Время существования активированного
комплекса ничтожно мало. Комплекс обладает максимумом потенциальной энер-
гии (рис. 18), вследствие чего он неустойчив (на энергетической кривой ему со-
ответствует максимум).
Кислотность и основность. Согласно классическим представлениям (Брен-
стед — Лоури) кислотами называют вещества, являющиеся донорами протонов,
основаниями — акцепторы протонов. В органической химии широко пользуются
понятиями кислот и оснований по Льюису. В соответствии с этим понятием кис-
лота — это ионы или молекулы с незаполненной электронной конфигурацией,
которые могут присоединять электронную пару с образованием ковалентной свя-
зи (акцепторы электронов, электрофильные реагенты: Н+; BF3; А1С13; NO2;
+
Вг+; R — С == О и др.). Основания Льюиса — ионы или молекулы с заполненной
электронной конфигурацией, способные отдавать электронную пару с образова-
нием ковалентной связи (доноры электронов, нуклеофильные реагенты): NH3,
НО; R3N; Н2Ь; ROH; F“; С1~; Br~; I“; CN~.
В органической химии, так же как и в неорганической, широко применяется
понятие константы диссоциации /Са (константы кислотности):
ХН Н+ + Х“; Яа
[Н+] [XI
[НХ]
Поскольку для органических кислот меньше единицы (для СН3СООН
/Са = 1,76* 10'6), часто вместо нее употребляют величину рКа; p^a = —IgAa-
Кислотность и основность органических соединений, так же как и другие хими-
ческие свойства, зависят от строения молекул.
Классификация органических соединений. Органические соеди-
нения принято делить на несколько больших групп, в зависимости
от строения углеродного скелета молекулы (схема 1).
Схема 1. Классификация
органических соединений
В молекулах ациклических или
алифатических соединений (иногда
их называют соединениями жирно-
го ряда) атомы углерода соединены
в «раскрытые» (неразветвленные
или разветвленные) цепи, не содержа-
щие колец или циклов. Цикличе-
ские соединения делятся на две
группы: карбоциклические, содержащие циклы из атомов угле-
рода, и гетероциклические, в построении циклов которых участ-
вуют кроме углерода атомы других элементов — кислород, азот,
сера («гетерос» — по-гречески другой); каждая из названных групп
делится на классы. Самый простой класс органических соедине-
ний — углеводороды. Замещая водороды в их молекулах на функ-
циональные (характеристические) группы, можно получить разно-
образные производные углеводородов. Функциональные группы об-
ладают характерной реакционной способностью, определяют хими-
ческое поведение и принадлежность соединения к определяемому
классу (табл. 3).
40
Таблица 3. Основные классы производных углеводородов
Класс Функциональная (характеристическая) группа Примеры представителей
Г алогенпроизводные F, Cl, Вг, I Галоген С2Н5С1 — этилхлорид
Оксисоединения (спирты, фенолы) —ОН Оксигруппа (гид- роксил) С2Н5ОН — этиловый спирт С6Н5ОН — фенол
О и
Оксосоединения (карбониль- ные соединения — альде- гиды, кетоны) ^>с=о Оксо-группа (кар- бонил) II СН3—С—Н — уксусный альдегид О II СН3—С—СН3 — ацетон
О II С6Н3—С—СН3 — ацетофенон
Карбоновые кислоты _с/° \он Карбоксил СН3—С\ — уксусная кисло; а ХОН
Нитросоединения -КЮ2 Нитрогруппа СН3—СН2—NO2 — нитроэтан
Амины —nh2 Аминогруппа СН3—СН2—NH2 — этиламин
Сульфокислоты -SO3H Сульфогруппа СбН5—SO3H — бензолсульфокислота
г аздел первый
Углеводороды
Углеводороды — наиболее простые по составу
органические соединения. Их молекулы построены из атомов толь-
ко двух элементов — углерода и водорода. Общая формула CnHm.
Они различаются по строению углеродного скелета и характеру
связей между атомами углерода (схема 2). По первому признаку их
делят на ациклические (алифатические) углеводороды, молекулы
Схема 2. Классификация углеводородов
которых построены из открытых углерод-углеродных цепочек,
например гексан и изогексан:
СН3
I
сн3—сн2—сн2—сн2—сн2—сн3 сн3—сн—сн2—сн2—сн3
гексан изогексан
и циклические (карбоциклические) углеводороды.
Карбоциклические углеводороды, обладающие особыми свойст-
вами («ароматический характер»), получили название ароматических,
например:
42
н
с
н—н
I II
н—с с—н
н
н н
с с
// \/ч.
н—с с с—н
I II I
н—с с с—н
/\ //
с с
I I
н н
бензол
нафталин
Другие карбоциклические углеводороды, например циклогексан,
называются алициклическими:
СН£
Н2С СН2
I I
Н2С сн2
циклогексан
По характеру связей между углеродными атомами углеводороды
могут быть насыщенные, или предельные (алканы), и ненасыщенные
(непредельные). Последние могут содержать разное количество двой-
ных (алкены, алкадиены, циклоалкены и др.), тройных (алкины,
циклоалкины и др.) связей или те и другие одновременно:
СНз—СН=СН2
пропилен
(пропен)
СН3
I
сн2=с—сн=сн2
изопрен
(2-метил-1,3-бутадиен)
сн
НоС ^сн
I I
Н2С сн2
циклогексен
сн3—с==сн
метил ацетилен
(пропин)
сн3
I
сн3—с=сн—сн2—с=сн
2-метил-2-гексен-5-ин
Ациклические (алифатические)
углеводороды
алканы
Строение, понятие о гомологическом ряде, изомерия. Алканы —
алифатические углеводороды, в молекуле которых атомы углерода
связаны между собой и с атомами водорода ординарной связью (ог-
связь). Отсюда и другое их название — предельные, или насыщенные,
углеводороды. Родоначальник и простейший представитель алка-
43
нов — метан СН4. В молекуле метана, как и в молекулах других ал-
канов, атом углерода находится в состоянии $р3-гибридизации.
Общая формула соединений этого ряда СпН2п+2. Каждый после-
дующий его представитель отличается от предыдущего на группу СН2
(метиленовая группа, табл. 4). Такой ряд родственных органических
соединений с однотипной структурой, близкими химическими и за-
кономерно изменяющимися физическими свойствами называется го-
мологическим рядом; члены этого ряда — гомологами; группа СН2,
на которую отличаются два соседних гомолога, — гомологической
разностью состава.
Введение в органическую химию понятия гомологии позволило
систематизировать громадное число существующих соединений и
чрезвычайно упростило ее изучение. Достаточно знать свойства не-
большого числа наиболее типичных представителей гомологического
ряда, чтобы получить общее представление о химическом поведении
остальных гомологов.
Гомологический ряд алканов по названию его первого представи-
теля часто называют рядом метана. Три первых соединения этого ряда
не имеют изомеров. Начиная с бутана, наблюдается явление изомерии,
т. е. существование нескольких соединений с одинаковым качествен-
ным и количественным составом и одинаковой молекулярной массой,
но различными физическими и химическими свойствами.
Строение бутана С4Ню может быть представлено с помощью двух
формул:
сн3—сн2—сн2—сн3 сн3—сн—сн3
сн3
«-бутан изобутан
Такой вид изомерии называют структурной изомерией (в данном слу-
чае— изомерия углеродного скелета). Углеводороды с неразветвлен-
ной углеродной цепью называют углеводородами «нормального» стро-
ения (н-бутан). С увеличением числа углеродных атомов в молекуле
алкана число изомеров быстро возрастает; так, для углеводорода
С5Н12 можно написать формулы трех изомеров:
СН3 сн3
I I
СН3—СН2—СН2—СН2—СН3 СН3—СН—СН2—СН3 н3с—с—сн3
СН3
н-пентан изопентан неопентан
Гексан (С6) имеет 5 изомеров; декан (Сю) — 75, эйкозан (С20) —
336 319.
Структурные формулы, понятие о первичном, вторичном и тре-
тичном атомах углерода. Приведенные формулы изомеров бутана
и пентана называют структурными. Они показывают не только какие
атомы и в каком количестве входят в молекулу данного соединения,
но отражают порядок и характер связей между ними. Различают пол-
ную, или развернутую, структурную формулу
44
Таблица 4. Алканы (гомологический ряд метана)
Название Формула Температура плавления, °C Температура кипения, °C Плотность р, Г«СМ“ 3 Показатель преломления,
Метан сн4 —182,5 — 161,5 0,415 (—164°С) —
Этан СН3-СН3 —183,2 —88,6 0 ,546 (—88°С) —
Пропан сн3-сн2-сн3 — 189,9 —42,2 0,585 (—44°С) —
«-Бутан СН3-(СН2)2-СН3 — 138,3 —0,5 0,600 1,3543 (— 13°С)
Изобутан СН3-СН (СН3)-СН3 — 159,4 — 11,7 0,603 (19°С) 1,3514 (—25°С)
«-Пентан СН3—(СН2)3—сн3 — 129,7 36,1 0,631 (26°С) 1,3570 (16°С)
«-Гексан СН3-(СН2)4-СН3 —95,3 68,7 0,659 1,3750
«-Гептан СН3-(СН2)5-СН3 —90,6 98,4 0,684 1,3876 *
«-Октан СН3—(СН2)6—сн3 —56,8 125,7 0,704 1,3975
«-Нонан СН3-(СН2),-СН3 —53,5 150,8 0,717 1,4054
«-Декан СН3-(СН2)8-СН3 —29,7 174,1 0,730 1,4120
«-Гексадекан СН3—(CH2)i4—сн3 18,2 286,8 0,775 1,4345
«-Эйкозан СН3-(СН2)18-СН8 36,8 342,7 0,778 (37°С) 1,4340 ;43°С)
«-Триаконтан СН3-(СН2)28-СН3 66,1 446,4 0,778 (70°С) 1,4536
н н
н н—с —н н н—с —н н
I I I I I
н— с--------с------с--------с-с —н
I I I I I
Н Н Н Н—с —н н
н
изооктан
и краткую, или звеньевую:
6СН3
сн3 —сн—сн2—с—сн3
5 4 3 2 | 1
сн3
8
изооктан
В последнее время стали использовать еще более сокращенный
способ изображения структурных формул, при котором указывают
только связи, соединяющие углеродные атомы:
изооктан
В изооктане имеется четыре типа углеродных атомов: атомы
1, 5, 6, 7, 8 связаны только с одним углеродным атомом — такие атомы
углерода называют первичными, атом 3 с двумя — вторичный атом,
атом 4 с тремя — третичный углеродный атом. Углеродный атом 2 на-
зывается четвертичным. Соответственно первичными, вторичными и
третичными называются и связанные с ними атомы водорода.
Номенклатура. Существует несколько способов наименования
органических соединений: тривиальные (исторические) названия, ра-
циональная и систематическая номенклатуры.
Тривиальные названия обычно связаны с источниками, первыми
способами получения веществ, именами ученых или являются слу-
чайными. Они не говорят о структуре молекулы и в большинстве слу-
чаев возникли в начальный период развития химии. Названия орга-
нических соединений по рациональной и систематической номенклату-
рах указывают не только вид и число атомов, входящих в его состав,
но и дают представления о структуре молекулы.
Рациональная номенклатура, получившая развитие во второй по-
ловине XIX в., обычно рассматривает соединение как производное от
наиболее простого («типичного») представителя данного гомологиче-
ского ряда, образованного заменой атомов водорода на другие груп-
пы. Она нашла применение для наименования относительно простых
органических соединений.
46
Наиболее удобной, дающей возможность назвать любое соединение,
является систематическая номенклатура органических соединений.
В учебнике она будет использована в соответствии с правилами
ИЮПАК [правила Комиссии по номенклатуре органических соеди-
нений при Международном союзе Чистой и Прикладной химии —
International Union of Pure and Applied Chemistry — сокращенно
IUPAC (ИЮПАК)].
Первые четыре представителя алканов имеют случайные названия:
метан, этан, пропан, бутан. По существу тривиальными можно считать
и названия следующих алканов, хотя они являются производными
греческих числительных, соответствующих числу углеродных атомов
в молекуле алкана [за исключением нонана и ундекана, корни названия
которых латинские (табл. 4)1, общим для всех гомологов является
окончание «ан». Эти названия не дают представления о строении алка-
нов (нормальная, разветвленная цепь и т. д.) и поэтому однозначно
могут быть использованы только для наименования алканов нормаль-
ного строения. Для наименования алканов с разветвленной углерод-
ной цепью необходимо знать названия углеводородных радикалов —
алкилов, частиц, условно выделенных из молекулы углеводорода от-
нятием одного атома водорода. Их название получают заменой окон-
чания «ан» соответствующего алкана на «ил». Отсюда и их групповое
название «алкилы». Общая формула алкилов СлН2л+1. В формулах
органических соединений алкилы в общем виде обозначаются Aik
или чаще R. Учитывая, что от алканов, начиная от пропана, можно
оторвать атомы водорода от первичного и вторичного, а от изобутана
и многих последующих углеводородов — и от третичного углерод-
ного атома, алкилы, содержащие одинаковое количество атомов угле-
рода, могут иметь разное название, а в случае алкилов с разветвлен-
ной цепочкой оно может быть достаточно сложным. Наименования
наиболее распространенных алкилов приведены на с. 48.
По рациональной номенклатуре предельные углеводороды с раз-
ветвленной углеродной цепью рассматриваются как производные мё-
тана, образованные замещением атомов водорода в его молекуле на
соответствующие радикалы. Для того чтобы назвать по рациональ-
ной номенклатуре приведенный ниже углеводород тетрадекан СиН30
СН3 СН3 СН3
I I I
Н3С—НС---С----CHg---G--СН3
нс—сн2—сн3 'сн3
сн3
мысленно выбираем «центральный» атом углерода (соединенный с мак-
симальным количеством других атомов углерода) так, чтобы можно было
однозначно назвать связанные с ним радикалы. При построении на-
звания алкана радикалы располагают по старшинству, указывая ко-
личество однотипных (ди-, три-, тетра- и т. д.). Название завершает-
ся наименованием основного соединения — метана. Приведенный вы-
ше алкан СиН30 называется метилизопропилвторбутилнеопентил-
47
Радикал Метил Фор мула СН3-
Этил СН3СН2-
я-Пропил СН3СН2СН2—
Изопропил CH3CHCH3 1
н-Бутил СН3СН2СН2СН2-
Изобутил Вторичный бутил (в/пор-бутил) СН3СН(СН3)-СН2— СН3СН2СН-СН3
Третичный бутил (/прет-бутил) сн3 СНз-С— 1 сн3
Радикал Формула
w-Пентил сн3—сн2—сн2-сн,- сн2—
Изопентил сн3-сн—сн2—сн2— СНз сн3
Неопентил СН3—С-СН2— 1 сн3 СНз
Третичный пентил (трет-пентил) 1 сн3—сн2-с— СНз
«-Гексил СН3-СН2-СН2-СН2-СН.? сн2—
Изогексил сн3-сн-сн2-сн2-сн2— 1 СН3
метаном. Рациональная номенклатура используется обычно для на-
именования только простых алканов.
По номенклатуре ИЮПАК для наименования алканов нормаль-
ного строения (табл. 4) и небольшого числа с разветвленной цепью
СН3—СН—СН3
СН3
изобутан
сн3—сн—сн2—сн3
I
сн3
изопентан
сн3
I
сн3—с—сн3
СН3
неопентан
сн3—сн—сн2—сн2—сн3
I
сн3
изогексан
сохраняются старые традиционные названия. Родовое название гомо-
логического ряда предельных углеводородов — алканы, окончание
«ан».
Для наименования алканов, имеющих разветвленную углеродную
цепь, их рассматривают как производные предельных углеводородов
нормального строения, в молекуле которых вместо атомов водорода
введены соответствующие радикалы. Для построения названия таких
алканов выбирают самую длинную, имеющую максимальное число
заместителей («главную») цепь, нумеруя в ней атомы углерода цифра-
ми 1, 2, 3, 4 и т. д. Направление нумерации выбирают так, чтобы циф-
ры, указывающие положение боковых цепей, были наименьшими, это
соблюдается независимо от природы заместителей. При построении
названия первыми располагают наиболее простые заместители: метил,
этил, пропил и т. д. Перед названием радикала ставят номер угле-
родного атома главной цепи, с которым он связан, цифру от названия
отделяют черточкой. Количество одинаковых заместителей показы-
вают умножающим префиксом ди-, три-, тетра- и т. д., а номера угле-
родных атомов главной цепи, с которыми они связаны, располагают по
возрастающей величине и ставят перед названием радикалов. Послед-
ним называется углеводород главной цепи. Например, алкан
СН3 СН3
I I
сн3 сн2 сн-сн3
I I I
сн3 —с---сн— с—сн2—сн—сн2—сн3
1 2 | 3 4 | 5 6 | 7 8
сн3 сн3 сн3
по номенклатуре ИЮПАК называется 2,2,4,6-тетраметил-3-этил-4-
изопропилоктан.
Источники и способы получения. Алканы широко распространены
в природе, это важные компоненты нефти, природных и попутных
газов. Содержание их в нефти отечественного происхождения колеб-
лется от 30 до 89%. Низшие алканы, главным образом метан, этан,
пропан, — основные компоненты природных газов, причем доля ме-
тана достигает 97—98%. Смеси высокоплавких твердых алканов со
49
значительной молекулярной массой встречаются в природе в виде ми-
нерала озокерита, который после специальной обработки и очистки
может быть превращен в церезин—воскообразный продукт, приме-
няющийся в промышленности. Алканы с числом атомов углерода от 20
до 30 входят в состав восковых оболочек плодов, семян, листьев.
Основными источниками промышленного получения алканов яв-
ляются нефть, природные или попутные газы, каменный уголь, слан-
цы, твердые парафины, а также продукты, получаемые при их хими-
ческой переработке. Из нефти алканы выделяют фракционной пере-
гонкой или получают разнообразными методами химической пере-
работки (см. с. 145); из каменного угля — гидрированием. Обычно
используют низкосортные бурые угли. Тонко измельченный и сме-
шанный с тяжелым маслом уголь обрабатывают водородом при тем-
пературе 400—600° С и давлении 202 • 105 — 707 • 105 Па в при-
сутствии катализатора (оксиды железа). Образуется смесь, состоя-
щая из большого числа разнообразных соединений; из нее после
фракционирования, а в отдельных случаях — дополнительной об-
работки — получают бензины, смазочные масла и другие промышлен-
ные продукты. В последнее время, особенно в странах, бедных нефтью,
распространение нашел синтез углеводородов из оксида углерода и
водорода. Источники — генераторный газ, водяной газ и синтез-газ,
получаемые при переработке каменного угля или природного газа.
Так, синтез-газ может быть получен из метана и его ближайших гомо-
логов при взаимодействии с водяным паром, диоксидом углерода или
кислородом (t = 750—900°С):
СН4 + Н2О СО + ЗН2; СН44- СО2 2СО + 2Н2; СН4 + 0,5О2 СО + 2Н2
В качестве катализатора применяют никель, нанесенный на оксид
алюминия, или магнезит, с добавками небольших количеств меди,
оксидов магния или хрома. Для синтеза алканов применяют очищен-
ную смесь оксида углерода и водорода (Г.2); кобальт-торий-магниевый,
железо-медный или сплавные железные катализаторы (/ = 170—
-320° С):
п СО + (2м + 1) Н2 -> Сл Н2п+2 + п Н2О
Образовавшаяся смесь алканов называется «синтином».
В лаборатории алканы получают из соединений с меньшим числом
углеродных атомов, чем в синтезируемом продукте, с тем же числом
атомов углерода путем замены заместителя на водород и из соединений
с большим числом углеродных атомов в результате разрушения угле-
родного скелета молекулы исходного вещества. Основные лабора-
торные способы получения метана и его гомологов следующие.
Получение алканов из соединений с мень-
шим числом атомов углерода (синтетические способы).
Реакция Вюрца, Взаимодействие металлов с моногалогенпроиз-
водными алканов:
СН31 + СНУ1 + 2Na СН3—СН3 + 2Na
мс тилиодид этан
50
Открыта в 1855 г. французским ученым Вюрцем. Наш соотечест-
венник П. П. Шорыгин установил, что конечный продукт образуется
в результате следующих промежуточных превращений: а) образова-
ние металлорганического соединения
CH3I + 2Na -> CH3Na + Nal
метил ио дид метили атрий
б) синтез алкана
CH3Na + CH3I -> СН3—СН3 + Nal
этан
Кроме натрия можно использовать литий, цинк, магний и некото-
рые другие металлы. Этой реакцией пользуются для синтеза алканов
с четным числом углеродных атомов в молекуле из первичных гало-
геналкилов (атом галогена находится у первичного углеродного ато-
ма). Если в реакции участвуют различные галогенпроизводные, об-
разуется смесь алканов, например:
------- СН3—СН3
СН3 +сн3—СН2—СН21 4- 2Na---------------> СН3—СН2—СН2—СН3
—2NaI
-----_> СН3—(СН2)4—сн3
Разделить эту смесь на отдельные компоненты трудно. Галогеналкилы
по своей реакционной способности в реакции Вюрца располагаются
в следующем порядке: R—I>R—Br>R—Cl. Третичные галоген-
производные в этих условиях образуют алкены (с. 70).
Электролиз солей одноосновных органических кислот. Этот метод
был предложен немецким ученым Кольбе (1894) и получил название
метода Кольбе;
zO
/7 электролиз
2R—С--------------> R— R + 2МеОН + 2СО2 + Н2
хОМе
соль карбоновой
кислоты
Так, электролиз водного раствора натриевой соли капроновой кис-
лоты протекает следующим образом:
на аноде
,0 -------> СН3— (CH2)S—СН3 + 2СО2
сн3—(СН2)4—С + 2Н2О ^.тролиз.
\0Na ^^2NaOH + H2f
На аноде в качестве промежуточных продуктов образуются пере-
кисные соединения и свободные радикалы:
О
II
q СН3—(СН2)4—с—о
2СН3—(СНД4—С -> О ------->
\г)- II
и СН3—(СН2)4—С—О
51
о
II
СН3—(СН2)4—С—О. СН3—(СН2)3—СН2 . + СО2
о
II
СН3—(СН2)4—С—о. сНз—(СН2)з—СН2 . + СО2
-> СН3 (СН2)8—СН3
Этим способом могут быть получены алканы с четным числом ато-
мов углерода в молекуле.
Получение алканов из соединений с тем же
числом атомов углерода. Получение (восстановление)
из производных предельных углеводородов (галогенпроизводных, спир-
тов, кислот, металлорганических соединений). При получении алканов
из этих производных необходимо заменить соответствующую группу
на атомы водорода (удаление заместителя). Иногда это удается легко,
например действием воды:
/СН3
Zr/ + 2Н0Н -> 2СН4 + Zn (ОН)2
ХСН3
цинкдиметил метан
CH3CH2MgI + НОН 2С2Нб + Mg(OH)I
этил магнийио дид этан
В других случаях требуется применение специальных восстано-
вителей или катализаторов (например, LiAlH4). В качестве восстано-
вителя при получении алканов из иодпроизводных алканов приме-
няется иодистоводородная кислота при нагревании (Бертло, 1868):
СН3—СН2—СН2—СН2—СН2—CH2I + HI ->
1-иодгексан
-> СН3—СН2—СН2—СН2—СН2—СН3 + 12
гексан
Восстановление галогенпроизводных алканов может быть проведено
водородом, образовавшимся при взаимодействии металла с кисло-
той:
Zn; НС1
СН3—СН—СН3--------> СН3—СН—СН3
Вг Н
2-бромпропан пропан
Восстановление молекулярным водородом возможно только в
присутствии катализаторов (платина, палладий, никель). Восста-
новление спиртов и кислот будет рассмотрено на с. 218, 219.
Гидрирование непредельных соединений. Алканы могут быть полу-
чены присоединением водорода к непредельным углеводородам в
присутствии катализатора (платина, палладий, никель):
52
Н2; кат.
Н3С—СН2—СН2—СН2—СН=СН2.
1-гексен
Н3С-(СН2)4-СН3
гексан
Н2; кат.
С2Н2 > С2Нб
ацетилен этан
Эта реакция будет подробно рассмотрена в следующих разделах.
Получение алканов из соединений с боль-
шим числом углеродных атомов. Сплавление солей
одноосновных предельных кислот с едкими щелочами (реакция декар-
боксилирования):
СН3—С + NaOH СН4 + Na2CO3
^ONa
уксуснокислый метан
натрий
Физические свойства. Четыре первых представителя ряда метана — газо-
образные вещества, начиная с пентана (С5) до гексадекана (С1б) углеводороды
нормального строения — жидкости, с С17 и выше — твердые вещества. В ряду
метана для алканов нормального строения по мере роста молекулярной массы
(см. табл. 4) наблюдается увеличение температур кипения и плавления, а также
плотности. Разница в температурах кипения соседних гомологов у нормальных
алканов С6—С10 составляет 20—30° и постепенно она уменьшается до 15° у угле-
водородов С15—С2о. Алканы с разветвленной цепью углеродных атомов кипят
при более низких температурах по сравнению с алканами нормального строения;
с увеличением числа заместителей, превращением молекул в более разветвлен-
ные разница в температурах кипения алканов нормального и разветвленного
строения возрастает. Эта же закономерность наблюдается и для плотности. Это
хорошо видно из табл. 5.
Указанные закономерности становятся понятными, если вспомнить, что для
перехода жидкого вещества в газообразное необходимо преодолеть межмолеку-
лярные взаимодействия. На температуру плавления алканов в кристаллическом
состоянии большое влияние оказывает строение молекул, их способность «упа-
ковываться» в кристаллы. Поэтому температура плавления приведенного ниже
2,2-диметилбутана —98,2°С» а 2,3-диметил-
бутана—1356С.
Все алканы легче воды, их плотность ( 'lSx'
не превышает 0,8 г«см“3.
Алканы практически нерастворимы в V-У \___/
воде, но хорошо растворимы в органичес-
ких растворителях. Метан, этан и высшие Рис. 19. Строение цепочки ал-
гомологи не имеют запаха, средние облада- кана
ют запахом бензина. В молекулах алканов
цепочки углеродных атомов имеют зигза-
гообразную форму (рис. 19).Угол между валентностями составляет 109°28';
центры углеродных атомов расположены друг от друга на расстоянии 0,154
нм, расстояние от центра углеродного атома до центра атома водорода 0,11 нм.
Химические свойства. Кислоты (серная, азотная), щелочи (гидр-
оксиды натрия и калия), окислители, металлы, в том числе и щелоч-
ные, при обычных условиях на алканы не действуют. Отсюда и воз-
никло их старое название «парафины» от латинского «parum affinis» —
лишенные сродства.
Алканы не способны к реакциям присоединения, так как в их мо-
53
Таблица 5. Сравнительная характеристика физических свойств гексанов
Название Формула Темпера- тура кипения, °C Темпера- тура плав- ления, °C Плотность р, г-см~3
н-Гексан СН3—(СН2)4—СН3 68,7 —95,3 0,659
2-Метилпешан сн3—сн—сн2—сн2-сн3 1 сн3 60,0 — 153,7 0,654
З-Метилпентан СН3—СН —сн—сн,—сн3 1 сн3 63,0 -118 0,664
2,3-Диметилбу- тан сн3—сн—сн—сн3 1 1 сн3 сн3 сн3 58/) — 135,1 0,668 (при 17°С)
2,2-Диметилбу- тан 1 сн3—с—сн2—сн3 1 сн3 49,7 —98,2 0,649
лекулах все связи насыщены. В то же время на солнечном свету
они легко вступают во взаимодействие с хлором и другими галогена-
ми; в паровой фазе нитруются, а при повышенных температурах окис-
ляются и проявляют способность к разложению (термическое разло-
жение). Следовательно, для алканов характерны реакции замещения,
разложения, окисления.
Причина химической инертности алканов в особенности строения
их молекул. В молекулах алканов содержатся только о-связи (С—С
и С—Н), для которых характерна малая полярность и поляризуе-
мость. Поэтому для предельных углеводородов в первую очередь ти-
пичны реакции, идущие по радикальному типу. Реакции же, иду-
щие по ионному типу, присущи соединениям с полярными или легко
поляризующимися связями. Для гомолитического разрыва связей С—С
и С—Н с высокой энергией (339,4—347,8 и 398,0—414,8 кДж/моль
соответственно) требуются повышенные температуры или излучение,
богатое энергией.
Взаимодействие с галогенами. На свету, в темноте при нагрева-
нии (250—400° С) или в присутствии катализаторов (хлориды меди,
сурьмы, олова или иод) газообразный хлор последовательно заме-
щает в молекуле метана все четыре атома водорода с образованием
галогенпроизводных:
СН4 + С12 -> СН3С1 4- НС 1; СН3С1 + С1у -> СН2С12 + НС1
метан хлормста.) дихлорметан
СН2С12 + С12 -> СНС13 + НС1; СНС13 + С12 СС14 + НС1
хлороформ четырех-
хлор истый
углерод
54
Бромирование алканов протекает менее энергично, чем хлориро-
вание, взаимодействие с иодом осуществить не удается (реакция эндо-
термична, обратима). Фтор взаимодействует с алканами со взрывом,
что делает невозможным, без принятия специальных мер, их прямое
фторирование. Следовательно, в реакциях с метаном и его гомолога-
ми галогены располагаются по своей реакционной способности в сле-
дующем порядке:
F > Cl > Вг > (I)
не реагирует
Этот порядок сохраняется и при взаимодействии галогенов с боль-
шинством других органических соединений.
Галогенирование алканов под действием света или высоких темпе-
ратур протекает по цепному радикальному механизму и является
типичным примером реакции радикального замещения (S н):
Хлорирование метана начинается с распада под влиянием УФ
излучения (фотохимическое хлорирование) или повышенных темпе-
ратур (термическое хлорирование) молекулы хлора (инициирование
цепи; I):
Ь; t°
Cl : Cl----Cl - + С1. (I)
Атом хлора атакует молекулу метана с образованием частицы СН3,
несущей один неспаренный электрон (свободный радикал):
С1. +СН4 -> СН3.4-НС1 (II)
метил
При взаимодействии метила со второй молекулой хлора образу-
ются хлорметан и новый атом — хлор:
сн3 + С1: С1 СН3С1 + СЬ (III)
который со следующей молекулой метана образует новый свободный
радикал метил и хлороводород и т. д. Такие реакции называются
цепными.
Этап (реакции II, III), в ходе которого происходит исчезновение
одних и появление других свободных радикалов, получил название
продолжение или развитие цепи. Процесс продолжается до тех пор,
пока не прекратят свое существование свободные радикалы, веду-
щие эту же цепь превращений («обрыв» цепи, реакции IV; V; VI):
Сн; + С1 СН3С1 (IV); С1 • 4-С1. Cl2 (V);
сн’ + сн’-> СН3—СН3 (VI)
Для получения продуктов хлорирования метана со значительным
55
выходом развитие цепи должно проходить с большей скоростью, чем
ее обрыв, а следовательно, концентрация свободных радикалов долж-
на быть невысокой по сравнению с концентрацией других молекул.
Реакции радикального замещения инициируются свободными ра-
дикалами или веществами, легко их образующими (инициаторами),
протекают с высокими скоростями и могут тормозиться или полностью
Рис. 20. Энергетическая диаграмма реакции ради-
кального хлорирования метана
прекращаться при внесении небольших количеств веществ, активно
взаимодействующих с радикалами (ингибиторы).
Возможность осуществления, скорость и направление реакций
радикального замещения алканов (S R) определяются их энергетикой,
прочностью С—Н-связей в молекуле алкана, устойчивостью (ста-
бильностью) образующихся в качестве промежуточных продуктов
свободных радикалов, характером заместителей и рядом других
факторов. Остановимся на некоторых из этих вопросов подробнее, так
Таблица 6. Тепловые эффекты галогенирования метана ДЯ, кДж*моль-1
Д/7, кДж-моль-1, при действии
Стадия г2 С1? Вг? 12
1. Г2 > 2 г. 2. СН3Н + Г- ->СН3.+НГ з. сн3-ч-г2 -> сн3г + г- 4-135,0 — 134,1 —293,3 4-243,0 —4,19 -96,37 4-192,7 4-62,8 —87,9 4-150,8 4-138,3 —75,4
S стадий 24-3 —427,4 — 100,6 —25,1 + Ь2,8
56
как многие из существующих тут закономерностей носят общий ха-
рактер.
Реакция (I) эндотермична, ее тепловой эффект ЛЯ равен затра-
чиваемой энергии (табл. 6, рис. 20). Для осуществления реакции (II)
должно произойти столкновение атома хлора с молекулой метана, об-
ладающей необходимой энергией активации £а и соответствующей
ориентацией. Тепловой эффект реакции ЛЯ будет равен разности
начальной и конечной энергии системы. Превращение исходных ве-
ществ в продукты реакции происходит через переходное состояние,
которому соответствует максимум потенциальной энергии системы
(рис. 20):
исходные вещества
(тетраэдрический уг-
лерод: sp3)
переходное состояние (углерод
постепенно становится триго-
нальным)
продукты реакции
(тригональный
углерод; sp2)
Образование алкильного радикала является лимитирующей ста-
дией и определяет скорость SR реакции. Энергия диссоциации С—Н-
связи ЛЯ зависит от характера углеродного атома (первичный, вто-
ричный, третичный; табл. 7). Поэтому при галогенировании алканов
легче всего замещаются атомы водорода у третичного, труднее у
вторичного и с еще большим трудом у первичного атома углерода.
Последнее место в этом ряду занимает метан.
Таблица 7. Энергия диссоциации Д/f, активации Еа и относительная
реакционная способность С—Н-связей в реакциях радикального галогенирования
Разрывающаяся связь С—н Энергия диссоциации С—Н-св язи АН, кД ж/моль Энергия активации Ё&, кДж/моль Относительная реак- ционная способность С—Н-связи
СЬ Вг- С12 Вг2
CHS—Н 427,4 16,8 75,4
сн3—сн2—н СНз 406,4 4,2 54,5 1 1
\сн—н СНз СНз 93,9 2,0 43,6 3,9 8,2
сн3—с—н СНз 381,3 1,3 31,4 5,1 1600
По мере роста активности заместителя и повышения температуры
реакции селективность замещения снижается (F<Cl<Br). Поэтому
на практике при галогенировании алканов образуется сложная смесь
57
продуктов, это связано с суммарным действием всех перечисленных
ранее факторов. Так, при галогенировании изобутана образуются:
сн3
I '
СН3—СН—СН3
изобутан
СН3
CHj
С12; ftv; 25°С > —CH—CH2CI + СН3—С—СН3
галогени- 1-хлор-2-метилпро- 1 пан (64%) С1 2-хлор-2-метил-
рование пропан (36%) СН3 СН3 -ВГг:. ?\;„.!27°C_^ СН-,—CHCH-Rr 4- CH»—CH—СН
Ьбром-2-метил- I
пропан (1%) Вг
2-бром-2-метил-
пропан (99%)
Кроме того, нужно помнить, что вероятность замещения девяти ато-
мов водорода при первичных углеродных атомах больше, чем заме-
щение одного водородного атома у третичного углерода. Хлориро-
вание в присутствии катализаторов протекает по цепному ионному
механизму:
С1:С1 + А1С13 -> А1С14 + С1+; С1+ + Н:СН3 -> СН3+ НС1
СНз + С1:С1 -> СН3С1 + С1+ и т. д.
В лабораторной практике обычно не применяют прямое галоге-
нирование для получения галогенпроизводных алканов, в технике
значительное применение нашло галогенирование метана для синтеза
СН3С1; СН2С12; СНС13 и СС14.
Нитрование. При замене атомов водорода в молекуле алканов на
нитрогруппу —NO2 образуются нитросоединения. Процесс называ-
ется нитрованием. При обычной температуре азотная кислота не дей-
ствует на алканы, при нагревании — окисляет. М. И. Коновалов в
1889 г. предложил методы нитрования алканов разбавленной
(13%-ной) азотной кислотой при температуре 130—140° С и повышен-
ном давлении:
СН3 СН3
СН3— CH—СН2—СН2—CH3 + HO1NO2 -> СН3 —С—СН2—СН2—СН3
no2
2-метилпентан 2-нитро-2-метил пентан
Практическое значение приобрело нитрование газообразных алка-
нов парами азотной кислоты или оксидами азота (парофазное нитро-
вание). Процесс в зависимости от строения алкана, характера нит-
рующего агента ведется при температурах 200—500° С и давлении
до 15 • 105 Па.
Так же, как при галогенировании алканов, нитрогруппа легче
замещает водород у третичного атома углерода, труднее — у вторич-
ного и с еще большей трудностью — у первичного. С повышением
58
температуры селективность резко уменьшается. При парофазном ни-
тровании наряду с нитропроизводными исходного углеводорода об-
разуются нитросоединения с меньшим числом атомов углерода в мо-
лекуле вследствие деструкции (разрушения) углеродной цепи — де-
структивное нитрование. Оно протекает более интенсивно с увели-
чением длины углеродной цепочки и повышением температуры. В ре-
зультате образуется сложная смесь нитросоединений:
2-нитропропан
нитроэтан
Нитрование сопровождается частичным окислением алканов, с по-
вышением температуры содержание продуктов окисления возрастает.
Нитрование алканов протекает по свободнорадикальному механизму:
HNO3 -> но • Ч- NO2. RH + НО» R. 4- Н2О
R. + HNO3 -> RNO + НО» RH + NO2» -> R. + HNO2
Сульфирование. Серная кислота при обычной температуре не
взаимодействует с алканами, при нагревании действует как окисли-
тель. Сульфируются предельные углеводороды, содержащие не менее
восьми атомов углерода, при этом атом водорода в молекуле алкана
замещается на сульфогруппу SO3H:
59
НС
С8Н17Н + ^>S02 -> C8H17SO2OH + Н20
но
октан сульфооктан
Практически получение сульфокислот и их производных стало
возможным после открытия реакций сульфохлорирования и сульфо-
окисления.
Соли сульфокислот с 10—18 атомами углерода, широко применяю-
щиеся в качестве моющих средств (см. с. 416), могут быть получены
сульфохлорированием керосиновых фракций нефти:
/1М
с„н2„+2 + SO2 + С12----> C„H2„+1SO2CI + НС1
алкилсульфохлорид
C„H2n+1SO2CI + 2NaOH -> C„H2n+iSO2ONa + NaCl + H2O
алкилсульфонат
Сульфохлорирование проводят при комнатной температуре, бар-
ботируя диоксид серы и хлор через жидкие алканы при облучении
ультрафиолетовым светом. Реакция протекает по цепному радикаль-
ному механизму. Легче всего замещается водород у вторичного угле-
родного атома, труднее у первичного и практически не участвует в
реакции из-за пространственных затруднений водород у третичного
углеродного атома.
При сульфоокислении алканов реакция протекает по свободно-
радикальному механизму; при этом образуется смесь сульфокислот,
содержащих сульфогруппы главным образом у вторичных углеродных
атомов:
СлН2Л+2 4" so2 4" 0,5 О2 —> СлН2Л+1 SO2OH
Реакция сопровождается частичным окислением алканов.
Окисление. Предельные углеводороды при комнатной температуре
очень стойки к действию обычных окислителей (КМпОг, КгСг2О7).
При высокой температуре алканы сгорают с образованием диоксида
углерода, воды и выделением большого количества теплоты (полное
окисление):
СлН2Л+2 4- (Зп 4- 1)О2 -> пСО2 4- («4- 1) Н2О
В последние годы получили промышленное применение разнооб-
разные методы неполного окисления парафиновых углеводородов
при средних температурах в жидкой или газообразной фазе чистым
кислородом или кислородом воздуха (аутоокисление) обычно в при-
сутствии катализаторов или с помощью других окислителей (МпОг)»
В результате образуются спирты, альдегиды, кетоны, кислоты:
60
Неполное окисление может проходить без разрыва и с разрывом
углеродной цепи. В первом случае продукты реакции содержат столь-
ко же атомов углерода, сколько их было в молекуле исходного алкана,
во втором случае — меньше. Неполное окисление алканов — цепной
радикальный процесс с вырожденными разветвлениями. Теоретиче-
ские основы этих процессов сформулированы в работах А. Н. Баха,
Энглера, Н. Н. Семенова, Н. М. Эмануэля и других ученых. Реак-
ция протекает за счет образования при окислении алканов углеводо-
родных и алкилперекисных радикалов (неразветвленная реакция)
и за счет радикалов, образующихся при распаде перекисей и гидро-
перекисей (вырожденное разветвление), рождающих новые главные
цепи. Реакция инициируется с помощью специальных веществ или
за счет углеводородных радикалов, возникающих при окислении
алканов кислородом воздуха при повышенной температуре. Общая
схема реакций в последнем случае будет иметь следующий
вид:
RH + О2 -> R- -г НОО- (I) инициирование
R* + О2 ROO (II); ROO + RH -> ROOH 4- R. (Ill) развитие цепи
и т. д.
Алкилгидроперекиси ROOH, возникающие при взаимодействии
перекисных радикалов ROO* с молекулой алкана, являются первыми
(первичными) продуктами окисления. С увеличением числа угле-
родных атомов в алканах их стойкость к окислителям сни-
жается.
В соответствии с ранее описанными закономерностями при взаимо-
действии перекисных радикалов с я-алканами образуются в основном
вторичные, с изоалканами — третичные гидроперекиси:
Ri
I
R—СН—R15 R-С—R2
I I
О—ОН о—он
Алкилперекиси в ходе реакции частично распадаются (вырож-
денное разветвление цепей) с образованием радикалов, инициирующих
новые цепи превращений (II—VI):
R—ООН -> RO* + *ОН (IV) инициирование
RO- + RH -> ROH + R. (V); R. + О2 -> ROO* (II) ]
> развитие цепи
ROO.+ RH -> ROOH+R. (Ill): .OH + RH -> R.+Н2О (VI)J
В результате получаются вторичные продукты окисления — спир-
ты, альдегиды (кетоны), кислоты, часто с меньшей молекулярной
массой, чем исходный алкан, а также непредельные соединения. Так,
при деструктивном окислении бутана образуются следующие
продукты:
61
о
II
-> сн3он + сн3—сн2—с—н
метанол
СНз-СН,-СН2-СН3----> СН3—СН2—СН2ОН
1-пропанол
пропаналь
+ НС
\н
муравьиный
альдегид
СгНзОН+СНз-с'7
\н
этанол уксусный альдегид
Для получения требуемых соединений подбираются соответствующие
условия проведения реакции с максимальным выходом нужного про-
дукта.
Особое значение имеет окисление алканов в высокомолекулярные
карбоновые кислоты, которые затем используются для производства
мыла и других моющих средств. Исходным сырьем для получения
синтетических карбоновых кислот служит смесь парафинов, основная
масса которых представлена углеводородами С16—С28. Окисление
проводят кислородом воздуха с использованием соединений мар-
ганца при температуре 100—110° С. При этом образуются кислоты
с числом атомов углерода от Q до С20. Наибольшую ценность в про-
мышленности имеют кислоты С7—С9 и С10—С16. Карбоновые кислоты
С10—С16 применяются для мыловарения, получения синтетических
моющих средств, производства консистентных смазок, в производстве
синтетического каучука, в промышленности строительных материа-
лов (линолеум, асбестосмоляные плиты, заменители битума). Основ-
ной потребитель кислот С7—С9 — промышленные предприятия, про-
изводящие синтетические спирты, используемые в качестве пласти-
фикаторов морозостойких пластмасс.
Термическое разложение. Алканы, как и большинство органических
соединений, устойчивы только при сравнительно невысоких темпера-
турах. При повышении температуры они разлагаются с разрывом
связей С—С (реакции расщепления) и С—Н (реакции дегидри-
рования). Химические процессы, происходящие при термическом раз-
ложении углеводородов, называются крекингом и имеют большое
народнохозяйственное значение.
Крекинг может быть осуществлен без катализатора (термический
крекинг) и с его участием (каталитический или термокаталитический
крекинг). Термическое разложение при более высоких температурах
(700—800° С) в технике называется пиролизом. Пиролиз приводит к
более глубоким изменениям алканов и частичному образованию угле-
рода. При термическом разложении алканов разрыв углеродной цепи
и дегидрирование сопровождаются изомеризацией и циклизацией ал-
канов.
Характер образующихся при крекинге продуктов зависит от стро-
ения алкана и термического воздействия (температуры, продолжи-
62
тельности нагрева и т. д.), а также от наличия и вида катализатора.
Так, при крекинге н-бутана могут образовываться
СН3
сн2
I
сн2
СН3
-* сн3—сн3 + сн2=сн2
— сн2=сн—сн3 + сн4
СН2=СН—СН2—СН3 4- н2
СН2=СН—СН=СН2 + 2Н2
сн3—сн3 + сн4 + с
2СН=СН + ЗН2
Механизмы термического и каталитического крекинга различны.
При термическом крекинге происходит гомолитическое расщепление
С—С-связи с образованием свободных радикалов:
СН3 (СН2)б сн3 —сн3сн2сн2. + сн3сн2сн2сн2сн2.
свободные углеводородные радикалы
Свободные радикалы вследствие высокой реакционной способности
существуют непродолжительное время. Продолжительность жизни
наиболее устойчивых метильных и этильных радикалов 10"3—10’4 с.
Сложные радикалы превращаются в более простые радикалы и ал-
кены:
R—СН2СН—СН3 -> R. 4-СН2=СН—СН3
I
1
пропен
Этот процесс может продолжаться с образованием в конечном итоге
наиболее простых метильных и этильных радикалов, алкенов к ато-
марного (моно) водорода
₽ -----> RP 4- СН2=СН2
।
1 этилен
Ri—СН—СН2«-----
LL,
Н ------>RX—СН=СН2 4- Н.
алкен
Образовавшиеся метильные и этильные радикалы реагируют с моле-
кулами алкана:
сн3. 4- R—СН2—СН2—СН3 ->СН4 4- R—сн2—СН—СН3
Образовавшийся радикал вступает в дальнейшие превращения. При
соединении двух радикалов образуются алканы более сложного строе-
ния (рекомбинация, 1) или алканы и алкены (диспропорционирова-
ние, II):
------> R-CH2—СН2—Ri (I)
R. 4-Ri—СН2—СН2.--
------> RH4-R1—СН=СН2 (II)
алкан алкен
63
Распад свободных радикалов, как правило, идет по p-связи по от-
ношению к углеродному атому, имеющему неспаренный электрон.
При каталитическом крекинге разрушение молекулы алканов
происходит гетеролитически с образованием ионов карбония (см.
с. 74).
Наибольшей термической устойчивостью обладает метан (до 800°С);
с ростом длины и увеличением разветвленности углеродной цепочки
термостойкость молекулы алканов снижается.
Отдельные представители. Применение. Простейший представи-
тель алканов — метан СН4. Другое название метана — болотный
газ; оно связано с его образованием при гниении растений на дне болот.
Метан, встречающийся в угольных шахтах, где скопления его могут
привести к взрывам, получил название рудничного газа. Это основной
компонент природных газов (до 98%), в значительных количествах
метан присутствует в газах нефтепереработки. Он широко применя-
ется как топливо в технике и быту, является важным сырьем в хи-
мической промышленности (схема 3).
Схема 3. Промышленное использование метана
Смеси пропана и бутана используются в качестве топлива и сырья
для получения спиртов, альдегидов, кетонов, кислот, нитропарафи-
нов, пропилена, бутадиена. Пентаны идут на образование изопрена
QHS.
64
В результате переработки алканов получаются диены, ацетилен.
}Кидкие алканы применяются в качестве моторного топлива, высшие
парафины — как сырье для химической переработки. В последние
годы газообразные алканы (метан, этан, пропан, бутан) и жидкие
алканы нормального строения с 10—20 атомами углерода в молекуле
используются для получения микробиологическим способом белко-
вых препаратов, аминокислот, липидов.
АЛКЕНЫ
Алкенами, или этиленовыми углеводородами, называются соеди-
нения, содержащие в молекуле двойную связь ^с=С^ , поэтому
они относятся к группе непредельных алифатических углеводородов.
Общая формула гомологического ряда СПН2П, т. е. алкены содержат на
два атома водорода меньше, чем алканы с тем же числом атомов угле-
рода. Старое название соединений этого ряда — олефины. Голланд-
ские химики обнаружили, что этилен при соединении с хлором об-
разует маслянистое вещество, поэтому он получил название масло-
родного газа (по-латински oleum — «масло»).
Гомологический ряд, строение, номенклатура, изомерия. Гомоло-
гический ряд алкенов начинается с этилена С2Н4. В молекуле этилена
атомы углерода находятся во 2-м валентном состоянии ($р2-гибри-
дизация), т. е. из четырех валентных орбиталей (одна s- и три р-орби-
тали гибридизованы только три (s- и две р-). Поэтому в молекуле эти-
лена пять a-связей, лежащих в одной плоскости, и одна л-связь, об-
разованная негибридизованными р-орбиталями, оси которых лежат
в плоскости, перпендикулярной плоскости расположения о-связей,
образующих молекулу этилена (см. рис. 15). Следовательно, двойная
связь между атомами углерода образована и о-, и л-связью. Расстоя-
ние между углеродными атомами, связанными двойной связью, равно
0,134 нм, валентные углы — 120°. Наличие в молекуле алкенов двой-
ной связи — отличительная особенность этой группы углеводородов
и определяет их поведение в химических реакциях. •
Химические формулы и названия первых представителей ряда
этилена приведены в табл. 8.
По рациональной номенклатуре олефины рассматриваются как
производные этилена, в молекуле которого один или несколько ато-
мов водорода заменены углеводородными радикалами. Так, например,
СН2=СН—СН2—СН3 будет называться этилэтилен.
В отдельных случаях требуется уточнить положение радикалов,
что достигается указанием на симметричность или несимметричность
молекулы:
1 2
СН3—С=СН.2
СН3
несимм. диметилэтилен
По номенклатуре ИЮПАК родовое название углеводородов с
3—682 65
Таблица 8. Алкены (гомологический ряд этилена)
Название Формула Температура плавления, °C Температура кипения, °C Плотновть р, г/см3 Показатель преломления 20 nD
Этилен, этен сн2=сн2 —169,4 —103,8 0,570 (при —103,8°С) 1,3630 (—103,8°С)
Пропилен, пропен сн3-сн=сн2 —185,2 —47,0 0,609 (при —47°С) 1,3567 (—70°С)
1-Бутен сн2=сн—сн2—сн3 —139,3 —6,3 0,740 (при — 110°С) 1,3792
2-Булен} цис- сн3 СНз /с=<\ — 139,3 3,7 0,724 (при —78,5°С) 1,3930 (—25°С)
транс- Изобутилен, 2-метилпро- II н Н СН3 /с==с\ СНз Н СН3—с=сн2 — 105,8 — 140,7 0,9 —6,6 0,604 0,695 (при —70°С) 1,3848 (—25°С) 1,3926 (—25°С)
пен 1-Пентен СНз сн3—сн2—сн2—сн=сн2 — 138,0 30,2 0,640 1,3714
1-Гексен СНз (СН2)3СН=СН2 — 139,8 63,5 0,674 1,3877
1-Гептен СНз (СН2)4—сн=сн2 — 119 93,6 0,697 1,3998
1 -Октен СН3(СН2)5-СН=СН2 — 102 122,0 0,715 1,4090
двойной связью — «алкены». Для наименования алкенов с разветв-
ленной углеродной цепочкой выбирают самую длинную углеродную
цепь, включая в ее состав ненасыщенные углеродные атомы, давая
ей название соответствующего алкана с заменой окончания «ан» на
«ен». Нумерацию проводят таким образом, чтобы ненасыщенные угле-
роды имели наиболее низкие номера. В названии указывается номер
первого ненасыщенного атома углерода; в остальном поступают так
же, как и при наименовании алканов:
СН3 С2Н5 СН3
I I I
сн3—с=сн — сн— сн—сн2—сн3
1 2 3 4 5 6 7
2,5-Диметил-4-этил-2-гептен
Сохраняются тривиальные названия для гомологов:
сн2=сн2 Н3С—сн=сн2
этилен
проп илен
сн3
н3с—с=сн2
изобутилен
Одновалентные радикалы, образованные
чание «енил»:
из алкенов, имеют окон-
3 2 1 4 3 2 1
сн3—сн=сн— сн3—сн=сн—сн2—
1-проп енил 2-бутенил
Сохраняются тривиальные названия
для следующих радикалов:
СН2=СН-
винил
(этенил)
3 2 1
Н2С=СН—СН2—
аллил
(2-пропенил)
сн3
2 I I
сн2= с—
изопропенил
(1-мети лэ тенил)
Структурная изомерия в ряду алкенов связана со строением угле-
родного скелета и положением двойной связи:
СН2=СН—СН2-СН2—СН3 СН3—СН=СН—СН2—сн3
1-пентен 2-пентен
сн3 сн3 сн3
I I I
сн2 =с—сн2—сн3 сн2—сн —сн—сн3 сн3—сн=с—сн3
2-метил-1-бутен 3-метил-1-бутен 2-метил-2-бутен
В ряду алкенов встречается еще один вид изомерии — геометриче-
ская изомерия, которая является частным случаем стерео-, или про-
странственной изомерии (см. с. 109), связанной с существованием
соединений, различающихся пространственным расположением входя-
щих в их состав групп. В случае геометрической изомерии, иначе на-
зываемой цис-трансизомерией, пространственные изомеры отлича-
ются расположением заместителей у ненасыщенных атомов углерода
относительно плоскости двойной связи. Например, в молекуле
2-бутена СН3—СН=СН—СН3 метильные группы могут быть распо-
ложены по одну сторону двойной связи (цисформа) и по разные сто-
роны от двойной связи (трансформа):
3* €7
цисформа
(цис-2-бутен)
н сн3
II
с
Нз^ \
трансформа
(транс-2-бутен)
Существование геометрической изомерии вызвано особенностью двой-
ной связи, свободное вращение вокруг которой невозможно, так как
это приведет к нарушению л-молекулярной орбитали и разрыву л-
связи (см. рис. 15). Это потребует значительной затраты энергии —
порядка 251,4 кДж/моль.
Цис- и трансизомеры существенно отличаются по своим физиче-
ским и некоторым химическим свойствам (см. табл. 8). Трансизомеры
по сравнению с цисизомерами обычно имеют более высокие темпера-
туры плавления, более низкие температуры кипения, меньшую плот-
ность. Электрические моменты диполей трансформы равны 0 или име-
ют небольшую величину в отличие от дипольных моментов молекул
цисконфигурации. Это различие широко используется для установ-
ления пространственного строения изомеров. Цис- и трансизомеры
под действием ультрафиолетового излучения, повышенных температур
или некоторых химических реагентов способны превращаться друг
в друга. Трансизомеры обычно отличаются большей устойчивостью,
чем цисизомеры.
Способы получения. В природе алкены встречаются в небольших
количествах. В промышленном масштабе их получают главным об-
разом при переработке нефти, природных и попутных газов. Они
могут быть также выделены из продуктов химической переработки ка-
менного угля (коксовый газ).
В ходе переработки нефти и газа, при термическом и каталитиче-
ском крекингах, пиролизе образуется сложная смесь продуктов (ал-
канов, алкенов, цикланов, ароматических углеводородов и т. д.),
состав которой зависит от исходного сырья, температуры, времени
контакта, давления, катализатора и т. д. Для получения низкомоле-
кулярных алкенов (С2—С5) применяют пиролиз (/ = 700—800° С)
газообразных алканов, выделенных из природных, попутных газов и
газов нефтепереработки и жидкого нефтяного сырья. Выход олефинов
достигает 50%, главным образом этилена и пропилена. При термичес-;
ком крекинге (t = 450—550° С, р = 20 • 105—70 • 105 Па) с исполь-^
зованием в качестве сырья керосина, лигроина, мазута (см. с. 146) .
выход алкенов незначителен. При парофазном крекинге (/ = 550—
—600° С, р = 3 • 105 —5 • 10б Па) получают до 35% от исходного;
сырья газообразных продуктов, наполовину состоящих из алкенов.
Газы, полученные при каталитическом крекинге, содержат до 30%
пропилена и бутиленов. '
Среди химических процессов, протекающих при переработке нефти<
и газа, следует выделить дегидрирование (реакцию отщепления водо*1
рода и образования ненасыщенных соединений):
68
СН3—СН3 ----> СН2=СН2
—н2
Дегидрирование протекает быстрее при применении специальных,
избирательно (селективно) действующих катализаторов (селектив-
ные катализаторы), ускоряющих только этот процесс. Широко при-
меняется дегидрирование для получения этилена из этана, н-бутилена
из н-бутана и изоамилена из изопентана!
СНз—сн2—сн2—сн3 —> сн3—сн2—сн=сн2
-н2
н-бутан 1-бутен
СН3—СН—СН2—СН3------->- CHg-CH—CH=CHS
I -н» I
СНз СНз
2-метилбутан 3-метил-1-бутен
Высшие 1-алкены (С10—С20) с четным числом атомов углерода по-
лучают полимеризацией (см. с. 82) этилена в присутствии алюминий-
органических катализаторов.
Существует ряд лабораторных способов синтеза алкенов. В боль-
шинстве их в молекуле отнятием заместителей, расположенных у
соседних углеродных атомов, создается двойная связь:
II II
-с— с-------> —с = с—
I I -Ав
А В
Эта реакция называется реакцией элиминирования (Е). Частным ее
случаем является описанная выше реакция дегидрирования. В за-
висимости от строения исходного соединения, характера заместителей
подбирают и условия реакций (температура, катализаторы и т. д.).
Отнятие воды от спиртов (дегидратация спиртов). Спирты при
нагревании в присутствии водоотнимающих средств (серная, фосфор-
ная кислоты) или при пропускании их паров через оксид алюминия,
хлорид цинка теряют молекулу воды с образованием олефина. Этот
метод в силу простоты, доступности сырья не потерял своего значения
и в настоящее время для получения в присутствии кислоты этилена
из этилового спирта:
H2SO4
СН2—СН2-----^СН2=СН2
I I -Н2о
Н ОН этилен
этиловый спирт
В качестве промежуточного продукта образуется этилсерная кис-
лота:
СН3—СН2—ОН + HOSO2OH СНз—СН2—OSO2OH + Н2О
При нагревании она разлагается с образованием алкена:
t°
СН3СН2—OSO2OH--------сн9=сн2
-H2SO4
этилсерная кислота этилен
69
При дегидратации спиртов более сложного строения водород отщепля-
ется от наименее гидрогенизированного (связанного с наименьшим
количеством водорода) углеродного атома (правило Зайцева):
СН3 СН3
СН3—СН2—СН—с—сн3 —сн3 — сн2—сн=с—сн3
| I -н,о
ОН н
2 метил-3-пентанол 2-метил-2-пентен
Спирты различного строения дегидратируются неодинаково: легче
всего отщепляется гидроксильная группа, расположенная у третич-
ного углеродного атома, труднее — у вторичного и значительно труд-
нее — у первичного.
Получение из галогенпроизводных. Алкены могут быть получены
отнятием галогенводородов (дегидрогалогенирование) от галоген-
алкилов с помощью спиртовых растворов щелочей или твердой ще-
лочи:
СН3 СН3
СН2 — С—СН3 + NaOH—^СН2=С—СНз + NaCl + Н2О
Н С1
Галогеналкилы различного строения с неодинаковой легкостью пре-
вращаются в непредельные соединения: легче всего отщепляют моле-
кулу галогенводорода третичные галогеналкилы, труднее — первич-
ные, вторичные занимают промежуточное положение.
Олефины могут быть получены отщеплением (при действии Zn, Mg
и некоторых других металлов) двух атомов галогена, расположенных
у соседних углеродных атомов:
СН3—СН2—СН—СН—СН3 + Zn -> СН3—СН2—СН=СН—СН3 + ZnBr2
I I
Вг Вг
Получение из ацетиленовых соединений. Частичное (селективное)
гидрирование алкинов приводит к олефинам:
Н2; кат.
R—C = C-Rj------> R—CH=CH-R1
Отличительной особенностью этой реакции является возможность
образования двух изомеровз цис- и транс-. Подбирая условия реакции,
тип катализатора, ее удается направить в сторону предпочтительного
образования одного из них (стереоселективная реакция).
Физические свойства. Первые три гомолога ряда этилена — газы (табл. 8)»
начиная с пентена до олефина С17 — жидкости, с С18 — твердые вещества. Гомо-
логи нормального строения кипят при температуре более высокой, чем их изоме-
ры разветвленного строения. Олефины мало растворимы в воде, плотность их
меньше единицы и возрастаете увеличением молекулярной массы. Алкены мало-
полярны, их электрические моменты диполей невелики, они легко поляризуют-
ся.
70
Химические свойства. Химическое поведение алкенов связано с
наличием в их молекуле двойной связи и способностью присоединять
другие атомы и группы. Ненасыщенные атомы углерода в молекуле
алкенов связаны между собой о- и л-связями. л-Связь менее прочная,
чем о-связь. Для разрыва о-связи требуется 341,9 кДж/моль, а двой-
ной связи — 611,7 кДж/моль, т. е. меньше, чем для разрыва двух
о-связей — 695,5 кДж/моль, следовательно, на разрыв л-связи требу-
ется 611,7—341,9 = 269,8 кДж/моль. Поэтому двойная углерод-
углеродная связь обладает способностью разрываться в определен-
ных условиях как бы наполовину, при этом атомы углерода, присоеди-
няя новые группы, становятся насыщенными:
тг-связь А В
/ t \ / t \
a-связь а-связь
Для этиленовых углеводородов наиболее характерны реакции при-
соединения; они легко окисляются и полимеризуются.
При рассмотрении алкенов читатели впервые сталкиваются с на-
личием в молекуле органического соединения функциональной (ха-
рактеристической) группы, определяющей его химические свойства.
На активность двойной связи влияют строение углеводородных ра-
дикалов, наличие и характер заместителей.
Реакции присоединения. Реакции присоединения по
двойной углерод-углеродной связи в зависимости от природы частиц,
которые обеспечивают их осуществление, протекают или по электро-
фильному (АЕ), или по радикальному (А д) механизму. Необходимо
помнить, что электроны, образующие л-связь, более доступны дейст-
вию атакующего реагента, чем электроны a-связи, так как они как бы
«вытолкнуты» наружу. Поэтому в химических превращениях алкены
выступают в качестве доноров электронов (л-оснований). Естественно,
что реагенты, имеющие незаполненные валентные орбитали (электро-
фильные), взаимодействуют с олефинами наиболее эффективно. Ре-
акции с нуклеофильными реагентами (имеющими заполненные ва-
лентные орбитали) для олефинов не характерны. Реакции электро-
фильного (А Б) и нуклеофильного (A N) присоединения можно рас-
сматривать в качестве частного случая кислотно-основных реакций,
в основе которых лежит теория Льюиса (см. с. 40). В реакциях при-
соединения кислоты Льюиса выступают в роли электрофильных ре-
агентов, основания — нуклеофильных.
Присоединение галогенов. Алкены очень легко присоединяют га-
логены с образованием дигалогенпроизводных, содержащих два атома
галогена у соседних углеродных атомов (вицинальные дигалогенпро-
изводные):
СН2=СН2 + Вг2 СН2Вг—СН2Вг
этилен 1,2-дибромэтан
Реакция протекает легко в растворителе, темноте, при комнатной
температуре. Она начинается с электрофильной атаки молекулой брома
71
двойной связи. Под влиянием л-электронного облака олефина моле-
—S - 6
кула галогена поляризуется, образуется наведенный диполь Вгч-Вг,
положительный конец которого выступает как электрофильный ре-
агент и захватывает л-электроны двойной связи с образованием не-
устойчивого л-комплекса. Затем л-связь и связь бром—бром гетеро-
литически разрываются. Бром за счет электронов л-связи образует
обычную о-связь с одним из ненасыщенных углеродных атомов, дру-
гой углеродный атом, лишившись электронов, заряжается положи-
тельно, в результате образуется карбкатион (а-комплекс). Возможно
образование из л-комплекса иона бромония за счет перекрытия сво-
бодной р-орбитали атома углерода с неподеленной парой электронов
брома:
быстро 1 медленно
сн2=сн2 + Вг2 сн2фсн2 -< ...............
л-комплекс
I
сн2—сн2 + Вг~ или H2C~ СН2 + Bf*
Вг:-^ 'в/
электрофильная
> атака
карбкатион ион бромония
((^-комплекс) (бромониевый катион)
II III
Карбкатион или ион бромония атакуются анионом брома с обра-
зованием 1,2-дибромэтана:
Ж быстро ч
СН2— СН2 + Вг”----------> сн2—сн2
| | | нуклеофильная атака
Вг Вг Вг
карбкатион 1,2-дибромэтан
Таким же образом идет присоединение по месту разрыва двойной
связи HBr, H2SO4, НгО и других неорганических веществ.
Первая стадия — электрофильная атака этилена бромом с обра-
зованием карбкатиона или иона бромония протекает относительно
медленно, вторая крайне быстро. Поэтому именно скорость первой ста-
дии определяет скорость всей реакции (лимитирующая стадия).
Из галогенов с олефинами наиболее энергично взаимодействует
фтор (со взрывом), труднее всего—иод (реакция практически не идет).
Введение электронодонорных заместителей в молекулу алкена \ско-
ряет, а электроноакцепторных — замедляет присоединение. Так, от-
носительная скорость присоединения брома (метанол, 25° С) в ряду
этилена и некоторых его производных изменяется следующим образом:
Алкен . . СН2=СН2 С2Н5—СН=СН2 СН3 С2Н5 Н3С СН3 СН2=СНВг
II \ /
С = С С=С
Н Н dH3 \н3
Относительная
скорость . . 1 97 4,2-103 93-104 0,04
72
Ускоряет присоединение и применение смешанных галогенидов (СНГ
С1Вг), молекула которых поляризована. Присоединение брома (обес-
цвечивание бромной воды) — качественная реакция на двойную связь.
Для количественного определения двойной связи удобно использо-
вать стандартный раствор бромата калия — бромида калия, кото-
рый смешивается с алкеном, и к смеси добавляют серную кислоту.
После окончания реакции добавляют иодид калия (в избытке). Вы-
делившийся иод оттитровывают тиосульфатом. Галогенирование ал-
кенов широко применяется в промышленности и в аналитической,
химии.
Присоединение галогенводородов:
СН2=СН2 + НВг------> СН3—СН2Вг
этилен бромэтан
В случае несимметрично построенных олефинов водород предпочти-
тельнее присоединяется к наиболее гидрогенизированному из нена-
сыщенных атомов углерода, галоген — к другому ненасыщенному
углеродному атому (правило Марковникова):
СН3—СН=СН2 + НС1---------> СН3-СНС1-СН3
пропилен 2-хлорпропан
Избирательное присоединение водорода является следствием взаим-
ного влияния атомов в молекулах органических соединений. Углерод
является более электроотрицательным атомом, чем водород, поэтому
электронная плотность С—Н-связей в метильной группе будет не-
сколько смещена в его сторону, что, естественно, приведет к повы-
шению у него электронной плотности (BJ и, в свою очередь, вызовет
ее повышение в связи С3—Сг (+/-эффект):
н
Электроны, осуществляющие эту связь, приобретают способность
взаимодействовать с электронами л-связи. В результате этого проис-
ходит смещение электронной плотности л-связи в сторону углеродно-
го атома Сх, л-связь поляризуется. Поэтому у атома углерода С2 будет
наблюдаться некоторая нехватка электронной плотности (+М, а У
атома углерода Q — некоторый избыток (—Метильная группа
выступает в качестве донора электронов. Такими донорами электро-
нов являются и другие алкильные группы (см. с. 37).
При взаимодействии пропилена с галогенводородом протон Н+ в
первую очередь будет присоединяться к углеродному атому Сь не-
сущему частичный отрицательный заряд, т. е. к наиболее гидрогени-
зированному-
+3 —3 + Вг-
сн3—сн = сн2 + Н*------СН3—СН—СН3--------СН3—СНВг—сн3
пропилен карбкатион 2-бромпропан
73
Предпочтительное образование при взаимодействии пропилена с бро^
мистым водородом 2-бромпропана (до 90%), а не 1-бромпропана свя-:
зано и с тем, что из двух возможных карбкатионов I и II >
-------> СНоСН—сн3
н+
СН3—СН=СН2 +
-----> сн3—сн2 —сн2
(I)
(II)
образование катиона I энергетически более выгодно. Он образуется
быстрее и более устойчив, так как его положительный заряд у 2-го угле-
родного атома частично компенсируется индуктивным эффектом двух
метильных радикалов, а в катионе II—только одним радикалом.
В общем случае направление присоединения определяется относи-
тельной реакционной способностью алкенов (см. с. 72), быстротой
образования соответствующего иона, его устойчивостью.
По легкости образования и устойчивости карбониевые ионы рас-
полагаются в такой последовательности:
СН3-С—СН3 > сн3—сн—сн3 > сн3 —сн2 > сн3
сн3
Из галогенводородных кислот наиболее легко присоединяется HI,
остальные галогенводороды располагаются по этому признаку в та-
кой последовательности:
НВг > НС1 > HF
Правило Марковникова соблюдается только при гетеролитическом
механизме присоединения галогенводородов к алкенам (Ля), если
присоединение протекает по радикальному механизму (например, в
присутствии перекиси), порядок присоединения обратный (эффект
Хараша, или «перекисный эффект»):
СН3—СН=СН2-!1®!_
в отсутствие
перекисей
в присутствии
гц cuRr___гц по правилу
СН3-СНВг-СН3 Марковникова
перекисей
СН3—СН2—СН2Вг
не по правилу
Марковникова
Из галогенводородов присоединяется по радикальному типу (Лн)
практически только НВг. Реакция протекает по следующей схеме:
2НВг + О2 -> 2Вг • + Н2О2
Н2О2 + НВг Вг • + [Н3О2] •
инициирование
СН3—СН=СН2 + Вг •
пропен
СН3—СНВг—сн2.
свободный радикал I
-> СН3—СН—СН2Вг
свободный радикал II
рост цепи
74
Из двух свободных радикалов (I и11), которые могут образовывать-
ся при взаимодействии пропена с атомом брома, радикал II более ус-
тойчив, так как свободный электрон у второго углеродного атома
вступает во взаимодействие с электронными облаками пяти соседних
связей С—Н; а в радикале I — только с одним. Затем свободный ра-
дикал II вступает во взаимодействие с молекулой бромводорода с об-
разованием 1 -бромпропана:
СН3—СН—СН2Вг + НВг СН3—СН2—СН2Вг + Вг • рост цепи И т. Д.
свободный радикал II 1-бромпропан
Гидратация (присоединение воды). При присоединении воды к
олефинам образуются спирты:
R—СН=СН2 + НОН -> R—СН—СН3
ОН
Водород направляется к наиболее гидрогенизированному атому угле-
рода. В промышленности осуществляется прямая гидратация (ката-
лизаторы— фосфорная кислота, фосфаты, оксид алюминия; повы-
шенная температура):
СН,=СН, + нон сн3—сн,он
Способность олефинов к гидратации возрастает с увеличением длины
углеродной цепочки (до известных пределов) и количества алкильных
групп у атомов углерода, связанных двойной связью. Ниже приве-
дены относительные скорости реакций для некоторых олефинов:
СН3
н3с—сн=сн2 сн3—с=сн2
1 8000
СН3
С2Н5—с=сн2
10 000
Присоединение серной кислоты. При взаимодействии алкенов с
серной кислотой водород присоединяется к наиболее гидрогенизиро-
ванному углеродному атому, HSO4 — к другому (правило Марков-
никова), образуя связь углерод — кислород:
СН3—СН=СН2
пропен
H2SO4
----> СН3—СН—СН3
OSO3H
кислый изопропил сульфат
Реакция протекает легко при пропускании газообразного алкена
через концентрированную кислоту или при перемешивании смеси
алкена с кислотой. При разбавлении алкилсерных кислот водой с
последующим нагреванием образуются спирты (сернокислотная гид-
ратация):
75
СН3
СН3—С + Н,0 сн3—сн—сн3 + H,so4
I I
OSO3H он
изопропиловый •
спирт
Образование галогенгидринов. При взаимодействии хлора или брома
с алкенами в присутствии воды образуются галогенгидрины. Галоген
присоединяется к наиболее гидрогенизированному атому углерода
(Лг-реакция; С1+—электрофильный реагент):
С12 + Н2О НОС1 + НС1
3-|- 5— ---Ь
СН3—СН=СН2 + НОС1 -> СН3—СН—СН2С1
пропен |
он
Алкилирование алкенов. В присутствии катализаторов H2SO4,.
HF, А1С13 и BF3 алкены присоединяют алканы (алкилирование алке-
нов):
СН3 СН3 СН3 СНз
| | H2SO4 (КОНЦ.) I |
СНз—С=СН2 + Н—С—СН3---------------> СНз—СН—СН2—С—СН3
2-метилпропен I I
Сг13 СН3
2-метилпропен 2, 2, 4-триметилпентан
Гидрирование. Алкены в присутствии катализаторов (платина,,
палладий, никель) легко присоединяют водород, превращаясь в пре-
дельные углеводороды:
Ni
R—СН=СН2 + Н2 -> R—СН2—СН3
В отсутствие катализатора даже водород в момент выделения с
алкенами не взаимодействует.
На примере реакций гидрирования
алкенов рассмотрим явление гетероген-
ного катализа. Скорость гидрирования
зависит от вида и активности катали-
затора, условий гидрирования и строе-
ния алкенов. На платине и палладии
гидрирование успешно идет при комнат-
ной температуре, на никеле — при наг-
ревании. Роль катализатора заключает-
ся в понижении энергии активации и,
следовательно, значительном увеличе-
нии скорости реакции при невысоких
ход реакции температурах. Катализатор не оказыва-
Рис. 21. Энергетическая диа-
грамма реакции гидрирования
алкенов в отсутствие (/) и в
присутствии (2) катализатора
ет влияние на суммарное изменение
энергии всей системы (рис. 21), а изме-
няет реакционный путь. Устойчивость
алкенов к гидрированию возрастает с
76
увеличением степени замещения у ненасыщенных углеводородов:
R2C=CR2 > R2C=CHR > R2C=CH2 > RCH=CHR > RCH=CH2 > CH2=CH2
Гидрирование ненасыщенных связей широко применяется в технике,
лабораторной и аналитической практике. В пищевой промышлен-
ности большое значение имеет гидрирование жиров.
Много сделали для изучения гидрирования наши отечественные
ученые С. А. Фокин, С. В. Лебедев, Н. Д. Зелинский и зарубежные
исследователи Сабатье, Сендеран, Вильштеттер.
Реакции окисления. Окисление олефинов. Олефины легко
окисляются. Направление окисления и характер образующихся про-
дуктов зависит от строения алкенов, вида окислителя и условий окис-
ления. При окислении разбавленным раствором перманганата калия
на холоду в щелочной среде (окисление по Вагнеру) образуются гли-
коли (см. с. 178).
ЗСН3—СН=СН2 + 2КМпО4 + 4Н2О ЗСН3—СН—СН2 + 2MnO2 + 2КОН
пропилен он он
1, 2-пропиленгликоль
При более энергичном окислении олефинов (азотная кислота, пер-
манганат калия в кислой среде, хромовая кислота) происходит разрыв
молекулы по двойной связи с образованием кислот и кетонов:
сн3—сн=сн—сн3
2-бутен
хромовая
кислота
------> 2СН3—СООН
tO] уксусная
кислота
хромовая
СН3— С=СН—СН3 СНз—С=О + СНзСООН
I [°] I
СНз СН3
2-метил-2-бутен ацетон уксусная
кислота
Анализируя продукты окисления, можно сделать заключение о стро-
ении исходного алкена.
При окислении олефинов кислородом воздуха в присутствии сере-
бряных катализаторов образуются окиси олефинов:
Ag; 200—300°С
2СН2=СН2 + О2-------------->
этилен
2СН2—СН2
окись этилена
Это важный промышленный способ получения окиси этилена.
Окисление надкислотами. Реакция Прилежаева. При действии
надкислот на алкены они количественно превращаются в эпоксиды:
77
L
z°
R—CH=CH2 + CH3—C< -* R—CH—CH2 + CH3COOH
алкен X00H \ /
aUliYCti
окись алкена
(эпоксид)
Озонирование алкенов (озонолиз). Для установления строения ал*
кенов используется реакция озонирования:
/°\
СН3—С—с—сн2—сн3 + 03 -> Н3С—с с—сн2—сн3
II I \ /I
СНз Н НС3 О—О Н
2-метил-2-пентен озонид
Образовавшийся озонид (неустойчивое, легко взрывающееся ве-
щество) разлагают водой и, анализируя продукты реакции, устанав-
ливают строение исходного алкена:
/°\ .о
/ \ Н2О;
Н3С—С С—СН2—СНз----------> СН3—С=0 + СН3—СН2С\
|\ /| -н.о, | \н
Н3С 0—0 н сн3
озонид ацетон пропионовый альдегид
Озонирование является примером реакции расщепления алкенов.
Полимеризация алкенов. Полимеризацией назы-
вается соединение одинаковых молекул (мономеров) с образованием
нового, сложного вещества с большой молекулярной массой (полимер).
Если в образовании полимера участвуют различные мономеры, про-
цесс называется сополимеризацией. Количество молекул мономера п,
соединившихся при образовании полимера, называется степенью по-
лимеризации. Практически полимерные материалы состоят из смеси
молекул, построенных из различного числа мономеров, поэтому под
молекулярной массой полимера понимают его среднюю молекуляр-
ную массу. Получение полимеров всегда состоит из двух этапов: син-
тез мономеров (эти вопросы разбираются при рассмотрении соответ-
ствующих классов соединений) и образование из них полимеров (по-
лимеризация).
Из олефинов в качестве мономеров наиболее широкое применение
нашли этилен, пропилен, бутилены:
иСН2=СН2 -> [— СН2—СН2—]л
этилен полиэтилен
лСН3—СН=СН2 ->
пропилен
—СН—СН2— 1
СНз 1
полипропилен
Свойства полиолефинов во многом зависят от способа полимери-
зации. Различают линейную (цепную) и ступенчатую (миграционную)
полимеризацию (схема 4). В первом случае промежуточные продукты
реакции не удается изолировать, а конечный продукт отличается вы-
78
Схема 4. Классификация полимеризации алкенов
сокой молекулярной массой. Во втором случае процесс протекает сту-
пенчато, с постепенным нарастанием молекулярной массы, а промежу-
точные продукты могут быть выделены. Образовавшийся полимер
обычно состоит из небольшого числа молекул мономера.
Полимеризация может быть проведена по ионному или радикаль-
ному пути. Примером ступенчатой ионной полимеризации может
служить синтез диизобутилена (Бутлеров, 1878):
СН3 сн, НзС СНз
н+ | сн3—с=сн2 I |
СН3—С=СН2-----------> СН3—С+--------------> СН3—С—СН2—С—СН3
I 60%-на я I I + —Н+
СН3 Нг5О‘ сн3 Н3С
изобутилен
СН3 СН3 СН3
|| | Н2; кат.
-> СНз—С—СН2—С=СН2 + СН3—С—СН=С—СНз —>
I I I
СН3 СН3 СН3
80%20%
диизобутилен
79
СН3
-> СН3—С—СН2—СН—сн3
СНз СНз
2, 2, 4-триметилпентан (изооктан)
В присутствии 60%-ной серной кислоты реакция ограничивается
образованием димера. При гидрировании изобутилена образуется
2,2,4-триметилпентан (изооктан). Реакция имеет практическое зна-
чение для получения высокооктанового моторного топлива.
Широкое распространение для промышленного получения поли-
олефинов с большой молекулярной массой нашла цепная полимери-
зация, осуществляемая по свободнорадикальному механизму. Реак-
ция начинается с активирования какой-либо молекулы под влиянием
температуры, света, радиационного излучения, инициаторов (пер-
оксид, азо- и диазосоединения и др.), которая вызывает появление
новых радикалов и полимеризацию большого числа других молекул,
с которыми они сталкиваются. Первая фаза протекает относительно
медленно, вторая — практически мгновенно. При цепной радикаль-
ной полимеризации почти невозможно выделение промежуточных
продуктов с небольшой степенью полимеризаций — димеров, тримеров
и т. д. Различают три главные стадии процесса.
1. Инициирование (образование свободного радикала)
hv
м -> м.
На практике инициирование чаще всего осуществляется с помощью
инициаторов, распадающихся в условиях реакции на свободные ра-
дикалы.
2. Рост цепи. Первоначально образовавшийся мономерный ра-
дикал (М-), соединяясь с молекулами олефинов, образует новые ра-
дикалы с большой молекулярной массой (макрорадикалы )*
М . + М М2* + М -> Мз
Процесс протекает крайне быстро (несколько секунд).
3. Обрыв цепи. На этой стадии мономерный радикал превращает-
ся в макромолекулу полимера:
м; +М. -> Мп
Обрыв цепи может происходить вследствие взаимодействия двух
макрорадикалов (рекомбинация), диспропорционирования (образов
вание из макрорадикала предельного и непредельного соединения!
и взаимодействия макрорадикала с инициатором полимеризации.
Так, радикальная полимеризация пропилена может быть представлен
на следующим образом.
1. Инициирование цепи
RO-OR -> 2RO •
2. Зарождение и рост цепи
80
-632
_ сн,=сн—сн.
RO. + СН2=СН -> RO—СН2—СН. —!-►
СН3 СНз
пСН,=СН—сн,
RO—СН,—СН—СН3—СН --------► RO— /СН,—СН \ — СН,—СН •
11 I । I ।
СН8 СНз СН3/„ СН3
3. Обрыв цепи
RO— /СН,—СН\—сн,—СН
\ СН,'„ СН,
RO-/CH -СН \—СН,—СН.
I I
\ СН8/Л СН3
-------;---------------► RO—/СН2—СН СН2—СН—СН—CH2-/CH-CH2\-OR
(рекомбинация)--I I ) I I II I
\ СНз Л СНз СНз \СН3 )п
СН.
-----------_ RO—/сн9—сн \-CH2-CH2+RO-/CH2-CH \—сн=сн
диспропорционирование I I / I III I
\ СНз/я СНз \ СН3/л СНз
----------522---------- RO—/СН2—СН \—СН,—СН—OR
( АнзЛ СНз
со
Радикальная полимеризация сопровождается нежелательными
процессами (разветвление цепи), полученные продукты обладают
худшими механическими свойствами.
Цепная полимеризация, осуществляемая через образование а
качестве движущих ее частиц ионов, получила название ионной поли-
меризации и в зависимости от знака заряда (иона) делится на катион-
ную и анионную полимеризацию. Катализаторы инициирования ка-
тионной полимеризации — H2SC>4, BF3, А1С13, SnCk, TiCk; анион-
ной — основания, щелочные металлы, металлорганические соедине-
ния. Механизм катионной полимеризации с участием кислот Льюиса
может быть представлен следующим образом: на первом этапе катали-
затор TiCk реагирует с сокатализатором НА, образуя комплексное
соединение:
НА 4- TiCl4 -> fATiCIJ~H+
Затем протон присоединяется к мономеру (инициирование) с образо-
ванием карбкатиона, и начинается рост цепи:
Н+ + п сн8=сн—СН, +
СН2=СН СН3—СН--------->СН3—СН—/СН2—СН СН2—СН
। I III
СН3 СН3 СН3\ СН3/п-I СН3
На заключительном этапе происходит обрыв цепи:
СНз—СН—/СН2—СН \ — СН2—СН + [ATiCU]- -*•
I । ।
СН3 \ СНз/н-1 СН3
-> СН3—СН—/СН2—СН \—СН=СН + [ATiC]4]-H+
I I I
СН3 \ СН3 //г-1 СН3
Катионная цепная полимеризация проводится при низких темпера-
турах, в растворителе (например, углеводороде). По активности апро-
тонные катализаторы располагаются следующим образом:
BF3 > AICI3 > TiCl4 > SnCl4
Примером анионной полимеризации может служить полимериза-
ция этилена по методу Циглера — Натта. В качестве катализатора
применяются комплексы, содержащие триэтилалюминий (С2Н5)3А1,
хлорид титана и металлический литий. В процессе реакции молекула
этилена разрывает поляризованную связь и становится
между атомами металла и углерода:
Me—СН2—СН3 Me—СН2—СН2—СН2—СН3 и т. д.
СН2=СН2 СН2=СН2
Это становится возможным в результате образования при взаимо-
действии (СН3СН2)3А1 и TiCh комплекса, состоящего из TiCl3, на
82
поверхности которого адсорбируется триэтилалюминий, а связь С—А!
поляризуется.
Полимеры, полученные полимеризаций по ионному типу, отли-
чаются хорошими механическими свойствами, высокими температу-
рами плавления и могут иметь определенное расположение замести-
телей в пространстве — стереорегулярные полимеры, что существен-
но влияет на их свойства. Так, полипропилен /—СИ2—СН—\ м0.
\ СН3 Л
жет быть получен в трех формах: изотактический стереорегулярный
полипропилен (все метильные группы расположены по одну сторону
цепи, рис. 22, а); синдиотактический стереорегулярный полипропи-
лен (метильные группы располагаются в пространстве в определенном
порядке по разные стороны углеродной цепи, рис. 22, б) и атактиче-
ский (не обладающий регулярной стереоконфигурацией) полимер,
(рис. 22, в).
Полиолефины широко используют в народном хозяйстве. Поли-
этилен — для изоляции электропроводов, изготовления пленок, труб,
посуды. Полипропилен, не уступая полиэтилену в химической стой-
кости, превосходит его по прочности на разрыв, более стоек к высоким
температурам; применяется для изготовления изоляции, деталей ра-
диоаппаратуры и машин; труб, сетей, канатов, фильтрующих тканей.
Теломеризация. Особым видом полимеризации является сополи-
меризация этилена с четыреххлористым углеродом, получившая на-
4* 83
звание теломеризации. Реакция инициируется перекисными ради-
калами (R* ), образующимися при распаде перекисных соединений:
R.+CCU -> RC1 + СС13-
(перекис-
ный радикал)
Образовавшийся радикал СС13* вызывает цепную полимеризации*
этилена:
СС13 • + СН2-СН2 -> СС13СН2—СН2 . + мСН2=СН2 -> СС13 (СН2—СН2).л+1
При взаимодействии радикала СС13(СН2—СНг)^ с четыреххло-
ристым углеродом наступает обрыв цепи с образованием теломера:
СС13 (СН2—CH2)n+i + СС14 -> СС13 (СН2—СН2)л+1 С1 + СС13 •
Образовавшийся радикал СС13* начинает создание новой цепи.
Вещества, участвующие в теломеризации и обрывающие рост поли-
мерной цепи, называются телогенами. Степень полимеризации tt
обычно не превышает 10—15. Полученные тетрахлоралканы при-
меняются для синтеза хлорзамещенных и аминозамещенных кислот,,
а на их основе — ряда других соединений:
H2O; H2SO4
Cl—(СН2—СН2)Л—СС13---------С1—(СН2—СН2)Л—СООН ->
хлорзамещенные карбоновые
кислоты
NH3
---> NH2—(СН2—СН2)п—соон
аминокислоты
Реакции замещения. При высокотемпературном хло-
рировании пропилена происходит замещение атомов водорода, при-
чем хлор вступает в a-положение к двойной связи (аллильное поло-
жение):
С12
СН2=СН—СН3-------> СН2=СН—СН2С1 + НС1
500° с
пропилен аллилхлорид
Реакция протекает по свободнорадикальному механизму. Естест-.
венно, возникает вопрос, почему в молекуле, где имеется активная
функциональная группа (\c=C<Q, присоединяющая хлор при ком-
натной температуре, может быть осуществлено замещение водоро-
дов на атомы галогена только при высокой температуре. При высоко-
температурном взаимодействии хлора (или брома) с гомологами эти-
лена могут происходить оба свободнорадикальных процесса — при-
соединение (Afi) и замещение (Sfi). Однако при прохождении этих
процессов на первой стадии образуются два неодинаковых по своей
устойчивости радикала. Если радикал I при высоких температурах
очень быстро распадается (превращаясь в исходное вещество и 1,2-
дихлорпропан), то аллильный радикал II обладает повышенной ус-
тойчивостью (мезомерный эффект) и реагирует с молекулой хлора,
образуя аллилхлорид:
84
СН3—СН=СН2 + о.
Cl
C1S I
—CH3—CH—CH2C1-> CH3—CH—CH2C1 (AR)
I» 2-ди хлор пропан
Cl
Cl? I
---> CH2—CH=CH2 -> CH2—CH=CH2 (SR)
—HC1 11 ал лил хлорид
Поэтому реакция практически смещена в сторону образования аллил-
хлорида. Водороды, расположенные у различных углеродных атомов
(СН2=СН—Н—винильный, СНг^СН—СНг—Н — аллильный и т. д.),
замещаются с неодинаковой легкостью и могут быть по этому признаку
расположены в такой последовательности:
аллильный > третичный > вторичный > первичный > СН3 • > винильный
Отдельные представители. Применение. Высокая реакционная
способность, доступность и низкая стоимость исходного сырья пре-
вратили олефины в один из наиболее важных классов органических
соединений, широко применяемых в разнообразных промышленных
синтезах. Из большого числа гомологов особую ценность для промыш-
ленности представляют первые четыре.
Этилен С2Н4. Этилен — газ, без цвета и запаха, мало растворим
в воде. С воздухом образует взрывоопасные смеси. Является исход-
ным веществом для получения разнообразных продуктов органического
синтеза, имеющих первостепенное народнохозяйственное значение
(схема 5). Большая его часть расходуется на получение полиэтилена,
окиси этилена, этилового спирта. Промышленные пути получения —
выделение из продуктов переработки нефтяного сырья, коксо-
вого газа, дегидрирование этана.
Пропилен СН3—СН^ СНо. Второй по значению гомолог ряда эти-
лена. Получается из продуктов переработки нефти, пиролизом бутана
(совместно с этиленом), дегидрированием пропана. Является исход-
ным сырьем для получения изопропилового спирта, полипропилена,
окиси пропилена и ряда других продуктов (схема 6).
Бутилены С4Н8. Получают из продуктов переработки нефтяного
сырья (бутан — бутеновая фракция). Изобутилен — исходное ве-
щество в синтезе изооктана и полиизобутилена; я-бутены — сырье
для получения дивинила.
АЛКИНЫ
Алкинами, или ацетиленовыми углеводородами, называются со-
единения, содержащие в молекуле тройную связь —С=С—. Общая
формула алкинов СЛН2П_2. Они содержат в молекуле на четыре атома
водорода меньше, чем алканы, и на два, чем алкены с тем же числом
углеродных атомов, и изомерны алкадиенам (см. с. 97).
Ряд органических соединений с одинаковым числом атомов угле-
рода в молекуле и тождественными функциональными группами, но
с возрастающей ненасыщенностью, т. е. с увеличивающимся числом
85
Схема 5. Промышленное использование этилена
полимери-
---------(--СН2—СН2—)п
зация
полиэтилен
СЪ-------поли мери -
---------► СН2С1—СН2С1 СНС1=СН2 > [-СНС1-СН2-]П
зация
дихлорэтан винил--полихлорвинил
хлорид
ЕС!
---------* СН3-СН2С1
хлористый этил
НОС!
---------► СН2С1СН2ОН . * СН2=СН—СН=СН2--К каучук
этиленхлоргидринI-----------------------------------дивинил
Н,О
---------> СН3—СН2ОН—
этиловый спирт
H,SO4
СН3—СН2—OSO3H-------
этилсерная
кислота
•СН3—сн2—ОН
этиловый спирт
С2Н 5—О—С2О5
NH,
-----> сн3—сн2—nh2
диэтиловый эфир
с,н4---
этила мин
С<Н4 полиме- Г
------->СвН5СН2СН3 СвН5-СН=СН2 > —СН —сн2—
ризация I
этилбензол стирол г-г* и
L CeH{j п
полистирол
---------горение---------► сварка, резка металлов
------------------------->. СН2-СН2
окись этилена
О,
------СН3—СНО----------->- сн3—соон
уксусный----------------уксусная
альдегид кислота
СО + Hs
СН3-СН2-С
пропионовый альдегид
СО + Н,О
---------► СН3—СН2—СООН
пропионовая кислота
СС14
----------- СН2С1(СН2)ПСС13 — синтетические волокна и полимерные'
материалы
86
Схема 6. Промышленное использование пропилена
полимеризация
—СН2—СН—
i
СНз
полипропилен
Н2О
ОН
СНз—сн—сн3
изопропиловый
спирт
СН2С1СНС1СН3
1,2-ДИХлорпропан
СН2=СН—СН2С1
аллилхлорид
—> Н2С—сн—сн3
окись пропилена
СН2=СН—СН3
пропилен
О
II
СНз—С—СН3
ацетон
—>сн2=сн—сно
акролеин
---СНз—сн-сно
СН3 '
--Св — изомасляный альдегид
^0
----> СНз—СН2—СН2—с
хн
масляный альдегид
СО 4- Н2О
--------------► сн3—СН—соон
I
СНз
изомасляная кислота
С,Н.
--------------► СвН5-СН(СН3)2
изопроп илбензол
акрилонитрил
87
Таблица 9. Алкины (гомологический ряд ацетилена)
Название Формула Т емпера тура плавления. ®С Температура кипения, °C Плотность р, Г’СМ”3 Показатель преломления,
Ацетилен , этин нс=сн —80,8 -83,6 0,613 (—80°С) —
Метилацетилен, пропин сн3—с=сн — 104,7 —23,3 0,690 (—40°С) 1,3747 (при т. кип.)
Этилацетилен, 1-бутин СН = С—СН2—СНз -122,5 8,6 0,668 1,3962 (при 0°С)
Диметилацетилен, 2-бу- тин СНз—С=С—СН3 —28,0 27,2 0,693 1,3921
Пропилацетилен , 1 -пен- тин сн3—сн2—сн2—с=сн —98,0 39,7 0,695 1,3852
Метилэтилацетилен, 2-пентин СН3-СН2—С==С-СН3 — 101,0 55,5 0,712 1,4039
кратных связей между атомами углерода, называется изологическим
рядом. Изологи отличаются между собой на четное количество атомов
водорода. Например, изологический ряд этана исчерпывается тремя
членами:
CjHe С2Н4 С2Н2
этан этилен ацетилен
По мере укрупнения молекулы увеличивается возможность об-
разования кратных связей и, следовательно, число членов изологи-
ческого ряда. Аналогичные соотношения наблюдаются у карбоцикли-
ческих и гетероциклических соединений.
Гомологический ряд, строение, номенклатура, изомерия. Первый
представитель гомологического ряда алкинов — ацетилен СН=СН,
в молекуле которого, так же как и у других гомологов, ненасыщен-
ные атомы углерода связаны между собой тройной связью и нахо-
дятся в третьем валентном состоянии (sp-гибридизация), т. е. у них
из четырех орбиталей (одна s и три р) гибридизованы только две ($ и
р). Из трех связей между ненасыщенными атомами углерода одна
о- и две л-связи. В молекуле ацетилена все четыре атома расположены
на одной прямой, углы между а-связями — 180° (см. с. 29). Длина
связи С—С в молекуле ацетилена равна 0,120 нм, расстояние между
атомами углерода и водорода С—Н 0,106 нм. Энергия тройной связи
равна 829,6 кДж/моль и отличается от суммы энергий трех ординар-
ных связей: энергия связи С—Н в молекуле ацетилена 506,9 кДж/моль.
Формулы и названия основных гомологов приведены в табл. 9.
При наименовании алкинов тривиальное название практически
сохранилось только для первого гомолога — ацетилена. По рацио-
нальной номенклатуре алкины рассматриваются как производные
ацетилена, в котором один или оба водорода заменены на алкилы:
СН3—С=С-СН2—СН3 СН3—С=С—СН—СН3
метилэтилацетилен I
сн3
метилизоп ропилгцетилен
По номенклатуре ИЮПАК родовым названием углеводородов с трой-
ной связью являются алкины. Для наименования алкинов по ИЮПАК
выбирают самую длинную цепь, содержащую тройную связь, оконча-
ние «ан» в названии соответствующего алкана заменяется на «ин»,
перед названием цифрой указывается номер первого ненасыщенного
атома углерода. Цепь углеродных атомов в молекуле алкина нуме-
руют таким образом, чтобы ненасыщенные атомы углерода обознача-
лись наиболее низкими номерами. В остальном поступают так же, как
и при наименовании алканов:
5 4 3 2 1
СН3—СН—С = С—СН3 4-метил-2-пентин
СН3
Углеводородные радикалы, содержащие тройную связь, называются
алкинильными группами. Наиболее часто встречающиеся радикалы
имеют тривиальные названия:
89
Н—С = С--------этинил
Н—С=С—СН2 — пропаргил
Структурная изомерия в ряду алкинов связана со строением угле-
родного скелета и положением тройной связи:
НС = С—СН—СН3 3-метил-1 -бутин
I
сн3
НС = С—СН2—СН2—СН3 1-пентин
СН3—С = С—СН2—СН3 2-пентин
Молекула ацетилена имеет линейное строение, геометрическая
изомерия у алкинов отсутствует.
Способы получения. Наибольшее промышленное значение из ал-
кинов имеет ацетилен, получаемый в технике разложением карбида
кальция водой или термической деструкцией углеводородов.
Получение ацетилена из карбида кальция (Вёлер, 1862):
СаС2 + 2Н2О -> НС==СН + Са(ОН)2
карбид ацетилен
кальция
По этому способу, хотя он и требует значительного расхода электро-
энергии для получения СаСг, получают основное количество ацетиле-
на для синтезов и сварочных работ.
Получение ацетилена термическим разложением углеводородов.
Основное сырье: алканы Сх—С4, бензины:
1500—-1600°С
2СН4-----------> СН==СН + ЗНа
метан
1200°С
С2Н6-----> СН = СН + 2Н2
этан
Получение из метана требует наибольшей затраты энергии, но
это компенсируется его доступностью и низкой стоимостью. Высокие
температуры, необходимые для пиролиза, могут быть достигнуты
частичным сжиганием исходного сырья (окислительный пиролиз):
7СН4 + ЗО4 2СН = СН + С + 2СО + 8Н2 + 4Н2О
метан
Сейчас разработаны способы получения ацетилена пиролизом при тем-
пературе 20 000°С (получение в «плазме») с выходом до 80%.
Получение из дигалогенпроизводных алканов:
Н Вг
С2Н5—С—С—СН3 + 2NaOH -> С2Н5—С=С—СН3 + 2NaBr + 2Н2О
| | (спирт, р -р) 2-бутин
Вг Н
2, 3-дибромпентан (I)
90
H Cl
I I
CH3—С—С—H + 2NaOH -> CH3-C=C-H + 2NaCl + 2Нг0
I | (спирт, p-p) 1-пропин
H Cl
1,1 -дихлорпропан
(II)
Дегидрогалогенирующие средства: твердые NaOH; КОН; спирто-
вые растворы щелочей, NHaNa. Вицинальные галогенпроизводные
алканов (I) синтезируют из алкенов, следовательно, этим путем мо-
жет быть осуществлен переход от двойной связи к тройной.
Геминальные дигалогеналканы (II) получают из карбонилсодер-
жащих соединений; используя эти превращения, можно перейти от
альдегидов и кетонов к алканам:
PCI* NaOH
R—СН2—С.-------------> R—СН2—СНС12 -------> R—С==Н
ХН геминальный (спирт. алкин
дигалогеналкан Р*Р)
альдегид
Получение алкинов алкилированием металлорганических произ-
водных ацетилена (см. с. 95).
Физические свойства. Гомологи С2—С4 (за исключением СН3—СН2С = СН) —
газы; Сб—С15 — жидкости; С15 и выше — твердые вещества (табл. 9). У алкинов
наблюдаются те же закономерности изменения температур кипения и плавления,
что и у алкенов. Положение тройной связи существенно влияет на температуру
кипения (1-пентин кипит при температуре +39,6°С; 2-пентин — при 56,2°С).
Химические свойства. Химическое поведение алкинов связано с на-
личием в их молекуле тройной связи и особенностями ее строения.
Для алкинов характерны реакции присоединения, протекающие сту-
пенчато сначала с образованием алкенов (или их производных), затем
алканов. Можно было бы предположить, что алкины, содержащие в
молекуле две л-связи, присоединяют электрофильные реагенты зна-
чительно легче, чем алкены. На практике это не подтверждается.
В молекулах алкинов ненасыщенные (s/7-гибридизованные) угле-
родные атомы расположены ближе друг к другу, чем в молекуле ал-
кенов (0,12 и 0,134 нм соответственно), и обладают повышенной элект-
роотрицательностью. л-Электроны тройной связи несколько втянуты
внутрь молекулы, поэтому положительно заряженные атомы углеро-
да меньше экранированы с внешней стороны; поляризуемость л-элект-
ронов тройной связи уменьшена. Отсюда алкины труднее, чем алкены,
присоединяют электрофильные реагенты и легче — нуклеофиль-
ные.
Реакции замещения SlV связаны с кислотными свойствами 1-ал-
кинов и их способностью образовывать ацетилениды. Алкины легко
полимеризуются и изомеризуются, вступают в реакции конденсации,
окисляются.
Реакции присоединения. Алкины вступают в реакции
электрофильного (Лг), нуклеофильного (Лдг) и радикального при-
соединения (Лн).
91
Гидрирование алкинов. Водород в присутствии катализаторов (пла-
тины, палладия, никеля) восстанавливает алкины в алканы. На пер-
вом этапе образуется алкен, который затем восстанавливается в ал-
кан:
сн=сн + Н2 —СН2=СН2 4- Н2 —сн3—сн3
ацетилен этилен этан
Присоединение галогенов
СН==СН + Вг ->- СНВг=СНВг + Вг2 -> СНВг2—СНВг2
ацетилен 1,2-дибром этан 1, I, 2, 2,-тетрабромэтан
Присоединение галогенов к тройной связи происходит с меньшей ско-
ростью, чем присоединение к алкенам. На промежуточном этапе об-
разуются /пранс-изомеры.
Присоединение галогенводородов (Лг). Присоединение галоген-
водородов к алкинам происходит по правилу Марковникова, но менее
активно, чем к алкенам. Реакция протекает в две стадии:
на на
СН==СН —>- СН2=СНС1 > СН3—СНС12
ацетилен винилхлорид 1,1-дихлорэтан
Реакция применяется для получения винилхлорида.
Присоединение воды (реакция гидратации). В присутствии серной
кислоты и сульфата двухвалентной ртути алкины легко присоединя-
ют воду с образованием уксусного альдегида:
сн==сн + н2о
H2SO4+HgSO4
он
виниловый .уксусный
альдегид
Образовавшийся на первом этапе реакции виниловый спирт не-
устойчив и перегруппировывается в уксусный альдегид (правило
Эльтекова). Эта реакция, открытая в 1884 г. М. Г. Кучеровым и на-
званная его именем, имеет промышленное значение. Присоединение
воды к гомологам ацетилена происходит в соответствии с правилом
Марковникова:
H2SO4+HgSO4
ацетон
сн3—с=сн + н2о
метилацетилен
Присоединение спиртов (реакция Фаворского и Шостаковского):
КОН (ТВ)
сн=сн + нос,н5 ----------сн2=сн—о—сн2—сн3
ацетилен этиловый 200°С) этилвиниловый эфир
спирт
Реакция протекает в присутствии едких щелочей с образованием
алкилвиниловых эфиров.
92
Присоединение карбоновых кислот. В присутствии Н3РО4 ацетилен
способен присоединять карбоновые кислоты с образованием сложных
виниловых эфиров:
Н Н
нс==сн + н—о—с—сн3 —+ о
II / \ II
о н о—с—сн3
уксусная кислота уксусновиниловый эфир
(винилацетат)
Реакция имеет важное практическое значение для получения ви-
нилацетата (см. с. 96).
Присоединение синильной кислоты.
кат.
СН = СН + HCN-------> СН2=СН—CN
ацетилен акрилонитрил
Реакция протекает при участии в качестве катализатора раство-
ра, содержащего хлорид меди, продукт реакции — акрилонитрил
имеет важное промышленное значение (см. с. 96).
Присоединение оксида углерода и спиртов (реакция Ренке). Реакция
идет в присутствии катализатора (карбонилы металлов):
О
Ni (СО)4
Н-С=С-Н + СО + СН3ОН----------------->- Н2С=СН—Ск
метилакрилат 'ОСН3
Реакция применяется для получения эфиров акриловой кислоты.
Полимеризация алкинов. Ацетилен легко вступает в
реакцию полимеризации с образованием, в зависимости от условий
реакции и природы катализаторов, разнообразных продуктов, широко
применяющихся в качестве сырья для синтеза каучуков, пластических
масс, химических волокон, пленкообразующих материалов.
Образование бензола (Зелинский, Казанский):
450°С
ЗСН = СН----------------> С6Нв
ацетилен активированный уголь бензол
Образование винилацетилена. При взаимодействии двух молекул
ацетилена в присутствии водного раствора хлоридов меди к аммония
получают винилацетилен и дивинилацетилен:
2НС=СН -> сн = с—сн=сн2; зсн==сн -> сн2=сн—с===с—сн=сн2
ацетилен винилацетилен ацетилен дивинилацетилен
При присоединении к винилацетилену хлороводорода образуется
хлоропрен, мономер, применяемый в производстве полихлоропрена.
СН2=СН—С = СН +НС1 СН2=СС1—сн=сн2
винилацетилен хлоропрен
93
Образование купрена:
250°С
пСН = СН —(С2Н2)Л
медь купрен
Купрен — желтый аморфный порошок с малой теплопроводностью,
применяется в качестве изоляционного материала.
Реакции ацетиленового атома водорода.
В молекуле алкинов (СН=СН; R—С=СН) атом водорода у ненасыщен-
ного углеродного атома обладает способностью отрываться в при-
сутствии сильных оснований в виде протона. Алкины с концевой аце-
тиленовой группой проявляют кислотные свойства.
Образование ацетиленидов. При взаимодействии ацетилена с ам-
миачным раствором оксида серебра выпадает осадок серого цвета —
ацетиленид серебра:
НС=СН + 2 [Ag(NH3)2] ОН AgC==CAg + 4NH3 + 2Н2О
ацетиленид
серебра
Аналогично протекает реакция с аммиачным раствором хлорида
меди (I):
СН3—С=СН + [Cu(NH3)2] ОН -> СН3—C=CCu + 2NH3 + Н2О
ацетиленид меди
В чем же причина кислотных свойств алкинов? Связь между не-
насыщенным углеродным атомом и атомом водорода (=С—Н) осу-
ществляется в результате перекрытия sp-орбитали углеродного атома
и s-орбитали атома водорода. sp-Орбиталь обладает повышенным s-ха-
рактером по сравнению с sp2- и $р3-орбиталями (доля s-орбитали в
образовании sp-орбитали 50%), что повышает электроотрицательность
углеродного атома:
_]_3 —5 —5 -j-5
н->с=с н
Образующийся при отрыве водорода анион обладает значительной
стабильностью: R—С=С—Н + В" ВН + R—С==С~, следовательно,
/?2
К = kt/k2 будет тем больше, чем меньше k2. Поэтому связь С—Н в ал-
кинах приобретает способность гетеролитически разрываться с от-
щеплением протона (кислотные свойства).
Ацетилениды серебра и меди применяются для разнообразных син-
тезов, в сухом состоянии они крайне взрывоопасны. Образование аце-
тиленидов серебра — качественная реакция на тройную связь.
HO^CNa и HC^CMgl используются для получения гомологов аце-
тилена:
СН = СН + NaNH2 —RHCe=C№ + NH3
ацетиленид
натрия
НС=С№ + C2H6Br -> НС=С—С2Н5 + NaBr
этилацетилен
94
НС=СН + CH3MgI -> HC=CMgI 4- сн4
ацетилены дма г нийиод
HC==CMgI + С2н51 -> НС==С-СД + Mgl2
этилацетилен
Взаимодействие с альдегидами и кетонами. При взаимодействии
альдегидов с ацетиленом образуются вторичные спирты (алкинол ы);
лО
НС=С—Н + сн3—c<f
\н
уксусный
альдегид
CuC-CCu
ОН
СН3—С—С=СН
Н
алкинол
действие второй молекулы уксусного альдегида приводит к образо-
ванию алкиндиола:
ОН
;О
СН3—С—С = СН + сн3—сС
CuC-CCu
ОН он
I I
сн3—С—С = С—С—СН3
н н
ал к ин диол
Н
При действии кетонов образуются третичные спирты:
СН3
| кон (ТВ.)
Н—С=С—Н + с—сн3 -------->
сн3 сн3 сн3
I (CH8)SC=O | |
H—Chee С—С—СН3----------* СН3—С—С = С—С—СН3
I I I
ОН он он
алкинол алкиндиол
Окисление алкинов. Алкины более стойки к действию
обычных окислителей, чем алкены. Окисление перманганатом приво-
дит к разрыву С—С-связи и образованию карбоновых кислот:
Ю]
СН3—С=С—СН3
диметилацетилен
О о
II II
СН3—С—С—СНз
дикетон
[О], Н2О
/0
2СН3—С
\он
уксусная кислота
Особое значение в технике имеет горение ацетилена. В ходе реак-
ции выделяется большое количество теплоты:
2СН = СН 4- 50, -> 4СО2 4- 2Н2О — Д/7
Изомеризация алкинов (реакция Фаворского):
КОН (спирт. р-р)
СН3—СН2—С=СН ------------► сн3—с=с—сн3
95
Схема 7. Промышленное использование ацетилена
96
Реакция протекает при нагревании в присутствии спиртовых раство-
ров щелочей.
Отдельные представители. Применение. Ацетилен. Из алкинов
особое значение в промышленности имеет ацетилен. Простота получе-
ния, относительно низкая стоимость, химическая активность, разно-
образие реакций, в которых он способен участвовать, сделали ацетилен
важнейшим исходным соединением для многочисленных промышлен-
ных синтезов (схема 7). Ацетилен широко используется для резки и
сварки металлов, так как при его сгорании выделяется большое ко-
личество теплоты, а температура ацетиленкислородного пламени пре-
вышает 3000°С.
Ацетилен — бесцветный газ, без запаха, хорошо растворимый
во многих органических растворителях. Смеси ацетилена с кислоро-
дом и воздухом, особенно при повышенном давлении, исключительно
взрывоопасны, поэтому при работе с ним нужно проявлять крайнюю
осторожность. Ацетилен хранят и транспортируют в специальных
баллонах, заполненных пористыми материалами (активированный
уголь, алюмосиликаты), пропитанными ацетоном.
АЛКАДИЕНЫ
Диеновые углеводороды (алкадиены) содержат в молекуле две
двойные связи. Общая формула СПН2П_2.
Классификация, номенклатура, изомерия. Способы получения и
особенно свойства диенов зависят от взаимного расположения двой-
ных связей, поэтому диены принято классифицировать по этому
признаку, деля их на три группы.
1. Диены, у которых две двойные связи находятся у одного атома
углерода и содержат группировку /С=С=С\ (аллен и его гомо-
логи). Они называются диенами с кумулированными или алленовыми
связями по названию аллена — первого гомолога, содержащего эту
систему.
II. Диены, у которых двойные связи разделены одной простой
связью. Они содержат группировку ^С = С— С=С^ и называются
диенами с сопряженными или конъюгированными связями.
III. Диены, в молекуле которых двойные связи разделены двумя
\ 1 I / ГЛ
и более простыми связями: рс = С—(СН2)Л—С = С^. Они называются
диенами с изолированными связями. Формулы и названия наиболее
важных диенов приведены в табл. 10.
По ИЮПАК допускается употреблять тривиальные названия для
некоторых диенов: аллен, изопрен. В отдельных случаях при постро-
ении названия симметричных диенов их рассматривают как состоящие
из двух радикалов: дивинил, диаллил (табл. 10). По ИЮПАК наиме-
нование диенов строится по тому же прйнципу, что и ’алкенов. Перед
окончанием «ен» ставится приставка «ди» (два), указывающая на ко-
личество двойных связей в молекуле. Этот принцип общий. Если в
97
Т з б л r п я JO, Алкалиеноаые угленопорплы
Формула Название Температура плавления, °C Температура кипения, °C ПЛОТНОСТЬ р, г-см“3 Показатель преломления 20 nD
тривиальное по ИЮПАК
С кумулированными связями (1,2-диены)
сн2=с=сн, сн3—сн=с=сн2 Аллен Метилаллен Пропадиен 1,2* Бутадиен — 146,0 — 136,3 —32,0 4-10,3 0,662 (при т. кип.) 0,676 (при 0сС) 1,4168 (при т. кип.) 1,4206 (при 1°С)
С сопряженными связями (1,3-диены)
сч X ©1 О X II 0 X II и X 1 V X 1 Я? и <_>—5 и II е> <N X X (J о Дивинил Изопрен 1,3-Бу та диен 2-Метил-1,3-бута- диен — 108,9 — 145,9 —4,5 +34,0 0,658 (при 0°С) 0,6810 1,4292 (при —25°С) 1,4219
С изолированными двойными связями 1
СН2=СН—СН2-СН=СН2 Дивинилметан 1,4-Пентадиен — 148,3 +25,9 0,6608 1,3888
сн2=сн—сн2—сн2—сн=сн2 Диаллил 1,5-Гексадиен — 140,7 +59,9 0,6929 1,4042
молекуле находятся три двойные связи, то перед окончанием «ен»
ставится приставка «три» (триен); четыре — «тетра» — тетраен и т. д.
Это положение справедливо и по отношению к соединениям, содер-
жащим несколько тройных связей: алкадиина, алкатетраина и т. д.
По этому же принципу строится и название соединений, содержащих
одновременно двойные и тройные связи: алкенины, алкантриенины,
алкандиендиины и т. д. Нумерация углеродной цепи начинается с того
конца, к которому ближе расположена двойная связь:
СН3
СН3 СН3 СН2
I I I
СНз — С=С------С=СН—СНз
1 2 3 4 5 6
2,3-диметил-4-этил-2,4-гексадиен
СН3
сн3 — с=сн—СН—с= с—сн2—сн3
1 2 3 4,5 6 7 8
СНз
2,4-диметил-2-октен-5-ин
СН3 СНз
I I
СН3СН= с—с=с—сн2—с=сн
1 2 3 4,5 6 7 8
СНз
3,4,5-триметил-2,4-октадиен-7-ин
Диены с кумулированными двойными связями (1,2-диены, аллен и
его гомологи) пока не имеют большого практического применения.
Диены с изолированными связями ведут себя в химических реакциях
так же, как и алкены, с той разницей, что в реакциях могут участво-
вать одна или обе двойные связи. Наибольший интерес в химическом
отношении и наибольшее значение в химической промышленности
имеют диены с сопряженными двойными связями. 1,3-Бутадиен, изо-
прен — мономеры, из которых построены важнейшие природные и
синтетические материалы. Поэтому основное внимание будет уделено
именно этой группе диенов.
Способы получения. Получение 1,3 - бутадиена. Из
этилового спирта. В 1910 г. С. В. Лебедев предложил синтез 1,3-бута-
диена из этилового спирта в присутствии смешанных катализаторов.
В 1928 г. руководимый им коллектив химиков стал победителем меж-
дународного конкурса по разработке промышленного получения син-
тетического каучука, объявленного Высшим Советом Народного Хо-
зяйства СССР. В 1932 г. в нашей стране уже начали работать первые
в мире заводы по производству синтетического каучука из 1,3-бута-
диена. Синтез осуществляется по следующей схеме:
Al2O8+ZnO
2СН3—СН2—ОН---------> СН2=СН—СН=СН2 4- Н2 + Н2О
этиловый спирт 1,3-бутадиен
39
По-видимому, реакция протекает через образование следующих про-
межуточных продуктов:
С2Н5ОН----> СН3—С дегидрирование
-н‘ \н
этанол уксусный альдегид
2СН3—С--------> СН3—СН—СН2—С
уксусный альдегид альдоль
/н,
сн3—сн—сн2—с >- сн3—снсн2—сн2
ОН Х'Н он он
альдольная
конденсация
гидрирование
альдоль диол
сн3—сн—сн2—сн2 —сн2=сн—сн=сн2
| | -2Н2О
он он
дегидратация
диол
Применение AI2O3 и ZnO в качестве катализаторов позволяет осущест-
вить перечисленные процессы в ходе одного цикла при повышенной
температуре. Процесс сопровождается образованием в качестве по-
бочных продуктов ряда других соединений. В настоящее время этот
исторически важный способ имеет ограниченное применение, его
целесообразно применять только при использовании в качестве сырья
дешевого синтетического этилового спирта.
Из бутана и бутенов. Каталитическое дегидрирование я-бутана
и н-бутенов с образованием 1,3-бутадиена — наиболее важный и пер-
спективный способ его получения. В промышленности осуществляется
в двух видах: двухступенчатое дегидрирование н-бутана и односту-
пенчатое дегидрирование н-бутана и н-бутенов. Сырье — бутановая
фракция, выделяемая из продуктов нефтепереработки или попутных
газов; н-бутены — из газов деструктивной переработки нефтяного
сырья.
Двухступенчатое дегидрирование н-бутана:
кат. 1
сн3—сн2—сн2—сн3------
—Н2
м-бутан
сн2=сн—сн2—сн3—
1-бутен кат .2
сн3—сн=сн—сн3 — н‘
2-бутен (66%)
СН2-СН—сн=сн2
1,3-бутадиен
^катализатор 1 — оксид хрома, нанесенный на оксид алюминия;
* катализатор 2 — оксид железа и хрома).
2. Одноступенчатое дегидрирование н-бутана:
100
сн3—сн2—сн2—сн3-------► сн2=сн—сн=сн2
««бутан —2Нг 1,3-бут .1Д иен
(катализатор — смесь оксидов алюминия и хрома). Получение 1,3-бута-
диена по одноступенчатой схеме осуществляется по более простой
технологии и требует меньших энергетических затрат, чем по двух-
ступенчатой, и поэтому экономически выгоднее.
Получение 1,3-бутадиена из н-бутенов. Обычно осуществляется
по технологической схеме второй части двухступенчатого метода.
Получение изопрена. В настоящее время отсутствует
единый общепринятый метод получения изопрена. В каждом конкрет-
ном случае выбор метода связан с наличием сырья, энергетическими
ресурсами и т. д. Наиболее перспективными способами являются:
получение изопрена дегидрированием изопентана и изопентенов и
синтез из пропилена.
Дегидрирование изопентана и изопентенов. Сырье: н-пентан, изо-
меризованный в изопентан, изопентан газового бензина, изоамилены,
выделенные из продуктов деструкции бензина:
сн3
Н3С—СН
сн2
с!н3
525—600°С
сн3
—-сн2 =с—снсн3—
2-мет ил-1 -бутен
СН3
| 560—600°С
СНз— С=СН—СН3 --------------
2-метил-2-бутен (60%)
СН2=С—СН=СН2
изопрен
2- мети л бутан
СН3
—->сн2=сн—СН—СН3—
3-мет ил-1-бутен
Дегидрирование протекает в присутствии (в качестве катализа-
торов) оксидов хрома, молибдена или вольфрама, нанесенных на оксид
алюминия.
Возможно одностадийное дегидрирование изопентана в изопрен:
СНз СНз
| 560°С |
СН3—СН—СН2—СН3-------НЗН2= С—СН=СН2
Синтез изопрена из пропилена. Первая стадия — образование
димера:
1^б_200°С
2СН2=СН—СН3-----------------------> СН2=С—СН2—СН2—СН3
пропилен I
СН3
2-метил-1-пентен (димер)
Димеризацию проводят при давлении 200-105 Па; катализатор —
трипропилалюминий.
Изомеризация 2-метил-1 -пентена в 2-метил-2-пентен протекает
при температуре 150—300°С в присутствии фосфорной кислоты на
-алюмосиликате:
101
CH2=C—CH2—CH2—CHS сн3—с=сн—сн2—сн3
СНз СНз
2-мет ил-1-вентен 2-метил-2-пентен
Заключительный этап — образование изопрена (температура
650—750°С, катализатор — бромид водорода):
СНз
сн3—с=сн—сн2-сн3--------► сн2=с—сн=сн2
—CFL I
2-тетил-2-пентен I
СН3
изопрен
Химические свойства диенов с сопряженными двойными связями.
1,3-Диены отличаются рядом особенностей, которые обусловлены элек-
тронным строением. В молекуле 1,3-бутадиена все атомы углерода
Рис. 23. Схематическое изображение строения молекулы ди-
винила
находятся в состоянии $р2-гибридизации и лежат в одной плоскости
а четыре параллельные друг другу орбитали л-электронов, располо-
женные перпендикулярно плоскости, в которой лежат ядра атомов
углерода, объединены в одну молекулярную орбиталь (рис. 23).
Образование общей молекулярной орбитали является следствием
взаимодействия между собой двух сопряженных двойных связей.
В этом случае отдельные пары л-электронов не закреплены за опре-
деленными связями, а до известной степени распределены (делокали-
зованы) по всей электронной системе, т. е. по всем находящимся в
сопряжении связям, с образованием общего электронного облака.
Максимальная электронная плотность возникает между Q и Сг, С$
и С< атомами углерода, меньшая — между С2 и С3. Атомы С2 и С3,
кроме о-связи, как бы дополнительно связаны между собой. Вследствие
этого расстояние между Q—С2 и С3—С* больше, чем между ненасы-
щенными атомами углерода в алкенах, а между С2—С3 короче простой
связи в алкенах. Длина связей в первом случае равна (нм) 0,136
(длина обычной двойной связи — 0,134); во втором 0,146 (длина обыч-
ной С—С связи 0,154).
Такое взаимодействие двух соседних л-связей называется стати-
ческим эффектом сопряжения. Эффект сопряжения сказывается и на
энергетическом состоянии молекулы: внутренняя энергия молекулы
на 14,7 кДж/моль ниже, чем можно ожидать при отсутствии этого
эффекта. Таким образом, диены с сопряженными двойными связями
102
более устойчивы по сравнению с диенами с кумулированными и изоли-
рованными двойными связями. Иногда для написания формул соеди-
нений с сопряженными двойными связями пользуются следующим
изображением: СНгтСНтСН^СН?. В этой формуле с помощью пунк-
тирных линий пытаются показать выравненность распределения
электронной плотности между углеродными атомами. Приведенные
особенности строения 1,3-диенов и определяют их свойства. Они ха-
рактеризуются пониженной теплотой сгорания, гидрирования. От-
личительной особенностью сопряженных диенов является возникнове-
ние динамического эффекта сопряжения — перераспределение элект-
ронного облака молекулы под влиянием атакующего реагента.
Реакции присоединения. У диенов с сопряженными
связями эти реакции могут протекать по двум направлениям: а) по
месту разрыва одной из двойных связей (вторая остается незатрону-
той)— так называемое 1,2-присоединение; б) по концевым углерод-
ным атомам системы сопряжения с образованием новой двойной связи
в положении С2—С3 — так называемое 1,4-присоединение:
ci.
СН2=СН—СН=СН2--------
1,3-бутадиен
1,2-присоединение
------------------->- сн2о—chci—сн=сн2
1,2-дихлор-3-бутен
1,4-присоединение
-------------------->СН2С1— СН=СН—СН2С1
1,4-дихлор-2-бутен
Выход 1,2- и 1,4-продуктов зависит от характера присоединяющегося
вещества и условий взаимодействия.
При присоединении хлора к 1,3-бутадиену образуется половина,
а при присоединении брома — 2/з 1,4-изомера от общего количества
продукта. Аналогично идет присоединение галогенводородов:
1,2-присоединение
сн2=сн—сн=сн2---
1,3-бутадиен
1,4-присоединение
СН3—CHCI — сн= сн2
2-хлор-3-бутев
СН2С1—СН—СН—сн3
1-хлор-2-бутен
Если присоединение НВг идет при —80°С, то образуется 80% продук-
та присоединения в положение 1,2 и 20% в 1,4; при 40°С картина об-
ратная.
Присоединение водорода «в момент выделения» идет в 1,4-поло-
жение, в присутствии катализатора — в 1,2- и 3,4-.
СН2=СН—СН=СН2
1,3-бут адиен
Hf/Ni
--->• СН3—СН2—СН2—СН3
и-бутан
----СН3—СН==СН—СНз
2-бутен
Возможность двоякого 1,2- и 1,4-присоединения к диенам с сопря--
108
женными связями будет понятна, если рассмотреть механизм этой ре-
акции на примере присоединения хлора:
СН2=СН—СН=СН2 + С12 —
---сн2=сн—сн=^сн2
ж-комплекс (1)
1 I >4 4 ♦ ’ 2 >4 i
CH2C1—CH—CH=CH2 —----*-СН2С1— сн=сн-сн2 + сГ
(2а) (26)
или
СН2С1— С1(^СН^СН2 (3)
CH2C1—CHC1—СН=СН2 СН2С1—СН=СН—СН2С1
1,2-дихлор-З-бутен 1,4-дихлор-2-бу1ен
Реакция начинается с электрофильной атаки реагентом л-электронов
одной из двойных связей, в результате чего образуется л-комплекс
(1). Этот комплекс быстро превращается в сопряженный карбкатион,
имеющий положительный заряд на Сг и С* (2а и 26). Наиболее устой-
чивым состоянием будет промежуточное между 2а и 26. Это обозна-
чают в виде двусторонней стрелки, соединяющей обе структуры. Та-
кие структуры, которые нельзя описать одной обычной формулой,
а распределение электронной плотности в них является промежуточ-
ным между несколькими (двумя и более) изображениями, называются
мезомерными. В нашем примере состояние 2а и 26 описывает
мезомерный аллильный карбкатион, а формулы 2а и 26 называют гра-
ничными формулами. Этот же мезомерный катион можно описать,
формулой (3).
Образованием мезомерного карбкатиона завершается первая ста-
дия механизма электрофильного присоединения хлора к 1,3-бута-
диену. На второй стадии анион хлора атакует один из электрондефи-
цитных атомов углерода (Сг или С4). В результате образуется смесь
продуктов 1,2- и 1,4-присоединения. По аналогичному механизму
идет присоединение брома, галоген водородов и каталитическое гид-
рирование.
Кроме электрофильных реагентов диены способны присоединять
и свободные радикалы в 1,2- и 1,4-положении, проявляя при этом
большую реакционную способность, чем алкены.
Диеновые синтезы. Для синтеза разнообразных органи-
ческих веществ и количественного определения 1,3-диенов имеет
большое значение присоединение к ним в положение 1,4 соединений,
содержащих активную двойную связь, или диенофилов. Эти реакции
носят название диеновых синтезов или синтезов Дильса—Альдера.
В общем виде они имеют следующий вид:
104
диен
диенофил
циклоалкен
В качестве диенофилов используют различные соединения, содержа
щие /С=с/, —С==С— или —Ы=М-связи. Этилен и его гомологи —
слабые диенофилы. Реакция протекает быстрее, если двойная связь
активирована электроноакцепторными группировками. Хорошими
диенофилами являются, например, акрилонитрил СНг=СН—CN. ак-
ролеин СНг=СН—СНО, малеиновый ангидрид:
/СН‘2
СН
I
СН
\зн2
1,3-бутадиен
малеиновый ангидрид аддукт
Полимеризация диенов. В присутствии ряда реаген-
тов на свету 1,3-диены полимеризуются. Полимеризация диенов с
сопряженными двойными связями может идти по типу 1,2- или 1,4-
присоединения. В последнем случае возможно образование цис- и
трансизомеров:
СН=СН2
1,2-
пСН2=СН-СН=СН2
1,3-бутадиен
1,4-ЦИС-
1,4-транс -
с=с
L—Н2С СН2— Ап
г—Н2С Н
с=с
Н СН2— Ап
полибутадиены различного
строения
Реакция полимеризации диенов имеет важное народнохозяйствен-
ное значение.
Отдельные представители. Применение. \,3-Бутадцен.
СН2=СН—СН=СН2 — бесцветный газ с резким запахом. В про-
мышленности получают из этилового спирта, н-бутана, бутиленов и
ацетилена. Один из основных мономеров для получения каучука и
латексов (см. с. 106) применяется также для синтеза других промыш-
ленных продуктов, в том числе перхлордивинила — средства для
борьбы с филоксерой на виноградных плантациях.
Н Н
105
Изопрен СН2—С—СН—СН2— бесцветная жидкость. Является струк-
СН3
турным компонентом большой группы важнейших природных соеди-
нений: природного каучука, терпенов (см. с. 425), каротиноидов
(см. с. 430). Является одним из основных мономеров при полу-
чении синтетического каучука. Главные способы промышленного
получения: дегидрирование изопентана и изопентенов, получение
из пропилена, синтез из ацетилена.
Каучук. Натуральный каучук представляет собой высокоэластич-
ную массу, добываемую из сока растений — каучуконосов. В промыш-
ленном масштабе каучук получают из сока дерева гевеи, произраста-
ющего в тропических странах (Южйая Америка, Индонезия, Малайя,
Шри Ланка). Для извлечения каучука млечный сок (латекс), полу-
ченный подсечкой деревьев, коагулируют, добавляя органические
кислоты, прокатывают в листы и сушат. Состав полученного продукта
(С6Н8)П. После обработки сырого каучука серой получают эластич-
ную резину. Каучук представляет собой полимер со средней молеку-
лярной массой 300 000—400 000, содержащий остатки изопрена в цис-
форме:
В связи с высокой потребностью промышленности в каучуке большая
часть его получается синтетическим путем. Для получения синтети-
ческого каучука в качестве мономеров используют 1,3-бутадиен, изо-
прен, хлоропрен, стирол, кремнийорганические соединения. Используя
разные мономеры, меняя условия полимеризации, получают син-
тетический каучук, не уступающий, а по некоторым свойствам превос-
ходящий природный.
Карбоциклические углеводороды
ЦИКЛОАЛКАНЫ
Циклоалканы, или цикланы, — углеводороды, содержащие в мо-
лекуле циклы (кольца), построенные из атомов углерода (карбоцик-
лические соединения), связанных между собой о-связью. Из рассмот-
ренной ранее классификации органических соединений (см. с. 42)
следует, что цикланы входят в состав алициклических соединений.
Общая формула циклоалканов СПН2П. Следовательно, молекулы цик-
ланов, не имеющие заместителей, состоят из связанных между собой
и замкнутых в кольца групп СНа (метиленовая группа); отсюда и дру-
гое их название — полиметиленовые соединения.
Гомологический ряд, номенклатура, изомерия. В табл. И приве-
дены формулы и названия наиболее важных цикланов, а также ши-
роко применяющиеся упрощенные формулы, при написании которых
подразумевается, что атомы углерода находятся в вершинах много-
106
Таблица 11. Циклоалканы
Название Формула Температура плавления, °C Температура кипения, СС Плотность р, Г-СМ"3 Показатель преломления «20 nD
Циклопропан; триме- тилен CsHe; / — 126,6 —32,8 0,720 (при —79СС) 1,3799 (—42,5СС)
Циклобутан; тетра ме- тилен С4Н8; — —90,0 12,9 0,700 1,3752 (при 0°С)
Циклопентан; пентаме- тилен С5Н1О: /~ \ —93,3 49,5 0,745 1,4067
Циклогексан, гексамети- лен С6Н12 \ 1 6,5 81,4 0,778 1,4262
Циклооктан, октаметилен с8н, | Ч| 14,3 148 (при 464,17-102 Па) 0,830 1,4563
угольника и с ними связаны (если отсутствуют заместители) по два
водородных
атома:
циклогексан
По рациональной номенклатуре цикланы рассматриваются как
замкнутые цепочки, построенные из групп СН2. По номенклатуре
ИЮПАК цикланы имеют те же названия, что и алканы с тем же числом
атомов углерода, но с прибавлением префикса «цикло». Если в цикле
замещено больше одного атома водорода на алкилы, то указывается
номер углеродного атома цикла, у которого расположены заместители.
При этом нумерация должна быть произведена таким образом, чтобы
все атомы цикла, несущие боковые цепи, получили наименьшие но-
мера:
Структурная изомерия цикланов связана с различной величиной
циклов (1, II), видом и строением заместителей (изомерия радикалов*
III, IV, V, VI) и их расположением (VII, VIII):
СН3
сн2—сн2—сн3
I
-СИ —СН3
метилциклопетан
пропилциклопропаи 1-метил-2-этилциклояропан
<111) (IV)
1, 2, 3-триметилциклопропан
СН3
СН—СН3
изопр онилцикло-
пропан
(VI)
Н3с СН3
1, 2-диметилцикло-
бутан
(VII)
-------------СН3
1, 3-диметилцикло-
бутан
(VIII,
108
Для цикланов характерны различные виды стереоизомерии (геомет-
рическая, оптическая, поворотная изомерия). Геометрическая изо-
мерия возникает при наличии в цикле нескольких заместителей,
расположенных у различных атомов углерода:
Применяется несколько способов написания изомеров, считают,,
что плоскость цикла расположена перпендикулярно к плоскости чер-
тежа, а заместители расположены снизу или сверху плоскости (I);
или располагают цикл в плоскости чертежа, а положение замести-
телей над ней или под ней указывают затемненными и светлыми тре-
угольниками (II):
сн3 сн3
цис- форма
(И)
/СНз
СН3
транс-форма
О поворотной изомерии циклов см. с. 113.
Источники и способы получения. Цикланы и их производные ши-
роко представлены в природе. Они входят в состав нефти, эфирных
масел и некоторых других природных соединений. Нефть является
основным природным источником цикланов с пятью и шестью атомами
углерода в цикле, содержание которых в нефти отдельных место-
рождений превышает 50%. Это дало основание Марковникову на-
звать этот класс соединений нафтенами («нафта» — по-гречески нефть).
В эфирных маслах (см. с. 425) представлены главным образом цикли-
ческие углеводороды с десятью атомами углерода (терпены) и их раз-
нообразные производные. В растительных и животных тканях широко
распространены производные гексаоксициклогексана—инозиты.
Синтетическим путем цикланы получают или из алифатических
соединений (создание цикла, циклизация), либо из производных цик-
ланов. При синтезе циклов часто получают не углеводород, а его про-
изводные, поэтому на практике два эти метода (циклизация и полу-
чение цикланов из производных) объединяются.
Получение из дигалогенпроизводных алканов'.
109
•CH2Br
CHj + Zn-
^СНгВг
+ ZnBr,
1. З-дибромпропан
циклопропан
Для отнятия галогенов используется натрий или цинковая пыль
в спиртовом растворе. Способ особенно удобен для получения трех-
41 четырехчленных циклов; циклы, содержащие пять атомов углерода
-и более, этим методом синтезируются с трудом.
Прямая циклизация алканов’.
СН3 —С—СН2—СН—сн3
I I
сн3 сн3
2, 2» 4-триметилпентан
1,1, 3-триметилциклопентан
Получение гидрированием ароматических углеводородов (см. с. 118):
Каталитическое гидрирование бензола и его гомологов — наиболее
важный способ получения циклогексана и его производных. В качестве
катализаторов применяют никель, платину, палладий. Температура
гидрирования зависит от строения гидрируемого соединения, вила
катализатора и т. д.
Взаимодействие дигалогенпроизводных с динатриймалоновым эфи-
ром (см. с. 231). Реакция применяется для получения трех—шести-
членных циклов.
Декарбоксилирование солей двухосновных кислот. Используют
соли кислот, содержащих шесть атомов углерода и более: '
сн2-сн2-сч
ХО
^Са------►
/ — СаСО,
о
сн2—сн2—с/
%
циклопен.
тан он
+ 4Н
циклопентан
адипиновокислый кальций
Для получения кетонов можно использовать сухую перегонку
двухосновных кислот с оксидами металлов (МпО; ВаО). Метод при-
меняется для получения циклов, содержащих пять атомов углерода
и более в молекуле.
Физические свойства. Характеристика цикланов приведена в табл. 11. Для
«их наблюдаются те же закономерности в изменении физических свойств, что и
ПО
для других углеводородов. Отличие состоит в более высоких температурах кипе-
ния и плавления, чем у соответствующих алканов.
Химические свойства. Цикланы, содержащие различные по раз-
меру циклы, неодинаково ведут себя в химических реакциях. Соеди-
нения с трех- и четырехчленными циклами отличаются повышенной
реакционной способностью, для них характерны реакции присоеди-
нения. Углеводороды с циклами из пяти и более атомов углерода бо-
лее устойчивы, для них характерны реакции замещения. Большое зна-
чение в химии цикланов играют реакции, связанные с сужением и рас-
ширением циклов (превращение, или изомеризация, циклов).
Действие галогенов:
циклопропан
ССЦ
----* Вг—СН2—СН2—СН2—Вг (I)
1, 3-дибромпропан
+ НВг
бром циклопентан
(11)
циклопентан
В первом случае реакция (I) сопровождается разрывом цикла и
присоединением атомов брома. Она протекает главным образом по
ионному (А Е) механизму. Скорость присоединения к малым циклам
уменьшается в ряду 12>Вг2>С1г, а также с переходом от трех- к
четырехчленному циклу. Хлор при взаимодействии уже с циклопро-
паном частично вступает в реакцию замещения. Взаимодействие с
галогенами пятичленных и содержащих большее количество атомов
углерода циклов не сопровождается разрывом углерод-углеродных
связей, а приводит к реакциям замещения (реакция II), которая про-
текает по цепному свободнорадикальному механизму S R. Скорость
ее, а также направление (до известной степени) зависят от характера
галогена и строения циклана.
Действие галогенводородов:
/\ 4- НВг
СН3—СН2—СН2Вг
С цикланами, содержащими пять атомов углерода и более в цикле,
галогенводороды не взаимодействуют. Реакция протекает главным!
образом по ионному механизму с соблюдением правила Марковникова.
Действие водорода (гидрогенолиз). В присутствии катализаторов
(платины, палладия, никеля) водород присоединяется к циклопропа-
ну с образованием пропана:
/\ + н,— ->сн3—сн2—сн3
С увеличением размеров цикла его устойчивость возрастает и для
осуществления гидрогенолиза требуются все более высокие темпера-
туры. Начиная с циклогексана, цикланы не способны к реакциям гид-
рогенолиза.
Окисление цикланов. Цикланы, даже циклопропан и циклобутан,
довольно стойки к действию окислителей. При повышенных темпера-
111
турах, под действием сильных окислителей происходит разрыв цикла
с образованием двухосновных кислот с тем же числом атомов углерода
в молекуле:
циклогексан
СН2—СН2—СООН
сн2—сн2—соон
адипиновая кислота
Превращение циклов. В присутствии катализаторов (сильные
кислоты, кислоты Льюиса) может происходить взаимное превращение
циклов (расширение или сужение). При расширении циклов атомы
углерода боковой цепи участвуют в создании нового, большего по
размерам цикла; при сужении, наоборот, углеродные атомы цикла
переходят в боковую цепь. Для реакции часто используют не угле-
водороды, а их производные. Процесс сопровождается разнообразными
структурными превращениями, частичным окислением и т. д.
1. Действие иодводородной кислоты (сужение цикла):
метил циклопентан
2. Действие азотистой кислоты (Н. Я. Демьянов):
циклогексанол
-----—СН2ОН
циклопентилкар-
бинол
3. Действие хлорида алюминия (Зелинский и др.). При действии
хлорида алюминия на алкилцкклобутаны при низких температурах
образуется циклопентан или его производные, затем производные
циклогексана. При нагревании циклогексана и его производных они
превращаются в производные циклопентана:
R
A1CU
на холоду
Ri
А1С1,
на холоду
112
где R; Rii R2; R3 — углеводородные радикалы, содержащие разное
количество атомов углерода.
Из приведенных реакций видно, что химическое поведение цик-
ланов тесно связано с размерами циклов. Вначале считали, что все
атомы в цикле расположены в одной плоскости. Из этого следовало,
что валентные углы в 3-членном цикле равны 60, в 4-членном — 90,
в 5-членном — 108, в 6-членном — 120° и т. д. В 1885 г. Байер вы-
сказал предположение, что чем больше отклонение валентного угла
в цикле от угла в тетраэдре (109°28'), тем больше напряженность и
меньше устойчивость цикла. Мерой напряженности он предложил
считать половину разности между углом в тетраэдре и валентным
углом того или иного цикла (а):
Название
Угол отклонения
Название
Угол отклонения а
а
Циклопропан . . Ц-24О44'
Циклобутан . . . +9°44'
Циклопентан . . -j-0°44'
Циклогексан . . . —5° 16'
Циклогептан . . . —9°33'
Циклооктан . . . —15°51
Полученные цифры хорошо объясняли химическое поведение
циклопропана, циклобутана, циклопентана, но противоречили экс-
периментальным данным о стойкости средних и больших циклов.
Объяснение этому было дано после открытия поворотной изомерии
органических соединений.
Поворотная изомерия. Поворотная изомерия в молекуле органи-
ческих соединений является результатом не вполне свободного (ог-
раниченно свободного) вращения групп атомов молекулы вокруг
о-связей. Возникающие при этом поворотные изомеры получили на-
звание конформеров (или конформаций). Конформеры обладают раз-
ным запасом свободной энергии, а следовательно, различна и вероят-
ность существования молекулы в виде того или другого конформера.
Поясним это на примере уже изученного соединения этана. В моле-
куле этана СН3—СН3 одна метильная группа может поворачиваться
относительно другой, не нарушая при этом порядка связей, валент-
ных углов и длины связей,
заторможенная заслонённая
(типа „козел")
заторможенная заслонённая
(с пунктирными линиями
и „клиньями")
образуя различные поворотные изомеры (конформеры). Переходя из
одного конформера в другой молекулы этана преодолевают опреде-
5—682 ИЗ
ленный энергетический «барьер». Две крайние конформации, они по-
лучили название «заслоненная» и «заторможенная», будут максималь-
но отличаться друг от друга по запасу энергии (около 12,&
кДж/моль) (рис. 24). При рассмотрении молекулы вдоль оси, соединяю-
щей два углеродных атома в «заслоненной» конформации, атомы во-
дорода у одного углеродного атома будут закрывать водородные ато-
Рис. 24. Зависимость потенци-
альной энергии этана от угла
поворота вокруг простой угле-
род-углеродной связи:
заторможенная (Л), заслоненная
(В) конформации
Рис. 25. Проекция Ньюмена
для этана:
заторможенная (а), заслоненная (б)
конформации
мы, расположенные у другого. В «заторможенной» (шахматной) кон-
формации атомы водорода, расположенные у первого (ближнего к.
зрителю) углеродного атома, будут находиться между водородами
второго углеродного атома. Это хорошо видно при применении проек-
ций Ньюмена (рис. 25). Заторможенная конформация энергетически
более выгодна.
В простых органических соединениях (алканы) вследствие малого-
энергетического барьера переход из одной конформационной формы
в другую осуществляется легко, выделить конформеры в чистом виде
не удается. В большинсте случаев при работе с органическими вещест-
вами приходится иметь дело со смесью конформационных модифика-
ций, в которых преобладает наиболее энергетически выгодная форма.
В сложных молекулах, например в цикланах, конформационное
состояние молекулы существенно влияет на ее реакционную способ-
ность. Цикланы характеризуются различным пространственным стро-
ением циклов. Молекула циклопропана представляет собой жесткую
систему, все три атома углерода которой лежат в одной плоскости.
Как известно, прочность связи между углеродными атомами зависит
от полноты перекрывания гибридизованных облаков электронов.
В случае о-связи (например, в алканах) максимальное перекрытие
гибридизованных облаков осуществляется по прямой, соединяющей
два атома. Валентный угол между связями 109°28'. В молекуле цикло-
пропана о-связи отличаются от обычных, характерных для атомов угле-
рода, находящихся в состоянии 5р3-гибридизации. Это связано с тем,.
114
что максимумы перекрывания электронных облаков углерод-угле-
родных связей в циклопропане находятся не на прямой, соединяющей
эти атомы, а расположены снаружи (изогнутые связи) (рис. 26). Та-
кое явление приводит к уменьшению напряжения, так как угол между
связями увеличивается от 60 до 106°, но сами связи приобретают час-
тично ненасыщенный характер и получили название «банановых».
По прочности связи они зани-
мают промежуточное положение
между о- и л-связями. Поведе-
ние циклопропана в химических
реакциях объясняется понижен-
ной устойчивостью цикла, что
связано с неполным перекрыва-
нием $р3-орбиталей углеродных
атомов. Уменьшение устойчивос-
ти цикла хорошо видно при
сравнении теплот сгорания Рис. 26. Строение циклопропана (а);
(табл. 12). «банановые» связи (б)
Отклонение от нормального
(109°28') значения валентных уг-
лов наблюдается и в молекуле циклобутана, что сказывается и на его
химическом поведении. Однако на поведение более сложных циклов
большее влияние оказывает строение поворотных изомеров. Рассмот-
Таблица 12. Характеристика цикланов
Соединение Число атомов углерода Теплота сгорания циклана, кДж/моль на одну группу СН2 Избыток энергии по сравнению с циклогексаном
Циклопропан 3 698,0 38,5 •
Циклобутан 4 686,7 27,2
Циклопентан 5 666,4 5,5
Циклогексан 6 659,5 0,0
Циклогептан 7 663,3 3,8
Циклооктан 8 664,5 5,0
Циклононан 9 666,4 5,5
Циклодекан 10 664,5 4,6
Для ациклических соединений — 659,5 —
рим поворотную изомерию циклов на примере одного из главных пред-
ставителей цикланов — циклогексана. Избыточный запас энергии в
молекуле цикланов (напряжение) возникает: а) в результате откло-
нения валентных углов от нормального валентного угла (109°28'), так
называемое байеровское или угловое (ангулярное) напряжение;
б) отклонения атомов от наиболее выгодной «заторможенной» кон-
формации, в результате чего возникает напряжение, получившее на-
звание напряжения заслонения (торсионного); в) взаимодействия
непосредственно не связанных между собой атомов или групп атомов
5* Р5
(отталкивание или притягивание), что существенно влияет на кон-
формацию молекул. Совершенно естественно, что циклогексан, как
и любое другое соединение, стремится принять такую пространст-
венную конфигурацию, при которой число всех этих факторов было бы
наименьшим, т. е. чтобы внутренняя энергия молекулы была мини-
мальной.
Если молекула циклогексана будет иметь плоское строение, то
валентные углы между атомами углерода будут отличаться от нор-
мальных на 5°16', а следовательно, в молекуле будет иметь место угло-
вое напряжение. Атомы водорода при этом будут находиться в
невыгодном заслоненном положении, что будет приводить к значи-
тельному торсионному напряжению. Понятно, что молекула цикло-
гексана стремится принять такую конфигурацию, в которой бы эти
напряжения отсутствовали и молекула была бы более устойчивой.
Для циклогексана существует две конформации без углового напря-
жения. Валентные углы в них равны 109°28'. Они получили названия
«кресла» и «ванны».:
кресло ванна
В конформации «ванна» четыре атома углерода (2, 3, 5, 6) лежат
в одной плоскости, а два (1 и 4) — возвышаются над ней. Конформа-
ция ванны приблизительно на 28,9 кДж/моль менее устойчива, чем
конформация кресла.
В более выгодной конформации кресла атомы углерода расположе-
ны по три в двух параллельных плоскостях, перпендикулярно к ко-
торым проходит ось симметрии третьего порядка 00; расстояние
между плоскостями равно примерно 0,05 нм (рис. 27). В этой кон-
Рис. 27. Циклогексан—конформация «крес-
ло»
116
формации циклогексан имеет 12 С—Н-связей, которые можно разде-
лить на две группы. Шесть из них параллельны оси симметрии и друг
другу и чередуются «вверх—вниз». Они называются аксиальными свя-
зями и обозначаются а. Шесть других связей направлены радиально
от кольца к периферии молекулы, называются экваториальными свя-
зями и обозначаются е (они направлены под углом 109°28' к оси сим-
метрии). В случае замещенных циклогексана возникает вопрос, в
каком положении будет находиться заместитель (отличный от водо-
родного атома) — в аксиальном или экваториальном?
Рассмотрим в качестве простейшего примера мети л циклогексан.
Поскольку известен только один метилциклогексан, значит, возможны
три предположения: а) метильная группа расположена в аксиальной
связи; б) она расположена в экваториальной связи; в) обе возмож-
ности вероятны, если две формы так быстро превращаются одна в
другую, что их нельзя разделить. На самом деле оказывается верным
именно третье предположение — цикл быстро переходит из одной
формы кресла в другую, при этом один из концов кресла поднимается,
а другой опускается и все заместители, находившиеся в экваториаль-
ном положении, становятся аксиальными, и наоборот:
la 1а
Это явление получило название инверсии цикла, а его скорость —
частоты инверсии. Для циклогексана инверсия происходит настоль-
ко быстро, что молекула совершает около 106 превращений в секун-
ду, проходя при этом через энергетический барьер величиной при-
мерно 46,09 кДж. Метилциклогексан в равновесной смеси аксиаль-
ной и экваториальной форм содержится преимущественно в эквато-
риальной форме, как и большинство других монозамещенных цик-
логексана, имеющих иные «тяжелые» заместители.
Для циклопентана вследствие минимального углового напряжения
в наибольшей степени подходит плоская форма кольца. Однако в
действительности молекула циклопентана не является плоской, так
как в такой структуре все атомы водорода находятся в заслоненном
состоянии, что уменьшает стабильность молекулы примерно на
41,9 кДж. Для того чтобы снизить торсионные напряжения, цикло-
пентан принимает неплоскую конформацию, хотя это и приводит
к возникновению небольшого углового напряжения.
Ряд фактов убедительно доказывает, что молекула циклобутана
не является плоской, а быстро превращается из одной «складчатой»
конформации в другую:
117
Как и в случае циклопентана, торсионный эффект частично ослаб-
ляется за счет небольшого углового напряжения.
Циклы, содержащие С7—С12, также имеют торсионное напряже-
ние и поэтому менее устойчивы, чем циклогексан. Циклы с очень
большим числом звеньев — «макроциклы» приближаются по своей
устойчивости к циклогексану.
Отдельные представители. Применение. Циклопропан С3Н6 —
обладает наркотическим действием, применяется в хирургии; цикло-
пентан СбН10 используется в разнообразных синтезах и в качестве
добавки, улучшающей качество моторного топлива; циклогексан
С6Н12 — для получения адипиновой кислоты, циклогексанола и
циклогексанона (см. с. 112, 161, 194).
Арены
Арены, или ароматические углеводороды, — соединения, содер-
жащие в молекуле особую циклическую группировку из шести атомов
углерода, которая называется бензольной группировкой (бензольное
ядро):
С—
С—
бензольное ядро
Название углеводородов этой группы (бензола, нафталина, антра-
цена и их многочисленных и разнообразных производных) «аромати-
ческие соединения» — случайное и сегодня потеряло свой первона-
чальный смысл. Действительно, первые открытые соединения или
обладали специфическим, иногда приятным запахом, или были вы-
делены из природных сильно пахнущих продуктов. Но количество «аро-
матных» веществ среди многочисленных известных соединений этой
группы невелико. В то же время наблюдается ряд особенностей в стро-
ении, физических свойствах и химическом поведении этих веществ,
связанных с наличием в молекуле бензольных группировок.
Различают одноядерные (одна бензольная группировка в молекуле)
и многоядерные ароматические углеводороды, содержащие две или
более бензольных группировок. В молекулах аренов в качестве бо-
ковых цепей могут находиться углеводородные радикалы с неразветв-
118
ленной или разветвленной углеродной цепочкой, а также содержащие
двойные и тройные связи и циклические группировки:
этилбензол
винилбензол (стирол)
Следовательно, арены могут содержать в молекуле наряду с аро-
матическими ядрами разнообразные по строению алифатические цепи,
а также включать в состав молекулы другие (не содержащие ядер
бензола) циклические группировки. Естественно, что это обстоятель-
ство не может не сказаться на номенклатуре, изомерии, физических
свойствах и химическом поведении аренов.
ОДНОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
Гомологический ряд, строение, номенклатура, изомерия. Пер-
вый и один из наиболее важных представителей гомологического ряда
одноядерных ароматических углеводородов — бензол С6Н6. Отсюда
и общее название гомологического ряда — ряд бензола. Общая фор-
мула гомологического ряда бензола СпН2п_б. Наиболее важные го-
мологи и их характеристика приведены в табл. 13. Химическая
формула бензола была предложена еще в 1865 г. немецким ученым Ке-
куле, однако споры о строении бензола продолжались до самого пос-
леднего времени. По мнению Кекуле, молекула бензола представляет
собой шестичленный цикл, состоящий из атомов углерода, соединен-
ных чередующимися одинарными и двойными связями. Каждый атом
углерода связан с одним атомом водорода, т. е. бензол является 1,3,5-
Циклогексантриеном:
119
н
с
Н—\с—н
I II
Н—с с—н
/
с
I
н
бензол
(формула Кекуле)
Формула Кекуле правильно отражает элементный состав, соот-
ношение углерода и водорода и равноценность всех атомов водорода
в молекуле бензола. Однозамещенные бензола при отсутствии изо-
мерии радикалов (например, С6Н5СН3 — толуол, С6Н5Вг — бром-
бензол) не имеют изомеров. В то же время формула Кекуле не отве-
чает на вопрос, почему для бензола, содержащего в молекуле три
двойные связи (ненасыщенное соединение), характерны в первую
очередь реакции замещения, протекающие в мягких условиях. Поче-
му он не обесцвечивает бромную воду (качественная реакция на двой-
ную связь)? Не объясняет формула Кекуле и большую устойчивость
бензольного ядра к действию окислителей и высоких температур.
Кроме того, согласно этой формуле должны существовать два изомера
у 1,2-дизамещенных бензола, что противоречит экспериментальным
данным. Ответы на эти вопросы были получены в результате приме-
нения в исследовании бензола наряду с химическими физических ме-
тодов.
По современным представлениям атомы углерода в молекуле бен-
зола образуют правильный шестиугольник со сторонами 0,140 нм
(напоминаем, что длина простой ^С—С^ связи 0,154, двойной —
0,134 нм). Каждый атом углерода связан с одним атомом водорода,
длина С—Н-связи 0,109 нм. Углы, образованные связями Н—С—С
и С—С—С, равны 120° (рис. 28, а). Молекула бензола неполярна.
Все атомы углерода и водорода лежат в одной плоскости. Атомы угле-
рода в молекуле бензола находятся во втором валентном состоянии
(s/Агибридизация), т. е. из четырех (одна s- и три р-орбитали) гибри-
а
Рис. 28. Молекула бензола
120
Таблица 13. Арены (гомологический ряд бензола)
Формула Наименование Температура плавления, °C Температура кипения, °C Плотность р, г«см~3 Показатель преломления
тривиальное по ИЮПАК
С6Н„ Бензол Бензол +5,4 80,1 0,878 1,5011
СвН5СН3 Толуол Метилбензол —95,0 110,6 0,867 1,4969
СвН4(СН3)2 Ксилол
орто-Ксилол 1,2-Диметилбензол —25,2 144,4 0,880 1,5055
мета-Ксилол 1,3-Диметилбензол —47,9 139,1 0,864 1,4972
пара-Ксилол 1 -4-Диметилб ензол 13,3 138,4 0,861 1,4958
С6Н5С2Н5 Этилбензол Этилбензол —95,0 136,2 0,8670 1,4959
СвН5С3Н7 Пропил бензол «-Пропилбензол —99,5 159,2 0,8620 1,4920
CflH:CH(CHs)2 Кумол Изопропилбензол —96,0 152,4 0,8618 1,4915
С6Н4(СН3)(С3Н7) лара-Цимол 1 -Метил-4-изопропил- бензол —67,2 177,1 0,8573 1,4909
дизованы только три (s и 2р) орбитали. Следовательно, каждый атом
углерода связан с соседними углеродными атомами и атомом водорода
о-связями, затрачивая на это 3 электрона. Четвертый электрон не
закреплен за определенным атомом углерода. Шесть незакрепленных
(делокализованных) р-электронов образуют единую устойчивую зам-
кнутую циклическую л-систему (рис. 28, б), дополнительно связывая
углеродные атомы, электронное облако которой расположено под
и над бензольным кольцом. Поэтому в молекуле бензола, по сущест-
ву, нет ни одинарных, ни двойных связей.
Для бензола характерно явление кругового сопряжения; необхо-
димое условие его — параллельность орбиталей р-электронов, обра-
зующих л-систему. Реальная молекула бензола обладает меньшей
на 150,8 кДж/моль энергией (энергия сопряжения, резонанса) по срав-
нению с энергией молекулы, для которой справедлива формула Ке-
куле (1, 2, 3-циклогексатриен). Поэтому углерод-углеродные связи
в молекуле бензола обладают повышенной прочностью. Отсюда и
особенности в физических свойствах и химическом поведении бензола
и его гомологов. В химическом отношении — это, в первую очередь, ус-
тойчивость бензольного ядра к повышенным температурам, окисли-
телям, инертность по сравнению с олефинами в реакциях присоеди-
нения, легкость протекания и своеобразие реакций электрофильного
замещения (нитрования, сульфирования, галогенирования и т. д.).
Рядом особенностей, как будет показано, обладают производные
бензола. Совокупность всех этих свойств получила название «арома-
тический характер».
Указанные особенности строения бензола затрудняют написание
его структурной формулы. В последнее время для обозначения бен-
зола используют формулы, в которых с помощью стрелок, пунктир-
ных линий или окружности, вписанной в шестиугольник, стремятся
показать выравненность электронных плотностей между атомами
углерода:
Хотя эти формулы более точно отражают некоторые особенности
строения бензола, формула Кекуле продолжает широко применяться.
Однако, используя ее, нельзя забывать о ее недостатках.
Для наименования ароматических соединений часто применяются
исторические названия: бензол, толуол, стирол, кумол. По номен-
клатуре ИЮПАК ароматические углеводороды рассматриваются как
производные бензола, положение заместителей указывают цифрами,
учитывая, что номера атомов углерода, у которых расположены за-
местители, были наименьшими:
122
метилбензол
(толуол)
1-метил-З-этилбензол
В случае двух заместителей вместо цифр можно пользоваться
приставками 1,2—орто (р-)\ 1,3—мета (л/-); 1,4—пара (п-):
1,2-диметилбензол 1,3-диметилбензол
(орто-ксилол) (мета-ксилол)
1,4-диметилбензол
(пара-ксилол)
Для наименования ароматических углеводородов, имеющих слож-
ную боковую цепь или несколько бензольных ядер, их рассматри-
вают как производные алифатических углеводородов, содержащих ра-
дикал СвН5 — фенил (Ph):
1.2-дифенилэтан
Общее название ароматических углеводородов — арены, общее
название радикалов — арилы (Аг). Наиболее распространенные арилы:
С6Н5— —фенил (от старинного названия бензола «фен»)
С6Н4^ —фенилен (о-, ж-, п-)
СН3—СбН4-----толилы (о-, м-, п-)
С6Н6СН2- — бензил Ph—СН2 —
с6н5сн/ —бензилиден или рь-сн/
С6н5с^ — бензенил Ph—
Однозамещенные бензола не имеют изомеров. Изомерия ди- и поли-
замещенных бензолов связана с различным расположением замести-
телей (например, о-, ж-, п-) и их строением.
Источники и способы получения. Основные источники получения
ароматических углеводородов — нефть и продукты сухой перегонки
(коксования) каменного угля. Выделение ароматических углеводо-
родов из продуктов коксования каменных углей — наиболее старый
и до 50-х годов основной способ их получения. При нагревании выше
1000°С без доступа воздуха (сухая перегонка) уголь разлагается с
образованием твердых (кокс), жидких (каменноугольная смола, амми-
ачная вода) и газообразных (газы коксования) продуктов перегонки.
133
Кокс — твердая, пористая масса, состоящая в основном из углерода,
применяется в больших количествах в металлургии. Газы коксования*
содержащие водород, метан, оксид и диоксид углерода, азот, этиле*
новые и ароматические углеводороды после отделения последних ис-
пользуются в качестве топлива. Из аммиачной воды получают аммиак
и соли аммония. Каменноугольная смола — буро-черная масло-
подобная масса, с резким запахом, содержит большое количество
органических соединений различной природы. Выход смолы около
3,0%. На первом этапе переработки каменноугольную смолу пере-
гоняют, обычно собирая 4 фракции (табл. 14).
Таблица 14. Основные фракции каменноугольной смолы
Фракции Температура, °C Выход, % Состав
Легкое масло До 180 0,5—2-,0 Бензол, толуол, ксилолы, циклопентадиен, гетеро- циклы
Среднее масло 180—240 12 Фенол, нафталин, крезолы
Тяжелое масло 240—270 10 Нафталин и его производные
Антраценовое масло 270—360 20 Антрацен, фенантрен
Остаток от перегонки — 60% от массы дегтя — называется пеком.
Это твердая, размягчающаяся при нагревании масса темного цвета.
Из перечисленных фракций разнообразными приемами выделяются
индивидуальные органические соединения.
В последние годы коксохимическая промышленность как источ-
ник ароматических углеводородов уступила первое место нефти.
Содержание ароматических углеводородов в нефти сильно колеблется.
И хотя в отдельных случаях оно достигает 60%, основное количество
ароматических углеводородов получается из нее при химической
переработке (ароматизация нефти), главным образом пиролизе и ка-
талитическом риформинге, в ходе которого протекают реакции де-
гидрирования и дегидроциклизации (см. с. 146).
Кроме этого, получение ароматических углеводородов может быть
осуществлено дегидрированием циклопарафинов (Зелинский) с при-
менением в качестве катализатора платины на оксиде алюминия:
+ зн2
и дегидроциклизацией парафинов (Б. А. Казанский, А. Ф. Платэ,
В. Л. Молдавский) (катализаторы — оксиды хрома, молибдена, ва-
надия):
сн3—(сн2)4—сн3 —
124
Синтетические способы получения. Полиме-
ризация ацетилена (см. с. 93).
Взаимодействие ароматических и алифатических галогенпроиз-
водных с металлическим натрием (реакции Вюрца—Фиттига):
СбН5Вг + С4Н9Вг 4- 2Na---> СбН5—С4Н9
бром- бутилбро- —2NaBr бутилбензол
бензол мид
Алкилирование ароматических углеводородов галогенпроизводными
и олефинами (реакция Фриделя—Крафтса, см. с. 129).
Физические свойства. Характеристика аренов приведена в табл. 13. Арома-
тические углеводороды — жидкости или твердые вещества с высокими показа-
телями преломления, легче воды и практически в ней нерастворимы. Хорошо
растворяются в органических растворителях. Горят ярким коптящим пламенем.
Температуры кипения и плавления аренов сильно зависят от их строения. При
работе с ароматическими веществами необходимо помнить, что они в большинст-
ве случаев токсичны.
Химические свойства. В молекулах одноядерных ароматических
углеводородов различают две части: ароматическое ядро и боковую
цепь. Боковых цепей может быть несколько, они могут отличаться
строением (прямые, разветвленные углерод-углеродные цепи), со-
держать различное количество по-разному расположенных ненасы-
щенных связей (двойные, тройные) и т. д. Химические свойства одно-
ядерных ароматических углеводородов в общем случае определяются
именно наличием и особенностями химического поведения этих двух
компонентов молекулы, а также их взаимным влиянием друг на дру-
га. Для ароматического ядра вследствие особенностей, его строения
характерны, в первую очередь, реакции электрофильного замещения
(5^); реакции присоединения, окисления протекают значительно труд-
нее. Поведение боковой цепи зависит от ее строения. Нередко одни и
те же реагенты могут взаимодействовать и с ароматическим ядром,
и с боковой цепочкой. В этих случаях направление атаки будет за-
висеть от условий реакции. Различным будет и механизм однотипных
реакций (замещения, присоединения и т. д., табл. 15).
Т А б л и ца 15. Механизм однотипных реакций
Часть молекулы Тип разрыва связи Промежу- точная частица Механизм реакции
Реакции замещения
Бензол, ароматическое ядро Гетеролиз
в алкилбензолах
Насыщенная боковая цепь в Гомолиз
алкилбензолах
Карбкатион Ионный, нецепной, SE
Цепной, радикальный,
Радикал
Реакции присоединения
Бензол, ароматическое ядро Гомолиз Радикал
в алкилбензолах
Ненасыщенная боковая цепь Гетеролиз Карбкатион
в алкилбензолах
Гомолитический,
Ионный, нецепной, А&
125
Реакция замещения. Из реакций замещения для арома-
тических соединений особое значение имеют реакции электрофиль-
ного замещения, с помощью которых получают важные в промышлен-
ном отношении производные бензола:
нитрование
сульфирование
хлорирование
алкилирование
по Фриделю-Крафтсу
ацилирование
по Фриделю-Крафтсу
Нитрование бензола. Под действием концентрированной азотной
кислоты или смеси концентрированных азотной и серной кислот («нит-
рующая смесь») атомы водорода бензольного ядра замещаются на
нитрогруппу:
сбн6 + hono2 —> c6h5no2 + Н2О
бензол нитробензол
Нитрование бензола — типичная реакция электрофильного за-
мещения, поэтому на ее примере будет рассмотрен механизм этих пре-
вращений. Электрофильное замещение в ароматическом ряду — про-
цесс, протекающий в две стадии. Реакции предшествует превращение
молекулы реагента с образованием электрофильной частицы (в на-
+
шем случае — катион нитрония NO2) и аниона:
+ -
HNO3 + 2H2SO4^NO2 + 2HSO4 + Н3О+
нитроний-
катион
Катион нитрония притягивается секстетом л-электронов молекулы
бензола с образованием л-комплекса. Затем происходит разрушение
секстета, два из шести электронов, закрепляясь (локализуясь) у од-,
ного из углеродных атомов, образуют о-связь с заместителем. Этот атом?
углерода переходит из sp2- в $р3-состояние. Оставшиеся 4 электрон^
распределяются между пятью атомами углерода бензольного ядра^
образуется фенониевый катион (о-комплекс) — реально существую-
щее соединение, образование которого доказано:
к -комплекс
а -комплекс
126
На этом первом этапе реакция напоминает реакцию присоединения к
алкенам (см. с. 72). Ив том и в другом случае образуется карбоние-
вый ион. Однако на второй стадии образовавшийся о-комплекс, яв-
ляясь сильной протонной карбокислотой, _при участии основания
легко теряет протон (он соединяется с HSO7, образуя молекулу сер-
ной кислоты), переходя в ароматическое соединение:
о -комплекс
ли1робензол
Отщепление протона с восстановлением ароматичности (обра-
зование нитробензола) энергетически выгоднее, чем взаимодействие
между промежуточным катионом (о-ком-
плексом) и анионом (HSOr), которое
наблюдается в случае реакции с алкена-
ми. Энергетическая диаграмма реакции
электрофильного замещения в бензоль-
ном ядре приведена на рис. 29. Для об-
разования л-комплекса (точка 1) нуж-
но преодолеть энергию активации Afj,
а о-комплекса (точка 2) — АЕ2', отщепле-
ние протона и образование нитробензо-
ла требует преодоления энергетического
барьера, равного Д£3. Энергия актива-
ции реакции электрофильного замеще-
ния Е а.
Первая стадия — атака бензола но-
ч-
ном нитрония NO2 с образованием а-
комплекса — протекает медленно, вто-
рая — отщепление протона — быстро.
Поэтому первая стадия является лими-
тирующей и определяет скорость реак-
ции электрофильного замещения.
Рис. 29. Энергетическая диаг-
рамма реакции электрофиль-
ною замещения (нитрование)
в бензольном ядре
Нитрование ароматических углеводородов осуществляется в мяг-
ких условиях. При введении второй и третьей нитрогрупп (мета-
положение) требуются более жесткие условия:
HNO3: H?SO4
бен юл
HNO3, H2SO4
-Н2О
NO2
1,3-динитробензол
Н.\О<; H2SO4 _
-н2о
no2
1,3,5-тринитробензол
127
Сульфирование. При действии на бензол и его гомологи концентра
рованной серной кислоты или олеума атомы водорода в бензольном
ядре замещаются на сульфогруппу. Реакция сульфирования обра-
тима:
бензолсульфокислота
Сульфированию предшествует образование электрофильного аген-
та SO3H+ (гидросульфониевый ион):
3H2SO4 -> Н3О+ + 2HSCV + SO3H+
В дальнейшем реакция протекает через следующие стадии:
о-комплекс сульфокислота
Н4' + HSOT^^l H2SO4
Еще более активным электрофильным реагентом является триоксид
серы, в котором имеется дефицит электронной плотности на атоме
серы O=^Z° ;
О~
биполярный
ион,
а-комплекс
медленно
медленно
сульфокислота
Галогенирование. В присутствии катализатора (А1С13, А1Вг3, FeCl3,
FeBr3. ZnCh) происходит замещение атомов водорода бензольного ядра
на атомы галогена:
А1Вг3
Вг + НВг.
бромбензод
Роль катализатора заключается в создании положительного иона
галогена или поляризации связи галоген-галоген в молекуле галогена
и создания тем самым необходимых условий для электрофильного за-
мещения:
128
Cl2 + FeCl3 '^~~Г FeCl4 + Cl+
а-комплекс
медленно
быстро
Возможно, что свободный ион С1+ не образуется как кинетиче-
ски независимая частица, хлорид железа лишь поляризует молекулу
хлора:
С1
|&- а+
С1—Fe—Cl... Cl
I
Cl
Хлор замещает водород в ароматическом ядре активнее брома,
осуществить иодирование и фторирование практически не удается»
в первом случае из-за недостаточной активности иода, во втором, на-
оборот, из-за чрезмерной активности фтора.
В отсутствие катализатора, при нагревании или под действием
света галогены замещают атомы водорода в боковой цепи предпочти-
тельно в a-положение. Реакция протекает по радикальному меха-
низму:
Cl2 h‘->* 2С1 •
ci
а-хлорэтилбензол
Алкилирование. Действуя на бензол галогеналкилами в присутст-
вии безводного хлорида алюминия (реакция Фриделя—Крафтса,
1877), удается ввести в молекулу бензола алкильную группу (реак-
ция алкилирования):
+ СН—СН2С1
бензол
безв А1С1 з
этилбензол
129
Реакция алкилирования ароматических углеводородов так же,
как и приведенные ранее реакции нитрования, сульфирования и гало-
генирования, протекает по Se -механизму. Роль катализатора заклю-
чается в создании атакующего электрофильного агента (карбониевый
ион или полярный комплекс). В качестве катализатора применя-
ются кислоты Льюиса. По убывающей активности в этой реакции они
располагаются в следующий ряд:
А1Вг3 > А1С13 > SbCl5 > FeCl3 > SnCl4
Механизм реакции алкилирования галогеналкилами может быть
представлен следующим образом:
С1
S+ В— I
СН3—СН2С1 + А1С13 СН3—СН2 . • -С1 • • • Al—CI
I
С1
поляризованный комплекс
или (если для упрощения записи предположить поляризацию с об-
разованием ионов):
СН3—СН2С1 + А1С13 AlClf + СН3—СН+
карбкатион
к-комплекс
о-комплекс
этилбензол
В присутствии кислот H2SO4; Н3РО4; HF, BF3 бензол и его гомо-
логи алкилируются алкенами:
изопропил бен зол
(кумол)
Механизм может быть представлен следующим образом:
к -комплекс
д-комплекс
130
При алкилировании бензола первичными галогеналкилами в про-
дуктах реакции обнаруживаются изомеры, содержащие как первич-
ный, так и вторичный (а иногда и третичный) алкильный заместитель^
например:
+ сн3—сн2—СН2С1
зо%
70%
Это может происходить вследствие перехода менее устойчивого
первичного карбониевого иона (образующегося под влиянием ката-
лизатора) в более устойчивый вторичный (а при соответствующих
структурах — в третичный) карбкатион.
Ацилирование. При действии на ароматические углеводороды хлор-
ангидридов или ангидридов кислот (см. с. 236, 238) удается ввести
в бензольное ядро ацилы (см. с. 235); катализаторы — кислоты
Льюиса. Реакция не сопровождается перегруппировками в радикале.
Процесс обычно ограничивается введением одного заместителя
(моноацил производные):
О О
I! &+ II &-
СН3С—С1 + А1С13 ч± СН3—С-•-СЬ • • А1С13
хлорангидрид
уксусной
кислоты
о
6+11
СН3—С.„С1...А1(С1)3
метилфенилкетон
(ацетофенон)
Ацилирование можно рассматривать как частный случай реакции
Фриделя—Крафтса.
Завершая рассмотрение реакций электрофильного замещения, не-
обходимо остановиться на так называемом правиле ориентации (или
замещения) в бензольном ядре. Реакции замещения после вхождения
в бензольное ядро одного заместителя и образования монопроизвод-
ных бензола на этом не останавливаются, замещение может продол-
жаться с образованием ди-, три- и более замещенных бензола. Кроме
этого, в реакциях могут участвовать гомологи бензола и его произ-
водные. Естественно, возникает вопрос, в какое положение войдет
второй заместитель, будет ли затруднено его вхождение или, наоборот,
131
оно может быть осуществлено с большей скоростью, при более мягких
условиях?
Экспериментальным путе^м было установлено, что положение,
в которое вступает второй заместитель, зависит от природы первого
заместителя, вида действующего реагента (электрофильный или нук-
леофильный) и условий проведения реакции. Первые два условия иг-
рают особенно важную роль. Поясним это на примере реакции нит-
рования. При нитровании монопроизводных бензола в зависимости
от характера уже имеющегося заместителя X нитрогруппа будет
предпочтительно вступать в орто-, пара- или метаположения:
no2
параизомер
Это видно из данных, приведенных в табл. 16.
Таблица 16. Продукты реакции нитрования монопроизводных бензола
Заместитель (X) Ориентация, % Относительная реакционная способность (по отношению к бензолу)
орто- мета- пара-
СН3 56,5 3,5 40 24
С(СН3)3 12,0 8,5 79,5 15,7
С1 29,6 0,9 68,9 0,033
no2 6,4 93,2 0,3 IO"7
Заместители в зависимости от того, в какое положение они на-
правляют вхождение второго заместителя, делятся на две группы:
заместители первого и второго рода.
В реакциях электрофильного замещения заместители первого рода
направляют вхождение второго заместителя преимущественно в орто-
и параположения (орто- и пара-ориентанты):
ортоизомер параизомер
где Xj — заместитель первого рода; Y — электрофильный реагент.
132
к заместителям первого рода относятся группы (расположены в по-
рядке возрастания ориентирующего влияния): галогены <
<—СН3<—CH2R<CR3<OR<OH<NH2<NHR<NR2 и др.
Заместители второго рода направляют (ориентируют) вхождение
второго заместителя в реакциях электрофильного замещения пре-
имущественно в метаположение (л/епга-ориентанты):
метаизомер
где Хи — заместитель второго рода; Y — электрофильный реагент.
К ним относятся (в порядке возрастания ориентирующего эффекта):
—COOR<—СООН<—СНО<—SO3H<—CN<—NO2 и др.
С чем связано ориентирующее влияние находящегося в бензоль-
ном ядре заместителя? Как уже указывалось, в молекуле бензола
существует равномерное распределение электронной плотности. Вве-
дение заместителя нарушает эту равномерность. Неравномерное рас-
пределение электронной плотности в бензольном ядре у монопроиз-
водных бензола — следствие суммарного действия двух эффектов:
индуктивного эффекта (±/) и эффекта сопряжения, или мезомер-
ного эффекта (±Л1). Рассмотрим это на примере двух соединений:
оксибензола (фенола, I) и нитробензола (II), в первом случае это
заместитель первого рода (группа ОН), во втором — второго рода
(группа NO2).
В молекуле фенола электроны связи С—О смещены к атому кисло-
рода, так как он обладает большей электроотрицательностью, созда-
ется диполь с частичным отрицательным зарядом на атоме кислорода
(—/, I). В то же время свободная неподеленная электронная пара ато-
ма кислорода вступает в сопряжение с л-электронами бензольного яд-
ра (+М, I). В молекуле фенола оба эффекта (—I и +Af) оказывают
противоположное действие, но в момент реакции динамический эф-
фект сопряжения (мезомерный эффект), значительно превосходит
индуктивный эффект, в результате электронная плотность у атома
кислорода понижается, сокращается длина связи С—О, а электрон-
ная плотность в ядре возрастает, особенно в орто- и пара-положениях:
•п—И
I
Следовательно, заместители первого рода — группировки атомов,
способные отдавать электроны бензольному ядру (электронодонорные),
при этом электронная плотность в ядре возрастает, особенно в орто-
и пара-положениях. Естественно, что при атаке молекулы монозаме-
133
щенного бензола электрофильными реагентами они и занимают пред-
почтительно эти положения, причем их вхождение происходит легче,,
чем при замещении в бензоле.
Рассмотрим второй пример. В молекуле нитробензола (II) электрон-
ная плотность связи С—N будет смещена в сторону атома азота (—/).
В самой нитрогруппе имеется семиполярная связь (см. с. 270), в ре-
зультате чего возникает сопряжение, направленное от бензольного
ядра в сторону нитрогруппы (—Л4). Таким образом, и индуктивный
эффект, и эффект сопряжения в этом случае направлены в одну сто-
рону:
_ + п
О—N=C
Введение нитрогруппы приводит к снижению электронной плотности
ароматического кольца по сравнению с бензолом.
Следовательно, заместители второго рода принимают (оттягива-
ют) электроны от бензольного ядра (электроноакцепторные). Элект-
ронная плотность в ядре уменьшается. Электрофильный заместитель
предпочтительно вступает в мета-положение, и вхождение его в этом
случае затруднено по сравнению с бензолом. Распределение элект-
ронной плотности в бензольном ядре является только одной из пред-
посылок, влияющих на скорость и направление реакций замещения.
Большое значение имеет устойчивость образующегося а-комплекса,
которая зависит от энергии активации переходного состояния и воз-
можности участия введенного заместителя в делокализации заряда
образующегося иона.
Реакции присоединения. Ароматические соединения
с трудом вступают в реакции присоединения. Присоединение галогенов.
Под влиянием ультрафиолетового излучения к молекуле бензола при-
соединяется три молекулы хлора с образованием гексахлор цикло-
гексана (гексахлорен):
бензол гексахлорциклогексан
Присоединение водорода (см. с. ПО).
Окисление и дегидрирование ароматичес-
ких углеводородов. Бензольное ядро отличается исключи-
тельной стойкостью к действию окислителей KMnCU в кислой среде,
пероксида водорода, хромовой смеси и т. д. При действии окислите-
134
лей на гомологи бензола окисляются боковые цепи с образованием аро-
матических альдегидов и кислот, причем число образовавшихся кар-
бонильных или карбоксильных групп равно числу боковых цепей:
толуол
бензойная кислота
При очень энергичном окислении происходит разрушение бензоль-
ного ядра. Так, в присутствии катализатора, содержащего смесь
оксидов ванадия и молибдена, при температуре 350—450°С бензол
окисляется кислородом воздуха с образованием малеинового ан-
гидрида:
Дегидрирование алкилбензолов. Реакция дегид-
рирования алкилбензолов широко применяется для получения сти-
рола:
CHj—сн3
этилбензол
Сг2О3‘А12О3; 6ОО°С
сн=сн2
стирол
Реакции замещения и присоединения — примеры химических
превращений бензольного ядра. Реакция окисления, дегидрирования
алкилбензолов — реакции боковой цепи. Если для реакций арома-
тического ядра характерны процессы, протекающие через стадию об-
разования карбкатиона и требующие для своего осуществления ка-
тализаторов или условий для образования электрофильных реаген-
тов (реакции электрофильного замещения), то реакции боковой цепи,
как это уже указывалось, проходят с участием свободных радикалов
или в условиях, способствующих их образованию. Рассмотрим броми-
рование этилбензола:
(s«)
(S£)
Если бромирование осуществлять в присутствии катализаторов,
без освещения, замещаются атомы водорода бензольного ядра (I путь,
135
Sr); при нагревании, под действием света, в отсутствие катализаторов
замещаются атомы водорода алкильных радикалов (II путь, S й).
В первом случае атакующей частицей является ион брома, во вто-
ром — атом (свободный радикал) брома. Все эти положения справед-
ливы и для других реакций замещения (нитрования, сульфирования и
т. д.). При замещении в боковую цепь заместитель предпочтительно
направляется в а-положение:
Реакции нуклеофильного замещения в данном учебнике не рассматри-
ваются.
Отдельные представители. Применение. Бензол С6Н6 — жид-
кость с характерным запахом, почти нерастворима в воде. Получает-
ся при сухой перегонке каменного угля и химической переработке
нефти. Важнейший представитель ароматических углеводородов (схе-
ма 8).
Толуол (метилбензол) С6Н5СН3 применяется для получения бен-
зойной кислоты (см. с. 222), сахарина.
// им о л (параметилизопропилбензол)
является основой для получения многих эфирных масел (см. с. 428).
Стирол (винилбензол) — приятно пахнущая жидкость:
сн=сн
2
Получают дегидрированием этилбензола в присутствии катализатора.
Исходный мономер для получения полистирола.
Полистирол — бесцветное, стеклоподобное твердое вещество. От-
личается стойкостью к воде, кислотам, щелочам. Получается полиме-
ризацией стирола:
136
Схема в. Промышленное использование бензола
фенилэтиловый спирт
Широко применяется в радиотехнике, бытовой промышленности, хи-
мии для получения «пенопласта» и разнообразных производных поли-
стирола путем сополимеризации с другими мономерами (бутадиеном,
дивинилбензолом и др.).
МНОГОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ
СОЕДИНЕНИЯ
Многоядерными ароматическими соединениями называются ве-
щества, содержащие в молекуле два или более связанных между собой
бензольных ядер. Различают многоядерные соединения с неконденси-
137
рованными (изолированными) ядрами — дифенил, трифенилметан и
конденсированными — нафталин:
Многоядерные ароматические углеводороды с
неконденсированными (изолированными) ядрами
В молекулах этих соединений бензольные ядра могут быть со-
единены или непосредственно, или с помощью цепочки, состоящей из
различного количества атомов углерода. Наибольшее значение имеют
дифенил, дифенилметан, трифенилметан и разнообразные производ-
ные последнего.
Дифенил — кристаллическое вещество, /пл = 70°С, получается
при пропускании паров бензола через расплавленный свинец, а также
выделяется из каменноугольной смолы:
2СбнД с6Н5-С6Н5 + Н2
В химическом отношении дифенил напоминает бензол. Применяется
в качестве устойчивого высококипящего (/кип = 254°С) теплоносите-
ля и для получения красителей.
Дифенилметан и трифенилметан получают по реакции Фриде-
ля—Крафтса, например:
138
Это кристаллические вещества, используются в синтезе разно-
образных органических соединений, многие из которых являются кра-
сителями. Красители трифенилметанового ряда: фуксин, фенол-
фталеин и др.
Многоядерные ароматические
углеводороды с конденсированными ядрами
В молекулах этих соединений бензольные ядра имеют общие угле-
родные атомы. Важнейшими представителями являются нафталин,
антрацен, фенантрен.
Нафталин
Состав нафталина установлен
ение — Эрленмейером (1866):
А. А. Воскресенским (1858), стро-
а а
а а
По современным представлениям молекула нафталина имеет пло-
ское строение, а облако л-электронов распределено в ней менее равно-
мерно, чем в бензоле.
Сравнивая длины связей в молекуле нафталина (нм) с длиной оди-
нарных и двойных связей, видно, что они занимают промежуточное
положение. Различают а-положение (1, 4, 5, 8 атомы углерода) и 13-
положение (2, 3, 6, 7 атомы углерода), поэтому для нафталина су-
ществуют а- и р-монопроизводные.
Способы получения и свойства нафталина. Нафталин — бесцветное
кристаллическое вещество, /пл = 80°С, /Кип - 218°С, летуч, обла-
дает сильным характерным запахом. Хорошо растворим в органиче-
ских растворителях. Основной промышленный способ получения —
выделение из каменноугольной смолы. Большая его часть сосредото-
чена во фракциях среднего и тяжелого масел (табл. 14).
Химические свойства. Нафталин проявляет ароматический харак-
тер, но легче, чем бензол, вступает в реакции присоединения и за-
мещения.
Реакции замещения. При этих реакциях предпочтительно получа-
ют а-производные:
139
галогенирование Cl2
Cl
4C12
Cl Cl
Cl
cr
Cl Cl
перхлорнафталин
H2SO4;80°C
^\Л
I II I
а-хлорнафталин
SO3H
сульфи-
рование
H2SO4;
160°C
а-нафталинсульфокислота
нитрование HNOS
а-нитронафталин
Гидрирование нафталина
Нафталин гидрируется легче, чем бензол. Тетралин и декалин
применяются в качестве органических растворителей.
Окисление нафталина. При энергичном окислении нафталина
(350—450°С) образуется фталевый ангидрид, катализатор—оксид
ванадия V2O5:
140
нафталин
фталевый ангидрид
Нафталин используется для получения фталевого ангидрида,
фталевой кислоты и других производных.
Антрацен
Молекула антрацена С14Н10 имеет
представлена формулой
плоское строение и может быть
Различают а-положения (1, 4, 5, 8); ^-положения (2, 3, 6, 7) и у-поло-
жения (9, 10), последние иногда называют мезоположениями.
Получают кристаллизацией из каменноугольной смолы. Антра-
цен — бесцветное, кристаллическое вещество, /пЛ = 213°С, он легко
вступает в реакции замещения (заместитель легче всего вступает в
мезоположение) и присоединения. При окислении антрацена в при-
сутствии солей ванадия и молибденовой кислоты образуется антра-
хинон:
О
II
Q
[O1; 375—500°С
I ц ।
II
о
антрацен антрахинон
Из антрацена кроме антрахинона получают разнообразные кра-
сители.
Фенантрен
Фенантрен С14Н10
141
Выделяется из каменноугольной смолы. Бесцветное, кристалличес^
кое вещество; /пл = 100°С. Фенантреновая структура входит в сос-
тав молекул многих биологически активных соединений: стероидов,
витаминов, гормонов.
Высшие полициклические углеводороды
Высшие полициклические углеводороды могут быть построены
из большого числа бензольных ядер, связанных между собой различ-
ным образом:
гексацен
Основной источник их получения — каменноугольная смола. Кро-
ме того, они образуются в качестве побочных продуктов при пиро-
лизе нефти и газа. Входят в состав некоторых лекарственных соеди-
нений и могут служить сырьем для получения красителей. Отдель-
ные представители полициклических углеводородов обладают канцеро-
генным действием, т. е. способны вызывать рак (от лат. cancer —
рак).
НЕБЕНЗОИДНЫЕ СОЕДИНЕНИЯ
С АРОМАТИЧЕСКИМИ СВОЙСТВАМИ
При описании аренов особое внимание было уделено рассмотре-
нию совокупности их физических и химических свойств, которые
обычно определяют общим понятием — ароматический характер.
Под ароматическим характером, как мы указывали, понимается ус-
тойчивость соединений к действию окислителей, легкость протекания
142
реакций электрофильного замещения и др. Соединения ароматиче-
ского характера с большим трудом присоединяют другие атомы и
группы. Производные ароматических углеводородов, в которых за-
мещающие группы связаны с углеродными атомами бензольных ядер,
обладают рядом характерных свойств: повышенной кислотностью гид-
роксильной группы, пониженной основностью аминогруппы, малой
подвижностью атомов галогена. Указанные свойства связаны с особен-
ностями строения бензольных колец: планарностью молекулы, ве-
личинами валентных углов (120°), наличием секстета из л-электро-
нов и т. д.
В настоящее время установлено, что ароматические свойства при-
сущи не только соединениям, в состав молекул которых входят ядра
бензола, а всем соединениям, содержащим циклическую систему дело-
кализованных л-электронов, число которых должно быть равно
(4/г+2), где п = 0, 1, 2, 3 и т. д. Для бензола п = 1, число сопря-
женных электронов 6; для нафталина п = 2, число сопряженных
электронов 10, для фенантрена соответственно 3 и 14. Это положение
получило название «правило Хюккеля». В настоящем разделе учеб-
ника будут рассмотрены только такие небензоидные ароматические
соединения, молекулы которых построены из атомов углерода. Гетеро-
циклические ароматические соединения будут описаны в разделе ге-
тероциклов (см. с. 309).
Ароматические системы с пятичленными
циклами
Если от молекулы циклопентадиена (I) отнять протон, то образу-
ется циклопентадиенильный анион (II), обладающий ароматически-
ми свойствами:
I II
Ароматический характер циклопентадиенил-аниона станет понятным,
если рассмотреть его строение. Кольцо циклопентадиенила — пра-
вильный плоский пятиугольник с углами, равными 108°. Атомы угле-
рода находятся в состоянии $р2-гибридизации. Четыре атома углерода
имеют по одному негибридизованному л-электрону; кроме этого, один
углеродный атом имеет еще два неподеленных электрона (оставшиеся
после отщепления протона). Эти шесть электронов образуют единый
секстет сопряженных л-электронов, что и придает молекуле арома-
тические свойства. Условно строение иона может быть представлено
следующим образом:
143
Для циклопентадиенил-иона достаточно легко протекают реакции
с электрофильными реагентами. Особый интерес представляют егс
соединения с ионами металлов группы железа — металлоцены:
циклопента-
диенилнатрий
+ FeCl2
-NaCl
циклопента-
•диенилжелезо
(ферроцен)
Молекула ферроцена имеет форму сэндвича (бутерброда): атом
железа находится между двумя плоскими пятичленными циклами и
связан со всеми 10 углеродными атомами. Расстояние между атомами
углерода 0,14 нм. Ферроцен — кристаллическое вещество оранже-
вого цвета, /пЛ = 173°С. Вступает в реакции электрофильного за-
мещения.
Ароматические системы с семичленными
циклами
Примером этой группы соединений является циклогептатриенил-
катион (тропилий-ион), который образуется при отнятии водорода
с двумя электронами (гидридный анион) от циклогептатриена:
тропилий- ион
Тропилиевый катион — стабильный карбониевый ион, в котором
имеется секстет из л-электронов, равномерно распределенных между
семью углеродными атомами. Его отличительная особенность — спо-
собность реагировать с нуклеофильными реагентами:
Азулен
Молекулу азулена можно рассматривать как систему, состоящую
из двух конденсированных ионов — циклопентадиенильного и тро-
144
пилия. Пятичленное и семичленное кольца имеют по шесть делокали-
зованных электронов (два электрона общие для двух колец):
азулен
Азулен — кристаллическое вещество синего цвета, обладающее
ароматическим характером. Его производные входят в состав эфирных
масел некоторых растений.
НЕФТЬ И ЕЕ ПЕРЕРАБОТКА
Нефть — жидкое, горючее полезное ископаемое, состоящее из
углеводородов (алканов, циклоалканов, ароматических углеводоро-
дов) с примесью воды с растворенными в ней солями, и соединений,
содержащих азот, серу, кислород. Состав нефти различных месторож-
дений неодинаков: в одних случаях преобладают алканы, в других —
цикланы или ароматические углеводороды. Хотя вопрос о происхож-
дении нефти нельзя считать окончательно решенным, однако, по мне-
нию большинства ученых, она образовалась в результате сложных
химических и биохимических превращений из остатков микроскопи-
ческих растений и животных, накопившихся в течение многих миллио-
нов лет на дне лагун и морей.
Основное количество разведанных запасов нефти находится на тер-
ритории СССР, стран Ближнего и Среднего Востока, Северной к Юж-
ной Америки (США, Канада, Венесуэла), Румынии, в Северном море.
По запасам нефти СССР занимает первое место. В 1985 г. добы-
ча нефти в СССР (с газовым конденсатом) должна достичь
620—645 млн. т.
Из нефтяных пластов, расположенных на разной глубине, нефть
поступает на поверхность под давлением либо растворенных в ней га-
зов (фонтанный способ), либо специально закачиваемой воды, либо
с помощью насосов.
В свежей нефти растворены попутные газы и твердые парафины,
которые отделяются и используются в нефтехимической промыш-
ленности.
Физические свойства. Нефть — жидкость от светло-желтого до черного
цвета, легче воды, с высокой теплотворной способностью 43 995 — 46 090 кДж/м3.
Переработка нефти. После специальной подготовки (удаления воды,
солей) нефть направляется на простую перегонку. Это основной про-
цесс первоначальной ее обработки. Нефть нагревают до кипения и
отбирают фракции (погоны), кипящие в определенных температурных
интервалах. Количество отбираемых фракций зависит от состава нефти
и путей их дальнейшего использования.
Продукты, полученные при первичной переработке нефти, ис-
пользуются в качестве топлива в двигателях и являются сырьем для
6—682
145
Таблица 17. Основные фракции перегонки нефти
Наименование фракции Температура кипения, °C Примерный состав Использование
Бензиновая фракция До 180 С5 С12 Моторное топливо
Керосиновая фракция 180—300 С9 С16 Тракторное топливо, топли- во для реактивных двига- телей , бытовое назначе- ние, сырье для крекинга
Соляровые масла До 360 С12 ^20 Дизельное топливо, смазоч- ные масла, сырье для кре- кинга
Мазут (нефтяной остаток) Выше 360 Сырье для крекинга, полу- чение парафина, смазоч- ных масел, гудрона
дальнейшей переработки. Стремясь увеличить количество получае-
мого из нефти бензина (после прямой перегонки образуется 10—20%),
в ходе вторичной переработки высококипящие фракции и мазут под-
вергают крекингу для частичного разрушения (деструкции) «тяжелых
молекул». Впервые крекинг нефтяного сырья был осуществлен рус-
ским ученым, сотрудником Петербургского технологического инсти-
тута А. А. Летним, а первый проект промышленной установки для
крекинга разработал в 1891 г. известный русский инженер В. Г. Шу-
хов.
В зависимости от исходного сырья и поставленных целей приме-
няют различные разновидности крекинга, которые объединены в две
большие группы: термический крекинг (проводится без катализатора)
и термокаталитический крекинг. Термический крекинг осуществля-
ют при температуре 460—560° С и атмосферном или повышенном дав-
лении (до 1,01 • 107 Па). Крекинг при более высоких температурах
(700—800° С) получил название пиролиза.
Каталитический крекинг проводят в присутствии алюмосиликатов
или платины. В ходе крекинга происходит разложение сложных угле-
водородов с образованием соединений с меньшей молекулярной массой:
C3SH38 -> с9н20 + снь
Одновременно протекают реакции изомеризации, циклизации,
уплотнения. В результате образуется смесь разнообразных продук-
тов, состав которой сильно зависит от условий крекинга.
При повышении температуры возрастает выход газообразных про-
дуктов и ароматических углеводородов. Осуществляя вторичную пе-
реработку нефти, важно не только увеличить выход бензина (он мо-
жет быть доведен до 60—70% от массы сырья), но и повысить его ка-
чество. Качество бензина сильно зависит от входящих в его состав угле-
водородов.
Эталоном хорошего бензина является изооктан (2,2,4-триметил:
пентан):
146
сн8
СНз—СН2—СН—сн3
СН8 СНз
Поэтому качество бензина принято оценивать с помощью «октановых»
чисел. Для изооктана оно равно 100. Октановые числа парафинов изо-
строения выше, чем нормального строения; еще выше они у олефинов,
цикланов и особенно у ароматических углеводородов.
Превращение низкосортных бензинов в высококачественные (с
высоким октановым числом) осуществляется с помощью риформинга.
При этом происходит изомеризация алканов и образование аромати-
ческих соединений. Для повышения качества бензинов применяют
разнообразные добавки, например раствор тетраэтилсвинца (С2Н5)4РЬ
в этилбромиДе С2Н5Вг.
Отдельные компоненты нефти, полученные в результате ее первич-
ной и вторичной переработки, и в первую очередь алканы Q—С5,
этилен, пропилен, ацетилен, циклогексан, бензол, толуол, являются
важнейшим сырьем для синтеза многочисленных органических сое-
динений, в том числе каучука, пластических масс, моющих средств и др.
6*
Раздел второй
Функциональные
производные углеводородов
При замещении атомов водорода в углеводоро-
дах на функциональные (характеристические) группы получаются
их разнообразные производные: галогенпроизводные, окси производ-
ные, альдегиды и кетоны, кислоты и т. д. Соединения, содержащие од-
нотипные функциональные группы, называются соединениями с од-
нородными функциями (иногда полифункциональными); разные —
соединениями со смешанными функциями (гетерофункциональными).
ГАЛОГЕНПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Производные углеводородов, содержащие вместо одного или не-
скольких атомов водорода галогены: фтор, хлор, бром, иод, назы-
ваются галогенпроизводными. В зависимости от характера угле-
водорода, в молекулу которого введен галоген, их делят на насыщен-
ные (галогеналкилы, I), ненасыщенные (II), ароматические (III; IV).
В молекулах последних галогены могут находиться в ядре (III) или
в боковой цепи (IV). По числу атомов галогена, содержащихся в мо-
лекуле, они делятся на моно-(1; II; III; IV) и полигалогенпроизвод-
ные: [ди- (V); три- (VI) и т. д.]
о -хлортолуол • бензилхлорид
(Ш) (IV)
CH3—СН2Вг
этилбромид
(I)
СН2==СН—СН2С1
аллилхдорид
(1-хлор-2-пропен)
(П)
СН2С1—СН2С1 снг3 СС14
1,2-дихлорэтан йодоформ четырёххлористый
углерод
(V) (VI) (VII)
Номенклатура. Изомерия. По рациональной номенклатуре на-
звание галогенпроизводных образуют из названия углеводородного
радикала и галогена, в необходимых случаях указывают положение
последнего:
С1
I
С2Н5Вг СН3—СН—СН2—СН3 СН2=СНС1 С6Н5СН.С1
тилбромид втор-бутилхлорид винилхлорид бензилхлорид
По номенклатуре ИЮПАК положение атома галогена указывают
148
Таблица 18. Галогенпроизводные углеводородов
Соединение Формула Темпера- тура ки- пения, °C Темпера- тура плавле- ния, °C Плотность р, г «см”3
Хлормечан СН3С1 —24 —97 0,920
Хлорэтан С2Н5С1 13,1 — 139 0,910
Дихлормета н СН2С12 40,2 —96,5 1,325
1,2-Дихлорэтан СН2С1—СН2С1 83,7 —35,7 1,252
Хлороформ СНС!3 61,2 —63,6 1,498
Четыреххлористый угле- СС14 76,5 —22,9 1,593
род Винилхлорид СН2=СНС1 — 14 —59,7 0,969
Аллилхлорид СН2=СН—СН2С1 46 —137 (при 13°С)
Хлорбензол С6Н5С1 131,7 —45,2 1,106
БензилхлорнД С6Н5СН.,С1 179,3 —41,1 —
цифрой, которая вместе с его названием располагается перед наиме-
нованием углеводородного радикала. Нумерацию атомов углерода
главной цепи начинают с того конца, к которому ближе расположен
атом галогена:
СН3 СН3 С1
I I I
СН3—СН—СН—СН2—СН—сн3
6 5 4 3 2 1
2-хлор-4, 5-диметилгексан
СН3
5 4| 3 2 1
СНз—С=С—СН2—СН2С1
СНз
1-хлор-З, 4-диметил-3-пентен
В отдельных случаях применяются тривиальные названия: хлоро-
форм СНС13; йодоформ СН13. Изомерия в ряду галогенпроизводных
связана с особенностями строения углеродного скелета, положением
атомов галогена: л
СН3—СН2—СН2—СН2С1
Ьхлорбутан
С1
СН3—С—СН3 СН3—СН—СН2—С1
I I
СНз СНз
2-хлор-2-метилпропан 1-хлор -2-метилпропан
и различным расположением атомов и групп в пространстве (цис-,
трансизомерия; оптическая изомерия):
цисформа
Хс=с/
С'Г 'сНз
трансформа
Способы получения. 1. Замещение атомов водорода в углеводоро-
дах на галоген. В результате галогенирования алканов, цикланов, бен-
зола и его гомологов, алкенов происходит замещение атомов водорода
149
на галоген. В промышленности широко применяются хлорирование
метана. Создавая определенные условия, реакцию направляют пре-
имущественно в сторону образования одного из компонентов (СН3С1,
СН2С12, СНС13, ССЬ). Этот же способ нашел применение и для синтеза
этилхлорида С2Н5С1. В то же время прямым хлорированием алканов
часто не удается получить индивидуальное соединение, а выделение
нужного хлорпроизводного из образовавшейся смеси веществ обычно
является трудной задачей.
Бензол и его гомологи хлорируются легче, чем алканы. На холоду
и в присутствии катализаторов (А1С13, FeCl3) галоген замещает атомы
водорода в бензольном ядре, а при нагревании или на свету в
отсутствие катализатора замещаются атомы водорода в боковой цепи:
бензилхлорид бензилиден- бензотрихлорид
хлорид
При проведении реакции в газовой среде (пропускание хлора в
парах кипящего толуола) могут быть замещены все три атома водорода
метильной группы (реакция протекает по радикальному механизму).
Если катализатор обладает достаточной активностью (А1С13), все
шесть атомов водорода в молекуле бензола могут быть замещены на
хлор:
А1С13
СбНб + 6С12 —> СбС1б 4- 6НС1
бензол гексахлор-
бензол
Реакция протекает по ионному механизму (см. с. 128).
Под действием ультрафиолетового излучения в отсутствие ката-
лизатора к молекуле бензола может присоединяться 3 молекулы хлора
или брома:
СбН6 4- ЗС12 СбН6С16
гексахлорциклогексан
(гексахлоран)
Это основной промышленный способ получения гексахлорана.
При высоких температурах (400—60Ээ С) удается заместить атомы
водорода в алкенах, не нарушая двойной связи:
436°С
СН2=СН2 4- С12----------> СН2=СНС1 4- НС1
избыток
этилена винилхлорид
150
500—550°С
СН2=СН—СН3 + С12-----------> СН2=СН—СН2С1 + НС1
пропилен аллилхлорид
Все особенности приведенных реакций были рассмотрены раньше.
Присоединение галогенов к ненасыщенным углеводородам'.
СН2=СН2 + С12 -> СН2С1—СН2С1
этилен 1, 2-дихлорэтан
Механизм этой реакции разобран (см. с. 72). В промышленности в
качестве сырья применяются получаемые при химической перера-
ботке нефти этиленсодержащие фракции.
Присоединение галогенводородов к ненасыщенным углеводородам
(гидрогалогенирование\ см. с. 73).
СН2=СН2 + НС1 -> СН3—СН2С1
этилен этилхлорид
СН==СН + НС1 -> СН2=СНС1
ацетилен винилхлорид
Гидрохлорирование обычно проводят в жидкой фазе (например, вза-
имодействие этилена с хлороводородом проводится в этилхлориде,
катализатор — хлорид алюминия). Гидрохлорирование — важный
промышленный способ получения этил- и винилхлорида.
Получение галогенпроизводных из снмртов. Для получения гало-
генпроизводных алканов применяются спирты (см. с. 159). При дей-
ствии на них РС15, SOC12 и галогенводородов гидроксильная группа
в молекуле спирта может быть замещена на атом галогена:
С2Н5ОН + РС15 -> С2Н5С1 + НО + РОС13
этиловый этилхлорид
спирт
С2Н5ОН + SOC12 С2Н5С1 + SO2 f + НС11
Г; СаС12
C2H5OH + HC1--------> C2H5C1 + H2O
Для увеличения выхода при использовании галогенводородов
необходимо применять водоотнимающие средства (например, СаС12),
которые связывают образующуюся воду и предотвращают гидролиз
полученных галогеналкилов.
Физические свойства. Физические свойства галогенпроизводных зависят
от строения радикала, вида и количества атомов галогена в молекуле (табл. 18).
С увеличением длины углеродной цепочки и с переходом от фтора к иоду темпе-
ратура кипения и плотность моногалогенпроизводных возрастают. Дигало-
генпроизводные —тяжелые масла или твердые вещества. Все галогенпроизвод-
ные практически нерастворимы в воде, хорошо растворимы в органических раст-
ворителях, большинство имеют специфический, часто резкий запах, раздражаю-
щий слизистую оболочку, некоторые обладают анестезирующим действием
(СН2С12, СНС13), токсичны, являются антисептиками (СН13).
Химические свойства. Большинство галогенпроизводных угле-
водородов — весьма реакционноспособные соединения, широко при-
меняемые в разнообразных синтезах. Наибольшее значение для со-
единений этого класса имеют реакции замещения (S2v) и отщепления
151
(элиминирования, Е). Примером взаимодействия галогеналкилов с
металлами является реакция Вюрца и Вюрца — Фиттига, а также
реакции с магнием (см. с. 261).
Особую группу превращений галогенпроизводных углеводородов
составляют реакции восстановления молекулярным водородом с уча-
стием катализатора:
№
RX + Н2 -> RH + НХ
и водородом в момент его выделения:
СН3 СН3
I Zn; НС] |
СНз—С—СН3--------> СНз—С—СН3
Вг Н
Реакции замещения. Примерами реакций замещения
могут служить следующие превращения:
+NaI ru I
метилиодид + NaOH CH ГЛТ-J
ru pi С.Г13С/Г1 метиловый спирт -f-NaC-N CH CM
СГ13СЛ > C1I3C.IN ацетонитрил +N;i, > си MH
метиламин -f-Na NO, СИ NJ О
> ^Пз1\1<72 нитрометан
Повышенная способность алкилгалогенидов к реакциям замещения
связана с особенностями строения их молекулы. Например, в моле-
куле СН3С1 ковалентная связь между атомом углерода и галогеном
вследствие большей электроотрицательности атома галогена поля-
ризована (—/-эффект), электронная плотность у атома углерода по-
нижается:
Н
I +S —5
Н->С->С1
I
н
В результате он легко атакуется молекулами или ионами, могу-
щими предоставлять свою электронную пару (ОН~; I"; CN”; NO2;
NH3; Н2О):
152
нуклеофильное замещение (5^)
Реакционная способность галогенпроизводных в реакциях нуклео-
фильного замещения зависит от строения углеводородного радикала,
природы галогена, вида атакующего нуклеофильного агента; большую
роль играют также условия реакции (температура, наличие или от-
сутствие катализатора) и характер растворителя, в среде которого
взаимодействуют вещества. Все эти факторы влияют и на механизм
реакции.
Типичным примером реакций этого типа является взаимодействие
галогеналкила со щелочью:
R—Cl + NaOH -> ROH + NaCl
алкилхлорид спирт
По современным представлениям реакции нуклеофильного заме-
щения могут протекать по двум механизмам; они обозначаются
(II) и Sjv2 (I). В случае Sjv, (I) скорость реакции зависит как от
концентрации алкилгалогенида (R—X), так и от концентрации иона
гидроксила (ОН"):
V=K[RX][OH-], S„2 (I)
где К — константа скорости реакции, л-моль”1 •с'1, т. е. скорость
реакции зависит от концентрации обоих реагентов и описывается ки-
нетическим уравнением второго порядка.
Классическим примером реакции Saz2 является образование ме-
тилового спирта из метилхлорида под действием щелочи:
СН3С1 4- NaOH -> СН3ОН + NaCl
метилхлорид метиловый
спирт
о = К [СН3С1] [ОН-].
В случае (II) скорость реакции зависит от концентрации
только одного из реагирующих веществ (R—X) и не зависит от кон-
центрации другого (ОН"):
с^Я^-Х], (II)
Реакция описывается кинетическим уравнением первого порядка.
К таким реакциям относится образование третичного бутилового
спирта:
СН3 СН3
СН3—С—СН3 + NaOH -> СН3—С—СН3 + NaBr
Вг ОН
2-бром-2-метилпропан 2-метил-2-пр©панел
(трет-бутил бромид) (mpem-бутилшвый спирт}
Остановимся на механизме каждой из этих реакций в отдельности.
153
При" взаимодействии метилхлорида с едким натром (Здд) гидроксил-
анион атакует атом углерода в молекуле метилхлорида с обратной
стороны по отношению к атому хлора:
н
н :о: —► н^с о:
н
Если ион гидроксила обладает необходимой энергией, то он при-
ближается настолько к молекуле метилхлорида, что между ними воз-
никает определенное взаимодействие, в результате связь между ме-
тильной группой и хлором постепенно ослабляется. Наступает mo-
г. ент, когда гидроксильная группа и углеродный атом метильной
1 руппы оказываются связанными между собой, а связь углерода с
хлором еще не разорвана окончательно. Такая структура получила
название промежуточного или переходного состояния (II). Затем
между кислородом гидроксильной группы и углеродом метильной
труппы возникает ковалентная связь, осуществляемая электронной
карой, предоставленной кислородом, а связь между углеродом и
хлором разрывается (III):
но
н\
I—c:ci
sp3
н н
А5'2
но:с—н + сг
jAh
I II III
активированный
комплекс
По мере приближения к углеродному атому отрицательно заря-
женной гидроксильной группы она оказывает все большее влияние
на атомы водорода метильной группы — отталкивая их. В результате
этого водородные атомы постепенно раздвигаются и в переходном со-
стоянии а; ом углерода и три водородных атома располагаются в од-
ной плоскости. Угол между связями Н—С—Н становится равным
120°. В молекуле СН3ОН они снова приобретают «нормальную» кон-
фигурацию с углом 109°, но противоположную первоначальной. От-
дельные авторы сравнивают этот процесс с «выворачиванием зонтика
при сильном ветре» (Р. Моррисон).
В реакциях S,vQ реакционная способность галогенпроизводных
определяется главным образом пространственными факторами и
уменьшается с увеличением числа заместителей у атома углерода,
связанного с галогеном:
СН3Х > первичный >> вторичный Д> третичный
Иначе может быть представлен механизм реакции S^. Так, ме-
ханизм взаимодействия в рассмотренной реакции третичного бутил-
бромида со щелочью выглядит следующим образом:
154
f
H3C H3C сн3
'Х медленно \ S
Н3С>^С—Вг ---------с+ + Вг
Н3С сн3
СН,
Н3С СН; _ / 3
\ ' оыстро Т
е+ + он -----------*- но—с—сн3
I \
СН3 СНз J
Отрыв брома с образованием карбкатиона и аниона брома проте-
кает медленно, а образование молекулы спирта (ионный процесс) —
быстро. Естественно, что первая стадия определяет скорость реакции
(лимитирующая стадия). Она осуществляется за счет сольватации
образовавшихся ионов молекулами растворителя, поэтому скорость
реакции зависит только от концентрации алкилгалогенида. Карбоние-
вый катион имеет плоское строение. Реакционная способность гало-
генпроизводыых в реакциях нуклеофильного замещения изме-
няется в обратном порядке в отличие от Syy2:
третичный > вторичный » первичный > СН3
На практике реакция замещения S^2 характерна для СН3Х и
первичных галогенпроизводных, а — для третичных. В случае
вторичных галогенпроизводных наблюдается смешанный механизм
реакции.
Активность галогенпроизводных возрастает с увеличением моле-
кулярной массы галогена: атом иода более подвижен, чем хлора. Это
становится понятным при рассмотрении характеристик связей и их
способности к поляризации:
Длина, нм Энергия, кДж/моль . . Электрический момент ди- С—F 0,138 428,2 С-С1 0,177 331,8 С—Вг 0,194 279,0 С-1 0,213 219,5
поля (СН3—X), Кл-мХ Х10“30 1 5,98 6,25 6,08 5,51
Поляризуемость .... малая — —> 60ЛЫ1
По мере увеличения размера атома уменьшается прочность связи
между ядром и периферическими электронами, участвующими в об-
разовании связи, и она легче поляризуется. Легкость отщепления
галогенов в S.v-реакциях падает в ряду
I- > Вг- > Cl- > F-
Если атом галогена находится у ненасыщенного углеродного атома
или связан с атомом углерода бензольного ядра, например в моле-
кулах винилхлорида (СН2=СНС1) или хлорбензола (С6Н5С1), которые
находятся в состоянии 5р2-гибридизации, то в результате сопряжения
между свободными электронами атома галогена и л-электронами двой-
ной связи или ароматического секстета бензольного ядра его подвиж-
ность резко снижается (+/W-эффект). Если же атом галогена находится
у атома углерода в a-положении к двойной связи или к углеродному
атому бензольного ядра, например в аллилхлориде (СН2=СН—СН2С1)
или бензилхлориде (С6Н5СН2С1), то он обладает повышенной реак-
151
ционной способностью. Реакции замещения атомов галогена в «ал-
лильном» и «бензильном» положениях (S/vJ идут с высокими скорос-
тями. Повышенная реакционная способность этих соединений в ре-
акциях замещения связана с более легкой диссоциацией связи С—X
и дополнительной стабилизацией катиона:
Атом галогена, удаленный от двойной углерод-углеродной связи
или бензольного кольца, по своему поведению не отличается от га-
логенпроизводных алканов (например, СНг^СН—(СНг)4—СНг—С1;
С6Н5—СНг—СН2—СНг—О). При проведении реакции нуклеофиль-
ного замещения подбирают такой растворитель, в котором раство-
рялись бы оба реагирующих вещества и который способствовал бы
разрыву связи С—X.
Используя реакцию нуклеофильного замещения, можно получить
из галогенпроизводных не только спирты (см. с. 159), но и органиче-
ские соединения многих других классов (нитрилы, амины, нитро-
соединения). Во всех этих реакциях в молекулу нового вещества вво-
дится алкил (например, метил— СН3), поэтому галогенпроизводные
являются алкилирующими реагентами.
Отщепление галогенводородов (элиминирование;
Е). Галогеналкилы под действием спиртовых растворов щелочей от-
щепляют галогенводороды с образованием непредельных соединений:
(NaOH, спирт, р-р)
СН3—СН2С1-----------> сн2=сн2
этилхлорид — НС1 этилен
В ходе реакции после образования карбкатиона его стабилизация
происходит за счет отщепления протона от a-ненасыщенного угле-
родного атома под влиянием гидроксил-аниона и образованием двой-
ной связи:
сн2—сн2 —► сн2—сн2 + Н2О + С1~
н:о:
Образование магнийорганических соеди-
нений (см. с. 261). Реакция протекает в среде безводного диэтило-
вого эфира С2Н5ОС2Н5:
эфир
СН3Вг + Mg---> CH3MgBr
Фторпроизводные алканов. Фторпроизводные алканов по способам полу*
чения, свойствам и использованию резко отличаются от других галогенпроиз-
водных, поэтому их описание целесообразно выделить в специальный раздел.
Номенклатура фторпроизводных аналогична номенклатуре других галоген-
производных. Частично замещенные фтором углеводороды называют по общим
для всех галогенпроизводных правилам. Для наименования полностью фториро-
156
ванных соединений (перфторуглероды) к названию углеводорода прибавляют
приставку перфтор, например C2F6 — перфторэтан.
Прямое фторирование углеводородов вследствие высокой химической актив-
ности фтора затруднено. В результате выделения большого количества теплоты
происходит расщепление углеродного скелета молекулы. Реакция часто сопро-
вождается взрывом. Поэтому для получения фторпроизводных были разработаны
особые методы.
Действие фторидов металлов на хлор-, бром- и иод производные углеводородов.
Этот способ удобен для получения монофторпроизводных:
С2Н5С1 4- AgF -> C2H5F + AgCl
Фторирование углеводородов соединениями, легко отдающими фтор (CoF3;
MnF4; SbF5). Разбавленные азотом пары углеводорода пропускают над CoF3,
получают фторпроизводное. Образующийся CoF2 восстанавливают в CoF3, обра-
батывая фтором:
200—350°С
С7Н1б + 32CoF3---------> С7Н16 + 32CoF2 + 16HF
гептан перфторгептан
250°С
2CoF2 + F2-----> 2CoF3
Прямое фторирование углеводородов. В настоящее время осуществляют пря-
мое фторирование углеводородов в присутствии азота:
азот
СН4 + 4F2----> CF4 4- 4HF
метан четырех-
фтористый
углерод
Свойства. Физические свойства и химическое поведение фторпроизводных
зависят от количества атомов фтора, содержащихся в молекуле. Температуры
кипения перфторуглеродов незначительно отличаются от температуры кипения
алканов с тем же числом атомов углерода, но резко отличаются от температуры
кипения других галогенпроизводных.
Монофторпроизводные, особенно вторичные и третичные, нестойки, легко
отщепляют фторводород:
C8H17F -> С8Н16 4- HF
токсичны.
Дифтор производные более устойчивы и мало токсичны.
Полностью фторированные соединения отличаются исключительной терми-
ческой и химической стойкостью. Большинство перфторсоединений не разлага-
ется при нагревании до 500°С, CF4 — до 300°С; стойки к действию кислот, щело-
чей, окислителей, не токсичны. Перфторсоединения применяются в качестве
инертных растворителей, смазочных масел, для производства пластических масс.
Отдельные представители. Метилхлорид СН3С1. Промышленный
способ получения — хлорирование метана. Применяется в качестве
метилирующего средства, растворителя, хладоагента в холодильных
установках, для получения кремнийорганических соединений, тетра-
метилсвинца.
Этилхлорид С2Н5С1. Промышленные способы получения: хлори-
рование этана, гидрохлорирование этилена. Применяется для произ-
водства тетраэтилсвинца (этилирующее средство), в качестве анесте-
зирующего вещества, растворителя.
Дихлорэтан CH2CI—CH2CI. Получают хлорированием этилена.
Используется в качестве растворителя, в разнообразных органиче-
157
ских синтезах, для получения винилхлорида, янтарной кислоты, эти-
лендиамина, этиленгликоля, каучука:
... —С2н4—S—S—С9Н4—S—S—-С2Н4—S—S—с2н4— ...
II II " II II II II
S S S S S S
полисульфидный каучук
для экстракции жиров и восков; обезжиривания меха, шерсти, метал-
лических изделий перед хромированием и никелированием.
Хлороформ СНС13. В промышленности получают хлорированием
метана и действием Са(ОС1)2 на этиловый спирт:
4Са(ОС1)2 4- 2С2Н5ОН 2СНС!3 + (НСОО),Са 4- 2Са(ОН)2 4~ СаС12 4~ 2Н2О
Употребляется для получения фреонов, тефлона, в качестве ор-
ганического растворителя. При стоянии в' присутствии кислорода
воздуха разлагается с образованием фосгена (COCI2), являющегося
отравляющим веществом (ОВ).
Четыреххлористый углерод CCta. Применяют для синтеза фреонов
и других соединений, в качестве растворителя (в том числе и в быто-
вой химии). Как растворитель CCU обладает существенным преиму-
ществом перед многими другими — он негорюч. Поэтому использу-
ется для наполнения огнетушителей специального назначения.
Винилхлорид СН2=СНС1. Мономер используется при синтезе важ-
ного полимерного материала — поливинилхлорида:
пСН2=СНС1 -> Г—СН2—СН—1
I
С1 \п
поливинилхлорид
Из поливинилхлорида изготовляют электроизоляционные материалы
(пленки, изоляционные трубки), трубы, пленки, упаковочные мате-
риалы, заменители кожи, линолеум. После специальной обработки
поливинилхлорид употребляется для производства лаков, лечебного
белья. Винилхлорид получают гидрохлорированием ацетилена, хло-
рированием этилена, дегидрохлорированием дихлорэтана.
Аллилхлорид СН2=СН—CH2CI. Исходное сырье для производства
глицерина, аллилового спирта. Получается высокотемпературным
хлорированием пропилена.
Хлорбензол С6Н5С1. Получают хлорированием бензола. Исходное
сырье для синтеза разнообразных производных бензола.
Дифтордихлорметан CF2CI2. Является представителем группы
галогенпроизводных, содержащих атомы фтора и хлора — фреонов
(фреон-12). Газ, без цвета, запаха и вкуса (/кип = —30°, /пл =
= —155" С). Синтезируется из четыреххлористого углерода в при-
сутствии SbCh:
SbCl6
CCi4 4- 2HF--> CC12F2 4- 2HC1
фреон 12
Широко применяется в качестве хладоагента в холодильных установ-
-158
ках и растворителя инсектицидов. Не токсичен, не взрывоопасен и
не вызывает коррозии металлов.
Тетрафторэтилен CF2=CF2. Газ, без цвета и запаха, мало токси-
чен. Получают пиролизом хлордифторметана:
650°С
2CHF2C1 -----> CF2=CF2 + 2НС1
Является исходным сырьем для полимера тефлона, фторопласта-4,
отличающегося крайне высокой химической стойкостью. Практически
на него не действуют никакие вещества, за исключением щелочных
металлов.
Понятие об инсектицидах. Некоторые галогенпроизводные: 4,4'-дихлор ди-
фенилтрихлорметилметан (ДДТ); гексахлорциклогексан (гексахлоран) и другие
относятся к так называемым инсектицидам — химическим веществам, применяе-
мым для борьбы с вредными насекомыми. Инсектициды в виде порошков или
растворов вносятся в почву вместе с семенами или распыляются с самолетов.
Они явлются эффективным средством борьбы с вредителями сельскохозяйст-
венных растений, резко сокращая потери урожая. В то же время их применение
требует большой осторожности, так как они могут уничтожить полезную микро-
флору, а попадая в организм животных и человека, могут оказывать вредное
действие.
Кислородсодержащие производные
углеводородов
Многочисленную и исключительно важную группу органических
соединений составляют кислородсодержащие производные углеводо-
родов: спирты, фенолы, простые эфиры, органические перекиси, аль-
дегиды и кетоны, органические кислоты и их многочисленные функцио-
нальные производные (сложные эфиры; ангидриды кислот и т. д.).
ГИДРОКСИ Л СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ
УГЛЕВОДОРОДОВ (СПИРТЫ, ФЕНОЛЫ, НАФТОЛЫ)
Гидроксильная (оксигруппа) группа ОН является функциональ-
ной группой двух важных классов соединений — спиртов и фенолов.
В фенолах она связана непосредственно с атомом углерода аромати-
ческого ядра, в спиртах — с любым другим углеводородным атомом:
феноль
а-нафтол
спирты
этиловый спирт (1)
СН2—СН СН2ОН циклогексанол (3) бензиловый спирт (4)
аллиловый спирт (2)
159-
В зависимости от углеводородного остатка спирты бывают пре-
дельными (насыщенными, 1), непредельными (ненасыщенными, 2),
циклическими (3), ароматическими (4). В зависимости от количества
гидроксильных групп спирты и фенолы делятся на одноатомные (I а,б),
двухатомные (II а, б), трехатомные (III а, б), многоатомные (IV а, б):
I а. сн3он
метанол
о -крезол
Па. СН2—СН2
I I
он он
этиленгликоль
III а. СН2—СН2—СН2
ОН ОН ОН
глицерин
флороглюцин
сорбит
Одноатомные спирты
Общая формула R—ОН или Аг—(СН2)П—ОН; CnH2«_i—ОН, где
R — насыщенный или ненасыщенный радикал алифатического ряда;
Аг — ароматический радикал; — алициклический радикал.
Одноатомные спирты, являющиеся производными гетероцикличе-
ских соединений, будут рассмотрены в другом разделе (см. с. 318),
оксипроизводные ароматических соединений, группа «ОН» в молекуле
которых связана с углеродным атомом бензольного ядра, — в разделе
«Фенолы». В табл. 19 приведены некоторые характеристики ряда
важнейших одноатомных спиртов и фенолов.
В зависимости от характера углеродного атома, с которым связана
гидроксильная группа в молекуле спирта, различают: первичные
спирты — гидроксильная группа расположена у первичного атома
углерода (1); вторичные — гидроксильная группа находится у вто-
ричного углеродного атома (2) и третичные спирты — окси группа
расположена у третичного углеродного атома (3):
СН3—СН2—СН2—ОН (1)
пропиловый спирт
он
I
СН3—СН—СН3 (2)
изопропиловый спирт
он
I
СНз—С—СН3 (3)
I
СН3
трет-бутиловый спирт
Номенклатура, изомерия. При наименовании отдельных спиртов
широко применяются исторические названия: древесный спиртСН3ОН,
винный спирт С2Н5ОН. По рациональной номенклатуре алифатические
спирты с прямой углеродной цепочкой называют по наименованию
Й60
Таблица 19. Физические свойства некоторых наиболее важных
представителей одноатомных оксисоединений
Название Формула 1 Темпера- тура плав- ления, °C Темпера- тура ки- пения, еС Плот- ность р, г-см~3 Показа- тель пре- ломления
Метиловый спирт (метанол) СН3ОН —97,8 64,7 0,792 1,3286
Этиловый спирт (этанол) СНз—СН2—ОН — 114 78,3 0,789 1,3614
Пропиловый спирт (1-пропанол) СН3—СН.>—сн2—он -127,0 97,2 0,804 1,3856
Изопропиловый спирт СН3—СН—СН3 —88,5 82,4 0,786 1,3771
(2-пропанол) Бутиловый спирт (1-бутанол) 1 он СН3-(СН2)2-СН2-ОН —89,6 117,5 0,810 1,3993
Амиловый спирт (1-пентанол) СН3— (СН2)3-СН.ОН —78,2 137,8 0,814 1,4100
Изоамиловый спирт СН3—СН—СНо—СН2—ОН — 117 132,1 0,813 1,4058
(3-метил-1 -бутанол) Аллиловый СПИр! 1 СНз СН2=СН—СН он — 129 96,9 0,854
Цикл о гекса нол свнпон —24 161,5 0,962 —
Бензиловый спирт с6н5сн2он — 15,3 205,4 1,046 —
Фенол С6Н5ОН +43 181,2 1,054 1,5425
о-Крезол С6Н4(СН3)ОН 30,8 190,9 (при 45°С) 1,048 (при 40°С) 1,5443
ж-Крезол СвН4(СН3)ОН 11,9 201,2 1,034 (при 20°С) 1,5432
п-Крезол С6Н4(СН3)ОН 35,5 201,1 1,034 (при 15°С) 1,5361
(при 17°С)
радикала (I), а с разветвленной цепочкой рассматривают как произ-
водные простейшего метилового спирта — карбинола (II). По номен-
клатуре ИЮПАК наличие группы ОН в молекуле спирта обозначается
окончанием «ол» с указанием номера углеродного атома, у которого
она расположена; последний обязательно включается в главную цепь.
Нумерацию атомов углерода начинают с того конца цепи, к которой
ближе расположена группа ОН (III, IV, V):
СН3—СН2—ОН
этиловый спирт;
этанол
(I)
он
сн3—С—сн3
СН3
триметилкарбинол;
2-метил-2-пропанол
(П)
3 2 1
СН3—СН2—СН2—ОН
пропиловый спирт;
1-пропанол
(П1)
161
СН3
i
СН3—СН=СН—СН—СН2—ОН
5 4 3 2 1
2-метил-З-пентен- 1-ол
(IV)
Изомерия спиртов связана со строением радикала (изомерия угле-
родного скелета) и с положением гидроксила (изомерия положения).
Например, соединения I а, б, в изомерны друг другу, так же как
соединения II а, б, в и III а, б, в, соответственно:
ОН
I а. СН3—СН2—СН2—СН?ОН б. СН3—СН—СН2ОН в. СН3—СН2—СН—СН3
1-бутанол I 2-бутанол
сн3
2-мет ил-1 -пропанол
II а. СН3—СН=СН—СН2ОН б. СН2==СН—СН2—СН2—ОН
2-бутен-1-ол З-буген-1-ол
ОН
в. сн2=сн—сн—сн3
З-бутен-2-ол
циклобутанол метилциклопропанол циклопропилкарби .@л
В ряду одноатомных спиртов может иметь место различное про-
странственное расположение групп (стереоизомерия).
Способы получения. Одноатомные спирты в природе в свободном
состоянии встречаются в небольших количествах, но их производные
(в первую очередь—простые эфиры, см. с. 190, и сложные эфиры,
см. с. 239) не только широко распространены, но в отдельных слу-
чаях используются в качестве промышленных источников получения
спиртов. Остатки метилового спирта (метоксигруппы ОСН3) являются
структурными компонентами лигнина. Высокомолекулярные одно-
атомные спирты (С16—С30) участвуют в образовании восков (см. с. 402).
При их омылении они выделяются в свободном состоянии. Это важ-
ный промышленный способ получения спиртов с числом атомов угле-
рода в молекуле от 16 до 30.
Присоединение воды к алкенам (гидратация алкенов). При при-
соединении воды к этилену образуется первичный спирт, при гидра-
тации его гомологов в соответствии с правилом Марковникова — вто-
ричные или третичные спирты:
сн2=сн2 + Н2О -> сн3—СН — он
этилен этиловый спирт
162
СН3—СН=СН2 + Н2О -> СН3—СН—СН3
ОН
пропилен изопропиловый спирт
он
СНз—С=СН2 + Н,0 -> сн3—с—сн3
I I
СНз СНз
2- метилпропен 2- метил-2- пропанол
В промышленности гидратация олефинов проводится двумя спо-
собами: при участии серной кислоты (сернокислотная гидратация)
и прямая гидратация. С увеличением длины углеродной цепочки (до
известных пределов) скорость реакции возрастает, алканы с разветв-
ленной углеродной цепочкой реагируют быстрее, чем олефины нор-
мального строения. Труднее всего реагирует с водой этилен.
Сернокислотная гидратация алкенов протекает в две стадии:
СН2=СН2 4- HOSO2OH -> СН3—СН2—OSO2OH
этилен этилсерная кислота
СН3—СН2—OSO2OH + Н2О -> СНз—СН2ОН 4- h2so4
зтиловый спирт
Прямую гидратацию алкенов проводят в одну стадию, применяя
в качестве катализатора фосфорную кислоту или оксид алюминия:
А12О3
СН2=СН2 + Н2О-----> СН3—СН2ОН
этиле. этиловый спирт
Гидратация этилена проходит при температуре 300—350° С, дав-
лении 100 • 10б Па, катализатор — смесь фосфорной и вольфрамо-
вой кислот, нанесенных на силикагель.
Гидратация алкенов — важнейший промышленный способ полу-
чения ряда спиртов, позволяющий использовать дешевое и доступное
сырье — газы крекинга. Прямая гидратация алкенов экономически
более выгодна, чем сернокислотная, проведение которой связано со
значительным расходом труднорегенерируемой серной кислоты.
Восстановление альдегидов и кетонов. При восстановлении аль-
дегидов образуются первичные спирты, кетонов — вторичные (см.
с. 160):
z° 2Н
СНо—С< —> СН3-СН2ОН
\н
уксусный этиловый спирт
альдегид
2Н
СНд—С—СН3 -> СН3—СН—СН3
II I
О он
ацетон изопропиловый спирт
Гидролиз галогенпроизводных (см. с. 153).
163
Получение спиртов из оксида углерода и водорода. Ранее уже рас-
сматривалось получение углеводородов из оксида углерода и водо-
рода. В ходе этой реакции образуются разнообразные кислородсо-
держащие соединения, в первую очередь этиловый спирт. Меняя
условия и катализаторы, реакцию можно направить в сторону боль-
шего обра звания метилового спирта или высших спиртов.
Получение спиртов действием металлорганических соединений
на альдегиды и кетоны (реакции Бутлерова, Тищенко, Вагнера, Зай-
цева, см. с. 263).
Получение спиртов брожением. Этот метод широко применяется
для получения этилового и бутилового спиртов (см. с. 173, 176).
Гидролиз сложных эфиров. Применяется в промышленности для
образования некоторых высокомолекулярных спиртов из восков (см.
с. 402):
О О
II нон II
R—С—О—Ri-------> R—С—ОН + RL—ОН
сложный э Ьир карбоновая высокомолеку-
кислота лярный спирт
Оксосинтез. При оксосинтезе спирты образуются в результате при-
соединения к алкенам оксида углерода и водорода с последующим
восстановлением образующихся карбонильных соединений:
СН3
| [Со (СО)4]2
СНЧ—СН—СН=СН2 + СО + Н9---------->
3-мети ч -1 - бутен
СН3 О
I II
СНз—СН—СН2—СН2—С—н
4- метил пен та на ль
СН3
-> сн3—сн—сн—сн3
ь°
\н
2, 3-диметилбутаналь
СН3
-> СН3—СН—СН?—СН2—СН2ОН
4-метил-1-пентанол
СН3—сн— сн—сн2он
СНз СНз
2, З-диметил-1-бутанол
Реакция протекает при температуре 120—140эС, давлении (200—
250) • 105 Па.
Получение спиртов восстановлением карбоновых кислот. Этим ме-
тодом могут быть синтезированы высокомолекулярные спирты из
карбоновых кислот, полученных в результате окисления алканов или
омыления жиров:
восст.
R—СООН -----> R—СН2ОН
Восстановление может быть осуществлено молекулярным водоро-
дом в присутствии катализаторов или с помощью специальных вос-
164
становителей (LiAlHi). В первом случае удобнее проводить восста-
новление не самих кислот, а их сложных эфиров:
zO Н,; Ni
RC<f ---------> RCH2OH + RXOH
\ORj
сложный э^ир
Физические свойства. Спирты нормального строения с числом атомов угле-
рода в цепи доСп — жидкости; содержащие более 11 атомов углерода — твердые
вещества. Легче воды, без цвета. До С3 с резким «спиртовым» запахом; С4 — Сб с
резким, неприятным запахом («сивушный запах»); твердые — без запаха. Пер-
вые гомологи хорошо растворимы в воде, по мере увеличения молекулярной
массы растворимость падает. Низшие спирты легче воспламеняются и горят бес-
цветным пламенем. Спирты разветвленного строения кипят при более высокой
температуре, чем спирты нормального строения, причем температура кипения
третичных спиртов выше, чем вторичных (см. табл. 12).
При сравнении температуры кипения спиртов с температурами кипения
представителей других классов органических соединений обнаруживается, что
спирты кипят при более высоких температурах, например:
t кип., °G
СН3—СН2—ОН (этиловый спирт).............-]-78,3
СН3—СН2—CI (этилхлорид)..................4-12,4
СН3—О—СН3 (диметиловый эфир)..............—23,7
Это объясняется тем, что молекулы спирта ассоциируют с помощью водородных
связей.
Многие спирты ядовиты — 10—20 мл метилового спирта вызывает слепоту,
большие количества — смерть.
Химические свойства. Химическое поведение спиртов определя-
ется, в первую очередь, наличием в их молекуле функциональной
группы ОН. В их разнообразных химических превращениях особое
место принадлежит двум типам реакции замещения, протекающим
с разрывом связи С—О и О—Н:
Н
R—С—О—Н
Н
В то же время для спиртов характерны реакции отщепления (эли-
минирования), приводящие к образованию двойной связи; реакции
окисления и дегидрирования. Положение гидроксильной группы (у
первичного, вторичного или третичного атомов углерода), молеку-
лярная масса существенно влияют на химическое поведение спиртов.
Реакции, протекающие с разрывом связи
О—Н. Образование алкоголятов. Спирты — чрезвычайно слабые кис-
лоты (рКа~ 18, для воды р/(а 16), однако, взаимодействуя с металла-
ми (К, Na, Mg, Al), образуют алкоголяты — твердые вещества бе-
лого цвета:
2СН3—СН2—ОН + 2Na -> 2СН3—СН2—ONa + Н._, f
этиловый спирт этилат н*трия
Алкоголяты, полученные из метилового спирта, называются мети-
латами (например, метилат натрия CH3Na), из этилового — этила-
165
тами. В присутствии воды они гидролизуются с образованием спирта
и щелочи:
СН3—СН2—ONa + НОН СН3—СН2—ОН + NaOH
По строению алкоголяты можно рассматривать как соли щелочей и
очень слабых кислот. Их спиртовые растворы обладают сильно ос-
новными свойствами. Сами спирты не меняют окраски индикаторов,
но в определенных условиях проявляют или очень слабые щелочные,
или кислотные свойства, являясь тем самым амфотерными соедине-
ниями. Образование алкоголятов — пример кислотных свойств спир-
тов. Строение углеводородного радикала существенно влияет на под-
вижность атома водорода гидроксильной группы — легче всего за-
мещается атом водорода в метаноле; труднее — в третичных спиртах:
Ri
СН3ОН > R — СН,—ОН > R —сн—R! > R —С—R о.
I I
ОН он
Образование метилата и этилата натрия происходит бурно, по
мере роста молекулярной массы спирты менее активно вступают в
реакцию с металлами. Образование алкоголятов высокомолекуляр-
ных спиртов происходит только при нагревании. Алкоголяты широко
применяются в разнообразных синтезах для введения в молекулу
алкоксилыюй группы OR.
Взаимодействие спиртов с магнийорганическими соединениями
(Л. А. Чугаев, Ф. В. Церевитинов). При действии на спирты магний-
органических соединений образуются углеводороды и производные
спиртов — алкоголяты:
С2Н5ОН + CH3MgI -> t СН4 + C3H5OMgI
этиловый метилгсагниа- этоксимагний-
^пирт иодид иодид
Реакцию используют для определения количества гидроксильных
групп в молекуле спирта (по объему образовавшегося углеводорода).
Образование сложных эфиров. Реакция этерификации. Взаимо-
действие спиртов с кислотами (органическими и неорганическими)
приводит к образованию производных кислот, называемых сложными
эфирами:
О о
II I н+1 Н
СНз—СН2ОН + НО—С—СН3 СН3—С— О—СН2—СНз + Н2О
этиловый уксусная уксусноэтиловый эфир
спирт кислота (этилацетат)
СН3ОН +
НОХ /О
на холоду
СН3ОЧ /О
н—oz
+ н20
метиловый
спирт
монометилсульфат
СН3ОН+
СНз—Оч ,0
н—с/ Ж
на холоду
СНз—О О
\s^ +Н20
СН3—0/Хо
диметилсульфат
166
Реакция получила название реакции этерификации от латинского
aether — эфир. Скорость этерификации возрастает с увеличением силы
кислоты и зависит от природы спирта (Б. Н. Меншуткин): первичные
спирты реагируют быстрее вторичных, последние быстрее третичных
(СН3ОН> первичные > вторичные>третичные спирты). Реакцион-
ная способность карбоновых кислот в реакциях этерификации может
быть представлена следующим рядом:
НСООН > СН3СООН > RCH.COOH > R2CHCOOH > R3CCOOH
Этерификация спиртов органическими (карбоновыми) кислотами
ускоряется добавлением сильных минеральных кислот. Реакция об-
ратима, обратная реакция называется гидролизом. Гидролиз сложных
эфиров, проводимый водными растворами щелочей, называется омы-
лением (см. с. 240).
Легче, чем с кислотами, спирты образуют сложные эфиры при вза-
имодействии с хлорангидридами и ангидридами кислот (см. с. 236):
О
z° II
СНз—С/ + СН3ОН СН3—С—ОСН3 + НС1
ХС1 уксуснометиловый эфир
ацетилхлорид
/О
СНз—С<^
/О + СН3ОН
СН3—С.х
уксусный
'ангидрид
О о
I! II
СН3—С—ОСНз + СН3С—ОН
уксус нометиловый
эфир
Механизм реакции этерификации следующий: вначале протон (ката-
лизатор) присоединяется к карбонильному кислороду карбоксильной
группы, образуется карбкатион:
.8-
сн3—с'
сн3-
о—н
I
-с—о—н
Далее происходит нуклеофильная атака молекулой спирта (имекЯцей
неподеленные электронные пары на атоме кислорода) карбкатиона
(что приводит к образованию оксониевого иона):
О—Н
сн3—с—о—н +
+
с2нб
I
:О:
I
Н
О—н
сн3—с—о—н
I
+0—С2Нб
I
н
Ион обратимо теряет молекулу воды с образованием нового карб-
катиона:
167
он
I
сн3—С—он
I
с2н5—о—н
+
он
I
сн3—с+ + Н2О
с2н5-о
Последний, отщепляя протон,
превращается в сложный эфир:
ОН О
I II
сн3—с+ ОН3-С-ОС2Н5 + н+
| уксусноэтиловый
С2Н5—О эфир
Выделяющаяся при этерификации молекула воды образовалась
из гидроксила кислоты и водорода спирта (а не наоборот). Это было
установлено методом «меченых атомов». В молекулу спирта был вве-
ден изотоп кислорода 18О, который затем был обнаружен в молекуле
сложного эфира:
О О
СН3-1*О- Н + Н-0 -С— R R—С—18О—СН3 + Н2О
«Меченые соединения» широко применяются в химии, биологии,
физике для разнообразных исследований.
Реакции, протекающие с разрывом связи
С—ОН. Замещение гидроксильной группы в молекуле спирта на атом
галогена. Эти реакции уже были рассмотрены в разделе галогенпроиз-
водных. Для замещения гидроксила на галоген применяют галоген-
водородные кислоты, галогениды фосфора (РСк, РС13; Р13; РОС13)
или тионилхлорид (SOCI2):
С2Н5ОН 4- НС) С2Н5С1 + Н2О
этиловый этилхлорид
спирт
С2Н5ОН 4- РС15 -> С>Н5С1 + РОС13 + НС1
ЗС2Н5ОН 4- РОС1з -> ЗС2Н5С1 + РО(ОН)3
ЗС2Н5ОН 4- Р13 ЗС2Н51 + Р(ОН)3
С2Н5ОН 4- SOC12 -> С2Н5С1 4- но + so2
Наиболее удобно для замещения гидроксильной группы использовать
тионилхлорид; применение галогенидов фосфора осложняется обра-
зованием побочных продуктов. Образующаяся при взаимодействии
спиртов с галогенводородами вода разлагает алкилгалогенид на спирт
и галогенводород; реакция обратима.
Успешному взаимодействию спиртов с галогенводородами способ-
ствует присутствие сильных кислот, наличие катализаторов (хлори-
ды цинка, алюминия, кальция) минимальное содержание воды в спир-
те (происходит гидролиз галогеналкилов) Спирты по реакционной
способности при взаимодействии с галогенводородами располагаются
в следующий ряд: аллиловый > бензиловый>третичный>вторич-
168
ный>первичный>СН3ОН. Реакционная способность кислот снижает-
ся при переходе от HI к НС1: HI > НВг > НС1. В случае третичных
спиртов замещение гидроксильной группы на атом галогена протекает
по S/угмеханизму через образование протонированного спирта
(ROHo):
СН3х
снз-с—о—н
сн3/
сн3 сн3 сн3
I + -Н2О | + ГВг-1 |
СН3—С---О—Н 7----» СН3—С+-----> СН3—С—Вг
II I I
сн3 н сн3 сн3
Реакция может сопровождаться изменением строения алкильного
радикала (перегруппировка), и атом галогена не всегда оказывается
связанным с тем углеродным атомом, у которого находилась гид-
роксильная группа. В случае первичных спиртов реакция протекает
по Здго-механизму и не сопровождается изменением строения алкиль-
ного радикала.
Дегидратация спиртов. Дегидратация, или дегидратирование,
(отнятие воды) от молекулы спирта приводит к образованию этилено-
вых углеводородов (1) или простых эфиров (2):
СН3—СН2—ОН -> СН2=СН2 + Н2О (1)
этиловый спирт этилен
СН3—СН2—О Н + НО —СН2—СН3 -> СН3—СН2—О—СН2—СН3 + Н2О (2)
диэтиловый эфир
При образовании молекулы воды водород, в соответствии с прави-
лом Зайцева, всегда отнимается от наименее гидрогенизированного
атома углерода, находящегося по соседству с углеродным атомом,
несущим гидроксильную группу:
ОН Н
I I
СН3—СН—СН—СН3 -» СН3—СН=СН—СН3 + Н2О
2-бутанол 2-бутен
Дегидратацию проводят с помощью водоотнимающих средств (серная
и ортофосфорная кислоты, оксид алюминия, силикагель, хлорид
цинка) при нагревании. Температура дегидратации первичных и вто-
ричных спиртов 200—350° С. Из двух возможных реакций 1 и 2 пер-
вая требует7 более высоких температур. Первичные спирты дегидра-
тируются труднее вторичных, легче всего отщепляется молекула воды
от третичных спиртов.
Механизм реакции 1:
СН3 СН3 СН3 СН3 СН3 СН3
I I +н+ II -н2о | | -н+
СН3—С--С—Н СНз—С—С—Н ^z=2 СН3—С---С—Н
II II I *
Н НО Н +О—Н н
I
н
169
СНз
I
* СН3-С=СН-СН3
Реакция сопровождается перегруппировками.
Механизм реакции 2
+н+ + /Н +
С2Н5ОН СН3-СН2-СН3 СН2 + Н2О
ХН
СН3—СН2—О—Н + СН3—СН2, СН3—СН2—6—СН2—СН3 ->
н
-> СН3—сн2—о—сн2—сн3 + н+
Окисление спиртов. При окислении первичных спиртов образуются
альдегиды; вторичных — кетоны; окисление третичных спиртов со-
провождается разрывом углеродной цепи обычно около атома угле-
рода, несущего гидроксильную группу.
Альдегиды могут подвергаться дальнейшему окислению с обра-
зованием карбоновых кислот с тем же числом углеродных атомов.
Окисление кетонов сопровождается разрывом углеродной цепи:
К2Сг2О7: о
H2SO4 КМпО4 или КоСг2О7
R—СН2ОН---------> R—С\ --------------:----> R—СООН
О2/кат. ХН
первичный спирт альдегид карбоновая кислота
КМпО4
R.
R—СН—ОН---
СгО3
вторичный
спирт
о
II
R—С—Ri -> смесь карбоновых кислот
кетон
Образование различных продуктов при окислении первичных, вто-
ричных и третичных спиртов дает возможность использовать эту ре-
акцию для установления строения спирта.
Окислители: хромовая смесь, перманганат калия, кислород в при-
сутствии металлической меди. Окисление спиртов ведут при нагре-
вании, оптимальные температуры зависят от строения спирта и окис-
ляющей способности примененного окислителя. Первичные и вто-
ричные спирты окисляются легче, чем третичные.
Дегидрирование спиртов. При пропускании паров спирта над де-
гидрирующим катализатором (раскаленная медь, железо, никель)
первичные и вторичные спирты теряют два атома водорода (дегидри-
рование), при этом первичные спирты превращаются в альдегиды,
вторичные — в кетоны:
170
Си
сн3—сн2он —сн3—сно + н2
этиловый спирт 250°С уксусный
альдег ид
Си
СН3—СН—СН3---------> СН3—С—СН3 + Н2
| 250° С ||
ОН о
и зо п ро п ило вый ацетон
сп ip г
Отдельные представители. Метиловый спирт, метанол СН3ОН.
Прозрачная бесцветная жидкость со слабым спиртовььм запахом.
Смешивается с водой, ацетолом, спиртами в любых соотношениях.
Ядовит.
Основные способы промышленного получения. 1. Синтез из ок-
сида углерода и водорода:
СО -Ь 2Н2 СН3ОН
Температура реакции 300—400° С, повышенное давление, ката-
лизатор — оксид цинка и хрома.
2. Неполное окисление метана и его гомологов:.
[О]
СН4 —> СН3ОН
3. Некоторое количество метилового спирта образуется при сухой
перегонке древесины; отсюда и одно из названий метанола — дре-
весный спирт. Это наиболее старый способ его получения.
Метиловый спирт — один из важнейших продуктов химической
промышленности, широко применяется в качестве растворителя и
полупродукта при синтезе многих органических соединений. Основ-
ные пути промышленного использования метанола приведены на схе-
ме 9. Особенно большие количества метанола расходуются для произ-
водства формальдегида и в реакциях метилирования. Метанол —
один из исходных продуктов в синтезе метионина — важной амино-
кислоты (см. с. 295), стимулирующей рост птиц.
Этиловый спирт, этанол СН3—СНг—ОН. Жидкость без цвета,
со «спиртовым» запахом, жгучая на вкус. Смешивается с водой в лю-
бых соотношениях. Смесь, содержащая 96,6 6 спирта и 3,4% воды,
перегоняется совместно (такие растворы называются азеотропными
смесями), отсюда и ее название «спирт-ректификат». Спирт-ректифи-
кат широко применяется в пищевой и других отраслях промышлен-
ности. Получить чистый «обезвоженный» этанол удается только с по-
мощью1 водоотнимающих средств, например прокаленным сульфатом
меди (лабораторный метод), перегонкой смеси спирта, воды и бензола.
В технике и лабораторной практике концентрация этилового спир-
та равна объемной доле, выраженной в процентах или градусах.
Концентрацию спирта в градусах определяют ареометром (спирто-
метром), градуированным специальным образом.
В промышленности этанол получают гидратацией этилена, бро-
жением пищевого сырья, гидролизом растительных материалов, пере-
работкой сульфитного щелока.
171
Схема 9. Синтезы на основе метилового спирта
неполное окисление нсно формальдегид
4-НС1 СН3С1 метилхлорид
*
4-NH3 ch3nh2
метиламин
+SO3 (CH3O)2SO2 диметилсульфат
СН3ОН метиловый спирт +со+н2 > СН3СООН уксусная кислота
Н-СН^СН СН3—о—сн=сн2 винилметиловый эфир
+СННСН-4-СО сн2=сн—соо—сн3
метиловый эфир акриловой кислоты
Ч- CeH6NH2 CeH5N(CH8)2 диметила нилин
+h2s - CH3SN метилмеркаптан
172
1. Гидратация этилена (см. с. 75).
2. Превращение простых углеводов — гексоз (см. с. 362), глав-
ным образом наиболее распространенной глюкозы С6Н12О6 (см. с. 362),
в присутствии дрожжей или выделенных из них биологических ка-
тализаторов — ферментов. Суммарно спиртовое брожение выража-
ется следующим уравнением:
СбН12О6 -> 2СО2 + 2С2Н5ОН
гексоза этиловый спирт
Кроме этилового спирта и диоксида углерода также образуются, но
в значительно меньших количествах бутиловый и амиловый спирты,
янтарная кислота и другие вещества. Спиртовое брожение использу-
ется не только для получения спирта, этот процесс лежит в основе
многих пищевых производств: виноделия, пивоварения.
Получение этилового спирта с помощью спиртового брожения мо-
жет быть осуществлено различными путями: а) брожением пищевого
сырья; б) гидролизом растительных материалов; в) переработкой
сульфитного щелока.
а. Брожение пищевого сырья. В качестве сырья используют крах-
малсодержащие продукты: картофель, зерно кукурузы, ячменя,
пшеницы, ржи, проса, овса (содержание крахмала в них колеблется
от 18 до 60%), отход сахарного производства — мелассу (содержит
до 60% сахарозы, см. с. 377). Картофель, зерно после очистки и уда-
ления оболочек (в небходимых случаях) разрыхляют и разваривают,
превращая в однородную жидкую массу, в которую вводят солод —
измельченное и высушенное проросшее зерно ячменя. Солод содер-
жит высокоактивные ферменты — амилазы, способные осахаривать
крахмалсодержащее сырье:
60°С
(С6Н10О5)Л НоО > т С12Н220п
крахмал мальтоза
Иногда вместо солода применяют специальным образом получен-
ные ферментные препараты. Продукт, образовавшийся после осаха-
ривания, охлаждают и вводят в него дрожжи, содержащие фермент
мальтозу, фермент вызывает гидролиз мальтозы с образованием двух
молекул глюкозы:
мальтаза
С12Н22О11 + Н2О > 2С6Н12Ос
мальтоза глюкоза
Глюкозосодержащий раствор под влиянием ферментов дрожжей в ре-
зультате сложных биохимических процессов превращается в бражку,
содержащую до 10% этанола, который отгоняют и очищают. Выход эти-
лового спирта из одной тонны исходного сырья зависит от его вида,
качества, особенностей технологии и колеблется от 90 до 360 л.
Остаток после отгонки этанола (кубовый остаток) содержит си-
вушное масло — ядовитую маслянистую жидкость крайне неприят-
ного запаха («сивушный запах»). В состав сивушного масла входят
спирты С3—С5. Основные компоненты: 1-пропанол (7%); 2-метил-1-
пропанол (24%); З-метил-1 -бутанол и 2-метил-1-бутанол.
173
б. Экономически выгодно получение этилового спирта из отходов
переработки древесины — опилок и щепы (гидролиз растительных
материалов). Процесс состоит из двух стадий: гидролиза содержащей-
ся в них целлюлозы
(СбН10О5)Л п Н2О —► п С6Н12Об
целлюлоза глюкоза
♦
и сбраживания раствора, содержащего глюкозу с образованием эти-
лового спирта. Гидролиз проводят растворами минеральных кислот
при нагревании. Из одной тонны сухой древесины получают 160—
200 л «гидролизного» этилового спирта, применяемого для техни-
ческих целей. При комплексной переработке образовавшихся гидро-
лизатов кроме этилового спирта получается значительное количество
диоксида углерода, кормовых дрожжей, фурфурола (см. с. 312),
лигнина и других ценных продуктов.
в. Этиловый спирт получают из сульфитных щелоков — отходов,
образующихся при производстве целлюлозы в целлюлозно-бумажной
промышленности. Сульфитные щелоки содержат значительное коли-
чество гексоз, которые после специальной обработки могут сбражи-
ваться .
Из описанных способов получения этанола экономически наибо-
лее выгоден способ гидратации этилена, особенно прямая гидратация.
Стоимость этилового спирта, получаемого из пищевого сырья, наи-
большая, гидролизного — ниже. Самый дешевый этанол получают
синтетическим путем, к нему по стоимости приближается этанол,
получаемый из сульфитного щелока. Необходимо помнить, что при
получении 1 т этанола гидратацией этилена удается сэкономить до
4 т зерна или 12 т картофеля, которые могут быть использованы для
пищевых целей.
Этиловый спирт — один из основных продуктов, выпускаемых хи-
мической и пищевой промышленностью (схема 10). Этанол использу-
ется в производстве синтетического каучука, этилацетата, диэтило-
. / zP \ т?
вого эфира, хлораля СС13—C<f и в качестве растворителя. Ьоль-
\ ХН/
шие количества этанола, образующегося при брожении пищевого
сырья, идут в пищевую промышленность для получения спиртных на-
питков (водки, ликеров и т. д.), парфюмерную для изготовления ду-
хов, одеколона и медицинскую промышленность.
Пропиловые спирты. 1-Пропанол (н-пропиловый спирт)
СН3—СНг—СН2—ОН — бесцветная жидкость с характерным запахом.
Хорошо растворим в воде и многих органических растворителях.
Пропанол выделяют из сивушного масла и используют для получения
пропионового альдегида. 2-Пропанол, изопропиловый спирт
СН3—СН—СН3 — жидкость без цвета, со спиртовым запахом. Хорошо
ОН
растворим в воде. Получается гидратацией пропилена. Применяется
в качестве растворителя и для синтезов разнообразных химических
продуктов.
174
Схема 10. Синтезы на основе этилового спирта
175
Бутиловые спирты. 1-Бутанол, н-бутиловый спирт
СН3—(СНг)2—СН-2—ОН. Получается совместно с ацетоном при аце-
тонобутиловом брожении углеводов. Применяется в качестве раство-
рителя лаков и красок, для синтеза сложных эфиров, масляной кис-
лоты.
Бутиловые спирты с разветвленной углеродной цепочкой получают
гидратацией бутиленов, за исключением 2-метил-1 -пропанола
СН3—СН—СН2—ОН, который выделяют из сивушных масел или
СН3
синтезируют из оксида углерода и водорода. Все они применяются
для разнообразных синтезов и в качестве растворителей.
Амиловые спирты С&НПОН. Получают из пентанов С5Н12, выделяе-
мых из легких погонов нефти, хлорированием и последующим омы-
лением:
С5Н12 + С12 -> С5НИС1 + НС1
С5НПС1 + NaOH -> C5H1JLOH + NaCl
или из амиленов (С5Н10), содержащихся в продуктах крекинга и пиро-
лиза алканов, сернокислотной гидратацией:
н2о
СНз—С=СН—СН3 --------> (СН3)2С—СН2—СН3
I (H2so4) |
СН3 ОН
З-метил-1-бутанол и 2-метил-1-бутанол выделяют из сивушных масел.
Первый является исходным продуктом для получения уксусноизо-
амилового эфира, вещества с приятным «грушевым» запахом, исполь-
зуемого в приготовлении конфет, фруктовых вод, в качестве раство-
рителя и для разнообразных синтезов.
Высшие спирты (см. с. 402). Циклогексанол С6НПОН. Бесцветная
жидкость со слабым запахом. Плохо растворим в воде. Получают в
промышленности гидрированием фенола (см. с. 188). Применяется в
синтезе адипиновой кислоты, в качестве растворителя масел, жиров,
синтетических смол и как добавка при производстве синтетических
моющих средств и хозяйственного мыла.
Виниловый спирт СНг^СН—ОН. В свободном состоянии не су-
ществует. В тех случаях, когда в ходе реакции он образуется, проис-
ходит его превращение в альдегид. Аналогичная изомеризация (в
альдегиды или кетоны) наблюдается для всех ненасыщенных спиртов
с гидроксильной группой, расположенной у атома углерода с двой-
ной связью, — «винильное положение» (правило Эльтекова):
\ । /Р
}С=С—ОН —с—с<
/ | | хн
Такое превращение дает дополнительный выигрыш в энергии в
91,8 кДж/моль, что приводит к повышению устойчивости всей систе-
мы.
176
Поливиниловый спирт СН2—СН—1 полимер винилового спир-
L Ап
та — белый порошок, растворимый в воде. Получают омылением по-
ливинилацетата. Применяют в производстве синтетических волокон
и лекарственных препаратов.
Аллиловый спирт СНг—СН—СНг—ОН. Простейший представи-
тель одноатомных ненасыщенных спиртов. Жидкость с резким запа-
хом, хорошо растворимая в воде. Получается хлорированием про-
пилена и щелочным гидролизом образовавшегося аллилхлорида:
500°С
СН2=СН—СН3 + С12-------> СН2=СН—СН2С1 + НС!
пропилен аллилхлорид
50—1С0°С
СН2=СН—СН2С1 + Н2о-------> СН2=СН—СН2ОН + НС1
Аллиловый спирт обладает высокой реакционной способностью,
содержит в молекуле две функциональные группы: двойную связь
и гидроксильную группу. Для него характерны реакции, которые
были рассмотрены ранее для каждой из этих групп:
СН2=СН—СН2ОН-
аллиловый спирт
+ СН3—СН2—сн2—ОН
1 -пропанол
———> СН2С1—СНС1—СН..ОН
2, З-дихлор-1-пропанол
/О
__> сн2=сн—с/
\н
акролеин
ОН C1 он
2-ХЛОР-1,3-пропандиол
Нго-ю^ сн сн_сн
I I I
он он он
глицерин
Аллиловый спирт применяется для получения глицерина, в произ*
водстве смол, парфюмерной промышленности.
Бензиловый спирт С6Н5—СНгОН. Бесцветная жидкость с прият-
ным запахом, плохо растворим в воде. Широко распространен в при-
роде, входит в состав эфирных масел, принимает участие в формирова-
нии запаха цветов, составной компонент ароматообразующих веществ
пшеничного хлеба. В промышленности получают омылением бензил-
хлорида:
c6h5ch2ci + н2о----> С6Н5СН2ОН
7—682
177
Химические свойства бензилового спирта связаны с наличием в
его молекуле оксигруппы и бензольного ядра — вступает во все ре-
акции, характерные для первичных спиртов и производных бензола.
Близкое соседство бензольного ядра и оксигруппы приводит к повы-
шенной подвижности последней в реакциях замещения. Применяется
в парфюмерной промышленности.
Многоатомные спирты
Многоатомными называются спирты, содержащие в молекуле две
гидроксильные группы и более. В зависимости от количества гидро-
ксильных групп они делятся на двухатомные (две группы ОН), или
диолы; трехатомные, или триолы и т. д. Двухатомные спирты назы-
вают также гликолями.
У одного атома углерода в молекуле многоатомного спирта может
содержаться не более одной гидроксильной группы. Спирты, содер-
жащие две или три геминальные гидроксильные группы (у одного С-
атома: 1,1-диолы; 1,1,1-триолы), выделить в свободном состоянии
не удается, в момент образования они отщепляют воду и превращаются
в альдегиды, кетоны или карбоновые кислоты:
2NaOH
СН3—СНС12--------
—2NaCl
СН3—СН
/ОН
1, 1-дихлор-
этан
СН3-С^
уксусный
альдегид
С1 С1
сн3—с—сн3
2, 2-дихлорпропан
2NaOH
—2NaCl
ОН он
о
II
СН3—С—СН3
ацетон
3NaOH
СНз—СН2—СС13------------
- 3NaCl
1,1, 1-трихлорпропан
ОН"
СН3—СН2—С\°Н
он
сн3—сн2—с
^он
пропионовая кислота
Эфиры 1,1-диолов (ацетали) выделены в свободном состоянии; они
достаточно устойчивы (см. с. 201).
Номенклатура. Изомерия. Изомерия многоатомных спиртов обус-
ловлена строением углеродного скелета и различиями в положении
гидроксильных групп, а также их пространственным расположением.
По ИЮПАК для наименования многоатомных спиртов к окончанию
«ол» прибавляют приставку «ди», «три» и т. д., соответствующую ко-
личеству гидроксильных групп, положение которых указывается циф-
рами. Для наименования отдельных спиртов широко применяются
исторические названия:
СН2—СН2
I I
ОН он
1, 2-этандиол
(этиленгликоль)
СН3—СН—СН2—СН2
ОН ОН
1, 3-бутандиол
сн2—сн—сн2
I I I
он он он
1, 2, 3-пропантриол
(глицерин)
178
но он
1,2-цис-циклогександиол
По рациональной номенклатуре наименование диолов строится как произ-
водное от названия соответствующего этиленового углеводорода с добавлением
слова «гликоль» — этиленгликоль, пропиленгликоль и т. д. Как правило, этот
способ применяется для наименования диолов простого строения, у которых ок-
сигруппа расположена у соседних углеродных атомов (а-гликоли). Диолы, со-
держащие гидроксильные группы у соседних третичных углеродных атомов
получили название пинаконов:
СН3 СН3
/I I \
СН3 ОН ОН СН3
2, 3-ди метил-2, 3-бутандиол
(пинакон)
Способы получения. Получают многоатомные спирты теми же спо-
собами, что и одноатомные, с той лишь разницей, что в молекулу вво-
дится не одна, а несколько гидроксильных групп.
Гидролиз галогенпроизводных'.
2NaOH
СН3—СН—СН2—СН9--------> СН3—СН—СН2—СН2
| | * —2NaCl | |
Cl Cl он он
1, 3-дихлорбутан 1, 3-бутандиол
Гидроксилирование алкенов (реакция Вагнера)!
[О], НоО
СНз—СН=СН2 -------СН3—СН—СН2
п₽опилеп он он
I, 2-пропандиол
В качестве окислителей применяется щелочной раствор КМпО4
или Н2О2.
Получение из а-оксидов
Н2О; Н+
СН3—СН—СН2--------> СНз—СН—СН2
'о'/ ОН он
окись пропилена 1, 2-пропандиол
При гидратации а-оксидов обычно образуются гликоли.
Отдельные представители этой группы соединений получают ори-
гинальными методами:
z° н2
СН = СН + 2НС< НО—СН2—С=С—СН2—ОН ->
ХН
ацетилен муравьиный
альдегид
179
у*
но—сн2—сн2—сн2—сн2—он
1, 4-бутандиол
Физические свойства. По своим физическим свойствам низшие гликоли —
хорошо растворимые в воде вязкие жидкости, многие из которых обладают слад-
ким вкусом. Введение в молекулу второй гидроксильной группы сильно сказы-
вается на свойствах спиртов: диолы кипят при более высоких температурах, чем
одноатомные спирты, что связано с появлением дополнительных водородных свя-
зей.
Химические свойства. Химические свойства многоатомных спиртов
близки к свойствам одноатомных. В реакциях может участвовать
одна или несколько гидроксильных групп.
Повышенная подвижность атомов водорода гидроксильных групп
(рК& СНгОН—СН2ОН равна 14,7) — особенность многоатомных
спиртов. Они превращаются в алкоголяты не только при взаимодей-
ствии с натрием или калием, но и с гидроксидами тяжелых металлов
образуя комплексные соединения:
Н
I
Н2С—ОН НО—СН2 Н2С—Оч .0—сн2
| + Cu(OH)2 + I -> | /Си/ I + 2Н2О
Н2С—ОН НО—СН2 Н2С—Oz \0—сн2
этиленгликоль I
н
комплексное соединение
Окисление гликолей:
СН2—ОН [О]
СН2—ОН
этиленгликоль
Z° [О]
но—сн2—с/ —>
\н
гликолевый
альдегид
глиоксаль
[О]
Я
но—сн2—с<
хон
гликолевая кислота
гл иоксиловая
но/
щавелевая
кислота кислота
Реакция протекает легко, по продукту реакции до известной сте-
пени можно судить о строении гликолей.
Получение сложных эфиров кислот:
О
II
СН2—ОН сндсоон СН2—О—С—СН3 сн8соон
СН2—ОН СН2ОН -н2сГ
этиленгликоль неполный эфир
О
II
СН2—о—с—сн3
сн2—о—с—сн3
II
о
полный эфир
Дегидратация гликолей (пинаколиновая перегруппировка). Гли-
коли, отщепляя воду (в присутствии кислот или хлорида цинка),
превращаются в разнообразные соединения, характер которых зави-
180
сит от строения гликоля, условий реакции. Отнятие воды может про-
текать внутримолекулярно и межмолекулярно:
СН—СН2 ,0
| I---------► сн3-с/
ОН ОН —н,о \н
этиленгликоль уксусный альдегид
Механизм этой реакции может быть представлен следующим об-
разом:
сн2—он н*
I ---------
CH2—Q4
сн—он —н2о
I +
сн—о—н
н
-н +
При отнятии воды от молекулы пинакона мигрирует не водород,
а метильная группа (вместе с парой электронов) (пинаколиновая пере-
группировка):
сн3 сн3 сн3 сн3
I I 1.1 -н2о
СНз—С---С—ОН '*" сщ—с------С—ОН
• I I II
о—Н СНз Н—О—Н СНз
пинакон
СН3 О
I II
сн3—с—с—сн,
Ан
пинаколин
Отнятие воды от гликолей, гидроксильные группы которых раз-
делены двумя углеродными атомами (у-гликоли), приводит к образо-
ванию внутренних циклических эфиров:
НО—СН2—СН2—СН2—СН2—ОН
1, 4-бутандиол
(7-гликоль)
тетрагидрофуран
(7-окись)
а от р-гликолей — к образованию непредельных спиртов:
НО—сн2—сн2—сн2—ОН----------> сн2=сн—сн2—он
, — н,о
1, 3-пропандиол аллиловый спирт
(3-гликоль)
он он
Отдельные представители. 1,2-Этандиол, этиленгликоль СН2—СН2.
Простейший и наиболее важный представитель гликолей (эти соеди-
181
нения названы так за свой сладкий вкус — от греческого гликос —
сладкий). Бесцветная, вязкая жидкость, без запаха.
Основной промышленный способ получения этиленгликоля —
гидратация окиси этилена, которую проводят избытком воды в при-
сутствии серной или фосфорной кислот при нагревании (50—100° С):
(Н+)
СН2—СН2 + Н2О —> СН2—СН2
\ / II
О он он
окись этилена этиленгликоль
Этиленгликоль — важный продукт химической промышленности.
Широкое применение нашли разнообразные эфиры этиленгликоля.
Диоксан — простой циклический эфир этиленгликоля, получают при
перегонке последнего с серной кислотой:
ОН но О
Н.С^ ^СНз СН,
I + L------------------” 1 I '
НоСх /сн? - 2Н2О Н.С сн,
\н но \о/
этиленгликоль диоксан
используется в качестве растворителя.
Эфиры этиленгликоля и двухосновных кислот идут на изготовле-
ние пленкообразующих веществ и синтетических волокон. Этилен-
гликоль употребляется в текстильной, косметической, табачной про-
мышленностях в качестве гигроскопического вещества. Водные раст-
воры этиленгликоля замерзают при температурах ниже нуля (50%-ный
при —37 С), что позволяет применять их для приготовления анти-
фризов — замерзающих при низких температурах жидкостей, ис-
пользуемых для охлаждения двигателей внутреннего сгорания.
Глицерин, 1, 2, З-Пропантриол СН2—СН—СН2. Единственный
ОН ОН ОН
практически важный представитель трехатомных спиртов. Сиропо-
образная сладкая жидкость, без цвета, хорошо растворимая в воде.
Водные растворы глицерина замерзают при низких температурах (67%-
ный раствор при —46,5° С); применяются в качестве антифризов.
Глицерин широко распространен в природе, является основным
спиртом, участвующим в построении молекул различных групп ли-
пидов (см. с. 390). В промышленности глицерин получают гидроли-
зом ацилглицеринов (триглицеридов):
О
II
СН2—О—С—R
I О СН2ОН
СН—О—С—Rj + ЗН2О -> СН—ОН + R—СООН + Rj—СООН + R2—СООН
10 । т гчтт карбеновые кислоты
у СН2ОН
сн2—о—с—r2
триглицерид глицерин
182
Это старый способ, не потерявший своего значения и в настоящее
время. Однако получение глицерина гидролизом триглицеридов свя-
зано с использованием для технических целей больших количеств
ценных пищевых продуктов — жиров и масел, поэтому этот способ
постепенно заменяется синтетическим, основным сырьем в котором
является пропилен:
450—500°С
СН2=СН—СН3 + С12---------> СН2=СН—СН2С1 + НС1
пропилен аллилхлорид
150—160сС
СН2=СН—СН2С1 + NaOH-----------> СН2=СН—СН2ОН + №С1
аллиловый спирт
-> СН2С1— снон—СН2ОН
СН2=СН—СН2ОН + НОС1—
-> СН2ОН—СНС1—СН2ОН
монохлоргидрины глицерина
СН2С1— СНОН—СН2ОН 150°С сн2—сн—сн2
+ 2NaOH---------> I I I
CH2OH—СНС1—СН2ОН —2NaCi ОН он он
глицерин
Глицерин широко применяется во многих отраслях промышлен-
ности. Основное его количество расходуется для получения произ-
водных — глифталевых смол, нитроглицерина. Глифталевые смолы
(глифтали) — полиэфиры глицерина и фталевой кислоты — применя-
ются для изготовления лаков. Глицеринтринитрат или, как его на-
зывают в технике, «нитроглицерин»* — сложный эфир глицерина и
азотной кислоты — получают действием азотной кислоты в присутст-
вии серной кислоты на глицерин:
СН2—ОН сн2—ono2
I H2so4 |
СН—ОН + 3HONO2----------> сн—ono2 + ЗН2О
сн2—он сн2—ono2
глицерин глицеринтринитрат
(нитроглицерин)
Глицеринтринитрат — взрывчатое вещество огромной силы (взры-
вается от легкого толчка). Для взрывных работ часто применяется
смесь, состоящая из 75% нитроглицерина и 25% инфузорной земли,
называемая динамитом, которая безопаснее в работе.
В пищевой промышленности глицерин используется для приго-
товления ликеров и безалкогольных напитков, в бумажной и коже-
венной — для предохранения материала от высыхания. Он входит в
состав многих косметических препаратов и широко применяется в
качестве смягчающего кожу вещества.
* Нитроглицерин не является нитросоединением. Нитросоединения содер-
жат в молекуле группу NO2, азот которой непосредственно связан с углеродным
атомом (см. с. 270).
183
Ксилит СН2ОН(СНОН)3СН2ОН и сорбит СН2ОН(СНОН)4СН2ОН.
Получаются восстановлением соответствующих моносахаридов:
первый — ксилозы (см. с. 369), второй — глюкозы (см. с. 363).
Сорбит широко распространен в природе, входит в состав многих
плодов и ягод (в плодах рябины до 7% сорбита). Ксилит и сорбит
применяются больными диабетом в качестве заменителя сахара, для
приготовления кондитерских изделий, напитков, зубных паст.
Сорбит — первый полупродукт при производстве аскорбиновой кис-
лоты (витамина С, см. с. 371).
Фенолы, нафтолы
Особую группу составляют оксисоединения, гидроксильная груп-
па которых связана с атомами углерода бензольного ядра, и окси-
производные нафталина. Первые называются фенолами, вторые —
нафтолами:
л -крезол
(4-окситолуол)
фенол-
(оксибензол)
гидрохинон
(л-диоксибензол)
пирокатехин
(о-диоксибензол)
пирогаллол
ДДЗ-триоксибензол)
флороглю ЦИН
(1,3,5- триоксибензол)
ОН
а-нафтол
(1-нафгол)
/?-нафтол
(2-нафюл)
Изомерия фенолов и нафтолов связана с различным положением гид-
роксильных групп, строением и положением заместителей. Фенолы,
кроме этого, могут быть изомерны ароматическим спиртам:
184
о-крезол
бензиловый спирт
Способы получения фенолов и нафтолов. Получение фенола сов-
местно с ацетоном (кумольный способ). Исходное сырье — бензол и
пропилен:
бензол
СН2=СН—СН3
Н3РО4
изопропилбензол
(кумол)
гидроперекись
изопропилбензола
пропилен
1Н1
ацетон
Этот метод, разработанный в 1949 г. советскими учеными
П. Г. Сергеевым, Б. Д. Кружаловым, Р. Ю. Удрисом и М. Е. Нем-
цовым, экономически наиболее выгоден: он позволяет получить два
ценных продукта — фенол и ацетон — при использовании дешевого
сырья — пропан-пропиленовой фракции газов крекинга нефти.
Получение из бензолсулъфокислот (см. с. 268).
Получение фенола парофазным каталитическим гидролизом бензол-
хлорида. Сначала из бензола окислительным хлорированием полу-
чают бензолхлорид. Катализатор — хлориды меди и железа, нанесен-
ные на оксид алюминия. Затем бензолхлорид гидролизуют с образо-
ванием фенола:
бензол
4- HC1
4- 0,5 О2
235—245°С
-И2О
400—420°С
-Н2О
4- HC1
Выделение из каменноугольной смолы. Не потерял своего значения
и старый способ получения фенолов — выделение их из побочных
продуктов коксования каменных углей. При этом каменноугольную
смолу обрабатывают растворами щелочи и выделяют фенолы из полу-
ченных продуктов кислотой с последующей их очисткой:
NaOH H2SO4
С2Н5ОН ------> C6H5ONa----------> С6Н5ОН
—НаО ж —NaHSO4
фенол а фенолят * фенол
натрия
Получение из солей соответствующих сульфокислот и хлорфенолов
двух- и многоатомных фенолов и нафтолов (см. с 268).
185
Получение из солей диазония (см. с. 288):
CgHsNgCl- + Н20 -> С„Н6ОН + N2 + НС1
Физические свойства. Фенолы и нафтолы — кристаллические вещества с
характерным запахом, трудно растворимые в воде. Отличаются более высокими
температурами кипения по сравнению с соответствующими спиртами. Например,
температура кипения (°C) фенола С6Н5ОН 181, а циклогексанола С6НцОН —161.
Химические свойства. Химическое поведение фенолов и нафтолов
связано с наличием в их молекуле оксигруппы и ароматического ядра.
Они вступают во многие реакции, характерные для этих групп. В то
же время в химическом поведении фенолов и нафтолов наблюдается
ряд особенностей, вызванных взаимным влиянием этих групп. На-
пример, фенолы обладают более сильными кислотными свойствами
(р/<а С6НбОН 9,7), чем спирты, и способны вступать в реакции со
щелочами. Это связано с тем, что неподеленная электронная пара ато-
ма кислорода оксигруппы вступает во взаимодействие с л-электро-
нами бензольного ядра (+М-эффект), электронная плотность смеща-
ется и атом водорода оксигруппы приобретает большую подвиж-
ность:
При введении в молекулу фенола электроноакцепторных замести-
телей (галогены, NO2; СНО; CN и др.) подвижность атома водорода
оксигруппы возрастает, кислотность повышается; электронодонорныг
заместители (СН3; С2Н5) уменьшают кислотность фенолов.
Взаимодействие с водными растворами щелочей. Образуются соли
фенолов — феноляты:
С6Н5ОН + NaOH -> C6H5ONa + Н2О
фенол фенолят
натрия
С FeCl3 фенолы дают комплексные соединения — феноляты желе-
за. Растворы производных одноатомных фенолов имеют интенсивную
фиолетовую окраску, крезолов — окрашены в голубой цвет. Реакция
с FeCl3 — качественная реакция на фенолы.
Образование простых эфиров. При взаимодействии
фенолов с алкилхлоридами, диметилсульфатом, хлоруксусной кислотой
в присутствии водных растворов щелочи при нагревании образуются
простые эфиры:
186
+C2H$I
-Nal; H20
4-(CH3)2SO4
- CH3OSO3Na
NaOH (водный p-p)
Ч-Cl CHnCQOH
-NaCl
—NaCl
аллилфениловый эфир
Образование сложных эфиров. Взаимодействие фе-
нолов с хлорангидридами и ангидридами кислот приводит к образо-
ванию сложных эфиров:
уксуснофени левый
эфир
Реакции, связанные с заменой гидроксильной группы в молекуле
фенола (например, взаимодействия с РСВ), протекают значительно
труднее, чем у спиртов.
Реакции замещения в бензольном кольце. Гидроксильная группа —
заместитель первого рода, поэтому реакции электрофильного заме-
щения в бензольном ядре протекают с фенолами значительно легче,
чел с бензолом, а заместители направляются в орто- и параполо-
жение;
187
он
Восстановление фенола. При гидрировании фенола образуется
циклогексанол. Реакцию проводят при 150° С и давлении 20 • 105 Па,
катализатор — металлический никель, нанесенный на оксид алюми-
ния. Это основной промышленный способ получения циклогексанола:
циклогексанол
Окисление фенолов. Фенолы (особенно триалкил- и многоатомные
фенолы и нафтолы) легко окисляются с образованием разнообразных
продуктов окисления. Легкая окисляемость позволяет использовать
их в качестве антиокислителей (антиоксидантов). Внесение небольших
количеств антиоксидантов замедляет окисление молекулярным кисло-
родом углеводородов, альдегидов и кетонов, липидов. Антиокисли-
тельные свойства фенолов связаны с их способностью реагировать
с перекисными радикалами RO2, ведущими цепь окисления, с обра-
зованием в результате этого малоактивных радикалов, не способных
продолжать цепь окисления:
гидрохинон перекисной малоактивный
радикал радикал
хинон
Антиокислители фенольной природы широко применяются для
стабилизации бензинов, замедления старения каучуков. В пищевой
промышленности антиоксиданты фенольной природы применяются
для сохранения жиров и масел, сухого молока, кондитерских изделий,
рыбных и мясных продуктов, пищеконцентратов. Наибольшее рас-
пространение получили З-трет-бутил-4-оксианизол, 3,5-дитрет-бутил-
4-окситолуол и нордигидрогваяретовая кислота:
нордигидрогваяретовая кислота.
Отдельные представители. Фзнол, оксибензол, карболовая кислота
С6Н5ОН. Бесцветное кристаллическое вещество с характерным запа-
хом, краснеет на воздухе вследствие окисления, вызывает ожоги при
попадании на кожу. Ядовит. Качественная реакция — фиолетовое
окрашивание с FeCl3. Получение см. с. 185. Один из важнейших
продуктов химической промышленности, используется для получе-
ния фенолформальдегидных смол, циклогексанола, пикриновой кис-
лоты (см. с. 188), красителей, лекарственных препаратов, душистых
веществ. Водные растворы фенола применяются в качестве антисеп-
тика.
Крезолы, Получаются из каменноугольной смолы. Применяются
для изготовления искусственных смол.
Ди- и трифенолы, нафтолы и их производные. Гидрохинон и пи-
рогаллол применяются в фотографии; пирогаллол используется также
при определении содержания кислорода в газовых смесях; нафтолы —
для получения синтетических красителей.
189
ПРОСТЫЕ ЭФИРЫ
Простыми эфирами называются соединения с общей формулой R—О—Rt;
Аг—О—R; Аг—О—Аг, где R, R4, Аг — углеводородные радикалы. Их можно
рассматривать как продукты замещения атома водорода в молекуле оксисоеди-
нения на радикал.
Номенклатура, изомерия. Для наименования эфиров могут применяться
тривиальные названия (серный эфир; фенетол), но в большинстве случаев назва-
ние эфиров складывается из наименования радикалов, с добавлением слова эфир:
диметиловый эфир, диэтиловый эфир, метилэтиловый эфир. По ИЮПАК для на-
именования простых эфиров к названию углеводорода прибавляют название
алкокси (RO) или арилокси группы (АгО). За основу названия выбирают ради-
кал с большим числом атомов углерода:
СН3—О—СН3 СН3—О—С2Н5
диметиловый эфир, метилэтиловый эфир,
метоксиметан метоксиэтан
с2н5-о-с2н5 С6Н5-О-С6Н8
серный эфир, дифениловый эфир,
диэтиловый эфир, феноксибензол
этоксиэтан
Изомерия эфиров связана с составом и строением радикалов.
Способы получения, Межмолекулярная дегидратация спиртов (см. с. 169).
Взаимодействие алкоголятов с залоге налкилами:
С2Н5—О—Na + С2Н51 -> С2Н5—О—С2Н5 + Nal
этилат натрия этилиодид диэтиловый эфир
Взаимодействие ацетилена со спиртами. Эта реакция была описана ранее
(см. с. 92), она применяется для получения простых алкилвиниловых эфиров.
Физические свойства. Первые два представителя: диметиловый и метилэти-
ловый эфиры — газы: начиная с диэтилового эфира — жидкости (табл. 20).
Таблица 20. Простые эфиры
Название Формула Темпера- тура плавле- ния, °C Темпера- тура ки- пения, °C Плотность Р, г-см-3
Диметиловый эфир СН3—О—СН3 — 140 —24 0,661
Диэтил овый эфир С2Н5-О-С2Н5 — 116 34,6 0,714
Ди-н-пропиловый эфир СзН7 О С3Н7 — 122 91 0,736
Диизоприп иловый эфир СН3 —60 69 0,735
(СНз—СН)2О
Дивинил^вый эфир (СН2=СН),0 — 35 0,773
Метилфениловый эфир, с6н5—о—сн3 —37,3 154 0,994
анизол
Эшлфениловый эфир, фе- С6Н5—О—С2Н5 —33 172 —
нетол
Дифениловый эфир с«н5-о-с6н5 27 259 1,072
1,4-Диоксан о "о Н,7 101,4 1,033
Те гра гидрофуран Xх \ — 108 66 0,888
190
Температура кипения эфиров ниже температур кипения соответствующих спир-
тов, так как их молекулы не ассоциированы за счет водородных связей. Они об-
ладают приятным (эфирным) запахом, большинство плохо растворимо в воде,
хорошо во многих органических растворителях.
Химические свойства. Простые эфиры — устойчивые органические соеди-
нения. Они не гидролизуются, в обычных условиях не вступают во взаимодейст-
вие с металлическим натрием, на них не действуют щелочи и большинство кислот.
Взаимодействие с иодоводородом. При нагревании с иодоводородом простые
эфиры разлагаются с образованием спирта и галогеналкила:
С2Н5—О—С2Н5 + HI -> С2Н5ОН + С2Нб1
диэтиловый эфир этилиодид
Присоединение сильных, кислот. Эфиры растворяются в кислотах с выделе-
нием теплоты:
^2^5 \ С2Нб<
>0 + НС1 >0 • НС1
С2Н/ с2н5/
Образовавшиеся соединения можно рассматривать как соли оксония. В мо-
лекуле оксониевых соединений атом кислорода связан с помощью ковалентных
связей с углеводородными радикалами и ионной связью с анионом:
С2И5\
:о: + н—ci —•-
c2h5zV_>
ci
диэтилоксоний хлорид
Образование пероксидов. На свету при хранении простые эфиры окисляются
с образованием перекисных соединений:
О2
R—СН2—О—Ri -> R—СН—О—Ri
О—ОН
Образовавшиеся перекиси (главным образом в виде гидроперекисей) представ-
ляют большую опасность при работе с эфирами, так как они могут быть причи-
ной взрыва.
Отдельные представители. Диэтиловый эфир, этоксиэтан, серный эфир
С2Н5—О—С2Н5. Бесцветная подвижная жидкость с эфирным запахом, горюч.
Широко применяется в качестве органического растворителя, обладает анесте-
зирующим действием.
Эфиры фенолов: С6Н5—О—СН3 — анизол; СеН5—ОС2Н5 — фенетол — вы-
сококипящие жидкости с приятным запахом, применяются в парфюмерной про-
мышленности.
Диоксан — простой циклический эфир этиленгликоля. Получение см.с.
182. Применяется в качестве органического растворителя.
Эпоксиды
Эпоксидами, или эпокисями, называются простые эфиры циклического стро-
ения с а-окисным кольцом:
191
Отличаются высокой реакционной способностью. Наиболее важный представи-
тель эпоксидов — окись этилена.
Окись этилена СН2—СН2. Летучая жидкость со специфическим запахом.
ох
Хорошо растворима в воде, огнеопасна, ядовита. Получают прямым окисле-
нием этилена в присутствии серебряного катализатора:
Ag; 250°С
2СН2=СН2 + 02--------> 2СН2—СН2
Вырабатывается в больших количествах и широко используется в различных
синтезах (получение антифризов, моющих средств, пластических масс, эмульга-
торов). Применяется в качестве пестицида.
Органические перекисные соединения
При замещении атомов водорода в молекуле перекиси водорода Н—О—О—Н
на углеводородные радикалы получают гидроперекиси (замещен один атом во-
дорода) и перекиси (замещено два атома водорода):
О—ОН R—О—О—Ri
гидроперекись перекись
Перекисные соединения получаются при аутоокислении углеводородов,
реакция протекает по свободнорадикальному механизму. Гидроперекиси обла-
дают кислотными свойствами, при взаимодействии с растворами щелочей обра-
зуют соли. Перекиси и гидроперекиси относятся к сильным окислителям. Работа
с ними требует особой осторожности ввиду их взрывоопасности.
ОКСОСОЕДИНЕНИЯ (АЛЬДЕГИДЫ И КЕТОНЫ)
Альдегидами и кетонами называются соединения, содержащие в
молекуле оксо- или карбонильную группу \с=0. Отсюда другое
их название — карбонильные соединения. В молекуле альдегидов
атом углерода карбонильной группы связан с углеводородным ради-
/£
калом и атомом водорода —C<f , в молекуле кетонов — с двумя
\н
О
li
углеводородными радикалами R—С—Rx. Содержащаяся в молекуле
альдегидов группа —С< называется альдегидной группой и рас-
сматривается как функциональная группа; группа ^>С=0 в моле-
куле кетонов, называемая кетогруппой, является функциональной
группой кетонов.
В зависимости от характера углеводородных радикалов, входящих
в молекулу альдегидов и кетонов, различают предельные, непредель-
ные циклические, ароматические и гетероциклические оксосоедине-
ния. Кетоны могут быть, кроме этого, смешанного типа: радикалы,
192
с которыми связана кетогруппа, могут принадлежать к разным клас-
сам углеводородов. По количеству карбонильных групп различают
монокарбонильные, дикарбонильные и поликарбонильные соединения:
СН3—С<
ХН
О
II
СН3—С—СН3
уксусный альдегид, ацетон, пропанон
ацетальдегид, этаналь
л0
сн,=с—cZ
I ХН
сн3
метакриловый альдегид,
2- метилпропеналь
О
акролеин,
2-пропеналь
циклопен-
танон
z0
свн5-с<
бензойный
альдегид
О
II
С6Н5—С—С6Н5
дифенилкетон,
бензофенон
о
II
с6н5—с-сн3
метилфенил кетон*
ацетофенон
О
II
СН3—С—СН=СН2
м етилвинил кетон,
З-бутен-2-он
глиоксаль
О о
II II
сн3—с—с—сн3
диацетил
Номенклатура. Изомерия. Альдегиды часто называют по наимено-
ванию кислот, в которые они превращаются при окислении (уксусный
альдегид). По рациональной номенклатуре при наименовании кетонов
называют углеводородные радикалы и добавляют слово кетон (ди-
метилкетон). По ИЮПАК в основе наименования альдегидов и кетонов
лежит название углеводорода с тем же числом углеродных атомов,
включая углеродный атом карбонильной группы, в случае альдеги-
дов к нему добавляют окончание «аль» (бутаналь), кетонов — «он» с
указанием номера углеродного атома, связанного с кислородом (2-
пентанон). Нумерацию углеродной цепи начинают с того конца, к
которому ближе расположен карбонильный углерод. В остальном по-
рядок наименования остается общим.
Изомерия оксосоединений определяется строением углеводород-
ных радикалов:
4 3 2
СН3—СН2—СН,
бутаналь
(масляный альдегид)
О
СН3—СН2—СН2—С—СН3
5 4 3 2 1
2-пентанон
(метилпропилкетон)
193
Таблица 21. Альдегиды и кетоны
Название Формула Темпера- тура плав- ления, °C Темпера- тура ки- пения, °C Плотность Г‘СМ“3 Показа- тель пре- л омления л20 nD
Муравьиный альдегид, фор- мальдегид Н-С< я о —92 — 19,2 0,815
Уксусный аль- дегид , аце- тальдегид о 1 о I о — 122,6 +20,8 0,781 1,3316
Пропионовый альдегид, пропаналь /О сн3-сн2— —81 +49,1 0,807 1,3636
Масляный аль- дегид , бута- наль сн3- (СН2)2-С< о X —97 +75 0,817 1,3843
Бензальдегид С6Н5 -с<н /О сн—c<f хн
Акролеин, про- пеналь сн2= —56 —88 178,1 +52,5 1,046 0,841 1,5459 1,3998
Кротоновый альдегид сн3—сн=сн—с< О X —76,5 4-Ю4 0,859 —
Ацетон, пропа- нон СН3- -с—сн3 II О -95,3 56,1 0,781 1,3602
Циклогексанон / —31,2 155,4 0,946 —
Бензофенон С6Н5- о II -с-с(л5 48 306 — —
Метилфенилке- тон, ацето- фенон Свн6- О II -С—СН3 19,8 202,0 1,028 1,5342
Глиоксаль я о /О 3—с< хн 15 50,4 1,140 —
/он О
Салициловый альдегид >4 1,6 197 1,169 —
Анисовый аль- дегид сн3—с ЧН 2,5 248 1,123 —
194
СН,—СН—С/
I ХН
О
II
сн3—сн2—с—сн9—сн3
диметилуксусный
альдегид,
метилпропаналь
(изомасляный
альдегид)
3-пентанон (диэтилкетон)
СН3—СН—С—СН3
I II
СНз О
З-метил-2-бутанон
(метилизопропилкетон)
Наиболее важные представители альдегидов и кетонов приведены
в табл. 21.
Способы получения. Окисление спиртов (см. с. 170).
Дегидрирование спиртов (см. с. 170).
Восстановление карбоновых кислот в альдегиды. Обычно восста-
навливают не сами карбоновые кислоты, а их производные (восста-
новление по Розенмунду):
В качестве катализаторов применяют отравленный серой палла-
дий, нанесенный на BaSO4. Восстановление производных кислот мо-
жет быть осуществлено и с помощью соединений типа LiAlH(OR)3
при низкой температуре:
—С—С1
L1A1H (ОСДМз
хлорангидрид
циклогексанкарбоно-
вой кислоты
Окисление ароматических углеводородов, содержащих метильную
группу в боковой цепи. В большинстве случаев окисляют метилбензол
или его производные:
С.Н5СН3 -> CJRC<f
ХН
Окислитель СгО3 в присутствии уксусного ангидрида.
Разложение солей карбоновых кислот. При сухой перегонке каль-
циевых и бариевых солей карбоновых кислот образуются кетоны (если
соль муравьиной кислоты, то альдегид). Строение образовавшегося
кетона зависит от строения соли: если кислотные остатки одинаковы,
образуется симметрично построенный кетон, если соль двухосновной
кислоты— циклический:
О
II
СН3—С—Оч
>Са
СН?—С—СК
II
О
о
II
сн3—С—СН3 СаСО3
пропанон
уксуснокислый
кальций
195
о
II
СН,—СН2—С—Ох
I >Ва
СН2—СН2—С—О'
II
о
285—290°С
ВаСО?
Кетоны и уксусный альдегид могут быть также получены при про-
пускании паров кислот (t = 400—500° С) над катализаторами, со-
держащими оксиды кальция, бария, марганца, цинка (кетонизация
кислот):
О
40П°С ||
СНзСООН + нсоон------------>- сн3—сч + сн3—с—сн3 + со2 + Н2О
кат.
уксусная муравьиная
кислота кислота
Гидролиз дигалогенпроизводных. При гидролизе геминальных ди-
галогенпроизводных, содержащих два атома галогена у крайнего
углеродного атома, образуются альдегиды: в других случаях — ке-
тоны:
СН3—СНС12 + 2Н2О
I, 1-дихлорэтан
/ОН"
сн3—с—н
ХОН_
СНз—Сх
ацетальдегид
С1 С1
СНз—С—СН3 + 2Н2О
2, 2-Дихлорпропан
—2НС1
НО
_СН3—С—СНз.
о
II
СН3—С—СН3
ацетон
ОН 1
С6Н5СНС1, + 2Н2О---------->
„ “ — 2HCI
бензилиденхлорид
/О
----- C6H3-cf
—Н2О \тт
бензальдегид
Синтез уксусного альдегида и кетонов из ацетилена и его гомо-
логов. Реакция Кучерова (см. с. 92).
Оксосинтез. Альдегиды могут быть получены в результате при-
соединения СО и Н*2 к олефинам. Реакция протекает в присутствии
никелевых и кобальтовых катализаторов при 100—200° С
(101—252,5)10* Па:
196
R—CH=CH2 + CO + H2—
zO
R—CH2—CH2—C<f
XH
альдегид нормального
строения
____-> R—CH—CH3
I /О
C<
XH
альдегид изостроения
В реакцию вступают все олефины. Это важный промышленный
способ синтеза альдегидов, позволяющий использовать дешевое сы-
рье — продукты химической переработки нефти.
Получение из гликолей с помощью пинаколиновой перегруппировки:
R R2 R R2 R R2
I I +h+ | | ||
Ri—C—C—R3------> Rr-C—C—R3 -----> Rj-C-C—R3----->
II I + I -H*° I + -H+
он он он о—H OH
I
H
R1
—> R—C—C—R3
OH r2
Для синтеза ароматических альдегидов и кетонов кроме уже при-
веденных способов применяются специфические методы. Остановимся
на двух наиболее характерных из них.
Прямое введение альдегидной группы (реакция Гапгтермана —
Коха):
толуол
I-толуиловый
альдегид
Получение ароматических кетонов по реакции Фриделя— Крафтса:
бензол
+ С1—С—СН3
ацетилхлорид
А1С13
метилфенилкетон
197
Получение циклических кетонов (циклоалканонов). При восстанов-
лении фенолов молекулярным водородом в присутствии никеля под
давлением образуется циклогексанол, который при окислении или
дегидрировании (пропускают над мелкораздробленной медью, t =
== 250—280° С) переходит в циклогексанон:
н
зн2
фенол
циклогексанол
циклогексанон
Физические свойства. Температуры плавления и кипения наиболее важных
представителей альдегидов и кетонов приведены в табл. 21. Муравьиный альде-
гид — газ, остальные альдегиды и кетоны — жидкости или твердые вещества.
Температуры кипения альдегидов и кетонов ниже, чем у спиртов и кислот с тем
же числом атомов углерода в молекуле в связи с отсутствием межмолекулярных
водородных связей Так, температура кипения «-бутанола (°C) 117, масляного
альдегида 75,6, а масляной кислоты 165°С). Кетоны кипят при более высоких
температурах, чем соответствующие альдегиды. Оксосоединения с разветвленной
цепью углеродных атомов имеют более низкие температуры кипения, чем оксо-
соединения нормального строения.
Низшие альдегиды — соединения с резким специфическим запахом, причем
у альдегидов более резкий запах, чем у кетонов. Высокомолекулярные альдеги-
ды (С1о и С12) обладают приятным запахом, применяются в парфюмерии. Альде-
гиды и некоторые кетоны участвуют в формировании вкуса и аромата хлеба, вина
и ряда других пищевых продуктов. Альдегиды и кетоны легче воды, многие из
них хорошо в ней растворимы.
Химические свойства. Альдегиды и кетоны принадлежат к числу
наиболее реакционноспособных органических соединений, причем
альдегиды активнее кетонов. Высокая активность оксосоединений
связана с наличием в их молекуле карбонильной группы и особенно-
стями ее строения. В карбонильной группе связь между атомами угле-
рода и кислорода осуществляется двумя парами электронов и со-
стоит из о- и л-связей. Вследствие большей электроотрицательности
атома кислорода электронная плотность л-связи смещена в его на-
правлении:
">С=О
1,22 нм
Электрический момент диполя карбонильной группы 9,0 • 10-30 Кл-м.
В результате этого у атома кислорода карбонильной группы создается
повышенная электронная плотность, у атома углерода ее недостаток.
Атом углерода карбонильной группы приобретает электрофильные
свойства и активно взаимодействует с нуклеофильными реагентами.
Наоборот, атом кислорода становится нуклеофильным, поэтому спо-
собен взаимодействовать с электрофильными реагентами. Необхо-
димо также помнить, что атом углерода карбонильной группы нахо-
дится в состоянии $р2-гибридизации, и часть молекулы, содержащая
карбонильную группу, является плоской (атомы кислорода, карбо-
нильный углерод и два непосредственно с ним связанных атома лежат
в одной плоскости),
198
r_12°o
120°Qc==O
Rf 120°
поэтому она доступна для реагентов, расположенных сверху и снизу
от нее. Введение в углеводородный радикал заместителя, снижающе-
го электронную плотность у атома углерода карбонильной группы,
увеличивает его способность взаимодействовать с нуклеофильными
реагентами, и наоборот, электронодонорные заместители, повышаю-
щие электронную плотность у углеродного атома, снижают ее. Имен-
но этим объясняется большая реакционная способность альдегидов и
кетонов алифатического ряда по сравнению с оксосоединениями аро-
матического ряда.
На реакционную способность оксогруппы влияет не только харак-
тер заместителей, но и их объем. С его увеличением реакционная
способность альдегидов и кетонов снижается, поэтому альдегиды, кар-
бонильная группа которых связана с одним радикалом и атомом водо-
рода, обладают большей реакционной способностью, чем кетоны:
Н Н СН3 СН3 СН3
с=о > с=о > с=о > ''сн—с—сн
I I I / II \
Н СНз СНз СНз О СНз
Карбонильная группа увеличивает кислотность атомов водорода в
а-положении.
Для альдегидов и кетонов характерны следующие виды реакций:
присоединения, замещения, окисления, конденсации и полимериза-
ции.
Реакции присоединения (Ad N). В ходе реакции при-
соединения атОхМ углерода карбонильной группы взаимодействует с
нуклеофильным реагентом (I), двойная связь разрывается с образо-
ванием аниона (II), который, взаимодействуя с протоном, дает окси-
соединения:
оксосое- нуклео-
динение фильный
реагент (I),
\ Небыстро)
с —О -------------R—I
оксипроизводные
” Ш) (III)
Примером реакций присоединения, протекающих по Ad N механизму,
может служить взаимодействие оксосоединений с синильной кисло-
той, спиртами, гидросульфитом натрия и другими реагентами.
Восстановление альдегидов и кетонов. При восстановлении альде-
гидов образуются первичные спирты, кетонов — вторичные:
/°
сн3-с<_
+ н2 сн3—сн2он
этиловый спирт
199
о он
II I
сн3—с—сн3 + н2 ->- сн3—сн—сн3
ацетон изопропиловый спирт
Реакции протекают в присутствии никеля, кобальта, платины,
палладия. Возможно восстановление LiAlBU или водородом в момент
выделения.
Присоединение синильной кислоты (цианид-иона). Реакция про-
текает в присутствии основания; роль гидроксил-иона заключается
в образовании при его взаимодействии с цианистым водородом цианид-
иона, являющегося активным нуклеофилом. В отсутствие основания
реакция практически не происходит (основной катализ):
Н—C=N -h OH" :C=N + Н2О
цианиц-
ион
6- О-”
г+р _ _ I
сн,—с' + :c=n =: СН,—с—н
pH . 3 I
1---------1 C=N
уксусный цианид-
альдегид -ион
О- ОН
I I
сн,—с—н + н,о —•- сн,—с + он
I I
C=N C=N
нитрил а-оксипропионовой
кислоты
На практике для получения циангидрина синильную кислоту добав-
ляют к смеси цианистого натрия и карбонильного соединения. Реак-
ция применяется для получения нитрилов а-окси- и а-аминокислот.
Присоединение гидросульфита натрия (NaHSO3):
ONa ОН ONa
Io __ II
:s—он сн3—с—s=o
О
Из кетонов в эту реакцию вступают только метилкетоны сн3-—С—R
и некоторые кетоны ациклического ряда (например, циклогексанон
/ \=о Реакция обратима, при добавлении кислоты или ос-
нования образуются исходные продукты. Гидросульфитные соедине-
ния — кристаллические бесцветные вещества. Реакция применяется
для выделения и количественного определения оксосоединений. В
пищевой промышленности этим методом устанавливают содержание
оксосоединений в веществах, обусловливающих вкус и аромат многих
пищевых продуктов.
Присоединение металла рганических соединений (см. с. 263).
200
Действие аммиака на альдегиды и кетоны. При взаимодействии
альдегидов с аммиаком образуются альдегидоаммиаки:
NH3 HNH NH
CH3—C^ +\nh3 CH3— c—0“ CH3— C—OH —-CH-CH + H2o
H J, I ацетальдимин
Полученный альдимин легко полимеризуется с образованием аль-
дегидаммиака:
СН3
СН
HN^NH
I I
ЗСН3—CH=NH -> Н3С—НС СН—сн3
\н
Кетоны в этих условиях не взаимодействуют с аммиаком. В более
жестких условиях образуются сложные продукты взаимодействия.
Присоединение спиртов. При взаимодействии альдегидов со спир-
тами образуются ацетали, которые можно рассматривать как простые
эфиры, образованные диолом, содержащим две гидроксильные группы
у одного углеродного атома, и двумя молекулами спирта. В качестве
промежуточного продукта образуется полуацеталь:
О
сн3—+ с2н5он
он о—с2н5
I слон I
СНз—С—ОС2Н5 1--* СНз—С—ОС2Н5
полуацеталь
ацеталь
Реакция протекает при небольшом нагревании, в присутствии сле-
дов минеральной кислоты. Для получения аналогичных производных
кетонов—кеталей применяются специальные методы. Ацетали в отли-
чие от простых эфиров легко разлагаются кислотами на исходные
продукты. Ацетали — приятно пахнущие бесцветные жидкости, хо-
рошо растворимые во многих органических растворителях и плохо —
в воде. Они играют большую роль в процессах биосинтеза сложных
органических соединений, участвуют в формировании запаха ряда
пищевых продуктов. Состав ацеталей вина существенно влияет на его
букет.
Реакции замещения. В реакциях замещения альдегиды
и кетоны обменивают атом кислорода на другие атомы. Взаимодейст-
вие с пентахлоридом и бромидом фосфора'.
/Р
СН3—С< + РС15 -> СН3—СНС12 + Р0С13
ацетальдегид 1, 1-дихлорэтан
201
О Cl Cl
II \/
CH3—С—CH3 + РС15 -* СНз-C—СН3 + РОС13
ацетон 2, 2-дихлорпропан
Взаимодействие с гидроксиламином'.
СН3—+ H2N—ОН -> СН3—C=N—ОН + Н2О
Н гидроксиламин
альдоксим
О N—ОН
II II
СНз—С—СН3 + H2N—ОН -> СН3—С—СНз + Н2О
кетоксим
В реакции применяется солянокислый гидроксиламин NH2OH-HC1,
и выделяющаяся соляная кислота может быть оттитрована, что ис-
пользуется для количественного определения альдегидов и кетонов.
Взаимодействие с гидразином и его производными. Альдегиды и
кетоны с одной молекулой гидразина H2N—NH2 образуют сначала
гидразоны, затем азины:
о
0 сн3-с<^
СНз—с/ + H2N—NH2------------- СНз—C=N—NH2-----------
\Н -н2о | -н2о
ацетальдегид гидразин Н
гидразон
-> СН3—C=N—N=C—СН3
I I
н н
альдазин
О
II
сн3—с—сн3 + h2n—nh2---------
3 2 2 -Н2О
ацетон гидразин
О
II
СНз—с-сн3
---> СН3—C=N—NH2----------->- СН3—C=N—N=C—СН3
I -н*° I I
СНз CH3 СН3
гидразон кетазин
Из производных гидразина для реакции с альдегидами к кетонами ис-
пользуют фенилгидразин С6Н5—NH—NH2; 2,4-динитрофенилгидра-
/°
зин (O2N)2C6H3—NH—NH2; семикарбазид H2N—NH—С . Обра-
\nH2
зующиеся продукты — обычно кристаллические вещества с харак-
терными температурами плавления, что позволяет использовать эту
реакцию для выделения, очистки и разделения альдегидов и кетонов.
202
Отнесение рассмотренных реакций к реакциям замещения условно.
По существу это реакции присоединения (Ad N), в которых участвуют
слабые нуклеофилы NH3; NH2—ОН; NH2—NH2 и другие производ-
ные гидразина с последующим отщеплением от образовавшегося про-
дукта молекулы воды.
Окисление альдегидов и кетонов. Взаимодейст-
вие со слабыми окислителями. Альдегиды легко окисляются даже при
действии слабых окислителей, с образованием одноосновных карбо-
новых кислот с тем же числом атомов углерода-
Z Z
СН3-С + 2 [Ag (NH3)2] + ОН- -> СН3-С + 4NH3 + Н2О + 2Ag
\н \)Н
ацетальдегид уксусная кислота
Серебро при этом тонким слоем покрывает стенки колбы, поэтому
окисление альдегидов аммиачным раствором оксида серебра получило
название «реакции серебряного зеркала». Она используется для из-
готовления елочных украшений и зеркал, является качественной ре-
акцией на альдегиды. Для окисления альдегидов применяется еще
фелингова жидкость (реактив Фелинга, см. с. 358).
Окисление кетонов протекает с трудом при взаимодействии с силь-
ными окислителями при нагревании и сопровождается разрывом угле-
родной цепи по обеим сторонам карбонильной группы (I; II) с обра-
зованием, в общем случае, смеси из четырех кислот (правило Вагнера
и Попова):
I II
I II : [О1 + Н2О
сн3—сн2—с — СН?—сн2—-----------
! i сн2
= i I
СН3
3-гептанон
- СНз-С
\н
уксусная кислота
СНз—сн,—сн2—сн.,—с
\эн
валериановая кислота
Z
- СНз—сн2—с
\эН
пропионовая кислота
масляная кислота
По образовавшимся продуктам реакции можно судить о строении
кетона.
Реакция Канниццаро. Молекула альдегида в присутствии кон-
центрированных щелочей может окисляться за счет другой молекулы
203
альдегида, которая при этом восстанавливается до спирта:
свн5-с + с6н5—с + кон —> свн5—с + СеН5—СН2ОН
\н \н \ок
бензойный альдегид соль бензойной бензиловый спирт
кислоты
Реакция Тищенко. Алифатические альдегиды под действием эти-
лата алюминия в неводной среде образуют сложные эфиры:
О /О
/ (С2Н5О)3А1 II
СН3—С 4-СН3—С------------> СН3—С—О—СН2—СН3
\н \н
ацетальдегид уксусноэтиловый эфир
Реакции альдегидов и кетонов, связанные
с подвижностью атомов водорода в а-п вложе-
нии. Атомы водорода в молекулах альдегидов и кетонов, находя-
щиеся в a-положении к карбонильной группе, обладают большой под-
вижностью (протонизированы) вследствие снижения электронной плот-
ности у углеродного атома карбонильной группы (эффект сопряжения
и индуктивный эффект) и в присутствии оснований взаимодействуют
с галогенами с образованием а-галогенпроизводных:
Z
СН3—С -f- С12 -> СН2С1—С + НС1
\н \н
уксусный альдегид хлор этана ль
О о
II II •
СН3—С—СНз + Вг2 -> СН2Вг—С—СН3-|-НВг
ацетон бромпропанон
Реакция протекает под влиянием нуклеофильных реагентов через
промежуточное образование аниона:
О
II
СН3—С—СН3 + ОН" СНз—С~СН2 + Н.О
О
анион
СНз—с—СН2 + Вг2 -> СН3—С—СН2Вг + Вг’
Первая стадия протекает медленно (лимитирующая стадия), вторая —
быстро. Структура аниона может быть представлена двумя гранич-
ными формулами:
О О’
II _ I
СН3—С—СН2 и СН3—С=СН2
204
Реакции уплотнения. Альдегиды под влиянием щелочей и карбо-
натов щелочных металлов способны вступать в реакции уплотнения за
счет подвижных атомов водорода в a-положении (поликонденсация и
полимеризация). На 1-м этапе под действием щелочей молекула аль-
дегида отщепляет протон, превращаясь в карбанион, который всту-
пает в реакцию со второй молекулой альдегида:
Н2О
карбанион
Обр азовавшийся димер содержит две функциональные группы и
/ \
поэтому получил название альдоль I —с — аль; ОН—ол |, а вид уплот-
\ \н '
нения — альдольной конденсации. В 1861 г. с помощью альдольной
конденсации Бутлеров синтетическим путем впервые получил соеди-
нения, относящиеся к классу углеводов:
Н\
С=О Са (ОН); С6Н12Об
н/
формальдегид гексоза
Кротоновая конденсация. Взаимодействие альдегидов при на-
гревании может идти с потерей молекулы воды. По аналогии с обра-
зовавшимся простейшим продуктом этот вид конденсации получил
название кротоновой конденсации:
/£> /Р
/7 <* /Z
СН3—С + СН3—С----------->
\т_—Н2О
ацетальдегид
/0
СН3—СН=СН—С
\н
кротоновый альдегид
Подобно ацетальдегиду в реакцию кротоновой конденсации вступают
и другие альдегиды, имеющие в a-положении к карбонильной группе
водородные атомы. Реакция проходит через стадию альдольной кон-
денсации и позволяет перейти от предельных альдегидов к непре-
дельным:
205
/Г « он- I
R—СН2—С +R—СН2—С------------> R—СН2—СН—СН —С -------->
\н \н J \н
к
альдоль
----> R—СН2—СН=С—С
-н’° I \н
R Н
непредельный альдегид
Низкомолекулярные кетоны также способны вступать в реакции
альдольной и кротоновой конденсации:
0 0 ОН О
II « II ОН- | ||
сн3—с+нсн2— с—сн3-------->сн3—с—сн?—с—сн3 -*
СН3 сн3
4-окси-4-метил-2-пентанон
----> СН3—С=сн—С—СН3
--н2о | ||
СН3 О
окись мезитила
Взаимодействие альдегидов с фенолами. Фенолформальдегидные
смолы. В кислой и щелочной среде альдегиды вступают во взаимо-
действие с фенолами с образованием фенолоспиртов:
Группа ОН — заместитель 1-го рода, поэтому молекула формаль-
дегида направляется преимущественно в орто- и параположения. При
нагревании фенолоспирты конденсируются с образованием, в зависи-
мости от условий, различных продуктов:
206
Продукт с молекулярной массой до 1000, называемый ризолом, ис-
пользуют в качестве лаков и для приготовления пресс-порошков, в
состав которых входят разнообразные наполнители: древесная мука,
стеклянное волокно, графит и другие материалы. После тщательной
обработки (прессования) получают готовый продукт, обладающий
в зависимости от наполнителя и условий обработки различными свой-
ствами.
Отдельные представители. Применение. Муравьиный альдегид,
формальдегид, метаналь Н—С . Бесцветный газ с резким запахом.
хн
Хорошо растворим в воде. 40%-ный водный раствор называется фор-
малином. Ядовит. При полимеризации с аммиаком образует уротро-
пин C6H12N4. В промышленности формальдегид получают неполным
окислением метана при температуре 600° С:
[О]
сн4----> сн2о + Н2О
(окислители — оксиды азота) и окислительным дегидрированием ме-
тилового спирта в присутствии металлических (медь, серебро) или
оксидных катализаторов:
СН3ОН + 0,5 о?—> н—с + Н2О
ХН
Формальдегид — важный продукт органического синтеза. При-
меняется в производстве полимерных материалов, мономеров каучука,
диолов, фармацевтических препаратов. Формалин используется для
протравливания семян перед посевами, для дубления кожи, хранения
анатомических препаратов. Уротропин применяется в качестве ле-
карственного вещества и для консервирования икры.
Уксусный альдегид, ацетальдегид, этаналь СН3—С<^ . Летучая
жидкость с резким запахом. Смешивается с водой и многими орга-
ническими растворителями во всех отношениях. Ядовит. Получают
по реакции Кучерова, дегидрированием этилового спирта, окисле-
нием этилена:
СН2=СН2 + 0,5 On сн3—cZ
ХН
Катализатор — раствор хлорида меди или палладия. Уксусный аль-
дегид полимеризуется в тример | СН3—cZ I . Применяется для
\ ХН/3
получения пластических масс, уксусной кислоты, уксусного ангид-
рида, этилового спирта, молочной кислоты и других продуктов.
/°
Бензойный альдегид, бензальдегид С6Н5—. Маслянистая жид-
хн
207
кость с запахом горького миндаля. Получают окислением толуола в
присутствии оксидов металлов:
толуол
02:240°
бензойный
альдегид
или омылением бензилиденхлорида С6Н&СНС12. Применяется для син-
теза красителей.
О
Л II
Ацетон, оиметилкетон, пропанон СН3—С—СН3. Бесцветная жид-
кость со специфическим запахом. С водой и многими органическими
растворителями смешивается во всех отношениях. Горюч. Получа-
ется при сухой перегонке древесины, ацетонобутиловым брожением
углеводов. Наиболее экономически выгодные способы: кумольный и
дегидрирование изопропилового спирта. Ацетон — один из важней-
ших органических растворителей, широко применяющийся во мно-
гих отраслях промышленности. Большая реакционная способность
ацетона позволяет широко использовать его в разнообразных синтезах.
Циклогексанон / ^Рименяется для получения капро-
лактама (полупродукт при производстве капрона) и адипиновой кис-
лоты (см. с. 233).
О
Метилфенилкетпон, ацетофенон СН3—С—-С6Н5. Используется для
ароматизации мыла.
Ванилин — производное ароматических альдегидов:
В качестве ароматизатора употребляется при производстве конди-
терских изделий, безалкогольных напитков.
В молекуле ненасыщенных оксосоединений присутствуют две ре-
О
акционноспособные группировки — карбонильная группа с и двой-
ная связь Группировки и их взаимное положение опре-
208
деляют химическое поведение этих соединений. Две ненасыщенные
связи =С и ХчС=О могут находиться у одного углеродного ато-
ма с=С=О (кумулированные связи), могут быть разделены про-
и, наконец, могут
первой группы
стой связью с=С—С=О (сопряженные связи)
быть удалены друг от друга. Представителем
является кетен СНг=С=О, второй — акролеин
Соединения третьей группы обладают всеми свойствами оксосоедине-
ний и алкенов.
Акролеин СН2=СН—С<^ . Бесцветная жидкость с резким, раз-
дражающим запахом, /Кип = 56° С. Ограниченно растворим в воде.
Акролеин может быть получен методом альдольной конденсации
при температуре 300° С в присутствии силиката натрия, нанесенного
на силикагель:
/Р
нсно + СН3—СС -> СН2 —СНо—------------------>сн2=сн—с<
\н I ХН -н2о Хн
он
формаль- ацетальдегид p-оксипропионовый акролеин
дегид альдегид
и каталитическом окислении пропилена:
СН2=СН—СН3
пропилен
акролеин
+ о2 СН2=СН—С
+Н2о
Используется в синтезе глицерина, акрилонитрила (СНг=СН—CN).
Кетен СН2=С==О. Газ, ядовит, /КИп = —41° С, /пЛ = —134,6° С.
Получается пиролизом ацетона:
О
II
СН3—С—СН3 -> СН2=С=О + сн4
Чрезвычайно реакционноспособен. Применяется для получения ук-
сусной кислоты, уксусного ангидрида, сорбиновой кислоты (см. с. 227)
и других продуктов:
8—682
209
Н20
-----------------СН3—СООН
уксусная кислота
z0
сн3соон СНз с\о
СН3—
уксусный ангидрид
сн3—сн=сн—сс
\н
-----------------> СН3—СН=СН—СН=СН—СООН
сорбиновая кислота
КАРБОНОВЫЕ КИСЛОТЫ
Карбоновыми кислотами называются соединения, содержащие в
молекуле карбоксильную группу —С<^ . Карбоксильная группа
\ОН
определяет основность кислоты, поэтому в зависимости от количества
карбоксильных групп различают одноосновные (одна карбоксильная
группа, I; II; III; IV), двухосновные (две карбоксильные группы, V)
и многоосновные кислоты (VI). Карбоновые кислоты бывают алифати-
ческие (насыщенные и ненасыщенные), карбоксильная группа в ко-
торых связана с алифатическим радикалом (I; II; III; IV); алицикли-
ческие (IV), ароматические (III) и т. д.:
СН3—СН2—СООН
пропионовая кислота (I)
СООН
сн2
I
СООН
СН2=СН—СООН СбН5СООН
акриловая кислота (II) бензойная
кислота (III)
С6НПСООН
циклогексанка рбо-
новая кислота (IV/
СООН
НООС—СН2—СН—СН2—СООН
1, 2, З-пентантрикарбоновая кислота (VI)
малоновая кислота (V)
Одноосновные насыщенные и ароматические
карбоновые кислоты
Общая формула одноосновных карбоновых кислот
z°
матических Аг—С<^ , где R и Аг— углеводородные радикалы.
ХОН
Старое название алифатических карбоновых кислот — жирные
, аро-
ЮН
кислоты, или кислоты жирного ряда, так как многие представители
этой группы соединений впервые были выделены из жиров. Наиболее
важные представители приведены в табл. 22.
210
Таблица 22. Одноосновные карбоновые кислоты (С^Щп^СООН; АгСООН)
Название Формула Температура пла вления, °C Температура кипения, °C Показатели пре- ломления РКа (Н,О; 20°С)
Муравьиная кислота-, метановая н—соон 8,4 100,7 1,3714 3,75
Уксусная кислота, этановая сн3—соон 16,6 118,1 1,3720 4,76
Пропионовая кислота, пропа- новая сн3—сн2—соон —22,0 141,0 1,3874 4,87
Масляная кислота, бутановая СН8—(СН2)2—соон —7,9 163,5 1,3980 4,82
Валериановая кислота» пента- новая СН8—(СН2)з—СООН —34,5 186,3 1,4085 (25°С) 4,86
Капроновая кислота 5 гексановая СН8—(СН2)4—соон —3,9 205,8 1,4163 (25°С) 4,88
Лауриновая кислота, додекано- вая СНз—(СН2)10—соон 44,2 298,9 1,4191 (80°С) —
Пальмитиновая кислота» гек- садекановая СНз—(СН2)14—соон 64,0 390 »0 (с раз- ложением) 1,4309 (70°С) —
Стеариновая кислота» ектаде- ка новая СН3—(СН2)1е—соон 69,4 360,0 1,4335 (70°С) —
Бензойная кислота СвН6СООН 122,0 249,0 1,5397 4,17
Изомерия. Номенклатура. Изомерия насыщенных карбоновых
кислот определяется строением углеводородного радикала; аромати-
ческих — строением радикала и положение^м карбоксильной группы.
Для наименования карбоновых кислот широко применяются триви-
альные названия, которые часто связаны с первыми источниками их
получения: муравьиная, уксусная, стеариновая. По ИЮПАК в основе
наименования кислот положено название углеводорода с тем же чис-
лом атомов углерода, к которому прибавляется окончание «овая»,
нумерация начинается с атома углерода карбоксильной
группы:
4 3 2 1
СН3—СН2—СН2—СООН
СНз
3-метилбутановая кислота
Нахождение в природе. Способы получения. Карбоновые кислоты
широко распространены в природе как в свободном состоянии, так
и в виде структурных компонентов ряда природных органических
соединений. Несмотря на это их получение из природных источников,
как правило, большого практического значения не имеет, за исклю-
чением кислот С6 и С30 (см. с. 211, 394). Основное количество карбо-
новых кислот получают синтетическими способами.
Окисление органических соединений. Окисление алканов, алкенов,
ароматических углеводородов, спиртов, альдегидов и кетонов с об-
разованием карбоновых кислот было подробно рассмотрено ранее.
Эти методы имеют большое практическое значение и широко применя-
ются в технике и лабораторной практике.
Оксосинтез. Карбоновые кислоты могут быть получены при взаимо-
действии алкенов с оксидом углерода и водой:
R—СН2—СН=СН2 + СО + Н2О-----
алкен
R—СН2—СН2—СН2—СООН
СООН
I
R—СН2—СН—СН3
карбоновые кислоты
Реакция протекает при температуре 50—100° С, давлении до
10 • 105 Па, катализатор — сильные кислоты (H2SO4, Н3РО<).
Промышленное получение карбоновых кислот оксосинтезом мо-
жет быть осуществлено и в две стадии: синтез альдегидов из алкенов,
оксида углерода и водорода (см. с. 196) и последующее окисление их
в кислоты.
Гидролиз нитрилов. При нагревании нитрилов (R— C=N) и
Аг—C=N с водными растворами щелочей или кислот они гидроли-
зуются, образуя кислоты и аммиак:
212
СН3С1 + NaCN -------►
J —NaCl
метилхлорид
ацетонитрил
HCI
СН3—СГ + NH4C1
ХОН
уксусная
кислота
NaOH
сн3—с' + NH4OH
xONa
уксуснокислый
натрий
CH3—C=N или С°Н~К
н2о
амид уксусной
кислоты
+ NaCN ——1
—NaCl
фенилацетонитрил
+Н2О[н+] или COH~],t
амид фенилуксусной
кислоты
бензилхлорид
НС1
+ NH4C1
Н2О
NaOH
фенилуксусная
кислота
фенилуксуснокислый
натрий
Гидролиз тригалогенпроизводных углеводородов, содержащих ато-
мы галогена у одного углеродного атома:
CbH5CCl3 + 2НОН----------* С6Н5СООН + ЗНС1
бензотрихлорид бензойная кислота
Гидролиз сложных эфиров (триацилглицеринов, см. с. 393). Ре-
акция широко применяется для получения высокомолекулярных
(С14—С22) карбоновых кислот, имеющих, как правило, прямую угле-
родную цепочку; гидролиз протекает ступенчато:
О
СН2—О—С—R
СН—ОН
О
Н,О
[Н+]; [он-] СН—О—С—Ri + RCOOH--
СН—О—С—Rj + НОН-----------
I или липаза
о
сн2—о—с—r2
сн2—о—с—r2
о
триацилглицер ин
о
диацилглице^ин
213
сн2—он сн2—он
I н2о СН-ОН + RiCOOH
СН—О—С—Ri + R2COOH-------> I
|| сн2—он
О глицерин
СН2—он
мо но ацил гл иц ери н
(RCOOH; RjCOOH; R2COOH — карбоновые кислоты).
Получение из металлорганических соединений (см. с. 261). При
действии на магнийорганические соединения (реактив Гриньяра) ди-
оксида углерода образуются смешанные соли карбоновых кислот, ко-
торые при разложении минеральными кислотами превращаются в кар-
боновые кислоты:
R—Mgl + СО2-------> R—С<
X)MgI
реактив смешанная соль
Гриньяра
zO
R—С< 4- НС1-----------> R—СООН + MgCll
^OMgl
карбоновая
кислота
Декарбоксилирование двухосновных кислот (см. с. 230).
Синтез с помощью малонового эфира (см. с. 231).
Физические свойства. Первые три кислоты (Ct—С3) — подвижные жидкос-
ти с резким запахом, начиная с С4 (масляная кислота) — маслянистые жидкости
с крайне неприятным запахом (запах прогорклого сливочного масла). С С1о —
твердые вещества, без цвета и запаха (см. табл. 22). Низкомолекулярные кислоты
хорошо растворимы в воде, с увеличением молекулярной массы растворимость
резко падает. На физические свойства карбоновых кислот большое влияние ока-
зывает их способность к ассоциации с образованием линейных или циклических
димеров за счет водородных связей. (Энергия водородной связи ~ 29,33 кДж/моль;
длина связи 1,74 нм.)
R^C\ //C~R
Ю—h..«oz
0,100
циклический димер
R R
I I
С С
\ 0,100 0,174 \
О О — Н--0 О—Н
линейный димер
Способностью к ассоциации и прочностью водородных связей объясняется
относительно высокая температура кипения кислот по сравнению не только с
углеводородами, имеющими столько же атомов углерода в молекуле, но и со
спиртами.
Химические свойства. Химическое поведение кислот, в первую
очередь, связано с наличием в их молекуле карбоксильной группы, а
также со строением углеводородного радикала. Рассмотрим распре-
деление электронной плотности в карбоксильной группе молекулы не-
214
диссоциированной карбоновой кислоты. Электронная плотность л-
связи в группе \с=О смещена в сторону атома кислорода. Вслед-
ствие этого у атома углерода создается недостаток электронной плот-
ности и он притягивает к себе неподеленные электронные пары атома
кислорода гидроксильной группы. В результате электронная плот-
ность О—Н-связи смещается в сторону атома кислорода, водород ста-
новится подвижным и приобретает способность отщепляться в виде
протона:
R—С
<5 +
В водном растворе карбоновая кислота диссоциирует с образованием
карбоксилат-аниона и иона гидроксония:
zO
R—С< 4- Н2О R—С< + Н3О+
хо—н хг
карбоновая карбоксилат-
кислота анион
В образующемся карбоксилат-анионе отрицательный заряд не лока-
лизуется на одном атоме кислорода:
III
Таким образом, карбоксилат-ион является мезомерным ионом.
Делокализация электронов сопровождается выигрышем энергии,
поэтому мезомерная структура (III) энергетически более выгодна,
чем каждая из граничных структур (I и II). Выигрыш энергии («мезо-
мерная стабилизация») при образовании карбоксилат-аниона объяс-
няет причину диссоциации карбоновых кислот: карбоксилат-анион
обладает меньшей энергией, а следовательно, более устойчив, чем сама
карбоновая кислота. Мезомерная структура карбоксилат-аниона под-
тверждается данными рентгеноструктурного анализа: в молекуле му-
равьиной кислоты одна связь углерод—кислород имеет длину 0,136 нм
(простая связь), а другая — 0,123 нм (двойная связь), в формиате
натрия обе связи одинаковы — 0,127 нм.
Одноосновные карбоновые кислоты, за исключением муравьиной
кислоты, — слабые кислоты (см. табл. 22). Степень диссоциации их
в водных растворах невелика (/<а ~ 10"5; р/<а = 4,7—5) и зависит
от характера радикала, связанного с карбоксильной группой. Алкиль-
ные радикалы, обладающие +/-эффектом, понижают кислотность (I).
Введение электроноакцепторных атомов или групп (обладающих
—/-эффектом) в радикал, особенно в соседнее положение к карбо-
ксильной группе, увеличивают силу кислот (II):
215
CHj—
сн2
/*о
г+v
X—CH2-«-Cv
Группы с —/-эффектом: NRaCORCF; I<Br<CKF; а также не-
насыщенные группировки и ядро бензола:
CR=CR2 < СвН5 < С = CR.
Группы с 4-/-эффектом:
сн3<сн2—сн3 < с—сн
^Нз
уСН3
С—СН3 ;
\зн3
увеличением числа атомов углерода в радикале кислоты константы
диссоциации кислот уменьшаются. Влияние заместителей на кислот-
ность хорошо видно при сравнении рКа уксусной и хлоруксусной
кислот:
Кислота рКа
СН3СООН 4,76
уксусная
СН2С1СООН 2,85
хлоруксусная
Кислота рКа
СНС12СООН 1,25
дихлор уксусная
СС13СООН 0,66
грихлоруксусная
Пониженная электронная плотность у углеродного атома карбоксиль-
ной группы (+8) вызывает смещение электронной плотности связи
С—С и ее понижение (+8J у углеродного атома, находящегося в а-
положении по отношению к карбоксильной группе. Атомы водорода,
связанные с этим атомом углерода, приобретают повышенную по-
движность:
н Со5‘
R-?—с\
I Ло—н
н
а
Основные химические превращения карбоновых кислот связаны с
проявлением кислотных свойств (образование солей), заменой группы
ОН в карбоксильной группе кислоты на разнообразные группировки
(С1; Вг; NH2 и др.) с образованием функциональных производных
карбоновых кислот (см. с. 235); реакциями углеводородных радикалов;
декарбоксилированием кислот. Бензольные ядра ароматических кар-
боновых кислот участвуют во многих превращениях, характерных
для аренов, причем карбоксильная группа выступает в роли мета-
ориентанта (заместитель 2-го рода). В определенных условиях кар-
боксильная группа способна восстанавливаться в альдегидную или
спиртовую группу.
Кислотность карбоновых кислот. Диссоциация
карбоновых кислот (см. с. 215).
Образование солей. Карбоновые кислоты при взаимодействии с
216
щелочными металлами, оксидами и гидроксидами,
аммиаком образуют соли:
карбонатами,
2СН3СООН + Са----------> (СН3СОО)2Са + Н2 f
уксусная уксуснокислый
кислота кальций
CHjCOOH + NaOH -------CH3COONa 4- Н2О
уксуснокислый
натрий
^^хСООН [Tjj + NaHCO3 — ^C^/COONa + СО2 4- HjO
бензойная кислота бензоат натрия
СН3СООН + NH3 -------CH3COONH4
уксуснокислый
аммоний
Соли карбоновых кислот легко гидролизуются, растворы солей ме-
таллов первой и второй группы имеют щелочную реакцию.
Реакции по карбонильному углероду. В ходе
этих реакций нуклеофил атакует карбонильный углерод с последую-
щим разрывом С—О-СВЯЗИ;
8—
R С/0
к—
рон
Z
нуклеофил
В зависимости от характера реагента образуются сложные эфиры,
ангидриды, галогенангидриды, происходит восстановление карбо-
новых кислот гидридами.
Образование сложных эфиров. Реакция этерификации. При взаимо-
действии карбоновых кислот со спиртами в присутствии сильных ми-
неральных кислот образуются сложные эфиры карбоновых кислот:
/ н + /
R—С + R,OH R—С' + Н2О
ЮН 'ORi
карбоновая сложный
кислота эфир
СООН
бензойная,
кислота
+ сн3он
ООСНз
метилбензоат
+ Н2О
метанол
Механизм реакции этерификации может быть представлен в следую-
щем виде:
5—
R ‘с/0
К—L.
карбоновая протонированная
кислота кислота (1)
Иретон, присоединившись к карбонильному кислороду, активирует
углеродный атом карбонила по отношению к нуклеофилам. Вместо
частичного положительного заряда 8+ в карбоновой кислоте появля-
ется целый положительный заряд в протонированной кислоте (I). За-
тем он атакуется молекулой спирта (нуклеофил):
+ /ОН .. /ОН
R—С< + R'— О—Н R—Г z
п
Образовавшийся промежуточный продукт (II) может быстро и обра-
тимо превращаться в продукт Па:
ОН он
R—С—OH R—С—ОН2
I I +
R'—О—Н R'-O
+
II II а
Отрыв молекулы воды, а затем потеря протона приводит к сложному
эфиру:
/ОН /° +
R—С\ R—С—OR' + Н3О
сложный эфир
R'—О +
II а
Реакция обратима, катализатор — ион водорода — необходим и для
обратной реакции — гидролиза сложного эфира. Скорость реакции
зависит от строения спирта и силы кислоты (см. с. 166) и уменьшается
с увеличением размеров R и R' вследствие пространственных затруд-
нений.
Образование ангидридов (см. с. 238).
Образование хлорангидридов (см. с. 236).
Восстановление карбоновых кислот. В лабораторных условиях кар-
боновые кислоты восстанавливаются в спирты под действием алюми-
нийгидрида лития LiAlHi:
zO
4RC/ + 3LiAlH4 -> [(RCH2O)4A1] Li + 2LiA102 + 4H2
XOH алкоголят
карбоновая
кислота
[(RCH2O)4A1] Li + 4НС1 -> 4R—СН2ОН + А1С1, + LiCl
спирт
218
Карбоновые кислоты восстанавливаются (с трудом!) каталитиче-
ски или действием натрия в спирте. В промышленности часто восста-
навливают их сложные эфиры. Реакция применяется для получения
из высокомолекулярных карбоновых кислот, образующихся при
гидролизе жиров или окислении парафинов, высокомолекулярных
спиртов.
Реакции замещения в углеводородном ра-
дикале кислоты. Галогенирование алифатических кислот в а-
положение. Карбоновые кислоты хлорируются и бромируются. В при-
сутствии небольших количеств фосфора; РС13; иода замещение проис-
ходит исключительно в а-положение:
zO РС13 zO
r—сн2—c<f + Cl2 ----> r—сна—c<f + на
\он хон
карбоновая кислота хлорзамещенная кислота
Реакция протекает по ионному механизму. При свободнорадикальном
галогенировании избирательность замещения (a-положение) не соб-
людается.
Замещение атомов водорода бензольного ядра в ароматических кар-
боновых кислотах. Группа СООН является заместителем 2-го рода,
направляя вхождение заместителя в реакциях электрофильного
замещения в метаположение:
NOj
бензойная -м-нитробензойнаЯ
кислои кислота
Вхождение группы NO2 затруднено, реакция протекает при нагре-
вании.
Окисление карбоновых кислот. Одноосновные карбоновые кислоты,
за исключением муравьиной кислоты, устойчивы к действию окисли-
телей. Относительно легко окисляются карбоновые кислоты, имею-
щие третичный углеродный атом в a-положении, при этом образуются
а-оксикислоты:
Н ОН
1 /Р Г°1 1
СН3—С—С -------------> СН3—С—
СН3 Хч°Н СН3 0Н
изомасляная кислота а-оксимасляная кислота
В биологических системах под действием ферментов (оксидазы) окис-
ляется (J-углеродный атом (Р-окисление):
,0
R-CH2—СН2—C<f
\он
О; оксидаза
R—СН—СН
I
ОН
карбоновая кислота
р-оксикарбоновая кислота
319;
Декарбоксилирование карбоновых, кислот. Сплавление солей кар-
боновых кислот со щелочами с образованием алканов было рассмотре-
но ранее, аналогично ведут себя соли ароматических кислот:
i°
C6H5COONa + NaOH -> СбНб + Na2CO3
бензоат натрия бензол
Сухая перегонка солей карбоновых кислот (см. с. 195).
Отдельные представители. Муравьиная кислота НСООН. Содер-
жится в соке крапивы, выделениях муравьев, хвое, фруктах. Жид-
кость с резким запахом, при попадании на кожу вызывает ожоги (кра-
пивные ожоги). Относится к средним по силе кислотам (р/<а 3,7);
соли и эфиры муравьиной кислоты — формиаты. Основной техни-
ческий способ ее получения — пропускание оксида углерода через
увлажненный гидроксид натрия:
120—150°С
СО + NaOH -----------> HCOONa
10,ЫО6 Па
2HCOONa + H2SO4 2HCOOH + Na2SO4
Муравьиная кислота вступает во все реакции, характерные для
карбоновых кислот; в то же время она обладает рядом особенностей,
связанных с присутствием в ее молекуле атома водорода вместо угле-
водородного радикала. В отличие от других карбоновых кислот она
легко окисляется с образованием воды и диоксида углерода
[О]
НСООН —> Н2О + СО2
что позволяет применять ее в качестве восстановителя. В пищевой
промышленности это свойство муравьиной кислоты используется
при получении никелевых катализаторов, в частности для гидроге-
низации жиров (см. с. 415):
200°С
(HCOO)2Ni----> Ni + СО. + Н2
При нагревании с концентрированной серной кислотой муравьи-
ная кислота разлагается:
HsSO4
НСООН-------> со + Н2О
Муравьиная кислота применяется в текстильной промышленности
для протравливания тканей при крашении, для дубления кожи, в
пищевой — для дезинфекции бродильных чанов и бочек, в медицин-
ской — для получения «муравьиного спирта», в химической — для
синтеза щавелевой кислоты.
Уксусная кислота СН3—СООН. Бесцветная жидкость с резким
специфическим запахом. Безводная уксусная кислота называется
«ледяной», так как при замерзании (+16,6° С) внешне она напоминает
лед. Уксусная кислота смешивается с водой и многими органическими
растворителями в любых соотношениях. Она широко распространена
в природе как в свободном состоянии, так и в виде производных (глав-
220
ним образом сложных эфиров). Составляет до 85% всех органических
кислот в зерне пшеницы и кукурузы. Соли и эфиры уксусной кислоты
называются ацетатами.
Старый способ получения уксусной кислоты, известный людям
еще в глубокой древности, — уксуснокислое брожение водных раст-
воров этилового спирта. Он не потерял своего значения и в настоящее
время для получения «пищевой» уксусной кислоты. При этом этило-
вый спирт окисляется в уксусную кислоту с помощью особой группы
т^икроорганизмов, называемых уксуснокислыми бактериями. Сум-
марное уравнение реакции брожения имеет следующий вид:
СН3—СН2ОН + О2 -> СН3СООН + Н2О
Концентрация образовавшегося таким образом «уксуса» — 5—7%,
из него перегонкой выделяют 70—80%-ную уксусную эссенцию.
Для получения уксуса используют продукты брожения виноград-
ного и фруктового соков, пиво. Не потерял своего значения и другой
способ — сухая перегонка отходов деревоперерабатывающей про-
мышленности. Образующийся при этом наряду с древесным дегтем
водный слой содержит до 10% уксусной кислоты, откуда она выделя-
ется в виде уксуснокислого кальция. При обработке его серной кис-
лотой получается древесный уксус. Основное количество уксусной
кислоты вырабатывают, однако, синтетическим путем. Основные пути:
а) каталитическое окисление ацетальдегида:
,0
2СН3—+ О2 -> 2СН3—СООН
чн
ацетальдегид уксусная кислота
б) синтез из метанола и оксида углерода:
сн3он + со -> СН3СООН
Катализатор — трифторид бора, фосфорная кислота, хлориды цинка
и меди. Температура реакции 200—300°С, давление до 1010 • 105 Па;
в) в СССР разработан процесс окисления н-бутана при темпера-
туре 145° С, и давлении 50,5 • 105 Па с выходом уксусной кислоты
до 80%.
Уксусная кислота — один из важнейших химических продуктов,
применяется в качестве растворителя, при производстве искусствен-
ных волокон (ацетатный шелк), красителей, сложных эфиров, лекар-
ственных препаратов. Широкое применение нашла уксусная кислота
в пищевой промышленности в качестве консервирующего средства при
производстве маринадов и как вкусовая добавка.
Масляная, бутановая кислота СН3(СН2)2СООН. Жидкость со спе-
цифическим запахом. Содержится в виде глицеридов в сливочном и
некоторых других маслах. Получают маслянокислым брожением угле-
водосодержащего сырья и окислением н-бутилового спирта. Соли и
эфиры — бутираты.
Высшие жирные кислоты алифатического ряда. Среди них особое
значение принадлежит жирным кислотам С12—С18 с четным числом
221
углеродных атомов- СцН^СООН — лауриновая (додекановая);
С15Н27СООН — миристиновая (тетрадекановая); С13Н31СООН —
пальмитиновая (гексадекановая) и С17Н35СООН — стеариновая (ок-
тадекановая) кислоты. Они широко представлены в природе как в сво-
бодном состоянии, так и в виде структурных компонентов различных
групп липидов (см. с. 390). Получаются главным образом гидролизом
жиров, окислением углеводородов.
Бензойная кислота С6Н5СООН. Типичный и наиболее важный
представитель одноосновных ароматических кислот. Белое кристал-
лическое вещество. Распространена в природе в свободном состоянии
и в виде производных. Получают окислением толуола при 140°С (ка-
тализатор — соли кобальта):
У\/сн> /\/соон
I || + 1,50, - | || + Н.О
толуол бензойная кислота
Степень диссоциации ароматических кислот выше, чем кислот
алифатического ряда; бензойная кислота значительно сильнее, чем
уксусная. Она вступает в те же реакции, что и уксусная, а кроме
того, в реакции, характерные для аренов. Соли и эфиры бензойной
кислоты — бензоаты.
Бензойная кислота и ее производные используются в синтезе кра-
сителей, фармацевтических препаратов. Сложные эфиры бензойной
кислоты и одноатомных спиртов (от метилового до амилового) идут на
изготовление духов и одеколонов. Широкое применение бензойная кис-
лота и ее натриевая соль нашли в пищевой промышленности для кон-
сервации пищевых продуктов. Именно присутствием бензойной кислоты
в мякоти клюквы, брусники, рябины (наряду с некоторыми другими
соединениями) объясняется их способность к длительному хранению.
Одноосновные ненасыщенные кислоты
В молекуле одноосновной карбоновой кислоты может содержаться
одна или несколько двойных или тройных связей*. Общая формула
мононенасыщенных одноосновных карбоновых кислот С^Н^^СООН,
диненасыщенных СПН2П_3СООН и т. д. Важнейшие представители
этой группы соединений приведены в табл. 23.
Изомерия. Номенклатура. Структурная изомерия одноосновных
ненасыщенных кислот связана со строением углеродного скелета и
положениехМ двойных связей. У мононенасыщенных кислот двойная
связь может занимать различное положение по отношению к кар-
боксильной группе, у ди- и полиненасыщенных одноосновных кислот
— еще и различное положение по отношению друг к другу. Для не-
* Кислоты, содержащие в радикале тройные сьрзи, в книге не рассматрива-
ются.
222 Ф
Таблица 23, Одноосновные ненасыщенные карбоновые кислоты
Название Формула Температура плавления, °C Температура кипения, Показатель преломления „20 nD Р*а (Н2О| 20°С) х
Акриловая кислота, 2-пропеновая СН2=СН—СООН 13,0 142 1,4224 4*26
Кротоновая кислота, 2-бутеновая сн3-сн==сн—соон 72 189 1,4456 —
Винилуксусная кис- лота , 3-бутеновая сн2=сн—сн2—соон —39 163 1,4257 (15°С) 4,69
Метакриловая кисло- та, 2-метил-2-про- пеновая сн2=с—соон СНз 16 163 1,4314 — -
Олеиновая кисло1а СН3(СН2)7СН=СН(СН2)7СООН 14 223 (при 13,3-102 Па) 1,4582 —
Сорбиновая кислота СН3СН=СН—сн=снсоон 134 2z8 — —
го ьо СО Линолевая кислота Линоленовая кислота Коричная кислота (транс) с17н31соон С17Н29СООН СвН5—СН=СН—соон —5,2 —11 До —11,3 133 230 (при 21,28-102Па) 232 (при 28,6-102 Па) 300 1,4699 1,4800 4,44
А
насыщенных кислот существует
цис-, транс-) изомерия:
Н—С—СН3
II
Н—С—СООН
•цис-к рото нов а я
кислота
пространственная (геометрическая
СН3—С—Н
II
н—с—соон
транс-кротонов ая
кислота
При наименовании ненасыщенных кислот широко применяются
тривиальные названия (акриловая, сорбиновая кислота). По номен-
клатуре ИЮПАК при построении названия кислоты сохраняются
те же принципы, что и при наименовании алкенов и кислот:
СН3—СН=СН—СООН 2-бутеновая кислота
Нумерация начинается с атома углерода карбоксильной группы.
Способы получения. Для получения ненасыщенных карбоновых
кислот обычно используют соединение, уже содержащее одну из функ-
циональных групп: карбоксильную группу или двойную связь. В пер-
вом случае (присутствует карбоксильная группа) в молекуле создают
вторую функциональную группу (двойная связь); во втором случае
(присутствует двойная связь) создают карбоксильную группу. Многие
из ненасыщенных кислот, главным образом высокомолекулярные кис-
лоты, содержащие от 12 до 22 атомов углерода, одну или несколько
двойных связей и имеющие определенную пространственную конфигу-
рацию, получают гидролизом липидов (см. с. 390). Отдельные из не-
насыщенных карбоновых кислот получают оригинальными методами.
1. Получение из галогенкарбоновых кислот (дегидрогалогениро-
вание). При действии спиртовых растворов щелочей на галогензаме-
щенные карбоновые кислоты
СН3—СН2—СН—СООН + 2NaOH -> СН3—СН=СН—СООН + NaBr + Н2О
| 2-бутеновая кислота
Вг
2-бромбутановая кислота
получают ненасыщенные карбоновые кислоты. Особенно легко де-
гидрогалогенируются а-галогенкарбоновые кислоты.
2. Дегалогенирование дигалогензамещенных карбоновых кислот,
содержащих атомы галогена у соседних углеродных атомов:
СН2Вг—СНВг—СООН + Zn -> СН2=СН—СООН + ZnBr2
2, 3-дибромпропановая акриловая кислота
кислота
3. Дегидратация р-оксикислот (см. с. 245).
В реакциях 1, 2, 3 создается двойная связь при наличии карбоксиль-
ной группы.
4. Окисление непредельных альдегидов:
z° [О] /уО
Н2С=СН—С" ~7/
ХН
акролеин акриловая кислота
224
5л Получение непредельных карбоновых кислот из алкенов;
cis
СН2=СН—СН3 —СН2=СН—СН2С1 + НС1
пропен 450°С аллилхлорид
СН2=СН—СН2 Cl + KCN -> СН2=СН—СН2—CN + КС1
нитрил винилуксусной
кислоты
2Н2О
СН2=СН—СН—CN > СН2=СН—СН2—СООН
3-бутеновая кислота
В реакциях 4, 5 в молекуле непредельного соединения создается
карбоксильная группа.
6. Конденсация Кневенагеля. При взаимодействии малонового
эфира (см. с. 231) и карбонилсодержащих соединений в присутствии
пиридина за счет конденсации кротонового типа получаются a, (J-
мононенасыщенные кислоты:
СООС2Н5 СН3 СН3
I \ c8hbn \
СН2 + С=О--------------------------------> С=СН—СООН
| / —Н2О; —2С2НбОН; —СОя /
СООС2Н5 СН3 СН3
малоновый З-метил-2-бутеновая
эфир кислота
Физические свойства. Ненасыщенные одноосновные кислоты — жидкости»
или твердые вещества с большей плотностью, чем соответствующие ненасыщен-
ные кислоты; низшие — со специфическим запахом, растворимы в воде; высшие
без запаха и нерастворимы в воде.
Химические свойства. Химическое поведение ненасыщенных одно-
основных кислот связано с наличием в их молекуле двух активных
группировок: карбоксильной группы и двойной связи. Они вступают
во все реакции, характерные для кислот (образование солей, сложных
эфиров, хлор ан гидридов и т. д.), а в связи с наличием двойной связи —
в реакции присоединения, окисления, полимеризации.
Введение в молекулу двойной связи увеличивает силу кислоты,
поэтому степень диссоциации одноосновных ненасыщенных кислот
выше, чем соответствующих насыщенных (—/-эффект). Влияние двой-
ной связи снижается по мере удаления ее от карбоксильной группы,
наиболее сильно —/-эффект проявляется в аф-непредельных кис-
лотах. В то же время карбоксильная группа, являясь сильным ак-
цептором электронов, снижает активность двойной связи в реакциях,
электрофильного присоединения и повышает в реакциях нуклеофиль-
ного присоединения.
Кислоты, в молекуле которых двойная связь находится между
2-м и 3-м углеродными атомами, отличаются некоторыми особенностя-
ми в своем химическом поведении из-за сопряжения между двойной,
связью и карбоксильной группой. Например, при присоединении га-
логенводородов водород направляется к наименее гидрогенизирован-^
ному атому углерода (1,4-присоединение):
225
+8 z—ч ро”5 >>
СН2=СН—С + НС1 —-- сн2а—сн2—с
хон ХОН
акриловая кислота
3-хлорпропановая кислота
Это происходит и при присоединении воды, спирта, аммиака:
H2SO4; 100°С
СН2=СН—СООН + Н2о -------------> сн2—сн—соон
акриловая кислота | |
ОН Н
Р-оксипропионов ая
кислота
Отдельные представители. Акриловая, пропеновая кислота
СН»=СН—СООН. Бесцветная жидкость с резким запахом. Смешива-
ется с водой в любых соотношениях. Получают гидролизом акрило-
нитрила:
2H2O; [Н+]
СН2=СН—CN----------* СН2=СН—СООН + NH3
акрилонитрил акриловая кислота
взаимодействием ацетилена с оксидом углерода и водой (реакция Рен-
кэ):
сн=сн + СО + Н2О -> СН2=СН—соон
акриловая кислота
Из окиси этилена и синильной кислоты:
[Н+]; Н2О
Н2С—СН2 + HCN -> H2C—СН2—CN---------->
о он
окись этиленциангидрин
этилена
-> НОС—сн2—СООН
‘I
он
t°
----> сн2=сн—соон
Н2О акриловая
кислота
₽-оксипропионовая
кислота
Широкое применение нашли эфиры акриловой кислоты, полимери
задней которых получают ценные полимерные материалы (органиче-
ское стекло).
Метакриловая, метилпропеновая кислота СН2=С—СООН.
СН3
Бесцветная жидкость. Получают в промышленности из ацетона:
Н3СЧ
>С=О + HCN ->
H3CZ
ацетонциангидрин
HSO; (H2SO4)
ацетон
226
-н,о
а-оксиизомасляная
кислота
сн2=с—соон
СНз
метакриловая
кислота
Большое техническое значение имеет метиловый эфир метакри-
ловой кислоты (метилметакрилат), полимеризацией которого полу-
чают «органическое стекло» (плексиглас):
СООСНз
I
п СН2=С
I
СНз
метилметакрилат
I
—СН,—С—
I
сн3
СООСНз-|
полиметил метакр плат
п
Сорбиновая, 2,^-гексадиеновая кислота
СН3—СН=СН—СН=СН—СООН. Кристаллическое вещество. Бу-
дучи безвредной, является отличным антисептиком. Применяется
для консервирования мясных и рыбных изделий, сыра, фруктовых
компотов, плодоягодных пюре, соков, овощей.
Олеиновая, 9-октадеценовая кислота
СНз—(СН2)7—СН=СН—(СН2)7—СООН
цис
Маслянистая жидкость без цвета и запаха. Широко распростра-
нена в природе, входит в состав почти всех жиров. Природная оле-
иновая кислота является цис-изомером. Во многих жирах ее содержа-
ние доходит до десятков процентов, а в некоторых является основным
компонентом. Так, в масле арахиса ее содержание (%) составляет
50—80, в оливковом — 70—85. Получают гидролизом жиров (см.
с. 396) с последующим выделением ее из образовавшейся смеси кис-
лот. Олеиновая кислота является не только важнейшим компонентом
пищи, но и находит широкое применение в технике. Олеиновая кис-
лота используется в качестве пластификатора при производстве
пластмасс, для производства мыла, в аналитической химии.
Линолевая, 9,12-октадекадиеновая кислота
СН3—(СН2)4—СН=СН—СН2—СН=СН—(СН2)7—соон
цис цис
Светло-желтая маслянистая жидкость, нерастворима в воде, хо-
рошо растворима в ряде органических растворителей. Природная,
линолевая кислота является цисизомером. Очень широко распростра-
нена в природе, входит в состав триглицеридов, фосфолипидов (см.
с. 404), многих масел и жиров. Содержание ее в подсолнечном и ку-
курузном маслах достигает 60—70%. Масла, содержащие линолевую
кислоту, могут применяться в производстве олиф, красок и лаков.
Принадлежит к числу незаменимых жирных кислот, так как человек
и большинство животных лишены способности ее синтезировать. По-
этому для полноценного питания линолевая кислота должна входить,
в состав рациона.
227
Линоленовая, 9,12,1 Ъ-октадекатриеновая кислота
СН3—СН2—СН=СН—СН2—СН=СН—СН2—СН=СН—(СН2)7—СООН
цис цис цис
Бесцветная жидкость, входит в состав глицеридов многих жиров и
масел. В льняном масле ее содержание достигает 30—50%. Принад-
лежит к числу незаменимых для человека и животных жирных кислот.
Двухосновные кислоты
В молекуле двухосновных (дикарбоновых) кислот содержатся две
карбоксильные группы. В зависимости от строения остальной час-
ти молекулы различают насыщенные, ненасыщенные, ароматические
и другие двухосновные кислоты. Наиболее важные представители
приведены в табл. 24.
Изомерия. Номенклатура. Структурная изомерия насыщенных
алифатических двухосновных карбоновых кислот обусловлена строе-
нием углеродного скелета, ненасыщенных — строением скелета и по-
ложением двойных связей. У ненасыщенных кислот имеет место гео-
метрическая изомерия:
Н—С—СООН ноос—С—н
II II
Н—С—СООН Н—С—СООН
малеиновая кислота фумаровая кислота
(цисизомер) (трансизомер)
У ароматических двухосновных кислот изомерия также связана со
взаимным положением карбоксильных групп в бензольном ядре:
I II
\/\зоон
фталевая
СООН
I II
\/\соон
изофталевая
СООН
терефталевая
.Для наименования двухосновных кислот широко применяются три-
виальные названия (щавелевая, малеиновая, фталевая и др.), по
ИЮПАК родовое окончание «диовая».
Способы получения. Для получения двухосновных кислот исполь-
зуют принципиально те же методы, что и для одноосновных, с той
разницей, что в молекуле необходимо создать не одну, а две кар-,
боксильные группы (окисление диолов, гидролиз динитрилов, окис-'
ление оксикислот с первичной спиртовой группой). '
Физические свойства. Двухосновные кислоты — бесцветные кристалличес-
кие вещества. Кислоты счетным числом углеродных атомов имеют более высокие’
температуры плавления, чем кислоты с нечетным числом углеродных атомов.
Двухосновные кислоты хорошо растворимы в воде, растворимость уменьшается Ci
увеличением молекулярной массы. Кислоты с нечетным числом углеродных ато-|
228 i
Таблица 24. Двухосновные карбоновые кислоты
Название Формула Температура плавления °C (Н2О; 20°О) 4 Р*а а2
Щавелевая кислота, этандиовая ноос—соон 189,5 1,27 4,27
Малоновая кислота, пропандиовая ноос—сн2—соон 135,6 2,86 5,70
Янтарная кислота, бутандиовая ноос—сн2—сн2—соон 185 4,21 5,64
Глутаровая кислота, пентандиовая НООС—(СН2)з—соон 97,5 4,34 5,27
Адипиновая кислота, гександиовая НООС—(СН2)4—соон 152 4,41 5,28
Малеиновая кислота (цис-) Фумаровая кислота (транс-) Фталевая кислота (бензол-1,2-ди- карбоновая) ноос—сн=сн—соон ноос—сн-сн—соон /\/соон 1 II 130,5 287 200 1,92 3,02 2,58 6,23 4,38 5,62
Изофталевая кислота (бензол-1,3- дикарбоновая) 1 IL соон соон 348 3,62 4,60
Терефталевая кислота (бензол-1,4- дикарбоновая) соон соон 0 300 (возгоняется) 3,54 4,46
соон
мов растворяются лучше, чем соседние гомологи с четным числом углеродный
атомов. ’
Химические свойства. В водных растворах двухосновные кислоты
последовательно диссоциируют на ионы:
kt /О
НООС—(СНгЬ—соон + Н2О НООС— (СНХ—СС —F Н3о+
^о
I
НООС—(СН2)д—+ Н2О
^о-
- + н30+
где kx и k2 — константы диссоциации первого и второго карбоксила
соответственно.
Двухосновные кислоты сильнее одноосновных; kr дикарбоновых.,
кислот почти в два раза выше, чем у соответствующих монокарбоно-.
вых кислот, так как вторая карбоксильная группа, уступая в качестве
акцептора электронов, способствует ионизации первой карбоксиль-
ной группы. Второй протон отщепляется труднее (&х > fe>), карбокси-
лат-анион (1) уменьшает способность к диссоциации второй карбо-
ксильной группы. Степень диссоциации двухосновных кислот снижав
ется с увеличением молекулярной массы.
Двухосновные кислоты вступают во все химические реакции, ха-
рактерные для одноосновных кислот: образование солей, сложных'-
эфиров, ангидридов, галогенангидридов и т. д. В реакциях можег
участвовать одна или обе карбоксильные группы. В то же время ди-;
карбоновые кислоты алифатического ряда С2—С5 обладают рядом'
особенностей в своем химическом поведении, связанных с располо-*
жением карбоксильных групп относительно друг друга. "'j
Кислоты, у которых карбоксильные группы находятся близком
друг к другу (щавелевая, малоновая, алкилзамещенные последней)^
легко декарбоксилируются при нагревании, превращаясь в моно-н
карбоновые кислоты:
150°С
НООС—СООН--------> НСООН + со2
щавелевая муравьиная
кислота кислота
140°С
НООС—сн2—соон-------> сн3соон + со2
малоновая кислота уксусная кислота
R 1 /°
НООС—С—СООН -> R—СН—СООН + СО2 1
1 Ri дизамещенная мало- новая кислота 1 Ri дизамещенная ук- сусная кислота
Дикарбоновые кислоты, у которых карбоксильные группы нахо-1
230
дятся в положениях 1,4 и 1,5, при нагревании образовывают внут-
ренние циклические ангидриды (циклодегидратация):
СН2—СООН
СН2—СООН — Н2О
янтарная кислота
сн2—c<f
сн2—с<
^о
янтарный
ангидрид
Это связано с изогнутым строением углеродной цепочки «клешня»,
(карбоксильные группы близко подходят друг к другу, рис. 30) и
устойчивостью образующихся пяти- или шестичленных циклов. Во-
Рис. 30. Схема построения четырех- (а) и пяти-
членных (б) углеродных цепей
дородные атомы метиленовой группы малоновой кислоты или ее ди-
этилового эфира (малоновый эфир) обладают большой подвижностью
(протонизированы) и могут быть заменены на алкилы и некоторые
другие группы. Эта особенность, а также возможность декарбоксили-
рования образующихся соединений широко используется для син-
теза карбоновых кислот (синтезы с малоновым эфиром, или синтезы
Конрада):
СООС2Н5
НСН + C2H5ONa (спирт, р-р) -> [СН(СООС2Н5)2]~ Na* + С2НбОН
| алкоголят натриймалоновый э^ир
СООС2Н5 натрия
малоновый
эфир
Образовавшийся анион [СН(СООС2Н5)2]' вследствие эффекта сопря-
жения (мезомерный эффект) обладает повышенной устойчивостью (ме-
зомерный анион) и является сильным нуклеофильным реагентом.
Мезомерный эффект может быть показан или с помощью формулы с
делокализованными электронами (I), или набором граничных струк-
тур (II) с локализованныхм зарядом:
231
ОС2Н6
. с—о
H-Z -
с^о
I
OC2HS
мезомерный анион
На следующем этапе натриймалоновый эфир взаимодействует с
алкилгалогенидом:
СООС2Н5
[СН(СООС2Н6)21~ Na+ + R—Cl ->R—С—Н + NaCl
I
СООС2Н5
моноалкилмало’
новый эфир
Образовавшийся продукт гидролизуют и декарбоксилируют:
СООС2Н5 соон
| гидролиз; [Н] |
R—С—Н + 2Н2О---------------> R—С—Н + 2С2Н5ОН
I I
СООС2Н5 соон
мо ноалкилмало-
новая кислота
СООН
i t°
R—С—Н -> R-CH2—СООН + СО2
гл ГАТТ алкилуксусная
CUUtl кислота
Кроме алкилгалогенидов в синтезах с малоновым эфиром могут
быть использованы и эфиры а-галогензамещенных кислот. Использо-
вание синтезов с малоновым эфиром для получения непредельных
кислот было описано ранее (реакция Кневенагеля).
Двухосновные кислоты, содержащие двойные связи и ароматиче-
ское ядро, вступают также в реакции, характерные для алкенов и
производных бензола.,
Отдельные представители. Щавелевая, этандиовая кислота
НООС—СООН. Бесцветное кристаллическое вещество, хорошо раст-
воримое в воде. Обычно существует в виде кристаллогидрата, содер-
жащего две молекулы воды. Чрезвычайно широко распространена в
растениях главным образом в виде кальциевых солей. Соли и эфиры
щавелевой кислоты называют оксалатами. В большом количестве при*
сутствует в щавеле, кислице. Получают из формиата натрия:
360сС
2HCOONa------> NaOOC—COONa 4- Н2
NaOOC—COONa + H2SO4 НООС—СООН + Na2SO4
232
Щавелевая кислота легко окисляется, это используют в аналити-
ческой химии для установления титра перманганата калия:
[О]
НООС—СООН >- 2СО2 + Н2О
Щавелевая кислота и ее соли широко применяются в промыш-
ленности. При ситцепечатании — как протрава, в деревообделочной
промышленности для обработки орехового и красного дерева. Ис-
пользуется также для осаждения редких металлов, в качестве ката-
лизатора в реакциях поликонденсации.
Малоновая, пропандиовая кислота НООС—СН2—СООН. Бесцвет-
ное кристаллическое вещество. Содержится в свекольном соке. Полу-
чается из монохлоруксусной кислоты:
KCN 2Н2О
С1— СН2—СООН----> CN—СН2—СООН -----> НООС—СН2—СООН
-КС1 —nh3
монохлоруксусная малоновая кислота
кислота
Широко применяется в разнообразных синтезах (витаминов Вь Вг,
барбитуровой кислоты) диэтиловый эфир малоновой кислоты.
Адипиновая, гександиовая кислота НООС—(СНг)<—СООН.
Бесцветное кристаллическое вещество. Ограниченно растворима в
воде. Содержится в свекольном соке. Получается окислением цикло-
гексанола, циклогексанона или их смеси азотной кислотой при тем-
пературе 55—80° С и давлении до ЮНО5 Па:
ОН
I
j/44) HNOS
I I------> НООС—(СН2)4— соон
Адипиновая кислота — в техническом отношении важнейшая
насыщенная дикарбоновая кислота. Применяется в производстве
синтетических смол, для синтеза пластификаторов, найлона. В пище-
вой промышленности употребляется в качестве заменителя лимонной
кислоты.
Малеиновая и фумаровая кислоты НООС—СН=СН—СООН. Наи-
более простые и важные из двухосновных ненасыщенных кислот. Яв-
ляются геометрическими изомерами: малеиновая кислота — цис-,
фумаровая — трансизомер. Фумаровая кислота обнаружена в лишай-
никах, грибах, мышцах животных; малеиновая кислота в природе не
встречается. Вступают в большинство реакций, характерных для эти-
леновых соединений (присоединения, окисления, полимеризации) и
карбоновых кислот (образование солей, эфиров). Сильно отличаются
друг от друга по физическим свойствам (температура плавления ма-
леиновой кислоты 130° С, фумаровой 258° С), растворимости в воде,
степени диссоциации и некоторым химическим свойствам. Так, от-
нятием воды от молекулы малеиновой кислоты получают малеиновый
ангидрид: фумаровый ангидрид из-за удаленности карбоксильных
групп не образуется:
233
н—с—с/°
II Х0
н-с—c<f
\он
-> н2о +
/ОН 2 Н-С-С'
н-с-с<
малеиновая кислота
о
малеиновый ангидрид
В химическом отношении малеиновая кислота более активна: под
влиянием брома, иода, азотистой кислоты она переходит в фумаровую,
которая более устойчива.
Малеиновую кислоту в промышленности получают окислением
бензола и используют в пищевой промышленности для стабилизации
жиров, масел, сухого молока. Широкое применение для синтеза поли-
мерных материалов и яблочной кислоты нашел ангидрид малеино-
вой кислоты.
Фталевые кислоты С6Н4(СООН)г. Ранее указывалось, что сущест-
вуют три бензолди карбоновые кислоты, отличающиеся взаимным по-
ложением карбоксильных групп. Фталевые кислоты — кристалли-
ческие вещества, плохо растворимые в воде. Получаются окислением
о-, м-, n-ксилолов; фталевая кислота (о-изомер)—окислением наф-
талина:
а соон
соон
4- 2СО2 + Н2О
фталевая кислота
Фталевые кислоты проявляют все свойства кислот (образование
солей, эфиров, амидов и других производных) и вступают во многие
реакции, характерные для дизамещенных бензола. В отличие от других
изомеров фталевая кислота (о-изомер) легко отщепляет воду с обра-
зованием фталевого ангидрида
-Н2О
фталевый ангидрид
а соон
соон
фталевая кислота
который применяется для получения эфиров фталевой кислоты, ис-
пользуемых в качестве пластификаторов, красителей и глифталей —
полиэфирных смол на основе фталевой кислоты и глицерина:
о
II
—с
0 *
111
C-L-O—сн— СН(ОН)—СН2-гО—
/ I
остаток фталевой остаток
кислоты глицерина
234
Терефталевая кислота (n-изомер) применяется, в производстве
полиэтилентерефтала — сырья для изготовления волокна лавсан:
терефталевая
кислота
ООН 4- но—сн—СНу-ОН
этиленгликоль
О
<д
еоосн—сн2—о—)л
лавсан
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
КАРВОНОВЫХ КИСЛОТ
При замене гидроксила в карбоксильной группе карбоновой кис-
лоты на атомы галогена (X=F; Cl; Вг), группы OCOR; OR!; NH2 об-
разуются разнобразные функциональные производные кислот: галоген-
ангидриды, ангидриды, сложные эфиры, амиды кислот:
галогенанг идриды
амид кислоты
карбоновая
кислота
ангидрид кислоты
сложный эфир
Одновалентный кислотный остаток, образовавшийся после удаления
гидроксильной группы, называется кислотным радикалом или аци-
лом:
/О
R—с/
ацил
Ниже приведены названия некоторых ацильных остатков:
,0
Н—С—
формил
СН3—С—
ацетил
СН3—(СН2)2—С—
бутирил
,0
СН3—СН2—С—
пропионил
СН3—(СН^з—С—
валерил
235
К производным кислот относят и их соли (см. с. 216). Производные
кислот в зависимости оф строения ацила могут быть алифатическими,
(насыщенными и ненасыщенными), ароматическими, содержать цик-
лические группировки. Карбоновые кислоты и их производные иногда
называют ацильными соединениями. Основные химические превра-
щения ацильных соединений связаны с заменой группы А на другие
группировки, а их реакционная способность зависит от электроотри-
цательности этой группы (+М - и —1-эффект группы А):
Чем больше 8+ на атоме углерода карбоксильной группы, тем легче
взаимодействует функциональное производное R—с нуклео-
^А
фильным реагентом. В общем виде реакция имеет следующий вид:
, У° ° __
R—С R—С—А R—С.
♦ ХА | XZ
:z z
где Z — нуклеофильный реагент (НОН; NH3; ROH). По легкости эли-
минирования заместителя А производные карбоновых кислот рас-
полагаются в следующий ряд:
> л
> 3°
" ' о
eF
,0
nh2
OR
Во всех этих превращениях функциональные производные выступают
в качестве ацилирующих агентов, а реакции, в ходе которых проис-
ходит введение в молекулу ацильного остатка, — реакций ацилиро-
вания. Кроме реакций ацилирования они способны восстанавливаться,
галогенироваться. Отдельные классы производных участвуют в ха-
рактерных для них превращениях.
Галогенангидриды карбоновых кислот
Общая формула галоген ан гидридов карбоновых кислот R—с<^ ,
где X — фтор, хлор, бром, иод. Название их строится по принципу
галогеналкилов: называется ацил и атом галогена, например
О О
II II
Н—С—С1 — формилхлорид; сн3—С—С1 — ацетилхлорид и т.д.
236
Получение. Галогенангидриды кислот образуются при действии
галогенидов фосфора (РС15, РС1з), тионилхлорида на кислоты или их
ангидриды:
СН3СН2—с/ + РС15 ->
хон
пропионовая кислота
,0
СН3-(СН2)2-С<
Х)Н
СНз—СН2—С< + РОС15 + НС11
ХС1
пропионилхлорид
zO
+ SOC12 -> СН3(СН2)2-С< + SO2t + НС11
ХС1
бутановая кислота
бут ирилхлорид
Свойства. Галогенангидриды — жидкости или твердые вещества
с резким запахом, разлагаются водой («дымят» на воздухе):
ацетилхлорид
+ Н20 -> сн3—
+ НС1
уксусная кислота
Галогенангидриды — наиболее реакционноспособные производные
карбоновых кислот. Для них характерны реакции нуклеофильного
замещения, связанные с обменом атома галогена на другие атомы и
группы и введение в молекулу реагирующего с ним вещества ацила
(реакции ацилирования). Хлорангидриды ароматического ряда менее-
активны, чем алифатического.
Реакции ацилирования
гидролиз
алкоголиз
аммонолиз
ацидолиз
Восстановление хлорангидридов
/О 4Н; LiAlH4
СН3—СН2—с/ ---------------->- СН3—СН2—СН2ОН + НС1
ХС1 1-пропанол
пропионилхлорид
237
/°
Отдельные представители. Бензоилхлорад c6H5C<f . Жидкость
о 4(31
с резким запахом, /КИп = 197° С, /пЛ = —1 С, применяется для вве-
/О
дения в молекулу бензоила с6Н5С<^ .
Ацетилхлорид сн3—с/ . Жидкость, /КИп = 5ГС, /Пл =
ХС1
= —112°С. Применяется для введения в молекулы органических
О
соединений ацетила сн3—.
>С1
Фосген о=С<^ . Полный хлорангидрид угольной кислоты, от-
^С1
равляющее вещество (ОВ). Применяется для разнообразных синтезов^
СО + С12
+ 4NH,
С,Н5ОН
активиро-
ванный
уголь
фосген
h2n4
V=o
H2N'
мочевина
z
Cl—С"^
ХОС2Н5
этиловый эфир хлор-
мура вьинэй кислоты
Ангидриды карбоновых кислот
Получаются при отнятии молекулы воды от двух молекул карбо-
новой кислоты или взаимодействием хлорангидридов g солями этих
КИСЛОТ;
о
z° II
СН8—C<f + NaO—С—СН3------>
NCI -NaCl
СНз-С^
/°
СНз-сС
ацетилхлорид ацетат натрия
уксусный ангидрид
Ангидриды кислот применяются в реакциях ацилирования, однако
они менее активны, чем хлорангидриды:
zO NH,
Сщ____q/z -------------> CH3CONH2 + CH3COONH4 (аммонолиз)
\q ацетамид
гм / °
CHg ppi г)н II
\0 СН3С—ОСН3 + СНзСООН (алкоголиз)
уксусный ангидрид метилацетат
238
сн3-с^°
Основные представители. Уксусный ангидрид О. Резко
СН3-с/
пахнущая бесцветная жидкость. /КИп = 139,5° С, /пл = —73°С.
В промышленности получается из кетена и уксусной кислоты*
СН3—С<^
сн2=с=о + СНзСООН -> о
кечен уксусная кислота СН3__
^0
Широко применяется в качестве ацетилирующего агента.
Сложные эфиры карбоновых кислот
Сложные эфиры можно представить как производные карбоновых,
кислот, в которых ацильная группа связана с алкоксильной группой!
О
II
ацил алкок-
сильна я
группа
Номенклатура. Изомерия. По рациональной номенклатуре наиме-
нование эфира строится из двух частей: названия кислоты и спирто-
вого радикала. По ИЮПАК для наименования сложных эфиров к
названию кислоты прибавляют название спиртового радикала, из-
меняя при этом в названии кислоты окончание «овая» на «оат». Ниже
приведены примеры названий некоторых сложных эфиров:
zO этиловый эфир муравьиной кислоты, этилформиат,
Н—C<f этил мета ноат
\О-С2Н5
zO метиловый эфир уксусной кислоты, метилацетат,
СН3—С? метилэтаноат
^0—СН3
О
|| метиловый эфир 2-бром-З-бутеновой
СН2=СН—СНВг—С—ОСН3 кислоты, метил-2-бромбутен-З-оат
Изомерия сложных эфиров определяется изомерией кислотных и спир-
товых остатков.
Нахождение в природе. Способы получения. Сложные эфиры ши-
роко представлены в природе, но обычно в небольших количествах.
Они участвуют в разнообразных процессах, протекающих в живом
организме, являются ароматобразующими компонентами ряда рас-
тений. В значительных количествах в природе представлены только
сложные эфиры высокомолекулярных спиртов и кислот — воски;
эфиры эфирных масел.
239
Синтетические способы получения.
1. Реакция этерификации (см. с. 217).
2. Взаимодействие хлорангидридов и ангидридов кислот со спир-
тами или фенолами:
бензоилхлорид
NaOH
этилбензоаг
(СН}СО)2О +
уксусный
ангидрид
фенилацетат
Физические свойства. Сложные эфиры низкомолекулярых и среднемоле-
кулярных кислот и спиртов — жидкости с приятным фруктовым запахом, высо-
комолекулярных — твердые вещества без запаха. Плохо (низшие гомологи) или
совсем нерастворимы в воде, хорошо растворимы во многих органических раство-
рителях.
Химические свойства. Сложные эфиры вступают во все реакции
нуклеофильного замещения, описанные ранее для ангидридов и хлор^
«ангидридов кислот, проявляя при этом меньшую активность.
Гидролиз. Это наиболее важная реакция сложных эфиров:
/О +н2о о
R—C<f ~------> R-C< + RTOH
^ORi —н2о ^ОН
Гидролиз ускоряется в присутствии кислот, щелочей и ферментов.
Гидролиз сложных эфиров с помощью ферментов (эстераз) играет боль-
шую роль в сложных биохимических процессах, протекающих в жи-
вом организме.
Переэтерификация. Под переэтерификацией понимают замену
спиртового остатка в молекуле сложного эфира на другой спиртовой
остаток, происходящую при нагревании эфиров со спиртами. Реакция
ускоряется в кислой или щелочной среде:
О
z° II
R—С< + R2OH-----> R—С—OR2 + RiOH
xORi
Восстановление сложных эфиров. При восстановлении образуются
две молекулы спирта:
О
II [Н]
R—С—ORj------> R—СН2ОН + RjOH
Восстановители: натрий в кипящем спирте, алюмогидрид лития.
✓О
Отдельные представители. Этилформиат н—
ХОС2Н5
Жидкость, /Кип ~ 55°С, обладает запахом рома, применяется в пи-
щевой промышленности в качестве ромовой эссенции.
240
V t
У ксусноэтиловыи эфир, этилацетат сн3—c<f . /Кип =
ХОС2Н5
78°С, применяется в качестве растворителя, для синтеза разно-
образных химических препаратов.
Уксусноизоамиловый эфир, изоамилацетат СН3—C<f
хосбнп
Жидкость с запахом груши, /Кип — 139°С. Применяется в пищевой
промышленности в качестве грушевой эссенции.
Масляноэтиловый эфир, этилбутират СН3— (СН2)2—сХ
ХОС2Н5
Прозрачная жидкость с запахом ананаса, /Кип = 121°С. Приме-
няется в качестве ананасовой эссенции.
Особую группу сложных эфиров составляют сложные эфиры ми-
неральных кислот, представителем которых является диметилсуль-
фат (CH3)2SO4. Получают действием олеума на метиловый спирт:
SO3 +2СН3ОН----->- (CH3)2SO4 + Н2О
Диметилсульфат используют как метилирующее средство (введение
в молекулу метильной группы). Сильно ядовит.
оксикислоты
Оксикислотами называются производные углеводородов, содер-
жащие в молекуле два вида функциональных групп: гидроксильные
О
ОН и карбоксильные —с—ОН- В зависимости от количества кар-
боксильных групп различают одно-, двух- и многоосновные оксикис-
лоты, а от числа гидроксильных групп (включая гидроксильную груп-
пу, входящую в состав карбоксильной группы) — двух-, трех- и
многоатомные оксикислоты.
По строению углеводородного радикала, с которым связаны окси-
и карбоксильные группы, оксикислоты делятся на алифатические
(насыщенные и ненасыщенные), ароматические, гетероциклические
и т. д.:
НО—СН2—СООН гликолевая, оксиуксусная (одноосновная, двухатомная
оксикислота)
ОН
I
СН3—СН—СООН молочная (одноосновная, Двухатомная оксикислота)
НООС—СН—СН2—СООН яблочная (двухосновная, трехатомная оксикислота)
ОН
Ноос—снон—снон—соон винная (двухосновная, четырехатомная оксикис-
лота)
9-682
241
соон
галловая (одноосновная, четырехатомная арома-
тическая оксикислота)
но^^у ОН
ОН
ОН
НООС—СН2—С—СН2—СООН лимонная (трехосновная, четырехатомная окси-
| кислота)
СООН
Структурная изомерия, номенклатура. Структурная изомерия
оксикислот вызывается особенностями строения углеродного скелета
(изомерия скелета), положением функциональных групп и их взаим-
ным расположением. В зависимости от положения оксигруппы по
отношению к карбоксильной различают а-, 0-, у- т. д. оксикислоты:
7 р а
I I I
—С—С— с—соон
I I I
При наименовании оксикислот широко применяются тривиальные
названия: молочная, лимонная, винная.
По рациональной номенклатуре в основе наименования лежит название со-
ответствующей кислоты, положение гидроксильной группы указывается буква-
ми греческого алфавита а, 0, у:
ОН
4 3 | 2 1
сн3—сн—сн2—соон
3-®ксимасляная кислота
По ИЮПАК наименование оксикислоты строят, прибавляя при-
ставку окси (диокси, триокси и т. д.) к систематическому названию
кислоты. При нумерации атомов углерода углерод-углеродной цепи
номер один присваивается атому углерода карбоксильной группы.
Приведенную выше оксикислоту следует назвать 3-оксибутановая
кислота.
Способы получения. Оксикислоты получают синтетическим путем
из кислот (или их производных) введением в молекулу оксигруппы;
из спиртов введением карбоксильной группы, из альдегидов и кето-
нов — через нитрилы кислот. Для получения некоторых кислот (мо-
лочной, лимонной) широко применяются биохимические методы.
1. Гидролиз галогензамещенных кислот. Этот способ удобен для
получения а-оксикислот из-за доступности а-галогенпроизводных
кислот:
242
Н+; t°
СН3—СНС1—СООН + НОН--------->СН3—СН—СООН+НС1
I
ОН
2-хлорпропановая кислота 2>оксипропановая кислота
2. Гидратация непредельных кислот:
3- h2so4
сн2==сн—с—он + н2о —* сн2—сн2—с—он
4
он
акриловая кислота 3-оксипропановая кислота
Присоединение воды к а-, p-непредельным кислотам вследствие
эффекта сопряжения (1,4-присоединения) идет не по правилу Марков-
никова: гидроксил направляется к более удаленному от карбоксиль-
ной группы ненасыщенному атому углерода.
3. Получение из альдегидов и кетонов (циангидринный, окси-
нитрильный синтез):
ОН ОН
zO HCN I 2H,O I
СНз—С< ------> СН3— С—CN-------> СН3—СН—СООН
ХН | “NHa
Н
уксусный альдегид нитрил-а-окси- молочн я кислота
пропионовой кислоты
Этот способ применяется для получения а-оксикислот.
4. Получение Р-оксикислот. Реакция Реформатского. При взаимо-
действии альдегидов или кетонов с а-бромзамещенными эфирами кар-
боновых кислот в присутствии металлического цинка образуются
производные P-эфиров карбоновых кислот. Гидролизом последних мо-
гут быть получены эфиры кислот или сами кислоты:
СН3 О
I II* эфир
СН3—С=О + ВгСН2С—ОС2Н5 + Zn------------
ацетон этилбромацет т
сн3 О сн3 о
I II Н,О; [Н+] I ||
СН3—С—СН2 —СОС2Н6----------► СНз— G—СН2—С—ОС2На
OZnBr ОН
этиловы й э фир-₽-©кеииз®валериановой
кислоты
Физические свойства. Оксикислоты — бесцветные вязкие жидкости или
кристаллические вещества. Низшие хорошо растворимы в воде, с ростом молеку-
лярной массы растворимость уменьшается.
Химические свойства. Химическое поведение оксикислот, как и
Других гетерофункциональных соединений, т. е. соединений, со-
держащих в молекуле разные функциональные группы, определяется
природой этих групп. Оксикислоты вступают в большинство химиче-
243
ских реакций, характерных для кислот (наличие группы СООН)
и спиртов (присутствие группы ОН). Строение углеводородного ра-
дикала также сказывается на их химическом поведении — аромати-
ческие оксикислоты вступают во многие превращения, характерные
для соответствующих производных бензола. В одних химических
реакциях каждая из указанных функциональных групп может участ-
вовать независимо друг от друга, в других ход реакции и характер
образующихся продуктов зависит от их взаимного влияния, в не-
которых случаях окси- и карбоксильная группы могут взаимодей-
ствовать между собой.
Кислотные свойства. Введение в молекулу кислоты оксигруппы
повышает силу кислоты: оксикислоты — более сильные, чем соот-
ветствующие им карбоновые кислоты.
Они могут образовывать соли, сложные эфиры, амиды, галоген-
ангидриды. В реакции с РС15 может взаимодействовать и спиртовая
группа.
Спиртовые свойства. Возможно окисление вторичной спиртовой
группы с образованием кетокислоты (1); замена спиртовой группы на
атом галогена (2); образование простых эфиров (3); сложных эфиров
(4):
О
[О] II ^сн3— с—соон (1)
НС1
— -> сн3—сн—соон 1 (2)
он 1 С1
СНз— СН—соон (CH.hSO. -> сн3—сн—соон (3)
1 осн3
С17Н 35СОС1
— > сн3—сн—соон (4)
ОСОС17Н35
Отличие в поведении а-, р-, у- и Ъ-оксикислот.
а. Дегидратация а-оксикислот. При нагревании а-оксикислот вы-
деляется вода и образуется циклический сложный эфир — лактид:
а
сн3—сн—оГннб’:—с==о
I .......’ | - 2Н2О
°=cH*PKZS° ~ сн~сн3-----
а
сн3—сн—о—с=о
о=с —о — сн—СНз
лактид
Лактиды — кристаллические вещества, разлагающиеся при кипячении
в присутствии кислот и щелочей на исходные продукты.
б. Р-Оксикислоты при нагревании теряют воду с образованием не-
предельных кислот:
244
СН3— СН—СН,—соон------>- сн3—сн=сн—соон
| -Н,о
он
р-оксимасляная кислота кротоновая кислота
в. у- и 8-Оксикислоты при нагревании образуют внутренние слож-
ные эфиры — лактоны:
т ₽ « /°
СНз—сн—сн2—сн2—соон ——-> сн3—сн—сн2—сн2—с
у-оксивалериановая кислота лактон 7-оксивалериановой кислоты
Стереохимия углерода. В органической химии известны два ос-
новных вида изомерии — структурная и пространственная. Многочис-
ленные виды структурной изомерии были уже подробно рассмотрены
в предыдущих разделах. К пространственной изомерии относятся:
1) геометрическая (цис- и трансизомерия); 2) оптическая и 3) поворот-
ная изомерия. Раздел органической химии, изучающий пространствен-
ное строение молекул, его влияние на свойства веществ, на направле-
ние и скорость органических реакций, называется стереохимией (от
греческого слова stereos — пространственный). Первый из перечислен-
ных видов пространственной изомерии — геометрическая изомерия—
был рассмотрен в разделе алкенов, а также карбоновых кислот (фума-
ровая и малеиновая кислоты) и в некоторых других разделах.
Оптическая изомерия. В 1815 г. французский химик Био обна-
ружил, что некоторые органические вещества в жидком и растворенном
состояниях способны вращать плоскость плоскополяризованного света.
Ранее эта особенность была отмечена для кристаллов кварца — при
прохождении через них поляризованного луча происходит вращение
плоскости поляризации, причем одни кристаллы вращают ее вправо,
другие — влево. В 1848 г. Л. Пастер обнаружил эту способность у
винных кислот. Это явление, названное оптической активностью, полу-
чило объяснение в работах Вант-Гоффа и Ле Беля. Вант-Гофф высказал
предположение, что оптическая активность органических соединений
связана с наличием в их молекуле асимметрического атома углерода
т. е. углеродного атома, связанного с четырьмя различными замес-
тителями. Для большинства оптически активных соединений это пред-
положение оказалось справедливым. Так, в молекуле молочной кис-
лоты содержится атом углерода (он отмечен звездочкой С*), связанный
с четырьмя различными группами:
Н О
I х/
н3с—с*—с
I \
он юн
молочная кислота
Если представить, что он находится в центре тетраэдра («/^-гибридиза-
ция), то группы Н3С; Н; ОН; СООН будут находиться в вершинах
(углах) тетраэдра. Наличие асимметрического атома углерода в моле-
245
Г
Рис. 31. Пространственные изомеры молочной кислоты
куле молочной кислоты приводит к образованию двух пространствен-
ных изомеров (рис. 31), отличающихся друг от друга как несимметрич-
ный предмет от своего изображения в зеркале (как левая и правая ру-
ка). Отсюда и другое название оптической изомерии — зеркальная
изомерия. Подобные пространственные изомеры называются зеркаль-
ными изомерами (энантиомерами) или оптическими антиподами.
Молочная кислота существует в виде двух пространственных изо-
меров — энантиомеров (оптических антиподов), каждый из которых
проявляет оптическую активность — обладает способностью вращать
плоскость поляризованного света. Один из изомеров — правовращаю-
щий, другой — левовращающий.
Оптическая неактивная модификация, состоящая из равных коли-
честв двух (лево- и правовращающих) антиподов, называется рацема-
том. Рацематы образуются в ходе химической реакции, когда вероят-
ность образования каждого из оптических антиподов одинакова. Ра-
цемат может представлять смесь двух антиподов или их молекулярных
соединений (истинные рацематы).
По большинству химических и физических свойств оптические ан-
типоды не отличаются друг от друга. Различие состоит лишь в том, что
один из них лево-, другой правовращающий, и в их способности крис-
таллизоваться в энантиоморфные формы.
Рацемические соединения (истинные рацематы) отличаются по сво-
им физическим свойствам (температуре плавления, плотности и т. д.)
от энантиомеров.
Для пространственного изображения оптических антиподов кроме
тетраэдрических моделей широко применяются проекционные форму-
лы, названные по имени ученого, предложившего их, формулами Фи-
шера:
СООН
Н------------ОН
СООН
НО— ------н
СНз
СНз
зеркало
246
При пользовании формулами Фишера нужно помнить, что верх-
няя и нижняя группы (для молочной кислоты —СООН и —СН3) ле-
я<ат за плоскостью чертежа, а боковые (в нашем случае Н и ОН) —
перед плоскостью чертежа. Асимметрический атом углерода находится
в плоскости чертежа на пересечении прямых, связывающих эти груп-
Рис. 32. Поляриметр
пы. Такие модели при вращении их в плоскости чертежа нельзя сов-
местить. При их написании обычно главную (наиболее окисленную)
функциональную группу располагают наверху, а основную — угле-
родную цепь — вертикально.
В последнее время молекулы органических соединений, проявляю-
щих оптическую активность и способных существовать в виде пары
энантиомеров, принято называть хиральными (от греческого слова
«хиро» — рука). Хиральные молекулы не имеют ни центра, ни плос-
костей симметрии, но могут иметь ось симметрии. Если молекулы не
имеют и оси симметрии, то они являются асимметрическими, т. е. не
все хиральные молекулы полностью асимметричны. Асимметричес-
кий атом получил название хирального атома или хирального
центра.
Для измерения оптической активности применяется прибор, на-
званный поляриметром (рис. 32). Свет от источника /, проходя через
поляризатор 2, в качестве которого употребляется неподвижно ук-
репленная призма Николя, поляризуется. Поляризованный луч,
попадая на анализатор (вторую призму Николя 4), может полностью
проходить через нее, когда оси кристаллов обеих призм строго парал-
лельны, или полностью задерживаться, если они повернуты на 90°.
Для наглядности прохождение поляризованного света через призму
Николя (рис. 33,а) часто сравнивают с прохождением лезвия ножа че-
рез книгу: если они расположены в одной плоскости, нож проходит,
если книгу повернуть на 90°, нет (рис. 33,6).
Для измерения оптической активности исследуемое вещество в жид-
ком состоянии или в виде раствора помещают в поляриметрическую
247
Рис. 33. Прохождение поляризованного све-
та через призму Николя
трубку 3 (см. рис. 32), которую устанавливают между поляризатором
и анализатором. Если исследуемое вещество оптически неактивно, то
световой луч, проходя через него, не изменяя плоскости поляризации,
попадает в анализатор. Если оно активно, то изменяется плоскость
поляризации, и для прохождения луча через анализатор его нужно
повернуть на угол, соответствующий вращению находящегося в трубке
вещества. Величину этого угла измеряют с помощью круговой шкалы
5 (см. рис. 32). Измеренное вращение зависит от природы вещест-
ва, концентрации, длины трубки, длины волны источника света, тем-
пературы измерения, вида растворителя и обычно пересчитывается в
удельное вращение [а]. Удельным вращением называется угол враще-
ния плоскости поляризации жидкостью или раствором, наблюдаемый
при толщине слоя 1 дм и концентрации исследуемого вещества 1 г/мл,
найденный при определенный температуре. Удельное вращение для
жидкости вычисляют по формуле
[а]х = a/(/d); для растворов [а]' а100/(/С),
где а — угол вращения плоскости поляризации изучаемым веществом
в трубке длиной / при плотности вещества d\ С — объемная концент-
рация (количество граммов оптически активного вещества в 100 мл
раствора). Величина оптического вращения зависит, как уже указыва-
лось, от длины волны света X, который был использован при измере-
нии. Обычно используют свет натриевой лампы с длиной волны 589
нм.
Разобранный пример стереоизомерии молочной кислоты наиболее
простой. Большей сложностью отличается стереоизомерия винной кис-
лоты НООС — СНОН—СНОН — СООН. В молекуле винной кис-
лоты имеется два асимметрических атома. В таких случаях количество
оптически активных изомеров N определяется по формуле N = 2п,
где п — количество асимметрических атомов углерода. Таким образом,
для винной кислоты должно существовать четыре оптически активных
изомера — две пары оптических антиподов:
248
соон
соон
соон
соон
‘ ОН но [4 Н— он но н
н 11 пи
и ОН НО н но н н пн
П - пи С/П
( соон ( СООН ( СООН СООН
1 < 2 3 4
Каждый асимметрический атом в молекуле винной кислоты может
обеспечивать, или левое (—), или правое (+) вращение. Возможны
следующие комбинации:
1) + 2) - 3) + 4) —
— + + —
При рассмотрении приведенных формул можно убедиться, что
структуры 3 и 4 являются оптическими антиподами, а 1 и 2 при пово-
роте на 180°С в плоскости чертежа совпадают, т. е. они соответствуют
конфигурации одной молекулы, получившей название мезовинной кис-
лоты. Мезовинная кислота не обладает оптической активностью вслед-
ствие внутримолекулярной компенсации. Следовательно, для винных
кислот возможно существование двух оптических антиподов (3 и 4),
их рацемической смеси и неактивной мезовинной кислоты.
Если сравнивать стереоизомеры 3 и 4 с мезоформой, то окажется,
что они имеют одинаковое расположение групп Н и ОН у одного асим-
метрического атома углерода и разное — у другого, т. е. не составля-
ют друг с другом пару оптических антиподов. Такие стереоизомеры
называются диастереомерами. Они отличаются друг от друга не толь-
ко по величине угла вращения, но и по физическим и химическим свой-
ствам. Если два одинаковых боковых заместителя у соединений с дву-
мя асимметрическими атомами в стандартной проекционной формуле
(формуле Фишера) расположены по одну сторону (справа или слева)
от главной оси, то они называются эритроформами; по разные — трео-
формами. Так, для хлоряблочной кислоты стереоизомерные формы 1 и
2 будут эритроформами, а 3—4 — СООН треоформами: СООН 1 соон
СООН
х_т он но н н он НО - ц
П н с/п 11VJ ci ГТ Q] \-zfT н Н- - ГТ Г]
Г1 < ci ci СООН < соон соон ( СООН
1 ! 2 3 4
эритроформы трео формы
49
У винной кислоты треоизомерами являются изомеры 3 и 4.
Оптически активные соединения, имеющие родственную конфигу-
рацию у асимметрического углеродного атома, относятся к одному и
тому же стереохимическому ряду: D или L. Русским ученым Розано-
вым было предложено соединение, содержащее гидроксил в проекци-
онных формулах Фишера справа от асимметрического атома углерода,
относить к Р-ряду, слева — к L-ряду. Символы D и L не имеют отно-
шения к знаку вращения.
Вопрос об абсолютной конфигурации оптически активных соедине-
ний, т. е. о действительном расположении групп в пространстве, в те-
чение длительного времени не имел своего разрешения. С помощью
разработанных в последние годы физических методов исследования
была установлена абсолютная конфигурация ряда органических сое-
динений.
Синтетическим путем и в ходе многих биохимических процессов
часто получаются рацемические смеси, разделение которых на оптичес-
кие антиподы, учитывая, что их физические и химические свойства
одинаковы, представляют трудную задачу. Наиболее эффективными
являются три метода разделения.
1. Физический метод. При кристаллизации рацемических смесей
некоторых антиподов образуются кристаллы, отличающиеся друг от
друга, как несимметрический предмет от своего зеркального изображе-
ния. Они могут быть разделены механическим путем. Этот метод был
открыт Пастером и применен для разделения неактивной винной кис-
лоты в виде ее натриево-аммониевой соли.
2. Биохимический метод. Основан на способности отдельных видов
микроорганизмов предпочтительней поглощать из рацемической смеси
один из антиподов.
3. Химический метод. Используется способность оптических анти-
подов превращаться в ходе химических реакций при взаимодействии с
оптически активным реагентом в диастереоизомеры, которые сущест-
венно отличаются по своим свойствам (в частности, по растворимости):
----------------------------------------------------(+) DA DB
(+) DA и (—) LA + DB -> (+)DA-DB + (—) LA DB— '--------------------*
-----> (—) LA DB
рацемат оптически смесь диастереомеров
активный
реагент
---------- (+) DA + (-) LA
выделение
антиподов
Отдельные представители. Молочная, а-оксипропионовая кис-
лота СН3—СИОН—СООН. Существует в природе, в виде двух опти-
ческих антиподов и рацемата. Мясомолочная кислота (+), правовра-
щающая. Кристаллическое вещество [а]^5 = 4-3,82°. Содержится в
мышечном соке. Левовращающая (—), обладает теми же свойствами,
что и правовращающая = —3,82°.
Молочная кислота брожения (±) — рацемат. Гигроскопичная си-
250
ропообразная жидкость. Наиболее важный способ получения — мо-
лочнокислое брожение:
С6Н12О----> 2СН3—СНОН—СООН
гексоза молочная кислота
При молочнокислом брожении сахаров обычно образуется рацеми-
ческая форма молочной кислоты. Молочнокислое брожение играет
очень большую роль в производстве молочнокислых продуктов (прос-
токваши, ацидофилина, кефира, кумыса), при изготовлении жидких
дрожжей для хлебопечения, квашения капусты, огурцов, силосовании
кормов.
Молочную кислоту получают синтетическим путем из уксусного
альдегида:
/О HCN 2Н2О
СН3—С<-------> СН3—СН—CN------> СН3—СН-СООН
ХН | -nh8 I
ОН он
уксусный молочная кислота
ангидрид
или омылением а-хлорпропионовой кислоты СН3—СНС1—СООН.
Молочная кислота используется в кожевенной промышленности
при обработке кож, текстильной в качестве протравы и в медицине.
Очень широкое применение она нашла в пищевой промышленности при
изготовлении конфет, безалкогольных напитков. Эфиры молочной
кислоты СН3—СН—СООН употребляются в качестве улучшающих
ОСОС17Н35
добавок в хлебопечении, при производстве кондитерских изделий, мо-
роженого.
^блочная, оксиянтарная кислота НООС—СНг—СНОН—СООН.
Встречается в виде двух оптических антиподов и рацемата. В расти-
тельном мире широко распространена левовращающая L-яблочная
кислота. Является одной из главных органических кислот в яблоках,
рябине, груше, кизиле, махорке (до 6,5%). Бесцветное кристалличес-
кое вещество [а]^ = —2,3°. Получают гидролизом бромянтарной кис-
лоты:
н2о
НООС—СН2—СНВг—СООН------> НООС—сн2—СНОН—СООН
—НВг
или гидратацией фумаровой или малеиновой кислот при 200°С;
НООС—СН-СН—СООН + н2о -------> ноос—сн—сн2—соон
он
Левовращающую яблочную кислоту выделяют из плодов рябины.
Применяется в пищевой промышленности в производстве конди-
терских изделий и безалкогольных напитков.
Винная, диоксиянтарная кислота НООС—СНОН — СНОН —
— СООН. Встречается в виде двух оптических антиподов, рацемата и
Мезовинной кислоты (табл. 25).
251
Таблица 25. Свойства винных кислот
Наименование Температура плавления, °C а, в воде
Винная (левовращающая; L-кислота) 170 — 12
Виннокаменная (правовращающая; D-кис- лота) Рацемат (виноградная кислота) 170 + 12
206 Неактивная
Мезовинная 140 »
В природе встречается правовращающая (D-винная) кислота, по-
лучившая название винно-каменной, так как она впервые была выде-
лена из «винного камня» — отхода, получаемого при изготовлении и
выдержке виноградных вин и представляющего собой кислую калие-
вую соль НООС — СНОН — СНОН — COOK. Получают винную кис-
лоту из «винного камня» или синтетическим путем.
Исследования винных кислот сыграли выдающуюся роль в изуче-
нии явления изомерии. В 1830 г. Берцелиус, изучая данные кислоты,
ввел понятие изомерии. В 50-х годах прошлого века Пастер, работая
с винными кислотами, выяснил природу рацематов и методы их раз-
деления.
Винная кислота в пищевой промышленности используется при из-
готовлении фруктовых вод, химических разрыхлителей теста, в анали-
тической химии для определения ионов калия, в медицине. Широкое
применение нашли и соли винно-каменной кислоты. Калиево-натрие-
вая соль NaOOC — СНОН — СНОН — СООК-4НгО, получившая
название сегнетовой соли, является одним из компонентов фелинговой
жидкости, которая используется для качественного и количественного
определения восстанавливающих веществ: альдегидов, сахаров (см.
с. 357). Сегнетовая соль употребляется и в радиотехнике (пьезокрис-
таллы).
Лимонная кислота (трехосновная, четырехатомная оксикислота)
СООН
СООН—сн2—с—сн2—соон
но
Основная органическая кислота плодов цитрусовых, в лимонах ее
содержание достигает 9% на сухую массу. Кристаллическое вещество,
хорошо растворима в воде. Лимонную кислоту получают из листьев
махорки, лимонов и биохимическим путем с помощью гриба Aspergil-
lus niger. Применяется в пищевой промышленности при производстве
конфет, кремов, безалкогольных напитков, в текстильной промышлен-
ности при крашении тканей и медицине.
Рицинолевая кислота
252
Н3С—(СН2)5—СН—СН2—СН=СН—(СН2)7—соон
он
Мононенасыщенная, одноосновная, двухатомная оксикислота. Явля-
ется основной кислотой касторового масла (80—95%). Касторовое мас-
ло широко используется в технике в качестве смазывающего вещества,
а в медицине как слабительное.
Оксибензойная кислота С6Н4(ОН)(СООН). Существует в виде трех
изомеров (о,;и,п-кислоты), важнейшим является о-оксибензойная, или
салициловая кислота:
он
СООН
Бесцветное кристаллическое вещество, трудно растворяется в воде.
Получается действием диоксида углерода на фенолят натрия (реакция
Кольбе — Шмидта):
фенолят
натрия
сал ицилат
натрия
салициловая
кислота
Применяется в аналитической химии, медицине, анилокрасочной и
пищевой промышленности, при консервировании фруктов. Производ-
ные салициловой кислоты являются лечебными препаратами:
Галловая, 3,4,5-триоксибензойная кислота
соон
но
он
Кристаллическое вещество, хорошо растворимое в кипящей воде.
В природе галловая кислота встречается как в свободном, так и связан-
ном состоянии, обычно в растениях, содержащих дубящие вещества
(чернильные орешки, дубовая кора, листья чая). Получают гидроли-
зом танина. Применяется для получения пирогаллола, образующегося
при ее нагревании, синтеза красителей, в аналитической химии.
253
ОКСОКИСЛОТЫ (АЛЬДЕГИДО- И КЕТОКИСЛОТЫ)
Альдегида- и кетокислотами называются гетерофункциональные
соединения, содержащие в молекуле две функциональные группы:
кислотную (карбоксильную) и альдегидную или кетонную.
В зависимости от положения альдегидо- и кетогруппы по отноше-
нию к карбоксилу различают а-, (3-, у- и т. д. альдегидо- и кетокислоты:
О О
II II
н—С—СООН сн3—с—соон
а-альдегидокислота а-кетокислота
о
II
сн3—с—сн2—соон
3-кетокислота
Номенклатура, изомерия. Для наименования оксокислот часто
применяют тривиальные названия:
О
II
сн3—с—соон
пировиноградная
кислота
О
II
сн3—с—сн2—соон
ацетоуксусная кислота
о
II
сн3—С—СН2—сн2—соон
левулиновая кислота
По номенклатуре ИЮПАК главная цепь, содержащая обе функцио-
нальные группы, нумеруется, начиная с карбоксильной группы, по-
ложение же альдегидной или кетонной группы обозначается соответст-
вующей цифрой с добавлением приставки «оксо». Например:
Sc—СН2—сн2—соон
Н/
4-оксобутановая кислота
О
сн3—с—сн2—соон
З-оксобутановая кислота
Получение, свойства. Оксокислоты можно получать либо введением
карбонильной группы в карбоксилсодержащее соединение, либо,
наоборот, введением карбоксильной группы в оксосоединение. Наи-
более общими способами получения являются следующие.
Гидролиз дигалогензамещенных кислот:
2Н?О
CHCU-СООН -------5
—2НС1
дихлврукеусная
кислота
г ОН
I
СН—СООН
I
Loh
^>с—соон
глиоксиловая кислота
о
н
Окисление оксикислот:
[О]
СН3—СН—соон —> сн3—с—соон
ОН
молочная кислота
О
пировиноградная кислота
254
Окисление гликолей:
[О] [О]
СН2—СН2 —>- СН2—СООН —> НС—СООН
III II
он он он о
этиленгликоль гликолевая глиоксиловая
кислота кислота
Омыление оксонитрилов*.
/£> KCN >0 2Н2О
R—C<f-------> R—С<----------> R—С—соон
ХС1 -КС! \CN -NH ||
о
ацилхлорид оксенитрил а-оксфкислата
Оксокислоты проявляют химические свойства, характерные для обеих
функциональных групп.
Основные представители. Глиоксиловая кислота %С—СООН,
W
единственно возможная а-альдегидокислота. Существует в виде крис-
таллогидрата, содержащего одну молекулу воды. Глиоксиловая кисло-
та распространена в природе, особенно в большом количестве она со-
держится в незрелых фруктах. Играет важную роль в обменных про-
цессах, протекающих в растениях и микроорганизмах. Она вступает в
реакции, типичные для альдегидов и кетонов.
О
Пировиноградная кислота СН3—С—СООН. Наиболее важный
представитель кетокислот. Бесцветная жидкость с запахом, напоми-
нающим запах уксусной кислоты, /кип = 165°С, /пл = 13,6°С. Смеши-
вается с водой во всех отношениях. Широко распространена в природе,
является важнейшим промежуточным продуктом при распаде углево-
дов в растениях, в спиртовом и молочнокислом брожении. Получается
пиролизом виноградной кислоты, отсюда и ее тривиальное название—
пировиноградная кислота:
t° г п
НООС—СН—СН—СООН--------------->- сн2=с—соон -> сн3—с—соон
I | -СО2; —-Н2О | II
он он L он о
виноградная кислота
пировиноградная
к ислота
Близкое расположение в пространстве обеих функциональных
групп отражается на химических свойствах пировиноградной кислоты:
как кислота она сильнее уксусной, а как кетон она значительно актив-
нее обычных кетонов. Типичными реакциями а-кетокислот являются
их декарбоксилирование под действием разбавленной серной кислоты
при слабом нагревании и декарбонилирование при нагревании с кон-
центрированной серной кислотой:
разб. H2SO4
CHS—с—соон—
II
о
;О
+ со2
н
/>°
КОНЦ. СН3—С< + со
\он
255
о
Ацетоуксусная кислота СН3—- С—СН2—СООН 36—37°С. Обра-
зуется за счет (J-окисления при метаболизме жирных кислот. В сво-
бодном виде представляет собой сиропообразную жидкость. Как и
другие р-оксокислоты, ацетоуксусная кислота очень неустойчива и
легко декарбоксилируется с образованием ацетона:
СН3СОСН2СООН сн3сосн3 + со2
Соли, эфиры и другие производные ацетоуксусной кислоты вполне
устойчивы.
Ацетоуксусный эфир (этиловый эфир ацетоуксусной кислоты)
СН3—С—СН2—СООС2Н5. Бесцветная приятно пахнущая жидкость,
О
*кип = 18ГС. Из всех производных ацетоуксусной кислоты ее этило-
вый эфир имеет особо важное значение как в разработке теоретичес-
ких представлений в органической химии, так и в препаративном отно-
шении.
Получают его сложноэфирной конденсацией (конденсация Кляйзе-
на) — взаимодействием двух молекул этилового эфира уксусной кис-
лоты в присутствии металлического натрия, его алкоголята, амида или
гидрида:
C2H5ONa
СН3—С—ОС2Н5 + СН3—С—ОС2Н5-------> СН3—С—СН2—С—ОС2Н5
Механизм сложноэфирной конденсации напоминает механизм аль-
дольной конденсации (см. с. 205). Первая стадия — образование
аниона этилацетата под действием сильного основания (например, эти-
лата натрия):
С2Н5О 4- Н:—СН2—:СН2—С< + С2Н5ОН
i хос2н5 хос2н5
Являясь очень сильным нуклеофилом, этот карбанион атакует кар-
бонильный атом углерода второй молекулы этилацетата:
о о о
сн3—сА-—+ :сн2— cf = СН,—С—СН2— С—ОС2Н5
ХОС2Н5 '-' ОС2Н5 I
На завершающей стадии происходит отщепление этилат-иона (ката-
лизатор) и образование ацетоуксусного эфира:
О- О
I II Z*
СНз—С—СН2-С< * СН3—с-сн2—+ С2Н5О-
| ХОС2Н5 ХОС2Н5
OCjHb
256
Ацетоуксусный эфир — классический пример органического соеди-
нения, для которого характерно явление таутомерии — подвижной,,
динамической изомерии. Одна из изомерных форм ацетоуксусного эфи-
ра содержит кетогруппу (кетофэрма); другая — гидроксильную группу
у ненасыщенного углеродного атома (енольная форма); отсюда и назва-
ние этого вида таутомерии — кето-енольная таутомерия:
О ОН
II I
СН3—С—СН2—СООС2Н5 СН3—С=СН—СООС2Нб
кетонная форма енольная форма
Такое превращение возможно из-за наличия в кетонной форме
СН2-группы, расположенной между двумя СО-группами, в результате
чего водороды в этой метиленовой группе приобретают значительную
подвижность. Енольная же форма обладает определенной стабиль-
ностью (по сравнению с виниловыми спиртами) вследствие возникаю-
щего сопряжения:
ОН О
I II
СН3—С=СН—С—ОС2Н5
Равновесие между кетонной и енольной формами ацетоуксусного
эфира зависит от растворителя и температуры. При обычных условиях
соотношение кетонной и енольной форм составляет примерно 93 : 7.
При сильном охлаждении эфира (до—78°С) можно выделить чистую
кетонную форму (кристаллическое вещество. /Пл = —38°С). Чистая
енольная форма может быть получена путем перегонки (она кипит
ниже кетонной формы имеет /пл = —44°С). Однако при стоянии лю-
бой из этих форм постепенно происходит превращение их друг в друга.
В химическом отношении ацетоуксусный эфир способен давать ре-
акции как для кетонной, так и для енольной формы.
Реакции кетонной формы.
Восстановление водородом в момент выделения'.
О ОН
II 2Н |
СНз—С—СН2СООС2Н5------> СНз—СН2СООС2Н5
Присоединение синильной кислоты'.
О НО CN
II HCN \/
СНз—С—СН2СООС2Н5------> СН3—С—СН2—СООС2Н5
Взаимодействие с гидросульфитом натрия'.
О НО SO3?Ja
|| NaHSO, \/
СН3—С—СН3СООС2Н5------> СН3—С—СН2—СООС2Н5
Реакция с гидроксиламином'.
О N—ОН ОС2Н5
II nh2oh II I
СН3—С—СН2СООС2Н5------> СНз—С—СН2—С=О------------
-;.2о -CtHftOH
ОКСИ ft.
257
N—О
CH3—\=О
^сн^
метилоксазолон
Образующийся вначале оксим неустойчив и, теряя молекулу спир-
та, легко превращается в гетероциклическое соединение — метилок-
сазолон.
Реакция с фенилгидразином:
II NH2—NH-С«Н8
СН3—С—СН2СООС2Н5---------->-
—н2о
N—NH—С6Н5
—С2Н6ОН
N-N-CBH5
-> сн3—с \=о
метилфенилпиразолон
Как и в предыдущем случае, реакция заканчивается образованием
гетероциклического соединения (метилфенилпиразолон).
Реакции енольной формы. Для ацетоуксусного эфира
характерна качественная реакция на енолы — образование красно-
фиолетового окрашивания при взаимодействии с раствором хлорида
железа (III) (за счет сложных внутрикомплексных соединений железа).
Реакция присоединения брома по двойной связи. Происходит быстро
на холоду, а образующийся дибромид, легко теряя бромистый водород,
превращается в производное кетонной формы:
Вг Вг
Вг2 | |
СН3—С=СН—СООС2Н5 СН3—С СН—СООС2Н5-------------->
I I -НВг
ОН он
-> СНз—С—СН—СООС2Н5
11 I
О Вг
Ацилирование енольного гидроксила. Действием ацетил хлорида в
пиридине получают соответствующее О-ацетилпроизводное ацетоуксус-
ного эфира:
СН3СОС1
СН3—С=СН—СООС2Нб--------> СН3—С=СН—СООС2Нб
I пиридин |
он о—со—сн3
Действие пентахлорида фосфора:
РС15
СНз—С—СН—СООС2Н5--------> СНо—С=СН—СООС2Н5
ОН С1
258
Образование натрийацетоуксусного эфира. Происходит при взаимо-
действии с металлическим натрием, алкоголятом натрия или щелочью:
C2H6ONa
СНз—С=СН—СООС2Н5------------> сн3—с=сн—СООС2Н5
I -с2н6он I
ОН ONa
натрийацетоуксусный эфир
Натрийацетоуксусный эфир имеет широкое применение в органи-
ческом синтезе для получения различных соединений. При алкилиро-
вании и ацилировании натрийацетоуксусного эфира наблюдаются ре-
акции с переносом реакционного центра (А. Н. Несмеянов), в резуль-
тате чего вместо ожидаемых О-алкил- или О-ацилпроизводных образу-
ются соответствующие С-производные (алкил- и ацил ацетоуксусные
эфиры):
СН3 > СН3—С—СН—СООС2Н5 алкилирование || О
СН3—С=СН—СООС2Н5—
О—Na COCH3 CH3COCI СН3—С—СН—СООС2Н5 ацилирование q
Алкилированные и ацилированные производные ацетоуксусного
эфира представляют интерес в качестве исходных веществ для даль-
нейших превращений. Большое значение имеют реакции кетонного и
кислотного расщепления этих соединений, а также расщепление са-
мого ацетоуксусного эфира.
Кислотное расщепление ацетоуксусного эфира и его алкильных
и ацильных производных осуществляется действием концентрирован-
ного спиртового раствора щелочи и приводит к образованию различ-
ных карбоновых кислот:
СН3—С—1— СН2—С—О—•—С2Н5
Hi Hi
О : О • -> 2СН3СООК + С2Н5ОН
К—О—i—Н К—:О—Н
R
| 2NaOH
СН3—С—СН—СООС2Н5 -----> СН3—С—ONa + R—СН2—COONa + С2Н5ОН
259
CO-- R
| 2NaOH
CH3—C—CH—COOC2H5------> CH3C—ONa + R—CO—CH2—COONa + C2H5OH
Кетонное расщепление осуществляется действием разбавленных
растворов щелочей или кислот: происходит вначале омыление эфира,
в затем — декарбоксилирование образующейся |3-кетокислоты и обра-
зование метил кетонов:
он-
СН3—С—СН2—СООС2Н5 + но—н------*
II -с2н5он
О
СН3—С—СН2—СООН -> СН3—С—СН3 4- СО2
R R
I он- I ......
СН3—С—СН—СООС2Н5 4- НОН-------> СН3—С—СН—: СОО :Н ->
II -С2НбОН || ....’
О О
-> СН3-С—СН2—R 4- СО2
СО—R СО—R
| +НОН; ОН- |
сн3—С—СН—СООС2Н5--------> -> сн3—с—сн—соон ->
II -С2Н6ОН ||
о о
СН3С—СН2—С—R 4- со2
Таким образом, в результате кетонного и кислотного расщепления
ацетоуксусного эфира и его алкильных и ацильных производных мо-
гут быть получены уксусная кислота и ее гомологи, ацетон и его гомо-
логи, дикетоны, кетокислоты и т. д.
Раздел третий
Элементорганические
соединения
Молекулы большинства рассмотренных органи-
ческих соединений (углеводородов, спиртов, альдегидов, кетонов и
кислот) состоят из атомов небольшого числа элементов — органогенов:
С, Н, О, N, S, С1, Вг, I и Р (в виде фосфатов). Однако атомы углерода
обладают способностью соединяться практически со всеми элементами
периодической системы Д. И. Менделеева. Органические соединения,
в молекуле которых атом углерода непосредственно связан с атомами
этих элементов, получили названия элементорганических соединений.
Первыми изученными элементорганическими соединениями были сое-
динения углерода с металлами — металлорганические соединения.
По мере расширения наших знаний возникли разделы органической
химии, посвященные органическим соединениям кремния, серы, фос-
фора и т. д.
МЕТАЛЛОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Химия металлорганических соединений получила большое разви-
тие в последнее время. Это относится, в первую очередь, к соединениям
магния, алюминия, лития, которые из-за исключительно высокой ре-
акционной способности могут быть использованы промышленностью
для синтеза самых разнообразных веществ. Ниже приведены примеры
названий металлорганических соединений: C2H5MgBr — этилмагний-
бромид; Zn(C2H5)2 — диэтилцинк; СН2 = CHLi — виниллитий;
QHsMgBr — фенилмагнийбромид; А1(С2Н5)3 — триэтилалюми-
ний и т. д.
Способы получения. Взаимодействие металлов с галогеналкилами.
Этот метод применим к металлам со средней реакционной способностью
Li, Mg, Al и Zn.
СН3Вг + 2Li -> CH3Li + LiBr
метиллитий
СН31 + Mg - CH3MgI
метилмагний-
иодид
C2H5I + Zn -> C2H5ZnI
ЭТИЛЦИНКЙОДИД
3RX + 2A1 -> RA1X2 + R2A1X
Большую роль в этих реакциях играет растворитель. Так, магний-
органические соединения можно получать в абсолютно сухом диэтило-
261
вом эфире, в третичных аминах (например, диметиланилин в петролей-
ном эфире) и тетрагидрофуране. Алкильные соединения лития получа-
ют обычно в бензоле, так как они с эфиром медленно, но реагируют.
Фениллитий получают в эфире. Смешанные алюминийорганические
соединения типа RA1X2 hR2A1X получают без растворителя, исполь-
зуя в качестве катализаторов А1С13 или 12.
Действие сплавов на галогеналкилы. В случае очень реакционноспо-
собных металлов, например натрия, нельзя использовать взаимодейст-
вие его с галогеналкилом, так как основной процесс сопровождается
побочным — реакцией Вюрца:
ЦГ + 2Na------> RNa + Rf------> R—R
- Nar
—Nar
Сплавы же Na с мало реакционноспособными тяжелыми металлами,
которые непосредственно не взаимодействуют с галогенпроизводными
углеводородов, легко вступают в реакцию с последним:
4PbNa + 4С2Н5Вг -> Pb(C2H5)4 + 4NaBr + ЗРЬ
сплав
Получение алюминийорганических соединений (реакция Циглера).
Триалкильные соединения алюминия получают взаимодействием оле-
финов, водорода и мелкораздробленного металла:
6СН2=СН2 + 8Н2 + 2А1 -> 2(С2Н5)з А1
Реакция имеет большое значение благодаря широкому промышлен-
ному использованию алюминийорганических соединений, особенно
при полимеризации алкенов.
Химические свойства. Металлорганические соединения обладают
большой химической активностью. Связь С—Me поляризована, при-
чем металл является положительным концом диполя, углерод —
отрицательным:
\8-
—С Me
Реакционная способность металлорганических соединений сильно
зависит от характера металла: она наиболее велика у соединений ще-
лочных металлов и мала у тяжелых металлов.
Реакции металлорганических соединений — это типичные гетеро-
литические реакции.
Реакции замещения. Образование углеводородов. При вза-
имодействии металлорганических соединений с водой (1), спиртами (2),
кислотами (3), аммиаком, первичными и вторичными аминами (4) —
соединениями, содержащими «подвижный водород», образуется угле-
водород:
н—о—Н4- СНз-J—Mgl СН4 + Mgl(OH)
метилмариий* метан
ИОД
262
С?Н5°:Н.±.С?4з—j—CH4 + C2H5OMgI (2)
СН3СОО-ну+уc7h5—MgBr -> C6H6 + CH3COOMgBr (3)
H2N—:’h ’+W^-Mgl -> CH4 + NH2MgI (4)
(CH3)2 NjH + CH~3i—Mgl -> CH4 4- (CH3)2NMgI
Реакции (2)—(4) используются в аналитической химии как метод
определения активного водорода по Чугаеву — Церевитинову.
Взаимодействие магнийорганических соединений с ацетиленом (см.
с. 95).
Реакции присоединения. Металлорганические соединения легко
присоединяются к поляризованным кратным связям С=О, C=N,
C=N, C=S и др. Наибольший интерес представляют реакции присо-
единения к оксогруппе:
OMgT
рс==о + R—Mg—Г ----------- /С R
' Углеводородный радикал магнийорганического соединения присое-
диняется к карбонильному углероду, а остальная часть молекулы —
к атому кислорода; при разложении образовавшихся продуктов водой
получаются спирты. Подбирая соответствующие карбонилсодержащие
соединения, синтезируют первичные, вторичные и третичные спирты
(реакции Тищенко, Вагнера, Зайцева, Бутлерова):
н2о
Н2С=О + CH3MgI CH3CH2OMgI —> СН3СН2ОН + Mgl (ОН)
этиловый
спирт
n OMgl ОН
/, I Н,О I
Н3С—С<^ + CH3MgI СН3—СН--------> СНз—СН—СН3 + Mgl (ОН)
Н СН3
втор-пропиловый спирт
О OMgl он
/ I н2о |
СН3—С + CH3MgI -> СНз—С—СН3------► СНз—С—СН3 + Mgl (ОН)
\гн I I
СН3 СН3 СН3
mpem-бутиловый спирт
О OZnCl ОН
/ | н2о |
СН3—С + Zn(CH3)2 -> СН3—С—СН3--------> СН3—С—СН3 + Zn(OH)Cl
\ I I
С1 СН3 СН3
С помощью последней реакции Бутлеровым впервые были получе-
ны третичные спирты. Бутлеров и его ученики применяли для синтеза
спиртов цинкорганические соединения. Позднее работами французе-
263
кого химика Гриньяра было показано, что целесообразнее использо-
вать в таких синтезах магнийорганические соединения.
К этому же типу реакций относится и реакция металлорганических
соединений с диоксидом углерода, приводящая к получению карбоно-
вых кислот:
z° HCl zO
R—Me + CO2 R—C< ---------> R-C<f + MeCl
^OMe 4)H
Органические соединения цинка и магния. Цинкорганические со-
единения относятся к числу первых синтезированных металлоргани-
ческих соединений (Франкланд, 1894). Получаются действием гало-
генпроизводных на цинковые стружки или медь-цинковый сплав.
Цинкорганические соединения — низкокипящие жидкости, без цвета,
самовоспламеняющиеся на воздухе, что требует проведения работы с
ними в среде инертного газа.
Магнийорганические соединения имеют наибольшее значение сре-
ди органических соединений металлов II группы. Обычно их получают
с помощью реакции Гриньяра— взаимодействием галогеналкилов
с магнием в среде сухого органического растворителя (см. с. 261).
Наилучшие результаты получаются при использовании первичных
галогеналкилов. Синтезы с магнийорганическими соединениями про-
водят без их предварительного выделения из растворов, в которых они
получены, добавляя к ним второй компонент реакции. Магнийоргани-
ческие соединения широко применяются для разнообразных синтезов
и аналитических исследований.
Алюминийорганические соединения. Получают по реакции Циг-
лера (см. с. 261). Алюминийорганические соединения — бесцветные,
вязкие жидкости, самовоспламеняются на воздухе. Широкое приме-
нение в разнообразных синтезах нашел триэтил алюминий. Он исполь-
зуется для получения полиолефинов (особенно полиэтилена низкого
давления), каучука. Особый интерес представляет применение три-
алкилалюминиевых производных для синтеза высших спиртов. Снача-
ла получают высшие триалкилалюминиевые соединения из этилена
(избыток) и низшего триалкилалюминия (обычно триэтилалюминия):
R3A1 + 3nCH2=CH2 -> [R(CH2—-СН2)Л]3 А1
Полученные соединения окисляются кислородом в алкоголяты,
которые при разложении водой дают высший спирт и гидроксид алюми-
ния. При действии же диоксида углерода на высшие алюминийтриал-
килы можно получить соответствующие высшие жирные кислоты:
О2 ЗН2О
-> [R(CH2—СН2)ЛО]3 А1------> R(CH2—СН2)Л ОН
— А1(ОН)з
алкоголях высший спирт
СО2 ЗН2О
-> [R(CH2—СН2)„ СОО]3 А1 ----->- R(CH2—СН2)„СООН
— А1(ОН)3
высшая жипная кислота
Высшие жирные спирты и кислоты применяются для синтеза мою-
[R(CH2—СН2)Л]3 А1-
264
щих средств. Кислоты, кроме того, представляют интерес для получе-
ния синтетических пищевых жиров, так как они имеют неразветвлен-
ную цепь углеродных атомов, как и в природных жирах.
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Хотя кремний, так же как и углерод, находится в четвертой группе
периодической системы элементов, его соединения значительно отли-
чаются от соединений последнего. Основными группами кремнийорга-
нических соединений являются: производные силана SiH4(H3Si—SiH3
— дисилан, Si(CH3)4 — тетраметилсилан); гидроксильные производные
силана (R3SiOH— триалкилсиланол, RSi(OH)3— алкилсилантриол);
аминопроизводные силана (силазаны) i(CH3)3SiNH2 — триметилсила-
зан, (CH3)3Si — NH — Si(CH3)3 — гексаметилдисилазан]; соединения,
содержащие группу Si — О — Si, — силоксаны [(CH3)3SiOSi(CH3)3
— гексаметилдисилоксан)].
Для синтеза кремнийорганических соединений используют тетра-
хлорид кремния (получают действием хлора на кремний):
SiCl4 + LiAlH4 -> SiH4 + LiA1C14
тетра- силан
хлорид
RMgr RMgT RMgr
SiCl4 + RMgr -> RSiCl3--► R2SiCl2 -> R3SiCl---->- R4Si
Тетраметилсилан Si(CH3)4 — жидкость; /КИп = 26°C, по химичес-
ким свойствам несколько напоминает алканы. Хлорпроизводные —
алкилхлорсиланы легко гидролизуются водой.
Гидроксильные производные (силанолы) легко теряют воду:
2(С2Н5)3 Si—ОН -> (С2Н5)3—О—Si(C2H5)3 + Н2О
В последнее время разнообразные продукты полимеризации крем-
нийорганических соединений применяются в качестве электроизоля-
ционных, смазочных и каучукоподобных материалов.
СЕРОСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ
СОЕДИНЕНИЯ
В органических соединениях сера проявляет валентность, равную
двум (восстановленная сера), четырем и шести (окисленная сера). К со-
единениям первой группы относятся тиоспирты и тиоэфиры, ко второй
группе — сульфоксиды, сульфиновые кислоты, сульфоны и сульфо-
кислоты:
R—S—Н
тиоспирт
о
R—S-R f
R—S—Ri
тиоэфир сульфоксиды
о
t
R—S—ОН
сульфиновые кислоты
о о
t t
R—S—R. R—S—OH
о о
•ульфоны сульфокислоты
265
Тиоспирты, тиоэфиры. Тиоспирты (тиолы, меркаптаны) и тиоэфиры
(диалкилсульфиды, диарилсульфиды) можно рассматривать как произ-
водные сероводорода Н—S—Н. В случае тиоспиртов в молекулу вве-
ден один углеводородный радикал, в случае тиоэфиров — два. Тио-
спирты называют следующим образом: к названию радикала добавля-
ют слово «меркаптан» или формируют название, как в случае спиртов,
заменяя окончания «ол» на «тиол» (ИЮПАК). Для наименования тио-
эфиров к названию радикалов добавляют слово «сульфид»:
CH3SH — метилмеркашан; метантиол
C2H5SH — этилмеркаптан; эгантиол
С2Н5—S—С2Н5 — диэтилсульфид
Тиолы и тиоэфиры получают при взаимодействии галогеналкилов
и спиртов с сероводородом и сульфидами:
С2Н51 + KSH -> KI + C2H5SH
метилиодид этантиол
оксид тория
с2н5он + H2S----------> C2H5SH + Н2О
этиловый спирт С этантиол
2C2H5I + K2S -> 2KI + (C2H5)2S
этилиодид диэтилсульфид
Тиолы — низкокипящие жидкости с резким, крайне неприятным
запахом, тиоэфиры — нейтральные жидкости, кипят при более высо-
ких температурах, труднорастворимы в воде.
По химическим свойствам тиолы приближаются к сероводороду
(образование солей — меркаптидов RSMe), проявляя в то же время
некоторые свойства спиртов (взаимодействие с ацил хлоридами). При
окислении тиолов в мягких условиях образуются дисульфиды, при
действии сильных окислителей — сульфокислоты:
[О]
C2H5SH---
[О]
С2Н5—s—S—с2н5
ди этилдисульфид
С2Н5—SO2OH
этансульфокислота
Тиолы добавляют к природному газу для обнаружения по запаху
его утечки в жилых и производственных помещениях.
Иприт (Р,[3-дихлордиэтилсульфид) О—(СН2)2—S—(СН2)2—С1 яв-
ляется отравляющим веществом (ОВ).
К соединениям, содержащим восстановленную серу, могут быть от-
несены два важных в практическом отношении вещества: сероуглерод
и тиомочевина.
Сероуглерод S=C=S. Жидкость с неприятным запахом, /Ккп =
= 46,3°С. Очень горюч, токсичен. Сероуглерод нерастворим в воде,
хорошо растворим в органических растворителях. Получается при
266
взаимодействии серы с углеродом (температура 850—900°С) или из
метана:
СН4 + 4S CS2 + 2Н2
Температура реакции 600°С, катализатор — оксиды кремния или алю-
миния. Применяется в качестве растворителя, в производстве вискоз-
ного шелка, каучука. Является фунгицидом.
S
Тиомочевина, тиокарбамид h2N—С—NH2. Белое кристалличес-
кое вещество, хорошо растворимое в воде. Применяют в синтезе ряда
лекарственных препаратов, пластмасс, а также в качестве ростового
вещества.
Сульфокислоты. Из органических соединений окисленной серы наи-
более важными являются сульфокислоты — соединения, содержащие
в молекуле сульфогруппу SO3H, атом серы которой связан с углеродом.
Получение и свойства сульфокислот алифатического ряда было рас-
смотрено ранее (см. с. 60). В настоящем разделе познакомимся с суль-
фокислотами ароматического ряда.
Основным способом получения ароматических сульфокислот явля-
ется реакция сульфирования соответствующих углеводородов:
Z\
I || + h2so4 -* | || + нго
Сульфирование проводят олеумом на холоду или концентрированной
серной кислотой при нагревании. Для введения второй сульфогруппы
температуру повышают до 240°С, третьей — до 300°С. Сульфирование
— типичная реакция электрофильного замещения, электрофильным
агентом является гидросульфит (ион SO3H+) или триоксид серы SO3.
Сульфокислоты отделяют от серной кислоты, используя различия в
растворимости их кальциевых и бариевых солей.
Ароматические сульфокислоты — бесцветные кристаллические ве-
щества, хорошо растворимы в воде, гигроскопичны.
Сульфокислоты относятся к сильным кислотам. Для них характер-
ны три группы реакций — реакции в самой сульфогруппе, замещения
сульфогруппы и реакции бензольного ядра:
^\4/SO3H /\/SO3Na
| || + NaOH -* | || + H2O
натриевая соль
бензолсульфокислоты
Соли сульфокислот используют для получения ряда других соеди-
нений. Например, сплавлением солей сульфокислоты с едкими щелоча-
ми с последующим разложением образующихся фенолятов кислотой
получают фенолы. Это один из важных промышленных методов полу-
чения фенолов:
267
+ H2so4
При обработке солей сульфокислоты пентахлоридом фосфора по-
лучают сульфохлориды:
сульфохлорид
+ POClg + NaCl
Сульфохлориды — реакционноспособные вещества, реагируют со
спиртами и фенолами с образованием эфиров сульфокислот, а с аммиа-
ком дают амиды сульфокислот (сульфамиды).
Сплавление солей сульфокислот с цианистым калием приводит к
нитрилам:
Гидролиз сульфокислот. При действии на сульфокис-
лоту перегретого пара образуются соответствующий углеводород и
серная кислота:
Сульфогруппа относится к заместителям второго рода и в реакциях
электрофильного замещения ориентирует вхождение новых замести-
телей в метаположение.
Сульфокислоты применяются в технике для получения красителей,
лекарственных препаратов. Введением сульфогруппы в ароматическое
ядро часто пользуются для повышения растворимости соединений в
воде. Хлорангидриды сульфокислот используют для получения эфи-
ров, амидов ит. п., производные амидов — при изготовлении лекарст-
венных препаратов.
268
Часть
2
Азотсодержащие
органические
соединения
Гетеро-
циклические
соединения
Биоорганические
соединения
и некоторые
соединения
изопреноидного
характера
Раздел четвертый
Азотсодержащие
органические соединения
Органические соединения, содержащие в мо-
лекуле атомы азота, широко представлены в
природе. К ним относятся белковые вещества,
многие важнейшие физиологически активные сое-
динения, полимерные материалы, красители,
лекарственные препараты.
К группе более простых по строению азотсо-
держащих органических соединений, в которых
атом азота связан непосредственно с атомом уг-
лерода, можно отнести следующие:
1) нитрозосоединения R—N=O
2) нитросоединения R—
3) амины: первичные R—NH2
вторичные R—NH—R'
третичные R—N—R*
4) диазосоединения ArN2X
5) азосоединения ArN2Ar'
6) азиды (алкил- или арилазиды) RN3
7) гидразины (алкил или арилгидразины)
R_NH—NH2
8) некоторые производные карбоновых кис-
лот:
а) амиды R—с—NH2
б) нитрилы R—C=N
в) азиды (ацилазиды) R—CN3
г) гидразиды (ацилгидразины) R—С—NH—NHa
9) соединения со смешанными функциями, та-
кие, как аминоспирты, аминофенолы, аминокис-
лоты, нитрокислоты, аминосахара и т. д.
269
НИТРОСОЕДИНЕНИЯ
Определение, строение, номенклатура, изомерия. Нитросоедине-
ниями называются вещества, содержащие в молекуле нитрогруппу
(—NO2), азот которой непосредственно связан с атомом углерода. В
состав молекулы органического соединения могут входить одна или
несколько нитрогрупп. В зависимости от строения радикала, с кото-
рым связана нитрогруппа, различают алифатические (насыщенные и
ненасыщенные), алициклические, ароматические и гетероцикличес-
кие нитросоединения. С другой стороны, нитрогруппа в молекуле
нитросоединения может быть соединена с первичным, вторичным или
третичным атомами углерода, образуя, соответственно, первичные,
вторичные и третичные нитросоединения. Строение нитрогруппы от-
личается рядом особенностей, влияющих на физические и химические
свойства нитросоединений. Атом азота на своем внешнем электронном
уровне имеет четыре орбитали, на которых могут располагаться не
более восьми электронов. Поэтому формула нитрогруппы, как остат-
ка азотной кислоты, содержащая десятиэлектронную группировку
Чтобы не нарушать правила октета, следует допустить, что в
нитрогруппе имеется семиполярная связь между азотом и кислоро-
дом:
—N или N
чо’ ХО*
Физико-химическими методами исследований установлено, однако,
что оба атома кислорода в нитрогруппе абсолютно равноценны, по-
этому на самом деле истинное строение нитрогруппы следует изобра-
жать в виде мезомерных структур
что более точно передает строение группы с выравненной электронной
плотностью у обоих атомов кислорода:
Для наименования нитросоединений к названию соответствующего
углеводорода прибавляют приставку «нитро», например:
270
СН3
СН3—СН—сн—сн3
no2
2-нитро-З-метилбутан
CH3—N02
нитрометан
(3-нитро-
нафталины
Изомерия нитросоединений связана со строением углеродной це-
почки и положением нитрогруппы. Нитросоединения изомерны эфи-
рам азотистой кислоты:
R—NO2
нитросоединение
R—О—N=O
эфир азотистой кислоты
Способы получения. Основным способом получения нитросоеди-
нений является реакция нитрования. Нитрование может проводиться
азотной кислотой, нитрующей смесью (смесь азотной и серной кислот),
оксидами азота. В зависимости от строения исходных веществ, условий
нитрования, характера нитрующего агента в результате реакции по-
лучаются несколько различные продукты. Общая схема реакции имеет
следующий вид:
R—Н
или
Аг—Н
нитрование
R—NO2
или
ArNO2
Нитрование алканов и цикланов может быть осуществлено по реак-
ции Коновалова или в газовой фазе (парофазное нитрование, см. с. 58).
Нитрование ароматических соединений — типичный пример реакций
электрофильного замещения (см. с. 126). Нитрогруппа является замес-
тителем второго рода, поэтому вторая и третья нитрогруппы вступают в
метаположения. Если получение нитробензола может быть успешно
осуществлено при температуре 30—40°С, то введение второй нитро-
группы требует более высоких температур: 90—100°С, а третьей 130—
150°С, а также сильного нитрующего агента.
Для получения первичных или вторичных алифатических нитро-
соединений можно использовать взаимодействие галогеналкилов с
271
uz
Таблица 25. Физические свойства некоторых нитросоединений
Название Формулы Температура плавления, °C Температура кипения, °C Относительная плотность d20 4 Показатель преломления Электрический момент диполя, Кл-м -10”30
Нитрометан CH3NO2 —28,6 101,0 1,138 — 10,59
Нитроэтан CH3CHoN02 —90 114,8 1,051 — 10,65
Нитробензол no2 5,7 210,9 1,2030 1,5529 13,26
2-Нитротолуол Z ^-no2 —3,2 222,3 1,1630 1,5474 12,32
СН3
/ S—no2 16 232 1,1571 1,5475 13,93
3-Нитротолуол
СЙз
4-Нитротолуол CH3—NO2 52 238 1,1286 1,5346 (при 62,5еС) 14,83
I j3-Динитробензол Nof 90 303 1,571 (d°) — 12,62
NO^
18 3 5-Тринитробензол 04 О z 1 z\ 122 разл. 1,688 — 2,67
нитритами серебра или натрия (в диметилформамиде или диметилсуль
фоксиде, обладающих большой сольватирующей способностью). В ка-
честве побочных продуктов образуются эфиры азотистой кислоты, ко-
торые можно легко отделить, так как они имеют значительно более
низкие температуры кипения, а также в отличие от нитроалкилов легко
гидролизуются водой:
-------> C2H5NO2
С2НаВг + AgNO2------- нитроэтан
—AgBr
этилбромид н q
-------> С2Н5—О—NO —С2Н5ОН
(-HNO,)
этилнитрит
Третичные нитросоединения этим путем получить не удается.
Физические свойства. Нитросоединения жирного ряда — высококипящие
жидкости, с приятным запахом, мало или совершенно не растворимые в воде.
Плотность первых четырех представителей гомологического ряда алканов (до
нитробутана) больше единицы, последующих — меньше единицы. Ароматичес-
кие нитросоединения — жидкости или твердые вещества, с запахом горького
миндаля, ядовиты. Нитросоединения обладают большим электрическим момен-
том диполя. Повышенная полярность нитросоединений, высокие температуры
кипения и плавления (например, по сравнению с изомерными им эфирами азо-
тистой кислоты) связаны с наличием в их молекуле семиполярной связи
(табл. 25)
Химические свойства. Химическое поведение нитросоединений,
определяется наличием в молекуле нитрогруппы и ее особенностями,
а также строением углеводородного радикала и их взаимным влия-
нием.
Восстановление нитросоединений. При восстановлении нитросоеди-
нений образуются первичные амины. Особенно большое промышленное
значение имеет восстановление ароматических нитросоединений:
6Н
C6H5NO2 ----->-C6H5NH2
—2Н8О
нитробензол анилин
В зависимости от условий восстановления (в кислой, щелочной или
нейтральной средах) и характера восстановителя в ходе реакции об-
разуются различные промежуточные продукты, многие из которых
нашли широкое применение в технике. В кислой или нейтральной
среде:
нитробензол
фенилгидроксиламин анилин
В щелочной среде:
10—682
273
фенилгидроксиламин
конден-
сация
2Н
—Н2О
Любые промежуточные продукты восстановления нитросоединений
могут быть получены также электролитическим путем при соответству-
ющих режимах электролиза.
Действие щелочей на нитросоединения. При введении в молекулу
углеводорода нитрогруппа вследствие ее электроноакцепторных
свойств резко повышает подвижность атомов водорода в а-положении.
Первичные и вторичные нитросоединения приобретают способность
растворяться в щелочах с образованием солей. При действии кислоты
образуется нитросоединение в аци-нитроформе, которая затем пере-
ходит в нитроформу:
а NaOH /ONa
R—СН2—---------->R—CH—N->0 соль аци-формы
0 1HC1
нитросоединен ие i
R—CH=N—ОН
\
О
аци- нитроформа
Взаимное превращение двух форм нитросоединений является типичным
примером динамической изомерии (таутомерии):
а /О /ОН
R—СН2—Nf R—CH=N<
ЧО ЧО
нитроформа аци-нитро форма
Действие азотистой кислоты на нитросоединения. При взаимодей-
274
ствии первичных нитросоединений с азотистой кислотой образуются
цитроловые кислоты, вторичных — псевдонитролы:
R— СН,—NO2 + НО—N=O---------> R—СН—NO R—C=N—ОН
-Н,о | |
первичное нитросоединение
нитроловые кислоты
no2
О I
R—СН—NO, + НО—N=O-----------► R—С—NO
| -н,о |
R R
вторичное нитросоединение псевдонитролы
Третичные нитросоединения (отсутствуют а-водородные атомы) с
азотистой кислотой не реагируют.
Щелочные соли нитроловых кислот и их растворы окрашены в ярко-
красный цвет, растворы псевдонитролов в эфире и хлороформе — в си-
ний цвет. Эта реакция позволяет различить первичные, вторичные
и третичные нитросоединения.
Конденсация с альдегидами. Наличие подвижных а-водородных ато-
мов в первичных и вторичных нитросоединениях делает возможным
их конденсацию с альдегидами по альдольно-кротоновому типу:
ОН
а (ОН”) |
с6н5сн=о + CH3NO2------>С6Н5СН—CH2NO2-------►
-НгО
(i - нитро - а - фе ни л-
этиловый спирт
----> с6н5сн=сн—no2
нитростирол
Реакции бензольного ядра ароматических нитросоединений. Нитро-
группа предпочтительно ориентирует вхождение второго заместителя
в реакциях электрофильного замещения в метаположение. Реакции за-
мещения в этом случае протекают гораздо труднее, чем для самих угле-
водородов:
275
Ю*
При наличии в бензольном ядре нескольких нитрогрупп, обладаю-
щих электроноакцепторными свойствами, ядро в орто- и параположе-
ниях становится доступным для нуклеофильных реагентов:
NO2 no2
I I
__MJJ
UH,N—OH(C2H5ONa) I II 1Nn2
NO I H
^2 no2
1,3-динитробензол 2,4-динитроанилин
В случае тринитросоединений водородные атомы в ортоположении
обладают еще большей подвижностью, что сказывается и на легкости
окисления. Так, тринитробензол окисляется в щелочной среде слабыми
окислителями (например, железосинеродистым калием), превращаясь
в тринитрофенол (пикриновая кислота):
NO2 no2
K3Fe(CN)e
I
no2
тринитробензол
[О]
ОН
NO2 I no2
\У\/
V
NO2
пикриновая кислота
Применение. Нитропарафины применяются в промышленности в
качестве растворителей, добавок к дизельным топливам, снижающим
температуру их воспламенения, при производстве взрывчатых веществ,
пластмасс, в реактивной технике, в качестве полупродуктов в синтезе
аминов, альдегидов и кетонов, жирных кислот. Ароматические нитро-
соединения широко применяются для получения красителей, пласт-
масс, душистых и взрывчатых веществ.
Отдельные представители. Нитрометан СН3—NO2. Жидкость,
tКип = 101,0°С. Применяется в качестве растворителя. Хлорированием
нитрометана получают трихлорнитрометан (хлорпикрин) CHCI3NO2,
который применяется для борьбы с грызунами в хлебохранили-
щах и складах и в разнообразных синтезах. Хлорпикрин токсичен.
Нитроэтан С2Н5—NO2. Жидкость, /КИп = П4,8°С. Применяется
для получения гидроксиламина (действием серной кислоты), одновре-
менно образуется уксусная кислота:
н2о
сн3—сн2—no2--------> сн3—соон + nh2oh
(H2SO4)
нитроэтан уксусная гидроксиламин
кислота
Нитроциклогексан C6HnNO2. Жидкость, /кип = 205°С. Получают
нитрованием циклогексана. Применяется в качестве полупродукта в
синтезе капролактама.
Нитробензол C6H5NO2. Жидкость желтоватого цвета, с запахом
276
горького миндаля, гКИп = 210,9°С. Плохо растворим в воде и хорошо
во многих органических растворителях. Исходный продукт в производ-
стве анилина (см. с. 273), широко применяется в анилинокрасочной,
парфюмерной, фармацевтической промышленностях.
no2
Тринитротолуол, тол, тротил O2N—СН3. Твердое
КЮ2
вещество, /пЛ = 80°С. Получают прямым нитрованием толуола нит-
рующей смесью:
Является одним из самых распространенных детонирующих взрывча-
тых веществ.
NO..
а-Нитронафталин Твердое вещество, /пЛ = 6ГС. Полу-
чают нитрованием нафталина. Используют для получения а-нафти-
ламина — исходного вещества для синтеза ряда производных нафтали-
на и азокрасителей.
АМИНЫ
Амины можно рассматривать как производные аммиака, в молекуле
которого атомы водорода замещены на углеводородные радикалы.
Однозамещенные производные аммиака называются первичными, двух-
замещенные — вторичными, трехзамещенные — третичными аминами:
NH3
аммиак
R—NH2 R—NH
I
R
первичный амин вторичный амин
R—N—R
I
R
третичный амин
Таким образом, первичные амины содержат аминогруппу —NH2,
вторичные — иминогруппу ^NH, третичные — лишь третичный атом
азота. Известны также соединения с четвертичным атомом азота: чет-
вертичный гидроксид аммония и его соли:
277
г R
I
R— N—R
R
OH-
четвертичный аммоний-
гидроксид
четвертичные аммоние-
вые соли
В зависимости от строения радикала различают алифатические
(насыщенные и ненасыщенные), алициклические, ароматические (со-
держащие аминогруппу в ядре или в боковой цепи) и гетероцикличес-
кие амины.
Номенклатура. Изомерия. По рациональной номенклатуре назва-
ния аминов образуются от названия углеводородных радикалов, со-
единенных с атомом азота, и суффикса «амин»:
СН3
СН3—NH2 CH3-NH-CH3 ch3-n—сн3
метиламин диметиламин триметиламин
Амины, содержащие ароматические радикалы, а также амины с не-
сколькими аминогруппами рассматриваются, обычно, как аминопроиз-
водные углеводородов:
NH2
N, N-диметиламиноб енз ол
(N, N-диметиланилин)
аминобензол (фени-
ла мин, анилин)
nh2— сн2—сн2—nh2
1,2-диаминоэтан
Многие амины имеют тривиальные названия (табл. 26). Для наиме-
нования аминов пользуются номенклатурой ИЮПАК, например:
3 2 1 2 1
СН3 — СН—СН2—NH2 СН3—NH—СН- СН3
I I3
СН3 СН3
1-амино-2-метилпропан 2-метиламинопропан
1 2
СН3—N—СН2—СН3
СН3
дим етиламиноэтан
Изомерия аминов зависит от изомерии радикалов. Для аминов воз-
можна метамерия.
Способы получения. Восстановление нитросоединений (реакция
Зинина). В 1842 г. И. Н. Зинин восстановлениехм нитробензола суль-
фидом аммония впервые синтетическим путем получил анилин. Реак-
ция сыграла выдающуюся роль в создании анилинокрасочной промыш-
ленности и получении разнообразных органических красителей синтети-
ческим путем. В настоящее время восстановление нитробензола в тех-
нике осуществляется двумя путями:
278
а) восстановлением нитробензола железными стружками в присут-
ствии раствора хлорида железа (III):
4CeH5NO2 + 9Fe + 4Н2О--------» 4CeH5NH2 + 3Fe3O4
нитробензол анилин
б) каталитическим гидрированием нитробензола молекулярным
водородом:
CeH5NO2 —К" C6H5NH2 + 2Н2О
нитробензол анилин
Реакция протекает при температуре 270—300°С, давлении до
15,2-104 Па в присутствии металлов или их оксидов (например, меди,
нанесенной на диоксид кремния).
Алкилирование аммиака (реакция Гофмана). При действии аммиака
на галогеналкилы атомы водорода в аммиаке постепенно замещаются
на углеводородные радикалы:
NH3 + СН31 -------->- [CH3NH3]+ I" —CH3NH2 + NH4I
метилиодид метиламмонийиодид метиламин
NH3
CH3NH2 + CH3I---------> [(CH3)2NH2]+I-------HCH3)2NH + NH4I
ди метила ммонийиодид диметиламин
NH3
(CH3)2NH + CH3I---------> [(CH3)3NH]+I------->(CH3)3N + NH4I
триметиламмоний- триметил-
иэдид амин
(CH3)3N + CH3I--------> [(CH3)4N]+I-
тетраметиламмоний-
иодид
Алкилирование аммиака является примером реакций нуклеофиль-
ного замещения: атом углерода в иодистом метиле вследствие поляриза-
ции связи С8+->- F” имеет частичный положительный заряд и нук-
леофильно атакуется аммиаком, азот которого отдает на образование
связи неподеленную электронную пару.
Для получения низших алкиламинов (Сг—С<) иногда прибегают к
алкилированию аминов не галогеналкилами, а спиртами:
350°С; А12О3
R—ОН + NH3----------> R—NH2 + Н2О
2R—ОН + NH3------> R2NH + 2Н2О
3R—ОН + NH3------> R3N + ЗН2О
Смесь аминов разделяют фракционной перегонкой на эффективных ко-
лонках.
Получение первичных аминов из амидов кислот (перегруппировка
Гофмана). При расщеплении амидов гипохлоритами или гипоброми-
тами протекают реакции, которые можно выразить суммарным урав-
нением:
279
о
II
R—С—NH2 + NaOBr--------* R—NH2 + CO2 + NaBr
амид карбоновой первичный
кислоты амин
Восстановление нитрилов и изонитрилов. Восстановление нитрилов
при помощи металлического натрия в спирте или водородом в присут-
ствии палладиевого катализатора приводит к первичным аминам:
2Н2
R—C=N--------> R—СН2—NH2
нитрил первичный амин
Изонитрилы при восстановлении образуют вторичные амины:
2Н2
R—N^C у R—NH—СН3
изонитрил вторичный амин
Физические свойства. Простейшие амины алифатического ряда — бесцвет-
ные, газообразные, горючие вещества, растворимые в воде. Обладают явно выра-
женным запахом испорченной рыбы. Растворимость аминов в воде падает с уве-
личением молекулярной массы. Ароматические амины представляют собой бес-
цветные жидкости или твердые вещества, обладающие неприятным запахом и
весьма ядовитые. В табл. 26 приведены характеристики некоторых аминов.
Химические свойства. Химическое поведение аминов определяется
наличием в их молекуле аминогруппы. На внешней электронной обо-
лочке атома азота имеется пять электронов. В молекуле амина так
же, как в молекуле аммиака, атом азота затрачивает на образование
трех ковалентных связей три электрона, а два электрона остаются
свободными:
CH3:N: Н
Н
Наличие свободной электронной пары у атома азота дает ему воз-
можность присоединять протон, поэтому амины, подобно аммиаку,
проявляют основные свойства: образуют гидроксиды, соли. В этих
реакциях амины, отдавая электронную пару на образование новой свя-
зи, ведут себя как нуклеофильные реагенты.
Образование гидроксидов. Водные растворы низших аминов имеют
щелочную реакцию, окрашивают красный лакмус в синий цвет вслед-
ствие образования алкилзамещенных гидроксидов аммония:
СН3—NH2 + Н2О [СН3—-NH3J+OH~
метиламин гидроксид метиламмония
Высшие амины, плохо растворимые в воде, не дают щелочной ре-
акции на лакмус.
На основные свойства аминов влияет строение радикала, с которы-
ми связана аминогруппа. Алифатические амины обладают большими
основными свойствами, чем ароматические. Это объясняется тем, что
свободная электронная пара атома азота в ароматическом амине на-
ходится в сопряжении с л-электронами бензольного ядра:
280
Таблица 26. Физические свойства некоторых аминов
Название Формулы Температура плавления, °C Температура кипения, °C Относительная плотность ^20 4 Показатель 20 преломления пр кв (25°С)
Метиламин ch3-nh2 —92,5 —6,5 0,769 (^°) 1,432 (при 17,5°С) 1,8-10-5
Диметиламин (CH3)2nh —96 7,4 0,680 (О 1,350 (при 17°С) 9,6-10-*
Триметиламин (CH3)3N — 124 3,5 0,662 К5) — 4,4-10~*
Этиламин сн3-сн2—nh2 —80,6 16,6 0,706 И) — 5,6-10~4
Диэтиламин (СН3—CH2)2NH —50 55,8 0,7056 1,3873 (при 18°С) —
Триэтиламин (СНз-CH2)3N — 115 89,5 0,723 1,4003 —
«-Пропиламин СНз-СНз—сн2—nh2 —83 48,7 0,719 1,3900 (при 16,6° С) —
Этилендиамин (1,2-диа- миноэтан) nh2—сн2—сн2—nh2 8,5 116,5 — — —
Название Формулы
Анилин (аминобензил) О и Z I to
N-Метиланилин (N-метиламинобензол) NH—СН3
N, N-Диметиланилин (N, N-диметиламинобен- зол) { ">-N(CH3)2
о-Толуидин (2-аминотолуол) <N К Z । 5 /\/
л<-Толуидин (3-аминотолуол) nh2 СН3
л-Толуидин (4-аминотолуол) н3с—nh2
Окончание табл. 26
Температура плавления, °C Температура кипения, °C Относительная плотность .20 d4 Показатель 20 преломления пр Кв (25°С)
—6,2 184,4 1,022 1,5863 3,82-10-10
—57 196,3 0,986 1,5702 (при 2 ГС) 5-10-1°
2,5 192,5 0,955 1,5582 1,15.10-»
—27,7 199,7 1,004 1,5728 2,47. IO-10
—43,6 203,2 0,989 1,5711 (при 22°С) 4,92-IO"10
—43,7 200,4 1,046 1,5532 (при 59°С) 1,18-10-»
Такое распределение электронной плотности снижает способность
атома азота удерживать протон, т. е. приводит к уменьшению основных
свойств.
Образование солей. С минеральными кислотами амины образуют
соли. Реакция образования таких алкил аммониевых солей подобна
той, которая происходит между аммиаком и минеральной кислотой:
Н
H:N: + НС1
Н
аммиак
Н V
H:N:H С1“
Н _
аммонийхлорид
СН3
H:N: + НС1
Н
метиламин
СН3
CH3:N: + HNO3
Н
- СНз-I
± H:N:H
_ Н _
метиламмонийхлорид
сг
СН3 -1
± CH3:N:H
Н _
диметиламмонийнитрат
no;
Под действием более сильного основания соли аминов вновь обра-
зуют свободные амины:
NaOH
[R2NH2]+C1“--->- r2nh
—NaCl
-Н2О
Реакция алкилирования. Алкилгалогениды взаимодействуют с пер-
вичными, вторичными и третичными аминами, образуя соответствую-
щие соли (см. с. 279, реакция Гофмана).
Реакция ацилирования. Первичные и вторичные амины при взаимо-
действии с хлорангидридами, ангидридами или сложными эфирами
образуют алкиламиды кислот:
-HCI
(R'CO)2O
R—NH2---------------f----
алкила мин —R'COOH
О
R'-C
\OR'
- R'OH
О
II
R—NH—С—R'
алкиламид
283
Третичные амины не вступают в эту реакцию.
Взаимодействие с азотистой кислотой. Первичные алифатические
амины реагируют с холодным водным растворохм азотистой кислоты,
давая алкилдиазониевые соли. Эти соли чрезвычайно неустойчивы и
разлагаются с образованием смеси продуктов:
НО—N=O
сн3—сн2—сн2—NH2--------> [СН3—СН2—CH2N* ] +
—н«о
пропиламин пропилдиазониевый ион
—N2 + Н2О
----[СН3—СН2—СН2]-------
СН3СН2СН2ОН
СН3СН(ОН)СН3
СН3СН=СН2
(СН2)3
Первичные ароматические амины при взаимодействии с азотистой
кислотой в присутствии сильных минеральных кислот образуют соли
диазония (см. с. 288).
Вторичные амины, как алифатические, так и ароматические, и пре-
вращаются в нитрозоамины — соединения, содержащие характеристи-
ческую (нитрозо) группу —N=O:
R4
>NH + НО—N=O —~?
D'/ —н2о
нитрозоамин
вторичный
амин
Третичные алифатические амины с азотистой кислотой образуют
непрочные соли, распадающиеся при нагревании с образованием слож-
ных смесей органических продуктов (в том числе альдегидов и кето-
нов).
Третичные ароматические амины дают п-нитрозосоединения:
НО—N=O-
п-нитрозо-N, N-диметиланилин
Последняя реакция представляет собой обычное электрофильное
замещение в бензольном ядре, сильно активированном электронодо-
норной диметиламиногруппой.
Реакция бензольного ядра в ароматических аминах. Аминогруппа
является заместителем первого рода, она повышает электронную плот-
ность в ядре бензола, особенно в орто- и параположениях, и облегчает
вхождение заместителей в реакциях электрофильного замещения.
Применение. Амины применяются в качестве селективных раство-
рителей, в производстве красителей, поверхностно-активных веществ,
фармацевтических препаратов. Алкиламины используются при произ-
водстве ускорителей вулканизации каучука, инсектицидов. В промыш-
284
ленности полимерных материалов большую роль играют диаминпроиз-
водные углеводородов.
Отдельные представители. Моноамины алифатического ряда. Мо-
но- , ди- и триметиламины используются в производстве красителей,
фармацевтических препаратов. Этиламин применяется при крашении
тканей, в медицине, как компонент ракетного топлива.
1,6-Гексаметилендиамин NH2—(СН2)в—NH2. Твердое вещество, по-
лучается из адипиновой кислоты:
2NH3
НООС—(СН2)4—СООН ------->• H4NOOC—(СН2)4—coonh4 ------->
-2Н2О
адипиновая кислота диаммонийадипат
-----> H2N—СО—(СН2)4—со—nh2---------->
—2Н2О
диамид адипиновой кислоты
8Н/кат
----> CN—(СН2)4—CN--------> H2N— [СН2]6—NH2
динитрил адипиновой 1,6-гексаметилендиамин
кислоты
Гексаметиленди амин играет большую роль в производстве полимер-
ных материалов, в молекуле которых мономеры соединены амидными
группами —NH—СО—, получивших название полиамидов. Примером
полиамидов является продукт поликонденсации гексаметилендиамина
и двухосновной карбоновой кислоты (адипиновой) — найлон:
/гНООС—(СН2)4—СООН + nNH2—(СН2)е—NH2--------->
—2пН2О
адипиновая кислота гексаметилендиамин
----> [—СО—(СН2)4—СО—NH—(СН2)6—NH—]л
найлон
и продукт полимеризации капролактама — капрон:
СН2—СН2
I I
п СН2 СН2 -
сн2 с=о
капролактам
—NH—(СН2)5 — С—NH—(СН2)5—С— \п
капрон
Полиамиды, выпускаемые во многих странах под различными на-
званиями, из-за высокой прочности, упругости и химической устой-
чивости нашли широчайшее применение для изготовления техничес-
ких и бытовых тканей, лент, сетей и т. д.
Аминобензол, анилин C6H5NH2, /КИп = 184,4°С; /пл = —6,2°С.
Бесцветная жидкость с характерным запахом. Получается восстанов-
лением нитробензола (см. с. 279). Легко бромируется, сульфируется,
окисляется. Аминогруппа— сильный электронодонорный заместитель,
повышающий электронную плотность в ароматическом ядре. Бромиро-
вание анилина происходит легко уже при действии бромной воды,
давая триброманилин:
285
Нагревание анилина с концентрированной серной кислотой при-
водит к образованию сульфаниловой кислоты, являющейся важным
полупродуктом в синтезе ряда красителей и лекарственных веществ:
NH2
сульфаниловая кислота
Анилин окисляется уже на воздухе, что проявляется в образовании
желто-бурой окраски. При окислении анилина хлорной известью появ-
ляется фиолетовое окрашивание (качественная реакция на анилин).
Азотная кислота действует на анилин тоже как окислитель. Поэтому
перед нитрованием проводят «защиту» аминогруппы анилина чаще все-
го ацетильным остатком:
(СН8СО)2О
-СНзСООН
Введенная ацетильная защита не меняет ориентирующего влияния
заместителя и при действии на ацетанилид нитрующей смеси образу-
ются n-нитроацетанилид (в основном) и о-нитроацетанилид (в неболь-
шом количестве):
Для получения нитроанилинов необходимо снять введенную «за-*
щиту», что достигается кислотным гидролизом:
286
п-нитроанилин
Анилин — один из важнейших продуктов современной органичес-
кой химии. Применяется в производстве красителей, фармацевтичес-
ких препаратов, вспомогательных веществ для резиновой промышлен-
ности, получения полимерных материалов.
ПОНЯТИЕ О ДИАЗО- И АЗОСОЕДИНЕНИЯХ
В диазосоединениях ArN2X атомы азота связаны с ароматическим
радикалом и неорганическим остатком Х(С1“, Вг* NO3, HSO4, BF4,
ОН").
Азосоединения содержат группировку —N=N—(азогруппа), свя-
занную с двумя радикалами. В зависимости от характера радикала ди-
азо- и азосоединения могут быть алифатического, ароматического или
гетероциклического ряда. Наибольший интерес представляют аромати-
ческие диазо- и азосоединения благодаря разнообразному применению
в промышленных синтезах.
Диазосоединения
Строение. Изомерия. Диазосоединения существуют в нескольких
формах:
[Аг—N+ = N] Cl-
соли диазония
[Аг—N=N—ОН
диазогидраты
[Аг—N+ = N] ОН-
гидроксиды диазония
Аг—N=N—ОМе
диазотаты
Рассмотрим взаимные превращения различных форм диазосоеди-
нений (таутомерия) на примере простейших веществ:
NaOH
[C6H5N+ = NJ Cl- ~ СбН5—N=N—ONa
на
фен и лдиа зон ийх лор ид
НС1 AgOH
фенилди азотат натрия
НС1
(разб)
NaOH
изомеризац ия
[C6H5N+ = N] ОН* t * CeH5—N=N—ОН
гидроксид фенил- фенилдиазогидрат
диазония
В кислых средах диазосоединения существуют в виде солей диазо-
ния, в щелочных — в виде диазотатов, а в средах, близких к нейт-
287
ральной, — в виде изомерных гидроксидов диазония и диазогидратов.
Из всех форм диазосоединений наибольший интерес представляют
соли диазония, поэтому подробнее остановимся на рассмотрении имен-
но этих соединений.
Способы получения. Основной способ получения солей диазо-
ния — реакция диазотирования: действие азотистой кислоты на соль
первичного ароматического амина в кислой среде. Азотистую кислоту
получают непосредственно в процессе диазотирования, действуя со-
ляной кислотой на нитрит натрия. Весь процесс проводят при ох-
лаждении, применяя избыток соляной кислоты:
c6h5nh2 + НС1-----> C6H5NH2-HC1
анилин солянокислый анилин
NaNO2 + НС1-------> HNO2 ± NaCl
C6H5NH2-HC1 + HNO2------> [C6H5N=N]+Cr + 2H2O
фенилдиазонийхлорид
Свойства. Соли диазония в сухом состоянии легко взрываются,
поэтому пользуются обычно их растворами, полученными в результа-
те реакции диазотирования.
Превращения диазосоединений могут быть объединены в две груп-
пы: реакции, протекающие с выделением азота, и реакции, протекаю-
щие без выделения азота.
Реакции с выделением азота (обмен диазогруппы на
другие группировки). Получение фенолов'.
+ X- + Н2О------* Аг—ОН + N2 + НХ
фенол
Реакция гидролиза солей диазония медленно протекает в ледяном
растворе солей диазония. Она является причиной, по которой соли
диазония следует немедленно использовать после их приготовления,
так как при повышении температуры реакция может стать главной.
Замещение диазогруппы на хлор, бром и CN-группу. Реакция Зайд-
мейера (катализаторы — соответствующие соли одновалентной меди):
ГАгЫ!
288
Гидролиз нитрилов приводит к карбоновым кислотам. Таким об-
разом, синтез нитрилов из солей диазония — хороший способ превра-
щения нитросоединений в карбоновые кислоты, например:
hno8
h2so4
л-нитротолуол
NaNO2
НС1
n -толуидин
n- толуиловая
кислота
Этот путь синтеза ароматических карбоновых кислот более универ-
сален, чем карбоксилирование реактива Гриньяра и окисление боко-
вых цепей.
Замещение диазогруппы на иод:
KI
[ArN=N]+Cr -----> Ari + N2 + КС1
Реакция идет при простом смешении реагентов.
Обмен диазогруппы на фтор:
hbf4
[C6H5N=N]+C1-------> [C6H5N = N]+ BF7
фенилдиа зонийборфторид
, _ осторожное нагревание
[C6H5N=N] +bf4---------------------> C6H5F+N2 + BF3
Борфториды диазония довольно устойчивы, при нагревании в сухом
виде разлагаются с образованием арилфторидов.
Образование металлорганических соединений (реакция Несмеянова).
При взаимодействии солей диазония с галогенидами металлов (ртуть,
сурьма, висмут, олово) в присутствии медного порошка образуются
смешанные металлорганические соединения:
[C6H5N = N]+Cr + HgCl24- 2Cu ---> C6H5Hg—Cl + N2 + Cu2Cl2
Взаимодействие солей диазония co спиртами. Реакция идет в двух
направлениях: спирт окисляется в альдегид, соль диазония восстанав-
ливается до углеводорода, а диазогруппа обменивается на алкокси-
группу и образуется простой эфир фенола:
289
Г CeH5N 1 +
III СГ
N
СгНаОН
,0
C6He + сн3с<^ + n2
с6н5-о-с2н5 + n2 + НС1
фенетол
Реакции, идущие без выделения азота. К та-
ким реакциям относятся восстановление диазосоединений и азосоче-
тание. Восстановление солей диазония. Приводит к соответствующим
гидразинам, например:
Г C6H5-N 1+ Cl-
lll
L NJ
фенилдиазо нийх лор ид
С6Н5—NH—NH2 + НС1
фен илгидразин
Восстановление ведут с помощью дихлорида олова или сульфида
натрия.
Реакция азосочетания. Это основной способ получения азосоедине-
ний. Соли арилдиазониев (слабые электрофилы) вступают во взаимо-
действие с производными бензола, имеющими сильные электроно-
донорные группировки, такие, как фенолы и амины. Атака электрофила
всегда происходит почти исключительно в параположение к гид-
роксилу (в фенолах) или аминогруппе (в аминах). Если параположение
занято, то атака направляется в ортоположение. Первичный аромати-
ческий амин, из которого была получена соответствующая соль диазо-
ния, называют диазосоставляющим (диазокомпонентом) реакции азосо-
четания. Амин или фенол, сочетающийся с солью диазония, называют
азосоставляющим реакции азосочетания.
Азосочетание фенолов проводят в слабощелочной среде (отрица-
тельно заряженный кислород фенолят-аниона активирует ароматиче-
ское ядро сильнее, чем гидроксил самого фенола, что важно для после-
дующей Se -атаки катионом диазония):
Первичные и вторичные ароматические амины сочетаются с солями
диазония в нейтральных или слабокислых средах по атому азота, об-
разуя диазоаминосоединения (1,3-диарилтриазены), которые при под-
кислении перегруппировываются в аминоазосоединения:
290
X—N ci'+ NH,—X X-->
\=/ III \=/ -на
N
фенилдиазонийхлорид анилин
диазоа минобензол (1,3-дифенилтриазен)
аминоазобензол
Третичные ароматические амины сочетаются с солями диазония в>
кислых средах:
n-диметила миноазобензол
Активность азосоставляющих в реакциях азосочетания увеличива-
ется с увеличением электронной плотности бензольного ядра. Это про-
является в соединениях (многоатомные фенолы, аминофенолы, поли-
амины), являющихся наиболее активными азокомпонентами. С другой,
стороны, активность диазосоставляющих растет с возрастанием поло-
жительного заряда на катионе диазония, чему в значительной мере
способствует наличие в ароматическом ядре диазокомпонентов силь-
ных электроноакцепторных заместителей.
Азосоединения играют большую роль в производстве красителей,
многие из которых являются их производными.
По современным представлениям окраска органических соединений
связана с наличием в их молекуле особых группировок, получивших
название хромофорных групп или хромофоров (носителей цветности,
от греческих слов «хромое» — цвет, «форос» — носитель). Характер-
ная структурная особенность хромофоров — наличие у них группиро-
вок, содержащих двойные связи. К этим группам относятся:
\ к /О ч /
—N=N— >С=О )N=O — Nf >С=С<
/ / / \
азогруппа оксогруппа нитрозогруппа нитрогруппа этинил
Некоторые из них являются сильными хромофорами (азогруппа),
другие — очень слабыми (оксогруппа). К сложным хромофорным груп-
291
пам относится система сопряженных двойных связей, например хи~
ноидные структуры:
Для превращения окрашенного вещества в краситель в его молеку-
лу вводят специальные группировки — ауксохромы (от греческого
ауксо — увеличение). Эти группы не только увеличивают интенсив-
ность окраски, но и обусловливают сродство окрашенного вещества к
материалу. Ими являются аминогруппы, оксигруппы, сульфогруппы,
карбоксилы и некоторые другие.
Примером азокрасителей может служить паракрасный, применяе-
мый для окраски хлопчатобумажных тканей:
ОН
O2N—N = N — X \
Используемые в промышленности азокрасители содержат часто
две азогруппы и более. Например, кислотный сине-черный краси-
тель, применяемый для крашения шерстяных тканей:
ОН NH2
Окраска некоторых азокрасителей способна сильно изменяться в
зависимости от pH среды. Такие азокрасители применяются в качестве
индикаторов. Одним из наиболее распространенных индикаторов явля-
ется метиловый оранжевый (гелиантин), получающийся при сочетании
диазобензолсульфокислоты (диазосоставляющий) с диметиланилином
(азосоставляющий):
диазобензолсульфок ислота диметиланилин
(внутренняя соль)
292
метиловый оранжевый
В щелочной и нейтральной средах гелиантин имеет желтый цвет,
в кислой среде он приобретает розово-красную окраску. Изменение
окраски вызывается изменением хромофора. В нейтральной и щелоч-
ной среде хромофором является азогруппа —N = N—, в кислой воз-
никает новый хромофор — хиноидное ядро. Реакция обратима:
СН3
+Н
желтый метиловый оранжевый
розово-красный метиловый оранжевый
он-
Аминокислоты
Аминокислотами называются соединения, содержащие в молекуле
два типа функциональных групп: аминогруппу NH2 и карбоксиль-
ную группу СООН. Следовательно, аминокислоты относятся к гетеро-
функциональным органическим соединениям.
В зависимости от количества функциональных групп различают:
моноаминомонокарбоновые, моноаминодикарбоновые, диаминомоно-
карбоновые кислоты и т. д., например:
СН3—СН—СООН НООС—СНо—СН—СООН
I “ I
nh2 nh2
моноаминомонокарбоновая моноаминодикарбоновая
кислота кислота
NH0—(СН2)4—СН—СООН
I
nh2
диаминомонокарбоновая кислота
В зависимости от положения аминогруппы по отношению к карбо-
ксильной группе различают а-, р-, у-аминокислоты, а от строения угле-
водородного радикала, связанного с амино- и карбоксильными группа-
ми, — алифатические, ароматические и гетероциклические аминокис-
лоты. В специальную группу выделяют аминокислоты, содержащие
Дополнительно окси- и тиольные группы.
Аминокислоты играют исключительно важную роль в жизни живот-
ных и растительных организмов, являясь теми кирпичиками, из кото-
293
рых построены молекулы важнейшего природного полимера — белка.
Поэтому широко распространено деление аминокислот, предложенное
специалистами в области биоорганической химии, на аминокислоты
природные (обнаруженные в растительных и животных организмах)
и синтетические, получаемые в лабораториях и не имеющие природных
аналогов. Природные аминокислоты, в свою очередь, делятся на про-
теиногенные (входящие в состав белков) и непротеиногенные (не вхо-
дящие в состав белков). Среди природных аминокислот выделяют груп-
пу так называемых незаменимых аминокислот, которые не могут синте-
зироваться в животном организме и организме человека.
В настоящее время в природных объектах обнаружено свыше 150
аминокислот, 26 из них входят в состав белков, 10 аминокислот явля-
ются незаменимыми. Важнейшие аминокислоты приведены в табл. 27.
Номенклатура. Изомерия. Для наименования аминокислот ши-
роко применяются тривиальные названия. По рациональной номенкла-
туре аминокислоты рассматривают как производные соответствующих
карбоновых кислот, положение аминогруппы указывается буквами
а, р, 7 и т. д. (табл. 27). По ИЮПАК для наименования аминокислот
группу NH2 называют приставкой «амино», указывая цифрой номер
углеродного атома, с которым она связана, затем следует название
соответствующей кислоты.
Для аминокислот характерна структурная изомерия, связанная с
особенностями строения углеродного скелета и взаимным расположе-
нием функциональных групп, и пространственная — оптическая изо-
мерия, связанная с наличием асимметричных углеродных атомов.
Все природные аминокислоты, за исключением аминоуксусной кисло-
ты, содержат один или несколько асимметрических атомов углерода и,,
следовательно, могут существовать в виде нескольких стереоизомеров,
количество которых определяется по формуле, приведенной ранее.
Каждой паре оптических антиподов соответствует один рацемат. Раз-
личают D- и L-ряды аминокислот:
СООН СООН
I I
H2N—С—н н—с—nh2
I I
сн3 СНз
L-конфигурация D-конфигурация
L-(+)-аланин D-(—)-аланин
Все входящие в состав белков аминокислоты (протеиногенные)'
представляют собой L-формы; £)-формы встречаются в природе редко.
Способы получения. Гидролиз белков под влиянием ферментов^
кислот или щелочей. Этим способом можно получить любую из кислот,,
входящую в состав белков. Однако разделение образовавшейся при гид-
ролизе смеси аминокислот представляет собой трудную задачу.
Практическое значение имеет гидролиз биомассы дрожжей, выра-
щенных на углеводородсодержащем сырье, и включающей до 40%
белков. Сырьем для получения биомассы микробиологическим путем
могут служить также диоксид углерода, спирт, парафины нефти,
природный газ, отходы деревоперерабатывающей промышленности.
294
Таблица 27. Наиболее важные представители аминокислот
Название Формула Условные со- кращения названий (русские и и латинские) Температу- ра плав- ления, °C
Природные I. М аминокислоты, входящие в соста оноаминомонокарбоновые кислот ь в белков >1
Глицин, гликокол, ами- ноуксусная кислота NH2—СН2—СООН Гли; Gly 292
а-Аланин, а-аминопропио- новая кислота СН3—СН—СООН 1 nh2 Ала; Ala 297
Валин, 7-аминоизовале- риан овая кислота сн3—сн—сн—соон 1 1 СНз NH, Вал; Vai 315
Лейцин, а-амино-7-метил- валериановая кислота СН3—СН—СН2—СН—СООН 1 1 СН3 NH2 Лей; Leu 337
Изолейцин, а-амино-?- метилвалериановая кис- лота СНз—сн2—сн—сн—соон СНз NH2 Иле; He 284
Фенилаланин, а-амино-₽- фенилпропионовая кис- лота II. С6Н5—СН2—СН—соон 1 nh2 Моноаминодикарбоновые кислоты Фал; Phen 278
Аспарагиновая кислота, аминоянтарная кислота НООС—СН—СН2—СООН nh2 Асп; Asp 251
Глутаминовая кислота, а-аминоглутаровая кис- лога III. НООС—сн—сн2—сн2—соон 1 nh2 Диаминомонокарбоновые кислоть Глу; л Glu 248
Лизин, а,е-диаминокап- роновая кислота NH2—(СН2)4—СН—СООН nh2 IV. Окси-и тиолоаминокислоты Лиз; Lys 224
Серин, а-амино-3-окси- пропионовая кислота НО—СН2—СН—СООН nh2 Сер; Ser 228
Треонин, а-амино-|3-окси- масляная кислота СНз—сн—сн—соон 1 1 он nh2 Тре; Thr 251 (раз- ложение)
Цистеин, а-амино-^-тио- пропионовая кислота сн2—сн—соон SH nh2 Цис; Cys 178
Метионин, а-амино-7- метилтиомасляная кис- лота СН3—S—сн2—сн2—сн—соон nh2 Мет; Met —
295
Продолжение табл. 2Т
Название Формула Условные сокращения названий (русские и латинские) Темпера, тура плавления °C
Тирозин, а-амино-^-(п-ок- сифенил)пропионовая кислота НООС—СН—СН2—он nh2 Тир; Туг —
Гетероциклические аминокислоты
Пролин, а-пирролидин- карбоновая кислота Н2С—СН2 Про; Pro —
Н2С 1 сн—соон
Триптофан, а-амино-?- N Н i II 1; сн2—сн—соон Три; Try 382
индолилпропионовая кислота 1 II 1 н nh2
Аминокислоты, не входящие в состав белков
э-Аминобензойная кисло-
та (антраниловая кис-
лота)
145
n-Аминобензойная кис-
лота
СООН
^X'-NH2
и
187
Полученные из белковых гидролизатов аминокислоты разделяют
методами ионообменной хроматографии, электрофореза и газожидкост-
ной хроматографии.
Действие аммиака на галогензамещенные кислоты (аминирование,
аммонолиз). Реакцию осуществляют при взаимодействии водного,
спиртового или жидкого аммиака обычно на а-галогензамещенные кис-
лоты (как наиболее доступные). Аминокислоты в аммиачной среде пре-
вращаются в свои аммониевые соли:
296
Cl nh2
I I /°
CH2—COOH + 3NH3 -► H2C—c<f + NH4C1
\0NH4
монохлоруксусная
кислота
аммониевая соль
аминоуксусной
кислоты
Получение из циангидринов (оксинитрилов). Важным методом
получения а-аминокислот является действие аммиака на циангидрины
альдегидов и кетонов с последующим омылением аминонитрила в ами-
нокислоту:
ОН
zO | NH3
СН3—cZ + HCN -> СНз—СН—CN------------►
ХН оксинитрил —Н,О
nh2
I 2Н2О
-> СНз—СН—CN--------1
- NHa
аминонитрил
nh2
сн3—СН—соон
аминокислота
Зелинский предложил вместо синильной кислоты и аммиака дейст-
вовать на карбонильные соединения цианистым аммонием, образую-
щимся при смешении водных растворов хлорида аммония и цианида
калия:
NH2 nh2
zO NH.CN I 2НгО I
CH3—C<f---------> CH3—C—CN--> CH3—C—COOH
XCH3 -н2о 1 —nh8 I
CH3 CH3
аминонитрил
Присоединение аммиака к непредельным кислотам:
акриловая(кислота
X)
:nh3 #
—сн2—сн2—с;
I хон
NH2
0-аминопропионовая кислота
Присоединение аммиака происходит не в соответствии с правилом
Марковникова.
Конденсация альдегидов с малоновой кислотой и аммиаком (получе-
ние ^-аминокислот по методу В. М. Родионова):
NH2
zO /СООН I
R—С< + Н2С< + NH3 -> R—СН—СН2—СООН + Н2О + СО2
ХН хзоон
альдегид малоновая ^-аминокислота
кислота
297
Возможно, здесь первоначально происходит отщепление воды за
счет карбонильного кислорода и подвижных атомов водорода малоно-
вой кислоты с образованием ненасыщенной кислоты, к которой затем
присоединяется аммиак. По другим представлениям, в основе реакции
лежит конденсация малоновой кислоты с альдегидом по типу альдоль-
ной конденсации с образованием промежуточного продукта (1), кото-
рый затем аминируется в производное (2), после декарбоксилирования
которого образуется р-аминокислота:
zO
СН3- С< +
ХН
/СООН
нсн
уксусный малоновая
альдегид кислота
он соон
I I NH3
СНз—сн—сн -------->
| -н2о
соон
(1)
nh2 соон
I I
-> сн3—сн—сн--------->
I - С02
соон
(2)
NH2
сн3—сн—сн2—соон
₽-а1УИномасляная кислота
Восстановление нитрокислот. Важным способом синтеза аромати-
ческих аминокислот является восстановление нитрокислот. В зависи-
мости от последовательности химических превращений образуются
орто-, мета- и параизомеры. Например, из толуола можно получить все
изомерные нитробензойные кислоты, восстановление которых приводит
к соответствующим аминокислотам:
n-нитробензой- л-аминобензой-
ная кислота на я кислота
:98
[О]
м-нитробен-
зойная кислота
СООН
‘ Ц-NH,
л-аминобензой-
ная кислота
Микробиологический синтез. В промышленности многие аминокис-
лоты получают микробиологическим путем, выращивая на специаль-
ной питательной среде микроорганизмы, продуцирующие в процессе
своей жизнедеятельности определенные аминокислоты. Таким путем
получают, например, лизин и глутаминовую кислоту.
Кроме приведенных общих методов для синтеза отдельных амино-
кислот существуют свои, экономически выгодные и нашедшие не толь-
ко лабораторное, но и промышленное применение.
Физические свойства. Аминокислоты — бесцветные, кристаллические ве-
щества с высокими температурами плавления (табл. 27). Плавятся с разложени-
ем, нелетучи. Они хорошо растворимы в воде и плохо во многих органических
растворителях.
В большинстве случаев аминокислоты D-ряда сладкие на вкус, L-ряда —
горькие или безвкусные.
Высокие температуры плавления аминокислот, их плохая растворимость в
органических растворителях и хорошая растворимость в воде, нелетучесть и не-
которые другие, не типичные для органических соединений свойства связаны с
особенностями строения их молекул. В молекуле свободной аминокислоты амино-
группа, обладающая основными свойствами, вступает во внутримолекулярное
взаимодействие с кислотным карбоксилом, образуя внутреннюю соль (биполяр-
ный ион). Строение ее может быть описано следующей формулой:
ЙН3—(СН2)д—соо-
Химические свойства. Две функциональные группы NH2 и СООН,
содержащиеся в молекуле аминокислоты, определяют их химическое
поведение.
Амфотерный характер аминокислот. Изоэлектрическая точка.
Имея в молекуле одновременно кислотную и основную группу, амино-
кислоты в водных растворах ведут себя как типичные амфотерные сое-
динения. В кислых растворах они проявляют основные свойства, реа-
гируя как основания, в щелочных — как кислоты, давая соответствен-
но две группы солей:
NH3—(СН2)3—COO- + НС1 -> [NH3—(СН2)3—СООН]+СИ
NH3— (СН2)3—СОО“ + NaOH [NH2~(CH2)3—СОО]“ Na+ + Н2О
Благодаря своей амфотерности аминокислоты в живом организме
играют роль буферных веществ, поддерживающих определенную кон-
центрацию водородных ионов. Буферные растворы, полученные при
взаимодействии аминокислот с сильными основаниями, находят широ-
кое применение в биохимии. Соли аминокислот с минеральными кисло-
тами (соляной или серной) в большинстве случаев лучше растворимы
в воде, чем свободные аминокислоты. Соли с органическими кислотами
299
(пикриновой и другими) труднорастворимы в воде и используются дд^
идентификации и разделения аминокислот.
Если в водном растворе аминокислоты пропускать электрический
ток, то в зависимости от pH среды она может мигрировать к аноду или
катоду. Те узкие значения pH, при которых аминокислота, оставаясь
электронейтральной, не мигрирует под влиянием электрического тока
ни к аноду, ни к катоду, называют изоэлектрической точкой:
ОН” (pH выше, чем в NH3—СН2—СОО" Н+ (pH ниже, чем в
изоэлектрической точке) изоэлектрическая точка; изоэлектрической точке)
аминокислота не мигри-
рует ни к катоду, ни
к аноду
NH2—СН2—COO” NH3—СН2—СООН
наличие аминогруппы, наличие недиссоциирован-
аминокислота ведет себя ной карбоксильной груп-
как анион — мигрирует пы, аминокислота ведет
к аноду себя как катион — миг-
рирует к катоду
Образование комплексов с металлами. Почти все а-аминокислоты
образуют комплексы с ионами двухвалентных металлов. Самыми устой-
чивыми являются комплексные соли меди, образующиеся в результате
взаимодействия с гидроксидом меди (II). Комплексы представляют со-
бой ярко окрашенные в синий цвет хелатные соединения:
2СН2—СООН О=С-----Оч у NH2—сн2
I + Си(ОН)2-----> I >Cu< I
NH2 —н2О Н2С—NH/ ХО-----С=О
Водные растворы таких комплексных солей в отличие от растворов-
солей соответствующих металлов обладают очень низкой электропро-
водимостью.
Реакции аминокислот, связанные с наличи-
ем карбоксильной группы.
Образование солей (см. с. 299).
Образование сложных эфиров:
NH3—СН2—COO” + С2Н5ОН + НС1 -> [NH3—СН2—СООС2Н5]+ С1” + Н.П
аминоуксусная соль этилового эфира
кислота аминоуксусной кислоты
2 [H3N—СН2—СООС2Н5]+Cl” +Ag2O 2NH2—СН2—СООС2Н5 + 2AgCi 4- Н2О
этиловый эфир аминоуксус-
ной кислоты
Метиловые и этиловые эфиры аминокислот — жидкости, легко пере-
гоняющиеся в вакууме, поэтому эта реакция применяется для разделе-
ния смеси аминокислот.
Образование галогенангидридов. Реакцию проводят, действуя пяти-
хлоридом фосфора на аминокислоту в кислой среде:
300
. НС1 PCI5
йНз—CH2—COO--------->- [NH3—CH2—COOHJ+ Cl~----->
—HC1
- РОС];
z* г
NH3—CH2—C<f Cl-
XC1J
соль хлорангидрида
аминоуксусной кислоты
Г
\ С1“
xnhJ
Cl- + NH3 -------->
НС1
При взаимодействии хлорангидрида аминокислоты с аммиаком об-
разуется амид кислоты:
Г /°
H3N—СН2—C<f
Х21
соль амида аминоуксус ной
кислоты
Декарбоксилирование. При нагревании аминокислот в твердом сос-
тоянии с высококипящими растворителями (или с баритовой водой)
происходит декарбоксилирование с образованием соответствующих
аминов:
г
сн3—сн—соон -> сн3—сн2—nh2 + со2
I т этиламин
nh2
аминопропионовая смесь
Аналогичный процесс гнилостного расщепления белков происходит
во время пищеварения под действием ферментов, выделяемых микроор-
ганизмами, населяющими кишечник.
Реакции, связанные с наличием аминогруп-
п ы. Образование солей (см. с. 299).
Ацилирование аминокислот. Аминогруппу аминокислот можно под-
вергнуть ацилированию хлорангидридами и ангидридами кислот уже
при комнатной температуре:
NH3—сн2—соо~+ \СН3—С— /2 О ->
уксусный ангидрид
о ®
J3 II
СН3—С—NH—сн2 —с—он + СН3СООН
ацетиламиноуксусная кислота
Ацильные производные аминокислот широко используются при изу-
чении последовательности их в белках и синтезе пептидов.
Алкилирование аминокислот. При алкилировании аминокислот га-
логеналкилами образуются моно-, ди- и триалкилпроизводные. Триал-
килпроизводные представляют собой четвертичные аммониевые осно-
вания, внутренние соли которых называются бетаинами:
, H3rt—СН2—С00- -Д. R'NH2—сн2—соо-
R'l , + R'l , +
---> R2NH—СН2—СОО- —> R3N—СН2—СОО’
бетаин аминоуксусной
кислоты
301
Ряд алкилированных аминокислот нашел применение при изуце,
иии первичной структуры белков и синтезе пептидов.
Действие азотистой кислоты на аминокислоты. При действии
азотистой кислоты на аминокислоты, содержащие первичную амино-
группу, выделяется азот и образуется оксикислота, т. е. аминокислоты
ведут себя в этой реакции как первичные алифатические амины:
R—СН—СООН + HNO2 R—СН—СООН + N2 + Н2О
nh2 он
Эта реакция лежит в основе количественного определения аминокислот
по Ван-Сляйку газометрическим или манометрическим способом.
Реакции, связанные с наличием и взаимным
влиянием амино- и карбоксильной групп.
Отношение аминокислот к нагреванию.
1. При нагревании а-аминокислот образуются циклические амиды,
называемые дикетопиперазинами:
а-аминопропионовая
кислота
сн3—сн—с=о
I I
_______ NH NH
-2Нг°_О=С—СН—СН3
2, 5-диметил-З, 6-дикето-
пиперазин
2. При нагревании ^-аминокислот отщепляется аммиак и образуется
непредельная кислота:
СН2—СН—СООН > СН2=СН—СООН
|| NH8 акриловая кислота
;nh2" н]
Р-аминопропионовая
кислота
3. При нагревании у- и S-аминокислот образуются внутренние цик-
лические амиды — лактамы:
СН2—СН2
H2N—СН2—СН2—СН2—СООН---------> I I
7-аминомасляная кислота ^*0 СН^ ^С О
NH
лактам у-аминомасля-
ной кислоты
Пептиды
Аминокислоты, входящие в состав белковой молекулы, связаны меж-
ду собой по типу амидной связи. Пептиды — это амиды, образующиеся
в результате взаимодействия аминогрупп и карбоксилов аминокислот.
Амидная связь —NH—СО— в таких соединениях называется пептид-
ной связью.
В зависимости от числа аминокислотных остатков в молекуле раз-
802
дичают ди-, три-, тетрапептиды и т. д. вплоть до полипептидов. Услов-
но считают вещества с молекулярной массой до 10 000 полипептидами,,
а с большей — белками, например:
nh2—СН2—СО—NH—сн2—СООН
дипептид
NH2—СН2—СО—NH—СН—СО—NH—СН—СООН
I I
СН3 СН2-СбН5
трипептид
NH2—СН—СО / NH—СНСО \ —NH—СН—СООН
I I ।
R \ R /п R
полипептид
Последовательность соединения остатков аминокислот в молекуле
пептида влияет на его свойства. Так, при взаимодействии двух различ-
ных аминокислот (условно обозначим их через А и В) можно получить
два дипептида: АВ и ВА, различающихся по своим физическим и хи-
мическим свойствам. Например, при взаимодействии гликокола и ала-
нина могут образоваться дипептиды:
NH2—СН2—СО—NH—СН—СООН
СН3
глицилаланин (гли-ала)
СН3—СН—СО—NH—сн2—соон
nh2
аланилглицин (ала-гли)
Названия полипептидов производятся от названия кислотного*
остатка аминокислоты, прореагировавшей карбоксильной группой
(ацильный радикал), и названия кислоты, реагирующей своей амино-
группой.
Полипептиды встречаются в организмах животных и человека,
являясь, чаще всего, продуктами распада белков. Таков, например,
трипептид глутатион, выделенный первоначально из мускульной ткани,
а затем обнаруженный во многих живых клетках.
Ряд пептидов играет важную биологическую роль. К ним можно’
отнести, например, декапептид, известный как грамицидин С — анти-
биотическое вещество, гормоны гипофиза окситоцин, вазопрессин и
адренокортикотропный гормон. Важнейшим пептидом является инсу-
лин — гормон поджелудочной железы, снижающий содержание саха-
ра в крови. Недостаток инсулина вызывает сахарный диабет.
Изучение пептидов проводилось главным образом как необходимая
ступень при изучении белков. Таким образом, сформировались два
аспекта химии пептидов: установление их строения и подход к их лабо-
раторному синтезу. Для выяснения структуры конкретного пептида
необходимо знать, какие аминокислоты входят в его состав, сколько
имеется аминокислот каждого вида, какова последовательность соеди-
нения аминокислот. Ответить на эти вопросы помогают чисто химичес-
кие методы в сочетании с современными физико-химическими метода-
ми анализа.
Пептиды заданного строения невозможно синтезировать прямой
конденсацией а-аминокислот, так как уже при реакции только двух
303
различных аминокислот образуются четыре различных дипептида.
Так, при конденсации глицина и аланина могут быть получены липец-
гиды ала-ала, гли-гли, ала-гли и гли-ала. Для направленного синтеза
пептидов заданного строения пользуются обычно следующими прие-
мами:
а) защита аминогруппы предыдущего компонента:
H2N—СН—СООН Z—NH—СН—СООН
R R
б) защита карбоксила последующего компонента:
NH2—СН—СООН nh2—сн—соох
R' R'
в) активация карбоксила предыдущего компонента (чаще) или ами-
ногруппы последующего компонента (реже):
Z—NH—СН—СООН Z—NH—СН—С—А
I I 11
R R О
или
NH2—СН—СООХ -> A'—NH—СН—СООХ
R' R'
г) конденсация полученных производных:
Z—NH—СН—СО—А + NH2—СН—СООХ -----
I I “НА
R R'
Z—NH—СН—СО—NH—СН—СООХ
R R'
или
Z—NH—СН—СООН + A'—NH—СН—СООХ ->
I I
R R'
Z—NH—СН—СО—NH—СН—СООН
R L
д) снятие защиты, получение необходимого пептида:
Z—NH—СН—СО—NH—СН—СООХ -> NH2—СН—СО—NH—СН—СООН
II II
R R' R R'
Защитные группировки должны, как обычно, удовлетворять двум
требованиям: легко вводиться и легко удаляться. На практике разра-
ботан ряд методов активации и защиты функциональных групп. Все
они имеют свои преимущества и недостатки.
304
Значение аминокислот. Исключительная роль аминокислот в
жизнедеятельности живого организма связана в первую очередь с тем,
что они входят в состав белков. При попадании в желудочно-кишеч-
ный тракт белки пищи под действием ферментов распадаются на сос-
тавляющие их аминокислоты, которые затем используются организмом
для построения своих собственных белков — тканей, крови, кожи,
волос и т. д.
Следует отметить, что организм человека обладает способностью
синтезировать только некоторые из нужных ему аминокислот. Имеется
ряд незаменимых аминокислот, которые организм построить не мо-
жет и должен получать с пищей. К ним относятся: лизин, лейцин,
нзолейцин, метионин, фенилаланин, триптофан, треонин, валин, гис-
тидин, аргинин.
Среднесуточная потребность взрослого человека в аминокислотах
(кг) приведена ниже:
Аминокислота Потребность Аминокислота Потребность
Триптофан .... 0,001 Пролин 0,005
Лейцин 0,005 Гистидин .... 0,002
Аргинин 0,006 Тирозин 0,004
Цистин 0,003 Аланин 0,008
Изолейцин .... Валин 0,004 0,004 Серин Глутаминовая ки- 0,003
Треонин 0,003 слота 0,0016
Лизин Метионин .... 0,004 0,003 Гликокол .... Аспарагиновая кис- 0,003
Фенилаланин . . . 0,003 лота 0,006
При нехватке одной из аминокислот в белке он становится недос-
таточно полноценным и не полностью усваивается организмом. Поэто-
му аминокислоты, полученные синтетическим или микробиологичес-
ким путем, должны добавляться в пищу для повышения ее полноцен-
ности. С этой целью широко используется, например, лизин, которо-
го недостает в растительной пище (белке пшеницы). Смеси аминокислот
применяются в медицине для питания больных. Отдельные аминокис-
лоты используют в разнообразных синтезах, в аналитической химии,
некоторые аминокислоты применяются в пищевой промышленности в
качестве вкусовых добавок, например мононатриевая соль глутами-
новой кислоты, придающая продукту вкус и запах куриного
бульона.
Из аминокислот, не входящих в состав белков, следует отметить
о-аминобензойную (антраниловую) кислоту, которая применяется в
У\/соон
I II
\//\nh2
антраниловая кислота
синтезе красителей и лекарственных препаратов, пищевой промышлен-
ности.
<1—682
305
АМИДЫ КИСЛОТ
Амидами кислот называются продукты замещения гидроксильной
группы в карбоксиле на группу NH2.
Название амидов слагается из двух частей: слова амид и названия
кислоты или ацила*
СН3—С<
XNH
амид уксусной кислоты;
ацетамид
у)
сен5-с<
xnh2
амид бензойной кислоты;
бензамид
ацетамид
Способы получения. Действие амми ака на хлорангидриды кислот
(см. с. 237).
Гидролиз нитрилов (см. с. 308).
Сухая перегонка аммониевых солей карбоновых кислот:
СН3—C<f -> СН3—С< + Н2О
X)NH4 'NH2
уксуснокислый
«ммоний
Физические свойства. Амиды кислот — твердые вещества, за исключением
формамида, являющегося жидкостью; ограниченно растворимы в воде, раствори-
мость уменьшается с увеличением молекулярной массы. Растворы имеют нейт-
ральную реакцию.
Химические свойства. В отличие от аминов и аммиака амиды почти <
не обладают основными свойствами. Они образуют соли только с силь-
ными кислотами, которые легко разлагаются водой с выделением ис-
ходного амида:
ацетамид
+НС1
СН3—С
Z 1
\nh2.
HCI
хлорводородный ацетамид
При гидролизе амидов образуются кислоты:
+ Н2О
ацетамид
zO
------- сн3—с<
-NH. \ОН
уксусная кислота
Нагревание амидов с водоотнимающими средствами приводит к
образованию соответствующих нитрилов:
zO г
СН3—С<----------> СН3—С= N + Н,0
NNH2 рА
ацетамид ацетонитрил
Восстановлением амидов получают первичные амины с тем же ко-
личеством углеродных атомов:
zO 4 [Н]
R-C<------------> R—СН2— NH2
xNH2 (~н2О)
306
Отдельные представители. Ацетамид СН8—с/ применяется
\nh2
в бумажной и кожевенной промышленности.' О
Карбамид, мочевина, диамид угольной кислоты NH2—С—NH2.
Кристаллическое вещество без цвета и запаха. Хорошо растворимо в
воде. Конечный продукт азотистого обмена млекопитающих. Один из
важных продуктов химической промышленности. Карбамид получает-
ся в больших количествах в промышленном масштабе из диоксида уг-
лерода и аммиака:
О
г, р II
СО2 + 2NH3 H2N—COONH4 —H3N—С—NH2
карбамат аммония мочевина
Применяется для получения полимеров, синтеза лекарственных
соединений, в качестве азотного удобрения, производства гербицидов,
а также кормового азота.
НИТРИЛЫ
Нитрилы можно рассматривать как продукты замещения водоро-
да в молекуле цианистоводородной кислоты (HC=N) на алкилы. С дру-
гой стороны, нитрилы являются производными карбоновых кислот.
Общая формула нитрилов R-C=N. Углеводородный радикал
в молекуле нитрилов может быть насыщенным, ненасыщенным, арома-
тическим и т. д. Название нитрилов строится из названия соответст-
вующей кислоты и слова нитрил:
СН3—С = N СН2=СН—С N
нитрил уксусной кислоты, нитрил акриловой кислоты,
ацетонитрил экрилонитрил
Получение. Нитрилы получают из амидов карбоновых кислот при
нагревании с водоотнимающими средствами:
z° г
R—---------->- R—C=N + Н2О
^NH2
Важным способохм получения нитрилов является взаимодействие
галогензамещенных углеводородов с цианидом калия:
R—Cl + KCN -> R—C=N 4- КС1
Ароматические нитрилы, кроме того, могут быть получены сплавле-
нием солей сульфокислот с цианидом калия:
бензонитрил
307
11»
Химические свойства. Все нитрилы при гидролизе превращаются
в соответствующие кислоты. На практике гидролиз осуществляют
либо в кислой, либо в щелочной среде, получая карбоновую кислоту
в свободном виде или в виде ее соли:
R-<H + nh4ci
,^aQH..^ R—+ NH3 |
XONa
Осторожным омылением нитрилов можно получить вначале амид,
который при дальнейшем гидролизе образует соответствующую кар-
боновую кислоту:
н2о
R—C=N ---:
R—с/
о
nh2
н2о
-NH3
кислота
нитрил
амид
Восстановление нитрилов, как и восстановление амидов, приво-
дит к соответствующим первичным аминам:
4 [Н]
R—C=N-------> R—СН2—NH2
Отдельные представители. Ацетонитрил CH3CN. Жидкость,
<кип = 82°С. Применяется в качестве растворителя многих органичес-
ких соединений и неорганических солей. Токсичен, как и прочие нит-
рилы.
Акрилонитрил СН2=СН—CN. Жидкость, /Кип = 78°С. Обладает
высокой химической активностью и используется в связи с этим в мно-
гочисленных лабораториях и промышленных синтезах (реакции циан-
этилирования). Является мономером в производстве полимернык ма-
териалов и синтетического каучука.
Раздел пятый
Г етероциклические
соединения
Гетероциклическими соединениями называются
вещества, в молекуле которых имеются циклы, содержащие наряду с
атомами углерода атомы других элементов (гетероатомы).
Выделение гетероциклических соединений в особую группу связа-
но с рядом особенностей их химических свойств и все увеличивающимся
их значением. Производные гетероциклических соединений имеют
важное физиологическое значение; к ним относятся нуклеиновые кис-
лоты, хлорофилл, ряд витаминов, алкалоиды. Многие гетероцикли-
ческие соединения широко применяются как лекарственные препараты.
Особое значение имеют соединения, содержащие в молекуле пяти- и
шестичленные циклы, а также азот, кислород и серу в качестве гетеро-
атомов. Легкость образования циклов с такими гетероатомами можно
объяснить, с одной стороны, тем, что валентные углы между связями у
этих атомов мало отличаются от валентных углов атома углерода в сос-
тоянии гибридизации sp3 (109°) и sp2 (120°). С другой стороны, эти ато-
мы имеют сравнительно небольшие атомные объемы, близкие к объему
метиленовой группы. В связи с этим включение гетероатомов N, О, S
вместо групп —СН2— и —СН= в циклическую группировку мало ска-
зывается на напряжении цикла и почти не изменяет общую геометрию
молекулы.
Гетероциклические соединения классифицируют по числу атомов в
цикле, а также по числу и индивидуальности гетероатома. В особую
группу обычно выделяют гетероциклы с конденсированными ядрами.
При наименовании гетероциклов широко пользуются три-
виальными названиями, например фуран, пиридин, пиримидин, индол,
хинолин и т. д.:
4 (т, 4
1 N N
. 1 1
Фуран
пиридин пиримидин
индол
309
Как видно из приведенных формул, нумерация в гетероциклах всег-
да начинается с гетероатома. Для простых систем возможны обозна-
чения положения с помощью букв греческого алфавита: а (рядом с ге-
тероатомом) р и у (по мере удаления от гетероатома).
Свойства гетероциклов определяются не только имеющимися ге-
тероатомами, но и в значительной мере зависят от характера связей в
цикле. Гетероциклы, не содержащие кратных связей, как правило, по
химическим и физическим свойствам похожи на соответствующие ацик-
лические соединения. Например, реакции диэтилового эфира сходны с
реакциями тетрагидрофурана, а пиперидина — с диэтиламином:
СН3 СН3
СН2 СН2
сн2—сн2
I I
сн2 сн2
диэтиловый
эфир
тетрагидрофуран
СН3 СН3
I I
снг сн2
\гн
диэтила мин
сн2—сн2
сн2 сн2
\н
пирролидин
Ряд насыщенных гетероциклических соединений, таких, как окись
этилена, тетрагидрофуран, диоксан, ангидриды двухосновных карбо-
новых кислот и некоторые другие, представляют собой производные
соединений жирного ряда. Поэтому химия таких соединений рассмат-
ривается в соответствующих разделах, посвященных изучению соеди-
нений алифатического ряда.
Существует, однако, и другая, очень обширная группа гетероцик-
лов, имеющих сопряженные системы кратных связей. Такие гетероцик-
лы напоминают своей устойчивостью и химическими свойствами бен-
зол и его производные и получили название ароматических гетероцик-
лических соединений.
Ароматические гетероциклические
соединения
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ОДНИМ ГЕТЕ-
РОАТОМОМ
Строение. Важнейшими пятичленными
гетероатомом являются фуран, тиофен и
гетероциклами с одним
пиррол:
фуран
Известно, что циклическая система обладает ароматическими свой-
ствами лишь тогда, когда она имеет плоское строение, (4п + 2)
делокализованных л-электронов (правило Хюккеля) и содержит не-
прерывную цепь сопряжения. Все названные пятичленные гетероцик-
310
лы отвечают этим требованиям — имеют плоское строение и содержат
ароматический секстет л-электронов, из которых 4л-электрона постав-
ляется четырьмя атомами углерода, а еще два — свободной парой
электронов соответствующего гетероатома:
н
Из трех элементов — кислород, азот и сера — кислород более
электроотрицательный элемент. По сравнению с азотом кислород силь-
нее удерживает свои электроны, следовательно, в фуране участие па-
ры неподеленных электронов гетероатома в ароматическом сопряжении
слабее, чем в пирроле. Резко выраженный ароматический характер
тиофена обусловлен прежде всего слабой электроотрицательностью
серы (меньшей, чем электроотрицательность азота и кислорода). Аро-
матический характер этих соединений подтверждается относительно
высокими энергиями сопряжения: 92,2 кДж/моль у фурана, 100,6
кДж/моль у пиррола и 117,3 кДж/моль у тиофена (у бензола — 150,1
кДж/моль).
Источники получения фурана, тиофена и пиррола. Получение
из 1,4-дикарбонилсодержащих соединений. Это один из наиболее об-
щих способов синтеза пятичленных ароматических гетероциклов с од-
ним гетероатомом. 1,4-Дикарбонильное соединение может быть дике-
тоном, ди карбоновой кислотой или кетокислотой. При нагревании этих
соединений с дегидратирующим агентом (СаС12, H2SO4, Р2О5 и др.)
образуются производные фурана; при проведении дегидратации в сре-
де аммиака — производные пиррола; в присутствии фосфорных соеди-
нений серы — производные тиофена. Реакция дегидратации протека-
ет, по-видимому, через образование диенола:
СН---СН
1,4-дикарбонильное соединение
диенол
NH3j(—2Н2О)
СН—СН
II II
R—С С—R
H2SO4 I (—2Н2О)
P2S6 I (-2Н,О)
производные производны!
фурана тиофена
производные
пиррола
311
Получение фурана из слизевой кислоты. При сухой перегонке сли-
зевой кислоты образуется пирослизевая кислота, при декарбоксили-
ровании которой (при нагревании в запаянных трубках) получается
фуран:
?НО:—СН----НС — ЮН
н~с
Гб=с
юн
c-i” J
|\соон----------->
"ppQ —ЗН2О; —СО2
ню
пирослизевая фуран
кислота
слизевая к..слота
Получение фурана из фурфурола. Фурфурол (от латинского furfur —
отруби) — наиболее доступное производное фурана, получаемое в
промышленных масштабах из сырья, богатого пентозанами (отруби,
кочерыжки, шелуха подсолнечных и ячменных семян). Гидролизом их
получают сначала пентозы, которые затем претерпевают дегидратацию
с циклизацией и превращаются в фурфурол. Декарбонилированием
фурфурола получают фуран:
пентоза
Получение тиофена в промышленности. Образуется из бутана и па-
ров серы:
700°С
СН3—СН2—СН2—СН3 + 4S ----
Получение пиррола из ацетилена и аммиака:
пиррол
312
Взаимопревращения фурана, тиофена и пиррола. Между фураном,
тиофеном и пирролом существует взаимосвязь, открытая советским
ученым Ю. К. Юрьевым:
Эти превращения названных гетероциклов друг в друга происходят
при температуре 450°С в присутствии триоксида алюминия.
Физические свойства. Фуран — бесцветная жидкость, /кип = 32°С, с запа-
хом, напоминающим запах хлороформа. Электрический момент диполя равен
2,3-1О“30 Кл»м. Тиофен —бесцветная жидкость, /кип = 84®С. Электрический
момент диполя равен 1,7*10"30 Кл«м. Пиррол —бесцветная жидкость, /кип =
= 130°С. Электрический момент диполя равен 5,9* 10“30 Кл*м.
Химические свойства. 1. Реакция электрофильного
замещения. Наиболее характерными реакциями фурана, тиофе-
на и пиррола, как и других ароматических соединений, являются ре-
акции электрофильного замещения (Sf). Эти соединения подвергаются
нитрованию, сульфированию, галогенированию, ацилированию и т. д.
Во всех этих реакциях пятичленные гетероциклы ведут себя активнее
бензола, напоминая своей реакционной способностью ароматические
амины и фенолы. Электрофильное замещение происходит в положе-
ние 2.
Галогенирование. При бромировании фурана на холоду, вероятно,
происходит сначала присоединение молекулы брома. Образующееся
промежуточное вещество (которое не удалось выделить) отщепляет
молекулу бромистого водорода и превращается в 2-бромфуран:
Вг2
на холоду
2-бромфуран
Для галогенирования тиофена можно использовать бром или хлор
при низкой температуре, при этом могут образовываться как моно-,
так и полигалогентиофены. Более избирательно действует сульфурил
хлорид SO2C12:
313
При иодировании пиррола образуется тетраиодпиррол — иодол,
являющийся хорошим антисептиком. При действии сульфурилхлорида
на холоду образуется 2-хлорпиррол:
Сульфирование. Фуран и пиррол значительно легче, чем тиофен,
окисляются и осмоляются при действии минеральных кислот. Осмо-
ляющее действие кислот заключается в том, что вначале происходит
протонизация фурана по кислороду:
фуран
При этом пара электронов, участвующая в образовании ароматичес-
кого секстета, выключается из сопряжения с двойными связями, аро-
матичность нарушается и образующийся оксониевый ион, обладающий
свойствами диена с сопряженными двойными связями, легко полиме-
ризуется и осмоляется. Аналогично ведет себя и пиррол. Поэтому суль-
фировать фуран и пиррол удается только пиридинсульфотриоксидом
(А. П. Терентьев) | |j * s°3» или c&HsN • SO3:
у C6H,N-SO3
-SO3H + C5H5N
пиридин
2-фурансульфо.
кислота
314
/ \ C.H.N-SO. Л \
у--------> y-SO3H + c5h5n
^hT ^ьг
I I
H H
2-пиррол-
сульфокис-
лота
Повышенная устойчивость тиофена позволяет сульфировать его
серной кислотой. В отличие от бензола он сульфируется уже на холо-
ду-
H2SO<
на холоду
2-тиофенсульфо-
кислота
C6H6N-SO3
Нитрование. Хотя тиофен значительно устойчивее к действию ми-
неральных кислот, чем фуран и пиррол, однако он все же окисляется
азотной кислотой с разрушением молекулы. Поэтому для нитрования
тиофена, как и для нитрования фурана и пиррола, используют обычно
ацетилнитрат СН3—С—ONO2 (смесь уксусного ангидрида и азотной
II
кислоты): О
А
Л \ CH3COONO2 л \
<f >--------------> /—NO2 + СН3СООН
\ О / пиридин \ о /
2-нитрофуран
Аналогично получают 2-нитропиррол и 2-нитротиофен;
ch3coono2
(СН.СО)2О; 5°С
2-нитропиррол
2-нитротиофен
Ацилирование. Реакцию ацилирования по Фриделю — Крафтсу
проводят в случае фурана и пиррола, используя вместо трихло-
рида алюминия более мягкие катализаторы (SnCl2, SnCl4, ZnCl2,
BF3):
315
—С—СН3 + СН3СООН
2-ацетил фуран
(СН3СО)2О
250°С
X X—с—сн3 + СНзСООН
н
2-ацет ил пиррол
2-ацилтиофен
2. Реакции гидрирования. Гидрирование фурана, тио-
фена и пиррола приводит к насыщенным гетероциклическим системам,
лишенным ароматических свойств. Фуран гидрируется над нике-
левым или платиновым катализатором до тетрагидрофурана
(ТГФ):
Тетрагидрофуран — ценный растворитель. Кольцо ТГФ лежит в ос-
нове пятичленных циклических форм углеводов (фуранозы).
Тиофен восстанавливается действием металлического натрия в
аммиаке или спирте (никелевые и платиновые катализаторы при гидри-
ровании отравляются тиофеном и конечным продуктом его гидрирова-
ния — тиофаном):
\ s / (C,H5OH+Na)
тиофан
Тиофан и его гомологи содержатся в сернистых нефтях.
Электрохимическое восстановление пиррола может привести к про-
дукту неполного гидрирования — пирролину. Каталитическим вос-
становлением в присутствии никелевого или палладиевого катализато-
ров можно получить пирролидин:
316
Другие реакции пятичленных гетероцик-
лических соединений.
1. Фурановое кольцо чувствительно к действию окислителей. Уже
на воздухе фуран самоокисляется, одновременно полимеризуясь. Его
можно окислить до малеинового ангидрида:
воздух
V2O6; 320°С
малеиновый ангидрид
2. Наибольшая непредельность фурана по сравнению с другими пя-
тичленными гетероциклами проявляется в реакции с малеиновым ан-
гидридом. Здесь фуран ведет себя не как ароматическое соединение,
а как диен с сопряженной системой двойных связей, вступая в реак-
цию диенового синтеза:
Тиофен и пиррол такой реакции не дают.
3. Атом азота в пирроле формально напоминает атом азота во вто-
ричных аминах:
н
пиррол
вторичный амин
Однако в отличие от вторичных аминов пиррол практически не обла-
дает основными свойствами. Он, например, не способен присоединять
галогенпроизводные углеводородов с образованием четвертичных аммо-
ниевых солей. Происходит это потому, что л-электроны атома азота
втянуты в общий ароматический секстет электронов, вследствие чего
атом азота не способен присоединять протон. Мало того, пиррол прояв-
317
ляет слабо кислые свойства: атом водорода в NH-группе может заме,
щаться на натрий и калий при взаимодействии с названными металла-
ми или концентрированным раствором КОН. Этим пиррол напоминает
скорее фенол. Образующийся пирролкалий (или натрий) легко гидро-
лизуется с выделением пиррола:
пирролкалий
Реакцией образования металлических производных пиррола поль-
зуются для получения гомологов пиррола. Образующиеся при взаимо-
действии их с галогеналкилами N-алкилпирролы при нагревании изо-
меризуются в а-алкилпирролы:
-KI
N-алкилпиррол
4. Одним из важнейших пятичленных гетероциклических соедине-
ний является альдегид фурфурол (см. с. 312). Химические свойства
фурфурола напоминают химические свойства типичного ароматичес-
кого альдегида — бензальдегида. Подобно бензальдегиду фурфурол
вступает в реакцию Канниццаро:
кон
—СН2ОН
Конденсация, идущая по типу бензоиновой, приводит к образованию
фуроина:
KCN
X СН—С—X \
0 ОН О 0
фуроин
При окислении фурфурола образуется пирослизевая кислота, при вос-
становлении — фуриловый спирт:
—СН2ОН
фуриловый спирт
[Н]
[О]
фурфурол
пирослизевая кислота
Важнейшие природные производные пиррола. Производные пир-
рола широко распространены в природе, к их числу относятся такие
важные соединения, как хлорофилл, гемоглобин, гетероциклические
18
аминокислоты, витамин В12. Многие из этих соединений содержат в
молекуле особую группировку из четырех пиррольных ядер, называе-
мую ядром порфина:
При замещении атомов водорода в ^-положении пиррольных колец
на радикалы образуются порфирины. Многие важные природные сое-
динения являются металлическими производными порфиринов. Пиг-
менты крови человека и животных — гемоглобины, содержат атом
железа, являясь хромопротеидами. Их молекула состоит из двух час-
тей — белковой части (глобинов) и простетической — гема:
Зеленые пигменты растений — хлорофиллы, являются магнийпро-
изводными, в их молекуле атомы азота связаны с магнием:
S19
Хлорофиллу принадлежит выдающаяся роль в фотосинтезе — ус.
воении углекислого газа зелеными растениями за счет световой энер-
гии с образованием органического вещества. Фотосинтез является ос-
новным процессом образования органического вещества на
Земле.
Конденсированные системы, содержащие
пятичленные гетероциклы с одним гетероатомом
Существует ряд гетероциклических систем, в которых гетероциклы
сконденсированы с одним или двумя карбоциклическими кольцами.
При комбинации рассмотренных нами пятичленных гетероциклов с од-
ним бензольным ядром образуются следующие конденсированные ге-
тероциклические системы:
бензофуран, или кумарон
бензотиофен, или тионафтен
Наибольший интерес представляет индол.
Группа индола
Название индол получил в связи с тем, что впервые был получен из
природного синего красителя индиго (Байер, 1866). Он найден также в
каменноугольной смоле, образуется при гниении белков. Содержится
в цветах жасмина и апельсина.
Способы получения. 1. Один из наиболее простых методов, раз*
работанный А. Е. Чичибабиным (1915), заключается в конденсации
анилина и ацетилена:
2. Наиболее старый и
(сам индол этим методом
общий метод получения гомологов индола
получается с трудом), открытый Фишером
320
(1886), — нагревание фенилгидразонов с дихлоридом цинка, серной'
или полифосфорной кислотой:
фенилгидразон пропионового
альдегида
У\ СН3
фенилгидразон ацетона
2-метилиндол
Физические свойства. Индол — бесцветное кристаллическое вещество,
/пл — 52,5°, /кип = 254—255°С. Обладает в больших концентрациях неприят-
ным фекальным запахом. Однако в небольших концентрациях пахнет жасмином
и используется в парфюмерии при изготовлении некоторых духов.
Химические свойства. Реакции индола и его простых производных
напоминают реакции пиррола.
1. Индол обладает слабоосновными свойствами и в то же время сла-
бокислыми, образуя индолкалий:
2. В отличие от пиррола большинство реакций электрофильного
замещения индола идет преимущественно в Р- (исключение — реакция
сульфирования), если же оно занято — то в а-положение;
321
2,3-дигидроиндол
2-индолсульфокислота
3. Большое значение имеет получение магнийорганических соедине-
ний индола:
CH8Mgl
-сн4
RI
р-алкилиндол
Если на N-магнийиодиндол действовать сначала хлорацетонитрилом,
а затем омылять образующийся нитрил, образуется 3-индолилуксусная
кислота (гетероауксин):
I
Mg]
322
Гетероауксин — кристаллическое вещество, /пЛ — 164°С. Это гор-
мон роста растений (влияет на рост корней).
К производным индола относится аминокислота триптофан (см.
с. 296) и известный с глубокой древности прочный краситель индиго.
Его получали окислением производного индола — индоксила, содер-
жащегося в виде гликозида в некоторых растениях, встречающихся в
Восточной Азии. Индоксил существует в виде двух таутомерных форм:
При его окислении образуется индиго:
Для синтеза индиго индоксил можно получить конденсацией антрани-
ловой и хлоруксусной кислоты:
+ С1—СН2—СООН
хлоруксусная кислота
антраниловая
кислота
(-НС1)
(-Н3С>
фенилглицин-о-карбоновая
кислота
индоксиловая кислота
индоксил
Индиго, или иначе индиготин, — синий порошок, плавится с раз-
ложением при 390°С. Это очень прочное вещество, почти нерастворимое
в обычных растворителях, в кислотах и щелочах. Перед применением
индиго в крашении его восстанавливают в щелочных растворах в дие-
нол — белое индиго:
323
Белое индиго благодаря наличию двух енольных гидроксилов хо-
рошо растворимо в щелочах. Окрашиваемую ткань погружают в бак
(куб) со щелочным раствором белого индиго, а затем вынимают: кис-
лород воздуха окисляет белое индиго в синее, которое прочно удержи-
вается на волокне. Такой метод крашения получил название кубового.
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ДВУМЯ
ГЕТЕРОАТОМАМИ
Представители этой группы соединений содержат в цикле два гете-
роатома, если один из них — атом азота, то общее название таких сое-
динений — азолы:
Производные их являются важными в физиологическом отношении
соединениями и лекарственными препаратами.
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
Шестичленные гетероциклы с одним атомом азота
Шестичленные гетероциклы могут содержать один или не-
сколько гетероатомов, а также быть сконденсированными, например,
с бензольным ядром. Из шестичленных гетероциклов ароматического
характера с одним гетероатомом наибольший интерес представляют
соединения, содержащие атом азота:
пиридин
хинолин
изохинолин
324
Группа пиридина
Строение. По строению пиридин внешне напоминает бензол, в мо-
лекуле которого одна группа ==СН— замещена атомом азота:
4Т
1
Свободная пара электронов атома азота не взаимодействует с л-элект-
ронами кольца: ароматический секстет л-электронов образован пятью
электронами углеродных атомов и одним — атома азота. Пиридин име-
ет плоское строение. Атом азота связан с двумя соседними углеродными
атомами $р2-гибридизованными связями. Межатомные расстояния С—С
равны между собой и практически равны длине связи С—С в бензоле
0,139 нм. Расстояние С—N меньше — 0,134 нм. Однако атом азота в
пиридине более электроотрицателен, чем группа =СН— в бензоле.
Это приводит к тому, что электронная плотность у атома азота пириди-
нового кольца больше, чем у атомов углерода, которые несут дробный
положительный заряд. Неравномерное распределение электронной
плотности в молекуле проявляется в сравнительно большом электри-
ческом моменте диполя пиридина, составляющем 7,3-10"30 Кл-м.
Источники получения. 1. Пиридин вместе со своими гомологами
(монометилпиридины — пиколины, диметилпиридины — лутидины,
триметилпиридины — коллидины) содержится в каменноугольном
дегте, в костном масле. Получают обычно из каменноугольного дегтя.
2. Пиридин был получен Рамзаем (1877) по видоизмененному синте-
зу Бертло — пропусканием смеси ацетилена с синильной кислотой через
раскаленную трубку:
HC=N
Пропуская через бензол азот, возбужденный электромагнитным полем
высокой частоты, Б. И. Михайлов констатировал образование пири-
дина. Однако оба эти синтеза не имеют препаративного значения, так
как дают ничтожный выход продукта.
3. Гомологи пиридина могут быть получены последующим реакци-
ям: а) р-пиколин — конденсацией акролеина с аммиаком:
СН.
/
СН сн=сн2
II + I
сн2 сн
%
NH#
| || +2Н2О
'Ч/
пиколин
325
б) 2-метил-5-этилпиридин— конденсацией ацетилена с аммиаков
в присутствии катализатора:
4CH=CH+NH3
или из уксусного альдегида и аммиака при наличии ацетата аммо-
ния:
4СН3—С\ +NH3
Физические свойства. Пиридин — бесцветная жидкость с резким пеприят-
_____ 4- 1 1 КОГ Г ъ „ ,_______
ным запахом, /кип — 115°С. С водой и многими органическими растворителями
смешивается во всех отношениях.
Химические свойства. Ароматический характер пиридина выра-
жается очень ярко. Он не разрушается дихроматом калия и азотной
кислотой; термически устойчив. Особый химический характер пириди-
на обусловлен своеобразным распределением электронной плотности
вследствие повышенной электроотрицательности гетероатома. Элект-
ронная плотность в ядре пиридина оказывается пониженной, причем
в а- и у-положениях значительно больше, чем в Р-:
0.96
N
В результате этого реакции замещения в л-дефицитных гетероциклах
с нуклеофильными реагентами идут легче, чем реакции электрофиль-
ного замещения. Электроноакцепторное влияние атома азота способст-
вует этим реакциям, так что становится возможной быстрая атака
нуклеофилом одного из атомов углерода гетероцикла. Реакции элект-
рофильного замещения всегда идут в положение 3, так как оно меньше
всего обеднено электронами и имеет наибольшую (после атома азота)
электронную плотность.
Основность пиридина. Пиридин обладает основными свойствами за
счет свободной электронной пары гетероатома. Водные растворы пири-
дина окрашивают лакмус в синий цвет. Однако пиридин более слабое
основание, чем третичные алифатические амины:
326
Дв= 1,07.10”®
4-H+
(CH3)3N (CH3)3N —H; KB = 6,3-10”6.
—H+
С сильными минеральными кислотами пиридин дает хорошо кри-
сталлизующиеся соли:
C5H5N + НО------[C6H6NH]+Cr
С галогеналкилами пиридин ведет себя подобно третичным аминам,
присоединяя их и образуя соли N-алкилпиридиния, которые при на-
гревании изомеризуются в производные 2-алкилпиридиния:
| || 4- СН31
иодид
иод ид
Реакции нуклеофильного замещения (Sn). а. При нагревании пириди-
на до 130°С в растворе ксилола с амидом натрия с хорошим выходом
образуется 2-аминопиридин (Чичибабин, О. А. Зейде):
^N/^NHNa
+ NaOH
^n/\nh2
2-аминопиридин
б. При действии паров пиридина (температура около 400°С) на сухое
едкое кали образуется 2-оксипиридин:
I || + кон
г^\ +-
I II +кн
327
I II + H2o—> | || + KOH
\/\ж 'Sj/'^oh
2-оксипиридив
в. Более сильные нуклеофилы, такие, как металлорганические сое-
динения щелочных металлов, действуют на пиридин в более мягких
условиях:
'W ^n/\c6H5
2-фенилп иридии
Реакции электрофильного замещения (SE). Пиридин значительно
труднее вступает в реакции с электрофильными реагентами по сравне-
нию с бензолом и даже нитробензолом. Лишь в жестких условиях и с
плохим выходом его удается нитровать, сульфировать и бромирэвать.
+ +
Сильные электрофильные реагенты (NO2; SO3H) действуют в кислой
среде, причем пиридин превращается в соль, атом азота становится
аммониевым — положительно заряженным и потому еще более элект-
роноакцепторным, пассивируя ядро в еще большей степени:
Второй заместитель этими реакциями практически ввести не удает-
ся. Алкилирование по Фриделю—Крафтсу не происходит.
Восстановление. При каталитическом или электрохимическое! вос-
становлении пиридина образуется пиперидин — гетероциклическое
соединение неароматического характера:
\ б [н] //\
N I
Н
пиперидив
328
Производные пиридина. 1. При конденсации 2-метилпиридина с
формальдегидом и последующей дегидратации образующегося р-(2-
0иридил)этанола получают 2-винилпиридин, применяемый для сополи-
Меризации с бутадиеном при промышленном производстве специаль-
ных видов каучука:
у0 _
I II + н-\ —* I II
4\’/\сн3 хн ^n/\ch2—СН2ОН
м
=сн2
2-винилпиридин
2. Ядро пиридина содержится в молекуле витамина РР, являющего-
ся амидом никотиновой (p-пиридинкарбоновой) кислоты:
никотиновая (3-пиридин
карбоновая кислота)
витамин РР (никоти-
намид)
Витамин РР применяется для лечения пеллагры и некоторых дру-
гих заболеваний.
3. Алкалоиды — это группа азотистых соединений, обладающих
основными свойствами и встречающихся преимущественно в растени-
ях. Очень часто алкалоиды обладают сильным физиологическим или
фармакологическим действием. Ряд известных алкалоидов содержит
ядро пиридина и пиперидина. К ним относятся никотин, анабазин и
др. Никотин в виде солей лимонной и яблочной кислот в большом
количестве (до 3%) содержится в листьях и корнях табака. Это один из
наиболее ядовитых алкалоидов, 40 мг никотина является смертельной
дозой для человека. Применяют никотин в качестве инсектицида в
садоводстве. При окислении никотина образуется никотиновая кисло-
та:
[О]
никотиновая кислота
Анабазин — изомер никотина:
329
W h
Содержится в табаке и в азиатском растении Anabasis aphylla. Широко
применяется в сельском хозяйстве в качестве инсектицида, более силь-
ного, чем никотин.
Шестичленные гетероциклы с двумя атомами
азота
К ним относятся:
4
пиридазин
пиразин
Пиридазины в природе не найдены. Получаются синтетически. Пи-
ридазиновое кольцо очень устойчиво к окислению. Не поддается элект-
рофильному замещению.
Пиразины можно получить самопроизвольной конденсацией а-
аминокетонов. Сам пиразин — значительно более слабое основание,
чем пиридин. К электрофильному замещению склонен еще меньше,
чем пиридин.
Пиримидин — важнейший из шестичленных гетероциклов с двумя
атомами азота. Пиримидиновое ядро входит в многочисленные природ-
ные соединения, среди которых находятся витамины, коферменты,
нуклеиновые кислоты.
Пиримидин можно синтезировать через барбитуровую кислоту,
которую сначала превращают с помощью пентахлорида фосфора в
2,4,6-трихлорпиримидин, восстановлением которого иодоводородом
получают пиримидин:
II
С—ОС2Н5
Н2С
ОС2Н5
C2H6ONa
- 2С2Н6ОН
II
С—NH
барбитуровая кислота
мочевина
малоновый эфир
330
таутомерия
2,4, 6-трихлорпиримидин пиримидин
При гидролизе нуклеиновых
изводных пиримидина:
кислот получаются три важных про
цитозин
Главной таутомерной формой этих соединений (по данным преиму-
щественно физических исследований) следует считать правую. Левая
присутствует только в растворах и притом в весьма незначительном
количестве. К пиримидиновым производным принадлежат лучшие из
сульфамидных препаратов (сульфазин, сульфадимезин и др.).
Витамин Вх (тиамин) тоже содержит пиримидиновый цикл:
СН3 + уНг
N4s_CI- Сн2_/Ч N
ОН—СН2—СН2 s/ II I
И^СНз
Отсутствие витамина Вх в пище, например при питании шлифованным
331
рисом (тиамин содержится в рисовых отрубях), вызывает известное
на Востоке нервное заболевание бери-бери. Суточная потребность а
витамине Вх человека — 4 мг. Необходимость в нем связана с тем, что
витамин Вх (в виде пирофосфата) входит в структуру фермента карбо-
ксилазы, катализирующего некоторые стадии метаболизма сахаров а
организме (отщепление от пировиноградной кислоты).
Азотистые бигетероциклы
Группа пурина
пурин
Производные пурина имеют исключительное биологическое значение
прежде всего потому, что входят наряду с пиримидиновыми основа-
ниями в структуру нуклеиновых кислот и имеют, таким образом, отно?
шение к программированию синтеза белков и к явлениям наследствен-
ности. К таким соединениям относится аденозинтрифосфат — перенос-
чик энергии в биохимических реакциях и фосфорилирующий агент.
Мочевая кислота служит одним из ключевых соединений в синтезе
пуриновых производных (2,§,8-триоксипурин). Играет у птиц и репти-
лий роль вещества, выводящего из организма избыток азота (как мо-
чевина у млекопитающих):
ОН
мочевая кислота
Аденин, 6-аминопурин, является одним из наиболее распростра-
ненных пуриновых оснований:
В связанном виде содержится в нуклеиновых кислотах и легко
332
получается при их гидролизе. В свободном состоянии встречается в не-
которых растениях (чай, сахарная свекла, хмель), а также в грибах.
В значительных количествах содержится в дрожжах, бактериях, орга-
нах животных (мускулах, желтом теле, печени), моче. В больших коли-
чествах аденин выделяют из чая и мелассы.
Гуанин, 2-амино-6-оксипурин, сопутствует аденину во многих расте-
ниях и органах животных:
ОН
Значительные количества содержатся в чешуе и коже рыб, пресмыка-
ющихся и амфибий. Часто своеобразный переливчатый блеск их по-
кровов обусловлен присутствием выкристаллизовавшегося гуанина.
Он также может быть получен при гидролизе нуклеиновых кислот.
Алкалоиды кофеин и теобромин — также производные пурина.
Теобромин — алкалоид, содержащийся в бобах какао (до 1,8%):
О СН3
Хорошо растворим только в горячей воде; /пЛ = 35ГС. Применяется
в качестве одного из сильнейших мочегонных средств. При метилиро-
вании превращается в кофеин:
Кофеин — алкалоид, содержащийся в зернах кофе (~1 %), в листь-
ях чая (до 5%), в орехах кола (3%) и других тропических растениях.
Он является возбудителем центральной нервной системы и стимулято-
ром сердечной деятельности, широко применяемым в медицине. Необ-
ходимый для этого кофеин извлекают из отходов чая и кофе. В промыш-
ленных масштабах получают из мочевой кислоты.
333
ШЕСТИЧЛЕННЫЕ КИСЛОРОДСОДЕРЖАЩИЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
НЕАРОМАТИЧЕСКОГО ХАРАКТЕРА
Приведенные соединения представляют интерес вследствие того,
что их производные имеют большое химическое, физиологическое и
промышленное значение.
Сам а-пирон мало интересен. Значительно большее значение имеет
его производное с ортоконденсированным кольцом — кумарин. Он най-
ден во многих растениях, особенно в больших количествах содержится
в бобах тонка и ясменнике. Вероятно, кумарин в растениях находится
в виде гликозида, этим можно объяснить, что запах кумарина часто
появляется только во время высыхания растений (например, у сена),
которое сопровождается гидролизом. В промышленности кумарин по-
лучают по реакции Перкина — конденсацией салицилового альдегида
с уксусным ангидридом в присутствии ацетата натрия:
(СН3СО)2О
CH3COONa
Кумарин кристаллизуется в бесцветные кристаллы с приятным
запахом (запах ясменника и сена), /пЛ 67°С. Он находит практичес-
кое применение в качестве отдушки для печенья, лимонадов, мыл и
табаков, а также компонента фруктовых эссенций и т. д.
Как и в случае а-пирона, имеет большее значение не сам у-пирон,
а его производные — хромоны и флавоны — 2,3-бензо-у-пирон назы-
вается хромоном:
334
Этот скелет лежит в основе многих природных веществ. К ним отно-
сится группа витаминов Е (токоферолов). Впервые эта группа витами-
нов была выделена из масла ростков пшеницы.
Сейчас известно семь веществ, близких по строению, обладающих
действием витамина Е, например:
СН3 О
НО I II
сн3
сн3 сн3 СНз
I I I
(СН2)3-СН-(СН2)3-СН-ЧСН2)3-СН-СН3
сн3
3-токоферол
Особенно распространены в растительном мире производные 2-фе-
нилхромона (флавона):
О
флавон
Природные производные флавона — оксипроизводные, содержащие
гидроксилы в положениях 5—7 и иногда в положениях 3, 3' и 4'.
Такие соединения окрашены в желтый цвет и представляют собой
природные красители, содержащиеся в желтых цветах. Примерами
могут служить лутеолин — красящее вещество почек тополя и резе-
ды и кверцетин — вещество, широко распространенное в желтых цве-
тах:
335
он о
ОН о
лутеолин
кверцетин
В растениях они находятся в виде гликозидов. Один из гликозидов
кверцетина — рутин, вещество, относящееся к группе витаминов Р,
при отсутствии которых в пище увеличивается проницаемость крове-
носных сосудов, что ведет к «пурпуровой болезни» — множественным
точечным кровоизлияниям.
При восстановлении оксифлавонов можно получить антоцианидины.
Так, кварцетин дает при этом цианидин — типичный представитель
антоцианидинов — соединений, содержащих оксониевый кислород,
входящий в ароматический цикл:
цианидин
Антоцианидины — относительно прочные твердые соли, интенсивно
окрашенные В виде гликозидов они являются атоцианами (антоциано-
выми красителями), окрашивающими цветы, ягоды и овощи (по-гре-
чески антоциан — краска цветов).
НУКЛЕИНОВЫЕ КИСЛОТЫ. СТРОЕНИЕ И
БИОЛОГИЧЕСКАЯ РОЛЬ
В каждой живой клетке содержатся нуклеопротеиды — сложнейшие вещест-
ва, состоящие из белков, соединенных с природными биополимерами другого ти-
па— нуклеиновыми кислотами. Связь белка (обладающего основными свойст-
вами) с нуклеиновыми кислотами осуществляется ва счет солеобразных и водороД-
336
пых связей. Ее легко разрушить путем солевой коагуляции белка, а нуклеиновые
кислоты выделить в свободном состоянии. Нуклеиновые кислоты — высокомолеку-
лярные соединения, с молекулярной массой от 200 тысяч до нескольких
миллионов. При полном гидролизе нуклеиновых кислот образуется смесь азотсо-
держащих гетероциклических оснований (пуриновых и пиримидиновых), пентоз
и фосфорной кислоты. Подобно тому как основой скелета белка является поли-
пептидная цепь (полиамидная), основой скелета нуклеиновых кислот служит
полинуклеотидная цепь (полиэфирная):
•——----- пентоза —О—Р—О— пентоза —О—Р—------------~
I I
он он
При мягком гидролизе нуклеиновых кислот можно выделить структурные
единицы, называемые нуклеотидами. Они содержат пуриновое или пиримидино-
вое основание, пентозу и фосфорную кислоту. При отщеплении от нуклеотида
фосфорной кислоты образуется нуклеозид — соединение гетероциклического ос-
нования с пентозой.
Нуклеиновые кислоты делятся на два больших класса: рибонуклеиновые
кислоты (РНК) и дезоксирибонуклеиновые кислоты (ДНК)- В состав РНК в ка-
честве пентозы всегда входит £)-рибоза, а в качестве гетероциклических основа-
ний — цитозин, урацил; аденин и гуанин. В состав ДНК входит 2-дезокси-£>-ри-
боза, а также цитозин, тимин, аденин и гуанин.
Остатки сахара находятся в фуранозной форме. В нуклеозидах соот-
ветствующий моносахарид связан атомом Q (fJ-связь) с атомом N3
пиримидинового или N9 пуринового кольца. В РНК обнаружены сле-
дующие нуклеозиды:
nh2 < с 5’ ° > сн2он сн2он 4'<Гн Н/1' К_н н У н Ч—< н н Ч К ; он он он он цитидиц уридин В ДНК обнаружены i Vй" 1 j 5'СН2ОН 3 СН2ОН 4\н нут н н Х| у н н V- к н он он он ОН аденозин гуанозин дезоксирибонуклеозиды общей формулы: 5' СН2ОН К 4'<Н НЛ' н I*н он н
12—682
337
(R — остаток цитозина, тимина, аденина или гуанина.)
В нуклеотидах фосфорная кислота этерифицирует гидроксил у
С3 или С5 остатка сахара у нуклеозида, например:
З'-уридинмонофосфаТ
З'-цитозиндезоксирибонуклеотид
Построение полинуклеотидных цепей молекул РНК и ДНК из
нуклеотидов осуществляется путем образования фосфорэфирных свя-
зей между гидроксилами С3 и С5 нуклеозида. В качестве примера мож-
но привести структурную единицу молекулы РНК со следующей по-
следовательностью гетероциклических оснований — аденин, урацил7
гуанин, цитозин:
338
Одной из важнейших проблем, связанных с синтезом белка в живой
клетке, является вопрос о том, что заставляет аминокислоты, входящие
в состав белка, соединяться между собой в строгой последовательности,
характерной для каждого типа белка. С этим тесно связан вопрос, ка-
ким образом информация о последовательности аминокислот передает-
ся каждому новому поколению клеток. К настоящему времени совер-
шенно четко установлено, что ДНК служит источником информации
для развития клетки, включая синтез необходимых ферментов и такое
самокопирование, которое необходимо для воспроизведения путем
клеточного деления. Молекулы ДНК, находящиеся в ядрах клеток,
содержат всю информацию наследственности.
Отдельные ДНК различаются между собой соотношением различ-
ных оснований (аденин, гуанин, цитозин, тимин) и последовательно-
стью соединения соответствующих нуклеотидных фрагментов (первич-
ная структура). Современные представления о вторичной структуре
ДНК базируются на исследовании Д. Уотсона и Ф. Крика. На основа-
нии данных рентгеноструктурного анализа и факта определенной
упорядоченности структуры ДНК различного происхождения был сде-
лан вывод, что вторичная структура ДНК представляет собой а-спи-
раль, состоящую из двух полинуклеотидных цепей, закрученных
одна вокруг другой и вокруг общей для обеих цепей оси (рис. 34).
При этом основания ориентированы перпендикулярно к оси спирали.
Рис. 34. Схематическое
изображение двойной
спирали двух полинукле-
отидных цепей молеку-
лы ДНК
Рис. 35. Взаимодействие
пар оснований в двой-
ной спирали ДНК:
А, Т, Г, Ц — соответствую-
щие основания; С — 2-дезок-
он-О-рибоза; Р — фосфатная
группа
12*
339
На один виток двойной спирали ДНК приходится 10 оснований в каж-
дой из полинуклеотидных цепей. Обе полинуклеотидные цепи удержи-
ваются и стабилизируются за счет водородных связей и вандервааль-
совых взаимодействий между основаниями. При этом друг против дру-
га располагаются так называемые комплементарные пары оснований:
тимин и аденин, цитозин и гуанин (точнее — их остатки):
остаток остаток аденина
гимина
На рис. 35 показано взаимодействие пар оснований в двойной спи-
рали (встречающихся в молекуле ДНК в соотношении 1:1).
В длинной цепи ДНК возможно огромное число вариантов сочета-
ния различных нуклеотидов, что позволяет одной молекуле ДНК со-
держать большое количество разнообразной информации. Участки
цепи ДНК со строго определенной последовательностью нуклеотидов,
по сути дела, представляют собой «гены», ответственные за передачу
того или иного наследственного признака. Сами ДНК не играют роли
непосредственного участника синтеза белков, поскольку в большинст-
ве случаев этот синтез идет вне ядра клетки — в цитоплазме, где нет
ДНК. Следовательно, генетическая информация должна передаваться
от ДНК каким-либо другим веществам, которые доставляли бы ее от
ядра к тем участкам в цитоплазме, где происходит синтез белка. Роль
таких веществ выполняют РНК. Существует три типа РНК: информа-
ционные РНК (несут информацию о том, какой именно белок должен
синтезироваться в конкретной клетке); рибосомальные РНК (осущест-
вляют в рибосомах синтез белка); транспортные РНК (доставляют
отдельные аминокислоты в рибосомы, к месту синтеза белка).
Раздел шестой
Биоорганические соединения
и некоторые соединения
изопреноидного характера
Углеводы
Углеводы и их производные уже давно занимают
исключительное место в технике и повседневной жизни человека.
Успехи биоорганической химии позволили оценить подлинную
роль углеводов и в самом процессе жизнедеятельности. Разнообразные
производные углеводов, находясь в составе клеток любого животного
организма, выполняют роль конструкционного материала, субстратов,
регуляторов специфических биохимических реакций, а также постав-
щиков энергии. В соединении с липидами, белками, в составе нуклеи-
новых кислот углеводы образуют сложные высокомолекулярные комп-
лексы, лежащие в основе субклеточных структур и представляющие
собой основу живой материи.
Будучи первичными продуктами фотосинтеза, углеводы являются
первыми органическими веществами в кругообороте углерода в при-
роде и, таким образом, служат как бы мостом между минеральными и
органическими соединениями. Углеводы широко распространены в
природе, особенно в растительном мире: до 80% сухого вещества рас-
тений приходится на долю углеводов. Содержание их в живых организ-
мах — около 20% сухого вещества.
Название «углеводы» возникло на основании анализа первых из-
вестных представителей этой группы соединений. Оказалось, что эти
вещества состоят из углерода, водорода и кислорода, причем соотно-
шение числа атомов водорода и кислорода у них такое же, как в воде:
на два атома водорода — один атом кислорода. Таким образом, их
рассматривали как соединение углерода с водой. Термин «углеводы»
был предложен профессором Дерптского (Юрьевского) университета
К. Шмидтом. В дальнейшем стали известны многие углеводы, не отве-
чающие этому условию, однако название «углеводы» до сих пор оста-
ется общепринятым.
Классификация углеводов. Большой класс углеводов можно клас-
сифицировать в соответствии со следующей схемой:
34)
Углеводы
сложные углеводы
(полисахариды, полиозы)
простые углеводы
(моносахариды, мо-
нозы)
альдозы
кетозы
I
I
низкомолекуляр-
ные , сахароподоб-
ные
(олигосахариды)
I
высокомолекуляр-
ные,
несахаропсдобные
триозы (С3)
тетрозы (С4)
пентозы (Св)
восстанав- невосстанав-
ливающие ливающие
гомополи- гетерэпо-
сахариды лисахариды
гексозы (Сб)
Все углеводы подразделяют на две группы: простые углеводы (или
простые сахара) и сложные углеводы (или сложные сахара).
Простыми углеводами (моносахаридами, или монозами) называют-
ся такие углеводы, которые не способны гидролизоваться с образова-
нием более простых углеводов. Большинство этих веществ имеет сос-
тав, соответствующий общей формуле СпН2пОп, т. е. число атомов
углерода у них равно числу атомов кислорода. Если моносахарид со-
держит альдегидную группу, то его называют альдозой, если кетонную
— кетозой. В зависимости от числа углеродных атомов в молекуле
моносахарида различают триозы (С3), тетрозы (Сл), пентозы (Cs), гек-
созы (С6).
Сложными углеводами (полисахаридами, или полиозами) называ-
ют такие углеводы, которые способны гидролизоваться с образованием
простых углеводов. Общая формула этих веществ CmH2nOn, т. е. число
атомов углерода у них не равно числу атомов кислорода.
К сложным углеводам относятся довольно разнообразные по сво-
им свойствам вещества, и поэтому их делят на две подгруппы: низко-
молекулярные, сахароподобные полисахариды (олигосахариды) и вы-
сокомолекулярные, несахароподобные полисахариды.
Олигосахариды при гидролизе распадаются на 2—8 молекул моно-
сахаридов («олигос» — по-гречески — немногий). По своему строению
олигосахариды могут быть восстанавливающими и невосстанавливаю-
щими.
Высокомолекулярные полисахариды при гидролизе распадаются
на очень большое число моносахаридов. В их составе может содержать-
ся до нескольких десятков тысяч моноз. Полисахариды, построенные
342
из остатков одного и того же простого сахара, называются гомополи-
сахаридами. Гетерополисахариды построены из остатков различных
моносахаридов.
МОНОСАХАРИДЫ
Строение. Важнейшими и типичными представителями моносаха-
ридов являются глюкоза (виноградный сахар) и фруктоза (фруктовый
сахар). Они изомерны друг другу и имеют молекулярную формулу
С6Н12О6. Изучение структуры этих моносахаридов помогло в значи-
тельной степени изучению строения и свойств других представителей
этого класса. Ниже приведены некоторые факты, позволяющие уста-
новить структуру глюкозы и фруктозы.
При энергичном восстановлении глюкозы иодистым водородом был
получен 2-иодгексан СН3—СН2—СН2—СН2—CHI—СН3 — вещество с
неразветвленной цепью углеродных атомов, а значит, и сама глюкоза
имеет неразветвленную цепь. Наличие сильной восстанавливающей
способности у глюкозы, легкость ее окисления (например, бромной во-
дой), происходящего без разрыва углерод-углеродной связи, дает ос-
нование считать, что глюкоза содержит альдегидную группу:
(С5НиО5) + НОВг -> (СвНиОб) + НВг
глюкоза глюконовая кислота
Важным подтверждением наличия в глюкозе альдегидной группы
является присоединение синильной кислоты с последующим омыле-
нием нитрила и восстановлением образовавшейся глюкогептоновой
кислоты иодистым водородом до эн антовой кислоты:
HCN 2Н2О
(С5НиОб)С^-----> (С5НпОб)СН------->
н \он
глюкоза
HI
(С6НиОб) СН—СООН СН3—(СН2)5—соон
н-энантовая кислота
глюкогептоновая кислота
Учитывая образование глюкозой сложных эфиров с органическими
кислотами, Вертело (1860) заключил, что глюкоза является многоатом-
ным спиртом. В 1869 г. А. А. Колли показал, что глюкоза представля-
ет собой пятиатомный спирт. Из всего сказанного следует, что глюкоза
представляет собой пятиатомный альдегидоспирт следующего строе-
ния:
сн2он—снон—снон—снон—снон—с=о
н
Фруктоза при восстановлении иодистым водородом тоже дает 2-иод-
343
гексан, т. е. она также имеет неразветвленную цепь углеродных атомов
и присоединяет синильную кислоту, следовательно, содержит карбо-
нильную группу. Однако окисление фруктозы происходит с разрывом
углеродной цепи:
ООО О
[О] II II II II
сен1206 —> с—с + с—снон—снон—с
фруктоза 1,11 I
у он он он он
щавелевая винная кислота
кислота
а следовательно, фруктоза является кетозой. Как и глюкоза, фруктоза
реагирует с пятью молекулами уксусного ангидрида, образуя 5 слож-
ноэфирных группировок, т. е. она содержит 5 гидроксильных групп.
Таким образом, фруктоза представляет собой пятиатомный кетоспирт:
О
II
сн2он—с—снон—снон—снон—сн2он
Моносахариды, содержащие, подобно глюкозе, альдегидную груп-
пу, получили название альдоз; моносахариды, содержащие кетонную
группу, стали называть кетозами.
Чтобы подчеркнуть, что в состав моносахарида входит альдегидная
группа и что этот моносахарид содержит, например, шесть углеродных
атомов, объединяют названия в слово «альдогексоза» (аналогично кето-
гексоза, альдопентоза и т. д.).
Итак, по своему химическому строению моносахариды представля-
ют собой многоатомные альдегидо- или кетоспирты.
Стереохимия. Пространственные конфигурации моносахаридов,
D- и L-ряды. Моносахариды содержат асимметрические углеродные
атомы. Альдотетрозы, например, имеют два асимметрических углерод-
ных атома, альдопентозы —три, альдогексозы — четыре. В результате
этого у моносахаридов имеется большое число стереоизомеров. Для
альдогексозы, имеющей четыре асимметрических атома углерода, чис-
ло стереоизомеров, согласно формуле N = 2Л, будет равно 24 = 16.
У альдогексоз (так же, как и у кетогексоз) половина оптически дея-
тельных стереоизомеров является антиподами другой половины. Та-
ким образом, 16 стереоизомеров альдогексоз образуют 8 пар антиподов.
Например, природному моносахариду £)-глюкозе соответствует синте-
тически полученный антипод А-глюкоза:
1 н ,0 '^<н
Н—2С*—он но—2С*—н
но—зс*—н 1 н—зс*—ОН
Н—4С*—он j но—4с*—н
Н—5С*—он 1 но—5С—н
6СН2ОН 1 6СН2ОН
D-глюкоза L-глюкоза
344
Нумерацию цепи принято вести, начиная от альдегидной группы в
альдозах, а в кетозах — начиная от того конца цепи, к которому ближе
кетогруппа. Определение взаимного расположения Н- и ОН-групп у
каждого асимметрического центра представляет трудную задачу.
В 1906 г. А. М. Розанов предложил судить о принадлежности того или
иного моносахарида к D- или L-ряду по расположению в пространстве
атомных групп у последнего асимметрического углеродного атома (он
же — предпоследний атом в цепи): если конфигурация совпадает с
конфигурацией £)-глицеринового альдегида, то моносахарид относят
к D-ряду, если конфигурация совпадает с конфигурацией L-глицери-
нового альдегида, то моносахарид принадлежит к L-ряду:
СНО
I
Н—с*—он
СН2ОН
D- глицериновый альдегид
(СНОН)„СНО
I
н—с*—он
с!н2он
D-ряд
СНО
НО—с*—н
I
СН2ОН
L-глицериновый альдегид
(СНОН)ЛСНО
НО—с*—н
СН2ОН
Ь-ряд
Таким образом, весь стереохимический D-ряд моносахаридов —
до гексоз включительно, можно изобразить схемой 11.
Антиподами (зеркальными изображениями) каждого из представ-
ленных в схеме моносахаридов являются соответствующие моносаха-
риды L-ряда.
Следует заметить, что обозначения «О» или «L» вовсе не служат
указанием на направление вращения плоскости поляризации раство-
рами сахаров. Например, природная D-фруктоза вращает плоскость
поляризации влево. Для указания направления вращения использу-
ют знаки «+» (правое вращение) и «—» (левое вращение) так же, как
это принято ранее в оксикислотах (см. с. 250). Тогда природная фрук-
тоза и глюкоза будут обозначаться: D(—) — фруктоза; £>(+) — глюко-
за.
Циклическая структура моносахаридов. Хотя альдегидные и ке-
тонные формулы моносахаридов хорошо объясняли многие свойства
этих веществ, однако постепенно накапливались факты, которые эти-
ми формулами не могли быть объяснены.
1. Отсутствие некоторых реакций на альдегидную группу. Оказа-
лось, что моносахариды, давая ряд реакций на альдегидную группу
(окисление до СООН, реакция с синильной кислотой, восстановление
аммиачного раствора оксида серебра, реакции с фелинговой жидко-
стью), в то же время не дают в обычных условиях некоторых альдегид-
ных реакций (не реагируют с гидросульфитом натрия, не дают окраши-
вания с фуксинсернистой кислотой и т. д.).
2. Мутаротация. Все моносахариды обладают оптической актив-
ностью, однако угол вращения плоскости поляризации свежеприготов-
34Ь
Схема 11. £)-ряд альдоз
н-
сно
H-4-OH
ll-
>н
Н-|-ОН
СН2ОН
D -рибоза
СНО
--ОН
Hl
н
он
H-f-OH
сн2он
D -аллоза
СНО
Н-|-ОН
СНОН
D -глицериновый альдегид
сно
СН2ОН
D-эритроза
Ho-
СНО
--H
•н
СН2ОН
D-арабиноза *
н
СНО
--ОН
сно
но—н
сно
нД-он
но—н
СНО
--Н
сно
н-
►и
IH
но
•И
ён2он
D-альтроза
н—он
Н-|-ОН
СН2ОН
D-треоза
Н----ОН
СН2ОН
D- ксилоза
н—он
сно
Н-4-ОН
сно
но—н
но—н
но—н
сно
но4-н
но—н
Н—|—ОН
СН2ОН
D-ликсоза
сно
н—он
Hl
СНО
—Н
HO--H
но—н
но- -н
Н— -ОН
СН2ОН £н2он
н-
он
н-
--он
СН2ОН Сн2он
н-
•н
СН2ОН СН2ОН
D-глюкоза D-манноза D-гулоза D-идоза D-галактоза D-талоза
н
н
н
н
ленных растворов моносахаридов по мере стояния изменяется, пока не
достигнет некоторой постоянной величины. Это изменение угла вра-
щения, или изменение оптической активности, растворов сахаров при:
стоянии получило название мутаротации. Например, для О-глюкозы
это изменение происходит от + Ю6 до + 52,5°, что изображают
обычно так: + 106°+52,5°.
3. Количество диастереомерных моносахаридов. Если исходить из
формул Фишера, то для альдогексоз должно существовать 16 диасте-
реомеров. Все они были найдены в природе или синтетически получены
Фишером. Однако оказалось, что каждый из этих моносахаридов спо-
собен существовать в двух совершенно индивидуальных кристалличес-’
ких формах с различными константами, в том числе с различными уг-
лами вращения. Так, например, существует две О-глюкозы с углами^
вращения +106 и +22,5°. Характерно, что при растворении любой из"
них всегда получается раствор, угол вращения которого меняется вслед-
ствие мутаротации и постепенно достигает для обеих одной и той ж&
всегда строго постоянной величины, которая для глюкозы составляет
+52,5°. Таким образом, число реально существующих альдоз вдвое,
превышает число предсказанных. Удвоение числа диастереомерных^
форм говорит о наличии в молекуле моносахаридов еще одного допол-
нительного асимметрического углеродного атома.
4. Особые свойства одной из гидроксильных групп. Еще одно серм
езное противоречие оксикарбонильным формулам Фишера заключает^
346
г
ся в том, что одна из гидроксильных групп моносахаридов резко отли-
чается по своим свойствам от других гидроксилов. Так, например, при
нагревании моносахаридов со спиртами в присутствии небольшого ко-
личества сухого хлорида водорода происходит образование хорошо
кристаллизующихся веществ — гликозидов (см. с. 359). Гликозиды в
присутствии разбавленных минеральных кислот очень легко гидро-
лизуются, причем из одной молекулы гликозида образуется одна моле-
кула моносахарида и одна молекула спирта. Если исходить из форму-
лы Фишера, то в глюкозе присутствует 5 гидроксилов, каждый из ко-
торых мог бы дать соединения типа простого эфира. Почему же только
один гидроксил участвует в образовании гликозида? Далее, если обра-
зовалось вещество, относящееся к простым эфирам, то почему оно так
легко подвергается гидролизу, тогда как основное свойство простых
эфиров — это стойкость к гидролизу?
Все эти факты нашли объяснение, когда было доказано, что каждый
моносахарид может существовать в виде ряда таутомерных форм. В
растворах моносахаридов наряду с альдегидными и кетонными форма-
ми всегда содержатся циклические таутомерные формы.
Еще А. А. Колли в 1870 г. предложил для глюкозы циклическую
полуацетальную формулу с трехчленным окисным кольцом:
/О
СНоОН—снон—снон—снон—сн—с<
1-0-1°"
Наличием подобного кольца он объяснял отсутствие некоторых альде-
гидных реакций у глюкозы.
Позже, в 1883 г., Толленс предложил формулу с пятичленным окис-
ным кольцом:
/Н
сн2он—снон—сн—снон—снон—с<
I о I °".
Циклические формулы называют также окисными или полуацеталь-
ными. Образование циклической формы из альдегидной можно пред-
ставить следующим образом. Как известно, альдегиды легко присое-
диняют спирты, превращаясь в полуацетали (см. с. 201):
альдегид полуацеталь
Альдегидная группа моносахарида, расположенная в пространстве
близко к одной из гидроксильных групп, взаимодействуя с ней, обра-
зует внутренний полуацеталь:
347
альдегидная форма
^сн2он
циклическая форма
(окисная, полуацетальная)
Образование циклических форм объясняет все выше перечисленные
факты, которые нельзя было объяснить альдегидными формулами:
1. То, что моносахариды не дают в обычных условиях некоторых
альдегидных реакций, объясняется тем, что в водных растворах моно-
сахаридов преобладают циклические формы. Альдегидные же формы
находятся в небольших количествах, в отдельных случаях составляют
несколько десятых долей %. Впоследствии оказалось, что могут быть
созданы условия, в которых эти реакции возможно осуществить.
2. Явление мутаротации: в циклической форме моносахарида по-
является дополнительно еще один асимметрический углеродный атом,
входивший ранее в карбонильную группу. В зависимости от располо-
жения гидроксила у этого нового асимметрического центра различают
так называемые а- и (3-формы (или аномеры): если гидроксил у вновь
образовавшегося асимметрического атома углерода расположен по од-
ну сторону от углеродной цепи с гидроксилом, определяющим принад-
лежность к D- или L-ряду (или по одну сторону с окисным кольцом,
образованным из этого гидроксила), то такая форма называется а-
формой (а-аномером); если же полуацетальный гидроксил располага-
ется по другую сторону углеродной цепи от гидроксила,определяюще-
го принадлежность к D- или L-ряду (или по другую сторону от окис-
ного кольца, образованного из этого гидроксила), то такая форма
называется (3-формой (или (3-аномером):
н—с—он
но—с—н о
J
н—с—он
—-г
полуацетальные,
или гликозидные
гидроксилы
Н—С—ОН
но—с-—Н о
J
н—с—он
сн2он
0, D-глюкоза
аг, D-глюкоза.
В кристаллическом виде моносахариды существуют в цикличес-
кой форме и являются устойчивыми соединениями, во многих слу-
чаях они могут быть выделены в виде двух аномеров. Однако при раст-
ворении индивидуального аномера в воде быстро наступает таутомер-
ное превращение, приводящее к образованию равновесной смеси
нескольких форм моносахаридов. Внешне это проявляется в изме-
нении угла вращения сахара.
348
3. Удвоение числа диастереомеров становится также объяснимым
появлением нового асимметрического углеродного атома. В цикличес-
кой форме альдогексозы имеют уже пять асимметрических атомов уг-
лерода, следовательно, образуется 25, или 32, диастереомера, а альдо-
пентозы образуют, соответственно, 24, или 16, диастереомеров.
4. Образование гликозидов с участием одного гидроксила сахара
также хорошо объясняется существованием циклической формы. Если
у альдегидной формы монозы все гидроксилы одинаковы — это спир-
товые гидроксилы, то у циклической формы есть один гидроксил, рез-
ко отличающийся от других. Это гидроксил, образовавшийся из альде-
гидной или кетонной группы, который называется полуацетальным или
гликозидным гидроксилом. Он более реакционноспособен, чем осталь-
ные спиртовые гидроксилы, поэтому легко вступает в реакцию со спир-
том, образуя гликозид (или циклический полуацеталь), также легко спо-
собный гидролизоваться в кислой среде (в отличие от простых эфиров):
Н ОН
--------
Н—С—ОН
I н+
НО—С—Н О + R'OH ^2
I
н—с—он
н—с------
СН2ОН
Н OR'
'с—-----
I
н—с—он
НО—С—н о
I
н—с—он
I
н—с-------
СН2ОН
Если гликозид образован из a-формы сахара, то это а-гликозид, а
если (3-, то Р-гликозид.
Таким образом, циклические формы сахаров объяснили все суще-
ствовавшие до их открытия противоречия.
Характер окисных колец. Кольцо циклической формы сахара мо-
жет быть шестичленным и пятичленным. Окисные формы моноса-
харидов с полным правом можно рассматривать как производные
соответствующих гетероциклических соединений (см. с. 310, 334):
шестичленные как производные пирана, а пятичленные как производ-
ные фурана (точнее как производные тетрагидропирана и тетрагидро-
фурана соответственно). Формы моносахаридов, содержащие шестич-
ленные циклы, называют пиранозами, а пятичленные — фуранозами:
349
СН2ОН
с=о
I
но—с—н
I
н—с—он
Н—с—он
сн2он
D- фруктоза
сн,он
I 2
С-ОН
I
но—с—н
I
н—с—он о
н—с—он
сн2-----
a, D-фруктопираноза
СН2ОН
I
но—с------
I
но—с—н
I С
Н—С—ОН I
СН2ОН
3, D-фруктофураноза
В 20-х годах нынешнего столетия Хеуорзс предложил новый спо-
соб условного перспективного изображения структур моносахаридов, .
очень наглядно показывающий взаимное пространственное располо-
жение гидроксильных групп. Согласно его предложению, цикличес-
кую форму моносахарида условно считают плоской. Для изображения,
на бумаге ее мысленно располагают таким образом, чтобы кислород-
ный атом пиранозного кольца находился на наибольшем расстоянии
от глаза наблюдателя справа (у фуранозного кольца — посредине),
а углеродная цепь была бы обращена выпуклой стороной к наблюдате-
лю. Обычно часть молекулы, приближенную к наблюдателю, пока-
зывают жирной линией. Нумерацию цепи ведут по часовой стрелке,,
начиная справа от атома кислорода:
нумерация;
пираноза
Заместители, располагающиеся в формуле Толленса справа от угле-и
родной цепи, помещаются снизу, под условно плоским кольцом фор-
мулы Хеуорзса. Заместители же, находящиеся слева от углеродной,
цепи в формуле Толленса, помещаются сверху, над условно плоским,
кольцом формулы Хеуорзса. Концевую (последнюю по нумерации)
СНгОН-группу в углеродной цепи располагают над плоскостью коль-
ца в формуле Хеуорзса, если атом, с которым она связана, имеет £>-
конфигурацию, и снизу — если он имеет L-конфигурацию. Ниже
представлены формулы Толленса и Хеуорзса для а-О-глюкопиранозЫ;
и p-D-фруктофуранозы:
1
н—с-
н2он
а - D-глюкопираноза
350
fi -D -фруктофураноза
Таким образом, в формулах Хеуорзса полуацетальный гидроксил
и концевая СНгОН-группа располагаются у а-аномеров по разные
стороны условно плоского кольца молекулы, а у 0-аномеров — по
одну сторону.
Таутомерия моносахаридов в растворах. Сахара были исторически
одними из первых веществ, для которых наблюдалось явление тауто-
мерии. Таутомерия сахаров — это так называемая кольчатоцепная
или оксо-окси-таутомерия. Моносахариды, в зависимости от условий
реакции и примененных реагентов, реагируют в одной из таутомер-
ных форм: пиранозной, фуранозной или ациклической:
a-D -глю кофураноза
форма /?-/)-глюкофураноза
таутомерные формы
D -глю козы
Существование таутомерии для моносахаридов подтверждено экс-
периментально путем исследования их оптической активности, а так-
же с помощью ЯМР- и ИК-спектроскопии. Явление мутаротации свя-
зано со взаимными превращениями таутомерных форм моносахарида и
установлением равновесия между ними. Положение равновесия
зависит от структуры и стереохимии моносахарида, но не зависит от
исходной таутомерной формы данного сахара. Так, свежеприготов-
ленные водные растворы а- и 0-О-глюкозы имеют удельное враще-
ние [аЬ = +106° и 22,5° соответственно. С течением времени удельное
вращение первого падает, а второго возрастает, в обоих случаях до-
стигая постоянной величины +52,5°.
Понятие о конформационной изомерии. Для сахаров в циклическом
виде возможен еще один вид изомерии — конформационная изомерия,
351
связанная с расположением в пространстве углеродных атомов шести-
членного цикла. Для пиранозного кольца из-за его несимметричнос-
ти, связанной с наличием в нем кислорода, возможно большее число
конформаций, чем для циклогексана. Предпочтительность той или
иной конформации определяется соотношением размеров и числа за-
местителей реального сахара и их пространственным расположением.
Результаты рентгеноструктурных исследований моносахаридов пока-
зали, что пиранозы существуют в виде кресловидной конформации,
причем такой, в которой наибольшее число объемных заместителей
расположено экваториально. Переход от формулы Хеуорзса к конфор-
мации кресла для пиранозы начинают с атома кислорода окис-
ного кольца, изображая его в верхней правой точке кресловидной
формы:
превращается в
Для а-аномеров гидроксил у Сх располагается в аксиальном (а}
положении, для Р-аномеров — в экваториальном (е):
Местоположение остальных заместителей определяется их распо-
ложением в цис- или трансположении к гидроксилу у Сх:
превращается в
Поскольку молекулы циклопентанов почти плоские, формулы Хеу-
орзса достаточно хорошо изображают конформационные формы фура-
ноз.
Способы получения. Гидролиз ди- и полисахаридов. Один из ос-
новных способов получения моносахаридов, имеющий большое прак-
352
тическое значение. Гидролиз происходит под действием кислот или
ферментов:
С12Н22Ои + Н2О -> 2СеН12О6
дисахарид моносахарид
(С6Н10О5)л -р Н2О —► п СбН12Ое
полисахарид
Альдольная конденсация. Первое синтетическое сахаристое вещест-
во было получено Бутлеровым альдольной конденсацией формальде-
гида в присутствии гидроксида кальция:
н—с-н
Са (ОН) 2 О
Н2С + Н—С—Н--------> Н2С—СН--------> Н2С—СН—СН
II II I II II II
0 0 он о он он о
Н2С—СН—СН + НСН—СН—СН Н2С—(СН2)4—сн
I I II I I II II II
он он о он он о он он о
гексоза
Полученный густой сироп содержал довольно сложную смесь са-
харистых веществ. Он был назван Бутлеровым «метиленитаном».
Впоследствии Фишером из такого сиропа, содержащего смесь гексоз,
была выделена оптически недеятельная гексоза, представляющая
собой, как оказалось, рацемическую смесь природной £)-фруктозы и
ее не встречающегося в природе антипода — L-фруктозы.
Оксинитрильный синтез. Пользуясь этим методом, разработанным
еще в 80-х годах прошлого века, можно удлинять углеродную цепь и
переходить от тетроз к пентозам, от пентоз к гексозам и т. д.
Рассмотрим схему получения гептоз из глюкозы:
cZ CN CN с//°
н— хн * —он н— * —ОН ИО- * —н н— \nh2 —он
НО— н— * HCN —Н > * —он н— но— + X о х * 1 * 1 Н- НО— * гидролиз —он > * —н 1 1 1 о —ОН + —н
н— * —он н— * —он Н— * —он н— —он
( ЗНоОН н— * —он СН2ОН Н— * —ОН 2Н2ОН^ н— —он СН2ОН.
D-глюкоза
(гексоза) диастереомерные оксинитрилы
(циангидрины)
353
Ю
;O
HO—
'\nh2
—н
H—
—OH
ва (OH)2
H—
HO—
H—
—OH
H—
—OH
CH2OH
но—
Н—
^OBa,
—OH
H—
—OH
HO—
H—
—OH
H—
-OH
но-
^ОВ31д
—Н
H—
—OH
HO—
H—
—OH
H—
—OH
CH2OH
СН2ОН
разделение диастереомеров
\эва]
с/°
НО—
н—
—он
н—
—он
—он
H2SO<
Н—
—он
[Н+]
н—
0Н ° [Н]
н—
—он
но—
но—
но—
но—
н—
—он
н—
—он
н—
н—
—он
н—
—он
н—
—он
н—
—он
н—
—он
СН2ОН
сн2он
СН2ОН
СН2ОН
гептоза
—H
—H
—H
о
—н
—н
—н
—н
—н
—н
На начальной стадии действием синильной кислоты на глюкозу
получают смесь диастереомерных оксинитрилов (циангидринов). Обыч-
но не проводят их прямого омыления до соответствующих кислот, а
получают сначала амиды. Смесь диастереомерных амидов действием
гидроксида бария (или кальция) переводят в соответствующие соли.
На этой стадии проводят дробную кристаллизацию и таким обра-
зом разделяют смесь диастереомеров на отдельные компоненты. Каж-
дую из солей действием серной кислоты далее переводят в кислоту,
из которой при нагревании получают лактон, восстановлением
которого амальгамой натрия в слабокислой среде получают
гептозу.
Метод укорачивания цепи. Для получения моносахаридов можно
применять так называемые методы укорачивания углеродной цепи.
Эти методы используются для получения некоторых малодоступных
сахаров. Они основаны на окислении моносахаридов, содержащих
большее количество углеродных атомов, чем в нужном моносаха-
риде.
Наиболее старый метод — распад по Руффу, в силу своей простоты
применяется и до сих пор. Он основан на окислении альдозы перок-
сидом водорода в присутствии солей (чаще всего ацетата) трехвалент-
ного железа. В результате окисления углеродный атом карбонильной
354
- К
группы моносахарида отщепляется в виде углекислоты, а атом, сле-
дующий за ним в цепи, окисляется до альдегидной группы:
Н---ОН
но---н
но---н
н---он
Н2О 2
Fe(OCOCH8)3
н---он
н---он
н---ОН
СН2ОН
D-глюкоза
СН2ОН
D-арабиноза
Механизм этого превращения до конца не выяснен.
Физические свойства. Моносахариды представляют собой твердые крис-.
таллические вещества. Все они гигроскопичны, хорошо растворимы в воде, лег-
ко образуют сиропы, из которых их выделить в кристаллическом виде часто бы-
вает трудно. В спирте моносахариды растворяются плохо, в эфире совсем не.
растворяются. Растворы моносахаридов имеют нейтральную реакцию на лакмус
и обычно обладают сладким вкусом. Сладость разных моносахаридов весьма раз,-.
лична. Например, фруктоза приблизительно в три раза слаще глюкозы. Раство-
ры моносахаридов обладают оптической активностью, для них характерно явле-.
ние мутаротации.
Химические свойства. Каждый представитель группы моносаха-.
ридов в растворах существует одновременно в виде нескольких тауто-
мерных форм, поэтому они могут проявлять свойства спиртов, карбо-
нильных соединений и циклических полуацеталей. Рассматривая раз-
личные реакции сахаров, мы будем приводить формулу лишь той тау-.
томерной формы, которая непосредственно участвует в данной реак-
ции.
1. Окисление. Моносахариды легко окисляются, причем в зависи-
мости от условий получаются разнообразные продукты. Как альде-
гидная, так и концевая первичная спиртовая группа альдоз может
быть окислена до карбоксильных групп. При использовании слабых^
окислителей типа бромной воды, гипохлоритов, разбавленной азотной
кислоты окисляется только альдегидная группа и образуются альдо-
новые кислоты. Более сильные окислители, такие, как концентриро-
ванная азотная кислота, окисляют как альдегидную, так и первичную
спиртовую группу, приводя к образованию двухосновных окси-
кислот, так называемых сахарных кислот. Если предварительно за-
щитить альдегидную группу (например, превращением альдозы в гли-
козид), то становится возможным избирательное окисление первичной
спиртовой группы и образуются уроновые кислоты. Ниже
приведена схема возможных направлений окисления альдоз:
355
сильные
окислители
----с/ — (СНОН)„—СН2ОН-------
ХН а льдоза
слабые
окислители
С< —(СНОН)Л—сн,он
х>н
альдоновые кислоты
>С—(СНОН)Л—
Hz хон
сахарные кислоты
уроновые кислоты
Кетозы не окисляются слабыми окислителями. При действии силь-
ных окислителей происходит расщепление молекул.
Восстановление. Осторожное восстановление моносахаридов веде!
к образованию соответствующих многоатомных спиртов — глицитое
.(тетритов, пентитов, гекситов и т. д.):
zO
Cf сн,он сн,он
|ХН I I
(СНОН)Я Л ^НОН)„ [Н] с=0
СН2ОН СН2ОН (СНОН)т
СН2ОН
альдоза многоатомные кетоза
спирты
(рлициты)
Восстановление удобно проводить боргидридом натрия (NaBHi),
•поскольку этот реагент, как и исходный моносахарид, растворим
в воде, и процесс можно вести в водном растворе.
Действие щелочей. Приводит к изомеризации моноз в смесь саха-
ров, отличающихся конфигурацией первого и второго атомов углерод
да. Так, из глюкозы, маннозы или фруктозы получается равновесная
смесь этих трех сахаров. Процесс проходит через стадию образования
лромежуточного продукта — ендиола:
он
н---он
с—он
но---н
н----он
н----он
СН2ОН
D-глюкоза
но----н
н-----он
н-----он
СН2ОН
ендиол
СН2ОН
D-манноза
СН2ОН
2С=О
но—н
н--он
н—он
СН2ОН
D-фрукто за
Подобные превращения могут претерпевать все моносахариды.
Происходящее при этом изменение конфигурации у второго углерод-
ного атома альдоз называется эпимеризацией, а альдозы, отличаю-
щиеся конфигурацией только у второго углеродного атома, — эпи-
мерами.
Реакция «серебряного зеркала». Подобно обычным альдегидам, аль-
дозы легко дают реакцию «серебряного зеркала» с аммиачным раство-
ром оксида серебра (реактив Толленса). Этот реактив готовят, смеши-
вая растворы едкого натра и нитрата серебра, в результате чего выпа-
дает в осадок оксид серебра:
2А2+4-2ОН- -> Ag2O | + Н2О
При осторожном добавлении водного раствора аммиака осадок раство-
ряется и образуется ион Ag(NH3)2. Добавлением альдозы к реактиву
Толленса осаждают металлическое серебро («серебряное зеркало»):
Ag(NH3)2 + альдоза альдоновая кислота + Ag |
Кетозы тоже дают положительную, реакцию с этим реактивом. Объяс-
няется это частичным превращением кетоз в соответствующие альдозы
под действием щелочной среды (аммиак).
Реакция с фелинговой жидкостью. Реактив Фелинга (фелингова
жидкость) готовят, смешивая равные объемы водного раствора суль-
357
фата меди и щелочного раствора натрий-калиевой соли винной кис-
лоты. При нагревании раствора в присутствии альдозы выпадает крас-
ный осадок СшО. Реакцию используют для количественного определе-
ния сахаров:
сГ° с//°
рок Н. |\ок
Н—С—Оч уО—с
I >С< I
Н—С—О' ХО—с
(СНОН)„
I
СН2ОН
+ н20 ->
I
ONa
с=о
ONa
альдоза
,0
с<
| хон
(СНОН)Л + Cu2O I +4
СН2ОН
альдоновая
кислота
COOK
I
снон
I
снон
COONa
Кетозы, изомеризуясь под действием щелочной среды реактива Фе-
линга в соответствующие альдозы, дают положительную реакцию с
фелинговой жидкостью.
Реакция с синильной кислотой. Была подробно рассмотрена ранее
(оксинитрильный синтез, см. с. 353).
Взаимодействие с фенилгидразином (см. с. 202). В результате реак-
ции моносахаридов с избытком фенилгидразина образуются соедине-
ния, содержащие два остатка фенилгидразина на молекулу сахара,.,
так называемые озазоны. Сначала моносахарид реагирует с фенилгид-
разином, давая фен ил гидразон; вслед за этим группа —СН—ОН,.
соседняя с первоначальной альдегидной функцией, окисляется до кар-
бонильной группы (механизм этого процесса недостаточно ясен); на-
конец, третья молекула фенилгидразина реагирует с вновь образовав-:
шейся карбонильной группой:
•с^° 1CH=N—NH—С6Нб
1 I 1 CeH5NHNH2 2СНОН > I -Н2О R альдоза . 2СНОН CeHbNH—NH2
R фенилгидразон —NH3; —CeH5NHa
1CH=N—NH—С6Н5
2С=О
R
CH=N—NH—CeH5
C=N—NH—С6Н5
R
c.h6nhnh2
—н2о
фенилозазен
358
Озазоны представляют собой кристаллические вещества, окрашен-
ные в желтый цвет. Их можно использовать для выделения и иденти-
фикации сахаров. Поскольку реакция образования затрагивает толь-
ко углероды Сх и С2 моносахаридов, то, следовательно, эпимерные
альдозы и изомерные им кетозы дают один и тот же озазон, например:
CH=N—NH—С6Н5 C=N—NH—СЙН5 Д О
"\н
н— —ОН НО— —н но— —Н
но— —Н ЗСвНЛНХНг Н— -он 3CeH»NHNHf НО— —н
н— —ОН Н— —ОН Н— —он
н— —ОН Н— —он
с :н2он ( :н2он ( :н2он
D-глюкоза фенилозазон D-манноза
| 3C6H5NHNH2
СН2ОН
С=О
НО—н
н---он
н---он
сн2он
D-фруктоза
При действии соляной кислоты озазоны гидролизуются в озоны.
Однако чаще используется расщепление озонов нагреванием с бенз-
альдегидом, поскольку реакция протекает с лучшим выходом и одно-
временно связывается выделяющийся фенилгидразин:
CH=N—NH—СбН5 СНО
I 2С«НбСНО |
C=N—NH—СбН5 ---------------------> С=О
I —2СвН5СН—N—NH—CeH& |
R R
озазон озон
При восстановлении озонов амальгамой натрия в слабокислой сре-
де происходит избирательное восстановление альдегидной группы до
первичной спиртовой группы. Таким методом можно осуществлять
переход от альдоз через озазоны и озоны к кетозам.
Алкилирование. При алкилировании сахара реагируют в цикличес-
кой (пол у ацетильной) таутомерной форме. Гликозидный гидроксил
легко взаимодействует даже с метиловым спиртом в присутствии хло-
рида водорода. Спиртовые гидроксилы в этих условиях не затрагива-
ются; для их алкилирования необходимы более сильные алкилирую-
щие агенты, например алкилгалогениды (в присутствии оксида
серебра):
359
CHjOH
При действии на полностью алкилированные моносахариды раз-
бавленных минеральных кислот происходит гидролиз только у перво-
го углеродного атома (полуацеталь), остальные метоксильные груп-
пы не гидролизуются (простые эфиры):
лентаметил-ос-Г>-глюко-
пиранозид
СН3ОН +
2,3,4,6-тетраметил- а-Р-глю^
копираноза
Ацилирование. При действии ацилирующих агентов (например,
ангидридов или галогенангидридов кислот) происходит образование
сложных эфиров по всем гидроксильным группам:
пентаацетил-а- £»-глюкопираноза
Определение размера окисного кольца моносахаридов методом ис-
черпывающего метилирования (метод Хеуорзса). Этот метод основан
на различии химических свойств полуацетального и спиртового гид-
роксилов. Последовательность операций лучше всего рассмотреть на
конкретном примере — доказательстве размера окисного кольца
£>-глюкозы:
260
Р-глюкопираноза .£>-глюкофурано за
Н2О(Н+) 1н2О(Н+)
триметоксиглутаровая кислота
диметоксиянтарная кислота
Вначале глюкоза подвергается исчерпывающему метилированию,
например, действием йодистого метила в присутствии оксида серебра.
Полученное производное содержит пять метоксильных групп. При про-
ведении гидролиза омыление происходит только у первого атома уг-
лерода (полуацетальный гидроксил), четыре другие метоксильные
группировки сохраняются (образованы спиртовыми гидроксилами).
Полученная после гидролиза тетраметилглюкоза, содержащая сво-
бодный полуацетальный гидроксил, может, следовательно, существо-
вать и в открытой, ациклической форме. При окислении такого
соединения, например, концентрированной азотной кислотой должны
окисляться до карбоксильной группы только карбонильные группы и
углеродные атомы, содержащие свободный гидроксил: Сэ — у пира-
нозной формы (левая часть схемы) и С4 — у фуранозной формы (пра-
вая часть схемы). Если глюкоза имеет пиранозное кольцо, то на по-
361
следней стадии этого метода, после окисления, должна образоваться^
триметоксиглутаровая кислота; в случае фуранозного кольца продук-
том окисления должна быть диметоксиянтарная кислота. В действи-
тельности при проведении всех этих реакций в конечном счете была
получена только триметоксиглутаровая кислота, что совершенно од-
нозначно доказывало наличие пиранозного кольца в глюкозе.
С помощью метода Хеуорзса были определены размеры окисных
колец всех пентоз и гексоз. Он применим и для решения вопроса о
строении других углеводов, особенно полисахаридов.
Брожение гексоз. Брожением называется сложный процесс расщеп-
ления моносахаридов под влиянием различных микроорганизмов, в
большинстве случаев сопровождающийся выделением газообразных
продуктов (СО2, Н2 и др.) и приводящий в конечном итоге к образова-
нию таких веществ, как спирт, молочная кислота и т. д.
Различают разные виды брожения: а) спиртовое брожение:
зимаза
С6Н12Ов---> 2С2Н5ОН + 2СО2
б) маслянокислое брожениег
Свн12ов -> СН3—СН2—СН2—СООН 4- 2СО3 + 2На
масляная кислота
в) молочнокислое брожение:
С,н12о„ -> 2СН3СН(ОН)СООН
молочная кислота
В этом случае получают смесь стереоизомеров со слабым левым или
правым вращением.
Лимоннокислое брожение:
о он о
[О] II | II
с6н1206 —> С—СН2—С—СН2—С + 2Н2О
он соон он
лимонная кислота
Существуют и другие виды брожения. Не все моносахариды сбра-
живаются одинаково — галактоза, например, сбраживается труднее
глюкозы.
Процессы брожения играют важную роль не только в повседнев-
ной жизни, но и широко используются в промышленности.
В организмах высших животных непрерывно протекают процессы
биохимического расщепления и синтеза моносахаридов. При мышеч-
ном сокращении в результате расщепления углеводов образуется мо-
лочная кислота. Этот процесс называется гликолизом.
Дегидратация с циклизацией. Пентозы при нагревании с разбав-
ленной соляной или серной кислотой образуют фурфурол (см<
с. 312):
S62
iHOi—сн—сн—!бн
i i I I I
i Hi-CH Cr—iH
.. i iV-
j OH НЮ c=o
t°, (H+)
— зн2о
CH—CH
II II
CH c—c
о
фурфурол
пентоза
Гексозы в этих условиях циклизуются с образованием а-окси-
метилфурфурола, который в отличие от фурфурола нестоек и лег-
ко гидролизуется с образованием левулиновой и муравьиной
кислот:
Н
ОН !
Н:
НОН2С О:Н~‘
Г; (Н+)
— ЗН2О
НО—СН
а-оксиметилфурфурол
гексоза
СН3—С—СН,—СН2—СООН + НСООН
II
О
левулиновая кислота муравьиная
кислота
Фурфурол можно обнаружить при помощи цветной реакции с фло-
роглюцином (1,3,5-триоксибензолом) и таким образом отличить
глюкозы от гексоз.
Отдельные представители. Гексозы, D-Глюкоза
и—-он
но-— н
Д-глюкоза
6сн2он
363
/)(+)-Глюкоза (виноградный сахар, декстроза) очень широко рас-
пространена в природе, содержится в соке винограда и других сладких
плодов, в небольших количествах — в организмах животных и че-
ловека. Она входит в состав важнейших полисахаридов, таких, как
тростниковый сахар, молочный сахар. Важнейшие высокомолекуляр-
ные полисахариды — крахмал, гликоген (животный крахмал) и
клетчатка — целиком состоят из остатков глюкозы, соединенных
друг с другом различным способом.
Главным способом технического получения глюкозы является гид-
ролиз крахмала и клетчатки. Гидролиз крахмала с помощью разбав-
ленных минеральных кислот ведется в автоклавах при повышенном
давлении и 150сС. В зависимости от продолжительности процесса
гидролиза образуется либо патока (содержит 32—40% глюкозы),
либо техническая глюкоза (65—99% глюкозы).
Глюкозу для медицинских целей получают путем очистки — пере-
кристаллизации — технической глюкозы из водных или водноспир-
товых растворов.
Применяется глюкоза в пищевой промышленности как заменитель
тростникового сахара. Глюкоза используется в текстильном и некото-
рых других производствах в качестве восстановителя. В медицине
чистая глюкоза применяется в виде растворов для введения в кровь
при ряде заболеваний, в производстве таблеток и т. д. Из глюкозы
получают витамин С.
При восстановлении глюкозы образуется шестиатомный спирт
сорбит, при окислении бромной водой — глюконовая кислота, при
окислении концентрированной азотной кислотой — глюкаровая* (са-
харная) кислота:
СНоОН с/° с/°
н— -он н— Х)Н -он н— х>н —он
но— —н но— —н но— —н
н— —он н— —он н— —он
н— —он н— —он н— —он /ОН
СН2ОН СН2ОН с ^0
сорбит глюконовая кислота глюкаровая кислота
£>-Галактоза вместе с глюкозой входит в состав некоторых глико-
зидов и полисахаридов; остатки галактозы — в состав сложнейших
биополимеров-ганглиозидов, или гликосфинголипидов. Они обнару-
жены в нервных узлах (ганглиях) человека и животных и содержатся
также в ткани мозга, в селезенке и эритроцитах.
* По номенклатуре ИЮПАК название отдельных сахарных кислот етреится
из названия соответствующего моносахарида путем замены окончания «оза»
окончанием «аровая кислотаэ, например: глюкаровая, маннаровая и т. з
364
D -Галактоза
z
\н
н-он
но—н
но-----н
н----он
СН2ОН
/)(+)-галактоза
а - D -галактопираноза
Н ОН
fi-D -галактопираноза
Получают галактозу в основном гидролизом молочного сахара^
При восстановлении галактозы образуется недеятельный спирт дуль-
цит. При окислении галактоза дает сначала галактоновую, а затем—
двухосновную слизевую (галактаровую) кислоту:
СН2ОН
н---он
НО-----Н ------
но----н
н----он
СН2ОН
D-галактоза
[И].
[OJ.
но----н
но----н
н----он
СН2ОН
дульцит
НО-------Н [°1
н---он
но---н
НО---н
н----он
СН2ОН
н---он
галактоновая
кислота
слизевая (галактаро-
вая) кислота
365
D-манноза
н—он
н—он
сн2он
Р(+)-манноза
СН2ОН
Н/Г-—°\ОН
|/Н \|
|\ОН НО/
НО Ч--—КН
н .н
P~D -маннопираноза
.Манноза сравнительно мало распространена в природе. Она со-
держится в ячмене, корке апельсина. Получают ее гидролизом поли-
сахарида маннана, содержащегося в грибах и некоторых дрожжах.
Как видно из формул, манноза отличается от глюкозы конфигурацией
заместителей только у второго углеродного атома, т. е. она является
эпимером глюкозы. Поскольку эпимеры дают одинаковые озазоны,
то озазон маннозы идентичен озазону глюкозы (см. с. 359).
При восстановлении маннозы образуется спирт маннит (содержит-
ся в грибах, маслинах, жасмине), при окислении — манноновая кисло-
та:
/О
c<f
Х)Н
НО- —н
НО— —н ?О]
н— —он
н— —ОН
( :н2он
D-манноновая
кислота
zO
// сн9он
но— —н но— —Н
но— -Н но— —Н
н— —он н— —ОН
н— —он н— —ОН
с ;н2он ( >Н2ОН
D-манноза D-маннит
D-Фруктоза
н—он
сн2он
О(-)-фруктоза а-£)(-)-фруктопираноза , /?-£>(-)-фруктофураноза
D(—)-Фруктоза (фруктовый сахар,
левулеза) в свободном состоя
366
нии содержится во фруктах, меде. Входит в состав многих сложных
сахаров, например тростникового сахара, из которого может быть по-
лучена гидролизом. Образует высокомолекулярный полисахарид,
инулин, содержащийся в некоторых растениях. Фруктозу получают
также из инулина. При восстановлении фруктозы образуются сорбит
и маннит: СН2ОН СН2ОН СН2ОН
2=0 н— * —он но— ♦ —н
НО— * —н * [Н] но— * —н * —он + но— Е X О * 1 * 1
Н— —ОН н— н—
н— * -ОН н— * —он н— ♦ —он
с О-фр :н2он уктоза с D-c :н2он орбит с D-j :н2он ианнит
При окислении фруктоза дает винную и щавелевую кислоту:
СН2ОН 1 с=о zO
но— —н ( н ХОН ОН
Ю] + V/1 1
Н— —он и он
н— ( Реакция с фенилгид —он :н2он разином \он щавелевая кислота приводит К ( винная образован У° ^Н кислота ию озазона:
СН2ОН 1 с=о сн2он 1 C=N—NH—С6Н5
но— —н HO- —н
CeH6NHNH2 c«h5nhnh2
т т гчтт . ij он
rd— —ин * — н2о л— - NH8; — CeHbNH,
н— —ОН Н— —ОН
с :н2он С :н2он
с/° |\н фенилгидразон CH=N—NH—С6Н5 C=N—NH—СеН5
C=N—NH—СвНб но Н C«HbNHNH, но Н Н он
н он H ОН СН2ОН — н2о н он СН2ОН озазон
367
Пентозы. Пентозы встречаются в природных условиях главным
образом как составные части молекул более сложно построенных
веществ, например сложных полисахаридов, называющихся пенто-
занами, а также растительных камедей. Пентозы в значительном ко-
личестве (10—15%) содержатся в древесине, соломе и т. д.
Арабиноза
D( )-арабиноза L (+)-арабиноза /?-£-арабофураноза
В природе преимущественно встречается Ц+)-арабиноза. Она со-
держится в вишневом клее, свекле и аравийской камеди (гуммиара-
бик), откуда ее и получают. Из свеклы арабиноза получается вместе
с глюкозой, которую удаляют брожением.
При восстановлении арабинозы получают многоатомный спирт
арабит (встречается в лишайнике), при окислении — арабоновую
кислоту.
D-Рибоза
£)-рибоза a-D-рибопираноза D-рибофураноза
£>-Рибоза играет значительную биологическую роль. Она входит в
состав рибонуклеиновых кислот и некоторых других биологически
важных соединений. Получают рибозу эпимеризацией арабинозы. При
восстановлении рибозы образуется недеятельный спирт рибит, при
окислении — рибоновая кислота.
D-Ксилоза
Ж+)-ксилоза а-2)-ксилопираноза
В природе распространена £>(+)-ксилоза. Она образуется при
гидролизе полисахарида ксилозана, находящегося в соломе, отрубях,
древесине, шелухе подсолнечника. Гидролизаты, содержащие кси-
лозу, используются для выращивания некоторых видов дрожжей,
которые в качестве белкового источника применяются для кормления
сельскохозяйственных животных.
368
При восстановлении ксилозы получают спирт ксилит, при окисле-
нии — ксилоновую кислоту:
СН2ОН
н---он
НО---Н
н---он
но---н
н---он
СН2ОН
Н-----он
СН2ОН
ксилит
[О]
н---он
НО---н
н------он
СН2ОН
ксилоновая кислота
Некоторые природные соединения — произ-
водные моносахаридов. Фосфорные эфиры. Из фосфатов
сахаров, играющих важную роль в фотосинтезе, большое значение
имеют 1,6-дифосфат фруктозы и 1-фосфат глюкозы:
1-фосфат-Д-глюкопиранозы
Гликозиды. Сахара в природе часто встречаются в виде гликози-
дов (циклических ацеталей). При кислотном или ферментативном гид-
ролизе они расщепляются с образованием сахара и неуглеводного ос-
татка — агликона.
Агликоны часто представляют собой оксисоединения жирного и
ароматического рядов. Они нередко являются органическими ядами.
Многие гликозиды применяются в медицине. Интересно отметить, что
подавляющее большинство известных природных гликозидов — это
Р-гликозиды.
Гликозид амигдалин содержится в семенах горького миндаля,
косточках персика, абрикосов, слив, вишен, яблок, груш, листьях
лавровишни и т. д.
13—682 369
Под действием кислот амигдалин распадается на две молекулы глюко-
зы, молекулу бензойного альдегида' и синильной кислоты.
Гликозид ванилина содержится (около 2%) в стручках ванили:
.ванилин-^-Р-глюкопиранозид
Гликозид синигрин — гликозид семян горчицы и корней хрена.
Калиевая соль синигрина под влиянием находящегося в семенах гор-
чицы фермента распадается на глюкозу, аллилгорчичное масло и кис-
лый сернистокислый калий:
о—S\ yJoso3K
с\
хсн— сн=сн2
гидролиз
глюкоза + S=C=N—СН2—СН=СН2 + KHSO3
аллилгорчичное масло
Витамин С. Большое значение для человека имеет витамин С,
или аскорбиновая кислота, являющаяся противоцинготным факто-
ром. Витамин С широко распространен в природе; он содержится в
лимонах, апельсинах, черной смородине, свежей капусте, плодах ши-
повника, луке, картофеле, красном перце и т. д. Суточная потреб-
ность взрослого человека в витамине С больше, чем в других витами-
нах, и составляет около 100 мг. Недостаточность в нем вызывает цингу
(скорбут, кровотечение десен, выпадение зубов), а также повышен-:
ную восприимчивость к инфекционным заболеваниям. В настоящее
время в СССР производят витамин С в больших количествах в про-
мышленном масштабе. Сырьем для производства служит глюкоза^
которую сначала восстанавливают в D-сорбит. Ферментативно окис-
ляя /5-сорбит, получают L-сорбозу, которую после ацетонировани#
с целью защиты соответствующих гидроксильных групп окисляют
перманганатом калия в 2-кето-А-гулоновую кислоту. В присутствии
разбавленных кислот происходит лактонизация и енолизация с об*-
разованием L-аскорбиновой кислоты-
370
СН2ОН
н----он
ферментативное
НО-----Н окисление
СН2ОН
н—он н—он
сн2он сн2он
D-глю коза D-сорбит
НО----н
н---он
с=о
<[н2он
или
сн2он
но—н
СН2ОН
Л-сорбоза
СН2ОН
СН2ОН
НО-----Н 2СН3СОСН3
| Н-----ОН ~2Н2°
I-------н
сн2он
2-кето-Л-
£-аскорбиновая кислота
ПОЛИСАХАРИДЫ
Олигосахариды (сахароподобные полисахариды).
Дисахариды
В зависимости от числа молекул простых сахаров, образующихся
при гидролизе молекулы олигосахарида, различают* дисахариды,
гидролизующиеся на две молекулы монозы; трисахариды, гидроли-
зующиеся на три молекулы монозы, и т. д. Наибольшее значение име-
ют дисахариды.
Строение. Все дисахариды можно рассматривать как гликозиды,
образованные из циклической формы моносахарида путем замещения
атома водорода в полуацетальном гидроксиле остатком другой моле-
13
371
кулы моносахарида, что хорошо видно из сравнения общей форму*
лы гликозида и дисахарида:
гликозид
дисахарид
Остатки двух молекул моносахарида в дисахариде могут быть оди-
наковыми или разными.
Если одна молекула моносахарида всегда участвует в построении
молекулы дисахарида своим полуацетальным гидроксилом, то другая
может соединяться либо полуацетальным, либо одним из спиртовых
гидроксилов. В последнем случае в молекуле дисахарида будет оста-
ваться свободным один полуацетальный гидроксил. Отсутствие или
наличие в молекуле дисахарида полуацетального гидроксила отра-
жается на свойствах дисахаридов.
В случае дисахарида, построенного за счет объединения моноса-
харидов полуацетальными гидроксилами, строение характеризуется
закрепленными циклическими формами. Альдегидная группа без
гидролиза такого дисахарида образоваться не может, поэтому он не
обладает восстанавливающими свойствами и называется невосстанав-
ливающим дисахаридом:
невосстанавливающий дисахарид
В случае же дисахарида, построенного с участием одного полуаце-
тального и одного спиртового гидроксила, в молекуле сохраняется’
один полуацетальный гидроксил. В таком дисахариде циклическая,
форма одного остатка моносахарида не является закрепленной, она
может перейти в таутомерную альдегидную форму, дисахарид облада-
ет восстанавливающими свойствами и называется восстанавливающим;
дисахаридом:
372
восстанавливающий дисахарид
Получение. Практически дисахариды получают из природных ис-
точников, хотя существуют и синтетические методы получения. При
извлечении олигосахаридов из природных источников обычно приме-
няют водную экстракцию. Основной задачей при этом является выде-
ление индивидуальных соединений из сложной смеси моно- и олигоса-
харидов. Для этой цели разработан ряд методов, основанных на хро-
матографии.
Физические свойства. Дисахариды — типичные сахароподобные полиса-
хариды. Это твердые вещества или некристаллизующиеся сиропы, хорошо раст-
воримые в воде. Как аморфные, так и кристаллические дисахариды обычно пла-
вятся в некотором интервале температур и, как правило, с разложением.
Химические свойства. Химическое поведение дисахаридов скла-
дывается из свойств гидроксильных групп, восстанавливающего зве-
на (у восстанавливающих дисахаридов) и свойств гликозидов. Буду-
чи по своему строению гликозидами, дисахариды легко гидролизуют-
ся разбавленными минеральными кислотами и сравнительно стойки к
щелочному гидролизу. Кислотный гидролиз приводит к расщепле-
нию гликозидной связи и образованию моносахаридов. Восстанавли-
вающие дисахариды проявляют реакции, характерные для карбониль-
ной группы в такой же степени, как это свойственно соответствующим
моносахаридам: они мутаротируют в растворах, восстанавливают фе-
лингову жидкость, окисляются до альдоновых кислот, восстанавлива-
ются до многоатомных спиртов, образуют озазоны, присоединяют
синильную кислоту. Дисахариды являются многоатомными спирта-
ми, а поэтому они, подобно моносахаридам, растворяют гидроксид
меди с образованием синего окрашивания.
Отдельные представители. Мальтоза (солодовый сахар). Название
возникло в связи со способами получения мальтозы из крахмала при
действии солода (maltum по-латыни — солод). В результате гидро-
лиза мальтоза расщепляется на две молекулы глюкозы:
373
C12H220h + Н2О - 2СвН12О„
мальтоза глюкоза
Обладает восстанавливающими свойствами, следовательно, имеет один
свободный полуацетальный гидроксил. В молекуле мальтозы кисло-
родный мостик, связывающий два остатка глюкозы, соединяет первый
атом углерода одного остатка с четвертым атомом углерода другого
остатка глюкозы*-
4-(а-Р-глюкопиранозидо)-£)-глюкопираноза
Мальтоза кристаллизуется из водных и водно-спиртовых раство-
ров с одной молекулой воды С^Н^Оц-Н2О. Водные растворы мута-
ротируют. Удельное вращение после завершения мутаротации [a]D =
= 136°.
Солодовый сахар, являясь промежуточным продуктом при гидро-
лизе крахмала, в то же время широко распространен в растительных
и животных организмах. Он значительно менее сладок, чем тростни-
ковый сахар (в 0,6 раза при одинаковых концентрациях).
Мальтоза — типичный восстанавливающий дисахарид: восстанав-
ливает фелингову жидкость, дает гидразон и озазон. При осторожном
окислении мальтозы образуется одноосновная мальтобионовая кисло-
та:
мальтоза
* Обозначением ~ Н, ОН пользуются, когда речь идет либо о смеси аномй^
ров, либо когда аномерная конфигурация сахара не установлена, либо когД$
конкретная аномерная конфигурация не имеет значения в какой-либо конкрет*'
ной реакции.
374
Одним из распространенных методов, применяющихся для изуче-
ния строения олигосахаридов, является метод метилирования. Рас-
смотрим его применительно к мальтозе. Вначале олигосахарид под-
вергают метилированию, например, действием диметилсульфата в
щелочной среде, в результате получают октаметилпроизводное. По-
следующий кислотный гидролиз приводит к расщеплению только
лишь гликозидных связей, причем образуются метилзамещенные
двух молекул моносахаридов, в случае мальтозы — 2,3,4,6-тетраме-
тилглюкоза и 2,3,6-триметилглюкоза:
2.3,6-триметилглюкоза
2,3,4,6-тетраметилглюкоза
При осторожном окислении восстанавливающих дисахаридов ти-
па мальтозы образуются соединения, называемые альдобионовыми
кислотами.
Целлобиоза. Это дисахарид, образующийся при гидролизе целлю-
лозы (клетчатки). Целлобиоза, как и мальтоза, построена из двух
остатков глюкозы, но соединенных не а-1,4-, а Р-1,4-гликозидной
связью:
375
4-( li-D -глюкопиранозидо)-£>-глюкопираноза
Кристаллическая целлобиоза представляет собой вещество с /цл =
= 225°С, растворы ее обладают мутаротацией, причем [a]D меняется
от ±14,2 до ±34,6°. Целлобиоза обладает всеми свойствами восста-
навливающих сахаров.
Лактоза (молочный сахар). Название возникло в связи с получе-
нием из молока (по-латыни lactum). При гидролизе лактозы образуют-
ся глюкоза и галактоза. Лактоза обладает восстанавливающими свой-
ствами, следовательно, в ее молекуле имеется один свободный полу-
ацетальный гидроксил. Этот гидроксил принадлежит остатку глюкозы,
а кислородный мостик соединяет первый атом углерода (fJ-аномер)
остатка галактазы с четвертым атомом углерода остатка глюкозы:
остаток галактозы остаток глюкозы
4-(Д -D -галактопиранозидо)- D - глюкопираноза
В коровьем молоке содержится 4—5,5%, в женском 5,5—8,4%
лактозы. Лактоза в отличие от других сахаров не гигроскопична,
поэтому она не отсыревает и плохо растворяется в воде. Молочный
сахар применяется как фармацевтический препарат и как питание для
грудных детей.
Лактоза кристаллизуется с одной молекулой воды, температура
плавления гидрата 202°С.
Водные растворы лактозы мутаротируют. После завершения мута-
ротации устанавливается удельное вращение [<%1d = 52,2°. Лактоза
в 4 или 5 раз менее сладка, чем сахароза.
Сахароза (тростниковый, или свекловичный, сахар). Названия воз-
никли в связи с ее получением либо из сахарной свеклы, либо из са-
харного тростника. Тростниковый сахар был известен за много сто-
летий до нашей эры, но лишь в середине XVIII в. был обнаружен в
сахарной свекле и только в начале XIX в. был получен в производст-
венных условиях. Сахароза широко распространена в растительном
мире. Листья и семена всегда содержат небольшие количества трост-
никового сахара. Он находится также в плодах (абрикосах, персиках,
груше, ананасе). Его много в кленовом, пальмовом соке, кукурузе.
Эго наиболее известный и широко применяемый сахар. При гидро-^
лизе из него образуются глюкоза и фруктоза. Сахароза — невосста^
376
навливающий сахар, у нее нет свободного полуацетального гидро-
ксила. Сахароза гидролизуется легче, чем многие другие дисахариды.
Это связано с тем, что один из остатков моносахаридов, входящих в
его молекулу — остаток фруктозы, находится в фуранозной форме.
Глюкоза участвует в образовании сахарозы в а-аномерной конфигу-
рации, а фруктоза в Р-:
2 -(а-Л-глюкопиранозидо)- 0 - Р-фруктофуранозид
Сахароза кристаллизуется без воды, = 184,5°С. Удельное
вращение водного раствора сахарозы [alD = 66,5°. Растворы сахаро-
зы не обладают мутаротацией. При гидролизе сахароза превращает-
ся в смесь равных количеств глюкозы и фруктозы. Фруктоза характе-
ризуется более сильным левым вращением, чем глюкоза правым,
1ос]£> = —92° и [ос]/) = 52,5° соответственно, поэтому раствор гидро-
лизованной сахарозы имеет левое вращение. В связи с изменением
в процессе гидролиза правого вращения раствора на левое гидролиз
сахарозы получил название инверсии (инверсия — обращение). Смесь
равных количеств глюкозы и фруктозы, получающаяся в результате
инверсии тростникового сахара, называется инвертным сахаром.
Природным инвертным сахаром является мед, состоящий в основном
из глюкозы и фруктозы.
Сахарозу производят в промышленных огромных масштабах.
В нашей стране ее получают из сахарной свеклы, которая содержит
16—20% сахарозы (сахарный тростник содержит 14—26% сахарозы).
Промытую свеклу измельчают и в аппаратах (диффузорах) многократ-
но извлекают сахарозу водой, нагретой до 80°С. Полученную жид-
кость, содержащую сахарозу с большим количеством примесей, обра-
батывают известью, которая осаждает ряд органических кислот, бел-
ковые и некоторые другие вещества. Часть извести расходуется на
образование сахарозой растворимых в холодной воде кальциевых са-
харатов.
Сахараты, оставшиеся в растворе после фильтрации от всех при-
месей, разрушают диоксидом углерода, получая свободную сахарозу
и карбонат кальция. Осадок углекислого кальция отделяют фильтра-
цией, фильтрат после дополнительной очистки упаривают в вакууме
до получения кашицеобразной массы. Выделившиеся кристаллы са-
харозы отделяют центрифугированием. Таким образом, получают
сырой сахарный песок, имеющий желтоватый цвет и маточный раствор
бурого цвета, дающий еще некоторое количество сахарозы и некрис-
таллизующийся сироп — свекловичную патоку, или мелассу. Сахар-
ный песок очищают (рафинируют) и получают в готовом виде.
Грегалоза (микоза, грибной сахар). Впервые была найдена в спо-
377
рынье, затем во многих грибах и некоторых растениях. При гидроли-
зе трегалоза распадается на две молекулы глюкозы.
Трегалоза относится к невосстанавливающим дисахаридам, обра-
зованным за счет полуацетальных гидроксилов двух молекул a-D-
глюкопиранозы:
l-(a D-r iwкопирацрзидо)-0-D-глюкопиранозид
Высокомолекулярные (несахароподобные)
полисахариды
Высшие полисахариды содержат сотни и тысячи остатков моноса-
харидов, в связи с чем строение их долго оставалось неизученным.
Только в последние десятилетия химикам удалось достичь больших
успехов в выяснении строения этих соединений.
1. Все полисахариды представляют собой длинные цепи, в кото-
рых остатки моносахаридов связаны между собой кислородными мос-
тиками.
2. Эти кислородные мостики образованы за счет полуацетального
гидроксила предыдущего остатка моносахарида со спиртовым гидро-
ксилом последующего. Например, образование высокомолекулярно-
го полисахарида из молекулы гексозы можно представить следующим
образом:
Полиозная цепь, следовательно, представляет по своему строению
полигликозид.
3. Полигликозидные цепи, образующие молекулу полисахаридов,
могут быть неразветвленными и разветвленными.
4. В основе полимерной цепи полиозы может лежать только один
моносахарид (гомополисахариды) или различные (гетерополиса-
хариды).
Важнейшими гомополисахаридами, состоящими из остатков глю-
козы, являются крахмал, гликоген и клетчатка (или целлюлоза).
378
Крахмал
Крахмал образуется при фотосинтезе в зеленых листьях в виде
зерен, которые легко обнаружить в микроскоп, используя реакцию
с иодом: крахмальные зерна окрашиваются в синий или сине-черный
цвет. В листьях же крахмал расщепляется на олиго- или моносахари-
ды, которые, перемещаясь в подземные клубни или зерна злаков,
снова превращаются в крахмальные зерна. Наибольшее содержание
крахмала в следующих культурах (%): рис (зерно) — 62—82; куку-
руза (зерно)— 65—75; пшеница (зерно) 57—75; картофель (клубни) —
12—24.
Свойства. Внешний вид крахмала хорошо известен: это белый
аморфный порошок, похожий на муку (отсюда и название «картофель-
ная мука»). Крахмал всегда содержит 10—20% воды, удалить кото-
рую можно медленным нагреванием (во избежание расщепления его
гигантских молекул на более короткие полисахариды) до 100—110°С.
Высушенный крахмал очень гигроскопичен.
Из крахмала можно приготовить коллоидный водный раствор —
крахмальный клейстер, для чего суспензию крахмала в небольшом ко-
личестве воды необходимо постепенно, при хорошем перемешивании
выливать в кипящую воду. Настаивание крахмала с горячей водой
вызывает набухание крахмальных зерен, причем небольшая часть по-
лисахаридов переходит в раствор. Растворы крахмала вращают плос-
кость поляризации вправо: [а]р = +195°.
Хорошо известна очень чувствительная цветная реакция крахма-
ла с иодом. Характерно, что возникающее при этом синее окрашивание
исчезает при кипячении и вновь появляется при охлаждении. На иод-
крахмальной реакции основано широкое применение крахмала в ана-
литической химии — в качестве индикатора в иодометрии.
Крахмал в обычных условиях и при действии обычных реагентов
не обнаруживает восстанавливающей способности: он не восстанав-
ливает ни фелингову жидкость, ни гидроксид меди, ни аммиачный
раствор оксида серебра. Однако более чувствительные окисляющие
реактивы (динитросалициловая кислота) позволяют обнаружить в
крахмале ничтожное количество свободных альдегидных групп.
Использование таких реактивов лежит в основе химического метода
определения степени полимеризации и молекулярной массы полисаха-
ридов крахмала.
Используя методы исчерпывающего метилирования и ацетилиро-
вания можно обнаружить, что среднее звено молекулы крахмала со-
держит три свободных спиртовых гидроксила (получены «триметил-
крахмал» и «триацетилкрахмал»).
Быстрое нагревание крахмала приводит к расщеплению полиса-
харидной цепи на более короткие фрагменты — декстрины. Состав
крахмала и декстринов один и тот же, но степень полимеризации крах-
мала больше, чем декстрина. Декстринизацию можно проводить,
используя ферментативный или кислотный гидролиз крахмала, кото-
рые в обоих случаях можно выразить следующим образом:
372
(СбН10О5)л —► (C6H10O5)x —*• C12H22O11 CgH12O6
крахмал ряд декстринов мальтоза глюкоза
По мере убывания величины молекулы (уменьшение степени поли-
меризации) декстрины дают с иодом окрашивание от сине-фиолетового,
фиолетового, красно-фиолетового, красно-оранжевого, оранжевого
до желтого цвета. Слабо декстринизированный крахмал, дающий с
иодом еще синий цвет, но лучше обычного крахмала растворяющийся
в воде, называется растворимым крахмалом.
При хлебопечении крахмал муки частично превращается в дек-
стрины, которые более растворимы в воде, а потому легче переварива-
ются и усваиваются. Обычно ферменты пищеварительных соков при
достаточно низких температурах гидролизуют крахмал до глюкозы
(как и минеральные кислоты при длительном кипячении).
Фракции крахмала и их строение. Крахмал не является однород-
ным веществом: он представляет собой смесь полисахаридов, постро-
енных по двум различным типам, поэтому крахмал можно разделить
на две основные фракции: амилозу и амилопектин. Для разделения
крахмальных фракций предложено много способов. Простейшим из
них является извлечение амилозы горячей (80°С) водой, в которой она
растворима. Амилопектин при этом только разбухает и остается в зер-
нах. Из водного раствора амилозу можно получить упариванием воды
или осаждением спиртом. Содержание амилозы и амилопектина в
крахмале различного происхождения колеблется от 15—25% амилозы
до 75—85% амилопектина.
Амилоза. Молекулы амилозы представляют собой неразветвлен-
ные (или очень мало разветвленные) полисахариды со степенью поли-
меризации 1000—6000, что соответствует молекулярной массе 16 000—
1 000 000. Средняя молекулярная масса амилозы картофеля около
400 000, кукурузной и рисовой — между 100 000 и 200 000.
Все глюкозные остатки связаны между собой а = 1,4-глюкозид-
ной связью. Приблизительно строение амилозы может быть вы-
ражено следующей формулой:
Как видно из формулы, подавляющее большинство остатков глюкозы
содержит три свободные гидроксильные группы: у второго, третьего и
шестого атомов углерода. Свободный полуацетальный гидроксил в
молекуле амилозы находится лишь у одного из глюкозных остатков в
самом «начале» цепи. В связи с этим восстанавливающая способность
380
амилозы ничтожна. При неполном гидролизе амилозы образуется ди-
сахарид мальтоза, имеющий также а-1,4-глюкозидную связь. Строение
амилозы было доказано методом исчерпывающего метилирования.
Водные растворы амилозы очень нестойки. Через некоторое время
амилоза выпадает в виде нерастворимого осадка (ретроградация).
С иодом амилоза дает чисто синее окрашивание. По данным рентгено-
Рис. 36. Комплекс амилозы с иодом
структурного анализа молекула амилозы свернута в спираль. Внутри
спиралевидной молекулы остается канал диаметром около 5 нм, в ко-
тором могут располагаться подходящие по размеру молекулы, образуя
особого типа комплексы — так называемые соединения включения.
Одним из них является соединение амилозы с иодом, окрашенное в си-
ний цвет (рис. 36).
Амилопектин. Подобно амилозе, амилопектин построен из молекул
глюкозы, но цепи в амилопектине сильно разветвлены. Большинство
глюкозных остатков имеют, как и в амилозе, а-1,4-глюкозидные свя-
зи, но в точках ветвлений присутствует 1,6-глюкозидная связь. Меж-
ду точками ответвления в основной цепи располагаются 20—25 глю-
козных остатков:
Цепи из глюкозных остатков в молекуле амилопектина многократ-
но ветвятся. «Ветвистая» структура амилопектина подтверждается
данными ферментативного расщепления этого полисахарида. Физико-
химические методы исследования амилопектина свидетельствуют о
сферической форме его молекул.
381
Общее число глюкозных остатков в молекуле амилопектина значи-
тельно выше, чем в молекулах амилозы. Молекулярная масса амило-
пектина порядка 106 — это значит, что степень полимеризации равна
примерно 6000.
В точках ветвления остатки глюкозы в амилопектине имеют по
два свободных гидроксила (у С2 и С3). Однако большинство глюкоз-
ных остатков в амилопектине, так же как в амилозе, имеет по три сво-
бодные гидроксильные группы (у С2, С3 и С6). Доля свободных альде-
гидных групп в амилопектине ничтожна, поэтому он, как и амилоза,
практически не обладает восстанавливающей способностью. С иодом
дает фиолетовое окрашивание.
Применение крахмала. Крахмал является одним из основных
компонентов пищевых продуктов. Мука, хлеб, картофель, крупы от-
носятся к главным источникам углеродов в нашем питании. Очищен-
ный крахмал применяют в производстве кондитерских и кулинарных
изделий, колбас. В текстильной промышленности он используется
для производства шихты, апретуры, загустителей к краскам. Крах-
мал применяется в спичечной, бумажной, полиграфической, косме-
тической промышленности, в переплетном деле. В медицине и фарма-
кологии крахмал идет на приготовление присыпок, паст (густых мазей),
а также необходим в производстве таблеток и т. д.
Подвергая крахмал кислотному гидролизу, можно получить глю-
козу в виде чистого кристаллического препарата или в виде патоки —
окрашенного некристаллизующегося сиропа. Патока широко исполь-
зуется в кондитерской промышленности. В последние годы появилось
производство так называемых модифицированных крахмалов, под-
вергшихся специальной обработке или содержащих улучшающие их
свойства добавки. Модифицированные крахмалы широко применяют-
ся в различных отраслях промышленности.
Гликоген
Гликоген, или животный крахмал, является запасным полисаха-
ридом животных. Он является одним из важнейших источников энер-
гии для животного организма. Все биологические процессы сопровож-
даются гликолизом — расщеплением гликогена и образованием
молочной кислоты. Животный крахмал содержится во всех клетках жи-
вотного организма, особенно много его в печени (10—20%) и мышеч-
ных тканях (до 40%). Он содержится также в некоторых низших рас-
тениях, например дрожжах и грибах.
Гликоген можно выделить из животных тканей путем обработки
их 5—10 %-ной трихлоруксусной кислотой на холоду с последующим
осаждением спиртом.
Гликоген — белый аморфный порошок, хорошо растворяющийся
даже в холодной воде с образованием опалесцирующих растворов.
Растворы гликогена имеют правое вращение, удельное вращение их
очень близко вращению крахмала: [аЬ = +196°.
Гликоген легко гидролизуется кислотами и ферментами, образуя в
382
качестве промежуточных веществ декстрины и мальтозу, а при пол-
ном гидролизе — глюкозу.
Молекулы гликогена построены по тому же принципу, что и моле-
кулы амилопектина (наличие а = 1,4- и 1,6-глюкозидных связей).
От амилопектина гликоген отличается лишь тем, что у него больше бо-
ковых ветвей (разветвления повторяются через каждые 8—16 остатков
глюкозы), вследствие чего молекула гликогена более плотная; общее
число остатков глюкозы в молекуле гликогена выше, чем в молекулах
амилопектина. Молекулярная масса гликогена исчисляется миллио-
нами, степень полимеризации 2 500—25 000. С иодом растворы глико-
гена дают окрашивание от винно-красного до красно-бурого в зависи-
мости от происхождения гликогена, вида животного и других условий.
Окрашивание иодом исчезает при кипячении и вновь появляется при
охлаждении.
Клетчатка, или целлюлоза
Клетчатка (С6Н10О5)п является высокомолекулярным полисахари-
дом, входящим в виде главной составной части в растительные клетки —
отсюда и название. Другое название — целлюлоза — того же про-
исхождения, так как по-латыни cellula означает «клеточка». В раз-
личных видах древесины содержание целлюлозы колеблется от 50 до
70%. Важнейшими сопутствующими ей веществами являются лигнин
(сложное вещество ароматического характера), гемицеллюлозы, пен-
тозаны, пектиновые и некоторые другие вещества. Волокна хлопка,
льна и конопли состоят также преимущественно из клетчатки.
Строение. Целлюлоза — гомополисахарид, построенный из ос-
татков глюкозы, соединенных между собой 0-1,4-связями. Это под-
тверждается тем, что при частичном гидролизе клетчатки образуется
дисахарид целлобиоза, имеющий, как известно, тоже 0-1,4-глюкозид-
ную связь. В общем виде строение целлюлозы можно выразить форму-
лой
Степень полимеризации п в клетчатке в среднем равна 6 000—
12 000, что соответствует молекулярной массе 1 000 000 — 2 000 000.
При очистке целлюлозы получают технические ее образцы с молекуляр-
ной массой 50 000—150 000. Большинство фрагментов глюкозы в
клетчатке содержит три свободных гидроксила (у Сг, С3 и С6).
Получение. При получении чистой целлюлозы растительные ма-
териалы (хлопок-сырец, древесная щепа и др.) сначала обезжиривают
органическими растворителями, а затем удаляют сопутствующие
вещества растворами щелочи (натронный и сульфатный способы) или
383
гидросульфита кальция (сульфитный способ) при 120—160°С при не-'
большом давлении. Эти способы основаны на высокой химической
стойкости целлюлозы, которая не взаимодействует с упомянутыми реа-
гентами, в то время как сопутствующие ей вещества при этом раство-
ряются.
Физические и химические свойства. Помимо химической стой-
кости, целлюлоза обладает и большой механической прочностью. Она
растворима лишь в ограниченном числе растворителей, таких, как:
1) реактив Швейцера — раствор гидроксида меди в концентрирован-
ном аммиаке («медноаммиачный комплекс»); 2) солянокислые растворы
хлорида цинка и некоторых других солей; 3) концентрированные ми-
неральные кислоты: соляная (41—42 'о НС1), серная (выше 72%) и
фосфорная.
Целлюлоза обладает слабой оптической активностью: [a]D ==
= —3,21° (реактив Швейцера).
Гидролиз целлюлозы происходит под влиянием кислотных катали-
заторов, к действию щелочей глюкозидные связи оказываются очень
стойкими. Чтобы провести полный гидролиз клетчатки до глюкозы,
ее сначала растворяют в серной кислоте, разбавляют раствор водой и
кипятят продолжительное время:
(СеН10О5)я 4“ л Н2о —► п С6Н12О6
клетчатка глюкоза
Кислотный гидролиз клетчатки имеет большое техническое значе-
ние: он служит основой производства этилового спирта (гидролиз-
ный спирт), получаемого гидролизом отходов древесины с последую-
щим сбраживанием образующейся глюкозы.
Как и другие высшие полисахариды, целлюлоза не обладает вос-
станавливающей способностью.
При действии окислителей (озон, пероксид водорода, хлорнова-
тистая кислота и ее соли, разбавленная азотная кислота, перманганат
калия и другие) получаются сложные смеси продуктов окисления,
носящие название оксицеллюлоз.
Химические свойства целлюлозы определяются прежде всего при-
сутствием гидроксильных групп. Главными реакциями целлюлозы
как многоатомного спирта являются образование алкоголятов и эфи-
ров целлюлозы. Действуя металлическим натрием, можно получить
тринатрийалкоголят целлюлозы [C6H7O2(ONa)3]n.
В отличие от низших спиртов целлюлоза при обработке концентри-
рованными растворами едких щелочей образует прочные соединения
(алкалицеллюлоза), имеющие строение алкоголятов:
[С6Н7О2 (ОН)3]Я + п NaOH -> [СбН7О2 (ОН)2 ONa]„ + п Н2О
алкалицеллюлоза
Этот процесс получил название мерсеризации. При этом происходит
набухание волокна и повышается его восприимчивость к красителям.
Алкалицеллюлоза при действии воды распадается снова на целлюлозу
и едкую щелочь:
[СбН7О2(ОН)2 ONaJrt 4- мН2О [С6Н7О2(ОН)3]Я nNaOH
384
Регенерированная целлюлоза имеет уже другую физическую струк-
туру.
Мерсеризацию хлопчатобумажных тканей широко применяют для
придания им лучшей окрашиваемости и улучшения внешнего вида.
Щелочная целлюлоза имеет также большое значение в качестве проме-
жуточного продукта при производстве вискозного волокна.
Будучи многоатомным спиртом, клетчатка способна образовы-
вать простые и сложные эфиры. В зависимости от условий реакции
можно получить производные по одному, двум или всем трем гидро-
ксилам (на одно среднее звено остатка глюкозы).
Общим способом получения простых эфиров клетчатки является
взаимодействие алкалицеллюлозы с алкилгалогенидами или алкил-
сульфатами в избытке щелочи:
[C6H7O2(OH)2ONa]n + ЗпС2Н5С1 + 2п NaOH -> [С6Н7О2(ОС2Н5)3]Л +
+ Зл NaCl + Зп Н2О
или
[С6Н7О2(ОН);, ONa]^ + Зл (СН3)2 SO4 + 2л NaOH ->
-> [СбН7О2 (ОСН3)3]Л + 3nCH3NaSO4 + 2лН2О
Среди простых эфиров целлюлозы наибольшее применение в тех-
нике имеют метил-, этил- и бензил целлюлозы. При метилировании
целлюлоза приобретает некоторую растворимость в воде. Метилцел-
люлоза используется главным образом как загуститель (взамен крах-
мала) в текстильной, косметической и пищевой промышленности.
Этилцеллюлоза обладает высокой механической и химической проч-
ностью, термо- и морозостойкостью. Она идет на производство
пластмасс, пленок и лаков.
Из сложных эфиров целлюлозы наибольший практический интерес
представляют азотнокислые, уксуснокислые эфиры и ксантогенат
целлюлозы.
Азотнокислые эфиры (нитраты) целлюлозы получают действием
на нее смесью азотной и серной кислот. Все нитраты целлюлозы горю-
чи, а некоторые — взрывчаты. Максимальное число остатков азотной
кислоты, которое можно ввести в клетчатку, равно трем на каждое
звено глюкозы:
[С6Н7О2 (ОН)3]Л + 3nHNO3 [C6H7O2(ONO2)3]n + 3n Н2О
Продукт полной этерификации — тринитрат целлюлозы — должен
содержать в соответствии с формулой 14,1% азота. На практике по-
лучают продукт с несколько меньшим содержанием азота (12,5—13,5%),
известный в технике под названием «пироксилин». Пирокси-
лин — детонирующее взрывчатое вещество большой силы, это дро-
бящее взрывчатое вещество. Используется в подрывных работах.
Пироксилин нерастворим ни в воде, ни в спирте, ни в эфире; такие
растворители, как ацетон и уксусноамиловый эфир, желатинируют
пироксилин, т. е. пироксилин в этих растворителях набухает и об-
разует густую студенистую массу. Из этой массы прессуют ленты
различной толщины и размеров, которые при высыхании могут быть
385
применены в качестве бездымного пороха. Он сгорает медленнее и,
•сгорая, не дает дыма. Пироксилин желатинируется также при помо-
щи нитроглицерина. Образующийся «гремучий студень» представляет
собой особый вид динамита и также применяется в подрывном деле.
Продукт нитрования клетчатки, содержащий 10% азота, отвечает
по составу динитрату целлюлозы: в технике такой продукт известен
под названием «коллоксилин». При действии на него смеси спирта и
эфира образуется вязкий раствор, так называемый коллодий. Колло-
дий — густая, клейкая жидкость, оставляющая по высыхании блес-
тящую пленку коллоксилина. Коллодий применяется в медицине
вместо пластыря, чтобы закрыть небольшие порезы. Если к колло-
дию добавить камфору и испарить растворитель, то остается гибкая
пленка — целлулоид.
Исторически — это первый известный тип пластмасс. Еще с прош-
лого века целлулоид получил широкое применение как удобный тер-
мопластический материал для производства многих изделий. В осо-
бенности важно использование целлулоида в производстве киноплен-
ки и нитролаков. Серьезным недостатком этого материала является
его горючесть.
При действии на целлюлозу смеси уксусного ангидрида, уксусной
кислоты и серной кислоты или дихлорида цинка (последние играют
роль катализаторов) образуется триацетат целлюлозы:
[C6H7O2(OH)3]n + Зп (СН3СО)2 О [СбН7О2(ОСОСН3)3]л + ЗпСН3СООН
Триацетилцеллюлозу называют первичным ацетатом. Неполное аце-
тилирование целлюлозы или частичный гидролиз первичного ацетата
приводит к вторичному ацетату:
[С6Н7О2(ОСОСН3)3]Л + пН2О-+ [С6Н7О2 (ОСОСН3)2 ОН]л + пСН3СООН
Ацетилцеллюлоза идет на изготовление ацетатного шелка. Из полу-
ченных двух типов ацетилцеллюлозы готовят растворы, причем пер-
вичный ацетат растворяют обычно в смеси C112CI2 и спирта, а вторич-
ный ацетат — в ацетоне. Вязкие прядильные растворы продавливают
через колпачки с тонкими отверстиями — фильеры (сухое прядение),
далее растворители в токе нагретого воздуха испаряются, а струйки
раствора превращаются в тончайшие нити из ацетилцеллюлозы —
ацетатный шелк. Ацетилцеллюлоза используется в производстве тка^
ней, трикотажа, негорючей кинопленки, пластмасс, электроизоляций.^
Общий принцип, лежащий в основе получения ацетатного волоки#,
(растворение, затем формование нитей), можно осуществлять иначе:*
большое техническое значение имеет вискозный способ. Сущность^
этого процесса — образование растворимого в воде ксантогената цел-
люлозы при действии на нее сероуглерода и щелочи:
3nCS2; 3nNaOH Г / zS \ '
[С6Н7О2(ОН)3]л----------->- СбН7О2 о—С<^ I 4-ЗпН2О
целлюлоза L \ xSNa/3Jn
ксантогенат целлюлозы
Образовавшееся соединение представляет собой натриевую cojiif
сложного эфира дитиоугольной (ксантогеновой) кислоты. Ксантогенать^
386
целлюлозы растворяются в воде или в разбавленной щелочи, об-
разуя так называемые вискозные растворы. Вискозный раствор по-
дают на прядильную машину, с помощью дозирующего насоса про-
давливают через фильеры в «прядильную ванну», содержащую раз-
бавленную серную кислоту и некоторые сульфаты (мокрое прядение).
Под действием кислоты ксантогенатные группы отщепляются и
целлюлоза регенерируется, образуя гладкую нить вискозного*
шелка:
С6Н,О2 {О—c/S ) + Зп H2SO4 -* [С6Н7О2 (ОН)3]„ + 3n CS2 + 3n NaHSO4
\ SNa /3_ п
Вискозный шелк — самый дешевый, так как его можно произво-
дить из сравнительно мало очищенной клетчатки и с применением
не очень дорогих реактивов. Вискозная нить, но более толстая и «на-
рубленная» на мелкие куски представляет собой штапельное волок-
но, из которого получают ткани, заменяющие хлопчатобумажные.
Если вискозу вместо фильер продавливать через узкие щели, то полу-
чается прозрачная пленка — хорошо известный всем целло-
фан.
Первые промышленные способы химической переработки целлю-
лозы возникли в связи с развитием бумажной промышленности.
Бумага — это тонкий слой волокон клетчатки, спрессованных и про-
клеенных для создания механической прочности, а также гладкой
поверхности для предотвращения растекания чернил. Первоначально*
для изготовления бумаги употребляли растительное сырье, из кото-
рого чисто механически можно было получить необходимые волокна:
стебли риса, хлопка, использовались также собираемые у населения
изношенные ткани (тряпки). Однако по мере развития книгопечата-
ния перечисленных источников сырья стало не хватать для удовлет-
ворения растущей потребности в бумаге. Особенно много бумаги
расходуется для печатания газет, причем вопрос о качестве (белизне,
прочности, долговечности) для газетной бумаги значения не имеет.
Зная, что древесина примерно на 50% состоит из клетчатки, к бумаж-
ной массе стали добавлять размолотую древесину. Такая бумага не
прочна и быстро желтеет, особенно на свету. Для того чтобы улуч-
шить качество древесных добавок к бумажной массе, были предло-
жены различные способы химической очистки древесины, позволяющие
получить из нее более или менее чистую целлюлозу, освобожденную
от сопутствующих веществ — лигнина, смол и т. д. Для выде-
ления целлюлозы было предложено несколько способов. По сульфит-
ному способу измельченную древесину «варят» под давлением с гидро-
сульфитом кальция. При этом сопутствующие вещества растворяют-
ся и освобожденную от примесей целлюлозу отделяют фильтрованием.
Образующиеся сульфитные щелока являются в бумажном производ-
стве отходами. Однако вследствие того, что они содержат наряду с
другими веществами способные к брожению моносахариды, их исполь-
зуют как сырье для получения этилового спирта (см. с. 174).
387
Пектиновые вещества, растительные камеди и :
слизи
Пектиновые вещества очень широко распространены в природе;
они встречаются во многих растениях, в том числе и в водорослях.
Известны растворимые пектиновые вещества и нерастворимые (прото-
пектин).
Растворимые пектиновые вещества содержатся преимущественно
в растительных соках, плодах, ягодах, корнеплодах и могут быть
осаждены спиртом из водных растворов.
Протопектин находится в основном в межклеточном веществе и
первичной стенке растущих растительных клеток.
Пектиновые вещества известны более 100 лет, однако основные
черты их строения выяснены сравнительно недавно. Основу всех пек-
тиновых веществ составляет полигалактуроновая (пектовая) кислота,
построенная из остатков £>-галактуроновой кислоты, соединенных
<х-1,4-гликозидными связями:
В растениях эта кислота содержится не в свободном виде: часть ее
карбоксилов этерифицирована метиловым спиртом, а часть сущест-
вует в виде солей.
Многочисленными исследованиями доказано, что, как правило,
пектиновые вещества являются гетерополисахаридами, причем в по-
строении основной цепи наряду с D-галактуроновой кислотой участ-
вуют и нейтральные моносахариды: L-арабиноза, £>-галактоза и L-
рамноза.
Протопектин — сложный комплекс, в образовании которого наря-
ду с пектиновыми веществами участвуют целлюлоза, ионы кальция,
магния, фосфорной кислоты.
Пектиновые вещества легко образуют студни с сахарами и кисло-
388
тами, что очень широко используется в пищевой промышленности для
изготовления желе и мармеладов.
Пектиновые вещества играют полезную роль в организме челове-
ка: они регулируют работу кишечника, являются противоядием при
некоторых отравлениях (например, ртутных) и т. д.
Камеди — вещества, выделяющиеся в виде прозрачных густею-
щих масс при повреждении растений, механическом ранении или па-
тологических процессах, вызываемых бактериями или грибками. Из
выделенной растениями аморфной массы можно извлечь камеди
щелочью с последующим осаждением кислотой. Это гидрофильные ве-
щества, в большинстве случаев хорошо растворимые в воде с образо-
ванием клейких растворов. Таковы аравийская камедь, или гуммиара-
бик, получаемая из сенегальской акации, камедь тракаганта, вишне-
вый клей и др. Камеди являются гетерополисахаридами, состоящими
из нескольких моносахаридов, среди которых может быть одна или
несколько уроновых кислот.
Слизями называют полисахариды, родственные камедям, но при-
сутствующие в неповрежденных растениях. Их источником служат
кора, корни, листья, семена. Слизи являются продуктами нормально-
го метаболизма растений и служат либо пищевым резервом, либо ве-
ществами, удерживающими воду. Среди слизей встречается много
нейтральных гетеро- и гомополисахаридов.
Липиды
Липидами называется сложная смесь органических веществ, вы-
деляемых из объектов растительного, животного и микробиологичес-
кого происхождения. Липиды обладают близкими физико-химичес-
кими свойствами, в первую очередь нерастворимостью в водей хоро-
шей растворимостью в ряде органических растворителей (диэтиловом
эфире, бензине, бензоле, хлороформе и др.). Липиды широко распро-
странены в природе. Являясь обязательной составной частью каждой
клетки, они вместе с углеводами и белками образуют основную массу
органических веществ всех живых организмов. Липиды выполняют
роль структурных компонентов клетки, запасных и защитных ве-
ществ.
Их состав исключительно сложен и зависит от источника получе-
ния (растения, животные, микроорганизмы), его состояния, методов
выделения и многих других факторов. Сложность состава, разнооб-
разие входящих в липиды компонентов, относящихся к различным
классам органических соединений, — причина того, что до настоящего
времени отсутствует единое, принятое всеми определение этой груп-
пы биоорганических соединений и их строгая научная классификация.
Большинство исследователей относят к липидам природные про-
изводные высших жирных кислот, спиртов и альдегидов, связанные
сложноэфирной, простой эфирной, амидной и гликозидной связями.
К липидам относятся сложные эфиры трехатомного спирта глицерина
и высокомолекулярных жирных кислот — ацилглицерины (глице-
риды), воски, фосфо- и гликолипиды, сфинголипиды. В группу липи-
389
дов иногда включают жирорастворимые пигменты (каротиноиды,
хлорофиллы), стерины, жирорастворимые витамины (А, Е, D, К),
продукты незавершенного биосинтеза липидов и вещества, образовав-
шиеся в результате разнообразных превращений липидов.
Классификация, природные источники и функции липидов. Как
уже указывалось, единая классификация липидов отсутствует. Наибо-
лее целесообразно классифицировать липиды в зависимости от их хи-
мической природы, биологических функций (структурные, запасные и
защитные липиды), а также по отношению к некоторым реагентам
например, к щелочам.
По химическому составу липиды обычно делят на две группы:
простые и сложные. Простые липиды не содержат азота, фосфора и
серы. К ним относятся главным образом нейтральные липиды, являю-
щиеся производными высших жирных кислот, одно-, двух- и много-
атомных спиртов, альдегидов (ацилглицерины, эфиры диолов, воски,
алкильные липиды, плазмалогены), а также их структурные компо-
ненты (спирты, карбоновые кислоты).
В состав сложных липидов входят фосфолипиды и сфинголипиды.
Фосфолипиды — соединения, при гидролизе которых образуются
наряду со спиртами (чаще всего глицерином) и высокомолекуляр-
ными жирными кислотами фосфорная кислота, азотистые основания,
аминокислоты и ряд других соединений. Сфинголипиды содержат
сфингазиновые основания, являющиеся длинноцепными аминодиолами.
В состав простых и сложных липидов могут входить гликолипиды,,
содержащие в качестве структурных компонентов углеводные фраг-
менты.
Иногда в самостоятельные группы липидов выделяют жирораство-
римые пигменты, стерины, жирорастворимые витамины. Некоторые
из этих соединений могут быть отнесены к группе простых (нейтраль-
ных) липидов, другие — сложных.
По другой классификации липиды в зависимости от их отношения
к щелочам делят на две большие группы: омыляемые и неомыляемые.
К группе омыляемых липидов относятся простые и сложные липиды,
которые при взаимодействии со щелочами гидролизуются с образова-
нием солей высокомолекулярных кислот, получивших название «мы-
ла». К группе неомыляемых липидов относятся соединения, не
подвергающиеся щелочному гидролизу (стерины, жирорастворимые
витамины, простые эфиры и т. д.).
По своим функциям в живом организме липиды делятся на струк-
турные и запасные. Структурные липиды (главным образом фосфоли-
пиды) образуют сложные комплексы с белками (липопротеиды, см.
С. 423) и углеводами, из которых построены мембраны клетки и кле-
точных структур, и участвуют в разнообразных процессах, протекаю-
щих в клетке. Кроме фосфолипидов в состав структурных липидов
входят глико-, сульфо- и некоторые другие липиды.
Запасные липиды (в основном ацилглицерины) являются энерге-
тическим резервом организма и участвуют в обменных процессах.
В растениях они накапливаются главным образом в плодах и семенах
(табл. 28).
390
Таблица 28. Содержание липидов в семенах и плодах растении
Культура Содержание липидов, % Культура Содержание липидов, %
Подсолнечник (семянка) Хлопчатник (семена) Соя > Лен » Арахис (ядро) Маслины (мякоть) Конопля (семена) Тунг (ядро плода) Рапс (семена) Сурепка » Горчица » Клещевина » 30—58 20—29 15—25 30—48 50—61 28—50 32—38 48—66 45—48 29—48 25—49 35—59 Пшеница (зерновка) Рожь » Кукуруза » Рис » Овес » Просо » Г речиха » Арбуз (семена) Какао (бобы) Кокосовая пальма (копра) Кедр (ядро ореха) 2,7 2,5 5,6 2,9 7,2 4,5 3,8 14—45 49—57 65—72 26—28
у животных и рыб — в подкожных жировых тканях и тканях, окру-
жающих внутренние органы, а также печени, мозговой и нервной тка-
нях. Содержание их зависит от многих факторов (вида, возраста,
питания и т. д.) и в отдельных случаях составляет 95—97% всех вы-
деляемых липидов.
Особую группу по своим функциям в живом организме составляют
защитные липиды растений — воски и их производные, покрывающие
поверхность листьев, семян и плодов.
Значительное количество липидов находится в молоке животных
(%): дельфин — 46, кит — 45, слон — 20, коза — 4,8, корова —
3,6. Активные «создатели» липидов — некоторые микроорганизмы,
содержание липидов в которых может достигать 20—50% от биомассы.
В живом организме липиды выполняют разнообразные функции.
Им принадлежит важная роль в формировании и старении организма,
в деятельности его защитных механизмов. Запасные липиды — ак-
кумуляторы химической энергии и используются организмом при не-
достатке питания и заболеваниях. Подкожные жировые ткани предо-
храняют животных от охлаждения, а внутренние органы — от меха-
нических повреждений.
Липиды являются важным компонентом пищи, во многом опреде-
ляя ее пищевую ценность и вкусовое достоинство. Исключительно
велика роль липидов в разнообразных процессах пищевой техноло-
гии. Порча зерна и продуктов его переработки при хранении (про-
горкание) в первую очередь связана с изменением его липидного комп-
лекса. Липиды, выделенные из ряда растений и животных, — основное
сырье для получения важнейших пищевых и технических про-
дуктов (растительного масла, животных жиров, в том числе сливоч-
ного масла, маргарина, глицерина, жирных кислот и др.).
В настоящее время основное количество липидов получают из при-
родных объектов.
В технике липиды, выделенные разнообразными методами, полу-
чили название сырого жира или просто жира, хотя они содержат все
391
компоненты липидов. Жиры, добываемые из растительного сырья,
называются растительными маслами. В настоящем разделе будут рас-
смотрены только основные группы липидов.
ПРОСТЫЕ ЛИПИДЫ
Глицериды (ацилглицернны)
Определение. Глицеридами (ацилглицеринами) называются слож-
ные эфиры глицерина и высокомолекулярных карбоновых кислот.
Они составляют основную массу липидов (в отдельных случаях до-
95—97%) и являются по существу жирами. Глицериды — наиболее
распространенный и важный компонент простых нейтральных
липидов.
В состав жиров в основном входят триглицериды, но присутству-
ют ди- и моноглицериды:
О О
I! II
СН2—О—С—R СН2—О—С—R
СН—О—С—О
II II
О СН—О—С—Ri
I
сн2—о—с—r2 сн2—он
II
о
триглицерид 1, 2-диглицерид
(триацилглицерин) (1, 2-диацилглицерин)
(R, R1? R2 — углеводородные радикалы).
СН2—ОН
I
СН—О—С—Ri
СН2—ОН
2-моноглицерид
(2-ацилглицерин)
Карбоновые (жирные) кислоты глицеридов
В жирах обнаружено свыше четырехсот карбоновых кислот раз-
личного строения. Однако большинство из них присутствует лишь в;
незначительном количестве. Наиболее распространенные в жирах
кислоты (основные кислоты жиров) содержат от 12 до 18 атомов угле-
рода (табл. 29): они часто называются жирными кислотами. В состав,
многих жиров входят в небольшом количестве низкомолекулярные
кислоты (С2—С10). Кислоты с числом атомов углерода выше 24 присут-
ствуют в восках.
Молекулы большинства кислот, входящих в состав жиров, постро-
ены из неразветвленных углерод-углеродных цепей, содержащих чет-
ное число углеродных атомов. Кислоты, содержащие нечетное число
атомов углерода, имеющие разветвленную углеродную цепочку или
содержащие циклические фрагменты, присутствуют в незначительных
количествах. Исключение составляют липиды некоторых микроорга-
низмов. В состав глицеридов наиболее распространенных жиров в
значительном количестве входят ненасыщенные кислоты, содержащие
1—3 двойные связи: олеиновая, линолевая и линоленовая (табл. 29).
В жирах животных присутствует арахидоновая кислота, содержащая
четыре двойные связи, в жирах рыб и морских животных обнаружены
392
кислоты с пятью, шестью и более двойными связями. Большинство
ненасыщенных кислот липидов имеет цисконфигурацию, двойные
связи у них изолированы или разделены метиленовой (—СН2—) груп-
пой. Кислоты, включающие окси-, кето-, эпоксигруппы, в большинстве
природных объектов встречаются в незначительных количествах, ис-
ключением является рицинолевая кислота (12-окси-9-октадеценовая
кислота, табл. 29), содержание которой достигает в касторовом масле
85%. В основном насыщенные и ненасыщенные кислоты триглицери-
дов растений, произрастающих в СССР, состоят из 18 углеродных ато-
мов.
Таблица 29. Основные карбеновые (жирные) кислоты жиров
Кислота Число углеродных атомов Формула
Насыщенные кислоты
Лауриновая 12 СН3(СН2)10СООН
Миристиновая 14 СН3(СН2)12СООН
Пальмитиновая 16 СН3(СН2)14СООН
Стеариновая 18 СН3(СН2)1бСООН
Арахиновая 20 СН3(СН2)18СООН
Бегеновая 22 СН3(СН2)20СООН
Церотиновая* 26 СН3(СН2)21СООН
Монтановая* 28 СН3(СН2)2бСООН
Мелиссиновая 30 СН3(СН2)28СООН
Ненасыщенные кислоты
Олеиновая
Эруковая
Линолевая
Линоленовая
Арахидоновая
СН3— СН2)7—СН=СН—(СН2)7—СООН
СН3—(СН2)7—СН=СН—(СН2)П—СООН
СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН
СНз—(СН2—СН=СН)з—(СН2)7—соон
СН3(СН2)з(СН2СН=СН)4(СН2)3СООН
Рицинолевая
Оксикислоты
СН3—(СН2)5—СНОН—СН2—СН=СН—
—(СН2)7—соон
Циклические кислоты
Чалмугровая
(СН2)12—соон
* Встречаются в восках.
Большинство природных жиров и масел представляет собой смесь
глицеридов, отличающихся сочетанием относительно небольшого чис-
ла жирных кислот. Учитывая, что одним из структурных компонентов
393
всегда является глицерин, свойства масел обусловливаются составом
и расположением ацилов кислот. Несмотря на относительно неболь-
шое число основных кислот (5—8), участвующих в образовании гли-
церидов, количество возможных триглицеридов может быть значи-
тельным:
Количество разных жирных кислот в
жире.......................... 5 6 7 8 9 10
Количество возможных глицеридов . 75 126 196 285 405 550
Поэтому состав жиров достаточно сложен.
Природные жиры содержат главным образом смешанные триглице-
риды, включающие остатки различных кислот. По ненасыщенности
их делят на четыре группы: GSU2 — мононасыщенные; GS2U — ди-
насыщенные; GU3 — ненасыщенные и GS3 — тринасыщенные; где
G — остаток глицерина; S — остаток насыщенной, и U — ненасы-
щенной кислоты.
В природных растительных триглицеридах 1-е и 3-е положения
заняты предпочтительно остатками насыщенных кислот, 2-е — нена-
сыщенной.
Изомерия, номенклатура, установление строения. Изомерия три-
глицеридов связана с различным положением (1,2,3)-ацилов в моле-
куле триглицерида, их различным строением, положением двойных
связей: возможна также цис- и трансизомерия. В глицеридах имеет
место оптическая изомерия, связанная с появлением в молекуле гли-
церида асимметрического углеродного атома (2), при наличии раз-
личных заместителей в 1-м и 3-м положениях:
О
II
Н2С—О—С—R
О
II
нс*—О—С—Rx
Н2С—О-С—R2
II
О
Для наименования глицеридов чаще всего применяется следующий
принцип: нумеруются атомы углерода 1,2,3 и указывается положение
заместителей с добавлением слова глицерин.
Для строения глицеридов определяют положение заместителей с
помощью избирательного ферментативного гидролиза, а затем общими
методами их строение.
Физические свойства. Триглицериды — жидкости или твердые вещества
без цвета, вкуса и запаха, нерастворимы в воде, но хорошо растворимы в орга-
нических растворителях.
Плотность триглицеридов 900—980 кг/м3 (при 15°С). По современным пред-
ставлениям кислотные остатки триглицеридов могут располагаться в форме
кресла (2), вилки (/) или стержня (3) (рис. 37). Триглицериды в твердом состоя-
нии существуют в виде нескольких кристаллических полиморфных форм.
394
Рис. 37. Возможные конфигурации молекул в кристаллах
триглицеридов
Химические свойства. Глицериды вступают во все химические
реакции, характерные для сложных эфиров, однако в их химиче-
ском поведении имеется ряд особенностей, связанных со строением
жирных кислот и глицерина.
Гидролиз триглицеридов. Под влиянием фермента липазы, кислот,
щелочей или сульфокислот триглицериды гидролизуются с образова-
нием сначала ди-, а затем моноглицеридов и в конечном итоге — жир-
ных кислот и глицерина. Гидролиз сильно ускоряется, если его про-
водить при 220—260°С и (25—60)-105 Па (безреактивное расщепление
жиров):
О
II
СН2—О—С—R СН2—ОН
СН—О—С— R] 4- НОН [н+]: (ОН~\ сн—О—С—R, + RCOOH Н,° г
I || или липаза || R2—СООН
I О о
сн2—о—с—r2
|] СН2—О—С—R2
о II
триглицерид
< тр иацилг лиц е р и н)
диглицерид (диацилглицерин)
сн2—ОН сн2—он
I Н2о |
сн—О—С—Ri---------------->- СН—ОН
|| -Ri-COOH |
О сн2—он
сн2—он
моноглииерид
(моноацилглицерин)
Результат полного гидролиза выражается схемой
О
II
СН2—О—С—R
| LH+]; [ОН-]
СН—О—С—Rj + ЗН2О-------->
Сн2—О—С—R2
395
сн2—он
I
—» СН—ОН + R—СООН + Ri—СООН + R2—СООН
I
сн2—он
Гидролиз триглицеридов широко применяется для получения жир-
ных кислот, глицерина, моно- и диглицеридов. Гидролитический рас-
пад жиров, липидов зерна, муки, крупы и других жиросодержащих
пищевых продуктов является одной из причин ухудшения их качества
и в конечном итоге порчи. Особенно ускоряется этот процесс с повы-
шением влажности хранящихся продуктов, температуры.
Переэтерификация. Глицериды в присутствии катализаторов (ме-
тилат и этилат натрия, натрий и калий, едкий натр) способны обмени-,
ваться (внутри- и межмолекулярно) ацилами; эта реакция получила
название переэтерификации:
О О
II II
СН2—О—С—R СН2—О—С—R
I | кат.
СН—О—С—R2 + СН—О—С—Rx----------->
СН2—О— С—R3 СН2—О—С—R
О
II
СН2—О—С—R
I
СН—О—С—Rx
СН2—О—R3
II
С
о
II
СН2—О—С—R
+ сн—о—с—r2
II
о
СН2—О—С—R
II
О
Переэтерификация приводит к изменению свойств жиров и масел,
поэтому она широко применяется в пищевой промышленности для по-
лучения продуктов с заданными свойствами.
Алкоголиз. Глицериды при нагревании со спиртами образуют соот-
ветствующие эфиры жирных кислот с высвобождением глицерина.
Такой обмен спиртов в сложных эфирах получил название алкоголиза:
О
II
СН2—О—С—R СН2—ОН О
I I II
СН—О—С—R + ЗСН3ОН СН—ОН + 3R—С—О—СН3
II I
О сн2—он
СН2—О—С—R
396
(катализаторы: едкий натр, алкоголяты щелочных металлов, серная
кислота, хлорид водорода). Алкоголиз применяется для промышлен-
ного и лабораторного получения сложных эфиров жирных кислот,
моно- и диглицеридов (глицеролиз).
Ацидолиз, При нагревании триглицеридов со свободными жирными
кислотами до 250—300°С происходит обмен ацилов:
О О
II II
СН2—О—С—R СН2—О—С—R
СН—О—С—R + 2Rj—СООН СНО—С—Rj + 2RCOOH
II II
О о
СН2—О—С- в СН2-О—С—Ri
Реакция протекает в присутствии серной кислоты, воды или три фто-
рида бора. Процесс ускоряется с увеличением силы свободной жирной
кислоты.
Алкоголиз и ацидолиз можно рассматривать как частные случаи
переэтерификации.
Рассмотренные реакции являются примером превращений, связан-
ных с наличием в молекуле триглицерида сложноэфирных групп. Не
меньшее значение имеют реакции глицеридов, связанные с превраще-
нием углеводородных радикалов.
Гидрогенизация глицеридов. Гидрирование жиров молекулярным
водородом в промышленности проводят при температурах 180—240°С
в присутствии никелевых и медно-никелевых катализаторов, как пра-
вило, при небольшом давлении. Подбирая соответствующие условия
реакции, удается осуществить этот процесс селективно (избиратель-
но), гидрируя сначала радикалы линоленовой кислоты до линолевой,
затем линолевой до олеиновой, а уже потом радикалы олеиновой (если
это необходимо) до стеариновой кислоты. Полученный продукт с за-
ранее заданными кислотным составом и свойствами называется сало-
масом:
О
II
СН2—О—С— (СН2)7—СН=СН—(СН2)7—СНз
СН—О—С—(СН2)7—СН=СН—СН2—СН=СН— (СН2)4—СН3 —
|| Ni; Си
О
СН2—О—С—(СН2)7—СН=СН—СН2—СН=СН—СН2—СН=СН—СН2—СН3
397
о
II
СН2—О—С—(СН2)7—СН=СН—(СНз),—сн3
I н2
----> СН—О—С—(CHJ,—СН=СН—СН2—СН=СН—(СН2)4—сн3-------->-
|| Ni; Си
О
СН2-О—С—(СН2)7—СН=СН—СН2—СН=СН—(CH2h—сн3
СН2—О—С—(СНг),—СН=СН—(СН2)7—СН3
о
н2
СН2—О—С— (СНг),—СН=СН— (СН2)7—СН3 ----->
II Ni; Си
о
СН3—О—С— (СН2)7—СН=СН—(СН2)7—сн3
II
о
о
II
СН2—О—С—Cj7 Н35
о
----> II
СН — О—С—С17Н35
СН2—о—с—с17н35
Окисление жиров. Жиры и масла, особенно содержащие радикалы
ненасыщенных жирных кислот, окисляются кислородом воздуха.
Начальными продуктами окисления являются разнообразные по стро-
ению перекиси и гидроперекиси. Они получили название первичных
продуктов окисления.
В результате сложных превращений перекисей образуются вторич-
ные продукты окисления: спирты, альдегиды, кетоны, кислоты с
углеродной цепочкой различной длины, а также их производные, в
частности продукты полимеризации.
Скорость, направление и глубина окисления зависят в первую оче-
редь от состава жиров и масел: с увеличением степени непредельности
жирных кислот, входящих в состав глицеридов, скорость их окисле-
ния возрастает. Кроме этого, на окисление влияют присутствие влаги,
следов металлов, температура, свет и интенсивность контакта с кис-
лородом воздуха и наличие антиокислителей (ингибиторов) — ве-
ществ, добавление которых приводит к обрыву цепей окисления. Сре-
ди них наибольшее значение имеют окислители фенольной природы:
бутилокситолуол, бутилоксианизол, пропилгаллаты. Из природных
антиокислителей наиболее активными являются токоферолы.
Накопление продуктов окисления в жирах и маслах приводит к
изменению их свойств и снижению пищевой ценности, а некоторые
398
из продуктов окисления оказывают вредное влияние на организм
Поэтому необходимо принимать меры по предотвращению или замедле-
нию окисления жиров и масел, используемых для пищевых целей.
В то же время процесс окисления может быть использован в технике
для получения продуктов с новыми положительными свойствами,
например образование защитных пленок при глубоком окислении и
полимеризации масел («высыхание», см. с. 416).
Производные глицерина с простой эфирной
связью (алкильные липиды; плазмалогены)
В группу нейтральных липидов с простой эфирной связью, содер-
жащих в качестве структурного компонента глицерин, входят: ал-
кильные липиды (производные высших спиртов алифатического ряда
I, II) и плазмалогены (1-алкенильноэфирные липиды), являющиеся,
производными высших жирных альдегидов (III):
СН2—OR О CH2OR
I II I
НО—С—Н Ri—С—О—С—Н О
I I II
СН2ОН сн2—о—с—r2
I II
О СН2—О—СН=СН—R
II I
Ri—С—О—С—Н О
I II
сн2—о—с—r2
III
(где R; Rf, R2 — углеводородные радикалы).
Большинство алкильных липидов присутствует в природных объ-
ектах в виде ди ацильных производных (II). В состав их молекул вхо-
дят высокомолекулярные спирты, главным образом с неразветвленной
цепочкой, содержащие четное число углеродных атомов в молекуле.
Наиболее распространены спирты алкильных липидов: гексадеканол,
октадеканол, 9-октадицен-1-ол (табл. 30).
Алкильные липиды обнаружены в растениях, животных и микро-
организмах. В наибольшем количестве они содержатся в липидах
морских животных.
В составе нейтральных плазмалогенов (альдегидогенные липиды)
присутствует винильноэфирная группировка —О—СН=СН—, по-
этому при их гидролизе образуются альдегиды. Плазмалогены обна-
ружены в липидах млекопитающих, молочном жире, желтке яйца,
липидах морских животных. Альдегиды, участвующие в построении
плазмалогенов, содержат обычно 16—18 атомов углерода и имеют не-
разветвленные углеродные цепочки. Ненасыщенные альдегиды пред-
ставлены моноенами; главный представитель — олеиновый альдегид,
СН3—(СН2)7—СН=СН—(СН2)7—•
При кислотном гидролизе плазмалогенов (I) образуются альдеги-
399
ды (II) и диацилглицерины (III); при щелочном — алкениловые эфиры
глицерина (IV) и высшие жирные кислоты (V, VI):
[Н+1
О СН2—О—СН=СН—R
Н I
rx—с —о—с—н О
I II
сн2—о - с—r2
I
---» R-CH?—С 4-
ъ
II
сн2—он
4- Rx—С—О—С—Н О
II I II
О СН2—о—с—r2
III
СН2—О—СН=СН—R
lQH~U но—с—н
сн2—он
+ Ri—СООН + R2—СООН
VI
Производные глицерина с простой эфирной связью не имеют промыш-
ленного применения, но они играют большую роль в разнообразных
процессах, протекающих в живом организме.
Диельные липиды
В последние годы в составе липидов обнаружены сложные (I),
простые (II) и 1-алкенильные (III) эфиры, структурным компонентом
которых является не глицерин, а двухатомные спирты (диолы) раз-
личного строения, они получили название диольных липидов:
О
II СН2—О—С—R СН2—О—R СН,—О—СН=СН—R
(СН,)» (СН,)„ 1
° 1 0 СН,—О—С—Rj
1 II 1 II II
СН2—О—С—Rt СН,—О—С—Rt О
I II III
(где R, Rt — углеводородные радикалы).
Из диолов чаще всего участвуют в построении диольных липидов
этиленгликоль, 1,2- и 1,3-пропандиолы, 1,4-бутандиол. В значитель-,
ном количестве диольные липиды присутствуют в липидах морских
организмов. По своим химическим свойствам они напоминают аналог
гичные производные глицерина.
400
Вески
Определение, классификация. Спирты восков. Восками называются
входящие в состав липидов сложные эфиры высокомолекулярных од-
ноосновных кислот и одноатомных высокомолекулярных спиртов
R—СН2—О—С—Rx, где R—СН2—О — остаток алифатического вы-
О
сокомолекулярного спирта; Rx—С— — остаток карбоновой (жирной)
кислоты.
В состав восков входят главным образом кислоты, содержащие
24—32 атома углерода, и спирты, углеродная цепочка которых состо-
ит из 14—30 атомов углерода (табл. 30).
Таблица 30. Основные спирты липидов
Историческое название ИЮПАК Число углерод- ных атомов Формула
Миристиловый 1-Тетрадеканол 14 СН3—(СН2)12—СН2ОН
Цетиловый 1-Гексадеканол 16 СН3—(СН2)14—СН2ОН
Стеариловый 1-Октадеканол 18 СН3-(СН2)1е-СН2ОН
Карнаубиловый 1-Тетракозанол 24 СН3-(СН2)22—СН2ОН
Цериловый 1-Гексакозанол 26 СН3-(СН2)24—СН2ОН
Мирициловый 1-Триакснтанол 30 СН3-(СН2)28—СН2ОН
Мелиссиловый 1-Гентриаконтанол 31 СН3—(СН2)29—СН2ОН
Олеиловый 9-Октадецен-Гол 18 СН3(СН2)7СН=СН(СН2)7СН2ОН
Выделенные из природных источников воски содержат значитель-
ное количество примесей свободных жирных кислот, спиртов, углево-
дородов.
В зависимости от происхождения различают растительные, живот-
ные, воски, вырабатываемые насекомыми, и ископаемые.
Нахождение в природе, способы получения. Воски широко рас-
пространены в природе. В растениях они покрывают тонким слоем
листья, стебли, стволы и плоды, предохраняя их от смачивания водой,
высыхания, действия микроорганизмов.
Из растительных восков промышленное значение имеют воски, по-
крывающие листья пальм (карнаубский воск), воски липидов риса и
подсолнечника. Из восков животного происхождения наибольшую роль
играют спермацет и спермацетовое масло, шерстяной жир; из восков
насекомых — пчелиный воск. Первые два продукта выделяют из мас-
лообразной массы, содержащейся в голове кашалота и в длинном ка-
нале, проходящем вдоль всего туловища. Твердый кристаллический
продукт белого цвета, состоящий главным образом из цетилового эфи-
ра пальмитиновой кислоты, называется спермацетом, а жидкий про-
14—682 401
дукт, оставшийся после его выделения, — спермацетовым маслом. Из
одного кашалота получают 3—5 т спермацетового жира.
«Шерстяной жир» — жиропот овечьей шерсти, от желтого до темно-
коричневого цвета, с резким неприятным запахом. После специаль-
ной обработки из него получают слабоокрашенный мазеобразный про-
дукт — ланолин — сложного химического состава, основной компо-
нент которого эфиры кислот С1о—С20 и спиртов С18—С20. В шерсти
овец содержится 5—10% «шерстяного жира».
Пчелиный воск получают из пчелиных сот вытапливанием или экс-
тракцией после удаления меда. Он состоит главным образом из эфиров
кислот Сгв—С30 и спиртов С44—C3i и содержит до 15% углеводородов.
Ископаемые воски (горный воск, воск бурых углей) включают до
70% сложных эфиров кислот и спиртов с числом атомов углерода
больше 24.
Свойства. Большая часть восков — твердые, упругие вещества,
нерастворимые в воде и при комнатной температуре довольно плохо
растворимые во многих органических растворителях. В химическом
отношении воски (особенно содержащие остатки насыщенных жирных
кислот) отличаются значительной стойкостью; с трудом омыляются,
стойки к действию кислорода воздуха.
Применение. Свойства восков определяют и пути их использо-
вания. Они применяются в качестве электроизоляционных покрытий,
являются важным исходным продуктом для получения разнообраз-
ных моющих средств, используются в косметике при изготовлении кре-
мов и помад. В бытовой химии воски применяются в качестве компонен-
тов лощильных составов, используемых для полировки паркетов,
мебели и в медицинской промышленности.
Гликолипиды
Гликолипидами называется большая и разнообразная группа нейт-
ральных липидов, в состав молекулы которых входят остатки моноз.
Гликолипиды широко распространены в растениях, животных и мик-
роорганизмах. Они выполняют функции структурных липидов, уча-
ствуя в построении мембран, им принадлежит важная роль в форми-
ровании клейковины пшеницы, определяющей хлебопекарное досто-
инство муки. Чаще всего в построении молекулы гликолипидов участ-
вуют D-галактоза, D-глюкоза, уроновые кислоты:
галактозил ди глицерид
глюкозилдиглицерид
402
(где R, Ri — углеводородные радикалы).
СЛОЖНЫЕ ЛИПИДЫ
Фосфолипиды
Определение, распространение в природе. Фосфолипидами назы-
вается наиболее разнообразная и важная группа сложных липидов,
структурными компонентами которых являются фосфорная кислота,
насыщенные и ненасыщенные жирные кислоты, альдегиды, спирты
(глицерин, диолы, миоинозит, сфиногозин, см. с. 408), азотистые ос-
нования (холин, этаноламин), аминокислоты, связанные между собой
сложной или простой эфирной или амидной связью.
Фосфолипиды являются обязательной составной частью растений
и животных; их содержание колеблется в широких пределах. Особен-
но много их в нервной и мозговой тканях (до 30%). Среднее содержание
фосфолипидов в зерне и семенах некоторых культур приведено ниже:
Культура
Соя..........
Хлопчатник .
Подсолнечник
Клещевина
Пшеница . .
Рожь . . . .
Содержание фосфоли-
пидов, %
1,8
1,7
0,7
0,3
0,54
0,6
Культура Содержание фосфоли-
пидов, %
Кукуруза . . . . 0,89
Рис....... 0,20
Просо..... 0,83
Гречиха... 0,93
Лен........ 0,6
Овес ...... 0,37
Роль фосфолипидов в жизнедеятельности живого организма чрез-
вычайно велика. Вместе с белками и другими соединениями они уча-
ствуют в построении мембран клеток и субклеточных структур, вы-
полняя роль «несущих конструкций», способствуют переносу хими-
ческих веществ, а также осуществляют другие функции в биохимичес-
ких процессах, протекающих в живом организме.
Основные группы
фосфолипидов
Фосфолипиды могут быть разделены на несколько групп, в зависи-
мости от строения «центрального» структурного фрагмента — полиола:
глицерофосфолипиды, диольные фосфолипиды, сфинголипиды. В по-
строении молекул первой группы соединений участвует глицерин,
второй — диолы, третьей — сфингозин. Представители первой и тре-
тьей групп наиболее распространены и играют особо важную роль
в живых организмах. Учитывая специфичность строения сфингозина,
14*
403
его производные часто выделяют в самостоятельную группу сложных
липидов.
Глицерофосфолипиды. Общая формула. Основные структурные ком-
поненты. Общая формула глицерофосфолипидов имеет следующий
вид:
СН2—О—R
RiO—C—н
I 0
I II
сн,—О—Р—ОХ
О"
где R, Ri — могут быть ацилами или R = —CH = R2, a R, — ацил;
Х=Н; -CH2-CH2N(CH3)3; -CH2-CH2NH(CH3)2; -СН2-СН-СООН
+ NH3
и другие остатки.
Следовательно, в молекуле фосфолипидов всегда имеются замести-
тели двух типов: гидрофильные и гидрофобные (см. с. 408). В качест-
Рис. 38. Строение молекул фосфоглицеридов
ве первого выступает аминопроизводное, второго — высокомолеку-
лярные ацилы или алкилы. Условно это можно представить следую-
щим образом (рис. 38). «Голова» молекулы представляет собой остаток
фосфорной кислоты с аминопроизводными, «хвосты» — ацильные или
алкильные радикалы.
Большая часть жирных кислот фосфолипидов содержит четное
число углеродных атомов, соединенных в неразветвленную цепь.
Однако содержание кислот изостроения и с нечетным числом углерод-
ных атомов выше, чем в простых, нейтральных липидах. Состав жир-
ных кислот фосфолипидов растений отличается повышенным содержа-
нием ненасыщенных кислот; животных — насыщенных.
Фосфолипиды, содержащие два ацильных радикала, значительно
больше распространены в природе, имеют большее народнохозяйст-
венное и биологическое значение, чем фосфорсодержащие плазмало-
гены.
Наиболее простое строение из глицерофосфолипидов имеют широ-
ко распространенные в природе фосфатидные (фосфатидиновые) кис*
лоты:
404
о
и
О СН2—О-С—R
II I
Rj—С— О—С—Н
I 0
I II
СН,—О—Р—ох
I
О-
монофосфатидная кислота
В растениях, животных тканях и микроорганизмах обнаружены
молекулы, построенные из нескольких остатков фосфатидных кислот:
О о-
II I
О СН2—О—С—R СН2—О—Р—О—сн2 о
II I I II I II
Rx—С—О — С—Н О’ СНОН О Н—С—О—С—R
СН2—О—Р—О—СН2 СН2—О—С—Rr
II II
о о
трирлицерофоефа т
Наиболее распространены среди фосфолипидов фосфатидил холины
и фосфатидилэтаноламины. В молекуле первых X представлен остат-
ком аминоспирта — холина [НО—СН2—СН2—N+(CH3)3](OH“):
О
О СН2—О—С—R
II I
Rj—с—о — с—н о
I II
сн2—О—Р—О—СН2—СН2—N*(CH3)3
0“
фосфатидилхолин (лецитин)
В фосфолипидах масличных семян и животных содержание фос-
фатидилхолинов достигает 30—50%.
В молекуле этаноламинфосфатидов (их ранее неправильно назы-
вали кефалинами) содержится остаток этаноламина НО—СН2—СН2—
NH2:
О
II
СН2—О—С—R
О
СН—О—С—Ri
О
II +
СН2—О—Р—О—СН2—СН2—NH3
О'
фосфатаднлэтанола м ин
405
Содержание фосфатидилэтаноламинов в фосфолипидах масличных
семян и животных тканях достигает 20—25%.
В фосфатидил инозитах «X» представлен остатком циклического
спирта — гексаоксициклогексана (инозит). Из восьми его возможных
изомеров в фосфатидил инозитах обнаружена только одна форма —
миоинозит:
гексаоксициклогексан
(миоинозит)
В молекулах фосфатидил инозитов может содержаться один (моно-
фосфатидил инозит) или несколько остатков миоинозита.
В природных фосфатидилинозитах в образовании связи с фосфа-
тидильным остатком участвует первый углеродный атом миоинозита
(I). Оксигруппы в миоинозите могут быть этерифицированы кислота-
ми различного строения. Фосфоинозитиды, в молекулах которых
спиртовые группы миоинозита (обычно в положении 4,5) этерифици-
рованы ортофосфорной кислотой, получили название полифосфоино-
зитидов:
СН2— о—с—R
трифосфоинозитид (II)
(где X = ОРО3Н2).
Особую группу фосфоинозитидов составляют сложные фосфоино-
зитиды, содержащие в молекуле остатки углеводов, аминокислоты и
некоторые другие соединения. Сложные фосфоинозитиды, содержащие
остатки моноз (главным образом галактозы, арабинозы), можно рас-
сматривать как сложные гликолипиды:
406
В состав другой группы фосфолипидов входит фосфатидил серин:
О
II
О СН2—О—С—R
II I
Rx—С—О — С—Н О
I II *
СН2—О—Р—О—СН2—СН— NH3
I I
О' соон
Среди продуктов его гидролиза обнаружена аминокислота серин:
НО—СН2—СН—СООН
NH2
В тканях живых организмов фосфатидилсерины присутствуют в виде
солей калия, натрия и магния.
Фосфорсодержащие плазмалогены. Эта группа соединений присутст-
вует главным образом в фосфолипидах животных и человека. На их
долю приходится до 30—40% фосфолипидов мозга, сердечной мышцы.
Обнаружены они в составе растений, микроорганизмов. Строение их
может быть представлено следующей формулой:
О СН2—О—СН=СН—R
II I
Ri—С—О—С—Н
I ?
СН2—О—Р—ох
О-
(где R, Ri — углеводородные радикалы; X — —СН2—СН2—N(CH3)S;
—СН2-СН2—ЙН8).
407
Воздух
жшш
--------Вода—
б
Рис. 39. Устойчивые системы фосфолипид — вода:
а — на границе вода — воздух, б — в воде
Свойства, получение, применение. Фосфолипиды — бесцветные
вещества, без запаха, хорошо растворимы в диэтиловом эфире, хлоро-
форме, плохо в ацетоне. В водных растворах молекулы фосфолипидов
вследствие поверхностной активности располагаются на поверхности,
образуя мономолекулярный слой, причем гидрофобные части молекулы
направлены наружу, гидрофильные — внутрь или образуют мицеллы
(рис. 39).
В промышленности фосфолипиды получают в качестве побочного
продукта при производстве растительных масел.
Фосфолипиды нашли широкое применение в медицине и в качест-
ве улучшителей и эмульгаторов в ряде отраслей пищевой промышлен-
ности (хлебопекарной, кондитерской, масложировой).
Сфинголипиды
Сфинголипидами называется группа сложных липидов, основой
молекулы которых являются алифатические аминоспирты, из которых
наиболее распространены сфингозин и церебрин:
СН3—(СН2)12—сн=сн—сн—сн—сн2он
он nh2
сфингозин
СНз- (СН2)12-СН2—СН-СН СН СН2ОН
он он nh2
церебрин (фитосфингозин)
С этим основным структурным стержнем молекулы связаны другие
структурные компоненты сфинголипидов: азотистые основания, фос-
форная кислота, жирные кислоты, аминокислоты, монозы.
Сфинголипиды делятся на две большие группы: фосфорсодержащие
сфинголипиды и гликосфинголипиды. Сфинголипиды широко представ-
лены в живом организме, участвуют в построении мембран, в сложных
процессах, связанных с нервной деятельностью животных. Интересно,
что содержание фосфорсодержащих сфинголипидов в организме жи-
вотных увеличивается по мере эволюции его нервной системы: у млеко-
питающих их до 10 % от суммы липидов, у рыб — 1—2%.
408
Фосфорсодержащие сфинголипиды. Наиболее распространены среди
этой группы соединения, содержащие остатки холина:
ОН О
I II +
СН3-(СН,)12—СН=СН—СН—СН—СН2—О—Р—О—СН2—СН2 N(CH3)3
NH—CR О-
II
о
цера мидфосфорилхо лин
(где R — углеводородный радикал).
В состав фосфорсодержащих сфинголипидов входят остатки толь-
ко насыщенных (пальмитиновая, стеариновая, лигноцериновая СН3—
(СН2)22—СООН) или моноеновых кислот [нервоновая СН3—(СН2)7—
—СН=СН —(СН2)13СООН]. Отличительной особенностью химичес-
кого поведения этой группы сфинголипидов является их относитель-
ная устойчивость к щелочному и кислотному гидролизу; по своим
физическим свойствам — это бесцветные кристаллические вещества,
обладающие оптической активностью.
Гликосфинголипиды. Эта группа включает несколько типов соеди-
нений: цереброзиды (I), ганглиозиды (II), сульфолипиды (III) и не-
которые другие:
СН2ОН но Оу Кон н Н 0 СН2ОН СН2ОН HOJroVon"<^r°V Кон нЛ К. ни у г—о—с н?—сн—с н—с н=с н—с13н27 Г Ан С—с23н4, Н || 4» Q церазин (1) СН2ОН СН2ОН кон ни о К он ни til
Н N— у Н Н/<| 1/ н Н N И н N V н Н OH / н NH-с-сн3н н он II О О н н. Л k N Н—с—С Н3 /н но\| II \ н/1 0 НООС \) К Н II—с—он н—с—он 1 сн2он ганглиозид (II) (моносиалоганглиозид) 1 О=с-с15н31
409
Н2ОН
H0/fi-------4
|\0SO3Na Н
Н
ОН
Н2—СН—СН—СН=СН—(сн2)1Г-сн3
NH ОН
о=<!:—сн2—с16н„
сульфатид (III)
(сульфат цереброзида)
Молекулы цереброзидов (I) состоят из остатков жирных кислот,
гексоз и сфингозина. Соединения этой группы входят в состав липидов
мозга. Сульфолипиды (III) обладают кислотными свойствами за счет
наличия в молекуле сульфогруппы, в живом организме присутствуют
в виде солей. Ганглиозиды (II) можно рассматривать как одну из
групп гликолипидов, молекулы их построены из моноз, аминосахаров,
сиаловой кислоты, сфингозина. Входят в состав липидов головного
мозга.
АНАЛИЗ ЛИПИДОВ
Анализ липидов и продуктов их превращения, учитывая особен-
ности их состава, является сложной задачей, требующей применения
наряду с химическими методами современных физико-химических
методов исследования (хроматографии, спектроскопии, рентгенострук-
турного анализа и т. д.).
Изучение липидов начинается с их выделения; наиболее эффектив-
но это удается осуществить, экстрагируя липиды органическими раст-
ворителями (диэтиловый эфир, бензин) или их смесями (хлороформ +
+ метанол).
В живом организме часть липидов связана с белками и углеводами,
образуя разнообразные по сложности и прочности комплексы и соеди-
нения. Растворители обладают неодинаковой способностью разрушать
эти комплексы и выделять липиды, поэтому состав липидов до извест-
ной степени зависит от выбора растворителя.
В практике пищевой промышленности состав и качество жиров и
масел характеризуют с помощью разнообразных «чисел», подразуме-
вая под ними расход определенных реагентов на реакции с жиром.
Наибольшее значение имеют числа: кислотное, омыления, иодное.:.
Кислотным числом называется показатель, характеризующий ко- f\
личество свободных жирных кислот, содержащихся в жире. Он вы-
ражается в миллиграммах гидроксида калия, затраченного на нейтра-
лизацию свободных жирных кислот, содержащихся в 1 г жира. Учи-
тывая, что хранение пищевых продуктов, содержащих жиры и масла,
всегда сопровождается гидролизом последних, по величине кислотно-
го числа можно, до известной степени, судить об их качестве. В за-
водской практике кислотное число используется при расчете коли-
чества щелочи, необходимого для щелочной рафинации жиров и масел.
Число омыления равно количеству миллиграммов гидроксида ка-
лия, необходимого для омыления глицеридов и нейтрализации сво-J
бодных жирных кислот в 1 г жира или масла:
410
C3H5(OCOR)3 + ЗКОН------► C3H5(OH)3 + 3RCOOK
триацилглицерин
По числу омылении можно судить о средней молекулярной массе
входящих в состав жира жирных кислот и определить количество
щелочи, необходимое для омыления жира.
Иодное число — показатель, характеризующий непредельность
жирных кислот, входящих в состав жира. Он выражается в процентах
иода, эквивалентного галогену, присоединяющемуся к 100 г жира.
Существует несколько методов определения иодного числа. Одним из
наиболее распространенных является бромометрический метод. При
этом применяется раствор брома в безводном метиловом спирте, на-
сыщенном бромидо?л натрия. Бром образует непрочное комплексное
соединение с NaBr:
NaBr + Вг2-----> NaBr-Br2
Отщепляясь, бром реагирует с ненасыщенными глицеридами:
О
II
СН2—О—С— (СН2)7—СН=СН—(СН2)7—сн3
о
II
сн—о—с—с17н35
СН2—О—С—С17Н35
II
о
+ NaBr-Br2----->
О Вг Вг
СН2—О—cL—(СН2)7— СН— СН-(СН2)7-сн3
+ NaBr
СН—о—с—с17н35
о
II
СН2—О—С—С17Н35
Количество непрореагировавшего брома определяют иодометрически:
NaBr-Br2 + 2KI---->• КВг + NaBr + Ь
I2 -|- 2Na2S2O3 --> Nal -j- Na2S4O6
Зная исходное количество брома, можно легко вычислить иодное
число жира.
Иодное число применяется для определения вида жира, способнос-
ти его к «высыханию», расчета потребного количества водорода на его
гидрогенизацию.
Величины двух последних констант для многих жиров, не подверг-
шихся разрушению, колеблются в незначительных пределах и харак-
теризуют вид жира и его качество (табл. 31).
Для детального изучения липидов их разделяют на отдельные клас-
Таблица 31. Основные виды жиров и масел
Жиры и масла Содержание и состав жирных кислот Характеристика
насыщен- ных ненасы- щенных основных* темпера- тура застывания число омыления иодное число
Соевое Арахисовое 14—20 20—21 75—86 79—80 Масла —18 —2 3 191—193 185—186 120—140 90—95,5
С2 С18 ^18 46—65 39—70
Хлопковое 22—30 75—78 45—56 2—4 191—198 101—116
С18 16—35
Подсолнечное 10—12 до 90 С?8 46—70 16—18 186-194 119—136
Рапсовое 2—6 94—98 Эруковая 0—10 167—181 94—103
11—56
^18 6—44 0—10 167—181 94-103
Оливковое 9—18 82—91 ^18 70—82 0—6 185—200 72—89
Кокосовое до 90 10 С?2 44—52 16—25 251—264 7—12
С14 13—18
Пальмовое 44—57 43—56 с° 39—47 31—41 196—210 52—58
С18 45—50
Пальмоядровое 79—83 17—21 с° ь16 10—19 19—24 240—257 15—20
Масло какао 58—60 40—42 ^18 23—25 21—27 192—196 34—38
с16 31—34
Льняное 6—9 91—94 С?8 41—63 18—27 191—195 175—190
Касторовое До 0,6 94—97 Рационолевая — 10—18 176—186 92—90
кислота 80—94
Тунговое 3—10 90—97 Элеостеарино- -2—17 185—197 145—176
вая кислота,
66—82
Животные жиры
Говяжий 45—60 43—52 ^18 43—44 24—29 30—38 190—200 32—47
Бараний 52—62 38—48 С18 36—42 32—45 192—198 31—46
С?8 25—31
Свиной 33—49 48—64 С18 34—44 22—32 193—200 46—66
25—32
Китовый 10—22 78—90 — 181—193 100—161
Кашалотовый — — 17—20 125—150 62—92
* Индекс 0, 1, 2, 3 —• указывает на количество двойных связей в молекуле кислоты
412
сы, каждый из которых подвергают дальнейшему исследованию.
В зависимости от особенностей изучаемого класса соединений приме-
няются различные методы, они подробно описаны в специальной лите-
ратуре.
ПОЛУЧЕНИЕ И ПЕРЕРАБОТКА ЖИРОВ
И МАСЕЛ
Основные источники получения жиров и масел. Растительные мас-
ла содержатся в плодах и семенах сои, арахиса, хлопка, подсолнечни-
ка, рапса, оливкового дерева, пальмы, льна, какао. Масла извлекают
также из маслосодержащих отходов некоторых производств: кукуруз-
ные зародыши, рисовая мучка, семена косточковых плодов, виноград-
ные косточки. В СССР растительное масло получают главным образом
из семян подсолнечника, хлопчатника, сои, льна, конопли.
Животные жиры содержатся в тканях крупного и мелкого рогатого
скота (говяжий и бараний жиры, свиной смалец), китов и кашалотов и
коровьем молоке.
Твердые жиры растительного происхождения (масло какао, паль-
мовые масла) отличаются относительно высоким содержанием насы-
щенных жирных кислот (лауриновой, миристиновой, пальмитиновой,
стеариновой); жидкие — ненасыщенных (олеиновой, линолевой).
По отношению к окисляющему действию кислорода жидкие расти-
тельные масла условно делятся на высыхающие, полувысыхающие,
невысыхающие. Животные жиры делятся на жиры наземных живот-
ных, молочные жиры и жиры морских млекопитающих и рыб.
В настоящее время производство растительных масел растет быст-
рее, чем животных жиров. Это связано с их большей физиологической
ценностью и экономической целесообразностью их производства.
Извлечение жиров. Основное количество масла извлекается (из
специально подготовленного растительного сырья) экстракционным
или форпрессово-экстракционным способом. Содержание его в остатке
(шроте) при использовании этих методов не превышает 1 %. Экстрак-
ция масла проводится бензином или гексаном при температуре 50—
55°С в специальном аппарате, получившем название экстрактор. При
извлечении масла из высокомасличного сырья часть его может быть
извлечена прессованием (отжим под повышенным давлением), а остав-
шееся количество экстракцией (форпрессово-экстракционный метод).
Качество масла, полученного прессовым методом, несколько выше,
чем экстракционного. В технике в зависимости от особенностей сырья
и аппаратурного оформления применяются различные варианты этих
методов.
Рафинация масел. Полученное «сырое» масло содержит значи-
тельное количество примесей, мешающих его дальнейшему использо-
ванию или придающих ему нежелательные вкусовые качества.
Очистка масла осуществляется с помощью целого комплекса разно-
образных физико-химических процессов и называется рафинацией.
Нерастворимые в масле примеси удаляют отстаиванием, а затем
фильтрацией. Фосфолипиды, примеси белковых и слизистых веществ
41;
удаляют гидратацией (обработка водой). Выделенные и очищенные
фосфолипиды являются ценным продуктом.
Свободные жирные кислоты, содержащиеся в сырых маслах, уда-
ляют обработкой щелочью (щелочная рафинация), образующиеся
при этом соли жирных кислот (мыла) отделяют вместе с увлеченными
примесями (соапсток). Так как при щелочной нейтрализации из масла
удаляют не только свободные жирные кислоты, но и разнообразные
примеси, оно при этом частично осветляется. Масло, прошедшее стадии
очистки, может обладать более интенсивной окраской и неприятным
запахом. В этих случаях для удаления красящих веществ его подвер-
гают адсорбционной очистке («отбелке») — обрабатывают порошкооб-
разными веществами (адсорбентами), способными поглощать (адсор-
бировать) окрашенные примеси. В качестве адсорбентов используются
природные глины, активизированный древесный уголь. Сильно пах-
нущие компоненты масла удаляют обработкой водяным паром под
глубоким вакуумом (дезодорация). Очищенное от примесей, «рафини-
рованное» масло является ценным пищевым продуктом, а также при-
меняется в пищевой промышленности в качестве сырья при гидрогени-
зации; получении маргарина, мыла.
Гидрогенизация жиров. В отдельных отраслях промышленности
(маргариновая, мыловаренная, кондитерская) в значительных коли-
чествах требуются твердые жиры. Эти потребности не могут быть удов-
летворены за счет ресурсов твердых животных и растительных жиров.
Превращение жидких растительных масел в твердые или полутвердые
жиры достигается гидрогенизацией.
Гидрирование жиров осуществляется электролитическим водоро-
дом при участии никелевых и медно-никелевых катализаторов при
температурах 180—220°С в аппаратах (автоклавах) непрерывного
действия при давлении (0,5—1,5)-105 Па. Меняя условия гидрогени-
зации, можно получить продукты (саломасы) разной степени «насы-
щенности», предназначаемые для различных целей Процесс гидроге-
низации осложняется рядом побочных реакций, в первую очередь изо-
меризацией (смещением двойных связей, переходом радикалов непол-
ностью гидрированных жирных кислот в трансформу). Эти процессы
существенно влияют на свойства и пищевую ценность готовых продук-
тов. Полученный при гидрогенизации саломас после удаления катали-
затора и рафинации используется в различных отраслях промышлен-
ности.
Переэтерификация и гидропереэтерификация жиров. В последнее
время в промышленности получила развитие переэтерификация жи-
ров, позволяющая из высокоплавких животных жиров и их смесей
с растительными жирами получить жиры с любым глицеридным сос-
тавом, а следовательно, с любыми свойствами. Переэтерификация
жиров проводится при температурах 50—90°С, в присутствии катали-
заторов метилата натрия, этилата натрия, сплава натрия и калия.
Полученные продукты не содержат, подобно гидрированным жирам,
нежелательных трансизомеров и обладают высокой биологической цен-
ностью. В отдельных случаях гидрогенизацию жиров проводят с
4J4
участием катализаторов, осуществляющих одновременно и их переэте-
рификацию (гидропереэтерификация).
Получение и переработка животных жиров. Основной способ по-
лучения животных жиров — вытапливание их из жиросодержащих
тканей. Различают мокрую вытопку (сырье в течение всего процесса
находится в непосредственном контакте с водой и паром) и сухую. В
отдельных случаях с целью повышения выхода жира на измельчен-
ную ткань воздействуют слабыми растворами щелочей и карбонатов
(NaOH; Na2CO3).
Высыхание жиров. Растительные масла, содержащие глицериды
полиненасыщенных жирных кислот, обладают повышенной спо-
собностью поглощать кислород воздуха, претерпевая при этом слож-
ные химические изменения. Этот процесс получил название высыха-
ния масел, а такие масла — высыхающими. Высыхание масел проте-
кает особенно быстро в присутствии катализаторов, называемых сик-
кативами. На практике это свойство высыхающих масел используется
для приготовления олифы. Олифой называют растительное масло, со-
держащее сиккативы, частично окисленное и полимеризованное в ре-
зультате нагревания при доступе воздуха. Олифы применяются в ка-
честве пленкообразователей для защиты металлических и деревянных
конструкций зданий и сооружений. Добавлением к олифам минераль-
ных пигментов (красок) получают масляные краски. Лучшее сырье
для получения олифы — льняное, конопляное, тунговое масла. В свя-
зи с ростом спроса на пленкообразующие материалы и необходимостью
сократить расход пищевых растительных масел для технических це-
лей в последние годы в большом количестве в качестве пленкообразо-
вателей применяют синтетические смолы. Особое распространение на-
шли глифгалевые и пентафталевые смолы, продукты полимеризации
фталевого ангидрида с глицерином или пентаэритритом.
ПОВЕРХНОСТНО-АКТИВНЫЕ И МОЮЩИЕ
ВЕЩЕСТВА
Поверхностно-активными веществами называются соединения, ад-
сорбирующиеся на поверхности раздела фаз и способные снижать
поверхностное натяжение. В состав молекулы этих соединений входят
две группы: гидрофобная и гидрофильная. У поверхностно-активных
веществ липидной природы гидрофобная группа обычно представлена
углеводородной цепочкой, состоящей из 10—20 атомов углерода,
гидрофильная — группами СООН, COONa, COO”, —SO3Na, —NH3.
В водных системах молекулы этих веществ располагаются в поверх-
ностном слое таким образом, что гидрофильная группа оказывается
обращенной к воде, а гидрофобная выталкивается из нее (см. рис. 39).
Все поверхностно-активные вещества могут быть разделены на два
класса: ионогенные соединения, которые при растворении в воде
диссоциируют на ионы, и неионогенные, не диссоциирующие на ионы
в водных растворах. В зависимости от характера продуктов, обра-
зующихся при диссоциации ионогенных соединений, различают анио-
415
ноактивные и катионоактивные. Большинство моющих средств отно-
сится к анионоактивным.
Анионоактивные — диссоциируют на длинноцепочечный анион и
катион (Na+, К+). К ним относятся мыла, алкилсульфаты, алкил-
сульфонаты, алкиларилсульфаты:
R—COONa R—СОСГ + Na+
соль жирной анион катион
кислоты
Катионоактивные образуют при диссоциации длинноцепочечный ка-
тион и анион:
[СбН5—CH2N (С2Н5)3]+СГ [C6H5CH2N (С2Н5)3]+ + сг
триэтилбензиламмонийхлорид катион анион
К неионогенным поверхностно-активным веществам относятся эфи-
ры пропиленгликоля, моно- и диглицериды.
Анионоактивные вещества. Мыла. Мылами называются соли вы-
сокомолекулярных карбоновых кислот, растворы которых обладают
моющим действием. Практическое значение имеют натриевые и калие-
вые соли кислот с 10—20 атомами углерода в молекуле. Для получе-
ния мыла исходный жир или смесь жирных кислот, в том числе полу-
ченных синтетическим путем, обрабатывают при кипячении раствором
едкого натра (варка мыла). При этом происходит гидролиз глицеридов
и нейтрализация образовавшихся жирных кислот. Полученный про-
дукт получил название мыльного клея. После его охлаждения и раз-
делки получают клеевое мыло. Для получения ядрового мыла к
мыльному клею добавляют концентрированный раствор поваренной
соли (высаливание); образуется два слоя, верхний — ядро после от-
деления, промывки и охлаждения является высококачественным мы-
лом. Свойства мыл зависят от состава жирных кислот; соли ненасы-
щенных кислот обладают большей растворимостью, чем насыщенных.
Жидкими мылами являются калиевые соли жирных кислот.
Моющее действие мыла (способность удалять находящиеся на по-
верхности загрязнения) связана с его поверхностно-активными свой-
ствами. Водные растворы мыл — коллоидные системы, содержащие
наряду с мылами жирные кислоты (R — СООН), которые образуются
в результате гидролиза, едкий натр, а также кислые мыла и ионы
RCOO" и Na+. В водных растворах мыла создают эмульсию, причем
углеводородные цепи обращены внутрь шариков эмульсии, а гидро-
фильные группы расположены снаружи. К отрицательно заряженной
поверхности таких образований притягиваются молекулы воды и мы-
ла. Образующиеся частицы называются мицеллами и имеют сложный
состав. При стирке белья или мытье рук молекулы мыла окружают
частицы «грязи», образуя вокруг нее оболочку. При определенных
условиях частицы грязи отрываются от поверхности и переходят в
водно-мыльную среду (солюбилизация), кроме этого, они могут под
действием мыла распадаться на более мелкие частицы и, прилипая к
пузырькам воздуха, переходить в пенную фазу (флотация). Все это
приводит к очистке поверхности от грязи.
Использование мыла в быту (стирка), технике (мытье тканей, су-
416
дов) снижается; они заменяются синтетическими моющими средствами
(детергентами). Это связано с их большей моющей способностью, по
сравнению с мылами (в 10—30 раз), низкой стоимостью, доступностью
сырья для изготовления и рядом их преимуществ над мылами. Напри-
мер, в жесткой воде, вследствие образования нерастворимых солей
магния, кальция и железа, моющее действие мыл снижается:
2R—COONa + Mg+ (R—COO)2Mg4 + 2Na+
Кроме этого, они в силу своих щелочных свойств оказывают нежела-
тельное действие на ряд тканей. Нужно также помнить, что для произ-
водства мыла используются пищевые жиры, что приводит к уменьше-
нию пищевых ресурсов.
Алкилсульфаты — соли эфиров серной кислоты, получаются по
следующей схеме:
парафины жирные кислоты -> жирные спирты -> соли алкилсульфатов
Они не образуют нерастворимых соединений с кальцием и магнием
и обладают прекрасной моющей способностью.
Алкилсульфонаты — соли жирных сульфокислот — получаются
сульфохлорированием алканов. Не теряют моющего действия в жест-
кой воде.
Неионогенные поверхностно-активные вещества. Представителем
этой группы являются эфиры полиэтиленгликоля
RCOO (СН2—СН2О)/гН R—COO (СН2—СН2О)Д OCRi
* моноэфир диэфир
моноглицериды Н—О—СН2—СНОН—СН2—OCOR и другие продук-
ты. Они широко применяются в различных отраслях пищевой про-
мышленности в качестве эмульгаторов и веществ, улучшающих ка-
чество пищевых продуктов.
Белковые вещества
Белками, или белковыми веществами (протеинами), называются
высокомолекулярные органические соединения, молекулы которых
построены из остатков а-аминокислот. Количество последних может
колебаться очень сильно и достигать иногда нескольких тысяч.
Белки играют исключительную роль в жизни живого организма,
выполняя весьма разнообразные функции. Из них состоит основная
масса протоплазмы клеток, они выполняют каталитические, строитель-
ные, энергетические, обменные, защитные и многие другие функции.
«Повсюду, где мы встречаем жизнь, мы находим, что она связана с
каким-либо белковым телом, и повсюду, где мы встречаем какое-либо
белковое тело, не находящееся в процессе разложения, мы без исклю-
чения встречаем и явления жизни», — писал Ф. Энгельс.* По сущест-
* Энгельс Ф. Анти-Дюринг. —Маркс K.f Энгельс Ф. Соч. 2-е изд.,
М., 1961, т. 20, с. 83.
417
ву вся деятельность организма (развитие, движение, распад и многое
другое) связана с белковыми веществами.
Все это дало основание Ф. Энгельсу еще в прошлом веке сделать
свое знаменитое заключение: «Жизнь есть способ существования
белковых тел...»*.
^Белками являются и многие важнейшие физиологически активные
соединения: ферменты, некоторые гормоны и антибиотики и другие
вещества.
Белки являются необходимой составной частью пищевого рацио-
на человека и животных. Пищевая ценность белков в первую очередь
определяется их аминокислотным составом. Несмотря на разнообра-
зие в строении и функциях, элементный состав белковых веществ ко-
леблется незначительно (в % на сухую массу): 50—55 С; 6,5—7,3 Н;
21,5—23,5 О; 15—17,5 N; 0—2,5 S. Некоторые белки содержат в не-
больших количествах фосфор, селен, металлы (железо, цинк, медь).
Количество белковых веществ в животных и растительных орга-
низмах неодинаково. Обычно в животных организмах белков больше,
чем в растениях. Неодинаковое количество их присутствует в отдель-
ных организмах и тканях. Так, содержание белков (в %) в мышцах
18—23, мозгу — 8—9, сердце 16—18, крови 7—8,5. В семенах расте-
ний находится от 5—20% белковых веществ. Особенно богаты белком
семена бобовых и масличных культур (до 20—35%). Различно содер-
жание белковых веществ и в пищевых продуктах. Главным источником
белков для человека являются (в %) мясо животных (до 20), рыба (до
18), молоко (до 3), сыр (до 22,5), пшеничный хлеб (до 8),*изделия из
круп (10).
Строение белков. Согласно общепринятой теории молекула белка
состоит из остатков а-аминокислот, связанных между собой пептидны-
ми связями:
Н О Н Н О
Впервые мысль о пептидных связях была высказана выдающимся
русским биохимиком А. Я. Данилевским (1838—1923). Состав и по-
следовательность расположения аминокислотных остатков в моле-
куле белка^огут быть различными, вследствие чего разнообразие
белков поистине безгранично.
Пептидные связи являются не единственными видами связей в бел-
ках. Отдельные пептидные цепи или их участки могут быть связаны
между собой дисульфидными (—S—S—), солевыми и водородными
связями. Солевые связи образуются между свободными аминогруппами
(например, N-концевая аминогруппа, расположенная на одном кон-
це полипептидной цепи или е-аминогруппы лизина) и свободными кис-
* Энгельс Ф. Анти-Дюринг.—Маркс К., Энгельс Ф. Соч. 2-е изд., т. 20,
с. 82.
418
лотными группами (С — концевая кар-
боксильная группа полипептидной це-
пи, свободные карбоксильные группы
двухосновных » аминокислот)Водород-
ные связи в белках могут ^возникать
между атомом кислорода карбонильной
группы и атомом водорода амидной
группы, а также за счет водорода ок-
сигрупп оксиаминокислот и кислорода
пептидных групп:
#В молекуле белка существуют также
и некоторые другие виды взаимодейст-
вия.
Если определение аминокислотного
состава белка может быть проведено от-
носительно быстро, то выяснение пос-
ледовательности соединения аминокис-
лотных .остатков — задача исключитель-
но сложная. ^Выдающимся достижением
в этой области было установление
структуры и синтез гормона инсулина
(Сенглер, 1963), состоящего из остат-
ков 51 аминокислоты и имеющего моле-
кулярную массу 5733 (рис. 40). Сочета-
нием из двух и трех букв на рисунке ус-
ловно обозначаются остатки аминокис-
лот.
w В настоящее время известны состав
и последовательность соединения ами-
нокислотных остатков пептидных цепей
ряда белков.
Различают первичную, вторичную,
третичную и четвертичную структуры
белковых молекул. Все белки, независи-
Рис. 40 Строение молекулы инсулина (зачерненным показаны дисульфидные связи)
Рис. 41. Вторичная структура белковой мо-
лекулы
мо от того, к какой группе
они относятся и какие функ-
ции выполняют, построены из
* относительно небольшого на-
бора (обычно 20) аминокис-
лот. Эти структурные компо-
ненты или, как их иногда на-
зывают, «строительные блоки»
расположены в различной, но
всегда строго определенной
для данного вида белка пос-
ледовательности. Под первич-
ной структурой белка пони-
мают вид, число и последова-
тельность аминокислотных ос-
татков в полипептидной цепи.
Поли пептидные цепи име-
ют спиралевидную или зиг-
загообразную конфигурацию,
которая поддерживается с
помощью водородных связей,
она называется вторичной
структурой белковой молекулы (рис. 41). Полипептидная спирале-
видная цепочка может быть по-разному расположена в пространст-
ве. Это пространственное расположение, или как иногда говорят
«упаковка», получило название третичной
структуры (рис. 42).
♦Следует отметить, что «свертывание» по-
липептидной цепи с образованием третич-
ной структуры происходит не хаотично, а
строго определенно, и любые изменения в
третичной структуре влияют на биологиче-
скую активность белков.
В последние годы сформулировано
представление о так называемой четвер-
тичной структуре белковой молекулы. В от-
дельных случаях молекулы состоят из не-
скольких полипептидных цепочек, связан-
Рис. 42. Молекулярная мо- ных межДУ собой водородными, ионными И
дель белка миоглобина некоторыми другими нековалентными ви-
дами связей. В определенных условиях
они способны диссоциировать на более
мелкие «субмолекулы», которые могут опять соединяться в перво-
начальную молекулу. Такое объединение нескольких частиц с тре-
тичной структурой получило название четвертичной структуры. Од-
нако далеко не у всех белков мы встречаем все четыре уровня струк*
турной организации.
Выделение и свойства белков. Белки экстрагируют из белоксо^
держащего материала водой, растворами солей, щелочей, кислот, во-
420
доспиртовыми растворами. Полученный таким образом продукт обыч-
но содержит значительное количество примесей. «Для дальнейшего
выделения и очистки белка раствор обрабатывают солями (высалива-
ние), насыщают спиртом или ацетоном, нейтрализуют. При этом выде-
ляется соответствующая фракция белка. Выделить белок в неизменном
состоянии очень трудно, для этого необходимо соблюдать целый ряд
условий: «низкая температура, определенная реакция среды и т. д.
Выделенные и очищенные белки имеют в большинстве случаев
вид белых порошков или сохраняют природную форму (например,
белки шерсти). Установлено, что белки по форме молекул могут быть
разделены на две группы: фибриллярные, или нитевидные и глобуляр-
ные, или шаровидные. Представителем первой группы является фибро-
ин шелка (он обычно сохраняет после выделения природную форму).
Ко второй группе относится большинство белков живых организ-
мов.
Многие белки растворимы в воде, щелочах, кислотах, но нераство-
римы в органических растворителях. В растворах белки представляют
собой высокомолекулярные коллоиды и обладают многими свойствами
коллоидных растворов.
Белки относятся к амфотерным электролитам, так как они содер-
жат аминные и карбоксильные группы:ев щелочном растворе прояв-
ляют кислотные свойства, в кислом — основные. Поэтому при пропус-
кании электрического тока через щелочной раствор белка его моле-
кулы будут двигаться к аноду, а при пропускании через кислый раст-
вор — к катоду. Однако при определенной для каждого белка реак-
ции среды количество положительных и отрицательных зарядов в
молекуле белка будет одинаковым, и белковые молекулы не будут
передвигаться ни к катоду, ни к аноду. Такое значение pH среды на-
зывается изоэлектрической точкой. Это одна из основных констант
белка.
^При действии на белок сильных кислот и щелочей, высоких тем-
ператур и некоторых других воздействиях они денатурируют. Денату-
рация белка — хорошо знакомый процесс, который можно, например,
наблюдать при варке куриного яйца. При денатурации нарушается
структура белка (вторичная, третичная, четвертичная), изменяются
его физико-химические свойства.
уПод действием кислот, щелочей или протеолитических ферментов
бёлковые вещества распадаются с образованием в конечном итоге сме-
си а-аминокислот. Гидролиз протекает ступенчато, с образованием
все более и более простых продуктов:
Белок альбумозы (пептоны) -> полипептиды -> дипептиды -> а-аминокислоты
Цветные реакции белков. Для белков характерны
некоторые цветные реакции, связанные с наличием в молекуле опреде-
ленных группировок и аминокислотных остатков.
Биуретовая реакция. Появление фиолетового окрашивания при об-
работке белка концентрированным раствором щелочи и насыщенным
раствором CuSCk. Связана с наличием в молекуле пептидных
связей.
421
Ксантопротеиновая реакция — появление желтой окраски в ре-
зультате действия на белки концентрированной азотной кислоты.
Реакция связана с наличием в белке ароматических колец.
Миллонова реакция — появление вишнево-красного окрашивания
при действии на белок Миллонова реактива (раствор нитрата ртути в
азотистой кислоте). Реакция объясняется присутствием в белке фе-
нольных группировок. *
Реакция на серу (сульфгидрильная реакция) — выпадение черного
осадка сульфида свинца при нагревании белка с раствором плюмби-
та (связана с наличием в белке сульфгидрильных групп).
• Реакция Адамкевича—появление фиолетового окрашивания при
добавлении к белку глиоксалевой и концентрированной серной кис-
лот. Связана с наличием индольных группировок.
Классификация белков. Белки подразделяются на протеины
(простые белки), состоящие только из остатков аминокислот, и слож-
ные белки (протеиды), которые при гидролизе дают аминокислоты и
вещества небелковой природы (фосфорную кислоту, углеводы, липиды,
гетероциклические соединения, нуклеиновые кислоты). Протеины
составляют основу запасных веществ семян, а протеиды — основу
ядер, протоплазмы клеток. Протеины и протеиды разделяются на ряд
подгрупп.
Протеины. Альбумины — белки, имеющие сравнительно не-
большую молекулярную массу, хорошо растворяются в воде. Из водя-
ных растворов высаливаются насыщенным раствором сернокислого
аммония, при нагревании свертываются (денатурируют). Белок яй-
ца — типичный представитель альбуминов. Многие из них получены
в кристаллическом состоянии.
Глобулины — белки, нерастворимые в чистой воде, но раствори-
мые в теплом 10%-ном NaCl. Чистый глобулин извлекают,^разбавляя
солевой раствор большим количеством воды. Глобулины — самые
распространенные белки, входят в состав мышечных волокон, крови,
молока, яиц, семян.
^Проламины незначительно растворимы в воде. Растворяются в
60—80 %-ном водном этиловом спирте. При гидролизе проламинов об-
разуется в большом количестве аминокислота пролин. Характерны
для семян злаков.
г Глютелины— растворимы только в 0,2%-ной щелочи. Найдены в
семенах пшеницы, риса, кукурузы.
к Протамины — обнаружены только в молоках рыб. Являются
сильными основаниями. Совершенно не содержат серы.
^Протеиноиды — нерастворимые белки, входящие в состав шел-
ка; волос, рогов, ногтей, копыт и сухожилий. Имеют нитевидную
(фибриллярную) форму молекул. Содержат серу.
Фосфопротеины — содержат фосфорную кислоту. Играют большую
роль в питании молодого ор^низма. ПримерОхМ их является казеин —
белок молока. *
х/7 р о т е и д ы. Сложные белки делятся на группы в зависимости
ог состава их небелковой части, которая называется простатической
группой. Липопротеиды — гидролизуются на простой белок и лини-
ды. Липопротеиды содержатся в большом количестве в составе зерен
хлорофилла и протоплазмы клеток.
Гликопротеиды — гидролизуются на простые белки и высокомоле-
кулярные углеводы. Не растворяются в воде, но растворяются в раз-
бавленных щелочах. Содержатся в различных слизистых выделениях
животных.
Хромопротеиды — гидролизуются на простые белки и красящие
вещества. Например, гемоглобин крови распадается на белок глобин и
сложное азотистое основание, содержащее железо.
Нуклеопротеиды — гидролизуются на простые белки (обычно про-
тамины, или гистоны) и нуклеиновые кислоты.
Использование белков. Исключительная роль белковых веществ
в животных организмах, ряд их ценных технических свойств превра-
тили белок в незаменимый продукт питания и важный вид сырья для
многих отраслей промышленности. Все возрастающую роль играет
производство^иологически активных белковых препаратов: ферментов,
гормонов, антисывороток, кровезаменителей.
Белок является самой важной и в то же время наиболее дефицит-
ной частью пищи. Суточная потребность в белке взрослого человека—
100—150 г. Основное качество пищевого белка — его способность
снабжать организм необходимым количеством аминокислот. По ами-
нокислотному составу пищевые белки делятся на две группы: полно-
ценные (содержащие все необходимые для организма животных и
человека аминокислоты) и неполноценные, в последних часть незаме-
нимых аминокислот или полностью отсутствует, или присутствует в
незначительном количестве.уОтсюда следует, что при организации
правильного питания нужно учитывать не только количество, но и ка-
чество белка^Более тур^ необходимое для человека количество белка
во многом завдсит от его1 качества. Главными и наиболее ценными ис-
точниками белка животного происхождения являются: молоко и мо-
лочные продукты (творог, сыр), яйца, рыба, мясо и получаемые из
них продукты. Источники белков растительного происхождения —
зерновые продукты (мука, крупа, макароны, бобовые). Овощи относи-
тельно бедны белком, но их следует учитывать как дополнительный
источник белков; кроме того, они способствуют лучшей усвояемости
белков.
, Несмотря на обилие и разнообразие источников пищевого белка,
население ряда стран испытывает острый белковый голод. Один из
путей устранения белкового дефицита — поиски новых их источни-
ков. В настоящее время проводятся исследования по более эффектив-
ному использованию дешевого растительного белка, применяемого
для кормления скота; получению белков из микробиологического
сырья. Последний путь особенно перспективный. Дешевые и доступные
сырьевые источники для производства кормовых дрожжей (например,
использование для их выращивания парафинов нефти), их высокая
продуктивность — за одни сутки 1 т дрожжей может дать до 100 т
потомства, высокое содержание и качество белка в биомассе привле-
кают к его микробиологическому синтезу пристальное внимание ис-
следователей.
423
Получение пищевого белка — одна из основных проблем в созда-
нии искусственной и синтетической пищи. «Одна из извечных и корен-
ных проблем человечества — обеспечение пищей. Я полагаю, — гово-
рил академик А. Н. Несмеянов, — что теперь химия созрела до такой
степени, что должна всключиться в помощь сельскому хозяйству в
коренном вопросе — производстве самой дорогой и дефицитной пищи —•
белковой, включиться непосредственно, а не только на вспомога-
тельных фронтах производства удобрений, инсектицидов, фунгици-
дов, гербицидов, консервантов и т. д.»*. Многое в этом направлении
уже делается.
Изопреноиды
Изопреноидами называются широко распространенные в природе
органические соединения, молекулы которых включают различное
число изопреновых группировок С5Н8 и имеют полиизопреновый ске-
лет. Общая формула углеводородов этой группы (С5Н8)П. К ним отно-
сятся такие важные соединения, как терпены, каротиноды, природ-
ный каучук. В группу изопреноидов наряду с углеводородами (С5Н8)П
входят их разнообразные кислородсодержащие производные с окси,
карбонильной, карбоксильной и другими функциональными группами.
Изопреноиды делят на группы по числу изопреновых остатков
(п = 2, 3, 4 и т. д.) и строению углеродного скелета (ациклические,
моно-, ди- и полициклические):
терпены СюН1б ,,(С5Н8)2 /г=2
сесквитерпены С15Н24 (С5Н8)3 /г=3
дитерпены С20Щ2 (С5Н8)4 /2=4
каротиноиды ^40^64 (CSH8)8 /2=8
политерпены — (С5н8)л —
Первые три группы углеводородов и их многочисленные кислород-
содержащие производные часто объединяются под общим названием—
терпеноиды.
ТЕРПЕНОИДЫ
Терпеноиды широко распространены в природе. Они входят в
состав эфирных масел, живицы, липидов животных тканей (сквален).
Монотерпены и их кислородсодержащие
производные
Из довольно многочисленных монотерпенов особое значение
имеют алифатические терпены (молекула их состоит из прямой угле-
род-углеродной цепи и содержит три двойные связи (I): моноцикличес-
* Вестник АН СССР, 1969, № 1, с. 27.
424
кие терпены (в молекуле содержится кольцо из атомов углерода и две
двойные связи, II, III) и бициклические терпены (в молекуле имеется
два кольца и двойная связь, IV, V). При написании терпеноидов ши-
роко применяются условные формулы (III, V):
с н3—с=с н—с н 2—с н2—с—с н=с н2
сн3 сн2
мирцен (I)
Алифатические терпены и их кислородсодер-
жащие производные
По своим физическим свойствам алифатические терпеноиды —
жидкости, легче воды, нерастворимые в ней и хорошо растворимые
во многих органических растворителях. Обладают приятным запахом.
Их химическое поведение определяется наличием в молекуле соот-
ветствующих функциональных групп. Наибольшее значение из них
имеют следующие соединения.
Мирцен СН3—С=СН—СН2—СН2—С—СН=СН2. Содержится в масле
СН3 СН2
хмеля.
Гераниол СН3—-С=СН—СН2—СН2—С=СН—СН2ОН. Является одним
СН3 СН3
из основных компонентов розового масла.
Цитраль СН3—С=СН—СН2—СН2—С=СН—С< . Одно из важ-
I I \
сн3 сн3 н
нейших соединений в группе алифатических терпеноидов. Рас-
пространен в эфирных маслах.
ОН
Линалоол СН3—С=СН—СН2—СН2—С—СН=СН2. Содержится в ко-
СН3 СН3
риандровом и померанцевом маслах. Обладает запахом ландыша.
425
Моноциклические терпены и их кислородсодер-
жащие производные
Большинство моноциклических терпенов можно рассматривать
как производные ментана, содержащие в молекуле две двойные свя-
зи — ментадиены:
сн
H2cf’>сн2
н2с<4>сн2
сн
I
сщ—сн—сн3
10 8 9
ментан
(1-метил-4- изопропилциклогексан)
Особое значение имеют четыре представителя этой группы соединений
лимонен, а-терпинеол, ментол и ментан:
Лимонен содержит один асимметрический атом углерода и может
существовать в двух оптически деятельных формах (£> и L) и оптиче-
ски недеятельной рацемической форме. Находится в масле цитрусо-
вых, укропа, тмина.
Ментол принадлежит к числу важнейших кислородсодержащих
производных моноциклических терпенов. Кристаллическое вещество
с приятным запахом. Основной компонент перечной мяты. Получается
гидрированием тимола:
зн
тимол
ментол
426
Применяется в'медицине как дезинфицирующее средство, в качест-
ве компонента зубной пасты.
а-Терпинеол — жидкость с запахом сирени. Используется в пар-
фюмерии.
Эфирные масла
Ациклические и моноциклические терпены в значительном ко-
личестве присутствуют в эфирных маслах (анисовом, розовом, гвоз-
дичном, тминном и др.). Эфирными маслами называют жидкие, аро-
матические, легко летучие смеси органических веществ вырабатывае-
мые растениями. Они включают до 1000 компонентов. Эфирные масла
хорошо растворимы в органических растворителях и нерастворимы в
воде. На воздухе под действием света и кислорода они осмоляются,
изменяют свой цвет и запах. В составе эфирных масел хвойных и цит-
русовых растений в большом количестве присутствуют терпеновые
углеводороды, в других — спирты и оксосоединения. Выделяют эфир-
ные масла из эфиромасличных растений отгонкой с водяным паром или
экстракцией органическими растворителями. Они широко применя-
ются в парфюмерной, косметической, медицинской и пищевой про-
мышленностях, а также являются сырьем для получения отдельных
органических соединений.
В СССР возделывается около 20 эфироносов, особое значение име-
ют кориандр, анис, тмин, мята перечная, лаванда, герань, роза.
Лимонное масло. Получают прессованием из корок лимона. Из
1000 штук лимонов получают до 500 г масла. Его главный компо-
нент — лимонен.
Мятное масло. Получают из мяты. Основной компонент — ментол.
Анисовое масло. Получают из растений семейства зонтичных. Ос-
новной компонент — эфир анетол:
ОСН3
СН=СН—СНд
анетол
Кориандровое масло — содержится в семенах кориандра. Основной
компонент — линалоол.
Бициклические терпены и их кислородсодержа-
щие производные
Среди бициклических производных особое значение имеют произ-
водные трех групп углеводородов: карана, пинана, камфана:
427
карай
пинан
камфан
Наиболее распространенным из этой группы соединений является
а-пинен, главная составная часть скипидара:
а-пинен
а-Пинен— жидкость с /кип = 156° С, запахом хвои. Получается
из скипидара. Применяется для получения камфоры, инсектицидов,
в качестве растворителя.
Скипидар извлекают из живицы, которая вытекает из надрезов на
стволах сосны. Живица — раствор канифоли (смесь смоляных кислот
с общей формулой Ci9H29COOH) в скипидаре (терпентиновое масло),
который состоит из смеси различных терпенов. Скипидар получают
из живицы отгонкой с водяным паром. Это бесцветная жидкость, легче
воды, смешивается с большинством органических растворителей.
Применяется в текстильной промышленности, лакокрасочной для
приготовления лаков, в парфюмерии, для синтеза камфоры:
камфора
Камфора — прозрачная, бесцветная, вязкая кристаллическая мас-
са, /пл = 175° С. Получается из камфорного дерева (Япония, Тай-
вань) отгонкой из измельченной древесины с водяным паром или син-
тетическим путем. Синтез камфоры проводят по следующей схеме:
428
СН3СООН_
(H2SO4)*
омыление
—СН3СООН
а-пинен камфен борнилацетат
295-310°С
v2o5
борнеол
камфора
Камфора применяется в качестве пластификатора при получении цел-
лулоида, в медицине как стимулятор сердечной деятельности.
Дитерпены и их производные
Из представителей этой группы изопреноидов важным является
фитол— непредельный спирт, составной компонент хлорофилла и
витамина К:
СН3—СН— (СН2)з—СН—(СН2)з—СН—(СН^—С=СН—СН2ОН
СН3 СН3 СН3 СН3
фитол
Каротиноиды
К Этой группе изопреноидов относятся углеводороды (С5Н8)8 и
их кислородсодержащие производные. Каротиноиды — красящие ве-
щества от желтого до ярко-красного цвета. Они широко распростра-
нены в природе; вместе с хлорофиллом содержатся не только в зеленых
частях растений, но и в семенах. В организм животных они попадают
с пищей. Природные каротиноиды можно рассматривать как произ-
водные ликопина, содержащегося в плодах томата, шиповника, яго-
дах, фруктах:
При замыкании одного или двух колец на концах молекулы лико-
пина образуются a-, [J- и у-каротины, отличающиеся количеством
и строением боковых колец:
429
СН3 СНз
3-каротин
Известно до 100 каротиноидов. Их можно разделить на углеводо-
роды, называемые каротинами, и их кислородсодержащие производ-
ные (спирты, альдегиды, эпоксиды, кислоты), часть которых объеди-
няется под общим названием «ксантофиллы».
Каротиноиды хорошо растворяются в большинстве органических
растворителей и не растворяются в воде. Некоторые каротиноиды яв-
ляются провитаминами А. Из одной молекулы p-каротина в животном
организме образуется две молекулы витамина А:
\/ I I СН2ОН
1 J
Витамин
420
Раздел седьмой
Выделение, очистка и
идентификация органических
соединений
ВЫДЕЛЕНИЕ И ОЧИСТКА ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Изучение свойств органических соединений не-
возможно без их выделения и очистки. В каждом конкретном случае
химик-органик решает эти задачи с учетом индивидуальных особен-
ностей исследуемых смесей. Так, в случае твердых семей используют
такие методы, которые основаны на различной растворимости органи-
ческих соединений, а также способности к перегонке с водяным паром
или возгонке. Жидкие смеси разделяют, как правило, с помощью пере-
гонки. Некоторые способы разделения применимы для любых сме-
сей, например экстракция. Но особенно универсальны методы хромато-
графии, позволяющие разделять смеси газов, жидкостей и твердых ве-
ществ. Методы хроматографии основаны на распределении компонен-
тов смесей между двумя фазами — неподвижной и подвижной (элюент),
протекающей через неподвижную.
Различают следующие виды хроматографии: в зависимости от сре-
ды— газовую, жидкостную или газожидкостную; от механизма раз-
деления — адсорбционную (молекулярную), ионообменную, оса-
дочную или распределительную и, наконец, от формы проведения
процесса — колоночную, тонкослойную, бумажную или капилляр-
ную.
В последние годы методы хроматографии получили широкое рас-
пространение благодаря своей универсальности и создания автомати-
ческих приборов — хроматографов.
Идентификация (опознание) неизвестного соединения. Иденти-
фикация неизвестного соединения выполняется по следующей схеме:
1) предварительные испытания; 2) определение ряда физических
констант: температур плавления, кипения, плотности, показателя пре-
ломления, молекулярной рефракции, оптической активности, моле-
кулярной массы и др.; 3) определение качественного элементного со-
става; 4) определение растворимости; 5) проведение реакций на функ-
циональные группы; 6) получение производных (одного или несколь-
ких) для окончательного установления структуры соединений.
Предварительные испытания. Отмечается агрегатное состояние,
цвет, запах и проводится прокаливание пробы. Обращают внимание,
является вещество твердым или жидким; присутствие окраски может
431
свидетельствовать о наличии в молекуле хромофорных групп; наличие
запаха может указать на некоторые определенные вещества. Харак-
терным «терпеновым» запахом обладают углеводороды терпенового ря-
да (камфен, пинен), а также циклогексан, пинаколин, трет-бут анол;
очень резкий запах у муравьиной и уксусной кислот; дурманяще-
сладковатый запах у галогенпроизводных; фенолы имеют запах «кар-
болки»; ароматические нитросоединения — запах горького миндаля;
неприятно-сладкий запах у изонитрилов; меркаптаны по запаху на-
поминают сероводород и т. п.
Прокаливание пробы. Навеску вещества (около 0,1 г) помещают
на крышку фарфорового тигля и подносят к краю пламени для опре-
деления воспламенимости. Затем крышку осторожно нагревают и,
наконец, сильно прокаливают. Отмечают при этом воспламеняемость,
характер пламени, а также плавления (для твердых веществ), запах
образующихся газов или паров, остаток после прокаливания.
При оценке полученных результатов следует иметь в виду, что
многие жидкие вещества при горении дают характерное пламя. Если
вещество воспламеняется, то в дальнейшем следует принимать необ-
ходимые меры предосторожности. В случаях, когда после прокалива-
ния получают минеральный остаток, следует провести качественный
элементный анализ.
Определение физических констант. Температура плавления. Плав-
ление — процесс перехода кристаллического твердого тела в жид-
кость — происходит при постоянной температуре, называемой тем-
пературой плавления. Температура плавления определяется приро-
дой тела и зависит от внешнего давления. Температурные пределы
плавления неизвестного вещества служат ценным указанием на сте-
пень чистоты соединения. Большая часть чистых соединений, имею-
щих кристаллическую структуру, плавится в пределах 0,5°. Если
наблюдаемая температура плавления лежит в больших пределах, то
вещество необходимо перекристаллизовать из соответствующего раст-
ворителя и вновь определить температуру плавления.
Следует иметь в виду, что ряд органических соединений, таких,
как аминокислоты, соли кислот или аминов и т. п., плавятся с раз-
ложением и имеют значительные температурные пределы плавления.
Естественно, что в таких случаях величиной температуры плавления
нельзя охарактеризовать степень чистоты органического соединения.
Температура кипения. Кипение — переход жидкости в пар не
только путем испарения со свободной поверхности, но и во всем объ-
еме вследствие непрерывного образования и роста в жидкой фазе
пузырьков насыщенного пара, внутри которых происходит испаре-
ние жидкости. При постоянном внешнем давлении кипение происхо-
дит при вполне определенной температуре, называемой температурой
кипения. Температура кипения — важнейшая физико-химическая
характеристика вещества. Она возрастает с увеличением внешнего
давления и достигает наивысшего значения в критической точке Тк»
которая определяет наивысшую возможную температуру кипения при
критическом давлении рк.
Температура кипения — важнейшая характеристика чистоты ор-
432
ганической жидкости. Обычно определение температуры кипения про-
водят в ходе очистки или разделения веществ методом дистилляции.
Существует определенная связь между строением соединения и его
температурой кипения: 1) температура кипения членов любого гомо-
логического ряда повышается в восходящем направлении ряда, но вли-
яние каждой последующей СН2-группы меньше; 2) при введении в мо-
лекулу заместителя, способствующего ассоциации жидкости, наб-
людается весьма заметное повышение температуры кипения; 3) на
температуру кипения оказывают влияние также разветвление цепи и
положение функциональной группы: чем более разветвлена углеводо-
родная цепь, тем ниже температура кипения (поскольку меньше ас-
социация).
Плотность, Плотность р — физическая величина, определяемая
для однородного вещества его массой в единице объема (в кг/м3). Плот-
ность, как правило, уменьшается с ростом температуры (вследствие
теплового расширения тел) и незначительно увеличивается с повыше-
нием давления. При агрегатных превращениях вещества плотность
изменяется скачком, причем при переходе из жидкого состояния в
твердое она обычно растет.
Поскольку плотность зависит от состава и строения вещества,
то ее определение весьма полезно как при оценке чистоты органиче-
ского соединения, так и при качественной идентификации вещества.
Простое наблюдение — является жидкость легче или тяжелее
воды — уже дает некоторое представление о структуре этого соеди-
нения. Как правило, соединения, содержащие несколько функцио-
нальных полярных групп, способствующих ассоциации жидкости,
имеют плотность большую, чем вода.
Коэффициент преломления. Коэффициент преломления света п —
постоянная величина для данного вещества, равная отношению сину-
сов угла падения а света на поверхность раздела двух сред и угла пре-
ломления р света:
п = sina/sin ₽.
Величина п зависит от длины волны света и температуры, поэтому ее
измерение проводят в монохроматическом свете при постоянной тем-
пературе. Определение коэффициента преломления жидкости поз-
воляет получить быструю информацию относительно строения химиче-
ского соединения или доказательство чистоты соединения на послед-
ней стадии его идентификации.
Коэффициент преломления находят с помощью специальных при-
боров — рефрактометров.
Кроме коэффициента преломления в органической химии исполь-
зуются и другие рефрактометрические константы, являющиеся про-
изводными от п, Для установления строения органических соедине-
ний наибольшее значение имеет молекулярная рефракция. Молеку-
лярная рефракция R — характерная рефрактометрическая констан-
та, мало или неизменяющаяся с температурой, давлением, агрегатным
состоянием вещества. Для вычисления R по показателю преломления
света п используют формулу Г. Лорентца и Л. Лоренца:
15—682
433
п2 — 1 М
= n2+2 Т ’
где р — плотность; М — молекулярная масса вещества; R имеет раз-
мерность см3/моль. Молекулярная рефракция непосредственно связана
с поляризуемостью ионов и молекул, т. е. со способностью их элект-
ронных оболочек деформироваться во внешнем электрическом поле
или электрическом поле других ионов и молекул.
В первом приближении молекулярная рефракция — аддитивная
величина, складывающаяся из атомных рефракций отдельных атомов
или радикалов, а также инкрементов, приписываемых определенным,
структурным элементам — кратным связям, напряженным 3- и 4-член-
ным циклам и т. д. Найденная экспериментально молекулярная реф-
ракция для многих соединений оказалась отличной от рассчитанной
по аддитивной схеме. Положительная разница этих величин, полу-
чившая название экзальтации молекулярной рефракции, часто свя-
зана с наличием в молекуле системы сопряженных связей. Отрица-
тельная разница, именуемая как депрессия молекулярной рефрак-
ции, как правило, обусловлена разветвленностью углеродного ске-
лета молекулы или наличием в цикле гетероатома.
До появления современных физических методов исследования мо-
лекулярная рефракция широко применялась в органической химии
для установления строения органических соединений. Имеются по-
дробные справочные данные величин атомных, структурных и группо-
вых рефракций, а также рефракций связей, что позволяет простыми
средствами разрешать различные структурные вопросы.
Оптическая активность — способность среды вызывать вращение
плоскости проходящего через нее поляризованного излучения (света).
Оптическая активность чистых жидкостей или растворов возникает
в тех случаях, когда молекула соответствующего соединения построе-
на асимметрично. Оптически активными могут быть и кристаллы, если
их структура характеризуется отсутствием симметрии. Вращение
плоскости поляризации называют правым (обозначают знаком +),
если оно происходит по часовой стрелке, и левым (—), если это вра-
щение против движения (для наблюдателя, смотрящего навстречу
хода лучей).
Угол <р поворота плоскости поляризации линейно зависит от тол-
щины b слоя активного вещества (или его раствора) и концентрации
С этого вещества:
Т = [а] дС,
где ср — угол вращения плоскости поляризованного света в градусах;
b — толщина слоя, дм; С — концентрация вещества, г/см3; [а] —
удельная оптическая активность, град/дм-г/см3.
Удельная оптическая активность зависит не только от природы
вещества, но и от агрегатного состояния, температуры, давления, типа
растворителя, длины волны поляризованного света и т. д.
Измерения угла вращения ф выполняют с помощью специальных,
приборов — поляриметров. Принципиальная схема визуального по-
ляриметра приведена на рис. 32.
434
Поляриметрия имеет большое значение в органической и биоло-
гической химии. На основании определения знака и величины вра-
щения плоскости поляризации можно судить о химическом строении
и пространственной конфигурации молекулы. Известно, например,
что в природе синтез белка происходит из левых оптических L-изоме-
ров аминокислот — 19 из 20 жизненно важных аминокислот оптиче-
ски активны. Физиологическое и биологическое действие оптических
изомеров часто совершенно различно. Удивительный факт преиму-
щественной роли одной из форм оптических L-изомеров в биологиче-
ских процессах не находит пока объяснения и может иметь важное
значение для понимания происхождения жизни на Земле.
Молекулярная масса. Определение молекулярной массы основано
на понижении температуры замерзания (криоскопия) или возраста-
нии температуры кипения раствора (эбуллиоскопия) по сравнению с
соответствующей температурой чистого растворителя. В том и в дру-
гом случае молекулярную массу вычисляют по уравнению
1000
Л1 =-------
т2Д/
где К — криоскопическая или эбуллиоскопическая постоянная раст-
ворителя, °C; — масса растворенного вещества, г; т2 — масса раст-
ворителя, г; AZ — изменение температуры замерзания или кипения
растворителя в присутствии исследуемого вещества, °C.
Определение молекулярной массы по формуле (1) возможно для
достаточно разбавленных растворов неэлектролитов. Если же раство-
ренное вещество диссоциирует, необходимо учитывать увеличение
числа молей i образовавшихся в растворе частиц:
* = l+a(n— 1),
(1)
где а — степень диссоциации; п — число частиц, на которые распада-
ется соединение при диссоциации; i — коэффициент Вант-Гоффа,
показывающий, во сколько раз возрастает число молей растворенного
вещества в результате диссоциации.
Для определения молекулярной массы методом криоскопии берут
определенное количество чистого растворителя и измеряют его тем-
пературу замерзания. Затем расплавляют растворитель, вносят в
него навеску исследуемого вещества и вновь измеряют температуру
отверждения, используя тот же термометр или термопару.
Точность определения молекулярной массы зависит от точности из-
мерения разности температур. Например, термометр Бекмана имеет
погрешность измерения Л/ = 0,002°; еще большую точность измере-
ния (на порядок) можно достигнуть, используя термопару.
При эбуллиоскопическом определении молекулярной массы из-
меряют температуру кипения чистого растворителя, вносят в него на-
веску изучаемого вещества и вновь измеряют температуру кипения
раствора с тем же термометром. Точность определения молекулярной
массы методом эбуллиоскопии (ошибка — несколько %) существен-
но ниже, чем методом криоскопии, — сказывается влияние колеба-
15* 435
ний внешнего давления, изменения концентрации раствора в резуль-
тате испарения растворителя и т. д.
Качественный элементный анализ. Обнаружение элементов, вхо-
дящих в состав органического соединения, осуществляют в два этапа:
1) вначале органическое вещество разлагают, в результате чего опре-
деляемый элемент выделяется в виде его неорганического соедине-
ния; 2) проводят качественное определение элемента известными ме-
тодами аналитической химии (неорганический анализ).
Основными методами разложения, применяемыми в элементном
анализе, являются окисление, восстановление или сочетание окис-
ления и восстановления. К наиболее эффективным и универсальным
способам разложения относятся способы, сопровождающиеся полной
деструкцией органического вещества: методы «сухого» и «мокрого» сож-
жения, сплавления с окислителями, пиролиз, восстановление ме-
таллами и др. В частности, если провести восстановление пробы ис-
следуемого вещества металлическим натрием, то содержащиеся в ор-
ганическом соединении элементы (С, Н, О, N, Hal) переходят в раст-
воримую форму NaHal, NaCN, Na2S, NaCNS. Полученную смесь ана-
лизируют методами качественного неорганического анализа.
Определение растворимости. Растворимость твердых и жидких
веществ в жидкостях может изменяться в широких пределах в зави-
симости от природы растворяемого вещества и растворителя. Извест-
но, что неполярные вещества лучше растворяются в неполярных раст-
ворителях, в то время как для полярных веществ более пригодны по-
лярные растворители.
Большое число органических растворителей, вошедших в лабора-
торную практику, позволяет во многих случаях подобрать раствори-
тели с избирательной (селективной) растворяющей способностью.
Известны данные растворимости органических соединений в таких
растворителях, как углеводороды, хлорпроизводные углеводородов»
спирты, простые и сложные эфиры, кетоны, нитросоединения, нитри-
лы, амиды, кислоты и др.
При оценке растворимости используют растворители или раство-
ры, химически взаимодействующие с растворяемым веществом (5 %-
ные растворы NaOH, NaHCO3, НС1, а также растворы концентрирован-
ной H2SO4 и 85%-ной Н3РО4). Определение растворимости органи-
ческого вещества как в органических растворителях, так и в раство-
рах позволяет сделать вывод о полярности и кислотно-основных свой-
ствах исследуемого соединения. При обсуждении растворимости орга-
нических соединений необходимо обратить внимание на поведение
органических соединений по отношению к воде и эфиру.
Растворимость в воде. В воде как полярном растворителе раство-
ряются те вещества, которые имеют полярные свойства. В свою оче-
редь полярные свойства определяются числом и природой функцио-
нальных групп, входящих в молекулу растворяемого вещества. Эти-
леновые, ацетиленовые связи и структуры типа бензола мало влияют
на полярность. Введение атомов галогена также не оказывает замет-
ного влияния на полярность, а увеличение при этом молекулярной
массы приводит к уменьшению растворимости в воде.
436
Наиболее полярными соединениями являются соли, поэтому они,
как правило, растворимы в воде. К веществам, также растворимым
в воде за счет полярных группировок, относят полиолы, сахар, амино-
спирты, оксикарбоновые кислоты, ди- и поликарбоновые кислоты,
амиды низших кислот, алифатические аминокислоты, сульфокислоты.
В пределах гомологических рядов для соединения g одной функ-
циональной группой растворимость в воде ограничивается членом
ряда, содержащим пять атомов углерода. Это происходит потому, что
по мере роста углеводородной неполярной части молекул гомологи-
ческого ряда ее полярная группа остается без изменения — в резуль-
тате уменьшается структурное сходство растворенного вещества и
растворителя.
Соединения с разветвленной углеродной цепью в воде более раст-
воримы, чем соответствующие соединения с нормальной цепью. На-
пример, триметил уксусная кислота растворима в воде, изовалериано-
вая — мало, а н-валериановая— нерастворима. В такой же после-
довательности изменяется растворимость тргт-бутилового спирта,
метилизопропилкарбинола и н-амилового спирта.
Растворимость в эфире. Присутствие в соединении сильно поляр-
ных групп является причиной весьма малой его растворимости в эфи-
ре, поскольку эфир является, по существу, неполярным раствори-
телем, приближаясь по своим свойствам к углеводородам. В эфире
нерастворимы соли аминов и органических кислот и другие электро-
литы, пониженной растворимостью обладают также соединения, име-
ющие более одной полярной функциональной группы. Растворимыми
в эфире и нерастворимыми в воде оказываются углеводороды и их
галогенпроизводные, простые и сложные эфиры, высшие спирты, выс-
шие альдегиды и кетоны, оксимы, средние и высшие карбоновые кис-
лоты, ароматические карбоновые кислоты, ангидриды кислот, лак-
тоны, высшие нитрилы и амиды кислот, фенолы, тиофенолы, высшие
амины, хиноны, азосоединения.
Если молекула содержит полярные и неполярные структурные
элементы, то они оказываются растворимыми как в воде, так и в эфи-
ре. К таким соединениям относятся низшие алифатические спирты,
низшие алифатические альдегиды и кетоны, низшие алифатические
нитрилы, амиды кислот и оксимы, низшие циклические простые эфиры
(тетрагидрофуран, диоксан), низшие и средние карбоновые кислоты,
окси- и кетокислоты, дикарбоновые кислоты, многоатомные амины,
пиридин и его гомологи, аминофенолы.
Нерастворимы в воде и эфире высокомолекулярные соединения
(крахмал, целлюлоза, белки и т. п.), высококонденсированные угле-
водороды, амиды высших кислот, антрахиноны, производные пурина,
некоторые аминокислоты (цистин, тирозин), сульфаниловая кислота,
высшие амины и сульфамиды. Как правило, эти соединения имеют вы-
сокую температуру плавления.
Растворимость в основаниях и кислотах. В растворах едкого натра
и гидрокарбоната натрия растворимы вещества, обладающие свой-
ствами достаточно сильных кислот: карбоновые, сульфоновые и суль-
финовые кислоты, некоторые замещенные фенолы. В растворах едкого
437
натра растворимы более слабые кислоты: фенолы, некоторые енолы,
имиды, первичные алифатические нитросоединения, незамещенные или
монозамещенные по азоту арилсульфамиды, оксимы, тиофенолы, мер-
каптаны.
В растворах хлороводородной кислоты растворимы вещества со
свойствами оснований — алифатические и ароматические амины. Амфо-
терные вещества растворяются как в кислотах, так и в щелочах. К та-
ким веществам относятся: аминокислоты, аминофенолы, аминосульфо-
новые и аминосульфиновые кислоты и др. Растворение в концентри-
рованной серной кислоте часто сопровождается разогреванием, вы-
делением газов и другими признаками протекания химических реак-
ций при растворении: ненасыщенные соединения превращаются в
водорастворимые эфиры серной кислоты; спирты этерифицируются
или дегидратируются; олефины могут полимеризоваться; некоторые
углеводороды сульфируются и т. п. В концентрированной серной кис-
лоте в отличие от сиропообразной фосфорной кислоты, помимо спир-
тов, альдегидов, алициклических кетонов, метилкетонов и сложных
эфиров, растворимы также многие хиноны, простые эфиры и непре-
дельные углеводороды.
К наиболее инертным с точки зрения растворимости относятся не-
насыщенные углеводороды жирного ряда, ароматические углеводороды
и галогенпроизводные этих углеводородов — они нерастворимы ни
в воде, ни в эфире, ни в растворах кислот и оснований.
Испытания на растворимость позволяют в ряде случаев получить
предварительную информацию о природе анализируемого соединения.
Проведение реакций на функциональные группы. Обнаружение
той или иной функциональной группы в органическом соединении
проводят после ряда предварительных испытаний (указывающих на
принадлежность соединения к тем или иным классам органических
соединений). При этом выбирают реактивы, которые обладают наи-
большей специфичностью к отдельным функциональным группам.
Аналитические реакции должны протекать достаточно быстро и сопро-
вождаться такими характерными изменениями, как выпадение осад-
ка, изменение цвета, появление характерного запаха или изменение
растворимости испытуемого соединения. Например, рекомендуют
при обнаружении ненасыщенных соединений реакцию с бромом, со-
провождающуюся его обесцвечиванием. Альдегиды и кетоны можно
обнаружить по образованию осадка динитрофенилгидразонов, обра-
зующихся при взаимодействии альдегидов и кетонов с динитрофенил-
гидразином. Первичные амины можно идентифицировать с помощью
изонитрильной реакции; первичные алифатические амины можно
отличить от ароматических путем диазотирования и азосочетания.
С раствором церийаммонийнитрата спирты образуют в водных раст-
ворах окрашенные комплексы красного цвета, фенолы — коричневый
или зеленовато-коричневый осадок.
Наличие в молекуле нескольких функциональных групп может
затруднить их обнаружение химическими реакциями. Более надеж-
ную информацию о строении вещества дает его инфракрасный спектр,
в котором по характеристическим полосам поглощения окончательно
438
устанавливают присутствие определенных функциональных групп.
Получение производных. Синтез производных представляет собой
заключительную стадию исследования, поскольку предварительные
испытания уже сузили круг возможных соединений до одного-двух
типов.
При выборе производного руководствуются следующими сообра-
жениями: 1) оно должно быть твердым веществом, легко очищаться,
желательный интервал температур плавления 50 4- 250° С; 2) ре-
акция, с помощью которой получают производные, должна давать
хороший выход вещества (производного); 3) свойства производного
(например, /пл) должны заметно отличаться от свойств исходных ве-
ществ (минимальная разность в /пл должна составлять 5°).
В качестве производных для спиртов рекомендуют фенил- и а-
нафтилуретаны; альдегидов и кетонов — фенилгидразоны; кислот,
ангидридов и хлорангидридов кислот — амиды и анилиды; первич-
ных и вторичных аминов — фенилтиомочевины; третичных аминов —
соли пикриновой, п-толуолсульфо- или платинохлорводородной
кислот; ароматических углеводородов—нитропроизводные; фенолов —
а-нафтилуретаны и т. п.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Установление химической структуры вещества может быть быстро
и эффективно выполнено с помощью современных физико-химических
методов анализа, таких, как газовая, жидкостная или тонкослойная
хроматография (для разделения сложных смесей), и спектральных
методов — инфракрасной спектроскопии, спектроскопии видимой и
ультрафиолетовой области спектра, ядерного магнитного резонанса,
масс-спектроскопии и других (для идентификации органических со-
единений).
Некоторые общие положения спектроскопии. Идентификация ор-
ганических соединений спектральными методами основана на взаимо-
действии исследуемого вещества с электромагнитным излучением.
Если вещество поглощает излучение, это приводит к изменению энер-
гии его молекулы:
he
ДЕ = /iv =-= h^c, (2)
А
где Е — изменение энергии, Дж; h — постоянная Планка (6,62 х
X 10-31 Дж/-с); с — скорость света, 2,998 • 108 м/с; v — частота
электромагнитных колебаний, с"1; А, — длина волны, м; со — волно-
вое число, см-1. Чем выше энергия излучения, тем меньше его длина
волны, больше частота и волновое число. Спектр электромагнитных
колебаний охватывает огромный диапазон энергий — более 22 по-
рядков.
Для органической химии очень важны ультрафиолетовая (УФ),
видимая и инфракрасная (ИК) области спектра и область коротких
439
радиоволн, используемых в спектроскопии ядерного магнитного резо-
нанса (ЯМР).
Оптическая спектроскопия. Оптические спектры занимают об-
ласть электромагнитных колебаний от далекого инфракрасного из-
лучения до рентгеновского. Наблюдения оптических спектров осу-
Рис. 43. Схема спектрального Прибора
ществимы с помощью прибора, принципиальная схема которого пред-
ставлена на рис. 43.
Луч от источника излучения /, пройдя через кювету с анализируе-
мым образцом 2, попадает на монохроматор 3. Монохроматор имеет
входную и выходную щели 4 и 4', фокусирующие линзы 5 и 5' и призму
или дифракционную решетку 6. Выйдя из щели 4, монохроматический
луч попадает на приемник излучения 7, который с помощью регистри-
рующего устройства 8 измеряет интенсивность излучения, прошед-
шего через образец.
Электронные спектры. Поглощение света в видимой и УФ-областях
спектра связано с переходом валентных электронов на орбитали воз-
бужденных состояний. В таких переходах могут участвовать а- и л-
электроны одинарных, и, соответственно, кратных связей и п-электро-
ны неподеленных пар гетероатомов. Поскольку электроны имеют
различную энергию, они могут быть возбуждены излучением с раз-
личной длиной волны. Из соотношения (2) следует, что чем меньше
разность энергий между основным и возбужденным состоянием, тем
. при больших длинах волн происхо-
---------------—т—--------а* дит поглощение излучения.
На рис. 44 схематично представ-
;; : лены различные типы переходов элек-
тронов в зависимости от энергети-
*е *о ческих уровней электронных орбита-
। i a г i лей. Из схемы следует, что наиболее
с к с к о легко возбуждаемы n-электроны гете-
* роатомов и л-электроны кратных свя-
| _]____________________п зей, в то время как о-электроны насы-
I щенных связей требуют для возбуж-
----------------------г, дения высоких энергий. Поэтому для
многих соединений электронные пере-
ходы соответствуют дальней (вакуум-
------------------------------------------------------ст ной) УФ-области спектра, в практике
Рис. 44. Различные типы перехо- РеДк0 используемой. В ближней УФ-
дов электронов------------области проявляют себя характеристи-
440
ческие группы и сопряженные системы. Такая особенность УФ-погло-
щения уменьшает информативность метода, но вместе с тем позволяет
определять функциональные группы в молекулах, сложность которых
изменяется в широких пределах.
Помимо положения полосы поглощения на шкале длин волн, дру-
гой важной характеристикой электронного спектра является интен-
сивность поглощения, которая зависит от вероятности электронного
перехода и полярности возбужденного состояния.
Интенсивность поглощения может быть выражена через коэффи-
циент пропускания т, определяемый в виде отношения Ф^/Фо, где
Ф, и Фо — поток монохроматического излучения, прошедшего и,
соответственно, падающего на поглощающую среду. Поглощение света
подчиняется закону Бера—Ламберта—Бугера:
Д= — ]gT = e/C,
где А — оптическая плотность (абсорбционность); b — длина пути
света в образце, см; С — концентрация вещества, моль/л; & — мо-
лярный коэффициент абсорбции, л/моль-см, характеризующий мо-
лярную поглощательную способность вещества. Молярный коэффици-
ент 8 зависит также от длины волны и температуры.
Электронный спектр представляет собой графическую зависимость
интенсивности поглощения т в % или А от длины волны % или час-
тоты V. В качестве меры интенсивности поглощения еще лучше ис-
пользовать величину 8 или ее десятичный логарифм 1g 8.
Полная энергия молекулы квантована и определяется суммой
энергий электронного £эл, колебательного ЕкоЛ и вращательного
Еър состояний. Величина этих энергий убывает в ряду Езл > £кол >
> ^вр-
При переходе электрона из основного состояния Ег в возбужден-
ное £г поглощается квант света, и возникающий при отдельном пере-
ходе спектр должен состоять из одиночной линии. Однако на элект-
ронные уровни Ег и £г накладываются колебательные и вращательные
подуровни. Множество последних, близость их расположения приво-
дят к тому, что отдельные полосы поглощения в электронных спект-
рах сливаются и получается широкая полоса поглощения. Лишь для
простых молекул в газовой фазе можно наблюдать при большом раз-
решении ряд полос, состоящий из большого числа узких линий.
В электронных спектрах проявляют себя ковалентно ненасыщен-
ные группы атомов— так называемые хромофоры. Полосы поглощения
(в области 200—800 нм) хромофоров вызваны, как правило, ил*-
или лл*-переходами. Например, если спектры сняты для растворов
соответствующих соединений в алифатических углеводородах, то кар-
бонильная группа имеет максимум поглощения 280 нм (1g е = 1,3),
нитрогруппа — 270 (1g 8 = 1,3), диазогруппа — 660 (1g е = 1,3),
нитрозогруппа — 350 (1g 8—1,1), группа \c=N— — 240 нм
(1g е = 2,2) и т. п.
Электронные спектры наиболее пригодны для обнаружения орга-
нических соединений, имеющих систему сопряженных кратных связей
или гетероатомы.
441
Метод ИК-спектроскопии. Никакие два соединения не могут дать
одинаковые ИК-спектры, поэтому идентификация органического со-
единения однозначно решается сравнением спектров неизвестного
вещества со спектром стандартного образца. Кроме того, ИК-спектр
содержит так называемые характеристические полосы поглощения,
которые относятся к некоторой, определенной группе атомов (функци-
ональным группам) и которые мало зависят от структуры остальной
части молекулы.
ИК-излучение поглощается органической молекулой и преобра-
зуется в зависимости от частоты излучения в энергию вращения (час-
тота менее 100 см-1) и энергию колебания (частота 10 000 — 100 см-1).
Это поглощение квантовано, поэому вращательный спектр молекул
состоит из дискретных линий, а колебательный — из полос, поскольку
каждое изменение колебательной энергии сопровождается изменениями
многочисленных дискретных состояний вращательной энергии. Для
идентификации органических соединений особенно важны колеба-
тельно-вращательные полосы в области 4000—650 см-1.
ИК-спектр, как правило, представляет собой зависимость интен-
сивности полос поглощения, выраженной либо через пропускание т,
либо через оптическую плотность Л, от длины волны в микрометрах
или волнового числа в обратных сантиметрах.
Молекулярные колебания могут быть 2 типов: валентные и дефор-
мационные. Валентное колебание наблюдается вдоль оси связи, когда
изменяется межатомное расстояние; при деформационном колебании
происходит изменение угла, образованного связями около общего ато-
ма, или движение группы атомов по отношению к остальной части
молекулы. Частота, при которой наблюдается полоса поглощения,
зависит от относительных масс атомов, геометрии молекулы и осо-
бенностей связей. Так, в области наиболее высоких частот (3600—
2800 см-1) находятся полосы поглощения групп, содержащих легкий
атом водорода (С—Н, N—Н, О—Н). Замена водорода на более тяже-
лый атом приводит к смещению полосы поглощения в длинноволновую
область: колебания связи С—С1 расположены в области 700—600 см-1.
Химические связи, обусловливающие «скелет» органической моле-
кулы связи (С—С, С=О, С—N), имеют поглощение в области 1500—
700 см-1 (область «отпечатков пальцев»). При увеличении кратности
связи происходит повышение частот поглощения:
Сьязь Частота, см"1
С—С, С=О, C=N, N=O 190Э— 1500
С=С, C=N 2300— 2000
На рис. 45 представлены типичные спектры бензола, толуола и
ксилола, из которых видно, что небольшие структурные изменения
молекул приводят к существенным изменениям в спектрах.
В настоящее время накоплен большой экспериментальный мате-
риал по ИК-спектрам органических соединений. Сведения об этих
спектрах нормированы и могут быть заложены в память ЭВМ, с по-
мощью которой выполняется автоматизированный поиск структуры
вещества по ИК-спектру исследуемого соединения.
44.2
4000 3000 2000 1600 1200 800 400 см1
4000 3000 2000 1600 1200 800 400 см1
Рис. 45. ИК-спектры бензола (7), толуола (2), ксилола (5)
Спектроскопия ядерного магнитного резонанса (ЯМР). Ядерный
магнитный резонанс — метод, в основе которого лежит резонансное
поглощение электромагнитных волн веществом в постоянном маг-
нитном поле. Явление ЯМР обусловлено наличием магнитного момента
р у ядер некоторых атомов, таких, как Н1, F19, С13, N14, О17, Р31, В11
и др. Основное уравнение ЯМР связывает частоту v поглощаемого
электромагнитного излучения с напряженностью магнитного поля
/7о:
V = --- ,
2к
где у — гидромагнитное отношение, характеризующее данный вид
ядер i = (/— спиновое квантовое число).
Для ЯМР наиболее удобны ядра атомов
со спиновым числом V2; такие ядра имеют не-
четный атомный номер и нечетное массовое
число (Н1, С13, F19, Р31). Во внешнем магнит-
ном поле Но ядра со спином могут нахо-
диться в двух энергетических состояниях, со-
ответствующих ориентации магнитного мо-
мента параллельно приложенному полю Но
7------т —1/2
/
/
Е.--------<
------т —+1/2
Рис. 46. Расщепление
энергетических уровней
ядер со спином V2 в
магнитном поле
443
(магнитное квантовое число т = +V2) и антипараллельно (маг-
нитное квантовое число т = —V2) (рис. 46). Энергия, отвечающая
переходам между магнитными энергетическими уровнями, соответст-
вует радиочастотной области спектра.
Для практического наблюдения ЯМР ампулу 5 с исследуемым ве-
ществом помещают в катушку радиочастотного генератора 4 (рис. 47).
Рис. 47. Блок-схема спектрометра ЛМР
Катушка с ампулой расположена в зазоре сильного магнита 6 перпен-
дикулярно направлению магнитного поля //о. Генератор развертки
магнитного поля 7 создает на катушках переменное поле Н± неболь-
шой величины (Н1 //о). При достижении резонанса сигнал от
детектора поступает в приемник 3 и через усилитель 2 регистрируется
с помощью самописца 1,
Спектр ЯМР высокого разрешения, регистрируемый при высокой
однородности магнитного поля, определяется магнитным взаимо-
действием ядра с электронной оболочкой всей молекулы. Окружаю-
щее ядро электроны как бы экранируют ядро, и поскольку ядра, на-
ходящиеся в химически различных положениях, экранируются неоди-
наково, резонанс таких ядер наблюдается при разных значениях на-
пряженности магнитного поля. Смещение резонансных частот хими-
чески эквивалентных ядер, пропорциональное полю Не, называется
химическим сдвигом. Величину химического сдвига 8 удобно выражать
в миллионных долях (м. д.):
н —
Ь =------— 106,
7/эт
где Н и /7ЭТ — напряженности магнитного поля при достижении ре-
зонанса соответственно для исследуемого образца и эталонного со-
единения.
В ЯМР-спектроскопии в качестве эталона часто используют тетра-
метилсилан, который дает единичную узкую линию.
На рис. 48 представлен вид спектров ЯМР некоторых спиртов,
из которых следует, что спектры имеют относительно простой вид. Чем
больше электронная плотность вокруг протонов, тем сильнее они экра-
44!
Рис. 48. ЯМР-спектры метилового (а), этило-
вого (б) и изопропилового (в) спиртов
(в СС14)
нированы и тем в более си-
льном поле находится сиг-
нал.
Интегральная интенсив-
ность поглощения химичес-
ки неэквивалентных прото-
нов пропорциональна чис-
лу протонов каждого типа.
Это уже дает важную ин-
формацию о строении мо-
лекулы. На спектрах ЯМР
также проявляется спин-
спи новое взаимодействие
протонов, которое можно
себе представить как не-
прямое взаимодействие
спинов протонов через на-
ходящиеся между ними
связывающие электроны.
В результате этого взаимо-
действия происходит рас-
щепление линий поглоще-
ния химически неэквива-
лентных протонов или, как
говорят, образование муль-
ти п летов. Мул ьти п л ет-
ность — неоценимая ин-
формация о строении орга-
нического соединения. Ос-
новные правила мультип-
летности сигналов и рас-
пределения интенсивностей
линий в мультиплетах сво-
дятся к следующему: чис-
ло линий в мультиплете,
обусловленном спин-спино-
вой связью с группой со-
держащей п магнитных ядер со спином V2, равно п+1, интенсив-
ность этих линий представлена ниже:
Вид сигнала
Синглет ........................
Дуплет .........................
Триплет ........................
Квартет (квадруплет)............
Квинтет ........................
Секстет .......................•
Число линий (« + 1) Распределение интенсивности
1 1
2 1:1
3 1:2:1
4 1 : 3 : 3 : 1
5 1:4:6:4:1
6 1:5:10:10:5:1
Как видно из рис. 48, правила мультиплетности сигналов хорошо
иллюстрируются на спектрах простейших спиртов.
445
Следует заметить, что расстояние между линиями в мультиплетах
характеризует энергию спин-спинового взаимодействия и не зависит
от напряженности магнитного поля Но.
ЯМР-спектроскопия имеет следующие особенности: 1) спектры
ЯМР (по сравнению с ИК-спектрами) имеют более простой вид и легче
интерпретируются, поскольку в молекуле фиксируются лишь хими-
чески неэквивалентные протоны; 2) интенсивности линий (площади
пиков) в спектрах ЯМР пропорциональны числу резонирующих ядер,
что широко используется для количественного анализа и изучения
структуры соединения.
Эти и некоторые другие особенности ЯМР-спектроскопии приво-
дят к тому, что наиболее оптимальной областью ее использования яв-
ляется определение структуры органических соединений, изучение
изомерии, пространственной геометрии молекул и количественный
элементный анализ.
Масс-спектрометрия. Масс-спектрометрия (масс-спектроскопия,
масс-спектрография) — метод исследования, основанный на опре-
делении массы (точнее, отношения массы к заряду и относительного
количества ионов, получаемых из исследуемого вещества).
В процессе масс-спектрометрического исследования вещество ис-
паряют, бомбардируют в вакууме пучком электронов и регистрируют
образовавшиеся положительные ионы, в том числе отдельные фраг-
менты (осколки) молекулы. Совокупность значений масс ионов и их
относительных содержаний называют масс-спектром.
Масс-спектрометр состоит из пяти осовных элементов: 1) системы
ввода пробы; 2) ионизационной камеры и ионно-оптической системы;
3) камеры анализатора и магнита; 4) коллектора ионов и усилителя;
5) регистрирующего устройства.
В результате взаимодействия молекул исследуемого вещества
с электронным пучком образуются положительно заряженные ионьц
которые под действием электростатического поля попадают в ионно-
оптическую систему, где ускоряются и фокусируются. Развертка
спектра достигается либо за счет магнитного поля, накладываемого
на камеру анализатора, либо за счет ускоряющего напряжения ионно-
оптической системы.
Разделение ионов достигается с помощью воздействия на пучок
ионов статических полей — электрического и магнитного. Откло-
нение заряженной частицы в статическом электрическом поле явля-
ется функцией тсЧе, где v — скорость иона; отклонение иона, дви-
жущегося в магнитном поле, является функцией mv/e\ поэтому
комбинированием этих двух типов полей можно разделить ионы по
величине т/е.
Основной характеристикой масс-спектрометра является его раз-
решающая способность (сила). Под разрешающей способностью по-
нимают разницу в величине mJe ионов, которую можно достаточно
четко обнаружить прибором. В приборах низкого разрешения полу-
чаются спектры единиц масс ионов, в то время как в приборах вы-
сокого разрешения — с регистрацией сотых и даже тысячных долей
единицы масс.
446
Масс-спектрометрия фиксирует результаты воздействия иони-
зирующего пучка электронов на органическую молекулу. Сущест-
вует ряд общих правил, позволяющих предсказывать появление в
спектре характерных пиков: 1) для неразветвленных соединений от-
относительная интенсивность молекулярного иона максимальна
и понижается по мере увеличения разветвленности; 2) в пределах
гомологического ряда с увеличением молекулярной массы относи-
тельная интенсивность молекулярного иона понижается; 3) распад
молекулы идет преимущественно по разветвленным звеньям угле-
водородной цепи молекулы; 4) двойные связи, циклические стр у к*
100
80
20
о
20 30 40 50 60 70 80 90 т/е
б
100 -
80 -
60 -
-М-(Н2О и
40 |-СН2=СН2)
М-(Н2О
и СН3)
м-сн5
20 I-
м-н2о
20 30 40 50 60 70 80 90 т/е
Рис. 49. Масс-спектры изомеров пентанола:
и — пентанол-1; б — пентанол-2; в — 2-метилбутанол-2
447
туры и особенно ароматические или гетероароматические циклы стаби-
лизируют молекулярный ион и таким образом повышают вероятность
его появления; 5) двойные связи способствуют аллильному распаду
с образованием аллильного карбониевого иона (СН2=СН—СН2);
6) связи С—С, следующие за гетероатомом, часто разрываются, ос-
тавляя заряд на фрагменте, содержащем гетероатом; 7) распад часто
сопровождается удалением небольших стабильных нейтральных мо-
лекул, таких, как оксид углерода, олефины, аммиак, сероводород,
цианистый водород, меркаптаны, кетоны или спирты.
Помимо образования фрагментов молекулы за счет простого раз-
рыва связей в молекулярном ионе возможно появление ионов из-за
внутримолекулярной атомной перегруппировки (например, мигра-
ции атомов водорода в молекулах, содержащих гетероатом).
В качестве примера, на рис. 49 приведены масс-спектры изомер-
ных пентанолов, из которых видна высокая характеристичность масс-
спектров. Степень характеристичности такова, что она позволяет
в случае индивидуальных веществ различать практически любые
химические соединения.
Количественный молекулярный масс-спектральный анализ ос-
нован на пропорциональности интенсивности всех линий масс-спект-
ра каждого из вещества его парциальному давлению в области иони-
зации. Суммарный масс-спектр смеси является поэтому аддитивным
наложением масс-спектров каждого из компонентов смеси. Молеку-
лярный масс-спектральный анализ получил значительное распростра-
нение в органической химии, охватывая практически все классы
органических соединений, в том числе полисахариды, пептиды, ал-
калоиды, стероиды, терпеноиды и другие биологически активные
соединения.
При анализе примесей чувствительности масс-спектрометрии мо-
жет быть очень велика—10"7—10"9 %. Ограничения в примене-
нии масс-спектрометрии связаны со сложностью и высокой стои-
мостью аппаратуры, превосходящей по стоимости ПК- и УФ-
спектрометры.
Хроматографические методы разделения, идентификация и коли-
чественное определение. Термин «хроматография» (chroma — по-гре-
чески цвет, краска) был предложен в 1904 г. русским ботаником
М. С. Цветом, показавшим, что при пропускании через слой сор-
бента растительных пигментов последние разделяются, образуя ряд
окрашенных зон. Отличительной чертой хроматографического раз-
деления и анализа смесей является распределение отдельных компо-
нентов между двумя фазами — неподвижной и подвижной (элюент),
протекающей через неподвижную.
При разделении и анализе органических соединений наиболь-
шее распространение получили методы газовой, жидкостной, моле-
кулярно-ситовой, бумажной, тонкослойной и ионообменной хрома-
тографии. В табл. 32 приведены основные характеристики этих ме-
тодов.
Как правило, наиболее эффективен в хроматографии так назы-
448
Таблица 32. Важнейшие виды хроматографии
Хроматография Подвижная фаза Неподвижная фаза Форма примене- ния Механизм разделения
Газожидкостная Газ Жидкость Колонка Распределительный
Газо-адсорбционная » Твердая » Адсорбционный
Жидкостно-жид- костная Жидкость Жидкость » Распределительный
Жидкостно-адсорб- ционная » Твердая Адсорбционный
Бумажная Бумага Полосы или листы Распределительный или адсорбцион- ный
Тонкослойная » Твердая Тонкий слой Адсорбционный
Ионообменная » » Колонка Ионный обмен
Молекулярно-сито- вая » » Молекулярно-сито- вый
ваемый элюентный анализ. Суть его в том, что, например, через
колонку, заполненную сорбентом, непрерывно пропускают элюент —
газ или жидкость. Подвижная фаза служит в этом случае только для
перемещения растворенного вещества анализируемой смеси, а раз-
деление происходит благодаря разному сродству определяемых
компонентов к неподвижной фазе. При однократном введении пробы
компоненты начинают перемещаться через колонку с различными ско-
ростями, образуя отдельные зоны сорбции. Выходная кривая (хро-
матограмма) будет представлять собой ряд отдельных колоколооб-
разных кривых — «пиков». Количество пиков равно числу компо-
нентов, если достигается полное их разделение, при этом время вы-
хода отдельного компонента из колонки («время удерживания»)
может быть качественной характеристикой вещества, а площадь под
пиком — его количественной характеристикой.
Газовая хроматография (газо-адсорбционная хроматография) —
вариант хроматографии, в котором разделение производится с по-
мощью подвижной газовой фазы, проходящей вместе с анализируе-
мой смесью над твердым сорбентом. В качестве сорбента используют
силикагели, алюмогели, молекулярные сита, пористые полимеры и
другие сорбенты, а в качестве газа-носителя (элюента) — гелий, ар-
гон или азот.
В газожидкостной хроматографии стационарной фазой служит
нелетучая жидкость (высококипящие углеводороды, сложные эфи-
ры, силоксаны и др.), нанесенная в виде пленки на твердый носитель
(например, цеолит, пемзу, огнеупорный кирпич, кизельгур). Испы-
туемую пробу вводят в колонку в виде пара, после чего компоненты,
имеющие разную растворимость в стационарной жидкой фазе, рас-
пределяются между этой фазой и газом-носителем по закону равно-
весия (закон Генри). Чем больше растворимость компонента в жидкой
фазе, тем больше время удерживания этого компонента.
449
Жидкостная хроматография начала необычайно быстро разви-
ться в последние 10—20 лет. Как и в случае газовой хроматогра-
мм, жидкостная хроматография существует в виде жидкостной ад-
сорбционной и жидкостной распределительной хроматографии. В жид-
костно-адсорбционной хроматографии используется различное срод-
ство компонентов смеси к твердому сорбенту, взятому в качестве
неподвижной фазы; в жидкостной распределительной хроматогра-
фии — различная растворимость (распределение) веществ между
подвижной и неподвижной жидкими фазами, удерживаемыми по-
ристым инертным носителем. Эффективность разделения в жидкост-
ной хроматографии в значительной степени зависит от соотношения
полярных и неполярных групп в растворенных веществах и двух
жидких фазах. В этом методе для целей идентификации используют
нанесение реагента способом напыления. Применение в жидкостной
хроматографии высокого давления сделали этот метод еще более
эффективным.
В ионообменной хроматографии используется различное срод-
ство ионов раствора к ионообменным центрам противоположной по-
лярности в неподвижной фазе (ионообменнике). Ионный обмен пред-
ставляет собой химический процесс.
Необходимо упомянуть еще об одном виде хроматографии — си-
товой хроматографии, называемой также гель-фильтрацией или
гельпроникающей хроматографией. Здесь применяют пористые твер-
дые материалы (гели) с определенным узким распределением пор по
диаметрам. Молекулы, эффективный диаметр которых больше, чем
диаметр пор, проходят через колонку быстрее, чем молекулы меньших
размеров, способных диффундировать внутрь пор: в порах перенос
вещества в направлении оси колонки отсутствует, и молекулы мень-
ших размеров движутся через колонку медленнее. В качестве сор-
бентов в ситовой хроматографии используют пористые стекла, моле-
кулярные сита, в том числе декстрановые гели (сефадексы), полиакри-
лоамидные гели, а также гидрофобные полимеры — полистиролы с
большей или меньшей степенью сшивки (порагель, стирагель и др.).
Области применения газовой, жидкостной и ситовой хроматографии
различны. Газовую хроматографию применяют для разделения летучих
соединений. Методом жидкостной хроматографии анализируют прежде
всего такие вещества, которые нельзя без разложения перевести в
газовую фазу (молекулярная масса от 100 до нескольких тысяч). На-
конец, вещества с большей молекулярной массой (от нескольких ты-
сяч до миллиона) могут быть разделены с помощью ситовой хромато-
графии.
Аппаратурное оформление, техника, методика работы в хромато-
графии чрезвычайно разнообразны. В простейшем случае (тонко-
слойная хроматография) адсорбент (силикагель, оксид алюминия)
тонким слоем (~0,25 мм) наносится на стеклянную пластину или
алюминиевую фольгу. Испытуемую пробу (С 200 мкг) в виде не-
большого пятна с помощью микропипетки или микрошприца (на 1 мкл)
наносят на нижнюю часть пластинки. После испарения растворителя
этот конец пластинки погружают в кювету с подвижной жидкой фа-
450
зой так, чтобы проба находилась над жидкостью. Под действием ка-
пиллярных сил жидкая фаза медленно поднимается вверх по пластине,
захватывая компоненты пробы и перемещая их с разной скоростью по
слою адсорбента. Процесс разделения заканчивают, когда передний
край подвижной фазы («фронт растворителя») почти достигнет верх-
него края пластинки. Обнаруже-
ние хроматографических зон про-
водят с помощью специальных ре-
агентов.
В тонкослойной хроматографии
идентификацию органического ве-
щества проводят по измерению па-
раметра удерживания — величины
/?у, которая представляет собой
отношение расстояния, пройденно-
го растворенным веществом к рас-
стоянию, пройденному фронтом
растворителя (рис. 50).
В адсорбционной хроматогра-
Рис. 50. Определение величины Rf в
тонкослойной хроматографии:
1 — линия старта; 2 — фронт растворите-
ля; 3 — определяемое вещество
фии чрезвычайно велика роль по-
движной фазы. Подвижная фаза — это не только растворитель. Чем
сильнее подвижная фаза взаимодействует с адсорбентом, тем меньше
взаимодействие адсорбента и растворенного вещества и тем быст-
рее растворенное вещество будет передвигаться через хроматографи-
ческий слой. Для любого данного адсорбента можно составить таб-
лицу, в которой растворители будут размещены в ряд по мере
возрастания их элюирующей способности (так называемые «элюотроп-
ные ряды»). Например, для силикагеля этот ряд таков: циклогексан,
гептан, пентан, четыреххлористый углерод, сероуглерод, бензолхло-
рид, этилбензол, толуол, бензол, 2-хлорпропан, хлороформ, нитро-
бензол, диизопропиловый эфир, диэтиловый эфир, этилацетат,.
2-бутанол, этанол, вода, ацетон, уксусная кислота, метанол.
Успешное проведение хроматографического разделения также за-
висит от правильного выбора адсорбента, активность которого опре-
деляется рядом факторов: «силой» активных центров, плотностью их
распределения на поверхности адсорбента, размерами частиц, содер-
жанием воды и др.
Простым видом хроматографического анализа является также
бумажная хроматография. Рольнеподвижной фазы здесь играет фильт-
ровальная бумага (целлюлоза), а подвижная фаза (растворитель) пере-
мещается в результате действия, в основном, капиллярных сил. Дви-
жение растворителя может быть снизу вверх (восходящая хромато-
графия) или сверху вниз (нисходящая хроматография). Для получения
бумажной хроматограммы нарезают полоски бумаги длиной 15—
30 см и шириной в несколько сантиметров. Отмечают линию старта
на одном из концов полоски, на которую наносят каплю испытуемого
раствора (или вещества). Ьсли необходимо, то испаряют раствори-
тель (анализируемой пробы) и полоску бумаги приводят в соприкос-
новение с подвижной фазой, используя герметичную камеру для
451
предотвращения испарения подвижной фазы. Хроматографирование
заканчивают, когда фронт подвижной фазы приблизится к противо-
положному концу бумажной полоски. Бумагу вынимают, сушат, а
хроматографические пятна проявляют специальными реагентами.
В методах газовой и газожидкостной хроматографии разделение
л анализ смесей веществ выполняют с помощью специальных пра-
вы ход
Рис. 51. Принципиальная схема газового хроматографа
боров — газовых хроматографов. Основными частями хроматографа
1рис. 51) являются система для ввода исследуемой пробы, хромато-
графическая колонка, детектор, системы термостатирования и ре-
гистрации. Газ-носитель подается в хроматографическую колонку из
баллона 1 через редуктор 2 и вентиль тонкой регулировки давления
<?, которое регистрируется точным манометром 4. Газ попадает в термо-
стат 10, где подогревается, и затем в устройство для ввода проб (ин-
жектор ) 5. Испытуемая жидкая проба вводится в инжектор с помощью
микрошприцов небольшого объема (1 мкл). Поскольку инжектор
помещен в термостат и имеет температуру, близкую к температуре ки-
пения определяемых компонентов, проба быстро испаряется,образую-
щиеся пары захватываются газом-носителем и подаются в хромато-
графическую колонку 6. После разделения в хроматографической
колонке отдельные компоненты пробы в виде бинарной смеси с газом-
носителем попадают в детектор 7 — устройство, реагирующее на
изменение состава протекающего газа. Получаемый сигнал усилива-
ется электронным преобразователем сигнала 8 и регистрируется само-
писцем 9.
Условия хроматографирования определяются типом детектора,
природой газа-носителя, адсорбента и жидкой фазы, скоростью элю-
ирования, температурой и другими факторами. В результате реализа-
ции этих условий получают хроматограмму, которая фиксирует из-
менение концентрации выходящих из хроматографической колонки
компонентов анализируемой смеси. На рис. 52 приведена типичная
хроматограмма разделения триметилсилильных производных сахаров
кукурузной патоки.
452
Рис. 52. Разделение триметилсилильных производных компо-
нентов сахаров кукурузной патоки методом газовой хрома-
тографии:
1 — левоглюкозан (1,ь-ат гидро-Р-Р-глюкопирагоза); 2 — уизомер-Р-глю-
козы; 3 — а-Р-глюкоза; 4 — Р-Р-глюкоза; 5 — фенил-Р-Р-глюкопирано-
зид (внутренний стандарт); 6 - мальтоза; 7 •—Р-целлобиоза; 8 — изо-
мальтоза; 9 — мальтотриоза; 10 мальтотетраоза
Наиболее распространены детекторы газовой хроматографии —
термокондуктометрические (катарометры) и ионизационные. Катаро-
метр — детектор по теплопроводности представляет собой термосоп-
ротивление, омываемое газовым потоком (элюентом). Его сопротив-
ление меняется в зависимости от температуры, которая, в свою оче-
редь, определяется теплопроводностью газовой среды. Последняя
различна для газа-носителя и бинарной смеси: газ-носитель + компо-
нент анализируемой смеси. Это позволяет фиксировать выходную
кривую смеси (хроматограмму). Для большей чувствительности та-
кого детектора его подключают по мостовой схеме Уитстона, исполь-
зуя две ячейки для измерения теплопроводности. Через одну из них
(измерительную) пропускают элюат из хроматографической колонки,
через другую (сравнительную) — чистый газ-носитель. Таким обра-
зом, детектор измеряет различие в теплопроводности чистого газа-
носителя и смеси газа-носителя с веществом, выходящим из хромато-
графической колонки.
Ионизационные детекторы более чувствительны, а в некоторых слу-
чаях (детекторы электронного захвата) и более селективны. Главной
частью ионизационных детекторов является ионизационная камера,
где происходит ионизация молекул, попадающих в нее с потоком газа-
носителя из хроматографической колонки. Ионизацию исследуемых
веществ или газа-носителя осуществляют в пламени водорода (пламен-
но-ионизационный детектор) под воздействием потока частиц радио-
активного источника, например никелевой фольги^ содержащей ||Ni
453
(детектор электронного захвата), и другими способами. Образующиеся
ионы под воздействием приложенного напряжения создают электри-
ческий ток, величина которого определенным образом связана с мас-
совой скоростью определяемого вещества.
Блок-схема хроматографа для жидкостной хроматографии пока-
зана на рис. 53. Подвижная фаза (растворитель) подается в хромате-
Рис. 53. Блок-схема жидкостного хроматографа высо-
кого давления
графическую колонку 7 из резервуара 1 с помощью насоса 2. Предо-
хранительный вентиль 3 необходим для целей безопасности системы,
а демпферирующее устройство 4 позволяет сгладить возможную не-
равномерность (зависит от типа насоса) подачи потока жидкости. Дав-
ление контролируют на входе в хроматографическую колонку 7 мано-
метром 5. Подвижная фаза подается в колонку 7 через устройство для
ввода пробы 6. Элюат из колонки 7 направляют в детектор S, подклю-
ченный к регистрирующему прибору 9, и затем в коллектор фракций
10, где можно собрать составные части исследуемой пробы. Устройство
для ввода пробы, колонку и детектор термостатируют (блок /7).
Идентификация органического соединения в методах газовой и
жидкостной колоночной хроматографии может быть выполнена с по-
мощью следующих параметров: времени удерживания t R и объема
удерживания V R анализируемого вещества. Время удерживания —
время от момента ввода пробы в хроматографическую колонку до мо-
мента выхода из нее максимальной концентрации определяемого ве-
щества. Объем удерживания — объем подвижной фазы, прошедшей
через хроматографическую колонку от момента ввода пробы до мо-
мента выхода максимальной концентрации определяемого вещества:
V R t rFoq, где Fo6 — объемная скорость подвижной фазы при
данной температуре и давлении на выходе из колонки. Объемы удер-
живания являются более точными характеристиками разделяемых
компонентов смеси.
Качественное исследование анализируемых смесей может быть вы-
полнено также с помощью детекторов различного принципа действия,
универсально или избирательно реагирующих на отдельные классы
химических соединений. Особенно эффективно для целей идентифика-
ции органических соединений использование в качестве детектора
масс-спектрометра, позволяющего проводить как определение моле-
кулярной массы, так и молекулярной структуры отдельных компо-
нентов анализируемой смеси.
Широко используют также для этих же целей аналитическую ре-
454
акционную хроматографию, в которой стадии разделения (хромато-
графия) и определения (физико-химический детектор) дополняют
проведением химических реакций в специальных микрореакторах, при-
мыкающих к хроматографической колонке или даже вне хромато-
графической системы, для получения более характерных производ-
ных определяемых органических соединений.
Количественный хроматографический анализ осуществим на основе
методов абсолютной калибровки, внутренней нормализации или внут-
реннего стандарта. Метод абсолютной калибровки заключается в
построении графической зависимости высоты или площади пика от со-
держания вещества в пробе.Метод внутренней нормализации преду-
сматривает отнесение площади или высоты пика к величине суммар-
ного сигнала детектора от всех компонентов пробы:
ct = f\ 100,
I )
\ 1 /
где pi — нормированный параметр хроматографического пика; {t —
нормированный множитель, учитывающий неодинаковую чувстви-
тельность детектора к определяемым веществам.
Метод внутреннего стандарта состоит в том, что к известному коли-
честву анализируемой пробы добавляют определенное количество не
содержащегося в нем соединения (стандартного вещества) и полу-
ченную смесь хроматографируют:
_Pi f i Чсг ‘ ЮО
Per Чем
где pi и рст — измеряемые параметры (высота пика h или площадь
пика S) хроматографических пиков; Д — нормированный множитель
для определяемого соединения относительно стандартного вещества;
7ст и 7см — взятые количества (в г, моль или см3) стандартного ве-
щества и полученной смеси.
ЛИТЕРАТУРА
1. Несмеянов А. Н., Несмеянов Н. А. Начала органической химии.— М.—Л.,
1974, кн. 1 и 2.
2. Моррисон Р., Бойд Р. Органическая химия. — М.: Мир, 1974.
3. Гауптман 3., Грефе Ю., Реманс X. Органическая химия. — М.: Химия,
1979.
4. Терней А. Современная органическая химия. —М.: Мир, 1981, т. 1 и 2.
5. Петров А. А., Бальяп X. В.. Трощенко А. Т. Органическая химия. —
М,: Высшая школа, 1981.
6. Грандберг И. А. Органическая химия. —М.: Высшая школа, 1980.
7. Сайкс П. Механизмы реакций в органической химии. —М.: Мир, 1977.
8. Я новская Л. А. Современные теоретические основы органической хи-
мии. — М.—Л.: Химия, 1978.
455
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агликоны 369
Адамкевича реакция 422
Адипиновая кислота 112, 233, 285
Азокрасители 292
Азолы 324
Азосоединения 290
Азосочетания реакция 290
Акриловая кислота 223, 224, 226, 297,
302
Акрилонитрил 93, 96, 226
Акролеин 177, 209
а-Аланин 294, 295, 303, 305
Алкалицеллюлоза 384, 385
Алкалоиды 329
Алкилсульфиды 266
Алкины 42, 85
Алкоголи см. Спирты
Алкоголиз 237, 396
Алкоголяты 105
Алкоксигруппа 190
Аллиловый спирт 161, 177, 181
Альдегидокислоты 254
Альдегиды 192
— номенклатура 193
— получение 195
— представители 207
— свойства 198
Альдогексозы 344, 346
Альдозы 342, 346
Альдоксим 202
Альдольная конденсация 205, 275, 353
Амигдалин 370
Амиды 306
Амиловые спирты
Амилоза 380
Амилопектин 381
о-Аминобензойная
305
/г-Аминобензойная
Аминогруппа 277,
Аминокислоты 293
— изомеры 294
— получение 294
— представители 295
— свойства 299
Амины 277 сл.
Ангидриды кислот 238
Анетол 427
Анизол 187, 190
Анилин 279, 282, 285
Аномеры 348
Антиподы 246
456
161, 176
кислота 296, 298,
кислота 296, 298
293
Антрахинон 141
Антраниловая кислота 296, 298, 305
Антрацен 141
L-Арабиноза 367
Арабит 368
Арахидоновая кислота 393
Арахиновая кислота 393
Арил 123
Асимметрический углеродный атом
245
Аспарагиновая кислота 295
Аспирин 253
Ауксохромы 292
Ацетали 201
Ацетальдегид см. Уксусный альдегид
Ацетамид 307
Ацетил 235
Ацетилен 85, 88, 90, 96
— строение связей 29
Ацетиленид серебра 94
Ацетиленистая медь 94
Ацетилцеллюлоза 386
Ацетон 92, 185, 193, 194, 208
Ацетонитрил 308
Ацетоуксусный эфир 256
Ацетофенон 131, 208
Ацидолиз 237, 396
Ацил 235
Бегеновая кислота 393
Белки 417
— классификация 422
Бензальдегид 204, 207
Бензамид 306
Бензил 123
Бензиловый спирт 177
Бензин 145
Бензойная кислота 134
Бензол 120, 121, 124, 136
Бензохинон 189
Бетаины 301
Биуретовая реакция 422
Болотный газ 64
Борнеол 429
Брожение 362
Бутадиен-1,3 98, 99, 100, 102—106
Бутан 44, 45, 61, 62, 63, 65
Бутандиовая кислота см. Янтарная
кислота
Бутил 47
Бутилен 85
Бутиловые спирты 176
Бутины 88
Бутлерова теория строения 11
Вагнера реакция 77
Валериановая кислота 211
Валин 295, 305
Ванилин 208.
Вёлера реакция 7
Винилацетилен 93
Виниловый спирт 92
Винная кислота 248, 249, 251
Вискоза 387
Витамины 3, 6
— А 430
— В! 331
— В12 312
— С 370
— Е 335
— РР 329
Водородная связь 16
Воски 401
Высыхание жиров 415
Вюрца реакции 50
Вюрца—Фиттига реакции 125
Газ природный 49
Галактоза 346, 365, 366
Галактоновая кислота 365, 366
Галловая кислота 242, 253
Галогенангидриды 236—238
Галогенпроизводные 148 сл.
Галактопираноза 365
Гаттермана—Коха реакция 197
Ганглиозид 365, 409
Гексаметилендиамин 285
Гексан 42, 45, 52, 54, 124
Гексахлоран 134, 159
Гексахлорциклогексан см. Гексахло-
ран
Гексозы 342, 363
Гемицеллюлоза 383
Гемоглобин 319
Гептан 45, 157
Гептоза 354
Гераниол 309
Гетероциклические соединения 309
Гетероциклы 309, 332, 334
— пятичленные 310
-----с двумя гетероатомами 324
-----одним гетероатомом 310, 320
—• шестичленные 324
-----с двумя гетероатомами 330
-------одним гетероатомом 324
Гибридизация 26
Гидразон 202
Гидрогенизация жиров 414
Гидропереэтерификация 414
Гидрохинон 184, 189
Гликоген 382
Гликозидный гидроксил 348, 349
Гликозиды 349, 369, 372
Гликокол 295, 305
Гликолипиды 402
Гликоли 178
Гликопротеиды 422
Глиоксимовая кислота 255
Глицерин 178, 182
— тринитрат 183
Глицериновые альдегиды 345
Глицериды 392, 397
Глобулины 422
Глутаровая кислота 229
Глутаминовая кислота 295, 305
Глюкоза 343—351, 363—383
Глюконовая кислота 364
Глюкопираноза 351, 361, 363
Глюкофураноза 351, 361
Глютелины 422
Гомологи 44
Гомологическая разность 44
Гомологический ряд 44
Гофмана реакция 279
Гриньяра реакции 261
Двойная связь 29, 65
Декстрины 380
Диазогидрат 287
Диазония хлорид 287
Диазония соли 288
Диазосоединения 287
Диазотирования реакции 287
Диастереомеры 249, 346, 349
Диацетилцеллюлоза 386
Дивинил см. Бутадиен-1,3
Диены 97
Дикетопиперазин 302
Дильса—Альдера синтез 104
Диметиламин 279, 281
«-Диметиламиноазобензол 291
Диметиловый эфир 190
Диметилсульфат 375, 385
Диоксан 182, 190
Диолы 180
Дисахариды 371—378
Дифенил 119, 138
Дифтордихлорметан 158
Дихлорэтан 157
Диэтиловый эфир 190
Диэтилсульфид 266
Дульцит 365
Ендиол 357
Енольная форма 257, 258
Жирные кислоты высокомолекуляр-
ные 211, 221, 223, 393
Жиры 392
— гидрогенизация 397, 414
Заместители первого и второго рода
132, 133
457
Зелинского—Казанского реакция 93
Зинина реакция 278
Изобутилен 66, 79
Изолейцин 295, 305
Изомерия И
— геометрическая 67, 109, 224
— динамическая 257
— конформационная ИЗ—118
— оптическая 109, 245, 294, 344
— поворотная 109, 113—118
— пространственная 67, 109, 224, 245,
294, 344
Изомеры 11
Изопрен 98, 101, 106
Изопреноиды 424
Имидазол 324
Инверсия 377
Инвертный сахар 377
Индиго 323
Индол 320
Индуктивный эффект 37, 216
Инозит 406
Инулин 367
Инсулин 303
Камеди 389
Каменноугольная смола 123, 124
Камфан 428
Камфора 428
Камфен 429
Канниццаро реакция 203
Капролактам 285
Капрон 285
Капроновая кислота 211
Каран 428
Карбид кальция 90
Карбонильная группа, электронное
строение 198, 199
Карбоновые кислоты см. Кислоты
Карбоциклические углеводороды 106
Каротиноиды 429
Каучук 106
Квантовые числа 16, 21
Керосин 146
Кетены 208—210
Кето-енолъная таутомерия 257
Кетозы 342, 344
Кетокислоты 254
Кетоксимы 202
Кетоны 192 сл.
Кислоты
— алифатические двухосновные 228
----- одноосновные непредельные 222
-----.— предельные 210
— фосфатиднце 404
Клетчатка 383
Коксование 123
Коллодий 386
Коллоксилин 386
Кольбе, метод получения алканов 51
Коновалова реакция 58
Конформации 109, 113—118
Крахмал 379
Крезолы 161, 184, 189
Крекинг-процесс 62, 68, 146
Кремнийорганические соединения 265
Кротоновая кислота 223, 224
Ксантогенаты клетчатки 386
Ксантопротеиновая реакция 421
Ксилит 368, 369
D-Ксилоза 368
Ксилолы 121
Кумол 121, 185
Купрен 94
Кучерова реакция 92
Лавсан 235
Лактамы 302
Лактиды 244
Лактоза 376
Лактоны 244
Ланолин 402
Лауриновая кислота 211, 393
Лебедева реакция 99
Лейцин 295, 305
Лецитины 405
Лигнин 383
Лизин 295, 305
Ликопин 429
Лимонен 425, 426
Лимонная кислота 242, 252
Линалоол 425
Линолевая кислота 223, 393
Линоленовая кислота 223, 393
Липазы 395
Липиды 389 сл.
— методы исследования 410
Липопротеиды 422
Мазут 146
Малеиновая кислота 228, 233
Малоновая кислота 229, 233
Мальтоза 373
Манноза 346, 366
Маннит 367
Марковникова правило 73, 74, 92
Масляноэтиловый эфир 241
Мезовинная кислота 249
Мелиссиновая кислота 393
Ментан 426
л<ета-Изомеры 123
Метакриловая кислота 223, 226
Металлорганические соединения 261
Метан 13, 15, 44, 45, 47, 50, 64, 90
Метанол см. Метиловый спирт
Метил 47
Метиламин 278, 281
Метилбензол см. Толуол
2-Метилбутадиен-1,3 см. Изопрен
Метиловый спирт 161, 171, 172
Метилоранж 292
458
Метилфенилкетон 208
Метилэтилацетилен 88
Метионин 295, 305
Меркаптаны 266
Меркаптиды 266
Мерсеризация 384
Миллонова реакция 422
Мирициловая кислота 393
Мирцен 425
Молочная кислота 241, 245, 250
Молочный сахар см. Лактоза
Моноамины 278, 281
Моносахариды 342, 343
Муравьиная кислота 211, 220
Мутаротация 345
Мыла 416
Мясомолочная кислота 250
Найлон 285
Нафталин 138—141
Нафтены 109
Нафтолы 184
Нефть 123, 145, 189
Никотин 329
Нитрилы 307
Нитробензол 272—276
Нитроглицерин 183
Нитрозоамины 284
Нитросоединения 270
Нитрофенол 188
Номенклатура 48
Нуклеиновые кислоты 336
Нуклеопротеиды 423
Озазоны 358, 366, 373
Озокерит 50
Окись этилена 77, 181, 192, 226
Оксигидрохинон 184
Оксикислоты 241 сл.
Оксимы 202
2-Оксипропановая кислота см. Молоч-
ная кислота
Оксокислоты 254
2-Оксопропановая кислота см. Пиро-
виноградная кислота
Оксосинтез 196
Октановое число 147
Олеиновая кислота 223, 393
Олефины 28, 65—85
Олигосахариды 373
Олифа 415
Оптическая изомерия 245
Оптические антиподы 246
Органические вещества, источники 9
-----классификация 40
Орто-изомеры 123
Пальмитиновая кислота 211, 393
77яра-изомеры 123
Парафины см. Углеводороды предель-
ные
Пектиновые вещества 388
Пентан 44, 45
Пентозаны 368
Пентозы 368
Пептидные связи 302, 418
Пептиды 302
Перекиси органические 192
Переэтерификация 396, 414
Пинан 428
а-Пинен 428
Пиразол 324
Пиран 334, 349
Пиранозы 349, 350
Пиридин 324—328
Пировиноградная кислота 254, 255
Пирогаллол 184
Пирокатехин 184
Пироксилин 386
Пиролиз нефти 146
Пирослизевая кислота 312, 318
Пиррол 310, 312—318
Поверхностно-активные вещества 415
---- неионогенные 417
Полиамиды 285
Поливинилацетат 177
Поливиниловый спирт 177
Поливинилхлорид 158
Полимеризация 78—83
Полиметилметакрилат 227
Полиметиленовые углеводороды см.
Циклоалканы
Полистирол 136, 137
Полисахариды 342, 371
Полиэтилен 78
Полуацетали 201
Поляриметр 247
Порфин 319
Принцип Паули 21
Проламины 422
Пролин 296, 305
Пропан 45, 52, 57
Пропен 66, 87
Пропил 47
Пропиловые спирты 174
Пропин 88
Пропионовая кислота 211
Протамины 422
Протеиды 422
Радикалы свободные 80
Рацематы 246
— разделение на антиподы 250
Резорцин 184
D-Рибоза 337, 346, 368
Рибонуклеиновые кислоты 337
Рибофураноза 337, 368
Риформинг нефти 146
Рицинолевая кислота 223, 252, 393
Родионова метод 297
Рудничный газ 64
459
Сесквитерпены 424
Синтез-газ 50
Синтин 50
Слизи 389
Соляровые масла 146
Сорбиновая кислота 223, 227
Сорбит 160, 364, 367
Спирт этиловый 161, 171, 173—175
Спирты
— многоатомные 178
— одноатомные 160
— вторичные 160
— изомерия 160, 178
— номенклатура 160, 178
— первичные 160
— получение 162, 179
— третичные 160
— химические свойства 165, 180
— строение 160, 178
Стеариновая кислота 211, 222, 393
Стекло органическое 227
Стереоизомерия 67, 109, 224, 245, 294,
344
Стерины 390
Сульфгидрильная реакция 422
Сульфогруппа 267
Сульфокислоты 265, 267
Сфингозин 408
Танин 253
Таутомерия см. Изомерия динамиче-
ская
Теория строения Бутлерова И
Терефталевая кислота 228, 229, 235
Терилен см. Лавсан
Терпены 424
Терпинеол 426
Тетрафторэтилен 159
Тимол 426
Тиолы см. Тиоспирты
Тиомочевина 267
Тиоспирты 266
Тиофен 310—316
Тиоэфиры 266
Тирозин 296, 305
Тищенко реакция 204
Толуол 121, 123, 136, 150
Треонин 295, 305
Триглицериды 392
Триозы 342
Триолы 178
Триптофан 296, 305, 323
Трисахариды 371
Тройная связь 29, 89
Тростниковый сахар см. Сахароза
Тротил 277
Углеводороды 42, 43, 65, 85, 97, 106,
118
— ароматические 118 сл.
— ацетиленовые 85 сл.
— диеновые 97
— классификация 42
— непредельные 65, 85, 97
— полиметиленовые 106
— предельные 43 сл.
— отдельные представители 64
— этиленовые см. Олефины
Уксусная кислота 211, 220
Уксусноизоамиловый эфир 241
Уксусноэтиловый эфир см. Этилацетат
Уксусный альдегид 193, 194, 207
Уксусный ангидрид 238, 239
Уроновые кислоты 356
Фелингов раствор 357
Фенантрен 141
Фенетол 190
Фенил 123
Фенилаланин 295, 305
Фенилгидразин 202, 290, 358
Фенол 159, 184
Фенолы 159, 184—189
Фенолформальдегидные смолы 206
Феноляты 186
Фитол 429
Фишера формулы 246
Флороглюцин 184
Формальдегид 194, 207
Формил 235
Фосген 238
Фосфолипиды 403
Фосфопротеины 422
Фотосинтез 379
Фреон-12 158
Фриделя—Крафтса реакция 126, 129—
131
Фруктоза 343, 344, 366, 367
Фталевая кислота 229, 234
Фталевый ангидрид 234
Фумаровая кислота 229, 233
Функциональные группы 41
Фуран 309—318
Фуранозы 349, 350
Хеуорзса формулы 350
Химическая связь
-----двойная 29, 65
-----диеновые 97
----- конъюгированные 97
----- кумулированные 97
-----сопряженные 97
----- ковалентная 12, 16
----- координационная 14
---------- полярность 14
------ семиполярная 14
------ тройная 85
------ электровалентная 14
------ энергия 14
Хлорбензол 149, 158
Хлорвинил 149, 150, 158
Хлористый аллил 149, 151, 158
460
Хлористый ацетил 238
Хлористый бензил 149
Хлористый бензоил 238
Хлоропрен 106
Хлорофилл 319
Хлороформ 149, 158
Холин 405, 409
Хромопротеиды 423
Хромофоры 54, 291
Целлобиоза 375
Целлюлоза см. Клетчатка
Целлулоид 386
Цереброзид 410
Церазин 409
Циклоалканы 106
Циклобутан 107
Циклогексан 107
Цикло-оксо-таутомерия 35t
Циклопентан 107
Циклопропан 107
Цис-транс-изомерия см. Геометриче-
ская изомерия
Цистеин 295
Цитраль 425
Чалмугровая кислота 393
Четыреххлористый углерод 54, 149,
158
Число иодное 411
Число кислотное 410
— омыления 410
Электронное облако 16
Электронное строение связи 16
Элементорганические соединения 261
Энергия образования связи 14
Эпоксиды 191
Эруковая кислота 393
Этаналь см. Уксусный альдегид
Этандиовая кислота см. Щавелевая
кислота
Этандиол см. Этиленгликоль
Этанол см. Этиловый спирт
Этаноламин 405
Этен см. Этилен
Этерификация 217
Этил 47
Этилацетат 241
Этилен 28, 65
Этиленгликоль 178
Этиловый спирт 161, 171, 173—175
Этилформиат 240
Этин см. Ацетилен
Этоксиэтан см. Диэтиловый эфир
Эфиры простые 190
— фосфоглицериновые 404
— фосфосфингозиновые 408
Яблочная кислота 251
Янтарная кислота 229
ОГЛАВЛЕНИЕ
Предисловие........................................................... 3
Введение ........................................................... 5
Часть 1. Углеводороды. Функциональные производные углеводородов. Эле-
менторганические соединения .......................................... 5
Раздел первый. Углеводороды.......................................... 42
Ациклические (алифатические) углеводороды........................ 43
Алканы .......................................................... 43
Алкены .......................................................... 65
Алкины .......................................................... 85
Алкадиены........................................................ 97
Карбоциклические углеводороды....................................106
Циклоалканы......................................................106
Арены.............................. . . .........................118
Одноядерные ароматические углеводороды...........................119
ААногоядерные ароматические соединения...........................137
Многоядерные ароматические углеводороды с неконденсированными
(изолированными) ядрами..........................................138
Многоядерные ароматические углеводороды с конденсированными яд-
рами ........................................................... 139
Высшие полициклические углеводороды............................142
Небензоидные соединения с ароматическими свойствами..............142
Ароматические системы с пятичленными циклами.....................143
Ароматические системы с семичленными циклами.....................144
Нефть и ее переработка...........................................145
Раздел второй. Функциональные производные углеводородов . . • 148
Галогенпроизводные углеводородов.................................148
Кислородсодержащие производные углеводородов.....................159
Гидроксилсодержащие производные углеводородов (спирты, фенолы,
нафтолы)........................................................ 159
Одноатомные спирты 160
Многоатомные спирты..............................................178
Фенолы, нафтолы..................................................184
Простые эфиры....................................................190
Оксосоединения (альдегиды и кетоны)............................. 192
Ненасыщенные карбонильные соединения. Кетены.....................208
Карбоновые кислоты ............................................. 210
Одноосновные насыщенные и ароматические карбоновые кислоты . . 210
Одноосновные ненасыщенные кислоты................................222
Двухосновные кислоты.............................................228
Функциональные производные карбоновых кислот ................... 235
Галогенангидриды карбоновых кислот.............................. 236
Ангидриды карбоновых кислот......................................238
Сложные эфиры карбоновых кислот..................................239
Окси кислоты ....................................................241
Оксокнслоты (альдегидо- и кетокислоты)...........................254
Раздел третий. Элементорганические соединения........................261
Металлорганические соединения .................................. 261
Кремнпйорганическпе соединения...................................265
Серосодержащие органические соединения.....................«... 265
Часть 2. Азотсодержащие органические соединения. Гетероциклические сое-
динения. Биоорганические соединения и некоторые соединения изопрено-
идного характера.....................................................269
Раздел четвертый. Азотсодержащие органические соединения . . . 269
462
Нитросоединения ..................................................270
Амины .........................................г . , ... 277
Понятие о диазо- и азосоединениях.................................287
Диазосоединения ..................................................287
Аминокислоты......................................................293
Пептиды...........................................................302
Амиды кислот......................................................306
Нитрилы...........................................................307
Раздел пятый. Гетероциклические соединения...........................309
Ароматические гетероциклические соединения....................... 310
Пятичленные гетероциклы с одним гетероатомом......................310
Конденсированные системы, содержащие пятичленные гетероциклы
с одним гетероатомом.............................................320
Пятичленные гетероциклы с двумя гетероатомами....................324
Шестичленные гетероциклы.........................................324
Шестичленные гетероциклы с одним атомом азота....................324
Шестичленные гетероциклы с двумя атомами азота...................330
Азотистые бигетероциклы .........................................332
Шестичленные кислородсодержащие гетероциклические соединения
неароматического характера.......................................334
Нуклеиновые кислоты. Строение и биологическая роль................336
Раздел шестой. Биоорганические соединения и некоторые соединения
изопреноидного характера .............................................. 341
Углеводы ......................................................341
Моносахариды .............................................343
Полисахариды...................................................371
Олигосахариды (сахароподобные полисахариды). Дисахариды ... 371
Высокомолекулярные (несахароподобные) полисахариды.............378
Липиды . .......................................................389
Простые липиды..................................................392
Глицериды (ацилглицерины) ......................................392
Карбоновые (жирные) кислоты глицеридов..........................392
Производные глицерина с простой эфирной связью (алкильные липиды;
плазмалогены) .................................................399
Диольные липиды.................................................406
Воски...........................................................401
Гликолипиды.....................................................402
Сложные липиды..................................................403
Фосфолипиды.....................................................403
Сфинголипиды ...................................................408
Анализ липидов ................................................ 410
Получение и переработка жиров и масел ......................... 413
Поверхностно-активные и моющие вещества.........................415
Белковые вещества...............................................417
Изопреноиды.....................................................424
Терпеноиды......................................................424
хМонотерпены и их кислородсодержащие производные................424
Алифатические терпены и их кислородсодержащие производные . . . 425
Моноциклические терпены и их кислородсодержащие производные . . 426
Эфирные масла...................................................427
Бациклические терпены и их кислородсодержащие производные . . . 427
Дптерпены и их производные......................................429
Каротиноиды.....................................................429
Раздел седьмой. Выделение, очистка и идентификация органических
соединений.............................................................431
Выделение и очистка органических соединений.........................431
Физико-химические методы исследования органических соединений . . 439
463
Алексей Петрович Нечаев,
Татьяна Владимировна Еременко
ОРГАНИЧЕСКАЯ ХИМИЯ
Зав. редакцией С. Ф. Кондрашкова. Редактор М. М. Пенкина. Мл. редактор Л. С. Макарки-
на. Технический редактор Т. Д. Гарина. Художественный редактор Т. М. Скворцова. Худож-
ник Ю. Д. Федичкин. Корректор С. К. Завьялова.
ИБ № 5320—5321
Изд. № Хим. — 685. Сдано в набор 17.08.84. Подп. в печать 22.03.85. Формат 60X90716. Бум.
ни. журн. Гарнитура литературная. Печать высокая. Объем 29 усл. печ. л. 58 усл. кр.-отт.
30,41 уч.-изд. л. Тираж 28 000 экз. Зак. № 682. Цена 1 р. 30 к.
Издательство «Высшая школа», 101430, Москва. ГСП-4, Неглинная ул. д. 29/14.
Ярославский полиграфкомбинат Союзполиграфпрома при Государственном комитете СССР
по делам издательств, полиграфии и книжной торговли. 150014, Ярославль, ул. Свободы, 97.
I p. 30 к