/







































































































































































































































































































































Author: Емельянова И.З.
Tags: плодоводство химия химические производства издательство лесная промышленность
Year: 1976




Text
И. 3. ЕМЕЛЬЯНОВА
ХИМИКО-ТЕХНИЧЕСКИЙ
КОНТРОЛЬ
ГИДРОЛИЗНЫХ ПРОИЗВОДСТВ
ИЗДАНИЕ 2-е, ПЕРЕРАБОТАННОЕ
Издательство
«ЛЕСНАЯ ПРОМЫШЛЕННОСТЬ»
Москва 1976
УДК 634.0.863:66.012.1
Химико-технический контроль гидролизных производств. Изд. 2-е, перс-
раб. Емельянова И. 3. М., «Лесная промышленность», 1976, с. 328.
В книге опнсаны методы анализов сырья, вспомогательных материалов,
полупродуктов и готовой продукции, которые применяют иа гидролизных
заводах и на заводах, перерабатывающих сульфитный щелок, при контроле
за технологическими процессами получения этилового спирта, кормовых
дрожжей, фурфурола и углекислоты. Изложены методы анализа сточных вод
на различных стадиях биологической очистки.
Книга предназначена для инженерно-технических работников и
исследователей, работающих на заводах и в научно-исследовательских институтах.
Она может быть использована в учебных заведениях при подготовке
инженеров и техников для микробиологической промышленности.
Табл. 26, ил. 72, библиогр.— 98 назв.
Рецензент главный технолог Бобруйского гидролнзного завода
М. А. 3 и н и н а
Ираида Захаровна Емельянова
ХИМИКО-ТЕХНИЧЕСКИЙ КОНТРОЛЬ ГИДРОЛИЗНЫХ ПРОИЗВОДСТВ
Издание второе, переработанное
Редактор издательства В. С. Баранова
Художественный редактор В. Н. Журавский
Технический редактор В. М. Волкова
Корректор Л. С. Безуглина
Переплет художника В. Д. Петухова
Сдано в набор З/Ш-1976 г. Подписано в печать 16/VIII-1976 г. Т-13788.
Формат 60Х90'/іб. Бумага типографская № 2. Усл. печ. л. 20,5. Уч.-изд. л. 22,87.
Тираж 2200 экз. Издат. № 185/75. Заказ 142. Цеиа 1 р. 38 к.
Издательство «Лесная промышленность», 101000, Москва, ул. Кирова, 40а
Ленинградская типография № 8 Союзполнграфпрома
при Государственном комитете Совета Министров СССР
по делам издательств, полиграфин и книжной торговли.
190000, Ленинград, Прачечный пер., 6
Е —^~93—76 © Издательство «Лесная промышленность», 1976
yjoi (Uli *— /Ъ
ВВЕДЕНИЕ
Гидролизными производствами в нашей стране принято
называть заводы, перерабатывающие моносахариды, которые
получают в результате гидролиза полисахаридов растительных
тканей. Существует два типа таких заводов — гидролизные
заводы микробиологической промышленности и заводы,
перерабатывающие сульфитный щелок, в целлюлозно-бумажной
промышленности [1].
Сырьем для гидролизных заводов является не только
древесина хвойных и лиственных пород, но и некоторые ткани
однолетних растений — подсолнечная лузга, кукурузная кочерыжка,
хлопковая шелуха, гуза-пая и др. После гидролиза всех легко-
и трудногидролизуемых полисахаридов, содержащихся в
перерабатываемом сырье, получают раствор моносахаридов (гидро-
лизат), раствор фурфурола (фурфурольный конденсат) и
лигнин в твердом состоянии.
Сульфитный щелок получают на целлюлозно-бумажных
комбинатах при сульфитной варке древесины хвойных и лиственных
пород с целью получения целлюлозы. Во время такой варки
легкогидролизуемые полисахариды (гемицеллюлозы) древесины
гидролизуются и переходят в раствор в виде моносахаридов.
Лигнин превращается в лигносульфонаты, которые тоже
переходят в раствор. После варки получают целлюлозу и
сульфитный щелок — раствор моносахаридов и лигносульфонатов,
который направляют на сульфитный завод для переработки [2].
Моносахариды, полученные в результате гидролиза
полисахаридов различных растительных тканей, состоят из гексоз и
пентоз. На заводах спирто-дрожжевого профиля из гексоз
получают этиловый спирт и углекислоту, а из пентоз — кормовые
дрожжи и фурфурол. На заводах дрожжевого профиля все
моносахариды — гексозы и пентозы — расходуются на
выращивание дрожжей.
Кроме обычных кормовых дрожжей, которые содержат
витамины группы В, заводы выпускают кормовые дрожжи,
обогащенные лизином и витамином D2. За последнее время в связи
с развитием животноводства на промышленной основе быстро
увеличивается спрос на кормовые дрожжи, которые являются
I* 3
обязательной витаминной добавкой в комбикорма домашних
животных. Для удовлетворения потребностей народного
хозяйства построены и строятся заводы дрожжевого профиля
большой мощности.
Характерная особенность гидролизных производств состоит
в том, что для получения разнообразных продуктов из
полисахаридов растительных тканей на этих заводах применяют
химические, биохимические и микробиологические процессы. Это
своеобразие технологии нашло свое отражение в выборе
методов анализа и в постановке контроля производства. Наряду
с химическими и физико-химическими методами используются
биохимические и микробиологические. Контроль за
технологическим процессом на гидролизных предприятиях разнообразен,
в этом его сложность и отличие от контроля на химических
предприятиях, где применяют только химические процессы.
На гидролизных предприятиях получают до 20 видов
готовой продукции. В настоящей книге освещены вопросы контроля
производства только этилового спирта, кормовых дрожжей,
фурфурола и углекислоты.
Организация работы центральной заводской лаборатории
Центральная заводская лаборатория (ЦЗЛ) является основной
структурной единицей завода; Начальник ЦЗЛ — инженер-технолог с помощью
цеховых лабораторий, мастерской контрольно-измерительных приборов (КИП)
и рабочих, обслуживающих технологические участки, организует выполнение
всех работ по контролю и учету качественных и количественных показателей
основных и вспомогательных цехов и завода.
Объем работ и штаты ЦЗЛ определяются в зависимости от количества
и разнообразия ассортимента готовой продукции, выпускаемой заводом. На
заводах большой мощности в состав ЦЗЛ входит следующие лаборатории
(или группы): аналитическая; микробиологическая; контроля качества сырья
и готовой продукции; научно-исследовательская, иногда и санитарная. На
заводах небольшой мощности обязанности этих лабораторий выполняют группы
или отдельные сотрудники. Цеховые лаборатории не входят в состав ЦЗЛ,
они являются структурными единицами цехов; центральная заводская
лаборатория осуществляет только методическое руководство ими.
Аналитическую лабораторию (группу) возглавляет химик-аналитик
(инженер). Сотрудники этой лаборатории осуществляют методическое
руководство всеми цеховыми лабораториями, проверяют работу лаборантов и проводят
с ними занятия; готовят реактивы, устанавливают концентрации титрованных
растворов, составляют таблицы для быстрого нахождения результатов
анализа и проверяют приборы, которыми пользуются в цеховых
лабораториях и в цехе; проводят калибровку мерной посуды, которой пользуются
в лабораториях, в цехе и на складе готовой продукции.
Микробиологическую лабораторию возглавляет микробиолог, имеющий
высшее специальное образование. Сотрудники этой лаборатории следят за
состоянием спиртовых и кормовых дрожжей и за появлением инфекции;
обнаружив заражение дрожжей посторонней микрофлорой, устанавливают источник
заражения и сразу организуют проведение дезинфекции; на время остановки
завода они проводят консервацию дрожжей; эпизодически выводят чистую
культуру спиртовых и кормовых дрожжей и делают ее подсев к работающим
дрожжам или полную замену старой культуры дрожжей новой; контролируют
сухие товарные дрожжи на содержание в иих жизнеспособных клеток; про-
веряют и инструктируют сотрудников цеховых лабораторий и рабочих,
выполняющих наблюдения за микробиологическими процессами; при биологической
очистке сточных вод завода микробиологи контролируют состояние
микрофлоры активного ила. Кроме того, сотрудники этой лаборатории проводят
научно-исследовательскую работу и принимают участие во всех
микробиологических работах иа заводе.
Лабораторию контроля за качеством материалов возглавляет инженер-
химик. Сотрудники этой лаборатории выполняют следующие работы: делают
анализы сырья и вспомогательных материалов, поступающих иа завод, для
определения соответствия их требованиям, стандартов и в случае поступления
недоброкачественных материалов составляют рекламации; следят за чистотой
тары, упаковкой и маркировкой готовой продукции, при отгрузке
потребителю отбирают средние пробы и делают анализ отгружаемой продукции,
составляют сертификаты, удостоверяющие ее качество; следят за соблюдением
правил хранения готовой продукции, сырья и вспомогательных материалов;
выполняют арбитражные анализы, делают разбор рекламаций на продукцию
предприятия н устанавливают причины и лиц, виновных в отправке
потребителю продукции, не соответствующей требованиям стандарта на нее;
участвуют в работах по подготовке и внедрению новых стандартов.
Научно-исследовательскую лабораторию (НИЛ) возглавляет иижеиер-химик
или микробиолог. Сотрудники этой лаборатории проводят исследовательские
работы с целью улучшения технологических процессов, повышения качества
выпускаемой продукции и устранения причин брака и потерь. Они участвуют
в пусконаладочных и аварийных работах, а также проводят исследования,
связанные с внедрением новой техники.
Санитарную лабораторию возглавляет инженер-химик. Совместно с
сотрудниками Госсанинспекции (или по их указанию) сотрудники этой
лаборатории делают анализы воздуха производственных помещений и атмосферного
воздуха, окружающего завод, с целью установления наличия в нем веществ
вредных для здоровья работающих. На основании полученных данных
определяют степень вредности условий труда, находят источники загрязнения
воздуха и дают рекомендации для снижения концентрации вредных веществ
в воздухе. При отсутствии очистных сооружений на заводе сотрудники
лаборатории систематически проводят анализ различных заводских стоков, чтобы
вовремя предупредить сброс в водоем сточных вод, имеющих загрязнение
выше установленной нормы.
Цеховые лаборатории проводят оперативный хнмико-технический и
микробиологический контроль за выполнением технологического регламента своего
цеха. Сотрудники лаборатории, делая анализы экспрессными методами в цехе,
более сложными методами — в лаборатории и по показаниям контрольно-
измерительных приборов, следят за соблюдением установленных параметров
ведения процесса, за состоянием спиртовых и кормовых дрожжей. В случае
обнаружения брака готовой продукции, потерь, инфекции и других нарушений
технологического регламента сразу информируют руководство цеха для
принятия срочных мер.
Контроль за качеством очистки воды, которую используют в котельной,
выполняют сотрудники лаборатории химводоочистки. На очистных
сооружениях сотрудники лаборатории определяют степень загрязнения поступающих
стоков, эффективность их очистки на каждой станции и качество очищенной
воды, направляемой в водоем.
Кроме перечисленных работ начальник ЦЗЛ с сотрудниками принимают
участие в работе отделов и цехов предприятия по изучению причин,
вызывающих выпуск брака, в разработке мероприятий для повышения качества
продукции; совместно с сотрудниками мастерской КИП контролируют
правильность показаний контрольно-измерительных приборов; ведут учет и
отчетность производства по утвержденным формам в установленные сроки.
Начальник ЦЗЛ дает заключения о забракованной продукции и о возможности
исправления брака, составляет месячные, квартальные и годовые химико-
технические отчеты по действующей форме и осуществляет руководство всеми
работами по контролю и учету на заводе.
О точности и достоверности результатов анализов
При вычислении качественных и количественных показателей работы
завода, цеха, бригады исходными даиными являются результаты анализов сырья,
полупродуктов и готовой продукции н результаты замеров их количеств. Для
правильной оценки вычисленных показателей и определения допустимых
пределов колебаний их значений надо знать не только точность анализа илн
замера, но также представлять, какова степень достоверности исходных данных.
Точность метода н достоверность результатов аналнзов — разные понятия,
хотя их часто необоснованно принимают за снноннмы. Точность метода — это
точность определения содержания интересующего компонента и том количестве
исследуемого материала, которое было взято на анализ, прн отсутствии в нем
мешающих веществ. Эту точность указывают в прописи метода. Она зависит
от специфичности и чувствительности реакции, лежащей в основе метода, от
воспроизводимости результатов аналнзов и от технического оформления
анализа — аппаратуры, способа наблюдения и фиксации конца реакции и т. п.
Она не зависит от правильности отбора пробы, ее подготовки и хранения, от
способа выражения результатов анализа и других причин.
Достоверность результатов аналнзов более широкое понятне. Она зависит
от всех погрешностей, которые могут быть прн определении интересующего
компонента во всей массе исследуемого материала. Это погрешности,
допускаемые при отборе и составлении средней пробы, прн разделке н хранении
пробы, погрешности нз-за присутствия в пробе веществ, мешающих проведению
анализа, нлн веществ, определяемых вместе с интересующим компонентом,
а также погрешности, допускаемые прн вычислении результата анализа за счет
применения условных н эмпирических коэффнцнентои и т. д.
Точность и достоверность взаимосвязаны. Достоверный результат анализа
не может быть получен неточным методом. Достоверность не может быть выше
точности. Но точные результаты анализа, полученные точным методом могут
быт недостоверными. Так, например, определение влажности древесного сырья
методом высушивания делают с точностью ±0,1% отн. Нет сомнения, что
в том количестве исследуемого сырья, которое было взято на анализ,
содержание влаги определили с упомянутой точностью. Но если анализ делали для
определения влажности нескольких десятков тонн древесного сырья различного
гранулометрического состава, пробу которого отбирали в течение смены,
потом осуществляли разделку пробы и анализировали, то достоверность
результатов анализа в этом случае будет значительно ниже точности метода. Другой
пример определения пеитозанов по фурфуролу. Несмотря иа то, что количество
полученного фурфурола определяют очень точно, достоверность результата
этого анализа всегда низкая, потому что иыход фурфурола нз пентоз
колеблется, в зависимости от их природы, в пределах 88—67% от теоретического
выхода.
Прн сведении баланса цеха или завода часто встречают затруднении —
баланс не сходится. Происходит это потому, что количество н качество
израсходованных материалов, полупродуктов, готовой продукции н отходов были
определены, может быть, с одинаковой точностью, но с различной степенью
достоверности.
ГЛАВА 1. ОТБОР ПРОБ
Для правильной оценки качества сырья, вспомогательных
материалов, полупродуктов, готовой продукции, отходов и
сточных вод очень важно правильно отобрать среднюю пробу,
которая по своему составу соответствовала бы составу всей массы
материала, подлежащего исследованию [3].
Средние пробы составляют из индивидуальных проб,
отобранных из различных мест массы материала. Индивидуальная
проба соответствует составу материала в точке отбора пробы.
Если материал находится в покое, то индивидуальные пробы
отбирают с его поверхности и на различной глубине. При
движении материала индивидуальные пробы отбирают из потока
периодически, через установленные промежутки времени. Смесь
всех индивидуальных проб, отобранных из партии материала,
называют первичной средней пробой. Обычно количество
первичной пробы бывает большим, поэтому для взятия из нее
лабораторной пробы, предназначенной для анализа, первичную пробу
сокращают.
Когда материал однородный, то составление средней пробы
из индивидуальных и отбор лабораторной пробы не
представляют трудностей. При неоднородном материале отбор средней
пробы иногда представляет большие трудности. Выбирая точки
отбора индивидуальных проб, надо учитывать возможные
расслоения материала при перевозке, изменения состава при
хранении и т. д. Перед отбором лабораторной пробы осуществляют
разделку первичной пробы, которая заключается в хорошем
перемешивании и измельчении первичной пробы.
Отбор проб сыпучих материалов
Отбор проб из отвалов. Пробы опилок, хлопковой
шелухи, подсолнечной лузги, кукурузной кочерыжки, торфа,
извести, суперфосфата и других материалов, находящихся в кучах
или штабелях, а также подсушенного активного ила с иловых
площадок отбирают методом вычерпывания. При выборе точек
отбора проб учитывают следующее.
Наружный слой материала обычно отлича-
ется от всей массы материала влажно- Г Л
стью. В сырую погоду он более влажен, /^
чем слои материала, находящиеся внут- /—
ри, в сухую — наоборот. Кроме того, на- _ , _
г і J r Рис. 1. Расположение то-
ружныи слои материала при длительном чек отбора проб из шта.
хранении выветривается и окисляется. беля
Внутренние слои материала часто
недоступны для отбора проб. Раскапывание кучи иногда не
допускается по условиям хранения материала, ибо оно способствует
проникновению воздуха во внутреннюю часть штабеля, что
может вызвать химические изменения материала, разогрев и
самовозгорание.
Для выбора точек отбора проб на поверхности штабеля
наносят взаимно перпендикулярные линии. Первую горизонталь
проводят на боковой поверхности на расстоянии 0,5 м от
основания штабеля, следующую горизонталь проводят на
расстоянии 0,5 м от первой и т. д. На этих горизонталях намечают
точки отбора на расстоянии 2 м друг от друга в шахматном
порядке (рис. 1). В каждой точке на глубине 0,5—0,7 м берут
лопатой или щупом порцию материала в количестве 0,5—1 кг.
При отборе пробы щупом его вдавливают в материал, затем
вытаскивают и опрокидывают. Если щуп имеет продольную щель,
то после вдавливания его вращают для заполнения материалом.
Отбор проб материалов из вагонов, которые
прибывают на завод, производят при разгрузке методом
ступенчатого вычерпывания. Сначала пробы отбирают с поверхности
в точках на трех продольных линиях, из которых одна проходит
посередине вагона, а две боковые отстоят от стенок вагона на
7б ширины вагона. Расстояние между точками отбора 90 см.
Из каждой точки отбирают по одной лопате. Затем снимают
слой материала толщиной 'Д высоты вагона и отбирают пробы
так же, как с поверхности. После этого снимают слой равный
половине высоты вагона и отбирают пробы как с предыдущих
слоев. Наконец, снимают слой в 5/б высоты вагона и снова
отбирают пробы. Для составления средней пробы вагона
смешивают одинаковые количества отобранных проб. Из
саморазгружающихся вагонов пробы отбирают в течение всего времени
разгрузки, периодически подставляя пробоотборник в удобных
местах потока материала.
Отбор проб материала с транспортера
производят автоматическим пробоотборником, который периодически
сгребает движущийся материал по всей ширине транспортера,
или при сбрасывании материала с транспортера, периодически
подставляя пробоотборник.
Отбор проб упакованных материалов
производят из нескольких тарных мест с соблюдением правил отбора
проб, указанных в стандарте на данный материал.
Отбор и составление среднесменных и
среднемесячных проб сырья. Пробы древесины, торфа,
растительных отходов сельского хозяйства производят
автоматическим пробоотборником или вручную с транспортерной ленты.
При автоматическом отборе разовые пробы отбирают через 10—
15 мин по 200—300 г, а при ручном отборе через каждые 2 ч
по 1 кг. Отобранные разовые пробы помещают в бидон с плотно
прилегающей крышкой. Смесь разовых проб, отобранных за
смену, является среднесменной пробой, ее количество должно
быть не менее 4 кг. Среднесменная проба сырья является
индивидуальной пробой при составлении среднемесячной пробы.
Для составления среднемесячной пробы берут среднесменные
пробы из опилок, щепы и коры в количестве пропорциональном
сырью, переработанному за смену. Например, за смену
переработано 50 т древесины, для составления среднемесячной пробы
берут 500 г. Гранулометрическим анализом установлено, что
сырье, переработанное за смену, состояло из опилок, щепы и
коры в соотношении 15:2:3. Следовательно, 500 г пробы,
отобранной для составления среднемесячной, должны содержать
375 г опилок, 50 г щепы и 75 г коры.
8
Среднемесячную пробу хранят в банках, закрытых пробками.
Герметичность не требуется, потому что результаты анализа
пересчитывают на абсолютно сухое вещество.
Разделка проб. Операции, которые надо произвести,
чтобы из первичной пробы, составляющей иногда несколько
сотен килограммов, отобрать лабораторную пробу в количестве
не более 0,5 кг, называют разделкой проб. Первичную пробу
отбирают в плотнозакрывающуюся тару, удобную для переноски.
До разделки пробу можно хранить не более 1 суток в
неотапливаемом помещении, в хорошо закрытых ящиках. Разделка
первичной пробы состоит из следующих операций: измельчения,
перемешивания и сокращения. Все операции надо делать быстро,
чтобы уменьшить время испарения влаги из пробы. Измельчать
необходимо все куски независимо от трудности измельчения.
Если количество пробы
небольшое, ее измельчают вручную,
прочным инструментом, чтобы
в пробу не попали кусочки
истираемых частей
инструментов. При большом количестве
первичной пробы куски
измельчают дробилками разного
типа. Измельчение
продолжают делать по мере
сокращения пробы. Чем меньше коли-
Рис. 2. Квартование первичной пробы
чество пробы, отбираемое при
сокращении, тем меньше должны быть размеры кусков.
Для перемешивания первичную пробу насыпают конической
горкой, высыпая каждую лопату на вершину горки. Затем весь
материал из собранной горки лопатой переносят на другое
место, также ссыпая его на вершину горки со всех сторон. После
тщательного перемешивания пробу сокращают методом
квартования (рис. 2). Горку материала 1 расплющивают 2 и делят
на четыре части 3. Два противоположных сектора отбирают,
перемешивают, снова собирают горку и производят квартование.
Так делают несколько раз. После последнего сокращения горку
расплющивают, делят на 16—20 квадратов 4 и из середины
квадратов, расположенных в шахматном порядке, совком отбирают
порцию пробы по всей глубине слоя. Порции пробы,
получаемые при делении пробы, должны быть одинаковы по величине
и составу.
Упаковка средней пробы. Отобранную пробу делят
на две части и помещают в плотнозакрывающиеся банки.
Банки с пробой, предназначенной для контрольного или
арбитражного анализа, завертывают в бумагу, обвязывают шпагатом,
концы которого пломбируют или опечатывают сургучной
печатью на картонной пластинке. На склянку со средней
пробой наклеивают этикетку с указанием наименования завода-
изготовителя, названия материала, номеров вагонов или цистерн,
номера накладной, даты отбора пробы, фамилий лиц,
отбиравших пробы, и, если надо, надпись «Огнеопасно», «Яд» и т. д.
Если проба предназначена для контроля за выполнением
технологического регламента на заводе, то пробу сразу передают
в лабораторию на анализ.
Отбор проб жидкостей
Жидкие материалы перемешиваются лучше, чем твердые,
поэтому отбор средних проб жидкостей осуществить легче.
Однако при отборе проб из больших емкостей надо иметь в виду,
что жидкости при стоянии расслаиваются.
Отбор проб из емкостей производят глубинными
пробоотборниками разнообразных конструкций, а также с
помощью краников, сделанных на различной высоте сборников.
На рис. 3 показаны четыре образца глубинных
пробоотборников. Первые два образца /, 2 предназначены для отбора сред-
ней пробы жидкости, которая нахо-
1 у дится в чане или в другой подобной
емкости. Они представляют собой
цилиндрические сосуды
вместимостью 1—2 л с грузом в дне для
облегчения погружения их в жидкости.
Пробоотборники отличаются между
собой приспособлениями для
ограничения скорости их заполнения.
В пробоотборнике 1 это узкое
горлышко, а в пробоотборнике 2—
крышка с отверстиями диаметром
3—5 мм. Для отбора средней пробы
пробоотборник на цепочке
равномерно опускают на дно емкости с жидкостью с такой скоростью,
чтобы он успел заполниться за время его погружения.
Контролируют по пузырькам воздуха, которые по мере заполнения
пробоотборника выходят на поверхность жидкости. Заполненный
пробоотборник поднимают и всю жидкость выливают из него
в чистую склянку. Отобранную среднюю пробу отправляют на
анализ.
Третий и четвертый образцы пробоотборников применяют
для отбора индивидуальных проб на различной высоте емкости
с жидкостью. Пробоотборник 3 представляет цилиндрический
сосуд вместимостью 1—2 л с подвижной крышкой, которую-
можно открывать в любой момент погружения пробоотборника,
регулируя при этом величину отверстия. При отборе пробы
к пробоотборнику прикрепляют рулетку, закрывают крышку и
на цепочке в опускают его в жидкость. Глубину погружения
определяют рулеткой. Во время погружения пробника цепочка а
должна висеть свободно, чтобы пробник был закрыт. На нуж-
10
Рис. 3. Глубинные
пробоотборники
ной глубине спуск пробоотборника приостанавливают и,
натягивая цепочку а, открывают его. Когда пробоотборник
заполнится жидкостью, о чем узнают по прекращению выделения
пузырьков воздуха, который выходит из него при заполнении
жидкостью, пробоотборник закрывают, ослабляя цепочку а.
Затем, держа натянутой цепочку б, пробоотборник за цепочку в
поднимают на поверхность, отобранную жидкость выливают
в сухую склянку и закрывают пробкой.
Пробоотборник 4 представляет собой длинную стеклянную
трубку, на одном конце которой имеется кран. Этим
пробоотборником отбирают индивидуальные пробы спирта, метанола и
других подобных продуктов из небольших емкостей. При
закрытом кране в емкость с жидкостью погружают трубку на нужную
глубину, а затем открывают кран. Когда трубка заполнится
жидкостью, кран закрывают и трубку вынимают из жидкости.
Отобранную жидкость из пробоотборника выливают в чистую
склянку и закрывают пробкой.
Для составления средней пробы отбирают несколько
индивидуальных проб из разных мест емкости. На каждую склянку
с пробой делают наклейку, в которой указывают координаты
отбора пробы. Все отобранные пробы отправляют в
лабораторию для составления средней пробы и анализа.
Отбор проб жидкостей из потока осуществляют
для контроля за ходом технологического процесса. Для анализа
отбирают разовую пробу или среднюю за определенный
промежуток времени. Разовую пробу отбирают с помощью пробоот-
борного крана, установленного на трубопроводе, или используют
открытый перелив жидкости из одной емкости в другую. Ее
состав соответствует составу жидкости, протекающей по
трубопроводу в момент отбора пробы. Среднюю пробу жидкости из
потока отбирают непрерывно в течение установленного времени
{сутки, смена) автоматическим пробоотборником или
составляют ее из разовых проб. В этом случае разовые пробы
отбирают несколько раз в строго установленное время в количествах
пропорциональных количеству жидкости, протекающей по
трубопроводу за время между отбором предыдущей и отбираемой
пробы. Анализ средней пробы характеризует состав жидкости,
прошедшей по трубопроводу за время, в течение которого
отбирали пробы. Для предохранения проб сахарных растворов
(нейтрализата, бражки и т. д.) от микробиологического
заражения при отборе среднесменных проб в них добавляют несколько
капель фенола или концентрированной серной кислоты.
При отборе средней пробы гидролизата после испарителя
надо учитывать, что концентрация сахара в нем и скорость
потока меняются очень часто в большом диапазоне. Отбор
средней пробы гидролизата из трубопровода надо осуществлять
непрерывно пробоотборником, обеспечивающим отбор количества
гидролизата, пропорциональный скорости потока, и быстрое
11
охлаждение гидролизата, чтобы предотвратить испарение. Если
все эти условия отбора нельзя выполнить, то пробу
гидролизата следует отбирать после инвертора.
Для автоматического отбора средней пробы нейтрализата
или субстрата, протекающего по трубопроводу, пользуются си-
фонно-поплавковым пробоотборником (рис. 4). Пробоотборник
[4] представляет собой цилиндрический сосуд емкостью 1—3 л,
внутри которого находится сифон 2 и отборник с поплавком.
Отборник состоит из воронки 1 (емкостью 6—8 мл), на конец
которой надета резиновая трубка, согнутая в кольцо. Другой
конец резиновой трубки соединен с отводной
стеклянной трубкой. На резиновую трубку
надет поплавок 3. Верхний край воронки
отборника на 10—15 мм ниже верхнего края
сифона. Кольцо резиновой трубки должно быть
такой величины, чтобы поплавок не
касался стенки сосуда. При заполнении сосуда
жидкостью поплавок должен свободно держаться
на ее поверхности. Верхний край кольца
резиновой трубки при этом будет находиться
над поверхностью жидкости.
Отбор проб сифонно-поплавковым
пробоотборником в приемник производится
периодически. Жидкость из трубопровода при
открытом кране в штуцере тонкой струей
поступает в пустой пробоотборник. Когда уровень
жидкости в нем достигнет края воронки,
пробоотборник заполнится, но жидкость не будет
выливаться, потому что в это время всплывет
поплавок и поднимет кольцо сливной
трубки выше поверхности жидкости. Жидкость
из трубопровода будет поступать в резервуар
пробоотборника до тех пор, пока ее
уровень не достигнет верхнего края
сифона. В результате действия сифона жидкость быстро
выльется из резервуара пробоотборника в канализацию или через
какую-нибудь емкость вернется в поток. Жидкость,
находящаяся в отборнике, выльется в приемник. После выливания
жидкости из отборника цикл отбора пробы повторяется
автоматически. Для устранения пены в резервуар пробоотборника
кладут несколько деревянных кубиков, смоченных пеногаси-
телем.
Отбор конденсатов паров с амо и спарени я
жидкостей, содержащих летучие вещества, производят с
помощью установки, показанной на рис. 5. Горячую жидкость
(барда, лютер и др.) из кубовой части колонны направляют
в испаритель-сепаратор 1, где пар отделяется от жидкости.
Через нижнее отверстие в испарителе жидкость вытекает в ка-
12
Рис. 4. Сифонно-
поплавковый
пробоотборник
нализацию. Пар идет вверх, проходит в холодильник 2,
конденсируется и стекает в приемник, которым при отборе разовой
пробы служит кружка, а при отборе среднесменной пробы —•
бутыль.
Отбор проб горячих жидкостей. Фурфурольный
конденсат, спиртовой конденсат и другие подобные жидкости
отбирают через холодильник, присоединенный к пробоотборному
крану на трубопроводе. Пробу горячей жидкости, которая в
трубопроводе находится под давлением, отбирают с помощью
установки (рис. 6). Поток жидкости, регулируемый игольчатым
вентилем 2, направляют в маленький испаритель 1, в котором
Барда
Г
Пар „
ХиЛакт
у
Рис. 5. Установка для
отбора проб конденсата
паров самоиспарения
жидкостей
Рис. 6. Установка для
отбора проб горячих
жидкостей
происходит снижение давления. Жидкость из испарителя
собирают в пробник (кружку). Так отбирают пробы горячего гидро-
лизата, предназначенные для экспрессного определения РВ
и серной кислоты на варочной площадке. Пробу для
лабораторного исследования отбирают следующим образом. Берут
сухую колбу вместимостью 1 л, взвешивают, наливают в нее
400—450 мл дистиллированной воды, снова взвешивают, а затем
в нее отбирают пробу гидролизата. На кран надевают
резиновую трубку, свободный конец ее опускают в приготовленную
колбу с водой, открывают кран и набирают приблизительно
200—300 мл гидролизата. Затем колбу с пробой охлаждают до
комнатной температуры, взвешивают, вычисляют разбавление
пробы при отборе и делают анализ.
Отбор проб на очистных сооружениях
Пробы сточной воды, поступающей на очистку,
отбирают из камеры гашения напора общезаводского стока с
глубины 0,5 м пробоотборником (рис. 7) вместимостью 250—300 мл
каждый час. Среднесуточную пробу поступившей воды
13
составляют из разовых проб, отбираемых каждый час в
количествах, пропорциональных скорости поступления сточной воды [5].
Пробы воды из водоемов отбирают батометром на
500—1000 м выше (по течению реки) спуска сточных вод и ниже
спуска в пункте наиболее полного смешения сточной и речной
воды на глубине 0,5 м.
Пробы отбросов с р е ш ето к отбирают через каждые
2 ч. Все отбросы с решеток собирают в ящик, который имеет
небольшие отверстия в дне и стенках, взвешивают и берут
совком среднюю пробу в количестве 0,5 кг.
Пробы осадков из песколовок и первичных
отстойников отбирают во время их выгрузки через каждые
5 мин ковшом по 100—150 мл. Из равных количеств
индивидуальных проб составляют среднюю пробу,
предназначенную для анализа.
Пробы смеси воды и активного
ила из аэротенков, преаэраторов и
вторичных отстойников отбирают на различной
высоте пробоотборником (см. рис. 7)
вместимостью 500 мл. Отобранную пробу
анализируют сразу.
Пробы подсушенного активного
и л а с иловых площадок отбирают щупом
методом вычерпывания (см. с. 7).
Транспортировка, хранение
и консервирование проб. Иногда
ник для° сточной места отбора проб находятся на расстоя-
воды и активного нии нескольких километров от лаборатории,
ила поэтому необходимо организовать
доставку проб при соблюдении правил их
хранения. Пробы воды сохраняют в посуде из полиэтилена или
из стекла пирекс с притертыми пробками. При транспортировке
их помещают в ящики с гнездами, стенки которых обиты
войлоком. Время хранения проб должно составлять не более
1 суток.
Лучшим способом консервации проб является хранение проб
при температуре около 3°С. Если нет возможности хранить
пробы при низкой температуре, то каждую пробу делят на три
части. Первую часть, предназначенную для определения окисляе-
мости, содержания минерального азота и общего азота,
консервируют, прибавляя к 1 л пробы 2 мл 25% -ного раствора серной
кислоты. Вторую часть пробы, предназначенную для анализа
на содержание взвешенных веществ, нитритов и нитратов,
консервируют, прибавляя к 1 л пробы 2 мл хлороформа. Третью
часть пробы, предназначенную для определения биологически
потребляемого кислорода (БПК.), до анализа сохраняют при
температуре 3°С, прибавлять к ней консервирующие вещества
нельзя.
14
Отбор проб газов
Пробы газов отбирают из газопроводов, из емкостей, где
они могут находиться под разным давлением, и из атмосферы.
Для санитарного надзора за состоянием воздушной среды и для
установления потерь газа при его получении или хранении
анализируют пробы воздуха, отобранные в производственных
помещениях и из атмосферы, окружающей предприятие. При отборе
проб газа пользуются пробоотборниками и приспособлениями
различных конструкций. Наиболее распространенные из них
показаны на рис. 8.
Отбор проб из
газопроводов делают с помощью
приспособления /, которое
представляет собой изогнутую
трубку, вставленную в газопровод на
глубину 7з его диаметра. На
конце трубки имеется кран. Для
отбора пробы к концу трубки
присоединяют газоанализатор или
пробоотборник и открывают
кран. Когда газ заполнит сосуд
прибора или пробоотборника,
кран закрывают и отобранный
газ анализируют.
Отбор проб из
баллонов. Газообразные продукты,
заключенные в емкости,
являются более однородными по своему
составу, чем жидкости, но при
хранении под давлением они
могут расслаиваться и выделять, например, воду, если она
является примесью в газе. Перед отбором пробы газа из баллона
для регулирования струи выходящего газа на штуцер баллона
навинчивают редукционный вентиль (редуктор). Пробу газа
отбирают в газоанализатор или пробоотборник. Давление газа,
выходящего из баллона при отборе пробы, не должно
превышать 0,1 атм.
Пробоотборники. Для отбора проб газа применяют
пробоотборники, основанные на засасывании газа в пустое
пространство или на пропускании газа через пробоотборник в
течение некоторого времени (см. рис. 8).
Аспиратор 2 представляет собой два сосуда А и Б
(стеклянные бутыли вместимостью 3—5 л), которые соединены
резиновой трубкой через нижние тубусы. Перед отбором пробы газа
сосуд А заполняют насыщенным раствором хлористого натрия
доверху и закрывают пробкой, в которую вставлена трубка
с краном а (кран открыт). Когда избыток раствора хлористого
Рис. 8. Пробоотборники для отбора
проб газов:
/ — приспособление для отбора проб
газа из трубопровода; 2 — аспиратор;
3 — эвакуированный сосуд; 4 —
пипетка для отбора проб газа
15
натрия, вытесненный пробкой из сосуда А, заполнит всю трубку
(до и после крана) кран а закрывают. Для отбора пробы газа
трубку соединяют с источником отбираемого газа и открывают
зажим б и кран а. Раствор хлористого натрия будет
переливаться в сосуд Б, а сосуд А будет заполняться отбираемым
газом. Отбор пробы прекращают после заполнения сосуда А
газом, закрывая кран а и зажим б. Для перемещения части (или
всего) отобранного газа в газоанализатор сосуд А ставят ниже
сосуда Б, соединяют его с газоотборной пипеткой прибора и
открывают кран а и зажим б. Жидкость из сосуда Б будет
переливаться в сосуд А, а вытесняемый газ будет заполнять
пипетку прибора. Когда отберут нужное количество газа, кран а
и зажим б закрывают и аспиратор отсоединяют от прибора.
Эвакуированный сосуд 3 представляет собой склянку
вместимостью от 0,5 до 10 л. Сосуд закрывается пробкой, в
которую вставлен тройник с краном. Перед отбором пробы насосом
из сосуда откачивают газ до разрежения 8—13 кПа F0—100 мм
рт. ст.). При отборе пробы газа один конец тройника соединяют
с источником газа, а другой с манометром. Когда давление в
сосуде достигнет.атмосферного давления, отбор пробы прекращают,
кран закрывают и сосуд отсоединяют от источника газа. Для
определения количества отобранного газа сосуд взвешивают до
и после отбора пробы. Извлекают газ из такого пробоотборника
путем засасывания в пипетку газоанализатора.
Пипетку 4 применяют для отбора небольшого количества
газа. На обоих концах пипетки имеются краны. Для отбора пробы
нижний конец пипетки соединяют с источником газа, а
верхний — с водоструйным насосом. Затем открывают оба крана и
через пипетку пропускают газ в течение 5—10 мин. Когда из
пипетки будет полностью вытеснен находящийся в ней воздух
и она заполнится отбираемым газом, отбор прекращают,
закрывают верхний кран, потом нижний и отсоединяют пипетку от
насоса и источника газа. Для перевода газа из пипетки в
газоанализатор ее верхний конец присоединяют к прибору, а пипетку
почти всю погружают в раствор хлористого натрия и открывают
оба крана. Раствор хлористого натрия, заполняя пипетку,
вытесняет из нее газ в газоанализатор. При массовых анализах
для удобства транспортировки проб делают батареи газовых
пипеток, которые устанавливают в одном штативе.
ГЛАВА 2. СЫРЬЕ
Состав растительных тканей
Древесина хвойных и лиственных пород, кукурузная
кочерыжка, подсолнечная лузга, хлопковая шелуха, гуза-пая и
другие подобные материалы, которые являются сырьем для
гидролизных производств, состоят из целлюлозы, гемицеллюлоз,
лигнина и экстрактивных веществ.
16
Целлюлоза — полисахарид высокой степени
полимеризации, макромолекулы которого построены из остатков ?-D-глю-
козы, соединенных глюкозидными связями 1—4 в длинные
цепочки. Эмпирическая формула целлюлозы (CeHioOsJn [6].
ноч ,в
V—
н—с—он
I
но—с—н о
н—с—он
н—с
н
он
н
он н
н
н
о он
сн2он
н он
он
сн2он
сн2он
сн2он
-1)-глюкоза
Н
н А а
|/ Л
н /ч
|/н- ЛгоП/он н
0 К он и Л |\н
х——/ н н N о
н
он
СУ ОН
л-2
Часть молекулы целлюлозы
СН2ОН
Макромолекулы целлюлозы образуют микрофибриллы —
тончайшие волокна, в которых они связаны силами
межмолекулярного взаимодействия (силами ван дер Ваальса) и водородными
связями. На некоторых участках микрофибрилл макромолекулы
находятся в строгом порядке, образуя кристаллиты, но в
большей части фибрилл макромолекулы расположены беспорядочно,
переплетаясь между собой и с макромолекулами других
полисахаридов и лигнина. Самая чистая целлюлоза, выделенная из
растительной ткани, содержит до 10% ксилана и маннана.
Целлюлоза — трудногидролизуемый полисахарид.
Гемицеллюлозы — легкогидролизуемые полисахариды
различные по составу и строению. В состав гемицеллюлоз
входят пентозаны, гексозаны, пектиновые вещества и смешанные
полисахариды. Степень полимеризации полисахаридов
гемицеллюлоз значительно ниже, чем у целлюлозы. Их макромолекулы
имеют линейное строение, иногда с боковыми ответвлениями 17].
2 Заказ № 142
17
Пентозаны являются главной составной частью гемицел-
люлоз многих растительных тканей. Они разнообразны по
своему составу и строению. Молекулы пентозан бывают в виде
линейных и разветвленных цепочек. Боковыми ответвлениями
являются остатки пентоз, метилпентоз и уроновых кислот.
Название пентозану дают по названию пентозы, из остатков
которой состоит основная цепочка его молекулы. В растительных
тканях, перерабатываемых гидролизными производствами,
содержится ксилан, арабан и смешанные пентозаны. Их
эмпирическая формула СНО^
ОН
нон2с
^-D-ксилоза (пираноза) ы-Ь-араЬиноза (фураноза)
Наиболее распространенным и изученным пентозаном
является ксилан. Молекулы ксилана различных растительных тканей
отличаются друг от друга. Часто они представляют собой
разветвленные цепочки. Основная цепочка состоит из остатков
?-D-ксилопиранозы, которые связаны между собой глюкозиднои
связью 1—4. Боковые ответвления состоят из остатков 4-метил-
D-глюкуроновой кислоты в положении 1 — 2 или 1 — 3, а также
из остатков ксилозы и арабинозы. Ксилан хвойных пород
древесины содержит около 20% остатков 4-метил-О-глюкуроновой
кислоты, а ксилан лиственных пород—10%. При гидролизе
ксилана образуется ?-D-ксилоза, глюкуроновая кислота и иногда
арабиноза
Н
н
н
о-»
Часть молекулы ксилана (араіїометоксиглюкуроноксилан)
18
Арабан является составной частью пектиновых веществ.
Его количество в гемицеллюлозах значительно меньше
количества ксилана. Молекула арабана, который встречается в
пектиновых веществах гемицеллюлоз древесины, представляет собой
разветвленную цепочку. Основная цепочка состоит из остатков
a-L-арабофуранозы, которые связаны между собой глюкозид-
ной связью 1—5. Боковые ответвления состоят тоже из
остатков a-L-арабинозы. Арабаны других растительных тканей
изучены мало.
Гексозаны являются одной из главных составных частей
гемицеллюлоз и в незначительных количествах находятся в
целлюлозе. Гексозаны — полисахариды, которые при гидролизе
дают гексозы. В отличие от целлюлозы они обладают
более низкой степенью полимеризации. Название
гексозанам дают по названию гексоз, из остатков которых состоит
основная цепочка полисахарида. В растительных тканях,
перерабатываемых гидролизными производствами, находят маннан,
галактан и смешанные полисахариды — глюкоманнан и галакто-
глюкоманнан. Их эмпирическая формула (СбНюО5)п- Гексозаны
различных растительных тканей по своему строению и составу
сильно отличаются друг от друга. Молекулы гексозанов бывают
в виде линейных и разветвленных цепочек. Некоторые
гексозаны, как маннан, содержат метоксильные, ацетильные и
карбоксильные группы. Строение гексозанов очень разнообразно
и мало изучено. Маннан находят в целлюлозе, полученной из
хвойных пород древесины, поэтому при определении содержания
маннана в образце растительной ткани интересуются
содержанием легкогидролизуемого маннана, трудногидролизуемого
маннана и общего содержания маннана в ней.
Пектиновые вещества—-сложный комплекс, в
котором цепочки полисахаридов—• арабана и галактана связаны
с цепочками пектиновой кислоты. Молекулы пектиновой кислоты
представляют собой прямые цепочки из остатков a-D-галакту-
роновой кислоты, которые примерно иа 75% метилированы
Н ОН СООСНз
или метилированная полигалактуронодая кислота
в положении 1—4. При гидролизе пектиновых веществ
образуется галактуроновая кислота, арабиноза и галактоза.
2* 19
В некоторых растительных тканях пектиновая кислота
присутствует в виде солей.
Гексоуроновые кислоты находятся в гемицеллюло-
зах растительных тканей в виде самостоятельных полимерных
соединений (пектиновая кислота) или в виде боковых
ответвлений полисахаридных молекул
V0 V
I I I
неон неон hoch
' І і І
HOCH HOCH HOCH
і f I
HOCH HCOH HCOH
- І І І
HCOH HCOH HCOH
і І І
COOH COOH COOH
D-галатуронодая JJ-елтуроноВая В-маннуронобая
кислота кислота кислота
В гидролизатах гемицеллюлоз различных растительных
тканей найдены галактуроновая, глюкуроновая и маннуроновая
кислоты. Содержание гексоуроновых кислот в растительных
тканях колеблется в больших пределах. В лиственных породах
древесины уроновых кислот в 2 раза больше, чем в хвойных. В
молодых растительных тканях и в коре их содержится значительно
больше, чем в древесине ствола. Особенно много уроновых
кислот в хлопковой шелухе и в подсолнечной лузге. Глюкуроно-
вую кислоту находят в ксилане многих растительных тканей.
Она в виде остатков 4-метил-о-глюкуроновой кислоты
составляет боковые ответвления от основной цепочки из остатков
ксилозы. Наиболее распространенной из гексоуроновых кислот
является галактуроновая кислота, которая входит в состав
пектиновых веществ.
Лигнин представляет собой смесь полимерных соединений,
в основе которых лежат ароматические вещества. У различных
растений природа ароматических звеньев лигнина неодинакова.
Лигнин древесины хвойных пород состоит из производных
пирокатехина, а лигнин древесины лиственных пород — из
производных пирокатехина и пирогаллола. Функциональными группами,
определяющими свойства лигнина, являются метоксильные и
гидроксильные группы — фенольные и спиртовые (в боковой це-
20
почке). Структура лигнина многообразна. В настоящее время
считают установленным, что лигнин и углеводы в растительных
тканях являются двумя взаимопроникающими системами, связь
между которыми не только механическая, но и химическая
[6, с. 80]. Часть лигнина находится в свободном состоянии, ее
можно извлекать органическими растворителями (спирт, диок-
сан). Большая же часть лигнина находится в связанном
состоянии, для ее извлечения из растительной ткани применяют
жесткие условия реакций, которые изменяют в какой-то мере
строение природных макромолекул лигнина.
Минеральные вещества входят в состав
растительных тканей. Их природа и количество зависят от вида
растительной ткани, условий произрастания и других причин.
Минеральные вещества разделяют на растворимые и нерастворимые
во время гидролиза полисахаридов. Растворимые минеральные
вещества состоят из солей калия, натрия и других металлов
с угольной, серной, соляной и кремневой кислотами.
Нерастворимые вещества, которые после гидролиза полисахаридов
остаются в лигнине, состоят из окислов кальция, магния, железа,
марганца и их солей с фосфорной и кремневой кислотами. При
определении качества сырья, предназначенного для гидролиза,
интересуются содержанием в нем растворимых минеральных
веществ, так как, переходя в раствор во время гидролиза, они
вступают в реакцию с серной кислотой, понижают ее
концентрацию, что замедляет процесс гидролиза.
Экстрактивные вещества присутствуют во всех
растительных тканях. Они очень разнообразны по составу и
количественному соотношению компонентов в зависимости не только
от природы, но и от микроструктуры растительной ткани, поэтому
их классифицируют на следующие группы: вещества,
экстрагируемые водой, вещества, экстрагируемые эфиром, спиртом и
другими органическими растворителями. При водной обработке
в раствор переходят пектиновые вещества, моно-, ди- и трисаха-
риды, некоторые минеральные вещества, красители, танниды,
циклические спирты, органические кислоты — уксусная и
муравьиная, а также некоторые вещества, экстрагируемые
органическими растворителями. При обработке растительной тканіг
органическими растворителями в раствор переходят смолы, воски,
масла, фенолы, терпены, алифатические и ароматические
углеводороды, стерины и некоторые вещества, экстрагируемые водой.
Содержание веществ, экстрагируемых водой, в древесине
ели и сосны 2—3%, в древесине дуба до 7%, а в лиственнице —
от 13 до 22%. В растительных тканях однолетних растений их
содержание колеблется от 3 до 13%. Содержание веществ,
экстрагируемых органическими растворителями, обычно
составляет не более 2%, но древесина хвойных пород содержит до
25% смол. В табїі. 1 приведены данные о составе некоторых
растительных тканей [8, 9, 10, 11, 12, 13].
21
Химический состав растительных
Название
растительной ткани
Древесина сосны
Древесина ели
-Древесина пихты
-Древесииа листвениицы
Древесина осины
Древесина березы
;Кора сосиы
Луб сосиы
Шишки сосны
Кора березы
Прикамбиальный слой
березы
Сок березы до инверсии
Сок березы после
инверсии
Сок оснны до инверсии
Сок осины после
инверсии
Прикамбиальный слой
осины
Одубииа древесины дуба
Одубина коры ивы
Кукурузная кочерыжка
Подсолнечная лузга
-Хлопковая шелуха
-Хлопковые коробочки
Туза-пая
'Овсяная шелуха
Рисовая шелуха
Стебель тростника
Виноградная лоза
Торф
Полисахариды
легко-
гидроли-
зуемые
17,84
17,30
14,93
17,82
20,29
26,54
12,40
23.30
20,57
22,52
29,26
0,55
Нет
0,81
Нет
21,15
20,79
12,35
37,94
7,06
4,90
17,57
20,55
34,72
18,10
21,05
27,14
17-40
трудио-
гидроли-
зуемые
47,66
48.02
44,22
38,57
44,04
39.40
14.20
13.90
36,30
17,95
18,01
Нет
„
*
я
34,57
37,35
31,85
33,39
32,00
34.30
37,43
38,32
28,62
29.05
38.90
30,84
2-18
Гексозы
Глюкоза
51,79
53,24
48,60
41.64
48.08
41,25
13.78
17,67
33,26
19.21
16.65
2.06
2.43
2,10
2,29
32,92
40.09
34,86
38,71
33,00
34,50
41,50
43,15
33,31
34.59
41,50
38.71
.8-24.8
Манноза
12,12
9,64
8,97
12.63
1.51
1.22
5,75
8,46
11,76
2.36
Нет
п
0,73
0.59
Следы
Нет
"Я
Следы
я
1.34
Нет
ж
0.54
1.38
0,6-3,5
Галактоза
2.03
1,02
0,76
2.48
0.83
1,31
1,02
2,04
4,51
1,16
11,88
Нет
я
п
7,13
1,67
1,36
2.06
1,02
0,50
1,39
2,52
1,26
1,02
0,78
1.94
0.2—4,2
Фруктоза
—
—
—
—
Нет
—
—
—
—
0,28
1.47
0,79
1.66
1.26
0,74
—
—
—
—
— ;
—
—
—
—
—
—
22
тканей (% от абс. сухого вещества)
ТАБЛИЦА I
Пеитозы
Ксилоза
5,34
5,01
4.30
3,43
17,83
24.15
2,06
0,79
5,02
13.26
10,96
Нет
Следы
Нет
0,15
8.85
17,58
8,36
33,80
9,10
15,20
14,22
12.05
32,77
15,62
20.50
17.41
2,2-6.1
Арабиноза
і.
0,
і
0.
0
0.
3
8
2
3
7
52
79
52
70
73
88
99
32
32
98
16
Нет
0
19
Нет
0
4
0
0
3
0
1
1
2
3
2
2
1
20
.40
.98
.84
77
90
.20
.42
,08
,24
.04
.25
.51
0-0.9
Пенто-
заны
8
18
24
6
12
4
95
56
57
76
24
80
Метил-
пентозы
Рамноза
0.04
Нет
»
Следы
—
—
—
—
0.34
Нет
¦
»
-
0.95
Нет
—
Нет
Следы
,
Нет
я
¦т
»
0,1-1.6
Уроно-
вые
кислоты
4
4
3
3
7
3
0
1
2
5
2
2
5
8
7
0
I
13
10
5
4
4
8
00
15
97
47
96
71
35
24
48
31
70
.98
,57
,89
.42
.23
,70
,78
,70
.98
,48
,93
,59
Зола
0
0
0
0,
0
0,
1
2
0
0
6
I
4
3
7
18
3
2
17
27
53
22
26
14
39
19
52
52
23
10
92
54
,56
.00
41
.60
Вещества,
экстрагируемые
водой
3,19
2,76
3.39
13,02
2,82
1,41
—
—
—
—
—
—
—
—
—
—
3,35
6,03
3,60
—
—
12.85
8,16
5,96
11,66
9,16
7.16
—¦
спиртом,
эфиром
1,78
0,94
0,70
0.82
0,82
0,91
—
—
—
—
—
—
—
—
—
0,72
3,19
0,30
—
—
1,70
1,04
0.33
2,07
0,49
0,85
¦
Меток-
силь-
ные
группы
4
5
4
5
5
3
1
2
2
3
3
2
2
2
4
32
62
01
70
60
75
94
59
47
07
98
,92
71
,17
36
Лиг
24
28
29
24
21
19
43
17
24
27
27
15
26
26
21
25
17
18
20
38
НИН
70
07
87"
61
81
74'
63
12
71-
51
65
18
00
00
29
63
.17
96
,8&
8S
10-25
Гидролиз полисахаридов
Гидролиз полисахаридов — основная реакция, по которой
изучают состав и строение растительной ткани, подлежащей
переработке, дают оценку сырью для полугения того или иного
продукта и выбирают оптимальный режим получения
моносахаридов. По скорости гидролиза полисахариды разделяют на
легко- и трудногидролизуемые. По количеству пентоз или
фурфурола в гидролизате выделяют так называемое пентозансодер-
жащее сырье, которое имеет повышенное содержание пентоз.
Это сырье целесообразно использовать для получения
фурфурола, кристаллической ксилозы, ксилита или ксилитана. По
содержанию общего количества гексоз (сбраживаемых Сахаров)
и отдельно из них глюкозы определяют целесообразность
использования сырья для получения этилового спирта и
кристаллической глюкозы. Для выращивания дрожжей одинаково
пригодны пентозные и гексозные сахара. При производстве
дрожжей важно знать суммарное потенциальное содержание
моносахаридов в сырье.
Все методы анализа сырья с целью установления состава,
строения н свойств полисахаридов основаны на определении
отдельных компонентов, содержащихся в гидролизатах
полисахаридов, полученных при различных условиях гидролиза.
Наиболее полную информацию о составе моносахаридов, олигосаха-
рпдов и уроновых кислот, присутствующих в гидролизатах,
получают тогда, когда делают анализ хроматографическим
методом на бумаге. Этим методом сахара и другие вещества
разделяют и выделяют из смеси с различными веществами в том виде
в каком они присутствуют в гидролизатах без изменений, а
затем определяют их количество. При анализе смесей углеводов
¦с помощью газожидкостной хроматографии сахара необходимо
сначала превратить в летучие производные, а затем провести их
разделение и определение. Без потерь перевести сахара в
летучие производные невозможно, поэтому результаты анализов
менее точны, но время, затрачиваемое на анализ этим методом,
значительно меньше [14].
Однако как бы точен ни был метод определения углеводов
в гидролизатах, надо всегда иметь в виду, что гидролиз
полисахаридов очень сложный процесс, в котором одновременно
протекает много реакций и образуется очень много разнообразных
продуктов, состав и свойства которых не установлены до сих
пор. При гидролизе гемицеллюлоз образуются моносахариды,
метилированные и ацетилированные пентозы и гексозы, уроно-
Бые кислоты и другие вещества. При гидролизе целлюлозы
сначала появляются продукты различной степени полимеризации —
декстрины, олигосахариды, моносахариды, а потом продукты
распада моносахаридов. Минеральные и экстрактивные вещества,
находящиеся в растительных тканях, в условиях гидролиза
24
тоже переходят в раствор. Таким образом, гидролизаты
представляют собой растворы различных моносахаридов, продуктов
их распада и других веществ, которые перешли в раствор при
гидролизе растительной ткани. При выборе оптимального
режима гидролиза руководствуются количеством моносахаридов,
найденных в полученных гидролизатах. Чем больше удалось
получить моносахаридов, тем меньше их разложилось, тем полнее
прошел гидролиз полисахаридов. Выход моносахаридов в
производственных условиях варки составляет 50—60% от
потенциально возможных. Следовательно, 40—50% моносахаридов
разлагаются. Некоторые продукты разложения моносахаридов
обладают токсическими свойствами по отношению дрожжей.
Они тормозят биохимические процессы. Смолоподобные
продукты разложения моносахаридов выпадают в осадок при
охлаждении гидролизата, вызывая карамелизацию аппаратуры.
Особенности процесса гидролиза полисахаридов надо учитывать
при анализе сырья и полупродуктов, а также при разработке
технологических режимов.
Состав сульфитного щелока
Сульфитный щелок представляет собой водный раствор
многих органических и минеральных веществ, которые образуются
при сульфитной варке целлюлозы. В зависимости от жесткости
режима варки и плотности загрузки древесины в котел
содержание сухих веществ в сульфитном щелоке колеблется от 10
до 20%, органических веществ от 9 до 15%. Минеральные
вещества составляют около 10% от сухих веществ.
Органическими веществами, присутствующими в сульфитном
щелоке, являются продукты гидролитического распада гемицел-
люлоз и продукты взаимодействия варочной кислоты с
лигнином. Во время варки сульфитной целлюлозы в результате
гидролитического распада гемицеллюлоз в раствор переходят: мо1
носахариды — глюкоза, манноза, галактоза, ксилоза, арабиноза;
метилпентозы; уроновые кислоты; метиловый спирт; продукты
неполного гидролиза — олигосахариды; продукты распада
моносахаридов — фурфурол, оксиметилфурфурол, метилфурфурол,
муравьиная кислота, левулиновая кислота и др. В результате
взаимодействия варочной кислоты с лигнином в раствор
переходят высокомолекулярные лигносульфоновые кислоты. Реакция
сернистой кислоты с моносахаридами приводит к образованию
альдегидбисульфитных соединений [2].
Минеральную часть сухих веществ сульфитного щелока
составляют минеральные вещества, содержащиеся в гемицеллюло-
зах и в лигнине древесины, и вещества, присутствующие в
варочной кислоте: сернистая кислота, сульфит кальция, магния,
аммония и натрия в зависимости от ее состава. Возможно
присутствие мышьяка и селена. При оценке сульфитного щелока
25
как сырья для получения кормовых дрожжей и этилового спирта
-основными показателями являются количество и состав Сахаров,
содержащихся в нем. В сульфитном щелоке определяют
концентрацию редуцирующих веществ (РВ) или концентрацию
редуцирующих Сахаров после удаления редуцирующих несахаров,
которых в сульфитном щелоке содержится до 25% от РВ. В
отдельных случаях, когда меняют сырье или режим варки
целлюлозы, делают анализ сульфитного щелока на содержании
каждого сахара хроматографическим методом. Кроме Сахаров,
-в сульфитном щелоке определяют содержание сухих веществ,
серы, свободной и связанной двуокиси серы (SO2), кальция,
магния, минерального азота и мышьяка.
Определение гранулометрического состава древесного сырья
Для определения качества древесного сырья по степени его
«змельчения и по содержанию в нем коры на гидролизных
заводах применяют метод, основанный на отделении опилок
просеиванием через сито и сортировке крупных частиц вручную.
Мешающие вещества. Кора в виде опилок.
Аппаратура. Сито с диаметром отверстий 6 мм.
Подготовка и хранение проб. Пробу до анализа хранят в
хорошо закрытом бидоне во избежание изменения влажности.
Ход анализа. Из средней пробы древесного сырья на анализ
берут около 2 кг. Взвешенную пробу просеивают через сито над
противнем. Отделенные опилки быстро собирают во взвешенную
банку, закрывают пробкой и взвешивают. Оставшуюся
древесину на сите сортируют вручную на щепу кондиционных и
некондиционных размеров, стружку и кору. Взвешиванием
определяют количество каждой фракции.
Расчет. Содержание в анализируемом сырье каждой фракции выражают
в процентах, вычисляя по следующей формуле:
х__ fllOO
~~ g
где X— содержание выделенной фракции в исследуемой пробе, %;
а—количество выделенной фракции, г; g — количество сырья, взятого на анализ, г.
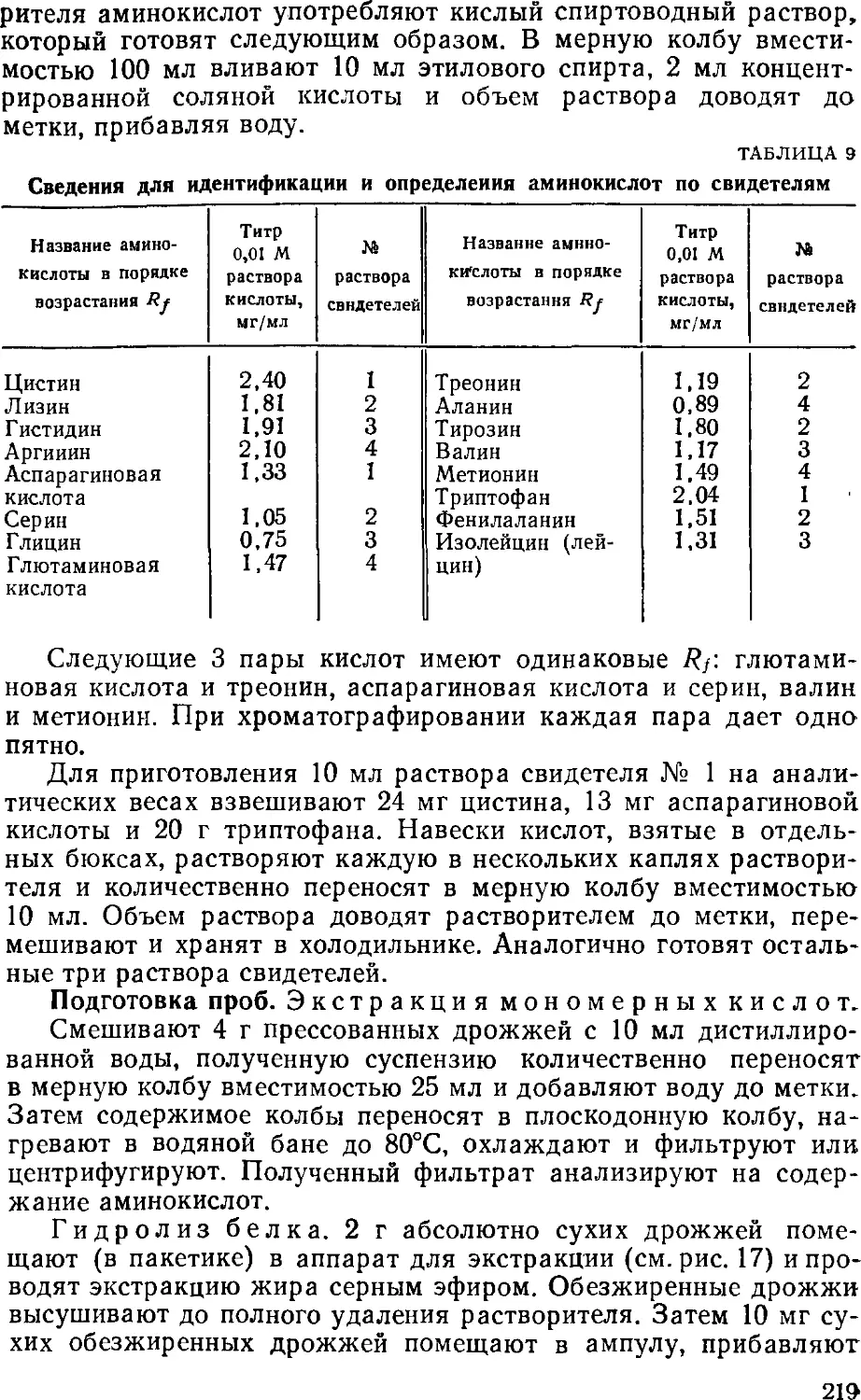
Относительное содержание фракций в пробе определяют с точностью
±3—5% отн. Точность определения коры значительно меньше, так как
измельченную кору определяют вместе с опилками.
Предостережение. Анализ надо проводить вдали от нагревательных
приборов, от тяги и открытых окон, чтобы уменьшить ошибки за счет испарения
влаги при сортировке сырья.
Определение содержания хвойных и ляствеиных пород древесины
Для анализа сырья на содержание в нем древесины
лиственных или хвойных пород применяют метод, основанный на
реакциях получения окрашенного соединения при действии надреве-
•26
сину последовательно перманганата калия, соляной кислоты и
аммиака. Хвойные породы дают желтое соединение, а
лиственные — красное. Окрашенную древесину сортируют по цвету и
определяют содержание каждой фракции.
Реактивы и их приготовление. 1. Перманганат калия (КМпО4)
1%-ный раствор. 2. Соляная кислота 12%-ный раствор. 3.
Аммиак 1%-ный раствор.
Ход анализа. Около 100 г анализируемой пробы помещают
в стакан вместимостью 500 мл и заливают 1%-ным раствором
перманганата калия. Через 2 мин раствор перманганата
сливают, пробу древесины хорошо промывают водой, а затем
заливают 12%-ным раствором соляной кислоты, ждут 2 мин,,
кислоту сливают и пробу хорошо промывают водой. Пробу,,
отмытую от кислоты, заливают 1%-ным раствором аммиака,
выдерживают 1 мин и раствор сливают. Древесина хвойных
пород при этом окрашивается в желтый цвет, а лиственных —
в красный. Мокрую пробу помещают на фильтровальную
бумагу для подсушивания, а затем сортируют на частицы разного'
цвета и каждую фракцию взвешивают.
Расчет. Содержание в анализируемой пробе хвойных н лиственных пород
древесины выражают в процентах и вычисляют по формуле
elOO
X:
g
где X — содержание хвойной (лиственной) древесины в пробе, %; а —
количество хвойной (лиственной) древесины, г; g — количество пробы, взятой на
анализ, г.
Содержание хвойных и лиственных пород в древесном сырье определяют
с точностью ±3—4%.
Экспрессный метод определения влажности сырья
Метод предназначен для быстрого определения влажности
в больших навесках проб сырья [15]. Метод основан на
высушивании анализируемого материала в токе горячего воздуха да
полного удаления влаги.
Анализируемая часть пробы должна быть однородна по
размерам частиц. Подсолнечную лузгу, хлопковую шелуху и
другие материалы, состоящие из частиц примерно одинакового
размера, анализируют без сортировки. Древесное сырье сортируют
на опилки, щепу и стружки, а затем каждую фракцию
анализируют отдельно. Влажность средней пробы древесного сырья
вычисляют на основании результатов определения
гранулометрического состава и влажности каждой фракции.
Мешающие вещества. Летучие компоненты растительных
тканей.
Аппаратура. Вентиляторный прибор.
Вентиляторный прибор (рис. 9) состоит из
центробежного вентилятора 1 с электромотором 2, электрокалори-
27
фера 3 и сушильной камеры 4. Центробежный вентилятор и
электромотор служат для засасывания воздуха в калорифер
через шторный затвор 5, которым регулируют количество воз-
у духа, поступающего в
прибор. Электрокалорифер,
предназначенный для
нагревания воздуха, подаваемого
в сушильную камеру,
состоит из каркаса фарфоровых
трубок с намотанной на них
нихромовой проволокой,
который помещен в
металлический кожух 6. Нагревание
воздуха до нужной
температуры регулируют реостатом,
смонтированным на панели,
и контролируют
термометром, помещенным перед
сушильной камерой. На
воронке из белой жести 7
находится сушильная камера,
представляющая собой
алюминиевую чашку, дно и
крышка 8 которой сделаны
из сетки. Сушильная камера
легко вставляется и
снимается с воронки прибора.
Температуру воздуха,
прошедшего через
анализируемую пробу, измеряют
термометром.
Подготовка и хранение
проб. Среднюю пробу сырья
хранят в бидоне, плотно
закрытом крышкой, или в
эксикаторе. Древесное сырье
перед анализом сортируют
на опилки, щепу и стружки,
определяют относительное
содержание каждой
фракции в пробе (см. с. 26).
Рис. 9. Вентиляторный прибор для
определения влажности сырья
Ход' анализа.
Вентиляторный прибор ставят в
вытяжной шкаф. Сушиль-
„..„ кямет снимают с прибора, помещают в нее около
20? г пробы и взвшиваютна технических весах с
точностью до 0 5 г Затем камеру с навеской ставят на воронку,
включают прибор в электрическую сеть и полностью открывают
28
шторный затвор. Во время сушки регулируют температуру
воздуха, подаваемого в сушильную камеру. В начале сушки
температура воздуха до камеры должна быть около П0°С,
а в конце—105°С. Степень высушивания ориентировочно
определяют по температуре воздуха, выходящего из сушильной
камеры. Когда температура воздуха, выходящего из камеры,
достигнет 101—102°С, камеру снимают с прибора, быстро
взвешивают на технических весах и снова ставят на включенный
прибор для контрольного досушивания. Через 10—15 мин
сушильную камеру с пробой снимают с прибора и снова
взвешивают. Еслн за последние 10—15 мин сушки количество пробы
изменится не более чем на 0,5 г, то анализ считают
законченным. Время высушивания для опилок составляет не более 1 ч,
для щепы 1,5—2 ч, в зависимости от размера ее частиц. После
каждого анализа камеру очищают от пробы специальной
щеточкой.
Расчет. В растительных материалах определяют относительную или
абсолютную влажность. При вычислении относительной влажности количество
воды, найденное в пробе, относят к количеству влажной пробы, взятой иа
анализ. При вычислении абсолютной влажности количество воды, найденное
в пробе, относят к количеству абсолютно сухой навески.
Относительную Л'отн или абсолютную Хаос влажность сырья выражают
в процентах, вычисляя по следующим формулам:
¦ 100, *абс= g~a • 100.
*отн
где g — количество сырья, взятого на анализ, до сушки, г; а — количество
сырья, взятого иа анализ, после сушки, г.
При анализе однородного сырья полученные результаты считают
влажностью средней пробы анализируемого сырья. При анализе смешанного сырья
влажность средней пробы Xcv вычисляют с учетом данных о
гранулометрическом составе по следующей формуле:
У
100 n 100 '
где X\ — влажность опилок, %; *2— влажность щепы, %; ai — содержание
опилок в средней пробе сырья, %; а2 — содержание щепы в средней пробе
сырья, %.
Влажность в однородном сырье определяют с точностью ±2—3% отн.
Предостережение. Сортировку пробы надо делать быстро, вдали от
нагревательных приборов, открытых окон и от тяги.
Определение влажности сырья различного гранулометрического состава
Для определения влажности сырья без сортировки его на
фракции частиц различного размера [16] применяют метод,
основанный на высушивании пробы материала в сушильном
шкафу в два приема. Сначала навеску исследуемого материала
около 300 г сушат до воздушносухого состояния, затем ее
измельчают, отбирают из нее среднюю пробу в количестве не
29
более 5 г и определяют влажность отобранной пробы,
высушивая ее до полного удаления влаги.
Мешающие вещества. Летучие вещества, содержащиеся
в пробе.
Аппаратура. Сушильный шкаф. Противень из белой жести.
Ход анализа. На противень помещают 200—300 г средней
пробы сырья и взвешивают на технических весах. Затем пробу
шпателем распределяют на противне ровным слоем высотой
не более 1 см и ставят в сушильный шкаф при температуре
105°С. Через 3—4 ч противень с пробой вынимают из
сушильного шкафа, охлаждают иа воздухе до комнатной температуры
и взвешивают. Так определяют влажность сырья в большой
навеске, высушенной до воздушносухого состояния.
Подсушенную пробу измельчают ножом до размеров опилок,
очень хорошо перемешивают, берут из нее среднюю пробу и
определяют ее влажность высушиванием до полного удаления
влаги при 105°С в сушильном шкафу или облучением
инфракрасной лампой, как описано ниже при определении влажности
измельченного сырья.
Расчет. Относительную влажность исходной пробы сырья X вычисляют
в процентах по формуле
go
где X — относительная влажность сырья, %; W — относительная влажность
сырья при подсушивании его до воздушносухого состояния, %; go —
количество исходной пробы, взятое для подсушивания до воздушносухого
состояния, г; g — количество исходной пробы после подсушивания, г.
Относительную влажность пробы при подсушивании ее до воздущносухого
состояния вычисляют в процентах по формуле
_._ (оо-а)ІОО
а0
где по — количество воздушносухой пробы, взятое на анализ, г; а —
количество воздушносухой пробы после высушивания до полного удаления влаги, г.
Влажность сырья определяют с точностью ±4—6% отн.
Определение влажности измельченного материала
Для определения влажности сырья и других материалов,
измельченных до размеров частиц 2—0,25 мм, применяют метод,
основанный на высушивании исследуемой пробы материала да
полного удаления влаги при нагревании его инфракрасными
лучами специальной лампы [17] или в сушильном шкафу при
температуре 105°С. Время высушивания навески пробы
инфракрасной лампой составляет 8—10 мин, а в сушильном шкафу — 3—5 ч.
Мешающие вещества. Летучие компоненты анализируемого»
материала.
30
Аппаратура. Установка для высушивания пробы
инфракрасной лампой или сушильный шкаф.
Установка для нагревания пробы инфракрасными
лучами, показанная на рис. 10, представляет собой лампу
накаливания /, у которой верхняя часть купола имеет зеркальную
поверхность, служащую рефлектором. Для защиты глаз от
яркого света верхняя часть лампы закрыта кожухом 2. Лампа
укреплена на штативе.
Ход анализа при высушивании инфракрасной лампой. В
предварительно высушенном и взвешенном алюминиевом бюксе 3
с крышкой (или в чашке Петри) взвешивают 10—12 г
измельченного материала с точностью до 0,01 г.
Бюкс с навеской помещают на асбест
или на фарфоровую подставку в
центре освещенного круга включенной
лампы. Крышку с бюкса убирают.
Расстояние между поверхностью, на которую
устанавливают бюкс с навеской, и
ближайшей к ней точкой лампы должно быть
10 см для лампы мощностью 500 Вт
и 7 см для лампы мощностью 250 Вт.
Продолжительность облучения навески
лампой мощностью 500 Вт 8 мин,
а 250 Вт— 10 мин. Во время облучения
пробу периодически перемешивают
стеклянной палочкой. После облучения
пробы бюкс закрывают крышкой, ставят на
металлическую плиту, охлаждают в
течение 4—5 мин и взвешивают. Затем
облучение пробы повторяют в течение 1 мин и снова
взвешивают. Если количество пробы изменилось не более, чем на 0,02 г,
то высушивание считают законченным.
Ход анализа при высушивании в сушильном шкафу. В
предварительно взвешенный высушенный стеклянный бюкс
помещают 2—3 г исследуемой пробы, закрывают его крышкой и
взвешивают с точностью ±0,0002 г. Затем бюкс с пробой
ставят в сушильный шкаф, открывают и оставляют при
температуре 105°С на 3 ч. Высушенную пробу в бюксе закрывают
крышкой в сушильном шкафу, бюкс переносят для охлаждения
в эксикатор с серной кислотой или с хлористым кальцием.
Охлажденный бюкс с пробой взвешивают и снова ставят в
сушильный шкаф для досушивания в течение 1 ч. Досушивание
повторяют до тех пор пока разница между двумя последующими
взвешиваниями не будет превышать 0,0003 г.
Расчет. Относительную влажность аналнзнруемой пробы вычисляют в
процентах по формуле (^-в) 100
Рис. 10. Установка для
высушивания материала
инфракрасной лампой
31
где X — влажность исследуемого материала, %; g — количество пробы, взятое
на анализ, до сушки, г; а — количество пробы после сушки, г.
Влажность определяют с точностью ±1—3% отн.
Определение минеральных веществ (золы)
Для определения растворимых минеральных веществ в сырье
гидролизных заводов (см. с. 21) предназначен метод, состоящий
из двух анализов: определения золы в исследуемом сырье и
определения золы в лигнине после удаления полисахаридов
путем гидролиза. По разности результатов этих анализов находят
содержание растворимых минеральных веществ в исследуемом
сырье, которые при гидролизе переходят в раствор.
Аппаратура. Муфельная печь. Установка для кипячения с
обратным холодильником.
Реактивы и их приготовление. 1. Перекись водорода 3%-ный
раствор. 2. Серная кислота 80%-ный раствор. 3. Индикатор
метиловый оранжевый 0,1%-ный раствор в воде.
Подготовка пробы. Исследуемую пробу в количестве около
10 г измельчают, хорошо перемешивают, если надо
подсушивают до воздушносухого состояния и хранят в плотно закрытой
склянке. Перед анализом в пробе определяют влажность
высушиванием в сушильном шкафу или инфракрасной лампой
(см. с. 30).
Ход анализа. 1. Определение суммы
растворимых и нерастворимых минеральных веществ.
Во взвешенный фарфоровый тигель берут навеску сырья 2—3 г
с точностью 0,0002 г и ставят его на включенную электрическую
плитку под тягой. Когда навеска сырья сгорит, тигель помещают
в муфельную печь и остаток прокаливают при температуре 600°С
в течение 3—4 ч для полного удаления углерода. Если через 4 ч
прокаливания зола будет иметь черные частицы или серый цвет,
то ее смачивают 3—5 каплями 3%-ного раствора перекиси
водорода и тигель ставят на включенную электроплитку для
испарения жидкости. Потом тигель с золой снова помещают в
муфельную печь, прокаливают в течение 1 ч при температуре 600"С
и переносят в эксикатор. После охлаждения до комнатной
температуры тигель с золой взвешивают и снова ставят в
муфельную печь для контрольного прокаливания на 1 ч. Если при этом
количество золы изменилось не более чем на 0,0003 г, то
прокаливание считают законченным.
2. Определение нерастворимых
минеральных веществ. Берут навеску воздушносухой пробы около 3 г,
отвешивая с точностью до 0,0002 г, помещают ее в фарфоровую
чашку, заливают 50 мл 80%-ной серной кислотой и хорошо
перемешивают стеклянной палочкой. Смесь выдерживают 4 ч,
перемешивая ее палочкой через 15—20 мин. Затем к смеси
добавляют немного воды и количественно переносят ее в коническую
колбу, расходуя при этом не более 500 мл воды. Колбу закры-
32
вают стеклом и ставят на кипящую водяную баню. Через 3 ч
содержимое колбы фильтруют через беззольный фильтр на
воронке Бюхнера. (Во время этой операции надо принять все меры
предосторожности, чтобы избежать потерь осадка!) Осадок
отмывают горячей водой от серной кислоты до отрицательной
реакции по метиловому оранжевому. Фильтр с осадком переносят
во взвешенный, прокаленный тигель, ставят на элекроплитку
для сжигания, а потом в муфельную печь для озоления. Далее
анализ ведут, как описано выше при определении золы в сырье.
Расчет. Содержание суммы растворимых и нерастворимых минеральных
веществ в абсолютно сухом сырье Х\ вычисляют в процентах по формуле
л\ =
где gi — количество абсолютно сухого сырья, взятое на сжигание, г; п\ —
количество золы после сжигания пробы, г.
Содержание нерастворимых минеральных веществ в абсолютно сухом
сырье А^ вычисляют в процентах по формуле
где #2 — количество абсолютно сухого сырья, взятое на гидролиз, г; а2 —
количество золы после сжигания лигнина, г.
Содержание растворимых минеральных веществ в абсолютно сухом
сырье X находят по формуле
Л ^= Л ] — Л 2.
Количество растворимых минеральных веществ определяют с точностью
±2—4% отн.
Эбулиостатический метод определения редуцирующих Сахаров
(метод Низовкина и Емельяновой)
Метод предназначен для определения редуцирующих саха-
ров в водных растворах. По первому варианту метода можно
анализировать бесцветные или слабоокрашенные растворы,
имеющие концентрацию редуцирующих Сахаров выше 0,08%.
По второму варианту можно анализировать темноокрашенные
растворы и растворы, имеющие концентрацию редуцирующих
Сахаров от 0,08 до 0,001% [18].
Метод основан на реакции восстановления двухвалентной
меди редуцирующими сахарами в щелочной среде при
кипячении в присутствии желтой кровяной соли. Образующаяся закись
меди, реагируя с желтой кровяной солью, дает хорошо
растворимое комплексное соединение, что позволяет проводить анализ
прямым титрованием. Индикатором конца реакции служит
метиленовая синяя, которая в окислительной среде имеет синюю
окраску, а в восстановительной среде она бесцветна.
Окисление редуцирующих Сахаров меднощелочным раствором
является сложным процессом, который состоит из нескольких сотен
3 Заказ № 142 33
реакций, протекающих одновременно [19]. Малейшие изменения
условий анализа, особенно соприкосновение реагирующей
жидкости с кислородом воздуха, оказывают сильное влияние на
количество восстановленной меди. Проведение анализа прямым
титрованием в эбулиостате обеспечивает стандартные легковос-
производимые условия для окисления Сахаров меднощелочным
раствором в токе водяного пара при отсутствии доступа
кислорода воздуха к реагирующей
жидкости. Это дает
возможность получать хорошо
воспроизводимые результаты.
Мешающие вещества.
Органические и неорганические
вещества, которые не являются
сахарами, но
восстанавливают медь в щелочном растворе
(фурфурол, сернистые
соединения и др.).
Аппаратура. Установка для
определения редуцирующих
веществ эбулиостатичсскпм
методом и два дозатора.
Установка для
определения
редуцирующих Сахаров, показанная
на рис. 11, состоит из прибора
эбулиостата, электроплитки
и бюретки на 10 мл с ценой
деления 0,1 мл. Прибор эбулио-
стат имеет внешний сосуд 1
и внутренний сосуд 2. Во
Рис. 11. Установка для определения внешНий сосуд которым явля-
редуцнрующих Сахаров эбулиостати- J^' ?
ческим методом- ется коническая колба
вместимостью 1 л, налита вода. Во
время титрования он служит
парообразователем. Внутренний сосуд, называемый эбулиоста-
том, является реакционным сосудом, в котором проводят
титрование (на рис. 12 он показан отдельно).
Для пропускания пара через реагирующую жидкость внутрь
эбулиостата впаяна трубка, нижний конец которой доходит
почти до дна, а вверху имеется боковое отверстие для выхода
пара во время титрования. На узкую часть эбулиостата надета
пробка, которая плотно вставляется во внешний сосуд. Чтобы
регулировать отвод избыточного пара из сосуда 1 во время
анализа в пробку вставлена стеклянная трубка, на которую надета
резиновая трубка с зажимом. На кончик бюретки надета
резиновая пробка. При опускании кончика бюретки в эбулиостат
пробка плотно закрывает верхнее отверстие, а маленькое боко-
вое отверстие остается открытым. Кончик бюретки во время
титрования должен быть ниже бокового отверстия, чтобы
образующаяся капля сахарного раствора не могла быть увлечена
паром в маленькое отверстие эбулиостата.
Для отмеривания первого и второго растворов служат
дозаторы (рис. 12). Дозатор б представляет собой два
сообщающихся сосуда, которые закрываются стеклянными кранами.
Большой сосуд вместимостью 100 мл предназначен для
хранения раствора сернокислой меди (первого раствора), а
маленький — является пипеткой для его отмеривания. Он имеет две
отметки на верхней и нижней трубках, внутренний диаметр
которых 1—2 мм. Вместимость
сосуда между метками 5 мл.
Для отмеривания первого
раствора открывают кран / и
заполняют сосуд-пипетку до верхней
метки. Затем кран / закрывают,
подставляют эбулиостат под кончик
дозатора и медленно открывают
кран 2. Когда жидкость
выливается из шарика, кран можно
открыть полностью, а в конце
выливания кран надо опять немного
прикрыть.
Дозатор в представляет собой
градуированную пипетку
вместимостью 5 мл, к верхнему концу
которой припаян тройник с
трехходовым краном 3. Боковой
отвод тройника свободен, а на
верхний — надета резиновая груша. Щелочной раствор сегнето-
вой соли (второй раствор) хранят в колбе вместимостью 50—
100 мл, закрытой резиновой пробкой. Для отмеривания 5 мл
второго раствора поворачивают кран 3 так, чтобы груша
сообщалась с пипеткой, а боковой отвод при этом был закрыт.
Нажимают грушу, погружают кончик дозатора во второй раствор
в колбе, медленно отпускают грушу, набирают нужное
количество раствора в пипетку (до метки), повертывают кран так,
чтобы груша сообщалась с воздухом через боковой отвод, а
пипетка была закрыта, убирают колбу со вторым раствором,
кончик дозатора опускают в эбулиостат с первым раствором, снова
повертывают кран и выпускают весь раствор в эбулиостат.
Реактивы и их приготовление. 1. Раствор сернокислой меди
(CuSO4) — 1%-ный раствор. Приготовление. 10 г CuSO4X
Х5Н2О ч. д. а. и 0,04 г метиленовой синей отвешивают на
аналитических весах, количественно переносят в мерную колбу
вместимостью 1 л с помощью дистиллированной воды, объем
раствора доводят до метки и перемешивают. 2. Щелочной раствор
3* 35
Рис. 12.
а — эбулиостат; б, в —дозаторы
сегнетовой соли и желтой кровяной соли — второй раствор.
Приготовление. 50 г сегнетовой соли помещают в стакан
и растворяют в 200—300 мл дистиллированной воды. В другом
стакане в 100 мл дистиллированной воды растворяют 4 г
желтой кровяной соли. В мерную колбу вместимостью 1 л вливают
150 мл 50% -ного раствора едкого натра и раствор сегнетовой
соли, раствор перемешивают, прибавляют раствор желтой
кровяной соли и воды до метки и снова перемешивают. 3. Глюкоза
марки «Медицинская» (С6Ні206 • Н2О). 4. Глюкоза ангидридная
(СєН^Об) перекристаллизованная из спирта.
Приготовление. 21 г глюкозы «Медицинская»
помещают в коническую колбу вместимостью 500 мл, вливают 300 мл
96% -ного этилового спирта, колбу закрывают пробкой, в
которую вставлен шариковый холодильник, и ставят на водяную
баню при температуре около 80°С. Для более полного
растворения глюкозы в спирте раствор кипятят в течение 1 ч, изредка
помешивая содержимое колбы покачиванием. Когда почти вся
глюкоза растворится в спирте, раствор быстро фильтруют
через бумажный фильтр в стакан. Отфильтрованный
пересыщенный раствор глюкозы в спирте оставляют в покое на 2—3 суток
при температуре 10—15°С в стакане, закрытом часовым стеклом.
При стоянии глюкоза выделяется из раствора в виде белых
кристалликов, образующих колонии сферической формы на стенках
и на дне стакана. Через 3 суток кристаллизацию считают
законченной. Маточный раствор сливают, кристаллы стеклянной
лопаточкой переносят в бюкс и сушат в вакуум-эксикаторе или
в вакуумной сушильной печке при температуре не выше 50°С до
полного удаления спирта. Высушенную глюкозу хранят в
закрытом бюксе в эксикаторе над серной кислотой.
5. Меднощелочной раствор готовят сливанием одинаковых
объемов первого и второго растворов перед каждым анализом.
Установка титра меднощел очно го раствора.
Титр меднощелочного раствора можно устанавливать по
любому интересующему редуцирующему сахару, который хотят
определять. При анализе гидролизата и сульфитного щелока титр
меднощелочного раствора принято устанавливать по глюкозе,
потому что главные составные части углеводов растительных
тканей — глюкоза и ксилоза — имеют одинаковую редуцирующую
способность. Титром меднощелочного раствора по глюкозе
называют количество миллиграммов глюкозы, которое расходуется на
восстановление 10 мл меднощелочного раствора при данных
условиях титрования. Изменение порядка прибавления сахарного
раствора к меднощелочному оказывает влияние на количество
сахара, идущее на восстановление меди, поэтому титр
меднощелочного раствора устанавливают для первого и второго
вариантов титрования отдельно.
Для установки титра на аналитических весах в закрытом
бюксе берут навеску безводной абсолютно сухой глюкозы око-
36
ло 0,1 гс точностью до 0,0002 г, растворяют ее в
дистиллированной воде, количественно переносят в мерную колбу
вместимостью 100 мл, объем раствора доводят до метки, прибавляя
дистиллированную воду, и перемешивают. Вычисляют
концентрацию приготовленного раствора, наливают его в бюретку и
проводят титрование 10 мл меднощелочного раствора в эбулиостате
по первому или второму варианту (см. «Ход анализа»). Делают
не менее трех параллельных титрований, расхождения в
результатах не должны превышать 0,03 мл A капля). Вычисляют
средний объем раствора глюкозы, необходимый для восстановления
меди в 10 мл меднощелочного раствора. Затем готовят другой
раствор из той же глюкозы, с ним повторяют титрование и
также вычисляют средний объем раствора глюкозы, пошедший
на анализ. На основании полученных данных при титровании
каждого глюкозного раствора вычисляют титр меднощелочного
раствора Т по формуле
T=CV,
где С — концентрация раствора глюкозы, мг/мл; V — объем раствора глюкозы,
пошедший на титрование, мл.
За окончательный результат берут среднее значение Т из
двух найденных значений.
Подготовка пробы. 1. При концентрации редуцирующих са-
харов более 0,13% пробу перед анализом разбавляют
дистиллированной водой в мерной колбе и анализируют по первому
варианту. 2. При концентрации редуцирующих Сахаров менее
0,001% пробу перед анализом упаривают в 5—10 раз в
выпарительной чашке на кипящей водяной бане и анализируют по
первому или по второму варианту. 3. Пробу, содержащую много
редуцирующих несахаров, анализируют после осаждения их
основным уксуснокислым свинцом (см. с. 58).
Ход анализа. Анализ светлых растворов,
содержащих 0,13—0,08% редуцирующих веществ.
(Первый вариант.) В сосуд / (см. рис. 11) наливают воду и ставят
его на электроплитку. Для равномерного образования пара, что
является обязательным условием правильного проведения
анализа, в сосуд / наливают водопроводную воду, которая
содержит растворенные газы, и на дно кладут кусочки разбитого
стеклянного фильтра или шамотного кирпича.
Бюретку споласкивают 2—3 раза анализируемым раствором
и, выливая при этом раствор через кран, добиваются полного
заполнения носика бюретки. (Пузырьки воздуха не
допускаются). Затем в бюретку наливают анализируемый раствор до
метки 0. Когда вода закипит, внутренний сосуд 2 (эбулиостат)
вынимают и в него из дозаторов наливают сначала 5 мл
первого раствора, а затем 5 мл второго раствора. Легкими
движениями перемешивают жидкость в эбулиостате и медленно
вставляют его в сосуд /. Затем в збулиостат вставляют кончик
37
бюретки с анализируемым раствором так, чтобы пробка, надетая
на бюретку, плотно закрыла верхнее отверстие, и ждут, когда
пар начнет проходить через меднощелочной раствор. Медноще-
лочной раствор нагревается до температуры кипения воды в
течение 0,5 мин. Если пар не будет долго проходить через
реагирующую жидкость, надо, подвинтив зажим, уменьшить его
выход через отводную трубку. Как только пар начнет проходить
через меднощелочной раствор, из бюретки в эбулиостат
вливают струйкой около 80—90% того количества сахарного
раствора, которое должно пойти на титрование. (Если
концентрация анализируемого раствора совершенно неизвестна, то
делают предварительное титрование, а потом окончательное).
Сахарный раствор прибавляют так, чтобы прохождение пара
через реагирующую жидкость не прекращалось. Когда
закончат прибавление сахарного раствора, перевертывают песочные
часы и ждут 2 мин. Затем снова прибавляют анализируемый
раствор из бюретки со скоростью 1 капля в 6—7 с. При
появлении ярко-желтой окраски реагирующей жидкости от
прибавления одной капли сахарного раствора анализ заканчивают,
и по бюретке измеряют объем сахарного раствора, пошедший на
титрование.
Анализ темных растворов и растворов,
содержащих редуцирующих веществ менее 0,08%•
(Второй вариант). Из чистой глюкозы с известной влажностью
готовят 100 мл приблизительно 0,1%-ного раствора. Точное
содержание глюкозы в растворе вычисляют, зная навеску и
влажность глюкозы или определяя его как при титровании медноще-
лочного раствора, титр которого известен. Полученным
раствором заполняют бюретку. В сосуд 1 наливают водопроводную
воду и ставят на электрическую плитку. Когда вода закипит,
в сосуд 2 из дозаторов вливают 5 мл первого раствора и 5 мл
второго раствора. После перемешивания жидкости в эбулиостате
к ней добавляют 5 мл анализируемого раствора, имеющего
концентрацию редуцирующих веществ меньше 0,05% или 1—2 мл
темного анализируемого раствора, окраска которого настолько
интенсивна, что затрудняет наблюдение изменения окраски ме-
тиленовой синей в конце титрования. Затем к жидкости в
эбулиостате добавляют такой объем раствора глюкозы из бюретки,
чтобы на дотитровывание после 2 мин кипячения оставалось не
более 0,5 мл. Величину объема раствора глюкозы, которой надо
добавить к меднощелочному раствору на холоду, определяют
предварительным титрованием.
После перемешивания жидкости эбулиостат вставляют в
сосуд /, закрывают его пробкой, надетой на кончик бюретки с
раствором глюкозы известной концентрации, и ждут. Когда пар
начнет проходить через жидкость, перевертывают песочные часы
на 2 мин, после чего начинают дотитровывание, прибавляя
раствор глюкозы со скоростью 1 капля в 6—7 с до появления жел-
38
той окраски. По бюретке определяют количество раствора
глюкозы, израсходованной на титрование.
Расчет. При анализе по первому варианту концентрацию редуцирующих
Сахаров в исследуемой пробе в пересчете на глюкозу находят по формуле
ТпШ
X = ¦
К1000
где X — концентрация редуцирующих Сахаров в исследуемой пробе, %; Т —
титр меднощелочного раствора по глюкозе, мг; V — объем анализируемого
раствора сахара, пошедшего на титрование, мл; п — разведение; 1000 —
перевод миллиграммов в граммы.
При анализе по второму варианту концентрацию редуцирующих
Сахаров в исследуемой пробе в пересчете на глюкозу вычисляют по следующей
формуле:
СУ) 100
#1000
где X — концентрация редуцирующих Сахаров в исследуемой пробе, %; Т —
титр меднощелочного раствора по глюкозе, мг; С — концентрация глюкозы
в растворе, которым вели дотитровывание, мг/мл; V — объем раствора
глюкозы, пошедший на дотитровывание, мл; g — объем исследуемой пробы,
взятый на анализ, мл.
Воспроизводимость результатов титрования в среднем из трех
параллельных определений составляет ±0,03 мл раствора сахара.
Предостережения. 1. В начале анализа, если быстро вставить холодный
эбулиостат с меднощелочным раствором в сосуд / и сразу закрыть его
пробкой, то в результате мгновенной конденсации пара давление понизится и
раствор частично или полностью перельется по внутренней трубке эбулиостата
в сосуд /. Обнаружить это можно, наблюдая за цветом воды в сосуде /.
От капли меднощелочного раствора она мутнеет, а от большего количества
станет грязно-синего цвета. Если будет обнаружен переброс меднощелочного
раствора, то анализ испорчен. 2. Для получения хорошо воспроизводимых
результатов анализа надо сахарный раствор в конце титрования прибавлять
всегда с одинаковой скоростью, потому что реакция окисления идет не
мгновенно [19]. 3. При определении количества смеси из нескольких Сахаров в
пересчете на глюкозу надо иметь в виду, что редуцирующая способность
некоторых Сахаров (галактозы, арабннозы) значительно отличается от
редуцирующей способности глюкозы (см. с. 91) [18].
Определение легко- и трудиогидролизуемых полисахаридов
(метод Кизеля и Семигановского)
Метод используют для определения легко- и трудногидроли-
зуемых полисахаридов в растительной ткани [20], а также для
определения потенциального содержания моносахаридов в сырье.
Потенциальным называют то количество моносахаридов,
которое можно получить при самых благоприятных лабораторных
условиях гидролиза полисахаридов растительной ткани.
Метод основан на определении количества моносахаридов,
образующихся из легкогидролизуемых полисахаридов
исследуемой растительной ткани при их гидролизе разбавленной соляной
кислотой при 100°С и на определении количества моносахаридов,
образующихся из трудногидролизуемых полисахаридов той же
39
растительной ткани при их гидролизе сначала
концентрированной серной кислотой при комнатной температуре, а потом
разбавленной серной кислотой при 100°С. Потенциальное
количество моносахаридов в сырье находят как сумму легко-
и трудногидролизуемых полисахаридов. Оно всегда меньше
теоретического на то количество моносахаридов, которое
разложилось во время гидролиза, и оно всегда выше того количества
моносахаридов, которое получают из такого
же сырья в производстве.
Мешающие вещества. Продукты распада
моносахаридов во время гидролиза — фурфу-
рол, оксиметилфурфурол и др., которые
реагируют с меднощелочным раствором при
определении Сахаров.
Аппаратура. 1. Установка для нагревания
реакционной смеси на водяной базе с
обратным холодильником (рис. 13). 2. Установка
для определения редуцирующих Сахаров эбу-
лиостатическим методом (см. рис. 11).
Реактивы и их приготовление. 1. Соляная
кислота 2%-ный раствор. 2. Серная кислота
80%-ный раствор. 3. Едкий натр 20%-ный
раствор. 4. Все реактивы для эбулиостатического
метода.
Подготовка проб. Исследуемую пробу
растительной ткани измельчают до опилок
размером 2—0,25 мм, подсушивают до воздушносу-
хого состояния и хранят в плотно закрытой
склянке. Перед анализом пробу хорошо
перемешивают и определяют ее влажность (см.
с. 30).
Ход анализа. Определение легко-
гидролизуемых полисахаридов.
Навеску растительной ткани около 5 г,
взвешенную с точностью ±0,0002 г, помещают в коническую
колбу вместимостью 500 мл, приливают 200 мл. 27о-ного
раствора соляной кислоты. Содержимое колбы перемешивают так,
чтобы все опилки были в кислоте, а затем колбу ставят на
кипящую водяную баню и закрывают пробкой, в которую
вставлен шариковый холодильник (см. рис. 13). Через 3 ч нагревание
прекращают, содержимое реакционной колбы фильтруют на
воронке Бюхнера через бумажный фильтр. Остаток на фильтре
(целлолигнин) промывают горячей водой до отрицательной
реакции на кислоту по метиловому оранжевому (фильтровальная
бумажка, смоченная раствором метилового оранжевого, в
присутствии кислоты розовеет от одной капли фильтрата) и
сохраняют для определения в нем трудногидролизуемых
полисахаридов. Фильтрат и промывные воды соединяют вместе в мерной
40
Рис. 13. Установка
для нагревания на
водяной бане с
обратным
холодильником
колбе вместимостью 500 мл и объем доводят дистиллированной
водой до метки. Полученный раствор гидролизата легкогидроли-
зуемых полисахаридов после тщательного перемешивания
анализируют на содержание редуцирующих веществ эбулиостатиче-
ским методом (см. с. 33).
Определение трудногидролизуемых
полисахаридов. Целлолигнин — остаток после гидролиза легкогидро-
лизуемых полисахаридов — переносят на часовое стекло,
подсушивают при 50—60 С до воздушносухого состояния,
количественно переносят в стакан или в фарфоровую чашку, заливают
40 мл 80%'-ной серной кислоты и тщательно перемешивают
стеклянной палочкой. Гидролиз целлолигнина концентрированной
серной кислотой продолжают 3 ч, тщательно перемешивая
реагирующую смесь через каждые 15—20 мин. За это время трудно-
гидролизуемые полисахариды превращаются в декстрины. Для
проведения инверсии декстринов к реагирующей смеси
прибавляют 20—30 мл воды и количественно переносят ее в коническую
колбу вместимостью 1 л, расходуя при этом около 600 мл
дистиллированной воды. Колбу с реагирующей смесью ставят на
кипящую водяную баню, закрывают пробкой, в которую вставлен
обратный холодильник (см. рис. 13) и нагревают в течение 5 ч.
Затем раствор образовавшихся моносахаридов фильтруют на
воронке с ситчатым дном через полотняный фильтр. Остаток
(лигнин) на фильтре промывают небольшими порциями горячей
воды до отрицательной реакции промывной воды на кислоту по
индикатору метиловому оранжевому.
Фильтр и промывные воды количественно переносят в мерную
колбу вместимостью 1 л, объем раствора доводят до метки,
прибавляя дистиллированную воду, и тщательно перемешивают.
В мерную колбу вместимостью 100 мл вливают 50 мл
(отмеривание пипеткой) полученного гидролизата и осторожно по
каплям при постоянном перемешивании прибавляют 10 мл 20%-ного
раствора едкого натра для нейтрализации серной кислоты.
После охлаждения нейтрализованного раствора до комнатной
температуры его объем доводят до метки, прибавляя
дистиллированную воду. Раствор хорошо перемешивают и анализируют
на содержание редуцирующих веществ эбулиостатическим
методом.
Расчет. Количество моносахаридов, которое можно получить при
гидролизе легкогидролизуемых (или трудногидролизуемых) полисахаридов,
вычисляют по формуле
Y _ С,Ул
где Хл — количество моносахаридов, которое получено при гидролизе
легкогидролизуемых (или трудногидролизуемых) полисахаридов, % от абсолютно
сухой анализируемой растительной ткани; Сл — концентрация моносахаридов
41
в полученном гидролизате легкогидролизуемых (или трудногидролизуемых!
полисахаридов, %; Vn — объем полученного гидролизата легкогидролизуемых
(или трудногидролизуемых) полисахаридов, мл; п.—-разбавление гидролизата
при его нейтрализации; g — навеска абсолютно сухой растительной ткаии,
взятая для гидролиза легкогидролизуемых полисахаридов, г.
Потенциальное количество моносахаридов в сырье, т. е. количество
моносахаридов, которое можно получить при гидролизе анализируемого сырья,
является суммой моносахаридов, найденных в гидролпзатах
легкогидролизуемых и трудногпдролизуемых полисахаридов.
Содержание легкогидролизуемых (или трудногидролизуемых)
полисахаридов Ял (или #т) в исследуемой растительной ткани, выраженное в
процентах, находят умножением количества моносахаридов на коэффициент К.
Вычисляемый на основании реакции гидролиза полисахаридов, при которой на
каждую молекулу образующегося моносахарида расходуется одна молекула
воды, коэффициент К зависит от состава гндролизуемых полисахаридов. При
гидролизе пентозанов он равен 0,88, а при гидролизе гексозанов—0,90. При
вычислении легкогидролизуемых полисахаридов древесины разных пород его
принято считать равным 0,89. Таким образом, содержание легкогидролизуемых
полисахаридов в анализируемой древесине будет равно Ял=0,89 Хл% от абс.
сухого сырья, а содержание трудногпдролизуемых полисахаридов Ят =
=0,90 Хт% от абс. сухого сырья.
Содержание полисахаридов и потенциальных моносахаридов в сырье
определяют с точностью ±3—5% отн.
Определение пентозанов (метод Толленса)
Метод предназначен для определения содержания пентозанов
в растительных материалах. Он может быть использован для
определения потенциального содержания фурфурола в сырье,
т. е. для определения максимального количества фурфурола,
которое можно получить из анализируемого сырья.
Метод основан на определении фурфурола, образующегося
при дегидратации пентоз, которые получаются при гидролизе
пентозанов в солянокислой среде при нагревании. В этих
условиях одновременно протекает гидролиз пентозанов, полиурони-
дов и гексозанов с образованием уроновых кислот и метилпен-
тоз. Уроновые кислоты, отщепляя углекислый газ (СО2),
переходят в пентозы, а затем в фурфурол. Метилпентозы после
дегидратации дают метилфурфурол. Во избежание разложения
образующийся форфурол вместе с метилфурфуролом сразу
отгоняют из реакционной смеси. В полученном конденсате
определяют количество фурфурола бромид-броматным
методом.
В основе бромид-броматного метода лежит реакция броми-
ровання фурфурола по месту разрыва двойной связи. Реакция
протекает по следующему уравнению:
42
НС СН ВгНС СН ВгНС СНВг
¦ | II ?| || ! |
|| II / 2 ? 2
о' чн о Чі V хВг чн
фурфурол Диоромірурсрурол Тетра5ром(рур<рурал
Полное бромирование фурфурола до тетрабромфурфурола
почти заканчивается через 1 ч, причем скорость реакции,
измеряемая количеством брома, израсходованным на бромирование,
сначала очень большая—за 4 мин расходуется 50% брома,
а потом она постепенно уменьшается. Остальные 50% брома
расходуются за 56 мин. Для анализа используют время броми-
рования 4 мин и считают, что реакция прошла до образования
дибромфурфурола.
Бром для бромирования получают при подкислении раствора
бромид-броматной смеси по следующей реакции:
5КВг+КВгО3+6НС1 -* ЗВг2+ЗН2О+6КС1.
Чтобы получить хорошо воспроизводимые данные анализа,
необходимо строго соблюдать время бромирования. Скорость
бромирования и в течение 5-й минуты остается достаточно
большой. Реакцию бромирования прерывают, добавляя в
реагирующую жидкость раствор йодистого калия, который сразу вступает
в реакцию со свободным бромом
2К1 + Вг2 —2КВг+12.
Выделившийся йод оттитровывают раствором гипосульфита
2Na2S2O3+12 — 2NaI+Na2S4O6.
Количество фурфурола находят по количеству
израсходованного брома, которое эквивалентно оттитрованному йоду.
Теоретический выход фурфурола из пентоз составляет 64%-.
Практический выход фурфурола зависит от природы пентоз и от
источника их получения. При дегидратации ксилозы выход фурфурола
колеблется в пределах 46—56%', при дегидратации арабинозы
он составляет 43—46%. При получении фурфурола из уроновых
кислот его выход составляет 21—29%'[21].
Мешающие вещества. Полиурониды и гексозаны. Метилфур-
фурол и другие летучие бромируемые вещества.
Аппаратура. Прибор для определения пентозанов.
Конические колбы на 500 мл с притертыми пробками.
43
Прибор для определения пентозанов (рис. 14) состоит из
реакционной колбы и холодильника, соединенных на шлифах
(резиновые соединения не допускаются). Колба закрывается
стеклянной пришлифованной пробкой с градуированной
воронкой.
Реактивы и их приготовление. 1. Соляная кислота 13,
15%-ный раствор. 2. Хлористый натрий, кристаллический. 3.
Уксуснокислый анилин. Приготовление. 9 мл ледяной
уксусной кислоты смешивают с 1 мл свежеперегонного анилина.
Анилин перегоняют на установке, собранной на шлифах, при этом
колбу с анилином нагревают на воздушной бане. 4. Бромид-
бромата калия 0,05 н. раствор, в 1 л
которого содержится 1,392 г бромноватокис-
лого калия (КВгОз) и 10 г бромистого
калия (КВг). 5. Едкий натр 1,58 н.
раствор. 6. Молибденовокислый аммоний,
2,5%-ный раствор. 7. Йодистый калий,
10%-ный раствор. 8. Гипосульфит, 0,05 н.
раствор. 9. Крахмал, 0,5%-ный раствор.
Подготовка проб. Пробу воздушносу-
хого сырья измельчают до размеров
опилок 2—0,2 мм, хорошо перемешивают
и хранят в плотно закрытой банке. Перед
анализом определяют влажность иссле-
Рис. 14. Прибор для дуемого сырья.
определения пентозанов Ход анализа. Около 2 г сырья,
содержащего примерно 0,1—0,2 г
пентозанов, взвешивают на аналитических весах,
количественно переносят его в реакционную колбу (см. рис. 14) и
заливают 100 мл 13,15%-ной соляной кислоты. Для поддержания
постоянной концентрации ионов хлора в реакционной жидкости
во время отгонки в колбу вносят 10 г кристаллического
хлористого натрия. С целью предотвращения пенообразования во
в,ремя реакции добавляют небольшой кусочек парафина, а для
создания условий равномерного кипения реагирующей
жидкости в нее помещают 1—2 кусочка пористого стекла (битый
стеклянный фильтр) или пемзы. Колбу закрывают, ставят на
электроплитку с асбестовой сеткой, соединяют с холодильником и
включают нагрев. Образующийся конденсат собирают в мерный
цилиндр вместимостью 25 мл. В воронку наливают 25 мл
соляной кислоты. Нагрев реагирующей смеси регулируют так, чтобы
за 10 мин получать 25 мл конденсата. После отгонки каждых
25 мл жидкости в колбу из воронки вливают 25 мл 13,15%-ной
соляной кислоты, а конденсат из цилиндра выливают в
мерную колбу вместимостью 500 мл. Перегонку ведут 100 мин,
а потом проверяют конец отгонки по реакции с
уксуснокислым анилином. Если фильтровальная бумажка,
смоченная уксуснокислым анилином, порозовеет при нанесении
44
на нее одной капли дистиллята из холодильника, то отгонку
фурфурола продолжают, повторяя проверку через каждые
10 мин. При полной отгонке фурфурола бумажка,
смоченная уксуснокислым анилином, при нанесении на нее капли
конденсата не розовеет, но иногда желтеет. Желтое соединение
дает метилфурфурол и другие производные фурфурола. Если нет
признаков розовой окраски с уксуснокислым анилином, то для
окончательной проверки бумажку нагревают в сушильном
шкафу при температуре 80—90°С в течение 2 мин. Отсутствие
розовой окраски бумажки после нагревания считают концом
отгонки фурфурола. Объем конденсата, собранного в мерную
колбу, доводят до метки, прибавляя 13,15%-ный раствор
соляной кислоты, хорошо перемешивают и анализируют на
содержание фурфурола. Берут 100 мл конденсата, вносят его в
коническую колбу вместимостью 500 мл и нейтрализуют, прибавляя
постепенно 200 мл 1,58 н. раствора едкого натра. Раствор
охлаждают до комнатной температуры, а потом к нему добавляют
10 мл 2,5%'-ного раствора молибденовокислого аммония и 25 мл
0,05 н. раствора бромид-бромата. Колбу закрывают стеклянной
пробкой (резиновая пробка не допускается). Содержимое колбы
перемешивают, колбу ставят на белую поверхность (бумагу) и
ждут когда начнет появляться желтая окраска раствора. Обычно
появление желтой окраски раствора, свидетельствующее о
появлении свободного брома в растворе, начинается через 15—
20 с. В момент появления желтой окраски перевертывают
песочные часы на 4 мин и ждут. По истечении 4 мин в колбу вливают
10 мл 10%-ного раствора йодистого калия и плотно закрывают
ее. Содержимое колбы осторожно перемешивают легким
движением колбы и снова выдерживают 4 мин (песочные часы). Через
4 мин выделившийся йод оттитровывают 0,05 н. раствором
гипосульфита натрия. Сначала раствор гипосульфита добавляют
быстро струей, чтобы предотвратить возможное улетучивание
йода. Когда раствор станет бледно-желтым, к нему добавляют
1—2 капли раствора крахмала и продолжают титрование,
добавляя раствор гипосульфита по каплям и перемешивая раствор
после прибавления каждой капли. Титрование считают
законченным, когда от одной прибавленной капли раствора гипосульфита
раствор в колбе станет совершенно бесцветным.
Аналогично проводят холостой опыт, в котором вместо 100 мл
конденсата берут 100 мл 13,15%-ной соляной кислоты.
Расчет. Содержание потенциального фурфурола в анализируемом сырье
вычисляют по следующей формуле:
„ (а - Ь) К0.0024 ¦ 100
¦Л = =-= ,
где X — содержание потенциального фурфурола в абсолютно сухой пробе
сырья, %; а —объем 0,05 н. раствора гипосульфита, израсходованного на
45
титрование в холостом опыте, мл; b — объем 0,05 н. раствора гипосульфита,
пошедшего на титрование прн анализе конденсата, мл; V — объем
полученного конденсата, мл; V\—объем конденсата, взятый на титрование, мл; g —
количество абсолютно сухой пробы, взятое иа анализ, г; 0,0024 — количество
фурфурола, соответствующее 1 мл 0,05 н. раствора гипосульфита натрия при
получении дибромфурфурола, г.
Содержание пентозанов в анализируемом сырье находят умножением
вычисленного количества фурфурола X на эмпирический коэффициент К.
Теоретический коэффициент для пересчета фурфурола в пентозаны равен
УИ„ 132
*-96—1>375'
где Мп — молекулярная масса звена пентозанов A32); Мф — молекулярная
масса фурфурола (96).
Учитывая, что практический выход фурфурола из пентоз зависит от их
природы, при вычислении результата анализа теоретический коэффициент Кт
для пересчета фурфурола в пентозаны делят иа эмпирически установленный
выход фурфурола из данных пентоз, который вычислен в процентах от
теоретического.
Если пентозаны анализируемого сырья состоят в основном из ксилана,
коэффициент для пересчета фурфурола в пентозаны будет равен частному от
деления теоретического коэффициента Кт на эмпирически установленный
выход фурфурола ИЗ КСИЛОЗЫ Кис-
Kr I.375 _
~7 ТМ563
При анализе растительной ткани, которая кроме ксилана содержит ара-
бан (например, древесина), коэффициент для пересчета фурфурола в
пентозаны принимают равным 1,71 и 1,88 [6, с. 100]. Отсутствие постоянного
коэффициента для пересчета фурфурола в пентозы делает нецелесообразным
введение поправки иа содержание уроновых кислот, предложенную некоторыми
исследователями [21, с. 422].
Прн определении фурфурола среднее отклонение из трех титрований
составляет ±0,05 мл раствора гипосульфита натрия.
Предостережение. Количество отогиаиного фурфурола зависит от
скорости перегонки, концентрации и объема кислоты в реакционной колбе, степени
измельчения анализируемой пробы и других условий, [6]. Для получения
воспроизводимых результатов надо точно выполнять все условия анализа,
описанные в методике.
Определение уроновых кислот (метод Волскон)
Метод предназначен для определения уроновых кислот
непосредственно в растительных тканях и в их гидролизатах,
полученных различными способами.
Метод основан на определении количества углекислого газа,
который образуется при декар'боксилировании гексоуроновых
кислот в солянокислой среде. При определении уроновых кислот
в сырье проводят гидролиз полисахаридов и декарбоксилирова-
ние образующихся уроновых кислот одновременно. При анализе
46
гидролизатов проводят только одну реакцию декарбоксилиро-
вания
Н ,0
(снонL
соон
неї
н о
(СНОН),
I 3
сн2он
СО
Іексуроновая Пентоза
кислота
Образующийся углекислый газ извлекают из реагирующей
смеси, пропуская через нее воздух, а затем улавливают,
пропуская газ через раствор гидроокиси бария (Ва(ОНJ).
Количество углекислого газа находят по количеству нейтрализованной
гидроокиси бария, которое определяют титрованием раствора
Ва(ОН)г 0,1 н. раствором соляной кислоты до и после опыта.
Рис. 15. Установка для определения уроновых кислот
Мешающие вещества. Углеводы в условиях декарбоксилиро-
вания уроновых кислот частично разлагаются с образованием
углекислого газа.
Аппаратура. Установка для определения уроновых кислот
(рис. 15). Установка для титрования. Установка для хранения
и отмеривания раствора гидроокиси бария (рис. 16).
Реактивы и их приготовление. 1. Соляная кислота, 0,1 н.
раствор и 12%-ный раствор. 2. Гидрат окиси бария (Ва(ОН)г),
0,12 н. раствор. Приготовление. 25—30 г Ва(ОНJ х. ч.
47
растворяют віл дистиллированной воды и оставляют стоять.
Через 12—16 ч прозрачный раствор сливают в склянку для
хранения и отмеривания приготовленного раствора (см. рис. 16).
3. Цинк гранулированный.
Подготовка проб. Пробу сырья измельчают до размеров
опилок 0,2—2 мм, подсушивают до воздушносухого состояния,
хорошо перемешивают и хранят в плотно закрытой склянке. Перед
анализом определяют ее влажность.
Ход анализа. В реакционную колбу / (см. рис. 15) помещают
5—10 г анализируемого сырья (или 20—25 мл гидролизата),
100 мл 12%-ного раствора соляной кислоты и
несколько кусочков пористого стекла для
равномерного кипения. Колбу присоединяют
к обратному холодильнику, который соединен
с сосудом 2, заполненным гранулированным
цинком для поглощения паров соляной
кислоты. Пипеткой (см. рис. 16) отмеривают 100 мл
0,12—0,14 н. раствора гидроокиси бария,
выливают их в трубку Базыриной 3, которую
сразу соединяют с промывной склянкой 4,
заполненной водой для поглощения остатков
соляной кислоты, и контрольной склянкой 5.
В контрольную склянку помещают 40—
50 мл прозрачного раствора гидроокиси бария,
который меняют при появлении помутнения.
(Отводы контрольной склянки всегда должны
быть закрыты кусочками каучука с пробками,
которые снимают только во время опыта).
Контрольную склянку присоединяют к
двугорлой склянке 6 и включают водоструйный насос
так, чтобы через систему проходил слабый
ток воздуха (один пузырек в 1 с). Скорость
воздуха регулируют винтовым зажимом
перед контрольной склянкой. Контрольную
продувку ведут в течение 15 мин. Если раствор гидроокиси бария
в трубке Базыриной мутнеет, то это значит, что в пробе есть
соли угольной кислоты. Для того чтобы учесть расход
гидроокиси бария на реакцию с солями угольной кислоты продувку
продолжают в течение 4 ч на холоду, затем раствор из трубки
Базыриной сливают в цилиндр, берут из него 20 мл и оттитро-
вывают 0,1 н. раствором соляной кислоты. Полученные данные
учитывают при расчете как расход раствора гидроокиси бария
на холостую пробу.
Если раствор гидроокиси бария после 15 мин продувки не
мутнеет, то включают колбонагреватель и кипятят реакционную
смесь в течение 4 ч, считая с момента начала кипения. Нагрев
не должен быть сильным. Через 4 ч выключают
колбонагреватель, закрывают винтовой зажим между реакционной колбой и
48
Рис. 16. Установка
для хранения и
отмеривания
раствора гидроокиси
бария
поглотителями с раствором едкого кали, закрывают зажимами
отводы до и после трубки Базыриной, отключают склянку 4 от
2 и контрольную склянку 5 от 6. Затем отключают трубку
Базыриной от системы, жидкость с осадком углекислого бария
(ВаСОз) быстро сливают в сухой цилиндр вместимостью 150 мл,
закрывают цилиндр пробкой и осадку ВаСО3 дают отстояться.
Берут 20 мл отстоявшегося прозрачного раствора гидроокиси
бария, переносят его пипеткой в сухую колбу и титруют 0,1 н.
раствором соляной кислоты в присутствии фенолфталеина.
На каждую партию реактивов делают холостой анализ (без
исследуемой пробы) по вышеописанной методике. При наличии
в пробе солей угольной кислоты за холостой анализ принимают
тот, который проводят при продувке пробы на холоду, как
указано выше.
Расчет. Содержание уроновых кислот в абсолютно сухой растительной
ткани, взятой для анализа, вычисляют по формуле
(Уо _ у) юр . 0,0097 • 100
Х~ 2б? "
где X — содержание уроновых кислот в абсолютно сухой пробе растительной
ткани, %; Vo — объем 0,1 н. НС1, израсходованный на титрование 20 мл 0,12 н.
Ва(ОНJ при анализе исследуемой растительной тканн, мл; V — объем 0,1 н.
НС1, израсходованный на титрование 20 мл 0,12 н. Ва(ОН)г при холостом
анализе, мл; 100 —объем 0,12 н. Ва(ОНJ. взятый на анализ, мл; 20—объем
0,12 н. Ва(ОНJ, взятый на титрование, мл; 0,0097 — количество уроновых
кислот, соответствующее 1 мл 0,1 н. раствора Ва(ОН)г г; g — количество
абсолютно сухой растительной ткани, взятое на анализ, г (или объем гидроли-
зата, взятый на анализ, мл).
При определении уроновых кислот среднее отклонение из трех титрований
не должно превышать ± 0,1 мл 0,1 н. раствора соляной кислоты.
Определение веществ, экстрагируемых водой
Для определения веществ, которые экстрагируются из
растительных тканей водой при разных температурах,
предназначен метод, основанный на извлечении из исследуемой
растительной ткани веществ, которые растворяются в воде при 25 и при
100°С. Количество извлеченных веществ находят взвешиванием
пробы до и после экстракции. Этими веществами являются
моносахариды, полисахариды низкой степени полимеризации,
минеральные вещества, белки, дубильные вещества, пектиновые
вещества, органические кислоты и другие вещества.
Мешающие вещества. Летучие вещества.
Аппаратура. Термостат. Установка для нагревания с
обратным холодильником (см. рис. 13).
Подготовка проб. Исследуемую растительную ткань
измельчают до размеров опилок 0,2—2 мм, хорошо перемешивают,
подсушивают до воздушносухого состояния и хранят в плотно
закрытой склянке. Перед анализом определяют влажность пробы
(см. с. 30).
4 Заказ № 142 49
Ход анализа. Берут около 2 г пробы, взвешивают ее с
точностью 0,0002 г, помещают в коническую колбу емкостью 250 мл
и приливают 100 мл дистиллированной воды. Если экстракцию
проводят при 25°С, то колбу закрывают пробкой и ставят в
термостат при 25°С. Экстрагирование проводят в течение 48 ч,
периодически перемешивая содержимое колбы.
При проведении экстракции при 100°С колбу с пробой
закрывают пробкой, в которую вставлен шариковый холодильник,
и ставят на кипящую водяную баню (см. рис. 13). Экстракцию
проводят в течение 3 ч, периодически перемешивая
содержимое колбы покачиванием. После экстракции опилки
количественно переносят на взвешенный стеклянный фильтр,
отфильтровывают, промывают 20—30 мл горячей воды и фильтр с
опилками ставят в сушильный шкаф. Через 3 ч сушки при 105°С
фильтр с пробой охлаждают в эксикаторе, взвешивают и снова
помещают в сушильный шкаф. Через 1 ч досушивания фильтр
с осадком опять взвешивают. Досушивание повторяют до тех
пор, пока разница в двух последующих взвешиваниях будет
составлять не более 0,0004 г.
Расчет. Содержание веществ, растворимых в воде в абсолютно сухом
сырье, вычисляют по формуле
Х- (g-gi)lOO
g
где X — содержание веществ, экстрагируемых водой при 25°С (или при 100"С),
в абсолютно сухом сырье, %; g— количество абс. сухих опилок до экстракции,
г; gt — количество абс. сухих опилок после экстракции, г.
Воспроизводимость результатов анализа в среднем из трех определений
составляет ±3—5% оти.
Определение веществ, экстрагируемых органическими растворителями
Для определения содержания веществ, экстрагируемых
различными органическими растворителями из растительных
тканей, предназначен метод, основанный на извлечении из
анализируемой растительной ткани веществ, растворимых в
применяемом растворителе (эфир, бензол, спирт, ацетон и др.), при
температуре кипения растворителя. Этими веществами являются
смолы, воски, масла и другие вещества (см. с. 21).
Количество извлекаемых веществ находят по изменению массы
абсолютно сухой пробы до и после экстракции. Наибольшее
количество экстрагируемых веществ находят при экстракции спирто-
бензольной смесью A :2).
Аппаратура. Аппарат Сокслета или аппарат Э-8.
Аппараты для экстракции (рис. 17) состоят из колбы,экстрактора
и обратного холодильника, которые собраны на шлифах.
В аппарате Э-8 экстрактор помещен в широкую трубку, которая
соединяет колбу с холодильником.
50
Реактивы и их приготовление. Органический растворитель —
спирт, или эфир, или спиртобензольная смесь.
Приготовление. Смешивают один объем этилового спирта 96° и два
объема бензола, вливая их в сухую колбу с пришлифованной
пробкой.
Подготовка проб. Исследуемую растительную ткань
измельчают до размеров опилок 0,2—2 мм, сушат до воздушносухого
состояния и хранят в плотно закрытой
склянке. Перед анализом определяют ее
влажность.
Ход анализа. Из фильтровальной
бумаги свертывают гильзу, взвешивают ее,
потом насыпают в нее 2—3 г пробы,
снова взвешивают с точностью 0,0002 г
и помещают в экстрактор. Уровень
опилок в гильзе должен быть на 1—2 см
ниже верхнего уровня сифона. Колбу
аппарата, высушенную при 105°С,
взвешивают, наливают в нее растворитель и
снова взвешивают. Количество
растворителя должно быть в 2—2,5 раза больше
емкости экстрактора аппарата.
Соединяют колбу с экстрактором и
холодильником, включают воду в холодильник,
а затем начинают нагрев, подставляя под
колбу водяную или песчаную баню в
зависимости от температуры кипения
растворителя. Во время нагревания пары
растворителя поднимаются в холодильник,
конденсируются и капают на опилки. Как рис \-] Аппараты для
только экстрактор наполнится раствори- экстракции:
телем до верхнего уровня сифона,
растворитель переливается в колбу. Затем
начинается следующий цикл извлечения
веществ из опилок свежей порцией растворителя. В аппарате
Сокслета экстракцию продолжают 6—8 ч, в аппарате Э-8
экстракцию заканчивают через 2—3 ч.
Во время экстракции нагрев растворителя регулируют так,
чтобы в течение 1 ч происходило 15—20 сливов в аппарате Э-8
и 5—8 сливов в аппарате Сокслета. Слишком слабое нагревание
тормозит экстракцию, сильное нагревание создает угрозу
улетучивания паров растворителя, что может привести к
прекращению слива растворителя через сифонную трубку.
После окончания экстракции гильзу с опилками вынимают и
отгоняют растворитель из экстракта. Когда растворитель
заполнит экстрактор на 1—2 см ниже верхнего уровня сифона,
аппарат разбирают, отогнанный растворитель выливают в
сухую склянку, аппарат снова собирают и продолжают отгонку
— аппарат Сокслета; 2 —
аппарат Э-8
4*
51
растворителя до тех пор, пока не останется 5—10 мл жидкости
в колбе. Затем колбу вынимают из аппарата, помещают в
сушильный шкаф и сушат при 105°С в течение 4—5 ч до полного
удаления растворителя.
Расчет. Содержимое веществ, экстрагированных органическим
растворителем, в абсолютно сухом сырье вычисляют по формуле
alOO
~ 8 '
где X — содержание веществ, экстрагированных органическим растворителем,
в абсолютно сухом анализируемом сырье, %; g— количество абсолютно сухого
сырья, взятое на анализ, до экстракции, г; а — количество экстрагированных
веществ, г.
Воспроизводимость результатов анализов в среднем из трех определений
составляет ±4—5% оти.
Предостережение. 1. Летучие экстрагируемые вещества, содержащиеся
в анализируемой пробе, и экстрагируемые вещества, разлагающиеся при
105°С, этим методом не определяются. 2. Количество и состав экстрагируемых
веществ зависит от применяемого растворителя.
Определение метоксильных групп (метод Фибека и Шваппаха)
Метод предназначен для определения метоксильных групп
[22, 23] в растительных тканях, в лигнине и в сульфитном
щелоке. Из всех метоксильных групп, содержащихся в древесине,
85—90%' входит в состав лигнина и только 10—15%
принадлежит метилированным углеводам и их производным. Метод
основан на реакции разложения простых эфиров
концентрированной йодистоводородной кислотой с образованием йодистого
метила по уравнению
ROCH3-f HI-* ROH-f CH3I.
Определение количества образующегося йодистого метила
основано на его реакции с бромом, которая протекает в растворе
уксуснокислого натрия (или калия) в ледяной уксусной кислоте
по следующему уравнению:
CH3I-f Br2-*CH3Br+IBr.
При наличии избытка брома образовавшийся бромистый йод
окисляется в йодноватую кислоту по уравнению
IBr+2Br2-f ЗН2О — НЮа+5НВг.
Избыток брома удаляют из реакционной жидкости
муравьиной кислотой, затем добавляют в нее йодистый калий,
подкисляют серной кислотой и йод, выделившийся из йодноватой
кислоты, титруют раствором гипосульфита
НЮ3+5Н1 —3I2-f3H2O
3I2+6Na2S2O3-~ 6NaI-f 3Na2S4O6.
52
Аппаратура. Прибор для определения метоксильных групп,
дрексели, двугорлая склянка, установка для титрования
и конические колбы вместимостью 250 мл с притертыми
пробками.
Установка, показанная на рис. 18, представляет собой
прибор для определения метоксильных групп, к которому (внизу)
присоединены регулятор давления газа, двугорлая склянка
и два дрекселя для промывки газа, пропускаемого через
прибор. Перед анализом в дрексель 2 наливают концентрированную
серную кислоту, в дрексель 5 —насыщенный раствор
двууглекислого натрия. Регулятором
давления служит
стеклянный тройник /,
вертикальный конец которого опущен
на 25 см в сосуд с водой.
Прибор для определения
метоксильных групп состоит из
реакционной колбы 4
(вместимостью 25—30 мл),
обратного холодильника 5, промы-
валки 6, двух дрекселей и
склянки с поглотителями,
которые соединены
шлифами. Реакционная колба
погружена в масляную баню
или в баню со сплавом Вуда.
Скорость потока газа,
пропускаемого через
прибор,измеряют счетчиком
пузырьков, которым может служить
любой дрексель с
жидкостью.
Реактивы и их
приготовление. 1. Гипосульфит натрия 0,1 н. раствор. 2. Уксуснокислый
калий (или натрий), 10%-ный раствор в ледяной уксусной
кислоте. 3. Йодистый калий, 10%-ный раствор. 4. Серная кислота,
разбавленная 1:9. 5. Уксуснокислый натрий, 22%-ный раствор.
6. Муравьиная кислота 90%-ная. 7. Суспензия красного фосфора,
содержащая 0,06 г красного фосфора в 100 мл воды или смесь
равных объемов 5%-ных растворов сернокислого кадмия и
гипосульфита. 8. Бром — свежеприготовленный раствор в ледяной
уксусной кислоте в присутствии уксуснокислого калия.
Приготовление. 100 г безводного уксуснокислого калия растворяют
в смеси 200 мл ледяной уксусной кислоты и 100 мл уксусного-
ангидрида. Берут 145 мл приготовленного раствора и в нем
растворяют 5 г брома. 9. Иодистоводородная кислота свежеперег-
нанная 57%-ная (плотность 1,7 г/см3, температура кипения
126—127°С). Приготовление. Перегонку йодистоводород-
5S
Рис. 18. Установка для определения
метоксильных групп
ной кислоты производят следующим образом. Кислоту
помещают в колбу, добавляют к ней небольшое количество E—10 г
на 500 мл кислоты) красного фосфора, колбу закрывают пробкой
с обратным холодильником и с трубкой для ввода углекислого
газа (СОг), а затем ставят на электроплитку или другой
нагревательный прибор и кипятят 20—30 мин в вытяжном шкафу.
Во время кипячения через кислоту непрерывно пропускают
углекислый газ. После кипячения кислоты с фосфором ее
охлаждают, переливают в перегонный стеклянный аппарат, собранный
на шлифах, и перегоняют, непрерывно пропуская через нее
углекислый газ. Иногда при перегонке образуется ядовитый газ фос-
фин (РН3). Во избежание несчастных случаев необходимо
следить за постоянным прохождением углекислого газа. Во время
отгонки и после отгонки не прекращать пропускание углекислого
газа через аппарат до его полного охлаждения. Очищенную
иодистоводородную кислоту хранят в небольшой темной склянке
с притертой пробкой, заплавленной парафином. Перед
заполнением склянки кислотой ее продувают углекислым газом. При
взятии части кислоты на анализ пространство над кислотой
в склянке заполняют углекислым газом для предохранения
разложения йодистоводородной кислоты под влиянием воздуха.
Иодистоводородная кислота образует с водой постоянно
кипящую (азеатропную) смесь. При температуре 126—127СС
перегоняется 57%-ная иодистоводородная кислота. На анализ нельзя
брать кислоту, концентрацией менее 56,5%. Контролем
может служить расход 0,1 н. раствора гипосульфита натрия
(Na2S3O3) в холостом опыте, он не должен превышать 0,5 мл,
в противном случае иодистоводородную кислоту необходимо
перегнать.
Подготовка проб. Исследуемую растительную ткань или
лигнин измельчают до размеров опилок 0,2—2 мм, подсушивают до
воздушносухого состояния и хранят в плотно закрытой склянке.
Перед анализом в пробе определяют влажность.
Ход анализа. Для улавливания свободного йода и йодисто-
водородной кислоты из углекислого газа, который содержит
йодистый метил, в промывалку 6 (см. рис. 18) наливают водную
суспензию красного фосфора или смесь равных объемов 5%-ных
растворов сернокислого кадмия и гипосульфита натрия. Уровень
жидкости в промывалке должен быть выше нижнего конца кол-
тіачка на 4—5 см.
Жидкость в промывалке меняют через 3—4 анализа. Для
улавливания йодистого метила берут 10 мл раствора
уксуснокислого калия в ледяной уксусной кислоте, добавляют к нему
10—12 капель брома и выливают 6 мл в дрексель 7 и 4 мл
в дрексель 8. В сосуд 9 наливают 10—20 мл водного раствора
уксуснокислого натрия, подкисленного муравьиной кислотой, для
поглощения брома, который увлекается током углекислого газа
из поглотителей.
54
В реакционную колбу помещают 0,07—0,09 г растительной
ткани или 0,03—0,04 г лигнина с известной влажностью,
взвешенные с точностью 0,0002 г. Затем в колбу вливают 5 мл йоди-
стоводородной кислоты, вносят около 0,2 г красного фосфора,
колбу присоединяют к холодильнику и нагревают на бане при
145°С. Для отгонки образующегося йодистого метила через
реагирующую жидкость все время пропускают равномерно
углекислый газ. Через 1 ч от начала кипения реакцию образования
йодистого метила и его отгонку считают законченными. Сосуды
с поглотителями отключают от прибора и содержимое их
переносят в коническую колбу вместимостью 250 мл, в которую
предварительно вливают 15 мл 22%>-ного раствора уксуснокислого
натрия. Сосуды несколько раз прополаскивают водой и
промывные воды выливают в ту же колбу. Общий объем жидкости,
собранной в колбе, должен составлять около 125 мл. Затем
в колбу вливают 0,5 мл 90%-ной муравьиной кислоты,
закрывают ее пробкой и содержимое хорошо перемешивают. Если
окраска жидкости от присутствия брома не исчезнет, то
прибавляют несколько миллилитров раствора уксуснокислого натрия.
Для проверки полноты связывания свободного брома к
раствору добавляют еще 12 капель муравьиной кислоты и ждут
2 мин. (Отсутствие свободного брома можно проверить по
индикатору метиловому красному. В отсутствие брома розовая
окраска раствора будет устойчивой). К раствору добавляют 10 мл
10%-ного раствора йодистого калия и 10 мл 10%-ного раствора
серной кислоты. Выделившийся йод оттитровывают 0,1 н.
раствором гипосульфита натрия до слабо-желтой окраски,
добавляют 0,5 мл 0,5%-ного раствора крахмала и продолжают
титровать до исчезновения синего окрашивания. Если к раствору был
добавлен метиловый красный для установления наличия брома,
то в конце титрования синяя окраска перейдет в розовую.
Расчет. Содержание метоксильных групп (ОСН3) в анализируемом
материале вычисляют по следующей формуле:
(а — б) 31,03 • 100
~ ?1000 • 6
где X — содержание метоксильиых групп в абсолютно сухом анализируемом
материале, %; а — объем 0,1 н. раствора гипосульфита натрия,
израсходованный иа титрование, мл; б — объем 0,1 и. раствора гипосульфита натрия,
израсходованный на холостой опыт, мл; 31,03 — молекулярная масса меток-
сильной группы; 6—число атомов йода, приходящееся на 1 метоксильную
группу в приведенных реакциях; g—количество абсолютно сухого материала,
взятое иа анализ, г.
Содержание метоксильных групп определяют с точностью ±0,01% от абс.
сухого материала.
Определение лигнииа по Класону (модификация Комарова)
Для определения лигнина в древесине хвойных и лиственных
пород предназначен метод, основанный на удалении из
растительной ткани углеводов и экстрактивных веществ [6, с. 85].
55
Остаток растительной ткани считают лигнином. Экстрактивные
вещества удаляют путем обработки пробы спиртобензольной
смесью в экстракционном аппарате (см. с. 50). Для удаления
полисахаридов их гидролизуют сначала 72%-ной серной
кислотой, а затем разбавленной кислотой при нагревании. Остающийся
лигнин высушивают до полного удаления влаги и вычисляют его
содержание в исследуемой абсолютно сухой растительной ткани.
Мешающие вещества. Экстрактивные и трудногидролизуемые
полисахариды.
Аппаратура. Аппарат для экстракции (см. рис. 17).
Установка для кипячения с обратным холодильником (см. рис. 13),
Стеклянный фильтр № 1.
Реактивы и их приготовление. 1. Спиртобензольная смесь
A :2 по объему) или серный эфир. 2. Серная кислота 72%-ная.
Подготовка пробы. Исследуемую растительную ткань
измельчают, просеивают, отбирают фракцию опилок размером 0,2—
2 мм и подсушивают до воздушносухого состояния. Перед
анализом определяют влажность пробы.
Ход анализа. Берут около 3 г (с точностью 0,0002 г) воз-
душносухих опилок, помещают их в аппарат для экстракции и
проводят экстракцию смол, жиров и других веществ
спиртобензольной смеси как описано на с. 51. Вычисляют содержание
экстрактивных веществ в анализируемой пробе.
Затем берут около 1 г (с точностью до 0,0002 г) обессмолен-
ной абсолютно сухой пробы, количественно переносят ее в
фарфоровую чашку, прибавляют 15 мл 72%-ной серной кислоты,
смесь тщательно размешивают и оставляют стоять на 3 ч при
комнатной температуре, периодически перемешивая. Через 3 ч
•смесь количественно с помощью 200 мл воды переносят в
коническую колбу вместимостью 500 мл, закрывают колбу пробкой,
в которую вставлен обратный холодильник, и ставят на
включенную электроплитку. Содержимое колбы кипятят в течение
1 ч и оставляют стоять на 1 сутки, чтобы частицы лигнина
укрупнились и осели на дно колбы. На следующий день осадок
лигнина из колбы количественно переносят в стеклянный
фильтр, фильтруют с помощью вакуума, промывают горячей
водой до отрицательной реакции на ион SO'4 по метиловому
оранжевому или по хлористому барию (ВаСЬ) и сушат в
сушильном шкафу при 105°С до полного удаления влаги. (Стеклянный
фильтр перед фильтрацией должен быть высушен при 105°С до
полного удаления влаги.)
Расчет. Содержание лигнина в абсолютно сухой исследуемой
растительной ткани в процентах вычисляют по формуле
Y_ а100A00-Л-экс)
g
где X — содержание лигннна в пробе, %; а — количество абсолютно сухого
лигнина, г; g — количество абсолютно сухой обессмоленной растительной
56
ткани, взятое на гидролиз, г; ЛЭкс — содержание экстрагируемых веществ.
в анализируемой пробе, %.
Лигнин определяют с точностью ±5—6% отн.
Предостережение. Состав экстрагированных веществ растительных тканей
разнообразен, поэтому нет единого метода предварительной обработки
растительной ткани перед гидролизом полисахаридов. При высоком содержании,
гаинидов в анализируемой растительной ткани дополнительно проводят
экстракцию водой при 25 и 100°С, а иногда и слабым раствором щелочи.
Определение редуцирующих Сахаров в сульфитном щелоке
Для определения содержания редуцирующих Сахаров в
сульфитном щелоке, бражке и других жидкостях после удаления из.
них редуцирующих несахаров, которыми являются лигносульфо-
наты и продукты распада моносахаридов, предназначен метод,,
основанный на определении редуцирующих веществ после
удаления редуцирующих несахаров осаждением их основным
уксуснокислым свинцом. Количество редуцирующих Сахаров,
составляет от 10 до 25% от общего количества редуцирующих
веществ [24]. Редуцирующие несахара при реакции с основным
уксуснокислым свинцом образуют труднорастворимые соединения»
которые выпадают в осадок. Осаждение несахаров проводят при
температуре 90—95°С, чтобы предотвратить выпадение в осадок
плюмбата маннозы, который может образоваться при избытке
уксуснокислого свинца. Осадок отделяют фильтрованием через
бумажный фильтр. В фильтрат, содержащий избыточный
основной уксуснокислый свинец и свинец, который образовал плюм-
бат маннозы, добавляют оксалат натрия. Свинец, находящийся
в растворе, во всех соединениях реагирует с оксалатом, образуя
труднорастворимый оксалат свинца, который выпадает в осадок:
Na2C2O4-f РЬ+ + — РЬС2О4.
Белый кристаллический осадок оксалата свинца
отфильтровывают через плотный или двойной бумажный фильтр и в
фильтрате определяют концентрацию редуцирующих Сахаров эбулио-
статическим методом (см. с. 33).
Мешающие вещества. Фурфурол, оксиметилфурфурол и
другие редуцирующие несахара, которые не осаждаются основным
уксуснокислым свинцом.
Аппаратура. Установка для определения редуцирующих
веществ эбулиостатическим методом (см. рис. 11).
Реактивы и их приготовление. 1. Оксалат натрия, насыщенный,
раствор. 2. Двууглекислая сода (NaHCO3) кристаллическая или
0,1 н. раствор едкого натра. 3. Основной уксуснокислый свинец,
насыщенный раствор. Приготовление. Берут 100 г окиси
свинца (РЬО) и 300 г уксуснокислого свинца (РЬ/СН3СОО/2),
тщательно перемешивают и растирают в фарфоровой ступке.
Затем ступку с содержимым ставят на кипящую водяную баню
и ждут, когда произойдет сплавление. Вовремя сплавления массу
мешать не надо. Через 40—60 мин смесь превращается всметано-
образную массу, покрытую коркой. Сплавление проверяют
57"
нажатием стеклянной палочки на корку, из-под которой при
полном сплавлении выступает белая сметанообразная масса. После
получения сплава его заливают 500 мл воды, количественно
переносят в мерную колбу вместимостью 1 л, объем раствора доводят
водой до метки, перемешивают, переливают в коническую «олбу,
закрывают пробкой и ставят в термостат при 30°С на 1 сутки.
Затем раствор отделяют от осадка, декантируя его через сифон
в склянку с нижним тубусом. Склянку закрывают пробкой,
в которую вставлена трубка с натронной известью. Надо
избегать соприкосновения приготовленного раствора с воздухом.
Углекислый газ, находящийся в воздухе, реагирует с основным
уксуснокислым свинцом, образуя углекислый свинец, который
выпадает в осадок. При этом концентрация свинца в растворе
понижается. 4. Все реактивы для определения редуцирующих
веществ эбулиостатическим методом (см. с. 35).
Подготовка проб. Сырой сульфитный щелок охлаждают до
комнатной температуры и нейтрализуют до pH 4, прибавляя
кристаллическую двууглекислую соду или 0,1 н. раствор едкого
натра.
Ход анализа. Определяют количество основного
уксуснокислого свинца, которое надо прибавлять к 10 мл пробы для
полного осаждения редуцирующих несахаров.
В стакан вместимостью 150—200 мл помещают 10 мл
нейтрализованного сульфитного щелока, добавляют 50 мл
дистиллированной воды, раствор перемешивают и из бюретки по каплям
прибавляют раствор основного уксуснокислого свинца до
прекращения образования осадка. Последнюю каплю добавляют по
стенке стакана. Если достигнута полнота осаждения, то на
стенке стакана не появится белый налет осадка. Для проверки
полноты осаждения нескольких капель осажденного щелока
отфильтровывают на часовое стекло и к фильтрату добавляют одну
каплю раствора основного уксуснокислого свинца. Если раствор
мутнеет сразу, то полнота осаждения не достигнута, осаждение
повторяют, добавляя к 10 мл сульфитного щелока большее на
0,5 мл количество раствора основного уксуснокислого свинца.
Если отфильтрованный раствор сульфитного щелока остается
прозрачным при добавлении к нему раствора основного
уксуснокислого свинца, то осаждение повторяют, добавляя к 10 мл
сульфитного щелока на 0,5 мл меньше раствора основного
уксуснокислого свинца. Таким образом устанавливают с точностью
±0,3 мл объем раствора основного уксуснокислого свинца,
который нужно добавить к 10 мл сульфитного щелока, чтобы
полностью осадить редуцирующие несахара.
Осаждение редуцирующих несахаров. В стакан
вместимостью 200—300 мл помещают 20 мл нейтрализованного
сульфитного щелока, добавляют к нему 100 мл дистиллированной
воды и нагревают до 95—98°С. К горячему сульфитному щелоку
при постоянном помешивании по каплям добавляют раствор
58
основного уксуснокислого свинца в количестве, необходимом для
достижения полноты осаждения, которое установлено
предварительным опытом. После охлаждения до комнатной температуры
содержимое стакана количественно переносят в мерную колбу
вместимостью 200 мл, прибавляя воду доводят объем раствора до
метки, перемешивают и фильтруют через складчатый бумажный
фильтр. Пипеткой отмеривают 50 мл полученного фильтрата,
переносят его в мерную колбу вместимостью 100 мл, прибавляют
10 мл насыщенного раствора щавелевокислого натрия и объем
раствора доводят дистиллированной водой до метки. Раствор
перемешивают и фильтруют через плотный или двойной фильтр.
В фильтрате определяют полноту осаждения свинца, прибавляя
несколько капель раствора щавелевокислого натрия к 1—3 мл
фильтрата. Если образуется белый осадок, значит полнота
осаждения свинца не достигнута. Опыт повторяют с большим
количеством щавелевокислого натрия для осаждения свинца из 50 мл
фильтрата после первого осаждения. Избыток щавелевокислого
натрия в растворе пробы не влияет на определение
редуцирующих Сахаров. Образовавшийся осадок отфильтровывают через
плотный фильтр и в фильтрате определяют редуцирующие
сахара эбулиостатическим методом по первому варианту.
При расчете учитывают разведение пробы при удалении
редуцирующих несахаров и свинца. Редуцирующие сахара при
отсутствии редуцирующих примесей в анализируемой жидкости
определяют с точностью ±2% отн.
Определение сухих веществ высушиванием на бумажных роликах
Для экспрессного определения сухих веществ в сульфитном-
щелоке, в упаренной последрожжевой бражке и в других
подобных растворах предназначен метод, основанный на
удалении воды из тонких слоев анализируемого раствора,
распределенного в капиллярах фильтровальной бумаги, при нагревании
в сушильном шкафу при 105°С. Особенностью сухих веществ,
содержащихся в сульфитном щелоке, является то, что при
высушивании они образуют вязкую массу и прочную поверхностную
пленку, препятствующую удалению последних порций воды.
Мешающие вещества. Органические вещества, которые при
105°С разлагаются или обугливаются.
Аппаратура. Сушильный шкаф. Бюксы. Эксикатор.
Подготовка проб. Горячий сульфитный щелок охлаждают до
комнатной температуры.
Ход анализа. Готовят бюксы с роликами из фильтровальной
бумаги. Вырезают полоски фильтровальной бумаги с
поперечным расположением волокон (см. с. 76) длиной 10—20 см и
шириной, которая на несколько миллиметров меньше высоты
бюкса. Полоску свертывают в ролик, ставят в бюкс и закрывают
крышкой. Крышка не должна касаться ролика. Когда приготовят
59-
необходимое количество бюксов с роликами, их ставят в
сушильный шкаф, открывают, высушивают до полного удаления
влаги при 105°С и до анализа хранят в эксикаторе над серной
кислотой.
Для параллельных анализов взвешивают два-три бюкса с
сухими роликами. На ролик в бюксе наливают 3—5 мл
сульфитного щелока, бюкс закрывают, взвешивают и ставят в
сушильный шкаф (крышку снимают). Через 3—4 ч бюкс
закрывают крышкой в сушильном шкафу, переносят в эксикатор для
охлаждения, а затем взвешивают и снова ставят в сушильный
шкаф. Так повторяют до тех пор, пока разница между
предыдущим и последующим взвешиванием не будет превышать 0,0003 г.
Расчет. Содержание сухих веществ в сульфитном щелоке выражают
в граммах на 100 г сульфитного щелока или в граммах на литр щелока
и вычисляют по формуле
й100 v ДІ000
Xl = —— или Х2 = —у— ,
где Xi —содержание сухих веществ в сульфитном щелоке, г/100 г; Х2 — тоже,
г/л; g—количество сульфитного щелока, взятого на анализ, г; а —
количество сухих веществ после высушивания сульфитного щелока, г; V — объем
сульфитного щелока, взятый на анализ, мл.
Сухие вещества, которые не разлагаются во времи высушивания при
105°С, определяют с точностью ±1,5—2% отн.
Предостережение. При анализе растворов, содержащих вещества,
которые разлагаются при 105°С (например, сахара), высушивать их следует при
более низкой температуре или в вакуум-эксикаторе.
Определение сухих веществ по плотности
Для экспрессного определения сухих веществ в сульфитном
щелоке предназначен метод, основанный на измерении
плотности сульфитного щелока ареометром. Результат анализа
находят по табл. 2, составленной на основании эмпирических данных,
полученных при определении сухих веществ в сульфитном
щелоке высушиванием проб при 105°С. Состав сухих веществ
зависит от состава варочной кислоты, от древесины и от режима
варки, поэтому данные табл, 2 надо эпизодически проверять.
Мешающие вещества. Жидкие вещества, растворенные в
сульфитном щелоке, которые имеют плотность отличную от 1,000.
Аппаратура. Ареометр. Цилиндр без носика. Термометр.
Подготовка проб. Горячий сырой сульфитный щелок
охлаждают до 20°С,
Ход анализа. Около 200 мл сульфитного щелока наливают
в цилиндр вместимостью 250 мл, опускают в него термометр,
а затем осторожно, держа за верхний конец над градуировкой,
погружают ареометр [25]. Погружение считается неправильным,
если у ареометра будет смочена шейка выше уровня жидкости
в цилиндре. В этом случае ареометр вынимают, моют водой, на-
.60
ТАБЛИЦА 2
Зависимость плотности от содержания сухих веществ в сульфитном щелоке
Плотность
при 20°С,
г/см3
I 012
1.015
1,023
1.032
1.037
1,045
Содержание сухих веществ,
г/л, при варке
жесткой
целлюлозы
25
35
52
70
81
97
мягкой
целлюлозы
85,6
Плотность
при 20°С,
г/см3
,052
.075
,116
,162
[,210
,263
1.320
Содержание сухих веществ«
г/л, прн варке
жесткой
целлюлозы
114,0
166,0
260,7
374,0
490.1
631.5
800.5
мягкой
пеллюлозы
120,8
178,2
282,4
392,1
513.4
658,0
834,2
сухо вытирают и повторяют его погружение, принимая нужные
предосторожности для получения правильных показаний.
Плотность по шкале ареометра измеряют по верхнему мениску (см.
рис. 35) при 20°С.
Расчет. Содержание сухих веществ в сульфитном щелоке в граммах на
литр находят по табл. 2.
Плотность ареометром определяют с точностью ±0,001 г/см3. Сухне
вещества в сульфитном щелоке определяют с точностью ±5% отн.
Определение общей SO2
Для определения в сульфитном щелоке свободной и
связанной SO2, присутствующей в виде сернистой кислоты H2SO3,
сульфита кальция CaSO3 и бисульфита кальция Са(Н5ОзЬ [26,
27] предназначен метод, основанный на реакциях окисления SO2
йодом до серной кислоты и сульфата кальция. Реакции
протекают по следующим уравнениям:
H2SO3+I2+H2O-*H2SO4+2HI;
CaSO3+I2+H2O-
Ca(HSO3J+2l2-f2H2O-
> CaSO4+2HI;
¦CaSO4-f-H2SO4+4HI.
Мешающие вещества. Органические вещества, реагирующие
с йодом при комнатной температуре.
Аппаратура. Установка для титрования.
Реактивы. 1. Йод, 0,01 н. раствор, готовят из фиксанала.
2. Крахмал, 0,5%-ный раствор.
Ход анализа. В коническую колбу вместимостью 250 мл
вливают 50 мл дистиллированной воды, 1 мл анализируемого
сульфитного щелока и 4—5 капель свежеприготовленного 0,5%-ного
раствора крахмала. Полученный раствор титруют, осторожно по
61
каплям прибавляя из бюретки 0,01 н. раствор йода до
появления устойчивой синей окраски.
Расчет. Содержание общей SO2 в сульфитном щелоке вычисляют по
формуле
v яО.00032 • 100
g
где X — концентрация общей SO2 в сульфитном щелоке, %; а — объем 0,01 г.
раствора йода, пошедший на титрование, мл; g — объем сульфитного щелока,
взятый на анализ, мл; 0,00032 — количество SO2, соответствующее 1 мл 0,01 н.
растовра йода, г.
Содержание общей SO2 определяют с точностью ±2% отн.
Определение легкоотщепляемой SO2
Для определения в сульфитном щелоке количества SO2,
связанной в альдегидбисульфитных соединениях, предназначен
метод, основанный на реакции разложения
альдегидбисульфитных соединений в щелочной среде. Образующуюся свободную
SO2 определяют титрованием раствором йода как общую SO2
(см. выше).
Мешающие вещества. Сернистая кислота, ее соли и
органические вещества, реагирующие с йодом.
Аппаратура. Установка для титрования.
Реактивы. 1. Иод, 0,01 н. раствор, готовят из фиксанала.
2. Едкий натр, 1 н. раствор.. 3. Соляная кислота, 1 н. раствор.
4. Крахмал, 0,5%-ный раствор.
Ход анализа. Берут 1 мл сульфитного щелока, разбавляют
его водой и титруют 0,01 н. раствором йода с индикатором
крахмал, чтобы окислить свободную и связанную SO2 до иона SO4.
Подготовленную пробу, в которой не осталось ионов SO3,
подщелачивают, добавляя 20 мл 1 н. NaOH, и ждут 15 мин. Затем
раствор подкисляют, вливая в него 25 мл 1 н. раствора соляной
кислоты, и титруют 0,01 н. раствором йода в присутствии
крахмала до появления синей окраски.
Расчет. Содержание легкоотщепляемой SO2 в сульфитном щелоке
вычисляют по формуле
„ (а - ft) 0,00032 • 100
g
где X— содержание легкоотщепляемой SO2 в анализируемой пробе, %; а —
объем 0,01 н. раствора йода, израсходованный на анализ, мл; Ь—объем
0,01 н. раствора йода, израсходованный на окисление свободной и связанной
SO2, мл; g — объем сульфитного щелока, взятый иа анализ, мл; 0,00032 —
количество SO2, соответствующее 1 мл 0,01 н. раствора йода, г.
Легкоотщепляемую SO2 определяют с точностью ±1—2% отн.
Определение моносульфита кальция
Для определения в сульфитном щелоке SO2, связанной
в сульфитах кальция CaSO3 и Ca(HSO3J, сумму которых
называют моносульфитом кальция [26], предназначен метод, осно-
62
ванный на реакции окисления иона SO3 до иона SO4 йодом
в кислой среде после удаления из анализируемого раствора
свободной сернистой кислоты путем выпаривания.
Мешающие вещества. Сернистая кислота и нелетучие
вещества, реагирующие с йодом.
Аппаратура. Установка для титрования и водяная баня.
Реактивы. 1. Йод, 0,01 н. раствор, готовят из фиксанала.
2. Крахмал, 0,5%-ный раствор.
Ход анализа. В фарфоровую чашечку вливают 1 мл
сульфитного щелока, ставят ее на кипящую водяную баню и
выпаривают щелок до полного исчезновения запаха SO2. Остаток
растворяют в воде, раствор количественно переносят в коническую
колбочку вместимостью 250 мл и титруют 0,01 н. раствором
йода в присутствии крахмала, до появления синей окраски
раствора.
Расчет. Содержание моносульфита кальция в сульфитном щелоке
выражают в процентах SO2 и вычисляют по формуле
v a0,00032 • 100
g
где X — содержание моносульфита в пробе, % SO2; a — объем 0,01 н.
раствора йода, израсходованный на анализ, мл; g — объем сульфитного щелока,
взятый на анализ, мл; 0,00032—количество SO2, соответствующее 1 мл 0,01 н.
раствора йода, г.
Воспроизводимость в среднем нз трех титрований ±0,05 мл 0,01 н.
раствора йода.
Определение свободной SO2
Свободной SO2 называют SO2, которая присутствует в
сульфитном щелоке в виде сернистой кислоты и бисульфита кальция.
Содержание свободной SO2 в сульфитном щелоке находят по
разности между содержанием общей SO2 и SO2 в виде
моносульфита.
Определение иона SO4 комплексометрическим методом
Для определения ионов SO4 в сульфитном щелоке, сусле,
сточной воде и других жидкостях предназначен метод,
основанный на выделении ионов SO4 из пробы осаждением их в виде
сернокислого бария в кислой среде. Количество выделенных
ионов SO4 определяют взвешиванием (см. с. 66) или
комплексометрическим методом. Полученный сернокислый барий
растворяют в титрованном растворе трилона Б [28], избыток которого
оттитровывают раствором хлористого магния. Количество
трилона Б, израсходованное на реакцию с барием, эквивалентно
количеству сульфат-иона. Оптимальное количество сульфат-
ионов, которое надо брать на анализ, 5—25 мг.
Мешающие вещества. Медь, сернистая кислота.
Аппаратура. Установка для удаления SO2 из сульфитного
щелока. Установка для титрования. Водяная баня.
63
Реактивы и их приготовление. Все реактивы готовят на
дважды дистиллированной воде, перегнанной в стеклянном
приборе, которая не должна содержать медь. 1. Азот или
углекислый газ в баллоне. 2. Соляная кислота плотностью 1,19 г/см3.
3. Азотнокислое серебро, 1%-ный раствор. 4. Хлористый барий,
0,05 н. раствор. 5. Хлористый магний, 0,05 н. раствор.
Приготовление. В дистиллированной воде растворяют 5,085 г MgCl2-
6Н2О и объем доводят в мерной колбе до 1 л. Поправочный
коэффициент к нормальности раствора устанавливают по точному
раствору трилона Б. 6. Аммиачный буферный раствор.
Приготовление. В мерную колбу вместимостью 1 л вливают 100 мл
20%-ного раствора хлористого аммония, 100 мл 25%-ного
аммиака и дистиллированную воду до метки на колбе. Раствор
хранят в склянке, у которой есть колпачок поверх пробки для
предохранения от улетучивания аммиака. 7. Аммиак, 9 н.
раствор. Приготовление, 67 мл 25%-ного раствора аммиака
разбавляют водой до 100 мл. 8. Сернокислый магний, 0,01 н.
раствор готовят из фиксанала, 9. Индикатор — хромоген
черный ЕТ-00. Приготовление. В 20 мл аммиачного буферного
раствора растворяют 0,5 г индикатора, раствор количественно
переносят в мерную колбу вместимостью 100 мл, и прибавляя
этиловый спирт, объем доводят до метки на колбе.
Приготовление сухого индикатора. Смешивают 0,25 г индикатора с 50 г
хлористого натрия и растирают в ступке. 10. Трилон Б, 0,005 н.
расгвор. Приготовление. В дистиллированной воде
растворяют 9,30 г трилона Б и объем раствора в мерной колбе
доводят до 1 л. Установка титра раствора трилона Б по раствору
сернокислого магния, приготовленного из фиксанала. В
коническую колбу вместимостью 250 мл вливают 50 мл 0,01 н.
раствора сернокислого магния, 50 мл дистиллированной воды (без
меди), 5 мл аммиачного буферного раствора и 5—7 капель
раствора индикатора (или около 0,1 г смеси сухого индикатора) и
титруют 0,05 н. раствором трилона Б при постоянном
перемешивании до изменения окраски на синюю с зеленым оттенком.
Отсчитывают по бюретке количество миллилитров а раствора
трилона Б, пошедшего на титрование, и по следующей формуле
вычисляют поправочный коэффициент К к нормальности
SO-0,01 J0
_ _ J_
Л аО.05 а '
Подготовка проб. Из анализируемого сульфитного щелока
удаляют SO2. Для этого в колбу вместимостью 250 мл (рис. 19)
вливают 100 мл дистиллированной воды, 10 мл пробы и 3 мл
соляной кислоты плотностью 1,19 г/см3 и вставляют барбатер,
который соединен через редуктор с баллоном углекислого газа
или азота. Колбу ставят на включенную электроплитку,
сульфитный щелок нагревают почти до кипения, а затем осторожно
64
открывают кран для подачи газа и в течение 1 ч пропускают
через него газ. После удаления SO2 раствор фильтруют, осадок
и фильтр промывают горячей водой, промывные воды
присоединяют к фильтрату и анализируют.
Ход анализа. Разбавленную пробу сульфитного щелока после
удаления SO2 помещают в колбу вместимостью 500 мл,
добавляют дистиллированную воду до объема раствора 200 мл,
подкисляют концентрированной соляной кислотой до кислой
реакции по метиловому оранжевому и нагревают до кипения. В
горячий раствор прибавляют осторожно по стенке 0,05 н. раствор
хлористого бария (ВаСЬ) до полного осаждения иона SO4
(обычно не более 25 мл). Окончание
реакции образования сернокислого
бария (BaSO4) определяют по
прозрачности раствора на границе соприкосновения
его с раствором хлористого бария.
Затем прибавляют 1—2 мл избытка
раствора ВаСЬ, колбу с реагирующей
жидкостью ставят на электроплитку, кипятят
10 мин и переносят колбу на водяную
баню. Через 1 ч нагревания жидкость
фильтруют, употребляя небольшой
беззольный фильтр «синяя лента», который
предварительно промывают горячей
водой. Осадок стараются оставить в колбе.
Потом осадок промывают 5—6 раз
дистиллированной водой, подогретой до 40—¦
50°С, при этом осадок сернокислого
бария на стенках колбы не трогают.
Промывную воду декантируют и фильтруют
через тот же фильтр. Часть осадка
сернокислого бария, попавшую на фильтр,
промывают подогретой водой 2—3 раза до отрицательной
реакции промывной воды на ион хлора (по AgNO3). Осадок на
фильтре переносят в колбу с осадком, вливают 5 мл 9 н.
раствора аммиака, фильтр разворачивают стеклянной палочкой и
расправляют на дне колбы. В колбу с осадком вливают раствор
трилона Б из расчета 6 мл 0,05 н. раствора трилона Б на
каждые 5 мг предполагаемого содержания сульфат-ионов в объеме
пробы, взятой на анализ. Содержимое колбы осторожно
нагревают на песчаной бане и кипятят до растворения осадка 3—
5 мин, держа колбу в наклонном положении, периодически
перемешивая жидкость качанием колбы. Раствор охлаждают,
приливают 50 мл дистиллированной воды, 5 мл аммиачного
буферного раствора, добавляют 5 капель раствора индикатора (или
0,1 г сухой индикаторной смеси) и избыток трилона Б после
реакции с сернокислым барием оттитровывают 0,05 н. раствором
хлористого магния до перехода синей окраски в лиловую.
тг
Рис. 19. Установка для
удаления SO2 из
сульфитного щелока
5 Заказ № 142
65
Расчет. Содержание сульфат-нона в сульфитном щелоке X выражают
в процентах SO4 нли SO2 и вычисляют по формуле
Х==_(а-ЬJА- 100
#1000
где а — объем 0,05 н. раствора трнлона Б, взятый иа анализ, мл; b — объем
0,05 н. раствора хлористого магния, израсходованный на титрование избытка
трнлона Б, мл; g — объем сульфитного щелока, взятый иа анализ, мл; 1 мл
0,05 и. раствора трнлона Б эквивалентен 2,4 мг иона SO4 нли 1,6 мг SO2.
Сульфат-нон определяют с точностью ±1—3% отн.
Определение серы
Для определения всей серы, связанной в органических и
минеральных веществах, находящихся в сульфитном щелоке,
сточной воде и других жидкостях предназначен метод, основанный
на окислении серусодержащих веществ бромом до получения
ионов SO4, количество которых определяют после осаждения
в виде сульфата бария взвешиванием или комплексометрическим
методом (см. с. 63).
Мешающие вещества. Двуокись серы, присутствующая в
воздухе лаборатории.
Аппаратура. Муфельная печь на 800°С. Эксикатор. Тигли.
Реактивы и их приготовление. 1. Бром или бромная вода,
5%-ный раствор в воде. 2. Едкий натр, 50%-ный раствор. 3.
Азотная кислота плотностью 1,19 г/см3. 4. Аммиак, 25%-ный раствор.
5. Азотнокислое серебро, 1%-ный раствор. 6. Хлористый барий,
10%-ный раствор. Перед анализом его разбавляют в 5 раз,
получая 2%-ный раствор. 7. Соляная кислота плотностью 1,19 г/см3.
8. Метиловый оранжевый, 0,1%-ный водный раствор.
Ход анализа. Окисление серусодержащих
веществ. В колбу Кьельдаля вместимостью 500 мл вливают
5 мл сульфитного щелока, добавляют к ним 1 мл 50% -ного
раствора едкого натра и 3—5 мл брома или 5 мл бромной воды
(при низком содержании серы в пробе). Колбу ставят на
кипящую водяную баню в вытяжной шкаф. Через 15 мин колбу
снимают с бани, охлаждают до комнатной температуры, к
реагирующей смеси прибавляют 15 мл концентрированной азотной
кислоты, закрывают колбу воронкой или втулкой и оставляют
стоять в течение 12 ч. Затем реакционную смесь в колбе
нагревают на электроплитке до кипения и кипятят 6 ч, не открывая
колбу. Через 6 ч кипячения из горла колбы вынимают воронку,
реакционную жидкость выпаривают досуха, колбу с осадком
охлаждают и, вливая в нее 5 мл концентрированной азотной
кислоты, снова нагревают и упаривают содержимое колбы
досуха. Обработку пробы азотной кислотой повторяют до
получения в колбе осадка белого цвета. В охлажденную колбу с
белым осадком добавляют 5 мл концентрированной соляной
кислоты и колбу снова нагревают для упаривания ее содержимого
66
досуха. Осадок соляной кислотой обрабатывают 2 раза. На этом
заканчивается окисление серы до ионов SO4.
Выделение серы в виде сульфата бария.
Осадок в колбе растворяют в теплой воде, подкисленной
соляной кислотой, и полученный раствор фильтруют в стакан.
Фильтрат нейтрализуют, добавляя 25%-ный раствор аммиака (по
метиловому оранжевому), а затем разбавляют, прибавляя к нему
250 мл воды. Раствор подкисляют, прибавляя 1 мл
концентрированной соляной кислоты, нагревают до кипения и проводят
осаждение ионов SO4, осторожно вливая 100 мл горячего
2%-ного раствора хлористого бария. Выпавший осадок
сернокислого бария состоит из очень мелких кристаллов, которые
проходят через фильтр во время фильтрования. Для увеличения
размеров кристаллов сернокислого бария раствор с осадком
в стакане нагревают на водяной бане в течение 2 ч и оставляют
стоять 16 ч при комнатной температуре. Из стакана осадок
количественно переносят на беззольный фильтр «синяя лента»,
вставленный в обычную воронку, промывают холодной водой до
отрицательной реакции промывной воды на хлор с
азотнокислым серебром B мл промывной воды должны оставаться
прозрачными после прибавления к ним 1—2 капель 1%-ного
раствора азотнокислого серебра).
Определение количества выделенной серы.
Фильтр с осадком переносят в тигель, который был
предварительно прокален до полного удаления влаги и хранился в
эксикаторе. Тигель с осадком сернокислого бария ставят в
сушильный шкаф для подсушивания, а потом переносят на
электроплитку для сжигания фильтра. (Не допускать пламени!)
После озоления фильтра тигель с осадком ставят в муфельную
печь для прокаливания при 600—800сС. Прокаливание ведут
3—4 ч. Периодически тигель вынимают из печи, охлаждают
в эксикаторе и взвешивают. Прокаливание считают законченным,
когда разница между предыдущим и последующим
взвешиваниями, отличающимися временем прокаливания на 1 ч, не будет
превышать 0,0003 г.
Расчет. Содержание серы в анализируемой пробе в процентах SO2
вычисляют по формуле
_ аО,2746 • 100
~ g
где X — содержание серы в пробе, °/о SO2; a — количество осадка
сернокислого бария, г; g— объем пробы, взятый на анализ, мл; 0,2746 — количество
SCb, соответствующее 1 г BaSCi, г.
Содержание серы определяют с точностью ±1—2% отн.
Определение SO2, связанной в лнгносульфоновом комплексе
Содержание SO2, связанной в лигносульфоновом комплексе,
непосредственно определить нельзя, поэтому его вычисляют как
5* 67
разность между содержанием всей серы в сульфитном щелоке,
выраженным в процентах SO2, и суммой общей SO2, легкоотщеп-
ляемой SO2 и SO4, выраженных в процентах SO2 [27].
Определение кальция и магния комплексометрическим методом
Для определения суммы кальция и магния в сульфитном
щелоке, гидролизатах, сточной воде и других жидкостях
предназначен метод, основанный на реакциях кальция и магния с кислой
динатриевой солью этилендиаминтетрауксусной кислоты (три-
лон Б)
Na2H2Ci0H12O8N2+Ca+ + —Na2CaCioHi208N2+2H + .
При этой реакции кальций и магний замещают два
незамещенных кислотных водородных атома в молекуле
этилендиаминтетрауксусной кислоты. Образуется растворимое в воде
комплексное соединение, которое разлагается в кислой среде, но
устойчиво в щелочной. Реакцию проводят на холоду при pH 12.
Индикатором служит кислый хром черный специальный (эрио-
хром черный Т), который в присутствии ионов кальция и магния
имеет красную окраску [28].
Мешающие вещества. Медь, стронций, барий.
Реактивы и их приготовление. 1. Дистиллированная вода,
дважды перегнанная в стеклянном аппарате. 2. Аммиачный
буферный раствор. Приготовление. В мерную колбу
вместимостью 1 л вливают 100 мл 20%-його раствора хлористого
аммония, 100 мл 25%-ного раствора аммиака, объем раствора
доводят до метки на колбе, прибавляя дважды дистиллированную
воду. Раствор хорошо перемешивают. 3. Индикатор — кислый
хром черный специальный (эриохром черный Т) употребляют
в виде раствора или в твердом состоянии. Приготовление
раствора. В 10 мл аммиачного буферного раствора
растворяют 0,5 г индикатора, переносят его в мерную колбу
вместимостью 100 мл и, прибавляя этиловый спирт, доводят объем
раствора до метки. Приготовление индикаторной
смеси. Для употребления индикатора в твердом виде готовят
его смесь с хлористым натрием: 0,2 г индикатора и 50 гх. ч.
хлористого натрия смешивают и хорошо растирают в фарфоровой
ступке. 4. Трилон Б — этилендиаминтетраацетат натрия, 0,05 н.
раствор. Приготовление. В дважды дистиллированной воде
растворяют 9,3061 г бигидрата или 8,4052 г безводного трилона
Б и объем раствора в мерной колбе доводят до 1 л. Если
раствор мутный, его фильтруют.
Установка титра. Титр раствора трилона Б
устанавливают по разным веществам — по окиси магния,
металлическому магнию, углекислому кальцию и по сернокислому магнию.
Сернокислый магний (MgSCU • 7Н2О) при хранении частично
68
теряет кристаллизационную воду, поэтому если хотят по нему
устанавливать титр раствора трилона Б, то перед анализом его
помещают на 24 ч в эксикатор над смесью из 5 частей сульфата
магния и 1 части воды. Для установки титра готовят 0,05 н.
раствор выбранного соединения. В коническую колбу
вместимостью 250 мл вливают 10 мл приготовленного раствора,
добавляют 50 мл дважды дистиллированной воды, 5 мл аммиачного
буферного раствора, 5—6 капель индикатора кислого хром
черного специального и титруют раствором трилона Б до перехода
винно-красной окраски раствора в синюю. Вычисляют
количество граммов магния, кальция или окиси кальция, которое
соответствует 1 мл раствора трилона Б, т. е. титр раствора
трилона Б по выбранному веществу.
Подготовка проб. При анализе сырого сульфитного щелока
его нейтрализуют до pH 3—4 по метиловому оранжевому.
Ход анализа. В коническую колбу вместимостью 250 мл
вливают 1 мл сульфитного щелока, 5 мл аммиачного буферного
раствора, 100 мл дважды дистиллированной воды и 1,5—2 мл
раствора индикатора или 0,1 г индикаторной смеси. Жидкость
в колбе перемешивают и титруют, прибавляя раствор трилона
Б из бюретки до перехода красно-коричневой окраски титруемоп
жидкости в зеленую или синюю.
Расчет. Содержание кальция и магния в сульфитном щелоке выражают
в процентах кальция или окиси кальция и вычисляют по формуле
X = TalOO.
где X — содержание кальция и магния, °/о Са или СаО; Г—титр раствора
трилона Б, т. е. количество граммов определяемого вещества,
соответствующее 1 мл раствора трилона Б; а — объем раствора трилона Б,
израсходованный на анализ, мл.
Воспроизводимость результатов титрования в среднем из трех
определений ± 0,05 мл раствора трилона Б.
Определение мышьяка колориметрическим методом
Этот метод предназначен для определения мышьяка в
органических и неорганических веществах [30]. Метод основан на
реакциях минеральных соединений трехвалентного мышьяка с
атомарным водородом (в момент выделения), в которых мышьяк
в присутствии катализатора — хлористого олова
восстанавливается до мышьяковистого водорода (АэНз). Сложные
органические соединения перед анализом минерализуют различными
способами, чтобы получить неорганические соединения
трехвалентного мышьяка. Минеральные вещества, содержащие
пятивалентный мышьяк, перед анализом специально обрабатывают,
чтобы мышьяк превратить в трехвалентный. Количество
образовавшегося мышьяковистого водорода (АэНз) определяют по
интенсивности окраски соединения, которое он дает при реакции
69
с бромной ртутью (HgBr2), протекающей по следующим
уравнениям:
AsH3+3HgBr2-~3HBr+As (HgBrK;
As (HgBrK+AsH3 -*- 3HBr+As2Hg3.
Мешающие вещества. Соединения, содержащие серу, сурьму,
и фосфор. Сера, восстанавливаясь до сероуглерода, реагирует
с бромной ртутью и образуется соединение, окрашенное в
желтый цвет. Сурьма при анализе дает бурое пятно на бромнортут-
ной бумажке.
Аппаратура. Прибор для определения мышьяка. Установка
для возгонки бромной ртути. Установка для минерализации.
Реактивы и их приготовление. 1. Основной стандартный
раствор мышьяка. Приготовление. 0,1320 г мышьяковистого
ангидрида (As2O3), (яд!) растворяют в 25 мл 1 н.
раствора едкого натра, раствор количественно
с помощью дистиллированной воды переносят
в мерную колбу вместимостью 1 л и его объем
доводят до метки на колбе. В 1 мл
приготовленного раствора содержится 100 мкг мышьяка.
2. Рабочий стандартный раствор мышьяка.
Приготовление. В мерную колбу вместимостью
Рис. 20. Установка для возгонки бромной ртути
100 мл вливают 1 мл основного стандартного раствора мышьяка
и добавляют дистиллированную воду до метки. В 1 мл
приготовленного раствора содержится 1 мкг мышьяка. Раствор
неустойчив, его готовят в день проведения анализа. 3. Серная
кислота плотностью 1,84 г/см3. 4. Серная кислота, разбавленная
1:8 (по объему), х. ч., без мышьяка. 5. Пергидроль (Н2О2),
30%-ный раствор. 6. Цинк гранулированный, без мышьяка.
7. Двухлористое олово х. ч. 8. Вата, пропитанная уксуснокислым
свинцом. Приготовление. Гигроскопическую вату
погружают в 5%-ный раствор уксуснокислого свинца, затем ее
вынимают, слегка отжимают между листами фильтровальной бумаги,
высушивают на воздухе и хранят в плотно закрытой склянке.
9. Бромнортутные бумажки. Приготовление. Бромную ртуть
очищают возгонкой. На дно стакана вместимостью 150—200 мл
помещают 10 г HgBr2. Стакан ставят на песчаную баню и
закрывают дном круглодонной колбы вместимостью 100 мл, через:
которую пропускают холодную водопроводную воду (рис. 20).
При нагревании бромная ртуть возгоняется и в виде белых
кристаллов / оседает на холодном дне колбы. Очищенную бромную
70
ртуть хранят в темной склянке, закрытой притертой пробкой.
Для приготовления бромнортутных бумажек берут беззольные
фильтры и погружают их на 1—2 мин в 5%-ный раствор
очищенной бромной ртути в этиловом спирте, вынимают пинцетом
и сушат на воздухе, положив на стеклянные палочки.
Высушенные фильтры разрезают на полоски размером 2X60 мм и хранят
в темной склянке с притертой пробкой. 10. Парафин, 5%-ный
раствор в петролейном эфире.
Приготовление стандартной шкалы. Для
проверки чистоты реактивов проводят холостой опыт. В сосуд 1
прибора (рис. 21) вливают 10 мл дистиллированной воды, 50 мл
H2SO4 A:8), 0,2 г хлористого олова и 2 г цинка. Сосуд
закрывают пробкой с насадкой 2, в которую
помещена вата, пропитанная уксуснокислым
свинцом, и вставлена бромнортутная бумажка 4.
Через 1,5 ч вынимают бромнортутную бумажку из
прибора. Если на ее конце, который был
вставлен в трубку, нет следов желтой окраски, то
считают примененные реактивы чистыми.
Для приготовления стандартной шкалы
проводят анализы по нижеприведенной методике
с десятью различными количествами мышьяка.
Основной стандартный раствор мышьяка раз-
Рис. 21. Прибор для определения мышьяка
<5авляют в 100 раз, получают раствор, который содержит 1 мкг
мышьяка в 1 мл. На анализ берут 1, 2, 3, ..., 10 мл
приготовленного раствора. В результате получают десять бромнортутных
бумажек, которые отличаются между собой величиной пятна и
интенсивностью его окраски от желтого до коричневого цвета.
Делают две-три серии таких анализов, чтобы установить
воспроизводимость. Отбирают наиболее характерно окрашенные
десять бумажек, величина и цвет пятен которых соответствует 1,
2, 3, ..., 10 мкг мышьяка, взятого на анализ. На неокрашенной
части бумажек простым карандашом пишут количество микро-
граммов мышьяка, которое дало эту окраску. Для
консервирования окраски бумажки опускают на мгновение в раствор
парафина в петролейном эфире, высушивают на воздухе и помещают
между двух стеклянных пластинок. Края пластинок заклеивают
бумагой. Шкала готова.
Подготовка проб. При анализе сульфитного щелока, чтобы
удалить серу, минерализацию проводят следующим образом.
В колбу Кьельдаля вместимостью 250 мл вливают 25 мл
сульфитного щелока и 25 мл концентрированной серной кислоты.
71
Колбу укрепляют в штативе в наклонном положении (см.
рис. 64), подставляют под нее электроплитку и включают
нагрев. В начале нагревания реакционная смесь сильно пенится,
поэтому нагрев должен быть слабым. После прекращения
вспенивания колбу охлаждают, добавляют в нее 15 мл пергидроли
и снова кипятят до обесцвечивания реакционной смеси.
Бесцветную жидкость охлаждают, количественно переносят в мерную
колбу вместимостью 200 мл и объем раствора доводят водой
до метки на колбе. Приготовленный раствор анализируют.
Ход анализа. Берут прибор для определения мышьяка
(рис. 21). Шарик 2 в нижней части насадки набивают ватой,
пропитанной уксуснокислым свинцом. В трубочку 3 (диаметром
2—2,5 мм), которая является верхней частью насадки, помещают
полоску бромнортутной бумажки 4 всегда на одну и ту же
глубину. Выступающий конец бумажки загибают, чтобы она не
перемещалась во время анализа. Обе части насадки соединяют,
хорошо притирая шлифы 5.
В реакционный сосуд 1 вместимостью 100 мл помещают 10—
20 мл исследуемого раствора, 50 мл серной кислоты A:8), 0,2 г
двухлористого олова и около 2 г цинка. После внесения цинка
сосуд быстро закрывают подготовленной насадкой, прибор
накрывают колпачком из черной бумаги или ставят в темное место.
Через 1,5 ч анализ считают законченным. Вынимают из
прибора бромнортутную бумажку и сравнивают ее окраску с
окраской полосок стандартной шкалы или с окраской полосок бромно-
ртутных бумажек, полученных в параллельных анализах,
поставленных с различными количествами эталонного раствора. Для
ускорения анализа проводят опыт сразу в нескольких приборах
с различным количеством исследуемого раствора.
При сравнении окраски бромнортутной бумажки с окрасками
полосок стандартной шкалы учитывают два фактора: площадь
окрашенной части бумажки и интенсивность окраски. Если
окраска бромнортутной бумажки не совпадает ни с одной из
окрасок стандартной шкалы, то берут среднее значение между
полосками близкими по окраске или повторяют анализ с
другим количеством исследуемой пробы.
Расчет. Содержание мышьяка в сульфитном щелоке вычисляют по
формуле
Y aVlOO
где X — концентрация мышьяка в сульфитном щелоке, %; а — количество
мышьяка, найденное в пробе сравнением окраски бромнортутной бумажки
со стандартной шкалой, мкг; g — объем сульфитного щелока, взятый для
минерализации, мл; V — объем раствора, полученного после минерализации
сульфитного щелока, мл; gi — объем раствора, полученного после
минерализации сульфитного щелока, помещенный в прибор, мл; 10е — перевод
микрограммов (мкг) в граммы (г).
Чувствительность реакции 0,5 мкг мышьяка. Точность определения
мышьяка ±15—25% оти.
72
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ РЕДУЦИРУЮЩИХ ВЕЩЕСТВ
Хроматографическое разделение веществ на бумаге
Теоретические сведения. Хроматографическое
разделение смесей веществ на бумаге относится к методам
распределительной хроматографии, основанным на различии величин
коэффициентов распределения анализируемых веществ между
подвижным и неподвижным растворителями. Подвижным
растворителем является смесь одного или нескольких веществ с водой.
Неподвижным растворителем является набухшая целлюлоза.
Целлюлоза набухает за счет воды, которая находится в
подвижном растворителе.
Коэффициент распределения вещества зависит от его
растворимости в подвижном растворителе и от адсорбции его на
целлюлозе. Механизм разделения смеси веществ с помощью
хроматографии на бумаге заключается в следующем. На пути
движения растворителя по бумаге помещают смесь веществ,
подлежащих разделению. Растворитель, достигнув участка бумаги, на
котором находится смесь веществ, начинает экстрагировать
отдельные вещества и передвигать их по бумаге с различной
скоростью. При движении по бумаге вещества группируются по
скорости их движения, образуя ряд отдельных групп, каждая
из которых содержит одно или несколько веществ. Величина
скорости движения веществ по бумаге обусловлена величиной
равнодействующей двух противоположно направленных сил:
силы сродства вещества к растворителю (растворимость) и силы
адсорбции вещества на набухшей целлюлозе. Чем выше
растворимость данного вещества в растворителе и чем быстрее
передвигается растворитель по бумаге, тем больше сила, двигающая
вещество в направлении движения растворителя. Сила
адсорбции вещества на целлюлозе тормозит это движение. Величина
и конфигурация молекул передвигаемых веществ оказывает
влияние на скорость движения вещества и его отделение от
других веществ. Коэффициенты распределения веществ различны
при употреблении различных растворителей, поэтому для хрома-
тографического разделения смесей различных веществ
необходимо выбирать растворитель, обеспечивающий наиболее полное
разделение веществ.
При разделении смеси бесцветных веществ для обнаружения
их на бумаге хроматограмму проявляют, обрабатывая ее
раствором вещества (проявителем), дающего окрашенное
соединение с разделенными веществами. По проявленной хроматограмме
определяют наличие интересующих компонентов и степень их
разделения. Отношение длины пути, который прошло вещество
по бумаге, к длине пути, который прошел растворитель по
бумаге за то же время в стандартных условиях хроматографиро-
вания, есть величина постоянная, специфичная для каждого
73
соединения при употреблении определенного растворителя. Ее
принято обозначать Rf.
Техника хроматографирования. Различают
четыре вида хроматографирования на бумаге: восходящая
хроматография, нисходящая, многократная и двумерная. При
восходящей хроматографии растворитель движется снизу вверх.
Нижний конец хроматограммы при этом погружен в
растворитель. При нисходящей хроматографии, наоборот, верхний конец
хроматограмм погружен в растворитель. Направление движения
растворителя по бумаге сверху вниз.
Многократная хроматография может быть нисходящей и
восходящей. Этот вид хроматографирования характеризуется пов-
торностью пропускания растворителя по бумаге в выбранном
направлении. После того как растворитель дойдет до конца
хроматограммы, ее вынимают, высушивают и хроматографирова-
ние повторяют. Многократное хроматографирование применяют
в том случае, когда надо разделить смесь веществ, имеющих
очень близкие Rf, или смесь веществ, образующих размытые
зоны — «хвосты» (например, при анализе сульфитных щелоков).
При таком хроматографировании получают компактные пятна
разделенных веществ.
Двумерная хроматография может быть нисходящей,
восходящей и многократной. Особенностью ее является применение
двух различных растворителей, которые пропускают по бумаге
поочередно во взаимно перпендикулярных направлениях. Для
получения двумерной хроматограммы берут лист бумаги и в
одном из ее углов наносят каплю анализируемого раствора,
просушивают пятно и хроматографируют, применяя первый
растворитель. Когда будет достигнуто нужное разделение на группы
веществ, хроматограмму высушивают и снова хроматографируют
со вторым растворителем, повернув хроматограмму на 90°. Этот
вид хроматографирования применяют для разделения смесей,
состоящих из 10—20 компонентов, имеющих близкие Rf
(например, при разделении смеси аминокислот).
Камеры. Камерой для хроматографирования может
служить эксикатор, банка с притертой крышкой, стеклянный
колпак на шлифованном стекле и стеклянные камеры с
каркасами из нержавеющей стали. Камеры должны герметически
закрываться. Эксикатор применяют при хроматографировании на
лопатках (рис. 22). Банку с притертой крышкой применяют при
восходящей и многократной хроматографии (рис. 23).
Стеклянный колпак, притертый к стеклу, применяют при восходящем или
нисходящем хроматографировании на длинных лентах при
количественном определении углеводов и других веществ (рис. 24).
Камеры из стекла в каркасе из нержавеющей стали применяют
для двумерной хроматографии (рис. 25).
Хроматографирование проводят при температуре 18—22°, помещая камеры в
специальные комнаты или в вытяжной шкаф.
74
п
Рис. 22. Установка для нисходящего хроматографи-
рования в эксикаторе:
I — заготовка хроматограммы; 2— хроматограмма раствора
трех Сахаров; 3 — хроматограмма пробы и свидетеля —
раствора двух Сахаров; 4 — хроматограмма раствора трех
олигосахаридов
л,
*-
и
S
"V
1
-О-
Рис. 23. Установка для восходящего хроматографиро-
вания в банке с подставкой:
/—подставка; 2— хроматограммы; 3 — растворитель;
4—заготовка хроматограммы; 5 — линия старта; 6 — надрез для
подвешивания хроматограммы; 7 — проявленная хроматограмма
Рис. 24. Установка для
нисходящего хроматографировання под
колпаком:
/ — растворитель; 2 — хроматограмма;
3 — приемник для растворителя; •* —
заготовка хроматограммы; 5 — линия
старта; 6 — проявленная хроматограмма
о о
о о о
о о
Рис. 25. Установка для нисходящего хро-
матографирования в камере для
получения двумерных хроматограмм:
/—хроматограмма; 2 — растворитель; 3 —
проявленная хроматограмма-свидетель после
пропускания одного растворителя; 4 —
проявленная хроматограмма-свидетель после двух
растворителей
Хроматограф и ческая бумага. При выборе сорта
хроматографнческой бумаги для разделения углеводов,
аминокислот и других веществ руководствуются результатами ее
испытания, которое проводят на чистых растворах смесей
интересующих веществ, применяя соответствующие растворители.
Скорость разделения смесей веществ зависит не только от сорта
хроматографической бумаги, но также и от направления в ней
волокон целлюлозы. Полоска хроматографической бумаги во
время анализа играет роль колонки из набухших волокон
целлюлозы. Между волокнами во время хроматографирования
движется растворитель с анализируемой смесью
веществ. Волокна целлюлозы в листе бумаги
расположены в определенном порядке,
поэтому в полоске бумаги их расположение
зависит от того, как она вырезана. Если
полоска вырезана так, что направление волокон
в ней продольное, т. е. совпадает с
направлением движения растворителя, то скорость
движения растворителя, а следовательно, и
скорость разделения анализируемой смеси
веществ на этой полоске будет больше, чем на
полоске с поперечным расположением
волокон. Площадь, занимаемая веществом,
выделенным во время хроматографирования, будет
больше, если разделение проводят на
полоске с продольным расположением волокон. Это
влияние структуры бумаги необходимо
учитывать при хроматографических анализах и
особенно при количественном определении
компонентов.
Определение направления
волокон целлюлозы в листе бумаги.
Из листа бумаги (одного из его углов) вырезают две полоски
шириной 1 см и длиной 10 см (рис. 26). Большим и
указательным пальцами правой руки зажимают по одному концу
обеих полос и придают им горизонтальное положение. По
степени прогиба полос определяют в них расположение волокон.
Полоска а почти горизонтальна, в ней волокна имеют продольное
расположение, а в полосе б — поперечное. Следовательно,
в листе бумаги волокна целлюлозы расположены как указано
стрелкой.
Заготовка хроматограмм. Для каждого вида
анализа из хроматографической бумаги вырезают кусочки разной
формы и различных размеров. При подготовке хроматограмм
для качественного анализа бумагу нарезают в виде лопаток
(см. рис. 22) с продольным или поперечным расположением
волокон различных размеров. Наиболее удобны лопатки размером:
а=1,5-^-3 см; б~Ю~15 см; в=10-н20 см и г=10-н20 см. Для
Рис. 26.
Расположение волокон
целлюлозы в листе
бумаги:
/ — лист бумаги; а —
полоска бумаги с рас-
ПОЛОЖЄЕІНЄМ ВОЛОКОН
в продольном
направлении; б — полоска
бумаги с поперечным
расположением
волокон
76
количественного определения из бумаги нарезают полоски с
продольным или поперечным расположением волокон различных
размеров. При заготовке хроматограмм для нисходящего хро-
матографирования лист хроматографической бумаги с известным
расположением волокон размечают простым карандашом на
полоски шириной 3 см и длиной 25—30 см и проводят линию
старта 5 на расстоянии 10 см от предполагаемого верхнего
края полос. На противоположной стороне листа проводят две
линии для обозначения размеров «язычков» (см. рис. 24).
Ширина «язычка» 7 полоски должна быть обязательно на 2—3 мм
больше, чем горлышко колбы, в которую он будет вставлен,
поэтому их вырезают при постановке полос на хроматографирова-
ние. При заготовке хроматограмм для восходящего хроматогра-
фирования нарезают полоски без «язычков» (см. рис. 23), но
с обозначением места отверстия 6 для подвешивания
хроматограмм и линии старта 5.
Хроматографическую бумагу и заготовки хроматограмм
хранят в конвертах из кальки или полиэтилена. Свертывать и
перегибать хроматографическую бумагу в листах и заготовках
нельзя.
Растворители. При выборе растворителя для
разделения смеси веществ руководствуются значениями Rf или Rx
анализируемых веществ:
г, путь, пройденный веществом
f путь, пройденный растворителем '
Q путь, пройденный веществом
х путь, пройденный одновременно другим контрольным
веществом (например, глюкозой)
Для разделения углеводов предложено много разных смесей
органических растворителей с водой. В табл, 3 приведены Rf
и Rx по глюкозе, т. е. /?гл для различных веществ при
употреблении различных растворителей. Чем больше отличаются
значения Rf или /?гл различных веществ, тем быстрее и лучше
проходит их разделение при хроматографировании.
Для приготовления растворителя в делительную воронку
вливают отмеренные объемы компонентов растворителя, смесь
энергично взбалтывают в течение 3—5 мин и дают постоять 20—
30 мин. Смеси некоторых растворителей не расслаиваются при
стоянии, поэтому их используют для хроматографирования сразу
после приготовления. Но есть смеси, которые расслаиваются при
стоянии, их надо выдержать до полного разделения жидкостей
в делительной воронке. Для хроматографирования используют
верхний или нижний слой согласно прописи анализа.
Смеси редуцирующих веществ в гидролизатах и сульфитных
щелоках, в которых содержатся 5—7 моносахаридов, оли-
госахариды, уроновые кислоты и другие вещества, разделяют
77
ТАБЛИЦА 3
Значение Rf и /?гл некоторых редуцирующих веществ
при различных растворителях
Название вещества
Галактоза
Глюкоза
Манноза
Фруктоза
Арабиноза
Кснлоза
Рамноза
Целлобиоза
Галактуроновая кислота
Оксиметилфурфурол
Растворители
этилацетат-
вода —
*/
0.15
0.18
0.22
0.23
0.25
0,33
0.47
0.11
0,10
0,90
- пиридин —
E:1:5)
«гл
0.83
1.0
1.22
1.28
1.39
1.83
2,61
0.60
0.55
—¦
и-бутаиол —
пиридин — вода
C:1: 0,5)
«/
0.13
0.16
0.20
0.19
0,25
0.20
—
—
—
я-бутанол —
уксусная
кислота — вода
D:1: 0,5)
Rf
0,12
0,13
0,17
0,18
0,19
0,21
—.
—
—
с помощью растворителя из этилацетата, пиридина и воды
E:1:5).
Приготовление растворителя из
этилацетата, пиридина и вод ы. Цилиндром отмеривают 150мл
этилацетата, 30 мл пиридина и выливают их в делительную
воронку на 500 мл. Смесь хорошо взбалтывают, прибавляют к ней
150 мл дистиллированной воды, снова взбалтывают в течение
2—3 мин и оставляют стоять для расслаивания. Когда верхний
слой жидкости станет прозрачным, нижний слой сливают, а
верхний используют для хроматографирования. Если при хранении
слитый верхний слой, предназначенный для
хроматографирования, станет мутным, его надо профильтровать через бумажный
фильтр. Мутный растворитель нельзя применять для анализа.
Изменение температуры в комнате, где хранится
приготовленный растворитель, иногда вызывает расслаивание смеси, на
дно оседают капли воды. Такой растворитель необходимо
хорошо взболтать и снова поставить в делительной воронке на
расслаивание. Если вода отделится, то ее сливают через кран,
а растворитель используют для анализа. Если же раствор
останется мутным, то его надо отфильтровать.
Проявители. Для проявления хроматограмм углеводов
применяют различные вещества, которые дают окрашенное
соединение с углеводами. В табл. 4 приведены составы растворов,
которые применяют для проявления углеводов.
Для проявления хроматограмм гидролизатов растительных
тканей, которые не содержат кетоз, наилучшим проявителем для
углеводов признан раствор анилинфталата. В случае необходи-
78
Проявители для углеводов
ТАБЛИЦА 4
Проявители
Состав
и способ приготовления
Что проявляется
Анилинфталат
а-Нафтол
Бензидин
Азотнокислое
серебро
Мочевина
1,66 г фталевой кислоты и 1 мл
свежеперегнанного анилина
растворяют в бутаноле или
этаноле. Объем раствора
доводят до 100 мл
растворителем. Раствор хранят в темной
склянке
Перед употреблением
смешивают равные объемы двух
растворов следующего состава:
первый раствор — 1%-ный
раствор а-нафтола в водонасы-
щенном бутаноле; второй
раствор— 10%-ный раствор орто-
фосфорной кислоты в водона-
сыщенном бутаноле
0,5 г бензидииа и 20 мл
ледяной уксусной кислоты
помещают в мерную колбу
вместимостью 100 мл и прибавляют
этанол до метки. Раствор
взбалтывают
Смешивают 25 мл 0,1 н. AgNOe,
18 мл 5 н. NH4OH и 7 мл
этилового спирта. Раствор
взбалтывают
5 г мочевины растворяют в
20 мл 1 н. НС1 и объем
раствора доводят до 100 мл,
прибавляя этанол
Альдозы, уроновые
кислоты, олигосахариды,
фурфурол, оксиметилфур-
фурол, метилированные
альдозы
Кетозы, диангидриды
дифруктозы
Углеводы
Углеводы
Кетозы
мости проверки анализируемого раствора на содержание в нем
кетоз используют раствор а-нафтола.
Проявление хроматограмм. Хроматограмму
просушивают на воздухе до полного удаления растворителя,
опрыскивают проявителем из пульверизатора или на мгновение
погружают в проявитель, отжимают между листами фильтровальной
бумаги и помещают в сушильный шкаф, который
предварительно нагревают до нужной температуры. Хроматограмму
в сушильном шкафу кладут на ребро так, чтобы она не
прикасалась к стенкам шкафа.
При проявлении хроматограмм а-нафтолом их выдерживают
при 100°С в течение 5 мин. Фруктоза с а-нафтолом дает
соединение зеленоватого цвета, сахароза и рафиноза — фиолетового.
Окраска пятен неустойчива, в течение суток она исчезает.
Проявление хроматограмм анилинфталатом проводят при
различной температуре и времени проявления. В табл. 5 приведены
79
условия проявления анилинфталатом различных редуцирующих
веществ и цвет образующихся соединений.
ТАБЛИЦА 5
Проявление хроматограмм анилинфталатом
Название вещества
Олигосахариды
и продукты
реверсии
Уроновые кислоты
Целлобиоза
Галактоза
Глюкоза
Манноза
Арабиноза
Ксилоза
Рамноза
Оксиметилфурфу-
рол
Фурфурол и
другие РВ, имеющие
Условия проявления
Температура,
105-130
50-70
100-130
100—105
100-105
100-105
70-80
70—80
100-105
50-70
50-70
Время,
мин
5-Ю
3-5
5-Ю
5
5
5
3-5
3-5
5
2
2
Цвет пятна
Коричневый
Желтый и
оранжевый
Коричневый
То же
Красный
То же
Коричневый
Желто-розовый
Розовый, желтый,
зеленый
Флюоресцируют в
ультрафиолетовом
свете
Нет
„
Да
Нет
Приемы идентификации веществ после хро-
м атографирования. Для установления природы веществ,
разделенных на бумажной хроматограмме, используют
различные свойства этих веществ: скорость движения по бумаге,
реакции с образованием окрашенных соединений и свидетелей-
спутников. Скорость движения «аждого вещества по бумаге при
употреблении определенного растворителя является постоянной
величиной. Зная Rf или Rx разделенных веществ, определяют
расположение того или иного вещества на хроматограмме.
Применяя различные проявители, устанавливают наличие
функциональных групп. Например, при разделении смеси кетоз и альдоз
две параллельные хроматограммы проявляют различными
проявителями, одну проявляют анилинфталатом, а другую а-нафто-
лом. При идентификации альдоз, разделенных на
хроматограмме, пользуются тем, что анилинфталат дает с гексозами и
пентозами соединения различного цвета.
Для установления природы вещества по свидетелю-спутнику
проводят одновременное хроматографирование пробы и смеси
пробы со свидетелем или одного свидетеля. Из хроматографиче-
ской бумаги вырезают лопатку 3 с широким хвостиком (см.
рис. 22) и делят его пополам вертикальной линией. На одну
половину наносят 0,01 мл пробы. На другую половину хвостика
наносят 0,01 мл пробы, пятно высушивают и в то же место
наносят 0,01 мл раствора известных веществ. Иногда на вторую
80
половину наносят только раствор известных веществ, служащий
свидетелем-спутником. Лопатку ставят на хроматографирование,
потом проявляют и сравнивают интенсивность окраски и цвет
пятен, полученных на хроматограмме пробы и на хроматограмме
пробы со свидетелем. Если на хроматограмме пробы со
свидетелем обнаруживают лишнее пятно, которого нет иа
хроматограмме анализируемой пробы, то, следовательно, в
анализируемой пробе нет того вещества, которое взято в качестве
свидетеля. Если же на хроматограмме со свидетелем обнаруживают
увеличение площади или интенсивности
окраски какого-либо пятна по сравнению
с аналогично расположенным пятном на
хроматограмме без свидетеля, то это
пятно соответствует тому веществу,
которое взято в качестве свидетеля.
Подготовка проб для хро-
матографирования. Кислые
пробы гидролизатов и сульфитных щелоков
перед анализом нейтрализуют содой до
pH 3—4. Гидролизаты, полученные при
гидролизе растительных тканей
концентрированной серной кислотой,
нейтрализуют сухим едким барием или
углекислым барием. Выпавший осадок
сернокислого бария удаляют из анализируемого
раствора фильтрованием или
декантацией после отстаивания.
Нейтрализованный гидролизат должен содержать 3—
5% редуцирующих веществ. Растворы,
содержащие менее 1 % редуцирующих
веществ, упаривают на водяной бане под
вакуумом до концентрации 2—3%. Если
упаривание раствора не желательно, то
на анализ берут соответственно большее количество пробы. Для
этого на хроматограмму наносят анализируемый раствор
несколько раз в одно и то же место по 0,01 мл, хорошо
просушивая пятно на бумаге после каждого нанесения пробы.
Микропипетка с приспособлением. Отмеривают
анализируемый раствор и наносят его на бумагу мнкропипеткой
со специальным приспособлением (рис. 27). Приспособление
состоит из металлического кожуха 2, в стенке которого имеется
микровинт .3. В середине кожуха помещена резиновая груша /,
надетая на верхний конец пипетки. Цена деления пипетки
0,001 мл.
В сухую чистую микропппетку набирают 0,01 мл
анализируемого раствора, погружая ее кончик в стаканчик с пробой. Если
в пипетку набралось раствора меньше 0,01 мл, то, ослабляя
давление на грушу поворотом микровинта, засасывают нужное
Рис. 27. Микрошшетка
с приспособлением
6 Заказ № 142
81
количество пробы. Если в пипетку набралось раствора больше
0,01 мл, то легким прикосновением кусочка фильтровальной
бумаги к отверстию в кончике пипетки убирают избыток раствора.
Внешнюю поверхность кончика пипетки, которая погружалась
в раствор, вытирают фильтровальной бумагой.
Отмеренный анализируемый раствор из пипетки на заготовку
хроматограммы переносят следующим образом. Держа
заготовку двумя руками (локти положить на стол) прикладывают
ее к отверстию микропипетки тем местом, куда надо нанести
анализируемый раствор, при этом весь отмеренный объем пробы
переходит на бумагу. Если часть раствора осталась в пипетке,
то повертывают микровинт так, чтобы увеличить давление на
грушу. Полученное пятно анализируемой жидкости сушат на
воздухе при комнатной температуре. При отмеривании объема
пробы пипеткой надо следить, чтобы не было пузырьков воздуха
или разрывов в отмеренном объеме жидкости.
Хроматографирование для качественных методов
Качественные методы, предназначенные для определения
наличия интересующих веществ в пробе с помощью хроматографии
на бумаге, являются не только экспрессными, но и массовыми,
позволяющими анализировать одновременно много проб. При
анализе проб, отобранных на различных стадиях
технологического процесса, качественный анализ становится
полуколичественным. Сравнение интенсивностей окрасок пятен на хромато-
граммах проб, отобранных в различное время, дает возможность
судить о скорости увеличения (при гидролизе) или уменьшения
(при биохимических процессах) концентрации тех или иных са-
харов. В зависимости от содержания редуцирующих веществ
в пробе применяют хроматографирование на лопатках из хро-
матографической бумаги или на полосках.
Хроматографирование на лопатках применяют в тех случаях,
когда число компонентов смеси, подлежащей разделению, больше
пяти, концентрация редуцирующих веществ выше 3% или
значения Rf компонентов смеси очень близки.
Вырезают заготовки хроматограмм в виде лопаток с
продольным расположением волокон. Лопатки должны иметь
следующие размеры: а = 2 см; 6 = 15 см; в = 20 см (см. рис. 22).
Хроматографирование проводят в эксикаторе. Растворителем
служит верхний слой смеси из этилацетата, пиридина и воды
в соотношении 5:1:5 (см. с. 78). Подготавливают пробы,
отмеривают и наносят их на хроматограммы, так как описано
выше. Хроматографируют одновременно четыре хроматограммы,.
на которые нанесены различные количества @,01, 0,02, 0,03 и
0,04 мл) анализируемой пробы. Когда растворитель дойдет
почти до конца хроматограммы, все хроматограммы вынимают
из камеры и просушивают на воздухе в вытяжном шкафу. Хро-
82
матограмму, на которую было нанесено 0,01 мл пробы,
проявляют анилинфталатом, как описано на с. 80. Остальные три
хроматограммы ставят на повторное хроматографирование. Так
повторяют еще 3 раза, проявляя одну хроматограмму после
каждого хроматографирования. Проявленные хроматограммы
расшифровывают, пользуясь данными табл. 3 и 5.
Хроматографирование на полосках применяют когда число
компонентов смеси, подлежащей разделению, меньше пяти,
концентрация редуцирующих веществ в пробе составляет от 0,1 до
3% и значения Rf компонентов смеси различны.
Для хроматографирования употребляют полоски размером
3x30 см. Подготавливают пробы и их наносят на
хроматограммы как описано на с. 81. Камерой для
хроматографирования служит банка с притертой крышкой. Поток растворителя —
восходящий (см. рис. 23). В банке диаметром 25 см
одновременно можно хроматографировать шесть-семь полосок. Каждую
пробу, подлежащую анализу, наносят на одну или несколько
полосок в различном количестве. Полоски с нанесенными на них
пробами (после просушки пятен проб) нанизывают на
стеклянную палочку на расстоянии 2—3 см друг от друга. Затем
палочку кладут на стеклянную подставку в камере, на дно которой
налит растворитель. Слой растворителя должен быть таким,
чтобы нижние концы хроматограмм погружались в него на 1—¦
2 см, а пятна проб были выше уровня растворителя не менее,
чем на 2 см. После погружения хроматограмм в растворитель
банку закрывают и ждут, когда растворитель поднимется по
хроматограммам почти до палочки, на которой они висят.
Открыв крышку банки вынимают палочку с нанизанными хрома-
тограммами, просушивают их и проявляют. Если требуется,
хроматографирование повторяют несколько раз и только потом
проявляют хроматограммы. Хроматограммы проявляют
анилинфталатом, а затем, пользуясь данными табл. 3 и 5, проводят
идентификацию веществ, выделенных из пробы.
Варьируя количество пробы, взятой на анализ,
многократностью хроматографирования и применяя свидетелей-спутников,
можно получать хроматограммы, на основании которых удается
установить наличие неизвестных веществ и веществ,
находящихся в пробах в исчезающе малых количествах.
Хроматографированне для количественных методов
Перед количественным хроматографическим анализом смесей
редуцирующих веществ проводят качественный анализ для
установления природы веществ, которые выделяют из смеси хрома-
тографированием на бумаге. Выделенными веществами могут
быть отдельные редуцирующие вещества или группы веществ,
обладающих близкими свойствами. Например, анализ гидроли-
зата делают для установления содержания в нем гексоз, пентоз,
6* 83
уроновых кислот, олигосахаридов или для определения
содержания в нем глюкозы, ксилозы, оксиметилфурфурола и т. д.
В зависимости от задачи, поставленной перед аналитиком,
выбирают нужные условия проведения хроматографирования и
методы количественного определения.
Хроматографирование гидролизатов и сульфитных щелоков
с целью отделения моносахаридов от других редуцирующих
веществ и одновременного разделения их между собой проводят
на длинных Cx45 см) полосках хроматографической бумаги,
применяя нисходящее хроматографирование при непрерывном
удалении растворителя, прошедшего через бумагу (см. рис.24).
Прикосновение нижнего конца хроматограммы к горлышку
колбочки, которая служит приемником, создает условия
непрерывного равномерного движения растворителя во время
хроматографирования, что необходимо для разделения веществ.
При хроматографировании гидролизатов применяют полоски
с продольным расположением волокон, а при анализе
сульфитных щелоков во избежание образования «хвостов» применяют
полоски с поперечным расположением волокон.
Для анализа одной пробы гидролизата или сульфитного
щелока берут 10—12 полосок бумаги. На одном конце каждой
полоски вырезают «язычок», ширина которого должна быть на 2—
4 мм больше ширины горлышка колбочки, служащей
приемником. На расстоянии 10 см от другого конца полоски на линии
старта, наносят исследуемый раствор, пользуясь специальной
пипеткой (см. рис. 27). При концентрации редуцирующих
веществ в анализируемой пробе около 3% иа
хроматографирование берут 0,01 мл, при более низкой концентрации количество
пробы на хроматографирование соответственно увеличивают
(см. с. 81). Полоски бумаги, после нанесения на них
анализируемого раствора, устанавливают в строго вертикальном
положении (см. рис. 24). Конец полоски, где нанесена проба,
загибают в блюдце так, чтобы пятио пробы отстояло от края
блюдца на расстоянии не менее чем 2 см. Второй конец полоски
вставляют в горлышко колбы-приемника так, чтобы края
«язычка», изгибаясь, плотно прилегали к стенкам горлышка.
В блюдце наливают растворитель—смесь этилацетат—пиридин—
вода 5:1:5 и все накрывают стеклянным колпаком.
Окружающая температура воздуха должна быть 18—20сС. Через 24 ч
хроматографирования снимают четыре полоски, высушивают их на
воздухе в вытяжном шкафу, одну из них проявляют, а три
полоски используют для определения ксилозы и рамнозы, которые
обычно за сутки хроматографирования хорошо отделяются от
других Сахаров. В блюдце вливают новую порцию растворителя,
закрывают колпаком все хроматограммы и продолжают хрома-
тографировать еще 25—30 ч. За 50—55 ч хроматографирования
разделяются арабиноза и манноза, за 80—90 ч — глюкоза и
галактоза. Если галактозы и олигосахарндов нет, то хроматогра-
84
фирование заканчивают через 2—2,5 суток. Хроматографирование-
считают законченным тогда, когда на проявленной хромато-
грамме расстояние между пятнами, которые соответствуют
разделенным сахарам, будет не менее 3 мм. В противном случае
хроматографирование продолжают с новой порцией
растворителя. Проявленную хроматограмму называют хроматограммой-
свидетелем. Непроявленные хроматограммы используют для
определения разделенных Сахаров.
Определение редуцирующих веществ эбулиостатическим
потенциометрическим методом
Метод предназначен для определения микроколичеств C00—
10 мкг) редуцирующих Сахаров и других редуцирующих веществ,
извлеченных из хроматограмм, а также для анализа водных
растворов редуцирующих Сахаров, имеющих концентрацию
около 0,01% (сточная вода и др.) [29].
Метод основан на реакции окисления редуцирующих веществ
меднощелочным раствором, которую проводят в эбулиостате
титрованием (см. с. 35). Особенность метода состоит в том, что
вместо цветного индикатора (метиленовая синяя) применяют
значительно более чувствительный способ обнаружения конца
титрования, основанный на измерении
окислительно-восстановительного потенциала платинового электрода, погруженного
в реагирующую жидкость. Платиновый электрод в этом случае
является индикаторным электродом. Для измерения его
потенциала составляют электрическую цепь из платинового электрода
и хлорсвинцовоамальгамного электрода. Электроды соединяют
электролитическим ключом и цепь замыкают на
милливольтметр. Хлорсвинцовоамальгамный электрод служит электродом
сравнения, он имеет определенный постоянный потенциал,
который равен потенциалу платинового электрода в момент конца
реакции. В начале титрования разность потенциалов электродов
максимальна. По мере добавления сахарного раствора разность
потенциалов уменьшается за счет увеличения потенциала
платинового электрода. В момент восстановления всей двухвалентной
меди до одновалентной происходит скачок
окислительно-восстановительного потенциала. Значение потенциала индикаторного
электрода становится равным потенциалу электрода сравнения
и продолжает увеличиваться. Стрелка милливольтметра в эта
время быстро проходит через нуль.
Мешающие вещества. При анализе растворов, не прошедших
хроматографического разделения, определению суммы
редуцирующих Сахаров мешает разнообразие их редуцирующих
способностей и присутствие посторонних редуцирующих веществ —
уроновых кислот, фурфурола и т. д. При анализе элюатов из
хроматограмм этих помех нет.
Аппаратура. Установка для извлечения Сахаров из непрояв-
ленных хроматограмм. Установка для потенциометрического
85
титрования, которая состоит из эбулиостата, вставленного в
коническую колбу, платинового электрода, электролитического
ключа, хлорсвинцовоамальгамного электрода и милливольтметра
на 150 мв (ток потребления 15 мка) со шкалой на 100 делений.
Дозаторы. Потенциометр. Насыщенный каломельный электрод.
Эбулиостат (рис. 28) представляет собой стеклянную
пробирку, внутрь которой впаяна трубка для отвода пара,
а вверху имеется отверстие / для выхода пара. Эбулиостат
вставляется в резиновую пробку снизу вверх. Чтобы во время анализа
он находился на определенном уровне погружения, на его стенке
имеется упор 2 утолще-
ние из стекла.
Дозаторы (см. рис.
12) для отмеривания
первого и второго растворов
имеют метки 2 мл.
Платиновый
электрод (рис. 29, а)
состоит из платиновой
пластинки /, впаянной в
стеклянную трубку. Контакт
платины с клеммой 4
осуществляется через ртуть 2 и
изолированную медную
проволоку 3,
погруженную в нее. Припой
медной проволоки к платине
недопустим.
Электролитический кл ю ч (рис. 29, б)
представляет собой
стеклянную трубку, имеющую расширение на одном конце. Узкий
конец закрыт пробкой, сделанной из рулончика полоски
фильтровальной бумаги. Трубка заполнена насыщенным раствором
хлористого калия.
Хлорсвинцовоамальгамный электрод.
Приготовление. В тигель помещают 10 г чистого
гранулированного свинца, 90 г чистой ртути и ставят его на электроплитку.
•Когда свинец растворится в ртути амальгаму охлаждают до
комнатной температуры, окисную пленку снимают стеклянной
палочкой, а блестящую амальгаму переливают в стаканчик и
заливают насыщенным раствором хлористого свинца, который
содержит кристаллы РЬСЬ. Затем измеряют потенциал
приготовленного электрода. В амальгаму электрода погружают
платиновую проволоку, второй конец которой присоединяют к
потенциометру. В раствор электрода погружают конец
электролитического ключа, которым соединяют его с насыщенным
каломельным электродом, присоединенным ко второму контакту по-
25
Рис. 28.
Эбулиостат
Рис.
трод
29. Платиновый элек-
а и электролитический
ключ б
86
тенциометра. Цепь замыкают и потенциометром измеряют
разность потенциалов. Если потенциал приготовленного электрода
по сравнению с насыщенным каломельным электродом меньше
420 мВ, то в раствор хлористого свинца добавляют 1—2 капли
1%-ного раствора хлористого калия и снова измеряют
потенциал. Прибавлением хлористого калия добиваются получения
потенциала электрода равного 420—440 мВ.
Приготовленный электрод переносят в специальный сосуд
(рис. 30) с платиновым контактом /. В расширенную часть
сосуда с клеммой 5 помещают амальгаму свинца 2 так, чтобы она
покрывала всю платину и не попала во внутреннюю трубку
сосуда. Внешний конец трубки закрывают пробкой 6, сделанной
из плотного рулончика полоски фильтровальной бумаги. Сосуд
и трубку заполняют раствором хлористого
свинца 4 с кристаллами 3, и закрывают пробкой.
В жидкости электрода не должно быть
пузырьков воздуха и уровень ее должен быть выше
верхнего конца трубочки.
Реактивы и их приготовление. 1. Первый
раствор в 1 л содержит 2 г сернокислой меди (х. ч.
CuSO4-5H2O). 2. Второй раствор в 1 л
содержит 50 г сегнетовой соли, 75 г едкого натра
и 0,8 г желтой кровяной соли. 3. Хлористый
калий, насыщенный раствор. 4. Глюкоза
ангидридная кристаллическая, приготовление см. на с. 38.
5. Глюкоза, 0,020—0,025%-ный раствор.
Приготовление. Раствор глюкозы, предназначенный
для титрования, готовят перед каждой серией
анализов путем разбавления 0,1—0,2%-ного раствора,
приготовленного из глюкозы марки «Медицинская». Точную
концентрацию приготовленного раствора определяют титрованием медно-
щелочного раствора, титр которого установлен по точной
навеске ангидридной абсолютно сухой глюкозы.
Установка титра медно ще л очного раствора,
Меднощелочным раствором называют смесь равных объемов
первого и второго растворов.
Титр меднощелочного раствора — это количество глюкозы
или другого редуцирующего вещества, которое в условиях
анализа необходимо для восстановления меди, содержащейся в 4 мл
меднощелочного раствора, выраженное в микрограммах (мкг).
При анализе смеси Сахаров принято титр устанавливать по
глюкозе и вычисление концентрации Сахаров делать в пересчете на
глюкозу. При анализе отдельных Сахаров, уроновых кислот и
других веществ после их выделения хроматографированием титр
меднощелочного раствора устанавливают по каждому
определяемому веществу. Так как у аналитика не всегда имеются
чистые вещества, которые он определяет, то в этом случае вместо
установки титра по разным веществам можно установить титр
87
Рис. 30 Хлор-
свинцово-
амальгамный
электрод
ло глюкозе и, пользуясь эмпирическим коэффициентом,
пересчитать его в титр по интересующему веществу. Значение
эмпирического коэффициента для некоторых веществ приведено при
расчете результата анализа.
Для установки титра берут навеску около 0,1 г ангидридной
глюкозы, растворяют ее в дистиллированной воде и объем
доводят до 500 мл. После тщательного перемешивания раствора
им споласкивают микробюретку и заполняют ее. В пустой эбу-
лиостат из бюретки вливают 2 мл раствора глюкозы, добавляют
по 2 мл первого и второго растворов, эбу-
лиостат вставляют в колбу с кипящей
водой и ведут титрование как описано ниже
(«Ход анализа»). Проводят не менее трех
титрований. Для вычисления титра берут
среднее значение объема раствора глюкозы,
пошедшего на титрование, и вычисляют по
формуле
.а
.5-
Т =
где Т — титр меднощелочиого раствора, мкг: а —
навеска глюкозы, г; Vo — объем приготовленного
раствора глюкозы, мл; V — объем раствора глюкозы,
пошедший иа титрование, мл.
Рис. 31. Определение расположения Сахаров на не-
проявленной хроматограмме с помощью основного
и дополнительных свидетелей:
/ — проявленная хроматограмма — основной свидетель; // —
частично проявленная хроматограмма; 1—7 — проявленные
кусочки хроматограммы — дополнительные свидетели; а, б,
а, г — зоны расположения Сахаров
Подготовка проб. Непроявленные хроматограммы,
предназначенные для определения отдельных Сахаров, хорошо
просушивают до полного удаления запаха растворителя, а затем,
сравнивая с хроматограммой-свидетелем, разрезают на кусочки,
каждый из которых должен содержать одно пятно. Вследствие
неравномерной плотности хроматографической бумаги
расположение Сахаров на разных хроматограммах одной и той же серии
не совпадает. Для уточнения расположения пятен на непро-
явленных хроматограммах пользуются дополнительными
свидетелями, которыми являются кусочки хроматограммы,
прилегающие к вырезанному пятну. На рис.31 показаны две параллельные
хроматограммы. Хроматограмма / полностью проявлена, она
является главным свидетелем. На ней имеется четыре пятна,
соответствующие расположению разделенных Сахаров — а, б,
в, г. Хроматограмма // проявлена частично. Заштрихованная
часть хроматограммы состоит из проявленных кусочков, которые
88
являются дополнительными свидетелями. Номера кусочков
указывают очередность их отрезания и проявления. На двух не-
проявленных кусочках этой хроматограммы расположены
сахар б и сахар г.
Применение дополнительных свидетелей при вырезании не-
проявленных пятен на хроматограммах дает полную гарантию
точного определения расположения разделенных веществ. После
разрезания нескольких хроматограмм непроявленные кусочки,
содержащие одно и то же вещество, помещают в отдельный
пакетик из кальки и хранят до анализа.
Ход анализа. Сахара из хроматограммы извлекают водой на
установке (рис. 32). Для элюации полоску / фильтровальной
бумаги размером ЗХЮ см всю смачивают, опуская на мгновение
в дистиллированную воду, и вешают на край чашки Петрн 6
так, чтобы один конец ее оставался в воде.
К другому концу полоски прикладывают
кромку кусочка хроматограммы 2, из
которого хотят извлечь сахар. Кусочек
хроматограммы приклеивается водой к бумаге,
вода начинает двигаться по нему, увлекая
сахар. Когда вода подойдет к нижней части
кусочка хроматограммы, берут покровное
стекло, на половине его поверхности
наносят воду в виде мелких капель и
приклеивают его с одной стороны хроматограммы.
Таким же приемом приклеивают второе
покровное стекло 3 к другой стороне ров из непроявленных
хроматограммы. Стекла плотно прижимают хроматограмм
друг к другу пальцами, чтобы между
ними было минимальное расстояние. Свободный конец стекол
вставляют в эбулиостат 4, находящийся в подставке 5. Всю
установку закрывают стеклянным колпаком, чтобы предотвратить
испарение воды из висящей бумаги. Элюат собирают в
эбулиостат, в котором определяют количество сахара, как описано
ниже. Первые капли элюата содержат наибольшее количество
сахара. Если количество извлекаемого сахара невелико, то в
эбулиостат достаточно собрать 5—8 капель жидкости. Если
количество Сахаров большое, судя по пятну на проявленной хромато-
грамме, то соответственно увеличивают время извлечения и
количество собираемой жидкости. После элюации кусочек
хроматограммы высушивают и проявляют для проверки полноты
извлечения сахара. Если сахар был извлечен не полностью, то на
кусочке хроматограммы будет пятно. В этом случае элюат
выливают прочь и извлекают сахар из другого кусочка, содержащего
тот же сахар.
Количество сахара, извлеченного из хроматограммы,
определяют на установке, показанной на рис. 33. К элюату в эбу-
лиостате /, который вставлен в пробку, приливают отмеренные
89
Рнс. 32. Установка
для извлечения саха-
дозаторами 2 мл первого раствора, 2 мл второго раствора и
перемешивают их легким круговым движением. Эбулиостат
вставляют на пробке в колбу с кипящей водой и закрывают его
пробкой, в которую заранее
вставлены электролитический ключ 3
и платиновый электрод 2, соединенный
с милливольтметром 5. В верхний
конец электролитического ключа
опускают конец электрода сравнения 4,
который соединен с милливольтметром.
В это время цепь замыкается, стрелка
милливольтметра отклоняется вправо.
Величина отклонения стрелки зависит
от количества сахара, которое было
извлечено из хроматограммы. Чем
больше отклонение стрелки
милливольтметра, тем больше сахара в эбу-
лиостате. В свободное отверстие в
пробке, закрывающей эбулиостат,
вставляют кончик микробюретки с 0,02%-
ным раствором глюкозы. Точную
концентрацию раствора глюкозы
определяют отдельным титрованием перед
анализом. Если стрелка
милливольтметра остановится на делении 30—35,
то ждут 2 мин по песочным часам.
Если стрелка остановится на большем
делении, то из бюретки по каплям
приливают в эбулиостат раствор
глюкозы до тех пор, пока стрелка дойдет
до деления 30 и ждут 2 мин, а затем
прибавляют раствор глюкозы из
бюретки со скоростью 0,005 мл/с.
Титрование заканчивают, когда
стрелка милливольтметра достигнет
нуля. По микробюретке определяют
объем раствора глюкозы, который
пошел на дотитровывание. После
каждого титрования платиновый электрод
и конец электролитического ключа
споласкивают сразу горячей водой из
промывалки. Затем электрод охлаждают на воздухе, погружают
его в 0,2 н. раствор соляной кислоты, вынимают и тщательно
моют холодной водой.
Расчет. 1. Концентрацию глюкозы в растворе, который применяют для
дотитровывания, вычисляют по формуле
Рис. 33. Установка для
определения редуцирующих
веществ эбулиостатнческим
потенциометр ическим методом
с =
т
V
'90
где С — концентрация глюкозы в растворе, мкг/мл; Т — титр меднощелочного
раствора, мкг глюкозы; V — объем раствора глюкозы, израсходованный на
анализ, мл.
2. Концентрацию сахара в анализируемом гидролизате или в сульфитном
щелоке вычисляют по формуле
у {Т-Са)К
Х= ?ЇОЇ
где X — концентрация сахара в пробе, %; Т — титр меднощелочного раствора
по глюкозе, мкг; С — концентрация глюкозы в растворе, который
употребляли для дотитровывания, мкг/мл; а —объем раствора глюкозы,
израсходованный на дотитровывание, мл; g —- объем анализируемого раствора, взятый
иа хроматографирование, мл; 104 — перевод микрограммов в проценты; К —
эмпирический коэффициент, показывающий, какое количество микрограммов
определяемого вещества эквивалентно 1 мкг глюкозы при восстановлении
меднощелочиого раствора в условиях анализа. Значение К для различных,
веществ:
Глюкоза 1,00 Галактуроновая кислота . 1,30
Галактоза 1,07 Оксиметилфурфурол . . . 0,70
Ксилоза 1,00 Фурфурол 0,45
Арабиноза 0,95
Минимальное количество сахара, которое можно определить, 10 мкг.
Точность определения ±2%, если на анализ берут не менее 100 мкг.
Предостережение. 1. Периодически надо проверять потенциал хлорсвин-
цовоамальгамного электрода. Иногда он увеличивается за счет диффузии
хлористого калия в раствор электрода из электролитического ключа. При
повышении потенциала электрода следует заменить (частично или полностью)
раствор над амальгамой свежим, при котором значение потенциала электрода
будет 420—440 мВ по сравнению с потенциалом насыщенного каломельного
электрода. 2. Надо следить за чистотой ртути в трубке платинового
электрода. Помутнение ртути и появление желто-зеленого налета или жидкости
над ртутью свидетельствует о наличии трещин в стекле в месте соединения
его с платиной. Неисправность платинового электрода недопустима при
анализе, его надо заменить. 3. Раствор глюкозы, из которого готовят раствор
для дотитровываиия, надо хранить в холодильнике. Срок хранения 3 суток.
Определение олигосахаридов
Для определения количества олигосахаридов, выделенных
из гидролизатов и сульфитных щелоков с помощью
хроматографии на бумаге предназначен метод, основанный на реакции
гидролиза олигосахаридов и определении количества
образовавшихся редуцирующих веществ эбулиостатическим потенцио-
метрическим методом.
Мешающие вещества. Редуцирующие вещества, имеющие Rf
близкое к Rf олигосахаридов.
Аппаратура. Установка для нисходящей хроматографии на
полосках бумаги (см. рис. 24). Все для эбулиостатического по-
тенциометрического метода определения редуцирующих веществ
(см. с. 85).
Реактивы и их приготовление. 1. Растворитель — смесь я-бу-
танола, пиридина и воды в соотношении 6:4:3 или смесь
91
ацетона, м-бутанола и воды 7:2:1. 2. Анилинфталат, раствор.
Приготовление см. на с. 79. 3. Серная кислота, 6 н. раствор.
4. Двууглекислый натрий, кристалл. 5. Все реактивы для эбу-
лиостатического потенциометрического метода (см. с. 85).
Ход анализа. Хроматографирование. Для выделения
олигосахаридов из смеси редуцирующих веществ гидролизатов
проводят хроматографирование на пяти-шести полосках
хроматограф ической бумаги CX30 см) с машинным расположением
волокон так же, как при разделении Сахаров. Растворителем
служит смесь из н-бутанола, пиридина и воды или смесь
ацетона, н-бутанола и воды. Количество растворителя, взятое для
хроматографирования, должно быть таким, чтобы его хватило
на хроматографирование в течение 50 ч при 20°С. После хро-
матографирования одну полоску проявляют анилинфталатом,
как описано на с. 79. Об относительной длине цепочки
олигосахарида судят по его Ягл- Для целлобиозы Ягл =
= 0,60-=-0,63. Чем меньше Rru, тем длиннее цепочка молекулы
.олигосахарида, тем больше остатков моноз содержится в его
молекуле.
Извлечение олигосахаридов из хромаю-
грамм. Пользуясь проявленной хроматограммой как
основным свидетелем, вырезают кусочки непроявленных хромато-
грамм, на которых находится один или все олигосахариды. Для
более точного определения границ расположения
олигосахарида пользуются дополнительными свидетелями (см. с. 88).
Из вырезанных кусочков хроматограмм олигосахариды
извлекают водой, как описано на с. 89. Раствор олигосахаридов
(элюат) собирают в мерный цилиндр вместимостью 3 мл. Для
полного извлечения олигосахарида достаточно собрать 1,5—
2 мл элюата. Кусочки хроматограмм после извлечения
олигосахаридов высушивают и проявляют, чтобы убедиться в
полноте элюирования.
Гидролиз олигосахаридов. Перед количественным
определением собранные 2 мл элюата олигосахарида из
цилиндра выливают в эбулиостат. Тем же цилиндром отмеривают
2 мл. 6 н. H2SO4 и тоже выливают в эбулиостат, который
закрывают пробкой и ставят в кипящую водяную баню. При
анализе олигосахаридов, имеющих RrJ1 больше 0,1, гидролиз
проводят в течение 60 мин. При анализе олигосахаридов, имеющих ./?гл
меньше 0,1, время гидролиза увеличивают до 120 мин. После
гидролиза эбулиостат вынимают из бани и осторожно всыпают
в него рассчитанное количество соды для полной
нейтрализации серной кислоты.
Анализ гидролизата. Количество моносахаридов,
образовавшихся из олигосахаридов во время гидролиза,
определяют эбулиостатическим потенциометрическим методом. В
эбулиостат с нейтрализованным раствором моносахаридов вливают
по 2 мл первого и второго растворов, перемешивают, вставляют
92
его в колбу с кипящей водой и проводят титрование раствором
глюкозы известной концентрации, как описано на с. 90.
Расчет, Концентрацию олигосахарида в анализируемом растворе
выражают в процентах моносахарида, который образуется при его гидролизе и
вычисляют по формуле
v (T-Ca)K
х
Обозначения см. с. 91.
Определение уроновых кислот
Для определения уроновых кислот в гидролизатах
хлопковой шелухи, подсолнечной лузги и других растительных тканей
предназначен метод, основанный на определении уроновых
кислот по их редуцирующей способности после их выделения с
помощью хроматографии на бумаге эбулиостатическнм потенцио-
метрическим методом. Количество уроновых кислот вычисляют
в пересчете на галактуроновую кислоту, редуцирующая
способность которой известна.
Мешающие вещества. Вещества, имеющие Rj близкое к Rf
уроновых кислот. Редуцирующая способность уроновых кислот
различна, что влияет на точность результата, если его
вычисляют в пересчете на галактуроновую кислоту.
Аппаратура. Установка для нисходящего хроматографирова-
ния (см. рис. 24). Все для эбулиостатического потенциометри-
ческого метода.
Реактивы и их приготовление. 1. Растворитель — смесь н-бу-
танола, пиридина и воды в соотношении 3:2:1.
Приготовление. В делительную воронку вливают три объема н-бута-
нола, два объема пиридина и один объем дистиллированной
воды. Смесь энергично взбалтывают в течение 1—2 мин. При
стоянии смесь не должна расслаиваться. 2. Все реактивы для
определения редуцирующих веществ эбулиостатическим потен-
циометрическим методом.
Ход анализа. Хроматографирование исследуемой пробы
проводят так же, как при количественном определении Сахаров
(см. с. 83), применяя растворитель — смесь бутанола, пиридина
и воды. Через 120—130 ч хроматографировання снимают одну
полоску, высушивают и проявляют анилинфталатом. На
основании проявленной хроматограммы определяют, достигнуто ли
нужное разделение уроновых кислот и отделение их от
углеводов. Если результат хроматографирования удовлетворяет
исследователя, то все хроматограммы снимают, сушат на воздухе
до полного удаления запаха растворителя и, пользуясь
дополнительными свидетелями, из хроматограмм вырезают кусочки,
содержащие одну или несколько уроновых кислот (см. рис. 31).
Извлекают уроновые кислоты из хроматограмм водой и
определяют их количество так, как описано на с. 89 для Сахаров.
93
Расчет. Содержание уроновых кислот в исследуемой пробе выражают
в процентах галактуроновой кислоты, для которой /(=1,30 (см. с. 91) н
вычисляют по формуле
у (Т-Са)К
Обозначения см. на с. 91.
При определении отдельной уроновой кислоты ее концентрацию находят
в пересчете на галактуроновую кислоту илн сначала определяют К нужной
кислоты, которое используют для вычисления ее концентрации.
Определение оксиметилфурфурола
Для определения оксиметилфурфурола и других
редуцирующих продуктов распада моносахаридов во время гидролиза,
которые имеют Rf отличное от Rf Сахаров предназначен метод,
основанный на выделении оксиметилфурфурола из гидролиза-
тов хроматографией на бумаге с последующим определением
его количества эбулиостатическим потенциометрическим
методом.
Мешающие вещества. Редуцирующие вещества, имеющие Rf
близкое к Rf оксиметилфурфурола.
Аппаратура. Камера для нисходящего хроматографирова-
ния. Все для эбулиостатического потенциометрического
определения редуцирующих веществ.
Реактивы. 1. Растворитель — смесь этилацетата, пиридина
и воды в соотношении 5:1:5 (см. с. 78). 2. Все реактивы для
эбулиостатического потенциометрического метода.
Ход анализа. Выделение оксиметилфурфурола и
редуцирующих веществ, имеющих /?/=1, проводят на пяти-шести полосках
Cx20 см) с поперечным расположением волокон под
колпаком, применяя нисходящее однократное хроматографирование.
В качестве растворителя применяют верхний слой смеси этил-
ацетат—пиридин—вода E:1:5) или другие растворители
(см. с. 77). Как только растворитель дойдет почти до конца
полосы, хроматографирование считают законченным. Хромато-
граммы снимают, сушат и одну из них проявляют анилинфта-
латом.
Элюирование и количественное определение выделенных
веществ проводят так, как описано на с. 89.
Расчет. Концентрацию оксиметнлфурфурола вычисляют в процентах по»
формуле
где
94
К=0,70.
Другие
у
обозначения
(Т-Са)К
?І0і
см. на с. 91.
Определение редуцирующих веществ колориметрическим методом
Метод предназначен для определения отдельных
редуцирующих веществ (сахаров, уроновых кислот и др.) после их
выделения из смеси хроматографированием на бумаге.
Метод основан на реакции редуцирующих веществ с анилин-
фталатом. После проявления хроматограмм окрашенные
продукты реакции элюируют ледяной уксусной кислотой и
растворы колориметрируют на колориметре ФЭК. Количество
определяемого вещества находят по калибровочной кривой,
составленной для этого вещества.
Аппаратура. Колориметр ФЭК или другой.
Реактивы и их приготовление. 1. Анилинфталат.
Приготовление. 1,66 г фталевой кислоты и 1 мл свежеперегнан-
ного анилина растворяют в бутаноле или этаноле, раствор
переносят в мерную колбу вместимостью 100 мл и объем доводят
растворителем до метки. Приготовленный раствор хорошо
перемешивают и хранят в склянке из темного стекла с притертой
пробкой. 2. Уксусная кислота ледяная или спиртовый раствор
¦соляной кислоты. Приготовление. Смешивают 29 мл
36%-ной соляной кислоты, 420 мл 95%-ного этилового спирта
и 51 мл дистиллированной воды. 3. Глюкоза безводная (см.
с. 36).
Построение калибровочной кривой. Готовят точно 1%-ный
раствор глюкозы (или другого вещества, которое хотят
определять). Он содержит 104 мкг/мл или 100 мкг глюкозы в 0,01 мл.
Из этого раствора разбавлением дистиллированной водой
готовят растворы, которые в 0,01 мл содержат 90, 80, 70, 60, 50,
40, 30, 20 и 10 мкг глюкозы. На ленту хроматографической
бумаги в разные места наносят по 0,01 мл каждого
приготовленного раствора. На другую ленту наносят разные количества
1%-ного раствора — 0,01; 0,02 мл и т. д., которые
соответственно содержат 100, 200 мкг и т. д. глюкозы. После высыхания
пятен растворов на воздухе ленты на мгновение погружают
в раствор анилинфталата, осушают между листов
фильтровальной бумаги и кладут на ребро в сушильный шкаф. При
температуре 100°С ленты выдерживают 10 мин. (Для других веществ
время и температура проявления указаны в табл. 5.) Из лент
вырезают окрашенные пятна, элюируют и определяют
оптическую плотность элюата, как описано ниже. На основании
полученных данных строят кривую зависимости оптической
плотности (интенсивности окраски) элюата от количества глюкозы,
взятой на реакцию с анилинфталатом, т. е. калибровочную
кривую.
Подготовка проб. После хроматографирования проявляют
одну хроматограмму, чтобы убедиться, что разделение
закончено.
95
Ход анализа. Все высушенные хроматограммы проявляют
анилинфталатом. Условия проявления хроматограмм зависят от
природы определяемого вещества. При определении пентоз и
уроновых кислот хроматограмму проявляют при температуре
70°С, при определении гексоз — при 100°С в течение 10 мин
(см. табл. 5). Из приведенных хроматограмм вырезают кусочки
с пятном интересующего вещества, каждый кусочек разрезают
на мелкие тонкие полосочки, которые кладут в пробирку и
заливают 8 мл ледяной уксусной кислоты или 8 мл спиртового
раствора соляной кислоты. Элюирование ведут 1 ч при
комнатной температуре. Через каждые 5—10 мин пробирки с
кусочками хроматограмм энергично взбалтывают для ускорения
элюации. Через 1 ч элюат осторожно сливают в подготовленную
сухую пробирку и измеряют его оптическую плотность на
фотоколориметре. Если пользуются фотоэлектроколориметром
ФЭК-М, то интенсивность окраски измеряют в кювете шириной
10 мм. Светофильтр подбирают в зависимости от цвета
анализируемого раствора, определяемого по табл. 17 (с. 294).
Расчет. Количество глюкозы в элюате, выраженное в микрограммах
(мкг), находят по калибровочной кривой для глюкозы. Концентрацию
глюкозы в анализируемой пробе, которую хроматографировали, вычисляют по
формуле
Y_ alOO
где X — концентрация глюкозы в пробе, %; а — количество глюкозы,
найденное в элюате, мкг; g — объем пробы, взятый для хроматографирования, мл.
Минимальное определяемое количество вещества зависит от его природы.
Для Сахаров оно составляет 10 мкг. Редуцирующие вещества определяют
с точностью ±10—15% оти.
Предостережение. 1. Потери определяемого вещества обусловлены
способом элюирования, который не гарантирует полноту извлечения окрашенного
соединения из бумаги, особенно при большом количестве извлекаемого
вещества. 2. Интенсивность окраски пятен на хроматограмме в значительной
степени зависит от времени и температуры проявления хроматограмм. 3. Нельзя
пользоваться калибровочной кривой, сделанной не для определяемого
вещества.
ГЛАВА 3. ВСПОМОГАТЕЛЬНЫЕ МАТЕРИАЛЫ
ВОДА
Определение жесткости воды комплексометрическим методом
Для определения общей, устранимой и постоянной
жесткости воды предназначен метод, основанный на определении
содержания ионов кальция и магния в пробе до и после
кипячения путем титрования раствором трилона Б с индикатором
хромоген черный ЕТ-00 или кислотный хром темно-синий.
Жесткостью воды называют некоторые ее свойства, которые
обусловлены наличием в ней солей кальция, магния, бария, же-
96
леза и других металлов. Степень жесткости воды
характеризуют содержанием в ней только ионов кальция и магния,
потому что ионов других металлов обычно очень мало.
Количество кальция и магния, найденное в пробе до
кипячения, называют общей жесткостью воды. Оно
соответствует количеству кальция и магния, которые связаны в кислые
и средние соли разных кислот. При кипячении воды
бикарбонаты кальция и магния переходят в плохо растворимые
карбонаты, которые выпадают в осадок. Понижение концентрации
кальция и магния в растворе за счет образования осадка
называют устранимой жесткостью, а количество кальция
и магния, остающееся в растворе после кипячения,—
постоянной жесткостью.
Трилон Б — динатриевая соль этилендиаминтетрауксусной
кислоты (см. с. 68) с катионами многих двух- и трехвалентных
металлов, как, например, кальция, магния, цинка, марганца,
железа (Fe2+ и Fe3+) и др. образует внутрикомплексные
соединения, прочность которых зависит от pH среды. Каждый катион
определяют при том значении pH, при котором этот катион
образует с трилоном Б соединение более прочное, чем с
индикатором. В этом случае при титровании пробы раствором три-
лона Б в точке эквивалентности изменяется окраска
индикатора.
Для определения кальция и магния при pH 9—12
применяют один из следующих индикаторов: хромоген черный ЕТ-00,
кислотный хром темно-синий и кислый хром черный
специальный (эриохром черный Т). Железо определяют при pH 1—2
с роданистым аммонием или сульфосалициловой кислотой.
С индикатором пурпуреатом аммония (мурексид) при pH в
пределах от 3,5 до 12 определяют медь, а при pH в пределах 12—
13 — кальций (см. с. 100).
Мешающие вещества. Щелочность выше 7 мг-экв/л. Медь,
цинк, марганец, масло.
Аппаратура. Установка для титрования. Конические колбы
вместимостью 250 мл.
Реактивы и их приготовление. 1. Дистиллированная вода
Н-катионированная или дважды перегнанная в стеклянном
приборе. Вода не должна содержать даже следов меди. Для
проверки воды на содержание меди к 100 мл воды прибавляют
1 мл аммиачного буферного раствора (см. ниже) и 5—7
капель индикатора кислотного хрома темно-синего. При полном
отсутствии меди раствор будет голубой с сиреневым оттенком.
Все реактивы готовят на этой воде. 2. Аммиачный буферный
раствор. Приготовление. В мерную колбу вместимостью
1 л вливают 100 мл 20%-ного хлористого аммония (х. г.),
100 мл 25%-ного аммиака и объем раствора доводят до 1 л,
прибавляя дистиллированную воду. 3. Индикатор один из трех:
7 Заказ № 142 97
хромоген черный ЕТ-00 кислотный хром темно-синий или
кислый хром черный специальный (эриохром черный Т).
Приготовление раствора. 0,5 г индикатора растворяют в 20 мл
аммиачного буферного раствора, прибавляют 80 мл этилового
спирта и раствор перемешивают. Раствор индикатора хромоген
черный ЕТ-00 надо готовить каждые 10 дней. Приготовление
индикаторной смеси. 0,2 г индикатора и 50 г хлористого натрия
(х. ч.) хорошо растирают в фарфоровой ступке. 4. Трилон Б
0,1 н.; 0,05 н.; 0,01 н. растворы. Приготовление. 0,1 н.
раствор получают, растворяя 18,6 г трилона Б в 1 л
дистиллированной воды (без меди). Мутный раствор фильтруют. 0,05 н.
раствор содержит 9,3 г/л, а 0,01 н.— 1,86 г/л трилона Б.
Установка титра раствора трилона Б по сернокислому
магнию. В коническую колбу вместимостью 250 мл вливают 100 мл
0,01 н. MgSO4, 5 мл аммиачного буферного раствора и 3—5
капель одного из индикаторов. Содержимое колбы перемешивают
и титруют приготовленным 0,1 н. раствором трилона Б до
отчетливого изменения цвета жидкости. Вычисляют количество
магния эквивалентное 1 мл приготовленного раствора. При
установке титра 0,01 н. раствора трилона Б на титрование берут
10 мл 0,01 н. раствора MgSO4, 90 мл воды, 5 мл аммиачного
буферного раствора и 3—5 капель раствора индикатора —
обязательно хрома темно-синего, потому что этот индикатор
обладает большей чувствительностью. 5. Сульфид натрия, 5%-ный
раствор. Хранят в опарафинированной склянке не более 14 дней.
6. Сернокислый магний, 0,01 н. раствор, приготовленный из фик-
санала или по навеске (см. с. 68). 7. Солянокислый гидроксила-
мин (NH2OH'HC1) 1%-ный раствор.
Подготовка проб. 1. Определяют pH и температуру воды.
Кислую воду нейтрализуют щелочью по метиловому
оранжевому, холодную воду подогревают до 20°С. 2. При содержании
железа и алюминия более 10 мг/л пробу перед анализом
разбавляют дистиллированной водой. 3. Воду, загрязненную
маслом, титруют при нагревании с индикатором хромом
темно-синим или эриохромом черным Т. Хромоген черный ЕТ-00 в
присутствии масла обесцвечивается. 4. Устанавливают наличие меди
в пробе по отсутствию отчетливой точки эквивалентности при
титровании пробы трилоном Б. 5. Наличие марганца в пробе
устанавливают следующим образом. К отмеренному количеству
пробы прибавляют все реактивы и, не титруя, дают постоять
1—2 мин. В присутствии марганца раствор становится серым,
титровать его нельзя.
Ход анализа. Определение общей жесткости.
В зависимости от жесткости воды для титрования применяют
растворы трилона Б различной концентрации. При жесткости
воды выше 20 мг-экв/л титруют 0,1 н. раствором трилона Б,
при жесткости от 0,5 до 20 мг-экв/л — 0,05 н. раствором и при
жесткости ниже 0,5 мг-экв/л — 0,01 н. раствором.
9S
В отсутствие ионов меди, цинка и марганца.
В коническую колбу вместимостью 250 мл вливают точно
отмеренное количество пробы, которое устанавливают
предварительным титрованием, добавляют дистиллированную воду в
таком количестве, чтобы объем жидкости в колбе стал 100 мл,
5 мл аммиачного буферного раствора и 5—6 капель
индикатора или 0,1—0,2 г индикаторной смеси. Содержимое колбы
перемешивают и медленно, по каплям, прибавляют из бюретки
раствор трилона Б. При изменении окраски раствора от одной
капли раствора трилона Б титрование заканчивают и по
бюретке определяют объем раствора, пошедший на титрование.
В присутствии ионов меди и цинка. В колбу
с пробой, разбавленной дистиллированной водой, как описано
выше, прибавляют 1 мл 5%-ного раствора сульфида натрия,
а затем аммиачный буферный раствор, индикатор и титруют
раствором трилона Б (см. выше). В присутствии цинка
титрование проходит без видимых отклонений, но результат анализа
завышен.
В присутствии марганца. В колбу с пробой,
разбавленной водой, прибавляют 3 капли раствора солянокислого гид-
роксиламина, а затем прибавляют аммиачный буферный
раствор и титруют раствором трилона Б как описано выше.
Определение постоянной жесткости. В колбу
вместимостью 500 мл наливают 250 мл пробы, закрывают ее
пробкой, в которую вставлен обратный холодильник, кипятят
в течение 1 ч, горячую воду отфильтровывают от осадка,
охлаждают до комнатной температуры, переливают в мерную колбу
вместимостью 250 мл и объем доводят до метки, прибавляя
дистиллированную воду. Пробу, освобожденную от бикарбонатов,
анализируют так же, как при определении общей жесткости.
Расчет. Жесткость воды в разных странах выражают в градусах
жесткости, значение которых различно. В СССР с 1951 г. жесткость воды
выражают в миллиграмм-эквивалентах на 1 л (мг-экв/л). 1 мг-экв/л равен
20,04 мг/л нонов кальция или 12,16 мг/л ионов магния. 1 мг-экв/л соответствует
2,804 немецкого градуса, 5,005 французского градуса, 3,511 английского
градуса, 50,045 американского градуса [30].
Общую и постоянную жесткость воды вычисляют по формуле
ан.ЮОО
g
где X — жесткость воды, мг-экв/л; а — объем раствора трилона Б,
израсходованный на титрование, мл; н.— нормальность раствора трилона Б; g —
объем пробы, взятый на анализ, мл.
Устранимую жесткость находят по разности между общей и постоянной
жесткостью.
Воспроизводимость в среднем из трех определений ±0,04 мл раствора
трилона Б.
Предостережение. При анализе умягченной воды, содержащей
незначительное количество кальция и магния, титрование проводят 0,01 н. раствором
трилона Б с индикатором хромом темно-синим, который обладает большей
чувствительностью, чем другие индикаторы.
7* 99
Определение кальция в присутствии магния
Для определения жесткости воды по кальцию предназначен
метод, основанный на реакции кальция с трилоном Б при
pH 12—13 в присутствии мурексида в качестве индикатора.
В этих условиях магний выпадает в осадок в виде гидроокиси
магния, а мурексид реагирует только на ионы кальция.
Мешающие вещества. Медь при концентрации выше 2 мг/л.
Щелочность выше 300 мг/л затрудняет наблюдение точки
эквивалентности.
Аппаратура. Установка для титрования. Конические колбы
вместимостью 250 мл.
Реактивы и их приготовление. 1. Едкий натр, 1 н. раствор.
2. Индикатор мурексид (аммонийная соль одноосновной
пурпуровой кислоты). Приготовление раствора. 0,15 г
мурексида растворяют в 100 мл абсолютного этиленгликоля.
Водный раствор мурексида неустойчив. Приготовление
индикаторной смеси. 0,20 г мурексида растирают
в ступке и смешивают с 100 г хлористого натрия. Смесь
устойчива. В точке эквивалентности цвет индикатора меняется от
розового до лилового. 3. Трилон Б, 0,1 н.; 0,01 н. растворы. 1 л 0,1 н.
раствора содержит 18,6 г трилона Б.
Подготовка проб. В пробе определяют щелочность, pH и
температуру. Кислую воду нейтрализуют по метиловому
оранжевому, холодную воду подогревают до комнатной
температуры. При щелочности воды выше 300 мг/л пробу разбавляют.
Ход анализа. В коническую колбу вместимостью 250 мл
вливают 10—100 мл пробы, в которых содержится 5—10 мг
кальция, добавляют дистиллированную воду без меди (см. с. 97)
в таком количестве, чтобы объем раствора стал 100 мл, и 5-6 мл
раствора едкого натра, которые обеспечили бы получение в
реакционной смеси pH 12—13. Раствор в колбе хорошо
перемешивают, вносят в него 0,1—0,2 г индикаторной смеси (или 1 —
2 капли раствора мурексида) и сразу начинают титровать,
прибавляя медленно при постоянном перемешивании раствор
трилона Б до изменения окраски реагирующей жидкости. Чтобы
удостовериться, что раствор больше не меняет окраску,
прибавляют 1—2 капли избытка раствора трилона Б и оставляют его
в качестве свидетеля. Проводят повторный анализ, в котором
конец титрования определяют более точно, сравнивая окраску
в точке эквивалентности с окраской свидетеля.
Расчет. Содержание кальция в пробе вычисляют по формуле
v ан.1000
g
где X — концентрация кальция в пробе, ыг-экв/л; а — объем раствора
трилона Б, израсходованный на анализ, мл; н. — нормальность раствора
трилона Б; g — объем пробы, взятый на анализ, мл.
100
Содержание магния вычисляют по разности между общей жесткостью
и содержанием кальция.
Воспроизводимость в среднем из трех определений ±0,06 мл раствора
трилона Б.
Предостережение. Индикатор мурексид неустойчив в щелочной среде,
поэтому титрование трилоном Б надо начинать сразу после прибавления
индикатора.
Определение хлора
Для определения ионов хлора в водопроводной воде и
в воде водоемов, которую используют для гидролиза и для
разбавления субстрата перед выращиванием дрожжей, при
содержании хлора не более 0,1 мг-экв/л предназначен метод,
основанный на реакции ионов хлора с азотнокислой ртутью [31].
2Cl' + Hg(NO3J — HgCl2.
Для определения количества хлора проводят титрование
анализируемой воды, подкисленной до pH 3—3,5, раствором
азотнокислой ртути в присутствии смешанного индикатора,
состоящего из дифенилкарбазона и бромфенола синего.
Мешающие вещества. Взвешенные и окрашенные вещества.
Аппаратура. Установка для титрования. Коническая колба
вместимостью 100 мл.
Реактивы и их приготовление. 1. Азотнокислая ртуть
Hg(N03J х.ч., 0,1 н. раствор. Приготовление. В
дистиллированной воде растворяют 16,68 г Hg(NC>3J* V2H2O,
добавляют 2 мл азотной кислоты плотностью 1,54 г/см3 и объем
раствора доводят водой до 1 л. Титр раствора азотнокислой ртути
устанавливают по навеске абсолютно сухого хлористого натрия.
2. Смешанный индикатор. Приготовление. 0,5 г
дифенилкарбазона и 0,05 г бромфенола синего растворяют в 100 мл
96%-ного этилового спирта. Раствор хранят в темной склянке,
он пригоден в течение 1 месяца. 3. Азотная кислота, 0,05 н.
раствор. 4. Едкий натр, 0,05 н. раствор.
Подготовка пробы. Мутную пробу фильтруют через
бумажный фильтр.
Ход анализа. В коническую колбу вместимостью 100 мл
вливают 50 мл анализируемой воды и прибавляют 10—15
капель смешанного индикатора. Если вода приобретает синюю
окраску (рН>4,4), то к ней добавляют по каплям 0,05 н.
раствор азотной кислоты до появления желтой окраски. Если вода
приобретает желтую окраску после добавления индикатора, то
к ней добавляют сначала 0,05 н. раствор едкого натра до
получения синей окраски, а затем по каплям прибавляют 0,05 н.
раствор азотной кислоты до получения желтой окраски pH 3,3.
После нейтрализации к воде желтого цвета прибавляют 0,5 мл
0,05 н. раствора азотной кислоты и титруют 0,1 и. раствором
азотнокислой ртути до появления фиолетовой окраски раствора.
101
Расчет. Содержание ионов хлора в воде вычисляют по формуле
х _ аЗ,55 • 1000
~ S
где X — концентрация ионов хлора в воде, мг/л; g — объем воды, взяты»
на анализ, мл; а — объем 0,1 н. раствора азотнокислой ртути,
израсходованного на анализ, мл; 3,55 — количество хлора, соответствующее 1 мл 0,1 н.
раствора азотнокислой ртути, мг.
Воспроизводимость в среднем из трех определений ±0,06 мл 0,1 н.
раствора азотнокислой ртути.
Предостережение. Результат анализа зависит от pH титруемого
раствора. Окраска индикатора появляется при pH ниже 3 в присутствии избытка
ионов ртути, а при pH выше 3,5 до окончания реакции.
Определение брома и йода
Для анализа воды, которую используют на гидролиз и
разбавление субстрата, предназначен метод, основанный на
окислении ионов брома и йода до образования ионов ВгО3 и Юз,
которые определяют йодометрическим методом [32]. При
определении суммы ионов брома и йода в качестве окислителя
применяют гипохлорит. Реакции окисления протекают по
следующим уравнениям:
Вг'+ЗСЮ' — ВгОз+ЗСГ;
Для удаления избытка гипохлорита натрия к реакционной
смеси добавляют муравьинокислый натрий
HClO+HCOONa-*CO2+H2O+NaCl.
К раствору, содержащему образовавшиеся ионы ВгОз и
Юз, добавляют йодистый калий и соляную кислоту.
Юз+51'+6Н- — 312+ЗН2О.
Выделившийся йод оттитровывают гипосульфитом натрия.
3I2+6Na2S2O3 — 6NaI-f-3Na2S4O6.
При определении только йода окисляют бромной водой.
Избыток окислителя разрушают муравьинокислым натрием.
Br2+HCOONa —¦ CO2+HBr+NaBr.
Образовавшиеся ионы Юз определяют титрованием
раствором гипосульфита. Оптимальное количество йода и брома
для анализа от 5 до 50 мкг.
102
Мешающие вещества. Кальций, магний и органические
вещества.
Аппаратура. Муфельная печь. Водяная баня. Установка для
титрования.
Реактивы и их приготовление. 1. Едкий калий, 1 н. и 2 н.
растворы. 2. Этиловый спирт (ректификат). 3. Соляная кислота
плотностью 1,19 г/см3 (х.ч.), без брома и йода.
Приготовление. К продажной соляной кислоте, содержащей бром или
йод, прибавляют небольшое количество хлората калия и
перегоняют ее. 4. Серная кислота плотностью 1,84 г/см3 (х.ч.).
5. Серная кислота, 0,1 н. и Зн. растворы. 6. Муравьинокислый
натрий, 20%-ный раствор. 7. Бромная вода. 8. Мочевина
(ч.д. а.), 0,5%-ный раствор. 9. Хлористый натрий без брома и
йода насыщенный раствор. Приготовление. Для получения
хлористого натрия свободного от брома и йода химически чистый
хлористый натрий перекристаллизовывают 2 раза. Первую
кристаллизацию проводят из раствора, подкисленного соляной
кислотой, которая предварительно была освобождена от брома
и йода перегонкой. Вторую кристаллизацию проводят
пропусканием газообразного хлористого водорода через насыщенный
раствор хлористого натрия. Хлористый водород получают, действуя
серной кислотой (х.ч) на хлористый натрий, который был 1 раз
перекристаллизован. Полученный хлористый водород сначала
очищают, а потом направляют в раствор хлористого натрия. Для
очистки от ионов брома хлористый водород пропускают через
раствор бихромата калия в концентрированной серной кислоте.
Для очистки от свободного брома и ионов йода хлористый
водород пропускают через металлическую ртуть. Осадок хлористого
натрия, выпадающий при пропускании хлористого водорода
через его раствор, отфильтровывают на стеклянном фильтре№4,
высушивают и прокаливают при 200—250°С. 10. Гипохлорит
калия или натрия, раствор. Приготовление. Через 100 мл
12%-ного раствора едкого натра (или едкого кали) медленно
пропускают газообразный хлор, который получают действием
соляной кислотой (без брома и йода) на перманганат калия.
Раствор едкого натра во время пропускания хлора охлаждают
смесью льда с солью до —7°С. Через 30 мин прекращают
пропускать хлор и отбирают пробу для определения щелочности
раствора, которая должна составлять 0,2—0,3 н. Раствор хранят
в склянке из темного стекла при температуре 16—18°С, тогда
его свойства не изменяются в течение года. Щелочность раствора
гипохлорита натрия определяют следующим образом. В
коническую колбу вместимостью 250 мл, соблюдая последовательность,
вливают 25 мл 0,1 н. раствора серной кислоты, 0,5 г йодистого
калия и 1 мл раствора гипохлорита. Выделившийся свободный
йод оттитровывают 0,1 н. раствором гипосульфита натрия в
присутствии крахмала. Затем к раствору добавляют несколько
капель метилового оранжевого и титруют остаток 0,1 н. раствора
103
серной кислоты 0,1 н. раствором едкого натра. Щелочность
раствора гипохлорита вычисляют по формуле
д —(ді —
х
g
где X — концентрация щелочи в растворе гипохлорита, (н. нормальность); а —
объем 0,1 н. раствора серной кислоты, взятый на анализ, мл; Oi — объем 0,1 н.
раствора гипосульфита, израсходованный на титрование, мл; а^ — объем 0,1 н.
раствора едкого натра, израсходованный на титрование, мл; g — объем
раствора гипохлорита, взятый на анализ, мл.
11. Борная кислота х. ч. насыщенный раствор. 12. Молибденово-
кислый аммоний ч. д. а., 10%-ный раствор. 13. Хромовокислый
калий, 0,05 н. раствор. 14. Гипосульфит натрия, 0,001 н. раствор,
свежеприготовленный разбавлением 0,05 н. раствора. 15.
Крахмал, 0,5%-ный раствор. 16. Йодистый калий х.ч., не
содержащий йодата и свободного йода.
Ход анализа. Удаление магния и органических
веществ. В стакан помещают 100 мл (или 500 мл в
зависимости от содержания брома и йода) пробы, отфильтровывают
и приливают 1 н. или 5н. раствор едкого кали, в 2—3 раза
превышающий количество, необходимое для полного осаждения
магния. Жидкость с осадком переносят в выпарительную
чашку вместимостью 200 мл и выпаривают на водяной бане до
тех пор, пока объем жидкости с осадком достигнет 50—
70 мл. Выпавший осадок отфильтровывают на стеклянном
фильтре № 4 и промывают 3—4 раза дистиллированной водой.
Фильтрат и промывные воды сливают в выпарительную чашку
(платиновую или кварцевую), упаривают досуха, а затем
прокаливают при 400сС.
Прокаленный осадок растворяют в горячей воде, раствор
фильтруют, осадок на фильтре промывают 3—4 раза горячей
водой. Фильтрат и промывные воды сливают в мерную колбу
вместимостью 50 мл и нейтрализуют, прибавляя по каплям
сначала Зн. раствор серной кислоты, а потом 0,1 н. раствор до
нейтральной реакции по лакмусовой бумажке. Каплю раствора,
взятую чистой стеклянной палочкой, наносят на лакмусовую
бумажку и по окраске мокрого пятна судят о pH раствора.
Раствор в колбе охлаждают до комнатной температуры,
объем его доводят дистиллированной водой до метки,
перемешивают, получают раствор А, а затем анализируют.
В том случае, когда выпаривание воды затрудняется
наличием большого количества солей, проводят экстракцию брома,
йода и их солей 96% -ным спиртом из упаренной смеси.
Загустевшую смесь воды и солей охлаждают, хорошо растирают для
получения однородной массы, приливают к ней 5 мл 96%-ного
этилового спирта и снова хорошо растирают. После отстоя
прозрачный раствор спирта сливают и обработку осадка спир-
104
том повторяют 5—7 раз. Спиртовый экстракт после добавления
1—2 капли 1 н. раствора едкого кали выпаривают на водяной
бане досуха, остаток прокаливают при 400°С, растворяют
в воде и раствор нейтрализуют, как описано выше.
Определение брома и йода. В коническую колбу
вместимостью 250 мл помещают 5 мл раствора, полученного
после удаления магния и органических веществ. Прибавляют
1 каплю 3 н. раствора серной кислоты, 1 мл насыщенного
раствора хлористого натрия, 1,5 мл насыщенного раствора борной
кислоты, 0,2 мл раствора гипохлорита и нагревают на кипящей
водяной бане в течение 5 мин. К горячему раствору добавляют
0,2 мл 20%-ного раствора муравьинокислого натрия, колбу
с раствором ставят на нагретую песчаную баню и замечают
время начала кипения. Через 3 мин кипячения колбу с
раствором охлаждают, прибавляют несколько кристалликов
йодистого калия, 1 мл 3 н. раствора серной кислоты и одну каплю
10%-ного раствора молибденовокислого аммония.
Выделившийся йод оттитровывают 0,01 н. раствором гипосульфита до
соломенного цвета раствора, добавляют 2—3 капли раствора
крахмала и титруют до исчезновения синей окраски раствора.
Параллельно делают холостой опыт, в котором вместо
анализируемого раствора вливают такое же количество
дистиллированной воды.
Определение йода. 1. В отсутствие нитритов.
Отмеренные пипеткой 10 мл раствора, полученного после удаления
магния и органических веществ, помещают в коническую колбу
вместимостью 250 мл, подкисляют 0,1 н. раствором серной
кислоты до оранжевой окраски метилового оранжевого,
прибавляют 0,2 мл свежеприготовленной насыщенной бромной воды и
нагревают его на электроплитке до кипения. Кипятить не надо.
В горячий раствор вносят 1—2 капли 20%-ного раствора
муравьинокислого натрия и ждут 2 мин. Затем раствор
охлаждают, прибавляют несколько кристалликов йодистого калия и
1 мл 3 н. раствора серной кислоты. Выделившийся йод
оттитровывают 0,001 н. раствором гипосульфита натрия, добавляя
в конце титрования раствор крахмала.
2. В присутствии нитритов. Нитриты мешают определению
йода, поэтому перед анализом их разрушают мочевиной или
азидом натрия.
Определяют содержание нитритов в растворе, полученном
после удаления магния и органических веществ (см. с. 279),
и вычисляют количество мочевины, которое нужно прибавить
к 10 мл анализируемого раствора, исходя из того, что на 1 мг
NO2 требуется 0,85 мг мочевины.
В коническую колбу вместимостью 100 мл вливают 10 мл
раствора, полученного после удаления магния и органических
веществ, прибавляют объем 0,5%-ного раствора мочевины,
который содержит количество мочевины, несколько превышающее
105
вычисленное, раствор подкисляют, добавляя 0,1 н. раствор
серной кислоты до появления оранжевой окраски метилового
оранжевого (выносная проба) и оставляют стоять. Через 30 мин
к раствору по каплям прибавляют бромную воду до
исчезновения желтой окраски. Дальнейший ход анализа такой же, как
для проб, не содержащих нитриты.
Расчет. 1. Содержание ионов йода в анализируемой воде вычисляют по
формуле
v 21,15eVil000
*1 = W
где Xi — концентрация ионов йода в воде, мкг/л; g — объем воды, взятый на
анализ, мл; а — объем 0,001 н. раствора гипосульфита натрия,
израсходованный на титрование, с учетом поправки на холостой опыт, мл; V — объем
раствора А, взятый для определения йода, мл; V\ — объем всего раствора А, мл;
21,15 — количество йода, эквивалентное 1 мл 0,001 н. раствора гипосульфита
натрия, мкг.
2. Содержание ионов брома в анализируемой воде вычисляют по формуле
v 13.32(^—^)^,1000
л2 = ¦
где Х% — концентрация ионов брома в воде, мкг/л; щ—объем 0,001 н.
раствора гипосульфита натрия, израсходованный на титрование при определении
суммы йода и брома, с учетом поправки на холостой опыт, мл; V% — объем
раствора А, взятый на определение суммы йода и брома, мл; 13,32 —
количество брома, эквивалентное 1 мл 0,001 н. раствора гипосульфита натрия, мкг.
Бром и йод определяют с точностью ±6% отн.
Определение фтора
Для анализа воды, которую подают на гидролиз и на
разбавление субстрата, используют метод, основанный на
колориметрической реакции ионов фтора с ализаринсульфонатом
тория [33].
Ализаринсульфонат тория (торийализариновый лак)
получают при взаимодействии ализаринсульфоната натрия
(ализарин S) с ионами тория по следующему уравнению.
О ОН
Th(NO3L—> Th(C14H7O7SL-
SO3Na
Торнйализариновый лак — соединение красного цвета,
реагируя с ионами фтора в условиях проведения анализа, превра-
106
щается в ализариносульфонат натрия — соединение желтого
цвета
Th (C!4H7O7SL+4F' — ThF4+4CnH7O7S\
Фтористый торий — труднорастворимая соль выпадает
в осадок. По изменению окраски реакционной смеси находят
количество фтора в анализируемой пробе.
Проведению реакции мешают многие вещества, поэтому
перед анализом выделяют фтор из его соединений, находящихся
в исследуемой воде, по методу Тананаева, который основан на
реакциях получения летучей кремнефтористоводородной
кислоты [34]
3CaF2+3H2SO4 — 3CaSO4+6HF;
6HF+SiO2-vH2SiF6+2H2O.
Образующуюся кремнефтористую
кислоту отгоняют и в отгоне
определяют фтор.
Аппаратура. Установка для
отгонки кремнефтористоводородной кислоты
с водяным паром (рис. 34).
Реактивы и их приготовление.
1. Серная кислота разбавленная 1:1.
2. Едкий натр, 0,1 н. раствор. 3.
Сернокислое серебро кристаллическое.
4. Кварцевый песок тонкоизмельчен-
ный. 5. Фтористый натрий,
стандартный раствор, содержащий в 1 л 10 мг ионов фтора. 6. Ализарин-
сульфонат натрия (Ci4H7O7SNa • Н2О), 0,00025 М раствор.
7. Азотнокислый торий (Th(NO3L), 0,001 М раствор. 8. Азотная
кислота, 025 н. раствор. 9. Буферный раствор.
Приготовление. Смешивают равные объемы 0,4 М раствора монохлорук-
сусной кислоты и 0,2 М раствора едкого натра.
Ход анализа. Выделение фтора. Отмеривают 1 л
анализируемой пробы воды. Часть ее помещают в перегонную
колбу (см. рис. 34), добавляют к ней 2 мл 0,1 н. раствора
едкого натра и упаривают. Остальную часть отмеренной воды
добавляют в перегонную колбу периодически, маленькими
порциями, по мере упаривания. Пробу выпаривают до получения
2—3 мл объема смеси воды и выпавших солей. Затем убирают
нагрев и в перегонную колбу вносят 0,1 г кварцевого песка,
25 мл раствора серной кислоты A : 1), перемешивают и
прибавляют сульфат серебра в количестве достаточном для полного
связывания ионов хлора. Смесь в колбе снова хорошо
перемешивают, колбу закрывают пробкой, в которую вставлены две
трубки. Конец одной трубки соединяют с холодильником,
нагревают содержимое колбы почти до кипения, соединяют конец
107
Рис. 34. Установка для
отгонки
кремнефтористоводородной кислоты
второй трубки с паровиком и проводят отгон образовавшейся
кремнефтористоводородной кислоты с водяным паром.
Собирают 100 мл конденсата, который содержит фтор из всех
соединений, находящихся віл анализируемой воды.
Определение фтора. В коническую колбу
вместимостью 200 мл помещают 10 мл полученного конденсата, который
содержит не более 0,1 мг фтора и прибавляют к нему 5 мл
0,00025 М раствора ализаринсульфоната натрия. Раствор
приобретает слабо-розовую окраску. Затем добавляют по каплям
0,25 н. раствор азотной кислоты до появления желтой окраски
раствора. К желтому раствору прибавляют 5 мл буферного
раствора и 5 мл 0,001 М раствора азотнокислого тория.
Одновременно готовят в таких же колбочках стандартную
шкалу, проводя анализы с различным количеством
стандартного раствора фтористого натрия. Через 30 мин визуально
сравнивают окраску раствора, полученного при анализе пробы,
с окраской растворов стандартной шкалы. Определяют
количество миллиграммов фтора в эталонном растворе стандартной
шкалы, интенсивность окраски которого совпадала с
интенсивностью окраски раствора пробы.
Расчет. Содержание фтора віл анализируемой воды вычисляют по
формуле
X = я10,
где X — концентрация фтора в исследуемой воде, ыкг/л; а — количество фтора
в эталонном растворе, интенсивность окраски которого совпала с окраской
пробы, мкг; 10 — коэффициент концентрирования при отгонке
кремнефтористоводородной кислоты.
Содержание фтора в воде определяют с точностью ±1—2% отн.
Определение масел
Для определения загрязненности воды маслом и жиром
делают качественную пробу с камфорой.
Ход анализа. В широкий стакан наливают 500 мл
анализируемой воды и дают ей спокойно постоять в течение 1 ч. Затем
маленький кусочек камфоры кладут пинцетом на поверхность
воды в стакане. Если вода не содержит масла и жира, то
кусочек камфоры будет быстро двигаться по ее поверхности,
описывая круги. В присутствии даже незначительных количеств
(следов) жира кусочек камфоры будет неподвижно лежать на
поверхности воды.
СЕРНАЯ КИСЛОТА
Определение моногидрата ареометром
Метод основан на зависимости величины плотности от
концентрации моногидрата в серной кислоте. Плотность серной
кислоты измеряют ареометром [25]. Концентрацию моногидрата
в серной кислоте находят по табл. 1 (с. 316).
108
1 -і
Аппаратура. Ареометр, отградуированный в пределах 1,4—
1,84 или 1,72—1,84. Термометр.
Ход анализа. В стеклянный цилиндр вместимостью 250 мл
наливают 200 мл исследуемой кислоты. Цилиндр должен иметь
диаметр на 10—20 мм больше, чем диаметр ареометра, чтобы
последний мог плавать не касаясь стенок его. Цилиндр ставят
на горизонтальную поверхность,
измеряют температуру кислоты термометром,
затем осторожно погружают в него
ареометр, который предварительно был
хорошо протерт мягкой тряпочкой.
Ареометр погружают дважды. Первое
погружение ареометра делают без соблюдения
особых предосторожностей для
ориентировочного определения плотности. Перед
вторым погружением ареометр
промывают водой, досуха протирают мягкой
тряпочкой, а потом погружают его в
кислоту, стараясь не смочить шейку
ареометра выше уровня, замеченного при
первом погружении. Когда ареометр
остановится, на его шкале замечают
деление, которое совпадает с нижним
мениском (рис. 35). При отсчете глаз
аналитика должен находиться в одной
горизонтальной плоскости с поверхностью
жидкости.
Анализируемая кислота, находящаяся в цилиндре, не
должна содержать пузырьков. Мениск должен быть вогнутым. При
выпуклом мениске, который указывает на наличие жира на
ареометре, определение повторяют, предварительно очистив
поверхность ареометра от загрязнений спиртом.
Расчет. Если температура кислоты во время измерения плотности
отличалась от 20°С, то сначала находят температурную поправку для плотности
в дополнении к табл. 1. При температуре кислоты выше 20°С поправку
прибавляют к значенню плотности, которая определена ареометром, прн
температуре кислоты ниже 20°С поправку отнимают. По плотности кислоты,
приведенной к 20°С после внесения температурной поправки, по табл. 1 находят
концентрацию моногидрата в серной кислоте, выраженную в граммах на
100 г или в граммах на 1 л.
Определение моногидрата титрованием
Метод основан на титровании навески кислоты раствором
щелочи с индикатором метиловый красный [35].
Аппаратура. Установка для титрования.
Ход анализа. Около 3 г исследуемой кислоты взвешивают
в закрытом бюксе с точностью 0,0002 г. В мерную колбу
вместимостью 500 мл вливают 250 мл дистиллированной воды.
Рис. 35. Установка для
измерения плотности
жидкости ареометром:
/ — по верхнему мениску; 2 —
по нижнему мениску
109
Затем осторожно, положив бюкс в воронку, вставленную
в горло колбы, выливают кислоту из бюкса, бюкс и воронку над
колбой споласкивают водой, расходуя 200—250 мл воды. Колбу
с раствором серной кислоты охлаждают до 20°С, объем раствора
доводят до метки дистиллированной водой и перемешивают.
В коническую колбу объемом 250 мл вливают 50 мл
приготовленного раствора кислоты, добавляют 2—3 капли индикатора
и титруют 0,5 н. раствором едкого натра до перехода окраски из
красной в желтую.
Расчет. Содержание моногидрата в серной кислоте вычисляют по
формуле
а0,02452К0100
Л — ^~р і
где X — концентрация моногидрата в серной кислоте, г/100 г; а — объем 0,5 и.
раствора едкого натра, израсходованный на анализ, мл; g — количество
анализируемой кислоты, взятое для разведения, г; Vo — объем разбавленной
кислоты, мл; V — объем разбавленной кислоты, взятый на титрование, мл;
0,02452 — количество серной кислоты в 1 мл 0,5 н. раствора, г.
Определение мышьяка
Мышьяк в серной кислоте определяют колориметрическим
методом в приборе, описанном на с. 69, применяя те же
реактивы. Отличие состоит в подготовке пробы к анализу. Для
перевода пятивалентного мышьяка в трехвалентный
исследуемую пробу разбавляют и кипятят.
Ход анализа. В стакан вместимостью 100 мл вливают 10 мл
дистиллированной воды и 5 мл анализируемой кислоты.
Раствор в стакане кипятят 5 мин, охлаждают и количественно
переносят в сосуд прибора, расходуя для этого 45 мл воды. Затем
в сосуд добавляют 0,2 г хлористого олова и 2 г цинка, прибор
закрывают подготовленной насадкой и ставят в темное место
на 1 ч. Дальнейший ход анализа см. на с. 69.
Расчет. Содержание мышьяка в серной кислоте вычисляют по формуле
alOO a
X
gdW '
где X — концентрация мышьяка в серной кислоте, %; а — количество
мышьяка, найденное по шкале окрасок, мкг; g — количество кислоты, взятое на
анализ, мл; d — плотность анализируемой серной кислоты, г/см3.
ИЗВЕСТКОВОЕ МОЛОКО
Суспензию извести в воде называют известковым молоком.
Качество известкового молока оценивают его активностью, т. е.
содержанием окиси кальция СаО,
ПО
Определение окиси кальция титрованием
Метод основан на реакции нейтрализации гидрата окиси
кальция соляной кислотой
Са (ОНJ+2НС1 — СаС12+2Н2О.
Мешающие вещества. Гидроокись магния.
Реактивы. 1. Соляная кислота, 1 н. раствор. 2.
Фенолфталеин, 1%-ный водно-спиртовой раствор.
Ход анализа. Пробу известкового молока тщательно
перемешивают, цилиндром отмеривают 25 мл, вливают их в мерную
колбу вместимостью 1 л, добавляют дистиллированную воду до
метки и перемешивают. В коническую колбу на 250 мл
помещают 50 мл полученного раствора, прибавляют 2—3 капли
фенолфталеина и титруют 1 н. раствором соляной кислоты при
постоянном помешивании. Когда исчезнет розовая окраска,
раствору дают постоять 3—5 мин для растворения кусочков СаО.
В это время снова появляется розовая окраска. Титрование
продолжают до тех пор, пока розовая окраска не будет
появляться при стоянии раствора в течение 3 мин.
Расчет. Активность известкового молока вычисляют по формуле
а0,028 . 1000 • 1000
Х-
ggl
где X — содержание СаО в известковом молоке, г/л; а — объем 1 н. раствора
соляной кислоты, израсходованный на титрование, мл; 0,028 — количество
СаО, эквивалентное 1 мл 1 н. раствора соляной кислоты, г; g— объем
известкового молока, взятый для разведения, мл; gi — объем известкового молока,
взятый для титрования, мл.
Определение окиси кальция ареометром
Метод основан на зависимости плотности известкового молока
от содержания в нем окиси кальция СаО. Плотность измеряют
ареометром.
Мешающие вещества. Окись магния, углекислые соли
кальция и магния и другие вещества, находящиеся в извести.
Аппаратура. Ареометр, отградуированный в интервале
плотностей 1 —1,4 г/см3 при 20°С. Термометр. Цилиндр без носика.
Ход анализа. В цилиндр без носика вместимостью 250 мл
вливают около 200 мл хорошо перемешанного анализируемого
известкового молока, ставят его на горизонтальную
поверхность и осторожно погружают в него ареометр. Диаметр
цилиндра должен быть таким, чтобы ареометр в нем плавал, не
задевая стенок. Сразу, как только ареометр перестанет
колебаться, определяют плотность известкового молока, замечая
деление шкалы ареометра, совпадающее с верхним мениском
(см. рис. 35). Измерение плотности надо проводить при 20°С
і і
быстро, чтобы молоко не успело расслоиться. В случае
заметного расслоения молока анализ повторяют. Перед измерением
плотности определяют температуру известкового молока в
цилиндре. Если она отличается от 20°С более чем на ±2°С, то
пробу в цилиндре соответственно охлаждают или нагревают
до 20°С, а только потом измеряют плотность.
Расчет. Содержание окиси кальция в известковом молоке находят по
плотности, пользуясь данными, приведенными в табл. II. с. 317. Концентрацию
окиси кальция определяют с точностью ±3—5% отн.
Определение магния
Магний определяют по разности между суммой кальция и
магния и содержанием кальция в пробе комплексометрическнм
методом.
Ход анализа. Сумму кальция и магния определяют
титрованием разбавленной пробы раствором трилона Б с индикатором
кислый хром черный, как описано на с. 99. Титрованием
той же разбавленной пробы раствором трилона Б с
индикатором мурексид определяют кальций, как описано на с. 100.
Расчет. Содержание магния в анализируемой пробе извести или
известкового молока вычисляют по формуле
v (Vi - V2) 0,002015А;л100
g
где X— содержание магния в пробе, г/100 г; V} — объем раствора трилона Б,
израсходованный на титрование кальция и магния, мл; V2 — объем раствора
трилона Б, израсходованный на титрование при определении кальция, мл; п —
разбавление; g— количество пробы, взятое на анализ, г; 0,002015 —
количество окиси магния, эквивалентное 1 мл 0,1 н. раствора трилона Б, г; К —
нормальность раствора трилона Б.
АММОФОС
Аммофос — смесь сернокислого аммония и суперфосфата.
Раствор аммофоса называют раствором питательных солей.
В нем определяют содержание азота, фосфора и мышьяка,
которые связаны в минеральных соединениях.
Определение азота
Метод предназначен для определения азота в растворе
аммофоса и может быть использован для определения
минерального азота в сульфитном щелоке, субстратах, в последрожже-
вой бражке и в сточной воде. Метод основан на реакции
аммонийных солей с гидроокисью магния в слабощелочной среде
при pH около 8
(NH,JSO4+Mg(OHJ —2NH3+MgSO4+2H2O.
112
Образующийся аммиак отгоняют в раствор борной кислоты,
а затем оттитровывают серной кислотой. Борная кислота в
водном растворе реагирует как слабая одноосновная кислота,
поэтому она хорошо поглощает аммиак. Соли борной кислоты
имеют щелочную реакцию в растворе. Эту особенность
используют для определения количества отогнанного аммиака по
количеству серной кислоты, которая пойдет на титрование с
индикатором, имеющим переход окраски при pH около 5, что
соответствует pH раствора борной кислоты.
Можно проводить отгонку аммиака в титрованный 0,1 н.
раствор серной кислоты и ее избыток, после нейтрализации
аммиаком, оттитровывать 0,1 н. раствором
едкого натра.
Мешающие вещества. Вещества,
обладающие кислыми свойствами. Их
соединение с Mg(OHJ понижает pH раствора,
что затрудняет отгонку аммиака.
Аппаратура. Установка для отгонки
аммиака (рис. 36) и установка для
титрования.
Реактивы и их приготовление. 1. Окись
магния (MgO) ч. д. а. крист. 2. Борная
кислота (Н3ВО3), раствор 20 г/л. 3.
Серная кислота, 0,1 н. раствор. 4. Едкий
натр, 0,1 н. раствор. 5. Индикатор один
из двух: бромфеноловый синий или
смешанный. Приготовление. 0,1 н.
бромфенолового синего растворяют
В 3 МЛ. 0,05 Н. раствора едкого натра И Рис. 36. Установка для
Объем раствора ДОВОДЯТ ДО 100 МЛ, при- отгонки аммиака
бавляя дистиллированную воду.
Смешанный индикатор готовят так: один объем 0,1%-ного
спиртового раствора метиленовой синей смешивают с двумя объемами
0,1%-ного спиртового раствора метилового красного. Интервал
перехода окраски смешанного индикатора в пределах pH 5,6—
5,8 от розовой к серо-зеленой.
Ход анализа. В реакционную колбу объемом 500 мл вливают
20 мл раствора аммофоса E0 мл субстрата или последрожже-
вой бражки), 150 мл дистиллированной воды и очень быстро
вносят 0,2 г окиси магния. Колбу сразу закрывают пробкой,
в которую вставлена трубка, и присоединяют к холодильнику.
Под колбу ставят электроплитку и включают нагрев.
Приемником конденсата служит коническая колба вместимостью
250 мл, в которую вливают 50—100 мл раствора борной
кислоты (или серной кислоты) из расчета 50 мл борной кислоты
на 1 мг азота в отгоне и прибавляют 5—7 капель индикатора.
Нижний конец холодильника погружают в кислоту. Если
холодильник без расширения на конце, то в начале отгонки его
8 Заказ № 142
ИЗ
конец должен только касаться жидкости, чтобы избежать
засасывания кислоты в холодильник.
Отгонку считают оконченной, когда красная лакмусовая
бумажка, смоченная каплей конденсата из холодильника, не
будет изменять свой цвет. По окончании отгонки аммиака
реакционную колбу отсоединяют от холодильника и его
внутреннюю трубку споласкивают дистиллированной водой над
приемником. Аммиак в борной кислоте оттитровывают 0,1 н.
раствором серной кислоты. Конец титрования устанавливают,
сравнивая окраску индикатора в пробе с окраской того же
индикатора в растворе свидетеля. Свидетелем является смесь
50 мл раствора борной кислоты и дистиллированной воды,
освобожденной от СО2 кипячением. Объем добавляемой воды
должен быть равен объему собранного конденсата. Количество
индикатора в свидетеле должно быть точно таким, как в пробе.
После отгонки аммиака проверяют pH реакционной жидкости
в колбе, он должен быть не ниже 7,5. Если pH ниже 7,5, то
анализ надо повторить, добавив больше окиси магния.
Расчет. Содержание аммиачного азота в анализируемом растворе
находят по формуле
х а\А • 1000
g
где X— концентрация аммиачного азота в пробе, мг/л; а — объем 0,1 н.
раствора серной кислоты, израсходованный на титрование аммиака, мл; g—
объем пробы, взятый для отгонки аммиака, мл; 1,4 — количество
миллиграммов аммиачного азота, эквивалентное 1 мл 0,1 н. раствора серной кислоты.
Чувствительность метода 0,08 мг азота в 50 мл отгона.
Воспроизводимость в среднем из трех определений ±0,06 мл 0,1 н. H2SO4.
Предостережение. Главными источниками ошибок являются потери
аммиака при отгонке и неполнота отгонки в результате снижения pH.
Определение фосфора
Для определения фосфора, содержащегося в минеральных
соединениях, которые находятся в растворе аммофоса,
субстрате, последрожжевой бражке и сточной воде, предназначен
метод, основанный на реакции осаждения фосфора в виде фос-
форномолибденовокислого аммония. Последний после отделения
от жидкости обрабатывают раствором едкого натра. Избыток
едкого натра оттитровывают соляной кислотой. По расходу
едкого натра на реакцию с фосфорномолибденовокислым
аммонием вычисляют содержание фосфора в анализируемой пробе.
Этот метод может быть использован и для определения
фосфора в органических соединениях после их минерализации
в тех случаях, когда нет колориметра, чтобы делать анализ
колориметрическим методом.
Аппаратура. Установка для титрования.
14
Реактивы и их приготовление. 1. Едкий натр, 0,1 н. раствор.
2. Соляная кислота, 0,1 н. раствор. 3. Фенолфталеин, 0,1%-ный
спиртовой раствор. 4. Молибденовая смесь.
Приготовление. В коническую колбу вместимостью 300 мл помещают
40 г молибденовокислого аммония и 120 мл 20%-ного раствора
аммиака. В другую коническую колбу вместимостью 750 мл
вливают 250 мл азотной кислоты плотностью 1,3—1,4 г/см3 и
100 мл дистиллированной воды. Небольшими порциями при
постоянном перемешивании и охлаждении вливают раствор
молибденовокислого аммония в раствор азотной кислоты. Если
полученный раствор окажется мутным, его фильтруют через
бумажный фильтр. Раствор хранят в склянке из темного стекла.
Употреблять раствор для анализа можно только через 3 недели
после его приготовления.
Ход анализа. В стакан вместимостью 100 мл вливают 10 мл
раствора аммофоса и 30—40 мл дистиллированной воды.
Раствор в стакане нагревают до 75°С. В другой стакан наливают
25 мл молибденовой смеси и нагревают ее до 50°С. Затем
раствор аммофоса перемешивают несколько секунд, чтобы охладить
перегретые стенки стакана выше уровня жидкости, и вливают
в него тонкой струей при постоянном помешивании подогретый
раствор молибденовой смеси. Реагирующую жидкость оставляют
стоять 3 ч при комнатной температуре. Выпавший осадок
количественно переносят в стеклянный фильтр, отфильтровывают,
промывают дистиллированной водой до отрицательной реакции
на кислоту по лакмусовой бумажке или по метиловому
оранжевому. Фильтр с осадком вставляют на пробке в чистую колбу,
приливают точно отмеренные бюреткой 5—10 мл 0,1 н. раствора
едкого натра для полного растворения осадка. Колбу
соединяют с вакуум-насосом, фильтр промывают 8—10 мл 0,1 н.
едкого натра, потом 10—20 мл воды до отрицательной реакции
на щелочь по фенолфталеину. Собранный раствор и промывные
воды титруют 0,1 н. раствором соляной кислоты с
фенолфталеином. По бюретке определяют количество 0,1 н. раствора едкого
натра, взятое для растворения осадка и на промывку фильтра,
и количество 0,1 н. раствора соляной кислоты, которое пошло на
титрование.
Расчет. Содержание фосфора в анализируемой пробе вычисляют по
формуле
v (а-б) 0,00155- 100
А. = •
g
где X — содержание фосфора в пробе, %; а — объем 0,1 н. раствора едкого
натра, взятый на анализ, мл; б — объем 0,1 н. раствора соляной кислоты,
пошедший иа титрование, мл; g — объем раствора аммофоса, взятый на
анализ, мл; 0,00155 — количество фосфора, эквивалентное 1 мл 0,1 н. раствора
соляной кислоты, г.
Воспроизводимость в среднем из трех определений ±0,05 мл 0,1 н.
раствора соляной кислоты.
8* 115
АММИАЧНАЯ ВОДА
Определение аммиака
Аммиак в аммиачной воде определяют по плотности или
титрованием.
1. Плотность аммиачной воды измеряют ареометром,
отградуированным в пределах 0,8—1,0 при 20°С по нижнему мениску,
как описано на с. 109. Концентрацию аммиака находят по
плотности, пользуясь данными, приведенными в табл. III,
с. 317. 2. Определение аммиака в аммиачной воде титрованием
основано на нейтрализации аммиака 0,1 н. раствором соляной
кислоты. При выполнении этого анализа необходимо принять
меры предосторожности, чтобы аммиак не мог улетучиться из
пробы, отмеренной для титрования.
Аппаратура. Установка для титрования.
Реактивы. 1. Соляная (или серная) кислота, 1 н. раствор.
2. Едкий натр, 1 н. раствор. 3. Индикатор — метиловый красный,
0,1%-ный спиртовый раствор.
Ход анализа. В коническую колбу вместимостью 250 мл
вливают 50 мл 1 н. раствора соляной кислоты и взвешивают.
С помощью дозатора в (см. рис. 12) отмеривают 1—2 мл
анализируемой аммиачной воды и сразу выливают в колбу с
кислотой. Реакционную колбу взвешивают, а затем избыток
кислоты, оставшийся после реакции с аммиаком, оттитровывают
1 н. раствором едкого натра, добавив 2—3 капли индикатора.
Расчет. Содержание аммиака в'анализируемой пробе вычисляют по
формуле
(й — б) 0,01703 • 100
Х-
g
где X — концентрация аммиака в пробе, %; а—количество 0,1 п. раствора
соляной кислоты, взятое на анализ, мл; б — количество 1 н. раствора едкого
натра, пошедшее иа анализ, мл; g — количество аммиачной воды, взятое на
титрование, г; 0,01703 — количество аммиака, эквивалентное 1 мл 1 и. раствора
соляной (илн серной) кислоты, г.
Аммиак определяют с точностью ±3—5% отн.
ЕДКИЙ НАТР
Определение едкого иатра
Метод основан на реакции нейтрализации едкого натра
соляной кислотой. Определению содержания едкого натра в
растворе или в твердом едком натре мешает сода (углекислый
натрий). Чтобы исключить ее влияние, перед титрованием
к раствору едкого натра прибавляют хлористый барий.
Образующийся углекислый барий выпадает в виде осадка, а
хлористый натрий не мешает анализу.
Аппаратура. Установка для титрования. Коническая колба
вместимостью 250 мл.
116
Реактивы. 1. Соляная кислота, 1 н. раствор. 2. Хлористый
барий, 10%-ный раствор. 3. Фенолфталеин, 1%-ный спиртовый-
раствор.
Ход анализа. Навеску твердого едкого натра (или его
раствора), взвешивают с точностью до 0,01 г и растворяют ее в
таком количестве дистиллированной воды, чтобы получить
примерно 4%-ный раствор. Объем раствора в мерной колбе
доводят до метки.
В коническую колбу вместимостью 250 мл вливают 25 мл
приготовленного раствора едкого натра и 20 мл 10%-ного
раствора хлористого бария (предварительно нейтрализованного
по фенолфталеину), раствор перемешивают и титруют 1 н.
раствором соляной кислоты с фенолфталеином в качестве
индикатора.
Расчет. Содержание едкого натра вычисляют по формуле
аО.О4лЮО
~ g
где X — концентрация едкого натра в анализируемой пробе, %; а — объем
1 н. раствора соляной кислоты, израсходованный на титрование, мл;
g—навеска пробы, взятая для разбавления, г; я — разбавление; 0,04 — количество-
едкого натра, эквивалентное 1 мл 1 н. раствора соляной кислоты, г.
Воспроизводимость в среднем из трех определений ±0,06 мл 1 н.
раствора соляной кислоты.
ГЛАВА 4. ПОЛУЧЕНИЕ И ОЧИСТКА РАСТВОРОВ МОНОСАХАРИДОВ
На гидролизных заводах, раствор моносахаридов получают
гидролизом полисахаридов растительных тканей, который
проводят разбавленной @,5—1%-ной) серной кислотой в гидро-
лизаппаратах. Во время варки в гидролизаппарат, в котором
находится сырье, подают серную кислоту и воду через
смеситель, а из гидролизаппарата отбирают гидролизат —
сернокислый раствор моносахаридов. После окончания гидролиза
полисахаридов подачу серной кислоты прекращают. Чтобы отмыть
моносахариды от лигнина в гидролизаппарат подают некоторое
время воду. После отмывки Сахаров сначала прекращают
подачу воды, а потом отбор гидролизата.
Для проведения гидролиза по заданному режиму при
отсутствии автоматического управления процессом контролируют:
1) количество воды, пара и серной кислоты, подаваемых в
гидролизаппарат; 2) температуру подаваемой воды и отбираемого
гидролизата; 3) перепад давления, т. е. разность давлений,
измеряемых манометрами, установленными на крышке гидролиз-
аппарата и на линии выдачи гидролизата до вентиля, и 4)
концентрацию серной кислоты и Сахаров в гидролизате. На
основании результатов анализа гидролизата на содержание серной
кислоты и сахара во время варки регулируют подачу серной
117
кислоты, температуру, скорость выдачи, устанавливают конец
варки и конец промывки.
Горячий гидролизат из гидролизаппарата идет в испаритель,
где он охлаждается за счет частичного испарения и
одновременно очищается от летучих примесей. Конденсат паров
самоиспарения содержит большую часть образовавшегося
фурфурола, его называют фурфурольным конденсатом. Для
определения уноса жидкости с парами в испарителе фурфурольный
конденсат иногда анализируют на содержание Сахаров.
После окончания варки анализируют лигнин и гидролизат
для оценки качества проведенного гидролиза. В лигнине
определяют содержание неотмытых моносахаридов и серной кислоты,
а также содержание оставшихся трудногидролизуемых
полисахаридов. Гидролизат анализируют на содержание
моносахаридов, на содержание продуктов распада моносахаридов —
фурфурола, оксиметилфурфурола, левулиновой и муравьиной
кислот—и на содержание продуктов неполного гидролиза —
олигосахаридов.
На гидролизных заводах для удаления серной кислоты из
гидролизата его нейтрализуют известковым молоком.
Образующийся гипс выпадает в осадок, который отделяют
отстаиванием. Если нейтрализат предназначен для получения этилового
спирта, то необходимо контролировать содержание гипса
в осветленном нейтрализате. Повышенная концентрация гипса
в сусле вызывает гипсацию бражных колонн. Если нейтрализат
предназначается для выращивания дрожжей, то контроль по
гипсу не нужен.
На заводах, перерабатывающих сульфитный щелок,
растворы моносахаридов (гидролиз гемицеллюлоз древесины)
получают во время варки целлюлозы. На завод поступает сырой
-сульфитный щелок, который содержит моносахариды, лигно-
сульфонаты и сернистую кислоту. Сырой сульфитный щелок
нейтрализуют известковым молоком. В осадок выпадает
сульфит кальция, который удаляют после отстаивания.
Одновременно с минеральными кислотами нейтрализуют часть
органических кислот.
В последнее время стали применять ступенчатую
нейтрализацию, при которой гидролизат или сульфитный щелок сначала
нейтрализуют известковым молоком до pH 3, затем донейтра-
лизовывают аммиаком. Иногда на дрожжевых заводах
нейтрализацию проводят только аммиаком.
Глубину нейтрализации гидролизатов и сульфитных
щелоков контролируют по общей кислотности и по pH, которые
определяют во время нейтрализации. Для определения потерь
редуцирующих веществ при нейтрализации со шламом его
анализируют на содержание сухих и редуцирующих веществ.
Осветленный нейтрализат анализируют на содержание различных
веществ в зависимости от дальнейшей переработки.
118
Определение сахара и серной кислоты экспрессным методом
Для быстрого определения концентрации Сахаров и серной
кислоты в горячем гидролизате на рабочем месте варщика
предназначен метод, основанный на измерении плотности и
электропроводности гидролизата [36]. Плотность измеряют
специальным ареометром, отградуированным в процентах сахара
при температуре 70°С. Электропроводность измеряют прибором
ЭМУ (электромагнитный указатель), шкала которого
отградуирована в процентах серной кислоты.
Мешающие вещества. Вещества,
растворенные в гидролизате, которые обладают
плотностью, отличающейся от плотности
воды, и изменяют электропроводность
гидролизата, обусловленную наличием серной
кислоты.
Аппаратура. Прибор РВ-мер (рис. 37).
Прибор РВ-мер для определения
концентрации сахара и серной кислоты в
горячем гидролизате устанавливают на
рабочем месте варщика. Он состоит из
измерительной ячейки, ареометра-поплавка /,
платиновых электродов 3, прибора ЭМУ 4
и термометра 2. Стеклянная
измерительная ячейка представляет собой два
сообщающихся сосуда вместимостью 150—
200 мл. Внизу ячейки имеется отвод,
на который надета резиновая трубка с
зажимом для спуска жидкости. В узком
сосуде, предназначенном для измерения
электропроводности, находятся два
платиновых электрода. Один из них
неподвижен, он впаян в стенку ячейки, а другой укрепленный на
штативе, подвижен. Оба электрода соединены гибкими проводами
с прибором ЭМУ через звонковую кнопку. Прибор ЭМУ имеет
две шкалы: нижняя шкала отградуирована в процентах серной
кислоты, а верхняя — в процентах РВ (редуцирующих веществ).
Во время анализа стрелка прибора указывает по нижней шкале
концентрацию серной кислоты в гидролизате, налитом в ячейку^
а по верхней — поправку к показаниям ареометра за счет
присутствия серной кислоты.
В широком сосуде ячейки определяют температуру и
плотность гидролизата. Ареометр-поплавок, которым измеряют
концентрацию Сахаров, отградуирован в процентах глюкозы
в пределах от 1 до 7% при 70°С, с учетом средних поправок на
сухие вещества (несахара), растворенные в гидролизатах. Цена
деления 0,5% глюкозы.
Рис. 37.
Прибор РВ-мер
Проверка прибора. Правильность показаний
ареометра проверяют сравнением результатов анализа гидролизата
на содержание Сахаров экспрессным методом с результатами
анализа той же пробы гидролизата на содержание
редуцирующих веществ эбулиостатическим методом. Если расхождения
результатов, полученных указанными методами, при анализе
5—10 проб различных гидролизатов в среднем будут не более
0,3% абс, то поплавком можно пользоваться. Если
расхождения будут более 0,4% абс, то надо сделать анализы 20—30 проб
гидролизатов, отобранных на различных стадиях варок, обоими
методами. На основании полученных данных вычислить
средние поправки для верхнего, среднего и нижнего делений шкалы
поплавка и пользоваться ими при анализах.
Правильность показаний прибора ЭМУ зависит от
расстояния между электродами, поэтому перед анализом ячейку и
электрод прочно укрепляют в штативе, а затем делают
проверку. Прибор ЭМУ включают в электрическую сеть
напряжением 220 В и ждут 15 мин. Затем в ячейку вливают 0,1 н.
@,49%-ный) раствор серной кислоты, замыкают цепь, нажимая
звонковую кнопку, и смотрят на показания прибора. При 20—
25°С прибор должен показывать по нижней шкале 0,52% H2SO4.
Если прибор ЭМУ показывает другую концентрацию кислоты,
то, передвигая вниз или вверх подвижный платиновый электрод
при замкнутой цепи (кнопка нажата), находят расстояние
между электродами, при котором стрелка прибора покажет 0,52%
H2SO4. Электрод хорошо укрепляют в штативе и снова
проверяют правильность показаний прибора. Малейшее
передвижение электрода после установки искажает результат анализа.
Разница между показанием прибора и концентрацией эталона,
составляющая 0,03%, включает в себя две поправки:
температурную поправку (анализ делают при 70°С) и поправку на
присутствие Сахаров и других веществ, влияющих на
электропроводность гидролизата.
Подготовка проб. Пробу анализируют сразу после отбора,
потому что анализ предназначен для оперативного контроля за
ходом варки.
Ход анализа. Прибор ЭМУ при постоянной эксплуатации
должен быть включен в электросеть все время. В ячейку
наливают анализируемую пробу горячего гидролизата. В широкий
сосуд ячейки осторожно опускают ареометр так, чтобы не
замочить его шейку, выступающую над уровнем жидкости. Замечают
показания термометра и деление на шкале ареометра,
совпадающее с верхним мениском гидролизата. Затем нажимают
звонковую кнопку и отсчитывают показания прибора ЭМУ по
нижней шкале — концентрацию серной кислоты в гидролизате, по
верхней — поправку на показания ареометра за счет
присутствия серной кислоты.
120
Расчет. Содержание Сахаров в гидролизате вычисляют по формуле
X = a — a\ + (t — 70H,1,
где X — концентрация Сахаров в гидролизате, %; о. — концентрация сахароа-
по показанию ареометра, %; at — поправка на показание ареометра за счет
присутствия серной кислоты, определяемая по верхней шкале прибора
ЭМУ, %; t — температура гндролизата вовремя анализа, "С; 70 — температура,
при которой градуирован ареометр, °С; 0,1 —температурная поправка на
показания ареометра, %.
Концентрацию сахара определяют с точностью ±0,2—0,3% аба, а
серную кислоту — ±0,02% абс.
Определение неотмытых редуцирующих веществ
и серной кислоты в лигнине
Для определения потерь моносахаридов с лигнином за счет
недостаточной его промывки в гидролизаппарате и количества
серной кислоты, оставшейся в лигнине, предназначен метод,
основанный на измерении количества редуцирующих веществ и
серной кислоты, которые переходят в раствор при промывке
лигнина горячей водой. Редуцирующие вещества определяют
эбулиостатическим методом, а кислоту титрованием щелочью.
Аппаратура. 1. Установка для титрования. 2. Воронка и
колба для фильтрования под вакуумом. 3. Установка для эбули-
остатического метода определения РВ (см. рис. И).
Реактивы. 1. Все реактивы для эбулиостатического метода
(см. с. 35). 2. Метиловый оранжевый, 0,1%-ный раствор.
3. Едкий натр, 0,1 н. раствор.
Подготовка проб. Пробу лигнина измельчают,
подсушивают до воздушносухого состояния и определяют влажность
(см. с. 31).
Ход анализа. В стакан вместимостью 500 мл помещают
50 г исследуемого лигнина, отвешенного с точностью ±0,05 г,
вливают 100 мл горячей дистиллированной воды, смесь хорошо
перемешивают стеклянной палочкой и оставляют стоять 5—
10 мин. Отстоявшийся раствор фильтруют через бумажный
фильтр на фарфоровой воронке под вакуумом. Осадок лигнина
в стакане снова заливают 100 мл горячей воды. Промывку
повторяют 3—4 раза до полного отсутствия кислоты по
индикатору метиловый оранжевый. Промывные воды количественно
переносят в мерную колбу вместимостью 500 мл, объем их
доводят до метки, прибавляя дистиллированную воду, и
перемешивают. Полученный раствор анализируют на содержание РВ
эбулиостатическим методом по второму варианту и на
содержание серной кислоты титрованием 0,1 н. раствором едкого
натра в присутствии индикатора метиловый оранжевый.
Расчет. 1. Содержание редуцирующих веществ в лигнине вычисляют по
формуле
v CV100
х
121'
где X— содержание редуцирующих веществ в абсолютно сухом лигнине, %;
•С — концентрация редуцирующих веществ в промывной воде, найденная
эбулиостатическим методом, %; V — объем промывной воды, мл; g — количество
абсолютно сухого лигнина, взятое на анализ, г.
2. Содержание серной кислоты в лигниие вычисляют по формуле
v a0,0049K100
л. — ¦
gg\
где X — содержание серной кислоты в абсолютно сухом лигиине, %; а —
объем 0,1 н. раствора едкого натра, израсходованный на титрование, мл;
g\ — объем промывной воды, взятый на титрование щелочью, мл; 0,0049 —
количество серной кислоты в 1 мл 0,1 н. раствора, г.
Определение олигосахаридов
Для определения олигосахаридов и декстринов в гидроли-
зате и в сульфитном щелоке предназначен метод, основанный
на определении редуцирующих веществ в пробе до и после
инверсии. Инверсию (гидролиз) низкополимерных углеводов
проводят 2—3%-ной серной кислотой при 100°С в течение 3 ч.
По увеличению концентрации редуцирующих веществ после
инверсии, которые определяют эбулиостатическим методом,
находят количество полимеров.
Мешающие вещества. Вещества, изменяющие свою
редуцирующую способность во время инверсии.
Аппаратура. Установка для кипячения с обратным
холодильником (см. рис. 13). Установка для определения РВ
эбулиостатическим методом (см. рис. 11).
Реактивы. 1. Серная кислота плотностью 1,84 г/см3. 2. Все
реактивы для эбулиостатического метода.
Ход анализа. Пробу гидролизата или сульфитного щелока
фильтруют через стеклянный фильтр, чтобы в анализируемой
жидкости не было волокон целлюлозы. В колбу вместимостью
250 мл вливают 100 мл отфильтрованной пробы и 1 мл
концентрированной серной кислоты. Колбу ставят на электроплитку,
плотно закрывают пробкой, в которую вставлен обратный
холодильник, включают нагрев и кипятят в течение 3 ч от начала
закипания. После инверсии содержимое колбы фильтруют и
анализируют. Содержание редуцирующих веществ
эбулиостатическим методом определяют в пробе до и после ее кипячения
с серной кислотой.
Расчет. Содержание полисахаридов в исследуемой пробе вычисляют
в процентах глюкозы по формуле
Х = А — Ах,
где Лі —концентрация редуцирующих веществ в пересчете на глюкозу в пробе
до инверсии, %; А—то же после инверсии, %•
322
Определение степени полноты гидролиза
Степенью полноты гидролиза называют количество
полисахаридов, перешедших в раствор во время варки, которое
выражают в процентах от потенциальных моносахаридов,
содержащихся в сырье. Для определения степени полноты гидролиза
предназначен метод, основанный на установлении
потенциального содержания моносахаридов в сырье (см. с. 39) и в
техническом лигнине, отмытом от моносахаридов.
Степень полноты гидролиза X вычисляют по формуле
п~л то,
X
п
где П — содержание потенциальных моносахаридов в абсолютно сухом
сырье, %; Л—количество моносахаридов, найденное в отмытом лигнине после
инверсии, % от абсолютно сухого сырья.
Определение общей кислотности
Для определения общей кислотности в гидролизатах и
сульфитных щелоках на рабочем месте нейтрализаторщицы
предназначен метод, основанный на реакции нейтрализации
минеральных и органических кислот едким
натром до pH 5,6—5,8. Величину общей
кислотности выражают в условных градусах
кислотности и обозначают К°. 1 градус
кислотности A К°) соответствует 1 мл
0,1 н. NaOH, израсходованного на
титрование 100 мл анализируемой
жидкости.
Аппаратура. Установка для
титрования в цеху (рис. 38). Установка
для титрования, которой
пользуются нейтрализаторщицы в цехе,
состоит из деревянной подставки, склянки
с титрованным раствором и бюретки. Для
быстрого заполнения бюретки до
отметки «0» применяют бюретку / с
приспособлением для автоматической установки
нулевого уровня жидкости. Чтобы
заполнить такую бюретку до отметки «0»,
достаточно нажать резиновую грушу 3.
Титрованный раствор заполнит бюретку
точно до «0», а его избыток
автоматически возвратится в бутыль. Для
лучшего, наблюдения за изменением окраски индикатора в
эквивалентной точке титрования внизу подставки вмонтирована
электрическая лампочка, которая освещает титруемый раствор сбоку
через матовое стекло 2.
Рис. 38. Установка для
титрования в цехе
123-
Реактивы и их приготовление. 1. Едкий натр, 0,1 н. раствор.
2. Смешанный индикатор по Андерсену. Приготовление.
Смешивают один объем 0,1%-ного спиртового раствора метиле-
новой синей с двумя объемами 0,1%-ного спиртового раствора
метилового красного. Окраска индикатора в кислой среде
розовая, в нейтральной — серо-зеленая, в щелочной — зеленая.
Переход окраски при pH 5,6—5,8. 3. Серная кислота, 0,01 н.
раствор. 4. Вода для разбавления пробы. Приготовление.
Дистиллированную воду в количестве 5—10 л (суточный
расход) наливают в склянку соответствующей емкости и
добавляют 5 мл смешанного индикатора. Если вода окрасится в
розовый цвет, то к ней прибавляют по каплям 0,01 н. раствор
едкого натра до появления зеленой окраски от одной капли
щелочи. Если вода будет иметь зеленую окраску после
прибавления индикатора, то ее нейтрализуют 0,01 н. раствором серной
кислоты до появления розовой окраски, а затем добавляют
1—2 капли 0,01 н. раствора едкого натра до появления зеленой
.окраски.
Ход анализа. В коническую колбу вместимостью 500 мл
вливают 200 мл воды для разбавления, 2 мл титруют 0,1 (или
-0,01) н. раствором едкого натра до появления зеленой окраски
жидкости. По бюретке определяют количество раствора едкого
натра, израсходованное на титрование.
Расчет. Кислотность анализируемой пробы выражают в градусах
кислотности и вычисляют по формуле
где X — кислотность пробы, К0; л — объем 0,1 н. раствора едкого натра,
израсходованный на титрование, мл; g — объем пробы, взятый на анализ, мл.
Воспроизводимость в среднем из трех определений ±0,06 мл 0,1 н.
раствора едкого натра.
Определение pH индикаторными бумажками со стандартной шкалой
Для быстрого определения pH в бесцветных и слабоокра-
шенных растворах предназначен метод, который основан на
сравнении окраски индикатора после погружения индикаторной
•бумажки в анализируемую жидкость с окраской полосок
стандартной шкалы.
Мешающие вещества. Красящие вещества.
Аппаратура. Набор индикаторных бумажек. Белая
фарфоровая пластинка.
Индикаторные бумажки выпускают для измерения
pH в пределах от 1 до 9 с различной точностью. В зависимости
от места расположения стандартной шкалы различают два типа
индикаторных бумажек (рис. 39). В типе / полоска
индикатора 1 и цветные полосы стандартной шкалы находятся на
одном и том же кусочке фильтровальной бумаги. На коробке или
124
на специальном вкладыше приведены значения pH для
стандартной шкалы. В типе // индикатор отделен от стандартной
шкалы, которая находится на внутренней стороне обложки.
При измерении pH в окрашенных жидкостях более точные
данные получают, применяя индикаторные бумажки типа /,
потому что в этом случае в анализируемую жидкость погружают
индикатор и стандартную шкалу
вместе. Изменение цвета
индикатора и цвета полос
стандартной шкалы за счет окраски
жидкости одинаково. При
употреблении индикаторных бумажек типа
// невозможно учесть степень
маскировки цвета индикатора
окраской анализируемой
жидкости.
Ход анализа. Из коробки
берут одну индикаторную бумажку
и на мгновение опускают ее
в анализируемую жидкость весь
конец бумажки, на котором
нанесен индикатор и стандартная
шкала (тип /) или только индикатор (тип //). Мокрую
бумажку кладут на белую фарфоровую пластинку или на белую
бумагу и визуально определяют, с цветом какой полосы
стандартной шкалы совпадает цвет индикатора. Если цвет
индикатора точно совпадает с цветом какой-нибудь полосы
стандартной шкалы, то pH анализируемой жидкости равен значению,
обозначенному на стандартной шкале. Если цвет индикатора
является промежуточным между цветом двух полос
стандартной шкалы, то pH анализируемой жидкости считают равным
¦средней величине значения pH, соответствующих этим полосам.
Рис. 39. Индикаторные бумажки со
стандартной шкалой
Определение pH потенциометрическим методом
Для измерения pH в водных растворах в пределах 1—13
с точностью ±0,1 предназначен метод, основанный на
способности некоторых электродов изменять свой потенциал в
зависимости от pH раствора, в который их погружают [37, 38, 39].
Такими электродами являются водородный, хингидронный,
стеклянный, сурьмяный и др. Для измерения pH раствора
составляют гальванический элемент из индикаторного электрода,
погруженного в анализируемый раствор, и электрода
сравнения, имеющего определенный постоянный потенциал. Электроды
соединяют электролитическим ключом. В качестве
индикаторного электрода для измерения pH в лабораторных условиях
применяют стеклянный электрод. Электродом сравнения
служит каломельный электрод. Разность потенциалов на концах
125
электродов такого элемента находится в прямой зависимости от
величины pH анализируемого раствора. Мембрана стеклянного
электрода обладает большим сопротивлением, поэтому ЭДС
элемента измеряют ламповым потенциометром,
отградуированным в единицах pH [40, 41, 42].
Многие заводы выпускают pH-метры со стеклянными
электродами разнообразных конструкций. К каждому
потенциометру прикладывают описание его устройства и
порядок работы на нем. Этими сведениями следует
руководствоваться при измерениях pH в
лаборатории.
Стеклянный эл~ектрод (рис. 40) состоит
из двух сосудов, вставленных один в другой.
Внешним сосудом служит трубка из специального
электропроводного стекла, на конце которого
имеется тонкостенный шарик /. В сосуд налит
0,1 н. раствор соляной кислоты, насыщенный
хлористым калием. Внутренним сосудом является
насыщенный каломельный электрод 2. В этом
сосуде находится ртуть 4, контакт 3 с которой
выведен в клемму стеклянного электрода.
Растворы обоих сосудов сообщаются через отверстие
диаметром 0,5 мм в верхней части внутреннего
сосуда.
Потенциал стеклянного электрода является
суммой трех потенциалов: потенциала,
возникающего на внешней поверхности стеклянного шарика,,
погруженного в исследуемый раствор, потенциала
на внутренней поверхности стеклянного шарика,
соприкасающегося с раствором соляной кислоты,
который имеет постоянный pH, и потенциала
каломельного электрода (внутреннего). Потенциал на
внутренней стенке шарика и потенциал
каломельного электрода постоянны, поэтому величина
потенциала стеклянного электрода зависит только от
величины потенциала на внешней поверхности
шарика, т. е. от величины pH анализируемого
раствора. Иногда внутренняя и внешняя поверхности
стеклянного шарика бывают различны по своим
электролитическим свойствам, поэтому возникает
потенциал асимметрии, который искажает результат измерения.
Эту ошибку измерения корректируют, проверяя показания
электрода при погружении его в буферные растворы с
известными pH.
Каломельный насыщенный электрод обычно
применяют в качестве электрода сравнения, потому что его
потенциал почти не изменяется во времени. На рис. 41
представлена конструкция каломельного электрода вместе с элек-
126
Рис. 40.
Стеклянный
электрод
тролитическим ключом. Электрод помещен в стеклянный сосуд,
в дно которого впаяна стеклянная трубка 5, а в боковую
стенку — платиновая проволока 4 с выведенным контактом
в клемму. Электрод состоит из ртути 3, покрытой слоем
кристаллической каломели 2 и слоем насыщенного раствора каломели
и хлористого калия /. Трубка 5, служащая электролитическим
ключом, заполнена агар-агаром, приготовленным на
насыщенном растворе хлористого калия. Потенциал электрода
возникает на поверхности металлической ртути, соприкасающейся
с насыщенным раствором каломели и хлористого
калия.
Насыщенный раствор хлористого калия
берут для того, чтобы уменьшить величину
диффузионного потенциала, возникающего на
границе соприкосновения жидкости, содержащейся
в каломельном полуэлементе, с жидкостью во
втором полуэлементе, и для того, чтобы
диффузия хлористого калия в испытуемый раствор не
снижала концентрацию хлористого калия в
каломельном полуэлементе. Растворимость
хлористого калия зависит от температуры, поэтому
величина потенциала каломельного электрода
тоже зависит от температуры. Эта
зависимость выражается формулой, по которой
величина потенциала каломельного электрода Ек=
= 0,2504 + 0,00065- (t— 18°C) по отношению к
потенциалу нормального водородного электрода,
при той же температуре.
Ртуть, употребляемую для зарядки
электрода, предварительно промывают слабой
азотной кислотой, потом дистиллированной
водой и сушат фильтровальной бумагой. Каломель перед
употреблением промывают 2 раза насыщенным раствором
хлористого калия для удаления сулемы. Для лучшего
контакта платины с ртутью платиновую проволоку электрода
перед зарядкой амальгамируют. В сосуд электрода наливают
2%-ный раствор азотнокислой ртути, слегка подкисленный
азотной кислотой, и опускают в него платиновую проволоку,
соединенную с положительным полюсом аккумулятора. Клемму
электрода присоединяют к отрицательному полюсу
аккумулятора B В). В течение 3—5 мин пропускают ток, затем сосуд
электрода хорошо промывают дистиллированной водой и
осуществляют зарядку электрода. Во избежание всползания
хлористого калия пробку и горлышко сосуда смазывают
вазелином. Электрод хранят, погрузив его конец в склянку с
раствором хлористого калия.
Рис. 41.
Каломельный
электрод
127
Определение pH при непрерывной нейтрализации гидролизата
Измеряют pH в потоке нейтрализата [43] элементом,
составленным из сурьмяного электрода и насыщенного каломельного
электрода (рис. 42). Сурьмяный электрод 1 изготовлен из
металлической чистой сурьмы в виде цилиндрика, запрессованного
в стержень из фторопласта. К сурьме припаян провод, который
вместе с фторопластовым
стержнем помещен в металлическую
трубку, укрепленную на крышке
нейтрализатора. Каломельный
электрод 2 — электрод
сравнения — находится в стеклянном
сосуде. Нижний конец трубки
сосуда электрода закрыт пробкой
из фильтровальной бумаги,
свернутой в плотный рулончик, и
погружен в сосуд
электролитического ключа 3. В дне сосуда
имеется длинная трубка, на которую
надета предохранительная
резиновая трубка 4, ее конец закрыт
пористым наконечником. Сосуд
и обе трубки заполнены
насыщенным раствором хлористого
калия.
Сурьмяно-каломельный
элемент подключен к электронному
потенциометру ЭПДЗ или
ЭПД12, шкала которого
отградуирована в единицах pH. Этим
потенциометром непрерывно
измеряют pH в потоке нейтрализата.
Одновременно показания
потенциометра с помощью электрического позиционного
устройства используются для автоматического регулирования подачи
известкового молока. Сурьмяный электрод надо чистить через
каждые 2—3 ч и периодически, 2—4 раза в сутки, проверять
показания потенциометра в лаборатории pH-метром со
стеклянным электродом.
Нейтрализатор
Рис. 42. Установка для измерения
pH прн непрерывной
нейтрализации
Определение серной и органических кислот
титрованием с различными индикаторами
Для определения отдельно серной кислоты и органических
кислот в гидролизатах предназначен метод, который основан
на реакции нейтрализации кислот щелочью. Сильные
кислоты-— серная и муравьиная — оттитровываются до pH 3—3,2
128
по индикатору метиловому оранжевому. Слабые органические
кислоты нейтрализуют до pH 8—8,2 по фенолфталеину.
Мешающие вещества. Соли слабых органических кислот,
которые образуются во время титрования, создающие буфер-
ность раствора. Определению серной кислоты мешает
муравьиная кислота.
Аппаратура. Установка для титрования.
Реактивы. 1. Едкий натр, 0,1 н. раствор. 2. Метиловый
оранжевый, 0,1%-ный водный раствор. 3. Фенолфталеин, 0,5%-ный
спиртовый раствор.
Подготовка проб. Гидролизат охлаждают до комнатной тем^
пературы и фильтруют через бумажный фильтр.
. Ход анализа. В коническую колбу вместимостью 250 мл
вливают 100 мл дистиллированной воды, 2 мл анализируемой
пробы и 2—3 капли метилового оранжевого. Полученный
раствор титруют 0,1 н. раствором едкого натра до появления
оранжевого окрашивания. Замечают количество раствора едкого
натра, пошедшее на титрование. Затем к оттитрованной
жидкости прибавляют 2—3 капли фенолфталеина и продолжают
титровать до появления розовой окраски раствора. Определяют
количество раствора едкого натра, пошедшее на титрование
с фенолфталеином.
Расчет. Концентрацию серной кислоты в гидролизате вычисляют по
формуле
v o0,0049 • 100
л. — ¦ ,
где X — концентрация серной кислоты в пробе, %; а — объем 0,1 н.
раствора едкого натра, израсходованный на титрование с метилоранжем, мл;
g — объем гидролизата, взятый на титрование, мл; 0,0049 — количество
серной кислоты, эквивалентное 1 мл 0,1 н. раствора едкого натра, г.
Концентрацию органических кислот в пробе вычисляют в пересчете на
уксусную кислоту по формуле
_ (g] — д) 0.006 • 100
'~~ g
где Xi —концентрация органических кислот в пересчете на уксусную кислоту, %;
п[ — объем 0,1 н. раствора едкого натра, израсходованный на титрование
с фенолфталеином, мл; 0,006—количество уксусной кислоты, эквивалентное
1 мл 0,1 н. раствора едкого натра, г.
Воспроизводимость в среднем нз трех определений ±0,1—0,15 мл 0,1 н.
раствора едкого натра.
Определение серной и органических кислот кондуктометрическим титрованием
Для определения серной и органических кислот в
гидролизате предназначен метод, который основан на измерении
электропроводности гидролизата во время нейтрализации кислот
раствором аммиака [44]. Электропроводность раствора при
нейтрализации сильных и слабых кислот изменяется по-разному.
9 Заказ № 142 129
При добавлении аммиака к смеси кислот сначала
нейтрализуются сильные кислоты — серная и муравьиная, что
сопровождается уменьшением электропроводности раствора. В
эквивалентной точке она достигает минимального значения. При
дальнейшем прибавлении аммиака нейтрализуются слабые
органические кислоты, что сопровождается повышением
электропроводности. В эквивалентной точке электропроводность
достигает максимального значения, дальнейшее прибавление
избытка аммиака не изменяет электропроводность.
Относительное значение электропроводности измеряют лого-
метром после прибавления каждой порции аммиака.
Полученные результаты изображают на графике в виде
кривой зависимости электропроводности от
количества прилитого аммиака, по которой
находят расход аммиака на нейтрализацию серной
кислоты и отдельно на нейтрализацию
органических кислот.
Мешающие вещества. Муравьиная кислота
мешает определению серной кислоты.
Аппаратура. Установка для кондуктометриче-
ского титрования.
Установка для кондукт о м етриче-
ского титрования (рис. 43) состоит из
логометра 4 и титровальной ячейки, в которую
опущены два платиновых электрода 2, ме-
Рис. 43. Установка для кондуктометрического титрования
шалка 1 и кончик бюретки. Логометр — электромагнитный
прибор переменного тока, предназначен для измерения
электропроводности раствора. Шкала прибора имеет 100 делений. Прибор
рассчитан на 220 В. Его показания почти не зависят от
колебаний питающего тока. Стрелка логометра не имеет
определенного положения, поэтому при отсутствии тока в цепи она
может находиться в любом месте шкалы.
Титровальной ячейкой служит стеклянный стакан. Мешалка
для перемешивания титруемого раствора приводится во
вращение мотором 3, укрепленным на штативе. Пластинчатые
платиновые электроды, соединенные с логометром проводами, для
сохранения постоянного расстояния между ними укреплены на
штативе в специальном держателе. Для периодической
проверки прибора в ячейку наливают 200 мл дистиллированной
воды, бюретку заполняют 2 н. раствором соляной кислоты,
включают логометр и мотор мешалки в электрическую сеть и
порциями по 0,5 мл прибавляют соляную кислоту из бюретки
в ячейку. После прибавления каждой порции записывают пока-
130
зания логометра. На основании полученных данных строят
график зависимости показаний логометра от количества
прилитой соляной кислоты. При правильной работе логометра на
графике получают прямую линию.
Реактивы. 1. Аммиак, 1 и. раствор. 2. Соляная кислота, 2н.
раствор. ,
Ход анализа. В ячейку вливают 100 мл гидролизата,
охлажденного до комнатной температуры, ставят ее иа подставку,
укрепленную на штативе, и поднимают так, чтобы электроды "и
мешалка были погружены в гидролизат. Бюретку заполняют
1 н. раствором аммиака и кончик ее помещают над ячейкой.
Логометр и мешалку включают в электрическую сеть и
начинают титрование. Раствор
аммиака прибавляют к гидролизату
порциями по 1 мл. После прибав-
ления каждой порции аммиака
записывают показание логометра.
Титрование прекращают тогда,
когда прибавление 3—4 порций
аммиака ие изменяют показания
логометра.
\
\
1
і
і
-
•Sei,
1
0
-о—
U в 12 16 20
1и. раствор аммиака, пя. '
Рис. 44. Кривые кондуктометриче-
ского титрования серной н
оргаиических кислот
Расчет. На основании полученных
данных строят график завнсямостн
показаний логометра от количества
прилитого аммиака, как показано на
рис. 44. Участок AB соответствует
изменению электропроводности прн
титровании серной кислоты, участок ВС —
при титровании органических кислот,
участок CD — прн добавлении избытка
аммиака. Количество 1 и. раствора
аммиака, пошедшее иа титрование серной кислоты, находят по абсциссе
точкно. Количество 1 и. раствора аммиака, израсходованное на титрование
серной н органической кислот, находят по абсциссе точки С. Разность между
значеннями абсцисс точек СнВ соответствует количеству аммиака,
израсходованному на титрование органических кислот.
Содержание серной кислоты или органических кислот в гидролизате
выражают в процентах серной кислоты и вычисляют по формуле
а0,049 • 100
" 1UQ •
где X — содержание серной (органических или их суммы) кислоты в
анализируемой пробе гидролиэата, %; а —объем 1 и. раствора аммиака, пошедший
иа титрование серной (органических нлн нх суммы) кислоты, мл; 0,049
—количество сериои кислоты в 1 мл 1 и. раствора, г.
Прн вычислении органических кислот в процентах уксусной кислоты
формула примет следующий вид:
X = 0,06а,
здесь 0,06 — количество уксусиой кислоты в 1 мл 1 и. раствора, г.
""°ржанне серной и органических кислот определяют с точностью
абс.
9*
131
Определение кальция кондуктометрическим титрованием
Для определения кальция в нейтрализате» бражке и барде,
где он содержится в виде солей серной и органических кислот,
предназначен метод, основанный на реакции осаждения
кальция в виде щавелевокислого кальция [44]
CaR+Na2C2O4-* CaC2O4+NaR.
v Во время титрования электропроводность титруемого
раствора остается почти без изменения до полного осаждения
кальция. При добавлении избытка щавелевокислого натрия
электропроводность начинает резко возрастать. Результат
анализа находят графическим методом.
Мешающие вещества. Сульфат-ионы.
Аппаратура. Установка для кондуктометрического
титрования (см. рис. 43).
Реактивы. 1. Уксуснокислый барий, 1 н. раствор.
Приготовление см. на с. 133. 2. Щавелевокислый натрий, 1 н. раствор.
Подготовка проб. Горячий нейтрализат фильтруют и
охлаждают до комнатной температуры.
Ход анализа. Определяют количество 1 н. раствора
уксуснокислого бария, которое необходимо прибавить к 100 мл
анализируемой пробы, чтобы полностью осадить сульфат-ионы.
С этой целью проводят анализ пробы на содержание гипса, как
описано на с. 133. Количество 1 н. раствора уксуснокислого
бария, израсходованное на реакцию с сульфат-ионами, которое
находят по графику, делят на 3, так как на титрование берут
300 мл пробы.
Определение кальция. В стакан, предназначенный
для титрования на установке, вливают 100 мл охлажденного
нейтрализата, включают мешалку, добавляют к пробе немного
кристаллического сернокислого бария, приливают
установленное количество 1 н. раствора уксуснокислого бария, которое
необходимо для полного осаждения сульфат-ионов (избыток
недопустим) и начинают титрование. К пробе прибавляют
1 н. раствор щавелевокислого натрия по 2 мл и замечают
показание логометра после прибавления каждой порции.
Титрование считают законченным, когда получат 4—5 показаний лого-
метра, указывающих на увеличение электропроводности от
прибавления каждой порции раствора щавелевокислого натрия.
Расчет. Полученные результаты наносят на график зависимости
показаний логометра от количества прилитого раствора щавелевокислого натрия.
Соединяя точки, получают две прямые линии (рис. 45), продолжения
которых пересекаются в точке, соответствующей концу титрования, т. е. моменту
полного осаждения кальция из раствора. Абсцисса этой точки указывает
количество щавелевокислого натрия, пошедшее иа осаждение всего кальция.
Концентрацию кальция в анализируемой пробе вычисляют по формуле
в0,02 • 100
X =
g
132
I
| го
Z it в
Растбор
а ю
где X — концентрация кальция в анализируемой пробе, %; а — объем 1 н.
раствора щавелевокислого натрия, пошедший на анализ, мл; g — объем пробы,
взятый на анализ, мл; 0,02 — количество кальция, соответствующее 1 мл 1 н.
раствора щавелевокислого натрия, г.
Кальций определяют с точностью ±2—3% отн.
Определение гипса кондуктометрнческим титрованием
Для определения концентрации гипса в нейтрализате и
спиртовой бражке предназначен метод, основанный на реакции
осаждения сульфат-ионов в виде сернокислого бария
CaSO4+Ва (СН3СООJ — BaSO4+ Ca (СН3СООJ.
При добавлении уксуснокислого бария к раствору гипса
электропроводность раствора почти не изменяется до полного
осаждения сульфат-ионов.
Прибавление избыточного количества
уксуснокислого бария вызывает резкое
повышение электропроводности раствора.
Результат анализа находят
графическим методом.
Мешающие вещества. Ионы
фосфорной и угольной кислот.
Аппаратура. Установка для кондук-
тометрического титрования (см. рис. 43).
Реактивы и их приготовление.
Уксуснокислый барий, 1 н.раствор.
Приготовление. Готовят
приблизительно 1 н. раствор. 127,5 г уксуснокислого
бария х. ч. растворяют в
дистиллированной воде, из которой удален углекислый газ путем
кипячения в течение 1—2 ч. Объем раствора доводят в мерной колбе
до 1 л, хорошо перемешивают и проверяют реакцию полученного
раствора лакмусовой бумажкой. Если реакция кислая, то
раствор нейтрализуют едким барием до получения нейтрального
раствора. Титр приготовленного раствора уксуснокислого бария
устанавливают кондуктометрическим титрованием раствора
сульфата натрия, который готовят путем нейтрализации 100 мл
0,1 н. раствора серной кислоты 1 н. раствором едкого натрия
в присутствии фенолфталеина. Полученный нейтральный
раствор сернокислого натрия подкисляют, добавляя к нему 1 мл
1 н. раствора уксусной кислоты, количественно переносят в
стакан для кондуктометрического титрования, объем раствора
доводят дистиллированной водой до 200 мл и оттитровывают
приготовленным раствором уксуснокислого бария.
Подготовка проб. Горячий нейтрализат фильтруют и
охлаждают до комнатной температуры.
Ход анализа. Мерным цилиндром отмеривают 300 мл ней-
трализата, выливают его в стакан для титрования, стакан
Рис. 45. Кривые
кондуктометрического титрования при
определении иона кальция
133
ставят на подставку в установке (см. рис. 43) и передвигают
так, чтобы электроды и мешалка оказались погруженными в
жидкость. Для ускорения кристаллизации сернокислого бария,
образующегося во время титрования, в качестве затравки
в пробу добавляют немного кристаллического сернокислого
бария, затем включают мешалку, записывают показания лого-
метра, прибавляют 1 мл уксуснокислого бария, ждут, когда
остановится стрелка логометра, и снова записывают показания
логометра. Раствор уксуснокислого бария прибавляют
порциями по 1 мл. После прибавления каждой порции записывают
показания логометра. Прибавление
раствора уксуснокислого бария
прекращают тогда, когда прильют 4—
5 порций, вызывающих увеличение
электропроводности титруемого
раствора.
Расчет. На основани полученных данных
строят график зависимости показании лого-
метра от количества прилитого уксуснокислого
бария. Получают две примые линии (рис. 46).
Точка пересечения продолжения этих линий
к'
/1
I
|
/
•**
г и в 8
соответствует моменту окончания титрования
Рис. 46. Кривые
кондуктометр ического титрования
при определении сульфат-
иона
Раствор В&(Сп3С00)г,МЛ сульфат-ионов. По абсциссе этой точки
находят количество уксуснокислого барня,
пошедшее иа реакцию с гипсом.
Концентрацию гипса и анализируемой пробе
иычисляют по формуле
х _ аО.068 • 100
~~ g
где X— концентрация гипса в анализируемой пробе, %; а—объем 1 н.
раствора уксуснокислого бария, пошедший иа титрование, мл; g — объем пробы,
взятый иа титрование, мл; 0,068 — количестио гнпса в 1 мл 1 н. раствора
CaSO4, г.
Содержание гипса определяют с точностью ±2—3% отн.
Определение свободных и связанных летучих оргаинческих кислот
Для определения отдельно свободных летучих органических
кислот, летучих органических кислот, связанных в соли,
и суммы свободных и связанных летучих кислот, которые
находятся в сульфитном щелоке, гидролизате, сусле, в сточной воде
и в других водных растворах, предназначен метод, основанный
на реакции нейтрализации летучих органических кислот
щелочью в присутствии индикатора фенолфталеина после их
отгонки из анализируемого раствора. Свободные летучие кислоты
отгоняют с водяным паром. Чтобы отогнать летучие
органические кислоты, связанные в соли, перед отгоном добавляют
фосфорную кислоту. Соли слабых кислот вступают в обменную
реакцию с фосфорной кислотой. Связанные органические
кислоты становятся свободными, их отгоняют с паром и титруют.
134
Количество связанных кислот можно определять в отгоне
титрованием или вычислять как разность между общим
количеством летучих органических кислот и количеством свободных
кислот. Аналогично общее количество летучих органических
кислот можно определять в отгоне или вычислять как сумму
свободных и связанных летучих органических кислот.
Мешающие вещества. Сернистая кислота, углекислота и
другие вещества, которые при нагревании разлагаются с
образованием SU2 или СОг.
Аппаратура. Установка для отгонки летучих веществ с
водяным паром (рис. 47). Установки для титрования йодом и
щелочью.
1
A
S3
Щ
nap
4-
Води
г
1/ \l
JE
Рис. 47. Установка для отгонки
летучих веществ с водяным
паром:
1 — изоляция асбестом; 2 — зажим
Реактивы и их приготовление. 1. Фосфорная кислота (H3PO4)
плотностью 1,12 г/см3. 2. Едкий натр, 0,1 н. раствор. 3. Иод,
0,01 н. раствор. 4. Индикатор фенолфталеин, 1%-ный раствор
в этиловом спирте. 5. Индикатор крахмал, 0,5%-ный раствор.
6. Синие лакмусовые бумажки.
Ход анализа. Летучие кислоты из пробы сульфитного щелока,
гидролизата или другого раствора отгоняют по-разному в
зависимости от поставленной задачи.
1. Определение свободных летучих кислот.
Для отгонки летучих кислот из пробы в колбу объемом 250 мл
(рис. 47) вливают 50 мл исследуемой жидкости, укрепляют ее
над электроплиткой и закрывают пробкой, в которую
вставлены барботер, отводная трубка и капельная воронка (вместо
термометра). Барботер соединяют с паровиком, но пар не
пропускают, кран 2 открыт. Отводную трубку соединяют с
холодильником. Для приема конденсата ставят мерную колбу
вместимостью 500—1000 мл. Когда содержимое колбы нагреется
до 90—95°С (появятся первые пузырьки), закрывают кран 2 и
отгоняют летучие кислоты с водяным паром. Отгонку считают
законченной, когда синяя лакмусовая бумажка, смоченная
каплей конденсата из холодильника, не изменит своего цвета.
135
Объем конденсата доводят до метки, прибавляя
дистиллированную воду, перемешивают и анализируют на содержание кислот
следующим образом.
В коническую колбу объемом 250 мл вливают 100 мл
конденсата и титруют 0,1 н. раствором едкого натра в присутствии
фенолфталеина до появления слабо-розовой окраски. По
бюретке определяют количество раствора едкого натра, пошедшее
на нейтрализацию кислот.
При анализе сульфитного щелока в отгоне кроме
органических кислот всегда содержится сернистая кислота. Для
введения поправки в результат анализа на SO2 в конденсате
определяют ее содержание. В коническую колбу вливают 100 мл
конденсата, титруют 0,01 н. раствором йода в присутствии
крахмала до появления синего окрашивания раствора (см. с. 61).
Результат анализа учитывают при вычислении концентрации
летучих органических кислот.
При анализе спиртовой бражки или последрожжевой
бражки, которые содержат очень много углекислого газа (СОг),
в конденсате может присутствовать углекислота. Для ее
удаления 100 мл конденсата помещают в колбу, закрывают пробкой,
в которую вставлен шариковый холодильник, и кипятят в
течение 30 мин. После удаления СОг, не открывая колбы, убирают
электроплитку, холодильник через верхний конец споласкивают
дистиллированной водой (воду собирают в ту же колбу).
Из колбы вынимают пробку с холодильником, содержимое
колбы охлаждают до комнатной температуры и титруют 0,1 н.
раствором едкого натра с фенолфталеином.
2. Определение связанных летучих
органических кислот. После отгона свободных кислот из пробы,
не разбирая установки и не отключая пара, ставят для приема
конденсата другую чистую мерную колбу. В реакционную
колбу через капельную воронку вливают 10 мл фосфорной
кислоты и продолжают отгон до отрицательной реакции на кислоту
по синей лакмусовой бумажке. Объем конденсата доводят до
метки на колбе, прибавляя дистиллированную воду,
перемешивают 100 мл конденсата и титруют 0,1 н. раствором едкого
натра.
При анализе сульфитного щелока конденсат проверяют на
присутствие сернистой кислоты, и если она обнаружена, ее
определяют, как описано выше. При анализе бражек и других
жидкостей, содержащих много углекислых солей, конденсат
проверяют на присутствие углекислоты, и если она обнаружена, ее
удаляют кипячением.
3. Определение суммы свободных и
связанных кислот. В реакционную колбу вливают 50 мл
анализируемой жидкости, колбу включают в установку для отгонки
с водяным паром, вливают через капельную воронку 10 мл
фосфорной кислоты (плотность 1,12 г/см3), содержимое колбы
133
подогревают, включают пар и ведут отгон кислот как описано
выше. Собранный конденсат анализируют на содержание
кислот, титруя 100 мл конденсата ОД н. раствором едкого натра.
При анализе сульфитного щелока в конденсате определяют
сернистую кислоту, а при анализе бражек из конденсата
удаляют углекислоту кипячением (см. выше).
Расчет. Содержание в анализируемой пробе свободных или связанных
летучих органических кислот, а также их сумму выражают в процентах ук-
сусиой кислоты и вычисляют по формуле
a0.006V100
Л. —
где X — концентрация свободных (связанных или их суммы) летучих
органических кислот в пересчете иа уксусную кислоту, %; а — объем 0,1 н. раствора
«дкого натра, пошедший на титрование, мл; V — объем конденсата,
собранного при отгоике свободных (связанных или их суммы) летучих кислот, мл;
Vi —объем конденсата свободных (связанных или их суммы) летучих кислот,
взятый на титрование, мл; g — объем пробы, взятый на анализ, мл; 0,006 —
количество уксусной кислоты, эквивалентное 1 мл 0,1 н. раствора едкого
натра, г.
При анализе сульфитного щелока вводится поправка на сернистую
кислоту.
Содержание летучих органических кислот вычисляют по следующей
формуле:
v (a-O.laOO.OOoVlOO
V\g
где 0,l?i — объем 0,1 н. раствора йода, пошедший на титрование
конденсата, мл. Остальные обозначения те же.
Воспроизводимость в среднем из трех титрований ±0,05 мл 0,1 н. NaOH.
Определение левулииовой кислоты хроматографическим методом
Для определения концентрации левулиновой кислоты в гид-
ролизате предназначен метод, основанный на реакции
левулиновой кислоты с фенолфталеинатом натрия после ее выделения
с помощью хроматографии на бумаге.
Аппаратура. Установка для хроматографирования на
полосках бумаги под колпаком (см. рис. 24). Микропипетка со
специальным приспособлением (см. рис. 27). Водяная баня.
Установка для элюирования (см. рис. 32). Микробюретка.
Реактивы и их приготовление. 1. Хроматографическая бумага
марки М или Б. 2. Растворитель — смесь н-бутанола и 1,5 н.
раствора аммиака A:3). 3. Проявитель — солянокислый
раствор 2,4-динитрофенилгидразина. 4. Фенолфталеинат натрия,
0,01 н. раствор. Получение фенолфталеината
натрия. В стакан вместимостью 200 мл вливают 100 мл 20%-ного
раствора углекислого натрия, всыпают 5 г фенолфталеина и
кипятят. Полученный раствор упаривают в выпарительной чашке
на водяной бане досуха. Осадок многократно обрабатывают
95%-ным этиловым спиртом для извлечения фенолфталеината
натрия. Спиртовий раствор фенолфталеината натрия выпари-
137
вают досуха, осадок обрабатывают спиртом и полученный
раствор вновь упаривают досуха. Сухой фенолфталеинат натрия
сохраняют в герметически закрытой склянке.
Приготовление 0,01 н. раствора ф е н о л ф т а л е и н а т а натрия.
0,35 г фенолфталеината натрия растворяют в дважды
перегнанной воде, освобожденной от углекислоты, объем раствора
доводят в мерной колбе до 1 л и оставляют стоять на сутки. Если
за это время выпадет осадок, его отфильтровывают. Для
предотвращения разложения раствора при длительном хранении
в него добавляют 2—3 капли 0,1 н. раствора щелочи. Титр
приготовленного раствора устанавливают по 0,01 н. раствору
соляной кислоты.
Подготовка проб. 1. Перед анализом пробу нейтрализуют
сначала известковым молоком до pH 5. Выпавший осадок
отфильтровывают и прибавляют раствор аммиака до pH 6—7.
2. Цилиндром отмеривают 100 мл нейтрализованного гидроли-
зата и 25—30 мл его наливают в фарфоровую чашку
вместимостью 50 мл. Чашку ставят на кипящую водяную баню для
упаривания. По мере упаривания жидкости в чашку из цилиндра
добавляют пробу. Когда 100 мл пробы будут упарены до объема
меньше 25 мл, их количественно переносят в мерную колбу
вместимостью 25 мл и объем доводят до метки, прибавляя
дистиллированную воду. Выпавший осадок отфильтровывают.
Фильтрат—анализируемая проба, упаренная в 4 раза.
Ход анализа. Хроматографирование. Для выделения
левулиновой кислоты применяют нисходящее
хроматографирование на полосках хроматографической бумаги 3x35 см (см.
с. 84). Отмеривают и наносят пробу на полоски бумаги
микропипеткой с приспособлением, как вписано на с. 81. Пробу
наносят на 5—7 полосок по 0,1—0,2 мл на каждую полосу в
несколько приемов. Полоски с пробой помещают в камеру и хро-
матографируют, употребляя в качестве растворителя смесь
н-бутанола и аммиака. Через 24 ч хроматографирование
прекращают. Хроматограммы вынимают из камеры, высушивают в тяге
и одну проявляют. Для проявления хроматограмму
опрыскивают солянокислым раствором 2,4-динитрофенилгидразина и
выдерживают 90 мин в сушильном шкафу при 80сС. На светлом
фоне хроматограммы при наличии левулиновой кислоты
обнаруживают оранжевое пятно. Элюацию левулиновой кислоты из не-
проявленных хроматограмм проводят, как описано на с. 89.
Элюат от нескольких хроматограмм соединяют вместе и титруют
из микробюретки 0,01 н. раствором фенолфталеината натрия.
Одновременно в качестве холостого опыта проводят элюацию-
из кусочка непроявленной хроматограммы, равного по своему
размеру кусочку хроматограммы с левулиновой кислотой,
который отрезают на участке без пятен между левулиновой
кислотой и фронтом растворителя. Полученный элюат также титруют
раствором фенолфталеината натрия.
133
Расчет. Содержание левулииовои кислоты в пробе вычисляют по формуле
(а-б) 0.00116- 100
nmg
X =
тде х концентрация левулииовои кислоты в пробе, %; а — объем 0,01 н.
раствора фенолфталенната натрия, пошедший иа титрование левулнновой
кислоты, мл; б — то же в холостом опыте; п — концентрирование пробы при
упаривании;' т — число хроматограмм, элюаты которых были объединены; g —
объем упаренной пробы, нанесенный на хроматограмму, мл; 0,00116 — титр
0,01 н. раствора левулиновой кислоты, г/мл.
Левулнновую кислоту определяют с точностью ±4—5% отн.
1'
Jl
го ы во ео im
Номер порции
Определение органических кислот хроматографическим методом
Для раздельного определения таких низкомолекулярных
жирных кислот, как муравьиная, уксусная, пропионовая и
масляная, в гидролизате, сульфитном щелоке, бражке и сточной воде
f45] предназначен метод,
основанный на выделении каждой
органической кислоты из смеси кислот
<; помощью хроматографии на
колонке, заполненной силикагелем,
и определении количества
выделенной кислоты титрованием
раствором щелочи. При анализе гидроли-
затов и сульфитных щелоков хрома-
тографируют конденсаты,
полученные после отгонки кислот из пробы
с помощью водяного пара (см.
с. 135).
Мешающие вещества. Летучие
минеральные кислоты.
Аппаратура. Установка для
отгонки летучих веществ с водяным
паром (см. рис. 47). Колонка для
хроматографирования (рис. 48).
Водяная баня.
Подготовка колонки для хр о м а то графиро-
вания. Для разделения кислот употребляют колонку высотой
45 см с внутренним диаметром 10—11 мм. Перед заполнением
колонки силикагель измельчают, просеивают, отбирают фракцию,
состоящую из зерен размером 0,15—0,18 мм (80—100 меш),
которую заливают Юн. раствором соляной кислоты, хорошо
перемешивают и оставляют стоять на сутки, после чего кислоту
сливают и силикагель промывают водой до отрицательной
реакции на кислоту по метиловому оранжевому. Промытый
силикагель помещают тонким слоем на стекло и сушат при 150°С
в сушильном шкафу в течение 8—10 ч. Таким же образом
регенерируют силикагель после его многократного использования.
Рнс. 48. Определение
органических кислот хромато-
графнческнм методом:
а — колонка для хроматографи-
роваиия; 6 — кривые элюации
кислот из искусственной смеси;
С, — муравьиная кислота; С2 —
уксусная кислота; Са — пропио-
иовая кислота; С, — масляная
кислота
139
Для загрузки колонки берут 10—12 г сухого силикагеля,
помещаемого в фарфоровую чашку и прибавляют к нему
небольшими порциями при постоянном помешивании 10—11 мл воды
до получения массы, частицы которой слипались бы между
собой, но не прилипали к стенкам чашки. Увлажненный силика-
гель сразу заливают бензолом для предохранения от влияния
воздуха и тщательно перемешивают.
На ситчатое дно колонки помещают кусок стеклянной ваты,
на которую небольшими порциями кладут приготовленную массу
увлажненного силикагеля с бензолом. Легким постукиванием
по стенке колонки уплотняют слой силикагеля и способствуют
удалению пузырьков воздуха. Высота слоя увлажненного
силикагеля должна составлять 30 см, он всегда должен быть покрыт
бензолом для предохранения от влияния воздуха. На силикагель
в колонке кладут кусок стеклянной ваты так, чтобы он
находился на 1—1,5 см выше уровня бензола, и на него помещают
0,5 г сухого силикагеля.
Реактивы и их приготовление. 1. Едкий натр, 1 н. раствор.
2. Серная кислота, 1 н, раствор. 3. Фосфорная кислота
плотностью 1,12 г/см3. 4. Бензол ч. д. а. 5. Растворы бутанола в
бензоле: 2%-ный Б2, 5%-ный Es, 10%-ный Б10, 15%-ный Bis»
20%-ный Б2о, 25%-ный Б25, 30%>-ный Б3о. 6. Этиловый спирт
нейтральный. 7. Едкое кали, 0,02 н. спиртовый раствор. 8.
Фенолфталеин, 1%-ный спиртовый раствор. 9. Силикагель марки КСК
№ 2 или № 5.
Подготовка проб. Летучие органические кислоты выделяют
из анализируемой пробы путем отгонки с водяным паром (см.
стр. 135).
Ход анализа. Для анализа берут такое количество пробы,
чтобы в нем содержалось 1—2 г летучих кислот в пересчете на
уксусную кислоту. Отгон нейтрализуют 1 н. раствором едкого
натра по фенолфталеину, добавляют избыток щелочи в
количестве 1—2 мл и выпаривают на водяной бане досуха. Сухой
остаток — натриевые соли летучих кислот растворяют в 1 н,
растворе серной кислоты, добавляя ее немножко больше, чем было
добавлено 1 н. раствора едкого натра до выпаривания. Раствор
количественно переносят в мерную колбу вместимостью 50 мл,
объем его доводят до метки, прибавляя дистиллированную воду,
и перемешивают (раствор I).
Для хроматографирования берут 1 мл полученного раствора I
летучих кислот и равномерно смачивают им слой сухого
силикагеля в колонке. Постукивая по колонке, добиваются опускания
ваты с силикагелем в слой бензола, который покрывает
увлажненный силикагель, и начинают хроматографирование. В
колонку через капельную воронку приливают растворитель с такой
скоростью, чтобы за 2—3 мин в приемнике собралось 2 мл
элюата. Приемники с отметкой 2 мл меняют как только в них
набирается 2 мл элюата. Собранный элюат количественно с по-
140
мощью 2—3 мл этилового спирта переносят в колбу объемом
25 мл и титруют 0,02 н. спиртовым раствором едкого кали в
присутствии фенолфталеина.
Сначала в колонку прибавляют чистый бензол, который
элюирует капроновую кислоту, а затем валериановую кислоту.
После окончания элюации упомянутых кислот, о чем
свидетельствует отсутствие кислот в элюате, в колонку прибавляют
раствор бутанола в бензоле Б2, который элюирует масляную
кислоту. Как только в последней порции элюата не будет кислоты,
растворитель Б2 заменяют растворителем Б5, который извлекает
пропионовую кислоту. После Б5 пропускают Бю, затем Біб,
которые не извлекают кислот. Их пропускание необходимо для
постепенного выравнивания концентраций растворителя. Затем
пропускают растворитель Б2о, который элюирует уксусную
кислоту. После окончания извлечения уксусной кислоты пропускают
растворитель Б25, который ничего не элюирует, а потом
растворитель Бзо, который элюирует муравьиную кислоту.
Таким образом, каждая кислота будет содержаться в
нескольких порциях элюата. Для нахождения ее количества надо
суммировать расход щелочи на титрование всех порций элюата,
которые ее содержат. За время анализа набирается до 100
порций элюата. Иногда объединяют первые порции элюата той или
иной кислоты для сокращения числа анализов. Для каждой
кислоты вычисляют количество элюата, в котором она была
найдена. Титрованием чистого растворителя, примененного для
элюации, определяют количество 0,02 н. раствора едкого кали,
которое при титровании элюата кислоты расходуется на
реакцию с растворителем (холостой опыт).
После окончания хроматографирования колонку сразу
подготавливают для проведения следующего анализа. Для этого
из колонки вынимают верхний слой ваты с силикагелем, а
оставшийся увлажненный силикагель промывают, пропуская через
него сначала по 3—5 мл каждого растворителя в порядке
убывания концентрации в них бутанола — Б25, Б2о, Б]5 и т. д., а
потом 25—30 мл бензола.
Расчет. Содержание каждой кислоты в анализируемой пробе вычисляют
по формуле
(а-бJМ5О- 100
где X — содержание кислоты в анализируемой пробе, %; а — объем 0,02 н.
KQH, израсходованный на титрование всех порций элюата, которые
содержали определяемую кислоту, мл; б — объем 0,02 н. КОН, израсходованный
на титрование чистого растворителя в количестве равном объему элюата
определяемой кислоты, мл; М — молекулярная масса определяемой кислоты;
g — объем анализируемой пробы, взятый для отгонки летучих кислот, мл;
ІО5 — количество определяемой кислоты, эквивалентное 1 мл 0,02 н. раствора
едкого кали, г; 50—объем раствора 1, мл; gi — объем раствора I, взятый
для хроматографирования, мл.
Летучие органические кислоты определяют с точностью ±5—10% отн-
141
Определение фурфурола
Для определения фурфурола в нейтрализате, субстрате и
барде предназначен метод, основанный на выделении
фурфурола из пробы отгонкой с водяным паром и определении его
в конденсате бромид-броматным методом (см. с. 42).
Мешающие вещества. Метилфурфурол и другие летучие бро-
мируемые вещества. Пентозы и уроновые кислоты, которые во
время отгонки частично разлагаются с образованием
фурфурола.
Аппаратура. Установка для отгонки летучих веществ с
водяным паром (см. рис. 47). Установка для титрования и
конические колбы вместимостью 500 мл с притертыми пробками.
Песочные часы на 4 мин.
Реактивы и их приготовление. 1. Соляная кислота, 12%-ный
раствор. 2. Аммоний молибденовокислый, 2,5%-ный раствор.
3. Бромид-бромат калия, 0,05 н. раствор. 1 л раствора содержит
1,392 г бромноватистокислого калия (КВгО3) и 10 г бромида
калия (КВг). Раствор хранят в склянке из темного стекла с
притертой пробкой. 4. Калий йодистый (KI), 10%-ный раствор
5. Серноватистокислый натрий (Na2S2O3), 0,01 н. раствор.
Приготовление. Раствор готовят из фиксанала, употребляя
дистиллированную воду, из которой углекислый газ удален
кипячением. 6. Крахмал, 0,5%-ный раствор. 7. Уксуснокислый
анилин. Приготовление. В капельницу вливают отмеренные
цилиндром 5 мл ледяной уксусной кислоты и 5 мл свежеперег-
нанного анилина. После хорошего перемешивания жидкостей
получают уксуснокислый анилин. Анилин перегонят в установке,
собранной на шлифах.
Подготовка проб. Пробы, содержащие пентозы или уроновые
кислоты перед анализом нейтрализуют до pH 6,5—7 содой.
Ход анализа. В круглодонную колбу вместимостью 500 мл
вливают 100 мл анализируемой жидкости, закрывают ее
пробкой, в которую вставлены барботер, отводная трубка и
термометр. Барботер соединяют с паровиком, а отводную трубку с
холодильником (см. рис, 47). Приемником конденсата служит
мерная колба вместимостью 250 мл. Жидкость в реакционной
колбе подогревают на электроплитке до 95—97°С, и, закрывая
винтовой зажим 2 направляют водяной пар в реакционную колбу.
Во время отгонки следят, чтобы уровень жидкости в колбе был
постоянным. Скорость отгонки поддерживают равной 80—90 мл
за 10 мин. Отгонку фурфурола продолжают до отрицательной
реакции конденсата на фурфурол (отсутствие розового
окрашивания от последней капли конденсата на фильтровальной
бумажке, смоченной уксуснокислым анилином). После окончания
отгонки объем конденсата доводят до метки дистиллированной
водой. Содержимое колбы тщательно перемешивают и
определяют в нем фурфурол бромид-броматным методом.
142
Цилиндром отмеривают 35 мл дистиллированной воды,
выливают их в коническую колбу вместимостью 250 мл, приливают
к ним 3 мл 12%-ной соляной кислоты, 25 мл конденсата, 2 мл
2,5%-ного раствора молибденовокислого аммония и 5 мл 0,05 н.
раствора бромид-бромата калия. Колбу закрывают притертой
пробкой, ставят на белую поверхность и замечают время
появления желтого окрашивания жидкости, которое обычно
наступает через 10—15 с. С момента появления желтого
окрашивания смесь должна стоять точно 4 мин (песочные часы). Затем
добавляют 2 мл 10%-ного раствора йодистого калия и
выдерживают еще 4 мин. Выделившийся йод оттитровывают 0,01 н.
раствором гипосульфита натрия до слабо-желтого окрашивания,
добавляют 2—3 капли раствора крахмала и продолжают
титровать до полного исчезновения синей окраски раствора.
Параллельно проводят холостой опыт. В коническую колбу вливают
60 мл дистиллированной воды, 3 мл 12%-ной соляной кислоты,
2 мл 2,5%-ного раствора молибденовокислого аммония и 5 мл
0,05 н. раствора бромид-бромата калия. Ждут 4 мин, добавляют
2 мл 10%-ного раствора йодистого калия и после 4 мин
ожидания титруют раствором гипосульфита натрия с индикатором
крахмал.
Расчет. Содержание фурфурола в исследуемой пробе вычисляют по
формуле
(а - б) У0.00048 . 100
х ш '
где X — содержание фурфурола в пробе, %; а — объем раствора
гипосульфита, израсходованный на титрование в холостом опыте, мл; б — объем
раствора гипосульфита, израсходованный на титрование пробы, мл; V — объем
конденсата, отогнанный из пробы, мл; g — объем пробы, взятый на анализ,
мл; gi — объем конденсата, взятый на титрование, мл; 0,00048—количество
фурфурола, эквивалентное 1 мл 0,01 н. раствора гипосульфита натрня, г.
Воспроизводимость в среднем из трех определений ±0,05 мл 0,01 и.
раствора гипосульфита натрия.
Предостережение. 1. Если проба содержит пентозы н уроновые кислоты,
то отгонять фурфурол надо при pH около 7. 2. Воздух помещения, где
делают анализ, не должен содержать фурфурол. Присутствие фурфурола в
воздухе определяют бумажкой, смоченной уксуснокислым анилином: оиа
розовеет, если в воздухе есть фурфурол 3. Температура в реакционной колбе во
время отгонки фурфурола должна быть не выше 102°С.
Определение показателей биологической доброкачественности
Биологическая доброкачественность нейтрализованного гид-
ролизата определяется по следующим показателям: 1)
содержанию редуцирующих несахаров в процентах от общих РВ; 2)
содержанию веществ, осаждающихся после продувки нейтрализата
воздухом, выраженное в миллилитрах на 1 л нейтрализата или
в миллилитрах на 100 г общих РВ; 3) содержанию бромируе-
мых веществ (в пересчете иа фурфурол) в процентах от
общих РВ.
143
Токсические вещества, находящиеся в гидролизатах, не
идентифицированы, поэтому оценку биологической
доброкачественности делают на основании результатов групповых анализов.
Значения показателей зависят от сырья, от режима гидролиза и
от степени очистки нейтрализатов. Нормативы на показатели
биологической доброкачественности не установлены. В качестве
примера можно привести только значение показателя
содержания редуцирующих несахаров. При переработке хвойных пород
древесины в гидролизате допустимо наличие редуцирующих
несахаров в количестве 5—6% от общих РВ. Увеличение их
концентрации до 10% приводит к снижению спир-
тообразующей способности дрожжей на 10—
20% и к понижению выхода кормовых
дрожжей на 15—25%.
Метод определения биологической
доброкачественности состоит из следующих
анализов: 1) определения редуцирующих веществ
эбулиостатическим методом до и после
осаждения редуцирующих несахаров; 2)
определения веществ, которые выпадают в осадок
после 3 ч отстоя осветленного нейтралнзата
продутого воздухом; 3) определения броми-
руемых веществ бромидброматным методом.
Чем ниже определяемые показатели, тем
выше биологическая доброкачественность
нейтрализатора.
Аппаратура. Установка для определения
редуцирующих веществ (см. рис. 11—12).
Установка для продувки нейтрализата воздухом
и отстойник Лысенко (рис. 49). Установка
для титрования.
Реактивы и их приготовление. 1. Основной уксуснокислый
свинец, насыщенный раствор. Приготовление см. на с. 57.
2. Растворы для эбулиостатического метода (см. с. 35). 3.
Щавелевокислый натрий, насыщенный раствор. 4. Соляная кислота,
12—13%-ный раствор. 5, Бромид-бромат калия, 0,05 н. раствор.
Приготовление. 1,392 г КВгО3 и 10 г КВг растворяют в
дистиллированной воде и объем доводят до 1 л в мерной колбе.
6. Аммоний молибденовокислый, 2,5%-ный раствор. 7. Калий
йодистый, 10%-ный раствор. 8. Гипосульфит натрия, 0,05 н.
раствор. 9. Крахмал, 0,5%-ный раствор.
Ход анализа. 1. Опр еделение редуцирующих
веществ (общихРВ). В мерную колбу вместимостью 200—250мл
вливают 10 мл нейтрализата, добавляют дистиллированную воду
до метки на колбе, раствор перемешивают, вливают в бюретку
и титруют им меднощелочной раствор в эбулиостате по I
варианту (см. с. 37). Вычисляют концентрацию общих РВ в
анализируемой пробе.
144
Рис. 49. Установка
для продувки
нейтрализата
воздухом / и отстойник
Лысенко 2
2. Определение редуцирующих веществ
после осаждения несахаров. В стакан вместимостью
150—200 мл вливают 10 мл нейтрализата и 50 мл
дистиллированной воды. Раствор нагревают до 90—95°С (до появления
первых пузырьков),снимают с электроплитки и при постоянном
перемешивании прибавляют к нему по каплям 1 мл раствора
основного уксуснокислого свинца. После охлаждения до комнатной
температуры содержимое стакана количественно переносят в
мерную колбу вместимостью 100 мл, объем раствора доводят до
метки дистиллированной водой, взбалтывают и фильтруют через
бумажный фильтр. Получают фильтрат I. Проверяют полноту
осаждения. На часовое стекло наливают 1 мл фильтрата I и
добавляют 1 каплю раствора основного уксуснокислого свинца.
Раствор должен оставаться прозрачным, в противном случае
надо повторить анализ, прибавив для осаждения 1,5 мл
раствора основного уксуснокислого свинца. В мерную колбу
вместимостью 100 мл вливают 50 мл фильтрата I и 5 мл раствора ок-
салата натрия. Объем раствора доводят до метки, прибавляя
дистиллированную воду. Выпавший осадок оксалата свинца
отфильтровывают и в фильтрате II проверяют полноту удаления
свинца. На часовое стекло наливают 1 мл фильтрата II и 3—5
капель раствора оксалата натрия. Раствор должен оставаться
прозрачным. Если в растворе появилась едва заметная муть,
осаждение надо повторить, добавив к фильтрату II большее
количество раствора оксалата натрия. Избыток оксалата
натрия не влияет не реакцию окисления редуцирующих веществ
меднощелочным раствором, а свинец изменяет результат
анализа.
В фильтрате II определяют содержание редуцирующих
веществ эбулиостатическим методом.
3. Определение оседающих веществ. В
делительную воронку вместимостью 1 л наливают 500 мл
анализируемой пробы, закрывают пробкой, в которую вставлен барботер
н отводная трубка (рис. 49). Барботером служит стеклянная
трубка, на конце которой имеется шарик с отверстиями.
Отводную трубку присоединяют к включенному водоструйному
насосу. Зажимом на тройнике регулируют вакуум, чтобы не
допустить большого пенения и возможных потерь пробы. Воздух
пропускают в течение 1 ч. Затем отключают насос, жидкость
хорошо перемешивают и переливают в отстойник. При
отстаивании отстойник периодически вращают в гнезде штатива, чтобы
осадок, осевший в конусную часть, переместился в
градуированный отросток. Через 3 ч отстоя замеряют объем осадка в
миллилитрах, умножают на 2 и получают количество веществ,
оседающих при отстое продутого нейтрализата в миллилитрах на литр
(мл/л). Определять эти вещества весовым методом нельзя,
потому что они состоят из жидких и твердых веществ. Жидкие
10 Заказ № 142 ]45
вещества проходят через фильтр, а некоторые твердые вещества
при высушивании при 105°С улетучиваются или обугливаются.
4. Определение бронируемых веществ
(летучих и нелетучих). В мерную колбу вместимостью 100 мл
вливают 2 мл нейтрализата, прибавляют дистиллированную воду до
метки на колбе. Раствор перемешивают. В коническую колбу
объемом 250 мл вливают 10 мл дистиллированной воды, 10 мл
приготовленного раствора нейтрализата, 5 мл соляной кислоты,
5 мл раствора молибденовокислого аммония и 10 мл раствора
бромид-бромата калия. Колбу закрывают стеклянной пробкой и
ставят в темное место на 24 ч. По истечении указанного времени
в колбу вливают 5 мл раствора йодистого калия, ждут 4 мин,
а затем выделившийся йод оттитровывают раствором
гипосульфита натрия в присутствии крахмала. Параллельно делают
холостой опыт, в котором вместо пробы вливают 10 мл воды.
Расчет. Содержание летучих и нелетучих бромируемых веществ в
анализируемой пробе вычисляют в пересчете на фурфурол по формуле
(а - б) 0.00024 • 5/гЮО
где X — содержанне бромируемых веществ в пробе, %; а — объем 0,05 н.
раствора гипосульфита, израсходованный на титрование в холостом опыте, мл;
6 — объем 0,05 н. раствора гипосульфита натрия, израсходованный на
титрование пробы, мл; g — объем разбавленной пробы, взятой на анализ, мл; п —
разведение; 0,00024 • 5 — количество фурфурола эквивалентное 1 мл 0,05 и.
раствора гипосульфита натрия при бромировании до тетрабромфурфу-
рола, г.
Показатель — содержанне редуцирующих несахаров в процентах от общнх
РВ вычисляют по формуле
где а — содержание общнх РВ в пробе, %; б— содержание общих РВ
в пробе после осаждении несахаров, %.
Показатель — содержание взвешенных веществ, осаждающихся после
продувки нейтралнзата воздухом в течение 1 ч, вычисляют в миллилитрах на 1 л
нейтрализата нли в миллилитрах на 100 г общих РВ по формуле
v\m
Х
~~ 10a '
где V — объем осадка, мл/л; а — концентрация общих РВ, %.
Показатель — содержанне бромнруемых веществ в пересчете на фурфурол
вычисляют в граммах на 100 г общих РВ по формуле
<яоо
где а — концентрация общих РВ, %; б — содержание бромнруемых вешеств
в пробе, % фурфурола.
146
ГЛАВА 5. ПОЛУЧЕНИЕ ЭТИЛОВОГО СПИРТА
Гидролизат и сульфитный щелок после нейтрализации,
осветления, охлаждения и добавки питательных солей называют
суслом. Гексозные сахара, содержащиеся в сусле, с помощью
микроорганизмов — спиртовых дрожжей — сбраживают на
этиловый спирт. В результате получают спиртовую бражку, из
которой в аппаратном отделении извлекают, очищают и
концентрируют этиловый спирт.
Процесс образования спирта очень сложен. Он состоит из
целого ряда биохимических превращений гексоз в клетках
дрожжей, поэтому принято писать суммарное уравнение протекающих
реакций, которое предложил Гей-Люссак:
С6Н12О6-*2С2Н5ОН+2СО2.
Скорость брожения и количество спирта, образующееся из
100 кг гексоз, зависят от качества сусла, от расы дрожжей, их
физиологического состояния и от наличия инфекции. Качество
сусла обусловливается концентрацией и составом гексоз, т. е.
сбраживаемых Сахаров, наличием питательных солей (фосфора,
азота, калия) и концентрацией токсических веществ, которые
угнетают дрожжи или снижают их спиртообразующую
способность. Кроме того, для нормальных условий жизнедеятельности
дрожжей сусло должно иметь определенные pH и температуру.
Контроль за работой бродильной батареи состоит в том, чтобы,
определив качество и количество дрожжей и сбраживаемых са-
харов в сусле, расчитать скорость потока сусла, при котором
будет обеспечен максимальный выход спирта из сброженного
сахара при полном отброде, и проследить за выполнением
установленного технологического регламента.
В отделении ректификации в бражных колоннах выделяют
спирт из бражки вместе с другими летучими веществами,
которые содержались в сусле (метанол) и образовались во время
брожения, как побочные продукты (сивушное масло, альдегиды,
эфиры и т. д.). Спиртовый конденсат после бражной колонны
направляют для очистки и укрепления на эпюрационную,
ректификационную и метанольную колонны. Контроль в
ректификационном отделении состоит в определении качества готового
спирта и спиртовых конденсатов на различных стадиях очистки,
а также в определении потерь спирта с бардой, лютером и через
воздушники.
Определение сбраживаемых Сахаров химическим методом
Для определения сбраживаемых Сахаров (гексоз) в
нейтрализованных гидролизатах и сульфитных щелоках [46]
предназначен метод, который основан на определении гексоз по разности
между количеством редуцирующих веществ, найденных после
10* 147
удаления редуцирующих несахаров, и количеством пентоз и
фурфурола. Редуцирующие несахара удаляют из пробы
осаждением основным уксуснокислым свинцом. При анализе гидролиза-
тов пентозы и фурфурол можно определять в пробе без удаления
редуцирующих несахаров. При анализе сульфитного щелока
пентозы и фурфурол надо определять обязательно после удаления
редуцирующих несахаров из пробы. Концентрацию пентоз и
фурфурола определяют бромид-броматным методом, а
редуцирующие вещества — эбулиостатическим методом.
Мешающие вещества. Редуцирующие несахара, уроновые
кислоты, летучие бромируемые вещества, а также фурфурол
в воздухе помещения, где делают анализ.
Аппаратура. Установка для эбулиостатического метода (см.
рис. II и 12). Установка для отгонки летучих веществ с водяным
паром (см. рис. 47). Установка для титрования.
Реактивы и их приготовление. 1. Основной уксуснокислый
свинец, насыщенный раствор. Приготовление см. на с. 57. 2.
Щавелевокислый натрий, насыщенный раствор. 3. Серная кислота
плотностью 1,84 г/см3. 4. Все реактивы для определения
редуцирующих веществ эбулиостатическим методом. 5. Все реактивы
для определения фурфурола бромид-броматным методом (см.
с. 44).
Подготовка проб. Пробы гидролизатов и сульфитных
щелоков нейтрализуют до pH 5,5—7,0 и определяют количество
раствора основного уксуснокислого свинца, которое необходимо для
полного осаждения редуцирующих несахаров из 10 мл пробы
(см. с. 58).
Ход анализа. 1. Осаждение редуцирующих
несахаров. В стакан вместимостью 300—500 мл наливают 50 мл
сульфитного щелока (или нейтрализованного гидролизата) и
200 мл дистиллированной воды. При анализе гидролизата воду
прибавлять не надо. Жидкость в стакане нагревают до 90—95°С
(до появления первых пузырьков) и по каплям прибавляют к ней
предварительно установленное количество раствора основного
уксуснокислого свинца до полного удаления осаждающихся
веществ. Количество раствора основного уксуснокислого свинца,
необходимое для полного осаждения редуцирующих несахаров,
составляет на 10 мл гидролизата 0,5—1,0 мл, а для сульфитных
щелоков оно колеблется в больших пределах, поэтому его надо
устанавливать для каждой анализируемой пробы.
Затем раствор с осадком количественно переносят в мерную
колбу вместимостью 500 мл, его объем доводят до метки,
прибавляя дистиллированную воду, перемешивают и фильтруют.
Получают фильтрат № 1. Перед анализом из фильтрата № 1
удаляют свинец, осаждая его оксалатом натрия.
2. Определение редуцирующих веществ после
осаждения редуцирующих несахаров. В мерную
колбу вместимостью 50 мл вливают 25 мл фильтрата № 1, при-
148
бавляют 7 мл насыщенного раствора оксалата натрия, объем
раствора доводят до метки дистиллированной водой,
перемешивают и фильтруют. В полученном фильтрате № 2 определяют
концентрацию редуцирующих веществ эбулиостатическим
методом по варианту I (см. с. 37). Полученный результат анализа
является суммарной концентрацией гексоз, пентоз и фурфурола,
которая выражена в процентах глюкозы, так как титр медноще-
лочного раствора установлен по глюкозе. Учитывая, что
редуцирующая способность фурфурола в условиях определения его
эбулиостатическим методом в 2 раза ниже, чем у глюкозы,
найденное количество редуцирующих веществ А будет выражаться
следующей суммой:
А = Г + Л + 0.5Ф, откуда Г = А — П — 0,5Ф, A)
где А — концентрация редуцирующих веществ в пробе после осаждения,
% глюкозы; Г — концентрация гексоз, % глюкозы; П — концентрация пентоз,
% глюкозы; Ф — концентрация фурфурола, %; 0,5—коэффициент для
перевода фурфурола в глюкозу.
3. Определение суммы пентоз и фурфурола.
В мерную колбу вместимостью 250 мл вливают 200 мл
фильтрата № 1, прибавляют насыщенный раствор оксалата натрия до
метки, раствор перемешивают и фильтруют. Получают
фильтрат № 3.
В колбу вместимостью 500 мл вливают 50 мл фильтрата №3
и осторожно по стенке прибавляют 90 мл серной кислоты
плотностью 1,84 г/см3, раствор хорошо перемешивают. Колбу
укрепляют в штативе, закрывают пробкой, в которую вставлены
барботер, отводная трубка и термометр. Барботер соединяют с
паровиком, а отводную трубку с холодильником (см. рис. 47).
Кран 2 открыт полностью. До пропускания пара содержимое
колбы нагревают на электроплитке до 80—90°С, затем
электроплитку опускают на 15—20 см ниже и включают пар, закрывая
кран 2. Приемником конденсата служит мерная колба
вместимостью 500 мл. Во время отгонки следят, чтобы уровень жидкости
в реакционной колбе оставался постоянным, а температура
реагирующей жидкости не превышала 105°С во избежание
разложения фурфурола, образующегося при дегидратации пентоз.
Скорость отгонки поддерживают равной приблизительно
8 мл/мин. Отгонку продолжают до отрицательной реакции
последней капли конденсата на фурфурол с уксуснокислым
анилином. (Если в конденсате есть фурфурол, то бумажка, смоченная
уксуснокислым анилином, порозовеет от одной капли конденсата
из холодильника, В этом случае отгонку продолжают.) После
окончания отгонки объем конденсата доводят до метки,
прибавляя дистиллированную воду, раствор тщательно перемешивают
и анализируют на содержание фурфурола бромид-броматным
методом.
14»
В коническую колбу вместимостью 250 мл вливают 10 мл
дистиллированной воды, 3 мл 12—13%-ной соляной кислоты,
50 мл конденсата, 2 мл 2,5%-ного раствора молибденовокислого
аммония и 5 мл 0,05 н. раствора бромид-бромата калия. Колбу
закрывают стеклянной пробкой, ее содержимое перемешивают и
ждут появления желтой окраски. Обычно желтая окраска
появляется через 10—15 с. Сразу перевертывают песочные часы и
ждут 4 мин. Затем к реакционной жидкости приливают 2 мл
10%-ного раствора йодистого калия, колбу закрывают и снова
ждут 4 мин, после чего выделившийся йод титруют раствором
гипосульфита натрия в присутствии крахмала. Параллельно
проводят холостой опыт, в котором вместо конденсата берут
50 мл дистиллированной воды.
Расчет. Содержание суммы пентоз и фурфурола вычисляют по формуле
в (а-бH,00092V100 • 1,035 /ох
*= gg\ ' ()
где В —концентрация пентоз и фурфурола, % пентоз; а — объел раствора ги-
лосульфита, пошедший иа титрснмние-в~хожкггом опыте, мл; б — объем
раствора гипосульфита, пошедший на титрование конденсата, мл; V — объем
конденсата, полученного при отгонке, мл; g — объем конденсата, взятый на
анализ, мл; gi — объем анализируемой пробы, взятый для отгонки фурфурола,
мл; 0,00092 — количество пентоз эквивалентное 1 мл 0,01 н. раствора
гипосульфита, г; 1,035 — эмпирический коэффициент.
Учитывая, что количество фурфурола эквивалентное 1 мл
0,01 н. раствора гипосульфита натрия равно 0,00048 г. т. е.
в 2 раза меньше произведения 0,00092-1,035, применяемого для
вычисления пентоз, результат анализа В является суммой пентоз
и удвоенного количества фурфурола, которое определяется
в этом анализе.
В = П + 2Ф. откуда П = В — 2Ф,
где П—содержание пентоз, %; Ф —содержание фурфурола, %; 2 —
коэффициент для пересчета фурфурола в пентозы.
4. Определение фурфурола. В колбу вместимостью
500 мл вливают 100 мл фильтрата № 3 (см. выше), закрывают ее
пробкой с двумя отводами (к паровику и к холодильнику) и
включают в установку для отгонки с водяным паром (см.
рис. 47). Приемником служит мерная колба вместимостью
250 мл. Перед включением пара содержимое колбы подогревают
до 90—95°С, Отгонку ведут до отрицательной реакции
конденсата с уксуснокислым анилином. Когда отгонка закончена,
объем конденсата доводят до метки дистиллированной водой,
раствор перемешивают и определяют в нем фурфурол бромид-
ароматным методом. Параллельно делают холостой опыт, в
котором вместо конденсата берут 20 мл дистиллированной воды.
150
Расчет. Содержание фурфурола в анализируемой пробе вычисляют па
формуле
ф (а-б) 0,00048 • 100
где Ф — концентрация фурфурола %; 0,00048 — количество фурфурола
эквивалентное 1 мл 0,01 н. раствора гипосульфита натрия, г. Остальные
обозначения см. выше.
Содержание гексоз Г, т. е. сбраживаемых Сахаров, в анализируемой пробе
вычисляют по формуле, в которой концентрацию пентоз Я выражают через
результат анализа В:
Г = А — В + І.5Ф.
Содержание сбраживаемых Сахаров определяют с точностью ±3—5% отн..
Определение сбраживаемых Сахаров биохимическим методом
Для определения сбраживаемых Сахаров в сусле,
поступающем в бродильную батарею, а также в гидролизате и
сульфитном щелоке предназначен метод, основанный на определении
количества редуцирующих веществ, исчезающих из анализируемой
пробы при спиртовом брожении, которое проводят в термостате
при +32°С в течение 10 ч.
Мешающие вещества. Редуцирующие несахара, изменяющие
свои редуцирующие свойства во время спиртового брожения.
Галактоза — трудносбраживаемая гексоза, которая во время
брожения в лабораторных условиях в течение 10 ч остается
почти без изменений. Посторонняя микрофлора —
уксуснокислые, молочнокислые, маслянокислые бактерии и другие
микроорганизмы, утилизирующие гексозы и спирт.
Аппаратура. Термостат. Установка для определения
редуцирующих веществ эбулиостатическим методом (см. рис. 11 и 12).
Реактивы и их приготовление. 1. Все реактивы для эбулио-
статического метода определения РВ (см. с. 35). 2. Свежие
дрожжи, взятые в цехе перед анализом, промытые на воронке
под вакуумом и отфильтрованные до 75%-ной влажности.
Подготовка проб. Пробы сусла анализируют сразу после
отбора во избежание бактериального заражения. Кислые пробьг
гидролизатов и сульфитных щелоков можно хранить. Перед
анализом пробы фильтруют, кислые пробы нейтрализуют до pH
4,4—4,6,
Ход анализа. В спиртовом цехе берут дрожжевую суспензию^
и микроскопированием определяют наличие в ней инфекции.
Если дрожжи чистые, то их отфильтровывают на воронке под;
вакуумом, промывают водой и отсасывают до получения
лепешки дрожжей, имеющей влажность около 75%. На
технических весах взвешивают 2 г дрожжей, взятых из середины
лепешки, кладут их в фарфоровую чашку и приливают к ним 5—
10 мл анализируемой пробы. Содержимое чашки тщательно
размешивают стеклянной палочкой и количественно переносят
151
б чистую простерилизованную колбу вместимостью 250 мл,
используя при этом 90—95 мл пробы, которая была
предварительно отмерена цилиндром. Колбу закрывают пробкой,
сделанной из плотно свернутой ваты, которая вместе с колбой была
простерилизована нагреванием в сушильном шкафу при
температуре 120—130°С, и ставят в термостат. Брожение ведут при
32°С (±Г) в течение 8 ч, изредка перемешивая жидкость, не
открывая колбы,
После постановки пробы на брожение сразу делают анализ
оставшейся пробы на содержание редуцирующих веществ эбу-
лиостатическим методом. Через 8 ч брожения из колбы берут
10 мл отбродившей жидкости, фильтруют и определяют в ней
концентрацию оставшихся редуцирующих веществ. Колбу
с бродящей жидкостью оставляют в термостате еще на 2 ч.
Через 10 ч от начала брожения из колбы отбирают снова 10 мл
жидкости, фильтруют и анализируют на содержание
редуцирующих веществ. Если концентрация редуцирующих веществ за 2 ч
дображивания не изменилась, то анализ считают законченным,
если она уменьшилась, то брожение продолжают еще 2 ч и снова
определяют РВ. После окончания брожения дрожжи опять
проверяют микроскопированием на присутствие инфекции. Если
будут обнаружены уксуснокислые бактерии или другие
микроорганизмы, то анализ повторяют.
Расчет. Содержание сбраживаемого сахара в анализируемой пробе
вычисляют по формуле
X=a-ff.
где X — концентрация сбраживаемого сахара, %; о — концентрация
редуцирующих веществ в пробе до брожения, %; б — концентрация редуцирующих
веществ в пробе после брожения, %.
Сбраживаемый сахар определяют с точностью ±5% отн.
Определение физиологического состояния дрожжей микроскопированием
На гидролизных и сульфитно-спиртовых заводах для
спиртового брожения используют три культуры дрожжей: Saccharomy-
ces cerevisiae — пылевидные дрожжи, имеющие шарообразную
или эллипсообразную форму, размножаются почкованием; Schi-
zosaccharomyces pombe — дрожжевые клетки, имеющие
палочковидную форму, размножаются делением; Saccharomyces vini —
ветвистые дрожжи, размножаются почкованием (рис. 50).
Для оценки качества дрожжей, находящихся в бродильной
батарее, и для оценки концентрации дрожжей, имеющих
шарообразную, эллипсовидную или палочковидную форму,
предназначен метод, основанный на определении физиологического
состояния дрожжей по относительному содержанию молодых,
зрелых, перезрелых, почкующихся, голодающих и мертвых клеток.
Количество различных дрожжевых клеток подсчитывают под
микроскопом с помощью камеры Тома.
152
Физиологическое состояние клеток определяют по внешним
признакам. Молодые клетки имеют еле заметную
оболочку, однородную протоплазму, вакуоли отсутствуют или очень
малы. Зрелые клетки имеют тонкую оболочку, заметно
зернистое строение протоплазмы, вакуоли средней величины.
Такие клетки имеют максимальную бродильную активность.
Перезрелые клетки имеют толстую оболочку, зернистую
протоплазму. Эти клетки содержат жир. Клетки в стадии
Рис. 50. Культуры спиртовых дрожжей:
— Saccharomyces cerevisiae; 2 — Saccharomyces
З — Schizosaccharomyces pombe
vini;
1
размножения — почкующиеся или делящиеся. Почкующие
клетки представляют собой сдвоенные клетки, одна из которых
нормальной величины, а другая — значительно меньше.
Маленькую клетку, не отделившуюся от большой материнской
клетки, называют почкой. Делящиеся
клетки представляют собой обычную
дрожжевую клетку, посредине которой
постепенно образуется граница раздела.
Голодающие клетки имеют размер
меньше обычных клеток, зернистую
протоплазму, вакуоли отсутствуют. В этих
клетках отсутствует гликоген и жир.
Meртвые клетки. Отмирание и
полное разложение (автолиз) дрожжевых
клеток происходит в несколько стадий.
Сначала протоплазма становится
крупнозернистой (комковатой), затем
разрушается оболочка и протоплазма
распадается на мелкие части. Мертвые клетки
под микроскопом видны как черные
клетки. Красящие вещества,
присутствующие в гидролизатах и сульфитных щелоках, окрашивают
мертвые клетки в бурый цвет.
Мешающие вещества. Ветвистые дрожжи и частицы твердых.
веществ, мешающие подсчету отдельных дрожжевых клеток.
Аппаратура. Микроскоп. Счетная камера Тома.
Счетная камера Тома (рис.51) представляет собой
толстое отшлифованное стекло 1 с тремя выступами. На
среднем выступе нанесена микроскопическая сетка 2. Расстояние
между смежными линиями сетки равно 0,05 мм, площадь сетки
равна 1 мм. Вся сетка разделена на 400 малых квадратов,
площадь каждого квадрата равна V4oo мм2 или У4-104 см2.
Толщина слоя жидкости, которая может поместиться между
поверхностью сетки и покровным стеклом, равна 0,1 мм. Объем
2
= = = =
Рис. 51. Счетная камера.
Тома
153,
* о О о •
о ° о° ° с°
0
жидкости между сеткой и покровным стеклом составляет 0,1 мм3,
а объем жидкости над каждым маленьким квадратом lUx
X Ю3 мм3. На сетке имеются дополнительные линии,
повторяющиеся через каждые четыре малых квадрата и делящие пополам
каждую пятую горизонтальную и вертикальную полосы,
образованные двумя соседними линиями. Эти дополнительные линии
нужны для лучшей ориентировки при подсчете дрожжевых
клеток.
Ход анализа. Испытуемую жидкость, содержащую дрожжи,
разбавляют так, чтобы в поле зрения микроокопа было видно
от 30 до 60 дрожжевых клеток. Одну каплю разбавленной
жидкости с дрожжами помещают на средний выступ поверхности
камеры Тома, где нанесена сетка и накрывают ее покровным
стеклом, которое плотно прижимают к
поверхности камеры дтгя удаления пузырьков
воздуха. Стекло притирают к крайним выступам
поверхности камеры до появления радужных
полос — колец Ньютона, свидетельствующих
о том, что толщина слоя жидкости между
покровным стеклом и средним выступом
на поверхности камеры, где нанесена сетка,
стала равной 0,1 мм. Покровное
стекло должно иметь абсолютно ровную
поверхность.
Счетную камеру помещают на предметный
столик микроскопа. Подсчет дрожжей
ведут при увеличении в 500—600 раз (объектив
№ 7, окуляр № 4). При этом увеличении
в поле зрения микроскопа помещается не менее 16 малых
квадратов, которые на рис. 51 обведены толстыми линиями.
Количество дрожжевых клеток, находящихся в 16 квадратах, считают
в четырех рядах по горизонтали в каждом квадрате. Подсчет
начинают с верхнего левого квадрата, обведенного толстой
линией на рис. 52. Дрожжевые клетки, которые пересекаются
нижней и левой сторонами квадрата, подсчитывают в смежных
квадратах. Для большей точности результатов анализа дрожжи
подсчитывают в пяти местах препарата, анализируя четыре
препарата.
Расчет. Результаты подсчетов записывают в определенном порядке и
вычисляют среднеарифметические величины содержания дрожжевых клеток
в 16 квадратах. Обычно ограничиваются подсчетом содержания в 16
квадратах всех клеток и отдельно — почкующихся и мертвых клеток. Содержание
почкующихся клеток выражают в процентах к количеству живых дрожжевых
клеток (разность между количеством всех клеток и количеством мертвых
клеток), а мертвых — к общему количеству дрожжевых клеток.
Содержание дрожжей в исследуемой пробе, выраженное в граммах на 1 л
(г/л) вычисляют по нижеприведенной формуле, исходя из предположения, что
в 1 г живых дрожжей, обладающих влажностью 75%, находится 1010
дрожжевых клеток, или предварительно определив количество дрожжевых клеток
154
Рис. 52. Поле
зрения под
микроскопом при подсчете
дрожжей в камере
Тома
в 1 г прессованных дрожжей, используемых на заводе, как описано ниже,
Anim An
Л = I6VJV 4ЛГ10 '
где X — содержание дрожжей 75%-ной влажности в анализируемой пробе,,
г/л; А — среднее число дрожжевых клеток, найденное в 16 квадратах; 16V=
I? l
= _—__ —объем жидкости в 16 квадратах, мл; V= -т—тщ —объем
жидкости в одном малом квадрате, мл; п — разбавление пробы перед анализом;
N — число дрожжевых клеток в 1 г дрожжей 75%-ной влажности.
Концентрацию дрожжей определяют с точностью ±3—5 % оти.
Определение наличия инфекции
Для установления инфекции в сусле, бражке, дрожжевой
суспензии и в водных растворах после промывки трубопроводов,
чанов и т. д. предназначен метод, основанный на
микроскопическом исследовании пробы, с целью установления наличия
посторонних микроорганизмов, которые утилизируют сахара или
этиловый спирт. На гидролизных и сульфитно-спиртовых
заводах дрожжи инфицируются чаще всего молочнокислыми и
уксуснокислыми бактериями и микодермой. На южных заводах
встречается заражение дрожжей сарцинами и маслянокислыми
бактериями (рис. 53).
Молочнокислые бактерии имеют форму палочек
различной величины до коккообразных, образующих группы или
цепочки. На заводах обычно находят дикие виды этих бактерий.
Они поселяются не только в растворах, содержащих сахара, на
и на различных частях аппаратуры, в помещениях, где
проливают сахарные растворы и редко проводят дезинфекцию.
Молочнокислые бактерии превращают гексозы в молочную кислоту,
которая угнетающе действует на спиртовые дрожжи.
Оптимальная температура развития молочнокислых бактерий 24—50°С,
Они анаэробны, спор не образуют.
Уксуснокислые бактерии аэробны, при больших
скоплениях на поверхности жидкости образуют розоватую пленку,,
пахнущую уксусной кислотой. Они имеют форму коротких
палочек, образующих цепочки. Эти бактерии превращают в
уксусную кислоту этиловый спирт, который образуется в
результате сбраживания гексоз дрожжами. Оптимальная температура-
развития уксуснокислых бактерий 23—24°С.
Микодерма — дикие дрожжи, или пленчатые грибы. Она
хорошо развивается в присутствии спирта в бражке при низкой
кислотности и при хорошем доступе воздуха. Микодерма окис-
ляет этиловый спирт до углекислоты и воды. При больших
скоплениях на поверхности жидкости она образует белоснежную'
пленку. Оптимальная температура для развития микодермы 30°С
Маслянокислые бактерии — палочковидной формы
превращают гексозы в масляную кислоту, они аэробны,
образуют споры. Оптимальная температура для их развития 25°С.
155:
С а р ц и н ы аэробны, они превращают гексозы в молочную
и уксусную кислоты. Оптимальная температура для их
развития 25°С.
Мешающие вещества. Взвешенные органические и
неорганические вещества.
Рис. 53. Дрожжи и бактерии:
/ — почкующиеся дрожжи; 2 — дрожжи и молочнокислые
бактерии; 3 — уксуснокислые бактерии; 4 — микодерма; 5 —
молочнокислые бактерии; 6 — маслянокислые бактерии
Реактивы. 1. Едкий натр, 0,1 н. раствор. 2. Соляная кислота,
1 н. раствор.
Ход анализа. Одну каплю исследуемого раствора наносят на
предметное стекло, накрывают ее покровным стеклом, которое
плотно прижимают, чтобы удалить избыток жидкости между
156
стеклами. Приготовленный препарат рассматривают в
нескольких местах под микроскопом при увеличении в 800—1000 раз
с иммерсией. Иногда исследуемая жидкость содержит
взвешенные вещества, которые по своей форме похожи на
микроорганизмы. Чтобы не ошибиться в определении наличия инфекции,
готовят еще два препарата: один с добавкой одной капли 0,1 н.
раствора едкого натра к одной капле препарата, а другой —
с добавкой одной капли 1 н. раствора соляной кислоты.
Органические вещества, присутствующие в анализируемой пробе,
растворяются в щелочи. Минеральными примесями обычно
является гипс, его растворяют в соляной кислоте. Рассматривая все
три препарата под микроскопом и сравнивая наблюдаемые
микробиологические картины с образцами различных дрожжей и
¦бактерий, определяют наличие инфекции и какой именно.
Результаты наблюдений выражают словами: «В поле зрения
микроскопа имеется одна, пять, десять.... много (каких?)
бактерий».
Определение количества дрожжевых клеток в 1 г дрожжей
75%-иой влажности
Для определения количества дрожжевых клеток, имеющих
форму шариков или палочек, в 1 г дрожжевой массы,
полученной фильтрованием дрожжевой суспензии под вакуумом,
предназначен метод, основанный на подсчете в камере Тома
количества дрожжевых клеток в дрожжевой суспензии известной
концентрации. Количество дрожжевых клеток в 1 г
прессованных дрожжей зависит от их размеров, которые определяются
расой дрожжей и условиями проведения процесса брожения
в цехе. Для разных рас дрожжей, используемых на заводах, оно
колеблется в больших пределах.
Аппаратура. Микроскоп и камера Тома (см. рис. 51).
Ход анализа. На фарфоровой воронке под вакуумом
фильтруют 500 мл дрожжевой суспензии через бумажный фильтр.
Фильтрование продолжают до полного удаления жидкости.
Отфильтрованные дрожжи называют прессованными, они содержат
75% влаги, удаляемой при их высушивании при 105°С. Из
середины полученной дрожжевой лепешки берут 1 г дрожжей,
помещают их в мерную колбу вместимостью 1 л, прибавляют
100 мл воды, тщательно размешивают дрожжи в воде, чтобы не
было комков, и приливают воду до метки. Полученную
суспензию тщательно перемешивают и определяют в ней содержание
дрожжевых клеток с помощью камеры Тома как описано на
с. 153. Три маленьких почки считают за одну дрожжевую клетку.
Расчет. Количество дрожжевых клеток, содержащихся в 1 г
анализируемых прессованных дрожжей, вычисляют по формуле
Л1000 А\ ¦ 1QS • 1Q3 А 09
16V ~ 16 . 1 - 4
157
где X—число дрожжевых клеток в 1 г анализируемых дрожжей; Л—число
16
дрожжевых клеток, найденное в объеме 16 квадратов; 16К= . .~s =4 ¦ 10~6 —
объем жидкости в 16 квадратах камеры Тома, мл.
Количество дрожжевых клеток в 1 г прессованных дрожжей определяют
с точностью ±4—5% оти.
Определение концентрации дрожжей в дрожжевых суспензиях
Концентрацию дрожжей любой расы, находящихся в
бродящей жидкости и в дрожжевой суспензии после сепарации,
определяют методом взвешивания дрожжей, отфильтрованых из
определенного объема суспензии.
Мешающие вещества. Взвешенные органические и
минеральные вещества (гипс и др.).
Аппаратура. Установка для фильтрования под вакуумом.
Ход анализа. В зависимости от предполагаемой концентрации
дрожжей 1000, или 500 или 100 мл хорошо перемешенной
дрожжевой суспензии фильтруют под вакуумом через два
одинаковых фильтра, положенных на фарфоровую воронку.
Отфильтрованные дрожжи промывают на воронке 2 раза, расходуя 50 мл
дистиллированной воды. После полного отсасывания жидкости
дрожжи с фильтрами вынимают из воронки, верхний фильтр
с дрожжами отделяют от нижнего, кладут на заранее
взвешенное стекло и взвешивают. Нижний фильтр кладут на другое
взвешенное стекло и тоже взвешивают.
Расчет. Концентрацию дрожжей в анализируемой пробе дрожжевой
суспензии вычисляют по формуле
„ А1000
Х = —.
где X — содержание дрожжей в пробе, г/л; А — количество отфильтрованных
дрожжей, г; g — объем дрожжевой суспензии, взятый на анализ, мл.
Концентрацию дрожжей в суспензии определяют с точностью ±5—7% отн.
Определение спиртообразующей способности дрожжей
Для определения спиртообразующей способности дрожжей,
отобранных из бродильных чанов, предназначен метод,
основанный на вычислении выхода спирта из 100 кг сброженного
сахара, когда брожение нейтрализованного гидролизата или
сульфитного щелока проводят анализируемыми дрожжами в
лабораторных условиях. Количество сброженного сахара находят по
разности концентраций редуцирующих веществ до и после
брожения, определяя их эбулиостатическим методом. Количество
образовавшегося спирта определяют флотационным или, более
точно, вакуум-нитритным методом. Согласно уравнению Гей-
Люссака теоретически достижимый выход спирта из 100 кг
сброженного сахара составляет 64,7 л при 20°С. Лучшие
производственные дрожжи на гидролизных заводах дают выход спирта
90—92% от теоретически возможного, т. е. 58—59 л из 100 кг
158
сброженного сахара. На сульфитно-спиртовых заводах выход
спирта в производственных условиях получают более высоким,
до 60—61 л из 100 кг сброженного сахара.
Мешающие вещества. Редуцирующие несахара, изменяющие
свою редуцирующую способность во время брожения. Трудно-
сбраживаемый сахар — галактоза. Летучие вещества, которые
имеют плотность отличающуюся от плотности воды, если спирт
определяют флотационным методом. Посторонние
микроорганизмы — инфекция.
Аппаратура. Термостат. Установка для эбулиостатического
метода (см. рис. 11 и 12). Установка для флотационного или ва-
куум-нитритного метода (см. рис. 55 или 56).
Реактивы. Все реактивы для определения редуцирующих
веществ эбулиостатическим методом и для определения спирта
флотационным методом или вакуум-нитритным методом.
Подготовка проб. Пробу дрожжей отбирают перед анализом.
Из бродильного чана берут такое количество бродящей
жидкости, которое содержит около 15 г дрожжей. Дрожжи
отфильтровывают через бумажный фильтр под вакуумом, промывают 50—
70 мл воды и отсасывают до получения лепешки прессованных
дрожжей. Пробу нейтрализованного гидролизата или
сульфитного щелока отбирают после введения питательных солей,
охлаждают до 30°С, фильтруют и сразу используют для анализа.
Хранить нейтрализат и дрожжи нельзя.
Ход анализа. В нейтрализате определяют концентрацию
редуцирующих веществ и спирта. На брожение берут 6 г дрожжей
и 300 мл нейтрализата. Брожение проводят в колбе вместимостью
500 мл, которая предварительно была простерилизована вместе
с ватной пробкой в сушильном шкафу при температуре 130°С
в течение 1 ч. В колбу для брожения вливают 250 мл
нейтрализата и подогревают до 30°С. В фарфоровую чашку кладут 6 г
дрожжей, приливают 50 мл нейтрализата, тщательно
размешивают стеклянной палочкой и полученную суспензию выливают
в колбу с подогретым нейтрализатом. Колбу закрывают ватной
пробкой и ставят в термостат, температура в котором 32°С
регулируется с точностью ± 1°С. Во время брожения жидкость
в колбе изредка перемешивают (не открывая колбы), чтобы
поднять осевшие дрожжи. Через 6 ч брожения отбирают пробу
1 в количестве 50—80 мл фильтруют и определяют в ней спирт.
Еще через 1 ч отбирают пробу 2, которую также анализируют
на содержание спирта. Если концентрация спирта в пробах
будет одинакова или в пробе 2 ниже, чем в пробе 1, то брожение
прекращают. Если концентрация спирта в пробе 2 выше, чем
в пробе 1, то брожение продолжают еще 1 ч и отбирают пробу 3,
в которой определяют спирт. Пробу, в которой обнаружена
наибольшая концентрация спирта, анализируют на содержание
редуцирующих веществ. Полученные данные используют для
вычисления спиртообразующей способности дрожжей.
159
Расчет. Спиртообразующую способность дрожжей вычисляют как выход
спирта из 100 кг сброженного сахара в литрах или в процентах от
теоретически возможного выхода по следующим формулам:
(С - с) 100 (С - с) 1С0 ¦ 100
А — а ' 1~ (А —а) 64,7
где X— выход спирта из 100 кг сброженного сахара, л; Xi — выход спирта
из 100 кг сброженного сахара, % от теоретического выхода; С —
концентрация спирта в бражке, %; с — концентрация спирта в сусле до брожения, %;
А—концентрация редуцирующих веществ до брожения, %; а—концентрация
редуцирующих веществ после брожения, %; 64,7 — теоретический выход
спирта из 100 кг сброженного сахара, л.
Спиртообразующую способность дрожжей определяют с точностью ±3—
5% оти.
Предостережение. Спнртообразующая способность дрожжей
характеризует физиологическое состояние дрожжевой массы в бродильных чанах,
поэтому при выполнении анализа надо особенно тщательно контролировать
появление посторонних микроорганизмов в бродящей жидкости.
Определение бродильной активности дрожжей
Бродильной активностью дрожжей называют количество
сбраживаемых Сахаров, которое 1 т дрожжей в течение 1 ч
превращает в спирт. Бродильная активность зависит от физиологи-
чеокого состояния дрожжей, их спиртообразующей способности
и от условий питания дрожжей. Приток сусла в головной чан
должен быть рассчитан так, чтобы дрожжи всегда были
обеспечены сбраживаемым сахаром, а в бражке, отбираемой
из последнего чана батареи, была максимальная
концентрация спирта при полном отсутствии сбраживаемых
Сахаров [47].
Бродильная активность является основным показателем
производительности бродильного отделения. При неритмичной
работе бродильная активность может колебаться в больших
пределах. В случае невозможности обеспечения дрожжей
необходимым количеством сахара надо из батареи изъять излишек
дрожжей. Бродильную активность дрожжей вычисляют на основании
данных анализов на содержание дрожжей в бродильной
батареи (или чане), анализа сусла на содержание сбраживаемого
сахара и замера скорости притока сусла при условии
получения максимальной концентрации спирта в бражке и отсутствия
гексоз.
Av
" ~ D100 '
где X — бродильная активность дрожжей, т. е. количество сахара
сбраживаемого 1 т дрожжей в течение 1 ч, т; А — концентрация сбраживаемого
сахара в сусле, %; v — скорость подачи сусла, м3/ч; D — количество дрожжей
в бродильной батарее, т.
160
Определение Сахаров экспрессным хроматографическим методом
Для определения наличия несбродивших гексоз в спиртовой
бражке [48], предназначен метод, основанный на реакции гексоз
с анилинфталатом после выделения их из пробы путем хрома-
тографирования на бумаге, В результате этой реакции
образуется соединение, которое на бумаге дает пятно
коричнево-зеленого цвета. Сравнивая интенсивность окраски и величину пятна
на хроматограмме с пятном свидетеля (см. с. 80), визуально
определяют содержание несбродивших Сахаров или их
отсутствие.
Аппаратура. Камера для хроматографирования (см. рис.22).
Микропипетка (см. рис. 27).
Реактивы и их приготовление. 1. Растворитель — смесь этил-
ацетат — пиридин — вода E:1:5). Приготовление см. на с. 78.
2. Проявитель — раствор анилинфта-
лата. Приготовление см. на с. 79.
Ход анализа. Хроматографирова-
ние проводят в эксикаторе на хромато-
графической бумаге, вырезанной в
виде лопаток (см. рис. 22) следующих
размеров: а = б= 10 см, в=45 см, г =
= 15 см. Отмеренные микропипеткой
0,005 мл или 0,01 мл пробы, наносят
на узкую часть лопатки на расстоянии
1 см от широкой части, как описано на
с. 81. Пятно высушивают на воздухе
и бумажную лопатку устанавливают
в эксикаторе, погружая ее узкий конец
на 1—2 см в чашку Петри, в которую налит растворитель.
Одновременно можно в одном эксикаторе поставить на хромато-
графирование четыре пробы. Через 45—60 мин, когда
растворитель дойдет почти до конца хроматограммы, хроматографирова-
ние прекращают. Хроматограмму вынимают из эксикатора,
просушивают при комнатной температуре до полного удаления
запаха пиридина и проявляют, погружая ее на мгновение в
проявитель. Для удаления избытка проявителя хроматограмму
слегка отжимают между листами фильтровальной бумаги и
помещают на 5 мин в сушильный шкаф, нагретый до 105—110°С.
На проявленной хроматограмме бражки всегда имеется
розовое пятно, соответствующее пентозам (рис. 54). Между розовым
пятном и местом нанесения пробы, в случае присутствия гексоз
в бражке имеется коричневато-зеленоватое пятно. При полном
отброде этого пятна на хроматограмме бражки нет. При наличии
пятна, соответствующего гексозам, можно определить их
концентрацию в бражке. Для этого готовят параллельно (или заранее)
хроматограммы растворов глюкозы различных концент-
траций от 0,5 до 0,02%. Сравнивая величину и интенсивность
Рис. 54. Хроматограммы:
/ — сусла; // — бражки; Г — гек-
созы — коричневое пятно; Я —
пентозы розовое пятно
II Заказ М> 142
161
окраски гексозного пятна на хроматограмме бражки с пятнами
хроматограмм глюкозных растворов, устанавливают визуально
концентрацию гексоз в анализируемой пробе.
Чувствительность реакции гексоз с анилинфталатом 5 мкг.
Определение метилового спирта колориметрическим методом
Для определения малых количеств метилового спирта (до
0,008%), присутствующего в водных растворах (гидролизат,
сульфитный щелок, сточная вода) и в растворах этилового
спирта [49], предназначен метод, основанный на реакции
окисления метилового спирта марганцовокислым калием до
формальдегида в присутствии фосфорной (или серной) кислоты:
5СН3ОН+2КМпО4+4Н3РО4-^5НСОН+2КН2РО4+
+2МпНРО4+ЗН2О.
Реакция не прекращается на образовании формальдегида,
а сразу идет дальше, формальдегид окисляется до муравьиной
и угольной кислоты. Чтобы прекратить окисление
образовавшегося формальдегида, к реагирующей смеси прибавляют
щавелевую кислоту, которая вступает в реакцию с оставшимся
марганцовокислым калием.
Количество образовавшегося формальдегида определяют по
его реакции с парафуксиносернистой кислотой, в результате
которой образуется окрашенное соединение. Сравнивая визуально
интенсивность окраски реакционной смеси с окрасками,
полученными с эталонными растворами, находят концентрацию
метилового спирта в анализируемой пробе.
Мешающие вещества. Формальдегид, метилацетат. Этиловый
спирт в этих условиях окисляется до уксусного альдегида,
который не мешает определению образовавшегося формальдегида.
Аппаратура. Набор колориметрических пробирок в штативе.
Установка для отгонки летучих веществ.
Реактивы и их приготовление. 1. Серная кислота х. ч. A:1).
2. Пирогаллол, 10%-ный раствор. 3. Спирт метиловый
синтетический. 4. Этиловый спирт ректификованный высшей очистки.
5. Окислительная смесь. Приготовление. В колбу
вместимостью 100 мл помещают 3 г марганцовокислого калия (КМпО4)
и 15 мл 85%-ной фосфорной кислоты (Н3РО4). Объем смеси
доводят до 100 мл, прибавляя дистиллированную воду. Срок
хранения смеси 14 дней. 6. Основной эталонный раствор метилового
спирта в воде. Приготовление. Растворы готовят при
температуре 20°С. В колбу вместимостью 50 мл вливают 20 мл
дистиллированной воды, 0,5 мл метанола, объем раствора доводят
до метки дистиллированной водой и перемешивают. В мерную
колбу вместимостью 500 мл вливают 10 мл приготовленного
1%-ного (по объему) раствора метанола, добавляют дистилли-
162
рованную воду до метки на колбе, получают основной
эталонный раствор, имеющий концентрацию 0,02% метанола, из
которого готовят рабочие эталонные растворы. 7. Рабочие
эталонные растворы метилового спирта в воде. Приготовление.
В шесть мерных колбочек вместимостью 50 імл из бюретки
вливают основной эталонный раствор метанола в воде в следующих
количествах: 45, 40, 35, 30, 25 и 20 мл. Объемы доводят до
метки, прибавляя дистиллированную воду. Полученные рабочие
эталонные растворы имеют соответственно следующие
концентрации метанола: 0,018; 0,016; 0,014; 0,012; 0,010 и 0,008%- Срок
хранения 1 месяц.
Подготовка проб. Ориентировочно определяют содержание
в пробе метанола и формальдегида. Для этого из 25 мл пробы
отгоняют в мерную колбу вместимостью 25 мл 10—15 мл и
объем конденсата доводят до метки на колбе, прибавляя воду.
К 5 мл конденсата прибавляют 5 мл парафуксиносернистой
кислоты. Если появится окрашивание, то в пробе есть
формальдегид. Приблизительную концентрацию метанола определяют,
делая анализ по нижеприведенной методике, сравнивая рабочие
растворы эталонов с концентрацией 0,02 и 0,008%- Оптимальная
концентрация метанола в пробе 0,020—0,008%. Если метанола
в пробе более 0,020%, то отгон разбавляют, а если меньше
0,008%, то при перегонке повышают его концентрацию в
конденсате.
Ход анализа. Отгонка метанола. В колбу вместимостью
100 мл вливают 50 мл пробы, закрывают пробкой, в которую
вставлен каплеотбойник. Колбу ставят на включенную
электроплитку и присоединяют к холодильнику. Конденсат собирают
в мерную колбу объемом 50 мл. Отгонку прекращают, когда
в приемник соберут 30—35 мл конденсата. Объем собранного
конденсата доводят до метки, прибавляя дистиллированную
воду.
Удаление формальдегида. Если в пробе есть
формальдегид, то его удаляют, обрабатывая конденсат
пирогаллолом. В круглодонную колбу вместимостью 50 мл вносят 10 мл
анализируемого раствора, туда же добавляют 10 мл 10%-ного
раствора пирогаллола и 5 мл концентрированной серной
кислоты. Колбу закрывают и оставляют на 10 мин. Затем из смеси
отгоняют 10 мл конденсата и в нем определяют метанол.
Определение метанола. Берут семь пробирок с
притертыми пробками из бесцветного стекла с плоским дном
вместимостью 20 мл. В шесть пробирок вносят по 5 мл рабочих
эталонных растворов и в одну — 5 мл конденсата анализируемой
пробы. Затем во все семь пробирок вливают по 4 капли
этилового спирта ректификата. (При анализе спиртовых бражек
этиловый спирт не добавляют.) Содержимое пробирок
перемешивают, вливают в каждую из них по 2 мл окислительной смеси
и сразу перемешивают встряхиванием пробирки. Через 10 мин
11* 163
в том же порядке приливают по 3 мл раствора щавелевой
кислоты. После обесцвечивания растворов добавляют в каждую
пробирку по 5 мл реактива парафуксиносернистой кислоты.
По истечение 15 мин визуально сравнивают окраски исследуемых
растворов с окрасками эталонных растворов метанола. В случае
очень слабых окрасок сравнение окрасок проводят через 1 ч.
Сравнением устанавливают, с каким эталоном совпадает или
близка окраска раствора, полученная при анализе пробы.
Расчет. Содержание метанола в анализируемом растворе вычисляют по
формуле
х
g '
где X — концентрация метанола в пробе, %; а — концентрация метанола
в конденсате, найденная по эталону, %; g — объем пробы, взятый для
отгона метанола, мл; К —объем собранного конденсата, мл.
Чувствительность реакции 15 мкг. Содержание метанола в пробе
определяют с точностью ±10—15% отн.
Определение этилового спирта флотационным методом
(метод Низовкнна и Королькова)
Для определения этилового спирта в бражке, барде и лю-
тере на гидролизных и сульфитно-спиртовых заводах [50, 51]
предназначен метод, основанный на зависимости плотности
растворов спирта от температуры и
концентрации спирта в растворе. Концентрацию
спирта в растворе измеряют специальным
поплавком, плотность которого известна.
Изменяя температуру анализируемого
раствора, изменяют его плотность, делая ее
равной плотности поплавка. По
температуре, при которой поплавок повиснет в
растворе, т. е. не будет всплывать наверх или
опускаться на дно, а будет оставаться в
покое на любом уровне в растворе, находят
концентрацию спирта в анализируемом
растворе по заранее вычисленной таблице
(паспорт поплавка).
Мешающие вещества. Летучие вещества,
имеющие плотность, отличающуюся от
плотности воды.
Аппаратура. Установка для отгонки
летучих веществ. Установка для определения
спирта флотационным методом с набором
поплавков. Песочные часы на 10 м.
Установка для определения спирта (рис. 55)
состоит из стеклянного стаканчика (высотой 60 мм и диаметром
30 мм), подвешенного на штативе, поплавка /, мешалки 2 и
термометра 3 со шкалой от 0 до 50°С с ценой деления 0,1°С.
Рис. 55. Установка для
определения спирта
флотационным
методом
164
Поплавок представляет собой пустотелый стеклянный шарик
диаметром около 10 мм с цветной маркировкой, который хранят
в отдельном футляре с обозначением номера поплавка. К
каждому поплавку прилагается паспорт, в котором приведена
таблица концентраций растворов спирта при различных
температурах повисання данного поплавка. При измерении различных
концентраций спирта пользуются различными поплавками. Для
заводов выпускают наборы из шести поплавков, которые
отличаются между собой плотностью. Температуры их повисання
в чистой воде находятся в пределах от 20 до 38°С.
Проверка поплавков. Соответствие показаний
поплавка с данными, приведенными в его паспорте, проверяют по
температуре его повисання в дистиллированной воде. С этой
целью поплавок хорошо промывают в крепкой серной кислоте
или в хромовой смеси, а затем в дистиллированной воде. Для
проверки поплавка употребляют дистиллированную воду,
предварительно прокипяченную в течение не менее 15 мин для
удаления растворенного воздуха, в противном случае на поплавке
при измерении температуры его повисання появляются мелкие,
еле заметные пузырьки, которые искажают результат измерения.
Чтобы температура воды в измерительном стаканчике, где
плавает поплавок, не изменялась во время анализа за счет
теплообмена с окружающим воздухом, его погружают в стакан
(вместимостью 300 мл) с водой, которая имеет температуру на 1 —
2°С выше температуры повисання поплавка в воде. Воду в обоих
стаканах хорошо перемешивают и дают температуре воды посте-
ненно снижаться. Если разность между температурой повисання
поплавка и температурой окружающей среды составляет около
10°С, то к показаниям термометра прибавляют поправку на
выступающий столбик ртути термометра, которая составляет 0,1°С,
Показания поплавка считают правильными, если найденная
температура его повисання в воде совпадает с данными,
приведенными в его паспорте, или отличается от них не более чем на
0,ГС.
Реактивы и их приготовление. 1. Едкий натр, 10%-ный
раствор. 2. Едкий натр, 50%-ный раствор. 3. Соляная кислота, 5 н.
раствор. 4. Бромид-броматная смесь, раствор, в 1 л которого
содержится 27,8 г бромноватокислого калия (КВгОз) и 112,9 г
бромистого калия (КВг). 5. Сернокислый гидразин
кристаллический.
Подготовка проб. 1. Перед анализом спирт отгоняют из
бражки, в которую добавлена щелочь, чтобы отделить его от
летучих кислот. В плоскодонную колбу вместимостью 250 мл
вливают 50 мл бражки, отмеренные пипеткой, добавляют 1 мл
10%-ного раствора едкого натра, колбу закрывают пробкой,
в которую вставлен каплеотбойник, соединенный с
холодильником, ставят ее на включенную электроплитку и начинают отгон.
Конденсат собирают в мерную колбу вместимостью 25 мл. Через
165
10—15 мин, когда в приемнике наберется 24—24,5 мл спиртового
конденсата, перегонку прекращают и объем конденсата в колбе
доводят до метки, прибавляя дистиллированную воду. На
чистых растворах спирта установлено, что при перегонке спирта из
слабых растворов требуется отогнать 50% объема раствора,
чтобы в отгон перешло 99,9% спирта. 2. Примеси летучих
веществ из спиртового конденсата удаляют превращением их в
нелетучие вещества и вторичной отгонкой спирта. Летучие
вещества превращают в нелетучие путем бромирования или
обработкой сернокислым гидразином. Бромирование. В
плоскодонную колбу вместимостью 250 мл помещают 50 мл спиртового
конденсата, отогнанного из 100 мл бражки, добавляют 2 мл 5 н.
раствора соляной кислоты и 1 мл раствора бромид-броматной
смеси. Содержимое колбы перемешивают, закрывают стеклянной
пробкой и оставляют стоять на 10 мин (песочные часы). Затем
в колбу вливают 1 мл насыщенного раствора едкого натра и
осуществляют отгонку как описано выше. Полученные 25 мл
конденсата анализируют на содержание спирта, учитывая, что
содержание спирта в этом отгоне в 4 раза выше, чем в исходной
пробе бражки. Обработка сернокислым
гидразином [52]. В колбу для перегонки вливают 50 мл спиртового
конденсата, добавляют 3 г сернокислого гидразина и отгоняют 25 мл
спиртового конденсата. 3. Барда и лютер содержат спирта
тысячные доли процента, чувствительность флотационного метода
только сотые доли процента, поэтому перед анализом надо
сконцентрировать конденсаты их паров не менее чем в 10 раз.
Концентрирование спирта в конденсате проводят при отборе пробы
специальным пробоотборником (см. рис. 5), которым отбирают
из колонны конденсат паров самоиспарения барды или лютера.
Экспериментально установлено, что концентрация спирта в
таких конденсатах в 10 раз больше, чем в испаряющейся
жидкости (барде или лютере). Конденсаты, отобранные
пробоотборником, перед анализом бромируют, как описано выше, и
отгоняют спиртовый конденсат, который анализируют.
Ход анализа. Температуру повисання поплавка определяют
вдали от нагревательных приборов и от тяги, чтобы избежать
влияния потоков воздуха различной температуры, которые могут
быть причиной неправильных результатов наблюдений.
В стакан вместимостью 30 мл выливают 25 мл отогнанного
спиртового конденсата, погружают в него термометр (только
шарик с ртутью), мешалку и поплавок. Выбор поплавка делают
на основании измерения температуры окружающего воздуха и
предполагаемой концентрации спирта в анализируемом отгоне.
Для удобства берут тот поплавок, который должен повиснуть
в анализируемом растворе при температуре, отличающейся от
температуры окружающего воздуха не более чем на 3СС. При
пользовании таким поплавком исключается влияние разницы
между температурой раствора и окружающего воздуха на ско-
166
рость и точность установления температуры повисання поплавка.
Температура раствора спирта, а следовательно, и его плотность
легко удерживаются на нужном значении почти постоянными
в течение времени наблюдения за повисанием поплавка.
Конвекционные токи в растворе, которые могут увлекать
поплавок в направлении своего движения, в этом случае
минимальны.
Поплавок вынимают из футляра с помощью мешалки и
кладут на чистую тряпочку. (Поплавок нельзя брать голыми
пальцами во избежание загрязнения его поверхности!). Если
поплавок, спущенный в исследуемый раствор, тонет, следовательно,
его плотность больше плотности раствора. Чтобы повысить
плотность раствора его охлаждают. Если поплавок всплывает в
исследуемом растворе, следовательно, его плотность меньше
плотности раствора. Чтобы понизить плотность раствора, его
нагревают. Охлаждают и нагревают стакан с исследуемым раствором
погружением на несколько секунд в холодную или теплую воду,
налитую в стакан вместимостью 200—300 мл. После удаления
стакана с водой наружную поверхность стакана с пробой
хорошо вытирают фильтровальной бумагой, содержимое стакана
перемешивают, замечают показания термометра и наблюдают
за движением поплавка. При наблюдении за поплавком следят,
чтобы на нем не было пузырьков воздуха. Температуру
подгоняют ступенчато. Если поплавок плавает на поверхности
раствора, то, заметив температуру раствора, его нагревают
приблизительно на 1°С при постепенном помешивании и снова
наблюдают за движением поплавка после успокоения раствора
в стакане. Если поплавок при новой температуре всплывает, то
раствор нагревают еще на 1°С. Если поплавок потонет,
следовательно, температура повисання поплавка находится между
значениями до и после нагревания. Затем продолжают изменять
температуру раствора на 0,2—0,3°С до тех пор, пока не найдут
температуры всплывания и погружения поплавка, которые будут
отличаться между собой не более чем на 0,2°С. За результат
определения берут среднее значение между этими
температурами и проверяют его, наблюдая повисание поплавка в растворе
при найденной температуре.
Температуру повисання поплавка определяют в течение не
более 10 мин. Увеличение времени анализа может привести
к неправильным результатам из-за возможного испарения
спирта из раствора. По окончании анализа поплавок вынимают
из раствора мешалкой, протирают его чистой тряпочкой или
фильтровальной бумагой и кладут в футляр. Во избежание
путаницы в обозначении номера поплавка перед анализом
проверяют его маркировку по паспорту.
Расчет. По таблице паспорта поплавка, которым делали анализ, находят
концентрацию спирта в анализируемом спиртовом конденсате, полученном из
пробы, в объемных процентах.
167
Если анализировали бражку (барду, лютер) после удаления летучих
примесей бромироваиием или сернокислым гидразином, то концентрацию спирта
вычисляют по формуле
Х--
10ft
где X—концентрация спирта в пробе, мл/100 мл; С — концентрация спирта
в конденсате, найденная по таблице в паспорте поплавка, мл/100 мл; k —
концентрирование спирта во время отгонок; 10 — концентрирование спирта при
отборе проб паров самоиспареиия барды или лютера.
Если бражку анализировали после первого отгона, ие удаляя летучие
примеси, то в формулу вводят эмпирическую поправку на их содержание.
Поправку на тяжелые примеси определяют одним из следующих двух
способов:
1. Анализируют 5—10 образцов бражки иа содержание спирта двумя
методами — флотационным и вакуум-иитритиым. Вычисляют среднюю
арифметическую концентрацию спирта, найденную при анализе всех проб, отдельно
флотационным методом и вакуум-иитритным методом (см. ниже), а затем
находят коэффициент К по формуле
Сф
где Св — средняя концентрация спирта в бражке, определенная вакуум-нит-
ритным методом, мл/100 мл; Сф—средняя концентрация спирта в бражке,
определенная флотационным методом, мл/100 мл.
В этом случае формула для вычисления концентрации спирта в бражке
будет иметь следующий вид:
х-тк-
2. Определяют концентрацию спирта в 10—20 пробах нейтрализата флота-
циоииым методом. Для этого анализируют конденсат летучих веществ после
их отгона из подщелоченной пробы иейтрализата. Найденная температура по-
вясания поплавка обычно бывает ниже температуры его повисання в чистой
воде. Это значит, что отогиаииые летучие вещества тяжелее спирта,
следовательно, концентрация спирта, определенная в их присутствии, будет ниже
истинной. По таблице паспорта поплавка находят значение концентрации спирта
в отгонах всех проб (оиа имеет отрицательный знак) и вычисляют
среднеарифметическое значение Ct. При такой поправке формула для вычисления
концентрации спирта в бражке примет следующий вид:
x
- С < -СП
-~r~\~k~)-
к
Температуру повисаиия поплавка измеряют с точностью ±0,1°С, прн этом
точность определения концентрации спирта в отсутствии мешающих примесей
составляет ±0,01% абс.
Предостережения. 1. Во время анализа надо уделить особое внимание
предотвращению потерь спирта прн его отгонке из пробы. Пробка,
закрывающая колбу, должна плотно входить в горло колбы. Надо иметь в виду, что
первые капли отгона содержат самое большое количество спирта. 2. Величина
поправочного коэффициента К или поправки Сі обусловлена количеством и
природой летучих веществ, присутствующих в бражке. Оиа изменяется в
зависимости от сырья и режима гидролиза, поэтому нельзя установить для всех
заводов одинаковую поправку. На одном и том же заводе поправку надо
устанавливать периодически при смеие сырья или изменении режима
гидролиза. 3. При ежедневном пользовании поплавком его надо через 1—2 дия
промывать хромовой смесью.
168
Определение этилового спирта вакуум-нитритным методом
Для определения этилового спирта в бражке, барде и лю-
тере спиртовых цехов и в сточной воде предназначен метод,
основанный на реакции взаимодействия спирта с азотистой
кислотой, в результате которой образуется этилиитрит (газ).
Реакция предложена Фишером и Шмидтом. Она протекает по
следующему уравнению:
С2Н5ОН+HNO2 -* Q>H5ONO2+H2O.
Азотистую кислоту получают при разложении азотнокислого
натрия соляной кислотой. Полученный этилнитрит промывают
щелочным раствором перманганата калия для
очистки от окислов азота, которые образовались во
время реакции, и для удаления следов
азотнокислого натрия. Промытый этилиитрит обрабатывают
одновременно серной кислотой и титроваииым
раствором пермаигаиата калия. При этом серная
кислота с этилиитритом образует свободную азотистую
кислоту, которая сразу окисляется перманганатом
калия до азотной. Для определения количества
пермангаиата калия, израсходованного на
окисление азотистой кислоты, которое эквивалентно
количеству этилового спирта, находят избыток
пермангаиата калия, окисляя им йодистый калий в кис-
Рис. 56. Установка для определения спирта вакуум-нитритным
методом
лой среде до йода. Выделившийся йод оттитровывают раствором
гипосульфита натрия.
Мешающие вещества. Спирты и летучие сернистые
соединения.
Аппаратура. Установка для определения спирта вакуум-инт-
ритиым методом (рис. 56). Установка для титрования.
Вакуумный насос.
Установка для определения спирта состоит из
стеклянного сосуда А, вставленного в колбу Бунзена
вместимостью 500—700 мл. Верхняя половина сосуда обмотана
асбестовым шнуром для изоляции тепла руки, которой держат сосуд во
время анализа (при встряхивании). Вместимость сосуда должна
быть обозначена на нем. Если обозначения иет, то перед
анализом определяют его емкость следующим образом. В сосуде
создают максимально возможный вакуум, а затем через воронку
заполняют его водой из мерного цилиндра. Объем воды,
пошедший на заполнение сосуда А, измеренный мерным цилиндром,
считают за объем сосуда А.
169
Реактивы и их приготовление. 1. Азотистокислий натрий х. ч.
(NaNO2), 50%-ный раствор. 2. Соляная кислота (НС1), раствор.
Приготовление. 55 мл НС1 плотностью 1,19 г/см3
разбавляют водой до объема 100 мл. 3. Едкий натр, 25%-ный раствор.
4. Марганцовокислый калий, 3%-ный раствор. 5. Серная кислота
(H2SO4), 10%-ный раствор. Приготовление. В мерную
колбу вместимостью 1 л вливают около 500 мл
дистиллированной воды, затем осторожно, медленно по стенке вливают 56,7 мл
H2SO4 плотностью 1,84 г/см3 и объем раствора доводят до 1 л,
прибавляя дистиллированную воду. 6. Марганцовокислый калий
(КМпО4), 0,02 н. раствор. Приготовление. Раствор готовят
из фиксанала или растворяя 0,64 г КМпО4 в 1 л
дистиллированной воды. Приготовленный раствор переливают в темную
склянку, оставляют на 7 суток, а затем устанавливают его титр
по 0,02 н. раствору гипосульфита натрия. 7. Гипосульфит натрия
(ЫагЭгОз), 0,02 н. раствор. Приготовление. Раствор готовят
из фиксанала или по навеске. 5 г гипосульфита натрия х. ч.
растворяют в 1 л дистиллированной воды, из которой удалена
углекислота кипячением в течение 1—2 ч. Полученный раствор
перемешивают и определяют его титр по хромпику или по йоду.
Раствор хранят в склянке из темного стекла, к которой
присоединена бюретка с приспособлением для автоматической
установки уровня раствора на нуле. Воздух в склянку должен
проходить через трубку с натронной известью. 8. йодистый калий,
10%-ный раствор. 9. Крахмал, 0,5%-ный раствор.
Приготовление. Растворяют смесь из 0,5 г крахмала и 10 мл воды и
выливают ее тонкой струей в 90 мл кипящей
дистиллированной воды, размешивают стеклянной палочкой и кипятят
2—3 мин.
Реактивы, употребляемые для анализа, кроме раствора
гипосульфита, хранят в склянках, закрытых резиновыми пробками,
в которые вставлены пипетки нужного объема. Верхнюю часть
пипеток закрывают резиновыми колпачками (резиновая трубка,
закрытая с одного конца пробкой). Растворы азотнокислого
натрия, соляной кислоты и щелочи хранят в склянках
вместимостью 200—250 мл, растворы перманганата и серной кислоты —
в склянках вместимостью 500 мл. Перед анализом склянки
с реактивами размещают на столе в порядке очередности
использования растворов.
Подготовка проб. Анализируемую пробу разбавляют так,
чтобы в ней содержалось спирта в пределах 0,005—0,150%.
Бражку, барду, лютер и конденсаты паров их самоиспарения
на гидролизных заводах анализируют без отгонки спирта. На
сульфитно-спиртовых заводах спирт в бражке и барде
обязательно определяют после отгонки. Лютер и конденсаты паров
самоиспарения барды и лютера анализируют без перегонки.
Ход анализа. В сосуде А создают вакуум 39,99—53,33 кПа
C00—400 мм рт. ст.) водоструйным насосом и присоединяют
170
к колбе Бунзена. При пользовании более мощными вакуумными
насосами на вакуумной линии насос—колба Бунзена ставят
тройник, чтобы регулировать величину вакуума. Зажимом,
надетым на резиновую трубку на отводе тройника, соединенного
с атмосферой, Г раз перед анализом для постоянного
пользования устанавливают величину притока воздуха. Если нет
манометра, то вакуум в сосуде А определяют, наливая в него воду
через воронку при закрытом нижнем кране. При нужном для
анализа вакууме вода должна заполнить сосуд наполовину его
объема. При большем или меньшем заполнении водой сосуда Л
увеличивают или уменьшают приток воздуха зажимом и
добиваются нужного результата. Затем в сосуд А последовательно
вводят через воронку 1 мл 50%-ного раствора азотистокислого
натрия, 10 мл анализируемого раствора и 1 мл 20%-ной соляной
кислоты. При вливании растворов в сосуд А верхний кран
открывают очень мало, чтобы пропустить жидкость и не впустить
воздух в сосуд. Сосуд А отсоединяют от насоса, снимают каучук
с колбы Бунзена, вынимают его из колбы и в течение 0,5 мин
энергично встряхивают для ускорения реакции образования
этилнитрита. После встряхивания в сосуде А над жидкостью
находится этилнитрит и окислы азота (побочные продукты
протекающих реакций). В сосуд А вводят 2 мл 25%-ного раствора
едкого натра, содержимое сосуда встряхивают 5 раз и сосуд
вставляют в колбу Бунзена. Колбу соединяют с насосом,
создают в ней вакуум больший, чем в реакционном сосуде, и
осторожно, открыв не полностью нижний кран сосуда, выпускают
из него жидкость. Во избежание потерь газа в трубке сосуда
оставляют столбик жидкости высотой 5 мм. Для промывки этил-
нитрита в сосуд А вводят через воронку одновременно 2 мл
25%-ного раствора едкого натра и 10 мл 3%-ного раствора
марганцовокислого калия. Сосуд вынимают из колбы и встряхивают
в течение 0,5 мин, снова вставляют в колбу, в которой создают
вакуум и, открывая не полностью нижний кран, удаляют из
сосуда щелочной раствор перманганата, оставляя столбик
раствора над краном. Таким же приемом промывают сосуд А 4 раза
дистиллированной водой от щелочного раствора перманганата.
В сосуд А, содержащий очищенный этилнитрит, вводят
последовательно 20 мл 0,02 н. раствора перманганата калия и 20 мл
10%-ного раствора серной кислоты. Сосуд встряхивают в
течение 1 мин (песочные часы), вставляют в чистую колбу Бунзена
вместимостью 250 мл и, открывая верхний кран, а потом
нижний, выливают жидкость из сосуда в колбу. Сосуд промывают
2 раза, расходуя по 10 мл дистиллированной воды, промывные
воды сливают в ту же колбу. К кислому раствору перманганата
калия в колбе сразу приливают 10 мл 10%-ного раствора
йодистого калия и выделившийся йод титруют 0,02 н. раствором
гипосульфита натрия. Вначале раствор гипосульфита натрия
приливают быстро до получения слабо-желтой окраски раствора.
171
Затем, добавив 0,5 мл раствора крахмала, продолжают
прибавлять раствор гипосульфита натрия по каплям при постоянном
перемешивании раствора до исчезновения синей окраски
титруемой жидкости.
Для каждой серии анализов делают холостой опыт, в
котором вместо 10 мл исследуемого раствора берут 10 мл
дистиллированной воды. Поправку холостого опыта вычисляют как
разность между объемом 0,02 н. раствора перманганата калия,
взятым на анализ, и объемом 0,02 н. раствора гипосульфита натрия,
пошедшего на титрование.
Расчет. Содержание спирта в анализируемой пробе вычисляют по
формуле
v (а — б — в) 0,000581
где X — концентрация спирта в пробе, % объемный; а — объем 0,02 н.
раствора пермаиганата калия, взятый на анализ, мл; б — объем 0,02 н. раствора
гипосульфита натрия, пошедший иа анализ, мл; в — поправка холостого опыта,
мл; и —разведение исходной пробы; 0,000581—количество этилового спирта
эквивалентное 1 мл 0,02 н. раствора перманганата калия, мл; Кі —
эмпирический поправочный коэффициент при отклонении температуры опыта от 20°С,
Кг — эмпирический поправочный коэффициент, учитывающий зависимость
результата анализа от объема сосуда А. К\ и /С2 находят по табл. IV, с. 319і;
10 — объем разбавленного исследуемого раствора, взятый на анализ, мл.
Концентрацию спирта определяют с точностью ±1—1,5% оти.
Предостережение. Анализ нельзя проводить при температуре
окружающего воздуха выше 25СС и при солнечном освещении рабочего места
аналитика.
Определение этилового спирта экспрессным методом
Для быстрого определения концентрации этилового спирта
в барде и в лютере на рабочем месте аппаратчика предназначен
метод, основанный на реакции окисления спирта двухромовокис-
лым калием в кислой среде, протекающей по уравнению
4-2K2SO4+llH2O.
При этой реакции шестивалентный хром восстанавливается
до трехвалентного, что сопровождается изменением окраски
раствора из желтой в зеленую. Интенсивность зеленой окраски
зависит от количества спирта в испытуемой пробе. Сравнивая
визуально полученную окраску раствора с окраской эталонов
стандартной шкалы, находят концентрацию спирта в
анализируемой пробе.
Мешающие вещества. Летучие окисляемые вещества.
Аппаратура. Набор колориметрических пробирок.
Реактивы и их приготовление. 1. Двухромовокислый калий
(К2СГ2О7), раствор в серной кислоте. Приготовление. В
горячей серной кислоте плотностью 1,84 г/см3 растворяют 1,80 г
172
К2СГ2О7. После охлаждения объем раствора доводят до 100 мл,
прибавляя ту же кислоту. 2. Эталонные растворы этилового
спирта в воде: раствор 1 содержит 0,20% спирта, раствор 2 —
0,24% и раствор 3 — 0,28%.
Подготовка пробы. Концентрацию спирта в барде и в лю-
тере определяют, анализируя конденсаты их паров
самоиспарения, которые отбирают, как описано на с. 12.
Ход анализа. Берут четыре сухие плоскодонные колбы
вместимостью 50 мл. В одной из них проводят анализ исследуемой
пробы, а в остальных — анализ эталонных растворов различной
концентрации для приготовления стандартной шкалы. В первую
колбу вливают 5 мл исследуемого конденсата паров
самоиспарения барды или лютера, во вторую — 5 мл раствора 1,
в третью — 5 мл раствора 2 и в четвертую — 5 мл раствора 3.
Затем в каждую колбу осторожно, по стенке, из бюретки
вливают по 2,5 мл раствора двухромовокислого калия в серной
кислоте. Содержимое колб перемешивают, закрывают стеклянными
пробками и оставляют на 5 мин.
Для сравнения полученных окрасок растворы из колб
переливают в колориметрические пробирки из бесцветного стекла,
одинакового диаметра с притертыми пробками, пробирки ставят
в штатив и визуально определяют, с окраской какого
эталонного раствора совпадает или наиболее близка окраска раствора
с исследуемой пробой.
Расчет. Концентрация спирта в приведенных эталонных растворах
рассчитана на основе экспериментальных данных о составе конденсатов паров
самонспарения барды и лютера. Установлено, что в конденсатах паров
самоиспарения концентрация спирта в 10 раз больше, чем в барде или лютере,
а концентрация окисляемых веществ, являющихся примесями, составляет
около 0,13%. Таким образом, концентрация спирта в эталонном растворе 1,
равная 0,20%, соответствует концентрации спирта в барде или лютере —
0,007%, в растворе 2 — 0,010% и в растворе 3 — 0,015%. Стандартную
шкалу эталонных растворов готовят 1 раз в смену, окраска не изменяется
в течение 8 ч.
Концентрацию этилового спирта определяют с точностью ±7—10% отн.
Предостережение. Величина поправки на окисляемые вещества меняется
в зависимости от сырья и от технологического режима, поэтому ее
периодически проверяют, делая анализы барды и лютера, а также их конденсатов
вакуум-нитритным методом.
Определение спирта стеклянным спиртомером
Для быстрого определения концентрации этилового спирта
в водных растворах [53, 54] предназначен метод, основанный на
измерении концентрации спирта ареометром, отградуированным
в объемных процентах спирта при 20°С. Если измерение
делают при другой температуре, то по официальной таблице
находят, какой концентрации соответствует показание
спиртомера [55].
173
Мешающие вещества. Вещества, имеющие плотность,
отличающуюся от плотности воды.
Аппаратура. Стеклянный спиртомер. Термометр с ценой
деления не более 0,5°С. Цилиндр без носика. Стеклянная
мешалка.
Стеклянный спиртомер представляет собой
ареометр, отградуированный в объемных процентах этилового спирта
при 20сС. В некоторых спиртомерах внутри находится
термометр. В зависимости от цены наименьшего деления, пределов
измерений и наличия термометра в спиртомере
установлены и выпускаются пять типов стеклянных
спиртомеров: А, Б, В, Г типы без термометра и
тип Д с термометром (рис. 57). Спиртомеры
каждого типа имеют номера, соответствующие
различным пределам измерений. Мешалкой служит
стеклянный стержень, один конец которого согнут в плоскую
спираль.
Ход анализа. Стеклянный цилиндр, спиртомер,
термометр и мешалку тщательно моют теплой водой,
насухо вытирают мягким полотенцем и
споласкивают анализируемым раствором спирта. В чистый ци-
линдр наливают 3Д объема исследуемого спирта,
перемешивают его мешалкой, дают жидкости успокоиться
и погружают в нее термометр. Осторожно, держа
спиртомер за верхний конец над шкалой, опускают
его в цилиндр до начала шкалы и выпускают из рук
так, чтобы он свободно погружался в жидкость под
действием своего веса. Стержень спиртомера не
должен быть смочен, больше, чем на 2—3 мм выше
V
Рис. 57. Стеклянный спиртомер с термометром
уровня жидкости. Через 2—3 мин, когда спиртомер примет
температуру анализируемой жидкости, определяют, на каком
делении уровень жидкости пересекает шкалу спиртомера. Если
линия уровня жидкости пересекает шкалу между делениями, то на
глаз определяют, какую часть деления надо прибавить к целым
делениям. Отсчет делают по нижнему мениску (см. рис. 35).
Затем определяют температуру анализируемого раствора и
вынимают из него спиртомер и термометр.
Расчет. При определении концентрации спирта при 20°С показание
спиртомера соответствует содержанию спирта в пробе в объемных процентах. При
определении показаний спиртомера при другой температуре результат анализа
находят по официальной табл. III для стеклянных спиртомеров [55].
Концентрацию спирта определят с точностью от 1% до 0,1% абс. в
зависимости от цены деления на спиртомере.
174
Определение этилового спирта металлическим спиртомером
Для определения крепости готового этилового спирта и
содержания спирта в чистых водных растворах любой
концентрации при температурах от —25 до +40°С [53, 56] предназначен
метод, основанный на измерении температуры и плотности
анализируемого раствора специальным ареометром —
металлическим спиртомером. Результаты анализа находят по
официальным таблицам [55] на основании показаний спиртомера
и термометра. Концентрацию этилового спирта в спиртоводных
растворах выражают в процентах по объему или в процентах
по массе. При высоком содержании этилового спирта в растворе
его концентрацию называют крепостью спирта,
а проценты по объему— градусами крепости.
Мешающие вещества. Все вещества, имеющие
плотность, отличающуюся от плотности воды.
Аппаратура. Металлический спиртомер.
Термометр с ценой деления 0,1°С. Цилиндр
вместимостью 250—500 мл диаметром не менее 60 мм.
Мешалка.
Металлический спиртомер (рис. 58)
представляет собой ареометр переменной массы.
Он состоит из полого шара с двумя стержнями:
верхний стержень прямоугольного сечения и ниж-
Рис. 58. Металлический спиртомер и гирька
ний — в виде конуса с круговым сечением. На верхнем
стержне нанесена условная шкала, имеющая 10 больших
делений, каждое из которых разделено на 10 делений. Ниже
отметки «0» поставлена цифра 100. На конце нижнего
стержня имеется грушевидный наконечник, служащий
балластом для обеспечения вертикального положения спиртомера,
плавающего в анализируемом растворе во время измерения.
К каждому спиртомеру прикладывается комплект из восьми
гирек разной массы с обозначениями 20, 30, 40, 50, 60, 70, 80, 90.
Гирьки навешивают на балласт выпуклой стороной для
изменения массы спиртомера, которое необходимо при измерении
различных плотностей спиртоводных растворов. Спиртомер и гирьки
позолочены для предотвращения коррозии. Их хранят в
специальном футляре. На спиртомере и гирьках поставлен номер
спиртомера. Гирьки, приложенные к одному спиртомеру, нельзя
использовать для измерения плотности другим спиртомером.
Каждый спиртомер подлежит государственной проверке, после
которой к спиртомеру прикладывается свидетельство с
указанием поправок на его показания.
175
Ход анализа. Стеклянный цилиндр, термометр и мешалку
тщательно моют теплой водой, насухо вытирают мягким
полотенцем и споласкивают анализируемым раствором спирта.
Спиртомер и гирьки обтирают, слегка касаясь позолоченной
поверхности сначала мягким полотенцем, смоченным спиртом,
а затем сухим полотенцем. Исследуемый спирт наливают в
стеклянный цилиндр на 3Д его объема, погружают в него термометр
и мешалку, жидкость перемешивают и дают ей успокоиться.
Берут спиртомер за верхнюю часть стержня (выше шкалы),
осторожно, держа его вертикально, погружают в спирт до
верхнего стержня и выпускают из рук, чтобы дальше он погружался
свободно. Если спиртомер погрузится так, что уровень
жидкости будет ниже шкалы, то его вынимают из спирта, на
балласт навешивают гирьку 90 и снова осторожно погружают
в спирт. Если уровень жидкости в цилиндре опять будет ниже
шкалы, то гирьку заменяют на более тяжелую с отметкой 80.
Так повторяют до тех пор, пока не подберут гирьку, при
которой уровень жидкости в цилиндре будет пересекать шкалу
спиртомера. Если спиртомер без гирьки, погружается так, что
уровень жидкости выше шкалы, то в этом случае измерение надо
проводить при более низкой температуре жидкости и
окружающей среды. Спиртомер, погруженный в исследуемый спирт,
должен свободно плавать в вертикальном положении, не касаясь
стенок цилиндра. На поверхности спиртомера или около гирьки,
подвешенной на его балласте, не должно быть пузырьков
воздуха. Если есть пузырьки, то их убирают, слегка встряхивая
спиртомер так, чтобы подвешенная гирька во время
встряхивания двигалась вверх и вниз по стержню. Верхний стержень
спиртомера при измерении не должен быть смочен более чем на
2—3 мм выше уровня жидкости (см. рис. 35).
После погружения спиртомера ждут 3—4 мин, чтобы он
принял температуру раствора, и определяют, на каком делении
уровень жидкости пересекает шкалу спиртомера. Отсчет ведут по
нижнему мениску. Глаза аналитика сначала должны быть ниже
уровня жидкости, чтобы видеть основание мениска
(поверхность жидкости) в виде эллипса. Постепенно поднимая голову,
аналитик наблюдает, как эллипс, суживаясь, превращается
в прямую линию, которая отчетливо проектируется на шкалу
спиртомера. Замечают деление шкалы, которое пересекает эта
линия. Если линия пересекает шкалу между делениями, то на
глаз определяют, какую часть деления надо прибавить к целым
делениям. При пользовании спиртомером без гирьки к отсчету
по шкале прибавляют число 100, указанное на стержне
спиртомера. При употреблении спиртомера с гирькой к отсчету по
шкале прибавляют число, указанное на гирьке. Например, если
уровень жидкости пересекает шкалу спиртомера без гирьки на
высоте 1,55 деления, то показание спиртомера равно 101,55.
Если уровень жидкости пересекает шкалу спиртомера, на кото-
176
рый повешена гирька 60, на высоте 9,75 деления, то показание
спиртомера равно 69,75. Считывают показания термометра,
погруженного в анализируемый раствор спирта. После измерений
спиртомер и гирьку сразу вытирают, слегка касаясь мягким
полотенцем их поверхности, и кладут в футляр.
Расчег. На основании результатов измерения температуры анализируемого
раствора и замера показаний спиртомера по официальным таблицам [55] иа-
ходят крепость спирта, т. е. концентрацию спирта в анализируемом растворе
в процентах по объему и, если надо, пересчитывают в проценты по массе или
вычисляют содержание безводного спирта в анализируемом растворе,
выражая его в литрах или килограммах. В табл. 6 приведен образец официальной
таблицы 4.
ТАБЛИЦА 6
Относительное содержание спирга в зависимости от показании
металлического спиртомера и температуры раствора (часть таблицы 4)
Температура,
°С
+30.0
29.5
29.0
28.5
Показания спиртомера
108,0
96,4
96.5
96.6
96,7
107,8
107,6
107,4
Содержание спирта при 20°С, % (по объему
96,3
96.4
96,5
96.6
96.2
96.3
96,4
96.5
96,1
96.0
95.9
107,2
96.0
96.9
В первой вертикальной графе дана температура жидкости во время
измерения в пределах от +40° до —25°С через каждые 0,5"С. В верхней
горизонтальной графе помещены показания металлического спиртомера в делениях
условной шкалы от ПО до 20 с интервалом 0,2. Цифры в остальных графах
обозначают объемное содержание спирта в расгворе при соответствующих
температурах и показаниях спиртомера. Для определения концентрации спирта
находят число на пересечении строки, соответствующей температуре
жидкости во время измерения, и столбца, соответствующего показанию
спиртомера. Например, при температуре анализируемой жидкости 29°С спиртомер
показал 107,6. Следовательно, концентрация спирта в анализируемом растворе
96,4% (по объему).
Проценты по объему показывают, сколько миллиметров абсолютного
A00%-ного) спирта находится в 100 мл раствора. Проценты (по. массе)
показывают, сколько граммов абсолютного спирта находится в 100 г его
раствора. Для перевода процентов (по массе) в проценты (по объему) и
наоборот пользуются следующим уравнением:
0.78927Л = Cd,
где 0,78927 — плотность безводного спирта при 20°С, г/см3; А — концентрация
спирта в растворе, % (по объему); С — концентрация спирта в растворе, %
(по массе); d — плотность водиоспиртового раствора при +20°С, г/см3.
Этиловый спирт определяют с точностью ±0,05% абс.
Определение этилового спирта окислительиым методом
Для определения этилового спирта в присутствии метанола,
эфиров и альдегидов в метанольной и эфироальдегидной
фракциях предназначен метод, основанный на реакции окисления
12 Заказ № 142
177
этанола двухромовокислым калием при нагревании в
сернокислой среде до уксусной кислоты.
3C2H5OH+2K2Cr2O7+8H2SO4 — 3CH3COOH+2K2SO4+
+2Cr2(SO4K+llH2O.
При этих условиях метанол окисляется до углекислого газа,
этилацетат и ацетальдегид — до уксусной кислоты. Углекислый
газ удаляют кипячением реакционной смеси с обратным
холодильником. Образовавшуюся уксусную кислоту отгоняют из
реакционной смеси и собранный конденсат титруют 0,1 н.
раствором едкого натра. Найденное количество уксусной кислоты
соответствует сумме этанола, ацетальдегида и этилацетата.
Отдельно в пробе определяют концентрацию эфиров методом
омыления и альдегиды методом с гидраксиламином, вычисляя
результаты анализа соответственно в пересчете на этилацетат
и на ацетальдегид. По разности между количеством
окисляемых, образующих уксусную кислоту, и суммой эфиров и
альдегидов находят содержание этилового спирта.
Мешающие вещества. Этилацетат, ацетальдегид.
Аппаратура. Установка для кипячения с обратным
холодильником (см. рис. 13). Установки для отгонки летучих веществ
и для титрования. Ареометр.
Реактивы и их приготовление. 1. Двухромовокислый калий,
10%-ный раствор. 2. Серная кислота, 40%-ный раствор. 3.
Едкий натр, 0,1 н. раствор. 4. Фенолфталеин, 1%-ный спиртовый
раствор. 5. Все реактивы для определения альдегидов и эфиров
по ГОСТ 10749 — 72.
Ход анализа. В мерную колбу вместимостью 200 мл вливают
50 мл дистиллированной воды, колбу с водой взвешивают с
точностью до 0,1 г, вливают в нее 5—10 мл исследуемой пробы,
снова взвешивают и объем раствора в колбе доводят водой до
метки. По разности результатов взвешиваний находят количество
пробы, взятое для разведения. В колбу вместимостью 250 мл
помещают 10 мл разбавленного раствора, добавляют 50 мл
10%-ного раствора двухромовокислого калия, 50 мл 40%-ного
раствора серной кислоты и несколько кусочков пемзы или
пористого стекла для создания равномерности кипения. Колбу
присоединяют к обратному холодильнику, ставят на электроплитку,
нагревают до кипения и кипятят 30 мин. Затем плитку
отставляют и колбе дают остыть в течение 10 мин. Холодильник
промывают над колбой, расходуя около 30 мл воды, колбу
отсоединяют от холодильника и сразу закрывают пробкой, в
которую вставлен каплеотбойник и капельная воронка вместимостью
300—350 мл. Колбу соединяют с холодильником установки для
отгона летучих веществ, включают электроплитку и начинают
отгон уксусной кислоты из реакционной смеси. Нагрев должен
быть таким, чтобы за 20 мин отгонялось 100 мл конденсата.
178
Приемником конденсата служит коническая колба вместимостью
500 мл. После отгонки 80 мл конденсата начинают добавлять
дистиллированную воду из капельной воронки, регулируя
количество капель так, чтобы уровень жидкости в колбе не снижался.
За 1 ч 20 мин собирают 400 мл конденсата, отгонку
прекращают и собранный конденсат титруют 0,1 н. раствором едкого
натра в присутствии фенолфталеина. Аналогично проводят
холостой опыт, в котором вместо пробы берут 10 мл
дистиллированной воды. Холостой опыт делают на каждую вновь
приготовленную партию реактивов.
Содержание эфиров и альдегидов в граммах на 100 мл
определяют в пробе по ГОСТ 1074—72. Для перевода полученных
результатов в граммы на 100 г определяют плотность фракции
ареометром и делают вычисление как описано на с. 179.
Расчет. Содержание окисляемых веществ, дающих уксусную кислоту,
вычисляют по формуле
X - (а ~ б>> °-0046 ' 20° ' 10°
где X — концентрация окисляемых веществ, дающих уксусную кислоту, %
(по массе); g— количество исследуемой пробы, взятое на анализ, г; а — объем
0,1 н. раствора NaOH, пошедший на титрование конденсата пробы, мл;
б— объем 0,1 н. раствора NaOH, пошедший на титрование в холостом опыте,
мл; 200 — объем разведеной анализируемой пробы, мл; gi—объем разведеной
пробы, взятый на анализ, мл; 0,0046 — количество окисляемых веществ
эквивалентное 1 мл 0,1 н. раствора едкого натра.
Содержание этанола в исследуемой пробе вычисляют по формуле
где X— концентрация этанола в исследуемой пробе, г/100 г;
Xt—концентрация суммы окисляемых веществ, дающих уксусную кислоту, г/100 г; Хэ —
концентрация эфиров в исследуемой пробе в пересчете на этилацетат, г/100 г;
Хц — концентрация альдегидов в исследуемой пробе в пересчете на ацеталь-
дегид, г/100 г; 1,04 — коэффициент, показывающий, во сколько раз
молекулярная масса эфиров и альдегидов меньше молекулярной массы этанола.
Этиловый спирт определяют с точностью ±5—7% отн.
Определение метилового спирта окислительным методом
Для определения концентрации метилового спирта в
метаноле и метанольной фракции, содержащей этанол, эфиры и
альдегиды, предназначен метод, основанный на реакции окисления
метилового спирта двухромовокислым калием при нагревании
в кислой среде до СО2.
CH3OH+K2Gr2O7+4H2SO4 — CO2+K2SO4-j-Cr2(SO4K+6H2O.
Образующийся углекислый газ отгоняют слабым током
воздуха и поглощают титрованным раствором гидроокиси бария
(Ва(ОНJ). Избыток гидроокиси бария оттитровывают соляной
кислотой и по количеству углекислоты, вступившей в реакцию
с Ва(ОНJ) находят содержание метанола в пробе. Уксусная
12* 179
кислота, образующаяся из этилового спирта, не мешает
определению, потому что при отгонке углекислого газа (СОJ она
конденсируется в обратном холодильнике и стекает в реакционную
колбу.
Мешающие вещества. Углекислый газ, находящийся в
воздухе помещения, где делают анализ. Вещества, которые в
условиях реакции окисления метанола образуют углекислый газ.
Аппаратура. Установка для определения метанола (рис. 59).
Установка для титрования.
Реактивы и их приготовление. 1. Двухромовокислый калий,
10%-ный раствор. 2. Серная кислота, 40%-ный раствор. 3. Гид-
Рис. 59. Установка для определения метанола
окислительным методом;
/ — трубка Базырииой; 2 — деталь
3 — отверстия
трубки Базыриной;
роокись бария, 0,12—0,14 н. раствор. Приготовление.
В 1 л дистиллированной воды, из которой углекислый газ
удален кипячением, растворяют 20—25 г гидроокиси бария
(Ва(ОН)г) х. ч. Растворение проводят при интенсивном
перемешивании, а затем раствору дают отстояться в течение 10—12 ч.
За это время углекислый барий (ВаСО3), присутствующий в
растворе, оседает на дно, прозрачный раствор сливают сифоном
в склянку установки для хранения баритовой воды (см. рис. 16).
4. Соляная кислота, 0,01 н. раствор. 5. Едкое кали, 50%-ный
раствор. 6. Фенолфталеин, 1%-ный спиртовый раствор.
Подготовка проб. Пробу разбавляют водой до содержания
метанола в пределах 0,1—0,7%. В мерную колбочку
вместимостью 100 мл наливают 5—10 мл дистиллированной воды,
колбочку взвешивают на аналитических весах, добавляют
исследуемую пробу в количестве 0,5 до 10 мл, в зависимости от
предполагаемого содержания метанола, колбу снова взвешивают. По
180
разности результатов взвешивания находят количество пробы,,
взятое для разбавления.
Ход анализа. В трубку Базыриной (см. рис. 59) вливают
100 мл раствора гидроокиси бария и сразу закрывают ее
отводные трубки, надевая на них резиновые трубочки, один конец
у которых закрыт кусочками стеклянных палочек. В
реакционную колбу вливают 50 мл 10%-ного раствора двухромовокис-
лого калия, 50 мл 40%-ной серной кислоты и 10 мл
разбавленной пробы. Колбу сразу закрывают пробкой, в которую
вставлены обратный холодильник и трубка, соединяющая ее
с поглотительными склянками, в которые налит раствор едкого
кали. С трубки Базыриной снимают пробки и один ее конец
соединяют с верхней частью обратного холодильника, а другой —
с контрольной склянкой, в которую налито 40—50 мл раствора
гидроокиси бария. Отводы контрольной склянки всегда должны
быть закрыты пробками, которые снимают только во время
опыта.
Контрольную склянку присоединяют к двугорлой склянке и
включают водоструйный насос или аспиратор таким образом,
чтобы через систему проходил слабый ток воздуха A пузырек
в секунду). Включают колбонагреватель и подогревают
реакционную колбу при очень слабом нагреве (подложить асбест
и приподнять колбу над ним на 4—5 см) в течение 40—50 мин
до закипания содержимого колбы. В это время интенсивно
выделяется углекислый газ, который поглощается баритовой водой
в трубке Базыриной. Как только жидкость закипит, асбест
убирают, реакционную колбу опускают на колбонагреватель,
реагирующую смесь кипятят еще 50 мин и заканчивают анализ
следующим образом. Выключают колбонагреватель, закрывают
винтовой зажим между реакционной колбой и поглотителями
с раствором едкого кали, закрывают зажимами отводы до и
после трубки Базыриной, отключают контрольную склянку от
двугорлой склянки, отключают трубку Базыриной от системы,
баритовую воду быстро сливают в сухой цилиндр вместимостью
150 мл, закрывают его пробкой и дают отстояться осадку
ВаСОз. Отмеривают пипеткой 20 мл отстоявшейся прозрачной
жидкости, помещают ее в сухую колбочку и сразу титруют
0,1 н. раствором соляной кислоты в присутствии фенолфталеина,,
до исчезновения розовой окраски от одной капли кислоты.
На каждую партию реактивов делают холостой опыт, в
котором на анализ по вышеописанной методике берут вместо
разведенной пробы фракции, 10 мл дистиллированной воды.
Расчет. Содержание метанола в исследуемой фракции вычисляют по фор-
муле v (я-бH.0016 • ІООпіОО
где X — концентрация метанола в пробе, г/100 г; а — объем 0,1 н. раствора
НС1, пошедший на титрование 20 мл раствора гидроокиси бария в холостом
181
¦опыте, мл; б — то же при анализе пробы, мл; п — разведение; g — количество
пробы, взятое для разведения, г; gi — объем разведенной пробы, взятый на
анализ, мл; 20 — объем раствора гидроокиси бария, взятый на титрование, мл;
100 — объем раствора гидроокиси бария, взятый для поглощения углекислого
газа, мл; 0,0016 — количество метанола эквивалентное 1 мл 0,1 н. раствора
гидроокиси бария, г.
Полученные данные анализа из процентов по массе в проценты по
объему переводят с помощью табл. V (см. с. 318).
Метанол определяют с точностью ±2—4% оти.
Предостережение. Перед анализом надо проверить герметичность уста-
ковки и следить, чтобы во время анализа не было подсоса воздуха,
содержащего углекислый газ.
ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ ИЛ ЭТИЛОВЫЙ СПИРТ И МЕТОДЫ
ЕГО ИСПЫТАНИЯ ПО СТАНДАРТАМ РАЗНЫХ СТРАН
Технические требования
Главными показателями качества этилового спирта,
которые приняты во всех странах, являются внешний вид и вкус
этилового спирта, его крепость, наличие кислот, эфиров,
альдегидов, сивушного масла и метанола [57, 59, 60, 62, 63, 65].
Другие показатели, как содержание фурфурола, окисляемость и
сухой остаток, приняты не для всех сортов спирта. Наличие ке-
тонов и растворимость в воде проверяют только в США.
Содержание серы и щелочи определяют только в СССР. Технические
требования, предъявляемые к качеству этилового спирта в
некоторых странах, приведены в табл. 7.
Одинаковые качественные показатели в разных странах
оценивают, используя неодинаковые методы испытания спирта.
Для сравнения методов испытания этилового спирта,
применяемых в нашей стране [61], с методами, применяемыми в Польше
[58], в Румынии [64] и в США [65], ниже приведены прописи
"методов испытания спирта по стандартам этих стран.
Определение крепости спирта
В СССР крепость спирта определяют металлическим
спиртомером (см. с. 175). В Польше и в Румынии пользуются
спиртомерами, отградуированными в объемных процентах, или
определяют плотность по воде пикнометрическим методом при 15° или
20°С и по официальным таблицам находят крепость спирта.
В США крепость спирта измеряют ареометром или определяют
плотность спирта по отношению к воздуху при 15,56°С, а затем
по таблицам находят концентрацию спирта.
Определение кислотности
Кислотность спирта определяют по стандартам разных стран
титрованием пробы спирта, разбавленной водой 1:1, раствором
¦едкого натра. Перед титрованием пробу кипятят с обратным
382
холодильником для удаления углекислоты, а затем титруют,
применяя индикатор фенолфталеин. В СССР титрование пробы
проводят с индикатором лакмусом в присутствии углекислоты,
которая не мешает определению кислотности в этих условиях
[66]. В 50%-ном спирте фенолфталеин меняет окраску при pH
выше 9, поэтому применение его в качестве индикатора
приводит к завышенным результатам анализа.
Определение сложных эфиров
Определение сложных эфиров по стандартам разных стран
основано на реакции омыления их едким натром. Сложные
эфиры представляют собой соединения, в которых атом
кислорода связан с одним спиртовым остатком (алкилом) и одним
кислотным остатком. По внешнему виду формулы их строение
имеет сходство со строением солей, в которых место металла
занимает углеводородный радикал. При действии едкой щелочи
из них образуется спирт и соль кислоты
C2H5-O-C-CH3+NaOH — C2H5OH+CH3COONa.
О
Реакция протекает в щелочной среде при кипячении
реагирующей жидкости. После омыления эфиров в щелочной среде
пробу титруют кислотой в присутствии фенолфталеина или
лакмуса. Затем находят количество щелочи, взятое на омыление,
которое эквивалентно количеству омыленных эфиров, и
вычисляют содержание эфиров в пробе в пересчете на уксусноэтило-
вый эфир. Определению сложных эфиров мешают альдегиды,
присутствующие в пробе. При содержании альдегидов более 0,5%
их удаляют, обрабатывая пробу солянокислым гидроксила-
мином.
Определение альдегидов
Альдегиды по стандартам разных стран определяют
различно. В Польше и в Румынии применяют колориметрический
метод, основанный на реакции альдегидов с фуксиносернистой
кислотой. В США делают только качественную реакцию по*
обесцвечиванию раствора перманганата калия. В СССР
применяют метод, основанный на реакции с гидроксиламином.
Ниже приводится колориметрический метод определения
альдегидов по стандарту Польши [58]. Метод основан на
способности альдегидов выделять из бесцветной фуксиносернистой
кислоты фуксин, который окрашивает реакционную смесь в
розовый цвет. Количество альдегидов находят сравнением
полученной окраски с окрасками эталонов, содержащих уксусный
альдегид известной концентрации.
183
Технические требования на этиловый
Показатель качества
Внешний вид
(прозрачность, цвет)
Запах
Вкус
Содержание этилового
спирта, % объемы., не
менее
Плотность при 15,56"С,
г/см3
Содержание кислот в
пересчете на уксусную
кислоту, мг/л, не более
Содержание эфиров
в пересчете на уксусно-
этиловый, мг/л, не
более
Содержание альдегидов,
мг/л, ие более
Содержание сухого
остатка, мг/л, не более
Содержание фурфурола,
мг/л
Содержание метанола,
% объемн.
Содержание сивушного
масла, % объемн. не
более
Содержание серы, мг/л
Проба на окисляемость,
мин, не менее
Проба на чистоту
Ацетон и другие кетоны,
язопропил и третичный
бутанол
Растворимость в воде
Содержание щелочи в
пересчете на NaOH, мг/л,
не более
Ректификат из пищевого сырья
Экстра
ГОСТ 5969 — 67
Высшей очистки
I сорт
Прозрачная бесцветная жидкость
Характерный для ректификованного этнлов
96.5
—
12
25
2
—
3
—
20
—
—
—
Свойственный спирту без
96,2
—
15
30
4
—
96
—
20
50
10
—
Не допускается
Должен выдержи
4
—
15
15
—
10
Должен выдержи
—
—
—
—
—
184
спирт по стандартам разных стран
СССР
Ректификат
Высший сорт
технический ГОСТ 18300
I сорт
без механических примесей
ого спирта, без
постороннего п
96.2
15
30
4
4
Выде
вать испытание
4
15
нать испытание
Отсутстнует
запаха посторонних
зивкуса
96
15
30
4
10
ржинает нспыт;
ние
То же
4
Отсутствует
15
—
10
— 72
II сорт
нещестн
96
20
50
10
15
15
10
—
15
Спирт этиловый технический
ГОСТ 17299 — 71
Марка
А
—
95
15
80
250
10
Отсутствует
0.10
0.10
Отсутстнует
—
—
Б
—
94
30
180
350
20
5
0.10
0.10
10
—
—
Показатель качества
Внешний внд
(прозрачность, цвет)
Запах
Вкус
Содержание этилового
спирта, % объемн., не
менее
Плотность при 15,56°С,
г/см3
Содержание кислот в
пересчете на уксусную
кислоту, мг/л, не более
Содержание эфиров в
пересчете на уксусноэтило-
вый, мг/л, не более
Содержание альдегидов,
мг/л, не более
Содержание сухого
остатка, мг/л, не более
Содержание фурфурола,
мг/л
Содержание метанола, %
объемн.
Содержание сивушного
масла, % объемн., не
более
¦Содержание серы, мг/л
Проба на окисляемость,
мин, ие менее
Проба на чистоту
Ацетон и другие кетоны,
изопропил и третичный
бутанол
Растворимость в воде
Содержание щелочи в
пересчете на NaOH, мг/л,
не более
Ректификат
обычный
Польша
из пищевого сырья PN 62/A-79522
высшей очистки
Бесцветная прозрачная жидкость
Свойственный спирту,
допускается слабый
запах побочных
дуктиь орижсг
Свойственный
96
—
20
50
50
10
0,10
0,005
—
20
—
—
—
—
про-
Свойственный
очищенному спирту, без
ощущения других запахов
ия
спирту без других вкусовых
ощущений
96,5
—
15
30
30
10
Не допускается
0,03
0,001
—
40
—
—
—
—
і
Примечание. Прочерк (—J означает, что этого показателя в стан
186
Продолжение табл. 7
Румыния
Ректификат
STAS 4523 - 54
Бесцветная
прозрачная
жидкость
Характерный
—
96
—
40
50
15
—
Следы
То же
0,005
15
—
—
США
Этиловый спирт О-Е-760-В 16/IV 1957
I
Класс А
99,5
0,7962
30
—
10
—
5
—
Класс В
п
Класс А
Класс Б
Бесцветная прозрачная жидкость
Свойственный спирту
То же
95
0,8158
30
—
—
10
—
99,4
0,7967
30
—
10
—
94,9
0,8162
30
—
10
—
Выдерживает испытание
То
5
Выдержива
То
же
5
;т испытания
же
5
—
дарте нет.
Реактивы и их приготовление. 1. Спирт этиловый
абсолютированный. 2. Спирт этиловый ректификат 96°. 3. Этиловый эфир
безводный. 4. Альдегидаммиак или уксусный альдегид. 5.
Серная кислота плотностью 1,84 г/см3. 6. Реактив Виллавеккиа.
Приготовление. В 150 мл горячей воды растворяют 1,5 г
основного фуксина. Раствор охлаждают, добавляют 100 мл
раствора бисульфита натрия плотностью 1,36 г/см3 C9%-ный) и
перемешивают. Добавляют 1 л дистиллированной воды,
осторожно по стенке приливают 15 мл серной кислоты плотностью
1,84 г/см3 и повторно перемешивают. Приготовленный реактив
Виллавеккиа бесцветный или слегка желтоватый, с резким
запахом сернистого газа. Реактив хранят в бутыле из темного
стекла. 7. Реактив Гирарда. Приготовление. 0,15 г
основного фуксина растворяют в 150 мл горячей воды. После
охлаждения в раствор добавляют 100 мл раствора бисульфита
натрия плотностью 1,3 г/см3 C2%-ный), перемешивают его,
добавляют 1 л воды, осторожно, по стенке, вливают 15 мл серной
кислоты плотностью 1,84 г/см3 и взбалтывают. Раствор хранят
в бутыле из темного стекла. 8. Эталонные растворы.
Приготовление. 1,331 г альдегидаммиака растворяют в 1 л 96° спирта.
Полученный раствор содержит 1 г ацетальдегида в 1 л
абсолютного спирта. Из этого раствора, разбавляя его спиртом
ректификатом 96°-ным, готовят растворы, содержащие в 1 л
раствора следующие количества уксусного альдегида: 0,01; 0,02;
0,03; 0,05; 0,07; 0,09 и 0,10 г. Растворы хранят в бутылях, на
которых обозначен номер бутыли и концентрация альдегида
в абсолютном спирте. При использовании уксусного альдегида
эталонные растворы готовят следующим образом. В ампулу,
взвешенную с точностью 0,0002 г, вливают 0,5 г уксусного
альдегида, ампулу быстро запаивают и снова взвешивают. Затем
ампулу помещают в бутыль вместимостью 500 мл, в которую
предварительно вливают 200 мл 96° спирта, и ампулу разбивают
стеклянной палочкой. К полученному раствору добавляют
96°-ного спирта столько, чтобы раствор содержал 1 г альдегида
в 1 л абсолютного спирта. Из полученного раствора готовят
серию эталонных растворов с различным содержанием
альдегида, как описано выше.
Ход анализа. Опр еде л ение альдегидов при
концентрации ниже 0,01 г/л. В колориметрическую пробирку
вливают 5 мл исследуемого спирта, в пять пробирок вливают по
5 мл эталонных растворов различных концентраций. Во все
пробирки добавляют дистиллированную воду до отметки 10 мл.
Затем в каждую пробирку вливают из бюретки по 4 мл
реактива Виллавеккиа, закрывают их притертыми пробками, сильно
встряхивают и ставят в штатив. Через 20 мин сравнивают
окраски растворов исследуемого спирта с эталонными
растворами. Для усиления интенсивности окраски можно прибавлять
по 2 мл реактива Виллавеккиа.
188
Определение альдегидов при концентрации
выше 0,01 г/л. Пробу спирта разбавляют до концентрации 50%
(по объему). Количество воды, необходимое для разбавления
спирта, находят по табл. 8. В колориметрическую пробирку
вливают 10 мл разбавленного до 50% (по объему) спирта, в
остальные пробирки вливают по 10 мл одного из эталонных
растворов, имеющих различные концентрации. В каждую пробирку
добавляют по 4 мл реактива Гирарда, пробирки закрывают
лробками, встряхивают и ставят в штатив. Через 20 мин,
сравнивая интенсивность окрасок растворов, устанавливают с
окраской какого эталона совпадает окраска, которую дал
анализируемый спирт. При более высоком содержании альдегидов
в пробе спирта перед анализом ее разбавляют 50°-ным спиртом.
ТАБЛИЦА 8
Количество миллилитров воды, которое надо добавить к 100 мл спирта
данной крепости, чтобы получить 50%-иыи спирт (при +15°С)
Крепость
спирта, %
по объему
100
99
98
97
Объем воды,
107,4
105,0
102.7
100,4
Крепость
спирта, %
по объему
96
95
94
93
Объем воды,
мл
98,1
95,8
93,6
91.4
Крепость
спирта, %
по объему
92
91
90
89
Объем воды,
мл
89.2
87.0
84.8
82.6
Расчет. Концентрация альдегидов в пробе равна концентрации альдегидов
в эталонном растворе умноженной на разбавление.
При содержании альдегидов в пробе меньше 0,01 г/л точиость определения
±0,0015 г/л, больше 0,01 г/л точность — ±0,005я г/л, где п—разбавление.
Определение метилового спирта
В большинстве стран определение метанола основано на
реакции получения окрашенного соединения при
взаимодействии метанола с фуксиносернистой кислотой. В США в качестве
стадартного принят метод, основанный на получении
окрашенного соединения с хромотроповой кислотой, который приводится
ниже [65]. Метод основан на реакции окисления метанола
перманганатом калия до формальдегида. Количество
образовавшегося формальдегида определяют по его реакции с
хромотроповой кислотой, в результате которой получается окрашенное
соединение. Сравнивая интенсивность окраски полученной
реакционной смеси с окраской эталона, определяют содержание
метанола в испытуемой пробе спирта.
Реактивы и их приготовление. 1. Перманганат калия, водный
раствор (КМпСч). Приготовление. В мерной колбе
вместимостью 100 мл в дистиллированной воде растворяют 3 г М
189
прибавляют 15 мл 85%-ной фосфорной кислоты (Н3РО4) и объем
раствора доводят водой до метки на колбе. Реактив пригоден
в течение 1 месяца. 2. Хромотроповая кислота, 5%-ный водный
раствор кислоты или ее натриевой соли. 3. Эталонный раствор —
0,1% (по объему) метанола в 95%-ном этиловом спирте.
Ход анализа. Порцию пробы спирта разбавляют
дистиллированной водой до содержания спирта 5—6%. В мерную колбу
вместимостью 50 мл вливают 2 мл раствора перманганата,
охлаждают в ледяной бане, прибавляют 1 мл разбавленной пробы
и дают стоять 30 мин в ледяной бане. Обесцвечивают
окисленную пробу, прибавляя небольшое количество сухого бисульфита
натрия. Затем добавляют 1 мл раствора хромотроповой кислоты
и, вращая колбу, прибавляют 15 мл серной кислоты. Колбу
помещают в горячую воду F0—75°С) на 15 мин, после чего
вынимают из воды, охлаждают до комнатной температуры и
доливают дистиллированной водой до метки. Сравнивают цвет
раствора, который дала проба с цветом, который получен с
эталонным раствором, проанализированным параллельно по той же
методике. Образец считают выдержавшим испытание, если
интенсивность его окраски меньше, чем у эталона. Присутствие
метилового спирта проявляется в пурпурной окраске раствора,
а его отсутствие — в светло-соломенной окраске или полном
отсутствии окраски раствора.
Определение окисляемости
Определение окисляемости спирта по стандартам разных
стран основано на измерении времени обесцвечивания раствора
марганцовокислого калия, добавленного к испытуемому спирту.
Ниже приводится определение окисляемости по стандарту США.
Ход анализа. 20 мл исследуемого спирта охлаждают до 15°С>
добавляют 0,1 мл 0,1 н. раствора перманганата калия и
оставляют на 15 мин. Розовая окраска не должна исчезать полностью.
Необходимо соблюдать следующие меры предосторожности:
не проводить опыты при ярком освещении; температура должна
быть 15—16°С; спирт не должен соприкасаться с корковыми
пробками; цилиндры должны быть абсолютно чистыми.
Определение сухого Остатка
Сухим остатком называют нелетучие вещества, которые
остаются после испарения спирта при температуре 100—105°С.
Сухими веществами, растворимыми в спирте, являются
минеральные вещества, которые попадают в спирт в результате
коррозии метанольнон колонны, трубопроводов и сборников спирта.
Нелетучие вещества по стандартам разных стран определяют
одинаково: определяют количество сухих веществ, остающихся
после испарения 100 мл исследуемого спирта при 100—1О5'"С.
190
Определение фурфурола
фурфурол в спирте определяют не во всех странах. В США
наличие фурфурола в спирте не проверяют. В большинстве
стран определяют содержание фурфурола в спирте, используя
методы, основанные на реакции с солянокислым или
уксуснокислым анилином, и ограничиваются заключением о том, что
анализируемый спирт выдержал испытание или не выдержал.
Определение сивушного масла
Сивушное масло — это смесь высших спиртов: амилового,
изоамилового, бутилового и др. По стандарту США сивушное
масло в этиловом спирте определяют по запаху. Смешивают
10 мл испытуемого спирта с 5 мл дистиллированной воды и 1 мл
чистого глицерина. Смесь испаряют, смачивая ею
фильтровальную бумагу. Если после испарения всей смеси бумага не будет
иметь постороннего запаха, то считают, что спирт выдержал
испытание.
По стандартам других стран сивушное масло в спирте
определяют колориметрическим методом, основанным на реакции
Комаровского, при которой в результате взаимодействия
высших спиртов с циклическим альдегидом и серной кислотой
образуется продукт красного цвета. В качестве циклического
альдегида в Польше и в Румынии применяют салициловый
альдегид, в СССР — фурфурол.
Определение веществ, нерастворимых в воде
Веществами, нерастворимыми в воде, являются бензол,
бензин, полимеры и другие вещества. В стандартах США, Польши
и СССР предусмотрены нормы их содержания в некоторых
сортах этилового спирта. Методы определения этих веществ
основаны на нерастворимости их в спирте, разбавленном водой.
В США определяют растворимость спирта в воде
сравнением прозрачности водноспиртового раствора с прозрачностью
чистой воды. Смешивают 15 мл испытуемого спирта с 45 мл
дистиллированной воды и оставляют на 1 ч. Визуально
сравнивают прозрачность полученного раствора с прозрачностью воды,
налитой в такой же сосуд. Если прозрачность разбавленного
спирта не отличается от прозрачности дистиллированной воды,
то считают, что спирт выдержал испытание.
По стандарту Польши определяют содержание бензола и
бензина. В колбу вместимостью 4 л вливают 1,5 л
исследуемого спирта и 1,5 л дистиллированной воды, закрывают ее
пробкой, в которую вставлен дефлегматор, соединенный с
холодильником. Выходное отверстие холодильника соединяют с
приемником (рис. 60). Колбу нагревают так, чтобы скорость отгонки
не превышала 30 капель в минуту. После отгонки 13 мл
191
^ конденсата (нижняя отметка на приемнике)
^ г дистилляцию прекращают и в приемник до-
бавляют насыщенный раствор поваренной со-
ли до тех пор пока уровень жидкости в при-
емнике не достигнет делений между 1—2 мл.
53пл Содержимое приемника хорошо перемеши-
) Ч вают и оставляют на 1 ч до расслаивания
* * жидкости. Верхний слой представляют
бензол и бензин, его объем измеряют по шкале
приемника и вычисляют содержание бензола
и бензина в исследуемом спирте в
миллилитрах на литр.
Рис. 60. Приемник конденсата для определения веществ,
нерастворимых в воде
Определение ацетона, других кетонов, изопропилового спирта
и третичного бутилового спирта
Эти компоненты определят только по стандарту США. Метод
определения основан на реакции кетонов, изопропилового и
третичного бутилового спиртов с желтой окисью ртути в кислой
среде, при которой образуется труднорастворимое соединение,
выпадающее в осадок.
Реактивы. Реактив Дениже. Пр иготов лен и е. Растворяют
5 г желтой окиси ртути в 60 мл серной кислоты A:2) и объем
раствора доводят до 100 мл, прибавляя дистиллированную воду.
Ход анализа. К 1 мл пробы спирта прибавляют 4 мл
дистиллированной воды и 10 мл реактива Дениже. Смесь нагревают
на водяной бане. Если через 2 мин нагревания осадка не будет,
то спирт выдержал испытание.
ГЛАВА 6. ПОЛУЧЕНИЕ КОРМОВЫХ ДРОЖЖЕЙ
При выращивании дрожжей контролируют их
физиологическое состояние, качество субстрата, подаваемого в дрожжера-
стильный чан, по содержанию в нем Сахаров, питательных
солей (азота, фосфора, калия) и вредных веществ, тормозящих
рост дрожжей. Жидкость, находящуюся в дрожжерастильном
чане, проверяют на содержание в ней кислорода и определяют
ее pH. Концентрацию дрожжей определяют в суспензии, которая
идет на сепарацию, и в бражке, направляемой в канализацию.
При выделении дрожжей из бражки контролируют степень
сгущения дрожжевой суспензии на различных стадиях
сепарирования и определяют потери дрожжей. При высушивании
дрожжевой суспензии контролируют влажность дрожжей и их фер-
192
ментативную активность (наличие живых клеток в товарных
дрожжах недопустимо). Высушенные дрожжи анализируют на
содержание сырого протеина, золы и витамина D2, если
выпускают облученные дрожжи. Эпизодически качество
выпускаемых товарных дрожжей надо характеризовать и по другим
интересным показателям, таким как содержание усвояемого белка,
содержание витаминов Вь В2, РР, которые приняты в
стандартах разных стран, но не предусмотрены в стандарте на
кормовые дрожжи в нашей стране.
Определение физиологического состояния дрожжей микроскопированием
Для определения активности размножения дрожжевых
клеток, состояния клеток — относительного содержания
почкующихся и мертвых клеток, однородности культуры дрожжей и
степени загрязнения посторонними дрожжами, грибами и
бактериями — предназначен метод, основанный на исследовании
внешнего вида дрожжевой массы под микроскопом.
Рост на солодовом сусле
Рост на стеклах обрастания
1 S 2 „.У J
Рнс. 61. Культуры кормовых дрожжей:
/ — Candida tropicalis; 2 — Torulopsis utilis; 3 — Trichosporon
На гидролизных заводах и на заводах, перерабатывающих
сульфитный щелок, в качестве основных культур дрожжевых
цехов используют дрожжи рода Candida и Torulopsis.
Спутниками основных культур являются дрожжеподобные грибы рода
Trichosporon (рис. 61). Род Candida содержит 30 видов, он
относится к группе неспорообразующих дрожжей, для которых
характерно образование псевдомицелия и бластоспор. Отдельные
виды этого рода определяются не только физиологическими
признаками, но так же и характером расположения мицелия и
бластоспор. Род Torulopsis имеет 22 вида, он отличается от рода
Candida отсутствием хорошо развитого псевдомицелия.
Псевдомицелием называют нити или цепочки, состоящие из удлиненных
13 Заказ № 142 193
клеток, образовавшихся путем почкования. Род Trichosporon
характеризуется наличием не только псевдомицелия, но и
истинного многоклеточного мицелия и артроспор, на которые
расчленяется мицелий. Артроспорами называют отдельные
цилиндрические и округленные членики мицелия, которые иногда
располагаются зигзагообразно.
Вид дрожжей определяют на основании их способности
ассимилировать и сбраживать различные сахара, усваивать этиловый
спирт и азот в различных соединениях, а также по форме и
размеру клетки, по росту на солодовом сусле и по характеру штриха
на солодовом агаре и т. п. По морфологическим и
физиологическим признакам дрожжи, используемые на заводах, относят
к видам Candida tropicalis и Torulopsis utilis. Candida tropicalis
ассимилирует и сбраживает глюкозу, мальтозу, сахарозу и
галактозу. Лактозу не ассимилирует и не сбраживает.
Азотнокислый калий не ассимилирует. Torulopsis utilis в отличие от
Candida tropicalis не ассимилирует галактозу, не сбраживает
мальтозу и галактозу, но ассимилирует азотнокислый калий.
Различные расы этих видов дрожжей, выделенные на разных заводах,
отличаются между собой активностью утилизации органических
веществ заводских сред, выносливостью по отношению
различных токсических веществ, присутствующих в этих средах, и
урожайностью, т. е. относительным выходом биомассы. Расы
одного и того же вида дрожжей различаются характером
почкования, размером клеток, формой колоний и штриха. Многие
расы дрожжей, растущих в виде одиночных клеток, при
культивировании в производственных условиях становятся мицелиаль-
ными расами.
К виду Candida tropicalis отнесены расы дрожжей,
выделенные на гидролизных заводах: Хорская-9, Саратовская-5, Тавдин-
ская-1, Лобвинская-2, Краснодарская-14 и расы дрожжей,
выделенные на заводах, перерабатывающих сульфитный щелок:
Сухонская-2, Выборгская-2, Приозерская-3, Сокольская-5, Сухон-
ская-4. К виду Torulopsis utilis отнесены расы дрожжей,
выделенные на гидролизных заводах: Канская-2, Красноярская-2
и расы дрожжей, выделенные на сульфитных заводах:
Соликамская-1, Выборгская-1 и др.
Дрожжами-спутниками или дрожжами-примесями, которые
часто встречаются на гидролизных заводах, являются дрожжи
Zygofabospora и Trichosporon, а также различные виды рода
Candida. Например, Candida arborca длительное время
применяется на Канском и Бирюсинском заводах. Выделены расы этого
вида Канская-3, Бирюсинская-3 и др. Кроме различных видов
дрожжей-спутников встречаются мицелиальные грибы рода
Aspergillius, Penicillium и др., а также бактерии.
По почкованию и нарастанию клеток в виде цепочек и
веточек судят об активности накопления дрожжей, об
обеспеченности дрожжей сахаром и кислородом, наличии вредных при-
194
месей и т. д. Для определения однородности культуры дрожжей
и наличия посторонних микроорганизмов учитывают
морфологические особенности культивируемых родов дрожжей,
возможность их перерождения, наличие дрожжей-спутников полезных
и вредных, а также наличие бактерий.
Определение чистоты культуры дрожжей
Для определения однородности дрожжевой массы,
культивируемой в дрожжерастильном чане, предназначен метод,
основанный на установлении примесей в основном виде дрожжей и
идентификации их путем высева на суслоагар. Каждый вид
дрожжей имеет свою характерную форму колоний, по которой
определяют наличие тех или иных микроорганизмов. Чистой
культурой дрожжей называют культуру, которая содержит
только один вид дрожжей.
Ход анализа. Из дрожжерастильного чана на различной
высоте отбирают пробы бражки, содержащей дрожжи. Пробы
бражки разбавляют стерильной водопроводной водой в 10—
50 раз в зависимости от концентрации дрожжей, и из них делают
рассев на поверхность агара в чашках Петри. Через 2—3 суток
выросшие колонии просматривают через лупу, отбирают чашки,
в которых колонии расположены изолированно друг от друга,
а затем подсчитывают количество колоний различного вида
дрожжей. На основании полученных данных вычисляют
процентное соотношение различных видов дрожжей и
устанавливают степень загрязненности производственной культуры
дрожжей.
Некоторые культуры дрожжей (Кр-9, Л-2, СД-5) рода
Candida размножаются в виде сильно разросшихся пучков или
веточек, состоящих из большого числа клеток. При разбавлении
пробы водой они не распадаются на отдельные клетки, поэтому
колонии таких дрожжей вырастают на агаре не из одной клетки,
как другие, а из нескольких клеток. При определении степени
зараженности по относительному количеству колоний в этом
случае можно сделать ошибку. Если высев на сусло-агар
показал наличие в дрожжерастильных чанах посторонних
дрожжей или грибов, то их выделяют из колоний высевая на
солодовое сусло или повторяя высев на сусло-агар, и проводят
с ними специальные исследования.
При длительном культивировании дрожжей в
производственных условиях они изменяются, приспосабливаясь к условиям
внешней среды. Физиологические изменения дрожжей влекут
за собой их морфологические изменения. Изменяется размер
клеток, урожайность, вид колоний, характер почкования и т. д.
Вновь образованные расы дрожжей выделяют в виде чистой
культуры, изучают их свойства в лабораторных условиях и дают
оценку о целесообразности культивирования выделенной расы.
13* 195
Определение кислорода полярографическим методом
Для определения кислорода, растворенного в субстрате,
барде и бражке на разных стадиях выращивания дрожжей [67,
68], предназначен метод, основанный на расшифровке
поляризационных кривых, которые получаются при восстановлении
и окислении веществ, находящихся в электролитической ячейке.
Одним электродом служит ртуть, падающая каплями из
капилляра в исследуемую жидкость, другим — насыщенный
каломельный электрод. Природа исследуемого вещества характеризуется
величиной потенциала окисления или восстановления.
Концентрация исследуемого вещества в растворе измеряется величиной
изменения силы тока или так называемой высотой волны.
Определение содержания кислорода в жидких средах
основано на его способности легко восстанавливаться, причем поля-
рограмма в этих случаях состоит из двух волн равной величины.
Первая волна обусловлена восстановлением кислорода до
перекиси водорода:
О2-(-2Н+-(-2е-»-Н2О2 (в кислой среде) или
О2-(-2Н2О-(-2е-»-2Н2О2 (в нейтральной и щелочной среде).
Вторая волна соответствует восстановлению перекиси
водорода до воды или до иона ОН:
Н2О2+2Н++2е->-2Н2О (в кислой среде) или
Н2О2+2е—>-2ОН~ (в щелочной среде).
Первая волна характеризуется резко выраженным
максимумом, расположенным в пределах до 1 В. По этой волне ведут
определение растворенного кислорода в субстратах и других
полупродуктах дрожжевого производства, хотя она легко
подавляется поверхностно-активными веществами, как например
дрожжевым автолизатом, раствором крахмала и др. Вести
определение кислорода при потенциале выше 1 В в субстратах
невозможно из-за наличия веществ, восстанавливающихся при
этом напряжении.
Для определения концентрации кислорода снимают полные
полярограммы или только три точки на потенциале 0,5—0,6 В,
которые принадлежат: 1-я — исходному раствору, 2-я —
исходному раствору после насыщения его кислородом в результате
продувки воздуха и 3-я — исходному раствору, из которого
удален кислород продувкой инертного газа (азот, водород,
углекислота). На рис. 62 показаны полярограммы барды сульфитно-
спиртового завода при различном содержании кислорода и
графический расчет концентрации кислорода по трем точкам. Цену
деления шкалы полярографа устанавливают экспериментально
и составляют калибровочные кривые или таблицы для выраже-
196
ния зависимости показаний прибора от концентрации кислорода
в жидкости в процентах или в миллиграммах на литр.
Мешающие вещества. Поверхностно-активные вещества и
вещества, имеющие окислительно-восстановительный потенциал
близкий к кислородному.
Аппаратура. Полярограф. Электролитическая ячейка с
капельным ртутным электродом и насыщенным каломельным
электродом. Медицинский шприц.
0,1 0,2 0,3 0.40,5 Ц60,7ф 0,9 1,0
Е.В
Рис. 62. Полярограммы барды при
различном содержании кислорода:
/ — подпрограмма исходной барды; 2 —
барда после насыщения кислородом;
3 — барда после удаления кислорода
Рис. 63. Принципиальная схема
установки для снятия полярограмм
Принципиальная схема установки для снятия полярограммы
показана на рис. 63. Описание работы на полярографе дается
в инструкции, которая прилагается к каждому прибору. Перед
анализом проводят подготовку ячейки и проверку установки
следующим образом. Трубку 4, соединяющую два сообщающихся
сосуда ячейки, заполняют агар-агаром, приготовленным на
насыщенном растворе хлористого калия. Заряжают каломельный
электрод б, как описано на с. 127. Систему капельного электрода /
заполняют чистой сухой ртутью, укрепляют на штативе и
поднимают воронку с ртутью 3 на такую высоту, чтобы ртуть капала
со скоростью 3—5 капель в секунду. В левый сосуд 5 наливают
насыщенный раствор хлористого калия, погружают в него конец
каломельного электрода, который присоединен к аноду. В
воронку с ртутью вставляют платиновый контакт, который
соединен с гальванометром 8. В правый сосуд 2 наливают 0,1 н.
197
раствор хлористого калия, включают полярограф и снимают
полярограмму. После проверки работы электрической цепи
полярограф выключают, правый сосуд освобождают и
промывают водой.
Реактивы и их приготовление. 1. Инертный газ — азот,
водород или углекислота в баллоне. 2. Фенол, раствор 80%-ный.
3 рТуТь чистая. Очистка ртути. Фильтровальной бумагой
удаляют влагу с поверхности ртути, а затем ее фильтруют
через чистый полотняный фильтр и помещают в делительную
воронку, которая установлена над стеклянной колонкой высотой
0,5—1 м и диаметром 20—25 мм. Колонка, имеющая внизу кран,
заполнена на 5/б высоты 5%-ным раствором азотной кислоты.
Открывают кран делительной воронки так, чтобы ртуть вытекала
и падала в азотную кислоту мелкими каплями. Во время
прохождения через столб азотной кислоты ртуть очищается от
примесей. Из колонки ртуть выливают в чашку, а потом снова в
воронку. Промывают ртуть азотной кислотой несколько раз, после
чего несколько раз промывают дистиллированной водой до
отрицательной реакции промывной воды на кислоту по
метиловому оранжевому. С очищенной ртути удаляют влагу
фильтровальной бумагой.
Подготовка проб. Перед анализом пробу субстрата,
отобранную из дрожжерастильного чана, обрабатывают фенолом для
остановки дыхания дрожжей. Берут медицинский шприц, ставят
поршень в положение минимального объема (до упора в шарик),
набирают раствор фенола в количестве, превышающем
свободный объем около стеклянного шарика. На иглу накалывают
лист бумаги, шприц перевертывают иглой вверх и выдавливают
из него воздух и избыточное количество фенола. Лист бумаги
предохраняет руки от фенола, который стекает по игле. Иглу и
корпус шприца тщательно протирают фильтровальной
бумагой, иглу опускают в сосуд с исследуемой пробой и плавным
движением поршня отбирают нужный объем пробы. Пузырьки
воздуха, попавшие в шприц с пробой, выдавливают, иглу
втыкают в корковую пробку, шприц несколько раз переворачивают
для смешивания пробы с фенолом, а затем содержимое шприца
выдавливают в правый сосуд ячейки.
Ход анализа. Реохорд 7 ставят на потенциал 0,6 В.
Переключатели полярографа ставят в положения, указанные в
инструкции к полярографу. Включают питание гальванометра от сети.
Перо самописца (если он есть) опускают на бумагу, включают
коробку скоростей и на протяжении 2—3 см ленты записывают
силу тока, которая соответствует концентрации кислорода
в пробе. Если нет самописца, то снимают показания по шкале
полярографа при различных потенциалах и вычерчивают
кривую или снимают показания полярографа только на потенциале
0,6 В. Затем приподнимают ртутный электрод, через боковую
трубку в исследуемую пробу вводят трубку от баллона с водо-
198
родом или другим инертным газом и ведут продувку до тех пор,
пока стрелка гальванометра не остановится на нуле или около
нуля. Для очистки водорода от примесей мышьяковистого
водорода и сероводорода, его пропускают через дрексели,
заполненные одни перманганатом калия, другие — щелочью. Ртуть
на дне правого сосуда выливают, ртутный электрод ставят на
место и кран на вводной трубке закрывают. Через некоторое
время стрелку и перо гальванометра устанавливают на
постоянном значении концентрации кислорода равном 0. Если снимают
полярографические кривые, то после продувки инертного газа
через исследуемую жидкость измеряют силу тока на различных
потенциалах и чертят кривую.
Аналогично проводят насыщение пробы кислородом воздуха,
используя насос или резиновую грушу. После 100%-ного
насыщения кислородом анализируемой пробы снимают показания
гальванометра на различный потенциалах или только на одном,
равном 0,6 В. На этом анализ считают законченным.
Гальванометр, самописец и полярограф выключают, воронку с ртутью
опускают, ячейку освобождают и промывают дистиллированной
водой.
Расчет. Степень насыщения исследуемой пробы кислородом определяют
по формуле
(я - б) 100
X -
—б
где X — степень насыщения кислородом, %; а — показания гальванометра для
анализируемого раствора на потенциале 0,6 В, мка; б — показания
гальванометра, после удаления кислорода на потенциале 0,6 В, мка; в — показания
гальванометра после насыщения кислородом на потенциале 0,6 В, мка.
Концентрацию кислорода в анализируемой пробе находят по заранее
сделанной градуировочной кривой зависимости концентрации кислорода от
степени насыщения раствора при +20°С.
Концентрацию кислорода, растворенного в жидкости, определяют с
точностью ±4—7% отн.
Определение Сахаров экспрессным методом
Для быстрого определения наличия Сахаров (гексоз и
пентоз) в бражке после выращивания дрожжей, в сточной воде и
других жидкостях, где их концентрация менее 0,2%,
предназначен метод, основанный на выделении Сахаров из смеси с
редуцирующими несахарами с помощью хроматографии на бумаге.
Одновременно можно анализировать 5—^10 проб. Наличие
Сахаров (гексоз и пентоз), выделенных на хроматограмме,
устанавливают по их реакции с анилинфталатом, в результате которой
они образуют окрашенные соединения. По интенсивности
окраски и величине пятен, полученных после проявления хромато-
грамм, путем сравнения их со свидетелями (эталонами) находят
концентрации отдельно гексоз и пентоз.
199
Мешающие вещества. Олигосахариды, уроновые кислоты.
Аппаратура. Камера для восходящего хроматографирования
на лентах (см. рис. 23). Микропипетка (см. рис. 27). Водяная
баня.
Реактивы и их приготовление. 1. Растворитель — смесь этил-
ацетата, пиридина, воды E:1:5). Приготовление см. на с. 78
2. Проявитель — анилинфталат. Приготовление см. на с. 78.
3. Хроматографическая бумага. Приготовление
заготовок хроматограмм. На листе хроматографической бумаги
определяют расположение волокон целлюлозы, как описано на
с. 76. Затем размечают лист на полоски шириной 2—3 см с
продольным расположением волокон. На расстоянии 4—5 см от
края проводят простым карандашом линию старта на всех
будущих полосках сразу. На расстоянии 25—30 см, в
зависимости от размеров камеры, проводят линию конца
хроматограмм. На расстоянии 1,5 см от конца противоположного линии
старта проводят линию, на которой после сделают надрез
крестом для получения отверстия, чтобы подвесить хроматограмму
(см. рис. 23). Размеченный лист разрезают на полоски, которые
хранят в пакетах из кальки или из полиэтилена. Свертывать и
перегибать хроматографическую бумагу в листах и заготовках
нельзя. 4. Эталонные растворы ксилозы и глюкозы, имеющие
концентрацию 0,2, 0,1 и 0,05%. Можно брать и другие
концентрации в зависимости от состава анализируемой пробы.
Подготовка проб. Если проба содержит менее 0,2%
редуцирующих веществ, то ее надо упарить в 2—5 раз или нанести на
хроматограмму соответственно больший объем. Для
упаривания 100 мл пробы отмеривают цилиндром, 25 мл из них
наливают в выпарительную чашку и ставят на кипящую
водяную баню. По мере испарения в чашку добавляют пробу
из цилиндра. Упаренную пробу количественно переносят
в мерную колбу вместимостью 25 или 10 мл и объем раствора
доводят до метки, прибавляя воду. Для одновременного
хроматографирования подготавливают 3—5 проб.
Ход анализа. При анализе пяти проб берут восемь
бумажных полосок (заготовок) и делают на них крестообразные
надрезы так, чтобы стеклянный стержень, на который их повесят,
плотно входил в отверстие. На конце полоски, который не будет
погружен в растворитель, простым карандашом пишут номер
пробы или эталонного раствора. Отмеривают и наносят пробу
на подготовленные полоски с помощью микропипетки с
приспособлением (см. рис. 27), как описано на с. 81. В середину линии
старта на три полоски наносят по 0,01 мл эталонных растворов
различных концентраций и на пять полосок наносят по 0,01 мл
упаренной анализируемой пробы. На каждую полоску наносят
только одну пробу или один эталонный раствор. Если проба не
упарена, то после высыхания пятна в то же место наносят еще
0,01 мл пробы. Так повторяют 2—5 раз вместо выпаривания
200
пробы. Пятна жидкостей на полосках просушивают на воздухе
при комнатной температуре.
Для хроматографирования в камеру, которой служит
стеклянная банка с хорошо пришлифованной крышкой, опускают
подставку для стержня и наливают в нее такое количество
прозрачного (мутный не допускается) растворителя, чтобы его слой
был высотой 3—5 см. На стеклянный стержень (палочку)
нанизывают через крестообразные разрезы восемь полосок на
расстоянии 3—5 см друг от друга и, держа стержень за середину,
осторожно вешают его на крючки подставки в банке. При этом
концы хроматограмм (полосок) должны погрузиться в
растворитель на 1—1,5 см. Банку закрывают. Во время
хроматографирования надо следить, чтобы ленты не соприкасались, а их
концы до окончания хроматографирования оставались
погруженными в растворитель на 1—1,5 см. Температура окружающего
воздуха должна быть 18—25°С. Когда растворитель дойдет по
полоске до стержня, банку открывают, стержень с хроматограм-
мами вынимают, банку закрывают, а стержень кладут на
подставку в вытяжном шкафу и хроматограммы сушат на воздухе
до полного удаления запаха растворителя. Если хотят получить
отделение не только гексоз от пентоз, но разделить смесь на
отдельные сахара, то проводят многократное хроматографиро-
вание следующим образом. Высушенные хроматограммы на
стержне снова ставят в банку с растворителем, ждут, когда
растворитель дойдет до стержня, вынимают и сушат их. Так
повторяют 2—5 раз.
Для проявления хроматограмм в чашку наливают
проявитель и на мгновение погружают в него одну хроматограмму,
держа ее за концы. Мокрую хроматограмму кладут между
листами фильтровальной бумаги, слегка проглаживают рукой для
удаления избытка проявителя и помещают в сушильный шкаф
при 100—105°С. Так проявляют все полученные хроматограммы
с пробами и с эталонами. В сушильном шкафу хроматограммы
не кладут, а ставят на ребро, изгибая их вкруг.Хроматограммы
не должны касаться стенок сушильного шкафа. Через 5—10 мин
хроматограммы вынимают из сушильного шкафа и
устанавливают состав редуцирующих веществ в пробе. Сравнивая
интенсивность и величину пятен, полученных на хроматограммах
проб, с пятнами на хроматограммах эталонов, устанавливают
содержание Сахаров в пробах.
Расчет. Определение наличия Сахаров в пробе. Если на
хроматограмме анализируемой пробы обнаружено пятно розового или
красного цвета, которое расположено приблизительно в середине хроматограммы,
то в пробе есть пентозы. Если на хроматограмме есть пятно коричневого
цвета, которое расположено вблизи пятна от нанесенной пробы, то в
анализируемой пробе есть гексозы. Для большей уверенности сравнивают цвета и
положения пятен на хроматограммах пробы и эталонов.
Определение концентрации Сахаров, обнаруженных в пробе, делают
сравнением величины и интенсивности пятен на хроматограмме пробы и на
201
хроматограммах эталонов. Визуально устанавливают, какой эталон дал пятно
одинаковое с пятном пробы. Концентрацию сахара в анализируемой пробе
вычисляют по формуле
где X — концентрация сахара в анализируемой пробе, %; а — концентрация
сахара в эталонном растворе, %; л— сгущение пробы при выпаривании или
число раз нанесения пробы на хроматограмму.
Чувствительность метода 5 мкг. Концентрацию Сахаров определяют с
точностью ±7—8% оти.
Определение фосфора колориметрическим методом (метод Бриггса)
Для определения фосфора, связанного в минеральных и
органических соединениях, предназначен метод, основанный на
реакциях получения молибденовой сини. Фосфор, связанный
в минеральных соединениях (раствор аммофоса), определяют
непосредственно в пробе или после ее озоления. Фосфор,
связанный в органических соединениях (дрожжи, активный
ил и др.), определяют после минерализации пробы, при этом
количество найденного фосфора составляет сумму фосфора
в минеральных и органических веществах, содержащихся в пробе
[69]. Фосфорсодержащие соединения обрабатывают молибдено-
вокислым аммонием в кислой среде в присутствии гидрохинона
и сульфита натрия. В результате образуется фосфорномолибде-
новая кислота
PO;-KNH4JMoO4-*H3PO4 • 12МоО3.
При действии сильных восстановителей (гидрохинона и
сульфита натрия) в кислой среде шестивалентный молибден
восстанавливается, образуя различно окрашенные соединения низших
степеней окисления. Соединения, которые соответствуют
формуле МопОзп-ж, где х может быть равен 1, 2, 3, имеют синюю
и фиолетовую окраску. Смесь этих соединений называют
молибденовой синью. Некоторые считают эту смесь за один продукт
и приписывают ему формулу Mo6Oi7 [70].
Количество образующихся окрашенных соединений
пропорционально количеству фосфора, присутствующего в реакционной
смеси. Интенсивность окраски раствора молибденовой синью
оценивают визуально, сравнивая с окраской растворов
стандартной шкалы, или с помощью фотоэлектроколориметра, к
показаниям которого имеется градуировочная кривая.
Мешающие вещества. Окрашенные и взвешенные вещества.
Аппаратура. Установка для минерализации пробы. Фото-
электроколориметр или набор пробирок A0 шт.) для
колориметрического анализа вместимостью 20 мл с притертыми
пробками из бесцветного стекла одинакового диаметра с отметкой
5 мл.
202
Фотоэлектроколориметр. Интенсивность окраски
растворов, которые получают при анализе, можно определять фо-
тоэлектроколориметрами различных марок. Прибор ФЭК.-М,
например, (рис. 64) основан на принципе уравнивания двух
световых пучков при помощи переменной щелевой диафрагмы Д.
Источником света служит лампа Л на 8 В, которая через
трансформатор и стабилизатор напряжения включается в сеть
переменного тока. Два световых пучка от лампы, выделенные с
помощью двух конденсаторов К\ и Кг, отражаются от зеркал Зі
и 32, проходят через фильтры Ф\ и Фг, а затем через кюветы
А\ и A2 и линзами проектируются на фотоэлементы Зі и 32.
Фотоэлементы включены в цепь с гальванометром так, что при
одинаковой интенсивности
световых пучков, падающих на
фотоэлементы, стрелка
гальванометра стоит на нуле.
Перед зеркалами Зі и 32
находятся теплозащитные стекла
Гі и Т2, которые, поглощая
инфракрасные лучи лампы,
предохраняют фотоэлементы от
нагревания.
При измерении на пути
правого светового пучка
помещают кювету с
исследуемым раствором, а кювету
с растворителем помещают на
пути левого светового пучка.
Щелевая диафрагма при этом
полностью открыта (левый
барабан устанавливают на
100 делении светопропускания).
Рис. 64. Принцнпальная схема фото-
электроколориметра
В результате поглощения света
анализируемым раствором луч света, падающий на
фотоэлемент 32, будет меньшей интенсивности, чем луч света,
падающий на фотоэлемент Зі. Стрелка гальванометра при этом
отклоняется от нуля. Для уравнивания интенсивности обоих
световых лучей на пути левого пучка вводят фотометрический клин В,
стрелка гальванометра встает на нуль, затем меняют
положение кювет. На пути правого пучка света кювету с исследуемым
раствором заменяют кюветой с растворителем. Гальванометр
опять покажет разницу интенсивностей лучей, потому что
увеличится интенсивность светового потока, падающего на
фотоэлемент Зг. Для уравнивания интенсивности луча, падающего
на фотоэлемент Зі, изменяют ширину щелевой диафрагмы Д,
которая фиксируется связанным с ней отсчетным барабаном Б,
отградуированным в единицах оптической плотности.
Реактивы и их приготовление. 1. Серная кислота плотностью
1,84 г/см3. 2. Сернокислая медь кристаллическая безводная.
203
3. Сернокислый натрий кристаллический безводный. 4. Едкий
натр, 30%-ный раствор. Приготовление. В фарфоровый
стакан вместимостью 200 мл вливают 50 мл дистиллированной
воды, а затем осторожно, маленькими кусочками, при
помешивании опускают в воду 30 г едкого натра. После растворения
едкого натра раствор охлаждают, количественно переносят
в мерную колбу вместимостью 100 мл и объем раствора доводят
водой до метки. 5. Индикатор — метиловый красный, 0,1%-ный
спиртовый раствор. 6. Молибденовокислый аммоний, 5%-ный
раствор в серной кислоте. Приготовление. Помещают 25 г
молибденовокислого аммония в колбу вместимостью 600—
700 мл, добавляют 200 мл дистиллированной воды и 200 мл
серной кислоты, разбавленной 1 :2. Когда молибденовокислый
аммоний растворится, раствор охлаждают, количественно
переносят в мерную колбу вместимостью 500 мл, объем доводят водой
до метки и тщательно перемешивают. 7. Гидрохинон
(СбН4(ОНJ), 1%-ный раствор. Приготовление. В стакан
вместимостью 500 мл помещают 10 г гидрохинона, добавляют
30 мл дистиллированной воды и 10 капель концентрированной
серной кислоты. После растворения гидрохинона раствор
количественно переносят в мерную колбу вместимостью 1 л и объем
доводят водой до метки. Раствор гидрохинона не устойчив, его
готовят перед употреблением. 8. Сернокислый натрий, 20%-ный
раствор. Приготовление. В мерную колбу вместимостью
100 мл помещают 20 г сульфита натрия, прибавляют 50 мл
дистиллированной воды, хорошо перемешивают до полного
растворения соли и объем раствора доводят водой до метки.
Раствор можно хранить не более 5 суток. 9. Основной эталонный
раствор однозамещенного фосфорнокислого калия (КН2РО4).
Приготовление. В ступке измельчают 2—3 г
однозамещенного фосфорнокислого калия ч. д. а. и высушивают до
полного удаления влаги. Около 1 г измельченного абсолютно сухого
КН2РО4 обвешивают с точностью 0,0002 г, количественно
переносят в мерную колбу вместимостью 500 мл и растворяют,
добавляя 400¦мл • дистиллированной воды. Затем в раствор
вливают I мл хлороформа для предохранения его от
бактериального заражения, прибавляют воды до метки и перемешивают.
Титр приготовленного раствора выражают в миллиграммах на
миллилитр фосфора (Р, мг/мл) или пятиокиси фосфора
(Р2О5 мг/мл) и вычисляют по формуле
I36V '
где Т — концентрация фосфора (или РгО5) в приготовленном растворе, мг/мл;
g — количество однозамещенного фосфорнокислого калия, взятое для
приготовления раствора, г; А — атомная масса фосфора, равная 31 (или
молекулярная масса,Р2О5=142); 136 — молекулярнаи масса однозамещенного
фосфорнокислого калия; V — объем приготовленного раствора, мл.
204
10. Рабочие эталонные раствора КН2РО4. Приготовление.
Основной эталонный раствор разбавляют дистиллированной
водой в различных соотношениях так, чтобы получить 15
растворов, содержащих различные количества фосфора 0,01, 0,02 ...
... и до 0,15 мг/мл.
Построение калибровочной кривой.
Анализируют 15 рабочих эталонных растворов, имеющих концентрации
фосфора в пределах от 0,01 до 0,15 мг/л. Каждый анализ
повторяют не менее 3 раз и из полученных результатов берут
среднее значение. Одновременно анализируют не больше трех-четы-
рех растворов, потому что окраска реакционной смеси
изменяется при долгом стоянии. Анализ растворов делают, как описано
ниже. Полученные данные наносят на график зависимости
оптической плотности реакционной смеси от концентрации фосфора
Р или пятиокиси фосфора Р2О5 в анализируемом растворе
в миллиграммах на литр и строят калибровочную кривую,
которой пользуются для нахождения результата анализа при
исследовании различных проб.
Подготовка проб. Определение фосфора,
связанного в минеральных соединениях. 1. Пробы водных
растворов суперфосфата или аммофоса, приготовленных для
добавления в нейтрализат, барду или сточную воду, перед
анализом фильтруют и разбавляют в 100—200 раз, чтобы получить
растворы, содержащие 60—120 мг/л фосфора. 2. Пробы
субстрата, бражки, барды и сточной воды на гидролизных заводах
перед анализом фильтруют и, не разбавляя, анализируют.
3. Пробы сильноокрашенных субстратов, бражки и барды,
полученных из сульфитного щелока, перед анализом озоляют
следующим образом. Отмеривают 50 мл отфильтрованной бражки,
10—20 мл из них вливают в фарфоровый тигель и ставят на
кипящую водяную баню. По мере выпаривания в тигель
доливают остальную часть отмеренной пробы. После выпаривания
воды сухой остаток в тигле ставят в муфельную печь при
температуре 300—400°С для сжигания органических веществ.
Периодически тигель с остатком вынимают из печи, охлаждают
в эксикаторе и взвешивают. Остаток прокаливают до тех пор
пока разница между двумя последующими взвешиваниями
будет составлять не более 0,0003 г. Золу в тигле растворяют,
приливая 1 мл концентрированной серной кислоты, полученный
раствор осторожно, по стенке, выливают в мерную колбу
вместимостью 100 мл, в которую предварительно было влито 50 мл
дистиллированной воды, тигель споласкивают водой, которую
выливают в ту же колбу. После охлаждения раствор
нейтрализуют 30%-ным раствором едкого натра по индикатору
метиловому красному до слабо-желтого цвета, объем раствора доводят
водой до метки и перемешивают. Если полученный раствор
мутный, то его фильтруют, а потом анализируют.
205
Определение фосфора, связанного в
органических соединениях. Пробы дрожжей, активного ила
и осадков, отобранных на различных стадиях очистки сточных
вод, анализируют после их минерализации. Если результат
анализа хотят знать в пересчете на сухое вещество, то определяют
влажность пробы. Для минерализации берут 0,5 г сухих дрожжей
(или активного ила и т. д.), на лодочке помещают в колбу
Кьельдаля вместимостью 250 мл, вливают 20 мл серной кислоты
плотностью 1,84 г/см3, добавляют 0,5 г сернокислой меди и 1 г
сернокислого натрия. Укрепив колбу в штативе над
электроплиткой наклонно (под тягой), включают слабый нагрев колбы
(рис. 65). В начале минерализации идет бурное выделение
газов, что сопровождается сильным вспениванием реакционной
смеси. Когда прекратится пенение, нагрев
реакционной смеси усиливают, опуская колбу на
электроплитку. Нагревание продолжают до
полного обесцвечивания реагирующей смеси. При
сжигании обращают внимание, чтобы на стенках
колбы не оставались обугленные частицы
реакционной смеси. Если они есть, то, осторожно
вращая колбу, смывают их в реагирующую
жидкость. Для гарантии полноты минерализации
пробы реагирующую жидкость после обесцвечи-
Рис 65 Уста- вания нагревают еще 30 мин. Затем нагрев вы-
новка для ми- ключают, колбу охлаждают, ее содержимое ко-
иерализации личественно переносят в мерную колбу вмести-
дрржжей (с^а" мостью 10 мл, объем раствора доводят до метки
быи нагрев} и перемешивают_ в колбу вместимостью 50 мл
помещают 25 мл полученного раствора,
нейтрализуют 30%-ным раствором едкого натра в присутствии
индикатора метилового красного до слабо-желтого цвета.
Раствор охлаждают, его объем доводят дистиллированной водой до
метки и перемешивают. Если раствор мутный, то его надо
отфильтровать перед анализом.
Ход анализа при визуальном колориметрироваиии.
Одновременно с анализом исследуемой пробы анализируют пять-десять
рабочих эталонных растворов выбранных концентраций для
приготовления стандартной шкалы. В приготовленные
пронумерованные пробирки вносят в каждую по 1 мл одного из выбранных
рабочих эталонных растворов и в одну пробирку вносят 1 мл
приготовленной пробы. Во все пробирки добавляют по 1 мл
раствора молибденовокислого аммония, 0,5 мл раствора
гидрохинона, 0,5 мл раствора сернистокислого натрия и объем
раствора в каждой пробирке доводят водой до 5 мл. Пробирки
закрывают пробками, а реагирующую смесь перемешивают
легким встряхиванием и оставляют на 30 мин. В том случае, когда
анализируют окрашенный раствор (субстрат, бражку и т. д.),
стандартную шкалу готовят, прибавляя 1 мл раствора, имею-
206
щего окраску, которая совпадает с окраской анализируемой
пробы, но который не содержит фосфор, например, барду или
нейтрализат до прибавления питательных солей. По истечении
30 мин сравнивают появившуюся окраску раствора в пробирке
с пробой с окрасками растворов стандартной шкалы. Находят
пробирку стандартной шкалы, в которой окраска раствора
совпадает с окраской раствора в пробирке с пробой.
Ход анализа при колориметрировании на ФЭК. Прибор
ФЭК-М включают в электрическую сеть и вставляют красные
светофильтры № 4. В пробирку помещают 1 мл исследуемого
раствора, приготовленного, как описано выше, приливают
к нему 1 мл раствора молибденовокислого аммония, 1 мл
гидрохинона и 2 мл раствора сульфита натрия, пробирку
закрывают пробкой, раствор перемешивают и оставляют на 20 мио.
Если анализируют бесцветный раствор, то в качестве
растворителя употребляют дистиллированную воду, а если —
окрашенный, то готовят растворитель, разбавляя анализируемый
раствор в 5 раз.
Переводят рукоятку регулировочного винта гальванометра,
расположенного на крышке прибора, в положение «открыто».
В случае отклонения стрелки гальванометра от нуля до
подключения к фотоэлементам его устанавливают на нуль с помощью
рукоятки на верхней крышке прибора. В кюветы № 5 наливают
в две растворитель, в третью — испытуемую пробу из пробирки
после выдерживания с реактивами в течение 20 мин. Индекс
левого барабана вращением «к себе» устанавливают на
нулевое деление шкалы оптической плотности. В правый держатель
помещают кювету с испытуемым раствором и кювету с
растворителем. Каждый держатель кювет имеет три гнезда для
установки кювет. Держатели кювет вращаются на оси и фиксируются
в трех положениях. В левый держатель помещают одну кювету
с растворителем. Вращая держатели кювет, в правый пучок
света помещают кювету с испытуемым раствором, а в левый
с растворителем. Гальванометр из положения «0» (выключен)
переключают в положение «1» (малая чувствительность) и,
вращая рукоятку фотометрических клиньев «Грубая настройка»,
устанавливают стрелку гальванометра на нуль. Затем
гальванометр переключают в положение «2» (большая
чувствительность) и снова стрелку приводят на нуль, вращая рукоятки
клиньев для точной настройки.
В правый пучок света вводят кювету с растворителем
вместо кюветы с исследуемой пробой и, вращая левый
измерительный барабан «от себя», стрелку гальванометра снова
устанавливают на нуль сначала при малой чувствительности, а потом
при большой. Величину оптической плотности исследуемого
раствора отсчитывают по красной шкале барабана.
Измерение повторяют несколько раз. Из полученных отсчетов
вычисляют среднеарифметическое значение оптической плотности
207
исследуемого раствора и по калибровочной кривой находят
содержание фосфора в растворе, взятом на анализ.
Расчет. 1. Содержание фосфора, связанного в минеральных соединениях,
в анализируемой пробе вычисляют по формуле
X = СліООО или X = С\п,
где X— концентрация фосфора (или Р2О5) в анализируемой пробе, мг/л; С —
концентрация фосфора (или Р2О5) в анализируемом растворе, найденная при
визуальном сравнении пробы с эталоном, мг/мл; Сі — концентрация фосфора
(или Р2О5) в анализируемом растворе, иайдеииая по калибровочной кривой,
мг/мл; п — разведение.
2. Содержание фосфора, связанного в органических и минеральных
соединениях пробы (дрожжи, активный ил), вычисляют в процентах от
абсолютно сухого вещества пробы по формуле
v CV100
X ¦-
?A-0,01 IF) '
где X — содержание фосфора в пробе абсолютно сухих дрожжей, %; С —
концентрация фосфора в анализируемом растворе, мг/мл; V — объем раствора
пробы после минерализации, мл; g — количество дрожжей, взятое для
минерализации, г; W—влажность анализируемых дрожжей, %.
Для пересчета количества фосфора в количество пятиокиси фосфора
(Р2О5) иайдеииое количество фосфора умножают иа коэффициент К.
142 =2.29.
' 62
где Afp и Alp 0 —молекулярные массы фосфора и Р2О5.
Концентрацию фосфора в пробе при визуальном колориметрироваиия
определяют с точностью ±7—12% отн., при колориметрироваиии иа ФЭК —
±4—5% оти.
Определение калия
Для определения калия в сусле, барде, последрожжевой
бражке и других жидкостях [71] предназначен метод,
основанный на обменной реакции калия с тетрафенилборатом натрия,
которая протекает в кислой среде по уравнению
K++Na [В (С6Н5L] - К [В (С6Н5L] +Na+.
Образующийся труднорастворимый тетраборат калия
выпадает в осадок, его отфильтровывают, высушивают до полного
удаления влаги и взвешивают. Затем вычисляют содержание
калия в пробе.
Мешающие вещества. Соли аммония и органические
вещества, которые осаждаются тетраборатом натрия.
Аппаратура. Установка для отгонки аммиака (см. рис. 36).
Стеклянный фильтр № 2. Сушильный шкаф.
Реактивы и их приготовление. 1. Тетраборат натрия, 3,5%-
ный раствор. 2. Уксусная кислота, 1%-ный и 10%-ный растворы.
3. Перекись водорода плотностью 1,12—1,13 г/см3 C0%-ная).
208
4. Серная кислота, 0,1 н. раствор. 5. Окись магния
кристаллическая. 6. Красная лакмусовая бумажка.
Подготовка проб. 1. Пробу фильтруют через бумажный
фильтр. 2. Из пробы удаляют аммонийные соли следующим
приемом. В плоскодонную колбу вместимостью 250 мл вливают
50 мл пробы, добавляют 100—150 мл дистиллированной воды,
0,8 г окиси магния, затем колбу включают в установку для
отгонки аммиака (см. рис. 36). Приемником служит коническая
колба вместимостью 250 мл, в которую влито 25—50 мл 0,1 н.
раствора серной кислоты. Конец отгонки аммиака устанавливают
красной лакмусовой бумажкой, которая не должна менять цвет
при смачивании каплей конденсата из холодильника.
(Собранный аммиак можно определить титрованием 0,1 н. раствором
едкого натра.) 3. Минерализация органических соединений.
Реакционную колбу после отгонки аммиака отключают от
перегонной установки, вливают в нее 10 мл 30%-ной перекиси
водорода, ставят на включенную электроплитку и кипятят.
Минерализацию заканчивают, когда объем реагирующей
жидкости уменьшится до 15—20 мл, при этом жидкость станет
бесцветной.
Ход анализа. Пробу, освобожденную от аммонийных солей
и органических веществ, отфильтровывают, осадок на фильтре
промывают дистиллированной водой, фильтрат и промывные
воды сливают в стакан вместимостью 50 мл, нагревают до
появления первых пузырьков (кипятить не надо!), добавляют 2 мл
10%-ного раствора уксусной кислоты, перемешивают и медленно,
при постоянном перемешивании прибавляют 8 мл 3,5%-ного
раствора тетрафенилбората натрия. Реакционную смесь
оставляют в покое. Через 15 мин содержимое стакана охлаждают до
комнатной температуры, фильтруют через стеклянный фильтр
№ 2 без вакуума. Предварительно взвешивают пустой сухой
фильтр. Для удаления остатков тетрафенилбората натрия осадок
на фильтре промывают 2—3 раза 1%-ным раствором уксусной
кислоты, расходуя не более 15 мл, а затем водой. Полноту
отмывки определяют следующим образом. В пробирку наливают
немного раствора хлористого калия, добавляют 1—2 последних
капли фильтрата, который вытекает из воронки, и 1—2 капли
уксусной кислоты. Если тетрафенилборат натрия отмыт
полностью, то жидкость будет прозрачной. Появление
незначительной мути свидетельствует о наличии тетрафенилбората в
фильтрате. В этом случае промывку надо продолжить.
Осадок тетрафенилбората калия, отмытый от
тетрафенилбората натрия, в стеклянном фильтре помещают в сушильный
шкаф и сушат до полного удаления влаги при температуре
115—120°С. Первое взвешивание делают через 1,5 ч сушки
после охлаждения фильтра в эксикаторе, а второе — через 1 ч
дополнительной сушки. Обычно этого времени достаточно для
полного удаления влаги из осадка.
14 Заказ № 142 209
Расчет. Содержание калия в анализируемой пробе выражают в
миллиграммах окиси калия (КгО) и вычисляют по формуле
„ а0.1314 . 1000
л. = —
g
где X — концентрация калия в анализируемой пробе в пересчете на КгО, мг/л;
а — количество осадка тетрафенилбората калия, мг; g — объем пробы, взятый
на анализ, мл; 0,1314 — количество окиси калия (КгО), эквивалентное 1 мг
тетрафенилбората калия. Содержание калия определяют с точностью ±3—7% отн.
Определение сырого протеина экспрессным методом
Для быстрого определения белка в прессованных и сухих
дрожжах применяются различные модификации метода
Кьельдаля. Окисление белка протекает в два этапа: окисление
углерода до углекислого газа (СО2) и превращение азота в
сульфат аммония (см. с. 227).
После просветления кипящей реагирующей смеси
заканчивается сгорание углерода, содержащегося в анализируемом
материале. Превращение освободившегося азота в сернокислый
аммоний, называемое минерализацией, требует дальнейшего
нагревания. В зависимости от природы анализируемого
материала процесс минерализации длится 16 ч и более. Точно
установить его окончание трудно из-за отсутствия внешних
признаков. Автору экспрессного метода [72] удалось найти состав
катализатора и способ нагревания реагирующей смеси, при
которых процессы окисления углерода и минерализации азота
происходят за 15 мин. Окончание минерализации фиксируется
четкими внешними признаками состояния реагирующей смеси.
Осветление смеси наступает внезапно. Перед завершением
реакции из гранул двуокиси кремния, находящихся в реагирующей
жидкости, восходит столб мелких пузырьков. Окончание
реакции характеризуется относительно спокойной поверхностью смеси.
Эти признаки позволяют легко и точно установить конец
реакции. Кроме того, условия минерализации, примененные в
экспрессном методе, дают возможность с большей точностью
определять устойчивые органические соединения, как например
никотиновую кислоту (гетероциклическое соединение) и
триптофан, которые содержатся в белке дрожжей. Их неполная
минерализация в условиях анализа по методу Кьельдаля является
причиной получения заниженных результатов анализа на
содержание белка в дрожжах. Никотиновая кислота согласно ее
формуле содержит 11,38% азота. При минерализации по методу
Кьельдаля с катализатором CuSO4 в ней находят 11,26% азота,
т. е. 98,94% от теоретического, а экспрессным методом— 11,29%
азота, т. е. 99,21%. Триптофан по формуле содержит 13,72%
азота. По методу Кьельдаля в нем находят 98,7% от
теоретического, а экспрессным методом — 99,7%.
210
Мешающие вещества. Органические и неорганические веще-
щества, содержащие азот.
Аппаратура. Установка для минерализации (см. рис. 65).
Установка для отгонки аммиака (см. рис. 36). Установка для
титрования или один из приборов для колориметрического
определения аммиака (с. 274).
Реактивы и их приготовление. 1. Серная кислота плотностью
1,84 г/см3. 2. Окись ртути красная без азота. 3. Силикагель,
гранулы не селенизированные. 4. Сульфат калия безводный
кристаллический. 5. Едкий натр, гранулы (без азота). 6. Борная
кислота, 4%-ный раствор. 7. Гипосульфит натрия,
кристаллический. 8. Цинк, пыль. 9. Серная кислота, 0,1 н. раствор. 10.
Смешанный индикатор. Приготовление. Пять объемов 0,2%-
ного спиртового раствора бромкрезола зеленого смешивают
с одним объемом 0,2%-ного спиртового раствора метилового
красного.
Подготовка проб. Определяют влажность анализируемой
пробы высушиванием ее при 105°С.
Ход анализа. Проверяют интенсивность нагрева
электроплитки при полном нагреве — проба кипячения: 250 мл воды
в колбе Кьельдаля вместимостью 500 мл должны нагреваться
до волнистого кипения за 4—5 мин.
В сухую колбу Кьельдаля вместимостью 500 мл помещают
0,5—1 г анализируемых дрожжей, отвешенных с точностью
±0,0002 г, добавляют 6 г гранул силикагеля, 1,3—1,5 г окиси
ртути и 12 г сульфата калия. Вращением колбы перемешивают
ее содержимое, добавляют 15 мл концентрированной серной
кислоты и снова перемешивают. Колбу укрепляют в штативе
над электроплиткой и включают слабый нагрев (см. рис. 65).
(Нельзя нагревать колбу выше уровня жидкости!) В течение
5 мин нагревания обычно заканчивается вспенивание
реагирующей жидкости. Затем включают полный нагрев и кипятят
жидкость в течение 8—12 мин. Через 10—14 мин от начала
нагревания реагирующая смесь полностью осветляется. Реакцию
прекращают, когда вид смеси в колбе не изменяется в течение
3—5 мин. Столб мелких пузырьков, восходящих из гранул
силикагеля, сменяется на крупные и редко образующиеся пузыри.
После окончания минерализации колбу охлаждают,
прибавляют около 200 мл дистиллированной воды и, вращая колбу для
ускорения растворения лепешки, быстро прибавляют реактивы
в следующем порядке: 25 г едкого натра, 5 г пятиводного
гипосульфита натрия и 0,5 г цинковой пыли. Колбу быстро
закрывают пробкой, в которую вставлена трубка, соединяющая ее с
холодильником (см. рис. 36). Включают электроплитку и
отгоняют аммиак. Приемником служит колба, в которую налито
75 мл раствора борной кислоты. Конец отгонки аммиака
определяют по красной лакмусовой бумажке или с реактивом Нес-
слера (см. с. 274). Количество аммиака, отогнанного в раствор
14* 211
борной кислоты, определяют титрованием его 0,1 н. раствором
серной кислоты в присутствии индикатора. Для более удобного
определения конца титрования пользуются сравнением окраски
титруемого раствора с окраской свидетеля. Свидетелем служит
дистиллированная вода, в которую добавлен индикатор в таком
же количестве, как в пробу. Вода налита в такую же точно
колбу, как колба для титрования.
Количество отогнанного аммиака можно определять
колориметрическим методом с реактивом Несслера, как описано на
с. 273.
Расчет. Количество сырого протеина, содержащегося в анализируемых
дрожжах, вычисляют по формуле
аО.ООН • 100 • 6,25
g(l-0,0lW) '
где X — содержание сырого протеина в пробе абсолютно сухих дрожжей, %;
а — объем 0,1 н. раствора серной кислоты, израсходованный на титрование,
мл; g —количество дрожжей, взятое на анализ, г; W — влажность дрожжей, %;
0,0014 — количество азота, эквивалентное 1 мл 0,1 н. раствора серной
кислоты, г; 6,25 — эмпирический коэффициент для пересчета азота в белок.
Содержание сырого протеина (белка) определяют с точностью ±3% отн.
Предостережение. 1. При анализе проб, содержащих азот, связанный
в минеральных соединениях, его надо определить отдельно и учесть при
вычислении результатов анализа. 2. Для правильного ведения анализа надо
тщательно регулировать нагрев и точно отмеривать реактивы. При анализе
некоторых материалов очень важно, чтобы в первые минуты нагрев проводить
постепенно ввиду высокой точки кипения окисляющей смеси. 3. Нельзя в
реакционную колбу добавлять с реактивами фильтровальную бумагу и другие
материалы.
Определение витамина D2 колориметрическим методом
Метод предназначен для определения витамина D2 в
дрожжах до и после облучения их ультрафиолетовым светом [73]. При
облучении дрожжей ультрафиолетовыми лучами находящийся
в них эргостерин превращается в витамин D2 (кальциферол или
эргокальциферол) по следующей схеме:
Эростерин-*-Лумистерин—»-Тахистерин—>¦ Витамин D2
Супрастерин I Супрастерин II
В облученных дрожжах наряду с витамином D2 находятся
все промежуточные продукты и продукты превращения
витамина D2 в супрастерины I и П. В чистом виде витамин D2
представляет собой бесцветные кристаллы, которые нерастворимы
в воде и хорошо растворимы в жирах [74].
Метод определения витамина Ь2 состоит из двух частей:
выделения витамина D2 из дрожжей и количественного определения
выделенного витамина D2. Для выделения витамина D2 из
дрожжей последние сначала обрабатывают щелочным спиртоводным
раствором. Витамин D2 при этом не изменяется. Для
предотвращения окисления витамина D2 кислородом воздуха эту опера-
212
цию проводят в токе азота, добавив к щелочному раствору
аскорбиновую кислоту или аскорбинат натрия. Из оставшихся
веществ, не перешедших в раствор, экстрагируют витамин Dz
вместе со стеринами. Затем стерины осаждают дигитонином,
образующиеся дигитониды отфильтровывают, из фильтрата
удаляют растворитель током углекислого газа, полученный сухой
остаток растворяют в дихлорэтане и анализируют на
содержание витамина Т>2.
Количественное определение витамина D2 основано на его
реакции с двухлористым оловом, при котором образуется
соединение, окрашенное в желто-розовый цвет, с максимумом
поглощения света в области 500 ммк. На фотоэлектроколориметре
устанавливают оптическую плотность исследуемого раствора и
по калибровочной кривой находят концентрацию витамина D2>
а затем вычисляют содержание витамина Ог в дрожжах.
Активность препаратов витаминов выражают в
международных единицах (м. е) или в интернациональных единицах (и. е).
1 м. е=1 и. е=0,025 мкг (микрограмм) чистого витамина.
Мешающие вещества. Стерины. Вещества неизвестного
строения, присутствующие в дрожжах, которые не отделяются от
витамина O2 при его экстракции и с двухлористым оловом дают
окрашенные соединения.
Аппаратура. Установка для нагревания в токе азота. Фото-
электроколориметр. Установка для отгона летучих веществ.
Водяная баня. Делительные воронки вместимостью 250 и 150 мл.
Пробирки с притертыми пробками вместимостью 15 мл. Часы
песочные на 1; 1,5 и 2 мин. Дрексели.
Реактивы и их приготовление. 1. Спирт этиловый ректификат
без альдегидов и сивушных масел. Приготовление. К 1 л
этилового спирта, находящегося в колбе, прибавляют 1 г
азотнокислого серебра, растворенного в 5 мл дистиллированной
воды, раствор перемешивают, колбу закрывают стеклянной или
корковой пробкой и оставляют стоять. Через 8 ч в спирт
добавляют 5 г едкого кали в виде спиртового раствора,
перемешивают и снова оставляют стоять. Через 24 ч спирт осторожно
сливают с осадка в перегонную колбу, добавляют к нему 500 г
негашеной извести и перегоняют на установке с дефлагматором.
Первые порции спиртового конденсата отбрасывают. Собирают
фракцию, которая отгоняется, когда температура паров спирта
на выходе из дефлегматора в холодильник равна 78,3°С.
Таким способом получают безальдегидный и бессивушный спирт.
2. Едкое кали, насыщенный спиртовый раствор. 3. Сернокислый
натрий безводный. 4. Бензол. 5. Углекислый газ в баллоне,
6. Дигитонин (С56Н92О29), 4%-ная водная суспензия. 7.
Аскорбиновая кислота или аскорбинат натрия. 8. Азот, очищенный от
кислорода. Очистка. Азот из баллона пропускают через два
последовательно соединенных дрекселя. В первом дрекселе
налит щелочной раствор пирогаллола, а во второй — дистиллиро-
215
ванная вода. 9. Серный эфир очищенный от перекисей.
Очистка. К 1 л эфира, помещенного в делительную воронку,
приливают 10 мл 40%-ного раствора едкого натра и 100 мл 4 % -ного
раствора перманганата калия, смесь энергично взбалтывают,
а затем дают постоять для расслаивания. Если водный слой
смеси (раствор перманганата) станет зеленым, то его сливают
и эфир обрабатывают новой порцией перманганата. После
расслаивания розовый (или красный) раствор перманганата,
прореагировавший с перекисями, выливают из воронки. Эфир в той
же воронке промывают несколько раз дистиллированной водой
до полного удаления щелочи (реакция с фенолфталеином).
Очищенный эфир сливают в колбу, сушат, добавляя к нему
безводный сернокислый натрий, и перегоняют. 10. Двухлористое олово
раствор. Приготовление. В пробирку с пришлифованной
пробкой помещают 100 мг двухлористого олова, прибавляют
10 мл смежеперегнанного хлористого ацетила, стеклянной
палочкой быстро раздавливают крупинки двухлористого олова,
закрывают пробирку пробкой и энергично встряхивают в течение
1 мин. Затем пробирку ставят в штатив и после отстаивания
в течение 2—3 мин жидкость фильтруют через двойной
складчатый фильтр в сухую пробирку и закрывают пришлифованной
пробкой. Раствор готовят перед употреблением. 11. Дихлорэтан
х. ч. Очистка. Дихлорэтан сушат, добавляя к нему безводный
сернокислый натрий, а затем перегоняют в токе углекислого
газа. Полученный бесцветный прозрачный конденсат
дихлорэтана не должен давать муть при добавлении 1 капли 20%-ного
раствора пятихлористой сурьмы в 10 мл дихлорэтана. 12.
Витамин D2 х.ч. кристаллический. Приготовление
стандартного раствора. В дихлорэтане растворяют 25 мл витамина
D2 и объем раствора в мерной колбе вместимостью 100 мл
доводят до метки, прибавляя дихлорэтан. В мерную колбу
вместимостью 10 мл помещают 1 мл приготовленного раствора и
объем его доводят до метки, прибавляя дихлорэтан. В 1 мл
полученного раствора содержится 0,025 мг витамина D2, или
1000 и. е. Этот раствор называют стандартным.
Построение калибровочной кривой. Анализируют
различные количества от 0,1 до 1 мл стандартного раствора
витамина D2. В пробирку вливают 0,1 (или 0,2, ..., 1 мл)
стандартного раствора витамина D2, объем доводят до 1 мл,
прибавляя дихлорэтан, затем добавляют 1 мл двухлористого олова и
через 3 мин измеряют оптическую плотность на фотоэлектроко-
лориметре с сине-зеленым фильтром при толщине слоя жидкости
в кювете 5 мм. В кювету для сравнения наливают чистый
дихлорэтан. На основании полученных данных строят график
зависимости оптической плотности раствора от количества
витамина D2, взятого на анализ, т. е. калибровочную кривую,
которой пользуются для вычисления содержания витамина D2
в анализируемой пробе.
214
Ход анализа. Выделение витамина D2 из
дрожжей. В колбу вместимостью 100 мл помещают 1 г сухих
дрожжей, прибавляют к ним 25 мл спиртового раствора едкого кали
и 40—50 мг аскорбиновой кислоты или аскорбината натрия.
Колбу ставят на кипящую водяную баню, закрывают пробкой,
в которую вставлены обратный холодильник и трубка для
подвода азота и нагревают в течение 1 ч, непрерывно пропуская
слабый ток азота. Затем содержимое колбы охлаждают до
комнатной температуры и количественно переносят в делительную
воронку вместимостью 150 мл, расходуя при этом 20 мл
дистиллированной воды. Для извлечения витамина D2 в делительную
воронку прибавляют 50 мл серного эфира, воронку закрывают,
энергично встряхивают 3—5 мин и ставят в штатив. После
расслаивания жидкостей нижний водный слой сливают в
реакционную колбу, а эфирный слой — в делительную воронку
вместимостью 250 мл. Водный слой снова выливают в воронку
вместимостью 150 мл, добавляют к нему 50 мл серного эфира
и повторяют экстракцию. Эту операцию делают еще 3 раза,
сливая эфирные растворы в одну воронку.
Для удаления щелочи собранный эфирный экстракт
промывают 4 раза дистиллированной водой, расходуя на каждое
промывание 40—50 мл воды. Промытый экстракт переливают
в колбу, добавляют 5—10 г безводного сернокислого натрия для
удаления воды из раствора и ждут 30 мин, изредка
перемешивая раствор. Сухой эфирный экстракт фильтруют через
бумажный фильтр и перегонную колбу, осадок промывают 50 мл
серного эфира, которые присоединяют к фильтрату. Для полного
удаления эфира через эфирный экстракт пропускают
углекислый газ. Полученный сухой остаток растворяют в 5 мл бензола,,
который затем так же отгоняют углекислым газом.
Сухой остаток после отгонки бензола содержит витамин D2
и стерины. Его растворяют в 10 мл дихлорэтана. Для удаления
стеринов берут 7 мл раствора, помещают их в пробирку с
притертой пробкой и прибавляют 0,5 мл 4%-ной водной суспензии
дигитонина, пробирку закрывают пробкой и взбалтывают 1 раз
в секунду в течение 10 мин. Выпавший осадок отфильтровывают,
получают фильтрат № 1. В чистую прибирку помещают 1 мл
фильтрата № 1, прибавляют 5 мл бензола, раствор
перемешивают и растворитель отгоняют током углекислого газа.
Полученный сухой остаток анализируют на содержание витамина D2.
Определение витамина D2. Сухой остаток в пробирке
растворяют в 2 мл дихлорэтана. К полученному раствору
прибавляют 1 мл раствора двухлористого олова в хлористом
ацетиле, пробирку закрывают, встряхивают 2—3 раза и оставляют
стоять. Через 3 мин определяют интенсивность появившейся:
окраски раствора с помощью фотоэлектроколориметра (см.
с. 207). Оптическую плотность раствора определяют с
сине-зеленым фильтром в кювете, в которой толщина слоя жидкости
215
¦5 мм. В кювету сравнения наливают чистый дихлорэтан.
Определив оптическую плотность раствора, по калибровочной кривой
находят соответствующую ей концентрацию витамина D2 в
анализируемом фильтрате № 1.
В необлученных дрожжах содержатся неизвестные вещества,
которые с двухлористым оловом дают тоже окрашенные
соединения, поэтому одновременно анализируют дрожжи до и после
облучения и по разности результатов двух анализов находят
содержание витамина Бг в облученных дрожжах.
Расчет. Содержание витамина D2 в анализируемой пробе облученных
дрожжей вычисляют по формуле
у ап g'n'
~~g гГ~'
где X—содержание витамина D2 в I г облученных дрожжей, и. е.; а, п\ —
количество витамина D2) найденное в I мл фильтрата № I по калибровочной
кривой при анализе облученных и необлученных дрожжей , и. е.; g, gi —
количество облученных и необлученных дрожжей, взятое иа анализ, г; п, П\ — раз-
ведеиие при анализе облученных и необлучеииых дрожжей.
Витамин D2 определяют с точностью ±7—15% отн.
Предостережение. Витамин D2 окисляется кислородом воздуха, на свету
он превращается в ядовитый токсистерии. Пробы облученных дрожжей надо
хранить в хорошо закрытых склянках из темного стекла в темном месте [74].
Определение мышьяка в дрожжах
В дрожжах и полупродуктах (субстрат, бражка и др.)
мышьяк определяют колориметрическим методом, как описано
на с. 69. Отличие в анализах различных проб состоит в
подготовке проб. На гидролизных заводах мышьяк определяют
в полупродуктах и в дрожжах без предварительной
минерализации. При анализе в сосуд прибора (см. рис. 21) помещают 10 мл
субстрата или 0,1 г сухих дрожжей. На сульфитных заводах
полупродукты перед анализом минерализуют так же, как
сульфитный щелок (см. с. 71), а дрожжи минерализуют, как
описано ниже.
Минерализация дрожжей. В колбу Кьельдаля
помещают 2 г сухих дрожжей, добавляют 15 мл концентрированной
серной кислоты, не содержащей мышьяк, и нагревают до
прекращения пенения. Затем колбу немного охлаждают, добавляют
в нее 15 мл пергидроли, снова нагревают и кипятят до
обесцвечивания жидкости в колбе. Полученный бесцветный раствор
охлаждают и количественно переносят в мерную колбу
вместимостью 100 мл, объем раствора доводят до метки водой и
перемешивают. На анализ берут 5—10 мл приготовленного раствора.
Расчет. Содержание мышьяка в дрожжах вычисляют по формуле
дя100
І '
где X—содержание мышьяка в дрожжах, мкг/100 г дрожжей; а —
количество мышьяка в пробе, найденное по стандартной шкале, мкг; g — количество
дрожжей, взятое на анализ, г; п — разведение пробы при минерализации.
216
Определение аминокислотного состава белка
Для определения качественного и количественного состава
аминокислот белка дрожжей, активного ила и других биомасс
предназначен метод, основанный на колориметрическом
определении количества каждой аминокислоты, выделенной
хроматографией на бумаге. Мономерные аминокислоты определяют
после их экстракции из пробы горячей водой. Полимерные
аминокислоты, составляющие макромолекулы белка,
предварительно гидролизуют [75]. Для выделения аминокислот проводят
нисходящее, многократное хроматографирование двумя
растворителями. Проявителем служит слабокислый раствор нингид-
рина, который с аминокислотами дает сине-фиолетовое
соединение (ДИДА) по уравнению
RCH(NH2)COOH+2
Аминокислота Нингидрин
Соединение ДИДА
сине-фиолетобого цбег,
Идентификацию аминокислот проводят с помощью
свидетелей и на основании известных значений Rf отдельных
аминокислот. При анализе дрожжей в гидролизатах белка находят почти
все аминокислоты. На хроматограммах таких
многокомпонентных смесей пятна аминокислот расположены очень близко,
поэтому для их идентификации делают параллельное
хроматографирование искусственных смесей аминокислот.
Количественное определение аминокислот основано на
реакции получения медного производного соединения ДИДА,
оранжево-красного цвета, которое имеет максимум поглощения при
530 ммк.
Растворы медного производного ДИДА, полученные при
анализе пробы и свидетеля, колориметрируют на фотоэлектроколо-
риметре и, определив их оптическую плотность, вычисляют
концентрацию аминокислоты в пробе. Этим методом нельзя
определить пролин и оксипролин.
Мешающие вещества. Жиры и другие подобные вещества.
21Т
Аппаратура. Аппарат для экстракции (см. рис. 17). Камера
для двумерного хроматографирования (см. рис. 25). Фотозлект-
роколориметр (см. рис. 64). Водяная баня. Эксикатор. Термостат.
Реактивы и их приготовление. 1. Трилон Б, 0,5%-ный
раствор. 2. Серный эфир. 3. Спирт этиловый ректификат. 4. Соляная
кислота плотностью 1,19 г/см3. 5. Нингидрин, 0,5%-ный раствор
в ацетоне. Этот раствор надо хранить в темной склянке с
пришлифованной пробкой в холодильнике. 6. Сернокислая медь
0,005 % -ный раствор в 75 % -ном этиловом спирте.
Приготовление. В мерной колбе вместимостью 100 мл растворяют в 22 мл
дистиллированной воды 5 мг пятиводной сернокислой меди
(CuSO4-5H2O) и объем доводят до метки, прибавляя 96% -ный
этиловый спирт. 7. Хроматографическая бумага марки Б.
Подготовка бумаги. Для удаления из хроматографической
бумаги всех веществ, которые мешают проведению анализа
аминокислот, ее обрабатывают раствором трилона Б. Лист
хроматографической бумаги разрезают пополам, погружают в банку
с 0,5%-ным раствором трилона Б и оставляют на 3 суток. Затем
бумагу вынимают из раствора трилона Б и погружают в
проточную водопроводную воду. После отмывки трилона Б бумагу
сушат в вытяжном шкафу при полном отсутствии даже следов
соляной кислоты. (Обязательно работать в резиновых
перчатках!) 8. Растворитель 1. Приготовление. Готовят смесь
н-бутанола, уксусной кислоты и воды в соотношении 4:1:5.
В делительную воронку вливают 40 мл м-бутанола, 10 мл
ледяной уксусной кислоты (обязательно 100%-ной) и 50 мл
дистиллированной воды. Смесь энергично взбалтывают в течение
2 мин и оставляют в покое. После расслаивания смеси сливают
нижний слой, захватывая немного и верхний, и ставят его в хро-
матографическую камеру. Верхний слой используют как
растворитель. 9. Растворитель П. Приготовление. Готовят
смесь н-бутанола, уксусной кислоты и воды в соотношении
8:3: 1. В делительную воронку вливают 40 мл н-бутанола, 15 мл
ледяной уксусной кислоты и 5 мл дистиллированной воды.
Смесь энергично взбалтывают в течение 2 мин и оставляют на
10 мин, при ее расслоении готовят новую смесь. Однородную
прозрачную жидкость используют в качестве растворителя.
10. Аминокислоты: цистин; лизин; гистидин; аргинин; глицин; ала-
нин; тирозин; триптофан; фенилаланин; лейцин или изолейцин;
аспарагиновая кислота или серии; глютаминовая кислота или
треонин; валин или метионин. Приготовление растворов
свидетелей. Готовят четыре раствора свидетелей, каждый
из которых должен содержать несколько аминокислот, имеющих
заметно отличающиеся Rf. Концентрация кислоты в растворе
должна быть равной 0,01 М. В табл. 9 приведены названия
аминокислот в порядке возрастания их скорости движения по
бумаге, т. е. Rf, титр аминокислоты в ее 0,01 М растворе и номер
свидетеля, в котором присутствует кислота. В качестве раство-
218
рителя аминокислот употребляют кислый спиртоводный раствор,
который готовят следующим образом. В мерную колбу
вместимостью 100 мл вливают 10 мл этилового спирта, 2 мл
концентрированной соляной кислоты и объем раствора доводят до
метки, прибавляя воду.
ТАБЛИЦА Э
Сведения для идентификации и определения аминокислот по свидетелям
Название
аминокислоты в порядке
возрастания Rj
Цистин
Лизин
Гистидин
Аргииин
Аспарагиновая
кислота
Серии
Глицин
Глютаминовая
кислота
Титр
0,01 М
раствора
кислоты,
мг/мл
2.40
1.81
1,91
2.10
1,33
1.05
0,75
1.47
раствора
свидетелей
1
2
3
4
1
2
3
4
Название амнно-
кіґслотьі в порядке
возрастания Rj
Треонин
Алании
Тирозин
Валин
Метионин
Триптофан
Фенилаланин
Изолейцин
(лейцин)
Титр
0,01 М
раствора
кислоты,
мг/мл
1,19
0.89
1.80
1.17
1.49
2.04
1,51
1,31
№
раствора
свидетелей
2
4
2
3
4
I
2
3
Следующие 3 пары кислот имеют одинаковые Rf.
глютаминовая кислота и треонин, аспарагиновая кислота и серии, валин
и метионин. При хроматографировании каждая пара дает одно
пятно.
Для приготовления 10 мл раствора свидетеля № 1 на
аналитических весах взвешивают 24 мг цистина, 13 мг аспарагиновой
кислоты и 20 г триптофана. Навески кислот, взятые в
отдельных бюксах, растворяют каждую в нескольких каплях
растворителя и количественно переносят в мерную колбу вместимостью
10 мл. Объем раствора доводят растворителем до метки,
перемешивают и хранят в холодильнике. Аналогично готовят
остальные три раствора свидетелей.
Подготовка проб. Экстракция мономерных кислот.
Смешивают 4 г прессованных дрожжей с 10 мл
дистиллированной воды, полученную суспензию количественно переносят
в мерную колбу вместимостью 25 мл и добавляют воду до метки.
Затем содержимое колбы переносят в плоскодонную колбу,
нагревают в водяной бане до 80°С, охлаждают и фильтруют или
центрифугируют. Полученный фильтрат анализируют на
содержание аминокислот.
Гидролиз белка. 2 г абсолютно сухих дрожжей
помещают (в пакетике) в аппарат для экстракции (см. рис. 17) и
проводят экстракцию жира серным эфиром. Обезжиренные дрожжи
высушивают до полного удаления растворителя. Затем 10 мг
сухих обезжиренных дрожжей помещают в ампулу, прибавляют
219
к ним 0,2 мл 6 н. раствора соляной кислоты (НС1), ампулу
запаивают и, поместив в термостат, выдерживают ее при
температуре 105—110°С 24 ч. После гидролиза белков содержимое
ампулы количественно переносят в фарфоровую чашку
вместимостью 25—50 мл или в тигель и выпаривают досуха. К сухому
остатку приливают 1 мл дистиллированной воды и снова
раствор упаривают досуха в вакуум-эксикаторе над твердым едким
натром. Так повторяют 3 раза для полного удаления соляной
кислоты. Полученный сухой остаток растворяют в 0,5 мл
растворителя для аминокислот, раствор фильтруют через
складчатый фильтр или вату.
Ход анализа. Хроматографирование. Для
качественного анализа хроматографирование проводят на бумаге,
вырезанной в виде больших лопаток (см. рис. 22). Камерой для хро-
матографирования служит эксикатор.
Для количественного анализа проводят нисходящее
многократное хроматографирование на листе хроматографической
бумаги. Одновременно хроматографируют испытуемый раствор
и растворы свидетелей. Микропипеткой (см. рис. 27) на линию
старта на расстоянии 3 см друг от друга наносят 0,01 мл
испытуемого раствора, затем четыре раствора свидетелей по 0,002—
0,005 мл и снова исследуемый раствор. Пятна нанесенных
растворов просушивают на воздухе, лист бумаги помещают в хро-
матографическую камеру, верхний конец листа погружают
в растворитель I, налитый в лодочку, камеру закрывают и ждут
когда растворитель дойдет до нижнего конца листа. Затем хро-
матограмму вынимают, просушивают в вытяжном шкафу и
снова помещают в хроматографическую камеру.
Хроматографирование растворителем I проводят 3 раза. Потом в лодочку
наливают растворитель II, хроматограмму просушивают до
полного удаления запаха растворителя I, повертывают ее на 90°.
устанавливают в камеру и 2 раза пропускают через нее
растворитель II. Хроматограмму вынимают из камеры, хорошо
высушивают на воздухе до полного удаления запаха растворителя,
опрыскивают ее раствором нингидрина из пульверизатора,
высушивают и на сутки кладут между двумя листами
фильтровальной бумаги в темное место.
Идентификация аминокислот. На проявленной хро-
матограмме сравнивают расположение пятен исследуемой пробы
с расположением пятен свидетелей и устанавливают, какой
кислоте принадлежит каждое пятно на хроматограмме пробы. Так
определяют качественный состав аминокислот, которые
содержатся в белке дрожжей.
Определение концентрации аминокислот.
Из хроматограммы пробы вырезают пятно аминокислоты и
соответствующее ему пятно известной аминокислоты из
хроматограммы свидетелей. Если фон хроматограммы окрашен, то для
контроля вырезают кусочек хроматограммы без пятна такого же
220
размера, как с пятнами. Каждый вырезанный кусочек хрома-
тограммы нарезают на мелкие кусочки, помещают в отдельную
пробирку и приливают к ним 5 мл раствора для экстракции
@,005%-ный раствор сернокислой меди в 75%-ном спирте).
Пробирки ставят на 1 ч в темное место. За это время проходит
реакция образования медного соединения ДИДА и его
растворение в спирте. Через 1 ч растворы в пробирках приобретают
оранжево-красный цвет. Растворы с кусочками хроматограмм
хорошо перемешивают, фильтруют, сливают в кюветы (толщина
жидкости 1 см) и на фотоэлектроколориметре с зеленым
светофильтром определяют оптическую плотность растворов. В
кювету сравнения в качестве растворителя наливают экстракт из
кусочков хроматограммы без пятна или, если фон хромато-
граммы белый, наливают чистый раствор для экстракции.
Расчет. Величина оптической плотности раствора при анализе
аминокислот зависит в сильной степени от условий хроматографирования (качества
бумаги, числа пропусканий растворителей и др.), поэтому для определения
концентрации аминокислоты в анализируемом растворе нельзя пользоваться
заранее сделанными калибровочными кривыми. Здесь пользуются
установленной зависимостью между концентрацией кислоты и оптической плотностью
раствора, которая в пределах концентраций аминокислот от 0,025 до 0,25 мкмоль
(мнкромолей) имеет линейный характер. Содержание каждой кислоты
в абсолютно сухих дрожжах, взятых на анализ, вычисляют по формуле
У _ кап
a\gg\
где X — содержание аминокислоты в 1 г абс. сухих дрожжей, мг; k —
количество аминокислоты в растворе свидетелей, которое взято на хроматогра-
фирование, мг; а — оптическая плотность раствора пятна из хроматограммы
пробы; й\ — оптическая плотность раствора пятна из хроматограммы
свидетелей; п — разведение пробы; g — количество абсолютно сухих дрожжей,
взятое на анализ, г; gi — объем раствора пробы, нанесенный на хроматограмму, мл.
Чувствительность реакции аминокислот с нингидрином 2—5 мкг. Точность
определения аминокислот ±5—15% отн.
Определение лизина микробиологическим методом
Для определения свободного лизина в кормовых препаратах
и в дрожжах, обогащенных лизином перед сушкой [76],
предназначен метод, основанный на использовании культуры Esche-
richia coli (E. coli). Концентрацию микроорганизмов после
выращивания определяют нефелометрическим методом или на
фотоэлектроколориметре. Результат анализа находят по
калибровочной кривой. Для анализа достаточно 5—10 мкг лизина.
Мешающие вещества. Белки и свободные аминокислоты не
мешают.
Аппаратура. Термостат. Автоклав. Один из следующих
приборов: нефелометр, мутномер (см. с. 290), набор
колориметрических пробирок или фотоэлектрический колориметр.
Реактивы и их приготовление. 1. Основная среда.
Приготовление. В стакан вместимостью 500 мл вливают 300 мл
221
дистиллированной воды и растворяют в ней следующие
реактивы: 7 г двузамещенного фосфорнокислого калия К2НРО4, 3 г одно-
замещенного фосфорнокислого калия КНгРО4, 0,5 г
лимоннокислого натрия С6Н5О7Ыа3 • 5НгО, 0,1 г сернокислого магния
MgSO4*7H2O, 1 г сернокислого аммония (NH^SO^ 2 г
глюкозы и 0,5 г хлористого натрия NaCl. Раствор количественно
переносят в мерную колбу вместимостью 1 л, объем доводят до
метки, прибавляя дистиллированную воду, перемешивают и
измеряют pH. Раствор должен иметь pH 6,9—7,0. Его хранят
в холодильнике. Срок хранения 5—10 дней. 2. Стандартный
раствор L-лизина гидрохлорида. В 1 мл водного раствора
содержится 500 мкг L-лизина гидрохлорида. Раствор хранят под
слоем толуола в холодильнике. Перед анализом раствор
разбавляют в 50 раз до концентрации лизина 10 мкг в 1 мл. 3.
Основная культура Escherichia coli (E. coli). Мутант Е. coli 432
выращивают на косяках мясо-пептонного агара при
температуре 37°С в течение 24 ч. 4. Посевной материал.
Приготовление. За сутки до анализа основную культуру Е. coli
пересевают в пробирки на среду из 5 мл основной среды и 5 мл
разбавленного стандартного раствора L-лизина гидрохлорида.
Выращивание ведут в термостате при 37°С. Через 20 ч
инкубации получают бактериальную суспензию. Для приготовления
посевного материала 1—2 капли бактериальной суспензии
разбавляют 10 мл физиологического раствора.
Подготовка пробы. 1. Определяют влажность пробы. 2. При
анализе сухих дрожжей или кормового концентрата проводят
экстракцию свободного лизина из пробы. В фарфоровой ступке
растирают 1 г пробы с 20—30 мл дистиллированной воды в
течение 5—10 мин. Полученный экстракт лизина
отфильтровывают, осадок на фильтре промывают водой, все сливают в
мерную колбу и разбавляют водой так, чтобы в 1 мл раствора
содержалось 10—20 мкг лизина.
Ход анализа. Одновременно с анализом экстракта
испытуемой пробы проводят анализ различных количеств стандартного
раствора L-лизина гидрохлорида, чтобы получить данные для
калибровочной кривой. Для приготовления стандартной шкалы
берут 15 пробирок. Каждый анализ делают в трех повторностях.
В первые три пробирки вливают по 5 мл воды, во вторые — по
1 мл раствора лизина A0 мкг/мл) и по 4 мл воды, в третьи —
по 2 мл раствора лизина и по 3 мл воды, в четвертые — по 3 мл
раствора лизина и по 2 мл воды и в пятые — по 4 мл раствора
лизина и по 1 мл воды. Во все пробирки вливают по 5 мл
основной среды. Таким же образом вместо стандартного раствора
лизина разливают экстракт испытуемой пробы. Все пробирки
закрывают ватными пробками и стерилизуют в автоклаве в
течение 20 мин. Затем пробирки охлаждают на воздухе, в каждую
вносят по 1 капле посевного материала, закрывают пробками и
ставят в термостат при 37°С. Через 24 ч выращивание бактерий;
222
заканчивают, пробирки вынимают из термостата, помещают
в автоклав, прогревают текучим паром в течение 15 мин. После
охлаждения измеряют степень мутности содержимого каждой
пробирки нефелометром или мутномером. При отсутствии этих
приборов мутность можно определять фотоэлектроколоримет-
ром при синем светофильтре или визуально в
колориметрических пробирках.
На основании данных анализа (средних из трех определений)
различных количеств стандартного раствора лизина строят
калибровочную кривую зависимости показаний прибора от
количества лизина, взятого на анализ. Определив показания прибора
при исследовании жидкостей с экстрактом пробы, по
калибровочной кривой находят количество лизина, которое было взято
на анализ экстракта в каждой пробирке.
Расчет. Содержание L-лизина в абсолютно сухой пробе вычисляют по
формуле
аУІООО
где X — содержание лизина в абс. сухой пробе, мг/кг; g — количество абс.
сухой пробы, взятое на экстракцию, г; V — объем экстракта, мл; v — объем
экстракта, взятый на анализ, мл; а — количество лизина в экстракте, взятом
на анализ, найденное по калибровочной кривой, мкг.
ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ НА СУХИЕ КОРМОВЫЕ ДРОЖЖИ И
МЕТОДЫ ИХ ИСПЫТАНИЯ ПО СТАНДАРТАМ РАЗНЫХ СТРАН
Технические требования
Главными показателями качества кормовых дрожжей во всех
странах принято считать содержание сырого протеина, влаги,
золы и органолептические свойства дрожжей (табл. 10). В
нашей стране, кроме этих показателей, нормируют крупность
гранул и содержание металломагнитных примесей [77]. В
Чехословакии нормируют содержание истинного белка, мышьяка,
тяжелых металлов и кислотность дрожжей [78—79]. В Польше
предъявляют особые требования к качеству белка. Там
нормируют содержание белка, усвояемого организмом животных,
и проверяют ферментативную активность дрожжей [80—81].
Для сравнения способов оценки качества дрожжей,
применяемых у нас и в других странах, ниже приведены методы
испытания кормовых дрожжей по стандартам этих стран.
Определение влажности и сухих веществ
Определение сухих веществ является ответственным
анализом, потому что в некоторых странах производственный план
предприятий, выпускающих дрожжи, и цены на дрожжи
рассчитывают на абсолютно сухие дрожжи (Чехословакия).
223
ТАБЛИЦА 10
Технические требования
Показатель качества
Внешний вид
Цвет
Запах
Вкус
Степень измельчения
Крупность для
гранулированных дрожжей:
диаметр гранул, мм,
в пределах
длина гранул, мм,
Не более
на сухие
кормовые
СССР
ГОСТ 20083 - 74
Группы
высшая
первая
вторая
третья
Порошок, чешуйки или гранулы
От светло-желтого до коричневого
Свойственный дрожжам, без
постороннего запаха
—
—
5,0-9,0
18
—
—
5,0-9,0
18
—
—
5,0-9,0
18
—
—
5,0-9,0
18
дрожжи по стандартам разных стран
ЧССР
он
к
ЧСН-56-6810*
Легкие хлопья или порошок,
который при хранении спрессовывается
От светло-желтого
до серого и
коричневого
Чястый, с
оттенком пряного, без
постороннего
запаха
Пряный, характе
—
—
—
От белого до
серого и
светло-коричневого
Пряный, без
постороннего запаха
эный для дрожжей
—
—
Польша
РА-59
А—79О06
Пластинки, крупа,
мука
От бронзового до
темно-бронзового
Своеобразный,
посторонние запахи
не допускаются
Своеобразный,
допустима горечь
Дрожжи должны
просеиваться через
сито с отверстиями
4 мм. Комки
должны легко
растираться
Дрожжи не
способны к брожению
10
48
Дрожжи лишены
ферментативной
способности
10
48
75
проход через сито с
отверстиями
диаметром 3 мм, %, не более
Биологическое состояние
(ферментативная
способность)
Содержание влаги, %, 10 10 10 10 9
не более
Содержание сырого про- 56 51 46 43 45
теина в пересчете на
сухое вещество, %, не
менее
Коэффициент
усвояемости белка, %, не менее
Содержание истинного — — — — 36 36
белка (по Бернштейну)
в пересчете на сухое
вещество, %, не менее
Содержание золы в пе- Ю 10 10 Ю 9 10 Ю
ресчете на сухое
вещество, %, не более
Кислотность на 100 г — — — — 1500 1000
а. с. дрожжей, мг КОН
Содержание мышьяка — — — — — Следы 300
на 100 г а. с. дрожжей,
мкг
Содержание тяжелых
металлов
Примечание. * — стандарт на кормовые дрожжи, полученные из пекарских или пивных дрожжей. Прочерк озна-
чает, что этого показателя в стандарте нет.
Влажность и содержание сухих веществ в дрожжах во всех
странах, кроме Советского Союза, определяют высушиванием
пробы дрожжей при 105°С. Для ускорения сушки прессованных
дрожжей и дрожжевого молока некоторые прибавляют
кварцевый песок или этиловый спирт (ЧССР).
Определение сухих веществ по стандарту
ЧССР [80, 81]. Чистый кварцевый песок высушивают в
фарфоровой или алюминиевой чашке при температуре 105—110°С.
Чашку охлаждают в эксикаторе и вместе с песком взвешивают.
Потом кладут в нее 3—5 г дрожжей, взятых из середины бруска,
и добавляют примерно 10 мл 96%-ного этанола. Содержимое
чашки хорошо растирают стеклянной палочкой и сушат
инфракрасными лучами лампы (при непрерывном перемешивании) до
тех пор, пока не исчезнут пары. Окончательно досушивают в
сушильном шкафу при температуре 105°С.
Содержание сухих веществ в дрожжах вычисляют по формуле
alOO
~ g '
где g — количество дрожжей до сушки, г; а — количество дрожжей после
сушки, г. Результат анализа вычисляют до 0,01 %.
Воспроизводимость в среднем из двух определений ±0,5% абс.
Определение ферментативной способности дрожжей
Сухие кормовые дрожжи, предназначенные для скармливания
животным, должны быть полностью лишены ферментативной
способности. Это условие оговорено в стандартах многих стран
без приведения метода испытания. В польском стандарте дан
метод проверки наличия ферментативной способности дрожжей.
Метод основан на измерении количества углекислого газа (СОг),
который выделяется, если дрожжи поместить в сахарный
раствор и выдержать их при температуре 30°С. Кормовые сухие
дрожжи за 24 ч пребывания в сахарном растворе не должны
бродить, углекислый газ не должен выделяться.
Ход анализа. В колбу вместимостью 750 мл вливают 400 мл
дистиллированной воды и нагревают ее в термостате до 30°С.
В выпарительную чашку вместимостью 100 мл помещают 10 г
сухих исследуемых дрожжей, слегка растертых в ступке,
приливают к ним из колбы 50 мл подогретой воды и хорошо
перемешивают. Из колбы в стакан отливают еще 50 мл подогретой
воды, а в колбу с 300 мл оставшейся подогретой воды всыпают
40 г сахарного песку. Когда сахар растворится, в реакционную
колбу выливают дрожжевую суспензию из чашки и закрывают
ее резиновой пробкой. В пробку вставлены термометр и
изогнутая стеклянная трубка для соединения воздушного
пространства реакционной колбы с воздушным пространством другой
колбы вместимостью 100 мл, в которую налито 80 мл воды. Колбу
226
с водой плотно (герметически) закрывают пробкой с двумя
трубками: короткой и длинной. Короткую трубку, конец
которой кончается на уровне пробки, соединяют с реакционной
колбой. У длинной трубки один конец доходит до дна колбы, а
второй — опускают в открытый цилиндр. Реакционную колбу
с дрожжами в сахарном растворе ставят в термостат на 24 ч
при 30°С. Колбу с водой и цилиндр ставят вблизи.
Если дрожжи имеют ферменты, в цилиндре появится вода,
вытесненная углекислым газом во время ферментации. Сухие
товарные кормовые дрожжи не должны иметь ферментативную
способность, цилиндр должен оставаться сухим во время
проведения анализа в течение суток.
Определение сырого протеина (метод Кьельдаля)
Содержание сырого протеина (общего белка) в дрожжах во
всех странах определяют по методу Кьельдаля или по одной из
многих его модификаций. Наиболее распространенная пропись
метода Кьельдаля приводится ниже. Метод основан на
определении количества азота, содержащегося в белке дрожжей.
Дрожжи обрабатывают серной кислотой при нагревании в
присутствии сернокислой меди в качестве катализатора и
сернокислого натрия как водоотнимающего вещества. Органические
вещества окисляются до углекислого газа и воды. Азот,
освобождающийся при разрушении органических веществ,
восстанавливается до аммиака, который связывается серной кислотой
в сернокислый аммоний. Реакции продолжаются более 16 ч.
Образовавшийся сернокислый аммоний разлагают щелочью,
выделяется аммиак, который отгоняют в титрованный раствор серной
кислоты (или в раствор борной кислоты), и ее избыток оттитро-
вывают раствором щелочи. Содержание сырого протеина
в дрожжах находят как произведение количества азота,
найденного в аммиаке, умноженное на эмпирический коэффициент 6,25.
Мешающие вещества. Соли аммония и органические
вещества, содержащие азот, которые не являются белком.
Аппаратура. Установки для минерализации дрожжей (см.
рис. 65), для отгонки аммиака (см. рис. 36) и для титрования.
Реактивы и их приготовление. 1. Серная кислота плотностью
1,84 г/см3. 2. Серная кислота, 0,1 н. раствор. 3. Едкий натр,
33%-ный раствор и 0,1 н. раствор. 4. Сернокислая медь
безводная ч. д. а. 5. Сернокислый натрий безводный. 6. Смешанный
индикатор. Приготовление. Смешивают один объем 0,1 %-ного
спиртового раствора метиленовой синей и два объема 0,1 %-ного
спиртового раствора метилового красного. 7. Тальк. 8.
Лакмусовые красные бумажки.
Ход анализа. Минерализация. В колбу Кьельдаля
помещают 0,5—0,6 г дрожжей (с известной влажностью),
отвешенных с точностью 0,0002 г, прибавляют 20 мл серной кислоты
15* 227
плотностью 1,84 г/см3, 0,5 г сернокислой меди и 1 г
сернокислого натрия. Колбу укрепляют в штативе в наклонном
положении над электроплиткой под тягой и включают слабое
нагревание. Горло колбы не плотно закрывают стеклянной втулкой или
воронкой. При нагревании реагирующей смеси сразу начинается
процесс окисления органических соединений, который
сопровождается бурным выделением газов, жидкость в колбе становится
черной. Постепенно содержимое колбы светлеет, выделение
газов уменьшается, в это время усиливают нагрев, опуская колбу
на плитку. Кипячение реагирующей жидкости продолжают в
течение 10—16 ч до полного сжигания углерода и превращения
азота в сернокислый аммоний. После обесцвечивания
реагирующей жидкости нагревание продолжают еще 30—40 мин, а потом
выключают нагрев и колбу с образовавшимся сплавом солей
охлаждают на воздухе.
Определение аммиака. После охлаждения в колбу
вливают 250 мл дистиллированной воды, растворяют лепешку
сплава, добавляют 10—15 г талька (для равномерности кипения
жидкости при отгонке аммиака), кусочек розовой лакмусовой
бумажки, затем быстро вливают расчетное количество 33%-ного
раствора едкого натра, колбу сразу закрывают пробкой с
трубкой, соединенной с холодильником, и включают нагрев (см.
рис. 36). Лакмусовая бумажка должна стать синей после
прибавления щелочи. Количество 33%-ного раствора едкого натра,
необходимое для нейтрализации серной кислоты и создания
щелочной среды, рассчитывают предварительно. Для
нейтрализации 20 мл серной кислоты, взятой для минерализации, надо
прибавить 100 мл 33%-ного раствора едкого натра. Конденсат из
холодильника собирают в коническую колбу вместимостью 250 мл,
в которую налито 30—50 мл 0,1 н. раствора серной кислоты и
несколько капель смешанного индикатора (или 100 мл раствора
борной кислоты см. с. 113). В начале отгонки аммиака нижний
конец холодильника погружают в приемник, а потом
приподнимают так, чтобы он касался поверхности кислоты.
Перед прекращением отгонки аммиака опускают приемник,
конец холодильника снаружи споласкивают водой из промы-
валки (вода стекает в приемник) и с помощью красной
лакмусовой бумажки проверяют наличие аммиака в последней капле
конденсата. Если бумажка не меняет цвета, то отгон аммиака
считают законченным. Собранный конденсат титруют 0,1 н.
раствором едкого натра до обесцвечивания раствора. Одна капля
избытка раствора щелочи дает зеленое окрашивание. Расход
0,1 н. раствора едкого натра на титрование эквивалентен
избытку кислоты в приемнике, который остался после реакции
отогнанного аммиака с кислотой. Если отгон аммиака
осуществляли в борную кислоту, то собранный конденсат титруют
0,1 н. раствором серной кислоты (H2SO4) в присутствии сме-
228
шанного индикатора. В этом случае количество кислоты,
пошедшее на титрование, эквивалентно количеству аммиака.
Расчет. Содержание сырого протеина в дрожжах вычисляют по формуле
(д- ff) 0,0014- 100
Х = g(\-Q?\W)
где х — концентрация сырого протеина в дрожжах, %; л — объем 0,1 н.
H2SO4, взятый на анализ, мл; б — объем 0,1 н. NaOH, израсходованный на
титрование, мл; g — количество дрожжей, взятое на анализ, г; W —
влажность анализируемых дрожжей, %; 0,0014 — количество азота, эквивалентное
1 мл 0,1 н. раствора едкого натра, мг; 6,25 — эмпирический коэффициент для
пересчета азота в белок.
Количество отогнанного аммиака определяют с точностью ±1—2% отн.
Точность определения сырого протеина зависит от наличия посторонних
азотсодержащих веществ и от того, насколько коэффициент 6,25 соответствует
составу анализируемого белка.
Предостережение. Если дрожжи выращивают на сульфитном щелоке,
полученном при варке целлюлозы на аммонийном основании, или
нейтрализацию гидролизатов и сульфитного щелока проводят аммиаком, то получают
дрожжи с высоким содержанием минеральносвязанного азота. В этих
случаях при определении сырого протеина в дрожжах надо вносить поправку
на содержание минерального азота, который определяют как описано на с. 112.
Определение белка по Бериштейну
Содержание истинного белка по Бернштейну определяют во
всех странах одинаково. Метод определения основан на
отделении истинного белка от других азотсодержащих веществ путем
осаждения его сульфатом меди в щелочной среде. При таком
разделении веществ в растворе остаются аминокислоты,
находящиеся в соке дрожжевых клеток, и азотсодержащие
вещества, которые находились в жидкости дрожжевой суспензии
перед плазмолизом или сушкой. В осадке определяют азот
методом Кьельдаля с применением селенового катализатора и,
пользуясь эмпирическим коэффициентом 6,25, вычисляют
содержание истинного белка в анализируемой пробе дрожжей. Ниже
приводится пропись метода по стандарту ЧССР.
Реактивы и их приготовление. 1. Сульфат меди, 6%-ный
раствор. 2. Едкий натр, 1,25%-ный раствор. 3. Серная кислота
плотностью 1,84 г/см3, без азота. 4. Селеновый катализатор.
Приготовление. Смешивают 250 г сульфата калия, 4 г
селена в порошке и 4 г безводного сульфата меди. 5.
Фенолфталеин, 1%-ный раствор. 6. Едкий натр, 0,25 н. раствор и 30%-ный
раствор. 7. Серная кислота, 0,25 н. раствор. 8. Ферроцианид
калия, раствор. 9. Индикатор Траши. Приготовление.
К 100 мл 0,1%-ного водного раствора метиленовой синей при
непрерывном перемешивании добавляют 15 мл 0,03%-ного
раствора метилового красного в 96%-ном этаноле. В кислой среде
такой индикатор приобретает красно-фиолетовый цвет, при pH
5,2 он бесцветен, а в щелочной среде он имеет зеленый цвет.
229
Ход анализа. Осаждение. В стакан вместимостью 400 мл
помещают 1 г сухих дрожжей или 20 мл суспензии прессованных
дрожжей B00 г/л) или 10 мл дрожжевого молока, прибавляют
100 мл воды и кипятят 2 мин. Затем в стакан вливают 25 мл
6%-ного раствора сульфата меди и при постоянном
помешивании добавляют 25 мл 1,25%-ного раствора едкого натра, при
этом белок выпадает в осадок. Раствору с осадком дают
постоять, а потом его фильтруют через двойной фильтр,
промывают горячей водой до тех пор, пока фильтрат по реакции
с ферроцианидом калия не покажет отсутствие меди. Осадок на
фильтре подсушивают на воздухе и анализируют на содержание
азота.
Минерализация. В колбу Кьельдаля помещают фильтр
с осадком, добавляют 25—30 мл серной кислоты плотностью
1,84 г/см3 и 1 г селенового катализатора. Колбу нагревают
сначала при слабом нагреве, а затем при сильном. Дальнейший ход
анализа такой же как при определении сырого протеина по
методу Кьельдаля. Содержание истинного белка в дрожжах
вычисляют по формуле, которая приведена на с. 229.
Определение усвояемого белка
Усвояемым белком называют ту часть сырого протеина,
которая растворяется в солянокислом растворе пепсина при
температуре 37—40°С. Пепсин — препарат, содержащий протео-
литический фермент, который выделяют из слизистой оболочки
свиных желудков. В 1%-ном растворе соляной кислоты при
температуре 37—40°С белок, который усваивается животными,
в присутствии пепсина переходит в раствор. Количество
усвояемого белка находят по разности между количеством сырого
протеина и количеством белка, который остается в остатке после
растворения усвояемого белка. Содержание усвояемого белка
выражают в процентах от сырого протеина и называют
коэффициентом усвояемости белка.
Реактивы и их приготовление. 1. Пепсин, отвечающий
требованиям фармакопеи. 2. Соляная кислота, 25%-ный раствор.
3. Все реактивы, применяемые для определения сырого протеина
(см. с. 227).
Ход анализа. В стакан или в колбу вместимостью 750 мл
помещают 2 г исследуемых дрожжей, прибавляют 480 мл воды, 1 г
пепсина и 10 г 25%-ной соляной кислоты, хорошо
перемешивают и ставят в термостат на 24 ч. Затем смесь фильтруют
через складчатый фильтр (фильтрат должен быть прозрачным)
и осадок на фильтре промывают дистиллированной водой.
Фильтр с осадком переносят в колбу Кьельдаля, прибавляют
20 мл серной кислоты плотностью 1,84 г/см3 проводят
минерализацию и определение аммиака, как описано на с. 229.
Параллельно в пробе дрожжей определяют содержание сырого
протеина.
230
Расчет. Сначала вычисляют содержание неусвояемого белка и всего белка
в пробе дрожжей по формуле, приведенной на с. 229. Затем вычисляют
коэффициент усвояемости белка, находящегося в пробе, по следующей формуле:
„ (А - В) 100
== л~ '
где X — коэффициент усвояемости белка, %; А-—содержание сырого
протеина в дрожжах, %; В — содержание неусвояемого белка в дрожжах, % •
Результаты двух анализов могут отличаться на более чем на 0,5% абс.
Определение кислотности
Кислотность дрожжей определяют титрованием водной
вытяжки дрожжей раствором щелочи с индикатором
фенолфталеином. Степень кислотности выражают в миллиграммах
уксусной кислоты или, как по стандарту ЧССР, в миллиграммах
едкого калия на 100 г абс. сухих дрожжей.
Ход анализа. В коническую колбу помещают 10 г сухих
дрожжей или 40 г прессованных дрожжей, приливают 200 мл
дистиллированной воды, смесь 30 мин взбалтывают, а затем
фильтруют. Дрожжевое молоко разбавляют в 2 раза,
взбалтывают и фильтруют. В колбу для титрования вливают 100 мл
фильтрата и титруют 0,1 н. раствором едкого натра или едкого
кали с фенолфталеином.
Расчет. Содержание кислот в дрожжах вычисляют по формуле
х апКЮО
где X — кислотность дрожжей, мг уксусной кислоты или мг едкого кали на
100 г абс. сухих дрожжей; а — объем 0,1 н. раствора щелочи,
израсходованный на титрование, мл; п — разведение; g—количество дрожжей, взятых на
анализ, г; влажность дрожжей, %; К=6 мг уксусной кислоты или 5,61 мг
едкого кали.
Определение гидрохлорида тиамина (витамина В,)
Определение витамина В і в дрожжах основано на получении
окрашенного соединения после облучения гидрохлорида
тиамина ультрафиолетовым светом. Сравнивая интенсивность
окраски полученного раствора с эталоном, находят концентрацию
витамина Bj в дрожжах.
Аппаратура. Установка для облучения ультрафиолетовым
светом.
Реактивы и их приготовление. 1. Соляная кислота, 0,001 н.
раствор. 2. Гидрохлорид тиамина. 3. Метиловый спирт. 4.
Едкий натр, 20%-ный раствор. 5. Ферроцианид калия, 1%-ный
раствор. 6. Бутиловый спирт. 7. Стандартный раствор тиамина.
Приготовление. В дистиллированной воде растворяют
100 мг гидрохлорида тиамина и объем в мерной колбе доводят
до 1 л. В мерную колбу вместимостью 1 л вливают 10 мл
231
приготовленного раствора, 100 мл 0,01 н. раствора соляной
кислоты и объем раствора доводят до 1 л, прибавляя
дистиллированную воду. В 1 мл приготовленного раствора содержится
0,001 мг витамина Bi.
Ход анализа. В коническую колбу помещают 100 г сухих
дрожжей и 100 мл 0,01 н. раствора соляной кислоты. Смесь
тщательно перемешивают в течение 15 мин, добавляют к ней
700 мл воды, взбалтывают еще 15 мин и фильтруют через
стеклянный фильтр. Осадок на фильтре промывают
дистиллированной водой. Фильтрат и промывные воды собирают в мерную
колбу вместимостью 1 л. Промывку осадка заканчивают, когда
объем фильтрата с промывной водой достигнет 1 л. Берут две
маленькие делительные воронки, в одну из них вливают 2 мл
стандартного раствора витамина Ві, а в другую — 2 мл
раствора, приготовленного из пробы дрожжей. В каждую воронку
вливают 2 мл метилового спирта, 1 мл 20%-ного раствора
едкого натра и 1 каплю 1%-ного раствора ферроцианида калия.
Содержимое воронок взбалтывают . в течение 2 мин, затем
в каждую воронку добавляют по 0,5 мл дистиллированной воды,
10 мл бутилового спирта, снова взбалтывают в течение 1 мин
и оставляют в покое для расслаивания. После разделения
жидкостей нижний, водный, слой выливают прочь, а верхний, спир-
товый слой из каждой воронки выливают в отдельную пробирку.
Пробирки должны быть из бесцветного стекла одинакового
диаметра. В темной комнате содержимое пробирок облучают
ультрафиолетовым светом. В присутствии гидрохлорида тиамина
образуется окрашенное соединение. Сравнивая окраску
раствора в пробирке, в которую был добавлен испытуемый
раствор, с окраской в пробирке с раствором гидрохлорида тиамина
известной концентрации (стандартный раствор), находят
содержание витамина Ві в анализируемой пробе.
Определение рибофлавина (витамина В2)
Определение витамина В2 в дрожжах основано на
получении окрашенного соединения рибофлавина. Сравнивая
интенсивность окрасок растворов, полученных при анализе пробы и
стандартного раствора, находят концентрацию рибофлавина
в пробе дрожжей.
Аппаратура. Фотоэлектроколориметр или пробирки Нес-
слера (колориметрические пробирки).
Реактивы и их приготовление. 1. Уксусная кислота
концентрированная. 2. Марганцовокислый калий, 5%-ный раствор.
3. Перекись водорода, 30%-ный раствор. 4. Раствор Видгиза.
Приготовление. В смеси из 20 мл концентрированной
серной кислоты и 100 мл воды растворяют 5 г желтой окиси ртути.
После охлаждения раствор фильтруют. 5. Стандартный раствор
рибофлавина. Приготовление. В дистиллированной воде
232
растворяют 10 мг рибофлавина и объем раствора доводят до
метки в мерной колбе вместимостью 100 мл. В мерную колбу
вместимостью 100 мл вливают 4 мл полученного раствора и
объем раствора доводят до метки, прибавляя
дистиллированную воду. В 1 мл приготовленного стандартного раствора
содержится 0,004 мг витамина В2.
Ход анализа. В коническую колбу помещают 10 г сухих
дрожжей и 50—60 мл теплой дистиллированной воды, смесь
взбалтывают и в течение 15 мин выдерживают на водяной бане
при температуре 60°С для полного извлечения витамина В2.
Затем смесь фильтруют через стеклянный фильтр, осадок на
фильтре промывают несколько раз горячей водой. Фильтрат
с промывными водами после охлаждения переносят в мерную
колбу вместимостью 100 мл и объем раствора доводят водой до
метки. К 10 мл полученного раствора прибавляют 1 мл
концентрированной уксусной кислоты и 0,5 мл 5%-ного раствора
перманганата калия. Раствор перемешивают в течение 2 мин,
добавляют к нему 0,25 мл перекиси водорода для получения
бесцветного раствора и фильтруют. В пробирку Несслера
помещают 5 мл фильтрата, добавляют 3 мл раствора Вингиза и
объем раствора доводят до метки. В присутствии рибофлавина
раствор окрашивается в желто-оранжевый цвет.
Параллельно проводят получение окрашенного соединения,
беря на анализ 1 мл стандартного раствора вместо 10 мл
пробы. Содержание рибофлавина в дрожжах определяют,
сравнивая окраски растворов, полученных при анализе пробы и
стандартного раствора. Сравнение делают визуально в
пробирках Несслера или на фотоэлектроколориметре.
Определение никотиновой кислоты (витамина РР)
Определение витамина РР в дрожжах основано на
получении окрашенного соединения при реакции никотиновой кислоты
с бромцианидом калия и n-аминометилсульфатом.
Интенсивность полученной окраски раствора определяют на
спектрофотометре и сравнивают с окраской, полученной при анализе
эталона.
Аппаратура. Спектрофотометр.
Реактивы и их приготовление. 1. Серная кислота, 0,1 н.
раствор. 2. Однозамещенный фосфат калия, 10%-ный раствор.
3. Бромцианид калия, раствор. Приготовление. В стакан
вместимостью 100 мл помещают 7,5 г бромида калия,
прибавляют 50 мл дистиллированной воды, охлаждают в ледяной
ванне и по каплям добавляют раствор цианида калия до
полного обесцвечивания раствора. Затем добавляют еще 1 мл
раствора цианида калия и объем доводят водой до 100 мл.
Приготовленный раствор хранят в склянке из темного стекла с
притертой пробкой не более 2 месяцев. 4. Метиловый раствор.
233
Приготовление. Готовят 5%-ныи раствор л-аминометио-
сульфата в 0,5 н. растворе соляной кислоты. Раствор хранят
в темноте. 5. Стандартный раствор витамина PP.
Приготовление. В мерную колбу вместимостью 500 мл вносят 0,5 г
чистой никотиновой кислоты, добавляют 50 мл 0,1 н. раствора
серной кислоты и объем полученного раствора доводят водой
до метки. В 1 мл приготовленного стандартного раствора
содержится 1 мг никотиновой кислоты. Раствор хранят при 0°С
не более 1 года.
Ход анализа. В коническую колбу помещают 10 г сухих
дрожжей и 50—60 мл горячей воды. Колбу ставят на водяную
баню и выдерживают при 60°С в течение 15 мин. Полученный
экстракт витамина РР отфильтровывают через стеклянный
фильтр, осадок на фильтре промывают несколько раз горячей
водой. После охлаждения фильтрат и промывные воды
вливают в мерную колбу вместимостью 100 мл, добавляют воду до
метки, раствор перемешивают и анализируют.
Берут две пробирки, в одну из них помещают 1 мл
стандартного раствора витамина РР, а в другую — 1 мл экстракта,
приготовленного из дрожжей. В каждую пробирку добавляют б мл
дистиллированной воды и 1 мл 10%-ного раствора однозаме-
щенного фосфата калия. Пробирки помещают в водяную баню
с температурой 70°С. После нагревания в каждую пробирку
добавляют 2 мл раствора бромцианида калия, содержимое
перемешивают вращательным движением и на 5 мин оставляют
их в водяной бане. Затем обе пробирки вынимают из водяной
бани, добавляют в каждую из них по 10 мл метилового
раствора и ставят в темное место на 1 ч. За это время растворы
приобретают золотистую окраску, интенсивность которой
измеряют спектрофотометром при светофильтре № 420. Сравнивая
интенсивность окрасок растворов в обеих пробирках, находят
концентрацию никотиновой кислоты в анализируемых дрожжах.
Определение свинца
Свинец в дрожжах определяют колориметрическим методом,
который основан на его реакции с сульфитом натрия. Перед
анализом дрожжи минерализуют. Освободившийся свинец
в щелочной среде с сульфитом натрия дает соединение бурого-
цвета. По интенсивности окраски полученного раствора находят
содержание свинца в дрожжах.
Аппаратура. Пробирки для колориметрирования.
Реактивы и их приготовление. 1. Серная кислота плотностью
1,84 г/см3. 2. Азотная кислота концентрированная. 3.
Уксуснокислый аммоний, насыщенный раствор в аммиаке. 4.
Цианистый калий, 10%-ный раствор. 5. Сульфит натрия (Na2SO3),
10%-ный раствор.
Ход анализа. В колбу Кьельдаля помещают 1—2 г
исследуемых дрожжей, добавляют смесь концентрированной серной и
234
азотной кислоты в соотношении 1 : 1 и нагревают. Когда раствор
станет почти бесцветным, добавляют 2—3 мл воды и 0,3—0,5 г
оксалата аммония, чтобы освободиться от окислов азота,
и снова нагревают 1—2 ч. После мокрого сжигания дрожжей
к остатку в колбе прибавляют порциями несколько раз раствор
ацетата аммония в аммиаке для извлечения свинца. Каждую
порцию экстракта фильтруют в пробирку Несслера, добавляют
1 мл 10%-ного раствора цианистого калия для предотвращения
влияния солей других металлов (железа, меди), объем
раствора доводят водой до 50 мл, прибавляют 2 капли 10%-ного
раствора сульфита натрия и хорошо перемешивают. В
присутствии свинца в пробе получают темный или бурый раствор
в зависимости от его количества. Сравнением окраски
полученного раствора с окраской растворов, полученных при анализе
растворов с известной концентрацией свинца, находят
содержание свинца в анализируемой пробе дрожжей.
ГЛАВА 7. ПОЛУЧЕНИЕ ФУРФУРОЛА
В основе получения фурфурола из пентозансодержащего
сырья лежат реакции гидролиза пентозанов и дегидратации
пентоз до фурфурола. При проведении фурфурольных варок
фурфурол из реакционной смеси отгоняют паром и получают
фурфурольный конденсат. Кроме того, фурфурол получают как
побочный продукт при гидролизе полисахаридов растительных
тканей в виде фурфурольного конденсата паров самоиспарения
гидролизатов в испарителях. Из фурфурольных конденсатов
на фурфурольнои колонне получают товарный фурфурол после
отделения его от примесей — воды, скипидара, летучих кислот
и других веществ.
Контроль в фурфурольном цехе заключается в выполнении
следующих анализов: определении потенциального содержания
фурфурола в перерабатываемом сырье, определении
концентрации фурфурола, скипидара и кислот в фурфурольном
конденсате, определении качества и количества полученного
товарного фурфурола по показателям ГОСТа и определении потерь
фурфурола.
Концентрацию фурфурола в его водных растворах и в
готовом фурфуроле определяют стандартным бромид-броматным
методом. Методы газожидкостной хроматографии, спектрофото-
метрические и полярографические пока не нашли
распространения [82].
Определение фурфурола экспрессным методом
Для быстрого определения фурфурола в укрепленном
конденсате пользуются методом, основанным на расслаивании не-
смешивающихся жидкостей, которые имеют различную плотность.
235
Фурфурольний конденсат при стоянии разделяется на два
слоя. Замеряют объем слоя жидкости, содержащей фурфурол,
и по специальным таблицам находят концентрацию фурфурола
в пробе конденсата.
Мешающие вещества. Вещества, которые не растворяются
в воде и обладают плотностью близкой к плотности фурфурола.
Аппаратура. Градуированные цилиндры вместимостью
250 мл.
Ход анализа. Пробу горячего укрепленного конденсата
охлаждают в колбе до 20°С. В цилиндр вливают 250 мл
охлажденного конденсата и ждут 10—15 мин. После расслаивания
жидкостей замеряют объем фурфурольного слоя и по табл. 11
находят содержание фурфурола в пробе.
ТАБЛИЦА 11
Содержание фурфурола в водно-фурфурольной
фракции в зависимости от объема фурфурольного
слоя, образующегося при стоянии 250 мл фракции
Объем
фурфурольного
слоя, мм
5
10
15
20
25
30
35
40
45
50
Концентрация
фурфурола, %
(по массе)
Ю.2
11.9
13.6
15,3
17.0
18.7
20,4
22,1
23,8
25.5
Объем
фурфурольного
слоя, мл
55
60
65
70
75
80
85
90
95
100
Концентрация
фурфурола, %
(по массе)
27,2
28,9
30.6
32,3
34,0
35,7
37,4
39.1
40,8
42.5
Концентрацию фурфурола определяют с точностью ±10% отн.
Определение кислотности
Для определения кислотности фурфурольного конденсата
пользуются методом, основанным на титровании кислот
раствором едкого натра в присутствии фенолфталеина. Кислотность
выражают в градусах кислотности.
Аппаратура. Установка для титрования.
Реактивы и их приготовление. 1. Едкий натр, 0,1 н. раствор.
2. Фенолфталеин, 0,1%-ный спиртовый раствор.
Ход анализа. Фурфурольний конденсат фильтруют. В
коническую колбу вместимостью 250 мл вливают 50 мл фильтрата,
добавляют 2—3 капли раствора фенолфталеина и титруют 0,1 н.
раствором едкого натра до появления бледно-розовой окраски
раствора.
236
Расчет. Содержание кислот в анализируемой пробе вычисляют по
формуле
~ g
где X — кислотность фурфурола, К° (градус кислотности); а — объем 0,1 н.
раствора едкого иатра, пошедший на титрование, мл; g — объем конденсата,
взятый иа анализ, мл; 1 К0 соответствует 1 мл 0,1 н. раствора щелочи,
которая расходуется иа титрование 100 мл пробы.
Воспроизводимость в среднем из трех титрований +0,05 мл 0,1 н.
раствора едкого иатра.
Определение скипидара экспрессным методом
Для определения скипидара в фурфуроле-сырце пользуются
методом основанным на измерении объема скипидара, который
отделяется от фурфурола при стоянии.
Мешающие вещества. Вещества, имеющие плотность,
близкую к плотности скипидара.
Аппаратура. Мерная колба вместимостью 500 мл с узким
горлом, наверху которого имеется градуировка от 490 до 500 мл
с интервалом 0,1 мл.
Ход анализа. В дистиллированной воде при 60—70°С
растворяют 10 мл анализируемого фурфурола-сырца. После
охлаждения раствор количественно переносят в мерную колбу
вместимостью 500 мл, на узком горле которой имеется градуировка.
Объем раствора доводят водой до 500 мл, перемешивают и
оставляют стоять до полного расслаивания жидкости. Через
30 мин измеряют объем верхнего скипидарного слоя.
Расчет. Содержание скипидара в пробе фурфурола-сырца вычисляют по
формуле
а0.89 . 100
где X — концентрация скипидара в фурфуроле-сырце, г/100 г; а — объем
скипидарного слоя, мл; g — объем фурфурола-сырца, взятый на анализ, мл;
d — плотность фурфурола-сырца, взятого иа анализ, г/см3; 0,89 — средняя
плотность скипидара, г/см3.
Воспроизводимость в средием из трех замеров объема скипидара ±0,05 мл.
Определение фурфурола бромид-броматным методом
Для определения фурфурола в товарном фурфуроле и
в его водных растворах предназначен метод, основанный на
реакции бромирования фурфурола до получения дибромфурфурола,
хотя реакция бромирования фурфурола продолжается до
образования тетрабромфурфурола (см. с. 43). Бромирование
фурфурола прерывают через 4 мин, добавляя йодистый калий,
который сразу вступает в реакцию со свободным бромом.
Расход брома на бромирование фурфурола в течение 4 мин
составляет 50% от расхода брома на полное бромирование фурфурола
до тетрабромфурфурола, поэтому принято считать, что за 4 мин
237
бромирования фурфурол превращается в дибромфурфурол. При
лнализе необходимо строго соблюдать время бромирования,
так как скорость бромирования в течение пятой минуты бро-
мнрования почти не уменьшается. Иод, выделившийся в
результате реакции брома с йодистым калием, оттитровывают
раствором гипосульфита натрия, его количество эквивалентно не-
прореагировавшему брому. По разности между количеством
брома, найденного в холостом опыте, и при анализе пробы находят
количество брома, пошедшего на бромирование фурфурола,
и вычисляют концентрацию фурфурола в анализируемой пробе.
Мешающие вещества. Летучие бромируемые вещества.
Аппаратура. Установка для титрования. Секундомер.
Конические колбы вместимостью 500 мл с притертыми пробками.
Реактивы и их приготовление. 1. Соляная кислота, 0,55%-ный
раствор. 2. Молибденовокислый аммоний, 2,5%-ный раствор.
3. Бромид-бромат калия, 0,05н. раствор. Приготовление.
В дистиллированной воде растворяют 1,392 г бромата калия
(КВгОз) и 10 г бромида калия (КВг). Объем раствора доводят
до 1 л в мерной колбе. Раствор хранят в склянке из темного
стекла с притертой пробкой. 4. Гипосульфит натрия 0,05 н.
раствор, приготовленный из фиксанала растворением в
дистиллированной воде, из которой углекислый газ удален кипячением.
5. Йодистый калий, 10%-ный раствор. 6. Крахмал, 0,5%-ный
раствор.
Подготовка проб. 1. Пробу фурфурола-сырца и товарного
фурфурола разбавляют в 500 раз. Навеску пробы 1—1,5 г,
взятую с точностью 0,0002 г, растворяют в воде и объем
раствора в мерной колбе доводят до 500 мл. Полученный раствор
фильтруют через бумажный фильтр. На анализ берут 5 мл
фильтрата. 2. Пробу лютера фильтруют через бумажный
фильтр. На анализ берут 20 мл фильтрата. 3. Пробу
фурфурольного конденсата фильтруют через бумажный фильтр.
На анализ берут 5 мл фильтрата.
Ход анализа. Анализируемую пробу в количестве,
указанном выше, помещают в коническую колбу вместимостью 500 мл
с притертой пробкой, прибавляют к ней 300 мл 0,55%-ного
раствора соляной кислоты, 10 мл 2,5%-ного раствора молибдено-
вокислого аммония и 25 мл 0,05 н. раствора бромид-бромата
калия. Колбу закрывают пробкой, ставят на лист белой
бумаги и наблюдают за появлением в реагирующей жидкости
свободного брома, который окрашивает раствор в
бледно-желтый цвет. С момента появления желтой окраски раствора ждут
4 мин (секундомер), а затем к реагирующей жидкости
приливают 10 мл раствора йодистого калия, колбу снова закрывают
и ждут еще 4 мин. За это время проходит обменная реакция
между свободным бромом и йодистым калием. Выделившийся
свободный йод окрашивает жидкость в темно-желтый цвет.
Иод оттитровывают раствором гипосульфита натрия до соло-
238
менно-желтой окраски раствора, прибавляют 0,5 мл раствора
крахмала и продолжают титровать до исчезновения синей
окраски. Параллельно проводят холостой опыт, в котором вместо
пробы берут такое же количество воды.
Расчет. Содержание фурфурола в фурфуроле-сырце и в товарном
фурфуроле вычисляют по формуле
(я — б) 0.0024 -500-100
Л. = ¦
gg\
где X— концентрация фурфурола в пробе, %; а — объем 0,05 н. ЫагБгОз,
пошедший на титрование в холостом опыте, мл; б — объем 0,05 и. ЫагЭгОз,
пошедший на титрование пробы, мл; g — навеска пробы фурфурола, взятая
для разведения, г; gi — объем разбавленной пробы, взятый на анализ, мл;
0,0024 — количество фурфурола, эквивалентное 1 мл 0,05 н. раствора
гипосульфита натрия, г; 500— объем разбавленной пробы, мл.
Содержание фурфурола в конденсате и в лютере вычисляют по формуле
„ (а-б) 0.0024 • 100
л =¦ •.
Обозначения те же.
Воспроизводимость при титровании в среднем из трех определений
составляет ±0,05 мл 0,05 и. раствора гипосульфита.
Предостережение. 1. При анализе фурфурола надо выдерживать время
бромирования 4 мин с точностью до 1 с. Увеличение времени бромирования
на 5 с приводит к завышению результата анализа на 1%. По этой причине
иногда в товарном фурфуроле находят фурфурола более 100%. 2. Не следует
делать одновременно два анализа. 3. Молибдеиовокислый аммоний является
катализатором, ускоряющим реакцию бромирования фурфурола, увеличение
его количества в реагирующей жидкости приводит к получению завышенных
результатов. Надо иметь в виду, что 2,5%-ный раствор молибденовокислого
аммония является насыщенным раствором при 20 С. Понижение температуры
в лаборатории может привести к выпадению кристаллов из раствора,
которые могут быть захвачены при всасывании раствора пипеткой, и таким
путем иа анализ будет взято катализатора больше, чем предусмотрено прописью
метода.
ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ НА ФУРФУРОЛ И МЕТОДЫ ЕГО ИСПЫТАНИЯ
ПО СТАНДАРТАМ РАЗНЫХ СТРАН
Технические требования на фурфурол
Показатели для определения качества фурфурола,
применяемые в некоторых странах [83, 84, 85], приведены в табл. 12.
Главными нормируемыми показателями являются плотность,
температура перегонки и кислотность. Содержание фурфурола
в товарном продукте определяют не во всех странах. По
румынскому стандарту не требуется определять содержание
фурфурола, но обязательно определение воды и золы, которые не
определяют ни у нас, ни в ГДР. Содержание веществ,
нерастворимых в воде, по румынскому стандарту не определяют.
239
ТАБЛИЦА 12
Показатели качества
Внешний вид
Плотность при 20иС,
г/см3, в пределах
Содержание фурфурола,
%, не менее
Фракционный состав:
температура начала
перегонки, °С, не
выше
температура
окончания перегонки, "С, не
выше
перегоняется до
температуры окончания
перегонки, %, не
менее
остаток, %. не более
Технические требования на фурфурол пс
СССР
ГОСТ 10437 - 71
I сорт
II сорт
Жидкость, не
содержащая взвешенных частиц,
от желтого до бурого
цвета, темнеющая при
хранении, с резким
запахом
1,159-1,161
99,5
149
165
98,5
1.0
1,150-1,161
94,0
—
165
97
3.0
> стандартам разных стран
ГДР
ТГЛ 15233 - 63
Для анализа
Чистейший
Чистый
Прозрачная бесцветная жидкость,
быстро приобретающая желтую,
потом коричневую окраску под
действием света и воздуха
1,158-1,160
99,0
—
—
—
—
98,5
—
—
—
1,156-1,162
98,0
—
—
—
—
Румыния
STAS
Тип R
Жидкость
от желтого
до
коричневого цвета
1,152-1,157
—
—
173
98,5
1.2
779 - 54
Тип N
Жидкость
темно-коричневого цвета
1,157-1,161
—
—
176
97
2,4
Пределы кипения, °С, не
менее 95% объема
Показатель преломления
Содержание органиче-
ских кислот в пересчете
на уксусную кислоту,
%, не более
Содержание сульфатов
Содержание примесей,
нерастворимых в воде
Выкипаемость до
температуры 158°С, %, не
более
Остаток после
прокаливания в виде сульфата,
°/о, не более
Фракционный состав:
до 148°С перегоняется,
% объемн., не более
до 155°С перегоняется,
% объемн., не более
до 165°С перегоняется,
% объемн., не менее
Содержание воды, %,
не более
Зола, %, не более
1,525-1,526
0,05
Отсутствуют
То же
1,517-1,524
0,10
Отсутствуют
То же
18
160-163
0,10
159,5-163.5
0.20
Проходит испытание
0.005
0.01
159-169
0,07
0.7
2.5
5
96
0.7
0.02
Примечание. Прочерк означает, что этого показателя в стандарте нет.
Для сравнения способов определения качества фурфурола,
применяемых у нас и в других странах, ниже приведены
методы испытания фурфурола по стандартам Румынии и ГДР.
Определение фурфурола с солянокислым гидроксиламином
По стандарту ГДР используется метод, основанный на
реакции фурфурола с солянокислым гидроксиламином,
протекающей по уравнению
С4Н3О • CHO+H2NOH • HCl-vC4H3O • CH = NOH-f HC1 +
2О.
Выделившуюся соляную кислоту оттитровывают раствором
едкого натра. Содержание фурфурола находят по количеству
едкого натра, израсходованного на титрование, которое
эквивалентно количеству фурфурола в анализируемой пробе.
Аппаратура. Установка для титрования.
Реактивы. 1. Солянокислый гидроксиламин, 10%-ный
раствор. 2. Едкий натр, 1 н. раствор. 3. Метиловый оранжевый,
0,1 %-ный спиртовый раствор.
Ход анализа. Около 1 г исследуемого фурфурола и 15 мл
дистиллированной воды вливают в коническую колбу и
закрывают притертой пробкой. Раствор перемешивают, прибавляют
к нему 25 мл раствора солянокислого гидроксиламина,
закрывают пробкой, хорошо перемешивают и через 5 мин титруют
1 н. раствором едкого натра, добавив 0,2 мл раствора
метилового оранжевого, до оранжево-желтой окраски раствора.
Параллельно проводят холостой опыт, в котором не прибавляют
фурфурол.
Расчет. Содержание фурфурола в пробе вычисляют по формуле
(а — ff) 0,09609 • 100
х- і '
где X—концентрация фурфурола в исследуемой пробе, %; а — объем 1 н.
NaOH, израсходованный на титрование при анализе пробы, мл; б—объем
1 н. NaOH, пошедший иа титрование в холостом опыте, мл; g — количество
фурфурола, взятое на анализ, г; 0,09609 — количество фурфурола,
эквивалентное 1 мл 1 н. раствора едкого натра, г. Воспроизводимость в среднем из трех
определений составляет ±0,05 мл 1 н. раствора едкого натра.
Проба на содержание веществ, нерастворимых в воде
Метод используется в стандарте ГДР.
Ход анализа. В 100 мл дистиллированной воды растворяют
5 мл фурфурола. Раствор переливают в колориметрическую
пробирку и визуально определяют его прозрачность. При ана-
242
лизе фурфурола марок «Для анализа» и «Чистейший» растворы
должны быть прозрачными, а при анализе фурфурола марки
«Чистый» допускается легкая муть.
Определение фракционного состава фурфурола
В разных странах этот показатель качества фурфурола
определяют по-разному. В СССР нормируют температуру
начала и конца разгонки, количество фракции, отгоняемой до
165°С, и количество остатка. В ГДР нормируют температуру,
при которой перегоняется 95% объема фурфурола, взятого на
анализ. В Румынии определяют количество трех фракций,
отгоняемых в разных температурных интервалах, и количество
остатка.
Определение остатка после прокаливания в виде сульфата
Метод используется в стандартах ГДР и Румынии.
Ход анализа. Около 50 г исследуемого фурфурола
выпаривают досуха в фарфоровом тигле, добавляют 1 мл серной
кислоты плотностью 1,84 г/см3, ставят его на песчаную баню,
нагревают до полного удаления газообразных продуктов, а затем
переносят в муфельную печь и прокаливают при 800°С.
Периодически тигель с осадком вынимают из муфельной печи, охлаждают
и взвешивают. Прокаливание считают законченным, когда
разница между двумя последующими взвешиваниями будет
составлять не более 0,0003 г.
Содержание сухого остатка в фурфуроле в виде сульфатов
вычисляют в процентах.
ГЛАВА 8. ПОЛУЧЕНИЕ ДВУОКИСИ УГЛЕРОДА
Двуокись углерода образуется при сбраживании гексозных
Сахаров дрожжами на этиловый спирт. Углекислый газ,
выделяющийся при спиртовом брожении, отбирают из бродильных
чанов и направляют в углекислотный цех для сгущения и
очистки. На различных стадиях сгущения и очистки газа от
примесей его проверяют на содержание двуокиси углерода,
воды, масел и окисляемых веществ — спиртов, эфиров,
альдегидов, органических кислот и минеральных кислот. Сжиженную
двуокись углерода гидролизные заводы выпускают трех марок:
сварочная I и II сорта, пищевая и сухой лед [87, 88]. Для
сравнения качества товарной двуокиси углерода, которую получают
на гидролизных заводах в нашей стране, с качественными
показателями на сжиженную двуокись углерода, выпускаемую
в других странах, в табл. 13 приведены технические требования
на сжиженную двуокись углерода в стальных баллонах по
стандартам ГДР и Японии [86—89].
16* 243
ТАБЛИЦА 13
Технические требования на сжиженную двуокись углерода по стандартам разных стран
Показатели качества
Цвет
Посторонние запах
и вкус
Содержание двуокиси
углерода (СО2), % объ-
емн., не менее
Содержание воды:
мг/100л газа, не более
% по массе, не более
Содержание
минеральных масел
Содержание сернистой
и азотистой кислот и
органических соедине-
СССР
ГОСТ 8050-64
сварочная
I сорт
—
Отсутствуют
99,5
—
Отсутствуют
То же
11 сорт
—
Отсутствуют
99,0
—
Отсутствуют
То же
в
пищевая
—
Отсутствуют
99,8
—
0,10
Отсутствуют
Отсутс
ГОСТ 12162-66
сухой лед
пищевой
технический
Белый с желтым
оттенком
Запах отсутствует,
вкус кислый
99.96
—
—
Отсутствуют
твуют
99.96
—
—
Следы
ГДР
тгл
2968-56
—
—
98
300
—
—
—
Япония
J. 1
№ 1
—
Отсутствуют
99
—
—
—
—
S. К. 1106-1931
—
Отсутствуют
99,5
—
0,05
—
—
№ 3
—
—
99,5
—
0.005
—
—
дов и органических
кислот)
Расход перманганата,
мг/100 л газа, не более
Содержание водяных
паров в углекислом
газе:
при нормальных
условиях (давление 760 мм
рт. ст., температура
20°С), т/и3, не более
или по точке росы, °С,
не выше
Содержание аммиака и
моноэтаноламина *
Содержание окиси
углерода (СО), % объемн.*,
не более
Содержание
сероводорода *
Содержание соляной
кислоты *
0,06
Минус 42
Отсутствуют
То же
0,515
Минус 24
Отсутствуют
То же
Отсутствуют
То же
* По этим показателям двуокись углерода, выпускаемую гидролизными заводами, не анализируют,
Примечание. Прочерк означает, что этого показателя в стандарте нет.
Определение двуокиси углерода
Для определения концентрации двуокиси углерода в газе,
•отбираемом из бродильных чанов, на различных стадиях его
сгущения и в готовой продукции предназначен метод,
основанный на реакции углекислого газа с едким кали
К2СО3.
Измеренный объем газа приводят в соприкосновение с
раствором едкого кали. После поглощения углекислого газа
щелочью измеряют объем оставшихся газов или объем
поглощенного СО2.
Мешающие вещества. Кислоты минеральные и органические,
вода и другие вещества, реагирующие со щелочью.
Аппаратура. Прибор для определения двуокиси углерода.
Прибор для определения двуокиси углерода (рис. 66)
представляет собой пипетку емкостью 100 мл,
которая изогнута под углом 90°. Оба
конца пипетки сужены и закрываются
притертыми кранами 1 и 2. На одном
конце пипетки имеется градуировка от 92
до 100 мл с ценой деления 0,1 мл. Ко
второму концу пипетки припаян сосуд с
воронкой вместимостью 125 мл, на котором
Рис. 66. Прибор для оп- имеется отметка 105 мл. Прибор укреп-
ределения двуокиси угле- лен на деревянной подставке.
рода Реактивы. Едкое кали, 30%-ный
раствор.
Ход анализа. Прибор ставят в вертикальное положение и
градуированный конец пипетки соединяют резиновой трубкой
с пробным краном на газопроводе или с редуктором баллона,
в котором находится анализируемый газ. Открывают сначала
оба крана пипетки, а затем осторожно кран на газопроводе или
на редукторе. Газ пропускают через прибор в течение 3—5 мин.
За это время воздух из пипетки будет полностью вытеснен и она
заполнится анализируемым газом. Прибор отключают,
закрывают кран 2, кран 1 и затем кран от газопровода или баллона.
Прибор отсоединяют от каучуковой трубки и устанавливают
на столе в горизонтальном положении. Для уравнивания
давления газа в пипетке с атмосферным давлением несколько раз
быстро открывают и закрывают кран 2. В приемный сосуд
наливают до метки 105 мл 30%-ного раствора едкого кали и
осторожно открывают кран 2 так, чтобы газ не прорывался из
пипетки в раствор. По мере поглощения углекислого газа
раствор едкого кали будет заполнять пипетку. Когда прекратится
поступление раствора едкого кали в пипетку, кран 2 закрывают,
прибор слегка встряхивают, чтобы добиться полного
поглощения углекислого газа, поворачивают его в вертикальное положе-
246
ниє так, чтобы кран 1 находился наверху, и по делениям на
конце пипетки определяют объем поглощенного углекислого
газа, который равен процентному содержанию двуокиси
углерода в исследуемом газе. В отсутствии мешающих веществ
содержание двуокиси углерода в газе определяют с точностью
±0,1% отн.
Определение воды по точке росы
Для определения воды в сжиженной двуокиси углерода
предназначен метод, основанный на определении температуры
конденсации паров воды, находящейся в исследуемом газе. Чем
ниже точка росы, тем меньше содержание воды в газе. Норми^
руется точка росы, которая соответствует
определенной допустимой концентрации воды
в газе.
Аппаратура. Гигрометр (рис. 67).
Конденсационный гигрометр
состоит из двух сосудов, вставленных один
в другой. Внешний, стеклянный, сосуд 3
имеет верхний и нижний тубусы, через которые
пропускают углекислый газ. Внутренний,
медный сосуд 4 имеет зеркальную внешнюю
поверхность, он герметически вставлен в
стеклянный сосуд.
Ход анализа. Нижний тубус внешнего
сосуда (см. рис. 67) соединяют резиновой
трубкой со штуцером вентиля редуктора на баллоне с исследуемым
газом, открывают вентиль так, чтобы через сосуд проходил
слабый ток газа. Во внутренний сосуд вливают 30 мл этилового
спирта и небольшими порциями добавляют сухой лед. Смесь 2
перемешивают термометром / и наблюдают за зеркальной
поверхностью сосуда. При появлении первых признаков
помутнения поверхности, что свидетельствует о конденсации паров воды,,
замечают температуру смеси во внутреннем сосуде. Эта
температура есть точка росы.
Определение воды
. Для определения воды в сжиженной двуокиси углерода
пользуются методом, основанным на поглощении воды
пятиокисью фосфора ( РгО5).
Измеренное количество анализируемого газа пропускают
через слой Р2О5 в трубке. Взвешивая трубку до и после
пропускания газа, находят концентрацию воды в нем.
Мешающие вещества. Вещества, реагирующие с пятиокисью
фосфора при комнатной температуре.
Аппаратура. Адсорбционная трубка, заполненная пятиокисью
фосфора.
Рис. 67. Гигрометр
247
Ход анализа. Отбирают 100 л газа из трубопровода или из
¦баллона, поставленного вертикально. Измеряют его объем,
давление и температуру. Затем газ пропускают через
адсорбционную трубку со скоростью 1 л/мин. Адсорбционную трубку
¦взвешивают до и после пропускания газа.
Расчет. Концентрацию воды в анализируемой пробе газа вычисляют по
¦формуле
где X — содержание воды в газе, г/л; а, б — масса трубки до и после
пропускания газа, г; V — объем пропущенного газа, приведенный к нормальным
условиям 101,324 кПа G60 мм рт. ст.) при 15°С, л.
Содержание воды в газе определяют с точностью ±1—2%.
Определение окисляемых веществ
Для определения окисляемых веществ в сжиженной
двуокиси углерода пользуются методом, основанным на окислении
перманганатом калия в нейтральной среде при комнатной
температуре органических соединений — спиртов, эфиров,
альдегидов и кислот, а также минеральных
кислот— сернистой и азотистой. Количество пер-
манганата, израсходованное на окисление,
находят титрованием щавелевой кислотой.
Аппаратура. Поглотитель Полежаева
(рис. 68).
Реактивы. 1. Перманганат калия, 0,01 н.
раствор. 2. Щавелевая кислота, 0,01 н.
раствор. 3. Серная кислота разбавленная 1 :3.
Ход анализа. В поглотитель Полежаева
наливают 20 мл 0,01 н. раствора пермангана-
та калия и 20 мл дистиллированной воды.
Баллон с исследуемым газом ставят в
вертикальное положение, редуктор соединяют
резиновой трубкой с поглотителем, открывают кран
и в течение 1 ч пропускают 25 л газа через
раствор перманганата калия в поглотителе.
После пропускания газа жидкость из
поглотителя количественно переносят в коническую
колбу вместимостью 250 мл, прибавляют из
бюретки 20 мл 0,01 н. раствора
щавелевой кислоты. Бесцветный раствор,
содержащий избыток щавелевой кислоты, равный израсходованному
количеству перманганата калия, оттитровывают 0,01 н. раствором
перманганата калия до устойчивой розовой окраски
раствора.
Рис. 68.
Поглотитель Полежаева:
1 — шлиф
.248
Расчет. Количество перманганате калия, которое пошло на окисление-
примесей, присутствующих в 100 л пробы двуокиси углерода, вычисляют по»
формуле
Х=* 0,316 -4V.
где X — количество перманганати калия, расходуемое на окисление примесей,,
содержащихся в 100 л анализируемого газа, мг; V — объем 0,01 н. КМпО4,-
израсходованный на окисление примесей в 25 л пробы газа, мл; 0,316 —
грамм-эквивалент марганцовокислого калия (КМпО4).
Холостым опытом устанавливают поправку на окисляемость воды н серной
кислоты, которые употребляют в анализе, и при расчете вычитают эту
поправку.
Проба иа содержание минеральных масел
Метод предназначен для качественного определения
наличия минеральных масел в сжиженной двуокиси углерода.
Ход анализа. Баллон с исследуемым газом кладут в
горизонтальное положение, на выпускной штуцер вентиля надевают
мешок из плотной ткани, открывают вентиль и выпускают га»
из баллона большой струей. При быстром расширении
углекислый газ превращается в лед. Кусочек полученного сухого льда
10—20 г кладут на чистую фильтровальную бумагу. Двуокись
углерода быстро испарится. Если в ней были минеральные
масла, то на бумаге будет жирное пятно.
ГЛАВА 9. ОЧИСТКА СТОЧНЫХ ВОД
В связи с быстрым развитием всех отраслей
промышленности значительно увеличивается количество очищенной воды,,
направляемой для водоснабжения населения, поэтому к ее
качеству требования повышаются с каждым годом [90, 91].
Очистку сточных вод осуществляют на городских станциях
и на очистных сооружениях, построенных для одного или
нескольких заводов. Задачами контроля при очистке стоков,
является: 1) установление, чем и в какой степени загрязнены-
поступающие сточные воды; 2) проверка эффективности работы
каждого агрегата очистных сооружений и обеспечение
заданных условий их работы; 3) проверка соответствия качества
очищенной воды установленным санитарным нормам для данного
водоема. Точки отбора проб и методы анализа стоков на
различных стадиях очистки выбирают в зависимости от их
загрязненности и способа очистки [5].
Санитарные условия спуска сточиых вод в водоемы
Требования, предъявляемые к составу воды, которую
спускают в водоемы, зависят от назначения воды водоема, от
размера водоема и его загрязненности, т. е. от категории водоема^
Основными показателями загрязненности считают следующие:
взвешенные и плавающие вещества, температура, pH, запах,
окраска, растворенный кислород, минеральный состав,
возбудители заболеваний, ядовитые вещества и радиоактивные вещества.
Санитарные условия спуска сточных вод в водоемы описаны
в книге [90] и документах Госсанинспекции.
Определение оседающих веществ
Для определения количества веществ, выпадающих в
осадок при различном времени отстоя пользуются методом,
основанным на измерении объема осадка, который образуется при
отстаивании воды в течение 1—2 ч.
Аппаратура. Отстойники Лысенко вместимостью 500—1000 мл,
цена деления в отростке 0,1 мл (см. рис. 49).
Ход анализа. Сразу после отбора пробу тщательно
перемешивают и наливают в четыре отстойника. Через каждые 10—
15 мин в течение 1—3 ч визуально определяют объем осадка,
который за это время осел в отростке отстойника. Чтобы
осадок не задерживался в конусной части отстойника, его
периодически вращают вокруг оси (не взбалтывать!).
Расчет. Концентрацию взвешенных веществ X, способных оседать на дно
¦отстойника за каждые 10—15 мин отстоя, выражают в миллилитрах на 1 л
(мл/л) и вычисляют по формуле'
2? _ а\ +0.2 ¦+• . . . ап
gn
au a,2, ..., an — объем осадков в 1, 2,..., и-м отстойниках, мл; gn — объем
сточной воды, налитой в п отстойников, мл.
На основании полученных данных строят кривую зависимости количества
осадка от времени отстоя, которую используют для расчета режима работы
отстойника. Объем оседающих веществ определяют с точностью ± 10—25%
отн. в зависимости от скорости уплотнения осадка.
Определение взвешенных веществ
Для определения концентрации твердых взвешенных
органических и неорганических веществ в пробе воды предназначен
метод, основанный на выделении взвешенных веществ путем
•фильтрования и определении их количества взвешиванием
после высушивания до полного удаления влаги при 105°С.
Содержание минеральных веществ в осадке определяют после его
озоления, а количество органических веществ в осадке находят
по разности между количествами сухих веществ и золы.
Жидкие взвешенные вещества не определяются.
Мешающие вещества. Взвешенные вещества, которые
разлагаются или улетучиваются при температуре сушки.
Аммонийные соли и другие вещества, разлагающиеся при прокаливании.
Аппаратура. Сушильный шкаф. Муфельная печь. Бюксы.
Тигли.
250
Ход анализа. 1 л анализируемой воды фильтруют на
воронке под вакуумом через плотный фильтр, который
предварительно высушивают (не сгибая!) до полного удаления влаги
при 105°С. Фильтр с осадком складывают, переносят в сухой
взвешенный бюкс и сушат в сушильном шкафу при 105°С до
полного удаления влаги. Высушивание считают законченным,
когда расхождение между двумя последующими
взвешиваниями не превышает 0,0003 г. Затем фильтр с осадком
перекладывают в тигель, сжигают его на электроплитке под тягой и
для прокаливания ставят в муфельную печь при 600°С. Во время
прокаливания наблюдают за изменением цвета осадка.
При большом количестве органических веществ осадок
сначала становится черным, иногда появляется запах жженных
перьев, а при незначительном количестве органических
веществ осадок сначала буреет. Если органических веществ
в осадке нет, то он сразу становится белым. Выделение бурых
паров в первые минуты прокаливания осадка наблюдается при
высоком содержании солей азотной кислоты. После озоления
тигель вынимают из печи, охлаждают в эксикаторе,
взвешивают и снова ставят в печь. Через 1 ч прокаливания
взвешивание повторяют. Прокаливание продолжают до тех пор пока
разница между двумя последующими взвешиваниями будет
составлять не более 0,0003 г.
Расчет. Количество миллиграммов осадка после сушки при 105°С равно-
содержанию абсолютно сухих взвешенных веществ в 1 л анализируемой пробы.
Состав взвешенных веществ характеризуют процентным содержанием
органических и неорганических веществ, которые вычислиют по формулам:.
где Хк—содержание минеральных веществ в абс. сухих взвешенных
веществах, г/100 г; а — количество золы, г; с — количество абс. сухих веществ, г;
Хо ¦— содержание органических веществ в абс. сухих взвешенных веществах,.
г/100 г.
Концентрацию взвешенных веществ определяют с точностью ±0,5 мг/л..
Предостережение. Сточные воды гидролизных заводов и ЦБК содержат
во взвешенном состоянии вязкие смолоподобные вещества, которые при
фильтрации сорбируются на фильтровальной бумаге, а во время высушивания при.
105°С частично обугливаются.
Определение сухого и прокаленного остатка
Для определения концентрации растворенных твердых
органических и неорганических веществ в анализируемой воде
предназначен метод, состоящий из двух анализов: 1) определения-
суммы органических и минеральных веществ (сухого остатка)
выпариванием отмеренного количества пробы на водяной бане-
с последующим высушиванием остатка при 105°С до полного-
удаления влаги; 2) определения минеральных веществ
(прокаленного остатка) прокаливанием сухого остатка при 600°С.
251:
По разности между сухим и прокаленным остатками находят
содержание органических веществ в пробе.
Мешающие вещества. Твердые вещества, которые
разлагаются или улетучиваются при 105°С. Минеральные вещества,
которые разлагаются при 600°С.
Аппаратура. Водяная баня. Сушильный шкаф. Муфельная
печь.
Ход анализа. Цилиндром отмеривают 100 мл
отфильтрованной исследуемой воды и часть ее наливают в тигель
вместимостью 50 мл, заполняя его на 3U объема. Предварительно тигель
взвешивают после высушивания при 105°С и после
прокаливания при 600°С. Тигель с пробой ставят на кипящую водяную
баню. По мере испарения анализируемой воды в тигель
добавляют воду из цилиндра. После выпаривания 100 мл пробы
тигель тщательно обмывают снаружи дистиллированной водой,
вытирают фильтровальной бумагой и ставят в сушильный шкаф
для высушивания осадка при 105°С. Через 3 ч тигель с осадком
вынимают из сушильного шкафа, охлаждают в эксикаторе,
взвешивают и снова ставят в сушильный шкаф. Высушивание
считают законченным, если расхождения между двумя
последующими взвешиваниями не более 0,0003 г. Тигель с сухим
остатком ставят в тигельную печь и прокаливают при 600°С. Когда
зола станет белой, тигель вынимают из печи, охлаждают
в эксикаторе, взвешивают и снова ставят в печь. Через каждый
час прокаливания тигель вынимают, охлаждают и взвешивают.
Прокаливание заканчивают, когда разница между двумя
последующими взвешиваниями будет не более 0,0003 г.
Расчет. Содержание сухого остатка и прокаленного остатка в
анализируемой воде выражают в миллиграммах на 1 л (мг/л) и вычисляют по
формуле X—aW, где а — количество осадка в тигле после высушивания при
105°С или после прокаливания при 600°С, мг.
Сухой и прокаленный остатки определяют с точностью ±0,5 мг/л.
Предостережения. 1. Часть органических веществ, растворенных в
сточной воде гидролизных заводов и ЦБК, при 105°С обугливается и
улетучивается. 2. Аммонийные соли в состав золы не входят, потому что при
прокаливании они разлагаются по следующему уравнению:
(NH4JSO4 — 2NH3 + SO3 + H2O.
Образующиеся аммиак, серный ангидрид н вода улетучиваются.
Определение щелочности
Для определения общей щелочности и щелочности по
фенолфталеину, или щелочного числа, предназначен метод,
основанный на титровании анализируемой воды раствором серной
или соляной кислоты с различными индикаторами. В воде,
имеющей pH выше 8, определяют оба показателя, а в воде,
имеющей pH ниже 8, определяют только общую щелочность.
252
При нейтрализации до pH 3,2 по метиловому оранжевому
количество кислоты, пошедшей на титрование, называют общей
щелочностью воды, оно соответствует количеству всех слабых
кислот (угольной, кремневой и др.).
Реакции протекают с кислыми и средними солями по
уравнениям:
Na2CO3+H2SO4-*Na2SO4+CO2+H2O;
Ca(HCO3J-f-H2SO4 — CaSO4+2CO2+2H2O;
Na2Si03+ H2SO4 — Na2SO4+H2SiO3.
При нейтрализации воды, имеющей pH выше 8, количество
кислоты, пошедшей на титрование с индикатором
фенолфталеином, называют щелочностью по фенолфталеину. В этом случае
средние соли угольной кислоты оттитровываются до
бикарбонатов, т. е. наполовину, соли кремневой кислоты — полностью и
соли фосфорной кислоты переходят в кислые. Реакции
протекают по следующим уравнениям:
2СаСО3+2НС1 —Са(НСО3J+СаС12;
Na2Si03+2HCl — H2Si08+2NaCl;
Na3PO4+HCl — Na2HPO4+NaCl.
Аппаратура. Установка для титрования.
Реактивы и их приготовление. 1. Серная или соляная
кислота, 0,1 н. или 0,01 н. раствор. 2. Фенолфталеин, 1%-ный
спиртовой раствор. 3. Метиловый оранжевый, 0,2%-ный раствор или
смешанный индикатор. Приготовление. 1 г метилового
оранжевого и 2,5 г индигокармина растворяют в 1 л воды.
Подготовка проб. Пробу фильтруют через бумажный фильтр
и определяют pH по бумажке, смоченной раствором
фенолфталеина.
Ход анализа. Вода имеет pH ниже 8. Берут две
одинаковые конические колбы вместимостью 250 мл. В одну колбу
вливают 100 мл дистиллированной воды, 1 каплю раствора
кислоты, которой будут титровать, и 3 капли индикатора. Полученная
окраска раствора служит свидетелем при определении конца
титрования во время анализа пробы. В другую колбу вливают
100 мл исследуемой воды, 3 капли индикатора и титруют 0,1 н.
раствором серной (соляной) кислоты до окраски раствора,
совпадающей с окраской свидетеля. Расход кислоты на титрование
соответствует общей щелочности пробы. Индикатором служит
метиловый оранжевый или смешанный индикатор.
Вода имеет pH выше 8. В колбу для титрования
вливают 100 мл анализируемой воды, 2—3 капли
фенолфталеина и титруют 0,1 н. раствором серной (соляной) кислоты до
253
обесцвечивания раствора. При низкой щелочности пробы
титрование проводят 0,01 н. раствором кислоты. Измеряют
количество кислоты, пошедшее на титрование, оно соответствует
щелочности по фенолфталеину или щелочному числу. Затем к
бесцветному раствору добавляют 3 капли смешанного индикатора
или метилового оранжевого и продолжают титровать кислотой
до появления окраски, совпадающей с окраской свидетеля.
Измеряют расход кислоты от начала титрования, он соответствует
общей щелочности.
Расчет. Общую щелочность Ха и щелочность воды по фенолфталеину Х$,
(щелочное число) выражают в миллиграмм-эквивалентах на 1 л воды
(мг-экв/л) 1 мл 1 н. раствора содержит 1 мг-экв.
При титровании 100 мл пробы 0,1 и. раствором кислоты формулы для
вычисления щелочности имеют следующий вид:
аО.1 • 1000 у 60,1 • 1000
Ао- ]00 , Лф- 100
где а — объем 0,1 и. раствора кислоты, пошедший на титрование с метиловым
оранжевым, мл; Ь — объем 0,1 н. раствора кислоты, пошедший на титрование
с фенолфталеином, мл.
Щелочность определяют с точностью ±0,05 мг-экв.
Определение окисляемости перманганатиой (метод Кубели)
Метод предназначен для быстрого определения количества
кислорода, необходимого для химического окисления легкоокис-
ляемых веществ, содержащихся в анализируемой воде. Этот
метод рекомендован Международным стандартом на воду [91]
для оценки загрязненности воды. Он используется почти во всех
странах. В отличие от ХПК (химически потребляемый кислород)
результаты анализа, полученные этим методом, называют окис-
ляемостью перманганатной. Чтобы яснее представить
химическую сущность этого понятия, в табл. 14 приведены данные
расхода кислорода на окисление некоторых веществ
перманганатом и теоретический расход кислорода, вычисленный по
формуле горения до образования углекислого газа (СОг) и воды
(НгО). Анализ данных таблицы показывает, что расход
кислорода при окислении перманганатом для различных веществ
сильно отличается от теоретического. Если фенол в условиях
анализа успевает окислиться полностью, расходуя при этом
кислорода больше теоретического, то уксусная, масляная и пропио-
новая кислоты почти не окисляются, а на окисление глюкозы
расходуется только 59% кислорода от теоретического.
Метод основан на реакции окисления органических и
неорганических веществ, содержащихся в анализируемой воде,
кислородом, который выделяется в результате разложения перманга-
ната калия в кислой среде при кипячении в присутствии
восстановителей:
254
После окончания окисления определяют количество перман-
ганата калия, которое не вступило в реакцию, титруя его
щавелевой кислотой
По разности между взятым и найденным количеством
перманганата калия вычисляют количество перманганата калия,
израсходованное на окисление веществ, содержащихся в пробе.
Окисляемость выражают в миллиграммах перманганата калия
или в миллиграммах кислорода на 1 л воды.
Мешающие вещества. Хлор при концентрации выше 300 мг/л.
Сернистый водород, соли закиси железа, нитриты и другие
минеральные вещества, которые окисляются перманганатом калия
на холоду.
Аппаратура. Установка для титрования.
Реактивы и их приготовление. 1. Перманганат калия, 0,1 н.
и 0,01 н. растворы. 2. Щавелевая кислота, 0,01 н. раствор.
3. Серная кислота. Приготовление. К 30 мл
дистиллированной воды осторожно, маленькими порциями, при постоянном
помешивании добавляют 10 мл серной кислоты плотностью
1,84 г/см3 и несколько капель 0,01 н. раствора перманганата
калия до появления устойчивой розовой окраски. Раствор
кипятят 30 мин. Если он обесцветится, то прибавляют еще
перманганата калия до бледно-розовой окраски раствора.
Подготовка проб. Анализируемую воду разбавляют
дистиллированной водой в зависимости от ее окисляемости. Воду,
поступающую на очистку, разбавляют в 5 раз, очищенную воду
разбавляют в 2 раза.
Ход анализа. В коническую колбу вместимостью 250 мл
вливают 75 мл дистиллированной воды, 5 мл раствора серной
кислоты, 10 мл 0,01 н. раствора перманганата калия и 10 мл
разбавленной пробы. Для равномерности кипения в
реагирующую жидкость кладут кусочек пористого стекла, колбу
закрывают маленькой воронкой, ставят на включенную электроплитку
и замечают время появления первых пузырьков (начало
кипения). Содержимое колбы кипятят 10 мин (песочные часы).
После окончания окисления колбу снимают с электроплитки,
вливают в нее 10 мл 0,01 н. раствора щавелевой кислоты и
горячую жидкость сразу титруют 0,01 н. раствором перманганата
калия до появления розовой окраски, не исчезающей в течение
1 мин. Затем определяют окисляемость дистиллированной воды,
которую употребляют для разбавления пробы и для анализа.
Параллельно проводят холостой опыт, в котором вместо 10 мл
разбавленной анализируемой воды к реактивам прибавляют
10 мл дистиллированной воды, кроме 75 мл. После окончания
холостого опыта, не выливая реакционной жидкости, проводят
проверку титра 0,01 н. КМпО4 по щавелевой кислоте следующим
255
образом. В колбу с горячей реакционной жидкостью
вливают 10 мл 0,01 н. раствора щавелевой кислоты и оттитровы-
вают ее 0,01 н. раствором перманганата калия до розового
окрашивания, не исчезающего в течение 1 мин. На основании
полученных данных вычисляют поправочный коэффициент для
пересчета концентрации рабочего раствора КЛІПО4 в точно
0,01 н. раствор К= —, где а — количество миллилитров
раствора перманганата калия, пошедшее на титрование. Если
анализируемая вода содержит много минеральных веществ,
которые окисляются перманганатом калия на холоду, то их
определяют отдельно. Анализ проводят как описано выше, но без
нагревания.
Расчет. Окисляемость анализируемой воды выражают количеством
кислорода или перманганата калия, которое расходуется на окисление веществ,
содержащихся віл пробы, и вычисляют по формуле
(А-а-б)К1№
Х = 1
где X—окисляемость анализируемой воды, мг/л; А — объем 0,01 н. КМпО4,
прибавленный к реагирующей жидкости в начале анализа и при титровании
избытка щавелевой кислоты, мл; а — объем 0,01 н. КМпС>4, пошедший на
титрование щавелевой кислоты, мл; б — объем 0,01 н. КМпО4, необходимый для
окисления веществ, содержащихся в дистиллированной воде, взятой для
анализа G5 мл) и прибавленной к реагирующей жидкости с разбавленной
пробой, мл; К — 0,32 мг перманганата калия или 0,08 мг кислорода; g — объем
неразбавленной пробы, взятый для титрования, мл.
Предостережения. 1. 0,01 н. раствор перманганата калия неустойчив,
поэтому его надо готовить через каждые 1—2 дня разведением 0,1 н. раствора
и проверять его титр по 0,01 н. раствору щавелевой кислоты, раствор которой
надо готовить ежемесячно. Хранят растворы перманганата калия в склянке
из темного стекла. 2. Реакционную колбу (коническая вместимостью 250 мл)
перед анализом надо обрабатывать раствором перманганата калия, затем
дистиллированной водой и употреблять только для определения окисляемости
перманганатиым методом.
Определение растворенного кислорода (метод Вииклера)
Метод предназначен для определения концентрации
кислорода в воде до и после ее очистки, а также в воде из водоемов.
Метод основан на йодометрическом определении кислорода,
израсходованного на окисление двухвалентного марганца в
четырехвалентный. Кислород, растворенный в анализируемой воде,
фиксируют, окисляя им двухвалентный марганец в
четырехвалентный в щелочной среде. Реакции протекают по следующим
уравнениям:
MnSO4+2NaOH -* Mn (OHJ+Na2SO4;
2Мп (ОНJ+ О2 -* 2Н2Мп03.
256
Белый осадок гидроокиси марганца быстро окисляется
кислородом, растворенным в воде, при этом образуется ортомарган-
цоватистая (Н4МпО4) или метамарганцоватистая кислота
(Н2МпОз), которая выпадает в виде коричневого осадка. В
кислой среде эти кистоты реагируют как гидроокиси. Когда весь
растворенный кислород прореагирует с марганцем, выпавший
осадок марганцоватистой кислоты растворяют в серной кислоте
в присутствии йодистого калия. Четырехвалентный марганец
восстанавливается йодом в двухвалентный по уравнению
Количество выделившегося свободного йода, эквивалентное
количеству кислорода в анализируемой воде, определяют
титрованием раствором гипосульфита натрия.
Мешающие вещества. Нитриты, двух- и
трехвалентное железо, различные органические вещества,
сульфиды, сульфиты, политионаты, гипохлориты,
взвешенные вещества и другие вещества, которые
поглощают или восстанавливают выделившийся
свободный йод на последней стадии реакции, или
окисляют йодид до свободного йода. Активный
ил и другие микроорганизмы, поглощающие
кислород из воды или сорбирующие свободный йод
при анализе. Рис. 69.
Аппаратура. Склянка для инкубации (рис. 69). Склянка для
Установка для титрования. инкубации:
/ Од
р
/. Определение Кислорода в ЧиСТОй воде. ''
С
р р кыпгчок 3-
Реактивы и их приготовление. 1. Сернокислый шлифы
марганец (MnSO4) раствор. Приготовление.
В дистиллированной воде растворяют 400 г MnSO4-2^0 и объем
раствора доводят до 1 л. При употреблении сернокислого
марганца, содержащего другое количество кристаллизационной
воды, делают соответствующий пересчет или готовят раствор,
имеющий плотность 1,270 г/см3 при 20°С. Приготовленный
раствор фильтруют и проверяют на содержание марганцоватистой
кислоты и солей окиси железа. Если при добавлении 2 мл
приготовленного раствора сернокислого марганца к 10—20 мл
подкисленного раствора йодистого калия после прибавления 1—2
капель крахмала раствор останется бесцветным, или появится очень
слабая синяя окраска, то этим раствором можно пользоваться.
При появлении интенсивного синего окрашивания раствор
выливают и готовят новый из другого сернокислого марганца. 2.
Щелочной райствор йодистого калия. Раствор не должан содержать
карбонаты, нитриты, йодаты и соли окиси железа, поэтому перед
его приготовлением очищают щелочь, которая всегда содержит
карбонаты, и проверяют чистоту йодистого калия. Очистка
едкого натра. В фарфоровый стакан вместимостью 1 л
17 Заказ № 142 257
наливают 450 мл дистиллированной воды и в нее осторожно,
кусочками, при постоянном перемешивании добавляют 750 г едкого
натра. Раствору дают немного остыть, переливают его в склянку,
закрывают резиновой пробкой и оставляют стоять 2 суток.
Углекислый натрий почти нерастворим в концентрированном
растворе едкого натра, поэтому при стоянии он выпадает в
осадок на дно. Поверхность раствора едкого натра за время
стояния покроется корочкой карбоната натрия, который образуется
в результате поглощения углекислого газа из воздуха над
раствором.
Совершенно прозрачную жидкость после отстоя с помощью
сифона переливают в другую чистую сухую склянку. Затем
делают качественную реакцию на присутствие нитритов (см. с. 279).
Если раствор едкого натра содержит нитриты, то их удаляют
следующим образом. Раствор едкого натра без карбоната
помещают в фарфоровый стакан, опускают в него 1 г сплава Де-
варда E0% меди, 45% алюминия и 5% цинка), раствор
кипятят в течение 1 ч и снова проверяют на содержание нитритов.
Если нитриты не все исчезли, то кипячение повторяют. После
удаления нитритов горячий раствор отстаивают до полного
осаждения образовавшегося осадка, прозрачный раствор сифоном
сливают в склянку, определяют его концентрацию ареометром
и используют для приготовления реактива.
Проверка йодистого калия. Реакция на
присутствие йодатов — кислый раствор йодистого калия не синеет при
добавлении 1 капли раствора крахмала, если нет йодатов. При
положительной реакции на йодаты реактив надо сменить.
Реакция на отсутствие веществ, поглощающих йод: кислый раствор
йодистого калия после добавления 1 капли 0,01 н. раствора
йода в присутствии крахмала становится синим. Если раствор
не посинеет от прибавления йода, то устанавливают, сколько
надо прибавить йода к раствору, содержащему 150 г йодистого
калия (КІ), чтобы он синел от прибавления 1 капли 0,01 н.
раствора йода. Для этого 1 г КІ растворяют в воде,
подкисленной серной кислотой и титруют 0,1 н. раствором йода (или
более концентрированным). Полученный результат используют
при приготовлении раствора.
Приготовление щелочного раствора. В мерную
колбу A л) вносят 150 г йодистого калия и 250 мл
дистиллированной воды. После растворения КІ, если надо, добавляют ранее
установленное количество йода, затем прибавляют раствор
едкого натра в таком количестве, которое содержит 500 г NaOH,
объем полученного раствора доводят дистиллированной водой
до 1 л и хорошо перемешивают. Раствор хранят в бутыли из
темного стекла.
3. Серная кислота разбавленная 2:3. 4. Гипосульфит натрия
0,01 н. раствор. Приготовление. Раствор готовят из фикса-
нала или по навеске из расчета 2,5 г/л чистого гипосульфита
258
натрия (Na2S2O3'5H2O). В присутствии двуокиси углерода
(СО2) гипосульфит натрия разлагается, поэтому воду,
предназначенную для приготовления раствора, кипятят в течение 2 ч
для полного удаления СО2, а затем, закрыв колбу пробкой
с хлоркальциевой трубкой, охлаждают до комнатной
температуры и используют для приготовления раствора. Раствор
гипосульфита натрия консервируют, прибавляя на 1 л раствора 4 мл
1 н. раствора едкого натра и 5 мл хлороформа. Раствор хранят
в склянке из темного стекла, закрытой пробкой, в которую
вставлены сифон для отбора раствора в бюретку и хлоркальцие-
вая трубка.
Установка титра. Несмотря на все предосторожности,
0,01 н. раствор гипосульфита натрия неустойчив, поэтому его
титр проверяют ежедневно. Готовят 0,01 н. раствор двухромово-
кислого калия (К2СГ2О7) из фиксанала или по навеске из
расчета 0,4902 г КгСг2О7 (перекристаллизованного и высушенного
при 150°С) в 1 л раствора. В коническую «олбу вместимостью
250 мл вливают 5 мл серной кислоты B:3), 3 мл
свежеприготовленного 10%-ного раствора йодистого калия и 20 мл 0,01 н.
раствора двухромовокислого калия. Колбу ставят в темное место
на 10 мин, затем добавляют 50 мл дистиллированной воды и
титруют раствором гипосульфита натрия в присутствии
крахмала до перехода синей окраски раствора в светло-зеленую.
Нормальность раствора гипосульфита натрия вычисляют по
формуле
_ 0.01 • 20
н* у •
где V — объем раствора гипосульфита натрия, пошедший на
титрование, мл.
5. Крахмал 0,5%-ный раствор. Приготовление. В
фарфоровой чашке смешивают 0,5 г растворимого крахмала с 5 мл
дистиллированной воды и выливают в стакан с кипящей водой
(95 мл) при постоянном перемешивании. Раствор доводят до
кипения, охлаждают, фильтруют и консервируют, прибавляя
0,125 г салициловой кислоты или толуол.
Подготовка проб. При отборе пробы воды надо замерить ее
температуру и атмосферное давление. Хранить пробу не
рекомендуется.
Ход анализа. Фиксирование растворенного кислорода
проводят в инкубационной склянке, вместимость которой при
закрытой пробке устанавливают предварительно весовым методом.
На анализ берут склянку вместимостью около 250 мл, наполняют
ее доверху исследуемой водой, закрывают пробкой, поверхность
склянки осушают фильтровальной бумагой и ставят ее в чашку
или в кристаллизатор. Если берут склянку вместимостью 100
или 350 мл, то соответственно изменяют количество
прибавляемых реактивов. Затем пробку вынимают, на дно склянки
17* 259
пипеткой осторожно вводят 1 мл раствора сернокислого
марганца и 1 мл щелочного раствора йодида калия, колбу
закрывают притертой пробкой, энергично перемешивают в течение 40—
50 с, ставят в чашку и ждут, когда осядет коричневый осадок.
Вылитые 2 мл воды учитывают при вычислении растворенного
кислорода. Потом склянку открывают, осторожно, по стенке, над
жидкостью вводят 3 мл серной кислоты B:3), быстро закрывают
пробкой и перемешивают содержимое склянки, перевертывая ее
вверх дном несколько раз. Вылитые 3 мл жидкости не
учитывают при расчете, потому что кислород, находящийся в них,
уже прореагировал с марганцем. После растворения осадка
жидкость, которая приобрела цвет выделившегося йода,
количественно переносят в колбу для титрования, прибавляют раствор
гипосульфита натрия до бледно-желтой окраски раствора
(количество оставшегося йода соответствует приблизительно 0,5 мл
раствора гипосульфита натрия), прибавляют 1 мл раствора
крахмала и титруют до первого исчезновения синей окраски.
2. Определение кислорода в воде, содержащей нитриты и
железо.
Для устранения влияния нитритов на определение кислорода
методом Винклера наиболее эффективным и удобным считалось
добавление азида натрия. В последнее время предложено
заменить дефицитный азид натрия сульфаниловой кислотой или
мочевиной при анализе воды, которая содержит нитритов до
15 мг/л. Влияние двух- и трехвалентного железа при
концентрации не выше 100 мг/л устраняют прибавлением фтористого
калия.
Реактивы. 1. Все реактивы, употребляемые при определении
кислорода в чистой воде. 2. Сульфаниловая кислота (или
мочевина), 40%-ный раствор. 3. Фтористый калий раствор,
содержащий 40 г KF-2H2O в 100 мл.
Ход анализа. Проводят фиксацию растворенного кислорода,
как описано выше при анализе чистой воды. После
отстаивания осадка в склянку вводят 0,3 мл 40%-ного раствора
сульфаниловой кислоты (или мочевины), 1 мл 40%-«ого раствора
фтористого калия и сразу добавляют кислоту для растворения
осадка. Далее анализ ведут, как описано при исследовании
чистой воды.
3. Определение кислорода в воде, содержащей сульфиты,
соли серноватистой кислоты, свободный хлор, хлорноватистую
кислоту и вещества, окисляемые йодом.
Реактивы. 1. Все реактивы для определения кислорода в
чистой воде. 2. Щелочно-гипохлоритный раствор 2 н.
Приготовление. Раствор готовят по навеске из товарного гипохлорита
натрия (NaCIO) 149 г/л или из приготовленного. Гипохлорит
натрия получают путем пропускания газообразного хлора через
2,1 н. раствор едкого натра при охлаждении реакционной смеси.
Хлор прекращают пропускать тогда, когда на 1 мл полученного
260
раствора после его подкисления и добавления к нему йодистого
калия при титровании гипосульфитом натрия будет
расходоваться 20 мл 0,1 н. раствора гипосульфита натрия (Na2S2O3)
(индикатор крахмал). 3. йодистый калий, 0,1 н. раствор. Для
повышения стойкости раствора к нему добавляют 10 мл 1 н.
раствора едкого натра на 1 л раствора. 4. Сернистокислый натрий,
0,1 н. свежеприготовленный раствор F,3 г/л ЫагЭОз). Раствор
окисляется на воздухе.
Ход анализа. Исследуемую воду обрабатывают гипохлори-
том натрия для окисления примесей. В коническую колбу
наливают 250 мл пробы воды, добавляют в нее 0,2 мл щелочно-ги-
похлоритного раствора, колбу закрывают и ее содержимое
перемешивают в течение 20—30 с. Затем в колбу вливают 1 мл
0,1 н. раствора йодистого калия, 1 мл раствора серной кислоты
A:9) и выделившийся йод оттитровывают 0,1 н. раствором
гипосульфита натрия в присутствии крахмала до исчезновения
синей окраски. К бесцветному раствору прибавляют 0,1 мл 0,1 н.
раствора йодистого калия для восстановления синей окраски,
раствор количественно переносят в инкубационную склянку и
определяют растворенный кислород, как описано выше при
анализе чистой воды. Для фиксации кислорода в этом анализе
добавляют 3 мл щелочного раствора йодистого калия вместо 1 мл,
как при исследовании чистой воды.
В случае анализа воды, содержащей свободный хлор и гипо-
хлориты, пробу не обрабатывают гипохлоритом натрия, а
прибавляют к ней 1 мл 0,1 н. раствора йодистого калия, 1 мл
серной кислоты A:9) и титруют 0,01 н. раствором гипосульфита
натрия, как при анализе чистой воды.
4. Определение кислорода в воде, содержащей активный
ил и другие микроорганизмы.
Реактивы. 1. Все реактивы для анализа чистой воды. 2.
Сернокислая медь, 10%-ный раствор. 3. Иод, 0,01 н. раствор.
Ход анализа. При исследовании воды из аэротенков, которая
содержит активный ил, надо во время отбора пробы воды
прекратить процесс потребления кислорода микроорганизмами. Для
этого перед отбором пробы на анализ определяют
концентрацию или в жидкости аэротенка, вычисляют количество 10% -ного
раствора сернокислой меди, которое надо влить в
пробоотборник перед его заполнением водой с активным илом из расчета
2,5 мл 10%-ного раствора сернокислой меди (CuSO4) на 1 г
сухого активного ила. Пробу отбирают в склянку вместимостью
0,5 л с пришлифованной пробкой. Сначала в склянку
вливают вычисленное количество раствора сернокислой меди, а
потом заполняют ее отбираемой пробой воды доверху и закрывают
пробкой. Воздуха над жидкостью в склянке не должно быть.
После оседания ила часть жидкости над ним с помощью сифона
переливают в инкубационную склянку и делают определение
кислорода, как описано при анализе чистой воды.
261
При определении кислорода в воде, которая содержит много
фито- или зоопланктона (например, вода из очистных прудов),
необходимо изменить порядок определения конца титрования
йода гипосульфитом натрия. Выделившийся свободный йод
сорбируется микроорганизмами и при титровании гипосульфитом
натрия он не успевает десорбироваться. Для ускорения
десорбции йода титрование проводят не до первого исчезновения синей
окраски, а прибавляют небольшой избыток раствора
гипосульфита натрия, который оттитровывают 0,01 н. раствором йода
до появления синей окраски титруемой жидкости.
Расчет. Содержание растворенного кислорода в анализируемой воде
вычисляют по формуле
а0,08 • 1000
где X — концентрация растворенного кислорода в пробе, мг/л; а — объем
0,01 н. Na2S2O3, израсходованный на титрование, мл; V—емкость склянки,
установленная при ее калибровке, мл; (V — 2) — объем воды, взятый иа
анализ, мл; 0,08 — количество кислорода, эквивалентное 1 мл 0,01 н. раствора
гипосульфита натрия, мг.
Растворимость кислорода в воде зависит от температуры и давления,
поэтому вычисляют процент насыщения воды кислородом, используя данные
табл. 14 [5].
.. „, концентрация Оо в пробе при t° ,_.
Насыщение, о/о = ^ К-= п ? -100.
растворимость Ог при ^°
Растворимость кислорода Ор при атмосферном давлении Р отличном от
760 мм рт. ст. вычисляют по формуле
ОР
"Р- 760 '
где О —растворимость кислорода при 101,324 кПа G60 мм рт. ст.).
Воспроизводимость в среднем из трех определений ±0,05 мл 0,01 н.
раствора гипосульфита натрия.
ТАБЛИЦА 14
Растворимость кислорода
760
Температура, °С
0
1
2
3
4
5
6
7
Растворимость,
мг/л
14,62
14,23
13,84
13,48
13,13
12,80
12.48
12,17
Температура, °С
8
9
10
11
12
13
14
15
в воде при различных температуре и давлении
мм рт. ст. (по Фоку) [5]
Растворимость,
мг/л
11,87
11,59
11,33
11,08
10,83
10,Є0
10.37
10,15
Температура, °С
16
17
18
19
20
21
22
23
Растворимость,
мг/л
9,95
9,74
9,54
9,35
9,17
8,99
8,83
8,68
Температура, °С
24
25
26
27
28
29
30
Растворимость,
мг/л
8,53
8,38
8,22
8,07
7,92
7,77
7,63
262
Определение биохимически потребляемого кислорода (БПК)
Для определения БПКб и БПКго в воде до и после ее очистки
предназначен метод, основанный на проведении биохимического
окисления органических веществ, содержащихся в
анализируемой воде при температуре 18—20°С в течение заданного
времени. Биохимически потребляемым кислородом (БПК)
называют то количество миллиграммов кислорода, которое тратят
аэробные бактерии за определенное время на минерализацию
органических веществ, содержащихся віл анализируемой воды.
Определяя концентрацию кислорода в испытуемой воде до
и после биохимического окисления, находят количество
израсходованного кислорода, т. е. БПК при соответствующем времени
инкубации, которое пишут как индекс. Процесс биохимического
окисления зависит от природы окисляемых веществ. В табл. 15
приведены данные о расходе кислорода на биохимическое
окисление некоторых органических веществ в сравнении с
теоретически вычисленным расходом кислорода по формуле сгорания
до углекислого газа и воды.
ТАБЛИЦА 15
Перманганатная
окисляем ості
> и БПК органических веществ
(по данным Шульце—Форстера и Гада) |92]
Название вещества
Муравьиная кислота
Уксусная кислота
Пропиоиовая кислота
Масляная кислота
Щавелевая кислота
Метиловый спирт
Этиловый спирт
Декстрины
Глюкоза
Феиол
Крахмал
Расход
КМпО(, мг
на 1000 мг
вещества
126
5
14
6
493
58
65
846
2355
9660
461
Теоретический
расход
кислорода, мг
на 1000 мг
вещества
250
1066
1513
1818
178
1500
20Э0
1185
970.
2383
1185
БПК, % от теоретического
расхода кислорода
5- дневный
34
66
86
63
56
64
65
44
60
71
57
20-ииевный
100
84
93
80
65
84
86
71
89
84
76
Как видно из данных табл. 15, микроорганизмы не доводят
окисление органических веществ до конца даже за 20 дней
инкубации. При удлинении времени инкубации скорость окисления
уменьшается.
Аппаратура. Склянки для инкубации калиброванные
вместимостью 150—250 мл с хорошо притертыми пробками и
колпачками (см. рис. 69). Термостат с терморегулятором температуры
в пределах ± 1°С.
263
Реактивы и их приготовление. 1. Разбавляющая вода.
Приготовление. Готовят отдельно четыре раствора: 1)
фосфатный буфер, имеющий pH 7,2; он содержит в 1 л раствора 8,5 г
однозамещенного фосфата калия (КН2РО4), 21,75 г двузамещен-
ного фосфата калия (К2НРО4), 33,4 г двузамещенного фосфата
натрия (Na2HPO4 • Н2О) и 1,7 г хлористого аммония (NH4C1);
2) раствор сернокислого магния, 22,5 г MgSO4'7H2O в 1 л;
3) раствор хлористого кальция, 27,5 г безводного СаС12 віл;
4) раствор хлористого железа, 0,25 г FeCl3-6H2O в 1 л. Затем
в мерную колбу вместимостью 1 л вливают по 1 мл каждого
приготовленного раствора и дистиллированную воду до метки
на колбе. Приготовленный раствор называют разбавляющей
водой, она должна содержать кислорода не менее 8 мг/л. 2.
Культура микроорганизмов. Приготовление. 1-й способ. Около
2 г сухой земли помещают в колбу вместимостью 250 мл,
прибавляют 100 мл воды, энергично взбалтывают в течение 5 мин
и фильтруют через бумажный фильтр. Фильтрат, содержащий
микроорганизмы из земли, называют почвенной болтушкой.
2-й способ. 10 мл сточной воды разбавляют водопроводной водой
1 :50, прибавляют 2—3 мл разбавляющей воды и оставляют
в открытом стакане на 2—3 дня, пока появится муть или пленка
на поверхности воды. Для дальнейшего развития
микроорганизмов к ним прибавляют еще 2—5 объемов разбавляющей воды,
ждут 1—2 дня, а потом жидкость, содержащую
микроорганизмы, употребляют на анализ. 3. Едкий натр, 1 н. раствор.
4. Серная кислота, 1 н. раствор. 5. Индикатор бромтимол синий.
6. Все реактивы для определения кислорода по методу Винк-
лера.
Подготовка проб. 1. Анализируемую воду нейтрализуют до
pH 7,0—7,2 с индикатором бромтимол синий. 2. В сточной воде
количество растворенного кислорода очень мало по сравнению
с количеством органических веществ, которые надо окислить,
поэтому перед анализом исследуемую воду разбавляют водой,
насыщенной кислородом при 20°С. Разбавление рассчитывают
так, чтобы кислорода, содержащегося в разбавляющей воде,
хватило на окисление органических веществ, взятых на анализ.
Убыль кислорода за 5* суток должна быть не менее 4 мг/л,
а его концентрация после инкубации не ниже 2 мг/л. Воду из
чистых водоемов анализируют без разбавления. Биологически
очищенную воду разбавляют от 1 :20 до 1:4. Бытовые сточные
воды разбавляют от 1:20 до 1:100. Промышленные сточные
воды до очистки разбавляют от 1 : 100 до 1 : 1000 и более.
Необходимое разбавление рассчитывают ориентировочно по
результатам определения окисляемости перманганатной экспрессным
методом Кубеля. Частное от деления окисляемости на 5
показывает, во сколько раз надо разбавить пробу.
Ход анализа. В мерную колбу вместимостью 1 л вливают
1 мл анализируемой воды (если она разбавлена 1 :100), добав-
264
ляют 2 мл воды с микрофлорой и разбавляющую воду A8—
20°С) до метки на колбе. После перемешивания раствору дают
постоять для выделения пузырьков воздуха и разливают в
колбочки для инкубации. Раствор должен иметь температуру 20°С
и pH около 7,5. Количество колб, котарые заполняют
приготовленным раствором, определяется задачей, стоящей перед
аналитиком. Если надо определить биологически потребляемый
кислород п раз через различные промежутки времени, то число
колб для анализа равно 2га+1. Колбы заполняют
анализируемой жидкостью доверху и плотно закрывают пробками.
В колпачки наливают ту же жидкость, перевертывают колбу
с жидкостью вверх дном, плотно вставляют горло колбы в
колпачок и, снова перевертывая, ставят ее на дно. Не допускается
наличие пузырьков воздуха в колбе под пробкой и в колпачке.
Поверхность колбы осушают фильтровальной бумагой. Одну из
склянок не закрывают колпачком, в ней определяют
концентрацию кислорода в пробе до инкубации методом Винклера.
Параллельно приготовляют контрольную пробу, которой
является разбавляющая вода с добавкой микрофлоры из расчета
2 мл/л. Контрольную пробу разливают так же, как пробу в п+
+1 чистых инкубационных колб. Колбы закрывают колпачками
с той же жидкостью, а одну колбу закрывают только пробкой
и в ней определяют концентрацию кислорода. Все колбы с
анализируемой водой и контрольной пробой ставят в термостат при
20сС. Через каждое заданное время инкубации вынимают две
колбы с анализируемой водой и одну с контрольной пробой.
В жидкости из колпачков определяют нитриты, а в жидкости
из колб — растворенный кислород. Если нитритов будет
обнаружено в количестве более 0,1 мг/л, то дальнейшее
биохимическое окисление не проводят, так как этот результат анализа
свидетельствует о начавшемся окислении азота.
Расчет. Количество кислорода, которое микроорганизмы истратили на
окисление органических веществ, содержащихся віл анализируемой воды,
за т суток инкубации (БПКт), находят как разность между количеством
исчезнувшего кислорода в жидкости с анализируемой водой и в контрольной
пробе:
где Со н С — концентрации кислорода в разбавленной анализируемой пробе
соответственно до и после инкубации, мг/л; /Со и К—концентрация
кислорода в контрольной пробе соответственно до и после инкубации, мг/л; п —
разбавление пробы.
Предостережение. 1. В воде, прошедшей биологическую очистку, сильно
развит процесс нитрификации, который продолжается при ее инкубации.
Количество кислорода, которое за время инкубации будет истрачено на
окисление азотистых соединений, может в несколько раз превысить количество
кислорода, израсходованного на окисление органических веществ. Чтобы
прекратить процесс нитрификации, надо на 1 л разбавляющей воды добавить
6 мл 0,05%-ного раствора метиленовой синей. 2. При выполнении анализа надо
особое внимание уделить чистоте инкубационных склянок. Перед
анализом их следует вымыть хромовой смесью и высушить при 150—170°С.
265
Определение константы скорости потреблення растворенного кислорода
Константу скорости потребления растворенного кислорода
при биохимическом окислении органических веществ,
находящихся в сточной воде, .определяют методом кратных сроков.
Этот метод заключается в том, что исследуемую воду
анализируют на содержание БПКп и БПКгп, где л = 3 или 5 суток, и,
допуская, что потребление кислорода микроорганизмами
является мономолекулярной реакцией, вычисляют константу по
формуле і БПКЯ
Л~~ / lg БПК2я-БПК„ "
Получаемые данные нельзя считать точными, но ими часто
пользуются в расчетных формулах. Константу определяют
ежемесячно.
Определение химически потребляемого кислорода (ХПК)
бихроматным методом
Метод предназначен для определения количества кислорода,
которое нужно истратить на окисление органических и
неорганических веществ, присутствующих в анализируемой воде.
Химически потребляемый кислород называют ХПК и выражают
в миллиграммах кислорода на 1 л испытуемой воды (мг/л).
Этот показатель характеризует загрязненность воды всеми
видами веществ, которые можно окислить. Величина ХПК зависит
от природы окисляемых веществ, от силы окислителя и от
условий окисления. В качестве окислителей применяют бихромат
калия в кислой среде, йодат калия в кислой среде и перманганат
калия в кислой или щелочной среде. Наибольшее количество
веществ окисляется бихроматом калия в присутствии
катализатора-— сернокислого серебра при длительном кипячении B ч).
В этих условиях многие органические вещества окисляются
полностью до углекислого газа, воды и элементарного азота.
Алифатические углеводороды, спирты и кислоты с неразветвленной
цепью атомов углерода окисляются на 85—95% только в
присутствии катализатора. Ароматические углеводороды и пиридин
совершенно не окисляются даже при катализаторе. Определение
ХПК бихроматным методом признано наиболее точным,
поэтому результат анализа называют ХПКполное [93]. Во многих
странах этот метод является стандартным [95]. Метод основан
на реакции окисления органических и неорганических веществ,
находящихся в анализируемой воде, бихроматом калия в
кислой среде в присутствии катализатора. Шестивалентный хром
восстанавливается при этом до трехвалентного:
Сг2ОГ+14Н+4-6е-*2Сг-+7Н2О.
Количество восстановленного хрома, которое
пропорционально количеству кислорода, израсходованного на окисление
266
веществ, содержащихся в испытуемой воде, находят по разности
между количествами бихромата калия, взятого на анализ и
оставшегося после реакции. Концентрацию бихромата калия
определяют титрованием раствором соли Мора.
Мешающие вещества. Хлориды.
Аппаратура. Установки для кипячения с обратным
холодильником и для титрования.
Реактивы и их приготовление. 1. Двухромовокислый калий
(К2СГ2О7), 0,25 н. раствор. 2. Серная кислота плотностью
1,84 г/см3 х. ч. 3. Сернокислое серебро кристаллическое. 4. Же-
лезистосернокислый аммоний (соль Мора), 0,25 н. раствор.
Приготовление. В дистиллированной воде растворяют 98 г
Fe(NH4J' (SCub* 6Н2О, прибавляют 20 мл концентрированной
серной кислоты, охлаждают и объем раствора доводят до 1 л
в мерной колбе. Ратвор соли Мора неустойчив, поэтому его титр
проверяют ежедневно по 0,25 н. раствору двухромовокислого
калия. В коническую колбу вливают 25 мл 0,25 н. К2СГ2О7,
250 мл дистиллированной воды, 20 мл концентрированной
серной кислоты, смесь охлаждают до комнатной температуры,
прибавляют 3—4 капли раствора Ферроина (или другой индикатор)
и титруют раствором соли Мора. Нормальность раствора соли
Мора находят по формуле
25. 0.25
н. = >
а
где а — объем раствора соли Мора, пошедший на титрование, мл.
5. Индикатор — один из следующих. 1. Кислота н-фенилантра-
ниловая. Приготовление. 0,25 г кислоты растворяют
в 12 мл 0,1 н. раствора едкого натра и разбавляют водой до
250 мл. 2. Раствор Ферроина. Приготовление. 1,485 г
1,10-фенантралина (одноводного) и 0,695 г сернокислого
закисного железа (FeSO4'7H2O) растворяют в воде и объем доводят
до 100 мл в мерной колбе. 3. Дифениламин 1 %-ный раствор в
концентрированной серной кислоте.
Подготовка проб. В пробе определяют ХПК экспрессным
методом и вычисляют необходимое разведение анализируемой воды
перед анализом. Разведение должно быть таким, чтобы 50 мл
разбавленной пробы восстанавливало не более 20 мл 0,25 н.
раствора двухромовокислого калия.
Взвешенные вещества, содержащиеся в пробе, не отделяют.
Если проба содержит легкоосаждающиеся вещества, то перед
разбавлением ее тщательно перемешивают в закрытой склянке,
заполненной пробой до пробки. В пробе определяют хлориды
(см. с. 101) или удаляют их одним из следующих способов:
1. Ионы хлора осаждают, прибавляя сульфат серебра,
выпавший осадок отделяют на центрифуге (фильтровать нельзя!).
2. В реакционную смесь добавляют повышенное (по
сравнению с вычисленным) количество катализатора — сернокислого
267
серебра, чтобы связать хлориды. 3. Анализ проводят без
катализатора, хлориды при этом окисляются полностью. Определив
их количество отдельно, вводят поправку при вычислении ХПК
в пробе.
Ход анализа. В плоскодонную колбу вместимостью 500 мл
(лучше из стекла пирекс) вливают 50 мл разбавленной пробы,
25 мл 0,25 н. раствора двухромового калня (К2СГ2О7), вносят
1 г сернокислого серебра (если нет хлоридов) и осторожно,
маленькими порциями, при постоянном перемешивании вливают
75 мл серной кислоты плотностью 1,84 г/см3. Содержимое колбы
тщательно перемешивают, добавляют кусочек пемзы для
равномерного кипения, колбу закрывают пробкой, в которую вставлен
обратный холодильник, и ставят на включенную электроплитку.
Замечают время начала кипения реакционной смеси и кипятят
ее 2 ч. Затем убирают плитку, охлаждают колбу с реакционной
жидкостью, холодильник промывают дистиллированной водой
B00 мл), собирая ее в ту же колбу, отсоединяют колбу,
добавляют в нее 3—4 капли раствора Ферроина (или 10—15 капель
раствора н-фенилантраниловой кислоты) и титруют 0,25 н.
раствором соли Мора. В конце титрования наблюдают резкое
изменение окраски титруемой жидкости с переходом через
голубовато-зеленую в красно-голубую. Аналогично проводят
холостой опыт, в котором вместо 50 мл исследуемой воды берут
50 мл дистиллированной и проводят ее окисление, как в опыте.
Расчет. ХПК — количество миллиграммов кислорода, израсходованное
на окисление веществ, содержащихся в 1 л анализируемой воды (мг/л),
вычисляют по формуле
(а - ff) 2 • 1000
где а — объем 0,25 н. раствора соли Мора, пошедший на титрование в
холостом опыте, мл; б — объем 0,25 н. раствора соли Мора, взятый на титрование
пробы, мл; g — объем пробы, израсходованный на анализ, с учетом
разбавления, мл; 2 — количество кислорода, эквивалентное 1 мл 0,25 н. раствора
соли Мора или двухромовокислого калия, мг.
Если в исследуемой воде содержится хлор, который не связывали
сернокислым серебром и не удаляли' перед анализом, то из найденного количества
ХПК вычитают количество кислорода, израсходованного на окисление хлора,
при вычислении его исходят из теоретического расчета: на 1 мг иона хлора
требуется 0,225 мг кислорода.
Воспроизводимость в среднем из трех определений ±0,02 мл раствора
соли Мора.
Предостережение. При высоком содержании хлоридов в пробе увеличение
количества катализатора (Ag2SO4), необходимое для их связывания, приводит
к получению завышенных результатов анализа за счет окисления хлора в
хлористом серебре до элементарного хлора.
Определение химически потребляемого кислорода (ХПК)
экспрессным методом
Метод предназначен для быстрого определения ХПК.
Метод основан на окислении органических и неорганических
веществ бихроматом калия (см. с. 266). Отличительной особен-
268
ностью метода является применение серной кислоты
повышенной концентрации, что позволяет окислить 78—82% всех
окисляемых веществ за 2 мин без дополнительного нагревания.
Результаты анализов стабильны. Если вода на данной очистной
станции идет приблизительно одинакового состава, то можно
установить поправочный коэффициент, позволяющий результат
анализа пересчитывать В ХПКполное И БПКб-
Мешающие вещества. Хлориды.
Аппаратура. Установка для титрования.
Реактивы. Все реактивы, которые применяют при
определении ХПКполное бихроматным методом (см. с. 267).
Подготовка пробы. Воду, имеющую ХПК выше 1000 мг/л,
разбавляют до ХПК ниже 500 мг/л и на анализ берут 5 мл.
При ХПК воды от 500 до 1000 мг/л на анализ берут 1 мл.
Ход анализа. В коническую колбу с притертой пробкой
вместимостью 100 мл вливают 1 или 5 мл анализируемой пробы,
добавляют 2,5 мл 0,25 н. раствора бихромата калия и при
помешивании струей вливают 15 мл концентрированной серной
кислоты плотностью 1,84 г/см3. Реагирующую жидкость, имеющую
температуру около 100сС за счет разбавления кислоты,
перемешивают и оставляют в покое на 2 мин (песочные часы). Затем
реакционную смесь охлаждают до комнатной температуры,
добавляют 20—30 мл дистиллированной воды, 10—15 капель
раствора н-фенилантраниловой кислоты и титруют 0,25 н.
раствором соли Мора.
Расчет. Вычисляют ХПК по формуле, приведенной в бихроматном методе
определения ХПК на с. 268. При вычислении поправки на хлориды
пользуются эмпирическим коэффициентом 0,15 мг кислорода на 1 мг иона хлора.
Воспроизводимость в среднем из трех определений ±0,02 мл раствора соли
Мора.
Определение химически потребляемого кислорода (ХПК)
йодатным методом
Метод предназначен для определения количества кислорода,
которое нужно израсходовать на окисление органических и
неорганических веществ, содержащихся в 1 л анализируемой воды.
Метод основан на окислении органических и неорганических
веществ кислородом, выделяющимся при разложении йодата
калия в кислой среде при 180—200°С в присутствии
восстановителей:
21Оз~+2Н+ — 12+Н2О+50.
Реакция окисления начинается при 170°С, йодат калия
разлагается при 275°С, поэтому температуру песочной бани
поддерживают в пределах 180—200°С. Органические и неорганические
вещества окисляются до углекислого газа, сульфата аммония,
серной и фосфорной кислот. Количество кислорода, которое
было израсходовано на окисление веществ, содержащихся
269
в анализируемой пробе, находят по количеству разложившегося
йодата калия. Количество йодата калия, которое берут на
анализ, определяют взвешиванием. Для определения количества
оставшегося йодата калия из реакционной жидкости отгоняют
йод, образовавшийся при разложении КЮ3, а затем проводят
реакцию с йодистым калием.
ЮГ+5Г+6Н+-*312+ЗН2О.
Количество выделившегося йода, эквивалентное количеству
йодата калия, определяют титрованием раствором гипосульфита.
Мешающие вещества. Хлориды.
Аппаратура. Установки для кипячения с обратным
холодильником (см. рис. 13) и для титрования. Песчаная баня.
Реактивы. 1. Йодат калия кристаллический ч. д. а. 2.
Серная кислота плотностью 1,84. 3-. Йодистый калий кристаллический
4. д. а. 4. Гипосульфит натрия, 0,1 н., 0,05 н. и 0,01 н. растворы.
5. Крахмал, 0,5% -ный раствор.
Подготовка проб. В пробе определяют хлориды и ХПК
экспрессным методом. На основании полученных данных
устанавливают кратность необходимого разбавления пробы перед
анализом. Количество йодата калия, которое надо взять на окисление
при различном значении ХПК в разбавленной пробе, и
концентрацию (нормальность) раствора гипосульфита натрия,
применяемого для титрования, находят используя нижеприведенные
сведения:
ХПК, мг/л .... 5—20 20-150 более 150
КЮ3. г 0,02-0,03 0.05-0.10 0.10-1.00
Na2S2O3. н . . . 0.01 0.05 0.10
Ход анализа. На аналитических весах в бюксе берут
необходимую навеску йодата калия с точностью 0,0003 г, которую
растворяют в 2—3 мл дистиллированной воды и количественно
переносят в реакционную круглодонную колбу, расходуя при этом
не более 10 мл воды. Затем в колбу вливают 10 мл
анализируемой воды и осторожно, по стенке, при постоянном помешивании,
вливают 30 мл концентрированной серной кислоты.
Содержимое колбы перемешивают, колбу соединяют с обратным
холодильником, ставят на песчаную баню и включают нагрев.
Песчаную баню нагревают медленно, наблюдая за реагирующей
жидкостью. Как только появятся фиолетовые пары йода
(следовательно, температура достигла 170°С), замечают время и
продолжают нагревать еще 30—60 мин до тех пор, когда
прекратится выделение паров йода и жидкость в колбе станет
бесцветной. На этом реакция окисления считается законченной. Во
время нагрева надо измерять температуру песчаной бани, она
должна быть 180—200°С.
Реакционную колбу отсоединяют от холодильника,
охлаждают до комнатной температуры и вливают в нее 75 мл дистил-
270
лированной воды. Если жидкость в колбе станет желтой,
следовательно она содержит свободный йод. Для удаления йода
содержимое колбы кипятят без холодильника до полного
¦обесцвечивания, а потом охлаждают. При ХПК пробы меньше
70 мг/л содержимое колбы количественно переносят в
коническую колбу вместимостью 250 мл с пришлифованной пробкой
и титруют, как описано ниже. Если ХПК пробы больше, то
содержимое колбы количественно переносят в мерную колбу
вместимостью 250 мл, объем доводят дистиллированной водой до
метки на колбе, перемешивают и для титрования берут 20—50 мл.
В коническую колбу объемом 250 мл вносят анализируемый
раствор, прибавляют 1 г кристаллического йодистого калия и
50 мл дистиллированной воды. Колбу закрывают стеклянной
пробкой и оставляют стоять 10—15 мин в темноте.
Выделившийся йод оттитровывают раствором гипосульфита натрия
выбранной концентрации сначала до желто-соломенной окраски,
ботом добавляют 1 мл 0,5%-ного раствора крахмала и дотитро-
вывают до полного обесцвечивания раствора. Параллельно
проводят холостой опыт, в котором вместо пробы берут 10 мл
дистиллированной воды.
Расчет. ХПК анализируемой воды (в мг/л) вычисляют по формуле
( Л '?' - а) ¦ 0,6667 • 1000
ХПК = ———
g
тде о — объем 0,1 н. Na2S2O3, израсходованный на титрование пробы, мл;
л' — объем 0,1 н. МагвгОз, пошедший на титрование в холостом опыте, мл;
А—количество йодата калия, взятое на анализ пробы, мг; А' — количество
йодата калия, взятое в холостом опыте, мг; g — объем пробы воды, взятый
на анализ, с учетом разбавления, мл; 0,6667 — количество кислорода,
эквивалентное 1 мл 0,1 н. раствора гипосульфита натрия, мг.
Если в пробе были хлориды, которые не удаляли перед анализом, но
-количество их определяли, то из полученного результата надо вычесть
поправку на хлориды, вычисляя ее на основании установленного соотношения:
на 1 мг иона хлора расходуется 0,225 мг кислорода. Воспроизводимость
ів среднем из трех определений ±0,05 мл раствора гипосульфита натрия.
Определение общего азота
Для определения всего азота, связанного в различных
органических и минеральных соединениях в виде аммиачного,
нитритного и нитратного, предназначен метод, основанный на
реакциях восстановления окисленного азота до аммиака с
последующим образованием сернокислого аммония, окисления
органических соединений до углекислого газа и минерализации
азота органических соединений до сернокислого аммония.
Полученный сернокислый аммоний разлагается щелочью,
образующийся аммиак отгоняют в раствор борной кислоты. Количество
отогнанного аммиака определяют титрованием серной кислоты
или колориметрическим методом с реактивом Несслера.
271
Оптимальное количество азота в пробе, взятой для анализа
составляет 2—6 мг.
Аппаратура. Установки для минерализации пробы (см.
рис. 65) и для отгонки аммиака (см. рис. 36). Установка для
титрования или один из приборов для колориметрического
определения аммиака (с. 273).
Реактивы. 1. Серная кислота плотностью 1,84 г/см3. 2.
Красная окись ртути (HgO) без азота. 3. Силикагель безводный.
4. Сульфат калия кристаллический без азота. 5. Фенол ч. д. а.
6. Цинковая пыль. 7. Борная кислота (Н3ВО3), раствор 20 г/л,
приготовленный на безаммиачной воде (см. с. 274). 8.
Гипосульфит натрия кристаллический. 9. Едкий натр, 33%-ный
раствор. 10. Серная кислота, 0,1 н. раствор. 11. Смешанный
индикатор (см. с. 211) или реактивы для колориметрического
определения аммиака (см. с. 273).
Ход анализа. Минерализация. В колбу Кьельдаля
вместимостью 500 мл вливают 100 мл анализируемой воды или 2 г
активного ила с известной влажностью, добавляют 6 гранул
силикагеля, 1,5 г окиси ртути и 12 г сульфата калия.
Содержимое колбы перемешивают, осторожно добавляют 15—20 мл
концентрированной серной кислоты и снова перемешивают. Для
восстановления нитратов и нитритов к реакционной жидкости
прибавляют 4% фенола по отношению к количеству серной
кислоты в колбе, ждут 5 мин, а потом маленькими порциями
добавляют 1—2 г цинковой пыли и перемешивают. В этих условиях
окислы азота восстанавливаются до аммиака. Колбу с
реакционной жидкостью после восстановления окислов азота укрепляют
в штативе над колбонагревателем и при небольшом нагреве
кипятят 10—15 мин до полного обесцвечивания реагирующей
смеси. Под полным нагревом подразумевают такую
интенсивность нагрева, при которой 250 мл воды, помещенные в колбу
Кьельдаля вместимостью 500 мл, за 4—5 мин нагреваются до
кипения. Пробу на интенсивность нагрева делают
предварительно. (Нагревать колбу выше уровня жидкости в ней нельзя.)
Определение аммиака. После минерализации колбу
с реакционной жидкостью охлаждают, добавляют 200 мл
безаммиачной воды и, вращая ее, растворяют твердый осадок
образовавшихся солей. К раствору в колбе прибавляют 5 г пятивод-
ного гипосульфита натрия, кусочек розовой лакмусовой бумажки
и 10—15 г талька для равномерного кипения. Далее в колбу
быстро вливают вычисленное количество 33%-ного раствора
едкого натра и сразу закрывают ее пробкой, в которую
вставлена трубка, соединенная с холодильником. На каждые 20 мл
концентрированной серной кислоты, взятой на минерализацию,
надо прилить 100 мл 33% -ного раствора едкого натра. Для
получения щелочной среды реакционной смеси нижний конец
холодильника погружают в раствор борной кислоты, налитый в
коническую колбу вместимостью 250 мл, включают нагрев колбы
272
и отгоняют аммиак. Конец отгонки определяют по красной
лакмусовой бумажке, она не должна менять окраску от капли
конденсата из холодильника. Во время отгонки во избежание
засасывания кислоты в холодильник его нижний конец
приподнимают так, чтобы он только касался поверхности кислоты
в приемнике. Для более полного поглощения аммиака
количество борной кислоты в приемнике конденсата должно быть в
избытке. На 1 мг отгоняемого аммиака надо брать 50 мл раствора
борной кислоты.
Аммиак, собранный в раствор борной кислоты, титруют
0,1 н. раствором серной кислоты, прибавив 4 капли смешанного
индикатора. Чтобы точнее определить конечную точку
титрования, пользуются сравнением окраски титруемой жидкости с
окраской свидетеля. Свидетелем служит окраска раствора,
приготовленного из 50 мл борной кислоты и дистиллированной воды,
взятой в объеме собранного конденсата, после добавления 4
капель смешанного индикатора. При колориметрическом
определении аммиака отбор конденсата делают порциями и
анализируют, как описано на с. 277.
Расчет. Концентрацию общего азота в исследуемой пробе вычисляют по
формуле
йі.4-1000
Л = »
g
где X — концентрация азота в пробе, мг/л; а — объем 0,1 н. раствора серной
кислоты, пошедший на анализ, мл; g — объем пробы, взятый на анализ, мл;
1,4 — количество азота, эквивалентное 1 мл 0,1 н. раствора серной кислоты, мг.
Чувствительность метода 0,08 мг азота в 50 мл конденсата. Точность
определения ±5—7% оти.
Определение аммиачного азота колориметрическим методом
Метод предназначен для определения свободного и
связанного аммиака в сточной воде на различных стадиях ее очистки
при содержании аммиака в количествах до 0,01 мг/л. Метод
основан на извлечении аммиака из анализируемой воды путем
его отгонки из щелочной среды и определении количества
аммиака по окраске соединения, которое он дает с реактивом
Несслера. Реакция протекает по следующему уравнению:
NH3+2(HgI2 • 2KI)+3KOH^NHg2I+7KI+3H2O.
Интенсивность окраски образовавшегося меркураммония
(NHg2l) определяют на фотоэлектроколориметре или визуально
сравнением с окраской эталонных растворов.
Мешающие вещества. Формальдегид, хлор, сульфиды, спирты,
альдегиды, ацетон, алифатические и ароматические амины,
органические хлорамины, которые отгоняются из пробы вместе
с аммиаком и, реагируя с реактивом Несслера, дают соединения,
окрашенные в желто-зеленый цвет, или осадок в виде мути.
18 Заказ № 142 273
50
Кальций, который реагирует с фосфатом буферного раствора
и выпадает в осадок в виде фосфата кальция, что приводит
к понижению pH реагирующей жидкости и затрудняет отгон
аммиака. Магний. Присутствие его следов в дистиллированной
воде образует осадок (муть) при реакции с реактивом Несслера.
Аппаратура. Установка для отгонки аммиака (см. рис. 36).
Фотоэлектроколориметр (см. рис. 64), или цилиндры Генера,
или цилиндры Несслера удлиненной формы вместимостью 50 мл,
у которых отметка 50 находится на одинаковой высоте (рис.70).
Реактивы и их приготовление. Все реактивы готовят на
безаммиачной воде и хранят в посуде из стекла пирекс.
1. Безаммиачная вода. Приготовление. 1-й пособ.
Дистиллированную воду подкисляют серной кислотой, добавляют
несколько кристалликов перманганата
калия и перегоняют. Операцию
повторяют дважды. 2-й способ.
Дистиллированную воду пропускают через
колонку (диаметром 5 см, высотой 500 см),
заполненную на 3U высоты катионитом
СБС или КУ-2. Приготовленную
безаммиачную воду проверяют на
содержание аммиака с реактивом Несслера.
Максимально допустимая концентрация
аммиака в воде 0,01 мг/л. 2. Фосфатный
буферный раствор, имеющий pH 7,4.
Приготовление. 14,3 г безводного
однозамещенного калия (КН2РО4) и 68,8 г безводного двузаме-
щенного фосфорнокислого калия (К2НРО4) растворяют в
безаммиачной воде и объем раствора в мерной колбе доводят до 1 л.
Раствор проверяют на содержание аммиака с реактивом
Несслера. 3. Борная кислота, раствор 20 г/л. 4. Серная кислота,
0,1 н. раствор. 5. Растворы для дехлорирования: раствор
гипосульфита натрия 3,5 г/л или раствор сернокислого натрия
0,9 г/л. 6. Окись магния х. ч. 7. Серная кислота плотностью
1,84 г/см3. 8. Едкий натр, 0,1 н. и 0,01 н. растворы. 9.
Углекислый свинец кристаллический. 10. Реактив Несслера.
Приготовление. 100 г безводной йодистой ртути (Hgl2) и 70 г
безводного йодистого калия (KI) растворяют в небольшом
количестве B00—300 мл) безаммиачной воды и медленно при
помешивании приливают к охлажденному раствору, содержащему
160 г едкого натра в 500 мл безаммиачной воды. Полученный
раствор выливают в мерную колбу объемом 1 л и добавляют
до метки безаммиачную воду. Приготовленный реактив
проверяют по реакции с аммиаком. Характерная окраска должна
появляться после прибавления реактива к раствору аммиака
в течение 10 мин. При очень малых количествах аммиака в
растворе реактив не должен давать осадка (помутнения) в течение
2 ч. Реактив устойчив в течение 1 года, если его хранят в посуде
Рис. 70. Цилиндры:
/ — Несслера; 2— Генера
274
из стекла пирекс, закрыв корковой пробкой, в темноте. 11.
Основной раствор хлористого аммония. Пр и готов л ение. В
безаммиачной воде растворяют 3,819 г безводного хлористого
аммония и объем раствора в мерной колбе доводят до 1 л. В 1 мл
полученного раствора содержится 1 мг азота или 1,22 мг
аммиака (NH3). 12. Разбавленный раствор хлористого аммония.
Приготовление. В мерной колбе разбавляют 10 мл
основного раствора хлористого аммония, прибавляя безаммиачную
воду до 1 л. В 1 мл полученного раствора содержится 0,01 мг
азота или 0,0122 мг NH3. 13. Хлороплатинат калия (KaPtCU),
раствор. Приготовление. 2 г хлороплатината калия
растворяют в 300—400 мл дистиллированной воды, прибавляют 100 мл
концентрированной соляной кислоты и объем доводят в мерной
колбе до 1 л. 14. Хлористый кобальт (СоС2), раствор.
Приготовление. 12 г кристаллического шестиводного хлористого
кобальта (СоС12 • 6Н2О) растворяют в 250 мл
дистиллированной воды, прибавляют 100 мл концентрировашгой соляной
кислоты и объем доводят в мерной колбе до 1 л.
Приготовление постоянных эталонов. Для
шкалы из постоянных эталонов готовят 15 растворов, пользуясь
данными, приведенными в табл. 16. В мерную колбочку
вместимостью 50 мл вливают указанные в таблице приблизительные
объемы хлороплатината калия B г/л K2PtCl6) и хлористого
кобальта A2 г/л СоС12-6Н2О), объем раствора доводят до метки,
прибавляя безаммиачную воду. Полученный раствор имеет
окраску, которая совпадает с окраской реакционной жидкости,
когда на анализ было взято количество азота, указанное в
таблице. Эти растворы называют постоянными, потому что они не
изменяются в течение нескольких месяцев, если их хранят в
закрытых сосудах в темноте. Приготовленные постоянные эталоны
проверяют и корректируют по рабочим эталонным растворам
(см. ниже). Это необходимо потому, что точного совпадения
с данными, приведенными в таблице, получить невозможяо.
Окраска зависит от качества реактива Несслера, а оценка
интенсивности окраски — от применяемого освещения и
чувствительности зрения аналитика.
ТАБЛИЦА 16
Азот,
мг
0.000
0,002
0.004
0,007
0,010
Сведения для приготовления
KjPtCle
мл
1.2
2.8
4,7
5,9
7,7
СоС12,
мл
0
0
0,1
0,2
0,5
Азот,
мг
0,014
0.017
0,020
0.025
0,030
KjPtCl6
мл
9.9
II.4
12,7
15,0
17.3
постоянных эталонов
CoCIj,
мл
1,1
1.7
2,2
3,3
4.5
Азот,
мг
0.035
0.040
0.045
0,050
0.060
K2PtCl„
мл
19,0
19.7
19,9
20,0
20,0
СоС12
мл
5,7
7,1
8.7
10.4
15,0
18*
275
Построение калибровочной кривой. Серию
эталонных растворов готовят разведением основного раствора
хлористого аммония, который содержит 1 мг азота в 1 мл.
Разведение делают в мерных колбах вместимостью 50 мл, у
которых выше метки есть пространство не менее 2 мл. В каждую
колбочку вливают один из следующих объемов основного
раствора: 0; 0,2; 0,4; 0,7; 1,0; 1,4; 1,7; 2,0; 2,5; 3,0; 3,5; 4,0; 4,5; 5,0;
6,0 мл и добавляют безаммиачную воду до метки на колбе.
Приготовленные растворы содержат азота соответственно: 0,200,
400, 700, 1000, 1400, 1700, 2000, 2500, ..., 6000 мкг. К каждому
из приготовленных эталонных растворов приливают 1 мл
реактива Несслера, содержимое колбочек перемешивают и через
10 мин измеряют интенсивность появившейся окраски фотоэлек-
троколориметром. Оптическую плотность измеряют при синем
светофильтре (?i=400-M25 нм) в кювете, имеющей расстояние
между стенками 1 см. На основании полученных данных строят
кривую зависимости показаний прибора от содержания азота
в эталонных растворах, которую называют калибровочной
кривой.
Для анализа проб воды с очень низким содержанием
аммиачного азота аналогично готовят серию рабочих эталонов из
разбавленного основного раствора хлористого аммония, 1 мл
которого содержит 10 мкг азота. Приготовленные растворы
анализируют и строят калибровочную кривую, которой пользуются при
анализах.
Подготовка проб. 1. Пробу воды анализируют сразу после
отбора или консервируют, прибавляя 1—2 мл
концентрированной серной кислоты. Хранят пробу в холодильнике. 2. Перед
отгонкой аммиака в пробе определяют pH, содержание хлора
и кальция, наличие формальдегида и сульфитов, и количество
0,1 н. или 0,01 н. щелочи (кислоты), которое нужно добавить
к 500 мл пробы, чтобы ее pH стало равным 6—7, а после
добавления 25 мл буферного раствора pH стало равным 7,4 и не
изменилось при отгонке аммиака. 3. При содержании кальция
в пробе более 250 мг/л при отгонке аммиака вместо буферного
раствора прибавляют окись магния, количество которой
устанавливают предварительно. 4. Воду, содержащую хлор, перед
отгонкой аммиака дехлорируют, прибавляя на 1 мг остаточного
хлора 2 мл одного из растворов для дехлорирования. 5. Для
устранения влияния сульфидов в реакционную колбу с пробой
вносят углекислый свинец. 6. Формальдегид удаляют из пробы
перед отгонкой аммиака путем кипячения пробы при pH 2—3.
Ход анализа. Подготовка установки. В плоскодонную колбу
вместимостью 1 л вливают 500 мл дистиллированной воды,
10 мл буферного раствора и вносят кусочек пемзы для
равномерности кипения. Колбу ставят на включенную электроплитку
и присоединяют к холодильнику. Перегонку воды проводят для
очистки установки от следов аммиака. Периодически собирае-
276
мый конденсат проверяют на содержание аммиака с реактивом
Несслера. Когда исчезнут следы аммиака -в конденсате,
реакционную колбу отсоединяют, выливают из нее воду (пемзу
оставляют) и начинают анализ.
Отгонка аммиака. В реакционную колбу вливают
500 мл или адекватное количество пробы, доведенное до 500 мл
разбавлением безаммиачной водой. При содержании
аммиачного азота 0,05 мг и ниже на анализ берут 1 л пробы. В
анализируемую пробу вносят, если надо (устанавливают
предварительно), раствор для дехлорирования, углекислый свинец и
щелочь (или кислоту). Затем в колбу вливают 25 мл фосфатного
буферного раствора (или окись магния), быстро закрывают ее
пробкой, в которой вставлен холодильник, и ставят на
включенную электроплитку. Отгонку ведут со скоростью 6—10 мл/мин.
Конденсат собирают отдельными порциями по 50 мл (или
большими) в мерные цилиндры или в мерные колбочки, в которые
предварительно наливают 50 мл борной кислоты. Чтобы
поглощение аммиака было полным в приемнике конденсата должен
быть избыток борной кислоты. Отгоняют 300 мл конденсата,
потом подставляют новый приемник, отгонку продолжают еще
2—3 мин и собранный конденсат анализируют с реактивом
Несслера. Если обнаружено отсутствие аммиака, то перегонку
считают законченной.
Колориметрирование. Объем каждой порции собранного
конденсата в мерной колбе доводят до метки, прибавляя
безаммиачную воду. На анализ берут 50 мл конденсата, прибавляют
к нему 1 мл реактива Несслера и через 10 мин (или 30 мин) ко-
лориметрируют одним из трех нижеприведенных способов.
Колориметрирование на ФЭКе. При реакции
аммиака с реактивом Несслера в зависимости от концентрации
аммиака получают растворы, имеющие окраску от желтой до
коричневой с сильным светопоглощением в широком диапазоне
длин волн. Нужную длину волны света получают с помощью
светофильтра. Светопропускание зависит от ширины кюветы. При
содержании аммиака 0,005—0,06 мг в 50 мл отгона, который
берут на анализ, желтую окраску колориметрируют при длине
волны 400—425 нм в кювете шириной 5 см. При содержании
аммиака 0,02—0,25 мг пользуются кюветой шириной 1 см.
Красновато-коричневую окраску раствора, соответствующую
содержанию аммиака 0,5 мг и более в 50 мл отгона, колориметрируют
при длине волны в пределах 450—500 нм.
К 50 мл отгона прибавляют 1 мл реактива Несслера,
перемешивают и раствор оставляют в покое. Через 10 мин измеряют
оптическую плотность реакционной смеси при выбранной длине
волны и светопропускаемости на ФЭКе. По калибровочной
кривой находят, какому количеству азота (или NH3) в 50 мл
анализируемого раствора, соответствует найденная оптическая
плотность раствора.
277
Колориметрирование в цилиндрах Несслера.
На анализ берут объем разбавленного отгона, который
содержит не более 60 мкг азота, вливают его в цилиндр Несслера,
прибавляют безаммиачной воды до отметки 50 мл и 1 мл
реактива Несслера. Через 30 мин сравнивают полученную окраску
с окрасками постоянных эталонов. На белый лист бумаги
ставят цилиндр с пробой и такие же цилиндры с эталонами. Смотря
сверху вниз через слой жидкости, подбирают эталон, окраска
которого совпадает с окраской пробы. Если такого эталона нет,
то выбирают два эталона с большей и меньшей интенсивностью,
чем интенсивность окраски пробы, и берут среднее значение
концентрации азота.
Колориметрирование в цилиндрах Генера
(см. с. 282). В мерную колбу вместимостью 50 мл вливают
выбранный (от 1 до 50 мл) объем разбавленного конденсата,
добавляют безаммиачную воду до метки на колбе, прибавляют 1 мл
реактива Несслера и раствор выливают в цилиндр Генера.
Через 30 мин в другой цилиндр Генера наливают подходящий
постоянный эталон и просматривая слои жидкости в цилиндрах
сверху вниз, уравнивают окраски, изменяя высоту столба
жидкости в том или другом цилиндре с помощью крана внизу.
Когда достигнуто полное совпадение интенсивностей окрасок
в обоих цилиндрах, замечают высоту столба жидкости в
каждом цилиндре. Холостой опыт, в котором вместо анализируемой
пробы берут безаммиачную воду и добавляют все реактивы
в том же количестве, как описано выше, проводят для
определения аммиачного азота, содержащегося в реактивах.
Найденное количество азота учитывают при вычислении результатов
анализа.
Расчет. Содержание аммиачного азота в каждой анализируемой порции
конденсата (Хи Хг. .. Хп) вычисляют при работе на ФЭКе или с цилиндрами
Несслера по формуле
х аУ
при работе с цилиндрами Генера по формуле
aV *
у
п
VilOOO
где Хп—количество аммиачного азота в анализируемой п-й порции
конденсата, мг; а — количество аммиачного азота, найденное по калибровочной
кривой или по эталону, мкг; V—объем анализируемой порции конденсата, мл;
У] — объем конденсата, взятый на анализ, мл; h — высота столба эталона
в цилиндре, см; h\ — высота столба реакционной жидкости с пробой в
цилиндре, см.
Концентрацию аммиачного азота (или аммиака) в анализируемой пробе
вычисляют (в мг/л) по формуле
278
M+Xi+...+XJ-Л
g
где А — содержание аммиака в реактивах, найденное в холостом опыте, мг,
g — объем пробы, взятый для отгонки аммиака, мл; Xi, X2 .... Хп — количество
аммиачного азота в первой, второй л-й порции конденсата, мг.
Минимальное количество азота в 50 мл отгона, которое может быть
обнаружено, составляет 0,001 мг или 1 мкг. Содержание азота в пробе
определяют с точностью ±3—5% отн.
Предостережения. 1. Для получения более точных результатов при коло-
риметрировании калибровочные кривые надо строить на основании анализа
эталонных растворов, которые провести через все этапы анализа, включая
перегонку. 2. Содержание аммиака в безаммиачиой воде и фосфатном буфере
обычно бывает около 0,01 мг/л, поэтому при определении микроколичеств
аммиака в пробе результат следует записывать с точностью до сотых долей
мг/л. 3. Анализ надо проводить в комнате при полном отсутствии аммиака
в воздухе. 4. Присутствие следов магния в дистиллированной воде при
реакции с реактивом Несслера образует муть; этого можно избежать, добавив
немного сегнетовой соли к реакционной жидкости. 5. Ионообменные смолы
имеют предел насыщения, поэтому ионообменную колонку периодически надо
проверять, делая анализ воды до и после колонки с реактивом Несслера.
6. В колориметрических методах надо особое внимание уделять оптической
чистоте кювет и цилиндров. 7. Реактивы лучше хранить в посуде из стекла
пирекс.
Определение нитритов колориметрическим методом (метод Грисса)
Метод предназначен для определения нитрит-ионов ()
в воде из аэротенков. Метод основан на реакциях
получения азокраски малинового цвета. Сначала, действуя на
нитриты сульфаниловой кислотой в уксуснокислой среде,
получают диазосоединение: ШОзСб^Шг + ШОг + СНзСООН-)-
-^HSO3C6H4N = NCH3COO + 2H2O. Затем к реакционной
жидкости добавляют а-нафтиламин (соль ароматического
амина). Между диазосоединением и а-нафтиламином
происходит обменная реакция, в результате которой получается азо-
краска: ШОзСб^^МСНзСОО + СюНтЫНг^ШОзСб^^
= NCioH6NH2+CH3COOH. По интенсивности окраски
реакционной жидкости, которую измеряют фотоэлектроколориметром или
визуально, находят концентрацию нитрит-ионов в
анализируемой жидкости.
Мешающие вещества. Сильные окислители, восстановители
и алифатические амины, реагирующие с нитритами с
выделением азота. Микроорганизмы, превращающие нитриты в
нитраты и аммиак.
Аппаратура. Один из следующих приборов: фотоэлектро-
колориметр, цилиндры Генера или цилиндры Несслера
(см. рис. 70).
Реактивы и их приготовление. 1. Дистиллированная вода без
нитритов. Приготовление. В перегонную колбу вливают
1 л дистиллированной воды, 1 мл серной кислоты плотностью
1,84 г/см3, 2 мл 5%-ного раствора сернокислого марганца и Змл
0,04%-ного раствора марганцовокислого калия. Колбу ставят
на электроплитку, соединяют с холодильником и ведут
279
перегонку. Полученный конденсат не содержит нитрит-ионов, era
употребляют для приготовления всех реактивов и для
разбавления реакционной смеси и пробы. 2. Сульфаниловая кислота
ч. д. а. раствор в 5 н. уксусной кислоте. Приготовление.
В мерную колбу объемом 1 л вливают 600 мл воды без
нитритов и 285 мл ледяной уксусной кислоты, добавляют в него 8 г
сульфаниловой кислоты, раствор перемешивают до растворения
кислоты и, прибавляя воду, доводят его объем до метки на
колбе. Раствор перемешивают. 3. a-нафтиламин ч. д. а., раствор
в 5 н. уксусной кислоте. Приготовление. В мерную колбу
вместимостью 1 л вливают 600 мл воды без нитритов, 285 мл
ледяной уксусной кислоты, добавляют 5 г a-нафтиламина,
раствор перемешивают до полного растворения a-нафтиламина и,.
прибавляя воду, доводят его объем до метки на колбе. Если
раствор мутный, его фильтруют. 4. Ацетат натрия, 2 М раствор.
Приготовление. В мерную колбу вместимостью 100 мл
вносят 16,4 г ацетата натрия (NaC2H3O2-3H2O) и воду без
нитритов до метки на колбе. Раствор перемешивают и, если надо,
фильтруют. 5. Гидроокись алюминия, суспензия.
Приготовление. В колбу вместимостью 2 л помещают 125 г алюмока-
лиевых (или алюмоаммонийных) квасцов, вливают 1 л воды без
нитритов, раствор нагревают до 60сС и прибавляют к нему
медленно при помешивании 55 мл концентрированного аммиака.
Образовавшийся белый хлопьевидный осадок гидроокиси
алюминия отстаивают, декантацией сливают воду и суспензию
переносят в бутыль объемом 5—7 л прибавляя воду. Гидроокись
очищают от хлоридов, нитритов, нитратов и аммиака путем
многократных промывок большим количеством воды без нитритов.
Промывную воду отделяют декантацией после отстоя. 6. Нитрит
натрия (NaNO2) ч. д. а. основной стандартный раствор.
Приготовление. В мерную колбу вместимостью 1 л вносят 0,4929 г
нитрита натрия, прибавляют воду без нитритов до метки на
колбе и перемешивают. В 1 мл приготовленного раствора
содержится 0,1 мг азота или 0,3286 мг нитрит-иона (NO2). Раствор
неустойчив. Для его консервации прибавляют 5 мл хлороформа
и при хранении следят, чтобы в растворе всегда был хлороформ.
7. Нитрит натрия стандартный рабочий раствор.
Приготовление. Разбавляют основной стандартный раствор в 200 раз.
В 1 мл такого раствора содержится 0,0005 мг азота или
0,001 643 мг нитрит-иона (NO2).
Построение калибровочной кривой. Делают
серию опытов, в каждом из которых берут различные количества
миллилитров стандартного раствора нитрита натрия—1, 2,
3, .. и т. д. Анализ проводят, как описано ниже. Полученные
окрашенные растворы фотоколориметрируют и на основании
полученных данных строят две кривые зависимости оптической
плотности (показаний прибора): одна — от содержания в реак-
280
Ционной жидкости нитритного азота (мг N), другая — от
содержания нитрит-иона (мг NO2).
Подготовка проб. 1. Сразу при отборе пробы воды в нее
вливают концентрированную серную кислоту до получения pH 2—3,
чтобы прекратить жизнедеятельность микроорганизмов. Анализ
консервированной пробы должен быть сделан в течение суток
(не более). 2. Мутную воду или окрашенную осветляют или
обесцвечивают. В колбу вливают 200 мл анализируемой пробы,
прибавляют 4 мл густой суспензии гидроокиси алюминия, смесь
тщательно перемешивают и ставят на отстой. После
расслаивания суспензии осветленную и обесцвеченную воду фильтруют,
первую порцию отбрасывают, прозрачную воду анализируют.
3. Измеряют pH подготовленной пробы и путем титрования
отдельной порции воды с индикатором бромтимолом синим
устанавливают, сколько надо прибавить кислоты или щелочи к
определенному объему анализируемой воды, чтобы ее pH стало
6,5—7,5.
Ход анализа. Когда совершенно неизвестна концентрация
нитритов, делают сразу несколько анализов с различным
количеством пробы от 0,1 до 10 мл, а затем повторяют анализ с
наиболее подходящим количеством пробы, которое содержит от 0,1
до 10 мкг азота. В мерную колбу вместимостью 100 мл (или
цилиндр Несслера) вносят установленное количество воды,
50 мл дистиллированной воды без нитритов и 1 мл раствора
сульфаниловой кислоты. Содержимое колбы перемешивают и
дают постоять 3—10 мин. Величина pH такого раствора должна
быть около 1,4. Затем в колбу добавляют 1 мл раствора а-наф-
тиламина, 1 мл ацетата натрия и объем раствора доводят до
метки, прибавляя воду без нитритов. Такой раствор имеет pH
2—2,5. Колбу закрывают пробкой, хорошо перемешивают,
ставят в термостат при температуре 20°С и выдерживают в
течение 10—20 мин. Интенсивность появившейся окраски
реакционной жидкости измеряют на фотоэлектроколориметре с зеленым
светофильтром в кювете с расстоянием между стенками 5 см.
По калибровочной кривой находят количество нитритов во
взятом объеме пробы. При визуальном определении интенсивности
окраски в цилиндре Несслера проводят параллельно несколько
анализов рабочих стандартных растворов с различной
концентрацией нитритов. Сравнивая полученную окраску от
исследуемой воды с окрасками, полученными при анализе стандартных
растворов, находят стандартный раствор, окраска от которого
совпала с окраской от пробы. При определении интенсивности
окраски реакционной жидкости в цилиндрах Генера (см. с. 282)
параллельно проводят анализ одного-двух рабочих стандартных
растворов и находят высоту столба в одном цилиндре
реакционной смеси пробы, а в другом — реакционной смеси
стандартного раствора, при которых интенсивность окрасок обоих
жидкостей совпадает.
281
Расчет. Концентрацию нитритов в анализируемой пробе X в
миллиграммах иа лнтр (мг/л) при работе на ФЭКе и с цилиндрами Несслера вычисляют
ио формуле
аІООО
?1000 '
при работе с цилиндрами Генера вычисляют по формуле
v cIOOO-100 h
Л = ¦
?1000 hx '
ігде g — объем исследуемой воды, взятый на анализ, мл; а — количество
нитритов, найденное по калибровочной кривой или при сравнении со
стандартом, мкг; с — концентрация нитритов в стандартном растворе, мкг/мл; 100 —
¦объем реакционной смеси при анализе пробы, мл; h — высота столба
реакционной жидкости стандарта, см; hi— высота столба реакционной жидкости
анализируемой воды, см.
Минимальная определяемая концентрация нитритного азота в пробе —
0,01 мг/л. Воспроизводимость в среднем из трех определений ±0,002 мг/л.
Предостережение. На результат анализа заметно влияет даже
незначительное изменение температуры реагирующей жидкости во время анализа.
.Для получения более точных данных надо все реактивы и воду без нитритов
хранить в термостате при 20°С.
Определение нитратов (метод Грандваля и Ляжу)
Для определения нитратов в сточной воде на различных
стадиях ее очистки [5] предназначен метод, основанный на реакции
нитратов с фенолсульфокислотой. Образующаяся при этом
пикриновая кислота (нитропроизводное фенола), реагируя со
щелочью, дает соль, окрашенную в желтый цвет. Измеряя
интенсивность окраски раствора полученного соединения фотоэлек-
троколориметром или с помощью других приборов, находят
количество азота, связанного в нитраты.
Мешающие вещества. Хлориды, окрашенные соединения, ще-
.лочность выше 0,2 мг-экв/л, нитриты.
Аппаратура. Один из следующих приборов: ФЭК, цилиндры
Несслера или Генера.
Комплект цилиндров Генера (см. рис. 70) состоит
из двух цилиндров, сделанных из бесцветного стекла с
абсолютно плоским прозрачным дном. Внизу цилиндра имеется тубус
с краном для спуска жидкости. Диаметр цилиндра 3 см. Ци-
.линдр отградуирован по высоте на 100 делений. Для
определения концентрации вещества в исследуемой жидкости по ее
-окраске проводят сравнение окраски пробы с окраской
стандартного раствора. Берут два цилиндра, в один наливают
исследуемую жидкость, а в другой — стандартный раствор до
определенной отметки (например, до 100). Под цилиндром укрепляют на
штативе два зеркальца и поворачивают их так, чтобы луч света
был направлен перпендикулярно на дно цилиндра.
Просматривают окраски жидкостей в обоих цилиндрах сверху вниз. Если
окраска в одном цилиндре больше, чем в другом, то из него
выливают жидкость через кран маленькими порциями. Изменяя
282
таким способом высоту столба жидкости в обоих цилиндра
добиваются полного совпадения интенсивностей окрасок в обоих
цилиндрах и измеряют высоту жидкости в каждом цилиндре.
Для вычисления концентрации красящего вещества в пробе
пользуются равенством
ah = arh
c'lc>
где а и ас — концентрация красящего вещества в приборе и в
стандартном растворе; h и hc — высота столба жидкости в цилиндре
с пробой и со стандартным раствором.
Реактивы и их приготовление. Все реактивы готовят из
бесцветных химически чистых веществ и хранят в посуде из стекла
пирекс. 1. Фенолсульфокислота. Приготовление. В колбу
вместимостью 250 мл помещают 25 г бесцветного чистого
фенола, перегнанного при 181°С, и 150 мл серной кислоты
плотностью 1,84 г/см3. Колбу закрывают пробкой с обратным
холодильником, ставят на водяную баню и нагревают в течение 6 ч..
Приготовленную кислоту хранят в склянке из темного стекла
с притертой пробкой в темном месте. 2. Едкое кали, 10 н.
раствор E60 г/л) или 10%-ный раствор аммиака плотностью-
0,96 г/см3. 3. Суспензия гидроокиси алюминия.
Приготовление см. с. 280. 4. Сернокислое серебро, раствор 4,397 г/л. 1 мл-
этого раствора эквивалентен 1 мл хлора. 5. Перекись водорода,.
30%-ный раствор. 6. Нитрат калия (KNO3), основной раствор-
содержит 0,722 г перекристаллизованного безводного KNO3 в 1 л-
раствора, 1 мл этого раствора содержит 0,1 мг азота.
Разбавленный основной раствор 1 : 9 содержит 0,01 мг азота в 1 мл..
7. Эталон нитрата калия. Приготовление. 50 мл основного'
раствора, разбавленного или неразбавленного (в зависимости^
от содержания азота в анализируемой пробе), помещают в
фарфоровую чашку и выпаривают на водяной бане досуха. После
охлаждения остаток растирают с 2 мл фенолсульфокислоты
в течение 10 мин. Затем прибавляют 20 мл воды, 3 мл раствора
едкого кали, размешивают и количественно переносят в мерную-
колбу вместимостью 500 мл. Объем раствора доводят до метки
на колбе, прибавляя дистиллированную воду. Вычисляют
концентрацию азота, которая соответствует полученной окраске
раствора.
Приготовление калибровочной кривой.
Данные для построения калибровочной кривой получают, делая
анализы со следующими количествами разбавленного и
неразбавленного основного раствора нитрата калия: 0; 0,1; 0,3; 0,5^
0,7; 1,0; 1,5; 2,0; 3,5; 6,0; 10; 15; 20 и 30 мл. Интенсивность
окраски растворов определяют измерением их оптической
плотности на фотоэлектроколориметре при условиях колориметриро-
вания, которые применяют при анализе проб. Затем строят
кривую зависимости оптической плотности (показания прибора)
283,
реакционной жидкости от содержания азота (или NO3) в
анализируемом растворе.
Подготовка проб. Анализируемая вода обычно содержит
микроорганизмы, которые ведут биохимические процессы с
образованием нитратов и нитритов, поэтому пробу анализируют сразу
после отбора или консервируют, прибавляя серную кислоту до
pH 1 и сохраняя ее в холодильнике при температуре 3—5°С.
Перед анализом в пробе определяют щелочность, цветность,
содержание ионов хлора, нитрит-ионов и приблизительно
нитрат-ионов. Исследуемую воду, имеющую щелочность выше 5°С,
нейтрализуют 0,1 н. раствором серной кислоты по метиловому
оранжевому. При слабой окраске обесцвечивание не проводят.
Маскирующее действие окраски воды компенсируют при коло-
риметрировании. При цветности воды выше 10 единиц (см.
с. 292) ее обесцвечивают. К 100 мл пробы прибавляют 3 мл
суспензии, содержащей около 0,1 г гидроокиси алюминия, смесь
встряхивают в течение 15 мин и оставляют стоять для
расслаивания. Обесцвеченную воду отфильтровывают от осадка,
первую порцию отбрасывают.
Если вода содержит ионы хлора, то титрованием 10 мл пробы
раствором сернокислого серебра (Ag2SO4) устанавливают, какое
количество раствора Ag2SC>4 надо добавить в то количество
пробы, которое возьмут на анализ, чтобы осадить весь хлор.
При содержании в пробе нитритного азота более 1 мг/л нитрит-
ион окисляют в нитрат-ион. К 100 мл пробы прибавляют 0,5 мл
перекиси водорода и кипятят 10 мин. После окисления
нитратный азот определяют вместе с нитритным, а при расчете вводят
поправку на содержание нитритного азота. Приблизительный
результат анализа пробы на нитрат-ионы (NO3) используют
для установления объема пробы, который надо взять на анализ.
Оптимальное количество нитратного азота для анализа
находится в пределах 0,01—0,5 мг.
Ход анализа. Отмеренное количество пробы помещают в
фарфоровую чашку и выпаривают на водяной бане досуха. Если
проба содержит хлор, то его удаляют до выпаривания пробы.
К отмеренному количеству пробы, которое берут на анализ,
прибавляют раствор сернокислого серебра в количестве,
необходимом для осаждения всех ионов хлора (оно установлено
предварительным титрованием). Выпавший осадок отфильтровывают
и хорошо промывают.
Фильтраты и промывные воды количественно переносят в
фарфоровую чашку и выпаривают досуха. Чашку с осадком
охлаждают, вливают в нее 1 мл фенолсульфокислоты, осадок
тщательно растирают стеклянной палочкой в течение 5—10 мин,
вливают в чашку 20 мл дистиллированной воды и при
помешивании добавляют 5—7 мл раствора аммиака или едкого кали
до получения максимальной окраски раствора. Выпавшие
хлопья гидроокисей отфильтровывают, фильтрат и промывные
284
воды собирают в мерную колбу вместимостью 100 мл, объем
раствора доводят водой до метки на колбе, перемешивают и ко*
лориметрируют.
При работе на ФЭКе предварительно устанавливают
условия колориметрирования — ширину кюветы и светофильтр,
используя сведения, приведенные в табл. 17. Затем определяют
оптическую плотность исследуемого раствора и по
калибровочной кривой находят количество азота в пробе, взятой на анализ..
При колориметрировании в цилиндрах Несслера параллельна
с пробой готовят несколько эталонных растворов. В одинаковые
цилиндры Несслера наливают различные приготовленные
эталоны и исследуемый раствор, ставят их на белую бумагу и, про-
сматривая растворы сверху вниз, выбирают эталон, окраска
которого совпадает с окраской пробы.
При колориметрировании в цилиндрах Генера в один из них
наливают эталон, в другой — пробу. Луч света с помощью
зеркала направляют перпендикулярно на дно цилиндра и
просматривают слои жидкостей в цилиндрах сверху вниз. Изменяя
высоту столба жидкости в каждом цилиндре находят высоту
столба эталона и пробы, при которых их окраски совпадают..
Расчет. Концентрацию нитратного азота в анализируемой пробе при
колориметрировании на ФЭКе, в цилиндрах Несслера или Генера вычисляют
по формулам соответственно:
X-J*™—Л;
S
v cVhim .
Л *= -~ А*
где X — концентрация нитратного азота, мг/л; а — количество нитратного
азота в пробе, взятой на анализ, найденное по калибровочной кривой или по,
сравнению с эталоном, мг; g — объем пробы, взятый на анализ, мл; с —
концентрация нитратного азота в эталоне, мг/мл; V=100—объем реакционной
смеси при анализе пробы, мл; h — высота столба раствора эталона, см; h\ —
высота столба исследуемого раствора пробы, см; А — поправка на нитритный-
азот, содержащийся в пробе, мг/л.
При отсутствии в пробе мешающих веществ нитратный азот определяют
с точностью ±2—3% оти.
Определение азота органических соединений
Для определения азота, связанного в органических
соединениях, находящихся в сточной воде, предназначен метод,
основанный на определении азота после удаления из пробы аммиака,
нитритов и нитратов. Аммиак удаляют отгонкой при pH 7,4..
Нитриты и нитраты в кислой среде при нагревании в
присутствии солей двухвалентного железа превращают в окислы азота,,
которые удаляются во время минерализации пробы вместе
с другими газами. Азот, связанный органическими соединениями,
определяют после минерализации, как описано на с. 211.
Аппаратура. Установки для минерализации (см. рис. 65) и
для отгонки аммиака (см. рис. 36).
285..
Реактивы. 1. Фосфатный буферный раствор. Приготовление
см. на с. 274. 2. Сернокислое железо (FeSO4-7H2O)
кристаллическое. 3. Красная окись ртути (HgO) без азота. 4. Силика-
гель безводный. 5. Сульфат калия кристаллический. 6. Едкий
натр, 33%-ный раствор. 7. Гипосульфит натрия кристаллический.
8. Серная кислота плотностью 1,84 г/см3 и 0,1 н. раствор. 9.
Смешанный индикатор. Приготовление см. с. 211.
Ход анализа. Отгонка аммиака. В колбу Кьельдаля
вместимостью 0,5—1 л вливают 300—500 мл исследуемой воды,
добавляют 20—25 мл фосфатного буферного раствора, колбу
закрывают пробкой и включают в установку для отгонки
аммиака. Отгоняют 200 мл конденсата и проверяют отсутствие
аммиака в последней капле конденсата с реактивом Неослера
или по лакмусовой бумажке.
Удаление нитритов и нитратов при
минерализации. В реакционную жидкость после отгонки
аммиака прибавляют кристаллический сульфат железа из расчета
80 мг соли на 1 мг нитритного и нитратного азота, содержимое
колбы перемешивают и добавляют реактивы для
минерализации— 6 гранул силикагеля, 1,5 г окиси ртути, 12 г сернокислого
калия и 15—20 мл концентрированной серной кислоты. Колбу
укрепляют в наклонном положении в штативе и при слабом
нагревании держат до прекращения пенения. В это время
образуются и улетают окислы азота. Затем включают полный нагрев,
жидкость в колбе кипятят 15 мин до полного обесцвечивания.
На этом минерализация заканчивается. Далее анализ ведут,
как описано на с. 272 или 228.
Определение фурфурола колориметрическим методом
Метод предназначен для определения микроколичеств
фурфурола, который содержится в сточной воде, поступающей на
очистные сооружения. Метод основан на реакции фурфурола
с уксуснокислым анилином, в результате которой образуется
соединение красного цвета
С4Н3О • CHO-f 2С6Н5 • NH2-*C4H3O-CH = C6H4 • NH2+H2O.
Образовавшееся соединение на свету разлагается. Для
повышения устойчивости к нему прибавляют щавелевую кислоту,
двузамещенный фосфат натрия и хлорид натрия. Концентрацию
фурфурола в пробе находят по интенсивности окраски
полученного раствора, которую определяют фотоэлектроколориметром
с желто-зеленым фильтром. Реакция специфична. Метилфурфу-
рол и оксиметилфурфурол не мешают.
Мешающие вещества. Пентозы и уроновые кислоты при
отгонке фурфурола из кислой среды частично разрушаются с
образованием фурфурола.
286
Аппаратура. Фотоэлектроколориметр. Установка для отгонки
летучих веществ с водяным паром (см. рис. 47). Пробирки
вместимостью 20 мл с пробками на шлифах.
Реактивы и их приготовление. 1, Анилин свежеперегнанный.
Приготовление. В колбу Вюрца вместимостью 50 мл
помещают 10—15 мл анилина, соединяют ее на шлифах с
холодильником длиной 15—20 см, укрепляют в штативе над
электроплиткой на высоте 1 см, включают нагрев и осуществляют
перегонку. Приемником служит мерный цилиндр на 10 мл. Первую
порцию отгона в количестве 2—3 мл выливают, а последующие
5 мл отгона используют для анализа. Остаток в перегонной
колбе выливают, а установку сразу моют ацетоном или спиртом.
2. Уксусная кислота ледяная. 3. Щавелевая кислота, 5%-ный
раствор. 4. Двузамещенный фосфат натрия, 10%-ный раствор.
5. Хлористый натрий, 20%-ный раствор. 6. Стандартный раствор
фурфурола. Приготовление. 10—15 мл фурфурола
перегоняют под вакуумом на установке, собранной на шлифах.
Собирают фракцию, которая отгоняется при 150—160°С. Около
0,3 г свежеперегнанного фурфурола вливают в бюкс с
дистиллированной водой и взвешивают. Предварительно взвешивают
бюкс с водой. По разности находят количество фурфурола
в бюксе. Раствор количественно переносят в мерную колбу на
1 л, объем доводят до метки, прибавляя дистиллированную
воду. После хорошего перемешивания берут 1 мл
приготовленного раствора, выливают его в мерную колбу вместимостью
100 мл и, прибавляя воду, доводят объем до метки. Полученный
раствор — стандартный раствор фурфурола — содержит 3 мкг
(микрограмма) фурфурола в 1 мл.
Построение калибровочной кривой. Делают
несколько анализов с различным количеством фурфурола: 0,3;
0,6; 0,9; ...; 5 мл стандартного раствора. Каждую порцию
стандартного раствора разбавляют водой до 5 мл и анализируют,
как описано ниже. На основании полученных данных строят
кривую зависимости оптической плотности (показания прибора)
от количества фурфурола, взятого на анализ.
Подготовка проб. Пробу нейтрализуют до pH 6,5—7,0.
Ход анализа. Отгонка фурфурола. В колбу
вместимостью 250 мл вливают 100 мл анализируемой воды, закрывают
ее пробкой, в которую вставлены две трубки — одна от
холодильника, другая от паровика. Колбу ставят на электроплитку,
нагревают до 80—90°С и включают водяной пар (см. рис. 47).
Приемником фурфурольного конденсата служит мерная колба
вместимостью 250 мл. Во время отгонки конденсат, вытекающий из
холодильника, периодически проверяют на содержание форфу-
рола по бумажке, смоченной уксуснокислым анилином. В
присутствии фурфурола бумажка порозовеет. После окончания
отгонки объем собранного конденсата доводят водой до метки
на колбе и анализируют.
287
Определение фурфурола. В пробирку объемом20мл
с притертой пробкой вливают 1 мл свежеперегнанного анилина,
10 мл ледяной уксусной кислоты, 2 мл раствора хлористого
натрия, 1 мл раствора щавелевой кислоты, 1 мл раствора двузаме-
щенного фосфорнокислого натрия, перемешивая содержимое
пробирки после прибавления каждого раствора. Полученную
реакционную смесь охлаждают в водяной бане при 20°С. Затем
в пробирку вливают 5 мл фурфурольного конденсата, снова
ставят ее в водяную баню в темном месте и выдерживают при
20°С в течение 1 ч. Полученный окрашенный раствор колори-
метрируют. Раствор наливают в кювету с расстоянием между
стенками 2 см и измеряют оптическую плотность раствора при
длине волны 518 нм, применяя зеленый светофильтр № 4.
Во вторую такую же кювету для проведения холостого опыта
наливают отдельно приготовленную смесь всех используемых
реактивов и 5 мл дистиллированной воды.
Расчет. Концентрацию фурфурола в анализируемой воде вычисляют по
формуле
х ggl •
где X — концентрация фурфурола в пробе, мг/л; а — количество фурфурола
в 5 мл конденсата, найденное по калибровочной кривой, мг; V — объем
фурфурольного конденсата, мл; g— объем фурфурольного конденсата, взятый на
анализ, мл; gi — объем анализируемой воды, взятой для отгонки
фурфурола, мл.
Чувствительность реакции — 1 мкг фурфурола. Фурфурол определяют
с точностью ±3—5% отн.
Предостережения. 1. Анализ надо проводить в комнате, где в воздухе нет
фурфурола. 2. При колориметрировании необходимо обращать особое
внимание на чистоту кювет, они должны быть прозрачны, без жировых пятен от
рук. Хорошо вымытые кюветы споласкивают спиртом и сушат при комнатной
температуре. Кюветы нельзя сушить при нагревании. После заполнения
кюветы жидкостью внешняя поверхность ее должна оставаться сухой: Касаться
пальцами можно только торцевой стороны кюветы. 3. При отгонке фурфурола
из слабощелочной среды получают заниженные результаты анализа за счет
разложения фурфурола. Если в пробе содержатся пеитозы и уроновые
кислоты, то при отгонке фурфурола из кислой среды получают завышенные
результаты анализа за счет их разложения до фурфурола.
Определение сульфат-иоиа экспрессным методом
Метод предназначен для быстрого определения сульфат-
ионов в сточной воде на различных стадиях ее очистки. Метод
основан на реакции образования сернокислого бария,
количество которого определяют нефелометрически.
Мешающие вещества. Взвешенные и сильно окрашенные
вещества.
Аппаратура. Один из следующих приборов: мутномер (см.
рис. 71), нефелометр или набор колориметрических пробирок
диаметром 14—15 мл с отметкой 10 мл на одинаковой высоте.
Реактивы и их приготовление. 1. Хлористый барий 5%-ный
288
раствор, приготовленный на дистиллированной воде без
углекислоты. Хранят раствор в склянке с сифоном (см. рис. 16).
2. Соляная кислота разбавленная 1:5. 3. Основной эталонный
раствор сернокислого калия 0,9071 г/л. В 1 мл этого раствора
содержится 0,5 мг сульфат-иона. 4. Рабочий эталонный раствор
сернокислого калия — основной раствор, разбавленный в 10 раз.
В 1 мл этого раствора содержится 0,05 мг сульфат-иона. 5.
Дистиллированная вода, из которой удалена углекислота
кипячением в течение 2 ч.
Подготовка проб. Перед анализом пробу фильтруют.
Ход анализа. Берут семь колориметрических пробирок.
В одну из них наливают 10 мл исследуемой воды, а в остальных
готовят эталоны. В три пробирки вливают основной эталонный
раствор в количествах: 1,6; 3,2 и 6,4 мл. В другие три пробирки
вливают 2, 4 и 8 мл рабочего эталонного раствора. Объем
эталонных растворов в пробирках доводят до 10 мл (метка на
пробирке), добавляя дистиллированную воду без углекислоты.
Во все семь пробирок вливают по 0,5 мл соляной кислоты,
растворы перемешивают, прибавляют к каждому по 2 мл 5% -ного
хлористого бария и снова перемешивают. Растворы во всех
пробирках при этом становятся мутными из-за выпавшего осадка
сернокислого бария (BaSO4). Чтобы определить количество
BaSO4, которое выпало в исследуемой воде, проводят сравнение
мутности воды с мутностью эталонных растворов и находят
эталон, мутность которого равна или близка к мутности пробы.
Сравнение мутностей делают визуально в тех же пробирках или
пользуются прибором — нефелометром или мутномером.
Расчет. Концентрацию сульфат-иона в пробе вычисляют по формуле
cVIOOO
X.
S
где X— концентрация сульфат-иоиа в анализируемой воде, мг/л;
с—концентрация сульфат-иона в основном или рабочем эталонном растворе, взятом на
анализ, мг/мл; V—объем эталонного раствора, взятый на анализ, мл; g —
объем пробы, взятый на анализ, мл.
При визуальной оценке мутности раствора точность определения ±7-=-
-s-10% оти. При определении мутности нефелометром или мутномером точность
составляет ±3-н5°/о отн.
Определение мутности
Для определения концентрации взвешенных веществ в
бесцветной или слабоокрашенной воде после очистки или из
водоемов предназначен метод, основанный на измерении мутности
воды путем сравнения с мутностью эталонных растворов.
Измерение делают нефелометром или мутномером. При отсутствии
прибора мутность можно определять визуально в
колориметрических пробирках, но с меньшей точностью. Мутность выражают
количеством миллиграммов взвешенных веществ в 1 л пробы
(мг/л).
ТО Заказ № 142 289
Мешающие вещества. Красящие вещества.
Аппаратура. Нефелометр или мутномер. Набор
колориметрических пробирок из бесцветного стекла с плоским дном с
отметкой 100 мл на одинаковой высоте.
Мутномер Сант-Льюиса (рис.71) является
упрощенной моделью нефелометра [95]. Он основан на эффекте Тиндаля,
который заключается в том, что луч света, падающий на
жидкость под углом 45°, рассеивается каждой взвешенной частицей.
Сравнивая интенсивность света, рассеянного пробой и эталоном,
находят концентрацию взвешенных веществ в анализируемой
жидкости. Прибор смонтирован в деревянном ящике /,
покрытом изнутри матовой черной краской. Источником света служит
электрическая лампа 100 Вт на блоке регулировки ее
положения. Вертикальная
металлическая перегородка 2 внутри ящика
имеет световое окно высотой
50 мм, длиной 150 мм (ширина
ящика). Нижняя кромка окна
расположена на 76 мм выше
центра лампы. Вверху ящика
имеется полка 3 с двумя
отверстиями диаметром немного
меньшим, чем диаметр
колориметрических пробирок, в которых
делают измерение. Верхняя кромка
отверстий скошена для
правильной установки донышек
пробирок. Колориметрические пробирки
должны быть из бесцветного
стекла с плоским дном и иметь
отметки 100 мл на одинаковой высоте.
Положение лампы в приборе
должно быть отрегулировано так, чтобы конец нити был
направлен вверх, при этом плоскость нити должна находиться
от перегородки на расстоянии 49 мм. При измерениях свет от
лампы проходит через окно в перегородке, а затем вверх по
диагонали через нижнюю часть пробирок. В глаз наблюдателя
попадает только свет, отраженный взвешенными частицами.
Реактивы и их приготовление. 1. Основной эталонный
раствор, имеющий мутность 100 мг/л. 2. Рабочие эталонные
растворы различной концентрации 0,05; 0,08; 0,1; 0,2; 0,4 и т. д.
до 2 мг/л. Приготовление эталонов. В качестве
диспергированного вещества можно применять каолин, инфузорную
землю, фуллерову глину и другие подобные вещества. Перед
приготовлением суспензии каолин растирают в ступке и
просеивают через шелковое сито с отверстиями 0,1 мм. В колбу
вместимостью 5 л помещают 25—30 г просеянного порошка,
вливают 3 л дистиллированной воды, очень хорошо перемеши-
290
Рис. 71. Мутномер Сант-Льюиса
вают и оставляют стоять на сутки. После отстоя отбирают
сифоном примерно 1,5—2 л жидкости, стараясь не взмучивать
осадок. Отобранную жидкость сливают в бутыль, а к оставшемуся
осадку приливают 2 л воды, хорошо взбалтывают и оставляют
стоять на сутки. Эту операцию извлечения частиц, не
оседающих в течение суток, повторяют 3—5 раз. Собранную суспензию
сливают в одну бутыль и оставляют на 3 суток, чтобы отделить
фракцию самых мелких частиц. Через 3 суток жидкость над
осадком удаляют с помощью сифона, а осадок, состоящий из
частиц, которые оседают за 3 суток (но не оседают за 1 сутки
отстоя!), используют для приготовления основного эталонного
раствора.
Сначала готовят мутный раствор, смешивая осадок с 10—
20 кратным объемом воды. Полученную суспензию анализируют
на содержание взвешенных веществ весовым методом, причем
количеством взвешенных веществ считают количество
прокаленного осадка, полученного после фильтрации мутного раствора
через беззольный фильтр. Из суспензии известной
концентрации готовят основной эталонный раствор, добавляя к нему воду
нулевой мутности. Количество мутного раствора (суспензии)
известной концентрации, которое нужно взять для
приготовления определенного объема раствора заданной концентрации,
вычисляют по формуле
Х= , где X — требуемый объем мутного раствора, концентрация
которого известна, мл; V — заданный объем приготовляемого эталона, мл; С —
заданная мутность пряготовляемого эталона, мг/л; с — концентрация
взвешенных веществ в исходном растворе, мг/л.
Если исходный мутный раствор, например, имеет концентрацию 220 мг/л,
то для приготовления 500 мл основного эталонного раствора, имеющего кон-
500 • 100
центрацию 100 мг/л, надо взять Х= —нд^— =227,3 мл исходного
раствора, количественно перенести их в мерную колбу вместимостью 500 мл и объем
раствора довести до метки на колбе, прибавляя воду нулевой мутности.
Концентрацию приготовленного основного эталонного
раствора проверяют весовым методом и готовят из него рабочие
эталоны нужных концентраций. Для разведения основного
эталона пользуются водой нулевой мутности, а его количество
нужное для приготовления эталона заданной концентрации
вычисляют по вышеприведенной формуле. Для сохранения эталонов
в течение нескольких месяцев их консервируют, прибавляя 1 мл
насыщенного раствора сулемы на 1 л эталона. Эталоны,
имеющие концентрацию ниже 0,2 мг/л, неустойчивы даже при
консервировании, поэтому их заменяют каждую декаду.
Подготовка проб. Анализируемую воду, имеющую мутность
более 2 мг/л, разбавляют водой нулевой мутности.
Ход анализа. Испытуемую воду тщательно перемешивают
взбалтыванием, дают постоять 3—5 мин для удаления пузырь-
19* 291
ков воздуха и наливают в измерительную ячейку прибора. При
использовании нефелометра измерительной ячейкой служит
кювета, ее устанавливают в приборе и измеряют мутность, как
указано в инструкции к нефелометру. При работе с мутномером
пробу наливают в колориметрическую пробирку до отметки
100 мл. Визуально оценивают степень мутности и подбирают
подходящий эталон, который после энергичного взбалтывания
наливают в другую колориметрическую пробирку до отметки
100 мл. При анализе окрашенной воды, имеющей цветность выше
30° платиново-кобальтовой шкалы, отверстие колориметрической
пробирки с эталоном закрывают компенсационным фильтром.
Так же поступают при измерении нефелометром. Обе пробирки
вставляют в отверстия на полочке прибора, ожидают 5—10 мин,
чтобы удалились пузырьки воздуха, включают лампу прибора
и сравнивают мутность пробы с эталоном. Если мутность пробы
меньше мутности взятого эталона, то определение повторяют
с эталоном меньшей мутности, и наоборот. Меняя эталоны в
колориметрической пробирке, находят эталон, который имеет
мутность равную мутности пробы.
Расчет. Если нет эталона, с мутностью которого совпала бы мутность
пробы, то вычисляют среднее значение мутности двух эталонов, обладающих
большей и меньшей мутностью. Полученный результат, выраженный в
миллиграммах на литр, считают мутностью анализируемой пробы. Для
получения более точных данных готовят рабочий эталон с вычисленной мутностью
и с ним сравнивают пробы в приборе. Если анализируют разбавленную пробу,
то найденную мутность, т. е. концентрацию взвешенных веществ, умножают
на разведение.
Когда состав взвешенных веществ в эталонах почти совпадает с составом
и степенью дисперсности взвешенных веществ в анализируемой пробе, точность
определения мутиости составляет ±0,01 мг/л.
Определение цветности
Истинную цветность воды определяют методом сравнения
окраски испытуемой воды с окраской эталонов шкалы цветности
с помощью фотоэлектроколориметра или визуально. Цвет воды
обусловлен находящимися в ней растворенными,
коллоидно-растворенными и взвешенными веществами. Окраску
растворенными и коллоидно-растворенными веществами называют
истинной цветностью. Цветность воды в присутствии взвешенных
веществ называют кажущейся. Интенсивность окраски воды
зависит не только от количества растворенных и
коллоидно-растворенных веществ, но также от красящей способности
растворенных веществ и от размеров мицелл коллоидно-растворенных
веществ, поэтому ее оценивают в градусах цветности по
платиново-кобальтовой шкале или по хромово-кобальтовой шкале.
Мешающие вещества. Взвешенные вещества.
Аппаратура. Фотоэлектроколориметр. Набор цилиндров Нес-
слера объемом 100 мл, одинаковые по высоте и диаметру.
Реактивы и их приготовление. Реактивы для платиново-ко-
292
бальтовой шкалы цветности. 1. Хлороплатинат калия ()
кристаллический. 2. Кобальт хлористый (СОСЬ • 6Н2О)
кристаллический. 3. Соляная кислота. Приготовление шкалы.
В дистиллированной воде растворяют 1,245 г хлороплатината
калия и 1,01 г хлористого кобальта. Раствор количественно
переносят в мерную колбу вместимостью 1 л, прибавляют 100 мл
концентрированной соляной кислоты и объем доводят до метки,
прибавляя дистиллированную воду нулевой цветности.
Полученный основной раствор имеет цветность 500°. Рабочие эталонные
растворы для шкалы готовят разбавлением основного раствора
водой нулевой цветности. Объем основного эталона X, который
нужно взять для приготовления 100 мл рабочего эталона
заданной концентрации, вычисляют по формуле
где с — заданная цветность приготовляемого эталона в
градусах цветности.
Вычисленный объем основного эталона отмеривают
пипеткой, выливают в цилиндр Несслера и добавляют воду нулевой
цветности до отметки 100 мл. Цилиндр закрывают стеклянной
пробкой, раствор перемешивают и ставят в штатив шкалы.
Аналогично готовят серию эталонов различной цветности в
пределах от 1 до 80°, которой пользуются для анализа.
Если нет реактивов для приготовления платиново-кобаль-
товой шкалы, то готовят хромово-кобальтовую шкалу.
Реактивы. 1. Двухромовокислый калий (К2СГ2О7). 2. Кобальт
сернокислый (CoS04). 3. Серная кислота плотностью 1,84 г/см3
х. ч. Приготовление шкалы. Готовят два раствора.
Раствор № 1 (основной раствор). В 100 мл бесцветной воды
растворяют 0,0875 г двухромовокислого калия. В других 100 мл воды
растворяют 2,00 г сернокислого кобальта. Оба раствора
количественно переносят в мерную колбу вместимостью 1 л,
прибавляют 1 мл серной кислоты и объем доводят до метки, прибавляя
дистиллированную воду нулевой цветности. Полученный раствор
после перемешивания имеет цветность 500°. Раствор № 2. 1 мл
серной кислоты разбавляют бесцветной водой до 1 л. Этот
раствор употребляют для разбавления раствора № 1 при
приготовлении растворов рабочих эталонов, которые готовят так же как
эталоны платиново-кобальтовой шкалы.
Приготовление калибровочной кривой.
Готовят серию эталонов, имеющих различную цветность в пределах
от 0 до 80°. На фотоэлектроколориметре определяют их
оптическую плотность в кювете с расстоянием между стенками 5 мм
(или другим). Светофильтр подбирают в зависимости от
видимой окраски раствора, пользуясь данными, приведенными
в табл. 17. На основе полученных результатов анализов строят
кривую зависимости оптической плотности (показание прибора)
293
ТАБЛИЦА IT
Цвет светофильтра для растворов с различной видимой окраской
Цвет раствора
Фиолетовый
Синий
Голубой
Зеленый
Цвет светофильтра
Оранжевый,
зеленый,
желто-зеленый
Желтый, красный
Светло-желтый,
оранжевый
Красный,
оранжевый, пурпурный
Цвет раствора
Желто-зеленый
Желтый
Оранжевый
Красный
Цвет светофильтра
Фиолетовый
Синий,
фиолетовый
Зеленый, голубой,.
фиолетовый
Синий, зеленый
от цветности раствора, выраженную в градусах цветности по
платиново-кобальтовой шкале.
Подготовка проб. Мутную исследуемую воду центрифугируют
в течение 1 ч до полного удаления взвешенных веществ. Пробу,
имеющую цветность выше 80°, разбавляют водой нулевой
мутности.
Ход анализа. При визуальном определении цветности 100 мл
исследуемой воды наливают в цилиндр Несслера одинаковый
по размерам с цилиндрами эталонов шкалы цветности. Цилиндр
ставят иа лист белой бумаги, рядом с ним ставят цилиндр
с эталоном. Просматривая воду сверху вниз, подбирают эталон,
окраска которого совпадает -с окраской исследуемой воды. При
определении цветности на фотоэлектроколориметре исследуемую-
воду наливают в кювету подобранной ширины и измеряют
оптическую плотность при выбранном светофильтре.
Расчет. Результат анализа выражают в градусах цветности с указанием
цвета раствора. При визуальном определении цветность исследуемой воды
равна цветности эталона, с окраской которого она совпала. Если проба была
разбавлена перед анализом, то ее цветность будет равна произведению
цветности эталона на разведение. При работе на ФЭКе цветность находят по
калибровочной кривой, учитывая разбавление пробы перед анализом.
Цветность определяют с точностью ±5—-20% отн.
Предостережение. Эталонные растворы надо хранить в темном месте.
Срок хранения 2—3 месяца. Основной эталон платиново-кобальтовын шкалы
сохраняется в течение 1 года.
Определение порогового числа цветности
Кратность разбавления, при которой достигается почти
полное исчезновение окраски исследуемой воды, есть пороговое
число. Пороговое число цветности очищенной воды перед
спуском в водоем определяют методом разбавления окрашенной
воды бесцветной водой до получения бесцветной разбавленной
пробы. Пороговое число показывает, во сколько раз надо
разбавить пробу, чтобы она стала бесцветной.
294
Мешающие вещества. Взвешенные вещества.
Аппаратура. Набор цилиндров Несслера вместимостью 50 мл.
Ход анализа. В цилиндры Несслера наливают различное
количество испытуемой воды 1, 2, 5, 10 и т. д. мл, добавляют бес-
дветную воду до 50 мл, перемешивают и сравнивают получен-
дые окраски с водой нулевой цветности, налитой в такой же
дилиндр. Для более точного определения степени разбавления
готовят еще два-три разбавления близкие к найденному.
Расчет. Пороговое число цветности п испытуемой воды вычисляют по
-формуле
V
П
хде V — объем разбавленной воды, мл; g — количество пробы, взятое на
разбавление, мл.
Определение запаха
Для определения запаха воды до и после ее очистки, а также
запаха воды из водоемов предназначен метод, основанный на
органолептическом определении характера запаха и его
интенсивности. Приблизительно интенсивность запаха оценивают по
пятибалльной шкале, а более точно — по пороговому числу.
Запах воды обусловлен присутствием пахнущих веществ,
источниками которых являются: вещества, содержащиеся в
канализационных стоках и промышленных отходах; вещества,
выделяемые некоторыми видами микроорганизмов — плесенью,
слизистыми, планктоном (свободноплавающие) и др.
Микроорганизмы выделяют ничтожно малые количества летучих
эфирных масел, которые сообщают воде различные неприятные
запахи. Определить характер запаха и дать ему количественную
оценку можно только органолептически. Часто характер запаха
вскрывает источник загрязнений или распространенность
определенных микроорганизмов. На основании данных о запахе
можно выбрать способ очистки.
Ход анализа. Определение характера запаха. Пробу воды
отбирают в чистую колбу без запаха с притертой пробкой,
заполняя ее на 2/з объема. Воду в колбе нагревают или
охлаждают до 15—20°С, встряхивают, открывают пробку и носом
делают легкое втягивание воздуха из колбы. Для оценки
характера запаха естественного происхождения — от почвы и от
живущих и умерших в воде организмов и растений — пользуются
данными, которые приняты в стандарте СССР и в
Международном стандарте (М. ст.) на питьевую воду [91]. Характер
запаха от промышленных стоков и от реагентов, применяемых для
очистки воды, называют по веществам, которыми пахнет вода.
Например, фенольный, хлорный и т. д.
В табл. 18 приведены характеристики наиболее часто
встречающихся запахов.
295
ТАБЛИЦА 18
Характеристика запахов естественного происхождения
Название запаха
Огуречный
Болотный
Древесный
Плесневелый
Навозный
Торфяной
Символ
СССР
Б
Д
П
М. ст.
Ас
М
дР
Ер
Характеристика
Огуречный, например,
S у nur а
Запах ила, тины
Запах мокрой древесины,
коры
Затхлый, застойный
Например, Anabaena
Запах горфа
Название запаха
Бальзамический
Гнилостный
Землистый
Рыбный
Травянистый
Растительный
Символ
СССР
Г
3
Р
Т
М. ст.
В
Ds
Е
Df
0
V
Характеристика
Запах герани, фиалки,
ваннли
Фекальный, запах
застоявшихся сточных вод
Прелый, гнилостный,
запах свежевспаханнои
земли
Запах рыбы, например,
Uroglenopsis, Dinobryon
Запах травы, сена
Запах корнеплодов
Оценку интенсивности запаха в баллах делают на основании
данных, приведенных в табл. 19.
ТАБЛИЦА 19
Оценка интенсивности запаха воды в баллах
Балл
0
I
2
3
4
5
Интенсивность
Нет
Очень слабый
Слабый
Заметный
Отчетливый
Очень сильный
Описательное определение
Запах не ощущается
Запах, который не может быть обнаружен
в обычных условиях, но обнаруживается
в лаборатории опытным аналитиком
Запах, не привлекающий внимание
потребителя, но обнаруживается, если обратить
на него внимание потребителя
Запах, который легко обнаруживается
Запах, привлекающий к себе внимание и
делающий воду неприятной для питья
Запах такой интенсивности, что вода
совершенно непригодна для питья
Результат анализа записывают, указывая интенсивность
цифрой баллов, а характер запаха символом. Например, запах
пробы 3 Д. Это значит — древесный запах легко
обнаруживаемый.
Предостережение. 1. Пробы воды для определения ее запаха надо
отбирать при минимально возможной аэрации, иначе запах может исчезнуть.
2. Определение запаха сероводорода надо делать сразу после взятия пробы,
потому что сероводород улетучивается или видоизменяется, вступая в
реакцию с кислородом, растворенным в воде, и с кислородом воздуха. 3. Воздух
в помещении, где определяется запах, должен быть абсолютно без запаха.
4. Сотрудник, производящий определение, не должен курить н принимать пищу
за 30 мин до анализа. Его одежда не должна пахнуть. 5. Чувство обоняния
индивидуально и подвержено усталости. Способность обнаруживать легкие
запахи слабеет и пропадает, если обонянием пользуются длительное время,
а также после воздействия сильного запаха. Определение запаха одним
сотрудником не должно быть длительным. 6. Запах воды, которую обрабатывают
пахнущими реагентами (например, хлорированием), определяют через 30 мин
после обработки.
Определение порогового числа запаха
Для решения вопроса о возможности сброса в водоем воды
после очистки или о необходимости дополнительной обработки
воды для удаления запаха, которая обычно является
дорогостоящей, надо иметь точные данные о степени загрязненности
воды пахнущими веществами. Оценка интенсивности запаха
в баллах в этих случаях недостаточна, поэтому используют
метод, основанный на разбавлении пахнущей воды водой,
свободной от запаха, до тех пор, пока запах разводимой воды будет
еле заметным. Кратность разведения, при которой достигается
почти полная потеря запаха исследуемой воды, называют
пороговым числом. Оно показывает, во сколько раз надо разбавить
297
анализируемую пахнущую воду водой без запаха, чтобы
разведенная вода обладала еле заметным запахом.
Аппаратура. Установка для получения воды без запаха.
Водяная баня. Колба вместимостью 500 мл с притертой
пробкой. У ста но в к а для получения воды без запаха.
Удаление запаха из воды основано на адсорбции пахнущих
веществ активированным углем. Установка состоит из бутыли
вместимостью 5 л, которая заполнена гранулированным
активированным углем D . ю меш) • ^а дн0 бутыли положен слой
крупного гравия высотой 2,5 см. Бутыль закрыта резиновой
пробкой с двумя стеклянными трубками. Через трубку, конец
которой доходит до дна, впускают воду, имеющую запах, а
через другую трубку, конец которой находится над углем,
отбирают воду без запаха.
Реактивы. Вода без запаха Приготовление. Со
скоростью 1 л/мин водопроводную воду пропускают через установку
для получения воды свободной от запаха.
Ход анализа. Анализируемую воду в колбе, закрытой
стеклянной пробкой, нагревают до 40°С и определяют интенсивность
запаха как описано на с. 295, оценивая ее по пятибалльной
шкале. Если запах едва различим, т. е. он имеет 1 балл
интенсивности, то его пороговое число будет равно 1. Если
интенсивность запаха воды оценивается более высоким баллом, то
сначала определяют приблизительное значение порогового числа,
а затем точное. Для определения приблизительного порогового
числа готовят разведения пахнущей воды в 2, 10, 20 раз водой
без запаха. В три конические колбы вместимостью 500 мл с
притертыми пробками вливают отмеренные цилиндром 100, 20,
10 мл анализируемой воды и в каждую добавляют воду без
запаха в количестве соответственно 100, 180 и 190 мл, чтобы объем
разбавленной воды был 200 мл. Колбы закрывают пробками.
Еще в одну колбу наливают 200 мл воды без запаха. Все
четыре колбы нагревают в термостате до 40°С и проводят
определение. Первой берут колбу с водой без запаха, взбалтывают ее
содержимое, открывают пробку и вдыхают воздух из колбы.
Следующей берут колбу с водой наибольшего разведения, т. е.
ту, в которую влито 10 мл анализируемой воды и 190 мл воды
без запаха. Если в этой воде будет обнаружен запах, который
оценивается выше 1 балла, то другие колбы с разведенной
водой можно не испытывать. В этом случае готовят новые
разведения в 50, 100, 200 раз, в которых определяют интенсивность
запаха по пятибалльной шкале.
Когда будет найдено приблизительное пороговое число, т. е.
разведение, при котором исчезнет запах анализируемой воды,
готовят серию из пяти разбавлений, начиная от разбавления,
имеющего определенный, но слабый запах, что соответствует
интенсивности в 2—3 балла, и кончая разбавлением, при кото-
298
ром запаха нет. Приготовленную разбавленную воду
анализируют так же, как описано выше, и находят то минимальное
разбавление, при котором интенсивность запаха разбавленной воды
оценивается не выше 1 балла. Цифровое выражение
разбавления и есть пороговое число.
Определение качества активного ила
Активный ил — биологический окислитель — применяют для
окисления органических и неорганических веществ при очистке
сточных вод [96].
Активный ил представляет собой желеобразную массу
различной формы и консистенции, в которую вкраплены
бактериальные клетки шаровидной и палочковидной формы. Состав
микроорганизмов активного ила очень разнообразен. В нем
встречаются от 15 до 35 видов представителей таких
систематических групп, как коловратки, черви, грибы, сидячие
инфузории и брюхоресничные, нитчатые бактерии, простейшие —
бесцветные флагеляты и др. Некоторые виды микроорганизмов,
присутствующих в активном иле, обладают характерными
морфологическими признаками. Они служат индикаторными
микроорганизмами, по состоянию которых определяют недостатки и
нарушения режима работы очистных сооружений, не делая
анализа воды.
Для определения качества активного ила и установления
причин, вызвавших ухудшение его работоспособности, делают
несколько анализов. Определяют внешний вид хлопка, время
и полноту его осаждения и скорость уплотнения осадка. Микро-
скопированием при малом увеличении (в 120 раз)
устанавливают групповой состав микроорганизмов ила (коловратки,
бактерии, инфузории, дрожжи, грибы, черви, простейшие и др.),
наличие волокон целлюлозы и плотность хлопков ила. Микро-
скопированием при большом увеличении (в 600 раз)
устанавливают наличие индикаторных микроорганизмов в иле и дают
характеристику их состояния по свойственным им признакам.
Бактерии и простейших характеризуют по форме, подвижности,
наличию зооглей, бактерий и микроколоний. Грибы
характеризуют ветвистостью, толщиной нитей, наличием перегородок и
включений. Дрожжи характеризуют формой клеток, наличием
вакуолей, зернистым строением протоплазмы и способом
размножения клеток — почкованием или делением. Для оценки
качества ила результаты наблюдений сравнивают с данными,
приведенными в табл. 20.
Ход анализа. В градуированный цилиндр наливают 100 мл
иловой воды, отобранной из аэротенков, или из других мест
очистных сооружений, замечают время и наблюдают за
осаждением хлопков активного ила. Внешний вид хлопков
характеризуют следующими признаками: размером (крупный, мелкий);
299
ТАБЛИЦА 20
Сведения для оценки качества активиого ила
Активный ил
Вид хлопка и воды
Состав и состояние микроорганизмов
активного ила
Хороший ил,
удовлетворительно
работающий в аэро-
тенках
Ил из
регенератора при
удовлетворительной
регенерации
Ил из
регенератора при глубокой
регенерации
Голодающий ил
Нитрифицирующий ил
Плотный
компактный хлопок. Ил
быстро осаждается
в виде крупных
тяжелых хлопков.
Вода над илом
прозрачная
Хлопок крупный,
хорошо
осаждается. Вода над
илом прозрачная
Хлопки ила
распадаются. В воде
неоседающая муть
Хлопки ила
прозрачные. Вода
содержит неосе-
дающую муть
Хлопок рыхлый
после осаждения
всплывает. Вода
над илом
непрозрачная
Большое разнообразие
простейших по видовому составу
при небольшом количественном
преобладании какого-либо из
видов. Редко встречаются
следующие виды: Zionotis, Ro-
dophrya, Vorticella microstoma,
жгутиковые и мелкие амебы.
Всегда присутствуют Aspidissa,
Pamphagus, Zooglea. Все
микроорганизмы подвижны
Количественное преобладание
Peritricha (Carchesium,
Vorticella convallaria, Opercullaria)
над плавающими инфузориями.
Количество организмов
Peritricha и Zooglea больше, чем
в иле из аэротенков. Все
организмы подвижны
Преобладание крупных
свободноплавающих инфузорий.
Vorticella и Opercullaria
увеличенных размеров
Простейшие постепенно
мельчают, становятся прозрачными,
пищеварительные вакуоли
исчезают. Инфузории и коловратки
постепенно превращаются в
цисты. В клетках нитчатых
серобактерий исчезает сера. Zooglea
становятся прозрачными.
Всегда присутствуют в
заметном количестве коловратки
Callidina, Rotaria и др.
Количественное преобладание
Peritricha (Vorticella convalaria,
Carchesium Arcella) и крупных
амеб. Сильно развиты Zooglea
ramigera. Иногда присутствуют
малощетинковые черви Acla-
soma. Совершенно отсутствуют
Chilodon, мелкие амебы и
бесцветные жгутиковые
300
Активный ил
Вид хлопка и воды
Состав и состояние микроорганизмов
активного ила
Перегруженный
ил
Хлопкн ила
темные, плотные. Ьо-
да с опалесцен-
цней
Ил при
недостатке кислорода
Хлопки ила
распадаются на более
мелкие. Вода
мутная
Большое количество
бесцветных жгутиковых, мелких амеб
н мелких инфузорий.
Небольшое разнообразие видов
микроорганизмов, преобладают
количественно 2—3 вида.
Иногда присутствуют в заметных
количествах Podophria, Chilo-
don, Nematodes, Vorticella mi-
crostoma, Opercularia, нитчатые
бактерии Cladotrix и Beggiatoa
Большое количество
разнообразных жгутиковых, из ннфу-
зорнй господствуют Рагатае-
cium caudatum, которая
обнаруживает большую
выносливость к недостатку кислорода
н способность оживленно
плавать в гниющем нле.
Неподвижные коловратки, застывшие
в вытянутом состоянии или
отмирающие OpercuIIaria, мелкие
неподвижные с замкнутым
ресничным диском. Особи
Vorticella, раздутые в виде шаров,
которые через некоторое время
лопаются н исчезают
цветом (темный, светлый или прозрачный), прочностью —
компактный, плотный, рыхлый или распадающийся на мелкие
хлопки). Через 15 мин отстоя определяют степень осаждения
ила (полное или частичное) и прозрачность воды над ними
(прозрачная, мутная или легкая опалесценция.) За следующие
15 мин отстоя определяют степень уплотнения осадка, т. е., как
изменяется объем ила при стоянии. Хороший ил полностью
осаждается за 15 мин, вода над ним прозрачная.
Микроскопирование при малом увеличении.
Для просмотра структуры хлопков ила препарат готовят на
предметном стекле с углублением, чтобы не раздавить хлопок.
При увеличении в 120 раз просматривают несколько полей
зрения, устанавливают, какие микроорганизмы встречаются, какие
из них преобладают, каков их внешний вид, подвижность и т. д.
(рис. 72). На основании полученных данных, используя
сведения, приведенные в табл. 20, делают заключение о
работоспособности анализируемого ила.
Если в активном иле обнаружены целлюлозные волокна, то
определяют их содержание подсчетом в камере Тома (см. с. 153).
301
GladothrLx
dichotoma
„„_„.„„ Oxytricha Stylonlchia Olkomonqs
нитчатые бактерии, рвіНопвііа, pustulata, mutabilis^
толщина нитей J-штн дл.вО-ЮОмкм дл.Ш0-220мкм дл.17мкм
PhilodLna RotatrLa
megalotrocha, macrocerLa,
д/і.0,11-0,27нм д/і.0,о8.-(Шпм
Opercularia
gromerata,
дл. 4-50МКМ
Coleps
unclnatus,
дл. SOmk/i
Podophrya fixa,
дл Ю-28МКМ
AspLdisca uostata,
дл. ЗО-^О
Opercularia coarctata,
ал. бОмкм
ClnetoQhilum Amoeba Umax
'-'On nargantaceum
Amoeba proteus оиамШ-ПООмкм дл.35-4-5нм
Рис. 72. Простейшие активного ила
MorUcellg
GOnvallarLa,
дл60-120пт
Vorticella
microstoma,
дл.бО-110мкм
Zooqlea Tokophrya
ramlgera quactrcpartda,
Aeolosoma
tenebrarum
TrLchophrya
EpLstLUs
дл. 240 MKM
ThlotfirLx nivea
серобактерии, _
толщина нитей
J-4 мкм
Synura ubetla.
duan 70MKM
?egglatoa alba.
Bodo qlobosus а
a-?glo6osus, o-?. edax,
в-?. caudatus
SphaerotLlus
natans
Ряс 72.
ArceUa vulgarLs,
диам.48-150мкн
Наличие 5—15 тысяч целлюлозных волокон в 1 г абсолютно
сухого активного ила считают нормальным при очистке сточных
вод, содержащих 50—75 тысяч волокон в 1 л. Повышение
содержания целлюлозных волокон в активном иле свидетельствует
о нарушении режима очистки воды.
Микроскопирование при большом
увеличении. Для просмотра состава микроорганизмов активного ила
с целью обнаружения индикаторных микроорганизмов
используют прижизненные препараты, фиксация и окраска
микроорганизмов не допускается. Просмотр ведут при увеличении в 600 раа
и более. Пользуясь данными, приведенными в табл. 21, которые
характеризуют поведение индикаторных микроорганизмов в
зависимости от условий среды, устанавливают причины
неудовлетворительной работы активного ила.
Определение концентрации активного ила
Концентрацию активного ила в иловой воде из аэротенков и
вторичных отстойников определяют двумя методами: по объему
после стояния анализируемой жидкости в течение 30 мин и по
массе сухих веществ активного ила после его высушивания
при 105°С.
Мешающие вещества. Волокна целлюлозы и другие
взвешенные вещества, которые не являются активным илом.
Аппаратура. Градуированные цилиндры вместимостью 100 мл.
Сушильный шкаф. Бюксы. Установка для фильтрования под
вакуумом.
Ход анализа. Определение концентрации ила
по объему. Пробу иловой воды хорошо перемешивают,
разливают по 100 мл в три градуированных цилиндра и оставляют
стоять. Через 30 мин замеряют объем ила, осевшего на дно, во
всех цилиндрах.
Определение концентрации ила по массе.
После определения объема осевшего ила берут один цилиндр,
взбалтывают его содержимое и фильтруют на воронке под
вакуумом через два одинаковых фильтра. Цилиндр споласкивают
водой, которую выливают в воронку. Затем оба фильтра
вынимают из воронки, нижний фильтр кладут в отдельный,
предварительно высушенный и взвешенный бюкс. Для высушивания
фильтров и осадка бюксы ставят в сушильный шкаф при 105°С.
Периодически бюксы, закрыв крышками, которые тоже сушат,
вынимают из шкафа, охлаждают в эксикаторе, взвешивают и
снова ставят в сушильный шкаф, сняв крышки. Высушивание
продолжают до тех пор, пока разница между двумя
последующими взвешиваниями не будет превышать 0,0003 г. По разности
результатов взвешиваний фильтра с осадком и фильтра без
осадка находят количество сухого ила.
304
ТАБЛИЦА 21
Характеристика иидикаториых микроорганизмов
Название
микроорганизмов
Характеристика
Мелкие амебы — Amoeba
limax, мелкие
реснитчатые Lionotus, сосущая
инфузория — Podophrya,
бесцветные жгутиковые
Крупные амебы —
Amoeba protens
Корненожки — Arcella
vulgaris
Прикрепленные
реснитчатые — Vorticella micro-
s torn a
Прикрепленные
реснитчатые— Vorticella convol-
laria, Corchesium
Прикрепленные
реснитчатые— Opercullaria. co-
arctota, 01. glomerata
Свободноплавающие
реснитчатые, мелкие и
крупные — Oxytrichia, Colpi-
dium, Stilonichia
Свободноплавающие
реснитчатые, коловратки —
Phylodina Notommota
В больших количествах присутствуют в иле
при недостатке растворенного кислорода и
в перегруженном иле. Единичные
экземпляры встречаются в хорошо работающем
иле
Развиваются в хорошо работающем иле
В большом количестве встречаются в
хорошо работающем иле
Развиваются в перегруженном иле и при
недостатке кислорода в небольших
количествах
Развиваются в заметных количествах в
нитрифицирующем иле. При регенерации ила
появление этих инфузорий свидетельствует
о восстановлении первоначальных свойств
ила. При недостатке кислорода отрывается
от стебелька и превращается в
свободноплавающую особую форму «телотрех»
с венчиком ресничек иа заднем конце
Всегда присутствуют в активном иле в
различных количествах. Количество особей
в колонии от 3 до 50 шт. Характерным
является состояние ресничного аппарата
особей. При хороших условиях — достаточном
количестве кислорода и отсутствии
токсических веществ в сточной воде — ресничная
зона раскрыта, движения ресничек
активные. При неблагоприятных условиях
ресничная зона замкнута. При увеличенной
нагрузке особи сжимаются и инцистируют.
Оптимальная концентрация окисляемых
органических веществ в сточной воде для них
от 30 до 90 мг/л (по кислороду). При
нарушении режима очистки (pH,
температура, состав сточной воды) особи
раздуваются и гибнут
Иногда развиваются в больших
количествах. Их внезапное исчезновение не
свидетельствует о нарушении режима работы
очистных сооружений
Всегда присутствуют в активном иле.
Перегрузка ила и недостаток кислорода на
них не влияют. Внезапное исчезновение
коловраток, появление мертвых и раздутых
особей свидетельствует о резком
нарушении технологического режима очистки —
сильном изменении pH сточной воды,
повышенном содержании SO2 и др.
20 Заказ № 142
305
Название
микроорганизмов
Характеристика
Цисты инфузорий
Брюхоресннтчатая
инфузория — Aspidisca
Свободноплавающая
инфузория — Paramaecium
caudatum
Черви — Nematocdes
(круглые черви)
Малощетинковые черви
Aeolosoma Nais
Бактерии — Zooglea,
Z. ramigera, Z. ulva, Z.
compacta
Нитчатые бактерии —
Sphaerotilus (Cladotrix),
S. dichotoma
Arcella pamphagus
Нитчатые
серобактерии— Beggiatoa alba,
Thiothris nivea
Грибы — p. Fusarium,
p. Penicillium, p. Asper-
gillius, Frichoderma
Дрожжи и дрожжепо-
добные грибы
Образуются при резком изменении состава
сточной воды или при недостатке
питательных веществ
Почти всегда присутствует в иле. В
зимних условиях сильное развитие аспидиски
при отсутствии мелких амеб
свидетельствует о хорошем качестве ила. В летнее
время ее развитие свидетельствует о
глубокой нитрификации
Встречается в разлагающемся иле с
большим количеством бактерий. Вынослива
к недостатку кислорода
Единичные экземпляры часто встречаются
в активном иле. Сильное развитие червей
свидетельствует о наличии застойных зон
в очистных сооружениях и недостаточном
аэрировании
Хорошо развиваются в илах с устойчивой
нитрификацией
Развиваются при низкой нагрузке
активного ила
Обычно встречаются в активном иле в
незначительных количествах. Они образуют
длинные нити при медленном потоке
жидкости и, наоборот, мельчают при сильном
перемешивании. В регенераторе
деградируют из-за механического измельчения.
При перегрузке ила, недостатке кислорода
и при pH 7,5—8,5 их количество сильно
возрастает и вызывает вспухание ила
В заметных количествах встречается в
хорошем работающем иле. Отмечалось их.
присутствие в иле, утратившем нитрифика-
циоиную способность
Развиваются при резких перегрузках
активного ила. Очищают промышленные
стоки, содержащие сероводород. При
недостатке кислорода они окисляют
сероводород до серы, которая откладывается в их
клетках. При избытке кислорода сера
исчезает из клеток, она окисляется до
сульфата, подкисляя воду. В бескислородной
среде они могут жить до 3 недель
Присутствуют в незначительных
количествах при нормальных условиях работы
очистных сооружений. При нарушениях
технологического процесса и при pH ниже 5,
грибы сильно развиваются и вызывают
вспухание активного ила. Ветвящиеся гифы
грибов могут выносить кислую воду, они
прикрепляются к бортам аэротенков
Развиваются при pH ниже 5. Их появление
свидетельствует о снижении эффекта
очистки. Они вызывают распыление хлопков,
ила
Расчет. Концентрацию активного ила X в анализируемой иловой воде
вычисляют в миллилитрах на литр (мл/л) или в миллиграммах на литр (мг/л)
по формулам:
„ , V1000 v , alOOO
X мл/л = или X мг/л = ,
g ' gi
где V — объем ила в трех цилиндрах, мл; g — объем иловой воды, взятый
Для отстаивания, мл; а — количество осадка абсолютно сухого ила, мг; g\ —
объем иловой воды, взятый иа фильтрование, мл.
Концентрацию активного ила обоими вариантами метода определяют
с точностью ±10—15% отн.
Определение илового индекса
Иловым индексом называют объем активного ила, в котором
содержится 1 г абсолютно сухих веществ. Иловый индекс
характеризует плотность хлопка активного ила. Хорошо работающий
ил имеет иловый индекс 50—100 мл/г. Повышение значения
илового индекса более 120 мл/г свидетельствует об ухудшении
структуры ила, которое может быть вызвано развитием
нитчатых бактерий или плесневелых грибов.
Для определения илового индекса определяют концентрацию
активного ила по объему и по массе, как описано выше.
Расчет. Иловый индекс вычисляют по формуле
г
^м
где X — иловый индекс, мл/г; Со — концентрация ила по объему, мл/л; См —
-концентрация ила по массе, г/л.
Динамика осаждения активного ила
Динамику осаждения ила, т. е. изменение скорости
осаждения ила во время отстоя, определяют замером объемов осадка,
который выпадает каждые 15 мин на протяжении 2 ч отстоя.
Ход анализа. Пробу иловой смеси, отобранную из аэротен-
ков (отстойников и т. д.), разбавляют водой до концентрации
активного ила приблизительно 3 г/л. В градуированный цилиндр
вместимостью 100 мл наливают 100 мл разбавленной пробы,
хорошо перемешивают и ставят на отстой. Каждые 15 мин
записывают объем выпавшего осадка. Наблюдения ведут в
течение 2 ч.
Расчет. Полученные данные представляют графически в виде кривой
зависимости объема осадка от времени отстоя или в виде таблицы.
Определение токсичности
Для оценки токсичности отдельных промышленных стоков
при биологической очистке пользуются методом, основанным
на определении состояния микроорганизмов активного ила и
20* 307
результата их работы на испытуемой и разбавляющей воде.
Определяют БПКб и растворенный кислород в контрольной и
анализируемой воде до и после аэрации в присутствии
активного ила.
Аппаратура. Высокие склянки с барбатерами для
аэрирования. Вакуумный насос.
Ход анализа. Анализируемую воду разбавляют
водопроводной водой в соотношениях 1 :25; 1 :50 и 1 : 100. К 200—300 мл
каждой разбавленной воды добавляют активный ил из расчета
2 г/л, наливают их в высокие сосуды, вставляют барботер и
аэрируют в течение 5 ч. Параллельно ставят контрольный опыт,
в котором активный ил помещают в разбавляющую воду.
Во всех склянках испытуемую воду анализируют до и после
аэрации на содержание растворенного кислорода, БПК5 и ХПК
экспрессным методом. Кроме того, после аэрирования микроско-
пированием устанавливают состояние индикаторных
микроорганизмов активного ила.
Расчет. Токсичность сточной воды оценивают путем сравнения данных,
полученных в опытах при различном разведении ее, с данными контрольного
опыта.
Определение витамина В12
Для определения витамина Віг в сухом активном иле,
который используют при приготовлении комбикормов домашним
животным [98], предназначен метод, основанный на способности
культуры Escherichia coli (E. Coli) штамм 113-3 расти только
в присутствии витамина Bi2. Чем выше концентрация витамина
В12, тем выше урожай Е. Coli. Для количественного определения
витамина Віг одновременно выращивают культуру Е. Coli
штамм 113-3 на анализируемой среде и на контрольных
средах, которые содержат различные известные количества
витамина Віг- После выращивания сравнивают мутность опытной
среды от развившихся Е. Coli с мутностью контрольных сред
и находят содержание витамина Віг в анализируемой пробе.
Мешающие вещества. Метионин, к нему чувствительны
Е. Coli штамм 113-3.
Аппаратура. Автоклав. Термостат. Холодильник. Водяная
баня. Пробирки биологические длиной 180 мм. Нефелометр,
или фотоэлектроколориметр, или мутномер (см. рис. 71).
Реактивы и их приготовление. 1. Калий фосфорнокислый од-
нозамещенный и двузамещенный. 2. Железо сернокислое закис-
ное. 3. Магний сернокислый. 4. Глицерин. 5. Агар
микробиологический. 6. /-Аспарагин. 7. Соляная кислота, 1 н. раствор и
20%-ный раствор. 8. Витамин Ві2 для медицинских целей,
стандартный раствор, который содержит в 1 мл 0,1 мкг витамина Ві2.
Раствор хранят в склянке из темного стекла с притертой
пробкой под слоем толуола в холодильнике. 9. Едкий натр, 0,2;
0,5 и 1,5 н. растворы. 10. Уголь активированный марки БАУ.
308
11. Толуол. 12. Натрий лимоннокислый трехзамещенный. 13.
Аммоний сернокислый. 14. Глюкоза. 15. Хлористый натрий.
16. Азотнокислый натрий. 17. Казеин технический, 10%-ный
казеиновый кислый гидролизат. Приготовление. В колбу
вместимостью 1 л помещают 100 г размолотого казеина,
добавляют 500 мл 20%-ной соляной кислоты и перемешивают. Колбу
закрывают пробкой, в которую вставлен обратный холодильник,
и нагревают на кипящей водяной бане в течение 5—8 ч, а потом
при слабом нагреве кипятят на электроплитке в течение 15—
20 ч. Из полученного гидролизата под вакуумом отгоняют
соляную кислоту до получения густого сиропа. Для более полного
извлечения соляной кислоты к сиропу в колбе прибавляют
300 мл дистиллированной воды и снова отгоняют до получения
густого сиропа. Так повторяют 2 раза. После отгонки кислоты
к сиропу прибавляют 100 мл дистиллированной воды, раствор
нейтрализуют до pH 3,5, добавляя 5 н. раствор едкого натра,
количественно переносят в мерную колбу вместимостью 1 л и
объем доводят до метки, прибавляя дистиллированную воду.
Полученный раствор осветляют активированным углем.
Раствор выливают в колбу вместимостью 1,5 л, прибавляют 20 г
активированного угля и периодически встряхивают в течение 1 ч.
Осветленный раствор фильтруют на воронке Бюхнера под
вакуумом. Если фильтрат желтый, то осветление повторяют с новой
порцией активированного угля до получения бесцветного или
бледно-желтого раствора. Осветленный гидролизат разливают
в колбы вместимостью 100 мл, стерилизуют в автоклаве при
давлении 0,1 МПа A кгс/см2) в течение 20 мин, охлаждают и
в каждую колбу вливают по 5 мл толуола. Раствор под слоем
толуола хранят в холодильнике. 18. Чистая культура Е. Coli
штамм 113-3. Приготовление. Чистую культуру Е. Coli
выращивают в пробирках на косяках из агара. В колбу
вместимостью 100 мл наливают 60—70 мл дистиллированной воды,
6 мл 10%-ного кислого казеинового гидролизата, добавляют
0,5 мг сернокислого закисного железа, 20 мг сернокислого
магния и 20 мг двузамещенного фосфорнокислого калия, раствор
хорошо перемешивают для полного растворения внесенных солей.
Отдельно в 5—10 мл дистиллированной воды, подкисленной
несколькими каплями раствора соляной кислоты, растворяют 20 мг
/-аспарагина и раствор вливают в ту же колбу. Раствор в колбе
нейтрализуют до pH 7, прибавляя 1 н. раствор едкого натра,
вливают в него 200 мг глицерина и объем доводят до метки,
прибавляя дистиллированную воду. Раствор выливают в
коническую колбу, прибавляют 1,5 агара и нагревают на водяной
бане до полного растворения агара. Затем в колбу вливают
раствор витамина Ві2, который содержит 10 мкг витамина, раствор
перемешивают, разливают по 5 мл в пробирки и стерилизуют
в автоклаве в течение 15 мин под давлением 0,1 МПа A кгс/см2).
После охлаждения пробирок на застывшие косяки агара
309
засевают культуру Е. Coli штамм 113-3, все пробирки ставят
в термостат и выдерживают при 37°С в течение 24 ч. Культуру
Е. Coli пересеивают 1 раз в месяц. 19. Основная среда.
Приготовление. В колбу вместимостью 1 л наливают 900 мл
дистиллированной воды и последовательно растворяют в ней
следующие вещества: двузамещенный фосфорнокислый калий, 7 г;
однозамещенный фосфорнокислый калий, 3 г; сернокислый
магний, 0,1 г; сернокислый аммоний, 1 г; натрий
лимоннокислый, 0,5 г; глюкоза, 2 г; азотнокислый натрий, 2 г; хлористый
натрий, 0,5 г. После растворения всех указанных реактивов
проверяют pH раствора и, если надо, прибавляют раствор едкого
натра, чтобы pH раствора стал 6,8—7,0. Объем раствора
доводят до метки, прибавляя воду.
Ход анализа. Приготовление посевной культуры.
Посевную культуру готовят за сутки до проведения опыта.
В две пробирки вливают по 10 мл основной среды и
стандартного раствора витамина Віг из расчета 0,025 мкг витамина на
1 мл среды. Раствор стерилизуют в автоклаве при давлении
0,5 кгс/см2 в течение 20 мин, охлаждают и в каждую пробирку
вносят чистую культуру Е. Coli штамм 113-3 с агаровой среды.
Пробирки помещают в термостат при 37°С и выдерживают 16—
20 ч. В результате выращивания культуры Е. Coli получают
бактериальную суспензию, которую разбавляют стерильной
основной средой, предварительно разведенной вдвое стерильной
дистиллированной водой, в отношении 1 капля суспензии на
10 мл раствора. Э кет р а кц и я витамина Віг и з
анализируемой пробы. Берут навеску 10—20 г хорошо
измельченного сухого активного ила, влажность которого известна,
помещают в коническую колбу, приливают 70—80 мл
дистиллированной воды и энергично взбалтывают. Для получения
устойчивой суспензии прибавляют 0,1—0,2 г азотнокислого натрия.
Измеряют pH раствора, прибавляют 1 н. раствор соляной
кислоты до получения pH среды 5,0—5,5 и нагревают суспензию на
кипящей водяной бане в течение 20 мин. Затем суспензию
охлаждают, количественно переносят в мерную колбу вместимостью
100 мл, нейтрализуют до pH 6,8, прибавляя 0,5 н. раствор
едкого натра, объем доводят до метки, приливая
дистиллированную воду, и фильтруют. Полученный экстракт прибавляют к
питательной среде в различных количествах. В десять пробирок
вносят по 5 мл основной среды и в каждые две пробирки
следующие количества экстракта: 1, 2, 3, 4 и 5 мл. Объем раствора
в каждой пробирке доводят до 10 мл, прибавляя
дистиллированную воду.
Приготовление эталонных растворов
витамина Віг. В день опыта стандартный раствор витамина Ві2,
который имеет концентрацию 0,1 мкг/мл, разбавляют
дистиллированной водой последовательно 1 :100 и 1 :20. Полученный
стандартный раствор, разбавленный в 2000 раз, употребляют для
310
приготовления эталонных растворов, на основании анализа
которых строят калибровочную кривую. Каждый эталонный
раствор готовят в трех пробирках. В 15 пробирок вливают по 5 мл
основной среды, прибавляют следующие количества
разбавленного стандартного раствора витамина Віг: О мл, 0,5 мл; 1,0 мл,
1,5 мл и 2 мл. Объем раствора в каждой пробирке доводят до
10 мл, прибавляя дистиллированную воду.
Выращивание культуры Е. Coli штамм 113-3.
Культуру выращивают одновременно в пробирках со средой,
в которую добавлено известное количество витамина Віг и со
средой, в которую добавлено различное количество испытуемого
экстракта. Все 25 пробирок A0 с пробой и 15 со стандартным
раствором) помещают в автоклав и стерилизуют в течение
20 мин при давлении 0,05 МПа @,5 кгс/см2). После охлаждения
в каждую пробирку вносят 1 каплю посевной культуры —
бактериальной взвеси — и ставят в термостат на 24 ч при 37°С. После
инкубации все пробирки помещают в кипящую водяную баню
или в автоклав, прогревают их в течение 15 мин и охлаждают.
Определение концентрации витамина Ві2.
Жидкость в пробирках после нагревания мутная от взвешенных
мертвых микроорганизмов культуры Е. Coli. Количество
микроорганизмов пропорциональное содержанию витамина Ві2 в среде,
на которой они росли, определяют нефелометрическим методом.
Если для определения мутности пользуются нефелометром или
фотоэлектрическим колориметром с синим фильтром, то
измеряют мутность жидкости в каждой пробирке. На основании
полученных результатов анализов эталонных растворов строят
калибровочную кривую зависимости показаний прибора от
концентрации витамина Ві2, по которой находят количество
витамина В!2 в экстракте пробы, взятом на выращивание Е. Coli.
Если полученные результаты измерения мутности жидкостей
с экстрактом не укладываются в пределы калибровочной
кривой, то их отбрасывают. Для расчета используют только те
данные, которые соответствуют данным кривой, полученным в этом
опыте.
При работе с мутномером (см. рис. 71) калибровочную
кривую не строят, а сравнивают мутность эталонных растворов
с мутностью жидкости в пробирках с различным количеством
экстракта пробы. Находят, с каким эталоном совпадает или
близка мутность жидкости в пробирке с определенным
количеством экстракта.
Расчет. Содержание витамина В12 в 100 г активного ила вычисляют по
формуле
vg
где X — концентрация витамина Bi2 в активном иле, мкг/100 г; а —
количество витамина Ві2, найденное в экстракте, взятом на выращивание культуры
311
E. Coli, мкг; V—объем экстракта, взятый на выращивание культуры Е. Coli, мл,
Vi — объем всего экстракта, полученный после экстрагирования навескн
активного ила, мл; g—количество абсолютно сухого активного нла, взятое
на экстракцию, г.
Содержание витамина Ві2 в активном иле определяют с точностью
±10—25% отн.
Предостережения. 1. Определение мутности колориметром не может быть
точным, поэтому его следует применять только в том случае, когда нет
возможности измерить мутность нефелометром. 2. Прн наличии метионнна
в пробе результаты получают завышенными. Для определения величины
поправки на метионнн надо делать контрольный опыт, в котором до выращн-
вання бактерий разрушить витамин Віг в экстракте. К 5 мл экстракта
прибавляют 5 мл 0,2 раствора едкого натра, кипятят 20 мнн с обратным
холодильником, охлаждают и нейтрализуют как пробу. Если будет наблюдаться
рост культуры Е. Coli на этой среде, то при вычислении концентрации
витамина В12 надо вычесть поправку на содержание метнонина.
Определение микроорганизмов из группы кишечной палочки
Метод обнаружения бактерий группы кишечной палочки
в сточной воде до и после ее очистки [95] основан на
визуальном определении газообразования в среде, где развиваются
бактерии группы кишечной палочки. Степень бактериального
заражения, т. е. количественную оценку зараженности воды дают,
определяя индекс НВЧ 1 л. Индекс НВЧ 1 л основной
показатель бактериологического заражения воды означает наиболее
вероятное число бактерий кишечной палочки, присутствующих
в 1 л исследуемой воды [97]. Иногда степень зараженности воды
выражают колититром, т. е. минимальным объемом воды, в
котором может содержаться хотя бы одна бактерия.
Аппаратура. Термостат. Пробирки микробиологические.
Реактивы. 1. Лактоза. 2. Мясной экстракт. 3. Пептон.
Приготовление лактозного бульона. Готовят
лактозный бульон двойной концентрации. В коническую колбу
наливают 700 мл дистиллированной воды, прибавляют 6 г
мясного экстракта и 10 г пептона. Смесь медленно нагревают на
водяной бане при постоянном помешивании до полного
растворения прибавленных веществ. Отдельно в 100 мл
дистиллированной воды растворяют 10 г лактозы. Раствор выливают в ту же
коническую колбу и добавляют дистиллированную воду до
объема всего раствора 1 л. В приготовленном лактозном бульоне
проверяют pH. Он должен быть 6,8—7,0.
Ход анализа. Пробу воды в склянке энергично взбалтывают
до получения наиболее равномерного распределения бактерий
и наливают по 10 мл в пять пробирок. Затем в каждую
пробирку вливают 10 мл лактозного бульона, закрывают их
ватными стерильными пробками и помещают в термостат при 35—
37°С. Число пробирок и количество пробы, которое берут на
анализ, можно варьировать, но при этом надо помнить, что
концентрация питательных веществ не должна быть ниже минимума,
обеспечивающего рост бактерий. Через 24 ч инкубации все про-
312
бирки просматривают на свету. При развитии бактерий жидкость
в пробирках замутится. Если есть газообразование в среде, то
при легком взбалтывании жидкости в пробирке появляется струя
мелких газовых пузырьков. Отбирают пробирки с
обнаруженным газообразованием, а остальные пробирки снова помещают
в термостат при 35—37°С на 24 ч. После 48 ч инкубации
просматривают пробирки на свету для обнаружения выделения газа
при легком взбалтывании жидкости.
Если во всех пробирках газообразование не было обнаружено после
48±3 ч инкубации, то это дает право считать, что в анализируемой воде нет
бактерий группы кишечной палочки. Пробирки, в которых была обнаружена
любая степень газообразования после 24 и 48 ч инкубация, должны быть
подвергнуты подтверждающему анализу.
Обоснование выбора анализов для контроля за работой очистных сооружений
Выбор анализов зависит от технологической схемы очистки
сточных вод. Ниже приводятся обоснования для выбора
анализов при биологическом способе очистки воды [5, 96].
Температура. Температуру сточной воды, поступающей
на очистные сооружения, надо определять, чтобы обеспечить
нормальную работу всех агрегатов. Температура влияет на
гидравлический режим работы сооружений. Концентрация
кислорода, растворенного в воде, зависит от температуры. При
микробиологических процессах строго регламентируется
температура воды в аэротенках и воды, поступающей в аэротенк.
Нарушение температурного режима может привести к гибели
микроорганизмов активного ила или к снижению их
работоспособности. Задержание взвешенных веществ в первичных
отстойниках увеличивается при повышении температуры с 7 до 25°С.
pH воды, поступающей на очистку, влияет на процессы
коагуляции и осаждения взвешенных веществ, а также на
биохимическое окисление. Отклонение от заданного pH вызывает
развитие посторонних микроорганизмов и может привести к
гибели микроорганизмов активного ила.
Оседающие вещества — показатель загрязненности
воды твердыми веществами. Он используется для расчета
эффективности работы песколовок и первичных отстойников.
Взвешенные вещества — показатель загрязненности
воды на различных стадиях ее очистки. Вода, поступающая
в отстойники, обычно содержит 150—350 мг/л взвешенных
веществ. После отстоя их концентрация снижается на 30—5Ои/о-
После биохимического окисления вода содержит 7—25 мг/л
взвешенных веществ.
Окисляемость перманганатная — нормируемый
показатель загрязненности воды легкоокисляемыми
органическими и неорганическими веществами. Вода, поступающая на
биологическую очистку, должна иметь окисляемость 50—80 мг/л.
313
и БПКго — нормируемые показатели загрязненности
воды веществами, которые поддаются биохимическому
окислению. Они указывают, какое количество кислорода нужно
микроорганизмам для окисления легко- и трудноокисляемых веществ,
содержащихся в 1 л воды, при инкубации в течение 5 и 20
суток. Вода, поступающая в аэротенк, должна иметь БПК.5 не
выше 400 мг/л. Вода после аэротенков и из вторичных
отстойников должна иметь БПКб не выше 25 мг/л.
ХПК — нормируемый показатель загрязненности воды
органическими и неорганическими веществами, которые могут быть
окислены самыми сильными окислителями — бихроматом калия
или йодом. ХПК воды до очистки должно быть 300—500 мг/л.
ХПК очищенной воды не должно превышать 200 мг/л.
Кислород, растворенный в воде,— нормируемый
показатель загрязненности воды органическими веществами.
Чем больше содержание органических веществ в воде, тем
быстрее понижается в ней концентрация кислорода. Очищенная вода
содержит кислорода от 2 до 6 мг/л [при 20°С и давлении
101, 324 кПа G60 мм рт. ст.)] в зависимости от степени чистоты.
Содержание растворенного кислорода, выраженное в процентах
к его растворимости в воде при данной температуре и давлении,
является главным показателем степени чистоты воды.
Аммиачный азот — показатель загрязнения воды
фекалиями.
Нитриты, нитраты и общий азот. Сопоставляя
результаты анализов воды до и после ее очистки на содержание
окисных форм азота и общего азота выясняют степень
напряженности окислительных процессов, протекающих в аэротенках,
обнаруживают явление нитрификации, которое вызывается
недостатком кислорода в очищаемой воде.
Фосфор и азот — биогенные элементы, необходимые для
жизнедеятельности микроорганизмов активного ила. Чем выше
БПК сточной воды, тем больше требуется биогенных элементов.
Для нормального течения биохимических процессов необходимо^
чтобы в воде, поступающей в аэротенки, на каждые 100 мгБПК
было 5 мг азота и 1 мг фосфора.
Токсичность воды и содержание некоторых вредных
веществ, например фурфурола, необходимо обнаруживать
своевременно, чтобы обеспечить нормальную работу
микроорганизмов активного ила.
Хлориды определяют для контроля за солевым составом
воды, который изменяется по ходу очистки, и для суждения
о согласованности анализируемых проб.
Исследование микроорганизмов активного
ила по его внешнему виду и микроскопированием необходимо
для поддержания регламентированных условий их работы,
обеспечивающих максимальное окисление веществ, загрязняющих
воду.
314
Органолептические свойства воды — цвет,
запах— показатели степени очистки воды.
Бактериологический анализ. Установлено, что,
если вода содержит число колибактерий (кишечная палочка)
ниже определенной нормы, то в ней нет патогенных организмов.
Задачей бактериологических анализов является определение
числа колибактерий в выбранном объеме пробы.
Микроорганизмы в воде распределяются неравномерно, поэтому количество
колибактерий, которое обнаруживают в пробах воды, выражают
статистически как наиболее вероятное число колибактерий в 1 л
исследуемой воды и обозначают индексом НВЧ 1 л. Если
индекс НВЧ 1 л не более 50, то следует провести обычную
обеззараживающую обработку воды. При более высоком значении
индекса НВЧ решают вопрос о применении специальных
приемов обеззараживания воды.
Радиоактивность воды с каждым годом становится
все более вероятным загрязнением. Международная
комиссия по защите от радиоактивного излучения (МКРЗ)
предложила Всемирной организации здравоохранения включить
в Международный стандарт на питьевую воду следующие
допустимые количества содержания радиоактивных веществ: альфа-
активность— 3 пКи/л и бета-активность — 30 пКи/л.
Определение радиоактивности позволяет установить источник
загрязнения и ее понижение за счет очистки.
СПРАВОЧНЫЕ ТАБЛИЦЫ
ТАБЛИЦА Г
Плотность
прн 20°С,
г/см3
1,005
1,012
1,018
1,025
1,032
1,038
1,045
1,052
1,059
1.066
1.073
1,080
1.087
1.095
1,102
1,109
1,117
1,124
I.I32
1,139
1,147
1,155
1,163
1,170
1,178
I.I86
1,194
1,202
1,210
1,219
1,227
1,235
1,243
1,252
Плотность водных
Содержание
г/100 г
I
2
3
4
5
6
7
8
9
10
П
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
H,SOt
г/л
10,05
20,24
30,55
41,00
51,59
62,31
73,17
84,18
95,32
106,60
118,00
129,60
141,40
153,30
165,30
177,50
189,90
202,40
215,00
227,90
240,90
254,10
267,40
280,90
294,60
308,40
322.40
336.60
351,00
365.60
380,30
395,20
410,30
425,50
Плотность
прн 20°С,
г/см3
1,260
1,268
1,277
1,286
1,294
1,303
1,312
1,321
1.329
[,338
1.348
1,357
,366
[,376
[,385
,395
.405
,415
[,425
[,435
[,445
[,456
,466
,477
,488
,498
,509
,520
.531
1,542
1,553
1,565
1,576
]
,587
растворов серной кислоты
Содержание
Н
г/100 г
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
.so.
г/л
441,00
456,60
472,50
488,50
504,70
521,10
537,80
554,60
571,60
588,90
606,40
624,20
642,20
660,40
678.80
697,60
716,50
735,70
755,10
774,90
794,90
815,20
835,70
856,50
877,60
899,00
920,60
942,40
964,50
986,90
1010,00
1033,00
1056,00
1079,00
Плотность
прн 20°С,
г/см3
1,599
1,611
1,622
1,634
1,646
1,657
1,669
1,681
1,693
1,704
1,716
1,727
1,738
1,749
1,759
1,769
1,779
1,787
1,795
1,802
1,809
1,814
1,819
1,824
1,828
1,8312
1,8337
1,8355
1,8364
1,8361
1,8342
1,8305
Содержание
Н
г./100 г
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
jSO4
г/л
1103,00
1127,00
1152,00
1176.00
1201,00
1226,00
1252,00
1278,00
1303,00
1329,00
1355,00
1382,00
1408,00
1434,00
1460,00
1486,00
1512,00
1537,00
1562,00
1586,00
1610,00
1633,00
1656,00
1678,00
1700,00
1721,00
1742,00
1762,00
1781,00
1790,00
1816,00
1831,00
Поправки плотности в зависимости от температуры
(в интервале температур 15—25°С)
Плотность
прн 20°С,
г/см3
1,01
1,04
1,07
I, II
1,15
1,22
Поправка
на 1°С, г/см3
0,0002
0,0003
0,0004
0,0005
0,0006
0.0007
Плотность
прн 20°С,
г/см3
1,42
1,56
1,70
1,77
1,84
Поправка на
1°С, г/см'
0,0008
0,0009
0,0010
0,0011
0,0012
ТАБЛИЦА II
Содержание СаО в известковом молоке различной плотности
Плотность
при 20°С,
г/см3
1.0085
1,0170
1,0245
1,0315
1,0390
1,0460
1,0535
1.0605
Содержание
СаО,
г/л
10
20
30
40
50
60
70
80
Плотность
при 20°С,
г/см3
1,1185
1,1255
1,1325
1.1400
1,1475
1,1545
1.1615
1.1685
Содержа-
НИЄ
СаО,
г/л
160
170
180
190
200
210
220
230
Плотность
при 20°С,
г/см3
1
,0675
1.0750
.0825
1,0895
1.0965
1.1040
1.1110
Содержа-
НИЄ
СаО,
г/л
90
100
ПО
120
130
140
150
Плотность
при 20°С,
г/см3
1,1760
1.1835
1.9050
1.1975
1.2050
1,2125
1,2195
Содержание
СаО,
г/л
240
250
260
270
280
290
300
ТАБЛИЦА III
Плотность,
при 20сС,
г/см3
0,994
0.990
0,981
0,973
0,965
0.958
0.950
0,943
Плотность водных
Содержание NH»
г/100 г
1
2
4
6
8
10
12
14
г/л
9.94
19.79
39.24
53.38
77.21
95.75
114.0
132.0
растворов
Плотность
при 20°С,
г/м3
0.936
0.930
0.923
0,916
0.910
0.904
0,898
0,892
аммиака
Содержание
г/100 г
16
18
20
22
24
26
28
30
NH,
г/л
149,8
167,3
184,6
201.6
218.4
235.0
251.4
267.6
Температурные поправки плотности водных растворов
аммиака (в интервале температур 15—25°С)
Плотность при
20°С, г/см3
0,99
0.95
Поправка на
1°С
0.0002
0.0003
Плотность при
20°С, г/см3
0.92
0.90
Поправка на
0.0005
0.0006
ТАБЛИЦА IV
Поправочные эмпирические коэффициенты для определения спирта
вакуум-нитрнтным методом
Значення К[ в зависимости от объема реакционного сосуда
Вместимость
реакционного
сосуда, мл
160
170
180
200
210
АГі при
20° С
1,506
1.461
1,400
1.380
1,362
Вместимость
реакционного
сосуда, мл
260
270
280
290
300
А", при
20°С
1,282
1,267
1,259
1,242
1.236
Вместимость
реакционного
со суда і мл
220
230
240
250
Кі прн
20°С
1,339
1.325
1,308
1,291
Вместимость
реакционного
сосуда, мл
310
320
330
340
Ki при
20°С
1.226
1,212
1,207
1.200
317
Значения Кг в зависимости от температуры опыта
Температура,
°С
14
15
16
17
18
19
20
Кг
1,06
1,05
1,04
1,03
1,02
1,01
1,00
Температура,
°С
21
22
23
24
25
26
27
Температура,
*. | »с
0,99
0,98
0,97
0,96
0,95
0,94
0,93
28
29
30
31
32
33
34
к,
0,92
0,91
0,90
0,89
0,88
0,87
0,86
Концещ
г/100
1
2
3
4
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
Плотность
рация метилового
спирта
г
мл/100 мл
1,26
2,51
3,76
5,01
6,25
7,49
8,72
9,95
11,17
12,40
13,61
14,83
16,04
17,25
18,45
19,65
20,85
22.04
23,23
24,42
25,60
26,77
27,95
29,12
30,29
31,45
32,61
33,76
34,91
36,05
37,19
38,33
39,45
40,58
водных растворов метилового спирта
Плотность
при 20°С
г/см3
0,9983
0,9968
0,9949
0,9932
0,9914
0,9898
0,9881
0,9865
0,9849
0,9833
0,9817
0,9802
0,9786
0,9771
0,9757
0,9742
0,9727
0,9713
0,9698
0,9683
0,9668
0,9653
0,9639
0,9624
0,9609
0,9593
0,9579
0,9563
0,9548
0,9532
0,9516
0,9500
0,9483
0,9467
Концентрация метилового
г/100 г
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
спирта
мл/100 мл'
41,70
42,82
43,92
45,03
46,13
47,22
48,30
49,39
50,46
51,53
52,59
53,65
54,70
55,76
56,80
57,83
58,85
59,86
60,88
61,88
62,88
63,89
64,87
65,85
66,83
67,80
68,76
69,72
70,66
71,59
72,53
73,46
74,37
75,27
Плотность
при 20°С
г/см3
0,9450
0,9433
0,9415
0,9398
0,9380
0,9362
0,9344
0,9326
0,9306
0,9289
0,9268
0,9250
0,9231
0,9213
0,9193
0,9173
0,9151
0,9130
0,9110
0,9089
0,9068
0,9048
0,9026
0,9004
0,8984
0,8962
0,8940
0,8918
0,8895
0,8872
0,8850
0,8827
0,8803
0,8779
Концентраті
г/100 г
69
70
71
72
73
74
75
76
77
78
79
80
80,5
81
81,5
82
82,5
83
83,5
84
84,5
85
85,5
86
86,5
87
87,5
j метилового
спирта
мл/100 мл
76,16
77,07
77,94
78,81
79,68
80,54
81,39
82,24
83,08
83,92
84,76
85,58
86,00
86,41
86,81
87,21
87,61
88,00
88,38
88,76
89,16
89,54
89,93
90,30
90,69
91,06
91,43
Плотность
при 20°С
г/см3
0,8754
0,8731
0,8706
0,8681
0,8657
0,8632
0,8607
0,8582
0,8557
0,8533
0,8509
0,8484
0,8473
0,8461
0,8448
0,8435
0,8422
0,8409
0,8395
0,8381
0,8368
0,8355
0,8342
0,8329
0,8315
0,8301
0,8287
Концентрация метилового
г/100 г
88
88,5
89
8&,5
90
90,5
91
91,5
92
92,5
93
93,5
94
94,5
95
95,5
96
96,5
97
97,5
98
98,5
99
99,5
100
спирта
мл/100 мл
91,79
92,16
92,52
92,88
93,24
93,60
93,96
94,31
94,67
95,02
95,37
95,71
94,06
96,41
96,75
97,09
97,41
97,74
98,08
98,41
98,73
99,06
99,39
99,70
100,00
Плотность
прн 20°С
г/см3
0,8273
0,8259
0,8245
0,8231
0,8217
0,8203
0,8189
0,8175
0,8161
0,8147
0,8133
0,8119
0,8105
0,8091
0,8077
0,8063
0,8048
0,8033
0,8019
0,8005
0,7990
0,7976
0,7962
0,7947
0,7931
СПИСОК ЛИТЕРАТУРЫ
1. Технология гидролизных производств. М., 1973. 403 с. Авт.г
В. И. Шарков, С. А. Сапотницкий, О. А. Дмитриева, И. Ф. Туманов.
2. Сапотницкий С. А. Использование сульфитных щелоков. М., 1965.
264 с.
3. Виноградов А. В. Организация заводских лабораторий в химической;
промышленности. М.—Л., 1948. 191 с.
4. Корольков И. И. Автоматический сифонно-поплавковый
пробоотборник.—«Гидр, пром. СССР», 1951, № 4, с. 20.
5. Методика проведения технологического контроля работы очистных,
сооружений городских канализаций. М., 1964. 178 с.
6. Практические работы по химии древесины и целлюлозы. М., 1965.
411 с. Авт.: А. В. Оболенская, В. П. Щеглов, Г. А. Аким и др.
7. Шарков В. И., Куйбина Н. И. Химия гемицеллюлоз. М., 1972. 439 с.
8. Шарков В. И., Емельянова И. 3., Соловьева Ю. П. Исследование
химического состава полисахаридов клеточных стенок некоторых древесных
тканей.—«Труды ЛТА им. С. М. Кирова», Л., 1960, вып. 91, с. 325—335.
9. О химическом составе некоторых растительных тканей.— «Сборник
трудов ВНИИГС», М.—Л., 1960, т. VIII, с. 7—20. Авт.: Н. И. Куйбинаг
Э. Н. Гвоздева, Ю. П. Соловьева, Ван Цзун-ли, В. И. Шарков.
10. О химическом составе кукурузной кочерыжки.— «Гидр, пром.», 1962,.
№ 2, с. 7—9. Авт.: В. И. Шарков, Н. И. Куйбина, Ю. П. Соловьева и др.
11. О химическом составе некоторых растительных тканей.— «Сборник.
319-,
трудов ВНИИГС», М, 1965, т. XIV, с. 7—12. Авт.: В. И. Шарков, Н. И. Куй-
•бина, Ю. П. Соловьева, Э. Н. Гвоздева.
12. Стрелков С. С. Углеводы торфа и пути их использования.— «Торф,
гром.», 1958, № 7, с. 7—8.
13. Терек С. Изучение легко- и трудногидролизуемых гемицеллюлоз
сельскохозяйственных растительных отходов. Автореферат диссертации на соиск.
учен. степ. канд. технич. наук. Л., 1959 (ЛТА им. С. М. Кирова). 86 с.
14. Оводов Ю. С. Газожидкостная хроматография углеводов. (Обзор).
АН СССР Сибирское отделение Дальневосточный филиал. Сер.
биоорганическая химия. Владивосток, 1970. 64 с.
15. Емельянова И. 3., Пети П. К. Объективный метод определения
влажности сырья.—«Гидр. пром. СССР», 1949, № 2, с. 13—14.
16. Агеев Л. М., Корольков С. И. Химико-технический контроль
гидролизного и сульфитно-спиртового производства. М.—Л., 1953. 400 с.
17. Любин Б. О., Глазкова Э. Л. Экспрессный метод определения
влажности сырья и лигнина.— «Гидр. пром. СССР», 1953, № 4, с. 10—11.
18. Низовкин В. К., Емельянова И. 3. Эбулиостатический метод
определения редуцирующих Сахаров.— «Журн. прикл. химии», 1959, т. 32, вып. 11,
с. 2516—2525.
19. Емельянова И. 3. Изучение реакции окисления моносахаридов в
щелочной среде и разработка нового метода определения редуцирующих саха-
ров. Автореферат диссертации на соиск. учен, степени канд. хим. наук. Л.,
1954 (Лен. Гос. университет). 130 с.
20. Комаров Ф. П. Руководство к лабораторным работам по химии
древесины и целлюлозы. Л., 1934. 215 с.
21. Уайз Л. Э., Джаи Э. С. Химия древесины. Т. II, М.—Л., 1960. 556 с.
22. Viebock F., Schwappach А. Новый метод количественного определения
метоксильных и этоксильных групп. А. Вег. 63 В, 1960, с. 2818—2819.
23. Viebock F., Brecher С. Новый метод количественного определения
метоксильных и этоксильных групп. Вег. 63, 1930, с. 3207—3208.
24. Буевский А. В., Емельянова И. 3. Уточнение методики определения
Сахаров в сульфитных щелоках.— «Сборник трудов ВНИИГС», 1952, т. IV,
с. 76—85.
25. ГОСТ 18995.1—73. Продукты химические органические. Методы
определения плотности жидкостей.
26. Швальбе К., Зибер Р. Химический производственный контроль в
целлюлозной и бумажной промышленности. Л., 1934. 460 с.
27. Долгалева А. А. Методы контроля сульфитцеллюлозного
производства. М., 1971. 344 с.
28. Лурье Ю. Ю. Справочник по аналитической химии. М., 1971. 454 с.
29. Емельянова И. 3., Батракова Т. А. Новый метод количественного
определения редуцирующих Сахаров в древесных гидролизатах и сульфитных
щелоках при помощи хроматографии на бумаге.— «Журн. анал. химии», 1958,
т. 13, № 1, с. 142—148.
30. ГОСТ 6055—51. Вода. Методы химического анализа. Единица
измерения жесткости.
31. Крюков П. А. и Номикос Л. И. Меркуриметрический метод
определения концентрации ионов хлора.— В кн.: Современные методы химического
анализа природной воды. М., 1955, с. 44—46.
32. Гончарова И. А. Определение ионов брома и йода в природных
водах.— В кн.: Современные методы химического анализа природной воды. М.,
1955, с. 80—86.
33. Зенян А. А. Колориметрический торийализариновый метод
определения ионов фтора.— В кн.: Современные методы химического анализа
природной воды. М., 1955, с. 99—102.
34. Тананаев И. В. Новый метод определения фтора в апатитах,
фосфоритах и суперфосфатах, «ЖПХ», 1932, № 5, с. 834—841.
35. ГОСТ 2184—67. Серная кислота техническая.
36. Емельянова И. 3., Батракова Т. А., Соловьева Ю. П. Экспрессный
320
метод определения сахара и серной кислоты в гидролизатах.— «Гидр, и лесо-
хим. пром.» 1956, № 8, с. 14—15.
37. Современные методы и приборы для определения состава, свойств,
и состояния веществ. М., 1962, вып. 1. 181 с.
38. Краткий справочник физико-химических величин. Под редакцией
К. П. Мищенко и А. А. Равделя. Л., 1974. 200 с. Составители-. Н. М. Барон„
Э. И. Квят, Е. А. Подгорная и др.
39. Кинетика электродных процессов. М., 1952. 316 с. Авт.: А. Н. Фрум-
кин, В. С. Богоикий, 3. А. Иофа, Б. Н. Кабанов.
40. Хорнед Г., Оуен Б. Физическая химия растворов электролитов. М.„
1952. 623 с.
41. Виноградова Е. Н. Методы определения концентрации водородных
ионов. М., 1956. 155 с.
42. Пчелин В. А. Измерение активности водородных ионов (pH),
окислительно-восстановительных потенциалов и потенциометрическое титрование. М.,
1955. 200 с.
43. Рязаицев Н. В. Регулирование pH в процессе непрерывной
нейтрализации гидролизата.— «Гидр, и лесохим. пром.», 1959, № 3, с. 9—11.
44. Низовкин В. К., Корольков И. И. Кондуктометрический метод
определения кальция в нейтрализованных древесных гидролизатах и сульфитных
щелоках (сусле, бражке, барде). Кондуктометрическое определение
сульфатов—«Труды ВНИИГС», Л., 1947, т. II, с. 151—181.
45. Лурье Ю. Ю., Алферова Л. А., Бондарева Т. Н. Раздельное
определение низкомолекулярных жирных кислот в сточных водах.— «Зав. лаб.»,.
1964, № 7, т. 30, с. 799—801.
46. Емельянова И. 3., Батракова Т. А. К вопросу об определении
сбраживаемого сахара.— «Гидр. пром. СССР», 1950, № 2, с. 10—11.
47. Райцева М. К. Сбраживание гидролизатов. М., 1972. 39 с.
48. Емельянова И. 3., Батракова Т. А. Экспрессный хроматографическин
метод определении конца брожения.— «Гидр, н лесохим. пром.», 1956, № 3„
с. 14—15.
49. Волская Л. П., Соловьева Ю. П. Уточнение метода определения
метилового спирта применительно к полупродуктам гидролизного и сульфитно-
спиртового производства.— «Сборник трудов ВНИИГС». М.—Л., 1952, т. IV,.
с. 140—147.
50. Низовкин В. К., Корольков И. И. Флотационный метод определения
спирта —«Гидр. пром. СССР», 1948, № 1, с. 9.
51. Низовкин В. К-, Корольков И. И. Определение спирта методом пови-
сания поплавка при контроле гидролизного и сульфитно-спиртового
производства.—«Сборник трудов ВНИИГС». М — Л., 1952, т. IV, с. 91—132.
52. Модифицированный флотационный метод определения спирта в
водных растворах, содержащих примеси.— «Гидр, и лесохим. пром.», 1974, № 4„
с. 15—17. Авт.: Морозова С. Ф„ Румянцева В. А., Соболева Г. А. и др.
53. ГОСТ 3639—61. Растворы водноспнртовые. Методы определения
содержания этилового спирта.
54. ГОСТ 3637—59. Спиртомеры стеклянные.
55. Таблицы для определения содержания этилового спирта в водно-
спиртовых растворах. М., 1972. 364 с.
56. ГОСТ 3638—53 *. Спиртомеры металлические.
,_ PN — 62 „
57. . 7Q599 Спирт ректификат. Польская норма.
PN
58. ¦ 7Qg98 Спирт. Методы испытаний. Польша.
59. ГОСТ 18300—72. Спирт этиловый ректификованный технический.
60. ГОСТ 17299—71. Спирт этиловый технический.
61. ГОСТ 10749—72. Спирт этиловый технический. Методы анализа.
62. ГОСТ 5962—67. Спирт этиловый ректификованный. Технические
требования.
63. STAS 4523—54. Спирт этиловый. Румыния.
21 Заказ № 142 391
Ы. STAS 184—54. Спирт этиловый. Методы испытания. Румыния.
65. О-Е-760 в 14/IV 1957. Спирт этиловый. США.
66. Емельянова И. 3., Георгиевская Г. А. Определение кислотности
этилового спирта.— «Гидр, и лесохим. пром.», 1959, № 8, с. 15—17.
67. Виноградова Е. Н., Галлай 3. А, Финогенова 3. М. Методы
полярографического и амперометрического анализа. М., 1963. 299 с.
68. Гейровский Я-, Кута Я. Основы полярографии. М., 1965. 559 с.
69. Немцова Н. П., Коротченко Ф. Е. Определение фосфора на фотоэлек-
троколориметре ФЭК—М.— «Гидр, и лесохим. пром.» 1962, № 1, с. 17—19.
70. Некрасов Б. Н. Основы общей химии. Т. II. М., 1973. 656 с.
71. Морозова С. Ф., Румянцева В. А. Определение калия в гидролизных
средах.— «Гидр, и лесохим. пром.», 1974, № 1, с. 11—13.
72. Perrin С. Н. Ускоренная модификация способа определения азота
по Кьельдалю. Analytical Chem. 1953, v. 25, № 6, pp. 968—971.
73. Вендт В. П., Беляевская В. В., Говсеева Н. Н. Количественное
определение витамина D2 в облученных дрожжах.— В кн.: Витаминные ресурсы
и их использование. М., 1963, № 6, с. 197—203.
74. Физер Л., Физер М. Химия природных соединений фенантренового
ряда. М.—Л., 1953. 656 с.
75. Пасхина Т. С. Количественное определение аминокислот при помощи
хроматографии на бумаге.— В кн.: Современные методы в биохимии. Т. 1.
М., 1964. с. 162—167.
76. Метод определения L-лизина в процессе ферментации и в готовых
продуктах.— В кн.: Продукты микробного синтеза. Рига, 1966. с. 61—67. Авт.:
Л. С. Куцева, Л. Я. Арешкина, Н. М. Клюева, Е. П. Скоробогатова.
77. ГОСТ 20083—74. Дрожжи кормовые.
78. Отраслевая норма —тт-. Кормовые дрожжи. Норма качества. ЧССР.
79. Чехословацкая государственная норма ЧСН 56-6810. Дрожжи.
80. Польская норма —-. ппл . Сушеные кормовые дрожжи.
81. Польская норма . _ 7qn0r ¦ Дрожжи. Методы испытаний.
82. Шиманская М. В., Славинская В. А. Аналитическое определение
фурфурола. Рига, 1961. 183 с.
83. ГОСТ 10437—71. Фурфурол технический.
84. STAS 1779—54. Фурфурол. Румыния.
85. Стандарт ГДР ТГЛ 15233—63. Фурфурол.
86. ГОСТ 8050—64. Углекислый газ сжиженный.
87. ГОСТ 12162—66. Сухой лед.
88. Стандарт ГДР ТГЛ 2968—56. Жидкая двуокись углерода в стальных
баллонах.
89. У. У. S. К. 1106—1961. Жидкая двуокись углерода. Япония.
90. Черкинский С. Н. Санитарные условия спуска сточных вод в водоемы.
М, 1971. 208 с.
91. Международный стандарт питьевой воды. М., 1973.
92. Мейн Ф., Штофф Г., Кольшюттер Г. Очистка промышленных сточных
вод. М., 1963. 646 с.
93. Лурье Ю. Ю., Рыбникова А. И. Химический анализ
производственных сточных вод. М., 1974. 335 с.
94. Лурье Ю. Ю-, Николаева 3. В. Ускоренные методы определения би-
хроматной окисляемости.— В кн.: Очистка промышленных сточных вод, М.,
1968, № 4, с. 191—193.
95. Кокс Ч. Р. Контроль за процессами обработки воды. М., 1965. 415 с.
96. Роговская Ц. И. Биохимический метод очистки производственных
•сточных вод. М., 1967. 139 с.
97. ГОСТ 18963—73. Методы санитарно-бактериологического анализа.
98. ГОСТ 18663—73. Витамин Ві2 кормовой.
322
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азот аммиачный 116, 273
— минеральный 112
— нитратный 282
— нитритный 279
— общий 271
— органических соединений 285
— очистка 213
Активность бродильная 160
— известкового молока ПО
Альдегиды определения 250
Алюминий гидроокись
приготовление 280
Аминокислоты 217, 220
Аммиак 228, 274
Аммофос 112
Аиилин перегонка 287
Анилинфталат 79, 80
Арабан 19
Ареометр 60, 109, 111, 119, 173, 175
Ацетон 192
Барда 13, 172
Белок 217 см. протеин сырой
— истинный 229
— усвояемый 230
БПК 263
Бром 102
Бромирование 42, 166
Бумага хроматограф. 76, 218
Бумажки индикаторные 124
Буфер приготовление 97, 274
Вещества бромируемые 146
— взвешенные 250
— летучие 134
— минеральные 21, 32
— нерастворимые в воде 255, 326
— окисляемые 190, 248, 254
— оседающие 145, 250
— пектиновые 19, 20
— редуцирующие 33, 39, 57, 85, 95
— сухие 59, 251
— экстрактивные 21, 49, 50
Витамин Ві 231
— В2 232
— В12 308
— D2212
— РР 233
Влажность сырья 27, 29, 30
— дрожжей 223
Вода 96, 247
— без аммиака 274
запаха 298
меди 97
нитритов 279
цвета 281
— биологически очищенная 264
— разбавляющая 264
— сточная 264
Гидролиз белка 219
21"
— казеина 309
— полисахаридов 3, 24, 39, 92, 122
— полнота 123
Гексоз а ны 19
Гексозы 3, 22, 39, 78, 80, 147, 151, 16Е
Гемицеллюлозы ,17
Гипс 133
Градус жесткости 98
— кислотности 237
— крепости 175
— цветности 293
Гипосульфит 258, 259
Двуокись углерода 243
Древесина 16, 22, 25, 26, 27, 29
Доброкачественность 143
Дозаторы 35
Достоверность 5
Дрожжи спиртовые 152, 155, 157, 158
— кормовые 193, 195
— обогащенные витамином D2 3, 212'
лизином 221
Жесткость воды 97
Запах 295, 297
Зола 32
Ил активный 299, 304, 307
Индекс нловый 307
— НВЧ 312, 315
Индикатор 68, 97
— анилин уксуснокислый 44
— кислота к-фенилантраниловая 267"
— крахмал 170, 259
— смешанный 211, 227, 253, 101
— по Андерсену 124
Раствор ферроина 267
Инфекция 155, 158
йод 102, 269
Калий 208
Кальций 68, 100, 132
— моносульфит 62
— окись 111
Камеры для хроматографии 74, 75
— Тома 153
Катализатор селеновый 229
Кислород биологически
потребляемый 263
— константа скорости потребления-
266
— растворенный 262
— химически потребляемый 266, 268:
269
Кислота борная 113, 228
— левулиновая 137
— летучая органическая 134
— органическая 129, 139
— серная 108, 119, 129
— сернистая 61, 62, 63, 67
— уроновая 20, 46, 93
Кислотность дрожжей 331
323
— общая 123
— спирта 182
— фурфурола 236
Колититр 312
Консервация проб 14
Ксилан 18
Ксилоза 18
Лампа инфракрасного излучения 31
Лигнии 20, 55
Лизин 221
Лютер 164
Магний 68, 112
Масла в воде 108
спирте 191
углекислоте 249
Метоксильные группы 52
Микроорганизмы 152, 155, 193, 221,
264, 299, 305, 307, 308, 312
Минерализация 206, 209, 211, 216,
227, 231, 274
Молоко дрожжевое 226
— известковое 110
Мутиомер 290
Мутность 288, 289, 311
Мышьяк 69, ПО, 216
Натрий гидроокись 116
очистка 257
— хлористый очистка 103
Нитраты 282
Нитриты 279
Окисляемость воды бихроматная 266,
268
йодатная 269
перманганатная 254
— спирта 190
Оксиметилфурфурол 94
Олигосахариды 91, 122
Осаждение редуцирующих несаха-
ров 58, 148
Остаток прокаленный 243, 251
— сухой 190
Отстойник Лысенко 144
Очистка азота 213
— воды 267
— дихлорэтана 214
— эфира 214
— этанола 213
— натрия едкого 257
хлористого 103
Пентозаны 18, 42
Пентозы 23, 42, 78, 80
Поглотитель Полежаева 248
Полисахариды 17, 18, 19, 22, 42
Проба отбор 6, 7, 10, 13
— разделка 9
— средние 8
Пробоотборники 10г 12, 13, 14, 15
Потенциометрия 85, 125, 128
Протеин сырой 210, 227
Проявитель 78, 79, 137, 218
.324
Проявление хроматограмм 79,220, 138
Растворитель 77, 78, 137, 218
Реактив Несслера 274
Редуцирующие вещества 33, 39, 57,
73, 83, 85, 91, 93, 95, 119, 121, 122,
143
Сахар 33, 57, 85, 95, 161, 199
— сбраживаемый 147, 151
Свидетель 80, 88, 218, 253, 273
Свинец определение 234
— основной уксуснокислый 57
Сера 66
Скипидар 237
Спирт метиловый 162, 179, 189
— этиловый 3, 147, 164, 169, 172, 173,
175, 177, 182
Спиртомер 173, 175
Способность редуцирующая 39, 91
— спиртообразующая 158
— ферментативная 226
Сульфат-ион 63, 243, 288
Титрование кондуктометрическое 129,
132, 133
— потенциометрическое 85
Токсичность 144, 307
Точность 5
Требования технические на двуокись
углерода 244
дрожжи 224
спирт 184
фурфурол 240
Трилои Б 68, 97, 135
Фосфор 114, 202
Фотоэлектроколориметр 203
Фтор 106
Фурфурол 42, 142, 191, 235, 239, 286
Хроматографироваиие в колонке 139
— иа бумаге 73, 137, 199, 217
Хлор 101
Хлориды удаление 267
ХПК 266, 268, 269
Число пороговое 292, 297
Цветность 292
Целлюлоза 17
Цилиндры Генера 274, 282
— Несслера 274
Эбулиостат 35, 86
Электроды каломельный 126
— платиновый 86, 119, 131
— ртутный капельный 197
— стеклянный 126
— сурьмяный 128
— хлорсвинцово-амальгамный 87
Электролитический ключ 86
Элюация 89, 96, 221
Эталон 204, 280, 287, 290, 292
Щелок сульфитный 3, 25, 57, 59,
60—69
Щелочность 252
Ячейка 119, 197
СОДЕРЖАНИЕ
Введение
Организация работы центральной заводской лаборатории . . . .
О точности и достоверности результатов анализов
Глава 1. Отбор проб
Отбор проб сыпучих материалов
Отбор проб жидкостей
Отбор проб на очистных сооружениях
Отбор проб газов
Глава 2. Сырье
Состав растительных тканей
Гидролиз полисахаридов
Состав сульфитного щелока
Определение гранулометрического состава древесного сырья . .
Определение содержания хвойных и лиственных пород
древесины
Экспрессный метод определения влажности сырья
Определение влажности сырья различного гранулометрического
состава
Определение влажности измельченного материала
Определение минеральных веществ (золы)
Эбулиостатический метод определения редуцирующих Сахаров
(метод Ннзовкина и Емельяновой)
Определение легко- н трудногидролизуемых полисахаридов
(метод Кизеля и Семнгановского)
Определение пентозанов (метод Толленса)
Определение уроновых кислот (метод Волской)
Определение веществ, экстрагируемых водой
Определение веществ, экстрагируемых органическими
растворителями
Определение метоксильных групп (метод Фибека и Шваппаха)
Определение лигнина по Класону (модификация Комарова) . . .
Определение редуцирующих Сахаров в сульфитном щелоке . .
Определение сухих веществ высушиванием на бумажных
роликах
Определение сухих веществ по плотности
Определение Общей SU2
Определение легкоотщепляемой SO2
Определение моносульфита кальция
Определение свободной SO2
Определение иона SO4 комплексометрическим методом
Определение серы
Определение SO2, связанной в лигносульфоновом комплексе . .
Определение кальция и магния комплексометрическим методом
Определение мышьяка колориметрическим методом
Хроматографическне методы определения редуцирующих веществ
Хроматографическое разделение веществ на бумаге
Хроматографирование для качественных методов
Хроматографирование для количественных методов
Определение редуцирующих веществ эбулиостатическим потен-
циометрическим методом
Определение олигосахаридов
Определение уроновых кислот
Определение оксиметилфурфурола
Определение редуцирующих веществ колориметрическим методом
Глава 3. Вспомогательные материалы 96
Вода 96
Определение жесткости воды комплексометрическим методом . . 96
Определение кальция в присутствии магния 100
Определение хлора 101
Определение брома и йода 102
Определение фтора 106
Определение масел 108
Серная кислота 108
Определение моногидрата ареометром 108
Определение моногидрата титрованием 109
Определение мышьяка ПО
Известковое молоко ПО
Определение окиси кальция титрованием 111
Определение окиси кальция ареометром 111
Определение магния 112
Аммофос 112
Определение азота 112
Определение фосфора 114
Аммиачная вода 116
Определение аммиака 116
Едкий натр 116
Определение едкого натра 116
Глава 4. Получение и очистка растворов моносахаридов 117
Определение сахара и серной кислоты экспрессным методом . . 119
Определение неотмытых редуцирующих веществ и серной
кислоты в лигнине 121
Определение олигосахаридов 122
Определение степени полноты гидролиза 123
Определение общей кислотности 123
Определение pH индикаторными бумажками со стандартной
шкалой 124
Определение pH потенциометрическим методом 125
Определение pH при непрерывной нейтрализации гидролнзата 128
Определение серной и органических кислот титрованием с
различными индикаторами 128
Определение серной и органических кислот кондуктометриче-
ским титрованием 129
Определение кальция кондуктометрическим титрованием .... 132
Определение гипса кондуктометрическим титрованием 133
Определение свободных и связанных летучих органических
кислот 134
Определение левулиновой кислоты хроматографическим методом 137
Определение органических кислот хроматографическим методом 139
Определение фурфурола 142
Определение показателей биологической доброкачественности . . 143
Глава 5. Получение этилового спирта 147
Определение сбраживамых Сахаров химическим методом .... 147
Определение сбраживаемых Сахаров биохимическим методом . . 151
Определение физиологического состояния дрожжей микроскопи-
рованием 152
Определение наличия инфекции 155
Определение количества дрожжевых клеток в 1 г дрожжей
75%-ной влажности 157
Определение концентрации дрожжей в дрожжевых суспензиях 158
Определение спиртообразующей способности дрожжей 158
326
Определение бродильной активности дрожжей 160
Определение Сахаров экспрессным хроматографическим методом 161
Определение метилового спирта колориметрическим методом . . 162
Определение этилового спирта флотационным методом (метод
Низовкина и Королькова) 164
Определение этилового спирта вакуум-нитритным методом ... 169
Определение этилового спирта экспрессным методом 172
Определение спирта стеклянным спиртомером 173
Определение этилового спирта металлическим спиртомером . . 175
Определение этилового спирта окислительным методом 177
Определение метилового спирта окислительным методом .... 179
Технические требования на этиловый спирт и методы его
испытания по стандартам разных стран 182
Технические требования 182
Определение крепости спирта 182
Определение кислотности 182
Определение сложных эфиров 183
Определение альдегидов 183
Определение метилового спирта 189
Определение окисляемое™ 190
Определение сухого остатка 190
Определение фурфурола 191
Определение сивушного масла 191
Определение веществ, нерастворимых в воде 191
Определение ацетона, других кетонов, изопропилового спирта и
третичного бутилового спирта 192
Глава 6. Получение кормовых дрожжей 192
Определение физиологического состояния дрожжей микроскопи-
рованием 193
Определение чистоты культуры дрожжей 195
Определение кислорода полярографическим методом 196
Определение Сахаров экспрессным методом 199
Определение фосфора колориметрическим методом (метод
Бриггса) 202
Определение калия 208
Определение сырого протеина экспрессным методом 210
Определение витамина D2 колориметрическим методом 212
Определение мышьяка в дрожжах 216
Определение аминокислотного состава белка 217
Определение лизина микробиологическим методом 221
Технические требования на сухие кормовые дрожжи и методы их
испытания по стандартам разных стран 223
Технические требования 223
Определение влажности и сухих веществ 223
Определение ферментативной способности дрожжей 226
Определение сырого протеина (метод Кьельдаля) 227
Определение белка по Бернштейиу 229
Определение усвояемого белка 230
Определение кислотности 231
Определение гидрохлорида тиамина (витамина ВО 231
Определение рибофлавина (витамина Вг) 232
Определение никотиновой кислоты (витамина РР) 233
Определение свинца 234
Глава 7. Получение фурфурола 235
Определение фурфурола экспрессным методом 235
Определение кислотности 236
Определение скипидара экспрессным методом 237
Определение фурфурола бромид-броматным методом 237
327
Технические требования иа фурфурол и методы его испытания по
стандартам разных стран 239
Технические требования иа фурфурол 239
Определение фурфурола с солянокислым гидроксиламином . . . 242
Проба иа содержание веществ, нерастворимых в воде 242
Определение фракционного состава фурфурола 243
Определение остатка после прокаливания в виде сульфата . . . 243
Глава 8. Получение двуокиси углерода 243
Определение двуокиси углерода 246
Определение воды по точке росы 247
Определение воды 247
Определение окисляемых веществ 248
Проба на содержание минеральных масел 249
Глава 9. Очистка сточных вод 249
Санитарные условия спуска сточных вод в водоемы 249
Определение оседающих веществ 250
Определение взвешенных веществ 250
Определение сухого и прокаленного остатка 251
Определение щелочности 252
Определение окисляемости перманганатиой (метод Кубеля) . . . 254
Определение растворенного кислорода (метод Винклера) .... 256
Определение биохимически потребляемого кислорода <ЪПК) . . 263
Определение константы скорости потребления растворенного
кислорода 266
Определение химически потребляемого кислорода (ХПК) бихро-
матным методом 266
Определение химически потребляемого кислорода (ХПК)
экспрессным методом 268
Определение химически потребляемого кислорода (ХПК) йодат-
ным методом 269
Определение общего азота 271
Определение аммиачного азота колориметрическим методом . . 273
Определение нитритов колориметрическим методом (метод
Грисса) 279
Определение нитратов (метод Грандваля и Ляжу) 282
Определение азота органических соединений 285
Определение фурфурола колориметрическим методом 286
Определение сульфат-иона экспрессным методом 288
Определение мутности 289
Определение цветности 292
Определение порогового числа цветности 294
Определение запаха 295
Определение порогового числа запаха 297
Определение качества активного ила 299
Определение концентрации активного ила 304
Определение илового индекса 307
Динамика осаждения активного ила 307
Определение токсичности 307
Определение витамина Віг 308
Определение микроорганизмов из группы кишечион палочки . . 312
Обоснование выбора анализов для контроля за работой
очистных сооружений 313
Справочные таблицы 316
Список литературы 319
Предметный указатель 323