/



















































































































































































































































































































































Text
МЕТОДЫ
БИОХИМИЧЕСКОГО
АНАЛИЗА
РАСТЕНИЙ
П65
681.4
УДК 584.19
В книге изложены проверенные и усовершенствованные ав-
тором методы биохимии и физиологии растений, необходимые
при исследовании влияния разных внешних и внутренних фак-
торов и условий питания на растение и его продуктивность.
Описаны методы определения элементов минерального питания
растений, активности некоторых ферментов, интенсивности фо-
тосинтеза и дыхания, транспирации, водного режима растений
и др. Особое внимание уделено ускорению, упрощению и уточ-
нению методов анализа. Описаны новые хроматографические ме-
тоды определения аминокислот, сахаров, дикарбоповых кислот,
в которых применяются новые составы растворителей, дающих
более совершенное разделение исследуемых веществ в одномерных
хроматограммах.
Рассчитана на физиологов, биохимиков, агрохимиков, пре-
подавателей и студентов сельскохозяйственных вузов, биологи-
ческих факультетов университетов и педагогических институтов.
Ответственный редактор
чл.-кор. АН УССР А. С. Оканенко
Рецензенты
д-р хим. наук И. В. Пятницкий,
д-р биол. наук Д. М. Гродзинский
Редакция физиологии, биохимии и медицины
п 21006—003
11М221(04)—76
386—76
(£) Издательство «Наукова думка», 1976 г.
ПРЕДИСЛОВИЕ
Как известно, методы биохимического анализа используются для контроля
качества продукции сельскохозяйственных культур и количественных изменений
в составе веществ растений, а также направленности биохимических “процессов,
происходящих в растениях во время роста и развития. Исследование растений
с помощью биохимических методов дает возможность рациональнее воздейство-
вать на культурные растения в желаемом направлении для получения наиболь-
шего количества продуктов высокого качества и оказывает помощь селекционерам
при выведении новых улучшенных сортов сельскохозяйственных культур. Эти
задачи могут быть успешно решены только при наличии достаточного количества
надежных и ускоренных методов анализа.
Создатель хроматографического метода М. С. Цвет еще в 1910 г. писал, что
существенным условием всякого плодотворного исследования является правильно
разработанная методика, но, к сожалению, часто методика — самая слабая сто-
рона научных исследований. Каждое поколение в своей главной массе ученически
наследует приемы предыдущего и, не подвергая их основательной критике, до-
вольствуясь тем, что они «общеприняты», применяет их для получения новых ре-
зультатов.
Во время бурного развития биологической науки некритическое отношение
к «общепринятым» методам особенно опасно, так как может появиться огромное
количество сомнительных результатов, приводящих к ложным выводам. Наблю-
дается и другая крайность, когда признаются только новые методы, а все старые
считаются непригодными. И в данном случае необходим не менее строгий крити-
ческий подход, чтобы избежать грубых ошибок.
Существует много старых надежных методов, которые уже много лет приме-
няются без изменений. Однако в результате успешного развития аналитической
химии появилось много более чувствительных реактивов, индикаторов и новых
более совершенных приемов работы, которые можно с большой пользой исполь-
зовать и в старых методах, сделав их современными и более совершенными.
В последнее время широкое распространение в биологической химии полу-
чила хроматография, которая используется для разделения органических веществ
близкой природы. Многие вещества во время хроматографирования претерпе-
ают существенные изменения вплоть до полного их исчезновения, поэтому не
могут быть учтены. Для определения этих веществ, а также проявления и опреде-
ления индивидуальных соединений после хроматографирования, необходимы
чувствительные и точные методы химического анализа.
3
В данном руководстве методы изложены в определенном плане и достаточно
подробно чтобы пользуясь ими можно было провести анализ не прибегая к дру-
гим литературным источникам. Часть описанных методов уже опубликована в
разных журналах, источники которых приводятся в списке литературы в конце
каждой главы, а большая часть методов публикуется впервые.
Мы не пытались дать изложение всех существующих биохимических методов,
а только те которые были разработаны или усовершенствованы и проверены в ла-
бораторной практике. Автор с признательностью примет все критические заме-
чания и пожелания, возникшие в процессе использования данных методов.
Глава I
ЭЛЕМЕНТЫ МИНЕРАЛЬНОГО ПИТАНИЯ РАСТЕНИЙ
ОПРЕДЕЛЕНИЕ ПРОЦЕНТНОГО СОДЕРЖАНИЯ ЗОЛЫ,
МАГНИЯ, КАЛЬЦИЯ И ЖЕЛЕЗА ИЗ ОДНОЙ НАВЕСКИ
Сущность метода. Определение этих элементов прово-
дится из одной навески, причем последнюю используют всю цели-
ком. Содержание золы определяют в чистом виде, без песка. Кальций
и магний осаждаются одновременно — кальций в виде щавелево-
кислой соли, а магний в виде магнийаммонийфосфата. Раствори-
мость щавелевокислой соли кальция меньше, чем фосфорнокислой,
а растворимость фосфорнокислого магнийаммония меньше, чем ща-
велевокислого магния. Реакции идут по следующим уравнениям:
СаС12 + Н2С2О4 4- 2NH4OH = СаС2О4 4- 2NH4C1 4- 2Н2О,
MgCl2 4- NH4H2PO4 + 2NH4OH = MgNH4PO4 4- 2NH4C1 4- 2H2O.
Железо остается в растворе, образуя с сульфосалициловой кис-
лотой впутрикомплексное соединение желтого цвета:
3HO3S С6Н3 (ОН) СООН 4- FeCl3 4- 3NH4OH -
= Н3
£00
HO3S С6Н3<
\ О
4- 3NH4C1 + ЗН2О.
Осадок кальция и магния переносят на фильтр и отмывают от
примесей. Фильтрат собирают в мерную колбу, доводят до опреде-
ленного объема и фотометрируют при 440 нм. Осадок помещают
в колбу для титрования, высушивают для удаления избытка аммиа-
ка и определяют сначала магний, а затем кальций объемным мето-
дом. Для определения магния осадок растворяют в титрованной сер-
ной кислоте:
Mg (NH4) РО4 4- H2SO4 = MgSO4 4- NH4H2PO4,
а избыток кислоты оттитровывают едким натром.
Фосфорную кислоту титруют как одноосновную кислоту, если
присутствует щавелевая кислота. В этом случае показатель титро-
вания выше 4,5 и теоретически равен 5,6. Наиболее подходящим
индикатором при титровании является метил рот, который лучше
применять в смеси с метиленовой синью. Раствор индикатора гото-
вят на дистиллированной воде без^пирта, так как последний ме-
шает дальнейшему определению кальция, реагируя с перманганатом.
5
Грамм-эквивалент магния—%—равен 20,16. Определяемые количес-
тва MgO от 3 до 25 мг.
После определения магния здесь же определяют кальций титро-
ванием щавелевой кислоты перманганатом калия в присутствии из-
бытка серной кислоты. Реакция идет при температуре 60—70° С по
следующему уравнению:
5СаС2О4 + 2КМпО4 + 8H2SO4 = K2SO4 + 2MnSO4 + 5CaSO4 +
+ ЮСО2 + 8H2O.
CaO
Грамм-эквивалент кальция —равен 28,04. Определяемые ко-
личества СаО от 4 до 35 мг.
Ход анализа. Среднюю пробу исследуемого вещества су-
шат при температуре не выше 100° С, затем измельчают до однород-
ного состояния и сохраняют в эксикаторе в бумажном пакете.
В предварительно прокаленном и взвешенном на аналитических ве-
сах тигле емкостью около 30 мл отвешивают на технохимических
весах такое количество анализируемого вещества, чтобы в нем на-
ходилось от 3 до 25 мг MgO и от 4 до 35 мг СаО (около 0,6 г листьев,
около 3 г корней и других менее богатых этими элементами органов
растений). Тигли с навесками помещают в сушильный шкаф и су-
шат в течение двух часов при температуре 100° С. Затем охлаждают
в эксикаторе, содержащем хлористый кальций в качестве осушите-
ля, и взвешивают на аналитических весах. Тигли помещают в му-
фельную печь и сжигают пробу при слабом нагреве (450—500° С).
Можно сжигать и на электрической плитке, если пробы накры-
вать асбестовым колпаком. Для этого из асбестового листа изготов-
ляют цилиндрическое кольцо высотой 10 см и диаметром, вмещаю-
щим всю металлическую спираль. Если асбестовый лист тонкий, то
его наматывают в три слоя. После того как тигли помещены на плит-
ку, на нее ставится асбестовое кольцо, которое сверху накрывается
двумя или тремя листами асбеста. Сжигание на плитке удобнее и
быстрее, чем в муфеле.
После сжигания остаток в тигле обрабатывают 1 мл дистилли-
рованной воды, высушивают и снова прокаливают 15—20 мин до
полного сгорания. В золе не должно быть заметно черных частичек
угля, в противном случае обработку водой и прокаливание повто-
ряют. Когда сжигание доведено до конца, тигли охлаждают и взве-
шивают на аналитических весах. Вычитая из полученного веса
тару тигля, находят вес золы с песком.
Для растворения золы и определения содержания песка в тигель
прибавляют 1 мл дистиллированной воды, 2 мл раствора соляной
кислоты (1 1), перемешивают, выпаривают досуха на воздушной
бане и подсушивают при 120—130° С для обезвоживания кремне-
кислоты. К сухому остатку в тигле прибавляют 2 мл раствора соляной
кислоты (1 1), 3 мл воды, перемешивают, нагревают и горячим
фильтруют через беззольный фильтр средней плотности диаметром
6
7 см в коническую колбу на 100—200 мл, или стакан такой же ем-
кости, промывая тигель и фильтр горячей водой (5 раз по 5 мл), да-
вая каждый раз раствору полностью стечь, и один раз в конце про-
мывают капельным способом, направляя капли на край фильтра.
Фильтр, на котором находится песок и кремневая кислота, поме-
щают в тот же тигель, высушивают, прокаливают, охлаждают и
взвешивают. Разница между полученным весом и весом пустого тиг-
ля дает содержание песка и кремневой кислоты в навеске. Из по-
лученных данных вычисляют содержание золы по формуле:
У _ 100 (а — Ь)
где X — содержание золы (в %); а — вес золы с песком и кремне-
вой кислотой (в г); b — вес песка и кремневой кислоты (в а); п —
абсолютно-сухая навеска (в а).
Раствор золы в соляной кислоте, отфильтрованный в кониче-
скую колбу, служит для определения железа, магния и кальция.
Для этого к нему прибавляют 10 лм фосфатно-щавелево-сульфоса-
лицилового реактива, нагревают до 70—80° С, снимают с плитки
и медленно помешивая нейтрализуют по каплям концентрирован-
ным раствором аммиака до появления мути. Затем раствор нагре-
вают в течение 5 мин, прибавляют по каплям при помешивании
еще 1 мл концентрированного аммиака и оставляют до тех пор, по-
ка температура снизится приблизительно до 40° С. Потом снова
прибавляют 5 мл концентрированного аммиака, колбу закрывают
часовым стеклом или воронкой, отверстие которой закрыто влаж-
ной ватой, и оставляют на ночь для полного осаждения магния.
На следующий день раствор фильтруют в мерную колбу на 100 мл
через плотный фильтр диаметром 7 см и промывают раствором ам-
миака (1 10) до тех пор, пока колба наполнится почти до метки.
Определение железа
Раствор в колбе доводят водой до метки, перемешивают и измеряют
оптическую плотность при 440 нм. Нулевым раствором служит во-
да. Из полученных результатов измерений находят на калибровоч-
ной кривой концентрацию Fe2O3 и вычисляют содержание железа
по следующей формуле:
у_ 0,0001 100С 0,01.с
А — ------------ — ------,
п п ’
где X — содержание Fe2O3 (в %); С — концентрация Fe2O3 в коло<
риметрируемом растворе, мкг/мл\ п — абсолютно-сухая навеска
(в г); 100 — общий объем исследуемого раствора железа (в мл)\
0,0001 — коэффициент для пересчета микрограммов в граммы и
проценты.
Построение калибровочной кривой. В мер-
ные колбы на 50 мл последовательно вливают от 1 до 10 мл
7
стандартного раствора железоаммиачных квасцов, содержащего 0,1 мг
Fe2O3 в 1 мл раствора. Затем добавляют 5 мл фосфатно-щавелево-
сульфосалицилового реактива, 3 мл концентрированного раствора
аммиака, доводят водой до метки и измеряют оптическую плотность
в кюветах на 10 или 20 мм с синим светофильтром при 440 нм.
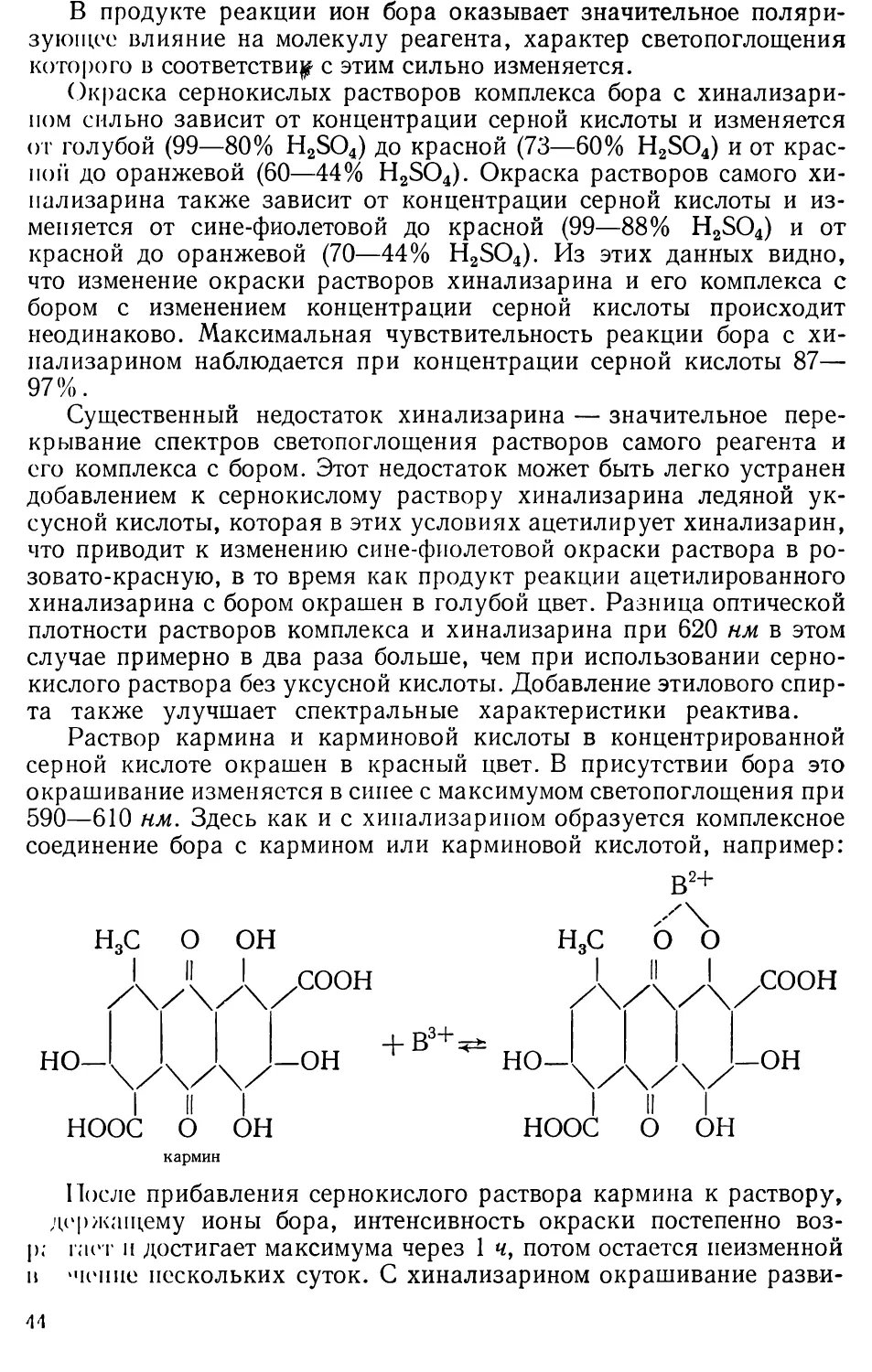
0|--1-1--1—Д--1--1-1--1--1-1
4 3 12 16 20
Концентрация Fe20j,мкг/мл
Рис. 1. Калибровочная кривая
для железа с сульфосалицило-
вой кислотой (светофильтр с мак-
симумом пропускания 440 юи):
На основании полученных результатов
измерений строят калибровочную кри-
вую (рис. 1).
Определяемые количества железа
от 2 до 20 мкг!мл Fe2O3.
Определение магния
Осадок магния и кальция, нахо-
дящийся на фильтре, продолжают
промывать раствором аммиака (1 10),
пока фильтрат перестанет давать
реакцию на хлор. Для проверки на
полноту промывания собирают в про-
бирку 5 мл фильтрата, подкисля-
ют азотной кислотой и прибавляют
1 каплю 1%-ного раствора азотнокис-
лого серебра. При полном промыва-
1 — толщина измеряемого слоя
IX) мм; 2 — толщина измеряемого
слоя 20 мм.
нии раствор в пробирке должен быть
прозрачным. После этого фильтр с
осадком вынимают из воронки, раз-
рывают надвое, помещают в колбу, в которой производилось
осаждение и сушат при 80° С до полного удаления влаги. Затем в
колбу вливают 20 мл дистиллированной воды, 20 мл 0,1 н. серной
кислоты и нагревают до кипения, размешивая и разминая крупин-
ки осадка стеклянной палочкой. После охлаждения палочку выни-
мают, споласкивая ее водой, прибавляют две капли 0,2%-ного рас-
твора метилрота и избыток кислоты титруют 0,1 н. раствором
едкого натра до перехода красного цвета индикатора в желтый. От-
дельно титруют 20 мл раствора серной кислоты 0,1 н. раствором
едкого натра и определяют количество щелочи, которое расходует-
ся на этот объем кислоты. Из полученных данных вычисляют со-
держание магния по следующей формуле:
2,016 К(а — Ь)
где X — содержание MgO (в %); К — нормальность раствора ед-
кого натра; а — объем раствора едкого натра, затраченный на
20 мл 0,1 н. серной кислоты (в мл)\ b — объем раствора едкого
натра, затраченный на исследуемый раствор (в мл)\ п — абсолют-
но-сухая навеска (в а); 2,016 — нормальный титр MgO, умножен-
ный на 100 для пересчета в проценты.
8
Определение кальция
После титрования магния в раствор прибавляют 5 мл серной
кислоты (1 9), нагревают до 70—75° С и титруют 0,05 н. раство-
ром перманганата до появления неисчезающей в течение 5—10 сек
розовой окраски. Содержание кальция вычисляют по формуле:
V 2,8 /< а
где X — содержание СаО (в %); К — нормальность раствора пер-
манганата калия; а — объем титрованного раствора перманганата
калия, затраченный при титровании (в мл); п — абсолютно-сухая
навеска (в г); 2,8 — нормальный титр СаО, умноженный на 100
для пересчета в проценты.
Реактивы. Фосфатно-щавелево-сульфосалициловый реактив. Отвешивают
30 г щавелевой кислоты, 30 г однозамещенного фосфата аммония и 30 г сульфо-
салициловой кислоты. Растворяют эти вещества в воде и доводят объем раствора
до 1 л.
Водный раствор метилрота. Отвешивают 0,1 г индикатора, растирают в ступ-
ке с 7,5 мл 0,05 н. раствора едкого натра и полученный раствор разбавляют водой
до 50 мл. Для приготовления смешанного индикатора добавляют 0,05 г метиле-
новой сини. В последнем случае титруют до грязно-синего цвета. Индикатор со-
храняют в темной склянке.
Стандартный раствор соли железа (III). Отвешивают 0,6040 г свежепере-
кристаллизованных воздушно-сухих железоаммонийных квасцов, растворяют
в небольшом количестве воды, переносят в мерную колбу на 1 л, прибавляют
20 мл серной кислоты (1 9), разбавляют водой до метки и перемешивают. 1 мл
полученного раствора содержит 0,1 мг Fe2O3.
Раствор соляной кислоты (1 1). Отмеривают 500 мл соляной кислоты
пл. 1,19, разбавляют водой до 1 л и перемешивают.
Раствор серной кислоты (1 9). Вливают в колбу 900 мл воды и прибавляют
при помешивании 100 мл серной кислоты пл. 1,84.
Раствор аммиака (1 : 10). К 100 мл концентрированного раствора аммиака
(пл. 0,91, 24—25% NH3) прибавляют 1 л воды, перемешивают и сохраняют
в ск\лянке с притертой пробкой.
Титрованный раствор серной кислоты (0,1 H.J. Приготавливают из фикса-
нала или из химически чистой серной кислоты. В последнем случае набирают
градуированной пипеткой 5,3 мл серной кислоты пл. 1,84, прибавляют к 200 мл
дистиллированной воды и разбавляют водой до 2 л.
Нормальность этого раствора устанавливают по буре. Отвешивают 1,9070 г
буры, растворяют в 60—70 мл воды, количественно переносят в мерную колбу
на 100 мл, разбавляют водой до метки и перемешивают. Набирают пипеткой
25 мл в колбу для титрования, прибавляют 1—2 капли раствора метилоранжа
и титруют серной кислотой до появления неисчезающей розовой окраски.
Из полученных данных вычисляют нормальность раствора кислоты по следующей
формуле:
/<и=-21ГЛ=0’004 а’
где Кк — нормальность титрованного раствора серной кислоты; а — объем рас-
твора серной кислоты, затраченный при титровании 25 мл раствора буры (в мл);
0,1 — нормальность раствора буры; 25 — объем раствора буры, взятый для ти-
трования серной кислотой (в мл).
Титрованный раствор едкого натра (0,1 hJ. Для приготовления титрован-
ного раствора едкого натра исходят из так называемой маслянистой щелочи,
которую готовят следующим образом: 100 г NaOH ч. д. а. растворяют в стакане
в 100 мл дистиллированной воды, охлаждают и переливают в цилиндр на 250 мл,
9
который закрывают пробкой и оставляют до полного оседания углекислого на-
трия. Из верхней прозрачной части раствора набирают пипеткой 6,5 мл масля-
нистой щелочи, выливают в мерную колбу на 1 л, разбавляют безуглекислой во-
дой до 1 л и перемешивают.
Нормальность раствора щелочи устанавливают по титрованному раствору
серной кислоты, титруя ею 25 мл щелочи в присутствии индикатора метилоранжа
или метилрота. Нормальность щелочи вычисляют по следующей формуле:
is __ а
Ащ — 25 ,
где /Сщ — нормальность титрованного раствора едкого натра; Кк — нормальность
титрованного раствора серной кислоты; а — объем серной кислоты, затраченный
при титровании щелочи (в мл)\ 25 — объем раствора едкого натра, взятый для
титрования серной кислотой (в мл).
Титрованный раствор перманганата калия (0,05 н.). Отвешивают 3,2 г
КМпО4, растворяют в горячей воде, разбавляют до 2 л, дают постоять 2—3 дня
и сливают прозрачный раствор или фильтруют через стеклянный фильтр. Нор-
мальность раствора перманганата калия устанавливают по щавелевой кислоте.
Для этого готовят 0,05 и. раствор последней. Отвешивают на аналитических ве-
сах 0,7875 г Н2С2О4 2Н2О, растворяют в воде, переносят в мерную колбу на
250 мл, доводят раствор водой до метки и перемешивают. 25 мл полученного
раствора отбирают пипеткой в колбу для титрования, прибавляют 5 мл раствора
серной кислоты (1 9), нагревают до 70—75° С и титруют раствором перманганата
калия до появления розовой окраски, не исчезающей в течение 10 сек. Последние
мл раствора надо доливать по каплям, прибавляя следующую каплю только тогда,
когда исчезнет окраска от предыдущей капли.
Нормальность раствора перманганата калия вычисляют по следующей фор-
муле:
0,05 25 _ 1,25
а а ’
где К — нормальность титрованного раствора перманганата калия; а — объем
раствора перманганата калия, затраченный при титровании щавелевой кислоты
(в мл)\ 0,05 — нормальность приготовленного раствора щавелевой кислоты;
25 — объем 0,05 н. раствора щавелевой кислоты, взятый для титрования.
КОМПЛЕКСОНОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
МАГНИЯ И КАЛЬЦИЯ
Сущность метода. После растворения золы в соляной
кислоте и удаления железа, алюминия, марганца и фосфорной
кислоты, в аликвотной части раствора титруют сумму кальция
и магния трилоном Б в присутствии индикатора хромогена черно-
го ЕТ-00. В другой части раствора определяют только магний пос-
ле удаления кальция в виде оксалата.
Двунатриевая соль этилендиаминтетрауксусной кислоты
(Na2H2C10H12O3N2 2Н2О) — трилон Б с кальцием, магнием и дру-
гими катионами образует .прочные комплексы. Реакция идет по
следующему уравнению:
Na2H2C10H12O8N2 + Са2+ = Na2CaC10H12O8N2 + 2Н+
372 2
Грамм-эквивалент трилона Б —— равен 186,1.
Комплексные соединения ионов кальция и магния с индика-
тором хромогеном черным менее устойчивы, чем комплексные со-
единения с трилоном Б. Поэтому при титровании трилоном Б в
ю
точке эквивалентности соединения с индикатором разрушаются,
освобождается свободный индикатор и наступает резкий переход
винно-красной окраски в синюю:
М§Л + Н2Я Mg/? + 2Н+ + А2-
где А — анион индикатора; — анион трилона Б.
Как видно из приведенного уравнения, на течение реакции
влияет кислотность раствора, поэтому для полноты взаимодействия
необходима щелочная среда, связывающая ионы водорода. Дис-
социация комплексных соединений также зависит от величины pH
раствора:
Ме2+ + Н2Т? ₽ МеЯ + 2Н+
Чем устойчивее комплексное соединение, тем ниже значение
pH полной диссоциации комплекса. При титровании кальция
и магния трилоном Б в присутствии хромогена черного ЕТ-00
создается среда с pH около 10, при которой комплексные соедине-
ния этих металлов с трилоном Б значительно прочнее, чем с инди-
катором. Алюминий и железо дают более прочные соединения с
индикатором, чем с трилоном Б, и это препятствует их титрова-
нию.
Хромоген черный ЕТ-00 в щелочном растворе очень чувстви-
телен к окислителям. В присутствии даже следов марганца (II),
каталитически ускоряющего окисление кислородом воздуха, ин-
дикатор быстро обесцвечивается. Фосфорная кислота в щелочном
растворе связывает кальций и магний, чем затрудняет титро-
вание; конец титрования растянут и не четкий. Поэтому все ме-
шающие вещества должны быть удалены из раствора. Чтобы сле-
ды марганца не мешали титрованию, к раствору перед прибавлением
индикатора приливают раствор гидроксиламина, который, являясь
сильцым восстановителем, служит защитой против окислителей.
Фосфорная кислота удаляется осаждением ионами трехвалентно-
го железа.
Ход анализа. В тигле емкостью 30 мл взвешивают опре-
деленное количество воздушно-сухого исследуемого материала:
0,5 а листьев и 2 а корней. В навеске должно содержаться от 2 до
20 мг MgO и от 3 до 28 мг СаО. Навеску высушивают до постоян-
ного веса и сжигают в муфеле при 400—500° С (или на плитке под
асбестовым колпаком). Остаток обрабатывают 1—2 мл дистил-
лированной воды, высушивают и снова прокаливают до полного
сгорания частичек угля. После сжигания и охлаждения прили-
вают в тигель 2 мл воды и 1 мл раствора соляной кислоты (1 1).
Содержимое тигеля нагревают до кипения и переносят в мерную
колбу на 50 мл. К полученному раствору приливают 2 мл 1%-ного
раствора хлорного железа, колбу ставят на плитку и снова на-
гревают почти до кипения. Затем раствор снимают с плитки и при-
бавляют при помешивании 2 мл аммиачного буферного раствора.
Содержимое колбы охлаждают, доводят до метки, перемешивают
и фильтруют в сухую колбу через сухой фильтр.
11
Определение суммы магния и кальция
Набирают пипеткой 20 мл фильтрата в колбу дл$Я титрования,
прибавляют 2 мл 1 %-кого раствора гидроксиламина, 50 мг смеси
индикатора хромогена черного ЕТ-00 с хлористым ^атрием, 2 мл
аммиачного буферного раствора и титруют 0,02 н. раствором три-
лона Б до перехода винно-красной окраски в синююх
Определение магния
Из остатка фильтрата набирают пипеткой 20 мл> выливают в
стакан или колбу и для осаждения кальция прибавляет 2 мл 10 % -
ного раствора оксалата калия. Содержимое перемешивают, до-
бавляют 0,5 мл аммиачного буферного раствора и с^ерез 20 мин
выделившийся осадок отфильтровывают через бумажный фильтр
в колбу для титрования, промывая колбу, в которой осуждали каль-
ций, и фильтр 25 мл воды.
После фильтрования к раствору прибавляют 2 мл 1%-ного
раствора гидроксиламина, 50 мг смеси индикатора хромогена чер-
ного ЕТ-00 с хлористым натрием, 2 мл аммиачного буферного
раствора и титруют 0,02 н. раствором трилона Б до перехода вин-
но-красной окраски индикатора в синюю.
Содержание окиси магния вычисляют по следующей формуле:
у 2,016 50 к а 5,04 К а
где X — содержание MgO (в %); X — нормальность раствора
трилона Б; а — количество раствора трилона Б, затраченное при
титровании магния (в мл)\ п — абсолютно-сухая навеска (в г);
2,016 — нормальный титр MgO, умноженный на 100 Для пересче-
та в проценты; 50 — общий объем исследуемого раствора золы
(в мл)\ 20 — объем исследуемого раствора, взятый для! титрования
магния трилоном Б (в мл).
Содержание окиси кальция вычисляется по формуле:
у_ 2,8 50 К(Ь — а) 7 К(Ь — д)
Г п 20 п
где Y — содержание СаО (в %); b — количество раствора трилона
Б, затраченное при титровании суммы кальция и магния (в мл)\
2,8 — нормальный титр СаО, умноженный на 100 для пересчета
в проценты.
Остальные показатели такие же, как в формуле, Приведенной
выше для вычисления содержания окиси магния.
Реактивы. Стандартный раствор сернокислого магния (0,02 nJ. Отвешивают
на аналитических весах 2,4650 г MgSO4-7H2O, растворяют в дистиллированной
воде, приливают 5 мл серной кислоты (1 9), раствор переносят в мерную колбу
на 1 л, доводят водой до метки и тщательно перемешивают.
Титрованный раствор трилона Б (0,02 н.). Отвешивают 3,7? г трилона Б,
растворяют его в 1 л дистиллированной воды и, если раствор мутней, фильтруют.
12
Нормальность раствора устанавливают по стандартному раствору сернокислого
магния. Для этого отбирают пипеткой 25 мл 0,02 н. раствора сернокислого магния,
прибавляют 3 мл аммиачного буфера, 50 мг смеси индикатора хромогена черного
ЕТ-00 с хлористым натрием и титруют раствором трилона Б до перехода окраски
из вишнево-красной в синюю. Нормальность трилона Б вычисляют по формуле:
„ 0,02 25
где К — нормальность раствора трилона Б; а — количество раствора трилона Б,
затраченное на титрование 25 мл 0,02 н. раствора сернокислого магния (в мл).
Индикатор хромоген черный ЕТ-00 или кислотный хром темно-синий
(1 100). Отвешивают 0,2 г индикатора ч. и 20 г хлористого натрия х. ч., высы-
пают в ступку и тщательно растирают. Сохраняют в темной склянке.
Аммиачный буферный раствор (pH 10). Отвешивают 55 г хлористого аммо-
ния х. ч., растворяют в 350 мл 25%-ного раствора аммиака и разбавляют дистил-
лированной водой до 1 л.
Раствор хлорного железа (1%-ный). Отвешивают 5 a FeCl3-6H2O, растворяют
в 200 мл воды, подкисляют 10 мл раствора соляной кислоты (1 : 1), разбавляют
водой до 500 мл и тщательно перемешивают.
Раствор оксалата калия (10%-ный). Отвешивают 50 г К2С2О4, растворяют
в воде, разбавляют до 500 мл, перемешивают и, если нужно, фильтруют.
Раствор сернокислого гидроксиламина (1%-ный). Отвешивают 1 г (NH2OH)2 •
• H2SO4, растворяют в воде и доводят до 100 мл.
Раствор соляной кислоты (1 1). (Приготовление см. на стр. 9).
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КАЛИЯ И НАТРИЯ
Сущность метода. Пробу исследуемого материала сжи-
гают и полученную золу растворяют в соляной кислоте, которую
затем нейтрализуют окисью цинка. При этом осаждается фосфор-
ная кислота и полуторные окислы. Фильтрат служит для опре-
деления калия и натрия. Ионы натрия дают с цинкуранилацета-
том труднорастворимый осадок:
(UO2)3 Zn (СН3СОО)8 + CH3COONa = (UO2)3 ZnNa (СН3СОО)9.
После промывания осадка этиловым спиртом входящий в его
состав цинк переводят в другое, менее растворимое соединение —
тетрароданомеркуриат цинка, действием тетрароданомеркуриата
калия:
(UO2)3 ZnNa (СН3СОО)# + К2 [Hg (CNS)J = Zn [Hg (CNS)J 4-
4- 3UO2 (CH3COO)2 4- 2CH3COOK 4- CH3COONa.
После центрифугирования осадка и промывания его растворяют
в соляной кислоте и окисляют роданид-ион титрованным раство-
ром йодата калия. При этом идет следующая реакция:
Zn [Hg (CNS)J + 6КЮ3 4- 8НС1 = 6НС1 4- 4HCN 4- HgCl2 4-
4- 3K2SO4 4- ZnSO4 4- 2H2O.
Избыток йодата калия определяют йодометрическим методом.
Для этого прибавляют избыток йодистого калия и выделившийся
йод титруют тиосульфатом натрия:
KJO3 4- 5KJ 4- 6НС1 = 6КС1 4- 3J2 4- ЗН2О,
J2 4- 2Na2S2O3 = 2NaJ 4- Na2S4O6.
13
На окисление роданид-иона расходуется только четыре заряда
йода, так как он из пятивалентного переходит в одновалентный:
J4’54-4e=J+1 Следовательно, на 1 атом натрия приходится 6
молекул KJO3 или всего 24 заряда. Отсюда грамм-эквивалент нат-
рия равен 0,958 и ----------1,291. Этот способ дает возможность
определять небольшие количества натрия (от 0,1 до 2 мг Na2O)
обычным объемным методом благодаря большому молекулярному
весу осадка Zn [Hg (CNS)J, содержащему четыре роданид-иона,
которые приходятся на 1 атом натрия.
Калий в нейтральной среде с кобальтинитритом натрия в при-
сутствии избытка ионов натрия дает осадок желтого цвета по сле-
дующей реакции:
2КС1 + Na3 [Со (NO2)6] = K2Na [Со (NO2)6] + 2NaCl.
Для сохранения состава осадка его промывают раствором,
содержащим ионы натрия и небольшое количество ионов калия.
Промытый осадок окисляется бихроматом калия в сернокислой
среде и растворяется. Реакция идет по следующему уравнению:
6K2Na [Со (NO2)6J + 1 1К2Сг2О7 + 41H2SO4 = 11 Сг2 (SO4)3 +
+ 6Со (NO3)2 + 6NaNO3 -|- 18KNO3 8K2SO4 -h 41Н2О.
Избыток бихромата калия титруется солью Мора в присутствии
индикатора фенилантраниловой кислоты или тиосульфатом нат-
рия после добавления йодистого калия. Бихромат калия в сер-
нокислой среде реагирует с сульфатом железа (II) соли Мора по
следующему уравнению:
К2Сг2О7 + 6FeSO4 + 7H2SO4 = Сг2 (SO4)3 + 3Fe2 (SO^ +
K2SO4 + 7H2O.
При йодометрическом определении избытка бихромата калия
добавляют йодистый калий, который вступает в реакцию с бихро-
матом калия, выделяя йод:
К2Сг2О7 + 6KJ + 7H2SO4 - 3J2 + Cr2 (SO4)3 + 4K2SO4 + 7H2O.
Выделившийся йод титруют раствором тиосульфата натрия в при-
сутствии раствора крахмала как индикатора:
J2 + 2Na2S2O3 = Na2S4Oe + 2NaJ.
Грамм-эквивалент калия равен 8,563 и ----------7J08. Опре-
деляемые количества от 0,5 до 10 мг К2О.
Ход анализа. Во взвешенном на аналитических весах
тигле на 30 мл отвешивают с точностью до 0,01 г такое количество
исследуемого материала, чтобы в нем содержалось от 0,25 до 4 мг
Na2O и от 2,5 до 50 мг К2О (0,4 г воздушно-сухих листьев, 1 г кор-
ней сахарной свеклы и других объектов с малым содержанием ще-
14
лочных металлов). Тигли с навесками помещают в сушильный шкаф
и сушат при температуре 105° С до постоянного веса. Затем тигли
помещают в муфельную печь или на плитку с открытой спиралью
под асбестовый колпак и сжигают при температуре не выше 600° С.
После сжигания тигли охлаждают и золу обрабатывают 1 мл дис-
тиллированной воды. При наличии темных частичек угля высуши-
вают на плитке и прокаливают еще 15—20 мин. После полного
сжигания золу в тигле смачивают 1 мл дистиллированной
воды, добавляют 10 капель раствора соляной кислоты (1 : 1),
нагревают до растворения золы, затем прибавляют еще 5 мл
воды, нагревают до кипения, прибавляют 1 мл 10 %-ной суспен-
зии окиси цинка, размешивают и кипятят 1—2 мин. При этом
с помощью окиси цинка осаждается вся фосфорная кислота, ме-
шающая определению натрия. Содержимое тигля переносят через
воронку в мерную колбу на 25 мд, доводят до метки, перемешивают
и фильтруют в сухую колбу через сухой фильтр, выбрасывая пер-
вую порцию фильтрата (3—4 мл). Затем 5 мл фильтрата отбирают
пипеткой в центрифужную пробирку для определения калия и
10 мл (при исследовании листьев сахарной свеклы — 3 мд) в фар-
форовую чашку емкостью 25—30 мл для определения натрия.
Определение натрия
Чашку ставят на водяную или воздушную баню и выпаривают
раствор досуха, не допуская прокаливания. После охлаждения
остаток в чашке смачивают 0,2 мл дистиллированной воды. Рас-
твор сливают в центрифужную пробирку, промывая стенки чашки
раствором цинк-уранилацетата 3 раза по 1 мл. Пробирку встряхи-
вают и оставляют на 1 ч для полноты осаждения осадка. Затем
центрифугируют, раствор сливают, а осадок промывают 1 раз раст-
вором цинкуранилацетата (1 мл) и 4 раза этиловым спиртом (по
2 мл), каждый раз центрифугируя и осторожно сливая раствор с
осадка по стеклянной палочке. К промытому осадку приливают
5 мл 10 %-ной уксусной кислоты и взбалтывают до его растворения.
Цинк, входящий в состав осадка, осаждают прибавлением 2 мл
0,1 н. раствора K2lHg (CNS)4L Через 20—30 мин отстаивания
осадок центрифугируют и промывают холодной водой 3 раза по
3 мл.
Промытый осадок Zn [Hg (CNS)4| растворяют в 5 мл соляной
кислоты (1 1) и выливают в колбу для титрования, промывая про-
бирку водой 4 раза по 2 мл. Большое количество воды в данном
случае вредит, поэтому следует прибавлять ее не более 10 мл. Затем
к раствору прибавляют пипеткой 10 мл 0,05 М раствора йодата
калия и перемешивают. Избыток йодата калия определяют с помо-
щью йодометрического метода. Для этого приливают 3 мл 20%-ко-
го раствора КJ и выделившийся йод титруют 0,1 н. раствором тио-
сульфата натрия в присутствии индикатора крахмала. После при-
бавления йодата калия раствор должен быть светло-желтого цвета,
15
что показывает наличие избытка йодата. Отдельно титруют 10 мл
0,05 М раствора KJO3 после прибавления 3 мл 20%-ного раствора
KJh 5 мл раствора соляной кислоты (1 1). В результате находят
количество тиосульфата, расходуемое на 10 мл йодата калия. Со-
держание натрия вычисляют по следующей формуле:
у _ 0,1291 25 к (а — Ь) _ 0,323 К (а — Ь)
Л 10/1 л
где X — содержание Na2O (в %); К — нормальность раствора тио-
сульфата натрия; а — объем титрованного раствора тиосульфата
натрия, затраченный на 10 мл йодата калия (в мл)\ b — объем ра-
створа тиосульфата натрия, затраченный на исследуемый раствор
(в мл)\ п — абсолютно-сухая навеска (в а); 25 — объем, в кото-
ром растворена зола из навески п (в мл)\ 10 — объем исследуе-
мого раствора, взятый для определения натрия (в мл)\ 0,1291 —
нормальный титр Na2O, умноженный на 100 для пересчета в про-
центы.
Определение калия
К 5 мл исследуемого раствора в центрифужной пробирке при-
бавляют 1 мл 10%-ного раствора кобальтнитрита натрия, содер-
жащего 20% NaNO2, перемешивают и оставляют на 2—3 ч для
полного выделения осадка. После этого центрифугируют и надоса-
дочную жидкость удаляют осторожным сливанием ее по стеклянной
палочке. Осадок получается плотный и взмучивания не происхо-
дит. К осадку приливают 5 мл промывной жидкости, содержащей
5% NaNO3 и 0,008% KNO3, осадок взмучивают и центрифу-
гируют. Прозрачный раствор над осадком сливают и промывают
осадок еще два раза, прибавляя каждый раз по 5 мл промывной
жидкости.
К промытому осадку приливают 3 мл 0,5 н. раствора бихрома-
та калия в серной кислоте и размешивают стеклянной палочкой
до полного растворения осадка. Раствор переносят в колбу для
титрования на 100 мл, смывая остаток 15 мл дистиллированной
воды. В дальнейшем избыток бйхромата калия можно определять
титрованием солью Мора или йодометрическим методом. В первом
случае к раствору прибавляют 4 капли 0,2%-ного раствора фе-
нилантраниловой кислоты и титруют 0,05 н. раствором соли Мора
до перехода вишнево-фиолетовой окраски в зеленую.
При определении избытка бихромата калия йодометрическим
методом к раствору в колбе прибавляют 30 мл дистиллированной
воды, 3 мл 20%-ного раствора йодистого калия, перемешивают,
и выделившийся йод титруют 0,05 н. раствором тиосульфата нат-
рия, прибавляя в качестве индикатора 1 мл 0,5%-ного раствора
крахмала.
В том и другом случае отдельно титруют 3 мл 0,5 н. раствора
бихромата после предварительного разбавления таким же коли-
Л6
чеством дистиллированной воды. Содержание калия вычисляют
по формуле:
у = 0,856 25 К(а — Ь) = 4,28 К(а — Ь)
5 п п ’
где X — содержание К2О (в %); К — нормальность титрованного
раствора соли Мора или тиосульфата натрия; а — объем раствора
соли Мора или тиосульфата натрия, израсходованный на 3 мл
0,5 н. раствора бихромата калия (в мл); b — объем раствора соли
Мора или тиосульфата натрия, израсходованный на избыток бих-
ромата калия (в мл); 25 — общий объем исследуемого раствора
(в мл); 5 — объем исследуемого раствора, взятый для осаждения
и определения калия (в мл); п — абсолютно-сухая навеска (в г);
0,856 —нормальный титр К2О, умноженный на 100 для пересчета
в проценты.
Реактивы. Раствор соли Мора (0,05 н.). 20 г соли Мора (NH4)2SO4* FeSO4
6Н2О взвешивают с точностью до 0,01 г, растворяют в дистиллирован-
ной воде, добавляют 20 мл концентрированной серной кислоты, разбавляют дис-
тиллированной водой до 1 л и перемешивают. Нормальность раствора устанавли-
вают по 0,1 н. раствору бихромата калия (см. стр. 138), и проверяют в день
определения калия.
Суспензия окиси цинка. 10 г окиси цинка х. ч., растирают в ступке с дистил-
лированной водой, прибавляя ее постепенно и доводя объем до 100 мл. Перед
набиранием необходимо тщательно взбалтывать.
Раствор уксусной кислоты (10%-ный). Отмеривают 100 мл ледяной уксусной
кислоты х. ч. и разбавляют до 1 л дистиллированной водой.
Цинкуранилацетат. В плоскодонную колбу па 1 л всыпают 53 г уксусно-
кислого уранила и 200 г уксуснокислого цинка, прибавляют 40 мл ледяной ук-
сусной кислоты и 700 мл дистиллированной воды, нагревают до растворения,
охлаждают, переносят в мерную колбу па 1 л, доводят водой до метки, переме-
шивают, дают постоять сутки, затем фильтруют и сохраняют в темной склянке.
Этиловый спирт (95%-иый). Применяется спирт ректификат.
Раствор тетрароданомеркуриата калия К2 [Hg (CNS)4J. Отвешивают 20 г
роданистого калия KCNS, растворяют в 200 мл воды в мерной колбе на 1 л,
затем добавляют 15 г HgSO4, взбалтывают до растворения, разбавляют водой до
метки и, если нужно, фильтруют.
Раствор йодата калия (0,05 М). Отвешивают на аналитических весах
10,7010 г KJO3 х. ч., растворяют в воде в мерной колбе на 1 л, разбавляют водой
до метки и хорошо перемешивают. На 10 мл этого раствора расходуется 30 мл
0,1 и. раствора тиосульфата натрия.
Раствор йодистого калия (20%-ный). Отвешивают 20 г KJ ч. д. а., растворяют
в воде, прибавляют 1 мл 2 н. раствора едкого натра, доводят водой до 100 мл
и сохраняют в темной склянке.
Титрованный раствор тиосульфата натрия (0, /н.). Отвешивают 25 г тио-
сульфата натрия Na2S2O3 5Н2О, растворяют в воде, в которой предварительно
растворено 0,2 г Na2CO3, затем объем доводят до 1 л водой, тщательно перемеши-
вают и устанавливают нормальность по бихромату калия. Для этого готовят 0,1 н.
раствор бихромата калия. Взвешивают 4,9035 г К2Сг2О7 х. ч., растворяют в воде,
переносят в мерную колбу на 1 л, доводят водой до метки и тщательно перемеши-
вают. Набирают пипеткой 25 мл этого раствора в колбу для титрования, прибав-
ляют 5 мл 20%-ного раствора йодистого калия, 10 мл серной кислоты (1 : 9) и тит-
руют тиосульфатом натрия в присутствии 1 мл 0,5%-ного раствора крахмала.
Нормальность раствора тиосульфата натрия вычисляют по формуле:
„ 25 0,1
2 6-2
17
где К — нормальность титрованного раствора тиосульфата; а — объем тиосуль-
фата, затраченный на 25 мл бихромата калия (в мл).
Раствор крахмала (0,5%-ный). Отвешивают 2,5 г растворимого крахмала
и 10 мг HgJ2 (для консервирования), растирают в фарфоровой ступке с небольшим
количеством воды и вливают в 500 мл кипящей воды, кипятят 1 мин и, если нуж-
но, фильтруют.
Регенерация урановых остатков. К фильтрату прибавляют при взбалтывании
концентрированный раствор аммиака до тех пор, пока прекратится выделение
желтого осадка. Последний отфильтровывают и промывают 2—3 раза раствором
аммиака (1 : 10). Осадок растворяют в 2 н. азотной кислоте и осаждают снова
избытком аммиака, фильтруют через воронку Бюхнера с отсасыванием, промывая
несколько раз разбавленным аммиаком. Полученную соль (NH4)2U2O7 высушивают
при 60—70° С. Затем из нее готовят реактив на натрий: в мерную колбу на 500 мл
помещают 20 г (NH4)2U2O7 и 100 г уксуснокислого цинка, добавляют 35 мл ледя-
ной уксусной кислоты и 300 мл воды, нагревают до растворения, охлаждают,
доводят до метки водой, перемешивают, дают постоять сутки, после чего фильт-
руют и сохраняют в темной склянке.
Раствор кобальтинитрита натрия (10%-ный). Отвешивают 10 г реактива
Na3 [Со (NO2)6], растворяют в 100 мл 20%-ного раствора NaNO2, фильтруют и
сохраняют в темной склянке.
Промывная жидкость (5%-ный NaNO3 и 0,008%-ный KNO3). Отвешивают
50 г химически чистого азотнокислого натрия, растворяют в воде, прибавляют
8 мл 1%-ного раствора азотнокислого калия и разбавляют водой до 1 л.
Раствор тиосульфата натрия (0,05 н.). Готовят из 0,1 н. титрованного
раствора тиосульфата натрия разбавлением в два раза водой (приготовление 0,1 н.
раствора тиосульфата см. на стр. 17). Приготовление остальных реактивов из-
ложено на стр. 9, 10, 138.
ОПРЕДЕЛЕНИЕ НАТРИЯ В ВИДЕ АНТИМОНАТА
Сущность метода. Исследуемый материал сжигают в
муфеле или на плитке под асбестовым колпаком, при этом зола
растворяется в соляной кислоте. Кальций, магний и другие ме-
шающие катионы удаляются осаждением оксихинолином и щавеле-
вой кислотой:
СаС12 + Н2С2О4 2NH4OH - СаС2О4 + 2NH4C1 + 2Н2О,
MgCl2 2C9H6NOH 2NH4OH = Mg (C9HGNO)2 + 2NH4C1 + 2H2O.
После отделения осадка центрифугированием избыток щаве-
левой кислоты и оксихинолина удаляют после выпаривания про-
каливанием, а в остатке осаждают натрий в виде антимоната:
NaCl + К [Sb (ОН)6] = Na [Sb (ОН)6] + КС1.
Осадок отмывают от избытка антимоната калия и растворяют
в соляной кислоте в присутствии йодистого калия, который реа-
гирует с сурьмой, выделяя йод:
Na [Sb (ОН)6] + 2KJ + 6НС1 = J2 + SbCl3 + NaCl + 2KC1 + 6H2O.
Выделившийся йод титруют 0,01 н. раствором тиосульфата
натрия. Грамм-эквивалент натрия -у- равен 11,5, а —----------15,5.
Растворимость антимоната натрия в воде довольно высокая —
около 40 мг в 100 мл воды, поэтому осадок следует промывать не
водой, а этиловым спиртом. Для промывания берут 40%-ный рас-
18
твор спирта в воде, так как чистый спирт не растворяет антимо-
нат калия, который необходимо отмыть. Антимонат натрия легко
образует пересыщенные растворы, из которых осаждение его
затруднено, поэтому осаждение натрия проводится действием
реактива на сухую соль натрия, выделившуюся на дне
чашки после выпаривания, что дает отличные результаты.
Антимонаты калия и натрия легко реагируют с кислотами,
образуя аморфный осадок ортосурьмяной кислоты:
Na [Sb (ОН)6] + НС1 = H3SbO4 + NaCl + 2Н2О.
Сильнощелочная среда также вредна, так как способствует
растворению осадка:
Na [Sb (OH)6J + 2NaOH = Na3SbO4 + 4H2O.
Отсюда следует, что осаждение натрия необходимо проводить
в нейтральной среде, но можно и в слабощелочной, что даже лучше,
так как нет опасности выделения осадка ортосурьмяной кислоты.
В данном методе после выпаривания и прокаливания образуют-
ся сухие соли щелочных металлов, частично карбонаты, имеющие
слабощелочную реакцию, вполне подходящую для осаждения
натрия. Антимонат калия дает осадки также и со щелочноземель-
ными металлами, которые перед осаждением натрия необходимо
полностью удалить с помощью оксихинолина и щавелевой кислоты.
Ход анализа. Навеску порошкообразного воздушно-
сухого материала, содержащего от 0,2 до 2,5 мг Na2O (1 г сухого
корня сахарной свеклы, 0,5 г листьев), помещают в тарированный
фарфоровый тигель и высушивают в сушильном шкафу при 100° С
до постоянного веса. Тигель ставят на электрическую плитку,
находящуюся в вытяжном шкафу, накрывают асбестовым колпа-
ком и сжигают около 1 ч. Затем тигель охлаждают, добавляют 5
капель дистиллированной воды, высушивают и снова прокаливают
на плитке под колпаком до полного сгорания частичек угля (в те-
чение 20—30 мин).
К золе в тигле прибавляют 2 мл дистиллированной воды, 1 мл
соляной кислоты (1 : 1), нагревают до кипения и содержимое тиг-
ля переносят в центрифужную пробирку на 20 мл, промывая
тигель дистиллированной водой (2 раза по 4 мл). К раствору
в пробирке приливают 2 мл 3%-ного раствора оксихиноли-
на в 2 %-пой щавелевой кислоте, добавляют 1 каплю раствора
метилоранжа и нейтрализуют концентрированным рас-
твором аммиака, добавляя его по каплям, при взбалтывании до
исчезновения красной окраски индикатора. Затем добавляют еще
5 капель аммиака, перемешивают, дают постоять 10 мин и центри-
фугируют 2—3 мин при 2,5—3000 об/мин. Прозрачный раствор
сливают в фарфоровую чашку диаметром 6 им, а к осадку прибав-
ляют 5 мл дистиллированной воды, перемешивают, центрифуги-
рую!' н раствор выливают в ту же чашку. Раствор выпари-
вают па воздушной бане досуха и остаток прокаливают сначала
19
при слабом, а затем при более сильном нагреве под асбестовым
колпаком на электроплитке с открытой спиралью до полного сго-
рания органического вещества.
После охлаждения в чашку прибавляют 1 мл дистиллирован-
ной воды, смачивают стенки чашки до растворения осадка, по-
мещают чашку на воздушную баню, выпаривают досуха и про-
каливают на плитке под колпаком в течение 15 мин. Чашке дают
охладиться и в нее осторожно по стенке вливают пипеткой 1 мл
раствора антимоната калия и поворачивают чашку так, чтобы смо-
чить весь осадок, не взмучивая его. Дают в таком виде постоять
15 мин, затем добавляют еще 1 мл реактива и оставляют на 30 мин,
время от времени покачивая чашку для перемешивания реактива,
но не очень сильно, чтобы не взболтать осадок. Такой способ
работы обеспечивает полноту осаждения натрия.
По истечении указанного времени, раствор в чашке переме-
шивают стеклянной палочкой и содержимое выливают в центри-
фужную пробирку с суженным пижним концом. При этом необхо-
димо возможно полнее перенести раствор, а не осадок антимоната
натрия, который может оставаться в чашке. Центрифугируют и
прозрачный раствор удаляют, сливая его по стеклянной палочке
как можно полнее. Осадок в чашке промывают 2;ил40%-ного рас-
твора этилового спирта, который затем переливают в центрифуж-
ную пробирку и центрифугируют. Затем раствор сливают, а оса-
док в чашке и в пробирке промывают еще 1 раз 2 мл 40%-ного
спирта.
К промытому осадку в чашке прибавляют 1 мл 20%-ного раство-
ра йодистого калия и 1 мл раствора соляной кислоты (1 1), раз-
мешивают стеклянной палочкой до растворения осадка и раствор
переносят в центрифужную пробирку, смывая остаток 2 мл соляной
кислоты (1 1). Выделившийся йод титруют 0,01 н. раствором тио-
сульфата натрия, прибавляя в конце титрования крахмал в ка-
честве индикатора. Если йода мало, титруют прямо в пробирке,
в противном случае раствор переносят в колбу.
1 мл 0,01 н. раствора тиосульфата соответствует 0,155 мг Na2O
или 0,115 мг Na. Содержание натрия в исследуемом материале
вычисляется по следующей формуле: "
v 1,55 К а
Л =-----------,
п ’
где X — содержание Na2O (в %); а — количество титрованного
раствора тиосульфата натрия, затраченное при титровании 4иода
(в мл)\ К — нормальность раствора тиосульфата натрия; п — аб-
солютно-сухая навеска (в г); 1,55 — нормальный титр Na2O,
умноженный на 100 для пересчета в проценты.
Осаждение натрия в чашках имеет неудобство — не видно об-
разования белого осадка на белом фоне чашки, поэтому трудно
судить о качестве и количестве его. Для устранения этого недо-
статка осаждение можно проводить в центрифужных пробирках,
20
хотя это несколько и замедляет ход анализа. В этом случае после
удаления оксихинолина и щавелевой кислоты прокаливанием
в чашку приливают 1 мл дистиллированной воды, растворяют
осадок и переносят раствор в центрифужную пробирку.
Чашку промывают (2 раза водой по 0,5 мл) и сливая промывные
воды в пробирку, которую в металлическом штативе помещают
в сушильный шкаф, при температуре 120° С выдерживают до пол-
ного высушивания. После этого охлаждают, добавляют по стен-
ке пробирки 1 мл раствора антимоната калия и не перемешивая
оставляют на 15 мин, затем добавляют еще 1 мл реактива,
перемешивают легким поворачиванием пробирки и оставляют
на 30 мин. Потом содержимое взбалтывают и центрифуги-
руют. Раствор осторожно сливают с осадка по стеклянной
палочке, а осадок промывают (2 раза по 2 мл 40%-ным раствором
спирта). К промытому осадку прибавляют раствор йодистого ка-
лия, соляную кислоту и титруют тиосульфатом натрия так, как
изложено выше.
Реактивы. Раствор антимоната калия. Нагревают 110 мл воды в колбе до
кипения, прибавляют 2 г пиросурьмянокислого калия K2H2Sb2O7 4Н2О, 3 мл
0,1 н. раствора КОН и кипятят до растворения. Нагревание прекращают, при-
бавляют около 20 мг Na [Sb (ОН)6 ] для получения насыщенного раствора этой
соли, взбалтывают, охлаждают и оставляют на ночь. На следующий день фильт-
руют через бумажный фильтр в полиэтиленовую посуду, где и сохраняют.
Необходимое количество соли Na [Sb (OH)G] получают следующим образом.
В центрифужную пробирку наливают 10 мл дистиллированной воды, прибавляют
0,2 г пиросурьмянокислого калия, ставят в стакан с кипящей водой и размеши-
вают до растворения соли. Затем приливают 1 мл 4%-ного раствора NaCl, взбал-
тывают, охлаждают под краном и через 10 мин центрифугируют. Раствор удаляют,
а осадок промывают 5 мл холодной дистиллированной воды. Осадок антимоната
натрия переносят в раствор антимоната калия, смывая его этим же раствором.
Реактив сохраняется в течение двух недель.
3%-ный раствор оксихинолина в 2%-ный щавелевой кислоте. Отвешивают
2 г Н2С2О4 2Н2О и 3 a C0H6NOH, всыпают в колбу, прибавляют 100 мл воды и
нагревают до растворения. Раствор охлаждают и переливают в темную склянку
для хранения.
Этиловый спирт (40%-ный). К 90 мл 95%-ного этилового спирта прибавляют
130 мл воды и перемешивают.
Приготовление остальных реактивов приведено на стр. 9, 17, 18.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ХЛОРА,
ФОСФОРА И МАРГАНЦА
Сущность метода. После озоления исследуемого ма-
териала и растворения золы в азотной кислоте хлор осаждают
азотнокислым серебром и раствор разбавляют до определенного
объема. В растворе после отделения его от осадка фильтрованием
определяют избыток ионов серебра титрованием йодистым калием.
Фосфор после осаждения его в виде фосфорномолибденовой кис-
лоты определяют объемным методом, а марганец — фотометриче-
ским персульфатным методом.
При сжигании органического вещества значительная часть
хлора теряется. Для избежания потерь исследуемую пробу
21
обрабатывают перед сжиганием раствором углекислого натрия или
раствором уксуснокислого бария. Последний является более под-
ходящим веществом, так как сжигание идет легче и при более низ-
кой температуре.
При титровании избытка раствора азотнокислого серебра
йодистым калием в качестве индикатора применяют крахмал в
присутствии нитрита натрия. Пока йодистый калий реагирует с
азотнокислым серебром:
KJ + AgNO3 = AgJ + KNO3
образуется труднорастворимый осадок йодистого серебра. В рас-
творе отсутствуют ионы йода, но после того как все ионы сереб-
ра свяжутся, в растворе появляются ионы йода, которые вступают
в реакцию с нитритом натрия, выделяя свободный йод:
2KJ + 2NaNO2 + 4HNO3 = J2 + 2KNO3 + 2NaNO3 + 2NO + 2H2O.
Выделившийся свободный йод с крахмалом дает ясно видимую
синюю окраску, поэтому можно без особых трудностей титро-
С1
вать 0,01 н. растворы. Грамм-эквивалент хлора — равен 35,46.
Обычно марганец в золе находится преимущественно в шести-
валентном состоянии, соединения которого окрашены в зеленый
цвет. При растворении в азотной кислоте он может переходить
в труднорастворимый осадок МпО2. Для растворения двуокиси
марганца прибавляют железный купорос. Реакция идет по сле-
дующему уравнению:
MnO2 + 2FeSO4 + 2H2SO4 = MnSO4 + Fe2 (SO4)3 + 2H2O.
В результате образуется ион двухвалентного марганца, кото-
рый затем окисляется персульфатом аммония в присутствии ио-
нов серебра как катализатора. В данном случае используется из-
быток серебра, остающийся после осаждения ионов хлора. Окис-
ление марганца идет до семивалентного состояния, соединения
которого окрашены в фиолетовый цвет:
2MnSO4 + 5 (NH4)2 S2O8+ 8Н2О = 2HMnO4 + 10NH4HSO4+ 2H2SO4.
Фосфорная кислота в азотнокислой среде при температуре 40—
50° С осаждается молибдатом аммония. Реакция идет по уравне-
нию:
Н3РО4 + 12 (NH4)2 МоО4 + 21HNO3 = (NH4)3 РО4 12МоО3 +
+ 21NH4NO3+ 12Н2О.
Выпавший желтый осадок отфильтровывают, отмывают от кисло-
ты и растворяют в оксалате калия:
2 (NH4)3 РО4 12МоО3 + 23К2С2О4 + 24Н2О = 2 (NH4)2 НРО4 +
+ (NH4)2MoO4 + 23К2МоО4 + 23Н2С2О4.
Образовавшуюся щавелевую кислоту титруют едким натром
в присутствии фенолфталеина. Как видно из последнего уравнения,
22
на 1 молекулу Р2О5 приходится 23 молекулы щавелевой кислоты
или 46 ионов водорода. Грамм-эквивалент фосфорного ангидри-
да равен 3,088, фосфора — 1,347.
Ход анализа. Отвешивают на технических весах от 1 до
3 г порошкообразного воздушно-сухого вещества (листьев берут
меньше, корней — больше) в предварительно взвешенном на ана-
литических весах тигле, который затем помещают в сушильный
шкаф и сушат при температуре 100° С до постоянного веса. К на-
веске в тигле приливают на каждый грамм вещества 2 мл 20%-ного
раствора уксуснокислого бария х. ч. и перемешивают стеклянной
палочкой. Палочку затем вынимают, вытирают кусочком беззоль-
ного фильтра, который бросают в тигель. Тигель ставят в сушиль-
ный шкаф, высушивают при 105° С и сжигают в муфеле или на
плитке под асбестовым колпаком. Черный остаток в тигле сма-
чивают 1 мл дистиллированной воды или 3%-ной перекиси водо-
рода, высушивают и снова прокаливают. Если содержимое тигля
темное, обработку водой повторяют и так поступают до тех пор,
пока произойдет полное сгорание угля.
После озоления и охлаждения в тигель добавляют 2 мл дистил-
лированной воды, перемешивают стеклянной палочкой, разминают
крупинки осадка и смесь выливают через воронку в мерную колбу
на 50 мл, споласкивая тигель дистиллированной водой (2 раза).
Затем набирают в мерный цилиндр 10 мл азотной кислоты (1 1),
3—4мл которой вливают в колбу, приподняв немного воронку, чтобы
дать свободный выход углекислоте, а остальной кислотой промывают
тигель, собирая промывные воды в мерную колбу. После того как
вся кислота перенесена в колбу, тигель и воронку промывают не-
большими порциями дистиллированной воды до тех пор, пока
раствора в колбе будет чуть больше половины. К раствору в колбе
прибавляют 2 капли 10%-ного свежеприготовленного раствора
FeSO4 7Н2О или соли Мора для растворения окислов марганца,
взбалтываютД—2 мин и осаждают ионы хлора 5 мл 0,1 н. раствора
азотнокислого серебра. Колбу ставят на плитку, нагревают до ки-
пения и кипятят 1 мин, затем охлаждают, доводят дистиллирован-
ной водой до метки, перемешивают и фильтруют в сухую колбу
через сухой обыкновенный фильтр диаметром 9 см, выбрасывая
первую порцию фильтрата (около 3 мл).
Из полученного фильтрата пипеткой на 10 мл отбирают в колбу
для титрования 20 мл избытка азотнокислого серебра для
определения хлора, 10 мл — в коническую колбу на 100 мл для
определения фосфорной кислоты и 10 мл — всухую пробирку для
определения марганца.
Определение хлора
К 20 мл фильтрата, предназначенного для определения хлора,
добавляют 2 мл полупроцентного раствора крахмала, одну каплю
1%-ного раствора NaNOa и титруют 0,01 н. раствором KJ до
23
появления неисчезающей голубой окраски. Количество раствора
йодистого калия, затраченного при титровании, вычитают из кон-
трольного титрования, которое осуществляется следующим обра-
зом. 6 мл уксуснокислого бария помещают в мерную колбу на 50 мл.
прибавляют 5 мл 0,1 н. раствора азотнокислого серебра, 10 мл азот-
ной кислоты (1 1), разбавляют водой, нагревают, охлаждают, доводят
водой до метки и фильтруют в сухую колбу через сухой фильтр,
выбрасывая первую порцию фильтра. 20 мл фильтрата титруют
0,01 н. раствором йодистого калия в присутствии 2 мл крахмала
и 1 капли 1%-ного раствора нитрита натрия.
Содержание хлора вычисляют по следующей формуле:
У _ 50 3,546 К(а — Ь)
где X — содержание хлора (в %); а — объем раствора йодистого
калия, затраченный на 20 мл контрольного раствора (в мл); b —
объем раствора йодистого калия, затраченный на 20 мл исследуе-
мого раствора (в мл); К — нормальность раствора йодистого ка-
лия; п — абсолютно-сухая навеска (в е); 50 — объем, в котором
растворена зола (в мл); 20 — объем фильтрата, взятый для опре-
деления хлора (в мл); 3,546 — нормальный титр хлора, умножен-
ный на 100 для пересчета в проценты.
Определяемые количества от 1 до 15 мг хлора в навеске.
Определение фосфора
Колбу с 10 мл фильтрата, предназначенного для определения
фосфора, накрывают воронкой, нагревают на плитке до начала
кипения, снимают с плитки и прибавляют 4 мл молибденовой жид-
кости. Раствор снова закрывают воронкой, периодически взбалты-
вают, пока он не охладится полностью и оставляют на 2 ч (лучше
на ночь) для полного выделения осадка. После отстаивания осадка
раствор фильтруют через плотный фильтр (синяя лента) диаметром
6—7 см. Осадок в колбе и на фильтре промывают 1%-ным раство-
ром KNO3, используя на каждое промывание 4—5 мл, j\q исчез-
новения кислой реакции в промывной жидкости (проба с метил-
оранжем), после чего фильтр вынимают из воронки,
разрывают надвое так, чтобы осадок был в одном месте. Ту часть
фильтра, где находится осадок, первой помещают в колбу, в ко-
торой производилось осаждение, а другой частью фильтра, прежде
чем поместить в колбу, вытирают горлышко колбы от приставшего
осадка.
Затем в колбу приливают 10 мл 10%-ного раствора окса-
лата калия и разминают палочкой фильтр для освобождения осад-
ка. Палочку вынимают, промывая ее горячей дистиллированной
водой. Колбу ставят на плитку и нагревают до растворения осад-
ка. Раствор охлаждают, прибавляют 2 капли 1%-ного раствора
фенолфталеина и титруют щавелевую кислоту 0,05 н. раствором
24
едкого натра до появления неисчезающей розовой окраски. Ко-
личество фосфора вычисляют по формуле:
v 50 0,3088 К а
Л ~ 10 п
где X — содержание Р2О5 (в %); а — объем раствора едкого нат-
ра, затраченный при титровании исследуемого раствора (в мл);
К — нормальность раствора едкого натра; п — абсолютно-сухая
навеска (в е); 50 — объем, в котором растворена зола навески (в
мл); 10 — объем исследуемого раствора, взятый для определения
фосфора (в мл); 0,3088 — нормальный титр Р2О5, умноженный
на 100 для пересчета в проценты.
Определяемые количества Р2О5 от 0,6 до 6 мг (в навеске от 1,5
до 15 мг).
Определение марганца
К Ю мл раствора в пробирке, предназначенного для определе-
ния марганца, прибавляют 0,5 г персульфата аммония, взбалтывают
и помещают в горячую воду на 5 мин. Раствору дают охладиться,
прибавляют 1 каплю 30%-ного раствора уксуснокислого натрия
и перемешивают.
В результате сжигания с уксуснокислым барием в растворе
находится ион бария, который с персульфатом аммония образует
осадок сернокислого бария. Взвесь отстаивают, а жидкость сли-
вают в сухую центрифужную пробирку и центрифугируют. Неболь-
шое количество осадка, которое находится во взвешенном со-
стоянии, при центрифугировании плотным слоем садится на дно
и дает возможность без затруднения слить прозрачный раствор
в кювету для фотометрирования. Оптическую плотность раствора
измеряют при 510—530 мм в кювете на 10 мм. Нулевым раствором
служит вода. Результаты измерений сравнивают с калибровочной
кривой и находят концентрацию марганца в колориметрируемом
растворе. Содержания марганца вычисляют по следующей формуле:
v _ 50 с __ с
Л 10 000 п ~ 200 п ’
где X — содержание МпО (в %); С — концентрация МпО в
колориметрируемом растворе (в мкг/мл); п — абсолютно-сухая
навеска (в г); 50 — объем, в котором растворена зола навески (в
мл); 10 000 — коэффициент для перевода микрограммов МпО в
граммы и пересчета их в проценты.
Определяемые количества от 2 до 30 мкг МпО в 1 мл раствора
(от 0,1 до 1,5 мг МпО в навеске).
Построение калибровочной кривой. Для
построения калибровочной кривой фотоколориметрируют раство-
ры парманганата калия, содержащие в 1 мл раствора 2, 3, 5, 7,
10, 15, 20 и 30 мкг МпО. Для приготовления этих растворов в
мерные колбы на 100 мл вливают 1,4; 2,1; 3,5; 4,9; 7,0; 10,6; 14,1;
17,6 и 21,1 мл 0,01 н. раствора КМпО4, разбавляют водой до метки
25
Концентрация МпО,мкг/мл
Рис. 2. Калибровочная кривая для марганца (све-
тофильтр с максимумом пропускания 540 нл«, кюве-
та с толщиной измеряемого слоя 10 мм).
и измеряют оптическую плотность раство-
ров при 510—530 нм в кювете на 10 мм.
На основании полученных данных строят
калибровочную кривую (рис. 2).
Примечание: Если определение
хлора не требуется, навеску сжигают без
прибавления уксуснокислого бария. В ос-
тальном ход анализа такой же.
Реактивы. Молибденовая жидкость. Отвешива-
ют 40 г молибденовокислого аммония, растворяют
в 250 мл воды и полученный раствор вливают при помешивании в 250 мл азотной
кислоты (1 1), прибавляют 70 г азотнокислого аммония и взбалтывают до рас-
творения, раствору дают отстояться и затем фильтруют. Сохраняют в склянке
с притертой пробкой. Раствор годен в течение одного месяца.
Титрованный раствор йодистого калия (0,01 н.). Приготовляется разбавле-
нием 0,1 н. раствора KJ. Для этого набирают пипеткой 50 мл 0,1 н. раствора йоди-
стого калия, вливают в мерную колбу на 500 мл, разбавляют дистиллированной
водой до метки и тщательно перемешивают. Для приготовления 0,1 н. раствора
отвешивают 16,6 г предварительно высушенного при 105° С KJ, растворяют в воде
в мерной колбе на 1 л, доводят до метки, перемешивают и устанавливают нормаль-
ность по 0,1 н. раствору азотнокислого серебра. Раствор йодистого калия сохра-
няют в темной склянке.
Для определения нормальности йодистого калия в колбу для титрования
набирают пипеткой 10 мл 0,1 н. титрованного раствора азотнокислого серебра,
прибавляют 1 мл азотной кислоты (1 1), 2 мл 0,5%-ного раствора крахмала,
1—2 капли 1%-ного раствора NaNO2 и титруют 0,1 н. раствором КД до появления
неисчезающей голубой окраски. Нормальность раствора KJ вычисляют по формуле:
а
где KJ — нормальность раствора KJ; К — нормальность раствора AgNO3; а —
объем раствора йодистого калия, затраченный при титровании 10 мл раствора
азотнокислого серебра (в мл)\ 10 — объем раствора азотнокислого серебра, взя-
тый для титрования (в мл).
Титрованный раствор азотнокислого серебра (0, 1 в.). Отвешивают 16,99 г
AgNO3, растворяют в воде, доводят объем до 1 л дистиллированной водой, пере-
мешивают и устанавливают нормальность по 0,1 н. раствору хлористого калия.
Для получения 0,1 н. раствора хлористого калия взвешивают 3,7275 г сухой
соли КС1 х. ч., растворяют в воде в мерной колбе на 500 мл, разбавляют водой
до метки и тщательно перемешивают.
Набирают 20 мл этого раствора в колбу для титрования, прибавляют 2 капли
азотной кислоты (1 1), 5 капель 2%-ного спиртового раствора дифенилкарбазона,
1 каплю 2%-ного раствора HgCl2 и титруют раствором AgNO3. В конце титруют
медленно при встряхивании, пока выпавший белый осадок не станет внезапно
фиолетовым, а раствор голубоватым. Нормальность раствора AgNO3 вычисляют
по формуле:
0,1 20
а ’
где К — нормальность раствора азотнокислого серебра; а — объем раствора
AgNO3, затраченный при титровании 20 мл 0,1 н. раствора хлористого калия
(в мл.).
26
Раствор уксуснокислого бария (20%-ный . Отвешивают 100 г соли
Ва (СН3СОО)2 х. ч., растворяют в воде и разбавляют до 500 мл. При отсутствии
этой соли ее можно приготовить из гидроокиси бария и уксусной кислоты.
Для этого отвешивают 124 г Ва (ОН)2 8Н2О, насыпают в стакан, прибавляют
350 мл дистиллированной воды, 47 мл ледяной уксусной кислоты и нагревают
при помешивании до растворения. Раствор фильтруют через обычный бумажный
фильтр, собирая фильтрат в мерную колбу на 500 мл. Объем раствора доводят
до 500 мл и перемешивают.
Раствор азотной кислоты (1 1). К 100 мл азотной кислоты пл. 1,4 г!см3
прибавляют 100 мл дистиллированной воды, перемешивают и сохраняют в/темной
склянке с притертой пробкой.
Раствор уксуснокислого натрия (30%-ный). Отвешивают 30 г уксуснокислого
натрия CH3COONa ЗН2О ч. д. а., растирают в ступке с 10 мг HgJ2, раство-
ряют в дистиллированной воде, доводят объем до 100 мл и фильтруют.
Раствор азотнокислого калия (1%-ный). 10 г соли KNO3 х. ч. растворяют
в дистиллированной воде и разбавляют до 1 л.
Раствор азотистокислого натрия (1%-ный). 1 г NaNO2 ч. д. а. растворяют
в 100 мл дистиллированной воды.
Титрованный раствор едкого натра (0,05 н.). Готовится из титрованного
0,1 н. раствора разбавлением его безуглекислой водой (в 2 раза). Углекислоту
удаляют из воды кипячением. Приготовление титрованного 0,1 н. раствора едкого
натра описано на стр. 9.
Раствор фенолфталеина (1%-ный). 1г индикатора растворяют в 60 мл
95%-ного этилового спирта и доливают дистиллированной водой до 100 мл.
Титрованный раствор перманганата калия (0, 01 н.). Готовят из 0,05 н.
раствора разбавлением в 5 раз. Набирают пипеткой 20 мл точно 0,05 н. раствора
перманганата, выливают в мерную колбу на 100 мл, доливают водой до метки
и перемешивают.
Железо сернокислое закисное FeSO4 7Н2О или соль Мора FeSO4 (NH4)2 SO4 •
• 6Н2О.
Персульфат аммония (NH4)2 S2O8.
Остальные реактивы, применяемые в методиках, приготовляются как опи-
сано па стр. 10, 18, 28.
ОПРЕДЕЛЕНИЕ ХЛОРА УСКОРЕННЫМ МЕТОДОМ
Хлор в растениях находится в виде хлорид-иона, поэтому лег-
ко извлекается даже водой. На основе этого свойства хлорид-иона
разработан простой метод быстрого определения его в растениях.
Сущность метода. Хлор извлекают из растительного
материала 1%-ным раствором азотнокислого калия при нагре-
вании. Белки и другие мешающие вещества удаляют с помощью
гидроокиси натрия и сернокислого цинка. После доведения до опреде-
ленного объема осадок отфильтровывают через бумажный фильтр и
в аликвотной части раствора хлор определяют агрентометриче-
ским или меркуриметрическим методом.
Ход анализа. Берут такую навеску сухого хорошо из-
мельченного исследуемого материала, чтобы в ней содержалось
от 0,6 до 10 мг хлора (0,3—0,5 г листьев сахарной свеклы, 0,2 г
черешков и 1 г корня). Навеску всыпают в колбу на 100 мл, добав-
ляют 25 мл 1%-ного раствора азотнокислого калия (можно и
NaNO3), помещают колбу в стакан с кипящей дистиллированной
водой и выдерживают 10 мин при периодическом взбалтывании.
После этого дают охладиться, добавляют 3 мл 1 н. раствора едко-
го натра, перемешивают и приливают 4 мл 10%-ного раствора
27
сернокислого цинка. Все содержимое переносят в мерную колбу на
50 мл, доводят водой до метки, тщательно перемешивают и филь-
труют в сухую колбу через сухой фильтр. Из фильтрата набирают
пипеткой 25 мл, выливают в колбу для титрования, прибавляют
2 капли раствора азотной кислоты (1 1), 1 каплю 2%-ного раство-
ра HgCl2, 5 капель 2%-ного раствора дифенилкарбазона и тит-
руют 0,01 н. раствором азотнокислого серебра до тех пор, пока
выпадающий белый осадок AgCl внезапно не окрасится в фиоле-
товый цвет, а раствор в голубовато-серый цвет.
Вместо азотнокислого серебра для титрования хлора можно
пользоваться азотнокислой окисной ртутью, которая в данном
случае даже лучше, так как не дает осадка и переход окраски лег-
че заметить. В этом случае к 25 мл фильтрата прибавляют 2 капли
раствора азотной кислоты (1 1), 5 капель 2%-ного раствора ди-
фенилкарбазона и титруют 0,01 н. раствором Hg (NO3)2 до появле-
ния неисчезающей розово-фиолетовой окраски раствора. Чтобы
легче было заметить начало перехода окраски, титруют со «сви-
детелем». Для приготовления «свидетеля» наливают в колбу для
титрования такое же количество исследуемого раствора, прибав-
ляют 2 капли азотной кислоты и 5 капель раствора дифенилкар-
базона. Этот раствор ставят рядом с титруемым. Титруют до яс-
ного изменения окраски по сравнению со «свидетелем».
Содержание хлора вычисляют по формуле:
V _ 50 3,546 К а _ 7,092 К • а
Л ~ 25 п ~ и
где X — содержание хлора (в %); а — объем титрованного раство-
ра AgNO3 или Hg (NO3)2, затраченный при титровании хлора
(в мл); К — нормальность раствора AgNO3 или Hg (NO3)2; п — аб-
солютно-сухая навеска (в г); 50 — общий объем вытяжки (в мл);
25 — объем вытяжки, взятый для титрования (в мл); 3,546 — нор-
мальный титр хлора, умноженный на 100 для пересчета в про-
центы.
Реактивы. Раствор азотнокислой окисной ртути (0,01 н.). Взвешивают на ана-
литических весах 1,083 г окиси ртути HgO х. ч., высыпают в стакан, прибавляют
2 мл 6 н. раствора азотной кислоты, растворяют, переливают раствор в мерную
колбу на 1 л, промывая стакан дистиллированной водой. Раствор в колбе дово-
дят водой до метки и перемешивают.
Раствор дифенилкарбазона (2%-ный). Взвешивают на технических весах
1 г дифенилкарбазона С6Н5—NH—NH—СО—N = N—С6Н5 и растворяют в
50 мл 95?4-ного этилового спирта.
Раствор'\сернокислого цинка (10%-ный). Отвешивают 100 г сернокислого
цинка ZnSO4 7Н2О, растворяют в дистиллированной воде, разбавляют до 1 л
и перемешивают.
Раствор едкого натра (1 н.). Отвешивают 40 г NaOH ч. д. а., растворяют
в прокипяченной дистиллированной воде и разбавляют этой же водой до 1 л. Со-
храняют в хорошо закрытой склянке.
Рat твор хлорной ртути (2% -ный). Отвешивают 1 г хлорной ртути HgCl2,
К1створяют в дистиллированной воде и разбавляют до 50 мл.
Put твор азотнокислого серебра (0, 01 н.). Готовится из 0,1 н. раствора раз-
ншлеписм дистиллированной водой в 10 раз.
28
ОПРЕДЕЛЕНИЕ ФОСФОРА II КАЛИЯ
В ОДНОЙ НАВЕСКЕ
Сущность метода. Исследуемое вещество сжигают в
присутствии азотнокислого цинка, что облегчает сгорание при
более низкой температуре. Большая часть органического вещества
окисляется за счет аниона азотной кислоты, входящего в состав цин-
ковой соли. Окисление органического вещества, например, глюкозы,
можно выразить следующим уравнением:
СвН12О6 + 6Zn (NO3)2 = 6СО2 + 6N2O3 + 6ZnO + 6Н2О.
Фосфат-ионы с ионами цинка образуют соединение, устойчи-
вое при высокой температуре. Калий извлекают раствором хло-
ристого натрия и определяют кобальтинитритным методом, а фос-
фор, после растворения осадка в кислоте, определяют объемным
или колориметрическим методом. Объемный метод изложен выше,
поэтому здесь более подробно описывается колориметрический
метод.
Фосфорная кислота с молибдатом аммония образует фосфор-
номолибденовую кислоту, которая легко восстанавливается мно-
гими восстановителями в синий гетерополикомплекс. Наиболее
подходящим восстановителем является ион двухвалентного
железа, который не взаимодействует с молибденом, не связан-
ным с фосфорной кислотой, в течение нескольких часов. Восста-
новление фосфорномолибденовой кислоты происходит по следую-
щему уравнению:
Н3 [Р (Mo3O10)J + 4FeSO4 + 2H2SO4 = Н3 [Р (Мо3О9ОН)4] +
+ 2Fe2 (SO4)3.
Реакция идет в кислой среде. Концентрация серной кислоты
должна быть от 0,5 до 1,0 н., так как в этих пределах достигается
максимальная интенсивность окраски. При концентрациях кис-
лоты ниже 0,4 и выше 1,2 н. интенсивность окраски сильно падает.
Восстановленный молибден имеет интенсивно синюю окраску,
которую фотометрируют при 660 нм. Так как развитие окраски
идет довольно быстро (10—15 мин) и интенсивность ее остается в
течение нескольких часов постоянной, можно пользоваться калиб-
ровочной кривой. При других восстановителях (хлористое оло-
во, гидрохинон, аскорбиновая кислота и др.) интенсивность окрас-
ки все время возрастает, вследствие восстановления молибдена,
не связанного с фосфорной кислотой, поэтому пользоваться калиб-
ровочной кривой невозможно.
Двухвалентное железо постепенно окисляется кислородом воз-
духа до трехвалентного. Этот процесс усиливается в щелочной
среде, поэтому раствор его соли готовят с сульфитом натрия и под-
кисляют серной кислотой, что предохраняет железо от окисления.
Двухвалентное железо в двойной соли с сульфатом аммония —
соли Мора Fe (NH4)2 (SO4)2 6Н2О — значительно труднее окис-
ляется кислородом воздуха, поэтому эта соль и применяется в
качестве восстановителя в данном методе.
29
Ход анализа. В предварительно взвешенном на аналити-
ческих весах тигле емкостью 30—40 мл отвешивают такое количество
растительного материала, чтобы в навеске содержалось от 2до 16лгг
К2() и от 0,6 до 6 мг Р2О5 (0,2—0,3 г воздушно-сухих листьев и
0,5 0,6 г корней сахарной свеклы). Тигель ставят в сушильный
шкаф и сушат при температуре 100° С до постоянного веса, если
не известна влажность материала. К навеске в тигле приливают
1 мл 5 н. раствора азотнокислого цинка, перемешивают стеклян-
ной палочкой, которую затем вынимают, смывая приставшие ча-
стички вещества и раствора каплями дистиллированной воды.
Тигель ставят на плитку в вытяжном шкафу и нагревают до пре-
кращения выделения бурых паров окислов азота. Затем накрывают
асбестовым колпаком и прокаливают в течение 15—20 мин. После
прокаливания тигель охлаждают, добавляют к остатку 2 мл азот-
ной кислоты (1 1), выпаривают на воздушной бане досуха и про-
каливают на плитке до удаления бурых паров окислов азота. Да-
лее тигель снимают с плитки, охлаждают, наливают пипеткой
10 мл 0,2 н. раствора хлористого натрия и содержимое хорошо
размешивают стеклянной палочкой, разминая крупинки. Разме-
шивание продолжают периодически в течение 10 мин и за-
тем фильтруют через сухой беззольный фильтр диаметром 6 см в
сухую колбу. В полученном растворе определяют калий. Для этого
набирают пипеткой 5 мл фильтрата, выливают в центрифужную
пробирку, прибавляют 1 мл 10%-ного раствора Na3Co (NO2)6,
перемешивают и оставляют на 2—4 ч (или на ночь). В дальнейшем
поступают так, как изложено выше (см. стр. 16). Содержание ка-
лия вычисляют по формуле:
v _ 0,856 10 К{д — Ь)
где X— содержание К2О (в /6); а — объем титрованного раство-
ра тиосульфата, израсходованный на 25 мл раствора перманга-
ната (в мл); b—объем раствора тиосульфата, израсходованный
на избыток перманганата при определении калия (в мл); К — нор-
мальность раствора тиосульфата натрия; п — абсолютно-сухая
навеска (в г); 10 — объем 0,2 и. раствора хлористого натрия, взя-
тый для извлечения калия (в мл); 5 — объем исследуемого раство-
ра, взятый в центрифужную пробирку для осаждения калия (в
мл); 0,856 — нормальный титр К2О, умноженный на 100 для пе-
ресчета результатов в проценты.
Определение фосфора объемным методом
Осадок, в котором находится фо:фат цинка, переводят в раствор.
Для этого осадок в тигле и на воронке сначала промывают 5 мл
дистиллированной воды, а затем воронку помещают в горлышко
конической колбы на 100 мл и растворяют осадок сначала в тигле в
4 мл азотной кислоты (1 5), а затем этим же раствором на филь-
1 ре, собирая фильтрат в колбу. Тигель и фильтр промывают еще 2 ра-
за по 4 мл азотной кислоты (1 5) и под конец один раз дистил-
лированной водой. В полученном растворе осаждают фосфор мо-
либденовой жидкостью и продолжают анализ дальше так, как это
изложено выше (см. стр. 24). Количество фосфора вычисляют по
формуле:
v 0,3088 а
где X — содержание Р2О5 (в %); К — нормальность раствора ед-
кого натра; а — объем раствора едкого натра, затраченный при
титровании исследуемого раствора (в мл); п — абсолютно-сухая
навеска; 0,3088 — нормальный титр Р2О5, умноженный на 100 для
пересчета в проценты. Определение количества Р2Об от 0,6 до 6 мг
в навеске.
Определение фосфора колориметрическим методом
Для колориметрического определения фосфора осадок фосфата
цинка и окиси цинка растворяют в серной кислоте. Для этого во-
ронку с осадком вставляют в горлышко мерной колбы на 50 мл и
осадок сначала в тигле, а затем на фильтре растворяют в 12 мл
раствора серной кислоты (1 9), собирая фильтрат в колбу. После
растворения осадка тигель и фильтр промывают 2 раза дистил-
лированной водой. Раствор в колбе доливают дистиллированной
водой до метки и тщательно перемешивают. Из этого раствора на-
бирают пипеткой 2 мл, вливают в сухую пробирку и прибавляют
6 мл 1 н. раствора серной кислоты. Если исследуемого раствора
берут не 2 мл, а больше или меньше, то соответственно уменьшают
или увеличивают объем прибавляемого раствора серной кислоты,
чтобы общий объем был 8 мл. Затем прибавляют 2 мл железо-мо-
либдатного реактива, перемешивают и через 15 мин измеряют
оптическую плотность раствора, используя светофильтр с макси-
мумом пропускания 660 нм в кювете на 10 мм. Раствором для
сравнения служит дистиллированная вода. Полученные данные
сравнивают с калибровочной кривой и вычисляют содержание фос-
фора по формуле:
х = 50 10 С = 0,025 С
л“ 10000 2 л ~ л
где X — содержание Р2О5 или Р (в %); С — концентрация Р2О5
или Р, найденная в колориметрируемом растворе (в мкг/мл); п —
абсолютно-сухая навеска (в г); 50 — общий объем исследуемого
раствора, в котором растворен осадок фосфата цинка (в мл); 10 —
объем колориметрируемого раствора (в мл); 2 — объем исследу-
емого раствора, взятый в пробирку для колориметрирования фос-
фора (в мл); 10 000 — коэффициент для перевода микрограммов
Р2О5 или Р в граммы и пересчета их в проценты.
Определяемые концентрации от 1 до 11 мкг/мл Р в колориметри-
руемом растворе при использовании кюветы с толщиной слоя 10 мм
и от 0,5 до 5,5 мкг/мл Р при использовании кюветы 20 мм.
31
Рис. 3. Калибровочная кривая для
фосфора с железо-молибдатным
реактивом (светофильтр с максиму-
мом пропускания 660 нм, кювета с
толщиной измеряемого слоя 10 л/л/).
Построение калибро-
вочной кривой. В су-
хие пробирки последовательно
вливают 0,5, 1,0, 1,5, 2,0, 2,5, 3,5,
4,5, 5,5 мл стандартного раствора,
содержащего 20 мкг!мл Р, добав-
ляют дистиллированную воду до
объема 6 мл, прибавляют 2 мл рас-
твора серной кислоты (1 9), 2 мл
железо-молибдатного реактива пе-
ремешивают и через 15 мин изме-
ряют оптическую плотность раство-
ра с красным светофильтром (мак-
симум пропускания 660 нм), в кюве-
те на 10 мм или в кювете на 20
мм для меньших количеств фосфо-
ра. Раствором сравнения служит
вода. Из полученных данных стро-
ят калибровочную кривую (рис. 3).
Реактивы. Стандартный раствор
фосфора. Отвешивают на аналитических
весах 0,2195 г высушенного при 105° С
до постоянного веса однозамещенного фосфата калия х. ч. КН2РО4, рас-
творяют в воде в мерной колбе на 250 мл, доливают дистиллированной водой до
метки и перемешивают. 1 мл этого раствора содержит 0,2 мг Р. Из этого раство-
ра готовят рабочий раствор разбавлением его в 10 раз дистиллированной водой.
Для этого набирают пипеткой 10 мл первоначального раствора, вливают в мерную
колбу на 100 мл, разбавляют дистиллированной водой до метки и тщательно пе-
ремешивают. 1 мл рабочего раствора содержит 20 мкг Р или 45,8 мкг Р2О5.
Железо-молибд а тный реактив. Отвешивают 12 г безводного сульфита натрия
или 24 г кристаллической соли (Na2SO3 7Н2О), всыпают в стакан, добавляют
2 г молибдата аммония и 100 мл 1 н. раствора серной кислоты. Размешивают до
полного растворения, затем добавляют 16 г соли Мора (Fc (NH4)2 (SO4)2 6Н2О),
растворяют и сохраняют в плотно закрытой склянке с притертой пробкой
не более 15 дней.
Раствор азотнокислого цинка (5 н.). К 100 мл азотной кислоты (1 1) при-
бавляют 21 г окиси цинка х. ч., размешивают до растворения и, если нужно,
фильтруют. Сохраняют в склянке с притертой пробкой.
Раствор хлористого натрия (0,2 н.). Отвешивают 10,7 г хлористого натрия
х. ч., растворяют в дистиллированной воде и разбавляют до 1 л.
Раствор серной кислоты (~ 1 н.). К 900 мл дистиллированной воды прибав-
ляют 27 мл серной кислоты пл. 1,84 и после охлаждения разбавляют до 1 л дистил-
лированной водой.
Раствор азотной кислоты (1: 5). К 500 мл дистиллированной воды приливают
100 мл азотной кислоты пл. 1,4 и перемешивают.
ОПРЕДЕЛЕНИЕ АЗОТА, ФОСФОРА И КАЛИЯ
В ОДНОЙ НАВЕСКЕ
Сущность метода. Навеску органического материала
сжигают в колбе Кьельдаля с помощью серной кислоты и огра-
ниченного количества хлорной кислоты в присутствии катализа-
тора молибдата натрия. Остаток, содержащий азот, фосфор и
калий, разбавляют дистиллированной водой до определенного
32
объема и в полученном растворе определяют азот хлораминным
методом, калий — объемным кобальтинитритным методом и фос-
фор — колориметрическим методом, в виде фосфорномолибдено-
вой сини.
Ход анализа. Отвешивают на аналитических весах та-
кое количество растительного материала, чтобы в навеске нахо-
дилось азота (N) и калия (К2О) от 2 до 16 мг и фосфора — от 0,6
до 6 мг на Р2О5 (например, 0,2 г листьев сахарной свеклы, по 0,5 г
на сухое вещество корней сахарной свеклы и веток яблони). На-
веску помещают в колбу Кьельдаля на 50—100 мл, прибавляют
0,3 мл 10%-кого раствора молибдата натрия, по 0,2 мл 42%-ной
хлорной кислоты на каждый 0,1 г сухой навески и, отмерив ци-
линдром, прибавляют концентрированную серную кислоту (3 мл
на навеску 0,2—0,3 г и 4 мл на навеску 0,5 г). Колбу встряхивают
до равномерного распределения кислоты, помещают на электро-
плитку и нагревают, регулируя нагрев так, чтобы не было силь-
ного вспенивания. Когда образование пены уменьшится и в колбе
появятся белые пары серной кислоты, колбу закрывают стеклянной
шариковой пробкой и продолжают нагревать на плитке с откры-
той спиралью, доводя раствор до кипения. Нагревание продол-
жают до полного просветления раствора.
После сжигания и охлаждения к содержимому колбы осторож-
но по стенке приливают 20 мл дистиллированной воды, переме-
шивают, выливают в мерную колбу на 50 мл, промывая неболь-
шими порциями воды. Раствор в колбе охлаждают, доводят водой
до метки и тщательно перемешивают. В этом растворе определяют
азот, фосфор и калий.
Определение азота t
Отбирают пипеткой 10 мл раствора, переносят в коническую
колбу на 200—250 мл и определяют азот хлораминным методом,
описанным ниже (см. стр. 72, 75).
Определение калия
Набирают пипеткой 25 мл раствора, вливают в фарфоровую
чашку и выпаривают на воздушной бане досуха. Чашку ставят на
плитку и нагревают до удаления белых паров серной кислоты (вы-
паривание проводится под вытяжным шкафом с хорошей тягой).
Для удаления аммонийных солей чашку накрывают асбестовым
колпаком и прокаливают на плитке еще 10—15 мин. После этого
чашку охлаждают, прибавляют 2 мл 3%-ного раствора нитрита
натрия (NaNO2), две капли ледяной уксусной кислоты и выпа-
ривают на водяной бане досуха. Водяной баней может служить
стакан с кипящей водой. После охлаждения, остаток в чашке рас-
творяют в 3 мл дистиллированной воды и переносят раствор в
центрифужную пробирку. Чашку промывают три раза по 1 мл,
3 6-2
33
присоединяя промывные воды к раствору в пробирке. В получен-
ном растворе, не обращая внимания на муть, осаждают калий,
прибавляя 1 мл 10%-ного раствора Na3 [Со (NO2)6L Раствор с
осадком оставляют на 1—2 ч для полного осаждения. В дальнейшем
поступают так, как это описано выше (см. стр. 16).
Определение фосфора
2 мл раствора помещают в пробирку, прибавляют одну каплю
раствора фенолфталеина и нейтрализуют серную кислоту 2 н. раст-
вором едкого натра до появления розово-красной окраски.
Появившуюся окраску обесцвечивают прибавлением 0,2 н. рас-
твора серной кислоты. Затем прибавляют 2 мл серной кислоты
(1 9), разбавляют дистиллированной водой до 8 мл, перемеши-
вают, добавляют 2 мл железо-молибдатного реактива, снова пе-
ремешивают и через 15 мин фотометрируют при 660 нм. В даль-
нейшем поступают так, как описано выше (см. стр. 31).
ОПРЕДЕЛЕНИЕ КАЛИЯ,
НЕОРГАНИЧЕСКОГО ФОСФОРА И НИТРАТОВ
В ВЫТЯЖКЕ ИЗ СВЕЖЕГО РАСТИТЕЛЬНОГО МАТЕРИАЛА
Сущность метода. Определяемые вещества извлекают
из пробы 1 %-ной уксусной кислотой и в полученной вытяжке опреде-
ляют калий — объемным кобальтинитритным или тетрафенилборат-
ным методом, неорганический фосфор — колориметрическим
методом, в виде фосфорномолибденовой сини и нитратный
азот — колориметрическим методом после восстановления его цин-
ком до нитрита.
Определение фосфорной кислоты колориметрическим методом
непосредственно в вытяжке из растительного материала не дает
достоверных результатов, так как органические вещества, при-
сутствующие в растворе, катализируют реакцию восстановления
молибдена, значительно усиливая интенсивность окраски, особен-
но, если применяют органические восстановители. При этом в за-
висимости от вида, возраста и условий произрастания растения
количество мешающих веществ и степень их влияния на реакцию
бывают разные. Поэтому изолирование фосфорной кислоты от ме-
шающих органических веществ является необходимым условием
достоверных результатов.
Нитраты восстанавливают металлическим цинком до нитритов
в слабокислой среде (pH 5,6), т. к. полнота восстановления за-
висит от pH раствора. Реакция идет по следующему уравнению:
NaNO3 + Zn + 2СН3СООН = NaNO2 + Zn (CH3COO)2 + H2O.
Полученную азотистую кислоту определяют с помощью реактива
Грисса, который с азотистой кислотой образует азокраситель крас-
34
ного цвета. Реакция идет по следующему уравнению:
C10H7NH2 + NH2CeH4SO3H + NaNO2 + СН3СООН -
а-нафтиламин сульфанило-
вая кислота
= NH.,C10He—N = N—C.H4SO3H + CH3COONa + 2Н2О.
диазосульфаниловая
кислота
Цинк оказывает влияние на последующую реакцию нитрита
с реактивом Грисса, поэтому после восстановления нитратов его
необходимо удалить фильтрованием. Концентрация уксусной кис-
лоты также очень влияет на скорость реакции нитрита с реактивом
Грисса. При концентрации уксусной кислоты более 30% реакция
заканчивается за 10 мин.
Ход анализа. Отвешивают на технических весах 1 г све-
жего растительного материала (корень, стебли), а при исследо-
вании листьев из них высекают пробочным сверлом такое количество
дисков, чтобы их общий вес был около 1 г. Если пробу берут на
месте произрастания растений, диски помещают в тарированные
стеклянные бюксы и затем взвешивают в лаборатории. Для инак-
тивации ферментов бюкс с навеской помещают в пары кипящей
воды на 8 мин. После этого навеску переносят в фарфоровую ступ-
ку и тщательно растирают с небольшим количеством 1 %-ного рас-
твора уксусной кислоты до получения однородной массы. Затем
суспензию из ступки количественно переносят в мерную колбу
на 25 мл, ступку промывают 1%-ной уксусной кислотой, доводя
этой же кислотой до объема 25 мл. Смесь тщательно перемешивают
и фильтруют в сухую колбу через сухой фильтр диаметром 7 см
средней плотности (белая лента).
Отбирают градуированной пипеткой емкостью 10 мл следую-
щие количества фильтрата: 10 мл в фарфоровую чашку для опре-
деления калия, 4 мл в пробирку для определения неорганического
фосфора и в другую пробирку от 0,2 до 4 мл для определения нит-
ратов.
Содержание нитратов в течение вегетации изменяется в боль-
шой степени, поэтому в зависимости от предполагаемого их коли-
чества берется различный объем исследуемого раствора.
Определение калия
Объемный кобальтинитритный метод. К Южл
фильтрата в фарфоровой чашке прибавляют 3 мл азотной кислоты
(1 1), и 0,2 мл 5 н. раствора азотнокислого цинка. Чашку поме-
щают на воздушную баню и выпаривают раствор в вытяжном шка-
фу досуха. К остатку прибавляют 1 мл азотной кислоты (1 1),
выпаривают досуха на воздушной бане, чашку переносят на эле-
ктроплитку, накрывают сверху асбестовым колпаком и прока-
ливают 10—15 мин. Обработку азотной кислотой повторяют, если
остаток после прокаливания будет иметь вместо желтого темный
3*
35
цвет. После сжигания в чашку прибавляют 1 мл 2 н. соляной кис-
лоты, размешивают и выпаривают на водяной бане досуха.
К остатку в чашке приливают 2 мл дистиллированной воды,
размешивают, сливают раствор в центрифужную пробирку; чашку
промывают три раза водой по 1 мл, добавляя промывные воды к
раствору в пробирке. Для осаждения калия в пробирку прили-
вают 1 мл 10%-кого раствора кобальтинитрита натрия и далее
поступают так, как изложено выше (см. стр. 16). Содержание ка-
лия вычисляют по формуле:
v_ 25 0,856 К (а — Ь)
А — 10 л
где X — содержание К2О (в %); 25 — общий объем уксуснокислой
вытяжки (в мл); 10 — объем вытяжки, взятый для определения
калия (вжл); 0,856 — нормальный титр К2О, умноженный на 100
для пересчета в проценты; /( — нормальность титрованного рас-
твора соли Мора или тиосульфата натрия; а — объем раствора
соли Мора или тиосульфата натрия, израсходованный на 3 мл
0,5 н. раствора бихромата калия (в мл); b — объем раствора Мора или
тиосульфата натрия, израсходованный на избыток бихромата
калия (в мл); п — навеска (в г).
Тетрафенилборатный метод. Определение ка-
лия можно также осуществить новым объемным тетрафенилбо-
ратным методом, который позволяет определять почти в два
раза меньшие количества калия, чем кобальтинитритный метод.
Сущность метода. Тетрафенилборат натрия в нейт-
ральных или слабокислых растворах образует с ионом калия оса-
док тетрафенилбората калия:
Na (В (СвН6)4] + К+ = К [В (C6H5)J + Na+
Кроме ионов калия реагент осаждает ионы аммония, рубидия и
цезия.
Определению не мешают сульфаты и фосфаты, соли натрия,
магния, кальция, цинка и кобальта. Ионы трехвалентного железа
маскируются фтористым натрием или трилоном Б.
Растворимость тетрафенилбората калия в воде заметна и соста-
вляет 0,58 мг в 100 мл дистиллированной воды при 20° С, поэтому
при определении небольших количеств калия необходимо про-
мывать осадок не дистиллированной водой, а насыщенным рас-
твором тетрафенилбората калия.
Осадок тетрафенилбората калия растворяется в уксуснокислом
растворе ацетата ртути:
к [В (CeH6)4J + 4Hg (СН3СОО)2 + ЗН2О = 4CH3COOHgC6H5 +
+ СН3СООК + ЗСН3СООН + Н3ВО3.
Если к полученному раствору прибавить раствор соляной кисло-
ты, выпадет осадок фенилмеркурхлорида:
CH3COOHgC6H6 + НС1 = ClHgC6H6 + СН3СООН.
36
Осадок отфильтровывают, а в растворе определяют избыток ионов
ртути титрованием йодистым калием с индикатором крахмалом
в присутствии нитрита натрия. Пока в растворе имеются ионы рту-
ти, выпадает красный осадок йодида ртути:
Hg2+ + 2KJ = HgJ2 + 2К+
Как только ионы ртути свяжутся, появляются свободные ионы
иода, которые вступают в реакцию с нитритом натрия. Выделив-
шийся свободный йод образует с крахмалом синюю окраску (см.
стр. 22).
Грамм-эквивалент -^Я-равен 5,888, а калия — 4,888.
Ход а на л и з а. К 10 мд фильтрата в фарфоровой чашке
прибавляют 2 мл азотной кислоты (1 : 1), помещают на воздуш-
ную баню и выпаривают до появления бурых паров. Затем чашку
ставят на водяную баню, добавляют 5 капель 30%-ной перекиси
водорода и выпаривают досуха. К остатку в чашке приливают 1 мл
азотной кислоты (1 1), 3 капли перекиси водорода, выпаривают
на водяной бане досуха и продолжают нагревание до прекращения
выделения бурых паров. После этого добавляют 10 капель раство-
ра соляной кислоты (1 1) и снова выпаривают до полного высы-
хания. К остатку в чашке приливают 2 мл дистиллированной во-
ды, размешивают до растворения, раствор сливают в центрифуж-
ную пробирку, а чашку промывают дистиллированной водой (2
раза по 1 мл), выливая ее в ту же пробирку. К раствору в пробир-
ке для осаждения калия постепенно при помешивании прибав-
ляют 2 мл 3%-ного раствора тетрафенил бората натрия и оставляют
на 10—15 мин. Затем центрифугируют, прозрачный раствор сли-
вают, а осадок промывают 2 раза промывной жидкостью по 5 мл.
При промывании поступают следующим образом: после сливания
жидкости с осадка к нему прибавляют 2—3 капли дистиллирован-
ной воды и энергично встряхивают пробирку; затем в центр про-
бирки направляют из пипетки струю промывной жидкости и быс-
тро всю ее выливают. При этом происходит перемешивание всего
осадка.
К промытому осадку приливают 5 мл 0,2 н. ацетата ртути, пе-
ремешивают стеклянной палочкой, прибавляют 3 мл 0,3 н. ра-
створа соляной кислоты, снова перемешивают и через 5 мин филь-
труют в сухую колбу через сухой фильтр диаметром 6 см. Затем
набирают пипеткой 4 мл фильтрата, выливают в колбу для тит-
рования, прибавляют 5 мл азотной кислоты (1 5), 20 мл дистил-
лированной воды, 1 мл 0,5%-кого раствора крахмала, 2 капли
1 %-кого раствора нитрита натрия и титруют 0,02 н. раствором
йодистого калия до перехода окраски в синий цвет на фоне ро-
зово-красного осадка HgJ2. Из полученных данных вычисляют
содержание калия по формуле:
У _ 25 0,589(^62 — 2^6)
37
где X — содержание К2О (в %); а — объем титрованного раствора
ацетата ртути, прибавленный к осадку тетрафенилбората калия
(в мл); b — объем титрованного раствора йодистого калия, затра-
ченный при титровании избытка ртути (в мл); Ki — нормальность
раствора ацетата ртути; К2 — нормальность раствора йодистого
калия; п — навеска (в г); 25 — общий объем вытяжки (в мл); 10 —
объем вытяжки, взятый для определения калия (в мл); 0,589 —
нормальный титр К2О, умноженный на 100 для пересчета в про-
центы; 2-------------(5 — объем раствора ацетата ртути, 3 —
объем раствора соляной кислоты, 4 — объем фильтрата, взятый
для титрования). Определяемые количества от 0,5 до 5 мкг К2О.
Определение неорганического фосфора
К 4 мл исследуемого раствора в пробирке прибавляют 8 капель
10%-ной суспензии окиси цинка, взбалтывают в течение 5 мин и
затем фильтруют через плотный фильтр диаметром 6 см. Осадок
промывают 1%-ным раствором уксуснокислого цинка (2 раза по
3 мл). В осадке на окиси цинка находится весь неорганический
фосфор. Воронку с осадком вставляют в чистую сухую пробир-
ку и осадок в первой пробирке и на фильтре растворяют в 4 мл
2 н. серной кислоты. Первую пробирку и фильтр промывают 4 мл
дистиллированной воды, доводя общий объем раствора во второй
пробирке до 8 мл. К полученному раствору прибавляют 2 мл же-
лезо-молибдатного реактива, перемешивают и через 15 мин из-
меряют оптическую плотность при 660 нм в кювете с толщиной
слоя 10 мм.
Если исследуемый раствор после прибавления серной кислоты
имеет слабую окраску, раствором для сравнения служит вода.
Если же раствор имеет заметную окраску, необходимо для сравне-
ния брать такой же раствор, как в опыте, но без железо-молиб-
датного реактива. Для этого берут 4 мл исследуемого раствора,
добавляют 8 капель суспензии окиси цинка, взбалтывают, филь-
труют, растворяют осадок в 4 мл 2 н. раствора серной кислоты и
разбавляют дистиллированной водой до объема 10 мл.
Полученные данные сравнивают с калибровочной кривой, на-
ходят концентрацию фосфора и вычисляют его содержание по фор-
муле:
х = 0,1 25 10 С = 6,25 С
4 • п п ’
где X — содержание неорганического фосфора (в мг Р на 100 г
исследуемого вещества); С — концентрация Р в колориметрируе-
мом растворе (в мкг/мл); п — навеска (в г); 25 — общий объем ук-
суснокислой вытяжки, мл; 4 — объем вытяжки, взятый для ко-
лориметрического определения фосфора, мл; 10 — объем коло-
риметрируемого раствора, мл; 0,1 — коэффициент для перевода
микрограммов в миллиграммы и пересчета их на 100 г вещества.
38
Определение нитратов
Восстановление нитратов цинком проводится в определенном
объеме раствора, поэтому исследуемого раствора должно быть
4 мл. В том случае, когда раствора будет меньше 4-х мл, его объем
в пробирке доводят до 4 мл 1%-ным раствором уксусной кислоты.
Затем к 4 мл раствора нитратов
прибавляют 4 мл 0,8 М раствора ДО’
уксуснокислого натрия, тщательно . /
перемешивают, прибавляют 10 мг g /
цинковой пыли и пробирку периоди- |ДО- /
чески встряхивают круговыми движе- | _ /
ниями в течение 10 мин со скоростью § /
1—2 встряхивания в мин. После этого /
раствор фильтруют в сухую пробирку & /
или колбочку через сухой беззольный | т /
фильтр диаметром 7 см. Из фильтрата /
набирают пипеткой 4 мл в сухую ' /
пробирку, прибавляют 1 мл реактива - /
на нитриты, перемешивают и через
10 мин измеряют оптическую плот-
ность раствора в кювете с толщиной
слоя 10 мм при зеленом светофильтре
с эффективной длиной волны 540 нм.
Раствором для сравнения служит оста-
ток фильтрата. Из полученных дан-
ных по калибровочной кривой на-
Q\--1--1--!--1--1-J--1-1--1--1
0,2 0,4 0,6 0,8 1,0
Концентрация N,мкг/мл
Рис. 4. Калибровочная кривая
для нитратного азота (свето-
фильтр с максимумом пропуска-
ния 540 нм, кювета с толщиной
измеряемого слоя 10 мм}.
ходят концентрацию нитратов в колориметрируемом растворе и
вычисляют содержание нитратного азота по формуле:
0,1 25 10 С = 25 С
bn bn
где X — содержание нитратного азота (в мг на 100 г исследуемого
вещества); С — концентрация нитратного азота в колориметри-
руемом растворе (в мкг1мл)\ b — объем уксуснокислой вытяж-
ки, взятый для восстановления цинком нитратов в нитриты (в
мл)-, п — навеска (в г); 25 — общий объем уксуснокислой вытяж-
ки (в мл)-, 10 — объем колориметрируемого раствора (в мл), умно-
женный па| 2, поскольку для определения берется только поло-
вина восстановленного цинком раствора; 0,1 — коэффициент для
перевода микрограммов в миллиграммы и пересчета их на 100 а
вещества.
Определяемые количества — от 0,25 до 2,5 мкг азота в 1 мл
исследуемого раствора (от 0,1 до 1 мкг!мл в колориметрируемом
растворе).
Построение калибровочной кривой. В су-
хие пробирки последовательно вливают 0,4, 0,8, 1,2, 1,6, 2,4, 3,2
и 4 мл стандартного раствора KNO3, содержащего 2,5 мкг N в 1 мл
39
раствора, добавляют 1%-ной уксусной кислоты до 4 мл и по 4 мл
0,8 М раствора уксуснокислого натрия, перемешивают, добавляют
по 10 мг цинковой пыли и периодически взбалтывают в течение
10 мин. После этого растворы фильтруют в сухие пробирки через
сухие фильтры. Из фильтратов набирают пипеткой по 4 мл в су-
хие пробирки, прибавляют по 1 мл реактива на нитриты, переме-
шивают и через 10 мин измеряют оптическую плотность раствора
в кювете с толщиной слоя 10 мм при 540 нм. Раствором для сравне-
ния служит вода. Из полученных данных строят калибровочную
кривую по образцу, приведенному на рис. 4.
Примечание. В этой же уксуснокислой вытяжке можно
определить и аминный азот. Для этого берут 0,5 мл вытяжки,
помещают в пробирку, добавляют 1,5 мл дистиллированной воды,
3 мл нингидринового реактива и далее продолжают так, как опи-
сано ниже (см. стр. 85).
Реактивы. Раствор тетрафенилбората натрия (3%-ный). Отвешивают 6 г
препарата, обычно содержащего около 50% Na (В (С6Н5)4], растворяют в 100 мл
дистиллированной воды, настаивают при взбалтывании 1—2 ч и затем фильтруют.
Тетрафенилборат натрия хорошо растворим в воде и переходит в раствор, а боль-
шая часть примесей нерастворима в воде и при фильтровании остается на фильтре.
Количество тетрафенилбората натрия в растворе проверяют следующим об-
разом. В центрифужную пробирку набирают 5 мл раствора хлористого калия,
содержащего 5 мг К2О, добавляют 1 мл раствора тетрафенилбората натрия, ^через
5 мин центрифугируют, промывают и определяют калий так, как описано выше
(см. стр. 37). Раствор годится, если найдено не менее 3 мг К2О.
Раствор реактива при хранении может помутнеть вследствие разложения:
Na [В (C6H6)J + 3H2O = CeH5B (OH)2 + 3CeHe + NaOH.
Кислая среда ускоряет разложение реактива, поэтому он должен иметь нейтраль-
ную реакцию. Раствор при хранении в холодильнике устойчив длительное время,
но лучше готовить его свежим на 2—3 дня, сохраняя в темпом и прохладном месте.
Промывная жидкость. К 5 мл раствора хлористого калия в центрифужной
пробирке, содержащим 5 мг К2О, приливают 2 мл 3%-ного раствора тстрафенил-
бората натрия, осадок отделяют центрифугированием и промывают 2—3 раза
дистиллированной водой по 10 мл. Промытый осадок взбалтывают с 2 л дистил-
лированной воды в течение 10 мин и в таком виде сохраняют. Перед работой
отфильтровывают необходимое количество раствора для промывания осадков.
Раствор ацетата ртути (0,2 н.). Отвешивают 10,83 г HgO, всыпают в ста-
кан, растворяют в 250 мл ледяной уксусной кислоты, раствор переносят в мерную
колбу на 500 мл, разбавляют дистиллированной водой до метки и тщательно
перемешивают.
Стандартный раствор нитрата калия. Отвешивают 0,1806 г KNO3 х. ч.,
растворяют в дистиллированной воде в мерной колбе на 500 мл, разбавляют во-
дой до метки и перемешивают. Набирают пипеткой 5 мл этого раствора в мерную
колбу на 100 мл, добавляют 1 мл ледяной уксусной кислоты, доводят дистилли-
рованной водой до метки и перемешивают. Раствор содержит 2,5 мкг азота в 1 мл
раствора.
Раствор ацетата натрия (0,8 М). Отвешивают 27,2 г CH3COONa • ЗН2О
х. ч., растворяют в дистиллированной воде и разбавляют до объема 250 мл. При
храпении добавляют 1 мл хлороформа.
Раствор соляной кислоты (0,3 н.). Набирают 25 мл соляной кислоты пл. 1,19,
разбавляют дистиллированной водой до 1 л и перемешивают.
Раствор уксусной кислоты (1%-ный). 10 мл ледяной уксусной кислоты раз-
бавляют дистиллированной водой до 1 л и перемешивают.
40
Реактив на нитриты (измененный реактив Грисса). Отвешивают 0,10 г аль-
фанафтиламина, всыпают в стакан на 100 мл, прибавляют 50 мл дистиллирован-
ной воды, нагревают до кипения и кипятят 1—2 мин. Нерастворившиеся фиоле-
товые жирные капли удаляют при помощи фильтровальной бумаги, нарезанной
полосками, а к прозрачному раствору прибавляют 0,25 г сульфаниловой кислоты
и размешивают стеклянной палочкой до полного ее растворения. Полученный
раствор выливают в мерную колбу на 100 мл, прибавляют 50 мл ледяной уксус-
ной кислоты, охлаждают, доводят дистиллированной водой объем до 100 мл
и тщательно перемешивают. Сохраняют в темном месте. Если при хранении по-
является розовая окраска, ее устраняют взбалтыванием с цинковой пылью и филь-
труют.
Очистка цинковой пыли от нитратов. 20 г цинковой пыли всыпают в стакан,
прибавляют 100 мл горячей 2%-ной уксусной кислоты и настаивают при помеши-
вании 30 мин. После этого фильтруют через воронку Бюхнера с отсасыванием,
промывают 50 мл 2%-ной уксусной кислоты, затем дистиллированной водой,
сушат при комнатной температуре на бумаге, высыпают в склянку с притертой
пробкой, где и сохраняют.
Раствор серной кислоты (2 и.). В мерную колбу на 500 мл наливают прибли-
зительно около половины колбы дистиллированной воды, прибавляют 27 мл
серной кислоты пл. 1,84, перемешивают, охлаждают, доливают дистиллированной
водой до метки и снова перемешивают.
Титрованный раствор йодистого калия (0,02 н.). Готовят из 0,1 н. раствора
KJ. Для этого набирают пипеткой 100 мл 0,1 н. раствора йодистого калия, выли-
вают в мерную колбу на 500 мл, разбавляют дистиллированной водой до метки
и тщательно перемешивают. Сохраняют в темной склянке.
Приготовление 0,1 н. раствора KJ и растворов остальных реактивов описано
выше (см. стр. 26, 32).
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ОБЩЕЙ СЕРЫ
Сущность метода. Органическое вещество растения
сжигают смесью азотной и хлорной кислот в присутствии ката-
лизатора молибдата натрия, а образующуюся серную кислоту
определяют хроматойодометрическим методом. Потеря серы ис-
ключается, так как окисление основной массы органического ве-
щества происходит в жидкой среде при температуре не превы-
шающей 100° С. В начале окислителем служит азотная кислота:
C3H7O2NS + 13HNO3 = H2SO4 + ЗСО2 + 6N2O3 + 2NO2 4- 9H2O.
Образующаяся серная кислота связывается молибдатом натрия:
H2SO4 + Na2MoO4 = Na2SO4 + Н2МоО4.
Сжигание на кипящей водяной бане продолжают до тех пор»
пока прекратится выделение бурых паров окислов азота. Даль-
нейшее сжигание проводят на воздушной бане до удаления бе-
лых паров хлорной кислоты. Вся операция сжигания занимает
около 1 ч. В остатке после сжигания серная кислота осаждается
хроматом бария:
H2SO4 + ВаСгО4 = BaSO4 + Н2СгО4.
Это уравнение показывает, что серная кислота заменяется экви-
валентным количеством хромовой кислоты, находящаяся в
растворе и может быть отделена от осадка сульфата и хромата ба-
рия центрифугированием или фильтрованием. Количество обра-
41
зовавшейся хромовой кислоты определяют йодометрическим методом:
2Н2СгО4 + 6KJ + 6H2SO4 = Сг2 (SO4)3 + 3J2 + 3K2SO4 + 8Н2О.
Выделившийся йод титруется тиосульфатом натрия:
2Na2S2O3 + J2 = Na2S4Oe + 2NaJ.
Грамм-эквивалент-^- равен 26,69, а серы -у— 10,69, так как
1 молекула серной кислоты вытесняет 1 молекулу хромовой кис-
лоты, которая выделяет 3 атома йода из KJ.
После сжигания растительного материала весь фосфор нахо-
дится в виде ортофосфорной кислоты, которая мешает определе-
нию сульфатов, так как при нейтрализации аммиаком она реагирует
с барием, образуя осадок фосфата бария:
ЗВа2+ + 2РО4“ = Ва3 (POJ,.
Чтобы избежать осаждения бария, в раствор добавляют избыток
хлористого кальция, который при нейтрализации аммиаком пер-
вый вступает в реакцию с фосфорной кислотой, образуя менее
растворимый осадок фосфата кальция:
2Н3РО4 + ЗСаС12 + 6NH4OH = Са3 (РО4)2 + 6NH4C1 + 6Н2О.
Хлористый кальций осаждает также и молибденовую кислоту:
СаС12 4- Na2MoO4 = СаМоО4 + 2NaCL
Ход анализа. Хорошо измельченную навеску исследуемо-
го растительного материала, содержащую от 0,5 до 5 мг серы (0,5 г
сухих листьев, 1 г зерна и корней сахарной свеклы помещают в
фарфоровую чашку, добавляют 0,5 мл 10%-ного раствора молиб-
дата натрия, 4 мл 42%-ного раствора хлорной кислоты и 10 мл
азотной кислоты пл. 1,4. Чашку ставят на кипящую водяную ба-
ню (можно на стакан с кипящей водой) и выпаривают до удаления
бурых паров окислов азота и полного растворения навески. После
этого добавляют еще 2 мл азотной кислоты пл. 1,4, чашку поме-
щают на воздушную баню, выпаривают досуха и удаления белых
паров хлорной кислоты. К охлажденному остатку приливают
5 мл 0,1 н. раствора соляной кислоты, тщательно перемешивают
стеклянной палочкой и выливают раствор в центрифужную про-
бирку, смывая остаток 5 мл дистиллированной воды. Пробирку
погружают в кипящую воду, прибавляют 2 мл 2,5%-ного раствора
хромата бария в 1 н. соляной кислоте и нагревают при размеши-
вании в течение 5 мин. Затем, продолжая нагревать, прибавляют
0,5 мл 25%-ного раствора хлористого кальция, размешивают и
нейтрализуют 25%-ным раствором аммиака, прибавляя его кап-
лями при размешивании до перехода окраски хрома в желтую. Можно
нейтрализовать в присутствии индикатора метилрота. После ней-
трализации пробирку вынимают из горячей воды и оставляют
до полного охлаждения раствора. Затем центрифугируют, раствор
сливают в колбу для титрования, а осадок промывают 1 раз 10 мл
дистиллированной воды, центрифугируют и присоединяют к ра-
створу в колбе. В полученном растворе находится эквивалентное
серной кислоте количество хромата, который и определяется йо-
дометрическим методом. Для этого прибавляют 5 мл раствора сер-
ной кислоты (1 4), 2 мл 20%-ного раствора йодистого калия и
титруют 0,01 н. раствором тиосульфата натрия в присутствии
индикатора крахмала. Из полученных данных вычисляют содер-
жание серы по формуле:
v 1,069 К а
где X — содержание серы (в %); а — количество 0,01 н. раствора
тиосульфата натрия, затраченное при титровании; п — абсолют-
но-сухая навеска (в г); 1,069 — количество серы, эквивалентное
1 мл нормального раствора хромата, умноженное на 100 для пере-
счета в проценты; К — нормальность раствора тиосульфата натрия.
Для пересчета на SO3 полученный результат умножают на коэф-
фициент 2,497.
Реактивы. Раствор хромата бария в соляной или хлорной кислоте (2,5% -ный).
К 5 г ВаСгО4 ч. д. а. в колбе добавляют 200 мл 1 н. раствора соляной кислоты
или 7%-ного раствора хлорной кислоты и нагревают до растворения хромата ба-
рия. Раствор охлаждают и сохраняют в темной склянке с притертой пробкой.
Серная кислота (1 4). К 400 мл дистиллированной воды в термостойкой по-
суде постепенно при помешивании прибавляют 100 мл серной кислоты пл. 1,84,
охлаждают и сохраняют в склянке со стеклянной притертой пробкой.
Титрованный раствор тиосульфата натрия (0,01 н.). Этот раствор готовится
из титрованного 0,1 н. раствора разбавлением. Для этого набирают пипеткой
50 мл 0,1 н. раствора, выливают в мерную колбу на 500 мл, разбавляют дистил-
лированной водой до метки и тщательно перемешивают. Приготовление 0,1 н.
раствора тиосульфата и других реактивов описано выше (см. стр. 17).
КОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ БОРА
Сущность метода. Наиболее подходящим методом оп-
ределения бора в растениях является колориметрический метод с
применением хинализарина или кармина.
Бор в концентрированной серной кислоте находится в виде
катиона В3+ (в гидросульфате бора — B(HSO4)3), а в растворах
серной кислоты меньшей концентрации — в виде иона борила ВО+
(в сульфате борила — (BO)2SO4). В связи с этим цветная реакция бора
с хинализарином (1, 2, 5, 3-тетраоксиантрахинон) в концентрирован-
ной серной кислоте может быть представлена следующим уравнением:
в2+
ОН О ОН ОН о \)
I II I I II I
он о он о
43
В продукте реакции ион бора оказывает значительное поляри-
зующее влияние на молекулу реагента, характер светопоглощения
которого в соответствие с этим сильно изменяется.
Окраска сернокислых растворов комплекса бора с хинализари-
ном сильно зависит от концентрации серной кислоты и изменяется
от голубой (99—80% H2SO4) до красной (73—60% H2SO4) и от крас-
ной до оранжевой (60—44% H2SO4). Окраска растворов самого хи-
нализарина также зависит от концентрации серной кислоты и из-
меняется от сине-фиолетовой до красной (99—88% H2SO4) и от
красной до оранжевой (70—44% H2SO4). Из этих данных видно,
что изменение окраски растворов хинализарина и его комплекса с
бором с изменением концентрации серной кислоты происходит
неодинаково. Максимальная чувствительность реакции бора с хи-
нализарином наблюдается при концентрации серной кислоты 87—
97%.
Существенный недостаток хинализарина — значительное пере-
крывание спектров светопоглощения растворов самого реагента и
его комплекса с бором. Этот недостаток может быть легко устранен
добавлением к сернокислому раствору хинализарина ледяной ук-
сусной кислоты, которая в этих условиях ацетилирует хинализарин,
что приводит к изменению сине-фиолетовой окраски раствора в ро-
зовато-красную, в то время как продукт реакции ацетилированного
хинализарина с бором окрашен в голубой цвет. Разница оптической
плотности растворов комплекса и хинализарина при 620 нм в этом
случае примерно в два раза больше, чем при использовании серно-
кислого раствора без уксусной кислоты. Добавление этилового спир-
та также улучшает спектральные характеристики реактива.
Раствор кармина и карминовой кислоты в концентрированной
серной кислоте окрашен в красный цвет. В присутствии бора это
окрашивание изменяется в синее с максимумом светопоглощения при
590—610 нм. Здесь как и с хинализарином образуется комплексное
соединение бора с кармином или карминовой кислотой, например:
Н3С О он
ААА/С00Н
H0~U\/U-0H
I II I
ноос о он
В2+
н3с 6 ^о
ААА/С00Н
H0-U\/\J-0H
I II I
ноос о он
кармин
После прибавления сернокислого раствора кармина к раствору,
держащему ионы бора, интенсивность окраски постепенно воз-
p.' га ст и достигает максимума через 1 ч, потом остается неизменной
в 'чение нескольких суток. С хинализарином окрашивание разви-
44
вается быстрее и достигает максимума уже через 20—25 мин. Ско-
рость развития окраски и ее интенсивность зависит от концентрации
серной кислоты. Скорость образования комплекса уменьшается,
а чувствительность реакции возрастает с увеличением концентра-
ции серной кислоты от 88 до 98%. Из этого следует, что нужно всег-
да работать с одинаковой концентрацией серной кислоты. Чув-
ствительность реакции ионов бора с кармином несколько выше, чем
с карминовой кислотой. Карминовую кислоту получают из коше-
нили, а кармин из карминовой кислоты кипячением ее водного раст-
вора, подкисленного соляной кислотой. При этом карминовая кис-
лота, являющаяся глюкозидным производным кармина, омыляется
с образованием кармина.
Кармин и карминовая кислота имеют по сравнению с хинализари-
ном некоторые недостатки, заключающиеся в более длительном раз-
витии окраски, меньшей устойчивости окрашенного комплекса и не-
сколько меньшей чувствительности реакции, но обладают одним
существенным преимуществом: отрицательное влияние многих при-
месей оказывается значительно меньшим.
Обычные элементы, присутствующие в золе (железо, алюминий,
кальций, магний, фосфор и хлор), не мешают реакции. Нитраты,
нитриты, перманганаты и другие окислители мешают реакции, так
как в очень кислой среде они окисляют кармин и хинализарин. При
небольшом количестве окислителей образуется сначала вещество,
окрашенное в фиолетовый цвет, похожее па соединение хинализари-
на и кармина с бором. При избытке окислителя происходит полное
разрушение реактива и раствор обесцвечивается. Фториды также
мешают этой реакции, так как образуют комплексное соединение
с бором, которое не реагирует с хинализарином и кармином.
Нитраты при сжигании органического вещества и прокаливании
разрушаются и не мешают определению бора. Фтор содержится в
растениях в незначительном количестве и не влияет на реакцию обра-
зования комплекса бора с хинализарином и кармином. Марганец
при прокаливании в присутствии карбонатов щелочных металлов
переходит в шестивалентный, а при подкислении в семивалентный
и как окислитель мешает определению бора. Потому его переводят в
осадок МпО2 и отфильтровывают.
При выпаривании растворов, содержащих бор, должна быть ще-
лочная среда, чтобы избежать потери бора, так как свободная бор-
ная кислота летуча. При работе в фарфоровой посуде для подще-
лачивания нужно брать гидроокись кальция, но не соду или щелочь,
которые образуют буру. Последняя во время прокаливания сплав-
ляется с эмалью и не переходит в раствор. В работе нельзя пользо-
ваться посудой из стекла, содержащего бор (молибденовое, пирекс
и др.), а нужно брать посуду из простого белого стекла или обык-
новенные склянки для реактивов.
Ход анализа. Отвешивают приблизительно 1 г воздушно-
сухого исследуемого вещества во взвешенном на аналитических ве-
сах тигле емкостью около 30 мл и сушат при 100—105° С до постоян-
45
ного веса, если неизвестна влажность. Тигли лучше брать платино-
вые, но можно пользоваться и фарфоровыми, если сжигать при тем-
пературе не выше 50£° С. К навеске в тигле приливают 2 мл 1 % -ного
раствора Na2CO3, перемешивают стеклянной палочкой, которую
затем вынимают и споласкивают 1—2 мл дистиллированной воды.
Тигель снова помещают в сушильный шкаф, высушивают при 105° С
и сжигают в муфельной печи при слабом нагреве или на плитке под
колпаком. Остаток в тигле смачивают 1 мл дистиллированной воды,
высушивают и снова прокаливают. Если зола темная от несгорев-
шего угля, обработку водой повторяют.
После сжигания и охлаждения в тигель прибавляют 5 мл ди-
стиллированной воды, 5 капель этилового спирта, размешивают
стеклянной палочкой, разминают крупинки и нагревают до кипе-
ния. При этом марганец переходит из шестивалентного в четырех-
валентный и осаждается в виде МпО2. После этого прибавляют 1 мл
1 н. раствора серной кислоты, перемешивают и выливают все из тиг-
ля через воронку в мерную колбу на 25 мл, тигель промывают ди-
стиллированной водой и доводят раствор в колбе до метки. Жид-
кость в колбе тщательно перемешивают и фильтруют через сухой
беззольный фильтр в сухую склянку па 100 мл. Из полученного
фильтрата набирают пипеткой 10 или 20 мл, в зависимости от коли-
чества бора, выливают в фарфоровую чашку, прибавляют 2 мл 1 % -но-
го раствора Na2CO3, помещают на кипящую водяную баню и вы-
паривают досуха. После охлаждения к остатку в чашке приливают
0,5 мл смеси (одна часть уксусной кислоты и одна часть этилового
спирта), смачивают весь сухой остаток, немедленно прибавляют 5 мл
0,03%-ного раствора кармина в концентрированной серной кисло-
те, тщательно перемешивают стеклянной палочкой и переливают
раствор в сухую пробирку. Чтобы избежать потери спиртово-уксус-
нокислой смеси за счет испарения, необходимо при серийных анали-
зах прибавлять ее сначала только в одну пробу, сюда же добавить
раствор кармина и после тщательного перемешивания перелить в су-
хую пробирку. Затем приступить к обработке спиртово-уксуснокис-
лой смесью и кармином следующей пробы. Когда все пробы будут
обработаны и растворы перенесены в пробирки, их оставляют на 1 ч
для развития окраски. После этого измеряют оптическую плотность
растворов при 610 нм в кювете с толщиной слоя 10 мм. Раствором
для сравнения служит раствор кармина в серной кислоте с добавкой
спиртово-уксуснокислой смеси в таком же соотношении, как и в опы-
те. Результаты измерений сравнивают с калибровочной кривой,
находят концентрацию бора в колориметрируемом растворе и вы-
считывают содержание его в исследуемом веществе по формуле:
у _ 0,1 25 5,5 С _ 13,75 С
В п В п ’
где X — содержание бора (в мг на 100 г исследуемого вещества);
С — концентрация бора, найденная в колориметрируемом растворе
(в мкг!мл)\ В — объем исследуемого раствора, взятого в чашку для
46
выпаривания и колориметрирования бора (в мл); п — абсолютно-
сухая навеска (в г); 25 — общий объем исследуемого раствора (в мл);
5,5 — объем колориметрируемого раствора бора (в мл); 0,1 — коэф-
фициент для перевода микрограммов в миллиграммы и пересчета
их на 100 г вещества.
Определение бора с хинализарином
При определении бора с хинализарином ход анализа такой же,
как и с кармином, только после выпаривания исследуемого раство-
ра в фарфоровой чашке к сухому остатку приливают 1 мл спирто-
во-уксуснокислой смеси, перемешивают стеклянной палочкой, не-
медленно прибавляют 5 мл 0,003%-ного раствора хинализарина в
концентрированной серной кислоте, тщательно перемешивают, пере-
ливают раствор в сухую пробирку и оставляют на 30 мин &ля раз-
вития окраски. При серийных анализах поступают так же, как и в
случае с кармином. Колориметрируют при 610 нм в кювете с толщи-
ной измеряемого слоя 10 мм. Раствором сравнения служит смесь,
состоящая из 5 мл 0,003%-ного раствора хинализарина и 1 мл спир-
тово-уксуснокислого раствора. Содержание бора вычисляют по
формуле:
Y _ 0,1 • 25 6 С _ 15 С
А ~ В п ~ В п ’
где 6 — объем колориметрируемого раствора (в мл).
Остальные обозначения такие же, как в методе с кармином.
Построение калибровочной кривой. В фар-
форовые чашки вливают последовательно 0,55, 1,1, 2,2, 3,3, 4,4 и
5,5 мл раствора буры или борной кислоты, содержащего 2 мкг бора
в 1 мл раствора, прибавляют 1 мл 1 %-ного раствора Na2CO3, выпа-
ривают на водяной бане досуха и охлаждают. В чашку прибавляют
0,5 мл смеси (одна часть уксусной кислоты и одна часть этилового
спирта), размешивают стеклянной палочкой, немедленно добавляют
5 мл 0,03%-ного раствора кармина, тщательно перемешивают и раст-
вор переливают в сухую пробирку. Затем эту операцию проводят
таким образом со всеми пробами. Растворы в пробирках оставляют
на 1 ч для полного развития окраски и после этого измеряют оп-
тическую плотность растворов при 610 нм в кювете с толщиной слоя
10 мм. Раствором для сравнения служит 0,03%-ный раствор кар-
мина в серной кислоте, содержащий смесь этилового спирта с ук-
сусной кислотой, как в опыте. Так как объем окрашенного раствора
равен 5,5 мл, то концентрация колориметрируемых растворов для
калибровочной кривой рассчитывается путем деления взятого в
чашку количества бора (в мкг) на 5,5. Полученные концентрации от-
кладывают на абсциссе, а оптические плотности на ординате и стро-
ят график (рис. 5).
При построении калибровочной кривой для хинализарина в
фарфоровые чашки вливают последовательно 0,6, 1,2, 2,4, 3,6, 4,8
47
Рис. 5. Калибровочная кривая
для бора (светофильтр с макси-
мумом пропускания 610 нм, кю-
вета с толщиной измеряемого
слоя 10 мм)'.
/ — с хинализарином; 2 — с кар-
мином.
и 6 мл раствора, содержащего 2 мкг бора в 1 мл, и выпаривают так
же, как при построении калибровочной кривой с кармином. После
выпаривания к сухому остатку в первой чашке прибавляют 1 мл
смеси (одна часть этилового спирта и
одна часть уксусной кислоты), пере-
мешивают стеклянной палочкой, до-
бавляют 5 мл 0,003%-ного раствора
хинализарина в серной кислоте, тща-
тельно перемешивают и раствор выли-
вают в сухую пробирку. Затем таким
же образом обрабатывают следующую
пробу. Когда все пробы будут пере-
несены в пробирки, их оставляют на
30 мин для полноты развития окрас-
ки. После этого измеряют оптическую
плотность растворов при 610 нм в
кювете с толщиной измеряемого слоя
мм. Раствором для сравнения слу-
жит смесь такого же количества ре-
активов, как в опыте, только без бора.
Объем окрашенного раствора равен
6 мл, поэтому концентрация колори-
метрируемых растворов для калибро-
вочной кривой рассчитывается путем деления взятых количеств
бора (в мкг) на 6 (рис. 5).
Реактивы. Раствор кармина (0,03%-ный). Взвешивают на аналитических
весах 0,15 г кармина, всыпают в мерную колбу на 500 мл, приливают 300 мл сер-
ной кислоты пл. 1,84 и взбалтывают до полного растворения кармина, затем доли-
вают до метки такой же кислотой, перемешивают и сохраняют в темном месте.
Чтобы очистить кармин от примесей, получают аммиачную соль кармина,
которой затем пользуются при приготовлении раствора. Для получения амми-
ачной соли берут 1 г кармина и растворяют его в 50 мл 10%-ного раствора ам-
миака. Раствор фильтруют через воронку Бюхнера с беззольным фильтром (синяя
лента), фильтрат выпаривают в фарфоровой чашке на водяной бане досуха и
сушат при 100° С в сушильном шкафу. Получается красно-бурый порошок —
C2iH19O11COONH4, из которого приготовляется 0,03%-ный раствор реактива в кон-
центрированной серной кислоте.
Раствор хинализарина (0,003%-ный). Взвешивают на аналитических весах
30 мг хинализарина, высыпают в мерную колбу на 1 л, наливают половину колбы
серной кислотой пл. 1,84, взбалтывают до растворения хинализарина, доводят
до метки концентрированной серной кислотой, перемешивают и сохраняют в тем-
ном месте с притертой стеклянной пробкой.
Стандартный раствор бора. Сначала готовят раствор буры, который содер-
жит 100 мкг бора в 1 мл. Для этого взвешивают на аналитических весах 0,4407 г
Na2B4O7- ЮН2О, переносят в мерную колбу на 500 мл, растворяют в дистиллиро-
ванной воде, доводят водой до метки и тщательно перемешивают.
Из этого раствора готовят рабочий стандартный раствор, разбавляя его в 50
раз дистиллированной водой. Набирают пипеткой 5 мл в мерную колбу на 250 мл,
доливают водой до метки и тщательно перемешивают. В 1 мл этого раствора со-
держится 2 мкг бора.
Смесь этилового спирта с уксусной кислотой (1 1). 50 мл ледяной уксусной
кислоты смешивают с 50 мл 95%-ного этилового спирта и сохраняют в склянке
со стеклянной притертой пробкой.
48
Раствор карбоната натрия (1%-ный). Взвешивают 10 г безводной соли —
Na2CO3, растворяют в дистиллированной воде в мерной колбе на 1 л, доливают
водой до метки и перемешивают.
Насыщенный раствор гидроокиси кальция. Отвешивают 3 г СаО, высыпают
в колбу, заливают 1 л дистиллированной воды, закрывают пробкой и взбалтывают
в течение 10 мин, затем фильтруют в реактивную склянку и сохраняют в хорошо
закрытой посуде без доступа углекислоты.
Приготовление остальных реактивов описано выше (см. стр. 32).
ОПРЕДЕЛЕНИЕ МЕДИ, ЦИНКА И КОБАЛЬТА
В ОДНОЙ НАВЕСКЕ
Сущность метода. После сжигания органического ве-
щества на плитке с открытой спиралью под колпаком из асбеста
или в муфельной печи золу растворяют в соляной кислоте с таким
расчетом, чтобы кислотность была около 0,05 н. Из полученного
раствора медь экстрагируется раствором дитизона в четыреххло-
ристом углероде. Экстракт выпаривают на водяной бане, а дитизон
и дитизонат меди разрушается азотной кислотой и перекисью во-
дорода. Медь определяют колориметрическим диэтилдитиокарба-
матным методом.
В водной фазе после извлечения меди остаются цинк и кобальт,
которые после подщелачивания раствора аммиаком, экстрагируют-
ся дитизоном. При обработке раствора дитизонатов 0,01 н. раство-
ром соляной кислоты цинк переходит в водную фазу и определяется
колориметрически после повторного экстрагирования его дитизо-
ном при pH 4,75. Кобальт после разрушения его дитизоиата азотной
кислотой и перекисью водорода определяется также колориметриче-
ским методом с иитрозо-Р-солыо.
Для аналитических целей используются растворы дитизона в
хлороформе и четыреххлористом углероде. При этом четырех-
хлористый углерод имеет преимущество, так как он менее летуч и
менее растворим в воде. Растворы дитизона в органических раство-
рителях зеленого цвета. При встряхивании раствора дитизона в четы-
реххлористом углероде или в хлороформе с водным раствором соли
металла образуется комплексное соединение металла с дитизоном —
дитизонат, растворимый в органическом растворителе:
NH—С6Н5\-
= N—С6Н5 ]2
В зависимости от того замещается в дитизоне один или оба водо-
родных иона, различают одно- и двузамещенные дитизонаты. В ана-
литической практике используются однозамещенные дитизонаты, в
большинстве случаев окрашенные в красный или красно-фиолето-
вый цвет. Одно- и двузамещенные дитизонаты находятся между собой
в подвижном равновесии и реакцию можно направить по желаемому
пути, создавая определенное pH и регулируя количество реагента.
Реакция образования дитизонатов обратима:
МеВодН. 4- 2Н2ДОрг. Me (НД)2орг -j- 2H^h.
4 6-2
49
Уравнение показывает, что на образование дитизонатов оказывает
влияние концентрация водородных ионов. Последним обстоятельст-
вом пользуются для разделения металлов с большим различием в
прочности комплексов^ Так медь, имеющая константу нестойкости
дитизоната— 1Д Ю-27, взаимодействует с дитизоном в сильно-
кислой среде и полностью извлекается органическими растворителя-
ми при pH 1._ Дитизонат цинка, константа нестойкости которого
равна 0,8 10 20, при pH 2 совсем не образуется, но его можно пол-
ностью извлечь из ацетатного буферного раствора при pH 4,75.
Кобальт начинает извлекаться только при pH 5,5. В цитратных
растворах цинк и кобальт полностью извлекаются только при
pH 8—9.
Так как дитизонаты металлов почти нерастворимы в водных раст-
ворах с определенным pH, а растворимость их в органических рас-
творителях больше в 10* — 107 раз, то величины коэффициентов экс-
тракции очень велики. С помощью небольшого объема раствора ди-
тизона в органическом растворителе можно извлечь очень малые
количества ионов металлов из большого объема водного раствора и
таким образом увеличить их концентрацию в тысячу и более раз.
Скорость образования дитизонатов зависит от свойств органическо-
го растворителя и главным образом от pH водного раствора.
В присутствии анионов, способных связывать металл в комплекс-
ное соединение (анионы винной, лимонной кислот и др.), реакция
образования дитизонатов замедляется и скорость экстракции умень-
шается. Образовавшийся дитизонат в органическом растворителе
можно разложить, встряхивая его с раствором кислоты. При разло-
жении кислотами выделяется свободный дитизон, который остает-
ся в органическом растворителе, а ион металла переходит в водную
фазу. Различные дитизонаты можно избирательно разлагать, при-
меняя различные величины pH. Так, дитизонат цинка легко разла-
гается 0,01 н. соляной кислотой, тогда как дитизонат меди и дити-
зонат кобальта при такой кислотности по разлагается.
Устойчивость дитизоната кобальта к кислотам возрастает с
увеличением продолжительности времени его хранения. Хотя дити-
зонат кобальта начинает образовываться только при pH 5,5, через
1 ч после получения он уже не поддается действию 0,01 н. соляной
кислоты, чем и пользуются для отделения ионов цинка, так как ди-
тизонат последнего независимо от времени хранения легко разру-
шается 0,01 н. соляной кислотой, причем цинк переходит в водную
фазу. Дитизонаты можно разлагать также щелочами. В этом слу-
чае в водную фазу переходит не только металл, но и дитизон в виде
дитизоната щелочного металла желтого цвета:
Me (НД)2орг. + 2ОНв-дн. Me (ОН)2вода + 2НД-д„.
Раствор дитизоната в четыреххлористом углероде в видимой об-
ласти спектра имеет два максимума поглощения — при 450 нм и
620 нм и минимум при 510 нм. Максимальное поглощение дитизоната
50
цинка находится при 535 нм, дитизоната меди при 545 нм и дитизо-
ната кобальта при 542 нм.
Устойчивость растворов отдельных дитизонатов в органических
растворителях во времени и под действием света различна. Раст-
воры дитизоната цинка в четыреххлористом углероде заметно не
разлагаются в течение нескольких дней как в темноте, так и на рас-
сеянном свету. Растворы дитизона в четыреххлористом углероде
и хлороформе под действием яркого света и высокой температуры
разлагаются с образованием желтого продукта окисления. Повышен-
ная температура действует сильнее, чем рассеянный свет. Пря-
мой солнечный свет быстро обесцвечивает раствор дитизона. Сла-
быми окислителями дитизон окисляется в дифенилтиокарбодиазои —
S=C (N=N—С6Н5)2. Это соединение не растворяется в водных раст-
ворах кислот и щелочей, но растворяется в хлороформе и четыреххло-
ристом углероде, образуя раствор желтого и коричневого цвета.
Трехвалентное железо окисляет дитизон обычно в щелочном раство-
ре, содержащем цитраты или тартраты. Окисление может быть пре-
дотвращено добавлением перед извлечением солянокислого гидро-
ксиламина.
Ионы меди в слабокислой или аммиачной среде реагируют с ди-
/N(C2H6)2
этилдитиокарбаматом натрия — S=G , образуя бурую кол-
^S—Na
лоидную суспензию диэтилдитиокарбамата меди:
<, S=C----S
I I
2 (С2Н5)2 N—С Х + Cu2+ = (С2Н6)2 N -> Си ч- N (С2Н5)2.
^SNa । । _ Q
О' — <5
Это соединение растворимо в амиловом спирте, амилацетате, ксило-
ле, хлороформе и четыреххлористом углероде.
Для экстракции комплекса меди лучше всего применять четырех-
хлористый углерод или хлороформ. Окраска экстрактов устойчива
в темноте. При действии света на раствор в четыреххлористом уг-
лероде происходит обесцвечивание раствора, поэтому все операции
следует производить при ослабленном дневном свете. Максимум све-
топоглощения диэтилдитиокарбамата меди находится при 435 нм.
Нитрозо-Р-соль (1-нитрозо-2нафтол-3,6-дисульфонат натрия)
NO
| ОН
х\/\/
NaO3S SO3Na
4:
51
образует с ионами кобальта в среде близкой к нейтральной комп-
лексное соединение, окрашенное в красный цвет. Один моль кобаль-
та связывает три моля реагента, при этом кобальт замещает водо-
род в гидроксильной группе и связывается с азотом нитрозо-груп-
пы ковалентной связью. Образовавшееся соединение устойчиво в
нейтральных и слабокислых растворах. Соединение не образуется
при pH меньше 2,5. При pH 6 реакция комплексообразования закан-
чивается в течение 5 мин при комнатной температуре. Верхний до-
пустимый предел pH составляет 8. Окрашенные соединения с нитро-
зо-Р-солью образуют также ионы меди, никеля и двухвалентного
железа.
Нитрозо-Р-соль растворяется в воде. Кислые растворы имеют
светло-желтую окраску, а щелочные — оранжевую. Поэтому после
образования соединения кобальта с нитрозо-Р-солью добавляют мине-
ральную кислоту для ослабления окраски реагента и разложения
комплексов других металлов, которые могут присутствовать в раство-
ре. В горячей 1 н. азотной кислоте разлагаются даже комплексы меди
и никеля. В этих условиях определению кобальта не мешают железо
(III), марганец, цинк, кадмий, свинец и олово. Окрашенное соеди-
нение кобальта устойчиво на свету в течение 1 ч. Максимум свето-
поглощения около 415 нм, но лучше измерения проводить при 490—
500 нм, так как при этой длине волны реагент обладает очень малым
светопоглощением.
Ход анализа. Взвешивают на технических весах 2 г
мелко измельченного воздушно-сухого вещества в предварительно
взвешенной платиновой или фарфоровой чашке диаметром 6 см и,
если неизвестна влажность, высушивают в термостате в течение 1—
2 ч при температуре 100° С. Охлаждают в эксикаторе и взвешивают
на аналитических весах. После этого чашку ставят на электриче-
скую плитку и нагревают до обугливания материала, а затем на-
крывают асбестовым колпаком и прокаливают до полного озоления.
Сжигание и озоление лучше проводить одновременно в нескольких
чашках. К остатку в чашке после охлаждения прибавляют 2 мл
дистиллированной воды, 2 мл азотной кислоты (1 : 1), выпаривают
на водяной бане досуха и прокаливают на плитке до удаления бурых
паров NO2. Охладив остаток, прибавляют 1 мл раствора соляной
кислоты (1 1), выпаривают на водяной бане досуха, прибавляют
2 мл 1 н. соляной кислоты, 5 мл дистиллированной воды, размеши-
вают и нагревают почти до кипения. Горячий раствор фильтруют
через фильтр с белой лентой диаметром 6—7 см, предварительно
промытый 0,1 н. соляной кислотой. Чашку и фильтр промывают
25 мл дистиллированной воды. Фильтрат переносят в делительную
воронку, смывая остаток 10 мл воды. Необходимо точно придержи-
ваться указанных объемов воды, чтобы в конце разбавления кон-
центрация кислоты была ~ 0,05 н.
52
Определение меди
К полученному солянокислому исследуемому раствору в дели-
тельной воронке прибавляют 2 мл 0,01 %-ного раствора дитизона
в четыреххлористом углероде и энергично взбалтывают в течение
2 мин. Дают фазам разделиться и сливают нижнюю органическую фа-
зу в другую делительную воронку. Обработку дитизоном повторяют
до тех пор, пока окраска его не перестанет изменяться. После этого
добавляют 2 мл четыреххлористого углерода х. ч. и извлекают оста-
ток дитизона, присоединяя его к первым экстрактам. Каплю раство-
ра дитизона в четыреххлористом углероде, плавающую на поверх-
ности водной фазы, удаляют постукиванием по делительной воронке
или легким ее встряхиванием.
Объединенные экстракты дитизоната меди промывают 10 мл
0,01 н. соляной кислоты, встряхивая около 1 мин. Фазу четырех-
хлористого углерода сливают в фарфоровую чашку, а водную фазу
присоединяют к основному раствору в делительной воронке. Для
разрушения дитизона и дитизоната меди в фарфоровую чашку прили-
вают 2 мл азотной кислоты (1 1) и выпаривают на водяной бане до
удаления четыреххлористого углерода. К оставшейся азотной кисло-
те добавляют 5 капель 30%-ной перекиси водорода и выпаривают
досуха. К остатку добавляют 0,5 мл азотной кислоты (1 1), 5 ка-
пель перекиси водорода и выпаривают досуха вторично. Остаток
смачивают 5 каплями разбавленного (1 1) раствора соляной кисло-
ты и снова выпаривают на водяной бане досуха. В чашку приливают
2 мл 10%-ного раствора лимоннокислого натрия, растворяют сухой
остаток и переливают раствор в делительную воронку, а чашку про-
мывают дистиллированной водой (2 раза по 5 л/л), выливая промыв-
ные воды в ту же воронку. К раствору прибавляют 1 мл 0,1 %-ного
раствора диэтилдитиокарбамата натрия, перемешивают, добавля-
ют 5 мл четыреххлористого углерода и взбалтывают в течение
2 мин. Слою органического растворителя дают отстояться так, чтобы
в нем не было капелек воды, высушивают конец воронки кусочком
фильтровальной бумаги, сливают экстракт в сухую пробирку и за-
крывают пробкой. Измеряют оптическую плотность раствора при
440 нм в кювете с толщиной слоя 10 мм. Раствором для сравнения
служит дистиллированная вода. Из полученных данных находят
по калибровочной кривой концентрацию меди и рассчитывают со-
держание ее в пробе по следующей формуле:
v 5 С
где X — содержание меди (в мг на 1 кг исследуемого вещества);
С — концентрация меди в колориметрируемом растворе (в мкг/мл);
п — абсолютно-сухая навеска (в а); 5 — объем колориметрируемого
раствора (в мл).
Экстракция цинка и кобальта. К раствору в
делительной воронке, оставшемуся после извлечения меди, прибав-
53
ляют5л*л 10%-ного раствора лимоннокислого натрия, 2 капли 0,1 % -
ного раствора индикатора крезолового красного и по каплям кон-
центрированный раствор аммиака до появления красно-фиолетовой
окраски (pH 9). Затем добавляют 5 мл 0,02%-ного раствора дити-
зона в четыреххлористом углероде, энергично взбалтывают, дают фа-
зам разделиться и выливают органическую фазу в другую делитель-
ную воронку. Далее извлечение ведут небольшими порциями раст-
вора дитизона (по 1 мл), пока последние не перестанут окрашиваться
в фиолетовый цвет и приобретут зеленый оттенок. После этого при-
ливают еще 2 мл четыреххлористого углерода, взбалтывают 1—2 мин,
дают отстояться и сливают органическую фазу, добавляя ее к пер-
воначальному экстракту. Объединенные экстракты оставляют на 1 ч,
затем промывают 10 мл дистиллированной воды и переносят в чис-
тую делительную воронку. Сюда же добавляют 10 мл 0,01 н. раствора
соляной кислоты и взбалтывают 2 мин, чтобы перевести весь цинк
в водную фазу. После разделения фаз четыреххлористый углерод
сливают в другую делительную воронку, где взбалтывают с 5 мл
свежего 0,01 н. раствора соляной кислоты. Кислотные вытяжки объ-
единяют, выливают в мерную колбу на 50 лы, смывая остаток раство-
ра дистиллированной водой, доводят до метки и тщательно переме-
шивают. В этом растворе находится весь цинк. Фазу четыреххло-
ристого углерода, содержащую дитизонат кобальта, выливают в
фарфоровую чашку для дальнейшего определения кобальта.
Определение цинка
5 мл раствора из мерной колбы на 50 мл выливают в делитель-
ную воронку, прибавляют 5 мл ацетатного буферного раствора (pH
4,75), 1 мл 10% -ного раствора тиосульфата натрия, 5 мл 0,001 % -ного
раствора дитизона в четыреххлористом углероде и взбалтывают
2 мин. Носик воронки вытирают фильтровальной бумагой для удале-
ния капель воды и сливают прозрачный нижний слой в сухую про-
бирку, закрывающуюся пробкой. К раствору в делительной ворон-
ке прибавляют снова 5 мл 0,001 %-ного раствора дитизона, взбал-
тывают и отделяют органическую фазу, присоединяя ее к первому
экстракту. В последнем случае должен быть некоторый избыток
дитизона, что видно по окраске, имеющей зеленоватый оттенок.
Если избытка дитизона нет (окраска красно-фиолетовая), то извле-
чение повторяют новой порцией дитизона. После полного извле-
чения объединенный экстракт перемешивают и измеряют его объем
и оптическую плотность при 490—510 нм в кювете с толщиной слоя
5 мм. Нулевым раствором служит 0,001 %-ный раствор дитизона
в четыреххлористом углероде. Следует избегать освещения раствора
дитизона и экстракта прямым солнечным светом.
Из полученных данных по калибровочной кривой находя^ кон-
центрацию цинка и вычисляют содержание его в исследуемом веще-
стве по формуле:
v 0,1 50 в с _ в с
А — ----~,
5 п п ’
54
где X — содержание цинка (в мг на 100 г исследуемого вещества);
50 — общий объем исследуемого раствора цинка (в мл); 5 — объем
исследуемого раствора цинка, взятого для извлечения дитизона
(в мл); В — объем колориметрируемого раствора дитизоната цинка
в четыреххлористом углероде (в мл); С — концентрация цинка в
колориметрируемом растворе (в мкг/мл); п — абсолютно-сухая на-
веска (в г); 0,1 — коэффициент для пересчета микрограммов в мил-
лиграммы на 100 г вещества.
Определение кобальта
К дитизоновому экстракту после обработки его 0,01 н. раство-
ром соляной кислоты, находящемуся в фарфоровой чашке, прили-
вают 2 мл азотной кислоты (1 1), одну каплю 2 н. раствора серной
кислоты, выпаривают на водяной бане досуха и прокаливают на
плитке. После охлаждения к остатку прибавляют 0,5 мл азотной кис-
лоты (1 1), 5 капель 30%-ной перекиси водорода и выпаривают на
водяной бане досуха. Обработку азотной кислотой и перекисью во-
дорода повторяют. К сухому остатку, который должен быть почти
бесцветным, добавляют 1 мл 10%-ного раствора лимоннокислого
натрия, растворяют остаток, добавляют 1 мл 0,1 %-ного раствора
нитрозо-Р-соли, перемешивают и оставляют на 10—15 мин (или
нагревают 1—2 мин). Затем добавляют 1 мл азотной кислоты (1
1), переносят содержимое чашки в градуированную пробирку (или
в мерный цилиндр) на 5 или 10 мл, промывают чашку небольшими
порциями дистиллированной воды, доводя объем раствора до 5 мл,
перемешивают и измеряют оптическую плотность при 490—500 нм
в кювете на 10 мм. Для сравнения используют раствор нитрозо-Р-
соли и азотной кислоты, взятых в таких же количествах, как и в
опыте, и разбавленных до 5 мл дистиллированной водой. Окрашен-
ные растворы защищают от прямого солнечного света. Из получен-
ных данных находят по калибровочной кривой концентрацию ко-
бальта в колориметрируемом растворе и рассчитывают содержание
его в исследуемом веществе по формуле:
v 0,1-5 С _ 0,5 С
где X — содержание кобальта (в мг на 100 а исследуемого вещества);
5 — объем колориметрируемого раствора (в мл); С — концентрация
кобальта в колориметрируемом растворе (в мкг/мл); п — абсолют-
но-сухая навеска (в г); 0,1 —переводной коэффициент микрограм-
мов в миллиграмм-проценты.
Построение калибровочной кривой для
определения меди. В серию делительных воронок наби-
рают последовательно 1, 2, 4, 6, 8 и 10 мл рабочего стандартного
раствора соли меди, содержащего 2 мкг Си в 1 мл, и доводят объем
дистиллированной водой до 20 мл. К растворам прибавляют по 2 мл
10% -ного раствора лимоннокислого натрия, 2 мл 0,1 % -ного раствора
55
диэтилдитиокарбамата натрия и 5 мл четыреххлористого угле-
рода, взбалтывают 2 мин и дают отстояться. Отделяют органическую
фазу в сухие пробирки, закрывающиеся пробкой. Измеряют опти-
ческую плотность растворов при 449 нм в кювете на 10 мм. При из-
мерении кювета должна быть хорошо закрыта, чтобы не происхо-
дило испарение растворителя. Из полученных данных строят ка-
либровочную кривую (рис. 6).
Построение калибровочной кривой для
определения кобальта. Набирают последовательно
Рис. 6. Калибровочная кривая
для меди с диэтилдитиокарбама-
том натрия (светофильтр с мак-
симумом пропускания 440 нм,
кювета с толщиной измеряемого
слоя 10 мм).
Рис. 7. Калибровочная кривая
для кобальта с нитрозо-Р-солью
(светофильтр с максимумом про-
пускания 490 нм, кювета с тол-
щиной измеряемого слоя 10 мм).
1, 2, 4, 6, 8 и 10 мл стандартного раствора соли кобальта, содержа-
щего 2 мкг Со в 1 мл, выливают в делительные воронки, доводят
объем дистиллированной водой до 10л/л, прибавляют по 5 мл 10%-но-
го раствора лимоннокислого натрия, по одной капле индикатора кре-
золового красного и по каплям раствор аммиака до появления крас-
ного окрашивания. Затем добавляют по 5 мл 0,01 % -ного раствора ди-
тизона в четыреххлористом углероде, встряхивают 2 мин, дают от-
стояться и отделяют органическую фазу в фарфоровые чашки. В дели-
тельные воронки приливают еще по 2 мл хлороформа, встряхивают
1 мин, дают отстояться и сливают органическую фазу в те же чашки.
К объединенным экстрактам добавляют по 2 мл азотной кислоты (1
1), по одной капле 2 н. серной кислоты, выпаривают на водяной бане
досуха и прокаливают на плитке. После охлаждения к остаткам
прибавляют по 0,5 мл азотной кислоты (1 1), по 5 капель перекиси
водорода и снова выпаривают на водяной бане досуха. К остаткам,
которые должны быть почти бесцветными, добавляют по 1 мл 10% -
56
Рис. 8. Калибровочная кривая
для цинка с дитизоном (свето-
фильтр с максимумом пропуска-
ния 490 нм, кювета с толщиной
измеряемого слоя 5 мм).
ного раствора лимоннокислого натрия, растворяют осадки, добав-
ляют по 1 мл 0,1 %-ного раствора нитрозо-Р-соли, перемешивают и
оставляют на 10—15 мин. Затем добавляют по 1 мл азотной кисло-
ты (1 1), переносят в градуированные пробирки, доводят до 5 мл
и измеряют оптическую плотность при 490—500 нм в кювете на 10 мм,
сравнивая с раствором, состоящим из 1 мл 0,1 % -ного раствора нитро-
зо-Р-соли и 1 мл азотной кислоты (1 1), разбавленных до 5 мл
дистиллированной водой. Окрашенные растворы защищают от света.
Из полученных данных строят кали-
бровочную кривую (рис. 7).
Построение калибро-
вочной кривой для опре-
деления цинка. В делитель-
ную воронку емкостью 100 мл поме-
щают 15 мл ацетатного буферного
раствора (pH 4,75), добавляют 5 мл
0,001 %-ного раствора дитизона в че-
тыреххлористом углероде, взбалты-
вают 2 мин для удаления следов ме-
таллов, реагирующих с дитизоном,
дают фазам разделиться и органиче-
скую фазу отбрасывают. Затем после-
довательно набирают градуированной
пипеткой 0,5, 1,0, 1,5, 2,0, 3,0, 4,0
и 5,0 мл стандартного раствора соли
цинка, содержащего 2 мкг Zn в 1 мл. С каждой из порций стандарт-
ного раствора осуществляют следующие операции. Очередную пор-
цию помещают в делительную воронку, содержащую очищенный бу-
ферный раствор, прибавляют 10 мл 0,001 % -ного раствора дитизона
в четыреххлористом углероде, взбалтывают 2 мин и оставляют на не-
которое время для разделения фаз. Носик воронки вытирают фильт-
ровальной бумагой, сливают прозрачный экстракт в сухую про-
бирку и закрывают ее пробкой. К оставшемуся в воронке буферному
раствору прибавляют новую, следующую порцию стандартного рас-
твора, 10мл 0,001 % -ного раствора дитизона, взбалтывают, дают от-
стояться и сливают органическую фазу в следующую пробирку.
Так поступают со всеми необходимыми для построения калибровоч-
ной кривой количествами стандартного раствора цинка. После этого
измеряют оптическую плотность всей серии окрашенных растворов
дитизоната цинка при 490—510 нм в кювете с толщиной слоя 5 мм.
Раствором для сравнения служит 0,001 %-ный раствор дитизона в
четыреххлористом углероде. Из полученных данных строят калиб-
ровочную кривую (рис. 8).
Примечание. После встряхивания часто образуется капля
четыреххлористого углерода, плавающая на поверхности водной фа-
зы. Для удаления ее с поверхности нужно легко встряхнуть верх-
нюю часть жидкости. В этом случае поверхностное натяжение на-
рушается и капля опускается вниз.
57
Реактивы. Стандартный раствор соли меди. Взвешивают на аналитических
весах 0,3930 г CuSO4 5Н2О, растворяют в дистиллированной воде, очищенной
дитизоном, выливают в мерную колбу на 500 мл, добавляют 1 мл раствора соля-
ной кислоты (1 1), доводят дистиллированной, очищенной дитизоном водой до
метки и перемешивают. 1 мл этого раствора содержит 0,2 мг Си.
Из этого раствора готовят рабочий раствор, разбавляя его в 100 раз. Отби-
рают пипеткой 5 мл в мерную колбу на 500 мл, доводят очищенной дитизоном во-«
дой до метки и тщательно перемешивают. 1 мл этого раствора содержит 2 мкг Си.
Стандартный раствор соли цинка. Взвешивают на аналитических весах
0,1 г металлического цинка или очищенной цинковой пыли, помещают в стакан,
приливают 5 мл серной кислоты (1 4), 5 мл дистиллированной воды, накрывают
часовым стеклом и нагревают до полного растворения. Раствор количественно
переносят в мерную колбу на 500 мл, доводят водой, очищенной дитизоном, до
метки и перемешивают. Вместо металлического цинка можно взять 0,4398 г соли
ZnSO4 7Н2О х. ч. и растворить в 500 мл дистиллированной воды. 1 мл этого
раствора содержит 0,2 мг Zn.
Из полученного раствора готовят рабочий раствор разбавлением в 100 раз.
Набирают пипеткой 5 мл, переносят в мерную колбу на 500 мл, доводят очищен-
ной дитизоном водой до метки и перемешивают. 1 мл этого раствора содержит
2 мкг Zn.
Стандартный раствор соли кобальта. Взвешивают на аналитических весах
0,2630 г безводной соли CoSO4, помещают в стакан, растворяют в воде, подкислен-
ной 1 мл раствора серной кислоты (1 4), переносят в мерную колбу на 500 мл,
разбавляют дистиллированной водой до метки и перемешивают. 1 мл полученного
раствора содержит 0,2 мг Со.
Из этого раствора разбавлением в 100 раз готовят рабочий раствор. Отби-
рают пипеткой 5 мл раствора в мерную колбу на 500 мл, доводят водой, очищенной
дитизоном, до метки и перемешивают. 1 мл этого раствора содержит 2 мкг Со.
Безводная соль CoSO4 готовится из кристаллогидрата CoSO4 7Н2О х. ч.
прокаливанием в муфельной печи при 500° С до постоянного веса. Безводную соль
сохраняют в хорошо закрытой склянке.
Дистиллированная вода, очищенная от следов металла. Особое внимание сле-
дует обратить на чистоту дистиллированной воды. Обыкновенную дистиллирован-
ную воду можно очистить дитизоном. Для этого к дистиллированной воде в дели-
тельной воронке прибавляют 0,01%-ный раствор дитизона в четыреххлористом
углероде из расчета 5 мл на 1 л воды, энергично взбалтывают в течение 3 мин,
дают отстояться и сливают органическую фазу. Воду фильтруют через беззоль-
ный фильтр, предварительно промытый 0,1 н. соляной кислотой. Первую часть
фильтрата выбрасывают, а остальную воду сохраняют в склянке с притертой
пробкой.
Ацетатный буферный раствор (pH 4,75). Растворяют в дистиллированной
воде 7,5 г уксуснокислого натрия CH3COONa ЗН2О и 2,7 мл ледяной уксус-
ной кислоты, затем разбавляют до 500 мл и перемешивают. Раствор очищают ди-
тизоном. Для этого раствор переносят в делительную воронку, добавляют 5 мл
0,01 %-ного раствора дитизона в четыреххлористом углероде и энергично взбалты-
вают 2—3 мин. Органическую фазу сливают, а водную, если нужно, обрабаты-
вают еще раз раствором дитизона. Затем водную фазу фильтруют через беззоль-
ный фильтр, предварительно промытый 0,1 н. соляной кислотой и очищенной во-
дой. Раствор сохраняют в склянке со стеклянной пробкой.
Раствор лимоннокислого натрия (10%-ный). Взвешивают 10 г трехзамещен-
ного лимоннокислого натрия C6H6O7Na3 5,5Н2О и 0,5 г лимонной кислоты,
растворяют в воде и разбавляют до 100 мл. Раствор переносят в делительную во-
ронку и удаляют ионы тяжелых металлов, встряхивая с небольшими порциями
0,01%-ного раствора дитизона в четырех хлор истом углероде до тех пор, пока
зеленая окраска не будет изменяться. Раствор фильтруют через бумажный фильтр,
предварительно промытый 0,1 н. соляной кислотой и очищенной водой, чтобы уда-
лить капли четыреххлористого углерода.
Раствор тиосульфата натрия (10%-ный). Растворяют в предварительно
прокипяченной и охлажденной дистиллированной воде 10 г Na2S2O3 5Н2О, до-
водят этой же водой до 100 мл и очищают дитизоном, встряхивая в делительной
58
воронке с небольшими порциями (по 2 мл) 0,01 %-ного раствора дитизона в четы-
реххлористом углероде, пока органическая фаза больше не будет окрашиваться
в яркий красный цвет. Часть дитизона переходит в водную фазу и окрашивает
ее в желтый цвет. Как только водная фаза окрасится в желтый цвет, а органиче-
ская будет иметь только слабый красный оттенок, очистку дитизоном прекращают.
Дитизон из водной фазы извлекают хлороформом, добавляя его по 2—3 мл и энер-
гично встряхивая в продолжение 2 мин. Извлечение повторяют до тех пор, пока
хлороформный экстракт больше не будет окрашиваться в зеленый цвет. После
этого раствор фильтруют через беззольный фильтр, предварительно промытый
0,1 н. соляной кислотой и очищенной водой. Сохраняют в темной склянке.
Раствор диэтилдитиокарбамата натрия (0,1 %-ный). Реактив представляет
собой белый кристаллический порошок, легко растворимый в воде. 0,1 г реакти-
ва C5H10NS2Na ЗН2О растворяют в 100 мл дистиллированной воды. Хранят
в темной склянке. Раствор устойчив в течение месяца.
Раствор нитрозо-Р-соли (0,1%-ный). Нитрозо-Р-соль — кристаллический
порошок золотисто-желтого цвета, растворимый в воде и спирте. Нейтральные
и кислые растворы имеют светло-желтую окраску, а щелочные — оранжевую.
0,1 г реактива C10H5O8NS2Na2 растворяют в 100 мл воды. Раствор при хране-
нии в темной склянке не изменяется в течение нескольких месяцев.
Раствор крезолового красного (0,1%-ный). Крезоловый красный — индика-
тор с интервалом pH перехода 7,2—8,8 от желтого к красному. Отвешивают 0,1 г
индикатора и растирают в ступке с 3 мл 0,1 н. раствора едкого натра, переносят
в мерную колбу на 100 мл, разбавляют дистиллированной водой до метки и пере-
мешивают.
Хлороформ. Имеющийся в продаже хлороформ ч. д. а. может быть исполь-
зован без всякой очистки. Для регенерации уже использованного хлороформа
его отделяют от водной фазы, промывают в делительной воронке концентрирован-
ной серной кислотой порциями по 5—10 мл до обесцвечивания. Затем встряхи-
вают с известью, помещают в колбу для отгонки и перегоняют на водяной бане,
собирая фракцию при температуре 61—61,5° С. В дистиллят добавляют 1% эти-
лового спирта по объему. Спирт предохраняет хлороформ от разложения.
Четырех хлористый углерод. Продажный четырех хлористый углерод ч. после
настаивания с несколькими каплями бромной воды энергично встряхивают в де-
лительной воронке с порциями по 5 мл 20% -ного раствора едкого натра, содержа-
щего солянокислый гидроксиламин (на 100 мл 20% -ного NaOH 0,8 г солянокислого
гидроксиламина). Встряхивание продолжают до тех пор, пока щелочь перестанет
окрашиваться в желтый цвет. После чего промывают один раз водой, переносят
в колбу для отгонки, добавляют гранулированный хлористый кальций и перего-
няют на водяной бане, собирая фракцию при температуре 76,5—77° С.
Имеющийся в продаже четыреххлористый углерод ч. д. а. может быть исполь-
зован без очистки. Для регенерации уже использованного четыреххлористого уг-
лерода его отделяют от водной фазы и, если жидкость мутная, фильтруют через
бумажный фильтр. Для разрушения дитизонатов встряхивают в делительной во-
ронке с концентрированной серной кислотой по 5—10 мл до тех пор, пока новая
порция кислоты останется бесцветной. Затем промывают дистиллированной во-
дой для удаления кислоты, встряхивают с 1%-ным раствором едкого натра,
снова промывают водой и перегоняют на водяной бане над окисью кальция.
Дитизон. Продажный препарат очищают перекристаллизацией:
а) 1 г дитизона растворяют в 50 мл хлороформа, фильтруют через бумажный
фильтр в стакан и прибавляют при помешивании 150 мл петролейного эфира.
При этом дитизон выпадает в виде темных сине-черных кристаллов. Фильтруют
и высушивают на воздухе или в вакуум-эксикаторе.
б) 1 г дитизона растворяют в 50 мл хлороформа и фильтруют через бумажный
фильтр в стакан. Раствор в стакане оставляют открытым в темном месте до тех
пор, пока испарится 2/3 объема хлороформа или помещают в вакуум-эксикатор
и выпаривают под разрежением до объема 10—15 мл. Из сконцентрированного
таким образом раствора дитизон выпадает в виде кристаллов. Раствор с осадком
фильтруют через пористый стеклянный фильтр № 2 или № 3, промывают стакан
и фильтр два раза четыреххлористым углеродом порциями по 5 мл, кристаллы
высушивают на воздухе или в вакуум-эксикаторе и сохраняют в темной склянке.
59
Дитизон, или дифенилтиокарбазон представляет собой кристаллический по-
рошок черно-фиолетового цвета, нерастворимый в воде. Очень мало растворим
в спирте и эфире. В 100 г хлороформа при комнатной температуре растворяется
около 2 г, а в 100 г четыреххлористого углерода — около 0,05 г реактива.
Раствор дитизона в четыреххлористом углероде (0,02%-ный). Отвешивают
20 мг очищенного препарата на аналитических весах, переносят в мерную колбу
на 100 мл, растворяют в четыреххлористом углероде, доводят этим же раствори-
телем до метки и перемешивают. Раствор сохраняют в темном месте при темпе-
ратуре 5° С. Из этого раствора в день использования готовят 0,01 %-ный и 0,001 %-
ный растворы разбавлением в 2 раза и в 10 раз четыреххлористым углеродом.
Аммиак. Продажный аммиак почти всегда содержит значительные количества
металлов группы дитизона, особенно цинка, свинца и меди. Поэтому аммиак не-
обходимо очистить от примесей. Очень чистый аммиак получают' 'методом изотер-
мической диффузии. Для этого в пустой эксикатор помещают два стакана —один
с продажным 25%-ным аммиаком, другой на 2/3 объема наполненный дважды пе-
регнанной водой и оставляют на 2—3 дня. Крышка эксикатора должна быть хо-
рошо притерта. Пары аммиака поглощаются водой до тех пор, пока не наступит
равновесие. Полученный раствор аммиака в бидистиллированной воде перели-
вают в склянку с притертой пробкой, где и сохраняют.
Азотная кислота. Концентрированную азотную кислоту пл. 1,40 наливают
в колбу аппарата Сокслета, ставят на плитку и отгоняют в верхнюю часть аппа-
рата объем жидкости, не достигающий верхней части сифона на 10—15 мм. Затем
охлаждают, снимают экстрактор и перегнанную кислоту выливают в склянку
с притертой пробкой.
Азотная кислота с водой образует постоянно кипящую смесь с температурой
кипения 121,9° С, содержащую 68,4% HNO3.
Соляная кислота. В соляной кислоте обычно содержится значительное коли-
чество цинка, ртути, свинца и других металлов, поэтому она требует очистки.
Соляная кислота с водой образует постоянно кипящую смесь с температурой ки-
пения 110° С, содержащую 20,2% НС1. Для очистки перегонкой берут соляную
кислоту пл. 1,19, разбавляют ее в два раза дистиллированной водой и перегоняют
в аппарате Сокслета аналогично азотной кислоте. Сохраняют в склянке с притер-
той пробкой.
Перегнанная в аппарате Сокслета соляная кислота имеет концентрацию около
6 н. Из этого раствора готовят растворы меньшей концентрации — 1 н., 0,1 н.
и 0,01 н. разбавлением очищенной дитизоном водой.
Очень чистую соляную кислоту можно получить методом изотермической диф-
фузии аналогично очистке аммиака.
ОПРЕДЕЛЕНИЕ МОЛИБДЕНА
Роданидный метод. Этот метод, с применением хлористого олова,
считается наилучшим вследствие избирательности и хорошей чувст-
вительности.
Сущность метода. В кислом растворе шестивалентный
молибден восстанавливается сильными восстановителями до пяти-
валентного, который в присутствии роданид-ионов образует соеди-
нение желтого или оранжевого цвета:
Мо5+ + 5CNS- Mo(CHS)B.
Эта реакция известна с 1863 г. и в настоящее время использу-
ется для колориметрического определения небольших количеств мо-
либдена. Окрашенное соединение — роданидный комплекс пяти-
валентного молибдена можно извлечь эфирами, спиртами и кетона-
ми. Удобно применять в качестве растворителя комплекса смесь
60
изоамилового спирта и четыреххлористого углерода (I 1), так как
эта смесь тяжелее воды и меньше извлекает роданид железа.
Для получения роданидных соединений пятивалентного молиб-
дена в качестве восстановителя шестивалентного молибдена и трех-
валентного железа используют хлористое олово, которое быстро вос-
станавливает железо до двухвалентного и обеспечивает максималь-
ное развитие окраски:
2FeCl3 + SnCl2 = 2FeCl2 + SnCl4,
2Мо6+О3 + SnCl2 + 2HC1 = 2HMo5+O3 + SnCl4.
Ионы трехвалентного железа повышают выход окрашенного со-
единения молибдена и его устойчивость. В присутствии ионов желе-
за образуется только пятивалентный молибден, что вызывает значи-
тельное увеличение интенсивности окраски. Присутствие меди необ-
ходимо, так как она способствует восстановлению трехвалентного
железа до двухвалентного. Этим достигается полное обесцвечива-
ние раствора, окрашенного роданидом железа.
Оптимальная концентрация раствора соляной кислоты при вос-
становлении хлористым оловом — 1 н. Концентрация роданистого
калия должна быть при реакции не ниже 1% , a SnCl2 не ниже 0,5%.
Трехвалентного железа должно быть в растворе 1—2 мг. Большее
содержание железа (до 10 мг) не меняет интенсивность окраски.
Необходимо строго придерживаться указанных в методике объемов
прибавляемых реактивов и воды.
Мешающее влияние титана при содержании его до 30 мг устра-
няют добавлением фтористого натрия. Если при определении молиб-
дена исследуемый раствор содержит ионы железа или меди, которые
повышают интенсивность окраски молибденового комплекса, то
необходимо и в стандартные растворы вводить эти ионы.
Ход анализа. Взвешивают 3—5 г воздушно-сухого ис-
следуемого вещества в фарфоровой или платиновой чашке диаметром
6 см, сжигают сначала на открытой плитке, затем под асбестовым
колпаком и прокаливают до озоления органического вещества.
Охлаждают, прибавляют к золе 2 мл дистиллированной воды, 2 мл
азотной кислоты (1 1), выпаривают на водяной или воздушной бане
досуха и прокаливают на плитке до удаления бурых паров окислов
азота. К остатку в чашке после охлаждения прибавляют 1—2 мл
раствора соляной кислоты (1 1) и выпаривают на водяной бане до-
суха. В чашку снова прибавляют 5 мл раствора соляной кислоты
(1 1), нагревают и горячим фильтруют через бумажный фильтр с
белой лентой диаметром 6—7 см, предварительно промытый 0,1 н.
соляной кислотой. Чашку и фильтр промывают 15 мл дистиллирован-
ной воды. Фильтрат переносят в делительную воронку, смывая ос-
таток 5 мл воды. К раствору в делительной воронке прибавляют одну
каплю 1 %-ного раствора сульфата меди, 1 мл 0,5%-ного раствора
хлорного железа, 2 мл 20%-ного раствора роданистого калия, 5 мл
61
смеси изоамилового спирта и четыреххлористого углерода (1 1) и
перемешивают. Добавляют 2 мл 10%-ного раствора SnCl2, взбалты-
вают до исчезновения красной окраски и появления оранжевой, остав-
ляют в покое на 1—2 мин, затем снова взбалтывают 0,5 мин и дают
отстояться в течение 2—3 мин. Отделившуюся органическую фазу
сливают в сухую пробирку. Если имеется муть, ее устраняют добав-
лением 10 капель изоамилового спирта и измеряют оптическую плат-
ность раствора при 440 нм в кювете на 10 мм. Раствором сравнения
служит экстракт из тех же реактивов, что и в опыте, только вместо
исследуемого раствора используют воду. Из полученных данных по
калибровочной кривой находят кон-
центрацию молибдена в колориме-
трируемом растворе и рассчитывают
содержание его в пробе по следующей
формуле:
v 5 С
Рис. 9. Калибровочная кривая
для молибдена с роданидом (све-
тофильтр с максимумом пропус-
кания 440 нм, кювета с толщи-
ной измеряемого слоя 10 мм).
где X — содержание молибдена (в мг
на 1 кг исследуемого вещества);
С — концентрация молибдена в ко-
лориметрируемом растворе (в мкг/мл);
п — абсолютно-сухая навеска (в а);
5—объем колориметрируемого рас-
твора (в мл).
Построение калибро-
вочной кривой. В де-
лительные воронки помещают последовательно 1, 2, 4, 6, 8 и 10 мл
стандартного раствора молибдена, содержащего 2 мкг молибдена в
1 мл, разбавляют дистиллированной водой до 18 мл, прибавляют по
5 мл раствора соляной кислоты (1 1), 2 мл раствора хлорного желе-
за (1 мг Fe в 1 мл раствора), по одной капле 1 %-ного раствора суль-
фата меди, 2 мл 20%-ного раствора роданистого калия, 5 мл смеси
изоамилового спирта и четыреххлористого углерода (1 1) и пере-
мешивают. Затем добавляют в каждую делительную воронку по 2 мл
10%-ного раствора SnCl2, взбалтывают до исчезновения красного
окрашивания и появления оранжевого, оставляют на 1—2 мин, снова
взбалтывают 0,5 мин и дают отстояться 2—3 мин. Органическую
фазу сливают в сухие пробирки и, если растворы мутные, добавля-
ют по 10 капель изоамилового спирта, перемешивают и измеряют
оптическую плотность растворов при 440 нм в кювете на 10 мм.
Раствором для сравнения служит органическая фаза контрольного
опыта, где вместо стандартного раствора молибдена используют
18 мл дистиллированной воды. Из полученных данных строят калиб-
ровочную кривую (рис. 9).
Реактивы. Стандартный раствор соли молибдена. Взвешивают 0,1500 г
МоО3, помещают в стакан, растворяют при нагревании в 20 мл 0,1 н. раствора
едкого натра, разбавляют дистиллированной водой приблизительно до 50 мл,
62
добавляют 10 мл 1 н. раствора соляной кислоты, переносят раствор в мерную колбу
на 500 мл, разбавляют дистиллированной водой до метки и тщательно переме-
шивают. 1 мл раствора содержит 0,2 мг Мо. Из этого раствора готовят рабочий
раствор разбавлением в сто раз. Для этого набирают пипеткой 2 мл раствора, по-
мещают в мерную колбу на 200 мл, доводят дистиллированной водой до метки
и тщательно перемешивают.
МоО3 получают из молибденовокислого аммония ч. д. а. прокаливанием в му-
фельной печи при 450—500° С в течение 45—60 мин. Препарат проверяют на пол-
ноту удаления аммиака с реактивом Несслера. 0,1 г МоО3 растворяют в 10 мл
0,2 н. раствора едкого натра и прибавляют одну каплю реактива Несслера.
При этом не должно быть желтого окрашивания, иначе прокаливание повто-
ряют.
Раствор роданистого калия (20%-ный). Взвешивают 20 г соли KCNS ч. д. а.,
растворяют в дистиллированной воде, доводят объем до 100 мл и, если нужно,
фильтруют. Раствор сохраняют в темной склянке.
Раствор хлорного железа. Взвешивают 2,5 г FeCl3 6Н2О, растворяют в ди-
стиллированной воде в мерной колбе на 500 мл, добавляют 10 мл раствора соля-
ной кислоты (1 1), разбавляют дистиллированной водой до метки и перемеши-
вают. 1 мл этого раствора содержит ~ 1 мг Fe.
Раствор хлористого олова (10%-ный). Отвешивают 5,3 г мелко измельченного
металлического олова, переносят в плоскодонную колбу на 200—250 мл, добав-
ляют 50 мл раствора соляной кислоты (1 : 1), две капли 4%-ного раствора серно-
кислой меди (для ускорения растворения) и нагревают до полного растворения
олова. Нагревание регулируют таким образом, чтобы кипение было слабым.
После растворения охлаждают, доводят водой объем до 100 мл и сохраняют в
склянке с притертой пробкой, в которую добавляют кусочек металлического
олова.
Если имеется чистый препарат хлористого олова, то можно приготовить рас-
твор следующим образом. 10 г соли SnCl2 2Н2О растворяют при нагревании
в 10 мл концентрированной соляной кислоты и разбавляют дистиллированной
водой до 100 мл. На дно склянки с раствором помещают кусочек металлического
олова.
Изоамиловый спирт и четыреххлористый углерод (1 1). К 100 мл изоами-
лового спирта ч. д. а. прибавляют 100 мл четыреххлористого углерода ч. д. а.
Смесь переносят в темную склянку с притертой пробкой и сохраняют в прохлад-
ном месте.
Кинетический метод. Скорость химической реакции при опреде-
ленной температуре зависит от концентрации реагирующих веществ.
Измеряя скорость реакции по изменению концентрации одного из
реагентов в единицу времени, можно определить концентрацию не-
известного реагента, если известна зависимость скорости реакции от
концентрации определяемого вещества. Особенно выгодно применять
этот метод для определения веществ, ускоряющих реакцию (ката-
лизаторов). В этом случае можно обнаружить следы каталитически
активных веществ.
Сущность метода. Изложенный выше роданидный метод
определения молибдена хотя и является одним из лучших методов,
однако недостаточно чувствителен при работе с материалом расти-
тельного происхождения, содержащим следы молибдена. Его можно
применять только при содержании молибдена от 2 до 20 мкг в на-
веске. При меньшем количестве молибдена в растениях нужно брать
большие навески (более 10 г), чтобы с уверенностью определить
концентрацию молибдена этим методом.
Более чувствительным методом является кинетический метод,
63
•основанный на каталитическом действии молибдена на реакцию
восстановления метиленовой сини гидразином:
N2H4 + 2
сг
метиленовая синь
ZNH\
= N2 + 2 (СН3)2 N • CeH3 С6Н3 N (СН3)2 + 2НС1.
\ s z
лейкосоединение
Каталитическое действие молибдена заключается в том, что в
кислой среде и при нагревании шестивалентный молибден быстро
восстанавливается гидразином в пятивалентный молибден:
N2H4 + 4Н2МоО4 = 4НМоО3 + N2 + 4Н2О.
Восстановленный молибден в свою очередь восстанавливает ме-
тиленовую синь в метиленовую белую (лейкоформу). Сам же пятива-
лентный молибден окисляется в шестивалентный, который снова
восстанавливается гидразином и этим ускоряет реакцию восстанов-
ления метиленовой сини. При нагревании метиленовая синь восста-
навливается гидразином и без молибдена, но значительно медленнее.
Поэтому нужно ставить контрольный опыт без молибдена, с ко-
торым и сравнивать исследуемый раствор. Необходимо строго соблю-
дать одни и те же условия при проведении реакции — температуру,
кислотность, концентрацию реагирующих веществ. В растворе долж-
ны отсутствовать каталитически активные примеси. Время в процес-
се реакции контролируют секундомером.
Для концентрирования молибдена его извлекают из исследуемого
раствора хлороформом в виде купроната или изоамиловым спиртом
в виде купфероната.
Купрон илиа-бензоиноксим — С6Н5- СНОН С (: NOH) С6Н5
образует с шестивалентным молибденом соединение белого цвета,
не растворимое в воде, но растворимое в хлороформе. Купронат мо-
либдена экстрагируется хлороформом из 0,3—1,5 н. растворов соля-
ной кислоты. В этих условиях медь и железо не экстрагируются.
Купферон —аммонийная соль нитрозофенилгидроксиламина
C6H5N (NO) ONH4 образует с шестивалентным молибденом в кис-
лой среде два соединения: СрМоО3 и Ср2МоО3 (Ср — молекула куп-
ферона), которые экстрагируются изоамиловым спиртом. Вместе с
молибденом экстрагируется и железо, поэтому их нужно разделять.
Молибден отделяется от железа водным раствором аммиака.
Ход анализа. Взвешивают 2—3 г (бобовых культур 1 г)
сухого исследуемого вещества в фарфоровой чашке диаметром 6 см
и сжигают сначала на открытой плитке, затем под асбестовым колпа-
•64
ком. Охлаждают, прибавляют 2 мл дистиллированной воды, 2 мл азот-
ной кислоты (1 1), выпаривают на водяной бане досуха и прокали-
вают на плитке до удаления бурых паров NO2. К остатку в чашке
после охлаждения прибавляют 1 мл раствора соляной кислоты
(1 1) и выпаривают на водяной бане досуха. Затем прибавляют 2 мл
дистиллированной воды, 3 мл раствора соляной кислоты (1 1), на-
гревают, перемешивают и горячим фильтруют через фильтр с белой
лентой диаметром 7 см, промывая чашку и фильтр 15 мл дистил-
лированной воды (порциями по 5 мл).
Фильтрат переносят в делительную во-
ронку, смывая остаток 15 мл воды, при-
бавляют 1 мл 2%-ного спиртового рас-
твора купрона, перемешивают, добавля-
ют 5 мл хлороформа и взбалтывают
1 мин. Дают отстояться и сливают ниж-
ний хлороформенный слой в фарфоровую
чашку диаметром 6 см, которую затем
ставят на водяную баню и выпаривают
досуха. К остатку приливают 1 мл азот-
ной кислоты (1 1), добавляют две капли
5 н. раствора азотнокислого цинка, вы-
паривают на воздушной бане досуха и
Рис. 10. Шариковые пробки:
а — шариковая пробка для про-
бирок; б — шариковая пробка
для колб Кьельдаля.
прокаливают на плитке. После охлажде-
ния к остатку прибавляют 0,5 мл азотной
кислоты (1 1), выпаривают досуха и про-
каливают на плитке под асбестовым
колпаком. Прокаливание продолжают до исчезновения темной
окраски и появления желтой, обусловленной окисью цинка. Чашку
снимают, охлаждают, добавляют 5 капель соляной кислоты (1 1)
и выпаривают досуха на водяной или воздушной бане. К остатку в
чашке после охлаждения прибавляют 1 мл раствора соляной кис-
лоты (1 1), растворяют осадок и раствор выливают в пробирку,
промывая чашку 2 мл дистиллированной воды.
После того как молибден извлечен из всех исследуемых проб
(при серийных определениях) и экстракты помещены в пробирки, к
ним прибавляют по 1 мл 0,016%-ного раствора метиленовой сини и
по 1 мл 2%-ного раствора сернокислого гидразина. Пробирки накры-
вают воронками или шариковыми стеклянными пробками (рис. 10),
ставят в алюминиевый штатив (рис. 11), а штатив погружают в кипя-
щую воду так, чтобы уровень раствора и пробирке был немного ни-
же уровня кипящей воды и выдерживают 10 мин. Затем штатив с про-
бирками вынимают и опускают в холодную воду. После охлаждения
фотометрируют при 610—660 нм в кювете на 5 мм, сравнивая с
раствором, состоящим из 1 мл 0,016%-ного раствора метиленовой
сини, 1 мл раствора соляной кислоты (1 1), 2 мл дистиллированной
воды и 1 мл 2%-ного раствора сернокислого гидразина, который
одновременно с исследуемыми растворами нагревается 10 мин в
кипящей воде. Измеряют разность оптических плотностей,
5 6-2
65
показывающую количество обесцвеченной метиленовой сини, что
пропорционально концентрации молибдена.
Одновременно с исследуемыми растворами в кипящую воду поме-
щают пробирку с контрольным раствором молибдена, который гото-
вят следующим образом. К 2 мл стандартного раствора соли молиб-
дена, содержащего 0,5 мкг/мл Мо, прибавляют 1 мл соляной кисло-
ты (1 1), 1 мл 0,016%-ного раствора метиленовой сини и 1 мл
Рис. 11. Штатив для пробирок с исследуемым
раствором, погружаемых в кипящую воду.
2%-ного раствора серно-
кислого гидразина. Наряду
с исследуемыми растворами
измеряют разность оптиче-
ской плотности и контроль-
ного раствора. Из получен-
ных данных рассчитывают
содержание молибдена по
следующей формуле:
х= Д + 0,1 (Д-0,5Дк)
" Дк
_ 1,1Д-0,05Дк
п Дк
где X — содержание молиб-
дена (в мг на 1 кг исследуе-
мого вещества); п — абсо-
лютно-сухая навеска (в г);
Д — разность оптической
плотности исследуемого
раствора и раствора метиле-
новой сини; Дк — разность оптической плотности контрольного
раствора и раствора метиленовой сини. Определяемые количества
молибдена от 0,2 до 2 мкг в навеске.
Примечание. При отсутствии купрона можно извлекать
молибден изоамиловым спиртом в виде соединения его с купферо-
ном. Для этого к раствору исследуемого вещества в делительной
воронке вместо раствора купрона прибавляют 2 мл 2%-ного водного
раствора купферона, перемешивают, добавляют 5 мл изоамилово-
го спирта и взбалтывают 1 мин. Фазам дают разделиться, водную
фазу отбрасывают, а к органической фазе, содержащей молибден
и железо, приливают 5 мл дистиллированной воды, 1 мл 2 н. раство-
ра аммиака и взбалтывают 1 мин. При этом молибден переходит в
водную фазу, которую после отстаивания сливают в фарфоровую
чашку и выпаривают на воздушной бане досуха. К остатку прили-
вают 1 мл азотной кислоты (1 1), две капли 5н. раствора азотно-
кислого цинка, выпаривают досуха и прокаливают на плитке до
посветления остатка (в горячем виде окраска бледно-желтая). После
охлаждения к остатку в чашке прибавляют 0,5 мл азотной кислоты
(1 1), выпаривают досуха и прокаливают на плитке. Затем до-
бавляют 5 капель соляной кислоты (1 : 1) и снова выпаривают досу-
66
ха на водяной или воздушной бане. В дальнейшем ход работы такой
же, как описано выше.
Реактивы. Раствор метиленовой сини. Взвешивают 0,16 г метиленовой сини
C16H18N3SC1 ЗН2О растворяют в дистиллированной воде и доводят объем до
100 мл. Полученный 0,16%-ный раствор используют для приготовления 0,016%-
ного раствора разбавлением в 10 раз. Для этого набирают пипеткой 10 мл 0,16%-
ного раствора, выливают в мерную колбу на 100 мл, разбавляют дистиллирован-
ной водой до метки и перемешивают.
Раствор сернокислого гидразина (2%-ный). Взвешивают 2 г N2H4 H2SO4,
растворяют в воде, доводят водой до 100 мл и перемешивают. Раствор сохраняют
в темном и прохладном месте.
Раствор купрона (2%-ный). Отвешивают 1 г купрона (а-бензоиноксима), пе-
реносят в мерную колбу на 50 мл, растворяют в этиловом спирте, доводят спиртом
до метки и перемешивают. Сохраняют в темном месте.
Раствор купферона (2%-ный). Отвешивают 2 г купферона СбН5 N(NO)
(ONH4), растворяют в дистиллированной воде, разбавляют до 100 мл и фильт-
руют. Для устойчивости в раствор купферона прибавляют 0,05 г фенацетина.
Раствор аммиака (~2 н.). 150 мл раствора аммиака пл. 0,90 разбавляют
дистиллированной водой до 1 л, перемешивают и сохраняют в склянке с притертой
пробкой.
ОПРЕДЕЛЕНИЕ ЙОДА
Йод, находящийся в растении в виде йодид-ионов, можно опреде-
лить после извлечения его из исследуемого материала дистилли-
рованной водой или растворами солей, что упрощает метод. При
определении общего содержания йода, включая и органические
соединения его, необходимо предварительно провести сжигание ис-
следуемого материала.
Сущность метода. Йод, находящийся в виде йодид-
ионов, извлекается из растительного материала 1%-ным раствором
алюминиевых квасцов в 1%-ной уксусной кислоте для более полно-
го отделения от белков и других коллоидов и осаждается из раство-
ра азотнокислым серебром вместе с ионами хлора, которые всегда
имеются в достаточном количестве. Растворимость хлорида серебра
значительно выше, чем йодида, поэтому весь йод осаждается вместе
с хлоридом серебра. После отделения и промывания осадка се-
ребро восстанавливается двухвалентным марганцем в щелочной'среде
до металлического, которое выпадает в осадок, а йод в виде йодид-
ионов переходит в раствор. Реакция идет по следующему уравне-
нию:
2AgJ + MnSO4 4NaOH = 2Ag + MnO2 + 2NaJ + Na2SO4 + 2H2O.
После удаления осадка центрифугированием, находящиеся в рас-
творе йодид-ионы окисляются перманганатом в нейтральной или
щелочной среде до йодата:
NaJ + 2КМпО4 + Н2О = NaJO3 + 2МпО2 4 2КОН.
Избыток перманганата при помощи двухвалентного марганца
осаждается в виде двуокиси марганца и удаляется центрифугирова-
5=
67
нием. Реакция идет по следующему уравнению:
2КМпО4 + 3MnSO4 + 4NaOH - 5MnO2 + 2Na2SO4 4-
+ K2SO4 + 2H2O.
Раствор, содержащий йодат, выпаривается на водяной бане до-
суха, остаток растворяется в дистиллированной воде и фильтруется
для удаления небольшого количества двуокиси маргайца, образо-
вавшегося во время выпаривания. В фильтрате определяется йод
фотометрическим йодокрахмальным методом. Определяемые коли-
чества — от 0,5 до 5 мкг йода в навеске.
Ход анализа. 5 г сухого хорошо растертого исследуемо-
го материала помещают в колбу на 250—300 мл, добавляют 100 мл
1 %-ного раствора уксусной кислоты, содержащей 1% алюминиевых
квасцов. Колбу погружают в кипящую воду и нагревают в течение
30 мин при периодическом взбалтывании. Затем фильтруют через
бумажный фильтр (красная лента) и осадок промывают два раза
по 25 мл 1%-ным раствором алюминиевых квасцов.
К фильтрату прибавляют 3 мл 0,1 н. раствора AgNO3, одну каплю
соляной кислоты (1 1), ставят колбу на плитку, нагревают раствор
до кипения и кипятят 1 мин. Раствору дают охладиться, переносят
суспензию в две центрифужные пробирки и центрифугируют при
3000 об/мин в течение 3—5 мин. После центрифугирования раствор
сливают, а в пробирки наливают новые порции суспензии и снова
центрифугируют. Под конец осадок переносят в одну пробирку и про-
мывают его 2 раза 0,5%-ным раствором алюминиевых квасцов и один
раз водой порциями по 5 мл.
К промытому осадку приливают 0,5 мл 10%-ного раствора сер-
нокислого марганца, перемешивают стеклянной палочкой, добав-
ляют 4 мл 0,2 н. раствора едкого натра, снова перемешивают, поме-
щают пробирку в горячую воду и нагревают 2 мин. Осадок хорошо
оседает, если имеется хотя бы небольшой избыток ионов Мп++
После охлаждения осадок отделяют центрифугированием. Раствор,
содержащий йодид, сливают в чистую центрифужную пробирку, а
осадок промывают один раз небольшим количеством горячей воды
(3 мл), центрифугируют и раствор присоединяют к первому центри-
фугату. Осадок, содержащий серебро, собирают, очищают и исполь-
зуют в дальнейшей работе по прописи, приведенной ниже (см. ре-
генерация серебра).
Для окисления йодид-ионов в йодат-ионы к раствору в пробирке
прибавляют 1 мл 0,1 н. раствора КМпО4 и 1 мл 0,2 н. раствора
NaOH, перемешивают и погружают пробирку в кипящую воду на
5 мин. Раствор должен быть окрашен в розово-фиолетовый цвет,
свидетельствующий об избытке перманганата, в противном случае
прибавляют еще несколько капель раствора перманганата. После
этого к еще горячему раствору прибавляют 1—2 капли 10%-ного
раствора сернокислого марганца, перемешивают, пока осядет вся
двуокись марганца и раствор сделается прозрачным. Затем центри-
68
Рис. 12. Калибровочная
кривая для йода (свето-
фильтр с максимумом про-
пускания 582 нм, кювета
с
фугируют и раствор сливают в фарфоровую чашку. Осадок промы-
вают 3 мл дистиллированной воды, центрифугируют и раствор сли-
вают в ту же чашку, которую потом помещают на водяную баню и
выпаривают раствор досуха. К сухому остатку приливают 1 мл ди-
стиллированной воды, перемешивают и фильтруют через маленький
бумажный фильтр в мерный цилиндр на 10 мл. Чашку и фильтр
промывают три раза водой порциями по 1 мл. Объем фильтрата в
мерном цилиндре доводят до 4 мл. К полученному раствору прибав-
ляют 0,5 мл 4 М раствора ортофосфорной кислоты, 0,2 мл 20%-ного
раствора KJ и 0,3 мл 0,5%-ного раствора
крахмала и перемешивают. Полученный
окрашенный в синий цвет раствор фотоме-
трируют при 580 нм в кювете на 10 мм.
сравнивая с контролем, в котором иссле-
дуемый раствор заменен водой. По резуль-
татам измерений оптической плотности
исследуемых растворов находят концентра-
цию йода, сравнивая с калибровочной кри-
вой, и вычисляют содержание йода в иссле-
дуемом материале по следующей формуле:
v 100 С 5 500 С
Л. — =-------,
п п *
где X — количество йода (в мкг в 100 г су-
хого вещества); С — концентрация йода в
фотометр ир уемом растворе (в мкг/мл)\
п — абсолютно-сухая навеска (в г); 5 —
объем фоториметрируемого раствора (в мл).
Построение калибровоч-
ной кривой. В пробирки поме-
щают последовательно 0,2, 0,4, 0,6, 0,8, 1,2, 1,6 и 2 мл
толщиной измеряемого
слоя 10 мм).
стандартного раствора KJO3, содержащего 2,5 мкг йода в 1 мл
раствора и добавляют дистиллированной воды до 4 мл. Затем при-
бавляют по 0,5 мл 4 М раствора Н3РО4, 0,2 мл 20%-ного раствора
KJ и по 0,3 мл 0,5%-ного раствора крахмала, перемешивают и из-
меряют оптическую плотность растворов при 580 нм в кювете на
10 мм. Раствором для сравнения служит контрольный раствор,
для проведения которого вместо раствора KJO3 берут 4 мл дистил-
лированной воды и все остальные реактивы. На основании получен-
ных данных строят калибровочную кривую (рис. 12).
Для определения общего содержания йода исследуемое вещество
необходимо сначала озолить и после растворения золы определить
йод по описанному выше методу. В этом случае навеску высушен-
ного и растертого исследуемого материала смешивают с карбонатами
калия и кальция в соотношении 1:1 1, помещают в тигель, сма-
чивают водой и высушивают в сушильном шкафу. Затем помещают
в муфельную печь и озоляют при 500° С. Полученную золу и карбо-
наты растворяют в разбавленной азотной кислоте (1 5), прибав-
ляя ее по 20 мл на каждый грамм навески. Раствор переносят в
69
колбу, смывая дистиллированной водой, и осаждают ионы йода азот-
нокислым серебром, поступая далее так, как описано выше.
Регенерация серебра. Осадок металлического сереб-
ра и перекиси марганца собирают в колбу. При достаточном коли-
честве осадка заливают смесью 0,4 М растворов серной и щавелевой
кислот (1 1). Содержимое колбы подогревают до 50—60° С. После
растворения перекиси марганца фильтруют и промывают осадок се-
ребра дистиллированной водой до удаления серной кислоты. Из
металлического серебра получают азотнокислое серебро следующим
образом. К осадку серебра прибавляют возможно малое количество
азотной кислоты (1 : 1) и полученный раствор выпаривают на во-
дяной бане досуха. Полученную соль сохраняют в темной склянке
и используют для приготовления 0,1 н. раствора AgNO3, применяе-
мого в данном методе.
Реактивы. Стандартный раствор йодата калия. Отвешивают 0,2108 г KJO3,
растворяют в дистиллированной воде в мерной колбе на 500 мл, разбавляют до
метки и перемешивают. 1 мл полученного раствора разбавляют дистиллирован-
ной водой до 100 мл, перемешивают и используют в работе. 1 мл полученного рас-
твора содержит 2,5 мкг йода в виде йодата (4,216 мкг KJOy).
Раствор алюмо-калиевых квасцов и уксусной кислоты (1%-ные). Отвешивают
10 г KAI (SO4)2 12Н3О, помещают в колбу па 1 л, растворяют в дистиллирован-
ной воде, приливают 10 мл уксусной кислоты х. ч., разбавляют дистиллирован-
ной водой до 1 л и перемешивают.
Раствор сернокислого марганца (10%-ный). 10 г сернокислого марганца
MnSO4 7Н2О растворяют в дистиллированной воде и разбавляют водой до
100 мл. _
Раствор едкого натра (~0,2 н.). Отвешивают 8 г NaOH х. ч., растворяют
в безуглекислой воде, разбавляя дистиллированной водой до 1 л. Раствор сохра-
няют в хорошо закрытой склянке.
Раствор перманганата калия (~0,1 н.). Отвешивают 3,2 г КМпО4, раство-
ряют в горячей воде и после охлаждения разбавляют дистиллированной водой
до 1 л.
Щавелево-сернокислый раствор (~0 2,М). Отвешивают 25,2 г Н2С2О4 2Н2О,
растворяют в 500 мл дистиллированной воды, приливают 20 мл концентрирован-
ной серной кислоты, разбавляют дистиллированной водой до 1 л и перемешивают.
Фосфорная кислста (4 М). Берут 136 мл фосфорной кислоты пл. 1, 7, разбав-
ляют дистиллированной водой до 500 мл и перемешивают.
Приготовление других реактивов, применяемых в этом методе, изложено
выше (см. стр. 9, 17, 26 ).
ЛИТЕРАТУРА
Бабко А. К., П'илипенко А. Т. Фотометрический анализ (Методы
определения неметаллов). М., «Химия», 1974.
Б у с е в А. И. Аналитическая химия молибдена. М., Изд-во АН СССР, 1962.
Веригина К. В., Добрицкая В. И. Методы определения микроэлемен-
тов в почвах и растениях. М., Изд-во АН СССР, 1958.
Коренман И. М. Аналитическая химия калия. М., «Наука», 1964.
Методы определения микроэлементов. М.—Л., Изд-во АН СССР, 1950.
Немодрук А. А., Каралова 3. К- Аналитическая химия бора. М.,
«Наука», 1964.
Пршибил Р. Комплексоны в химическом анализе. М., ИИЛ, 1955, 1960.
Починок X. Н. Определение зольных элементов в растениях. — В кн.:
Тр. н.-и. ин-та физиологии растений АН УССР, 13—14. Киев, Сельхозгиз,
1958, 159.
70
Ночи нок.Х. Н. Определение общей серы в сахарной свекле йодометрическим
методом.— Укр. хим. журн., 1951, 17, 3, 408.
Починок X. Н. Колориметричне визначення бору в рослинах та грунт!.—
В кн.: Праш Н.-д. in-ту ф1зюлогп рослин, вип. 21. Ки!в, Вид-во УАСГН,
1960, 85.
С е н д е л Е. Колориметрические методы определения следов металлов. М.,
«Мир», 1964.
Hertha Н. Taussky, and EphraimSchorr. Microcolorimetric me-
thod for the determination of inorganic phosphorus.— J. Biol. Chem., 1953,
202, 2, 675.
Глава II
АЗОТ И ЕГО СОЕДИНЕНИЯ
ОПРЕДЕЛЕНИЕ ОБЩЕГО И БЕЛКОВОГО АЗОТА
ХЛОРАМИННЫМ МЕТОДОМ
Сущность метода. Органические вещества сжигают
серной кислотой в присутствии катализатора сульфата меди или мо-
либдата натрия до углекислоты и воды. Азот при этом превращается
в аммиак и связывается серной кислотой:
2СН2 (NH2) СООН + 7H2SO4 = 4СО2 + 6SO2 + (NH4)2SO4 + 8Н2О.
гликоколь
Избыток серной кислоты нейтрализуется едким натром и азот ионов
аммония окисляется хлорамином. Реакция идет по следующему
уравнению:
,Na
(NH4)2 SO4 + ЗС6Н4 (СН3) SO2N< + 2NaOH =
ХС1
= ЗС6Н4 (СН3) SO2NH2 + N2 + Na2SO4 + NaCl + H2O.
rz 2N
Как видно из уравнения, грамм-эквивалент азота -g— равен
л сел 'т 3C7H7SO2NClNa Н2О о
4,669, а грамм-эквивалент хлорамина Т—-----------------122,8
с 3C6H5SO2NClNa ЗН2О 19О о
и хлорамина Б ——?------------------133,8.
В кислой среде хлорамин разлагается с выделением хлора:
С6Н4 (СН3) SO2NClNa + 2НС1 - С6Н4 (СН3) SO2NH2 + NaCl + Cl2.
Поэтому, во избежание потерь хлора реакцию следует проводить в
более щелочной области, но по мере подщелачивания раствора окис-
ление азота хлорамином замедляется. Эта реакция в щелочной об-
ласти ускоряется ионами брома, которые вступают в реакцию с
хлорамином, образуя свободный бром:
С6Н4 (СН3) SO2NClNa + 2КВг + H2SO4 =
= Вг2 + С6Н4 (СН3) SO2NH2 + K2SO4 + NaCl.
Бром в момент выделения более энергично вступает в реакцию,
окисляя азот:
(NH4)2 SO4 + ЗВг2 + 8NaOH = N2 + Na2SO4 + 6NaBr + 4H2O
72
Как видно из этих уравнений, первая реакция требует кислой
среды, а вторая — щелочной, следовательно, на скорость реакции
оказывает влияние концентрация водородных ионов; оптимальная
pH от 6,3 до 6,8. Реакция идет довольно медленно и для полного ее
окончания требуется не менее 25 мин.
Избыток хлорамина определяют йодометрическим методом. Реак-
ция идет по следующему уравнению:
,Na
СбН4 (СН3) SO2N< + 2KJ + Н2С2О4 = J2 + С6Н4 (СН3) so2nh2 +
\ci
+ К2С2О4 + NaCl.
Выделившийся йод титруется тиосульфатом натрия. Так как избы-
ток хлорамина во многих случаях приходится определять в присут-
ствии ионов меди, для подкисления применяется щавелевая кисло-
та, которая, связывая ионы меди, препятствует реакции ее с йо-
дистым калием. Это дает возможность определять хлорамин без
отделения ионов меди.
Наиболее длительной операцией при определении азота явля-
ется сжигание органических веществ серной кислотой. Для ускоре-
ния сжигания применяют различные катализаторы: соли меди,
ртути, селен и другие. В данном методе впервые применяется ката-
лизатор молибдат натрия в присутствии сульфата калия. Последний
повышает температуру кипения серной кислоты и этим ускоряет
реакцию. Добавка K2SO4 в количестве 0,2 г на каждый мл взятой
для сжигания серной кислоты уменьшает время сжигания почти на 1 ч
по сравнению с действием одного катализатора. Сжигание также
немного ускоряется при применении перекиси водорода. Очень быст-
ро идет сжигание со смесью серной и хлорной кислот, но послед-
няя, являясь сильным окислителем, вступает также в реакцию с
водородными соединениями азота, окисляя азот до свободного, что
дает существенные ошибки:
ЗНС1О4 + 4 (NH4)2SO4 = ЗНС1 + 4N2 + 4H2SO4 + 12Н2О.
Потерь азота не бывает, если хлорную кислоту берут в коли-
честве меньшем, чем нужно для полного окисления органических
веществ, которые затем сжигаются только с серной кислотой. Если
на 0,1 г сухого растительного материала брать 0,1 г НС1О4 (0,2 мл
40%-ной хлорной кислоты), то сжигается хлорной кислотой боль-
шая часть органических веществ без потерь азота. Остаток должен
быть еще темным или коричневым и в дальнейшем сжигается или
только с одной серной кислотой или с добавлением перекиси водо-
рода. Малейший избыток хлорной кислоты вреден и, если нет уве-
ренности в том, что не будет добавлен избыток хлорной кислоты,
лучше сжигание проводить без нее.
73
Определение общего азота
Ход анализа. Взвешивают сначала на технических, затем
на аналитических весах 0,2 г воздушно-сухого или 1 г сырого ис-
следуемого материала, содержащего от 1 до 9 мг азота, всыпают в
колбу Кьельдаля емкостью 30—50 мл так, чтобы вещество попало
на дно колбы. Для этого взвешивание производится на полоске
пергаментной бумаги, которой придается форма длинной лодочки.
В колбу приливают 0,3 мл 10%-ного раствора молибдата натрия,
4 капли 30%-ной перекиси водорода, прибавляют 0,4 г сульфата
калия, 2,5 мл концентрированной серной кислоты, перемешивают
и оставляют на 2—3 ч (или на ночь), затем ставят на электрическую
плитку и нагревают, регулируя нагрев так, чтобы пена не поднима-
лась слишком высоко. Когда образование пены ослабеет и в колбе
появятся белые пары серной кислоты, ее накрывают стеклянной
пробкой или воронкой и продолжают нагревание до просветления
раствора.
Сжигание заканчивается быстрее, если основную массу органи-
ческих веществ сжечь с помощью хлорной кислоты, а остаток с по-
мощью перекиси водорода. В этом случае поступают следующим
образом: к 0,2 г сухого исследуемого материала, находящегося в
колбе Кьельдаля, прибавляют 0,4 г сульфата калия, 0,3 мл 10%-ного
раствора молибдата натрия, 0,4 мл 40%-ного раствора хлорной
кислоты (или эквивалентное количество другой имеющейся кон-
центрации), 2,5 мл концентрированной серной кислоты, соблю-
дая эту последовательность в прибавлении реактивов. Смесь пе-
ремешивают и ставят на холодную электрическую плитку в наклон-
ном положении. Включают плитку и сжигают в течение 40—50 мин.
Затем колбу снимают с плитки, охлаждают 5 мин, прибавляют
3 капли 30%-ной перекиси водорода, ставят на плитку и сжигают
10—15 мин. Обработку перекисью водорода повторяют до полного
просветления. После последней обработки и просветления раствора
необходимо прокипятить еще 10 мин для полного разложения пере-
киси. После сжигания охлаждают и к раствору, содержащему кон-
центрированную серную кислоту, прибавляют осторожно по стенке
колбы 10 мл дистиллированной воды, раствор переносят в мерную
колбу на 50 мл, доводят водой до метки и тщательно перемешивают.
При ограниченном количестве исследуемого материала берут для
анализа 0,1 г сухого вещества и сжигают с 1,5 мл серной кислоты в
присутствии 0,2 мл 10%-ного раствора молибдата натрия и 0,3 г
сульфата калия. В этом случае остаток после сжигания разбавля-
ют до 25 мл.
Определение белкового азота
Ход анализа. Взвешивают на аналитических весах около
1 г сырого или 0,25 г сухого исследуемого растительного материала,
помещают в фарфоровую ступку, растирают с небольшим количест-
вом дистиллированной воды и переносят в стакан, промывая ступку
25 мл воды. Стакан ставят на плитку и нагревают почти до кипения.
Если исследуемый материал содержит большое количество крахмала,
нагревают 10 мин на водяной бане при температуре 40—50° С. Затем
к горячему раствору прибавляют 5 мл 3%-ного раствора сернокис-
лой меди, 5 мл 0,2 н. раствора едкого натра при перемешивании
и оставляют на 3 ч для отстаивания. После этого фильтруют через
беззольный фильтр диаметром 8—9 см (белая лента). Осадок де-
кантируют 2—3 раза горячей водой, затем переносят на фильтр и
промывают горячей водой до прекращения реакции на сульфат-ион
с ВаС12.
Промытый осадок с фильтром высушивают, помещают в колбу
Кьельдаля на 50 или 100 мл, прибавляют 0,4 г сульфата калия,
0,3 мл 10%-ного раствора молибдата натрия, 1 мл 40%-ного рас-
твора хлорной кислоты, 3 мл концентрированной серной кислоты,
ставят на плитку в наклонном положении и осторожно нагревают,
не допуская поднятия пены в горлышко колбы. Как только исчез-
нет пена и раствор закипит, накрывают колбу воронкой или шари-
ковой стеклянной пробкой и сжигают в течение 1 ч. Затем раствор
охлаждают на воздухе 5 мин, воронку снимают, прибавляют 3 кап-
ли 30 %-ной перекиси водорода, снова ставят колбу на плитку и на-
гревают 10—15 мин. Обработку перекисью водорода повторяют до
полного просветления раствора. После последней обработки пере-
кисью водорода кипятят не менее 10 мин. По окончании сжигания
колбу охлаждают на воздухе и прибавляют осторожно по стенке
10 мл дистиллированной воды. Раствор переносят в мерную колбу
на 50 мл, доводят объем водой до 50 мл и тщательно перемешивают.
В растворе, полученном после сжигания органических веществ,
определяют азот хлораминным методом. Для этого из исследуемо-
го раствора набирают пипеткой 20 мл при анализе корней, стеблей и
других объектов, содержащих менее 2% азота (в расчете на сухое
вещество) и 10 мл при анализе листьев, зерна и других объектов,
содержащих более 2% азота, и переносят в колбу для титрования.
Прибавляют одну каплю 0,1 %-ного раствора метилоранжа и ней-
трализуют 2 н. раствором NaOH до перехода окраски инди-
катора. Затем прибавляют 10 мл фосфатного буферного раствора
с pH 6,7, содержащего бромистый калий, перемешивают и по стен-
ке колбы приливают пипеткой 5 мл 0,12 н. раствора хлорамина.
Колбу накрывают часовым стеклом, раствор перемешивают круго-
вым движением и оставляют на 25 мин. После этого добавляют 2 мл
20%-ного раствора йодистого калия, 10 мл 8%-ного раствора щаве-
левой кислоты и выделившийся йод титруют 0,02 н. раствором тио-
сульфата натрия в присутствии индикатора крахмала, прибавляемо-
го в конце титрования. 1 мл 0,02 н. раствора тиосульфата натрия
соответствует 0,09338 мг азота.
Все реактивы должны быть проверены на содержание азота.
Для этого ставят контрольный опыт со всеми реактивами, применяе-
мыми в данном методе, проводя те же операции, что и при определе-
нии азота, только без исследуемого вещества. Контрольных опытов
75
ставят по два на каждую серию определений. Содержание азота
вычисляют по следующей формуле:
v 0,4669 К А(а — Ь)
А —---------------------,
v п
где X — содержание азота в расчете на абсолютно-сухое вещест-
во (в %); А — объем раствора, в котором растворена навеска после
сжигания (в мл); К — нормальность раствора тиосульфата натрия;
v — объем исследуемого раствора, взятый для окисления азота хлор-
амином (в мл); а — объем 0,02 н. раствора тиосульфата натрия,
затраченный на титрование контрольного раствора (в мл); b— объем
0,02 н. раствора тиосульфата натрия, затраченный на титрование
исследуемого раствора (в мл); п — абсолютно-сухая навеска (для
пересчета на абсолютно-сухое вещество необходимо определить влаж-
ность исследуемого материала высушиванием при 105° С до постоян-
ного веса), г; 0,4669 — нормальный титр азота, умноженный на
100 для пересчета в проценты.
Примечание. Разность (а — Ь) не должна превышать 22 мл.
Определяемые количества от 0,3 до 2 мг азота в титруемом растворе.
Если в навеске находится не более 2 мг азота, после сжигания рас-
твор не переносят в мерную колбу, а смывают в колбу для титрова-
ния, нейтрализуют и определяют азот так, как описано выше.
Реактивы. Раствор хлорамина (~0,12 н.). Отвешивают 15 г хлорамина Т,
или 16 г хлорамина Б, растворяют в дистиллированной воде, разбавляют водой
до 1 л, тщательно перемешивают и фильтруют через вату в темную склянку с при-
тертой стеклянной пробкой.
Дистиллированная вода (безаммиачная). Вода, применяемая в данном методе,
не должна содержать аммиак. Наличие аммиака в дистиллированной воде прове-
ряется реактивом Несслера. К 20 мл дистиллированной воды в пробирке прибав-
ляют одну каплю реактива Несслера, при этом не должно быть заметного желтого
окрашивания. Для удаления аммиака из дистиллированной воды ее кипятят в кол-
бе до тех пор, пока она перестанет давать реакцию на аммиак.
Раствор молибдата натрия (10%-ный). Отвешивают 10 г соли Na2MoO4
2Н2О, растворяют в 100 мл дистиллированной воды, прибавляют 0,5 мл 2 н.
раствора едкого натра, нагревают до кипения и кипятят 15 мин, затем охлаждают,
доводят безаммиачной водой до 100 мл и сохраняют в хорошо закрытой склянке
с притертой пробкой.
Раствор сернокислой меди (3%-ный). Отвешивают 30 г CuSO4 5Н2О, раство-
ряют в безаммиачной воде, разбавляют до 1 л, перемешивают и сохраняют в хо-
рошо закрытой склянке.
Раствор тиосульфата натрия (0,02 н.). Этот раствор готовится из титрован-
ного 0,1 н. раствора разбавлением в 5 раз водой. Приготовление 0,1 н. раствора
тиосульфата натрия описано выше (см. стр. 17).
Раствор едкого натра (~2н.). Отвешивают на технических весах 80 г NaOH
ч. д. а., растворяют в 1 л дистиллированной воды, нагревают до кипения и кипя-
тят 30 мин, затем охлаждают, доводят объем безаммиачной водой до 1 л, переме-
шивают и сохраняют в хорошо закрытой склянке. Этот раствор можно также
готовить из 20%-ного безаммиачного раствора едкого натра разбавлением в
2,5 раза безаммиачной водой.
Раствор щавелевой кислоты (8%-ный). Отвешивают 80 г Н2С2О4 2Н2О,
растворяют в горячей дистиллированной воде, разбавляют до 1 л и перемеши-
вают.
Сульфат калия (безаммиачный). К 200 г сульфата калия х. ч. в стакане ем-
костью 2 л прибавляют 1 л дистиллированной воды и 5 мл 20%-ного раствора
едкого натра. Смесь нагревают до кипения и кипятят до тех пор, пока раствор за-
76
мутится от выпадающей соли. Раствор переносят в фарфоровую чашку и выпари-
вают до небольшого объема (около 100 мл), затем охлаждают, прозрачный раствор
сливают, а остаток высушивают на воздушной бане при постоянном помешивании.
Сухую соль ссыпают в банку с притертой пробкой.
Фосфатный буферный раствор с бромистым калием (pH 6,7). Этот раствор
можно готовить по двум прописям: 1) отвешивают 52,8 г высушенной при 100° С
соли КН2РО4 и 100 а КВг, растворяют обе соли вместе в безаммиачной воде,
прибавляют 80 мл 2 н. раствора едкого натра, разбавляют до 1 л безаммиачной
водой, перемешивают и сохраняют в хорошо закрытой склянке; 2) отвешивают
60 г кристаллической, невыветрившейся соли Na2HPO4 12Н2О, высыпают в ста-
кан, прибавляют 30 г КН2РО4 и 100 г КВг. Растворяют все соли в безаммиачной
воде, доводят до 1 л, перемешивают и сохраняют в хорошо закрытой склянке.
Приготовление остальных реактивов описано выше (см. стр. 17, 70).
ОПРЕДЕЛЕНИЕ НЕБЕЛКОВОГО И БЕЛКОВОГО АЗОТА
ИЗ ОДНОЙ НАВЕСКИ
В небелковый азот входит азот растворимых в воде веществ:
аминокислот, амидов, аммиака и азотистых органических оснований.
Сущность метода. После растирания исследуемого мате-
риала с водой и перенесения в центрифужную пробирку белки осаж-
дают гидроокисью меди, отделяют центрифугированием и промыва-
ют дистиллированной водой. Раствор содержащий небелковый азот
помещают в колбу Кьельдаля, выпаривают, сжигают с серной кисло-
той и азот определяют хлораминным методом. Осадок, содержащий
белок, переносится в колбу Кьельдаля, сжигают с серной кислотой
и азот определяют хлораминным методом.
Ход а н а л и з а. Взвешивают сначала на технических,
затем на аналитических весах 0,25—0,5 г воздушно-сухого или 1—
2 г сырого исследуемого материала, содержащего не более 10 мг
суммарного количества азота. Навеску помещают в фарфоровую
ступку и растирают с 2 мл дистиллированной воды до однородной мас-
сы. Добавляют 3 мл 3%-ного раствора сульфата меди, размешивают
и переносят гомогенат в центрифужную пробирку, смывая остаток
8 мл воды. Пробирку помещают в кипящую воду и выдерживают в
течение 3 мин при периодическом перемешивании стеклянной палоч-
кой. Затем пробирку вынимают из кипящей воды, добавляют 3 мл
0,2 н. раствора едкого натра, размешивают, дают постоять 10 мин
и центрифугируют. Раствор сливают в колбу Кьельдаля на 100 мл,
а осадок промывают 2 раза горячей водой по 10 мл, добавляя к
промывной воде по 1 капле 3 %-ного раствора сульфата меди.
Определение небелкового азота
К объединенным экстрактам в колбе Кьельдаля прибавляют 2 мл
концентрированной серной кислоты, 0,3 мл 10%-ного раствора мо-
либдата натрия, 4 капли 30 %-ной перекиси водорода и выпаривают
на плитке до появления пены. Затем уменьшают нагрев так, чтобы
пена не попала в горлышко колбы и продолжают слабое нагревание
до появления белых паров серной кислоты. После этого колбу
закрывают стеклянной шариковой пробкой, усиливают нагрев и сжи-
гают до просветления раствора. К охлажденному остатку в колбе
77
прибавляют 10 мл дистиллированной воды, раствор переносят в
мерную колбу на 50 мл, смывая остаток водой, доводят объем во-
дой до 50 мл и перемешивают. Для определения азота набирают
25 мл раствора, выливают в колбу для титрования, прибавляют одну
каплю 0,1 %-ного раствора метилового оранжевого и охлаждая
нейтрализуют 2 н. раствором едкого натра до перехода окраски
индикатора в оранжево-желтую не допуская перетитровывания.
Затем добавляют 10 мл фосфатного буферного раствора с pH 6,7, со-
держащего бромид калия, 5 мл 0,12 н. раствора хлорамина и далее
поступают так, как описано выше при определении общего и бел-
кового азота. Для учета содержания азота в реактивах ставят конт-
рольный опыт. Из полученных данных вычисляют содержание не-
белкового азота по следующей формуле:
v 0,4669 50 К {а — Ь) _ 0,9338 К (а — Ь)
А 25 п ~ п
где X — содержание небелкового азота (в %); 50 — объем исследуе-
мого раствора (в мл)\ 25 — объем исследуемого раствора, взятый
для определения азота хлораминным методом (в мл)\ а — объем
0,02 н. раствора тиосульфата натрия, затраченный при титровании
контрольного раствора (в мл)\ b — объем 0,02 н. раствора тиосуль-
фата натрия, затраченный при титровании исследуемого раствора
(в мл)\ К — нормальность раствора тиосульфата натрия; п — на-
веска исследуемого вещества (в г).
Определение белкового азота
К осадку в пробирке, содержащему белковый азот, приливают
5 мл дистиллированной воды, размешивают и выливают в колбу
Кьельдаля на 100 мл, смывая остаток небольшими порциями воды.
К содержимому колбы прибавляют 0,3 мл 10%-ного раствора молиб-
дата натрия, 0,4 г сульфата калия, 3 мл концентрированной серной
кислоты, ставят на плитку и выпаривают при небольшом нагреве до
появления пены* Затем охлаждают, добавляют 4 капли 30 %-ной
перекиси водорода, помещают на плитку и осторожно нагревают,
не допуская поднятия пены в горлышко колбы. Как только исчез-
нет пена и раствор закипит, закрывают колбу шариковой стеклянной
пробкой и сжигают до просветления раствора. После сжигания и
охлаждения прибавляют по стенке колбы 10 мл дистиллированной
воды. Раствор переносят в мерную колбу на 50 мл, доводят объем
водой до метки и тщательно перемешивают.
Для определения азота набирают пипеткой 10 мл раствора, вы-
ливают в колбу для титрования, прибавляют 1 каплю 0,1 %-ного
раствора метилоранжа и нейтрализуют 2 н. раствором NaOH до пе-
рехода окраски индикатора в оранжево-желтую, избегая избытка
щелочи. Затем прибавляют 10 мл фосфатного буферного раствора
* Выпаривание лучше проводить в солевой бане — в растворе СаС1.?
(см. стр. 274).
78
с pH 6,7, содержащего КВг, 5 л/л 0,12 н. раствора хлорамина и далее
поступают так, как описано при определении общего и белкового
азота. Одновременно ставят контрольный опыт на содержание N
в реактивах. Содержание белкового азота вычисляют по формуле:
х 0,4669 50 К (а —6) 2,3345 К (а — Ь)
А ~ 10 п ~ п
где X — содержание белкового азота (в %).
Остальные обозначения такие же, как и в методе определения бел-
кового азота, изложенного выше.
ОПРЕДЕЛЕНИЕ ФРАКЦИОННОГО СОСТАВА
БЕЛКОВЫХ ВЕЩЕСТВ В РАСТЕНИЯХ
Все известные белки делятся на две группы: простые белки —
протеины, состоящие только из аминокислот и сложные белки —
протеиды, состоящие из простых белков в соединении с различными
небелковыми группами.
Простые белки по их различному отношению к растворителям —
воде, спирту и разбавленным растворам солей, кислот и щелочей
также делятся на группы.
Альбумины — хорошо растворимы в воде, в растворах
солей, кислот и щелочей. Эти белки составляют основную массу
белков протоплазмы клеток.
Глобулины — плохо растворимы в воде, ио хорошо раство-
ряются в слабых растворах солей, в разведенных растворах кислот
и щелочей. Из глобулинов состоит основная масса белков семян.
Г л и а д и н ы (проламины) — нерастворимы в воде, но
хорошо растворяются в 70—80%-ном этиловом спирте, разбавленных
кислотах и щелочах. Содержат большое количество глутаминовой
кислоты и пролина. Находятся в зернах злаков.
Глютелины — нерастворимы в воде и нейтральных раство-
рах солей, но легко растворяются в разбавленных щелочах и кисло-
тах. Содержатся в злаках.
Определение содержания различных групп белковых веществ
при соблюдении одинаковых условий анализа дает возможность ха-
рактеризовать биохимические процессы, связанные с превращением
белковых веществ в ходе роста и развития растений под влиянием
различных факторов.
Фракционный состав белков определяют в воздушно-сухих семе-
нах и в свежих вегетативных органах растений. При высушивании
растительного материала при плюсовой температуре происходят
нежелательные изменения в соотношении фракций белка, поэтому
берут для анализа или свежий растительный материал, или после
лиофильной сушки, когда вода удаляется в глубоком вакууме из
замороженного состояния.
Сущность метода. Белковые вещества извлекают по-
следовательно дистиллированной водой, затем 10%-ным раствором
79
хлористого натрия, далее 70—80%-ным этиловым спиртом и нако-
нец 0,2%-ным раствором щелочи. Экстракты собирают в отдельные
стаканчики и белки осаждают уксусной кислотой или сульфатом меди
и щелочью. Осадки белковых веществ отфильтровывают и сжигают
серной кислотой. В полученном растворе азот определяют хлорамин-
пым методом. Осаждение уксусной кислотой необходимо в том
случае, когда устанавливается аминокислотный состав исследуемой
фракции белка.
Ход анализа. Для анализа берут следующие количества
исследуемых веществ: 0,5 г воздушно-сухой муки зерна бобовых,
1 г пшеницы и других злаков, по 5 г клубней картофеля, свежих
листьев и стеблей. Взятую навеску тщательно измельчают и извле-
кают сначала водорастворимую фракцию белка, потом солераство-
римую, затем растворимую в спирте и наконец растворимую в ще-
лочи. В остатке содержатся сложные белки.
Водная фракция белков (альбумины)
Навеску помещают в фарфоровую ступку, добавляют немного
дистиллированной воды и растирают до однородной массы. Содержи-
мое ступки переносят в центрифужную пробирку, промывая ступку
водой с таким расчетом, чтобы на промывание расходовалось не
более 15 мл воды. Массу в пробирке перемешивают стеклянной па-
лочкой. Палочку споласкивают несколькими каплями воды, а рас-
твор центрифугируют в течение 3—5 мин при 2500 об/мин и про-
зрачный верхний слой сливают в стакан. К остатку в пробирке
прибавляют 15 мл дистиллированной воды, перемешивают 5 мин
палочкой, центрифугируют и раствор сливают в тот же стакан. Эту
операцию повторяют 3 раза.
Из полученного раствора осаждают чистый белок уксусной кисло-
той. Для этого прибавляют 10 капель ледяной уксусной кислоты
и нагревают на водяной бане 5—10 мин до выделения осадка. После
этого фильтруют через беззольный фильтр диаметром 7 см, соби-
рая весь осадок на фильтр, промывают 4 раза 1 %-ной уксусной
кислотой и фильтр с осадком высушивают в термостате.
Из полученного раствора можно осадить белок по Барнштейну.
Для этого раствор нагревают до 80—90° С, прибавляют 5 мл 3%-
ного раствора сульфата меди и затем при помешивании 5 мл 0,2 н.
раствора едкого натра. Осадку дают отстояться и фильтруют через
беззольный фильтр диаметром 8 см (белая лента). Осадок на фильт-
ре промывают 5 раз горячей водой, высушивают и определяют азот
хлораминным методом.
Солерастворимая фракция белков
(глобулины)
К остатку в центрифужной пробирке добавляют 15 мл 10%-ного
раствора хлористого натрия, перемешивают стеклянной палочкой
в течение 5 мин, после чего палочку споласкивают солевым раство-
80
ром. Содержимое пробирки центрифугируют и сливают раствор в
стакан. Так поступают 4 раза. Для осаждения белка прибавляют
10 капель ледяной уксусной кислоты и нагревают на водяной бане до
выпадения осадка. Осадок полностью переносят на фильтр при
помощи 1 %-ной уксусной кислоты, промывают 2 раза этой же кисло-
той и высушивают. Можно белок осадить также сульфатом меди и
щелочью, а азот определить хлораминным методом.
Растворимая в спирте фракция белков
(проламины)
Растворимые в спирте белки находятся в злаках (кукуруза,
ячмень, пшеница, рожь и др.) и отсутствуют в бобовых культурах,
поэтому при исследовании последних можно спиртовой вытяжки не
делать.
К остатку после извлечения раствором соли приливают 10 мл
70%-ного (по весу) этилового спирта, перемешивают палочкой и
смесь помещают в горячую воду при температуре 80° С на 5 мин,
все время помешивая. После этого пробирку вынимают, продолжая
периодическое перемешивание еще 5 мин, центрифугируют и рас-
твор сливают в стакан. К остатку приливают еще 10 мл 70%-ного
спирта, перемешивают, помещают в горячую воду на 5 мин и т. д.
Всего производят 4 извлечения.
Для осаждения белка спиртовый раствор разбавляют в два раза
водой, прибавляют 10 капель ледяной уксусной кислоты и нагре-
вают до кипения. Затем фильтруют, осадок переносят на фильтр
1%-ным раствором уксусной кислоты и высушивают. Здесь также
белок можно осадить по Барнштейну гидроокисью меди и опреде-
лить азот.
Растворимая в щелочи фракция белков
(глютелины)
К остатку в центрифужной пробирке после извлечения спиртом
прибавляют 15 мл 0,2%-ного раствора едкого натра. Содержимое
перемешивают в течение 5 мин стеклянной палочкой, центрифуги-
руют и сливают раствор в стакан. Так поступают 4 раза, соединяя
центрифугаты.
Для осаждения белка прибавляют 5 мл 10%-ного раствора три-
хлоруксусной кислоты, перемешивают и снова прибавляют по кап-
лям раствор этой же кислоты при энергичном размешивании до
появления осадка. Затем нагревают, фильтруют, осадок переносят
на фильтр с помощью 1%-ной уксусной кислоты и высушивают.
Для осаждения по Барнштейну к щелочному экстракту прибав-
ляют 2 мл 1 н. раствора серной кислоты для нейтрализации избытка
щелочи, перемешивают, прибавляют 5 мл 3%-ного раствора сульфа-
та меди, нагревают до 70—80° С, дают отстояться и фильтруют.
Осадок полностью переносят на фильтр с помощью горячей воды
и высушивают без промывания.
6 6-2
81
Труднорастворимые (сложные белки)
В остатке после извлечения различных фракций белков опреде-
ляют труднорастворимые белки. Для этого остаток из пробирки
переносят с помощью небольшого количества дистиллированной во-
ды в колбу Кьельдаля на 50—100 мл, прибавляют 0,4 г сульфата
калия, 0,3 мл 10%-ного раствора молибдата натрия и 3 мл концент-
рированной серной кислоты. Колбу помещают на плитку в наклон-
ном положении и нагревают до испарения воды и обугливания, не
допуская поднятия пены в горлышко колбы. Как только исчезнет
пена и раствор закипит, накрывают колбу воронкой или шариковой
пробкой и продолжают нагревание до полного просветления раство-
ра. Для ускорения процесса можно под конец сжигания применить
обработку перекисью водорода. Таким же образом сжигают все
фракции белков, поместив их с фильтрами в колбы Кьельдаля и
добавив такое же количество реактивов.
После сжигания содержимое колб Кьельдаля количественно пере-
носят в мерные колбы на 50 мл, доводят объем безаммиачной во-
дой до 50 мл, перемешивают и определяют азот хлораминным мето-
дом, как описано выше. Для определения азота в зависимости от
предполагаемого количества белков в данной фракции берут от 10
до 25 мл раствора. Содержание азота в каждой фракции белков
вычисляют по формуле:
Y _ 0,4669 50 К (а — Ь)
где X — содержание азота в данной фракции белков в расчете
на абсолютно-сухой вес исследуемого вещества (в %); 50 — общий
объем, в котором растворен остаток после сжигания фракции бел-
ка (в мл); v — объем исследуемого раствора, взятый для окисле-
ния азота хлорамином (в мл); п — абсолютно-сухая навеска ис-
следуемого вещества (для вычисления абсолютно-сухой навески
необходимо определить влажность исследуемого вещества высуши-
ванием его при 105° С до постоянного веса) (в г); а — объем 0,02 н.
раствора тиосульфата натрия, затраченный на контрольный опыт
(в мл); b — объем 0,02 н. раствора тиосульфата натрия, затрачен-
ный на исследуемый раствор (в мл); К — нормальность раствора
тиосульфата натрия.
Полученные результаты пересчитывают относительно суммарно-
го содержания азота во всех белковых фракциях, принимая его за
100, и таким образом получают относительное содержание фракций
белков:
v 100 а
где X — относительное содержание данной фракции белков (в %)
к общему содержанию белков; а — содержание азота данной фрак-
ции белков (в %) к исследуемому веществу, вычисленное по пре-
дыдущей формуле; с — сумма всех фракций белков (в %) к иссле-
дуемому веществу.
82
Реактивы. Раствор хлористого натрия (10%-ный). Отвешивают 100 г NaCl
яг. ч., растворяют в безаммиачной воде и разбавляют той же водой до 1 л.
Раствор этилового спирта (70%-ный). К 100 мл 95%-ного спирта прибавляют
27 мл безаммиачной воды, перемешивают и сохраняют в хорошо закрытой склянке.
Раствор едкого натра (0,2%-ный). Отвешивают 2 г NaOH х. ч., растворяют
в безаммиачной воде, разбавляют этой же водой до 1 л, перемешивают и сохраняют
в склянке без доступа воздуха.
Раствор трихлоруксусной кислоты (10%-ный). 100 г СС13СООН растворяют
в безаммиачной воде, разбавляют этой водой до 1 л и хорошо перемешивают.
Приготовление остальных реактивов описано выше (см. стр. 70, 76, 77).
ОПРЕДЕЛЕНИЕ АМИННОГО АЗОТА
НИНГИДРИНОВЫМ МЕТОДОМ
Сущность метода. Нингидрин — трикетогидринден-
гидрат с аминокислотами образует окрашенный в синий цвет дике-
тогидр инди л иденди кетогид р индамин :
/со\ /С0\
RCH (NH2) СООН + 2С6Н4< >С (ОН)2 = С6Н4< >СН—N =
\ссг \сох
,С(\
>С6Н4 +RCHO + СО2 + ЗН2О.
Чувствительность реакции в значительной степени зависит от
концентрации водородных ионов. Как сильнокислая, так и щелоч-
ная среда препятствуют реакции. Оптимальная кислотность раство-
ра при pH около 4,5. Кроме а-амипокислот с нингидрином дают окра-
шивание белки, пептоны и полипептиды.
Реакция идет в две фазы. Вначале происходит окислительное
дезаминирование аминокислоты и образование восстановленного нин-
гидрина — гидриндантина
ЛХХ ,СОх
СвН4< >С(ОН) О СН< )С6Н4
\сох ХССИ
без которого дальнейшая реакция не идет. Первой фазе реакции
мешает кислород воздуха, действующий как окислитель и препят-
ствующий образованию гидриндантина, поэтому в обычных усло-
виях реакция идет даже при нагревании очень медленно и мало-
чувствительна. Добавление готового гидриндантина к нингидрину
ускоряет реакцию. Гидриндантин трудно растворим в воде, поэтому
его растворяют в метил целлозольве или этилцеллозольве.
Можно получить гидриндантин в реакционной смеси в достаточ-
ном количестве во время реакции нингидрина с аминокислотой
добавлением восстановителей — SnCl2, KCN или аскорбиновой кис-
лоты. Присутствие пиридина и фенола значительно ускоряет реак-
цию и повышает чувствительность метода. Аскорбиновая кислота
является более подходящим восстановителем, чем цианистый калий:
6*
83
она способствует более быстрому развитию окраски и сохранению
ее при нагревании.
Метод с нингидрин-фенол-пиридиновым реактивом дает хорошие
результаты, но недостатком этого метода является то, что пири-
дин всегда загрязнен аммиаком, который также реагирует с реак-
тивом. Очистка пиридина от аммиака затруднительна, кроме того
пиридин обладает очень неприятным запахом, поэтому этот реактив
не получил широкого распространения.
Реактив Мура и Штейна — нингидрин-гидриндантин-целло-
зольв тоже имеет существенные недостатки. Его необходимо хра-
нить в атмосфере азота, и, несмотря на это, чувствительность реак-
тива катастрофически падает — на третие сутки равняется половине
первоначальной.
Значительным преимуществом обладает нингидриновый реактив
нового состава — раствор нингидрина в этиленгликоле с добавкой
бутанола и пропанола. Новый реактив не уступает старым по чувст-
вительности, но лишен их основных недостатков — неустойчивости
и неприятного ядовитого запаха. Значения цветового выхода продук-
тов реакции аминокислот с новым реактивом при pH 4,54 (в % к
цветовому выходу лейцина) следующие:
Аминокислота Выход окрашенно- го продук- та, % Аминокислота Выход окрашенно- го продук- та, %
Валин 105 Гистидин 99
Норвалин 105 Фенилаланин 99
Глутаминовая 104 Цистин 99
Гликоколь 102 у-Амино- масляная 99
Изолейцин 102 Аспарагино- вая 99
Норлейцин 102 Глутамин 96
Лизин 102 Триптофан 95
Метионин 102 Треонин 92
Серин 101 Тирозин 91
а-Аланин 100 Аспарагин 84
Аргинин 100 Цистеин 33
Лейцин 100 Р-Аланин 27
а-Амино- масляная 100 Аммиак 98
Как видно из приведенных данных, большая часть аминокислот
дает выход окрашенного продукта близкий к 100% (отклонения не
более 2%), 5 аминокислот дают отклонения до 5%, 2 аминокислоты
дают выход заниженный на 8—9% и только цистеин и |3-аланин
дают небольшой выход — всего 33% и 27% соответственно.
Ход анализа. Взвешивают 0,5 г свежего растительного
материала, содержащего от 50 до 500 л/кг аминного азота, растирают
в ступке с 5 мл 10%-ного раствора уксусной кислоты и разбавляют
водой до 100 мл. Смесь тщательно взбалтывают и фильтруют в су-
хую колбу через сухой фильтр, отбрасывая первую порцию фильтра-
84
та. Из остального фильтрата набирают пипеткой 2 мл, переносят
в сухую пробирку, прибавляют 3 мл нингидринового реактива,
0,1 мл 1 %-ного раствора аскорбиновой кислоты, перемешивают, за-
крывают пробирку воронкой или шариковой пробкой (см. рис. 10),
ставят в кипящую воду и нагревают 15 мин. Одновременно нагре-
вают и пробирку с контрольным раствором, состоящим из 2 мл
дистиллированной воды, 3 мл реактива и 0,1 мл раствора аскорбино-
вой кислоты. После нагревания пробирки вынимают из воды и дают
охладиться на воздухе в течение 15 мин при периодическом взбал-
тывании. Образовавшийся во время нагревания гидриндантин крас-
ного цвета при охлаждении и взбалтывании окисляется кислородом
воздуха и обесцвечивается. Окраска же продукта реакции нингидри-
на с аминокислотой сине-фиолетового цвета остается и делается бо-
лее заметной. После охлаждения объем окрашенного раствора дово-
дят 60%-ным спиртом до 5 мл и перемешивают.
Оптическую плотность окрашенного раствора измеряют в фото-
электроколориметре при 580 нм в кювете на 5 мм, сравнивая с
контрольным раствором. Из полученных измерений по калибровоч-
ной кривой находят концентрацию аминного азота в колориметри-
руемом растворе и вычисляют содержание его в исследуемом ве-
ществе по следующей формуле:
v__ 0,1 100 5 С __ 25 С
где X — количество аминного азота (в мг на 100 г исследуемого ве-
щества); С — концентрация аминного азота в колориметрируемом
растворе (в мкг/мл); 5 — объем колориметрируемого раствора (в мл);
п — навеска исследуемого вещества (в г); 100 — общий объем ис-
следуемого раствора (в мл); 2 — объем исследуемого раствора,
взятый для реакции с нингидриновым реактивом (в мл); 0,1 — коэф-
фициент для пересчета микрограммов в миллиграммы и на 100 г
вещества.
Определяемые количества от 0,2 до 2 мкг азота в \мл калориметри-
руемого раствора при измерении в кювете на 5 мм и от 0,1 до 1 мкг
в 1 мл при измерении в кювете на 10 мм. В последнем случае исполь-
зуют навеску в два раза меньшую.
Построение калибровочной кривой. В су-
хие пробирки набирают следующее количество стандартного рас-
твора аминокислоты — лейцина или а-аланина: 0,2, 0,4, 0,6, 0,8,
1,2, 1,6, 1,8, 2 мл раствора, содержащего 5 мкг азота в 1 мл, и дово-
дят безаммиачной водой объем в каждой пробирке до 2 мл. В одну
из пробирок (контрольную) набирают только 2 мл воды. Затем во все
пробирки прибавляют по 3 мл нингидринового реактива, по 0,1 мл
1 %-ного раствора аскорбиновой кислоты, растворы перемешивают и
переносят в специальный алюминиевый штатив для нагревания в
кипящей воде (см. рис. 11). Пробирки закрывают стеклянными ша-
риковыми пробками (см. рис. 10), штатив с пробирками опускают в
водяную баню с кипящей водой и нагревают 15 мин. После этого
85
штатив с пробирками вынимают, охлаждают в течение 15 мин при
периодическом взбалтывании, доводят объем растворов до 5 мл
прибавлением 60%-ного этилового спирта и измеряют оптическую
плотность при 580 нм в кювете на 5 мм и на 10 мм. Раствором
Концентрация аминного
азота, мкг/мл
Рис. 13. Калибровочная кривая
для аминного азота с нингидри-
новым реактивом (светофильтр с
максимумом пропускания 582 нм,
кювета с толщиной измеряемого
слоя 10 мм).
для сравнения служит раствор кон-
трольного опыта, не содержащий
азота. На основании полученных
данных строят калибровочную кри-
вую (рис. 13).
Реактивы. Нингидриновый реактив. От-
вешивают 1,2 г перекристаллизованного
нингидрина, помещают в коническую колбу,
прибавляют 15 мл н-пропилового спирта и
взбалтывают до растворения. Затем прибав-
ляют 30 мл н-бутилового спирта, 60 мл эти-
ленгликоля, перемешивают, добавляют 9 мл
4 и. ацетатного буферного раствора с pH 4,54,
снова тщательно перемешивают и сохраняют
в темной склянке в прохладном месте. Реак-
тив годен в течение 10 дней.
Ацетатный 4 п. буферный раствор
(pH ~ 4,54). Отвешивают 54,4 г соли
CHyCOONa ЗН2О х. ч., растворяют в 100 мл
дистиллированной воды в стакане, ставят
на плитку, нагревают до кипспия и кипятят
до тех пор, пока испарится не менее полови-
ны первоначального объема. Раствор немно-
го охлаждают, переносят в мерную колбу на
100 мл, прибавляют 30 мл ледяной уксусной
кислоты, разбавляют безаммиачной водой до
100 мл, перемешивают и сохраняют в хорошо
закрытой склянке.
Стандартный раствор аминокислоты. Аминокислоту предварительно вы-
сушивают в термостате при 80—90° С и сохраняют в эксикаторе. Для этого берут
лейцин или а-аланин достаточной чистоты. Отвешивают на аналитических весах
46,8 мг лейцина (или 31,8 мг а-аланина), растворяют в 10%-ном изопропиловом
спирте в мерной колбе на 100 мл, разбавляют этим же спиртом до метки и пере-
мешивают. Этот запасной раствор содержит 50 мкг азота в 1 мл. Для приготовле-
ния рабочего раствора набирают 5 мл этого раствора и разбавляют в мерной колбе
до 50 мл дистиллированной водой. В 1 мл находится 5 мкг азота.
Аскорбиновая кислота (1%-ный раствор). Отвешивают 0,1 г аскорбиновой
кислоты и растворяют в 10 мл безаммиачной воды. Готовится только на один день.
н-Бутиловый спирт, свободный от аммиака. Очищают перегонкой. В колбу
Вюрца наливают 500 мл СН3СН2СН2СН2ОН, прибавляют 1 мл 2 н. раствора сер-
ной кислоты, несколько бусинок (стеклянных или силикагеля) для избежания
толчков и отгоняют в дистилляционном аппарате до небольшого остатка (10—
15 мл), собирая фракции, кипящие при температуре 116—118° С. Отогнанный
спирт сохраняют в хорошо закрытой склянке в помещении, не содержащем паров
аммиака.
н-Пропиловый спирт, свободный от аммиака. Перегоняется также, как и бу-
тиловый спирт, с добавкой серной кислоты. Температура кипения 97,2° С.
Очистка этиленгликоля перегонкой. Продажный препарат очищается пере-
гонкой. Для этого в колбу Вюрца емкостью 750—1000 мл наливают 500 мл эти-
ленгликоля, прибавляют 1 мл 2 н. раствора серной кислоты, несколько бусинок,
закрывают пробкой с термометром на 250° С, колбу присоединяют к холодиль-
нику Либиха и отгоняют при температуре в парах 190—197° С до тех пор, пока
останется 20—30 мл. Если под конец перегонки остаток почернеет, перегонку не-
86
медленно прекращают. Перегнанный этиленгликоль сохраняется в склянке с при-
тертой пробкой в помещении, не содержащем паров аммиака.
Очистка нингидрина перекристаллизацией. Нингидрин (тр и кетогидрин де н-
гидрат) С9Н4О3 Н2О, — белый кристаллический порошок хорошо растворим
в горячей воде, щелочах и спирте. При хранении окрашивается в красноватый
цвет. Если качество продажного препарата вызывает сомнение, его можно легко
очистить перекристаллизацией. Для этого 10 г нингидрина растворяют при на-
гревании в 40 мл воды в стакане на 100 мл, добавляют 0,1 г аскорбиновой кислоты,
нагревают до кипения, кипятят 5 мин и горячим фильтруют через бумажный
фильтр диаметром 9 см в колбу на 100—150 мл. К фильтрату прибавляют 10 мл
раствора соляной кислоты (1 1), нагревают, добавляют 1 г активированного угля,
кипятят 3 мин и горячим фильтруют в стакан через бумажный фильтр, предвари-
тельно промытый 0,1 н. соляной кислотой. Фильтрат оставляют в открытом ста-
кане на ночь для кристаллизации. На второй день кристаллы нингидрина соби-
рают на стеклянный фильтр № 2 или № 3, жидкость отсасывают, а кристаллы про-
мывают 5 мл ледяной воды и сушат на бумажном листе при комнатной темпера-
туре. Препарат сохраняют в темной склянке.
ОПРЕДЕЛЕНИЕ АММИАЧНОГО И АМИДНОГО АЗОТА
И АРГИНИНА ТИМОЛ-ГИПОБРОМИТНЫМ МЕТОДОМ
Сущность метода. Гипобромит натрия реагирует с ам-
миаком, окисляя его до свободного азота:
2NH3 + 3NaBrO = 3NaBr + ЗН2О + N2.
В присутствии фенола взаимодействие происходит с образованием
индофенола, окрашенного в синий цвет:
NH3 + 3NaBrO + 2С0НбОН =
= 3NaBr+ ЗН2О + О=/ \= N—ОН.
Это соединение не экстрагируется органическими растворителями,
но если фенол заменить тимолом — СН3 (С3Н7) С6Н3ОН, то обра-
зуется окрашенное вещество — индотимол, которое легко
извлекается органическими растворителями. Реакция специфична,
так как за исключением некоторых аминов (гидразин, гидроксила-
мин), другие соединения, как органические, так и неорганические,
не мешают.
Чувствительность реакции зависит от порядка прибавления
бутанола и гипобромита. Если к раствору, содержащему азот
в виде ионов аммония и тимол, прибавить 1 мл н-бутанола,
тщательно взболтать и затем прибавить гипобромит, то на вы-
ход окрашенного продукта очень влияет способ прибавления
гипобромита (сразу или постепенно) и концентрация солей (NaCl
или Na2SO4). В присутствии солей чувствительность реакции
повышается в 10 раз. Максимальный выход окрашенного продукта
наблюдается в растворе, содержащем не менее 15% солей, при быст-
ром прибавлении всего количества гипобромита и немедленном взбал-
тывании. Если же раствор гипобромита прибавлять постепенно,
при взбалтывании, то выход окрашенного продукта получается
87
вдвое заниженным по сравнению с максимальным. С увеличением
концентрации солей до 15% интенсивность окраски возрастает.
При дальнейшем увеличении концентрации солей выход окрашен-
ного соединения остается постоянным. В присутствии хлористого
натрия чувствительность реакции выше, чем в присутствии серно-
кислого натрия.
Если к раствору, содержащему азот в виде ионов аммония и
тимол, прибавить гипобромит и затем извлечь окрашенное соеди-
нение н-бут^нолом, то на выход окрашенного продукта не влияют
присутствующие в растворе соли, а так же способ прибавления ги-
побромита. Однако такой способ работы понижает чувствительность
реакции более, чем в 5 раз. При содержании в растворе достаточного
количества ^зота (более 5 мкг), этот способ следует предпочесть,
так как в данном случае получают более стабильные результаты.
В этом случае не влияют примеси солей и меньше влияет загрязне-
ние реактивов следами аммиака.
Тимол с гипобромитом в отсутствии ионов аммония образует
соединение желтого цвета, которое экстрагируется я-бутанолом,
и поэтому в присутствии аммиака органическая фаза окрашивается
в зеленый ц13ет (смесь желтого и синего). Для того, чтобы желтое
окрашивание: не мешало колориметрическому определению азота,
соединение которого окрашено в синий цвет, измерение оптической
плотности проводится при 610—660 нм.
При содержании азота от 1 до 8 мкг в 16 мл раствора достаточно
присутствие 0,08 г тимола и 3 мл 0,34 н. раствора гипобромита. Из
всех аминокислот только аргинин при проведении реакции в при-
сутствии солей с предварительным добавлением н-бутанола дает
интенсивное желто-оранжевое окрашивание с максимумом погло-
щения при 470—477 нм. Это дает возможность определять аргинин
тимолгипобромитным методом в присутствии всех аминокислот,
находящихся в растительных экстрактах.
Аммиачный азот, так же как и аргинин, можно непосредственно
определять в вытяжке из растений, так как другие формы азота
не дают окрашенного соединения с тимолом при окислении гипо-
бромитом. Для определения амидного азота необходимо сначала про-
извести гидролиз амидов кислотой при нагревании. В результате
образуется аммонийная соль, которая затем уже реагирует с ти-
молом и гипобромитом:
2(CONH2CHNH2CH2COOH) + H2SO4 + 2Н2О =
аспарагин
2 (НООС CHNH2CH2COOH) + (NH4)2 so4.
аспарагиновая кислота
Для полного гидролиза аспарагина необходимо нагревание в ки-
пящей воде в течение 1,5 ч. Концентрация серной кислоты должна
быть не менее 2 н Гидролиз глутамина идет значительно быстрее.
Определяемые количества азота (в аммонийной форме) от 1 до
88
8 мкг в более чувствительном варианте и от 5 до 100 мкг в менее
чувствительном. Аргинина от 10 до 100 мкг.
X о д анализа. Из отобранной для анализа и измельченной
пробы отвешивают на технических весах 1 г листьев или других ор-
ганов свежих растений (0,5 г клубней картофеля), растирают в
ступке с 4%-ным раствором фосфорновольфрамовой кислоты, до-
бавляя ее из расчета 2 мл на каждый грамм навески. Полученную
массу переносят в мерную колбу на 50 мл, промывая ступку безам-
миачной водой. Содержимое колбы разбавляют дистиллированной
водой до 50 мл, взбалтывают и сейчас же фильтруют через обыкно-
венный бумажный фильтр, предварительно промытый два раза без-
аммиачной водой, в сухую колбу, выбрасывая первую порцию фильт-
рата (5—10 мл).
Определение аммиачного азота и аргинина
5 мл полученного фильтрата отбирают пипеткой в пробирку с
диаметром 20 мм и высотой 20 см, прибавляют 2 мл 4%-ного раство-
ра тимола, 8 мл смеси солей сернокислого и хлористого натрия,
1 мл н-бутанола и энергично взбалтывают. Затем из мерного ци-
линдра емкостью 10 мл быстро приливают 3 мл 0,34 н. раствора
гипобромита натрия, пробирку немедленно закрывают, перемешива-
ют раствор вначале двухкратным переворачиванием, далее энергич-
но взбалтывают в течение 5 сек и оставляют в покое на 15 мин для
окончания реакции. После этого прибавляют еще 3 мл н-бутанола,
тщательно перемешивают и дают отстояться в течение 10—15 мин.
Образовавшееся синее вещество — ипдотимол растворяется в
бутиловом спирте и концентрируется в верхнем слое, окрашивая
его в зеленый или голубой цвет в зависимости от количества азота
и желтого соединения, получающегося при окислении тимола и
аргинина бромом.
Нижний водный бесцветный слой необходимо удалить. Это мож-
но сделать с помощью делительной воронки или следующим обра-
зом. Берут пипетку на 20 мл, закрывают верхнее отверстие и вво-
дят нижний конец в раствор до самого дна пробирки, после чего
отсасывают водный слой, закрывают пипетку, вынимают ее и рас-
твор отбрасывают. Если в пипетку вместе с водным слоем попадает
некоторое количество спиртового окрашенного слоя, то в пипетке
фазы расслаиваются и их можно легко разделить, отбрасывая ниж-
ний бесцветный слой, а спиртовый слой присоединяя к экстракту.
Остаток водного слоя в пробирке удаляют с помощью стеклянной
трубочки с оттянутым концом или пипеткой на 1 мл аналогично от-
делению основной массы водного раствора.
Экстракт красителя, обычно немного мутный от капелек воды,
помещают в мерный цилиндр на 10 мл, для удаления мути прибав-
ляют 10 капель этилового спирта, разбавляют н-бутанолом до
5 мл, перемешивают и переливают в чистую сухую пробирку.
Полученный прозрачный зеленый раствор выливают в кювету с
89
толщиной слоя 10 мм и измеряют оптическую плотность при 660 нм
для определения азота и при 473 нм для определения аргинина.
Раствором для сравнения при определении азота служит вода, а
при определении аргинина — экстракт контрольного раствора.
Чтобы учесть загрязнения реактивов, параллельно проводится
определение азота в контрольном растворе, который обрабатывает-
ся так же, как исследуемый, но вместо исследуемого раствора в
колбу на 50 мл помещают безаммиачную воду, прибавляют в та-
ком же количестве фосфорновольфрамовую кислоту, раствор доводят
безаммиачной водой до 50 мл и фильтруют. Отбирают 5 мл фильт-
рата, переносят в пробирку, добавляют необходимые реактивы и
проводят все операции так же, как в опыте.
Из полученных данных по калибровочным кривым находят кон-
центрацию азота и аргинина и рассчитывают содержание их в ис-
следуемом веществе по следующим формулам:
У ОД 50 5 (С —а) = 5 (С — fl)
5' fi п ’
п
где X — содержание азота на 100 исследуемого вещества (в мг)\
Y — содержание аргинина на 100 г исследуемого вещества (в мг)\
50 — общий объем исследуемого раствора (в мл)\ 5' — объем иссле-
дуемого раствора, взятый для определения аммиачного азота и
аргинина (в мл)\ 5 — объем окрашенного колориметрируемого раство-
ра (влы); С— концентрация азота или аргинина в исследуемом рас-
творе (в мкг!мл)\ а — концентрация азота в контрольном растворе
(в мкг!мл)\ п — навеска исследуемого вещества (в а); 0,1 — коэф-
фициент для перевода микрограммов в миллиграммы и пересчета их
на 100 а вещества.
Определение аргинина
При небольшом содержании аргинина раствор концентрируют
выпариванием. Для этого отбирают 20 или 30 мл фильтрата, поме-
щают в фарфоровую чашку, прибавляют 0,2 М раствора ацетата нат-
рия из расчета 1 мл на 10 мл фильтрата, помещают на кипящую
водяную баню и выпаривают до небольшого объема (2—3 мл). Остав-
шийся раствор переносят в пробирку, промывая чашку дистилли-
рованной водой, и доводят объем до 5 мл. К раствору в пробирке
прибавляют 2 мл раствора тимола, 8 мл смеси солей хлористого и
сернокислого натрия и, если появляется осадок, его растворяют,
прибавляя по каплям 20 %-ный раствор едкого натра и избегая
избытка щелочи. Затем прибавляют 1 мл н-бутанола и далее посту-
пают, как это описано выше. Оптическую плотность измеряют при
473 нм, сравнивая с экстрактом контрольного раствора, т. е. хо-
лостой пробы. При вычислении учитывается взятый для анализа
объем исследуемого раствора.
90
Определение суммы
аммиачного и амидного азота
Для определения суммы аммиачного и амидного азота получен-
ный выше фильтрат разбавляют безаммиачной водой в 5 или 10 раз.
Затем отбирают 5 мл этого разбавленного раствора в пробирку
диаметром 20 мм и высотой 20 см, добавляют 1 мл 11 п. раствора
серной кислоты и перемешивают. Пробирку накрывают маленькой
воронкой и помещают в кипящую воду, не содержащую аммиак,
на 90 мин для гидролиза амидов. После этого раствор охлаждают,
прибавляют одну каплю 0,1 %-ного раствора фенолфталеина и при
охлаждении нейтрализуют 20%-ным раствором едкого натра до появ-
ления устойчивого розового окрашивания. Затем приливают 2 мл
4%-ного раствора тимола, прибавляют 6 мл смеси хлористого и
сернокислого натрия, перемешивают, и, если появляется муть,
ее растворяют одной каплей щелочи. К раствору прибавляют 1 мл
w-бутанола, взбалтывают и далее поступают так, как описано вы-
ше. Одновременно определяют азот в контрольном растворе, кото-
рым служит вода и все реактивы, применяющиеся в ходе анализа.
Раствором для сравнения служит бутанол. Из полученных резуль-
татов по калибровочной кривой находят концентрацию азота в ко-
лориметрируемом растворе и вычисляют сумму аммиачного и амид-
ного азота по следующей формуле:
v 0,1 50 5 (С — а) е _ 5 (С — а) е
гf "
5 п п
где X — содержание суммы аммиачного и амидного азота в 100 г
вещества (в мг); е — степень разбавления первоначального рас-
твора.
Значения остальных величин те же, что и в формуле, приведен-
ной выше для расчета содержания аммиачного азота.
Содержание амидного азота получают, вычитая аммиачный азот
из суммы аммиачного и амидного азота:
Х = А — В,
где X — содержание амидного азота в 100 г вещества (в мг); А —
суммарное содержание аммиачного и амидного азота в 100 г ве-
щества (в мг); В — содержание аммиачного азота в 100 г вещества
(в мг).
Построение калибровочной кривой для
определения аммиачного азота. Набирают в се-
рию пробирок 0,5, 1, 1,5, 2, 2,5, 3, 3,5 и 4 мл стандартного раствора
сернокислого или хлористого аммония, содержащего 2 мкг азота в
1 мл. Объем растворов доводят безаммиачной водой до 5 мл. В по-
следнюю, контрольную пробирку наливают 5 мл безаммиачной воды.
Затем прибавляют по 2 мл 4%-ного раствора тимола и после переме-
шивания по 8 мл раствора смеси солей сернокислого и хлористого
натрия, по 1 мл w-бутанола, энергично взбалтывают и далее посту-
91
пают так, как описано выше. Интенсивность окраски измеряют в
кювете с толщиной слоя 10 мм при 660 нм. Нулевым раствором слу-
жит экстракт контрольного раствора (последняя пробирка). На ос-
новании полученных данных строят калибровочную кривую (рис. 14).
Построение калибровочной кривой для
определения аргинина. Набирают последовательно в
азота,мкг/мл Концентрация аргининакг,/мл
Рис. 15. Калибровочная кривая
для аргинина (светофильтр с
максимумом пропускания
49U нм, кювета с толщиной из-
меряемого слоя 10 мм).
Рис. 14. Калибровочная кри-
вая для аммиачного азота в бо-
лее чувствительном методе (све-
тофильтр с максимумом пропу-
скания 660 нм, кювета с толщи-
ной измеряемого слоя 10 мм).
содержащего 20 мкг аргинина в 1 мл, и доводят объем безаммиачной
водой до 5 мл. В последнюю, контрольную пробирку, не содержащую
аргинина, прибавляют 5 мл дистиллированной воды. Затем в про-
бирки прибавляют по 2 мл 4%-ного раствора тимола, по 8 мл раство-
ра солей хлористого и сернокислого натрия и поступают дальше
так, как это указано выше.
Интенсивность окраски измеряют при 470—480 нм в кювете
10 мм. Раствором для сравнения служит экстракт контрольного рас-
твора, не содержащего аргинин. На основании полученных данных
строят калибровочную кривую (рис. 15).
Определение аммиачного и амидного азота
менее чувствительным методом
Если имеется достаточное количество материала, лучше брать
большую навеску и пользоваться менее чувствительным методом,
так как в этом случае количество азота в образце значительно пре-
вышает примеси его в реактивах, поэтому ошибка определения
уменьшается.
92
Ход анализа. Навеску 2 или 3 г исследуемого материала
растирают в ступке с 5 мл раствора фосфорновольфрамовой кислоты
и разбавляют безаммиачной водой до 50 мл. Раствор фильтруют в
сухую колбу через сухой фильтр, отбрасывая первую порцию фильт-
рата (5—10 мл). В фильтрате определяют аммиачный азот и сумму
аммиачного и амидного азота. По разности находят количество
амидного азота.
Определение аммиачного азота
10 мл фильтрата помещают в пробирку диаметром 20 мм и высо-
той 20 см, прибавляют 0,5 мл «-бутанола и взбалтывают до растворе-
ния спирта. Затем прибавляют 2 мл 4%-ного раствора тимола и
перемешивают. Если появляется муть или осадок, его растворяют
1—2 каплями 20%-ного раствора едкого натра. Далее к раствору
прибавляют 3 мл гипобромита натрия, тщательно перемешивают и
оставляют на 10 мин. После этого добавляют 3 мл «-бутанола, взбал-
тывают и оставляют на 10—15 мин для разделения слоев. Нижний
водный слой удаляют, отсасывая пипеткой, а к остатку, если он
мутный, добавляют 3 капли этилового спирта и перемешивают.
Спиртовый раствор, окрашенный в зеленый цвет, переносят в мер-
ный цилиндр на 10 мл, разбавляют «-бутанолом до 5 мл и измеряют
оптическую плотность раствора при 660 нм в кювете с толщиной слоя
10 мм. Раствором для сравнения служит бутанол. Одновременно
таким же путем определяют содержание аммиачного азота в конт-
рольном растворе, который не содержит исследуемого раствора,
но проводится через все стадии анализа и содержит то же количество
реактивов, что и остальные растворы. Для приготовления контроль-
ного раствора в мерную колбу на 50 мл помещают 5 мл фосфорно-
вольфрамовой кислоты, доводят безаммиачной водой до метки, пере-
мешивают и фильтруют. Отбирают 10 мл фильтрата и определяют
содержание аммиачного азота, чтобы внести поправку на загрязне-
ние реактивов. Из данных оптической плотности по калибровочной
кривой находят концентрацию азота в колориметрируемом раство-
ре и вычисляют содержание его в пробе по формуле, приведенной
выше (см. стр. 90).
Определение суммы
аммиачного и амидного азота
Набирают пипеткой 5 мл фильтрата, переносят в пробирку, при-
бавляют 1 мл 11 н. раствора серной кислоты, закрывают воронкой
или шариковой пробкой, помещают в кипящую воду, не содержа-
щую аммиак, и нагревают в течение 90 мин для гидролиза амидов.
Затем охлаждают и нейтрализуют 20%-ным раствором едкого натра
в присутствии фенолфталеина. При нейтрализации происходит
нагревание. Необходимо охлаждать раствор холодной водой, не
допуская повышения температуры. После этого прибавляют 0,5 мл
93
я-бутанола, взбалтывают, прибавляют 2 мл 4%-ного раствора ти-
мола, 3 мл гипобромита натрия и далее поступают также, как описа-
но выше при определении аммиачного азота. Из полученных дан-
ных вычисляют содержание суммы аммиачного и амидного азота по
формуле, приведенной выше (см. стр. 91).
Построение калибровочной кривой. В се-
рию пробирок помещают 0,5, 1, 2, 3, 4, 5, 6, 8 и 10 мл стандартного
Концентрация аммиачного азота,
мкг/мл
Рис. 16. Калибровочная кривая
для аммиачного азота в менее
чувствительном методе (свето-
фильтр с максимумом пропуска-
ния 660 нм, кювета с толщиной
измеряемого слоя 10 мм).
раствора, содержащего 10 мкг азота
в 1 мл, и разбавляют безаммиачной
водой до 10 мл. В последнюю пробир-
ку вносят 10 мл дистиллированной
воды. Этот раствор служит контролем
на реактивы и с ним сравнивают рас-
творы шкалы. В пробирки прибав-
ляют по 0,5 мл w-бутанола, взбалты-
вают, прибавляют по 2 мл раствора
тимола, поЗ мл раствора гипобромита
натрия и т. д., как описано выше при
определении аммиачного азота. На ос-
новании результатов измерений опти-
ческой плотности растворов строят
калибровочную кривую (рис. 16).
Определяемые количества — от 5 до
100 мкг азота.
Реактивы. Вода и реактивы почти всегда
загрязнены аммиаком или солями аммония,
поэтому они должны быть очищены от этих
нежелательных примесей и сохраняться в хо-
рошо закрытых склянках в помещениях, не
содержащих паров аммиака. Вследствие боль-
шой чувствительности метода во время рабо-
ты в лаборатории не должны проводиться анализы с использованием аммиака,
а также находиться реактивы, содержащие аммиак.
Вода дистиллированная без аммиака. Приготовление описано выше (см.
стр. 76).
Раствор едкого натра (20%-ный без аммиака). Отвешивают 200 г едкого
натра (х. ч. или ч. д. а.), растворяют в 1100 мл дистиллированной воды в боль-
шом стакане, который затем ставят на плитку, доводят до кипения и кипятят
40 мин. После этого раствор охлаждают и разбавляют безаммиачной водой до
1 л. Сохраняют в склянке, закрытой резиновой пробкой.
Раствор тимола (4%-ный). Взвешивают 4 г тимола с точностью до 0,01 г,
растирают в фарфоровой ступке с 8 мл 20%-ного раствора едкого натра. Раствор
переносят в мерную колбу на 100 мл, промывают ступку безаммиачной водой, до-
водят до метки и тщательно перемешивают. Сохраняют в темной склянке в про-
хладном месте не более трех дней.
Раствор гипобромита натрия (0,34 н.). 10 мл 20%-ного раствора^едкого
натра помещают в мерный цилиндр на 100 мл, приливают насыщенной бромной
воды до объема 100 мл и перемешивают.
Для определения концентрации брома отбирают пипеткой 5 мл раствора ги-
побромита (всасывают с помощью груши) и помещают в колбу для титрования,
содержащую 5 мл 20%-ного раствора KJ. Затем приливают 5 мл 2 н. раствора
серной кислоты, перемешивают и титруют 0,1 н. раствором тиосульфата натрия
в присутствии крахмала. Должно расходоваться не менее 17 мл 0,1 н. раствора
94
тиосульфата. Если раствор окажется крепче, его разбавляют 0,5 н. раствором
NaOH, определяя необходимое количество раствора щелочи по следующей фор-
муле:
где X — объем 0,5 н. раствора NaOH, который необходимо прибавить для разбав-
ления раствора гипобромита натрия (в мл); А — объем 0,1 н. раствора тиосуль-
фата натрия, затраченный на титрование 5 мл раствора гипобромита (в мл); В —
объем разбавляемого раствора гипобромита (в мл); 17 — объем 0,1 н. раствора
тиосульфата натрия, эквивалентный 5 мл 0,34 н. гипобромита натрия (в мл).
Раствор сохраняют в темной склянке в прохладном месте.
Для приготовления бромной воды в склянку со стеклянной пробкой нали-
вают 50—100 мл брома, 1 л дистиллированной воды и энергично взбалтывают
в течение 5—10 мин. При приготовлении раствора гипобромита натрия применяют
свеженасыщенную бромную воду.
н-Бутиловый спирт, свободный от аммиака. Очистка продажного н-бутило-
вого спирта от аммиака описана выше (см. стр. 86).
Отработанный бутиловый спирт очищают следующим образом. На каждые
500 мл спирта прибавляют 25 г безводного сульфата натрия и настаивают при
взбалтывании в течение 1 ч. Затем жидкость сливают, прибавляют 2 мл раствора
серной кислоты (1 9) и отгоняют, собирая фракцию, кипящую при температуре
выше 100° С.
Раствор смеси хлористого и сернокислого натрия. Отвешивают 140 г Na2SO4
ЮН2О или 60 г безводной соли и 280 г NaCl (х. ч. или ч. д. а.), помещают в
стакан, прибавляют 1100 мл дистиллированной воды, 2 мл 20%-ного раствора
едкого натра, нагревают до кипения и кипятят 40 мин. Раствор охлаждают, пере-
носят в мерную колбу на 1 л, промывают стакан безаммиачной водой, доливают
до метки этой же водой и перемешивают. Если раствор мутный, его фильтруют
через бумажный фильтр, предварительно промытый безаммиачной водой. Раствор
сохраняют в хорошо закрытой склянке.
Раствор фосфор ново л ьф ромовой кислоты (4%-ный) с NaCl. Отвешивают на
технических весах 4 г фосфорновольфрамовой кислоты и растворяют в 50 мл
безаммиачной воды. Затем взвешивают 10 г хлористого натрия х. ч. и тоже рас-
творяют в 50 мл безаммиачной воды. Оба раствора сливают вместе, перемешивают
и сохраняют в хорошо закрытой склянке.
Раствор фенолфталеина (0,1%-ный). 0,1 г фенолфталеина растворяют в
60 мл этилового спирта и доводят безаммиачной водой до 100 мл.
Серная кислота (~ 11 н.). Вливают 310 мл концентрированной, очищенной
перегонкой, серной кислоты в 500 мл безаммиачной воды, перемешивают, охлаж-
дают и доводят водой до 1 л. Сохраняют в склянке с хорошо притертой пробкой.
Концентрированную серную кислоту очищают от ионов аммония перегонкой из
реторты, которую помещают на плитку, обложив вокруг асбестом, чем достига-
ется равномерность обогрева.
Раствор ацетата натрия (0,2М). Отвешивают 2,72 г CH3COONa ЗН2О,
растворяют в безаммиачной воде, доводят объем до 100 мл и сохраняют в хорошо
закрытой склянке.
Стандартный раствор азота (аммонийного). Взвешивают на аналитических
весах 0,4719 г сернокислого аммония или 0,3821 г хлористого аммония х. ч.,
растворяют в безаммиачной воде в мерной колбе на 500 мл, доводят дистиллиро-
ванной водой до метки и перемешивают. 1 мл этого раствора содержит 10 мкг
азота.
Стандартный раствор аргинина. Взвешивают на аналитических весах 12,1 мг
солянокислого аргинина, растворяют в безаммиачной воде в мерной колбе на
500 мл, доливают водой до метки и перемешивают. 1 мл этого раствора содержит
20 мкг аргинина. Раствор готовится свежим на один день.
95
ОПРЕДЕЛЕНИЕ НИТРАТОВ В РАСТЕНИЯХ
Сущность метода. Нитраты извлекаются из свежего
растительного материала 1%-ной уксусной кислотой. В получен-
ном растворе нитраты восстанавливаются цинком при pH 5,6 до
нитритов, которые затем определяются колориметрическим методом
с а-нафтил амином и сульфаниловой кислотой (см. стр. 39).
Ход анализа. Отвешивают такое количество свежесо-
бранного растительного материала, чтобы в навеске содержалось
от 10 до 100 мкг нитратного азота (около 1 г сырого вещества), поме-
щают в фарфоровую ступку и растирают с 1%-ным раствором ук-
сусной кислоты. Растертую массу переносят в мерную колбу на 25 мл
с помощью 1 %-ной уксусной кислоты, доводя объем до метки. После
тщательного перемешивания фильтруют в сухую колбу через сухой
фильтр диаметром 7 см (белая лента). Из полученного фильтрата
отбирают пипеткой от 0,5 до 4 мл (в зависимости от предполагае-
мого количества нитратов), переносят в сухую пробирку с внутрен-
ним диаметром 16—18 мм, разбавляют 1%-ной уксусной кислотой
до 4 мл, приливают 4 мл 0,8 М раствора ацетата натрия и перемеши-
вают. С помощью мерки прибавляют 10 мг цинковой пыли и про-
бирку периодически встряхивают круговыми движениями в тече-
ние 10 мин со скоростью 1—2 встряхивания в 1 мин. После этого
раствор фильтруют в сухую пробирку или колбочку через сухой
беззольный фильтр диаметром 7 см. Из полученного фильтрата наби-
рают пипеткой 4 мл, выливают в сухую пробирку, прибавляют 1 мл
реактива на нитриты, перемешивают и через 10 мин колориметри-
руют при 540 нм в кювете на 10 мм, сравнивая с контрольным раство-
ром, который получается разбавлением 2 мл исследуемого раствора
дистиллированной водой до 5 мл.
По полученным данным с помощью калибровочной кривой нахо-
дят концентрацию азота и вычисляют его содержание в исследуе-
мом веществе по следующей формуле:
v 0,1 25 5 С 25 С
Уь Л — •
0,5 v п vn
где X — содержание нитратного азота на 100 г исследуемого веще-
ства (в мгу, С — концентрация нитратного азота в колориметри-
руемом растворе (в мкг!мл)\ 25 — общий объем исследуемой вытяж-
ки (в мл)\ v — объем вытяжки, взятый для восстановления цинком
нитратов в нитриты (в мл)\ 5 — объем колориметрируемого раство-
ра (в мл)\ 0,5 — часть восстановленного раствора, взятая для коло-
риметрирования; п — навеска исследуемого вещества (в г); 0,1 —
коэффициент для перевода микрограммов в миллиграммы и пересче-
та их на 100 г вещества.
Определяемые концентрации — от 0,1 до 1 мкг!мл колориметри-
руемого раствора. Количество нитратного азота в исследуемом рас-
творе, взятом для восстановления, должно быть от 1 до 10 мкг.
Приготовление реактивов и построение калибровочной кривой
описано в главе I (см. стр. 39—41).
96
ОПРЕДЕЛЕНИЕ МОЧЕВИНЫ
Сущность метода. Исследуемое вещество растирается
с фосфатным буферным раствором pH 7,0 и разбавляется до опре-
деленного объема. К определенной части этого раствора прибавляют
раствор фермента уреазы и выдерживают 10 мин при температуре
40° С. При этом идет гидролиз мочевины и азот полностью переходит
в аммиачную форму:
(NH2)2 СО + 2Н2О = (NH4)2 СО3.
После осаждения белков фосфорновольфрамовой кислотой и цен-
трифугирования азот определяют тимол-гипобромитным методом.
Ход анализа. Навеску исследуемого вещества, содержа-
щую от 0,1 мг до 1 мг мочевины (например, 2 г свежих листьев фа-
соли), растирают с 2 мл фосфатного буферного раствора pH 7 до
однородной массы, переносят в мерный цилиндр на 50 мл, разбавля-
ют водой до 50 мл и перемешивают. Набирают 10 мл полученной
суспензии, помещают в центрифужную пробирку, прибавляют 2 мл
раствора уреазы, перемешивают и погружают пробирку на 10 мин
в воду, нагретую до 45° С, не давая ей охладиться ниже 40° С.
После этого добавляют 2 мл фосфорновольфрамовой кислоты, пере-
мешивают и через 3—5 мин центрифугируют 3 мин при 2500 обIмин.
Прозрачный раствор сливают в сухую пробирку. Набирают пипет-
кой 10 мл этого раствора, переносят в пробирку диаметром 18—
20 мм и определяют азот тимол-гипобромитным методом. Для этого
к раствору прибавляют 0,5 мл «-бутилового спирта, взбалтывают до
растворения спирта, затем добавляют 2 мл раствора тимола и пере-
мешивают. Если появляется муть или осадок, его растворяют при-
бавлением 1—2 капель 20%-ного раствора NaOH. К раствору при-
бавляют 3 мл гипобромита натрия, перемешивают и оставляют на
10 мин. После этого добавляют 4 мл бутилового спирта, взбалтывают
и оставляют на 10—15 мин для разделения фаз. Нижний водный слой
удаляют, отсасывая его небольшой стеклянной пипеткой с резино-
вым колпачком, нажатием которого создается разрежение. После
полного удаления нижнего водного слоя, окрашенный раствор
помещают в мерный цилиндр на 5 или 10 мл, прибавляют 10 капель
этилового спирта для устранения мути и доводят объем бутиловым
спиртом до 5 мл. Прозрачный раствор переносят в сухую пробирку
и оптическую плотность измеряют при 660 нм в кювете с рабочей
длиной 10 мм, сравнивая с контрольным раствором.
Контрольный раствор готовят следующим образом. Берут такое
количество исследуемого вещества, которое равно х/5 части ранее
взятой навески и растирают его с 2 мл фосфорновольфрамовой кис-
лоты. Растертую массу переносят в центрифужную пробирку, смы-
вая остаток 10 мл безаммиачной воды, добавляют 2 мл раствора
уреазы, перемешивают и центрифугируют. Прозрачный раствор
сливают в сухую пробирку, набирают пипеткой 10 мл, выливают в
другую чистую пробирку, куда добавляют 0,5 мл бутилового спирта,
7 6-2
97
взбалтывают, прибавляют раствор тимола и продолжают далее
так, как описано выше при определении азота в мочевине.
Из полученных данных по калибровочной кривой находят кон-
центрацию азота в колориметрируемом растворе и вычисляют со-
держание мочевины в исследуемом веществе по следующей форму-
ле:
х _ 0,2143 50 14 5 С _ 7,5 С
Л 10 10' п ~ п
где X — содержание мочевины (в мг на 100 г исследуемого вещества);
50 — общий объем исследуемого раствора (в мл); 10 — объем ис-
следуемого раствора, взятый для разложения мочевины уреазой
(в мл); 10'— объем центрифугата, взятый для определения азота
(в мл); 14 — объем раствора после прибавления к 10 мл исследуе-
мого раствора уреазы и фосфорновольфрамовой кислоты (в мл);
5 — объем колориметрируемого раствора (в мл); С — концентрация
азота в колориметрируемом растворе (в мкг/мл); п — навеска ис-
следуемого вещества (в г); 0,2143 — коэффициент перевода мкг
азота в мг мочевины и пересчета ее на 100 г вещества.
Определяемые количества от 20 до 200 мкг мочевины в колори-
метрируемом растворе. Построение калибровочной кривой осущест-
вляется так же, как описано при определении аммиачного азота
тимол-гипобромитным методом (см. стр. 94, рис. 16).
Реактивы. Раствор уреазы, 5 г тонко растертой муки бобов сои помещают
в колбу емкостью 200—250 мл, добавляют 100 мл 30%-ного раствора этилового
спирта (35 мл 95% -ного спирта смешивают с 65 мл воды) и взбалтывают в течение
15 мин. Дают осадку осесть и центрифугируют. Центрифугат, даже если он мут-
ный, сливают в склянку и сохраняют на холоду. Фермент при температуре 1—3° С
сохраняется в течение месяца, а при комнатной температуре всего неделю.
Фосфатный буферный раствор с трилоном Б (pH 7). Отвешивают 6,8 г
КН2РО4 х. ч. и 0,9 г трилона Б, растворяют в безаммиачной воде, прибавляют
30 мл 1 н. раствора NaOH, разбавляют безаммиачной водой до 250 мл, добавляют
1 мл хлороформа и сохраняют в хороню закрытой склянке.
Приготовление остальных реактивов описано при определении аммиачного
азота тимол-гипобромитным методом (см. стр. 94, 95).
ОПРЕДЕЛЕНИЕ ГЛУТАТИОНА
Глутатион является трипептидом, состоящим из трех амино-
кислот: глутаминовой, цистеина и гликоколя. Его формула:
НООС CHCH2CH2CONHCHCONHCH2COOH.
I I
nh2 ch2sh
При кислотном гидролизе глутатион распадается на составляю-
щие его аминокислоты. Глутатион играет важную роль в окислитель-
но-восстановительных процессах, что обусловлено свойством
сульфгидрильных групп—SH— легко окисляться, превращаясь
в дисульфидную форму —S—S—.
98
Сущность метода. лутатион в слабощелочной среде
с ацетатом ртути дает осадок глутатионата ртути:
2C10H16OeN3SH + Hg (СН3СОО)2 = Hg (C10HieOeN3S)s + СН3СООН.
Осадок промывается водой и растворяется в соляной кислоте:
Hg (C10HieOeN3S)2 + 2НС1 = HgCl2 + 2C10H10O0N,SH.
Раствор отделяется от нерастворимых в воде веществ, и глу-
татион, перешедший в раствор, титруется 0,001 н. раствором йо-
дата калия в присутствии йодистого калия и крахмала:
C10H16O6N3S
6C10H16O6N3SH + KJO3 + 5KJ + 6НС1 =3 | + 6КС1 +
+ 6HJ + ЗН2О.
Йодистый калий прибавляется в избытке для связывания ионов
ртути в растворимое бесцветное комплексное соединение:
HgCl2 + 4KJ = К2 [HgJ4],
~ 6C10H16O6N3SN о
Грамм-эквивалент глутатиона —-°- - — равен 307,3.
Ход анализа. Отвешивают 4 г сырого растительного мате-
риала и тщательно растирают в ступке с 2 мл 0,3 н. раствора уксус-
нокислой ртути и 2 мл 30 %-ного раствора уксуснокислого натрия.
Гомогенат из ступки переносят в центрифужную пробирку, смы-
вая остаток 10 мл дистиллированной воды, перемешивают стеклян-
ной палочкой и оставляют па 10 мин для полноты осаждения. После
этого центрифугируют, раствор отбрасывают, а осадок 2 раза про-
мывают водой порциями по 10 мл при перемешивании.
В осадке вместе с другими веществами находится глутатион,
который растворяют в соляной кислоте. Для этого к остатку в
пробирке приливают 10 мл 1 н. раствора соляной кислоты и пере-
мешивают стеклянной палочкой в течение 5 мин. Затем добавляют
1 мл 20%-ного раствора йодистого калия, перемешивают и центри-
фугируют. Центрифугат переносят в колбу для титрования емкос-
тью 100 мл, а осадок промывают 10 мл воды при перемешивании.
После центрифугирования раствор присоединяют к первому центри-
фугату.
К полученному раствору добавляют 0,5 мл раствора крахма-
ла и титруют 0,001 н. раствором KJO3 до появления неисчезающей
синей окраски. 1 мл 0,001 н. раствора KJO3 соответствует 0,307 мг
глутатиона. Содержание глутатиона вычисляют по формуле:
v _ 30 700 а к
где X — содержание глутатиона (в мг на 100 г исследуемого вещест-
ва); а — объем 0,001 н. раствора йодата калия, затраченный при
титровании глутатиона (в мл)\ К — нормальность раствора йода-
та калия; п — навеска исследуемого вещества (в г); 30 700 —
99
нормальный титр глутатиона (в мг), умноженный на 100 для пере-
счета на 100 г вещества.
Реактивы. Раствор уксуснокислой окисной ртути (0,3 н.). Отвешивают
16,25 г HgO и растворяют при нагревании в 100 мл 20%-ного раствора уксусной
кислоты. Полученный раствор разбавляют дистиллированной водой до 500 мл.
Раствор йодата калия (0,001 н.). Готовится из 0,1 н. раствора разбавлением
дистиллированной водой. Для этого отбирают пипеткой 5 мл 0,1 н. раствора KJO3,
помещают в мерную колбу на 500 мл, разбавляют водой до метки и перемешивают.
Приготовление остальных реактивов приведено выше (стр. 17, 18, 27, 138).
ОПРЕДЕЛЕНИЕ АМИНОКИСЛОТ
С ПОМОЩЬЮ ХРОМАТОГРАФИИ НА БУМАГЕ
Сущность метода. Свободные аминокислоты извле-
кают из растительного материала слабым раствором уксусной кисло-
ты и очищают от примесей с помощью ионообменных смол. Элюат
концентрируют до определенного объема, наносят на хроматогра-
фическую бумагу и хроматографируют. Для полного разделения всех
аминокислот необходимо выполнять 7—9 одномерных хроматограмм
с различными растворителями. Растворители подбирают таким обра-
зом, чтобы в каждой хроматограмме четко разделить определенные
аминокислоты, причем разные в зависимости от растворителя.
В табл. 1 приведены номера смесей растворителей и отделяемые
ими аминокислоты. Цифрами обозначены четко отделяемые ами-
нокислоты и последовательность их движения на хроматрграмме.
Отделенные аминокислоты определяются колориметрическим нин-
гидриновым методом. Анализ осуществляют в два этапа:
1. Хроматограмму после разделения и высушивания опрыскивают
раствором нингидрина. По истечении времени развития окраски
пятно извлекают спиртовым раствором сульфата меди, который да-
ет с продуктом реакции более стабильное медное производное,
окрашенное в оранжево-красный цвет с максимумом поглощения
около 500 нм. Этим способом можно определить следующие амино-
кислоты: а-аланин, а-аминомасляную кислоту, аргинин, валин,
глутаминовую кислоту, изолейцин, лейцин, лизин, норвалин, нор-
лейцин, серин, треонин, тирозин, фенилаланин. Последние две
аминокислоты на бумаге дают более слабую окраску (в 1,3—1,4
раза), но определение их еще возможно с удовлетворительной точ-
ностью при внесении соответствующей поправки. Применение этого
способа выгодно, потому что аммиак не мешает определению, так
как не реагирует с нингидрином в присутствии уксусной кислоты,
находящейся в реактиве.
2. Остальные аминокислоты после хроматографирования, высуши-
вания хроматограмм и обработки 0,01 н. раствором гидроокиси ка-
лия для удаления аммиака, извлекают водой и определяют коло-
риметрическим нингидриновым методом так, как описано при опре-
делении аминного азота (см. стр. 83).
Если имеется хроматографическая бумага, однородная по порис-
тости и толщине, с весовым различием единицы площади, не пре-
100
вышающим 2%, то можно определить содержание аминокислот по
площади или по весу окрашенного пятна, что значительно проще,
но менее точно. Для определения аминокислотного состава белков
их сначала выделяют одним из способов, описанных выше. Полу-
Таблица 1
Схема одномерных хроматограмм с указанием отделяемых аминокислот
и последовательности их движения в определенном растворителе * *
Растворитель
Аминокислота
а-Аланин
р-Аланин
а-Аминомасляная
кислота
у-Аминомасляная
кислота
Аргинин
Аспарагин
Аспарагиновая
кислота
Валин
Гистидин
Гликоколь
Глутамин
Глутаминовая
кислота
Изолейцин
Лейцин
Лизин
Метионин
Норвалин
Норлейцин
Пролин
Серин
Тирозин
Треонин
Триптофан
Фенилаланин
Цистин
* Против отделяемой аминокислоты стоит число, показывающее место, занимае-
мое ею в ряду. Цифрами показана последовательность движения отделяемых амино-
кислот.
ченные чистые белки подвергают гидролизу 6 н. соляной кислотой
или гидроокисью бария. При гидролизе кислотой аспарагин и глу-
тамин превращаются в аспарагиновую и глутаминовую кислоты, а
триптофан разрушается, поэтому эти соединения определяются после
щелочного гидролиза.
Ход анализа. Для определения свободных аминокислот
берут от 2 до 4 г свежего растительного материала Гили соответствую-
щее количество сухого), содержащего 4—5 мг аминного азота, рас-
тирают в ступке с 5 мл 5%-ного раствора уксусной кислоты до
101.
однородной массы, переносят с помощью дистиллированной воды в
мерную колбу на 50 мл, доводят объем водой до 50 мл, перемеши-
вают и фильтруют в сухую колбу через сухой фильтр. В фильтрате
определяют концентрацию аминного азота. Для этого 1 мл фильт-
рата помещают в мерную колбу на 25 мл и разбавляют дистиллиро-
ванной водой до метки. В полученном растворе определяют амин-
ный азот колориметрическим методом, как изложено выше (см.
стр. 85), и таким образом находят содержание его в 1 мл перво-
начального исследуемого раствора, а затем в 20 мл, взятых для
дальнейшей работы.
Если исследуемый объект содержит небольшое количество во-
дорастворимых примесей, мешающих хроматографированию (саха-
ров, органических кислот и др.), как например клубни картофеля,
из раствора берут две порции по 20 мл, переносят в фарфоровые
чашки, которые затем ставят на водяную баню и выпаривают рас-
творы досуха. Остаток в одной из чашек растворяют в 0,05 н. соля-
ной кислоте, добавляя такой объем, чтобы концентрация аминного
азота в растворе была 2 мг!мл. Остаток во второй чашке растворяют
в 10%-ном растворе изопропанола, добавляя его в таком же объ-
еме, как и раствор соляной кислоты.
В первом растворе определяют цистин, тирозин и другие амино-
кислоты, которые хорошо растворимы в соляной кислоте, кроме
триптофана, аспарагина, аспарагиновой кислоты, глутамина и глу-
таминовой кислоты. Во втором растворе можно определить все
аминокислоты, кроме цистина и тирозина, которые трудно раствори-
мы в воде.
Если объект исследования содержит большое количество саха-
ров, органических кислот и других водорастворимых веществ, то
их отделяют пропусканием уксуснокислой вытяжки через ионо-
обменную колонку, содержащую сильнокислотные катиониты КУ-2,
СДВ-ЗТ, дауэкс-50, амберлит ИР-120 и др.
Приготовление колонки. Колонка представляет
собой стеклянную трубку длиной 20—30 см, оттянутую внизу. На
расстоянии 1—2 см от нижнего широкого конца впаивается стек-
лянный фильтр № 1 или № 2, служащий опорой для смолы. Фильтр
может быть заменен плотным слоем стеклянной ваты. Сверху колон-
ка имеет резервуар емкостью 30—40 мл.
Смолу перед наполнением колонок подвергают очистке. Ее за-
ливают 4 н. раствором соляной кислоты и оставляют на 2—3 ч.
Затем отмывают от соляной кислоты дистиллированной водой, де-
кантацией до слабокислой реакции (pH 6). Конец промывания ус-
танавливают индикатором метиловым красным, до перехода цвета
индикатора в желтый. Промытую смолу суспендируют в воде и за-
ливают в колонку. Смолу в колонке промывают несколько раз 2 н.
соляной кислотой до тех пор, пока вытекающий раствор будет бес-
цветным. Остаток кислоты вымывают водой до pH 6. Слой смолы в
колонке должен быть 15—20 см, степень измельчения частиц 0,2—
0,3 мм (50—70 меш).
102
Через подготовленную таким образом колонку пропускают 20
или 40 мл уксуснокислой вытяжки со скоростью вытекания 10 ка-
пель в 1 мин. При этом аминокислоты адсорбируются, а сахара и
органические кислоты остаются в растворе. После того как весь
раствор пропущен через колонку, ее промывают водой до удаления
сахаров. Для проверки на полноту промывания проводят реакцию
с альфанафтолом. В пробирку прибавляют 5 капель исследуемого
раствора, 3 капли 5%-ного свежеприготовленного спиртового рас-
твора альфанафтола и 1 мл концентрированной серной кислоты
пл. 1,84. После взбалтывания в присутствии сахаров появляется
розово-красное окрашивание. Далее проводят элюирование амино-
кислот 2 н. раствором аммиака, пропуская 50—60 мл его и промы-
вая остатки водой. Раствор собирают в фарфоровую чашку и выпа-
ривают на водяной бане досуха. Остаток растворяют в воде или
10%-ном изопропаноле, прибавляя такое количество растворителя,
чтобы концентрация аминного азота в растворе была 2 мг!мл.
Для регенерации колонки через нее пропускают 2 н. раствор
соляной кислоты до тех пор, пока стекающий раствор не станет
резко кислым. Затем отмывают кислоту водой до pH 6.
При исследовании аминокислотного состава белков их извле-
кают соответствующими растворителями и осаждают уксусной кис-
лотой, как описано выше (см. стр. 80, 81). Осадки белков промы-
вают этиловым спиртом и эфиром. Полученные препараты подвер-
гают кислотному и щелочному гидролизу. Для кислотного гидролиза
берут 50—100-кратный избыток 6 п. раствора соляной кислоты,
или 8 н. раствора серной кислоты, или 57%-пой йодистоводород-
ной, или смесь соляной и муравьиной кислот, а для щелочного —
5 н. раствора едкого натра или 1—4 н. раствора гидроокиси бария.
Продолжительность гидролиза от 16 до 24 ч при температуре 100—
120° С.
Длительное воздействие довольно больших концентраций силь-
ных электролитов при высокой температуре оказывает влияние на
аминокислоты, вызывая их потери. При этом степень разрушения
отличается в зависимости от стойкости аминокислоты к данному
реактиву. Присутствие примесей, в особенности сахаров, ведет
к повышенному образованию гуминовых соединений, что приводит
к значительным потерям аминокислот. Кислотный гидролиз в при-
сутствии сильных восстановителей (SnCl2, TiCl2) или в атмосфере
азота уменьшает потери аминокислот. Потери также уменьшают-
ся при гидролизе смесью соляной к муравьиной кислот.
Некоторые аминокислоты (триптофан, амиды) разрушаются силь-
нее кислотой, а другие (цистин, цистеин, серин, треонин) — ще-
лочью. Поэтому для удовлетворительного анализа всех аминокислот
необходимо проводить гидролиз белков не только кислотой, но и
щелочью.
юз
Кислотный гидролиз белков
Чаще всего гидролиз осуществляют 6 н. раствором соляной
кислоты, а при определении цистина и других легко разрушаемых
аминокислот пользуются смесью равных объемов 6 н. раствора соля-
ной кислоты и 90 %-ного раствора муравьиной кислоты.
Для гидролиза берут 20 мг чистого препарата белков, всыпают
в стеклянную пробирку диаметром 8—10 мм, прибавляют 1 мл гид-
ролизуемого раствора — 6 н. соляной кислоты или смеси 6 н. соля-
ной кислоты и 90 %-ной муравьиной кислоты (1 : 1). Пробирку запаи-
вают и помещают в кипящую воду на 20—24 ч. После окончания гид-
ролиза пробирку вскрывают и содержимое переносят в стаканчик,
смывая остаток безаммиачной водой (3 раза порциями по 0,5 мл).
Стаканчик с раствором помещают в вакуум-эксикатор, содержащий
едкий натр для поглощения кислоты, и выпаривают досуха, созда-
вая разрежение. Остаток растворяют в 1 мл дистиллированной
воды, переносят в центрифужную пробирку, прибавляют одну
каплю 80%-ного водного раствора фенола, встряхивают и центрифу-
гируют. Прозрачный раствор сливают в сухую пробирку и исполь-
зуют для определения аминокислот хроматографическим методом.
После кислотного гидролиза определяют следующие аминокисло-
ты: аланин, аргинин, аспарагиновую кислоту, валин, гистидин,
гликоколь (глицин), глутаминовую кислоту, изолейцин, лейцин,
лизин, метионин, пролин, серин, тирозин, треонин, фенилаланин
и цистин. Все эти аминокислоты можно разделить с помощью одно-
мерных хроматограмм с системами растворителей: 1, 2, 3, 4, 6 и 7.
Щелочной гидролиз белков
Гидролиз щелочью применяется главным образом при определе-
нии триптофана, который в щелочной среде более устойчив, чем
в кислой. Вообще же щелочной гидролиз приводит к большему раз-
рушению аминокислот, чем кислотный. Щелочи не только способст-
вуют потерям при гидролизе аминокислот, но часто вызывают ка-
чественные изменения в составе гидролизата. Например, серин
расщепляется на гликоколь, аланин, аммиак и пировиноградную
кислоту, треонин — на гликоколь и аминомасляную кислоту, цис-
тин — на тиомолочную кислоту, сероводород и аммиак, аргинин —
на орнитин и цитрулин. Щелочной гидролиз вызывает также раце-
мизацию аминокислот.
Щелочной гидролиз белков лучше всего проводить методом
Дрезе и Рейта. По этому методу 20 мг чистых белков помещают
в пробирку из щелочеупорного стекла, добавляют 0,1 г крахмала,
предварительно промытого 0,01 н. соляной кислотой, и 0,6 г гид-
роокиси бария — Ва (ОН)2 8Н2О, кристаллы которого не долж-
ны содержать углекислых солей. В смесь добавляют 0,5 мл без-
аммиачной воды, пробирку запаивают, помещают в этиленгликоль
104
или 80%-ный раствор хлористого кальция и нагревают при темпера-
туре 120° С в течение 15 ч.
После окончания гидролиза пробирку вскрывают и содержимое
смывают 4 мл дистиллированной воды в центрифужную пробирку.
Прибавляют одну каплю раствора фенолфталеина и нейтрализуют
1 н. раствором серной кислоты до обесцвечивания индикатора. Затем
добавляют каплями насыщенный раствор гидроокиси бария до
появления розовой окраски индикатора, перемешивают и центри-
фугируют. Раствор переносят в фарфоровую чашку, а осадок про-
мывают водой (2 раза порциями по 5 мл), присоединяя центрифу-
гаты к раствору в чашке. Чашку помещают на водяную баню и
выпаривают раствор досуха. Остаток растворяют в 1 мл дистилли-
рованной воды (или 10%-ного изопропанола, если раствор необ-
ходимо сохранять) и используют для определения аминокислот
хроматографическим методом.
После щелочного гидролиза определяется триптофан, аспарагин,
глутамин, аспарагиновая и глутаминовая кислоты, а также тиро-
зин и фенилаланин.
Рис. 17. Расположение ограничи-
тельных вырезов на бумаге для
хроматографии аминокислот:
а — зоны нанесения исследуемого рас-
твора; б — зоны нанесения стандарт-
ного раствора.
Хроматографирование аминокислот
Необходимо помнить, что получению компактных, хорошо отде-
ляемых пятен препятствуют примеси посторонних веществ, присут-
ствующие в исходном материале, реактивах и бумаге. Многие загряз-
нения, в особенности катионы тя-
желых металлов (железа, меди),
ускоряют разрушительные процес-
сы во время хроматографирования
и способствуют появлению «бород».
Поэтому бумагу и реактивы необхо-
димо очищать. Растворители очи-
щают перегонкой, а бумагу промы-
ванием кислотой. Важное значение
имеет также структура бумаги, ее
однородность и пористость.
Промытую кислотой хромато-
графическую бумагу марки «М»
или «С» с различием по толщине
около 2% разрезают на полосы дли-
ной 58 см и шириной 16,5 см. На
расстоянии 7 см от верхнего края
полосы проводят графитовым карандашом стартовую линию и разме-
чают на ней границы нанесения растворов исследуемых веществ.
Метки помещают от левого края полосы на таком расстоянии: 15 мм,
30 мм, 45 мм, 60 мм, 75 мм, 105 мм, 120 мм и 150 мм. Затем лез-
вием бритвы делают по этим меткам 8 вертикальных вырезов шири-
ной 2 мм и длиной 20 мм так, чтобы стартовая линия была посре-
дине (рис. 17). Для каждой камеры готовят две такие полосы.
105
После этого на стартовую линию наносят исследуемый и стан-
дартный растворы в следующих местах и количествах. Между 1-м
н 2-м вырезами, начиная от левого края, наносят в виде полоски
0,01 мл (2 раза по 0,005 мл), а между 5-м и 6-м вырезами 0,02 мл
(2 раза по 0,01 мл) испытуемого раствора. Между 3-м и 4-м вырезами
0,01 мл, а между 7-м и 8-м вырезами 0,02 мл стандартного раствора
аминокислот, отмечая номер стандарта на хроматограмме.
Таблица 2
Смеси растворителей для получения хроматограмм аминокислот
№ каме Растворители Соотношение
1 1 1 н-Бутанол — уксусная кислота — вода 5:1:2
2 Бутилацстат — ацетон — уксусная кис-
лота — вода 3 : 3 : 2 : 1
3 Пиридин — изоамиловый спирт— вода 4:1 1
4 Третичный бутанол — н-бутапол — вода 3:1 1
5 Изоамиловый спирт — я-бутапол — ук-
сусная кислота — вода 20 10:8:9
6 н-Пропанол — изоамиловый спирт— ук-
сусная кислота — вода G 6 2:3
7 н-Пропанол — этанол — пирофосфатный
буфер (pH 8,9) 2:1:1
8 н-Пропанол — ацетат натрия (2%-ный) 7 : 3
9 Вода — уксусная кислота 19: 1
Для анализа всех аминокислот готовят 9 пар бумажных полос,
нумеруя их по порядковому номеру стандартных смесей аминокис-
лот. Для седьмой пары берут бумагу, пропитанную пирофосфатным
буфером, а для восьмой пары — бумагу, пропитанную ацетатом нат-
рия. Для всех остальных хроматограмм используют бумагу, промы-
тую смесью щавелевой кислоты и оксихинолина. Один и тот же
исследуемый раствор наносят на все хроматограммы, а стандарт-
ные растворы наносят разные (только те, которые могут быть опре-
делены и делятся в данном растворителе). Состав аминокислот и их
содержание в стандартных растворах указаны в реактивах (см.
стр. 113). При этом номер стандартного раствора соответствует
номеру растворителя, в котором аминокислоты разделяются. После
нанесения растворов аминокислот на стартовую линию в промежут-
ках между ними наносят контрольные пятна индикаторов, по кото-
рым судят о движении аминокислот на хроматограмме.
Для хроматографирования полосы соединяют вверху по две с
помощью двух стеклянных палочек, скрепленных резиновыми коль-
цами. Верхний край скрепленных полос опускают в ванночку, а
полоски загибают по обе стороны ванночки в камеру. В ванночки
наливают по 40—50 мл растворителей, состав которых приведен
в табл. 2. Немного растворителя наливают на дно камеры, которую
106
затем плотно закрывают крышкой и оставляют на время, указанное
в описании получения хроматограмм.
П олучение хроматограммы 1. Смешивают 35 мл
«-бутанола с 7 мл уксусной кислоты и 14 мл дистиллированной во-
ды, размешивают, выливают в ванночку камеры № 1 и пропускают в
течение 48 ч, затем хроматограмму вынимают из камеры, высушива-
ют, помещают в камеру № 2 и снова пропускают в течение 48 ч
следующую смесь растворителей: бутилацетата — 21 мл, ацетона —
21 мл, уксусной кислоты — 14 мл и дистиллированной воды —
7 мл. Ориентиром служит пятно красителя — оранж-Ж, которое
должно опуститься до половины хроматограммы. Здесь отделяются
и определяются а-аланин, гликоколь, аргинин, гистидин, лизин и
цистин.
П олучение хроматограммы 2. Смешивают 21 мл
бути л ацетата, 21 мл ацетона, 14 мл уксусной кислоты и 7 мл ди-
стиллированной воды, размешивают, выливают в ванночку камеры
№ 2 и пропускают до тех пор, пока пятно метилоранжа окажется
на 20 см выше нижнего края хроматограммы. Здесь отделяются
фенилаланин, а-аминомасляная кислота, тирозин, а-аланин и
треонин.
Получение хр оматограммы 3. Смешивают 40 мл
пиридина, 10 мл изоамилового спирта и 10 мл дистиллированной воды,
выливают в ванночку камеры № 3 и пропускают до тех пор, пока
пятно оранжа-Ж опустится до нижнего края хроматограммы (около
48 ч). Здесь отделяются аминокислоты а-аминомасляная кислота,
треонин, а-аланин, серии и глутаминовая кислота.
Получение хр оматограммы 4. Смешивают 30 мл
третичного бутилового спирта, 10 мл «-бутанола и 10 мл дистил-
лированной воды, выливают в ванночку камеры № 4 и пропускают
до тех пор, пока пятно метилоранжа окажется на расстоянии 15 см
выше нижнего края хроматограммы. Здесь отделяются норлейцин,
лейцин, изолейцин и глутамин.
Получение хр оматограммы 5. Смешивают 20 мл
изоамилового спирта, 10 мл «-бутанола, 8 мл уксусной кислоты и 9 мл
дистиллированной воды, выливают в ванночку камеры № 5 и про-
пускают, пока пятно индикатора крезолрот опустится в самый
низ хроматограммы. Здесь отделяются фенилаланин, норвалин,
у-аминомасляная кислота и а-аланин.
Пол учение хр оматограммы 6. Смешивают 18 мл
«-пропанола, 18 мл изоамилового спирта, 6 мл уксусной кислоты и
9 мл дистиллированной воды, выливают в ванночку камеры № 6 и
пропускают до тех пор, пока пятно метилоранжа окажется на 6—8 см
выше нижнего края хроматограммы. Здесь отделяются валин, метио-
нин и цистин.
Получение хроматограммы 7. Смешивают 24 мл
«-пропанола, 12 мл этанола и 12 мл пирофосфатного буферного рас-
твора с pH 8,9, выливают в ванночку № 7 и пропускают до опуска-
ния пятна метилоранжа в самый низ хроматограммы. В данном
107
случае хроматографирование проводится на бумаге предварительно
пропитанной пирофосфатным буферным раствором и затем высушен-
ной. Здесь отделяются пролин, р-аланин, аспарагиновая кислота.
Получение хроматограммы 8. Смешивают 35 мл
к-пропанола и 15 мл 2%-ного раствора уксуснокислого натрия,
выливают в ванночку камеры № 8 и пропускают до тех пор, пока
пятно метилоранжа окажется в самом низу хроматограммы (около
48 ч), Хроматографирование проводят на бумаге предварительно
пропитанной 2%-ным раствором уксуснокислого натрия и высу-
шенной. Здесь отделяются пролин, аспарагин, аспарагиновая и глу-
таминовая кислота.
Получение хр оматограммы 9. Смешивают 57 мл
дистиллированной воды и 3 мл уксусной кислоты, выливают в ван-
ночку камеры № 9 и пропускают до тех пор, пока пятно метилоран-
жа окажется на 10 см выше нижнего края хроматограммы (около
5 ч). Здесь отделяется триптофан.
Получение хроматограммы 10. Пропускают сна-
чала первый растворитель: я-бутанол — уксусная кислота — вода
(30 6 12), смесь выливают в ванночку камеры № 1 и пропускают
в течение 48 ч. Затем, после высушивания, хроматограмму опрыски-
вают из пульверизатора боратным буферным раствором с pH 8,0
до равномерного впитывания раствора, избегая чрезмерного увлаж-
нения. После высушивания пропускают в камере № 8 второй рас-
творитель: 35 мл w-пропанола и 15 мл боратного буферного раствора
с pH 8,0. Второй растворитель пропускают до тех пор, пока пятно
метилоранжа опустится в самый низ хроматограммы (около 40 ч).
Здесь отделяются у-аминомасляная кислота, метионин, а-аланин,
треонин, аргинин, лизин, аспарагиновая кислота и цистин.
Получение хроматограммы 11. Пропускают сна-
чала первый растворитель: к-бутанол — уксусная кислота — вода
(30 6 12) в камере № 1 в течение 40 ч, затем после высушивания
хроматограмму помещают в камеру № 2 и пропускают в течение
40 ч второй растворитель: бутилацетат — ацетон — уксусная кис-
лота — вода (21 21 14 7). После этого хроматограмму высу-
шивают и опрыскивают из пульверизатора равномерным слоем бо-
ратного буферного раствора с pH 8,0, избегая чрезмерного увлаж-
нения. Хроматограмму высушивают, помещают в камеру № 8 и
пропускают в течение 40 ч третий растворитель: ^-пропанол —
боратный буфер с pH 8 (35 15). Ориентиром служит пятно оран-
жа-Ж, которое в конце хроматографирования должно находиться на
20 см выше нижнего края хроматограммы. Здесь хорошо отделяют-
ся метионин, треонин, валин, гистидин, аргинин, лизин, аспараги-
новая кислота и дистин.
Получение хроматограммы 12. Смешивают 30 мл
третичного бутанола, 10 мл «-бутанола и 10 мл 2%-ного раствора
ацетата натрия, вылирают в ванночку камеры № 4 и пропускают
до тех пор, пока пятно метилоранжа опустится в самый низ хрома-
тограммы (около 7 суток). Хроматографирование проводится на
108
бумаге, пропитанной 2%-ным раствором ацетата натрия. Здесь
отделяются норлейцин, лейцин, метионин, тирозин и а-аминомасля-
ная кислота.
Получение хроматограммы 13. Смешивают 36 мл
н-бутанола, 15 мл уксусной кислоты и 6 мл дистиллированной воды,
выливают в ванночку камеры № 1 и пропускают до тех пор, пока
пятно метилоранжа опустится до половины хроматограммы (около
60 ч). Здесь отделяются метионин, а-аминомасляная кислота, трео-
нин, глутаминовая кислота, глутамин, цистин.
Получение хроматограммы 14. Смешивают 21 мл
изопропанола, 21 мл бутилацетата, 12 мл уксусной кислоты и 6 мл
дистиллированной воды, выливают в ванночку и пропускают до тех
пор, пока пятно метилоранжа опустится в самый низ хроматограм-
мы. Здесь отделяются изолейцин, а-аминомасляная кислота,
у-аминомасляная кислота и а-аланин.
Для определения 8 незаменимых аминокислот необходимо полу-
чить следующие 5 хроматограмм: 4, 5, 6, 9 и 10. После хромато-
графирования полоски бумаги вынимают из камер, подвешивают за
края палочек в вытяжном шкафу и высушивают при комнатной тем-
пературе. В дальнейшем анализ проводят различными, приведен-
ными ниже методами.
Колориметрический метод
с использованием соединений меди
Высушенные хроматограммы опрыскивают 0,5%-ным раствором
нингидрина в ацетоне и оставляют на 16—20 ч при комнатной темпе-
ратуре для полного развития окраски. Затем окрашенные участки,
занятые аминокислотами, обводят карандашом, нумеруют и от-
мечают название аминокислоты и количество нанесенного на хро-
матограмму раствора. Обведенные пятна вырезают, складывают гар-
мошкой, сворачивают и помещают в сухую пробирку, в которую
приливают 5 мл 0,02%-ного раствора сернокислой меди в 50%-ном
этиловом спирте. Настаивают при периодическом взбалтывании до
тех пор, пока будет извлечено все окрашенное вещество и бумага
станет белой (требуется около 60 мин). После этого окрашенный в
оранжево-красный цвет раствор переносят в кювету на 10 мм и
фотометрируют при 490—510 нм. Результаты измерений оптической
плотности подставляют в приведенную формулу и вычисляют содер-
жание аминного азота в объеме раствора, нанесенного на хромато-
грамму:
/у [ у \ __ (К1 Къ) [(^б ”Ь *М) (Кб + Км)] [ / ТУ I ту \
(Аг + Х2)-------------(K7=KJ------------ + +
где Xi + Х2 — количество аминного азота в большом и малом пятне
исследуемого вещества (в мкг); — количество аминного азота
в большом пятне стандарта (в мкг); К2 — количество аминного
азота в малом пятне стандарта (в мкг); Кб — оптическая плот-
ность раствора, полученного из большого пятна стандарта; Км —
109
о тическая плотность раствора, полученного из малого пятна стан-
дарта; Х6 — оптическая плотность раствора, полученного из боль-
шого пятна исследуемого вещества; Хм — оптическая плотность рас-
твора, полученного из малого пятна исследуемого вещества.
Можно вместо количества аминного азота в Ki и К2 дать соот-
ветствующее количество аминокислоты, тогда и результаты будут
выражаться в весовых единицах аминокислоты.
Колориметрический нингидриновый метод
Этим способом можно определить почти все аминокислоты. Кро-
ме тех, что приведены в предыдущем методе, еще и следующие:
аспарагин, у-аминомасляную кислоту, аспарагиновую кислоту,
гистидин, гликоколь, глутамин, метионин, триптофан и цистин.
Определению мешает аммиак, который обычно присутствует в бу-
маге и должен быть удален до извлечения аминокислот. Чтобы уста-
новить расположение аминокислот на хроматограмме, ее проявляют
не полностью, а только до слабой окраски.
При анализе этим способом высушенные хроматограммы опрыс-
кивают 0,02%-пым раствором нингидрина и оставляют до проявле-
ния аминокислот (лучше на ночь). Контуры пятен обводят каран-
дашом, нумеруют, отмечают названия аминокислот и нанесенные
количества раствора, а для стандарта и весовое количество. Затем
хроматограммы опрыскивают 0,01 н. раствором КОН, высуши-
вают, помещают в эксикатор, на дне которого находится концентри-
рованная серная кислота и оставляют на ночь для полного удаления
аммиака. После этого пятна вырезают, складывают в виде гармошки,
сворачивают, помещают в сухие пробирки, приливают по 2 мл
безаммиачной воды и настаивают при периодическом взбалтывании
10 мин. Затем добавляют 3 мл нингидринового реактива и настаива-
ют при взбалтывании еще 5 мин. Растворы сливают в сухие про-
бирки, прибавляют по 0,1 мл 1 %-пого раствора аскорбиновой кисло-
ты, закрывают пробирки шариковыми пробками и ставят в кипя-
щую воду на 15 мин. Одновременно нагревают контрольную пробу,
приготовленную из такого же участка неокрашенной бумаги верх-
ней части хроматограммы. После охлаждения на воздухе при перио-
дическом взбалтывании растворы фотометрируют при 580 нм в кю-
вете на 10 мм. сравнивая с контрольным раствором. Результаты из-
мерений подставляют в приведенную выше формулу и вычисляют
содержание аминного азота в нанесенном на хроматограмму объеме
исследуемого раствора.
Определение по весу бумаги,
занятой пятном
Для этого метода пригодны только те виды бумаги, которые
имеют хорошую однородную структуру с отклонением веса едини-
цы площади бумаги, не превышающим 2% (Ватман, Шлейхер и
ПО
Шюлль, некоторые сорта из ГДР). Этим методом можно определять
не только аминокислоты, но и сахара, органические кислоты и дру-
гие вещества, проявленные любым способом. Аминокислоты можно
проявлять не только нингидрином, но и изатином, который для
некоторых аминокислот — пролина, тирозина, гистидина, аргини-
на, фенилаланина, триптофана, цистина — является более чувст-
вительным реактивом, чем нингидрин, и дает другую окраску пятен.
Избыток изатина с бумаги удаляют водой.
Хроматограммы после хроматографирования и высушивания
опрыскивают 0,5%-ным раствором нингидрина или 0,2%-ным раство-
ром изатина. Высушенные хроматограммы, обработанные нингид-
рином, оставляют на 10—20 ч при комнатной температуре для раз-
вития окраски, а хроматограммы, обработанные изатином, поме-
щают в сушильный шкаф и выдерживают 10 мин при температуре
100° С. После этого хроматограмму промывают дистиллированной
водой до удаления желтой окраски, обусловленной избытком иза-
тина, и снова высушивают.
Проявленную сухую хроматограмму помещают на освещенное сни-
зу стекло и обводят контуры пятен карандашом. Пятна нумеруют,
регистрируют, вырезают и взвешивают на аналитических весах.
По полученному весу вычисляют содержание аминокислот в пятнах,
подставляя его в приведенную выше формулу вместо оптической
плотности. Вместо Хб и Хи — вес большого и малого пятна, полу-
ченных из исследуемого раствора, а вместо /<б и /(м — вес боль-
шого и малого пятна, полученных из стандартного раствора той же
аминокислоты.
В дальнейшем расчет можно вести на содержание исследуемой
аминокислоты (в мг%), па содержание азота (в % к общему коли-
честву аминного азота), на содержание белка (в % к общему коли-
честву белков).
Для окончательного расчета содержания аминокислот в иссле-
дуемом материале полученные результаты (в мкг аминного азота)
м
пересчитывают на содержание аминокислоты умножением на ,
а мкг переводят в мг и на 100 г вещества, пользуясь следующей фор-
мулой:
у_ (Xi + A) М а 50 100 0,595 (Xj+ Х2) М а
Y ~ N п 0,03 20 1000 “ п
где Y —содержание аминокислоты в исследуемом веществе
(в мг%); М — молекулярный вес аминокислоты; N — атомный вес
азота; Х± + Х2 — количество аминного азота, найденное в большом
и малом пятне (в мкг); а — объем исследуемого раствора, получен-
ный после растворения остатка выпаренного экстракта (в мл);
0,03 — объем исследуемого раствора, нанесенный на хроматограм-
му в большом и малом пятне (в мл); 50 — общий объем экстракта
из навески (в мл); 20 — объем экстракта, взятый для выпаривания
(в мл); п — навеска исследуемого вещества (в г).
111
Для расчета содержания азота аминокислоты (в % к общему
количеству аминного азота) необходимо определить содержание его
в исследуемом растворе по изложенному выше методу (см. стр. 85)
и вычисляют количество его в объеме раствора, нанесенного на
хроматограмму. Это количество принимают за 100 и к нему относят
найденное количество аминокислоты на хроматограмме:
у _ (Х^х2) 100
где Y± — содержание аминокислоты (в % аминного азота); А —
общее содержание аминного азота в нанесенном на хроматограмму
объеме исследуемого раствора (0,03 мл) (в мкг).
Остальные обозначения такие же, как и в приведенной выше
формуле.
Для расчета процентного содержания аминокислоты в данном
препарате белка найденное количество аминного азота пересчиты-
М
вают на аминокислоту умножением на , полученный результат
переводят в мг и выражают (в % к исследуемому белку):
У _ (Xjl + XJ м а 100 (Xj + X2) М а
Y 2 14 /г -0,03 1000 ’ 4,2 п
где Y2 — содержание аминокислоты в белке (в %); п — навеска
белка, взятая для гидролиза (в мг).
Остальные обозначения такие, как в формуле на стр. 111.
Промывание бумаги
Для промывания бумаги приготовляют 1%-ный раствор щавеле-
вой кислоты, содержащий 0,1% о-оксихинолина (10 г щавелевой
кислоты и 1 г о-оксихинолина на 1 л раствора). В ванночку под-
ходящего размера помещают 4—5 листов хроматографической бума-
ги, заливают 1 л промывного раствора и выдерживают 1 ч при перио-
дическом покачивании ванночки. Затем раствор сливают, бумагу
заливают таким же количеством воды и выдерживают при покачи-
вании ванночки 10 мин. Промывают водой до удаления кислоты.
После 7-го промывания делают пробу с метилоранжем: в один ста-
кан набирают промывную жидкость, в другой — воду и прибавляют
по одной капле раствор метилоранжа. Промывают до одинаковой
окраски.
После промывания один конец бумаги зажимают зажимом-
прищепкой и подвешивают для высыхания. Бумагу можно оставить
и в ванночке в наклонном положении до полного высыхания. Не-
сколько листов промытой бумаги пропитывают пирофосфатным бу-
ферным раствором и несколько листов — 2%-ным раствором ук-
суснокислого натрия (для 7 и 8 хроматограмм).
Реактивы. Раствор уксуснокислого натрия (2%-ный). Отвешивают 20 г
CH3COONa ЗН2О и 1 г трилона Б, растворяют в дистиллированной воде, дово-
дят объем водой до 1 л и перемешивают.
112
Пирофосфатный буферный раствор (pH 8,9). Отвешивают 22,3 г Na4P2O7 •
ЮН2О, 6,6 г NaCl, растворяют в дистиллированной воде, добавляют 5,5 мл
1 н. раствора соляной кислоты и разбавляют водой до 1 л. Сохраняют в хорошо
закрытой склянке.
Боратный буферный раствор (pH 8,0). Отвешивают 3,65 г борной кислоты,
3,65 г хлористого калия и 0,15 г трилона Б, растворяют в дистиллированной воде,
добавляют 50 мл 0,1 н. раствора едкого натра, разбавляют водой до 500 мл, пере-
мешивают и сохраняют в хорошо закрытой склянке.
Раствор сернокислой меди (0,02%-ный). Отвешивают 0,04 г CuSO4 5Н2О,
растворяют в 100 мл дистиллированной воды, добавляют 100 мл 95%-ного эти-
лового спирта и перемешивают. Раствор служит для извлечения из бумаги окра-
шенного соединения аминокислоты с нингидрином.
Раствор едкого калия в этаноле (0,01 н.). Берут 10 мл 0,1 н. раствора КОН,
выливают в мерную колбу на 100 мл, доливают до метки 95%-ного этилового
спирта и перемешивают.
Раствор нингидрина (0,5%-ный). Отвешивают 0,5 г нингидрина, растворяют
в 100 мл ацетона, прибавляют 4 мл ледяной уксусной кислоты, перемешивают
и сохраняют в темной склянке. Нингидрин можно растворить также в смеси аце-
тона и этилового спирта (1 1). Раствор служит для проявления аминокислот
на хроматограммах.
Раствор нингидрина (0,02%-ный). Отвешивают 0,04 г нингидрина, раство-
ряют в 100 мл 95%-ного этилового спирта, прибавляют 8 мл ледяной уксус-
ной кислоты, добавляют 100 мл ацетона, перемешивают и сохраняют в темной
хорошо закрытой склянке. Раствор служит для частичного проявления ами-
нокислот.
Раствор изатина (0,2%-ный). Отвешивают 0,2 г изатина, растворяют в 100 мл
этилового или н-бутилового спирта, прибавляют 4 мл ледяной уксусной кислоты,
перемешивают и сохраняют в хорошо закрытой склянке. Раствор служит для
проявления аминокислот на хроматограмме и в первую очередь пролина.
Растворители: ацетон, бутилацетат, н-бутанол, третичный бутанол, изоами-
ловый спирт, изопропанол, н-пропанол, пиридин и этанол должны быть очищены
перегонкой. Для этого в колбу Вюрца наливают 500 мл растворителя, прибавляют
1 мл 4 н. раствора серной кислоты, несколько стеклянных бусинок или капилля-
ров, чтобы избежать толчков, и отгоняют (под вытяжным шкафом) в дистилля-
ционном аппарате до небольшого остатка (15—20 мл). Отогнанные растворы со-
храняют в хорошо закрытой склянке.
Приготовление стандартных растворов аминокислот. Для каждой хромато-
граммы готовится свой стандартный раствор, состоящий из смеси аминокислот,
которые делятся в данном растворителе. В 1 мл стандартного раствора содержится
по 100 мкг аминного азота каждой аминокислоты. Ниже приведены весовые ко-
личества аминокислот для 14 стандартных смесей, которые помещаются в мерные
колбы на 25 мл и растворяются в 10%-ном изопропиловом спирте. К смесям, в со-
став которых входит цистин или тирозин (№ 1, 2, 6, 10, 11 и 12), сначала
прибавляют 0,4 мл соляной кислоты (1 ^1), а затем растворяют в 10%-ном изо-
пропаноле. Номера стандартных растворов соответствуют номерам хроматограмм,
на которые они наносятся: Вес, мг № смеси Аминокислота Вес, мг
№ смеси Аминокислота
1 а-Аланин 15,9 а-Аланин 15,9
Гликоколь 13,4 Треонин 21,2
Аргинин 37,7
Гистидин 34,3 3 а-Аминомасля-
Лизин 32,7 ная кислота 18,4
Цистин 42,9 Треонин 21,2
2 Фенилаланин 28,9 а-Аланин 15,9
а-Аминомасля- Серин 18,8
ная кислота 18,4 Глутаминовая
Тирозин 32,3 кислота 26,3
8:6-2
113
№ смес Аминокислота Вес, мг № смеси Аминокислота Вес, мг
4 Норлейцин 23,4 Лизин 32.7
Лейцин 23,4 Аспарагиновая
Изолейцин 23,4 кислота 23,8
Глутамин 26,1 Цистин 42,9
5 28,9 11 Метионин 26,6
Фенилаланин Треонин 21,2
Норвалин 20,9 Валин 20,9
у-Аминомасля- 18.4 Г истидин 34,3
ная кислота Аргинин 37,7
а-Аланин 15,9 Лизин Аспарагиновая 32,7
20,9 кислота 23,8
6 Валин Цистин 42,9
Метионин 26,6
Цистин 42,9 12 Норлейцин 23,4
Лейцин 23,4
Метионин 26,6
7 Пролин 20,6 Тирозин 32.3
Р-Аланин 15,9 а-Аминомасля-
Аспарагиновая ная кислота 18,4
кислота 23,8 13 Метионин 26,6
8 Аспарагин 23,6 а-Аминомасля- ная кислота 18,4
Пролин 20,6 Треонин Глутаминовая 21,2
кислота 26,3
9 Триптофан 36,4 Глутамин 26,1
10 у-Аминомасля- 14 Изолейцин 23,4
ная кислота 18,4 а-Аминомасля-
Метионин 26,6 ная кислота 18,4
а-Аланин 15,9 у-Аминомасля-
Треонин 21,2 ная кислота 18,4
Аргинин 37,7 а-Аланин 15,9
ЛИТЕРАТУРА
Андреева Т. Ф., Осипова О. П. Количественное определение свобод-
ных и связанных аминокислот листьев при помощи хроматографии на бу-
маге.— В кн.: Методика количественной бумажной хроматографии сахаров,
органических кислот и аминокислот у растений. М.—Л., Изд-во АН СССР,
1962, 59.
Б а й л и Дж. Методы химии белков. М., «Мир», 1965.
Ермаков А. И., Арасимович В. В., Смирнова-Иконнико-
ва М. И., М у р р и И. К- Методы биохимического исследования растений.
Л., «Колос», 1972.
Зайцева Г Н., Тюленева Н. Н. Количественное определение амино-
кислот на хроматограммах посредством образования медных производных с
нингидрином.— Лабор. дело, 1958, 3, 24.
Пасхина Т. С. Количественное определение аминокислот при помощи хро-
матографии на бумаге.— В кн.: Современные методы в биохимии, 1. М., «Ме-
дицина», 1964, 162.
Починок X. Н., Берштейн Б. И. Колориметрический нингидриновый
метод определения аминного азота в растениях.— Укр. 6ioxiM. журн.,
1956, 28, 3, 354.
114
Починок X. Н. Определение аммиачного и амидного азота и аргинина в
растениях тимол-гипобромитным методом.— В кн.: Труды первой респуб-
ликанской научной конференции физиологов и биохимиков Молдавии. Ки-
шинев, «Картя Молдовеняска». 1964, 117.
ПочинокХ. Н. Микрометод определения общего и белкового азота в растени-
ях и общего азота в почве хлораминным методом.— В кп.: Питание и удобре-
ние растений. Киев, «Наукова думка», 1965, 202.
ПочинокХ. Н. Колориметрический метод определения нитратов в растениях
и почве.— Питание и удобрение растений. Киев, «Наукова думка», 1965,
211.
Починок X. Н. Определение в растениях аминного азота нингидриновым
реактивом нового состава.— Физиол. и биохим. культурных растений, 1972,
4, 1, 101.
Смирнова-Иконникова М. И., Веселова Е. П. Фракционный
состав белков семян зерновых бобовых культур.— ДАН СССР, 1951, 77,
6, 1071.
М о о г е S., S t е i n W. Н. A modified ninhydrin reagent for the photometric
determination of amino acide and related compounds.— J. Biol. Chem., 1954,
211, 907.
Глава III
УГЛЕВОДЫ
ОПРЕДЕЛЕНИЕ ГЛЮКОЗЫ, ФРУКТОЗЫ И САХАРОЗЫ
В РАСТЕНИЯХ ИЗ ОДНОЙ НАВЕСКИ
Сущность метода. После извлечения сахаров водой и
осаждения белков в фильтрате определяют сумму восстанавливаю-
щих сахаров—глюкозу, фруктозу и мальтозу йодометрическим
методом. Эти сахара в щелочной среде при нагревании восстанавли-
вают двухвалентную медь до одновалентной, которая выпадает в виде
Си2О.
Полной пропорциональности между количеством сахаров и за-
кисью меди не наблюдается. С увеличением количества сахара за-
киси меди выделяется больше, чем следует по реакции. Поэтому
нельзя пользоваться одним коэффициентом для пересчета восста-
новленной меди на сахар. Для этой цели может служить эмпириче-
ская формула, выведенная из экспериментальных данных.
Образовавшуюся закись меди определяют йодометрическим мето-
дом. Для этого меднощелочный реактив на сахара содержит KJO3
и KJ, которые в щелочной среде не реагируют между собой и не
мешают реакции ионов меди с сахаром. При подкислении раствора
выделяется йод:
KJO3 + 5KJ + 3H2SO4 = 3J2 + 3K2SO4 + 3H2O,
который в присутствии щавелевой кислоты вступает в реакцию с
закисью меди:
Cu2O Н2С2О4 = СиС2О4 CuJ2 Ц- Н2О.
Избыток йода титруют 0,01 н. раствором тиосульфата натрия в
присутствии крахмала:
2Na2S2O3 + J, = Na2S4O6 + 2NaJ.
В части исследуемого раствора после проведения гидролиза са-
харозы щавелевой кислотой
^12^22^11 + Н2О = 2С6Н12Об
определяют сумму сахаров и по разности сахарозу.
В другой части исследуемого раствора определяют фруктозу
после окисления глюкозы йодом в щелочной среде:
RCOH + J2 + 3NaOH = RCOONa + 2NaJ + 2H2O.
Наиболее полное окисление глюкозы происходит тогда, когда
нормальность раствора щелочи в 1,6 раза больше, чем йода, т. е.
116
когда соотношение между концентрациями йода и щелочи близко к
соотношению в приведенном выше уравнении.
Йод, оставшийся в растворе после окисления глюкозы, частично
разрушает фруктозу. Чем меньше содержание глюкозы, тем больше
йода остается в растворе, тем сильнее воздействие его на фруктозу
и наоборот. Зная содержание глюкозы, можно вводить поправку
с учетом разрушающего действия йода. С увеличением количества
фруктозы на единицу сахара образуется больше закиси меди. Учи-
тывая эти факторы, на основании экспериментальных данных по-
лучена эмпирическая формула, с помощью которой по образовав-
шейся закиси меди можно довольно точно вычислить содержание
фруктозы в присутствии различного количества глюкозы.
Ход анализа. Берут навеску исследуемого вещества с та-
ким расчетом, чтобы в ней было не больше 50 мг моносахаридов и
100 мг суммы сахаров (корня сахарной свеклы в зрелом состоянии
0,5 г, листьев сахарной свеклы, картофеля и пшеницы весной 3—
5 г, озимой пшеницы осенью 1—2 г), насыпают в стеклянный или
алюминиевый бюкс, закрывают крышкой и помещают в пары кипя-
щей воды на 5—10 мин для инактивации ферментов. Для этого над
кипящей водой укрепляют металлическую сетку, на которую и ста-
вят бюкс. Сосуд с кипящей водой на время инактивирования фер-
ментов закрывают крышкой. После этого пробу переносят в ступку,
растирают с небольшим количеством дистиллированной воды и коли-
чественно переносят в мерную колбу на 100 мл. Для осаждения белков
и других мешающих веществ прибавляют на каждый грамм сырой
навески по 1 мл 1 н. раствора едкого натра и по 1,2 мл 10%-ного
раствора сернокислого цинка. Если окраска исследуемого раствора
не препятствует наблюдению перехода цвета индикатора, то после
добавки необходимого количества 1 н. раствора NaOH, прибав-
ляют такой же объем 10%-ного раствора сернокислого цинка,
1—2 капли раствора фенолфталеина и добавляют по каплям при
взбалтывании раствор сернокислого цинка до обесцвечивания фе-
нолфталеина. После этого раствор в колбе доводят дистиллированной
водой до метки, тщательно перемешивают и фильтруют в сухую
колбу через сухой фильтр, выбрасывая первую порцию фильтрата.
При исследовании объектов, содержащих большое количество
крахмала (например, клубни картофеля), навеску не обрабатывают
паром, а растирают с 1 мл 1 н. раствора Na2CO3 и при осаждении
белков прибавляют на 1 мл 1 н. раствора едкого натра меньше.
В фильтрате определяют: восстанавливающие сахара (глюкозу,
фруктозу и мальтозу), сумму сахаров (моно- и дисахаридов).
Определение восстанавливающих сахаров
В пробирки для определения сахаров диаметром 30 мм и высо-
той 22 см, помещенные в алюминиевый штатив (см. рис. 11), налива-
ют пипеткой по 10 мл фильтрата, прибавляют по 10 мл медно-щелоч-
ного реактива, содержащего KJ и KJO3, и растворы перемешивают.
117
Пробирки закрывают воронками или шариковыми пробками, шта-
тив погружают в кипящую воду, отмечая время начала нагревания.
При. нагревании образуется осадок закиси меди красного цвета.
Через 15 мин после начала нагревания штатив с пробирками выни-
мают и охлаждают погружением в холодную воду. Количество осад-
Т а б л и ц а 3
Значения факторов для вычисления содержания сахаров
[248 — (а — &)] (а — Ь)
1000
а—Ь, мл Десятые доли миллилитров
0 | 0,1 0.2 0.3 0,4 | 0,5 | 1 0,6 | 1 | | 0.8 | 0,9
0 0,02 0,05 0,07 0,10 0,12 0,15 0,17 0,20 0,22
1 0,25 0,27 0,30 0,32 0,35 0,37 0,39 0,42 0,44 0,47
2 0,49 0,52 0,54 0,57 0,59 0,61 0,64 0,66 0,69 0,71
3 0,74 0,76 0,78 0,81 0,83 0,86 0,88 0,90 0,93 0,95
4 0,98 1,00 1,02 1,05 1,07 1,10 1,12 1,14 1,17 1,19
5 1,22 1,24 1,26 1,29 1,31 1,33 1,36 1,38 1,40 1,43
6 1,46 1,48 1,50 1,52 1,55 1,57 1,59 1,62 1,64 1,66
7 1,69 1,71 1,73 1,76 1,78 1,80 1,83 1,85 1,87 1,90
8 1,92 1,94 1,97 1,99 2.01 2,04 2,06 2,08 2,10 2,13
9 2,15 2,17 2,20 2,22 2,24 2,27 2,29 2,31 2,33 2,36
10 2,38 2,40 2,43 2,45 2,47 2,49 2,52 2,54 2,56 2,58
И 2,61 2,63 2,65 2,68 2,70 2,72 2,74 2,77 2,79 2,81
12 2,83 2,85 2,88 2,90 2,92 2,94 2,97 2,99 3,01 3,03
13 3,06 3,08 3,10 3,12 3,14 3,17 3,19 3,21 3,23 3,25
14 3,28 3,30 3,32 3,34 3,36 3,39 3,41 3,43 3,45 3,47
15 3,50 3,52 3,54 3,56 3,58 3,60 3,63 3,65 3,67 3,69
16 3,71 3,73 3,76 3,78 3,80 3,82 3,84 3,86 3,88 3,91
17 3,93 3,95 3,97 3,99 4,01 4,03 4,06 4,08 4,10 4,12
18 4,14 4,16 4,18 4,20 4,23 4,25 4,27 4,29 4,31 4,33
19 4,35 4,37 4,39 4,41 4,44 4,46 4,48 4,50 4,52 4,54
20 4,56 4,58 4,60 4,62 4,64 4,66 4,68 4,71 4,73 4,75
ка закиси меди определяют йодометрическим методом. Для этого
в пробирку прибавляют 5 мл смеси щавелевой и серной кислот
(прибавлять смесь кислот нужно постепенно при взбалтывании).
Вначале происходит нейтрализация раствора, затем выделяется йод,
который в присутствии щавелевой кислоты вступает в реакцию с за-
кисью меди. Когда вся закись меди растворится, остаток йода тит-
руют 0,01 н. раствором тиосульфата натрия в присутствии 0,5 мл
0,5%-ного раствора крахмала.
Параллельно проводится контрольный опыт без нагревания, для
которого берут 10 мл медно-щелочного реактива, прибавляют 5 мл
смеси щавелевой и серной кислот и 10 мл исследуемого раствора.
Выделившийся йод титруют 0,01 н. раствором тиосульфата натрия
в присутствии крахмала до первого исчезновения синей окраски.
Иногда синяя окраска через некоторое время появляется вновь.
На это не обращают внимания и дальше не титруют, так как появ-
ление окраски вызвано окислением ионов йода кислородом воздуха.
118
Из результатов титрования вычисляют содержание восстанавли-
вающих сахаров по следующей формуле:
х = А [248 — (д — 6)] (а — Ь) 0,1 A f
v п 10 000 v п
где X—количество восстанавливающих сахаров (в %); А—общий
объем исследуемого раствора (в мл); v — объем исследуемого раство-
ра, взятый для анализа (в мл); а — объем 0,01 н. раствора тиосуль-
фата натрия, затраченный при титровании контрольного раствора
(в мл); b—объем 0,01 н. раствора тиосульфата натрия, затрачен-
ный при титровании исследуемого раствора (в мл); п — навеска
исследуемого вещества (в г); [248 — (а — Ь)] — количество инверт-
ного сахара, соответствующее 1 мл 0,01 н. раствора тиосульфата
натрия (в мкг); 10000 — коэффициент для перевода микрограммов
сахара в граммы и пересчета результата в проценты; f — фактор,
равный --------—----------- , величины фактора приведены в
табл. 3.
Определение фруктозы
25 мл исследуемого раствора отбирают пипеткой в мерную колбу
на 50 мл, прибавляют одну каплю 0,1 %-ного раствора фенолфталеи-
на и нейтрализуют 0,4 н. раствором едкого натра, прибавляя его по
каплям до появления розовой окраски. Затем прибавляют 5 мл
0,05 н. раствора йода и 1 мл 0,4 н. раствора едкого натра, перемеши-
вают и оставляют па 10 мин. При этом происходит окисление глю-
козы йодом по приведенному выше уравнению. Через 10 мин при-
бавляют 1 мл 0,4 н. раствора соляной кислоты и выделившийся йод
обесцвечивают свежеприготовленным 0,05 н. раствором сульфита
натрия, приливая его из бюретки до соломенно-желтого цвета рас-
твора. Под конец добавляют 4 капли раствора крахмала и титруют
до исчезновения синей окраски. Реакция идет по следующему урав-
нению:
J2 + Na2SO3 + Н2О = Na2SO4 + 2HJ.
Так как реакция идет с образованием сильной йодистоводо-
родной кислоты, нужно сразу же нейтрализовать раствор щелочью,
чтобы избежать гидролиза дисахаров. Для этого после обесцвечи-
вания синей окраски прибавляют одну каплю раствора фенолфталеи-
на и нейтрализуют 0,4 н. раствором едкого натра. Объем раствора,
доводят дистиллированной водой до 50 мл и тщательно перемеши-
вают.
Для определения фруктозы отбирают 20 мл полученного раствора
выливают в пробирку для определения сахаров, прибавляют 10 мл
медно-щелочного реактива и перемешивают. Раствор нагревают в
кипящей водяной бане 15 мин. Дальше поступают так же, как опи-
сано выше при определении восстанавливающих сахаров. Из, полу-
119
ченных результатов вычисляют содержание руктозы по следующей
формуле:
I 220 'I
2 А [99 (а — с) — (а — Ь)] 259 — (а — с) Ч-
уг _ I CL — С I
Л v п 1 000 000
где X — содержание фруктозы (в %); А — общий объем исследуе-
мого раствора (в мл); v — объем раствора, взятый в пробирку для
определения фруктозы (в мл); 2 — фактор разбавления раствора
после окисления глюкозы йодом (50/25); а — объем 0,01 н. раство-
ра тиосульфата натрия, затраченный при титровании контроль-
ного раствора (в мл); b— объем 0,01 н. раствора тиосульфата нат-
рия, затраченный при определении восстанавливающих сахаров
(в мл); с — объем 0,01 н. раствора тиосульфата натрия, затрачен-
ный при определении фруктозы (в мл); п — навеска исследуемого
вещества (в г); 1000 000 — коэффициент перевода микрограммов в
граммы и проценты.
Из полученных результатов определения восстанавливающих
сахаров и фруктозы можно вычислить содержание глюкозы, если
отсутствует мальтоза, по формуле:
Г - В — Ф,
где Г — количество глюкозы (в %); В — количество восстанавли-
вающих сахаров (в %); Ф — количество фруктозы (в %).
Определение суммы сахаров
Из остатка исследуемого раствора, в котором определялись
восстанавливающие сахара, набирают пипеткой 25 мл, помещают в
мерную колбу на 50 мл, прибавляют 2 мл 8%-ного раствора щаве-
левой кислоты, погружают колбу в кипящую воду и выдерживают в
течение 10 мин при температуре кипения воды (при анализе кор-
неплодов сахарной свеклы время гидролиза увеличивается до
15 мин). При кислотном гидролизе сахарозы образуется глюкоза и
фруктоза, которые определяются как восстанавливающие сахара.
После гидролиза раствор охлаждают, прибавляют каплю раствора
фенолфталеина, нейтрализуют 1 н. раствором едкого натра до сла-
бо-розовой окраски раствора. При нейтрализации нескольких
образцов сначала определяют необходимый объем на одном образце.
Затем прибавляют в остальные щелочи на 0,2 мл меньше и после
перемешивания нейтрализуют 0,4 н. раствором едкого натра до
появления розовой окраски. Нейтрализованный раствор доводят
дистиллированной водой до метки и тщательно перемешивают. От-
бирают 10 мл полученного раствора в пробирку для определения са-
харов, прибавляют 10 мл медно-щелочного реактива и далее по-
ступают так, как описано при определении восстанавливающих саха-
ров. Из полученных данных вычисляют сумму сахаров по формуле:
х_ 2 Л[248 — (а — 6)] (а — Ь) = 0,2 A f
v • п 10 ООО “ v п
120
где X — содержание суммы сахаров в расчете на глюкозу (в %);
А — общий объем исследуемого раствора (в мл); v — объем раствора,
взятый для определения суммы сахаров (в мл); 2------------фактор
разбавления раствора после гидролиза; а — объем 0,01 н. раствора
тиосульфата натрия, затраченный при титровании контрольного
раствора (в мл); b — объем 0,01 н. раствора тиосульфата натрия,
затраченный при определении суммы сахаров (в мл); f — фактор,
„ [248 — (а — 6)] (а — Ь)
равный -------~1ооо—1> величины которого приведены в
табл. 3; п — навеска исследуемого вещества (в г).
Количество сахарозы рассчитывают по формуле:
X = 0,95 (С —В),
где X — содержание сахарозы в исследуемом веществе (в %); С —
содержание суммы сахаров в расчете на глюкозу (в %); В — содержа-
ние восстанавливающих сахаров в расчете на глюкозу (в %).
Реактивы. Медно-щелочный реактив. 75 г безводного карбоната натрия
Na2CO3, растворяют в 400 мл дистиллированной воды в колбе на 1 4. В другой
колбе растворяют в 400 мл дистиллированной воды 12 г сернокислой меди
CuSO4 5Н2О и 21,5 а винной кислоты. Затем этот раствор постепенно при взбал-
тывании вливают в раствор соды так, чтобы избежать выделения углекислоты.
К раствору добавляют 0,890 г KJO3 и 8 г KJ и взбалтывают до растворения со-
лей. Полученный раствор переносят в мерную колбу на 1 4, разбавляют дистил-
лированной водой до метки и перемешивают. Раствор переливают в двухлитро-
вую колбу, закрывают воронкой, помещают в водяную баню с холодной водой,
нагревают до закипания воды в бане и кипятят 5 мин. После этого оставляют на
ночь для охлаждения и отстаивания. На следующий день прозрачный раствор
сливают в склянку для хранения.
Смесь щавелевой и серной кислот. Отвешивают 60 г щавелевой кислоты, рас-
творяют в 800 мл дистиллированной воды и добавляют 70 мл концентрированной
серной кислоты пл. 1,84. Раствор разбавляют дистиллированной водой до 1 л.
Раствор едкого натра (0,4 н.). Готовят из 1 н. раствора разбавлением в
2,5 раза безуглекислой водой.
Раствор соляной кислоты (0,4 н.). 33 мл соляной кислоты пл. 1,19, растворяют
в дистиллированной воде в мерной колбе на 1 4, доливают водой до метки и пере-
мешивают. Этот раствор должен точно соответствовать 0,4 н. раствору NaOH,
что достигается титрованием и соответствующим разбавлением раствора НС1.
Раствор йода (0,05 н.). Отвешивают 6,35 г кристаллического йода и 13 г KJ,
помещают в ступку, растирают, прибавляют 20 мл дистиллированной воды и рас-
тирают до полного растворения йода. Раствор переносят в мерную колбу на 1 4,
промывают ступку дистиллированной водой, доводят объем до 1 4, перемешивают
и сохраняют в хорошо закрытой склянке с притертой пробкой.
Сульфит натрия (0,05 н.). Взвешивают 0,63 г Na2SO3 или 1,26 г Na2SO3
•7Н2О, растворяют в дистиллированной воде, разбавляют до 200 мл и переме-
шивают. Раствор готовится на 2—3 дня.
Приготовление остальных реактивов описано выше (см. стр. 28, 49, 76).
ОПРЕДЕЛЕНИЕ ГЛЮКОЗЫ, ФРУКТОЗЫ И САХАРОЗЫ
ЖЕЛЕЗОСИНЕРОДИСТЫМ КАЛИЕМ
Сущность метода. Железосинеродистый калий в ще-
лочной среде восстанавливают редуцирующими сахарами до же-
лезистосинеродистого калия. При этом на 1 моль глюкозы или
121
фруктозы расходуется 6 моль железосинеродистого калия:
6K3Fe (CN)6 + С6Н12О6 + ЗКОН = 6K4Fe (CN)6 + ЗС2Н4О3 + ЗН2О.
Хотя эта реакция неспецифична, так как железосинеродистый
калий окисляет многие органические вещества, не являющиеся са-
харами, метод может быть полезен во многих работах по биохимиче-
скому исследованию растений.
Избыток железосинеродистого калия определяют йодометриче-
ским методом:
2K3Fe (CN)6 + 2KJ + 3ZnSO4 = J2 + K2Zn3 [Fe (CN)6]2 + 3K2SO4,
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
В присутствии смеси солей Na2HPO4 и Na2CO3 железосинеродис-
тый калий почти одинаково интенсивно реагирует с фруктозой при
55° С и 100° С, в то время как глюкоза реагирует при 100° С ин-
тенсивно, а при 55° С очень слабо. Были найдены следующие темпе-
ратурные коэффициенты: для фруктозы — 1,16; для глюкозы
Г 50
—------10,8, где Ф1оо и Г1оО — количество K3Fe (CN)6, восстанов-
ленное фруктозой и глюкозой при 100° С в течение 15 мин; Ф55 и
Г55—количество K3Fe (CN)6, восстановленное фруктозой и глюко-
зой при 55° С в течение 30 мин.
Определив количество железосинеродистого калия, восстановлен-
ного исследуемым раствором, содержащим глюкозу и фруктозу,
при температуре 55° С и 100° С, можно, пользуясь температурными
коэффициентами, подсчитать количество фруктозы и глюкозы в
смеси.
Объем 0,005 н. раствора тиосульфата натрия (в мл), соответствую-
щий фруктозе, вычисляют по формуле:
Вф- (a~i\71°io,8 ~6) 1,12(а —£>) —0,104(а —с),
объем, соответствующий глюкозе, по следующей формуле:
Вг = (а — с) — 1,16 Вф = 1,12(а — с)— 1,30(а — Ь).
Так как с изменением концентрации сахара изменяется и соот-
ношение — объем тиосульфата натрия сахар, необходимо при
вычислении содержания сахара учитывать эти изменения введением
соответствующих поправок. С поправками формулы приобретают
следующий вид:
ch — 166 4-(а — Ь) р __ 145 4~ (д — с) &
1000 ф’ 1000 г’
где Ф — количество фруктозы в смеси (в мг); Г — количество глю-
козы в смеси (в мг); В$ — объем 0,005 н. раствора тиосульфата
натрия, соответствующий фруктозе (в мл); ВГ — объем 0,005 н.
раствора тиосульфата натрия, соответствующий глюкозе (в мл);
а — объем 0,005 н. раствора тиосульфата натрия, затраченный при
122
титровании реактива (в мл); b — объем 0,005 н. раствора тиосуль-
фата натрия, затраченный при титровании избытка реактива после
реакции при 55° С (в мл); с — объем 0,005 н. раствора тиосульфата
натрия, затраченный при титровании избытка реактива после реак-
ции при 100° (в мл).
Объемный йодометрический метод
Ход анализа. Навеску исследуемого вещества берут с та-
ким расчетом, чтобы в ней было не более 50 мг моносахаридов и 100 г
суммы сахаров (листьев сахарной свеклы и пшеницы весной —
3—5 г, озимой пшеницы осенью 1—2 г). Чем старее листья, тем
меньше навеска. Измельченный исследуемый материал взвешивают
в стеклянном или алюминиевом бюксе, который закрывают крыш-
кой и помещают в пары кипящей воды на 5—10 мин для инактива-
ции ферментов.
После этого пробу переносят в ступку, растирают и смывают
дистиллированной водой в мерную колбу на 100 мл, доводят объем
до 70—80 мл. Для осаждения белков и других мешающих веществ
прибавляют на каждый грамм навески 1 мл 1 н. раствора едкого
натра (на сырое вещество). После перемешивания добавляют 10%-
ный раствор сернокислого цинка в количестве, превышающем объем
прибавленной щелочи в 1,2 раза. Общий объем в колбе доводят
дистиллированной водой до 100 мл, перемешивают, дают отстоять-
ся и фильтруют в сухую колбу через сухой складчатый фильтр,
выбрасывая первую порцию фильтрата.
Определение содержания фруктозы и глюкозы
В две пробирки диаметром 18—20 мл набирают пипеткой по 5 мл
фильтрата, добавляют по 5 мл железосинеродистого реактива, пере-
мешивают, закрывают пробирки шариковыми пробками и помещают
одну пробирку в кипящую воду на 15 мин, а другую — в воду, нагре-
тую до температуры 55° С и выдерживают при этой температуре
30 мин. После чего пробирки охлаждают, растворы переносят в кол-
бы для титрования, добавляют по 1 мл 20%-ного раствора йодистого
калия, по 5 мл 3%-ного раствора сернокислого цинка в уксусной
кислоте и титруют выделившийся йод 0,005 н. раствором тиосульфа-
та натрия в присутствии крахмала до исчезновения синей окраски.
Отдельно титруют 5 мл реактива, что служит контролем. Из полу-
ченных данных вычисляют содержание глюкозы и фруктозы по фор-
мулам:
у _ А [1,12(0 —с) —1,3(а — 6)][145 + (а — с)1
Лг v п 10 000
v _ А [1,12(а — 6) — 0,104(а — f)][166 + (0 — 6)]
' ф v п 10 000
у _ А [143 + (а — с)] (0 —с)
Лм~" V п 10 000
123
где Хг — содержание глюкозы в исследуемом материале (в %);
Хф — содержание фруктозы в исследуемом материале (в %); Хм —
содержание моносахаридов (в %); А — общий объем исследуемого
раствора (в мл); v — объем исследуемого раствора, взятый для тит-
рования (в мл); п — навеска исследуемого вещества (в г); а — объем
0,005 н. раствора тиосульфата натрия, затраченный при титровании
реактива (в мл); b — объем 0,005 н. раствора тиосульфата натрия,
затраченный при титровании избытка реактива после реакции при
55° С (в мл); с — объем 0,005 н. раствора тиосульфата натрия, за-
траченный при титровании избытка реактива после реакции при
100° С (в мл).
Определение суммы сахаров и сахарозы
25 мл исследуемого раствора (фильтрат после отделения белков
и мешающих веществ) переносят в мерную колбу на 50 мл, прибав-
ляют 1 мл 1 н. раствора соляной кислоты, перемешивают, помещают
колбу в кипящую воду и выдерживают в течение 10 мин. После это-
го охлаждают, прибавляют каплю раствора фенолфталеина и нейтра-
лизуют 1 н. раствором едкого натра до появления розовой окраски,
избегая избытка щелочи. Объем раствора доводят дистиллирован-
ной водой до 50 мл и перемешивают. Из полученного раствора наби-
рают пипеткой 5 мл, где должно быть от 0,3 до 3 мг суммы сахаров,
вносят в пробирку, добавляют 5 мл железосинеродистого реактива,
закрывают шариковой пробкой и ставят в кипящую воду на 15 мин.
Затем охлаждают, погружая пробирку в холодную воду, переносят
раствор в колбу для титрования, добавляют 1 мл 20%-ного раствора
йодистого калия, 5 мл 3%-ного раствора сернокислого цинка и
титруют 0,005 н. раствором тиосульфата натрия в присутствии
крахмала. Из полученных данных вычисляют содержание суммы са-
харов в пересчете на глюкозу по формуле:
7 А [143+ (а — с)] (а — с)
L v п 5000
Количество сахарозы вычисляется по следующей формуле:
X = 0,95 (Z — В),
где X — содержание сахарозы в исследуемом веществе (в %); Z —
содержание суммы сахаров в расчете на глюкозу (в %); В — содер-
жание восстанавливающих сахаров в расчете на глюкозу (в %).
Остальные обозначения такие же, как в приведенной выше фор-
муле.
Колориметрический метод
Содержание глюкозы и фруктозы можно определить в меньшем
количестве материала, если пользоваться колориметрическим мето-
дом. В этом случае навеску берут в пять раз меньше, чем в объем-
ном методе. Извлечение сахаров и осаждение белков производят так
124
же, как и в объемном методе. После фильтрования необходимо из
раствора удалить остаток соли цинка. Для этого к фильтрату при-
бавляют 0,2 г смеси Na2CO3 и Na2HPO4 12Н2О (1 : 1), взбалтыва-
ют и фильтруют в сухую колбу через сухой фильтр. Из полученного
прозрачного раствора набирают в две пробирки по 2 мл, содержащих
от 0,04 до 0,2 мг моносахаридов, прибавляют побрил раствора желе-
зосинеродистого калия, перемешивают, закрывают шариковой проб-
кой и помещают одну пробирку в кипящую воду на 15 мин, а дру-
гую в воду с температурой 55° С на 30 мин. После этого охлаждают,
растворы переносят в мерные колбы на 50 мл, добавляют по 0,1 мл
8%-ного раствора щавелевой кислоты, по 5 мл 2 н. раствора серной
кислоты, взбалтывают до прекращения выделения пузырьков уг-
лекислого газа, разбавляют дистиллированной водой до метки и
перемешивают. Перед фотоколориметрированием прибавляют с ин-
тервалом в 1—2 мин по одной капле насыщенного раствора FeCl3,
перемешивают и точно через 10 мин после прибавления хлорного
железа фотометрируют в кювете с толщиной слоя 1 см при 610 нм.
Вместо хлорного железа, образующего с железистосинеродистым
калием берлинскую лазурь интенсивно синего цвета, можно восполь-
зоваться мышьяково-молибденовым реактивом Нельсона, молибден
которого восстанавливается железистосинеродистым калием в синий
гитерополикомплекс. В этом случае весь ход анализа остается тем же,
но вместо хлорного железа перед доведением объема до 50 мл
прибавляют по 2 мл мышьяково-молибденового реактива, разбавля-
ют дистиллированной водой до 50 мл и через 20—30 мин фотометри-
руют при 660—740 нм в кювете на 10 или 20 мм. Интенсивность
окраски в данном случае слабее в два раза по сравнению с хлорным
железом. Определяемые концентрации сахаров от 0,2 до 4 мкг/мл] в
колориметрируемом растворе.
Из полученных данных вычисляют оптическую плотность для
растворов фруктозы и глюкозы по следующим формулам:
Ф55= 1,12Е55 —0,104Е100,
Г100= 1,12Е100 — 1,ЗЕ55,
где Ф55 — оптическаягплотность раствора фруктозы при температу-
ре 55° С; Г1оо — оптическая плотность раствора глюкозы при темпе-
ратуре 100°С; Е65—оптическая плотность, полученная при фото-
метрировании раствора, реагировавшего при 55° С; Е100—опти-
ческая плотность, полученная при фотометрировании раствора,
реагировавшего при 100° С.
По вычисленным значениям оптической плотности находят, поль-
зуясь калибровочными графиками, количество сахара, отвечающее
этим величинам, и рассчитывают содержание его в исследуемом
веществе по следующей формуле:
х = 50 4 С _ А С
10 000 v п 200 v п ’
125
где X — содержание сахара в исследуемомГвеществе (в %); А —
общее количество исследуемого раствора (в мл); v — количество ис-
следуемого раствора, взятое для колориметрического определения са-
хара (в мл); С — концентрация сахара, найденная в колориметри-
руемом растворе (в мкг/мл); п — навеска исследуемого вещества
(в г); 50 — объем колориметрируемого раствора (в мл); 10 000
коэффициент для пересчета микрограммов в граммы и проценты.
Рис. 18. Калибровочные кривые
для глюкозы и фруктозы в же-
лезосинеродистом методе с хлор-
ным железом (светофильтр с
максимумом пропускания 610 кж,
кювета с толщиной измеряемого
слоя 10 мм):
юкоза при 100° С; 2— фрук-
тоза при 55° С.
Концентрация сахара, мкг/мл
Рис. 19. Калибровочные кривые
для глюкозы и фруктозы в же-
лезосинеродистом методе с мы-
шьяково-молибдатным реактивом
(светофильтр с максимумом про-
пускания 660 нм, кювета с тол-
щиной измеряемого слоя 20 мм):
/ — глюкоза при 100° С; 2 — фрук-
тоза при 55° С.
Построение калибровочной кривой. В се-
рию пробирок набирают 0,2, 0,4, 0,8, 1,2, 1,6 и 2 мл стандартного
раствора сахара — глюкозы или фруктозы, содержащего 0,1 мг
в 1 мл, добавляют дистиллированной воды до 2 мл и 5 мл железо-
синеродистого реактива. Пробирки с глюкозой ставят в кипящую
воду на 15 мин, а пробирки с фруктозой в воду, нагретую до 55° С
на 30 мин. Затем охлаждают, погружая пробирки в холодную воду,
растворы переносят в мерные колбы на 50 мл, добавляют по 0,1 мл
8% -ного раствора щавелевой кислоты, по 5 мл раствора серной кис-
лоты, взбалтывают до прекращения выделения пузырьков газа,
разбавляют водой до метки и перемешивают. Перед колориметриро-
ванием прибавляют по одной капле насыщенного раствора хлорного
железа, перемешивают и через 10 мин фотометрируют в кювете с
толщиной слоя 10 мм при 610 нм.
На основании полученных данных строят калибровочную кривую
(рис. 18).
126
Для построения калибровочной кривой с мышьяково-молибде-
новым реактивом вместо хлорного железа добавляют по 2 мл реак-
тива и через 20—30 мин фотометрируют при 660—740 нм. На осно-
вании полученных данных строят калибровочную кривую (рис. 19).
Реактивы. Железосинеродистый реактив. Отвешивают 0,60 г K3Fe (CN)6,
90 г Na2HPO4 12Н2О и 64 г Na2CO3. Растворяют все эти соли в 800 мл дистил-
лированной воды при нагревании, затем охлаждают, доводят объем раствора до
1 л и перемешивают. Сохраняют в темной склянке.
Раствор сернокислого цинка в уксусной кислоте (3%-ный). Отвешивают 30 г
ZnSO4 7Н2О, растворяют в 500 мл дистиллированной воды, добавляют 130 мл
ледяной уксусной кислоты, разбавляют водой до 1 л и перемешивают. Сохраняют
в склянке с притертой пробкой.
Мышьяково-молибденовый реактив. 25 г молибдата аммония и 3 г мышьяково-
кислого натрия Na2HAsO4 7Й2О, растворяют в 450 мл дистиллированной
воды при слабом нагревании, добавляют 21 мл концентрированной серной кислоты,
дают охладиться, доводят объем до 500 мл и сохраняют в темной склянке.
ОПРЕДЕЛЕНИЕ САХАРОВ
ПОСЛЕ ХРОМАТОГРАФИЧЕСКОГО РАЗДЕЛЕНИЯ ИХ НА БУМАГЕ
Количественное определение сахаров после хроматографического
разделения их даетвозможность определить содержание индивидуаль-
ных сахаров при совместном их присутствии в растительном материа-
ле. Если в исследуемом образце находятся только три сахара —
глюкоза, фруктоза и сахароза, то лучше определять содержание их
приведенным выше химическим методом, так как хроматографиче-
ский метод значительно длительнее.
Сущность метода. Сахара извлекаются из раститель-
ного материала 70%-пым этиловым спиртом или дистиллированной
водой. Полученный раствор сгущают выпариванием и очищают от
белков и пигментов. Остаток растворяют в 1 мл дистиллированной
воды (или 70%-ного спирта) и некоторую часть этого раствора нано-
сят в трех точках на одном уровне на полоску хроматографической
бумаги. После 48-часового хроматографирования бумажную ленту
высушивают и разрезают вдоль на три полоски. Сахара проявляются
на двух крайних полосках: на одной — резорциновым проявителем,
на другой — анилиновым или бензидиновым проявителем. По
обнаруженным пятнам сахаров устанавливают местонахождение их
на средней полоске, соответствующие участки которой отмечают ка-
рандашом и вырезают ножницами. Бумажные вырезки, содержащие
сахара, помещаются в сухие пробирки и сахара извлекают водой.
В полученных растворах сахара определяют с помощью колориметри-
ческих методов, сахарозу, фруктозу, раффинозу, сорбозу и другие
кетозы — молибденовым методом, а глюкозу, мальтозу, рибозу, рам-
нозу, галактозу и другие альдозы — медно-щелочным методом.
Ход анализа. Извлечение сахаров 70%- н ы м
этиловым спиртом. Отвешивают 1,5 г свежего исследуе-
мого материала (листья или другие органы растений), помещают в
ступку и тщательно растирают с 0,1 г MgCO3, и 14 мл 70%-ного эти-
лового спирта. Если навеска сухая, берут 15 мл спирта. Растертую
127
Рис. 20. Расположение ограни-
чительных вырезов на бумаге
для хроматографии сахаров:
а — зоны нанесения исследуемого
раствора.
массу переносят в центрифужную про ирку и центрифугируют.
Прозрачный раствор сливают в сухую пробирку, набирают пипеткой
10 мл, что соответствует 1 г навески, переносят в фарфоровую чашку
и выпаривают на водяной бане досуха. К остатку в чашке после
охлаждения приливают 1 мл дистиллированной воды и размешивают
1—2 мин. Жидкость переносят в небольшую центрифужную про-
бирку, центрифугируют и прозрач-
ный раствор сливают в сухую про-
бирку.
Извлечение сахаров
водой. Дистиллированная вода из-
влекает большее количество сахаров,
включая олигосахариды. Навеску 1,5 г
свежего растительного материала рас-
тирают в ступке с 0,1 г углекислого
магния,2 мл 10%-ного раствора серно-
кислого цинка и 12 мл дистиллирован-
ной воды. Суспензию переносят в цен-
трифужную пробирку, центрифугиру-
ют и набирают пипеткой 10 мл про-
зрачного раствора, что соответствует
1 г навески, выливая его в другую цен-
трифужную пробирку. К раствору при-
бавляют одну каплю фенолфталеина и добавляют по каплям насыщен-
ный раствор едкого бария до появления слабо-розовой окраски. При
этом избыток сернокислого цинка осаждается вместе с барием и рас-
твор очищается от солей:
ZnSO4 + Ba (ОН)2 = BaSO4 + Zn (ОН)2.
Осадок отделяют центрифугированием, раствор полностью сливают
в фарфоровую чашку, остаток промывают 3 мл дистиллированной во-
ды, центрифугируют и промывную жидкость добавляют к раствору
в чашке. Раствор выпаривают на водяной бане досуха, затем охлаж-
дают, остаток растворяют в 1 мл 70%-ного этилового спирта или
воды и раствор сливают в сухую пробирку.
Хроматографирование. Из хроматографической бу-
маги марки «С» или «М» вырезают ленты длиной 52 см и шириной
15 см. В верхней части ленты (на 7 см ниже верхнего края) отделяют
три зоны нанесения исследуемого раствора с помощью вырезов длиной
20 мм и шириной 3 мм. Для этого ленту помещают на фильтроваль-
ную бумагу и простым карандашом намечают места вырезов, прово-
дя линии длиной 20 мм на следующем расстоянии от левого края
ленты: 7—10 мм, 40—43 мм, 57—60 мм, 90—93 мм, 107—НО мм,
140—143 мм (всего 12 линий). По намеченным линиям делают выре-
зы с помощью лезвия безопасной бритвы так, как это показано на
рис. 20.
После того, как ленты будут готовы, наносят исследуемый рас-
твор. Для этого микропипеткой на 0,1 мл набирают 0,01 мл раствора
128
Рис. 21. Закрепление бу-
мажных полос в хромато-
графической камере с по-
мощью стеклянных пало-
чек:
а — стеклянные палочки;
б — хроматографическая ка-
мера с бумажными полосами
и наносят его в виде полоски длиной 30 мм между 1-м и 2-м выреза-
ми, затем такое же количество между 3-м и 4-м вырезами и между
5-м и 6-м вырезами. Пипетку с раствором нужно проводить с такой
скоростью, чтобы раствор распределялся равномерно по всему участ-
ку между вырезами. После высыхания бумаги снова наносят по
0,01 мл исследуемого раствора и так поступают до тех пор, пока будет
нанесено от 0,03 до 0,15 мг каждого из
определяемых сахаров (раффинозы от 0,05
до 0,3 мг).
После высыхания пятен складывают две
бумажные ленты вместе и в верхней части
скрепляют их с помощью двух стеклянных
палочек, соединенных по краям резиновыми
кольцами, которые вырезают из резиновой
трубки подходящего диаметра (рис. 21).
Здесь применяется нисходящая хромато-
графия. Камерой служит стеклянный ци-
линдр, в верхней части которого прикреп-
ляется ванночка с растворителем. Для
разделения сахаров можно применять сле-
дующие смеси растворителей: я-бутанол —
уксусная кислота — вода (6 : 2 3); w-бута-
нол — этанол — вода (6 3:2); я-бутанол —
пиридин — вода (6:2:2); я-бутанол —
диоксан — вода (4 4 1).
Растворитель наливается в ванночку
и немного в фарфоровую чашку или в
чашку Петри, помещенную на дно камеры.
Верхние концы бумажных ле11г, выходящие
из палочек, погружают в ванночку с рас-
творителем, а ленты помещают по обе сто-
роны ванночки (см. рис. 21). Камеру за-
крывают крышкой и оставляют на 48 ч,
давая возможность растворителю стекать с нижнего конца хромато-
граммы, который делают зубчатым. После хроматографирования бу-
мажные ленты вынимают из камеры за концы стеклянных палочек и
подвешивают за эти же палочки в вытяжном шкафу. Включают венти-
лятор и высушивают до удаления всего растворителя. Если пользо-
вались растворителями с пиридином или диоксаном, хроматограм-
мы дополнительно высушивают в сушильном шкафу при 100° С в
течение 30 мин.
После этого хроматограмму разрезают вдоль на три части, пред-
варительно проводя линии карандашомточномежду зонами нанесе-
ния сахаров. Крайние полоски (№ 1и№ 3) для установления место-
нахождения сахаров проявляют: одну резорциновым проявителем,
другую анилиновым или бензидиновым проявителем. Для этого их
опрыскивают из пульверизатора соответствующим проявителем,
высушивают в вытяжном шкафу и помещают в сушильный шкаф на
9 6-2
129
10 мин. С резорциновым проявителем выдерживают в шкафу 10 мин
при температуре 80° С, а с анилиновым или бензилиновым — при
температуре 90° С. При этом на месте расположения сахаров появ-
ляются окрашенные пятна. С резорцином реагируют фруктоза,
сахароза, раффиноза и другие кетозы, давая розово-красную окрас-
ку. С анилином образуют темно-серые пятна глюкоза, сахароза,
мальтоза и другие альдозы. С бензидином альдозы образуют коричне-
вые, а кетозы желтые пятна.
Сахара при применении данных растворителей располагаются
на хроматограмме в следующем порядке (табл. 4).
Таблица 4
Значения Rf сахаров
Сахара н-Бутанол: :уксусная кислота: :вода (6:2:3) «-Бутанол: .•этанол: :вода (6:3:2) «-Бутанол: .•пиридин: .-вода (6:2:2) н-Бутанол: :диоксан: :вода (4:4:1)
Раффиноза 0,08 0,06 0,06 0,06
Мальтоза 0,12 0,08 0,09 0,09
Сахароза 0,17 0,12 0,13 0,14
Галактоза 0,21 0,18 0,18 0,17
Глюкоза 0,23 0,19 0,21 0,22
Фруктоза 0,27 0,26 0,26 0,28
Рамноза 0,46 0,43 0,45 0,46
При работе с незнакомым объектом для установления состава
сахаров рядом с исследуемым раствором наносят смесь с известным
содержанием сахаров, например глюкозу, фруктозу и сахарозу,
по 0,01 мл 1%-ного раствора и после хроматографирования и про-
явления устанавливают наличие тех или иных сахаров путем
сравнения.
Колориметрическое определение саха-
ров. Сахара в элюате могут быть определены различными метода-
ми. Наиболее подходящими являются молибденовый метод для опре-
деления кетоз, которые в сернокислой среде восстанавливают шести-
валентный молибден до молибденовой сини и медно-щелочный метод
для определения альдоз, которые восстанавливают двухвалентную
медь до закиси меди. Последняя затем в кислой среде восстанавлива-
ет шестивалентный молибден до молибденовой сини. Полученный
синий раствор фотоколориметрируется при 660 нм, Эти реактивы на
сахара значительно лучше антронового реактива, так как превосхо-
дят его по чувствительности и не реагируют с волокнами бумаги.
Проявленные крайние полоски хроматограммы прикладывают к
средней непроявленной полоске и карандашом разделяют пятна най-
денных сахаров, проводя линии и через непроявленную полоску.
Отметив места расположения сахаров, нумеруют их и вырезают из
средней непроявленной полоски участки, соответствующие каждо-
му сахару. Бумажные вырезки хроматограммы разрезают в виде
130
гребешка, сворачивают в трубочку и опускают в сухие пробирки.
Для приготовления контрольного раствора вырезают в верхней части
хроматограммы (перед стартовой зоной) такой же по площади учас-
ток бумаги и помещают его в пробирку. В пробирки приливают пи-
петкой по 5 мл дистиллированной воды и настаивают при взбалты-
вании 5—10 мин. Из полученных экстрактов набирают пипеткой по
3 мл и выливают в сухие пробирки. К растворам, содержащим сор-
бозу, фруктозу, сахарозу, раффинозу и другие кетозы, прибавляют
по 2 мл раствора молибдата аммония в серной кислоте и перемешива-
ют. Закрывают пробирки стеклянными шариковыми пробками и
опускают в кипящую воду на 20 мин. После этого охлаждают и коло-
риметрируют в фотоэлектроколориметре при 660—700 нм в кювете
на 10 мм. Раствором для сравнения служит экстракт из бумаги сво-
бодной зоны с добавлением такого же количества реактивов и нагре-
ванием, как в опыте.
При определении глюкозы, галактозы, мальтозы, рибозы и рам-
нозы к порциям экстракта (3 мл) прибавляют по 1 мл медно-щелоч-
ного реактива, перемешивают, закрывают пробирки шариковыми
пробками и опускают в кипящую воду на 10 мин. После охлаждения
добавляют по 1 мл сернокислого раствора молибдата аммония, на-
правляя струю в центр пробирки, быстро перемешивают и остав-
ляют на 5 мин для окончания реакции восстановления. Затем энер-
гично взбалтывают до удаления углекислоты и колориметрируют
при 660—700 нм глюкозу, галактозу и рибозу в кювете на 5 мм, а
мальтозу и рамнозу в кювете на 10 мм. Раствором для сравнения
служит экстракт из бумаги без сахаров с добавлением такого же ко-
личества реактивов и нагреванием, как в опыте.
Полученные результаты сравнивают с калибровочной кривой,
находят концентрацию сахара в колориметрируемом растворе и вы-
числяют содержание его в исследуемом веществе по следующей
формуле:
v 0,1 5 а с 0,1667 а с
* 5
где X — содержание сахара (в мг на 100 а исследуемого вещества);
с — концентрация сахара в колориметрируемом растворе (в мкг/мл);
а — объем колориметрируемого раствора (в мл); в — объем иссле-
дуемого раствора, нанесенный на хроматограмму (в мл); 5 — общий
объем экстракта сахара, полученный из хроматограммы (в мл);
3 — объем экстракта сахара, взятый для колориметрического опре-
деления (в мл); 0,1 —коэффициент для пересчета микрограммов саха-
ра в миллиграммы и на 100 а вещества.
Построение калибровочных кривых для
сорбозы, фруктозы, сахарозы и раффинозы.
В сухие пробирки помещают различные количества раствора, со-
держащего 100 мкг исследуемого сахара в 1 мл: фруктозы и сорбо-
зы — 0,1, 0,2, 0,3, 0,4, 0,6 и 0,7 мл; сахарозы — 0,2, 0,4, 0,6, 0,8,
1,0 и 1,2 мл; раффинозы — 0,3, 0,5, 0,8, 1,2, 1,6 и 2,0 мл. Растворы
91
13Д
доводят дистиллированной водой до объема 3 мл. В контрольную
пробирку, не содержащую сахара, прибавляют 3 мл дистиллирован-
ной воды. Затем в пробирки добавляют по 2 мл 8%-ного раствора
молибдата аммония в 4 н. серной кислоте, перемешивают, пробирки
закрывают стеклянными шариковыми пробками, вставляют в шта-
тив, погружают в кипящую воду и нагревают 20 мин. Затем штатив с
Рис. 22. Калибровочные кривые
для сорбозы, фруктозы, сахарозы и
раффинозы с молибдат-сернокислым
реактивом (светофильтр с макси-
мумом пропускания 660 нм, кювета
с толщиной измеряемого слоя
10 мм):
1 — сорбоза; 2 — фруктоза; 3 — саха-
роза; 4 — раффиноза.
Рис. 23. Калибровочные кривые для
глюкозы, галактозы, мальтозы, ри-
бозы и рамнозы с медно-щелочным
реактивом (светофильтр с максиму-
мом пропускания 660 нм, кювета
для глюкозы, галактозы и рибозы
с толщиной измеряемого слоя 5 мм,
а для мальтозы и рамнозы —
10 мм):
1 — глюкоза; 2 — галактоза; 3 —
мальтоза; 4 — рибоза; 5 — рамноза.
пробирками вынимают, охлаждают растворы погружением в хо-
лодную воду и измеряют оптическую плотность при 660—700 нм в
кювете на 10 мм. На основании полученных данных строят калибро-
вочные кривые (рис. 22).
Построение калибровочных кривых для
глюкозы, мальтозы, рибозы, рамнозы и га-
лактозы. В сухие пробирки помещают различные количества
раствора, содержащего 100 мкг исследуемого сахара в 1 мл: глюко-
зы и галактозы — 0,1, 0,2, 0,3, 0,4, 0,6, 0,7, 0,8 и 0,9 мл; мальтозы,
рибозы и рамнозы — 0,2, 0,3, 0,4, 0,6, 0,8, 1,0 и 1,2 мл. Растворы до-
водят водой до 3 мл. В контрольную пробирку, не содержащую саха-
ра, прибавляют 3 мл дистиллированной воды. Затем во все пробирки
прибавляют по 1 мл медно-щелочного реактива, перемешивают,
закрывают стеклянными шариковыми пробками и погружают в ки-
132
пящую воду на 10 мин. После охлаждения приоавляют 1 мл молиб-
денового реактива, направляя струю в центр пробирки, немедлен-
но перемешивают и оставляют на 5 мин для окончания реакции вос-
становления. Затем энергично взбалтывают до удаления углекислоты
и измеряют оптическую плотность в фотоэлектроколориметре при
660—700 нм глюкозы и рибозы в кювете на 5 мм, а мальтозы и рам-
нозы в кювете на 10 мм. Раствором для сравнения служит контроль-
ный раствор, не содержащий сахара, но содержащий такое же коли-
чество реактивов, как опытный. На основании полученных данных
строят калибровочную кривую (рис. 23).
Реактивы. Насыщенный раствор едкого бария. Отвешивают 80 г Ва (ОН)2
8Н2О, помещают в 1 л горячей воды, взбалтывают до растворения, затем охлаж-
дают, закрывают пробкой и оставляют до просветления раствора. После этого
прозрачный раствор сливают в склянку и сохраняют в хорошо закупоренном виде.
Резорциновый проявитель. 0,5 г резорцина растворяют в 100 мл 95%-ного
этилового спирта и добавляют 2 мл раствора соляной кислоты (1 1). На хрома-
тограмме фруктоза, сахароза, раффиноза и другие кетозы дают с этим реактивом
розово-красные пятна после 10-минутного нагревания при 70—80° С.
Бензидиновый проявитель. 0,5 г бензидина растворяют в 100 мл 95%-ного
этилового спирта и добавляют 5 г трихлоруксусной кислоты. После опрыскивания
и 10-минутного нагревания при 90° С глюкоза и альдозы дают коричневые пятна,
фруктоза и сахароза — желтые пятна.
Анилиновый проявитель. 1 мл анилина растворяют в 100 мл 95%-ного этило-
вого спирта и добавляют 10 мл 80%-ной фосфорной кислоты. На хроматограмме
сахароза, глюкоза и др. альдозы дают с реактивом темно-коричневые пятна после
10-минутного нагревания при 90°
Молибдат-сернокислый реактив. Отвешивают 8 г молибденовокислого аммо-
ния, растворяют в 80 мл дистиллированной воды, затем добавляют 9 мл серной
кислоты пл. 1,84, охлаждают и разбавляют дистиллированной водой до 100 мл.
Сохраняют в темной склянке.
Медно-щелочный реактив для колориметрического определения сахаров. <Уг-
вешивают 75 г безводного карбоната натрия Na2CO3, растворяют в 400 мл
дистиллированной воды в колбе на 1 л и к этому раствору постепенно при взбал-
тывании, чтобы не выделялась углекислота, добавляют раствор, приготовленный
растворением 12 г сернокислой меди и 21,5 а винной кислоты в 400 мл дистилли-
рованной воды. Объем смешанного раствора доводят водой до 1 л и переливают
в двухлитровую колбу. Колбу закрывают воронкой, помещают в водяную баню
с холодной водой, нагревают до кипения воды в бане и кипятят 5 мин. После
этого оставляют на ночь и на следующий день прозрачный раствор сливают в
склянку для хранения.
Анилин-дифениламин-фосфорнокислый проявитель. 4%-ный раствор анилина
в этиловом спирте перед использованием смешивают с 4%-ным раствором дифе-
ниламина в этиловом спирте и фосфорной кислотой в соотношении 5 5 1. Для
проявления хроматограмму опрыскивают проявителем, слегка подсушивают на
воздухе, помещают в термостат и нагревают 10 мин при температуре 80° С. Это
универсальный проявитель. Сахара проявляются в виде синих и коричневых пя-
тен на почти бесцветном фоне.
Мочевина в качестве проявителя. 2 г мочевины растворяют в 10 мл 1 н. рас-
твор а^ол я ной кислоты, добавляют90 мл 95%-ного этилового спирта и перемеши-
вают. Хроматограмму проявляют при температуре 100° С в течение 10 мин. Ке-
тозы и олигосахариды, содержащие кетозы, дают синие пятна.
Приготовление 70%-ного спирта описано выше (см. стр. 83).
133
ОПРЕДЕЛЕНИЕ КРАХМАЛА
ОБЪЕМНЫМ И КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
Сущность метода. Метод основан на растворении крах-
мала при нагревании в 80 %-ном растворе азотнокислого кальция и
осаждении его из полученного раствора йодом. В присутствии йо-
дистого калия и азотнокислого кальция йод полностью осаждает
крахмал в виде темно-синего соединения, содержащего от 14 до
16% йода.
После центрифугирования и промывания осадка крахмал можно
определить двумя методами — объемным или колориметрическим.
В объемном методе крахмал окисляется бихроматом в присутствии
серной кислоты до углекислоты и воды:
4К2Сг2О7 + 66Н10О5 + 16H2SO4 = 6СО2 + 4K2SO4 + 4Cr2 (SO4)3 +
+ 21H2O.
Избыток бихромата оттитровывают 0,1 н. раствором соли Мора с ин-
дикатором фенилатраниловой кислотой или определяют йодометри-
ческим методом.
Сульфат железа (II) соли Мора реагирует с бихроматом калия
по следующему уравнению:
К2Сг2О7 + 6FeSO4 + 7H2SO4 = Cr2 (SO4)3 + 3Fe2 (SO4)3 + K2SO4 +
+ 7FLO.
В йодометрическом методе избыток бихромата калия реагирует
с йодистым калием, выделяя йод:
К2Сг2О7 + 6KJ + 7H2SO4 = 4K2SO4 + Cr2 (SO4)3 + 3J2 + 7H2O.
Выделившийся йод титруют раствором тиосульфата натрия в при-
сутствии крахмала как индикатора:
J2 + 2Na2S2O3 = Na2S4O6 + 2NaJ.
Грамм-эквивалент крахмала равен 6,75. Йод, адсорбирован-
ный крахмалом, практически не влияет на точность определения.
В колориметрическом методе осадок йодного крахмала растворяют
в гидроокиси натрия, разбавляют раствор дистиллированной водой
до определенного объема и проводят реакцию с йодом в кислой сре-
де. Оптическую плотность полученного синего раствора измеряют
при 610—660 нм и концентрацию крахмала в колориметрируемом
растворе находят по калибровочной кривой.
Объемный метод точнее колориметрического, но его можно при-
менять только при наличии в осадке крахмала без примесей других
органических веществ, например фруктозаны, осаждаемых йодом.
Определение крахмала в присутствии фруктозанов и декстринов
возможно только колориметрическим методом. Фруктозаны с йодом
дают осадок красно-бурого цвета и их можно, как и крахмал,
определять бихроматным методом.
134
Объемный бихроматный метод. Навеску иссле-
дуемого вещества, содержащую от 20 до 200 мг крахмала (около
3 г листьев, 1 г клубней картофеля и 0,25 г зерна), растирают в ступ-
ке с 5 мл 80%-ного раствора азотнокислого кальция. Растертую мас-
су переносят в коническую колбу на 100 мл, промывают ступку 10 мл
80%-ного раствора азотнокислого кальция и выливают раствор
в ту же колбу. Колбу закрывают воронкой, ставят на плитку, на-
гревают до кипения и кипятят при слабом нагреве 3—5 мин.
При этом крахмал переходит в коллоидальный раствор. Растворы
листьев и других частей растений, содержащие небольшое количест-
во крахмала, кипятят 3 мин, а большое количество крахмала —
5 мин, но не более, так как возможен нежелательный процесс гид-
ролиза крахмала.
После кипячения в колбу приливают 20 мл дистиллированной
воды, споласкивая ею воронку. После перемешивания суспензию
переносят в центрифужную пробирку, центрифугируют 2—3 мин
при 2000—3000 об/мин и сливают обычно мутный от коллоидаль-
ного состояния крахмала раствор в мерную колбу на 100 мл (или
50 мл при малом содержании крахмала). Если не вся суспензия вмес-
тилась в центрифужную пробирку, после центрифугирования первой
порции остаток центрифугируют в той же пробирке и центрифугат
присоединяют к первому в мерной колбе.
Колбу, в которой производилось кипячение, и осадок промывают
2—3 раза горячей водой порциями по 5—10 мл, центрифугируют и
присоединяют центрифугат к основному раствору в колбе. Раствор
в колбе разбавляют дистиллированной водой до метки, перемешива-
ют и используют для определения крахмала. Остаток после извле-
чения крахмала, находящийся в центрифужной пробирке, можно ис-
пользовать для определения гемицеллюлоз по методу изложенному
ниже.
Из полученного раствора крахмала набирают пипеткой 5 мл
(10 мл при анализе листьев), переносят в центрифужную пробирку,
в которую предварительно наливают 2 мл 0,5%-ного раствора йода,
перемешивают и оставляют на 15 мин для выделения осадка. При
этом осаждается йодокрахмальное соединение темно-синего цвета.
После этого центрифугируют, прозрачный раствор сливают как
можно полнее, а осадок промывают раствором, содержащим 5%
Са (NO3)2 и 0,01 % йода. Для этого к осадку прибавляют 5 мл рас-
твора для промывания, перемешивают стеклянной палочкой, которую
затем споласкивают этим же раствором. Промывание повторяют 3—
4 раза в зависимости от количества примесей. При анализе листьев
нужно промывать большее число раз. К промытому осадку в про-
бирке прибавляют пипеткой 5 мл ~ 0,5 н. раствора бихромата калия
в серной кислоте, перемешивают стеклянной палочкой, пробирку
вместе со стеклянной палочкой погружают в кипящую воду на
10 мин, время от времени размешивая раствор. После этого охлаж-
дают и раствор из пробирки переносят в колбу для титрования, смы-
вая остаток 10—15 мл дистиллированной воды. Раствор охлаждают,
135
прибавляют 3—5 капель 0,2%-ного раствора фенилантраниловой
кислоты и титруют 0,1 н. раствором соли Мора до перехода вишне-
во-фиолетовой окраски в зеленую.
В йодометрическом методе к раствору, содержащему избыток би-
хромата, помещенному в колбу, прибавляют 60 мл дистиллированной
воды для разбавления серной кислоты, охлаждают, добавляют 5 мл
20%-ного раствора йодистого калия и выделившийся йод титруют
0,1 н. раствором тиосульфата, прибавляя под конец 1 мл 0,5%-ного
раствора крахмала в качестве индикатора.
В том и другом методе отдельно титруется 5 мл 0,5 н. раствора
бихромата калия после предварительного разбавления таким же ко-
личеством воды, как и при титровании крахмала. На основании
полученных результатов вычисляют содержание крахмала по следую-
щей формуле:
У _ 0,675 В К(а — Ь)
где X — содержание крахмала (в %); В — общий объем исследуе-
мого раствора (в мл); V — объем исследуемого раствора, взятый
для осаждения крахмала йодом (в мл); а — объем 0,1 н. раствора соли
Мора (или тиосульфата натрия), затраченный при контрольном ти-
тровании бихромата (в мл); b — объем 0,1 н. раствора соли Мора
(или тиосульфата натрия), затраченный при определении крахмала
(в мл); К — нормальность раствора соли Мора (или тиосульфата),
п — навеска исследуемого вещества (в г); 0,675 — нормальный титр
крахмала, умноженный на 100 для пересчета в проценты. Определяе-
мые количества от 1 до 14 мг крахмала.
Колориметрический метод. Для колориметри-
ческого определения крахмала берут такую навеску исследуемого
материала, чтобы в ней находилось от 5 до 50 мг крахмала. Навеску
растирают в ступке с 5 мл 80%-ного раствора азотнокислого каль-
ция до однородной массы, которую затем переносят в коническую
колбу на 100 мл, смывая остаток 10 мл 80 %-ного раствора азотнокис-
лого кальция, и извлекают крахмал так, как это описано выше, в
объемном методе. После извлечения объем вытяжки доводят до 50
или 100 мл в зависимости от предполагаемого количества крахмала.
Набирают пипеткой 5 или 10 мл полученного раствора, переносят
в центрифужную пробирку, содержащую 2 мл 0,5%-ного раствора
йода, перемешивают и оставляют на 15 мин. После этого центрифу-
гируют, прозрачный раствор сливают, а осадок промывают два раза
5%-ным раствором азотнокислого кальция, содержащим 0,01 % йода.
К промытому осадку йодкрахмального соединения прибавляют
10 мл 0,1 н. раствора едкого натра, перемешивают и погружают
пробирку в кипящую воду на Ьмин. Раствор переносят в мерную кол-
бу на 50 мл, прибавляют 0,3 мл 0,5%-ного раствора йода, разбав-
ляют дистиллированной водой приблизительно до 40 мл, прибавляют
2 м; I п. раствора соляной кислоты, доводят водой до метки, пере-
ают и измеряют оптическую плотность раствора при 580—
610 нм в кювете на 10 мм. Полученные результаты сравнивают с
калибровочной кривой и вычисляют содержание крахмала по сле-
дующей формуле:
х _ 50 В - С __ ВС
Л ~~ 10 000 V п ~ 200 V п ’
Рис. 24. Калибровочная кривая
для крахмала (светофильтр с
максимумом пропускания
610 нм)’.
1 — толщина измеряемого слоя
10 мм; 2 — толщина измеряемого
слоя 5 мм.
где X — содержание крахмала (в %); В — общий объем исследуемо-
го раствора (в мл); V — объем исследуемого раствора, взятый для
осаждения крахмала йодом (в мл);
50 — объем окрашенного колориме-
трируемого раствора (в мл); С — кон-
центрация крахмала в колориметри-
руемом растворе (в мкг/мл); п — на-
веска исследуемого вещества (в г);
10 000 — коэффициент для перевода
микрограммов крахмала в граммы и
проценты.
Построение калибро-
вочной кривой. Берут 50 мг
очищенного крахмала, или соответ-
ствующую навеску неочищенного
крахмала, установив действительное
количество крахмала во взятом об-
разце описанным объемным методом.
Навеску растирают в ступке с 3 мл
80 %-ного раствора азотнокислого
кальция, переносят в коническую
колбу на 100 мл, промывая ступку
15 мл 80%-ного раствора азотнокис-
лого кальция, и кипятят 5 мин.
Раствор охлаждают, переносят в мер-
ную колбу на 50 мл и доводят дистилли-
рованной водой до метки. Из этого раствора, содержащего 1 мг крах-
мала в 1 мл, набирают в центрифужные пробирки 0,5, 1,2, 3, 4 и 5 мл,
добавляют до 5 мл 20%-ный раствор азотнокислого кальция, при-
бавляют по 2 мл 0,5%-ного раствора йода, перемешивают и остав-
ляют на 15 мин. После этого центрифугируют и осадок промывают
два раза 5%-ным раствором азотнокислого кальция. Растворяют
осадки в 10 мл 0,1 н. раствора NaOH при нагревании. Растворы
переносят в мерные колбы на 50 мл, прибавляют по 0,3 мл 0,5 %-
ного раствора йода, разбавляют дистиллированной водой до 40 мл,
прибавляют по 2 мл 1 н. раствора НО, доводят до метки и фото-
метрируют. На основании полученных данных строят калибровоч-
ную кривую (рис. 24).
Реактивы. Очистка крахмала. Необходимый для построения калибровочной
кривой очищенный крахмал готовят следующим образом. 1 г картофельного крах-
мала помещают в фарфоровую ступку и растирают с 10 мл дистиллированной
воды, прибавляя ее постепенно. Полученную суспензию переносят в колбу на
250 мл, прибавляют 15 мл 1 н. раствора едкого натра, накрывают воронкой и
137
пагоевакУГ до растворения крахмала. Горячий раствор фильтруют через вату в ста-
кац1 или цилиндр емкостью 250 мл. Затем охлаждают, прибавляют 100 мл 95% -
ного этил<>вого спирта, перемешивают и дают осадку крахмала осесть. Жидкость,
11аходящУюся над осадком, полностью сливают, а осадок промывают де-
кантацией два Раза порциями по 50 мл 60%-ного спирта. К остатку прибавляют
еще 25 мЛ 60%-ного спирта, 1 мл 1 н. раствора соляной кислоты, хорошо пере-
мешивают и фильтруют через стеклянный фильтр № 3 с отсасыванием или центри-
фугируют, промывая осадок 60%-ным спиртом до полного удаления ионов хлора
(проба с AgNO3). После этого крахмал переносят на бумагу, подсушивают на
воздухе переносят в стеклянный бюкс и сушат при 100° С до постоянного веса.
ПромывнУ10 жидкость, содержащую спирт, собирают и отгоняют при температуре
78—94° С- Количество спирта определяют по плотности.
Растер бихромата калия в серной кислоте (0,5 н.). Отвешивают 25 г К2Сг2О7
с точность10 До 0,01 г, растворяют в 250 мл дистиллированной воды в колбе ем-
костью не менее 2 л и постепенно при охлаждении приливают 800 мл концентри-
рованной H2SO4 пл. 1,84, тщательно перемешивают и после охлаждения перели-
вают в с#лянкУ с притертой стеклянной пробкой. Сохраняют в темном месте.
Раств°Р азотнокислого кальция (80%-ный). Отвешивают на технических
весах 200 г Са (NO3)2 4Н2О, растворяют в 70 мл дистиллированной воды в мер-
ном цилиНДРе» Доводят водой объем раствора до 250 мл и перемешивают. Из этого
раствора по мере надобности готовят разбавлением дистиллированной водой
20%-ный раствор и 5%-нын раствор с йодом, который применяется для промы-
вания осаДка йодистого крахмала. Для приготовления промывного раствора на-
бирают 10 МЛ 80%-ного раствора азотнокислого кальция, переносят в мерный
цилиндр, разбавляют дистиллированной водой до 160 мл, добавляют 3 мл 0,5%-
ного растЬ°Ра йода и перемешивают. Этот раствор готовят непосредственно перед
промыва1Фсм осадка, г о 0 .и
Раств°Р йода (0,5%-ный). Отвешивают 5 г кристаллического йода и 10 г KJ
с точность10 до 0,6^ помещают в ступку и растирают сначала сухую смесь, затем
с 10 мл дистиллированной воды до растворения всего йода. Раствор переносят
в мерную колбу на 1 л, разбавляют дистиллированной водой до метки, перемеши-
вают и сохраняют в хорошо закрытой склянке со стеклянной пробкой. у
Раств°Р соли Мора (0,1 н.). Отвешивают 40 г соли Мора, растворяют в ди-
стиллироьанк°й воде, добавляют 20 мл концентрированной серной кислоты, раз-
бавляют 0°Д°й до 1 л и перемешивают. Раствор* готовят на неделю, проверяя
титр в де!1Ь определения. Нормальность раствора соли Мора устанавливают по
0 1 н. рас^РУ бихромата калия. Для этого набирают пипеткой 25 мл 0,1 н. рас-
твора К2СГЛ» переносят в колбу для титрования, прибавляют 5 мл концентри-
рованной серной кислоты, 3—5 капель раствора фенилантраниловой кислоты
и титруют раствором соли Мора до перехода окраски из вишнево-фиолетовой в зе-
леную НаРмальность раствора соли Мора вычисляют по формуле:
„ 25 0,1
а ’
где ___цормальность раствора соли Мора; а — объем раствора соли Мора, за-
траченный при титровании бихромата калия (в лгл).
Раств°Р фенилантраниловой кислоты (0,2%-ный). Отвешивают 0,1 г фенил-
антранилов°й кислоты — СбН5—NH—СбН4СООН, растирают в ступке с 5 мл
0 1 н. расТв°Ра NaOH, переносят раствор в мерную колбу или цилиндр на 50 мл,
споласкиЬая стУпку дистиллированной водой. Раствор доводят водой до 50 мл
и перемеИ1Ивают- Сохраняют в темной склянке.
Pacntf°P соляной кислоты (~ 1 н.). Набирают 84 мл соляной кислоты пл. 1,19,
разбавляЮт Дистиллированной водой до 1 л и перемешивают.
ОПРЕДЕЛЕНИЕ КЛЕТЧАТКИ
Клетчаткз — высокомолекулярный полисахарид (С0Н2оО5)п, об-
pa iуклцЦЙ главную составную часть оболочек растительных клеток.
Чистая клетчаткам белое вещество без вкуса и запаха, волокнис-
138
того строения. Клетчатка нерастворима в воде, разбавленных кис-
лотах и щелочах, а также в органических растворителях. В состав
клеточных стенок входят также гемицеллюлозы и лигнин, которые
образуют основную часть клеточных стенок. Кроме этих соедине-
ний содержится еще некоторое количество смол, жиров, дубильных
и красящих веществ. Для определения клетчатки нужно удалить
все эти вещества, получив ее в чистом виде.
Сущность метода. Исследуемое вещество обрабатыва-
ют смесью уксусной и азотной кислот при нагревании. При этом
растворяется межклеточное вещество и клетчатка распадается на
отдельные волокна. Происходит также удаление лигнина, гемицел-
люлоз и других веществ. Идет гидролиз крахмала, пентозанов и дру-
гих веществ. После удаления примесей промыванием водой клет-
чатка окисляется бихроматом калия в сернокислой среде до угле-
кислоты и воды:
Q)H10O5 4~ 4К2Сг2О7 4~ 16H2SO4 = 6СО2 4~ 4Сг2 (SO4)3 4- 4K2SO4 4-
+ 21Н2О.
Избыток бихромата определяют титрованием солью Мора или йодо-
метрически. Грамм-эквивалент клетчатки -Сб^°°5 - равен 6,75.
Ход анализа. Исследуемый материал сушат, измельчают
и просеивают через сито с отверстиями 1 мм2. Из полученного по-
рошка берут навеску с таким расчетом, чтобы в ней находилось от
3 до 25 мг чистой клетчатки (0,05 г листьев и стеблей, 0,4 г зерна).
Навеску взвешивают на аналитических весах, переносят в центри-
фужную пробирку и приливают смесь уксусной и азотной кислот.
При навеске менее 0,1 г берут 5 мл смеси, а при навеске выше 0,1 г
берут 10 мл смеси уксусной и азотной кислот. Пробирку закрывают
стеклянной шариковой пробкой с палочкой и помещают в кипящую
воду на 25 мин, периодически размешивая. После этого центрифуги-
руют, прозрачный раствор осторожно сливают по палочке сколько
возможно, а к остатку приливают 10 мл' дистиллированной воды,
перемешивают, центрифугируют и раствор сливают. Промывают
водой 3 раза порциями по 10 мл. К промытому осадку клетчатки
приливают пипеткой 10 мл 0,5 н. раствора бихромата калия в сер-
ной кислоте, перемешивают стеклянной палочкой и пробирку поме-
щают в кипящую воду на 10 мин, периодически размешивая содер-
жимое стеклянной палочкой. После этого охлаждают и раствор выли-
вают в колбу для титрования емкостью 250 мл, смывая остаток
10—15 мл дистиллированной воды. Раствор охлаждают, прибавляют
5 капель 0,2%-ного раствора фенилантраниловой кислоты и титруют
0,1 н. раствором соли Мора до перехода вишнево-фиолетовой окрас-
ки в зеленую. Отдельно титруют 10 мл 0,5 н. раствора бихромата
калия, разбавив его 15 мл дистиллированной воды и охладив. Можно
остаток бихромата определить йодометрическим методом так, как
139
это описано при определении крахмала. На основании полученных
результатов находят содержание клетчатки по формуле:
У 0,675 К(а — Ь)
где X — содержание клетчатки (в %); Л — нормальность титрован-
ного раствора соли Мора (или тиосульфата); а — объем 0,1 н. рас-
твора соли Мора (или тиосульфата), затраченный при контрольном
титровании 10 мл бихромата (в мл); b — объем 0,1 н. раствора соли
Мора (или тиосульфата), затраченный при определении клетчатки
(в мл); п — навеска исследуемого вещества (в г); 0,675 — нормаль-
ный титр клетчатки, умноженный на 100.
Реактивы. Смесь азотной и уксусной кислот. На 100 мл 80%-ной уксусной
кислоты берут 10 мл азотной кислоты пл. 1,4, хорошо перемешивают и сохраняют
в склянке с притертой стеклянной пробкой.
Приготовление остальных реактивов описано выше (см. стр. 138).
ОПРЕДЕЛЕНИЕ ПЕКТИНОВЫХ ВЕЩЕСТВ
Пектиновые вещества находятся в клеточном соке плодов и ягод
в виде растворимого в воде пектина и клеточных оболочек и в виде
нерастворимого в воде соединения — протопектина. Пектин —
бесцветное вещество нейтральной реакции, растворимое в воде с
образованием коллоидного раствора, из которого осаждается спир-
том в виде студня. Очистка и исследование пектиновых веществ
сильно затруднены и поэтому химический состав их еще окончатель-
но не установлен. Исследователями из пектиновых веществ были
выделены следующие вещества: галактуроновая кислота, метиловый
спирт, уксусная кислота, арабиноза и галактоза.
В основном пектин представляет собой полисахарид, состоящий
из соединенных между собой остатков галактуроновой кислоты —
СНО (СНОН)4 СООН, большая часть карбоксильных групп кото-
рой этерифицирована метиловым спиртом, а меньшая часть замеще-
на металлами. При действии на пектин разбавленных щелочей или
фермента пектазы метоксильные группы легко отщепляются. При
этом образуется метиловый спирт и пектиновая кислота, которая
легко образует соли, называемые пектатами.
Пектиновая кислота представляет собой полигалактуроновую
кислоту — (С5Н7О4СООН)П и является слабой кислотой. В воде
растворяется мало, в спирте нерастворима. Щелочные соли пекти-
новой кислоты растЕоримы в воде, но осаждаются спиртом, а соли
щелочноземельных металлов в воде нерастворимы. Пектиновая кис-
лота из раствора легко осаждается в виде кальциевой соли, что
используется для количественного определения пектиновых ве-
ществ. Образующийся осадок пектата кальция, по нашим данным,
содержит 8,33% кальция. Исходя из этого молекулярный вес пекти-
новой кислоты равен 884, что соответствует следующей формуле —
^2(J*k)O22 (СООН)4. Очевидно, молекула пектиновой кислоты обра-
140
зуется из остатков 4 молекул галактуроновой кислоты и одной
молекулы галактозы:
4С5Н9ОбСООН + С6Н12О6 - 4Н2О = С2„Н40Ои (СООН)4.
галактуроновая галактоза пектиновая кислота
кислота
Грамм-эквивалент пектиновой кислоты равен 221.
Протопектин, являющийся кальциевой и магниевой солью более
или менее этерифицированной пектиновой кислоты, отлагается в
клеточных стенках и связан с целлюлозой. По мере созревания пло-
дов протопектин под воздействием фермента протопектазы превра-
щается в растворимый пектин. Протопектин переходит в пектин
также при кипячении с водой и разбавленными кислотами, в особен-
ности со щавелевой кислотой и ее аммонийной солью.
Наши опыты показали, что для аналитических целей протопек-
тин можно быстро извлечь, нагревая его с раствором соды, и еще
быстрее — с трилоном Б в щелочной среде. В этих условиях одно-
временно происходит отщепление метоксильных групп, образование
метилового спирта и растворимого в воде пектата натрия. Щелоч-
ноземельные металлы выпадают в виде карбонатов.
Сущность метода. Растворимый пектин, находящийся
в клеточном соке, извлекается теплой водой (50—60Q С) и омыляет-
ся едким натром. При этом происходит отщепление метоксильных
групп и образование метилового спирта и пектата натрия:
С26Н40О22 (СООСН3)3 СООН 4NaOH = С20Н40О22 (COONa)4 +
+ ЗСН3ОН Н.,0.
Из полученного раствора пектата натрия пектиновая кислота
осаждается хлористым кальцием в виде нерастворимой кальцие-
вой соли:
^26^40^22 (COONa)4 + 2СаС12 = С26Н40О22 (СОО)4Са2 + 4NaCL
После промывания осадка уксусной кислотой и водой от избытка
хлористого кальция пектат кальция разлагается углекислым натри-
ем при нагревании:
^-*2б^4о^22 (СОО)4 Са2 -р 2Na2CO3 = С26Н40О22 (COONa)4 -|- 2СаСО3.
При этом пектиновая кислота переходит в раствор в виде пек-
тата натрия, а кальций выпадает в виде углекислого кальция, не-
растворимого в воде. Осадок отделяется центрифугированием и раст-
воряется в уксусной или соляной кислоте:
СаСО3 + 2СН3СООН = Са (СН3СОО)2 + СО2 4- Н2О.
Если в исследуемом материале присутствует свободная щавеле-
вая кислота или ее растворимые в воде соли, то осадок СаСО3 рас-
творяется в уксусной кислоте, в которой оксалат кальция, находя-
щийся в осадке, нерастворим. В случае отсутствия щавелевой кис-
лоты осадок углекислого кальция растворяется в 0,1 н. соляной
кислоте. В осадке вместе с углекислым кальцием, кроме оксалата
141
кальция, находится небольшое количество белка, который нераство-
рим в кислоте. Этот осадок отделяют центрифугированием и в полу-
ченном растворе определяют кальций комплексонометрическим ме-
тодом с трилоном Б, который реагирует с кальцием по следующему
уравнению:
Са (СН3СОО)2 + Na2H2C10H12O8N2 = CaH2C10H12O8N2 + 2CH3COONa.
Избыток трилона Б оттитровывают сернокислым магнием. По най-
денному количеству кальция вычисляют содержание пектиновой
кислоты в пектине. В остатке после извлечения растворимого пек-
тина водой определяют содержание протопектина. Его извлекают
горячим раствором соды с небольшим количеством едкого натра.
При этом последовательно происходит выделение пектина и отщеп-
ление метоксильных групп с образованием натриевой соли свободной
пектиновой кислоты:
С26Н,10О22 (СООСН3)2 (СОО)2 Са Na2CO3 + 2NaOH =
= С2ВН10О22 (COONa)/ 2СН3ОН + СаСО3.
Извлечение в данном случае идет быстро, причем кальций пере-
водится в нерастворимый осадок, а пектиновая кислота в раствори-
мую в воде натриевую соль. В полученном растворе пектиновая
кислота осаждается в виде пектата кальция и определяется комплек-
сонометрическим методом по количеству связанного кальция. Здесь
также частично извлекаются белки, но они определению не мешают.
Ход анализа. Навеску исследуемого материала берут с
таким расчетом, чтобы в ней содержалось не более 0,09 г пектина или
протопектина. Обычно от 1 до 3 г сырого вещества (0,2—0,6 г сухо-
го вещества). Навеску растирают в ступке с 5 мл теплой дистилли-
рованной воды (60° С) до однородной массы. Содержимое переносят
в центрифужную пробирку емкостью 20 мл и остаток смывают 10 мл
теплой воды. Суспензию размешивают стеклянной палочкой и
настаивают в течение 5 мин. Затем центрифугируют при 2 000 об/мин
и раствор сливают в стакан на 50—100 мл, даже если он мутный.
Осадок промывают дважды теплой дистиллированной водой, прибав-
ляя ее по 10 мл, тщательно размешивая и центрифугируя. Промыв-
ные воды присоединяют к раствору в стакане. В полученном раство-
ре находится растворимый пектин, а в осадке — нерастворимый
пектин (протопектин).
Определение растворимого пектина
К вытяжке, содержащей растворимый пектин, добавляют 3 мл
2 н. раствора едкого натра и оставляют на ночь для омыления ме-
тилового эфира. Этот процесс можно ускорить. Для этого раствор
переливают в большую пробирку и погружают в кипящую воду на
8 мин, считая от мордента погружения. Концентрация NaOH в раство-
ре должна быть около 0,1 н. После омыления эфира к раствору пекта-*
та натрия прибавляют 1 мл ледяной уксусной кислоты, тщатель-
но перемешивают, добавляют 2 мл 25%-ного раствора хлористого
142
кальция, перемешивают, нагревают на плитке до кипения, охлаж-
дают в течение 10—15 мин и центрифугируют. Раствор сливают и от-
брасывают, а осадок промывают 4 раза 0,1 %-ным раствором уксус-
ной кислоты, прибавляя ее по 10 мл и каждый раз хорошо переме-
шивая палочкой.
К промытому осадку приливают 10 мл 1 %-ного раствора угле-
кислого натрия — Na2CO3, перемешивают и погружают пробирку
в кипящую воду на 5 мин, время от времени перемешивая стеклянной
палочкой. При этом пектат кальция разлагается: кальций выпадает
в виде СаСО3 и находится в осадке вместе с небольшим количеством
белка и оксалатом кальция (при содержании растворимой щавелевой
кислоты в исследуемом материале). Пектиновая кислота переходит
в раствор в виде пектата натрия. Осадок отделяют центрифугирова-
нием и промывают один раз 5 мл дистиллированной воды. Углекис-
лый кальций, находящийся в осадке, растворяют в уксусной кисло-
те или в соляной кислоте, если отсутствует щавелевая кислота.
Для этого к осадку в центрифужной пробирке приливают 10 мл
1%-ного раствора уксусной кислоты (или 10 мл 0,1 н. НС1), переме-
шивают, помещают пробирку в кипящую воду на 3 мин при частом
перемешивании и центрифугируют. Раствор сливают в колбу для
титрования на 100 мл, а осадок промывают один раз 10 мл дистил-
лированной воды, добавляя ее к первому раствору в колбе.
В растворе находится эквивалентное пектиновой кислоте коли-
чество кальция, которое определяют следующим образом. К раство-
ру прибавляют 5 мл 0,1 п. раствора трилона Б, 5 мл аммиачного бу-
ферного раствора, 50 мг индикатора хромогена черного ЕТ-00 и
титруют остаток трилона Б 0,02 н. раствором серпокислого магния
до перехода окраски индикатора в винно-красный цвет. Отдельно
титруют 5 мл 0,1 н. раствора трилона Б в присутствии аммиачного
буферного раствора 0,02 н. раствором сернокислого магния с
тем же индикатором. На основании полученных данных вычисляют
количество растворимого пектина по следующей формуле:
У _ 22,1 К(а — Ь)
где X — содержание пектиновой кислоты в исследуемом материале
(в %); К — нормальность титрованного раствора сернокислого
магния; а — объем 0,02 н. раствора сернокислого магния, затра-
ченный при титровании 5 мл 0,1 н. раствора трилона Б (в мл)\ b —
объем 0,02 н. раствора сернокислого магния, затраченный при титро-
вании кальция (в мл)\ п — навеска исследуемого вещества (в а);
22,1 — нормальный титр пектиновой кислоты, умноженный на 100
для пересчета в проценты.
Определение протопектина
К остатку в центрифужной пробирке после извлечения раствори-
мого пектина теплой дистиллированной водой приливают 15 мл
2%-ного раствора углекислого натрия, содержащего NaOH,
143
пробирку погружают в кипящую воду и нагревают 20 мин при частом
размешивании стеклянной палочкой. После этого центрифугируют и
раствор сливают в стакан на 50 или 100 мл. Остаток промывают
2 раза горячей дистиллированной водой порциями по 10 мл, разме-
шивая и центрифугируя.
Все центрифугаты сливают в стакан, в котором и осаждают
пектиновую кислоту. Для этого прибавляют 1 мл ледяной уксусной
кислоты, 2 мл 25%-ного раствора хлористого кальция, тщательно
перемешивают, ставят на плитку и нагревают при перемешивании
до начала кипения. Раствор охлаждают в течение 10—15 мин и цен-
трифугируют. Осадок промывают 4 раза 0,1 %-ной уксусной кисло-
той порциями по 10 мл. К промытому осадку пектата кальция
приливают 10 мл 1 %-ного раствора углекислого натрия, переме-
шивают и погружают пробирку в кипящую воду на 5 мин, перио-
дически перемешивая стеклянной палочкой. Осадок углекислого
кальция отделяют центрифугированием, раствор удаляют, а осадок
промывают один раз 5 мл дистиллированной воды. Затем к осадку
приливают 10 мл 0,1 н. раствора соляной кислоты, перемешивают
до растворения осадка СаСО3, выливают раствор в колбу для ти-
трования и остаток раствора смывают водой. Если есть нераство-
римый в НС1 темный хлопьевидный осадок, то его можно отцентри-
фугировать, прозрачный раствор слить в колбу для титрования,
а осадок промыть 10 мл дистиллированной воды, добавив промыв-
ную воду к раствору в колбе. К раствору в колбе прибавляют
3 мл аммиачного буферного раствора, 5 мл 0,1 н. раствора трилона Б,
индикатор и проводят титрование так, как это изложено выше. Со-
держание протопектина (в расчете на пектиновую кислоту) вычисля-
ется по той же формуле, что и содержание растворимого пектина.
ОПРЕДЕЛЕНИЕ СУММЫ ПЕКТИНА И ПРОТОПЕКТИНА
Сущность метода. Пектин и протопектин экстрагиру-
ются вместе с горячим 2%-ным раствором соды, содержащим не-
большое количество едкого натра. При этом идет не только извле-
чение пектина, но и отщепление метоксильных групп, так что в ре-
зультате получается свободная пектиновая кислота в виде натрие-
вой соли.
Пектиновую кислоту из уксуснокислого раствора осаждают
хлористым кальцием в виде пектата кальция, который затем отмы-
вают от избытка хлористого кальция уксусной кислотой и дистилли-
рованной водой. Количество кальция, связанное с пектиновой кисло-
той, определяют комплексонометрическим методом и пересчитывают
на пектиновую кислоту исходя из того факта, что пектат кальция
содержит 8,3% Са.
При извлечении пектиновых веществ этим методом частично из-
влекаются также белки, которые в дальнейшем сопутствуют пек-
тинам, но не реагируют с кальцием и поэтому не мешают определе-
нию пектина.
144
Ход анализа. Навеску исследуемого материала, в которой
должно быть не более 0,09 г пектиновых веществ (от 0,2 до 0,5 г су-
хого вещества или 1—2 г сырого), тщательно растирают в ступке
с 5 мл раствора, содержащего2% N2CO3 и0,16% NaOH. Суспензию
переносят в центрифужную пробирку на 20 мл и остаток смывают
щелочным раствором соды, расходуя на промывку 10 мл. Пробирку
помещают в кипящую воду и нагревают при помешивании в тече-
ние 20 мин. Затем центрифугируют, раствор сливают в небольшой
стакан, а остаток промывают два раза горячей водой, прибавляя
ее по 10 мл, тщательно перемешивая и центрифугируя. Промывные
воды присоединяют к первому центрифугату. В объединенном цен-
тр ифугате осаждают пектиновую кислоту. Для этого раствор под-
кисляют 1 мл ледяной уксусной кислоты, тщательно перемешивают,
добавляют 2 мл 25%-ного раствора хлористого кальция, снова пе-
ремешивают, ставят на плитку и нагревают при помешивании до
кипения. Затем стакан снимают с плитки, дают постоять около
10 мин при помешивании для удаления углекислоты и центрифуги-
руют. Раствор сливают и отбрасывают, а в пробирку, если не по-
мещается весь раствор, переносят остаток его и снова центрифуги-
руют. Раствор сливают с осадка и отбрасывают, а осадок промыва-
ют 4 раза 0,1%-ным раствором уксусной кислоты, смывая при этом
остатки осадка из стакана в пробирку, прибавляя ее по Ю^ли
каждый раз тщательно перемешивая палочкой.
К промытому осадку приливают 10 мл 1 %-ного раствора угле-
кислого натрия, перемешивают и погружают пробирку в кипящую
воду на 5 мин, периодически перемешивая стеклянной палочкой.
Затем центрифугируют, раствор сливают и отбрасывают, а осадок
промывают один раз 5 мл дистиллированной воды. К осадку при-
бавляют 10 мл 1%-ного раствора уксусной кислоты, перемешивают,
помещают в кипящую воду на 5 мин с частым перемешиванием стек-
лянной палочкой. После этого центрифугируют, раствор сливают
в колбу для титрования, осадок промывают 10 мл дистиллированной
воды, центрифугируют и промывную воду добавляют к раствору
в колбу. В полученном растворе определяют кальций комплексо-
нометрическим методом. Прибавляют 5 мл 0,1 н. раствора трилона
Б, 5 мл аммиачного буферного раствора, 50 мг хромогена черного
ЕТ-00 и титруют 0,02 н. раствором сернокислого магния до перехода
окраски индикатора в винно-красный цвет. Отдельно в тех же усло-
виях титруют 5 мл 0,1 н. раствора трилона Б раствором серно-
кислого магния. Из полученных данных вычисляют количе-
ство пектина в пересчете на пектиновую кислоту по форму-
ле, приведенной выше (см. метод определения растворимого пек-
тина).
Реактивы. Раствор углекислого натрия (2%) с едким натром (0,16 %).
Отвешивают 20 г безводной соли — Na2CO3, растворяют в дистиллированной
воде, добавляют 20 мл 2 н. раствора NaOH, разбавляют водой до 1 л, переме-
шивают и сохраняют в хорошо закрытой склянке.
Раствор хлористого кальция (25%-ный). Отвешивают 50 г кристаллической
10 6-2
145
соли СаС12 6Н2О, растворяют в дистиллированной воде и разбавляют до
200 мл.
Раствор соляной кислоты 0,1 н.). Набирают 8,5 мл соляной кислоты
пл. 1,19, разбавляют дистиллированной водой до 1 л и перемешивают.
Раствор трилона Б (~ 0,1 н.). Отвешивают 18,60 г высушенной при 80° С
соли Na2H2C10H12O8N2 2Н2О, растворяют в дистиллированной воде и разбавляют
до 1 л водой.
Раствор сернокислого магния (0,02 н.). Взвешивают на аналитических весах
2,4650 г MgSO4 7Н2О, растворяют в дистиллированной воде, переносят в мерную
колбу на 1 л, доводят водой до метки и перемешивают.
Приготовление остальных реактивов, применяемых в данном методе, описано
выше (см. стр. 12, 13, 49).
ОПРЕДЕЛЕНИЕ ГЕМИЦЕЛЛЮЛОЗ И ПЕНТОЗАНОВ
Гемицеллюлозы широко распространены в растениях., они со-
держатся в стенках клеток и являются промежуточным звеном между
целлюлозой и крахмалом. Они играют в растениях двоякую роль —
механическую и резервного вещества. Гемицеллюлозы исчезают
при прорастании семян. Устойчивость их к воздействию кислот
носит промежуточный характер между устойчивостью целлюлозы
и крахмала — они гидролизуются гораздо легче целлюлозы, но
труднее крахмала. При гидролизе гемицеллюлоз кроме d-глюкозы
образуются и другие сахара: d-маноза, d-галактоза, /-арабиноза
и /-ксилоза.
По продуктам гидролиза гемицеллюлозы делятся на гексозаны
и пентозаны. К первой группе принадлежат декстраны, маннаны,
галактаны, галактоманны и другие, ко второй — арабаны и ксиланы.
Встречаются также полиозы смешанного характера, состоящие из
гексоз и пентоз, например галактоарабаны. В растениях гексозаны
и пентозаны присутствуют большей частью одновременно. В геми-
целлюлозах, играющих роль резервного вещества, преобладают
гексозаны, а в гемицеллюлозах с механической функцией — пенто-
заны.
Большинство гемицеллюлоз нерастворимо в воде, а раствори-
мые дают лишь коллоидные растворы. Некоторые растворимы
в едких щелочах, например, пентозаны извлекаются из растений
разбавленными растворами щелочей и осаждаются кислотами и эти-
ловым спиртом.
Сущность метода. Крахмал растворяют в кипящем
80%-ном растворе азотнокислого кальция и отделяют центрифуги-
рованием. При этом удаляются и другие углеводы, растворимые
в воде, мешающие определению гемицеллюлоз. После промывания
остатка дистиллированной водой гемицеллюлозы гидролизуют 2 н.
соляной кислотой при температуре, близкой к кипению воды. Гид-
ролиз идет по следующим уравнениям:
(СбН10Об)п + пН2О = пС6Н12О6,
гексозаны гексоза
4" ПН2О = пС5Н10О6.
пентозаны пентоза
146
Использование более высокой концентрации соляной кислоты
дае^ возможность значительно уменьшить время гидролиза геми-
целлюлоз по сравнению с применявшейся до сих пор методикой.
Раствор сахаров, полученный в результате гидролиза гемицеллюлоз,
разбавляют до определенного объема и нейтрализуют едким натром.
В этом растворе общее количество сахара определяют медно-йо-
дометрическим методом.
При гидролизе пентозанов образуются пентозы — арабиноза,
ксилоза, рибоза, рамноза, которые определяют колориметрическим
методом с орциновым реактивом. Из найденного общего количества
редуцирующих сахаров рассчитывают содержание гемицеллюлоз,
а из общего количества пентоз — содержание пентозанов умноже-
нием на соответствующий коэффициент. Для определения отдель-
ных представителей гек-
соз и пентоз применя-
ется метод количествен-
ной бумажной хрома-
тографии.
Ход анализа.
80
25 25
Отвешивают на анали- рИс. 25. Шариковая пробка с палочкой,
тических весах 0,1—
0,2 г сухого растительного материала (или соответствующее
количество сырого), растирают в ступке с 5 мл 80%-ного раствора
азотнокислого кальция, смесь переносят в колбу на 100 мл, промы-
вают ступку 10 мл 80%-ного раствора азотнокислого кальция, при-
соединяя промывные воды к основному раствору. Колбу накры-
вают воронкой, ставят па плитку, нагревают до кипения и слабо
кипятят 5 мин. После этого колбу снимают с плитки, добавляют
20 мл дистиллированной воды, споласкивая воронку, перемешивают
и переносят в центрифужную пробирку столько раствора, сколько
поместится. Центрифугируют и раствор сливают в мерную колбу
на 100 мл, если определяется и крахмал, в противном случае раствор
отбрасывают. В пробирку наливают новую порцию жидкости, цен-
трифугируют, раствор сливают и поступают так до тех пор, пока
весь раствор будет отцентрифугирован. Осадок, содержащий
гемицеллюлозы, промывают 3 раза горячей водой порциями по
5 мл.
К промытому осадку в пробирке прибавляют 10 мл 2 н. раствора
соляной кислоты, закрывают шариковой пробкой с палочкой
(рис. 25), перемешивают, погружают в кипящую воду и нагревают
при слабом кипении в течение 45 мин, периодически размешивая.
После этого центрифугируют и раствор сливают в мерную колбу
на 50 или на 100 мл, в зависимости от количества сахаров. Остаток
промывают 2—3 раза горячей водой, прибавляя ее по 10 мл и присое-
диняя промывные воды к основному раствору. К полученному
раствору прибавляют одну каплю раствора фенолфталеина и ней-
трализуют 2 н. едким натром (около 10 мл) до появления розовой
окраски. Затем раствор разбавляют дистиллированной водой до
10;
147
метки, перемешивают и фильтруют в сухую колбу через сухой
фильтр, выбрасывая первую небольшую часть фильтрата.
Для определения сахаров из остатка фильтрата набирают пипет-
кой 5 или 10 мл, переносят в большую пробирку (диаметром 30 мм),
разбавляют дистиллированной водой до 10 мл, если брали 5 мл
исследуемого раствора, прибавляют 10 мл медно-щелочного реакти-
ва, закрывают воронкой или шариковой пробкой и ставят в кипя-
щую воду на 15 мин. После этого охлаждают, прибавляют 5 мл сме-
си щавелевой и серной кислот и титруют выделившийся йод 0,01 н.
раствором тиосульфата натрия. Подробнее определение восстанав-
ливающих сахаров описано выше (см. стр. 117). На основании
полученных данных вычисляют количество гемицеллюлоз по сле-
дующей формуле:
У = 0,9 Л[248 —(д —6)](д —6) _ 0,09 А е
10 000 v п v п
где X — содержание гемицеллюлоз (в %); А — общий объем ис-
следуемого раствора (в мл)-, v — объем исследуемого раствора, взя-
тый для определения сахаров (в мл); п — навеска исследуемого
вещества (в г); а — объем 0,01 и. раствора тиосульфата, затраченный
при титровании контрольного раствора (в мл); b — объем 0,01 н.
раствора тиосульфата, затраченный при титровании исследуемого
раствора (в мл); 0,9 — коэффициент для пересчета гексоз в гемицел-
люлозы; 248 — (а — Ь) — количество сахара, соответствующее
1 мл 0,01 н. раствора тиосульфата (в мкг); f — фактор, величины
которого приведены в табл. 3.
ОПРЕДЕЛЕНИЕ ПЕНТОЗ И ПЕНТОЗАНОВ
Из раствора, оставшегося после определения гемицеллюлоз,
набирают пипеткой 5 мл, переносят в мерную колбу на 50 мл, при-
бавляют каплями при встряхивании 2 н. раствор соляной кислоты
до обесцвечивания фенолфталеина, разбавляют дистиллированной
водой до метки и перемешивают. 1 мл этого раствора помещают
в обыкновенную пробирку, прибавляют 4 мл орцинового реактива,
закрывают пробирку шариковой пробкой и ставят в кипящую воду
на 15 мин. После этого охлаждают и измеряют оптическую плот-
ность при 610—660 нм в кювете 10 мм. На основании полученных
данных оптической плотности по калибровочной кривой находят
концентрацию пентозы в калориметрируемом растворе (в мкг/мл)
и рассчитывают содержание пентоз по следующей формуле:
у__ 50 В С а _____ В • С • а
Л ~ 5 п 10 000 1000 п 9
где X — содержание пентоз (в %); В — первоначальный объем
исследуемого раствора (в мл); 5 — объем первоначального раствора,
взятый для разбавления (в мл); 50 — объем исследуемого раствора
после разбавления (в мл); п — навеска исследуемого вещества
(в г); а — объем колориметрируемого раствора (в мл); С —
148
концентрация пентоз в колориметрируемом растворе (в мкг/мл)\
10 000 — коэффициент для перевода микрограммов в граммы и пере-
счета в проценты.
Цля вычисления количества пентозанов в исследуемом веществе
в процентах полученный результат умножают па коэффициент 0,88.
ОПРЕДЕЛЕНИЕ ОТДЕЛЬНЫХ ГЕКСОЗ И ПЕНТОЗ
Из первоначального раствора, оставшегося после определения
гемицеллюлоз, набирают 10 или 20 мл, переносят в фарфоровую
чашку, добавляют по каплям при перемешивании раствор 10%-пой
уксусной кислоты до обесцвечивания
фенолфталеина, помещают чашку на
водяную баню и выпаривают досуха.
После охлаждения остаток растворя-
ют в 1 мл дистиллированной воды,
раствор переносят в коническую цен-
трифужную пробирку, центрифуги-
руют и сливают прозрачный раствор
в чистую сухую пробирку. Из этого
раствора набирают микропипеткой
0,01 мл и наносят на ленту хрома-
тографической бумаги, подготовлен-
ной так, как описано выше в хрома-
тографическом методе определения
сахаров (см. стр. 128).
Бумажные ленты с нанесенными
сахарами помещают в камеру и про-
пускают растворитель я-бутанол —
уксусная кислота — вода (6 2 3).
В дальнейшем поступают, как указано
выше (см. стр. 129). Для определения
Рис. 26. Калибровочная кривая
для пентоз с орциновым реак-
тивом (светофильтр с максиму-
мом пропускания 660 нм);
1 — толщина измеряемого слоя
10 мм; 2 — толщина измеряемого
слоя 5 мм.
пентоз можно пользоваться орцино-
вым методом. В этом случае сахар из бумаги извлекают 3 мл дис-
тиллированной воды и для определения берут 1 мл экстракта.
Прежде, чем определять количество сахаров хроматографическим
методом, необходимо установить качественный состав исследуемых
сахаров. Для этого на хроматограмму рядом с исследуемым раство-
ром наносят растворы «свидетелей». Если в исследуемом растворе
обнаружено только одно вещество, определение следует проводить
более точным и быстрым объемным методом.
Построение калибровочной кривой для
пентоз. В сухие пробирки набирают 0,1, 0,2, 0,3, 0,4, 0,6,
0,8 и 1 мл стандартного раствора ксилозы, содержащего 100 мкг
сахара в 1 мл раствора, приливают дистиллированную воду до
1 мл и затем во все пробирки прибавляют по 4 мл орцинового реак-
тива. Растворы перемешивают, пробирки закрывают шариковыми
пробками и погружают в кипящую воду на 15 мин. После этого
149
охлаждают и измеряют оптическую плотность при 610—660 нм
в кювете па 10 мм. На основании полученных данных строят кали-
бровочную кривую по образцу (рис. 26).
Реактивы. Орциновый реактив. 0,2 г орцина СбН3 СН3 (ОН)2 раство-
нпот в 100 мл соляной кислоты (7:3). К полученному раствору прибавляют
2 капли насыщенного раствора FeCl3. Сохраняют в темном месте.
Стандартный раствор ксилозы (0,1 мг!мл). Отвешивают на аналитических
весах 50 мг ксилозы С5Н10О5, растворяют в дистиллированной воде в мерной
колбе на 500 мл, добавляют 5 мл 2 н. раствора соляной кислоты и разбавляют
водой до 500 мл. В 1 мл этого раствора содержится 100 мкг ксилозы.
Раствор соляной кислоты (2 н.). Отбирают 167 мл соляной кислоты пл. 1,19
в мерную колбу на 1 л, разбавляют дистиллированной водой до метки и пере-
мешивают. Приготовление остальных реактивов описано выше (см. стр. 76,
121, 138).
ОПРЕДЕЛЕНИЕ ЛИГНИНА
Лигнин — ароматическое вещество, пропитывающее клеточные
стенки одеревенелых тканей растений. В молодых растениях его
мало; с возрастом количество лигнина в тканях значительно повы-
шается. Он весьма устойчив к воздействию кислот. На этом основа-
но отделение его от других составных частей растения. Выделенный
лигнин представляет собой аморфное вещество коричневого цвета,
менее стойкое по отношению к окислителям и щелочам, чем целлю-
лоза. Лигнин растворяется в бисульфите натрия и сернистой кисло-
те. Основным структурным элементом лигнина является оксигидро-
конифериловый спирт:
ОСН3
I
НО— С6Н3—СН2—снон—СН2ОН.
По данным многих исследователей, полученным при изучении
различных препаратов лигнина, элементарный состав его колеблет-
ся между С10Н10О3 и СПН12О4.
Сущность метода. Лигнин отделяется от сопутствую-
щих веществ обработкой исследуемого материала 1%-ной уксусной
кислоты для удаления сахаров, органических кислот и других рас-
творимых веществ, затем ацетоном или смесью этилового спирта
и эфира (1 1) для удаления хлорофилла, липидов, смол и других
растворимых в органических растворителях веществ и 72%-ной
серной кислотой для удаления целлюлозы и гемицеллюлоз. После
промывания остатка дистиллированной водой от продуктов гидро-
лиза полученный лигнин окисляется бихроматом калия в присут-
ствии серной кислоты:
СИН12О4 + 8К2Сг2О7 + 32H2SO4 = 11СО2 + 8K2SO4 -ф
-ф 8Сг2 (SO4)3 -ф 38Н2О.
Избыток бихромата калия определяют титрованием его солью Мора
или йодометрическим методом.
Грамм-эквивалент лигнина равен 4,33 и был установ-
150
лен с помощью определения расхода бихромата калия на окисле-
ние препаратов лигнина, полученных из льна.
Ход анализа. Отвешивают па аналитических весах
0,05—0,1 г измельченного сухого или 0,2—0,5 г сырого исследуе-
мого материала (в навеске должно быть от 2 до 18 мг лигнина).
Сухой материал всыпают в центрифужную пробирку н добавляют
10 мл 1%-ного раствора уксусной кислоты. Сырой материал расти-
рают в ступке с 5 мл 1 %-ного раствора уксусной кислоты и перено-
сят в центрифужную пробирку, смывая остаток этой же кислотой.
Содержимое пробирки перемешивают в течение 5мин и центрифу-
гируют. Раствор сливают, а остаток промывают один раз 5 мл 1 % -пой
уксусной кислоты и затем 3 раза ацетоном (или смесью спирт —
эфир, 1 1), прибавляя его порциями по 3—4 мл и настаивая при
помешивании 3 мин. После этого осадок распределяют стеклянной
палочкой по стенкам пробирки, чтобы избежать выбрасывания,
помещают в горячую воду и нагревают до полного высыхания
в течение 5—10 мин. К сухому остатку в пробирках приливают
по 3 мл 72 %-ного раствора серной кислоты пл. 1,63 и перемешивают
стеклянной палочкой, разминая комочки до получения однородной
массы. Затем оставляют на 16 ч (обычно на ночь) при комнатной
температуре для растворения всей клетчатки.
На следующий день в пробирки прибавляют по 10 мл дистилли-
рованной воды, размешивают стеклянной палочкой и помещают
в кипящую воду на 5 мин. Раствор охлаждают, добавляют по 5 мл
дистиллированной воды, 0,5 мл 10%-ного раствора хлористого
бария и размешивают. Хлористый барий с серной кислотой образу-
ет тяжелый осадок BaSO4, который увлекает более легкий лигнин
и этим облегчает центрифугирование и удаление падосадочной жид-
кости. После этого центрифугируют, прозрачный раствор сливают
по стеклянной палочке, а осадок промывают 2 раза водой пор-
циями по 10 мл, тщательно перемешивая после прибавления каждой
порции воды. К промытому осадку лигнина прибавляют 10 мл
0,5 н. раствора бихромата калия в серной кислоте, помещают про-
бирку в кипящую воду и выдерживают 15 мин при периодическом
размешивании стеклянной палочкой. Затем охлаждают и все со-
держимое пробирки переносят в колбу для титрования, смывая
остаток 15—20 мл дистиллированной воды. К охлажденному рас-
твору добавляют 4—5 капель 0,2%-ного раствора фенилантраниловой
кислоты и титруют 0,1 н. раствором соли Мора до перехода вишнево-
фиолетовой окраски в зеленую. Для контрольного титрования берут
10 мл 0,5 н. раствора бихромата калия в серной кислоте, помещают
в колбу для титрования, прибавляют 15—20 мл дистиллированной
воды, охлаждают и титруют 0,1 н. раствором соли Мора в присут-
ствии индикатора фенилантраниловой кислоты.
Из полученных данных вычисляют содержание лигнина по фор-
муле:
У _ 0,433 К(а — Ь)
151
где X — содержание лигнина (в %); а — объем 0,1 н. раствора соли
Мора, затраченный при контрольном титровании 0,5н. раство-
ра бихромата калия (в мл)*, b — объем 0,1 н. раствора соли Мора,
затраченный при титровании избытка бихромата калия (в мл);
/< — нормальность титрованного раствора соли Мора; и — навес-
ка исследуемого вещества (в г); 0,433 — нормальный титр лигнина,
умноженный на 100.
Реактивы. Раствор серной кислоты (72%-ный). К 100 мл дистиллированной
воды приливают постепенно при охлаждении 170 мл концентрированной серной
кислоты. Раствор охлаждают до 20° С, измеряют ареометром плотность и, если
она отличается от величины 1,630, добавляют небольшими порциями концентри-
рованную серную кислоту или дистиллированную воду, пока плотность раствора
не достигнет нужной величины. Раствор сохраняют в склянке с притертой пробкой.
Приготовление остальных реактивов описано выше (см. стр. 40, 138).
ОПРЕДЕЛЕНИЕ ФРУКТОЗАНОВ
Фруктозаны представляют собой группу запасных полисаха-
ридов, в состав которых наряду с глюкозой входит и фруктоза.
Они широко распространены в растениях и являются их запасным
энергетическим фондом. Фруктозаны трудно растворимы в холодной
воде, хорошо растворимы в горячей воде и нерастворимы в спирте.
Они не обладают редуцирующей способностью, но легко гидроли-
зуются кислотой, образуя редуцирующие сахара.
Сущность метода. Исследуемый растительный мате-
риал обрабатывается 80%-ным этиловым спиртом для удаления
всех низкомолекулярных сахаров, которые могут мешать опреде-
лению фруктозанов. Из промытого спиртом остатка фруктозаны из-
влекают горячей водой и после осветления раствора определяют
колориметрическим методом с помощью молибдат-сернокислого
реактива.
Ход анализа. Отвешивают 0,5—1 г измельченного свежего
растительного материала, содержащего от 1 до 10 мг фруктозанов,
и растирают в ступке с 3 мл 95 %-ного этилового спирта до одно-
родной массы. При извлечении из сухого материала берут 3 мл
80%-ного спирта. Содержимое ступки переносят в центрифужную
пробирку с помощью 10 мл 80%-ного этилового спирта и настаивают
3 мин при периодическом перемешивании стеклянной палочкой.
После этого центрифугируют при 2000 об/мин, раствор сливают,
а осадок промывают еще 3 раза порциями по 5 мл 80%-ного спирта,
настаивая перед центрифугированием по 2—3 мин при перемеши-
вании. Спиртовые экстракты можно использовать для определения
низкомолекулярных сахаров или после перегонки спирт исполь-
зовать в работе.
Остаток, содержащий фруктозаны, переносят из пробирки в мер-
ную колбу на 50 мл с помощью 40 мл дистиллированной воды. Кол-
бу помещают в кипящую воду и нагревают в течение 30 мин. Затем
охлаждают, прибавляет 1 мл 1 н. раствора едкого натра, одну
152
каплю раствора фенолфталеина и прибавляют по каплям 10%-ный
раствор сернокислого цинка до обесцвечивания индикатора. Рас-
твор разбавляют дистиллированной водой до 50 мл, тщательно
перемешивают, дают отстояться, отбирают пипеткой из верхней
прозрачной части раствора 3 мл, содержащих от 30 до 200 мкг
фруктозанов, и выливают в сухую пробирку. Если в 3 мл находится
фруктозанов больше 200 мкг, то берут соответственно мспыпсе коли-
чество раствора и разбавляют дистиллированной водой до 3 мл.
К раствору в пробирке прибавляют 2 мл молибдат-сернокислого
реактива, перемешивают, закрывают пробирку стеклянной ша-
риковой пробкой и ставят в кипящую воду на 20 мин. После это-
го охлаждают, доводят объем водой до 5 мл и измеряют оптиче-
скую плотность раствора при 660 нм в кювете на 10 мм. Раствором
для сравнения служит вода. Содержание фруктозанов вычисляют
по формуле:
х = 50.5 С _ 0,00833 • С
Л 10 000 • п 3 п
где X — содержание фруктозанов (в % в расчете на раффинозу);
50 — объем исследуемого раствора фруктозанов (в мл); 3 — объем
исследуемого раствора, взятый для получения окрашенного раство-
ра (в мл); 5 — объем колориметрируемого раствора (в мл); С —
концентрация фруктозанов в колориметрируемом растворе (в мкг!мл,
в расчете на раффинозу); п —навеска исследуемого вещества (в г);
10 000 — переводной коэффициент микрограммов в граммы и про-
центы.
Калибровочную кривую строят так, как описано выше (см.
стр. 131), а приготовление реактивов (см. стр. 28, 133).
Если необходимо определить в отдельности альдозы и кетозы,
входящие в состав фруктозанов, то последние подвергают кислот-
ному гидролизу, а затем определяют отдельно содержание фрукто-
зы и глюкозы методами, изложенными выше. Для этого из фильтра-
та, содержащего фруктозаны и находящегося в мерной колбе на
50 мл, отбирают 25 мл, переносят в мерную колбу на 50 мл, прибав-
ляют 2мл 1 н. раствора соляной кислоты, помещают колбу в кипя-
щую воду и нагревают 20 мин. Затем охлаждают, прибавляют одну
каплю раствора фенолфталеина и нейтрализуют 1 н. раствором ед-
кого натра до появления розовой окраски. Объем раствора доводят
дистиллированной водой до 50 мл и перемешивают. В полученном
растворе определяют содержание фруктозы и глюкозы с железоси-
неродистым реактивом по методу, изложенному выше (см. стр. 123).
При использовании объемного метода берут навеску с таким рас-
четом, чтобы в ней находилось около 40 мг фруктозанов, а для коло-
риметрического метода навеска должна содержать от 6 до 10 мг
фруктозанов.
153
ПОЛЯРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ УГЛЕВОДОВ
Как известно, ряд веществ обладает оптической активностью,
т. е. способностью вращать на некоторый угол плоскость поля-
ризации проходящего через них поляризованного луча. Оптическая
активность органических веществ связана с наличием в молекуле
асимметрических атомов углерода. Угол поворота плоскости поля-
ризации пропорционален длине проходимого в активной среде
пути и пропорционален концентрации активного вещества, если
это вещество находится в растворенном состоянии.
Удельной вращательной способностью называют угол поворота
плоскости поляризации [а] при прохождении лучом пути длиной
в 1 дм в среде с концентрацией активного вещества 100 г в 100 мл
раствора.
При длине трубки I (в дм) и концентрации С (в г) в 100 мл рас-
твора угол поворота а плоскости поляризации выразится формулой:
Пропорциональность угла вращения и концентрации активного
вещества является основой для оптической сахариметрии: по вели-
чине угла вращения а определяют концентрацию сахара или крах-
мала, зная его удельную вращательную способность. Удельная
вращательная способность [cq^? наиболее распространенных угле-
водов следующая: сахарозы 4-66,5, глюкозы 4-52,5, фруктозы —
91,5, мальтозы 4-138,5, раффинозы 4-123,0, декстрина 4-194,8,
крахмала картофельного 4-195,4, кукурузного 4-184,6, пшенич-
ного 4-182,7, ржаного -+184,0, ячменного 4-181,5, овсяного
+ 181,3, просяного +171,4, гречневого+179,5 и крахмала рисово-
го +185,9.
Многие органические вещества, находящиеся в растениях, обла-
дают оптической активностью и могут вносить значительные ошиб-
ки при поляриметрическом определении углеводов. Например,
аминокислоты обладают правым вращением, белки — левым, неко-
торые органические кислоты вращают вправо, некоторые — влево.
Определение сахара в сахарной свекле
При поляриметрическом определении сахара в сахарной свекле
с осветлением раствора основным уксуснокислым свинцом ошибки
могут быть вызваны лишь наличием яблочной кислоты, аминокислот
и арабана. Из углеводов может иметь значение наличие раффинозы.
При тех небольших количествах мешающих веществ, которые встре-
чаются в сахарной свекле, суммарная ошибка не превышает 0,04%,
так как происходит взаимная компенсация оптических активностей
различных несахаров, присутствующих также в небольших коли-
чествах. Поэтому поляриметрический метод определения сахарозы
в сахарной свекле является быстрым и точным.
154
Специально приспособленные для определения сахарозы поляри-
метры, называемые сахариметрами, имеют международную сахар-
ную шкалу. Эта шкала дает отсчет 100 в том случае, когда в 100 мл
водного раствора содержится 26,00 г чистой сахарозы и раствор
поляриметрируется в трубке длиной 200 мм при 20° С. Навеска
26 г называется нормальной навеской, а поляриметрическая трубка
в 200 мм — нормальной трубкой.
Шкала сахариметра указывает непосредственно процент саха-
розы в исследуемом веществе, если взята нормальная навеска этого
вещества, раствор ее доведен до 100 мл и поляриметрия проведена
в трубке длиной 200 мм. Поэтому при поляриметрическом опреде-
лении сахара в сахарной свекле берут или нормальную навеску
вещества или кратные ей величины.
Ход анализа. Отбор средней пробы является чрезвы-
чайно важным делом. Самый точный анализ будет совершенно бес-
полезен, если для анализа взята проба, не соответствующая сред-
нему составу продукта. Различия в сахаристости между отдельными
корнями могут достигать значительной величины, особенно, если
корни имеют разную величину. Обычно большие корни имеют мень-
шую сахаристость, чем корни средней величины. Поэтому нужно сле-
дить, чтобы соотношение крупных, средних и мелких корней в пробе
было таким, как и во всей массе свеклы, из которой отбирается проба.
Чтобы выровнять различия в составе отдельных корней необходи-
мо брать пробу из многих корней. Опыт показал, что для анализа
нужно брать не менее 40 корней свеклы, чтобы сгладить влияние
колебаний в их составе.
Отобранную пробу корней измельчают на особых механических
терках, где из каждого корня вырезается сектор, или всю пробу
распиливают специальным набором циркулярных пил. Вместо
механической терки иногда (при небольшом количестве проб) приме-
няют обычную ручную терку (400 мм X 200 мм) из продырявлен-
ной жести. Так как измельчать целиком все корни нет необходи-
мости, их разрезают вдоль на две половинки. Используют только
одну половинку каждого корня, причем истирают ее вдоль наполо-
вину, что составляет четвертую часть корня.
Для селекции необходимо использовать среднюю, но небольшую
пробу для определения сахаристости, а главную массу корня со-
хранять для высадки на семена. В этом случае из корня наискось
высверливают небольшой цилиндр (свечу) диаметром 18 мм. Выре-
занную свечу превращают в тонко измельченную «шлифованную»
кашку путем продавливания через тонкое латунное сито, которое
служит дном цилиндра рычажного пресса. В шлифованной кашке
все клетки свеклы разрушены, поэтому сахар из нее легко извле-
кается холодной водой. Можно также брать пробу при помощи
сверла Кейль — Долле, что ускоряет анализ.
Полученную мезгу тщательно перемешивают и из нее берут на-
веску. К этой навеске прибавляют определенный объем раствора ос-
новного уксуснокислого свинца, который осаждает белки и другие
155
оптичсски-активные в щества, сахара же переходят в раствор.
Это извлечение сахара называется дигестия. Если сахар извлекают
при нагревании до температуры 80° С, процесс называется горячей
дигестией, если же извлечение сахара производится при обычной
комнатной температуре — холодной дигестией.
Горячая дигестия. Терки, измельчая свеклу, остав-
ляют много неразрушенных клеток, поэтому в данном случае нужно
пользоваться методом извлечения сахара при нагревании — горя-
чей дигестией. Полученную мезгу тщательно перемешивают и из
нее берут навеску в 26,00 г на технических весах в тарированную
фарфоровую, стеклянную или специальную нейзильберовую чашку,
из которой затем переносят в широкогорлую мерную колбу для ди-
гестии с меткой на 201,5 мл. Лишние 1,5 мл взяты для возмещения
объема, занятого нерастворимой частью мезги, а также осадком
от основного уксуснокислого свинца. Наливают в колбу до 4/5 ее
объема раствор основного уксуснокислого свинца и перемешивают.
Колбу ставят в воду, нагретую до 85° С, и оставляют в ней на 30 мин,
поддерживая температуру раствора в колбе в пределах 75—80° С
и часто взбалтывая ее содержимое. Для контроля за температурой
в раствор погружают термометр. Через полчаса колбу вынимают,
дают охладиться, доливают до метки раствор основного уксус-
нокислого свинца, тщательно перемешивают, фильтруют через
сухой фильтр в сухую колбу и поляриметрируют в 400-миллиметро-
вой трубке. Отсчет получают в процентах сахара в свекле. В случае
отсутствия специальных колб можно пользоваться обыкновенными
мерными колбами на 200 или на 100 мл. В колбу на 200 мл берут
навеску мезги 25,83 а, а в колбу на 100 мл навеску 12,91 г.
Холодная дигестия. Метод холодной дигестии са-
мый простой и быстрый, но он требует, чтобы все клетки были раз-
рушены, т. е. чтобы кашка была «шлифованной». Метод холодной
дигестии применяется главным образом при селекции сахарной
свеклы, где требуются массовые анализы отдельных корней. Он
выполняется в этом случае как микрометод с 1/4 нормальной навески
(6,5 г), к которой приливается 44,55 мл разбавленного раствора
основного уксуснокислого свинца. Поляриметрируют в 200-милли-
метровой трубке. Результат умножают на два. Объем прибавляе-
мого раствора основного уксуснокислого свинца получен на осног
вании допущения, что корнеплод сахарной свеклы содержит 91%
сока и 17% сахара.
Взвешивание мезги производят на технических весах с точностью
до 0,01 г. Навеску берут в тарированную лодочку или на кружок
из пергаментной бумаги определенных размеров и веса. Последний
способ является более удобным, так как перед добавлением осаж-
дающего реактива кружок с отвешенной мезгой вкладывают в ста-
канчик. К отвешенной пробе мезги (6,5 г), помещенной в стакан-
чик и снабженной этикеткой, приливают из автоматической пипетки
44,55 мл раствора основного уксуснокислого свинца. Особен-
ность автоматической пипетки — наличие двухходового крана,
156
с помощью которого пипетка наполняется, а при повороте крана
на 90° опорожняется через нижнюю трубку; при наполнении пипет-
ки излишек жидкости выливается через верхнюю боковую трубку
(рис. 27).
Дигестию продолжают при периодическом помешивании в те-
чение 45 мин. Полученный раствор фильтруют через простой бу-
мажный фильтр в сухую посуду. Фильтрат
наливают в 200-миллиметровую поляриза-
ционную трубку и поляриметрируют в
сахариметре. Отклонения в параллельных
пробах не должны превышать 0,2%. При
большом количестве определений лучше
пол ьзоваться пол я р иметр ической тр у б кой
непрерывного действия, обеспечивающей
более высокую производительность работы.
Такая трубка отличается от обычной тем,
что на одном конце ее сбоку прикреплена
воронка, а на другом конце—трубка для
сливания. Воронка и трубка сообщается с
главным каналом поляризационной трубки.
Раствор вливается в воронку, а избыток его
выливается через трубку для сливания на-
ружу. При отсутствии автоматической пи-
петки на 44,55 мл можно брать навеску
мезги 7,30 г и доливать 50 мл раствора
основного уксуснокислого свинца обычной
пипеткой.
Рис. 27 Автоматическая
пипетка для наливания
основного уксуснокислого
Реактивы. Раствор основного уксуснокислого свинца.
свинца. 300 г уксуснокислого свинца
РЬ (СН3СОО)2 ЗН2О растирают с 90 г свинцового глета РЬО в прису-
тствии 50 мл дистиллированной воды. Смесь переносят в фарфоровую чашку,
накрывают стеклом и нагревают на водяной бане до тех пор, пока первоначальная
желтая масса не приобретет белого или розово-белого цвета. При этом образуется
основная уксуснокислая соль 2РЬ (СН3СОО)2 РЬ (ОН)2. Затем при поме
шивании прибавляют 950 мл горячей воды, переносят в колбу, закрывают, взбал-
тывают и оставляют смесь стоять в теплом месте сутки. Раствор сливают с осадка
и сохраняют в плотно закрытой склянке. Этот раствор называется маточным.
Маточный раствор должен иметь плотность 1,235—1,240, окрашивать лакмусовую
бумажку в синий цвет и не окрашивать раствор фенолфталеина.
Из маточного раствора готовят рабочий раствор основного уксуснокислого
свинца. Для этого на каждый литр дистиллированной воды берут 25—28 мл
маточного раствора, перемешивают и дают постоять сутки в закрытой склянке.
Ускоренный поляриметрический метод
определения сахара в сахарной свекле
Сущность метода. В описанном выше методе поляри-
метрического определения сахара в сахарной свекле взвешивание
и дигестия занимают много времени, особенно при горячей дигестии.
В данном методе сахар определяют в соке сахарной свеклы, что
157
исключает взвешивание и дигестию. Этим заметно сокращается
время проведения анализа. Сущность метода заключается в том,
что мезга, полученная любым способом и отмеренная меркой, сме-
шивается с сухим свинцовым осветлителем, а необходимое количество
сока отделяется центрифугированием. Полученный сок поляримет-
рируют в микрополяризационной трубке. Шкала показывает объем-
ные проценты сахара — граммы в 100 мл сока.
Исходя из плотности сахарных растворов при 20° С, установлен-
ной с достаточной точностью, объемные проценты можно перевести
в весовые проценты по следующей формуле:
Р = 1 + 0.0037С ’ W
где Р — весовой процент сахара в соке (1 г сахара в 100 г сока);
С — объемный процент сахара в соке (1 г сахара в 100 мл сока).
Таким образом, в результате мы получаем содержание сахара
в соке сахарной свеклы в весовых процентах, что может быть до-
статочным для сравнительных целей. Производство же интересует
содержание сахара в свекле, а не в соке, поэтому в этом случае
полученный результат необходимо пересчитать в весовые проценты
сахара в свекле. Процентный состав основных веществ корня сахар-
ной свеклы, влияющих на анализ, можно выразить следующим
уравнением:
X + Н + Н2О = 100, (2)
где X — содержание сахара в корне (в %); Н — содержание сухих
несахаристых веществ (в %); Н2О — содержание воды в корне
(в %).
Процентное содержание сока в свекле будет равняться сумме
процентного содержания сахара и воды минус процент связанной во-
ды (by. X + Н2О — Ь. Отсюда в 100 г сока находится Р г сахара, а в
(X + Н2О — Ь) г сока находится X г сахара (где X — процент са-
хара в сахарной свекле). Откуда имеем:
у _ Р(Х + Н2О-Ь) п
л 100 •
Заменив Р в этом уравнении на значение его, взятое из уравне-
ния (1), и после преобразования получим:
X — (НгО Ь) /д\
100 —0,63С ’ W
Опытным путем найдено, что количество связанной воды в кор-
не сахарной свеклы в среднем равно 2%. Подставляя эту величину
в формулу (4), получим следующее уравнение:
у = С (Н2О — 2)
100 —0,63С • v 7
Это уравнение дает возможность вычислить процентное содержание
сахара в сахарной свекле по его количеству в соке и содержанию
158
воды в свекле. В том случае, когда в сахарной свекле определяется
процент сухого вещества, нет смысла определять содержание саха-
ра методом дигестии, так как определение сахара в свекле получа-
ется более точно и быстро по содержанию его в соке с учетом влаж-
ности корня. Такое определение является особенно правильным
при отборе более сахаристых корней с селекционной целью.
Если вместо процентного содержания воды в корне (Н2О) в урав-
нение (5) подставить его значение, взятое из уравнения (2), то после
преобразования получим:
С (100 — X — Н — 2) _ (98 —Я) С
Л “ 100 — 0,63С 100 + 0,37С W
Следовательно, для получения содержания сахара в свекле
из концентрации его в соке нужно знать содержание несахаристых
Рис. 29. Проточная поля-
риметрическая трубка' для
определения сахара в
соке.
веществ в корне. Многочисленные
наблюдения показали, что ко-
Рис. 28. Принадлежности для
поляриметрического опреде-
ления сахара в соке:
а — широкогорлая воронка для
наполнения патрона мезгой;
б — центрифужный стакан; в
патрон для мезги и фильтрова-
ния сока центрифугированием
личество несахаристых веществ в зрелых свежеубранных корнях
колеблется в пределах от 6 до 8%. При среднем содержании этих
веществ, равном 7%, получим следующую формулу для вычисле-
ния процентного содержания сахара в сахарной свекле, если из-
вестна его концентрация в соке:
91С
100 + 0,37С
(7)
Таким образом, в данном случае точность пересчета будет зави-
сеть от колебаний в содержании несахаров. При отклонении в со-
держании несахаров в сахарной свекле на один процент ошибка
пересчета будет составлять 0,2% сахара. Нужно иметь ввиду, что
и в методах с дигестией, где к навеске прибавляется определенное
количество раствора основного уксуснокислого свинца, тоже услов-
но принято содержание сока в сахарной свекле 91% и сахаристость
17%, поэтому колебания в их содержании вызывает аналогичную
ошибку.
Ход анализа. Мезгу получают любым из доступных спосо-
бов. При индивидуальном анализе небольшого количества корней мо-
жно приспособить небольшое сверло к дрели и, высверливая отверстие
159
в корне, получать достаточное количество мезги для анализа.
Из хорошо перемешанной исследуемой мезги отбирают с помощью
мерки 10—15 г и помещают в стакан. Сюда же прибавляют специаль-
ной меркой 0,4—0,5 г сухого свинцового осветлителя — тонкорастер-
того порошка Pb (NO3)2, тщательно перемешивают палочкой
и переносят эту смесь через широкогорлую воронку в патрон с сетча-
тым дном и фильтром (рис. 28). Через 2 мин после этого центрифуги-
руют, собирая прозрачный раствор в больший центрифужный патрон.
Получается 5—7 мл осветленного сока, который поляриметрируют
в обычном сахариметре в проточной поляриметрической трубке
длиной 52 мм и диаметром 5 мм (рис. 29). Шкала показывает
содержание сахара в соке в объемных процентах (вана 100мл сока).
Содержание сахара в соке в весовых процентах вычисляют
по формуле (1), а содержание в сахарной свекле по формуле (7). Для
расчета по формуле (5), по которой получаются самые точные резуль-
таты, необходимо определить содержание сухого вещества в мезге.
Для этого на технических весах отвешивают в стеклянном или алю-
миниевом бюксе 10 г мезги (берут две параллельные пробы), высуши-
вают при 105° С до постоянного веса и рассчитывают содержание
воды в процентах по следующей формуле:
Н2О = = 10а,
где а — сухой вес навески (в г).
Полученные данные подставляют в формулу (5) и вычисляют
содержание сахара в сахарной свекле. Можно также пользоваться
для этой цели номограммой (рис. 30).
Для пересчета содержания сахара на сухой вес, исходя из со-
держания его в соке, можно пользоваться номограммой (рис. 31)
или следующей формулой:
= С (98 - Ь)
(1 — 0,0063С) b ’
где Y — содержание сахара в сахарной свекле (в% сухого вещества);
С — содержание сахара в соке (в г на 100 мл); b — содержание су-
хого вещества в сахарной свекле (в %).
Реактивы. Азотнокислый свинец. Продажную соль Pb (NO3)2 растирают
в ступке в мелкий порошок и просеивают через шелковое сито. Сохраняют в сухом
месте.
При растворении солей в воде происходит некоторое увеличение объема жид-
кости. Это имеет значение при добавлении сухого осветлителя. Величина разжи-
жающего действия зависит от количества соли в растворе и от ее химического со-
става. Разжижающее действие уксуснокислого свинца почти в два раза больше,
чем азотнокислого. Поэтому для нашей цели лучше пользоваться последним,
хотя его осветляющее действие и несколько хуже, чем основного уксуснокислого
свинца. В условиях этого метода объем от прибавляемого осветлителя увеличи-
вается приблизительно на 0,5% при условии полного растворения соли. Это дает
ошибку при определении сахара около 0,1%, но в действительности ошибка будет
меньше, так как не вся соль растворяется в соке — часть ее выпадает вместе
с белком.
160
Рис. 30. Номограмма для определения содержания сахара в корне сахар-
ной свеклы по содержанию его в соке и влажности корня:
С — содержание сахара в соке, г на 100 мл; Н2О — содержание воды в корне
сахарной свеклы, %; X —содержание сахара в корне, % на сырое вещество.
11 G-2
с
У
Рис. 31. Номограмма для определения содержания сахара в корне сахарной
свеклы на сухое вещество:
С содержание сахара в соке, г на 100 мл; в — содержание сухого вещества в
корне, %; у — содержание сахара в корне, % на сухое вещество.
Определение крахмала поляриметрическим методом
Сущность метода. Крахмал переводят в раствор соля-
ной кислотой при нагревании. Посторонние оптически активные
вещества осаждают фосфорновольфрамовой кислотой, раствор дово-
дят до определенного объема и в поляриметре определяют угол
вращения поляризованного света. Удельное вращение fa]o для
крахмала различных культур, как это видно из приведенных данных,
имеет разное значение: от 171 до 195.
Ход анализа. Отвешивают 5 г средней пробы свежих
клубней картофеля, переносят в ступку и растирают с 2 мл 1 н.
раствора соляной кислоты до однородной массы. Затем прибавляют
еще 10 мл 1 н. раствора соляной кислоты, растирают еще некоторое
время и переносят в мерную колбу на 50 мл. Остаток смывают
15 мл дистиллированной воды, сливая в ту же колбу. Раствор по-
мещают в кипящую воду на 15 мин, периодически взбалтывая.
Затем охлаждают, прибавляют 2 мл 4%-ного раствора фосфорно-
вольфрамовой кислоты, доводят объем раствора дистиллированной
водой до 50 мл и фильтруют в сухую колбу через сухой фильтр.
Фильтрат поляриметрируют в сахариметре, пользуясь поляризацион-
ной трубкой длиной 200 мм. На основании полученных показаний
содержание крахмала вычисляют по следующей формуле:
v 50 100 0,3465 a n оо~~
А =-----1ПГ- .—0,8866 а,
195,4 и I ’ *
где X — содержание крахмала (в %); 50 — объем исследуемого
раствора (в мл)\ а — количество делений шкалы сахариметра;
I — длина поляризационной трубки (в дм\, п — навеска картофеля
(в а); 195,4 — удельное вращение картофельного крахмала (в [а]р);
0,3465 — коэффициент для пересчета показаний шкалы сахариметра
в градусы круговой шкалы поляриметра.
Для введения поправки на содержание других оптически актив-
ных веществ, например сахаров, берут 10 г картофельной мезги,
растирают в ступке с дистиллированной водой, переносят в колбу
на 50 мл, промывают ступку 25 мл воды, собирая промывные воды
в колбу. Сюда же прибавляют 4 мл 4%-ного раствора фосфорно-
вольфрамовой кислоты, доводят водой до метки, перемешивают и
фильтруют в сухую колбу. Отбирают пипеткой 25 мл фильтрата,
переносят в мерную колбу на 50 мл, прибавляют 2 мл 6 н. раствора
соляной кислоты, ставят в кипящую воду на 15 мин. Затем раствор
охлаждают, доводят дистиллированной водой до метки, перемеши-
вают и поляриметрируют в трубке на 200 мм. Показания поляри-
метра пересчитывают на содержание крахмала по приведенной вы-
ше формуле и эту величину вычитают из ранее полученных данных
содержания крахмала.
11*
163
Определение крахмала
в зерне пшеницы, ржи, ячменя, кукурузы
Берут 2 г муки исследуемого зерна, помещают в ступку и расти-
рают с 3 мл 1 н. раствора соляной кислоты, затем прибавляют еще
5 мл этой же кислоты, размешивают и переносят в мерную колбу
на 50 мл, промывая ступку 20 мл дистиллированной воды. Хорошо
взбалтывают и ставят в кипящую воду на 15 мин, периодически взбал-
тывая. После этого охлаждают, прибавляют 5 мл 4%-ногораствора
фосфорновольфрамовой кислоты, доливают водой до метки, переме-
шивают и через 5 мин фильтруют в сухую колбу через сухой фильтр.
Прозрачный фильтрат поляриметрируют в сахариметре, исполь-
зуя поляриметрическую трубку на 200 мм.
Содержание крахмала вычисляют по формуле:
У___ 50 34,65 а
Л = п I [а] ’
где X — содержание крахмала (в %); [а]— удельное вращение
крахмала данной культуры при температуре 20° С и линии D натри-
евого пламени; 34,65 — коэффициент пересчета шкалы сахари-
метра в градусы шкалы поляриметра, умноженный на 100 для пере-
счета в проценты. Остальные обозначения такие, как в приведенной
выше формуле.
Реактивы. Раствор фосфорновольфрамовой кислоты (4%-ный). Отвешивают
4 г фосфорновольфрамовой кислоты, растворяют в дистиллированной воде, раз-
бавляют водой до 100 мл и сохраняют в обыкновенной склянке.
ЛИТЕРАТУРА
Биохимические методы анализа растений. М., ИЛ, 1960.
Вечер А. С., Г о л и н с к а я Л. А., Л е в и ц к а я М. В. Определение
пектиновых веществ в картофеле.— Физиол. и биохим. культурных растений,
1973, 5, 5, 549.
Зайцева Г Н., Афанасьева Т. П. Количественное определение
углеводов методом нисходящей хроматографии на бумаге.— Биохимия, 1957,
22, вып. 6.
Кравченко С. Ф., Трухачева А. А. Технохимический контроль
кукурузно-крахмального производства. М., Пищепромиздат, 1952.
Починок X. Н. Микрометод определения в свекле сахарозы и восстанав-
ливающих сахаров.— Науч. зап. сахарной промышленности. Агрономический
вып., 1935, 3, 47.
Починок X. Н. Определение глюкозы, фруктозы и сахарозы в расте-
ниях из одной навески.— В кн.: Бюл. физиол. растений. Киев, Изд-во АН УССР,
1958, 27.
Починок X. Н. Определение крахмала в листьях и других органах рас-
тений.— В кн.: Тр. Ин-та физиол. растений АН УССР, 20. Киев, Изд-во АН УССР,
1959, 59.
Силин П. М. Химический контроль свеклосахарного производства. М.,
Пищепромиздат, 1959.
Von Horst Goring, Wolfgang Dreier. Eine einfache kolori-
metrische Methode zur simultanenbestimmung von Glukose, Fruktose und Saccha-
rose in pflanzlichen Geweben.— Flora, 1967, 5, A158, 549.
164
Глава IV
ФЕРМЕНТЫ
Ферменты — это специфические белковые вещества, обладаю-
щие мощным каталитическим действием, связанным со способностью
к специфическому активированию других веществ. Ферменты раз-
деляют на два больших класса — однокомпонентные, состоящие
исключительно из белка, и двухкомпонентные,состоящие из белковой
части и небелковой части, называемой простетической группой.
В простетическую группу часто входят витамины и металлы: желе-
зо, медь, кобальт, марганец, цинк и др.
Ферменты молекулярно гетерогенны. Они состоят из несколь-
ких молекулярных форм белка — изоферментов, которые катали-
зируют одну и ту же реакцию, но различаются рядом химических
свойств. Вещества, на которые действуют ферменты и которые ими
активируются, называются субстратами ферментов. Часто при
добавлении некоторых веществ, не принимающих участие в реак-
ции, скорость реакции уменьшается. Такие вещества называются
ингибиторами. Существуют специфические и неспецифические ин-
гибиторы. К неспецифическим ингибиторам относятся все осадители
белков — соли тяжелых металлов, таннин, трихлоруксусная кис-
лота и др. К специфическим относятся вещества, вступающие в ре-
акцию с активным центром фермента, например синильная кислота
ингибирует окислительные ферменты, связывая железо и медь,
входящие в простетическую группу.
Характерным свойством ферментов является их способность ка-
тализировать определенные химические реакции. Поэтому присут-
ствие фермента обнаруживается по протеканию катализируемой
им специфической реакции, а количество фермента определяется
по скорости реакции. Таким образом, измерение скорости специфи-
ческой реакции характеризует активность фермента и является
основным в методике исследования ферментов. При определении ак-
тивности ферментов принято следить за изменением начальной ско-
рости реакции под влиянием только одного из факторов, определяю-
щих скорость реакции, при сохранении всех остальных факторов
постоянными, так как скорость реакции уменьшается со временем.
Для получения линейной зависимости между количеством фер-
мента и скоростью реакции условия в реакционной смеси должны
поддерживаться постоянными в течение всего периода проведения
165
опыта. Реакцию следует проводить при определенной температуре,
и растворы всех компонентов должны быть доведены до той же
температуры прежде, чем в результате их смешения начнется реак-
ция. Температура не должна быть слишком высокой и величина
pH должна поддерживаться в пределах оптимального для действия
фермента значения. Раствор должен быть забуферирован в тех
случаях, когда в результате реакции образуется кислота. Скорость
реакции следует предварительно отрегулировать, подобрав такое
количество фермента, чтобы измерения могли быть проведены
с достаточной точностью.
Ферменты — относительно неустойчивые вещества. При небла-
гоприятных условиях они легко претерпевают денатурацию и инак-
тивирование. Работая с ферментами, следует прежде всего забо-
титься о том, чтобы не допустить их инактивирования. Следует из-
бегать действия температуры, превышающей 35—40° С, а также
кислых и щелочных растворов. Этиловый спирт, ацетон и другие
органические растворители инактивируют большинство ферментов
при комнатной температуре. Во многих случаях наблюдается мед-
ленное инактивирование ферментов при стоянии даже в наиболее
благоприятных условиях. Высушивание при комнатной температу-
ре вызывает полную инактивацию ферментов, но активность сохра-
няется, если высушивать в условиях высокого вакуума при низ-
кой температуре (в замороженном состоянии). Если раствор
фермента оставляют стоять продолжительное время, необходимо
принимать меры предосторожности от загрязнения бактериями и
подавлять их жизнедеятельность прибавлением антисептических
веществ.
Каталитическая активность ферментов характеризуется коли-
чеством прореагировавшего субстрата или количеством продукта
реакции за определенное время в присутствии данного весового
количества фермента. Она может быть выражена в различных еди-
ницах. Наиболее часто каталитическая активность ферментов
выражается числом микромолей субстрата, прореагировавшего за
1 мин, на определенное количество фермента или исследуемого ве-
щества.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ПЕРОКСИДАЗЫ
Пероксидаза — металлопротеин, содержащий железо в порфи-
риновом кольце. Она встречается во многих растениях, часто в от-
носительно высоких концентрациях, например в хрене. Пероксида-
за образует с перекисью водорода соединение, в котором перекись
активируется и становится способной окислять фенолы и аромати-
ческие амины, действуя как водородный акцептор. Перекись водоро-
да в присутствии пероксидазы окисляет о-фенилендиамин, лейко-
форму малахитового зеленого, бензидин, билирубин, пирогаллол,
гваякол, гидрохинон, катехин, о-, м- и n-крезол, тирозин и адре-
налин. Таким образом, пероксидаза относится к числу ферментов
166
с ограниченной специфичностью. У пероксидазы обнаружено до
20 изоэнзимов.
Пероксидаза обладает относительно низким молекулярным ве-
сом — около 40 000, и поэтому легко проходит через бумажный
фильтр. Пероксидаза инактивируется синильной кислотой, азидом
натрия, сероводородом, тиомочевиной, гидроксиламппом, окисью
азота, гидросульфитом натрия и избытком перекиси водорода. При
растирании растительных материалов с водой значительная часть
фермента находится в осадке и не переходит в раствор. Если же оса-
док обработать растворами солей щелочных или щелочноземельных
металлов, фермент полностью переходит в раствор.
Сущность метода. Исследуемую пробу свежесобран-
ного растения растирают в ступке с раствором азотнокислого или
хлористого кальция и после разбавления до определенного объема
и 30-минутного настаивания в фильтрате определяют активность
пероксидазы гваяколовым методом, который является наиболее
чувствительным и быстрым. Метод основан на способности гваякола
окисляться перекисью водорода при активном участии пероксидазы.
Гваякол при этом окисляется до тетрагваякохинона:
/ОН О • С6Н3 (ОСН3) СвН3 (ОСН3) О
4С6Н4 + 4Н2О2 = | +
^ОСНз О С6Н3(ОСН3) СвН3(ОСН3) о
- 8Н..О.
Раствор приобретает красно-коричневую окраску, интенсивность
которой через определенное время измеряют. Аналогично перок-
сидазе окисление гваякола могут катализировать также ионы се-
ребра, если окислителем служит персульфат аммония:
/ОН О С6Н3(ОСН3) С6Н3(ОСН3) о
4С6Н4 + 4 (NH4)2 S2O8 = j +
^ОСН3 О С6Н3(ОСН3) С6Н3(ОСН3) о
+ 4 (NH4)2SO4 + 4H2SO4.
Гваякол в нейтральном растворе медленно окисляется персуль-
фатом аммония до тетрогваякохинона. Азотнокислое серебро значи-
тельно ускоряет эту реакцию. При сравнении активности перокси-
дазы с активностью серебра при различной продолжительности ре-
акции и разной температуре было установлено, что концентрации
фермента и серебра, а также их активности совпадают при продол-
жительности реакции 15 мин и температуре 20° С. Поэтому по кали-
бровочной кривой, построенной для серебра, можно найти коли-
чество окисленного гваякола, а затем вычислить активность фер-
мента в микрограммах окисленного гваякола на 1 г исследуемого
вещества.
Ход анализа. Исследуемую пробу свежего растительного
материала измельчают, перемешивают и отвешивают 0,5 г. Навеску
167
переносят в фарфоровую ступку, прибавляют 2 мл 5%-ного
раствора азотнокислого или хлористого кальция и, если объект
имеет кислую реакцию, добавляют 0,05 г углекислого кальция и рас-
тирают в течение 3 мин. После этого приливают еще 5 мл 5%-но-
го раствора соли кальция, растирают в течение 1 мин и по-
лученную суспензию переносят в мерную колбу или мерный цилиндр
п\__I__I__i_I__I__।__1__।__I__I
и 0,05 0,10 0,15 ' 0.20 0.25
Концентрация Ад, мг/мл
Рис. 32. Калибровочная кривая
для каталитического действия
серебра на окисление гваякола
в течение 15 мин при 20° С (све-
тофильтр с максимумом пропус-
кания 440 нм, кювета с тол-
щиной измеряемого слоя 10 мм).
на 50 мл, промывая ступку раствором
азотнокислого или хлористого каль-
ция, которым доводят объем до 50 мл.
Содержимое колбы взбалтывают, дают
постоять 20—30 мин и фильтруют
в сухую колбу через сухой фильтр,
отбрасывая первую порцию фильтрата
(5—10 мл).
Из полученного фильтрата отби-
рают пипеткой 1 мл или более в за-
висимости от предполагаемой актив-
ности фермента (до 8 мл), переносят
в сухую пробирку, добавляют дистил-
лированную воду до 8 мл, 1 мл
0,3%-ного раствора гваякола и пере-
мешивают. Пробирку помещают в ван-
ну с температурой 20° С, и когда тем-
пература в контрольной пробирке
достигнет температуры ванны, прили-
вают 1 мл 0,05 н. раствора перекиси
водорода. После прибавления пере-
киси раствор немедленно переме-
шивают и оставляют в ванне.
Интенсивность появившейся окраски
измеряют точно через 15 мин после
прибавления перекиси водорода в фотоэлектроколориметре прг
440 нм в кювете с толщиной слоя \^мм. Раствором для сравнения
служит исследуемый фильтрат, взятый в таком же объеме, как и в
опыте, и разбавленный дистиллированной водой до 10 мл. Резуль-
таты сравнивают с калибровочной кривой, построенной для се-
ребра, и по соответствующему количеству серебра рассчитывают
активность пероксидазы по следующей формуле:
50 10 2,53 С = 84,3 С
15 а п ап
где А — активность пероксидазы при температуре 20° С (в мкмоль
гваякола окисленного в течение 1 мин при действии фермента, со-
держащегося в 1 г исследуемого вещества); С — концентрация се-
ребра (в мг!мл), найденная по калибровочной кривой; а — объем
исследуемого раствора, взятый для реакции с гваяколом (в мл)\
п — навеска исследуемого вещества (в г); 50 — объем вытяжки
из навески п (в мл)\ 10 — объем колориметрируемого раствора
168
(в мл)\ 15 — время действия фермента (в мин); 2,53 — количество
микромолей гваякола, окисляемого за 15 мин в присутствии 1 мг
серебра при 20° С.
Построение калибровочной кривой. В се-
рию пробирок набирают 0,1, 1, 2, 3, 4, 5 и 6 мл раствора азотнокисло-
го серебра, содержащего 0,5 мг Ag в 1 мл раствора, доливают дистил-
лированной водой до 8жл, прибавляют по 1 мл 0,3%-кого раствора
гваякола и перемешивают. Пробирки погружают в воду с темпе-
ратурой 20° С и прибавляют с промежутками в 1 мин по 1 мл 3%-ного
раствора персульфата аммония. Содержимое пробирки сразу после
прибавления персульфата аммония перемешивают и выдерживают
в воде при 20° С. Точно через 15 мин после прибавления персульфа-
та аммония в первой пробирке измеряют интенсивность окраски
раствора, а в остальных пробирках через такой же промежуток
времени с интервалом между измерениями в 1 мин, учитывая время
прибавления персульфата. Интенсивность окраски измеряют при
440 нм в кювете с толщиной слоя 10 мм. На основании полученных
измерений строят калибровочную кривую (рис. 32).
Реактивы. Раствор азотнокислого кальция (5%-ный). В качестве исходного
готовят 50%-ный запасной раствор. 500 г Са (NO3)2 4Н2О ч. д. а. растворяют
в 500 мл дистиллированной воды, разбавляют водой до 1 л и перемешивают. Из
этого раствора готовят 5%-ный раствор разбавлением водой в 10 раз.
Раствор гваякола (0,3%-ный). Если гваякол находится в твердом виде, его
предварительно расплавляют, погружая банку с реактивом в теплую воду. После
охлаждения набирают пипеткой 0,6 мл гваякола, приливают 100 мл дистилли-
рованной воды, находящейся в мерной колбе на 200 мл, взбалтывают до раство-
рения гваякола, разбавляют дистиллированной водой до 200 мл, перемешивают
и сохраняют в темной склянке.
Раствор перекиси водорода (0,05 н.). 0,5 мл 30%-ного раствора перекиси
водорода разбавляют дистиллированной водой до 150 мл. В этом растворе прове-
ряют содержание перекиси водорода следующим образом. Отбирают пипеткой
5 мл, помещают в колбу для титрования, прибавляют 5 мл 4 н. раствора серной
кислоты и титруют 0,05 н. раствором перманганата калия до появления неисче-
зающей розовой окраски. Должно расходоваться 5 мл раствора перманганата.
В случае большего или меньшего расхода перманганата раствор перекиси водо-
рода подгоняют к 0,05 н. разбавлением или добавлением пергидроля. Годность
раствора 2—3 дня.
Раствор персульфата аммония (3%-ный). Отвешивают 3 г неразложившейся
соли — (NH4)2S2O8 х. ч., растворяют в дистиллированной воде и разбавляют во-
дой до 100 мл< Раствор готовят свежим, т. к. срок годности одни сутки.
Стандартный раствор азотнокислого серебра. Отбирают чистые неразложив-
шиеся кристаллы AgNO3, отвешивают на аналитических весах 0,1968 г, растворяют
в дистиллированной воде в мерной колбе на 250 мл, доливают водой до метки,
перемешивают и сохраняют в темной склянке. 1 мл этого раствора содержит
0,5 мг серебра.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ПОЛИФЕНОЛОКСИДАЗЫ
Полифенолоксидаза является медьсодержащим ферментом (в нем
найдено приблизительно 0,17% меди). Она катализирует реакцию
окисления дифенолов кислородом воздуха, например о-дифенол
169
(пирокатехин) быстро превращается в о-хинон:
ОН О
I /ОН JI /О
2 + О2 = 2 +2Н2О.
Оптимальная кислотность действия фермента находится при pH
около 6,0. Образовавшийся ортохинон может быть снова восстанов-
лен аскорбиновой кислотой:
+ СН2—СН—СН—С = С—С = О =
II II
ОН он он он
Г-0—.
+ сн2—сн—сн—с—с—с = о.
II II II
ОН ОН 0 0
Таким образом, небольшое количество пирокатехина может быть
попеременно восстановлено и окислено много раз, действуя в ка-
честве промежуточного переносчика кислорода. При этом происхо-
дит непрерывное потребление кислорода и одновременное окисление
аскорбиновой кислоты в эквивалентных количествах. Полифенолок-
сидаза содержится в картофеле, сахарной свекле, яблоках и других
растениях. Полифенолоксидаза инактивируется синильной кис-
лотой и сероводородом. У полифенолоксидазы обнаружено 6 изо-
энзимов.
Сущность метода. Исследуемое вещество в виде водной
суспензии при pH около 6 в присутствии аскорбиновой кислоты
и пирокатехина встряхивают в течение 2 мин при температуре 20° С.
При этом идет окисление аскорбиновой кислоты. Точно через 2 мин
реакция прекращается добавлением метафосфорной кислоты и оста-
ток аскорбиновой кислоты определяют титрованием йодатом калия.
Из полученных данных находят количество окисленной аскор-
биновой кислоты и вычисляют активность фермента (в мкмоль
окисленной за 1 мин аскорбиновой кислоты на 1 г исследуемого
вещества).
Ход анализа. Отвешивают 1 г свежего растительного ма-
териала, растирают в фарфоровой ступке с дистиллированной водой,
переносят в мерную колбу или цилиндр на 50 мл и перемеши-
вают. Полученный раствор взбалтывают и, не давая осадку осесть, на-
бирают 10 мл суспензии, которую выливают в колбу для титрова-
170
ния на 250 мл. Прибавляют 1 мл фосфатного буферного раствора
с pH 6,4, приливают 5 мл 0,04 н. раствора аскорбиновой кислоты,
перемешивают и прибавляют 5 мл 0,2 %-ного раствора пирокатехина,
одновременно включая секундомер и встряхивая раствор. Для лучше-
го поступления кислорода воздуха раствор встряхивают равномерно
в течение 2 мин. Точно через 2 мин реакцию прекращают добавле-
нием 5 мл 5%-ного раствора мета- или ортофосфорной кислоты.
Необходимое количество кислоты отмеривают в чистый стакан-
чик еще до начала опыта. Перед началом опыта все растворы должны
быть доведены до температуры 20° С погружением в воду с этой
температурой. Остаток аскорбиновой кислоты определяют титро-
ванием 0,01 н. раствором йодата калия в присутствии 1 мл 0,5%-ного
раствора крахмала до появления неисчезающей синей окраски.
Одновременно проводят контрольное титрование. Для этого
набирают 10 мл суспензии в колбу для титрования, прибавляют
5 мл метафосфорной кислоты, 5 мл 0,04 н. раствора аскорбиновой
кислоты и титруют 0,01 н. раствором КJO3 в присутствии крахмала.
На основании полученных данных вычисляют активность полифенол-
оксидазы по следующей формуле:
д= 50 5 (а — Ь) _ 12,5 (а — Ь)
Л 10 п 2 п
где А — активность полифенолоксидазы (в мкмоль окисленной за
1 мин при 20° С аскорбиновой кислоты на 1 г исследуемого вещества);
50 — общий объем суспензии исследуемой ткани (в мл); п — навеска
исследуемого вещества (в г); 10 — объем суспензии, взятый в колбу
для определения активности фермента (в мл); 2 — время проведе-
ния реакции (в мин); а — объем 0,01 н. раствора KJO3, затраченный
на титрование контрольной пробы (в мл); b — объем 0,01 н. раствора
KJO3, затраченный на титрование исследуемой пробы (в мл); 5 —
коэффициент для пересчета миллилитров 0,01 н. раствора аскорби-
( 0,00088 Д
новой кислоты в микромоли —А * — = 5 .
\ U,UUUl/o /
Реактивы. Раствор пирокатехина (0,2% -ный). Отвешивают на технических
весах 0,2 г пирокатехина, растворяют в дистиллированной воде и разбавляют
водой до 100 мл. Раствор готовится на 1—2 дня и сохраняется в темной склянке
в прохладном месте.
Раствор йодата калия (0,01 н.). Отвешивают на аналитических весах 0,3566 г
KJO3, растворяют в дистиллированной воде в мерной колбе на 1 л, прибавляют
5 мл 1 н. раствора NaOH и 2 г KJ, растворяют, разбавляют дистиллированной
водой до метки, перемешивают и сохраняют в темной склянке.
Буферный раствор (pH 6,4). Отвешивают 5,44 г соли КН2РО4 х. ч., раство-
ряют в безуглекислой воде, добавляют 10 мл 1 н. раствора NaOH, разбавляют
такой же водой до 200 мл и сохраняют в хорошо закрытой склянке.
> Раствор аскорбиновой кислоты (~ 0,04 н.). Отвешивают на технических весах
0,35 г аскорбиновой кислоты, растворяют в дистиллированной воде, разбавляют
водой до 100 мл и перемешивают. Раствор готовят на 1 день.
Раствор метафосфорной кислоты (5%-ный). 50 г НРО3 растворяют в дистил-
лированной воде, затем разбавляют водой до 1 л, перемешивают и сохраняют
в склянке с притертой пробкой.
171
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ КАТАЛАЗЫ
Каталаза принадлежит к числу гемопротеидных ферментов, в
состав которых входит железо. Каталаза содержится в животных
и растительных организмах. Она катализирует расщепление переки-
си водорода на воду и молекулярный кислород. При этом атом железа
простетической группы подвергается попеременному окислению
и восстановлению. У каталазы обнаружено 5 изоэнзимов.
Перекись водорода способна в одинаковой степени легко отда-
вать и принимать электроны, т. е. служить то окислителем, то вос-
становителем (разница в окислительно-восстановительных потен-
циалах всего 0,30 в). Поэтому взаимодействие каталазы с перекисью
водорода идет в две стадии. В первой стадии реакции перекись
водорода служит окислителем:
R (Fe+2)2 + Н2О2 = R (Fe+3OH“)2.
Во второй стадии перекись водорода служит восстановителем:
R (Fe+3OH-)2 + Н2О2 = R (Fe+2)2 + 2Н2О + О2.
Второй стадии реакции способствуют образовавшиеся в первой
стадии гидроксил-ионы, которые выводят из сферы реакции ионы
водорода и тем самым понижают окислительный потенциал переки-
си водорода.
Суммируя эти две стадии получаем следующую реакцию:
2Н2О2 = 2Н2О + О2.
Так как каталаза принимает непосредственное участие в реак-
ции, то по количеству разложившейся перекиси водорода в едини-
цу времени можно судить о количестве фермента. Оптимальная
кислотность действия каталазы находится при pH около 7,0.
Многие растения имеют реакцию среды мало отличающуюся
от нейтральной, поэтому прибавление буферной смеси необяза-
тельно. В тех случаях, когда pH неизвестно или среда кислая, не-
обходимо растирать пробы с фосфатным буфером, имеющим pH 6,8—
7,0 или с углекислым кальцием (0,2—0,3 г), нейтрализующим кис-
лоты. Каталаза обладает высокой специфичностью — она действу-
ет только на перекись водорода. Каталаза инактивируется синиль-
ной кислотой, сероводородом, гидроксиламином и азидом натрия.
Она неустойчива в кислой среде и разрушается при pH ниже 3.
С увеличением температуры выше 10° С усиливается разрушитель-
ное действие перекиси водорода на каталазу. Растертые пробы не-
обходимо хранить при температуре ниже 10° С и не более 1 ч.
Сущность метода. Исследуемую ткань растирают
с водой или с фосфатным буфером pH 7 (или с СаСО3) и разбавляют
172
дистиллированной водой до определенного объема. Берут некоторую
часть суспензии и после доведения температуры до 20° С прибав-
ляют перекись водорода. Через 5 мин определяют остаток неразло-
жившейся перекиси водорода йодометрическим методом. Перекись
водорода в присутствии молибдата аммония как катализатора реа-
гирует с йодистым калием, выделяя йод, который затем титруют
тиосульфатом натрия:
Н2О2 + 2KJ + H2SO4 = J2 + K2SO4 + 2Н2О,
J2 + 2Na2S2O3 = 2NaJ + Na2S4Oe.
Титрованием контрольного раствора находят количество пере-
киси водорода до опыта. По разности результатов контрольного
опыта и опыта с образцом находят количество разложившейся
перекиси водорода, которое характеризует активность каталазы.
Ход анализа. Отбирают среднюю пробу исследуемого
материала и берут на технических весах навеску (1 г листьев и стеб-
лей и 2 г корней сахарной свеклы). Взятую навеску помещают в фар-
форовую ступку, прибавляют 2 мл дистиллированной воды, а в слу-
чае кислой реакции объектов 2 мл фосфатного буфера с pH 7 или
0,2 г СаСО3 и тщательно растирают. Содержимое ступки переносят
в мерную колбу на 100 мл, смывая водой. Объем доводят до 100 мл
и перемешивают. Суспензию энергично взбалтывают и, не давая
осадку осесть, набирают пипеткой 10 мл, переносят в большую про-
бирку диаметром 30 мм и высотой 20 см и разбавляют дистиллиро-
ванной водой до 20 мл. Пробирки помещают в металлический штатив
и погружают в воду с температурой 20° С. Когда температура в кон-
трольной пробирке, содержащей 20 мл воды, достигнет 20° С, при-
бавляют пипеткой 5 мл 0,1 н. раствора перекиси водорода, переме-
шивают и оставляют на 5 мин, контролируя время секундомером
от момента прибавления перекиси. Через 5 мин прибавляют 5 мл
раствора серной кислоты (1 9). Серная кислота прекращает дей-
ствие фермента. После этого приступают к титрованию остатка пере-
киси водорода. Для этого добавляют 1 мл 20%-ного раствора йодис-
того калия и в качестве катализатора 3 капли 10%-ного раствора
молибдата аммония или молибдата натрия. Выделившийся йод
титруют 0,02 н. раствором тиосульфата натрия в присутствии 1 мл
0,5%-ного раствора крахмала до неисчезающей синей окраски.
Параллельно проводится контрольное титрование. Для этого
берут такое же количество суспензии, как в опыте, помещают в про-
бирку, прибавляют 5 мл серной кислоты и перемешивают. При этом
фермент инактивируется. К смеси приливают 5 мл 0,1 н. раствора пе-
рекиси водорода, 1 мл 20%-ного раствора йодистого калия, 3 капли
молибдата аммония и титруют 0,02 н. раствором тиосульфата нат-
рия. Из полученных данных вычисляют активность каталазы по
следующей формуле:
100 2931 0,01 (а — b) + 1g j 586 0,01 (а — b) + 1g
10 п 5 ' п 1
173
где А — активность каталазы (в мкмоль перекиси водорода, разла-
гаемой 1 г исследуемого вещества в течение 1 мин при 20° С). Эти
величины пропорциональны такому количеству фермента, которое
в течение 5 мин разлагает половину добавленной перекиси водорода;
100 — общий объем суспензии исследуемого вещества (в мл)\ п —
навеска исследуемого вещества (в а); 10 — объем суспензии иссле-
дуемого вещества, взятый для определения активности фермента
(в мл\, 5 — время взаимодействия фермента с перекисью водорода
(в мину, а — объем 0,02 н. раствора тиосульфата натрия, затрачен-
ный на титрование контрольной пробы (в мл)\ b—объем 0,02 н.
раствора тиосульфата натрия, затраченный на титрование исследу-
емого раствора (в млу, 293 — коэффициент пересчета расходуемой
перекиси водорода в микромоли; (а — Ь) должно быть в пределах
от 8 до 23 мл при а, равном 25 — 26 мл.
Можно также пользоваться следующей менее сложной, но и
менее точной формулой:
А = 10' 100 (а -Ь) = 20 (а — Ь)
Я 10 п 5 “ п
где А — активность каталазы (в мкмоль перекиси водорода, разла-
гаемой 1 г исследуемого вещества в течение 1 мин\ (а — ft) должно
быть в пределах от 2 до 15 мл; 10' — коэффициент пересчета милли-
п па / 0,00034
литров 0,02 н. раствора перекиси водорода в микромоли п ’ m. =
\ UjUUUUu х
= ю).
Остальные обозначения такие же, как в приведенной выше фор-
муле.
Консервирование проб прибавлением толуола здесь не приме-
нимо, так как толуол катализирует реакцию окисления йодистого
калия кислородом воздуха и поэтому в присутствии толуола титро-
вание йода тиосульфатом натрия становится невозможным.
Для определения качества фермента определяют активность при
двух температурах, отличающихся на 10° С, например при 10°
и 20° С. Из полученных показателей активности вычисляют вели-
чину р, по следующей формуле:
н = 39 203 1g ,
А10
где р, — энергетический коэффициент (в г-кал); А20 — активность
фермента при 20° С; Л10 — активность фермента при 10° С. Чем
меньше jx, тем лучше качество фермента.
При определении активности каталазы в объектах, содержащих
большое количество крахмала (в клубнях картофеля, семенах
злаков и др.), необходимо перед определением дать крахмалу осесть
и для исследования брать раствор без осадка крахмала. В этом слу-
чае индикатор — раствор крахмала можно не прибавлять, так как
его обычно достаточно в растворе.
174
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
КАТАЛАЗЫ, ПОЛИФЕНОЛОКСИДАЗЫ И ПЕРОКСИДАЗЫ
ИЗ ОДНОЙ НАВЕСКИ
После определения активности каталазы в остатке исследуе-
мого раствора можно определить активность и некоторых других
ферментов — пероксидазы и полифенолоксидазы и, таким образом,
в одной навеске установить активность трех ферментов. В этом слу-
чае дальнейший ход анализа будет следующий. Остаток исследу-
емого раствора, после определения активности каталазы, энергич-
но взбалтывают и, не давая осадку осесть, отбирают 25 мл суспензии
в мерную колбу на 50 мл для определения активности пероксидазы,
разбавляют до метки 10% -ным раствором азотнокислого кальция,
перемешивают и оставляют на 30—40 мин при периодическом взбал-
тывании. Пока этот раствор настаивается определяют активность
полифенолоксидазы.
Определение активности полифенолоксидазы
Из остатка первоначального раствора после взбалтывания не-
медленно, не давая осесть осадку, набирают 10 мл суспензии в ко-
ническую колбу на 250 мл, прибавляют 1 мл фосфатного буферного
раствора с pH 6,4, 5 мл 0,04 н. раствора аскорбиновой кислоты,
перемешивают и добавляют 5 мл 0,2%-ного раствора пирокатехина,
одновременно включая секундомер и—встряхивая раствор. Через
2 мин прекращают действие фермента добавлением метафосфорной
кислоты и продолжают далее так, как описано выше (см. метод
определения активности полифенолоксидазы). После этого присту-
пают к определению активности пероксидазы.
Определение активности пероксидазы
Исследуемую суспензию, к которой добавлен раствор азотно-
кислого кальция, после 30—40 мин настаивания фильтруют в сухую
колбу через сухой фильтр и первую порцию фильтрата — 5—10 мл
отбрасывают. Из остальной части фильтрата набирают пипеткой
2 мл (при исследовании пшеницы — 0,5 мл), переносят в сухую
пробирку, разбавляют дистиллированной водой до 8 мл, прибавля-
ют 1 мл 0,3%-ного раствора гваякола и перемешивают. Пробирку
помещают в воду с температурой 20° С, приливают 1 мл 0,05 н.
раствора Н2О2, перемешивают и оставляют в воде, поддерживая
температуру 20° С. Через 15 мин после прибавления перекиси
водорода измеряют оптическую плотность при 440 нм в кювете
с толщиной слоя 10 мм. Раствором для сравнения служит исследу-
емый раствор, взятый в таком же количестве, как и в опыте, и раз-
бавленный водой до 10 мл. Полученные данные сравнивают с кали-
бровочной кривой и по найденному количеству серебра (в мг)
вычисляют активность пероксидазы по формуле, приведенной при
изложении метода определения активности пероксидазы.
175
Реактивы. Раствор перекиси водорода (0,1 н.). 3 мл пергидроля — 30% Н2О2
разбавляют дистиллированной водой до 500 мл и определяют содержание пере-
киси водорода в полученном растворе йодометрическим методом. На 5 мл этого
раствора должно расходоваться около 25 мл 0,02 н. раствора тиосульфата натрия.
Раствор сохраняют в темной склянке в прохладном месте не более недели. Из
этого раствора готовят 0,05 н. разбавлением в 2 раза дистиллированной водой.
Приготовление остальных реактивов описано выше (см. стр. 9, 17, 18, 76,
77, 169, 171).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ АСКОРБИНОКСИДАЗЫ
Фермент аскорбиноксидаза является медьсодержащим протеи-
ном. В нем найдено от 0,15 до 0,25% меди. Под воздействием аскор-
биноксидазы аскорбиновая кислота окисляется кислородом возду-
ха в дегидроаскорбиновую кислоту:
-----О
2 СН2—СН—СН—С = С—СО + О2 =
II II
он он он он
аскорбиновая
кислота
= 2 СН2—СН—СН-С—С—С = О + 2Н2О.
II II II
ОН ОН о о
дегидроаскорбиновая
кислота
Аскорбиноксидаза встречается во многих растениях — тыкве,
кабачках, огурцах, салате, петрушке, капусте, шпинате и др.
Наибольшая активность аскорбиноксидазы наблюдается при pH
около 6,0. Фермент инактивируется синильной кислотой, сероводо-
родом и окисью углерода.
Сущность метода. Исследуемое вещество в свежесобран-
ном виде растирают в ступке с дистиллированной водой. К опреде-
ленной части полученной суспензии добавляют фосфатный буфер
с pH ~ 6,4, аскорбиновую кислоту и смесь выдерживают при тем-
пературе 20° С в течение 10 мин при равномерном взбалтывании.
При этом происходит реакция окисления аскорбиновой кислоты.
Здесь применяют буфер с несколько более высоким pH, чем нужно,
так как аскорбиновая кислота понижает pH до нужной величины.
Через 10 мин реакция прекращается в результате добавления мета-
фосфорной кислоты и избыток аскорбиновой кислоты титруют
йодатом калия в присутствии йодистого калия и крахмала до по-
явления синей окраски. Йодат калия, попадая в кислую среду,
реагирует с йодистым калием, выделяя йод:
KJO3 + 5KJ + 6НРО3 = 3J2 + 6КРО3 + ЗН2О.
176
Выделившийся йод вступает в реакцию с аскорбиновой кислотой,
окисляя ее в дегидроаскорбиновую кислоту:
I—0—I
СН2—СН—СН—С = С—со + J2
II II
он он он он
I—0—I
= СН2—СН—СН—С—С—СО + 2HJ.
II II II
ОН ОН 0 0
Определив остаток аскорбиновой кислоты и зная количестве
аскорбиновой кислоты, которое было в начале опыта, по разности
находят количество окисленной аскорбиновой кислоты, характери-
зующее активность аскорбиноксидазы. Грамм-эквивалент аскор-
биновой кислоты —-у°в равен 88.
Ход анализа. Навеску (2 г листьев или 4 г клубней карто-
феля) растирают в ступке с дистиллированной водой, суспензию
переносят в мерную колбу или мерный цилиндр на 100 лы, разбав-
ляют водой до 100 мл и тщательно перемешивают. Затем суспензию
энергично взбалтывают и, не давая осадку осесть, отбирают 20 мл
в коническую колбу на 250 мл для определения активности аскор-
биноксидазы. Туда же добавляют 1 мл фосфатного буферного раство-
ра с pH 6,4, 5 мл 0,04 н. раствора аскорбиновой кислоты и взбал-
тывают в течение 10 мин на механической мешалке. После этого
реакцию прекращают добавлением 5 мл 5%-ного раствора мета-
или ортофосфорной кислоты. Остаток непрореагировавшей аскор-
биновой кислоты титруют 0,01 н. раствором йодата калия в присут-
ствии 1 мл 0,5%-ного раствора крахмала по появления неисче-
зающей синей окраски.
Параллельно проводят контрольное титрование исследуемого
раствора. Для этого набирают 20 мл исследуемой суспензии (после
предварительного взбалтывания) в коническую колбу, прибавляют
5 мл 5%-ного раствора мета- или ортофосфорной кислоты и переме-
шивают. Затем прибавляют 5 мл 0,04 н. раствора аскорбиновой кис-
лоты и титруют 0,01 н. раствором KJO3 в присутствии крахмала
до появления неисчезающей синей окраски.
На основании полученных данных вычисляют активность аскор-
биноксидазы по следующей формуле:
5 100 (а — Ь) _ 2,5 (а — Ь)
Я ~ 20 10 п “ п
где А — активность аскорбиноксидазы (в мкмоль аскорбиновой
кислоты, окисленной за 1 мин при 20°С на 1г исследуемого вещества);
100 — общий объем суспензии исследуемой ткани (в мл); 20 — объем
суспензии, взятый для определения активности аскорбиноксидазы
12 6-2
177
(в мл); 10 — время действия фермента (в мин); а — объем
0,01 н. раствора йодата калия, затраченный на титрование кон^
трольной пробы (в мл); b — объем 0,01 н. раствора йодата калия,
затраченный при титровании исследуемой пробы (в мл); 5 — коэф-
фициент пересчета миллилитров 0,01 н. раствора аскорбиновой
/ 0,00088
кислоты в микромоли г000017в " = 51; п — навеска исследуемого
вещества (в г).
Определение активности полифенолоксидазы
в этом же растворе
В оставшейся части суспензии исследуемого вещества можно
определить и активность полифенолоксидазы. Для этого исследуе-
мую жидкость энергично взбалтывают и, не давая осесть осадку,
отбирают 10 мл суспензии, переносят в коническую колбу на 250 мл,
добавляют 1 мл фосфатного буфера с pH 6,4, приливают 5 мл 0,04 н.
раствора аскорбиновой кислоты, перемешивают и прибавляют
5 мл 0,2%-ного раствора пирокатехина, одновременно включая
секундомер. Реакционную смесь встряхивают равномерно в течение
2 мин и точно через это время прекращают реакцию добавлением
5 мл 5%-ного раствора метафосфорной кислоты. Перед началом
опыта температура всех растворов должна быть доведена до 20° С,
погружением в воду с этой температурой. Остаток непрореагиро-
вавшей аскорбиновой кислоты определяют титрованием 0,01 н.
раствором KJO3 в присутствии 1 мл 0,5%-ного раствора крахмала
до появления неисчезающей синей окраски.
Параллельно проводят контрольное титрование. Для этого наби-
рают 10 мл суспензии, помещают в колбу для титрования, прибавляют
5 мл метафосфорной кислоты, 5 мл 0,04 н. раствора аскорбиновой
кислоты и титруют 0,01 н. раствором KJO3. Полученные данные
подставляют в формулу для вычисления активности полифенол-
оксидазы, учитывая, что общий объем суспензии исследуемого мате-
риала равен 100 мл. Таким путем рассчитывают активность поли-
фенолоксидазы (в мкмоль аскорбиновой кислоты, окисленной за
1 мин при 20° С с участием фермента, находящегося в 1 г исследу-
емого вещества).
Приготовление реактивов описано выше (см. стр. 18, 171).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ТИРОЗИНАЗЫ
Тирозиназа содержится в картофеле, сахарной свекле, злаках
и других растениях и является медьсодержащим белком. Она содер-
жит от 0,2 до 0,3% меди. Тирозиназа относится к числу типичных
оксидаз. Для ее действия необходимо присутствие свободного кис-
лорода. В основе каталитического действия фермента лежит изме-
нение валентности меди. Тирозиназа способствует окислению
тирозина, фенола, пирокатехина, л/-крезола, /г-крезола, пирогаллола
178
и адреналина. Оптимальная кислотность действия фермента лежит
при pH 6—8. Тирозиназа инактивируется синильной кислотой,
окисью углерода, сероводородом и диэтилдитиокарбаматом натрия.
При действии тирозиназы на тирозин в присутствии кислорода воз-
духа образуется меланин (черный пигмент) через диоксифенилала-
нин по схеме:
(НО) С6Н4СН2СН (NH2) СООН +
+ О (НО)2СбН3СН2СН (NH2) СООН + О ->
3, 4-диоксифенилаланин
->- CeH3O2CH2CH (NH2) СООН +
О=/\-------------NH
о=\А/соон
+ 02 -> НООС<\^\=О -> меланин
н — \/=0
промеланин
Сущность метода. Определение активности тирозиназы
состоит в том, что к определенному количеству водной вытяжки
из исследуемого материала прибавляют определенное количество
тирозина и выдерживается 4 ч в термостате при температуре 40° С.
При этом часть тирозина превращается в черный пигмент — мела-
нин. После этого меланин осаждается хлористым барием в присут-
ствии соды, фермент инактивируется нагреванием раствора до ки-
пения и осадок удаляют фильтрованием. В фильтрате определяют
остаток неокисленного тирозина броматометрическим методом. Для
этого прибавляют избыток 0,03 н. раствора бромат-бромидной смеси,
подкисляют соляной кислотой и оставляют на 10 мин. При этом
происходит бромирование тирозина:
КВгО3 + 5КВг + 6НС1 = ЗВг2 + 6КС1 + ЗН2О,
С6Н4 (ОН) СН2СН (NH2) СООН + ЗВг2 =
= СбНВг3 (ОН) СН2СН (NH2) СООН + ЗНВг.
Избыток брома титруют раствором метилоранжа, который обес-
цвечивается бромом. Реакция идет в основном по следующему урав-
нению:
(СН3)2 NC6H4N = NC6H4SO3Na +
+ 2Вг2-> (СН3)2 NC6H4NBr2 + Br2NC6H4SO3Na.
Титрование продолжают до появления неисчезающей красной
окраски, обусловленной избытком метилоранжа. Нормальность
раствора метилоранжа устанавливают по титрованному раствору
12* 7.9
бромат-бромида. Грамм-эквивалент бромата J 3 равен 27,83,
С6Н4 (ОН) СНоСН (NH2) соон п
грамм-эквивалент тирозина 4 v—-— ----------------------30,2.
Ход анализа. Отвешивают 2—3 г свежего растительного
материала и тщательно растирают в ступке с небольшим количеством
дистиллированной воды. Суспензию переносят“в мерную колбу
или мерный цилиндр на 50 мл, доводят водой до 50 мл, перемешива-
ют и дают осадку осесть. Если осадок плохо осаждается, центрифуги-
руют. Набирают пипеткой 10 мл прозрачной жидкости над осадком
в коническую колбу емкостью 250 мл, прибавляют 10 мл 0,05%-ного
раствора тирозина, 2—3 капли толуола, помещают в предваритель-
но нагретый до 40° С термостат и выдерживают при этой температу-
ре 4 ч. После этого к содержимому в колбе приливают 1 мл 5%-ного
раствора Na2CO3, 3 мл 10%-ного раствора хлористого бария, на-
гревают на плитке до кипения и фильтруют через бумажный фильтр
диаметром 7—8 см в колбу для титрования. Колбу, в которой про-
водилось осаждение, и осадок на фильтре промывают 3 раза горя-
чей водой порциями по 8—10 мл. Фильтрат охлаждают, добавляют
10 мл 0,03 н. бромат-бромидного раствора и 4 мл раствора соляной
кислоты (1 1), накрывают колбу часовым стеклом, перемешивают
и оставляют на 10 мин. Избыток брома титруют 0,2%-ным раство-
ром метилоранжа до появления неисчезающей красной окраски.
Чтобы избежать потери брома при титровании, необходимо после
прибавления очередной порции метилоранжа производить пере-
мешивание круговыми движениями колбы без энергичного взбалты-
вания.
Параллельно через все стадии анализа проводится контрольный
опыт. Для этого 10 мл исследуемого раствора помещают в колбу
для титрования, прибавляют 10 мл раствора тирозина, 25 мл дистил-
лированной воды, 10 мл бромат-бромидного раствора, 4 мл рас-
твора соляной кислоты (1 1), накрывают колбу часовым стеклом,
перемешивают и оставляют на 10 мин, после чего титруют метил-
оранжем. Активность тирозиназы вычисляют по формуле:
, _ 166,8 50 К(Ь — а) _ 208,5 К (Ь — а)
Я — 10 п 4
п
где А — активность тирозиназы (в мкмоль окисленного тирозина
за 1 ч на 1 г исследуемого вещества); 50 — общий объем исследуе-
мого раствора (в мл); 10 — объем исследуемого раствора, взятый
для определения активности фермента в (мл); 4 — время взаимо-
действия фермента с тирозином (в ч); а — объем раствора метил-
оранжа, затраченный при титровании контрольного раствора (в мл);
b — объем раствора метилоранжа, затраченный при титровании ис-
следуемого раствора (в мл); К — нормальность титрованного ра-
створа метилоранжа; п — навеска исследуемого вещества (в г);
166,8 — коэффициент пересчета миллилитров нормального раство-
ра тирозина в микромоли.
180
Реактивы. Раствор тирозина (0.05%). Отвешивают 0,1 г тирозина, помещают
в мерную колбу на 200 мл, добавляют 1,2 мл 5%-пого раствора Na2CO3, прили-
вают 150 мл дистиллированной воды, нагревают до растворения тирозина, затем
охлаждают, доводят водой до метки и перемешивают. Сохраняют в темной склянке
в прохладном месте.
Раствор бромат-бромида (0,03 н.). Отвешивают па аналитических весах
0,8349 г КВгО3, переносят в мерную колбу на 1 л, добавляют 10 г КВг, раство-
ряют в воде, разбавляют водой до 1 л и тщательно перемешивают.
Раствор метилового оранжевого (0,2%). Отвешивают на технических весах
2 г метилоранжа, помещают б колбу емкостью больше 1 л, приливают 1 л дистил-
лированной воды, взбалтывают до растворения и фильтруют. Нормальность при-
готовленного раствора метилоранжа устанавливают по бромату калия. Для
этого в колбу для титрования отбирают пипеткой 10 мл 0,03 н. раствора бромат-
бромида, добавляют 30 мл дистиллированной воды, 4 мл раствора соляной кислоты
(1 1) и титруют раствором метилового оранжевого до появления неисчезающей
красной окраски. Нормальность раствора метилоранжа вычисляют по формуле:
10 - 0,03 = 0,3
а а ’
где а — объем раствора метилоранжа, затраченный для титрования 10 мл раствора
бромат-бромида (в мл).
Раствор хлористого бария (10%-ный). 10 г ВаС12 2Н2О растворяют в ди-
стиллированной воде, разбавляют водой до 100 мл и перемешивают.
Раствор карбоната натрия (5%-ный). 50 г безводной соли Na2Co3 рас-
творяют в дистиллированной воде и доводят объем до 1л.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ САХАРАЗЫ
(ИНВЕРТАЗЫ)
Сахараза принадлежит к группе карбогидраз, расщепляющих
сложные углеводы на простые сахара. Сахараза известна еще
под названиями инвертазы и Р-фруктофуранозидазы, так как
действует на сахара, содержащие остаток fJ-фруктозы — саха-
розу, раффинозу, генцианозу и стахиозу. Оптимальная кислотность
действия фермента находится при pH 4,5 —5,5. Наиболее активно
этот фермент гидролизует сахарозу с образованием инвертного саха-
ра —глюкозы и фруктозы:
^12^22^11 4" Н2О = С6Н12О0 -]- СбН12Об.
глюкоза фруктоза
Сахараза содержится в растениях и микроорганизмах; наивысшие
концентрации встречаются в дрожжах. Сахараза проводит реакцию
гидролиза тростникового сахара почти в точности по уравнению
для мономолекулярной реакции. Фермент угнетается ионами тяже-
лых металлов (Ag+, Cu2+, Hg2+) и анилином.
Сущность метода. Исследуемое вещество после рас-
тирания с дистиллированной водой, доведения pH среды до 5 и до-
бавления сахарозы, настаивается в течение определенного време-
ни при температуре 40° С (от 2 до 16 ч в зависимости от активности
фермента). При этом происходит реакция гидролиза сахарозы
с образованием инвертного сахара. Количество образовавшихся
181
восстанавливающих сахаров определяют йодометрическим методом.
Количество восстанавливающих сахаров определяют также в исход-
ном растворе, в котором фермент инактивирован кипячением раство-
ра. Разность в содержании восстанавливающих сахаров в исследуе-
мой и контрольной пробе, отнесенная к весу исследуемого вещества,
с учетом времени действия фермента характеризует активность
сахаразы.
Ход анализа. Из исследуемой пробы листьев или других
органов растений отвешивают 1,00 г (при исследовании корнепло-
дов сахарной свеклы берут навеску 2,5 г) и растирают в ступке
с небольшим количеством дистиллированной воды до однородной
массы. Растертую массу переносят в мерную колбу на 50 мл, раз-
бавляют водой до метки и тщательно перемешивают. Из этой сус-
пензии после предварительного взбалтывания отбирают две пробы
по 10 мл, переносят в мерные колбы на 100 мл (или в колбы на 50 мл,
если берут меньшую навеску или активность фермента слабая).
Одну колбу, раствор которой в дальнейшем будет служить кон-
тролем, нагревают на плитке до кипения и слабо кипятят 1—2 мин
для инактивации ферментов. Затем охлаждают, прибавляют во все
колбы по 5 мл ацетатного буферного раствора с pH 5, по 10 мл
5%-ного свежеприготовленного раствора сахарозы, по 4 капли то-
луола и помещают в термостат, нагретый до 40° С, на 2 ч. Если
активность фермента слабая, как у корнеплодов сахарной свеклы,
инкубацию удлиняют до 8—16 ч. Затем колбы вынимают из термо-
стата и ко всем пробам прибавляют по 2,5 мл 1 н. раствора едкого
натра, по одной капле раствора фенолфталеина и добавляют по кап-
лям 10%-ный раствор сернокислого цинка до обесцвечивания инди-
катора (обычно расходуется около 1 мл раствора). В том случае,
если исследуемый раствор интенсивно окрашен в темный цвет,
1 мл сернокислого цинка прибавляют сразу, перемешивают и ос-
тавляют на некоторое время. Если осадок оседает легко с просвет-
лением раствора, то больше сернокислого цинка не добавляют.
В противном случае добавляют еще несколько капель раствора
сернокислого цинка. Объем раствора в колбах доводят дистилли-
рованной водой до метки, перемешивают и фильтруют в сухую
колбу через сухой фильтр. В полученном фильтрате определяют
содержание восстанавливающих сахаров. Для этого набирают
10 или 5 мл фильтрата, выливают в пробирку с диаметром 30 мм,
доводят объем водой до 10 мл (там, где брали 5 мл исследуемого рас-
твора), прибавляют 10 мл медно-щелочного реактива, перемешивают,
закрывают пробирки воронками или шариковыми пробками и шта-
тив с пробирками погружают в кипящую воду и выдерживают при на-
гревании 15 мин.
После этого штатив с пробирками вынимают и охлажда-
ют погружением в холодную воду. К охлажденным растворам
постепенно при взбалтывании прибавляют по 5 мл смеси щавелевой
и серной кислот. Когда вся закись меди растворится, остаток йода
титруют 0,01 н. раствором тиосульфата натрия в присутствии 0,5 мл
182
0,5%-ного раствора крахмала. Активность фермента вычисляют
по следующей формуле:
2» 50 100 0,00556(198 + а + b) (а — Ь) _ 0,278 (198 + а + Ь) (а — Ь)
Л ~ 10 10' t п “ tn
где А — активность сахаразы (в мкмоль инвертного сахара, обра-
зовавшегося в течение 1 ч на 1 г исследуемого вещества); 50 — об-
щий объем исследуемого раствора (в мл)\ 100 — объем раствора,
получаемый после гидролиза сазарозы (в мл)\ 10 — объем исследу-
емого раствора, взятый для определения активности фермента (в мл)\
10'— объем раствора, взятый для определения восстанавливающих
сахаров (в мл)\ t — продолжительностьдействияфермента на саха-
розу (в ч); п — навеска исследуемого вещества (в г); b — объем
0,01 н. раствора тиосульфата натрия, затраченный при титровании
исследуемого раствора (в мл)\ а — объем 0,01 н. раствора тиосуль-
фата натрия, затраченный при титровании контрольного раствора
(в мл)\ 0,00556 — коэффициент пересчета микрограммов в микро-
моли (—— = 0.00556); (198 +а + &) — количество ин-
вертного сахара г; мкг, соответствующее 1 мл 0,01 н. раствора тио-
сульфата натрия.
Реактивы. Ацетатный буферный раствор (pH 5). Можно готовить двумя
способами: 1) в мерную колбу на 100 мл помещают 29 мл 1 н. раствора едкого
натра, добавляют 2,5 мл ледяной уксусной кислоты, разбавляют дистиллирован-
ной водой до 100 мл и перемешивают; 2) 30 мл 0,2 н. раствора уксусной кисло-
ты смешивают с 70 мл 0,4 н. раствора уксуснокислого натрия.
Приготовление остальных реактивов изложено в методе определения сахаров
(см. стр. 17, 28, 43, 121).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ АМИЛАЗЫ
В соответствии с составом крахмала, состоящего из амилопектина
(Р-амилозы) и амилозы (а-амилозы), существуют и два фермента:
а-амилаза и р-амилаза.
В семенах злаков содержится свободная амилаза — Р-амилаза,
которая может быть экстрагирована водой, и связанная амилаза —
а-амилаза, которая водой не экстрагируется и неактивна. Вовре-
мя прорастания семян а-амилаза освобождается и переходит в ак-
тивное состояние. р-Амилаза разрушается нагреванием до темпера-
туры 70° С, а а-амилаза при этой температуре устойчива. Амилазы
расщепляют гликозидные связи в полисахаридах. а-Амилаза может
расщеплять гликозидные связи в середине полисахаридных цепей,
а Р-амилаза действует на нередуцирующий конец цепи, последо-
вательно отщепляя остатки мальтозы.
Сущность метода. Амилазы в итоге превращают ре-
зервный углевод — крахмал в дисахарид мальтозу, т. е. в такую
форму, в которой он может быть транспортирован и утилизирован:
(СбН±0Об)п + 0,5nH2O -> 0,5nC12H22Ou.
183
Мальтоза в щелочном растворе восстанавливает ионы меди и ее
можно определить таким же способом, как и моносахариды. По ко-
личеству образовавшейся мальтозы можно судить об активности
амилаз. Применяемые до настоящего времени методы определения
активности амилаз не учитывают действие фермента сахаразы, при-
сутствие которой может давать большие ошибки при исследовании
растительного материала, содержащего тростниковый сахар. Опи-
санный ниже метод исключает ошибку от влияния сахарозы.
Определение суммарной активности а и р-амилаз
В качестве примера приводится ход определения активности
амилаз в картофеле. При работе с другими объектами изменяется
только величина навески исследуемого материала: чем больше
предполагаемая активность фермента, тем меньше навеска.
Ход анализа. Пробу картофеля измельчают, отвешивают
2 г и растирают в ступке с 1 мл 0,2 М фосфатного буферного раствора
с pH 5,6 до однородной массы, затем прибавляют еще 6 мл буфер-
ного раствора, переносят в мерную колбу на 100 мл, промывая ступ-
ку дистиллированной водой и разбавляют водой до метки. Раствор
энергично перемешивают и, не давая раствору отстояться, отбирают
пипеткой по 25 мл суспензии в две мерные колбы на 50 мл. В каж-
дую прибавляют по 10 мл 2%-ного свежеприготовленного раствора
крахмала. Одну из колб (контрольную) помещают на плитку, раствор
доводят до кипения, кипятят 1 мин и охлаждают. Затем обе колбы
(исследуемую и контрольную) помещают в термостат при 40° С
на 2 ч. После этого колбы вынимают из термостата и прибавляют
к пробам по 4 мл 8%-ного раствора щавелевой кислоты, помещают
в кипящую водяную баню и выдерживают в течение 15 мин. При
этом гидролизуется вся сахароза и этим исключается влияние ин-
вертазы. После охлаждения растворы нейтрализуют 1 н. раствором
едкого натра в присутствии одной капли 0,1 %-ного раствора фенол-
фталеина. Содержимое колб разбавляют дистиллированной водой
до 50 мл и тщательно перемешивают. В растворах определяют вос-
станавливающие сахара. Для этого из верхней части отстоявшихся
растворов набирают по 10 мл, переносят в пробирки для определе-
ния сахаров, добавляют по 10 мл медно-щелочного реактива, пере-
мешивают, пробирки помещают в штатив, закрывают воронками
и штатив с пробирками погружают в кипящую воду на 15 мин.
Затем штатив с пробирками охлаждают погружением в холодную
воду. В пробирки постепенно при взбалтывании прибавляют по
5 мл смеси щавелевой и серной кислот и после растворения закиси
меди остаток йода титруют 0,01 н. раствором тиосульфата натрия.
В данном случае крахмал как индикатор не прибавляют, так как
его достаточно имеется в растворе.
Из полученных данных вычисляют активность амилазы по сле-
дующей формуле:
д = 100 50 [1,43 (а — Ь) + 0,96] = 14,3 (а — Ь) + 9,6
Л ~~ 25 10 • 2 п п
184
где А — активность амилазы (в мкмоль мальтозы, образовавшейся
в течение часа на 1 г исследуемого вещества); а — объем 0,01 н.
раствора тиосульфата натрия, затраченный при титровании кон-
трольного раствора (в мл)\ b—объем 0,01 и. раствора тиосульфата
натрия, затраченный при титровании исследуемого раствора (в мл)\
100 — общий объем исследуемого раствора (в мл)\ 50 — объем рас-
твора, полученный после ферментативного превращения крахмала
в мальтозу (в мл)\ 25 — объем исследуемого раствора, взятый для
определения активности фермента (в мл)\ 10 — объем раствора,
взятый для определения восстанавливающих сахаров (в мл)\ 2 —
продолжительность действия фермента (в ч); п — навеска иссле-
дуемого вещества (в а); 1,43 (а — Ь) 4-0,96 — количество мальтозы
(в мкмоль), найденное в 10 мл конечного исследуемого раствора.
Определение а-амилазы
В остатке первоначального раствора можно также определить
активность а-амилазы. Для этого исследуемый раствор энергично
взбалтывают и, не давая отстояться, набирают по 25 или 20 мл сус-
пензии в две мерные колбы на 50 мл. К растворам в колбах прибав-
ляют по 10 мл 2%-ного свежеприготовленного раствора крахмала.
Одну из колб (контрольную) ставят на плитку, доводят раствор
до кипения и кипятят 1 мин. Вторую колбу с исследуемым раство-
ром нагревают на водяной бане 15 мин при 70° С, поддерживая тем-
пературу в пределах 70° ± 0,5° С. Затем колбы быстро охлаждают
погружением в холодную воду и помещают в термостат при 40° С
в течение 2 часов. После этого колбы вынимают, исследуемый рас-
твор помещают на плитку, доводят до кипения и кипятят 1 мин.
После охлаждения растворы нейтрализуют 0,1 н. раствором едкого
натра в присутствии фенолфталеина, разбавляют дистиллированной
водой до метки и перемешивают. В растворе определяют восстанав-
ливающие сахара, как изложено выше, и вычисляют активность
а-амнлазы по приведенной выше формуле.
Реактивы. Раствор крахмала (2%-ный). 2 г растворимого крахмала расти-
рают в ступке с небольшим количеством дистиллированной воды, добавляя ее
до 20 мл. Полученную суспензию вливают при помешивании в 80 мл кипящей
воды, кипятят 1 мин, дают охладиться и разбавляют водой до 100 мл. Раствор
готовится свежим и годен в течение одного дня.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ПРОТЕАЗ
Протеолетические ферменты (протеазы) катализируют расщеп-
ление белков и гидролиз продуктов их распада до аминокислот.
Протеазы относятся к группе амидаз, т. е. ферментов, расщепляю-
щих амидные связи между СО- и NH-группами. В группу протеаз
входит большое количество ферментов, неодинаковых по своим свой-
ствам. Протеолетические ферменты растений резко отличаются
185
от иротсолетических ферментов животных. В растениях найдены
протеазы, например папаин, который расщепляет белки при сла-
бокислой реакции.
Протеазы типа папаина (или катепсина) активируются синиль-
ной кислотой, сероводородом, цистеином, глутатионом и другими
тиоловыми производными. Папаин (или катепсин) сам является тио-
ловым производным, в состав которого входят свободные SH-rpyn-
пы. При окислении фермента эти активные группы превращаются
в неактивные группы — S—S —, поэтому окислители являются инак-
тиваторами протеаз. Сероводород или глутатион вновь восстанав-
ливают неактивные группы до активных SH-rpynn.
Во ржи, пшенице и других растениях, а также в солоде встреча-
ется смесь ферментов: протеиназы (катепсины), действующие при
pH 5,0 и расщепляющие белки до полипептитов; полипептидазы,
действующие при pH 7,0 и гидролизующие промежуточные про-
дукты распада белков (полипептиды) до аминокислот и дипептидазы
действующие при pH 7,8 и расщепляющие дипептиды до аминокислот.
Протеазы действуют максимально на свой белок, а на другие белки
действуют значительно слабее, а иногда и совсем не действуют.
Сущность метода. Суспензию исследуемого материала
с pH 5,0, содержащую собственный белок, выдерживают в термоста-
те при температуре 40° С в течение 20 часов. После этого белки осаж-
дают фосфорновольфрамовой кислотой, раствор доводят до опреде-
ленного объема и фильтруют. В определенной части фильтрата оп-
ределяют содержание небелкового азота хлораминным методом.
В контрольной пробе, взятой до опыта, также определяют содержа-
ние небелкового азота. При разности находят приращение небелко-
вого (гидролизованного) азота, по которому и вычисляют активность
протеиназ. В остатке этих же растворов (исследуемых и контроль-
ных) определяют содержание аминного азота ингидриновым мето-
дом. По разности находят количество образовавшегося аминного
азота и по нему вычисляют активность пептидаз.
Ход определения активно с-т и протеиназ.
Из средней пробы исследуемого материала берут три навески по
0,8 г. Каждую навеску растирают с 2 мл дистиллированной воды
и переносят суспензию с помощью 10 мл воды в мерные колбы на
50 мл. Когда все навески будут растерты и перенесены в колбы,
к ним добавляют по 5 мл фосфатного буферного раствора с pH 5,0
и по 10 капель толуола. Две колбочки с исследуемыми растворами
закрывают пробками и ставят в термостат при 40° С на 20 часов.
В третью колбочку с контрольным раствором добавляют 3 мл
4%-ного раствора фосфорновольфрамовой кислоты, перемешивают,
разбавляют дистиллированной водой до 50 мл, снова перемешивают
и фильтруют. 25 мл полученного фильтрата переносят в колбу
Кьельдаля на 100 мл, прибавляют 0,4 г сульфата калия, 2 мл кон-
центрированной серной кислоты и 0,3 мл 10%-ного раствора молиб-
дата натрия. Смесь помещают на плитку, выпаривают на слабом
огне и сжигают, усиливая нагрев до просветления раствора.
186
Исследуемые растворы после 20-часовой инкубации вынимают
из термостата, прибавляют по 3 мл 4%-ного раствора фосфорно-
вольфрамовой кислоты, взбалтывают и через 5—10 мин разбавляют
безаммиачной водой до 50 мл, перемешивают и фильтруют через
сухие складчатые фильтры в сухие колбы. 25 мл прозрачного филь-
трата из каждой колбы помещают в колбы Кьсльдаля, прибавляют
по 0,4 г сульфата калия, по 0,3 мл 10%-ного раствора молибдата
натрия и по 2 мл концентрированной серной кислоты. Колбы ставят
на плитку в наклонном положении, выпаривают и сжигают до прос-
ветления раствора. В конце выпаривания нужно следить, чтобы
пена не поднималась выше горлышка колбы. В случае необходи-
мости уменьшают нагрев периодическим выключением плитки.
Как только вспенивание прекратится, колбы закрывают стеклян-
ными шариковыми пробками и усиливают нагрев до равномерного
кипения. После того как жидкость станет прозрачной и почти бес-
цветной (иногда с голубым оттенком), нагрев прекращают. Растворы
охлаждают и приливают по стенке колбы по 5 мл дистиллированной
воды. Растворы переносят в колбы для титрования емкостью 250—
300 мл, смывая остаток 20 мл воды. После этого нейтрализуют 2 н.
раствором едкого натра в присутствии метилоранжа до начала пере-
хода окраски индикатора в желтый цвет. Затем прибавляют по
10 мл фосфатного буферного раствора с pH 6,7, содержащего бро-
мистый калий, по 5 мл 0,12 н. раствора хлорамина, накрывают
стеклами, перемешивают и оставляют па 25 мин. После этого при-
бавляют по 3 мл 20%-ного раствора йодистого калия, по 5 мл раство-
ра серной кислоты (1 9) и выделившийся йод титруют 0,02 н. рас-
твором тиосульфата натрия в присутствии крахмала как индика-
тора. Также титруют после сжигания и контрольную пробу. Из
результатов титрования рассчитывают активность фермента по сле-
дующей формуле:
А = 333 50 к (а — Ь) 33,3 К (а - Ь)
Я 25 23 п ' п
где А — активность протеиназ (в мкмоль гидролизованного азота);
а — объем 0,02 н. раствора тиосульфата натрия, затраченный при
титровании контрольного раствора (в мл); b—объем 0,02 н. раст-
вора тиосульфата натрия, затраченный при титровании исследуемого
раствора (в мл); К. — нормальность раствора тиосульфата натрия;
п — навеска исследуемого вещества (в г); 50 — общий объем иссле-
дуемого раствора (в мл); 25 — объем исследуемого раствора, взя-
тый для сжигания и определения азота (в мл); 20 — продолжитель-
ность действия фермента (в ч); 333 — коэффициент пересчета весо-
вого количества азота, эквивалентного 1 мл 1 н. раствора тиосуль-
. / 0,004669
фата натрия, в микромоли I 0000014— = 3331.
Ход определения .активности пептидаз.
В остатке фильтрата после определения активности протеиназ
определяют содержание аминного азота. Для'этого отбирают 1 мл
187
фильтрата в пробирку, прибавляют 1 мл дистиллированной воды,
3 мл ипгидринового реактива, 0,1 мл 1 %-ного раствора аскорбино-
вой кислоты, перемешивают, закрывают стеклянной шариковой
пробкой и помещают в кипящую воду на 15 мин. Одновременно в ки-
пящую воду помещают пробирку с контрольным раствором, в ко-
торую вместо исследуемого раствора берут 2 мл дистиллированной
воды и прибавляют такое же количество реактива и аскорбиновой
кислоты, как в опыте. Через 15 мин пробирки вынимают, дают
охладиться на воздухе в течение 10—15 лшяпри периодическом взбал-
тывании и доводят объем водой до 5 мл. Оптическую плотность из-
меряют при 580 нм в кювете с толщиной слоя 5 мм, сравнивая с кон-
трольным раствором.
По полученным результатам с помощью калибровочной кривой
находят концентрацию аминного азота и рассчитывают активность
пептидаз по следующей формуле:
л 50 5 • 0,0714 (Ь — а) __ 0,892 (Ь — а)
1 20 и “ п
где А — активность пептидаз (в мкмоль аминного азота, образую-
щегося за 1 ч на 1 г исследуемого вещества); 50 — общий объем
исследуемого раствора (в мл)\ 1 — объем исследуемого раствора,
взятый для колориметрируемого раствора (в мл)\ 5 — объем коло-
риметрируемого раствора (в мл)\ 20 — время взаимодействия фер-
мента (в ч); b — концентрация аминного азота, найденная при ко-
лориметрировании исследуемого раствора (в мкг/мл}\ а — кон-
центрация аминного азота, найденная при колориметрировании
контрольного раствора (в мкг/мл)\ п — навеска исследуемого ве-
щества (в г); 0,0714 — коэффициент пересчета микрограммов азо-
та в микромоли.
Построение калибровочной кривой. В су-
хие пробирки последовательно наливают 0,2, 0,4, 0,6, 0,8, 1,2, 1,6,
1,8 и 2,0 мл стандартного раствора лейцина, содержащего 5 мкг
азота в 1 мл раствора и доводят объем безаммиачной водой до 2 мл.
В одну из пробирок наливают 2 мл такой же воды без аминокислоты
для получения контрольного раствора. Затем во все пробирки
приливают по 3 мл нингидринового реактива, по 1 мл 1 %-ного
раствора аскорбиновой кислоты, перемешивают, закрывают шарико-
выми пробками, помещают в кипящую воду и нагревают 15 мин.
После охлаждения в течение 10—15 мин с периодическим взбалты-
ванием доводят объем растворов водой до 5 мл и измеряют опти-
ческую плотность при 580 нм в кювете с толщиной слоя 5 мм. Раство-
ром для сравнения служит контрольный раствор, не содержащий
аминокислоты. На основании результатов измерений строят кали-
бровочную кривую (см. рис. 13).
Реактивы. Фосфатно-цитратный буферный раствор (pH 5,0). 5,1 г лимонной
кислоты — СбН8О7 -Н2О х. ч. и 9,2 г Na2HPO4 2Н2О (или 18,44 г Na2HPO4
12НоО) растворяют в безаммиачной воде в мерной колбе на 500 мл, разбавляют
188
безаммиачной водой до метки, перемешивают и сохраняют в хорошо закрытой
склянке.
Стандартный раствор аминокислоты. Отвешивают на аналитических весах
46,8 мг предварительно высушенной при 60° С аминокислоты лейцина или 31,8 мг
а-аланина, растворяют в 25%-ном этиловом спирте в мерной колбе на 100 мл,
разбавляют этим же раствором спирта до 100 мл и перемешивают. 1 мл получен-
ного раствора содержит 50 мкг азота. Из этого раствора готовят рабочий раствор
разбавлением дистиллированной водой в 10 раз. 1 мл рабочего раствора содержит
5 мкг азота.
Приготовление остальных реактивов описано при изложении соответствую-
щих методов (см. стр. 9, 17, 76, 77, 86, 164).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ УРЕАЗЫ
Уреаза принадлежит к ферментам группы амидаз. Она расщепля-
ет амид аминоугольной кислоты, мочевину, с образованием аммоний-
ной соли угольной кислоты:
(NH2)2 СО + 2Н2О = (NH4)2 СО3.
Уреаза содержится в микроорганизмах и во многих высших рас-
тениях, особенно большое количество ее содержится в бобовых
культурах, а также в семенах арбуза, кабачка и других тыквен-
ных. Действие уреазы строго специфично, поскольку она действу-
ет только на мочевину. Уреаза наиболее активна при pH 7. Она легко
инактивируется ионами тяжелых металлов (серебра, ртути, меди,
кадмия, свинца), кислотами при pH ниже 4, щелочами при pH
выше 8, а также боратами, фторидами, хинонами, формальдегидом
и окислителями. Защитное действие оказывают гуммиарабик, белки,
аминокислоты, синильная кислота, сероводород и трилон Б (от
ионов тяжелых металлов).
Сущность метода. Водную суспензию исследуемого
вещества в присутствии фосфатного буфера с pH 7, содержащего
трилон Б, смешивают с раствором мочевины и выдерживают 5 мин
при температуре 20° С. Действие фермента прекращают фосфорно-
вольфрамовой кислотой с одновременным осаждением белков. Ко-
личество образовавшегося аммиачного азота определяют колори-
метрическим тимол-гипобромитным методом. Активность уреазы
выражается в количестве микромолей аммиака, которое образуется
в течение 1 мин при 20° С под воздействием фермента, находящегося
в 1 г исследуемого вещества.
Ход анализа. Навеску 0,1 г измельченных бобов сои
или 0,5 г бобов люпина и других семян, содержащих уреазу, тща-
тельно растирают в фарфоровой ступке с Змл фосфатного буферного
раствора с pH 7,0. Кашицу переносят в мерный цилиндр на 50 мл,
смывая остаток кашицы дистиллированной водой и разбавляют
до 50 мл. Полученную суспензию перемешивают и, не давая осадку
осесть, отбирают пипеткой 1$млв пробирку, добавляют 2 мл 0,5% -но-
го раствора мочевины, перемешивают и оставляют на 5 мин при
20° С, измеряя время секундомером. Через 5 мин прибавляют 2 мл
4%-ного раствора фосфорновольфрамовой кислоты, перемешивают
189
и через 3—5 мин центрифугируют, а прозрачный раствор сливают
в сухую пробирку.
Для определения образовавшегося аммиачного азота набирают
от 1 до 10 мл центрифугата в зависимости от предполагаемого коли-
чества азота (при исследовании бобов сои 1 мл), помещают в про-
бирку с диаметром 18—20 мм, разбавляют дистиллированной водой
до 10 мл, прибавляют 0,5 мл я-бутилового спирта и взбалтывают
до растворения спирта. Затем приливают 2 мл 4%-ного раствора
тимола и перемешивают. Если появляется муть или осадок, его
растворяют 1—2 каплями 20%-ного раствора едкого натра. К смеси
прибавляют 3 мл гипобромита натрия, перемешивают переворачива-
нием пробирки и оставляют на 10 мин. После этого добавляют 3 мл
я-бутилового спирта, взбалтывают и оставляют на 10—15 мин
для разделения фаз. Нижний водный слой удаляют, отсасывая пипет-
кой. Окрашенный спиртовый раствор переносят в мерный цилиндр на
10 мл. Если раствор мутный, добавляют 10 капель 95%-ного этило-
вого спирта. Затем раствор разбавляют я-бутиловым спиртом до
5 мл, перемешивают и измеряют оптическую плотность при 660 нм
в кювете с толщиной слоя 10 мм, сравнивая с контрольным раство-
ром, который готовят следующим образом. В пробирку помещают
10 мл исследуемой суспензии, добавляют 2 мл раствора фосфорно-
вольфрамовой кислоты, 2 мл 0,5%-ного раствора мочевины, взбал-
тывают, через 5 мин центрифугируют и центрифугат сливают в
сухую пробирку. Отбирают такое же количество центрифугата как
в опыте, разбавляют до 10 мл дистиллированной водой и далее по-
ступают так же, как с исследуемым раствором.
Из данных оптической плотности по калибровочной кривой на-
ходят концентрацию азота (в мкг/мл) и рассчитывают активность
уреазы по следующей формуле:
А _ 50 14 0,071 5 С = 4,97 С
10 5' а п ап1
где А — активность уреазы (в мкмоль аммиачного азота, образовав-
шегося в течение 1 мин при 20° С на 1 г исследуемого вещества);
С — концентрация азота в колориметрируемом растворе (в мкг/мл);
5 — объем колориметрируемого раствора (в мл); 50 — общий объем
исследуемого раствора (в мл); 10 — объем исследуемого раствора,
взятый для взаимодействия с мочевиной (в мл); 14 — объем жидкости
после инкубации и осаждения белка фосфорновольфрамовой кисло-
той (в мл); 5'— продолжительность взаимодействия фермента с мо-
чевиной (в мин); а — объем центрифугата, взятый для определения
аммиачного азота (в мл); п — навеска исследуемого вещества (в г);
0,071 — коэффициент пересчета микрограммов азота в микромоли
(4- = 0,071).
Построение калибровочной кривой. Наби-
рают в пробирки последовательно 0,5, 1, 2, 3, 4, 5, 6, 8 и 10 мл
стандартного раствора сернокислого аммония, содержащего 10 мкг
190
азота в 1 мл раствора и доводят объем растворов безаммиачной водой
до 10 мл. В одну из пробирок прибавляют 10 мл такой же воды для
получения контрольного раствора. Затем во все пробирки прибав-
ляют по 0,5 мл //-бутанола, взбалтывают, прибавляют по 2 мл 4%-но-
го раствора тимола, перемешивают. Далее прибавляют по 3 мл
гипобромита натрия, снова перемешивают переворачиванием про-
бирок и оставляют на 10 мин. После этого добавляют по 3 мл бута-
нола, взбалтывают и оставляют, на 10—15 мин для разделения
фаз. Нижний водный слой удаляют, отсасывая пипеткой, а остаток,
окрашенный в зеленый цвет — спиртовый раствор, переносят в мер-
ный цилиндр на 5 или 10 мл. Растворы разбавляют бутанолом до
5 мл и измеряют оптическую плотность при 660 нм в кювете с тол-
щиной слоя 10 мм, сравнивая с контрольным раствором, вносящим
поправку на загрязнение реактивов. На основании полученных
данных строят калибровочную кривую (см. рис. 16).
Реактивы. Фосфатный буферный раствор с трилоном Б (pH 7). 6,8 г соли
КН2РО4 х. ч. и 0,9 г трилона Б растворяют в безаммиачной воде, добавляют
30 мл 1 н. раствора едкого натра, разбавляют безаммиачной водой до 500 мл,
добавляют 1 мл хлороформа, перемешивают и сохраняют в хорошо закрытой
склянке.
Раствор мочевины (0,5%-ный). 0,5 г мочевины (NH2)2CO, растворяют
в безаммиачной воде, доводят объем раствора до 100 мл и перемешивают.
Приготовление остальных реактивов описано при изложении тимолгипобро-
митного метода определения аммиачного азота (см. стр. 94—95).
ЛИТЕРАТУРА
Бах А. Н. Собрание трудов по химии и биохимии. М., Изд-во АН СССР, 1950.
Благовещенский А. В. Биохимия обмена азотсодержащих веществ ра-
стений. М., Изд-во АН СССР, 1958.
Диксон М., Уэбб Э. Ферменты. М., ИЛ, 1961.
К и з е л ь А. Р. Практическое руководство по биохимии растений. М.—Л.,
Биомедгиз, 1934.
Михлин Д. М., Броновицкая 3. С. Йодометрический метод определе-
ния полифенолоксидазы и пероксидазы.— Биохимия, 1949, 14, 4, 379.
Починок X. Н. К определению активности пероксидазы в растениях гвая-
коловым методом.— Научн. тр. Ин-та физиол. растений и агрохимии АН УССР,
1955, 9, 65.
Починок X. Н. Определение активности каталазы йодометрическим мето-
дом.— Научн. тр. Ин-та физиол. растений и агрохимии АН УССР, 1956,
11, 92.
Починок X. Н. Определение активности ферментов сахаразы и амилазы.—
В кн.: Бюл. по физиол. растений, вып. 4. Киев, Изд-во АН УССР, 1958, 42.
Самнер Дж.,Сомерс Г. Ф. Химия ферментов и методы их исследования.
М., ИЛ, 1948.
Ферменты. Современные достижения энзимологии (под редакцией А. Н. Баха
и В. А. Энгельгардта). М., Изд-во АН СССР, 1940.
Глава V
ПИГМЕНТЫ ПЛАСТИД
В пластидах зеленого листа находится целый ряд очень важных
пигментов, участвующих в процессе фотосинтеза: зеленые — х л о -
рофиллы и желтые — каротиноиды. 'Последние подразделя-
ются на каротины (углеводороды) и ксантофиллы (кис-
лородсодержащие производные). Из зеленых пигментов высших
растений главным оптически деятельным компонентом является
сине-зеленый хлорофилл a (C55H72O5N4Mg, мол. вес 893) и желто-
зеленый хлорофилл b (C56H70O6N4Mg, мол. вес 907), содержащиеся
в растениях в большинстве случаев в соотношении 3 1.
Хлорофиллы растворяются в спирте, хлороформе, ацетоне, серо-
углероде, эфире и других органических растворителях. В воде
хлорофиллы образуют коллоидные растворы. Спиртовые растворы
хлорофилла а имеют сине-зеленый цвет и пурпурно-красную флу-
оресценцию, растворы хлорофилла Ь — желто-зеленый цвет и буро-
коричневую флуоресценцию. Ацетоном, эфиром, хлороформом
извлекают из свежих и сухих листьев все пигменты. Этиловым спир-
том извлекают все пигменты полностью, кроме каротина. Петролей-
ным эфиром и бензином не извлекают пигменты из свежих листьев.
Из высушенных листьев извлекают полностью весь каротин и час-
тично другие пигменты. Если к петролейному эфиру добавить 10%
этилового спирта, то извлекается и хлорофилл. Бензол, ксилол, то-
луол и сероуглерод занимают промежуточное положение между по-
лярными и неполярными растворителями.
Хлорофиллы представляют собой сложные эфиры хлорофиллина—
(C32H30ON4Mg) (СООН)2, у которого водородный атом в одной
карбонильной группе замещен остатком метилового спирта, а в дру-
гой — остатком непредельного одноатомного спирта фитола —
(С20Н39ОН). Основной частью молекулы хлорофилла является тетра-
пирольное кольцо хлорина. Хлорофилл образует ряд производных,
которые в зависимости от строения получили следующие названия:
фитины, отличающиеся от хлорофиллов только отсутствием
магния; ф о р б и д ы, не содержащие магния и фитола; х л о -
рофиллиды, представляющие собой остаток хлорофилла
после удаления фитола, и филлины — вещества, содержащие
магний, кольцо циклопентана и два подвижных атома водорода.
192
Хлорофилл легко реагирует с кислотами. Слабые растворы кис-
лот отщепляют магний и превращают хлорофилл в феофитины а
и Ь:
СООСН3 СООСН3
(C32HS0ON4Mg) + 2НС1 = (C32H32ON4) ' + MgCl2.
СООС20Н39 СООС..()Н39
хлорофилл а феофитин
Аналогично из хлорофилла b получается феофитин Ь. Феофитины
легко растворяются в этиловом эфире, хлороформе, труднее в эти-
ловом спирте и петролейном эфире. Растворы феофитинов бурого
цвета. На колонке феофитин а черно-синего цвета, а феофитин b тем-
но-коричневого цвета.
Концентрированная соляная кислота, кроме магния, очень
быстро отщепляет фитол с образованием феофорбидов:
СООСН3 СООСН3
4- Н2О = (C32H32ON4) 4“ С20Н39ОН.
СООС20Н39 соон
феофитин а феофорбид а фитол
На колонке кольцо феофорбидов находится вверху и окрашено
в коричнево-зеленоватый цвет. Кристаллический феофорбид а сине-
черного цвета, а феофорбид b серо-черного цвета. Феофорбиды лег-
ко растворяются в хлороформе, ацетоне и пиридине, трудно —
в эфире. При действии фермента хлорофиллазы в спиртовом или
ацетоновом растворе происходит отщепление фитола от молекулы
хлорофилла и образование хлорофиллиды а и Ь:
соосн3 соосн3
(C32H30ON4Mg) + С2Н5ОН = (C32H30ON4Mg) +
СООС20Н39 СООС2Н6
хлорофилл а этилхлорофиллид а
4“ С20Н39ОН.
фитол
Под воздействием щелочи происходит омыление хлорофилла
с образованием свободного фитола, метилового спирта и двухоснов-
ных кислот — хлорофиллинов а и Ь:
СООСН3 COONa
(C32H30ON4Mg) + 2NaOH = (C32H30ON4Mg) +
COOC20H39 COONa
хлорофилл а натриевая соль
хлорофиллина а
+ с20н39он + СН3ОН.
фитол
Хлорофиллины обладают такой же окраской, как и хлорофиллы,
и мало отличаются от последних по спектрам поглощения. Вследствие
сложности химического строения молекул хлорофиллов растворы
13 6-2
193
их неустойчивы даже при комнатной температуре. Особенно быс-
тро идет разложение на свету. Под влиянием света растворы хло-
рофиллов сначала буреют, затем обесцвечиваются, причем на раз-
ложение пигментов наиболее сильно действуют красные лучи.
Хлорофиллы а и b обладают характерными спектрами поглощения
с двумя основными максимумами поглощения — в красной и сине-
фиолетовой областях спектра. Поглощение в синих лучах является
более сильным, чем в красных. Наблюдается небольшое смещение
максимумов поглощения в зависимости от растворителя. Так, раст-
воры хлорофилла а в этиловом эфире имеют максимумы при 660,0
и 427,5 нм, хлорофилла Ь при 642,5 и 452,5 нм, а в ацетоне — соот-
ветственно при 662,0 и 429,0 нм и при 644,0 и 455,0 нм.
Каротиноиды — обширная группа природных красящих
веществ, спутников хлорофилла, которые можно рассматривать
как производные каротина. Каждый каротиноид имеет характерную
кривую абсорбции света в видимой чарти спектра. Цвет каротинои-
дов обусловлен наличием в молекуле ненасыщенной углеродной цепи
со значительным числом (от 7 до 13) двойных сопряженных связей.
С увеличением числа двойных связей цвет пигмента от желтого
приближается к красному. К каротиноидам относятся широко рас-
пространенный в растениях оранжево-желтый пигмент каротин,
ненасыщенный углеводород С40Нб6. Известны три главные изо-
мерные формы каротина: а-, Р- и у-каротины. В растениях самым
распространенным из каротинов является Р-каротин, затем а-каро-
тин. В корнеплодах моркови встречается также у-каротин. Кароти-
ны растворяются в жирах и их растворителях. Хорошо растворимы
каротины в сероуглероде, бензоле, хлороформе, бензине, петро-
лейном и этиловом эфире. В спиртах почти не растворяются. Мак-
симумы поглощения раствора каротина в бензине лежат в преде-
лах 447,5—483,5 нм.
Среди оксикаротиноидов или ксантофиллов в зеленых растениях
преобладающим в количественном отношении является лютеин —
С40Н56О2 (около 60%), затем виолаксантин — С40Нб6О4 (около 20%)
в таком же, примерно, количестве неоксантин — С40Н5бО3 и неболь-
шое количество (около 2%) зеоксантина — изомера лютеина С40Н56О2.
Ксантофиллы хорошо растворимы в хлороформе, бензоле, ацетоне,
этиловом эфире и сероуглероде. В этаноле и метаноле растворяются
несколько хуже, а в петролейном эфире почти нерастворимы.
В растениях каротиноиды находятся в хромопластах — пласти-
дах плодов, лепестков цветов и других частей растений, а также
вместе с хлорофиллом в хлоропластах листьев и других зеленых
частей растений. В плодах томатов содержится изомер каротина
ликопин (С40Нб6), в плодах шиповника — рубиксантин (С40Н56О),
в плодах перца — капсантин (С40Нб6О3) и др. Выделение каротинои-
дов в чистом виде и их изучение стало возможным благодаря приме-
нению хроматографического метода, разработанного в 1906 г. вы-
дающимся русским ученым М. С. Цветом. Так как пигменты пластид
участвуют в фотосинтезе и играют определенную роль в процессах
194
Рис. 33. Прибор для
фильтрования с отсасыва-
нием и для выпаривания
органических растворите-
лей при температуре более
низкой, чем температура
кипения их.
роста и развития растении, исследование состава и количества пиг-
ментов в растениях в различных условиях их развития, а также
роли пигментов в биохимических процессах, происходящих в расте-
ниях, имеет большой научный и практический интерес.
Здесь описаны некоторые наиболее приемлемые методы опреде-
ления пигментов, надежность которых проверена длительной прак-
тикой.
ОПРЕДЕЛЕНИЕ СУММЫ ХЛОРОФИЛЛОВ а + b
ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
Листья растений для определения хлорофиллов необходимо ис-
пользовать в свежем виде. Основная опасность при извлечении сум-
мы хлорофиллов состоит в частичном гидролизе их в хлорофиллиды
под действием хлорофиллазы, которая ак-
тивна в водно-ацетоновых и спиртовых
растворах. Оптимум действия хлорофилла-
зы находится при pH 5,9. Видимый спектр
хлорофиллида почти тождествен спектру
хлорофилла, поэтому при фотоколориме-
трическом определении хлорофиллов дей-
ствие хлорофиллазы не сказывается на
результатах. У многих растений возможна
частичная феофитизация хлорофиллов при
контакте с кислым клеточным соком. По-
этому при растирании для нейтрализации
кислот прибавляется углекислый магний.
Сущность метода. Среднюю
пробу свежих листьев растирают с этило-
вым спиртом и углекислым магнием (для
нейтрализации кислот). Хлорофилл извле-
кают этиловым спиртом, раствор доводят
до определенного объема и фотометрируют
при 640—660 нм. Полученную величину
оптической плотности сравнивают с кали-
бровочной кривой и по найденной концен-
трации хлорофилла в растворе вычисляют
содержание его в исследуемом образце.
Калибровочную кривую приготавливают
из раствора хлорофилла известной кон-
центрации.
Ход анализа. 0,5 г средней про-
бы исследуемых листьев отвешивают в бюксе и растирают в ступке
с 0,05 г углекислого магния и 5 мл 95-градусного спирта. Содер-
жимое ступки помещают на стеклянный фильтр № 3 (или № 4)
в приборе для отсасывания (рис. 33) и раствор отсасывают.
Остаток в ступке и на фильтре промывают небольшими пор-
циями 80%-ного этилового спирта до полного извлечения хло-
рофилла. Раствор переносят в мерную колбу на 25 мл, разбав-
ляют спиртом до 25 мл, перемешивают и измеряют оптическую
13*
195
плотность прозрачного раствора в фотоэлектроколориметре при
610—660 нм в кювете с толщиной слоя 10 л/л/. Результаты изме-
рений сравнивают с калибровочной кривой, находят концен-
трацию хлорофилла и вычисляют содержание его в образце по сле-
дующей формуле:
0,1 25 С _ 2,5 С
~ П — п ’
где X — содержание хлорофилла (в мг на 100 а вещества); С — кон-
центрация хлорофилла в колориметрируемом растворе (в мкг/мл);
п — навеска исследуемого вещества (в г); 25 — общий объем экс-
тракта хлорофилла в (в мл); 0,1 — коэффициент пересчета микро-
граммов хлорофилла в граммы и на 100 а вещества.
Реактивы. Магний углекислый ч. д. а.
Этиловый спирт (95%-ный).
Этиловый спирт (80%-ный). Готовят из 95%-ного’ спирта. Для этого к 100 мл
95%-ного спирта прибавляют 21 мл дистиллированной воды и перемешивают.
Отработанный этиловый спирт отгоняют с дефлегматором, измеряют плотность
и концентрацию перегнанного спирта, который используют затем в дальнейшей
работе.
Выделение суммы хлорофиллов (а+Ь)
Сущность метода. Так как получение хлорофилла
в кристаллическом состоянии и сохранение его в чистом виде до-
вольно затруднительно вследствие сложности очистки и неустой-
чивости при хранении, лучше пользоваться раствором хлорофилла,
концентрацию которого можно определить химическим анализом.
Содержание хлорофилла лучше всего определять по азоту, входя-
щему в состав его молекулы.
Хлорофилл зеленых листьев после растирания их с безводным
сульфатом натрия для подсушивания извлекается этиловым эфиром,
который затем удаляется. Сухой хлорофилл промывают на колонке
из углекислого магния толуолом и петролейным эфиром и таким
образом очищают от каротиноидов, феофитинов и липидов. Чистый
хлорофилл извлекают из колонки этиловым спиртом и раствор
доводят до определенного объема. Часть полученного раствора ис-
пользуют для приготовления шкалы разбавлением в разной степени
спиртом. В растворах шкалы измеряют оптическую плотность в фо-
тоэлектроколориметре при 610—660 нм. В другой части раствора
определяют концентрацию хлорофилла по содержанию азота, ко-
торую определяют хлораминным методом. Грамм-эквивалент сум-
мы хлорофиллов (а 4- Ь) равен 74,75.
Ход работы. У свежих, только что собранных листьев
смородины или крапивы удаляют крупные жилки. Листья измель-
чают и отвешивают 10 г полученной массы. Навеску помещают в
фарфоровую ступку и растирают с 0,5 г углекислого магния и 20 г
безводного сульфата натрия до тех пор, пока содержимое ступки
превратится в сухой порошок. Этот порошок переносят на стеклян-
196
ный фильтр № 3 (или № 4) и извлекают хлорофилл 40—50 мл этило-
вого эфира, прибавляя его по 10 мл и медленно отсасывая. Не сле-
дует добиваться полного извлечения хлорофилла. Эфирный раствор
наливают в ступку, добавляют 15 г безводного сульфата натрия
и под вытяжным шкафом растирают до испарения всего эфира
и получения сухого зеленого порошка. Последний переносят в ко-
лонку, содержащую 15 г углекислого магния, уплотняют и промы-
вают толуолом до перемещения хлорофилла из слоя сульфата нат-
рия в слой углекислого магния, на что требуется 50—60 мл толуола.
Зону, находящуюся выше зеленой зоны хлорофилла удаляют, а ос-
тавшийся слой хлорофилла промывают два раза петролейиым эфи-
ром, отсасывая его полностью. Зеленую зону осторожно вынимают,
переносят на стеклянный фильтр № 3 и хлорофилл извлекают
25 мл 95%-ного этилового спирта, отсасывая раствор в экстрак-
тор. Экстракт переносят в колбочку на 25 мл и доводят
спиртом до метки. Из части этого раствора готовят шкалу.
Для этого отбирают пипеткой 5 мл, помещают в мерную колбу
на 25 мл, доводят 80%-ным этиловым спиртом до 25 мл и перемеши-
вают. Набирают градуированной пипеткой 0,25, 0,5, 1,0, 1,5, 2,0,
3,0, 4 и 5 мл этого раствора выливают в сухие пробирки и доводят
объем 80%-ным спиртом до 5 мл. Растворы перемешивают и изме-
ряют оптическую плотность при 610—660 нм в кюветах с толщиной
слоя 5 и 10 мм.
Для определения концентрации хлорофилла в исходном растворе
остаток этого раствора — (20 мл) количественно переносят в колбу
Кьельдаля на 50—100 мл, добавляют 2 мл концентрированной сер-
ной кислоты, 0,4 г сульфата калия и 0,3 мл 10%-ного раствора
молибдата натрия. Колбу опускают в кипящую воду и выпаривают
весь спирт. Затем колбу помещают на плитку в наклонном положе-
нии и нагревают до появления белых паров серной кислоты. После
этого колбу закрывают стеклянной шариковой пробкой или ворон-
кой и продолжают нагревать до полного сжигания органических
веществ. Когда раствор полностью обесцветится и станет прозрач-
ным, колбу охлаждают, прибавляют по стенке 5 мл воды и содержи-
мое переносят в коническую колбу, смывая остаток раствора 20 мл
дистиллированной воды. К полученному раствору прибавляют
каплю метилоранжа и нейтрализуют 20%-ным раствором едкого
натра до изменения цвета индикатора в оранжево-желтый. Затем
прибавляют 10 мл фосфатного буферного раствора с pH 6,7, содер-
жащего бромистый калий, перемешивают и по стенке колбы при-
ливают пипеткой 5 мл 0,12 н. раствора хлорамина. Колбу накры-
вают часовым стеклом, раствор перемешивают круговым движением
колбы и оставляют на 25 мин. После этого прибавляют 2 мл 20% -но-
го раствора йодистого калия, 5 мл 4 н. раствора серной кислоты
и выделившийся йод титруют 0,02 н. раствором тиосульфата натрия
в присутствии крахмала.
Параллельно проводят контрольный опыт, для чего в колбу
для титрования наливают 2 мл концентрированной серной кислоты,
197
прибавляют 20 мл воды, каплю метилового оранжевого, нейтрали-
зуют 20%-ным раствором едкого натра, прибавляют 10 мл буфер-
ного раствора, 5 мл 0,12 н. раствора хлорамина и оставляют на
25 мин. Затем добавляют 2 мл 20%-ного раствора йодистого калия,
Концентрация хлорофилла 6(&6)t мкг/мл
Рис. 34. Калибровочная кривая для
суммы хлорофиллов (а+ Ь) в этило-
вом спирте (светофильтр с максиму-
мом пропускания 610 нм)‘.
5 мл 4 н. раствора серной кислоты
и титруют выделившийся йод 0,02 н.
раствором тиосульфата натрия.
Из полученных данных вычис-
ляют концентрацию хлорофилла в
исходном растворе по формуле:
X = 1495 (^-&) = 74J5 (а _ ь\
где X — концентрация суммы хло-
рофиллов (а + Ь) (в мкг/мл); а —
объем 0,02 н.- раствора тиосульфата
патрия* затраченный при титрова-
нии контрольного раствора (в мл);
b — объем 0,02 н. раствора тиосуль-
фата натрия, затраченный при ти-
тровании исследуемого раствора
(в мл); 20 — объем исследуемого
раствора хлорофилла (в мл); 1495 —
количество хлорофилла, соответ-
ствующее 1 мл 0,02 н. раствора тио-
сульфата (в мкг).
Концентрация хлорофилла в растворах стандартной шкалы вы-
' толщина измеряемого слоя 10 мм\
2 *- толщина измеряемого слоя 5 мм.
числяется по следующей формуле:
X 5 • а __ X - а
25-5' “ 25
где С — концентрация хлорофилла в растворах шкалы (в мкг/мл);
X — концентрация хлорофилла в исходном растворе (в мкг/мл);
5 — объем исходного раствора хлорофилла, взятый для приготов-
ления рабочего раствора (в мл); 25 — объем рабочего раствора
в (мл); а — объем рабочего раствора хлорофилла, взятый для при-
готовления шкалы (в мл); 5' — объем растворов шкалы (в мл).
Затем строят калибровочную кривую (рис. 34).
Реактивы. Растворители употребляются марки х. ч. или ч. д. а. Приготовле-
ние остальных реактивов описано при изложении хлораминного метода опреде-
ления азота (см. стр. 76, 77).
ОПРЕДЕЛЕНИЕ КАРОТИНА,
СУММЫ ХЛОРОФИЛЛОВ (а+&) И СУММЫ КСАНТОФИЛЛОВ
ИЗ ОДНОЙ НАВЕСКИ
Сущность метода. Из навески свежих листьев, рас-
тертых с безводным сульфатом натрия, извлекают бензином каротин,
раствор которого доводят до определенного объема и фотометрируют.
198
Остальные пигменты извлекают этиловым спиртом. После разбав-
ления до определенного объема в полученном растворе определяют
содержание суммы хлорофиллов фотометрированием при 610—
660 нм. Затем хлорофилл омыляют и извлекают ксантофиллы хло-
роформом. Хлороформный раствор разбавляют до определенного
объема и определяют концентрацию ксантофиллов фотометрирова-
нием при 440 нм.
Ход анализа. 0,5 г исследуемых листьев растирают в ступке
с 2 г безводного сульфата натрия и 0,5 г углекислого магния. После
растирания смесь оставляют на 5 мин для подсушивания. Порошок
переносят в адсорбционную колонку. Колонки готовят из стек-
лянного фильтра № 3 с диаметром 25 мм, на который насыпают
10 г хорошо растертой смеси оксалата натрия и талька (1 1) и уплот-
няют. Подсушенную навеску высыпают в колонку, уплотняют
палочкой и покрывают ватным тампоном. Ступку споласкивают
5 мл бензина, которые выливают в колонку. Затем включают насос
и отсасывают со скоростью 50—60 капель в 1 мин. Колонку промы-
вают порциями бензина по 3 мл до полного извлечения нижней оран-
жевой полосы каротина, которая находится впереди. За ней следует
желтая полоса ксантофиллов, а выше — синяя полоса хлорофилла
а. Раствор каротина переносят в мерный цилиндр и доводят бензи-
ном до определенного объема (10 или 15 мл). Интенсивность окраски
полученного раствора каротина фотометрируют при 450 нм. При
использовании фотоэлектроколориметра ФЭК-56 применяют свето-
фильтр № 4 с эффективной длиной волны 440 нм. Полученные данные
сравнивают с калибровочной кривой, находят концентрацию каро-
тина и вычисляют содержание каротина по следующей формуле:
v 0,1 Л С
А — ----------,
п ’
где X — содержание каротина (в мг на 100 а вещества); С — кон-
центрация каротина в исследуемом растворе (в мкг/мл); А — объем
исследуемого раствора каротина (в мл); п — навеска исследуемого
вещества (в а); 0,1 — коэффициент пересчета микрограммов в мил-
лиграммы на 100 г вещества.
Определение суммы хлорофиллов («+&)
После отделения каротина остаток бензина в колонке отсасывают
и отбрасывают, а оставшиеся пигменты извлекают 15 мл 90%-ного
этилового спирта, который прибавляется небольшими порциями.
Если в ступке, в которой растиралась навеска, осталось некоторое
количество пигментов, то ее споласкивают спиртом и затем
сливают раствор в колонку. Колонку промывают до полного вымы-
вания всех пигментов. Спиртовую вытяжку переносят в мер-
ный цилиндр, доводят 90%-ным спиртом до 15 или 20 мл и измеряют
оптическую плотность при 610—660 нм в кювете с толщиной слоя
5 мм. Полученный результат сравнивают с калибровочной кривой
199
и рассчитывают содержание хлорофилла по формуле:
v _ 0,1 А С
где X — содержание хлорофилла (а + Ь) (в мг на 100 г вещества);
С — концентрация хлорофилла в колориметрируемом растворе (в
мкг/мл)-, А — объем исследуемого раствора хлорофилла (в мл);
п — навеска исследуемого вещества (в г); 0,1— коэффициент
пересчета микрограммов в миллиграммы на 100 г вещества.
Определение суммы ксантофиллов
Весь раствор пигментов после колориметрирования хлорофилла
переносят в делительную воронку с помощью 5 мл 90%-ного спирта,
добавляют 1 мл 20%-ного раствора NaOH, перемешивают и остав-
ляют в темноте на 2—3 ч для омыления хлорофилла. После этого
прибавляют 5 мл хлороформа, перемешивают, добавляют 10—15 мл
дистиллированной воды (половину объема спиртового раствора),
встряхивают 30 мин и дают фазам разделиться. В нижний хлоро-
формный слой переходят ксантофиллы, а в верхний спиртово-водный
слой — омыленный хлорофилл. Нижний слой отделяют, помещают
в мерный цилиндр, разбавляют 95%-ным спиртом до 15 или 20 мл*
перемешивают и измеряют оптическую плотность при 440 нм в кю-
вете с толщиной слоя 5 или 10 мм. Полученный результат сравни-
вают с калибровочной кривой и вычисляют содержание ксантофил-
лов (в мг на 100 г вещества) по формуле для расчета содержания
хлорофилла, где вместо С подставляют концентрацию ксантофилла
(в мкг/мл), найденную по калибровочной кривой.
Реактивы. Бензин (температура кипения от 50° до 120° С). Из авиацион-
ного бензина отгоняют фракцию, кипящую в пределах 50—120° С.
Этиловый спирт (90%-ный). К 100 мл 95%-ного спирта ректификата добав-
ляют 6,4 мл дистиллированной воды и перемешивают.
Тальк — силикат магния 4 SiO2 3MgO Н2О. Используется медицинский
препарат в виде мельчайшего порошка. Перед применением тальк высушивают
в сушильном шкафу 1—2 ч при температуре 105° С.
Раствор едкого натра (20%-ный). Приготовление (см. стр. 94).
Хлороформ. Используется медицинский препарат.
Сульфат и оксалат натрия. Применяют химически чистые соли.
Магний углекислый, (х. ч. или ч. д. а.).
ПОЛУЧЕНИЕ ЧИСТОГО КАРОТИНА
Сущность метода. Каротин извлекают из сухого
порошка моркови петролейным эфиром. После омыления примесей
и промывания дистиллированной водой раствор каротина в петро-
лейном эфире сгущается и каротин перекристаллизовывается из
смеси петролейного эфира и метилового спирта. Из этой смеси
растворителей перекристаллизацию проводят 2 раза и 1 раз из эти-
лового спирта с ацетоном. Чистый каротин растворяют в бензине
и из небольшой части полученного раствора готовят шкалу с раз-
200
личной концентрацией каротина, оптическую плотность которой
измеряют при 440 нм. В другой части раствора определяют концен-
трацию каротина химическим путем. Для этого растворитель уда-
ляют выпариванием, а каротин сжигают с бихроматом калия в сер-
ной кислоте. Реакция идет по следующему уравнению:
С40Н56 + 36К2Сг2О7 + 144H2SO4 = 40СО2 - 36K2SO4
+ 36Сг2 (SO4)3 + 172Н2О.
По остатку бихромата, который определяют йодометрическим
методом, или солью Мора судят о количестве каротина. Грамм-эк-
вивалент каротина С42°^66 равен 2,485. Метод дает точные результаты,
при условии отсутствия других органических веществ.
Ход анализа. 150 г свежей моркови измельчают на терке
и высушивают при 60—70° С досуха. Сухую морковь растирают
в ступке или измельчают в кофейной мельнице. 25 г порошка поме-
щают в воронку диаметром 4 см со стеклянным фильтром № 3, ко-
торую присоединяют к прибору для отсасывания (см. рис. 33).
Порошок уплотняют палочкой и каротин извлекают 50 мл петро-
лейного эфира, прибавляя его небольшими порциями (первый раз
20 мл, затем по 5 мл). Раствор переносят в делительную воронку,
добавляют 10 мл метилового или этилового спирта, 1 мл 20%-ного-
раствора едкого натра, взбалтывают и оставляют па 3 ч при частом
периодическом взбалтывании (или на ночь). При этом происходит
омыление сложных эфиров пигментов. После омыления раствор,
каротина в летролейиом эфире необходимо отмыть от щелочи и водо-
растворимых веществ. Для этого к раствору в делительной воронке
добавляют 20 мл дистиллированной воды, встряхивают и после раз-
деления фаз сливают нижний водный слой, а оставшийся эфирный
раствор промывают 2 раза 0,1 н. раствором едкого натра, прибавляя
его по 20 мл. Далее продолжают промывать водой до полного удале-
ния щелочи (проба с фенолфталеином).
После промывания влажный раствор каротина просушивают,
пропуская через колонку, содержащую внизу 1 г углекислого маг-
ния, а вверху 10 г безводного сульфата натрия. Колонку, после
пропускания раствора каротина, промывают 2 раза петролейным
эфиром порциями по 5 мл, при этом промывная жидкость присоединя-
ется к основному раствору каротина. В подсушенный раствор каро-
тина в петролейном эфире, находящийся в экстракторе, погружают
стеклянную трубку, верх которой присоединен к нижиему концу
стеклянного фильтра. Через фильтр и раствор с помощью насоса
протягивается воздух до тех пор, пока объем раствора уменьшится
до 10 мл (см. рис. 33). При протягивании воздуха сосуд с раствором
опускают в стакан с дистиллированной водой комнатной температу-
ры. Когда объем раствора уменьшится до 10 мл, к нему добавляют
при взбалтывании метиловый спирт: сначала 10 мл, потом по 5 мл
до полного растворения петролейного эфира и образования одно-
родной жидкости. Затем включают насос и продолжают протягивать
201
воздух через раствор, пока останется небольшой объем жид-
кости (около 5 мл). При этом каротин выпадает в осадок оранжево-
красного цвета, который отфильтровывают через стеклянный фильтр
№ 3 и промывают 15 мл смеси метилового спирта и петролейного
эфира (2 : 1), промывая сначала пробирку, в которой осаждался
каротин. Промытый осадок каротина на фильтре растворяют в 10 мл
этилового эфира, после чего промывают фильтр 10 мл петролейного
эфира, собирая весь раствор в прибор для отсасывания. Добавля-
ют к раствору 10 мл метилового спирта, перемешивают и про-
тягивают воздух через раствор до тех пор, пока останется
не более 5 мл жидкости. Выпавший осадок каротина отфильтровы-
вают через стеклянный фильтр №3 и промывают 15 мл смеси мети-
лового спирта и петролейного эфира (2 : 1).
Осадок на фильтре снова растворяют в 15 мл этилового эфира,
собирая |раствор в прибор для отсасывания, добавляют 10 мл
80%-ного ацетона и протягивают воздух через раствор до выпадания
осадка каротина. Когда объем раствора уменьшится до 5—8 мл,
фильтруют через стеклянный фильтр № 3 и промывают осадок 10 мл
80%-пого ацетона, промывая сначала прибор для отсасывания,
в котором прежде находился осадок каротина. Такая трехкратная
перекристаллизация необходима для полного удаления сопутствую-
щих каротину примесей липоидного характера, которые могут ме-
шать точному определению каротина. Затем каротин растворяют
в 5 мл этилового эфира и раствор переносят в мерный цилиндр.
Нерастворившийся остаток растворяют в 10 мл бензина. Перено-
сят весь раствор в мерный цилиндр и доводят объем бензином до
15 мл. 1 мл этого раствора отбирают в мерную колбу на 25 мл и го-
товят рабочий раствор, необходимый для приготовления шкалы
с различной концентрацией каротина. Кроме того, в две пробирки
диаметром 20—25 мм и высотой 12 см помещают по 5 мл этого раство*
ра для определения содержания каротина.
Определение содержания каротина
в исходном растворе
Пробирки, содержащие по 5 мл раствора каротина, помещают
в теплую воду и выпаривают содержимое досуха при помешивании
стеклянной палочкой в вытяжном шкафу. К остатку прибавляют
по 5 мл 0,5 н. раствора бихромата калия в серной кислоте, погру-
жают пробирки в кипящую воду и нагревают 10 мин, время от
времени перемешивая раствор стеклянной палочкой, которая все
время должна быть в пробирке. После охлаждения раствор из
пробирки переносят в колбу для титрования, промывая пробирку
дистиллированной водой. Полученный раствор разбавляют водой
до 100 мл, охлаждают, добавляют 5 мл 20% -ного раствора йодистого
калия и выделившийся йод титруют 0,1 н. раствором тиосульфата
натрия в присутствии крахмала. Отдельно (таким же способом)
титруют 5 мл 0,5 н. раствора бихромата калия, который служит
202
контролем. Избыток бихромата можно также оттитровать солью
Мора, как описано выше (см. стр. 138, 139).
На основании полученных данных вычисляют концентрацию
каротина по следующей формуле:
X = 248,4 (д - Ь) = 49 ?
натрия, затраченный на
Концентрами я каротина,мкг/мл
Рис. 35. Калибровочная кри-
вая для каротина (свето-
фильтр с максимумом про-
440 нм, кювета с
измеряемого слоя
5 мм).
где X — концентрация каротина в исходном растворе (в мкг/мл);
а — объем 0,1 н. раствора тиосульфата
титрование контрольного раствора (в мл);
Ь — объем 0,1 н. раствора тиосульфата
натрия, затраченный на титрование ис-
следуемого раствора (в мл); 5 — объем
раствора каротина, взятый для определе-
ния (в л/л); 248,4 — количество каротина,
соответствующее 1 мл 0,1 н. раствора
тиосульфата (в мкг).
Построение калибровоч-
ной кривой. 1 мл исходного рас-
твора каротина доводят бензином до
метки в мерной колбе на 25 мл, пере-
мешивают и используют полученный
рабочий раствор для приготовления
шкалы. Для этого 0,25, 0,5, 1,0, 1,5, 2, 3
и 4 мл раствора каротина помещают в
сухие пробирки, разбавляют до 5 мл,
перемешивают и измеряют оптическую
плотность растворов при 440 нм в кювете
с толщиной слоя 5 мм или 10 мм для ис-
следования объектов с малым содержа-
пускания
толщиной
нием каротина.
Зная содержание каротина в исходном растворе, вычисляют кон-
центрации каротина в растворах шкалы по формуле:
X а
125
Р___ X 1 а
С 5 25
где С — концентрация каротина в растворах шкалы (в мкг/мл);
X — концентрация каротина в исходном растворе (в мкг/мл); а —
объем рабочего раствора каротина, взятый для приготовления шка-
лы (в мл); 1 — объем исходного раствора каротина, взятый для при-
готовления рабочего раствора (в мл); 25 — объем рабочего раствора
каротина (в мл); 5 — объем, до которого разбавлен раствор а при
приготовлении шкалы (в мл).
Из полученных данных строят калибровочную кривую для ка-
ротина (рис. 35). Этой кривой можно пользоваться и при опреде-
лении ксантофиллов, вводя поправку: концентрацию каротина, по-
лученную по калибровочной кривой, умножают на следующие коэф-
фициенты: при определении суммы ксантофиллов — на 1,06,
виолаксантина — на 1,15, неоксантина — на 1,14 и лютеина —
на 1,04.
203
ПОЛУЧЕНИЕ КСАНТОФИЛЛА — ЛЮТЕИНА
Сущность метода. Ксантофиллы извлекают из листьев
смесью этилового спирта и эфира. Хлорофилл омыляется щелочью
ц вымывается водой. Ксантофиллы остаются в эфирном слое, из
которого пигменты переводят на безводный сульфат натрия, испа-
ряя эфир при размешивании соли. Полученный порошок отмывают
от каротина петролейным эфиром, а ксантофиллы, адсорбированные
на сульфате натрия, помещают в колонку с углекислым магнием, где
лютеин отмывается толуолом от липидов и других ксантофиллов.
Оранжевый слой лютеина отделяют и растворяют в смеси этилового
спирта и эфира. Раствор доводят до определенного объема и ис-
пользуют для приготовления шкалы и определения концентрации
лютеина химическим методом с помощью бихромата калия в серно-
кислотном растворе. Реакция идет по следующему уравнению:
ЗС40Н50О2 + Ю6К2Сг2О7 + 424H2SO4 - 120СО2 + 106K2SO4 +
+ 106Сг2 (SO4)3 + 508Н2О.
Отсюда грамм-эквивалент лютеина ~ЗС4030°°2 равен 2,683.
Ход работы. Отвешивают 10 г свежевысушенных при 50—
60° С и превращенных в порошок листьев крапивы, клевера или
сахарной свеклы, помещают в ступку и растирают с 1 г углекислого
магния, и 20 а безводного сульфата натрия. Смесь переносят в во-
ронку диаметром 40 л/л/ со стеклянным фильтром № 3, уплотняют
стеклянной палочкой и промывают 40 л/л петролейного эфира или
бензина для удаления каротина. Раствор полностью отсасывают
и удаляют, а из остатка извлекают ксантофиллы и другие пигменты
100 л/л смеси этилового спирта и эфира (1 1), прибавляя ее по
5—10 л/л и не добиваясь полного извлечения пигментов. Экстракт
переносят в делительную воронку емкостью 500 л/л, прибавляют
2 л/л 20%-ного раствора едкого натра, перемешивают и оставляют
на 3 ч или на ночь для омыления хлорофилла. Затем добавляют
10 мл этилового эфира, 100 л/л дистиллированной воды, встряхивают
1 мин и дают постоять 5 мин для разделения фаз. В верхнем, эфир-
ном слое находятся каротиноиды и часть продуктов разложения хло-
рофилла, а в нижнем, спиртово-водном слое находится омыленный
хлорофилл и другие водорастворимые вещества. Нижний слой пере-
носят в другую длительную воронку, прибавляют 20 мл эфира,
встряхивают, дают фазам разделиться и нижний слой отбрасывают,
а верхний эфирный раствор присоединяют к первому экстракту.
Раствор каротиноидов в эфире отмывают от хлорофилла сначала
3 раза 0,1 н. раствором NaOH, прибавляя его по 10 мл слегка встря-
хивая, затем дистиллированной водой до полного удаления щелочи.
Воду прибавляют порциями по 10—15 мл и только после четырех
промываний начинают пробовать на полноту промывания фенол-
фталеином. Промывание прекращают, как только промывная жид-
кость перестанет окрашиваться фенолфталеином.
204
Эфирный раствор каротиноидов помещают в ступку, прибавля-
ют 10 г безводного сульфата натрия и растирают под вытяжным шка-
фом до полного испарения эфира. Полученный порошок переносят
в колонку, содержащую 20 г безводного сульфата натрия. Для ко-
лонки используют воронку диаметром 25 л/л/ со стеклянным филь-
тром № 3. Добавленный порошок уплотняют стеклянной палочкой
и промывают петролейным эфиром или бензином, прибавляя раство-
ритель по 5 мл до полного вымывания каротина, фсофитипов и дру-
гих примесей, на что требуется от 5 до 6 промываний. Затем верх-
ний желтый или желто-зеленый слой, содержащий ксантофиллы,
извлекают из колонки стеклянной палочкой и переносят порошок
на другой стеклянный фильтр, содержащий 5 г углекислого магния,
хорошо растертого в ступке и уплотненного на фильтре. Уплотнение
углекислого магния достигается следующим образом: фильтр
№ 3 с пробкой вставляют в пробирку для отсасывания, включают
насос, всыпают растертый углекислый магний и уплотняют палоч-
кой. Порошок с ксантофиллами, перенесенный на фильтр, уплот-
няют палочкой и промывают толуолом, прибавляя его по 5 мл
и отсасывая со скоростью 60 капель в 1 мин до тех пор, пока идущие
впереди пигменты золотисто-желтого цвета не вымоются полностью,
а оранжевый слой лютеина отделится от зеленоватой верхней зоны.
После этого толуол отсасывают и верхнюю зеленую зону удаляют
полностью и даже часть оранжевого слоя, если присутствуют хотя
бы следы зеленого. Оставшийся чистый лютеин оранжевого цвета
растворяют в 10 мл смеси этилового спирта и эфира (1 1), прибав-
ляя ее небольшими порциями и собирая раствор после отсасывания
в мерный цилиндр. Раствор разбавляют 95%-ным спиртом до 10 мл.
1 мл полученного раствора переносят в мерную колбу на 25 мл,
разбавляют спиртом и используют для приготовления растворов
шкалы, а 8 мл помещают в пробирку для определения концентра-
ции лютеина.
Определение содержания лютеина
в исходном растворе
Пробирку, содержащую 8 мл раствора лютеина, ставят в горячую
воду и выпаривают раствор досуха. При выпаривании необходимо
перемешивать раствор стеклянной палочкой и регулировать тем-
пературу воды таким образом, чтобы не было бурного кипения.
К остатку после выпаривания добавляют 5 мл 0,5 н. раствора би-
хромата калия в серной кислоте, размешивают стеклянной палочкой,
оставляя ее в пробирке. Пробирку погружают в кипящую воду
и нагревают 10 мин, время от времени перемешивая раствор палоч-
кой. После охлаждения раствор из пробирки переносят в колбу
для титрования и разбавляют дистиллированной водой до 100 мл.
Раствор охлаждают, прибавляют 5 мл 20%-ного раствора йодистого
калия и выделившийся йод титруют 0,1 н. раствором тиосульфата на-
трия. Отдельно титруют таким же способом 5 мл раствора бихромата
205
1,0
^0,6
^Ot2
0 в
Концентрация лютеинсрмке/мл
Рис.?36. Калибровочная кри-
вая для ксантофиллов (све-
тофильтр с максимумом про-
пускания 440 нм, кювета с
толщиной измеряемого слоя
5 мм).
калия, который служит контролем. з результатов титрования
вычисляют содержание лютеина по следующей формуле:
X = 268,3- (а~А = 33,54 . (д _ Ь),
О
где X — концентрация лютеина в исходном растворе (в мкг/мл);
8 _ объем исходного раствора лютеина, взятый для определения
его содержания (в мл); а — объем 0,1 н.
раствора тиосульфата натрия, затрачен-
ный на титрование контрольного раство-
ра (в мл); Ь — объем 0,1 н. раствора
тиосульфата натрия, затраченный на
титрование исследуемого раствора (в мл);
268,3 — количество лютеина, соответ-
ствующее 1 мл 0,1 н. раствора тиосуль-
фата натрия (в мкг).
Построение калибровоч-
ной кривой. 1 мл раствора лю-
теина в мерной колбе на 25 мл разбав-
ляют 95%-ным спиртом до метки, пере-
мешивают и используют для приготов-
ления растворов шкалы. Для этого 0,25,
0,5, 1,0, 1,5, 2, 3 и 4 мл разбавленного
раствора лютеина помещают в сухие
пробирки и доводят этиловым спиртом
до 5 мл. Растворы перемешивают и из-
меряют оптическую плотность при 440 нм
в кювете с толщиной слоя 5 мм или
на 10 мм, если необходимо. Из результатов измерений оптической
плотности и концентраций лютеина в растворах шкалы строят ка-
либровочную кривую (рис. 36).
Концентрацию лютеина в растворах шкалы вычисляют
по формуле:
__ К 1 CL К CL
5~25 = 125
где С — концентрация лютеина в растворах шкалы (в мкг/мл);
X — концентрация лютеина в исходном растворе (в мкг/мл); 1 —
объем исходного раствора лютеина, взятый для получения разбав-
ленного раствора (в мл); 25 — объем разбавленного раствора (в мл);
а — объем разбавленного раствора лютеина, взятый при приготов-
лении шкалы (в мл); 5 — объем фотометрируемых растворов (в мл).
Получение лютеина в чистом виде осуществляется легче, чем
каротина, поэтому можно построить калибровочную кривую для лю-
теина и затем использовать ее при определении каротина и других
каротиноидов, умножая найденный результат на соответствующие
коэффициенты; при определении каротина — на 0,96, суммы ксан-
тофиллов— на 1,02, неоксантина — на 1,10 и виолаксантина —
на 1,11.
206
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПИГМЕНТОВ
С ПОМОЩЬЮ ХРОМАТОГРАФИИ ПА БУМАГЕ
При определении пигментов с помощью хроматографии на бумаге
следует учитывать возможность гидролиза хлорофиллов в хлоро-
филлиды под действием хлорофилазы, особенно у растений, обла-
дающих высокой активностью ее, например у сахарной свеклы.
У многих растений с кислым клеточным соком возможна частичная
феофитизация хлорофиллов. Во время хроматографирования состав
пигментов может измениться вследствие окисления кислородом
воздуха и взаимодействия с растворителями. Поэтому следует ис-
ключить действие этих факторов фиксированием пробы низкой тем-
пературой, нейтрализацией кислот углекислым магнием и исполь-
зованием для хроматографического разделения неполярных раст-
ворителей.
Сущность метода. Навеска средней пробы листьев
растирается в ступке с небольшим количеством углекислого маг-
ния, сухим льдом и смесью ацетона с этиловым спиртом (3 1).
Пигменты извлекают на стеклянном фильтре этим же растворите-
лем и раствор доводят до определенного объема. Часть полученного
раствора наносят на бумагу и после хроматографирования разде-
ленные пигменты извлекают из бумаги подходящими растворите-
лями и фотоколориметрируют. Сравнив полученные результаты
с калибровочными кривыми определенных пигментов, находят их
концентрации и содержание в исследуемом образце.
Для разделения пигментов па бумаге необходимо пользоваться
такими растворителями, которые в процессе хроматографирования
не оказывают разрушающего действия на пигменты и не способству-
ют окислению их кислородом воздуха (диоксан, толуол и др.).
Целесообразно использовать следующие смеси растворителей.
1. Бензин (температура кипения 40—100° С) 10 частей, ацетон
3 части по объему. При применении этого растворителя пигменты
имеют следующие Rf: хлорофилл b — 0,13; хлорофилл а — 0,27;
ксантофиллы — 0,30, 0,36, 0,53; феофитины — 0,73 и каротин —
0,98.
2. Бензин (температура кипения 40—120° С) 30 частей, ацетон
8 частей и пропиловый спирт 0,1 части по объему. При применении
этого растворителя пигменты имеют следующие Rf: хлорофилл b —
0,20; хлорофилл а — 0,46; ксантофиллы — 0,48, 0,53 и 0,69; фео-
фитины — 0,83 и каротин — 0,98.
Ход анализа. 1г средней пробы листьев отвешивают
с точностью до 0,01 г и растирают с 0,05 г углекислого магния, ку-
сочком сухого льда и 5 мл смеси ацетона с этиловым спиртом (3 1).
Массу переносят на стеклянный фильтр № 3 или № 4. Жидкость
отсасывают, а остаток промывают охлажденной смесью ацетона
со спиртом, предварительно смывая пигменты со стенок ступки,
в которой производилось растирание навески. Растворитель при-
бавляют небольшими порциями (по 2 мл) до полного вымывания пиг-
207
ментов. Фильтрат переносят в мерный цилиндр, доводят объем до
10 или 15 м/г, перемешивают и переливают в сухую пробирку.
0,5 мл полученного раствора наносят в виде горизонтальной полос-
ки на ленту хроматографической бумаги между двумя вырезами
в верхней части. Во время нанесения раствора на бумагу ее обду-
вают вентилятором для ускорения высыхания. Если раствор раз-
бавлен до 15 мл, наносят еще 0,5 мл исследуемого раствора и дают
высохнуть. При работе следует избегать интенсивного освещения.
Перед нанесением раствора пигментов хроматографическую
бумагу соответствующим образом подготавливают. Для этого
из хроматографической бумаги (Ленинградская «М»), предвари-
тельно промытой 0,1 н. раствором соляной кислоты, затем водой
и высушенной при обыкновенной температуре, вырезают полосы
длиной 50 см и шириной 10 см. В верхней части бумажной ленты
с двух сторон — на расстоянии 10 мм от правого и левого края и на
расстоянии 7—8 см от верхнего края делают лезвием безопасной
бритвы ограничительные вырезьгдлиной 20 мм и шириной 2 мм так,
как это описано ниже (см. стр. 246).
Приготовленные таким образом две бумажные полоски закреп-
ляют между двумя стеклянными палочками и помещают в хромато-
графическую камеру — стеклянный цилиндр с притертой крышкой.
В верхней части камеры находится ванночка, в которую наливают
30—40 мл растворителя: бензин — ацетон (10 3) или бензин — аце-
тон — пропиловый спирт (30 : 8 0,1), в который погружают верх-
ние концы бумажных лент. На дно цилиндра ставят чашку, в кото-
рую наливают растворитель такого же состава, как и в ванночку.
Камеру закрывают крышкой и затемняют черным колпаком. Раст-
воритель пропускают в течение 2—3 ч, пока фронт растворителя
опустится на 36—38 см от места нанесения пигментов и произойдет
полное разделение пигментов. После этого хроматограмму вынимают
из камеры и дают возможность быстро подсохнуть в вытяжном шкафу
при включенном вентиляторе. Вырезают ножницами окрашен-
ные зоны пигментов, складывают вырезки в виде гармошки, свора-
чивают и помещают в сухие пробирки, отмечая исследуемые веще-
ства номерами. Из вырезок каротин извлекают бензином с темпе-
ратурой кипения 40—120° С, а остальные пигменты 80%-ным
этиловым спиртом. Для этого в пробирки с бумажными вырезками,
содержащими пигменты, прибавляют по 5 мл растворителя, про-
бирки закрывают пробками и взбалтывают до полного извлечения
пигментов. В полученных растворах измеряют оптическую плотность
с помощью фотоэлектроколориметра. Каротин и ксантофиллы
фотометрируют при 440—450 нм, хлорофилл а при 660 нм, а хло-
рофилл в при 642 нм. Полученные данные сравнивают с калибровоч-
ными кривыми определяемых пигментов, находят концентрации
исследуемых веществ и вычисляют содержание пигментов по сле-
дующей формуле:
х = 0,1 5 4 С = 0,5 А С
ап а • п ’
1208
где X — содержание пигмента (в мг на 100 г исследуемого вещества);
А—объем экстракта пигментов из навески (в мл); а — объем
экстракта пигментов, нанесенный на хроматограмму (в мл); 5 —
объем колориметрируемого раствора пигмента (в мл); С — концентра-
ция пигмента, найденная по калибровочной кривой (в мкг/мл);
п — навеска исследуемого вещества (в г); 0,1 — коэффициент
пересчета микрограммов в миллиграммы и на 100 г вещества.
При работе с пигментами следует работать на слабом рассеянном
свету. Все операции с пигментами нужно осуществлять как можно
быстрее, а все получаемые растворы затемнять.
Реактивы. Этиловый спирт (80%-ный). К 100 мл 95%-ного спирта прили-
вают 21 мл дистиллированной воды и перемешивают.
Бензин (температура кипения 40—120° С). Из авиационного бензина отго-
няют фракцию, кипящую от 40 до 120° С. Так же поступают при получении фрак-
ции 40—100° С. Остальные органические растворители очищаются перегонкой.
Хроматографическая бумага. Хроматографическую бумагу моют в кювете
0,1 н. раствором соляной кислоты, затем дистиллированной водой до удаления
ионов хлора. После этого бумагу оставляют в кювете до полного высыхания.
ВЫДЕЛЕНИЕ ХЛОРОФИЛЛА А И ХЛОРОФИЛЛА В
Сущность метода. Листья растираются с безводным
сульфатом натрия для обезвоживания и пигменты извлекают этило-
вым эфиром, который затем удаляют, а пигменты переводят на без-
водный сульфат натрия. Каротин, феофитины и большую часть
ксантофиллов удаляют промыванием толуолом па колонке из угле-
кислого магния. Зону суммы хлорофиллов извлекают из колонки
и растворяют в ацетоне, который затем удаляют выпариванием
на сульфате натрия. Хлорофилл а отделяется от хлорофилла b
на колонке из сахара. Зоны хлорофилла а и хлорофилла Ь извлекают
из колонки отдельно и экстрагируют этиловым спиртом. Из полу-
ченных растворов готовят шкалу для хлорофилла а и хлорофилла Ь.
После измерения оптической плотности все растворы хлорофилла
а объединяют в делительной воронке, хлорофилл извлекают хло-
лоформом и сжигают по Кьельдалю для определения азота. По содер-
жанию азота устанавливают концентрацию хлорофилла а в раство-
рах шкалы. Таким же образом поступают с растворами хлорофилла
Ь. Установив концентрацию исходного раствора хлорофилла, вы-
числяют концентрацию его во всех растворах шкалы и из получен-
ных данных строят калибровочные кривые.
Ход работы. Из свежих листьев смородины, крапивы
или других растений удаляют крупные жилки. Листья измельча-
ют и отвешивают 15 г полученной массы. Навеску помещают в ступ-
ку, прибавляют 1 г углекислого магния, 30 г безводного сульфата
натрия и растирают до превращения содержимого в однородный
зеленый порошок. Сухой порошок переносят на стеклянный фильтр
№ 3, уплотняют палочкой и извлекают пигменты 60 мл этилового
эфира, прибавляя его по 10 мл и отсасывая. Эфирный раствор пиг-
ментов переносят в ступку, содержащую 15 г безводного сульфата
14 6-2
209
натрия, и под вытяжным шкафом перемешивают до испарения всего
эфира и превращения содержимого в сухой зеленый порошок. По-
следний растирают в ступке и переносят на колонку, содержащую
15 г углекислого магния, уплотняют и промывают толуолом до
вымывания суммы хлорофиллов из зоны сульфата натрия и пере-
мещения его на углекислый магний, на что расходуется 50—60 мл
толуола. Зону, находящуюся выше зеленой зоны суммы хлорофил-
лов, удаляют, а хлорофиллы на колонке промывают еще 2 раза
петролейным эфиром порциями по 10 мл и отсасывают жидкость
полностью. Зеленую верхнюю зону, содержащую хлорофиллы,
осторожно снимают, переносят на стеклянный фильтр № 3 и извле-
кают их как можно меньшим количеством ацетона, прибавляя его
по 3—5 мл.
Ацетоновый раствор хлорофиллов помещают в ступку, добав-
ляют 15 г безводного сульфата натрия и размешивают под вытяжным
шкафом до полного испарения ацетона^ Сухой зеленый порошок
растирают в ступке и переносят на колонку из сахарозы. Колонку
готовят следующим образом. Сахарозу высушивают в сушильном
шкафу при температуре 100° С, растирают в ступке и просеивают
через шелковое сито. Отвешивают 100 г полученной сахарной пудры,
растирают в ступке с 2 г углекислого магния, прибавляя ее к MgCO3
постепенно. Этой смесью наполняют колонку, длина которой должна
быть не менее 20 см, а диаметр 30—35 мм. На дно колонки перед на-
полнением вставляется тампон, состоящий из стеклянного шарика,
окруженного гигроскопической ватой. Сахарную пудру вносят
небольшими порциями и хорошо уплотняют палочкой при одно-
временном отсасывании насосом. В готовую колонку насыпают
порошок сульфата натрия с суммой хлорофиллов, уплотняют па-
лочкой и промывают бензином или петролейным эфиром. При этом
пигменты разделяются: первым движется каротин желто-оранжево-
го цвета, за ним ксантофиллы и хлорофилл а. Когда слой хлорофиллов
опустится ниже слоя сульфата натрия, отсасывание прекращают
и весь слой сульфата натрия удаляют. Затем включают насос и ко-
лонку промывают петролейным эфиром, содержащим 0,4% пропило-
вого спирта до тех пор, пока хлорофилл а (синяя зона) не опустится
в самый низ колонки, а хлорофилл b отделится достаточно хорошо.
Обычно между зонами хлорофиллов находится желтая зона одного
из ксантофиллов.
После разделения хлорофиллов растворитель отсасывают пол-
ностью и выключают насос. Верхнюю зону желто-зеленого цвета,
не содержащую хлорофилл Ь, удаляют, а нижележащую зеленую
зону хлорофилла b извлекают, переносят на стеклянный фильтр
№ 3, уплотняют и извлекают хлорофилл b 15 мл 95%-ного этилового
спирта, прибавляя его небольшими порциями и отсасывая экстракт
в чистую пробирку. Полученный раствор разбавляют в мерном ци-
линдре 95%-ным спиртом до 20 мл и перемешивают.
В нижней части колонки находится хлорофилл а, а выше зоны
хлорофилла а — небольшая часть ксантофиллов. Желтые пигменты
210
можно удалить частично, так как
они не мешают определению хлоро-
филла. Для извлечения хлорофилла
а в колонку с (оставшейся нижней зо-
ной приливают 20 мл 95%-ного эти-
лового спирта и полностью отсасыва-
ют. Раствор хлорофилла а переносят
в мерную колбу на 25 мл, разбав-
ляют спиртом до метки и перемеши-
вают. Из полученных растворов хло-
рофилла а и хлорофилла b готовят
растворы с различной концентрацией
для построения калибровочных кри-
вых.
Концентрация хлорофиллам кг/мл
Рис. 37. Калибровочная кривая
для хлорофиллов а (I) и b (II) в
этиловом спирте (светофильтр
для хлорофилла а с максимумом
пропускания 660 нм и для хло-
рофилла b — 642 нм)'.
1 — толщина измеряемого слоя
10 мм; 2 — толщина измеряемого
слоя 5 мм.
Построение калибро-
вочных кривых. Для приго-
товления шкалы хлорофилла а поме-
щают в сухие пробирки 0,25, 0,5, 1,0,
1,5, 2,0 и 2,5 мл первоначального
раствора и разбавляют 95%-ным спир-
том до 5 мл. Для шкалы хлорофилла b
набирают пипеткой 0,25, 0,5, 1,0, 1,5,
2,о, 3,0 и 4,0 мл первоначального
спиртового раствора хлорофилла переносят в сухие пробирки
и добавляют 95%-ный спирт до объема 5 мл. Измеряют оптическую
плотность полученных растворов при 642 нм (хлорофилл Ь) и
660 нм (хлорофилл а) в кюветах с толщиной слоя 5 и 10 мм.
Концентрацию хлорофилла в растворах шкалы вычисляют по
формуле:
с— х А
G 5“
где С — концентрация хлорофилла в данном растворе (в мкг/мл);
А — объем первоначального раствора хлорофилла, взятый для
приготовления шкалы (в мл); X — концентрация хлорофилла в пер-
воначальном растворе (в мкг/мл); 5 — объем фотометрируемого
раствора шкалы (в мл).
Полученные данные концентрации оптической плотности нано-
сят на график и получают калибровочные кривые для хлорофилла
а и хлорофилла b (рис. 37).
Определение содержания хлорофиллаanb
в растворе
После измерения оптической плотности все растворы шкалы
хлорофилла а и остаток исходного раствора переносят в делительную
воронку, промывая все пробирки и колбу самым слабым раствором
шкалы, а под конец смывают остатки 5 мл спирта. То же самое
делают и с растворами хлорофилла Ь, собирая их в другую
14*
211
делительную воронку. К объединенному раствору одного из хлоро-
филлов в делительной воронке прибавляют 20 мл хлороформа, 50 мл
дистиллированной воды, 5 мл 10%-ного раствора хлористого на-
трия, встряхивают 30 мин, дают отстояться 3—5 мин и нижний слой
хлороформа, содержащий хлорофилл, сливают в чистую колбочку.
Верхний, спиртово-водный слой удаляют и воронку промывают
дистиллированной водой. Раствор хлорофилла из колбочки поме-
щают в ту же воронку, добавляют 50 мл промывной жидкости, со-
стоящей из спирта, дистиллированной воды и 10%-ного раствора
NaCl (2 : 2 1), встряхивают 30 мин и дают слоям разделиться. Ниж-
ний слой, содержащий хлорофилл, отделяют и переносят в колбу
Кьельдаля на 50—100 мл. В раствор добавляют 2 мл 4 н. раствора
серной кислоты, колбу ставят в стакан с кипящей водой и выпари-
вают раствор почти досуха. Затем добавляют 2 мл концентрирован-
ной серной кислоты, 0,4 г сульфата калия, 0,3 мл 10%-ного раствора
молибдата натрия, помещают па плитку в наклонном положении
и сжигают до полного просветления.
После сжигания и охлаждения прибавляют 5 мл дистиллирован-
ной воды и переносят раствор в коническую колбу, промывая колбу
Кьельдаля 20 мл воды. К раствору прибавляют каплю метилоран-
жа и нейтрализуют 20%-ным раствором едкого натра до перехода
цвета индикатора из красного в желто-оранжевый. Затем прибав-
ляют 10 мл фосфатного буферного раствора с pH 6,7, содержащего
бромистый калий, перемешивают и приливают по стенке колбы
5 мл 0,12 н. раствора хлорамина. Колбу накрывают часовым стек-
лом, раствор перемешивают круговым движением колбы и оставляют
на 25 мин. После этого прибавляют 3 мл 20%-ного раствора йодис-
того калия, 5 мл раствора серной кислоты (1 9) и выделившийся
йод титруют 0,02 н. раствором тиосульфата натрия в присутствии
крахмала.
Параллельно проводят контрольный опыт. В колбу для титро-
вания берут 2 мл концентрированной серной кислоты, прибавляют
20 мл дистиллированной воды, каплю метилоранжа, нейтрализуют
20%-ной щелочью, прибавляют 10 мл буфера, 5 мл 0,12 н. раствора
хлорамина и через 25 мин прибавляют 2 мл 20%-ного раствора йо-
дистого калия, 5 мл раствора серной кислоты (1 9) и титруют
выделившийся йод 0,02 н. раствором тиосульфата натрия. Концен-
трацию хлорофилла а в растворе, из которого готовится шкала,
вычисляют по следующей формуле:
1489 (и —а)
Ла ” А
а концентрация хлорофилла b по формуле:
v 1512 (v — b)
в ’
где Ха — концентрация хлорофилла а в первоначальном растворе
(в мкг1мл}\ Хь — концентрация хлорофилла b в первоначальном
212
растворе (в мкг/мл); v — объем 0,02 н. раствора тиосульфата натрия,
затраченный при титровании контрольного раствора (в мл); а —
объем 0,02 н. раствора тиосульфата натрия, затраченный при титро-
вании раствора хлорофилла а (в мл); b — объем 0,02 и. раствора
тиосульфата натрия, затраченный при титровании раствора хлоро-
филла b (в мл); А — объем первоначального раствора хлорофилла а
(в мл); В — объем первоначального раствора хлорофилла b (в мл);
1489 — количество хлорофилла а, соответствующее 1 мл 0,02 н.
раствора тиосульфата натрия (в мкг); 1512 — количество хлорофил-
ла &, соответствующее 1 мл 0,02 н. раствора тиосульфата натрия
(в мкг).
Реактивы. Сахароза. Применяют химически чистый препарат, который вы-
сушивают при 100° С, растирают в ступке и просеивают через шелковое сито.
Промывная смесь (спирт — вода—10%-ный NaCl). Смешивают 40 мл
95%-ного этилового спирта, 40 мл дистиллированной воды и 20 мл 10%-ного рас-
твора хлористого натрия. Приготовление остальных реактивов описано выше (см.
стр. 9, 17, 76, 77).
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
ХЛОРОФИЛЛА Л, ХЛОРОФИЛЛА В К КАРОТИНОИДОВ
Сущность метода. Хлорофиллы а и b имеют различные
спектры поглощения, которые пересекаются при 440 и 648 нм.
Так как в точках пересечения кривых светопоглощения интенсив-
ность поглощения не зависит от изменения в относительных коли-
чествах обеих форм, для определения содержания хлорофиллов
в растениях выбирают длины воли, при которых спектры поглоще-
ния хлорофиллов а и Ь пересекаются. В неочищенных экстрактах
из растительных объектов кроме хлорофиллов содержатся кароти-
ноиды, которые интенсивно поглощают свет при 440 нм и не погло-
щают при 648 нм. Поэтому для определения суммы хлорофиллов
а и b пользуются последней длиной волны, при которой интенсив-
ность окраски пропорциональна концентрации хлорофиллов даже
в присутствии каротиноидов.
Так как хлорофиллы а и b и каротиноиды имеют максимумы
поглощения при разных длинах волн, их можно легко определить
в одном растворе спектрофотометрическим методом. Количественное
определение состава смеси поглощающих соединений основывается
на том, что полное погашение любой смеси равно сумме отдельных
погашений ее компонентов при той же длине волны:
Вт = ^1 +
причем каждая из парциальных оптических плотностей равна:
d = E I С,
где Е — молярный коэффициент погашения вещества при данной
длине волны; / — толщина поглощающего слоя (в см); С — кон-
центрация поглощающего вещества (в моль/л).
Если раствор содержит п окрашенных веществ, проводят п неза-
висимых измерений оптической плотности при п различных длинах
213
волн. В результате получают систему линейных уравнений. При
наличии в растворе двух окрашенных веществ с различными спек-
трами поглощения (например, хлорофилл а и хлорофилл Ь), опре-
деляют концентрации Сг и С2 растворов при двух длинах волн
по уравнениям:
Di = Е\а Са + Е\ь Сь;
D2 = E2a Са + Е2ь Сь, (1)
где D± и D2 — измеренные экспериментально оптические плотности
смеси веществ при длинах волн Ах и А2; Е\ и Е% — молярные коэф-
фициенты погашения хлорофилла а при длинах волн Ах и А2; Е\ь
и Еъь — молярные коэффициенты погашения хлорофилла b при дли-
нах волн А4 и А2; Са и Сь — концентрации поглощающих веществ
(в моль!л).
Значения молярных коэффициентов погашения либо берут из таб-
лиц, либо определяют экспериментально следующим образом. При-
готавливают чистые растворы каждого из исследуемых веществ
с известной концентрацией и определяют оптическую плотность их
при тех длинах волн, при которых будет проводиться спектрофото-
метрирование смеси этих веществ. Зная оптическую плотность при
данной длине волны, концентрацию раствора (в моль/л) и толщину
поглощающего слоя, вычисляют молярный коэффициент погашения
по уравнению:
где Ег — молярный коэффициент погашения данного вещества при
длине волны Dx — оптическая плотность при длине волны Ах
(должна быть в пределах от 0,2 до 1,0); С — концентрация исследу-
емого вещества (в молъ/лу, I — толщина поглощающего слоя (в см).
Полученные значения Е подставляют в систему уравнений (1)
и решают ее относительно концентраций поглощающих веществ:
Г —_____________ь_____ .
D2-E2 D±
р __ а____________а_____
Ь \ ЕН-^а \ ’
Максимум поглощения в красной области спектра хлорофилла а
находится при 662 нм, а максимум поглощения в красной области
хлорофилла b — при 644 нм. Определив молярные коэффициенты
погашения хлорофилла а и хлорофилла b при указанных длинах
волн и решив уравнения (2), можно получить следующие уравнения
для вычисления концентраций этих пигментов:
Cfl = 9,78 Dqq2 0,99
Cb — 21,43 • T?e44 —- 4,65 Z)662; (3)
Са + = 5,13 E)qq2 + 20,44 Т)644,
214
где Са — концентрация хлорофилла а (в мкг/мл); 'Сь — концентра-
ция хлорофилла b (в мкг/мл); DGG2 — оптическая плотность смеси
при 662 нм и толщине слоя 1 см; DG^ — оптическая плотность
смеси при 644 нм и толщине слоя 1 см.
Таким образом, при исследовании пигментов листьев можно в
ацетоновой вытяжке определить хлорофилл а и хлорофилл Ь, изме-
рив оптическую плотность раствора при 644 и 662 нм. Спектрофото-
метрическое определение суммы каротиноидов в присутствии хлоро-
филла можно также свести к двум компонентам, измерив оптическую
плотность смеси при 440 нм (максимум поглощения каротиноидов
и место пересечения кривых поглощения хлорофилла а и хлоро-
филла Ь) и определив концентрацию суммы хлорофиллов по урав-
нению (3). Содержание каротиноидов рассчитывают по уравнениям:
Cfe = 4,7 D44o 0,27
Ck = 4,7 D44o - (1,38 De62 + 5,48 D644),
где Ck — концентрация каротиноидов (в мкг/мл); Z)440 — опти
ческая плотность смеси при 440 нм и толщине слоя 1 см; С^Ь) —
концентрация суммы хлорофиллов, вычисленная по уравнению (3).
Ход анализа. Из усредненной пробы листьев отвеши-
вают 0,1 или 0,2 г в зависимости от содержания хлорофилла и рас-
тирают в ступке с 0,05 г MgCO3 и 5 мл 90%-ного ацетона. Содержи-
мое ступки переносят на стеклянный фильтр № 3 и извлекают пиг-
менты, добавляя небольшие количества 90%-ного ацетона (поЗ—
5 мл) и отсасывая раствор со скоростью 100—120 капель в 1 мин.
Как только все пигменты будут извлечены, раствор переносят в мер-
ную колбу на 25 мл, доводят ацетоном до метки, промывая прибор
для отсасывания, перемешивают и измеряют оптическую плотность
раствора на спектрофотометре СФ-5 или СФ-4 при 662 нм, 644 нм
и 440 нм в кювете с толщиной слоя 1 см.
Полученные величины подставляют в формулы (3) и (4) и вычис-
ляют концентрации хлорофилла а, хлорофилла b и каротиноидов.
Исходя из найденных концентраций пигментов, рассчитывают
содержание их в исследуемом образце по следующей формуле:
v 0,1 С А
где X — содержание пигмента в исследуемом образце (в мг на 100 г
вещества); С — концентрация пигмента (в мкг/мл); А — объем
экстракта пигментов (в мл); п — навеска исследуемого вещества
(в г); 0,1 — коэффициент пересчета концентрации пигмента (в мг/мл
и на 100 г вещества).
При использовании двухволнового спектрофотометрического
метода анализа смесей следует считаться с тем, что вычисления по
формулам часто дают величины, значительно отклоняющиеся
от истинного содержания пигментов. Основной причиной расхожде-
ний является несоответствие действительного положения макси-
мумов поглощения хлорофиллов в исследуемых растворах положе-
215
нию, предусмотренному в формулах. Ошибка в измерении в 1 нм
по сравнению с истинным положением максимума поглощения при-
водит к отклонению для хлорофилла а на 2%, а для хлорофилла b—
на 19% от истинного содержания.
Смещение длин волн кварцевого спектрофотометра только вслед-
ствие его нагревания во время работы может достигать 2 нм, стек-
лянного — 0,5 нм. Поэтому для работы с хлорофиллом лучше поль-
зоваться спектрофотометром СФ-5. Используемый для измерений
спектрофотометр должен быть хорошо откалиброван. Для данного
растворителя используют только определенно установленную для
него формулу. В лучшем случае ошибки при спектрофотометри-
ческом определении хлорофиллов в смеси составляют для хлоро-
филла а ± 2—3% и для хлорофилла b ± 20%.
Реактивы. Ацетон (90%-ный). К 90 мл свежеперегнанного ацетона (из пре-
парата ч. д. а.) прибавляют 10 мл дистиллированной воды и перемешивают.
ОПРЕДЕЛЕНИЕ ПРОЧНОСТИ СВЯЗИ
ХЛОРОФИЛЛА С БЕЛКАМИ
Сущность метода. Отношение пигментов фитохромо-
протеидных комплексов к растворителям зависит от растворимости
их в этих растворителях и от способности растворителей вызывать
денатурацию комплексов. Неполярные растворители — бензин,
петролейный эфир и гексан не извлекают хлорофилл из ненару-
шенных его комплексов с белками, но даже небольшие примеси по-
лярных растворителей к неполярным, например спирта к петролей-
ному эфиру, приводят к частичному извлечению хлорофилла.
Полное извлечение хлорофилла из листьев может быть достигнуто
уже при 2%-ной концентрации спирта в петролейном эфире.
После денатурации протеидного комплекса (высокой температу-
рой, ультразвуком, ультрафиолетовым облучением) хлорофилл
начинает частично извлекаться и неполярными растворителями. Это
происходит вследствие нарушения связи хлорофилла с белками
и растворения его в липоидной части комплекса, который извлека-
ется неполярными растворителями. Таким образом, по количеству
хлорофилла, извлекаемого неполярными растворителями, можно
судить о прочности связи хлорофилла с белками. Небольшая при-
месь полярного растворителя, например спирта, к неполярному уве-
личивает количество извлекаемого хлорофилла. С увеличением кон-
центрации спирта в петролейном эфире степень извлечения хло-
рофилла растет. Характер кривых извлечения хлорофилла смесью
полярных и неполярных растворителей показывает, что в листьях
возможно существование двух форм хлорофилл-белковых комплек-
сов. Одна форма менее прочная, из которой хлорофилл извлекают
при более низких концентрациях спирта в петролейном эфире,
и другая более прочная, из которой хлорофилл извлекают при более
высоких концентрациях спирта. По степени извлечения хлорофилла
0,4%-ным раствором спирта в петролейном эфире или бензине
216
можно судить о количестве менее прочно связанных компонентов.
По степени же извлечения 0,8%-ным раствором спирта в петролейном:
эфире или бензине можно судить о количестве и более прочно свя-
занных комплексов.
Для обезвоживания тканей используют сульфат натрия, но в ми-
нимальном количестве, так как он обладает некоторыми адсорбцион-
ными свойствами и может влиять па степень извлечения пигментов.
Ход анализа. Из средней пробы исследуемых листьев
берут 4 навески по 0,2 г, помещают в ступки и добавляют к ним
по 0,4 г безводного сульфата натрия и по 0,05 г углекислого магния.
Пробы растирают до полного высушивания. Затем одну навеску
растирают с 10 мл бензина (температура кипения 40—100° С),
содержащего 0,2% этилового спирта, другую с 10 мл бензина, со-
держащего 0,4% этилового спирта, третью с 10 мл бензина, содержа-
щего 0,8% этилового спирта, и четвертую с 10 мл бензина, содержаще-
го 1,2% этилового спирта. Содержимое ступок переносят на стеклян-
ные фильтры № 3, растворы отсасывают, а остатки промывают
соответствующими растворами порциями по 3 мл, отсасывая про-
мывную жидкость до тех пор, пока наберется 10 мл фильтрата. За-
тем измеряют оптическую плотность полученных растворов при
640—660 нм в кювете с толщиной слоя 10 мм. Полученные данные
сравнивают с калибровочной кривой и вычисляют количество из-
влеченного хлорофилла по следующей формуле:
М С Л,
где М — количество хлорофилла, извлеченного из данной навески
(в мкг)’, С — концентрация хлорофилла в исследуемом растворе
(в мкг/мл)’, А — объем исследуемого раствора хлорофилла (в мл).
Оставшийся на фильтре нерастворенный хлорофилл извлекают
10 мл 90%-ного этилового спирта, прибавляя его небольшими пор-
циями до полного извлечения хлорофилла. Объем экстракта дово-
дят спиртом до 10 мл и измеряют оптическую плотность раствора
при 640—660 нм в кювете с толщиной слоя 10 мм. Полученный
результат сравнивают с калибровочной кривой и вычисляют ко-
личество неизвлеченного хлорофилла (в мкг).
Из полученных данных вычисляют прочность связи хлорофилла
с белками при извлечении растворителями с разным составом после-
дующей формуле:
v _ X 100
Y ~ М + Х ’
где Y — прочность связи хлорофилла с белково-липоидным ком-
плексом по отношению к смесям растворителей (в % неизвлекаемо-
го хлорофилла); М — количество хлорофилла, извлекаемого смесью
полярного и неполярного растворителей (в мкг)’, X — количество хло-
рофилла, оставшегося после извлечения смесью растворителей (в мкг).
При исследовании прочности связи отдельно для хлорофилла а
и хлорофилла b поступают следующим образом. Из средней пробы
листьев берут пять навесок по 0,1 г, помещают в ступки, добавляют
217
по 0,2 г безводного сульфата натрия и растирают досуха. За-
тем одну навеску растирают с 10 мл бензина, содержащего 0,2%
спирта, фильтруют через стеклянный фильтр № 3 с отсасыванием,
осадок на фильтре промывают тем же растворителем 3—4 раза
порциями по 3 мл и промывную жидкость отсасывают. Три следую-
щие навески растирают с такими растворителями: одну — с бензином,
содержащим 0,4% спирта, другую — с бензином, содержащим 0,8%
спирта, и третью — с бензином, содержащим 1,2% спирта. Фильтру-
ют через стеклянный фильтр и промывают соответствующими рас-
творителями порциями по 3 мл до получения примерно 10 мл
фильтрата, который затем отбрасывают.
Оставшийся на фильтре неизвлеченный хлорофилл извлекают
20 мл 90%-ного ацетона, доводят объем экстракта до 20 мл и изме-
ряют оптическую плотность при 662 и 644 нм в кювете с толщиной
слоя 10 мм. Так поступают с остатками всех проб и затем вычисля-
ют концентрации неизвлеченного хлорофилла а и хлорофилла b
во всех пробах по формуле (3) (см. стр. 214).
Для определения общего содержания хлорофилла а и b в исследу-
емом образце извлекают хлорофилл из пятой навески 20 мл 90 %-но-
го ацетона, разбавляют раствор ацетоном до 20 мл и измеряют
оптическую плотность при 662 и 644 ммк. Полученные величины
подставляют в формулу (3) и вычисляют общие концентрации хло-
рофиллов а и Ь.
По найденным концентрациям рассчитывают показатели проч-
ности связи для хлорофилла а и хлорофилла &, используя формулу:
У _ С 100
У “ X
где Y — прочность связи хлорофилла с белково-липоидным комплек-
сом (в % неизвлекаемого хлорофилла а или Ь). Вычисляют для бен-
зина с 0,2%-ным, 0,4%-ным, 0,8%-ным и 1,2%-ным содержанием
спирта; С — концентрация неизвлекаемого хлорофилла а или b
в (мкг/мл); X — общая концентрация хлорофиллов а или b (в мкг/мл).
ЛИТЕРАТУРА
Аэров И. Л., Лихолат Д. А. Доп. АН УРСР, 1966, 12, 1599.
Бажанова Н. В., Маслова Т. Г., Попова И. А., Попова О. Ф.,
Сапожников Д. И., Эйдельман 3. М. Пигменты пластид зеле-
ных растений и методика их исследования. М.—Л., «Наука»,' 1964.
Годнее Т. Н. Строение хлорофилла и методы его количественного определе-
ния. Минск, Изд-во АН БССР, 1952.
Годнев Т. Н. К методике определения пигментов в хлоропластах растений.—
Физиол. раст., 1965, 12, 3.
Евстигнеев В. Б., Прохорова Л. И. Об определении хлорофиллов
а и b в смеси без разделения компонентов.— Биохимия, 1968, 33, 2, 286.
Лебедев С. И. Физиологическая роль каротина в растении. Киев, Изд-во
АН УССР, 1953.
Цвет М. С. Хроматографический адсорбционный анализ. М., 1946.
Шлык А. А. О спектрофотометрическом определении хлорофиллов а и Ь.— Био-
химия, 1968, 33, 2, 275.
N у b о m N.— Hereditas, 1955, 41, 483.
D. von W e t t s t e i n.— Experimental cell Research, 1957, 12, 427—506.
Глава VI
ОРГАНИЧЕСКИЕ КИСЛОТЫ
Органические кислоты широко распространены в растениях; они
играют важную роль в их метаболизме. Винная, лимонная, щавеле-
вая, яблочная, янтарная и другие кислоты находятся в растениях
в свободном состоянии и в виде солей калия, натрия и кальция.
Кроме щавелевой кислоты, встречающейся в виде труднораство-
римого оксалата кальция, органические кислоты или их соли встре-
чаются почти исключительно в растворимой форме.
При анализе органических кислот следует различать общую
кислотность или общее содержание анионов кислоты (свободной
и связанной) и концентрацию свободной кислоты или титруемую
кислотность. Свободные кислоты, кислые и нейтральные соли
находятся в подвижном равновесии, определяемом концентрацией
водородных ионов в растворе. При титровании щелочью учитыва-
ются только свободные кислоты и кислые соли. В различных орга-
нах растений органические кислоты распределены неравномерно.
В плодах и ягодах преобладают свободные кислоты (от 0,2 до 3%).
В остальных частях растений свободные органические кислоты
находятся обычно в небольших концентрациях. В листьях кислоты
содержатся, главным образом, в виде солей. Содержание органи-
ческих кислот в растениях подвергается суточным и сезонным из-
менениям. В соке растений кислотность значительно возрастает
ночью и уменьшается снова в течение дня. Постепенное старение
листьев вызывает значительные изменения в их кислотном метабо-
лизме, при этом происходит постепенная нейтрализация кислот
основаниями, поглощаемыми из почвы.
Существуют видовые и сортовые различия в накоплении кислот
растениями. В зависимости от условий выращивания культурных
растений кислотность плодов и овощей заметно изменяется. В про-
цессе созревания и хранения плодов и овощей происходят качествен-
ные и количественные изменения в составе органических кислот.
Так, в незрелых плодах и молодых листьях обычно содержится
янтарная кислота, в зрелых же плодах и стареющих листьях преиму-
щественно яблочная, винная и лимонная кислоты. В молодых листь-
ях щавеля, ревеня, шпината накапливаются яблочная и лимон-
ная кислоты, а в старых листьях преобладает щавелевая кислота.
Количественное определение и разделение органических кислот
219
основано на физико-химических свойствах солей калия, кальция,
бария, свинца и ртути, а также на окислительно-восстановительных
свойствах кислот.
Определяемые кислоты можно экстрагировать из свежей, замо-
роженной или же высушенной растительной ткани. Чтобы не про-
исходило нежелательных изменений в составе кислот при высуши-
вании исследуемого[ материала, необходимо избегать повышенной тем-
пературы. Следует применять: лиофильную сушку, сушку при ком-
натной температуре в вакуум-эксикаторе над сильными осушителями
(фосфорный ангидрид, серная кислота) и сушку с безводным сер-
нокислым натрием при температуре не выше 40° С.
ОПРЕДЕЛЕНИЕ СУММЫ ОРГАНИЧЕСКИХ КИСЛОТ
Сущность метода. Кислоты, находящиеся в растениях
в свободном состоянии или в виде растворимых и нерастворимых
в воде солей, извлекают раствором азотной кислоты в 70%-ном эти-
ловом спирте. При этом пектиновые вещества и другие коллоиды
остаются в осадке. В полученном растворе азотную кислоту ней-
трализуют едким натром и органические кислоты осаждают уксус-
нокислым свинцом. Осаждение идет в 70 %-ном растворе этилового
спирта, подкисленного уксусной кислотой для предотвращения обра-
зования основных солей. В 70%-ном спирте свинцовые соли орга-
нических кислот практически нерастворимы. После отделения осад-
ка центрифугированием и промывания 70%-ным спиртом для удале-
ния избытка свинца осадок обрабатывается карбонатом натрия. При
этом образуется нерастворимый в воде углекислый свинец, а орга-
нические кислоты и пигменты в виде натриевых солей переходят
в раствор:
РЬЛ + Na2CO3 = РЬСО3 + На2Л,
где Л — анион кислоты.
После центрифугирования и удаления раствора осадок раство-
ряется в соляной кислоте:
РЬСО3 + 2НС1 = РЬС12 + СО2 + Н2О.
В растворе находится эквивалентное кислотам количество
ионов свинца, которое определяется комплексонометрическим мето-
дом. Для этого добавляют в избытке трилон Б, который реагирует
с ионами свинца, образуя прочное комплексное соединение:
РЬС12 + Na2H2C10H12O3N2 + 2NH4OH = Na2PbC10H12O3N2 +
+ 2NH4C1 + 2H2O.
Остаток трилона Б титруют сернокислым магнием:
Na2H2C10H12O3N2 + MgSO4 +2NH4OH = Na2MgC10H12O3N2 +
+ (NH4)2 SO4 + 2H2O.
220
Суммарное количество ди- и трикарбоновых кислот выражают
через яблочную или лимонную кислоту. Грамм-эквивалент яблоч-
134 » 384,24 ал пл
нои кислоты —£— равен 67, а лимонной------------ 64,04.
Ход анализа. Отвешивают такое количество раститель-
ного материала, чтобы в нем находилось от 5 до 30 мг суммы кислот
(1—2 г свежего или 0,3—0,5 г сухого вещества), и навеску расти-
рают в ступке сначала с 0,5 г силикагеля или песка, а затем с 1 мл
2 н. раствора азотной кислоты и 5 мл 70%-ного этилового спирта.
При работе с мягкими легко растирающимися тканями силикагель
можно не прибавлять. Растертую массу переносят в центрифуж-
ную пробирку, смывая остаток 10 мл 70%-ного спирта. Тщательно
перемешивают стеклянной палочкой и настаивают в течение 15—
20 мин при частом перемешивании. Затем центрифугируют, раствор
сливают в стакан на 50 мл, а к осадку прибавляют 5 мл 70%-ного
спирта и настаивают 5 мин при частом размешивании. После этого
центрифугируют, раствор сливают в стакан, а осадок промывают
еще один раз 5 мл 95%-ного этилового спирта, добавляя центрифу-
гат к раствору в стакане. Для нейтрализации азотной кислоты
прибавляют одну каплю раствора фенолфталеина и добавляют по кап-
лям при размешивании 2 н. раствор NaOH до появления розово-крас-
ной окраски индикатора. В том случае, когда исследуемый раствор
окрашен, то для нейтрализации азотной кислоты прибавляют 1 мл
2 н. раствора едкого патра. После нейтрализации азотной кислоты
добавляют 0,3 то ледяной уксусной кислоты и 1 мл 20%-пого раство-
ра уксуснокислого свинца, размешивают и оставляют па 30 мин
для выделения осадка. Затем центрифугируют, раствор сливают
в склянку с отработанным спиртом, а стакан и осадок, находящийся
в центрифужной пробирке, промывают 2 раза 70%-ным спиртом
порциями по 5 мл. При этом весь осадок переносят в пробирку.
К промытому осадку в пробирке приливают 10 мл 1 %-ного
раствора углекислого натрия и размешивают стеклянной палочкой
в течение 5 мин, пока разложится весь осадок и свинец перейдет
в углекислую соль, которую затем отделяют центрифугированием.
Прозрачный раствор отбрасывают, а к осадку приливают 10 мл
0,1 н. раствора соляной кислоты, размешивают и выливают в колбу
для титрования на 100 мл, смывая остаток дистиллированной водой.
В полученном растворе свинец определяют комплексонометриче-
ским методом. Для этого добавляют 3 мл аммиачного буферного
раствора, 5 мл 0,1 н. раствора трилона Б, 50 мг индикатора хромо-
гена черного ЕТ-00 и титруют0,02 н. раствором сернокислого маг-
ния до перехода сине-зеленой окраски в винно-красную. Отдельно
титруют 5 мл 0,1 н. раствора трилона Б 0,02 н. сернокислым маг-
нием. Из полученных данных вычисляют сумму кислот по формуле:
х = 6,7 (а — Ь) К
п ’
где X — содержание суммы кислот (в %) в расчете на яблочную
кислоту; а — объем 0,02 н. раствора сернокислого магния,
221
затраченный на 5 мл 0,1 н. раствора трилона Б (в мл)\ b — объем
0,02 н. раствора сернокислого магния, затраченный при определении
кислот (в мл)\ К — нормальность раствора сернокислого магния;
п — навеска исследуемого вещества (в г); 6,7 — нормальный титр
яблочной кислоты, умноженный на 100 для пересчета в проценты.
Реактивы. Раствор этилового спирта (70%-ный). К 100 мл 95%-ного эти-
лового спирта прибавляют 39 мл дистиллированной воды и перемешивают.
Раствор уксуснокислого свинца (20%-ный). Отвешивают 20 г уксуснокислого
свинца РЬ (СН3СОО)2 ЗН2О, растворяют в 80 мл дистиллированной воды,
добавляют 1 мл ледяной уксусной кислоты и разбавляют такой же водой до 100 мл.
Раствор азотной кислоты (2 н.). 127 мл азотной кислоты пл. 1,42 разбавляют
дистиллированной водой до 1 л и перемешивают.
Раствор соляной кислоты (0,1 н.). Готовится из 1 н. раствора разбавлением
в 10 раз дистиллированной водой. Приготовление других реактивов см. на
стр. 12, 13, 76, 146.
ОПРЕДЕЛЕНИЕ СВОБОДНЫХ ОРГАНИЧЕСКИХ КИСЛОТ
Сущность метода. Свободные органические кислоты
можно определить йодометрическим методом, используя реакцию:
KJO3 + 5KJ + 6НД - 3J2 + 6КЛ + ЗН2О,
где А — анион кислоты.
К анализируемому раствору, содержащему свободные кислоты,
прибавляют в избытке нейтральный раствор йодид-йодата калия.
Выделившийся йод титруют раствором тиосульфата натрия. Эта
реакция в растворах с большой концентрацией ионов водорода
проходит моментально, но замедляется с понижением концентрации
ионов водорода, поэтому слабые кислоты этим способом непосред-
ственно определять нельзя, так как концентрация ионов водорода
в конце титрования недостаточна для течения реакции.
Анионы органических оксикислот образуют комплексные со-
единения с ионами кальция, бария, магния и цинка и в присутствии
этих катионов ведут себя как сильные кислоты. Винную, лимонную,
молочную и яблочную кислоты можно определить количественно,
если к раствору этих кислот прибавить избыток хлористого бария
или хлористого кальция, а затем йодид и йодат калия. Через 30 мин
реакция заканчивается и выделившийся йод можно оттитровать
тиосульфатом. Этот метод неприменим для определения таких ор-
ганических кислот, как уксусная, бензойная и янтарная, потому
что эти кислоты не содержат гидроксильных групп и не образуют
прочных комплексных соединений с ионами щелочноземельных ме-
таллов.
Если к раствору кислоты, содержащему йодат и йодид калия,
прибавить в избытке титрованный раствор тиосульфата натрия и
через некоторое время оттитровать избыток тиосульфата раствором
йода, то можно определить не только винную, лимонную, щавеле-
вую и яблочную, но и янтарную и бензойную кислоты. В присутствии
тиосульфата натрия концентрация йода в растворе настолько умень-
шается, что реакция проходит довольно быстро даже при очень
малой концентрации ионов водорода.
222
Определение свободных кислот в объектах,
не содержащих янтарной кислоты
Отвешивают 5 г свежего растительного материала и растирают
в фарфоровой ступке с 5 мл 10%-ного раствора хлористого бария.
Растертую массу переносят в центрифужную пробирку, остаток
смывают дистиллированной водой, перемешивают и центрифугируют.
Центрифугат сливают в мерную колбу на 50 мл, К остатку в про-
бирке приливают 1 мл раствора хлористого бария, 10 мл дистилли-
рованной воды, перемешивают и снова центрифугируют. Раствор
присоединяют к первому центрифугату, а осадок промывают еще
один раз водой, присоединяя промывные воды к основному раствору.
Объем доводят водой до 50 мл и перемешивают. Затем набирают
пипеткой 10 мл раствора или больше, в зависимости от возможного
количества кислот, помещают в колбу для титрования, добавляют
5 мл 10%-ного раствора хлористого бария, 5 мл 0,1 н. раствора
йодид-йодатного раствора, перемешивают, закрывают колбу пробкой
и оставляют на 30 мин. После этого прибавляют 0,5 мл раствора
крахмала и титруют 0,01 н. раствором тиосульфата до исчезновения
синей окраски. Содержание свободных кислот в пересчете на яб-
лочную кислоту вычисляют по формуле:
х = 6,7 50 ft а = 335 К а
п b п • b ’
где X — содержание свободных кислот (в %) в пересчете на яблоч-
ную кислоту; 50 — общий объем исследуемого раствора (в мл);
b — объем исследуемого раствора, взятый для титрования (в мл);
а — объем 0,01 н. раствора тиосульфата натрия, затраченный при
титровании (в мл); К — нормальность титрованного раствора тио-
сульфата; п — навеска исследуемого вещества (в г); 6,7 — нормаль-
ный титр яблочной кислоты, умноженный на 100 для пересчета в
проценты.
Определение свободных кислот
в присутствии янтарной кислоты
Получение вытяжки из исследуемого материала производится
так же, как при определении свободных кислот в объектах, не со-
держащих янтарной кислоты. Необходимое количество получен-
ного раствора, содержащее от 2 до 20 мг свободных кислот в пере-
счете на яблочную кислоту, переносят в колбу для титрования,
прибавляют 5 мл 10%-ного раствора хлористого бария, 5 мл 0,1 н.
йодид-йодатного раствора, 5 мл 0,1 н. раствора тиосульфата натрия
и оставляют на 20 мин. После этого прибавляют 0,5 мл раствора
крахмала и титруют 0,02 н. раствором йода до неисчезающей синей
окраски. Содержание кислот вычисляют по формуле:
х = 6,7 50 (0,5 — а) __ 335 (0,5 — К а)
n-b п b *
223
где X — содержание свободных кислот (в %) в пересчете на яб-
лочную кислоту; 0,5 — объем 1 н. титрованного раствора тиосуль-
фата натрия, прибавленный к исследуемому раствору (в мл)\ а —
объем титрованного раствора йода, затраченный при титровании
избытка тиосульфата натрия (в мл); К — нормальность титрованно-
го раствора йода.
Остальные обозначения такие же, как при расчете содержания
свободной кислоты в объектах, не содержащих янтарной кислоты.
Реактивы. Йодид-йодатный раствор (0,1 nJ. Отвешивают 3,567 г KJO3
•и 40 г KJ, растворяют в дистиллированной воде, разбавляют такой же водой
до 1 л в мерной колбе, перемешивают и сохраняют в темной склянке.
Титрованный раствор йода (0,02 н.). Готовится из 0,1 н. раствора йода раз-
бавлением в 5 раз дистиллированной водой. Для приготовления 0,1 н. раствора
отвешивают в бюксе 12,691 г йода ч. д. а. и переносят в мерную колбу емкостью
1 л, добавляют 26 г KJ, 35 мл воды и встряхивают до полного растворения йода.
После этого разбавляют водой до метки и перемешивают. Для приготовления
0,02 н. раствора отбирают пипеткой 50 мл 0,1 н. раствора, помещают в мерную
колбу па 250 мл, доводят водой до метки и перемешивают.
ОПРЕДЕЛЕНИЕ ЛИМОННОЙ КИСЛОТЫ
Лимонная кислота содержится в плодах и ягодах (от 0,2 до 3%),
особенно много ее в лимонах (6—8%). Она также содержится в та-
баке и махорке (от 0,5 до 7%, в расчете на сухое вещество). Только
виноград, рябина и кизил не содержат лимонной кислоты.
Сущность метода. Лимонную кислоту извлекают раз-
бавленной серной кислотой и в полученном растворе окисляют
перманганатом калия в присутствии бромистого калия до образо-
вания пентабромацетона, который затем извлекают хлороформом
и разлагают сульфитом натрия. Количество бромида, эквивалентное
лимонной кислоте, определяют аргентометрическим или меркуро-
метрическим методом.
Перманганат калия при комнатной температуре окисляет ли-
монную кислоту в ацетондикарбоновую кислоту:
СН2СООН
СН2СООН
/ОН |
5С< + 2КМпО4 4- 3H2SO4 = 5СО +’ K2SO4 +
|ХСООН |
I CH2COOH
CH2COOH
4- 2MnSO4 4- 5СО2 4- 8Н2О.
Одновременно перманганат калия окисляет бромид до свободного
брома:
2КМпО4 4- ЮКВг 4- 8H2SO4 = 6K2SO4 4- 2MnSO4 4- 5Вг2,
224
который в момент выделения энергично бромирует ацетондикарбо-
новую кислоту, образуя пентабромацетои:
СН2СООН СНВг2
I I
СО + 5Вг2 = СО + 2СО2 + 5НВг.
I I
СН2СООН СВг3
Этой реакции не мешают винная, яблочная и щавелевая кислоты.
Во время реакции марганец частично восстанавливается до четы-
рехвалентного и выпадает в виде бурого осадка двуокиси марганца:
2КМпО4 + 6КВг + 4H2SO4 = 3Br2 + 4K2SO4 + 2МпО2 + 4Н2О.
Осадок в дальнейшем мешает работе, поэтому его переводят в рас-
твор с помощью сернокислого закисного железа или соли Мора:
MnO2 + 2FeSO4 + 2H2SO4 = Fe2 (SO4)3 + MnSO4 -F 2H2O.
После обесцвечивания остается белый осадок или муть пента-
бромацетона, который плохо растворим в воде и поэтому находится
в осадке. Пентабромацетон легко растворяется в спирте, петролей-
ном эфире, хлороформе и других органических растворителях.
Осадок пентабромацетона можно отфильтровать или отцентрифу-
гировать, промыть ледяной водой и определить весовым или объем-
ным методом. Но можно пентабромацетои извлечь органическим
растворителем, не смешивающимся с водой — петролейным эфи-
ром или хлороформом.
Раствор пентабромацетона в спирте или хлороформе обладает
окислительными свойствами, вступает в реакцию с йодидами, выде-
ляя йод. Эта реакция лежит в основе йодометрического метода опре-
деления лимонной кислоты. Метод неточен, так как реакция про-
текает медленно, требует нагревания и не доходит до конца. В за-
висимости от температуры и продолжительности нагревания выде-
ляется разное количество йода. Пентабромацетои при температуре
кипения медленно разлагается щелочью, образуя бромид и соли
муравьиной, глиоксиловой и других кислот. На основе этой реак-
ции был предложен один из методов определения лимонной кислоты.
Но так как здесь происходит не одна, а несколько реакций, то со-
отношение между пентабромацетоном и щелочью приходится опре-
делять опытным путем, что усложняет метод и дает значительные
отклонения.
Вместо этих не очень точных и длительных методов разработан
более быстрый и точный метод, в котором пентабромацетои извле-
кают хлороформом и разлагают сульфитом натрия. Здесь реакция
идет быстро и до конца, поэтому количество образующегося броми-
да эквивалентно количеству пентабромацетона и лимонной кислоты.
15 62
225
Реакция идет по следующему уравнению:
СНВг2 СН3
I I
СО + 5Na2SO3 + 5Н2О = 5NaBr + 5NaHSO4 + СО.
I I
СВг3 СН3
Образовавшийся в результате реакции бромид натрия определя-
ют аргентометрическим или меркурометрическим методом:
NaBr + AgNO3 = AgBr + NaNO3,
2NaBr + Hg2 (NO3)2 = Hg2Br2 +2NaNO3.
Индикатором служит эозин в смеси с метиленовой синью или
роданид железа. Для устранения влияния сульфита натрия перед
титрованием прибавляют персульфат аммония.
При использовании роданида железа в качестве индикатора суль-
фит натрия мешает титрованию, особенно в меркуриметрическом
методе, так как ртуть восстанавливается до металлической. В этом
случае необходимо сульфит разрушить перманганатом калия:
5Na2SO3 + 2КМпО4 + 6HNO3 = 5Na2SO4 2KNO3 +
+ 2Mn (NO3)2 + 3H2O.
Небольшое количество сульфита не мешает, поэтому избыток
перманганата обесцвечивают одной или двумя каплями раствора
сульфита натрия. Грамм-эквивалент лимонной кислоты
СзН4 (ОН) (СООН)з оо ло
—-------------— в этом методе равен 38,42.
Ход анализа. Берут такую навеску исследуемого материала,
чтобы в ней находилось от 5 до 40 мг лимонной кислоты (воздушно-
сухой махорки 0,5 г, табака и листьев сахарной свеклы по 1 г, све-
жих ягод около 1 г), тщательно растирают в ступке с 1 мл раствора
серной кислоты (1 : 9). К растертой массе приливают 5 мл дистилли-
рованной воды, снова растирают и переносят все содержимое ступки
в мерную колбу на 25 мл, смывая остаток водой. Добавляют 2 мл
4%-ного раствора фосфорновольфрамовой кислоты и взбалтывают
в течение 5 мин. Затем доводят объем дистиллированной водой
до метки, перемешивают и оставляют на некоторое время для про-
светления раствора (можно также оставить на ночь). После этого
фильтруют в сухую колбу через сухой складчатый фильтр. Из полу-
ченного фильтрата отбирают пипеткой 5 мл (во взятом объеме долж-
но быть 1—8 мг лимонной кислоты), помещают в пробирку диа-
метром около 20 мм, прибавляют 1 мл раствора серной кислоты
(1 : 9), 1 мл 12%-ного раствора бромистого калия и перемешивают.
Затем добавляют 2 мл 4%-ного раствора перманганата калия, пере-
мешивают и оставляют в вытяжном шкафу на 10 мин при перио-
дическом встряхивании пробирки. В результате реакции раствор
приобретает коричневую окраску от выделившейся двуокиси
марганца. Если раствор обесцвечивается, прибавляют еще 0,5 мл
22$
4%-ного раствора перманганата. Затем для растворения двуокиси
марганца прибавляют 2 мл 20%-ного раствора сернокислого за-
кисного железа (или соли Мора). Если после взбалтывания рас-
твор не обесцвечивается, прибавляют по каплям раствор сернокисло-
го закисного железа до полного обесцвечивания. После этого при-
бавляют еще 1 мл раствора сернокислого закисного железа для вос-
становления избытка брома и содержимое пробирки переносят
в делительную воронку емкостью 100 мл, споласкивая пробирку
дистиллированной водой.
Для извлечения образовавшегося пентабромацетопа в делитель-
ную воронку приливают 5 мл хлороформа, предварительно промыв
пробирку хлороформом, взбалтывают 1 мин, затем дают постоять
2—3 мин для отделения хлороформа и сливают нижний слой в ко-
ническую колбу на 100 мл. При этом нужно тщательно отделять
хлороформ от остальной жидкости, чтобы ни одна капля водного
раствора не попала в колбу. К оставшемуся в делительной воронке
раствору прибавляют еще 5 мл хлороформа, взбалтывают 1 мин, дают
отстояться и сливают нижний слой, тщательно отделяя его от вод-
ного слоя, в колбу с хлороформным экстрактом. В том случае, когда
исследуемый раствор содержит муть пентабромацетона, при извле-
чении хлороформом в нем образуются устойчивые пузырьки, кото-
рые трудно удалить. В этом случае хлороформ отделяют вместе
с пузырьками, а затем промывают дистиллированной водой для уда-
ления бромидов. Для этого после извлечения и отделения нижнего
слоя делительную воронку освобождают от раствора и промывают
водой. Затем находящийся в колбе хлороформный раствор помеща-
ют снова в делительную воронку. Колбу промывают 20 мл воды,
которые также вносят в делительную воронку. Воронку с жидко-
стью встряхивают 1 мин, дают слоям разделиться в течение 3 мин
и сливают нижний слой в ту же колбу даже при наличии пу-
зырьков.
К полученному раствору пентабромацетона в хлороформе, нахо-
дящемуся в колбе, прибавляют 1 мл 10%-ного раствора сульфита
натрия. Колбу ставят на кипящую водяную баню в вытяжном
шкафу и нагревают до удаления хлороформа. Затем добавляют
20 мл дистиллированной воды, раствор переносят па плитку, на-
гревают до кипения и кипятят 2 мин. При этом происходит разло-
жение пентабромацетона и образование бромида натрия. В получен-
ном растворе бромид определяется аргентометрическим методом.
Для этого после охлаждения раствора прибавляют 2 мл 20%-пого
раствора персульфата аммония, две капли индикатора эозина с ме-
тиленовой синью и титруют 0,01 н. раствором азотнокислого сере-
бра до перехода окраски в розово-фиолетовую.
Можно также в качестве индикатора пользоваться роданидом
железа. В этом случае к охлажденному водному раствору в колбе,
содержащему бромид и сульфит натрия, приливают 1 мл азотной
кислоты (1 1) и по каплям при взбалтывании 4%-ный раствор пер-
манганата калия до появления неисчезающей желтой или розовой
15*
227
окраски, которую затем обесцвечивают добавлением I—2 капель
10%-ного раствора сульфита натрия. После этого прибавляют
еще 2 мл азотной кислоты ( 1 : 1), 1 мл 20 %-ного раствора железо-
аммонийных квасцов, 1 мл 0,01 н. раствора роданистого калия
и окрашенный в красный цвет раствор титруют 0,01 н. раствором
азотнокислого серебра до обесцвечивания индикатора. Вместо
азотнокислого серебра в этом случае можно пользоваться также
азотнокислой закисной ртутью, титруя 0,01 н. раствором Hg2 (NO3)2
до полного исчезновения окраски роданистого железа. Для опре-
деления поправки на индикатор к уже оттитрованному исследуе-
мому раствору прибавляют 1 мл 0,01 н. раствора роданистого калия
и титруют 0,01 н. раствором AgNO3 или Hg2 (NO3)2 до обесцвечи-
вания. Результат титрования служит поправкой и вычитается
из объема раствора азотнокислого серебра или азотнокислой закис-
ной ртути, израсходованного при титровании исследуемого рас-
твора.
Содержание лимонной кислоты рассчитывают по формуле:
v 3,842 • v К а
Л = -----------.-----,
п b
где X — содержание безводной лимонной кислоты в исследуемом
веществе (в %); v — общий объем вытяжки (в мл)\ b — объем вы-
тяжки, взятый для определения лимонной кислоты (в мл); а —
объем титрованного раствора азотнокислого серебра или Hg2 (NO3)2,
затраченный при титровании (в мл); К — нормальность титрован-
ного раствора AgNO3 или Hg2 (NO3)2; п — навеска исследуемого
вещества (в г); 3,842 — нормальный титр лимонной кислоты, ум-
ноженный на 100 для пересчета в проценты.
Реактивы. Раствор бромистого калия (12%-ный). 12 г КВг растворяют в ди-
стиллированной воде и разбавляют водой до 100 мл.
Раствор перманганата калия (4%-ный). Отвешивают 4 г соли КМпО4 и рас-
творяют в 100 мл дистиллированной горячей воды.
Раствор сернокислого закисного железа или соли Мора (20%-ный). К 50 г
FeSO4 7Н2О или Fe (NH4)2 (SO4)2 6Н2О добавляют 200 мл дистиллированной
воды, 5 мл серной кислоты пл. 1,84, растворяют и разбавляют водой до 250 мл.
Раствор годен в течение недели.
Раствор сульфита натрия (10%-ный). 10 г безводной соли Na2SO3 или
20 г Na2SO3 7Н2О растворяют в дистиллированной воде и доводят объем рас-
твора водой до 100 мл. Раствор годен в течение недели.
Железо-аммонийные квасцы (20%-ный раствор). Отвешивают 20 г соли
Fe (NH4) (SO4)2 12Н2О, помещают в стакан, добавляют 70 мл дистиллированной
воды, 10 мл азотной кислоты (1 1), нагревают до растворения соли, охлаждают,
разбавляют водой до 100 мл и перемешивают.
Раствор азотнокислого серебра (0,05 п.). Отвешивают на аналитических
весах 4,25 г неразложившейся соли AgNO3, предварительно высушенной при
100° С, растворяют в дистиллированной воде и разбавляют водой в мерной колбе
до 500 мл. Раствор сохраняют в темной склянке. Из этого раствора приготавли-
вают 0,01 н. раствор разбавлением водой в 5 раз.
Раствор роданистого калия (~ 0,1 н.). 1 г высушенной соли KCNS или
0,8 г NH4CNS растворяют в дистиллированной воде и разбавляют водой до 100 мл.
Из этого раствора готовят 0,01 к. раствор разбавлением водой в 10 раз.
228
Раствор эозина с метиленовой синью. Отвешивают 0,5 г натриевой соли эозина
и 0,2 г метиленовой сини, растирают в дистиллированной воде и разбавляют во-
дой до 100 мл.
Ртуть азотнокислая закисная (0,01 п.). Отвешивают 3 г Hg2 (NO3)2 2Н2О,
помещают в стакан, прибавляют 10 мл азотной кислоты (1 1) и 100 мл дистилли-
рованной воды, растворяют и разбавляют водой до 1 л. Раствор переносят в тем-
ную склянку, добавляют 10 капель металлической ртути и сохраняют в хорошо
закрытой склянке.
В случае отсутствия азотнокислой закисной ртути раствор ее можно приго-
товить из металлической ртути следующим образом. Отвешивают в стакане 15 г
металлической ртути, прибавляют 10 мл дистиллированной воды и 14 мл азотной
кислоты (1 1). Стакан ставят на кипящую водяную баню и нагревают, пока
растворится большая часть ртути. Затем раствор и остаток ртути переносят в мер-
ную колбу на 100 мл и оставляют на ночь. На второй день разбавляют водой до
100 мл и сохраняют в темном месте. Получается приблизительно 0,5 н. раствор.
Из этого раствора готовят рабочий 0,01 н. раствор. Для этого берут 10 мл 0,5 н.
раствора, вносят в мерную колбу на 500 мл, добавляют 5 мл азотной кислоты
(1 1), разбавляют водой до 500 мл, перемешивают, переливают в темную склянку
и добавляют 5 капель металлической ртути.
Нормальность полученного раствора устанавливают по 0,01 н. раствору бро-
мистого калия. Для этого взвешивают на аналитических весах 0,595 г предвари-
тельно высушенной соли КВг х. ч., растворяют в воде в мерной колбе на 500 мл,
разбавляют водой до метки и перемешивают. Отбирают пипеткой 20 мл этого
раствора в колбу для титрования, добавляют 1 мл азотной кислоты (1 1), 1 мл
20%-ного раствора железо-аммонийных квасцов, 1 мл 0,01 н. раствора роданистого
калия и окрашенный в красный цвет раствор титруют раствором азотнокислой
закисной ртути до полного обесцвечивания. Из затраченного объема вычитают
1 мл, который расходуется на 1 мл 0,01 п. раствора роданистого калия. Нормаль-
ность вычисляют по следующей формуле:
0,01 20
' а ’
где К — нормальность раствора азотнокислой закиси ртути; а — объем раствора
азотнокислой закиси ртути, затраченный при титровании 20 мл 0,01 н. раствора
КВг (в мл).
Нормальность азотнокислой закиси ртути периодически проверяют по бро-
мистому калию.
Приготовление остальных реактивов описано выше (см. стр. 9, 27, 1G4).
ОПРЕДЕЛЕНИЕ ВИННОЙ И ЯБЛОЧНОЙ КИСЛОТ
В ВИНОГРАДЕ
Винная кислота (d-винная) содержится в значительном коли-
честве только в винограде. Некоторые ягоды — красная сморо-
дина, крыжовник, брусника содержат небольшое количество вин-
ной кислоты (0,04—0,06%). В виде следов винная кислота обна-
ружена в черешне, землянике, айве, абрикосах и некоторых сортах
слив (ренклод). Кислая калиевая соль винной кислоты (КНС4Н4Ов)
труднорастворима в воде и нерастворима в спирте. Еще меньше
растворима средняя кристаллическая соль кальция — СаС4Н4О0.
Яблочная кислота содержится почти во всех растениях и пло-
дах, в том числе и в винограде. Кальциевая и бариевая соли яб-
лочной кислоты более растворимы в воде, чем соли винной и лимон-
ной кислот. В спирте же эти соли яблочной кислоты нерастворимы.
Еще меньше растворима ее свинцовая соль (РЬС4Н4ОВ).
Сущность метода. Находящиеся в винограде яблочная
и винная кислоты извлекают раствором уксусной кислоты. Экстракт
229
доводят до определенного объема и после фильтрования в аликвотной
части раствора, содержащей не более 25 мг каждой кислоты,
определяют сначала винную, затем яблочную кислоты. Для осаж-
дения и отделения винной кислоты раствор выпаривают на водяной
бане в присутствии ионов калия досуха, остаток подкисляют ук-
сусной кислотой и добавляют 50 %-ный раствор этилового спирта.
При этом выпадает кристаллический осадок кислого виннокислого
калия:
СНОНСООН СНОНСООК
| + СН3СООК = | + СН3СООН.
СНОНСООН СНОНСООН
В растворе остается яблочная кислота. Осадок кислого виннокисло-
го калия промывают 50%-ным спиртом и обрабатывают хлористым
кальцием. Образуется кристаллический осадок виннокислого каль-
ция:
КНС4Н4О0 СаС12 NH4OH - СаС4Н4О0 + КС1 + NH4C1 + Н2О.
Осадок виннокислого кальция промывают 50%-ным спиртом и
растворяют в соляной кислоте:
СаС4Н4Об + 2НС1 = Н2С4Н4Ов + СаС12.
Кальций определяют комплексонометрическим методом: титро-
ванием 0,01 н. раствором трилона Б в аммиачной среде:
Na2H2C10H12O3N2 + СаС12 + 2NH4OH = Na2CaC10H12O3N2 +
+ 2NH4C1 + 2Н2О.
Грамм-эквивалент винной кислоты С4^6°6 равен 75.
В спиртовом растворе после отделения винной кислоты нахо-
дится яблочная кислота, которую осаждают уксуснокислым свин-
цом в растворе, содержащем 70% этилового спирта:
СН (ОН) СООН СН (ОН)СООк
| + Pb (СН3СОО)2 - I >РЬ + 2СН3СООН.
СН2СООН СН2СОО z
Осадок яблочнокислого свинца после промывания 70%-ным
спиртом разлагается содой:
РЬС4Н4Об + Na2CO3 - Na2C4H4OB + PbCO3.
После удаления раствора осадок углекислого свинца растворяют
в соляной кислоте:
РЬСО3 + 2НС1 = РЬС12 + СО2 + Н2О.
Свинец определяют комплексонометрическим методом, добавляя
избыток трилона Б, с которым образуется прочное комплексное со-
единение:
РЬС12 + Na2H2C10H12O3N2 + 2NH4OH = Na2PbC10H12O3N2 +
+ 2NH4C1 + 2H2O.
230
Остаток трилона Б титруют сернокислым магнием:
Na2H2C10H12O3N2 + MgSO4 + 2NH4OH - Na2MgCJ0H12O3N2 +
+ (NH4)2SO4 -I- 2H2O.
Грамм-эквивалент яблочной кислоты 2 равен 67.
Ход анализа. Взвешивают 5 г винограда н растирают
в ступке с 10 мл 1%-ного раствора уксусной кислоты. Растертую
массу переносят в мерную колбу на 50 мл, смывая остаток дистил-
лированной водой. Доводят 1%-ной уксусной кислотой до метки,
тщательно перемешивают и фильтруют в сухую колбу через сухой
складчатый фильтр, отбрасывая первую порцию фильтрата (около
5 мл). Из остального фильтрата набирают пипеткой 20 мл, перено-
сят в фарфоровую чашку, добавляют 1 мл 10%-ного раствора угле-
кислого калия, 0,1 мл ледяной уксусной кислоты и полностью
выпаривают на водяной бане. Остаток обычно имеет полужидкую
консистенцию. После охлаждения к остатку добавляют 0,2 мл ле-
дяной уксусной кислоты, размешивают, добавляют 0,5 мл 50%-ного
этилового спирта и оставляют при периодическом перемешивании
на 5 мин. Затем раствор помещают в центрифужную пробирку,
а остаток смывают 50%-ным этиловым спиртом порциями по 0,5 мл,
присоединяя промывную жидкость к основному раствору. Промы-
вают 3—4 раза. Жидкость в пробирке оставляют на 10 мин при пе-
риодическом взбалтывании и затем центрифугируют. Раствор сли-
вают в стакан на 50 мл, а осадок промывают 1 раз 2 мл 50%-ного
спирта, добавляя промывную жидкость к раствору в стакане.
К осадку в пробирке прибавляют 0,2 мл 25%-ного раствора хло-
ристого кальция, встряхивают, помещают на несколько секунд
в кипящую воду и продолжают встряхивать до появления кристал-
лического осадка виннокислого кальция. Затем добавляют постепен-
но при взбалтывании четыре капли аммиачного буферного раствора,
2 мл 50%-ного этилового спирта, встряхивают и оставляют на 5 мин.
После этого центрифугируют и раствор сливают в стакан к ранее
полученной промывной жидкости, а осадок промывают два раза
50%-ным спиртом порциями по 3 мл, добавляя промывную жидкость
к раствору в стакане. В полученном осадке виннокислого кальция
определяют винную кислоту, а в растворе — яблочную кислоту.
Определение винной кислоты
К осадку в пробирке прибавляют 5 мл 0,1 н. раствора соляной
кислоты, перемешивают до растворения осадка и раствор выли-
вают в колбу для титрования емкостью 100 мл, смывая остаток
раствора дистиллированной водой. Прибавляют к раствору в кол-
бе 2 мл аммиачного буферного раствора, 3 мл раствора комплексо-
ната магния, 50 мг индикатора хромогена черного ЕТ-00 и титруют
0,01 н. раствором трилона Б до перехода окраски индикатора в си-
231
ний цвет. Из полученных данных рассчитывают содержание винной
кислоты по следующей формуле:
х = 7,5 50 К а = 18,75 К а
20 и п ’
где X — содержание винной кислоты (в %); 50 — общий объем
исследуемого раствора (в мл)\ 20 — объем исследуемого раствора,
взятый для определения (в мл)\а— объем титрованного раствора
трилона Б, затраченный при титровании (в мл)\ К — нормальность
раствора трилона Б; п — навеска исследуемого вещества (в г);
7,5 — нормальный титр винной кислоты, умноженный на 100 для
пересчета в проценты.
Определение яблочной кислоты
К раствору в стакане, содержащему яблочную кислоту и 50%-ный
спирт, прибавляют 0,5 мл 20%-ного раствора уксуснокислого
свинца, 10 мл 95%-ного этилового спирта, перемешивают и остав-
ляют на 30 мин для выделения осадка. Затем содержимое стакана
переносят в центрифужную пробирку, центрифугируют и раствор
сливают в склянку для отработанного спирта. Стакан и осадок про-
мывают 2 раза 70%-ным спиртом порциями по 3 мл. К промытому
осадку в пробирке приливают 10 мл 1%-ного раствора Na2CO3,
размешивают стеклянной палочкой в течение 5 мин для разложения
осадка и центрифугируют. Раствор удаляют, а к осадку приливают
10 мл 0,1 н. раствора соляной кислоты, размешивают и раствор пере-
носят в колбу для титрования, смывая остаток дистиллированной
водой. Добавляют 3 мл аммиачного буферного раствора, 5 мл 0,1 н.
раствора трилона Б, 50 мг хромогена черного ЕТ-00 и титруют
0,02 н. раствором сернокислого магния до перехода окраски инди-
катора в винно-красную. Отдельно титруют 5 мл 0,1 н. раствора
трилона Б сернокислым магнием. Из полученных данных вычис-
ляют содержание яблочной кислоты по формуле:
х = 6,7 50 К(а — Ь) __ 16,75 К(а — Ь)
20 п п 9
где X —содержание яблочной кислоты (в %); а — объем 0,02 н.
раствора сернокислого магния, затраченный на 5 мл 0,1 н. раствора
трилона Б (в мл)\ b — объем 0,02 н. раствора сернокислого магния,
затраченный при определении яблочной кислоты (в мл)\ К — нор-
мальность раствора сернокислого магния; п — навеска исследуемо-
го вещества (в г); 50 — общий объем исследуемого раствора (в мл);
20 — объем исследуемого раствора, взятый для определения яб-
лочной кислоты (в мл)-, 6,7 — нормальный титр яблочной кислоты,
умноженный на 100 для пересчета в проценты.
Реактивы. Углекислый калий (10%-ный раствор). 25 г К2СО3 растворяют
в дистиллированной воде и разбавляют водой до 250 мл.
Насыщенный раствор комплексоната магния. Взвешивают 25 г MgSO4 7Н2О,
растворяют в 50 мл дистиллированной воды. В другом стакане 38 г трилона Б
232
растворяют при нагревании в 250 мл воды. К горячему раствору трилона Б при-
ливают ранее приготовленный раствор сульфата магния, перемешивают до выпа-
дения осадка и оставляют при периодическом помешивании па 2 ч или на ночь.
После отстаивания раствор сливают, а осадок промывают декантацией 2 раза пор-
циями воды по 50 мл. Промытый осадок переносят в склянку па 1 л, смывая его
водой до наполнения склянки, хорошо взбалтывают и оставляют. При работе бе-
рут пипеткой прозрачный раствор. По мерс расходования добавляют воду и взбал-
тывают до насыщения.
Титрованный раствор трилона Б (0,01 н.). Готовят из 0,02 н. раствора разба-
влением водой в 2 раза. Приготовление 0,02 н. титрованного раствора описано вы-
ше (см. стр. 12).
Этиловый спирт (50%-ный). К 100 мл 95%-ного этилового спирта прибав-
ляют 96 мл воды и перемешивают.
Приготовление остальных реактивов описано выше (см. стр. 13, 40, 49, 83,
146, 222).
ОПРЕДЕЛЕНИЕ ЛИМОННОЙ И ЯБЛОЧНОЙ КИСЛОТ
ПРИ СОВМЕСТНОМ ПРИСУТСТВИИ
Сущность метода. В большинстве плодов и ягод при-
сутствуют только лимонная и яблочная кислоты. В данном случае
органические кислоты извлекают 70% -ным этиловым спиртом, под-
кисленным уксусной кислотой. Экстракт доводят до определенного
объема, в части которого определяют лимонную кислоту в виде
пентабромацетона так, как изложено выше в методе определения
лимонной кислоты. В другой части раствора определяют сумму
лимонной и яблочной кислот осаждением их в виде свинцовых солей
и определением свинца комплсксопометрическим методом. По най-
денному количеству свинца определяют сумму лимонной и яблоч-
ной кислот, а по разности находят содержание яблочной кислоты.
Ход анализа. Берут от 1 до 3 г свежего или соответствую-
щее количество сухого исследуемого материала с содержанием кис-
лот не более 40 мг и тщательно растирают в ступке сначала с 0,5 г
силикагеля, затем с 0,5 мл ледяной уксусной кислоты и 5 мл 70%-но-
го этилового спирта. Растертую массу переносят в центрифужную
пробирку, смывая остаток 10 мл 70%-ного спирта. Настаивают
при частом размешивании 15 мин и центрифугируют. Раствор сли-
вают в мерную колбу на 25 мл, а осадок промывают 2 раза 70%-ным
спиртом порциями по 5 мл. Раствор в колбе доводят 70 %-ным спир-
том до метки и перемешивают.
Определение лимонной кислоты
Из раствора кислот набирают сухой пипеткой 5 мл, выливают
в фарфоровую чашку и выпаривают на водяной бане досуха. Оста-
ток растворяют в 2 мл дистиллированной воды, переносят в про-
бирку диаметром 20 мм, смывая остаток 4 мл воды порциями по 2 мл.
К раствору прибавляют 1 мл раствора серной кислоты ( 1 9),
1 мл 12%-ного раствора бромистого калия, 2 мл 4%-ного раство-
ра КМпО4 и далее поступают так, как изложено выше в методе
233
определения лимонной кислоты. Содержание лимонной кислоты
вычисляется по формуле:
у 3,842 25 К - а = 19,21 # а
5 п п ’
где Y — содержание безводной лимонной кислоты (в %); а — объем
титрованного раствора азотнокислого серебра или Hg2 (NO3)2,
затраченный при титровании (в мл)\ К — нормальность титрован-
ного раствора AgNO3 или Hg2 (NO3)2; п — навеска исследуемого
вещества (в а); 25 — объем исследуемого раствора кислот (в мл)\
5 — объем исследуемого раствора, взятый для определения лимон-
ной кислоты (в мл)\ 3,842 — нормальный титр лимонной кислоты,
умноженный на 100 для пересчета в проценты.
Определение суммы лимонной и яблочной кислот
Остаток раствора лимонной и яблочной кислот (20 мл) перено-
сят в стакан, промывая колбу 5 мл 95%-ного спирта. Для осажде-
ния кислот прибавляют 1 мл 20%-ного раствора уксуснокислого
свинца, перемешивают и оставляют па 30 мин для выделения осадка.
Затем раствор и осадок из стакана переносят в центрифужную про-
бирку, центрифугируют, раствор сливают в склянку для отработан-
ного спирта, а осадок промывают 2 раза 70%-ным спиртом порциями
по 5 мл. К промытому осадку в пробирке приливают 10 мл 1 %-ного
раствора Na2CO3, размешивают стеклянной палочкой в течение 5 мин
для разложения осадка и центрифугируют. Раствор удаляют,
а к осадку карбоната свинца приливают 10 мл 0,1 н. раствора соля-
ной кислоты, размешивают стеклянной палочкой и раствор перено-
сят в колбу для титрования, смывая остаток дистиллированной во-
дой. Добавляют 3 мл аммиачного буферного раствора, 5 мл 0,1 н.
раствора трилона Б, 50 мг хромогена черного ЕТ-00 и титруют
0,02 н. раствором сернокислого магния до перехода окраски индика-
тора в винно-красную. Отдельно титруют 5 мл 0,1 н. раствора три-
лона Б сернокислым магнием. Из полученных данных вычисляют
содержание суммы лимонной и яблочной кислот в пересчете на ли-
монную кислоту по следующей формуле:
с = 6,4 25 К(а — Ь) _ 8 К(а — Ь)
20 п п '
где С — содержание суммы лимонной и яблочной кислот на лимон-
ную кислоту (в %); а — объем 0,02 н. раствора сернокислого магния,
затраченный на 5 мл 0,1 н. раствора трилона Б (в мл)\ b — объем
0,02 н. раствора сернокислого магния, затраченный при определении
кислот (в мл)\ К — нормальность раствора сернокислого магния;
п — навеска исследуемого вещества (в а); 25 — объем исследуемо-
го раствора лимонной и яблочной кислот (в мл)\ 20 — объем иссле-
дуемого раствора, взятый для определения суммы лимонной и яб-
лочной кислот (в мл)\ 6,4 — нормальный титр лимонной кислоты,
умноженный на 100 для пересчета в проценты.
234
Содержание яблочной кислоты вычисляют по следующему урав-
нению:
X = 67 (С~ Y) = 1,05 (С — У),
где X — содержание яблочной кислоты в исследуемом веществе
(в %); С — содержание суммы лимонной и яблочной кислот (в %);
Y — содержание лимонной кислоты (в %); 67 — г-же яблочной
кислоты; 64 — г-экв лимонной кислоты.
Приготовление реактивов, применяемых в данном методе, опи-
сано выше (см. стр. 13, 222, 228, 229).
ОПРЕДЕЛЕНИЕ ЩАВЕЛЕВОЙ КИСЛОТЫ
Щавелевая кислота встречается главным образом в кислице
и щавеле в виде кислой калиевой соли, а в большинстве других рас-
тений — в виде оксалата кальция. Наличие свободной щавелевой
кислоты в растениях точно не доказано. В небольшом количестве
она находится в малине, черной и красной смородине, землянике,
вишне и груше — от 0,01% до 0,05%. В растениях можно опреде-
лить водорастворимые оксалаты — нейтральные и кислые соли
калия, и нерастворимые в воде оксалаты — кальциевую соль.
Сущность метода. Растворимые соли щавелевой кис-
лоты извлекают теплой водой, белки удаляют из раствора уксусной
кислотой и щавелевую кислоту осаждают хлористым кальцием:
К2С2О4 СаС12 СаС2О4 + 2КС1.
После отделения осадка центрифугированием и промывания его
водой осадок растворяют в серной кислоте и титруют щавелевую
кислоту перманганатом калия в присутствии сульфата марганца, ко-
торый ускоряет реакцию и снижает окислительный потенциал пер-
манганата, уменьшая ошибку, обусловленную органическими при-
месями:
5Н2С2О4 + 2КМпО4 + 3H2SO4 = K2SO4 + 2MnSO4 + ЮСО2 + 8Н2О.
В остатке после извлечения водорастворимых солей щавелевой
кислоты определяют содержание оксалата кальция. Его растворяют
в соляной кислоте и осаждают щавелевую кислоту хлористым каль-
цием в аммиачной среде для отделения от растворимых в соляной кис-
лоте примесей. После промывания осадка дистиллированной водой
и растворения его в серной кислоте количество щавелевой кислоты
определяют перманганатометрическим методом.
н С О
Грамм-эквивалент безводной щавелевой кислоты —2 % 4- равен 45.
Определение водорастворимых солей
щавелевой кислоты
Отвешивают 0,4—1 г тщательно растертого сухого исследуемого
материала (или соответствующее количество свежего материала), по-
мещают в фарфоровую ступку и растирают с 5 мл дистиллированной
235
воды до однородной массы. Содержимое ступки переносят в центри-
фужную пробирку емкостью 20 мл, промывая ступку 10 мл воды и
присоединяя промывную жидкость к основному раствору. Пробирку
помещают в воду, нагретую до температуры 60°C, и выдерживают
10 мин при частом перемешивании. Затем центрифугируют и проз-
рачный раствор сливают в стакан емкостью 50 мл. Остаток обрабаты-
вают теплой водой еще 2 раза порциями по 10 мл, каждый раз переме-
шивая в течение 5 мин и центрифугируя. Центрифугаты объединяют.
Полученный раствор нагревают в стакане до кипения, снимают
с плитки, прибавляют 10 капель ледяной уксусной кислоты, пере-
мешивают и центрифугируют для удаления осадка. Прозрачный рас-
твор сливают в чистый стакан. Осадок в стакане и в пробирке про-
мывают 5 мл 1 %-ного раствора уксусной кислоты, центрифугируют
и промывную жидкость добавляют к раствору в стакане.
В полученном растворе осаждают щавелевую кислоту. Для этого
раствор нагревают почти до кипения, добавляют 2 мл 25 %-ного рас-
твора хлористого кальция и продолжают нагревать на слабом огне
при помешивании 5 мин. Затем стакан снимают с плитки и оставляют
на 2 ч или па ночь для полного осаждения. После этого раствор отде-
ляют от осадка центрифугированием. Раствор отбрасывают, а
осадок, помещенный в центрифужную пробирку, промывают водой
3 раза порциями по 5 мл, предварительно промывая стакан, в ко-
тором производилось осаждение.
Промытый осадок растворяют в 5 мл раствора серной кислоты
(1 :9). Раствор переносят в колбу для титрования, смывая остаток
20 мл дистиллированной воды. К раствору прибавляют 1 мл 10%-но-
го раствора сернокислого марганца, нагревают до 60—70° С и тит-
руют 0,01 н. раствором перманганата калия до появления розовой
окраски, не исчезающей в течение нескольких секунд.
Количество щавелевой кислоты в растворимых в воде оксалатах
вычисляют по следующей формуле:
v 4,5 К а
Л — ---------- ,
п '
где X — содержание безводной щавелевой кислоты в водораствори-
мых оксалатах в расчете на сухое вещество (в %); а — объем титро-
ванного раствора перманганата калия, затраченный при титровании
щавелевой кислоты (в мл)’, X — нормальность раствора перманга-
ната калия; п — навеска сухого исследуемого вещества (в а); 4,5 —
нормальный титр безводной щавелевой кислоты, умноженный на
100 для перерасчета в проценты.
Определение оксалата кальция
К остатку исследуемого вещества в центрифужной пробирке при-
бавляют 1 мл раствора соляной кислоты (1 : 1) и 15 мл дистиллиро-
ванной воды, перемешивают и помещают в горячую воду (около 70° С)
на 10 мин при периодическом перемешивании. Затем центрифуги-
236
руют и раствор сливают в стакан емкостью 50 мл. Остаток обраба-
тывают еще 2 раза 0,1 н. соляной кислотой порциями по 10 мл при
нагревании в течение 5 мин на водяной бане и при помешивании.
После каждой обработки соляной кислотой центрифугируют и кис-
лотные вытяжки объединяют.
Для осаждения щавелевой кислоты к полученному солянокисло-
му раствору прибавляют 2 мл 25% -ного раствора хлористого кальция,
нагревают до 70—80° С, прибавляют одну каплю метилоранжа и по
каплям добавляют концентрированный раствор гидроокиси аммо-
ния до появления мути. Раствор нагревают 1—2 мин при помешива-
нии для укрупнения кристаллов, затем, продолжая нагревание,
добавляют по каплям раствор аммиака до тех пор, пока окраска
индикатора изменится в желтую. После этого раствор нагревают
еще 5—10 мин и оставляют на 2 ч или на ночь.
После отстаивания центрифугируют, раствор удаляют, а осадок
промывают 1 раз 10 мл дистиллированной воды. После удаления на-
досадочной жидкости к осадку прибавляют 10 мл 2 н. раствора сер-
ной кислоты, размешивают, нагревают в кипящей воде 3—5 мин при
размешивании, центрифугируют и раствор выливают в колбу для
титрования. Остаток промывают еще 1 раз 10 мл 2 н. серной кислоты,
центрифугируют и раствор добавляют к первому в колбу. Раствор
нагревают до 60—70° С, добавляют 1 мл 10%-ного раствора серно-
кислого марганца и титруют 0,01 н. раствором КМпО4 до появления
розовой окраски, не исчезающей в течение нескольких секунд. 1 мл
0,01 н. раствора перманганата калия соответствует 0,45 мг Н2С2О4.
Количество щавелевой кислоты, связанной с ионами кальция, вы-
числяется по той же формуле, что и количество щавелевой кислоты,
находящейся в растениях в виде растворимых в воде солей. Для пе-
ресчета на оксалат кальция полученный результат умножают на
1,422. Определяемые количества —от 2 до 20 мг Н2С2О4. Если необ-
ходимо определить общее содержание растворимых и нерастворимых
в воде соединений щавелевой кислоты, извлечение водой не произ-
водят, а извлекают все соли щавелевой кислоты одновременно со-
ляной кислотой, как при определении оксалата кальция.
Приготовление реактивов, применяемых в данном методе, опи-
сано выше (см. стр. 9, 27, 41, 70, 146).
ОПРЕДЕЛЕНИЕ ЯНТАРНОЙ КИСЛОТЫ
Янтарная кислота встречается во многих растениях. В небольших
количествах она находится в незрелых вишнях, крыжовнике, виног-
раде, абрикосах, а также в смородине, черешне, яблоках. Кальцие-
вые и бариевые соли янтарной кислоты значительно более раствори-
мы в воде, чем соли других органических кислот. Бариевая соль
практически нерастворима в 70 %-ном этиловом спирте. Перманганат
калия при комнатной температуре не окисляет янтарную кислоту.
Сущность метода. Янтарную и другие органические
кислоты извлекают из растительного материала уксусной кислотой
237
в присутствии ацетата бария. Определенную часть вытяжки выпари-
вают па водяной бане досуха. Остаток промывают 70%-ным спиртом
от сахаров и других растворимых соединений. Осадок, состоящий
из бариевых солей органических кислот, растворяют в воде и обра-
батывают перманганатом калия. При этом все кислоты, кроме янтар-
ной, окисляются до углекислого газа и воды. Избыток перманга-
ната восстанавливается муравьиной кислотой:
2КМпО4 + 5НСООН = ЗСО2 + 2MnO2 + 2НСООК + 4Н2О.
Осадок двуокиси марганца удаляют центрифугированием, а рас-
твор выпаривают досуха. При этом образуется бариевая соль янтар-
ной кислоты, которая промывается 70%-ным спиртом от избытка ба-
рия. Промытый янтарнокислый барий растворяется в соляной кис-
лоте:
СН2СООХ СН2СООН
| )Ва 2НС1 = ВаС12 + | + Н2О.
СНХХХХ СН2СООН
Образуется эквивалентное количество ионов бария, которые осаж-
даются бихроматом калия в присутствии уксуснокислого натрия:
2ВаС12 + К2Сг2О7 + 2CH3COONa + Н2О = 2BaCrO4 + 2КС1 +
+ 2СН3СООН + 2NaCl.
Осадок хромата бария промывают водой, растворяют в соляной
кислоте и обрабатывают йодистым калием:
2BaCrO4 + 16НС1 + 6KJ = 3J2 + 2ВаС12 + 2СгС13 + 6КС1 + 8Н2О.
Выделившийся йод титруют тиосульфатом натрия. Грамм-эквива-
лент янтарной кислоты -С4^6^ равен 39,33.
Ход анализа. Взвешивают 5 г свежего измельченного рас-
тительного материала и растирают в ступке с 5 мл 1 %-ной уксусной
кислоты и 2 мл 20%-ного раствора уксуснокислого бария. Растер-
тую массу переносят в центрифужную пробирку, смывая остаток
1 %-ной уксусной кислотой в ту же пробирку, перемешивают стеклян-
ной палочкой и центрифугируют. Раствор сливают в мерную кол-
бу на 50 мл, а остаток промывают 2 раза 1 %-ной уксусной кислотой
порциями по 10—15 мл, при 5-минутном настаивании и тщательном
перемешивании. Центрифугаты присоединяют к раствору в колбе,
доводят объем дистиллированной водой до 50 мл, перемешивают, от-
бирают пипеткой 25 мл, помещают в фарфоровую чашку и выпаривают
на водяной бане досуха. Остаток в чашке, в котором должно быть от
2 до 18 мг янтарной кислоты, промывают 2 раза 70%-ным спиртом
порциями по 3—5 мл, с перенесением осадка в центрифужную про-
бирку и центрифугированием. Раствор удаляют, а осадок в чашке и
пробирке высушивают от остатка спирта на водяной бане, предва-
рительно распределив осадок на стенках пробирки, чтобы избежать
выбрасывания осадка. После высушивания осадок растворяют в
238
8 мл дистиллированной воды, добавляя ее по 2 мл сначала в чашку,
потом из чашки в пробирку.
К водному раствору в пробирке прибавляют 2 мл 4%-ного рас-
твора перманганата калия, перемешивают и оставляют на 40 мин.
После этого добавляют 1 мл 10%-ного раствора муравьиной кисло-
ты, перемешивают и оставляют еще па 10 мин. Затем центрифуги-
руют и раствор сливают в фарфоровую чашку, а осадок промывают
1 раз 3 мл воды. В чашку добавляют 0,2мл 20%-пого раствора ацета-
та бария и 2 капли 2%-ного раствора солянокислого гидроксиламина
для устранения окислительных процессов. Раствор выпаривают па
водяной бане досуха. После охлаждения осадок переносят в центри-
фужную пробирку с помощью 4 мл 70%-ного этилового спирта, цен-
трифугируют и раствор удаляют, а осадок промывают 2 раза 70%-ным
спиртом, прибавляя его по 3 мл и перед центрифугированием энер-
гично встряхивая с осадком. Для того, чтобы взболтать плотно осев-
ший осадок, необходимо прибавить 2 капли спирта и энергично
встряхнуть до взмучивания осадка, после этого прибавить осталь-
ной спирт и перемешать.
Осадок янтарнокислого бария растворяют в 3 мл 0,1 н. раствора
соляной кислоты, обрабатывая ею сначала чашку. Остаток кислоты
смывают 2 мл дистиллированной воды, присоединяя промывную жид-
кость к раствору. Далее к раствору в пробирке прибавляют 1 мл
0,5 н. раствора бихромата калия, 2 мл 30%-ного раствора уксусно-
кислого натрия, перемешивают и помещают пробирку в стакан с
горячей водой. Воду в стакане предварительно нагревают до кипения
и снимают с плитки. Пробирку с раствором оставляют в горячей во-
де на 5 мин. После этого пробирку вынимают из горячей воды, раст-
вор охлаждают и центрифугируют. Центрифугат сливают, а осадок
промывают 2 раза водой, прибавляя ее по 5 мл.
Промытый осадок ВаСгО4 растворяют в 10 мл 1н. раствора со-
ляной кислоты, переносят в колбу для титрования, смывая остаток
раствора дистиллированной водой. К раствору в колбе добавляют
1 мл 20%-ного раствора йодистого калия и выделившийся йод тит-
руют 0,01 н. раствором тиосульфата натрия в присутствии индика-
тора крахмала. Содержание янтарной кислоты вычисляют по следую-
щей формуле:
__________________________ 3,93 К cl В
п v ’
где X — содержание янтарной кислоты (в %); В — общий объем
исследуемого раствора (в мл); v — объем исследуемого раствора,
взятый для определения янтарной кислоты (в мл); а — объем титро-
ванного раствора тиосульфата натрия, затраченный при титровании
выделившегося йода (в мл); К — нормальность раствора тиосуль-
фата натрия; п — навеска исследуемого вещества (в г); 3,93 — нор-
мальный титр янтарной кислоты, умноженный на 100 для пересчета
в проценты.
Реактивы. Раствор бихромата калия (0,5 н.). 24,51 г К2Сг2О7 растворяют
в дистиллированной воде, разбавляют водой до 1 л и тщательно перемешивают.
239
Муравьиная кислота (10%-ный раствор). 41 мл муравьиной кислоты
пл. 1,22 разбавляют водой до 500 мл и перемешивают.
Гидроксиламин солянокислый (2%-ный раствор). Взвешивают 2 г соляно-
кислого гидроксиламина NH2OH НС1 ч. д. а., растворяют в дистиллирован-
ной воде и разбавляют водой до 100 мл.
Приготовление остальных реактивов описано выше (см. стр. 17,27, 138, 222,
228).
ОПРЕДЕЛЕНИЕ АСКОРБИНОВОЙ КИСЛОТЫ
РТУТНЫМ МЕТОДОМ
Этот метод дает возможность определять аскорбиновую кислоту
в самых разнообразных объектах — в бесцветных и в интенсивно
окрашенных (ягодах, плодах и др.).
Сущность метода. Аскорбиновая кислота в слабощелоч-
ной, нейтральной или слабокислой среде (pH 2,5—9) вступает в реак-
цию с хлорной ртутью, восстанавливая ее до одновалентной. При этом
образуется трудпорастворимый осадок хлористой ртути (каломель):
C6H8Oe 2HgCl2 = C0HeO0 + Hg2Cl2 + 2НС1.
После удаления избытка хлорной ртути и других соединений со-
ляной кислотой осадок каломели окисляется бромом и переводится
в раствор в виде хлорида двухвалентной ртути:
3Hg2Cl2 + КВгО3 + 5КВг + 6НС1 = 3HgBr2 + 3HgCl2 +
+ 6КС1 + ЗН2О.
Образовавшуюся двухвалентную ртуть титруют в аммиачной сре-
де трилоном Б в присутствии небольшого количества комплексо-
ната магния и индикатора хромогена черного ЕТ-00:
Na2H2C10H12O3N2 + HgCl2 = Na2HgCi0H]2O3N2 + 2НС1.
Грамм-эквивалент аскорбиновой кислоты .Сб^°6 в данном слу-
чае равен 44.
Некоторые вещества, находящиеся в растениях, могут в нейт-
ральной и слабощелочной среде восстанавливать двухвалентную
ртуть до одновалентной и этим мешать определению аскорбиновой
кислоты. Поэтому при определении аскорбиновой кислоты обработку
исследуемого материала хлорной ртутью следует проводить в сла-
бокислой среде, в которой реагирует только аскорбиновая кислота.
Ход анализа. Берут такую навеску исследуемого вещества,
чтобы в ней находилось от 0,2 до 2 мг аскорбиновой кислоты (0,1 г
шиповника, 0,5—1 г смородины, 2 г зеленых листьев, 5 г клубней
картофеля). Навеску помещают в ступку, добавляют 5 мл 1 %-ного
раствора уксусной кислоты, 2 мл 4 %-ного раствора HgCl2 и расти-
рают до однородной массы. Если исследуемые ягоды или плоды со-
держат свободные органические кислоты, уксусную кислоту не при-
бавляют, а вместо нее прибавляют дистиллированную воду.
Растертую массу переносят в центрифужную пробирку емкостью
20 мл, смывая остаток 10 мл дистиллированной воды, перемешивают
240
стеклянной палочкой и оставляют на 10 мин для полноты осаждения.
Затем центрифугируют (3 мин при 3000 об/мин), раствор отбрасы-
вают, даже если он мутноват. В осадке находится эквивалентное
аскорбиновой кислоте количество каломели (Hg2CL). Осадок промы-
вают 3 раза 1 н. раствором соляной кислоты, прибавляя ее по 10 мл
и каждый раз перемешивая стеклянной палочкой. К промытому осад-
ку в центрифужной пробирке приливают 10 мл 0,1 н. раствора бро-
мат-бромидного реактива и 2 мл 1 и. раствора соляной кислоты.
Содержимое перемешивают стеклянной палочкой в течение 5 мин, за-
тем центрифугируют и сливают центрифугат в колбу для титрования
на 100 мл. К остатку в пробирке прибавляют 10 мл дистиллирован-
ной воды,, перемешивают, центрифугируют и промывную жидкость
присоединяют к первому центрифугату. Колбу с раствором помеща-
ют на плитку в вытяжном шкафу, нагревают до кипения и кипятя'!
до удаления брома, на что требуется 2—3 мин. Затем колбу с раст-
вором охлаждают водой под краном, добавляют 3 мл аммиачного
буферного раствора, 5 мл насыщенного раствора комплексоната маг-
ния, 50 мг индикатора хромогена черного ЕТ-00 и титруют 0,002 н.
раствором трилона Б до перехода окраски индикатора в синюю. 1 мл
0,002 н. раствора трилона Б соответствует 0,088 мг аскорбиновой
кислоты.
Из полученных данных вычисляют содержание аскорбиновой
кислоты по формуле:
v 4400 а к
где X — содержание аскорбиновой кислоты (в мг па 100 г исследуе-
мого вещества); а — объем титрованного раствора трилона Б, за-
траченный при титровании исследуемого раствора (в мл)-, К — нор-
мальность раствора трилона Б; п — навеска исследуемого вещест-
ва (в г); 4400 — нормальный титр аскорбиновой кислоты (в мг,
умноженный на 100 для пересчета на 100 г вещества).
Реактивы. Раствор бромат-бромида (0,1 н.). Отвешивают 2,784 г КВгО3
и 20 г КВг, растворяют в дистиллированной воде и разбавляют водой до 1 л.
Раствор хлорной ртути (4%-ный). 4 г HgCl2 растворяют в дистиллированной
воде и разбавляют водой до 100 мл.
Титрованный раствор трилона Б (0,002 н.). Готовят из 0,02 н. раствора раз-
бавлением в 10 раз дистиллированной водой. Для этого 50 мл 0,02 н. раствора
трилона Б переносят в мерную колбу на 500 мл и разбавляют водой до метки.
Приготовление 0,02 н. раствора трилона Б и других растворов описано выше
(см. стр. 12, 13, 40, 138 и 232).
ОПРЕДЕЛЕНИЕ ДИ-И ТРИКАРБОНОВЫХ КИСЛОТ
С ПОМОЩЬЮ ХРОМАТОГРАФИИ НА БУМАГЕ
Сущность метода. Органические кислоты, кроме щаве-
левой, извлекают из исследуемого материала раствором уксусной
кислоты, отделяют центрифугированием и полученный раствор дово-
дят водой до определенного объема. В меньшей части раствора оп-
ределяют сумму органических кислот хромат-йодометричеоким
16 6-2
241
методом, а в большей части раствора определяют отдельные кислоты
после хроматографического разделения их на бумаге. Для этого рас-
твор выпаривают с уксуснокислым барием досуха, при этом органи-
ческие кислоты образуют с ионами бария средние соли, нераство-
римые в 70 %-ном этиловом спирте:
Ва (СН3СОО)2 + Н2Л = ВаЛ + 2СН3СООН,
где Л — анион кислоты.
Избыток уксуснокислого бария, сахара и другие растворимые веще-
ства отмываются 70%-ным этиловым спиртом. Полученные чистые
бариевые соли органических кислот разлагаются эквивалентным
количеством серной кислоты:
ВаЛ + H2SO4 = BaSO4 + Н2Л,
где Л — анион органической кислоты.
Образовавшийся осадок сернокислого бария отделяют центри-
фугированием, а раствор, содержащий свободные органические кис-
лоты, выпаривают па водяной бане. Остаток растворяют в таком ко-
личестве воды, чтобы получился 4%-ный раствор кислот в пересчете
на яблочную кислоту. После хроматографирования и проявления
окрашенные зоны кислот вырезают и кислоты извлекают дистил-
лированной водой, подкисленной уксусной кислотой. В растворах
после выпаривания с уксуснокислым барием определяют кислоты
бихромат-йодометрическим методом. Щавелевую кислоту, связан-
ную с ионами кальция, этим методом не учитывают. Хроматогра-
фирование ее связано со значительными потерями, поэтому опре-
деление щавелевой кислоты и ее солей нужно проводить химиче-
ским методом (см. стр. 235).
Ход анализа. Взвешивают такое количество раститель-
ного материала, чтобы в нем находилось от 50 до 100 мг суммы орга-
нических кислот (сухого вещества от 0,4 до 1,5 г и сырого от 2 до 6 г).
Навеску растирают в ступке с 10 мл 1%-ного раствора уксусной
кислоты и полученную массу переносят в центрифужную пробирку,
смывая остаток 1%-ной уксусной кислотой. Пробирку помещают в
горячую воду и нагревают 5—10 мин при периодическом перемешива-
нии содержимого. Затем центрифугируют и раствор сливают в мер-
ную колбу на 50 мл. К остатку приливают 10 мл 1%-ной уксусной
кислоты, перемешивают, настаивают при размешивании 5 мин и цен-
трифугируют. Раствор сливают в мерную колбу, а остаток промы-
вают еще 2 раза, прибавляя по 10 мл 1 %-ной уксусной кислоты. Про-
мывную жидкость присоединяют к раствору. Объем вытяжки дово-
дят дистиллированной водой до 50 мл и перемешивают.
Определение суммы органических кислот
Из полученного раствора кислот набирают пипеткой 5 мл, пере-
носят в фарфоровую чашку, добавляют 0,3 мл 20%-ного раствора
уксуснокислого бария и выпаривают раствор на водяной бане досуха.
242
Сухой остаток промывают 70%-ным этиловым спиртом. Для этого
в чашку прибавляют 2 мл 70%-пого спирта, перемешивают стек-
лянной палочкой до однородной массы и полученную взвесь сливают
в центрифужную пробирку, смывая остаток 70%-ным спиртом 2 раза
порциями по 2 мл. Затем пробирку с содержимым встряхивают в те-
чение 2 мин и центрифугируют. Прозрачный раствор удаляют, сли-
вая в склянку с отработанным спиртом, а осадок промывают еще
2 раза 70%-ным спиртом, прибавляя его по Злы и предварительно
споласкивая чашку. К промытому осадку прибавляют 10 мл 2 и. ра-
створа азотной кислоты, размешивают помещают в кипящую воду на
3 мин, центрифугируют и раствор сливают в фарфоровую чашку.
К остатку в пробирке прибавляют 2 мл 2 н. азотной кислоты и 10 мл
дистиллированной воды, размешивают и центрифугируют. Раствор
выливают в фарфоровую чашку, присоединяя к первому, добавляют
1 мл 42%-ной хлорной кислоты и раствор выпаривают на водяной
бане до малого объема (около 1 мл). Затем чашку помещают на воз-
душную баню и выпаривают досуха и до удаления хлорной кислоты.
После охлаждения к остатку в чашке добавляют 2 мл азотной кис-
лоты (1 1), смачивая стенки, помещают на кипящую водяную баню
и выпаривают досуха. Остаток растворяют в 5 мл 0,1 н. НС1, пере-
носят в центрифужную пробирку, смывая остаток 2 мл дистиллиро-
ванной воды. К полученному раствору прибавляют 1 мл 0,5 н. рас-
твора бихромата калия, 2 мл 30%-ного раствора уксуснокислого
натрия и перемешивают. При этом барий осаждается в виде хрома-
та бария:
2ВаС12 + К2Сг2О7 + 2CH3COONa Н2О = 2BaCrO4 + 2KCI +
+ 2NaCl + 2СН3СООН.
Пробирку с реакционной смесью ставят в стакан с горячей водой
на 3—5 мин для укрупнения осадка. Затем пробирку вынимают из
горячей воды, дают постоять 10—15 мин, центрифугируют, раствор
сливают, а осадок промывают водой 2 раза порциями по 5 мл. Оса-
док ВаСгО4 растворяют в 10 мл 1 н. раствора соляной кислоты, пере-
носят в колбу для титрования емкостью 100 мл, смывая остаток рас-
твора дистиллированной водой. К раствору в колбе прибавляют 1 мл
20%-ного раствора йодистого калия. При этом выделяется йод:
2ВаСгО4 + 16HCI + 6KJ = 2СгС13 + 2ВаС12 + 6КС1 + 3J2 - 8Н2О.
Выделившийся йод титруют 0,01 н. раствором тиосульфата натрия
в присутствии крахмала до исчезновения синей окраски:
J2 + 2Na2S2O3 = 2NaJ + Na9S4O„.
Результат титрования используют для вычисления количества 0,2 н.
раствора серной кислоты, необходимого для разложения бариевых
солей органических кислот, а также количества воды для раство-
рения свободных кислот. Содержание суммы органических кис-
16*
243
лот, кроме щавелевой кислоты, связанной с кальцием, вычисляют
по формуле:
v 4,47 В к а
где X — содержание суммы кислот (в % в расчете на яблочную кис-
лоту); В — общий объем экстракта (в мл); v —объем экстракта,
взятый для определения суммы кислот (в мл); а — объем 0,01 н. рас-
твора тиосульфата натрия, затраченный при титровании (в мл); К —
нормальность раствора тиосульфата натрия; п — навеска исследуе-
мого вещества (в г); 4,47 — нормальный титр яблочной кислоты,
умноженный на 100.
Получение 4°/о’ного раствора свободных органических кислот
Из остатка исследуемого раствора набирают 40 мл, переносят в
фарфоровую чашку диаметром 8 см, добавляют 2 мл 20%-ного рас-
твора уксуснокислого бария и выпаривают раствор на кипящей во-
дяной бане досуха. Сухой остаток промывают 70%-ным этиловым
спиртом, чтобы удалить сахара и другие растворимые примеси. Для
этого в чашку приливают 0,5 мл дистиллированной воды, перемеши-
вают стеклянной палочкой до однородной массы, прибавляют 5 мл
70%-ного спирта, снова перемешивают и переносят раствор с осад-
ком в центрифужную пробирку. Остаток осадка смывают в пробирку
70%-ным спиртом 2 раза порциями по 5 мл, затем раствор с осадком
перемешивают в течение 5 мин стеклянной палочкой и центрифуги-
руют. Раствор сливают, а осадок промывают еще 2 раза 70% -ным
спиртом порциями по 5 мл, споласкивая сначала чашку. При промы-
вании содержимое пробирки тщательно перемешивают стеклянной
палочкой в течение 3 мин. Затем палочку вынимают и смывают части-
цы осадка несколькими каплями 70%-ного спирта. Промывную жид-
кость отделяют от осадка центрифугированием. К промытому осад-
ку приливают 10 мл 1 %-ного раствора уксусной кислоты, перемеши-
вают стеклянной палочкой несколько минут и прибавляют при
помешивании рассчитанное количество 0,2 н. раствора серной
кислоты.
Количество 0,2 н. раствора серной кислоты, которое необходимо
прибавить к осадку для разложения бариевых солей органических
кислот, вычисляют по формуле:
Х = 40 ° к„,"№7 =26.7-а К.
где X — объем 0,2 н. раствора серной кислоты (в мл); а — объем
титрованного раствора тиосульфата натрия, затраченный при опре-
делении суммы органических кислот (в мл); К — нормальность тит-
рованного раствора тиосульфата натрия; 5 — объем исследуемого
раствора, взятый для определения суммы органических кислот (в мл);
40 — объем исследуемого раствора, взятый для получения осадка
бариевых солей органических кислот (в мл); 0,2 — нормальность
244
раствора серной кислоты; 0,667 — коэффициент перевода эквивален-
та органических кислот по хрому на эквивалент по барию.
После прибавления раствора серной кислоты пробирку с реакци-
онной смесью помещают в горячую воду и в течение 5мин тщательно
перемешивают раствор с осадком стеклянной палочкой. Затем ох-
лаждают и центрифугируют. Если осадок плохо отделяется от раст-
вора, прибавляют 3—5 мл 95%-ного этилового спирта. Прозрач-
ный раствор сливают в фарфоровую чашку, а осадок промывают
1 раз 5 мл 1%-ной уксусной кислотой, центрифугируют и раствор
присоединяют к первому цептрифугату. Чашку помещают на кипя-
щую водяную баню и содержимое выпаривают досуха. После охлаж-
дения к сухому остатку приливают такое количество дистиллирован-
ной воды, чтобы получился 4%-ный раствор кислот в пересчете па
яблочную кислоту, и перемешивают стеклянной палочкой до раство-
рения кислот. Переливают содержимое в сухую центрифужную про-
бирку, центрифугируют, сливают прозрачный раствор в сухую про-
бирку и закрывают пробкой.
Количество воды, которое необходимо для получения 4 %-ного
раствора органических кислот, вычисляют по формуле:
Y = -40 а К 4,47 = 8,94 а К,
5 4
где Y — объем дистиллированной воды, который нужно добавить к
остатку в чашке, чтобы получить 4%-пый раствор органических
кислот в пересчете на яблочную кислоту (в мл}\ 4—концентрация
получаемого раствора кислот (в %); 4,47 нормальный титр яблоч-
ной кислоты, умноженный на 100.
Остальные показатели тс же, что и в приведенной выше формуле
для расчета объема 0,2 н. раствора серной кислоты, используемой
при разложении бариевых солей органических кислот.
Качественная хроматография
Для установления качественного состава органических кислот
проводят хроматографирование на бумаге исследуемого раствора
одновременно с растворами «свидетелей». Из листа хроматографи-
ческой бумаги (марки «М» или «С»), промытой соляной или щавеле-
вой кислотой, вырезают ленты шириной 12 см и длиной 52 см. На
7 см ниже верхнего края ленты проводят стартовую линию. На ней
ставят точки на расстоянии 1,5 см от левого и правого края ленты,
а внутри на расстоянии 3 см от первых точек еще 2 точки — все-
го 4 точки. На средние точки наносят капилляром исследуемый рас-
твор свободных кислот, а на две крайних точки наносят раствор сме-
си кислот «свидетелей» (приблизительно по 0,005 мл). Приготовле-
ние описано ниже (см. стр. 253).
Две такие ленты соединяют вместе стеклянными палочками, на
концы которых одеты резиновые кольца, и помещают в камеру, по-
гружая верхнюю часть ленты в ванночку с растворителем. Ленты за-
гибают и опускают по обе стороны ванночки в камеру. В ванночку
245
наливают 30 мл растворителя: амиловый спирт — ацетон — 45%-ная
муравьиная кислота (12 3 5) или н-бутиловый спирт — ацетон —
45%-пая муравьиная кислота (5 1 2). Камеру закрывают крыш-
кой и оставляют до тех пор, пока фронт растворителя опустится в
самый низ бумажной ленты, на что требуется 20—24 ч. После этого
хроматограммы вынимают из камеры и подвешивают в вытяжном
шкафу. После высыхания их оставляют на ночь для полного удале-
ния муравьиной кислоты. На второй день хроматограммы проявляют
с помощью 0,005%-ного раствора индикатора нафтилового крас-
ного, который наносят на бумагу пульверизатором. Кислоты об-
разуют пятна красно-фиолетового цвета на желтом фоне.
В растворителе с амиловым спиртом кислоты движутся в такой
последовательности и с такими значениями 7?/: фумаровая (0,76),
янтарная (0,60}, щавелевая (0,50), яблочная (0,36), лимонная (0,28),
винная (0,18). В растворителе с «-бутиловым спиртом последователь-
ность та же, a Rf следующая: фумаровая — 0,79, янтарная — 0,67,
щавелевая — 0,60, яблочная — 0,47, лимонная — 0,39, винная —
0,28.
Количественный анализ кислот
методом хроматографии па бумаге
Из хроматографической бумаги (марки «М»), промытой щавеле-
вой или соляной кислотой, вырезают ленты длиной 52 см и шириной
13 см. В верхней части ленты на 7 см ниже верхнего края отделяют
зону нанесения исследуемого раствора
двумя вырезами длиной 20 мм и шири-
ной 2 мм, расположенными вертикально
по краям зоны нанесения и отстоящими
на расстоянии 10 мм от левого и правого
края ленты. Сначала намечают каранда-
шом места вырезов и линию старта.
Затем ленту помещают на двойной лист
фильтровальной бумаги и лезвием безо-
пасной бритвы делают вырезы (рис. 38).
После этого на стартовую линию наносят
в два приема 0,1 мл исследуемого рас-
твора кислот. Для этого микропипеткой
набирают 0,05 мл раствора, внешний
конец пипетки вытирают фильтровальной
бумагой и, коснувшись концом пипетки
начала стартовой линии, ведут по ней с
такой скоростью, чтобы раствор распре-
делился равномерно по всей длине линии. Растворитель удаляют
с бумаги высушиванием, а на стартовую линию наносят еще 0,05 мл
исследуемого раствора. Для каждого анализа готовят две такие
бумажные ленты, которые после нанесения исследуемого раствора
складывают вместе и скрепляют в верхней части с помощью двух
стеклянных палочек, соединенных по краям резиновыми кольцами,
Рис. 38. Расположение огра-
ничительных вырезов на бу-
маге для хроматографии ор-
ганических кислот:
а — зона нанесения исследуемо-
го раствора; б — ограничитель-
ные вырезы.
246
изготовленными из резиновых трубок. Стеклянные палочки закреп-
ляют выше стартовой линии (на 2—3 см ниже верхнего края бумаж-
ных лент).
В ванночку, находящуюся в хроматографической камере, нали-
вают около 35 мл растворителя (амиловый спирт — ацетон —
45%-ная муравьиная кислота или к-бутпловый спирт — ацетон —
45%-ная муравьиная кислота) и небольшое количество его наливают
в чашку, которая находится па дне камеры. Верхнюю часть бумаж-
ных лент, выходящую из палочек, погружают в ванночку с раствори-
телем, а бумажные ленты загибают с одной и с другой стороны ванноч-
ки, опуская их в камеру, которую затем закрывают крышкой, и остав-
ляют на 20—25 ч. После того как растворитель опустится в самый
низ бумажной полосы, хроматограмму вынимают из камеры, при-
касаясь только к стеклянным палочкам, и подвешивают в вытяж-
ном шкафу за концы палочек. Включают вентилятор, высушивают
до полного удаления растворителя и затем оставляют в вытяжном
шкафу при комнатной температуре до следующего дня для оконча-
тельного удаления муравьиной кислоты. На следующий день хрома-
тограмму опрыскивают 0,005%-ным раствором индикатора нафти-
лового красного и высушивают. Зоны, занятые кислотами, окра-
шиваются в красно-фиолетовый цвет на желтом фоне бумаги.
Определение содержания отдельных кислот
После идентификации кислот их нумеруют и вырезают окрашен-
ные зоны. Вырезки с одинаковыми кислотами объединяют из двух
хроматограмм, складывают гармошкой, сворачивают и помещают в
пробирку. В пробирки с вырезками приливают по 15 мл 1%-ного
раствора уксусной кислоты и настаивают 30 мин при частом взбал-
тывании. После этого растворы сливают в фарфоровые чашки, а бу-
магу в пробирках промывают еще 2 раза дистиллированной водой,
прибавляя ее по 10 мл и настаивая в течение 10—15 мин. Промыв-
ные воды добавляют в чашку.
К вытяжкам кислот в чашках приливают по 0,2 мл 20%-ного
раствора уксуснокислого бария и выпаривают на водяной бане до-
суха. Сухой остаток промывают 70%-ным этиловым спиртом, чтобы
удалить избыток уксуснокислого бария. Для этого в чашки прили-
вают по 2 мл 70%-ного спирта, перемешивают стеклянной палочкой
и сливают раствор вместе с осадком в центрифужные пробирки.
В чашки приливают еще по 2 мл 70 %-ного спирта и после перемеши-
вания присоединяют к растворам в центрифужных пробирках. Содер-
жимое пробирок взбалтывают в течение 3 мин, затем центрифугируют,
растворы сливают в склянку с отработанным спиртом, а осадки про-
мывают еще 2 раза, прибавляя по 3 мл 70%-ного спирта и предва-
рительно споласкивая чашки. К промытым осадкам прибавляют по
3 мл 0,1 н. раствора соляной кислоты, споласкивая ею сначала чаш-
ку. К полученным растворам приливают по 1 мл 0,5 н. раствора би-
хромата калия, растворы перемешивают, добавляют по 2 мл 30 %-ного
247
раствора уксуснокислого натрия, снова перемешивают, погружают
пробирки в горячую воду и оставляют на 5 мин. Затем пробирки вы-
нимают из горячей воды и оставляют на 10—15 мин. После охлаж-
дения центрифугируют, раствор сливают, а осадок промывают 2 ра-
за дистиллированной водой порциями по 5 мл.
К промытым осадкам хромата бария приливают по 10 мл 1 н.
раствора соляной кислоты, перемешивают до растворения и перено-
сят растворы в колбы для титрования, смывая остатки водой. В кол-
бы прибавляют по 1 мл 20%-ного раствора йодистого калия и тит-
руют выделившийся йод 0,01 н. раствором тиосульфата натрия в при-
сутствии крахмала до исчезновения синей окраски. Из результатов
титрования вычисляют содержание кислот по следующей формуле:
У___ 0,1 50 Э /С a v ______ 0,125 Э К a v
40 b п b п ’
где X — содержание данной кислоты в исследуемом материале (в %);
Э — г-экв определяемой кислоты: винной — 50, лимонной (безвод-
ной) — 42,7, щавелевой (безводной) — 30,1, яблочной — 44,7, ян-
тарной — 39,4, малоновой — 34,7, фумаровой — 38,7; К — нормаль-
ность титрованного раствора тиосульфата натрия; а — объем тит-
рованного раствора тиосульфата натрия, затраченный при определе-
нии данной кислоты (в мл); v — объем 4%-иого раствора свобод-
ных кислот, полученный из 40 мл исследуемого раствора (в мл); b —
объем 4%-ного раствора свободных кислот, нанесенный на хрома-
тограмму (на две бумажные ленты) (в мл); п — навеска исследуемого
вещества (в г); 50 — общий объем исследуемого раствора (в мл);
40 — объем исследуемого раствора, взятый для получения 4%-ного
раствора свободных кислот; 0,1 — коэффициент пересчета милли-
граммов в проценты.
Реактивы. Растворители для хроматографирования кислот. В стакане сме-
шивают 24 мл амилового спирта с 6 мл ацетона и 10 мл 45%-ного водного раствора
муравьиной кислоты. Перемешивают стеклянной палочкой до получения однород-
ного раствора. Реактивы применяют х. ч. или очищают перегонкой. Количество
растворителя рассчитано на одну хроматограмму. Близкий по свойствам раство-
ритель готовят следующим образом: 25 мл н-бутилового спирта, 5 мл ацетона
и 10 мл 45%-ного раствора муравьиной кислоты смешивают до получения одно-
родной жидкости. Раствор 45%-ной муравьиной кислоты готовят из более креп-
ких растворов. Количество более крепкого раствора муравьиной кислоты, необ-
ходимое для приготовления 100 мл 45%-ного раствора, вычисляют по формуле:
х_ 1,109 45 100 4990
— da d а ’
где X — объем разбавляемой кислоты (в мл); а — концентрация разбавляемого
раствора кислоты (в вес. %); d — плотность разбавляемого раствора кислоты.
Найденный объем более крепкого раствора кислоты помещают в мерную
колбу на 100 мл, разбавляют дистиллированной водой до метки и тщательно пе-
ремешивают.
Растворы индикаторов для проявления хроматограмм кислот.
1. 0,1 г нафтилового красного растворяют в 200 мл 95%-ного этилового
спирта. Перед употреблением этот раствор разбавляют в 10 раз 50%-ным спир-
том. Полученный раствор является хорошим проявителем: на желтом фоне об-
разуются красно-фиолетовые зоны, занятые кислотами. Переход окраски в ин-
тервале pH от 3,7 до 5,0.
248
2. 0,1 г диметилового желтого и 0,2 г бромфенолового синего растворяют
в 120 мл 95%-ного этилового спирта, добавляют 1 мл 2 п. раствора гидроокиси
натрия и разбавляют дистиллированной водой до 200 мл. Из этого раствора го-
товят проявитель разбавлением в 10 раз 50%-ным спиртом. Этот раствор прояв-
ляет янтарную кислоту не очень четко. На зслсновато-сипем фоне появляются
пятна кислот от желтого до красного цвета в зависимости от силы кислот. Пере-
ход окраски в интервале pH от 2,9 до 4,6.
Раствор серной кислоты (0,2 п.). Сначала готовят —2 н. раствор серной
кислоты. Для этого к 0,5 л дистиллированной воды приливают 56 мл серной'
кислоты пл. 1,84, перемешивают, охлаждают, разбавляют водой до 1 л и снова
перемешивают. Этот раствор разбавляют в 10 раз водой. Нормальность получен-
ного раствора устанавливают по бурс с индикатором метилоранжем или метило-
вым красным. Концентрацию доводят до 0,2 п. разбавлением водой.
ОПРЕДЕЛЕНИЕ ДИ- И ТРИКАРБОНОВЫХ КИСЛОТ
МЕТОДОМ ХРОМАТОГРАФИИ НА БУМАГЕ
ПО ПЛОЩАДИ ПЯТЕН
Сущность метода. Органические кислоты извлекают
1%-ной уксусной кислотой. Из плодов и ягод, содержащих значи-
тельное количество пектиновых веществ, кислоты извлекают 70 % -
ным этиловым спиртом, подкисленным уксусной кислотой. Щаве-
левая кислота, связанная с кальцием, не переходит в раствор и в этом
методе ее не определяют. Из полученного раствора кислоты осаж-
дают уксуснокислым свинцом и отделяют центрифугированием. Оса-
док обрабатывают карбонатом натрия. При этом кислоты переходят
в раствор в виде натриевых солей, а свинец в виде карбоната остает-
ся в осадке:
РЬД Na2CO3 - РЬСО;| - №2Д,
где А — анион органической кислоты.
После отделения осадка центрифугированием и промывания рас-
твор натриевых сэлей органических кислот пропускают через катио-
нит, в котором происходит обмен ионов натрия на ион водорода и
кислоты выделяются в свободном состоянии. Раствор доводят до
определенного объема и аликвотную часть его титруют 0,02 н. раство-
ром едкого натра для определения суммы кислот. Остаток раствора
выпаривают досуха и осадок растворяют в таком количестве воды,
чтобы получился 2%-ный раствор кислот в пересчете па яблочную
кислоту. Этот раствор наносят на бумагу и хроматографируют. После
проявления хроматограмм и идентификации кислот окрашенные
пятна вырезают и измеряют площадь или взвешивают на аналити-
ческих весах. Из полученных данных вычисляют содержание от-
дельных кислот, пользуясь соответствующими формулами, приве-
денными ниже.
Ход анализа. Берут такую навеску исследуемого материа-
ла, чтобы в ней находилось от 20 до 50 мг суммы кислот (свежего ма-
териала от 2 до 5 г и сухого от 0,5 до 1,5 г) и растирают с 1%-ной
уксусной кислотой или 70 %-ным спиртом в зависимости от наличия
пектинов.
249-
Извлечение 1 % -ной уксусной кислотой
При исследовании листьев, стеблей, корней навеску растирают
с 5 мл 1%-ной уксусной кислоты. В случае анализа одревесневших
тканей их предварительно растирают с пемзой, песком или силика-
гелем, добавляя 0,5—1 г. Растертую массу переносят в мерную кол-
бу на 50 мл с помощью 1 %-ной уксусной кислоты, доводя объем до
3/4 объема колбы. Колбу с содержимым помещают в кипящую воду
и нагревают при периодическом взбалтывании 10 мин. Затем колбу
вынимают из воды, охлаждают, доводят объем 1 %-ной уксусной кис-
лотой до метки, тщательно перемешивают и фильтруют через склад-
чатый бумажный фильтр в сухую колбу. Из фильтрата набирают
25 лм, помещают в фарфоровую чашку, добавляют 1 мл 20 %-ного рас-
твора уксуснокислого свинца и выпаривают на водяной бане досуха.
К остатку прибавляют 3 мл 70%-ного раствора этилового спирта,
размешивают стеклянной палочкой и переносят в центрифужную про-
бирку, смывая остаток 3 мл 70%-ного спирта. Размешивают, цент-
рифугируют, раствор сливают в склянку для отработанного спирта,
а осадок промывают 2 раза 70 %-ным этиловым спиртом порциями по
3 мл.
Извлечение спиртом
Навеску свежих плодов или ягод растирают в ступке сначала с
0,5 мл ледяной уксусной кислоты, затем с 5 мл 70 %-ного этилового
спирта. Растертую массу переносят в центрифужную пробирку,
смывая остаток 10 мл 70%-ного спирта. Перемешивают и настаивают
10 мин при частом перемешивании. После этого центрифугируют, рас-
твор сливают в стакан, а осадок промывают 2 раза 70 %-ным спиртом
порциями по 5 мл, с 5-минутным настаиванием и последующим цент-
рифугированием. К объединенному экстракту в стакане прибавляют
1 мл 20 %-ного раствора уксуснокислого свинца, 5 мл 95 %-ного этило-
вого спирта, перемешивают и оставляют на 30 мин для выделения
осадка. Затем раствор и осадок из стакана переносят в центрифужную
пробирку, центрифугируют, раствор сливают в склянку для отрабо-
танного спирта, а осадок промывают 1 раз 5 мл 70%-ного спирта.
Извлечение кислот
из осадка карбонатом натрия
К промытому осадку в центрифужной пробирке, содержащему
свинцовые соли органических кислот, добавляют 1 мл 5%-ного рас-
твора Na2CO3, 10 мл дистиллированной воды, размешивают стеклян-
ной палочкой в течение 5 мин для разложения осадка и затем цен-
трифугируют. Раствор сливают в стакан, а осадок промывают 1 раз
5 мл воды, присоединяя центрифугат к первому в стакане. Получен-
ный раствор натриевых солей органических кислот пропускают че-
рез колонку с катионитом, собирая фильтрат в мерную колбу на 50 мл.
250
Приготовление колонки
Используют колонку диаметром 1,5—2,5 гл* и длиной 20—30 см,
со стеклянным фильтром № 1 или № 2. Колонку заполняют Юг ка-
тионита КУ-1 или КУ-2 (16—50 мош), который перед этим замачива-
ют в 2 н. соляной кислоте в течение не менее 2 ч при периодическом
перемешивании. Затем кислоту сливают, а смолу промывают декан-
тацией один раз водой и переносят в стеклянную колонку. Через
колонку сначала пропускают 50 мл дистиллированной воды, которой
дают стечь до уровня смолы, потом 20 2 и. раствора соляной кисло-
ты. После этого смолу промывают дистиллированной водой до тех нор,
пока вытекающий раствор будет иметь слабокислую реакцию (pH 6,
метилрот, добавленный к фильтру, должен окрашивать его в желтый
цвет). В таком виде колонка готова к употреблению.
Получение 2 % -ного раствора свободных кислот
Исследуемый раствор натриевых солей органических кислот про-
пускают через колонку со скоростью 10 капель в 1 мин или фильтру-
ют со слабым отсасыванием повторно несколько раз (3—^пропус-
кая раствор через катионит. Колонку с остатком раствора промывают
дистиллированной водой до полного вымывания всех кислот, про-
водя пробу с метилротом. Вытекающий раствор и промывные воды
собирают в мерную колбу на 50 мл, доводя объем до метки. Раствор в
колбе тщательно перемешивают, отбирают 10 мл в колбу для титро-
вания, нагревают на плитке до кипения для удаления углекислоты,
охлаждают и титруют 0,02 н. раствором едкого натра в присутствии
фенолфталеина. Остаток исследуемого раствора (40 мл) помещают
в фарфоровую чашку и выпаривают на водяной бане досуха. Остаток
растворяют в таком количестве воды, чтобы получился 2%-ный раст-
вор суммы кислот в пересчете на яблочную кислоту. Необходимое
количество воды, которое надо прибавить к остатку, вычисляют по
следующей формуле:
X = ---^-2 а =13,4 /< а,
где X — объем воды, необходимый для растворения кислот (в мл);
К — нормальность раствора едкого натра; а — объем 0,02 п. раст-
вора едкого натра, затраченный при титровании (в мл); 10 — объем
исследуемого раствора, взятый для титрования (влы); 40 — объем
исследуемого раствора, взятый для выпаривания (в мл); 2 — необ-
ходимая концентрация кислот в растворе (в %); 6, 7 — нормальный
титр яблочной кислоты, умноженный на 100 для пересчета в проценты.
Хроматографирование
Для хроматографирования берут бумагу, имеющую однородную
структуру с отклонением веса единицы площади бумагу не превышаю-
щим 2%. Бумагу предварительно промывают соляной или щавелевой
251
кислотой и дистиллированной водой. Из промытой и высушенной
бумаги вырезают полоски шириной 16,5 см и длиной 58 см. Перед на-
несением исследуемого раствора на полоски бумаги на них отмечают
стартовые линии и делают вырезы так, как это описано в методе
определения аминокислоте помощью хроматографии на бумаге (см.
стр. 105). 0,01 мл исследуемого раствора наносят между 1-м и 2-м вы-
резами и 0,02 мл между 5-м и 6-м вырезами. 0,01 мл стандартного ра-
створа наносят между 3-м и 4-м вырезами и 0,02 мл между 7-м и 8-м
вырезами таким же способом, как при хроматографировании амино-
кислот.
После нанесения растворов и высушивания пятен две полоски
бумаги соединяются вместе вверху на расстоянии 2—2,5 см от края с
помощью двух стеклянных палочек, скрепленных резиновыми коль-
цами. Верхний край бумажных полосок опускают в ванночку и по-
лоски загибают по обе стороны ванночки в камеру. В ванночку на-
ливают 35 мл растворителя: амиловый спирт — ацетон — 45%-ная
муравьиная кислота (12 3 5) пли н-бутанол — ацетон — 45%-ная
муравьиная кислота (5 1 2). Небольшое количество растворите-
ля наливают на дно камеры, которую затем плотно закрывают крыш-
кой и оставляют на 20—24 ч, пока фронт растворителя окажется око-
ло нижнего края ленты.
После хроматографирования бумагу вынимают из камеры и под-
вешивают в вытяжном шкафу, цепляя за края палочек. Включают
вентилятор и сушат до удаления растворителя. Затем оставляют на
воздухе на 16—20 ч для удаления муравьиной кислоты. После этого
хроматограммы опрыскивают из пульверизатора 0,005%-ным раст-
вором нафтилового красного. Кислоты образуют красно-фиолетовые
пятна на желтом фоне. После идентификации кислот их нумеруют,
регистрируют, вырезают окрашенные зоны и взвешивают на анали-
тических весах или измеряют площади пятен. Из полученных данных
вычисляют содержание кислоты на хроматограмме по формуле:
х + х = (К1 ~ К2) [(*6 + Хм) - (Кб + Км)] + к +
1 2 Кб Км 1 2’
где + Х2 — суммарное количество кислоты в большом и малом
пятне (в мг)\ Ki — количество кислоты в большом пятне стандарта
(в мг)\ К* — количество кислоты в малом пятне стандарта (в мг)\
Кб — вес бумажной вырезки большого пятна стандарта (в а); Км —
вес бумажной вырезки малого пятна стандарта (в а); Хб — вес вы-
резки большого пятна определяемой кислоты (в а); Хм — вес вырез-
ки малого пятна определяемой кислоты (в а).
Содержание кислоты в исследуемом материале вычисляют по фор-
муле:
У = 0,1 50- а (Хх + Х2) = 4,17 а (Хх + Х2)
Г 0,03^40 -п п
где Y — содержание органической кислоты в исследуемом материа-
ле (в %); Xj. + Х2 — суммарное количество кислоты в большом и
252
малом пятне, вычисленное с помощью предыдущей формулы (в мг);
а — объем 2%-ного раствора кислот, полученный при растворении
остатка после выпаривания 40мл экстракта (в мл); 50 — общий объем
раствора кислот (в мл); 40 — объем исследуемого раствора, взятый
для выпаривания и получения 2%-пого раствора кислот (в мл);
0,03 — объем 2%-ного раствора кислот, нанесенный на хромато-
грамму (в большом и малом пятне) (в мл); п — навеска исследуемого
вещества (в г). Если кислоты извлекаются 1%-ной уксусной кисло-
той, то вес навески уменьшается вдвое, так как используется толь-
ко половина вытяжки; 0,1 — коэффициент для пересчета микрограм-
мов в граммы и проценты.
Промывание бумаги
для хроматографирования кислот
При хроматографировании органических кислот примеси метал-
лов, находящиеся в бумаге (в основном железо), могут давать клеш-
невидные комплексы с кислотами, изменяя их Rf, что влечет за со-
бой появление вместо одного нескольких пятен. Для удаления при-
месей (тяжелые металлы) бумагу промывают соляной или щавелевой
кислотой. Последняя имеет некоторое преимущество, так как обра-
зует с железом комплексное соединение, растворимое в воде.
Для промывания берут 2—3 листа хроматографической бумаги,
помещают в ванночку размером 52 X 64 см и приливают 1 л 0,2 н.
раствора соляной кислоты или 2%-ного раствора щавелевой кисло-
ты. В этом растворе бумагу выдерживают около 1 ч при периодиче-
ском покачивании ванночки. После этого раствор сливают, а бумагу
промывают?—8 раз дистиллированной водой, прибавляя ее по 1 л
и выдерживая при покачивании ванночки, в течение 10—15 мин.
Полноту промывания проверяют метилоранжем — к 100 мл про-
мывной воды в стакане прибавляют 2 капли 0,1 %-ного раствора
метилоранжа и сравнивают окраску с таким же количеством ди-
стиллированной воды и таким же количеством метилоранжа. После
промывания ванночку с бумагой оставляют на воздухе в наклонном
положении до высыхания.
Реактивы. Раствор едкого натра (0,02 н.). Готовят из 0,1 н. раствора едкого
натра разбавлением в 5 раз безуглекислой водой. Приготовление 0,1 н. раствора
NaOH описано выше (см. стр. 9).
Стандартный раствор органических кислот (свидетелей). Взвешивают на
аналитических весах следующие количества кислот: винной, яблочной и янтар-
ной по 0,2500 г, лимонной С3Н4(ОН)(СООН)3 Н2О — 0,2734 г, щавелевой
Н2С2О4 2Н2О — 0,3500 г, фумаровой — 0,1250 г. Все навески помещают в мер-
ную колбу на 25 мл, растворяют в 5%-ный муравьиной кислоте, доводят до метки
этим же раствором кислоты и тщательно перемешивают. В 1 мл раствора содер-
жится по 10 мг всех кислот, кроме фумаровой, которой содержится вдвое меньше
(5 мг в 1 мл). Ограниченная растворимость фумаровой кислоты в холодной воде
не позволяет приготовить более крепкий раствор.
ЛИТЕРАТУРА
Беннет-Кларк Т. А. Роль органических кислот в обмене растений. М.—Л.,
Б иомедгиз, 1938.
253
К о л ьтгоф И. М.» Белчер Р., Стенгер В. А. Матсуяма Дж.
Объемный анализ, 3. М., Госхимиздат, 1961.
К о м е т и а н и П. А., С т р у а Г. Микройодометрическое определение лимон-
ной кислоты.— Биохимия, 1948, 13, 1, 23.
Лебедева А. П., П о ч и н о к X. Н. К определению форм кальция и щаве-
левой кислоты в листьях сахарной свеклы.— Науч. зап. сахарной промыш-
ленности (агр.), 1934, 4—5, 31.
Подклето.в Н. Е. Определение малых количеств винной кислоты.— Вино-
делие и виноградарство СССР, 1951, 10, 19.
Пятницкий М. П. К методике определения яблочной, лимонной и щавеле-
вой кислот в табаке.— В кн.: Сборник работ по химии табака, 2, вып. 80.
Краснодар, 1931, 23.
Сапожникова Е. В. Методы определения щавелевой и винной кислоты.—
Тр. прикл. бот., ген. и селекции, 1934, сер. 3, 5, 255.
СмирновА. П., Колесник М. Л. Определение лимонной кислоты в ма-
хорке методом весового учета пентабромацетона, извлекаемого петролейным
эфиром.— В кн.: Сборник работ по химии табака и махорки, 9, вып. 145.
Краснодар, 1948, 159.
Сотников Е. И. К вопросу об определении органических кислот.— ДАН
СССР, 1933, 6, 83.
Солдатенков С. В., Мазурова Т. А. Анализ органических кислот
растений методом ионообменных смол и хроматографии на бумаге.— В кн.:
Методика количественной бумажной хроматографии сахаров, органических
кислот и аминокислот у растений. М.—Л., Изд. АН СССР, 1962, 27.
Уринсон Р. П. Метод определения лимонной кислоты.— Тр. прикл. бот.,
ген. и селекции, 1934, сер. 3, 5, 261.
Церевитинов Ф. В. Химия и товароведение свежих плодов и овощей.
М., Госторгиздат, 1949, 176.
Ш м у к А. А. К методике определения лимонной и яблочной кислот в табаке
и махорке.— В кн.: Сборник работ по химии табака, 5. Краснодар, 1934, 71.
Глава VII
ФОСФОРНЫЕ СОЕДИНЕНИЯ
И ЛИПИДЫ
В растениях большая часть фосфора находится в виде органичес-
ких соединений и только некоторая часть в виде неорганических фос-
фатов, количество которых меняется в зависимости от условий про-
израстания растений.
Фосфорорганические соединения растений могут быть разде-
лены на следующие основные группы:
Гексозофосфаты — гексозофосфорные эфиры (глюкозо-1-фосфат,
глюкозо-6-фосфат, глюкозо-1, 6-дифосфат, фруктозо-6-фосфат, фрук-
тозе-1, 6-дифосфат и др.).
Фосфатиды (или фосфолипиды) — глицериды, содержащие оста-
ток фосфорной кислоты и связанные с ним азотистые основания (хо-
лин, коламин), или глицерин, или инозитол. Общая формула фосфа-
тидов:
СИ, —OCORr
I
СН—ocor2
I
СН2—О—Р—он
где CORX и COR2 — остатки различных жирных кислот; В — остаток
азотистых оснований, или глицерина, или инозитола.
Фосфатиды, содержащие холин — (CH3)3N(OH)CH2CH2OH, назы-
ваются лецитинами, а содержащие коламин — CH2OHCH2NH2,
кефалинами. Кроме лецитинов и кефалинов в высших растениях
найдены: фосфатидил сер ин, фосфатидилглицерол, дифосфатидилгли-
цёрол и фосфатидилинозитол. Многие растительные фосфатиды со-
держат глюкозу, галактозу, пентозу.
Нуклеотиды — фосфорные эфиры нуклеозидов, в которых оста-
ток фосфорной кислоты присоединен сложноэфирной связью к 5-му,
3-му или 2-му атомам углерода рибозы или дезоксирибозы. Например,
255
формула аденозиндифосфорной кислоты (АДФ) имеет следующий
вид:
N=C—NH2
I I
НС C—N. ____________п____ О О
и и Чен | | и и
N—С—N^--------СН (СНОН)2 СНСН2—О—Р—О—Р—ОН.
I I
ОН он
Нуклеотиды могут быть получены путем мягкого химического
(кислотного или щелочного) или ферментативного гидролиза нуклеи-
новых кислот. Большое значение имеют ди- и трифосфорные
производственные нуклеозидов (АДФ и АТФ), которые содержат
в своем составе высокоэргические фосфорные связи. АДФ
и АТФ легко переходят друг в друга. Отщепление фосфата от АДФ
и'двух фосфатов от АТФ происходит легко при семиминутном гид-
ролизе в 1 и. растворе соляной кислоты при 100° С. Приростом
«семиминутного фосфора» пользуются для определения количества
АДФ и АТФ и чистоты их препаратов при отсутствии других легко
гидролизующихся фосфорорганических соединений.
Нуклеиновые кислоты — высокомолекулярные органические сое-
динения, полимеры, содержащие 15—16% азота и 8,5—9% фосфо-
ра. Они состоят из большого числа нуклеотидов, соединенных меж-
ду собой фосфорноэфирной связью. Каждый нуклеотид представляет
собой соединение пуринового или пиримидинового азотистого осно-
вания с углеводным компонентом (рибозой или дезоксирибозой) и
фосфорной кислотой. Последняя, образуя сложные эфиры с гидрок-
сильными группами 5-го атома углеводного компонента одного нук-
леотида и 3-го атома моносахарида другого нуклеотида, соединяет
нуклеотиды между собой в цепь молекулы полинуклеотида или
нуклеиновой кислоты.
Имеется два основных типа нуклеиновых кислот: рибонуклеино-
вые кислоты (РНК), содержащие аденин, гуанин, цитозин, урацил,
рибозу и фосфорную кислоту и дезоксирибонуклеиновые кислоты
(ДНК), содержащие дезоксирибозу вместо рибозы и тимин вместо
урацила. Нуклеиновые кислоты — нестойкие соединения: под воздей-
ствием кислот, щелочей, окислительных агентов, а также нагрева-
ния они распадаются, теряя свои природные биологические свойст-
ва и образуя нуклеотиды. Нуклеиновые кислоты легко образуют со-
ли и комплексы с ионами металлов и органическими основаниями, в
том числе и с белками. Нуклеиновые кислоты растворяются в щелоч-
ных растворах и в 10%-ном растворе хлористого натрия.
Фитин — кальциево-магниевая соль инозитфосфорной кислоты,
С6Нб (ОН2РО3)6. Широко распространен в растительном мире. В не-
которых органах растений большая часть фосфора сосредоточена
в фитине. Так, в зерне злаков и бобовых от 50 до 70 % всего фосфора
находится в фитине. Фитин в воде нерастворим, растворяется в
кислотах и выпадает в осадок при нейтрализации кислот.
.256
Липиды высших растений делятся на четыре основных группы:
жиры, воска, глицерофосфатиды (фосфатиды) и гликолипиды.
Жиры (нейтральные липиды) по своему химическому строению
представляют собой смесь сложных эфиров трсхатомиого спирта гли-
церина и высокомолекулярных жирных кислот и построены по типу:
СН2—OCORj
I
СН—ocor2
I
СН2—OCOR3,
где Rlt R2, R3 обозначают радикалы жирных кислот.
В состав жиров в основном входят следующие жирные кислоты:
насыщенные, т. е. не содержащие двойных связей — пальмитиновая
(С]5н 31СООН) и стеариновая (С17Н3бСООН), и ненасыщенные— олеи-
новая (С17Н33СООН), линолевая (С17Н31СООН), линоленовая
(С17Н29СООН). Глицериды гидролизуются щелочами с образованием
свободных жирных кислот и глицерина. Такой же гидролиз проис-
ходит и при влиянии фермента липазы, которая содержится в самих
жировых веществах.
Гликолипиды. К гликолипидам относятся липиды, в которых
глицерин соединен гликозидной связью с каким-нибудь сахаром.
Они являются основными компонентами зеленых растений. Наибо-
лее важными представителями гликолипидов являются мопогалак-
тозилдиглицерид и дигалактозплдпглицерид, имеющие следующее
общее строение:
CH2OCORi
I
CHOCOR,
СН2ОВ,
где Rj и R2 — остатки жирных кислот, обычно линоленовой кислоты;
В — остаток галактозы.
Все липиды в воде не растворяются, а растворяются в эфире, хло-
роформе, бензоле, сероуглероде, менее растворимы в петролейном
эфире и в кипящем спирте.
ОПРЕДЕЛЕНИЕ
НЕОРГАНИЧЕСКОГО И МАКРОЭРГИЧЕСКОГО ФОСФОРА,
ГЕКСОЗОФОСФАТОВ, ФОСФОЛИПИДОВ, НУКЛЕИНОВЫХ КИСЛОТ
И ФИТИНА В ОДНОЙ НАВЕСКЕ
Для количественного определения групп фосфорорганических
соединений применяют различные растворители и осадители, разде-
ляющие их. После сжигания органических веществ смесью хлорной
и серной кислот фосфор определяютколориметрическим методом в ви-
де молибденовой сини. В растениях находятся весьма активные фос-
фатазы, что может привести к значительному гидролизу фосфорных
17 ’6-2
257
эфиров. Поэтому необходимо быстро инактивировать фосфата-
зы обработкой исследуемого материала кипящим спиртом или горя-
чим водяным паром.
Сущность метода. После инактивации ферментов фосфаты,
фитин, гексозофосфаты, АДФ и АТФ извлекают из свежего раститель-
ного материала 1%-ной уксусной кислотой. АДФ и АТФ осаждают
активированным углем, а фосфаты и фитин — окисью цинка. Мак-
роэргический фосфор (АДФ и АТФ) определяют после 10-минут-
ного гидролиза 1 н. соляной кислотой при нагревании на водяной
бане. Фосфаты и фитин отделяются вместе с окисью цинка и раство-
ряются в серной кислоте. В полученном растворе фосфаты опреде-
ляют колориметрическим методом. Затем в новой порции раствора
фитин сжигают хлорной кислотой и суммарное содержание фосфатов
определяют также колориметрическим методом. Концентрацию фити-
на устанавливают по разности. После осаждения фитина и фосфатов
окисью цинка и отделения их фильтрованием в полученном растворе
определяют фосфор гсксозофосфатов, которые предварительно сжи-
гают хлорной кислотой. В остатке исследуемого растительного ма-
териала после извлечения растворимых в 1%-ной уксусной кислоте
веществ определяют фосфор фосфолипидов извлечением их смесью
этилового спирта и этилового эфира и последующим сжиганием хлор-
ной кислотой. Остаток после извлечения фосфолипидов используют
для определения фосфора нуклеиновых кислот. Для этого остаток
обрабатывают при нагревании 5 %-ной хлорной кислотой, которая
и растворяет нуклеиновые кислоты. После сжигания фосфор опре-
деляют колориметрическим методом.
Ход анализа. Взвешивают 1,56 г с точностью до 0,01 г све-
жего растительного материала и инактивируют ферменты кипящим
спиртом или горячим водяным паром. Для обработки паром навес-
ку переносят в бюкс, закрывают его и помещают в водяную баню
над кипящей водой; баню закрывают крышкой и продолжают обработку
в течение 5 мин. После этого бюкс вынимают и охлаждают на возду-
хе. Для обработки кипящим спиртом в пробирку диаметром 20 мм
наливают 8 мл 80%-ного этилового спирта, погружают пробирку
в кипящую воду и, как только спирт закипит, помещают в пробирку
грубо измельченную навеску и кипятят 2 мин, затем охлаждают.
Пробу, обработанную тем или иным способом, переносят в фар-
форовую ступку, добавляют 5 мл 1 %-ного раствора уксусной кисло-
ты, промывая ею бюкс или пробирку, и растирают до однородной
массы. Суспензию переносят в центрифужную пробирку, промывая
ступку 1%-ной уксусной кислотой 2 раза порциями по 5 мл. Содер-
жимое пробирки перемешивают стеклянной палочкой и настаивают
в течение 10 мин, периодически помешивая. Затем центрифугируют
и раствор сливают в мерную колбу или цилиндр емкостью 25 мл.
К остатку приливают 5 мл 1 %-ной уксусной кислоты, перемешивают
периодически в течение 5 мин, центрифугируют и раствор присоеди-
няют к первому. Остаток еще раз обрабатывают 5 мл 1 %-ного раст-
вора уксусной кислоты. Объем экстрактов доводят дистиллирован-
258
ной водой до 25 мл и перемешивают. Остаток сохраняют для опреде-
ления фосфолипидов и нуклеиновых кислот, а в растворе определяют
неорганические фосфаты, макроэргический <|)осфор, фитин и фосфор
гексозофосфорных эфиров.
16 мл раствора, что соответствует 1 г исследуемого вещества, по-
мещают в центрифужную пробирку, добавляют 0,1 г активированно-
го угля и взбалтывают 5 мин. При этом уголь адсорбирует АДФ и
АТФ, а в растворе остаются неорганический фосфат, фитин и фосфор-
ные эфиры сахаров. Уголь отделяют центрифугированием, а раствор
сливают в сухую колбочку, даже если наверху находится небольшое
количество угля. Осадок угля используют для определения макро-
эргического фосфора, а центрифугат — для определения неорганиче-
ского фосфора, фитина и фосфорных эфиров сахаров. Для этого отби-
рают 8 мл раствора, что соответствует 0,5 г вещества, выливают в
пробирку, добавляют 10 капель 10%-ной суспензии окиси цинка,
взбалтывают 3 мин, добавляют 0,2 мл 2 н. раствора NaOH, взбалты-
вают еще 2 мин и фильтруют через плотный фильтр (синяя лента)
диаметром 7 см. Осадок промывают 1%-ным раствором уксуснокис-
лого цинка 2 раза порциями по 4 мл, собирая фильтрат в фарфоровую
чашку. В осадке на окиси цинка находятся неорганические фосфаты
и фитин, а в растворе фосфорные эфиры сахаров.
Определение фосфора гексозофосфорных эфиров
К фильтрату в чашке добавляют 2 мл 5%-ного раствора хлор-
ной кислоты, 0,8 мл 10 н. серной кислоты и раствор выпаривают на
водяной бане до небольшого объема (0,5—1 мл). Затем чашку поме-
щают на плитку и выпаривают при слабом нагреве (при температуре
около 180° С) до тех пор, пока потемневший сначала раствор не ста-
нет светлым. После просветления раствора сжигание немедленно
прекращают, не допуская дальнейшего выпаривания. К охлажден-
ному раствору приливают 5 мл дистиллированной воды, помещают
чашку на кипящую водяную баню и нагревают 5 мин, затем охлаж-
дают, раствор переносят в мерный цилиндр на 10 мл, смывая остаток
водой, доводят объем водой до 8 мл и полученный раствор перелива-
ют в пробирку. К раствору прибавляют 2 мл железо-молибдатного
реактива, перемешивают и через 15 мин фотометрируют при 660 нм
в кювете с толщиной слоя 10 или 20 мм. Из данных измерений опти-
ческой плотности по калибровочной кривой находят концентрацию
фосфора и затем вычисляют содержание его в фосфорных эфирах са-
харов по формуле:
С1
где X — содержание фосфора (в мг на 100 г исследуемого веще-
ства); С — концентрация фосфора в колориметрируемом растворе
(в мкг/мл); 0,5 — навеска исследуемого вещества, отвечающая взято-
му количеству раствора; 10 — объем колориметрируемого раствора
(в мл); 0,1 — коэффициент перевода микрограммов в милиграммы
на 100 г вещества.
17* 259»
Определение неорганического фосфора
Осадок окиси цинка в пробирке и на фильтре, содержащий неор-
ганический фосфор и фитин, растворяют в Юли 1 н. раствора серной
кислоты, наливая ее в пробирку и пропуская несколько раз через
фильтр. Из полученного раствора набирают пипеткой две порции по
4 мл и переносят в две пробирки. В одной порции раствора определя-
ют неорганический фосфор. Для этого к раствору в первой пробирке
прибавляют 1 мл железо-молибдатного реактива, перемешивают и че-
рез 15 мин фотометрируют при 660 нм в кювете с толщиной слоя
10 мм. Из полученных данных находят по калибровочной кривой
концентрацию фосфора и вычисляют содержание его по формуле:
у-__ 0,1 10 5 С ___ ос г*
Л ~~ 4 0,5 ~
где X — содержание фосфора (в мг на 100 г исследуемого вещества).
Остальные обозначения ясны из хода определения и предыдущей
формулы.
Определение фосфора фитина
К 4 мл другой порции раствора, помещенного в широкую пробир-
ку и содержащего неорганический фосфат и фитин, прибавляют 1 мл
5%-ного раствора хлорной кислоты. Пробирку ставят на плитку
в наклонном положении, выпаривают и сжигают при слабом нагре-
ве (170—180° С) до просветления раствора. После охлаждения при-
ливают 3 мл дистиллированной воды. Пробирку помещают в кипящую
воду на 5 мин, охлаждают, переносят раствор в мерный цилиндр,
доводят объем водой до 4 мл и раствор выливают обратно в пробирку.
К раствору добавляют 1 мл железо-молибдатного реактива, переме-
шивают и через 15 мин фотометрируют при 660 нм в кювете с толщи-
ной слоя 10 мм. Из полученных данных находят концентрацию фос-
фора и вычисляют содержание суммы неорганического фосфора и
фосфора фитина по формуле:
у__ 0J 10 5 С _____п г р
4.0,5 -АО-С,
где Y — содержание суммы неорганического фосфора и фитина (в мг
на 100 г исследуемого вещества); С — концентрация фосфора в коло-
риметрируемом растворе (в мкг/мл).
Остальные обозначения такие же, как в предыдущих формулах.
Содержание фосфора фитина находят по разности:
W = Y — X,
где W — содержание фосфора фитина (в мг на 100 г вещества); Y —
суммарное содержание фосфора фитина и неорганического фосфора
(в мг на 100 г вещества); X — содержание неорганического фосфора
(в мг на 100 г вещества).
Для пересчета фосфора фитина на фитин, CeH6O24P6Ca5Mg, полу-
ченное количество фосфора умножают на коэффициент 4,688.
260
Определение макроэргического фосфора
Уголь, оставшийся в центрифужной пробирке и содержащий
АДФ и АТФ, промывают водой 2 раза порциями по 5 мл. Для этого
приливают в пробирку 5 мл дистиллированной воды, перемешивают,
центрифугируют и осторожно сливают жидкость с осадка. Чтобы
избежать потерь угля, наливают поверх воды перед центри-
фугированием 1 мл этилового спирта. После промывания в центри-
фужную пробирку с углем приливают 8 мл 1 п. раствора соляной
кислоты, перемешивают стеклянной палочкой и погружают пробир-
ку в кипящую воду на 10 мин, периодически помешивая содержимое.
Затем центрифугируют и раствор сливают в колбу емкостью 50—
100 мл, даже если сверху плавают частички угля, которые в
дальнейшем не мешают. Уголь промывают 1 раз дистиллированной
водой (5 мл), центрифугируют и промывную жидкость присоединяют
к раствору.
В растворе находится фосфорная кислота, образовавшаяся в
результате гидролиза пирофосфатных связей АДФ и АТФ. Раствор
нейтрализует 2 н. раствором едкого натра, прибавляя его с некоторым
недостатком: на 0,2 мл меньше, чем нужно для полной нейтрализа-
ции. Необходимое количество щелочи устанавливают отдельным
титрованием. Для этого в колбу для титрования набирают 8 мл 1 н.
раствора соляной кислоты, добавляют одну каплю раствора фенол-
фталеина и титруют из бюретки 2 и. раствором едкого натра до появ-
ления розовой окраски и стараясь пс псрстптровать. Полученный
результат уменьшают па 0,2 мл и этот объем щелочи прибавляют к
исследуемому раствору. После прибавления щелочи к раствору добав-
ляют 10 капель 10%-ной суспензии окиси цинка, колбу помещают на
плитку, нагревают раствор до кипения и кипятят 1 мин. Раствор еще
горячим фильтруют через плотный фильтр диаметром 7 см и осадок
в колбе и на фильтре промывают 1 раз 5 мл 1 %-ного раствора уксус-
нокислого цинка. Раствор выбрасывают, а осадок растворяют в колбе
и на фильтре в 8 мл 1 н. раствора серной кислоты, пропуская се не-
сколько раз через фильтр, пока растворится весь осадок.
Раствор собирают в сухую колбочку, набирают 4 мл, переносят
в сухую пробирку, добавляют 1 мл железо-молибдатного реактива,
перемешивают и через 15 мин фотометрируют при 660 нм в кювете
с толщиной слоя 10 мм. Из полученных измерений по калибровочной
кривой находят концентрацию фосфора и рассчитывают содержание
его в исследуемом веществе по формуле:
v_ 0,1 8 5 С г
Л- i 4
где X — содержание макроэргического фосфора (в мг на 100 г иссле-
дуемого вещества); С — концентрация фосфора в колориметрируе-
мом растворе (в мкг/мл); 8 — объем 1 н. раствора серной кислоты,
взятый для растворения окиси цинка (в мл)', 4 — объем раствора,
взятый для колориметрического определения фосфора (в мл)', 5 —
261
объем колориметрируемого раствора (в мл)\ 1 — навеска исследуе-
мого вещества, соответствующая раствору, взятому для извлечения
макроэргического фосфора (в а); 0,1 — коэффициент перевода мик-
рограммов в миллиграммы на 100 г вещества.
Определение фосфолипидов и гликолипидов
Из остатка исследуемого вещества после экстрагирования 1 %-ной
уксусной кислотой извлекают липиды. Для этого приливают 5 мл
ацетона, перемешивают стеклянной палочкой и настаивают 5 мин
при помешивании. Палочку споласкивают ацетоном, раствор с осад-
ком центрифугируют и раствор сливают в мерную колбу на 25 мл.
Осадок промывают ацетоном еще 1 раз. Затем остаток заливают 5 мл
смеси этилового спирта и этилового эфира (1 I), перемешивают в
течение 5 мин и центрифугируют. Остаток экстрагируют еще 2 раза
смесью спирта и эфира порциями по 5 мл. Все центрифугаты объеди-
няют в колбе, доводят объем до 25 мл и тщательно перемешивают. Из
полученного раствора набирают сухой пипеткой две порции по 2,5 мл
и помещают в фарфоровые чашки диаметром 6 см. Одна порция слу-
жит для определения суммы гликолипидов, а другая — для определе-
ния галактолипидов. Остальной раствор (20 м/z) переносят в колбу
Кьельдаля для определения фосфолипидов.
Определение фосфора фосфолипидов
Колбу Кьельдаля с раствором липидов ставят в теплую воду с
температурой 50—60° С и выпаривают эфир. Далее температуру во-
ды постепенно повышают до кипения и выпаривают содержимое кол-
бы почти досуха. К остатку приливают 2 мл 10 н. раствора серной
кислоты и 0,5 мл 50%-ной хлорной кислоты. Если есть хлорная
кислота другой концентрации, берут такое ее количество, чтобы во
взятом объеме находилось 0,25 г чистой кислоты. Колбу помещают
на плитку и содержимое сжигают при слабом нагреве (не вы-
ше 200° С) до просветления раствора. После сжигания и охлажде-
ния прибавляют 10 мл дистиллированной воды и помещают колбу
в кипящую воду на 5 мин для гидролиза пирофосфатов и удаления
свободного хлора. После охлаждения раствор переносят в мерный
цилиндр на 25 мл. споласкивая колбу водой. Объем раствора в
цилиндре доводят водой до 20 мл и перемешивают. Из этого раствора
набирают пипеткой 4 мл, выливают в сухую пробирку, добавляют
1 мл железо-молибдатного реактива, перемешивают и через 15 мин
измеряют оптическую плотность раствора при 660 нм в кювете с
толщиной слоя 10 мм. Результаты измерений сравнивают с калибро-
вочной кривой, находят концентрацию фосфора и вычисляют содер-
жание его по следующей формуле:
у _ 0,1 25 5 С _ 3,125 С
4 п п ’
262
где X — содержание фосфора в фосфолипидах (в мг на 100 г вещест-
ва); С — концентрация фосфора в колориметрируемом растворе (в
мкг/мл); 25 — объем экстракта (в мл); 4 — объем раствора, взятый
для колориметрического определения фосфора (в мл); 5 — объем
колориметрируемого раствора (в мл); п — навеска исследуемого ве-
щества (в г); 0,1 —коэффициент перевода микрограммов в милли-
граммы на 100 г вещества.
Определение суммы сахаров гликолипидов
Гликолипиды — липиды, в которых глицерин соединен гликозид-
ной связью с каким-либо сахаром: галактозой, трегалозой, араби-
нозой, глюкозой и др. Поэтому о количестве гликолипидов можно
судить по содержанию в них сахаров, которые освобождаются после
гидролиза. После щелочного гидролиза можно определить только
восстанавливающие сахара, а после последующего кислотного гид-
ролиза и дисахариды, получая сумму сахаров.
Для определения суммы сахаров в гликолипидах 2,5 мл исследуе-
мого раствора, находящегося в фарфоровой чашке, выпаривают на
водяной бане досуха. Остаток растворяют в 2 мл 0,1 н. раствора ед-
кого натра и раствор переливают в центрифужную пробирку, а чаш-
ку промывают 3 раза 0,1 н. раствором NaOH, прибавляя его по 1 мл
и сливая промывную жидкость в пробирку. Пробирку погружают в
кипящую воду и выдерживают 10 мин, затем добавляют 1 мл 1 н. рас-
твора соляной кислоты и продолжают нагревать еще 10 мин. Затем
охлаждают, прибавляют 1 мл 1 н. раствора едкого натра, 1 каплю
раствора фенолфталеина и добавляют по каплям 10%-ный раствор
сернокислого цинка до обесцвечивания окраски фенолфталеина.
Раствор переносят в мерный цилиндр, разбавляют дистиллированной
водой до 10 мл, переливают снова в центрифужную пробирку, центри-
фугируют и прозрачный раствор сливают в сухую пробирку. 3 мл
этого раствора выливают в пробирку, добавляют 1 мл меднощелоч-
ного реактива для колориметрического определения сахаров, пере-
мешивают, закрывают пробирку шариковой пробкой и помещают
в кипящую воду на 10 мин. После охлаждения добавляют 1 мл серно-
кислого раствора молибдата аммония, перемешивают и оставляют
на 5 мин. Затем энергично взбалтывают до удаления углекислоты и
фотометрируют при 660 нм в кювете с толщиной слоя 5 или 10 мм в
зависимости от интенсивности окраски. Раствором для сравнения
служит вода с добавлен и ем такого же количества реактивов, как при
анализе исследуемого раствора. Полученные результаты сравнивают
с калибровочной кривой, находят концентрацию сахаров в пересче-
те на галактозу и вычисляют их содержание в гликолипидах по
формуле:
у = 0,1 25 10 5 С = 16,67 С
Л — 2,5 3 п ~~ п
где X — содержание суммы сахаров в пересчете на галактозу в глико-
липидах (в мг на 100 г вещества); 10 — объем раствора, полученный
263
после гидролиза (в мл); 2,5 — объем первоначального раствора, взя-
тый для определения суммы сахаров гликолипидов; 3 — объем конеч-
ного раствора, взятый для колориметрического определения сахаров
(в мл).
Остальные обозначения такие же, как в предыдущей формуле.
Определение моносахаридов в гликолипидах
(галактозы)
2,5 мл исследуемого раствора, находящегося во второй чашке,
выпаривают на водяной бане досуха. Остаток переносят в центри-
фужную пробирку с помощью 5 мл 0,1 н. раствора едкого натра.
Пробирку погружают в кипящую воду на 10 мин для гидролиза ли-
пидов. Затем охлаждают, добавляют 2 мл дистиллированной воды,
1 каплю раствора фенолфталеина и по каплям 10%-ный раствор
сернокислого цинка до обесцвечивания фенолфталеина. Раствор пе-
реливают в мерный цилиндр на 10 мл, разбавляют водой до 10 мл и
выливают снова в центрифужную пробирку. Центрифугируют и про-
зрачный раствор сливают в сухую пробирку. 3 мл этого раствора пе-
реносят в чистую пробирку, прибавляют 1 мл меднощелочного реакти-
ва, перемешивают, закрывают пробирку шариковой пробкой и поме-
щают в кипящую воду на 10 мин. После охлаждения добавляют 1 мл
сернокислого раствора молибдата аммония, перемешивают и остав-
ляют на 5 мин. Затем энергично взбалтывают до удаления углекисло-
ты и фотометрируют при 660 нм в кювете с толщиной слоя 5 или
10 мм в зависимости от интенсивности окраски.Раствором для сравне-
ния служит вода (3 мл) и такое же количество реактивов, как в опыте.
Полученные результаты сравнивают с калибровочной кривой, нахо-
дят концентрацию сахаров в расчете на галактозу и вычисляют их
содержание в галактолипидах по формуле, применяющейся для
определения сахаров гликолипидов.
Количество дисахаридов (трегалозы и др.), связанных с липида-
ми, вычисляется по разности:
Т = С — М,
где Т — количество дисахаридов (в мг на 100 г вещества); С — ко-
личество суммы ди- и моносахаридов (в мг на 100 г вещества); М —
количество моносахаридов в расчете на галактозу (в мг на 100 г ве-
щества).
Определение фосфора нуклеиновых кислот
Остаток после извлечения липидов, находящийся в центрифуж-
ной пробирке, промывают еще 1 раз 5 мл 50%-ного этилового спирта
и центрифугируют. Раствор сливают, а к осадку приливают 10 мл
5%-ного раствора хлорной кислоты и перемешивают стеклянной
палочкой. Пробирку с раствором помещают в горячую воду, где
выдерживают 20 мин при температуре 70° С, периодически переме-
шивая стеклянной палочкой. После этого центрифугируют и раствор
264
сливают в мерную колбу на 25 мл. Остаток в пробирке снова зали-
вают 10 мл 5%-ной хлорной кислоты, настаивают 20 мин при 70° С
с периодическим помешиванием, центрифугируют и раствор присо-
единяют к основному раствору. Остаток промывают еще 1 раз 5 мл
5%-ной хлорной кислоты и нагревают при 70° С в течение 5 мин.
Объединенные экстракты доводят дистиллированной водой до 25 мл
и перемешивают. Набирают пипеткой 5 мл этого раствора, поме-
щают в небольшую колбу Кьельдаля и прибавляют 0,7 мл К) н. рас-
твора серной кислоты. Колбу с раствором ставят на плитку, раствор
выпаривают и сжигают до просветления при слабом нагреве. Раствор
охлаждают и приливают 4 мл воды. Колбу погружают в кипящую
воду, выдерживают 5 мин, затем охлаждают и содержимое переносят
в мерный цилиндр на 10 мл. Колбу промывают водой 3 раза порциями
по 1 мл, присоединяя промывные воды к раствору в цилиндре, и до-
водят объем этого раствора водой до 8 мл. После перемешивания
набирают пипеткой 4 мл, помещают в сухую пробирку, прибавляют
1 мл железо-молибдатного реактива, перемешивают и через 15 мин
фотометрируют при 660 нм в кювете с толщиной слоя 10 мм. По ка-
либровочной кривой находят концентрацию фосфора и вычисляют
содержание его в нуклеиновых кислотах по следующей формуле:
v 0,1 25 8 5 С 5 С
Л = ------г--Л------- — ------
5 4 п п
где X — содержание фосфора нуклеиновых кислот (в мг па 100 г ве-
щества).
Остальные обозначения такие же, как в предыдущих формулах.
Реактивы. Раствор уксуснокислого цинка (1%-ный). 10 г уксуснокислого
цинка Zn (СН3СОО)2 2Н2О х. ч. растворяют в дистиллированной воде, раз-
бавляют водой до 1 л и перемешивают.
Раствор хлорной кислоты (5%-ный). Хлорная кислота бывает различной кон-
центрации — от 30 до 70% НС1О4. Имеющуюся кислоту разбавляют дистиллиро-
ванной водой с таким расчетом, чтобы в растворе находилось 5 г основного веще-
ства (НС1О4) в 100 мл раствора.
Активированный уголь. Можно использовать активированный уголь марки
БАУ или других имеющихся марок с величиной частиц 0,5—1 мм. Активирован-
ный уголь всегда содержит фосфаты, которые должны быть удалены прежде, чем
применять уголь для адсорбции АДФ и АТФ, так как возможны большие ошибки
при определении макроэргического фосфора. Для очистки продажного активи-
рованного угля берут 50 г фракции 0,5—1 мм, помещают в стакан, приливают
200 мл 2 н. раствора соляной кислоты, стакан ставят на плитку, закрывают ча-
совым стеклом, нагревают раствор до кипения и кипятят 20 мин. Затем фильтруют
через воронку Бюхнера с отсасыванием и промывают дистиллированной водой
3 раза порциями по 50 мл. Уголь переносят снова в стакан, прибавляют 200 мл
2 н. соляной кислоты, кипятят 20 мин и снова фильтруют и промывают водой
3 раза порциями по 50 мл. Эту операцию в общей сложности повторяют 4 раза.
В конце уголь, находящийся на фильтре, промывают дистиллированной водой
до исчезновения кислой реакции (проба с метилоранжем). Промытый уголь пере-
носят на лист фильтровальной бумаги и высушивают на воздухе. Сохраняют
в стеклянной банке с притертой пробкой.
Уголь испытывают на содержание фосфора следующим образом: 0,5 г угля
помещают в пробирку, прибавляют 10 мл 1 н. раствора соляной кислоты, переме-
шивают, пробирку погружают в кипящую воду и выдерживают 10 мин. Затем
фильтруют, собирают фильтрат в фарфоровую чашку и выпаривают досуха. Если
265
остаток темный, добавляют 0,2 мл 5 н. раствора азотнокислого цинка, высуши-
вают и прокаливают на плитке. К сгоревшему остатку прибавляют 4 мл 1 н. рас-
твора серной кислоты и перемешивают. Сливают раствор в пробирку, прибавляют
1 мл железо-молибдатного реактива и через 15 мин наблюдают окраску раствора
и измеряют оптическую плотность при 660 нм. При этом не должно быть заметной
голубой окраски, которая приводила бы к ошибке при определении микроэрги-
ческого фосфора. Измерение оптической плотности при наличии слабой окраски
дает возможность вносить поправку при определении фосфора.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ РНК И ДНК
Сущность метода. В остатке растительного материала
после извлечения водорастворимых веществ и липидов, как это опи-
сано в предыдущем методе, находятся нуклеиновые кислоты в виде
нуклеопротеидов. Последние извлекают раствором хлористого нат-
рия и после очистки от нежелательных примесей в полученном рас-
творе определяют отдельно РНК и ДНК по содержанию в них угле-
водов: рибозы и дезоксирибозы.
Из раствора в хлористом натрии РНК и ДНК в отдельных пробах
осаждают гидроокисью цинка и полученные осадки промывают ук-
суснокислым цинком. Осадок РНК растворяют в едком натре и об-
разовавшуюся рибозу определяют колориметрическим методом по
реакции с орцином, который реагирует с рибозой, образуя продукт,
окрашенный в сине-зеленый цвет. Осадок ДНК растворяют в уксус-
ной кислоте, затем осаждают и промывают этиловым спиртом. После
удаления спирта высушиванием осадок ДНК гидролизуют 5%-ной
хлорной кислотой. В полученном растворе содержание ДНК опре-
деляют по содержанию дезоксирибозы колориметрическим методом
с дифениламином, который реагирует с дезоксирибозой с образова-
нием окрашенного в синий цвет соединения (реакция Дише).
Ход анализа. В остатке после извлечения и определения
кислоторастворимых фосфорноорганических соединений и липидов
определяют отдельно содержание РНК и ДНК. Осадок промывают
еще 1 раз 50%-ным этиловым спиртом порцией 5—10 мл для удале-
ния эфира, центрифугируют и раствор сливают. К осадку приливают
10 мл 10%-ного раствора хлористого натрия, содержащего 0,5%
лимоннокислого натрия. Пробирку закрывают шариковой пробкой с
палочкой, погружают в кипящую воду и настаивают 20 мин при пе-
риодическом перемешивании палочкой. Затем центрифугируют и рас-
твор сливают в мерную колбу на 25 мл. Остаток экстрагируют еще
2 раза, прибавляя по 8 мл раствора хлористого натрия и нагревая в
кипящей воде по 20 мин. Экстракты объединяют, доводят объем во-
дой до 25 мл и перемешивают.
Если в исследуемом материале определяют только РНК и ДНК,
то навеску (1—1,5 г) свежего растительного материала, как и в мето-
де определения фосфора нуклеиновых кислот, сначала обрабатывают
1%-ной уксусной кислотой, ацетоном и спиртово-эфирной смесью, а
потом извлекают РНК и ДНК 10%-ным раствором хлористого нат-
рия.
266
Определение РПК
6 мл раствора нуклеиновых кислот в хлористом натрии помещают
в центрифужную пробирку, добавляют 0,5 мл 1 и. раствора едкого
натра и 1 каплю раствора фенолфталеина. Затем прибавляют по кап-
лям 10%-ный раствор сернокислого цинка до обесцвечивания окрас-
ки индикатора, раствор перемешивают и через 5 мин центрифугируют
при 2000 об/мин. Жидкость сливают, а осадок, содержащий РНК,
промывают 1 раз Злы 1%-ного раствора уксуснокислого цинка. Пос-
ле центрифугирования и удаления раствора к осадку прибавляют
1 каплю насыщенного раствора хлористого железа, перемешивают
стеклянной палочкой, приливают 3 мл 70%-ного этилового спирта,
промывая при этом палочку. После периодического встряхивания в
течение 3 мин центрифугируют и раствор сливают, удаляя последние
капли прикосновением фильтровальной бумаги. К остатку в пробир-
ке приливают 1,5 мл 0,4 н. раствора едкого натра, содержащего 2%
хлористого натрия, и перемешивают стеклянной палочкой в течение
5 мин. Затем центрифугируют и раствор РНК сливают в сухую про-
бирку. К остатку приливают еще 1,5 мл 0,4 н. раствора NaOH, содер-
жащего NaCl, перемешивают той же палочкой в течение 5 мин, центри-
фугируют и раствор добавляют к первому. Полученный раствор РНК
перемешивают, набирают 1 мл, помещают в сухую пробирку, добав-
ляют 4 мл орцинового реактива, закрывают пробирку шариковой
пробкой и помещают в кипящую воду на 15 мин. Раствор охлаждают,
измеряют его объем, разбавляют до 5 мл дистиллированной водой и
фотометрируют при 610—660 нм в кювете с толщиной слоя 10 мм.
Раствором для сравнения служит орциновый реактив. Из получен-
ных данных находят по калибровочной кривой концентрацию РНК
в колориметрируемом растворе и вычисляют содержание ее в иссле-
дуемом веществе по формуле:
х = 0,1 25 3 5 С = 6,25 С
6 п п ’
где X — содержание РНК в исследуемом веществе (в мг на 100 а);
25 — объем исследуемого раствора РНК (в мл); 6— объем исследуе-
мого раствора, взятый для осаждения и определения РНК (в мл);
5 — объем колориметрируемого раствора (в мл); 3 — объем раствора
РНК, полученный после растворения ее в 0,4 н.растворе NaOH (в мл);
С — концентрация РНК в колориметрируемом растворе (в мкг/мл);
п — навеска исследуемого вещества (в г); 0,1 — коэффициент пере-
вода микрограммов в миллиграммы на 100 а вещества.
Определение ДНК
Для определения ДНК 10 мл раствора нуклеиновых кислот в
хлористом натрии помещают в центрифужную пробирку, прибавляют
1 мл 1 н. раствора едкого натра, одну каплю раствора фенолфталеи-
на и прибавляют по каплям при взбалтывании 10%-ный раствор
сернокислого цинка до обесцвечивания окраски индикатора.
267
Раствор перемешивают и оставляют на 5 мин. Затем центрифугируют
и раствор удаляют, а осадок промывают 1 раз 3 мл 1 %-ного раствора
уксуснокислого цинка. После центрифугирования и удаления рас-
твора к осадку, содержащему ДНК, приливают 0,4 мл 20%-ной
уксусной кислоты, перемешивают стеклянной палочкой до растворе-
ния осадка и прибавляют для осаждения ДНК 4 мл 95%-ного этило-
вого спирта, споласкивая при этом палочку. Раствор перемешивают
встряхиванием пробирки и оставляют на 5 мин для полноты осажде-
ния. После этого центрифугируют, раствор сливают, а осадок про-
мывают 1 раз 3 мл 70%-ного спирта. Осадок высушивают погруже-
нием пробирки в горячую воду, нагретую до 80—100° С. К сухому
остатку после охлаждения добавляют 2 мл 5%-ного раствора хлор-
ной кислоты и подвергают ДНК гидролизу, нагревая реакционную
смесь горячей водой с температурой 90° С в течение 15 мин. Затем
раствор охлаждают, прибавляют 0,1 мл раствора катализатора, 4 мл
дифениламинового реактива, измеряют объем раствора и разбавляют
5%-ной хлорной кислотой до 6 мл. Осадок отделяют центрифугиро-
ванием, прозрачный раствор сливают в сухую пробирку, которую
оставляют при температуре 30° С на 6 чили при комнатной темпера-
туре (18—20° С) на ночь. После этого интенсивность окраски раство-
ра измеряют при 600—610 нм в кювете с толщиной слоя 10 мм. Рас-
твором для сравнения служит вода с таким же количеством дифе-
ниламинового реактива, как в опыте. Результаты сравнивают с
калибровочной кривой и вычисляют содержание ДНК по формуле:
У _ 0,1 25 6 С _ 1,5 С
Л 10 п “ п
где X — содержание ДНК в исследуемом веществе (в мг на 100 а);
10 — объем исследуемого раствора, взятый для определения ДНК
(в мл)\ С — концентрация ДНК в колориметрируемом растворе
(в мкг!мл)\ 6 — объем колориметрируемого раствора (в мл).
Остальные обозначения такие же, как в приведенной выше фор-
муле для РНК.
Построение калибровочной кривой для
определения РНК. В сухие пробирки помещают 0,1, 0,2,
0,3, 0,4, 0,6, 0,8 и 1 мл стандартного раствора, содержащего 0,5 мг
РНК в 1 мл и доливают 0,4 н. paciвором NaOH до 1 мл. Затем при-
бавляют во все пробирки по 4 мл орцинового реактива, закрывают
пробирки шариковыми пробками и ставят в кипящую воду на 15 мин.
После этого растворы охлаждают, измеряют их объем, разбавляют
дистиллированной водой до 5 мл и фотометрируют при 610—660 нм
в кювете с толщиной слоя 10 мм. Раствором для сравнения служит
смесь (1 мл дистиллированной воды и 4 мл орцинового реактива). Из
полученных данных строят калибровочную кривую (рис. 39).
Построение калибровочной кривой для
определения ДНК. Для приготовления шкалы в сухие про-
бирки наливают 0,2, 0,4, 0,6, 0,8, 1,2, 1,6и2^стандартного раство-
ра, содержащего 0,24 мг ДНК в 1 мл раствора и разбавляют 5 % -ной
268
хлорной кислотой до 2 мл. В одну из пробирок (контрольную) вместо
стандартного раствора ДНК наливают 2 мл 5%-ной хлорной кис-
лоты. Затем во все пробирки прибавляют по 0,1 мл раствора катали-
затора и по 4 мл дифениламинового реактива, перемешивают и поме-
щают пробирки в воду с температурой 30° С на 6 ч. После этого изме-
Рис. 39. Калибровочная кривая
для РНК (светофильтр с макси-
мумом пропускания 610 нм,
кювета с толщиной измеряемого
слоя 10 мм).
Рис. 40. Калибровочные кривые
для ДНК (светофильтр с макси
мумом пропускания 610 нм,
кювета с толщиной измеряемого
слоя 10 мм):
1— продолжительное।ь реакции 6 ч;
2 — продолжительность реакции
24 ч при комнатной температуре.
ряют оптическую плотность окрашенного раствора при 600—610 нм
в кювете с толщиной слоя 10 мм, сравнивая с контрольным раство-
ром. Из полученных данных строят калибровочную кривую (рис. 40).
Реактивы. Стандартный раствор РНК. Взвешивают па аналитических
весах 0,0125 г очищенного сухого препарата рибонуклеиновой кислоты, раство-
ряют в 10 мл 0,4 н. NaOH, содержащего NaCl, переносят раствор в мерную колбу
на 25 мл, смывая остаток и разбавляя до метки тем же раствором едкого натра
и перемешивают. 1 мл полученного раствора содержит 0,5 мг РНК.
Вместо РНК можно приготовить стандартный раствор из рибозы. Для этого
взвешивают на аналитических весах 0,1000 г рибозы, растворяют в дистиллиро-
ванной воде в мерной колбе на 100 мл, доводят водой до метки и перемешивают.
Из этого раствора готовят рабочий раствор разбавлением в 10 раз водой. Рабочий
раствор содержит 0,1 мг рибозы в 1 мл раствора и соответствует 0,5 мг РНК.
При приготовлении шкалы берут такие же количества рабочего раствора рибозы,
как и стандартного раствора РНК.
Очистка РНК. 0,05 г продажного препарата РНК помещают в центрифужную
пробирку, добавляют 3 мл 1 %-ного раствора уксусной кислоты, перемешивают
стеклянной палочкой и центрифугируют. Раствор сливают, а остаток РНК еще
раз промывают 1%-ной уксусной кислотой и раствор удаляют как можно полнее.
К осадку приливают 4 мл 10%-ного раствора хлористого натрия, содержащего
цитрат натрия, пробирку помещают в горячую воду, нагретую до 80° С, и пере-
мешивают содержимое стеклянной палочкой до растворения осадка. Затем раствор
охлаждают погружением пробирки в холодную воду, прибавляют 0,2 мл 5%-ного
269
раствора хлорной кислоты, встряхивают и центрифугируют. Мутный раствор
сливают с плотного осадка в чистую центрифужную пробирку. К этому раствору
прибавляют 10 мл 95%-ного этилового спирта и перемешивают. Осадку РНК дают
осесть в течение 10 мин и затем центрифугируют. Раствор сливают, а осадок РНК
промывают 3 раза 70%-ным этиловым спиртом, прибавляя его по 3 мл, и четвер-
тый раз 95%-ным спиртом. Остаток высушивают погружением пробирки в горя-
чую воду, нагретую до 80° С. Сухой препарат переносят в пробирку, закрывают
пробкой и сохраняют в темном и прохладном месте.
Определение содержания РНК в стандартном растворе. 1 мл стандартного
раствора, содержащего 0,5 мг данного препарата РНК, помещают в пробирку
диаметром 20 мм, прибавляют 0,1 мл 50%-ной хлорной кислоты и 0,8 мл 10 н.
раствора серной кислоты. Пробирку ставят на плитку в наклонном положении
и выпаривают раствор при слабом нагреве до потемнения. Затем усиливают на-
грев fпочти до кипения и нагревают до просветления раствора. После этого
охлаждают, приливают 5 мл дистиллированной воды и нагревают на кипящей во-
дяной бане 5 мин. Далее охлаждают, доводят объем раствора водой до 8 мл, при-
бавляют 2 мл железо-молибдатного реактива, перемешивают и через 15 мин фо-
тометрируют при 660 нм в кювете с толщиной слоя 10 мм. Из полученных данных
по калибровочной кривой находят концентрацию фосфора в окрашенном растворе
и вычисляют количество рибонуклеиновой кислоты во взятом количестве стандарт-
ного раствора по следующей формуле:
где X — содержание РНК в 500 мкг препарата (в мкг)\ С — концентрация фос-
фора в колориметрируемом растворе (в мкг!мл)\ 10 — объем колориметрируемого
раствора (в мл)\ 9’—процентное содержание фосфора в чистой РНК.
Содержание чистой РНК в стандартных растворах вычисляют по формуле:
С X
С 500 ’
где С' — концентрация чистой РНК в стандартном растворе (в мкг!мл)\ С — кон-
центрация данного препарата РНК в растворе (в мкг!мл)\ X — содержание чистой
РНК в 500 мкг препарата (в мкг).
Стандартный раствор ДНК. Взвешивают на аналитических весах 0,0120 г
чистого препарата ДНК, переносят в пробирку, прибавляют 10 мл 5%-ного рас-
твора хлорной кислоты и нагревают в горячей воде при 90° С в течение 15 мин.
Раствор охлаждают, переносят в мерную колбу на 50 мл, разбавляют 5%-ной
хлорной кислотой до метки и перемешивают. 1 мл полученного раствора содер-
жит 0,24 мг ДНК.
Очистку продажного препарата ДНК проводят аналогично очистке РНК.
Определение содержания ДНК в стандартном растворе. 2 мл стандартного
раствора ДНК помещают в пробирку и прибавляют 0,8 мл 10 н. раствора серной
кислоты. Пробирку ставят на плитку в наклонном положении и слабо нагревают
до потемнения раствора, затем усиливают нагрев почти до кипения и продолжают
нагревать до просветления раствора. Раствор охлаждают, добавляют 5 мл воды,
пробирку погружают в кипящую воду и нагревают 5 мин. Раствору дают охла-
диться, затем переносят в мерный цилиндр на 10 мл, разбавляют водой до 8 мл
и помещают снова в пробирку. К этому раствору прибавляют 2 мл железо-молиб-
датного реактива, перемешивают и через 15 мин фотометрируют при 660 нм в кю-
вете с толщиной слоя 10 мм. Из полученных данных находят по калибровочной
кривой концентрацию фосфора в окрашенном растворе (С) и вычисляют содержа-
ние ДНК во взятом количестве стандартного раствора по следующей формуле:
X = 100 9 2° С = 108,5 С,
где X — содержание ДНК в 480 мкг препарата (в мкг)-, 9,22 — процентное содер-
жание фосфора в чистой ДНК.
270
Содержание чистой ДНК в стандартных растворах вычисляют по формуле:
г, _ С X
С “ 480 ’
где С' — концентрация чистой ДНК в стандартном растворе (в мкг/мл); С —
концентрация данного препарата ДНК в растворе (в мкг/мл); X — содержание
чистой ДНК в 480 мкг препарата (в мкг).
Раствор хлористого натрия с цитратом натрия (10%-ный). Отвешивают
50 г хлористого натрия ч. д. а. и 2,5 г трехзамещенного лимоннокислого натрия
Na3C6H6O7 5,5Н2О, растворяют в дистиллированной воде, разбавляют водой
до 500 мл и перемешивают.
Раствор едкого натра с хлористым натрием (—0,4 и.). 16 г едкого натра
и 20 г хлористого натрия х. ч. растворяют в дистиллированной воде, доводят объем
раствора водой до 1 л и перемешивают. Сохраняют в хорошо закрытой склянке.
Уксусная кислота (20%-ная). 200 мл ледяной уксусной кислоты растворяют
в дистиллированной воде, разбавляют водой до 1 л и перемешивают.
Дифениламиновый реактив на ДНК. 1,5 г дифениламина ч. д. а. растворяют
в 100 мл перегнанной ледяной уксусной кислоты и добавляют 0,5 мл серной кис-
лоты пл. 1,84. Раствор перемешивают и сохраняют в темной склянке в холодном
месте.
Раствор катализатора. К 5 мл 0,1%-ного раствора сернокислой меди при-
бавляют 0,2 мл 16%-ного раствора уксусного альдегида и перемешивают. Раствор
сохраняется не более недели. Для приготовления 16%-ного раствора уксусного
альдегида ампулу, в которой обычно находится 160 г альдегида, охлаждают в воде
со льдом. Охлажденную ампулу вскрывают и приливают содержимое к 600 мл
охлажденной льдом воды. Раствор разбавляют до 1 л водой и перемешивают.
Сохраняют в хорошо закрытой склянке в темпом прохладном месте. При отсут-
ствии уксусного альдегида его можно заменить формальдегидом. В этом случае
к 5 мл 0,1 %-ного раствора сернокислой меди добавляют 0,1 мл 40%-ного раствора
формальдегида и тщательно перемешивают.
Приготовление других реактивов, применяемых в этих методах, описано выше
(см. стр. 9, 17, 28, 32, 40, 43, 76, 83, 133, 138, 150, 196, 265).
ХРОМАТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ЖИРА
В МАСЛИЧНЫХ СЕМЕНАХ
Этот метод требует небольшого количества исследуемого материа-
ла: 10—20 мг, который содержит 2—11 мг жира, поэтому нужно
очень тщательно отбирать среднюю пробу. Его можно использовать
для определения жира в небольшой части семени с селекционной
целью.
Сущность метода. Раствор бихромата калия в серной
кислоте при температуре 100—125° С окисляет все углеводы и жиры
до угольной кислоты и воды:
СвН10Ов 4- 4К2Сг2О7 + 16H2SO4 = 6СО3 + 4Сг2 (SO4)3 +
+ 4K2SO4 + 21Н2О,
ЗС3Н5 (CO2C17H3i)3 + 157К2Сг2О7 + 628H2SO4 = 171СО2 +
+ 157Cr2 (SO4)3 + 157K2SO4 + 775Н2О.
Грамм-эквивалент крахмала, целлюлозы, гемицеллюлоз и дру-
гих углеводов с молекулой (СвН10Ов)х равен 6,75, дисахаридов —
7,13, моносахаридов — 7,51, жиров — около 2,8. Белки даже при
125Q С окисляются не полностью, а только на 70%, так как некоторые
271
аминокислоты (гликоколь, аланин) не окисляются бихроматом ка-
лия. Азот в процессе взаимодействия переходит в аммиачную форму
и связывается серной кислотой. Поэтому грамм-эквивалент белков
по величине приближается к грамм-эквиваленту углеводов и на-
ходится в пределах 6,5—6,7. В обезжиренном остатке семян маслич-
ных культур, кроме углеводов и белков, находятся еще фитин (око-
ло 2%), зола (около 5%) и небольшое количество других труднооки-
сляемых веществ, поэтому грамм-эквивалент обезжиренного остатка
несколько выше, чем углеводов и белков, и находится в пределах
7,55—7,85.
Из изложенного видно, что грамм-эквивалент жиров резко от-
личается от средней величины грамм-эквивалентов других веществ,
присутствующих в семенах (в 2,75 раза). Используя это обстоятель-
ство, можно, окисляя органические вещества бихроматом калия в
сернокислой среде, определить содержание жиров с погрешностью
±1%, если содержание их в семепах не ниже 15%. Реакцию
окисления проводят при температуре 125° С. Повышать температуру
выше этой величины не рекомендуется, так как выше 130° С идет
реакция саморазложения бихромата калия, сопровождающаяся вы-
делением кислорода. С повышением температуры скорость разло-
жения бихромата возрастает:
2К2Сг2О7 + 8H2SO4 -> 2K2SO4 + 2Cr2 (SO4)3 + 3O2 + 8H2O.
После сжигания органических веществ, которое заканчивается
через 15 мин, избыток бихромата калия определяют титрованием
солью Мора. Содержание жиров вычисляют по формуле, которая вы-
водится из системы уравнений:
X + Y = Н и 0>0077 + 0>0028 =
где X — содержание обезжиренного остатка в навеске (в г); У — со-
держание жиров в навеске (в г); Н — навеска окисляемого материа-
ла (в г); В — объем 1 н. раствора бихромата калия, израсходован-
ный на окисление взятой навески (в мл); 0,0028 — количество жи-
ров, окисляемых 1 мл 1 н. раствора бихромата калия (в г); 0,0077 —
количество обезжиренного остатка, окисляемого 1 мл 1 н. раствора
би хромата калия (в г).
Проверка этого метода на семенах подсолнечника, льна, конопли,
рыжика и сои показала, что результаты близки к величинам, полу-
ченным экстракционным методом с применением диэтилового эфира.
Погрешность ±1% абсолютных.
Ход анализа. Отвешивают на аналитических весах 20 мг
измельченных воздушно-сухих семян или 16мг ядер семян, помещают
в ступку, прибавляют 2 мл 0,5 н. раствора бихромата калия в серной
кислоте и растирают до растворения навески. Полученный раство.р"
переносят в пробирку диаметром 28—30 мм, смывая остаток 4 м i
72 %-ной серной кислоты, прибавляя ее порциями по 1 мл. В про-
бирку приливают пипеткой еще 10 мл 0,5 н. раствора бихромата ка-
272
лия в серной кислоте. Раствор перемешивают и оставляют на ночь
для осуществления гидролиза клетчатки и белка. На следующий
день пробирку встряхивают и погружают в раствор хлористого каль-
ция, нагретый до 125° С. После того как температура снова подни-
мется до 125° С, выдерживают раствор при этой температуре 15лшя,
регулируя нагрев так, чтобы отклонения температуры не превыша-
ли ±1° За время нагревания пробирку 2—3 раза встряхивают для
перемешивания раствора. Одновременно с исследуемым раствором
нагреванию подвергается и контрольный раствор, содержащий толь-
ко бихромат калия и серную кислоту в таком же количестве, как
в опыте.
После нагревания пробирки вынимают и погружают в теплую во-
ду, чтобы смыть хлористый кальций. Затем растворы охлаждают и
переливают в колбу для титрования, смывая остаток небольшим ко-
личеством воды. Остаток непрореагировавшего бихромата титруют
0,1 н. раствором соли Мора в присутствии 4—5 капель раствора фе-
нилантраниловой кислоты до перехода вишнево-красной окраски в
зеленую.
В исследуемом материале определяют также содержание влаги.
Для этого взвешивают в бюксе на аналитических весах 1 г исследуе-
мого материала и высушивают при 100° С до постоянного веса. Из
полученных данных вычисляют содержание жиров в семенах по
формуле:
= о>44 (а- /;)/< , <
Л п(1—0,01С) Э'’1,
где X — содержание жиров в сухом исследуемом материале (в %);
п — навеска исследуемого вещества (в г); а — объем раствора соли
Мора, затраченный на титрование контрольного раствора (в мл);
b — объем раствора соли Мора, затраченный при титровании остатка
бихромата калия после окисления навески (в мл); К — нормальность
раствора соли Мора; С — влажность исследуемого материала (в %).
Этим методом можно определить содержание жиров и после пред-
варительного извлечения их органическими растворителями. Резуль-
таты получаются точнее и можно исследовать семена с меньшим со-
держанием жиров. Для этого берут навеску исследуемого материа-
ла (обычно о^оло 25 мг), чтобы в ней находилось от 2 до 11 мг жира,
помещают в ступку и растирают с 2 мл ацетона. Растертую массу пе-
реносят с помощью 5 мл ацетона в центрифужную пробирку, закры-
вают ее пробкой и настаивают при взбалтывании 10 мин. Затем
центрифугируют, раствор сливают в пробирку с диаметром 28—30 мм,
а осадок обрабатывают 2 раза ацетоном порциями по 3 мл, настаи-
вают 5 мин и центрифугируют.
Пробирку, содержащую ацетоновые экстракты, погружают в го-
рячую воду с температурой 60—80° С и выпаривают до удаления
всего ацетона. К остатку, содержащему жир, добавляют 10 мл 0,5 н.
раствора бихромата калия в серной кислоте и пробирку погру-
жают в кипящую воду на 15 мин. После этого раствор охлаждают,
18 6-2 273
переносят в колбу для титрования и дальше поступают так, как это
изложено выше. Из полученных данных вычисляют содержание
жира в исследуемом материале по формуле:
v 0,28 (а — Ь) К
Л = -------------- ,
и
где X — содержание жира в исследуемом материале (в %); 0,28 —
количество жиров, окисляемое 1 мл 1 н. раствора бихромата калия,
умноженное на 100 для пересчета в проценты.
Остальные обозначения такие же, как в предыдущей формуле.
Если исследуемый материал содержит сахара, которые также изв-
лекаются ацетоном, то жиры экстрагируют хлороформом.
Реактивы. Раствор хлористого кальция для солевой бани. К 200 г кристалли-
ческой соли СаС12 6Н2О добавляют 22 мл дистиллированной воды и нагревают
до растворения. Температура кипения раствора должна быть не ниже 125° С.
Температуру кипения регулируют добавлением воды или выпариванием раствора.
Во время работы концентрация раствора возрастает и раствор кипит при более
высокой температуре. Для понижения температуры кипения концентрацию рас-
твора уменьшают добавлением воды.
Приготовление других реактивов, применяемых в данном методе, описано
ранее при изложении метода определения крахмала и лигнина (см. стр. 138,
152)
ЛИТЕРАТУРА
Кулаева О. Н., Попова Э. А. Количественное определение нуклеино-
вых кислот в листьях растений.— Физиол. раст., 1965, 12, 3, 558.
Нечаева Е. П. К методике определения нуклеиновых кислот в молодых
растениях.— Физиол. раст., 1966, 13, 5, 919.
Павлинова О. А., Афанасьева Г. П. Кислоторастворимые нуклео-
тиды и фосфорилированные сахара проводящих тканей сахарной свеклы.—
Физиол. раст., 1962, 9, 2, 133.
Яковенко Г. М., М и х н о А. И. Метод выделения и разделения по клас-
сам липидов листьев и хлоропластов растений.— Физиол. и биохим. культур-
ных растений, 1971, 3, 6, 651.
Bergkvist R. The Acid-soluble nucleotides of Wheat plant.— Acta Chemica
Scandinavica, 1956, 10, 8, 1303.
Burton К-A study of the conditions and mechanism of the Diphenyl amine reac-
tion for the collorimetric estimation of Deoxyribonucleic acid.— Bioch. J.
1956, 62, 2, 315.
De D eken-Grenson M., De Deken R. H. Elimination of substaces in-
tercering with nucleic acid estimation. Bioch. et Biophys. acta, 1959, 31,
1, 195.
Глава VIII
ФИЗИОЛОГИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЕ ИНТЕНСИВНОСТИ ФОТОСИНТЕЗА
При фотосинтезе под воздействием лучей видимого солнечного
спектра осуществляется реакция превращения углекислоты и воды
в богатые энергией органические вещества:
nCO2 + nH2O + (hv) -> (СН2О) п + пО2 — п! 12 ккал.
Это уравнение показывает только конечный результат. В дейст-
вительности процесс фотосинтеза состоит из многочисленных реак-
ций двух типов — световых и темновых. Первичные, световые, реак-
ции фотосинтеза — усвоение углекислоты, воды, солнечной энергии
и накопление ассимилятов являются ведущими процессами в созда-
нии урожая.
Для сознательного и рационального управления ходом накопле-
ния урожая необходимо обладать обширным экспериментальным
материалом, характеризующим фотосинтетическую деятельность рас-
тений в зависимости от внешних факторов и условий питания. Для
решения этих и многих других задач необходимо располагать раз-
личными методами учета интенсивности фотосинтеза в естественных
условиях, пригодных как для исследования отдельных листьев, так
и целых растений, а также посева в целом. Так как скорость хими-
ческой реакции можно характеризовать изменением концентрации
одного из реагирующих или образующихся веществ в единицу вре-
мени, в основе всех методов определения скорости фотосинтеза ле-
жит суммарное уравнение фотосинтеза. Теоретически можно контро-
лировать изменение количества любого вещества, входящего в
уравнение, но на практике это не всегда возможно. Так, учесть то
небольшое количество воды, которое усваивается в процессе фотосин-
теза, обычными методами невозможно вследствие большого содержа-
ния ее в растении и огромного расхода на транспирацию.
Распространение получили методы, основанные на учете коли-
чества выделяющегося кислорода, количества усваивающегося угле-
кислого газа и количества образующихся органических веществ. Так
как кислород и углекислый газ являются газами, методы их опреде-
ления называются газометрическими.
Наиболее точным и быстрым методом определения скорости фо-
тосинтеза является метод учета ассимилированного углекислого га-
за, так как на фоне небольшого содержания его в воздухе (около
18*
275
0,03% ио объему) легко можно обнаружить даже самое малое коли-
чество этого газа, использованное в фотосинтезе. По содержанию
углекислого газа также возможна непрерывная запись скорости про-
цесса фотосинтеза. При исследовании фотосинтеза необходимо учи-
тывать процесс дыхания растений, направленный в сторону про-
тивоположную фотосинтезу. В процессе дыхания поглощается кис-
лород, выделяется углекислый газ и большое количество энергии,
используемой для течения многочисленных физиологических и био-
химических процессов. Дыхание у растений происходит все время,
тогда как фотосинтез — только на свету. В то же время скорость фо-
тосинтеза значительно выше, чем дыхания. Во всех обычных методах
определения интенсивности фотосинтеза его учитывают как сумму
двух процессов — дыхания и фотосинтеза и называют наблюда-
емый фотосинтез. Для определения истинного фо-
тосинтеза необходимо установить также интенсивность дыха-
ния у помещенных в темноту листьев и затем внести поправку на
дыхание. Но такой метод определения истинного фотосинтеза не явля-
ется точным, так как скорость выделения углекислоты на свету не
соответствует темновому дыханию.
По приемам работы методы, основанные на учете количества асси-
милированного углекислого газа, делятся на две группы: определе-
ние интенсивности фотосинтеза в замкнутых системах; определение
интенсивности фотосинтеза в открытых системах.
В замкнутых системах листья или ветки растения помещают в
прозрачные камеры и выдерживают определенное время на свету,
затем анализируют оставшийся воздух и по уменьшению концентра-
ции углекислого газа судят о скорости фотосинтеза. В открытых сис-
темах через камеру с листом или растением непрерывно протягивают
естественный атмосферный воздух, который непрерывно или через
определенный промежуток времени анализируют на содержание
углекислого газа при входе в камеру и на выходе из камеры. По
уменьшению содержания углекислого газа в воздухе судят о ско-
рости фотосинтеза.
Замкнутые системы значительно хуже открытых систем, так как
наличие ограниченного количества воздуха создает ненормальные
условия для фотосинтеза вследствие перегрева листьев и быстрого
обеднения воздуха углекислым газом. Открытые проточные системы
больше всего приближаются к естественным условиям и тем больше,
чем быстрее ток воздуха через камеру. Ограничивающими факторами
скорости потока воздуха через камеру являются поглотители и ана-
лизаторы углекислого газа. Минимальная скорость потока воздуха
через камеру должна быть такой, чтобы не было запотевания камеры.
Эту скорость можно вычислить по следующей формуле:
х _ М 1667
Л “ В (100 — Т) ’
где X — минимальная скорость потока воздуха (в л!мин на 1 дм2
листа); М — транспирация (в г на 1 дм2 листа за 1 ч); В — абсолют-
276
ная влажность воздуха при температуре определения транспи ации
(в г/ж3); Т — относительная влажность воздуха во время определе-
/ о/\ лаап 1000 100 II
ния транспирации (в %); 1667=-----------коэффициент пересчета
транспирации в минуты, а кубометры в литры и устранения обозна-
чения в процентах.
Например, для сахарной свеклы в одной серии опытов при тем-
пературе воздуха 26° С величина транспирации в среднем равна
5 г/дм2 ч. Относительная влажность воздуха 55%. Абсолютная
влажность воздуха при 26° С, найденная в таблице, 24 г/мл Отсюда,
по приведенной формуле находят необходимую минимальную ско-
рость потока воздуха через камеру:
5 1667 _ _ л 2
24 (100 — 55) “ 7,7 л!мин дм •
Необходимую скорость тока воздуха через камеру также вы-
числяют учитывая максимально возможную интенсивность фотосин-
теза данного растения. Так, для сахарной свеклы максимальная ско-
рость ассимиляции углекислого газа равна около 48 мг/дм2 ч.
Чтобы обеднение воздуха углекислым газом не превышало 20%, нуж-
но в течение 1 ч через камеру пропустить 240 жгСО2 или около 480 л
воздуха. Поэтому скорость потока воздуха через камеру, в которой
находится лист площадью 1 дм2, равна 480/60 = 8 л/мин, что очень
близко к ранее вычисленной минимальной скорости. Для кукуру-
зы с более интенсивной ассимиляцией углекислого газа и скорость
потока должна быть выше. Так, при интенсивности фотосинтеза
60 мг/дм2* ч скорость потока воздуха должна быть не менее 10 л/мин
на 1 дм2 листа. Найденные величины минимальной скорости тока
воздуха через камеру (8—10 л/мин на 1 дм2), хотя и обеспечи-
вают нормальное течение фотосинтеза, еще недостаточны для пони-
жения температуры листа в камере до температуры листьев вне каме-
ры. Поэтому для предотвращения чрезмерного повышения темпера-
туры применяют другие способы: используют специальные камеры
с охлаждением, экраны из веществ, поглощающих инфракрасную
радиацию, и др.
При сравнительных исследованиях фотосинтеза газометриче-
ским методом в естественных условиях необходимо соблюдать сле-
дующие условия:
1. Измерение скорости фотосинтеза должно проводиться одновре-
менно во всех вариантах опыта и обязательно с параллельными опре-
делениями. Необходимо осуществлять максимально возможное чис-
ло параллельных определений и не менее двух.
2. При работе с листовыми камерами необходимо брать для иссле-
дования листья одного яруса и ориентировать их на солнце так,
чтобы освещенность всех камер была одинаковой.
3. Экспозиция должна быть достаточно длительной — не менее
30—60 мин, так как при постоянно меняющихся факторах внешней
среды короткие экспозиции — 1—3 мин не могут правильно отразить
277
фотосинтетическую деятельность растении по образованию и на-
коплению органических веществ. Обычно в первые минуты после
заключения листа в камеру происходит быстрое нарастание темпе-
ратуры и лист не успевает приспособиться к новым условиям. При
длительных же экспозициях постепенно устанавливается темпера-
турное равновесие, лист успевает адаптироваться и продолжает
нормально функционировать в новых условиях. С увеличением вре-
мени экспозиции возрастает количество ассимилированной углекис-
лоты и точность определения ее при использовании щелочных пог-
лотителей становится выше, поэтому даже при слабом фотосинтезе
можно достигнуть удовлетворительной точности, увеличив экспо-
зицию.
4. Во время определения интенсивности фотосинтеза необходимо
обеспечить достаточный ток воздуха через камеру (не менее 8— 10 л
в 1 мин на 1 дм2 площади листа), чтобы избежать углекислотного го-
лодания, запотевания камер и чрезмерного повышения температуры.
5. Во всех камерах необходимо во время определения измерять
температуру листьев, чтобы не допустить перегрева. Сравнительное
исследование фотосинтеза можно проводить при любых температу-
рах ниже 36°С. Очень важно, чтобы температура во всех камерах была
одинаковой и не превышала 36°С. Можно исключить перегрев листьев,
применяя специальные камеры или экраны, поглощающие инфра-
красную радиацию. В качестве последних можно использовать плос-
кие кюветы, наполненные 2,5%-ным раствором СиС12.
6. Определения интенсивности фотосинтеза должны сопровож-
даться измерением падающей радиации (с помощью фотоинтеграто-
ров или пиранометров), температуры и влажности воздуха (с помо-
щью психрометров).
ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ
ФОТОСИНТЕЗА В ЕСТЕСТВЕННЫХ УСЛОВИЯХ
С ПОМОЩЬЮ ПОЛЕВОГО ПРИБОРА (ПФС)
Сущность метода. При исследовании интенсивности
фотосинтеза поток воздуха, проходящий через камеру с листом с
определенной скоростью, пропускают через сосуд, содержащий титро-
ванный раствор гидроокиси бария с индикатором тимолфталеином.
Углекислый газ, который остается в воздухе после осуществления
фотосинтеза, поглощается раствором гидроокиси бария:
СО2 + Ва (ОН)2 = ВаСО3 + Н2О.
Одновременно через такое же количество раствора гидроокиси
бария пропускают с той же скоростью воздух, который не проходит
через камеру с листом. И воздух, участвующий в фотосинтезе и не
участвующий, пропускают до тех пор, пока почти вся гидроокись
бария будет нейтрализована углекислым газом. Чем меньше концен-
трация СО2 в воздухе, тем больше объем его, необходимый для нейт-
рализации раствора Ва (ОН)2. Поэтому для нейтрализации требует-
278
ся меньше воздуха, не участвующего в процессе фотосинтеза, и
больше воздуха, проходящего через камеру с листом. Измерив объем
воздуха в опыте с растением и в контрольном опыте, продолжитель-
ность фотосинтеза, площадь листа и объем рабочего раствора щаве-
левой кислоты, который расходуется на титрование такого же объе-
ма раствора Ва (ОН)2, как в по-
глотителе, рассчитывают интен-
сивность фотосинтеза.
Основная ошибка при нейтра-
лизации гидроокиси бария угле-
кислым газом воздуха связана
с концом нейтрализации, когда
концентрация щелочи сильно
снижена и углекислый газ погло-
щается не полностью. Для умень-
шения этой ошибки в качестве
индикатора применяется тимол-
фталеин, интервал перехода ко-
торого лежит в щелочной области
(9,4—10,6 pH). Даже при полном
обесцвечивании тимолфталеина
(pH 9,4) концентрация оставше-
гося количества гидроокиси
бария равна 2,5 10“5 н., что
еще обеспечивает полноту по-
глощения углекислоты. Чтобы ос-
таток неоттитрованного Ва (ОН)2
не вносил большой ошибки, ~
* ’ Рис. 41. Установка для определения
нужно пользоваться более креп- интенсивности фотосинтеза в полевых
кими растворами, превышающи- условиях (НФС),
ми не менее чем в 100 раз остав-
шуюся концентрацию. Если применяют 0,008 н. и 0,01 н. растворы,
ошибки будут от—0,25% до—0,31%, что вполне приемлемо при
определении содержания СО2в воздухе. При определении же интен-
сивности фотосинтеза эта ошибка не имеет существенного значения,
так как количество ассимилированной углекислоты определяется
по разности величин, имеющих одинаковую ошибку.
Для определения интенсивности фотосинтеза в полевых условиях
используют специальную установку, которая состоит из камер, спа-
ренных аспираторов и поглотителей углекислоты, смонтированных
на штативе. Общий вид установки показан на рис. 41. На одном
штативе монтируется четыре аспиратора. Такая группировка аспи-
раторов очень удобна: она дает возможность распределить приборы
по числу опытных участков. При этом исследование фотосинтеза про-
водят одновременно по всем вариантам опыта с необходимым коли-
чеством параллельных определений. Прибор приспособлен для по-
левых условий. Он не боится воды и может работать даже во время
дождя. Один человек может работать с 4—5 приборами. Этими же
279
приборами можно исследовать интенсивность дыхания, помещая
листья или растения в темные камеры. Можно также проводить одно-
временное определение интенсивности фотосинтеза и транспирации,
если воздух, прошедший через камеру с листом, сначала пропустить
через осушитель, а затем через поглотитель углекислоты. Таким пу-
тем можно изучить взаимосвязь фотосинтеза и транспирации. Вес
прибора около 18 кг.
Камеры. В полевых условиях наиболее удобны камеры-при-
щепки. В установке могут применяться камеры двух типов: камеру-
прищепку закрытого типа на 10 см2 исследуемой площади листа
Рис. 42. Камера-прищепка закрытого типа на 10 см2 (а) и камера-
прищепка полуоткрытого типа на 10 см2 (б).
(рис. 42, а) изготовляют из органического стекла толщиной 6 мм и
состоит она из двух одинаковых рамок, соединенных между собой с
помощью двух дюралиевых вилок и пружины. Стороны рамок, приле-
гающие к листу, обклеивают поролоном или пористой резиной. В двух
противоположных стенках каждой рамки имеется по два каналь-
ных отверстия для входа и выхода воздуха. От этих каналов внутрь
камер отходят по 3 малых отверстия с диаметром 2 мм. К входным
и выходным отверстиям прикреплены штуцера длиной 25 мм и
диаметром 6 мм, которые с помощью тройника и резиновой трубки
соединяются между собой попарно. Таким образом верхнее отделе-
ние камеры соединяется с нижним. Штуцера выходят в сторону руч-
ки крепления камеры, что наиболее удобно. Верхнюю и нижнюю на-
280
ружные стороны рамок обклеивают целлофаном или ацетобутират-
целлюлозной пленкой.
Камеру-прищепку полуоткрытого т и и а (рис. 42, б) так-
же изготавливают из органического стекла, рамки делают двойными
по площади. Одну половину такой рамки покрывают пленкой, а дру-
гую половину оставляют открытой. Против закрытой части верхней
рамки находится открытая часть
-и— нижней рамки и наоборот. Каналы
420
Рис. 43. Поглотители углекислоты Рис. 44. Аспиратор емкостью G л.
из воздуха (стеклянный фильтр):
“ “«а и отверстия находятся только в
раствора щелочи; о — поглотитель на
100 мл рабочего раствора щелочи. ЗЗКрЫТЫХ ЧЗСТЯХ КамерЫ. При
накладывании камеры на лист
он закрывается только с одной стороны, а другая остается откры-
той, что дает возможность листу нормально транспирировать.
Поэтому температура исследуемого листа мало отличается от темпе'
ратуры листьев вне камеры. Фотосинтез исследуют только в закры-
той поверхности листа с обеих сторон его. Опыты показали, что
работа с этой камерой дает результаты значительно более близкие
к действительным, чем работа с другими камерами.
281
Поглотители углекислоты. В установке используются поглоти-
тели, снабженные стеклянными пористыми пластинками № 1 или
2. Поглотитель (рис. 43) представляет собой стеклянную трубку с
внутренним диаметром 40 мм для 100 мл рабочего раствора и 30 мм
для 50 мл раствора, которая имеет вверху шарообразное расширение,
оканчивающееся выходной трубкой 8—10 мм в диаметре. В нижней
Рис. 46. Водяной мано-
метр.
40
части широкой трубки впаяна пористая стеклянная пластинка (№ 1
или № 2), ниже которой трубка оттянута до диаметра 7—8 мм, и к
этому концу припаяна стеклянная трубочка диаметром 6—7 мм и
длиной 25—30 мм. Конец трубочки соединен с помощью резиновой
трубки с изогнутой стеклянной трубкой такого же диаметра и дли-
ной 200 мм. Последняя в верхней части имеет резиновое кольцо,
вышекотороготрубка прикреплена к поглотителю с помощью резинки.
Готовые поглотители вставлены в гнезда штатива (см. рис. 41) меж-
ду стойками прибора по два. При изготовлении поглотителей необ-
ходимо применять стекло, устойчивое к щелочам («нейтральное» или
№ 23). «Молибденовое» стекло не пригодно, так как содержит боль-
шое количество борной кислоты, которая легко извлекается щелочью.
Аспираторы. Для протягивания воздуха и одновременного изме-
рения его объема служат аспираторы. Емкость аспираторов в уста-
новке кустарного производства 5—6 л и изготовляются они из оцин-
282
кованного железа. В портативной установке емкость аспираторов 3 л
и изготовляются они из латуни или из нержавеющей стали. Каждый
аспиратор (рис. 44) снабжен водомерной трубкой, изготовленной из
оргстекла. На верхнюю трубку аспиратора привинчивается накид-
ная гайка с внутренним штуцером, на который одевается клапан
Бунзена, изготовленный из резиновой трубки. Клапан необходим,
чтобы предохранить поглотители от попадания воды из аспиратора
и для регулирования направления тока воздуха. Такой же клапан на-
ходится на трубке для выхода воздуха, которая расположена против
водомерной трубки вверху аспиратора. Аспираторы монтируются
по четыре на одном штативе. Штатив может быть металлическим или
деревянным. Деревянный штатив состоит из двух параллельных брус-
ков, имеющих по два Т-образных паза, в которых скользят такой же
формы металлические планки, прикрепленные к аспираторам (рис. 45).
Металлические планки в верхней части прикрепляются к тросу, пе-
рекинутому через блок. Длина троса должна быть такой, чтобы при
нахождении одного аспиратора вверху другой был внизу на расстоя-
нии 10—15 см от нижнего бруска. Металлический штатив состоит из
дюралюминиевых труб диаметром 30 мм. С двух сторон каждой вер-
тикальной трубки натягивают тросы, по которым скользят аспирато-
ры (см. рис. 41).
После изготовления и монтажа аспираторов их проверяют на гер-
метичность с помощью водного манометра, который имеет специаль-
ную конструкцию, пригодную для полевых условий (рис. 46). Мано-
метр состоит из двух стеклянных трубок разного диаметра. В верх-
ней части более широкой трубки выдувают шарик, выше которого
делают боковое отверстие и впаивают короткую стеклянную трубку.
Длинную более узкую трубку вставляют внутрь широкой и верхние
края их сваривают. Манометр прикрепляют к деревянному штативу,
на который наклеивают шкалу из миллиметровой бумаги. Манометр
заполняют водой до шарика и устанавливают уровень против нуле-
вого деления шкалы.
Для определения герметичности с боковой трубки верхней час-
ти аспиратора снимают клапан Бунзена и присоединяют к ней с по-
мощью резиновой трубки манометр. Затем через тройник отсасывают
из аспиратора воздух до тех пор, пока уровень воды во внутренней
трубке манометра опустится в самый низ. Трубку на тройнике зажи-
мают винтовым зажимом. При герметичности вода в манометре не
должна подниматься. Проверенные аспираторы наполняют водой и
калибруют. Аспираторы емкостью 6 л калибруют с точностью до
0,05 л, а емкостью 3 л — с точностью до 0,02 л. Калибрование прово-
дят путем измерения объема вытекающей воды мерными колбами и
цилиндрами. Деления и количество литров наносят краской, несмы-
вающейся водой. Общий объем аспиратора устанавливают путем
взвешивания или измерения объема всей воды, находящейся в аспи-
раторе.
283
Подготовка аппаратуры
и определение интенсивности фотосинтеза
Новые или бывшие долгое время в работе поглотители моют 2 н.
раствором соляной кислоты, затем водопроводной водой, наливая
ее через верхнее отверстие и выливая через нижнее. Под конец спо-
ласкивают несколько раз дис-
тиллированной водой. После
этого в поглотитель наливают
немного рабочего раствора гидро-
окиси бария с тимолфталеином,
окрашенного в синий цвет, и
энергично взбалтывают. Если
раствор обесцвечивается, его вы-
ливают и наливают новую пор-
цию. Промывание раствором
гидроокиси бария продолжают
Рис. 47. Приспособление
для выливания отработан-
ного раствора гидроокиси
бария без доступа угле-
кислого газа.
Рис. 48. Установка для
наливания рабочего рас-
твора щелочи в поглоти-
тели с помощью полуав-
томатической пипетки.
до тех пор, пока прекратится обесцвечивание. Затем раствор выли-
вают как можно полнее, высасывая остаток с помощью приспособле-
ния (рис. 47). Если пользоваться приспособлением, воздух, поступаю-
щий в поглотитель, предварительно проходит через натронную из-
весть для удаления углекислого газа. После промывания трубки на
концах поглотителя закрывают зажимами, отсоединяют и закрывают
стеклянными палочками, Перед опытом в поглотители наливают с
284
помощью полуавтоматической пипетки рабочий раствор (гидроокись
бария с тимолфталеином) (рис. 48). Работа проводится следующим
образом. После наполнения пипетки па 50 или 100 мл берут поглоти-
тель, левой рукой зажимают верхнюю трубку, вынимают стеклян-
ную палочку, вставляют кончик пинетки в поглотитель до расшире-
ния и, придерживая левой рукой за резиновую трубку так, чтобы
был выход воздуха из поглотителя, выливают раствор Ва (ОН)2 из
пипетки до метки, находящейся па нижней трубке пинетки.
Вынув пипетку из поглотителя, зажимают резиновую трубку и
закрывают ее стеклянной палочкой. Наполненные поглотители встав-
ляют в гнезда штатива, присоединяют верхним концом к аспираторам
и протягивают воздух до обесцвечивания. Затем поглотители отсое-
диняют, закрывают отверстия зажимами Мора и выливают отрабо-
танный раствор с помощью приспособления (см. рис. 47) следующим
образом. Содержимое поглотителя энергично взбалтывают, перево-
рачивают поглотитель и присоединяют верхним концом к стеклянной
трубке, предназначенной для слива отработанного раствора в колбу,
а нижним концом к трубке с натронной известью. Зажим открывают
и отсасывают раствор в колбу с помощью резиновой трубки, созда-
вая разрежение. После этого зажимают обе резиновые трубки погло-
тителя зажимами Мора, начиная снизу. Поглотитель отсоединяют,
закрывают трубки стеклянными палочками и снимают зажимы.
В таком виде поглотитель готов к следующему наполнению титрован-
ным раствором Ва (ОН)2. После внесения отмеренного объема раство-
ра гидроокиси бария (50 или 100 мл) поглотитель может быть исполь-
зован для определения интенсивности фотосинтеза. В дальнейшем
поглотитель может быть использован без промывания до 100 раз.
Приборы перевозят в разобранном виде. На месте работы их со-
бирают и устанавливают на стыке опытных участков так, чтобы один
человек мог работать, определяя интенсивность фотосинтеза на двух
или трех участках одновременно. Каждый вариант опыта исследуют
с помощью одного или двух приборов. После сборки всех приборов
и проверки их герметичности один из каждой пары аспираторов (ниж-
ний) заполняют водой через верхнее отверстие с помощью резинового
шланга. Отверстие герметически закрывают, завинчивая накидную
гайку. Закрывают винтовой зажим (рис. 49,/) и наполненный аспи-
ратор перетягивают вверх и закрепляют. Поглотители заполняют
рабочим раствором гидроокиси бария, вставляют в гнезда штатива
и присоединяют верхними концами к аспираторам, а нижними к ка-
мерам. При этом резиновая трубка на нижнем конце поглотителя
должна быть зажата зажимом Мора (2), чтобы раствор из поглоти-
теля не попал в трубку. Камеру укрепляют на металлическом стерж-
не возле исследуемого листа, ориентировав ее на солнце. В таком
виде установка готова к работе.
Когда все приборы подготовлены к работе, открывают немного
зажим (7) на трубке, соединяющей аспираторы, и дают вытекать воде,
чтобы создать разрежение в системе. При этом дополнительно про-
веряют герметичность прибора. Если прибор герметичен, пузырьки
285
воздуха, появившиеся в поглотителе, исчезают и вытекание воды
из аспиратора прекращается. После этого снимают зажим (2) и, регу-
лируя винтовым зажимом (/), устанавливают небольшую скорость
тока воздуха, наблюдая за пеной в поглотителе. Как только все при-
боры будут установлены приблизительно на одинаковую скорость 1
тока воздуха, одевают камеры на исследуемые листья и отмечают
Рис. 49. Схема определения интенсивности фотосинтеза с помо-
щью аспираторной установки (обозначения в тексте).
время начала опыта. Затем устанавливают максимальную скорость
тока воздуха через поглотители так, чтобы пена поднялась до шари-
ка. Когда вытечет вся вода из аспиратора и пена в поглотителе ося-
дет, немедленно перетягивают аспираторы, чтобы поток воздуха в
камере не прекращался. При этом нужно следить за тем, чтобы пена
во всех поглотителях перед перетягиванием спадала до одного уров-
ня. При этом благодаря клапанам (3) ток воздуха продолжается.
Опыт продолжают до тех пор, пока в одном из поглотителей (кро-
ме контрольного, который обесцвечивается раньше) раствор начинает
заметно бледнеть. Тогда камеры снимают с листьев в такой же после-
довательности, как одевали их вначале, и отмечают время окончания
286
опыта. После этого раствор в поглотителях дотитровывают углекис-
лым газом воздуха, протягивая его с уменьшенной скоростью до обес-
цвечивания раствора. При дотитровапии скорость протягиваемого
воздуха снижается настолько, чтобы в нижней части поглотителя на-
ходился слой раствора без пены. Это улучшает наблюдение окраски.
Как только раствор обесцветится, закрывают зажим (/) и измеряют
разрежение в приборе с помощью компенсационного манометра, опи-
санного ниже (см. рис. 55), а также объем воздуха в последнем аспи-
раторе по водомерной трубке. Параллельно с опытными проводят
контрольное определение углекислого газа в воздухе.
Количество прошедшего через поглотитель воздуха находят пу-
тем умножения объема меньшего аспиратора в паре на число полных
выливаний и добавления объема, измеренного с помощью водомерной
трубки при последнем неполном выливании. Вычисленные объемы
приводят к нормальным условиям по формуле:
у. = Vt г°
0 It ’
где Vo — объем воздуха, приведенный к нормальным условиям (в л);
Vt — объем протянутого через поглотитель воздуха (в л); /0 — дли-
на столбика воздуха в манометре при 760 мм рт. ст. и 4° С; lt —
длина столбика воздуха в манометре при данных условиях (в мм).
Затем рассчитывают интенсивность фотосинтеза по формуле:
ф _ 2640 а (с — Ь)
Ф “ b t р
где Ф — интенсивность фотосинтеза (в мг/дм2 ч СО2); а — объем
0,02 н. раствора щавелевой кислоты, затраченный при титровании та-
кого же объема рабочего раствора Ва(ОН)2, как в поглотителе (100 или
50 мл) (в мл)\с — объем воздуха, протянутый через поглотитель в опы-
те с растением, приведенный к нормальным условиям (в л); b — объем
воздуха, протянутый через поглотитель в контрольном опыте (без рас-
тения), приведенный к нормальным условиям (в л); t — продолжи-
тельность фотосинтеза (в мин); р — исследуемая площадь листа
(в см2); 2640 = 0,44 X 60 X 100 —коэффициент пересчета результата
в мг/дм2 ч (0,44 — титр 0,02 н. водного раствора углекислого га-
за; 60 — количество минут в часе; 100 — количество см2 в дм2).
После определения фотосинтеза из поглотителей удаляют весь
раствор, наполняют их новой порцией раствора и используют в тот
же день. Если нужно исследовать фотосинтез на следующий день,
отработанный раствор оставляют в поглотителях до следующего дня
и удаляют его только перед наполнением свежим раствором.
Если нужно знать содержание углекислоты в воздухе во время
опыта, концентрацию ее вычисляют изданных контрольного опыта
по формуле:
v 22 a N
А “ b ’
где X — содержание углекислоты в воздухе, при данных условиях
(в мг/л); а — объем титрованного раствора кислоты, затраченный
287
при титровании такого же объема рабочего раствора Ва (0Н)2, как
в поглотителе (в мл); N — нормальность титрованного раствора кис-
лоты; b — объем воздуха, прошедший через поглотитель в контроль-
ном опыте, приведенный к нормальным условиям (в л); 22 —
нормальный титр водного раствора углекислого газа (в мг).
Для определения содержания углекислого газа в воздухе, приве-
денном к нормальным условиям, необходимо привести к нормальным
условиям объем воздуха, поступившего в аспираторы за время опы-
та. Для этого с помощью компенсационного манометра измеряют при
данных условиях длину столбика воздуха (lt) и затем вычисляют
содержание углекислого газа по следующей формуле:
у__ 22 a N • It
У ГТ0 ’
где Y — содержание углекислого газа в воздухе при нормальных
условиях (в мг/л); lt — длина столбика воздуха в трубке манометра
при данной температуре п давлении (в мм); lQ — длина столбика воз-
духа в трубке манометра при 0° и давлении 760 мм рт. ст. (в мм).
Для получения содержания углекислого газа в воздухе (в % по
объему) пользуются формулой:
__ X (или У)
1,98 10 ’
где X (или Y) — содержание углекислого газа в воздухе при данных
(или нормальных) условиях (в мг/л); 1,98 — плотность углекислого
газа при нормальных условиях (в г/л); 0,1 —коэффициент пересчета
миллиграммов в граммы и проценты.
Одновременное определение
интенсивности фотосинтеза и транспирации
Для определения интенсивности фотосинтеза и транспирации
воздух, прошедший через камеру с листом, пропускают сначала че-
рез поглотитель паров воды, а затем уже через поглотитель углекис-
лого газа. Поглотитель паров воды состоит из двух трубок (рис. 50),
которые наполняют гранулированным безводным хлористым каль-
цием. Входное и выходное отверстия трубок в нерабочем состоянии
соединяют резиновой трубочкой необходимой длины. Комплекты,
состоящие из двух трубок, нумеруют и взвешивают. При присоеди-
нении осушающих трубок к листовой камере и поглотителю углекис-
лоты резиновую трубку отсоединяют от второй осушающей трубки
и присоединяют к камере, а к осушителю с другой стороны присоеди-
няют поглотитель углекислого газа. Поглотители, осушающие воз-
дух, необходимо присоединять как можно ближе к листовым каме-
рам, чтобы предупредить конденсацию паров воды в проводящей си-
стеме.
После присоединения к аспираторам поглотителей углекислоты,
поглотителей паров воды и листовых камер работа в основном про-
288
Рис. 50. Поглотитель
пиров воды из воздуха:
а — резервувр для соби-
рания образуемого рас-
твора СаСЬ.
водится так же, как и при определении интенсивности фотосинтеза.
В данном случае необходимо создавать максимально возможный ток
воздуха через камеру с листом, чтобы избежать запотевания. Для
увеличения продолжительности опыта пользуются более крепким
раствором (0,01 н.) гидроокиси бария. Здесь лучше использовать
камеры-прищепки на 10 см2 исследуемой площади листа, допускаю-
щие достаточный ток воздуха, исключающий запотевание. Одновре-
менно проводят контрольный опыт па содержа-
ние углекислоты и паров воды в воздухе.
После окончания опыта комплекты осушаю-
щих трубок отсоединяют от камер, затем закры-
вают резиновой трубкой и взвешивают с
точностью до 0,001 г. Находят разницу в весе
и по полученным данным вычисляют скорость
транспирации по следующей формуле:
т == 60 с)
b • t • р 9
где Т — транспирация (в Н2О г/дм2 ч)\ а —
прибавка веса трубок в опыте (в a); g— при-
бавка веса трубок в контрольном опыте (в г);
b — объем воздуха, прошедший в контрольном
опыте через осушитель, приведенный к нор-
мальным условиям (в л); с — объем воздуха,
прошедший в опыте через камеру и осушитель,
приведенный к нормальным условиям (в л);
t — продолжительность опыта (в мин); р —
исследуемая площадь листа (в дм2); 60 — ми-
нуты для пересчета в часы.
Интенсивность фотосинтеза в этом случае вычисляется так же,
как без определения скорости транспирации по формуле, приведен-
ной выше. Из полученных величин интенсивности фотосинтеза и
транспирации вычисляют транспирационный коэффициент фото-
синтеза по формуле:
ТКФ = Т ф000 ,
где ТКФ (транспирационный коэффициент фотосинтеза) — коли-
чество испаренных весовых единиц воды на одну весовую единицу
усвоенного углекислого газа при фотосинтезе; Т — транспирация
(в г!дм2 ч Н2О);Ф — интенсивность фотосинтеза (в мг/дм2 чСО2).
Определение интенсивности дыхания
При определении интенсивности дыхания нужно исследовать пло-
щадь листьев в 10—30 раз большую, чем при определении
интенсивности фотосинтеза, так как скорость дыхания значительно
меньше, чем скорость фотосинтеза. Перед исследованием процесса
19 6-2
289
дыхания листья заключают в кассеты из капроновых нитей и остав-
ляют в естественных условиях освещения, а поглотители углекисло-
ты с помощью резиновых трубок соединяют с темными футлярами
(рис. 51). После подготовки приборов таким же образом, как при
исследовании фотосинтеза, одевают на листья, находящиеся в кассе-
тах, темные футляры и отмечают время начала опыта. Как только,
хотя бы в одном из поглотителей, раствор начнет обесцвечиваться»
футляры снимают с листьев, отмечают время конца опыта и нейтра-
Рис. 51. Камера для определения интенсивности фотосинтеза и дыхания
листьев, не отделенных от растения:
а — рамка с капроновыми нитями и непрозрачной нижней частью; б — футляр
прозрачный для определения интенсивности фотосинтеза и непрозрачный для
определения интенсивности дыхания.
лизуют растворы в поглотителях воздухом до обесцвечивания. Здесь
пользуются 0,01 н. раствором Ва (ОН)2. При работе с большими по-
глотителями, содержащими 100 мл 0,01 н. раствора гидроокиси бария,
для исследования берут листья с площадью от 1 до 4 дм\ а при рабо-
те с малыми поглотителями, содержащими 50 мл раствора, берут
листья или растения с общей площадью от 1 до 2 дм2.
Если исследуются целые растения с общей площадью листьев вы-
ше 4 дм2, параллельно нужно создавать холостой ток воздуха, ско-
рость которого вычисляется по следующей формуле:
К = 0,5(0 — 3),
где V — скорость тока воздуха через камеру (в л/мин); D — иссле-
дуемая площадь листьев (в дм2).
Интенсивность дыхания вычисляют по той же формуле, что и ин-
тенсивность фотосинтеза, только объем воздуха, прошедшего в опы-
те через камеры с листьями, вычитают из объема, прошедшего в кон-
трольном опыте, и формула приобретает следующий вид:
у___ 2640 а (Ь — с)
Y b t р ’
290
где Y — интенсивность дыхания, выраженная интенсивностью выде-
ления СО2, мг/дм2 ч. Остальные обозначения такие же, как в фор-
муле для расчета интенсивности фотосинтеза.
Так как дыхание в сильной степени зависит от температуры, из-
мерение температуры листа в камере обязательно. 11ри исследовании
дыхания целых растений с применением холостого параллельного
тока воздуха количество последнего учитывают с помощью реометра,
а интенсивность дыхания в этом случае вычисляют по формуле:
У___ 2640 а (Ь — с) (с -j- и)
b t с р ’
где v — объем воздуха, прошедший через камеру мимо поглотителя
за время t (холостой ток) (в л). Остальные обозначения, как при
описании формул для расчета интенсивности фотосинтеза и интен-
сивности дыхания.
МНОГОКАНАЛЬНАЯ УСТАНОВКА
ДЛЯ ОПРЕДЕЛЕНИЯ ИНТЕНСИВНОСТИ ФОТОСИНТЕЗА
В СТАЦИОНАРНЫХ УСЛОВИЯХ
При наличии электрического тока можно заменить аспираторы
одним насосом и, измеряя реометрами скорость воздушного потока,
определить объем воздуха, затраченный на проведение эксперимен-
та. Здесь дается описание уста-
новки на 14 и 20 каналов изме-
рений, применяемой в Институте
физиологии растений АН УССР.
Для измерения скорости тока
воздуха и его объема применя-
ются реометры со шкалой до
1,2 л/мин. Обычно прохождение
пузырьков воздуха в поглотите-
лях вызывает колебания пока-
заний реометров, затрудняющие
точные отсчеты. Этот недостаток
может быть ликвидирован вве-
дениемв систему дополнительных
капилляров диаметром 0,8 мм,
которые выполняют роль демп-
феров, сглаживая пульсации.
Рис. 52. Схема одного канала много-
канальной установки:
1 — камера для листа; 2 — поглотитель
углекислоты; 3 — силикагелевый осуши-
тель воздуха; 4 — капилляр; б — реометр;
6 — игольчатый кран; 7 — коллектор;
8 — вентиль для закрывания холостого
хода воздуха.
Точность измерения при скорости тока воздуха 0,9—1,0 л!мин рав-
на ± 2%. Поглотители углекислоты такие же, как в полевой уста-
новке. Принципиальная схема одного канала показана на рис. 52.
Анализируемый воздух проходит через поглотитель углекислоты,
капилляр реометра, игольчатый кран для точной регулировки расхо-
да воздуха, дополнительный стабилизирующий капилляр, а затем
через трубку распределителя всасывается насосом.
Установка рассчитана на определение интенсивности фотосинтеза
листьев площадью 10—15 см2. Реометры, стабилизирующие капил-
19*
291
ляры и игольчатые краны, объединены в один общий блок и помеще-
ны в футляр (рис. 53). Для отсасывания воздуха может служить ком-
прессорновакуумный насос производительностью 20 л/мин или мас-
ляный насос производительностью 50 л/мин.
Ход работы. Перед началом работы, до подключения по-
глотителей к блоку реометров, включается насос и с помощью иголь-
чатого крана в реометрах устанавливаются уровни, соответствую-
Рис. 53. Многоканальная установка для определения интенсивности фото-
синтеза.
щие одинаковой скорости потока воздуха (0,9 или 1,0 л/мин).
Учитывая, что подключение поглотителей к реометрам вызывает не-
которое падение их показаний, при предварительной регулировке
реометров уровни следует устанавливать на 0,1—0,2 л!мин выше то-
го значения, при котором предполагается производить опыт (см.
рис. 52).
Поглотители, в которые предварительно наливают по 100 мл 0,02 н.
раствора NaOH, подключаются к блоку реометров верхней частью
и к камерам нижней частью. Трубки на входе поглотителей закрыва-
ют зажимами Мора и включают насос при открытом холостом ходе.
Затем надевают камеры-прищепки на исследуемые листья и снимают
зажимы на входе поглотителей. Началом определения фотосинтеза
является закрытие холостого хода (рис. 52, <?), вызывающее почти
мгновенное включение тока воздуха через систему. Сразу же
после включения тока воздуха производится точная регулировка
его расхода по показаниям реометров. Расход во всех каналах уста-
навливается одинаковым (0,9 или 1 л/мин). В ходе опыта нужно сле-
дить за показаниями реометров, при необходимости регулируя их.
292
Во время ра оты измеряют разрежение в каждом канале с помощью
компенсационного манометра, который включается между поглоти-
телем и реометром. Показания манометра служат для приведения
объема воздуха к нормальным условиям. После 30—50-минутной
экспозиции поток воздуха через поглотители прекращают, откры-
Рис. 54. Установка для титрования щелочи из поглотителей без доступа углекис-
лого газа (обозначения в тексте).
вая холостой ход и отмечая время окончания опыта. После этого ка-
меры снимают с листьев. На вход поглотителей одевают зажимы,
чтобы не вылился раствор, и насос выключают.
Поглотители отсоединяют и остаток щелочи титруют 0,02 н. раст-
вором щавелевой кислоты. Титровать нужно без доступа углекис-
лого газа воздуха. Для этого собирают специальную установку, к ко-
торой присоединяют поглотитель во время титрования (рис. 54). Ра-
бота ведется следующим образом. Бюретку наполняют титрованным
0,02 н. раствором щавелевой кислоты и мениск устанавливают на ну-
левом делении. Если в пипетке (/) находится раствор, его выпускают
до нижнего отростка (2), затем открывают кран (3) и оттягивают рас-
твор из поглотителя через трубку (4), наполняя нижнюю часть пипет-
ки до расширения, после чего закрывают кран (5). С помощью зажи-
ма (5) или крана раствор выпускают до отростка (2). Так делают 2
раза для того, чтобы вытеснить предыдущий раствор и заполнить
новым раствором всю нижнюю часть пипетки. После этого присоеди-
293
няют сухую коническую колбу, в которую предварительно добавляют
2 капли 1%-ного раствора фенолфталеина и 5 мл 5%-ного раствора
хлористого бария для осаждения карбонат-ионов. Колбу продувают
1 мин воздухом, не содержащим углекислоты, с помощью груши (6).
Воздух выходит через клапан Бунзена (7). Затем пипетку наполня-
ют исследуемым раствором до верхнего отверстия (6) и закрывают
кран (2). Раствор из пипетки с помощью зажима (5) выливают до
метки (9) в колбу и титруют кислотой до обесцвечивания окрас-
ки индикатора. Если в поглотителе находится 100 мл раствора
едкого натра, для титрования отбирают 80 мл, так как некоторая
часть раствора расходуется на промывание пипетки. Из получен-
ных данных вычисляют интенсивность фотосинтеза по следующей
формуле:
где Ф — интенсивность фотосинтеза (в мг/дм2 ч СО2); а — объем
0,02 и. раствора щавелевой кислоты, затраченный на титрование
80мл раствора NaOH в опытном поглотителе (в мл)', Ь — объем 0,02 н.
раствора щавелевой кислоты, затраченный на титрование 80 мл раст-
вора NaOH в контрольном поглотителе (в мл)\ t — продолжитель-
ность опыта (в мин)\ р — исследуемая площадь листа (в см2)', с —
объем 0,02 н. раствора щавелевой кислоты, затраченный на титрова-
ние 80 мл первоначального раствора NaOH (в мл)', Ка — поправка
на разрежение в опытном поглотителе; Кь — поправка на разреже-
оопп 22 60 100 100 0,02
ние в контрольном поглотителе; 3300 = -----------------------
отношение произведения г-экб углекислого газа на 60 мин, 100 см2,
100 мл раствора, находящегося в поглотителе и нормальности
раствора щавелевой кислоты к 80 мл раствора NaOH, взятого
для титрования.
Разрежение измеряют с помощью компенсационного манометра,
который присоединяется между поглотителем и реометром к отвод-
ной трубке перекрытой зажимом. После снятия зажима уровень во-
ды в трубках манометра уравнивают передвижением подвижной
трубки и фиксируют показание шкалы, соответствующее уровню во-
ды. Компенсационный манометр можно изготовить следующим обра-
зом. Запаянную с одного конца стеклянную трубку с внутренним
диаметром 6—7 мм и длиной 550 мм прикрепляют к вертикальному
бруску деревянного штатива и рядом с трубкой наклеивают бумаж-
ную миллиметровую шкалу или линейку, начиная от 350 до 520 мм,
ведя отсчет от верхнего запаянного конца трубки. В трубку вливают
немного дистиллированной воды так, чтобы высота ее столба была
около 100 мм, и соединяют резиновой трубкой с другой, наполовину
наполненной водой стеклянной трубкой длиной 320 мм и диаметром
8—10 мм. Последнюю прикрепляют к деревянному бруску штатива
рядом с первой трубкой так, чтобы она свободно передвигалась на
294
150 мм вверх и вниз. На открытый верхний конец трубки надевают
резиновую трубку длиной около 0,5 м, в конце которой находится
стеклянная трубочка длиной около 5 см (рис. 55).
Когда манометр готов, на шкале находят нулевую точку данного
манометра, т. е. объем, занимаемый находящимся в трубке воздухом
при 0° С и давлении 760 мм рт. ст. Для этого подвижную трубку
устанавливают так, чтобы вода в обеих труб-
ках была на одном уровне, и измеряют по
шкале длину столба воздуха в неподвижной
трубке в мм (Z/). Затем измеряют темпера-
туру окружающего воздуха и атмосферное
давление. Из полученных данных вычисля-
ют нулевую точку (/0) по следующей фор-
муле:
= (Р-Г) lt
° 760(1 + 0,00366 Р) ’
умножают полученный
где /0 — длина столба воздуха в трубке при
0° С и давлении 760 мм рт. ст. (в мм)\
lt — длина столба воздуха в трубке при тем-
пературе/0 и давлении Р (в мм)\ Р — атмос-
ферное давление (в мм рт. ст. в данное
время); W — упругость водяного пара в
воздухе при температуре t° (в мм рт. ст.)\
t° — температура воздуха (в ° С в данное
время).
Для приведения пропущенного через
поглотитель объема воздуха к нормальным
условиям его соединяют с компенсацион-
ным манометром, измеряют длину столба
воздуха в данных условиях и вычисляют
поправочный коэффициент, на который и
в опыте объем. Поправочный коэффициент вычисляют по формуле:
где /0 — показания манометра при нормальных условиях (нулевая
точка); lt — показания манометра при измерении давления в данных,
условиях
При работе с многоканальной установкой можно использовать
также титрование раствора гидроокиси бария углекислым газом
воздуха в присутствии тимолфталеина. Рабочая схема установки и
начало опыта такие же, как при объемном методе. Во время опыта
скорость тока воздуха через поглотитель должна быть одинаковой
(0,8—1 л/мин). Как только раствор хотя бы в одном из поглотите-
лей начнет бледнеть, прекращают ток воздуха через поглотители,
отмечают время окончания опыта и снимают камеры с листьев.
295
Затем к поглотителям на выходе поочередно подключают газосчет-
чик ГСБ-400 (точность измерения ± 0,5%) и с помощью мембранно-
го насоса, изготовляемого мастерскими института рентгенологии
в г. Харькове, протягивают воздух с небольшой скоростью до обес-
цвечивания раствора. Записывают объем воздуха, прошедший
через счетчик, отсоединяют его и присоединяют к другому погло-
тителю, с которым поступают так же, как с первым. Так последо-
вательно нейтрализуют растворы во всех поглотителях. Из по-
лученных данных вычисляют интенсивность фотосинтеза по сле-
дующей формуле:
2640 a[(v-t /<1 + Вх)-(ц t /<2 + В2)]
t p(v-t К2 + В2)
где Ф — интенсивность фотосинтеза (в мг/дм2 ч СО2); а — объем
0,02 н. раствора кислоты, затраченный при титровании 100 мл ра-
бочего раствора Ва (ОН)2 (в мл)\ р — исследуемая площадь листа
(в см2)\ t — продолжительность прохождения воздуха через камеру
с листом (в мин)-, v — скорость тока воздуха через камеру
с листом (в л/мин); Вг —дополнительный объем воздуха в опыте,
прошедший через счетчик (в л); В2 — дополнительный объем воз-
духа в контрольном опыте (в л); — поправка на разрежение в
опыте; Л2 — поправка на разрежение в контрольном опыте; 2640 —
22 60 100 0,02 — произведение мг-экв углекислого газа на
60 мин, 100 см2 и нормальности раствора щавелевой кислоты.
Реактивы. Рабочий раствор гидроокиси бария. Для больших поглотителей
емкостью 100 мл применяют 0,008 н. раствор, а для меньших поглотителей ем-
костью 50 мл — 0,01 н. раствор. Эти концентрации рассчитаны на 30—40 мин
работы. При необходимости уменьшить экспозицию можно приготовить более
разбавленный раствор, а при необходимости увеличить экспозицию можно при-
готовить более крепкий раствор.
Для приготовления 0,008 н. раствора взвешивают 14 а, а для 0,01 н. раство-
ра — 17 г кристаллического Ва (ОН)2 8Н2О, помещают в стакан, добавляют
5 г ВаС12 * 2Н2О, приливают 500 мл дистиллированной воды, нагревают при по-
мешивании до растворения веществ, доводят до кипения. Затем дают отстояться
осадку ВаСО3 и сливают полученный раствор в 10 л прокипяченной дистиллиро-
ванной воды. К этому раствору прибавляют 100 мл 0,1 %-ного спиртового раствора
тимолфталеина и 30 мл «-бутилового спирта, тщательно перемешивают, закры-
вают пробкой и оставляют на сутки для просветления раствора. Прозрачный
раствор сливают сифоном в бутыль, предназначенный для наполнения поглоти-
телей. Для установки титра отбирают 100 или 50 мл раствора и титруют 0,02 н.
щавелевой кислотой до обесцвечивания окраски индикатора.
Раствор едкого натра (0,02 н.). Сначала готовят свободную от карбонатов мас-
лянистую щелочь. Для этого 500 г NaOH ч. д. а. растворяют в 500 мл дистилли-
рованной воды, раствор переносят в высокий цилиндр, закрывают пробкой и ос-
тавляют до осаждения карбоната натрия. В растворе определяют концентрацию
щелочи титрованием кислотой. Из этого раствора готовят с помощью разбавления
приблизительно 0,02 н. раствор. Для определения концентрации полученного
раствора его титруют 0,02 н. раствором щавелевой кислоты в присутствии 5 мл
5%-ного раствора хлористого бария и индикатора фенолфталеина. Для приго-
товления 0,02 н. раствора щавелевой кислоты отвешивают на аналитических
весах 1,26 г Н2С2О4 2Н2О, растворяют в воде и разбавляют водой до 1 л.
296
ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ ФОТОСИНТЕЗА
ЦЕЛЫХ БОЛЬШИХ ЛИСТЬЕВ И ЦЕЛЫХ РАСТЕНИЙ
При исследовании дневного хода фотосинтеза возникает необ-
ходимость изучать не только отдельные участки листьев, ио и це-
лые листья и даже целые растения, заключая их в соответствующие
камеры, что необходимо для оценки фотосинтетической активности
отдельных листьев и целых растений в природных условиях, хотя
это и является более трудной задачей. В этом случае работа имеет
специфические особенности. Так как скорость тока воздуха через
поглотитель ограничена и допускает работу только с маленькими
листьями общей площадью до 40 см2, или с камерами-прищепками па
10—15 см2, при исследовании больших целых листьев и растений
такой метод анализа всего пропущенного через камеру воздуха не
годится. В этом случае необходимо применять более универсальный
метод: создавать два параллельных потока воздуха через камеру,
один из которых пропускать через поглотитель углекислого газа
и анализировать любым точным методом, а другой протягивать
мимо поглотителя (холостой ход).
Исследование целых листьев
Для определения интенсивности фотосинтеза целых листьев их
заключают в кассеты-рамки, имеющие форму исследуемого листа,
между двумя рядами капроновых питой, чтобы лист не прикасался
4 6
Рис. 56. Камера для листьев злаков:
а — рамка с капроновыми нитями, б — футляр.
к стенкам камеры и свободно омывался воздухом во время работы
(см. рис. 51, 56). В нижней стенке рамки по всей ее длине проходит
входное отверстие диаметром 5 мм, от которого выведены внутрь на
одинаковом расстоянии друг от друга 8—10 отверстий диаметром
297
2 мм. Па входе имеется штуцер из оргстекла. С противоположной
стороны штуцера находится держатель из дюраля, за который с по-
мощью лапки кассету прикрепляют к металлическому стержню, вот-
кнутому в землю возле исследуемого растения. В начале опыта на
рамку с листом одевают плотно прилегающий прозрачный футляр,
боковые стенки которого изготовлены из оргстекла толщиной
4—5 мм, а верхняя и нижняя — из ацетобутиратцеллюлозной плен-
ки или целлофана. В верхней части футляра для отсасывания воз-
духа имеется дюралевый штуцер,
на который одевают пропарафи-
нированную резиновую трубку
соответствующего диаметра.
Ход работы. Собирается
установка, состоящая из насоса
Рис. 57. Схема установки для опре-
деления интенсивности фотосинтеза
целых листьев:
1 — камера для листа; 2 — реометр; 3 —
аспираторная установка; 4 — зажим Гоф-
мана; 5 — игольчатый кран; 6 — зажим
для перекрывания холостого хода насоса;
7 — зажим Мора.
Рис. 58. Схема установки для опреде-
ления интенсивности фотосинтеза це-
лых листьев с использованием много-
канальной установки:
1 — камера для листа; 2 — поглотитель
углекислоты; 3 — силикагелевый осуши-
тель воздуха; 4 — капилляр; 5 — реометр;
6 — игольчатый кран; 7 — коллектор, 8 —
вентиль для закрывания холостого хода
насоса; 9 — основной поток воздуха через
камеру мимо поглотителя.
или пылесоса, реометров (ротаметров или газовых счетчиков), листо-
вых камер и приборов для определения углекислого газа (рис. 57, 58).
После того как смонтирована вся установка и выбраны для иссле-
дования листья, включают насос при открытом зажиме (рис. 57,
6) и, регулируя этим зажимом, устанавливают необходимую ско-
рость холостого потока воздуха. Эту скорость находят, учитывая
площадь исследуемого листа, которую определяют приблизительно
умножением длины листа на ширину и на поправочный коэффициент
(для сахарной свеклы — 0,66, для кукурузы — 0,76). Скорость
холостого потока воздуха через камеру вычисляют по формуле:
Х = А D — V,
где X — скорость холостого потока воздуха (в л/мин)', D — пло-
щадь исследуемого листа (в дм2)\ V — скорость потока воздуха
через поглотитель (в л/мин); А—минимально необходимая для
298
данной культуры скорость потока воздуха через камеру (в л/мин
на 1 дм2, для сахарной свеклы и кукурузы от 8 до 10 л/мин).
Обеспечив необходимую скорость холостого потока воздуха,
запускают аспираторную установку. Для этого ослабляют зажимы
(рис. 57, 4), снимают зажимы (рис. 57, 7), пускают воздушный по-
ток через поглотители, немедленно одевают футляры па листья,
находящиеся в кассетах, и замечают время начала опыта. Устанав-
ливают определенную, максимальную скорость тока воздуха через
поглотитель, поддерживая ее в течение всего опыта. Как только раст-
вор хотя бы в одном из поглотителей начнет бледнеть, снимают фут-
ляры с листьев и отмечают время окончания опыта. Выключают
насос и раствор в поглотителях дотитровывают атмосферным воз-
духом, просасывая его аспираторами. Затем измеряют объемы воз-
духа, пропущенные через поглотители, приводят их к нормальным
условиям умножением на поправочные коэффициенты и вычисляют
интенсивность фотосинтеза по формуле:
ф _ 2640 а (с — Ь) (с v t)
с • b t р ’
где Ф — интенсивность фотосинтеза (в мг/дм2 ч СО2); а — объем
0,02 н. раствора щавелевой кислоты, затраченный на 100 или 50 мл
рабочего раствора Ва (ОН)2 (в мл); с — объем воздуха, протянутый
через поглотитель в опыте (в л); Ь — объем воздуха, протянутый
через поглотитель в контрольном опыте (в л); v — скорость холос-
того тока воздуха через камеру с листом во время опыта (в л/мин);
t — продолжительность опыта (в мин); р — площадь исследуемого
листа (в см2); 2640 — 22 60 100 0,02 — произведение мг-экв
углекислого газа на 60 мин, 100 см2 и нормальности раствора ща-
велевой кислоты.
Исследование целых растений
Необходимость исследования фотосинтеза целого растения вы-
звана тем, что углекислый газ воздуха ассимилируют не только
листья, но также стебли и другие зеленые органы растений. Ин-
тенсивность фотосинтеза целых растений определяют по такой же
схеме, что и фотосинтез целых листьев, но вместо листовых при-
меняются камеры, заключающие всю надземную часть растения.
Камеры небольших размеров изготавливают из органического
стекла в виде цилиндров или домиков разных размеров в зависи-
мости от величины и формы исследуемых растений. Для высоких
растений, таких как кукуруза, камеры лучше всего составлять из
отдельных секций, состоящих из рамок, сваренных из металличе-
ских трубок, размером 50 X 50 X 50 см и устроенных так, что их
можно наращивать вверх до необходимой высоты (рис. 59). На гото-
вый каркас камеры накладывают ацетобутиратцеллюлозную пленку
и склеивают ее ацетоном.
Небольшая камера имеет вверху трубку, через которую отса-
сывается воздух. Для отсоса воздуха внутри большой камеры
299
помещают трубку диаметром 3—4 см с высотой, близкой к высоте ка-
меры. Она закрепляется в подставке и имеет выход внизу, а по всей
длине в камере имеются отверстия, через которые отсасывается ис-
следуемый воздух. Для перемешивания воздуха в камере в нее по-
мещают вентилятор, число оборотов которого регулируется авто-
Рис. 59. Камеры для растений:
а — камера для работы с небольшими растениями злаков; б — подставка к злако-
вой камере; в — камера для растений сахарной свеклы; г — камера для рас-
тений кукурузы.
трансформатором ЛАТР-1. Для получения правильных данных
надземную часть растения изолируют от почвы, выделяющей угле-
кислоту. Для этого применяют подставки, на которые устанавли-
ваются камеры. Для небольших камер подставки можно изготовить
из оргстекла с соответствующим вырезом для стебля. Узкие и длин-
ные камеры не обязательно ставить на землю. Их можно фикси-
ровать на расстоянии 8—10 см от почвы и брать с этой же высоты
воздух для контрольного определения содержания углекислоты.
Подставки для больших камер (рис. 60) состоят из металлической
рамки (а) с углублением для камеры и внутреннего деревянного
вкладыша (б), состоящего из двух половинок с вырезом в центре
для стебля. Деревянные доски пропитывают парафином при нагре-
вании или олифой с последующей окраской. В углубление рамки
300
помещают полоску из губчатой резины или поролона. Перед опы-
том металлическую рамку одевают на растение п закрепляют на
почве так, чтобы растение было в центре. Затем вырезают из поли-
этиленовой пленки круг с вырезом в центре для растения и разре-
зают его в одном месте. Полученное из пленки кольцо одевают через
разрез на растение и прижимают плотно к стеблю н почве. После
этого вставляют деревянные вкладыши и все щели амазывают пла-
стилином. В углу подставки помещают небольшой вентилятор, на-
правляя лопасти вверх на растение.
Ход работы. Установку для определения интенсивности фо-
тосинтеза целых растений собирают по такой же схеме, как при оп-
Рис. 60. Подставка к камере для растения:
а — металлическая рамка; б — деревянная доска.
ределении фотосинтеза целых листьев (см. рис. 57, 58). Но вместо
реометров в холостом токе воздуха используют разные расходо-
меры, рассчитанные на регистрацию необходимой скорости потока,
а вместо камер для листьев используют камеры для целых растений.
В качестве расходомера для воздушного потока до 100 л/мин лучше
всего использовать переносные газовые счетчики типа ГКФ-6,
которые при регистрации от 5 до 100 л/мин дают погрешность в
пределах ±1%. Для протягивания воздуха через камеры с
растениями можно использовать пылесосы. Количество воздуха,
просасываемого через камеру, можно регулировать до некоторой
степени подачей разной величины напряжения электрического тока
с помощью автотрансформатора. Например, используя пылесос
типа «Уралец» и изменяя напряжение от 40 до 220 в, можно полу-
чить ток воздуха от 170 880 л/мин. С увеличением напряжения па
1 в скорость тока воздуха возрастает на 4 л/мин, следовательно,
умножением данного напряжения на 4 можно грубо определить
скорость воздушного потока. Более точную регулировку скорости
производят с помощью зажимов (рис. 57, 5, 6).
После того как вся установка собрана, включают пылесос с
помощью автотрансформатора ЛАТР-1, поворачивая ручку пере-
ключателя и регулируя зажимами (6 и 5) до необходимой скорости
холостого тока воздуха. Эту скорость находят по формуле, приве-
денной при описании исследования фотосинтеза целого листа, под-
ставляя вместо площади одного листа суммарную площадь всех
листьев исследуемого растения. Затем на аспираторной установке
ослабляют зажим (4), снимают зажим (7) и устанавливают скорость
301
тока воздуха через поглотители (0,8—1,0 л/мин), замечая при этом
время начала опыта. Как только раствор в поглотителях начнет
бледнеть, пылесос и поглотители выключают, а камеру немедленно
снимают с растения, замечая время окончания опыта. Далее записы-
вают показания счетчиков и раствор в поглотителях дотитровывают
углекислым газом воздуха. Рассчитывают количество протянутого
воздуха и приводят объемы к нормальным условиям умножением
на соответствующие коэффициенты. Определяют скорость холостого
потока во время опыта делением израсходованного объема воздуха
на время отсчета газосчетчика. На основании полученных данных
вычисляют интенсивность фотосинтеза по формуле, используемой
при исследовании фотосинтеза у целых листьев, подставляя вместо
площади одного листа сумму площадей листьев всего растения.
Необходимо помнить, что продолжительность опыта — это время
протягивания воздуха через поглотители из камер с растениями.
При работе с небольшими растениями (площадь листьев до 8 дти2)
можно для измерения скорости холостого потока воздуха пользо-
ваться реометрами или ротаметрами, дающими погрешность ±3%.
Анализ воздуха в камере необходимо проводить не менее чем двумя
параллельными определениями. Работа с целыми растениями зна-
чительно сложнее, чем с ограниченной площадью листьев или целым
одним листом, поэтому количество одновременно проводимых опре-
делений ограничено многими обстоятельствами. Здесь изложен ход
работы с одним растением, который дает возможность по такому же
типу проводить работу с двумя и тремя растениями одновременно,
проводя сравнительные исследования.
Можно также при работе с целыми растениями вместо аспира-
торной установки для протягивания исследуемого воздуха через
поглотители воспользоваться насосом и реометрами по типу много-
канальной установки. В этом случае во всех каналах во время опыта
измеряют разрежение компенсационным манометром. Вычисление
интенсивности фотосинтеза проводят по следующей формуле:
26,4 Кг t + BJ-(V2 К2 / + В2)](Кз Кз + Ki)
V1 t (V2 K2 t + B2)
где Ф — интенсивность фотосинтеза целым растением (в мг/дм2 ч
СО2); — скорость прохождения воздуха через поглотитель в
опыте (в л/мин); У2 — скорость прохождения воздуха через погло-
титель в контроле (в л/мин); У3 — скорость прохождения воздуха
в холостом потоке (в л/мин); — поправка на разрежение в потоке
через поглотитель в опыте; /С2 — поправка на разрежение в потоке
через поглотитель в контроле; К3 — поправка на разрежение воз-
духа в холостом потоке; Вг — дополнительный объем воздуха в
опыте при дотитровывании раствора Ва (ОН)2;В2 —дополнительный
объем воздуха в контроле при дотитровывании раствора Ва (ОН)2;
t — продолжительность опыта (в мин); 26,4 = 22 60 0,02 —
произведение мг-экв углекислого газа на 60 мин и нормальность раст-
вора щавелевой кислоты; а — объем 0,02 н. раствора щавелевой
302
кислоты, израсходованный при титровании такого же объема рабо-
чего раствора Ва (ОН)2, как в поглотителе (в мл).
Для получения интенсивности фотосинтеза полученный резуль-
тат для целого растения делят па площадь всех листьев растения (в
дм2). Все резиновые трубки, используемые в установке, необходимо
пропарафинировать, что увеличивает срок их годности и уменьшает
адсорбцию газов. Для этого куски резиновых трубок закрывают с
обоих концов зажимами, чтобы внутрь не попал парафин, свора-
чивают кольцами и опускают в расплавленный парафин (80—90° С)
на 5—10 мин. Затем вынимают, дают парафину стечь и остаток его
вытирают фильтровальной бумагой.
КОЭФФИЦИЕНТ ИСПОЛЬЗОВАНИЯ
СОЛНЕЧНОЙ ЭНЕРГИИ ПРИ ФОТОСИНТЕЗЕ
При усвоении в процессе фотосинтеза 44 г двуокиси углерода и
восстановлении ее до углеводов с образованием 30 г продуктов фо-
тосинтеза связывается 112 ккал световой энергии. Следовательно,
соотношение между весом ассимилированной углекислоты и ко-
личеством усвоенной энергии (в кал/мг СО2) равно 1 2,55. Это соот-
ношение дает возможность определить процент использованной в
процессе фотосинтеза энергии солнечной радиации (КФ), если из-
вестны интенсивность фотосинтеза и количество падающей радиа-
ции. Вычисление проводят по формуле:
КФ _ ,
где КФ — коэффициент использования солнечной энергии при фото-
синтезе (в % от данной радиации, суммарной или ФАР, падающей
или поглощенной); Ф — интенсивность фотосинтеза (в мг/дм2 • ч
СО2); Q — падающая солнечная радиация (суммарная или ФАР,
2 55 100
падающая или поглощенная в кал/см2 мин\ 0,0425 = —gg----
коэффициент пересчета приходящей радиации на 1 дм2 в 1 ч и энер-
гии усвоения углекислоты в проценты, 2,55 — количество кало-
рий использованных при усвоении 1 мг СО2.
ОПРЕДЕЛЕНИЕ СТЕПЕНИ
ВЛИЯНИЯ РАЗЛИЧНЫХ ВЕЩЕСТВ НА ТЕЧЕНИЕ
ОСНОВНЫХ ФИЗИОЛОГИЧЕСКИХ ПРОЦЕССОВ
Для выяснения степени воздействия химических веществ (пи-
тательных элементов, стимуляторов роста, гербицидов, аптитран-
спирантов и др.) на интенсивность фотосинтеза, дыхания и тран-
спирации пользуются следующими методами работы в зависимости
от способа применения испытуемых веществ.
1. Исследуемое вещество в определенной концентрации нано-
сится на половину листа до черешка. Эта половина листа является
303
его опытной частью. Другую половину листа опрыскивают водой и
она служит контролем. При опрыскивании одной половины листа
другую часть листа закрывают бумагой для защиты от возможного
попадания наносимой жидкости. Такой способ исследования обычно
применяют в том случае, когда растение имеет большие листья.
Через некоторое время, в зависимости от поставленной задачи,
определяют интенсивность фотосинтеза и транспирации для обеих
половинок листа, пользуясь камерами-прищепками на 10 cjj2 и
ориентируя их на солнце. Развитие действия вещества во времени
можно проследить, периодически исследуя интенсивность фотосин-
теза и транспирации в течение длительного времени. Из полученных
данных можно вычислить следующие характеристики:
Показатель влияния данного вещества на интенсивность фото-
синтеза:
Пф-100(-^- 1).
где Пф — показатель отклонения величины интенсивности фотосин-
теза опытной части листа от контрольной, принятой за 100. Поло-
жительные величины показывают на сколько процентов возросла,
а отрицательные — на сколько процентов снизилась скорость фо-
тосинтеза по сравнению с контролем; Фо — интенсивность фотосин-
теза обработанной половины листа (в мг/дм2 ч СО2); Фк — ин-
тенсивность фотосинтеза контрольной половины листа (в мг/дм2 ч
СО2).
Показатель влияния данного вещества на транспирационный
коэффициент фотосинтеза:
п’-|0°ННг-*)•
где Пт — показатель отклонения в расходе воды на усвоение еди-
ницы углекислого газа (в % к контролю). Отрицательные величины
показывают снижение транспирационного коэффициента фотосин-
теза, а положительные — повышение; То — транспирация обра-
ботанной половины листа (в г/дм2 ч)\ Тк — транспирация кон-
трольной половины листа (в г/дм2 ч).
Чем выше интенсивность фотосинтеза и чем больше снижается
транспирационный коэффициент фотосинтеза опытной половины
листа по сравнению с контрольной, тем эффективнее действие анти-
транспиранта. Поэтому для более полной характеристики действия
антитранспиранта вычисляют коэффициент эффективности, включаю-
щий оба вышеприведенных показателя:
где Ка — показатель эффективности действия антитранспиранта,
выраженный (в % к контролю). Положительные величины пока-
зывают степень снижения транспирационного коэффициента фото-
синтеза и повышения интенсивности фотосинтеза. Отрицательные —
304
степень угнетения фотосинтеза и возрастания транспирационного
коэффициента фотосинтеза.
В случае испытания химических веществ па гербицидные свойст-
ва, связанные с угнетением фотосинтеза и транспирации, из резуль-
татов определения интенсивности этих процессов у опытной и конт-
рольной частей листа вычисляют показатели, характеризующие
эффективность действия данного вещества:
г,_ loofi-i),
где Гф — показатель влияния гербицида на интенсивность фото-
синтеза (в % к контролю). Положительные величины показывают
степень угнетения, отрицательные — степень стимуляции; Гт —
показатель влияния гербицида на интенсивность транспирации
(в % к контролю). Положительные величины показывают степень
угнетения транспирации, отрицательные — степень стимуляции.
Чем выше эти показатели, тем эффективнее действие вещества,
поэтому можно пользоваться суммарным коэффициентом, который
можно вычислить по формуле:
Кр = Гф+Гт = 100[2-(-g- + -А)],
где Кг — показатель эффективности угнетения гербицидов интен-
сивности фотосинтеза и транспирации (в % к контролю). Положи-
тельные величины показывают степень угнетающего действия и
могут иметь значения от 0 до 200. Отрицательные величины показы-
вают степень стимулирования.
2. Так как исследуемые растения и листья могут отличаться по
величине интенсивности фотосинтеза и транспирации, для получения
более точных результатов поступают следующим образом. Предва-
рительно пометив исследуемые листья или растения цветными нит-
ками, определяют у них интенсивность фотосинтеза и транспира-
ции. Затем опытные листья или растения обрабатывают испытуемым
веществом и через определенное время снова проводят определение
интенсивности фотосинтеза и транспирации у тех же листьев или
растений. Из полученных данных вычисляют показатели Пф, Пт,
Ка и Кг по формулам, учитывающим предварительные определения
интенсивности фотосинтеза и транспирации:
Пф= 100ык--й.
ф фк Ио )'
эо (-Т®.
\ А К
фр Ик
Фк Ио
К, = 100 [2
I \
Ка
Фк Вк и0______. \
Фо Во ик
То Фк Вк Ио
тк Фо Во Ик
Ик I Тр * вк
Ио Тк Вр
Ц
20 6-2
305
где IIK — интенсивность фотосинтеза контрольного листа или
растения (в мг/дм2 ч СО2, предварительное определение);
И() - интенсивность фотосинтеза опытного листа или растения (в
мг/дм2 ч СО2, предварительное определение); Вк — транспирация
контрольного листа или растения (в г/дм2 ч, предварительное
определение).; Во — транспирация опытного листа (в г/дм2 ч, пред-
варительное определение).
Остальные обозначения такие же, как в формулах для вычисле-
ния показателей Пф, Пт, Ка и Кг.
При нанесении испытуемых веществ на целые листья или расте-
ния, кроме интенсивности фотосинтеза и транспирации, можно ис-
следовать так же интенсивность дыхания, заключив исследуемые
листья в темные камеры. Вычисления в этом случае производят по
формуле для расчета Пф, где вместо данных по фотосинтезу под-
ставляются данные, характеризующие дыхание.
ОПРЕДЕЛЕНИЕ СКОРОСТИ ПРОДВИЖЕНИЯ
УГНЕТАЮЩЕГО ДЕЙСТВИЯ ГЕРБИЦИДОВ
В ТКАНЯХ РАСТЕНИЙ
Зная интенсивность фотосинтеза, можно определить скорость
распространения угнетающего действия гербицида в необработан-
ные ткани растения из обработанной его части. Для этого предва-
рительно у двух листьев одного и того же растения определяют
интенсивность фотосинтеза. Затем одну половину опытного листа
или некоторую часть его обрабатывают испытуемым веществом и
через определенное время (1, 2, 4 ч и т. д.) снова проводят опреде-
ление интенсивности фотосинтеза в контрольном листе и в обеих
частях опытного листа (обработанной и необработанной). На осно-
вании полученных данных вычисляют скорость распространения
угнетения фотосинтеза по следующей формуле:
Кп = 100 (-"° ~ М,
п Ио Фк — Ик Фо )
где Кп — показатель скорости проникновения гербицидного дейст-
вия в необработанную часть листа (в %) от величины угнетения фо-
тосинтеза в обработанной части листа за данное время, характери-
зуется величинами от 0 до 100; Ио, Ик — интенсивность фотосинтеза
в опытном и контрольном листе до обработки гербицидом (в мг/дм2
ч СО2); Фк — интенсивность фотосинтеза в контрольном листе
после обработки гербицидом опытного листа (в мг/дм2 ч СО2);
Фо — интенсивность фотосинтеза в опытной части листа после об-
работки ее гербицидом (в мг/дм2 ч СО2); Фн — интенсивность
фотосинтеза в необработанной части листа после обработки второй
части гербицидом (в мг/дм2 ч СО2).
306
ИСПОЛЬЗОВАНИЕ ОПТИКО-АКУСТИЧЕСКИХ ГАЗОАНАЛИЗАТОРОВ
ДЛЯ ОПРЕДЕЛЕНИЯ ИНТЕНСИВНОСТИ ФОТОСИНТЕЗА
И ДЫХАНИЯ РАСТЕНИИ
Оптические методы газового анализа принадлежат к числу
наиболее избирательных и чувствительных. Чрезвычайно важным
является оптико-акустический метод, избирательность которого в
противоположность обычным спектроскопическим методам дости-
гается без спектрального разложения, а за счет применения селек-
тивныхоптико-акустических приемни-
ков, использующих специфичность
инфракрасных спектров поглощения
веществ.
Сущность метода. Опти-
ко-акустический метод измерения кон-
центрации углекислого газа в воздухе
основан на поглощении им прерывис-
той инфракрасной радиации и преоб-
разовании энергии акустических коле-
баний в электрическую. Оптическая
схема измерения в отечественных
оптико-акустических газоанализато-
рах основана на дифференциальном
методе, сущность которого сводится к
определению разности значений изме-
ряемой величины и известной величи-
ны, служащей мерой сравнения.
Для изучения фотосинтеза расте-
ний в естественных условиях пригод-
Рис. 61. Оптическая схема огг-
тико-акустического газоанализа-
тора:
1 — стабилизатор; 2 блок пита-
ния; .7 мотор; 4 отражатель;
5 — из л чат(*Л1>. 6 — обтюратор;
7 — сравнительная камера; 8 —
рабочий камера; .9 газ; 10 —
электронный СПМО111ИПУ1ЦИЙ потен-
циометр; 11 — измерительная ка-
мера; 12 — усилитель с выпрями-
телем.
излучения являются две
ны оптико-акустические газоанализа-
торы типа ОА 2210 с дифференциаль-
ной оптической схемой (рис. 61).
Схема состоит из двух идентичных
оптических каналов, дифференциаль-
ного лучеприемника и двух источни-
ков инфракрасной радиации, излуче-
ние которых направляется с по-
мощью вогнутых зеркал в оптические
каналы. Источниками инфракрасного
накаленные спирали, выполненные из жаропрочного сплава. Потоки
радиации прерываются обтюратором с частотой 5 гц. Прерывание
происходит всегда в одной и той же фазе. В правом канале системы
находится рабочая камера, через которую непрерывно протекает
исследуемая газовая смесь. В левом канале расположена сравни-
тельная камера, которую обычно заполняют азотом или чистым
воздухом и герметически закрывают. Пройдя газовые камеры, оба
потока радиации попадают в лучеприемное устройство. Оно состоит
из двух камер-цилиндров, в которых поглощаются потоки радиации,
20*
307
и мерной камеры, разделенной упругой мембраной микрофона
на две половины, каждая из которых соединена с соответствую-
щим лучеприемным цилиндром. Лучеприемные цилиндры гермети-
чески закрыты пластинами, пропускающими инфракрасную радиа-
цию. Лучеприемное устройство заполняется газовой смесью изме-
ряемого компонента с азотом воздуха.
Поток инфракрасных лучей при прохождении через рабочую
камеру частично поглощается поступающей на анализ газовой
Рис. 62. Принципиальная измеритель-
ная схема потенциометра ЭПП-09.
происходит. Таким образом, в
лучеприемные цилиндры посту-
пают потоки излучений, разность
значений которых зависит от
концентрации измеряемого газа,
протекающего через рабочую
камеру. Под воздействием пре-
рывистого излучения в луче-
Рис. 63. Блок-схема установки для
определения интенсивности фотосин-
теза и дыхания оптико-акустическим
газоанализатором:
/ — камеры для листьев; 2 — газовый
переключатель; 3 — осушитель воздуха
с хлористым кальцием и фосфорным ангид-
ридом; 4 — газоанализатор; 5 — блок рео-
метров с игольчатыми кранами; 6 — иголь-
чатый кран; 7 — форвакуумный насос; 8 —
трубка с зажимом для подключения мано-
метра; 9 — водяной манометр; 10 — трубки
для записи нулевого показания газоана-
лизатора.
приемных цилиндрах возни-
кают периодические колебания температур заполняющего их газа и,
соответственно, его давления. Давление газа преобразуется конден-
саторным микрофоном в напряжение переменного тока, которое
усиливается и регистрируется. Шкала отградуирована в объемных
процентах углекислоты. Для записи хода фотосинтеза применяют
газоанализаторы со шкалой от 0 до 0,05% СО2 по объему. Для умень-
шения относительной погрешности измерений и для непосредствен-
ной записи разности концентраций СО2 в воздухе до камеры и по-
сле камеры с листом лучше всего работать по открытой дифференци-
альной схеме. Для этого газоанализатор перестраивают так, чтобы
можно было пропускать воздух и через сравнительную камеру.
Перестройка газоанализатора несложна. В сравнительной камере
устанавливают два штуцера точно так же, как это сделано в рабочей
камере. При работе через сравнительную камеру протягивают на-
ружный воздух, отбирая его при входе в листовую камеру, а через
рабочую камеру воздух, прошедший через камеру с листом.
При таком способе измерения чувствительность газоанализатора
308
можно повысить до 0,0001% СО2 по объему. Это достигается под-
ключением его к электронному потенциометру типа ЭПП-09 2М
со шкалой 0—1,5мв. Можно также использовать электронный потен-
циометр типа ЭПП-09 со шкалой 0—10 мв, если повысить его чувст-
вительность в пять раз путем уменьшения сопротивления шунта
Rn в мостовой схеме (рис. 62). Выбор сопротивления шунта произ-
водится эмпирически с применением магазина сопротивлений. Чувст-
вительность потенциометра проверяется с источником напряжения
ИРН-53. Все новые сопротивления должны быть изготовлены из
манганина и достаточно состарены. Старение сопротивлений до-
стигается трехкратным повторением 12-часового цикла нагревания в
термостате при температуре 120° С и охлаждением между цикла-
ми. На чувствительность прибора оказывают влияние присутст-
вующие в воздухе пары воды. Для того чтобы обеспечить возмож-
ность измерять разность концентраций углекислого газа в двух
потоках воздуха с погрешностью не более 0,0001 % по объему, необ-
ходимо концентрацию водяных паров в анализируемом воздухе
уменьшить не менее, чем в сто раз. Для этой цели одного хлористо-
го кальция недостаточно и нужно воздух, осушенный хлористым
кальцием, пропустить через поглотитель с фосфорным ангид-
ридом, обладающим большой осушающей способностью.
Блок-схема установки для определения интенсивности фотосин-
теза и дыхания с помощью оптико-акустического газоанализатора
показана на рис. 63. Анализируемый воздух, протягиваемый из
камер (/) с исследуемыми листьями и вне камер с помощью форва-
куумного насоса (7), проходит через осушающий блок (<?) с СаС12 и
Р2О5 и попадает в сравнительную п рабочую камеры газоанализа-
тора (4). Расход воздуха через камеры газоанализатора регулирует-
ся и измеряется с помощью блока реометров с игольчатыми венти-
лями (5). На блок-схеме в одном из каналов показан манометр (9)
и игольчатый вентиль (6) для градуировки газоанализатора.
С помощью газового переключателя (2) рабочую камеру газоанали-
затора можно автоматически или вручную через определенные про-
межутки времени переключать с одной камеры с листом на другую
и таким образом записывать поочередно данные, характеризующие
несколько листьев или растений. При этом должен быть обеспечен
непрерывный и равномерный ток воздуха через все листовые камеры
как во время записи, так и в промежутках между ними. Для умень-
шения инерции прибора емкость всей системы газоанализатора долж-
на быть минимальной. В качестве листовых камер используют каме-
ры-прищепки на 10 см*, а также камеры для целых листьев с допол-
нительным холостым потоком воздуха, скорость которого должна
быть не менее 6 л/дм2 мин.
Ход работы. Прибор включают за 2 ч до начала работы для
прогрева и проверяют герметичность установки водяным манометром.
Перед началом измерений запускают насос, исследуемые листья
помещают в листовые камеры и протягивают наружный воздух
в течение 30—60 мин, чтобы листья адаптировались в условиях
309
опыта. Если исследуется влияние интенсивности света, листья выдер-
живают в камерах около 1 ч при той же интенсивности света, при
которой будут проводиться измерения, чтобы избежать искажаю-
щего влияния индукционных переходных явлений. Во время записи
характеристик интенсивности фотосинтеза или дыхания растений
периодически (не реже одного раза в течение 1 ч записи) регистри-
руют нулевое положение прибора, так как оно постепенно изменяет-
ся во времени вследствие различных причин. Дрейф нуля велик у
непрогретого прибора и увеличивается с ухудшением качества осу-
шителей. Для записи нулевого положения через обе камеры газо-
анализатора пропускают наружный воздух, взятый из одного места
в пространстве. Чтобы уменьшить разброс показаний прибора,
нужно принимать меры по уменьшению колебаний концентрации
СО2 в точках отбора воздуха для анализа. Для этого исследуемые
растения нужно обдувать вентиляторами и избегать присутствия
людей вблизи исследуемых растений. Скорость тока воздуха в из-
мерительных камерах газоанализатора должна быть во время ра-
боты строго одинаковой и такой же, как при градуировке. Разре-
жение воздуха в обоих каналах должно быть также одинаковым,
что проверяется водяным манометром, который подключается к
трубкам (5) на входе в камеры газоанализатора.
Интенсивность фотосинтеза вычисляют по формуле:
S
где Ф — интенсивность фотосинтеза (в мг/дм2 ч СО2); L — объем-
ная скорость тока воздуха через камеру (в л/час)\ N — показание
прибора при записи фотосинтеза; No — показание прибора при
записи нулевого положения; К — переводной множитель прибора
(в мг/л СО2на 1 деление); S — исследуемая площадь листа в камере
(в дм2).
Градуировка прибора. Градуировку газоанализа-
тора нужно производить периодически, определяя переводной мно-
житель /С, который вычисляется по формуле:
К___ АС
ДА/ ’
где АС — разность концентрации СО2 (в мг/л)\ &N — линейная
разность показаний прибора.
Наиболее простой метод градуировки газоанализатора состоит
в следующем. Разность концентрации СО2 (АС) получают при про-
пускании через обе камеры газоанализатора воздуха с постоянным
содержанием СО2 из одного места в пространстве, создавая в одной
из камер определенное разрежение. Его получают с помощью иголь-
чатого вентиля (6), установленного перед осушительным блоком
одной из камер газоанализатора. Разрежение измеряют водяным
манометром (см. рис. 46). Воздух с постоянным содержанием угле-
кислого газа, необходимый для калибровки газоанализатора, по-
лучают нагнетанием его компрессором в резиновую оболочку. Со-
310
держание углекислого газа в этом воздухе определяют с помощью
аспираторной установки. Затем этот воздух используют для калиб-
ровки газоанализатора. Разность концентраций углекислого газа
вычисляют по формуле:
1,98 10 0,0735 Р С
“ (1 +0,00367 /) 760" "(1
где ДС — разность концентраций СО2 (в л/«’/л); Р разность дав-
ления в камерах газоанализатора (в мм вод. ст., измеряемая водя-
ным манометром); С — концентрация С()2 в воздухе, используе-
мом для калибровки (в % но объему), приведенная к нормальным
условиям; t — температура воздуха в ° С; 1,98 — плотность угле-
кислого газа при нормальных условиях (в г!л)\ 10 — коэффициент
пересчета объемных процентов в мл/л\ 0,0735 — коэффициент
пересчета мм вод. ст. в мм рт. ст.
ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ
ДЫХАНИЯ И ДЫХАТЕЛЬНОГО КОЭФФИЦИЕНТА
ОТДЕЛЬНЫХ ОРГАНОВ РАСТЕНИЯ
Процесс дыхания является важнейшим источником энергии
растений, в котором происходит окисление органических веществ
кислородом воздуха (аэробное дыхание) и выделение двуокиси уг-
лерода, что для углеводов выражается следующим суммарным
уравнением:
(СН2О) п + пО2 -> иСО2 пН2О + /г112 ккал.
Если процесс дыхания происходит в точном соответствии с при-
веденным уравнением, отношение объемов выделяемого углекисло-
го газа и поглощаемого кислорода, которое называют дыхательным
коэффициентом, равняется единице. Однако очень часто дыхатель-
ный коэффициент значительно отклоняется от единицы. Это свиде-
тельствует о многообразии биохимических процессов. Отклонения
могут наблюдаться вследствие неполного окисления углеводов или
окисления других соединений более или менее богатых кислородом,
а также анаэробного дыхания, например:
СбН12Об -> 2СН3СНо0Н + 2СО, + 25 ккал.
При потреблении большего количества кислорода на образова-
ние органических кислот или окисление жирных кислот коэффи-
циент меньше единицы, а при образовании жиров из углеводов или
дыхании за счет соединеййй богатых кислородом, например орга-
нических кислот, коэффициент выше единицы.
Чтобы определить интенсивность дыхания и дыхательного коэф-
фициента, необходимо учитывать не только количество выделяемо-
го углекислого газа, но и количество поглощаемого кислорода. Эти
определения можно осуществить только при наличии специальных
приборов или хорошо притертых вакуумных эксикаторов и бюреток
311
на 100 мл, что более доступно. В последнем случае прибор состоит
из вакуумного эксикатора емкостью от 2 до 5 л, в крышку которого
вставлена пробка с двумя стеклянными трубками. Одна трубка изо-
гнутая и короткая, а другая прямая, доходящая до дна эксикатора,
с резиновой трубочкой на конце (рис. 64). Снаружи на концы сте-
клянных трубок одевают резиновые трубочки длиной 5 см, которые
перекрываются винтовыми зажимами.
Для измерения объема поглощенного кислорода служит прибор,
состоящий из бюретки на 50 мл с оттянутыми концами и стеклянной
трубки в 1,2—1,3 раза большего диаметра, чем бюретка, и на 10—
15 см длиннее ее. Нижний конец трубки оттягивается до образова-
ния трубочки диаметром 6—7 мм. Этот конец стеклянной трубки и
нижний конец бюретки соединяют с помощью резиновой трубки,
длина которой равна длине бюретки. На верхний изогнутый конец
бюретки также одевают резиновую трубку со стеклянным наконеч-
ником. Бюретку и стеклянную трубку прикрепляют к деревянному
штативу так, чтобы они могли свободно двигаться вверх и вниз,
но не опускаться самопроизвольно. В трубку наливают 50 мл воды.
Общий вид и размеры прибора показаны па рис. 65. Выделяющаяся
во время опыта двуокись углерода поглощается раствором гидро-
окиси бария и определяется титрованием избытка щелочи кислотой.
Для сравнительного изучения процесса дыхания нужно иметь до-
статочное количество хорошо притертых одинаковых эксикаторов.
Ход работы. Отвешивают от 10 до 30 г свежих листьев
или других органов растений и помещают в сухой эксикатор на дыр?
чатый вкладыш (рис. 64). Эксикатор закрывают крышкой, смазан-
ной вазелином. После того как все пробы помещены в эксикаторы,
а эксикаторы плотно закрыты крышками, все эксикаторы с пробами
и один контрольный (без пробы) присоединяют к вакуумному насо-
су и продувают наружный воздух в течение 5—10 мин одновременно
через все эксикаторы. Затем эксикаторы отсоединяют, набирают в
полуавтоматическую пипетку (см. рис. 48) 100 мл 0,05 н. Ва (ОН)2,
вставляют кончик пипетки в резиновую трубочку длинной стеклян-
ной трубки (рис. 64, а), находящейся в эксикаторе, и выливают рас-
твор. Если навеска исследуемого материала меньше 25 г, берут 50 мл
раствора Ва (ОН)2. После этого резиновые трубочки (см. рис. 64,
а, б) перекрывают винтовыми зажимами, на эксикатор одевают тем-
ный колпак и замечают время начала определения интенсивности
дыхания в данной пробе. После того как во все эксикаторы, не ис-
ключая контрольный, прибавили раствор гидроокиси бария и за-
крыли темными колпаками, пробы оставляют на 4 ч, время от време-
ни покачивая эксикаторы для перемешивания растворов.
Через 4 ч после начала опыта с каждой из проб поступают сле-
дующим образом. Снимают темный колпак с эксикатора и измеряют
количество потребленного кислорода с помощью прибора (рис. 65).
Для этого бюретку фиксируют в верхнем положении, а трубку с
водой опускают до тех пор, пока уровень воды в бюретке опустится
и установится на 5 мл выше крайнего нижнего деления, которое
312
отмечают как начальное. После этого верхнюю часть бюретки при-
соединяют к резиновой трубке (б) эксикатора и открывают винтовой
зажим. Передвижением бюретки вниз, а трубки с водой вверх до-
биваются одинакового положения уровней воды в обеих трубках и
измеряют объем прибавившейся воды в бюретке. Этот объем равняет-
ся объему потребленного кислорода. Затем отмечают 'температуру
воздуха и атмосферное давление.
Так поступают с пробами во всех
эксикаторах, включая контрольный.
Затем определяют количество
выделившейся во время дыхания
двуокиси углерода. Для этого бе-
рут пипетку Мора на 50 мл (или
на 25 мл при 50 мл Ва (ОН)2,
вставляют в резиновую трубку (а)
Рис. 64. Эксикатор, приспособленный для
определения интенсивности дыхания:
Рис. 65. Прибор для из-
мерения объема потреб-
ленного кислорода при
дыхании:
а — уравнительная стеклян-
ная трубка; б — трубка для
присоединения к эксикатору;
в — бюретка.
а — трубка для наливания раствора гидрооки-
си бария и отбора пробы раствора после опы-
та; б — трубка для присоединения прибора,
измеряющего объем потребленного кислорода.
и открывают зажим. К трубке (б) присоединяют трубку с нат-
ронной известью, открывают второй зажим и всасывают раствор в
пипетку выше метки. Пипетку вынимают, лишний раствор удаляют,
а оставшийся точный объем выливают в сухую колбу для титрования
и плотно закрывают пробкой. Так поступают со всеми пробами.
Каждый раствор титруют 0,05 н. раствором щавелевой кислоты в
присутствии фенолфталеина, пользуясь приспособлением для пре-
дохранения от углекислого газа воздуха (см. рис. 54).
Из полученных данных выделяют следующие характеристики:
1. Интенсивность дыхания рассчитывают по количеству выделив-
шегося углекислого газа по формуле:
п 100 22 А К (а — Ь) 1111 А К (а — Ь)
Uc°2 “ 1,98 v п t
V п • t
313
где Dca\ — интенсивность дыхания (в мл выделившегося СО2 на
100 г исследуемого материала за 1 ч); А —объем 0,05 н. раствора
Ва (011)2, взятый в эксикатор в начале опыта (в мл); v — объем рас-
твора Ва (ОН)2, взятый для титрования после опыта (в мл); t — про-
должительность опыта (в ч); п — навеска исследуемого вещества
(в г); а — объем титрованного раствора щавелевой кислоты, затра-
ченный при титровании Ва (ОН)2 в контрольном эксикаторе (в мл);
b — объем титрованного раствора щавелевой кислоты, затраченный
Таблица 5
/ 273,2 (В — W) \
Значение фактора f = I ~76() ^73 2 /Оу I
х. В мм t° С 720 730 740 750 760 770
14 0,886 0,899 0,911 0,924 0,936 0,949
16 0,878 0,890 0,903 0,915 0,928 0,940
18 0,870 0,882 0,895 0,907 0,919 0,932
20 0,862 0,874 0,886 0,899 0,911 0,923
22 0,853 0,865 0,877 0,889 0,901 0,914
24 0,845 0,857 0,869 0,881 0,893 0,905
26 0,836 0,848 0,860 0,872 0,884 0,896
28 0,827 0,839 0,851 0,863 0,875 0,887
30 0,818 0,829 0,841 0,853 0,865 0,877
32 0,809 0,821 0,833 0,844 0,856 0,868
34 0,799 0,811 0,823 0,834 0,846 0,858
при титровании Ва (ОН2) в опыте (в мл); К — нормальность рас-
твора щавелевой кислоты; 22 — г-экв углекислого газа в реак-
ции с Ва (ОН)2; 1,98 — плотность углекислого газа при нормаль-
ных условиях (в г/л); 100 — пересчет на 100 г вещества.
Для получения интенсивности дыхания (в мг СО2 на 100 г ис-
следуемого вещества за 1 ч) полученный выше результат умножают
на 1,98.
2. Интенсивность дыхания по поглощению кислорода можно рас-
считать по формуле:
п (Ло - Лк) f 100
=-----------—t-------’
где Dq2 — интенсивность дыхания (в мл поглощенного О2 на 100 г
исследуемого вещества за 1 ч); п — навеска исследуемого вещества
(в г); t — продолжительность опыта (в ч); До — объем кислорода,
измеренный в опыте при атмосферном давлении В и температуре
t° (в мл); Ак — объем кислорода, измеренный в контрольном опыте
при атмосферном давлении В и температуре /° (может быть как
положительная, так и отрицательная величина) (в мл); f — фактор
пересчета объема газа на объем в нормальных условиях —
273,2 (В — W) ,W7 *
тал/97Q о г Л (W— давление паров воды; В — барометрическое
/□U \£(j— t J
давление в мм рт. ст.).
314
Величины факторов приведены в табл. 5.
3. Дыхательный коэффициент вычисляют ио формуле:
где Рсо2 — интенсивность дыхания (в мл углекислоты за 1 ч);
Dq2 — интенсивность дыхания (в мл кислорода за 1 ч).
Для изучения динамики дыхания во времени в начале опыта при-
соединяют бюретку прибора для измерения объема газа к эксикато-
ру (к трубке б), устанавливают нулевое положение и через опреде-
ленное время измеряют количество поглощенного кислорода.
Реактивы. Раствор гидроокиси бария (0,05 п.). 40 г кристаллического ве-
щества Ва (ОН)2 8Н2О растворяют при нагревании в 500 мл дистиллированной
воды. Полученный раствор приливают к 4,5 л прокипяченной воды, перемешивают,
закрывают пробкой и дают отстояться. Прозрачный раствор сливают в бутыль,
снабженный полуавтоматической пипеткой.
Раствор щавелевой кислоты (0,05 н.). Отвешивают на аналитических весах
3,15 г Н2С2О4 2Н2О, растворяют в дистиллированной воде, разбавляют водой
до 1 л и тщательно перемешивают.
ХРОМАТОМЕТРИЧЕСКИЙ МЕТОД
ОПРЕДЕЛЕНИЯ ТЕПЛОТЫ СГОРАНИЯ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ РАСТЕНИЙ
Важным показателем энергетической оценки растительного ма-
териала наряду с другими данными биохимического анализа яв-
ляется теплота сгорания. Теплотой сгорания органических веществ
называется количество теплоты, выделяющееся при полном сгорании
единицы веса исследуемого вещества при условии, что углерод
превращается в угольный ангидрид, водород — в воду, сера — в
серный ангидрид, а азот выделяется в свободном виде. Теплота
сгорания выражается в малых калориях на грамм вещества (кал/г) или
в больших калориях — на килограмм вещества (ккал/кг). Теплота сго-
рания растительного материала необходима при определении
коэффициента использования солнечной радиации, а также при
установлении энергетической эффективности фотосинтетической дея-
тельности растений и энергетической ценности отдельных органов.
Теплота сгорания органических веществ определяется путем
сжигания навески растительного вещества в специальном приборе —
калориметре. Это наиболее точный метод, но длительный, требую-
щий специальных условий. Теплота сгорания органических веществ
может быть приближенно вычислена из данных элементарного
анализа. Одна из формул, предложенная Д. И. Менделеевым для
этой цели, имеет следующий вид:
Q = 81 С 4- 300 /7 — 26(0 — 5),
где Q — теплота Црорания (в кал/г)\ С, Н, О, 5 — содержание уг-
лерода, водорода, кислорода и серы (в %).
Определение содержания этих элементов в растительном материа-
ле еще более сложно, чем определение теплоты сгорания в калори-
315
метре, поэтому все аналогичные формулы не получили практи-
ческого применения. Ниже приводится метод мокрого сжигания
органических веществ, который значительно проще, требует меньше
времени и осуществляется обычными средствами без специальной
аппаратуры.
Сущность метода. Анализ многочисленных данных теп-
лоты сгорания органических веществ показывает, что при сжига-
нии углеводов на каждый затраченный грамм-эквивалент кислорода
выделяется около 28 ккал тепла, а при сжигании жиров, белков и
органических кислот — около 26 ккал тепла, Поэтому, если уста-
новить количество грамм-эквивалентов кислорода, необходимого
для сжигания весовой единицы данного органического вещества,
можно вычислить теплоту его сгорания. Для определения коли-
чества грамм-эквивалентов кислорода можно воспользоваться реак-
цией окисления органических веществ растений бихроматом калия
в серной кислоте:
С12Н22ОП - 8К2Сг2О7 32H2SO4 = 12СО2 - 8Сг2 (SO4)3 + 8K2SO4 +
+ 43Н2О 4- 1350 ккал.
Из этого уравнения видно, что для окисления грамм-молекулы са-
харозы расходуется 48 грамм-эквивалентов бихромата калия.
Отсюда теплота сгорания одного грамм-эквивалента сахарозы сле-
дующая:
1350 оо .
О = л6— = 28,1 ккал.
48
Моносахариды, крахмал, целлюлоза и другие углеводы дают такие
же величины.
Для окисления грамм-молекулы пальмитиновой кислоты тре-
буется 92 грамм-эквивалента бихромата калия:
ЗСН3 (СН2)14 СООН + 46К2Сг2О7 + 184H2SO4 = 48СО2 +
+ 46Cr2 (SO4)3 + 46K2SO4 + 232Н2О + 7195 ккал.
Отсюда теплота сгорания одного грамм-эквивалента кислоты
равна:
2398 .
Q = - gg— = 26,1 ккал.
Для окисления грамм-молекулы лейцина требуется 33 грамм-эк-
вивалента бихромата калия, отсюда теплота сгорания одного грамм-
эквивалента лейцина равна:
Q = = 25,9 ккал.
Исходя из этих величин, можно легко определить теплоту сгорания
растительного материала, окисляя его раствором бихромата калия
в серной кислоте. Углеводы, жиры и некоторые органические кис-
лоты окисляются бихроматом калия полностью до углекислоты и
воды. Белки же окисляются только на 75%, поэтому необходимо
316
знать их содержание для введения поправки. В формулу вводится
также поправка на содержание жиров и других, близких по кало-
рийности веществ, которые пс определяются. Таким образом, вы-
полнив два химических анализа — окисление бихроматом калия и
общий азот, можно довольно точно определить теплоту сгорания рас-
тительного материала.
Ход анализа. Взвешивают на аналитических весах 25—
26 мг воздушно-сухого измельченного материала (масличных семян
16—17 мг), помещают в ступку и растирают до однородной массы с
2 мл 0,5 н. раствора бихромата калия в серной кислоте. Получен-
ный раствор переносят в пробирку диаметром 28—30 мм, смывая
остаток 4 мл 72%-ной серной кислоты, прибавляя ее порциями по
1 мл. В пробирку приливают из пипетки еще 10 мл раствора бихро-
мата калия в серной кислоте, перемешивают и оставляют на ночь-
На второй день пробирку помещают в раствор хлористого кальция,
нагретый до 125° С, и после того как температура снова поднимется
до 125° С, выдерживают при этой температуре 15 мин, регулируя
нагревание так, чтобы отклонения температуры не превышали
± 1° С. Во время нагревания пробирку периодически встряхивают.
Одновременно нагревают пробирку с контрольным раствором, со-
стоящим из такого же количества бихромата калия и 72%-ной сер-
ной кислоты, как в пробирке с исследуемым раствором.
После нагревания пробирки опускают в теплую воду для смы-
вания хлористого кальция с наружных стенок пробирок, затем
охлаждают и растворы переливают в колбы для титрования, смы-
вая остаток небольшим количеством воды. После повторного охлаж-
дения остаток бихромата калия титруют 0,1 и. раствором соли
Мора в присутствии 5 капель феиилантраниловой кислоты до пере-
хода вишнево-красной окраски раствора в зеленую. В части ис-
следуемого материала определяют общий азот хлораминным методом
и влажность высушиванием при 100° С до постоянного веса. Из
полученных данных вычисляют теплоту сгорания по следующей
формуле:
с “ z -йГо - +95л'+4б°'
где Q — теплота сгорания (в кал/г); а — объем титрованного рас-
твора соли Мора, затраченный при титровании контрольного рас-
твора (в мл); b — объем раствора соли Мора, затраченный при тит-
ровании остатка бихромата калия после сжигания (в мл); К. — нор-
мальность раствора соли Мора; С — влажность исследуемого
материала (в %); N— содержание общего азота в исследуемом мате-
риале (в %); 24,8 — количество ккал на 1 г-же кислорода с поправ-
кой 460 кал!г; п — навеска исследуемого вещества (в г); 95 N —
поправка на 25% неокисленного белка с содержанием 16% азота.
В полученную величину теплоты сгорания можно внести поправ-
ку на содержание золы, определив ее описанным ранее методом
(см. стр. 6,7).
317
Теплоту сгорания с учетом содержания золы вычисляют по фор-
муле:
п _ 100 Q
— 100 — g ’
где Qi — теплота сгорания (в кал/г) с поправкой на содержание
золы; g — содержание золы в исследуемом материале (в % от су-
хого вещества).
Реактивы. Приготовление реактивов описано при определении жиров в мас-
личных семенах (см. стр. 76, 138, 152, 274).
МЕТОДЫ ИССЛЕДОВАНИЯ
ВОДНОГО РЕЖИМА РАСТЕНИЙ
При достаточном снабжении растений водой, покрывающей
расход ее на транспирацию, органы растения содержат оптимальное
количество воды и все процессы в растении идут нормально. Если
количество воды, поступающее из корней, не покрывает расход ее
на транспирацию, это приводит к отрицательному водному балансу
и листья в разной степени обедняются водой, что сказывается на
скорости биохимических и физиологических процессов.
Вода в растении связана с помощью водородных связей с ве-
ществами, обладающими гидрофильными свойствами. Прочность
этой связи зависит от физико-химических свойств веществ. Так, на
поверхности белка и других коллоидов вода удерживается с боль-
шой прочностью: на расстоянии 1 ммк от поверхности белка вода
имеет структуру льда, а на расстоянии 10 ммк — подвижность ее
ограничена. Связанная коллоидами вода отличается рядом физи-
ческих и химических свойств по сравнению с водой обычных раство-
ров. В зависимости от степени обеспечения растения водой коли-
чество ее, связанное коллоидами клетки и раствором клеточного
сока, может быть различным.
Клеточный сок отделен от чистого растворителя — воды клеточ-
ной оболочкой, проницаемой, главным образом, только для воды.
Так как химический потенциал воды в растворе ниже, чем в чистой
воде, возникает односторонняя диффузия воды в клетку. Этот про-
цесс называется осмосом, а сила, вызывающая осмос, от-
несенная к единице поверхности мембраны, называется осмотичес-
ким давлением и выражается в атмосферах или барах (106 дин/см2).
В клеточном соке, содержащем различные вещества, осмотическое
давление определяется суммой молекулярных концентраций от-
дельных компонентов — молекул, ионов и коллоидных частиц.
На разности осмотических давлений основан обмен веществ между
клеткой и окружающей ее средой.
Ниже изложены методы определения некоторых показателей,
характеризующих водный режим растений.
318
Определение водного дефицита листа
При недостаточном поступлении воды в листья наблюдается
водный дефицит. С возрастанием водного дефицита наступает мо-
мент, когда начинается снижение скорости процесса фотосинтеза.
Начало снижения интенсивности фотосинтеза у различных растений
наступает при разных величинах водного дефицита листьев. Так, у
кукурузы и яблони при 8—9%, а у сахарной свеклы при 16% вод-
ного дефицита. С дальнейшим возрастанием водного дефицита
интенсивность фотосинтеза продолжает падать и доходить до нуля
при водном дефиците 18—20% у кукурузы ц яблони и 36—40% — у
сахарной свеклы.
Водный дефицит обычно выражают в процентах от содержания
воды в насыщенных ею листьях. Определение этого показателя
связано с высушиванием пробы, что вызывает затруднения при ра-
боте в полевых условиях. Можно выражать водный дефицит также
в процентах от сырого веса насыщенных водой листьев. В этом случае
пробу высушивать не нужно и определение возможно в полевых усло-
виях. Хотя показатели при таком способе определения получаются
несколько ниже, порядок величин остается таким же. Поэтому
полученными величинами можно пользоваться на практике для
решения вопросов о влиянии водного дефицита па фотосинтез и о
сроках полива растений.
Сущность метода. Отобранные в виде дисков пробы
листьев после взвешивания насыщаются определенное время водой.
После вторичного взвешивания и определения количества погло-
щенной воды ее выражают в процентах от веса пробы, насыщенной
водой. Можно также выражать количество поглощенной воды в
процентах от общего содержания воды в насыщенной водой пробе.
В последнем случае после взвешивания пробы, насыщенной водой,
ее высушивают в сушильном шкафу и после взвешивания сухого
остатка по разности находят общее количество воды в насыщенной
водой пробе.
С увеличением времени пребывания пробы листьев в воде коли-
чество всасываемой воды возрастает. Процесс поглощения воды
можно разделить на две фазы: первая связана с устранением пассив-
ного водного дефицита, а вторая представляет собой дополнительное
поглощение воды, обусловленное продолжающимся осмосом и
поступлением воды в межклеточное пространство. Опыты показали,
что при 30-минутном насыщении пробы водой с небольшим водным
дефицитом количество ее становится близким к содержанию воды
в листьях после насыщения их через черешок, когда попадание во-
ды в межклеточное пространство исключено. С увеличением вод-
ного дефицита время, необходимое для насыщения ткани водой,
возрастает. Таким образом, в зависимости от времени пребывания
пробы в воде, могут получиться разные величины водного дефицита,
поэтому нужно всегда придерживаться одного метода работы, вре-
мени насыщения и температуры воды.
319
Ход работы. В предварительно взвешенные на аналити-
ческих весах стеклянные бюксы отбирают 12 дисков из исследуемых
листьев с помощью пробочного сверла диаметром 16—18 мм. Для
этого под лист, не отделенный от растения, подкладывают резиновую
пробку и вращением сверла с легким нажатием высекают диски,
собирая их внутри сверла. Для характеристики целого растения
диски для пробы высекают из листьев разных ярусов. Полученные
диски выталкивают карандашом в бюкс, который закрывают крыш-
кой, и сохраняют в тени до конца отбора всех проб. После этого
пробы отправляют в лабораторию и взвешивают на демпферных
аналитических весах с интервалами 2—3 мин. Взвешивание на ана-
литических весах можно проводить и в полевых условиях. После
каждого взвешивания пробу немедленно высыпают из бюкса в ста-
кан, содержащий 30—40 мл воды с температурой 15—20° С. После
перемешивания диски размещают на поверхности воды, стакан на-
крывают стеклом и оставляют в покое па 90 мин (время необходимое
для насыщения листа водой и с более высоким водным дефицитом).
Точно через 90 мин после опускания данной пробы в воду диски вы-
нимают с помощью сетчатой пластинки, помещают на фильтроваль-
ную бумагу отдельно каждый дискисверху прижимают другим лис-
том фильтровальной бумаги. После снятия всех капель воды диски
переносят в тот же бюкс, в котором они были до набухания, закрыва-
ют крышкой и оставляют до тех пор, пока все пробы будут вынуты
из воды. Затем бюксы с пробами взвешивают и из полученных дан-
ных вычисляют водный дефицит листьев по следующей формуле:
Y _ 100 (6 —д)
где X — водный дефицит листа (в % к весу насыщенного водой
листа при 90-минутном насыщении); а — вес пробы до насыщения
ее водой (в г); b — вес пробы после насыщения ее водой (в г). Если
имеются подходящие условия, можно после взвешивания пробы,
насыщенной водой, высушить ее в сушильном шкафу при 100 ° С до
постоянного веса и из полученных данных вычислить водный дефи-
цит листа в долях общего количества воды в листе при полном его
насыщении:
= 100(6 — д)
1 (6-п) ’
где — водный дефицит листа (в % к весу воды в насыщенном лис-
те при 90-минутном насыщении); п— содержание сухого вещества
в навеске (в г).
Определение коллоидно-связанной
и осмотически-связанной воды
Сущность метода.В основе метода лежит представление
о том, что коллоидно-связанная вода с упорядоченной структурой
не является растворителем при смешивании с раствором сахара.
320
В этом растворе распределяется только не связанная с коллоидами
вода. Смешав часть исследуемого материала с раствором сахара,
а другую часть с чистой водой, и определив концентрации получен-
ных растворов, можно на основании полученных данных составить
систему уравнений и вывести формулы для вычисления очень важ-
ных показателей, характеризующих водный режим исследуемого
материала: содержание коллоидно-связанной воды, осмотически-свя-
занной воды, концентрацию клеточного сока, осмотическое давле-
ние клеточного сока, количество воды, связанное единицей коллои-
дов> и количество воды, приходящееся на единицу растворенных
веществ клеточного сока.
Приведенные ниже формулы выведены на основании следующих
рассуждений: в навеске ткани и, содержащей а (% общей воды)
и х (% связанной воды), количество свободной воды d следующее:
, _ п(а — х)
100
В той же навеске п количество растворенных в воде веществ равно:
г___________________________ п Ь(а — х)
G 100(100 —г?) ’
где С — содержание растворимых в воде веществ в навеске (в г);
b — содержание растворимых в воде веществ (в % к клеточному
соку).
Если к определенному количеству исследуемого материала
(nJ после его измельчения добавить некоторое количество воды
(Н) и перемешать, вода распределится в клеточном соке, и концен-
трация его уменьшится в соответствии с количеством прибавленной
воды. После определения концентрации растворенных веществ в
полученном растворе (bj можно рассчитать содержание их в кле-
точном соке по следующей формуле:
р _ Hl by (а — х) . Н by
100(100 — by) 100 — by •
При добавлении к навеске исследуемого вещества (л2) некоторо-
го количества раствора сахарозы с концентрацией более высокой,
чем концентрация клеточного сока, будет происходить перераспре-
деление воды из клеточного сока к более крепкому раствору са-
харозы до наступления равновесия. Определив концентрацию
полученного раствора (&2), можно и в этом случае вычислить со-
держание растворенных веществ в клеточном соке но формуле:
г п2 Ь2 (а — х) А Ь2
Ь 100(100 —Ь2) 100 — b2 9
где А — количество воды, извлеченной раствором сахара.
Из правых частей двух последних формул составляется уравне-
ние, в которое вместо А подставляется его значение, получаемое
из следующего уравнения:
д _ (Ьо — Ь2)
ь2 ’
21 6-2
321
где S — количество раствора сахарозы, прибавленное к навеске
(в г).
После преобразований получается формула для расчета содер-
жапия коллоидно-связанной воды, приведенная ниже (см. стр. 323).
Ход анализа определения коллоидно-свя-
занной и осмотически-связанной воды в лис-
тьях и других сочных тканях. Отбирают пробу
свежих листьев или других частей растения весом около 7 г.
После измельчения пробы ножом и перемешивания помещают часть
пробы около 2 г в тарированный бюкс, взвешивают на аналитичес-
ких весах и сушат в сушильном шкафу при температуре 100° С до
постоянного веса. Из полученных данных вычисляют содержание
общей воды по формуле:
„ _ 100(6 —п)
где а — содержание общей воды в пробе (в %); b— вес сырой на-
вески (в г); п — содержание сухого вещества в навеске (в г).
Из другой части пробы отвешивают 2 г с точностью до 0,01 г,
помещают в тарированный бюкс, взвешивают, прибавляют 3 мл
25%-ного раствора сахарозы, закрывают крышкой и снова взве-
шивают. После этого содержимое бюкса перемешивают маленькой
стеклянной палочкой по длине, не превышающей высоты бюкса,
закрывают крышкой вместе с палочкой и оставляют на 20—24 ч
при периодическом размешивании до наступления равновесия.
В полученном растворе определяют концентрацию сахарозы с
помощью рефрактометра.
Остаток пробы тщательно растирают в фарфоровой ступке и
из полученной однородной массы отвешивают 2 г с точностью до
0,01 г, поместив навеску в тарированный бюкс. К навеске в бюксе
прибавляют 3 мл дистиллированной воды, закрывают крышкой и
взвешивают. Содержимое бюкса перемешивают маленькой стеклян-
ной палочкой, оставляют ее в бюксе и закрывают крышкой. Настаи-
вают в течение 2 ч при периодическом размешивании палочкой.
После этого содержимое бюкса переносят в центрифужную пробир-
ку, центрифугируют при 2—3000 об/мин и жидкость сливают с
осадка в сухую центрифужную пробирку. Так как в большинстве
случаев этот центрифугат мутный и окрашен в зеленый цвет, что
затрудняет рефрактометрирование, к нему добавляют 0,3 мл хлоро-
форма, взбалтывают, закрывают пробирку резиновой пробкой и
оставляют на ночь для осаждения коллоидов. На следующий день
раствор центрифугируют и из верхней осветленной части центри-
фугата берут 1—2 капли раствора и определяют его концентрацию
с помощью рефрактометра.
Ход анализа определения коллоидно-свя-
занной и осмотически-связанной воды в дре-
весных растениях. Исследование побегов древесных ра-
стений имеет свои особенности, так как они менее сочны и одре-
весневшие ткани измельчаются с трудом. Отобранную пробу весом
322
около 7 г, измельченную ножом или ножницами, перемешивают, по-
мещают частьпробы около 2 г в тарированный бюкс, взвешивают на
аналитических весах и сушат в сушильном шкафу при темпера-
туре 100° С до постоянного веса. Содержание общей воды вычис-
ляют по приведенной выше формуле. Остаток пробы (около 5 г)
немного растирают в фарфоровой ступке н из полученной массы
отвешивают 2 г с точностью до 0,01 г, помещая навеску в тариро-
ванный стеклянный бюкс. Прибавляют 5 мл 40%-ного раствора
сахарозы, закрывают крышкой и взвешивают. 11осле этого содер-
жимое бюкса перемешивают маленькой стеклянной палочкой, ко-
торую оставляют в бюксе, закрывают его крышкой и оставляют
на 20—24 ч, периодически размешивая палочкой.
Из остатка пробы отвешивают 2 г, помещают в фарфоровую ступку,
добавляют 0,5—1 г чистого песка, тщательно растирают, перено-
сят все содержимое ступки в бюкс, взвешивают, добавляют 5 мл
дистиллированной воды и снова взвешивают. Перемешивают ма-
ленькой стеклянной палочкой, закрывают и оставляют на 3—4 ч
(или на ночь), периодически размешивая.
После настаивания содержимое бюксов переносят в центрифуж-
ные пробирки, центрифугируют и из верхнего прозрачного слоя
берут 1—2 капли раствора и определяют его концентрацию с по-
мощью рефрактометра. Из полученных данных вычисляют содер-
жание коллоидно-связанной, нерастворяющей воды по формуле;
х = S(b{}~b2)(\W-bx) I-/L. // fr,(100 —6.2)
где X — количество коллоидно-связанной воды (в % к исследуе-
мому веществу); Н — количество воды, прибавленное к навеске
п± (в г); 5 — количество раствора сахарозы, прибавленное к на-
веске п2 (в г); — навеска исследуемого материала, к которой
добавлена вода (в г); п2 — навеска исследуемого материала, к ко-
торой добавлен раствор сахарозы (в г); Ьо — содержание сахарозы в
исходном растворе (по рефрактометру) (в %); — концентрация
раствора, полученного после настаивания навески nt с водой (в %);
Ь2 — концентрация раствора, полученного после настаивания на-
вески п2 с раствором сахарозы (в %); а — содержание общей воды в
исследуемом материале (в %).
Количество осмотически-связанной воды в процентах к иссле-
дуемому материалу вычисляется по формуле:
Y = а — х,
где а — содержание общей воды; х — содержание коллоидно-свя-
занной воды (в % к исследуемому веществу).
Полученные данные дают возможность вычислить содержание
водорастворимых веществ в клеточном соке по следующей формуле:
с = Н b2 + nt S(b0 — b2)]
Н b^ -j- zij S (6g — b2) ’
21
323
где С — концентрация раствора клеточного сока в расчете на са-
харозу (в %).
Зная концентрацию клеточного сока, можно вычислить вели-
чину осмотического давления его раствора по формулам:
В = С (0,01С + 0,72); при С 3—18,
В = С (0,02С 4- 0,54); при С 18 — 35,5,
В = С(0,04С —0,17); при С 35,5 — 50,
где В — осмотическое давление (в барах, 10е дин/см2).
На основании полученных экспериментальных данных можно
также установить количество коллоидно-связанной воды, удержи-
ваемой единицей нерастворимых в воде коллоидов по следующей
^формуле:
у _ _______________
2 100— (а + С) ’
и количество осмотически-связаппой воды, приходящееся на едини-
цу растворенных в соке веществ, по формуле:
у ___ а X
z 2 — с
где Х2 — количество коллоидно-связанной воды, удерживаемое 1 г
сухих нерастворимых в воде коллоидов (в г); У2 — количество ос-
мотически-связанной воды, приходящееся на 1 г растворенных в
клеточном соке веществ (в г); С — содержание водорастворимых
веществ в клеточном соке в расчете на сахарозу (в %); X — коли-
чество коллоидно-связанной воды (в %); а — количество общей
воды (в %).
Реактивы. Раствор сахарозы (25%-ный). 25 г сахарозы растворяют в 75 мл
дистиллированной воды и определяют концентрацию сахарозы рефрактометром,
проверяя ее перед каждым опытом.
Раствор сахарозы (40% -ный). 40 г сахарозы растворяют в 60 мл дистилли-
рованной воды и определяют концентрацию сахарозы рефрактометром.
ЛИТЕРАТУРА
Бойцова А. В., Игнатьев Б. Д., Унтилова А. Е. Метод опреде-
ления интенсивности дыхания растительных объектов.— Физиол. и биохим.
культурных растений, 1971, 3, 6, 657.
Власюк П. А., Гуляев Б. И., Оканенко А. С., Мануильс-
кий В. Д. Установка для автоматической записи поглощения СО2 растения-
ми.— Физиол. и биохим. культурных растений, 1971, 3, 1, 93.
Вознесенский В. Л., Заленский О. В., Семихатова О. А. Ме-
тоды исследования фотосинтеза и дыхания растений. М., «Наука», 1965.
Гуляев Б. И., Ману и л ьски й В. Д., Оканенко А. С. Оценка по-
грешностей измерения интенсивности фотосинтеза газометрическим методом.—
Физиол. и биохим. культурных растений, 1970, 2, 1, 34.
Г у с с в Н. А. О характеристике состояния воды в растении.— Физиол. расте-
ний, 1962, 9, 4.
Д у м а н с к и й А. В. Учение о коллоидах. М.— Л., Госхимиздат, 1948.
324
Оканен ко Л. С., Г у л я е в Б. И., Починок X. Н. Многоканальная
установка для измерения интенсивности фотосинтеза.— В кн.: Фотосинтез
и продуктивность растений, Рига, «Зипатне», 1965, 103.
Починок X. Н. Установка для газометрического определения фотосипте:
в естественных условиях.— Физиол. растений, 1958, 5, 2, 200.
Починок X. Н. Определение интенсивности фотосинтеза у целых листьев
и целых растений.— В кн.: Науч. тр. Укр. ин-та физиол. растений, 16. Киев,
Госсельхозиздат УССР, 1959, 89.
Починок X. Н. Интенсивность фотосинтеза и дыхания у кукурузы, сахар-
ной свеклы и яблони в связи с водным дефицитом листа.— В кн.: Фотосинтез
и продуктивность растений. Киев, «Наукова думка», 1965, 153.
Починок X. Н. Влияние гербицидов и антитранспирантов на интенсивность
фотосинтеза и транспирации растений.— Физиол. и биохим. культурных
растений. 1971, 3, 5, 538.
Слейчер Р. Водный режим растений. М., «Мир», 1970.
Толмачев И. М. Новый респирационный аппарат.— Тр. ин-та физиол. рас-
тений им. К. А. Тимирязева АН СССР, 1950, 7, 1, 265.
Тооминг X. Г., Гуляев Б. И. Методика измерения фотосинтетически
активной радиации. М., «Наука» 1968.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азот
определение амидного 87
— аминного 83
— аммиачного 87
— белкового 72, 74, 77
— небелкового 77
— общего 32, 72, 74
аминный, стандартный раствор 86
— построение калибровочной кри-
вой 85
аммиачный, стандартный раствор 95
— построение калибровочной кри-
вой 91, 94, 190
Азотная кислота, очищенная 60
раствор 1 1 27
— 1 5 32
— 2 н. 222
смесь с уксусной кислотой 140
Альбумины, выделение 80
Алюмо-калиевые квасцы, раствор в
1%-ной уксусной кислоте 70
Амилазы, определение активности
------суммарное 184
------а-амилазы 185
Аминокислоты
определение хроматографическое 100
----с медным производным 109
-------нингидрином ПО
----по весу бумаги ПО
выход окрашенного продукта 84
калибровочная кривая 86
стандартные растворы 86, 113, 189
Аммиак, очищенный 60
раствор 1 10 9
— 2 н. 67
Аммиачный буферный раствор pH 10
13
Аргинин, определение 87, 90
калибровочная кривая 92
стандартный раствор 95
Аскорбиновая кислота, определение
240
раствор 1% -ный 86
— 0,04 н. 171
Аскорбиноксидаза
определение активности 176
326
Аспираторы 281
Бария гидроокись
раствор 0,01—0,008 н. 296
— 0,05 н. 315
— насыщенный 133
Барий уксуснокислый, 20%-ный 27
хлористый, 10%-ный 181
хромат, 2,5%-ный в НС1 43
Белки
гидролиз кислотный 104
— щелочный 104
определение 77
сложные 82
фракционный состав 79
Бензин, t° кипения 50—120° 200
Бор
определение с кармином 43
---- хинализарином 47
калибровочная кривая 47
стандартный раствор 48
Бромат-бромид
раствор 0,03 н. 181
— 0,1 н. 241
Бумага хроматографическая
промывание 112, 209, 253
н-Бутиловый спирт
без аммиака 86
Буферный раствор
аммиачный pH 10 13
ацетатный pH 4,5 86, 183
— pH 4,75 58
боратный pH 8 113
пирофосфатный pH 8,9 113
фосфатный pH 6,4 171
— pH 7,0 98, 191
с КВг pH 6,7 77
фосфатно-цитратный pH 5,0 188
Винная кислота, определение
в винограде 229
Вода
очищенная 58
безаммиачная 76
безуглекислая 27
определение коллоидно-связанной
320
— осмотически-связанной 320
Водный дефицит листа
определение 319
Водный режим растения
исследование 318
Восстанавливающие сахара
определение 117
Галактоза, определение 264
Гваякол, 0,3% -ный 169
Гексозофосфорные эфиры
определение 259
Гексозы
определение 149
калибровочная кривая 132
Гемицеллюлоза, определение 146
Гербициды, определение скорости
распространения угнетения фото-
синтеза 306
Гидразин сернокислый, 2%-ный 67
Гидроксиламин сернокислый, 1%-ный
13
солянокислый 2%-ный 240
Глиадины, выделение 81
Гликолипиды, определение 263, 264
Глобулины, выделение 80
Глутаминовая кислота, определение
Глутатион, определение 98
Глюкоза
определение 116, 121
построение калибровочной кривой
126, 132
Глютелины, выделение 81
Дезоксирибонуклеиновая
определение 266, 267
построение калибровочной
268
стандартный раствор 270
Дигестия
горячая 156
холодная 156
Дитизон
перекристаллизация 59
раствор в СС14 60
Ди- и трикарбоновые кислоты
извлечение 70%-ным спиртом 250
— 1%-ной уксусной кислотой 250
определение хроматографическое
246
— — по площади пятен 249
Дифениламиновый реактив на ДНК
271
Дифенил карбазон 2%-ный 28
Дыхание
определение в листьях неотделенных
от растения 289
— дыхательного коэффициента 311
Едкий натр, см. натрия гидроокись
Железо
определение 5, 7
построение калибровочной кривой 7
стандартный раствор 9
сернокислое закисное, 20%«ный ра
створ 228
хлорное, 1%-ный раствор
— 0,5%-ный раствор 63
Железо-аммонийные квасны,
раствор 228
Железо-молибдатный ре;
Железосинеродистый рс;
Жир, определение хром;
436
Зола, определение 5
Изатин, 0,2%-ный раствор ИЗ
Изоамиловый спирт с СО.
63
Инвертаза, определение
181
Йод
определение 67
построение калибровочной
69
стандартный раствор 70
раствор 0,02 н. 224
— 0,05 н. 121
— 0,5%-ный 138
Йодид-йодат, 0,1 н. раствор 224
Калибровочная кривая для
глюкозы, мальтозы, рибозы, рам-
нозы и галактозы 132
сорбозы, фруктозы, сахарозы
фннозы 132
Калий
определение .тинитритом 13,
16, 35
тетрафепилборатом 36
зотнокислый, 1%-ный раствор 27
стандартный раствор 40
антимонат 21
бихромат, раствор 0,1 17
-----0,5 н. 239
-----0,5 н. в серной кислоте 138
Калий
бромистый, 12%-ный раствор 228
гидроокись, 0,01 н. в этаноле 113
йодат, 0,05 М. 17
— 0,01 н. 171
— 0,001 н. 100
— стандартный раствор 70
йодистый, 20%-ный раствор 17
— 0,01 н. раствор 26
— 0,02 н. раствор 41
оксалат, 10%-ный раствор 13
перманганат, 4%-ный раствор 228
— 0,1 н. 70
— 0,05 н. 10
— 0,01 н. 27
роданистый, 20%-ный раствор 63
— 0,1 н. 228
сернокислый безаммиачный 76
углекислый, 10%-ный раствор 232
Кальций
327
определение комплексонометри-
ческое 10
нермаиганатометрическое 5, 9
азотнокислый, 80%-ный раствор
138
— 5%-ный раствор 138, 169
гидроокись, насыщенный раствор 49
оксадат, определение 236
хлористый, 25%-ный раствор 146
— раствор для солевой бани 274
Камеры
листовые 290, 297
прищепки 280
для растений 300
Каталаза, определение активности 171
Катализатор реакции на ДНК 271
Кармин, 0,03%-ный раствор 48
Каротин
определение фотоколориметрическое
199
— спектрофотоколориметричсское
213
получение 200
построение калибровочной кривой
203
Клетчатка, определение 138
Кобальт
определение 49, 55
построение калибровочной кривой
56
стандартный раствор 58
Колонка ионообменная 102
Крахмал
2% -ный раствор 185
0,5%-ный раствор 18
определение колориметрическим ме-
тодом 136
— поляриметрическим методом 163
— хроматометрическим методом 135
очистка 137
построение калибровочной кривой
137
Крезоловый красный, 0,1%-ный ра-
створ 59
Ксантофиллы
определение 200
построение калибровочной кривой
206
Ксилоза
определение 149
стандартный раствор 150
Купрон, 2%-ный раствор 67
Купферон, 2%-ный раствор 67
Лигнин, определение 150
Лимонная кислота, определение 224
Липиды, определение 262
Лютеин
определение 205
построение калибровочной кривой
206
Магний
комплексонат 232
определение ацидиметрическое 5, 8
— комплексонометрическое 10—12
сернокислый, 0,02 н. раствор 12,
146
Мальтоза, определение 130
Манометр
водяной 282
компенсационный 295
Марганец
определение 21, 25
построение калибровочной кривой
25
сернокислый, 10%-ный раствор 70
Медно-щелочный реактив на сахаре
для объемного метода 121
— колориметрического метода 133
Медь
определение 49, 53
построение калибровочной кривой
55
стандартный раствор 58
серпокислая, 3%-ный раствор 76
— 0,02%-ный раствор 113
Метафосфорная кислота, 5%-ный ра-
створ 171
Метиленовая синь, 0,16%-ный рас-
твор 67
Метилоранж, 0,2%-ный раствор 181
Метилрот, водный 0,2%-ный 9
Молибден
определение роданидным методом 60
— кинетическим методом 63
построение калибровочной кривой
62
стандартный раствор 62
Молибдат-сернокислый реактив 133
Молибденовая жидкость 26
Мора соль
0,05 н. раствор 17
0,1 н. раствор 138
Мочевина
0,5%-ный раствор 191
определение 97
Муравьиная кислота, 10%-ный рас-
твор 240
Мышьяково-молибдатный реактив 127
Натрий
азотнокислый, 1%-ный раствор 27
гидроокись, 20%-ный 94
— 2 н. 76
— 1 н. 28
— 0,4 н. 121
— с NaCl 271
— 0,2 н. 70
— 0,1 н. 9
— 0,05 н. 27
— 0,02 н. 253
— 0,2%-ный 83
328
гипобромид, 0, 4 н. 94
диэтилдитиокарбамат, 0,1 %-ный 59
кобальтинитрит, 10%-ный 18
лимоннокислый, 10%-ный 58
молибдат, 10%-ный 76
определение с антимонатом 18
---цинкуранилацетатом 13
тетрафенилборат, 3% -ный 40
тиосульфат, 10%-ный 58
— 0,1 н. 17
— 0,05 н. 18
— 0,02 н. 76
— 0,01 н. 43
сульфит, 0,05 н. 121
— 10%-ный^28
углекислый, 5%-ный 181
— 1%-ный 49
— 2%-ный с NaOH 145
уксуснокислый, 30%-ный 27
— 2%-ный 112
— 0,8 М 40
— 0,2 М 95
хлористый, 10%-ный с цитратом 271
— 0,2 н. 32
— с Na2SO4 95
Нингидрин, 0,5%-ный раствор 113
0,02%-ный 113'
очистка перекристаллизацией 87
Нингидриновый реактив 86
Нитраты
определение 34, 96
построение калибровочной кривой
39
Нитрозо-Р-соль, 0,1%-пый раствор 59
Оксихинолин, 3%-ный раствор в 2%-
ной
щавелевой кислоте 21
Олово хлористое, 10%-ный раствор 63
Органические кислоты
определение суммы 220, 242
— свободных 222
---в отсутствие янтарной кисло-
ты 223
--------присутствии янтарной кис-
лоты 223
— хроматографическое 246
стандартные растворы 253
Орциновый реактив 150
Пектиновые вещества
определение растворимых 142
— суммы 144
Пентозаны, определение 148
Пентозы, определение 148
построение калибровочной кривой
149
Перекись водорода
0,05 н. раствор 169
0,1 н. раствор 176
Пероксидаза
определение активности 166
построение калибровочной кривой
169
1 leiiTii да:
187
11ерсульфат аммонии,
169
1 liii'MeiiThi пластид
определение фото колориметр и чес кое
195
— спектрофотометрическое 213
— хроматографическое 207
Пипетка автоматическая 157
Пирокатехин, 0,2%-ный раствор 171
11оглотители
паров воды из воздуха 289
углекислоты 281
Полипептидаза, определение актив-
ности 186
Полифенолоксидаза
определение активности 169
Прибор
для фильтрования с отсасыванием
195
— измерения объема кислорода
313
Принадлежности для поляриметри-
ческого определения сахара 159
Промывная жидкость для калия в ко-
бальтинитритном методе 18
тетрафепплборатпом методе 40
я-Пропиловый спирт без аммиака 86
Протеаза, определение активности 185
11р()теиплза, определение активности
186
Протопектин, определение 143
Проявители
для аминокислот 113
— органических кислот 248, 249
— сахаров 133
Рамноза, определение 132
Растворители для хроматографии
аминокислот 106J
органических кислот 248
пигментов 208
сахаров 129
Раффиноза, определение 130» IH'
Реактив на нитриты 41
Рибоза, определение 130, IM
Рибонуклеиновая кислоте
определение 266
очистка 269
построение калиб|
268
стандартный рАСТМ
Ртуть |
азотнокислая МИМ
— окисная, 0.0| Ш
Ртуть Д
— уксу< покиОЛМЬ 0,3 н. 100
- 0.2 н 40
Кривой
0,01 н. 229
хлорная, 4 %-ный раствор 241
— 2%-ный раствор 28
Сахара гликолипидов, определение 263
Сахараза (инвертаза)
определение активности 181
ах ар
определение в сахарной свекле 154
— ускоренным методом 157
Сахароза, 25%-ный и 40%-ный рас-
творы 324
определение 116, 154
Сахара
определение объемным методом 117,
121
колориметрическим 124, 131, 132
хроматографическим 127
Свинец уксуснокислый, 20%-ный рас-
твор 222
— основной 157
Сера общая, определение 41
Серебро азотнокислое
0,1 н. раствор 26
0,05 н. раствор 228
0,01 п. раствор 28
стандартный раствор 169
регенерация 70
Серная кислота
раствор 1 : 4 и 1 :9 43, 9
— 72%-ный 152
— И н. 95
— 2 н. 41
— 1 н. 32
— 0,2 н. 249
— 0,1 н. 9
Соль Мора
0,05 н. раствор 17
0,1 н. раствор 138
Соляная кислота
очищенная 60
раствор 1:19
— 2 н. 150
— 1 н. 138
— 0,4 н. 121
— 0,3 н. 40
— 0,1 н. 146
Сорбоза, определение 131
Стандартный раствор органических
кислот 253
Сульфит натрия
10%-ный раствор 228
0,05 н. раствор 121
Теплота сгорания органических ве-
ществ, определение 315
Тетрароданомеркуриат калия 17
Тимол, 4%-ный раствор 94
Тирозин, 0,05%-ный раствор 181
Тирозиназа
определение активности 178
Транспирация, определение 288
Трилон Б
0,1 н. раствор 146
0,02 н. раствор 12
0,01 н. раствор 233
0,002 н. раствор 241
Трихлоруксусная кислота, 10%-ный
раствор 83
Трубка поляриметрическая для сока
159
Углеводы определяемые поляримет-
рически 154
Уксусная кислота
20%-ный раствор 271
10% -ный раствор 17
1%-ный раствор 40
Урановые остатки, регенерация 18
Уреаза
приготовление раствора 98
определение активности 189
Установка для определения интен-
сивности фотосинтеза
в полевых условиях (ПФС) 279
многоканальная 291
Фенилаптраниловая кислота
0,2%-ный раствор 138
Фенолфталеин
1%-ный раствор 27
0,1 %-ный раствор 95
Фитин, определение 260
Фосфатно-щавелево-сульфосалици-
ловый реактив 9
Фосфолипиды, определение 262
Фосфор
определение объемным методом 21,
24
— колориметрическим методом 29,
31
— макроэргического 261
— неорганического 260
— нуклеиновых кислот 264
— эфиров сахаров 259
построение калибровочной кривой
32
стандартный раствор 32
Фосфорная кислота, 4 М раствор 70
Фосфорновольфрамовая кислота
4%-ный раствор с NaCl 95
---без NaCl 164
Фотосинтез, определение интенсивнос-
ти
в естественных условиях 278
целых листьев 297
растений 299
оптико-акустическим газоанализа-
тором 307
Фруктоза
определение объемным методом 116,
121
— хроматографическим методом 127
построение калибровочной кривой
126, 131
330
Фруктозаны, определение 152
Хинализарин, 0,003%-ный раствор 48
Хлор, определение 21, 27
Хлорамин, 0,12 п. раствор 76
Хлорная кислота, 5%-ный раствор
265
Хлорофилл
определение фотоколориметричсским
методом суммы 195, 198, 199
----- хлорофилла а 212
--------b 212
— спектрофотоколориметричсским
методом хлорофилла а 213
--------6 213
построение калибровочной кривой
суммы 496—198
хлорофилла а 211
— b 211
прочность связи с белками 216
Хроматографирование аминокислот
105
Хромоген черный ЕТ-00 с NaCl,
1 : 100 13
Цинк
определение 49, 54
построение калибровочной кривой 57
стандартный раствор 58
азотнокислый, 5 н. раствор 32
окись, 10%-ная суспензия 17
сернокислый, 10%-пый раствор 28
— 3%-ный раствор в уксусной кис-
лоте 127
0,2 М
дыхания
229
уксуснокислый, 1%-ный раствор 265
Цинковая пыль, очистка 41
Ципкурапилацетлт, реактив на Na 17
Шариковая пробка 65
с палочкой М7
атив для аспираторов 282
для пробирок погружаемых в
ВОДу 66
Щавелевая кислота
определение 235
8% -ный раствор 76
6%-ный раствор в серной
121
Ща вел е во -ее р I ю к ис л ы й р;
70
Эксикатор для определения
313
Эозин с метиленовой синыо
Этиленгликоль, очистка 86
Этиловый спирт
95%-ный 17
90%-ный 200
80%-ный 196
70% -ный 83
50%-ный 233
40%-ный 21
с уксусной кислотой 1 1 48
Яблочная кислота
определение в винограде 232
— в присутствии лимонной кислоты
233
Янтарная кислота, определение 237
ОГЛАВЛЕНИЕ
Предисловие 3
Глава 1. Элементы минерального питания растений
Определение процентного содержания золы, магния, кальция
и железа из одной навески . . 5
Комплексономстрическое определение магния и кальция 10
Определение содержания калия и натрия 13
Определение натрия в виде антимоната 18
Определение содержания хлора, фосфора и марганца 21
Определение хлора ускоренным методом 27
Определение фосфора и калия в одной навеске . 29
Определение азота, фосфора и калия в одной навеске 32
Определение калия, неорганического фосфора и нитратов в вы-
тяжке из свежего растительного материала 34
Определение содержания общей серы 41
Колориметрический метод определения бора . 43
Определение меди, цинка и кобальта в одной навеске 49
Определение молибдена 60
Определение йода 67
Глава II. Азот и его соединения 72
Определение общего и белкового азота хлораминным методом 72
Определение небелкового и белкового азота из одной навески 77
Определение фракционного состава белковых веществ в расте-
ниях 79
Определение аминного азота нингидриновым методом 83
Определение аммиачного и амидного азота и аргинина тимол-ги-
побромитным методом 87
Определение нитратов в растениях 96
Определение мочевины 97
Определение глутатиона . . 98
Определение аминокислот с помощью хроматографии на бумаге 100
Глава III. Углеводы 116
Определение глюкозы, фруктозы и сахарозы в растениях из одной
навески ... 116
Определение глюкозы, фруктозы и сахарозы железосинеродистым
калием . ... 121
Определение сахаров после хроматографического разделения их
на бумаге 127
Определение крахмала объемным и колориметрическим методом 134
Определение клетчатки 138
Определение пектиновых веществ...........................140
332
Определение суммы пектина и протопектина 144
Определение гемицеллюлоз и пентозанов 146
Определение пентоз и пентозанов 148
Определение отдельных гексоз н нснто: 149
Определение лигнина 150
Определение фруктозанов 152
Поляриметрический метод определения углеводов 154
Глава IV. Ферменты 165
Определение активности пероксидазы 166
Определение активности нолифенолоксида: 169
Определение активности каталазы 172
^Определение активности каталазы, нолифенолоксида: и
ксидазы из одной навески . 175
Определение активности аскорбиноксидазы 176
Определение активности тирозиназы . 178
Определение активности сахаразы (инвертазы) 181
Определение активности амилазы 183
Определение активности протеаз 185
Определение активности уреазы 189
Глава V. Пигменты пластид 192
Определение суммы хлорофиллов (а +6) фотоколориметрическим
методом . . 195
Определение каротина, суммы хлорофиллов (а + Ь) и суммы
ксантофиллов из одной навески 198
Получение чистого каротина 200
Получение ксантофилла — лютеина 204
Количественное определение пигментов с помощью хроматогра-
фии на бумаге . . . 207
Выделение хлорофилла а и хлорофилла b . 209
Спектрофотометрический метод определения хлорофилла а, хло-
рофилла b и каротиноидов 213
Определение прочности связи хлорофилла с белками 216
Глава VI. Органические кислоты 219
Определение суммы органических кислот 220
Определение свободных органических кислот 222
Определение лимонной кислоты 224
Определение винной и яблочной кислот в винограде 229
Определение лимонной и яблочной кислот при соименном при-
сутствии . 233
Определение щавелевой кислоты 235
Определение янтарной кислоты 237
Определение аскор*биновой кислоты ртутным методом 240
Определение ди- и трикарбоновых кислот с помощью хромато-
графии на бумаге . 241
Определение ди- и трикарбоновых кислот методом хроматогра-
фии на бумаге по площади пятен 249
Глава VII. Фосфорные соединения и липиды 255
Определение неорганического и макроэргического фосфора, гек-
созофосфатов, фосфолипидов, нуклеиновых кислот и фитина в од-
ной навеске 257
Определение содержания РНК и ДНК 266
Хроматометрический метод определения жира в масличных се-
менах .................................................... 271
333
Глава VIII. Физиологические методы 275
Исследование интенсивности фотосинтеза..................... 275
Определение интенсивности фотосинтеза в естественных усло-
виях с помощью полевого прибора (ПФС) 278
Многоканальная установка для определения интенсивности
фотосинтеза в стационарных условиях . 291
Определение интенсивности фотосинтеза целых больших листьев
и целых растений . . . . . 297
Коэффициент использования солнечной энергии при фотосинтезе 303
Определение степени влияния различных веществ на течение ос-
новных физиологических процессов . . 303
Определение скорости продвижения угнетающего действия гер-
бицидов в тканях растений 306
Использование оптико-акустических газоанализаторов для
определения интенсивности фотосинтеза и дыхания растений 307
Определение интенсивности дыхания и дыхательного коэффициен-
та отдельных органов растения . . 311
Хроматометрический метод определения теплоты сгорания ор-
ганических веществ растений . . 315
Методы исследования водного режима растений 318
Предметный указатель........................................326
Харитон Николаевич Починок
МЕТОДЫ
БИОХИМИЧЕСКОГО АНАЛИЗА РАСТЕНИЙ
Печатается по постановлению ученого совета Института физиологии
растений АН УССР
Редактор Т. М. Осинская. Художественный редактор Р. И. Калыш. Оформле-
ние художника А. Г. Комяхова. Технический редактор Б. А. Пиковскан
Корректоры А. С. Улезко, Л. Г. Бузиашвили.
Сдано в набор 31. J. 1975 г. Подписано к печати 25. XII. 1975 г. БФ 02731. NHi II ы
№ 303. Тираж 2400. Бумага № 1, 60x90i/ie- Печ. физ. листов 21,0. Уел. .«ни ... 'I '•
Учетно-изд. листов 20,98. Цена 2 руб. 25 коп.
Издательство «Наукова думка», Киев, Репин;),
Отпечатано с матриц Головного предприятия республиканского upon....и......
динения «Полиграфкнига» Госкомиздата УССР, г. Киев, ул. Донжгнко. » u<i ......
книжной фабрике «Коммунист» республиканского произволе i нс................ ♦
лиграфкнига» Госкомиздата УССР, Харьков, ул ....... II
ИЗДАТЕЛЬСТВО «НАУКОВА ДУМКА»
В 1976 году ВЫПУСТИТ В СВЕТ КНИГИ:
Корневое минеральное питание
и продуктивность растений.
Язык русский. 15 л. Цепа 1 руб. 66 коп.
В монографии освещаются результаты исследований в области
минерального питания культурных растений в связи с их продуктив-
ностью и учетом особенностей их выращивания па Украине. Рассмат-
риваются различные аспекты азотистого, фосфорного, калийного,
серного и магниевого питания растений, а также вопросы питания
овощных культур в условиях закрытого грунта и гидропоники.
Рассчитана на научных сотрудников, физиологов и биохимиков
растений, агробиологов, преподавателей и студентов биологических
факультетов вузов.
Микроэлементы в обмене веществ растений.
Язык русский. 15 л. Цена 1 руб. 66 коп.
В монографии представлены многолетние итоговые работы по
вопросам участия микроэлементов в отдельных звеньях обмена ве-
ществ. Основное внимание уделено изучению специфического дей-
ствия микроэлементов (марганца, бора, молибдена, цинка, лития) в
обмене нуклеиновых кислот, белков, таких надмолекулярных комплек-
сов высшего порядка как ДНП, а также активаторов и ингибиторов
роста, витаминов.
Рассчитана на научных сотрудников по физиологии и биохимии
растений, преподавателей и студентов биологических факультетов уни-
верситетов, сельскохозяйственных и педагогических институтов.
Предварительные заказы на эти книги принимают все магазины
книготорга, потребительской кооперации, «Книга — почтой», а также
непосредственно книжный магазин «Наукова думка» (252001 Киев -/,
ул. Кирова, 4), который после выхода книг из печати вышлет их
по Вашему адресу наложенным платежом.