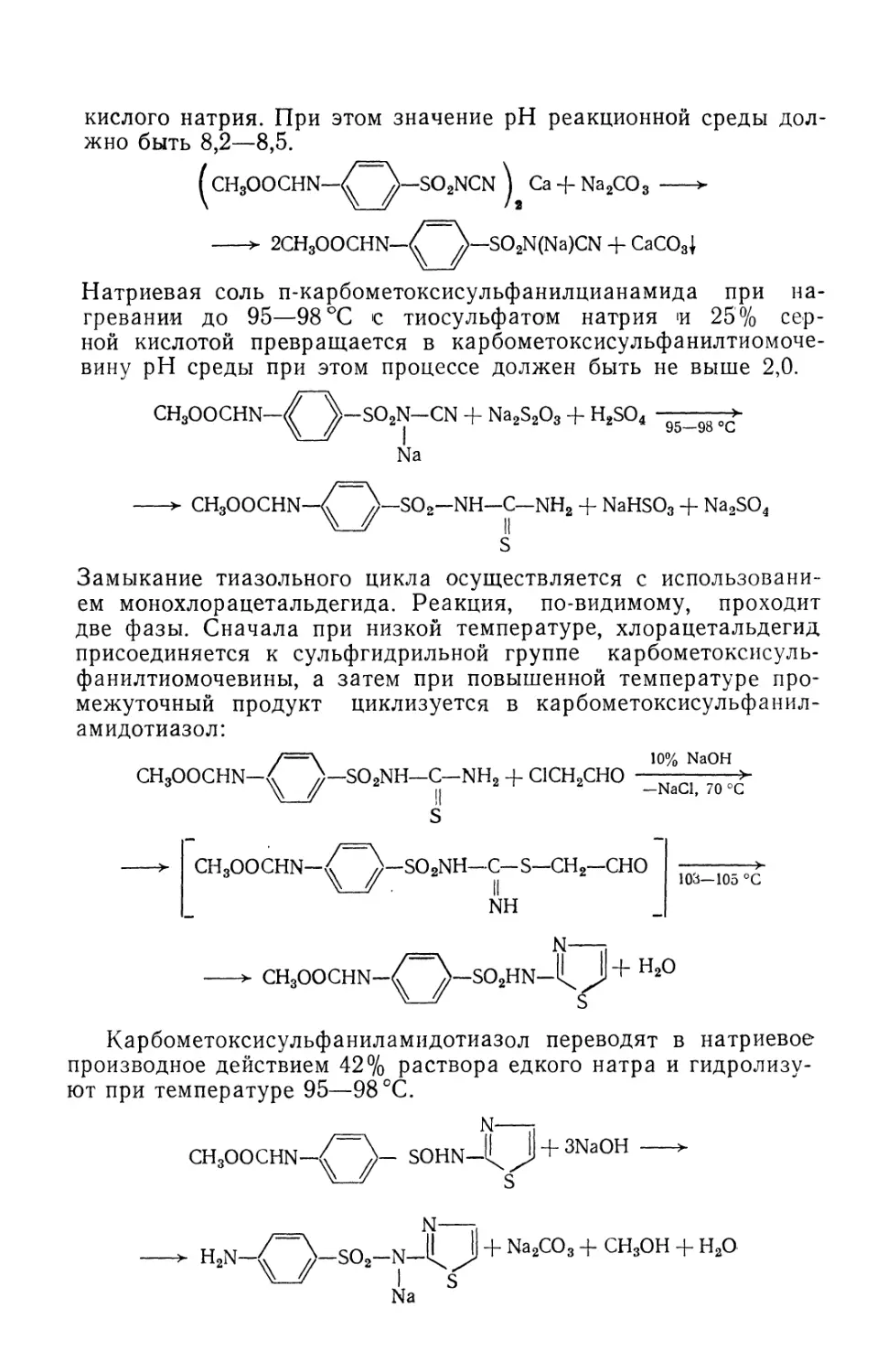
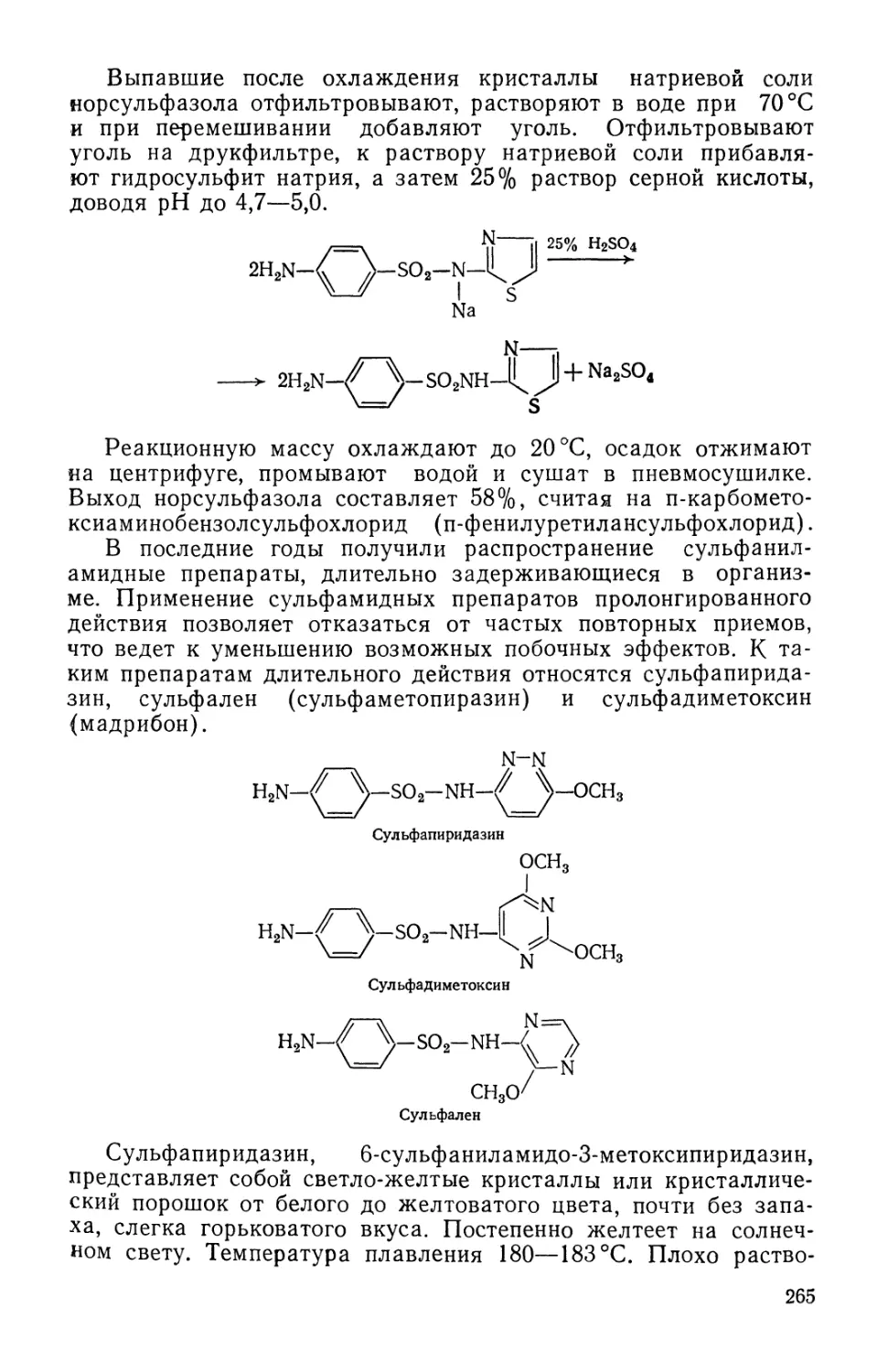
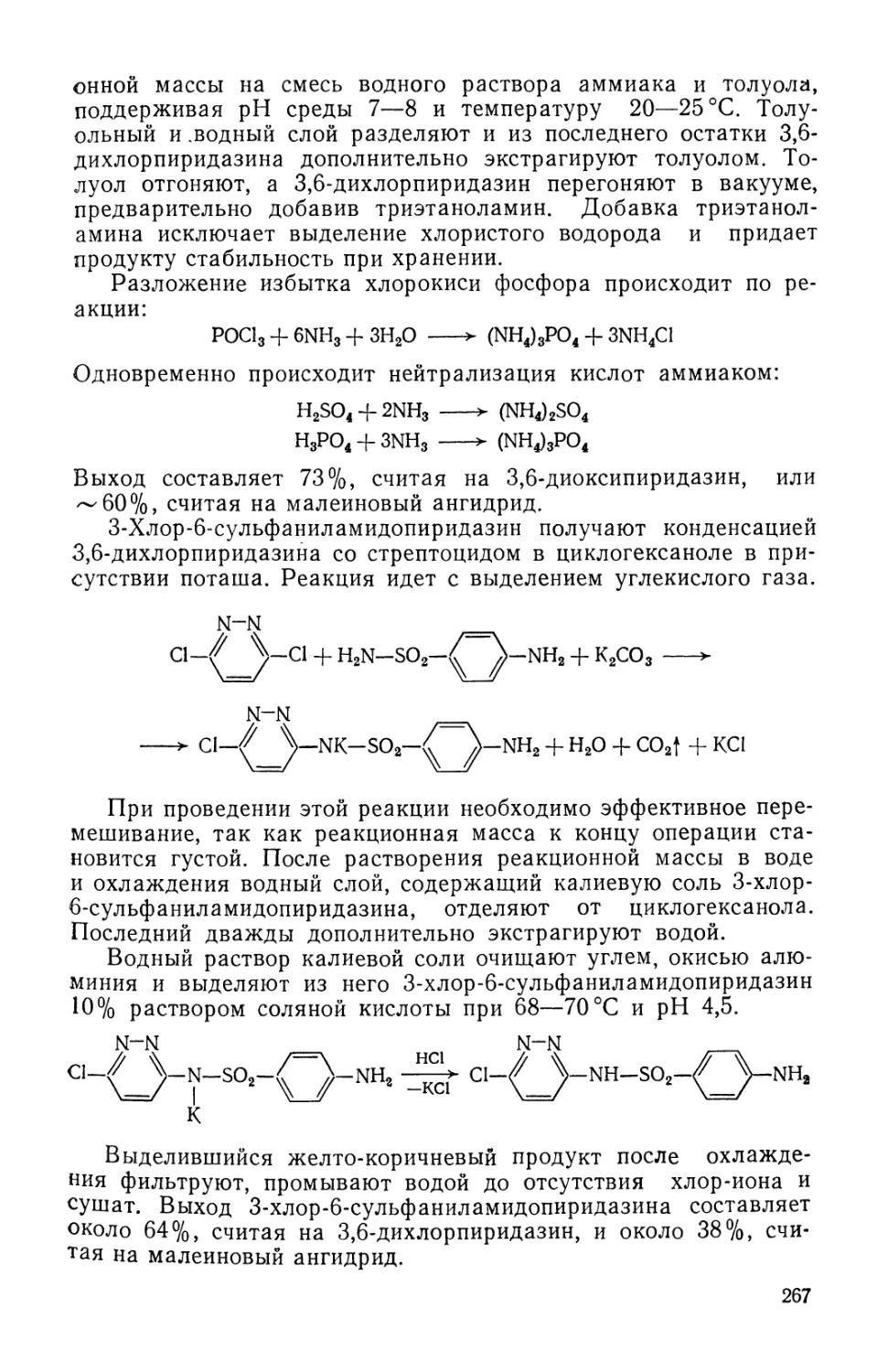
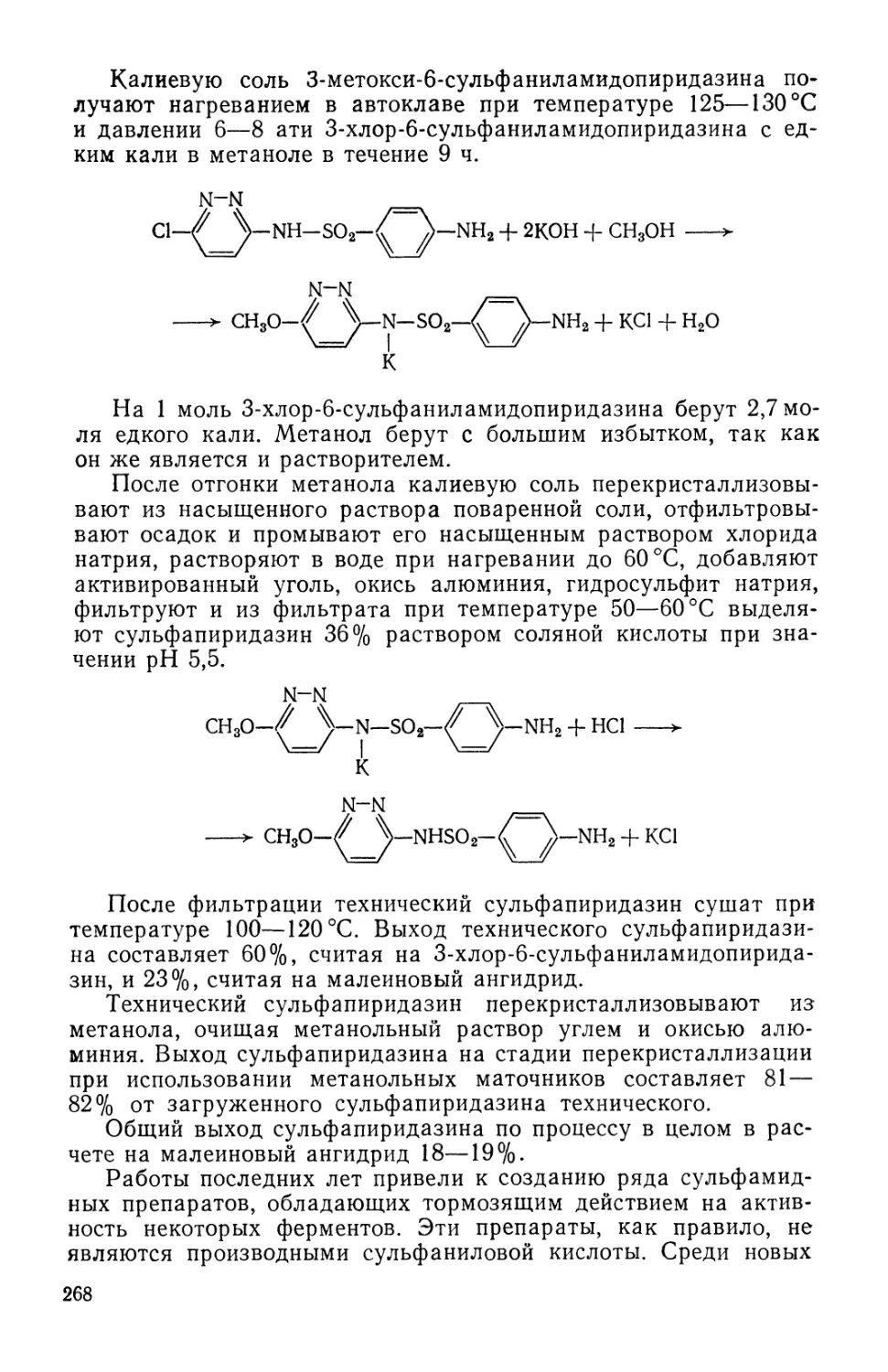
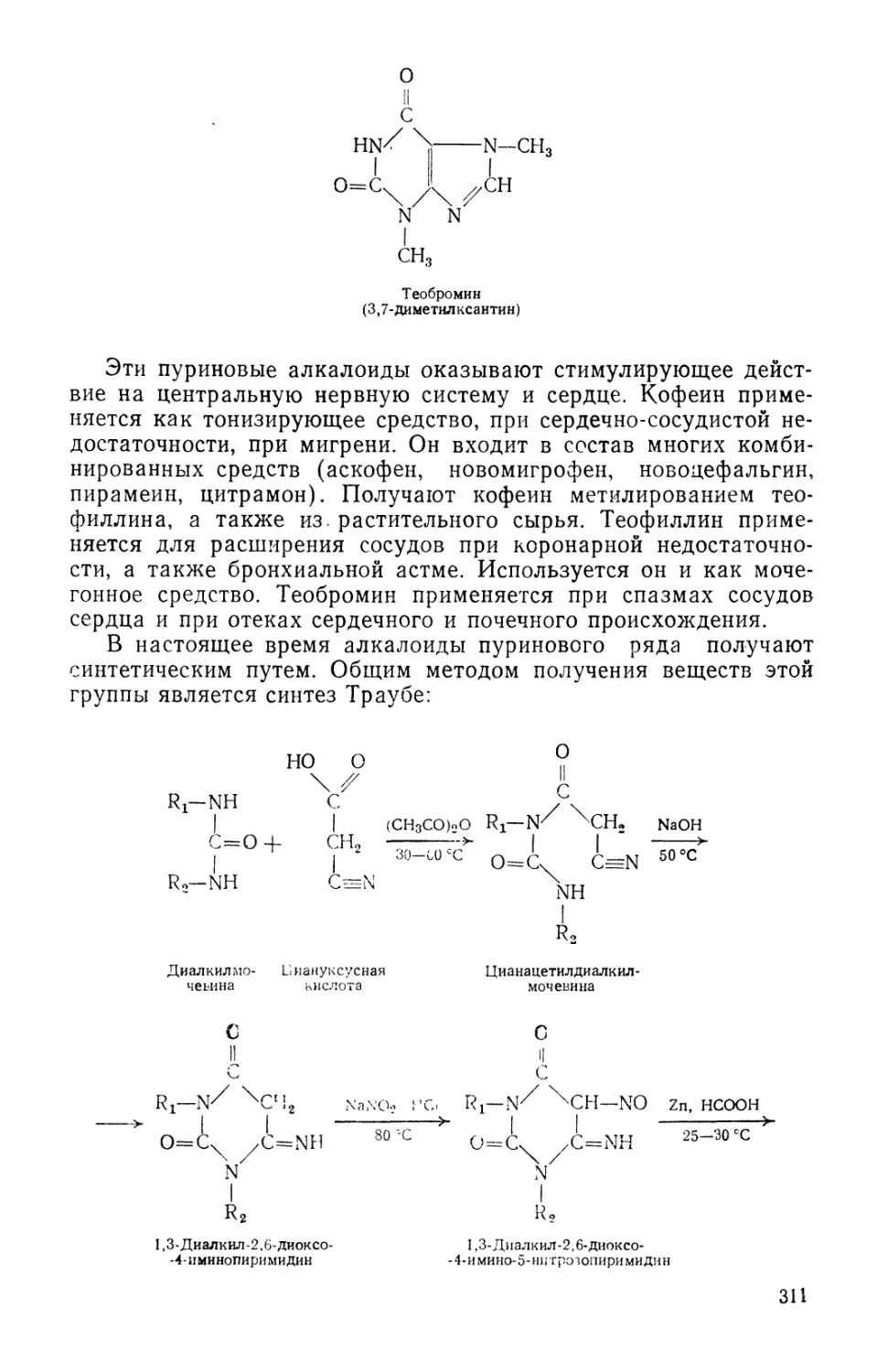
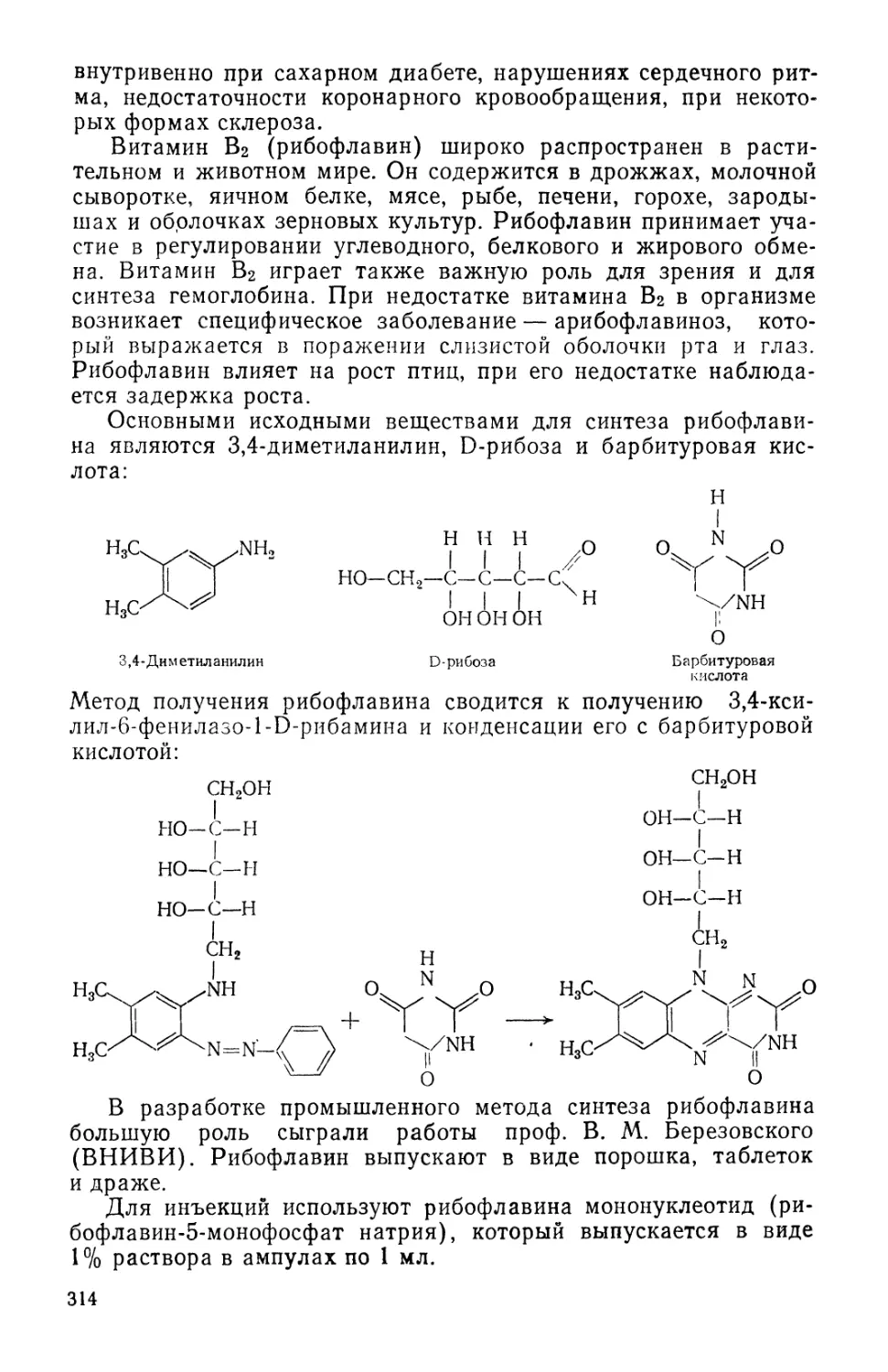
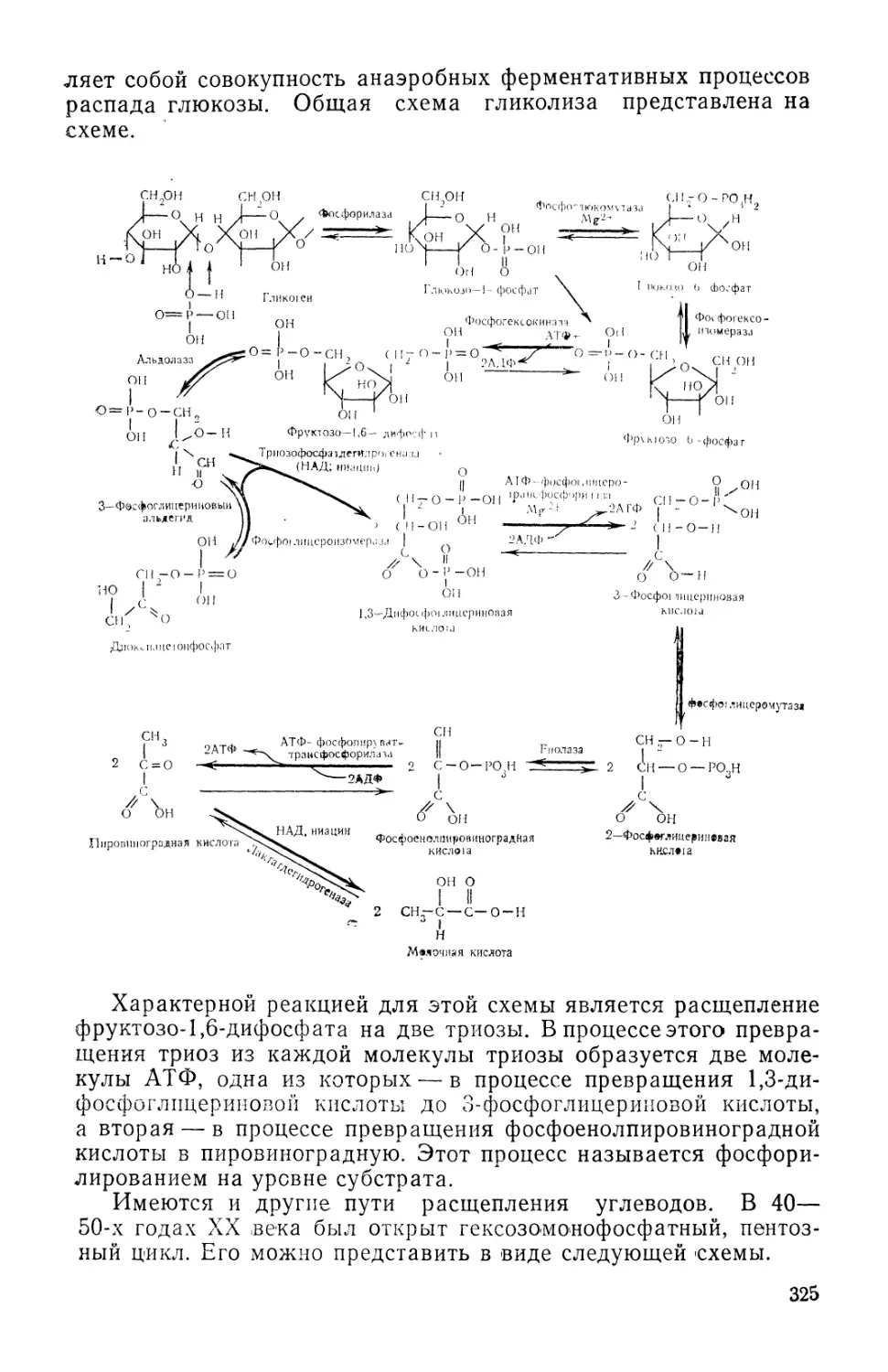
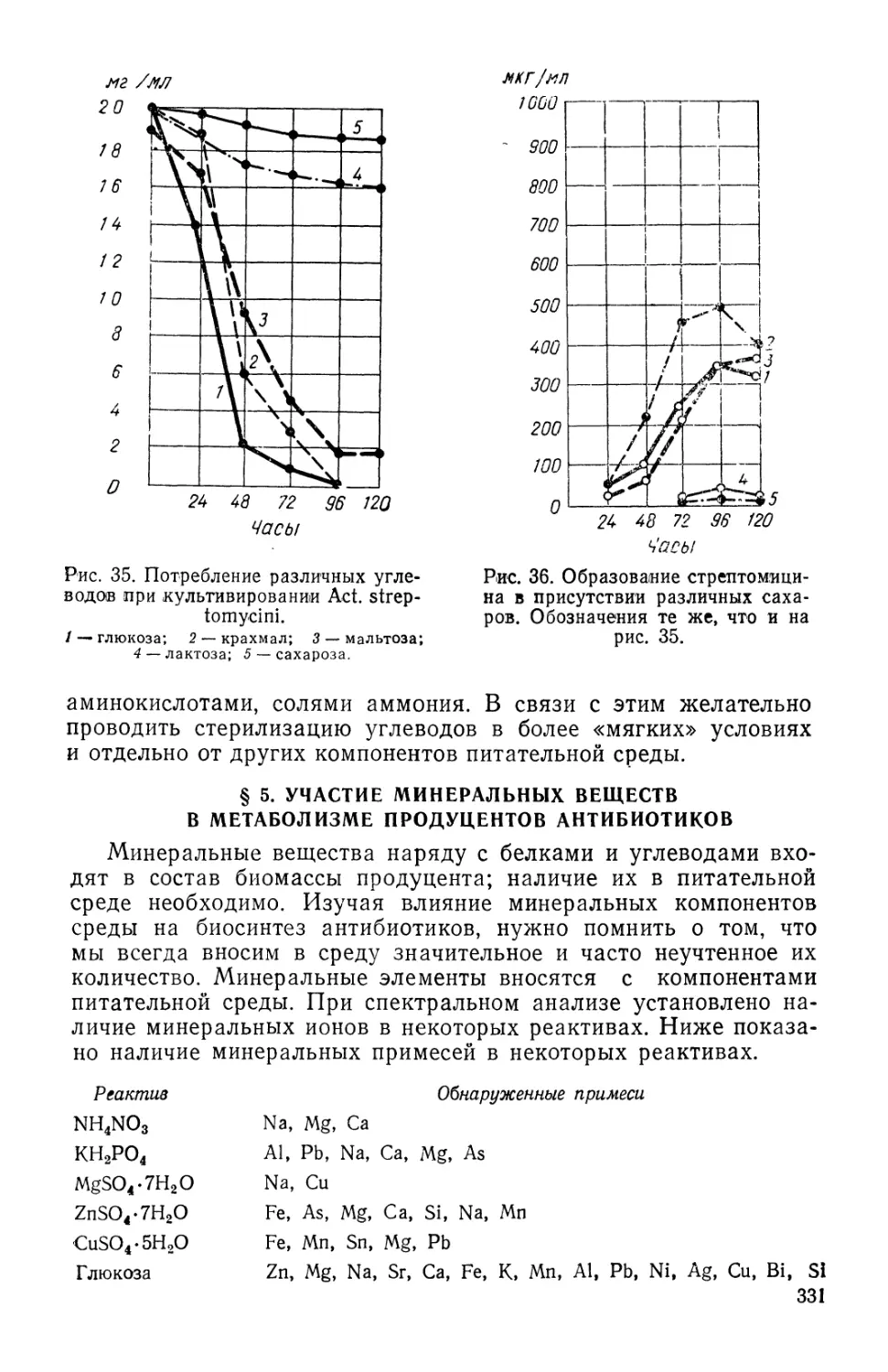
/



























































































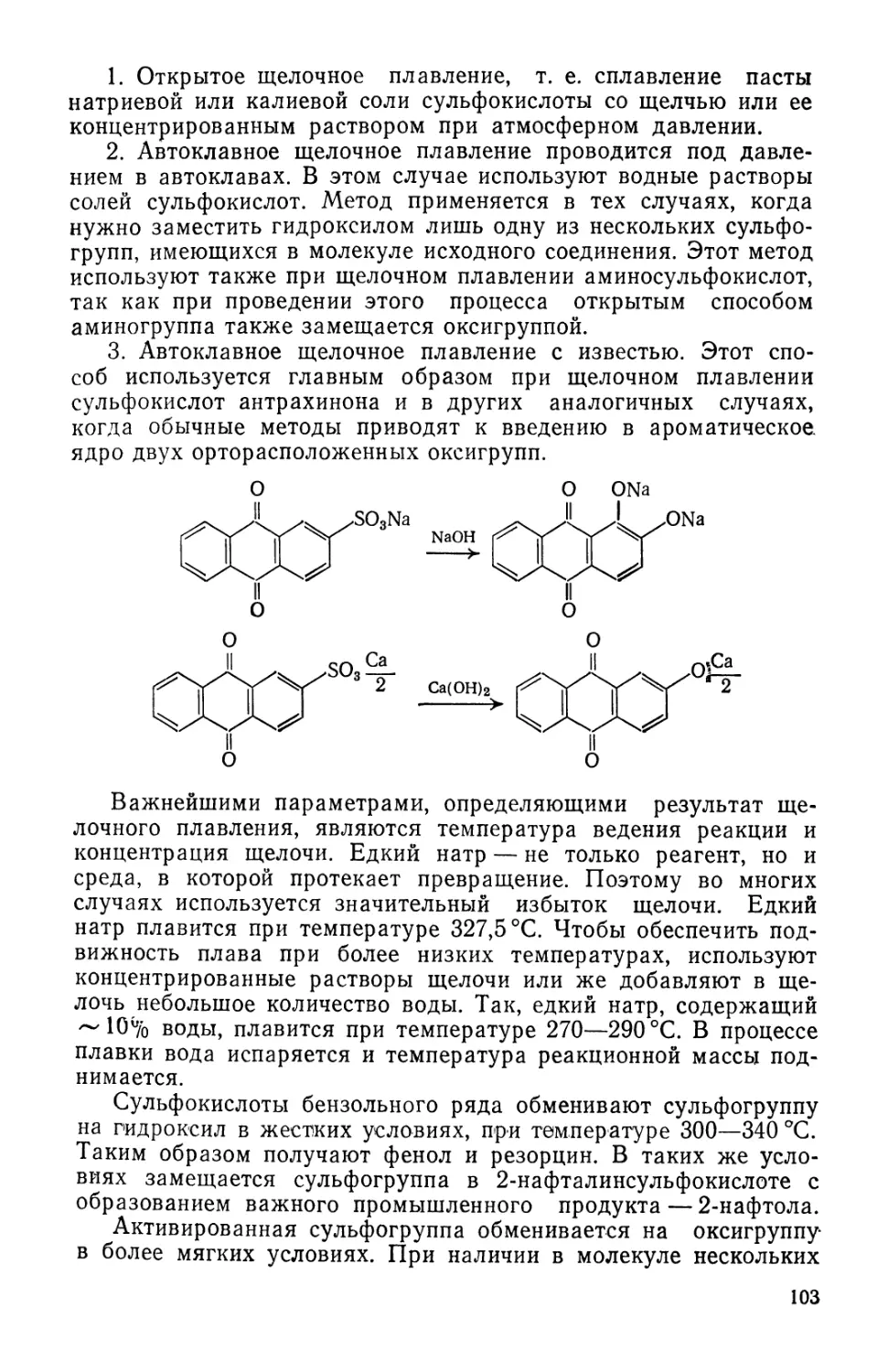





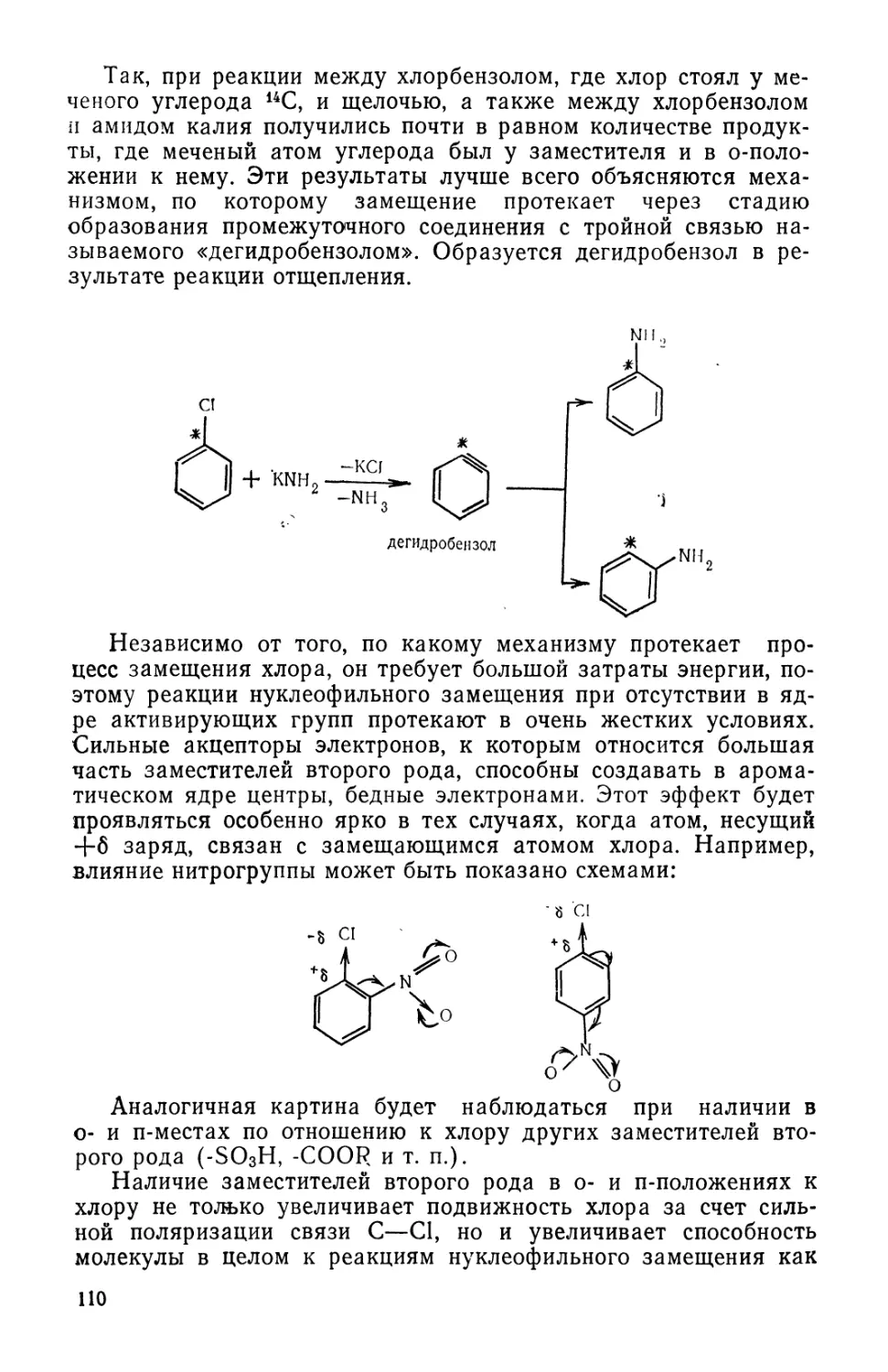





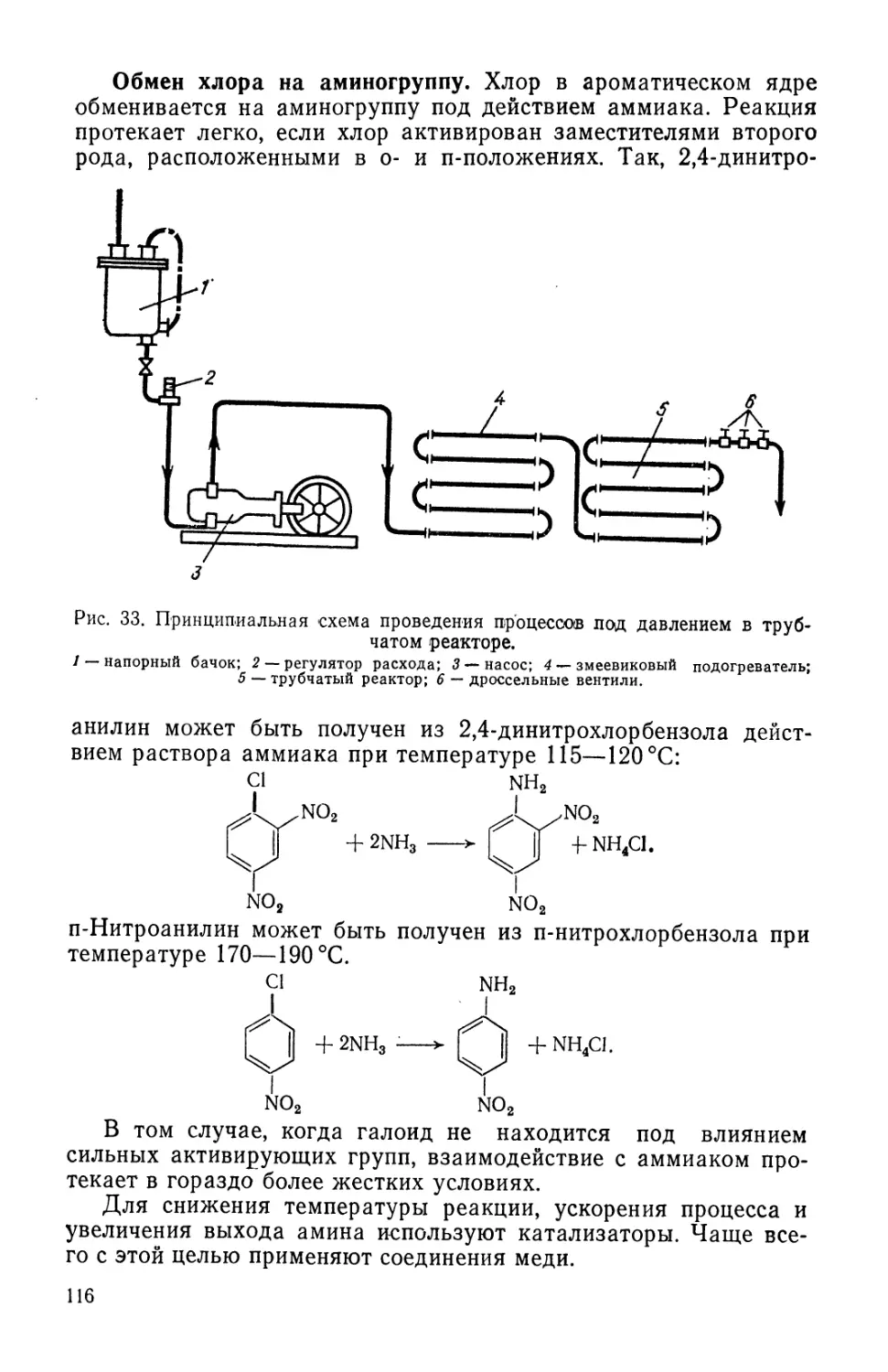
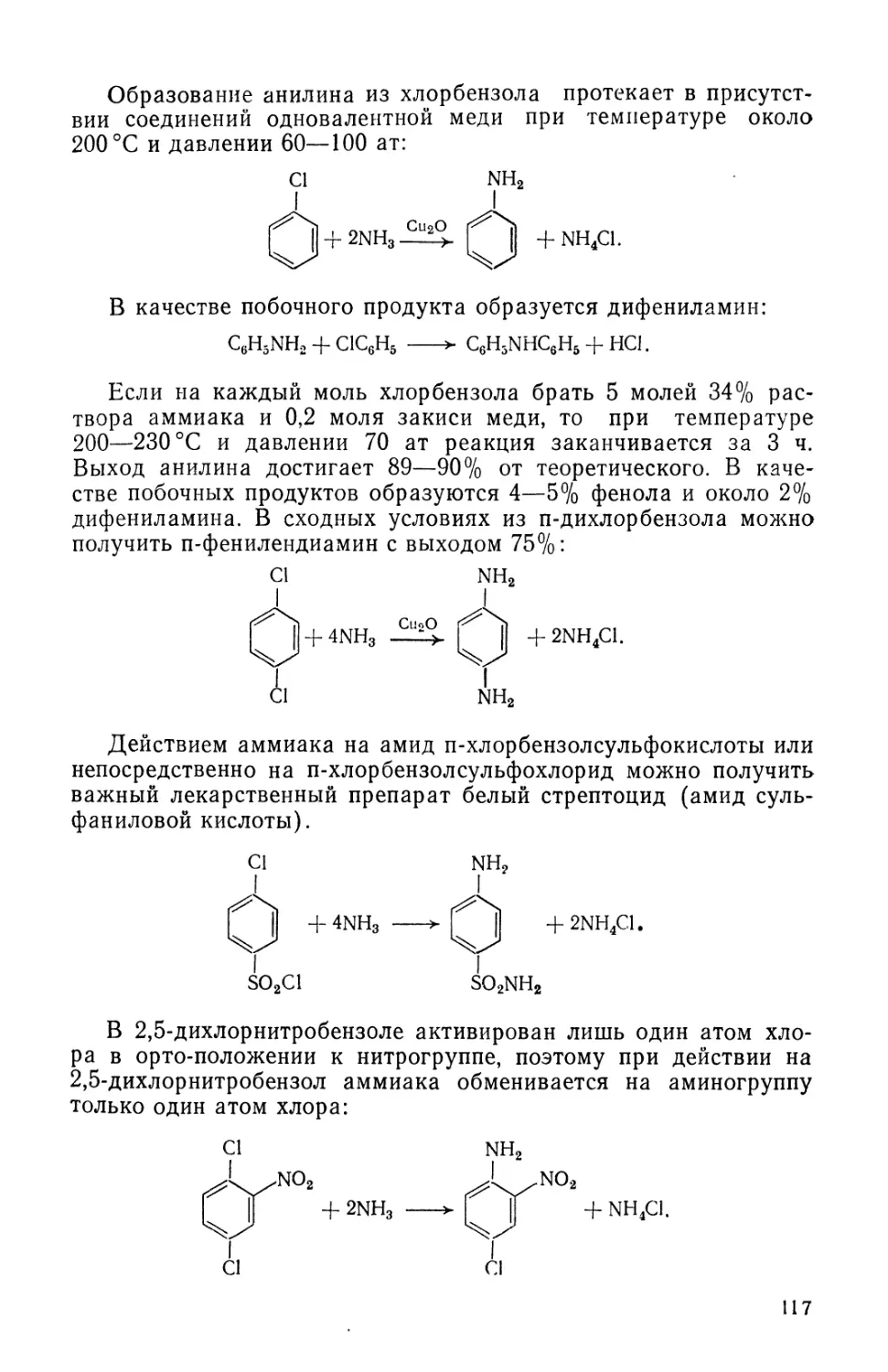





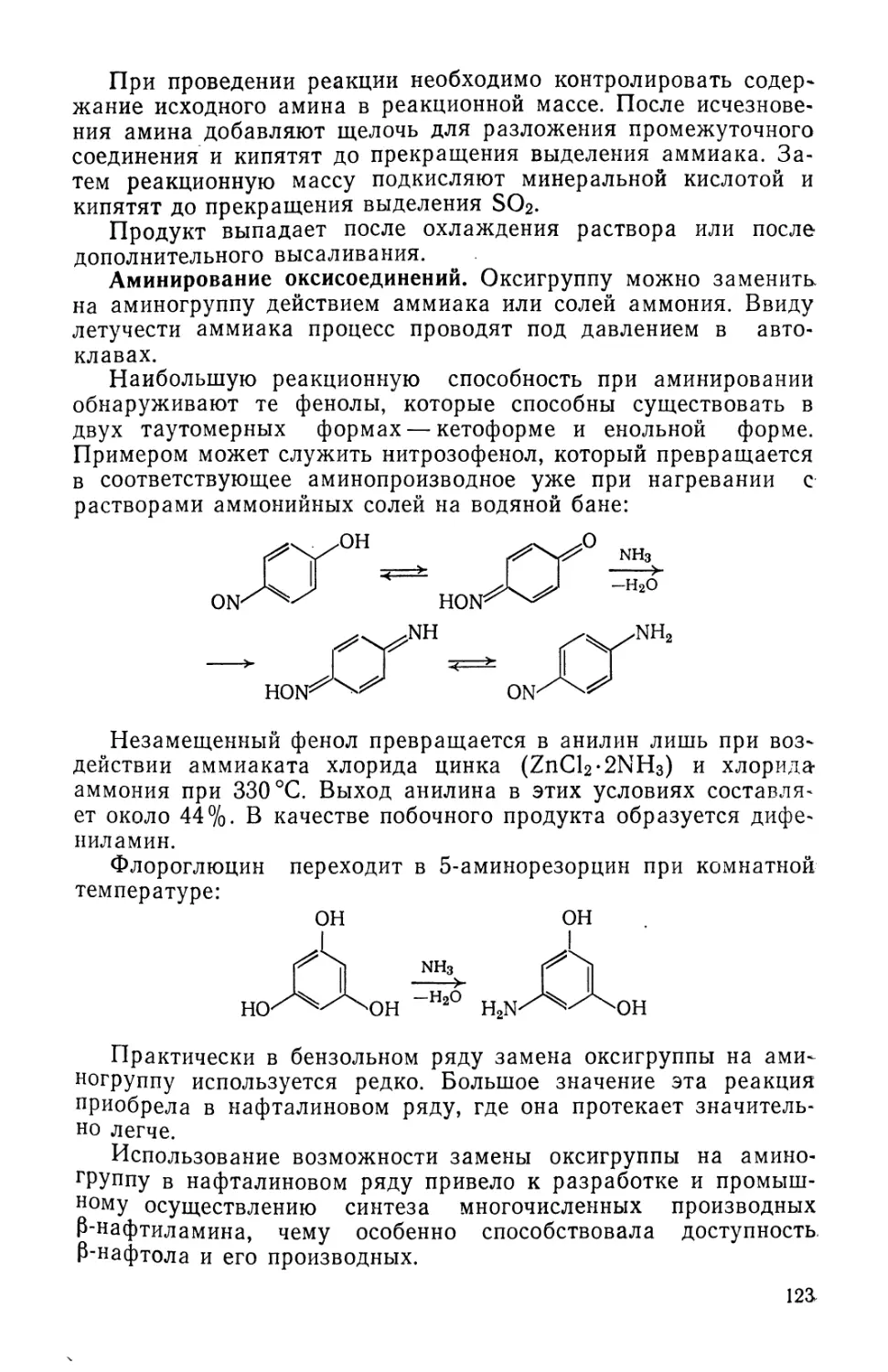
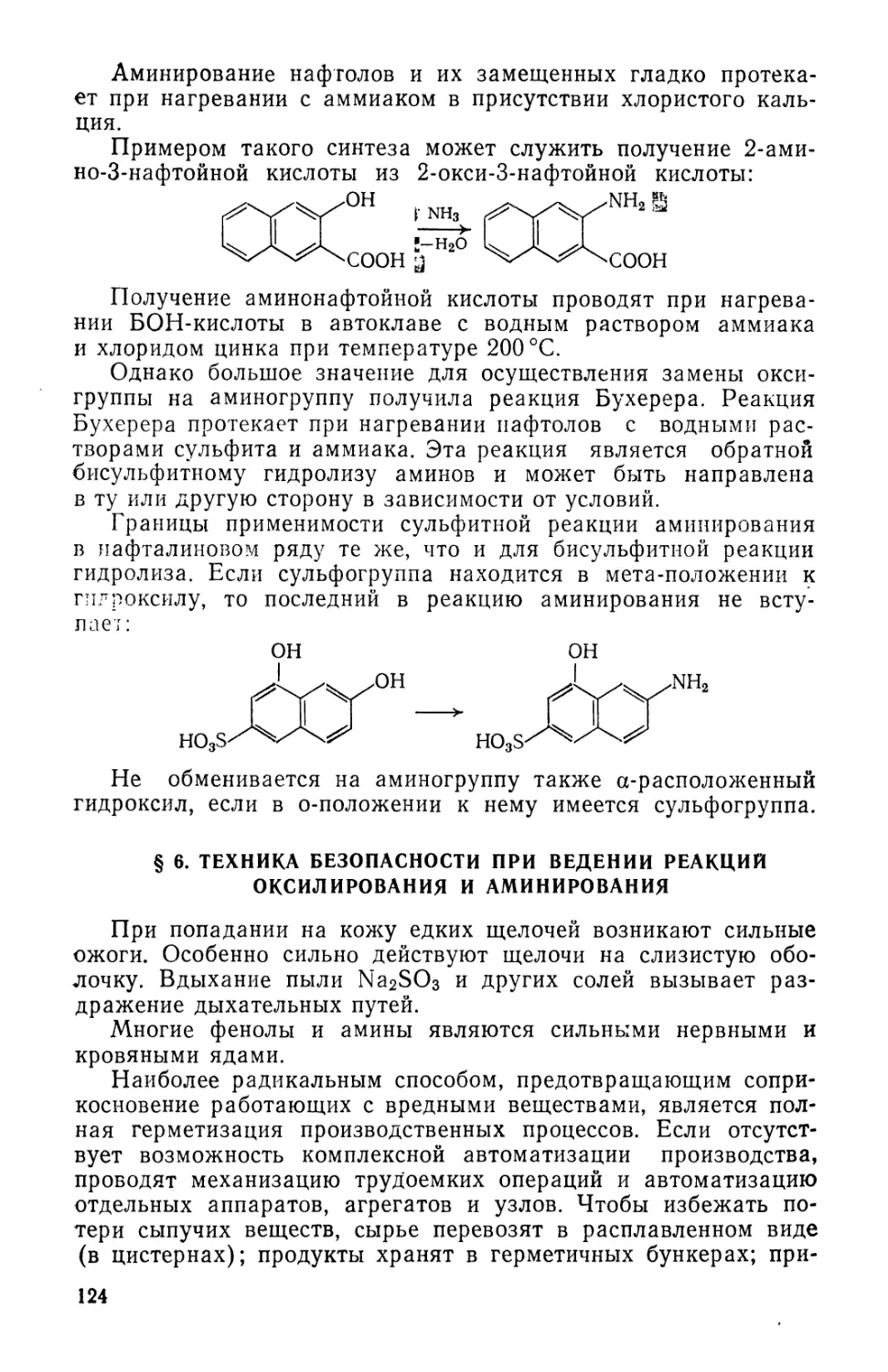








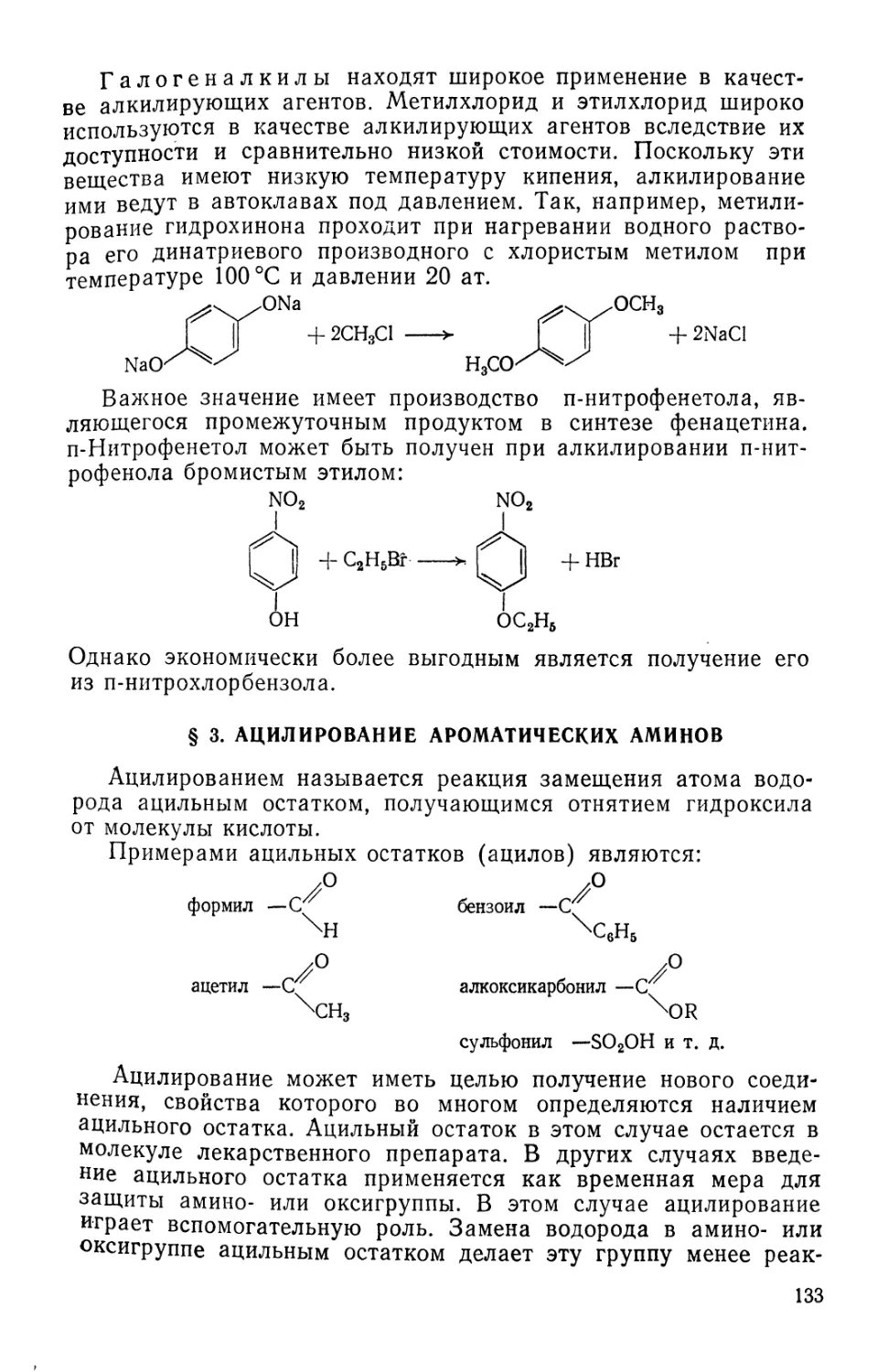


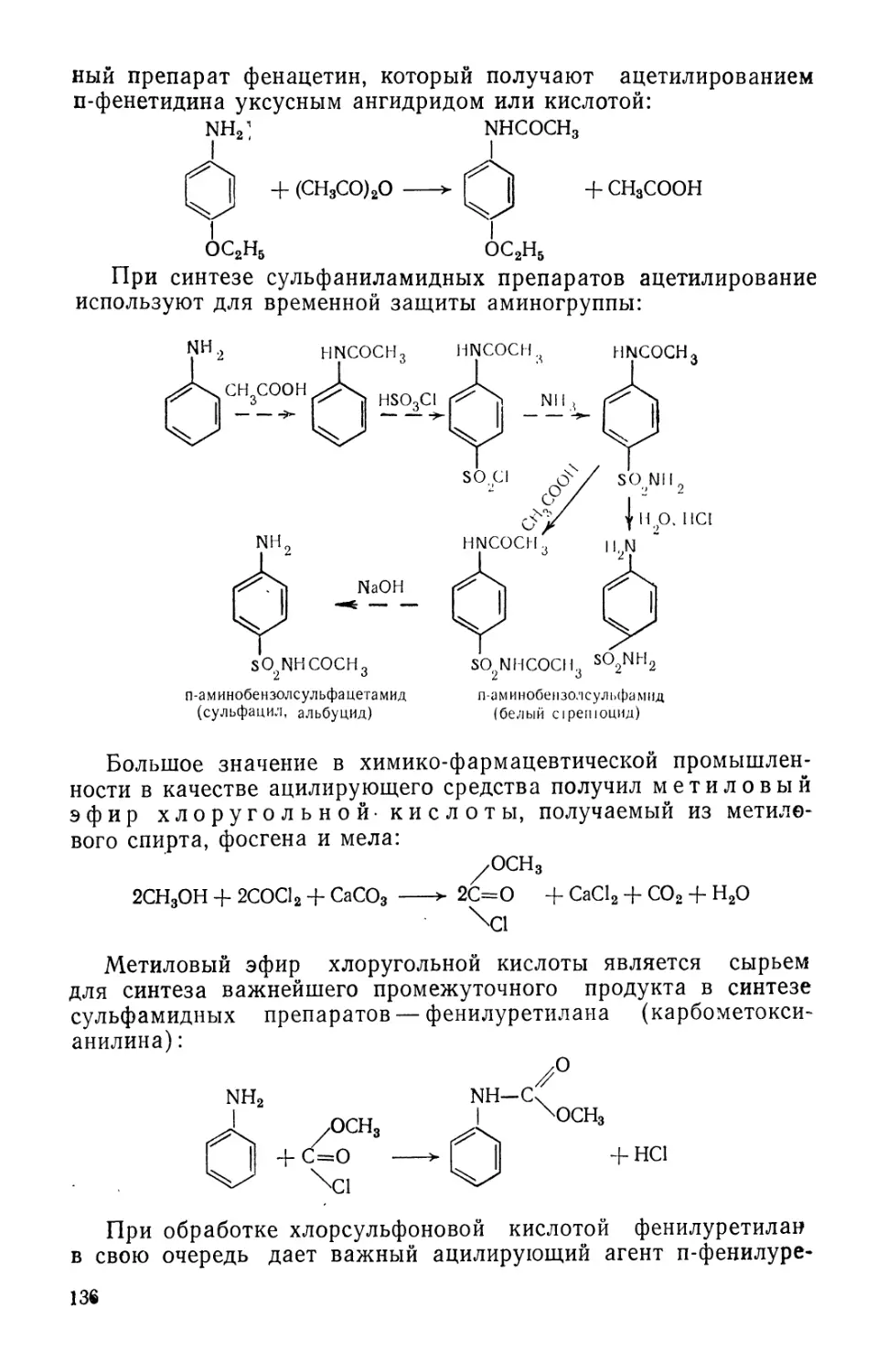

















































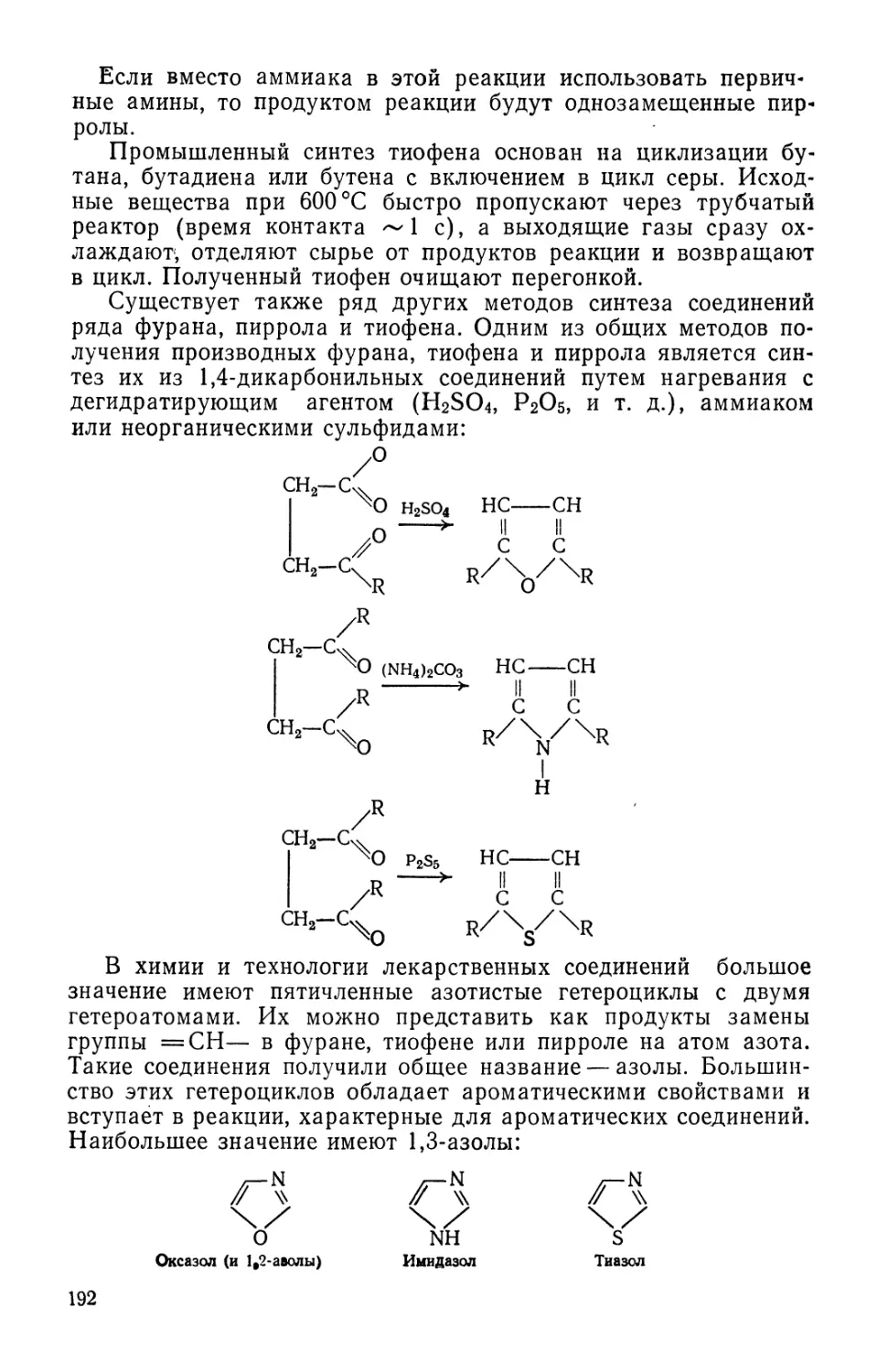





















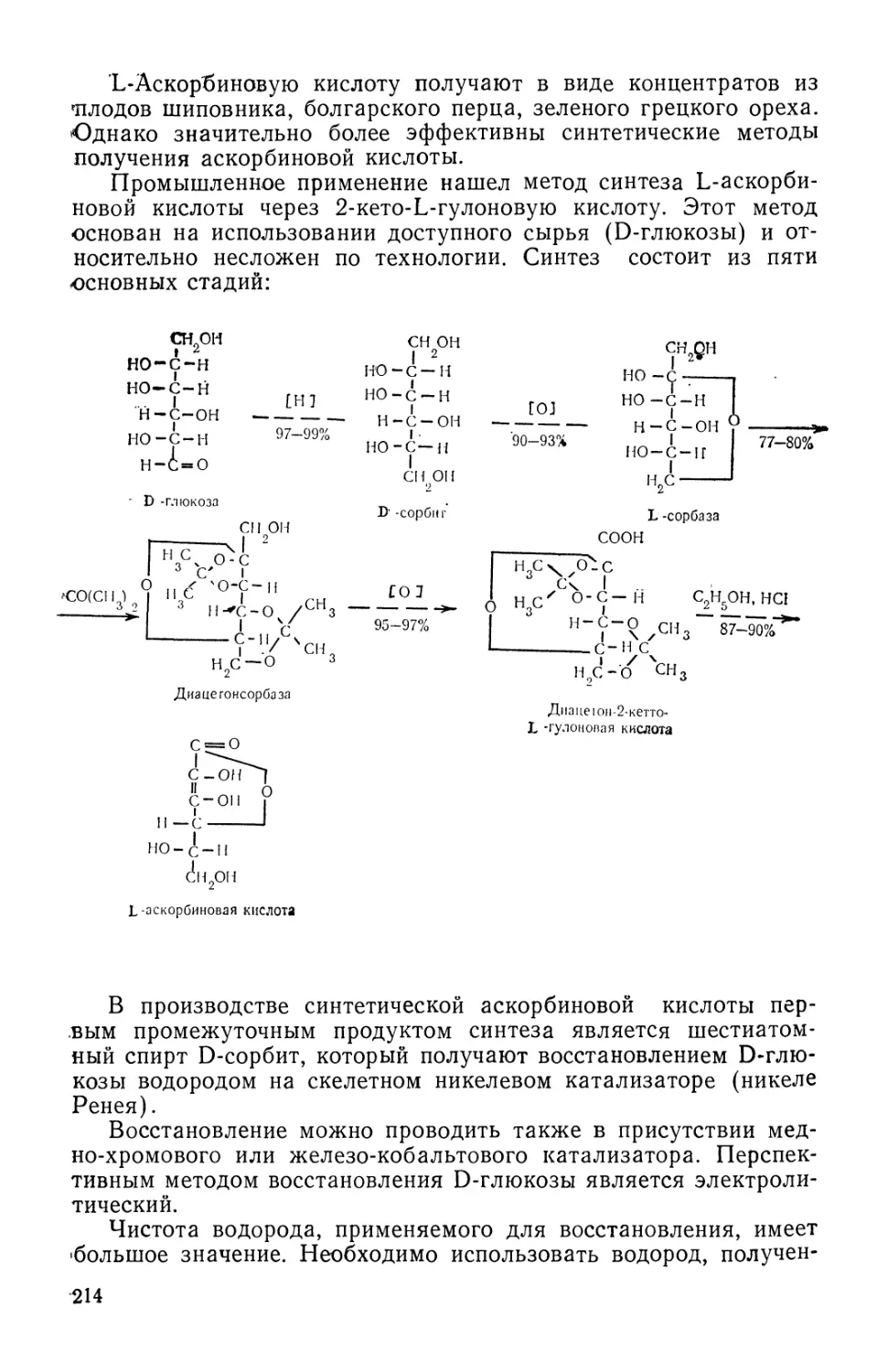








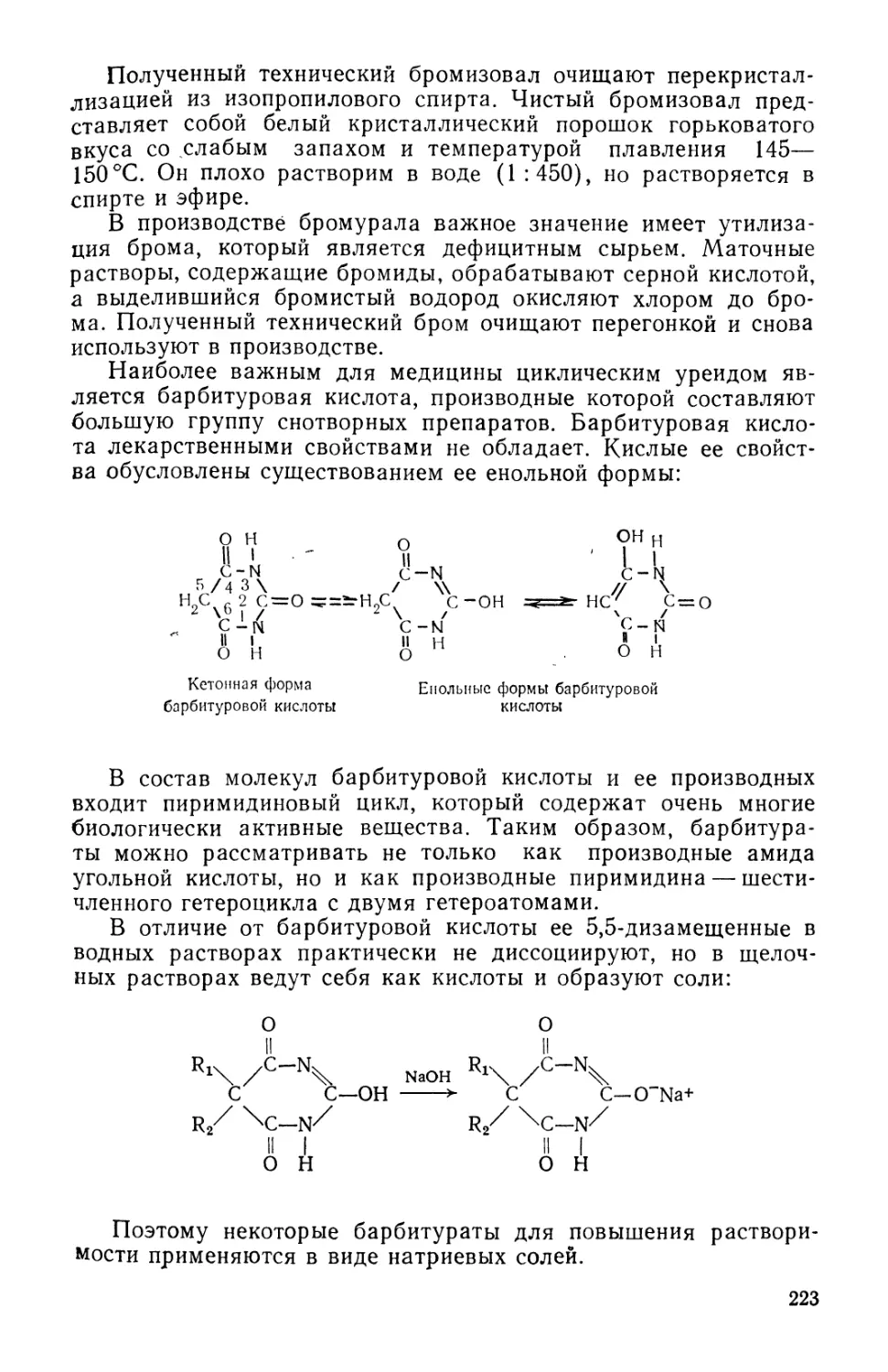







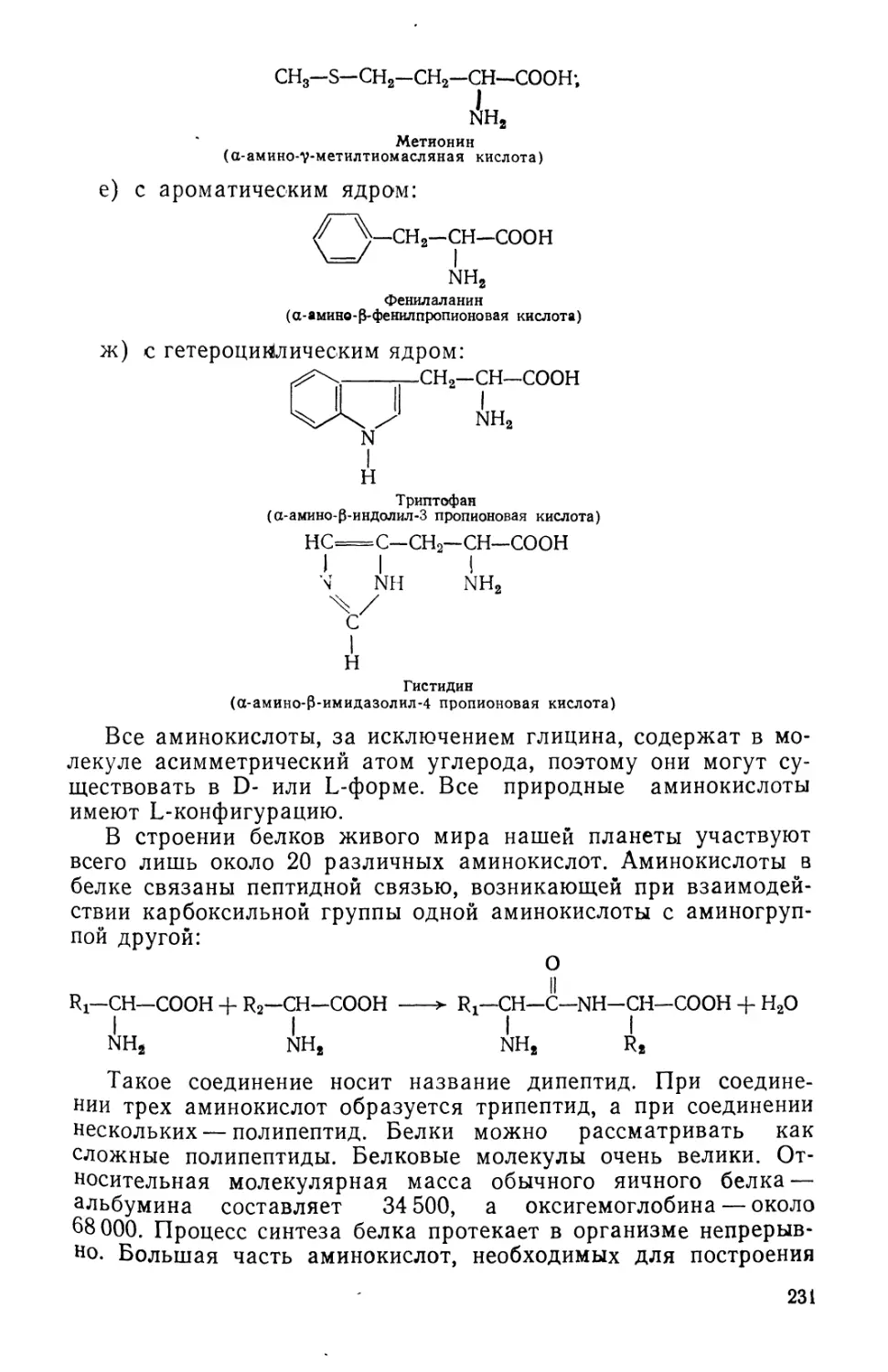



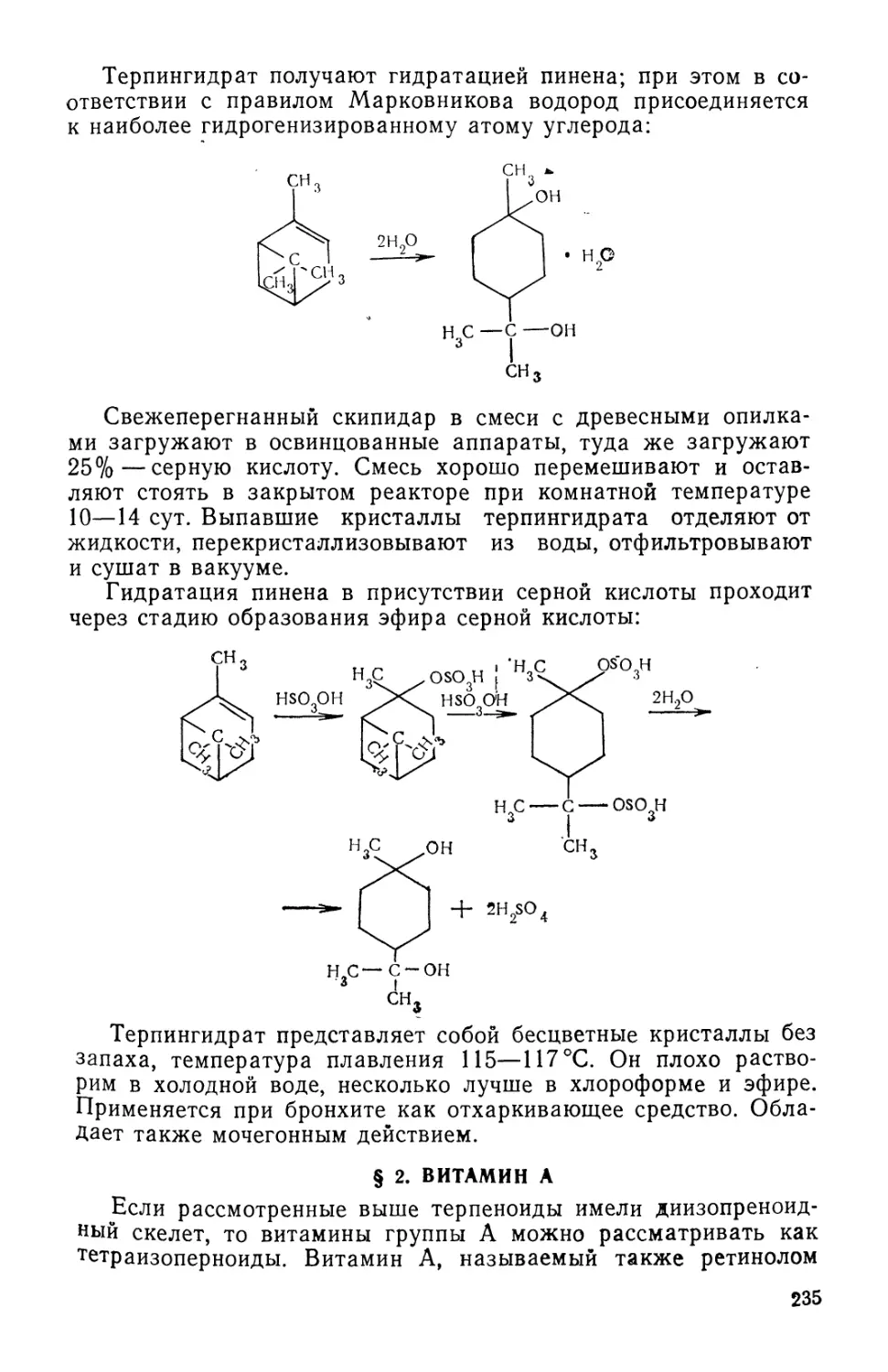

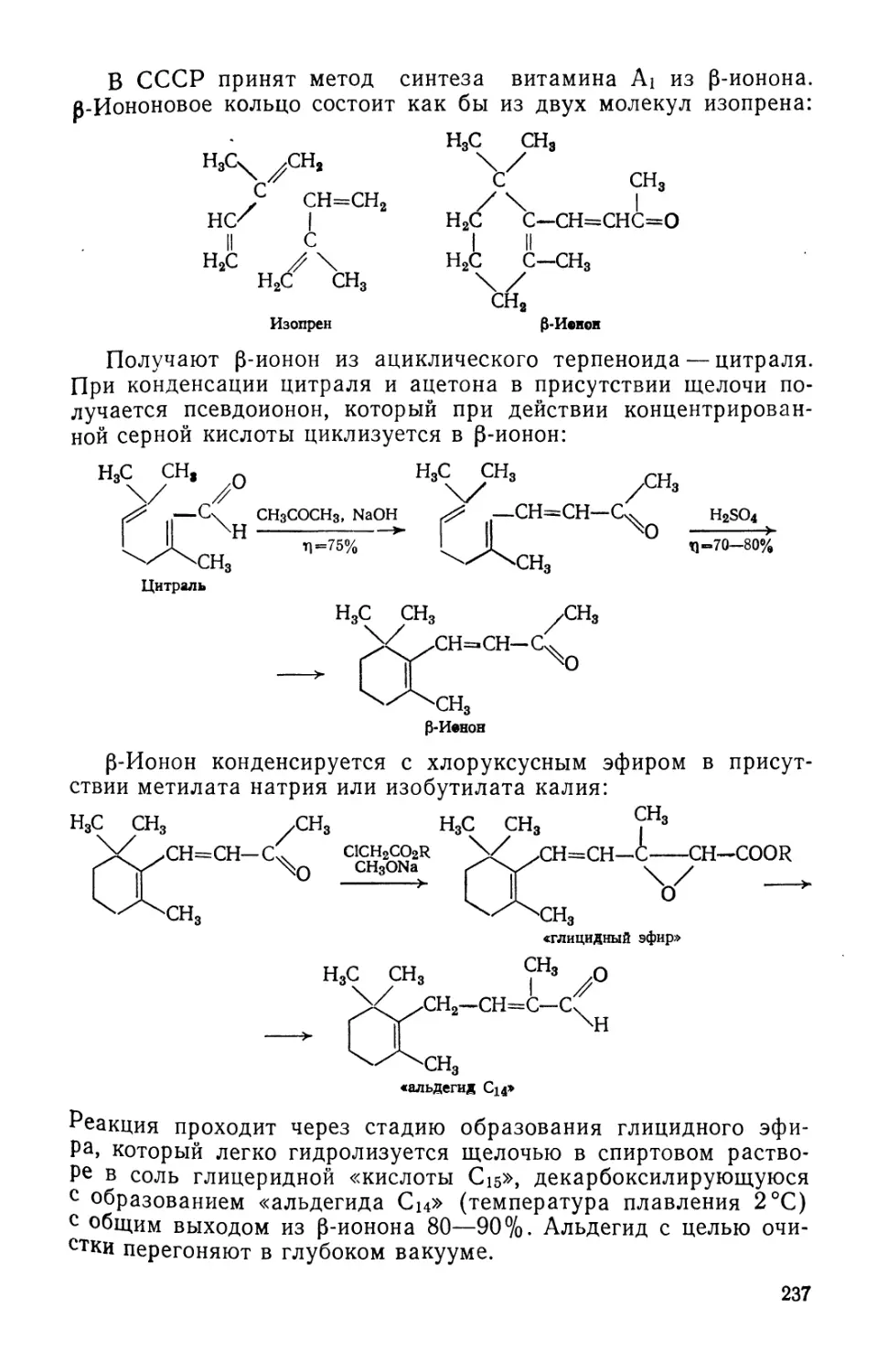
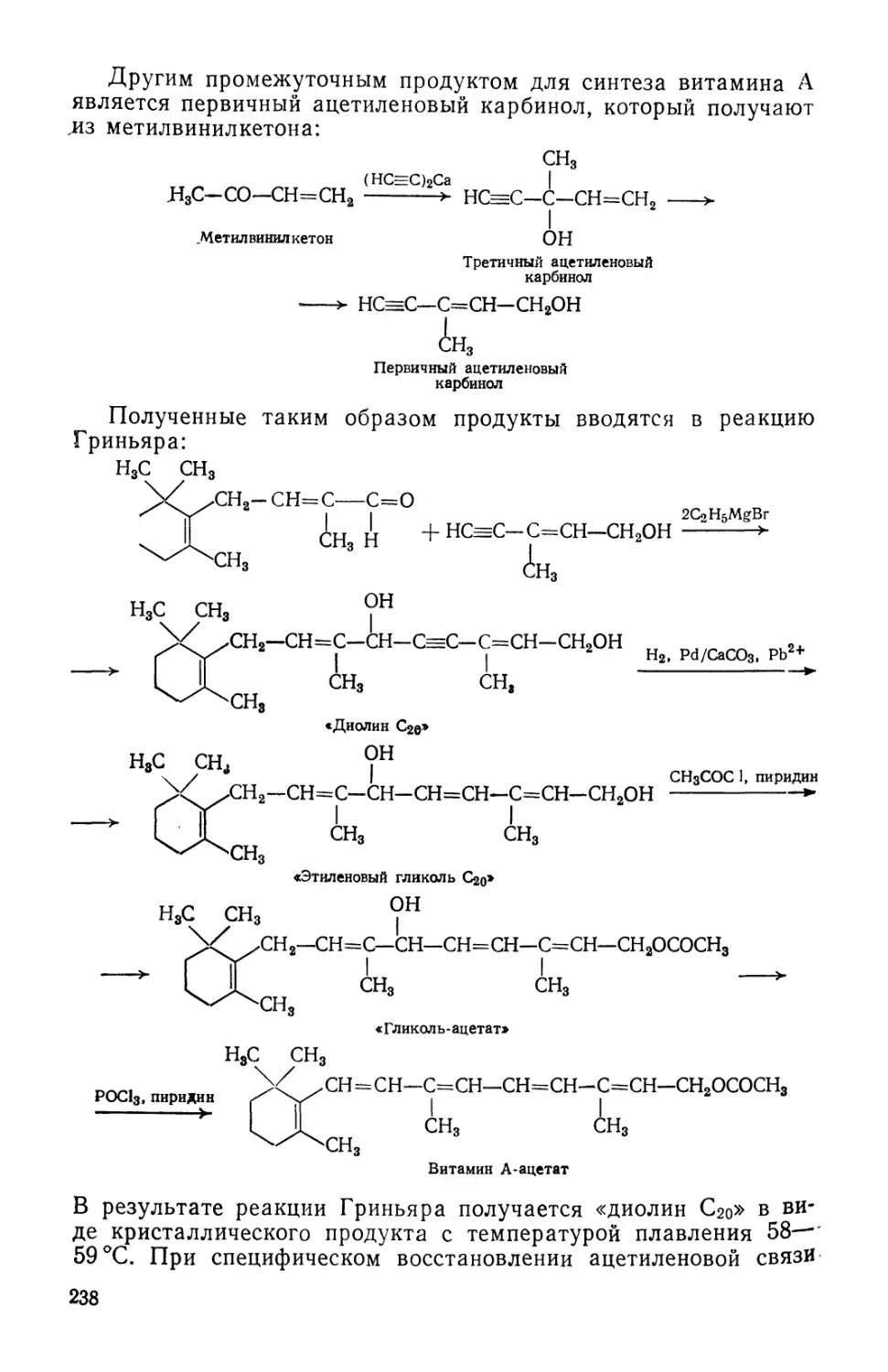



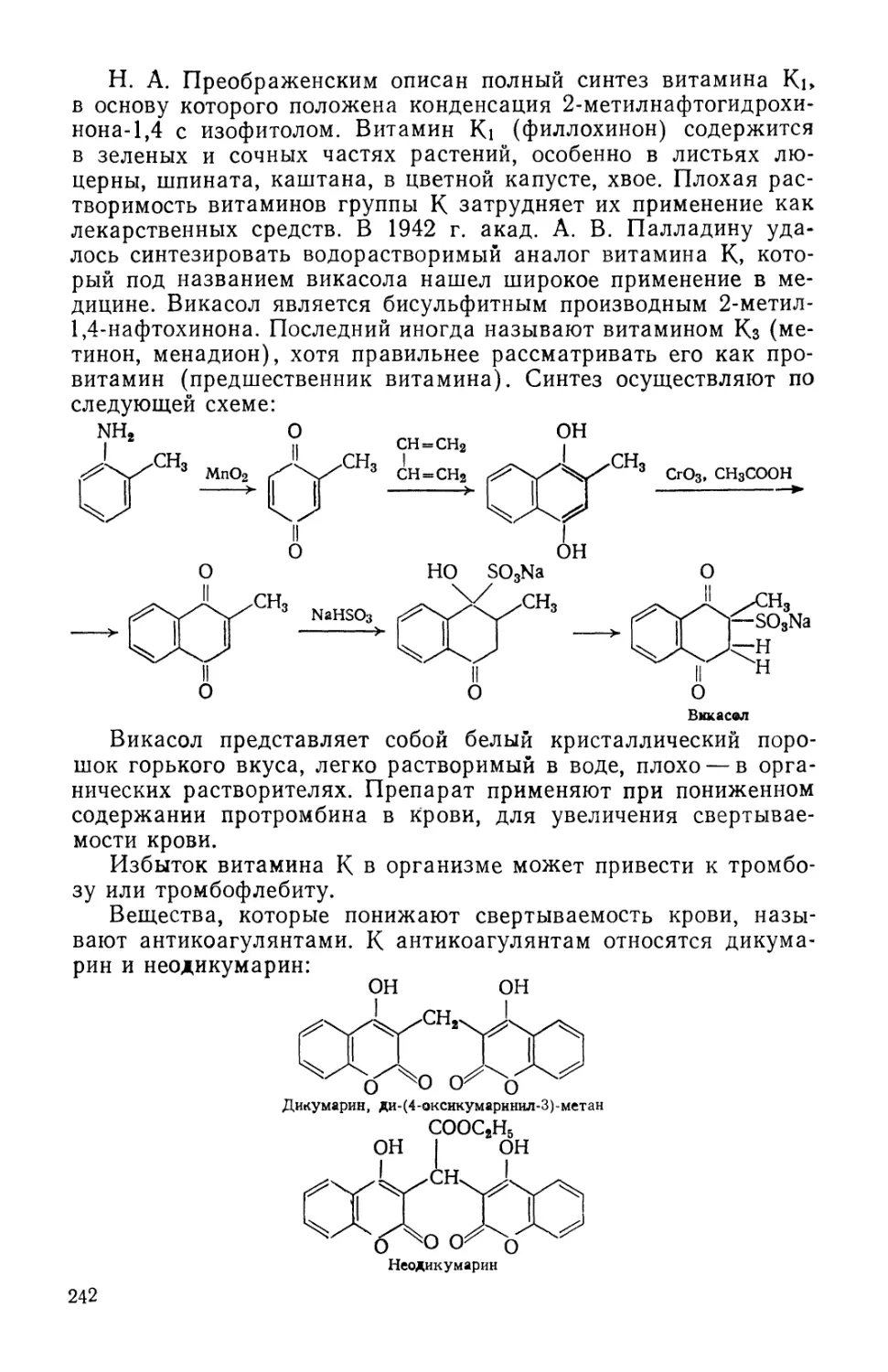


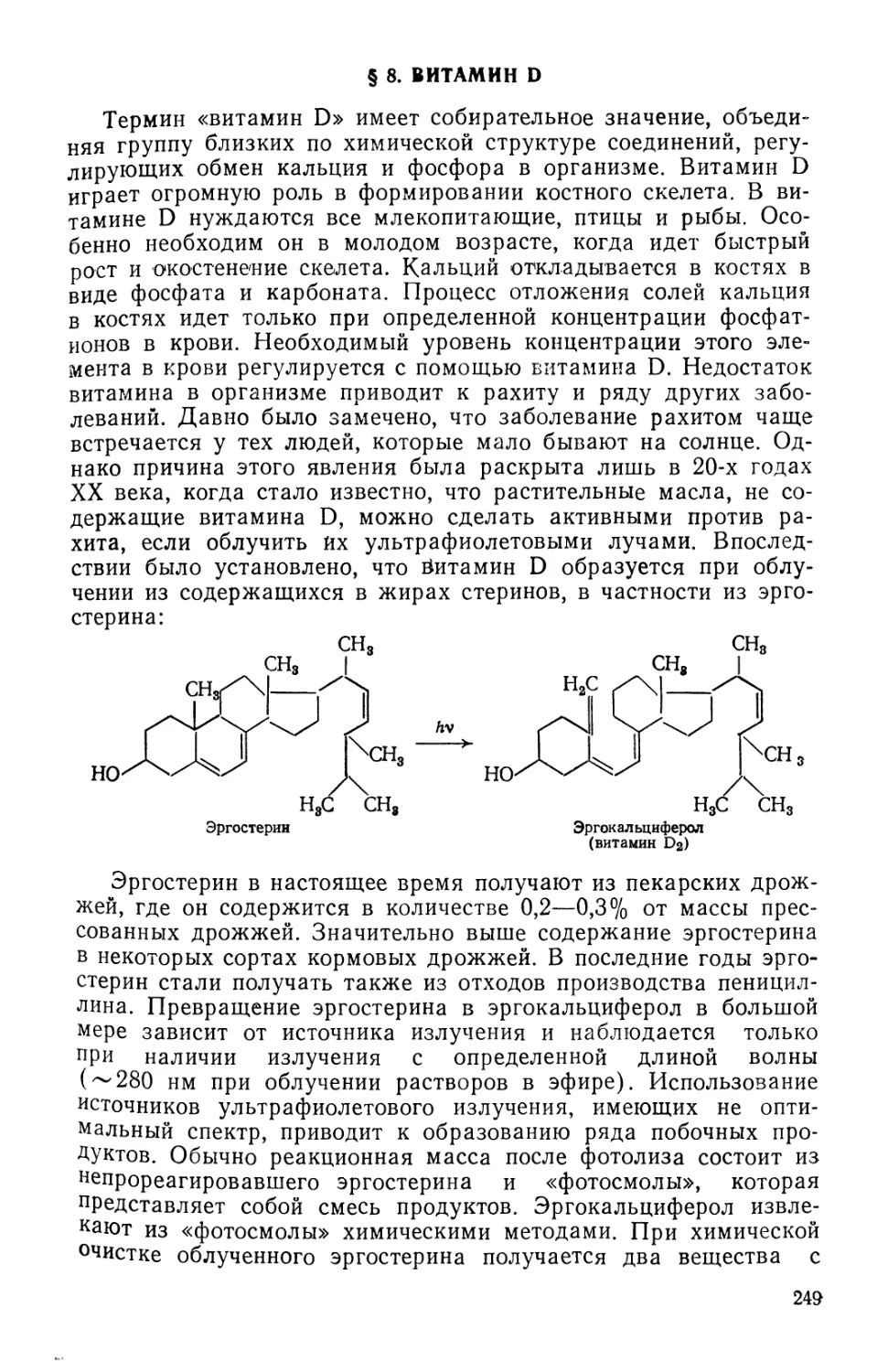




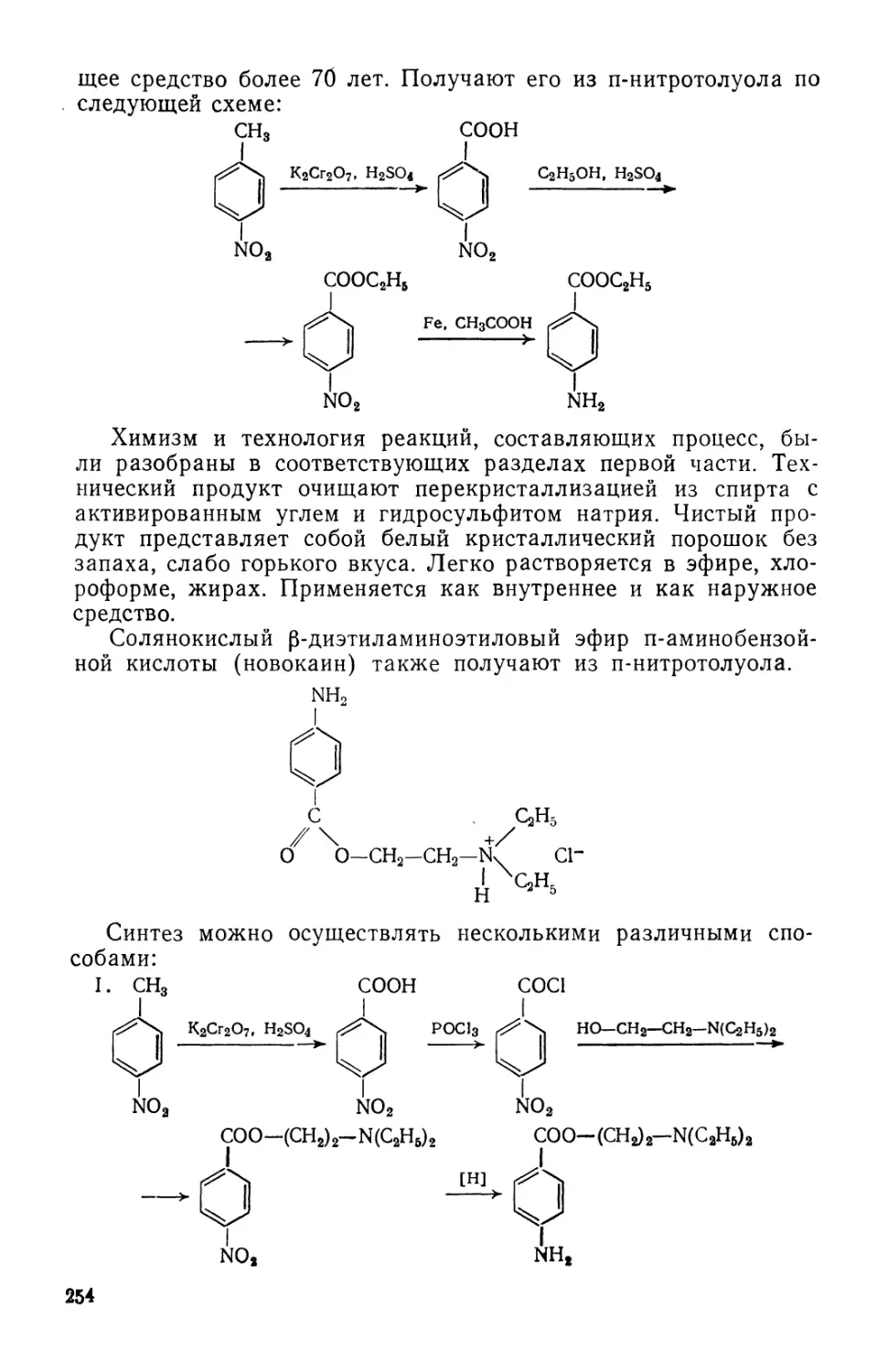
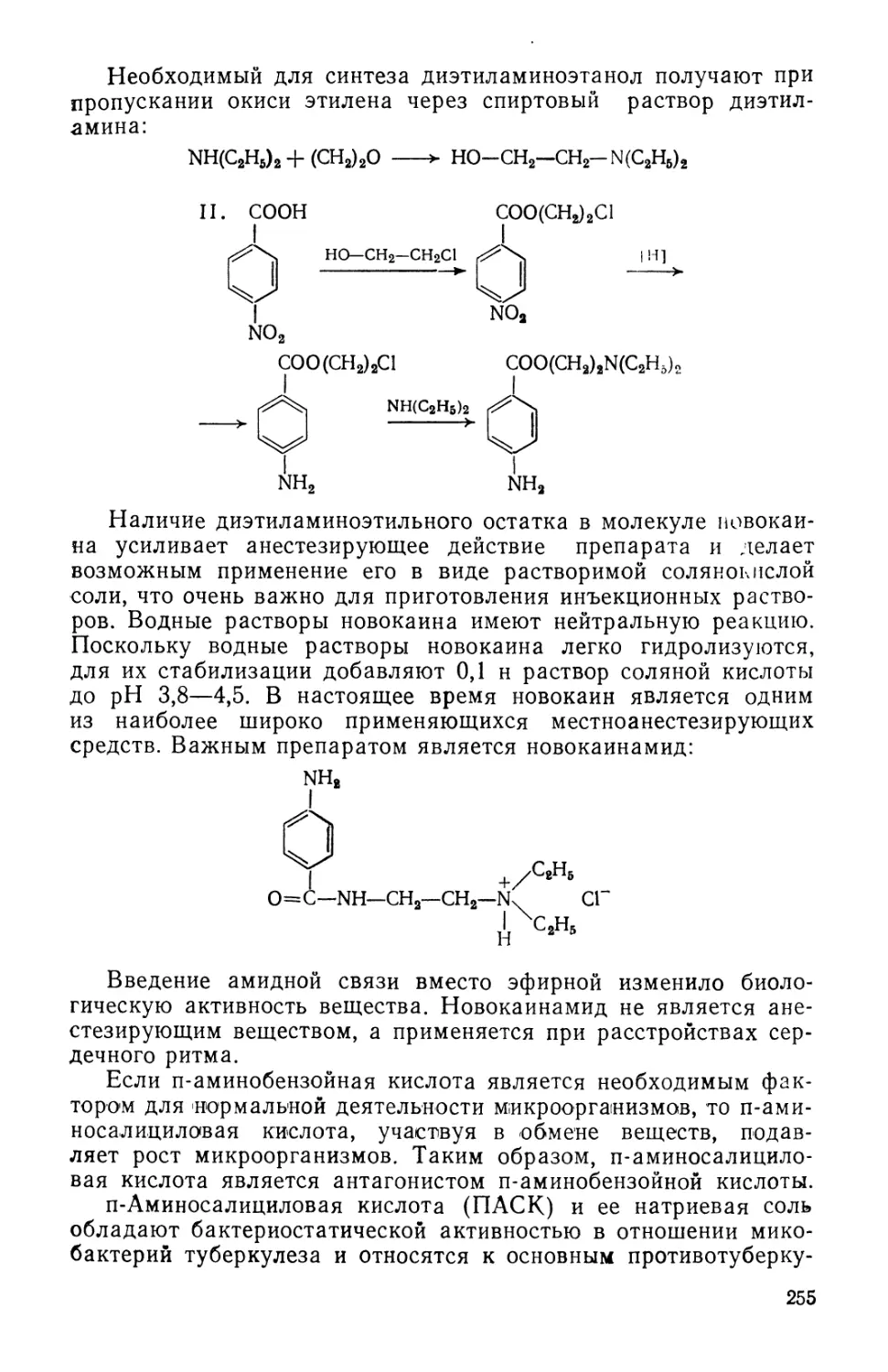




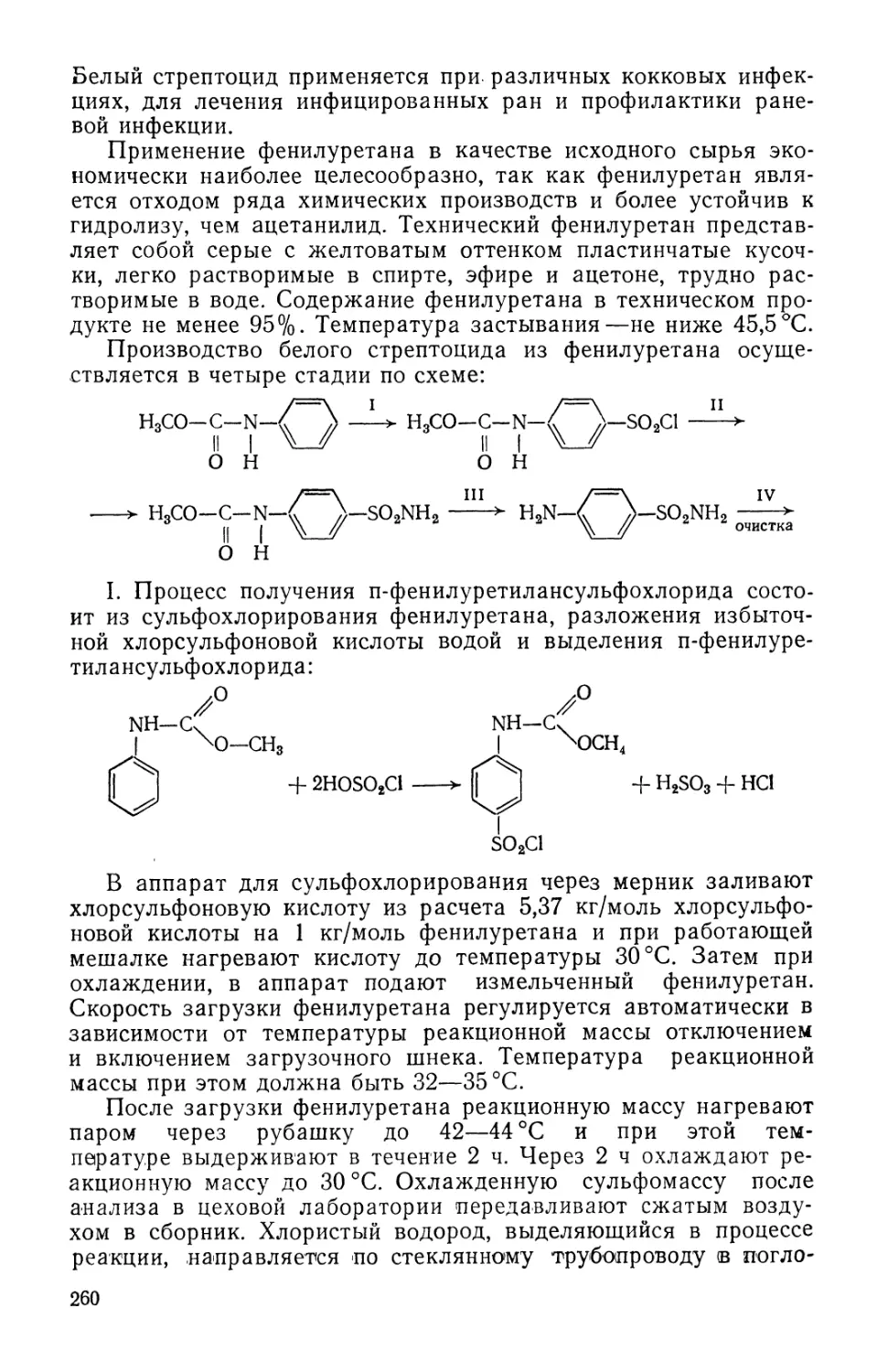




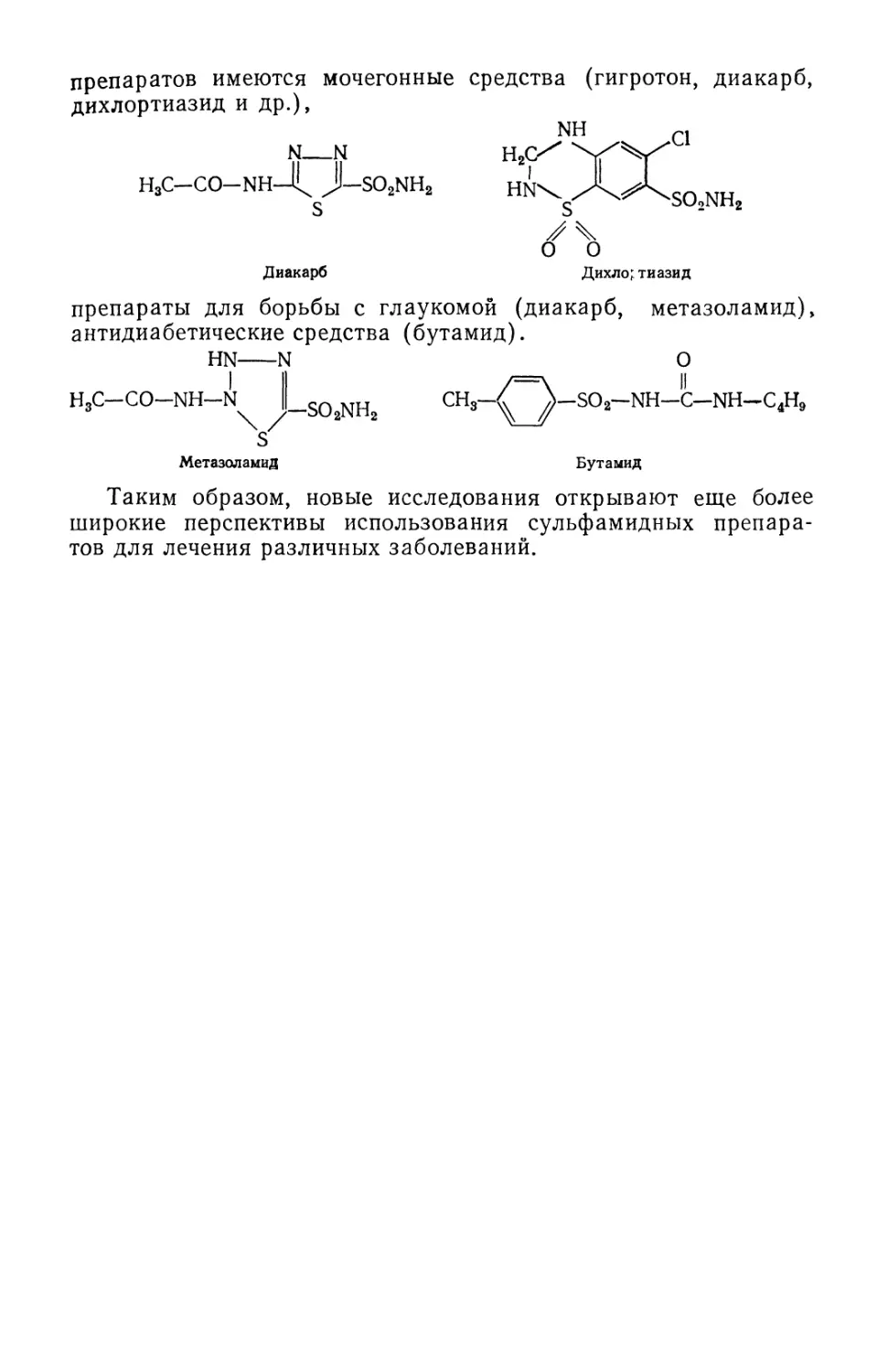

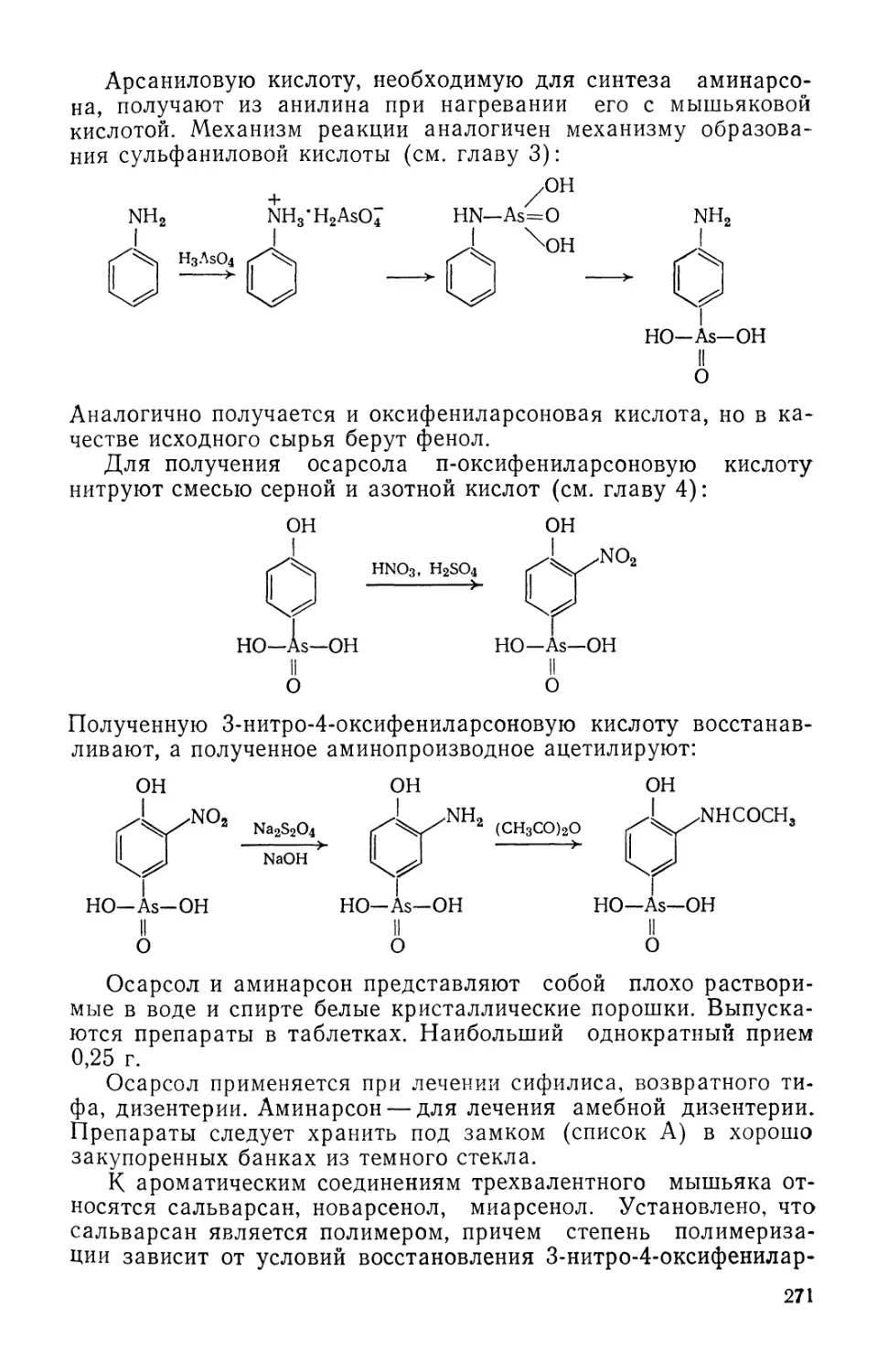





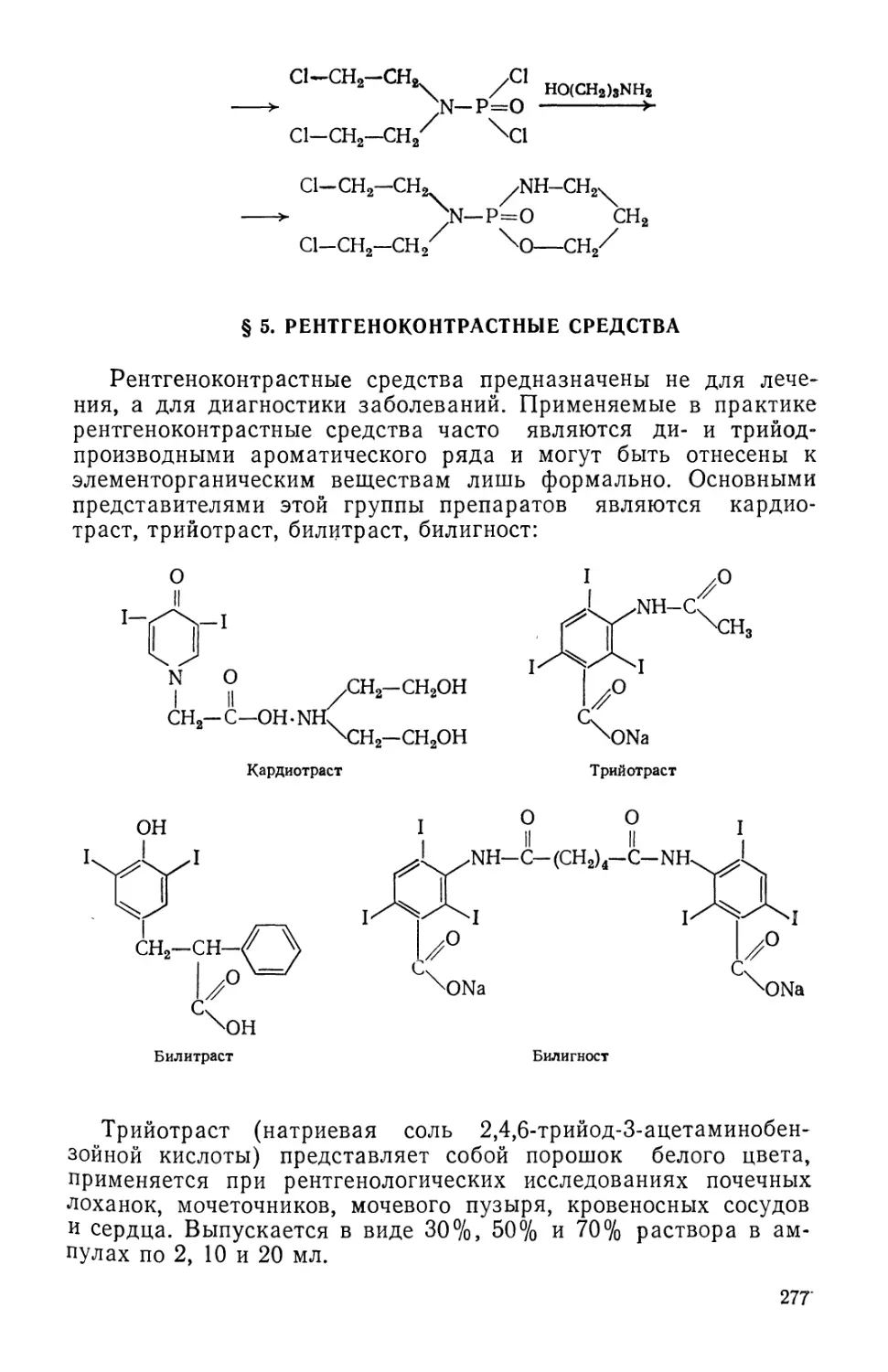




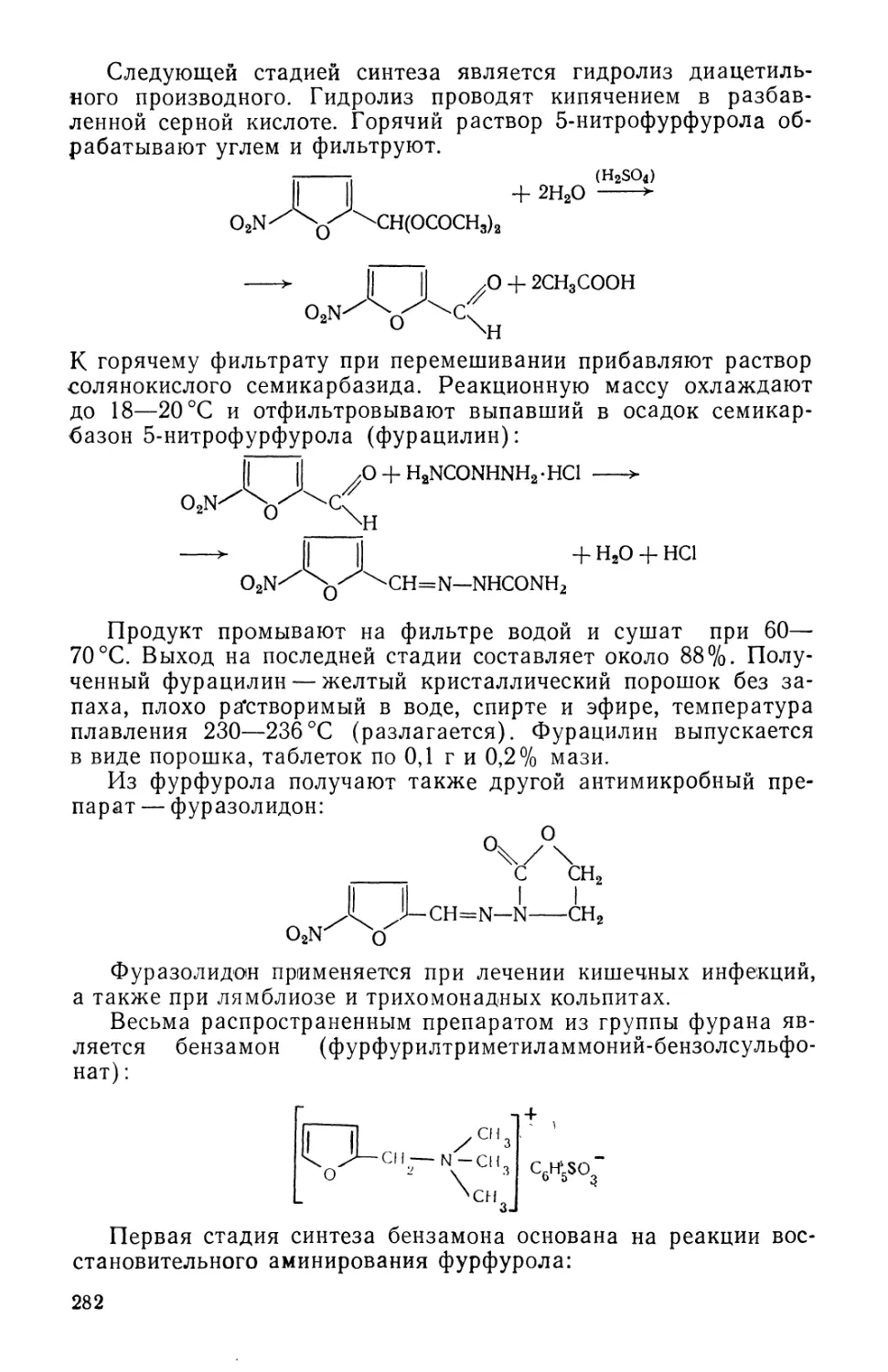


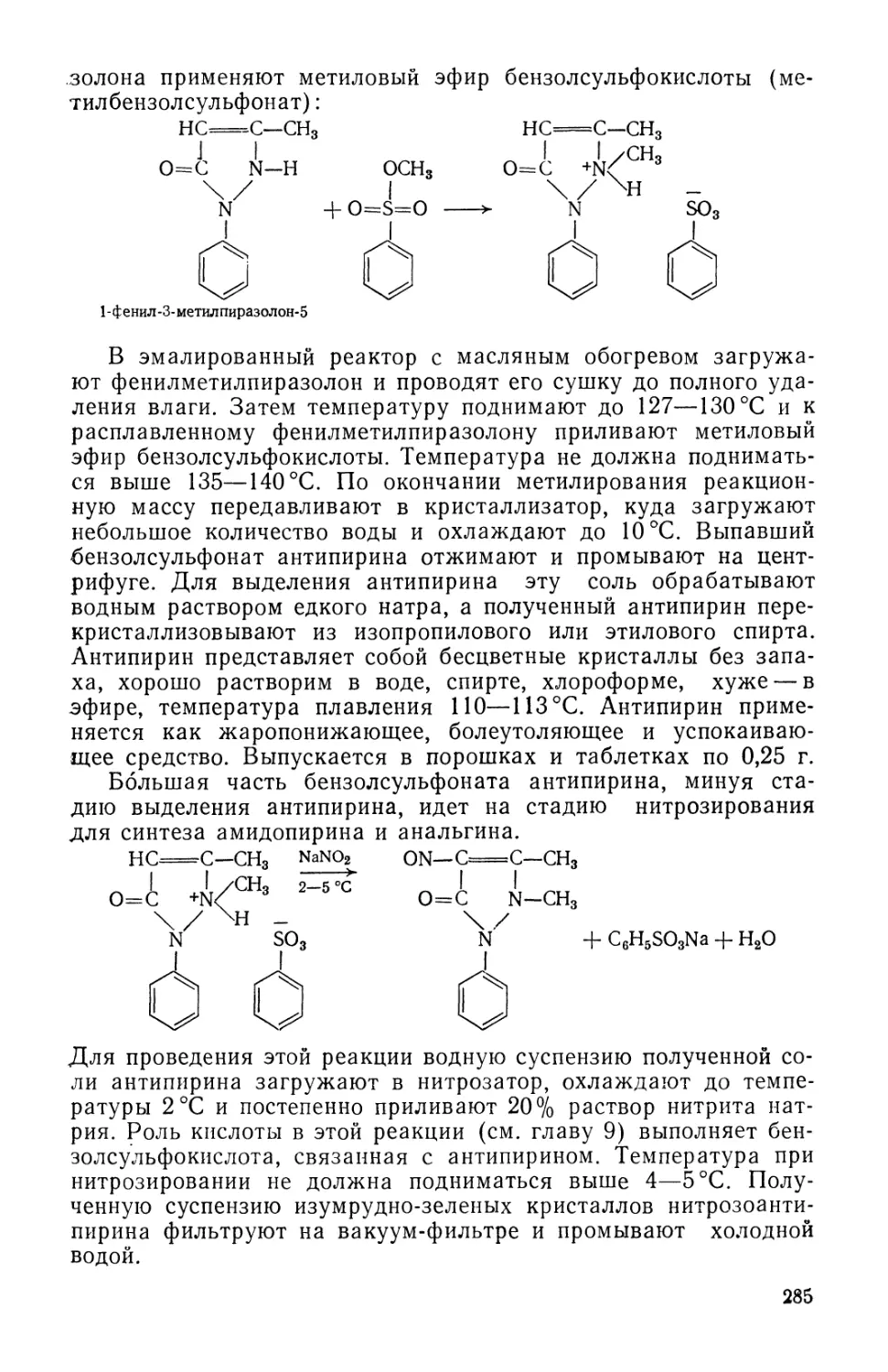


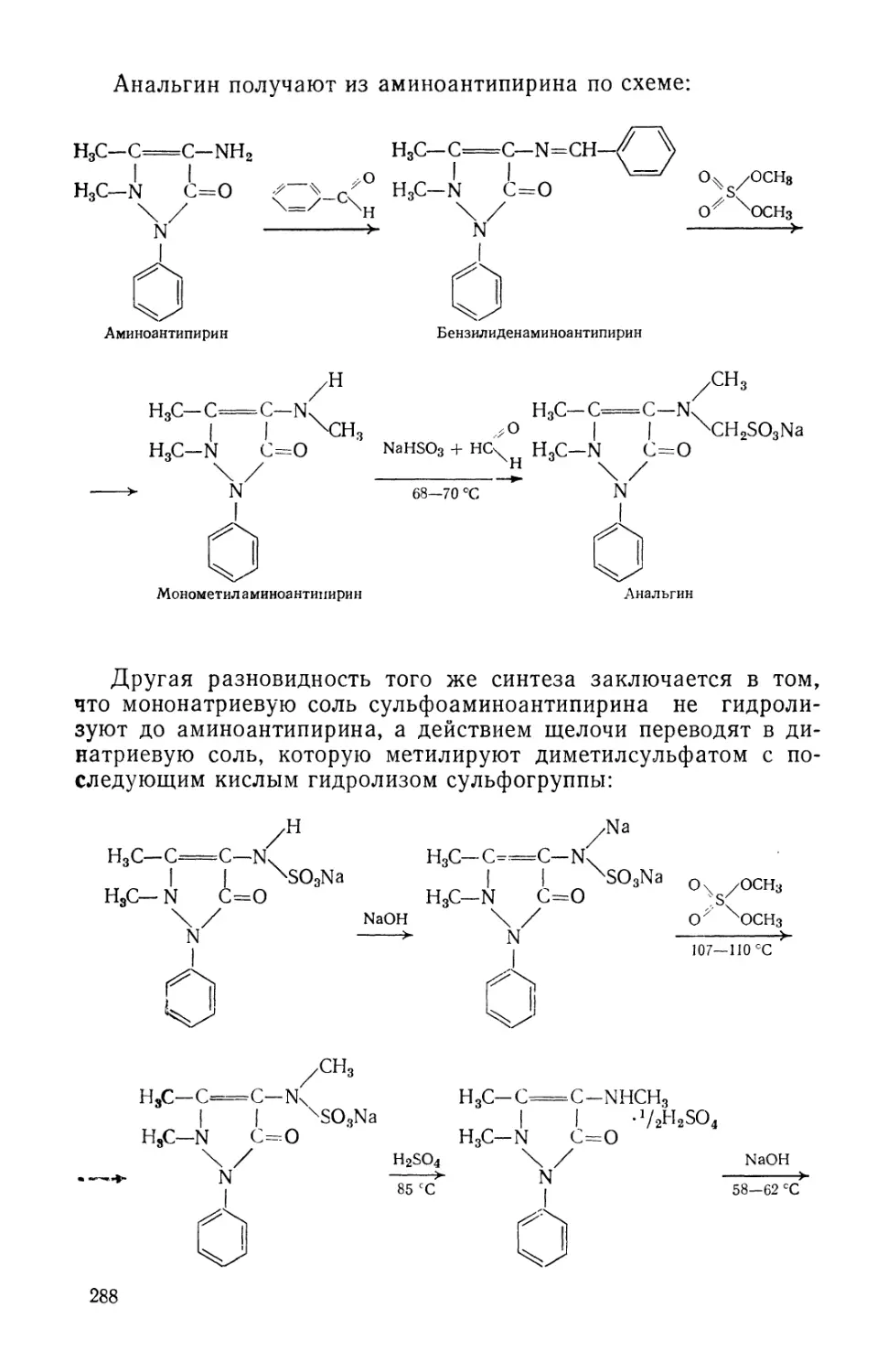



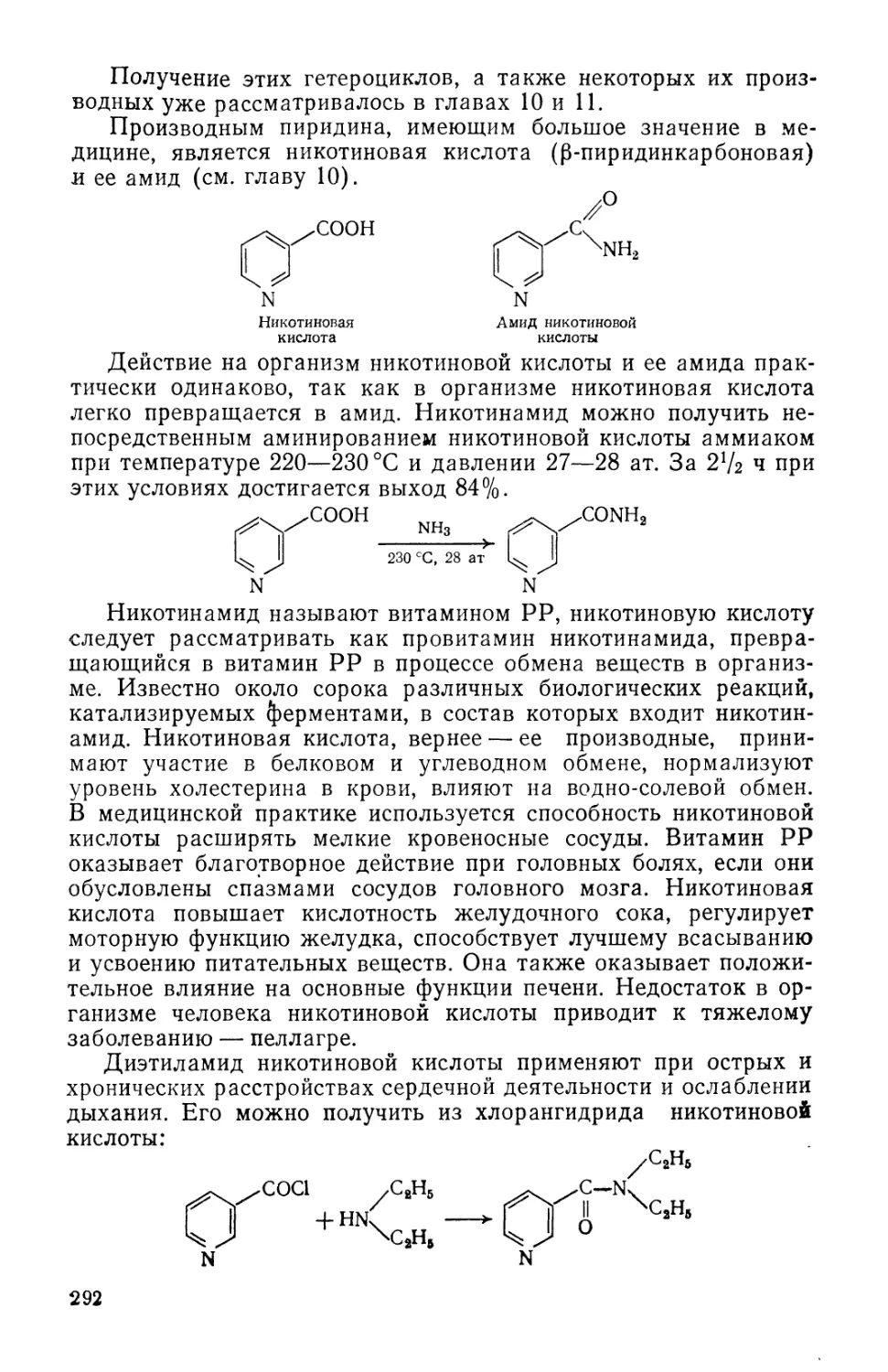


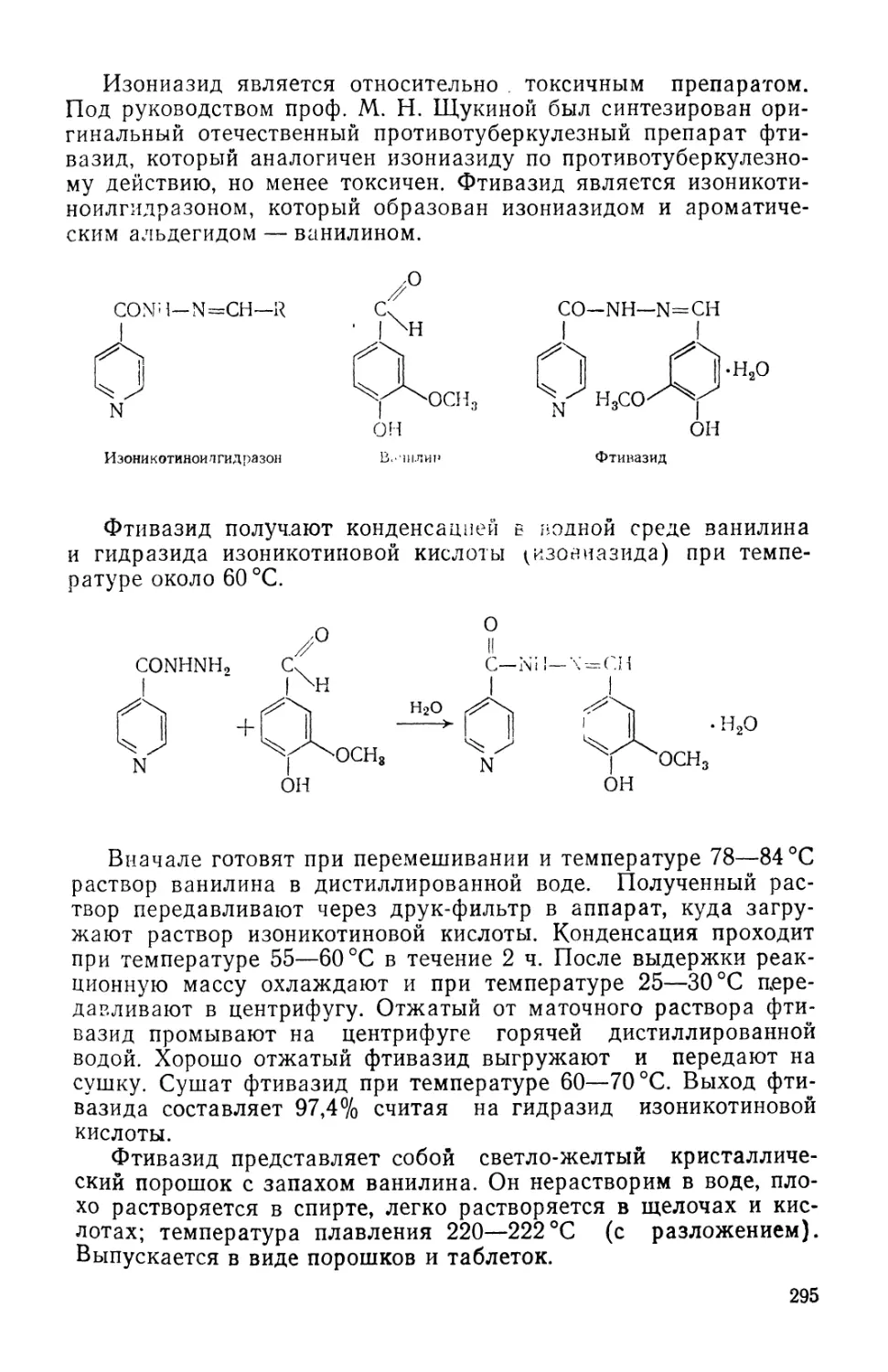
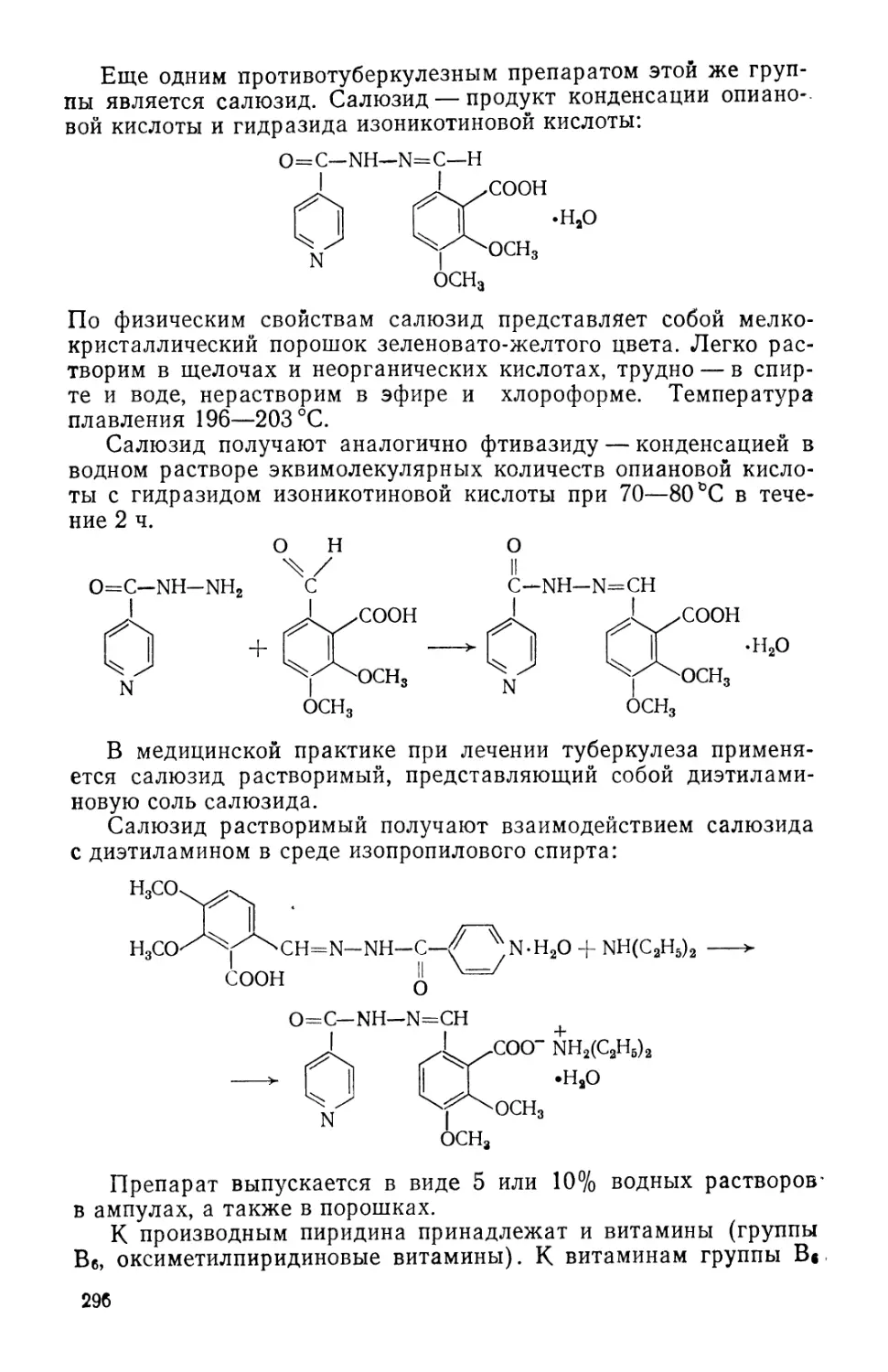

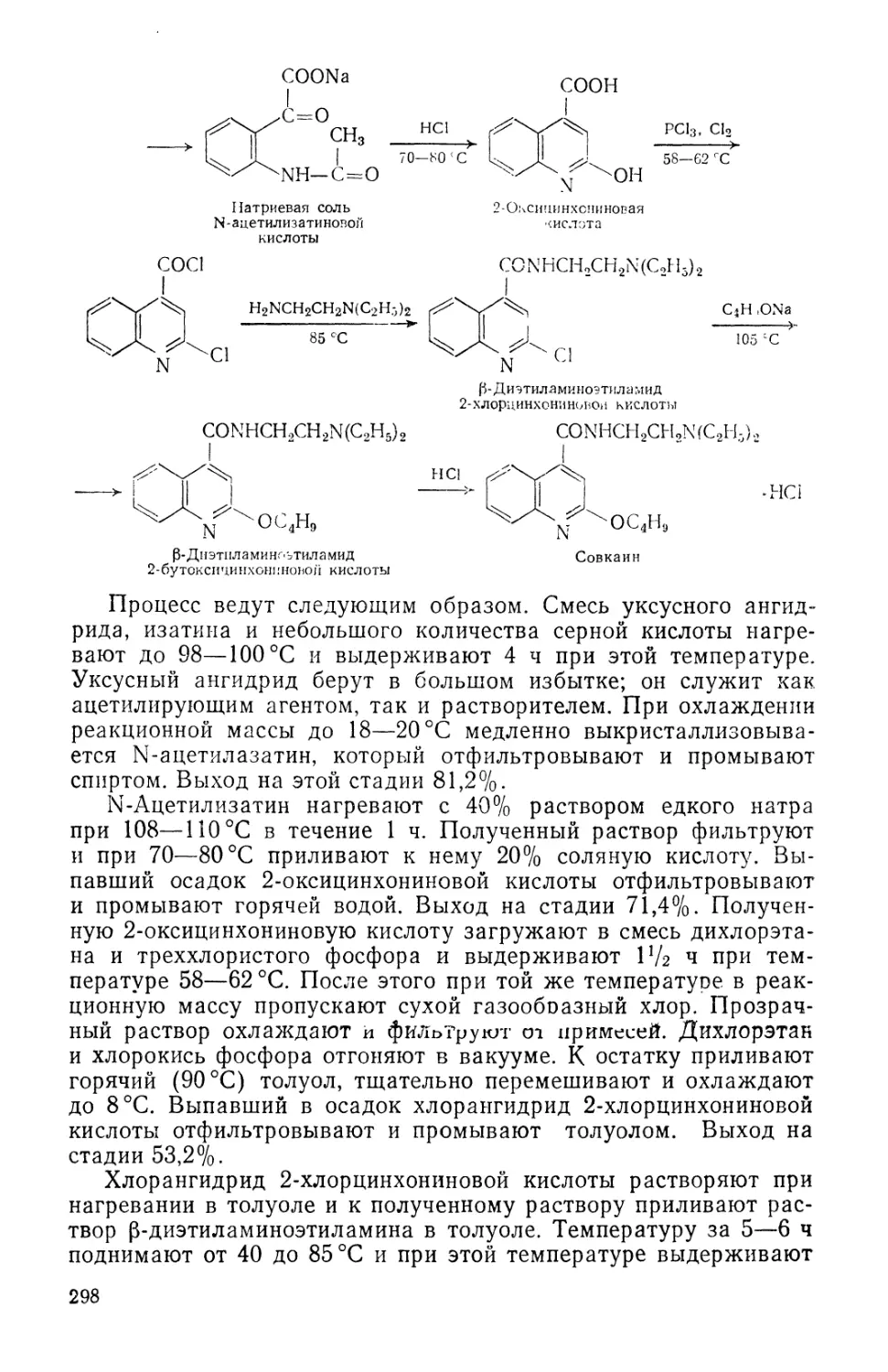

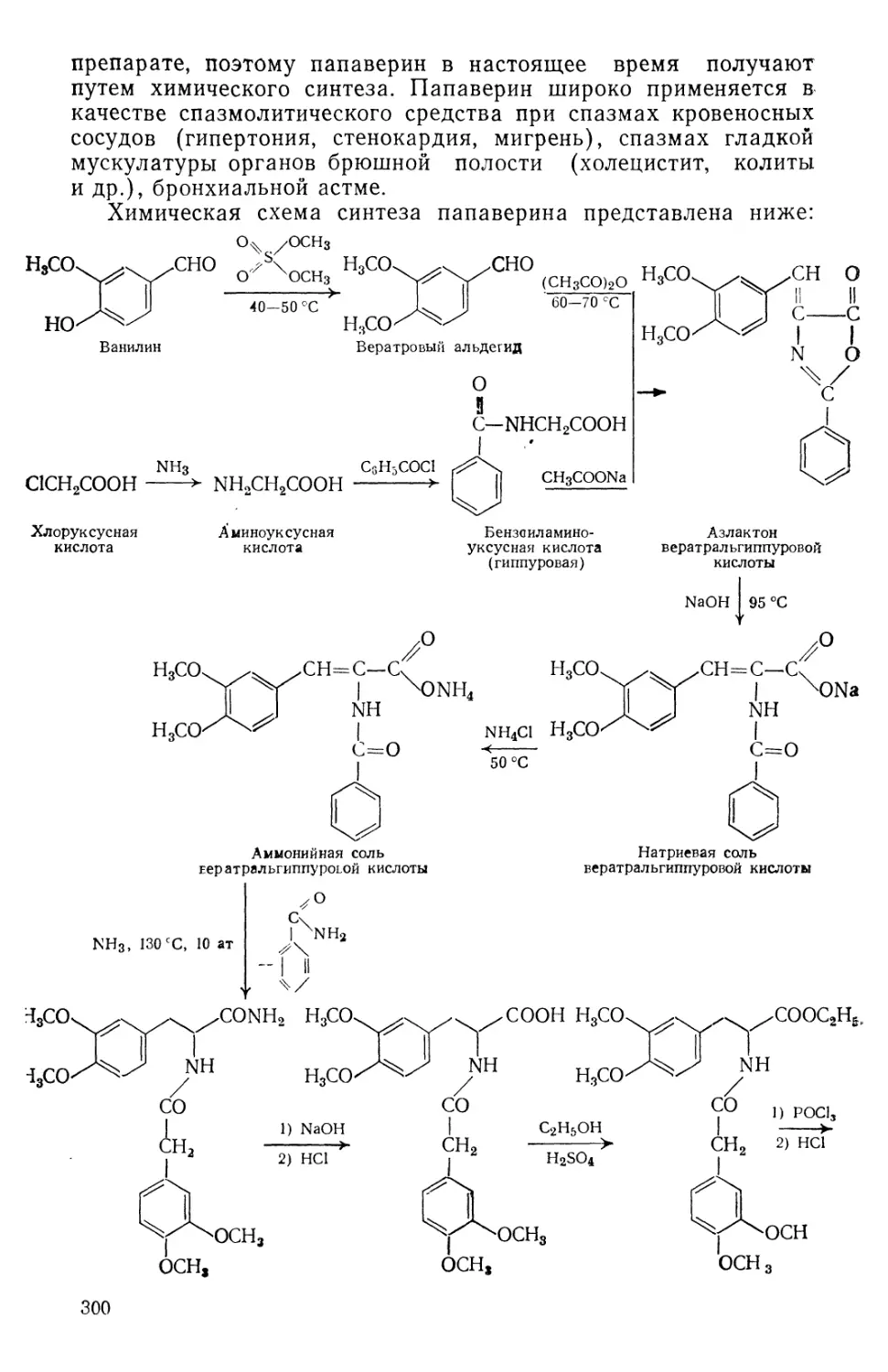









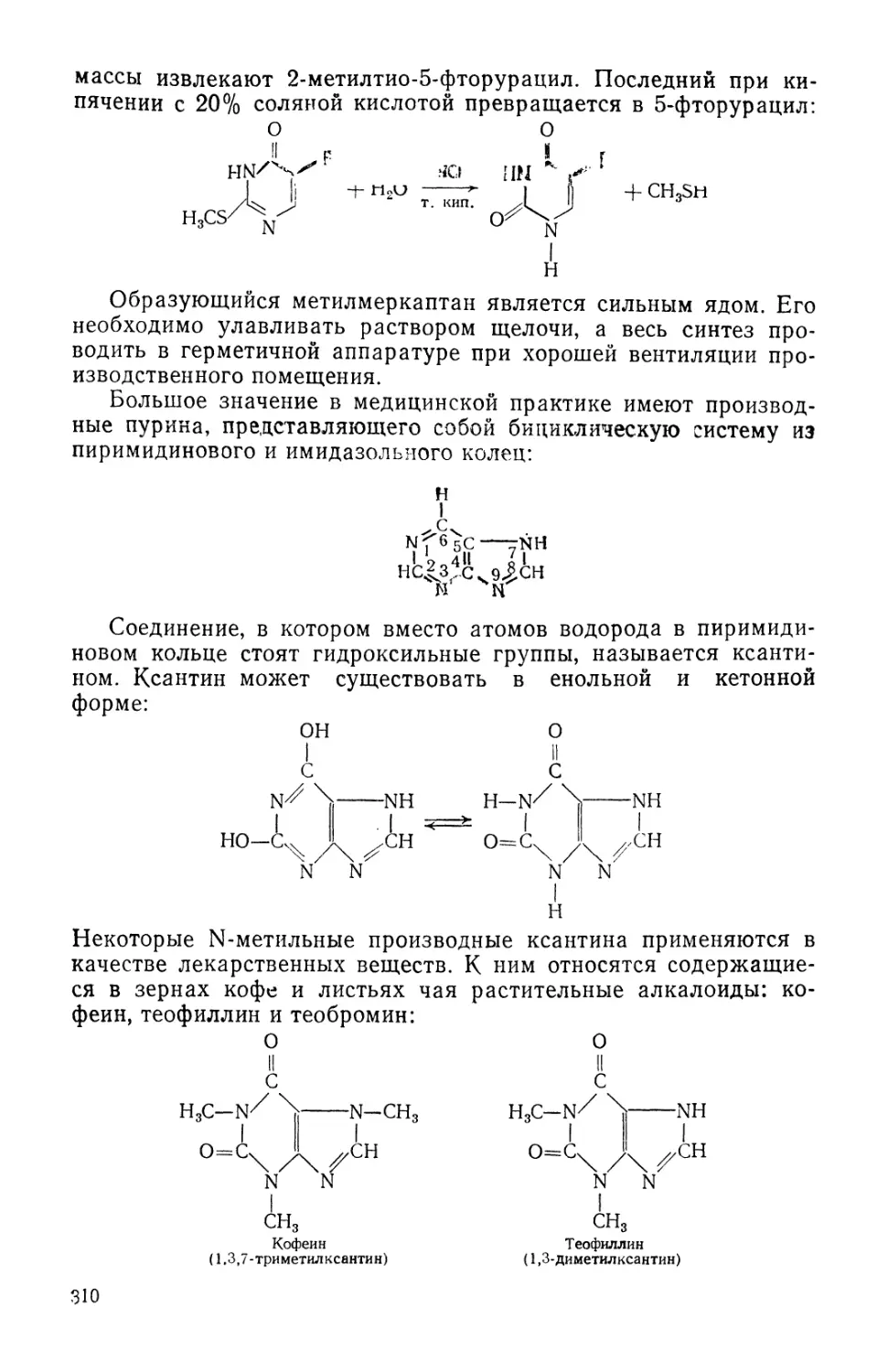
























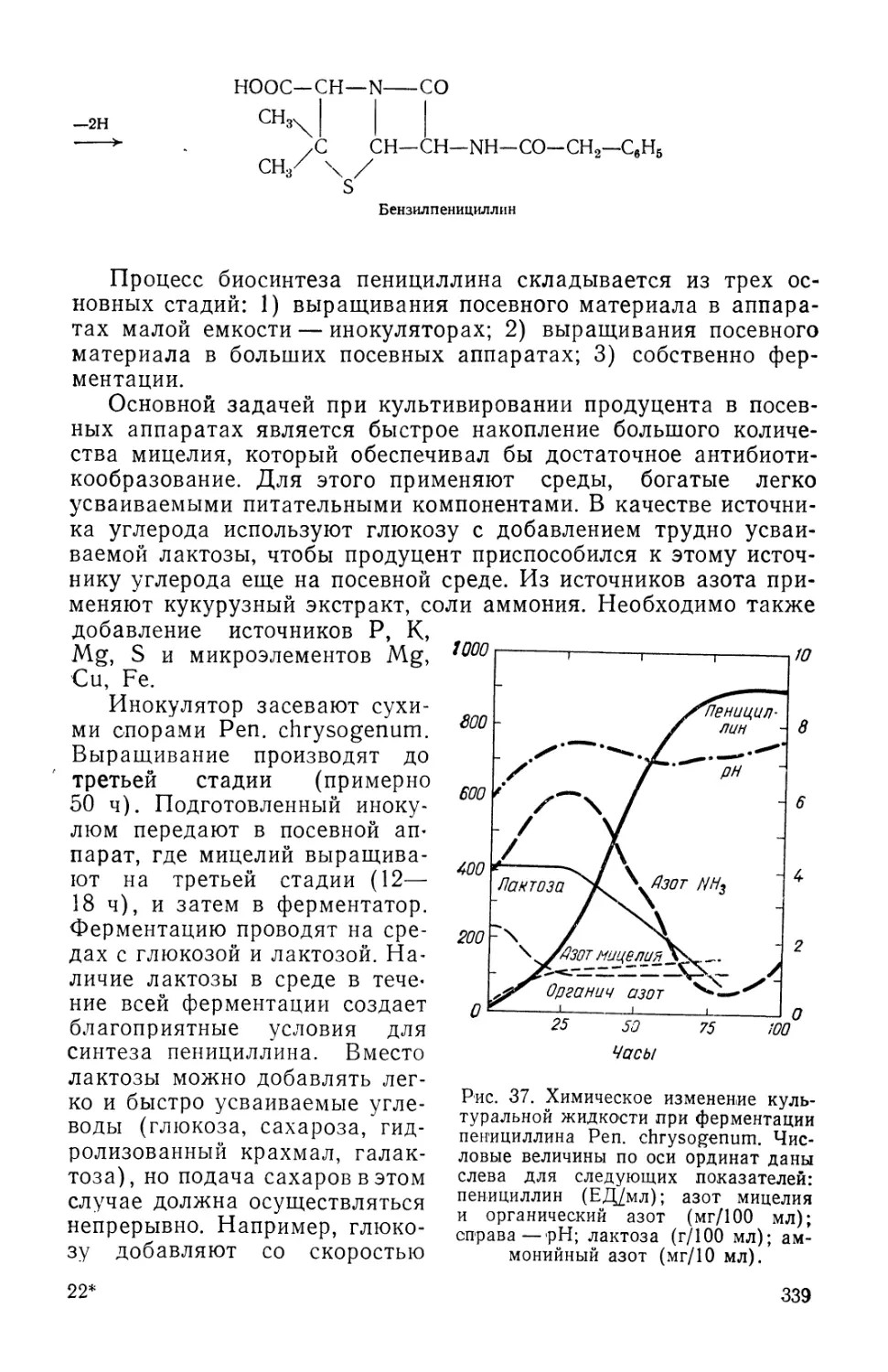







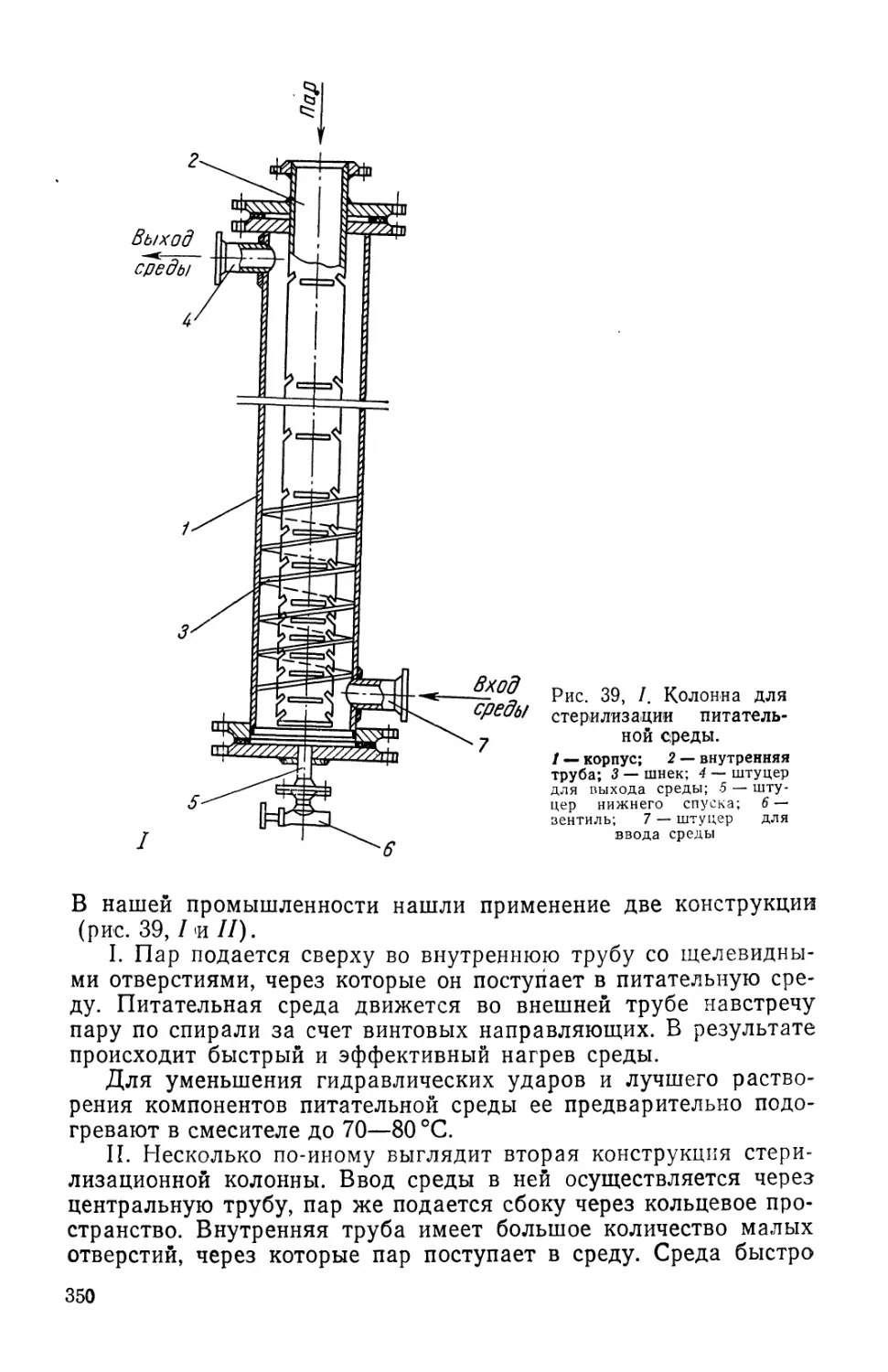

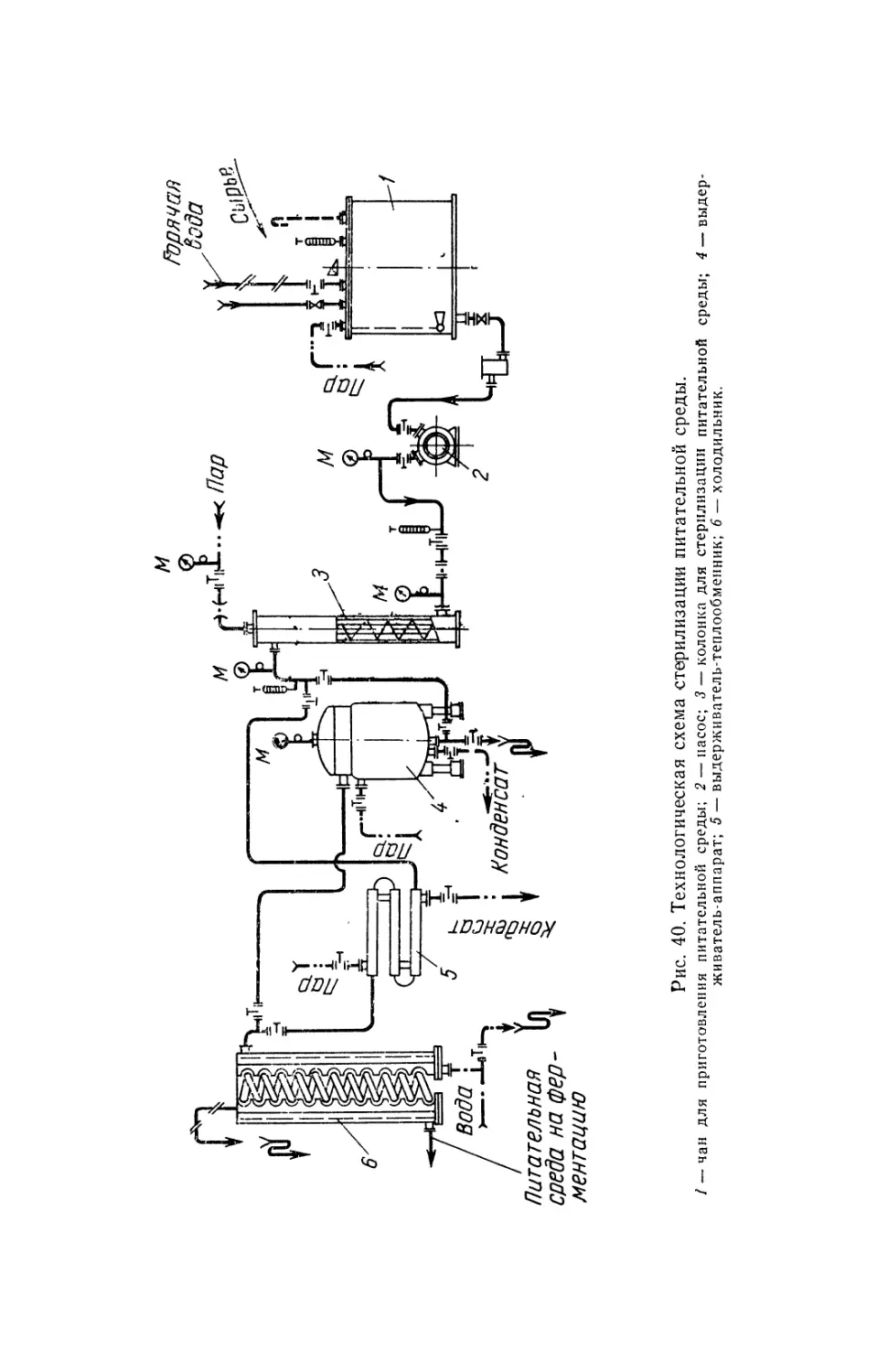









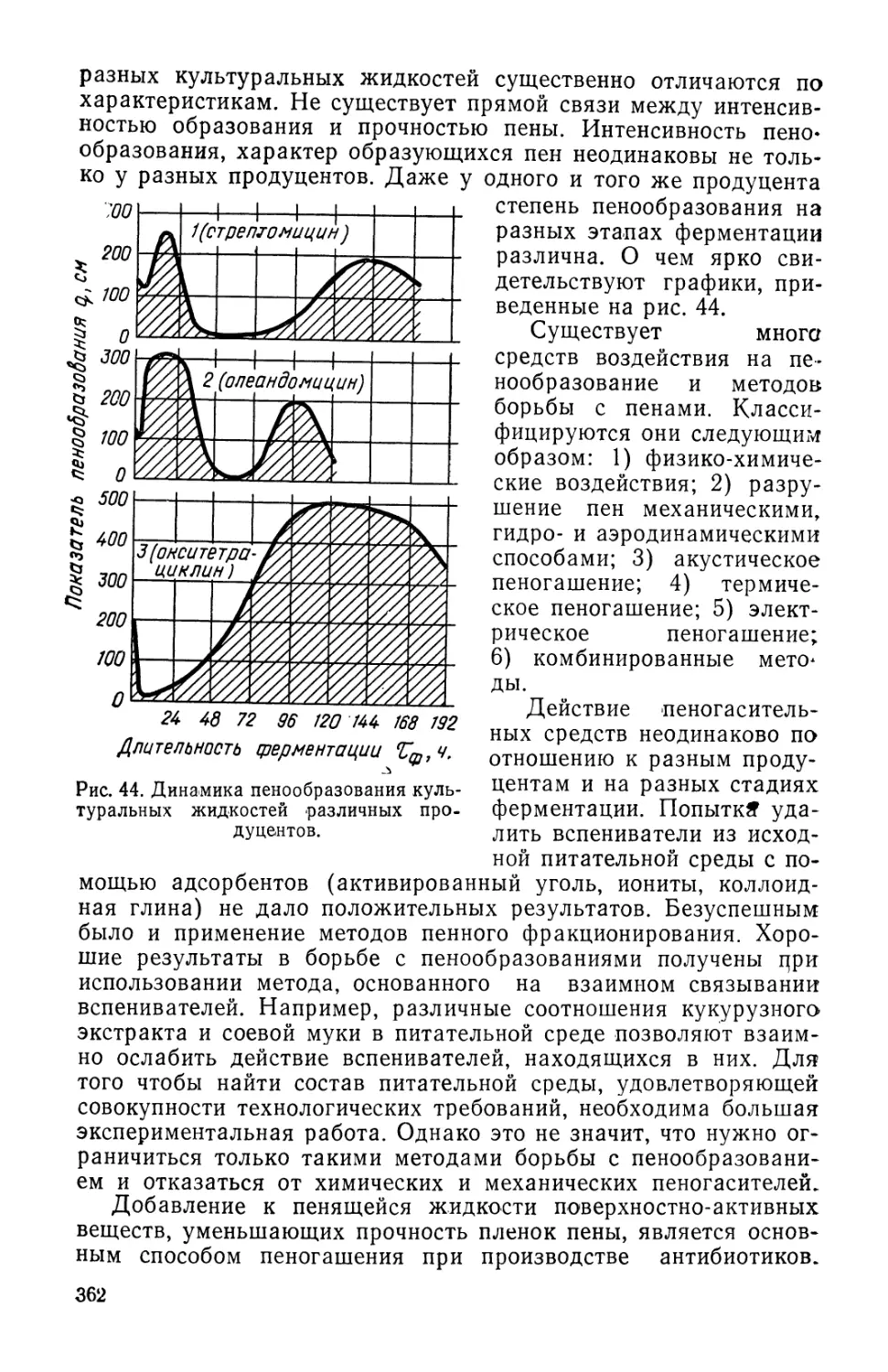


















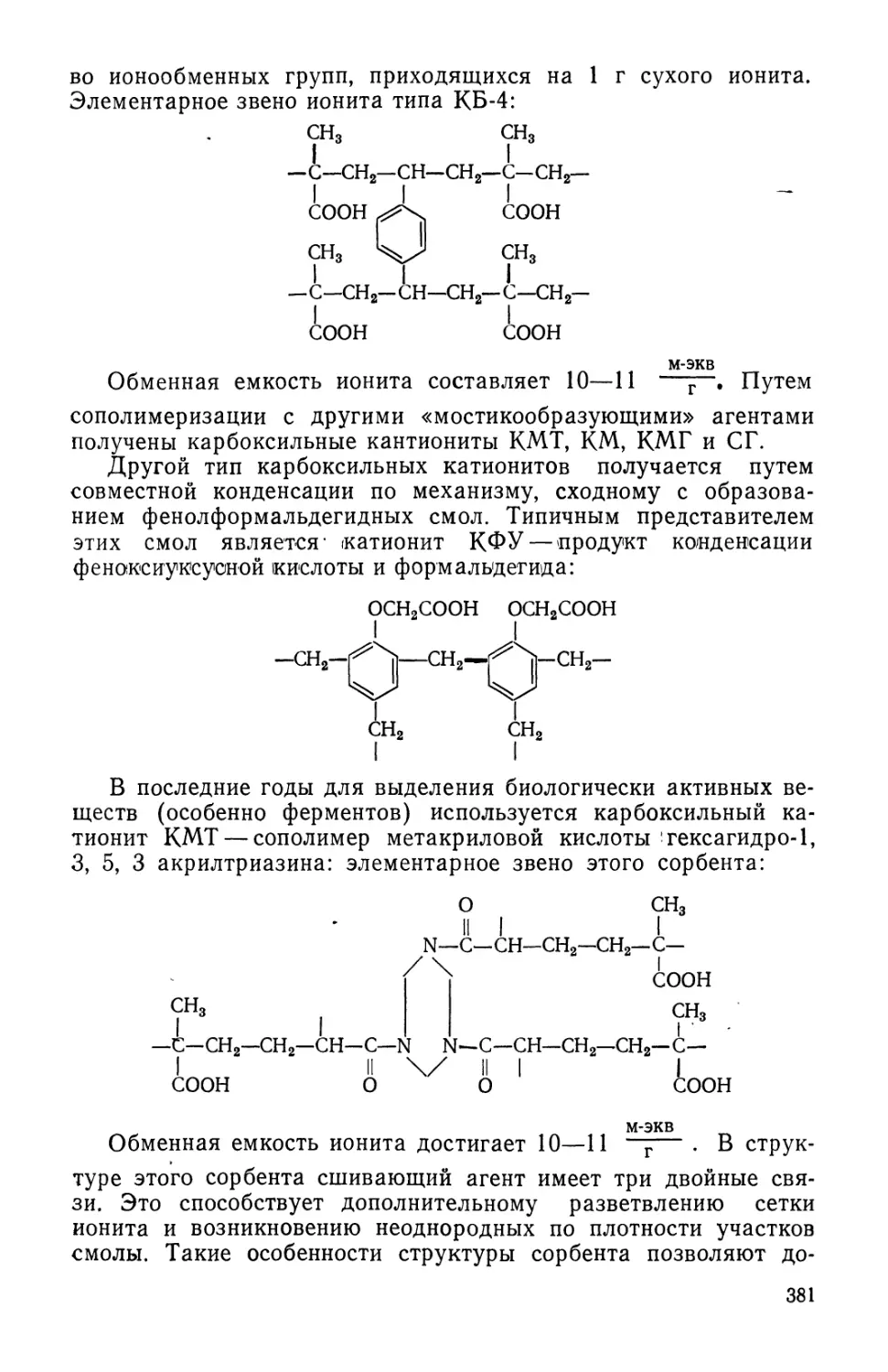


























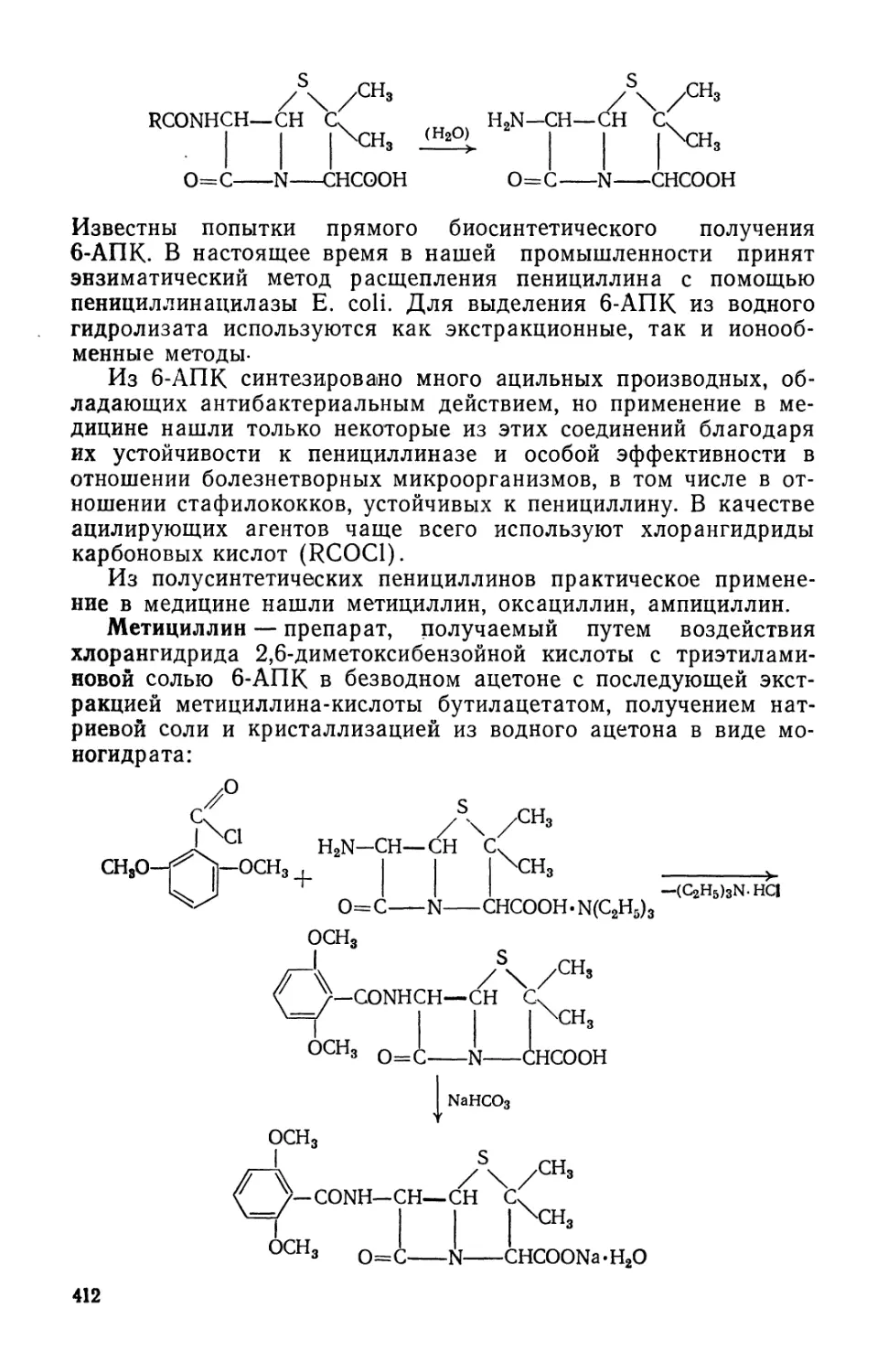
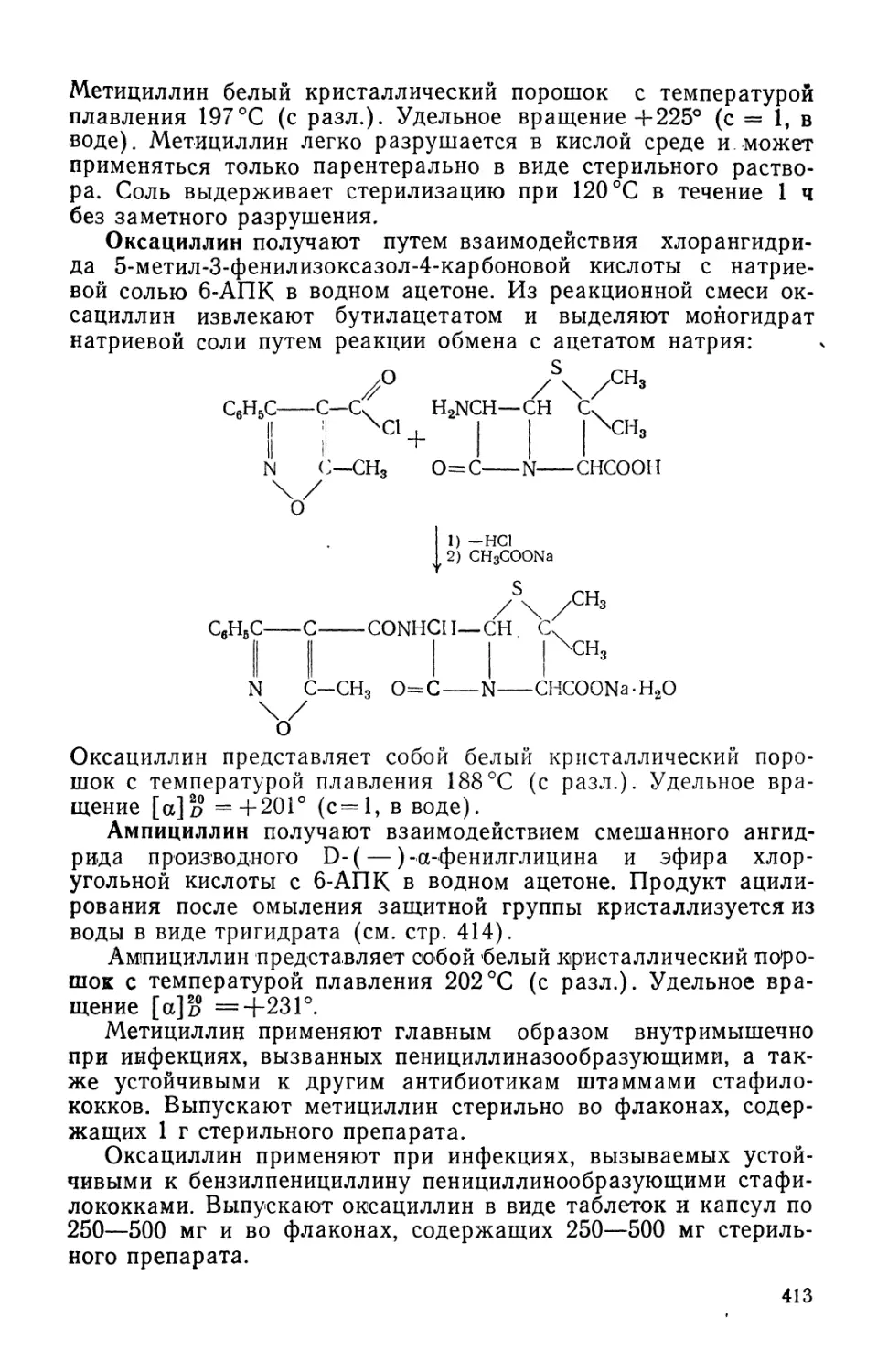

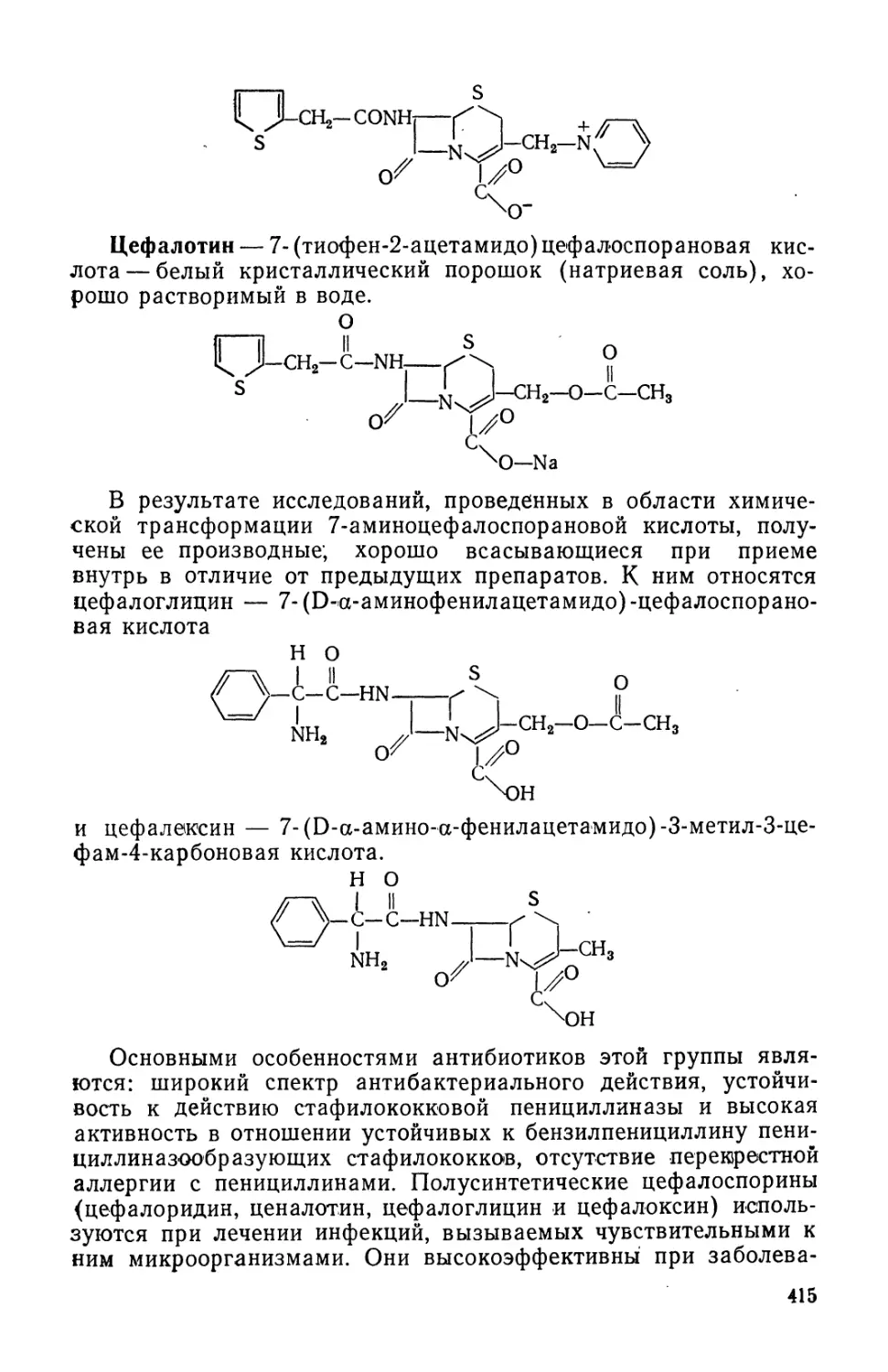




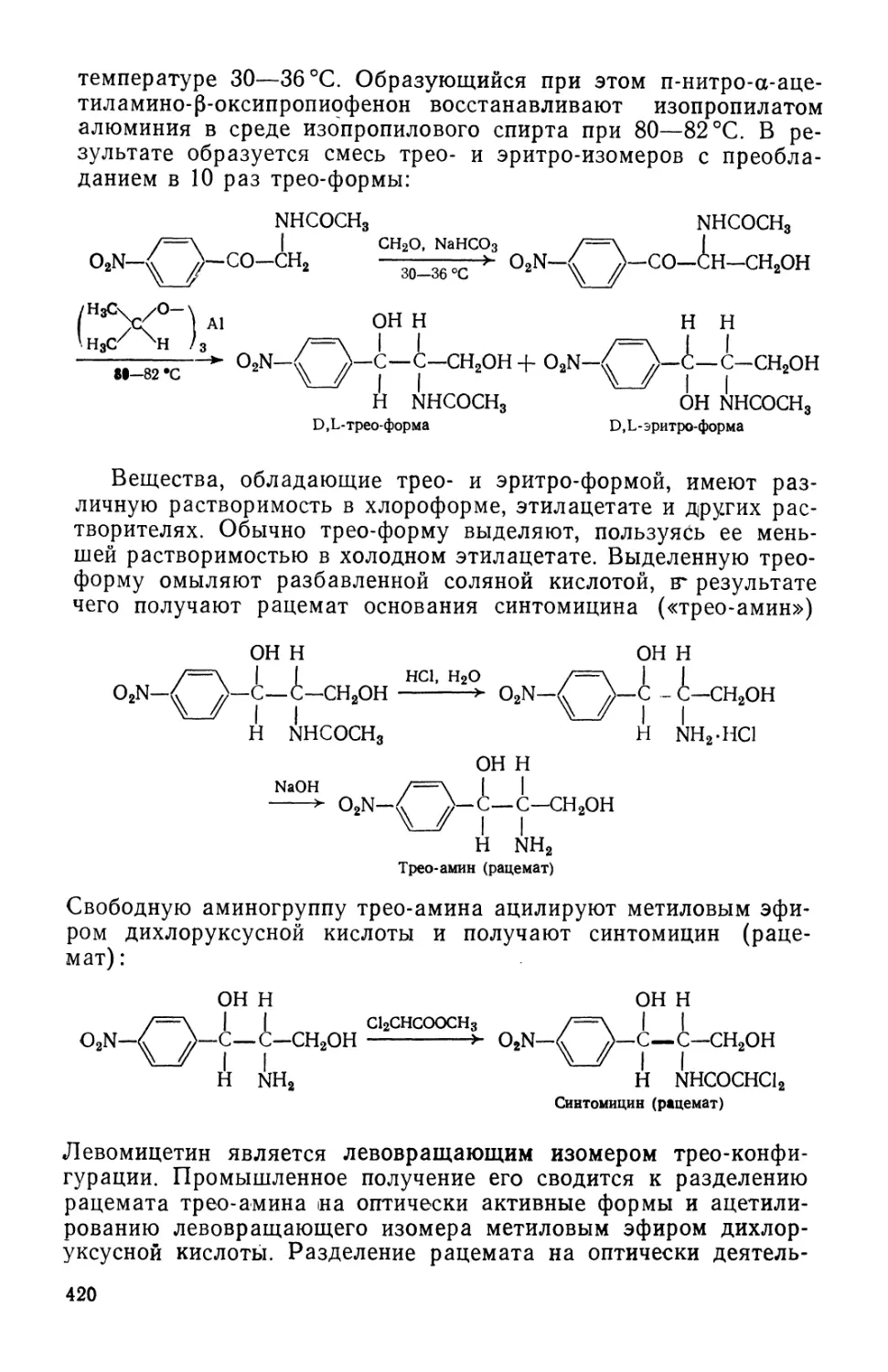

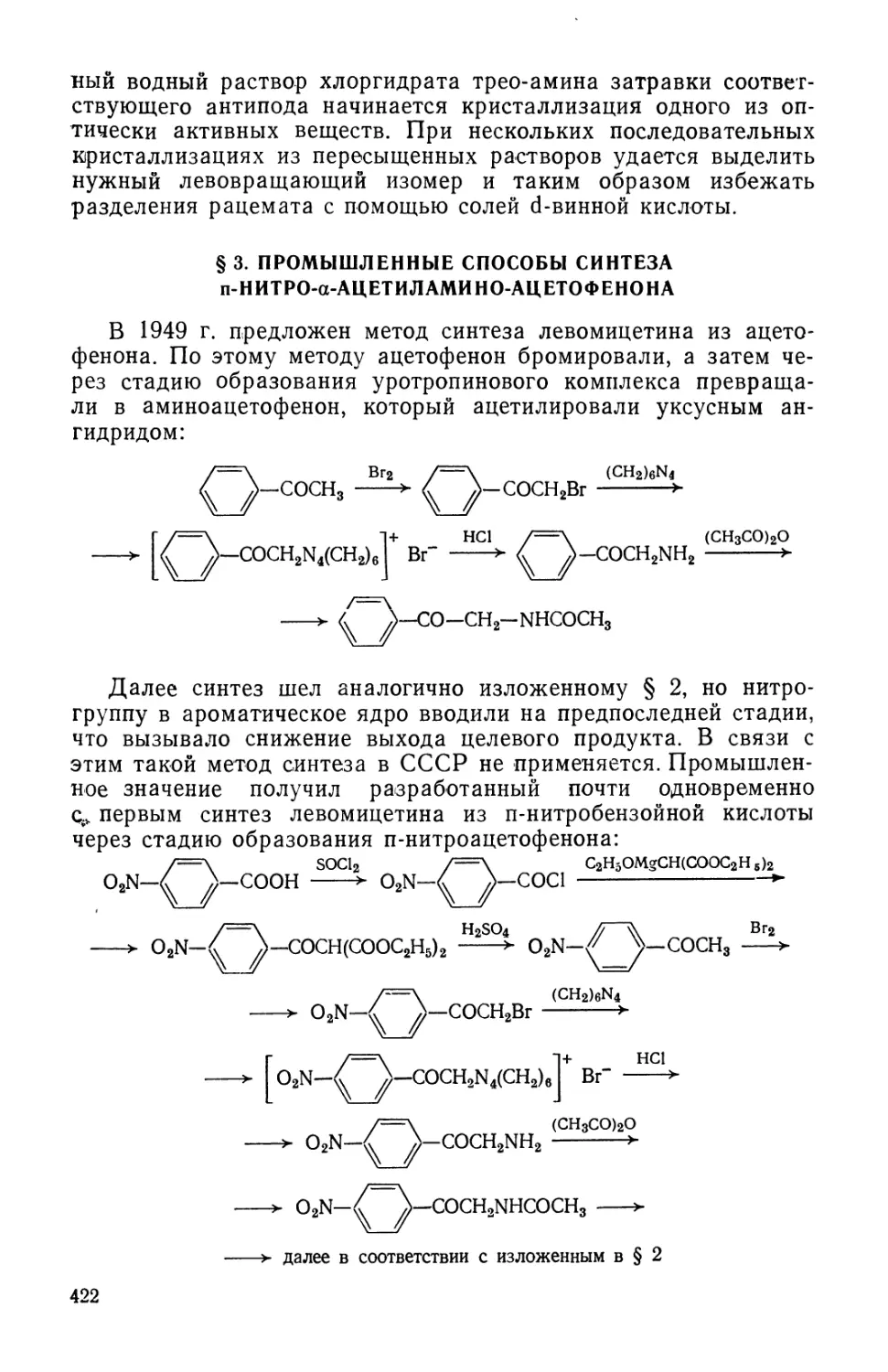
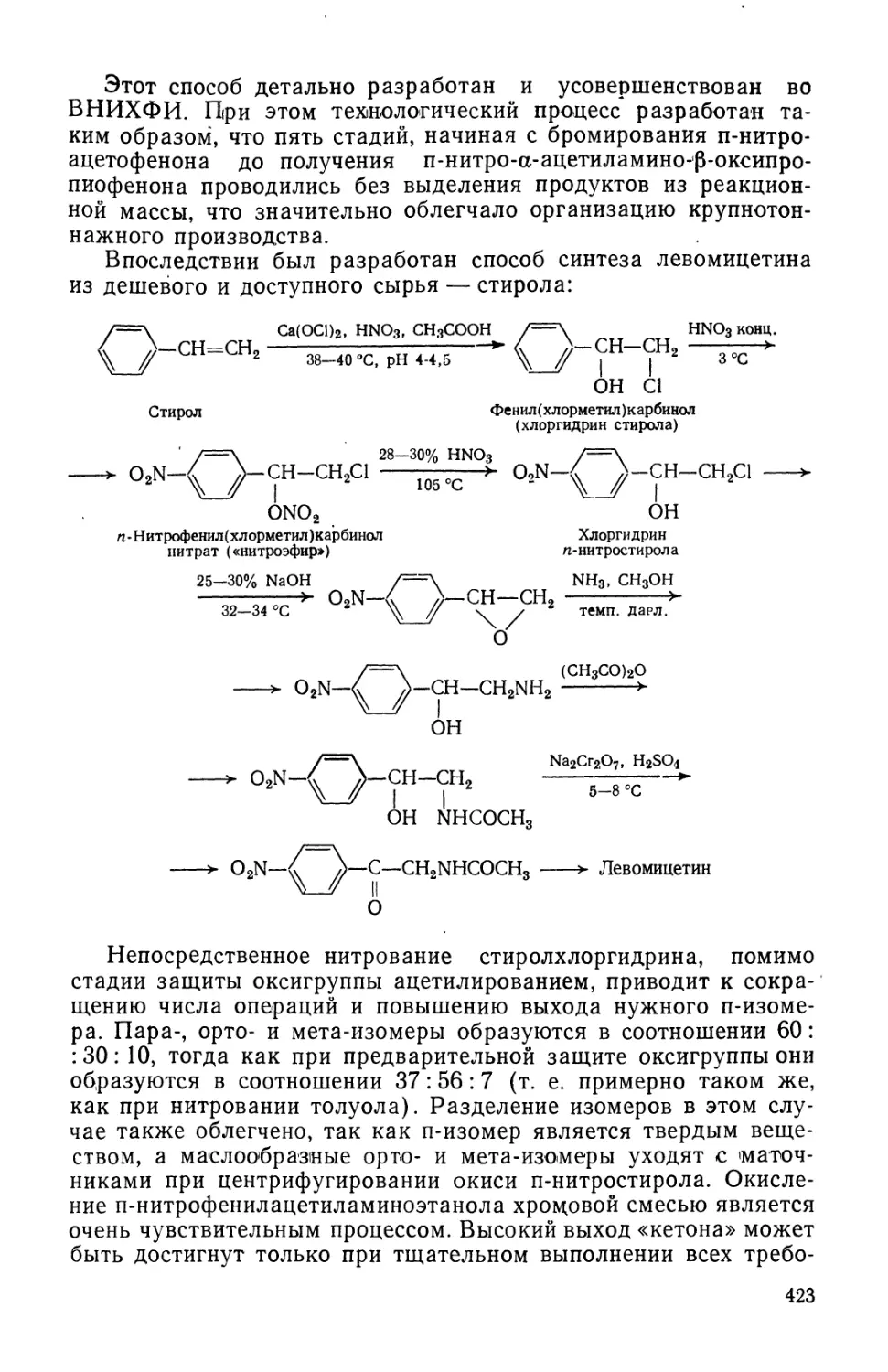












Text
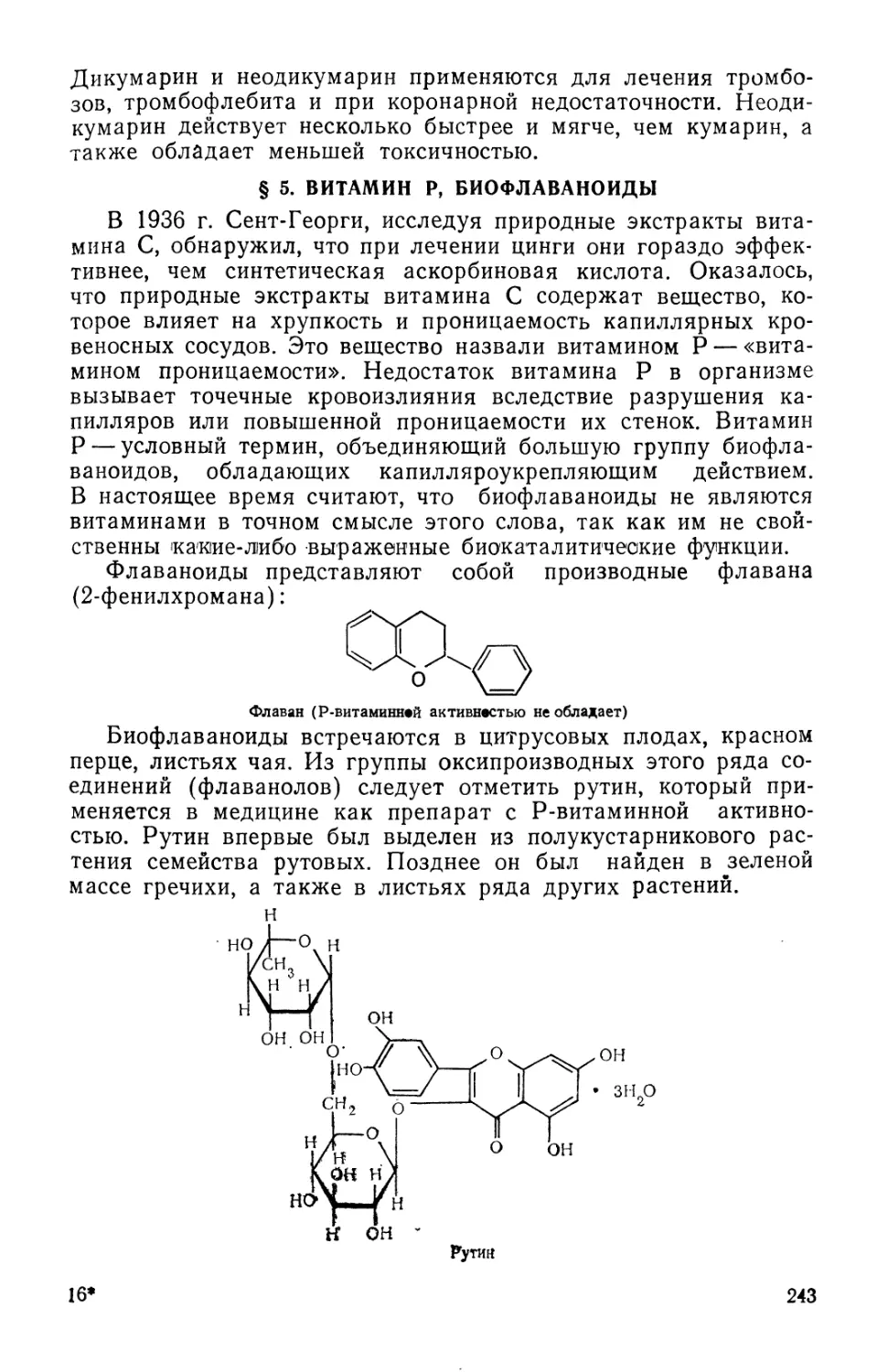
fiB-OACCET. ЙЯВОРОЬьСВЛ
ТЕХНОЛОГИЯ
ХИМИКО-
ФАРМАЦЕВТИЧЕСКИХ
ПРЕПАРАТОВ
И АНТИБИОТИКОВ
Б. В. ПАССЕТ, В. Я. ВОРОБЬЕВА
ТЕХНОЛОГИЯ
ХИМИКО-
ФАРМАЦЕВТИЧЕСКИХ
ПРЕПАРАТОВ
И АНТИБИОТИКОВ
Допущено Министерством высшего и среднего
специального образования СССР в качестве
учебного пособия для учащихся средних
специальных учебных заведений
Москва «Медицина» 1977
УДК 615.2.3.012@75.8)
Б. В. Пассет, В. Я. Воробьева. Технология химико-фармацевтических пре-
препаратов и антибиотиков. М., «Медицина», 1977, 430 с, с ил.
В учебнике описано производство синтетических лекарственных веществ, ви-
витаминов, антибиотиков, а также промежуточных продуктов для них.
Учебник состоит из трех частей.
В первой части дается характеристика сырья, используемого в химико-фар-
химико-фармацевтической промышленности, общая характеристика реакций, применяю-
применяющихся в синтезе лекарственных веществ, и подробный разбор химии, техно-
технологии и аппаратурного оформления важнейших методов синтеза промежу-
промежуточных продуктов для лекарственных веществ, витаминов и антибиотиков.
Вторая часть посвящена описанию технологии производства синтетических
лекарственных веществ и витаминов. В основу изложения материала поло-
положена химическая классификация лекарственных веществ.
Третья часть посвящена разбору технологии антибиотиков.
Учебник написан в соответствии с программой, утвержденной Министерством
высшего и среднего специального образования, и предназначен для учащих-
учащихся средних специальных учебных заведений.
В учебнике 57 рис., 3 табл.
Рецензенты:
зав. лабораторией синтеза смешанных органических соединений Всесоюзного
научно-исследовательского химико-фармацевтического института им. С. Орд-
Орджоникидзе, доктор химических наук, профессор Л. Н. Яхонтов
зав. кафедрой химии и технологии тонких органических соединений Москов-
Московского института тонкой химической технологии им. М. В. Ломоносова, доктор
химических иаук, профессор Р. П. Евстигнеева
„ 50700—145
П — 37 76 © Издательство «Медицина» Москва. 1977
039@1)—77
ПРЕДИСЛОВИЕ
Развитие химико-фармацевтической промышленности и пере-
перестройка учебных планов и программ по специальности: «Техно-
«Технология химико-фармацевтических препаратов и антибиотиков»
побудили авторов написать учебник, предназначенный для уча-
учащихся средних специальных учебных заведений, который при
небольшом объеме отражал бы современное состояние науки
и техники и соответствовал бы новой программе курса.
Техники-технологи подготавливаются для работы в химико-
фармацевтической промышленности на должностях старшего
аппаратчика, бригадира, мастера, начальника смены отделения
или цеха, контролера отдела технического контроля, лаборанта
цеховой или заводской лаборатории, а также лаборатории науч-
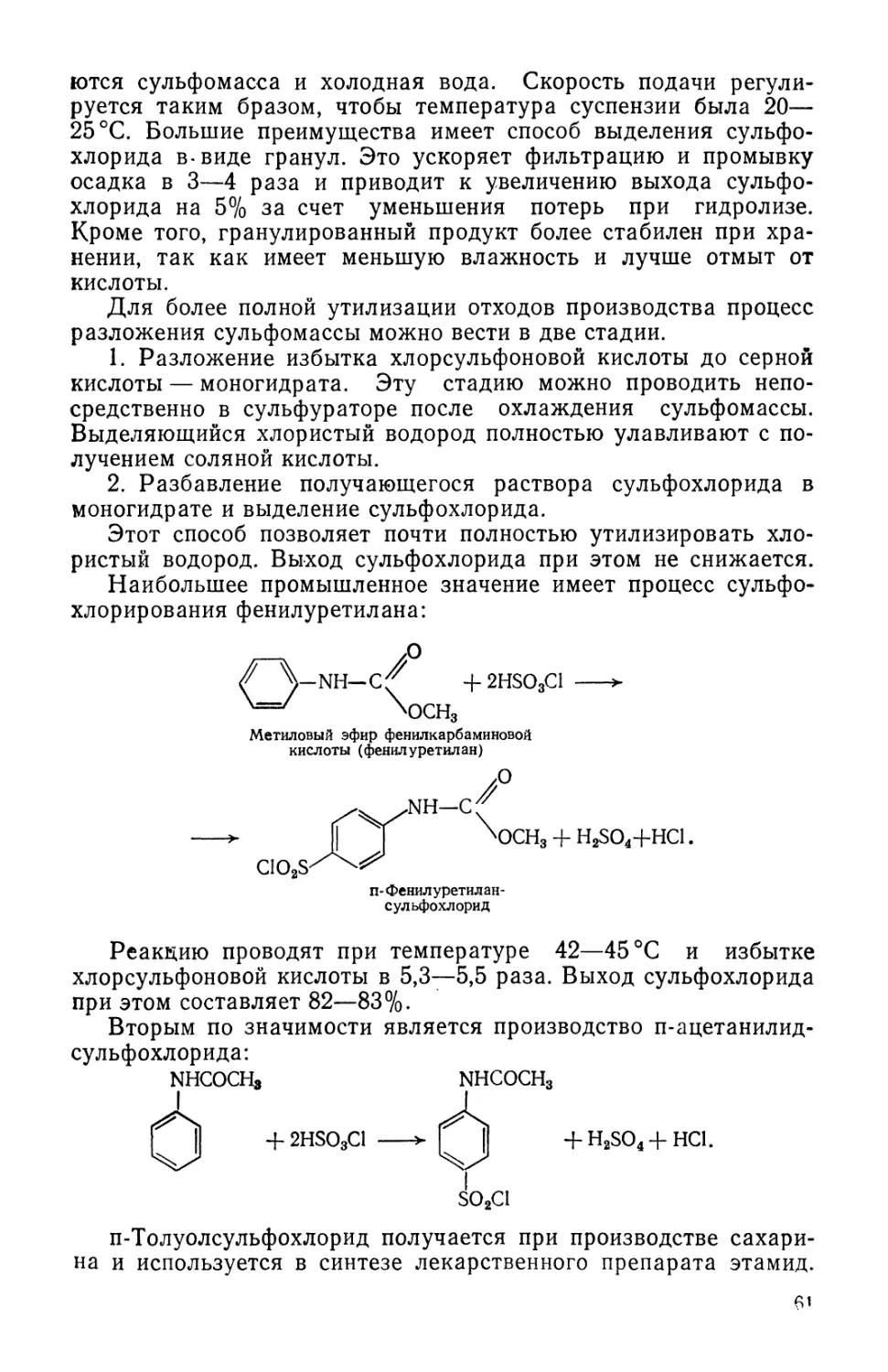
научно-исследовательского института.
Новая учебная программа по предмету «Технология химико-
фармацевтических препаратов и антибиотиков», построенная на
основе химической классификации лекарственных соединений,
в значительно большей мере, чем старая программа, способст-
способствует формированию у учащихся знаний и навыков, необходимых
им в будущей работе.
В первой части учебника изложены основные вопросы химии
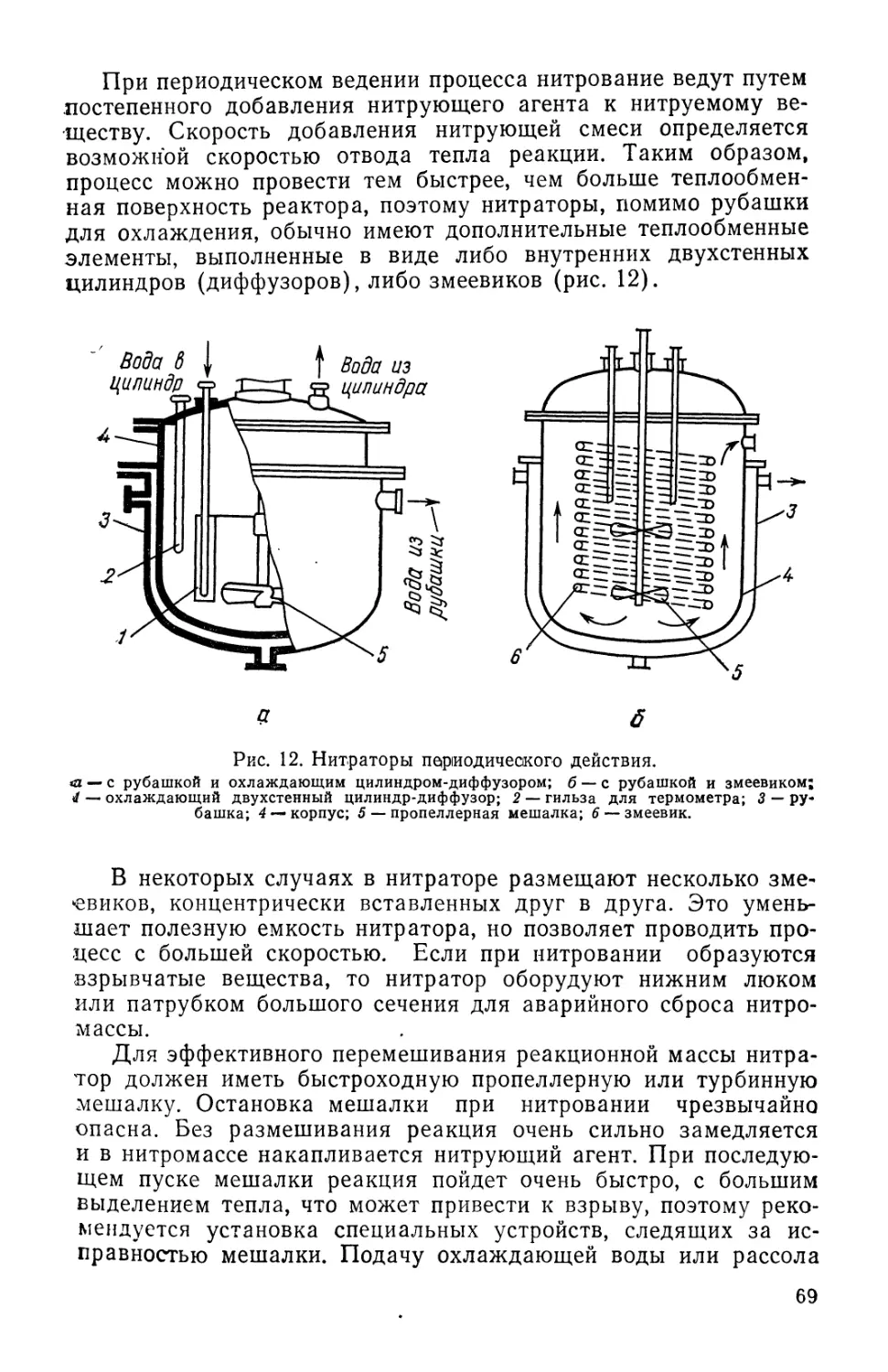
и технологии типовых процессов синтеза лекарственных веществ
и промежуточных продуктов для них. Рассмотрена также типо-
типовая аппаратура для этих процессов. Во второй части рассмат-
рассматриваются вопросы химии и технологии синтетических лекарст-
лекарственных веществ и витаминов. Вопросы аппаратурного оформле-
оформления затрагиваются здесь в меньшей мере, поскольку о типовых
решениях говорится в первой части. Значительное внимание
уделяется физиологической активности и области применения
рассматриваемых соединений. Третья часть учебника посвящена
технологии антибиотиков. Поскольку технология антибиотиков
отличается от технологии синтетических лекарственных веществ,
в этой части подробно рассматриваются типовые операции мик-
микробного синтеза и аппаратура для их осуществления.
Введение, часть первая и вторая, а также глава 21 третьей
части написаны доктором химических наук проф. Б. В. Пассе-
том, третья часть (за исключением главы 21)—кандидатам тех-
технических наук доц. В. Я. Воробьевой.
При написании учебника авторы стремились отразить новей-
новейшие достижения в области химии и технологии лекарственных
веществ, а также изложить весь программный материал в воз-
возможно более компактной форме. При изложении технологии
большое внимание уделялось вопросам техники безопасности и
охраны труда, защиты окружающей среды.
Авторы понимают, что при изложении столь большого
и сложного материала в учебнике малого объема неизбежны
определенные погрешности и с благодарностью примут все заме-
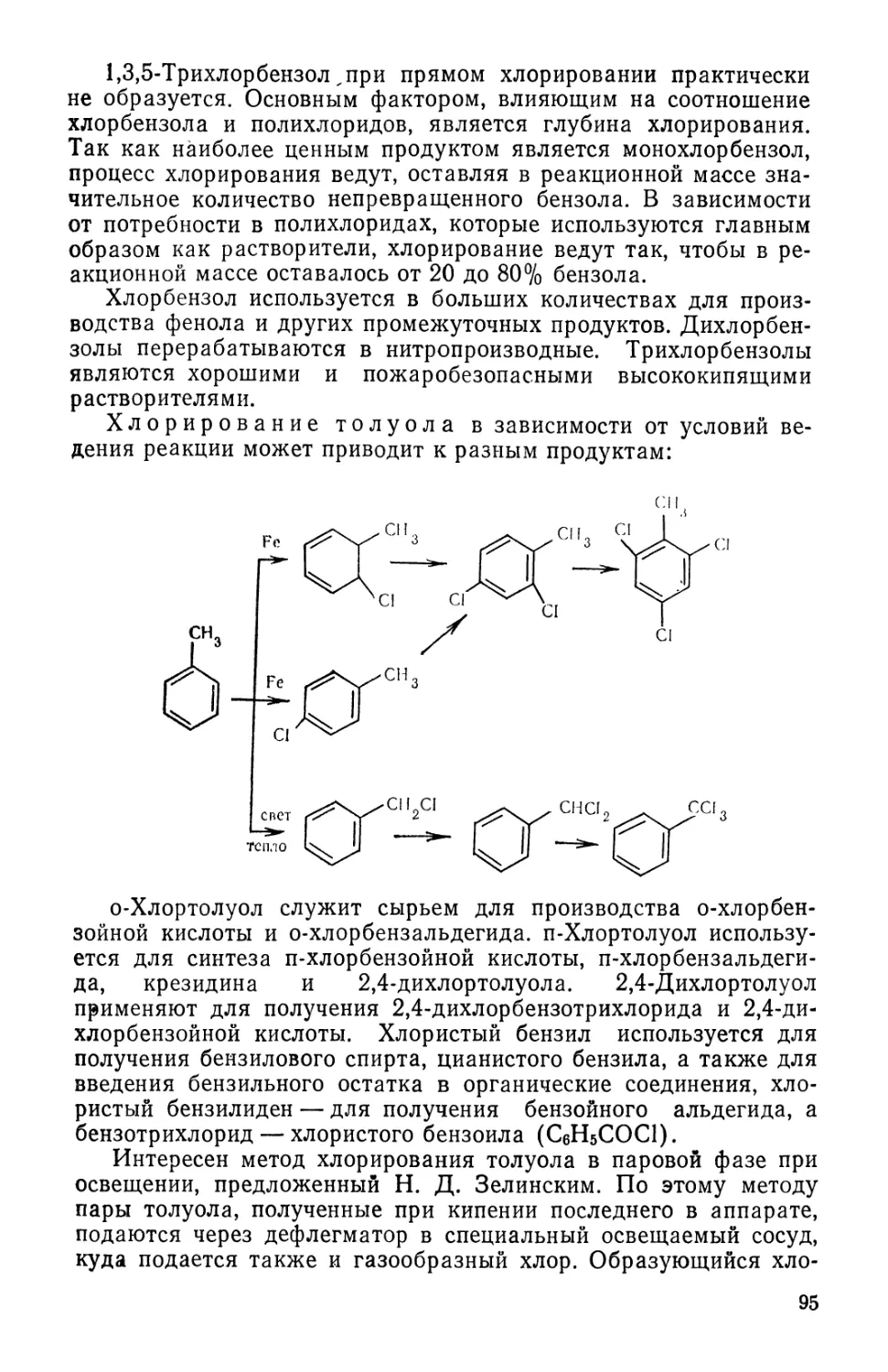
замечания и указания, направленные на улучшение этой книги.
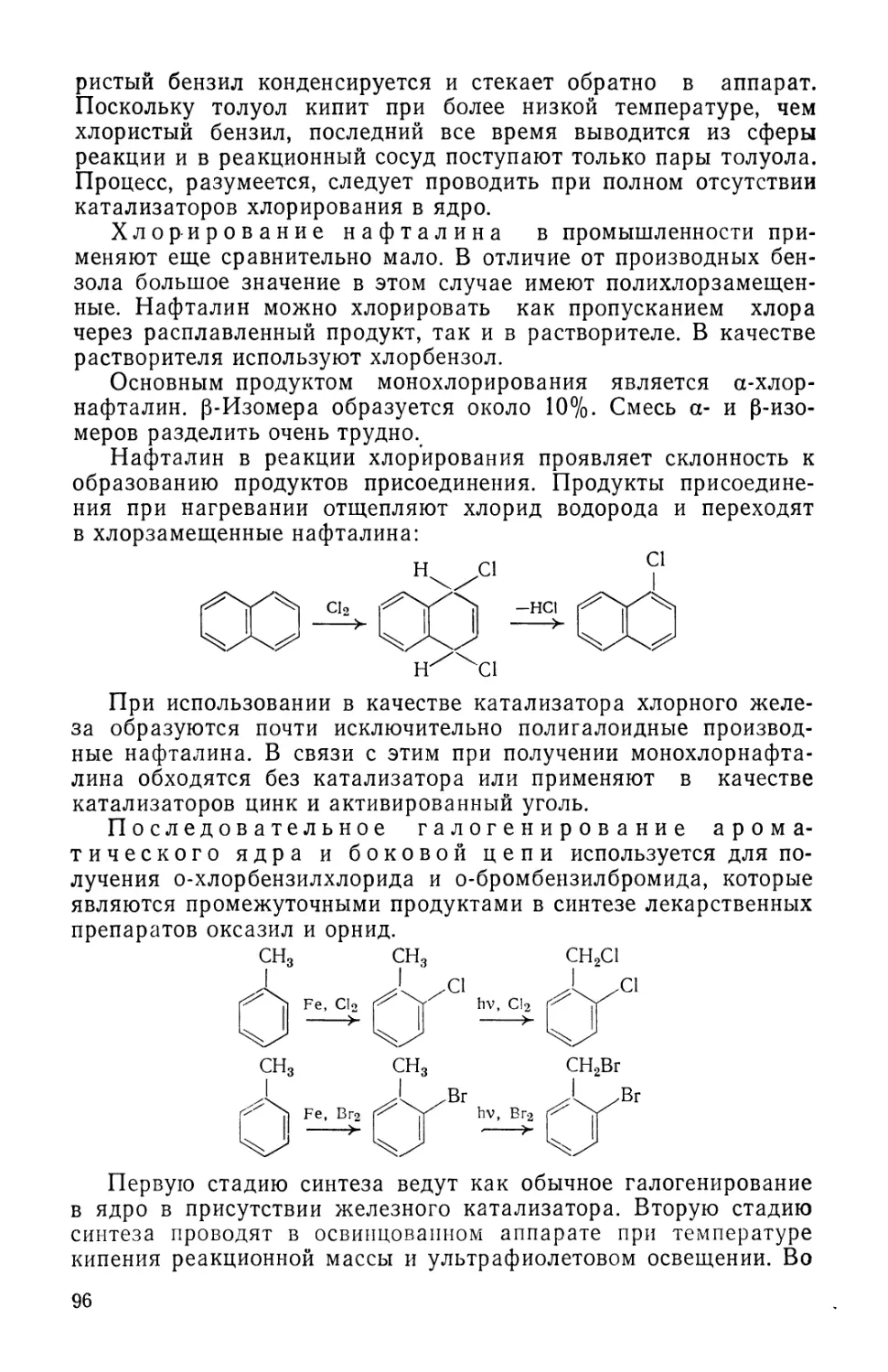
Авторы
ВВЕДЕНИЕ
За последние годы в химической технологии лекарственных
веществ достигнуты большие успехи. Появились новые препара-
препараты, новые методы синтеза химико-фармацевтических препаратов
и антибиотиков, открыты новые закономерности между их строе-
строением и биологической активностью, разработаны методы ана-
анализа, позволяющие следить за ходом синтеза лекарственного
вещества и управлять этим синтезом таким образом, чтобы по-
получить нужный продукт с высоким выходом и необходимого
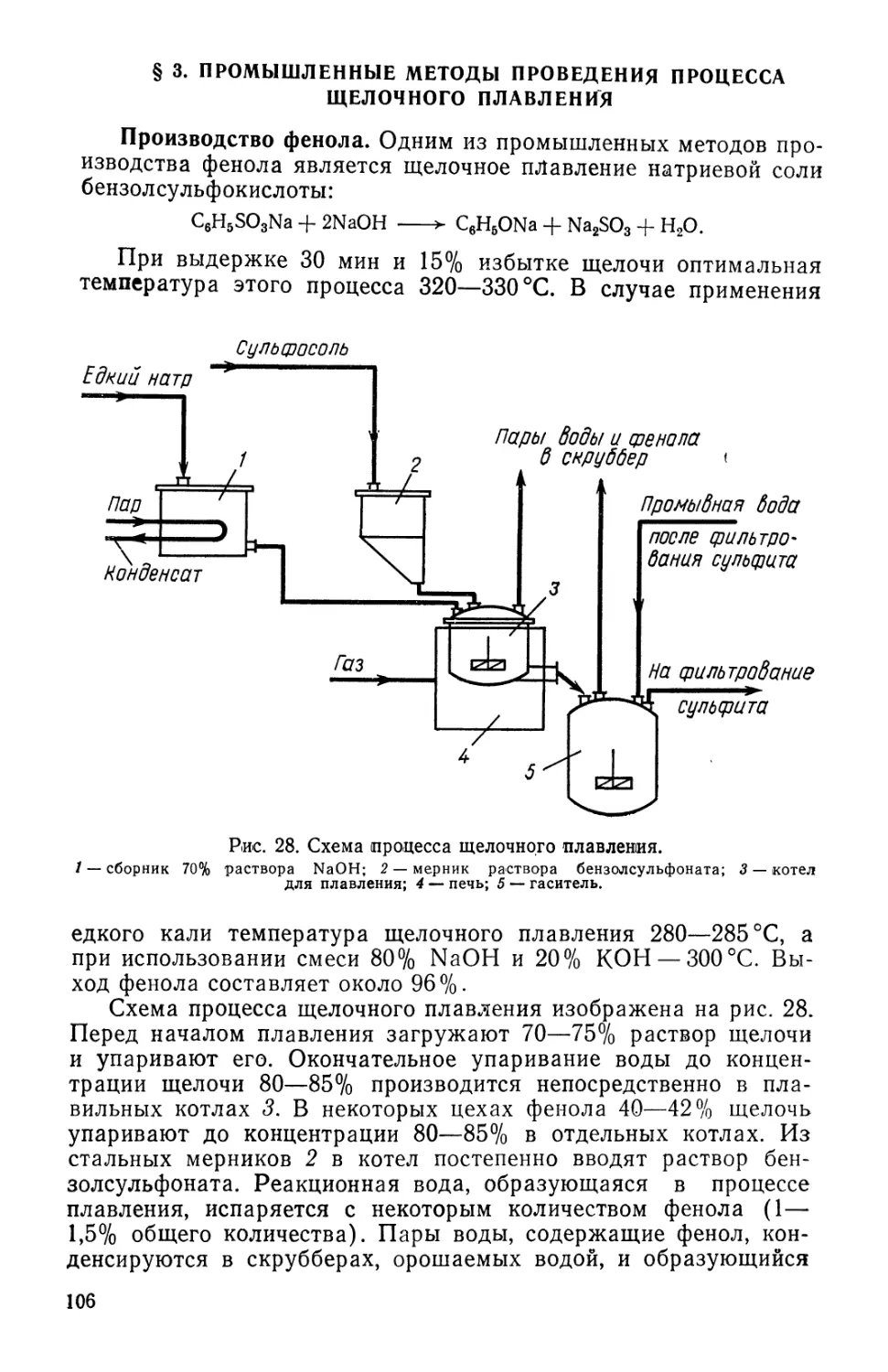
качества.
Однако для того, чтобы правильно оценить современное со-
состояние технологии химико-фармацевтических препаратов и
антибиотиков, нужно хотя бы в общих чертах знать историю
возникновения и развития этой отрасли техники. Современная
технология лекарственных средств своими корнями уходит в да-
далекое прошлое. Разумеется, тогда поиск веществ, облегчающих
страдания больного, был чисто эмпирическим и очень долгое
время все лекарственные вещества были естественного проис-
происхождения. Прогресс в области поиска новых лекарственных
средств был очень медленным и мог ускориться лишь вместе
с успехами науки и техники. Возрастающая потребность обще-
общества в лекарственных средствах, развитие естественных наук
способствовали созданию новых лекарственных средств. Суще-
Существует большое число лекарственных средств растительного,
минерального и животного происхождения, которые были изве-
известны за сотни, а иногда и за тысячи лет до того, как было
расшифровано их строение и предложен метод синтеза. Так,
трава ма-хуан описана в травнике Шень-Нуня (около 3000 лет
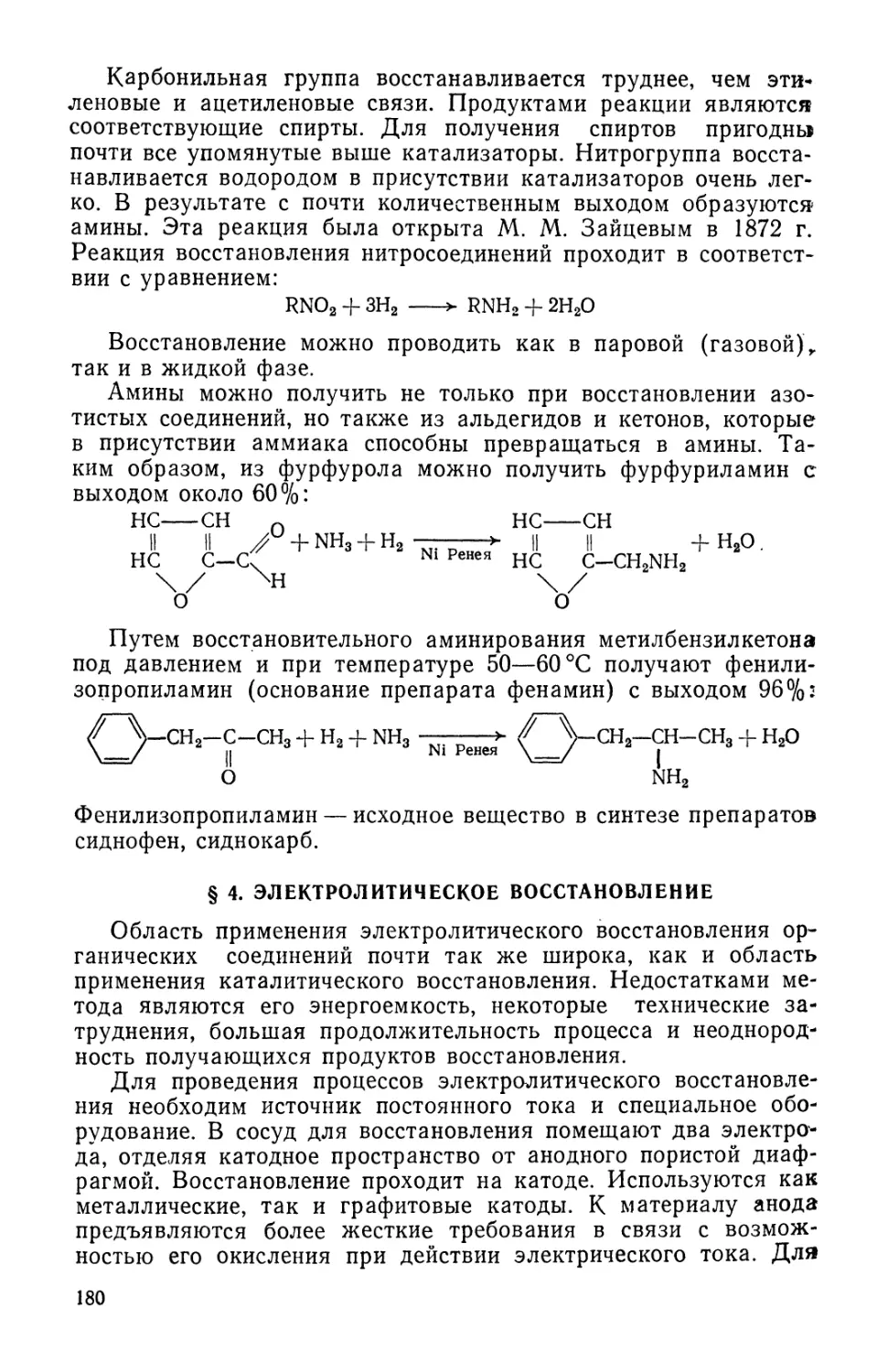
до н.э.), однако действующее начало этого лекарственного ра-
растения— эфедрин — было выделено лишь в 1887 г., первый пол-
полный синтез этого вещества был осуществлен в 1925 г., а фарма-
фармакологические свойства эфедрина основательно исследованы
лишь в 1928 г.
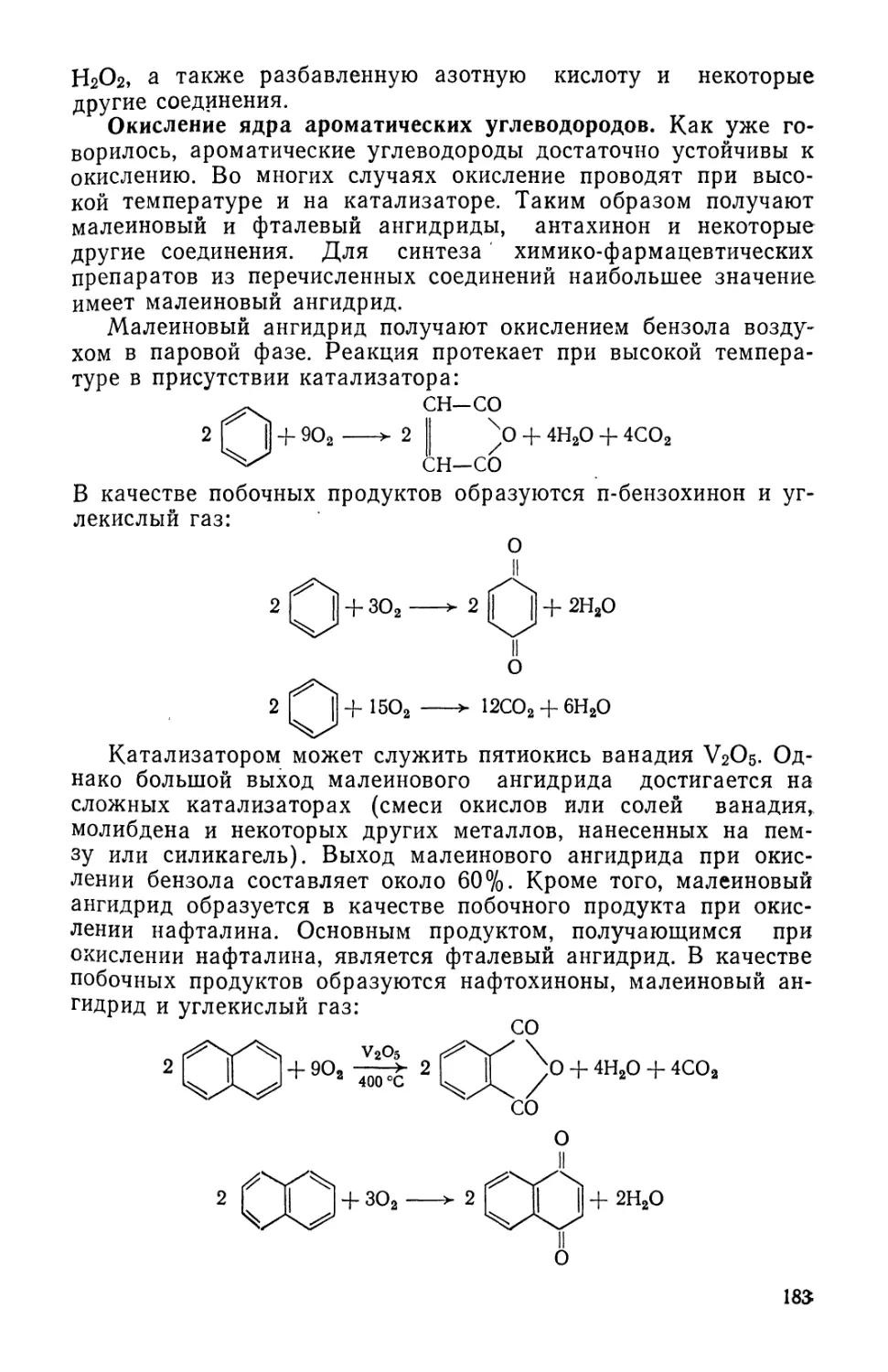
По дошедшим до нас документам можно судить о том, что
медицина и связанное с ее развитием искусство приготовления
лекарств получили наибольшее развитие в Древнем Египте, Ин-
Индии и Китае. Заслуживает внимания папирус Эберса, найденный
в Египте и относящийся к XVII веку до н.э. Он содержит около
800 рецептов различных лекарств. В Древнем Египте правом
приготовления лекарств пользовались лишь представители вер-
верховной касты жрецов. Египтяне умели готовить отвары, настои,
пилюли, мази, пластыри, примочки. Для приготовления этих
средств египтяне применяли ароматические смолы, обладающие
антисептическими свойствами, квасцы, серу, ртутные соедине-
соединения, твердые и жидкие испражнения животных.
Второй по значению является фармация Древней Индии.
Знаменитый врач древности Сушрута (V—VI век до н. э.) опи-
описал около 760 лекарственных растений. Индусы широко ис-
использовали для лечения болезней различные травы, коренья,
плоды, продукты животного происхождения (молоко, кровь,
жир) и различные минералы, из которых наибольшее значение
придавали соединениям ртути.
Перечень лекарственных средств, применявшихся в древнем
Китае, также весьма богат веществами растительного и живот-
животного происхождения. Из растительных средств использовались
лимонник, ревень, имбирь, чай, лук, чеснок, перец, гвоздика
и др. Китайцы применяли для лечения заболеваний препараты,
получаемые из пант, мускуса и т. п.
Египетская, индусская и китайская медицина оказали боль-
большое влияние на развитие фармации и медицины в Древней Гре-
Греции (Гиппократ, 460—377 г. до н. э.) и в древнем Рмме. Круп-
Крупнейшим представителем римской фармации являлся врач Клав-
Клавдий Гален A31—201 г.), который систематизировал накопивший-
накопившийся до него обширный материал о ядах, противоядиях и действии
на человека известных тогда лекарств. В качестве лекарственных
препаратов Гален широко применял извлечения из растительных
материалов. Клавдий Гален впервые ввел понятие о действую-
действующих веществах.
Позже, в период алхимии (IV—XVI века) все более значи-
значительную роль при получении лекарств начал играть химический
эксперимент. В этот период получили развитие важнейшие ме-
методы препаративной химии (перегонка, кристаллизация, пере-
переосаждение, фильтрование и т. п.), которые впоследствии поло-
положили начало химической технологии. В XIV—XV веках широкое
применение в качестве лекарств находят химические препараты.
Возникшие аптеки совмещают функции торгового и производст-
производственного учреждения, а также научно-исследовательской лабо-
лаборатории.
Однако на первом месте по-прежнему остаются лекарствен-
лекарственные вещества естественного происхождения.
Смеси веществ, извлекаемые из природного сырья, широко
применяются в медицине и до настоящего времени. Такие пре-
препараты часто называют галеновыми.
Галеновые препараты — это обычно не отдельные химиче-
химические вещества, а комплексные препараты, получаемые путем спе-
специальной обработки растительного или животного сырья
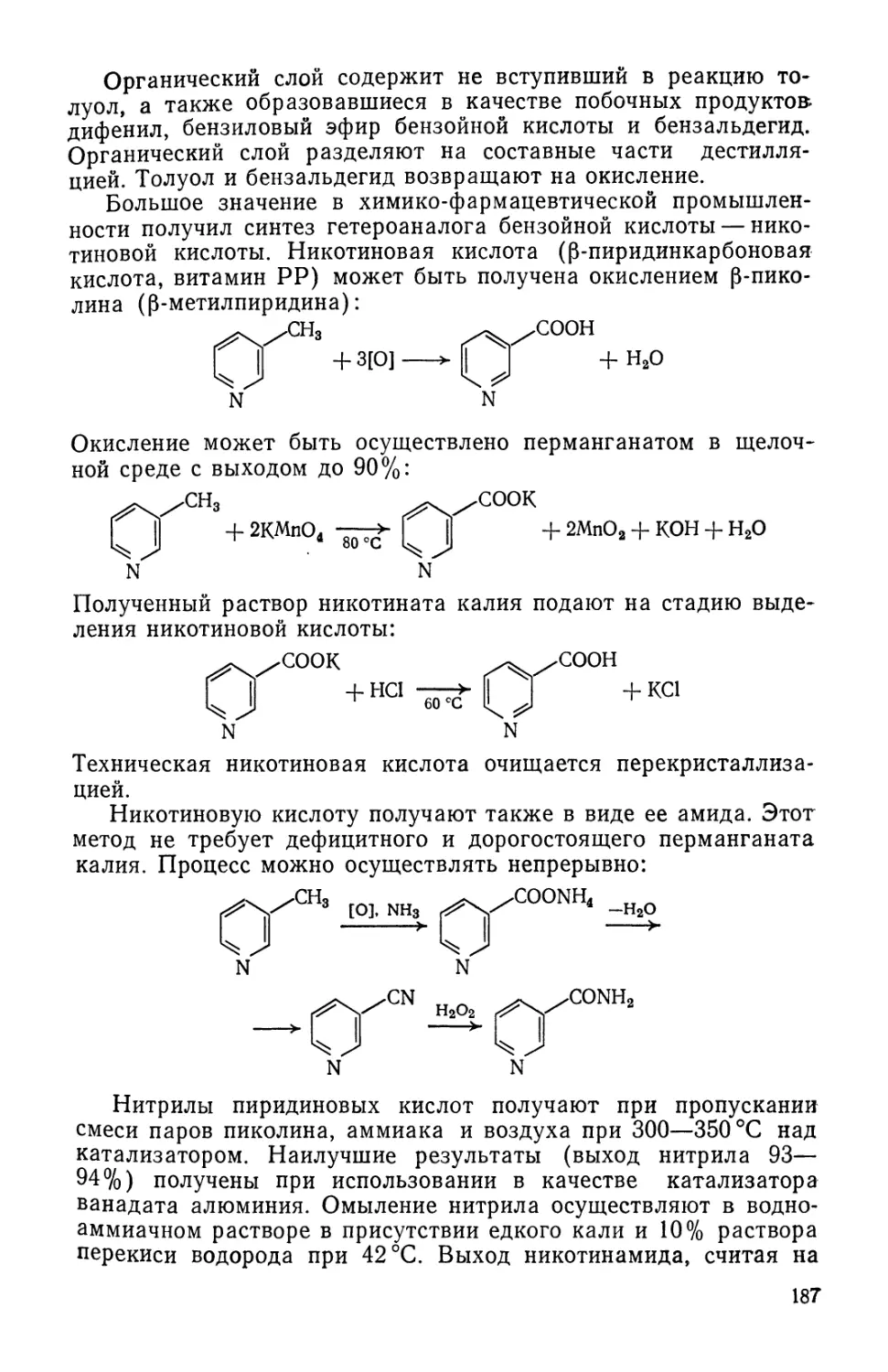
с целью максимального извлечения активного начала и освобож-
дения от балластных веществ. Надо полагать, что и в будущем
значительную часть лекарственных препаратов будут получать
выделением биологически активных веществ, содержащихся
в растительном, животном и минеральном природном сырье.
Основные причины этого заключаются в том, что точный состав
и строение лекарственных веществ растительного и животного
происхождения во многих случаях неизвестны; синтез природ-
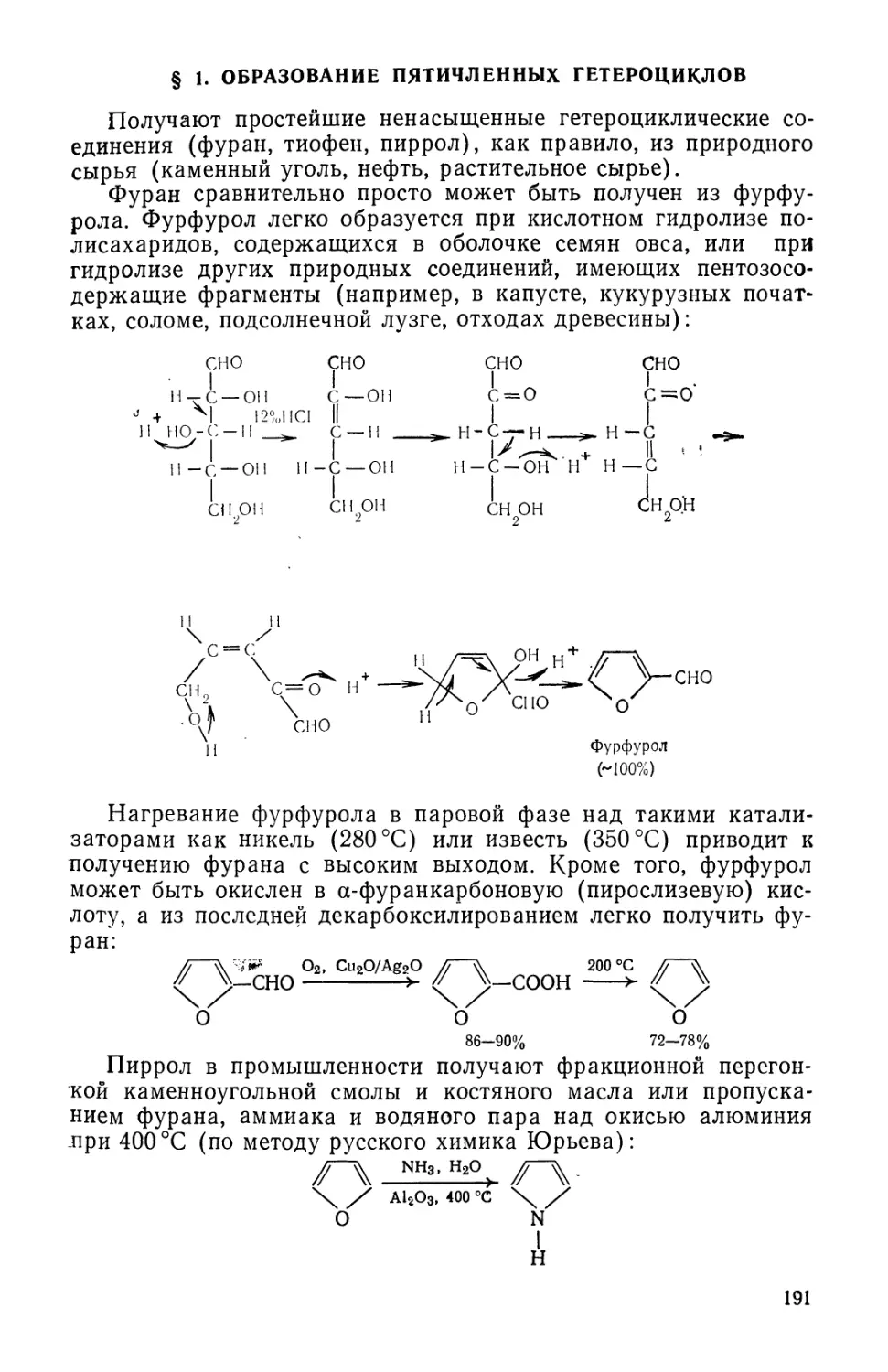
природных веществ часто оказывается дороже, чем извлечение их из
естественного сырья, и, наконец, химия и технология галеновых
и фитохимических препаратов развивается столь же быстрыми
темпами, что и химия и технология синтетических лекарствен-
лекарственных веществ и антибиотиков.
В России народная медицина существовала издавна. Име-
Имелись многочисленные «травники» и «зеленники», которые пред-
представляли собой сборники рецептов. Первоначальными состави-
составителями и распространителями подобных рецептурных сборников
были, вероятно, волхвы (жрецы). Летописные свидетельства об
этом относятся к началу X века н. э. В 1091 г. епископ Ефрем
Переяславский организовал при монастырях первые больницы,
которые обслуживали преимущественно представителей правя-
правящего сословия. В монастырях же были созданы первые рукописи
с наставлением о лечении больных и приготовлении простейших
лекарственных средств. В древней Руси не было разделения
труда между врачом и аптекарем. Врач не только лечил боль-
больного, но и сам изготавливал лекарства.
В XVI веке в Москве, Киеве, Архангельске, Пскове, Новго-
Новгороде, Туле и в ряде других крупных городов появились зеленные
дворы и огороды для выращивания некоторых видов лекарст-
лекарственных растений.
В 1581 г. была организована первая аптека для царской
семьи и приближенных лиц. Получение лекарственной помощи
из царской аптеки не членами царской семьи было связано с
большими трудностями даже для высшего дворянства. Первая
государственная аптека для обслуживания населения была от-
открыта лишь в 1673 г.
В первой половине XVII века был создан высший админист-
административный орган, ведавший медицинским делом в России — «Ап-
«Аптекарский приказ». В обязанности аптекарского'приказа входи-
входили организация медицинского обслуживания царской семьи
и приближенных к царскому двору лиц, медицинское обслужи-
обслуживание армии, охрана страны от эпидемических заболеваний, под-
подготовка медицинских кадров, приглашение из-за рубежа меди-
медицинских и аптечных работников, заготовка лекарственных расте-
растений, наблюдение за торговлей лекарствами и др. Аптекарский
приказ положил начало систематическому и планомерному изу-
изучению отечественного лекарственного сырья и его запасов.
В 1673 г. в России была введена государственная монополия на
торговлю лекарственными средствами.
Первые российские аптеки также выполняли функции науч-
ных лабораторий, где наряду с аптекарями работали так назы-
называемые аптекарские химики (алхимисты). Одним из первых
русских алхимистов был Тихон Ананьин G0-е годы XVII века).
В немалой степени способствовал развитию фармацевтического
дела в России Петр I, по прямому указанию которого были соз-
созданы Петербургский и Московский аптекарские огороды, где
разводились лекарственные растения и функционировали лабо-
лаборатории для переработки сырья. Создателем первой научной
химической лаборатории в России был М. В. Ломоносов A748).
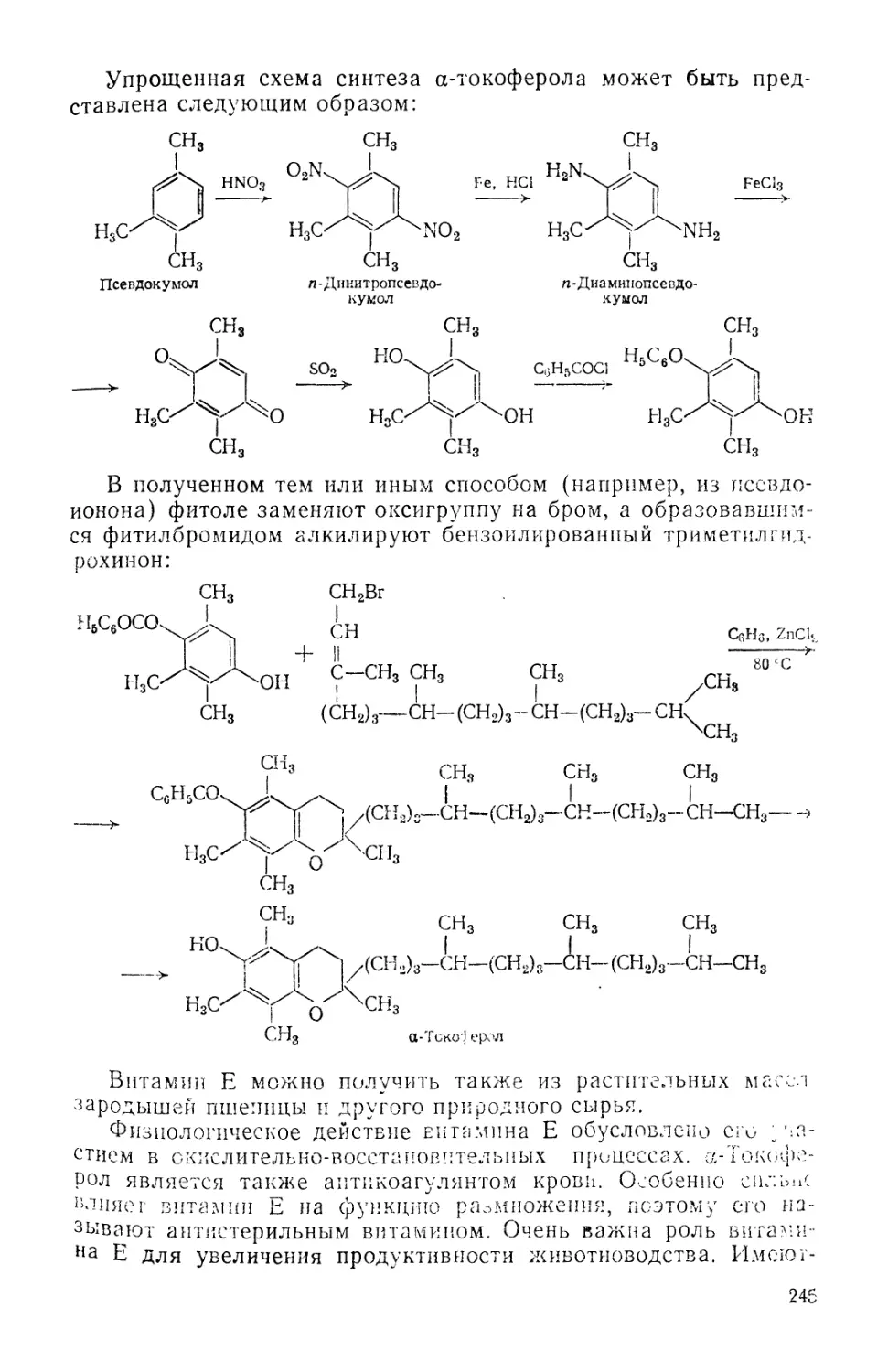
Начатое им дело продолжили Т. Е. Ловиц A757—1804) и
В. М. Севергин A765—1825). Последний опубликовал книгу па
фармацевтическому анализу, которая явилась первым отечест-
отечественным руководством по фармацевтической химии.
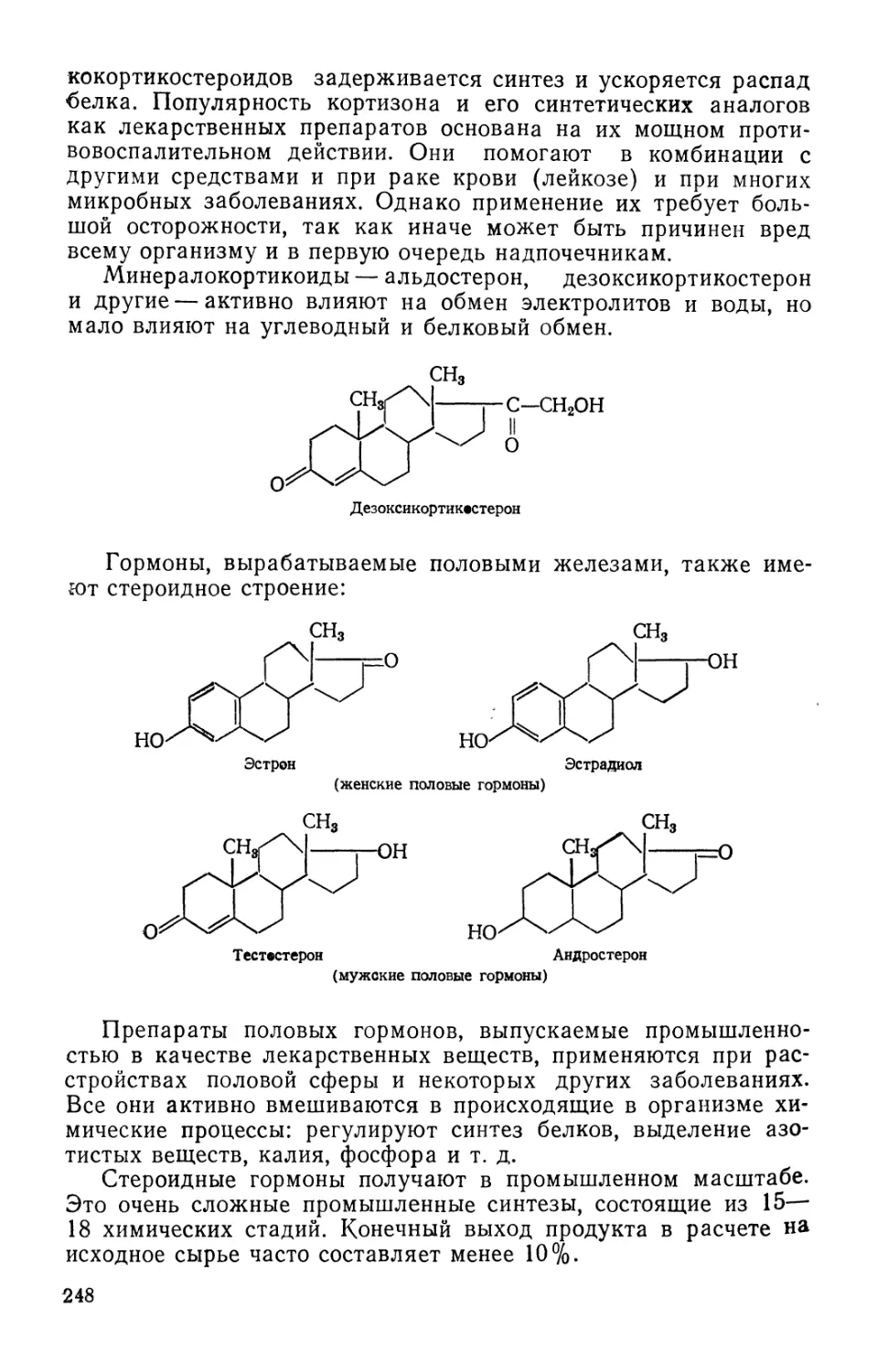
Развитие химии лекарственных веществ тесно связано с раз-
развитием органической химии и в особенности с развитием химии
природных и синтетических красителей, а также таких отраслей
знания, как биология .и медицина. Вторая половина XIX и нача-
начало XX века характеризуются параллельным и, можно сказать,
совместным развитием химии и технологии красителей и химии
и технологии лекарственных веществ. В настоящее время техно-
технологию таких сложных органических соединений, как красители^
лекарственные соединения (в том числе алкалоиды, гормоны,
витамины и антибиотики), душистые вещества, органические
фотореактивы и сложные органические препараты, применяемые
в различных областях народного хозяйства (антиоксиданты,
эмульгаторы, гербициды и т. п.), объединяют в одну дисципли-
дисциплину, называемую «тонкой химической технологией». Вместе с тем
имеется особенность, выделяющая химию и технологию лекар-
лекарственных веществ из этого комплекса. Главная особенность за-
заключается в том, что лекарственные вещества предназначены
для введения внутрь организма человека. Это обусловливает
особые требования к чистоте получаемых веществ, к отсутствию-
в них токсичных примесей.
Для поиска новых лекарственных веществ можно идти не-
несколькими путями, одним из которых являются изучение при-
природных соединений, обладающих сильным биологическим дей-
действием, и создание подобных им веществ с учетом взаимосвязи
между строением и биологической активностью. Другой путь
создания новых лекарственных веществ — поиск веществ, обна-
обнаруживающих сильную биологическую активность среди всех
синтезированных новых соединений (так называемый скрининг).
При этом аналогии с природными веществами не обязательны.
Наконец, третий путь — это изучение продуктов превращения
лекарственного вещества в организме (эти продукты называют-
называются метаболитами) и создание новых лекарственных веществ на
основе знаний об этих превращениях и о механизме действияг
известных препаратов на организм. Одним из наиболее перепек-
тивных путей создания новых лекарственных средств является
поиск, основанный на знании биохимии болезни и ее возбуди-
возбудителей.
После Великой Октябрьской социалистической революции
в нашей стране создаются все условия для развития химии и
технологии лекарственных веществ и химико-фармацевтической
промышленности. Уже в 1920 г. был организован Всесоюзный
научно-исследовательский химико-фармацевтический институт
имени С. Орджоникидзе (ВНИХФИ).
Благодаря работам ВНИХФИ наша страна была освобож-
освобождена от импорта йода (О. О. Магидсон, А. А. Байчиков,
Б. Н. Денисович, Д. А. Шапошников) и получила целый ряд
ценных отечественных препаратов, например, противомалярий-
противомалярийные (О. Ю. Магидсон, М. В. Рубцов, И. Т. Струков), сульфанил-
сульфаниламидные (О. Ю. Магидсон, М. В. Рубцов, А. М. Григоровский,
И. Я. Постовский), противотуберкулезные (М. Н. Щукина,
М. В. Рубцов, И. X. Фельдман), мышьякорганические соедине-
соединения (Г. Кирхгоф, М. Крафт), стероидные гормоны (В. И. Мак-
Максимов, О. С. Мадаева, Н. Н. Суворов). Уже с первых лет суще-
существования института ё нем проводилась огромная работа по
исследованию алкалоидов (акад. А. П. Орехов, Л. М. Уткин,
Е. С. Головчинская, К. А. Чхиквадзе).
ВНИХФИ успешно ведет работу в области технологии и по-
поиска психотропных средств, сердечно-сосудистых препаратов,
химиотерапевтических средств для лечения инфекционных за-
заболеваний и злокачественных новообразований.
В 1931 г. был создан Всесоюзный институт лекарственных и
ароматических растений, в настоящее время — Всесоюзный ин-
институт лекарственных растений (ВИЛР). В 1940 г. организован
Центральный аптечный научно-исследовательский институт
(ДАНИИ).
Большая научно-исследовательская работа по синтезу анти-
антибиотиков проводится во Всесоюзном научно-исследовательском
институте антибиотиков (ВНИИА). За сравнительно короткий
срок было открыто более 1000 антибиотиков, у нескольких сотен
из них выяснено строение, разработан и осуществлен синтез мно-
многих из них.
Огромная работа проводится во Всесоюзном научно-исследо-
научно-исследовательском витаминном институте (ВНИВИ). Коллективом ин-
института под руководством ведущих ученых (Н. А. Преображен-
Преображенский, В. М. Березовский, Г. И. Самохвалов, Е. С. Жданович,
В. А. Яковлев) были разработаны промышленные методы син-
синтеза витаминов А, В4, В2, С, пантотеновой, фолиевой кислот
и ряда других витаминных препаратов. Только за 5 лет (с 1959
по 1963 г.) во ВНИХФИ, ВНИВИ, ВНИИА и ВИЛР было раз-
разработано и внедрено в производство более 100 новых лекарст-
лекарственных препаратов.
Создание новых лекарственных средств и разработка их тех-
нологии — дело очень трудное. Над каждым новым эффектив-
эффективным лекарственным средством должны трудиться сотни специа-
специалистов разного профиля — химики-синтетики, физико-химики,
химики-аналитики, биологи, фармацевты, фармакологи, биохи-
биохимики, токсикологи, клиницисты, большое число специалистов в
области информации, прогнозирования, рекламы, сбыта, эконо-
экономики, санитарии и т. д. и т. п.
В настоящее время в мире насчитывается более 4000 орга-
органических химических веществ, предложенных в качестве ле-
лекарств, а общее количество препаратов, если считать препара-
препараты природного происхождения и комбинированные средства, до-
достигло нескольких десятков тысяч, причем большинство из них
разработаны за последние десятилетия. За 40 лет A930—1970)
выполнено почти в 30 раз больше научно-исследовательских ра-
работ по поискам лекарственных веществ, чем за всю историю
цивилизации до 1930 г. В то же время по физике, химии или ма-
математике рост научных исследований за этот же период был
всего лишь четырех-пятикратным.
Среди трудностей поиска лекарственных препаратов можно
выделить основные.
Для лечения целого ряда заболеваний найдены настолько вы-
высокоэффективные препараты, что синтезировать вещества, обла-
обладающие заметным преимуществом, очень трудно.
Современные новые лекарственные препараты являются про-
продуктами очень сложных многостадийных синтезов. На синтез
каждого нового соединения затрачивается много времени и
средств. Известно, что для нахождения одного препарата нужно
провести изучение от 500 до 3000 химических соединений. Ос-
Осложняют выпуск нового препарата и большие требования к ток-
токсикологии, требуются данные по фармакодинамике нового пре-
препарата, очень высоки требования к чистоте препарата, которая
контролируется современными физико-химическими методами.
Однако поиск новых препаратов не становится от этого менее
упорным. И в СССР и во многих зарубежных странах ассигно-
ассигнования на научно-исследовательские работы растут как в абсо-
абсолютном денежном выражении, так и в процентах к прибылям.
Среди причин интенсификации поиска новых лекарственных
препаратов главными являются следующие. Общий прогресс
биохимии, физиологии, гистологии и других биологических наук
открывает новые пути воздействия при лечении болезней, в ча-
частности, появилась возможность создания препаратов для про-
профилактики заболеваний или лечения их на самых ранних ста-
стадиях. Увеличение контингента лиц пожилого и престарелого
возраста приводит к необходимости поиска специальных так на-
называемых щадящих средств лекарственной терапии. Снижение
детской смертности приводит к появлению значительного про-
процента детей с врожденными дефектами, которые требуют посто-
постоянной поддерживающей терапии. В результате прогресса меди-
10
цинских наук появилась потребность в лекарствах при заболе-
заболеваниях, которые раньше считали не поддающимися
химиотерапии (новообразования, нервно-психические заболева-
заболевания и др.).
Достижения науки привели к необходимости создания спе-
специальных биологически активных препаратов — для человека
в космосе, для работы с радиацией, для решения проблемы пере-
пересадки органов и т. д. Успехи диагностики требуют создания но-
новых диагностических средств (рентгеноконтрастные вещества
и т. д.).
Долгое применение лекарств вызывает появление устойчивых
к этим лекарствам форм болезнетворных бактерий. Это требует
создания новых лекарств с новым механизмом действия.
Интенсивные поиски новых лекарственных средств в нашей
стране проводятся в ряде академических и прикладных инсти-
институтов, а также во многих вузах, к которым относится и Ленин-
Ленинградский химико-фармацевтический институт (ЛХФИ). Построе-
Построены новые научно-производственные базы в Купавне, Новокуз-
Новокузнецке, Свердловске. За годы девятой пятилетки значительно
возросли централизованные расходы на научные исследования
и экспериментальные работы в области синтеза лекарственных
препаратов.
В нашей стране создается государственная система регистра-
регистрации и биологических испытаний синтезируемых в СССР хими-
химических соединений. Создан Научно-исследовательский институт
по биологическим испытаниям химических соединений.
Таким образом, за годы Советской власти в нашей стране
была создана мощная научная база, которая может обеспечить
выполнение обширных планов развития химико-фармацевтиче-
химико-фармацевтической науки и промышленности.
Важным средством для повышения эффективности научных
исследований и внедрения в практику полученных результатов
является организация научно-производственных объединений,
а также использование других форм сближения науки и произ-
производства. Институты химико-фармацевтической промышленности
имеют большой опыт комплексного планирования научных ис-
исследований с привлечением центральных заводских лабораторий
и опытных цехов заводов. Важной стороной деятельности инсти-
институтов является повышение квалификации заводских работников,
повышение научного и методического уровня работы заводских
лабораторий.
Сближению науки с производством способствует введенная
теперь система комплексного и сквозного планирования разви-
развития науки и техники: при планировании заданий по новой тех-
технике предусматривается участие всех учреждений и предприя-
предприятий, связанных с выполнением работ. Проведено упорядочение
сети научно-исследовательских и проектных организаций, а так-
также укрепление опытно-промышленных баз институтов и пред-
11
приятии. Разработан план создания новых филиалов институтов
на базе действующих химико-фармацевтических предприятий.
Основными задачами этих филиалов будет внедрение новой хи-
химической технологии и аппаратурного оформления процессов,
разработка мер и средств техники безопасности и промышлен-
промышленной санитарии, методов очистки промышленных стоков, выбро-
выбросов в атмосферу и решение других технологических задач.
Огромную роль в ускорении технического прогресса в хими-
химико-фармацевтической промышленности должна сыграть комп-
комплексная автоматизация и механизация производства. В настоя-
настоящее время актуальным является переход от автоматизации ста-
стадий и операций к созданию комплексного автоматизированного
производства, работающего в оптимальном режиме. Накоплен-
Накопленный научный и производственный опыт делают эту задачу реаль-
реальной и выполнимой для целого ряда производств синтетических
лекарственных веществ, антибиотиков, фитопрепаратов и вита-
витаминов.
В осуществлении технического перевооружения химико-фар-
химико-фармацевтической промышленности важная роль принадлежит Го-
Государственному институту проектирования медицинской про-
промышленности (Гидромедпром) и его филиалам, которые совме-
совместно с отраслевыми институтами осуществляют проектирование
будущих предприятий. Новые проекты содержат перспективные
творческие решения, учитывающие не только современные, но
и.будущие достижения химической технологии, химического
машиностроения, средств автоматизации и механизации.
Современная химико-фармацевтическая промышленность
имеет ряд особенностей, которые определяют ее развитие. Пер-
Первой особенностью химико-фармацевтической промышленности,
о которой уже упоминалось, являются высокие требования к чи-
чистоте выпускаемой продукции. От той части медикаментов, кото-
которая предназначена для подкожных, внутримышечных и внутри-
внутривенных инъекций, помимо высокой химической чистоты, требует-
требуется полная стерильность. На микробную загрязненность должны
проверяться также таблетированные препараты.
Вторая особенность химико-фармацевтической промышленно-
промышленности заключается в сравнительно небольшом объеме производст-
производства большей части лекарственных средств. Только относительно
небольшое число лекарственных препаратов используется для
лечения разнообразных заболеваний, а потому выпускается в
большом количестве. К их числу относятся сульфаниламидные,
салициловые, противотуберкулезные препараты, анальгетики,
барбитураты, а также некоторые антибиотики.
Химико-фармацевтические производства, как правило, харак-
характеризуются большим удельным расходом сырья и материалов,
что объясняется многостадийностью и сложностью синтеза ле-
лекарственных препаратов. В табл. 1 приводится структура затрат,
из которой видно, что для синтетических лекарственных препа-
12
Таблица
Структура затрат по химико-фармацевтической промышленности
Элементы затрат
Сырье и материалы за вычетом стоимости утили-
утилизируемых отходов
Топливо
Энергия
Заработная плата
Отчисления на социальное страхование
Амортизация основных средств
Прочие расходы
Производство, % к итогу
синтетических
химико* фарма-
фармацевтических
препаратов
84,6
1,1
0,9
9,6
0,8
1,4
1,6
антибиотиков
58,5
"l,6
7,5
22,5
1,9
4,8
3,2
ратов удельный вес стоимости сырья в общей себестоимости со-
составляет более 80%, а для антибиотиков — около 60%. Это де-
делает особенно необходимой постоянную работу по совершенст-
совершенствованию технологии, повышению выходов по стадиям и утили-
утилизации отходов производства.
Химико-фармацевтическая промышленность характеризуется
сравнительно быстрым обновлением номенклатуры лекарствен-
лекарственных средств. Благодаря этой особенности, а также малым объе-
объемам производства в химико-фармацевтической промышленности
широкое распространение получили совмещенные схемы произ-
производства, позволяющие быстро переходить от получения одного
препарата к выпуску другого. Характерной особенностью хими-
химико-фармацевтической промышленности является также то, что
все выпускаемые ею вещества должны быть переработаны в го-
готовые лекарственные формы.
Большие качественные и количественные изменения произо-
произошли в химико-фармацевтической промышленности СССР за го-
годы девятой пятилетки. Прирост продукции за 1971—1974 гг. со-
составил около 55% против запланированных 47,4%. В 1,5 раза
увеличился выпуск антибиотиков, преимущественно полусинте-
полусинтетических. Производство антибиотиков, обладающих противо-
противоопухолевым действием, увеличилось в 2 раза. Более чем на 20%
увеличился выпуск противомикробных синтетических лекарст-
лекарственных средств. Сульфаниламидных препаратов сейчас выпу-
выпускается на 26% больше, чем в 1971 г., а производство витаминов
превышает уровень 1970 г. в 1,9 раза. За прошедшее пятилетие
освоен выпуск более 180 новых лекарственных средств. Для мно-
многих препаратов разработаны и внедрены в промышленное про-
производство новые, более эффективные и удобные в применении
лекарственные формы. За годы девятой пятилетки число меди-
медикаментов, потребность в которых удовлетворяется полностью,
13
возросло более чем на 300 наименований. Появилась возмож-
возможность увеличить поставку ряда лекарственных препаратов на
экспорт. Так, например, ib 1975 г. была увеличена продажа на
экспорт кофеина синтетического, новокаина, теофиллина, пени-
пенициллина, ампициллина, левомвдетина и др. Вступили в строй но-
новые цеха (по производству химико-фармацевтических препаратов
на Ереванском, Ленинградском, Одесском и других заводах.
Важнейшей задачей медицинской промышленности остается
повышение технического уровня производства. За 1971 —1975 гг.
проведено более 280 мероприятий, способствующих совершенст-
совершенствованию технологии химико-фармацевтических препаратов, и
более 180 — по механизации и автоматизации производственных
процессов. В результате такой модернизации производства на
механизированный и автоматизированный труд переведены за
5 лет более 20 тыс. человек, на 5 тыс. человек больше, чем на-
намечалось. Проведенные мероприятия позволили увеличить про-
производительность труда в медицинской промышленности за годы
девятой пятилетки на 57,1% против 45,8% по плану. Объем про-
производства химико-фармацевтических препаратов за это же вре-
время увеличился на 86% при плане 82,6%.
За годы предстоящей десятой пятилетки планируется значи-
значительно увеличить производство амидопирина, ацетилсалициловой
кислоты (аспирина), сульфадимезина, антибиотиков, инсулина,
гепарина, витаминов A, Bi, B2.
Одной из основных задач, стоящих перед медицинской про-
промышленностью в десятой пятилетке, является улучшение каче-
качества продукции. В 1976—1980 гг. наша промышленность выпу-
выпустит большое число новых лекарственных средств для
профилактики и лечения нервно-психических заболеваний, тубер-
туберкулеза, виру1СР1ых, острых бактериальных, инфекционных, сердеч-
сердечно-сосудистых и грибковых заболеваний. Большие работы
намечено провести в области совершенствования крупнотоннаж-
крупнотоннажных производств, в частности, намечено внедрить новые техно-
технологические процессы в производствах аспирина, витаминов А,
Е, С, пенициллина, тетрациклина, окситетрациклина, эритроми-
эритромицина, полусинтетических антибиотиков.
В десятой пятилетке планируется увеличить выпуск продук-
продукции медицинской промышленности на 44—46% при увеличении
производительности труда на 36—37%. Большие задачи ста-
ставятся по повышению эффективности научных исследований и
быстрому внедрению их в практику. Разрабатываются научные
основы технологии с преимущественным использованием замкну-
замкнутых циклов.
Намечено переработать X издание Государственной фармако-
фармакопеи и выпустить XI ее издание, куда войдет описание более
500 лекарственных препаратов, по своему качеству превосходя-
превосходящих зарубежные образцы. Сроки годности выпускаемых лекар-
лекарственных препаратов будут увеличены.
Часть I
ТЕХНОЛОГИЧЕСКИЕ МЕТОДЫ ПРОИЗВОДСТВА
ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ
Глава 1
СЫРЬЕ ДЛЯ ХИМИКО-ФАРМАЦЕВТИЧЕСКОЙ
ПРОМЫШЛЕННОСТИ
§ 1. ИСТОЧНИКИ СЫРЬЯ ДЛЯ ХИМИКО-ФАРМАЦЕВТИЧЕСКОЙ
ПРОМЫШЛЕННОСТИ
В промышленном производстве химико-фармацевтических
препаратов используют разнообразное сырье, получаемое как из
растительных и животных материалов, так и путем химическо-
химического синтеза. О роли растительного сырья в производстве галено-
галеновых и других химико-фармацевтических препаратов говорилось
выше. Ресурсы растительного сырья в СССР огромны. Боль-
Большие работы в настоящее время проводятся по выращива-
выращиванию наиболее ценных видов дикорастущих лекарственных
растений (например, женьшеня) на промышленных плантациях,
что должно значительно увеличить обеспеченность промышлен-
промышленности этими дефицитными видами сырья и снизить себестоимость
продукции.
В химико-фармацевтической промышленности используют
также животное сырье. Например, тиреоидин получают из щито-
щитовидной железы, гистидин — из крови животных, адреналин — из
надпочечников, инсулин — из поджелудочной железы и т. д.
Наиболее распространенным сырьем является химическое.
Минеральное сырье применяют для производства неорганиче-
неорганических солей (NaBr, КВг, KMnO4, BaSO4, HgCl2, Nal, KI и др.),
а также в качестве реактивов для проведения различных пре-
превращений органических соединений.
Для производства минерального сырья используются преи-
преимущественно рудные ископаемые. Химико-фармацевтическая
промышленность использует большие количества минеральных
кислот и щелочей, производимых химической промышленностью.
Исходное органическое сырье поставляет коксохимическая, неф-
нефтехимическая, анилинокрасочная промышленность и промышлен-
промышленность основного органического синтеза.
Значительную долю органического сырья занимают углево-
углеводороды ароматического ряда, получаемые из продуктов коксо-
коксования каменного угля и при переработке нефти.
15
Состав летучих продуктов коксования зависит от качества
исходного угля и режима коксования. Основными летучими про-
продуктами коксования являются каменноугольная смола, сырой
бензол, аммиак, вода и сероводород. Каменноугольная смола
представляет собой темно-бурую или черную вязкую жидкость
(плотность 1180—1230 кг/м3), состоящую из сложной смеси угле*
водородов и гетероциклических соединений. Разгонка каменно-
каменноугольной смолы производится на специальных установках.
Наиболее производительными являются смолоразгонные уста-
установки непрерывного действия. Из масел, получаемых при раз-
разгонке каменноугольной смолы, выделяют индивидуальные ве-
вещества, после чего масла поступают на дальнейшее использо-
использование.
Например, тяжелое масло применяется для улавливания бен-
бензола. Из легкого масла фракционной разгонкой получают бен-
бензол, толуол, ксилол, пиридин. Из фенольного масла фенолы извле-
извлекают раствором едкого натра. Растворимые в воде феноляты
нейтрализуются кислотами или углекислым газом. Из нафтали-
нафталинового масла путем кристаллизации выделяют сырой нафталин,
который очищают далее сублимацией. При кристаллизации
антраценового масла выпадает смолистая масса зеленого цвета,
состоящая из смеси антрацена, карбазола и фенантрена. Эту
массу отфильтровывают, а затем разделяют на составные части
путем химической обработки и дистилляции.
Получающийся при улавливании летучих продуктов коксо-
коксования каменного угля сырой бензол представляет собой легко-
легкоподвижную, резко пахнущую жидкость (плотность 880—
890 кг/м3), состоящую преимущественно из ароматических угле-
углеводородов. Сырой бензол содержит также небольшие количест-
количества тиофена, пиридина и других соединений. Индивидуальные
чистые вещества могут быть выделены из сырого бензола или
из его фракций ректификацией.
Рост потребности в ароматических углеводородах привел в
последнее время к увеличению их производства из нефти. Аро-
Ароматические углеводороды получают из бензинов прямой пере-
перегонки нефти и крекинг-бензинов, а также из продуктов пиро-
пиролиза керосина и других нефтяных дистиллятов. Для производ-
производства ароматических углеводородов применяют также процессы
ароматизации нефтяного сырья, основанные на дегидрировании
нафтеновых углеводородов и дегидроциклизации парафинов
и олефинов. В качестве катализаторов применяют окислы хрома,
алюминия, титана и других металлов, осажденные на носителях.
Процессам дегидрирования и дегидроциклизации (дегидроаро-
матизации) подвергают не только индивидуальные углеводоро-
углеводороды, но и сложные смеси их — нефтяные фракции.
Сочетание этих процессов на практике применяется в различ-
различном технологическом оформлении (платформинг, гидроформинг
и другие).
16
§ 2. ХАРАКТЕРИСТИКА НЕКОТОРЫХ ВИДОВ ОСНОВНОГО
И ВСПОМОГАТЕЛЬНОГО СЫРЬЯ
Бензол С6Н6 жидкий при обычной температуре углеводо-
углеводород; температура кипения 80,1 °С, температура плавления 5,5 °С,
плотность при комнатной температуре 880 кг/м3.
Технический бензол содержит небольшое количество приме-
примесей, главные из которых — сероуглерод, тиофен и неароматиче-
неароматические углеводороды. Бензол нефтяного происхождения обычно
тиофена не содержит. Для некоторых целей бензол, лишенный
тиофена, имеет значительные преимущества. Для определения
содержания тиофена в бензоле используют колориметрический
метод анализа. Бензол находит широкое применение в производ-
производстве промежуточных продуктов: для получения нитросоедине-
ний, сульфокислот и галогенпроизводных, а также в качестве
растворителя при проведении различных реакций.
Бензол относится к легковоспламеняющимся жидкостям
(температура вспышки 16 °С). Пары бензола в смеси с воздухом
образуют взрывоопасные смеси (пределы взрываемости 1,4—
7,1%). Бензол является токсичным веществом. Предельно допу-
допустимая концентрация (ПДК) паров бензола в производственном
помещении 20 мг/м3. При отравлении в первую очередь пора-
поражается центральная нервная система и кроветворные органы.
Толуол СбНбСН3 ближайший гомолог бензола, температу-
температура кипения 110,6°С, плотность 870 кг/м3.
Примесями в техническом толуоле, так же как и в техниче-
техническом бензоле, могут быть серосодержащие соединения. Приме-
Применяется толуол преимущественно для получения нитропродуктов,
галогенпроизводных и в меньшей степени для получения сульфо-
сульфокислот, а также как растворитель. Толуол — легковоспламеняю-
легковоспламеняющаяся жидкость (температура вспышки 5°С), образует взрыво-
взрывоопасные смеси с воздухом (пределы взрываемости 1,3—7%). То-
Толуол действует на нервную систему сильнее, чем бензол. Силь-
Сильнее сказывается и раздражающее действие паров. Значительно
слабее, чем бензол, влияет на кроветворение. Предельно допу-
допустимая концентрация для толуола 50 мг/м3.
Ксилолы С6Н4(СН3J. Технический ксилол является
смесью всех трех изомеров (орто-, мета- и пара-) с преоблада-
преобладанием мета-изомера F0—70%). Технический ксилол содержит в
качестве примеси этилбензол. Технический ксилол кипит в ин-
интервале 136,5°—141,5 °С, плотность его 860—870 кг/м3. Темпера-
Температура вспышки ксилола около 20 °С. Технический ксилол приме-
применяется для получения нитропродуктов, а также в качестве
растворителя. По токсичности ксилол сходен с бензолом и то-
толуолом. ПДК для него 50 мг/м3. о-Ксилол может быть отделен
от смеси мета- и пара-изомеров ректификацией. п-Ксилол выде-
выделяется из смеси с мета-изомером вымораживанием.
Метиловый спирт СН3ОН — легковоспламеняющаяся
жидкость, пары его образуют взрывоопасные смеси с воздухом.
2-976 17
Пределы взрываемое™ для метилового спирта 6,0—34,7%. Тем-
Температура кипения метилового спирта 64,7 °С. Метиловый спирт
является сильным нервным и сосудистым ядом. При отравлении
им характерны поражение зрительного нерва и сетчатки глаза,
возможен смертельный исход. Смертельная доза для взрослого
человека — 30 г. Предельно допустимая концентрация паров Аме-
тилового спирта в производственных помещениях 50 мг/м3.
Транспортируются спирты в железнодорожных цистернах и в
бочках. Хранение метилового спирта на прицеховой площадке
не допускается, отпуск его цеху со склада производят только
в количестве, необходимом для очередной загрузки, а слив из
бочек в приемную цеховую емкость — только в присутствии от-
ответственного лица, выделенного цеховой администрацией.
Этиловый спирт С2Н5ОН также является легковоспла-
легковоспламеняющейся жидкостью. Пределы взрываемости: 3,6—19%. Тем-
Температура кипения 78,4 °С, ПДК—1000 мг/м3. Оказывает на че-
человека наркотическое действие. Перевозится в цистернах и
стальных бочках.
Хлор СЦ и фосген СОСЦ при нормальной температуре
являются газами (для хлора температура кипения — 34 °С, для
фосгена — 8,2 °С). Их перевозят в жидком виде под давлением
в специальных стальных цистернах и баллонах (хлор). Сами
эти вещества негорючи и невзрывоопасны. Однако если в хлоре
содержится примесь водорода в количестве более 10% (по
объему), то такая смесь взрывоопасна. Хлор и фосген очень
токсичны. В случае вдыхания этих газов возможен смертельный
исход. ПДК для хлора 1 мг/м3, для фосгена 0,5 мг/м3.
Бром Вг2 в обычных условиях представляет собой буро-
красную тяжелую жидкость (плотность 3120 кг/м3) с температу-
температурой плавления — 7,3 °С и температурой кипения 59 °С. Его транс-
транспортируют'в стеклянных бутылях с притертыми пробками.
Очень летуч. Пары его токсичны и их действие на организм ана-
аналогично действию хлора (ПДК 1 мг/м3). Попадание на кожу
жидкого брома приводит к долго не заживающим ожогам.
Серная кислота H2SO4 является очень распространен-
распространенным неорганическим сырьем. Выпускается в нескольких товар-
товарных формах: купоросное масло (92,5% серная кислота) — жид-
жидкость с температурой застывания —30,3 °С; моногидрат A00%
серная кислота)—жидкость с температурой застывания
10,45 °С, получается смешением купоросного масла и олеума;
олеум 20% B0% раствор серного ангидрида в моногидрате) —
жидкость с температурой застывания—11°С. Олеум 65%
имеет температуру плавления —0,35 °С.
Серная кислота является негорючим продуктом, однако при
смешении олеума с такими органическими веществами, как ке-
керосин, масло и др. (например, при наливании в бутыли, загряз-
загрязненные этими веществами), возможен взрыв. Концентрирован-
Концентрированная серная кислота и олеум при попадании на тело вызывают
18
тяжелые ожоги. Серный ангидрид, растворенный в олеуме, вы-
выделяется из него на воздухе, образуя туман, сильно раздражаю-
раздражающий дыхательные пути и поражающий легкие. ПДК паров сер-
серной кислоты и серного ангидрида — 1 мг/м3.
Соляная НС1 и хлорсульфоновая HSO3C1 кисло-
кислоты негорючи и невзрывоопасны, но при попадании воды в хлор-
сульфоновую кислоту возможен ее выброс или разрыв сосуда,
в котором хранится продукт. На воздухе обе кислоты выделяют
хлористый водород, раздражающий слизистые оболочки и верх-
верхние дыхательные пути. Хлорсульфоновая кислота при попадании
на кожу вызывает тяжелые ожоги аналогично серной кислоте.
ПДК для хлористого водорода 5 мг/м3.
Азотная кислота HNO3 и меланж (HNO3+H2SO4) — негорю-
негорючие и невзрывчатые жидкости. В промышленности применяют
концентрированную азотную кислоту (плотность 1495—
1500 кг/м3) с концентрацией HNO3 96—98% и разбавленную
(плотность 1300—1370 кг/м3) с концентрацией 50—60%.
Меланж, кроме азотной, обычно содержит 7—10% серной
кислоты. При смешении концентрированной азотной кислоты
с некоторыми органическими веществами возможны сильные
взрывы и загорание. Попадание кислоты и меланжа на кожу
вызывает ожоги. Основное токсическое действие азотной кисло-
кислоты определяется выделяющимися из нее окислами азота, кото-
которые вызывают сильное раздражение дыхательных путей, а в тя-
тяжелых случаях — отек легких, возможны смертельные исходы,
ПДК для окислов азота — 5 мг/м3.
Едкий натр (каустическая сода) NaOH, едкое кали
КОН, карбонат натрия (кальцинированная сода) Na2CO3.
Каустическая сода, выпускаемая в виде твердого плава, со-
содержащего около 92% NaOH, или в чешуированном виде, транс-
транспортируется в стальных барабанах. Выпускаемый также 42%
водный раствор едкого натра перевозят в стальных железнодо-
железнодорожных цистернах или в бочках. Кальцинированная сода транс-
транспортируется в бумажных мешках, а также в специальных кон-
контейнерах. Все эти продукты негорючи и невзрывоопасны. При
длительной работе с кальцинированной содой могут возникнуть
экземы. Едкий натр (каустическая сода) и едкое кали при по-
попадании на кожу вызывают тяжелые поражения, особенно опас-
опасные для глаз.
Сульфид натрия Na2S применяется для восстановле-
восстановления, например, при получении аминов из нитросоединений. Вы-
Выпускается в виде твердого плава, содержащего 62—65% основ-
основного вещества. Хранится и транспортируется в стальных герме-
герметичных барабанах. Действие на кожу и на слизистые оболочки
аналогично действию щелочи.
При спуске растворов сульфида натрия в канализационную
систему, содержащую кислые сточные воды, образуется серово-
сероводород, который может проникнуть в производственные и быто-
2* 19
вые помещения. Сероводород образуется также при гидролизе
растворов сернистого натрия.
Сероводород H2S является сильным ядом, его ПДК
10 мг/м3. При содержании сероводорода в воздухе более
1000 мг/м3 в результате паралича дыхательного центра и сердца
мгновенно наступает смерть.
Нитрит натрия ЫаЫОг широко используется в качестве
реагента (в кислой среде) в реакциях диазотирования и нитро-
зирования. Хранится и транспортируется в крафт-мешках. Нит-
Нитрит натрия — сильный яд. Взаимодействие с кислотами приво-
приводит к образованию нестойкой азотистой кислоты и ядовитых
окислов азота. При взаимодействии нитрита натрия с некоторы-
некоторыми химикатами, например с влажной цинковой пылью, возмож-
возможно обильное выделение окислов азота и даже воспламенение
мешков и других горючих материалов.
Как видно из приведенных кратких характеристик, многие
виды сырья для химико-фармацевтической промышленности яв-
являются токсичными, огнеопасными и взрывоопасными вещест-
веществами.
Знание свойств перерабатываемых и получающихся веществ,
физико-химических особенностей процесса, умение предвидеть
и устранить опасности и вредное воздействие производства, точ-
точное выполнение правил техники безопасности и соблюдение мер
личной защиты позволяют предотвратить отравления, профес-
профессиональные заболевания и аварии.
На долю несчастных случаев, вызванных чисто технически-
техническими причинами, приходится только 15%. Большая часть несчаст-
несчастных случаев (~85%) происходит по организационным причи-
причинам. Следовательно, обеспечение должного порядка на произ-
производстве, повышение производственной дисциплины и культуры
труда приводят к резкому уменьшению аварий и несчастных
случаев. Это положение подтверждается практикой работы луч-
лучших предприятий.
Борьба с профессиональными отравлениями и заболеваниями
на заводах медицинской промышленности проводится в следую-
следующих основных направлениях:
1) тщательное соблюдение технологического режима, преду-
предусмотренного производственным регламентом и производственны-
производственными инструкциями, а также соблюдение соответствующих инст-
инструкций по технике безопасности и промышленной санитарии;
2) рационализация технологических процессов и аппаратур-
аппаратурного оформления, приводящая к снижению выделения в воздух
производственных помещений вредных газов, паров, пыли и
устранению непосредственного контакта работающих с вредны-
вредными веществами;
3) герметизация аппаратуры и коммуникаций, обеспечение
эффективной работы приточной и вытяжной вентиляции в цехах
и складах;
20
4) правильная организация складского хозяйства; механи-
механизация загрузки и выгрузки химического сырья; правильная
транспортировка продуктов;
5) замена высокотоксичных веществ менее токсичными; сни-
снижение концентрации токсичных веществ в воздухе за счет изме-
изменения их агрегатного состояния или применения специальных
выпускных форм (гранул, чешуек, паст и т. п.);
6) правильная организация отбора рабочих для производств,
где ведутся работы с токсичными веществами, с учетом состоя-
состояния здоровья и индивидуальной склонности к дерматитам, ал-
аллергическим заболеваниям и т. п.;
7) организация разъяснительной и просветительной работы
с целью глубокого усвоения правил техники безопасности и под-
поднятия культуры труда.
§ 3. СТАНДАРТЫ И ТЕХНИЧЕСКИЕ УСЛОВИЯ
В СССР все изделия народного хозяйства должны отвечать
определенным установленным для них требованиям. Эти требо-
требования изложены в специальных документах, которые в зависи-
зависимости от вида продукции и ее назначения называются стандар-
стандартами или техническими условиями.
Стандарты могут быть общесоюзными (ОСТ) и государст-
государственными общесоюзными (ГОСТ). Они составляются для про-
продуктов, изготовляемых в одной отрасли и потребляемых в других
отраслях народного хозяйства.
Как уже указывалось, все выпускаемые в СССР химико-фар-
химико-фармацевтические препараты должны отвечать требованиям Госу-
Государственной Фармакопеи СССР, которая является Государствен-
Государственным стандартом.
Для продуктов, выпускаемых и потребляемых в пределах
одной отрасли промышленности, обычно составляют технические
условия (ТУ).
Каждый стандарт или технические условия имеют определен-
определенный номер; год выпуска стандарта или ТУ указывается двумя
последними цифрами в номере. В связи с постоянным повыше-
повышением требований к качеству продукции стандарты и технические
условия периодически пересматривают с целью установления
более высоких технических требований.
Стандарты и технические условия на продукты химической
промышленности обычно включают название и формулу продук-
продукта, технические требования к его качеству, методы испытания,
способы упаковки и маркировку. Качество продукта обычно
характеризуется внешним видом, определенной концентрацией
основного вещества и примесей и физическими свойствами (тем-
(температурой кипения, плавления, плотностью, растворимостью
и т. п.).
21
На таре, в которой упакован продукт (бочка, ящик, мешок
и т. п.), имеются специальные обозначения, которые называются
маркировкой. В маркировке указывается название продук-
продукта и завода-изготовителя, номер стандарта, вид и сорт продукта,
его вес. Применение сырья с неясной маркировкой категорически
запрещено, так как ошибка в этом случае может привести не
только к порче продукта, но и к серьезной аварии или травме.
При выпуске опытных партий новых лекарственных препара-
препаратов в качестве временного стандарта используют временную
фармакопейную статью.
Глава 2
ОСНОВНЫЕ ХИМИЧЕСКИЕ РЕАКЦИИ, ЛЕЖАЩИЕ
В ОСНОВЕ СИНТЕЗА ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
§ 1. ОБЩАЯ ХАРАКТЕРИСТИКА ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Как уже отмечалось (см. с. 10, 13), химическая технология ле-
лекарственных веществ является совокупностью большого числа
химических и физико-химических процессов, отличающихся друг
от друга как по существу осуществляемых технологических про-
процессов, так и по аппаратурному оформлению и масштабам.
Практически нет ни одного приема химической техники, ни од-
одного аппарата, которые не применялись бы в химико-фармацев-
химико-фармацевтической промышленности. Тем не менее химическая технология
лекарственных веществ представляет собой единое целое,
объединенное общностью сырья, общностью методов переработ-
переработки этого сырья и, главное, общностью целей производства.
Очевидно, что специалист в химико-фармацевтической про-
промышленности должен обладать глубокими знаниями, особенно
в области органической химии и аппаратурного оформления про-
процессов тонкого органического синтеза.
Быстрое обновление номенклатуры лекарственных средств,
сравнительно небольшой объем и высокий материальный индекс
производства делают совершенно необходимым для каждого ра-
работника химико-фармацевтической промышленности знание хи-
химических и физических свойств перерабатываемых веществ
и физико-химических особенностей процесса, так как только при
этом условии возможно постоянное совершенствование сущест-
существующих и освоение новых производств.
В части знания химических и физических свойств перераба-
перерабатываемых веществ для химика-технолога наибольшее значение
имеет понимание связи реакционной способности и строения
перерабатываемых веществ, что позволяет ему предвидеть воз-
возможные направления реакции, а также решать вопрос о замене
одного вида сырья другим.
Знание физико-химических особенностей процесса позволяет
выбрать оптимальные условия для его проведения, обеспечиваю-
обеспечивающие максимальный выход и наилучшее качество готового про-
продукта. В этой части особое значение имеет знание механизма,
23
по которому осуществляется химическое превращение, и законов
изменения скорости реакции, что в конечном итоге определяет
степень превращения сырья в целевой продукт.
Под механизмом реакции понимают определенную последо-
последовательность элементарных стадий (превращений), через кото-
которые должны пройти исходные вещества, чтобы превратиться
в конечные продукты. Очевидно, что каждая «элементарная»
стадия является более простой реакцией, чем суммарная, а сум-
сумма эффектов всех этих стадий дает полный, или суммарный, эф-
эффект процесса. В некоторых случаях оказывается невозможным
определить химизм каждой элементарной стадии. Однако зна-
знание только части «элементарных» стадий может существенно
облегчить понимание основных свойств процесса в целом.
Глубина превращения характеризует степень превращения
исходных веществ в продукты реакции. Применительно к слож-
сложным реакциям следует различать глубину превращения, выход
и селективность. Для расчетов следует пользоваться мольными
концентрациями. Приведенные ниже зависимости справедливы
для реакций, протекающих без изменения объема реакционной
массы. В противном случае вместо концентраций следует брать
соответствующие количества (моль) веществ. Глубина превра-
превращения, как уже было сказано, характеризует степень превраще-
превращения сырья в продукты реакции. Она может быть рассчитана как
отношение изменения количества сырья в реакционной массе
к начальному количеству сырья:
t Со — Ск
* =—с~—»
где Со и Ск — начальная и конечная молярная концентрация
исходного вещества.
В отличие от глубины превращения выход есть отношение
количества полученного целевого продукта (СПр) к исходному
количеству сырья:
4=-%*..
Если процесс протекает с образованием ряда побочных про-
продуктов, то для характеристики такого процесса удобно пользо-
пользоваться понятием селективность. Селективность есть отношение
количества полученного целевого продукта к количеству превра-
превращенного сырья:
Ф Спр выход целевого продукта в %
Со — Ск ~ глубина превращения в %
Очевидно, что выход равен произведению селективности и
глубины превращения:
■Ч=ФЕ.
Общую схему синтеза лекарственных веществ принято под-
подразделять на два этапа — синтез промежуточных продуктов
24
и синтез лекарственного вещества из промежуточных продуктов.
Многие исходные вещества и промежуточные продукты имеют
ароматический характер.
§ 2. ОБЩИЕ СВЕДЕНИЯ О СТРОЕНИИ ПРОМЕЖУТОЧНЫХ
ПРОДУКТОВ. ПОНЯТИЕ ОБ АРОМАТИЧНОСТИ
В основе очень многих ароматических соединений лежит бен-
бензол. Важнейшей отличительной особенностью ароматических со-
соединений является высокая химическая устойчивость, сохраняю-
сохраняющаяся даже в условиях пиролиза.
В отличие от ненасыщенных со-
соединений для ароматических уг-
углеводородов свойственны реак-
реакции замещения, но не присоеди-
присоединения. Фенолы значительно отли-
отличаются от спиртов, обладая явно
выраженными кислотными свой-
свойствами.
В ароматических галогенпро-
изводных галоген в ядре значи-
значительно менее подвижен, чем в
алифатических соединениях и
т. д.
Циклическая формула (I)
бензола была впервые предложе-
предложена Кекуле в 1865 г.
Формула (I) допускала воз-
возможность у дизамещенных суще-
существования изомеров типа 1а и 16, что не соответствовало дейст-
действительности.
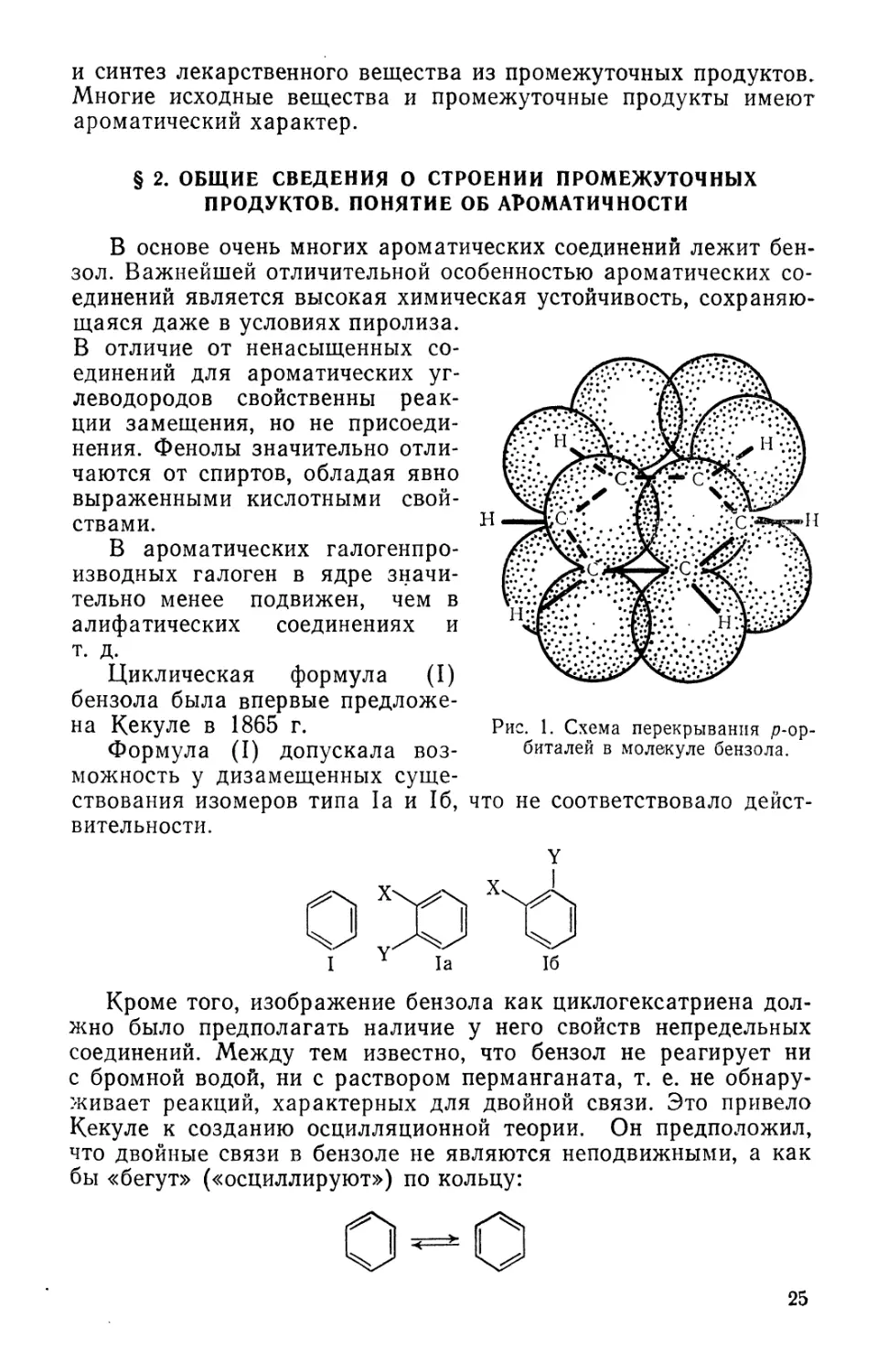
Рис. 1. Схема перекрывания р-ор-
биталей в молекуле бензола.
1а
16
Кроме того, изображение бензола как циклогексатриена дол-
должно было предполагать наличие у него свойств непредельных
соединений. Между тем известно, что бензол не реагирует ни
с бромной водой, ни с раствором перманганата, т. е. не обнару-
обнаруживает реакций, характерных для двойной связи. Это привело
Кекуле к созданию осцилляционной теории. Он предположил,
что двойные связи в бензоле не являются неподвижными, а как
бы «бегут» («осциллируют») по кольцу:
25
Эта гипотеза согласовывалась с результатами озонирования
бензола и его производных. (Эти реакции рассматривались в
курсе органической химии).
Исследование легкости гидрирования изолированной двойной
связи (циклогексен), двойной связи в сопряженной системе (цик-
логексадиен) и двойной связи в молекуле бензола позволило
сделать вывод о том, что разрушение замкнутой системы чере-
чередующихся двойных и простых связей в молекуле бензола при-
приводит к потере им его особых ароматических свойств.
Следовательно, со-
состояние я-электронов в
бензольном ядре являет-
является состоянием особой
устойчивости, обуслов-
обусловленным специальным со-
сопряжением связей этого
ядра. Мерой этой устой-
устойчивости является энергия
сопряжения (или энергия
стабилизации) бензола,
которую можно рассмат-
рассматривать- как разность внут-
внутренних энергий бензола
и гипотетического (т. е. в
действительности не су-
Рис. 2. Схематическое изображение молеку-
молекулы бензола.
ществующего) циклогек-
сатриена. Для бензола
эта величина равна 36
ккал/моль.
Особая устойчивость и энергетическая выгодность существо-
существования ароматических соединений объясняется наличием у них
циклического зт-электронного облака, образующегося в резуль-
результате перекрывания орбиталей р-электронов всех атомов кольца
(рис. 1). Такое представление о строении бензола объясняет рав-
равноценность всех шести углеродных атомов и всех углерод-угле-
углерод-углеродных связей — их длина 0,139 нм A,39 А) и наличие только
трех изомеров среди дизамещенных бензола (рис. 2).
Для образования общего для всей молекулы зт-электронного
облака недостаточно одного чередования простых и двойных
связей в молекуле циклического ненасыщенного соединения.
Необходимо, чтобы молекула была плоской, т. е. все атомы угле-
углерода, составляющие цикл, лежали в одной плоскости. Так, на-
например, молекула циклоактатетраена (С8Н8) не является пло-
плоской (II), а имеет форму «ванны» (III).
26
Это приводит к тому, что орбитали р-электронов соседних ато-
атомов не перекрываются и единое для всей молекулы я-электрон-
ное облако, не может образоваться. Циклооктатетраен является
типичным непредельным соединением — длины связей в молеку-
молекуле поочередно равны 0,154 и 0,133 нм A,54 и 1,33 А), т. е. они
являются обычными простыми и двойными связями.
Немецкий ученый Хюккель показал, что ароматическими мо-
могут быть лишь такие плоские циклические ненасыщенные соеди-
соединения, в молекуле которых имеется 4п+2 я-электронов, где п
натуральный ряд, т. е. при п = 1, 2, 3... число я-электронов равно
2, 6, 10, 14.... Это значит, что могут существовать ароматические
соединения, молекулы которых имеют единое я-электронное
облако, образованное двумя, шестью, десятью, четырнадцатью
и т. д. электронами.
Примерами могут служить:
.Цикле-
Л рвпенилкатмн
Циклооктадеканонаен
Таким образом, ароматическими являются такие ненасыщен-
ненасыщенные циклические соединения, у которых все атомы цикла прини-
принимают участие в образовании единой сопряженной системы,
а я-электроны этой системы образуют замкнутую электронную
оболочку.
В технологии лекарственных соединений большое значение
имеют ароматические гетероциклические соединения (см. гла-
главу 11).
Для изображения строения ароматических соединений поль-
пользуются несколькими способами. Обычно при написании уравне-
уравнений реакций ароматическое кольцо того или иного соединения
изображают в виде многоугольника с чередующимися простыми
и двойными связями. Если хотят подчеркнуть выравненность
связей в ароматическом соединении, то пользуются символами:
или
27
Следует, однако, помнить, что ни одна формула не может отра-
отразить все многообразие свойств реальной молекулы, так же как
нельзя объяснить устройство сложной детали, дав ее изображе-
изображение лишь в одной проекции. Поэтому в конкретных случаях при-
прибегают к такому изображению строения химического соедине-
соединения, которое подчеркивает специфичность поведения его в рас-
рассматриваемом случае.
§ 3. ТРИ ОСНОВНЫХ ТИПА РЕАКЦИЙ
В ТЕХНОЛОГИИ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ
Так как химико-фармацевтическая промышленность произ-
производит очень большое число промежуточных продуктов, химико-
фармацевтических препаратов, витаминов и антибиотиков, то
изучение технологии каждого из этих продуктов в отдельности
очень затруднительно. Поэтому при изучении технологии проме-
промежуточных продуктов их целесообразно классифицировать не по
составу и строению, а по химическим реакциям, используемым
для их получения.
Наиболее важными реакциями в промышленном синтезе ор-
органических полупродуктов и лекарственных веществ являются
следующие: 1) сульфирование и сульфохлорирование; 2) нитро-
нитрование; 3) галогенирование; 4) восстановление и окисление;
5) оксилирование и аминирование; 6) диазотирование и нитро-
зирование; 7) алкилирование и ацилирование аминов, спиртов
и фенолов; 8) алкилирование, алкоксилирование и ацилирование
атома углерода, в частности, в ядре; 9) образование циклов,
в частности, гетероциклов.
Все методы химической переработки сырья, используемые
в производстве промежуточных продуктов, можно разбить на
три основные группы.
К первой группе следует отнести реакции замещения атомов
водорода в ядре ароматических Сбедшений теми или иными груп-
группами атомов, называемыми заместителями. В эту группу входят
реакции 1—3. Введение заместителей в молекулу органического
соединения может преследовать две цели: а) придание промежу-
промежуточному продукту каких-то новых специфических свойств, сохра-
сохраняющихся и необходимых в конечном химико-фармацевтическом
препарате, или б) временное придание промежуточному продук-
продукту свойств, необходимых для его дальнейшего превраще-
превращения в другой промежуточный продукт или лекарственное
вещество.
Ко второй группе относят реакции превращения уже имею-
имеющихся в молекуле органического соединения заместителей
(групп) в другие с целью придания ему новых свойств или изме-
изменения его реакционной способности (реакции 4—6).
К третьей группе относятся реакции, сопровождающиеся из-
изменением углеродного скелета молекулы (реакции 9).
28
Некоторые реакции относятся одновременно к двум группам.
Реакции, принадлежащие к различным группам, тесно связаны
между сабой. Таж, аминогруппа (реакции 4) ;во млогах случаях
может быть тпюлучена только через нитросоединение (реакция 2),
а оксигруппа (реакции 5) — через сульфопроизводное (реак-
(реакции 1).
§ 4. НУКЛЕОФИЛЬНОЕ,
ЭЛЕКТРОФИЛЬНОЕ И РАДИКАЛЬНОЕ ЗАМЕЩЕНИЕ
Практически любая элементарная реакция сводится к элек-
электронному взаимодействию двух реагирующих частиц. При этом
одна из частиц может обладать либо избытком, либо недостат-
недостатком электронов и в соответствии с этим притягиваться к центрам
с противоположным «частичным зарядом». Существование таких
электроноизбыточных и электрононедостаточных центров в мо-
молекуле обусловлено взаимным влиянием атомов и способностью
химической связи к поляризации. Идея о взаимном влиянии
атомов в молекуле была высказана и обоснована великим рус-
русским химиком А. М. Бутлеровым.
Такое рассмотрение химических реакций лежит в основе раз-
разделения химических веществ на нуклеофильные и электро-
фильные.
К числу нуклеофильных можно отнести все молекулы
и анионы, склонные к отдаче электронов. К таким веществам
относятся многие соединения с атомами /jsjv , —S—, —О—,
а также ароматические и этиленовые углеводороды, содержащие
подвижные свободные электронные пары и я-электроны:
А
N-R, R-S-R, R-O—R, R-O-H и т. д.
Сюда же относятся отрицательно заряженные ионы:
ОН-, RO-, CN-, С1-, RCOO- и т. д.
К электрофильным относятся положительно заряженные
ионы (NO2, SO3H+, NO+, Вг+ и пр.) и поляризованные молеку-
молекулы, у которых более активен центр, бедный электронами, на-
например,
i 8+
— с — с —
Разделение химических веществ на электрофильные и нуклео-
нуклеофильные привело к классификации реакций на нуклеофильные
и электрофильные. Эта классификация оказалась особенно
29
удобной для реакций замещения. Схематически реакции элек-
трофильного и нуклеофильного замещения атома водорода могут
быть изображены следующим образом:
RH + X+ >■ RX + H+ (здесь Х+— электрофильный реагент);
RH + Y" >■ RY + H" (здесь Y~— нуклеофильный реагент).
Примерами реакций электрофильного замещения могут слу-
служить сульфирование, нитрование, алкилирование и галогениро-
вание ароматических соединений:
АгН + SO3H+ >- ArSO3H + Н+;
АгН + NOJ >■ ArNO2 + Н+;
АгН + С1+ >- АгС1 + Н+ит, д.
Примерами реакций нуклеофильного замещения являются
реакции замены сульфогруппы, диазогруппы, галогена в арома-
ароматическом ядре и некоторые другие:
АгН + NaNH2 ^ArNHa + NaH;
ArSO3H + OH- >• ArOH + SO3H-;
ArCl + OH- > АгОН + Аг- и т. д.
При рассмотрении реакций ароматического замещения воз-
возникают три основных вопроса: а) что представляет собой ата-
атакующая частица; б) как протекает замещение; в) как влияют
на реакцию другие группы (заместители), уже имеющиеся в ис-
исходном соединении.
Протеканию химической реакции замещения предшествует
образование электрофильной или нуклеофильной атакующей ча-
частицы:
2H2SO4 ч=£ HSO3+ + HSOr + Н2О
HNO3 + H2SO4 т—»- NOt + Н3О
+
CH3CI + AICI3 *=* СН£ + А1С1Г
NaOH 7—^Na+ + OH~
NaNH2 ^=fc NHa" + Na+ и т. д.
В большинстве случаев в ароматическом ряду замещение
проходит через стадию образования промежуточного комплекса
+ Y —
30
Промежуточный (переходный) комплекс обладает большей
энергией, чем исходная молекула и конечный продукт, а пото-
потому является неустойчивым. Трудность обнаружения этого комп-
комплекса в реакционной массе связана не только с его неустойчи-
неустойчивостью, но и с соотношением скоростей первой и второй ступеней
реакций. Например, при нитровании скорость второй ступени
всегда много больше скорости первой ступени, поэтому концен-
концентрация промежуточной частицы чрезвычайно мала и обнаружить
ее очень трудно.
N0* ^
z медленно
Общая скорость химического процесса всегда определяется
скоростью протекания наиболее медленной его стадии.
Реакции замещения могут протекать не только по ионному
(гетеролитическому) механизму, который был изложен выше,
но и по радикальному механизму. При радикальном, или гомо-
литическом, разрыве связи каждая из образовавшихся частиц
получает по одному электрону. Гомолитическая, или радикаль-
радикальная, диссоциация молекулы всегда приводит к нейтральным ча-
частицам — радикалам, содержащим один неспаренный электрон.
Образовавшиеся радикалы могут атаковать молекулы, образуя
новые молекулы и новые радикалы. Реакция радикального за-
замещения водорода может быть изображена следующим образом:
RH + Z- >- RZ + H*
Типичным примером радикального замещения является гало-
геннрование боковой цепи ароматических углеводородов:
С1-С1 >■ СГ+С1';
С6НбСН3 + СГ * ССН5СН2-+НС1;
СбН5СН2' +С12 > СбН5СН2С1 + СГ.
Радикальные реакции имеют ряд особенностей, которые
позволяют легко распознать их. Они ускоряются при облучении
ультрафиолетовыми лучами, так как последние часто вызывают
гемолитический распад молекул. Катализаторами радикальных
реакций могут служить перекиси и другие вещества, распадаю-
распадающиеся с образованием радикалов. Любое соединение, способное
реагировать с активными свободными радикалами с образова-
образованием неактивных веществ, будет служить ингибитором радикаль-
радикальной реакции, т. е. замедлять ее.
Радикальные реакции являются обычно цепными, так как
каждая атака радикалом свободной молекулы приводит к обра-
образованию нового радикала. И, наконец, радикальные реакции ча-
31
сто являются автокаталитическими и имеют индукционный пе-
период.
В противоположность радикальным гетеролитические (элек-
трофильные и нуклеофильные) реакции не подвержены влиянию
света и свободных радикалов, они не являются цепными и не
имеют индукционного периода. Эти реакции часто катализиру-
катализируются кислотами и основаниями. Большое влияние на протекание
таких реакций оказывает полярность среды (характер раствори-
растворителя) .
До недавнего времени пе?рвый вопрос, который ставили перед
собой химики при изучении механизма реакций, состоял в том,
каким образом разрывается химическая связь в реакционном
центре молекулы. При этом предполагалось, что возможны лишь
два варианта диссоциации связи — ионный (гетеролитический)
и радикальный (гомолитический). Однако в последние годы со-
советскими химиками была подробно изучена третья группа ре-
реакций, которые получили название реакций с одноэлектрон-
ным переносом.
Электроны более подвижны, чем атомы или группы атомов.
Поэтому перестройке реагирующих молекул может предшество-
предшествовать перенос электронов от одной молекулы к другой и лишь
затем, как следствие этого обмена, — перемещение значительно
менее подвижных ядер и частей молекул.
Допустим, что при столкновении двух одинаковых молекул
АВ произошел перенос одного электрона с одной молекулы на
другую. В результате должны возникнуть две новые частицы
анион-радикал (АВ)~ и катион-радикал (АВ)#+. Эти частицы
обладают одновременно свойствами как ионов, так и радикалов,
так как они несут электрический заряд и обладают неспаренным
электроном. Так, например, бензол в определенных условиях
«снимает» электрон со щелочного металла, превращаясь в ани-
анион-радикал бензола:
C6H6 + Na +=+ CeH5-Na+
Как правило, ион-радикалы очень активны и быстро всту-
вступают в дальнейшие химические превращения. Распад ион-ра-
ион-радикала приводит к одновременному возникновению иона и ра-
радикала:
(А—В)"- >- А- + В' (или А* +В-);
(А—В)* + > А+ + В' (или А' + В+).
Таким образом, существуют реакции, которые не являются
ни чисто ионными, ни чисто радикальными, так как в одном ак-
акте могут образовываться одновременно и радикалы и ионы.
Схематически это можно изобразить так:
A-5-B + X-i-Y > A++B'+X-
(Стрелка показывает перенос электрона.)
32
Ион-радикал может вступать в дальнейшие превращения, не
распадаясь предварительно на ион и радикал. Так, например,
проходит реакция кетонов с магнийорганическими соедине-
соединениями:
к-
R.TMnlfal
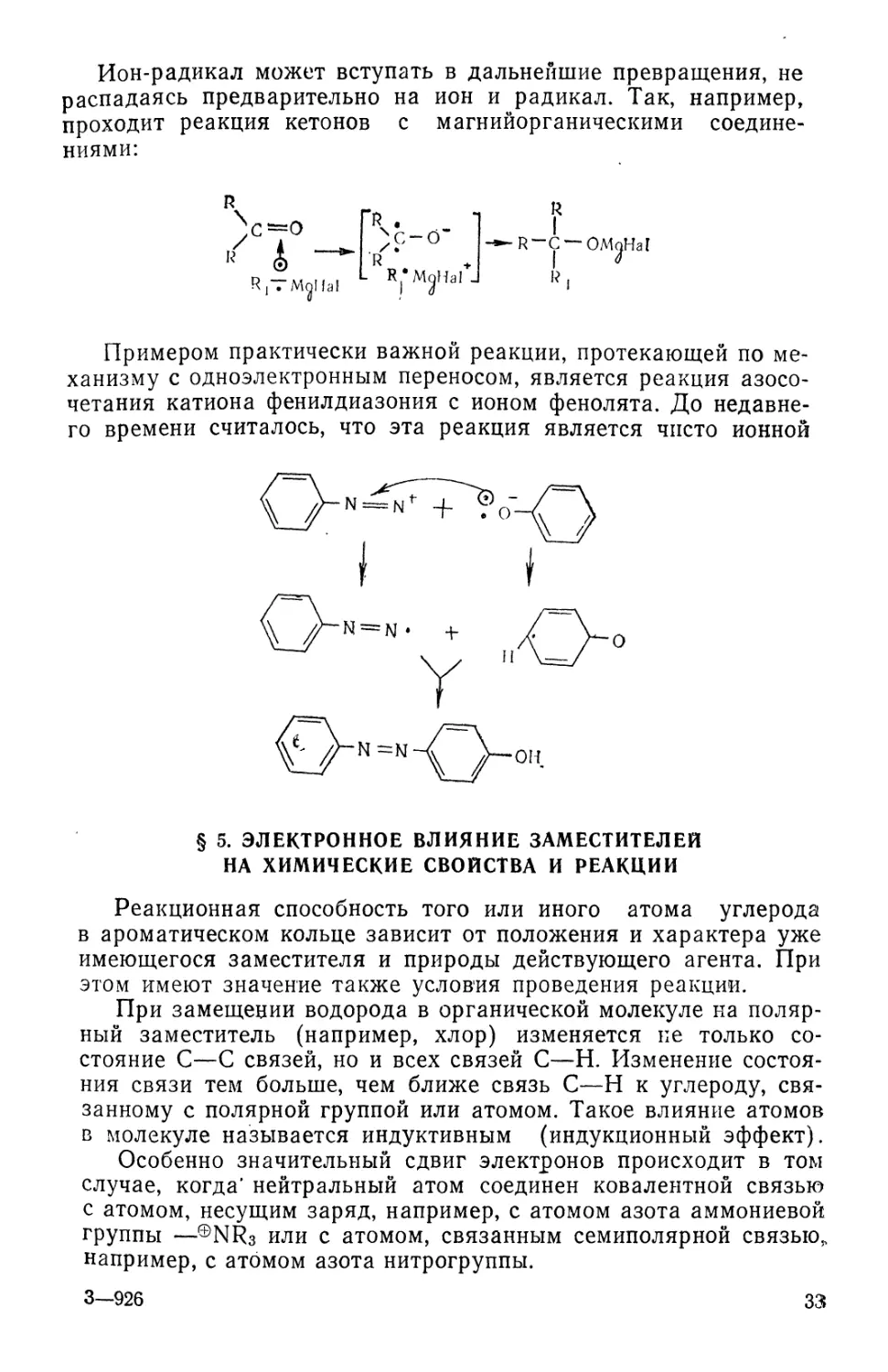
Примером практически важной реакции, протекающей по ме-
механизму с одноэлектронным переносом, является реакция азосо-
четания катиона фенилдиазония с ионом фенолята. До недавне-
недавнего времени считалось, что эта реакция является чисто ионной
он
§ 5. ЭЛЕКТРОННОЕ ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ
НА ХИМИЧЕСКИЕ СВОЙСТВА И РЕАКЦИИ
Реакционная способность того или иного атома углерода
в ароматическом кольце зависит от положения и характера уже
имеющегося заместителя и природы действующего агента. При
этом имеют значение также условия проведения реакции.
При замещении водорода в органической молекуле на поляр-
полярный заместитель (например, хлор) изменяется не только со-
состояние С—С связей, но и всех связей С—Н. Изменение состоя-
состояния связи тем больше, чем ближе связь С—Н к углероду, свя-
связанному с полярной группой или атомом. Такое влияние атомов
в молекуле называется индуктивным (индукционный эффект).
Особенно значительный сдвиг электронов происходит в том
случае, когда' нейтральный атом соединен ковалентной связью
с атомом, несущим заряд, например, с атомом азота аммониевой
группы —®NR3 или с атомом, связанным семиполярнои связью,,
например, с атомом азота нитрогруппы.
3—926 33
Индукционный эффект является основным электронным эф-
эффектом для насыщенных соединений.
Для того чтобы оценить индуктивное влияние различных за-
заместителей, можно сравнить константы диссоциации замещен-
замещенных уксусных кислот. Чем сильнее притягивает заместитель
электроны, тем больше константа диссоциации. Ниже приведены
константы диссоциации замещенных уксусных кислот при 25 °С.
Ка-Ю5
СН3СООН 1,82
С6Н5СН2СООН 5,03
СН3ОСН2СООН 33,5
I—СН2СООН . . .75
Вг-СН2СООН 138
С1-СН2СООН 155
F—СН2СООН 217
N=C—СН2СООН ... 356
С12СНСООН 5 140
С13С-СООН . . . . 121 000.
Индукционный эффект быстро затухает с удалением замести-
заместителя от реакционного центра. Это положение подтверждается
сравнением констант диссоциации масляной кислоты и трех изо-
изомерных хлормасляных кислот:
Ка-Ю4
Масляная кислота 0,15
а-Хлормасляная кислота . . . .14,0
Р-Хлормасляная » .... 0,89
у-Хлормасляная » .... 0,26.
В связи с наличием у атомов хлора, брома, йода и фтора
большого электроноакцепторного индукционного эффекта гало-
генозамещенные (На!) ароматические соединения проявляют
меньшую активность в реакциях электрофильного замещения по
сравнению с незамещенными углеводородами.
Важнейшим свойством я-связи является ее высокая подвиж-
подвижность. я-Связь поляризуется значительно легче и сильнее, чем
а-связь. Молекулы с чередующимися простыми и двойными
связями обладают рядом специфических особенностей. Анало-
Аналогичными особенностями отличаются соединения, в которых ка-
какой-либо атом, имеющий неподеленную пару электронов, нахо-
находится рядом с углеродным атомом, стоящим у двойной связи.
Эта электронная пара принимает участие в общей цепи сопря-
сопряжения (эффект сопряжения), вследствие чего происходит изме-
изменение распределения электронной плотности в молекуле.
Таким взаимодействием неподеленных пар электронов атома
кислорода с электронами бензольного ядра (эффектом сопряже-
34
ния) объясняется большая кислотность фенолов по сравнению
со спиртами и увеличение этой кислотности при наличии в ядре
электрофильных заместителей.
Эффект* сопряжения передается в основном в орто- и пара-
положения ароматического ядра, что иллюстрируется схемой:
Поскольку заместители могут обладать как индукционным
эффектом, так и эффектом сопряжения, при определении реак-
реакционной способности соединения следует учитывать сумму
электронных взаимодействий. Так, индуктивное влияние таких
заместителей первого рода, как оксигруппа, аминогруппа и атом
хлора, приводит к появлению на атомах углерода ароматическо-
ароматического кольца положительных зарядов. Однако участие неподелен-
ных пар электронов кислорода, азота и хлора в сопряжении
приводит к обогащению орто- и пара-положений ароматического
ядра электронами. При этом у окси- и аминогруппы эффект со-
сопряжения больше индуктивного эффекта и поэтому атомы угле-
углерода в орто- и пара-положениях к заместителю имеют некото-
некоторый отрицательный заряд, тогда как атомы углерода в мета-
положении заряжены положительно.
Другое распределение зарядов наблюдается в том случае,
когда заместителем является атом галогена (например, хлора).
В этом случае индукционный эффект значительно больше эф-
эффекта сопряжения и все ядро имеет некоторый положительный
заряд. Благодаря эффекту сопряжения этот заряд в орто- и
пара-положениях меньше, чем в мета-положении к заместителю.
Соли ароматических аминов обладают совершенно иной ре-
реакционной способностью, нежели свободные амины, так как в
катионе фениламмония все атомы углерода заряжены положи-
положительно и при том особенно сильно в орто- и пара-положениях.
3* 35
Аналогичная картина наблюдается при наличии в ядре других
заместителей второго рода, например, нитрогруппы.
Для того чтобы эффект сопряжения передавался в полной
мере, молекула соединения должна быть плоской (копланар-
ной). Нарушение копланарности молекулы приводит к отсутст-
отсутствию перекрывания облаков я-электронов и невозможности пере-
передачи электронных влияний. Из-
Известна, например, высокая реак-
реакционная способность пара-поло-
пара-положения N-диметиланилина по от-
отношению к электрофшгьным ре-
реагентам. Если, однако, в О-по-
ложения к диметиламиновой
группе ввести 2 метильные груп-
группы, то такой N-диметилксилидин
уже практически не реагирует с
азотистой кислотой. Причина за-
заключается в том, что в молекуле
диметилксилидина метильные
группы, расположенные в орто-
положениях, заставляют диме-
тиламинную группу повернуться
и тем самым выключают ее из
сопряжения с ядром (рис. 3).
Принято разделять все заместители в ароматическом кольце
на две группы: заместители первого рода (электронодонорные)
и заместители второго рода (электроноакцепторные).
К заместителям первого рода относятся следующие группи-
группировки атомов: —ОН, —OR, —OCOR, —SH, —SR, —NH2,
—NHR, —NR2, —NHCOR, —N=R—, —CH3, —CH2R, —CR3,
-F, -Cl, -Br, -I.
К заместителям второго рода относятся такие группы, как
—SO3H, —NO2, —СНО, —СООН, —COOR, —COR, —CN,
—СС13, —NHa, —NRs и т. п.
Заместители первого рода облегчают реакции ароматических
соединений с электрофильными реагентами и ориентируют при
этом новый заместитель в орто- и пара-положения. Заместители
второго рода затрудняют реакции ароматических соединений
электрофильными реагентами, и ориентируют в реакциях заме-
Рис. 3. Схематическое изобра-
изображение молекулы диметилксили-
диметилксилидина.
щения водорода новый заместитель в мета-положение. Реакции
замещения с нуклеофильными реагентами эти заместители об-
облегчают, направляя их в о- и п-положения.
Сформулированные правила отражают лишь главное направ-
направление реакции. Практически почти во всех случаях образуется
смесь изомеров, в которой, однако, преобладают те продукты,
которые образуются в соответствии с правилами ориентации.
Ориентация вступающего в ядро заместителя обусловлена
распределением электронной плотности в ароматическом ядре.
Как реакционная способность, так и ориентация зависят от
относительных скоростей реакций. Если говорят, что метильная
группа активирует кольцо, то это означает, что наличие метиль-
ной группы заставляет кольцо реагировать быстрее, чем реаги-
реагирует бензол. Эта группа вызывает орто- и пара-ориентацию при
электрофильном замещении атома водорода в ядре, т. е. в ее
присутствии замещение в орто- и пара-положения происходит
значительно быстрее, чем в мета-положение.
Сформулированные правила ориентации пригодны только
в случае гетеролитических замещений. Что касается замещений
в ароматическом ядре по гомолитическому (радикальному) ме-
механизму, то они протекают по иным законам.
Глава 3
СУЛЬФИРОВАНИЕ И СУЛЬФОХЛОРИРОВАНИЕ
§ 1. ОБЩИЕ СВЕДЕНИЯ
Сульфированием называют процесс замещения атома водо-
водорода в органическом соединении сульфогруппой —SO3H, осу-
осуществляемый путем обработки веществ различными сульфирую-
сульфирующими агентами.
Исходными продуктами при сульфировании служат главным
образом ароматические углеводороды, их амино- и окси-произ-
водные, а в ряде случаев и сами лекарственные вещества.
В качестве сульфирующих агентов применяются концентри-
концентрированная серная кислота, олеум, хлорсульфоновая кислота
(HSO3C1).
Сульфирование является одним из наиболее распространен-
распространенных процессов в тонком органическом синтезе. Введение сульфо-
группы в молекулу органического соединения может преследо-
преследовать различные цели. Сульфогруппа может вводиться с целью
либо придания соединению большей растворимости, либо даль-
дальнейшей замены сульфогруппы на гидроксил, аминогруппу и т. п.
Число водородных атомов, замещаемых сульфогруппой, мо-
может быть различно. Соединения с одной сульфогруппой в моле-
молекуле называются моносульфокислотами, с двумя — дисульфокис-
лотами, с тремя — трисульфокислотами, с четырьмя — тетра-
сульфокислотами.
Сульфокислоты — это, как правило, твердые кристаллические
вещества, хорошо растворимые в воде и обладающие ярко выра-
выраженным кислотным характером. По силе ароматические сульфо-
сульфокислоты близки к минеральным кислотам. В разбавленных вод-
водных растворах они практически полностью диссоциированы.
Тепловой эффект реакции нейтрализации ароматических суль-
фокислот такой же, как и тепловой эффект нейтрализации сер-
серной кислоты, и составляет 13,9 ккал/г-экв E8,1 кДж/г-экв).
RSO3H+NaO№-^RSO3Na+H2O+58,l кДж/г-экв.
Как правило, в результате сульфирования получается не
одна сульфокислота, а смесь сульфокислот, отличающихся
между собой количеством сульфогрупп.
Реакции сульфирования углеводородов серной кислотой или:
олеумом» можно представить в виде следующей схемы:
RHX + xH2SO4 -<—>• R(SO3H)X + хН20
38
или
RHX + xSO3 ^ R(SO3H)X,
где RHX — углерод, х — число атомов водорода, замещаемых
сульфогруппами, R(SOsH)i — продукт сульфирования (сульфо-
кислота).
Введение сульфогруппы в бензольное или нафталиновое яд-
ядро затрудняет их дальнейшее сульфирование. Моносульфокис-
лоты сульфируются труднее исходных углеводородов, дисульфо-
кислоты — труднее моносульфокислот. Поэтому при тщательном
подборе условий сульфирования (температуры, длительности
процесса, концентрации сульфирующего агента) удается полу-
получать сульфомассы с преимущественным содержанием желаемых
сульфокислот.
Сульфокислоты являются промежуточными продуктами в
синтезе ряда химико-фармацевтических препаратов (диазолин,
амидопирин и др.). В ряде случаев сульфогруппа входит в со-
состав лекарственного препарата, придавая ему большую раство-
растворимость (сульфобутадион, гемитиамин, анальгин). Большое
внимание привлекли к себе сульфопроизводные в связи с тем,
что сульфогруппы были обнаружены в составе природных био-
биологически активных веществ, в частности, в природном анти-
антикоагулянте крови гепарине. Установлено, что многие поверхност-
поверхностно-активные вещества (ПАВ), содержащие сульфогруппу
(в частности, производные сульфоянтарной кислоты), обладают
сильным антимикробным действием. На основе таких ПАВ гото-
готовят лекарственные шампуни для лечения себореи и других кож-
кожных заболеваний. Некоторые сульфо-ПАВ способны усиливать
действие антибиотиков и сульфаниламидных препаратов.
Особое значение в синтезе химико-фармацевтических препа-
препаратов имеет реакция сульфохлорирования, приводящая к обра-
образованию сульфохлоридов RSO2CI, являющихся важнейшими про-
промежуточными продуктами в производстве многочисленных суль-
сульфаниламидных препаратов.
§ 2. МЕХАНИЗМ РЕАКЦИИ СУЛЬФИРОВАНИЯ
'Сульфирование ароматических соединений является реакцией
электрофильного замещения и протекает через стадию образова-
образования промежуточного а-комплекса:
Существенным отличием этой реакции от других реакций
электрофильного замещения является ее обратимость.
39
Установлено, что истинными сульфирующими агентами (ата-
(атакующими частицами) являются серный ангидрид SO3 н сульфу-
рилий-катион П5Оз, образующиеся при диссоциации концентри-
концентрированной серной кислоты по уравнениям:
2H2SO4 z$=±. SO3 + Н3О+ + KSOJ
2H2SO4 <=*■ HaSOt + HSO4"
3H2SO4 +=± HSO3 + H3O+ + 2HSO7.
Понятно поэтому, что разбавленная серная кислота не может
сульфировать органические соединения, так как в присутствии
воды она диссоциирует по кислотному механизму:
H2SO4 + Н2О :*=£ Н3О+ + HSO7.
Так как при сульфировании серной кислотой выделяется вода
RH + H2SO4 ^=±: RSO3H + Н2О,
то способность серной кислоты диссоциировать с образованием
сульфирующих агентов (SO3 и HSOJ) по мере прохождения
процесса уменьшается. Вследствие этого для сульфирования
всегда берут избыток серной кислоты.
В зависимости от условий ведения процесса сульфирования
(температура, концентрация, полярность среды и т. п.) скорость
его может быть пропорциональна концентрации серной кислоты
в первой, второй и даже третьей степени. Таким образом, при-
приведенный в начале этого раздела механизм реакции является
самым простым, но не единственно возможным. Во многих слу-
случаях, в частности, в олеуме и в неводных растворителях, реак-
реакция может протекать через стадию образования пиросульфо-
кислоты:
SOJSOH' SOJI
Как уже отмечалось, сульфирование является процессом об-
обратимым. Обратный сульфированию процесс называют десуль-
фированием. Десульфирование является кислотно-катализируе-
кислотно-катализируемым процессом. Скорость его зависит от кислотности среды, т. е.
от наличия в ней ионов Н3О+.
Опытным путем установлено, что чем легче образуется суль-
фокислота, тем легче она и десульфируется. Этим свойством
сульфокислот часто пользуются для гашетки л ра^де\™еки;т про-
продуктов реакции.
При сульфировании олеумом, а также иеводными раство-
растворами серного ангидрида обратимость реакции мала и дпт прак-
практических расчетов ею можно пренебречь.
40
$ 3. УСЛОВИЯ ПРОВЕДЕНИЯ ПРОЦЕССОВ СУЛЬФИРОВАНИЯ
Т емпература оказывает большое влияние на ход процес-
процесса сульфирования. Изменение температуры ведения процесса
сказывается не только на скорости реакции, но и па характере
получающихся продуктов. Так, например, сульфирование нафта-
нафталина при 60ГС приводит к образованию а-сульфокислоты, a
сульфирование при 160 СС — к образованию |3-с\льфокнслоты в
качестве основного продукта. Сульфирование фенола серной
кислотой при комнатной температуре дает преимущественно
орто-сульфохислоту, а при 100 °С получается в основном
п-пзомер.
Температура, при которой ледутся процессы сульфирования,
в зависимости от химических свойств исходных веществ и жела-
желательного направления реакции колеблется в пределах от —10
до + 180°С. Б связи с этим в некоторых случаях требуется подо-
подогрев реакционной массы, а в некоторых — охлаждение.
Поскольку процесс сульфирования, как правило, может быть
описан рядом последовательных и параллельных реакций, то для
получения максимального выхода целевого продукта большое
значение имеет не только температура, но и продолжительность
ведения процесса.
Катализаторы в процессах сульфирования используются
сравнительно редко и роль их изучена недостаточно.
Среди катализаторов сульфирования наиболее интересны
ртуть и ее соли, позволяющие изменить место вхождения суль-
фогруппы при сульфировании олеумом соединений с мета-ориен-
тирующими заместителями. Так, сульфирование антрахинона
в отсутствие ртути приводит к образованию C-сульфокислоты,
а в присутствии ртути образуется а-сульфокислота.
Действие солей ртути основано на промежуточном образо-
образовании ртутно-органнческого соединения, а положение атома рту-
ртути в этом соединении определяет место вхождения сульфогруп-
пы в ароматическое ядро.
В некоторых случаях сульфирование ведут в присутствии
сульфатов щелочных металлов. Такие добавки снижают окис-4
лительное действие серной кислоты или серного ангидрида.
Скорость сульфирования при этом уменьшается, так как при
появлении в реакционной массе дополнительного количества
бисульфат-ионов (HSO7) равновесия
2H2SO4 <=> H3SO4+HSO7,
2H2SO4 <—>• SO3 + II3O+ + HSO7
смещаются влево и концентрации сульфирующих агентов умень-
уменьшаются.
Концентраци к серной кислоты оказывает решающее
влияние на ход процесса сульфирования. С уменьшением кон-
концентрации серной кислоты скорость сульфирования ре:ко сии-
41
жается. Предельную, или критическую, концентрацию серной
кислоты, при которой она еще способна сульфировать соедине-
соединение, называют «я-сульфйрования». При сульфировании концен-
концентрация серной кислоты по мере прохождения процесса снижает-
снижается до величины я-сульфирования, после чего процесс останавли-
останавливается. Практически в конце сульфирования концентрация БОз
в отработанной серной кислоте должна быть выше я-сульфиро-
я-сульфирования, так как в противном случае скорость сульфирования бу-
будет настолько мала, что проведение процесса окажется невоз-
невозможным. С другой стороны, большой избыток серной кислоты
при сульфировании нежелателен, так как приводит к увеличе-
увеличению расхода сырья и удорожанию продукта. Таким образом,
концентрация отбросной кислоты при сульфировании должна
превышать величину я-сульфирования лишь настолько, чтобы
обеспечить достаточную скорость в конце процесса.
Минимальное количество серной кислоты или олеума, тре-
требующееся для моносульфирования 1 кмоль органического веще-
вещества, может быть вычислено по формуле:
^> 80 A00 — лс)
°——iс=г^->
где С — концентрация исходного сульфирующего агента, вы-
выраженная в % SO3, яс—я-сульфирования.
Из формулы видно, что чем выше концентрация исходной
кислоты, тем меньше ее понадобится для ведения процесса. Од-
Однако следует помнить, что при повышении концентрации исход-
исходной кислоты (или олеума) возможно возникновение вторичных
реакций (образование полисульфокислот, сульфонов и т. д.).
От выбора концентрации сульфирующего агента зависит так-
также место вхождения сульфогруппы в ароматическое ядро. На-
Например, для получения а-нафталинсульфокислоты рациональнее
применять более концентрированную кислоту, чтобы предотвра-
предотвратить ее десульфирование. При получении же C-нафталинсульфо-
кислоты следует брать серную кислоту меньшей концентрации,
чтобы способствовать этим гидролизу побочного а-изомера
(C-изомер в этих условиях не десульфируется).
§ 4. ОСНОВНЫЕ СПОСОБЫ СУЛЬФИРОВАНИЯ
Сульфирование серной кислотой или олеумом. Для сульфи-
сульфирования серной кислотой в технике чаще всего применяют купо-
купоросное масло с содержанием 92—93% H2SO4 или моногидрат —
98—100% H2SO4.
Незамещенные углеводороды (бензол, нафталин) сульфиру-
сульфируются концентрированной серной кислотой при нагревании.
При сульфировании замещенных углеводородов заместители
первого рода облегчают вступление сульфогруппы в ядро, а за-
заместители второго рода — затрудняют его.
42
Введение второй сульфогруппы происходит значительно труд-
труднее, так как —SO3H является заместителем второго рода. По-
Поэтому процесс сульфирования до ди- и трисульфокислот ведут
обычно в две стадии. Сначала в относительно мягких условиях
вводят одну сульфогруппу, а затем, не выделяя моносульфокис-
лоту из реакционной массы, в жестких условиях (олеум, высо-
высокая температура) вводят вторую сульфогруппу.
При получении полисульфокислот, а также при сульфирова-
сульфировании углеводородов, имеющих в ароматическом ядре заместите-
Ки с по та 6 расходную емкость
Рис. 4. Схема типовой установки для смешения кислот.
/ — штуцеры для наполнения; 2 — оросительный холодильник; 3 — поддон; 4 — штуцеры
для отбора проб и замера уровня кислоты; 5 — погружной насос; 6 — люк.
ли второго рода, часто применяют олеум, т. е. раствор серного
ангидрида в серной кислоте. Чаще всего используют такие сорта
олеума, которые имеют достаточно высокую концентрацию БОз
и в то же время не затвердевают при обычной температуре.
Такими свойствами обладает олеум с концентрацией свободного
SO3 до 25% и 60—65% олеум. Эти сорта удобно транспортиро-
транспортировать и отмерять, тогда как олеум с концентрацией SO3 около
40% является в обычных условиях твердым веществом и тре-
требует длительного разогревания для превращения его в жидкость.
Олеум и серную кислоту нестандартных концентраций готовят
на месте путем смешивания стандартного сырья в необходимых
пропорциях. Схема типовой установки для смешения кислот
изображена на рис. 4.
На рис. 5 изображен аппарат, используемый для смешения
кислот в производствах небольшой мощности.
Условия смешения и разбавления кислот весьма несложны.
Для получения кислоты требуемой концентрации достаточно
тщательно перемешать рассчитанные и отмеренные количества
43
более концентрированной и менее концентрированной кислот
(или кислоты и воды). При смешении кислот выделяется тепло,
количество которого тем больше, чем больше разность концен-
концентраций кислот до и после смешения. Поэтому смешение кислот
должно вестись при охлаждении таким образом, чтобы темпера-
Рис. 5. Схема аппарата для смешения (кислот (а) и способы крепления змее-
змеевика (б).
/ — освинцованная или эмалированная мешалка; 2 — свинцовый змеевик; 3 — кислото-
кислотоупорное защитное покрытие; 4 — стальной сварной корпус; 5 — труба для передавлива-
ния.
тура в массе не превышала 50—60 °С, так как при более высокой
температуре усиливается коррозия, а при смешении олеума про-
происходит также испарение серного ангидрида.
Для защиты от коррозии аппаратуру для смешения кислот
обычно футеруют кислотоупорной керамической плиткой.
Сама операция сульфирования проводится в аппаратах, ко-
которые называют сульфураторами. Поскольку концентрированная
серная кислота и олеум не взаимодействуют с черными метал-
металлами, сульфураторы изготавливают из стали или чугуна. Для
нагрева или охлаждения реакционной массы сульфураторы
снабжены рубашками. Если реакционная масса имеет малую
44
вязкость (что обычно бывает при сульфировании жидких ве-
веществ), то перемешивание производят с помощью пропеллерной
мешалки .(рис. 6).
Если же реакционная масса вязкая (что бывает при сульфи-
сульфировании твердых веществ), то устанавливают якорную мешал-
мешалку (рис. 7).
Рис. 6. Сульфуратор для суль- Рис. 7. Сульфуратор для сульфирования при
ф:ирования при невязких реак-
реакционных массах.
/ — пропеллерная мешалка с элек-
электроприводом; 2 — люк; 3 — штуцер
для загрузки; 4 — труба для пе,ре-
давливания сульфомассы; 5 — кор-
корпус; 6 — гильза для термометра или
термопары; 7 —рубашка.
вязких реакционных массах.
/ — якорная мешалка; 2 — режущее приспособле-
приспособление; 3 — съемная труба для передавливания; 4 —
стальной или чугунный корпус; 5 — рубашка.
Порядок прибавления компонентов реакционной смеси мо-
может быть различным в зависимости от свойств исходного про-
продукта и температуры сульфирования.
По окончании загрузки всего количества сульфирующего
агента реакционную массу выдерживают, не прекращая переме-
перемешивания, при заданной температуре.
За процессом сульфирования следят, отбирая пробы от смеси
и проверяя растворимость их в воде или растворе соды. Иногда
определяют количество свободной серной кислоты в реакционной
массе.
Отбор средней пробы сульфомассы, особенно охлажденной,
требует большого внимания ввиду возможной ее неоднород-
неоднородности.
45
Сульфирование газообразным серным ангидридом. Основным
недостатком сульфирования серной кислотой является большой
непроизводительный расход последней. Одним из способов уст-
устранения потерь сульфирующего агента является использование
серного ангидрида. При сульфировании серным ангидридом во-
вода в результате реакции не образуется, а потому сульфирование
может быть осуществлено стехиометрическим количеством суль-
сульфирующего агента или при малом избытке его.
Вследствие большой активности серный ангидрид может
быть применен непосредственно лишь для сульфирования ве-
веществ, содержащих заместители второго рода.
Примером использования этого метода сульфирования в про-
промышленности может служить получение п-нитро-о-толуолсуль-
фокислоты:
Сульфирование п-нитротолуола ведут, барботируя газообраз-
газообразный серный ангидрид через раствор п-нитротолуола в моногид-
моногидрате или слабом олеуме. Расход сульфирующего агента при
этом существенно сокращается, а выход продукта увеличивается
на 5% по сравнению с выходом, наблюдаемым при сульфирова-
сульфировании 25% олеумом. Аналогичным способом можно сульфировать
нитробензол. Выход м-нитробензолсульфокислоты при этом уве-
увеличивается на 15% по сравцению с получаемым при сульфиро-
сульфировании олеумом.
В некоторых случаях при сульфировании серным ангидридом
исходное вещество растворяют не в моногидрате, а в дихлор-
дихлорэтане или ином инертном растворителе.
Недостатком сульфирования серным ангидридом является
невозможность его использования при вязких реакционных мас-
массах, а также образование в качестве побочных продуктов суль-
фонов R—SO2—R.
Сульфирование в парах. Другим способом уменьшения рас-
расхода серной кислоты при сульфировании является удаление об-
образующейся воды из сферы реакции.
Удаление воды из реакционной массы может быть осуществ-
осуществлено при пропускании паров сульфируемого вещества через сер-
серную кислоту, нагретую до температуры кипения исходного про-
продукта. Проходя через слой серной кислоты, пары частично суль-
сульфируются с образованием сульфокислот, остающихся в
реакционной массе. Непросульфировавшийся продукт, уходя из
46
аппарата, увлекает с собой реакционную воду
определяемом уравнением:
в количестве,
где Gc — масса паров непросульфировавшегося сырья, GB —
масса увлекаемой им воды, Мс — молекулярная масса сульфи-
сульфируемого продукта, Рс — упругость паров сульфируемого веще-
вещества при температуре сульфирования, Рв — упругость паров во-
воды над раствором ее в серной кислоте при заданных темпера-
температуре и концентрации раствора.
Благодаря удалению такого
количества воды из реакционной
массы расход серной кислоты
снижается на величину G = ,r_c, >
где С — начальная концентрация
серной кислоты в процентах SO3.
В производственных услови-
условиях отгонка воды с парами суль-
сульфируемого вещества не встреча-
встречает затруднений. Жидкости, полу-
- чаемые в результате конденса-
конденсации паров, расслаиваются в спе-
специальном отстойнике, после чего
исходный продукт возвращается
в цикл.
При сульфировании бензола
этот способ проведения процесса
позволил уменьшить расход сер-
серной кислоты в 2,5 раза. Получа-
Получающаяся при этом сульфомасса
содержит лишь 3—4% свободной
серной кислоты.
Способ сульфирования в па-
парах применим только в тех слу-
случаях, когда сульфируемое веще-
вещество имеет относительно низкую
температуру кипения, а концентрация серной кислоты, выражен-
выраженная в процентах SO3, не превышает 75—80%.
Наиболее удобны для проведения описываемого процесса ап-
аппараты барботажного типа. Типовая конструкция такого аппа-
аппарата изображена на рис. 8.
Сульфирование методом «запекания». Для получения арома-
ароматических п-аминосульфокислот применяется метод, получивший
название сульфирование запеканием. Метод основан на способ-
способности кислых сульфатов первичных ароматических аминов пере-
Рис. 8. Реактор для сульфирова-
сульфирования © парах.
1 — турбинная мешалка с электропри-
электроприводом; 2 — барботер; 3— труба для
подвода паров сульфируемого вещест-
вещества; 4 — штуцер для отвода паров воды
и
47
группировываться в п-аминосульфокислоты при нагревании до
температуры порядка 180 °С.
HO3S
Метод позволяет использовать для сульфирования эквимоле-
эквимолекулярное количество серной кислоты.
Поскольку для получения кислого сульфата амина может
быть использована разбавленная серная кислота, аппарат, в ко-
котором проводят эту реакцию, должен быть защищен от корро-
коррозии (футерован). При использовании концентрированной серной
кислоты эту операцию можно проводить в обычных сульфура-
торах.
Вторая стадия процесса — «запекание» кристаллического би-
бисульфата амина — раньше проводилась либо на противнях в спе-
специальных печах, либо в чугунных аппаратах, снабженных мощ-
мощными сошниковыми мешалками. Обогрев в обоих случаях велся
топочными газами. Отсюда произошло и название метода — «за-
«запекание».
Советскими исследователями разработан более совершенный
метод перегруппировки солей аминов в среде полихлоридов бен-
бензола. Перегруппировку сернокислой соли амина проводят в сре-
среде технического о-дихлорбензола при 175—180 °С в чугунных
котлах, снабженных якорными мешалками и обогреваемыми па-
парами высококипящего органического теплоносителя (ВОТ).
(см. рис. 7). Выделяющаяся при реакции вода отгоняется в виде
азеотропной смеси с дихлорбензолом. Такой способ ведения про-
процесса позволяет сократить затраты ручного труда, уменьшить
контакт обслуживающего персонала с токсичными аминами,
а также создает условия для точного соблюдения оптимальных
параметров ведения процесса, что приводит к повышению выхо-
выхода продукта.
Таким способом в Советском Союзе получают сульфанило-
вую и нафтионовую кислоты:
KH
Сульфаниловая кислота Нафтионовал кислота
§ 5. ОСНОВНЫЕ СПОСОБЫ ВЫДЕЛЕНИЯ СУЛЬФОКИСЛОТ
Выбор метода выделения сульфокислоты зависит главным
образом от ее свойств.
Некоторые сульфокислоты хорошо растворимы в концентри-
концентрированной серной кислоте и плохо растворимы в разбавленной.
Такие сульфокислоты выделяют простым разбавлением сульфо-
сульфомассы водой. Поскольку при разбавлении серной кислоты выде-
выделяется большое количество тепла, массу следует охлаждать.
В большинстве случаев, однако, сульфокислоты выделяются
в виде солей. В качестве нейтрализующих агентов применяют
кальцинированную соду, сульфит натрия, мел и известь. Кроме
нейтрализации и разбавления водой для выделения солей суль-
фокислот применяют высаливание поваренной солью и сульфа-
сульфатом натрия.
Нейтрализация сульфомассы содой и сульфитом натрия. При
нейтрализации сульфомассы содой или сульфитом натрия про-
происходят следующие основные реакции:
а) 2RSO3H + Na2CO3 >■ 2RSO3Na + Н2О + СО2;
H2SO4 + Na2CO3 > Na2SO4 + H2O + CO2;
б) 2RSO3H + Na2SO3 > 2RSO3Na + H2O + SO2;
H2SO4 + Na2SO3 >• Na2SO4 + H2O + SO2.
Образующиеся в результате нейтрализации сульфомассы нат-
натриевые соли сульфокислот могут находиться либо в растворе,
либо в осадке. Это зависит от концентрации раствора и соот-
соотношения растворимостей сульфата натрия и натриевой соли
сульфокислоты. Нейтрализация сульфомассы содой экономиче-
экономически менее выгодна, а потому используется реже, нежели нейтра-
нейтрализация сульфитом.
Нейтрализация сульфитом особенно выгодна в тех производ-
производствах, где комбинируются процессы сульфирования с процесса-
процессами щелочного плавления. В этих случаях используемый для ней-
нейтрализации сульфомассы сульфит является отходом процесса
щелочного плавления сульфосоли, а получающийся при нейтра-
нейтрализации сульфомассы сернистый газ может быть использован
для нейтрализации щелочного плава. Такой метод нейтрализа-
нейтрализации широко применяется в производстве фенола.
Нейтрализацию обычно проводят в аппарате, который пред-
представляет собой стальной котел, футерованный кислотоупорной
плиткой или гомогенно освинцованный. Для отвода тепла внутри
котла размещен свинцовый змеевик. Реакционная масса пере-
перемешивается рамной или лопастной мешалкой. В верхней части
аппарата помещается перфорированная труба, через которую
подается пар для пеногашения.
Сульфомассу постепенно, при работающей мешалке, загру-
загружают на предварительно нагретый до 90—95°С раствор сульфи-
4—926 49
та. При несоблюдении режима загрузки может произойти вы-
выброс реакционной массы из нейтрализатора.
После загрузки рассчитанного количества сульфомассы со-
содержимое нейтрализатора кипятят для полного удаления сер-
сернистого газа.
В тех производствах, где процессы сульфирования комбини-
комбинируются с процессами щелочного плавления и сернистый газ с
сульфитом натрия используется в качестве нейтрализующих
средств, устраивают специальные отделения для улавливания
сернистого газа. Принципиально производство может работать
и без отделения улавливания сернистого газа, но в этом случае
аппараты для подкисления щелочного плава смогут работать
лишь в тот момент, когда работает нейтрализатор сульфомас-
сульфомассы. Такая жесткая связь аппаратов крайне нежелательна, так
как почти всегда приводит к нарушениям графика работы про-
производства. Отделение улавливания сернистого газа служит свое-
своеобразным буфером, позволяющим увязывать работу отделения
сульфирования с работой отделения щелочного плавления. Од-
Однако организация отделения сернистого газа экономически целе-
целесообразна лишь в производствах большой мощности, так как
требует больших капитальных вложений. Следует также иметь
в виду, что при этом возникает ряд проблем, связанных с защи-
защитой окружающей среды от вредного воздействия сернистого
газа.
Основной аппаратурой отделения сернистого газа являются
газгольдеры, которые служат для кратковременного хранения
сернистого газа.
Схемы установки непрерывного действия для нейтрализации
сульфомассы сульфитом натрия описана на с. 58. Эта установка
является типовой и может быть использована не только в про-
производстве бензолсульфокислоты, но и при других процессах
сульфирования.
Нейтрализация сульфомассы мелом или известью. Мел и из-
известь — наиболее дешевое сырье для нейтрализации сульфомас-
сульфомассы. Нейтрализация мелом и известью протекает в соответствии
с уравнениями:
а) 2RSO3H + CaCO3 >- (RSO3JCa + Н2О + СО2;
H2SO4 + СаСО3 >■ CaSO4 + Н2О + СО2;
б) 2RSO3H + Са(ОНJ >• (RSO3JCa + 2Н2О;
H2SO4 + Са(ОНJ >- CaSO4 + 2H2O.
Как видно из уравнений, нейтрализация сульфомассы мелом
и известью приводит к одним и тем же основным продуктам.
Однако при нейтрализации сульфомассы известью не образует-
образуется углекислый газ, что существенно облегчает ведение процесса
на этой стадии. С другой стороны, мел является более дешевым
сырьем и при нейтрализации мелом сульфат кальция образует-
50
ся в более легко фильтрующейся форме. Вследствие этого мел
используют в качестве нейтрализующего агента чаще, чем из-
известь.
Успешное осуществление процесса нейтрализации сульфомас-
сы мелом или известью зависит от точности поддержания тем-
температуры и кислотности реакционной массы на заданном уров-
уровне. В зависимости от температуры ведения процесса и кислот-
кислотности среды может образовываться сульфат кальция, содержа-
содержащий то или иное количество молекул кристаллизационной воды.
Наиболее хорошо фильтрующаяся форма соответствует кристал-
кристаллогидрату CaSO4-2H2O (гипс). Гипс образуется при температу-
туре 60—65 °С и слабокислой среде. При нарушении этих усло-
условий осадок получается в плохо фильтрующейся форме, что за-
затрудняет дальнейший процесс выделения солей сульфокислот.
Для проведения нейтрализации сульфомассы мелом приме-
применяются стальные футерованные аппараты с мешалками.
В процессе нейтрализации сульфомассы выделяется значи-
значительное количество тепла, за счет которого температура реак-
реакционной массы поддерживается на уровне 60—65 °С. Температу-
Температура в этом случае может регулироваться только скоростью за-
загрузки компонентов. Размещение теплообменных элементов вну-
внутри реактора (змеевик) нерационально вследствие наличия в
аппарате довольно густой суспензии. Применение же рубашки
невозможно из-за толстого слоя футеровки.
Для поддержания кислотности реакционной массы на задан-
заданном уровне следует сульфомассу и меловую суспензию подавать
в аппарат одновременно в определенном соотношении. После
отделения гипса фильтрованием и промывки его получают про-
прозрачный раствор кальциевых солей сульфокислот. Кальциевые
соли можно выделить при упаривании этого раствора. Однако,
как правило, кальциевые соли предварительно переводят в нат-
натриевые, обрабатывая раствор содой:
(RSO3JCa + Na2CO3 > 2RSO3Na + СаСО3.
Для уменьшения потерь продукта промывные воды, получаю-
получающиеся при промывке осадков, используют для приготовления
меловой суспензии.
Выделение натриевых и кальциевых сульфокислот высали-
высаливанием. Полученные соли сульфокислот можно выделить из рас-
раствора упариванием последнего. Однако такой метод требует
больших энергетических затрат и, кроме того, может пройти ча-
частичное десульфирование образовавшейся сульфокислоты, поэто-
поэтому часто для выделения натриевых и кальциевых солей сульфо-
сульфокислот пользуются методом высаливания. Высаливанию можно
подвергать как разбавленные водой сульфомассы, так и полу-
полученные в результате нейтрализации сульфомасс растворы солей
сульфокислот. В качестве высаливающих агентов используют
чаще всего поваренную соль или сульфат натрия. Высаливание
4* 51
ведут в стальных футерованных котлах с мешалками. Наличие
в растворе большой концентрации одноименных ионов приводит
к уменьшению растворимости соли сульфокислоты и выпадению
ее в осадок.
Поваренная соль является наиболее дешевым соединением,
однако широкое применение ее для высаливания ограничивается
большим корродирующим действием, обусловленным одновре-
одновременным присутствием серной и соляной кислот при высаливании
кислых сульфомасс. Этот недостаток устраняется при высалива-
высаливании посредством сульфата натрия.
§ 6. СУЛЬФИРОВАНИЕ ВАЖНЕЙШИХ АРОМАТИЧЕСКИХ
СОЕДИНЕНИЙ
Сульфирование бензола. Сульфирование бензола протекает
по схеме:
SO3H
SO3H
Бензол Бензолсульфо- Бензол -л*-дисульфо-
кислота кислота
Бензолсульфокислоту получают путем сульфирова-
сульфирования бензола купоросным маслом или моногидратом, который бе-
берется в двойном избытке B моля серной кислоты на 1 моль бен-
бензола). Сульфирование начинают при температуре 60 °С, а закан-
заканчивают при 105°С. Наиболее низкий расход серной кис-
кислоты достигается при сульфировании в парах. Содержание
свободной серной кислоты в сульфомассе в конце процесса со-
составляет 4%.
Бензолсульфокислота является исходным продуктом для син-
синтеза фенола по сульфурационному способу. Она служит также
для синтеза бензол-м-дисульфокислоты и ряда других промежу-
промежуточных продуктов.
Бензол-м-дисул ьфо к и с л о т а получается при сульфи-
сульфировании моносульфокислоты олеумом при 80—90 °С. В случае,
когда исходным продуктом является бензол, сульфирование сна-
сначала ведут при температуре 40—45 °С 20% олеумом, а затем
продолжают при 80—90 °С, используя 65% олеум. По окончании
сульфирования серную кислоту и сульфокислоты переводят
в кальциевые соли. Гипс отделяют фильтрованием, а раствори-
растворимую кальциевую соль бензол-м-дисульфокислоты переводят в
натриевую. Натриевая соль выделяется при выпаривании рас-
раствора досуха. Она служит исходным продуктом для синтеза
резорцина.
52
Синтез некоторых других практически важных сульфокислог
бензольного ряда представлен на схеме:
100% H2SO4; 90-100 °С
Хлорбензол
С1
20% олеум; 105—120 °С
Хлорбензол-п-сульфокислота
С1
SO3H
NOa
п-Нитрохлорбензол
1 -Нитро-4-хлорбензол-
-3-сульфокислота
СН3
100% H2SO4;
110-120 °С
SO3H
п-Толуолсульфокислота
СН3
65% олеум;
125 СС
Толуол
о-Толуолсульфокислота
(побочный продукт)
100% H2SO4; 100—110 °С
2,4-Толуолди-
сульфокислота
SOoH
Фенол
2,4- Фенолдисульфокислота
Сульфирование нафталина. Сульфирование нафталина пред-
ставляет собой очень сложный процесс, который может быть
изображен схемой (с. 55).
При образовании моносульфокислот нафталина в первую-
очередь образуется а-нафталинсульфокислота. Однако а-суль-
фокислота легко дecyльфиpyeтcяJ в результате чего в сульфо-
массе начинает накапливаться р-изомер. Возможна также пря-
прямая изомеризация а-сульфокислоты в р-сульфокислоту. Для по-
получения максимального количества а-изомера процесс следует
вести при низкой температуре, высокой концентрации серной
кислоты (чтобы избежать гидролиза) и короткое время. Практи-
53
чески синтез сс-нафталинсульфокислоты осуществляют, сульфи-
сульфируя нафталин моногидратом при 60 °С.
Для получения р-нафталинсульфокислоты применяют 92—
96% серную кислоту. Сульфирование ведут при 160 °С. р-Суль-
фокислота нафталина служит основным сырьем для синтеза
крупнотоннажного продукта р-нафтола.
Сульфирование а-сульфокислоты нафталина при низкой тем-
температуре приводит к 1,5-дисульфокислоте. В качестве побочного
продукта при этом получается 1,6-дисульфокислота. Наиболь-
Наибольший выход 1,5-дисульфокислоты составляет 60—70% от теоре-
теоретического. При повышении температуры выход 1,5-дисульфокис-
1,5-дисульфокислоты резко падает вследствие гидролиза а-сульфогрупп.
1,5-Дисульфокислота нафталина используется в синтезе лекар-
лекарственного препарата диазолин.
I Олеум, 150-250°С ^ H03S-
§ 7. ПРИМЕРЫ СУЛЬФИРОВАНИЯ В ПРОМЫШЛЕННОСТИ.
ПРОИЗВОДСТВО НАТРИЕВОЙ СОЛИ БЕНЗОЛСУЛЬФОКИСЛОТЫ
Наиболее экономичным методом производства бензолсульфо-
кислоты является сульфирование бензола в парах. Технологиче-
Технологическая схема сульфирования изображена на рис. 9. Бензол из
хранилища насосом подается во внутренние трубы испарителя
типа «труба в трубе». Во внешние трубы испарителя (в рубаш-
рубашку) подается греющий пар давлением 8 ати. В испарителе бен-
бензол испаряется, а пары его перегреваются до температуры
150—160 °С. Давление паров бензола на выходе из испарителя
54
0,5—0,8 ати. Перегретые пары бензола поступают через барбо-
тер в сульфуратор, в который предварительно загружают 94—
96% серную кислоту. Пары бензола, не вступившие в реакцию
сульфирования, захватывают воду, выделившуюся в результате
сульфирования части подаваемого бензола, и поступают в свин-
свинцовый змеевик конденсатора обратного бензола. Сконденсиро-
Сконденсировавшаяся и охладившаяся до температуры 30—35 °С смесь бен-
бензола и воды стекает в отстойник непрерывного действия, где
Сульцюwacca
наотдувку г
бензола
Раствор
На он
Вода,
содержащая
Обратный
бензол
Отоадотаннйя
елочь
Рис. 9. Схема сульфирования бензола в парах.
1 — испаритель бензола типа «труба в трубе»; 2 — брызгоуловитель; 3 — холодильник-
конденсатор; 4 — нейтрализатор; 5 — отстойник; 6 — сульфуратор.
расслаивается на составные части. Бензол после промывки рас-
раствором щелочи возвращается в испаритель и далее на сульфи-
сульфирование. Производительность используемого в производстве
змеевикового конденсатора составляет 120 кг/ч-м2 водно-бен-
водно-бензольной эмульсии.
В течение первых 2 ч сульфирования скорость реакции вели-
велика и температура поддерживается за счет экзотермичности про-
процесса. Поддержание температуры на заданном уровне произво-
производят за счет регулирования температуры подаваемых паров бен-
бензола. Температура реакционной массы в этот период должна
быть 160—170 °С. В последующий период тепла реакции недо-
55
статочно для компенсации тепловых потерь и сульфураторы обо-
обогревают паром, поддерживая температуру на уровне 150—160 °С.
Образующаяся сульфомасса к концу сульфирования содержит
3—4% свободной серной кислоты. Общая длительность процес-
процесса 9—10 ч.
Срок службы сульфураторов периодического действия 2—
года. Наибольшая коррозия наблюдается в верхней части
Водяной
пар
Пары бензола
на, конденса-
конденсацию
* Сульфомасса на
нейтоализаиию
Рис. 10. Схема отдувки бензола.
/ — сборник сульфомассы; 2 — погружной насос; 3 — колонна для отдувки; 4 — холодиль-
иик-конденсатор; 5 — каплеуловитель; 6 — ловушка; 7 — вакуум-насос; 8 — адсорбер;
9 — гидравлический затвор.
аппарата. В связи с этим целесообразно корпус реактора делать
из двух царг, чтобы можно было заменять верхнюю по мере
износа, не демонтируя аппарат.
Кроме непрореагировавшей серной кислоты и сульфокислот,
сульфомасса после реакции содержит 1,5—2% бензола. Выделе-
Выделение бензола из сульфомассы позволяет уменьшить его расход на
20—25 кг на 1 т фенола. Отдувка бензола из сульфомассы про-
производится следующим образом (рис. 10).
Сульфомасса из сборника подается погружгхым насосом в
обогреваемую паром чугунную колонну. В колонне с помощью
вакуум-насоса создается разрежение, вследствие чего бензол
десорбируется из реакционной массы и, проходя через конденса-
56
тор, каплеотделитель и ловушку, поступает в адсорбер, запол-
заполненный активированным углем. Адсорбционную колонну перио-
периодически продувают острым паром, который уносит поглощенный
углем бензол. Смесь паров конденсируется, конденсат расслаи-
расслаивается и бензол возвращают в производство.
Отдутая сульфомасса поступает на нейтрализацию сульфи-
сульфитом. Нейтрализацию проводят непрерывным способом, схема
которого приведена на рис. 11. Сульфомассу и раствор сульфита
натрия параллельно в определенном соотношении загружают в
стальной футерованный кислотоупорной плиткой нейтрализатор.
По широкому перетоку реакционная масса, содержащая значи-
значительное количество сернистого газа, поступает в колонну для
Бензолсульшоиисло та
Вода
'$02 на
разложение
ере ноля та
Конденсат
Сультсоль
на пладку Конденсат
Рис. 11. Схема нейтрализации сульфомассы сульфитом натрия в производстве
бензолсульфокислоты.
/ — сборник раствора сульфосоли; 2 — гидравлический затвор; 3 — нейтрализатор; 4 — ре-
регулятор подачи сульфомассы; 5 — сборник сульфомассы; 5 —регулятор подачи сульфита;
7 — каплеуловитель; 8 — холодильники-конденсаторы; 9 — регулятор температуры; 10 —
сборник сульфита; 11 — насосы; 12 — рН-метр; 13 — колонна для отдувки.
отдувки и стекает по насадке вниз. Противотоком снизу подает-
подается водяной пар, который «отдувает» сернистый газ от раствора
натриевой соли бензолсульфокислоты.
Освобожденный от сернистого газа раствор продукта через
гидравлический затвор поступает в сборник.
Сернистый газ через эмалированный каплеуловитель и игури-
товый пакетный конденсатор подается в отделение для выде-
выделения фенола.
Получаемый после нейтрализации раствор содержит около
50% натриевой соли бензолсульфокислоты, около 4% сульфата
натрия, 0,7% сульфонов, 0,5% динатриевой соли бензол-м-ди-
сульфокислоты и 45% воды. Этот раствор направляется на ще-
щелочное плавление для получения фенола. Сухая натриевая соль
бензолсульфокислоты получается при упаривании раствора.
57
§ 8. КОНТРОЛЬ СУЛЬФИРОВАНИЯ
И ХАРАКТЕРИСТИКА ПРОДУКТОВ
Конец процесса сульфирования в производстве определяют
по исчезновению специфического запаха углеводорода в пробе
сульфомассы (сульфирование нитробензола, п-нитротолуола,
п-нитрохлорбензола), по полной растворимости сульфомассы
в воде, по достижению заданного значения концентрации сво-
свободной серной кислоты в сульфомассе (например, при сульфи-
сульфировании бензола в парах), по прекращению выделения паров
при сульфировании методом запекания (сульфирование анилина,
а-нафтилямина).
Помимо такой чисто качественной оценки, практика произ-
производства сульфокислот требует точного аналитического контроля
процесса сульфирования. Обычно определяют общее количество
сульфокислот в реакционной массе, соотношение моно-, ди- и
трисульфокислот, а также количество смол и сульфонов.
Общее количество сульфокислот определяют по разности ме-
между общим содержанием кислот, определяемым титрованием
щелочью, и содержанием серной кислоты, определяемым весо-
весовым методом в виде BaSO4. Содержание сульфонов может быть
определено полярографически.
Моно-, ди- и трисульфокислоты можно разделить методом
бумажной хроматографии. Последующее количественное опреде-
определение компонентов в каждой зоне хроматограммы проводится
спектрофотометрическим или полярографическим методом. Кро-
Кроме того, для анализа разработан ряд реакций, специфичных для
индивидуальных сульфокислот. Особенно широко применяется
качественное и количественное определение сульфокислот в ви-
виде солей с металлами или органическими основаниями типа
бензидина, фенилгидразина и т. п.
Сульфокислоты, за исключением отдельных представителей,
не имеют характерной температуры плавления, поэтому характе-
характеристика чистоты продукта часто затруднена. Для идентифика-
идентификации сульфокислот часто пользуются их производными, которые
легко очищаются и обладают характерными температурами
плавления. К таким производным в первую очередь относятся
сульфохлориды (RSO2CI) и сульфамиды (RSO2NH2).
§ 9. СУЛЬФОХЛОРИРОВАНИЕ. МЕХАНИЗМ РЕАКЦИИ
При взаимодействии эквимолекулярного количества хлор-
сульфоновой кислоты с ароматическими углеводородами обра-
образуются сульфокислоты:
RH + HSO3C1 > RSO3H + HCI.
Сульфирующее действие хлорсульфоновой кислоты объясня-
объясняется ее диссоциацией по схеме:
HSO3C1 4—+ SOj + HCl.
58
Большого распространения процесс сульфирования хлор-
сульфоновой кислотой не получил.
Очень большое значение в химико-фармацевтической про-
промышленности имеет процесс сульфохлорирования, т. е. процесс
образования хлорангидридов сульфокислот (сульфохлоридов)
при взаимодействии ароматического соединения с 4—5-кратным
избытком хлорсульфоновой кислоты:
RH + 2HSO3C1 1—»• RSO2C1 + НС1 + H2SO4.
Большой избыток хлорсульфоновой кислоты необходим для:
предотвращения снижения выхода сульфохлорида при протека-
протекании обратимой реакции превращения сульфокислоты в сульфо-
хлорид:
RSO3H + HSOgCl *=± RSO2C1 + H2SO4.
Значение реакции сульфохлорирования в химико-фармацев-
химико-фармацевтической промышленности обусловлено тем, что сульфохлориды
являются важными промежуточными продуктами в синтезе
сульфамидных препаратов. В зависимости от условий проведе-
проведения реакции и наличия заместителей в ароматическом кольце
процесс сульфохлорирования может протекать как через стадию
образования сульфокислоты, так и минуя эту стадию:
Прямое сульфохлорирование возможно благодаря образова-
образованию катиона SO2C1+ при диссоциации хлорсульфоновой кислоты
по уравнению:
3HSO3C1 -<—* SO2C1+ + H3O+ + 2SO3Ci-.
В качестве побочных продуктов при сульфохлорировании по-
помимо сульфокислот могут образовываться сульфоны:
RSO2C1 + RH > RSO2R + НС1.
§ 10. УСЛОВИЯ И СПОСОБЫ ПРОВЕДЕНИЯ
СУЛЬФОХЛОРИРОВАНИЯ
Процесс сульфохлорирования можно вести как непрерывным,
так и периодическим способом. Периодический метод применя-
применяется значительно чаще. Аппараты для проведения сульфохлори-
сульфохлорирования практически не отличаются от сульфураторов. Следует,
однако, иметь в виду, что выделение хлористого водорода в про-
процессе сульфохлорирования создает повышенную опасность кор-
59
розии оборудования, поэтому применение эмалированных аппа-
аппаратов предпочтительно. Кроме того, использование змеевиков в
таких аппаратах опасно, так как при их разъедании и попада-
яии воды в сульфомассу возможен выброс последней вследствие
^бурной реакции хлорсульфоновой кислоты с водой:
HSO3C1 + Н2О >■ H2SO4 + НС1.
Выделяющийся в процессе реакции хлористый водород улав-
улавливают водой в поглотительной системе.
Сульфохлориды, как правило, не растворяются или плохо
растворяются в холодной воде, поэтому их выделяют при вы-
выливании сульфомассы на лед или на воду, охлаждаемую льдом
или рассолом. Перегрев может привести к гидролизу сульфохло-
сульфохлорида:
RSO2CI + Н2О >- RSO3H + HC1.
Осадок отфильтровывают, тщательно промывают и отжи-
отживают на вакуум-фильтре.
Для уменьшения избытка хлорсульфоновой кислоты предла-
предлагались различные способы. Так, добавление к сульфомассе хло-
хлорида натрия при производстве бензолсульфохлорида привело
к значительному увеличению выхода сульфохлорида и уменьше-
уменьшению количества сульфонов. Причина этого заключается в том,
что образующаяся при реакции сульфохлорирования серная кис-
кислота частично связывается NaCl:
H2SO4 + 2NaCi ч=> Na2SO4 + 2НС1.
и равновесие сдвигается в сторону образования сульфохлорида.
К уменьшению избытка хлорсульфоновой кислоты приводит
также добавление к реакционной массе РС13, РС15, Р2О5. Этот
способ практического применения не нашел вследствие высокой
стоимости перечисленных реагентов. Предложено также прово-
проводить сульфохлорирование в среде инертного органического рас-
растворителя (четыреххлористый углерод, дихлорэтан). Образую-
Образующийся сульфохлорид хорошо растворяется в органическом рас-
растворителе и выводится таким образом из сферы реакции.
Большое значение имеет правильный выбор температуры и
времени ведения реакции. В большинстве случаев сульфохлори-
сульфохлорирование ведут при температуре 40—50 °С.
Имеет значение также технологический режим выделения
сульфохлорида из сульфомассы. Поскольку сульфохлориды гид-
•ролизуются, процесс выделения следует проводить при низкой
температуре и возможно более быстро. Предлагалось выделение
сульфохлорида из реакционной массы проводить непрерывным
способом в аппарате колонного типа с мешалкой. Через штуце-
штуцеры, находящиеся в нижней части аппарата, непрерывно пода-
♦60
ются сульфомасса и холодная вода. Скорость подачи регули-
регулируется таким бразом, чтобы температура суспензии была 20—
25 °С. Большие преимущества имеет способ выделения сульфо-
хлорида в« виде гранул. Это ускоряет фильтрацию и промывку
осадка в 3—4 раза и приводит к увеличению выхода сульфо-
хлорида на 5% за счет уменьшения потерь при гидролизе.
Кроме того, гранулированный продукт более стабилен при хра-
хранении, так как имеет меньшую влажность и лучше отмыт от
кислоты.
Для более полной утилизации отходов производства процесс
разложения сульфомассы можно вести в две стадии.
1. Разложение избытка хлорсульфоновой кислоты до серной
кислоты — моногидрата. Эту стадию можно проводить непо-
непосредственно в сульфураторе после охлаждения сульфомассы.
Выделяющийся хлористый водород полностью улавливают с по-
получением соляной кислоты.
2. Разбавление получающегося раствора сульфохлорида в
моногидрате и выделение сульфохлорида.
Этот способ позволяет почти полностью утилизировать хло-
хлористый водород. Выход сульфохлорида при этом не снижается.
Наибольшее промышленное значение имеет процесс сульфо-
хлорирования фенилуретилана:
+2HSO3C1
Метиловый эфир фенилкарбаминовой
кислоты (фенилуретилан)
+ H2SO4+HC1.
п- Фенил уретил ан-
сульфохлорид
Реакцию проводят при температуре 42—45 °С и избытке
хлорсульфоновой кислоты в 5,3—5,5 раза. Выход сульфохлорида
при этом составляет 82—83%.
Вторым по значимости является производство п-ацетанилид-
сульфохлорида:
NHCOCHs NHCOCHs
+ 2HSO3C1 > I +H2SO4 + HC1.
п-Толуолсульфохлорид получается при производстве сахари-
сахарина и используется в синтезе лекарственного препарата этамид.
§ 11. ТЕХНИКА БЕЗОПАСНОСТИ ПРИ ПРОВЕДЕНИИ
ПРОЦЕССОВ СУЛЬФИРОВАНИЯ И СУЛЬФОХЛОРИРОВАНИЯ
Многие углеводороды, которые подвергаются сульфированию
и сульфохлорированию, являются токсичными веществами. Осо-
Особенную опасность представляют процессы сульфирования в па-
парах, так как в этом случае вероятность возникновения токсиче-
токсических и взрывоопасных концентраций сульфируемых веществ при
нарушении герметичности аппаратуры является наибольшей.
Продукты сульфирования представляют значительно мень-
меньшую опасность, чем исходное сырье, как в смысле токсичности,
так и в отношении опасности взрыва и пожара.
Серный ангидрид, сернистый газ и хлористый водород дейст-
действуют раздражающе на слизистые оболочки. При попадании на
кожу серная, хлорсульфоновая кислота и олеум могут вызвать
серьезные ожоги. Вдыхание пыли Na2CO3, Na2SO3, Na2SO4 и
других солей вызывает сильное раздражение дыхательных
путей.
Наиболее действенными способами, предотвращающими со-
соприкосновение работающих с вредными веществами, являются
полная герметизация аппаратуры, а также механизация и авто-
автоматизация производственных процессов. Если отсутствует воз-
возможность комплексной механизации процесса, то механизация
проводится для отдельных узлов и аппаратов. В каждом случае
особое внимание обращают на ликвидацию источников проник-
проникновения паров вредных жидкостей и газов в рабочие помещения.
Для перекачивания вредных и опасных жидкостей вместо
обычных насосов применяют бессальниковые насосы (погруж-
(погружные, с электромагнитным приводом, с газовым уплотнением
сальника и др.), изготовленные из материалов, стойких к кор-
коррозии. Для транспортирования вредных и опасных жидкостей
используют трубопроводы с усиленными соединениями и надеж-
надежными зажимами прокладок. Открытые люки для отбора проб
из аппаратов заменяют герметическими пробоотборниками с за-
засасыванием проб вакуумом или автоматическими аналитически-
аналитическими приборами.
Вентиляционные устройства должны обеспечивать в рабочих
помещениях 4—6-кратный обмен воздуха за час. В отдельных
помещениях, где проводятся особенно вредные операции, крат-
кратность обмена воздуха должна составлять 8—12 в час. Все аппа-
аппараты, из которых возможно выделение вредных газов, обору-
оборудуются местной вентиляцией.
Производственные помещения, в которых перерабатываются
легковоспламеняющиеся жидкости, оборудуются в соответствии
с противопожарными нормами специальным взрывобезопасным
электрооборудованием и средствами для тушения пожара. Для
работающих с кислыми растворами и реакционными массами
нормами предусматривается выдача спецодежды из шерстяной
62
ткани. Такая ткань мало разрушается кислотами и поэтому за-
защищает тело рабочего от действия случайно попавших на одеж-
одежду кислых паров и жидкостей.
Большое значение для безопасности труда имеет покрой
спецодежды. Работающим с кислыми растворами выдаются
куртки и брюки. Применение комбинезонов в этом случае про-
противопоказано, так как в аварийном случае может возникнуть не-
необходимость быстро снять одежду. На работах, связанных с
выделением пыли (сушка и упаковка сульфокислот), нормами
предусмотрена выдача комбинезонов.
Глава 4
НИТРОВАНИЕ
§ 1. ОБЩИЕ СВЕДЕНИЯ
Нитрованием называют процесс замещения атома водорода
в органическом соединении нитрогруппой — NO2, осуществляе-
осуществляемый путем обработки нитруемых веществ различными нитрую-
нитрующими агентами. Исходными продуктами при нитровании служат
углеводороды ароматического ряда (бензол, толуол, нафталин
и т. д.), а также их замещенные (сульфокислоты, хлорпроизвод-
ные, амино- и оксипроизводные). В качестве нитрующих аген-
агентов применяют смесь азотной и серной кислот (меланж), азот-
азотную кислоту, смесь азотной и уксусной кислот, селитру в смеси
с серной кислотой.
Нитрование принадлежит к числу важнейших процессов хи-
химико-фармацевтической промышленности. Широкое применение
ароматических нитросоединений в синтезе лекарственных ве-
веществ объясняется высокой реакционной способностью нитро-
группы и получаемой ее восстановлением аминогруппы.
Нитросоединения биологически активны, многие из них ток-
токсичны. При наличии в молекуле карбоксильной или окси-
группы токсичность нитросоединения уменьшается. Среди нитро-
нитросоединений есть важные лекарственные вещества, например,
левомицетин, фурацилин и др.
Аналогично сульфокислотам различают мононитросоедине-
ния, динитросоединения и тринитросоединения. Полинитросоеди-
нения обладают взрывчатыми свойствами.
Нитросоединения представляют собой нерастворимые в воде
жидкости или твердые вещества желтоватого цвета. Ароматиче-
Ароматические нитросоединения имеют нейтральный характер, если в мо-
молекуле нет других заместителей, которые способны сообщить
соединению кислые или основные свойства.
В общем виде реакцию нитрования можно изобразить сле-
следующим образом:
RHX + HNO3 > R(NO2)X -i- xH2O.
Нитрогруппа является сильным заместителем второго рода.
Направляющее действие ее выражено значительно сильнее, чем
направляющее действие сульфогруппы, что объясняется боль-
64
шим эффектом сопряжения и индукционным эффектом. Моно-
нитросоединения нитруются до динитросоединений в значитель-
значительно более жестких условиях, чем исходные углеводороды — до
мононитроСоединений. Благодаря этому мононитропродукты по-
получаются без примеси полинитросоединений.
§ 2. МЕХАНИЗМ РЕАКЦИИ
Нитрование ароматических соединений является типичной
реакцией электрофильного замещения:
В отличие от сульфирования реакция нитрования необрати-
необратима. Опыты с дейтерированными соединениями показали, что
медленной стадией является образование промежуточного комп-
комплекса. Распад же его протекает практически мгновенно. Таким
образом, общая скорость процесса определяется первой его ста-
стадией.
Установлено, что активной частицей в реакциях нитрования
является нитроний-катион NO2, образующийся при диссоциации
концентрированной азотной кислоты по схеме:
2HNO3 -<—>- NO2 + N0^ + Н20.
В присутствии серной кислоты образование нитроний-катио-
на объясняется уравнением:
HNO3 + 2H2SO4 7—>■ NOj + Н3О+ + 2HS0J.
Поскольку серная кислота сильнее азотной, последняя в при-
присутствии серной кислоты ведет себя как основание, расщепляясь
на гидроксил и нитроний-катион. Увеличение концентрации воды
в реакционной массе приводит к сдвигу равновесия влево и
уменьшению нитрующей активности смеси.
В отличие от сульфирования нитрование в большинстве слу-
случаев является гетерогенным процессом. Установлено, что реак-
реакция идет практически только в кислотном слое. Скорость реак-
реакции зависит от состава кислотного слоя и от температуры. При
повышении температуры на 10 °С скорость реакции увеличивает-
увеличивается примерно вдвое. При плохом перемешивании, не обеспечива-
обеспечивающем создание однородной эмульсии, скорость реакции снижает-
снижается вследствие уменьшения концентрации нитруемого вещества
в кислотном слое.
Известны случаи нитрования разбавленной азотной кисло-
кислотой. Метод применяется сравнительно редко. Разбавленной
азотной кислотой нитруют некоторые соединения, имеющие в
5—926
65
ядре заместители первого рода (например, фенол). Механизм
нитрования в этом случае иной, так как нитроний-катиона в ре-
реакционной массе мало или нет совсем.
А. И. Титов на большой группе ароматических соединений
показал, что нитрование азотной кислотой с плотностью 1,4
и ниже идет только в присутствии азотистой кислоты. Реакция
в этом случае протекает по радикальному механизму:
HNO2 + HNO3 > N2O4 + Н2О;
N2O4 —»• 2NCY
RH + NO2' > R"+HNO2
R' + NCV > RNO2*
В отсутствие азотистой кислоты разбавленная азотная кис-
кислота нитрующим действием не обладает.
§ 3. УСЛОВИЯ ПРОВЕДЕНИЯ ПРОЦЕССОВ НИТРОВАНИЯ
Температура. Как правило, нитрование ведут при низ-
низких температурах, так как только в этом случае процесс проте-
протекает гладко и с высоким выходом. Если вести процесс при по-
повышенной температуре, то скорость реакции будет столь вели-
велика, что выделяющееся тепло нельзя будет отвести. Это приведет
к еще большему повышению температуры, возрастанию скорости
процесса и в конечном итоге к выбросу реакционной массы или
взрыву. Вторая причина ведения реакции при низкой темпера-
температуре заключается в том, что при повышенных температурах про-
проявляется окислительное действие азотной кислоты. Изменение
температуры нитрования приводит также к изменению соотно-
соотношения получающихся изомеров.
Время реакции. При интенсивном перемешивании реак-
реакционной массы процесс нитрования протекает с большой скоро-
скоростью. Практически время ведения процесса искусственно увели-
увеличивают, добавляя нитрующую смесь к исходному продукту
с такой скоростью, чтобы поддерживать температуру реакцион-
реакционной массы на заданном уровне.
Во многих случаях нитрование может быть проведено так
скоро, как скоро мы сможем отводить тепло реакции. Этот факт
имеет решающее значение при выборе конструкции реакторов.
Характер нитруемого продукта. Скорость про-
процесса нитрования, так же как и скорость сульфирования, сильно
зависит от характера и расположения заместителей в ядре аро-
ароматического соединения. Так, например, введение нитрогруппы
в ароматическое ядро снижает скорость вхождения второй нит-
нитрогруппы в тех же условиях в миллион раз. По своему влиянию
на скорость процесса нитрования заместители могут быть рас-
расположены в следующий ряд:
Замедление Ускорение
^ >
NO2 < SO3H < СООН < СГ<СН3 < ОСН3 < ОС,Нб < ОН.
166
Хлор и группы, расположенные левее его, замедляют реак-
реакцию и тем больше, чем дальше от хлора они стоят. Группы,
расположенные вправо от хлора, ускоряют реакцию, и тем боль-
больше, чем правее они находятся.
Ориентация нитрогруппы имеет некоторые особенности. Нит-
рогруппа охотнее, чем сульфогруппа, вступает в орто-положение
к уже имеющемуся заместителю первого рода.
При нитровании пара- и орто-оксисульфокислот нитрогруппа
замещает не только водород ядра, но и сульфогруппу:
.ОН
NO2
•ОН
HO3S
•ОН
HNO3
HO3S-
Характер нитрующего агента оказывает большое
влияние на условия ведения процесса. Если при нитровании кон-
концентрированной азотной кислотой или нитрующей смесью про-
процесс ведут при низкой температуре и интенсивном охлаждении
реакционной массы, то нитрование разбавленной азотной кисло-
кислотой (окислами азота) следует проводить при повышенной тем-
температуре с подогревом реакционной массы. Количество азотной
кислоты в нитрующей смеси обычно несколько больше теоре-
теоретического. В зависимости от характера нитруемого соединения
избыток может составлять от 1% до 10%.
Соотношение количеств серной кислоты и воды в нитрующих
смесях должно быть строго определенным, так как от этого со-
соотношения самым существенным образом зависит ход нитрова-
нитрования. Изменения в соотношении количеств серной кислоты и воды
приводят к значительному снижению выходов. Отношение коли-
количества серной кислоты к общему количеству воды, находящейся
в реакционной массе после нитрования, называется числом водо-
отнятия или дегидратирующей способностью и является важной
характеристикой процесса.
Катализаторы в процессах нитрования практически не
используются. Однако известно, что в присутствии ртути и ее
солей нитрование азотной кислотой приводит к получению поли-
нитрофенолов. Таким способом из бензола можно получить ди-
нитрофенол и пикриновую кислоту:
NO2
•ОН
Азотная кислота в этом случае выступает не только как нит-
нитрующий агент, но и как окислитель. Из бензойной кислоты при
67
этом получается 2,4,6-тринитро-З-оксибензойная кислота, из то-
толуола — 2,4,6-тринитро-м-крезол, из хлорбензола — 2,4,6-тринит-
ро-3-хлорбензол.
При нитровании нитрующей смесью окисления ароматическо-
ароматического кольца не наблюдается. В этом случае ртуть и ее соли лишь
несколько ускоряют обычное нитрование.
Интенсивность перемешивания гетерогенной реакционной
массы имеет большое значение. Исследование процесса нитрова-
нитрования бензола в лабораторном модельном реакторе показало, что
при увеличении числа оборотов мешалки с 500 до 1500 в минуту
скорость нитрования увеличилась в 20 раз. Дальнейшее увели-
увеличение числа оборотов на скорость процесса не влияло.
§ 4. ОСНОВНЫЕ СПОСОБЫ НИТРОВАНИЯ
Процессу нитрования предшествует приготовление нитрую-
нитрующей смеси. Любая производственная схема смешения кислот
включает аппаратуру для отмеривания, аппаратуру для смеше-
смешения и аппаратуру для хранения приготовленных смесей.
В качестве смесителя может быть использован любой аппа-
аппарат, конструкция которого обеспечивает интенсивное перемеши-
перемешивание жидкости и отвод тепла, выделяющегося в результате
смешения кислот. Одной из распространенных конструкций сме-
смесителя периодического действия является аппарат, изображен-
изображенный на рис. 5.
При большой мощности производства используют установку
для смешения кислот, изображенную на рис. 4 (с. 44).
Установка снабжена выносным оросительным теплообменни-
теплообменником, что позволяет поддерживать температуру смеси на задан-
заданном уровне при большой производительности системы. Интен-
Интенсивное перемешивание компонентов осуществляется с помощью
погружного насоса.
В некоторых случаях перемешивание осуществляют путем
барботирования воздуха через реакционную массу. Следует, од-
однако, иметь в виду, что применение смесителей с механическими
мешалками или циркуляционными насосами более рационально,
так как воздух, барботируя через смесь кислот, вносит с собой
влагу и выдувает из смеси некоторое количество окислов азота,
снижая концентрацию азотной кислоты. Кроме того, барботеры
весьма быстро разъедаются и выходят из строя.
Повышение температуры смеси выше 35—40 °С может приве-
привести к весьма неприятным последствиям. Высокая температура
приводит к частичному разложению азотной кислоты с образо-
образованием окислов азота. При использовании для приготовления
нитрующей смеси отработанной кислоты, содержащей неболь-
небольшое количество нитропродуктов, повышение температуры может
привести к взрыву. Наконец, высокая температура смеси приво-
приводит к повышенной коррозии аппаратуры.
68
При периодическом ведении процесса нитрование ведут путем
постепенного добавления нитрующего агента к нитруемому ве-
веществу. Скорость добавления нитрующей смеси определяется
возможной скоростью отвода тепла реакции. Таким образом,
процесс можно провести тем быстрее, чем больше теплообмен-
ная поверхность реактора, поэтому нитраторы, помимо рубашки
для охлаждения, обычно имеют дополнительные теплообменные
элементы, выполненные в виде либо внутренних двухстенных
цилиндров (диффузоров), либо змеевиков (рис. 12).
Вода 6
цилиндр
Вода из
цилиндра
Рис. 12. Нитраторы периодического действия.
«а — с рубашкой и охлаждающим цилиндром-диффузором; б — с рубашкой и змеевиком;
4 — охлаждающий двухстенный цилиндр-диффузор; 2 — гильза для термометра; 3 — ру-
рубашка; 4 — корпус; 5 — пропеллерная мешалка; 6 — змеевик.
В некоторых случаях в нитраторе размещают несколько зме-
змеевиков, концентрически вставленных друг в друга. Это умень-
уменьшает полезную емкость нитратора, но позволяет проводить про-
процесс с большей скоростью. Если при нитровании образуются
взрывчатые вещества, то нитратор оборудуют нижним люком
или патрубком большого сечения для аварийного сброса нитро-
массы.
Для эффективного перемешивания реакционной массы нитра-
нитратор должен иметь быстроходную пропеллерную или турбинную
мешалку. Остановка мешалки при нитровании чрезвычайно
опасна. Без размешивания реакция очень сильно замедляется
и в нитромассе накапливается нитрующий агент. При последую-
последующем пуске мешалки реакция пойдет очень быстро, с большим
выделением тепла, что может привести к взрыву, поэтому реко-
рекомендуется установка специальных устройств, следящих за ис-
исправностью мешалки. Подачу охлаждающей воды или рассола
69
во внутренний змеевик целесообразно вести, засасывая их с по-
помощью вакуума, чтобы в случае течи змеевика вода не попадала
в реакционную массу.
При нитровании вязких реакционных масс, что имеет место
при нитровании сульфокислот, использование внутренних змее-
змеевиков и охлаждающих элементов нецелесообразно, так как на
них может происходить кристаллизация реакционной массы.
В этом случае для проведения нитрования используют типовые
сульфураторы с рубашкой и якорной мешалкой. В связи с тем
что поверхность теплообмена в этом случае мала, процесс идет
очень долго. Что касается интенсивности перемешивания, то в
данном случае она не играет решающей роли, так как нитро-
сульфопродукты и исходные сульфокислоты растворимы в нит-
нитрующей смеси и реакционная масса является гомогенной.
Поскольку процессы нитрования в большинстве случаев про-
протекают с большой скоростью, а реакционные массы, как прави-
правило, имеют малую вязкость, процессы нитрования целесообразно
проводить в реакторах непрерывного действия. Это уменьшает
количество нитропродуктов, одновременно находящихся в аппа-
аппарате, и тем самым значительно увеличивает безопасность про-
процесса. Кроме того, при непрерывном ведении нитрования ликви-
ликвидируются непроизводительные затраты времени на загрузку, вы-
выгрузку, осмотр аппарата, предварительное охлаждение
продуктов и т. п. Это позволяет увеличить производительность
аппаратуры в 10—15 раз.
Один из распространенных нитраторов непрерывного дейст-
действия для нитрования ароматических углеводородов изображен
на рис. 13.
Нитратор представляет собой цилиндрический сосуд, внутри
которого размещена трубчатка, а в центральной широкой тру-
трубе— пропеллерная мешалка, работающая как осевой насос.
В межтрубном пространстве циркулирует охлаждающий рассол.
Нитруемый продукт подается сверху в центральную трубу, за
время прохода по которой он предварительно охлаждается. Вни-
Внизу при выходе из центральной циркуляционной трубы сырье ин-
интенсивно перемешивается с подаваемой снизу нитрующей смесью
и эмульсия поднимается вверх по охлаждаемым трубкам. Длина
трубчатки и производительность реактора рассчитываются та-
таким образом, чтобы за время прохода по трубчатке реакционная
масса успела прореагировать. Такой реактор работает в режи-
режиме, близком к режиму полного вытеснения. При изготовлении
и эксплуатации таких реакторов особое внимание следует обра-
обращать на плотность соединения труб с трубной решеткой. Нару-
Нарушение герметичности недопустимо, так как может явиться при-
причиной проникновения хладоагента в реакционную массу.
Недостатком конструкции является также наличие патрубка в
нижнем днище, что для аппаратов, заполненных агрессивными
жидкостями, нежелательно.
7§
Исходный ,а
углеводород %
Хладагент
Высокой производительностью отличаются также нитраторы
непрерывного действия, теплообменная поверхность которых вы-
выполнена в виде змеевиков (см. рис. 12). Витки змеевика образуют
как бы стакан, играющий роль диффузора, в который непрерыв-
непрерывно подают исходные компоненты. Реакционная масса отводится
через боковой штуцер. Поскольку уровень верхнего вцтка змее-
змеевика располагают несколько
ниже уровня жидкости, часть
реакционной массы не выхо-
выходит через боковой патрубок
после одного цикла, а засасы-
засасывается на рецикл во внутрен-
внутренний змеевик. Доля реакцион-
реакционной массы, пошедшей на ре- иитролро-
циркуляцию, и кратность ре- %%гиан%я
циркуляции при заданной про- кислота
изводительности системы зави-
зависят от насосного действия ме-
мешалки, которое пропорцио-
пропорционально скорости ее вращения.
При необходимости в реак-
реакторах описываемого типа уста-
устанавливают не один, а несколь-
несколько змеевиков разных диамет-
диаметров, расположенных концент-
концентрически один в другом.
В СВЯЗИ С проведением ре- Хладагент
акции нитрования в условиях
гетерогенной среды наиболь-
наибольшее применение для непрерыв-
непрерывного ведения процессов нитро-
нитрования получили реакторы пол-
пол- Нитрую-
Нитрующий агент
Рис. 13. Нитратор непрерывного дей-
действия.
— пропеллерная мешалка; 2 — трубчатка;
ного Перемешивания. ПрОЦеСС «?~корпус; *4--кран для регулирования
мононитрования в большинст- подачи нитРУюи*его агента-
ве случаев может быть осуще-
осуществлен в одну ступень. В отдельных случаях с целью уменьше-
уменьшения объема аппаратов процесс осуществляют в каскаде из двух
реакторов. Для получения полинитросоединений непрерывным
способом применяют каскад реакторов полного перемешивания
или секционный реактор. В этом случае в каждой ступени кас-
каскада поддерживаются условия, оптимальные для данной стадии
ведения процесса.
§ 5. ОСНОВНЫЕ МЕТОДЫ ВЫДЕЛЕНИЯ НИТРОПРОДУКТОВ
Процесс выделения нитропродуктов из реакцинной массы
складывается из отстаивания продукта, его нейтрализации и
промывки, дальнейшей кристаллизации, дистилляции или грану-
71
лирования. Отработанную кислоту подвергают денитрации и
концентрированию с целью дальнейшего использования в про-
производстве.
Отстаивание или сепарация реакционной массы применяется
для отделения нитропродукта от отработанной кислоты. Если
нитропродукт является в обычных условиях жидкостью, то от-
отстаивание проводят при температуре производственного поме-
помещения, если же он — твердое вещество, то отстаивают при повы-
Тяжелая
жидкость
Рис. 14. Отстойник периодиче-
периодического действия.
/ — стальной футерованный кор-
корпус; 2 — кран; 3 — смотровой фо-
фонарь.
Рис. 15. Отстойник непрерывного
действия.
шенной температуре, чтобы продукт находился в расплаве. При
этом реакционную массу иногда предварительно разбавляют
водой. Сепарацию можно проводить как в отстойниках периоди-
периодического, так и непрерывного действия. Типовой отстойник перио-
периодического действия изображен на рис. 14.
Это обычный стальной котел с коническим днищем и смотро-
смотровым фонарем. Благодаря наличию смотрового фонаря легко
установить конец истечения тяжелой жидкости и переключением
кранов направить легкую жидкость в сборники или хранилища.
При отделении застывающих нитропродуктов отстойники
снабжают рубашками или змеевиками для обогрева. В некото-
некоторых случаях для предохранения от коррозии отстойники футе-
футеруют кислотоупорной плиткой или освинцовывают.
72
При непрерывном ведении процесса нитрования используют
отстойники непрерывного действия (рис. 15). Эмульсия, посту-
поступающая на расслаивание, вводится в аппарат через штуцер, рас-
расположенный в средней его части. Высоту расположения входно-
входного штуцера рассчитывают в зависимости от соотношения объе-
объемов легкой и тяжелой фаз таким образом, чтобы линия ввода
эмульсии находилась на уровне границы раздела фаз. Легкая
жидкость поднимается вверх и удаляется из аппарата через
верхний штуцер. Тяжелая жидкость направляется вниз и выво-
Нитропродукт, бодо
нитропродунт
Рис. 16. Установка периодического действия для промывки нитропродуктов.
i — аппарат для промывки; 2 — мешалка; 3 — гидравлический затвор; 4 — ловушка; 5 —
трубы для передавливания.
дится из аппарата через нижний штуцер. Высоту «утки» для вы-
вывода тяжелой жидкости рассчитывают в зависимости от соотно-
соотношения плотностей расслаиваемых фаз. Общая высота отстойника
зависит от времени расслаивания и заданной производительно-
производительности аппарата. В некоторых случаях для более четкого расслаи-
расслаивания фаз отстойник непрерывного действия заполняют на-
насадкой.
Нитропродукты, отделенные от отработанной кислоты, содер-
содержат небольшое количество захваченных или растворенных кис-
кислот, поэтому следующей стадией обработки реакционной массы
является стадия нейтрализации и промывки нитропродукта.
Нейтрализацию ведут разбавленным раствором соды или ам-
аммиака. Дальнейшую промывку проводят водой. Для нейтрали-
7а
^—It
Кислый нитропродунт
Вода
Промытый
нитропро-
нитропродк
зации и промывки нитропродуктов используют однотипные ап-
аппараты.
Агрегат для периодической промывки нитропродукта изобра-
изображен на рис. 16. Он состоит из стального футерованного аппарата
с мешалкой и двумя трубами для передавливания и отстойника.
В аппарат загружают нитропродукт и воду (или раствор соды)»
перемешивают до получения однородной эмульсии, затем от-
отстаивают при выключенной мешалке до полного расслаивания
массы. По короткой трубке вод-
водный слой передавливают в от-
отстойник-ловушку, где происходит
дополнительное расслаивание за-
захваченного нитропродукта. Нит-
Нитропродукт передавливают па
длинной давильной трубе в
сборник или загружают в аппа-
аппарат воду и проводят повторную»
промывку.
Непрерывный процесс нейтра-
нейтрализации и промывки проводят
либо в системе, состоящей из ре-
реактора полного перемешивания
и отстойника непрерывного дей-
действия, либо в аппарате колонно-
колонного типа (рис. 17). В верхнюк>
часть аппарата вводят кислый
продукт (жидкость большей плотности), в нижнюю — вода
(жидкость меньшей плотности). В среднюю часть аппарата по-
подают раствор соды. Нитропродукт, движущийся через аппарат
сверху вниз, встречается сначала с раствором соды, а затем от-
отмывается от продуктов нейтрализации водой и выводится через
нижний штуцер. Промывные воды удаляются через верхний
штуцер. Ь j
Нейтрализованные и промытые жидкие нитропродукты очи-
очищают при необходимости перегонкой. Легкозастывающие нитро-
нитропродукты подвергают кристаллизации, гранулированию и сушке.
Гранулированием называется процесс отвердения расплав-
расплавленных веществ с образованием мелких частиц (гранул), напо-
напоминающих по форме крупу. Образование гранул происходит при
интенсивном перемешивании расплавленного нитропродукта и
холодной воды. Гранулирование частиц проводят в обычных ап-
аппаратах с пропеллерными мешалками. В этом случае в аппарат,
в котором находится большое количество холодной воды, при
работающей мешалке подают тонкой струей расплавленный
нитропродукт. Образовавшиеся гранулы отфильтровывают на
яутч-фильтре.
Более эффективно гранулирование с помощью приспособле-
приспособления, показанного на рис. 18. Приспособление это состоит из тру-
Р.ис. 17. Аппарат непрерывного
действия для нейтрализации и про-
промывки нитропродуктов.
74
Расплавленный
нитро продукт
m
бы, согнутой в виде кольца и снабженной соплами, куда пода-
подается холодная вода. В место пересечения струй воды тонкой
струей подается расплавленный нитропродукт. В зоне, обозна-
обозначенной на рисунке пунктиром, происходит интенсивное смеше-
смешение нитропродукта с водой и образование гранул. Гранулиро-
Гранулированный продукт легко отфильтровывается от воды.
Часть нитропродукта остается в отработанной кислоте, от-
отделяемой при сепарации реакционной массы. Это не только при-
приводит к потерям целевого про-
продукта, но и затрудняет утилиза-
утилизацию отработанной кислоты. В
связи с этим проводится экстрак-
экстракция нитропродукта из отработан-
отработанной кислоты.
Экстракцию обычно проводят
исходным нитруемым вещест-
веществом. Нитропродукт, содержащий-
содержащийся в кислотном слое, переходит
вследствие большей растворимо-
растворимости в органический слой. Если
отработанная смесь содержит
азотную кислоту, то параллель-
параллельно происходит частичное нитро-
нитрование экстрагента за счет остат-
остатков азотной кислоты. После раз-
разделения в отстойнике отработан-
отработанную кислоту направляют на кон-
концентрирование и денитрацию, а
раствор нитропродукта В НИТруе- рис. 18. Приспособление для гра-
мом веществе загружают в нит- нулирова-ния нитропродуктов.
paTOD. / — труба для подвода холодной воды;
* г-; ' ^ 2 — сопло для подвода расплавленно-
ПОЛучеННаЯ ОТраООТаННаЯ КИС- го нитропродукта; 3 — кольцеобразный
ЛОТа СОДерЖИТ НебОЛЬШОе КОЛИ- коллектор с соплами;^-сопла для хо-
чество органических примесей,
следы азотной кислоты и окислы азота, преимущественно свя-
связанные в виде нитрозилсерной кислоты. Кроме того, отработан-
отработанная кислота разбавлена водой, выделившейся при реакции нит-
нитрования.
Денитрацию и концентрирование отработанной кислоты про-
проводят на установке, схема которой представлена на рис. 19.
Отработанную кислоту, которая обычно содержит 68—75% сер-
серной кислоты, непрерывно подают в верхнюю часть скруббера.
Стекая вниз по насадке, она нагревается горячими парами и
газами, поступающими из реторты. Скруббер для нагревания
отработанной кислоты представляет собой стальную футеро-
футерованную диабазовой плиткой колонну, заполненную насадкой.
Нагретая кислота стекает в нижнюю часть скруббера, а оттуда
поступает в реторту, где нагревается до температуры 270—
75
280 °С. При этом происходит окисление органических примесей,
десорбция окислов азота и упаривание воды, содержащейся в-
кислоте. Реторта обогревается топочными газами. Она представ-
представляет собой литой аппарат из легированного чугуна. Крышка
аппарата защищена от коррозии слоем диабазовой замазки.
Больше всего подвержена коррозии та часть аппарата, которая
расположена на границе жидкой и газовой фаз. Эту часть аппа-
аппарата футеруют ферросилидовыми плитками. В крышке аппарата:
Отработанная
кислота
Купоросное
наело
Рис. 19. Схема денитрации и концентрирования отработанной кислоты.
/ — печь; 2 — реторта; 3 — скруббер; 4 — промывная колонна; 5 — холодильник.
имеются два штуцера. Через один из них поступает подогретая*
в скруббере кислота, а через другой — в скруббер поступают
горячие газы и пары. Сконцентрированная и освободившаяся от
примесей серная кислота непрерывно через штуцер в боковой
стенке проходит в холодильник, изготовленный из легированного-
чугуна, где не только охлаждается, но и освобождается от шла-
шлама, выпадющего при охлаждении кислоты. Средний срок служ-
службы реторт, скрубберов и холодильников составляет всего около*
1 года, так как они работают в очень жестких условиях. Газы
и пары, прошедшие через скруббер, поступают в промывнук>
колонну, орошаемую водой. Промытые газы удаляются в атмо-
атмосферу, а промывная вода — в канализацию кислых стоков.
76
§ 6. ПРИМЕРЫ НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Нитрование бензола. Нитрование бензола протекает по
схеме:
NOo
SNO2
Нитробензол м-Динитробензол
Для получения нитробензола берут нитрующую смесь
в количестве, соответствующем 0,97—1,01 моля азотной кислоты
на 1 моль бензола. Нитрование протекает гладко при 40—50 °С.
Концентрация отработанной серной кислоты должна быть не
ниже 69%.
Бензол можно нитровать и азотной кислотой (без серной).
Концентрация азотной кислоты должна быть около 60%. Сни-
Снижение концентрации азотной кислоты до 50% приводит к очень
сильному замедлению течения реакции. Образующуюся при нит-
нитровании воду можно отгонять в виде азеотропной смеси с избыт-
избытком бензола. Азеотропная отгонка воды с бензолом в вакууме
предложена и для случая нитрования бензола смесью азотной
и серной кислот. При проведении процесса таким образом по-
получающаяся отработанная кислота пригодна для повторного
применения.
Нитробензол является одним из важнейших продуктов орга-
органического синтеза. Около 90% нитробензола расходуется на про-
производство анилина.
Динитробензол получается при нитровании нитробензо-
нитробензола в более жестких условиях—при температуре 90°С, концен-
концентрация отработанной серной кислоты не менее 86%. При нитро-
нитровании образуется смесь трех изомеров: 90% — мета-, 8—9% —
орто- и 1—2% пара-динитробензола. Для выделения основного
продукта — метадинитробензола смесь обрабатывают горячим
F5—70°) водным раствором сульфита натрия. При этом орто-
и пара-изомеры превращаются в водорастворимые натриевые
соли сульфокислоты нитробензола и легко отделяются от основ-
основного продукта:
NO2
+ NaNOa
SO3Na
Динитробензол применяется для получения м-нитроанилина,
м-фенилендиамина, а также м-дихлорбензола.
Тринитробензол прямым нитрованием бензола не полу-
получают вследствие малой скорости реакции и низкого выхода.
77
Нитрование толуола. Толуол нитруется значительно легче
бензола. Нитрование до мононитропродукта ведут при темпера-
температуре 35—40 °С.
СНЯ
O2N
NO2
•СН3
56%
39%
5%
N0,
Смесь изомеров разделяют многократной фракционной пере-
перегонкой в вакууме или, пользуясь их различной растворимостью,
в органических растворителях.
Из нитротолуолов получают толуидины, толидины, нитробен-
зойные кислоты и другие продукты. п-Нитротолуол является ис-
исходным сырьем для синтеза ряда анестетиков.
При дальнейшем нитровании смеси нитротолуолов при 75—
80°С получается смесь изомеров с преобладанием 2,4-д и нит-
нитротолуол а. В смеси содержится также заметное количество
2,6-динитротолуола. Чистый 2,4-динитротолуол получают из сме-
смеси кристаллизацией.
Из тринитротолуолов наиболее важным является 2,4,6-три-
нитротолуол (тротил), имеющий большое значение как взрывча-
взрывчатое вещество. Для нитрования динитротолуолов в тринитрото-
тринитротолуол применяют безводную смесь серной и азотной кислот
с двойным избытком азотной кислоты по сравнению с теорети-
теоретически необходимым. Нитрование ведут при температуре около
100 °С.
Нитрование нафталина. Основной особенностью нитрования
нафталина является то, что нитрогруппа вступает почти исклю-
исключительно в а-положение:
Количество р-изомера, образующегося при нитровании нафтали-
нафталина, не превышает 5%. Вторая нитрогруппа также вступает в
а-положение, причем 1,8-изомера образуется почти в два раза
больше, чем 1,5-динитронафталина. Для разделения 1,5 и 1,8-ди-
78
нитронафталинов пользуются их различной растворимостью в
серной кислоте и в дихлорэтане.
Нитрование п-оксифениларсоновой кислоты является важ-
важным процессом в синтезе осарсола и некоторых других лекар-
лекарственных препаратов. В результате реакции получают 3-нитро-
4-оксифениларсоновую кислоту:
ОН ОН
-, он
о=а/
юн
Нитрующая смесь содержит 43—44% серной кислоты,
22—23% азотной кислоты и 33—35% воды. Сухую оксифенил-
арсоновую кислоту предварительно растворяют в серной кислоте.
Готовый продукт очищают перекристаллизацией из воды и су-
сушат при 55—60 °С.
§ 7. ПРОИЗВОДСТВО НИТРОБЕНЗОЛА НЕПРЕРЫВНЫМ МЕТОДОМ
Схема непрерывного нитрования бензола приведена на
рис. 20. Нитратор представляет собой стальной аппарат с ру-
рубашкой и двумя охлаждающими змеевиками. Змеевик меньшего
диаметра имеет почти сомкнутые витки и работает как охлаж-
охлаждаемый диффузор. Нитратор снабжен двумя мешалками — тур-
турбинной и пропеллерной, — посаженными на один вал. Верхняя
мешалка (турбинная) служит для интенсивного смешения по-
подаваемых на нее из мерников бензола, нитросмеси и отрабо-
отработанной кислоты. Нижняя мешалка (пропеллерная) работает как
осевой насос, прокачивая нитромассу. Подача бензола и нитро-
нитросмеси автоматически регулируется по заданному соотношению,
а количество отработанной кислоты — по температуре в реак-
реакторе, которая поддерживается на уровне 65—68 °С. Реактор ра-
работает в режиме полного перемешивания. При установившемся
процессе концентрация нитробензола в нем постоянная и состав-
составляет величину около 5%. Из нитратора реакционная масса по-
подается в спиральные теплообменники. Первый из них служит
своего рода «дозревателем». В нем реакция доходит практиче-
практически до конца. В нитраторе расходуется около 90% азотной кис-
кислоты, введенной с нитросмесью, а в спиральном холодильнике
9—9,5%. Таким образом, 99—99,5% всей загруженной азотной
кислоты вступает в реакцию. Во втором холодильнике реакци-
реакционная масса охлаждается до температуры 30 °С. Из спирального
холодильника реакционная масса поступает в отстойник непре-
непрерывного действия, где происходит разделение нитробензола и
отработанной кислоты. Так как разность плотностей нитробен-
79
зола и отработанной кислоты велика, расслаивание происходит
быстро (время пребывания реакционной массы в отстойнике
5—10 мин). Наиболее эффективны горизонтальные отстойники.
В расчете на 1 т товарного нитробензола из производствен-
производственного цикла выводится 0,9—1 т 70—73% отработанной серной
кислоты, содержащей 1,5—2,2% нитробензола и 0,25—0,5%
азотной кислоты. Перед утилизацией этой кислоты из нее экс-
экстрагируют нитробензол. Экстракцию ведут бензолом, который
частично нитруется азотной кислотой, содержащейся в отрабо-
. Нитрующая смесь
nl _с
Рис. 20. Схема непрерывного нитрования бензола.
/ — емкость для нитробензола; 2 — напорные бачки; 3 — отстойник непрерывного дейст-
действия; 4 — сборник отработанной кислоты; 5 — спиральные холодильники; 6 — нитратор не-
непрерывного действия.
тайной серной кислоте. После экстракции и разделения слоев
содержание нитробензола в отработанной кислоте падает до
0,1—0,15%, а азотной кислоты — до 0,01—0,03%. Бензол со сле-
следами нитробензола подается в хранилище, а из него в нитратор.
Технический нитробензол промывают слабой аммиачной во-
водой и чистой водой.
Очевидно, что получение нитробензола по непрерывному ме-
методу с высокой производительностью системы и с высоким ка-
качеством продукта возможно лишь при надежной ав-
автоматизации всего процесса. Для регулирования соот-
соотношения подаваемых бензола и нитросмеси используются авто-
автоматические регуляторы расхода жидкости в сочетании с регули-
регулирующими клапанами. Подача отработанной серной кислоты ре-
регулируется автоматически по температуре в нитраторе. Подача
80
воды в рубашку и змеевики нитратора регулируется по темпера-
температуре охлаждающей воды на выходе, а подача воды в спиральные
холодильники — по температуре реакционной массы в них. При
неисправности мешалки в нитраторе или прекращении подачи
воды в охлаждающие элементы, прекращается подача бензола
и пптросмеси, а затем отработанной кислоты и весь агрегат
останавливается.
Контроль водной промывки осуществляется по электропро-
электропроводности промывной воды.
Основная арматура, элементы теплообмена, мешалки и на-
насосы в производстве нитробензола выполнены из стали
1Х18Н9Т. Остальная аппаратура изготовлена из обычной угле-
углеродистой стали, защищенной в тех местах, где возможна кор-
коррозия, кислотоупорными плитками на диабазовой замазке.
§ 8. КОНТРОЛЬ НИТРОВАНИЯ И ХАРАКТЕРИСТИКА ПРОДУКТОВ
Конец реакции нитрования определяют химическим анали-
анализом содержания нитропродуктов в реакционной массе. В неко-
некоторых случаях (например, при нитровании бензола, нафталина
и некоторых других соединений) косвенным указанием на окон-
окончание реакции может служить исчезновение специфического
запаха исходного продукта. Кроме того, для определения конца
реакции измеряют плотность нитросоединения (при нитровании
бензола, толуола, хлорбензола), температуру застывания реак-
реакционной массы (производство динитробензола, динитрохлорбен-
зола, а-нитронафталина), содержание HNO3 в отработанной
кислоте.
Для количественного определения содержания нитросоеди-
неиий в продукте обычно восстанавливают нитрогруппу в уксус-
уксуснокислом растворе цинком или в солянокислом растворе хлори-
хлоридом олова(II) с последующим количественным определением
аминогруппы диазотированием.
Хорошим методом количественного определения нитрогруппы
является титрование хлоридом титана. Реакция протекает по
уравнению:
RNO2 + 6TiC13 -I- 6HC1 > RNH2 + 6TiCl4 + 2R>0.
Титр хлорида титана предварительно устанавливают по раство-
раствору соли желсза(Ш), обычно железоаммопийных квасцов, инди-
индикатором служит роданид калия.
Чистоту полученных нитросоединений контролируют также
по точке плавления (застывания).
Сумму азотной и азотистой кислот определяют нитрометром
Лунге, а азотистую кислоту отдельно — титрованием КМ1Ю4.
В отработанной кислоте титрованием раствором щелочи опреде-
определяют общее содержание кислот.
6-926 81
§ 9. ТЕХНИКА БЕЗОПАСНОСТИ
Все ароматические нитросоединения в большей или меньшей
степени ядовиты. Динитросоединения, кроме отравляющего дей-
действия на организм, вызывают раздражение кожи, дерматиты и
экземы. Нитросоединения опасны в пожарном отношении, а ди-
нитропродукты часто относятся к числу взрывчатых веществ.
Основным способом защиты работающих в производстве аро-
ароматических нитросоединений является полная герметизация обо-
оборудования, которая обеспечивается применением аппаратов с
надежным уплотнением крышек и сальников, установкой герме-
герметичных пробоотборников или автоматических приборов для ана-
анализа реакционной массы, а также бессальниковой арматуры
и насосов. Особое внимание должно быть обращено на пред-
предотвращение смешения жидкостей, в результате взаимодействия
которых могут выделиться опасные вещества. Так, например,
нельзя объединять канализационные трубопроводы для стока
отработанной азотной кислоты и органических жидкостей, изве-
известны случаи взрывов при поладании азотной кислоты в ук-
уксусную.
Выделение вредных веществ в атмосферу возможно в случае
попадания воды в азотную кислоту или смесь, содержащую
азотную кислоту. При этом выделяется большое количество
окислов азота. При вдыхании окислов азота может наступить
отек легких со смертельным исходом. С целью предотвращения
таких случаев следует все погружные охлаждающие элементы
в аппаратах, содержащих азотную кислоту, изготавливать из
кислотоупорных материалов, а также устанавливать сигнализа-
сигнализаторы появления кислоты в технической воде.
При накоплении непрореагировавшей азотной кислоты в нит-
раторе возможны хлопки, выбросы и даже взрывы. В связи
с этим необходимо блокировать работу мешалки с подачей нит-
росмеси, а также прекращать подачу нитросмеси при повышении
температуры в реакторе выше установленной.
Непрерывное осуществление процессов нитрования, как пра-
правило, обеспечивает большую безопасность вследствие снижения
концентрации азотной кислоты в реакционной массе. Все аппа-
аппараты, из которых возможно выделение окислов азота, должны
быть оборудованы вентиляцией.
Значительную опасность в производстве ароматических нит-
нитросоединений представляют процессы самопроизвольного раз-
разложения некоторых продуктов при повышении температуры до
170 °С. К таким соединениям относятся сульфокислоты нитро-
толуолов и нитрохлорбензолов, нитробензолсульфокислота, нит-
робензойные кислоты.
Большую опасность представляют также нагретые до высо-
высокой температуры кубовые жидкости, остающиеся от перегонки
нитротолуолов, нитрохлорбензолов и других соединений. При
82
попадании в эти кубовые остатки воздуха возможны вспышки
и даже взрывы. Аппаратура, в которой содержатся горячие ку-
кубовые остатки, перед вскрытием должна быть охлаждена или
заполнена* инертным газом. Смолистые остатки от перегонки
нитропродуктов способны самовозгораться. Поэтому их не сле-
следует хранить и накапливать.
Работать в цехах по производству нитросоединений можно
лишь в специальной исправной и чистой одежде. После работы
необходимо тщательно вымыть руки и принять теплый душ.
Горячий душ противопоказан, так как горячая вода усиливает
проникновение токсических веществ в организм через кожу.
Прием пищи в производственных помещениях недопустим.
Признаками отравления нитросоединениями являются поси-
яение губ, носа, ушей, кончиков пальцев, а также головная боль,
головокружение и тошнота.
Глава 5
ГАЛОГЕНИРОВАНИЕ
§ 1. ОБЩИЕ СВЕДЕНИЯ
Галогенированием, или галоидированием, называют реакцию
введения атома галогена в молекулу органического соединения.
Галогенирование, в зависимости от природы исходного вещества
и условий проведения процесса, может протекать как реакция
замещения атома водорода в молекуле органического соедине-
соединения или как реакция присоединения галогена. Галогенирование
является одной из наиболее распространенных и важных реак-
реакций в синтезе лекарственных веществ.
Галогенпроизводные — биологически активные соединения.
Галогенпроизводные алифатического ряда обладают наркотиче-
наркотическими свойствами, а ароматического — часто проявляют анти-
антимикробные свойства. Галогенпроизводные токсичны. Токсич-
Токсичность увеличивается с увеличением числа атомов галогена в мо-
молекуле соединения. В ряде случаев токсичность галогенпроиз-
водных используется при получении препаратов для борьбы
с вредителями сельского хозяйства.
Галогенпроизводные часто используются в синтезе лекарст-
лекарственных веществ как промежуточные продукты. Атом галогена
в алифатической части молекулы легко замещается нуклеофиль-
ными агентами. Галоген в ароматическом ядре вступает в эти
реакции в значительно более жестких условиях. Подвижность
галогена возрастает при наличии в орто- и пара-положениях
к нему электроноакцепторных заместителей. Хлорирование яв-
является значительно более распространенным процессом, чем
бромирование и йодирование. Это объясняется, прежде всего,
значительно более высокой стоимостью брома и йода по сравне-
сравнению с хлором. Прямое фторирование практически не исполь-
используется вследствие очень высокого теплового эффекта реакции
и ряда других технологических трудностей. Фторпроизводные
получают через диазосоединения или заменой атома хлора на
фтор при действии HF, KF или SbF5.
В отличие от реакций нитрования и сульфирования наличие
в молекуле ароматического соединения атома галогена лишь не-
незначительно затрудняет дальнейшее замещение. Поэтому одно-
одновременно с образованием моногалоидного производного при
84
хлорировании происходит образование дя- и три-галогензаме-
щенных.
Уравнение галогенирования в общем виде можно изобразить
так:
RHX + хГал2 > ИГалх + хНГал (замещение)
С=С + Гал2 > С(Гал)—С (Гал)(присоединение).
§ 2. МЕХАНИЗМ РЕАКЦИИ
В зависимости от условий ведения процесса галогенирование
может протекать различно.
Хлорирование в ароматическое ядро идет в присутствии ка-
катализатора железа и его солей. Активность взятого в качестве
катализатора хлорирования железа проявляется лишь после
того, как оно под действием хлора переходит в хлорное железо,
растворяющееся в хлорируемом соединении. Таким образом,
хлорирование в присутствии железа протекает как гомогенно-
каталитический процесс. В этом случае реакция протекает как
электрофильное замещение:
2Ге +ЗСГ0
■ 2ГсС1.
FcCI3
С1
+РсСГ+ПСГ
Хлорирование в ядро может катализироваться не только же-
железом, но также серной кислотой и йодом. В этих случаях обра-
образование катиона хлора протекает по следующим схемам:
H2SO4 + С12
+ HC1;
12 + С12
СП т—
1+ + С12 -<
=> 2C1I;
C1- + I+
* C1I + C1+
В отсутствие катализаторов, способствующих образованию
катиона хлора, но под влиянием ультрафиолетового излучения
(специальные лампы или солнечный свет) незамещенные арома-
ароматические соединения способны присоединять хлор.
85
В случае бензола образуется гексахлорциклогексан, который
при нагревании теряет 3 молекулы хлористого водорода и пре-
превращается в трихлорбензол:
С1 С1
i i
Ск
hv
-ЗС12 >-
С1
I-3HC1
ъ
С1
С1
Если в отсутствие катализаторов на свету подвергать хлори-
хлорированию ароматические соединения, имеющие боковую цепь, то
происходит замещение водорода в боковой цепи, а ароматическое
ядро в этом случае не затрагивается:
СН3 СН2С1 СНС12 СС13
Толуол
Хлористый
бензил
Хлористый
бензин идеи
Реакция в этом случае протекает по радикальному меха-
механизму:
hv
С1-С1 >- Cl'+Cl'
RH + C1- >- R'+HC1
R# + Cl2 > RC1 + СГ и т. д.
§ 3. УСЛОВИЯ ПРОВЕДЕНИЯ
ПРОЦЕССОВ ГАЛОГЕНИРОВАНИЯ
Температура оказывает значительное влияние на ход
процессов галогенирования. При повышении температуры реак-
реакция существенно ускоряется и как следствие этого образуется
большее число полигалогенпроизводных, поэтому хлорирование
в ядро с целью получения монохлорпроизводных предпочтитель-
предпочтительно вести при сравнительно низкой температуре B0—70°С).
В ряде случаев изменение температуры процесса приводит к из-
изменению ориентации входящего атома хлора. Так, например,
при хлорировании хлорбензола образуются в основном орто- и
пара-дихлорбензолы. Мета-изомера при хлорировании в обыч-
обычных условиях образуется 4—5%. При проведении же этой реак-
реакции в газовой фазе при температуре 450—600 °С без катализа-
катализатора образуется преимущественно мета-дихлорбензол. Такое
изменение ориентации в этом случае объясняется большей энер-
энергией активации замещения в мета-положение, а также измене-
изменением механизма реакции при высокой температуре от электро-
фильн©г* замещения к радикальному.
86
Поскольку реакция хлорирования протекает быстро и с вы*
делением большого количества тепла, для поддержания задан-
заданной температуры процесса в большинстве случаев необходима
охлаждение реакционной массы. Тепловой эффект бромирования
значительно меньше, поэтому при бромировании часто необхо-
необходим подогрев реакционной массы. Йодирование является реак-
реакцией эндотермической и для его проведения всегда необходим
подвод тепла.
Катализаторы в процессах галогенирования играют
весьма важную роль, так как определяют не только скорость,
но и направление процесса. Для галогенирования в ароматиче-
ароматическое ядро в промышленности в качестве катализатора исполь-
используют почти исключительно железо. Для проведения процесса
достаточно прибавки 1% от веса хлорируемого соединения же-
железных стружек или опилок. При использовании хлорного желе-
железа это количество может быть уменьшено до 0,5%, а при приме-
применении в качестве катализатора йода — до 0,1%.
При проведении хлорирования в боковую цепь присутствие
железа недопустимо, поэтому следует обращать особое внимание
на тщательную изоляцию стальных частей аппарата от реакци-
реакционной массы. Если хлорируемое сырье при температуре хлори-
хлорирования является твердым веществом, то хлорирование ведут
в растворе или в расплаве.
Природа растворителя влияет на скорость процесса.
Все жидкие вещества, которые можно применять в качестве рас-
растворителей при хлорировании твердых веществ можно разде-
разделить на три группы: а) вещества, не способные реагировать с
хлором (четыреххлористый углерод); б) вещества, которые не
реагируют с хлором, но активируют прохождение реакции (сер-
(серная кислота); в) вещества, способные реагировать с хлором
(хлорбензол, нитробензол).
Практика показала, что целесообразнее всего вести хлори-
хлорирование в растворителях, относящихся к группам б и в. При
использовании растворителей группы в выбирают такие, кото-
которые реагируют с хлором значительно медленнее основного
сырья (например, смесь полихлоридов бензола). Если для хло-
хлорирования брать вещества, содержащие воду, то эта вода будет
растворять выделяющийся при реакции хлорид водорода, обра-
образуя соляную кислоту. Это приводит к быстрому износу оборудо-
оборудования, в связи с чем исходные вещества перед хлорированием
высушивают.
§ 4. ОСНОВНЫЕ СТАДИИ ПРОЦЕССА ХЛОРИРОВАНИЯ
Процесс хлорирования можно разбить на следующие основ-
основные стадии: а) подготовка хлорирующего агента; б) подготовка
хлорируемого сырья; в) хлорирование; г) обработка продуктов
хлорирования; д) утилизация и очистка отходящих газов.
87
Подготовка хлорирующего агента складывается
из операций испарения и осушки хлора. Как правило, хлор
поступает на производство в баллонах, где он находится в жид-
жидком состоянии. Применяют также хлор из специальных бочек
и цистерн — при очень большом расходе хлора. Хлорный баллон
представляет собой прочный стальной сосуд со сферическим
днищем, вентилем и внутренней сифонной трубкой, доходящей
почти до дна сосуда.
Способ испарения хлора зависит от размера его потребления.
При очень малом расходе достаточно перевернуть баллон венти-
Хлор из цистерны
Хгор из баллонод
Теплая бода
Рис. 21. Схема испарительной станции.
/ — водяная баня; 2 — баллоны с хлором; 3 — коллектор; 4 — манометр; 5 — предохрани-
предохранительный клапан; 6 — коллектор газообразного хлора; 7 —испаритель трубчатого типа.
лем вниз так, чтобы конец сифонной трубки оказался в газо-
газовом пространстве. Таким способом можно получать небольшие
количества газообразного хлора при условии, что вентиль от-
открыт лишь настолько, чтобы скорость выпуска газообразного
хлора не превышала скорости его испарения.
При большем потреблении хлора требуется устройство спе-
специального обогреваемого испарителя. Простейший испаритель
можно сделать из пустого хлорного баллона, обогреваемого па-
паровой баней. При большом расходе хлора несколько хлорных
баллонов устанавливают на весах в вытяжном шкафу и соеди-
соединяют через коллектор с испарителем хлора. Жидкий хлор, выхо-
выходящий из баллонов, поступает в испаритель, где испаряется за
счет тепла, подводимого с греющим паром, и в газообразном со-
состоянии подается на осушку. Схема испарительной станции
представлена на рис. 21.
Скорость подачи хлора определяют по ротаметру. Преиму-
Преимуществом ротаметров перед многими другими приборами явля-
88
Серная
кислота
Хлор
сухой
ются простота устройства, надежность в эксплуатации, равно-
равномерность шкалы и низкая стоимость. Гидравлическое сопротив-
сопротивление ротаметра незначительно и не зависит от величины рас-
расхода.
Газообразный хлор передают на осушивание. В качестве осу-
осушающего агента используют концентрированную серную кисло-
кислоту. Простая установка для осушения хлора изображена на
рис. 22. Устройство аппарата
ясно из рисунка. Внутренний
цилиндр имеет ложное днище,
на котором размещается кера-
керамическая или стеклянная на-
насадка. При осушении хлор пе-
передавливает серную кислоту во
внутренний цилиндр и барбо-
тирует через нее. Внутренний
цилиндр при этом работает как
насадочная колонна. Капельки
серной кислоты, захваченные
хлором, отделяются в ловушке,
а отработанная серная кислота
периодически удаляется через
нижний штуцер.
В крупнотоннажных произ-
производствах осушение хлора осу-
осуществляется в керамических
колоннах, заполненных насад-
насадкой, которая сверху орошается
серной кислотой. Осушаемый
хлор подается в нижнюю часть
керамической колонны. Высу-
Высушенный хлор проходит брызго-
уловитель и направляется в хлоратор. Серная кислота циркули-
циркулирует в системе: абсорбционная колонна — сборник — циркуляци-
циркуляционный насос. По мере разбавления серная кислота выкачивает-
выкачивается из сборника на регенерацию, а сборник заполняют концент-
концентрированной кислотой.
Осушение хлорируемых углеводородов при
небольшой производительности системы может осуществляться
в стальных футерованных аппаратах с коническим днищем и
мешалкой. Для осушения жидкого сырья применяют гранули-
гранулированный или плавленый хлористый кальций, окись кальция,
едкий натр и едкое кали. В коническую часть аппарата загру-
загружают, например, безводный хлорид кальция, заливают осушае-
осушаемое вещество и включают мешалку. После непродолжительного
перемешивания A—2 ч) суспензии дают отстояться, осушенный
продукт сливают через боковой штуцер, а отработанный хлорид
кальция — через нижний. Хлорид кальция может поглощать
Отработан-
Отработанная серная
кислота
Рис. 22. Схема осушки хлора в осу-
осушителе с насадкой.
/ — ловушка; 2 — корпус осушителя; 3 —
цилиндр с насадкой; 4 — решетка.
89
влагу в количестве, равном своей массе, что соответствует об-
образованию шестиводного гидрата СаС^-бЫгО. Практически
загрузку хлорида кальция рассчитывают на поглощение 30—
50% влаги от его массы.
При большой производительности используют установку для
осушки с циркуляционным насосом. Установка (рис. 23) состоит
из колонны, заполненной осушителем, отстойника, контрольного
бачка и насоса. Обычно 10-кратная циркуляция жидкости при-
На хлориро-
■ >
боние
Хлор —*
Бензол
Отходящие
газы
3
На осушму
Рис. 23. Установка для осушки хло-
хлорируемого жидкого сырья.
1 — колонна с осушителем; 2 — отстойник;
3 — контрольный бачок; 4 — насос.
Хлорбензол
Рис. 24. Хлоратор периодического
действия.
1 — корпус; 2 — рубашка; 3 — колпак;
4 — охлаждаемый стакан; 5 — полки
для катализатора; 6 — указатель уров-
уровня.
водит к практически полному обезвоживанию. По окончании
осушки краны перекрывают и всю жидкость направляют в от-
отстойник, где она отстаивается от мелких частиц хлорида каль-
кальция, а затем перекачивают в сборник или хлоратор.
Некоторые углеводороды (бензол, толуол) осушают методом
азеотропной отгонки воды.
Хлорирование проводят в аппаратах, называемых хло-
хлораторами или абсорберами. Хлораторы для хлорирования в аро-
ароматическое ядро, помимо развитой теплообменной поверхности,
90
$тхаёящие газы
должны иметь приспособления для размещения катализатора.
Одна из конструкций хлоратора периодического действия пред-
представлена на рис. 24. Этот аппарат состоит из чугунного цилинд-
цилиндрического корпуса, конического днища, крышки и верхнего кол-
колпака. Аппарат имеет стальную рубашку для охлаждения реак-
реакционной массы. Внутри абсорбера
размещен охлаждаемый двухстенный
цилиндр, на котором укреплены полки
для катализатора. Перед началом опе-
операции на полках хлоратора помещают
катализатор в виде связок железных
лент. После этого в аппарат загружа-
загружают хлорируемый углеводород, включа-
включают охлаждение и начинают подачу
хлора через барботер, расположенный
в центральной части реактора.
Хлорид водорода и непрореагиро-
вавший хлор удаляются через штуцер
в верхнем колпаке аппарата. По окон-
окончании процесса из конусной части ап-
аппарата через нижний слив удаляют
шлам и скопившееся там небольшое
количество соляной кислоты, а затем
выгружают хлорированную массу.
Существенным недостатком такой
конструкции является недостаточная
поверхность теплообмена, что застав-
заставляет искусственно затягивать процесс
хлорирования, подавая в аппарат
хлор с небольшой скоростью.
Иначе решен вопрос отвода тепла
в хлораторах непрерывного действия
(рис. 25). Действие аппарата основа-
основано на том, что хлорирование ведут при
температуре кипения хлорируемого
продукта в избытке последнего. При
этом тепло реакции отводится за счет
испарения избытка исходного продук-
продукта, пары которого после охлаждения
и конденсации могут быть возвращены
в цикл.
Хлоратор непрерывного действия
состоит из стальной колонны, футеро-
футерованной изнутри диабазовой плиткой. В нижней части колонны
имеются патрубки для ввода хлорируемого сырья, хлора и для
очистки реактора. Над этими патрубками расположена решетка
и люк для выгрузки насадки. В качестве насадки, которой за-
заполнена колонна, используются смесь керамических и стальных
Хлор
5ензол
Рис. 25. Хлоратор непрерыв-
непрерывного действия.
/ — футерованная колонна; 2 —
верхний колпак; 3 — гильза для
термопары; 4 — люк для вы-
выгрузки насадки.
колец размером 25X25 или 50X50 мм. Стальные кольца слу-
служат источником образования хлорного железа, которое является
катализатором процесса хлорирования в ядро. Верхняя часть
хлоратора расширена. Она служит для отделения прохлориро-
ванной жидкости от отходящих газов. Прохлорированная жид-
жидкость выводится через патрубок, расположенный у дна расши-
расширенной части, а смесь паров исходного продукта и хлористого
водорода отводится сверху из аппарата.
Весь корпус аппарата изнутри футерован, а все патрубки
имеют керамические вставки. Благодаря этому коррозия прак-
практически отсутствует. Большой перегрев реакционной массы не-
невозможен, так как хлорирование идет при температуре кипения.
Аппарат работает в режиме полного вытеснения, что обеспечи-
обеспечивает максимальный выход монохлорпроизводных. Активность
катализатора можно варьировать, изменяя соотношение количе-
количества керамических и стальных колец в насадке.
Процесс хлорирования боковой цепи должен проводиться при
полном отсутствии железа. Материалом для изготовления аппа-
аппаратуры в этом случае может служить освинцованная сталь,
эмалированный чугун, кислотостойкие неметаллические мате-
материалы или стекло. Процесс проводят при температуре кипения
реакционной массы и избыточное тепло отводится с парами
кипящей жидкости, которая конденсируется в обратном холо-
холодильнике и стекает в реактор.
Помимо барботажных аппаратов для хлорирования в боко-
боковую цепь, используют также аппараты колонного типа. В этом
случае хлоратором служит колонна, изготовленная из освинцо-
освинцованной стали или керамики и заполненная керамической или
стеклянной насадкой. Хлорируемое сырье из напорного бака по-
подается в верхнюю часть колонны, а хлор вводится противотоком
снизу. Хлорированная масса удаляется через гидразатвор из
нижней части колонны, а хлористый водород и пары хлорируе-
хлорируемой жидкости поступают из верхней части колонны в обратный
холодильник. Здесь пары жидкости конденсируются и конденсат
стекает в колонну. Хлористый водород, освобожденный от паров
сырья, направляется на абсорбцию.
Твердые вещества, как уже было сказано, хлорируют в рас-
растворе. Катализатор в этих случаях, как правило, не требуется,
поскольку в качестве растворителей используют вещества, акти-
активирующие процесс. Реактор представляет собой чугунный котел
со стальной рубашкой, снабженный пропеллерной мешалкой
и кольцевым барботером. Практика показала, что труба, под-
подводящая хлор к барботеру, а также вал мешалки быстро разру-
разрушаются на границе раздела жидкой и газовой фаз. Коррозия
стенок чугунного аппарата значительно меньше и составляет
около 1 мм в год. Для защиты от коррозии вал мешалки и тру-
трубу барботера покрывают слоем диабазовой замазки и одновре-
одновременно обматывают стеклотканью, которая удерживает замазку
92
и хорошо противостоит действию концентрированной серной
кислоты.
Обработка продуктов хлорирования заключает-
заключается в разделении и очистке веществ, составляющих реакционную
массу, получаемую в результате хлорирования. Часто для этого
пользуются ректификацией смеси. В общем случае число ректи-
ректификационных колонн в установке непрерывного действия долж-
должГаш
Р.ис. 26. Установка периодического действия для абсорбции хлористого водо-
водорода из отходящих газов. /
/ — первый абсорбер; 2 — гуммированный вентилятор; 3 — сборник соляной кислоты-
4 — насос; 5 — холодильник; 6 — второй абсорбер.
но быть на единицу меньше числа компонентов. При малом тон-
тоннаже продукта столь сложная система не является экономически
целесообразной, и очистку продукта (фракционированная раз-
разгонка) проводят периодически по общепринятой схеме.
Очистка отходящих газов заключается в абсорбции
хлорида водорода и выделении хлорируемого сырья из газовой
смеси. Выделение хлорируемого сырья предшествует процессу
абсорбции хлористого водорода.
Для выделения органических веществ из отходящих газов
пользуются конденсационным или сорбционным методом. Кон-
Конденсационный метод применяется при большом содержании
паров вещества в газе. Вследствие большой агрессивности среды
93
(HC1) конденсацию рекомендуется проводить в графитовых или
эмалированных теплообменниках. При использовании сорбцион-
ного метода пары органических веществ поглощаются из отхо-
отходящих газов растворителями. Сорбционный метод очистки при-
применяется при малом содержании органических веществ в газо-
газовой смеси.
Аппаратурное оформление абсорбции хлористого водорода
определяется масштабами производства.
В производствах, работающих по периодическому методу,
когда отходящие газы получаются в нескольких периодически
действующих аппаратах и состав их непостоянен, используют
установку, изображенную на рис. 26. Хвостовые газы подаются
гуммированным вентилятором в нижнюю часть футерованного
насадочного абсорбера. Насадка абсорбера орошается водой,
циркулирующей в замкнутом контуре абсорбер — сборник —
насос — холодильник. Циркуляция поглотительного раствора ве-
ведется по получении соляной кислоты стандартной концентрации
C1%), после чего полученную соляную кислоту передают для
использования в производстве, а сборник заполняют чистой
водой.
Второй абсорбер служит для промывки водой отходящих га-
газов перед выбросом их в атмосферу. Промывные воды из вто-
второго абсорбера после нейтрализации сбрасываются в канали-
канализацию.
Для утилизации хвостовых газов в производствах, работаю-
работающих по непрерывному методу, когда состав и количество отхо-
отходящих газов постоянны, целесообразно использовать установки
непрерывного действия.
§ 5. ПРАКТИЧЕСКИ ВАЖНЫЕ ПРИМЕРЫ ГАЛОГЕНИРОВАНИЯ
Хлорирование бензола в присутствии катализатора
протекает по схеме:
CI
\ CI
94
1,3,5-Трихлорбензол ,при прямом хлорировании практически
не образуется. Основным фактором, влияющим на соотношение
хлорбензола и полихлоридов, является глубина хлорирования.
Так как наиболее ценным продуктом является монохлорбензол,
процесс хлорирования ведут, оставляя в реакционной массе зна-
значительное количество непревращенного бензола. В зависимости
от потребности в полихлоридах, которые используются главным
образом как растворители, хлорирование ведут так, чтобы в ре-
реакционной массе оставалось от 20 до 80% бензола.
Хлорбензол используется в больших количествах для произ-
производства фенола и других промежуточных продуктов. Дихлорбен-
золы перерабатываются в нитропроизводные. Трихлорбензолы
являются хорошими и пожаробезопасными высококипящими
растворителями.
Хлорирование толуола в зависимости от условий ве-
ведения реакции может приводит к разным продуктам:
Fc
о-Хлортолуол служит сырьем для производства о-хлорбен-
зойной кислоты и о-хлорбензальдегида. п-Хлортолуол использу-
используется для синтеза п-хлорбензойной кислоты, п-хлорбензальдеги-
да, крезидина и 2,4-дихлортолуола. 2,4-Дихлортолуол
применяют для получения 2,4-дихлорбензотрихлорида и 2,4-ди-
хлорбензойной кислоты. Хлористый бензил используется для
получения бензилового спирта, цианистого бензила, а также для
введения бензильного остатка в органические соединения, хло-
хлористый бензилиден — для получения бензойного альдегида, а
бензотрихлорид — хлористого бензоила (С6Н5СОС1).
Интересен метод хлорирования толуола в паровой фазе при
освещении, предложенный Н. Д. Зелинским. По этому методу
пары толуола, полученные при кипении последнего в аппарате,
подаются через дефлегматор в специальный освещаемый сосуд,
куда подается также и газообразный хлор. Образующийся хло-
95
ристый бензил конденсируется и стекает обратно в аппарат.
Поскольку толуол кипит при более низкой температуре, чем
хлористый бензил, последний все время выводится из сферы
реакции и в реакционный сосуд поступают только пары толуола.
Процесс, разумеется, следует проводить при полном отсутствии
катализаторов хлорирования в ядро.
Хлорирование нафталина в промышленности при-
применяют еще сравнительно мало. В отличие от производных бен-
бензола большое значение в этом случае имеют полихлорзамещен-
ные. Нафталин можно хлорировать как пропусканием хлора
через расплавленный продукт, так и в растворителе. В качестве
растворителя используют хлорбензол.
Основным продуктом монохлорирования является а-хлор-
нафталин. (З-Изомера образуется около 10%. Смесь а- и р-изо-
меров разделить очень трудно.
Нафталин в реакции хлорирования проявляет склонность к
образованию продуктов присоединения. Продукты присоедине-
присоединения при нагревании отщепляют хлорид водорода и переходят
в хлорзамещенные нафталина:
С1
Н
При использовании в качестве катализатора хлорного желе-
железа образуются почти исключительно полигалоидные производ-
производные нафталина. В связи с этим при получении монохлорнафта-
лина обходятся без катализатора или применяют в качестве
катализаторов цинк и активированный уголь.
Последовательное галогенирование арома-
ароматического ядра и боковой цепи используется для по-
получения о-хлорбензилхлорида и о-бромбензилбромида, которые
являются промежуточными продуктами в синтезе лекарственных
препаратов оксазил и орнид.
CHoCl
Первую стадию синтеза ведут как обычное галогенирование
в ядро в присутствии железного катализатора. Вторую стадию
синтеза проводят в освинцованном аппарате при температуре
кипения реакционной массы и ультрафиолетовом освещении. Во
96
избежание получения производных хлористого и бромистого
бензилидена второе галогенирование ведут при недостатке гало-
галогена против стехиометрического.
Галогенирование непредельных соединений
широко используется в синтезах целого ряда лекарственных ве-
веществ. К таким процессам относится бромирование аллилсуль-
фоната натрия жидким бромом в производстве унитиола:
СНЯ=СН—CH2SO3Na + Вг2
CHoBr—CHBr-CH2SO3Na.
§ 6. ПРОМЫШЛЕННОЕ ПРОИЗВОДСТВО ХЛОРБЕНЗОЛА
НЕПРЕРЫВНЫМ МЕТОДОМ
В связи с недостатками периодического производства хлор-
хлорбензола, главными из которых являются сложность и малая
производительность хлораторов — абсорберов, в настоящее вре-
время для получения моно- _
хлорбензола почти везде ВыЬ
переходят на установки
непрерывного действия.
Реактор для получения
хлорбензола непрерыв-
непрерывным хлорированием из-
избытка бензола был под-
подробно рассмотрен на с. 91
(см. рис. 25). Принципи-
Принципиальная схема непрерывно-
непрерывного хлорирования бензола
при кипении реакционной
массы приведена на рис.
27.
В условиях непрерыв-
непрерывного процесса реакцион-
реакционная масса представляет
:обой газо-паро-жидкост-
ную эмульсию плотно-
плотностью 200—300 кг/м3. Ре- Хлор №
актор работает в режимег—О
бензол
^ис- ^7. Схема непрерывного хлорирования
, бензола.
/ — колонна с катализатором: 2 — конденсаторы-
холодильники; 3 — приемник обратного бензола;
4 - холодильник.
актор работает в режиме
полного вытеснения, бла-
благодаря чему достигается
мольное соотношение
ХЛОрОеНЗОЛа К ДИХЛОрбеН-
ЗОЛу В ПрОХЛОрирОВаННОЙ
массе, близкое к 40.
Благодаря отводу тепла за счет испарения бензола произ-
производительность реактора непрерывного действия с рабочим объе-
объемом 1,7 м3 в 16 раз выше производительности хлоратора-абсор-
хлоратора-абсорбера периодического действия объемом 7 м3 (см. рис. 24).
7-926
На 1 т хлорбензола в колонну хлоратора непрерывного дей-
действия подается 700 кг хлора под давлением 1,2—1,3 ати и около
4200 кг бензола. В процессе хлорирования за счет теплоты реак-
реакции испаряется около 1450 кг бензола, а хлор почти нацело всту-
вступает в реакцию. Объем реакционной массы за счет паров,
образующихся при кипении бензола, увеличивается более чем
в 3 раза, а масса жидкости уменьшается с 4200 кг до 3100 кг.
Хлорированную жидкость, уходящую из верхней части реак-
реактора, охлаждают в теплообменнике и после нейтрализации под-
подвергают ректификации; смесь паров бензола и хлорида водоро-
водорода охлаждается в других теплообменниках, откуда сконденсиро-
сконденсированный бензол вновь попадает на хлорирование, а хлорид
водорода поступает на абсорбцию.
При большом масштабе производства исходный бензол осу-
осушают методом азеотропной отгонки воды.
Перевод производства хлорбензола на непрерывный метод
позволил почти в 2 раза снизить стоимость строительства цеха
хлорбензола мощностью 2 т готового продукта в час. Эксплуа-
Эксплуатационные затраты снизились более чем на 10%.
§ 7. ТЕХНОЛОГИЧЕСКИЕ ОСОБЕННОСТИ ПРОЦЕССОВ
БРОМИРОВАНИЯ
Как уже указывалось, процессы бромирования не сопровож-
сопровождаются выделением столь большого количества тепла, как про-
процессы хлорирования, поэтому при бромировании величина
теплообменной поверхности реактора, как правило, не лимити-
лимитирует скорость ведения процесса. С другой стороны, бром —
весьма активный корродирующий агент, поэтому аппараты для
бромирования обычно эмалируют или снабжают никелевыми
вкладышами. Бром является дефицитным сырьем. Улавливание
выделяющегося в процессе реакции бромида водорода не явля-
является достаточной мерой для утилизации последнего, так как по-
потребность в бромистоводородной кислоте невелика. Для исполь-
использования всего брома, загружаемого в реактор, можно проводить
бромирование в присутствии окислителей. Например:
6RH + ЗВг2 + КСЮ3 >■ 6RBr + КС1 + ЗН2О.
Окислителем может служить также хлор. В этих случаях в ка-
качестве бромирующего агента можно использовать смесь броми-
бромистого и бромноватистого натрия (бромид-броматную смесь), ко-
которая получается при действии брома на едкий натр:
6NaOH + Br2 >■ 5NaBr + NaBrO3
В этом случае процесс бромирования протекает по уравнению:
6RH + NaBrOg + SNaBr + ЗС12 > 6RBr + 6NaCl + ЗН2О.
Поскольку КС1О3, NaBrO3 и NaBr применяются в виде водных
растворов, наличие в реакционной массе воды может приводить
98
к протеканию целого ряда побочных процессов. В связи с этим
бромирование, как правило, ведут молекулярным бромом, а вы-
выделяющийся бромистый водород улавливают и регенерируют из
него бром действием окислителей:
2НВг + С12 >- 2НС1 + Вг2.
В некоторых случаях экономически целесообразно улавливание
бромида водорода щелочными растворами и выделение из них
товарного бромида натрия.
Для проведения процессов бромирования практический инте-
интерес представляет применение брома в виде газовой бромовоз-
душной смеси, жидкого брома или раствора брома в инертных
растворителях.
Сложным местом при проведении процессов бромирования
является механизация загрузки брома.
§ 8. КОНТРОЛЬ ПРОЦЕССОВ
Ход реакции хлорирования, как правило, контролируют по
изменению плотности реакционной массы. Скорость подачи хло-
хлора регулируют в зависимости от температуры реакционной мас-
массы. Расход хлора контролируют по массе баллонов. Галогено-
производные имеют характерные температуры кипения и плав-
плавления. Для некоторых галогенпроизводных разработан также
метод полярографического анализа. В некоторых случаях иден-
идентификацию продуктов проводят превращением их в сульфохло-
риды действием хлорсульфоновой кислоты.
Количественно галоген определяют нагреванием вещества
в запаянной трубке с крепкой азотной кислотой и AgNO3 (метод
Кариуса), либо прокаливанием исследуемого соединения с чи-
чистой окисью кальция (метод Либиха). Известны и другие мето-
методы определения содержания галогенов.
§ 9. ТЕХНИКА БЕЗОПАСНОСТИ
Галогенопроизводные ароматического ряда являются токсич-
токсичными веществами. Они обладают наркотическими свойствами
и в то же время местным раздражающим действием. Некоторые
галогенопроизводные могут вызывать экзему и другие кожные
заболевания.
Замещение хлором водорода в боковой цепи приводит к по*
лучению продуктов, очень сильно раздражающих дыхательные
пути и глаза.
Токсичность галогенопроизводных повышается с увеличением
числа атомов галогена в молекуле. В связи с этим, помимо гер-
герметизации оборудования, большое внимание необходимо уде-
уделить механизации всех ручных операций.
7* 99
При ручной разгрузке или выгрузке галогенопроизводных,
а также при отборе проб из аппарата необходимо соблюдать
все меры предосторожности, чтобы исключить попадание токсич-
токсичных продуктов на тело. Работать следует только в спецодежде;
обязательно надевать рукавицы при открывании люков, отборе
проб, загрузке и выгрузке продуктов и т. п.
Меры предосторожности при работе с веществами, подле-
подлежащими галогенированию, излагались в предыдущих главах
учебника.
Через неплотности в аппаратуре и трубопроводах в производ-
производственное помещение может выделяться хлор или бром, которые
оказывают сильное отравляющее действие на организм. Кроме
того, при нарушении технологического режима, хлор и бром мо-
могут содержаться в хвостовых газах. Хлор может выделяться
также из баллонов при нарушении герметичности вентиля.
В связи с этим особое внимание обращается ча герметизацию
аппаратов, в которых проводится работа с хлором или газооб-
газообразным бромом. Баллоны с хлором размещают в специальных
боксах или в шкафах с сильной вытяжной вентиляцией. Поме-
Помещения, где ведется галогенирование, оборудуют общей и мест-
местной приточно-вытяжной вентиляцией с повышенной кратностью
воздухообмена. Хвостовые газы после утилизации перед выбро-
выбросом в атмосферу следует пропускать через щелочные скрубберы.
Перевод производства на непрерывный метод обеспечивает
оздоровление условий труда и комплексную механизацию и
автоматизацию всех процессов.
Гл а в а 6
ОКСИЛИРОВАНИЕ И АМИНИРОВАНИЕ
Оксилированием и аминированием называют реакции введе-
введения в молекулу органического соединения окси- и аминогруппы,
протекающие по механизму нуклеофильного замещения. Таким
образом, образование оксигруппы при окислении органического
соединения не является оксилированием, а образование амино-
аминогруппы при восстановлении нитросоединений нельзя называть
процессом аминирования.
Как окси-, так и аминосоединения являются чрезвычайно
важными в химии лекарственных веществ. Многие лекарствен-
лекарственные вещества, как мы увидим в дальнейшем, имеют в своем
составе окси- и аминогруппы. Кроме того, синтез многих лекар-
лекарственных веществ и витаминов протекает через стадию образо-
образования окси- и аминопроизводных.
§ 1. МЕХАНИЗМ РЕАКЦИИ
В отличие от электрофильных и радикальных реакций, при
нуклеофильном замещении атакующая частица является доно-
донором электронов. Она может быть анионом или нейтральной мо-
молекулой. При этом реакция протекает по схеме:
Так как водород в виде отрицательного гидрид-иона обра-
образуется только в присутствии окислителя, реакции нуклеофиль-
нуклеофильного замещения водорода в ароматическом ядре идут лишь в
жестких условиях и сопровождаются побочными окислительно-
восстановительными процессами. В связи с этим в большей ча-
части практически важных реакций нуклеофильного замещения в
качестве исходных веществ используются сульфо-, галогено-
101
и некоторые другие производные, способные отщеплять в этих
реакциях стабильный анион
где Y- — анион SO3H~, C1", ОН" и т. д.
Отрицательно заряженные комплексы менее стабильны, чем
положительно заряженные, поэтому реакции нуклеофильного
замещения протекают в более жестких условиях, чем реакции
электрофильного замещения. Электроноакцепторные заместите-
заместители (заместители второго рода), особенно, если они находятся
в орто- и пара-положениях, увеличивают стабильность образую-
образующегося промежуточного комплекса и значительно облегчают
прохождение нуклеофильной реакции.
§ 2. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ СУЛЬФОГРУППЫ
Нуклеофильное вытеснение сульфогруппы является важней-
важнейшим способом синтеза окси- и аминосоединений в ароматиче-
ароматическом ряду. Особенно часто эта реакция используется для про-
промышленного получения оксисоединений. Поскольку процесс
обычно проводят при взаимодействии со щелочью при высокой
температуре, то он известен также под названием реакции ще-
щелочного плавления.
Обычно для щелочного плавления применяют растворы суль-
фокислот или их пасты, получающиеся при выделении солей
сульфокивлот. В качестве щелочи обычно используют едкий
натр. Едкое кали как более дорогое сырье используют лишь в
тех случаях, когда едкий натр оказывается недостаточно реак-
ционноспособным. В некоторых случаях используют смесь едко-
едкого натра и едкого кали. Такая смесь имеет более низкую
температуру плавления, чем каждая из взятых щелочей. Взаи-
Взаимодействие протекает по уравнению:
RSO3Na + 2NaOH > RONa + Na2SO3 + H2O.
Наличие в технической щелочи значительных примесей ми-
минеральных солей приводит к образованию в плаве комков, за-
затрудняющих перемешивание и значительно ухудшающих каче-
качество продукта вследствие местных перегревов реакционной
массы. Содержание в щелочи минеральных солей поэтому не
должно превышать 10%. Примесь хлората натрия в щелочи не-
недопустима, так как она может вызвать возгорание плава.
Щелочное плавление в промышленности осуществляется тре-
тремя способами.
102
1. Открытое щелочное плавление, т. е. сплавление пасты
натриевой или калиевой соли сульфокислоты со щелчью или ее
концентрированным раствором при атмосферном давлении.
2. Автоклавное щелочное плавление проводится под давле-
давлением в автоклавах. В этом случае используют водные растворы
солей сульфокислот. Метод применяется в тех случаях, когда
нужно заместить гидроксилом лишь одну из нескольких сульфо-
групп, имеющихся в молекуле исходного соединения. Этот метод
используют также при щелочном плавлении аминосульфокислот,
так как при проведении этого процесса открытым способом
аминогруппа также замещается оксигруппой.
3. Автоклавное щелочное плавление с известью. Этот спо-
способ используется главным образом при щелочном плавлении
сульфокислот антрахинона и в других аналогичных случаях,
когда обычные методы приводят к введению в ароматическое
ядро двух орторасположенных оксигрупп.
О
Важнейшими параметрами, определяющими результат ще-
щелочного плавления, являются температура ведения реакции и
концентрация щелочи. Едкий натр — не только реагент, но и
среда, в которой протекает превращение. Поэтому во многих
случаях используется значительный избыток щелочи. Едкий
натр плавится при температуре 327,5 °С. Чтобы обеспечить под-
подвижность плава при более низких температурах, используют
концентрированные растворы щелочи или же добавляют в ще-
щелочь небольшое количество воды. Так, едкий натр, содержащий
~10% воды, плавится при температуре 270—290 °С. В процессе
плавки вода испаряется и температура реакционной массы под-
поднимается.
Сульфокислоты бензольного ряда обменивают сульфогруппу
на ридроксил в жестких условиях, при температуре 300—340 °С.
Таким образом получают фенол и резорцин. В таких же усло-
условиях замещается сульфогруппа в 2-нафталинсульфокислоте с
образованием важного промышленного продукта — 2-нафтола.
Активированная сульфогруппа обменивается на оксигруппу
в более мягких условиях. При наличии в молекуле нескольких
103
сульфогрупп можно подобрать температуру процесса и концен-
концентрацию щелочи таким образом, что замещаться будет только
наиболее активированная сульфогруппа. Электронодонорные за-
заместители в о- и п-положениях к сульфогруппе будут затруднять
реакцию щелочного плавления, а электроноакцепторные — об-
облегчать. Нитросульфокислоты и хлорсульфокислоты ароматиче-
ароматического ряда в реакцих щелочного плавления не используют вслед-
вследствие образования значительного количества побочных про-
продуктов.
Обработка готового щелочного плава включает нейтрализа-
нейтрализацию избыточной щелочи, отделение сульфита, выделение и очи-
очистку оксисоединения.
Готовый щелочной плав разбавляют водой. Эту операцию
называют гашением щелочного плава. Для выделения сульфита
натрия в твердом состоянии полученную массу нагревают до
80—90 °С, а затем отфильтровывают и промывают на фильтре
выпавший сульфит. Оксисоединение выделяют при подкислении
щелочных маточников. Таким методом пользуются, например,
при получении фенола и 2-нафтола.
В тех случаях, когда сульфит отделяют в виде раствора,
плав разбавляют большим количеством воды, достаточным для
полного растворения. Избыток щелочи нейтрализуют кислотой.
Нейтрализацию ведут до полного выделения оксисоединения,
которое отделяют от раствора сульфита отстаиванием или филь-
фильтрацией.
В некоторых случаях более выгодным оказывается проводить
разложение сульфита кислотой:
Na2SO3 + 2НС1 > 2NaCl + SO2 + Н2О.
Для полного удаления сернистого газа из реакционной массы
процесс проводят при 80—100 °С и хорошем перемешивании.
Выделяющийся сернистый газ поглощают раствором щелочи
таким образом, чтобы получить сульфит натрия, который ис-
используется в качестве сырья во многих производствах.
Аппаратура в процессах щелочного плавления работает
в очень тяжелых условиях. Установлено, что расплавленные
щелочи в присутствии органических веществ агрессивно дейст-
действуют на черные металлы. Металл покрывается мельчайшими
трещинами межкристаллитной коррозии и становится хрупким.
По механическим свойствам и коррозионной стойкости в среде
концентрированных щелочей при высокой температуре лучшим
металлом является никель. Однако из чистого никеля аппарату-
аппаратуру делают редко, так как она получается слишком дорогой.
Обычно котлы для щелочного плавления изготавливают из ле-
легированного чугуна или легированной стали, содержащей ни-
никель, хром и молибден в качестве легирующих присадок. Никель
придает чугуну жаростойкость и улучшает щелочеустойчивость.
Хром способствует сохранению прочности при повышенных тем-
пературах, а также сообщает чугуну устойчивость к межкри-
сталлитной коррозии. Молибден способствует повышению изно-
износостойкости и прочности чугуна, а также сохранению прочности
чугуна при температуре выше 350 °С. Однако даже при исполь-
использовании легированных чугунов срок службы плавильных котлов
не превышает 2—3 лет. Вследствие необходимости поддержания
высокой температуры обогрев плавильных котлов обычно ведет-
ведется топочными газами. В районах с дешевой электроэнергией
можно использовать также электрообогрев. Для предотвраще-
предотвращения пригорания щелочного плава при подвижных плавах ис-
используют пропеллерные мешалки, а при вязких — якорные. Для
щелочной плавки под давлением применяются обычные авто-
автоклавы.
Гашение плава проводят в стальных котлах с рамными или
лопастными мешалками. Аппараты для подкисления разбавлен-
разбавленного плава обязательно должны быть защищены от кислотной
коррозии — футерованы или освинцованы.
Замещение сульфогруппы аминогруппой протекает при
сплавлении натриевой соли сульфокислоты с амидом натрия:
RSO3Na -j- NaNH2 > RNH2 + Na2SO3.
Большого практического применения эта реакция не получила.
Замена активированной сульфогруппы может быть проведена
при обработке водным раствором аммиака под давлением:
RSO3N* + 2NH8
-> RNH2 + NaNH4SO8.
Примером может служить промышленное получение амино-
антрахинонов из соответствующих сульфокислот:
О SO3K
II I
+ KNH4SO8.
В тех случаях, когда сульфогруппа обладает особенно высокой
нуклеофильной подвижностью, например:
О SO3Na
II
)С—SO3Na,.
О SO3Na
взаимодействие с аммиаком проходит при комнатной темпера-
температуре и атмосферном давлении.
105
§ 3. ПРОМЫШЛЕННЫЕ МЕТОДЫ ПРОВЕДЕНИЯ ПРОЦЕССА
ЩЕЛОЧНОГО ПЛАВЛЕНИЯ
Производство фенола. Одним из промышленных методов про-
производства фенола является щелочное плавление натриевой соли
бензолсульфокислоты:
C6H5SO3Na + 2NaOH > C6H6ONa + Na2SO3 + H2O.
При выдержке 30 мин и 15% избытке щелочи оптимальная
температура этого процесса 320—330 °С. В случае применения
Едкий натр
Сулыросоль
Пары воды и фенола
в скруббер
Конденсат
Промывная бода
после фильтро*
бани я сульфита
На фильтрование
Рис. 28. Схема процесса щелочного плавления.
/ — сборник 70% раствора NaOH; 2 — мерник раствора бензолсульфоната; 3 — котел
для плавления; 4 — печь; 5 — гаситель.
едкого кали температура щелочного плавления 280—285 °С, а
при использовании смеси 80% NaOH и 20% КОН — 300°С. Вы-
Выход фенола составляет около 96%.
Схема процесса щелочного плавления изображена на рис. 28.
Перед началом плавления загружают 70—75% раствор щелочи
и упаривают его. Окончательное упаривание воды до концен-
концентрации щелочи 80—85% производится непосредственно в пла-
плавильных котлах 3. В некоторых цехах фенола 40—42% щелочь
упаривают до концентрации 80—85% в отдельных котлах. Из
стальных мерников 2 в котел постепенно вводят раствор бен-
бензолсульфоната. Реакционная вода, образующаяся в процессе
плавления, испаряется с некоторым количеством фенола A—
1,5% общего количества). Пары воды, содержащие фенол, кон-
конденсируются в скрубберах, орошаемых водой, и образующийся
106
конденсат передают на выделение фенола. По окончании опера-
операции щелочной плав сливают в наполненный водой стальной га-
гаситель 5, снабженный мешалкой. Пары воды, выделяющиеся из
гасителя, содержат часть фенола и фенолята. Их конденсируют
и конденсат присоединяют к фенольным водам. Выход фенола
на стадии плавления — 96% от теоретического. При улавлива-
улавливании фенола из паров, выделяющихся в плавильном котле, выход
на стадии плавления можно повысить до 97—98%.
Выход на стадии гашения 99% от теоретического. При улав-
улавливании фенола из паров, выделяющихся из гасителя, выход
может достигать 99,5—99,8%.
В плаве, поступающем на гашение, содержится 30—34% фе-
фенолята натрия, 41—44% сульфита натрия и 2,5—3,5% едкого
натра. При гашении в гасителе фенолят натрия и избыток
едкого натра растворяются в воде, а сульфит остается в осадке.
Гашение и последующее фильтрование проводят при темпера-
температуре выше 40—50 °С, чтобы предотвратить образование кристал-
кристаллов водного сульфита натрия.
Суспензию сульфита в растворе фенолята подают из гаси-
гасителей в приемник с поднимающейся мешалкой. После декан-
декантации раствор фенолята сливают в сборники. Затем мешалку
опускают и размешивают сульфит. Размешиваемая суспензия
поступает на горизонтальные полуавтоматические центрифуги.
Отфильтрованный сульфит промывают и выгружают. Раствор
фенолята дополнительно отстаивается в отстойнике. Промыв-
Промывные воды из сборника направляют в гаситель,
используя их для гашения вместо воды.
Отходный сульфит, содержащий 81—83% сульфита натрия,
применяется без очистки в кожевенной и целлюлозной промыш-
промышленности. Из раствора отходного сульфита в воде можно полу-
получить также безводный сульфит, отвечающий техническим тре-
требованиям, предъявляемым к фотореагенту. Очищенный безвод-
безводный сульфит используется для получения ЫагБгОз.
Разложение раствора фенолята натрия осуществляется по
непрерывной схеме. Раствор фенолята натрия поступает в
стальной футерованный разлагатель со змеевиком и мешалкой.
Через этот аппарат барботирует сернистый газ, образующийся
при нейтрализации. Проходя через слой раствора фенолята
натрия, SO2 нейтрализует фенолят, при этом образуются суль-
сульфит натрия и фенол.
Очистку сырого фенола осуществляют перегонкой в вакууме.
При этом выделяются следующие фракции: фенольная вода, со-
содержащая до 8% фенола, жидкий фенол, содержащий до 15%
воды, товарный фенол, первичная смола.
Перевод на непрерывный процесс стадий разложения фено-
фенолята и перегонки фенола повышает производительность обору-
оборудования и снижает расходы на амортизацию и обслуживание.
При непрерывной перегонке, кроме того, удается уменьшить ко-
107
личество образующегося жидкого фенола и обеспечить более
полное извлечение фенола из первичной смолы. Организация
непрерывного процесса щелочного плавления связана с боль-
большими трудностями. Эти трудности обусловлены высокой темпе-
температурой реакции, большой вязкостью и легкой окисляемостью
плава, а также трудностью подвода тепла к реакционной массе.
Производство резорцина. Метод промышленного производст-
производства резорцина зависит от объема производства. При малом
объеме производства однородную порошкообразную смесь из
м-дисульфоната бензола и едкого натра засыпают в чугунные
трубки, имеющие дно на одном конце и плотную крышку на
другом. Эти трубки помещают в печь, где поддерживается тем-
температура 320—350 °С. Благодаря небольшой толщине слоя ре-
реакционной массы отпадает необходимость в ее перемешивании.
Видоизменением этого метода является запекание реакционной
смеси в тонком слое на противне. Для предохранения плава от
окисления запекание проводится при непрерывном пропускании
над противнями водяного пара (под «паровой подушкой»).
Однако пр!и больших масштабах производства проведение
процесса описанным выше способом нерационально. Щелочное
плавление в этом случае проводят в стальном литом котле ем-
емкостью около 1 м3. Котел снабжен мощной F4 кВт) лопастной
мешалкой, укрепленной в подпятнике и вращающейся со ско-
скоростью 20 об/мин. Выгрузка плава производится через широкое
выгрузочное отверстие в днище котла. Обогрев котла электри-
электрический. Особая трудность аппаратурного оформления процесса
получения резорцина заключается в том, что в процессе щелоч-
щелочного плавления несколько раз меняется консистенция реакцион-
реакционной массы. Загружаемая в аппарат порошкообразная смесь при
210—220 °С превращается в вязкую пластичную массу, которая
разжижается при температуре выше 220 °С; при температуре
290 °С масса снова густеет и снова превращается в порошко-
порошкообразный твердый продукт, который при 310 °С превращается
в тестообразную массу, а при 340 °С — затвердевает. Эти изме-
изменения вязкости реакционной массы обусловлены протеканием
процесса в несколько стадий. Вначале замещается одна сульфо-
группа и из реакционной массы отгоняется выделившаяся вода,
а затем происходит замещение второй сульфогруппы и отгонка
образовавшейся воды:
SO3Na SO3Na
+ 2NaOH >■ Г |[ + Na2SO3 + Н2О.
SO3Na ^^
SO3Na ONa
2NaOH > | II + NaaSO3 + H2O.
•ONa
108
Резорцин, в отличие от фенола, хорошо растворим в воде и
в водных растворах солей, а потому не осаждается при подкис-
лении растворенного в воде плава. В связи с этим после отде-
отделения сульфита из нейтрализованного раствора плава резорцин
извлекают экстракцией органическими растворителями.
§ 4. ОБМЕН ХЛОРА НА ОКСИ- И АМИНОГРУППУ
Атом хлора в ароматических соединениях можно заместить
на различные группы атомов.
Впервые возможность замены хлора на другие заместители
была открыта в 1870 г. русскими учеными А. Н. Энгельгардтом
и П. А. Лачиновым, осуществившими синтез динитроанилина и
динитрофенола из динитрохлорбензола, а также п-нитроанили-
на и п-нитрофенола из п-нитрохлорбензола. Впоследствии было
найдено, что хлор может быть замещен на другие группы не
только в хлорнитросоединениях, но и в других ароматических
хлорпроизводных. Число различных заместителей, на которые
может быть замещен хлор в ароматическом ядре, довольчо ве-
велико. Однако важнейшими из них являются окси- и аминогруп-
аминогруппа, а также производные этих групп. В общем виде реакция за-
замены хлора на другие заместители может быть изображена
следующим образам:
RC1 + НХ > RX + НС1,
где Х= -ОН, -OAlk, -NH2, -NHR и т. д.
В некоторых случаях, с целью связывания хлористого водо-
водорода, в реакцию предпочитают вводить вместо реагента НХ со-
соответствующее производное щелочного или щелочноземельного
металла МеХ:
RC1 + МеХ >■ RX + MeCl.
Реакции замены хлора относятся к реакциям нуклеофильно-
го замещения. Однако рассмотренный в начале этой главы
классический механизм нуклеофильного замещения в данном
случае не является главным.
Исследование реакций замещения галоида в ароматическом
ядре с помощью меченого углерода показало, что замещающая
группа становится не только к тому атому, с которым был свя-
связан галоид, но в равной степени и к соседнему атому углерода:
ОН
50%
109
Так, при реакции между хлорбензолом, где хлор стоял у ме-
меченого углерода 14С, и щелочью, а также между хлорбензолом
и амидом калия получились почти в равном количестве продук-
продукты, где меченый атом углерода был у заместителя и в о-поло-
жении к нему. Эти результаты лучше всего объясняются меха-
механизмом, по которому замещение протекает через стадию
образования промежуточного соединения с тройной связью на-
называемого «дегидробензолом». Образуется дегидробензол в ре-
результате реакции отщепления.
N11.,
дегидробензол
Независимо от того, по какому механизму протекает про-
процесс замещения хлора, он требует большой затраты энергии, по-
поэтому реакции нуклеофильного замещения при отсутствии в яд-
ядре активирующих групп протекают в очень жестких условиях.
Сильные акцепторы электронов, к которым относится большая
часть заместителей второго рода, способны создавать в арома-
ароматическом ядре центры, бедные электронами. Этот эффект будет
проявляться особенно ярко в тех случаях, когда атом, несущий
+8 заряд, связан с замещающимся атомом хлора. Например,
влияние нитрогруппы может быть показано схемами:
- ^ ел
-Ъ CI
Аналогичная картина будет наблюдаться при наличии в
о- и п-местах по отношению к хлору других заместителей вто-
второго рода (-SO3H, -COOR и т. п.).
Наличие заместителей второго рода в о- и п-положениях к
хлору не только увеличивает подвижность хлора за счет силь-
сильной поляризации связи С—С1, но и увеличивает способность
молекулы в целом к реакциям нуклеофильного замещения как
110
за счет наличия +6 заряда на реакционноспособном атоме угле-
углерода, так и за счет большей устойчивости промежуточного от-
отрицательно заряженного соединения. Очевидно, что при этом
реакция в большей мере будет протекать по классическому ме-
механизму нуклеофильного замещения.
Подвижность хлора в ядре значительно возрастает при уве-
увеличении числа заместителей второго рода в о- и п-положении к
галоиду. Так, замена хлора на оксигруппу в хлорбензоле про-
протекает лишь при температуре 350—400 °С и давлении выше
300 ат под действием 15% раствора щелочи. При наличии в ядре
одной нитрогруппы в о- или п-положении к хлору позволяет
снизить температуру реакции до 130—160°С, а давление до
2—6 ат. 2,4-Динитрохлбрбензол превращается в 2,4-динитрофе-
нол при нагревании с раствором едкого натра или даже соды
при обычном давлении, а 2,4,6-тринитрохлорбензол обменивает
хлор на гидроксил при нагревании с водой.
Влияние различных заместителей второго рода на подвиж-
подвижность хлора неравноценно. Влияние групп -NO2, -SO3H,,
-СООН, расположенных в о- и п-тюложениях к галоиду, опре-
определяется рядом: NO2>SO3H>COOH. Значительно увеличи-
увеличивает подвижность хлора в ароматическом ядре находящаяся в о-
и п-положении к нему —№R3-rpynna.
Заместители, уменьшающие положительный заряд связанно-
связанного с хлором атома углерода, то есть большая часть заместите-
заместителей первого рода при нахождении их в о- и п-положении к
галоиду, затрудняют замещение хлора.
Реакции обмена хлора на другие заместители первого рода
активируются соединениями меди и мышьяка.
Замещение хлора в боковой цепи протекает значительно лег-
легче, чем в ядре (при отсутствии активации). Реакция в этом слу-
случае идет по ионному механизму:
медленно
R-CH2-C1 > R—СН"£ + С1-;
быстро
R-CHt + X- ►■ R-CH2—X,
При переходе от ароматических углеводородов к их гетероцик-
гетероциклическим аналогам — пиридину и хинолину — нуклеофильная
подвижность галоида повышается, так как гетероатомы в этом
случае проявляют электроноакцепторное действие. Например,
в 2- и 4-хлорпиридинах атом хлора значительно подвижнее, чем
в хлорбензоле:
111
4-Галогенпиридины активнее 2-галогензамещенных. Более низ-
низкая реакционная способность 3-изомеров может быть иллюстри-
иллюстрирована примером замещения брома в 3-бромпиридине под дей-
действием аммиака. В этом случае аминирование удается осущест-
осуществить только в присутствии медных солей, как и в аналогичной
реакции с хлорбензолом:
CH3ONa
Вг
N
С1
NH3;
220 °С
CfjH3SH
100 °С
88%
93%
Х'инол'шш, замещенные галогеном в положениях 3,5,6,7,8,
являются устойчивыми соединениями, обладающими типичной
для галогенпроизводных бензола реакционной способностью по
отношению к нуклеофильным агентам. В отличие от них 2- и
4-галогенхинолины легко вступают в реакции замещения:
н2о
120 °С
о
С1
н
SO3Na
NaHSO3
N
N
За исключением небольшого числа веществ с подвижным
атомом хлора, замена галоида на другие заместители происхо-
происходит при температуре выше 100 °С. Поскольку эти процессы, как
правило, проводят в водных или спиртовых растворах, для до-
достижения необходимой температуры приходится вести реакцию
под давлением. При проведении этих процессов по непрерывно-
непрерывному методу используют аппараты змеевикового типа («трубчат-
(«трубчатки»). При периодическом методе используют автоклавы.
К автоклавам относят реакторы, работающие под дав-
давлением свыше 6 ат. Они представляют собой изготовленные из
легированной стали котлы со сферическими днищами и крышка-
ми. Поскольку автоклавы являются дорогостоящей аппаратурой,
защита их от коррозии особенно важна. С целью защиты от
коррозии- в корпус автоклава вставляют вкладыш, изготовлен-
изготовленный из металла, устойчивого к коррозии в условиях производст-
производства. Зазор между вкладышем и телом автоклава A0—20 мм)
заполняют свинцово-сурьмяным сплавом.
Рис. 29. Литой автоклав с чугунным Рис. 30. Автоклав с храмоникелевым
эмалированным вкладышем.
/ — гильза для термометра; 2 — труба для
передавливания; 3 — крышка; 4 — мешал-
мешалка; 5 — вкладыш; 6 — корпус.
вкладышем.
/ — крышка, облицованная нержавеющей
сталью; 2 — корпус; 3 — вкладыш из нер-
нержавеющей стали; 4 — паровая рубашка;
5 — труба для передавливания; 6 — вал с
мешалками.
В автоклавах, рассчитанных на работу под высоким давле-
давлением, устанавливают вкладыши, изготовленные из высоколе-
высоколегированных сталей и сплавов. При работе под низким давлением
можно использовать чугунный эмалированный вкладыш.
При проведении процесса замены галоида в ядре, как пра-
правило, требуется хорошее перемешивание реакционной массы,
поэтому автоклавы снабжаются механическими мешалками.
Для сохранения герметичности аппарата при большой скорости
вращения вала мешалки обычный одинарный неохлаждаемый
сальник непригоден. В этом случае используют двойные саль-
8-926
113
ники с охлаждением. По принципу устройства двойной сальник
может рассматриваться как два последовательно соединенных
простых сальника, причем нажимная втулка нижнего сальника
одновременно является корпусом верхнего. В качестве уплот-
уплотняющих прокладок в автоклавах низкого давления используют
паранит, а в автоклавах сред-
среднего и высокого давления —
медь, алюминий и его сплавы
и реже свинец.
Разновидностей конструк-
конструкций автоклавов имеется много.
Остановимся на некоторых из
них.
На рис. 29 представлен ли-
литой автоклав с чугунным эма-
эмалированным вкладышем емко-
емкостью 3 м3. Аппарат снабжен
трубой для передавливания ре-
реакционной массы, гильзой для
термометра или термопары и
пропеллерной мешалкой. Кор-
Корпус заглубленного сальника
имеет рубашку для охлажде-
охлаждения водой.
Автоклав рассматриваемой
конструкции обогревается то-
топочными газами. Возможно ис-
использование также электро-
электрообогрева.
В производстве п-нитроани-
лина из п-нитрохлорбензола
используется автоклав с хро-
моникелевым вкладышем и
двумя пропеллерными мешал-
мешалками, расположенными на од-
одном валу (рис. 30). Автоклав
имеет рубашку, предназначен-
предназначенную для обогрева паром высокого давления. На крышке аппа-
аппарата имеется люк для осмотра автоклава. Штуцеры в крышке и
крышка облицованы хромоникелевой сталью.
В некоторых случаях автоклавы изготавливают из легиро-
легированных сталей, отличающихся не только прочностью, но и зна-
значительной устойчивостью к коррозии. Такие автоклавы несколь-
несколько дороже, но значительно более удобны в эксплуатации, так
как не требуют какой-либо дополнительной защиты от коррозии.
Примером такого реактора служит автоклав с байонетовым за-
затвором (емкость 3,3 м3, рабочее давление 50 ат), изготовленный
из хромоникельмолибденовой стали (рис. 31). Эта сталь устой-
114
Рис. 31. Автоклав с байонетньш за-
затвором.
/ — крышка с байонетньш затвором
2 — рубашка; 3 — корпус; 4 — мешалка.
чива к коррозии под действием хлорида аммония, обладает
хорошими механическими свойствами и окалиностойкостью.
Автоклавы этого типа удобно использовать при проведении
процессов замены хлора на аминогруппу под действием ам-
аммиака.
Большой интерес представляет байонетовый затвор, образо-
образованный приливами на корпусе и выступами на крышке автокла-
автоклава. При повороте крышки вы-
выступы крышки заходят под
приливы корпуса и образуют
надежное крепление. Уплотне-
Уплотнение между корпусом и крыш-
крышкой создается с помощью коль-
кольца Т-образного сечения. Для
ремонта мешалки и осмотра
автоклава в крышке имеется
овальный люк.
В случае обогрева топочны-
топочными газами удобно использо-
использовать автоклавы бутылочного
типа (рис. 32). Такие автокла-
автоклавы отливаются из высоколеги-
высоколегированных сталей, обладающих
жаростойкостью и сопротивле-
сопротивлением к деформации в горячем
состоянии. Кроме того, эти ста-
стали должны сохранять постоян-
постоянную структуру металла при
многолетней эксплуатации в
жестких условиях. К материа-
материалу, из которого изготавливают
автоклавы бутылочного типа,
предъявляются также требова-
требования высокой устойчивости к
химической коррозии, поскольку форма автоклава затрудняет
размещение в нем вкладыша. Благодаря тому что в автоклаве
бутылочного типа крышка имеет значительно меньшую поверх-
поверхность, чем в рассмотренных ранее конструкциях, облегчается
герметизация реактора.
При непрерывном методе производства применяются аппа-
аппараты трубчатого типа, работающие по принципу полного вытес-
вытеснения.
Реакторы могут быть выполнены в виде змеевиков либо
смонтированы из прямых отрезков цельнотянутых стальных
труб, соединенных друг с другом ретурбендами при помощи
фланцев и болтов, с линзовым уплотнением соединений. Прин-
Принципиальная схема проведения процессов замены галоида в аппа-
аппаратах такого типа приведена на рис. 33.
Рис. 32. Автоклав бутылочного типа.
/_ крышка; 2 — вал с мешалкой; 3 — кор-
корпус; 4 — труба для передавливания.
8*
115
Обмен хлора на аминогруппу. Хлор в ароматическом ядре
обменивается на аминогруппу под действием аммиака. Реакция
протекает легко, если хлор активирован заместителями второго
рода, расположенными в о- и п-положениях. Так, 2,4-динитро-
Рис. 33. Принципиальная схема проведения процессов под давлением в труб-
трубчатом реакторе.
1 — напорный бачок; 2— регулятор расхода; 3—насос; 4— змеевиковый подогреватель;
5 — трубчатый реактор; 6 — дроссельные вентили.
анилин может быть получен из 2,4-динитрохлорбензола дейст-
действием раствора аммиака при температуре 115—120 °С:
Cl NH2
I NO2
• 2NH3 > I If f NH4C1.
I
NO2 NO2
п-Нитроанилин может быть получен из п-нитрохлорбензола при
температуре 170—190 °С.
Cl NH2
+2NH3
NH4C1.
NO2 NO2
В том случае, когда галоид не находится под влиянием
сильных активирующих групп, взаимодействие с аммиаком про-
протекает в гораздо более жестких условиях.
Для снижения температуры реакции, ускорения процесса и
увеличения выхода амина используют катализаторы. Чаще все-
всего с этой целью применяют соединения меди.
116
Образование анилина из хлорбензола протекает в присутст-
присутствии соединений одновалентной меди при температуре около
200 °С и давлении 60—100 ат:
NH«
+ NH4C1.
В качестве побочного продукта образуется дифениламин:
C6H5NH2 + C1C6H5 » C6H5NHC6H5 + HC1.
Если на каждый моль хлорбензола брать 5 молей 34% рас-
раствора аммиака и 0,2 моля закиси меди, то при температуре
200—230 °С и давлении 70 ат реакция заканчивается за 3 ч.
Выход анилина достигает 89—90% от теоретического. В каче-
качестве побочных продуктов образуются 4—5% фенола и около 2%
дифениламина. В сходных условиях из п-дихлорбензола можно
получить п-фенилендиамин с выходом 75%:
Cl NH2
J
2NH4C1.
Действием аммиака на амид п-хлорбензолсульфокислоты или
непосредственно на п-хлорбензолсульфохлорид можно получить
важный лекарственный препарат белый стрептоцид (амид суль-
фаниловой кислоты).
4NH3 > fj + 2NH4C1.
SO2NH2
В 2,5-дихлорнитробензоле активирован лишь один атом хло-
хлора в орто-положении к нитрогруппе, поэтому при действии на
2,5-дихлорнитробензол аммиака обменивается на аминогруппу
только один атом хлора:
С1 Ш2
1 NO2 .1 NO2
+ 2NH3 > Г J +NH4C1.
Cl Cl
117
Аналогично ведут себя и другие полигалоидные соединения,
у которых лишь один атом хлора находится в сопряжении с за-
заместителем второго рода:
При проведении реакций аминирования хлорпроизводных на
1 моль исходного соединения берут обычно от 6 до 18 молей
водного раствора аммиака.
Обмен хлора на окси- и алкоксигруппы. Замена хлора на
оксигруппу в молекуле хлорбензола протекает в жестких усло-
условиях. Процесс этот, однако, имеет огромное промышленное зна-
значение, так как более половины фенола, получаемого в Советском
Союзе, производится из хлорбензола.
Без катализатора процесс может быть осуществлен под дей-
действием 15—20% раствора едкого натра на хлорбензол при тем-
температуре 360—400 °С и давлении свыше 300 ат. В столь жестких
условиях сталь корродирует под действием щелочи, поэтому ре-
реактор защищают от коррозии никелевой обкладкой. Процесс
можно провести и при нормальном давлении, если пропускать
смесь паров воды и хлорбензола через нагретый до высокой
температуры катализатор
С6Н5С1 + Н2О > С6Н5ОН + НС1.
Д. В. Тищенко установил, что оптимальная температура гид-
гидролиза 550—600 °С. Наилучшие результаты получаются при ис-
использовании в качестве катализатора хлористого магния, нане-
нанесенного на силикагель и активированного соединениями меди.
Активность катализатора снижается довольно быстро. Регенера-
Регенерация его проводится нагреванием в токе воздуха при 600—700 °С.
В присутствии катализатора процесс можно осуществить в
более мягких условиях и в жидкой фазе (под давлением). Так,
если вкладыш автоклава сделать из меди, то процесс получения
фенола из хлорбензола можно провести при 300 °С с 2,5 моля
NaOH на 1 моль хлорбензола. В этих условиях за 10 ч дости-
достигается превращение хлорбензола в фенол на 92%. При отсутст-
отсутствии катализатора реакция в этих условиях проходит лишь на
35—39%.
При интенсивном перемешивании реакционной массы и высо-
высокой температуре C50—370 °С, 200 ат) процесс сильно ускоряет-
ускоряется. Реакция в этом случае проходит за несколько минут. Это
П8
создает предпосылки для непрерывного ведения процесса полу-
получения фенола из хлорбензола в трубчатом реакторе из меди или
из стальных труб, выложенных медью.
До 1927 г. фенол получали только по сульфурацион-ному спо-
способу:
C6H6 + H2SO4 > C6H5SO3H + H2O;
2C6H5SO3H + Na2SO3 >■ 2C6H6SO3Na + SO2 + H2O;
C6H5SO3Na + 2NaOH ^ C6H5ONa + Na2SO3 + H2O;
2CeH6ONa + SO2 + H2O > C6H5OH + Na2SO3.
В 1927 г. впервые в промышленном масштабе был осущест-
осуществлен синтез фенола через хлорбензол (способ Дау):
С6Н6 + С12 > С6Н5С1 + НС1
С6Н5С1 + 2NaOH >- C6H6ONa + NaCl + Н2О
C5H6ONa + HCl > C6H5OH + NaCl.
Несколько позднее был разработан способ профазного гид-
гидролиза хлорбензола. Этот способ оказалось очень выгодным
соединить с методом окислительного хлорирования бензола:
2С6Н6 + 2НС1 + О2 > 2С6Н5С1 + 2Н2О
С6НбС1 + Н2О ^ С6Н5ОН + НС1.
Именно этим способом перерабатывают хлорбензол в фенол в
Советском Союзе.
Наиболее перспективным является разработанный в Совет-
Советском Союзе П. Г. Сергеевым, Б. Д. Кружаловым и Р. Ю Удри-
сом кумольный способ синтеза фенола:
С6Н6 + СН2=СН-СН3 > С6Н5-СН(СН3J;
С6Н5-СН(СН3J + О2 > С6Н5-С(СН3JООН;
С6Н5-С(СН3JООН у С6Н5ОН + (СН3JСО.
В СССР до 1949 г. фенол получали только по сульфураци-
онному способу (щелочным плавлением натриевой соли бензол-
сульфокислоты). В 1949 г. в СССР был пущен первый в мире
цех синтеза фенола кумольным методом.
Проведенное сравнение технико-экономических показателей
различных способов получения фенола показало, что организо-
организовывать производство п© способу Дау нерационально, так как
расходуемый по этому способу едкий натр превращается в ре-
результате процесса в поваренную соль, утилизировать которую
невозможно. Что касается способа парофазного гидролиза
хлорбензола, то он был положен в основу при строительстве це-
цеха по производству фенола в СССР- Получаемый по этому спо-
способу фенол имеет себестоимость несколько ниже получаемого
по сульфурационному способу, однако капитальные вложения
на строительство цеха по получению фенола парофазным гид-
гидролизом хлорбензола на 85% выше, чем на строительство цеха
119
той же мощности по сульфурационному способу производства
фенола.
Получение фенола через хлорбензол осуществлено в Совет-
Советском Союзе по следующей схеме. Воздух и пары бензола на-
нагреваются в перегревателе до 270—300 °С, смешиваются с па-
парами 20% соляной кислоты и подаются в конвертор хлорирова-
хлорирования. В конверторе хлорирования около 10% бензола превра-
превращается в хлорбензол. Хлорбензол конденсируется и подается
на разгонку, а бензол возвращается в цикл. Затем пары хлор-
хлорбензола и воды перегревают до температуры 420 °С и пропус-
пропускают через конвертор гидролиза, где 10% хлорбензола превра-
превращается в фенол. Непрореагировавший хлорбензол улавливают
и возвращают на стадию приготовления парофазной смеси для
гидролиза, а хлористый водород улавливают и направляют на
стадию окислительного хлорирования бензола.
В полихлорпроизводных бензола при действии на них спир-
спиртовой щелочи в автоклаве можно заместить лишь, один атом
хлора. Таким образом могут быть получены п-хлорфенол
01 СНзОН.КаОН
190—195 °С, давление
С1
и пентахлорфенол
С1 ОН
V
ci
130-140 °С, давление J
a
Пентахлорфенол является антисептиком, а п-хлорфенол ис-
используется как сырье для многих органических синтезов.
В ряде случаев при действии на хлорпроизводные аромати-
ароматических соединений спиртовыми растворами щелочей образуются
не окси-, а алкоксипроизводные:
RC1 + HOAlk + NaOH >- ROAlk + NaCl + H2O.
Особенно легко в эту реакцию вступают нитрохлорпроиз-
водные.
Для успешного проведения этого процесса надо использо-
использовать спиртовые растворы щелочей, не содержащие воды. Еще
лучшие результаты получаются при использовании спиртовых
растворов алкоголятов. Замена хлора на алкоксигруппу в аро-
ароматических хлорнитросоединениях принадлежит к числу про-
процессов, требующих самого тщательного соблюдения условий
ведения реакции. При отклонении от оптимальных условий ре-
реакция осложняется побочными процессами. Важными вещест-
веществами, получаемыми этим способом, являются нитроанизолы.
120
Нитроанизолы получаются при нагревании в автоклаве при
105—110°С соответствующих нитрохлорбензолов с 80—90% ме-
метиловым спиртом и щелочью в течение 4—5 ч.
С1 ОСН3
1 .NO2 J\/m*
+ СН3ОН + NaOH > |Т + NaCl + H2O.
По окончании процесса избыток щелочи нейтрализуют сер-
серной кислотой, спирт отгоняют, а сырой нитроанизол промывают
сначала разбавленным раствором щелочи, а потом водой для
отделения побочно образующегося хлорфенола. Технический
продукт перегоняют с водным паром. о-Нитроанизол служит
для получения о-анизидина, а последний используется для син-
синтеза гваякола и лекарственных препаратов на его основе. Ана-
Аналогично из п-нитрохлорбензола и этилового спирта можно полу-
получить п-нитрофенетол, служащий сырьем для синтеза важного
лекарственного препарата — фенацетина.
Замещением хлора на метоксигруппу получают сульфанила-
сульфаниламид— сульфален:
N п N
СН3ОН
Г/—^
NHSO
2-/~\-
§ 5. ВЗАИМНЫЕ ПРЕВРАЩЕНИЯ АМИНО-
И ОКСИСОЕДИНЕНИЙ
Превращения аминов в оксисоединения и обратные процес-
процессы могут быть осуществлены различными способами и с раз-
различной легкостью в зависимости от строения и свойств этих
соединений. При наличии таких способов синтеза аминосоеди-
кения можно было бы получать, исходя из сульфокислот, а ок-
сипроизводные — из нитропродуктов:
RNO2 >• RNH2 -<—> ROH < RSO3H.
Превращение аминов в оксипроизводные. Превращение ами-
аминогруппы в оксигруппу может быть осуществлено тремя основ-
основными способами:
а) диазотированием аминогруппы и последующим разложе-
разложением образовавшегося диазосоединения:
NaNO2, H2SO4 H2O
RNH2 у- RN2HSO5 >- ROH + N2 + HSO4;
б) кислотным гидролизом амина:
H2SO4
RNH2 + H2O =*==*: ROH+NH8;
- 121:
в) действием на амин солей сернистой кислоты:
RNHo
NaHSO3
Реакция диазотирования ароматических аминов и свойства
образующихся диазосоединений будут подробно рассмотрены
в главе 9. Диазосоединения являются очень неустойчивыми и
реакционноспособными соединениями. При нагревании в вод-
водных или водноспиртовых растворах они легко разлагаются с
образованием оксисоединений и выделением азота.
Практически более важным методом для ароматических сое-
соединений является кислотный гидролиз аминосоединений.
Гидролиз амина до фенола протекает только в достаточно
жестких условиях. В качестве гидролизующего средства пред-
предложено использовать водные растворы фосфорной кислоты.
Реакция идет при температуре 350 °С и давлении 200 ат.
Н3РО4
C6H5NH2 + H2O >• C6H5OH + NH3.
Амины нафталинового ряда гидролизуются значительно
легче. В нафтиламин- и аминонафтолсульфокислотах амино-
аминогруппа практически полностью гидролизуется при нагревании
исходного продукта с 3% раствором серной кислоты в автокла-
автоклаве в течение 2 ч при температуре 180—190 °С.
При использовании в качестве гидролизующих агентов рас-
твора бисульфита гидролиз аминогруппы проходит при темпе-
температуре около 100°С. Это позволяет использовать для проведе-
проведения реакции обычные аппараты вместо автоклавов. Из произ-
производных бензола к Этой реакции способными оказались лишь
м-диамино- и м-оксиаминопроизводные.
В нафталиновом ряду обработка бисульфитом позволяет пе-
переводить в оксипроизводные как а-, так и р-нафтиламин. Не
поддаются переработке этим методом лишь те сульфокислоты
нафтиламинов, у которых сульфо- и аминогруппы находятся в
метаположении друг к другу. Процесс проводят при кипячении
аминосоединений с раствором бисульфита, который берут в
большом избытке, в реакторе, снабженном обратным холодиль-
холодильником. Реакция протекает через стадию образования бисульфит-
ного производного:
NH2
NH
NaHSO3
H
NaOH
NaO3S'
^-SO3Na nhT
NaO3S'
Ю.Н
122
При проведении реакции необходимо контролировать содер-
содержание исходного амина в реакционной массе. После исчезнове-
исчезновения амина добавляют щелочь для разложения промежуточного
соединения и кипятят до прекращения выделения аммиака. За-
Затем реакционную массу подкисляют минеральной кислотой и
кипятят до прекращения выделения SO2.
Продукт выпадает после охлаждения раствора или после
дополнительного высаливания.
Аминирование оксисоединений. Оксигруппу можно заменить
на аминогруппу действием аммиака или солей аммония. Ввиду
летучести аммиака процесс проводят под давлением в авто-
автоклавах.
Наибольшую реакционную способность при аминировании
обнаруживают те фенолы, которые способны существовать в
двух таутомерных формах — кетоформе и енольной форме.
Примером может служить нитрозофенол, который превращается
в соответствующее аминопроизводное уже при нагревании с
растворами аммонийных солей на водяной бане:
.он
Незамещенный фенол превращается в анилин лишь при воз-
воздействии аммиаката хлорида цинка (ZnCl2-2NH3) и хлорида-
аммония при 330 °С. Выход анилина в этих условиях составля-
составляет около 44%. В качестве побочного продукта образуется дифе-
дифениламин.
Флороглюцин переходит в 5-аминорезорцин при кохмнатной
температуре:
ОН ОН
HoN
Практически в бензольном ряду замена оксигруппы на ами-
аминогруппу используется редко. Большое значение эта реакция
приобрела в нафталиновом ряду, где она протекает значитель-
значительно легче.
Использование возможности замены оксигруппы на амино-
аминогруппу в нафталиновом ряду привело к разработке и промыш-
ному осуществлению синтеза многочисленных производных
Р-нафтиламина, чему особенно способствовала доступность.
Р-нафтола и его производных.
123,
Аминирование нафтолов и их замещенных гладко протека-
протекает при нагревании с аммиаком в присутствии хлористого каль-
кальция.
Примером такого синтеза может служить получение 2-ами-
но-3-нафтойной кислоты из 2-окси-З-нафтойной кислоты:
-соон у
Получение аминонафтойной кислоты проводят при нагрева-
нагревании БОН-кислоты в автоклаве с водным раствором аммиака
и хлоридом цинка при температуре 200 °С.
Однако большое значение для осуществления замены окси-
группы на аминогруппу получила реакция Бухерера. Реакция
Бухерера протекает при нагревании нафтолов с водными рас-
растворами сульфита и аммиака. Эта реакция является обратной
бисульфитному гидролизу аминов и может быть направлена
в ту или другую сторону в зависимости от условий.
Границы применимости сульфитной реакции аминирования
в нафталиновом ряду те же, что и для бисульфитной реакции
гидролиза. Если сульфогруппа находится в мета-положении к
гплроксилу, то последний в реакцию аминирования не всту-
вступает:
ОН ОН
„ОН
HO3S
Не обменивается на аминогруппу также а-расположенный
гидроксил, если в о-положении к нему имеется сульфогруппа.
§ 6. ТЕХНИКА БЕЗОПАСНОСТИ ПРИ ВЕДЕНИИ РЕАКЦИЙ
ОКСИЛИРОВАНИЯ И АМИНИРОВАНИЯ
При попадании на кожу едких щелочей возникают сильные
ожоги. Особенно сильно действуют щелочи на слизистую обо-
оболочку. Вдыхание пыли Na2SO3 и других солей вызывает раз-
раздражение дыхательных путей.
Многие фенолы и амины являются сильными нервными и
кровяными ядами.
Наиболее радикальным способом, предотвращающим сопри-
соприкосновение работающих с вредными веществами, является пол-
полная герметизация производственных процессов. Если отсутст-
отсутствует возможность комплексной автоматизации производства,
проводят механизацию трудоемких операций и автоматизацию
отдельных аппаратов, агрегатов и узлов. Чтобы избежать по-
потери сыпучих веществ, сырье перевозят в расплавленном виде
(в цистернах); продукты хранят в герметичных бункерах; при-
124
меняют пневмотранспорт. Чтобы предотвратить выделение вред-
вредных веществ при ручной загрузке сырья, загрузку производят
из контейнеров.
Вентиляционные установки должны обеспечивать в произ-
производственных помещениях 4—6-кратный обмен воздуха за 1 ч.
В отдельных помещениях, где размещаются аппараты для про-
проведения наиболее вредных операций, воздухообмен должен со-
составлять 8—12 объемов за 1 ч. Все аппараты, из которых воз-
возможно выделение вредных газов, оборудуются местной венти-
вентиляцией.
При проведении реакций замещения хлора в ароматическом
ядре процесс часто приходится вести под повышенным давле-
давлением в автоклавах. Автоклавное отделение должно быть изо-
изолировано от остальных капитальными стенами. Помещения, где
установлены автоклавы, должны иметь повышенный объем,
большую площадь окон и легко сбрасываемое покрытие.
Перед началом эксплуатации автоклава необходима самая
тщательная проверка всех его деталей. При положительном
результате осмотра аппарат подвергают гидравлическому испы-
испытанию на прочность в соответствии с «правилами устройства и
безопасной эксплуатации сосудов, работающих под давлением».
Согласно действующему законодательству каждый работник на
участке, где находится автоклав или другой аппарат, работа-
работающий под давлением, должен быть специально обучен. К рабо-
работе на автоклавах допускаются только лица, сдавшие экзамен
специальной комиссии и имеющие соответствующее удостовере-
удостоверение; каждый год проводится проверка их знаний. Все автокла-
автоклавы эксплуатируются в соответствии с правилами, утвержден-
утвержденными Госгортехнадзором. На каждый аппарат заводят специ-
специальную котельную книгу, куда заносят все данные об аппара-
аппарате и условиях его эксплуатации, а также сведения о проведен-
проведенных ремонтах и испытаниях.
Контроль за процессами в автоклавах проводят по темпера-
температуре и давлению. Для регулирования режима автоклавы обо-
оборудуются запорными вентилями, предохранительными клапана-
клапанами, манометрами, термометрами и т. п.
Манометр, установленный на автоклаве, должен быть обя-
обязательно дублирован. При несовпадении показаний обоих ма-
манометров аппарат не может быть допущен к эксплуатации до
проверки и замены неисправного прибора. Для наблюдения за
Давлением в процессах аминирования применяют специальные
аммиачные манометры, которые имеют устройство, предохраня-
предохраняющее от коррозии медные части прибора.
Предохранительные клапаны должны быть отрегулированы
таким образом, чтобы они срабатывали при повышении рабоче-
рабочего давления. Пружинные предохранительные клапаны устанав-
устанавливают на одном из штуцеров крышки автоклава в количестве
не менее двух штук. Выводами от предохранительных клапанов
125
служат специальные трубопроводы, выходящие из здания на-
наружу.
Для безопасной эксплуатации автоклавов важно, чтобы со-
соблюдалась определенная температурная зависимость между
давлением и температурой в аппарате. Если процесс будет про-
проходить ненормально и установленная зависимость не будет соб-
соблюдаться, надо немедленно приостановить нагрев, уменьшить
давление и выяснить причину несоответствия.
Большое значение имеет подготовка аппарата перед нача-
началом операции. Особое внимание следует обращать на герметич-
герметичность аппарата, правильность укладки прокладок и уплотне-
уплотнений, равномерную затяжку болтов. Если при работе автоклава
обнаруживается пропуск газа или подтекание жидкости, необ-
необходимо немедленно снять давление, а затем устранить неис-
неисправность. Подтягивание болтов или других крепящих устройств
на аппарате, находящемся под давлением, крайне опасно. От-
Открывать люк после передавливания содержимого аппарата или
спуска давления можно только после того, как работающий
убедится в полном отсутствии в аппарате избыточного давле-
давления (стрелки манометров должны стоять на нуле).
Помимо приведенных особенностей безопасной эксплуата-
эксплуатации аппаратов при проведении в них процессов обмена хлора
на другие заместители, существует ряд общих правил по тех-
технике безопасности, которые необходимо соблюдать при прове-
проведении любых химических процессов.
При проведении процессов аминирования и алкоксилирова-
ния следует иметь в виду, что аммиак и спирты образуют с воз-
воздухом взрыво- и пожароопасные смеси. Кроме того, аммиак
и спирты (особенно метиловый!) являются ядовитыми вещест-
веществами. Сильными ядами являются также многие нитросоедине-
ния, амины и хлорпроизводные, применяющиеся в качестве ис-
исходных веществ или получающиеся при проведении процессов
обмена хлора на амино-, оксигруппу и другие заместители.
Все работающие должны систематически знакомиться со
всей документацией, имеющей отношение к их работе. Хорошее
знание инструкций, правил и норм и точное их выполнение га-
гарантирует работающим безопасность их труда.
Гл ава7
АЛКИЛИРОВАНИЕ И АЦИЛИРОВАНИЕ
АМИНОВ И ОКСИСОЕДИНЕНИЙ
В химии и технологии лекарственных соединений большое
значение имеют реакции электрофильного замещения атома во-
водорода в амино- и оксигруппах. К этим реакциям в первую оче-
очередь относятся реакции алкилирования и ацилирования амино-
и оксисоединений. Помимо большого значения для промышлен-
промышленного и лабораторного синтеза, эти реакции играют большую
роль в жизнедеятельности организма.
Все указанные реакции проходят через промежуточную
стадию присоединения электрофильной частицы (Х+) к атому
азота аминогруппы или к атому кислорода оксигруппы с обра-
образованием в качестве промежуточного продукта аммониевого или
оксониевого 'иона:
/ /
RNH2 + Х+ > R—N-X >- R-N< + Н+
\н ЧХ
+ /Н -
ROH + Х+ >■ R—О< >■ R-OX + Н+.
Х
Чем основнее амин, тем активнее он будет вступать в реак-
реакции алкилирования и ацилирования. Поскольку ароматические
амины значительно менее основны, чем алифатические, алки-
лирование и ацилирование их протекает медленнее, чем али-
алифатических.
В неионизированном виде фенолы являются значительно ме-
менее нуклеофильными соединениями, чем амины, поэтому для по-
повышения их реакционной способности алкилирование и ацили-
ацилирование их предпочитают проводить в щелочной среде, где об-
образуются активные фенолятные ионы.
Реакции алкилирования и ацилирования в этой главе изла-
излагаются применительно к ароматическим амино- и оксисоедине-
ниям. Следует, однако, иметь в виду, что эти реакции примени-
применимы и для соответствующих алифатических соединений. Приме-
127
ры таких процессов приведены во второй части учебника, при
описании синтезов конкретных лекарственных веществ и вита-
витаминов.
§ 1. АЛКИЛИРОВАНИЕ АМИНОВ
Для алкилирования ароматических аминов применяются
спирты, галоидные алкилы, эфиры.
В общем виде реакция алкилирования аминов должна быть
изображена следующим образом:
RNH2 + AlkX > RNHAlk + НХ
RNHAlk + AlkX >- RN(AlkJ + HX,
где X — одновалентный заместитель, связанный с алкилом (на-
(например галоид), a R — ароматический остаток.
Таким образом, для реакции алкилирования характерно об-
образование как вторичного, так и третичного аминов, и в резуль-
результате алкилирования в большинстве случаев получается смесь
продуктов.
Реакция алкилирования принадлежит к числу реакций элек-
трофильного замещения, где в качестве электрофильного аген-
агента в случае мономолекулярного механизма выступает катион
Alk+. Однако известны случаи, когда алкилирование протекает
как реакция присоединения (например, с окисью этилена, с не-
непредельными соединениями).
Введение алкильных остатков в аминогруппу несколько уси-
усиливает ее электронодонорное влияние на ароматическое ядро.
Алкилирование ароматических аминов спир-
спиртами обычно проводят в присутствии минеральных кислот, из
которых чаще всего используют серную и соляную. Серную кис-
кислоту обычно загружают из расчета 0,05—0,3 моля на 1 моль
амина. Соляную кислоту добавляют в большем количестве, до-
доходящем иногда до 1 моля на 1 моль амина.
Каталитическая роль кислоты заключается в том, что она
протонирует спирт, в результате чего образуется активный ал-
+
коксониевый катион А1ЮН2, который легко дегидратируется,
давая карбониевый катион, вступающий в реакцию с аромати-
ароматическим амином:
AlkOH + Н+ ч—* AlkOH2
А1кОН2 <—> А1к+ + Н2О
RNH2 + А1к+ =<=> RNH2Alk > RNHAlk + Н+.
Спирт для алкилирования берется в избытке. При получе-
получении третичных аминов этот избыток больше (до 160% от тео-
теоретического), при получении вторичных — меньше.
Алкилирование спиртами проводят в автоклавах под давле-
давлением выше 30 ат и температуре 180—220 °С. Например, диме-
128
тиланилин получают при" нагревании 3,2 моля метилового спир-
спирта и 0,1 моля серной кислоты на каждый моль анилина при
температуре 205—215 °С и давлении около 30 ат в течение 6 ч:
,NH2 ^N^NtCHJ,
+ 2СН3ОН >• |Т + Н2О
В качестве побочного продукта образуется некоторое коли-
количество соли четвертичного аммониевого основания, для разло-
разложения которого реакционную массу нагревают в автоклаве с
раствором едкого натра:
N(
Диэтиланилин получают нагреванием солянокислого анили-
анилина с тремя молями этилового спирта под давлением при темпе-
температуре 180—200 °С. Однако при этом получается смесь продук-
продуктов, содержащая значительное количество моноэтиланилина.
В присутствии бромистоводородной кислоты процесс приво-
приводит к образованию диэтиланилина. Природа минеральной кис-
кислоты заметно влияет на скорость протекания реакции.
Алкилирование ароматических аминов спиртами может про-
протекать также и в паровой фазе при температуре 300—400 °С
при использовании в качестве катализатора окиси алюминия.
Алкилирование простыми эфирами осуществля-
осуществляют пропусканием смеси паров амина и эфира при температуре
250—350 °С через катализатор (А12О3, ThO2, TiO2, ZrO2).
Большой практический интерес представляет использование
в качестве метилирующего средства метилового эфира, так как
последний получается в качестве отхода при производстве ме-
метилового спирта:
АЬО3
CeH6NH2 + 2(CH3JO 230_295O^ C6H5N(CH3J + 2CH3OH.
При промышленном осуществлении этого процесса избыток
паров метилового эфира смешивают в испарителе с парами
анилина. Смесь паров поступает в контактный аппарат трубча-
трубчатого типа, где на 94—96% превращается в диметиланилин.
После отделения метанола смесь аминов с метиловым эфиром
поступает во второй контактный аппарат, после которого сте-
степень превращения анилина в диметиланилин достигает 99,5—
99,6% от теоретического. Общий выход диметиланилина с уче-
учетом потерь на других стадиях производства составляет 97,6%.
В качестве катализатора используется активная окись алюми-
алюминия. Катализатор работает без замены 5 лет. Этого удалось
Достичь благодаря применению испарителя с циркуляцией ани-
анилина при неполном его испарении. Установка производитель-
9-926 129
ностью 5000 т диметиланилина в год автоматизирована и об-
обслуживается всего двумя рабочими в смену. Коррозия в произ-
производстве диметиланилина парофазным методом практически от-
отсутствует, а потому вся аппаратура выполнена из обычной угле-
углеродистой стали.
Алкилирование сложными эфирами, например
диметилсульфатом (CH3JSC>4, и эфирами ароматических суль-
фокислот проходит по схеме:
RNH2 + CH3OSO2OCH3 >■ RNHCH3 + CH3OSO2OH;
RNH2 + CH3OSO3Na >• RNHCH3 + NaHSO4;
RNH2 + AlkOSO2R' >- RNHAlk + R'SO3H.
Аналогично могут быть получены соответствующие третич-
третичные амины.
Диметилсульфат широко используется -в химико-фармацев-
химико-фармацевтической промышленности в качестве метилирующего агента
для получения целого ряда препаратов (триметина, анальгина,
метилкофеина и т. д.). Недостатком диметилсульфата является
его большая токсичность, поэтому ib ряде случаев более рацио-
рациональным является использование ib качестве метилирующего
средства метилового эфира бензолсульфокислоты. Метили-
Метилирование метиловым эфиром бензолсульфокислоты используется
при промышленном получении антипирина, гексония Б и бен-
замона.
Алкилирование галогеналкилами применяется
в тех случаях, когда галоидный алкил доступнее соответству-
соответствующего спирта или когда реакционная способность спирта в ре-
реакции алкилирования значительно ниже реакционной способ-
способности галоидного алкила.
Большое значение в качестве алкилирующих агентов имеют
хлористый бензил (С6Н5СН2С1), монохлоруксусная кислота
(С1СН2СООН) и этиленхлоргидрин (С1СН2СН2ОН).
При реакции алкилирования в этих случаях выделяется хло-
хлористый водород
RNH2 + AlkCl >- RNHAlk + НС1,
и добавление веществ, связывающих кислоту, ускоряет процесс
Связывающим кислоту агентом может быть сам амин, такие
вещества, как сода, известь, карбонат кальция, едкий натр и
т. п.
Температура реакции обычно не превышает 100 °С. Поэто-
Поэтому в большинстве случаев процесс можно вести при атмосфер-
атмосферном давлении в аппарате с обратным холодильником. Однако
при работе с ннзкокипящими веществами (СН3С1, C2H5CI) ал-
алкилирование ведут в автоклавах. Реагенты обычно берут в сте-
хиометрическом соотношении. С помощью галогеналкилов мож-
можно получать несимметричные третичные амины, а также разде-
130
лять смеси третичных и вторичных аминов, полученные при ал-
килировании спиртами:
C6H5NHa + С2Н5ОН >• C6H5NHC2H5 + Н2О;
C6H5NHCaH6 + С6Н5СН2С1 > C6H5N + HC1.
Vh2c6h5
Полученный бензилэтиланили'н легко отделяется от диэтил-
анилина, а образующийся в качестве побочного продукта при
алкилировании анилина этиловым спиртом моноэтиланилин, та-
таким образом, полностью используется.
Этилирование анилина хлористым этилом в присутствии из-
извести при температуре 125°С под давлением 10—12 ати в тече-
течение 12 ч приводит к образованию преимущественно диэтилани-
лина. Введение этанольного остатка может быть достигнуто дей-
действием водного раствора этиленхлоргидрина на ароматический
амин:
RNH2 + С1СН2СН2ОН > RNHCH2CH2OH + HC1;
хн2сн2он
RNHCH2CH2OH + С1СН2СН2ОН > RN
\:н2сн2он
Чаще оксиэтильные производные получают не через этилен-
хлоргидрин, а действием на амин окиси этилена:
RNH2 + СН2 СН2 >- RNHCH2CH2OH;
О
RNHCH2CH2OH + СН2 СН2 >■ RN(CH2CHaOHJ.
О
Для получения монозамещенного производного реакцию ве-
ведут в большом избытке амина при температуре ниже 100 °С в
присутствии воды.
Для введения двух оксиэтильных остатков берут неболь-
небольшой избыток окиси этилена и реакцию проводят при темпера-
температуре 120—140 °С и давлении 5—6 ати. Соединения этого типа
имеют большое значение в синтезе противораковых препаратов,
так как образующаяся ди-оксиэтиламинная группа при дейст-
действии тионилхлорида легко превращается в ди-хлорэтиламинную
группу, обусловливающую противоопухолевое действие вещества.
Так как смесь окиси этилена с воздухом взрывчата, реак-
реакцию следует вести при полном отсутствии воздуха, что достига-
достигается продувкой аппарата азотом.
§ 2. АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ ОКСИСОЕДИНЕНИЙ
Алкилирование ароматических оксисоединений имеет гораз-
гораздо меньшее значение, чем алкилирование аминов, поскольку
соответствующие алкоксипроизводные удобнее получать через
9* 131
хлорзамещенные. Для проведения алкилирования оксигруппы
могут быть использованы спирты, алкильные эфиры серной кис-
кислоты и сульфокислот, галогеналкилы.
Алкилирование оксигруппы действием спир-
спирта в присутствии минеральной кислоты используется довольно
редко и применяется главным образом для получения алкокси-
производных нафталинового и антраценового ряда. Принципи-
Принципиально этим способом можно получить нитроанизол из нитрофе-
нитрофенола. Однако в основу промышленного метода была положена
реакция замены хлора в п-нитрохлорбензоле действием раство-
раствора щелочи в метиловом спирте (см. с. 121).
Алкилирование оксисоединений эфирами
серной кислоты и ароматических сульфокис-
сульфокислот имеет значительно большее значение.
Реакция метилирования диметилсульфатом протекает по
следующим уравнениям:
RONa + CH3OSO2OCH3 >• ROCH3 + CH3OSO2ONa;
RONa + CH3OSO2ONa > ROCH3 + Na2SO4.
Процесс ведут в щелочной среде. Первая из этих реакций
протекает легко при температуре ниже 100 °С. Вторая реакция
протекает в гораздо более жестких условиях и часто проводит-
проводится в автоклаве под небольшим давлением. В связи с этим при
метилировании диметилсульфатом обычно используют лишь од-
одну метильную группу. При метилировании фенола можно ис-
использовать примерно на 90% и обе метальные группы диметил-
диметилсульфата, если проводить реакцию при 100 °С в течение 5 ч, за-
загружая на^ 2 моля фенола 1 моль диметилсульфата и 3 моля
едкого натра в небольшом количестве (около 2 молей) воды.
Диметилсульфат используют в качестве метилирующего
средства при синтезе метиловых эфиров гидрохинона, оксиди-
фениламина, м-крезола, о-нитро-п-крезола и др.
ОН ОСН3
„NO2
(CH3JSO4
Ч
Этим же способом можно получать о- и п-нитроанизолы из
о- и п-нитрофенолов. Недостатком этого метода является ток-
токсичность диметилсульфата и относительно высокая его стои-
стоимость.
Алкилирование фенолов посредством эфиров ароматических
сульфокислот протекает гладко при кипячении с обратным хо-
холодильником смеси фенолята и соответствующего арилсульфо-
нового эфира. В качестве растворителя используют полихлори-
полихлориды бензола. Реакция протекает по уравнению:
RONa + R'SO2OAlk >■ ROAlk + R'SO3Na.
132
Галогеналкилы находят широкое применение в качест-
качестве алкилирующих агентов. Метилхлорид и этилхлорид широко
используются в качестве алкилирующих агентов вследствие их
доступности и сравнительно низкой стоимости. Поскольку эти
вещества имеют низкую температуру кипения, алкилирование
ими ведут в автоклавах под давлением. Так, например, метили-
метилирование гидрохинона проходит при нагревании водного раство-
раствора его динатриевого производного с хлористым метилом при
температуре 100 °С и давлении 20 ат.
.ONa
+ 2СН8С1 >• | || +2NaCl
Важное значение имеет производство п-нитрофенетола, яв-
являющегося промежуточным продуктом в синтезе фенацетина.
п-Нитрофенетол может быть получен при алкилировании п-нит-
рофенола бромистым этилом:
NO2 NO2
+ C2H5Rf * I у + НВг
ос2нб
Однако экономически более выгодным является получение его
из п-нитрохлорбензола.
§ 3. АЦИЛИРОВАНИЕ АРОМАТИЧЕСКИХ АМИНОВ
Ацилированием называется реакция замещения атома водо-
водорода ацильным остатком, получающимся отнятием гидроксила
от молекулы кислоты.
Примерами ацильных остатков (ацилов) являются:
формил —С^ бензоил —С
ацетил —-С алкоксикарбонил — (
3 X)R
сульфонил —SO2OH и т. д.
Ацилирование может иметь целью получение нового соеди-
соединения, свойства которого во многом определяются наличием
ацильного остатка. Ацильный остаток в этом случае остается в
молекуле лекарственного препарата. В других случаях введе-
введение ацильного остатка применяется как временная мера для
защиты амино- или оксигруппы. В этом случае ацилирование
играет вспомогательную роль. Замена водорода в амино- или
оксигруппе ацильным остатком делает эту группу менее реак-
133
ционноспособной и позволяет осуществлять такие превращения
ацилированного продукта, которые при наличии свободной ами-
но- или оксигруппы были бы невозможны. После проведения
этих превращений ацилокси- или ациламиногруппу снова прев-
превращают в окси- или аминогруппу.
В качестве ацилирующих агентов применяются кислоты, их
ангидриды, хлорангидриды, а в некоторых случаях также эфи-
ры и амиды кислот.
Скорость реакции ацилирования и условия ее проведения в
значительной мере зависят от природы ацилирующего
агента. Очевидно, что чем больше положительный заряд на
активном атоме углерода ацилирующего агента, тем легче и
быстрее пойдет реакция
Х\ jP
R-NH2+ /С—R' >- R-NH—Cf +HX.
Если рассматривать ацилирующий агент
где Х=—ОН, —С1, —О—С^ , и т. п., то чем больше элек-
электроотрицательность X, тем труднее могут быть смещены от не-
него электроны по направлению к ацильному радикалу и тем
больший положительный заряд будет на карбонильном углеро-
углероде. Таким образом, хлорангидриды (Х = —С1) должны обладать
наибольшей ацилирующей способностью, ангидриды кислот
°
(Х= —О—С\ ) должны быть более слабыми агентами,
органические кислоты (Х=—ОН) должны ацилировать еще сла-
слабее, а сложные эфиры (Х=—OR2) должны обладать наимень-
наименьшей активностью. Эти теоретические рассуждения хорошо сог-
согласуются с экспериментальными данными.
Реакция ацилирования аминов карбоновыми
кислотами является обратимой:
cf -<—>■ RNHC^ +H2O.
Обратимостью этой реакции пользуются для «снятия» ациль-
ной группы в тех случаях, когда ацилирование проводится для
временной защиты аминогруппы (например, при реакции нитро-
нитрования). В случае ацилирования аминов кислотой для сдвига
134
равновесия вправо целесообразно применять избыток кислоты
или выводить образующуюся воду из сферы реакции (напри-
(например, отгонять или связывать водоотнимающими средствами).
Реакции ацилирования хлорангидридами и ангидридами кис-
кислот необратимы, поэтому реагенты можно брать в стехиоме-
трических соотношениях.
формилирование аминов обычно проводят при нагре-
нагревании амина с избытком муравьиной кислоты. В качестве при-
примера можно привести формилирование анилина:
+ НСООН >- Г JJ Н + Н2О.
Реакцию проводят в избытке муравьиной кислоты при нагрева-
нагревании до 150°С. Образующаяся при реакции вода отгоняется вмес-
вместе с избытком муравьиной кислоты. Окончательную отгонку
остатков муравьиной кислоты проводят в вакууме.
Для формилирования можно применять не муравьиную кис-
кислоту, а ее амид (формамид), который получают из окиси уг-
углерода и аммиака:
СО + NH3 >■ HCONH2,
RNH2 + HCONH2 >- RNHCOH + NH3,
Ацетилироваиие анилина и его гомологов
можно вести уксусной кислотой с добавкой бензола. Образую-
Образующаяся по мере прохождения реакции вода отгоняется в виде азе-
отропной смеси с бензолом. Это позволяет проводить реакцию
с почти количественным выходом. Ацчлирование уксусной кис-
кислотой обычно ведут при температуре ПО—115°С. В технике
часто используют не только «ледяную» A00%), но и 80% ук-
уксусную кислоту, которую берут с 50% избытком.
Более энергичным ацилирующим агентом является уксусный
ангидрид.
RNH2 + (СН3СОJО > RNHCOCH3 + CH3COOH.
Уксусный ангидрид применяется для ацетилирования арома-
ароматических аминов в тех случаях, когда ацилирование уксусной
кислотой проходит медленно или продукт образуется с малым
выходом. Ацетилирование уксусным ангидридом ускоряется при
добавлении к реакционной массе небольших количеств серной,
фосфорной или хлорной кислоты.
Ацетилирование широко применяется в химико-фармацевти-
химико-фармацевтической промышленности как для получения лекарственных пре-
препаратов, так и для синтеза промежуточных продуктов. Так, да-
даже ацетанилид C6H5NHCOCH3 долгое время применялся в ка-
качестве жаропонижающего средства под названием антифебрина.
Ьольшое значение и до настоящего времени имеет лекарствен-
135
ный препарат фенацетин, который получают ацетилированием
п-фенетидина уксусным ангидридом или кислотой:
NH,1 NHCOCH3
I
+ (СН3СОJО
+ СНдСООН
ОС2Н5 ОС2Н5
При синтезе сульфаниламидных препаратов ацетилирование
используют для временной защиты аминогруппы:
NH,
HNCOCH
HNCOCH
HNCOCH3
P. I1CI
**^ л ц • • ^-* >^ ^-* | I Q
п-аминобензолсульфацетамид
(сульфацил, альбуцид)
SO2NHCOCH3 SO2NH2
п-ам и нобензол сульфамид
(белый сфепюцид)
Большое значение в химико-фармацевтической промышлен-
промышленности в качестве ацилирующего средства получил метиловый
эфир хлоругольной- кислоты, получаемый из метиле-
вого спирта, фосгена и мела:
•ОСН3
2СН3ОН + 2СОС12 + СаСОз >■ 2С=О + СаС12 + СО2 + Н2О
\
Метиловый эфир хлоругольной кислоты является сырьем
для синтеза важнейшего промежуточного продукта в синтезе
сульфамидных препаратов — фенилуретилаиа (карбометокси-
анилина):
/О
NH2
/OCHg
' C\c°i
NH—С
I
\
ОСНз
+ НС1
При обработке хлорсульфоновой кислотой фенилуретилан
в свою очередь дает важный ацилирующий агент п-фенилуре-
136
тилансульфохлорид (хлорангидрид карбометоксисульфаниловой
кислоты)
,0 лО
NH-c/ NH- "
I ЧОСН3
^^ + 2HSO3C1 ■
SO2C1
Из ацильных остатков ароматического ряда наибольшее зна-
значение имеет бензоил — СОС6Н5. Введение бензоила обычно осу-
ществляется при помощи хлористого бензоила
СбНбСОС1. Взаимодействие с хлористым бензоилом проводят
либо в среде инертного растворителя, либо в водной среде в
присутствии соды, щелочи, мела или других веществ, связыва-
связывающих выделяющийся хлористый водород:
RNH2 + С6Н5СОС1 > RNHCOCeH6 + HCI ^
Применяемый избыток хлористого бензоила составляет 10—
15%. Температура реакции обычно ниже 100 °С.
В качестве ацилирующего агента применяют также п-н и-
тробензоилхлорид. Нитрогруппу затем можно восстано-
восстановить и получить ценные промежуточные продукты для синтеза
лекарственных веществ.
Для получения весьма важных производных мочевины аро-
ароматические амины ацилируют фосгеном.
2RNH2 + СОС12 >- RNHCONHR + 2НС1.
Фосген является сильным отравляющим веществом и работа
с ним требует особых предосторожностей.
Большое значение имеют ариламиды ацетоуксус-
ной, бензоилуксусной и п-нитробензоилуксус-
ной кислот. Однако сами кислоты для ацилирования аминов
не применяются и получение ариламидов проводят с помощью
эфира соответствующей кетокарбоновой кислоты:
RCOCH2COOC2H5 + R'NH2 > RCOCHaCONHR' +С2НбОН.
Процесс проводят при нагревании эфира кетокарбоновой кис-
кислоты с амином в хлорбензоле или ксилоле. Образующийся при
реакции спирт непрерывно отгоняется из реакционной массы.
Процесс следует вести в эмалированной или алюминиевой аппа-
аппаратуре, так как железо отрицательно влияет на ход реакции.
Для получения ариламидов ацетоуксусной кислоты экономи-
экономически целесообразно использовать дикетен:
СН2=С-СН2
| | + RNH2 > CH3COCH2CONHR.
Ацилирование дикетеном проводится при низкой температу-
температуре в органических растворителях (бензоле, ацетоне) или в вод-
водных растворах.
137
Гидролиз ацильных производных проводят при
нагревании или кипячении с 5—10% раствором щелочи или с
разбавленными минеральными кислотами. Количество щелочи
берут несколько больше, чем необходимо для нейтрализации
кислоты, образующейся при гидролизе:
RNHCOCH3 + H2O >- RNH2 + CH3COOH.
§ 4. АЦИЛИРОВАНИЕ ОКСИГРУППЫ
Ацилирование оксигруппы в ароматических соединениях
проводится значительно реже, чем ацилирование аминогруппы.
В качестве ацилирующих агентов применяются те же веще-
вещества, что и при ацилировании аминогруппы. Поскольку реакция
с оксисоединениями идет менее энергично, для связывания вы-
выделяющейся воды или хлористого водорода необходимо приме-
применять соответствующие реагенты.
В тех случаях, 'когда ацилирование проводится кислотой, для
связывания выделяющейся воды применяют треххлористый фос-
фосфор (РСЬ) или хлорокись фосфора (РОСЬ). Возможно, что в
присутствии этих реагентов реакция протекает через стадию
образования хлорангидрида кислоты. Примером ацилирования
фенола может служить получение салола:
+ PC13
соон
о
■ Н3РО4 + ЗНС1
он
Часто для ацилирования оксисоединений используют ангид-
ангидриды и хлорангидриды кислот. Так, ацилированием салицило-
салициловой кислоты уксусным ангидридом получают аспирин (ацетил-
(ацетилсалициловую кислоту):
О
II
н ^.о-с-сн3
+ (СН3СОJО >■ Г jT +CH3COOH.
соон ^/^соон
Ацилирование фенолов уксусным ангидридом можно прово-
проводить также и в щелочном водном растворе при температуре
около 0°С, так как уксусный ангидрид на холоду реагирует с
оксисоединениями со значительно большей скоростью, нежели
с водой.
При ацилировании хлорангидридами кислот для связывания
выделяющегося хлористого водорода к реакционной массе до-
138
бавляют едкий натр, соду или поташ. Например, фенол в бен-
бензольном растворе в присутствии избытка поташа образует с хло-
хлористым бензоилом фенилбензойный эфир:
СбН5ОН + СбН5СОС1 > СвН5СООСвНв + НС1.
В качестве ацилирующих средств используют также кетен,
дикетен и фосген.
§ 5. ПРОИЗВОДСТВО ФЕНАЦЕТИНА
Фенацетин применяется как жаропонижающее, болеутоля-
болеутоляющее средство, он обладает некоторыми антиневралгическими
свойствами.
Фенацетин выпускается в виде таблеток и порошков, а так-
также в сочетании с аспирином, кофеином входит в состав пенталь-
гина. Фенацетин хорошо всасывается в желудочно-кишечном
тракте и оказывает действие через 15—30 мин. В организме фе-
фенацетин расщепляется на п-фенетидин и п-аминофенол, кото-
который обезвреживается в печени и выводится из организма. По
анальгезирующему действию фенацетин уступает амидопирину,
но превосходит аспирин.
Процесс производства фенацетина состоит из стадий ацети-
лирования п-фенетидина, выделения, доацетилирования и очист-
очистки полученного технического фенацетина.
Процесс ацетилирования состоит в замещении водорода ами-
аминогруппы ацетильной группой под действием уксусной кислоты.
Ацетилирование ведут 50—55% уксусной кислотой с 16,5% из-
избытком от теории при температуре 112—115 °С. Избыточную
уксусную кислоту отгоняют вместе с образующейся в процессе
реакции водой до содержания в плаве кислоты 5—7%. Остаточ-
Остаточная кислотность 5—7% необходима для обеспечения хорошей
растворимости окрашенных веществ и удаления их с маточника-
маточниками на стадии выделения технического фенацетина.
После ацетилирования уксусной кислотой остается 2—3%
непрореагировавшего п-фенетидина, который доацетилируют ук-
уксусным ангидридом после разбавления плава кипящей водой.
Полученный технический фенацетин охлаждают до 20—25 °С,
отфильтровывают на нутч-фильтрах, отмывают водой от уксус-
уксусной кислоты и следов п-фенетидина.
Очистка заключается в обработке горячего водного раство-
раствора фенацетина активированным углем для удаления окрашен-
окрашенных веществ и других примесей, фильтрации через друк-фильтр
для отделения отработанного угля; осветлении раствора фена-
фенацетина гидросульфитом натрия и кристаллизации фенацетина
из очищенного раствора при температуре 25—30 °С. Выпавшие
кристаллы фенацетина отфильтровывают на центрифуге, про-
промывают сначала водопроводной, а затем дистиллированной во-
139
дой, сушат при температуре 120—134 °С, просеивают и отправ-
отправляют в цех фасовки.
Описание технологического процесса. Техно-
Технологическая схема производства фенацетина приведена на рис.
34. Процесс состоит из следующих основных стадий.
1. Ацетилирование п-фенетидина. В ацетилятор /
загружают через мерник 2 отгоны уксусной кислоты из сборни-
сборника 7 и из ловушки паров уксусной кислоты 4. В аппарате 1 от-
отгоны уксусной кислоты перемешивают в течение 10—15 мин и
отбирают пробу для определения процентного содержания ук-
уксусной кислоты. Затем в ацетилятор загружают через мерник 2
98% уксусную кислоту. В аппарате ее перемешивают с отго-
отгонами в течение 10—15 мин и отбирают пробу для определения
концентрации кислоты. Концентрация должна быть в пределах
49—55%.
Кислоту в аппарате 1 нагревают при перемешивании до
102—105 °С. В нагретую уксусную кислоту загружают в несколь-
несколько приемов при перемешивании из мерника 3 п-фенетидин. По
окончании слива п-фенетидина реакционную массу нагревают
до температуры 102—105 °С и выдерживают при этой темпера-
температуре около 2 ч.
По окончании выдержки отгоняют уксусную кислоту. Пер-
Первые отгоны с содержанием до 15% уксусной кислоты сливают
в нейтрализатор, куда подают аммиачную воду для нейтрали-
нейтрализации уксусной кислоты, а затем нейтральный раствор сливают
в канализацию. Отгоны с содержанием уксусной кислоты 15%
и более собирают в сборник 7. Отгонку ведут до тех пор, пока
температура в реакционной массе не повысится до 112—115°С.
При температуре реакционной массы 112—115°С автомати-
автоматически прекращается нагрев, холодильник переключают на «об-
«обратный» и через мерник 2 загружают в аппарат / дополнитель-
дополнительное количество 98% уксусной кислоты, снова доводят темпера-
температуру реакционной массы до 112—115 °С и выдерживают при
этой температуре 24 ч. Температура во время выдержки под-
поддерживается автоматически. По окончании выдержки прекра-
прекращается нагрев реакционной массы и она самоохлаждается до
температуры 108—110 °С. Затем отбирают пробу на определе-
определение процентного содержания остаточного п-фенетидина и ук-
уксусной кислоты. Процесс ацетилирования считается закончен-
законченным при остаточном содержании п-фенетидина не выше 3% и
уксусной кислоты 8—13%. Если остаточного п-фенетидина бо-
более 3%, то ацетилирование продолжают еще 8—12 ч (в зависи-
зависимости от результатов анализа) и снова отбирают пробу на ко-
конец ацетилирования.
По окончании процесса ацетилирования из реакционной мас-
массы вновь отгоняют уксусную кислоту в сборник 7, установив
автоматический регулятор температуры на 118°С. После окон-
окончания отгонки уксусной кислоты при температуре 118°С регу-
140
лятор температуры устанавливают на 115 °С во избежание ос-
моления реакционной массы и отгонку кислоты продолжают
острым паром до содержания остаточной уксусной кислоты в
массе 5—7%.
После окончания отгонки уксусной кислоты и отбора пробы
реакционную массу передавливают при температуре 110—116 °С
в аппарат 8 для доацетилирования и выделения технического фе-
фенацетина.
2. Доацетилировние, разбавление и выделе-
выделение технического фенацетина. В аппарат 8 (рис. 34)
загружают рассчитанное количество воды. По достижении опре-
определенного уровня срабатывает электронный сигнализатор и по-
подача воды прекращается. Передавливание плана из аппарата 1
в аппарат 8 производят сжатым воздухом при температуре мас-
массы ПО—116°С по предварительно прогретому трубопроводу
и при работающих мешалках.
При температуре 90 °С производят доацетилирование оста-
остаточного п-фенетидина уксусным ангидридом, который сливают
тонкой струей из мерника 10. Количество уксусного ангидрида,
необходимого для доацетилиравашш, зависит от содержания ос-
остаточного п-фенетидина. Засасывание уксусного ангидрида в
мерник и слив его в аппарат производятся автоматически.
После слива уксусного ангидрида перемешивают массу в те-
течение 1 ч отбирают пробу на конец доацетилирования. Доаце-
Доацетилирование считается законченным, если содержание остаточ-
остаточного п-фенетидина в пробе не превышает 0,2%, а остаточная
кислотность составляет 4—5%. Если остаточного п-фенетидина
в пробе больше, чем 0,2%, а остаточная кислотность ниже 4—
5%, то в реактор добавляют уксусный ангидрид. При кислот-
кислотности выше 5% уксусный ангидрид не добавляют во избежание
потери фенацетина с маточником. После охлаждения до 20—
25 °С реакционную массу фильтруют на нутч-фильтре. Маточ-
Маточник собирают в нейтрализаторе 6 и после нейтрализации амми-
аммиачной водой сливают в канализацию. Технический фенацетин
промывают на фильтре водой от уксусной кислоты и п-фенети-
п-фенетидина.
По окончании промывки технический фенацетин отжимают
от промывных вод и выгружают через разгрузочный люк в спе-
специальную емкость для технического фенацетина, откуда он
пневмотранспортером засасывается в промежуточный бункер
15 на стадию очистки.
3. Очистка фенацетина.
В растворитель 19 заливают воду или маточник, нагретые до
температуры 90 °С. Прилив маточника производят автоматиче-
автоматически. Из смесителя 18 в аппарат 19 загружают смесь техническо-
технического фенацетина с углем и водой. После загрузки в растворитель
массу перемешивают в течение 10 мин, а затем отбирают про-
пробу. Отфильтрованный раствор с индикатором .бромтимол-синим
141
конденсат в канализацию
40
40
41
В цех цмсобкш,
Рис. 34. Технологическая схема производства фенацетина.
должен иметь зеленое окрашивание с синеватым оттенком, что
соответствует рН 6,8—6,6. Массу нагревают до 100—102 °С и
выдерживают при этой температуре 30—40 мин. Затем передав-
передавливают реакционную массу через предварительно нагретый
друк-фильтр 20 в кристаллизатор 22. Температура массы во вре-
время передавливания 95—100 °С. Промывку угля на друк-фильтре
ведут маточником, нагретым до температуры 85—95 °С. Отфиль-
Отфильтрованный и промытый уголь смывают с друк-фильтра водой.
В кристаллизатор загружают гидросульфит натрия и рас-
раствор охлаждают водой до температуры 20—30 °С. После оконча-
окончания охлаждения автоматически прекращают подачу охлажда-
охлаждающей воды, включают центрифугу, открывают клапан на линии
слива реакционной массы из кристаллизатора и клапан на линии
слива охлаждающей воды. Воду из рубашки кристаллизатора
сливают для того, чтобы при следующем заполнении кристал-
кристаллизатора не происходила кристаллизация продукта на стенке
аппарата из-за большого перепада температур у стенки.
Маточники с центрифуг 24 поступают самотеком в сборник
маточников 25, а из него центробежным насосом 26 подают в
сборники маточников 27. Заполнение сборников маточником и
нагрев маточников производят автоматически. Кристаллы фе-
фенацетина на центрифуге промывают водой.
Выгрузка осадка осуществляется механически. С помощью
пневмотранспортной установки фенацетин засасывают в прием-
приемный бункер 28. Из приемного бункера фенацетин загружают
в бункер 29, из которого шнекам фенацетин передается в воз-
воздуховод. Одновременно в воздуховод вентилятором подается
очищенный через фильтр и нагретый в калорифере 32 воздух
с температурой 120—135 °С. В токе горячего воздуха фенацетин
высыхает и поступает в циклон 34. Из циклона сухой продукт
через смеситель со шнеком 35 поступает на протирочное сито
36 и далее в приемный бункер 37. Увлажненный воздух выходит
из боковой части циклона в рукавный фильтр 30 для очистки
от пыли. На выходе воздуха из рукавного фильтра установлен
вентилятор среднего давления, работающий для подсоса воз-
воздуха из воздуховода, поэтому в сушилке создается вакуум
150—200 мм рт. ст.
§ 6. ТЕХНИКА БЕЗОПАСНОСТИ ПРИ ПРОВЕДЕНИИ
ПРОЦЕССОВ АЛКИЛИРОВАНИЯ И АЦИЛИРОВАНИЯ
Процессы алкилирования и ацилирования во многих случа-
случаях проводятся при повышенном давлении в автоклавах. Общие
требования по технике безопасности при проведении таких про-
процессов были изложены на с. 125—126. Помимо этого, следует
иметь в виду, что проведение процессов алкилирования и аци-
ацилирования связано с применением и получением веществ, обла-
обладающих высокой токсичностью. Многие ароматические амины
142
являются сильными кровяными ядами, а также действуют на
центральную нервную систему. Токсичны также многие оксисо-
единения.
Окись этилена является наркотиком с сильной специфиче-
специфической ядовитостью. Имеются указания на то, что окись этилена
в организме реагирует с аминогруппами белков.
Метиловый спирт является сильным нервным и сосудистым
ядом с резко выраженным кумулятивным действием. Особую
ядовитость метилового спирта связывают с образованием из
него в организме высокотоксичных формальдегида и муравьи-
муравьиной кислоты. При любом способе попадания в организм ме-
метиловый спирт в первую очередь поражает зрительный нерв и
сетчатку глаза. Сильным токсичным действием обладают так-
также фенол, этиленхлоргидрин и некоторые другие вещества, ис-
использующиеся в процессах алкилирования и ацилирования.
Многие из используемых в процессах алкилирования ве-
веществ (бензол, метиловый и этиловый спирты, окись этилена и
др.) образуют взрыво- и пожароопасные смеси с воздухом.
При проведении фосгенирования необходимо иметь в виду,
что фосген в 3,5 раза тяжелее воздуха, поэтому в случае утечки
он может заполнять приямки и другие низкие места в помеще-
помещении и довольно долго держаться там, создавая опасность отрав-
отравления людей, находящихся в помещении. Предельно допусти-
допустимая концентрация фосгена в воздухе производственных поме-
помещений 0,5 мг/м3.
Присоединение емкости с фосгеном к газовому трубопрово-
трубопроводу и особенно отсоединение его являются опасными операция-
операциями, так как в трубопроводе может остаться фосген, который при
развинчивании соединений попадает в этом случае в помещение.
В тех случаях, когда по технологическим условиям процесс про-
проводится под повышенным давлением и не в щелочной среде,
полная герметичность оборудования приобретает особое значе-
значение.
При проведении процесса в вакууме нельзя пользоваться об-
общецеховыми или общезаводскими вакуумными коммуникация-
коммуникациями. Эти коммуникации нельзя даже кратковременно присоеди-
присоединять к системе фосгенирования. В помещениях, где имеется обо-
оборудование, связанное с фосгенированием, система вентиляции
должна быть рассчитана таким образом, чтобы количество воз-
Духа, удаляемого вытяжной системой, было больше количества
воздуха, нагнетаемого приточной вентиляцией. Во всех произ-
производственных помещениях необходимо иметь аварийную аммиач-
аммиачную систему для нейтрализации газообразного фосгена.
Все работающие в отделении фосгенирования должны иметь
противогазы марки В и уметь ими пользоваться. Такие работы,
как отсоединение баллона от трубопровода, отбор проб, подтя-
подтягивание сальника на баллоне с фосгеном, проводятся только в
противогазе. При появлении запаха фосгена (напоминает за-
143
пах гнилых яблок) все находящиеся в помещении должны не-
немедленно надеть противогазы.
В связи с вредностью и опасностью ведения процессов алки-
лирования и ацилирования комплексная механизация и авто-
автоматизация этих производств имеют первостепенное значение.
Большое внимание уделяется также вентиляции производствен-
производственных помещений.
Острые отравления обычно наблюдаются в случае наруше-
нарушения работающими требований техники безопасности. Отравле-
Отравление может наступить в результате загрязнения тела токсичны-
токсичными веществами при чистке аппаратуры и коммуникаций без до-
достаточных мер предосторожности, при выгрузке и упаковке в
тару, а также при нарушении герметичности аппаратуры.
При попадании вредных веществ на кожу необходимо тот-
тотчас же смыть их теплой водой, а при значительном загрязнении
рабочей одежды — переодеться. Летом следует особенно тща-
тщательно оберегать тело от загрязнения, чаще менять рабочую
одежду и белье, работать в исправных рукавицах и перчатках,
следить за бесперебойной работой вентиляции. После работы
с вредными веществами необходимо принять теплый душ и
переодеться в чистую одежду, которая хранится отдельно от ра-
рабочей (в другом шкафчике или помещении).
Глава 8
АЛКИЛИРОВАНИЕ И АЦИЛИРОВАНИЕ
УГЛЕВОДОРОДОВ
Эта глава охватывает большую группу важных реакций
электрофильного замещения, общей чертой которых является
то, что активным реагентом в них является карбониевый ион
или поляризованная катионоидная частица, имеющая положи-
положительный заряд на атоме углерода. Электронодонорные замести-
заместители значительно облегчают протекание С-алкилирование и
С-ацилирования, а электроноакцепторные — затрудняют.
§ 1. МЕХАНИЗМ РЕАКЦИИ
В тех случаях, когда для алкилирования (С-алкилирования)
используются алкилгалогениды или непредельные углеводоро-
углеводороды, а для реакции ацилирования (С-ацилирования)—хлоран-
гидриды или ангидриды кислот эти реакции ведут в присутст-
присутствии катализаторов Фриделя — Крафтса, представляющих собой
безводные галогениды ряда металлов, например А1С1з, В1Вг3,
FeCl3, SnCl4, SbCb, ZnCb. Из приведенных катализаторов в
промышленной практике наибольшее применение получил хло-
хлорид алюминия.
Реакцию Фриделя—Крафтса можно рассматривать как свое-
своеобразную реакцию электрофильного замещения, где электро-
фильный реагент образуется по схеме:
RC1 + А1С13 —* R+ + А1С1Г;
RCOC1 + А1С18 <=* RC+ + A1C17
II
О
При проведении реакции с хлоридом алюминия применяют
избыток одного из жидких реагентов. В некоторых случаях ре-
реакцию проводят в инертном растворителе, например в нитро-
нитробензоле. Порядок смещения реагентов может быть различным
в зависимости от проводимой реакции.
Большое значение имеет правильный выбор температуры
реакции. Реакцию начинают при невысокой температуре и под-
поддерживают ее путем охлаждения реакционной массы. К концу
процесса температуру реакционной массы поднимают.
10-926 145
Образование карбониевых ионов из непредельных углеводо*
родов проходит по схеме:
СН3-СН + А1С13 -<—>- СНд-СН
II I -
СН2 СН2А1С13
При алкилировании ароматических соединений спиртами в
качестве катализаторов часто используют сильные кислоты.
R-OH + НХ 1—> R+ + Н2О + X"
Дальнейшее взаимодействие карбониевых ионов с аромати-
ароматическими соединениями идет по обычной схеме электрофильно-
го замещения. В тех случаях, когда реакции проводят в при-
присутствии катализаторов Фриделя — Крафтса, следует вести их
при возможно более низкой температуре, так как нагревание
может вызвать нежелательные процессы изомеризации. Напри-
Например, при нагревании ксилолов с катализаторами Фриделя —
Крафтса образуется смесь бензола, толуола, ксилолов и триме-
тилбензолов. При изомеризации по межмолекулярному механиз-
механизму одновременно может происходить и изомеризация перемеща-
перемещающейся алкильной группы:
^СН2СН2СНз
^ "
При проведении реакции С-ацилирования с использованием
катализаторов Фриделя — Крафтса механизм активации не-
несколько отличается от механизма активации при алкилирова-
алкилировании. Хлорид алюминия в этом случае, очевидно, присоединяет-
присоединяется по карбонильному кислороду:
О
II +
R-C + AlClg •<—* R—C-O-AICI3
I I
X X
Далее процесс проходит с отщеплением аниона и образова-
образованием ионной пары:
R-C-O-AlCig =*=> [R-C=O]A1C13X-
X
Очевидно, что реагент здесь менее активен, чем при алкили-
алкилировании.
R
I
I ^\/C=O..J
| + С=О...А1С13 ^ fOl +HX
X
146
В тех случаях, когда в качестве реагента используется ан-
ангидрид кислоты и в результате взаимодействия получается кар-
боновая кислота, ее гидроксильной группой связывается еще 1
моль хлорида алюминия. Таким образом, в этом случае для
проведения реакции необходимо не менее 2 молей А1С13.
Алкилирование парафиновых углеводородов происходит при
нагревании их с непредельными углеводородами под давлени-
давлением при высокой температуре. Процесс этот может протекать
также под влиянием А1С1з в присутствии НС1 или BF3.
СН3ч
;С-СН2 + НС(СН3)з > (СН3)зС-СН2-СН(СН3J
сн/
Под влиянием А1С13 к непредельным углеводородам могут при-
присоединяться хлорангидриды кислот:
СНЯ
СНя
Ч;=сн2 + R—co-ci
А1С13
ев
3\
:c-ch2-co-r
снч
Подобная реакция может быть проведена и с ацетиленовыми
углеводородами. В этом случае образуются C-хлорвинил-алкил-
кетоны:
А1С13
СН£ееСН + СН-СО-С1 >• С1СН-СН—СО-СНд
При действии на этиловый эфир малоновой кислоты металличе-
металлического натрия или алкоголята натрия один или оба (при избыт-
избытке натрия) водорода метиленовой группы замещаются на ато-
атомы натрия. При действии на натриймалоновый эфир галоидных
алкилов происходит замещение натрия на алкил или другой
радикал, ранее связанный с галогеном:
ОС2Н5
На
'ОС2Н5
<+ RCL iH —R +NaCI
Так можно получать замещенные малоновые эфиры, а после их
гидролиза — кислоты. Декарбоксилированием можно удалить
ОДин из карбоксилов и перейти к замещенным уксусной кис-
кислоты.
147
Реакции С-алкилирования и С-ацилирования находят широ-
широкое применение в синтезах лекарственных веществ. Конкретные
примеры таких синтезов будут рассмотрены во второй части
учебника.
§ 2. ПРИМЕРЫ РЕАКЦИЙ АЛКИЛИРОВАНИЯ
И АЦИЛИРОВАНИЯ УГЛЕВОДОРОДОВ
Алкилирование углеводородов спиртами
используется сравнительно редко. В качестве конденсирующих
средств при этом могут быть использованы хлорид алюминия,
серная и фосфорная кислоты. Реакция эта нашла применение
для синтеза поверхностно-активных веществ. Так, при алкили-
ровании нафталина бутиловым спиртом в среде серной кислоты
при 80—90 °С получают бутилнафталинсульфокислоту, извест-
известную также под названием «смачиватель НБ». При этом парал-
параллельно с алкилированием протекает сульфирование нафталина:
H2SO4 + СН3СН2СН(ОН)СН3 >
> CH3CH2CH(CH3)C10H6SO3H + H2O
Вторичные спирты в этих реакциях обладают большей реак-
реакционной способностью, чем первичные, а третичные — большей,
чем вторичные. Реакция может проводиться также в паровой
фазе в присутствии катализаторов (алюмосиликат, фосфорная
кислота на пемзе).
Значительно чаще алкилирование проводят алкилгалоге-
нидами. Реакцию проводят в безводном инертном раствори-
растворителе (например, нитробензоле) или в избытке алкилируемого
углеводорода (например, бензола):
С6Нб + С2НбС1 -^ С6НбС2Н5 + НС1
Реакция Фриделя—Крафтса — один из широко применяемых
методов синтеза.
В случае первичных галогеналкилов с тремя и более ато-
атомами углерода хлористый алюминий вызывает изомеризацию,
которая заключается в перемещении атома галогена к третич-
третичному, а при отсутствии такового — к вторичному атому углерода.
Вследствие этого галогеналкил присоединяется к ароматичес-
ароматическому углеводороду не концевым, в вторичным или третичным
углеродным атомом.
А1СЦ А1С13> СбНб
СН3СН2СНоС1 ►■ СН3СНС1СН3 >■ СбН5СН(СН3J;
А1С13 А1С13, С6Н6
(СН3JСН-СН2С1 >- (СН3KСС1 >■ СбН5-С(СН3K
При использовании в качестве алкилирующих агентов ди- и
тригалоидных соединений последние связывают соответственно
148
два или три ароматических радикала. Таким образом можно
получить дифенилметан, трифеыилметан и их производные:
А1С1з
СН2С12 + 2С6Нб >- СН2(СвНБ)а+2НС1;
А1С13
СНС13 + ЗС6Нб ►■ СН(СбН5K + ЗНС1
Дифенилметан и трифенилметац можно получить также из
хлористого бензила или хлористого бензилидена:
А1С13
QH5CH2C1+C6H6 > С6Н5СН2С6Н5 + НС1;
AlCla
С6НбСНС12 -{- 2С6Нв >• С6Н5СН(С6Н5J + НС1
Одной из стадий в производстве димедрола является:
А1С13
СС14 + 2С6Нб ►■ (С6Н5JСС12 + 2НС1
При помощи этого метода присоединить 4 фенильных ядра
к одному атому углерода нельзя
AlCIs
СС14 + ЗС6Н6 ►• (С6Н5KСС1 + ЗНС1
Аналогично галоидным производным ведут себя в реакции
Фриделя—Крафтса непредельные углеводороды эти-
этиленового ряда (алкены). Так, в присутствии хлорида алю-
алюминия из этилена и бензола получается этилбензол:
Сб^б -f- СН2=СН2 >• СбН5С2Н5
Этилбензол служит исходным продуктом для синтеза стиро-
стирола, который широко используется в промышленности полимер-
полимерных материалов. При реакции между бензолом и пропиленом
в присутствии хлористого алюминия образуется изопропилбен-
зол (кумол):
А1С13
СбН6 -f- СН2=СН—СН3 *" С6Н5СН(СНзJ
В связи с доступностью изопропилбензола (кумола) и высо-
высоким выходом продукта кумольный метод синтеза фенола приоб-
приобретает все большее значение.
Производство фенола кумольным методом заключается в
окислении кумола до гидроперекиси и далее разложении ее до
фенола и ацетона.
/СН3 /СНз
СНЧ m НО-О-С^
1ХСН3
+ °2110оС, Зат
Окисление изопропилбензола проводят воздухом в барботаж-
ной колонне из нержавеющей стали при давлении 3 ат и темпе-
149
ратуре 110°С. Жидкие продукты, содержащие 27—30% гидро-
гидроперекиси изопропилбензола, направляют для выделения изопро-
пилбензола в насадочную колонну, работающую при остаточ-
остаточном давлении 30 мм рт. ст. Здесь отгоняется основное количест-
количество изопропилбензола. Кубовый остаток, содержащий 70—75%
гидроперекиси изопропилбензола, перегоняют еще раз в ректи-
ректификационной колонне при остаточном давлении 10 мм рт. ст.
Полученную 90—92% гидроперекись изопропилбензола разла-
разлагают при 40—60°С 60% серной кислотой в реакторе с мешалкой
и рубашкой для охлаждения:
/СН3
НО-О—Сч ОН
]ХСН3 |
*" Г j) + СНзСОСНз
Реакционную массу нейтрализуют и передают на ректификацию
для выделения фенола и ацетона.
К этой же группе реакций следует отнести многочисленные
реакции с альдегидами и кетонами.
Конденсация формальдегида с фенолом приводит к получе-
получению фенолформальдегидных смол:
ОН - ОН ОН
/гСН2О + (/г+1)С6Н5ОН
-/гН2О
При пропускании хлористого водорода через смесь арома-
ароматического углеводорода и формалина в присутствии хлорида
цинка образуются производные хлористого бензила:
СН2О + Н3О+ -<
СН2ОН
■ СН2ОН + Н2О
RH + СН2ОН >- RCH2+ + Н2О
RCH2+ + СГ >■ RCH2C1
Эту реакцию иногда называют хлорметилированием. Полу-
Получение хлористого бензила хлорметилированием бензола эконо-
экономичнее хлорирования толуола. Другим преимуществом этого
способа является полное отсутствие в готовом продукте приме-
примесей веществ, содержащих атом хлора в ароматическом ядре, что
важно для производства фенилацетамида, предназначенного
для получения пенициллина.
В других условиях реакция с формальдегидом приводит к
образованию производных ди- и трифенилметана:
СН2О + Н3О+ =*
RH + СН2ОН -
RH + RCH2OH
CH2OH -
RCH2OH
> RCH2R
Н2О;
Н2О
150
Например:
H2SO4
NH2
Cl
HCl
-CH2O
+ сн2о
+ CClgCHO
хлораль
Формилхлорид НСОС1 является нестабильным соединением.
Однако соответствующий ему ион может быть использован при
пропускании в смесь ароматического соединения, хлорида алю-
алюминия и полухлористой меди безводных окиси углерода и хло-
хлористого водорода (ацилирование по Гаттерману — Коху):
А1С13
С6Нб + СО + НС1 ►■ С6Н5СНО + НС1
Другим методом синтеза альдегидов является использова-
использование амидов муравьиной кислоты (диметилформамида) в при-
присутствии хлорокиси фосфора (реакция Вильсмайера):
(CH3JN-C;f +POCI3
(CH3JN-C~O-P-C1
(CH3JN-C-O-P-C1
н
+ НС1
сг
Надо иметь в виду, что реакцию Вильсмайера можно про-
провести лишь с соединениями, обладающими высокой реакцион-
реакционной способностью. С бензолом и даже с нафталином она не про-
проходит.
151
Значительно большее значение имеет реакция Реймера и
Тимана, позволяющая синтезировать оксиальдегиды:
кон
О + НС1 + Н2О
+ снс1з
Активной частицей в этой реакции является соединение двух-
двухвалентного углерода — дихлоркарбен, который образуется при
взаимодействии хлороформа со щелочью:
СНС13 + КОН >- СС12 + КС1 + Н2О
Большая реакционная способность ароматического кольца
фенолятов позволяет получать оксикислоты при взаимодей-
взаимодействии безводных фенолятов с двуокисью углерода (реакция
Кольбе—Шмидта). Реакцию проводят в автоклаве, снабжен-
снабженном мощной лопастной мешалкой и специальной рубашкой для
обогрева паром высокого давления. Безводный фенолят натрия
нагревают до 180 °С и под давлением вводят в автоклав дву-
двуокись углерода. При этом образуется натриевая соль салицило-
салициловой кислоты:
ONa
+СО2
ОН
I
а
COONa
В настоящее время считают, что реакция проходит через ста-
стадию образования а-комплекса:
ONa
ОН
+ г/
COONa
В присутствии воды фенолят полностью диссоциирует, а по-
потому реакция не проходит.
Кроме фенолов, в эту реакцию вступают также аминофено-
лы. Таким образом, из м-аминофенола получают противотубер-
противотуберкулезный препарат — п-аминосалициловую кислоту (ПАСК):
ОН ОН
1, соон
+ со2—> Г ]
152
Практический интерес представляет синтез сложных
ароматических кетонов из хлорангидридов ароматиче-
ароматических кислот и ароматических углеводородов:
* СОС1
А1С13
Образующиеся 1,5- и 1,8-дибензоилнафталины разделяют,
пользуясь различной растворимостью их в хлорбензоле.
Большое промышленное значение имеет синтез бензоил-
бензойных кислот из фталевого ангидрида и арОхМатиче-
ских углеводородов в присутствии хлористого алюминия:
СО СООН
^4 Aids
Хлорид алюминия для проведения этой реакции берется в
количестве 2—2,5 моль на моль фталевого ангидрида. Так же,
как бензол, с фталевым ангидридом могут взаимодействовать
хлорбензол, толуол, нафталин и т. д.
о-Бензоилбензойная кислота и ее замещенные служат ис-
исходными продуктами ~для синтеза антрахинона и его
производных. Замыкание антрахинонового кольца является ре-
реакцией внутримолекулярного ацилирования и протекает в кис-
кислой среде:
ОН О
I /ОН I!
СООН +(Х С
+-н2о
Ацилирование фталевым ангидридом используется при синтезе
фенолфталеина, применяющегося не только как индикатор, но
и в качестве слабительного средства (пурген). Процесс прово-
153
дят в присутствии хлористого цинка при температуре 100—
105°С:
+н2о
§ 3. ПРОИЗВОДСТВО САЛИЦИЛОВОЙ КИСЛОТЫ
Салициловая кислота (о-оксибензойная) является одним из
важнейших продуктов химико-фармацевтической промышлен-
промышленности. Сама салициловая кислота имеет весьма ограниченное
применение в медицинской практике. Однако на основе сали-
салициловой кислоты производятся такие многотоннажные препара-
препараты, как ацетилсалициловая кислота (аспирин), фенилсалицилат
(салол), метилсалицилат и другие.
Как уже указывалось, салициловую кислоту получают кар-
боксилированием фенолята натрия. Приготовленный и предва-
предварительно упаренный до концентрации 60—70% раствор феноля-
фенолята натрия загружают в автоклав-карбонизатор. Аппарат снаб-
снабжен мощной лопастной мешалкой специальной конструкции,
лопасти которой вращаются между стальными ножами, закреп-
закрепленными на стенках аппарата. Скорость вращения мешалки
0,1 об/с.
Выпарку раствора фенолята натрия производят под атмос-
атмосферным давлением при нагреве реакционной массы до 170—
185 °С. Пары воды и фенола отводятся в холодильники, где
конденсируются и стекают в сборники. После упарки основной
массы воды включают вакуум и при работающей мешалке про-
проводят окончательное досушивание фенолята.
После окончателной досушки фенолята в автоклав подают
углекислый газ. Карбоксилирование начинают при температуре
50—60 °С, постепенно повышая температуру до 180—185 °С (за
7—8 ч). Давление в аппарате при этом около 6 атм.
После окончания карбоксилирования и отгонки фенола ре-
реакционную массу разбавляют водой и передавливают в аппарат
для выделения. Нейтрализацию щелочного раствора салицила-
та натрия проводят серной кислотой. Поскольку при этом вы-
выделяется большое количество тепла, реакционную массу охлаж-
охлаждают водой с помощью змеевиков или рубашки. Полученную
суспензию салициловой кислоты фильтруют на центрифуге. Пос-
После сушки салициловая кислота может быть в случае необходи-
необходимости очищена сублимацией при температуре 150—152 °С.
Салициловая кислота вызывает коррозию металлов. Ей хо-
хорошо противостоят алюминий и нержавеющая сталь марки
154
1X18H9T. Из этих материалов изготавливают большую часть
оборудования, работающего в среде салициловой кислоты. В
производстве салициловой кислоты в больших количествах при-
применяют растворы фенола, которые также вызывают коррозию
черных металлов. Однако скорость коррозии в этой среде неве-
невелика, а потому допускается эксплуатация аппаратуры из обыч-
обычной углеродистой стали.
§ 4. ТЕХНИКА БЕЗОПАСНОСТИ
Мероприятия по технике безопасности и охране труда при
проведении процессов алкилирования и ацилирования углеводо-
углеводородов аналогичны технике безопасности и охране труда при
проведении процессов ацилирования и алкилирования амино-
и оксисоединений (см. главу 7). Дополнительно к указанному
в главе 7, § 6 следует отметить опасность процессов, проводимых
в присутствии хлорокиси фосфора и хлорида алюминия. Оба эти
вещества легко разлагаются водой с выделением хлористого во-
водорода. Попадание воды в аппарат, где проводятся процессы с
применением этих веществ, может вызвать выброс реакционной
массы. Вследствие легкости разложения под действием влаги
перечисленные соединения хранят и транспортируют в герметич-
герметичной
Глава 9
ДИАЗОТИРОВАНИЕ И НИТРОЗИРОВАНИЕ.
ПРЕВРАЩЕНИЕ ДИАЗОСОЕДИНЕНИЙ
Вследствие очень высокой реакционной способности диазо-
и китрозосоединения часто используются в качестве промежу-
промежуточных продуктов в синтезах химико-фармацевтических препа-
препаратов. В связи с этим реакции диазотирования и нитрозирова-
ния, а также реакции превращения диазосоединений являются
весьма важными в технологии лекарственных веществ.
§ 1. МЕХАНИЗМ РЕАКЦИИ
Реакции диазотирования и нитрозирования относятся к ре-
реакциям злектрофильного замещения. Общая схема реакции
диазотирования:
RNH2 + NaNO2 + 2НХ > RN2X + NaX + 2H2O;
(X = HSO4, Cl, Вг, СЮ4 и т. д.)
не дает представления о действительном механизме реакции.
В настоящее время доказано, что активными реагентами в этой
реакции являются катионы или катионоидные частицы, образу-
образующиеся при взаимодействии минеральной кислоты, в среде ко-
которой проводится реакция, с азотистой кислотой, которая в свою
очередь образуется при взаимодействии нитрита с минеральной
кислотой. Такими частицами могут быть катион нитрозония
NO+, галоидный нитрозил NOHal, азотистый ангидрид N2O3,
нитрозацидий-катион H2NO2"ir. Активность самой азотистой кис-
кислоты значительно меньше, чем активность перечисленных актив-
активных частиц. Образование активных частиц протекает в соот-
соответствии со следующими уравнениями:
NaNO2 + HX >- NaX + HNO2;
HNO2 •<—*• Н+ + ЫОг";
HNO2 + H+ -<—> H2NOt;
NOr + H2NOj *=± N2O3 + H2O;
Н2Щ" + НаГ •<—>■ NOHal + H2O; (Hal-галоид)
H2NOj t—* H2O + NO.
Соотношение количеств образующихся активных реагентов
зависит главным образом от кислотности среды и природы
156
минеральной кислоты. При проведении реакции в соляной кис-
кислоте основным диазотирующим реагентом является хлористый
нитрозил NOC1, а при проведении реакции в среде серной кис-
кислоты — азотистый ангидрид N2O3. Катион нитрозония N0+ в
заметных количествах образуется лишь в концентрированной
серной кислоте.
Взаимодействие первичных аминов с диазотирующими реа-
реагентами протекает по схеме:
H'VX
icmio / \ <R N быстро
4
W //
быстро
N—04
быстро /=
= N -S~ II О
Вторичные амины реагируют с образованием N-нитрозосоедине-
ний:
н
*ГТ/*
АГк О"
N-NO
N-Нитрозосоединение может изомеризоваться в орто- или па-
ра-ыитрозосоединение:
N—N0
Alk
■> ON-<^\-NH--Alk
Третичные амины образуют п-нитрозопроизводные сразу:
Г*
N(AIk) + N^ —^-(AIk) U
Nx ^
(AIkJN
157
Продукт реакции, как правило, выделяется в виде осадка
интенсивно желтого цвета, представляющего собой соль следу-
следующего строения:
А1кч+ _-
Лимитирующей стадией процесса диазотирования является
нитрозирование. Амин вступает в реакцию в виде свободного
основания. Поскольку амин находится в состоянии динамиче-
динамического равновесия с солью
RNH2 + Н+ +=> RNl£,
реакция идет только за счет той части амина, которая находится
в виде свободного основания.
§ 2. УСЛОВИЯ ПРОВЕДЕНИЯ РЕАКЦИИ
Основными факторами, определяющими протекание процес-
процесса диазотирования в водной среде, являются температура веде-
ведения процесса, кислотность среды и соотношение реагентов.
Температура. Обычно диазотирование проводят при низ-
низких температурах @—5°С). Необходимость использования низ-
низких температур обусловлена тем, что с возрастанием температу-
температуры резко увеличивается скорость разложения соли диазония,
что уменьшает выход целевого продукта. Кроме того, низкая
температура способствует увеличению растворимости азотистой
кислоты и, следовательно, уменьшается способность улетучива-
улетучивания нитрозных газов (окислов азота). В отдельных случаях,
когда диазосоединение устойчиво, диазотирование может прово-
проводиться и при более высокой температуре. Необходимо помнить,
что реакция является экзотермичной и требует интенсивного
отвода тепла.
Кислотность среды. Основным условием проведения
реакции является наличие не менее 2 эквивалентов минераль-
минеральной кислоты на 1 эквивалент амина. В большинстве случаев
при практическом диазотировании количество кислоты превы-
превышает теоретически рассчитанное на 7г эквивалента и более. Во
время диазотирования и в конце его раствор должен показы-
показывать отчетливую кислую реакцию на конго, что соответствует
значению рН<2. Необходимость применения избытка кислоты
вызвана следующими причинами.
1. При более высоких значениях рН равновесие между ами-
амином и его солью смещается в сторону образования свободного
амина, плохо растворимого в воде, что затрудняет проведение
реакции.
158
2. При высоких значениях рН образующаяся соль диазония
может реагировать с исходным амином, образуя побочный про-
продукт — диазоаминосоединение.
3. При небольшой концентрации ионов водорода реакцион-
носпособные диазотирующие частицы NO+, NOC1, H2NO2+,
N2O3 переходят в неактивные формы — свободную азотистую
кислоту HNO2 и нитрит-ион NO2~.
4. Избыток минеральной кислоты повышает устойчивость
раствора диазосоединения.
Концентрация нитрита. Обычно используют 10—20%
раствор нитрита. В противоположность кислоте нитрит натрия
не применяется в избытке, так как при правильном ведении
процесса реакция диазотирования является практически коли-
количественной. С другой стороны, избыток нитрита оказывает не-
неблагоприятное влияние на устойчивость растворов диазосоеди-
диазосоединений и способствует смолообразованию. При правильно про-
проведенном диазотировании реакционная масса должна давать
сразу после окончания процесса слабую реакцию на азотистую
кислоту (фиолетовое окрашивание иодкрахмальной бумаги).
Перемешивание. Процесс диазотирования требует точ-
точного поддержания температуры (интенсивный отвод тепла) и
точного соотношения реагентов, поэтому интенсивное перемеши-
перемешивание реакционной массы является необходимой предпосылкой
для достижения хорошего результата. Обычно для ведения
процессов диазотирования используют быстроходные пропеллер-
пропеллерные или турбинные мешалки.
Как уже указывалось, амины, не содержащие кислотных
групп, растворяют в кислоте. При этом образуются соли аминов,
однако в реакцию диазотирования вступает не соль, а свобод-
свободное основание, которое находится в равновесии с солью. Если
в молекуле амина имеются кислотные группы SO3H, COOH, то
для растворения добавляют NaOH или Na2CO3 и образуются
растворимые натриевые соли. При выливании такого щелочно-
щелочного раствора в кислоту амин выпадает в мелкодисперсном со-
состоянии и диазотирование, несмотря на гетерогенность среды,
проходит нацело. Амины, являющиеся очень слабыми основа-
основаниями (например, 2,4-динитроанилин), растворяют в 100%
H2SO4 и диазотируют нитрозилсерной кислотой HSO4NO, полу-
получаемой растворением сухого NaNO2 в 100% H2SO4.
§ 3. ПРЕВРАЩЕНИЯ ДИАЗОСОЕДИНЕНИЙ.
ЗАМЕНА ДИАЗОГРУППЫ
Значение диазосоединений в промышленной и лабораторной
практике обусловлено их высокой реакционной способностью.
Одной из важнейших реакций диазосоединений является р е-
акция азосочетания. Реакция азосочетания, в результате
которой образуются азосоединения, происходит между ди-
159
азосоединениями и азосоставляющими. В качестве азосо-
ставляющих чаще всего используют амины и фенолы. Способ-
Способность этих веществ к реакции азосочетания обусловлена нали-
наличием сильных электронодонорных групп ОН и NH2, которые со-
создают частичные отрицательные заряды в орто- и пара-положе-
пара-положениях ароматического ядра. В эти места и вступают катионы
диазония:
Азосочетание является реакцией электрофильного замеще-
замещения. Диазосоединения участвуют в этой реакции в форме катио-
катиона диазония. Фенолы и нафтолы вступают в сочетание в ионизи-
ионизированной форме (в форме фенолят-анионов), а амины — в ви-
виде свободных оснований RNH2. Соли аминов не вступают в
реакцию азосочетания, так как группа ЫНз+ является не доно-
донором, а акцептором электронов и дезактивирует ароматическое
ядро.
Сочетание с фенолами и нафтолами чаще всего проводят в
слабощелочной среде (раствор соды), где они находятся в ак-
активной фенолятной форме. Сильнощелочные растворы NaOH не-
неблагоприятны для азосочетания, так как переводят соли диазо-
диазония в неактивную форму (антидиазотаты). Сочетание с амина-
аминами лучше вести в слабокислой среде (разбавленная уксусная
кислота). В этих условиях значительная часть амина находит-
находится в основной форме.
Так же как и диазотирование, азосочетание ведут при низ-
низких температурах, чтобы избежать разложения соли диазония.
Реакция проходит практически нацело. Продукт реакции, как
правило, нерастворим и может быть выделен из реакционной
массы фильтрованием.
Диазосоединения легко вступают также в другие реакции.
Строение и свойства диазосоединений меняются в зависимости
от значения рН среды. В кислой среде диазосодинения находят-
находятся в форме полностью диссоциированных солей диазония
R—N=NX~ (X — анион кислоты). При нейтрализации кислого
раствора образуется диазогидрат R—N=N—ОН, а при дальней-
дальнейшем повышении рН — син-диазотат R—N = NO~Na+. Син-диазо-
тат в сильнощелочной среде (иногда при нагревании) изомери-
зуется в транс-форму, которая называется анти-диазотат. При
подкислении анти-диазотат превращается в нитрозамин, кото-
160
рый далее превращается в диазогидрат и соль диазония. Все
эти превращения могут быть пояснены схемой:
. R-NeeN
Соль
диазония
QH"
*н+
R-N=N— ОН
Диазогид-
рат
R-NH—NO +
Нитрозамин
ОН
сн+
Син-диазотат
|он-
1 нагревание
R\
N=NXo_
Анти-диазотат
При стоянии растворы диазосоединений разлагаются. Наи-
Наиболее стабильны сильнокислые растворы, где диазосоединения
находятся в форме солей диазония. Наименее стабильны рас-
растворы при рН = 8—10. В сухом виде диазосоединения взрывча-
взрывчаты. При разложении диазосоединений образуется большое число
различных веществ, состав и количество которых зависит от
среды, температуры, наличия в растворе других веществ и т. п.
При нагревании растворов солей диазония диазониевая
группа может замещаться в зависимости от условий проведе-
проведения процесса на гидроксил, галоген и другие нуклеофильные
группы. Эта реакция'является одной из немногих реакций ну-
клеофильного замещения, в ароматическом ряду протекающих
по мономолекулярному механизму (Sjvl). Схема этих реакций
может быть представлена в следующем виде:
R — Nss
ROM +H
-^- ROCM
^
Лимитирующей стадией является отщепление азота от ка-
катиона диазония. Вторая стадия быстрая и скорость ее не зави-
зависит от концентрации нуклеофила. В слабополярных раствори-
растворителях превращения солей диазония могут протекать по ради-
радикальному механизму.
К числу важнейших реакций рассмотренного типа относится
замещение диазониевой группы гидроксилом:
RN? + ОН" > ROH + Na
Реакцию проводят при кипячении водного сернокислого рас-
раствора соли диазония. В качестве катализатора используют мед-
медный купорос.
Диазониевая группа может быть замещена галогеном, если
реакцию проводить в растворе соответствующей галогенводо-
11-926
161
родной кислоты в присутствии закисной соли меди (C112CI2 или
CB)
RNj + СГ ►■ RC1 + N2.
Аналогично, в присутствии закисной соли меди, действием
на соль диазония N-aCN можно заменить диазониевую группу
на —CN:
Си2С12
RN£ + CN" ►■ RCN+N2.
В специальных условиях возможно замещение диазониевой
группы водородом (гидрид-ионом). Реакцию проводят в присут-
присутствии восстановителей (Na2HPO2, Zn в кислой среде и т. п.):
RN+ + H" > RH + N2.
Соли диазония способны восстанавливаться до гидразинов.
В лабораторной практике в качестве восстановителей можно
использовать SnCl2 в среде соляной кислоты. В промышленно-
промышленности используют более дешевый восстановитель — бисульфит
натрия. Таким образом получают фенилгидразин из солянокис-
солянокислого фенилдиазония:
Л ^ , NaHSO3 Г/ ^
Р у— N2Cr >• У J—NH-NH2.HC1.
Фенилгидразин используется в химико-фармацевтической
промышленности для синтеза ряда препаратов — производных
1-фенил-3-метил-5-пиразолона.
§ 4. ПРОИЗВОДСТВО ГВАЯКОЛА
Гваякол A-окси-2-метоксибензол) производится химико-
фармацевтической промышленностью и служит исходным сырь-
сырьем для производства ванилина, который в свою очередь исполь-
используется для синтеза противотуберкулезного препарата фтивазид.
Гваякол применяют как полупродукт в производстве папавери-
папаверина. Гваякол представляет собой бесцветные или слабожелтые
кристаллы, плавящиеся при 28—30 °С или прозрачную светло-
желтого цвета жидкость со специфическим ароматическим за-
запахом и острым вкусом. Температура кипения гваякола 200—
205°С, он сравнительно плохо растворяется в воде A:60), хо-
хорошо — в спирте, эфире, хлороформе и уксусной кислоте. Горит
коптящим пламенем, перегоняется с водным паром, негигроско-
негигроскопичен, пары гваякола в смеси с воздухом взрывоопасны.
В Советском Союзе гваякол получают из о-анизидина по
схеме:
N=N HSO7
OCHg
162
NaNO2, H2SO4
Имеются патентные данные, говорящие о том, что обе опе-
операции (диазотирование о-анизидина и разложение о-метокси-
фенилдиазония) можно совместить в одном аппарате при не-
непрерывном ведении процесса. Связано это с тем, что при тем-
температуре ~100°С скорость разложения соли диазония значи-
значительно больше скорости диазотирования о-анизидина. К кипя-
кипящему раствору медного купороса в разбавленной серной кисло-
кислоте, содержащему о-анизидин, добавляют раствор нитрита
натрия, через реакционную массу пропускают перегретый водя-
водяной пар, дистиллат отгоняют со скоростью равной скорости при-
ливания раствора нитрита натрия. Выход гваякола при этом
методе производства составляет 80—82%. Основное достоинст-
достоинство метода — короткая аппаратурная схема и относительно ма-
малые энергетические затраты (нет охлаждения на стадии диазо-
диазотирования). Недостатки — трудность регулирования процесса,
большое смолообразование, пенообразование, выделение окис-
окислов азота вследствие термического разложения азотистой кис-
кислоты.
В Советском Союзе гваякол получают раздельным диазоти-
рованием о-анизидина и разложением сернокислого раствора
о-метоксифенилдиазония в присутствии медного купороса.
Процесс производства гваякола состоит из 8 стадий, из кото-
которых две химические:
1) приготовление раствора NaNO2;
2) получение сернокислого раствора о-анизидина;
3) диазотирование о-анизидина;
4) приготовление катализатора;
5) разложение сернокислой соли о-диазометоксибензола;
6) экстракция гваякола из гваяколовых вод;
7) вакуум-перегонка технического гваякола;
8) кристаллизация и фильтрация перегнанного гваякола.
Технологический процесс получения гваяко-
гваякола. Приготовление 30% раствора нитрита натрия ведут в эма-
эмалированном аппарате, снабженном лопастной или пропеллер-
пропеллерной мешалкой. Сначала в аппарат загружают необходимое ко-
количество воды, а затем при работающей мешалке загружают
нитрит натрия в количестве, рассчитаннбм с учетом процентного
содержания 100% вещества в техническом продукте (обычно
~98%). Перемешивание продолжают ~10 мин, после чего от-
отбирают пробу для анализа. Анализируют содержание нитрита
обычно по удельному весу раствора, который должен быть
1210—1220 кг/м3 при 20°С, что соответствует 30% содержанию
NaNO2.
Аппарат для приготовления сернокислого раствора о-анизи*
Дина и диазотирования его представляет собой эмалированный
стальной или чугунный реактор с рубашкой и змеевиком из
кислотостойкой стали для охлаждения и быстроходной мешал-
мешалкой. Предварительно в аппарате приготавливают 15—16% ра<>
11* 1ба
твор H2SO4, смешивая воду и техническую серную кислоту. Пос-
После охлаждения полученного раствора до 17—20 °С, не прекра-
прекращая перемешивания и охлаждения, постепенно приливают
рассчитанное количество о-анизидина. Температура реакцион-
реакционной массы при этом не должна превышать 20—25 °С, кислот-
кислотность реакционной массы после окончания загрузки о-анизиди-
о-анизидина должна быть 8,5—9%, а количество свободной серной кисло-
кислоты 1,25—1,26 моля на 1 моль амина (т. е. ~2,5 г-экв на 1 г-экв
амина). Небольшую часть полученного раствора (около 10—
15 л) отсасывают или передавливают в мерник и в дальней-
дальнейшем используют для снятия избытка азотистой кислоты после
диазотирования.
В полученный сернокислый раствор о-анизидина тонкой
струей из мерника постепенно загружают раствор NaNO2, конт-
контролируя скорость загрузки по температуре, которая не должна
превышать 20—25 °С. После слива почти всего раствора нитри-
нитрита определяют наличие избытка азотистой кислоты в реакцион-
реакционной массе по индикатору цинк-йод-крахмалу. Капля диазорас-
диазораствора с каплей индикатора должна дать синее окрашивание.
Последние порции раствора нитрита прибавляют очень осто-
осторожно, все время проводя контроль за избытком азотистой кис-
кислоты. Если проба с цинк-йод-крахмалом покажет наличие в ра-
растворе избытка азотистой кислоты (синее окрашивание не ис-
исчезает по истечении 10 мин), то избыток азотистой кислоты
снимают оставленным для этого раствором о-анизидина. В те-
течение всего процесса диазотирования среда должна быть силь-
сильнокислой (проба на конго). После окончания диазотирования в
цеховой лаборатории проверяют остаточную кислотность рас-
раствора, которая должна составлять 2,5—3,5% объемных в перес-
пересчете на серную кислоту.
Перед передачей на стадию разложения диазораствор ох-
охлаждают до температуры 5—7°С для кристаллизации сульфа-
сульфата натрия, выдерживают при этой температуре 1 ч и отфильтро-
отфильтровывают выпавший в осадок сульфат.
Катализатор для разложения диазораствора готовят раство-
растворяя медный купорос в разбавленной серной кислоте. Концент-
Концентрация медного купороса должна быть 28—30%, а концентрация
серной кислоты—1,5—2%.
Полученный катализатор нагревают до кипения A02—
103 °С) и, поддерживая кипение, подают в аппарат диазорас-
диазораствор и гваяколовую воду (т. е. раствор гваякола в воде, получа-
получающийся в качестве отхода) в соотношении 1:2 по объему. Ко-
Количество отгоняемой из аппарата воды и гваякола (паров)
должно быть равно сумме загружаемых количеств диазораство-
диазораствора и гваяколовой воды, так как аппарат для разложения рабо-
работает при постоянном уровне реакционной массы, который под-
поддерживается автоматически. Во время отгонки гваякола темпе-
температура воды на выходе из холодильника должна быть 35-*-
164
45 °С во избежание кристаллизации гваякола в трубках холо-
холодильника и закупорки системы. Образовавшийся в холодильни-
холодильнике конденсат, содержащий 4—5% гваякола, стекает в отстойник
непрерывного действия, где разделяется на гваякол и гваяколо-
вую воду. Отгонку гваякола считают законченной, если в про-
пробе конденсата отсутствуют капли гваякола.
Экстракцию гваякола из гваяколовой воды ведут дихлор-
дихлорэтаном. Экстракцию проводят дважды, после чего содержание
гваякола в гваяколовой воде падает от 2 до 0,5%. После от-
отгонки дихлорэтана получают технический гваякол, который со-
соединяют с гваяколом отделенным от гваяколовой воды в отстой-
отстойнике непрерывного действия и подают для дальнейшей очистки
в аппарат для обезвоживания и вакуум-перегонки.
Обезвоживание проводят при температуре 50—70 °С и ва-
вакууме 500—550 мм рт. ст. и заканчивают при температуре в па-
парах 112—116°С. Обезвоженный гваякол перегоняют в вакуум-
перегонном аппарате при температуре 105—120 °С и остаточ-
остаточном давлении 10—15 мм рт. ст. Перегонку считают законченной
при падении температуры в парах. Следует отметить, что тем-
температура охлаждающей воды на выходе из холодильника долж-
должна быть 30—35°С. При более низкой температуре гваякол мо-
может закристаллизоваться и закупорить холодильник.
Кристаллизацию гваякола ведут в алюминиевом аппарате с
рубашкой для охлаждения и мешалкой. Перегнанный гваякол
охлаждают до 15—20 °С, выдерживают при этой температуре
для полной кристаллизации. Взвесь кристаллов гваякола в
гваяколовом маточнике через нижний спуск передают на цент-
центрифугу. Кристаллический гваякол выгружают в деревянные
ящики, выложенные черной светонепроницаемой бумагой и пер-
пергаментом. Гваяколовые маточники собирают и передают на ва-
вакуум-перегонку. Их можно также использовать для синтеза ва-
ванилина наряду с техническим гваяколом.
Используемые в производстве гваякола о-анизидин, дихлор-
дихлорэтан, нитрит натрия являются сильными ядами. о-Анизидин, ди-
дихлорэтан и гваякол образуют взрывоопасные смеси с воздухом.
Кроме того, при нарушении технологического режима и разгер-
разгерметизации аппаратуры в производственное помещение могут
проникнуть токсичные окислы азота, которые могут образовать-
образоваться как на стадии диазотирования, так и на стадии разложения.
Основными профилактическими мероприятиями, обеспечиваю-
обеспечивающими безопасность работы при производстве гваякола, являются
герметизация аппаратуры, исправная приточно-вытяжная вен-
вентиляция, обеспечение работающих предохранительными средст-
средствами (противогаз марки А, перчатки из стойкой резины, фар-
фартук, спецодежда, респиратор и т. п.), заземление аппаратуры и
применение неискрящих инструментов, систематическая провер-
проверка знания всеми работниками цеха правил техники безопасно-
безопасности и правил безопасного ведения процесса.
Глава 10
ВОССТАНОВЛЕНИЕ И ОКИСЛЕНИЕ
Восстановление и окисление — неразрывно связанные между
собой реакции. Если одно из веществ, участвующих в реакции,,
окисляется, то другое восстанавливается, поэтому отнесение од-
одних реакций к реакциям восстановления, а других — к реакци-
реакциям окисления чисто условное, связанное с технической направ-
направленностью реакции, с тем, какое вещество является в данном
случае целевым продуктом. При этом в названии всегда указы-
указывается интересующий нас объект реакции (например, реакция
восстановления нитросоединений или реакция окисления толуо-
толуола). Восстановлением называют процесс, в результате которого
атом или группа атомов приобретают электроны. Окисление
заключается в потере электронов.
Наряду с изменением числа электронов, которыми облада-
обладает данный атом или группа атомов в процессе окислительно-вос-
окислительно-восстановительных реакций изменяется и состав молекулы. Напри-
Например, при восстановлении двойной связи молекула приобретает
2 атома водорода:
Н Н
II \ /он
^.С=С >- -С—С— или —С=О >- X/ ,
II II I / \н
а при восстановлении спиртов или нитросоединений молекула
теряет атом кислорода:
Н Н 0
I I /Г
—С—ОН > —С—Н или -N4 > -N=0 >
I I
H H
Многочисленные методы восстановления и окисления можно
разделить на четыре группы: химические, каталитические, элек-
электролитические и биохимические (или микробиологические). Все
эти методы находят широкое применение в химико-фармацев-
химико-фармацевтической промышленности.
§ 1. ВОССТАНОВЛЕНИЕ МЕТАЛЛАМИ
Наиболее разнообразную группу реакций восстановления
занимают химические методы. Особенно часто в качестве вос-
восстановителей используются металлы и их соли.
Восстановление натрием. Известны три метода восстановле-
восстановления натрием: восстановление амальгамой натрия, восстановле-
166
иие натрием и спиртом, восстановление растворами натрия в
жидком аммиаке.
Восстановление амальгамой натрия. Высокая
реакционная способность натрия не позволяет применять его в
водной среде. Натрий в виде амальгамы (т. е. в виде раствора
в ртути) реагирует с водой значительно медленнее, что позволя-
позволяет использовать амальгаму натрия в качестве восстановителя.
Амальгама, содержащая меньше 1,25% натрия, является жид-
жидкой при комнатной температуре. При более высокой концентра-
концентрации натрия амальгама твердая и ее можно измельчать механи-
механически. Процесс растворения натрия в ртути ведут под слоем то-
.луола или в атмосфере азота, чтобы предохранить теплую
амальгаму от воздействия кислорода воздуха. Необходимо
иметь в виду, что процесс растворения натрия в ртути является
экзотермичным. Обычно получение амальгамы проводят при
40—50°С, получая таким образом 2—3% амальгаму. Наиболее
чистую амальгаму получают электролитическим методом. При
зтом ртуть служит катодом, а платиновый анод помещают в
раствор натриевой соли (обычно хлорида или бикарбоната нат-
натрия). Таким способом получают жидкую чистую и однородную
амальгаму, содержащую до 0,5% натрия.
Восстановление проводят в водной или спиртовой среде.
Органическое соединение может находиться в растворе или
в виде суспензии. В некоторых случаях применяют эмульгиру-
эмульгирующие средства или органические растворители, смешивающиеся
с водой и спиртом. К смеси постепенно добавляют амальгаму
в количестве на 25—40% превышающем теоретическое. Об
окончании процесса судят по полному разложению амальгамы
и выделении ртути, которую отделяют декантацией и промыва-
промывают водой или спиртом. Скорость, а иногда и состав продуктов
восстановления зависят от величины рН. Амальгама натрия
оказывает значительно более сильное восстанавливающее дей-
действие в щелочных средах. Точный и непрерывный контроль зна-
значения рН осуществляют потенциометрически. Величину рН ре-
регулируют пропусканием СО2.
В настоящее время общепринятым считается ионный меха-
механизм восстановления амальгамой натрия. Первой стадией ре-
реакции является присоединение 1 или 2 электронов к молекуле
восстанавливаемого соединения, которое происходит в адсорб-
адсорбционном слое на поверхности металла. Затем образующиеся
анионные радикалы или двухзарядные анионы соединяются с
протонами. Эти два варианта общего механизма восстановле-
восстановления можно представить следующими схемами (R — молекула
восстанавливаемого соединения):
+2e
R > R2- > RH"" > RH2;
R —X R" —> RH -^X RH"" —> RH2.
167
Этиленовые изолированные связи не восстанавливаются
амальгамой натрия, сопряженные же двойные связи легко под-
поддаются восстановлению. Ароматические соединения бензольно-
бензольного ядра восстанавливаются амальгамой натрия лишь в отдель-
отдельных случаях (например, фталевые кислоты). Ароматические
полициклические углеводороды легко восстанавливаются до ди-
или тетра-гидропроизводных. Легкость восстановления возра-
возрастает в следующем порядке: дифенил, трифенил, нафталин, фе-
нантрен, антрацен. Очень легко восстанавливается амальгамой
натрия карбонильная группа в альдегидах и кетонах. Восста-
Восстановление в этих случаях обычно идет до соответствующих спир-
спиртов. Гидроксильные группы на водород под действием амальга-
амальгамы натрия, как правило, не обмениваются. Карбоновые кисло-
кислоты также устойчивы к действию амальгамы. Нитро- и другие
способные к восстановлению азотсодержащие группы легко
восстанавливаются амальгамой натрия, но практического зна-
значения этот метод не получил вследствие наличия более доступ-
доступных восстановителей.
Восстановление натрием со спиртом. Этим ме-
методом, названным по имени его изобретателей методом Буво —
Блана, восстанавливают сложные эфиры карбоновых кислот до
соответствующих спиртов, восстановление ведут металлическим
натрием в среде абсолютного спирта. Суммарное уравнение ре-
реакции имеет вид:
RCOOC2H5 + 4Na + ЗС2НбОН > RCH2OH + 4C2H5ONa.
Метод Буво — Блана имеет большое значение для получения
высших алифатических спиртов, синтез которых другим путем
осуществить трудно. У многоосновных кислот при этом восста-
восстанавливаются только этерифицированные карбоксильные груп-
группы.
Способ восстановления сложных эфиров по этому методу за-
заключается в следующем.
К кипящей смеси сложного эфира и обычно пятикратного
количества абсолютного спирта (обязателен хорошо действую-
действующий обратный холодильник) добавляют кусочки или гранулы
металлического натрия, взятого в небольшом избытке. После
загрузки всего натрия и необходимой выдержки реакционную
массу выливают в воду для разложения образовавшихся алко-
голятов. Спирт отгоняют, а продукты реакции извлекают экст-
экстракцией. Тепловой эффект реакции очень высок A25 ккал/моль),
а потому отвод тепла является важной технической проблемой.
Воду в качестве хладоагента применять нельзя (металлический
натрий!). В ряде случаев весьма удобным является изменение
порядка загрузки реагентов, а именно прибавление спиртового
раствора эфира к металлическому натрию или его эмульсии в
инертном растворителе. Этиловый спирт можно заменить высши-
высшими спиртами, включая амиловый. Это позволяет поднять темпе-
168
ратуру процесса и облегчает задачу охлаждения реакционной
массы.
Выход продукта, как правило, не превышает 80%. Потери
обусловлены непосредственной реакцией между натрием и
спиртом. Выделяющийся водород неактивен, а образующиеся
алкоголяты вызывают нежелательные побочные реакции, что
приводит к образованию высококипящих побочных продуктов.
Восстановление натрием в жидком аммиаке.
Жидкий аммиак и низшие алифатические амины обладают спо-
способностью растворять щелочные металлы. Аммиак хотя и реа-
реагирует с натрием:
2NH3 + 2Na > 2NaNH2 + H2,
однако в отсутствие катализаторов и при наличии соединений,
способных восстанавливаться, этот процесс практического зна-
значения не имеет. Хорошая растворимость щелочных металлов
(кроме натрия, в этом методе можно применять также калий
и литий) в аммиаке позволяет работать в широком интервале
концентраций. Этот метод делает возможным восстановление
соединений, которые гидролизуются в воде. Схематически реак-
реакцию можно изобразить
R + Na + NH3 >- RH +
При восстановлении раствором натрия или других металлов в
жидком аммиаке необходимо иметь в виду, что температура
кипения раствора очень низка (—33,5 °С), поэтому для ведения
процесса необходима специальная герметичная аппаратура. При
повышенных температурах реакция должна проводиться в ав-
автоклаве. Для достижения большей поверхности контакта необ-
необходимо энергичное перемешивание. Эфиры реагируют с раство-
раствором натрия быстрее и с большим выходом, чем по методу Бу-
во — Блана. Галогенопроизводные легко реагируют с раствором
натрия в аммиаке. Реакция идет в двух направлениях:
2RX + 2Na + NH3 > RH + RNH2 + 2NaX;
RX + 2Na + NH3 > RH + NaNH2 + NaX.
Соединения ацетиленового ряда восстанавливаются по этому
методу до олефинов, и образующиеся продукты имеют исклю-
исключительно транс-шнфигурацию:
Na, NH3 H\ /R
R—C=C-R >• ;C=G
r/ \h
Моноалкилацетилены в обычных условиях не восстанавли-
восстанавливаются. Благодаря этому возможно избирательное восстановле-
восстановление соединений с изолированными тройными связями, из кото-
которых одна находится на конце цепи.
Восстановление оловом и его солями. Восстановление оло-
оловом и его хлоридом — простой и давно известный способ, на-
нашедший широкое применение в лабораторной практике. Особен-
169
но удобен он для превращения ароматических нитросоединений
в амины, которые находят широкое применение в качестве про-
промежуточных соединений при синтезе фармацевтических препа-
препаратов. Большого промышленного применения этот метод не на-
нашел по чисто экономическим соображениям.
Восстановление оловом. Методика восстановления
нитросоединений оловом проста. К раствору или суспензии ни-
тросоединения в концентрированной соляной кислоте, взятой в
небольшом избытке, прибавляют небольшими порциями грану-
гранулированное олово. Реакцию начинают осторожно нагревая на
водяной бане реакционную смесь после загрузки первой пор-
порции олова. Если олова загрузили слишком много, реакция мо-
может начаться слишком бурно и потребуется быстрое охлажде-
охлаждение реакционной массы для предотвращения ее выброса. Как
правило, реакцию ведут, поддерживая спокойное кипение реак-
реакционной массы (обратный холодильник) до получения прозрач-
прозрачного раствора. Затем быстро отфильтровывают непрореагиро-
вавшее олово через фильтр, устойчивый к действию соляной
кислоты (пористая керамика, стеклянная вата). Раствор, со-
содержащий амин в виде комплексной соли с хлористым или
хлорным оловом, подщелачивают аммиаком или щелочью. Ес-
Если продукт реакции перегоняется с водным паром, то его отго-
отгоняют прямо из реакционной массы. В других случаях амин эк-
экстрагируют подходящим растворителем.
Восстановление нитросоединений оловом можно описать
уравнением:
R-NO2 + 3Sn + 6HC1 > R-NH2
Восстановление протекает ступенчато через ряд промежу-
промежуточных соединений:
R-NO2 ► R-NO ь R-NHOH > R-NH2.
Образующийся первоначально хлорид олова (II) может
вступать во взаимодействие с нитросоединением в качестве вое-
тановителя:
R_NO2 -f 3SnCl2 + 6НС1 > R—NH2 + 3SnCl4 + 2H2O.
Восстановление хлоридом олова. Хлорид олова
(II) оказывает более сильное и специфическое восстанавлива-
восстанавливающее действие, чем металлическое олово и часто дает лучшие
результаты. Хлорид олова восстанавливает до. аминов также
азосоединения:
R_N=N-R + 2SnCl2 + 4HC1 > 2RNH2 + 2SnCl4.
Характерной особенностью хлорида олова является его рас-
растворимость в воде и в этиловом спирте, что позволяет вести вос-
восстановление в гомогенной среде. Благодаря этому хлорид оло-
олова восстанавливает быстро и с хорошими выходами и при низ-
низких температурах, хотя реакцию можно вести и при температуре
кипения. Проведение восстановления хлоридом олова не вы-
170
зывает особых затруднений и в основном не отличается от мето-
методики восстановления оловом. Порядок загрузки реагентов осо-
особой роли* не играет. Хлорид олова не восстанавливает гетеро-
гетероциклические группировки, благодаря чему его можно использо-
использовать для получения аминопроизводных хинолина, ксантона, фе-
ноксазина. Как правило, хлорид олова не вытесняет галоиды,
что дает возможность получать галоидариламины с хорошими
выходами. Наиболее важной особенностью хлорида олово (II)
является возможность избирательного восстановления нитро-
групп в полинитросоединениях. Наиболее отчетливо избиратель-
избирательность восстановления проявляется в тризамещенных производ-
производных бензола с двумя нитрогруппами в положениях 2 и 4 отно-
относительно третьего заместителя. Так, в 2,4-динитротолуоле хло-
хлорид олова восстанавливает нитрогруппу в положении 2, прак-
практически не затрагивая нитрогруппу в положении 4. Хлорид оло-
олова (II) восстанавливает группу диазония до гидразогруппы:
+ SnCl2, HC1
R-N=N СГ >- R-NH-NH2.HC1.
Регенерацию олова при восстановлении оловом или хлори-
хлоридом олова лучше всего осуществлять электролитическим осаж-
осаждением олова на катоде. При этом способе может быть достиг-
достигнут практически полный возврат олова в цикл. В лабораторной
практике иногда пользуются осаждением олова в виде нерас-
нерастворимого сульфида при пропускании сероводорода через реак-
реакционную массу.
Восстановление цинком. Восстановление цинком проводят
как в кислой, так и в щелочной среде. Восстановление цинком
в кислой среде аналогично восстановлению оловом. Однако в
отличие от восстановления оловом образующийся хлорид цин-
цинка ZnCb восстановительными свойствами не обладает. Промыш-
Промышленное значение получило восстановление цинком в щелочной
среде. Конечным продуктом восстановления ароматических ни-
тржюединений цинком в щелочной среде являются гидразоеое-
динения. Процесс может быть выражен общим уравнением:
2RNO2 + 5Zn + lONaOH >- R-NH-NH-R + 5Na2Zn02 + 4H2O.
Помимо собственной значимости, ароматические гидразосоеди-
нения важны тем, что они обладают способностью в кислой сре-
среде перегруппировываться в соответствующие производные
4,4/-диаминодифенила (бензидина):
R-NH-NH-R >■ H2N~-R-R-NH2.
Ци'нк обычно используют в виде цинковой пыли, так каж по-
последняя обладает большой удельной поверхностью. Необходи-
Необходимое для восстановления количество цинковой пыли на 10—15%
больше теоретически рассчитанного. Образующийся цинкат в
условиях реакции гидролизуется, давая гидроокись цинка:
Na2ZnO2 + 2H2O >■ Zn(OHJ + 2NaOH.
171
Таким образом, едкий натр в реакции практически не расхо-
расходуется, поэтому для проведения восстановления его берут 0,1—►
0,2 моля на 1 моль нитросоединения. Для успешного проведе-
проведения восстановления необходимо энергично перемешивать до-
довольно вязкую массу, в которую для уменьшения вязкости иног-
иногда добавляют органические растворители. При этом продукты
реакции переходят в раствор, что облегчает их выделение. Вы-
Высокая концентрация щелочи может привести к образованию
амина наряду с гидразосоединением. Однако при недостаточной
щелочности процесс идет медленно, поэтому процесс разделяют
на две стадии. Сначала ведут восстановление в концентриро-
концентрированной щелочи, причем цинковую пыль и щелочь строго дози-
дозируют. На этой стадии образуются азокси- и азосоединения. За-
Затем образовавшиеся соединения восстанавливают новой порцией
цинковой пыли, но уже в разбавленном растворе щелочи. Тем-
Температура на первой стадии 80—90 °С, а на второй 65—70 °С.
Гидразосоединение выпадает в виде осадка вместе с гидро-
гидроокисью цинка. Для растворения последней реакционную массу
осторожно подкисляют до нейтральной реакции и отделяют ги-
гидразосоединение от раствора цинковой соли фильтрованием.
Восстановление нитросоединений цинком в щелочной среде про-
проводят в стальных или чугунных аппаратах с якорной мешалкой
и рубашкой.
При восстановлении цинком в щелочной среде контролируют
температуру и рН среды. Образующиеся в процессе восстанов-
восстановления азосоединения имеют интенсивную окраску. При дальней-
дальнейшем восстановлении они дают неокрашенные гидразосоедине-
ния, поэтому реакцию прекращают сразу после обесцвечивания
раствора, так как дальнейшее восстановление может привести
к появлению аминов в реакционной массе.
Бензидиновую перегруппировку осуществляют в стальном
аппарате, футерованном кислотоупорной плиткой или в эмали-
эмалированном реакторе с мешалкой; перегруппировку гидразосоеди-
нения в бензидиновое основание — в кислой среде. Продукт в
виде соли выделяют высаливанием NaCl или Na2SO4 и фильтро-
фильтрованием полученного осадка. Следует иметь в виду, что многие
производные бензидина являются канцерогенными веществами,
что требует соблюдения особых мер предосторожности на этой
стадии.
При бензидиновой перегруппировке также контролируют
температуру реакционной массы и рН среды.
В присутствии кислот цинк восстанавливает хлорангидриды
арилсульфокислот до тиофенолов:
RSO2C1 >■ RSO2H > RSOH > RSH.
Подобно большинству других восстановителей цинк в кислой
среде восстанавливает карбонильные соединения до одно- или
двухатомных спиртов. Замещение же кислорода двумя атомами
172
водорода можно осуществить при помощи амальгамы цинка в
среде соляной кислоты (метод Клемменсена):
ZnHgx + HC1
R-CO-Ri ►■ R—CHa-Ri + HaO.
Восстановление амальгамой цинка применяется исключительно
для альдегидов и кетонов, спирты таким образом не восстанав-
восстанавливают.
Серьезной проблемой при восстановлении цинком и оловом
является очистка производственных сточных вод.
Восстановление железом. Восстановление ароматических нит-
росоединений до соответствующих аминов железом (чугунными
стружками) в присутствии электролитов является одним из рас-
распространенных промышленных методов. Теоретические основы
этого процесса были разработаны В. О. Лукашевичем, который
показал, что при восстановлении нитросоединений железом од-
одновременно протекает четыре реакции:
Fe + 2Н2О >- Fe(OHJ + 2Н;
6Fe(OHJ + 4Н2О + RNO2 ► 6Fe(OHK + RNH2;
RNO2 + 6H >- RNH2 + 2H2O;
Fe + 8Fe(OHK >• 3Fe3O4 + 12H2O.
Скорость процесса восстановления лимитируется первой ста-
стадией, которая аналогична процессу влажной коррозии железа.
В связи с этим (для ускорения процесса влажной коррозии же-
железа) восстановление ведут в среде электролита, и для ведения
реакции используют не железо, а чугунную стружку. Природа
и концентрация электролита играют большую роль. Наиболее
активным электролитом является хлорид аммония, менее актив-
активны хлорид железа (II), сульфат аммония, хлорид натрия и т.д.
Оптимальная концентрация электролита ~3 н. Электролиты
вводят в реакционную массу в готовом виде (хлорид аммония}
или получают в самом реакторе при протравливании чугунной
стружки соляной кислотой:
Fe + 2НС1 >- FeCl2 + 2Н.
Для восстановления лучше всего использовать стружку се-
серого чугуна, которая является отходом металлообрабатываю-
металлообрабатывающих производств. Активность серого чугуна объясняется возник-
возникновением в присутствии электролитов гальванических элемен-
элементов на границе раздела железо—графит. Вследствие зернисто-
зернистого строения серый чугун в процессе восстановления распадает-
распадается на мелкие частицы, что ведет к ускорению восстановления.
Скорость реакции восстановления сильно зависит также от рН
среды. При рН>12 процесс практически прекращается. Как
правило, восстановление железом ведут в слабокислой среде.
Чугунная стружка должна быть специально подготовлена
Для реакции. После измельчения и просева, а также удаления
пыли, чугунную стружку обезжиривают и протравливают не-
173
большим количеством соляной кислоты, что увеличивает ее ак-
активность вследствие образования хлоридов.
В производстве восстановление проводят в стальных или чу-
чугунных аппаратах (редукторах), футерованных диабазовой
ллиткой на кислотоупорной замазке и снабженных мешалкой
(лопастной или сошниковой) и барботером для подачи остро-
острого пара.
Процесс восстановления чугунной стружкой ведут при тем-
температуре кипения реакционной массы. Обогрев чаще всего про-
производят острым паром, который служит не только теплоносите-
теплоносителем, но также выводит образующийся амин из сферы реакции
(перегонка с водяным паром).
В большинстве случаев нитропродукт постепенно загружают
в нагретую до температуры кипения суспензию чугунной струж-
стружки в растворе электролита. Каждую следующую порцию вво-
вводят лишь после того, как прореагирует предыдущая. В некото-
некоторых случаях порядок загрузки может быть изменен. Если полу-
получающийся амин летуч с водяным паром, то часть его отгоняется
с водяным паром во время процесса. Смесь паров воды и амина
конденсируется в холодильнике, после чего поступает в отстой-
отстойник для разделения. Остальную часть амина отгонят с водя-
водяным паром после окончания процесса восстановления и подще-
лачивания реакционной массы. Иногда выделившийся амин
сифонируют после предварительного отстаивания реакционной
массы в редукторе. Амины, плохо перегоняющиеся с водяным
паром, экстрагируют из реакционной массы органическими ра-
растворителями.
Наряду с несомненными достоинствами, к которым относит-
относится простота технологии, дешевизна сырья, высокий выход це-
целевого продукта реакции, процесс восстановления чугунными
стружками имеет и ряд недостатков. Как уже говорилось, чу-
чугунные стружки являются отходом металлообрабатывающих
производств. С развитием технологии металлообработки коли-
количество отходов этого производства уменьшается, что приводит
к сокращению сырьевой базы для процесса восстановления по
этому методу. Условия восстановления в значительной мере за-
зависят от качества чугунной стружки, но поскольку эта стружка
поступает с других предприятий, где она является отходом, ка-
качество ее нестабильно. Наконец, самая большая проблема —
это утилизация образующегося шлама. Фильтрация и транс-
транспортировка плохо фильтрующегося тяжелого и содержащего
абразивные частицы осадка представляет серьезные трудности.
Принципиально возможно восстановление железного шлама до
железа в мартеновских печах, однако для металлургов этот
шлам является плохим сырьем, и металлургические заводы бе-
берут его неохотно. Добавка различных электролитов в реакцион-
реакционную массу может привести к изменению состава шлама и его
цвета. В ряде стран этим пользуются для получения из желез-
174
ного шлама минеральных пигментов желтого, оранжевого и ко-
коричневого цветов. В СССР этот метод применения не нашел,
так как наша страна обладает большими запасами природного
сырья для производства минеральных пигментов.
§ 2. ВОССТАНОВЛЕНИЕ СУЛЬФИДАМИ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Сульфиды щелочных металлов применяются для восстанов-
восстановления нитросоединении ароматического ряда до аминов. Впер-
Впервые сульфид для этой цели применил Н. Н. Зинин, получивший
анилин при восстановлении нитробензола сульфидом аммония.
В промышленной практике чаще применяют сульфид натрия
Na2S, сульфгидрат натрия NaHS и полисульфиды натрия
Na2Cn (n = 2, 3...). В ди- и тр'инитросоедииениях сернистые щело-
щелочи могут избирательно восстанавливать лишь одну нитрогруп-
пу, не затрагивая остальные. В нитроазосоединениях они восста-
восстанавливают нитрогруппу, не затрагивая азогруппы. В некоторых
случаях сернистые щелочи используют и для восстановле-
восстановления мононитросоединений до соответствующих аминов. Сернис-
Сернистые щелочи могут полностью восстановить нитрогруппы и в по-
линитросоединениях, но в этих случаях реакцию проводят в бо-
более жестких условиях, чем при частичном восстановлении. При
использовании сернистых щелочей в качестве восстановителей
продукт реакции выделяется легче, а технология восстановле-
восстановления проще, чем при использовании чугунной стружки. Значи-
Значительно меньше в этом случае также и коррозия оборудования.
Однако реакции с сернистыми щелочами сопровождаются выде-
выделением сероводорода, а обезвреживание сточных вод, содержа-
содержащих вредные соединения серы, вызывает серьезные труд-
трудности.
При восстановлении нитросоединении сульфидом натрия воз-
возрастает щелочность среды:
4RNO2 + 6Na2S + 7H2O > 4RNH2 + 3Na2S2O3 + 6NaOH.
Повышенная щелочность среды благоприятствует образова-
образованию азокрасителей, что снижает выход целевого продукта и
ухудшает его качество. Этот эффект особенно заметен при ча-
частичном восстановлении полинитросоединений, в связи с чем
сульфид натрия используют лишь при восстановлении мононит-
мононитросоединений до аминов (в частности, при восстановлении о-
и п-нитроанизолов до о- и п-анизидинов).
Восстановление сульфгидратом (гидросульфидом) натрия в
этом отношении значительно удобнее, так как щелочность сре-
среды во время реакции не увеличивается:
4RNO2 + 6NaHS + H2O >■ 4RNH2 + 3Na2S2O3.
Недостатком этого восстановителя является то, что гидро-
гидросульфид приходится приготавливать непосредственно перед ис-
175
пользованием, насыщая водный или спиртовой раствор едкого
натра рассчитанным количеством сероводорода.
При использовании дисульфида натрия Na2S2 реакция про-
протекает по уравнению:
RNO2 + Na2S2 + H2O > RNH2 + Na2S2O3.
Восстановление трисульфидом Na2S3 проходит с выделением
серы, освобождение от которой представляет значительные
трудности:
г + Na2S3 + Н2О > RNH2 + Na2S2O3 + S.
Полисульфиды натрия получают нагреванием раствора Na2S
с рассчитанным количеством серы:
— 1)S > Na2Sn.
Восстановление сернистыми щелочами проводится следую-
следующим образом. К нагретой эмульсии или суспензии нитросоеди-
нения в воде постепенно при сильном перемешивании прилива-
приливают 15—25% раствор сульфида. При исчерпывающем восста-
восстановлении избыток сульфида должен составлять 20—40% по
сравнению с теоретическим количеством, а при избирательном
восстановлении — не более нескольких процентов. Температура
реакционной массы 80—90 °С. В некоторых случаях реакцию
проводят при кипении реакционной массы. Длительность вос-
восстановления сильно зависит от температуры и обычно составля-
составляет несколько часов.
При восстановлении сернистыми щелочами среда не являет-
является коррозионной, поэтому в качестве редуктора может быть ис-
использован стальной или чугунный аппарат с рубашкой и про-
пропеллерной или турбинной мешалкой. Поскольку процесс прохо-
проходит при кипении реакционной массы и вследствие разложения
сульфидов может выделяться сероводород, редуктор должен
быть оборудован обратным холодильником и ловушкой для
улавливания сероводорода.
Твердые, нерастворимые в воде амины выделяют при филь-
фильтровании реакционной массы на нутч-фильтрах или центрифу-
центрифугах. Жидкие нерастворимые амины отстаивают в делительных
воронках. Растворимые в воде амины экстрагируют или выса-
высаливают.
Маточники перерабатывают следующим образом. Оставши-
Оставшиеся сульфиды окисляют до Na2S2O3 кислородом воздуха, полу-
полученный раствор тиосульфата упаривают и очищают активиро-
активированным углем, а из полученного горячего концентрированного
раствора при охлаждении выделяют технический тиосульфат,
который очищают перекристаллизацией и выпускают в качест-
качестве товарного продукта.
176
Восстановление ароматических нитросоединений сернистыми
щелочами применяется в производствах о- и п-анизидинов:
H3CO"
о-Анизидин получается также при восстановлении о-нитро-
анизола чугунной стружкой (выход 90—94%). Однако при этом
методе производства возникают значительные трудности в вы-
выделении продукта. При экстрагировании о-анизидина хлорбен-
хлорбензолом раствор плохо отстаивается от суспензии шлама, а по-
потери растворителя достигают 20%. Экстрагирование бензолом
дает лучшие результаты, но потери бензола все же велики
(~14%). При вакуум-отгонке о-анизидина из реакционной мас-
массы возникают трудности при конструировании перегонного ку-
куба, в котором к концу перегонки остается сухой шлам. Все эти
трудности отпадают при восстановлении о-нитроанизола сер-
сернистым натрием щт гидросульфидом натрия. Поэтому данный
метод применяется на производстве, несмотря на снижение вы-
выхода продукта до 86—87%. Этим же методом получают п-анизи-
дин, о- и п-фенетидины, аминосалициловые кислоты (из нитро-
салициловых кислот), м-нитроанилин (из м-динитробензола),
аминофенолы (из нитрофенолов), хлораминофенолы, пикрами-
новую кислоту, 4-нитро-2-аминофенол, 1-нафтиламин и др.
§ 3. КАТАЛИТИЧЕСКОЕ ВОССТАНОВЛЕНИЕ ВОДОРОДОМ
Метод восстановления органических соединений водородом
в присутствии катализаторов с каждым годом приобретает все
большее значение. Основными достоинствами этого метода яв-
являются высокая скорость процесса, чистота получаемых про-
продуктов и простота их выделения. К числу недостатков, препят-
препятствующих широкому распространению этого метода, следует
отнести относительную дефицитность применяемого для восста-
восстановления электролитического водорода и катализаторов, боль-
большую пожарно- и взрывоопасность процесса и необходимость ис-
использования во многих случаях автоклавоз.
Восстановление водородом проводят в присутствии катали-
катализаторов, т. е. веществ, ускоряющих реакцию. Направление реак-
реакции и степень восстановления органического соединения зави-
зависят как от условий ведения процесса, так и от природы и мето-
метода приготовления катализатора. Чаще других используют
металлические катализаторы, особенно металлы VIII группы
периодической системы элементов. Активность катализаторов
может увеличиваться в присутствии специальных добавок—ак-
добавок—активаторов. Для увеличения поверхности катализатора его час-
часто наносят на пористое вещество — носитель. В качестве носи-
носителей используют силикагель, пемзу, уголь и т. п.
12-926 177
Одним из наиболее распространенных является скелетный
никелевый катализатор — никель Ренея. Его приготавливают
путем обработки сплава никеля и алюминия раствором щелочи.
При этом алюминий выщелачивается из сплава в виде алюми-
алюмината, а никель частично превращается в гидрид никеля:
2Al3Ni + 6NaOH + 6H2O >- Ni2H + 6NaA102 + 17Н
Полученный катализатор имеет специфическую пористую
структуру (скелетный катализатор) и содержит большое коли-
количество сорбированного водорода. Никель Ренея пирофорен (спо-
(способен к самовозгоранию на воздухе), поэтому его хранят под
слоем воды. При длительном хранении катализатор теряет свою
активность.
Восстановление водородом в присутствии никеля Ренея про-
проводят под давлением в атоклавах, как правило, в спиртовой
среде. Поскольку катализатор имеет большую плотность и быст-
быстро оседает на дно реактора, необходимо хорошее перемеши-
перемешивание. После окончания восстановления, о котором судят по ко-
количеству поглощенного водорода, катализатор отделяют фильт-
фильтрованием, а спиртовый раствор восстановленного соединения
направляют на стадию отгонки спирта.
Кроме никеля Ренея, применяют и другие катализаторы на
основе никеля. Весьма распространенный непирофорный нике-
никелевый катализатор получают при разложении формиата нике-
никеля в среде парафина при температуре 250—270 °С:
Ni(HCOOJ.2H2O >- Ni + 2СО2 + Н2 + 2Н2О
Используют также медно-никелевые катализаторы, карбонат
никеля и т.д.
Никелевые катализаторы широко используются для восста-
восстановления ненасыщенных соединений, карбонильных соединений
и для восстановления нитросоединений до аминов. Глубина
восстановления зависит от условий ведения процесса и актив-
активности катализатора. Так, например, при восстановлении нитро-
нитробензола водородом в присутствии никеля Ренея под давлением
1 ати получают анилин, а при 95 ати — циклогексиламин:
NH2
1 ати
Н2
Ni Ренея
NH2
95 ати
130-150 °С
О
Наряду с никелевыми большое распространение получили
платиновые и палладиевые катализаторы. Широкое примене-
178
ние этих катализаторов ограничивается их дороговизной. Наи-
Наиболее часто применяют катализаторы из платины и палладия
высокой степени дисперсности в виде так называемой платино-
платиновой или палладиевой черни. Широко применяются платиновые
и палладиевые катализаторы, получаемые осаждением метал-
металлов на носители—активированный уголь, пемзу, кремнезем,
силикагель. Обычные платиновые и палладиевые катализаторы
обладают малой селективностью. Для изменения активности
катализаторов и увеличения их селективности используют раз-
различные добавки. Например, для избирательного восстановления
тройной связи до двойной применяют так называемый катали-
катализатор Линдлара, который представляет собой палладий, осаж-
осажденный на карбонате кальция с добавлением ацетата свинца.
Избирательность действия этого катализатора увеличивается в
присутствии хинолина. Используются также и более дешевые,
хотя и менее активные катализаторы на основе кобальта, хро-
хрома, меди и их соединений.
Водород в присутствии катализаторов реагирует почти со
всеми органическими соединениями, способными к восстанов-
восстановлению. Однако результаты зависят от правильного подбора все-
всего комплекса условий.
Среди ароматических углеводородов труднее всего восста-
восстанавливаются моноциклические соединения, причем в результате
можно получить только продукты полного восстановления. Про-
Проводят восстановление в присутствии палладиевых, платиновых
или никелевых катализаторов. Значительно легче восстанавли-
восстанавливаются ароматические соединения с конденсированными коль-
кольцами. Легкость восстановления увеличивается с возрастанием
числа колец. В зависимости от активности катализатора и ус-
условий ведения процесса получают продукты полного или частич-
частичного восстановления. Большинство функциональных групп (за-
(заместителей) восстанавливается легче, чем ароматическое коль-
кольцо, поэтому среди продуктов обычно не бывает алициклических
соединений.
Вследствие сходства ароматических и гетероциклических
соединений последние гидрируются в условиях, аналогичных ус-
условиям восстановления ароматических соединений. Частичное
восстановление гетероциклических соединений можно прово-
проводить, используя в качестве катализаторов медь или хромиты.
Значительно легче протекает восстановление двойных или
тройных связей в алифатических или алициклических соедине-
соединениях. В противоположность водороду в момент выделения ка-
каталитически активированный водород легче взаимодействует
с изолированными двойными связями, чем с сопряженными.
Очень чувствительны к действию активированного водорода со-
соединения ацетиленового ряда. Вначале они восстанавливаются
До этиленовых производных, а затем до насыщенных соедине-
соединений.
12* 179
Карбонильная группа восстанавливается труднее, чем эти-
этиленовые и ацетиленовые связи. Продуктами реакции являются
соответствующие спирты. Для получения спиртов пригодны
почти все упомянутые выше катализаторы. Нитрогруппа восста-
восстанавливается водородом в присутствии катализаторов очень лег-
легко. В результате с почти количественным выходом образуются*
амины. Эта реакция была открыта М. М. Зайцевым в 1872 г.
Реакция восстановления нитросоединений проходит в соответст-
соответствии с уравнением:
RNO2 + ЗН2 >■ RNH2 + 2Н2О
Восстановление можно проводить как в паровой (газовой)у
так и в жидкой фазе.
Амины можно получить не только при восстановлении азо-
азотистых соединений, но также из альдегидов и кетонов, которые
в присутствии аммиака способны превращаться в амины. Та-
Таким образом, из фурфурола можно получить фурфуриламин с
выходом около 60%:
НС СН 0 НС СН
II II /У ~\~ NHo -J- Но i *■ || || -J- НоО
ттр С ——С Ni Ренея ттр h pu xjtt
v Чн v ' "
Путем восстановительного аминирования метилбензилкетона
под давлением и при температуре 50—60 °С получают фенили-
зопропиламин (основание препарата фенамин) с выходом 96%:
С/~Ч_Т 1 тт i \ттт
—L.rl3 "Т" **2 Г" iN*~*'
II
О
Фенилизопропиламин — исходное вещество в синтезе препаратов
сиднофен, сиднокарб.
§ 4. ЭЛЕКТРОЛИТИЧЕСКОЕ ВОССТАНОВЛЕНИЕ
Область применения электролитического восстановления ор-
органических соединений почти так же широка, как и область
применения каталитического восстановления. Недостатками ме-
метода являются его энергоемкость, некоторые технические за-
затруднения, большая продолжительность процесса и неоднород-
неоднородность получающихся продуктов восстановления.
Для проведения процессов электролитического восстановле-
восстановления необходим источник постоянного тока и специальное обо-
оборудование. В сосуд для восстановления помещают два электро-
электрода, отделяя катодное пространство от анодного пористой диаф-
диафрагмой. Восстановление проходит на катоде. Используются как
металлические, так и графитовые катоды. К материалу анода
предъявляются более жесткие требования в связи с возмож-
возможностью его окисления при действии электрического тока. Для
180
равномерного поступления восстанавливаемого вещества к ка-
катоду необходима мешалка, так как без перемешивания диффу-
диффузия протекает слишком медленно. Поскольку органические сое-
соединения не могут подвергаться электролизу непосредственно,
так как они либо не диссоциируют, либо диссоциируют очень
слабо, в качестве электролитов используют неорганические кис-
кислоты или щелочи. Для прохождения реакции важно, чтобы вос-
восстанавливаемое вещество находилось в растворе или в виде
тонкой суспензии. Для этого к электролиту часто добавля'ют
органические растворители, смешивающиеся с водой, но не под-
подвергающиеся восстановлению. Подбор условий восстановления
для отдельных соединений обычно осуществляется опытным пу-
путем. В зависимости от способности вещества к восстановлению
подбирают соответствующие катод, плотность тока, раствори-
растворитель, температуру, концентрацию, длительность процесса и*
т. п.
Электролитический метод пригоден для восстановления с
целью получения как аминов, так и любого из промежуточных
продуктов восстановления нитропроизводных.
§ 5. ТЕХНИКА БЕЗОПАСНОСТИ ПРИ ПРОВЕДЕНИИ
ПРОЦЕССОВ ВОССТАНОВЛЕНИЯ
При проведении процессов восстановления особенно важно
соблюдение всех мер по технике безопасности, так как многие
виды сырья и продуктов восстановления могут быть источни-
источником пожара, взрыва или отравления.
В помещениях, где проводят восстановление щелочными
металлами, должны быть предусмотрены меры, исключающие-
возможность попадания воды на щелочной металл. В связи с
этим в таких помещениях недопустимо использование воды в
качестве тепло- или хладоносителя. При использовании в каче-
качестве восстановителя цинковой пыли необходимо помнить о
том, что последняя образует взрывоопасные смеси с воздухом,
поэтому загрузку цинковой пыли целесообразно производить в
виде суспензии.
При восстановлении сернистыми щелочами может выделять-
выделяться сероводород, который является сильным отравляющим ве-
веществом. Отравление сероводородом наступает уже при кон-
концентрации его в воздухе 0,15 г/м3. В связи с этим герметизация
аппаратуры, из которой может выделяться сероводород, долж-
должна быть настолько полной, чтобы в помещении не ощущался
запах H2S.
Особенную опасность в пожарном отношении представля-
представляют процессы восстановления водородом. Как известно, водород
горюч и образует взрывоопасные смеси с воздухом. Опасность,
взрыва или пожара увеличивается при использовании пирофор-
пирофорных катализаторов. В связи с этим приготовление и регенера-
181
дия таких катализаторов должны проводиться в отдельном по-
помещении. Необходимо следить, чтобы частички катализатора
не забрасывались на стенки реактора, так как при подсыхании
они способны к самовозгоранию. Может загореться и отрабо-
отработанный катализатор, поэтому его не отжимают на фильтре до-
досуха и транспортируют в виде суспензии в воде или спирте. В
тех случаях, когда это возможно, следует пользоваться непиро-
непирофорными катализаторами.
Передавливание взрывоопасных реакционных масс произво-
производят азотом или другим инертным газом.
Для предотвращения создания взрывоопасных концентра-
концентраций газов и паров легковоспламеняющихся жидкостей и ядови-
ядовитых газов в аварийных условиях, в опасных местах должны быть
установлены автоматические сигнализаторы с включением ава-
аварийной вентиляции. Противопожарные мероприятия в цехах
должны быть направлены на предотвращение пожара и взры-
взрыва, а также на их локализацию в случае возникновения.
Многие вещества, использующиеся или получающиеся в
процессах восстановления, являются вредными или ядовитыми.
Так, все ароматические амины в большей или меньшей степени
являются ядами для человека. Они являются нервными и кро-
кровяными ядами, вызывают дерматиты, экземы. Некоторые ами-
носоединения (нафтиламины, бензидин, дианизидин) являются
канцерогенными веществами. Для всех работающих в производ-
производстве ароматических аминов установлены сокращенный рабочий
день F ч) и дополнительный отпуск A2 дней).
Обслуживающий персонал допускают к работе только после
проверки знаний правил техники безопасности, технологических
регламентов и инструкций.
§ 6. ОКИСЛЕНИЕ
О связи процессов восстановления и окисления уже говори-
говорилось в начале этой главы. Дать единую схему для всех реакций
окисления невозможно, так как результат реакции в каждом
отдельном случае зависит от природы реагентов и условий ве-
ведения процесса. Окислителем обычно служит кислород и бога-
богатые кислородом соединения, которые в условиях реакции спо-
способны отдавать его другому соединению. В качестве окислите-
окислителей могут быть использованы также соединения элементов с пе-
переменной валентностью.
Наиболее дешевым и распространенным окислителем явля-
является воздух. В большинстве случаев окисление воздухом исполь-
используется при ведении процесса в присутствии катализатора. Часто
в качестве окислителей применяют гипохлорит натрия NaCIO,
дихромат калия К2СГ2О7 (хромпик), двуокись марганца МпОг
(пиролюзит), перманганат калия КМпО4, перекись водорода
182
H2O2, а также разбавленную азотную кислоту и некоторые
другие соединения.
Окисление ядра ароматических углеводородов. Как уже го-
говорилось, ароматические углеводороды достаточно устойчивы к
окислению. Во многих случаях окисление проводят при высо-
высокой температуре и на катализаторе. Таким образом получают
малеиновый и фталевый ангидриды, антахинон и некоторые
другие соединения. Для синтеза химико-фармацевтических
препаратов из перечисленных соединений наибольшее значение
имеет малеиновый ангидрид.
Малеиновый ангидрид получают окислением бензола возду-
воздухом в паровой фазе. Реакция протекает при высокой темпера-
температуре в присутствии катализатора:
.. сн-со
2 Г 1 + 9О2 > 2 II \) + 4Н2О + 4СО2
СН—СО
В качестве побочных продуктов образуются п-бензохинон и уг-
углекислый газ:
О
+2Н2О
О
12CO2 + 6H2O
Катализатором может служить пятиокись ванадия V2O5. Од-
Однако большой выход малеинового ангидрида достигается на
сложных катализаторах (смеси окислов или солей ванадия^
молибдена и некоторых других металлов, нанесенных на пем-
пемзу или силикагель). Выход малеинового ангидрида при окис-
окислении бензола составляет около 60%. Кроме того, малеиновый
ангидрид образуется в качестве побочного продукта при окис-
окислении нафталина. Основным продуктом, получающимся при
окислении нафталина, является фталевый ангидрид. В качестве
побочных продуктов образуются нафтохиноны, малеиновый ан-
ангидрид и углекислый газ:
СО
v2o5 ~^^\О + 4Н2О + 4СО2
+ 2Н2О
183
+ 9O2
сн-со
н-сб
О + 6СО2 + ЗН2О
12О2
10СО2 + 4Н2О
В связи с относительной дефицитностью нафталина как
сырья и доступностью о-ксилола (из нефти) многие страны
начали получать фталевый ангидрид из о-ксилола. Работы по
промышленному освоению производства фталевого ангидрида
окислением о-ксилола в широком масштабе ведутся и в Совет-
Советском Союзе. В будущем большая часть фталевого ангидрида
сбудет получаться из о-ксилола:
О
•сн3
+ зо2
О + ЗН2О
Получение антрахинона может быть осуществлено аналогич-
шо окислению нафталина в паровой фазе с использованием в
качестве катализатора окислов ванадия и молибдена. Кроме
того, антрацен окисляется в паровой фазе воздухом в присут-
присутствии ванадата железа, нанесенного на пемзу. Процесс прохо-
проходит при 320—390 °С. Выход антрахинона достигает 95—97%:
О
2Н2О
Практически используемым приемом окисления антрацена до
лнрахинона является применение бихромата натрия в среде
серной кислоты:
Na2Cr207 + 4H2SO4 >■
Cr2(SO4K
5H2O
Для проведения процесса тщательно измельченный антра-
ден смешивают при температуре около 100 °С с небольшим
юбъемом водного раствора двухромовокислого натрия. К полу-
4 84
ченной суспензии добавляют 50% серную кислоту и остальное
количество бихромата.
Получение альдегидов из толуола и его производных. Полу-
Получение альдегидов ароматического ряда окислением толуола vt
его производных является важным технологическим процессом*
в синтезе лекарственных веществ.
Для окисления метильной группы до альдегидной применя-
применяют двуокись марганца. Процесс ведут в серной кислоте. В тех
случаях, когда двуокись марганца берут в избытке, использу-
используют водную серную кислоту. Если двуокись марганца взята в
стехиометрическом соотношении, применяют избыток концент-
концентрированной серной кислоты.
С6Н5СН3 + 2MnO2 + 2H2SO4 > С6Н5СНО + 2MnSO4 + ЗН2О
Если процесс вести при температуре не выше 40 °С, то в
результате реакции образуется альдегид. Если же температуру
реакции поднять до 60—70 °С, то основным продуктом будет
бензойная кислота.
Бензальдегид и его производные можно получить из хлори-
хлористого бензила и его замещенных окислением бихроматом в ще-
щелочной среде:
ЗС6НбСН2С1 + К2Сг207 + NaOH >
>- ЗС6Н5СНО + Сг2О3 + 2Н2О + NaCl + 2KC1
При окислении толуола воздухом в жидкой фазе образуется
смесь продуктов, главным из которых является бензойная кис-
кислота (см. с. 186). Бензальдегид получается при этом в относи-
относительно небольших количествах.
Окисление толуола воздухом до бензальдегида может быть
осуществлено в газовой фазе. Смесь паров толуола и воздуха
пропускают над катализатором (смесь окислов урана, молибде-
молибдена и меди) при температуре 475—500 °С. Выход бензальдегида
достигает 85—90%.
Получение бензойной кислоты и ее аналогов. При окислений
толуола и его производных хромовой кислотой, азотной кисло-
кислотой, перманганатом калия и другими сильными окислителями
образуются соответствующие карбоновые кислоты. Окисление
хромпиком в серной кислоте применяется в производстве о- и
п-нитробензойных кислот:
СООН
К2Сг20, + 4H2SO4 —£■ Г | + K*SO4 + Cr^SOJ, + 5H2O
Установлено, что чем ниже температура реакции, тем выше
должна быть концентрация серной кислоты.
185
Образующийся при этом в качестве побочного продукта аль-
альдегид доокисляют перманганатом:
СНО COONa (или К)
J
KMnO4 + Na2CO3 —> Г 11 +МпО2
Бихроматы применяются в качестве окислителей не только
в среде серной кислоты, но и в нейтральных растворах. Так,
окисление толуола водным раствором бихромата при давлении
200 ат и температуре 305—315 °С приводит к получению соли
бензойной кислоты:
С6Н5СН3 + Na2Cr207 > C6H5COONa + Сг2О3 + NaOH + Н2О;
2NaOH + Na2Cr207 > 2Na2CrO4 + H2O;
G6H6CH3 + 2Na2Cr04 > C6H5COONa + Cr2O3 + 3NaOH
Поскольку хроматы реагируют значительно медленнее, чем
бихроматы, в реакционную массу целесообразно добавлять неко-
некоторое количество кислоты (например, бензойной) с тем, чтобы
нейтрализовать образующуюся щелочь. В этих же условиях
окисляются до соответствующих карбоновых кислот производ-
производные толуола.
Весьма удобным окислителем является перманганат калия.
Окисление обычно ведут постепенным добавлением твердого
перманганата к водному раствору или суспензии окисляемого
вещества. В тех случаях, когда выделяющаяся щелочь может
повредить продукту (например, способствовать омылению
ацильного производного), к реакционной массе добавляют суль-
сульфат магния. Таким образом получают 4-нитро-2-ацетаминобен-
зойную кислоту, 4-хлор-2-ацетаминобензойную кислоту и неко-
некоторые другие промежуточные продукты:
СН3 СООН
I.NHCOCH3 .1 NHCOCH3
Г jj + 2КМпО4 >■ Г jj + 2MnO2 + 2KOH
NO2 NO2
Окисление метильной группы до карбоксильной кислородом
воздуха требует применения катализаторов.
Окисление толуола до бензойной кислоты кислородом воз-
воздуха протекает при температуре 140 °С и давлении 3—4 ат в
присутствии нафтената кобальта.
Для разделения образующейся смеси продуктов реакцион-
реакционную массу обрабатывают раствором соды. Бензойная кислота
в виде бензоата натрия переходит в водный слой и затем мо-
может быть выделена из него подкислением.
186
Органический слой содержит не вступивший в реакцию то-
толуол, а также образовавшиеся в качестве побочных продуктов-
дифенил, бензиловый эфир бензойной кислоты и бензальдегид.
Органический слой разделяют на составные части дестилля-
цией. Толуол и бензальдегид возвращают на окисление.
Большое значение в химико-фармацевтической промышлен-
промышленности получил синтез гетероаналога бензойной кислоты — нико-
никотиновой кислоты. Никотиновая кислота (р-пиридинкарбоновая
кислота, витамин РР) может быть получена окислением р-пико-
лина (р-метилпиридина):
Г| +з[О]—>\\ Л +н2о
N N
Окисление может быть осуществлено перманганатом в щелоч-
щелочной среде с выходом до 90%:
О/СН3 f^\/C00K
+ 2КМпО4 -^ Г J + 2МпО2 + КОН + Н2О
N N
Полученный раствор никотината калия подают на стадию выде-
выделения никотиновой кислоты:
.COOK ^. /СООН
N N
Техническая никотиновая кислота очищается перекристаллиза-
перекристаллизацией.
Никотиновую кислоту получают также в виде ее амида. Этот
метод не требует дефицитного и дорогостоящего перманганата
калия. Процесс можно осуществлять непрерывно:
N
Нитрилы пиридиновых кислот получают при пропускании
смеси паров пиколина, аммиака и воздуха при 300—350 °С над
катализатором. Наилучшие результаты (выход нитрила 93—
94%) получены при использовании в качестве катализатора
ванадата алюминия. Омыление нитрила осуществляют в водно-
аммиачном растворе в присутствии едкого кали и 10% раствора
перекиси водорода при 42 °С. Выход никотинамида, считая на
187
.{5-пиколин 65%. Разработано также электролитическое окисле-
окисление р-пиколина.
Никотиновую кислоту можно также получить окислением
хинолина:
ХООН
[О]
"Ч ||
-СО2
также окислением 2-метил-5-этилпиридина (МЭП):
N НООС N N
При окислении у-пиколина перманганатом или пиролюзитом
образуется изоникотиновая (у-пиридинкарбоновая) кислота, ко-
которая является сырьем для синтеза противотуберкулезных пре-
препаратов изониазида и фтивазида
СН3 СООН
I
[О]
N N
V-пиколин Изоникотиновая
кислота
§ 7. ТЕХНИКА БЕЗОПАСНОСТИ ПРИ ПРОВЕДЕНИИ
ПРОЦЕССОВ ОКИСЛЕНИЯ
Многие процессы окисления, особенно при проведении их в
паровой фазе, являются взрыво- и пожароопасными. В связи
с этим на аппаратах, в которых перерабатываются взрывоопас-
взрывоопасные смеси, устанавливают предохранительные мембраны, а на
соответствующих трубопроводах — огневзрывопреградители. В
таких цехах действуют строгие противопожарные нормы. В ча-
частности, запрещено пользоваться стальным инструментом и хо-
ходить в обуви, подбитой железными гвоздями или подковами,
так как это может явиться причиной возникновения искры, а
следовательно привести к взрыву или пожару.
Некоторые вспомогательные процессы, связанные с опасно-
опасностью самовозгорания, проводят в токе азота.
Многие вещества, используемые в процессах окисления, яв-
являются вредными. При применении хромовых растворов в реак-
реакции окисления не следует применять выплавление хромпика па-
паром, так как при этом в воздух выделяется аэрозоль хромовых
соединений. Не следует допускать ручного разбивания хромпи-
хромпика, если он находится не в кристаллах, а в сплавленном виде.
В этом случае следует растворять его в чанах с ложным дном.
При применении в качестве окислителя азотной кислоты сле-
следует предусматривать очистку от окислов азота выхлопных га-
газов.
Гл ава 11
ОБРАЗОВАНИЕ ГЕТЕРОЦИКЛОВ
Гетероциклическими называются соединения, которые со-
содержат в молекуле кольцо, состоящее из атомов углерода и еще
какого-либо одного или нескольких элементов. Гетероцикличе-
Гетероциклические соединения имеют особое значение в химии и технологии
лекарственных веществ, так как очень многие из них явля-
являются биологически активными. Некоторые производные гетеро-
циклов встречаются в природе — в организмах животных и в
растениях.
Среди природных гетероциклических соединений большое
значение имеют некоторые производные пиррола (порфирины),
индола (триптофан, серотонин. некоторые алкалоиды), пириди-
пиридина (пиридиновые коферменты), пиримидина и пурина (витамин
Вь нуклеиновые кислоты, нуклеозиды), птеридина (фолиевая
кислота и рибофлавин) и др.
Простейшие представители гетероциклических соединений и
их свойства рассматривались в курсе органической химии. Конк-
Конкретные лекарственные соединения, являющиеся производными
гетероциклов, будут рассмотрены в главе 17. В данной главе
разбираются лишь основные вопросы синтеза гетероцикличес-
гетероциклических соединений, имеющих значение для химико-фармацевтиче-
химико-фармацевтической промышленности.
Если синтезы соединений бензольного ряда обычно основа-
основаны на превращениях легко доступных производных бензола и
лишь в очень редких случаях приходится синтезировать бен-
бензольное кольцо из алифатических соединений, то при получе-
получении гетероциклических соединений дело обстоит иначе. Здесь,
как правило, приходится получать путем химического синтеза и
само гетероциклическое кольцо. Однако, прежде чем присту-
приступить к синтезу гетероциклического соединения, следует рассмот-
рассмотреть возможность использования доступного исходного вещест-
вещества, уже содержащего нужный гетероцикл. В этом случае синтез
осуществляется так же, как и в ряду производных бензола, т. е.
путем введения заместителей или их превращения в другие
группы.
Так, например, в синтезе незаменимой аминокислоты —-^
триптофана — рационально исходить из замещенного индола
189
CH2N(CH3Kr
ЛШСОСН8
\ + Na+C-COOCHg
\хюсн3
N
H
/NHa
\соон
.NHCOCH^
CH2-C-COOCH8
\coochs
Если же простого и доступного гетероциклического соедине-
соединения не имеется, то приходится синтезировать гетероцикл, заме-
замещенный таким образом, чтобы его можно было превратить в
целевое соединение.
В последующих главах во второй части учебника мы будем
часто встречаться с такого рода синтезами. Яркий пример тако-
такого синтеза — получение витамина Вв.
Одним из распространенных методов образования различ-
различных циклов, в том числе и гетероциклов, являются реакции
внутримолекулярного алкилирования и ацилирования. Особен-
Особенно это относится к синтезу пяти- и шестичленных циклов, так
как образование последних не приводит к значительным напря-
напряжениям в цикле, а потому протекает относительно легко.
Наиболее типичными комбинациями реагентов, используе-
используемыми для получения гетероциклов с одним гетероатомом яв-
являются тип А и Б. Другие сочетания имеют гораздо меньшее
значение:
Y
Тип Л
Тип Б
По типу А образуются С—X связи, по типу Б — одна С—С
и одна С—X связь (пунктиром обозначены образующиеся
связи).
190
§ 1. ОБРАЗОВАНИЕ ПЯТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
Получают простейшие ненасыщенные гетероциклические со-
соединения (фуран, тиофен, пиррол), как правило, из природного
сырья (каменный уголь, нефть, растительное сырье).
Фуран сравнительно просто может быть получен из фурфу-
фурфурола. Фурфурол легко образуется при кислотном гидролизе по-
полисахаридов, содержащихся в оболочке семян овса, или при
гидролизе других природных соединений, имеющих пентозосо-
держащие фрагменты (например, в капусте, кукурузных почат-
початках, соломе, подсолнечной лузге, отходах древесины):
сно
и-гс —он
J + Ч| 12%НС1
11 НО-С-Н _^
п-с —он
сн2он
Чс = сХ
■\? сно и
И Фурфурол
Иоо%)
Нагревание фурфурола в паровой фазе над такими катали-
катализаторами как никель B80 °С) или известь C50 °С) приводит к
получению фурана с высоким выходом. Кроме того, фурфурол
может быть окислен в а-фуранкарбоновую (пирослизевую) кис-
кислоту, а из последней декарбоксилированием легко получить фу-
фуран:
/** О2, Cu2O/Ag?O Г/ ^ 200 °С
сно
0 0
86-90% 72—78%
Пиррол в промышленности получают фракционной перегон-
перегонкой каменноугольной смолы и костяного масла или пропуска-
пропусканием фурана, аммиака и водяного пара над окисью алюминия
-при 400°С (по методу русского химика Юрьева):
/ГЛ-
)
У А12О3, 400 °С \
О N
191
Если вместо аммиака в этой реакции использовать первич-
первичные амины, то продуктом реакции будут однозамещенные пир-
ролы.
Промышленный синтез тиофена основан на циклизации бу-
бутана, бутадиена или бутена с включением в цикл серы. Исход-
Исходные вещества при 600 °С быстро пропускают через трубчатый
реактор (время контакта ~1 с), а выходящие газы сразу ох-
охлаждают, отделяют сырье от продуктов реакции и возвращают
в цикл. Полученный тиофен очищают перегонкой.
Существует также ряд других методов синтеза соединений
ряда фурана, пиррола и тиофена. Одним из общих методов по-
получения производных фурана, тиофена и пиррола является син-
синтез их из 1,4-дикарбонильных соединений путем нагревания с
дегидратирующим агентом (H2SO4, P2O5, и т. д.), аммиаком
или неорганическими сульфидами:
сн9—
О p2s5 НС СН
R *" " II
с с
2~
В химии и технологии лекарственных соединений большое
значение имеют пятичленные азотистые гетероциклы с двумя
гетероатомами. Их можно представить как продукты замены
группы =СН— в фуране, тиофене или пирроле на атом азота.
Такие соединения получили общее название — азолы. Большин-
Большинство этих гетероциклов обладает ароматическими свойствами и
вступает в реакции, характерные для ароматических соединений.
Наибольшее значение имеют 1,3-азолы:
О NH
Оксаэол (и 1,2-аволы) Имидазол
192
O О О
О NH S
-Иаоксазол Пшрмоа Изотвмод
1,3-Азолы могут быть получены циклизацией 1,4-дикарбо-
нильных соединений:
Ч)СаН6
Н6С,О'
Этиловый эфир 4-Метад-б-этоксн-
N-формил а аланмна оксазол
Эта реакция используется в синтезе витамина В6. В синтезе
1,3-азолов исходят также из а-галогенкетонов. Подобный метод
применим для синтеза тиазолов и используется при получение
тиазольного компонента в синтезе витамина Вь
СН,—С=О Hn
СН2~СНаОСОСН3
\-Бром-'у-ацетооропилвцвтат Тноформемид 4-Метид-5-Э-ацвтоксв-
: этвдтнаэод
Практически важным методом получения бензимидазолов,
бензтиазолов и бензоксазолов является циклизация N-ацилиро-
ванных ароматических о-диаминов, о-аминофенолов и о-амино-
тиофенолов:
C-R
NH
2-Н-Бензимидазол
■—R
О'
2-К-Бензоксазол
2-R-BeH3THa3cvi
13—926 193
Таким образом, получают важное спазмолитическое и гипо-
гипотензивное средство дибазол:
с6н5сн,соон,
140-200 °С
Вместо фенилуксусной кислоты можно использовать фенил-
ацетамид СбН5СН2СОЫН2 или цианистый бензил С6Н5СН2СМ.
Аналогично этому из этилендиамина и а-нафтилуксусной кисло-
кислоты получают лекарственный препарат нафтизин:
СН2-Ш2 {~Л-СН2СООН
I + У
Vch2-/
Практически важным примером синтеза 1,2-азолов являет-
является получение 1-арил-3-метилпиразолонов-5 из амида ацетоуксус-
ной кислоты и ароматических гидразинов:
СН3
СО Н2С—С-СНз
| +H2N-NH-Ar >■ | ||
СН2 О=С N
CONH2 N
Аг
§ 2. ОБРАЗОВАНИЕ ШЕСТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
Важнейшим представителем шестичленных гетероциклов яв-
является пиридин. Пиридин и его алкилпроизводные выделяют из
каменноугольной смолы. Наличие доступного природного источ-
источника получения пиридина и алкилпиридинов особенно важно по-
потому, что в настоящее время еще не разработан метод синтеза,
пригодный для крупного промышленного производства. Для
производных пиридина известен целый ряд методов синтеза,
однако ряд производных удобно получать из алкилпиридинов.
Сочленение бензольного и пиридинового колец приводит к
образованию бициклических молекул хинолина и изохинолина
Изохинолин
Производные хинолина и изохинолина являются биологи-
биологически активными соединениями и широко используются в син-
194
тезе лекарственных веществ. Некоторые гомологи хинолина
имеют тривиальные названия, например, 2-метилхинолин назван
хинальдином, а 4-метилхинолин — лепидином.
Хинолин и изохинолин можно выделять в виде оснований
перегонкой или химическими методами из каменноугольной смо-
смолы, а также перегонкой или крекингом некоторых нефтей.
Аналогично пиридину хинолин и изохинолин являются сла-
слабыми основаниями и сравнительно устойчивы к химическому
воздействию. Обе системы являются ароматическими и в этом
отношении похожи на бензол и пиридин. Однако, так же как
и у нафталина, связи 1—2, 3—4, 5—6 и 7—8 несколько укоро-
укорочены и по характеру ближе к двойным, чем остальные связи.
Это сказывается на реакционной способности хинолина, изохи-
нолина и их производных.
Одним из распространенных методов синтеза хинолинов яв-
является реакция Скраупа. Она заключается во взаимодействии
первичных ароматических аминов, не содержащих лабильных
заместителей, с глицерином в присутствии концентрированной
серной кислоты и окислителя, в качестве которого используют
м-нитробензолсульфокислоту, SnCl4, кислород и др. Реакция
идет по схеме:
СН2ОН
Н-ОН
С
С
о=с-н
H2SO4
80-140 °С
2Н2О
СН2ОН
Глицерин
II
сн2
Акролеин
СН,
-н2о
[О]
Реакция Скраупа является частным случаем более общего
синтеза Дебнера—Миллера, заключающегося во взаимодействии
ароматических аминов со свободным о-положением с а,р-нена-
13* К&
сыщенными альдегидами (кротоновым, коричным и др.)- Реак-
Реакция проходит в кислой среде:
В этом синтезе могут быть использованы альдегиды или смесь
альдегидов, способные в условиях реакции (в кислой среде)
конденсироваться с образованием замещенных акролеинов:
/О (X
NHo
Окислителем в таких реакциях являются образующиеся в каче-
качестве побочных продуктов шиффовы основания, которые при этом
восстанавливаются в N-алкилариламины. Таким образом, в от-
отличие от метода Скраупа, где окислитель добавляется специаль-
специально, в синтезе Дебнера—Миллера окислитель образуется в реак-
реакционной смеси в результате побочной реакции между исходным
альдегидом и одним из промежуточных продуктов.
Изохинолин и его производные можно получить при цикло-
дегидратации N-ацилпроизводных |}-фенилэтиламинов с после-
последующим дегидрированием образующихся 3,4-дигидроизохино-
линов. Реакцию проводят в1 присутствии сильно кислотных ката-
катализаторов (Р2О5, РОС13, Н3РО4) при нагревании до 100—160 °С:
Ы-Ацетил-р-феыилэтштамин
2-Метил-3,4-ди-
гидроизохинсхлин
2-Метилизо-
хиыолин
На приведенных примерах видно, что реакции внутримолеку-
внутримолекулярного ацилирования и алкилирования играют важную роль
в образовании гетероциклов.
Наиболее важные химические свойства хинолинов и изохи-
нолинов заключаются в следующем.
Хинолины и изохинолины легко образуют соли с сильными
кислотами:
Г
196
Хинолины и изохинолины алкилируются алкилгалогенидами
по атому азота, например:
Хинолины и изохинолины с трудом вступают в реакции
электрофильного замещения. Так, хинолин бромируется в 3-по-
ложение при температуре 300 °С с выходом 3-бромпроизводного
25%.
N N
При температуре 450—500 °С бромирование идет по ради-
радикальному механизму и образуется 2-бромхинолин.
2- и 4-галогензамещенные хинолины легко вступают в реак-
реакции нуклеофильного замещения:
С1 ОН О
N
СО
N
В синтезе аминохинола применяется реакция:
Cl NHR
N ^ N
При действии амида натрия в инертном растворителе хино-
лин вступает в реакцию нуклеофильного замещения:
NeNHa
N N
При действии окислителей первым окисляется бензольное
кольцо, благодаря чему возможно получение никотиновой кис-
кислоты из хинолина:
.СООН ^ ^СООН
СООН
N
N
Часть II
ТЕХНОЛОГИЯ СИНТЕТИЧЕСКИХ ЛЕКАРСТВЕННЫХ
ВЕЩЕСТВ
Глава 12
НЕОРГАНИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
§ 1. ОБЩИЕ СВЕДЕНИЯ О НЕОРГАНИЧЕСКИХ
ЛЕКАРСТВЕННЫХ ВЕЩЕСТВАХ
И ИХ ЗНАЧЕНИИ В МЕДИЦИНЕ
Многие неорганические соединения издавна применялись в
медицине. Основная их часть неорганических лекарственных
соединений является солями. В числе неорганических лекарст-
лекарственных веществ имеются соединения элементов всех групп new
риодической системы.
Среди соединений элементов первой группы мы нахо-
находим гидрокарбонат натрия NaHCO3, который применяется при
повышенной кислотности желудочного сока и кишечника, при
изжоге. Гидрокарбонат натрия используют для приготовления*
микстур, растворов для полосканий, ингаляций и т. д.
Карбонат лития применяется в качестве мочегонного сред-
средства, соли лития применяют как психотропные средства. Суль-
Сульфат меди оказывает рвотное действие, применяется также как
антисептическое и вяжущее средство. s
Применение препаратов серебра в медицине основано на его
бактерицидных свойствах. Применение нитрата серебра связа-
связано с его способностью свертывать белки. Этим пользуются для
прижигания ран и язв.
Соединения элементов второй группы периодической
системы применяются в медицинских целях еще чаще. Интерес-
Интересно отметить, что по физиологическому действию соединения
магния и кальция являются антагонистами. Действие, оказывае-
оказываемое на организм соединениями магния, снимается соединениями
кальция и наоборот, соли магния снимают некоторые явления,
вызываемые соединениями кальция. Окись магния применяют
как слабительное средство при отравлении кислотами, она так-
также является составной частью противоядия от мышьяка. Суль-
Сульфат магния применяют в качестве слабительного, а при парен-
парентеральном введении он оказывает успокаивающее действие на
центральную нервную систему. В виде растворов его применя-
применяют так же, как спазмолитическое средство, при гипертониче-
198
ской болезни, для обезболивания родов, в качестве противосу-
дорожного средства и т. д.
Хлорид кальция используют в качестве кровеостанавлива-
ющего средства, а также в качестве противоядия при отравле-
отравлении солями магния. Сульфат кальция широко используется для
наложения гипсовых повязок, снятия слепков с челюстей в
стоматологии и т. п. Сульфат бария непроницаем для рентге-
рентгеновых лучей. Это его свойство используется при рентгенологи-
рентгенологическом исследовании желудка и кишечника.
Соединения цинка при внутреннем применении, как прави-
правило, ядовиты, однако их используют для паст, мазей, присыпок
и т. п.
Соединения ртути в качестве лекарственных веществ при-
применялись еще в глубокой древности. Соединения ртути приме-
применяются как антисептические, мочегонные и слабительные сред-
средства. Растворимые соли ртути ядовиты. Первая помощь при от-
отравлении солями ртути — принятие препарата «Противоядие от
металлов» или активированного угля. Одновременно следует
принимать молоко, слизистые отвары, жженую магнезию с во-
водой. Совершенно недопустимо применение поваренной соли ил.и
прием соленой пищи, так как при этом в организме может обра-
образоваться сильный яд — дихлорид ртути HgCl2 (сулема).
Из соединений элементов третьей группы в медицине
используют соединения бора (борная кислота, бура) и алюми-
алюминия (квасцы, гидроокись алюминия).
Из соединений элементов четвертой группы в меди-
медицинской практике используют активированный уголь и некото-
некоторые соединения свинца (для приготовления свинцовых пласты-
пластырей и примочек).
Значительно шире применяются соединения элементов п я-
той группы периодической системы. При туберкулезе лег-
легких для пневмоторакса используют чистый азот. Закись азота
используется в хирургической практике для наркоза. 1 % рас-
раствор нитрита натрия применяют как сосудорасширяющее сред-
средство.
Известно, что мышьяк и его соединения являются сильными
ядами. Однако в малых дозах соединения трех- и пятивалент-
пятивалентного мышьяка применяются как лекарственные средства.
Мышьяковистый ангидрид As2O3 применяется для лечения не-
некоторых кожных заболеваний, а также в стоматологической
практике для некротизации пульпы. Раствор арсената натрия
Na2HAsO4-7H2O применяется для стимулирования кроветворе-
кроветворения при малокровии и неврозах.
Соединения висмута обладают антисептическим действием.
Из соединений элементов шестой группы в медицин-
медицинской практике используются как дезинфицирующие средства
перекиси (перекись водорода, гидроперит, перекись магния),
широко применяются также соединения серы. Сера способству-
199
ет отложению гликогена в печени, а также снижает содержание
сахара в крови. Сера входит в состав сульфатов, о применении
которых уже говорилось. Сама сера в виде мазей применяется
для лечения некоторых кожных заболеваний. В виде сероводо-
сероводорода и сернистых щелочей сера входит в состав лечебных ми-
минеральных вод и грязей.
Входящие в седьмую группу периодической системы
галогены и их соединения также используются для приготовле-
приготовления лекарственных средств. Раствор хлора в воде используется
как дезинфицирующее средство; разбавленная соляная кислота
(с пепсином) применяется при недостаточной кислотности же-
лудочного сока; хлорид натрия в виде изотонического раствора
вводят под кожу или внутривенно при больших потерях жидко-
жидкости организмом. Широкое применение в медицине находят соли
бромистоводородной кислоты — бромиды натрия и калия, ко-
которые действуют успокаивающе на центральную нервную си-
систему.
Настойки йода применяют как антисептические средства при
воспалительных заболеваниях кожи. Йод применяется также
как внутреннее средство для профилактики атеросклероза.
Важное значение имеет йод для деятельности щитовидной же-
железы, участвуя в синтезе ее гормона — тироксина. Препараты
йода используют также при лечении подагры, ревматизма и
других заболеваний.
В седьмой группе периодической системы элементов располо-
расположен также марганец. Перманганат калия КМпО4 является
сильным окислителем и используется в медицине как дезинфи-
дезинфицирующее средство.
Из элементов восьмой группы периодической системы
элементов для медицинских целей наибольшее значение имеет
железо и его соединения. Препараты железа применяются при
анемии, малокровии, а также как кровоостанавливающее сред-
средство.
Таким образом, как мы видим, многие неорганические со-
соединения используются в качестве лекарственных веществ. Од-
Однако область применения неорганических лекарственных ве-
веществ все же недостаточно широка и, следовательно, их значе-
значение в медицине несравненно меньше, чем значение органиче-
органических лекарственных веществ.
§ 2. ПОЛУЧЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
БРОМА И ЙОДА
Проблему получения брома и йода в нашей стране удалось
разрешить только в годы Советской власти. В 1924—1925 гг.
коллектив сотрудников ВНИХФИ под руководством О. Ю. Ма-
Магидсона разработал промышленный метод получения йода из
буровых вод. Йод в буровых водах содержится в относитель-
200
но небольших концентрациях B0—40 мг/л). Однако запасы
этих вод велики и представляют собой экономически выгодный
источник-получения йода. Йод в буровых водах содержится в
форме йод-аниона, поэтому для перевода I- в молекулярный
йод буровые воды обрабатывают серной кислотой и нитритом
натрия:
2 Г + 2NaNO2 + H2SO4 > Ia + Н2О -f 2NO + Na2SO4.
Выделившийся йод адсорбируют активированным углем.
Уголь с адсорбированным на нем подом отделяют и передают
на стадию десорбции. Десорбцию проводят либо 10—15% Рас*
твором едкого натра или сульфитом:
3I2 + 6Na0H + 3 + 2
I2 + Na2SO3 -|- H2O > 2HI -f Na2SO4.
Концентрация йода на угле .достигает 40%. Вследствие этого
концентрация солей йода в растворе, полученном после десорб-
десорбции, во много раз больше, чем в буровых водах, и достигает 4%.
Очищенные растворы йодида окисляют хлоратом калия или
нитритом натрия в кислой среде:
6 Г + ClOJT + 6H+ > 312 + СГ + ЗН2О;
2Г + 2NO~ + 4H+ >■ I2 + 2NO + 2Н2О.
Окисление ведут при 40—50°С в течение нескольких часов. Вы-
Выделившийся свободный йод оседает на дне. Его отфильтровы-
отфильтровывают, промывают водой и прессуют в виде плиток по 5—7 кг.
Доброкачественность йода определяется отсутствием в нем ми-
минеральных примесей, а также механических загрязнений, на-
например, графита. Последний определяется путем растворения
растертого в порошок йода в тиосульфате.
I2 + 2Na2S2Os > 2NaI + Na2S4O6
Образование прозрачного раствора указывает на отсутствие не-
нерастворимых примесей. В настоящее время Советский Союз
полностью обеспечивает свою потребность в йоде и даже экс-
экспортирует его.
В медицинской практике используются 10% и 5% спирто-
спиртовые растворы йода. Раствор йода спиртовой 10% готовят пу-
путем растворения йода в 96% спирте, раствор спиртовой 5% —
на разбавленном спирте с добавлением 2% йодида калия.
Бром также можно получать из буровых вод способом во
мгогом аналогичном описанному. Кроме того, его получают
из рапы морских заливов. В морской воде бром содержится
главным образом в виде магниевых сОлей. В качестве окисли-
окислителя при выделении брома используют хлор:
MgBr2 f С12 > MgCl2 + Br2.
Для извлечения молекулярного брома пользуются способно-
способностью брома перегоняться с водяным паром. В качестве сырья
201
для получения брома могут быть использованы также различ-
различные минералы (карналлит, сильвинит), содержащие этот эле-
элемент.
Бром — важное сырье в синтезе многих лекарственных пре-
препаратов.
Бромиды и йодиды натрия и калия широко используются,
в медицинской практике и выпускаются в качестве фармако-
фармакопейных препаратов.
В промышленности бромиды и йодиды обычно получают че-
через соответствующие галоидные производные железа. При по-
получении бромидов железные стружки переводят сначала в рас-
растворимый бромид железа:
Fe + Br2 >- FeBr2,
а затем в бромисто-бромное железо:
3FeBr2 + Br2 >- Fe3Br8.
Полученный раствор бромисто-бромного железа упаривают и
добавляют к нему раствор карбоната натрия:
Fe3Br8 + 4Na2CO3 + 4Н2О >- 8NaBr + Fe(OHJ + 2Fe(OHK + 4CO2,
Осадок гидроокисей железа отфильтровывают. Из фильтрата
после упаривания выкристаллизовывается бромид натрия.
Иодид натрия получают аналогично:
Fe3I8 + 4Na2CO3 + 4Н2О >- 8NaI + Fe(OHJ + 2Fe(OHK + 4CO2.
Для получения калиевых солей вместо карбоната натрия берут
карбонат калия.
§ 3. ПОЛУЧЕНИЕ ПЕРМАНГАНАТА КАЛИЯ
Перманганат калия КМ11О4 в силу своих окислительных
свойств применяется в медицинской практике как антисептиче-
антисептическое средство. В растворах различной концентрации (от 0,01 да
0,5%) перманганат применяется как наружное средство для
промывания ран, полоскания горла, а также в гинекологиче-
гинекологической практике. 2—5% растворы перманганата калия применя-
применяются при ожогах кожи, 0,02—0,1% растворы используют для
промывания желудка при отравлениях.
Перманганат получают из природного минерала пиролюзи-
пиролюзита МпОг. При сплавлении пиролюзита со щелочью в присутст-
присутствии окислителей образуется темно-зеленый манганат калия
К2М11О4, в котором марганец шестивалентен:
2МпО2 + 4КОН + О2 —*• 2К2Мп04 + 2Н2О
Плавку ведут в чугунных котлах, обогреваемых топочными га-
газами (см. главу 6). Перемешивание реакционной массы осуще-
осуществляется якорными или скребковыми мешалками. В котел за-
загружают тонкоразмолотый высокосортный пиролюзит и 50%
202
раствор едкого кали. Температуру поддерживают в пределах
200—270 °С. При более высокой температуре манганат разла-
разлагается. Поскольку окисление осуществляется за счет воздуха
и происходит только на поверхности плава, процесс идет мед-
медленно и выход манганата относительно невысок F0—65%).
Выход манганата калия можно резко повысить, а время реак-
реакции сократить, если брать избыток едкого кали и продувать
через жидкий плав при 160—220 °С воздух или кислород. Недо-
Недостатком этого метода является необходимость применения
большого избытка щелочи и связанные с этим трудности даль-
дальнейшей переработки плава.
Окисление манганата в перманганат происходит уже при
кипячении раствора:
ЗК2МпО4 + 2Н2О >■ 2KMnO4 + MnO2 + 4KOH.
Однако значительно более выгодным является электрохимиче-
электрохимическое окисление манганата. При этом на аноде образуется
КМпО4
— е~ у- МпО7,
а на катоде водород. Анод изготавливают из никеля или леги-
легированной стали, а катод из меди. Электролиз ведут в стальных
цилиндрических ваниах, снабженных змеевиком и нижним спу-
спускным штуцером при 60 °С в течение 6—8 ч. По окончании эле-
электролиза электролит сливают в кристаллизатор, который ох-
охлаждается водой. Технический перманганат калия отфильтро-
отфильтровывают на центрифуге. Для получения фармакопейного про-
дукта с содержанием основного вещества не менее 99% техни-
технический перманганат перекристаллизовывают из горячей воды,
фильтруют и сушат при 80 °С.
Перманганат калия разлагается на свету, поэтому его сле-
следует хранить в помещениях и в упаковках, защищенных от дей-
действия прямых солнечных лучей. Недопустимо хранение перман-
ганата совместно с веществами, способными легко окисляться
(фосфор, сера, сахар, глицерин, активированный уголь и др.)>
так как это может привести к пожару или взрыву.
Глава 13
ЛЕКАРСТВЕННЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
АЛИФАТИЧЕСКОГО РЯДА
§ 1. ГАЛОГЕНПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
АЛИФАТИЧЕСКОГО РЯДА
Введение в молекулу алифатического соединения атома га-
галогена приводит к усилению биологической активности у этого
соединения и появлению у него наркотических свойств. Если ме-
метан СН4 не обладает наркотическими свойствами, то хлористый
метил СН3С1 уже оказывает на организм человека слабое нар-
наркотическое действие. В хлористом метилене СН2С12 это дейст-
действие выражено настолько сильно, что его рекомендовали как
средство для наркоза, особенно в тех случаях, когда использо-
использование хлороформа вызывает рвоту. Хлороформ СНС13 же в на-
настоящее время используют для наркоза при хирургических опе-
операциях. Четыреххлористый углерод ССЦ обладает еще более
сильным наркотическим действием, однако он слишком токси-
токсичен для того, чтобы его можно было применять в медицинских
целях.
Таким образом, увеличение числа атомов галогена в моле-
молекуле алифатического соединения приводит к усилению нарко-
наркотических свойств и токсичности этого соединения. Наркотиче-
Наркотическая активность бромпроизводных ниже, чем соответствующих
хлорпроизводных, а у йодпроизводных она еще ниже. Поэтому
йодоформ СШз находит применение в медицине уже не как
наркотическое средство, а как антисептик. Антисептическое
действие йодоформа объясняется его способностью легко окис-
окисляться с выделением свободного йода, который и оказывает
дезинфицирующее действие:
2СН18 + 4[О] ► 312 + СО + СО, + Н,О.
Этим объясняется также то, что йодоформ не действует на не-
неповрежденные ткани (нет окислителя). На поврежденных же
тканях действие йодоформа проявляется за счет окислительно-
окислительного фермента каталазы. Медленное течение реакции окисления
йодоформа обусловливает его длительное и относительно мяг-
мягкое антисептическое действие.
Хлороформ применяется для наружного употребления в ма-
мазях как местнообезболивающее и легкое раздражающее сред-
204
ство, часто его используют в химико-фармацевтической про-
промышленности как растворитель. К хлороформу для ингаляци-
ингаляционного наркоза при хирургических операциях предъявляются
особые требования в отношении чистоты препарата. Это связа-
связано, в частности, с тем, что при воздействии влаги на свету хло-
хлороформ медленно разлагается с выделением сильного яда —
фосгена:
2СНС13 + Оя ► 2СОС1а + 2НС1,
поэтому после тщательной очистки наркозный хлороформ ста-
стабилизируют добавлением 1 % спирта. В присутствии спирта
фосген переходит в безвредное соединение — диэтиловый эфир
угольной кислоты:
/CI /ОС8Н6
С=О + 2СаНвОН ► С=О + 2НС1
Vi \эс8н6
Хлороформ для наркоза выпускается в склянках из темно-
темного стекла по 50 мл, заполненных доверху и рассчитанных на од-
однократное употребление. Через каждые 6 мес хранения хлоро-
хлороформ для наркоза подвергается химическому анализу.
В промышленности хлороформ получают из спирта или аце-
ацетона при взаимодействии их с гипохлоритом кальция или хлор-
хлорной известью. Процесс проводят в стальном аппарате, снаб-
снабженном рубашкой для обогрева паром и охлаждения водой,
мешалкой и обратным холодильником. В реактор загружают
10% раствор гипохлорита кальция и этиловый спирт. Для на-
начала реакции смесь нагревают до 45 °С. Дальнейшее нагрева-
нагревание до кипения происходит за счет теплового эффекта проходя-
проходящей экзотермической реакции. Спокойное кипение реакционной
массы поддерживают попеременным пуском в рубашку пара и
охлаждающей воды. Образование хлороформа происходит в
три стадии. На первой стадии спирт окисляется до ацетальде-
гида:
2СаН6ОН + Са(ОС1J ► 2СН8-С-О + СаС18 + 2Н8О
Н
Образовавшийся ацетальдегид хлорируется до трихлорацеталь-
дегида (хлораля):
2СН8—С-0 + ЗСа(ОС1)8 ► 2СС1,С=О + ЗСа(ОН),,
а хлораль превращается в хлороформ и кальциевую соль му-
муравьиной кислоты (формиат кальция).
2СС13С-О + Са(ОН), ► 2СНС!8 + (НСОО)8Са
Суммарно реакцию можно выразить следующим уравнением:
С,Н6ОН + 4Са(ОС1)8 ► 2СНС1, + СаС1а + 2Са(ОН)8 + Са(НСОО)8 + Н8О
205
После выдержки холодильник переключают на прямой и от-
отгоняют хлороформ с примесью этилового спирта. Сырой хлоро-
хлороформ отмывают водой от спирта, а затем купоросным маслом
•от остатков влаги. Полученный технический хлороформ не го-
годится для медицинских целей. Для получения фармакопейного
препарата технический хлороформ загружают в эмалирован-
эмалированный аппарат с мешалкой, добавляют равное количество воды
и перемешивают в течение 11/2 ч. После отстаивания нижний,
-хлороформный слой сливают в другой эмалированный аппарат,
добавляют туда 0,2% (по массе) 40% раствора формалина и
половинное количество концентрированной серной кислоты, пе-
перемешивают около 1 ч и дают отстояться. Нижний слой (кис-
(кислотный) направляют на утилизацию, а хлороформ отгоняют че-
через конденсатор-холодильник в аппарат для нейтрализации.
Перегнанный хлороформ промывают 10% раствором щелочи и
еще раз перегоняют в ректификационной насадочной колонне.
Чем медленнее и равномернее ведется перегонка, тем лучше
качество хлороформа. Перегнанный хлороформ пропускают че-
через фильтр, заполненный прокаленным поташом, и собирают в
бутыли, емкостью 15 л, куда предварительно заливают тща-
тщательно очищенный этиловый спирт в количестве 1% от веса
хлороформа. Из каждой бутыли отбирают пробу для анализа
хлороформа и установления его сортности. В случае соответст-
соответствия требованиям Государственной фармакопеи хлороформ фа-
фасуют в 50 мл склянки из темного стекла и герметично закры-
закрывают их.
Необходимо отметить, что при производстве хлороформа
герметизация оборудования необходима не только для созда-
создания безопасных условий работы в цехе, но и потому, что при-
примесь воздуха окисляет парообразный хлороформ, ухудшает его'
качество.
Чистый хлороформ представляет собой бесцветную прозрач-
прозрачную жидкость с характерным запахом и жгучим вкусом. Плот-
Плотность хлороформа 1,486 г/см3, температура кипения 61,2 °С,
температура плавления —63,5 °С. Хлороформ плохо растворим в
воде, но смешивается во всех отношениях со спиртом, эфиром,
бензином.
Как уже упоминалось, хлороформ можно получать не толь-
только из спирта, но и из ацетона:
2СН3СОСН3 + ЗСа(ОС1J >- 2СС13СОСН3 + ЗСа(ОНJ;
2СС13СОСН3 + Са(ОНJ >- 2СНС13 +Са(ОСОСН3J;
2СН3СОСН3 + ЗСа(ОС1J > 2СНС13 + 2Са(ОНJ + Са(ОСОСН3J.
Вместо гипохлорита кальция может быть также использована
хлорная известь CaClOCl.
Спирт и ацетон служат исходным сырьем для получения
хлороформа и электролитическим способом; при этом электро-
206
лизу подвергают смесь спирта или ацетона с хлоридом калия
или натрия. Схема процесса может быть представлена следу-
следующим образом:
NaOH < Na+ <
Катод
С12 + 2NaOH >-
NaOCl NaOCi NOH
C2H5OH >• H3C— C=O >• C13C-C=O >- CHClg + HCOONa
H H
Методы получения йодоформа аналогичны описанным для
хлороформа. Исходным сырьем также служит этиловый спирт
или ацетон. В качестве окислителя используют гипойодит калия
или натрия (KOI, NaOI). Так как эти соли нестабильны при
хранении, их получают в процессе производства при действии
щелочи на йод:
I2 + NaOH > NaOI + Nal + Н2О.
Далее процесс идет по той же схеме, что и в случае получения
хлороформа.
В отличие от хлороформа йодоформ представляет собой
твердое кристаллическое вещество желтого цвета с характер-
характерным неприятным запахом. Температура плавления чистого
йодоформа 119°С. При нагревании выше 120 °С йодоформ раз-
разлагается. Плотность йодоформа равна 4,008 г/см3. Йодоформ
почти нерастворим в воде и хорошо растворим в этиловом спир-
спирте, эфире и ацетоне. Государственная фармакопея требует со-
содержания чистого йодоформа в препарате не менее 99%. Боль-
Большим недостатком йодоформа является его неприятный и очень
навязчивый запах. Порог ощущения его запаха 0,000006 мг/л.
Попытки устранить этот недостаток путем прибавления к йодо-
йодоформу других пахучих соединений (например, ментола) или
связывания йодоформа не дали желаемых результатов.
§ 2. СПИРТЫ И ПРОСТЫЕ ЭФ ИРЫ
Введение гидроксильной группы в молекулу углеводорода
увеличивает наркотические свойства вещества. Однако увели-
увеличение числа гидроксильных групп в молекуле приводит к умень-
уменьшению этих свойств. Например, у этилового спирта наркотиче-
наркотические свойства явно выражены, а у этиленгликоля, глицерина и
маннита они практически отсутствуют. Изомер маннита — сор-
сорбит применяется в качестве заменителя сахара при заболева-
заболевании диабетом, так как имеет сладкий вкус. Физиологическое
Действие и токсичность нормальных первичных спиртов возра-
возрастает с увеличением длины углеродной цепи до 6—8 атомов, а
затем падает. Особую токсичность метилового спирта объясня-
207
ют образованием из него в организме ядовитых формальдеги-
формальдегида и муравьиной кислоты. Метиловый спирт является сильным
нервным и сосудистым ядом с резко выраженным кумулятив-
кумулятивным действием. Предельно допустимая концентрация для ме-
метилового спирта 0,05 мг/л. Смертельная доза при приеме внутрь
30 мл. Этиловый спирт является наркотиком, вызывающим сна-
сначала возбуждение, а затем паралич центральной нервной си-
системы. При длительном хроническом воздействии этиловый
спирт может вызвать тяжелые органические заболевания нерв-
нервней системы, сердечно-сосудистые заболевания и т. д. Пропило-
выо спирты действуют на организм сходно с этиловым, но при
рг -.ой концентрации паров сильнее его. Физиологическая ак-
Ti.i - ость спиртов усиливается с разветвлением углеродной цепи.
Вторичные спирты обладают более сильным наркотическим дей-
действием, чем первичные, а третичные — более сильным, чем вто-
вторичные. Спирты, имеющие в молекуле кратные связи, являются,
клк правило, более сильными наркотиками, но при этом увели-
увеличивается и токсичность спирта.
Государственная фармакопея X содержит описания следую-
следующих препаратов этого класса соединений: спирт этиловый,
хлорбутанолгидрат, глицерин.
В медицинской практике этиловый спирт применяют как на-
наружное антисептическое и раздражающее средство для обти-
обтираний и компрессов. Этиловый спирт обладает хорошими де-
дезинфицирующими свойствами и применяется для дезинфекции
рук к хирургических инструментов. Спирт применяют в различ-
различных разведениях для приготовления настоек, экстрактов и дру-
других лекарственных форм. В фармацевтической практике кон-
.центрацию спирта выражают в объемных процентах (граду-
(градусах). Государственная фармакопея описывает следующие пре-
препараты этилового спирта: безводный (т. е. абсолютный) и вин-
винный этиловый спирт 95°, 90°, 70° и 40°.
Хлорбутанол гидрат A,1,1-трихлор-2-метилпропанол-2) при-
применяют внутрь в качестве противорвотного средства и как сно-
снотворное, а также в качестве наружного средства при лечении
ран, язв, воспалительных процессов.
Глицерин находит применение как основа для мазей и рас-
растворитель для некоторых лекарственных веществ.
Простые эфиры применяются для ингаляционного наркоза.
Как анестетики они во многом похожи на галогенпроизводные
алифатического ряда. С увеличением длины углеродной цепи
в алкильных группах эфиров наркотическое действие их на ор-
организм и токсичность возрастают. Из простых эфиров в меди-
медицинской практике применяется только этиловый эфир. Госу-
Государственная фармакопея X приводит два препарата этилового
эфира: эфир медицинский и эфир для наркоза, которые отлича-
отличаются лишь степенью очистки.
Этиловый эфир получают при взаимодействии этилового
208
спирта с серной кислотой. Реакция проходит через стадию об-
образования этилсерной кислоты:
с2н5он
СгНбОН + H2SO4 -j£g- C2H5OSOSH + с2Н5-О~С2Н5 + HaSO4
Для приготовления медицинского эфира используют спирт-
ректификат 96° и серную кислоту, полученную контактным спо-
способом (наиболее чистую). Процесс ведут в стальном освинцо-
освинцованном аппарате при температуре 130—140 °С; при этом отго-
отгоняются образующиеся вода и эфир, а также часть загруженного
этилового спирта. Подачу спирта в реактор, так же как и от-
отгонку смеси воды, спирта и эфира, осуществляют непрерывно.
При нарушении оптимального режима процесса может проис-
происходить разложение образующейся в качестве промежуточного
продукта этилсерной кислоты и образование ряда побочных
продуктов (этилена, уксусного альдегида и др.)- Образующую-
Образующуюся смесь паров эфира, воды и спирта пропускают через раствор
щелочи для нейтрализации примеси кислых продуктов и пода-
подают в ректификационную колонну. Полученный после ректифи-
ректификации эфир еще содержит ряд примесей и может применяться
лишь для наружных целей. Для получения наркозного эфира
необходима дополнительная очистка. Эта очистка заключается
в промывке водой, затем щелочным раствором перманганата
калия, повторной промывке водой, сушке прокаленным хлори-
хлоридом кальция и, наконец, обработке металлическим натрием для
удаления следов влаги, спирта, перекиси и ацетальдегида. Об-
Обработанный таким образом эфир еще раз 'перегоняют, собирая
фракцию при температуре 34—35 °С. Очищенный эфир после
пробы на отсутствие альдегидов фасуют в 100 мл склянки из
темного стекла.
Производство эфира огнеопасно и взрывоопасно. Пары эфи-
эфира и спирта образуют взрывоопасные смеси. Пределы врыво-
опасных концентраций эфира в смеси с воздухом составляют
1,85—40% (по объему). В цехе не должно быть источников
искрения. Все электрооборудование устанавливается только во
взрывобезопасном исполнении. Инструмент (гаечные ключи и
т. п.) должен быть бронзовым или омедненным. Обувь должна
быть без железных гвоздей и подковок. Для предотвращения
разряда статического электричества все оборудование и трубо-
трубопроводы должны быть заземлены, а влажность воздуха в по-
помещении не должна быть меньше 70%. При заполнении буты-
бутылей шланг должен быть снабжен алюминиевым наконечником,
а бутыль устанавливают на заземленном алюминиевом листе.
Скорость движения спирта и эфира по трубопроводам не долж-
должна превышать 2,5 м/с. Наливать эфир в сосуды нужно по трубе
наполнения, опущенной почти до дна сосуда, чтобы избежать
удара струи. Все воздушные линии должны быть снабжены
огне-взрывопреградителями (гравийными фильтрами).
И—926 209
§ 3. АЛЬДЕГИДЫ, АЛЬДЕГИДОКИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ
У альдегидов сохраняются присущие спиртам наркотические
свойства. Удлинение алкильного радикала приводит к увеличе-
увеличению токсичности параллельно с увеличением физиологической
активности. Наркотические свойства существенно возрастают
при введении в молекулу альдегида галогена или при переходе
к ненасыщенным соединениям. Введение в молекулу альдегида
карбоксильной группы резко снижает токсичность. Относитель-
Относительно малая токсичность наблюдается также у альдегидов, спо-
способных к образованию гидратной формы. Примером может слу-
служить хлоралгидрат:
хт
СС13-С-Н
который можно рассматривать как гидратную формулу хлора-
ля:
CCl3-C;f
Из отдельных представителей этого класса, использующих-
использующихся в медицинской практике и включенных в Государственную
фармакопею, следует отметить формальдегид, уротропин и хло-
хлоралгидрат.
Формальдегид СНгО выпускают в виде 40% водного раство-
раствора. Вследствие высокой токсичности, обусловленной способно-
способностью формальдегида свертывать белки, он применяется лишь в
качестве наружного дезинфицирующего средства. Так как рас-
раствор формальдегида придает тканям упругость, его используют
для консервации анатомических и биологических препаратов.
Получают формальдегид каталитическим окислением метилово-
метилового спирта:
СН3ОН + О2 -^-£ СН2О + Н2О
Гексаметилентетрамин (уротропин) представляет собой
продукт взаимодействия формальдегида с аммиаком. Уротро-
Уротропин не является альдегидом, но в сильнокислой среде при на-
нагревании распадается с образованием формальдегида:
Третичный азот сообщает гексаметилентетрамину основные
свойства, благодаря чему он может образовывать соли с кис-
210
лотами. Гексаметилентетрамин способен давать комплексные
соединения с неорганическими солями AgNO3, СаС12 и т. д. Его
используют в медицине в качестве дезинфицирующего средства
при заболевании мочевых путей.
Гексаметилентетрамин был получен в 1860 г. А. М. Бутле-
Бутлеровым, однако в медицинской практике его стали применять
только 35—40 лет спустя. Получают его из формальдегида и
аммиака как жидкофазным, так и парофазным методом. При
жидкофазном методе к 40% раствору формальдегида добавля-
добавляют небольшой избыток 25% раствора аммиака и реакционную
массу перемешивают при температуре 40—50 °С:
6СН2О + 4NH3 > (CH2NN4 + 6Н2О
Смесь очищают от примесей активированным углем, который
затем отфильтровывают. Фильтрат упаривают в вакууме до на-
начала кристаллизации. При охлаждении выпадают кристаллы
уротропина, которые отфильтровывают, промывают и сушат при
температуре 30—35 °С. С целью дополнительной очистки гек-
гексаметилентетрамин лерекристаллизовывают из спирта. При га-
газофазном методе производства смесь газообразного формальде-
формальдегида, аммиака и азота пропускают через горячий реактор. Об-
Образующаяся вода в виде пара уносится из реактора с азотом,
а уротропин оседает на дно реактора:
СН- ЗСН"О, NH,
СНО+- NH, *-CH=NH —-^HN NH
2 3HO 2 |
-н2о z | 1 -н2о
сн9
Дальнейшая очистка полученного таким путем продукта
аналогична описанной выше. При газофазном методе гексамети-
гексаметилентетрамин получается более высокого качества. Кроме того,
при этом методе расходуется значительно меньше энергии и вре-
времени на упаривание водных растворов.
Хлоралгидрат является успокаивающим и снотворным сред-
средством. В больших дозах он действует как наркотическое сред-
средство и применяется в психиатрической практике. Применение
препарата должно проходить обязательно под наблюдением
врача. Получают хлоралгидрат из этилового спирта по схеме:
Cl2, Fe /°Н Н2О, H,SO4 /0H
С2Н5ОН + С12 —- СС1з-С--Н -z^T CCI3-C-H
\ос2н5 \он
Н* 211
По химическим признакам к рассматриваемой группе сое-
соединений можно отнести также L-аскорбиновую кислоту (вита-
(витамин С), которая представляет собой у-лактон 2,3-дегидро-Ь-гу-
лоновой кислоты (у-лактон Ь-трео-2,3,4,5,6-пентаоксигексен-
2-овой кислоты).
. '.г
>-4-f-
Н
ОН ОН'
Ч
Молекула аскорбиновой кислоты содержит \-лактоновое кольцо
и группировку редуктона с двумя сопряженными двойными свя-
связями:
ОН НО
—с = с—с=о
I
Важнейшим свойством такой системы является способность к
обратимым окислительно-восстановительным превращениям:
ОН НО 0 0
+2H
В молекуле аскорбиновой кислоты имеется два асимметриче-
асимметрических атома углерода в положениях 4 и 5. Поэтому она можег
существовать в виде двух рацематов, составленных из четырех
оптических изомеров:
но он
сн2он
L -аскорбиновая кислота
ОН ОН

о'н-с-он
сн2он
D -аскорбиновая кис.ютв
НО-<
ОН он
он
сн2он
-а
НО OHl
он
он
L -изоаскорбиновая кислота D -изоаскорбиновая кислота
D-Аскорбиновая кислота витаминными свойствами не обладает,
а напротив, является антагонистом витамина С. Изоаскорбино-
вые кислоты также не обладают витаминной активностью.
212
Витамины являются необходимыми для жизни и незамени-
незаменимыми органическими веществами, которые играют роль биоло-
биологических катализаторов в жизненно важных процессах, входят
в состав ферментных систем, но, как правило, не синтезируют-
синтезируются в самом организме, а поступают в него извне. Практически
во всех витаминах содержится гидроксильная или карбониль-
карбонильная группа, способная превращаться в карбоксильную. Только
никотинамид не содержит гидроксильной группы, однако она
содержится в молекуле кофермента, в составе которого нико-
никотинамид участвует в обмене веществ. Витамины находятся в
тесной связи с гормонами, близки по химическому строению к
антибиотикам, алкалоидам и многочисленным лекарственным
веществам, поэтому их нельзя рассматривать в отрыве от мно-
многочисленных биологически активных органических соединений.
L-Аскорбиновая кислота (витамин С) является переносчи-
переносчиком водорода в некоторых ферментативных реакциях, протека-
протекающих в живой клетке. Недостаток витамина С в организме
приводит к заболеванию цингой — болезнью, характеризующей-
характеризующейся заболеванием десен, выпадением зубов, структурными изме-
изменениями хрящей и костей. Тяжелая форма цинги может приве-
привести к смертельному исходу. Кроме того, недостаток витамина
С приводит к потере аппетита, анемии, быстрой утомляемости*
кровотечениям, склонности к инфекционным и простудным за-
заболеваниям. Витамин С является одним из витаминов, потреб-
потребность в котором не покрывается за счет пищевых продуктов,
так как L-аскорбиновая кислота разрушается при нагревании
во время приготовления пищи, а также при длительном хране-
хранении содержащих ее продуктов. Благодаря этому аскорбиновую
кислоту широко применяют для витаминизации пищевых про-
продуктов.
Применение L-аскорбиновой кислоты в медицинских целях
способствует быстрому заживлению ран, срастанию костей,
излечению язвы желудка и двенадцатиперстной кишки, а также
рекомендуется при тромбофлебите, артериосклерозе, гепатите и
т. д.
Физиологическая потребность человека в витамине С состав-
составляет 70—100 мг в день. При тяжелом труде и для кормящих
матерей эта норма увеличивается до 200 мг в день. L-Аскорби-
новая кислота содержится в свежих овощах и фруктах. О со-
содержании витамина С в некоторых продуктах можно судить по
следующим данным (в мг на 100 г):
Плоды шиповника 1000—4500 Крыжовник 30—50
Грецкий орех (незрелый) 1000—1800 Шпинат 15—40
Облепиха северная 300—450 Редис 25—35
Перец болгарский 200—400 Малина 15—25
Смородина черная 100—400 Картофель 10—20
Земляника садовая 35—70 Щавель 12—14
Капуста белокочанная 25—65 Смородина красная 8—16
Лимон 55 Арбуз 5—10
213
L-Аскорбиновую кислоту получают в виде концентратов из
плодов шиповника, болгарского перца, зеленого грецкого ореха.
Однако значительно более эффективны синтетические методы
получения аскорбиновой кислоты.
Промышленное применение нашел метод синтеза L-аскорби-
новой кислоты через 2-кето-Ь-гулоновую кислоту. Этот метод
основан на использовании доступного сырья (D-глюкозы) и от-
относительно несложен по технологии. Синтез состоит из пяти
основных стадий:
сн2он
но-с-н
но-с-н
н-с-он
но-с-н
н-с«о
" D -глюкоза
сн2он
97-99%
СП ОН
НО
но-с-н
н-с-он
но-с— и
СН9ОН
D -сорбн г
CH2
с
ГОЛ
90-93%
но
но-с-н
н-с-он
„о-с-,г I 77~ш
L -сорбаза
СООН
Сч I
О - С — Н
I
н-с-о
с-н с
7
С2Н5ОН, HCI
з 87-90%
Диацегонсорбаза
Дпаиеюп-2-кетто-
L -гулонопая кислота
L-аскорбиновая кислота
В производстве синтетической аскорбиновой кислоты пер-
.вым промежуточным продуктом синтеза является шестиатом-
шестиатомный спирт D-сорбит, который получают восстановлением D-глю-
D-глюкозы водородом на скелетном никелевом катализаторе (никеле
Ренея).
Восстановление можно проводить также в присутствии мед-
но-хромового или железо-кобальтового катализатора. Перспек-
Перспективным методом восстановления D-глюкозы является электроли-
электролитический.
Чистота водорода, применяемого для восстановления, имеет
•большое значение. Необходимо использовать водород, получен-
214
ный электролитическим путем. Для восстановления (гидриро-
(гидрирования) применяют 50—55% раствор глюкозы в воде, который
приготовляют в специальном реакторе-растворителе, снабжен-
снабженном паровой рубашкой и мешалкой. Для создания рН раствора
8,0—8,4 к раствору глюкозы в воде добавляют раствор
Са(ОНJ. Гидрирование проводят в автоклавах при темпера-
температуре 120 °С и давлении около 100 ат в течение 1,5—2 ч. Ката-
Катализатор обычно загружают в количестве 5% от массы глю-
глюкозы.
Получающийся при гидрировании раствор D-сорбита необхо-
необходимо очистить от следов солей никеля и других тяжелых ме-
металлов, которые являются ядами для микроорганизмов, исполь-
используемых на следующей стадии синтеза. Очистку можно прово-
проводить с помощью ионообменных смол (например, КУ-2 или
ЭДЭ-10П) или же осаждать ионы тяжелых металлов в виде не-
нерастворимых карбонатов или фосфатов, обрабатывая раствор»
сорбита двузамещенным фосфатом натрия и карбонатом каль-
кальция (мелом) при 70—80 °С. Дополнительная очистка раствора
осуществляется также благодаря тому, что получающиеся
аморфные осадки нерастворимых солей адсорбируют из раство-
раствора нежелательные примеси и красящие вещества.
Очищенный раствор сорбита выпаривают в вакуум-аппара-
вакуум-аппарате при вакууме не ниже 650 мм рт. ст. до концентрации 95%.
Сгущенный сорбит растворяют в 2—3-кратном количестве 96%
этилового спирта при 78°С, а затем проводят кристаллизацию
при 18—20 °С. Выпавшие кристаллы сорбита отжимают и про-
промывают спиртом на центрифуге, а затем высушивают при тем-
температуре 35—40 °С. Таким образом получают чистый медицин-
медицинский сорбит.
На заводах СССР производят сгущенный технический сор-
сорбит. Для этого очищенный и упаренный, как это было указа-
указано выше, 95% раствор сорбита разливают в кюветы, смазан-
смазанные маслом какао. При застывании образуются твердые стек-
стекловидные плитки с содержанием сорбита в пересчете на сухое
вещество 99,4 %.
Для пищевых целей выпускают плитки массой по 100 и
200 г, а для технических целей — до 10 кг. Сорбит находит
применение не только как промежуточный продукт для синтеза
аскорбиновой юислоты.
Его применяют вместо сахара в питании больных диабетом,,
а также в качестве лечебного средства при холецистите, запорах,,
в дерматологии, офтальмологии.
Потребителями сорбита являются также косметическая, пи-
Щевая и некоторые другие отрасли промышленности.
Окисление D-сорбита в L-сорбозу может быть осуществле-
осуществлено как химическим, так и биохимическим путем. В СССР ис-
используют биохимическое окисление с применением уксуснокис-
уксуснокислых бактерий. Сорбоза является вторым промежуточным про-
21S
дуктом в синтезе аскорбиновой кислоты. Реакция протекает по
следующей схеме.
см2он сн2он
но —с —н но-* с j
но-с-н jra__^HO-4-H I
н-i— он ^ н — cj: — он9
но—с —н но—с—н
сн.рп н2с
. D -сорбит 1* -сорбоза
Биохимическое окисление сорбита в сорбозу является ре-
результатом жизнедеятельности аэробных, кетогенных, уксусно-
уксуснокислых бактерий, культивируемых на питательной среде, состо-
состоящей из D-сорбита и дрожжевого автолизата или экстракта.
К основным факторам, влияющим на процесс биохимического
окисления сорбита в сорбозу, относятся состав питательной сре-
среды, количество воздуха, подаваемого для аэрации питательной
среды, герметичность аппаратуры и недопустимость заражения
среды посторонней микрофлорой. Технологический процесс
окисления D-сорбита в L-сорбозу состоит из следующих ста-
стадий: приготовления посевной культуры, приготовления рабочей
культуры, проведения биохимического окисления в фермента-
ферментаторах, очистки L-сорбозы.
Питательная среда должна быть биологически активной,
т. we. содержать, кроме углеводов, азота и фосфора, биологиче-
биологические катализаторы (витамины и ферменты) и ряд минеральных
солей. Поэтому к раствору очищенного сорбита добавляют ак-
активный субстрат в виде дрожжевого автолизата или экстракта.
Вместо автолизата можно использовать и препарат В-комплекс.
Для культивирования уксуснокислых бактерий применяют кри-
кристаллический чистый сорбит и автолизат пекарских дрожжей.
Посев производят в пробирках в условиях, обеспечивающих
стерильность. Приготовленную посевную культуру исполь лют
для приготовления рабочей культуры. Вместо кристаллического
сорбита в этом случае используют очищенный раствор сорбита.
Приготовленную питательную среду разливают в колбы емко-
емкостью I л по 500 мл в каждую и стерилизуют в автоклаве, затем
производят в каждую колбу посев культуры из пробирок с вы-
выращенным посевным материалом. Колбы выдерживают в тер-
термостате в течение 24 ч, а затем культуру из литровых колб пе-
переводят в бутыли емкостью 20 л (из расчета 1 колбу на 1 бу-
бутыль), содержащие по 5 л питательной среды. Бутыли также
выдерживают в термостате, а затем выращенную в них куль-
культуру переводят в аппараты-инокуляторы. Аппараты предвари-
предварительно стерилизуют острым паром, а затем засасывают в них
216
вакуумом питательную среду, добавлением серной кислоты до-
доводят рН до значения ~4,5 и подают рабочую культуру. Глу-
Глубинную культуру выращивают при температуре 30—32 °С в те-
течение 12—24 ч, а затем стерильно передают в ферментаторы.
Окисление проводят глубинным способом в специальных ап-
аппаратах-ферментаторах, которые представляют собой верти-
вертикальные эмалированные реакторы, снабженные рубашками для
обогрева горячей водой, мешалками и барботерами для распы-
распыления воздуха. В стерилизованную и охлажденную питатель-
питательную среду переводят из инокуляторов рабочую культуру из рас-
расчета 8—10% к объему среды, после чего через реакционную
массу продувают предварительно очищенный воздух. Процесс
идет при 30 °С в течение 20—24 ч. Выход L-сорбозы составляет
93—95%. Полученный раствор сорбозы очищают активирован-
активированным углем, фильтруют и быстро упаривают в вакууме до кон-
концентрации ~90% и кристаллизуют при охлаждении.
Ацетонирование L-сорбозы проводят с целью защиты гидро-
ксильных групп при вторичных углеродных атомах от окисле-
окисления на последующей стадии — стадии окисления СН2ОН-группы.
Ацетонирование проводят в присутствии серной кислоты или
олеума в реакторе из кислотостойкой стали или эмалированном,
снабженном быстроходной мешалкой и рубашкой для охлаж-
охлаждения водой. Наиболее эффективным является ацетонирование
избытком ацетона в присутствии олеума:
Ф сн2он снон
I , . I 2
но-с-
но--с-но
но-с-н | I с-нД
г °
:чо-с-н
? * н-с-о ^сн,
Hi—1 ни' сн^
2
L -Сорбоза Лмичеюнсорбоча
В реактор загружают холодный (—5°С) ацетон и постепенно,
в течение 30 мин, олеум с концентрацией свободного БОз, рав-
равной 18%- Затем загружают всю порцию сорбозы и поддержи-
поддерживают температуру реакционной массы в пределах 12—15СС. Ос-
Остаточная концентрация сорбозы не должна превышать 2 г/л.
Реакционную массу охлаждают далее до —10—12 °С и переда-
передают на нейтрализацию раствором едкого натра. Температура во
время нейтрализации не должна подниматься выше 0сС. Реак-
Реакция среды после нейтрализации должна быть слабощелочной.
Сильнощелочная среда вызывает конденсацию ацетона с обра-
образованием окиси мезитила и ряда других продуктов, а кислая—
распад диацетонсорбозы. Из реакционной массы после удале-
удаления растворителя с водяным паром и отгонки окиси мезитила
217
из остатка выделяют диацетон-Ь-сорбозу B,3,4,6-ди-О-изопро-
пилиден-сс-Ь-сорбозу) высаливанием раствором едкого натра
•или экстракцией органическими растворителями.
Дальнейшее окисление диацетон-Ь-сорбозы до диацетон-2-
кето-Ь-гулоновой кислоты можно проводить с помощью различ-
различных окислителей: перманганатом калия, гипохлоритом натрия,
.азотной кислотой, а также каталитическими и электролитиче-
электролитическими методами. Окисление диацетонсорбозы гипохлоритом
натрия и кальция проводят при рН=10—12 в присутствии ката-
катализатора NiSO4, который в щелочной среде превращается в
Ni(OHJ. Температура реакций 65—68 °С. Выход продукта на
этой стадии достигает 92—93%.
сн2он
о-с-н
и-с-о сн3
— с-н^с
н2с-о Хсн3
Диацетон-Ь-сорбоза
NaCIO Са(СЮJ
NLSO4
-СООПа
9Х/
с-ну
с-о сн3
Натриевая соль
диацетон-2-кето-Ь
гулоновой кислош
Окисление может быть также осуществлено непрерывным
способом в колонне насадочного типа. Реагенты подают непре-
непрерывно в нижнюю часть колонны. Поскольку реакция экзотер-
мична, то необходимую температуру поддерживают, охлаждая
реакционную массу через рубашку. Реакционную массу фильт-
фильтруют для отделения катализатора, а затем выделяют гидрат
диацетон-2-кето-Ь-гулоновой кислоты при помощи НС1.
Предложен метод прямого окисления L-сорбозы до 2-кето-
L-гулоновой кислоты кислородом в присутствии платинового
или палладиевого катализатора. Однако он пока не нашел про-
промышленного применения.
Последняя стадия получения технической аскорбиновой кис-
кислоты заключается в действии на дихлорэтановый раствор диа-
цетонкетогулоновой кислоты спиртового раствора хлористого
водорода при 57—58 °С в течение 2 сут.
соо
н-н2о
ofc
' чо-с-н
С2Н.ОН, HCI
сн
Гидрат диацетон-2-кето-
'Ь-гудоно&он кислоты
« аскорбиновая
кислота
218
Выпадающую при охлаждении реакционной массы аскорби-
аскорбиновую кислоту отфильтровывают на центрифуге, хорошо про-
промывают дихлорэтаном или хлороформом (иногда спиртом) и су-
сушат в вихревой сушилке при 90—95 °С.
Полученную техническую L-аскорбиновую кислоту очищают
перекристаллизацией из водных или водноспиртовых раство-
растворов. Для уменьшения потерь маточные растворы используются-
многократно. Для осветления растворов используют активиро-
активированный уголь с гидросульфитом натрия. Для уменьшения по-
потерь при перекристаллизации рекомендуется ускорение техно-
технологических операций, так как аскорбиновая кислота в горячих:
водных растворах нестабильна. Выход на стадии очистки тех-
технической аскорбиновой кислоты составляет около 93%, а об-
общий выход в расчете на D-глюкозу— около 54%.
Аскорбиновая кислота представляет собой твердое кристал-
кристаллическое вещество белого цвета с температурой плавления
192°С (с разложением). Она хорошо растворима в воде и в
метиловом спирте, умеренно — в этиловом спирте, плохо —
в амиловом, практически нерастворима в эфире, бензине, бен-
бензоле и других неполярных растворителях. Сухая и чистая крис-
кристаллическая аскорбиновая кислота устойчива по отношению
к кислороду воздуха. В водных растворах в присутствии возду-
воздуха, особенно в щелочной или кислой среде, она быстро окисля-
окисляется.
Кроме чистой L-аскорбиновой кислоты, ее соли (аскорбина-
ты натрия, кальция и железа) и эфиры (пальмитат аскорбино-
аскорбиновой кислоты) применяются как в медицинской практике, так
и для витаминизации пищевых продуктов.
§ 4. КАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
Введение карбоксильной группы в молекулу вещества, как
правило, снижает его физиологическую активность и токсич-
токсичность. Применение самих органических кислот в качестве ле-
лекарственных средств крайне ограничено вследствие их малой
биологической активности. Производные жирных карбоновых
кислот широко распространены в природе и входят в состав
пищевых продуктов, в частности — жиров. Физиологическая
активность кислот с разветвленной углеродной цепью выше, чем?
У кислот нормального строения. Карбоновые кислоты находят
применение в фармацевтическом анализе. В медицинской прак-
практике используются некоторые соли органических кислот (аце-
(ацетат калия, лактаты кальция и железа, цитраты натрия и меди,,
глюконат кальция), их эфиры и амиды.
Биологическая активность непредельных карбоновых кис-
кислот значительно выше, как и у всех непредельных соединений.
Среди ненасыщенных высших жирных кислот три кислоты (ли-
нолевая, линоленовая и арахидоновая) не синтезируются в ор-
219
ганизме, но являются необходимыми для его нормальной жиз-
жизнедеятельности. Эти кислоты называют незаменимыми жирны-
жирными кислотами и иногда неправильно относят к группе витами-
витаминов под названием витамина F. Эти кислоты содержат по 18
или 20 атомов углерода и от двух до четырех изолированных
непредельных связей с полной цис-конфигурацией:
Н Н Н Н
сн
з(сн2;
с:
18СН:
12 \
2 С
/
н
с
\
н
9\
(С2Н2OСООН
Линолевая кислота (Ci8) —9,12-ди-цис-октадекадиеновая кис-
кислота.
н н н н н н
с=с
/ 15\ / 12\ / 9\
СН3СНа с С (СН2OСООН
/\ /\
н н н н
Линоленовая кислота (Ci8) —9,12,15-три-цис-октадекатриено-
вая кислота
н нн нн нн н
\/\/\/\/
с=с с=с с=с с=с
СН3(СН,L \' \' \' (СН^зСООН
/\ /\ /\
н н н н н н
Арахидоновая кислота (Сго) — 5,8,11,14 - тетра-цис-эйкозатетра-
еновая кислота.
Незаменимые жирные кислоты участвуют в животном орга-.
низме в окислении насыщенных жирных кислот, принимая уча-
участие тем самым в процессе усвоения жиров и в жировом обме-
обмене кожных покровов. Биологическая активность арахидоновой
кислоты выше биологической активности линолевой и линоле-.
новой кислот в 10 раз.
Линолевая и линоленовая кислоты содержатся в раститель-
растительных маслах в виде триглицеридов, а в некоторых животных про-:
дуктах в виде фосфолипидов. Арахидоновая кислота встречает-
встречается только в животных жирах. Полноценная пища должна иметь
в своем составе 0,1% арахидоновой кислоты или 1% линолевой
и линоленовой кислот.
В качестве лекарственных веществ нашли применение эфи-.
ры и амиды кислот. Большое значение имеют эфиры дифенил-
уксусной и дифенилпропионовой кислот с алкиламиноспиртами,:
220
На их основе созданы многочисленные спазмолитические сред-
средства (спазмолитин, апрофен и др.)- Общая формула этих соеди-
соединений:
QH; V /Alk
;с—с—о—сн2-сна—ы
Выпускаются они в виде солей (гидрохлоридов).
Особое место занимают органические производные угольной
кислоты, особенно производные амидов угольной кислоты, ко-
которые являются ценными лекарственными препаратами.
Угольной кислоте соответствует два амида — неполный (кар-
баминовая кислота) и полный (карбамид):
/ОН
► С=О
^NHa
Карбаминовая
кислота
/NH2
*. с=о
^NHa
Карбамид
(мочевина)
Эфиры карбаминовой кислоты называют уретанами. Многие
уретаны обладают снотворным действием, Из производных мо-
мочевины наибольшее значение для медицины имеют ацильные
производные. Ацильные производные мочевины были впервые
получены Н. Н. Зининым и названы им уреидами. Уреиды мо-
могут быть открытые (ациклические) и закрытые (циклические):
/NHa /NHa
С=О -fCHgCOOH ► С=О + НЯО
\ \h8
0
/NHa ^00H .NH-C/
С=О +СН8 ^0=0
4 ^ 4
Примерами лекарственных веществ, принадлежащих к груп-
группе открытых уреидов, могут служить бромизовал (бромурал) и
карбромал (адалин).
Карбромал, 1М-A-бром-1-этилбутирил) -мочевина, представ-
представляет собой открытый уреид, полученный конденсацией мочеви-
мочевины с диэтилбромуксусной кислотой:
Вг
NHg-CO—NH— СО-С-С2Н5
СЙН5
Карбромал оказывает на организм человека успокаивающее и
снотворное действие.
221
Бромизовал, N- A -бромизовалерианил) .-мочевина, является
уреидом одноосновной монобромизовалериановой кислоты:
уСН3
NH2-CO-NH—СО-СН—СН
Аг Хшз
Бромизовал (бромурал), так же как и карбромал, оказывает
успокаивающее и снотворное действие на организм.
Промышленный синтез бромизовала (бромурала) состоит
из следующих химических стадий. Действием на изовалериано-
вую кислоту треххлористого фосфора получают хлорангидрид
изовалериановой кислоты:
3(СН3JСНСН2СООН + РС13 > 3(СНзJСНСН2СОС1 + Н3РО3.
Температура во время реакции повышается от 40 до 98 °С (на
10° в 1 ч). Избыток треххлористого фосфора может реагиро-
реагировать с образованием метафосфористой кислоты:
~ 2Н3РО3 + РС13 > ЗНРО2 + ЗНС1,
поэтому реакцию прекращают после прекращения выделения
хлористого водорода, который улавливают. После отстаивания
реакционной массы более тяжелую фосфористую кислоту сли-
сливают через нижний штуцер. Полученный хлорангидрид изовале-
изовалериановой кислоты, не выгружая его из аппарата, бромируют
жидким бромом при 60 °С. При этом получают смесь хлоран-
гидрида а-бромизовалериановой кислоты и бромангидрида
этой же кислоты:
(СН3JСН—СН2—СОС1 + Вг2 >- (СН3JСН—СН—СОС1 + НВг;
Вг
(СН3JСН—СН-СОС1 + НВг > (СН3JСН—СН—COBr + HCl
Вг Вг
Для десорбции НС1 и НВг реакционную массу нагревают до*
90 °С и выдерживают несколько часов. Кислые газы улавлива-
улавливают раствором щелочи. В отделении бромирования должна быть
повышенная кратность воздухообмена, а также приняты дру-
другие меры по технике безопасности (см. главу 5).
Полученную в результате бромирования смесь хлорангидри-
да и бромангидрида обрабатывают мочевиной, которую загру-
загружают в аппарат постепенно, так как реакция ацилирования мо-
мочевины экзотермична. Температура за время реакции повыша-
повышается от 55 °С до 80 °С.
(СН3JСН—CHBr—COC1 + (СН3JСН—CHBr-COBr+ 4CO(NH2J >-
>• 2(CH3JCH-CHBr-CO— NH—CO-NH2 + CO(NH2J.HBr -f
+ CO(NH2J.HC1
После окончания реакции массу разбавляют водой и бромизо^
вал отфильтровывают и промывают на центрифуге.
222
Полученный технический бромизовал очищают перекристал-
перекристаллизацией из изопропилового спирта. Чистый бромизовал пред-
представляет собой белый кристаллический порошок горьковатого
вкуса со слабым запахом и температурой плавления 145—
150 °С. Он плохо растворим в воде A:450), но растворяется в
спирте и эфире.
В производстве бромурала важное значение имеет утилиза-
утилизация брома, который является дефицитным сырьем. Маточные
растворы, содержащие бромиды, обрабатывают серной кислотой,
а выделившийся бромистый водород окисляют хлором до бро-
брома. Полученный технический бром очищают перегонкой и снова
используют в производстве.
Наиболее важным для медицины циклическим уреидом яв-
является барбитуровая кислота, производные которой составляют
большую группу снотворных препаратов. Барбитуровая кисло-
кислота лекарственными свойствами не обладает. Кислые ее свойст-
свойства обусловлены существованием ее енольной формы:
он о он н
II ! II ' I I
НХ г 2 С=О 5?=brHXv С-ОН -^—*. НС/ С = о
b U 8 н .он
Кетомная форма Енольные формы барбитуровой
барбитуровой кислоты кислоты
В состав молекул барбитуровой кислоты и ее производных
входит пиримидиновый цикл, который содержат очень многие
биологически активные вещества. Таким образом, барбитура-
барбитураты можно рассматривать не только как производные амида
угольной кислоты, но и как производные пиримидина — шести-
членного гетероцикла с двумя гетероатомами.
В отличие от барбитуровой кислоты ее 5,5-дизамещенные в
водных растворах практически не диссоциируют, но в щелоч-
щелочных растворах ведут себя как кислоты и образуют соли:
ОН ОН
Поэтому некоторые барбитураты для повышения раствори-
растворимости применяются в виде натриевых солей.
223
Общая формула лекарственных производных барбитуровой
кислоты может быть изображена следующим образом:
О Н
С
r/
I!
С—
С
где Ri — чаще всего водород, a R2, Rs — алкил или арил. Наи-
Наиболее часто применяемые в медицине производные бартуровой
кислоты отечественного производства приведены в табл. 2.
Таблица 2
Препарат
Барбитал
Барбитал-натрий
Фенобарбитал
Гексенал
Барбамнл
Этаминал-натрий
*
Н
н
н
-сн8
н
н
-сан5
-с2н5
-с2н6
-СН3
-С2Н6
-с,н6
_сан6
-с2н6
-о
~w
-СН2-СН2—СН(СН3)а
-СН—СНа—СН2—СН3
1
сн,
Форма
Кислота
Натриевая соль
Кислота
Натриевая соль
» i
Связь строения производных барбитуровой кислоты с их
биологической активностью подробно изучена. При этом были
установлены следующие закономерности. Удлинение алифати-
алифатической цепи до 5—6 углеродных атомов у заместителя в по-
положении 5 барбитуровой кислоты приводит к увеличению фи-
физиологической активности. Дальнейшее >величение алкильного
радикала снижает снотворный эффект и выбывает судороги.
Замена этильного радикала в 5,5-диэтилпроизводном на фе-
нильный приводит к усилению снотворного эффекта. Замена
линейного алкильного заместителя на разв^ тгленный чсилиьает
; 41гггсть препарата, i .; ^ кращает время его действия.
Фармакологическая активность Сарбит\ратов усиливается так-
также при введении в молекулу непредельных заместителей кли
галогенов.
При введении алкнль^ых остатков в положении 1 или 3 дли-
длительность действия препарата уменьшается, но появляется воз-
возбуждающий эффект. Одновременное замещение атомов водоро-
водорода обеих имидных гглпп приводит к тому, что введение препа-
препарата животным вызывает у них судороги.
224
Алкилированные производные барбитуровой кислоты полу-
получают конденсацией мочевины с соответствующими алкилиро-
ванными эфирами малоновой кислоты:
С=О + С > О=С С + 2С2Н*ОН
Для целей синтеза применяют производные малонового эфира,
а не малоновой кислоты, так как последняя термически неус-
неустойчива и легко декарбоксилируется при нагревании. Введение
заместителей в малоновый эфир осуществляется действием ме-
металлического натрия и соответствующего бромистого алкила:
ггьг\г и СООСоНБ
/ХЮС2Н6 QHsOH, Na I СДО г |
} HfN "с
4 -н2 f
соос2нб соос2нб соос2нб
или действием цинка и йодистого алкила:
Zn + С2Нб1 > Zx/
СООС2Н6 СООС2Н5 СООС2Нб
| ZnIC2H5 I C2H5I |
сн2 ——-> нс-zni ——>■ нс-с2нб
СООС2Н6 СООС2Н5 СООС2Н6
Для замены второго водорода метиленовой группы на ал-
кильный заместитель, процесс повторяют.
Используют также другие пути синтеза производных барби-
барбитуровой кислоты, некоторые из которых будут рассматриваться
ниже на конкретных примерах.
Одним из наиболее распространенных препаратов этого ря-
ряда является барбитал E,5-диэтилбарбитуровая кислота). Бар-
битал (веронал) является одним из первых снотворных средств.
Впервые он был получен в 1881 г., в медицинской практике
применяется с 1905 г. Этот препарат получают по описанному
выше методу. Конденсацию диэтилового эфира диэтилмалоно-
вой кислоты с мочевиной проводят в присутствии алкоголята
натрия. В результате получается натриевая соль барбитала —
мединал:
н г гппг н иг СО—NH
С + CO(NH2J —i С C«-ONa
HbQ/ ^СООСЛ Н5С2/
15-926 225
При подкислении раствора мединала чистой соляной кислотой
в осадок выпадает барбитал:
нг CO-NH мг CO-NH
6 2\ / \ НС1 НбЧ\ / \
С С—ONa >- С С=О
Осадок отфильтровывают и перекристаллизовывают из дистил-
дистиллированной воды с обработкой активированным углем.
Чистый барбитал представляет собой белый кристалличе-
кристаллический порошок горьковатого вкуса, без запаха. Температура
плавления 191 °С. Барбитал применяется как снотворное сред-
средство в порошках и таблетках по 0,3 и 0,5 г.
Не менее важным препаратом этой группы является фено-
фенобарбитал E-фенил-5-этилбарбитуровая кислота). Синтез фе-
фенобарбитала (люминала) отличается от схемы получения бар-
битала, так как фенильный радикал не может быть введен в
малоновый эфир тем же способом, что и этильный. В качестве
исходного вещества для синтеза фенобарбитала берут хлори-
хлористый бензил и обрабатывают его цианидом калия в спиртовой
среде.
-СН2—Cl + KCN
Из полученного цианистого бензила, диэтилового эфира уголь-
угольной кислоты и этилата натрия получают натриевое производное
этилового эфира фенилциануксусной кислоты:
CeH5CH2CN + C2H5ONa > CeH5-CH-CN + С2Н5ОН
Na
Н u r гл CN
нвс2а
с=о
CeH5-C-CN + С=О > CeH6-C-Na + С2Н5ОН
ос2нб
Процесс проводят следующим образом. В стальной эмали-
эмалированный аппарат загружают абсолютно сухой бензол (проба
с изопропилатом алюминия) и металлический гранулированный
натрий. Вытесняют воздух из аппарата азотом, затем горячим
маслом через рубашку нагревают массу до 70 °С и загружают
из мерника абсолютный этиловый спирт. После окончания вы-
выделения водорода загружают рассчитанное количество диэтило-
диэтилового эфира угольной кислоты, нагревают массу до кипения и
начинают загрузку цианистого бензила. После выдержки отго-
отгоняют из аппарата смесь спирта с бензолом. Охлаждают содер-
содержимое аппарата и отжимают на закрытом вакуум-фильтре нат-
натриевое производное. После промывки сухим бензолом натрие-
226
вое производное растворяют в абсолютном этиловом спирте или
отгоне этилового спирта с бромистым этилом и алкилируют
бромистым этилом:
CN CN
СбНб-С—Na + С2НбВг >- ^С + NaBr
с=о 2 б с=о
ОС2Н* ОС2Нб
Реакцию проводят при температуре около 60 °С в течение
3 ч, затем производят отгонку спирта и избытка бромистого
этила, после чего реакционную массу сжатым азотом передав-
передавливают в отстойник периодического действия (отстойную во-
воронку), куда загружают воду (для растворения NaBr) и 10—
15% серную кислоту для создания рН 6—7. После отстаивания
слоев нижний сливают в сборник, а верхний — этиловый эфир
фенилэтилциануксуснои кислоты передают на вакуум-дистилля-
иию. Следующей стадией является получение натриевого про-
производного «цианиминосоединения» из этилового эфира фенил-
^гнлциануксусной кислоты, дициандиамида и метилата натрия:
C6H64/CN УН* 8_12ОС
С + С—NH-CN + CH3ONa >•
с2н/ \с-ос2н5 II н
О
NH CN
II I
х /С—N4
> С C=NH + СН3ОН + С2НбОН
с2нб/ \с—n/
II I
О Na
Реакцию проводят при температуре 8—12 °С при энергич-
энергичном перемешивании. После выдержки из реакционной массы
отгоняют смесь этилового и метилового спирта. Натриевое про-
производное не выделяют. При нагревании с разбавленной серной
кислотой образуется фенобарбитал:
NH CN
98-102 °С
Nv
C=
NH + 4H2SO4 + 8H2O
II I
О Na
15* 227
С
Полученный фенобарбитал отфильтровывают, промывают и
перекристаллизовывают из смеси ацетона с водой A:3). Су-
Сушат очищенный продукт при температуре 90 °С до содержания
влаги не более 0,5%.
Фенобарбитал является снотворным средством длительного
действия. Кроме того, он несколько снижает артериальное дав-
давление и приостанавливает припадки эпилепсии. Выпускается
фенобарбитал в порошках и таблетках по 0,05 и 0,1 г.
§ 5. АЛИФАТИЧЕСКИЕ АМИНЫ, АМИНОКИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ
Среди алифатических аминов особое значение для медицин-
медицинской практики имеют производные бис-(р-хлорэтил) амина, ко-
которые применяются в качестве противоопухолевых веществ.
Злокачественные опухоли — это заболевания, связанные с уси-
усиленным размножением клеток, приводящим к быстрому росту
новообразованной опухолевой ткани, которая давит на сосед-
соседние органы, прорастая сквозь ткани и дезорганизуя работу ор-
организма. Большую роль в возникновении злокачественных опу-
опухолей играет нарушение обмена нуклеиновых кислот. Различ-
Различные производные бис-(р-хлорэтил)-амина оказывают тормозя-
тормозящее действие на размножение клеток, блокируя их митотиче-
ское деление. Механизм действия лекарственных веществ этой
группы основан на их алкилирующем действии и способности
легко реагировать с нуклеиновыми кислотами, белками и фер-
ферментами. Эти вещества также легко взаимодействуют с нуклео-
протеидами (белок и нуклеиновая кислота) клеточных ядер
кроветворных тканей. Высокой чувствительностью к производ-
производным бис-(р-хлорэтил)-амина обладают ядра клеток опухолевой
ткани. Таким образом, вещества этой группы, с одной стороны,
способны тормозить рост опухолевой ткани, с другой — угнета-
угнетают процесс кроветворения и являются токсичными веществами.
Хлорэтиламины обладают высокой токсичностью и в дозах,
близких к лечебным, могут вызывать нежелательные побочные
явления (угнетение кроветворения, нарушение функций желу-
желудочно-кишечного тракта). В связи с этим при применении пре-
препаратов этого ряда следует соблюдать особую осторожность.
Однако в борьбе со злокачественными опухолями пока прихо-
приходится пользоваться и токсичными препаратами, которые губи-
губительно действуют не только на больные, но и на здоровые тка-
228
ни. При этом стараются всячески уменьшить отрицательное
действие этих препаратов, применяя различные химические
средства, цереливание крови и т. д. В этой области имеются оп-
определенные достижения. Например, химиотерапией лимфогра-
лимфогранулематоза (раковое заболевание крови) удается в ряде слу-
случаев продлить жизнь больных на 10 лет.
Представителем указанной группы лекарственных веществ
является новэмбихип, гидрохлорид 2-хлорпропил-бис-(р-хлор-
этил) амина. Синтезируют новэмбихин в соответствии со следу-
следующей схемой:
СНг—СН2
V /Ch2-ch8oh
СНз-СН—CH2NH2 >■ СН3—СН— СН2—N4 >■
I I х;н2-сн2он
С1 С1
ХН2-СН2С1
>- СНз-СН—СН2—N4 СГ
I | ХСН2-СН2С1
С1 Н
Новэмбихин
Обе стадии синтеза проводятся при нагревании в раствори-
растворителе. В качестве растворителя на первой химической стадии ис-
используют этиловый спирт, а на второй—1,2-дихлорэтан.
Новэмбихин вводят только внутривенно. Дозы должны быть
строго индивидуализированы в зависимости от заболевания, по-
получаемого эффекта и переносимости (обычно 5—10 мг).
Некоторые меркаптопроизводные алифатических аминов об-
обладают способностью оказывать профилактическое радиозащит-
радиозащитное действие при остром лучевом поражении, повышая устой-
устойчивость организма к действию ионизирующей радиации. Дей-
Действие аминотиолов основано на их способности уменьшать ко-
количество радикалов, образующихся в тканях при облучении, а
также на способности этих соединений взаимодействовать с не-
некоторыми ферментами и придавать им устойчивость по отно-
отношению к лучистой энергии. Примерами противолучевых средств
могут служить меркамин — солянокислый 2-меркаптоэтиламин
(HS—СН2СН2—NH2-HC1) и цистамин — солянокислый бис(р-
аминоэтил)дисульфид (H2NCH2CH2S—SCH2CH2NH2-2HC1).
Меркамин применяют внутривенно, цистамин выпускают в
виде таблеток. Препараты являются профилактическими и
должны применяться до облучения или на начальной его ста-
стадии (например, во время лечения злокачественных опухолей ра-
радиоактивными изотопами). На поздних стадиях тяжелой фор-
формы лучевой болезни препараты лечебного эффекта не дают.
Введение карбоксильной группы в молекулу амина резко
снижает токсичность соединения и придает ему иную биологи-
биологическую активность. Аминокислоты, т. е. вещества, содержащие
в молекуле одновременно карбоксильную и аминогруппы, игра-
229
ют важную роль в происходящих в организме биологических
процессах. а-Аминокислоты являются структурными элемента-
элементами белков, некоторые аминокислоты входят в состав витаминов,
различных заменителей крови. Аминокислоты можно разбить
на несколько групп в соответствии с их химическим строением:
а) с одной амино- и одной карбоксильной группой
СН2—СООН
NH2
Глицин
(ашшоуксусная
кислота)
сн3—сн-сн—соон
СН3 NH2
В алии
(а-аминоизовалериановая
кислота)
СНз-СН
СН3--СН-СООН
1
NH2
Алании
(а-аминопропивновая
кислота)
СН3—СН—СНз-СН-СООН
СН3 NHa
Лейцин
(а-аминоизокапроновая
кислота)
2-СН-СН-СООН
1 1
СН3 NH2
Изолейцин
(а-амино-Р-метилвалериановая кислота)
б) с одной амино-, одной окси- и одной карбоксильной груп-
группами:
СН3—СН-СН—СООН
NH2
Треонин
(а-амино-C-оксимасляная кислота)
в) с одной амино- и двумя карбоксильными группами:
НООС— СНа—СН2—СН—СООН;
NH2
Глютаминовая
(а-ашшоглутаровая кислота)
г) с двумя амино- и одной карбоксильной группами:
СН2—СН2—СН2—СН2—СН-СООН
NH2 NH2
Лизин
(а,е-диаминокапроновая кислота)
H2N-C-NH-CH2-CH2-CH2—СН— СООН;
II I
NH NH2
Аргинин
(а-амино-б- гуанидинвалериановая кислота)
д) серосодержащие аминокислоты:
НООС—СН—СН2—S—S—СН2—СН— СООН
I I
NH2 NH2
Цистин
(р,р-дити«-бис-а-амннопропионовая кислота)
230
CH3—S-CH2-CH2—CH—COOH;
NH2
Метионин
(а-амино-7-метилтиомасляная кислота)
е) с ароматическим ядром:
/~~У-СН2-СН-СООН
NH2
Фенил аланин
(а-амино-Р-фенилпропионовая кислота)
ж) с гетероциклическим ядром:
—СН2-СН--СООН
,У NH2
N
Н
Триптофан
(а-амино-З-индолил-3 пропионовая кислота)
нс=с-сн2—сн—соон
I I (
\ NH NH2
у
н
Гистидин
(а-амино-З-имидазолил-4 пропионовая кислота)
Все аминокислоты, за исключением глицина, содержат в мо-
молекуле асимметрический атом углерода, поэтому они могут су-
существовать в D- или L-форме. Все природные аминокислоты
имеют L-конфигурацию.
В строении белков живого мира нашей планеты участвуют
всего лишь около 20 различных аминокислот. Аминокислоты в
белке связаны пептидной связью, возникающей при взаимодей-
взаимодействии карбоксильной группы одной аминокислоты с аминогруп-
аминогруппой другой:
О
Rr-СН—СООН + R2—СН—СООН > Rx—СН—С—NH—СН—СООН + Н2О
NH2 NH2 NH2 R2
Такое соединение носит название дипептид. При соедине-
соединении трех аминокислот образуется трипептид, а при соединении
нескольких — полипептид. Белки можно рассматривать как
сложные полипептиды. Белковые молекулы очень велики. От-
Относительная молекулярная масса обычного яичного белка —
альбумина составляет 34 500, а оксигемоглобина — около
68 000. Процесс синтеза белка протекает в организме непрерыв-
непрерывно. Большая часть аминокислот, необходимых для построения
231
белковых молекул синтезируется организмом, однако некоторые
аминокислоты организм синтезировать не может. Эти амино-
аминокислоты, называемые незаменимыми, должны поступать в орга-
организм с пищей. Их восемь: валин, лейцин, изолейцин, треонин,
лизин, метионин, фенилаланин и триптофан. Некоторые ученые
относят к незаменимым аминокислотам также аргинин и гисти-
дин.
Некоторые аминокислоты выпускаются химико-фармацевти-
химико-фармацевтической промышленностью и используются в качестве лекарст-
лекарственных средств при различных заболеваниях. Так, например,
метионин применяют при заболеваниях печени, глютаминовую
кислоту — при различных психических расстройствах и т. д.
Глава 14
ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ АЛИЦИКЛИЧЕСКОГО
РЯДА
§ 1. ТЕРПЕНОИДЫ
В состав эфирных масел растительного происхождения вхо-
входят вещества алициклического ряда, которые можно формаль-
формально рассматривать как производные изопрена. При этом цикли-
циклические димеры изопрена получили название терпенов, а триме-
ры — сесквитерпенов. Их кислородные производные предложе-
предложено называть терпеноидами. Эфирные масла выделяют из расте-
растений путем перегонки с водяным паром или экстракцией раз-
различными органическими растворителями. Из циклических тер-
пеноидов фармацевтическими препаратами являются ментол,
терпингидрат, камфара и некоторые их производные. Терпенои-
ды обладают антисептическими и дезинфицирующими свойст-
свойствами. Многие терпены и терпеноиды благодаря приятному за-
запаху используются в парфюмерной промышленности.
Ментол A-метил-4-изопропилциклогексанол-3) известен уже
более 2000 лет.
Н3E
СН3
Он содержится в эфирном масле перечной мяты в свободном
состоянии и в виде эфира уксусной кислоты. Содержание мен-
ментола в эфирном масле европейских сортов мяты 40—50%, в
японских — 75—80 %.
Эфирное мятное масло с высоким содержанием ментола
G0—80%) подвергают ректификации, отбирая фракцию с тем-
температурой кипения 208—212 °С. Из этой фракции при охлаж-
охлаждении выкристаллизовывается ментол. Из эфирных масел с
меньшим содержанием ментола D0—60%) последний выделя-
выделяют химическим путем. Для этого эфирное масло нагревают с
борным ангидридом или борной кислотой; образуется борный
эфир ментола, который легко отделяется от остальных состав-
233
ных частей масла вследствие большой разницы в температурах
кипения:
4- В(ОНK-
он
о-
Полученный борный эфир ментола легко гидролизуется водой
при нагревании, поэтому в процессе перегонки его с водяным
паром отгоняется сразу ментол.
Ментол представляет собой кристаллическое вещество с
сильным специфическим запахом мяты, температура его плав-
плавления 42—45 °С, температура кипения 215—216 °С. Он почти
нерастворим в воде и хорошо растворим в спирте, эфире, уксус-
уксусной кислоте. Применяют ментол в качестве дезинфицирующего
средства в зубных порошках, полосканиях, мазях от насморка
и т. п. Ментол входит также в состав успокаивающих средств
против невралгических и ревматических болей.
25—30% раствор ментола в ментиловом эфире изовалериа-
новой кислоты широко известен под названием валидола:
СН3 СН3
СН,
Н3С СН3 Н8С СН3
Валидол представляет собой маслянистую прозрачную жид-
жидкость с запахом ментола. Препарат выпускается также в виде
таблеток, содержащих валидол @,06 г) и сахар. Применяется
в качестве успокаивающего и сосудорасширяющего средства
при стенокардии, неврозах, истерии, а также как противорвот-
ное средство при морской и воздушной болезни.
В качестве лекарственного вещества из терпеноидов исполь-
используется также двутретичный спирт — терпингидрат (а-ментан-
диол-1,8, моногидрат):
Н3С ОН
НдС-С-ОН
сн3
234
Терпингидрат получают гидратацией пинена; при этом в со-
соответствии с правилом Марковникова водород присоединяется
к наиболее гидрогенизированному атому углерода:
Свежеперегнанный скипидар в смеси с древесными опилка-
опилками загружают в освинцованные аппараты, туда же загружают
25%—серную кислоту. Смесь хорошо перемешивают и остав-
оставляют стоять в закрытом реакторе при комнатной температуре
10—14 сут. Выпавшие кристаллы терпингидрата отделяют от
жидкости, перекристаллизовывают из воды, отфильтровывают
и сушат в вакууме.
Гидратация пинена в присутствии серной кислоты проходит
через стадию образования эфира серной кислоты:
Терпингидрат представляет собой бесцветные кристаллы без
запаха, температура плавления 115—117°С. Он плохо раство-
растворим в холодной воде, несколько лучше в хлороформе и эфире.
Применяется при бронхите как отхаркивающее средство. Обла-
Обладает также мочегонным действием.
§ 2. ВИТАМИН А
Если рассмотренные выше терпеноиды имели диизопреноид-
ный скелет, то витамины группы А можно рассматривать как
тетраизоперноиды. Витамин А, называемый также ретинолом
235
или аксерофтолом, содержится только в животных тканях. Осо-
Особенно много его в печени животных и рыб, а также в рыбьем
жире и жире, добываемом из печени морских животных: дель-
дельфинов и китов. Источником витамина А для рыб служит планк-
планктон, состоящий из мельчайших рачков и водорослей. В растени-
растениях витамин А не найден. Однако провитамин А (каротин) ши-
широко распространен в растительном мире. Особенно богаты ка-
каротином листья шпината, крапивы, люцерны, одуванчика, ща-
щавеля, петрушки. Много каротина содержится в моркови и
тыкве.
В витамине А нуждаются все животные. Человек, а также
травоядные и всеядные животные могут удовлетворять пот-
потребность в витамине А как за счет поступления с пищей этого
витамина, так и за счет каротина, который в организме прев-
превращается в витамин А. Хищные звери и птицы, а также рыбы
не способны к синтезу витамина А из каротина. Потребность
человека в витамине А зависит от возраста. Наиболее высокая
потребность наблюдается в возрасте 16—20 лет. Усиленное по-
поступление в организм витамина А требуется при лечении ряда
инфекционных заболеваний. Установлено, что ретинол повыша-
повышает устойчивость организма к некоторым ядам и токсинам. От-
Отсутствие в пище витамина Ai (ретинола) или C-каротина при-
приводит к ксерофтальмии — высыханию роговицы глаз и слепоте,
похудению и общему плохому состоянию, а недостаток — к ос-
ослаблению сумеречного зрения (куриной слепоте). Последнее
связано с тем, что витамин А участвует в создании зрительного
пурпура родопсина, обусловливающего вечернее, сумеречное
зрение. Добавка витамина А в корм животных обеспечивает
быстрый рост животных и значительную экономию кормов на
единицу привеса.
Витамин А можно получать как из природного сырья (кито-
(китовый жир), так и путем синтеза. Существует несколько методов
синтеза витамина А\. Как уже указывалось, ретинол представ-
представляет собой тетраизопреноид. Структурная формула витамина
Ai была установлена в 1931 г. Каррером:
Н3С СНз1,1 ^ Н СНя
1
/Cvv Xs. Xsv ,СН2ОН
с с с с
зН н н н
Витамин А (ретинол, аксерофтол)
Биологически активной является транс-конфигурация вита-
витамина. Ретинол является ненасыщенным первичным спиртом.
Он образует сложные эфиры с кислотами, окисляется до аль-
альдегида. При каталитическом гидрировании ретинол присоеди-
присоединяет 5 молей водорода, что подтверждает наличие пяти двой-
двойных связей в его молекуле.
236
В СССР принят метод синтеза витамина Ai из р-ионона.
ft-Иононовое кольцо состоит как бы из двух молекул изопрена:
Н3С СН3
CH3
С—СН=СНС=О
н,с ../\ н2с с-сн3
> СН=СН2 / \ |
е' | н2с сснснс
н/
с I
н2с
сн3
Изопрен
Получают р-ионон из ациклического терпеноида — цитраля.
При конденсации цитраля и ацетона в присутствии щелочи по-
получается псевдоионон, который при действии концентрирован-
концентрированной серной кислоты циклизуется в р-ионон:
СН* ~ Н3С СН3 ли
^ ,,—Сч СНзСОСНз, NaOH ^ л—СН=СН—-Сч^ H2SO4
Ъ =70-80%
Цитраль
няс
'"So
СН3
Р-Ивнон
р-Ионон конденсируется с хлоруксусным эфиром в присут-
присутствии метилата натрия или изобутилата калия:
Н3С СН3 /СН3 Н3С СН3 ?Нз
cich2co2r \/ СН=СН-<С CH-COOR
CHgONa (^^rS \ /
О *
Н3С
3
«глицидный эфир»
сн3
«альдегид С14»
Реакция проходит через стадию образования глицидного эфи-
Ра, который легко гидролизуется щелочью в спиртовом раство-
Ре в соль глицеридной «кислоты Cis», декарбоксилирующуюся
с образованием «альдегида Си» (температура плавления 2°С)
с общим выходом из р-ионона 80—90%. Альдегид с целью очи-
очитки перегоняют в глубоком вакууме.
237
Другим промежуточным продуктом для синтеза витамина А
является первичный ацетиленовый карбинол, который получают
.из метилвинилкетона:
СН3
(НСЕЕСJСа I
л3с-со-сн=сн2 >- нс=с-с-сн=сн2 —>■
„Метил винил кетон ОН
Третичный ацетиленовый
карбинол
—> нс=с—с=сн-сн2он
СН3
Первичный ацетиленовый
карбинол
Полученные таким образом продукты вводятся в реакцию
Гриньяра:
Н3С СН3
Х/Сн2-сн=с-с=о Вг
| СН8 Н + НС=С-С=СН—СН2°Н *~
н3с сн3 Vм
ХН2-СН=С-СН-С=С-С=СН-СН2ОН
| | Н2, Ра/СаСОз»
СН3 СН, "
«Диолин Сге»
ОН
| СН3СОС 1, пиридин
—^£1=^—\^Г\—L.rl=L>rl—•L/=L/rl—L/il2Url ^
I I
СНз СН3
СН3
«Этиленовый гликоль Qo*
ОН
Н3С
\/.СН2--СН=С-СН--СН=СН
СН
L3
«Гликоль-ацетат»
Н3С СН3
;н=сн—с=сн—сн=сн-
СН3
РОС13, пиридин ><^СН=СН^С=СН^СН=СН^С=СН-СН2ОСОСН3
Витамин А-ацетат
В результате реакции Гриньяра получается «диолин С2о» в ви-
виде кристаллического продукта с температурой плавления 58—'
59 °С. При специфическом восстановлении ацетиленовой связи
238
до этиленовой в присутствии катализатора Линдлара (см. гла-
главу 10) получают «этиленовый гликоль С20». Первичную гидрок-
сильную группу гликоля обрабатывают хлорокисью фосфора в
присутствии пиридина для дегидратации и аллильной перегруп-
перегруппировки. В результате получают ретинол-ацетат с общим выхо-
выходом до 45%.
Чистый витамин А очень чувствителен к воздействию окис-
окислителей и к ультрафиолетовым лучам. Его эфиры обладают
значительно большей устойчивостью. Уксусный эфир витамина
А обладает биологической активностью и хорошо кристаллизу-
кристаллизуется, поэтому он принят в качестве международного стандарта.
Фармакопейным препаратом витамина А является масляный
раствор витамина А-ацетата (аксерофтол-ацетат).
§ 3. ЗАМЕНИТЕЛИ ПЛАЗМЫ КРОВИ
Установлено, что потеря небольших количеств крови (менее
450 г) не приносит человеку вреда. Благодаря хорошо сбалан-
сбалансированным процессам- кроветворения и кроверазрушения об-
общее количество крови в кровеносной системе восстанавливается
уже через сутки. Состав крови восстанавливается медленнее,
так как с кровью уходят ее живые клетки — эритроциты, лейко-
лейкоциты, тромбоциты. Считали, что причиной смерти при потере
крови является кислородное голодание, так как в кровяном
русле уменьшается количество переносчиков кислорода — эрит-
эритроцитов. В настоящее время установлено, что в большинстве
случаев при кровопотере смерть наступает вследствие резкого
падения кровяного давления в сосудах, так как при этом жиз-
жизненно важные органы плохо снабжаются кровью. В таких слу-
случаях достаточно ввести в кровеносные сосуды плазму крови или
кровезаменитель (то есть жидкость, которая восстанавливает
общую массу крови), и давление повышается, а вместе с тем
восстанавливается снабжение органов кровью. Естественным
источником такой жидкости является кровь донора, если она
имеет требуемую групповую принадлежность.
В связи с трудностями при заготовке и хранении большого
количества препаратов из натуральной крови проблема крове-
кровезаменителей имеет очень большое значение в медицине. В хи-
хирургической практике кровезамещающие и противошоковые
растворы применяются не только в предоперационном периоде,
Во время и после операций, но и с целью парентерального пи-
питания, борьбы с интоксикацией и осложнениями гнойной инфек-
Ции. Особое значение приобретают кровезаменители при лече-
лечении ожоговой болезни и при больших потерях крови.
^Высокоэффективным кровезаменителем противошокового
Действия является полиглюкин, который представляет собой
Стерильный коллоидный раствор среднемолекулярной фракции
частично гидролизованного декстрана (полисахарид). Получа-
239
ют его гидролизом нативного декстрана, синтезируемого из са-
сахарозы при помощи определенного штамма бактерий. В пост-
построении этого полимера участвует только глюкоза, входящая в
структуру сахарозы. Молекулы глюкозы соединяются друг с
другом связями между атомами углерода в положениях 1 и 6:
Полиглюкин представляет собой бесцветную или слегка
желтоватую жидкость. Средняя относительная молекулярная
масса 60000 (±10000), рН = 4,5—6,5. Благодаря относительно
большой молекулярной массе полиглюкин не проникает через
сосудистые мембраны и при введении в кровяное русло долго
в нем циркулирует. Вследствие высокого осмотического давле-
давления он хорошо удерживает жидкость в кровяном русле, что при-
приводит к быстрому повышению и сохранению кровяного давле-
давления. Полиглюкин не токсичен, применяется при операционном
и травматическом шоке, ожогах, острых кровопотерях.
Хорошо зарекомендовал себя в клинической практике син-
синтетический кровезаменитель поливинилпирролидон. Водно-соле-
Водно-солевой 6% раствор поливинилпирролидона с молекулярной массой
12 600±2700, содержащий также ионы натрия, калия, кальция,,
магния и хлора применяется в медицинской практике под назва-
названием гемодез.
—сн—сн —
Поливинилпирролидон
240
Гемодез — прозрачная жидкость желтого цвета с рН 5,2—7,0;
применяется для дезинтоксикации организма при токсических
формах острых желудочно-кишечных заболеваний, при ожого-
ожоговой болезни, при послеоперационной интоксикации, при инфек-
инфекционных заболеваниях. Механизм действия гемодеза обуслов-
обусловлен способностью поливинилпирролидона связывать токсины,
циркулирующие в крови, и быстро выводить их из организма.
Препарат быстро выводится почками (до 80% за 4 часа) и ча-
частично через кишечник.
Для профилактики и борьбы с шоком применяется также
белковый кровезаменитель БК-8, получаемый из белков плаз-
плазмы крови крупного рогатого скота. Имеется и ряд других кро-
кровезаменителей.
§ 4. ВИТАМИН К И ВЕЩЕСТВА, ВЛИЯЮЩИЕ
НА СВЕРТЫВАЕМОСТЬ КРОВИ
Свертывание крови является сложным и жизненно важным
процессом. При повреждении клеточных тканей из протромби-
протромбина плазмы крови в присутствии ионов кальция под влиянием
фермента тромбокиназы образуется специфический фермент
тромбин (не содержащийся в крови). Под влиянием этого фер-
фермента растворимый в крови белок фибриноген превращается в
нерастворимый фибрин. Из нитей и сгустков фибрина создается
основа тромба, закупоривающего сосуд. Пониженная свертыва-
свертываемость крови является опасной, так как даже незначительная
травма может привести к большой кровопотере. С другой сто-
стороны, образование тромба без внешних повреждений — тромбо-
тромбофлебит— является также тяжелым и трудно поддающимся ле-
лечению заболеванием.
Свертываемость крови регулируется в организме витамином
К (витамин коагуляции). Витамины группы К являются про-
производными 2-метил-1,4-нафтохинона:
О
о
2-Метил-1,4-нафтохинон
СН3 СН3 СНз
I—L>rl==V>—(v-<JLi2J3—^**—1^-^2/3—^^—V^-^2/3
и ' I
0 HgC-CH
СНз
Витамин Ki
B-Метил-3-фитнл-1,4-ыафтвхинон)
16-926 24J
Н. А. Преображенским описан полный синтез витамина Кь
в основу которого положена конденсация 2-метилнафтогидрохи-
нона-1,4 с изофитолом. Витамин Ki (филлохинон) содержится
в зеленых и сочных частях растений, особенно в листьях лю-
люцерны, шпината, каштана, в цветной капусте, хвое. Плохая рас-
растворимость витаминов группы К затрудняет их применение как
лекарственных средств. В 1942 г. акад. А. В. Палладину уда-
удалось синтезировать водорастворимый аналог витамина К, кото-
который под названием викасола нашел широкое применение в ме-
медицине. Викасол является бисульфитным производным 2-метил-
1,4-нафтохинона. Последний иногда называют витамином Кз (ме-
тинон, менадион), хотя правильнее рассматривать его как про-
провитамин (предшественник витамина). Синтез осуществляют по
следующей схеме:
NH2 О _ _ ОН
сн=сн2
СгОз, СНзСООН
Викасол представляет собой белый кристаллический поро-
порошок горького вкуса, легко растворимый в воде, плохо — в орга-
органических растворителях. Препарат применяют при пониженном
содержании протромбина в крови, для увеличения свертывае-
свертываемости крови.
Избыток витамина К в организме может привести к тромбо-
тромбозу или тромбофлебиту.
Вещества, которые понижают свертываемость крови, назы-
называют антикоагулянтами. К антикоагулянтам относятся дикума-
рин и неодикумарин:
ОН ОН
б N) о^ о
Дикумарин, Ди-D-оксикумарннил-3)-метан
СООС2Н5
ОН | ОН
Неодикумарин
242
Дикумарин и неодикумарин применяются для лечения тромбо-
тромбозов, тромбофлебита и при коронарной недостаточности. Неоди-
Неодикумарин действует несколько быстрее и мягче, чем кумарин, а
также обладает меньшей токсичностью.
§ 5. ВИТАМИН Р, БИОФЛАВАНОИДЫ
В 1936 г. Сент-Георги, исследуя природные экстракты вита-
витамина С, обнаружил, что при лечении цинги они гораздо эффек-
эффективнее, чем синтетическая аскорбиновая кислота. Оказалось,
что природные экстракты витамина С содержат вещество, ко-
которое влияет на хрупкость и проницаемость капиллярных кро-
кровеносных сосудов. Это вещество назвали витамином Р — «вита-
«витамином проницаемости». Недостаток витамина Р в организме
вызывает точечные кровоизлияния вследствие разрушения ка-
капилляров или повышенной проницаемости их стенок. Витамин
Р — условный термин, объединяющий большую группу биофла-
ваноидов, обладающих капилляроукрепляющим действием.
В настоящее время считают, что биофлаваноиды не являются
витаминами в точном смысле этого слова, так как им не свой-
свойственны какие-либо выраженные биокаталитичеокие функции.
Флаваноиды представляют собой производные флавана
B-фенилхромана):
Флаван (Р-внтаминн*й активнестью не обладает)
Биофлаваноиды встречаются в цитрусовых плодах, красном
перце, листьях чая. Из группы оксипроизводных этого ряда со-
соединений (флаванолов) следует отметить рутин, который при-
применяется в медицине как препарат с Р-витаминной активно-
активностью. Рутин впервые был выделен из полукустарникового рас-
растения семейства рутовых. Позднее он был найден в зеленой
массе гречихи, а также в листьях ряда других растений.
н
он
Рутин
16*
243
Рутин применяется не только для лечения заболеваний, свя-
связанных с нарушением функций капилляров, но для лечения по-
последствий обморожений. В 1962 г. проф. Н. А. Преображенский
(с сотрудниками) впервые опубликовали полный синтез рути-
рутина. Рутин представляет собой кристаллическое вещество жел-
желтого цвета, плохо растворимое в воде, удовлетворительно — в
спирте и пиридине. Препарат выпускается в порошках и таб-
таблетках совместно с аскорбиновой кислотой. Акад. А. А. Кур-
санов и проф. М. Н. Запрометов разработали технологию по-
получения катехинов из отходов чайного производства и провели
широкие клинические испытания. Новый препарат оказался бо-
более эффективным, чем рутин. Его успешно применяют при ле-
лечении подкожных кровоизлияний и кровотечений из слизистых
оболочек. Флаваноиды содержатся главным образом в расти-
растительных продуктах. Сырьем для промышленного производства
препаратов витамина Р в нашей стране служат зеленая масса
гречихи, бутоны софоры японской и отходы чайного производ-
производства.
§ 6. ТОКОФЕРОЛЫ (ВИТАМИНЫ ГРУППЫ Е)
Термин «витамин Е» является групповым обозначением ве-
веществ, обладающих биологической активностью, присущей а-то-
коферолу:
?** СН3 СН8
(СН2)з-СН-(СН2K-СН-(СН2)з-СН(СН8)о
^0
СН3 а-Токоферел
Все токоферолы (греч. «потомство несу») являются производ-
производными хромана (бензо-р-дигидропирана):
О
В положении 2 ядра хромана у токоферолов находятся мет иль-
ильная группа и боковая цепь изопреноидного характера — остаток
фитола. Таким образом, по химической структуре токоферолм
родственны биофлаваноидам и некоторым другим уже рассмот-
рассмотренным нами соединениям.
Токоферолы получают как из природных источников. гг.к п
синтетическим путем. Попытки заменить сложный фитольный
остаток на более простой приводили к получению веществ не
обладающих Е-витаминной активностью.
244
Упрощенная схема синтеза а-токоферола может быть пред-
представлена следующим образом:
CH3
СН3
I
Fe, HC! H^
Н,С
СН3
S
FeCls
|
СН3
Псевдокумол
СН3
п - Дикитропсевдо-
кумол
I
NH2
CH3
гс-Диаминопсевдо-
кумол
сн3
сня
СН,
«ЧА, so2 H0V^l c'^coa НбСб°Ч
HsC/^V^^O HoC-^^^^OH
В полученном тем или иным способом (например, из пссвдо-
ионона) фитоле заменяют оксигруппу на бром, а образовавшим-
образовавшимся фитилбромидом алкилируют бензоилированпый триметнлгид-
рохинон:
СН3 СН2Вг
Н5С6ОСО I I
I
СН
сня
11 + !i
НзС^^^^ОН С—СН3 СН3 у"з СНз
СН3 (СН2K—CH-(CH2K--CH-(CH2K-CHN
, ZnCl,
>•
80 СС
CfiHsCO.
I
СНд СНз СНз
| /(СН^г-СН-^Н^з-СН-ССН^з-СН-СНа— ->
о
но.
СН3
I
СН8
сн.
а-Тско1
СН,
Витамин Е можно получить также из растительных масчм
зародышей пшеницы и другого природного сырья.
Физиологическое действие Ептамина Е обусловлено его уча-
участием в окислительно-восстановительных процессах. а-Токофг-
рол является также аптпкоагулянтом крови. Особенно сиг.ьпс
г^ияег витамин Е на фу}!кцию размножения, поэтому его на-
называют антистерильным витамином. Очень важна роль Битами-
На Е для увеличения продуктивности животноводства. Имеюг-
246
ся сведения о взаимосвязи наличия витамина Е в организме и
процесса старения организма. Отсутствие витамина Е приводит
также к мышечной дистрофии.
§ 7. СТЕРОИДНЫЕ СОЕДИНЕНИЯ, ГОРМОНЫ
Среди биологически активных веществ, используемых в ме-
медицине, важное место занимают стероидные соединения. В осно-
основе стероидных соединений лежит тетрациклическая структура
циклопентанопергидрофенантрена (принятая нумерация угле-
углеродных атомов и обозначения ядер показаны на схеме):
Биологически активные стероиды часто содержат метальные
группы в положениях 10 и 13, различные заместители в поло-
положениях 3 и 17, а также кратные связи. Молекулы стероидов
имеют сложную трехмерную конфигурацию, которая не нахо-
находит отражения в принятом написании формул. Все узловые ато-
атомы углерода E, 8, 9, 10, 13, 14) асимметрические, поэтому сте-
стероидные соединения имеют большое число пространственных
изомеров.
Стероиды широко распространены в природе. В каждой
клетке животного организма содержится стероид холестерин.
Холестерин
Особенно много холестерина в мозговых тканях, в печени,
почках, а также в крови. В организме человека содержится до
140 г холестерина. Часть холестерина поступает с пищей, од-
однако большая его часть синтезируется в организме. Нарушение
холестеринового обмена — одна из причин атеросклероза, это
связано с отложениями на стенках сосудов холестерина и его
эфиров. Отличающийся от холестерина наличием у С24 допол-
дополнительной этильной группы бета-ситостерин является важным
лекарственным средством, регулирующим холестериновый об-
246
мен, он широко применяется для лечения больных атеросклеро-
атеросклерозом. Близкий по строению к холестерину эргостерин содержится
в дрожжах и служит источником витамина D (см. § 8).
Содержащиеся в желчи кислоты (желчные кислоты) также
относятся к стероидам. Эти вещества способствуют усвоению
жиров, а также связывают и выводят из организма некоторые
токсины (яды). Примером желчной кислоты может служить
холевая кислота:
он сн3
I СНЯ I
н,с
с
он оУ он
Холевая кислота
Среди стероидных соединений находятся многие сердечные
гликозиды. Сердечные гликозиды являются ядами раститель-
растительного происхождения. В небольших цозах эти вещества стиму-
стимулируют сердечную деятельность и широко используются в каче-
качестве лекарственных средств.
Многие гормоны имеют стероидную природу. Гормонами на-
называются вещества, выделяемые в кровь железами внутренней
секреции. Поступая в кровь в малых количествах, каждый гор-
гормон оказывает свое специфическое воздействие на внутритка-
внутритканевые процессы. Гормональные препараты применяются в ка-
качестве лекарственных средств в тех случаях, когда нарушена
нормальная деятельность той или иной железы и организму не-
необходимо возместить недостаток соответствующего гормона.
Очень часто гормональные препараты применяются для лече-
лечения других нарушений обмена веществ, а также при комплекс-
комплексном лечении инфекционных заболеваний. Гормоны коры надпо-
надпочечников — кортикостероиды — принято делить на две группы:
глюкокортикоиды и минералокортикоиды. К глюкокортикоидам
относятся кортизон и его производные (гидрокортизон, предни-
золон, преднизон и др.).
СНЯ ОН
—С-СН2ОН
II
X A J
о-
Кортизон
Глюкокортикостероиды активно влияют на углеводный и
белковый обмен, но менее активны в отношении водного и соле-
солевого обмена: Они способствуют накоплению гликогена в пече-
печени, повышают содержание сахара в крови, вызывают увеличе-
увеличение выделения с мочой связанного азота. Под влиянием глю-
247
кокортикостероидов задерживается синтез и ускоряется распад
белка. Популярность кортизона и его синтетических аналогов
как лекарственных препаратов основана на их мощном проти-
противовоспалительном действии. Они помогают в комбинации с
другими средствами и при раке крови (лейкозе) и при многих
микробных заболеваниях. Однако применение их требует боль-
большой осторожности, так как иначе может быть причинен вред
всему организму и в первую очередь надпочечникам.
Минералокортикоиды — альдостерон, дезоксикортикостерон
и другие — активно влияют на обмен электролитов и воды, но
мало влияют на углеводный и белковый обмен.
СН,
Дезоксикортик«стерон
Гормоны, вырабатываемые половыми железами, также име-
имеют стероидное строение:
НО"
Эстрон Эстрадиол
(женские половые гормоны)
НО
Тествстерон Андростерон
(мужские половые гормоны)
Препараты половых гормонов, выпускаемые промышленно-
промышленностью в качестве лекарственных веществ, применяются при рас-
расстройствах половой сферы и некоторых других заболеваниях.
Все они активно вмешиваются в происходящие в организме хи-
химические процессы: регулируют синтез белков, выделение азо-
азотистых веществ, калия, фосфора и т. д.
Стероидные гормоны получают в промышленном масштабе.
Это очень сложные промышленные синтезы, состоящие из 15—•
18 химических стадий. Конечный выход продукта в расчете на
исходное сырье часто составляет менее 10%.
248
§ 8. ВИТАМИН D
Термин «витамин D» имеет собирательное значение, объеди-
объединяя группу близких по химической структуре соединений, регу-
регулирующих обмен кальция и фосфора в организме. Витамин D
играет огромную роль в формировании костного скелета. В ви-
витамине D нуждаются все млекопитающие, птицы и рыбы. Осо-
Особенно необходим он в молодом возрасте, когда идет быстрый
рост и окостенение скелета. Кальций откладывается в костях в
виде фосфата и карбоната. Процесс отложения солей кальция
в костях идет только при определенной концентрации фосфат-
ионов в крови. Необходимый уровень концентрации этого эле-
элемента в крови регулируется с помощью витамина D. Недостаток
витамина в организме приводит к рахиту и ряду других забо-
заболеваний. Давно было замечено, что заболевание рахитом чаще
встречается у тех людей, которые мало бывают на солнце. Од-
Однако причина этого явления была раскрыта лишь в 20-х годах
XX века, когда стало известно, что растительные масла, не со-
содержащие витамина D, можно сделать активными против ра-
рахита, если облучить их ультрафиолетовыми лучами. Впослед-
Впоследствии было установлено, что йитамин D образуется при облу-
облучении из содержащихся в жирах стеринов, в частности из эрго-
стерина:
СН8 СН8
СН8 | СН8
|/N Н2С -
Н
Эргостерин Эргокальциферол
(витамин D2)
Эргостерин в настоящее время получают из пекарских дрож-
дрожжей, где он содержится в количестве 0,2—0,3% от массы прес-
прессованных дрожжей. Значительно выше содержание эргостерина
в некоторых сортах кормовых дрожжей. В последние годы эрго-
эргостерин стали получать также из отходов производства пеницил-
пенициллина. Превращение эргостерина в эргокальциферол в большой
мере зависит от источника излучения и наблюдается только
при наличии излучения с определенной длиной волны
(^280 нм при облучении растворов в эфире). Использование
источников ультрафиолетового излучения, имеющих не опти-
оптимальный спектр, приводит к образованию ряда побочных про-
продуктов. Обычно реакционная масса после фотолиза состоит из
непрореагировавшего эргостерина и «фотосмолы», которая
представляет собой смесь продуктов. Эргокальциферол извле-
извлекают из «фотосмолы» химическими методами. При химической
очистке облученного эргостерина получается два вещества с
249
разной витаминной активностью. Одно из них (с меньшей ак-
активностью) было названо витамином Di, а другое (эргокаль-
циферол) — витамином D2.
Из холестерина был получен витамин D3, названный холе-
кальциферолом. Активность его в 1,5 раза выше активности ви-
витамина D2. По химическому строению витамины группы D от-
отличаются друг от друга строением боковых цепей.
Витамин D содержится в небольших количествах в яичном
желтке, икре, в сливочном масле и молоке, в значительно боль-
больших количествах — вместе с витамином А в тресковой печени
и жире.
Выпускают витамин D в виде следующих препаратов: дра-
драже эргокальциферола (для профилактических целей), 0,125%
раствора эргокальциферола в масле (для лечебных целей),
0,5% раствора эргокальциферола в спирте (только для лечеб-
лечебных целей).
Витамин D вместе с витамином А содержится в так назы-
называемом «рыбьем жире». Препараты витамина D хранят в ус-
условиях, исключающих действие на них света и воздуха, так как
кислород воздуха окисляет витамин D, а на свету он превраща-
превращается в ядовитый токсистерин.
Глава 15
ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ
АРОМАТИЧЕСКОГО РЯДА
§ 1. ФЕНОЛЫ И ИХ ПРОИЗВОДНЫЕ
К классу фенолов относится большая группа лекарственных
веществ, обладающих антисептическими свойствами. Антисеп-
Антисептические свойства фенолов основаны на их способности сверты-
свертывать белки. При введении в молекулу фенола таких заместите-
заместителей, как алкил, алкоксил, галоген, усиливается бактерицидная
активность. Увеличение длины алкильной цепи или разветвле-
разветвление ее приводит к усилению антисептического действия. В отли-
отличие от алифатических спиртов при увеличении числа гидро-
ксильных групп в ароматическом ядре увеличивается токсич-
токсичность соединения.
Способы синтеза фенола излагались в первой части учеб-
учебника. Кристаллический фенол применяется в стоматологии. Го-
Государственная фармакопея требует содержания чистого фенола
в препарате не менее 98%. В медицинской практике применя-
применяется также 90% раствор фенола в воде, представляющий собой
бесцветную или розоватую жидкость с характерным запахом
фенола и кислой реакцией на лакмус. Разбавленные B—3%)
растворы фенола применяются для дезинфекции кожи рук и хи-
хирургических инструментов. Из-за кислой реакции фенол иногда
называют карболовой кислотой. Применяется фенол только как
наружное средство. Фенол оказывает на кожу раздражающее
и прижигающее действие, он легко всасывается через кожу и в
больших дозах может вызвать токсические явления.
В качестве антисептика используется также смесь о-, п- и м-
крезолов:
сн,
Раздражающее и токсическое действие крезолов на орга-
организм выражено слабее, что связано с их меньшей растворимо-
растворимостью.
251
Двухатомный фенол, резорцин
ОН
>Н
по токсичности близок к фенолу, но антисептические свойства
у него выражены сильнее. Резорцин находит применение при
лечении кожных заболеваний (наружно в виде паст и мазей).
Большое значение в медицине получило производное п-ами-
нофенола — фенацетин:
NH2 NHCOCHg
1
ОН ОС2Нб
я-Аминофенол Фенацетин
Технология синтеза фенацетина и область его применения
подробно излагались в первой части (глава 7, § 5). Принцип,
положенный в основу синтеза фенацетина, т. е. изучение про-
продуктов изменения анилина в организме и создание физиологи-
физиологически активных веществ путем преобразования молекулы ани-
анилина, вошел в литературу под названием «принцип фенацети-
фенацетина». Этот принцип, учитывающий пути обезвреживания в орга-
организме токсических веществ, находит широкое применение при
создании новых лекарственных средств.
§ 2. АРОМАТИЧЕСКИЕ КАРБОЛОВЫЕ КИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ
Введение карбоксильной группы в молекулу ароматического
соединения, как правило, снижает как токсичность, так и био-
биологическую активность вещества, поэтому сами карбоновые
кислоты редко используются в качестве лекарственных веществ.
Однако амино- и оксипроизводные карбоновых кислот широко
используются в медицине благодаря их высокой биологической
активности и относительно малой токсичности. Свободные аро-
ароматические карбоновые кислоты (например, бензойная) исполь-
используются как слабые антисептики, а их соли — как носители спе-
специфических катионов (см. главу 12).
Большое значение имеют лекарственные вещества на основе
салициловой кислоты, технология производства которой рас-
рассматривалась в главе 8. Способы получения некоторых произ-
производных салициловой кислоты (аспирина и салола) рассмотре-
рассмотрены в главе 7.
252
Фенилсалицилат (салол) является эфиром салициловой кис-
кислоты и фенола:
он
Фенилсалицилат (салол)
Фенилсалицилат применяется в порошках и таблетках при за-
заболеваниях кишечника. Действие препарата основано на том,
что в слабощелочном содержимом кишечника фенилсалицилат
гидролизуется на салициловую кислоту и фенол, которые уг-
угнетающе действуют на кишечную флору. Салициловая кислота
и фенол частично выделяются из организма почками и могут
оказывать дезинфицирующее действие в мочевых путях.
Действие ацетилсалициловой кислоты во многом сходно с
действием на^ организм салицилата натрия, что понятно, так
как ацетильное производное в желудочно-кишечном тракте
гидролизуется:
н2о r^v^
' Л + CHgCOOH
Ацетилсалициловую кислоту (аспирин) применяют внутрь
при мигрени, как жаропонижающее и болеутоляющее средство.
Она оказывает также противовоспалительное действие. В связи
с распространенностью и доступностью этого лекарственного
препарата следует отметить, что ацетилсалициловая кислота
может вызвать нежелательные побочные осложнения со сторо-
стороны желудка. Длительное применение аспирина может привести
к диспепсии и желудочным кровотечениям; кроме того, возмож-
возможны побочные явления, связанные с нарушением функций нерв-
нервной системы.
Большое значение в химии лекарственных соединений имеют
аминокислоты ароматического ряда, которые участвуют во мно-
многих жизненно важных процессах в качестве промежуточных ве-
веществ. В связи с этим п-аминобензойную кислоту рассматрива-
рассматривают как витамин (витамин Hi). п-Аминобензойная кислота яв-
является жизненно важным фактором для многих микроорганиз-
микроорганизмов, а также необходимым элементом для биосинтеза некото-
некоторых витаминов. Эфиры п-аминобензойной кислоты нашли
применение в медицине как лекарственные вещества с местно-
анестезирующим действием. Примерами таких веществ могут
служить новокаин и анестезин. Этиловый эфир п-аминобензой-
п-аминобензойной кислоты (анестезин) применяется как местноанестезирую-
253
щее средство более 70 лет. Получают его из п-нитротолуола по
следующей схеме:
СН3 СООН
К2Сг207, H2SO4 г^Ч С2Н5ОН, H2SO4
-О
NO2
СООС2Н6 СООС2Н5
Fe, CH3COOH
NO2 NH2
Химизм и технология реакций, составляющих процесс, бы-
были разобраны в соответствующих разделах первой части. Тех-
Технический продукт очищают перекристаллизацией из спирта с
активированным углем и гидросульфитом натрия. Чистый про-
продукт представляет собой белый кристаллический порошок без
запаха, слабо горького вкуса. Легко растворяется в эфире, хло-
хлороформе, жирах. Применяется как внутреннее и как наружное
средство.
Солянокислый р-диэтиламиноэтиловый эфир п-аминобензой-
ной кислоты (новокаин) также получают из п-нитротолуола.
NH2
I
I
С
j \
н
О O-CH2-CH2-N4 С1-
" ХС2Н5
Синтез можно осуществлять несколькими различными спо-
способами:
I. СН3 СООН СОС1
К2Сг207. H2SO4 ^\ РОС13 r^X HO-CH2-CH2-N(C2H5J
NO
2
COO-(CH2J-N(C2H6)9 COO-(CH2J-N(C2H6J
NO, NH,
254
Необходимый для синтеза диэтиламиноэтанол получают при
пропускании окиси этилена через спиртовый раствор диэтил-
амина:
Ш(С2НбJ + (СН2JО > HO-CH2-CH2-N(C2H6J
II. СООН
COO(CH2JC1 COO(CH2JN(C2H5J
NH(C2H6J
Наличие диэтиламиноэтильного остатка в молекуле новокаи-
новокаина усиливает анестезирующее действие препарата и делает
возможным применение его в виде растворимой солянокислой
соли, что очень важно для приготовления инъекционных раство-
растворов. Водные растворы новокаина имеют нейтральную реакцию.
Поскольку водные растворы новокаина легко гидролизуются,
для их стабилизации добавляют 0,1 н раствор соляной кислоты
до рН 3,8—4,5. В настоящее время новокаин является одним
из наиболее широко применяющихся местноанестезирующих
средств. Важным препаратом является новокаинамид:
NH2
0=C—NH—CH2—CH2—N' СГ
I i T-T
H
Введение амидной связи вместо эфирной изменило биоло-
биологическую активность вещества. Новокаинамид не является ане-
анестезирующим веществом, а применяется при расстройствах сер-
сердечного ритма.
Если п-аминобензойная кислота является необходимым фак-
фактором для нормальной деятельности микроорганизмов, то п-ами-
носалицилавая кислота, участвуя в обмене веществ, подав-
подавляет рост микроорганизмов. Таким образом, п-аминосалицило-
вая кислота является антагонистом п-аминобензойной кислоты.
п-Аминосалициловая кислота (ПАСК) и ее натриевая соль
обладают бактериостатической активностью в отношении мико-
бактерий туберкулеза и относятся к основным противотуберку-
255
лезным препаратам. При приеме внутрь ПАСК хорошо всасы-
всасывается и проникает в сыворотку крови и ткани внутренних ор-
органов. п-Аминосалициловую кислоту и ее соль часто применяют
в комбинации с другими противотуберкулезными препаратами.
Получают ПАСК из м-аминофенола. Первой стадией явля-
является карбоксилирование (см. главу 8):
COOK
ОН
CO2, KHCO3
103—105 CC
NH2
Реакция проходит под давлением при температуре 103—105°С.
Полученную калиевую соль п-аминосалициловой кислоты очи-
очищают переосаждением, продукты осмоления сорбируют активи-
активированным углем:
COOK СООН
+H2SO4
NH2
COOH
^OH
+ NaHCO3
NH2
+K2SO4;
+ CO2 + H2O
п-Аминосалицилат натрия представляет собой кристалличе-
кристаллический белый порошок с желтоватым или розоватым оттенком.
Легко растворим в воде, трудно — в спирте. Препарат выпуска-
выпускается также в виде гранул, состоящих из одной части п-амино-
салицилата натрия и 2 частей сахара, и в виде 3% водного рас-
раствора для инъекций.
§ 3. ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СУЛЬФОКИСЛОТ.
СУЛЬФАНИЛАМИДНЫЕ ПРЕПАРАТЫ
О значении ароматических сульфокислот и их производных
в технологии лекарственных веществ говорилось в главе 3.
Следует еще раз подчеркнуть, что введение сульфогруппы в мо-
молекулу ароматического соединения увеличивает растворимость
его в воде, снижает токсичность, расширяет терапевтический
эффект.
Особое значение для медицинской практики имеют производ-
производные амидов сульфокислот (хлорамины) и производные сульф-
сульфаниламида (сульфаниламидные препараты).
256
Препараты первой группы получают при замене атомами
хлора водородов аминогруппы в амидах сульфокислот:
/С1
SO2N4 SO2NC SO2N4
xci \ci \ci
Дихлорамин Б Хлорамин Т Дихлорамин Т
Хлорамины применяются в качестве антисептиков, дезинфи-
дезинфицирующих средств, они способны дезактивировать ряд отрав-
отравляющих веществ (ОВ).
Принцип действия хлораминов основан на том, что в водной
среде они гидролизуются с выделением хлорноватистой кисло-
кислоты, которая является сильным окислителем:
+ 2НОС1
SO2NH2
С12 + Н2О2
Н2О + [О]
Хлорамины в этом отношении сходны с гипохлоритами, но
имеют перед ними то преимущество, что при гидролизе хлор-
хлораминов не образуется щелочь, которая разъедает раневую по-
поверхность, в то время как при гидролизе гипохлоритов щелочь
образуется:
NaOCl + Н2О >■ NaOH + HOC1
Огромное значение в медицинской практике имеют сульфа-
сульфаниламидные препараты общей формулы:
О н
H2N—/~\-S—N4
О
Сульфаниламидные препараты были открыты в 30-х годах
XX века и быстро завоевали популярность. Работы по созда-
созданию новых сульфаниламидных препаратов продолжаются и в
наши дни, что привело к созданию целого ряда препаратов с
высокой эффективностью, малой токсичностью и большой дли-
длительностью действия. Специфичность действия сульфаниламид-
сульфаниламидных препаратов обеспечивается наличием в амидной группе то-
того или иного заместителя R. Ниже приводится перечень наибо-
17-926 257
лее распространенных сульфаниламидных препаратов с указа-
указанием строения заместителя R.
Препарат Заместитель R в сульф-
Стрептоцид белый
Сульфацил
Сульгин
Норсульфазол
амидной группе
Н
СО-СНз
N—II
II
/СН3
Сульфадимезин —<? \
Этазол
Сульфадиметоксин
хсн3
N—N
-1 J-C.H,
S
ОСН3
Д%. J-ОСНз
N
В настоящее время считают, что в основе действия сульфа-
сульфаниламидных препаратов лежит конкурентная борьба между п-
аминобензойной кислотой, необходимой для жизнедеятельности
микроорганизмов, и очень похожей на нее по структуре и раз-
размерам сульфаниловой кислотой, которая, вступая в реакции
вместо п-аминобензойной кислоты, задерживает рост бактерий.
H2N—^\- СООН H2N-/j>-SO2OH
л-Аминобензойная Сульфаниловая
кислота кислота
Таким образом, для того чтобы соединение обладало актив-
активностью, необходимо наличие сульфанильного радикала в мо-
молекуле сульфаниламида:
Это условие является обязательным, но не единственным. Эф-
Эффективность препарата и направленность его действия зависят
от строения молекулы в целом. Достаточно сказать, что из
25000 синтезированных и исследованных сульфаниламидов в
медицинскую практику вошло лишь около 40 препаратов.
258
Все сульфаниламидные препараты являются антимикробны-
антимикробными средствами, поскольку они нарушают процесс получения
микробами необходимых для их развития «ростовых факто-
факторов»— фолиевой кислоты и других веществ. Для получения те-
терапевтического эффекта необходимо принимать сульфанилами-
сульфаниламиды в дозах, достаточных для предотвращения возможности ис-
использования микробами п-аминобензойной кислоты, содержа-
содержащейся в тканях. С другой стороны, прием излишне больших
количеств сульфаниламидных препаратов может вызвать ряд.
осложнений.
В связи с широтой терапевтического действия и относитель-
относительно большими лечебными дозами производство сульфаниламид-
сульфаниламидных препаратов является одним из наиболее крупнотоннажных
производств синтетических лекарственных веществ.
Сульфаниламидные препараты представляют собой кристал-
кристаллические порошки белого или слегка желтоватого цвета, без
запаха. Сульфаниламиды плохо растворимы в воде, но хорошо
растворимы в ряде органических растворителей (спирт, ацетон)
и имеют характерные температуры плавления (в пределах
160—260 °С).
Основным сырьем для синтеза сульфаниламидных препара-
препаратов является хлорангидрид карбометоксисульфаниловой кисло-
кислоты, который чаще называют п-фенилуретилансульфохлоридом:
Н3С-О—С—NH—<( \—SO2C1
О
Основные закономерности синтеза п-фенилуретилансульфо-
хлорида, а также других ацилирующих реагентов, применяемых
для синтеза сульфаниламидных препаратов, и принципиальная:
схема получения последних были разобраны в главах 3 и 7. Ни-
Ниже приводится конкретная технология этих процессов.
Простейшим сульфаниламидным препаратом является п~
аминобензолсульфамид (сульфаниламид, стрептоцид белый).
Он представляет собой белый кристаллический порошок, без
запаха, практически не растворим в эфире и хлороформе, плохо
растворим в воде A:250), хорошо — в кипящей воде A:85),
разбавленной соляной кислоте, растворах едких щелочей и аце-
ацетона. Температура плавления 164—167 °С. Содержание стрепто-
стрептоцида белого в препарате должно быть не менее 99%.
Белый стрептоцид образует соли с кислотами (за счет ами-
аминогруппы в 4 положении) и со щелочами (за счет кислого про-
протона сульфамидной группы). Аминогруппа легко диазотирует-
ся, чем пользуются для аналитического определения препарата.
17* 269
Белый стрептоцид применяется при различных кокковых инфек-
инфекциях, для лечения инфицированных ран и профилактики ране-
раневой инфекции.
Применение фенилуретана в качестве исходного сырья эко-
экономически наиболее целесообразно, так как фенилуретан явля-
является отходом ряда химических производств и более устойчив к
гидролизу, чем ацетанилид. Технический фенилуретан представ-
представляет собой серые с желтоватым оттенком пластинчатые кусоч-
кусочки, легко растворимые в спирте, эфире и ацетоне, трудно рас-
растворимые в воде. Содержание фенилуретана в техническом про-
продукте не менее 95%. Температура застывания—не ниже 45,5°С.
Производство белого стрептоцида из фенилуретана осуще-
осуществляется в четыре стадии по схеме:
=\ i /=\ п
\_
О Н
( }SO2N2
\// очистка
-SO2NH2
\, // \ //
н
I. Процесс получения п-фенилуретилансульфохлорида состо-
состоит из сульфохлорирования фенилуретана, разложения избыточ-
избыточной хлорсульфоновой кислоты водой и выделения п-фенилуре-
п-фенилуретилансульфохлорида:
NH-cf NH-c/
Ч)—СН3 I NOCH4
+ 2HOSO2Ci v (fj + H2SO3 + HC1
SO2C1
В аппарат для сульфохлорирования через мерник заливают
хлорсульфоновую кислоту из расчета 5,37 кг/моль хлорсульфо-
хлорсульфоновой кислоты на 1 кг/моль фенилуретана и при работающей
мешалке нагревают кислоту до температуры 30 °С. Затем при
охлаждении, в аппарат подают измельченный фенилуретан.
Скорость загрузки фенилуретана регулируется автоматически в
зависимости от температуры реакционной массы отключением
и включением загрузочного шнека. Температура реакционной
массы при этом должна быть 32—35 °С.
После загрузки фенилуретана реакционную массу нагревают
паром через рубашку до 42—44 °С и при этой тем-
температуре выдерживают в течение 2 ч. Через 2 ч охлаждают ре-
реакционную массу до 30 °С. Охлажденную сульфомассу после
анализа в цеховой лаборатории передавливают сжатым возду-
воздухом в сборник. Хлористый водород, выделяющийся в процессе
реакции, направляется по стеклянному трубопроводу (в хюгло-
260
тительную систему. Избыток хлорсульфоновой кислоты разла-
разлагают водой:
SO/ + Н2О >• H2SO4 + HC1
В аппарат для разложения одновременно загружают воду и
сульфомассу. Реакцию разложения избытка хлорсульфоновой
кислоты водой проводят при 25—30 °С.
Суспензия выпадающего в осадок п-фенилуретилансульфо-
хлорида из аппарата поступает на нутч-фильтр. Пасту п-фенил-
уретилансульфохлорида отжимают на нутч-фильтре и промыва-
промывают водой до нейтральной реакции. Хлористый водород, образу-
образующийся в процессе разложения, направляется в поглотитель-
поглотительную колонну, где поглощается водой. Выход продукта состав-
составляет 83% от теоретического.
II. Следующей стадией является аминирование:
NH—с/ NH-c/
Х)СН3 | ХОСН3
+ 2NH3 > f| | + NH4C1
2Ci SO2NH2
Из мерника в аппарат для аминирования сливают 25% амми-
аммиачную воду и при включенной мешалке загружают 50% пасту
п-фенклуретилансульфохлорида. Значение рН среды при этом
должно быть 8—8,5. Затем пуском пара в рубашку аппарата
нагревают массу до 60 °С и при этой температуре выдерживают
3 ч. По окончании аминирования реакционную массу охлажда-
охлаждают водой до 35—40 °С и сжатым воздухом передавливают на
нутч-фильтр, где промывают водой от хлорида аммония. Вы-
Выход п-фенилуретилансульфамида составляет 95%, считая на п-
фенилуретилансульфохлорид.
III. Получение технического белого стрептоцида проводят
следующим образом. В реактор из мерника загружают раствор
едкого натра и при включенной мешалке загружают получен-
полученную пасту п-фенилуретилансульфамида. По окончании загруз-
загрузки массу размешивают в течение 30—40 мин, после чего нагре-
нагревают до 120°С и при этой температуре и давлении 1,8—2 ати
выдерживают 5 ч.
NH—Сч7 NH2
I ХОСН3 I
+ NaOH + Н2О > И ] + NaHCO3 + СН3ОН
SO2NH2 SO2NH2
261
По окончании выдержки охлаждают реакционную массу до
температуры 80 °С. После положительного анализа в цеховой
лаборатории на конец омыления, реакционную массу нагрева-
нагревают до 100 °С, передавливают в кристаллизатор и охлаждают до
20—22°С. По окончании кристаллизации суспензию техниче-
технического стрептоцида фильтруют на нутч-фильтре с поднимающей-
поднимающейся мешалкой. Выход технического стрептоцида составляет 88%,.
считая на п-фенилуретилансульфамид.
IV. Для очистки технического стрептоцида в растворитель
заливают воду, включают мешалку и засасывают суспензию
технического белого стрептоцида из нутч-фильтра. По оконча-
окончании загрузки нагревают суспензию до 80 °С, загружают активи-
активированный уголь и доводят рН среды до 6,5—6,7 соляной кисло-
кислотой. Для осаждения солей железа, находящихся в воде, зали-
заливают 98,5% уксусную кислоту. Затем нагревают реакцион-
реакционную массу до 100 °С и выдерживают в течение 2 ч. Передавли-
Передавливают через друкфильтр в -кристаллизатор, куда загружают так-
также гидросульфит натрия для обесцвечивания раствора. После ох-
охлаждения раствора до 25—28 °С массу фильтруют на нутч-
фильтре, центрифугируют и промывают дистиллированной во-
водой от хлоридов. Промытая и отжатая паста белого стрептоци-
стрептоцида подается в пневматическую сушилку. Кристаллы стрептоци-
стрептоцида отделяются от горячего воздуха в циклоне, откуда поступа-
поступают на фасовку. Выход белого стрептоцида составляет ~94%^
считая на технический белый стрептоцид.
Небольшая растворимость белого стрептоцида в воде пре-
препятствует его применению для инъекций. В связи с этим был
синтезирован растворимый стрептоцид, имеющий формулу
H2NO2S—{ \—NH—CH2—SO3Na
Препарат представляет собой белый кристаллический поро-
порошок хорошо растворимый в воде и практически нерастворимый
в эфире и хлороформе. Содержание основного вещества в пре-
препарате не менее 99%.
Получают растворимый белый стрептоцид конденсацией бе-
белого стрептоцида с бисульфитным производным формальдеги-
формальдегида:
н
Н-С=О + HSO2ONa
В процессе получения формальдегидбисульфита натрия значе-
значение рН среды должно быть 7,5—10. В реакционной массе не
допускается избытка формалина и бисульфита натрия, так как
избыток бисульфита натрия приводит к повышенному содержа-
262
нию золы в конечном продукте, а избыток формалина — к пло-
плохой растворимости стрептоцида в воде.
. NH2 HN—СН2—SO2ONa
но-сн2 ^
+ I —И | +н2о
S020Na \^
SO2NH2 SO2NH2
Реакция конденсации протекает при 70—75 °С и рН 4,2—4,4.
Выход растворимого белого стрептоцида, считая на белый
стрептоцид, составляет 77—78%.
К числу гетероциклических производных сульфаниламида
относится норсульфазол B-сульфаниламидотиазол).
H2N—\_J>—SO2NH-|1
S
Норсульфазол — белый или белый со слегка желтоватым от-
оттенком кристаллический порошок, без запаха, плохо раствори-
растворимый в воде, ацетоне и спирте. Практически не растворим ,в эфи-
эфире, растворим в минеральных кислотах и растворах едких и уг-
углекислых щелочей. Температура плавления 198—203 °С (с раз-
разложением).
Норсульфазол эффективен при инфекциях, вызванных гемо-
метическим стрептококком, пневмококком, гонококком, стафи-
стафилококком, а также кишечной палочкой.
В настоящее время известно несколько методов получения
норсульфазола. Эффективный метод получения норсульфазола
был разработан сотрудниками ВНИХФИ.
Исходным веществом для синтеза норсульфазола по этому
методу является цианамид кальция, который гидролизуется до
кислой кальциевой соли цианамида:
2Ca(N-C=N) + 2Н2О > Ca(HNCNJ + Ca(OHJ
К раствору кислой кальциевой соли при перемешивании и
охлаждении прибавляют п-фенилуретилансульфохлорид. Тем-
Температура смеси при этом должна быть не выше 25—30 °С. При
этой температуре и рН 9,5—9,8 при перемешивании реакцион-
реакционную массу выдерживают в течение 6 ч. Затем смесь нагревают
до 60 °С и перемешивают еще 2 ч. Смесь охлаждают до 20 °С,
отфильтровывают от осадка:
2CH3OOCHN-/~\-SO2Cl + Ca(HNCNJ
) Ca + СаС12
/2
Полученную кальциевую соль карбометоксисульфанилцианами-
да переводят в натриевую соль действием 10% раствора угле-
263
кислого натрия. При этом значение рН реакционной среды дол-
должно быть 8,2—8,5.
( CHgOOCHN—<T~Y-SO2NCN \ Са + Na2CO3 >•
> 2CH3OOCHN—<Г^—SO2N(Na)CN + CaCO3j
Натриевая соль п-карбометоксисульфанилцианамида при на-
нагревании до 95—98 °С с тиосульфатом натрия 'и 25% сер-
серной кислотой превращается в карбометоксисульфанилтиомоче-
вину рН среды при этом процессе должен быть не выше 2,0.
CH3OOCHN-<f~^--SO2N-CN + Na2S2O3 + H2SO4 "^—7^
Na
> CHgOOCHN—/ V-SO2-NH—C~NH2 + NaHSO3 + Na2SO4
\-J II
S
Замыкание тиазольного цикла осуществляется с использовани-
использованием монохлорацетальдегида. Реакция, по-видимому, проходит
две фазы. Сначала при низкой температуре, хлорацетальдегид
присоединяется к сульфгидрильной группе карбометоксисуль-
фанилтиомочевины, а затем при повышенной температуре про-
промежуточный продукт циклизуется в карбометоксисульфанил-
амидотиазол:
10% NaOH
CH3OOCHN-<4 /)—SO2NH—С—NH2 + С1СН2СНО
^/у j| —NaCl, 70 °С
CH3OOCHN-(v /)—SOaNH—С—S—CH2—CHO
/
NH
103-105 °С
Карбометоксисульфаниламидотиазол переводят в натриевое
производное действием 42% раствора едкого натра и гидролизу-
ют при температуре 95—98 °С.
N-
CH3OOCHN-
|
Na
,-N-U
Выпавшие после охлаждения кристаллы натриевой соли
норсульфазола отфильтровывают, растворяют в воде при 70 °С
и при перемешивании добавляют уголь. Отфильтровывают
уголь на друкфильтре, к раствору натриевой соли прибавля-
прибавляют гидросульфит натрия, а затем 25% раствор серной кислоты,
доводя рН до 4,7—5,0.
N 1| 25% H2SO4
S
Na
2H2N-
Реакционную массу охлаждают до 20 °С, осадок отжимают
на центрифуге, промывают водой и сушат в пневмосушилке.
Выход норсульфазола составляет 58%, считая на п-карбомето-
ксиаминобензолсульфохлорид (п-фенилуретилансульфохлорид).
В последние годы получили распространение сульфанил-
сульфаниламидные препараты, длительно задерживающиеся в организ-
организме. Применение сульфамидных препаратов пролонгированного
действия позволяет отказаться от частых повторных приемов,
что ведет к уменьшению возможных побочных эффектов. К та-
таким препаратам длительного действия относятся сульфапирида-
сульфапиридазин, сульфален (сульфаметопиразин) и сульфадиметоксин
(мадрибон).
N-N
—NH-/ \-ОСН3
Сульфапиридазин
ОСН3
М ""ОСНз
Сульфадиметоксин
N=\
HSN
Сульфален
Сульфапиридазин, 6-сульфаниламидо-З-метоксипиридазин,
представляет собой светло-желтые кристаллы или кристалличе-
кристаллический порошок от белого до желтоватого цвета, почти без запа-
запаха, слегка горьковатого вкуса. Постепенно желтеет на солнеч-
солнечном свету. Температура плавления 180—183 °С. Плохо раство-
265
рим в ацетоне, легко растворим в разбавленных кислотах и ще-
щелочах. Из щелочных растворов может быть выделен подкисле-
нием соляной кислотой до значения рН 6,0—5,5.
Сульфапиридазин применяется для лечения пневмонии,
бронхитов, тонзиллитов, фарингитов, гнойных отитов, дизенте-
дизентерии, энтероколитов, гнойных инфекций мочеполового тракта и
желчных путей, для профилактики послеоперационных инфек-
инфекций.
В литературе описано несколько способов получения суль-
фапиридазина. Наиболее простым и доступным является син-
синтез из 3,6-дихлорпиридазина. 3,6-Диоксипиридазин получают
кипячением водного раствора гидразин сульфата и малеиново-
го ангидрида в течение 3—4 ч.
НС—C<f „ кт N-N
II \ H2N юо *с // л
II О + | -H2SO4 >- HO-f Y-OH + H2SO4 + Н2О
H2N \—/
При этом малеиновый ангидрид берут с 5% избытком. Реак-
Реакционную массу охлаждают до 8—10 °С и выделившийся про-
продукт отфильтровывают на нутч-фильтре, промывают водой до
отсутствия гидразин сульфата в промывной воде и сушат при
100—120°С. Выход 3,6-диоксипиридазина составляет 87% от
теоретического, считая на гидразин сульфат.
Далее заменой оксигруппы в 3,6-диоксипиридазине на хлор
получают 3,6-дихлорпиридазин
N-N N-N
ЗНО— ( V-OH + 2РОС13 >■ ЗС1—f Y-C1 + 2Н3РО4
В сухой и чистый аппарат из мерника самотеком загружают
рассчитанное количество хлорокиси фосфора (на 1 моль 3,6-
диоксипиридазина 3,75 моля хлорокиси фосфора), включают
мешалку и через люк осторожно загружают 3,6-диоксипирида-
зин. Аппарат герметизируют, включают обратный холодильник
и осторожно из мерника приливают серную кислоту. Реакцион-
Реакционную массу в аппарате в течение 1 ч нагревают горячей водой
до температуры 62—68 °С и выдерживают при этой температу-
температуре в течение 40 мин. Эта реакция экзотермична и проходит с
выделением хлористого водорода, который через ловушку по-
поступает в колонну, орошаемую водой для поглощения хлори-
хлористого водорода. По окончании 40 минутной выдержки при 68 °С
реакционную массу в течение 2 ч нагревают до температуры
86 °С и выдерживают в течение 2 ч при температуре 86—
90 СС, затем реакционную массу охлаждают водой через ру-
рубашку до 20—25 °С и передавливают по частям в мерник реак-
реакционной массы. Разложение избытка РОС13 в массе проводят
при одновременном сливе водного раствора аммиака и реакци-
266
онной массы на смесь водного раствора аммиака и толуола,
поддерживая рН среды 7—8 и температуру 20—25 °С. Толу-
ольный и .водный слой разделяют и из последнего остатки 3,6-
дихлорпиридазина дополнительно экстрагируют толуолом. То-
Толуол отгоняют, а 3,6-дихлорпиридазин перегоняют в вакууме,
предварительно добавив триэтаноламин. Добавка триэтанол-
амина исключает выделение хлористого водорода и придает
продукту стабильность при хранении.
Разложение избытка хлорокиси фосфора происходит по ре-
реакции:
РОС13 + 6NH3 + ЗН2О > (NH4KPO4 + 3NH4C1
Одновременно происходит нейтрализация кислот аммиаком:
H2SO4 + 2NH3 > (NH4JSO4
Выход составляет 73%, считая на 3,6-диоксипиридазин, или
— 60%, считая на малеиновый ангидрид.
З-Хлор-6-сульфаниламидопиридазин получают конденсацией
3,6-дихлорпиридазина со стрептоцидом в циклогексаноле в при-
присутствии поташа. Реакция идет с выделением углекислого газа.
N-N ^=у^
С1-/ V-C1 + H2N-SO2-/^}--NH2 + К2СО3 >
N-N
> CI—/ \-NK-SO2-^~\-NH2 + Н2О + C02f + KC1
При проведении этой реакции необходимо эффективное пере-
перемешивание, так как реакционная масса к концу операции ста-
становится густой. После растворения реакционной массы в воде
и охлаждения водный слой, содержащий калиевую соль 3-хлор-
6-сульфаниламидопиридазина, отделяют от циклогексанола.
Последний дважды дополнительно экстрагируют водой.
Водный раствор калиевой соли очищают углем, окисью алю-
алюминия и выделяют из него З-хлор-6-сульфаниламидопиридазин
10% раствором соляной кислоты при 68—70 °С и рН 4,5.
N-N N-N
Выделившийся желто-коричневый продукт после охлажде-
охлаждения фильтруют, промывают водой до отсутствия хлор-иона и
сушат. Выход З-хлор-6-сульфаниламидопиридазина составляет
около 64%, считая на 3,6-дихлорпиридазин, и около 38%, счи-
считая на малеиновый ангидрид.
267
Калиевую соль З-метокси-6-сульфаниламидопиридазина по-
лучают нагреванием в автоклаве при температуре 125—130 °С
и давлении 6—8 ати З-хлор-6-сульфаниламидопиридазина с ед-
едким кали в метаноле в течение 9 ч.
N-N _
/ \/~j2КОН + СН3ОН
NH2 + KC1 + H2O
Ha 1 моль З-хлор-6-сульфаниламидопиридазина берут 2,7 мо-
моля едкого кали. Метанол берут с большим избытком, так как
он же является и растворителем.
После отгонки метанола калиевую соль перекристаллизовы-
вают из насыщенного раствора поваренной соли, отфильтровы-
отфильтровывают осадок и промывают его насыщенным раствором хлорида
натрия, растворяют в воде при нагревании до 60 °С, добавляют
активированный уголь, окись алюминия, гидросульфит натрия,
фильтруют и из фильтрата при температуре 50—60 °С выделя-
выделяют сульфапиридазин 36% раствором соляной кислоты при зна-
значении рН 5,5.
N-N
СН3О-/ V-N-SO2-/~~Y-NH2 + HC1 *
\=/ | \=/
К
--NH2 + КС1
После фильтрации технический сульфапиридазин сушат при
температуре 100—120 °С. Выход технического сульфапиридази-
сульфапиридазина составляет 60%, считая на З-хлор-6-сульфаниламидопирида-
зин, и 23%, считая на малеиновый ангидрид.
Технический сульфапиридазин перекристаллизовывают из
метанола, очищая метанольный раствор углем и окисью алю-
алюминия. Выход сульфапиридазина на стадии перекристаллизации
при использовании метанольных маточников составляет 81 —
82% от загруженного сульфапиридазина технического.
Общий выход сульфапиридазина по процессу в целом в рас-
расчете на малеиновый ангидрид 18—19%.
Работы последних лет привели к созданию ряда сульфамид-
сульфамидных препаратов, обладающих тормозящим действием на актив-
активность некоторых ферментов. Эти препараты, как правило, не
являются производными сульфаниловой кислоты. Среди новых
268
препаратов имеются мочегонные средства (гигротон, диакарб,
дихлортиазид и др.),
NH ni
N N
Н3С—CO-NH—(^ J—SO2NH2
S
Диакарб Дихло; тиазид
препараты для борьбы с глаукомой (диакарб, метазоламид),
антидиабетические средства (бутамид).
HN N О
Н3С—СО—NH—N ILso2NH2 СНз"" Ч~^—SOa—NH—С—NH—С4Н9
S
Метазоламид Бутамид
Таким образом, новые исследования открывают еще более
широкие перспективы использования сульфамидных препара-
препаратов для лечения различных заболеваний.
Глава 16
ЭЛЕМЕНТОРГАНИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ
ВЕЩЕСТВА
К элементорганическим соединениям относят вещества, со-
содержащие в молекуле, кроме атомов углерода, водорода, кис-
кислорода, азота и серы, атомы других элементов, связанные с уг-
углеродом. Известны лекарственные соединения, относимые к
этой группе, содержащие в молекуле атомы мышьяка, висмута,
ртути, фосфора и некоторых других элементов. Элементоргани-
ческие лекарственные соединения являются химиотерапевтиче-
скими средствами, обладающими, как правило, высокой токсич-
токсичностью, что позволяет применять их только под строгим вра-
врачебным контролем. Токсичность этих соединений накладывает
целый ряд особенностей на технологию их производства и спо-
способ хранения. Вместе с тем соединения этого ряда позволяют
вести борьбу с такими опасными и трудноизлечимыми заболе-
заболеваниями, как сифилис, возвратный тиф, дизентерия, тропиче-
тропическая малярия, злокачественные новообразования и др.
§ 1. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ МЫШЬЯКА
Неорганические соединения мышьяка и мышьяковые произ-
производные алифатического ряда не получили широкого примене-
применения в медицине вследствие их очень высокой токсичности. Ра-
Работы немецкого химика и биолога П. Эрлиха A854—1915)
привели к созданию целого ряда эффективных лекарственных
препаратов на основе ароматических соединений, содержащих
мышьяк. В СССР большие работы в этом направлении были
проделаны под руководством М. Я. Крафта во ВНИХФИ, где
был осуществлен синтез таких важных препаратов, как новар-
сенол, миарсенол, осарсол, аминарсон и других.
Среди ароматических соединений пятивалентного мышьяка
применение находят осарсол и аминарсон:
О О
НО—As— ОН НО—As—ОН
•NHCOCHg
NHCONH2
Лминарсвн
270
Арсаниловую кислоту, необходимую для синтеза аминарсо-
на, получают из анилина при нагревании его с мышьяковой
кислотой. Механизм реакции аналогичен механизму образова-
образования сульфаниловой кислоты (см. главу 3):
/он
NH3'H2As0r HN— As=O
HO-As-OH
Аналогично получается и оксифениларсоновая кислота, но в ка-
качестве исходного сырья берут фенол.
Для получения осарсола п-оксифениларсоновую кислоту
нитруют смесью серной и азотной кислот (см. главу 4):
О О
Полученную З-нитро-4-оксифениларсоновую кислоту восстанав-
восстанавливают, а полученное аминопроизводное ацетилируют:
ОН ОН ОН
1 „NO2
НО—As— ОН НО—As—ОН НО—As—ОН
II II II
0 0 0
Осарсол и аминарсон представляют собой плохо раствори-
растворимые в воде и спирте белые кристаллические порошки. Выпуска-
Выпускаются препараты в таблетках. Наибольший однократный прием
0,25 г.
Осарсол применяется при лечении сифилиса, возвратного ти-
тифа, дизентерии. Аминарсон — для лечения амебной дизентерии.
Препараты следует хранить под замком (список А) в хорошо
закупоренных банках из темного стекла.
К ароматическим соединениям трехвалентного мышьяка от-
относятся сальварсан, новарсенол, миарсенол. Установлено, что
сальварсан является полимером, причем степень полимериза-
полимеризации зависит от условий восстановления З-нитро-4-оксифенилар-
271
соновой кислоты. Иногда полимерную структуру сальварсана
изображают следующим образом:
он
МО
Nib
ОН
Сальварсан как лечебный препарат имеет много недостатков.
Он слишком токсичен, а также легко окисляется при хранении.
В нашей стране были синтезированы менее токсичные и более
стойкие к хранению препараты новарсенол и миарсенол
As As
NaOoS-CH2—NH
I
NH-CH2-SO3Na
OH OH
Миарсенол
Новарсенол и миарсенол применяются для лечения сифили-
сифилиса, возвратного тифа, тропической малярии. Новарсенол вводят
только внутривенно, а миарсенол — внутримышечно. Оба пре-
препарата хранятся под замком (список А).
§ 2. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ СУРЬМЫ И ВИСМУТА
Успех применения органических соединений мышьяка под-
подсказал химикам идею синтеза аналогичных препаратов других
элементов пятой группы периодической системы, в частности,
сурьмы и висмута. Аналогичные соединения были синтезирова-
синтезированы. Однако они оказались менее эффективными, чем соедине-
соединения мышьяка. Применение в медицине нашли препараты стиб-
амин, стибацетин, стибозан:
NH2 NHCOCHg NHCOCH3
I J XI
НО—Sb-ONa
I!
О
Стибамин
НО—Sb—ONa
II
О
Стибацетин
Из лекарственных соединений висмута следует отметить бисмо-
верол. Препарат представляет собой взвесь основной висмуто-
висмутовой соли моновисмутвинной кислоты в нейтрализованном пер-
персиковом масле:
хт
COO—Bi4
н-с—он
н—с-оч
I ol"—"Ог~1
coo/
Бисмоверол применяется в виде внутримышечных инъекций
при лечении сифилиса.
§ 3. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ РТУТИ
Ртутные соли органических кислот могут диссоциировать,
они токсичны и не нашли применения в медицине. Водораствори-
Водорастворимые органические соединения ртути являются диуретиками.
Они оказывают влияние на водно-солевой баланс между тка-
тканями и кровью. Примером таких соединений может служить
препарат меркузал.
ОСН3 /р
■CH,-Hg-O-cf
MvJH-G
-OCH2-COONa
Меркузал
Меркузал получают многостадийным синтезом из салицило-
салициловой кислоты, через стадии образования хлорангидрида ацето-
ксисалициловой кислоты (I), ее аллиламида, и 2-метокси-З-
ртутьоксипропиламидосалицилуксусной кислоты; последнюю
конденсируют с 5,5-диэтилбарбитуровой кислотой (барбиталом):
СО—NH-CH2—СН—СН2—Hg— О—Сч /Сч
-ОН
.^.соон ^ ifY^ J^
\*^och2cooh \^-осн2соон
XONH2 вг-сн2-сн=сн2
►
ОСН2СООН
.сош_сн2-сн=сн2 Hg@C0CH3)?
ОСН2СООН
18-926 273
HgCOCO
CO—NH—CH2—CH—CH2—Hg—О
2 Ь | CH3ONa, H2O
COCHo *"
ОСН2-СООН 3
ОСН
з
I 0=c\ /c
CONH—CH2-CH—CH2-HgOH xnh-c(on
OCH2—COONa
OCH3 /P
CO—NH—CH2-CH-CH2—Hg—O-c/ /C\
\MH_r/ \
4 C2H5
^0
-ОСН2—COONa
Меркузал вводят внутримышечно при отеках. Он является
очень сильным мочегонным средством. Уже при однократном
приеме меркузал а мочеотделение может доходить до 10 л в сут-
сутки.
Соединения ртути, нерастворимые в воде, но жирораствори-
жирорастворимые, лишены диуретических (мочегонных) свойств, но облада-
обладают противосифилитическим действием.
§ 4. ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Фосфорорганические соединения, как правило, являются
токсичными веществами. Многие из них применяются в качест-
качестве инсектицидов. Среди них имеются боевые отравляющие ве-
вещества (зарин), сильные комплексообразователи (амино- и по-
лиаминополифосфоновые кислоты).
Фосфорорганические соединения находят применение и в
качестве лекарственных средств. Например, препараты армии
и фосфакол эффективны при лечении глаукомы:
C2H5O/ 4)-<v />-NO2 C2H5/ XO
Фосфакол Армии
Для синтеза фосфакола взаимодействием треххлористого
фосфора с этиловым спиртом получают диэтилфосфористую
кислоту, при хлорировании которой образуется диэтилхлорфос-
фат. Последний с п-нитрофенолом образует фосфакол:
С2Н5ОН -^ (С2Н5ОJРОН (,5сС)С!!(+5^ (С2НЬОJРОС1
n-HOC6H4NO2 С2
274
Фосфакол представляет собой прозрачную, маслянистую
легко растворимую в спирте и эфире жидкость. Фосфакол вы-
выпускается в-склянках по 10 мл водного раствора с концентра-
концентрацией 0,02; 0,013 и 0,01% (список А). В неразведенном виде
фосфакол, так же как и большинство других фосфорорганиче-
фосфорорганических препаратов, сильно ядовит. Следует избегать попадания
этого и подобных ему препаратов на кожу и слизистые оболоч-
оболочки. При отравлениях этими веществами применяют специфиче-
специфические противоядия (атропин, апрофен, изонитрозин, дипиро-
ксим).
Особенно важно то, что среди фосфорорганических веществ
имеется большая группа соединений, обладающих противоопу-
противоопухолевой активностью. К этой группе лекарственных препаратов
относятся тиофосфамид (тиоТЭФ), дипин, тиодипин, бензотеф,
фторбензотеф и др.
Н2С СН2
H2f
л
/
н2
1 чсн2
N
/\
с—сн2
Тиофосфамид
N
F—<f\-C~NH—P—N
\=/ II II
0 0
Фторбензотэф
Перечисленные фосфорпроизводные этиленимина нарушают об-
обмен нуклеиновых кислот и блокируют митотическое деление
клеток, что приводит к торможению развития злокачественных
опухолей.
Получают соединения этой группы взаимодействием фосфор-
галоидных производных с этиленимином. Так, например, бен-
зотэф получают из амида бензойной кислоты, который последо-
последовательными обработками пятихлористым фосфором и водой
превращают в дихлорангидрид бензоиламидофосфорной кисло-
кислоты. При взаимодействии последнего с этиленимином в среде
бензола образуется бензотэф (диэтиленимид бензоиламидофос-
бензоиламидофосфорной кислоты):
Ci /?*
7 hn4 |
РС15. 2) Н2О | \СН2
CHCONHPC1
1) 5. ) 2 |
C6H5CONH2 >- C6H5CONHP—C1
II
О
оно гн
I II /
r
N
ii2C—■—Со2
Бензотэф
18* 275
Бензотэф — оригинальный отечественный противораковый
препарат алкилирующего действия, применяется внутривенно
при раке яичника, молочной железы, раке легкого с метастаза-
метастазами. Выпускается в ампулах, содержащих по 0,024 г стерильно-
стерильного порошка (список А).
Среди фосфорорганических соединений имеются гормональ-
гормональные препараты, которые также используются в онкологической
практике. Примером может служить фосфэстрол, который при-
применяется для лечения рака предстательной железы:
0 С2Н5 О
I! /г-\ I /г-\ II
NaO-P—О—V у- C=C-f V-0-P-ONa
1 \=/ I \=/ I
ONa C2H5 ONa
Фосфэстрол
Фосфэстрол был синтезирован с таким расчетом, чтобы пре-
препарат был неактивен при циркуляции в крови, но, попадая в
опухолевую ткань предстательной железы, он под влиянием со-
содержащейся в ней активной фосфатазы1 гидролизовался бы до
диэтилстильбэстрола, который блокировал бы разв-итие опухо-
опухолевой ткани. Таким образом, фосфэстрол может рассматривать-
рассматриваться как соединение, доставляющее активное вещество в опухо-
опухолевую ткань. Тот же принцип был положен в основу создания
другого противоопухолевого препарата — циклофосфана:
уСН2—СН2С1
H2C—NH N4
\ / XCH2-CH2C1
p
н2с-о чо
Циклофосфан
1 J24
н2с
Это вещество также выполняет «транспортную» роль, до-
доставляя активный бис-(р-хлорэтил)-амин в раковую ткань.
Циклофосфан является алкилирующим препаратом. Приме-
Применяется при лимфогранулематозе, лимфосаркоме, раке яичников,
раке молочной железы, раке легкого. Препарат относительно
малотоксичен.
Исходным веществом при синтезе циклофосфана служит ди-
этаноламин. При взаимодействии его с тионилхлоридом в сре-
среде хлороформа получают гидрохлорид ди-B-хлорэтил)-амина,
который при реакции с хлорокисью фосфора образует дихлорид
М,1\1-ди-B-хлорэтил)-фосфам1ида. Последний при взаимодействии
с З-аминопропанолом-1 дает циклофосфан:
но—сн2—сн2ч socl2 ci—сн2—сн2ч Р0С1з
;ш >■ ;nh-hci >■
НО—СН2—СН/ С1—СН2-СН/
1 Фосф|атаза — фермент, катализирующий гидролиз фосфорных эфиров.
Активность фосфатазы в раковых клетках значительно повышена.
276
с.-сн2-сн
Ч / HO(CH2)8NH2
хм-Р=О ►-
C1-CH2—CH
C1-CH2—CH,
Cl—CH,—CH
>
/NH-CH.
сн2
§ 5. РЕНТГЕНОКОНТРАСТНЫЕ СРЕДСТВА
Рентгеноконтрастные средства предназначены не для лече-
лечения, а для диагностики заболеваний. Применяемые в практике
рентгеноконтрастные средства часто являются ди- и трийод-
производными ароматического ряда и могут быть отнесены к
элементорганическим веществам лишь формально. Основными
представителями этой группы препаратов являются кардио-
траст, трийотраст, билитраст, билигност:
О
N О
/СН2-СН2ОН
СН2--С— 0H-NH4
х:н2-сн2он
Кардиотраст
Билигност
Трийотраст (натриевая соль 2,4,6-трийод-З-ацетаминобен-
зойной кислоты) представляет собой порошок белого цвета,
применяется при рентгенологических исследованиях почечных
лоханок, мочеточников, мочевого пузыря, кровеносных сосудов
и сердца. Выпускается в виде 30%, 50% и 70% раствора в ам-
ампулах по 2, 10 и 20 мл.
277"
Трийотраст-кислота образуется при йодировании м-амино-
бензойной кислоты хлористым йодом с последующим ацетили-
рованием продукта уксусным ангидридом:
СООН СООН СООН
I
IC1 *\/Ч^Х (СН3СОJО
NH2
Иодирование ведут в среде соляной кислоты при 60 °С в тече-
течение 3 ч. Осадок трийодаминобензойной кислоты отфильтровы-
отфильтровывают и промывают водой до отсутствия кислой реакции (по
конго). Для очистки полученного промежуточного продукта его
растворяют в разбавленном растворе едкого натра, а затем
осаждают соляной кислотой. Ацетилирование уксусным ангид-
ангидридом проводят в присутствии небольшого количества концент-
концентрированной серной кислоты при температуре 105—110° С и вы-
выдержке 2 ч. После охлаждения реакционной массы отфильтро-
отфильтровывают выпавшую в осадок 2,4,6-трийод-З-ацетиламинобензой-
ную кислоту. Для очистки полученную пасту трийотраст-кисло-
ты промывают водой, затем растворяют в 2,5% водном растворе
аммиака, кипятят с активированным углем, фильтруют в горя-
горячем состоянии и из полученного раствора 18% соляной кисло-
кислотой осаждают очищенный продукт. Осадок отфильтровывают,
промывают водой и сушат. Выход на стадии йодирования со-
составляет 60%, на стадии ацетилирования — 86,8%, температура
плавления трийотраст-кислоты 265—280 °С (разл).
2,4,6-Трийод-З-аминобензойная кислота является промежу-
промежуточным продуктом также при синтезе другого рентгеноконтра-
стного средства — билигноста. Билигност, динатриевая соль
•бис- C-карбокси-2,4,6-трийоданилида) адипиновой кислоты,
£ ^-NH-CO-(CH2L-CO-NH / \
NaOOCy Ч V xCOONa
применяется при рентгенографии желчных путей и желчного
пузыря. Выпускается в ампулах по 2, 10, 20 мл 20% раствора.
Билигност-'кислоту получают из трийодаминобензойной кис-
кислоты и дихлорангидрида адипиновой кислоты:
НООС-/ ^-1 + С1ОС(СН2LСОС1
278
о о
II II
1-4 j>-NH-C-(CH2L-C-NH-d „-
но—с/ ч v \c=o
II !
о но
Реакцию проводят в среде хлорбензола при кипении в тече-
течение 27г ч. Полученную билигност-кислоту переводят раствором
щелочи в динатриевую соль. Раствор динатриевой соли кипя-
кипятят с углем и сульфитом и фильтруют. Из фильтрата соляной
кислотой осаждают очищенную билигност-кислоту. Операцию
очистки повторяют. Дважды переосажденную билигаост-киелоту
промывают на фильтре спиртом и сушат. Температура плавления
полученного продукта 291—293°С (разлагается), выход 80%.
Глава 17
ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ
ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
На долю лекарственных веществ гетероциклического ряда
приходится приблизительно половина всех применяемых в ле-
лечебной практике препаратов. Некоторые из этих соединений
рассматривались в предыдущих главах, так как сложные мо-
молекулы их содержат в своем составе, кроме гетероциклического
ядра, различные алифатические или ароматические заместите-
заместители. Так, например, гетероциклические кольца входят в состав
молекул многих сульфаниламидных препаратов. Однако в тех
случаях, когда метод синтеза соединения определяется приро-
природой гетероцикла, входящего в молекулу, рассматривать техно-
технологию соответствующих лекарственных веществ целесообразнее
в соответствии с химической классификацией гетероцикличе-
гетероциклических соединений.
§ 1. ПРОИЗВОДНЫЕ ПЯТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
Наиболее важными и распространенными лекарственными
соединениями из группы пятичленных гетероциклов являются
производные фурана, пиразола и имидазола:
НС СН НС СН НС NH
II II II II II I
НС СН НС N НС СН
О N
v
Н
Фуран Пиразол Имидазол
Многие нитропроизводные фурана обладают антимикробным
действием. В некоторых случаях нитрофураны задерживают
рост микроорганизмов, устойчивых к сульфаниламидным препа-
препаратам. Применяют препараты нитрофуранового ряда чаще все-
всего в качестве антисептиков наружно, а иногда и для внутрен-
внутреннего приема при лечении ряда инфекционных заболеваний.
В исследованиях нитрофуранов и разработке технологии лекар-
лекарственных соединений этого ряда большую роль сыграли рабо-
работы советских ученых — акад. С. А. Гиллера и проф. Э. Ю. Гуд-
280
риниеце. Одним из важных лекарственных веществ этого ряда/
является фурацилин (семикарбазон 5-нитрофурфурола):
НС СН
II II
O2N-C C-CH=N-NHCONH2
Фурацилин
Фурацилин широко применяется в качестве наружного*
средства при лечении гнойно-воспалительных процессов и в ка-
качестве внутреннего средства для лечения бактериальной дизен-
дизентерии. Особое значение имеет фурацилин при лечении длитель-
длительно незаживающих ран, он применяется также и в ветеринар-
ветеринарной практике.
Фурацилин получают из фурфурола. Нитрование фурфуро-
фурфурола, так же как и нитрование фурана, проводят ацетилнитра-
том в среде уксусного ангидрида и уксусной кислоты. Альде-
Альдегидная группа фурфурола при этом ацилируется. Нитрование
проходит более гладко, если к реакционной массе добавляют
небольшое количество пиридина:
H2SO4
(СН3СОJО + HNO3 * CH3COONO2 + CH3COOH;
НС СН п (сн3соJо
II || /Г + CH3COONO2 + СН3СООН >
о
о
хлп fu II
^ Н II /О-С-СН3
O2N—С С—СНЧ + Н2О
\ / \о-с-сн3
о и
о
Процесс проводят следующим образом. К загруженной а*
нитратор смеси уксусного ангидрида и ледяной уксусной кис-
кислоты C:1) при перемешивании и охлаждении рассолом при-
приливают смесь азотной и серной кислот. Скорость прилива смеси
кислот регулируют таким образом, чтобы температура реакци-
реакционной массы не поднималась выше 7—10 °С. После окончания
загрузки кислот реакционную массу охлаждают до 0°С и по-
постепенно при температуре 0—5°С приливают фурфурол. После
выдержки в течение 40 мин реакционную массу разбавляют во-
Дой и при температуре не выше 35—40 °С медленно загружают
25% раствор едкого натра. По окончании загрузки, массу на-
нагревают до 50 °С и выдерживают при этой температуре 1 ч.
После охлаждения реакционной массы до 20°С выпавший оса-
Док 5-нитрофурфуролдиацетата отфильтровывают и промыва-
промывают водой. Выход на этой стадии 57—58%.
281
Следующей стадией синтеза является гидролиз диацетиль-
ного производного. Гидролиз проводят кипячением в разбав-
разбавленной серной кислоте. Горячий раствор 5-нитрофурфурола об-
обрабатывают углем и фильтруют.
(H2SO4)
+ 2Н2О >
О + 2СН3СООН
К горячему фильтрату при перемешивании прибавляют раствор
солянокислого семикарбазида. Реакционную массу охлаждают
до 18—20 °С и отфильтровывают выпавший в осадок семикар-
базон 5-нитрофурфурола (фурацилин):
—
^
HaNCONHNHg-HCl
О
и
+h2o + hci
=N—NHCONH2
Продукт промывают на фильтре водой и сушат при 60—
70 °С. Выход на последней стадии составляет около 88%. Полу-
Полученный фурацилин — желтый кристаллический порошок без за-
запаха, плохо ра'створимый в воде, спирте и эфире, температура
плавления 230—236°С (разлагается). Фурацилин выпускается
в виде порошка, таблеток по ОД г и 0,2% мази.
Из фурфурола получают также другой антимикробный пре-
препарат — фуразолидон:
О
СН2
I I
CH=N—N СН2
Фуразолидон применяется при лечении кишечных инфекций,
а также при лямблиозе и трихомонадных кольпитах.
Весьма распространенным препаратом из группы фурана яв-
является бензамон (фурфурилтриметиламмоний-бензолсульфо-
нат):
/снз
-СП — N-CH3
Первая стадия синтеза бензамона основана на реакции вос-
восстановительного аминирования фурфурола:
282
/0 *" " ||-CH2NH2
о \н о
Для получения фурфуриламина из фурфурола в смесь фурфу-
фурфурола, спиртового раствора аммиака и никеля Ренея пропускают
водород. Гидрирование длится 16—18 ч. Для реакции может
быть использован также жидкий аммиак. В этом случае про-
процесс проводят в автоклаве под давлением 50—90 ат и темпера-
температуре до 70 °С. Время процесса при этом резко уменьшается. По
окончании восстановительного аминирования катализатор с со-
соблюдением необходимых предосторожностей (см. главу 10) от-
отфильтровывают и промывают спиртом. Спирт отгоняют, а ос-
оставшийся фурфуриламин перегоняют в вакууме. Выход про-
продукта составляет около 55%. Для получения бензамона к экви-
эквимолекулярной смеси фурфуриламина и 10% водного раствора
поташа при перемешивании и температуре 25—26 °С медленно
добавляют метиловый эфир бензолсульфокислоты. Реакцион-
Реакционная масса должна быть все время щелочной, поэтому после за-
загрузки половины метилбензолсульфоната добавляют вторую
порцию раствора поташа:
_^ ^ IF"[I
V-CH2-N(CH^
По окончании загрузки реагентов реакционную массу раз-
размешивают 2 ч, а затем выдерживают еще 2 ч 35—40 °С и 1,5 ч
при 70 °С, добавляют активированный уголь, охлаждают и
фильтруют. Из упаренного фильтрата выделяют бензамон, ко-
который очищают перекристаллизацией из бутилового спирта.
Выход продукта с температурой плавления 132—134 °С состав-
составляет около 60% в расчете на фурфуриламин.
Бензамон представляет собой белый или желтоватый кри-
кристаллический порошок с характерным запахом амина, легко
растворяется в воде и спирте, плохо — в хлороформе, нераство-
нерастворим в эфире. Бензамон является оригинальным отечественным
препаратом, применяемым при лечении глаукомы. Выпускается
в виде 3 и 10% растворов и мазей.
Большое значение для медицинской практики имеют произ-
производные ряда пиразола. Пиразол имеет ароматический харак-
характер, а потому способен вступать в реакции замещения. Водород
у азота имеет слабый «кислый» характер, двойные связи в коль-
кольце могут полностью или частично гидрироваться. Лекарствен-
Лекарственные препараты, являющиеся производными пиразола, относят-
283
ся к двум основным группам: производным пиразолина, точ-
точнее— пиразолона-5, и производным пиразолидина.
/О
-" п г,
It i
1 NH || N || NH NH
N N N N
I к 1, Ъ
Пиразолон-5 Пиразол Пиразолин Пиразолидин
Для многих лекарственных веществ этого ряда характерно
наличие фенильного заместителя у азота в положении 1. Ха-
Характерным свойством производных пиразола является их спо-
способность к окислению, легкость которого зависит от характера
заместителей. Лекарственные вещества группы пиразола ока-
оказывают на организм жаропонижающее и болеутоляющее дей-
действие.
В строении, свойствах, биологической активности и способах
получения антипирина, амидопирина и анальгина много обще-
общего. Амидопирин (пирамидон) получают из антипирина, а аналь-
анальгин — из промежуточного продукта в синтезе амидопирина —
аминоантипирина:
CH3N^
НС=С-СН3 N—С=С—СН8
I I Сн/ | |
О=С N—СН3 О=С N-CH3
v v
Антипирин Амидопирин
Анальгин
При синтезе можно исходить из фенилгидразина и ацетоук-
сусного эфира (см. главу II), но поскольку 1-фенил-З-метилпи-
разолон-5 является крупнотоннажным продуктом, в качестве
исходного сырья в химико-фармацевтической промышленности
используют именно его. Для метилирования фенилметилпира-
284
золона применяют метиловый эфир бензолсульфокислоты (ме-
тилбензолсульфонат):
НС==С—СНз НС==С—СН3
О=С N-H ОСН3 О=С +n/CH3
\/ I \/\н -
N +O=S=O >- N SO3
I "
1-фенил-3-метилпиразолон-5
В эмалированный реактор с масляным обогревом загружа-
загружают фенилметилпиразолон и проводят его сушку до полного уда-
удаления влаги. Затем температуру поднимают до 127—130 °С и к
расплавленному фенилметилпиразолону приливают метиловый
эфир бензолсульфокислоты. Температура не должна поднимать-
подниматься выше 135—140 °С. По окончании метилирования реакцион-
реакционную массу передавливают в кристаллизатор, куда загружают
небольшое количество воды и охлаждают до 10 °С. Выпавший
бензолсульфонат антипирина отжимают и промывают на цент-
центрифуге. Для выделения антипирина эту соль обрабатывают
водным раствором едкого натра, а полученный антипирин пере-
кристаллизовывают из изопропилового или этилового спирта.
Антипирин представляет собой бесцветные кристаллы без запа-
запаха, хорошо растворим в воде, спирте, хлороформе, хуже — в
эфире, температура плавления ПО—113°С. Антипирин приме-
применяется как жаропонижающее, болеутоляющее и успокаиваю-
успокаивающее средство. Выпускается в порошках и таблетках по 0,25 г.
Большая часть бензолсульфоната антипирина, минуя ста-
стадию выделения антипирина, идет на стадию нитрозирования
для синтеза амидопирина и анальгина.
НС=С—СН3 NaNO2 ON—C=C—CH3
О=А 4/СНз ^ О=С N-CH3
\ / хн - \ /
N SO3 N + C6H5SO3Na + Н2О
Для проведения этой реакции водную суспензию полученной со-
соли антипирина загружают в нитрозатор, охлаждают до темпе-
температуры 2°С и постепенно приливают 20% раствор нитрита нат-
натрия. Роль кислоты в этой реакции (см. главу 9) выполняет бен-
золсульфокислота, связанная с антипирином. Температура при
нитрозировании не должна подниматься выше 4—5°С. Полу-
Полученную суспензию изумрудно-зеленых кристаллов нитрозоанти-
пирина фильтруют на вакуум-фильтре и промывают холодной
водой.
285
Восстановление нитрозоантипирина проводят смесью суль-
сульфита и бисульфита натрия
Н3С-С=С—NO H3C-C=C-NHSO3Na
N + Na2SO4
Сначала выдерживают реакционную массу 3 ч при температу-
температуре 22—25 °С, а затем 2—272 ч при температуре около 80 °С.
Образовавшуюся натриевую соль сульфоаминоантипирина
гидролизуют в присутствии муравьиной кислоты:
Н3С—С=С—NH—SO3Na H3C—C=C—NH2
| | -НСООН
Н3С—N С=О
Н3С—N C=O
\ / н2о, нсоон " \ /
N ►• N + NaHSO»
Муравьиная кислота реагирует при этом с непрореагировавши-
ми на предыдущей стадии сульфитом и бисульфитом, образуя
формиат натрия и сернистый газ:
Na2CO3 + 2НСООН >■ 2HCOONa+ СО2+ Н2О;
NaHSO3 + НСООН > HCOONa + SO2 + H2O.
Частично происходит восстановление сернистого газа до серо-
сероводорода и образование серы:
ЗНСООН + SO2 > H2S + 3CO2 + 2Н2О;
SO2 + 2H2S >■ 3S + 2Н2О.
Метилирование аминоантипирина проводят смесью фор-
формальдегида и муравьиной кислоты при температуре кипения
реакционной массы A00—105 °С):
Н3С—C=C-NH2
| | .НСООН
Н3С—N С=О
\ / сн2о, нсоон
N >-
286
/CHS
H3C--C=C-N4
I | \сн3 .нсоон
HgC—N C=O
CO2 + H2O
Полученную муравьинокислую соль амидопирина для выделе-
выделения свободного основания обрабатывают содой при 50 °С:
/
I I ХСН3
H3C-N C=O
\ /
« НСООН +Na2CO3
+ 2HCOONa + СО2 + Н2О
После нейтрализации амидопирин всплывает в виде масла. Очи-
Очищают амидопирин перекристаллизацией из изопропилового
спирта. Фармакопейный продукт представляет собой белый
кристаллический порошок слабо горького вкуса, без запаха.
Хорошо растворяется в воде, спирте, эфире.
Амидопирин оказывает жаропонижающее, болеутоляющее и
противовоспалительное действие. Он активнее антипирина. Его
применяют при головной боли, невралгиях, артритах, ревма-
ревматизме. Препарат выпускают в виде порошков и таблеток.
Амидопирин входит в состав многих комбинированных ле-
лекарственных средств (апикодин, новомигрофен, пентальгин, пи-
раминал, пирамеин, пирафен, веродон, амазол).
287
Анальгин получают из аминоантипирина по схеме:
H3C-C=C-NH2 H3C-C=C-N=CH-
II -oil
H3C-N C=0 *~~^-Ck H3C-N C=0
N >- N
Аминоантипирин Бензилиденаминоантипирин
Н3С—С=С—N4
I I 3 |
H3C-N C=O ' NaHSO3 + HC4 H3C-N
\ / Ц \
N 68—70 °C N
Монометил аминоантииирин Анальгин
Другая разновидность того же синтеза заключается в том,
что мононатриевую соль сульфоаминоантипирина не гидроли-
зуют до аминоантипирина, а действием щелочи переводят в ди-
натриевую соль, которую метилируют диметилсульфатом с по-
последующим кислым гидролизом сульфогруппы:
Н3С—C=C—N4 H3C~O-==C-N4
I I XSO3Na | | xSO3Na
H3C-N C=O H3C-N C=O
\ / NaOH \ / Oy XOCH3
N >■ N
107-110 CC
/CH3
HSC—C=C—Nx H3C— C=C—NHCH3
I I xSO3Na | | -V2H2SO4
H8C—N C=O H3C—N C=O
\ / H2SO4 \ / NaOH
85^ ^ 58-62 CC
288
/н
Н3С—С=С—Nx
I I ХСН3
H3C-N N=0
\/
N >- Анальгин
По активности и быстроте действия анальгин превосходит
антипирин и амидопирин. Хорошая растворимость дает возмож-
возможность широко пользоваться анальгином для парентерального
введения. Одновременное назначение анальгина и амидопирина
дает возможность получать быстрый (за счет быстрого поступ-
поступления в кровь анальгина) и длительный (за счет медленного
всасывания амидопирина) лечебный эффект. Применяют аналь-
анальгин при болях различного происхождения, лихорадочных состоя-
состояниях, ревматизме. Так же как и амидопирин, анальгин входит
в состав многих комбинированных препаратов (адофен, аналь-
фен, диафен, дикафен, кофадин, кофальгин, фенальгин и др.).
Из производных пиразолидина следует упомянуть бутадион:
0=С СН—СНо—СН2—СН2—СНо
N
По противовоспалительной активности бутадион значительно
превосходит амидопирин и производные салициловой кислоты.
Препарат применяют для лечения ревматизма в острой форме,
подагры, артритов.
Другую важную группу лекарственных соединений, являю-
являющихся производными пятичленных гетероциклов, составляют
производные имидазола.
В противоположность рассмотренной нами системе пиразола
изомерная ей система имидазола часто встречается в природных
биологически активных веществах — алкалоидах, витаминах,
нуклеиновых кислотах, аминокислотах.
)
f
н
н н
Пиразол Имидазол
19-926 289
Так же как в группе пиразола, определенное значение имели
производные продукта частичного восстановления пиразола—
пиразолина, в группе имидазола известны лекарственные веще-
вещества — производные имидазолина:
Н,С х
119С СН
н
Имидазо/шн
Наибольшее значение имеют производные имидазолина с заме-
заместителями в положении 2. В качестве примера можно привести
такие широко распространенные лекарственные средства, как
нафтизин (санорин), фентоламин pi др.
H2C-N/ >=\ HoC—NT/
| -HNOo I } | -HC1
H V__/ II
Нафти mti Фентоламин
Нафтизин оказывает длительное сосудосуживающее дейст-
действие, повышает артериальное давление, расширяет зрачки. В свя-
связи с сосудосуживающими свойствами при нанесении па слизи-
слизистые оболочки нафтизин оказывает противовоспалительное дей-
действие. Фентоламин, напротив, блокирует передачу сосудосужи-
сосудосуживающих импульсов, что приводит к расширению периферических
сосудов, снятию спазмов, улучшению кровоснабжения мышц,
кожи, слизистых оболочек. При применении этого препарата
происходит некоторое понижение артериального давления.
Наиболее распространенный путь синтеза двузамещенных
имидазолинов можно иллюстрировать схемой получения нафти-
нафтизина (см. также главу 11):
СНоСООН
сн
ПС1,
H...N—СН2—СН.?—NH2
t°
N-CH2
—CH2
H -НС1
NaOH
CH2—С
N—СН2
—СН2
н
СН2—Сх
HNO3 разбавл.
Н -HNO3
Вместо карбоновых кислот в подобных синтезах можно ис-
использовать их производные (эфиры, иминоэфиры, нитрилы
и т. п.). В частности, важный лекарственный препарат дибазол
290
B-бензил-бензимидазол) можно получать как из фенилуксус-
ной кислоты, так и из цианистого бензила. Поскольку циани-
цианистый бензил дешевле фенилуксусной кислоты, в производстве
используют последний способ, хотя дибазол из фенилуксусноГ
кислоты получается чшце и с более высоким ныхогом:
Дибазол является оригинальным отечественным препаратом.
Он обладает сосудорасширяющим, спазмолитическим и ;,<imeh
дивным действием, а также оказывает стимулирующее влияние
на функции спинного мозга. Применяют препарат при спазмах
кровеносных сосудов и гладкой мускулатуры внутренних орга-
органов (гипертонический криз, язвенная болезнь желудка и т. дЛ
а также при лечении нервных заболеваний.
5,6-Диметилбензимидазол входит в состав сложной молек\лы
гштамина Bi2, который влияет на кроветворную функцию ор. л~
нпзма, способствует нормализации функции печени, благоприят-
благоприятно влияет на регенерацию нервных волокон. Витамин Bi2 ос .ер-
жится во многих продуктах животного происхождения и г^чти
не встречается в растениях. Сырьем для получения вит? ми via B{2
служат отходы производства антибиотиков. Молекула витами-
витамина Bi2 является очень сложной (CosHgsCoNuOuP). В химиче-
химическом отношении молекула витамина Bjo напеминае! строение
гемина крови. Однако вместо атома железа в центре этой «ге-
миновой» части молекулы находится атом кобальта, .вязанный
с CN-группой. Отсюда и произошло одно из названий витамина
В12 — цианокобаламин.
н
i IX
N
5.6-Димет!'лСензи мда зол
!'^нмапче пу^леотидной части молекулы ■-. .амина В; 2)
§ 2. ПРОИЗВОДНЫЕ ШЕСТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
С ОДНИМ ГЕТЕРОЛТОЛ^ОМ
Из лекарственных соединений, содержащих в составе моле-
молекулы шестичленный цикл с одним гетероатомом, наибольшее
значение имеют производные пиридина и хинотина:
N N
Пиридин Хшюднн
19*
Получение этих гетероциклов, а также некоторых их произ-
производных уже рассматривалось в главах 10 и 11.
Производным пиридина, имеющим большое значение в ме-
медицине, является никотиновая кислота ((З-пиридинкарбоновая)
и ее амид (см. главу 10).
СООН
N N
Никотиновая Амид никотиновой
кислота кислоты
Действие на организм никотиновой кислоты и ее амида прак-
практически одинаково, так как в организме никотиновая кислота
легко превращается в амид. Никотинамид можно получить не-
непосредственным аминированием никотиновой кислоты аммиаком
при температуре 220—230 °С и давлении 27—28 ат. За 272 ч при
этих условиях достигается выход 84%.
'СООН NH3
230 СС, 28 ат
N N
Никотинамид называют витамином РР, никотиновую кислоту
следует рассматривать как провитамин никотинамида, превра-
превращающийся в витамин РР в процессе обмена веществ в организ-
организме. Известно около сорока различных биологических реакций,
катализируемых ферментами, в состав которых входит никотин-
никотинамид. Никотиновая кислота, вернее — ее производные, прини-
принимают участие в белковом и углеводном обмене, нормализуют
уровень холестерина в крови, влияют на водно-солевой обмен.
В медицинской практике используется способность никотиновой
кислоты расширять мелкие кровеносные сосуды. Витамин РР
оказывает благотворное действие при головных болях, если они
обусловлены спазмами сосудов головного мозга. Никотиновая
кислота повышает кислотность желудочного сока, регулирует
моторную функцию желудка, способствует лучшему всасыванию
и усвоению питательных веществ. Она также оказывает положи-
положительное влияние на основные функции печени. Недостаток в ор-
организме человека никотиновой кислоты приводит к тяжелому
заболеванию — пеллагре.
Диэтиламид никотиновой кислоты применяют при острых и
хронических расстройствах сердечной деятельности и ослаблении
дыхания. Его можно получить из хлорангидрида никотиновой
кислоты:
с н
О Л
N N
292
В производстве получение диэтиламида никотиновой кислоты
проводят нагреванием смеси никотиновой кислоты, диэтиламина
и хлорокиси фосфора, не выделяя хлорангидрид.
Фармацевтическим препаратом является 25% водный рас-
раствор диэтиламида никотиновой кислоты (кордиамин), представ-
представляющий собой жидкость слегка желтоватого цвета со своеобраз-
своеобразным запахом.
Не менее важное значение в медицине имеют производные
изоникотиновой (у-пиридинкарбоновой) кислоты. Некоторые
вещества этого ряда являются ценными химиотерапевтическими
средствами противотуберкулезного действия. Сотрудники
ВНИХФИ под руководством проф. М. Н. Щукиной провели
большую работу по созданию и внедрению в производство пре-
паратов-туберкулостатиков — производных изоникотиновой кис-
кислоты.
В настоящее время противотуберкулезные препараты приня-
принято делить на две группы: основные антибактериальные препара-
препараты (препараты первого ряда) и резервные антибактериальные
препараты (препараты второго ряда).
К препаратам первого ряда, являющимся основными химио-
химиотерапевтическими средствами для лечения разных форм тубер-
туберкулеза, относятся гидразид изоникотиновой кислоты (изониазид,
тубазид) и его производные — фтивазид, ларусан, салюзид рас-
растворимый, некоторые антибиотики (стрептомицин и др.), ПАСК
и ее производные.
Для лечения резистентных форм туберкулеза используют
препараты второго ряда — этоксид, тиоацетадон, салютизон,
этионамид, циклосерин, канамицин, флоримицин и др. Их ос-
основная особенность состоит в том, что они действуют на мико-
бактерии, ставшие устойчивыми к препаратам первого ряда.
Для получения стойкого лечебного эффекта и предупреждения
возможных рецидивов противотуберкулезные препараты долж-
должны применяться длительно, в среднем не менее года.
В молекулах ряда производных гидразида изоникотиновой
кислоты, обладающих противотуберкулезной активностью, со-
содержится группа:
Эта группа рассматривается как «транспортная», т. е. она спо-
способствует проникновению изоникотиновой кислоты через бакте-
бактериальную оболочку.
Одним из наиболее эффективных противотуберкулезных пре-
препаратов является изониазид (тубазид), являющийся гидразидом
изоникотиновой кислоты:
О
NT Ъ—С—NH~NH2
293
Изониазид представляет собой белый кристаллический поро-
порошок горьковатого вкуса легко растворимый в воде, трудно —
в спирте и в хлороформе, практически нерастворимый в эфире.
Содержание в препарате гидразида изоникотиновой кислоты не
ниже 98%. Температура плавления 170—174°С.
Изониазид обладает высокой противотуберкулезной активно-
активностью, применяется для лечения различных форм туберкулеза
легких у взрослых и детей, а также при туберкулезе гортани и
полости рта, слизистых оболочек, желудочно-кишечного тракта,
при костно-суставном туберкулезе, красной волчанке.
Микобактерии туберкулеза быстро приобретают устойчивость
к изониазиду. Однако при этом они сохраняют чувствительность
к стрептомицину и ПАСК, поэтому изониазид применяют в соче-
сочетании с этими противотуберкулезными средствами.
Основным путем производственного синтеза изониазида яв-
является получение его из у-пиколина, входящего в состав р-пико-
линовой фракции коксохимических оснований. Применяемый ме-
метод состоит в превращении содержащегося в ней у-пиколина в
метилольные производные:
СН3 СН2-СН2ОН СН(СН2ОНJ С(СН2ОНK
СН2О
15*5 U
NN NN
Метилольные производные окисляют азотной кислотой в изо-
никотиновую кислоту, которая представляет собой кристалличе-
кристаллическое вещество от светло-желтого до желто-розового цвета с тем-
температурой плавления 322—325 °С.
Отделяемая от метилольных производных смесь оснований,
содержащая р-пиколин и 2,6-лутидин, используется в производ-
производстве никотиновой кислоты.
Необходимый для синтеза изониазида гидразин-гидрат полу-
получают обработкой гидразин-сульфата окисью кальция при темпе-
температуре 80 °С:
H2N-NH2.H2SO4 + CaO ~^^ NH2-NH2.H2O + CaSO4
Затем в эмалированном аппарате ведут прямое ацилирование
гидразин-гидрата изоникотиновой кислотой при температуре
129—130 °С с азеотропной отгонкой воды:
О
II
СООН С—NH-NH2
N
Гидразид изоникотиновой кислоты кристаллизуется при охлаж-
охлаждении с выходом 85,6%.
294
Изониазид является относительно . токсичным препаратом.
Под руководством проф. М. Н. Щукиной был синтезирован ори-
оригинальный отечественный противотуберкулезный препарат фти-
вазид, который аналогичен изониазиду по противотуберкулезно-
противотуберкулезному действию, но менее токсичен. Фтивазид является изоникоти-
ноилгидразоном, который образован изониазидом и ароматиче-
ароматическим альдегидом — ванилином.
CON4-N=CH—R
Ч)СН3
Изоникотиноиагидразон В.-чплит»
Фтивазид получлют конденсацией в водной среде ванилина
и гидразида изоникотиновой кислоты ^кзоаиазида) при темпе-
температуре около 60 °С.
CONHNH
N
Вначале готовят при перемешивании и температуре 78—84 °С
раствор ванилина в дистиллированной воде. Полученный рас-
раствор передавливают через друк-фильтр в аппарат, куда загру-
загружают раствор изоникотиновой кислоты. Конденсация проходит
при температуре 55—60 °С в течение 2 ч. После выдержки реак-
реакционную массу охлаждают и при температуре 25—30 °С пере-
передавливают в центрифугу. Отжатый от маточного раствора фти-
фтивазид промывают на центрифуге горячей дистиллированной
водой. Хорошо отжатый фтивазид выгружают и передают на
сушку. Сушат фтивазид при температуре 60—70 °С. Выход фти-
вазида составляет 97,4% считая на гидразид изоникотиновой
кислоты.
Фтивазид представляет собой светло-желтый кристалличе-
кристаллический порошок с запахом ванилина. Он нерастворим в воде, пло-
плохо растворяется в спирте, легко растворяется в щелочах и кис-
кислотах; температура плавления 220—222°С (с разложением).
Выпускается в виде порошков и таблеток.
295
Еще одним противотуберкулезным препаратом этой же груп-
группы является салюзид. Салюзид — продукт конденсации опиано-.
вой кислоты и гидр азида изоникотиновой кислоты:
O=C-NH—N=C—H
нао
N
ОСН;
ОСН3
з
По физическим свойствам салюзид представляет собой мелко-
мелкокристаллический порошок зеленовато-желтого цвета. Легко рас-
растворим в щелочах и неорганических кислотах, трудно — в спир-
спирте и воде, нерастворим в эфире и хлороформе. Температура
плавления 196—203 °С.
Салюзид получают аналогично фтивазиду — конденсацией в
водном растворе эквимолекулярных количеств опиановой кисло-
кислоты с гидразидом изоникотиновой кислоты при 70—80ЬС в тече-
течение 2 ч.
ОН О
O=C-NH-NH2
•СООН
В медицинской практике при лечении туберкулеза применя-
применяется салюзид растворимый, представляющий собой диэтилами-
новую соль салюзида.
Салюзид растворимый получают взаимодействием салюзида
с диэтиламином в среде изопропилового спирта:
|^^ NH(CaH6J
СООН ^ ^^
O=C-NH—N=CH
NH2(CaH6J
N
Препарат выпускается в виде 5 или 10% водных растворов*
в ампулах, а также в порошках.
К производным пиридина принадлежат и витамины (группы
Вб, оксиметилпиридиновые витамины). К витаминам группы В«
296
принадлежат три вещества, коюрые в организме могут превра-
превращаться одно в другое:
СН2ОН СНО CH2NH2
J /СН2ОН HJ i
Н3С
N
Пиридоксин
(пиридоксол)
-2-Метил-3-окси-4,5-Диокси-
метилпиридин
N
Пиридоксаль
2-Метил-3-окси-4-формил-
-5-оксиметилпиридин
Н,С
N
Пиридоксамин
2-Метил-3-окси-4-амино-
метил-5-оксиметил-
пиридин
Фосфорные эфиры пиридоксина, пиридоксаля и пиридокс-
лмина в качестве коферментов входят в состав различных фер-
ферментов, катализирующих белковый обмен в организме. В этом
обмене особенно важную роль играет пиридоксаль-5-фосфат.
Витамин Вб обеспечивает синтез и обмен аминокислот в орга-
организме. Раньше витамин В6 (пиридоксин) получали из естест-
естественного сырья (дрожжей, рисовых отрубей). В настоящее время
пиридоксин получают путем многостадийного химического син-
синтеза. Фармакопейным препаратом витамина В6 является гидро-
гидрохлорид пиридоксина.
Весьма интересными для фармацевтической практики явля-
являются производные хинолина, среди которых имеется много цен-
ценных лекарственных препаратов.
Среди препаратов этой группы следует отметить активный
анестетик — совкаин, который применяют, в частности, для
спинномозговой анестезии.
CO-NH—СН2—CH2-N(C2H5),
•на
Совкаин
Совкаин получают из изатина, который ацилируют по ими-
ногруппе уксусным ангидридом. При обработке щелочью полу-
полученного ацильного производного получают в качестве промежу-
промежуточного продукта натриевую соль ацетилизатиновой кислоты,
которая в кислой среде превращается в 2-оксицинхониновую
кислоту. Последнюю переводят в хлорангидрид 2-хлорцинхони-
новой кислоты, которым ацилируют C-диэтиламиноэтиламид.
Полученное производное обрабатывают бутилатом натрия и пос-
после .подкисления получают совкаин.
N-Ацетнлнзатин
297
Натриевая соль
N-ацетилизатиновоп
кислоты
2-О:чСииинхсниновая
■<ислота
СОС1
)NHCH2CH2N(C2H3J
85 СС
QH ,ONa
105 СС
CONHCH2CH2N(C2H5J
ОС4Н9
Р-Диэтпламинг.ьтиламид
2-буТОКСИЦИНХОН1!НОЬОЛ КИСЛОТЫ
^ Cl
IN
Р-Диэтилдминоэтиламид
2-ХЛОрЦИНХОНННОЬОИ КИСЛОТЫ
CONHCH2CH2I\r(C2H-J
НС!
-на
Совкаин
Процесс ведут следующим образом. Смесь уксусного ангид-
ангидрида, изатина и небольшого количества серной кислоты нагре-
нагревают до 98—100 °С и выдерживают 4 ч при этой температуре.
Уксусный ангидрид берут в большом избытке; он служит как
ацетилирующим агентом, так и растворителем. При охлаждении
реакционной массы до 18—20 °С медленно выкристаллизовыва-
выкристаллизовывается N-ацетилазатин, который отфильтровывают и промывают
спиртом. Выход на этой стадии 81,2%.
N-Ацетилизатин нагревают с 40% раствором едкого натра
при 108—110 °С в течение 1 ч. Полученный раствор фильтруют
и при 70—80 °С приливают к нему 20% соляную кислоту. Вы-
Выпавший осадок 2-оксицинхониновой кислоты отфильтровывают
и промывают горячей водой. Выход на стадии 71,4%. Получен-
Полученную 2-оксицинхониновую кислоту загружают в смесь дихлорэта-
дихлорэтана и треххлористого фосфора и выдерживают 17г ч при тем-
температуре 58—62 °С. После этого при той же температуре в реак-
реакционную массу пропускают сухой газообоазный хлор. Прозрач-
Прозрачный раствор охлаждают и фильтруют oi примесей. Дихлорэтан
и хлорокись фосфора отгоняют в вакууме. К остатку приливают
горячий (90 °С) толуол, тщательно перемешивают и охлаждают
до 8°С. Выпавший в осадок хлорангидрид 2-хлорцинхониновой
кислоты отфильтровывают и промывают толуолом. Выход на
стадии 53,2%.
Хлорангидрид 2-хлорцинхониновой кислоты растворяют при
нагревании в толуоле и к полученному раствору приливают рас-
раствор |3-диэтиламиноэтиламина в толуоле. Температуру за 5—6 ч
поднимают от 40 до 85 °С и при этой температуре выдерживают
298
2 ч. При охлаждении реакционная масса разделяется на два
слоя, так как образующийся гидрохлорид р-диэтиламиноэтил-
амида 2гхлорцинхониновой кислоты нерастворим в толуоле. То-
луольный слой сливают, я продукт пястворяют в горячей воде,
фильтруют, горячий раствор охлаждают до 0—1 °С и подщела-
подщелачивают 7% раствором едкого кали. Выпавший осадок отфиль-
отфильтровывают и промывают ледяной водой. Выход на стадии 58,3%.
Реакцию C-диэтиламиноэтиламида 2-хлорцинхониновой кис-
кислоты с бутилатом натрия ведут в среде бутилового спирта при
105 °С в течение 5 ч. Раствор фильтруют и отгоняют в вакууме
бутиловый спирт. Осадок растворяют в бензоле при нагревании,
фильтруют и к фильтрату при хорошем перемешивании прили-
приливают разбавленную соляную кислоту. Продукт в виде гидро-
гидрохлорида переходит в водный слой. Очистка препарата основана
на том, что совкаин-основание, образующееся при подщелачи-
вании раствора гидрохлорида, растворим в эфире и может быть
экстрагирован им из водного слоя. Выход на стадии составляет
74,5%. Для получения фармакопейного препарата совкаин-ос-
совкаин-основание растворяют в спирте и через спиртовой раствор пропу-
пропускают газообразный хлористый водород. Полученный спиртовый
раствор гидрохлорида охлаждают до 0°С и продукт осаждают
добавлением эфира.
Другой препарат этого ряда — цинхофен (атофан) можно
полечить по реакции Скраупа или Дебнера. Оба эти метода
разбирались в главе 11.
СООН
I
Цинхофен B-фенилцинхониновая кислота) применяется при
лечении подагры.
Важным представителем лекарственных веществ производ-
производных изохинолина является папаверин.
Н3С(Х
ОСН<
осн
3
Папаверина гидрохлорид
Папаверин является природным алкалоидом. Однако запасы
растительного сырья не могут удовлетворить потребность в этом
299
препарате, поэтому папаверин в настоящее время получают
путем химического синтеза. Папаверин широко применяется в
качестве спазмолитического средства при спазмах кровеносных
сосудов (гипертония, стенокардия, мигрень), спазмах гладкой
мускулатуры органов брюшной полости (холецистит, колиты
и др.), бронхиальной астме.
Химическая схема синтеза папаверина представлена ниже:
Н8СО
НО
XJ
сио
О% /ОСН3
;\
ОСН3
Н3СО
40-50 °С
сно
(СН3СОJО
60-70 СС
Ванилин
Вератровый альдегид
О
!
NH3 C3H5COCI
С1СН2СООН > NH2CH2COOH >■
Хлоруксусная
кислота
C-NHCH2COOH
CH3COONa
Н,СО
Аминоуксусная
кислота
Бензоиламино-
уксусная кислота
(гиппуровая)
сн о
N О
v
i
Азлактон
вератральгиппуровой
кислоты
О
сн=с—с/
NH
с=о
НоСО
NH4
NH4CI
50 °С
Аммонийная соль
Еератральгиппуроьой кислоты
NaOH 95 °С
сн=с—с<
NH
С=О
Натриевая соль
вератральгиппуровой кислоты
/О
300
нхо
Н3СО
соон н3со
НС1
Н3СО
1) NaOH
2) СН3СООН
OCH3
соон
осн3
н3со
осн.
осн,
ОСН3
Папаверин-основание
Синтетический папаверин полностью идентичен природному.
§ 3. ПРОИЗВОДНЫЕ ШЕСТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
С ДВУМЯ ГЕТЕРОАТОМАМИ
Среди производных гетероциклических соединений с двумя
гетероатомами, имеющих ярко выраженную биологическую ак-
активность, большое значение имеют производные фенотиазина.
Фенотиазин не представляет интереса для медицинской
практики, хотя одно время он применялся как противоглистное
средство.
Однако производные фенотиазина, имеющие остаток третич-
третичного алифатического амина в положении 10, составляют боль-
большую группу нейтролептических средств. При подробном изуче-
изучении этих производных фенотиазина было установлено, что они
оказывают сильное влияние на центральную и периферическую
нервную систему. В поисках веществ, оказывающих еще более
активное и более избирательное влияние на функции централь-
центральной нервной системы, синтезировано много соединений, имею-
имеющих, кроме указанных заместителей в положении 10, замести-
заместитель в положении 2 (обычно —С1, —ОСН3, —CF3). Производ-
Производные фенотиазина оказывают успокаивающее действие при
состояниях сильного нервного возбуждения. Они проявляют
также антипсихотичеекое действие, снимая или уменьшая бред,
галлюцинации и другие болезненные психические явления.
301
Обычный метод синтеза лекарственных соединений этой груп-
группы заключается в алкилировании иминогруппы фенотиазина
хлорпроизводными соответствующего третичного амина:
S
Cl(CH2LN(AlkJ-HCl
'" " " '" .на
,А1к
v\lk
По внешнему виду все эти препараты представляют собой
белые (иногда с кремовым или голубоватым оттенком) кристал-
кристаллические порошки. Они хорошо растворимы в воде, спирте и
хлороформе и практически нерастворимы в эфире и бензоле.
Производные фенотиазина темнеют на свету, что связано с их
способностью к окислению, поэтому препараты этой группы хра-
хранят в банках из темного стекла с плотно закрывающимися
крышками (препараты гигроскопичны).
Важнейшим представителем этого ряда является аминазин —
гидрохлорид 2-хлор-10-C/-диметиламинопропил) — фенотиазина:
CHo-CH,-N4 -НС1
ХСН3
Исходным продуктом для синтеза аминазина и ряда других
сходных препаратов (этаперазин, хлорацизин, метеразин, три-
фтазин, френолон и др.) служит 2,4-дихлортолуол. 2,4-Дихлор-
толуол действием 57% азотной кислоты при температуре 130—
135 °С и 6—8 ат в течение 7—8 ч окисляют до 2,4-дихлорбензой-
2,4-дихлорбензойной кислоты.
НЗС YX нноз _ f ./СООН
JL Jl 130-135 °С, 6—8~ат JL J1
Выход 2,4-дихлорбензойной кислоты с учетом утилизации маточ-
маточных растворов составляет 95%.
Полученный 2,4-дихлорбензойной кислотой арилируют ани-
анилин:
NH2
I
С1
X
СООН
ноос
К2СО3, Си s
102-109 °С L
302
Для этого приготавливают водный раствор поташа (карбоната
калия), прибавляют к нему дихлорбензойную кислоту и реак-
реакционную массу перемешивают при 80—90 °С до прекращения
выделения углекислого газа. К полученному раствору калиевой
соли дихлорбензойной кислоты при 70—80 °С прибавляют ани-
анилин и свежеприготовленную медь (порошок), массу подогре-
подогревают и кипятят 6 часов при 102—109 °С, затем разбавляют водой
и отфильтровывают от меди. Горячий фильтрат очищают акти-
активированным углем и при подкислении соляной кислотой (до
рН 4,35—4,5) выделяют З-хлордифениламин-6-карбоновую кис-
кислоту. Выход промытой и высушенной кислоты составляет
82—83%.
Сухую З-хлордифенил-6-карбоновую кислоту декарбоксили-
руют при 180—200 °С при остаточном давлении 850-—
700 мм рт. ст. Затем повышают вакуум до 4—5 мм рт. ст. оста-
остаточного давления и отгоняют образовавшийся 3-хлордифенил-
амин. Выход на стадии составляет 90%.
ноос
о
N
1
Н
:w '
180-200 СС L
К
к
j
н
Смесь 3-хлордифениламина, серы и каталитического количества
йода перемешивают при 170—180 СС до прекращения выделения
сероводорода. Затем к реакционной массе добавляю! хлорбен-
хлорбензол и уголь, кипятят полчаса, фильтруют от угля и фильтрат
охлаждают. Выкристаллизовавшийся 2-хлорфенотназин отфиль-
отфильтровывают, промывают хлорбензолом и сппотсм и сушат при
100 °С.
Лла+s ^
Nil U
Вьтход на стадии с учетом регенерации маточников составляет
79%. Таким образом получают один из основных промежуточ-
промежуточных продуктов для синтеза аминазина.
Другой промежуточный продукт синтезируют следующим об-
образом. Каталитическим гидрированием этнленциангидринг по-
получают 3-аминопропанол:
Н2, NiRe
НО—СН2—СНо—CN — >• НО—СН0—СН,—СН2—NH2
2 " 95—100 СС 2-22
20-22 ат
303
Восстановление циан-группы проводят водородом при 95—100 GC
и 20—22 ат в спиртовом растворе в присутствии никеля Ренея,
Технология ведения процесса не отличается от обычной (см.
главу 10). Выход на стадии — 79,5%. Полученный у-аминопро-
панол прибавляют при перемешивании и температуре 30—40 °С
к 85% муравьиной кислоте, затем добавляют 37% формалин и
температуру поднимают до 90—95 °С:
HOCH2CH2CH2NH2 + 2СН2О + 2НСООН д()_95 сс>
>- HOCH2CH2CH2N(CH3J + 2СО2 + 2Н2О
Аналогичный метод алкилирования аминогруппы уже рас-
рассматривался нами при описании метилирования аминоантипи-
рина. Метилирование идет медленно, поэтому выдержка обычно
составляет 10—15 ч. После выдержки реакционную массу
охлаждают, нейтрализуют раствором едкого натра и дают от-
отстояться. Основная масса образовавшегося р-диметиламинопро-
панола собирается в верхнем слое. Ее отделяют от нижнего
водно-щелочного слоя. Часть диметиламинопропанола, раство-
растворившуюся в водно-щелочном слое, экстрагируют бензолом. Вы-
Выход 3-диметиламинопропанола составляет 88%. Замену гидр-
оксильной группы в диметиламинопропаноле на хлор проводят
при действии тионилхлорида, который берут - с избытком 5—
10%. Реакцию ведут в смеси хлорбензола и толуола A : 3) при
температуре кипения смеси:
СвН5С1, СбН5СН3
HOCH2CH2CH2N(CH3J + SOClo *•
л ч * кипение
>■ C1CH2CH2CH2N(CH3J.HC1 + SO2
После окончания процесса реакционную массу охлаждают и
загружают едкий натр и 2-хлорфенотиазин.
S
+ C1(CH2KN(CH3J + NaOH >-
Cl
СН2(
+ NaCl + H2O
^Cl
/CH3
,CH2CH2N4
ЧСН3
Смесь кипятят до прекращения выделения воды. Смесь раство-
растворителей отгоняют при остаточном давлении 300—400 мм рт. ст.,
а остаток перегоняют при 1 мм рт. ст. и температуре 210—226 °С.
304
Выход основания аминазина 91,2%. Полученное основание пере-
переводят в гидрохлорид.
НС1
•на
-а
/СН3 I
сн2сн2снХ ch2ch2ch2n(
хсн3 хсн3
Аминазин представляет собой белый со слабым кремовым
оттенком кристаллический порошок хорошо растворимый в воде,
спирте и хлороформе. В эфире и бензоле аминазин практически
нерастворим. Температура плавления 194—198 °С.
Аминазин является одним из основных представителей ней-
нейролептических средств. Кроме своеобразного успокаивающего
действия на нервную систему, препарат понижает температуру
тела, усиливает действие снотворных, наркотических, болеуто-
болеутоляющих и противосудорожных средств и снижает артериальное
давление. Аминазин широко применяется в психиатрии, хирур-
хирургии и акушерстве. Выпускается в виде таблеток (драже) по
0,025; 0,05; 0,1 г; 2,5% раствора в ампулах по 2 и 5 мл и 5% рас-
раствора в ампулах по 5 мл.
В связи с высокой токсичностью З-хлордифениламин-6-кар-
боновой кислоты, 3-хлордифениламина, 2-хлорфенотиазина, а
также аминазина-основания и аминазина (гидрохлорида) необ-
необходимо исключить возможность попадания их на кожу и слизи-
слизистые оболочки. Все операции по получению и переработке этих
веществ должны проводиться таким образом, чтобы исключить
прямой контакт с ними обслуживающего персонала. Аппарату-
Аппаратура должна быть тщательно герметизирована, а кратность воз-
воздухообмена в производственных помещениях повышена. Целесо-
Целесообразно применение специальных загрузочных устройств. После
работы обязателен прием душа и смена белья. При попадании
аминазина и других перечисленных веществ на руки или другие
части тела следует смыть их холодной, лучше слегка подкис-
подкисленной водой без мыла. При появлении зуда кожи, слизистых
оболочек глаз, отечности кожи ве>к, появлении сыти, снижении
артериального давления следует отстранить пострадавших от
контакта с аминазином и направить к врачу.
Ближайшим аналогом аминазина является пропазин, моле-
молекула которого отличается от молекулы аминазина отсутствием
атома хлора в положении 2:
S
:h2ch2ch2n(Ch3J.hci
Пропазин
20-926 305
Пропазин — препарат аналогичный по действиям аминазину.
Он менее токсичен и менее активен, чем аминазин.
Очень близким по строению к аминазину является препарат
этизин, отличающийся более короткой углеродной цепью в поло-
положении 10 и отсутствием атома хлора в положении 2:
CH2CH2N(CH3J-HC1
Этизин
Однако укорочение углеродной цепи на одну метиленовую груп-
группу приводит уже к существенному изменению биологической
активности вещества. Этизин применяют для лечения аллергиче-
аллергических заболеваний, а также используют в качестве снотворного.
Представителем другой подгруппы фенотиазиновых препа-
препаратов является этаперазин:
CH2—CH2-CH2-N
Этаперазин
—CH2-CH2-OH
Этаперазин применяют в психиатрии для лечения шизофре-
шизофрении, различных психозов и неврозов. В связи с сильным анти-
антипсихотическим действием этаперазин может применяться для
лечения больных, нечувствительных или малочувствительных к
действию аминазина. Этаперазин может также применяться для
лечения упорной бессонницы у больных с психическими или
нервными заболеваниями, а также для устранения чувства
страха, напряжения у таких больных. Этаперазин синтезируют
по следующей схеме:
сн2-сн2
VNH-6H2O
Пиперазин гексагидрат
в метаноле, ок. 70 СС
-*■ HN
—СНоСНоОН
С1(СН2KВг
в толуоле. 7О-8
Г C.(CH2K-N
Ы-(Р-оксиэтил)-пиперазин
S
N-CH2CH2OH
„ NaOH 2)
1-(р-оксиэтил)-4-G-хлорпропил)-
пиперазин
306
CH2CH2CH2—N' N-CH2CH2OH.2HC1
Этаперазин
Близким по действию и по строению к этаперазину является
препарат трифтазин:
N
•2КС1
CF,
сНоСн2сн2—n; :n-ch,
Трифтазин применяют для лечения различных видов шизофре-
шизофрении и других психических заболеваний, сопровождающихся бре-
бредом и галлюцинациями, при алкогольных психозах, неврозах и
других заболеваниях центральной нервной системы. Синтез три-
фтазина проводят по следующей схеме:
HNO3
Fe
CF3 40
H2SO4
"-50 °С
НС1 т. кип.
(CH3COJO
>
NO2
NH2
CuBr, Си т. кип.
NHCOCH3
N
Н
N
СОСНд
S
■CFS
на
^.
Т. КИП
CF,
12, 145—155 °С
С1(СН2KВг
NaNH2
N
(СН2KС1
CF3
N
Н
HN .N-C
98-100 °С
20*
307
Технология производства трифтазина здесь подробно не рас-
рассматривается, так как она складывается из процессов, подоб-
подобных тем, технология которых уже нами разбиралась ранее.
С лекарственными производными пиримидина мы уже неод-
неоднократно встречались. Пиримидиновый цикл входит в состав мо-
молекулы очень многих лекарственных препаратов (в частности,
в уже рассмотренные нами сульфаниламидные препараты, бар-
барбитураты и т. д.) и витаминов. В дополнение к уже сказанному
следует отметить, что среди производных пиримидина имеется
целая группа препаратов противоракового действия. В числе
таких препаратов в первую очередь нужно отметить производ-
производные урацила — метилурацил, фторурацил, допан и др.
О О
II
HN
N
Пиримидин
О
II
II
1
1
н
5-Фторурацил
Н
Урацил
<
О
II
"О
н
н
6-Метилурацил
(метацил)
.СН2СН2С1
f \:h2ch2ci
Допан
Метилурацил (метацил) применяют при лейкопении, а так-
также для лечения плохо заживающих ожогов, ран, переломов ко-
костей. Метилурацил служит исходным сырьем для синтеза препа-
препарата допан, который является оригинальным отечественным
противоопухолевым препаратом алкилирующего действия (см.
также главы 13 §5 и 16 §4). 5-Фторурацил относится к группе
антиметаболитов. Противоопухолевая активность препарата
объясняется его превращением в раковых клетках в вещества,
которые ингибируют фермент, принимающий участие в синтезе
дезоксирибонуклеиновой кислоты. Тем самым тормозится разви-
развитие раковой ткани. Применяют фторурацил при неоперабель-
308
яых формах рака желудка и толстого кишечника, а также при
злокачественных опухолях поджелудочной железы. Фторурацил
вводят внутривенно. Выпускается препарат в виде 5% раствора
натриевой соли в ампулах по 5 мл. Необходимо иметь в виду,
что фторурацил (как и большинство противораковых препара-
препаратов) обладает высокой токсичностью и угнетает кроветворение.
Технология получения фторурацила состоит из следующих
основных операций. К суспензии измельченного или гранулиро-
гранулированного металлического натрия в ксилоле при 45—50 °С при не-
непрерывном перемешивании прибавляют безводный этиловый
спирт. Реакционную массу нагревают до 65—75 °С и выдержи-
выдерживают при этой температуре 3 ч, после чего греют еще час при
95—100 °С.
2С2Н5ОН + 2Na > 2C2H5ONa + Н2.
К осажденной до 25 °С суспензии этилата натрия в ксилоле мед-
медленно прибавляют смесь этилфторацетата с этилформиатом:
FCH2COOC2H5 + НСООС2Н5 + C2H5ONa on >
> NaOCH=CFCOOC2H5 + (С2Н5JО + Н2О.
После длительной выдержки при комнатной температуре к реак-
реакционной массе добавляют метанол и полученный раствор без вы-
выделения натрийформилфторуксусного эфира используют для
дальнейшего синтеза. Помимо необходимых при работе с ме-
металлическим натрием предосторожностей (о чем уже неодно-
неоднократно говорилось), надо учитывать, что эфиры фторуксусной
кислоты являются сильным ядом. Как и при синтезе большин-
большинства противораковых препаратов, работать необходимо либо
в противогазе и противокислотном костюме, либо в скафандре
с поддувом воздуха, что удобнее и безопаснее. При этом забор
воздуха должен производиться из другого помещения, куда не
могут проникать вредные пары и газы.
К полученной на первой стадии реакционной массе добав-
добавляют S-метилизотиомочевину и полученную реакционную массу
кипятят 2 ч:
О О
II II
С С
NH2 H,C2O CF cH3ONa HN CF
I + II >■ I I! + NaOH + С2Н5ОН
с сн т-кип- /С сн
После отгонки в вакууме метилового спирта и ксилола, раз-
разбавления водой и подкисления до рН 4,0—5,0 из реакционной
309
массы извлекают 2-метилтио-5-фторурацил. Последний при ки-
кипячении с 20% соляной кислотой превращается в 5-фторурацил:
о О
1 Г
*С1 ПН *• f"
Образующийся метилмеркаптан является сильным ядом. Его
необходимо улавливать раствором щелочи, а весь синтез про-
проводить в герметичной аппаратуре при хорошей вентиляции про-
производственного помещения.
Большое значение в медицинской практике имеют производ-
производные пурина, представляющего собой бициклическую систему из
пиримидинового и имидазольного колец:
Соединение, в котором вместо атомов водорода в пиримиди-
новом кольце стоят гидроксильные группы, называется ксанти-
ном. Ксантин может существовать в енольной и кетонной
форме:
ОН О
А 4
NH H-n/ \ NH
НО г СИ О—Г '• , ГН
NN NN
I
н
Некоторые N-метильные производные ксантина применяются в
качестве лекарственных веществ. К ним относятся содержащие-
содержащиеся в зернах кофе и листьях чая растительные алкалоиды: ко-
кофеин, теофиллин и теобромин:
О О
N-CH3 H3C-N/ ^ NH
N
N N N N
Кофеин Теофиллин
A.3,7-триметил ксантин) A,3-диметилксантин)
310
Теобромин
C,7-диметилксантин)
Эти пуриновые алкалоиды оказывают стимулирующее дейст-
действие на центральную нервную систему и сердце. Кофеин приме-
применяется как тонизирующее средство, при сердечно-сосудистой не-
недостаточности, при мигрени. Он входит в состав многих комби-
комбинированных средств (аскофен, новомигрофен, новоцефальгин,
пирамеин, цитрамон). Получают кофеин метилированием тео-
филлина, а также из. растительного сырья. Теофиллин приме-
применяется для расширения сосудов при коронарной недостаточно-
недостаточности, а также бронхиальной астме. Используется он и как моче-
мочегонное средство. Теобромин применяется при спазмах сосудов
сердца и при отеках сердечного и почечного происхождения.
В настоящее время алкалоиды пуринового ряда получают
синтетическим путем. Общим методом получения веществ этой
группы является синтез Траубе:
Rx-NH
1
Ro-NH
Диалкилмо-
чеьина
С
II
НО О
\ //
с
| (СНзСОJО
Ниануксусная
кислота
О
II
с
Ri-N-7 \СН„ NaOH
| j
О=Сч C=N 50 с
NH
R2
Цианацетилдиалкил-
мочевина
С
II
с
Яг—n/ ^CMg Nai\o? ro Rx—n/ ^CH—NO zn, нсоон
O=C4 X=NH 8О'С O=Cv /C=NH 25-30 cc
N N/
I I
R2 Ro
1,3-Диалкил-2,6-Диоксо- 1,3-Диалкил-2,6-диоксо-
-4-нминопиримидин -4-имино-5-Ш1троюпиримидин
311
О О
II N
С С
^C—NHCHO нагрев Rr-N'' ^С NH
II |
N NN
к
1,3- Диалкил-2,6-диоксо- Диалкилксантнн
-4-амино-5-формиламинопиримидим
В числе производных шестичленных гетероциклов с двумя
гетероатомами, имеющих значение в медицинской практике, на-
находятся витамины групп Вь В2 и фолиевой кислоты.
-СН2—N —СН3 СГ(ВГ)
Витамин В! (тиамин)
ОН ОН ОН
I I I
СН2—СН—СН—СН—СН2ОН
нг N N
3N
о
Витамин В2 (рибофлавин)
ОН JO
Витамин В (фолиевая кислота)
Химический синтез этих сложных веществ осуществляется в
крупном промышленном масштабе. В связи с ограниченностью
объема курса технологии химико-фармацевтических препаратов
и антибиотиков программой не предусмотрено изучение техно-
технологии синтеза этих витаминов, поэтому мы ограничимся лишь
их общей характеристикой.
Витамин Bi содержится в дрожжах, зародышах и оболочках
пшеницы, овса, гречихи, а также в хлебе, изготовленном из му-
муки грубого помола. Он является составной частью кокарбокси-
лазы — кофермента, участвующего в углеводном обмене. По-
Поэтому недостаток витамина Bt в организме нарушает углевод-
углеводный обмен, что приводит к сердечным заболеваниям и заболева-
заболеваниям нервной системы. Витамин Bi оказывает влияние также
на белковый, жировой и водный обмен в организме.
312
Полное отсутствие в пище витамина Bi приводит к очень
тяжелому заболеванию — болезни бери-бери. При длительном
питании бедной этим витамином пищей возникает гиповитами-
гиповитаминоз, характеризующийся общим упадком сил, головными боля-
болями, бессонницей, болями в конечностях, расстройством сердеч-
сердечной и желудочно-кишечной деятельности. Витамин Bi приме-
применяют как лечебное средство не только при болезни бери-бери
и гиповитаминозе, но и при заболеваниях нервной системы, же-
желудочно-кишечного тракта, при стенокардии, некоторых кожных
заболеваниях и др.
Существует несколько методов синтеза витамина Bi, кото-
которые можно разбить на три группы. К первой группе относятся
методы, основанные на раздельном синтезе пиримидинового и
тиазолового компонентов с последующей их конденсацией:
.^V -А-<.Г1а(_Л Г"Ч-Т
Н,с-ЧА +
+ сг
CH,_N
Этот метод синтеза принят в СССР.
Другой путь получения витамина Bi заключается в постепен-
постепенном построении тиазольного цикла на предварительно синтези-
синтезированном пиримидиновом компоненте. Этот вариант синтеза ис-
используется, в частности, в японской промышленности. Третий
вариант — постепенное построение пиримидинового цикла на
тиазольном компоненте.
Витамин Bi (тиамин) выпускается как в виде хлорида (ти-
(тиамина хлорид), так и в виде бромида (тиамина бромид). И тот
и другой препарат выпускается в форме таблеток или драже и
в виде инъекционных растворов в ампулах. Кроме того, про-
промышленность производит дифосфорный эфир тиамина — кокар-
боксилазу. Для медицинского применения кокарбоксилаза вы-
выпускается в виде основания (тетрагидрат) и гидрохлорида:
СН2_м
Н Г/ ^NH Ч Jk II I
п^ N N 2 % ^СНа—СН9—О—Р—О—Р—О~.4Н«О (НС1)
I I
ОН ОН
Обе формы кокарбоксилазы аналогичны по действию. Биоло-
Биологические свойства кокарбоксилазы несколько отличаются от
свойств тиамина. Кокарбоксилазу вводят внутримышечно или
313
внутривенно при сахарном диабете, нарушениях сердечного рит-
ритма, недостаточности коронарного кровообращения, при некото-
некоторых формах склероза.
Витамин В2 (рибофлавин) широко распространен в расти-
растительном и животном мире. Он содержится в дрожжах, молочной
сыворотке, яичном белке, мясе, рыбе, печени, горохе, зароды-
зародышах и оболочках зерновых культур. Рибофлавин принимает уча-
участие в регулировании углеводного, белкового и жирового обме-
обмена. Витамин В2 играет также важную роль для зрения и для
синтеза гемоглобина. При недостатке витамина В2 в организме
возникает специфическое заболевание — арибофлавиноз, кото-
который выражается в поражении слизистой оболочки рта и глаз.
Рибофлавин влияет на рост птиц, при его недостатке наблюда-
наблюдается задержка роста.
Основными исходными веществами для синтеза рибофлави-
рибофлавина являются 3,4-диметиланилин, D-рибоза и барбитуровая кис-
кислота:
Н
I
НИН
—сн2—с—с—с—
I
он
но—сн2—с—с—с—с^ х
III хн Ч/nh
он он он г
о
3,4-Диметиланилин D-рибоза Барбитуровая
кислота
Метод получения рибофлавина сводится к получению 3,4-кси-
лил-6-фенилазо-1-1)-рибамина и конденсации его с барбитуровой
кислотой:
СН2ОН ,
I ^ н
НО— С—Н — —
I " н
НО—С—II , ~~
но-А-н он-с-н
NH О
о о
В разработке промышленного метода синтеза рибофлавина
большую роль сыграли работы проф. В. М. Березовского
(ВНИВИ). Рибофлавин выпускают в виде порошка, таблеток
и драже.
Для инъекций используют рибофлавина мононуклеотид (ри-
бофлавин-5-монофосфат натрия), который выпускается в виде
1% раствора в ампулах по 1 мл.
314
он он он о
III I!
СН2- СН- СН—СН—СН2—О- Р—ONa
N N n ОН
О
Рибофлавина мононуклеотид
Фолиевая кислота (витамин Вс, птероилглютаминовая кис-
кислота) является составной частью комплекса витаминов груп-
группы В. Она содержится в свежих овощах, а также в печени и
почках животных. Получается синтетическим путем. В организ-
организме человека синтезируется микрофлорой кишечника. Фолиевая
кислота является витамином, стимулирующим и регулирующим
процессы кроветворения. Кроме того, фолиевая кислота прини-
принимает участие в белковом обмене, катализируя синтез амино-
аминокислот. Фолиевую кислоту применяют для лечения различных
видов анемии, а также при заболеваниях крови, вызванных ра-
радиоактивным облучением.
В основе химической структуры витаминов группы фолие-
фолиевой кислоты лежит птеридиновое ядро, состоящее из пирими-
динового и пиразинового циклов.
N
N N
Птеридин
Птероилглютаминовая кислота является основным предста-
представителем этой группы веществ, содержащих остаток птеридина
и один или несколько остатков глютаминовой кислоты.
Получен сульфаниламидный аналог фолиевой кислоты, обла-
обладающий свойством понижать артериальное кровяное давление:
ОН
й—NH—^Л—SO2—NH—СН—СООН
СН2-СН2СООН
Фолиевая кислота выпускается в виде порошка и таблеток.
Часть III
ТЕХНОЛОГИЯ АНТИБИОТИКОВ
Глава 18
ТЕХНОЛОГИЯ БИОСИНТЕЗА АНТИБИОТИКОВ
И ПОЛУЧЕНИЯ НАТИВНОГО РАСТВОРА
С пояБлением первого антибиотика пенициллина началась
новая эра в медицине. Появилась возможность лечить многие
тяжелые заболевания.
В настоящее время антибиотики — наиболее активные сред-
средства современной химиотерапии инфекционных заболеваний.
С помощью антибиотиков многим людям спасена жизнь, у мно-
многих удалось достигнуть выздоровления или предупредить неред-
нередко опасные заболевания. Наряду с медицинской практикой
антибиотики широко применяются в других отраслях народного
хозяйства — в ветеринарии, животноводстве, сельском хозяйстве.
Они значительно уменьшают падеж молодняка (телята, ягнята,
поросята), способствуют повышению роста и массы птиц, разве-
разведению пушных зверей с высококачественным шерстяным покро-
покровом. Антибиотики широко используются в пищевой промышлен-
промышленности для консервирования различных продуктов. Как органи-
органические соединения они обогатили современную химию новыми
структурами, используя для изучения механизмов синтеза бел-
белков, нуклеиновых кислот и т. д. «Антибиотическими веществами
(антибиотиками) следует называть все продукты обмена любых
организмов, способные избирательно подавлять или убивать
микроорганизмы (бактерии, грибы, вирусы и др.)»- Из многих
известных определений это определение, данное М. М. Шемяки-
Шемякиным, А. С. Хохловым и др. A961), является самым общим и
наиболее правильно отражает роль и значение антибиотиков.
По химической природе антибиотики относятся к различным
классам химических соединений.
Антибиотики относятся к группе химиотерапевтических ле-
лекарственных средств, т. е. средств, действующих непосредствен-
непосредственно на возбудителей заболеваний. Эра антибиотиков началась с
открытия пенициллинов, применение которых оказалось чрезвы-
чрезвычайно успешным при лечении пневмонии, менингококкового ме-
менингита, сепсиса, ангин, гонореи, сифилиса и многих других за-
заболеваний, вызываемых грамположительными бактериями.
В медицинской практике пенициллины применяются в виде
инъекционных препаратов, кислотоустойчивые формы — внутрь
.316
в виде таблеток, реже как наружные средства в виде мазей.
Пеницилл'ины были открыты в 1940—1941 гг. и как лекарст-
лекарственные средства до сих пор не утратили значения. Получение
полусинтетических производных пенициллинов (оксациллин, ме-
тициллин, ампициллин и т. д.) значительно расширило область
их использования в медицине.
Несмотря на большие успехи, которыми ознаменовалось при-
применение пенициллинов, осталось значительное количество инфек-
инфекционных заболеваний, вызываемых грамотрицательными и кис-
кислотоустойчивыми бактериями, риккетсиями, грибами и виру-
вирусами. ^;
Однако уже в 1944 г. был описан стрептомицин, действую-
действующий на ряд грамотрицательных и кислотоустойчивых бактерий.
Особенно велико значение стрептомицина для лечения различ-
различных форм туберкулеза, в том числе туберкулезного менингита,
ранее приводившего к гибели практически всех больных.
Вслед за стрептомицином была выделена группа антибиоти-
антибиотиков полимиксинов с сильным избирательным действием на грам-
отрицателыше бактерии. Полимиксины до настоящего времени
широко применяются в медицинской практике, но только в каче-
качестве местных и внутриполостных средств. Более широкое исполь-
использование полимиксинов как общих средств для лечения заболе-
заболеваний, вызываемых грамотрицательными бактериями, ограничи-
ограничивается их высокой токсичностью.
Очень важным оказалось открытие в 1948—1950 гг. антибио-
антибиотиков широкого спектра действия — тетрациклинов (хлортетра-
циклин, окситетрациклин, тетрациклин). Они успешно
применяются в настоящее время главным образом в виде таб-
таблеток для лечения многих заболеваний, вызываемых грамполо-
жительными бактериями (пневмония, сепсис, менингит и др.),
а также многих инфекционных заболеваний, возбудителями ко-
которых являются грамотрицательные бактерии (чума, холера,
дизентерия, коклюш, туляремия, бруцеллез и др.), риккетсии
(сыпной тиф, марсельская лихорадка и т. д.) и некоторые круп-
крупные вирусы (трахома, вирусная пневмония, пситтакоз и др.).
Позднее, с появлением штаммов грамположительных бакте-
бактерий, устойчивых к пенициллинам, тетрациклинам, были выделе-
выделены новые антибиотики, действующие на резистентные штаммы
возбудителей заболеваний. Такими антибиотиками оказались
эритромицин A952), олеандомицин A954), новобиоцин A955)
и т. д. Вслед за указанными антибиотиками, а в некоторых
случаях и одновременно возникла группа антибиотиков, которая
применяется для лечения и профилактики кандидозов, вызывае-
вызываемых патогенными дрожжами, — нистатин A950), трихомицин
A952), амфотерицин В A956). Как противогрибковый препарат
находит некоторое применение гризеофульвин A959). Все выше-
вышеуказанные препараты антибиотиков назначаются в основном
внутрь в виде таблеток,
317
Помимо уже упомянутых соединений, для местного и внутри-
полостного применения, для лечения колибациллезов довольно
широко используются неомицины A949). Выпускаются они в
виде стерильных порошков во флаконах.
Все перечисленное свидетельствует о значительной эффектив-
эффективности антибиотиков при различных заболеваниях организма че-
человека.
Большинство антибиотиков получают путем биосинтеза с ис-
использованием микроорганизмов — актиномицетов, грибов, бак-
бактерий.
Основная задача процесса биосинтеза — подбор оптимальных
условий для развития продуцента и накопления антибиотика.
Биосинтез антибиотиков — двухфазный процесс. В течение пер-
первой фазы происходят быстрый рост и размножение мицелия или
бактериальных клеток. Культуральная жидкость в этот период
богата углеводами, азотом и неорганическим фосфором. Про-
Продукты обмена веществ микроорганизмов, в том числе антибио-
антибиотики, отсутствуют или находятся в крайне незначительных ко-
количествах.
Вторая фаза начинается с момента замедления роста культу-
культуры. Протекает она в культуральной жидкости, обогащенной
продуктами жизнедеятельности организма, с небольшим количе-
количеством углеводов и неорганического фосфора. В начале этой фа-
фазы мицелий обладает максимальной способностью к синтезу
антибиотика. Фазы отличаются по характеру и интенсивности
биохимических реакций. С учетом этих различий подбирают
условия, благоприятные для первой и второй фаз развития про-
продуцентов.
Одним из главных условий биосинтеза является обеспечение
продуцента всеми источниками питания — углеродом (С), азо-
азотом (N) и фосфором (Р). В качестве источников углерода чаще
всего используют углеводы. Источником азота могут быть раз-
разные вещества растительного и животного происхождения, со-
содержащие минеральный и органический N. Резкое различие в
условиях, благоприятных для роста продуцента, с одной сторо-
стороны, и в биосинтезе антибиотика — с другой, особенно четко про-
проявляется при добавлении в среду различных количеств фосфо-
фосфора. Оптимальное для биосинтеза количество фосфора зависит
от общей композиции среды, физиологических особенностей про-
продуцента, типа источника углерода и особенностей химической
структуры антибиотика. В последнее время все шире использу-
используются концентрированные среды. С повышением концентрации
питательных веществ в средах происходит увеличение биомас-
биомассы и возрастает потребность в кислороде. Очень важно, чтобы
в процессе биосинтеза максимально осуществлялся синтез
антибиотика и возникало как можно меньше побочных продук-
продуктов. Например, при биосинтезе тетрациклина на средах с добав-
добавлением йодидов, бромидов и, особенно, тиосоединений, подав-
318
ляется биосинтез хлортетрациклина, являющегося в данном
случае побочным продуктом. Уменьшение образования манози-
дострептомицина при биосинтезе стрептомицина добиваются
усилением аэрации. Процесс биосинтеза должен проходить так,
чтобы возникал преимущественно тот тип антибиотика, который
представляет больший интерес для медицинской практики. Это-
Этого добиваются введением в состав питательной среды так назы-
называемых предшественников. Например, введение фенилацетамида
при биосинтезе пенициллина способствует образованию пени-
пенициллина G, а феноксиуксусная кислота обусловливает синтез
пенициллина V. В целом введение при биосинтезе предшествен-
предшественника не только определяет и направляет синтез определенного
типа антибиотика, но и увеличивает выход.
§ 1. ПУТИ ИСПОЛЬЗОВАНИЯ МИНЕРАЛЬНЫХ
И ОРГАНИЧЕСКИХ АЗОТСОДЕРЖАЩИХ ВЕЩЕСТВ
МИКРООРГАНИЗМАМИ
Азот является постоянной составной частью мицелия грибов
и актиномицетов. В виде хитина он входит в состав оболочки
грибов. Из аминокислот состоят все белки. Содержится азот и
в составе пептидов, аминокислот, витаминов, ферментов, нук-
нуклеиновых кислот.
Количество азота в мицелии зависит от его содержания в
среде. При культивировании микроорганизмов на средах, обед-
обедненных азотом, количество его в мицелии составляет примерно
1%. При интенсивном использовании азота среды его количест-
количество в мицелии достигает 8%. Количество азотсодержащих компо-
компонентов в мицелии зависит от возраста культуры штамма и усло-
условий культивирования. Например, молодой мицелий Penicillium
chrysogenum (продуцент пенициллина) более богат белками,
чем старый. При старении количество небелкового азота обычно
повышается. Количество азота в мицелии продуцента, как пра-
правило, выше на средах без легкоуовояемых источников углерода,
т. е. при низком отношении N : С. При анализе белковых фрак-
фракций мицелия обнаружено, что количественный состав аминокис-
аминокислот белков меняется и зависит от условий культивирования,
фазы развития и штаммовой принадлежности. Наибольшее ко-
количество аминокислот приходится на аланин, серии, аргинин,
гистидин, глютаминовую кислоту (Actinomyces levoris). В на-
настоящее время еще нет обобщающих данных о связи между
аминокислотным составом белков продуцента и биосинтеза
антибиотика. В мицелии грибов и актиномицетов содержатся и
свободные аминокислоты. Количество их невелико у молодых
культур, а также уменьшается в момент интенсивного синтеза
антибиотика. На количественный и качественный состав свобод-
свободных аминокислот существенно влияет состав питательной среды.
Наибольшее воздействие оказывают органические кислоты, вхо-
319
дящйё в цикл дй- и трикарбоновых кислот. Наличие в среде
кетокислот, особенно пировиноградной и а-кетоглютаровой, при-
приводит к интенсивному синтезу внутри клетки соответствующих
аминокислот — а-аланина и глютаминовой кислоты. Углеводы
и аминокислоты менее заметно влияют на количественный со-
состав внутриклеточных свободных аминокислот. В процессе
голодания культуры, т. е. при недостатке азота в питательной
среде, но при достаточном содержании углеводов продуцент
использует азот свободных аминокислот.
В мицелии микроорганизмов содержатся также значитель-
значительные количества нуклеиновых кислот. Для микроорганизмов ха-
характерно высокое содержание нуклеиновых кислот по сравнению
с высшими организмами. Наибольшее количество нуклеиновых
кислот содержится в молодых культурах. Оба типа нуклеиновых
кислот (ДНК и РНК) обязательно присутствуют во всех без
исключения живых клетках и лишь вирусы содержат только
один из них (ДНК или РНК). Интенсивный синтез ДНК наблю-
наблюдается в лаг-фазе (т. е. в фазе подготовки клеток к делению).
В период интенсивного (логарифмического) и стационарного
роста культуры отмечается постоянство количества ДНК в
клетках. Содержание РНК подвержено более заметным колеба-
колебаниям. В лаг-фазе происходит интенсивный синтез РНК. Макси-
Максимальное количество РНК наблюдается в период наиболее ин-
интенсивного роста или непосредственно предшествует ему.
В периоды, когда скорость размножения клеток постоянна, со-
содержание РНК в расчете на одну клетку также приблизительно
постоянно.
Разнообразие и специфичность нуклеиновых кислот у микро-
микроорганизмов зависят от соотношения азотистых оснований, после-
последовательности расположения нуклеотидов, конфигурации макро-
макромолекулы. В различных типах ДНК может преобладать аденин
над гуанином и тимин над цитозином. Вследствие этого состав
ДНК у различных микроорганизмов может отличаться только
F+Ц /т. тт л
величиной отношения -дтгт (Г — гуанин, Ц — цитозин, А —
аденин, Т — тимин). Это отношение является показателем видо-
видовой специфичности. Видовая специфичность РНК не имеет та-
таких резко выраженных отличий, как ДНК, т. е. нуклеотидный
состав РНК у различных микроорганизмов близок. У всех изу-
изученных микроорганизмов в РНК преобладающим является
Г+Ц, т. е. показатель видовой специфичности РНК всегда
A+T ^A*
Очень важным вопросом процесса биосинтеза является обес-
обеспечение продуцента азотсодержащими компонентами питатель-
питательной среды с целью наибольшего выхода антибиотика.
Грибы и актиномицеты могут использовать органические и
неорганические азотсодержащие вещества. Из неорганических
320
источников азота чаще всего применяется сульфат аммония
(NH4JSO4. Он является наилучшим для биосинтеза окситетра-
циклина, стрептомицина и других антибиотиков. Нитраты ис-
используются реже. Из них чаще употребляется нитрат натрия —
NaNO3. Он хорошо усваивается продуцентом новобиоцина Act.
spheroides. Нитрат калия — KNO3 входит в состав среды для
Pen. nigricans (продуцент гризеофульвина). Основным источни-
источником азотного питания микроорганизмов являются азотсодержа-
азотсодержащие органические вещества: соевая мука, кукурузная мука, ку-
кукурузный экстракт, пептон, мясной экстракт и др. Учитывая
большие масштабы производства антибиотиков, экономически
целесообразно использовать соевую муку и кукурузный экстракт.
Соевая мука содержит в расчете на сухое вещество 40,5%
белков, 19,5% жиров, 29% безазотистых экстрактивных веществ,
5% клетчатки, 6% минеральных веществ.
Эти соотношения колеблются в зависимости от сорта сои,
места произрастания, способа обработки и хранения. Основной
белок соевой муки — глиценин. При гидролизе установлено на-
наличие в нем глицина,, валина, пролина, глютаминовой и аспа-
рагиновой кислот, аргинина, гистидина, лизина, триптофана, фе-
нилаланина. В безазотистых экстрактивных веществах обнару-
обнаружены: 1) углеводы, приблизительно 20%, из них в наибольшем
количестве сахароза, пентозаны — полисахариды, галактаны,
2) органические кислоты; 3) из минеральных веществ К, Na, Mg,
Fe, P, Са. В соевой муке содержатся витамины A, Bi, B2, С,
ферменты липаза, пероксидаза, каталаза, уреаза. Высокая
продуктивность питательных сред с соевой мукой связана с на-
наличием в ней больших количеств белка, жиров, ферментов и
витаминов. Исследования, проведенные с целью выяснить влия-
влияние отдельных составных частей соевой муки на антибиотикооб-
разование, показали, что, например, на биосинтез стрептомици-
стрептомицина наибольшее действие оказывают аминокислоты аргинин,
гистидин, лизин.
Другим часто используемым в промышленности источником
азотного питания является кукурузный экстракт. Его анализ
показывает следующее содержание (в расчете на сухое вещест-
вещество): общего азота 6,5—7,5%, в том числе азота белка 0,8—0,2%,
азота аминогрупп 2—2,5%, углеводов 6—10%, органических кис-
кислот 1,5—2,5%, минеральных веществ 2%.
В отличие от соевой муки кукурузный экстракт содержит зна-
значительно меньше белка. Азот включен в аминокислотные фраг-
фрагменты. Состав кукурузного экстракта колеблется в довольно ши-
широких пределах. Из составных частей кукурузного экстракта
наибольшее влияние на образование гризеофульвина оказывает
общее количество аминокислот и определенное соотношение
между свободными и связанными аминокислотами. Существен-
Существенным недостатком кукурузного экстракта является его нестан-
нестандартность. Это побуждает исследователей и производственников
21—926 321
искать другие азотсодержащие вещества. Например, в качестве
источника азотного питания может использоваться глютен —
отход крахмало-паточного производства. Он более стандартен,
богаче общим и белковым азотом, но намного беднее аминным
г.-;ите\? и неорганическим фосфором. В глютепе значительно
меньше витаминов. Как источники азотного питания могут ис-
использоваться экстракты хлопкового жмыха и жмыхов маслич-
масличных культур, например подсолнечного. По содержанию общего
и аминного азота они близки кукурузному экстракту.
В последние годы нашел применение препарат «фармамедия»
(желтоватый порошок), получаемый из зародышей семян хлоп-
хлопчатника. Его основной компонент — негидролизованный глобу-
глобулярный белок. Содержание белка в препарате колеблется от 55
до 60%. В его состав входит 18 аминокислот (лизин, гистидин,
аргинин, триптофан, аспарагиновая кислота, треонин и т. д.).
Препарат «профло» — частично обезжиренный порошок —
имеет такой же состав, как и предыдущий препарат, но в отли-
отличие от него относится к пищевым продуктам и содержит мень-
меньшее количество свободного госсипола.
Препарат «Профло ойл» — получают из семян хлопчатника
высшего качества. Содержание свободных жирных кислот в нем
не превышает 1%. Поскольку препарат представляет собой не-
неочищенное растительное масло, в его состав входят также ли-
пиды, фосфатиды, глицериды, токоферолы и другие вещества.
Препарат «фармамедия», добавляемый в среду в качестве
единственного или дополнительного источника азота, обеспечи-
обеспечивает рост различных микроорганизмов. Как показали экспери-
эксперименты и работа ряда предприятий, этот препарат обеспечивает
высокую и стабильную активность антибиотиков при их фермен-
ферментации. Препарат стандартен — выпускается в сухом виде.
Очень важно, чтобы в процессе ферментации выдерживалось
определенное соотношение N : С. Если в среде имеется избыток
аминокислот, то это угнетает синтез антибиотика не за счет
избытка азота, а вследствие того, что при дезаминировании на-
накапливается аммиак (его считают токсичным для продуцента).
Процесс дезаминирования нарушает диссоциацию нитрата ам-
аммония NH4NO3, и продуцент испытывает азотную недостаточ-
недостаточность.
В процессе биосинтеза количество азотсодержащих веществ
в среде изменяется по-разному у различных продуцентов, но об-
общим положением является уменьшение количества азота в пер-
первой фазе и увеличение во второй, когда начинается автолиз.
§ 2. ОСНОВНЫЕ ПУТИ ИСПОЛЬЗОВАНИЯ АЗОТА
МИКРООРГАНИЗМАМИ
Для того чтобы азот мог использоваться микроорганизмами,
его переводят в восстановленную форму. При добавлении в пи-
питательную среду нитратов происходит следующий процесс:
322
HNO3 —
Азотная
кислвта
—> HNO2
Азотистая
кислота
>• (HNOJ —
Гипонитрит
-> NH2OH —
Гидроксил-
амин
-*• NH,
Аммиак
Для осуществления этого процесса необходимо наличие фер-
фермента или ферментной системы, которая носит название нитрат-
редуктазы. Аммиак, получаемый в результате восстановления
нитратов или добавляемый в виде аммонийных солей, вступает
в реакцию с кетокислотами, образуемыми микроорганизмами из
углеводов. Этот путь аминирования кетокислот аммиаком пред-
представляет собой основной общий путь первичного построения ами-
аминокислот.
CHg-C-COOH H
6
hNHg —
->■ CH3—СН—СООН
NH2
Алании
У некоторых микроорганизмов обнаружен фермент глютаматде-
гидрогеназа, которая катализирует реакцию аминирования с
образованием глютаминовой кислоты:
НООС—СН2-СН2—С—СООН + NH3 >■ НООС-СН2-СН2—СН—СООН
II I
О NH2
У микроорганизмов, содержащих фермент аспартазу, аминиро-
вание происходит с образованием аспарагиновой кислоты:
НООС—СН=СН—СООН + NH8 >■ НООС-СН,—СН—СООН
NH2
При наличии у продуцента определенных энзиматических си-
систем синтез аминокислот может осуществляться также посредст-
посредством реакции переаминирования:
R—C—СООН + R'—СН—СООН +=± R—СН—СООН + R'—С—СООН
В 1 1 II
О NH2 NH2 О
Реакция переаминирования является обратимой.
Аминокислоты синтезируются с участием неорганических ис-
источников азота или образуют путем расщепления белков протес-
литическими ферментами.
В процессе биосинтеза происходит ряд ферментативных пре-
превращений аминокислот. В частности, они подвергаются дезами-
нированию. Основной путь окислительного дезаминирования —
обратная реакция по отношению к прямому восстановительному
аминированию.
21* 323
Вторым важным путем диссимиляции аминокислот является
декарбоксилирование, в результате которого образуются амины:
R—СН—СООН >- RCH2NH2 + СО2
NH2
У продуцентов антибиотиков обнаружено декарбоксилирова-
декарбоксилирование глютаминовой кислоты с образованием а- и у-аминомасля-
ной кислот. При биосинтезе стрептомицина наблюдается образо-
образование из гистидина гистамина и гистаминоподобных веществ.
N -~СН2—СН-СООН N _СН2—СН2-Ш
U и
U
N
А
Из минеральных азотсодержащих веществ в питательных
средах наиболее часто используют аммонийные соли серной, со-
соляной и азотной кислот. В случае, если в качестве источника
азота желательно иметь нитрат-ион, его вводят в виде нитрата
калия или натрия. Сульфат аммония оказывается пригодным
при биосинтезе почти всех антибиотиков. При биосинтезе хлор-
тетрациклина лучшим источником минерального азота оказался
хлорид аммония.
§ 3. ОСНОВНЫЕ ПУТИ МЕТАБОЛИЗМА УГЛЕВОДОВ
У ПРОДУЦЕНТОВ АНТИБИОТИКОВ
Углерод — постоянная составная часть протоплазмы, клеточ-
клеточной оболочки, ферментов и различных накапливающихся в клет-
клетках веществ. Углерод составляет 50% сухого веса мицелия.
В мицелии грибов и актиномицетов углеводы присутствуют в
виде полисахаридов, структура которых в подробностях неиз-
неизвестна. Продукты гидролиза полисахаридов состоят из большого
набора моносахаров. Глюкоза, манноза, галактоза содержатся
в наибольшем количестве, ксилоза, арабиноза, рамноза —
в меньшем. Содержание полисахаридов, а следовательно, и
моносахаров зависит от вида микроорганизма. Так, в полисаха-
полисахаридах штамма ЛС-1 (продуцент стрептомицина) больше манно-
зы, чем в полисахаридах штамма В-178. Из chrysogenum выде-
выделены два полисахарида: один состоит только из глюкозных
остатков, а в другом есть еще галактоза и манноза. Моносахара
входят в состав также тейхоевых кислот, которые найдены в
мицелии актиномицетов.
Известно несколько путей расщепления углеводов микроор-
микроорганизмами. Одним из них является гликолиз, который представ-
324
ляет собой совокупность анаэробных ферментативных процессов
распада глюкозы. Общая схема гликолиза представлена на
схеме.
сн0он снрн сн,он
-Он Н i—О / Фосфорилаза 1—0 Н
,он .X Хри ,Уу^=^ Кон
н-о
о
по
j| ■ он
О Н Гликоген
„ . СП-О-РО.Н
м?-+ . Л—о. ун
О=Р — ОН
он
О- Р-ОН
Off 6
Глюкозо-1-фосфат \
Фосфогексокина^а ^
ОН АТФ+- Oii
ОН
I 1юко:'.о () cbo^ct
Ф01 фогексо-
нчомераза
о=р-о-сн2 он
ОН 1^'°~^ Фруктозо— !,6- д
он
он
•]>р\мо-5о 0-фосфат
i\ -^.^ Трнозофосфаыегилрои-н;!.!.
I ГН ^S*»-.. /L! > И
Пировшюградная кис.
фвсфоглицеромутаз»
СН — О - Н
2 СН — О — РО..Н
С
Чон
ФосфоенолпировиноградНая 2—Фосфвглицеринввая
кисло!а кксл»1а
ОН О
I I!
2 СН —С —С—О-Н
Мелочная кислота
Характерной реакцией для этой схемы является расщепление
фруктозо-1,6-дифосфата на две триозы. В процессе этого превра-
превращения триоз из каждой молекулы триозы образуется две моле-
молекулы АТФ, одна из которых — в процессе превращения 1,3-ди-
фосфоглпцериновой кислоты до 3-фосфоглицериновой кислоты,
а вторая — в процессе превращения фосфоенолпировиноградной
кислоты в пировиноградную. Этот процесс называется фосфори-
лированием на уровне субстрата.
Имеются и другие пути расщепления углеводов. В 40—
50-х годах XX века был открыт гексозомомофосфатный, пентоз-
ный цикл. Его можно представить в виде следующей 'схемы.
325
Промежуточный ферме
НАДФ
носи 9
MC-J . Н2С01
^ ^^ носи
Г.1юкозо-С-фосфаг "НОСИ
^люкозофосфа! - НСОН
изомераза ^ НСОН
Фруктозодифосфатаза А,,,,,, 2" с3, , ,.2 з
• Л ФРУК' 030-6- фосфат Зрщрозо-4—фосфа i
ieoi;
поен неон
iic=o пеон неон
неон ПоСоро н,соро7'"
НСОН ^ Ксщу.юзо Г) -фосфат Рибо/юю 1-фосфат
Тра иске io,ia за
(|иамин11нрофосфа|)
Фрук гозодифосфат
НСОН I
I НОСИ
н2соро3- нсон
Глицеральде1ид—3- ,,Inu
фосфат Н(Г0Н
U НСОН
н2сон -- '
Н СОРО - - СедогептУлозо~7 —фосфат
чДиоксиацетонфосфат
Обмен глюкозо-6- фосфата по пентозофосфатному nyin
Продукты расщепления этого цикла принимают участие в
биосинтезе антибиотиков. Продуцент тетрациклина Act. aureofa-
ciens осуществляет расщепление углеводов питательной среды
по гексозомонофосфатному циклу.
Процесс диссимиляции углеводов у продуцента олеоандоми-
цина Act. antibioticus на первых этапах расщепления Сахаров
осуществляется через пентозный цикл, а с момента окисления
фосфотриоз в фосфоглицериновые кислоты в нем принимает
участие гликолитический цикл обмена. Продуцент эритромици-
эритромицина Act. erythreus не имеет фермента фосфофруктокиназы, обяза-
обязательного для гликолитического пути обмена углеводов.
Путь расщепления углеводов зависит от источника, исполь-
используемого для питания и развития продуцента. Например, проду-
продуцент неомицина Act. fradiae на среде с крахмалом после предва-
предварительного расщепления его до глюкозы в дальнейшем осуще-
осуществляет ее распад по пентозному циклу. Если же в среду
добавлять не крахмал, а глюкозу, то ее расщепление происходит
в основном по гликолитическому циклу. В зависимости от ис-
исходного углевода меняется антибиотическая продуктивность (на
среде с крахмалом — 2000 мкг/мл, на среде с глюкозой —
460 мкг/мл).
Путь, по которому происходит распад глюкозы в культурах
326
актиномицетов, может зависеть от наличия в питательной среде
некоторых компонентов. При высоких концентрациях фосфора
подавляется активность глюкозо-6-фосфатдегидрогеназы и рас-
распад глюкозы осуществляется преимущественно по пути ана-
анаэробного гликолиза. Высокие концентрации неорганического
фосфора тормозят расщепление углеводов и их усвоение. Рода-
Роданистый бензил угнетает распад глюкозы по пути анаэробного
гликолиза и активирует ее диссимиляцию по пентозному циклу.
При изучении трансформации глюкозы и пировиноградной кис-
кислоты грибами и актииомицетами — продуцентами антибиотиков
в среде обнаружены продукты цикла трикарбоновых кислот.
Оказалось, что цикл трикарбоновых кислот является главным
руслом, по которому происходит окисление основной части ком-
компонентов среды. Именно посредством этого цикла продукты об-
обмена углеводов, жиров и аминокислот окисляются до воды и уг-
углекислого газа. Ферменты, катализирующие реакции цикла три-
трикарбоновых кислот, имеются у многих микроорганизмов. Ряд
микроорганизмов могут использовать в качестве углерода дву-
углеродные соединения, например ацетаты. В этом случае отме-
отмечаются разновидности цикла трикарбоновых кислот. Например,
цикл глиоксиловой кислоты:
СН*—СООН
СН2—СООН снзсоок I
| >■ НО—С—СООН
СО-СООН |
СН.—СООН
Щавелевоуксусная
кислота
НО
СН2—СООН
|—СН—С
-СООН
Яблочная
кислота
Лимонная
кислота
сн2—соон
С—СООН
II
сн-соон
Цис-аконитовая
СН2—СООН
СН—СООН
НО—СН—СООН
Изолимонная кислота
О
|| СН2-СООН
СН—СООН + I
СН2-СООН
Глиоксиловая Янтарная
кислота кислота
327
Особенностью этого цикла являются две реакции, в резуль-
результате которых изолимонная кислота превращается в глиоксило-
вую при участии изоцитратлиазы и далее в яблочную кислоту
с помощью малатсинтазы. Остальные ферменты аналогичны
ферментам цикла Кребса.
В процессе культивирования микроорганизмов к «культураль-
ной жидкости накапливается много органических кислот. Коли-
Количество их зависит от вида используемых углеводов, степени
аэрации и физиологических особенностей продуцента. При раз-
развитии Act. aureofaciens (продуцент ТЦ и ХТЦI значительное
количество органических кислот образуется между 12 и 24 ч ро-
роста и снижается с уменьшением содержания углеводов в пита-
питательной среде. В культуре одного из продуцентов пенициллина
накапливаются значительные количества щавелевой кислоты,
что связывают с наличием в культуральной жидкости уксусной
кислоты.
О
II
СН2-ОН СН СООН
I I I
СНз-СООН > СООН - СООН > СООН
Гликолевая Глиоксило- Щагелевая
кислота вая кислота кислота
Имеется и другой путь образования щавелевой кислоты — че-
через янтарную и фурмаровую кислоты.
§ 4. ХАРАКТЕРИСТИКА ИСТОЧНИКОВ УГЛЕРОДА,
ПРИМЕНЯЕМЫХ В ПРОИЗВОДСТВЕ АНТИБИОТИКОВ
И ВЛИЯНИЕ ИХ НА БИОСИНТЕЗ
Углеводы являются одной из важнейших составных частей
сред для выращивания микроорганизмов. Продуценты антибио-
антибиотиков, как и многие микроорганизмы, могут потреблять различ-
различные углеводы, но наиболее часто в промышленности используют-
используются глюкоза, крахмал, гидрол, меласса.
Крахмал. Крахмал на 96—97% состоит из полисахаридов.
В нем содержится 0,2—0,7% минеральных веществ (в основном
фосфор). Обнаружены высшие жирные кислоты (пальметиновая,
стеариновая 0,6%). Углеводная часть крахмала состоит из ами-
амилазы — линейного полимера глюкозы и аминопектина — разветв-
разветвленного полимера глюкозы. Под действием ферментов а- и
р-амилазы крахмал расщепляется до мальтозы, а мальтоза даль-
дальше расщепляется с участием а-глюкозидазы до глюкозы.
Гидрол. В состав гидрола — отхода крахмало-паточного про-
производства входит до 50% глюкозы. Кроме нее имеется около
18% несбраживаемых Сахаров, представляющих собой продукты
неполного расщепления крахмала и вторичной полимеризации
1 Здесь и далее приняты сокращения: ТЦ — тетрациклин и ХТЦ — хлор-
тетрациклин.
328
глюкозы. В гидроле содержится некоторое количество органи-
органических кислот. Гидрол имеет рН~4,0, зольность не более 7%.
Основная часть зольных элементов — это хлорид натрия, обра-
образующийся при нейтрализации содой соляной кислоты, применяе-
применяемой для гидролиза крахмала, фосфор, магний, железо. Другие
минеральные элементы присутствуют в гидроле в минимальных
количествах. По внешнему виду гидрол представляет собой гус-
густой темный сироп с характерным запахом. Он является нестан-
нестандартным сырьем и должен проверяться в контрольных фермен-
тациях.
Меласса. Меласса — отход свеклосахарного производства.
Состав ее зависит от качества свеклы и методов переработки.
Средний состав мелассы: воды 18—25%, сухих веществ 75—
82%, в том числе сахарозы 45—50%. Из других органических
веществ в мелассе имеются кислоты (муравьиная, щавелевая,
янтарная, уксусная и пировиноградная, меланиноподобные ве-
вещества — продукты конденсации Сахаров и аминокислот, азоти-
азотистые соединения. Из минеральных веществ в мелассе содержатся
азотнокислые, сернокислые, хлористые, углекислые соли калия,
натрия, магния, железа, аммония. В небольших количествах в
состав мелассы входят фосфор, алюминий, медь.
Лактоза. В производстве пенициллина используют в качест-
качестве углеводного питания дисахарид — лактозу. Она содержится
в молоке и вырабатывается из молочной сыворотки, получаемой
при производстве сыров, творога и казеина. Сыворотку освобож-
освобождают от белков, упаривают до получения сиропа и в таком виде
используют при биосинтезе пенициллина. Лактоза-сырец содер-
содержит не менее 92% сахара и не более 3% воды, 2% золы и 1%
молочной кислоты; количество белков не превышает 2%.
При использовании концентрированных питательных сред в
них необходимо вводить повышенное количество различных ком-
компонентов. Компоненты питательной среды, являющиеся отхода-
отходами производства, подвергаются предварительной очистке (на-
(например, на ионообменных смолах). Такая очистка при производ-
производстве стрептомицина позволяет частично заменить глюкозу гид-
ролом.
Эффективность использования источников углерода обычно
оценивается экономическим коэффициентом, выражающим соот-
соотношение между массой получаемой биомассы и массой исполь-
использованного углевода. Микроорганизмы с высокой биохимической
активностью превращают приблизительно половину углеводов,
содержащихся в среде, в составные части клеток. В лаборатор-
лабораторных условиях этот коэффициент значительно ниже. Углерод, не
использованный на синтез клеточного материала, превращается
в углекислоту и промежуточные продукты, в том числе антибио-
антибиотики. В производстве стараются создать такие условия, при ко-
которых возможно большее количество углерода превращалось бы
в нужный продукт и как можно меньшее количество расходова-
расходовалось на синтез биомассы и углекислый газ.
329
На различных углеводных компонентах питательной среды
антабиотикообразование идет по-разному. У продуцентов суще-
существует определенная избирательность по отношению к углеводу,
входящему в состав среды. Антибиотики стрептомицин, ниста-
нистатин, мономицин образуются в большем количестве в присутствии
глюкозы. Для окситетрациклина, ТЦ, ХТЦ и неомицина более
благоприятны среды с крахмалом. В некоторых случаях более
высокий выход антибиотиков получали на средах со смесью глю-
глюкозы и крахмала (окситетрациклин, олеандомиции). При оценке
результатов влияния углеводов на синтез того или иного анти-
антибиотика необходимо учитывать штамм продуцента, его специ-
специфичность к углеводам и особенности метаболизма, обеспечиваю-
обеспечивающие накопление в среде нужного продукта. Очень важна общая
композиция среды. Одни и те же углеводы в синтетической и
комплексной среде могут дать совершенно разный выход анти-
антибиотика. Например, галактоза различными продуцентами стреп-
стрептомицина на соевой среде используется на 95%, а на синтетиче-
синтетической— на 15—25 или 90% в зависимости от штамма. Биосинтез
флоримицина на среде с сульфатом аммония более интенсивен
в присутствии глюкозы, чем крахмала. При замене сульфата
аммония нитратом натрия повышается выход флоримицина на
среде с крахмалом.
Различие в биосинтезе антибиотика обусловлено изменением
в обмене веществ в присутствии углеводов. Например, мицелий
Act. fradiae, выросший на среде с глюкозой, содержал почти
в 2 раза больше азота и значительно больше нуклеиновых кис»
лот, чем мицелий, выросший на крахмальной среде.
Биосинтез стрептомицина на среде ЛС-1 с глюкозой состав-
составлял 3440 мкг/мл, а на среде с крахмалом — всего 370 мкг/мл.
Низкую активность антибиотикообразования на среде с крах-
крахмалом, по-видимому, можно объяснить накоплением пировино-
градной кислоты. При оценке целесообразности использования
того или иного компонента среды важно помнить о продолжи-
продолжительности ферментации и следует стремиться к ее сокращению.
В случае очень интенсивного потребления углевода, как на-
наблюдается, например, при биосинтезе пенициллина (глюкоза
очень быстро окисляется продуцентом), в среду одновременно
с глюкозой добавляется лактоза — углевод, который окисляется
медленнее. Это позволяет обеспечивать структурным материа-
материалом построение молекулы пенициллина.
Образование антибиотиков при наличии различных углево-
углеводов в среде происходит неодинаково. Так, при биосинтезе стреп-
стрептомицина глюкоза потребляется более интенсивно, чем крахмал,
а антибиотикообразование выше на среде с крахмалом
(рис. 35, 36). На биосинтез антибиотиков в присутствии различ-
различных углеводов влияет стерилизация питательной среды. В жест-
жестких условиях стерилизации A,5—2 атм) в средах происходят
химические изменения, углеводы могут взаимодействовать с
330
мкг/мл
1000
96 120
Рис. 35. Потребление различных угле-
углеводов при культивированию Act. strep-
tomycini.
/ — глюкоза; 2 — крахмал; 3 — мальтоза;
4 — лактоза; 5 — сахароза.
2*+ 48 72 96 120
ч'асы
Рис. 36. Образование стрептомици-
стрептомицина в присутствии различных Саха-
Сахаров. Обозначения те же, что и на
рис. 35.
аминокислотами, солями аммония. В связи с этим желательно
проводить стерилизацию углеводов в более «мягких» условиях
и отдельно от других компонентов питательной среды.
§ 5. УЧАСТИЕ МИНЕРАЛЬНЫХ ВЕЩЕСТВ
В МЕТАБОЛИЗМЕ ПРОДУЦЕНТОВ АНТИБИОТИКОВ
Минеральные вещества наряду с белками и углеводами вхо-
входят в состав биомассы продуцента; наличие их в питательной
среде необходимо. Изучая влияние минеральных компонентов
среды на биосинтез антибиотиков, нужно помнить о том, что
мы всегда вносим в среду значительное и часто неучтенное их
количество. Минеральные элементы вносятся с компонентами
питательной среды. При спектральном анализе установлено на-
наличие минеральных ионов в некоторых реактивах. Ниже показа-
показано наличие минеральных примесей в некоторых реактивах.
Реактив Обнаруженные примеси
NH4NO3 Na, Mg, Ca
КН2РО4 Al, Pb, Na, Ca, Mg, As
MgSO4.7H2O Na, Cu
ZnSO4-7H2O Fe, As, Mg, Ca, Si, Na, Mn
CuSO4-5H2O Fe, Mn, Sn, Mg, Pb
Глюкоза Zn, Mg, Na, Sr, Ca, Fe, K, Mn, Al, Pb, Ni, Ag, Cu, Bi, Si
331
Элементами минерального питания, необходимыми для раз-
развития микроорганизмов в относительно больших количествах,
считаются Mg, Ca, S, К. Они должны присутствовать в пита-
питательной среде в концентрации 10~3—10~4 м/л. Элементы Fe, Си,
Zn, Mn и некоторые другие требуются в микроколичествах
A0~6—10~8 м/л). Значительное количество ионов содержится в
воде.
Физиологическое значение минеральных элементов различно.
Одним из их существенных свойств является влияние на физи-
физико-химическое состояние коллоидов протоплазмы. Под дейст-
действием ряда неорганических ионов поверхностный слой клетки
подвергается изменениям, которые сказываются в целом на
обмене веществ. Так, в присутствии хлорида натрия в среде при
биосинтезе стрептомицина изменяется проницаемость клеточной
мембраны, в результате чего антибиотик более легко переходит
из мицелия в культур альную жидкость.
Влияние микроэлементов на обмен веществ вызвано способ-
способностью их входить в состав энзиматических систем или активи-
активировать их, а от ферментных систем зависят скорость и направле-
направление биохимической реакции. В клетках растений, животных и
микроорганизмов многие органические соединения образуют
комплексы с металлами, которые отличаются по ряду свойств, в
том числе по устойчивости. Сами ионы металлов обладают ката-
каталитической активностью в отношении многих реакций, в частно-
частности ускоряют некоторые реакции в биологической системе.
Действие ионов металла обусловлено его свойствами, свойст-
свойствами ферментного белка и в целом свойствами комплекса фер-
фермент— металл.
Оптимальная концентрация минеральных веществ зависит от
ряда факторов: 1) цели культивирования (получение биомассы
или антибиотика); 2) наличия и соотношения других минераль-
минеральных элементов в питательной среде; 3) реакции питательной сре-
среды; 4) наличия хелатирующего агента; 5) количества посевного
материала.
В зависимости от цели культивирования можно изменить гра-
границы концентраций минеральных элементов. Концентрация, ко-
которая способствует накоплению биомассы, часто отличается от
той, которая необходима для образования антибиотиков. Напри-
Например, концентрация меди, обеспечивающая хороший рост проду-
продуцента, токсична для процессов образования пенициллина, а кон-
концентрация железа, необходимая для максимального образования
пенициллина, почти в 100 раз превышает концентрацию, которая
требуется для нормального роста продуцента.
Макро- и микроэлементы в питательной среде действуют
комплексно. Так, для действия ионов меди важно содержание
ионов железа. При повышении концентрации меди для опти-
оптимального ведения процесса необходимо повышение концентра-
концентрации и железа, поскольку оно как антагонист меди снижает ее
332
токсичность при высоких концентрациях. Оптимальное соотно-
соотношение меди и железа для различных микроорганизмов неодина-
неодинаково. Наилучший рост Aspergillum niger происходит при их
соотношении 1 : 4. Однако не только железо является антагони-
антагонистом меди. Например, в культурах дрожжей токсичность меди
снижается добавлением марганца. Оптимальное соотношение ме-
меди и железа зависит от реакции среды. Наибольшее снижение
токсичности наблюдается при рН 6-^-7. В кислой среде железо
не только не уменьшает, но даже увеличивает токсичность Си.
Границы оптимального уровня микроэлементов в среде можно
расширить добавлением хелатообразующих агентов. Это имеет
особое значение для развития молодых культур на синтетиче-
синтетических средах, поскольку в таких средах неуравновешенность эле-
элементов оказывается сильнее. По мере роста микроорганизмов
в среду выделяются различные продукты жизнедеятельности,
которые сами могут образовывать хелаты. Естественные суб-
субстраты (соевая мука, кукурузный экстракт), входящие в состав
питательной среды, обычно содержат все необходимые микро-
микроэлементы. В них входят также комплексообразующие соедине-
соединения, что устраняет проявление токсичности повышенного фона
металла. В связи с этим максимальный предел концентраций в
комплексных средах всегда выше, чем в синтетических.
§ 6. ЗНАЧЕНИЕ ОТДЕЛЬНЫХ ЭЛЕМЕНТОВ МИНЕРАЛЬНОГО
ПИТАНИЯ В ЖИЗНЕДЕЯТЕЛЬНОСТИ МИКРООРГАНИЗМОВ
Сера. Сера играет большую роль в структуре клеток, входит
в состав белков в виде серосодержащих аминокислот (цистин,
цистеин, метионин), в состав двух витаминов (биотин, тиамин)
и КоА1.
Источник серы в среде — неорганические сульфаты. В моле-
молекулу сера включается в восстановленном состоянии и, следова-
следовательно, она проходит ряд превращений:
SO4 " -» SO~ » SO2" > S 2О~ ~ > SH"
Тиосульфат — наилучший источник серы при биосинтезе пе-
пенициллина. Изменение количества серы в среде приводит к из-
изменению физиологических процессов в мицелии продуцента, а
также уровня образования пенициллина в культуральной жид-
жидкости. Сера является антагонистом фосфора при их совместном
воздействии на мицелий гриба. При повышенных концентрациях
фосфора в среде наблюдается развитие гиф мицелия, не проду-
продуцирующих пенициллин, а при добавлении к среде серы в избыт-
избытке наибольшее количество биомассы составляет продуктивный
мицелий.
1 КоА — кофермент ацилировалия—производное р-.меркаптоэтил амида
пантотеиовой кислоты.
333
Магний. Магний необходим для процессов окисления. Веду-
Ведущая роль его связана с гликолитическим циклом расщепления
углеводов. Магний активирует ферменты, деполимеризующие
мета- и пирофосфаты, а также АТФ у продуцента пенициллина
при рН 6,0. Магний необходим для биосинтеза стрептомицина.
Он стимулирует рост продуцента, способствует более полному
и быстрому использованию азотистых компонентов среды. По-
Потребность в магнии составляет 0,01—0,05% (в расчете на
MgSO4).
Кальций. Основная роль кальция заключается в нейтрализа-
нейтрализации выделяющихся органических кислот и поддержании кислот-
кислотности среды. Ионы Са+2 могут связывать фосфор. Кальций акти-
активирует ряд ферментов, например липазу. При наличии кальция
в среде наблюдается снижение лизиса некоторых бактериаль-
бактериальных клеток. Термоустойчивость бактериальных спор связана с
наличием в спорах дипиколиновой кислоты, которая в процессе
прорастания спор полностью исчезает. Ионы кальция играют
важную роль в синтезе этой кислоты и тем самым связаны с
термостабильностью спор.
Железо. Ионы железа относятся к микроэлементам и играют
каталитическую роль в жизнедеятельности микроорганизмов.
Недостаток или избыток железа в среде приводит к нарушению
тех или иных сторон метаболизма. Избыток железа тормозит
биосинтез стрептомицина и антибиотиков группы тетрациклина.
Оптимальной концентрацией железа для биосинтеза стрептоми-
стрептомицина является концентрация 0,005—0,25% (в расчете на FeSO4).
При больших концентрациях задерживается рост продуцента,
ухудшается потребление углеводов, фосфор среды связывается
в нерастворимый комплекс. Угнетающее действие железа на
биосинтез окситетрациклина связывают с присутствием в среде
жиров. Установлено, что в среде с различными маслами .его
угнетающее действие выражается более ярко, если масла содер-
содержат большое количество ненасыщенных жирных кислот (на-
(например, подсолнечное масло). Предполагают также, что угне-
угнетающее действие железа обусловлено образованием из органи-
органических кислот органических перекисей.
Цинк. Ион цинка влияет на углеводный, азотный и фосфор-
фосфорный обмен. В присутствии цинка возрастает экономический ко-
коэффициент, т. е. увеличивается отношение веса сухих веществ
мицелия к количеству усвоенных углеводов. Наличие цинка в
среде способствует биосинтезу ряда антибиотиков: стрептомици-
стрептомицина, пенициллина, неом'ицша и т. д. Количество необходимого
цинка в среде для биосинтеза стрептомицина зависит от среды
и штамма и колеблется в пределах 0,001—0,0001 % (в расчете на
ZnSO4). Недостаток цинка в среде замедляет биосинтез нео-
мицина.
Марганец. Марганец входит в состав многих ферментных си-
систем, в первую очередь кокарбоксилазы. Он стимулирует синтез
334
стрептомицина, пенициллина и других антибиотиков. Cthmj iii-
рующее действие на биосинтез стрептомицина связано с тем,
что марганец снижает угнетающее действие самого антибиотика
на дыхание продуцента.
§ 7. ЗНАЧЕНИЕ КИСЛОТНОСТИ И ДРУГИХ ХАРАКТЕРИСТИК
СРЕДЫ ДЛЯ ЖИЗНЕДЕЯТЕЛЬНОСТИ ПРОДУЦЕНТОВ
Кроме источников питания, для микроорганизмов необходи-
необходимы определенные физико-химические показатели окружающей
среды. В частности, большое значение для развития микроорга-
микроорганизмов имеет кислотность среды (рН). От нее зависят поступ-
поступление питательных веществ из среды в клетку, деятельность
ферментов, биосинтеза антибиотика. Известно, что для биосин-
биосинтеза пенициллина оптимальное значение рН 6,8—7,5, для стреп-
стрептомицина — рН 7—8,5. При рН ниже 6,0 уменьшается биосинтез
окситетрациклина. В процессе неуправляемого биосинтеза вели-
величина рН среды может существенно меняться. Это зависит от
вида микроорганизма, состава питательной среды. Например,
при неуправляемом биосинтезе пенициллина рИ среды повыша-
повышается, при биосинтезе стрептомицина — снижается. На изменение
рН в процессе питания влияют источники углерода и азота.
Высокие концентрации глюкозы в среде приводят к увеличению
кислотности среды за счет образования органических кислот.
Закисление сред наблюдается и при интенсивном потреблении
азота. Большинство продуцентов актиномицетов хорошо растут
при рН 6,5—7,5, но каждый продуцент имеет свой оптимум рН.
Так, для (Act. spheroides) оптимальным является рН 7,1—7,7;
при рН^80 происходит образование биологически неактивных
форм новобиоцина.
Регулирование кислотности среды производится с помощью
мела. Регулируя кислотность среды, мел влияет и на процесс
антибиотикообразования (табл.3).
Таблица 3
Влияние мела на биосинтез окситетрациклина (ОТЦ)
рН в конце
процесса
7,0
6,6
4,8
4,4
4,0
СаСО3, %
0,8
0,6
0,4
0,2
ОТЦ, мкг/мл
2360
2150
1390
285
24
Масса мицелия,
1,20
1,12
1,04
0,72
0,51
Неиспольчоганные
у г легоды, %
0,47
0,50
0,61
1,25
2,06
Уменьшение рН среды можно объяснить образованием боль-
большого количества органических кислот. Величина рН среды в
процессе ферментации должна способствовать синтезу фермеы-
335
тов, обеспечивающих синтез метаболитов, необходимых для
антибиотикообразования.
Регулировать рН среды можно также дробной подачей глю-
глюкозы в питательную среду. Например, разработан метод дроб-
дробной подачи глюкозы при биосинтезе гризеофульвина. В основу
метода положен принцип сохранения оптимального значения рН
в каждый данный момент ферментации. Использование глюкозы
или других компонентов питательной среды для регулирования
рН не всегда целесообразно, так как программа подачи глюко-
глюкозы как источника питания часто не соответствует использованию
ее как титрующего агента. Для регулирования рН среды ис-
используют также аммиак и углекислый газ. С их помощью про-
происходит очень мягкое изменение рН среды в небольших преде-
пределах. Вместо газообразного аммиака применяют аммиачную во-
воду. СО2 снижает эффективность аэрации и оказывает ингиби-
рующее действие на дыхание микроорганизма и следовательно
на биосинтез антибиотика. В производственных условиях для
регулирования рН среды используется иногда щелочь или
кислота.
Существенное значение на развитие микроорганизмов имеет
изменение окислительно-восстановительного потенциала (гН2).
Эта величина характеризует кислородно-водородное равновесие
в системе. Если рН среды указывает на соотношение между
концентрацией Н+ и ОН~ ионов, то гН2— между Н2 и О2. Кроме
того, гН2 характеризует суммарное окислительно-восстанови-
окислительно-восстановительное состояние, обусловленное не только концентрацией
(парциальное давление) кислорода и водорода, но и всеми при-
присутствующими окислителями и восстановителями. При исследо-
исследовании обменных процессов у грибов (Asp. niger) установлено,
что изменяя окислительно-восстановительные условия, можно
получить различные продукты метаболизма. Например, культуру
Asp. niger выращивали на синтетической среде при доступе О2,
а затем помещали в среду с N2 и Н2. При этом гН2 был равен 17,
углеводы окислялись до СО2 и Н2О. При помещении культуры
в атмосферу с N2 rH2 был равен 12—14. Культура при этом син-
синтезировала этиловый спирт. Ход изменения окислительно-восста-
окислительно-восстановительного потенциала у микроорганизмов имеет в основном
одинаковый характер. В первые часы роста в культуре обнару-
обнаруживается высокое значение rH2 (Pen. chrysogenum 10—15), за-
затем происходит снижение потенциала, и в конце ферментации
гН2 составляет 5ч-6. При биосинтезе стрептомицина у одного из
актиномицетов обнаружена зависимость между величиной гН2
и накоплением антибиотика.
Способность микроорганизмов к дыханию сопровождаемся
снижением гН2 в процессе развития. Это обусловлено специфи-
специфическими обменными процессами. При развитии микроорганиз-
микроорганизмов в культуральную жидкость выделяются восстановители,
снижающие потенциал. Так, продуцент тетрациклина Act.
336
aurecfaciens при развитии на среде с глицерином выделяет в
культуральную жидкость диоксиацетон. Как и многие другие ве-
вещества, имеющие одновременно карбонильные и спиртовые
группы, диоксиацетон в щелочной среде способен к образованию
этиленднольной группировки.
СН2-СО—СН2
I I
ОН
Такие этилендиолы были названы редуктонами. Они обла-
обладают очень сильными восстановительными свойствами. Биоло-
Биологический смысл образования восстановителей сводится к прояв-
проявлению у микроорганизмов приспособительного обмена. Восста-
Восстановители, легко связывая О2, тем самым предохраняют клетку
от избытка воздуха и таким образом регулируют доступ Ог в
клетку. Такое регулирование является оправданным, так как
чрезмерное аэрирование неблагоприятно для микроорганизмов.
Ряд ферментных систем активен только в восстановленном со-
состоянии (например КоА). Исследование изменения гНг при био-
биосинтезе позволяют сделать заключение о необходимости опреде-
определенных окислительно-восстановительных условий для микроор-
микроорганизмов. Знание этих условий открывает большие возможности
для регулирования процесса биосинтеза антибиотиков. Воздейст-
Воздействовать на окислительно-восстановительный потенциал можно,
меняя аэрацию или добавляя в питательную среду окислители
или восстановители. Это позволяет регулировать биосинтез
биологически активных соединений. Так, добавление в среду
восстановителя гипосульфита в определенных количествах поз-
позволяет увеличить выход витамина Bi2. При биосинтезе леворина
добавление гипосульфита позволяет снизить синтез леворина А
и повысить выход леворина В.
§ 8. БИОСИНТЕЗ ПЕНИЦИЛЛИНА
К пенициллинам относится группа близких по химическим
свойствам соединений, содержащих в своей структуре р-лактам-
ное и тиазолидиновое кольца.
Биосинтез пенициллина изучался с использованием меченых
соединений. На основании этих исследований высказано пред-
предположение, что при введении в питательную среду цистина, он
22—926 337
восстанавливается в цистеин, который, вероятно, и обеспечивает
синтез тиазолидинового кольца молекулы пенициллина. Из дру-
других аминокислот, которые обеспечивают структуру тиазолиди-
новой части молекулы, является валин. В ходе дальнейших ис-
исследований механизма биосинтеза пенициллина из мицелия Реп.
chrysogenum был выделен трипептид, состоящий из цистеина,
валина и 2-аминоадипиновой кислоты. В другой серии работ
показано, что при добавлении в среду меченого дипептида цис-
теинил-валина гриб использует его для построения пеницилли-
нового ядра без предварительного расщепления пептидной свя-
связи. Вероятно, этот дипептид образуется в мицелии в результате
реакции транспептидации и далее превращается в трипептид, ко-
который затем подвергается конденсации с образованием системы
пенициллинового бицикла.
Для получения определенного типа пенициллина необходимо
введение в среду предшественника. Синтез бензилпенициллина,
например, осуществляется при введении фенилуксусной кислоты
или фенилацетамида. Предшественники значительно стимули-
стимулируют общий выход пенициллина. С помощью их можно в 2 раза
и больше увеличить выход антибиотика.
Биосинтез молекулы бензилпенициллина можно представить
следующим образом:
СООН
HS—СН2—СН—NHa + НООС— СН2—С6Н5 >■
"—Н2О
1-Цистеин Фенилуксусная кислота
СООН
HS—СН2-СН— NHCOCH2QH5
ноос-с-он
(-Н2О)
CHo-
en/
Диметилпировиноградная кислота
НООС—С—NH—СО
СНз\ || CH-NH-CO-CH2-C6H5
СН3
SH
НООС-СН—NH—СО
/С СН2-СН—NH-CO—СН2~С6Н5
СНз \s/
338
НООС—СН—N СО
—2H
| |
С СН—СН—NH-CO-CH2—С6Н5
\ /
Бензилпенициллин
1000
10
Процесс биосинтеза пенициллина складывается из трех ос-
основных стадий: 1) выращивания посевного материала в аппара-
аппаратах малой емкости — инокуляторах; 2) выращивания посевного
материала в больших посевных аппаратах; 3) собственно фер-
ферментации.
Основной задачей при культивировании продуцента в посев-
посевных аппаратах является быстрое накопление большого количе-
количества мицелия, который обеспечивал бы достаточное антибиоти-
кообразование. Для этого применяют среды, богатые легко
усваиваемыми питательными компонентами. В качестве источни-
источника углерода используют глюкозу с добавлением трудно усваи-
усваиваемой лактозы, чтобы продуцент приспособился к этому источ-
источнику углерода еще на посевной среде. Из источников азота при-
применяют кукурузный экстракт, соли аммония. Необходимо также
добавление источников Р, К,
Mg, S и микроэлементов Mg,
Си, Fe.
Инокулятор засевают сухи-
сухими спорами Pen. chrysogenum.
Выращивание производят до
третьей стадии (примерно
50 ч). Подготовленный иноку-
люм передают в посевной ап-
аппарат, где мицелий выращива-
выращивают на третьей стадии A2—
18 ч), и затем в ферментатор.
Ферментацию проводят на сре-
средах с глюкозой и лактозой. На-
Наличие лактозы в среде в тече-
течение всей ферментации создает
благоприятные условия для
синтеза пенициллина. Вместо
лактозы можно добавлять лег-
легко и быстро усваиваемые угле-
углеводы (глюкоза, сахароза, гид-
ролизованный крахмал, галак-
юо
Рис. 37. Химическое изменение куль-
туральной жидкости при ферментации
пенициллина Pen. chrysogenum. Чис-
Числовые величины по оси ординат даны
тоза), но подача Сахаров в этом слева для следующих показателей:
пенициллин (ЕД/мл); аз
случае должна осуществляться
непрерывно. Например, глюко-
глюкозу добавляют со скоростью
у
пенициллин (ЕД/мл); азот мицелия
монийный азот (мг/10 мл).
22*
339
0,032% в час. Выход пенициллина получают выше на 15%, если
используют кукурузную среду, и на 65% —если синтетическую.
Химические изменения в культуральной жидкости представ-
представлены на рис. 37. Процесс ферментации можно разделить на две
фазы. Первая из них — фаза роста мицелия. В этот период ин-
интенсивно потребляется глюкоза, антибиотик образуется слабо.
Во второй фазе мицелий растет медленно, постепенно исполь-
используется лактоза, образование пенициллина достигает максимума.
Вторая фаза должна быть достаточно продолжительной. Если
же ферментация слишком продолжительна, а интенсивность об-
образования антибиотика при этом мала, то это экономически не-
нецелесообразно. Об окончании процесса ферментации судят по
исчезновению углеводов из питательной среды и прекращению
образования антибиотика. Для образования определенного типа
пенициллина в среду добавляют предшественник: для бензилпе-
нициллина фенилацетамид или фенилуксусную кислоту. Фенил-
уксусная кислота окисляется с первых же часов ферментации,
поэтому ее присутствие необходимо в течение всего процесса
ферментации. Предшественник желательно добавлять дробно,
так как большая концентрация его в среде оказывает токсиче-
токсическое действие на продуцент.
§ 9. БИОСИНТЕЗ ТЕТРАЦИКЛИНОВЫХ АНТИБИОТИКОВ
Продуцентом тетрациклина и хлортетрациклина является
Act. aureofaciens. В нашей стране ХТЦ имеет торговое название
биомицин, а за рубежом — ауреомицин и дуомицин. Синонимы
ТЦ — тетрациклин, стеклин, ахромицин, полициклин и т. д. ОТЦ
известен также под названием террамицин, продуцентом его
является Act. rimosus. В основе структуры тетрациклиновых
антибиотиков лежит полифункциональное гидронафтаценовое со-
соединение. ХТЦ является 7-хлорпроизводным, ОТЦ — 5-оксипро-
изводным. Синтез циклических соединений происходит с уча-
участием уксусной кислоты путем альдольной конденсации или
С-ацилирования (конденсация Кляйзена) по следующей схеме:
—зн2о
R-COOH + СН3СООН + СНдСООН + СН3СООН >
4-2Н
R—СОСН2СОСН2СОСН2СООН > RCOCH2CHOHCH2COCH2COOH
R COR R COOH
^l/COOH HO^ /i /OH J. /COOH I
ААон , и кЛ
он
340
При конденсации большего числа молекул уксусной кислоты
образуются более сложные производные.
В синтезе тетрациклиновых антибиотиков принимает участие
ацетил-КоА, -но в реакцию конденсации вступает его карбокси-
лированное производное — малонил-КоА. При использовании
меченой уксусной кислоты в качестве источника питания под-
подтвердилась справедливость приведенной выше схемы. В пользу
ацетатной теории синтеза тетрациклиновых антибиотиков гово-
говорит также подавление синтеза этих антибиотиков арсеннтами,
ингибирующими процесс превращения пировиноградной кислоты
в уксусную (т. е. в присутствии арсенитов не образуется уксус-
уксусной кислоты). Некоторые промежуточные этапы синтеза пока
не установлены. В культуральной жидкости обнаружены вещест-
вещества следующего строения:
СН, ОН
о2
CONH2
ОН ОН ОН
ОН ОН ОН ОН
6-Метил-1,3,10,11.12-пентагидрокси-
нафтацен-2-карбоксамид
он
он он о о
С1 СН
lnh3]
! 6н
он он 6 о
CONH2
С1 СН
О
341
окислительное
гидроксилирование
6-Ангидр#-7-хлортетрациклин
СНд СНд
ООН СНд N
II II II ^
[Н]
он о о о
5а,11а-Дегидр»-7-хлортетрациклин
он|
он о он о
хлортетрациклин
Представленная схема — сложный биохимический процесс.
Источником метальных групп служит метионин, обычно участ-
участвующий в реакциях метилирования в живых организмах. В виде
какого соединения вступает в реакцию хлор, неизвестно.
Было высказано предположение, что синтез тетрациклиновых
антибиотиков может происходить по пути, которому необходимы
хинная A,3,4,5-тетраоксициклогексанкарбоновая) и 3,4,5-триок-
сициклогексен-B)-карбоповая кислоты:
O и
он
342
coon
j
OH
На самом деле синтез тетрациклииов в их присутствии уве-
увеличивается, но показано, что в процессе синтеза они не участ-
участвуют. Ферментация ХЦ осуществляется в средах, в состав кото-
которых входят широко применяемые источники питания: кукуруз-
кукурузный экстракт, жмых, крахмал, сахароза. Использование глюко-
глюкозы сокращает цикл культивирования, но и уменьшает антибио-
тикообразование. В последнее время используют среды с 5—6%
крахмала. Для уменьшения вязкости питательной среды ее об-
обрабатывают оризииом, который частично расщепляет крахмал,
в результате чего последний легче усваивается. Введение ма-
масел для пеиогашепия должно быть ограничено, так как быст-
быстрое их расщепление может привести к накоплению в среде не-
непредельных жирных кислот, ингибирующих синтез тетрацик-
линов.
При развитии культуры в ферментаторе в первые 10 ч про-
происходит интенсивное накопление пировиноградной кислоты, нук-
нуклеиновых кислот, почти полностью используется фосфор. 'В те-
течение первых 2 сут потребляется крахмал. Биосинтез ХТЦ начи-
начинается в тот период, когда в среде наблюдается минимальное
количество источников азота и крахмала. В первые 2 сут рН
среды снижается с 7,2—6,6 до 6,8—6,1, затем постепенно в про-
процессе ферментации увеличивается и в конце достигает 8,0.
С целью активации ферментов пентозного цикла в среду добав-
добавляют роданистый бензил.
§ 10. БИОСИНТЕЗ ТЕТРАЦИКЛИНА
Биосинтез ТЦ осуществляется тем же продуцентом на пита-
питательной среде без ионов хлора. Процесс освобождения среды
от ионов хлора может осуществляться несколькими способами,
но возможно проведение ферментации на обычных средах, не
освобожденных от хлора, а с добавлением веществ, подавляю-
подавляющих биохимический процесс хлорирования. С этой целью в среду
добавляют бромиды и йодиды, но наиболее полное подавление
хлорирования наблюдается при добавлении в среду тиосоедине-
ний, чаще всего 2-меркаптобензтиазола (каптакс):
SH
34 3
Действие тиосоединений основано на том, что они связывают
медь, входящую в состав ферментов (оксидаз), ответственных
за процесс хлорирования. Предполагают также, что тиосоеднне-
ния способствуют более активному протеканию окислительно-
восстановительных реакций. В этих условиях ионы хлора уда-
удаляются из среды раньше, чем они вовлекаются в молекулу анти-
антибиотика.
В процессе биосинтеза ТЦ в культуральной жидкости может
образоваться 2-ацетил-2-декарбоксамидотетрациклин (АДТ).
Это биологически малоактивная примесь, которая ухудшает ка-
качество конечного продукта.
ом
АДГ
Синтез АДТ зависит от концентрации азота в ферментационной
среде. Отмечено также, что количество АДТ увеличивается при
высоких концентрациях каптакса (или других тиосоединений).
Роданистый бензил, добавляемый в среду, увеличивает выход
ТЦ на 35%, а биосинтез АДТ в 4—6 раз.
В соответствии с механизмом образования ТЦ синтез ТЦ и
АДТ расходится на первых этапах, а именно при синтезе мало-
нил-КоА.
§ 11. БИОСИНТЕЗ ОКСИТЕТРАЦИКЛИНА
Биосинтез ОТЦ проводят на средах, аналогичных по составу
средам, используемым для получения других тетрациклиновых
антибиотиков. Продуцентом является Act. rimosus. В первой фа-
фазе биосинтеза происходит интенсивный рост первичного мицелия
с гомогенной и базофильной протоплазмой с большим содержа-
содержанием белка и фосфорных соединений. Во второй фазе после рас-
распада первичного мицелия начинается прорастание обрывков
мицелия. Эти гифы мицелия второй генерации составляют ос-
основную массу мицелия до конца развития. Вторичный мицелий
вырастает в иных условиях среды, чем первичный. Он имеет сла-
слабо базофильную протоплазму с меньшим содержанием нуклеи-
нуклеиновых кислот и особенно РНК. Переход мицелия из первой
фазы во вторую связан с содержанием фосфора в среде и на-
наступает при полном его использовании. Изменяя концентрацию
фосфора в среде, можно регулировать соотношение первой и вто-
второй фаз. Продлить фазу антибиотикообразования можно, добав-
добавляя в питательную среду по ходу ферментации источники угле-
углерода и азота. По времени процесс биосинтеза окситетрациклина
более продолжителен, чем ХТЦ и ТЦ. Для синтеза ОТЦ суще-
3 44
стЕеныое значение имеет исходный рН среды и рН в ходе фер-
ферментации. Оптимальное значение рН в начале ферментации
g5 7, а в период биосинтеза — 6,0—6,8. В процессе фермента-
ферментации рН среды регулируют добавлением мела или аммиачной
воды.
Важным фактором для биосинтеза является аэрация культу-
культуры. Особенно велика потребность в Ог при интенсивном росте
первичного мицелия. Вторичный мицелий дышит менее интен-
12
Рис. 38. Динамика биохимических изменений в культуре Act. rimosus ЛСТ-П8
на синтетической среде.
/ — окситетрациклин; 2 — вес сухого мицелия; 3 — углеводы; 4— аммонийный азот;
5 — фосфор минеральных соединений; 6 — рН; 7—скорость накопления окситетрациклина.
сивно и потребление в Ог снижается. Ухудшение аэрации в боль-
большей степени влияет на образование антибиотика, чем на рост
биомассы. Кислород при антибиотикообразовании необходим
для гидроксилирования молекулы у 5-го и 6-го атома. В связи
с этим высокоактивные штаммы, продуцирующие ОТЦ, нужда-
нуждаются в лучшей аэрации, особенно на средах с большим процен-
процентом основных компонентов.
В условиях, благоприятных для синтеза ОТЦ, кроме него,
образуется до 12% 2-ацетил-2-декарбоксамидо-окситетрацикли-
на (АДОТ). Он менее активен, чем ОТЦ, в 10 раз. Предпола-
345
гают, что образование АДОТ связано с тем, что нарушается
процесс введения карбоксамидной группы. Одним из условий,
препятствующих этому процессу, является недостаток источни-
источников аминогрупп. Содержание АДОТ зависит также от количест-
количества в среде фосфора и кашалотового жира. Возможно, избыток
фосфора и кашалотового жира стимулирует рост продуцента и
создает благоприятные условия для большего использования
аминокислот и аминов в синтезе белков в ущерб биосинтезу
антибиотика. В промышленных условиях снижение количества
АДОТ достигается путем добавления в питательную среду боль-
больших количеств сульфата аммония. Динамика биохимических
изменений культуры Act. rimosus штамм ЛСТ-118 на синтетиче-
синтетической среде представлена на рис. 38.
§ 12. БИОСИНТЕЗ СТРЕПТОМИЦИНА
Биосинтез стрептомицина осуществляется с помощью проду-
продуцента Act. Streptomycini или Act. globisporys streptomycini, а за
рубежом — Streptomyces griseus.
Исследованиями Кароу показано, что меченый углерод глю-
глюкозы, добавленной в питательную среду, равномерно распреде-
распределяется между стрептозной и Ы-метил-Ь-глюкозамином; в гуани-
диновых остатках обнаруживаются лишь следы. Следовательно,
скелет стрептомицина происходит из углеводов питательной сре-
среды. Гуанидиновые группы молекулы антибиотика в большей
степени обеспечивают биологическую активность стрептомицина.
Удаление гуанидиновых группировок из молекулы антибиотика
или их превращение приводит к полной потере биологической
активности. При гидролизе стрептомицина в кислой среде моле-
молекула антибиотика распадается на два биологически неактивных
вещества — стрептидин и стрептобиозамин. Последний представ-
представляет собой дисахарид, в котором остаток стрептозы гликозидной
связью соединен с М-метил-Ь-глюкозаминам. В дигидрострепто-
мицине в стрептозной части молекулы содержится не альдегид-
альдегидная, а оксиметильная группа.
Стрептомицин
346
Наряду со стрептомицином при биосинтезе образуется манно-
зидстрептомицин. Он отличается от стрептомицина наличием
D-маннозы. Туанидиновые группы синтезируются при биосинте-
биосинтезе из соединений с большим отношением N : С. В некоторых
соединениях отношение N : С в 10 раз больше чем в белках.
В связи с этим образование стрептидиновых группировок стиму-
стимулируют такие соединения, как аргинин, гуанидин, мочевина
(в белках отношение N:C составляет 0,31, в аргинине — 0,78,
в гуанидине — 3,0, в мочевине — 2,0). Процесс, при котором об-
образуются гуанидиновые группы,
сводится к ферментному переносу амидиновых групп от моле-
молекулы донора к молекуле акцептора (процесс называется транс-
амидированием):
C^ rf R'NH2 > R'NH-Cf + RNH2
Донором амидиновой группы является аргинин. Предпола-
Предполагают, что акцептором этого процесса служит аминированный в
1-м и 3-м положении инозит или фосфорилированное производ-
производное инозита.
Как принято считать, ядро стрептидина образуется из глю-
глюкозы. Инозит, составляющий ядро молекулы стрептидина, явля-
является циклическим 6-атомным спиртом с той же эмпирической
формулой, что и глюкоза. Стимулирующее действие ряда комп-
комплексных сред (соевая мука и т. д.) способствует образованию
антибиотика за счет содержания инозита. Природные метаболи-
метаболиты инозита — хинная и шикимовая кислоты — также увеличи-
увеличивают биосинтез антибиотика. Имеются и другие гипотезы отно-
относительно образования инозита.
Основным материалом для биосинтеза этой группы антибио-
антибиотиков является глюкоза. Предполагают, что образование анти-
антибиотика происходит из глюкозы без разрыва цепочки. Одним из
промежуточных продуктов является глюкозамин. Отмечается
связь между накоплением глюкозамина и синтезом стрептоми-
стрептомицина. Метильная группа образуется при участии донатора ме-
метальной группы метионина. Стрептоза образуется из глюкозы.
Единой точки зрения по этому вопросу нет. Возможно, стрепто-
стрептоза образуется из глюкозы через ряд нуклеотидов.
Таким образом, для процесса ферментации стрептомицина
большое значение имеет углеводный состав питательной среды.
В качестве углеводного питания в промышленных условиях
чаще всего применяются глюкоза и крахмал. Выбор углевода
зависит от используемого штамма. Почти все штаммы продуцен-
продуцентов стрептомицина могут использовать животные жиры и расти-
347
тельные масла. Жиры способствуют увеличению биомассы, за-
замедляют использование углеводов, ускоряют потребление источ-
источников азота. Путь использования жиров, как и у большинства
микроорганизмов, — расщепление жиров на жирные кислоты и
глицерин, с последующим р-окислением жирных кислот. Из ор-
органических кислот стимулирующее действие на образование
стрептомицина оказывают молочная, пировиноградная и лимон-
лимонная кислоты.
Продуцент стрептомицина относится к микроорганизмам, об-
обладающим высокой протеолитической активностью. Это значит,
что он может расти на средах с белковым азотом (соевая мука,
жмыхи и т. д.). Белки гидролизуются до аминокислот и исполь-
используются продуцентом. Для биосинтеза стрептомицина аминокис-
аминокислоты неравноценны и по действию делятся на три группы:
1) аминокислоты, не влияющие на рост микроорганизмов, но
стимулирующие биосинтез антибиотика (аргинин, гастидин, ли-
лизин, в меньшей степени глицин, аланин, валин, фенилаланин);
2) аминокислоты, не оказывающие влияния на образование
антибиотика (аспарагин, серии, треонин, тирозин);
3) аминокислоты, подавляющие рост продуцента и тормозя-
тормозящие биосинтез антибиотика (триптофан, цистин).
Из неорганических источников азота для биосинтеза благо-
благоприятные соли аммония. Невозможность использования нитра-
нитратов связана с отсутствием у продуцента доноров водорода. При-
Присутствие нитрата в среде, содержащей кукурузный экстракт,
приводит к изменению всего процесса обмена, а концентрация
KNO3 0,25% препятствует вовлечению в обмен молочной кисло-
кислоты кукурузного экстракта. При концентрации KNO3 0,5% молоч-
молочная кислота используется интенсивно. При дальнейшем увели-
увеличении концентрации KNO3 в 2 раза продуценту приходится само-
самому синтезировать молочную кислоту.
Важное значение для продуцента стрептомицина имеет фос-
фосфор. При его недостатке замедляется использование углеводов,
азота, замедляется рост. При повышенном содержании фосфора
в мицелии появляется митохондрий, потребление углеводов уси-
усиливается, образование стрептомицина подавляется. Концентра-
Концентрация фосфора 0,04—0,07 мг/мл является оптимальной.
Различают две фазы развития продуцента стрептомицина:
1) интенсивный рост актиномицета, интенсивное использова-
использование основных компонентов среды, образование некоторого коли-
количества молочной кислоты, появление гомогенного мицелия с ба-
зофильной протоплазмой и высоким содержанием РНК;
2) более медленное потребление питательных веществ, тор-
торможение или прекращение синтеза РНК, снижение базофилии.
Основная масса клеток автолизируется. Обмен веществ в фазах
происходит по-разному. В молодых клетках в 2—3 раза больше
нуклеиновых кислот, содержание которых и определяет направ-
направление обмена веществ продуцента.
348
§ 13. ПРИГОТОВЛЕНИЕ И СТЕРИЛИЗАЦИЯ
ПИТАТЕЛЬНОЙ СРЕДЫ
Существенную роль в процессе биосинтеза играют приготов-
приготовление и стерилизация питательной среды. В зависимости от ком-
компонентного состава питательной среды методы приготовления
несколько отличаются. При приготовлении и стерилизации пи-
питательной среды очень важно сохранить в неизмененном виде ее
исходный состав. Чаще всего в промышленных условиях компо-
компоненты среды растворяют или суспендируют в подогретой воде
в одном аппарате или раздельно, а затем смешивают в аппара-
аппарате-смесителе с добавлением воды до требуемого объема. Как
правило, сначала готовят концентрат питательной среды, состав-
составляющий треть общего объема среды.
Для приготовления сред используют обычные цилиндриче-
цилиндрические аппараты с плоским или сферическим днищем. Тамие ци-
цилиндры снабжены рубашкой, змеевиком или просто барботером
для подогрева глухим или острым паром. Аппараты обеспечены
перемешивающим устройством и устройством для передачи сре-
среды !на стерилизацию.
Приготовленную питательную среду подвергают стерилиза-
стерилизации. Процесс стерилизации необходим для того, чтобы избавить-
избавиться от микроорганизмов, находящихся в питательной среде. При-
Применяют различные методы: ионизирующее излучение, ультра-
ультрафиолетовые лучи, ультразвук, рентгеновы лучи, обработку сред
химическими соединениями (окись этилена, перекись водорода,
р-пропиолактон и т. д.). Метод с нагреванием нашел промышлен-
промышленное применение в нашей стране.
Стерилизацию нагреванием можно осуществлять периодиче-
периодически и непрерывно. В настоящее время в отечественной промыш-
промышленности используются о'ба этих метода.
Периодический метод стерилизации в аппарате. Стерилиза-
Стерилизацию среды осуществляют подачей пара во все штуцеры аппара-
аппарата. Образующийся конденсат смешивают со средой. Среду на-
нагревают до определенной температуры и в зависимости от со-
состава выдерживают при этой температуре 30—40 мин. Затем
среду охлаждают водой, подаваемой в рубашку аппарата.
Метод отличается 'простотой, надежностью, аппарат стерили-
стерилизуют одновременно со средой. Недостатком является продолжи-
продолжительность пребывания среды при высокой температуре. Углево-
Углеводы при этом подвергаются карамелизации, что снижает качество
питательной среды. На среде, простерилизованной периодиче-
периодическим методом, выход пенициллина уменьшается на 10%. Второй
недостаток — повышенный расход пара. Периодический процесс
труднее подается автоматизации.
Непрерывный метод стерилизации. Непрерывный метод сте-
стерилизации питательной среды осуществляется в колоннах не-
непрерывного действия. Конструкции этих колонн разнообразны.
349
Рис. 39, /. Колонна для
стерилизации питатель-
питательной среды.
/ — корпус; 2 — внутренняя
труба; 3 — шнек; 4 — штуцер
для выхода среды; 5 — шту-
штуцер нижнего спуска; 6—
зентиль; 7 — штуцер для
ввода среды
В нашей промышленности нашли применение две конструкции
(рис. 39, /«//).
I. Пар подается сверху во внутреннюю трубу со щелевидны-
ми отверстиями, через которые он поступает в питательную сре-
среду. Питательная среда движется во внешней трубе навстречу
пару по спирали за счет винтовых направляющих. В результате
происходит быстрый и эффективный нагрев среды.
Для уменьшения гидравлических ударов и лучшего раство-
растворения компонентов питательной среды ее предварительно подо-
подогревают в смесителе до 70—80 °С.
II. Несколько по-иному выглядит вторая конструкция стери-
лизационной колонны. Ввод среды в ней осуществляется через
центральную трубу, пар же подается сбоку через кольцевое про-
пространство. Внутренняя труба имеет большое количество малых
отверстий, через которые пар поступает в среду. Среда быстро
350
Bwd среды
k----j--—•--1
Выход среды
Пар
099
Сеч. АОА
Рис. 39, //. Колонна для стерилизации питательной среды,
патрубок; 2 — распределитель пара; 3 — фланец; 4 — тройник; 5 — крышка; 6 — рас-
рассеивающий зонт; 7 — корпус колонны.
/Ърячая
дода
Питательная
среда на фер
ментацию
. ty I
Рис. 40. Технологическая схема стерилизации питательной среды.
— чан для приготовления питательной среды; 2 — иасос; 3 — колонка для стерилизации питательной среды; 4 — выдер-
живатель-аппарат; 5 — выдерживатель-теплообменник; 6 — холодильник.
и интенсивно нагревается с помощью рассеивающего зонта, по-
попадает в корпус колонны и затем — в выдерживатель. Режим
стерилизации зависит от состава питательной среды. Температу-
Температура пара, поступающего на стерилизацию, обычно поддерживает-
поддерживается около 5 ат.
При непрерывном методе.стерилизации среды концентрат пи-
питательной среды проходит через систему аппаратов, состоящих
из нагревателя, выдерживателя (аппарат для выдержки среды
Секцил
нагрева
Секцил
выдержки
двухтрубный хп
С w .
лластинчагь/й
с
Секция ох-
охлаждения
пластинчать/й
испаритель-
испарительная камера
Рис. 41. Различные типы секций непрерывных стерилизаторов.
с — стерилизуемая среда; п — пар; к — конденсат; в — вода.
при температуре стерилизации) и теплообменника для охлажде-
охлаждения среды до температуры ферментации. Технологическая схема
стерилизации среды приведена на рис. 40.
В качестве нагревателя среды могут служить аппараты раз-
различной конструкции, выполненные в виде колонны, парового
инжектора, пластинчатого или двухтрубного теплообменников.
Нагревательные аппараты должны обеспечивать быстрый на-
нагрев среды до нужной температуры. Из представленных на рис.
41 конструкций в этом плане наиболее удачной является паро-
паровой инжектор. Он обеспечивает быстрый нагрев среды, в нем
в отличие от нагревательной колонны отсутствуют гидравличе-
гидравлические удары и другой недостаток колонны — засорение отверстий
барботера осадками из питательной среды. Препятствием для
использования в качестве нагревателя теплообменников являет-
является трудность удаления из них отложений осадков солей.
23—92в
353
Основное значение выдерживателя — постоянство длитель-
длительности пребывания в нем каждого элементарного объема среды.
Выдерживатели могут быть емкостные и трубчатые. В емкост-
емкостном выдерживателе длительность пребывания питательной сре-
среды при постоянной объемной скорости протекания жидкости че-
через аппарат равна длительности заполнения аппарата. В вы-
держивателях такого типа из-за продольного смешения струй
длительность выдержки различна для разных частей жидкости.
Для устранения этого недостатка в аппаратах выдержки под
штуцером подачи среды устанавливают конические или сфери-
сферические отражатели, предусматривают установку сегментных по-
полок, подают питательную среду по касательной к стенке аппа-
аппарата или сверху вниз по трубе, расположенной по оси аппа-
аппарата.
При относительно высоких температурах стерилизации, ког-
когда емкость выдерживателя измеряется десятками литров, целе-
целесообразно использовать трубчатые выдерживатели. В них легко
регулировать объемную скорость и, следовательно, длительность
выдержки. Режим движения жидкости в трубе должен быть
развитым турбулентным (Re=104—106). Турбулентный ре-
режим в трубчатом выдерживателе улучшает теплоотдачу, что
позволяет проводить стерилизацию при более высокой темпе-
температуре.
Выбор теплообменника для охлаждения стерильной пита-
питательной среды обусловливается прежде всего максимальным со-
сохранением ее стерильности.
Расчет выдерживателя. Конструкция выдерживателя долж-
должна обеспечивать постоянство длительности пребывания в нем
каждого элементарного объема среды, содержащего одну спо-
спору. Для определения объема выдерживателя требуется знать
время выдержки, которое позволило бы обеспечить стерильность
выходящей из него среды. Время выдержки фактически равно
времени заполнения:
Время выдержки вычисляется по уравнению:
2,3 , N6
Т___ ■■ |гГ ■
ВЫД. ~~" IT lS \T 9
где No — исходное число микроорганизмов; N0 = C0' Уж-106;
Vm — объем стерилизуемой среды в м3; Со — концентрация мик-
микроорганизмов в 1 мл; обычно Со принимается равной
1700—2000 микроорганизмов в мл; N — вероятность выживания
микроорганизмов в стерильной среде; обычно микроорганизмы
выживают только в 1—3 из 100 операций стерилизации среды,
т. е. N=0,01—0,03; К—константа скорости отмирания микро-
354
организмов; определяется по температуре стерилизации среды
из таблицы1.
После нахождения времени выдержки (тВыд.) находят объем
выдерживаТеля:
v ВЫД. — КСр. ' Ь
где Топер. — время операции; Vcp.—объем питательной среды
после нагревательной колонки; к — коэффициент заполнения
выдерживателя.
Пример. Задано: Со = 2000 микроорганизмов в 1 мл, #=0,01, F,K=7 M3-Fcp.=
= VjK-f Уконденсата пара. ПринИМ-аеМ FCp. раВНЫМ 8 М3. (VCp. МОЖНО расСЧИ-
тать точно, зная продолжительность нагрева среды в нагревательной колонке»
объем среды до нагревательной колонки — Уж, температуру среды до и после
нагрева, параметры греющего пара.) Топер.=90 мин. & = 0,9.
'Р/асоадотрим цдеа варианта: I — *стерил. = 125°, II—*стерил.= 140°. Рассчи-
Рассчитать объем выдерживателя.
Решение
Вариант I A25°)
2,3 20007.10е _t
При 125° из таблицы К=4,57 мин-1. тВыд.= 4 57 g (ГШ =Ь,1мин.
8-7
Округляя в сторону увеличения, получаем Твыд. « 7 мин. Увы д. = о 9-90 ^
=0,69 м3. Округляя, получаем 7Выд. « 700 л.
Вариант II A40°)
2,3 2000-7-10е
При 140° из таблицы К « 104,8 мин". тВыд. ~-ю4 g '8 q~OT— :
Округляя в сторону увеличения, получаем Твыд'^1 мин.
=0,099 м3. Округляя, получаем Увыд. « 100 л.
Из известных конструкций теплообменников наибольшей
герметичностью обладают теплообменники типа «труба в тру-
бе». Несмотря на их громоздкость и недоступность внутренней
поверхности для механической очистки накипи, а также повы-
повышенный расход металла на 1 м2 теплообменной поверхности,
они находят самое широкое применение в промышленности.
Обычные кожухотрубные теплообменники не используются из-
за большого числа труб в трубных решетках и, следовательно,
опасности заражения питательной среды микроорганизмами из
охлаждающей воды.
Компактен и эффективен пластинчатый теплообменник. Он
отличается хорошей герметичностью, высоким коэффициентом
теплопередачи и большой поверхностью теплообмена. Он легко
разбирается для очистки. Имеется возможность регенерации
в нем 70—80% теплоты нагретой воды. Применение столь же эф-
эффективных спиральных теплообменников невозможно из-за пло-
плохого качества уплотнений между торцами листов и крышками,
1 «Микробиологическая промышленность», 1970, № 4.
23* 355
Стерилизация среды при значительной температуре A35°С
и выше) требует снятия избыточного давления после выдерж-
выдержки. Иногда это делается с помощью испарительной камеры. В
испарительной камере жидкость вскипает, давление уменьша-
уменьшается, и уже со сниженным давлением среда может проходить
через теплообменник в ферментатор.
§ 14. ОЧИСТКА ВОЗДУХА ОТ МИКРООРГАНИЗМОВ,
АЭРАЦИЯ КУЛЬТУРАЛЬНЫХ ЖИДКОСТЕЙ.
КОНСТРУКЦИЯ ФЕРМЕНТАТОРОВ
Выращивание микроорганизмов — продуцентов антибиотиков
происходит в аэробных условиях и, следовательно, требует боль-
больших количеств стерильного воздуха для аэрации. Атмосферный
воздух содержит значительное количество микроорганизмов, га-
газовые загрязнения органического и неорганического происхож-
происхождения, пары влаги и т. д. При сжатии воздуха в компрессорах
количество примесей увеличивается за счет механических вклю-
включений, а также смазочных масел.
Очистка воздуха от указанных примесей и стерилизация его
могут осуществляться различными методами: нагреванием до
высоких температур, облучением ультрафиолетовыми лучами,
электростатическим осаждением, пропусканием через растворы
антисептиков, фильтрацией через различные фильтрующие ма-
материалы. В промышленных условиях наиболее доступным и
экономичным оказался метод тонкой очистки воздуха пропус-
пропусканием его через зернистые и волокнистые материалы. Нередко
используются комбинированные фильтры, имеющие зернистые
и волокнистые материалы одновременно. В качестве зернистых
материалов чаще всего применяется активированный уголь. Во-
Волокнистые материалы разнообразны: стеклянная или шлаковая
вата, стеклянные срезы, базальтовое волокно (выдерживающее
высокую температуру, что удобно при стерилизации фильтра в
производстве). В последние годы широкое распространение в
промышленности для очистки воздуха нашла ткань Петрянова,
изготавливаемая из перхлорвинила, ацетилцеллюлозы
(ФПА-15) и полистирола (ФПС-15). Перспективным материа-
материалом для очистки воздуха является высокообъемный нетканый
фильтрующий материал из антимикробного целлюлозного во-
волокна. Предварительная стерилизация его не требуется. Микро-
Микроорганизмы, осевшие на его ^поверхности, гибнут.
Механизм очистки воздуха с помощью этих материалов от-
отличается от механической фильтрации. Очистка происходит од-
одновременно под действием ряда факторов: сил инерции, элект-
электростатического приближения, прямого захвата частиц, осажде-
осаждения под действием гравитационных сил и диффузии.
Механизм задержки микроорганизмов и других примесей
изучен для волокнистых материалов, на примере которых рас-
356
сматривается чаще всего процесс осаждения аэрозолей. При
соприкосновении аэрозольной частицы с волокном она задер-
живается^ на нем благодаря ван-дер-ваальсовым или электро-
электростатическим силам и в связи с этим удаляется из потока газа.
Это выражается количественно через коэффициент т]0. При
инерционном механизме осаждения частиц аэрозолей этот ко-
коэффициент приближенно пропорционален числу Стокса (Stk):
stk= 18Мв4'
где Vo — исходная скорость потока; d — диаметр частиц; рч —
плотность частиц; ц — вязкость; dB —диаметр волокна.
Из представленного выражения следует, что инерционное
осаждение усиливается с увеличением Vo диаметра аэрозоль-
аэрозольных частиц d, их плотности рч и уменьшается с увеличением
вязкости \х и диаметра волокна dB.
Микробные частицы благодаря своим малым размерам об-
обладают броуновским движением и диффундируют из потока в
случайных направлениях. Если этот процесс происходит вблизи
волокна, то может наступить осаждение частиц на волокне.
Эффект диффузионного осаждения в отличие от инерционного
усиливается с уменьшением скорости газа, так как в этом слу-
случае частица большее время находится вблизи волокна.
Выведено уравнение для коэффициента диффузного осаж-
осаждения:
2,3 / 2Рбр \*/з
^ [2B,002 — InReB)]V* \ Мв ) '
где ReB= —5-^>, a D6P=^—-г ; константа Больцмана, Т — аб-
солюТная температура; /?бр — коэффициент броуновской диф-
диффузии.
- В практике же для расчета ц011к (коэффициент диффузного
осаждения и касания) и г]0к (коэффициент осаждения касани-
касанием) используют более приближенный расчет Лэнгмюра.
В случае наличия у аэрозольных частиц и волокна электри-
электрического заряда, может наблюдаться осаждение частиц за счет
электростатического притяжения. Осаждение частиц при элек-
электростатическом притяжении может происходить на всей поверх-
поверхности волокна.
Коэффициент электростатического осаждения определяется
без вычисления предельных траекторий частиц.
Т]оэ = ^ ,
где пс — количество осаждаемых частиц в секунду; cn — кон-
концентрация части в единице объема потока до цилиндра; Vc =
= VodB расход газа через проекцию волокна единичной длины
на плоскость, перпендикулярную потоку.
357
Технологическая схема получения стерильного сжатого воз-
воздуха может быть осуществлена различными методами, но она
обязательно предусматривает следующие основные стадии.
1. Предварительная очистка воздуха от механических при-
примесей. Как правило, используются масляные фильтры из колец
Рашига, металлической стружки или иной насадки, смоченной
минеральным маслом.
2. Сжатие воздуха. При выборе компрессора следует отдать
предпочтение турбокомпрессорам. В них сжимаемый воздух не
загрязняется смазочным маслом. Давление сжатия -воздуха в
компрессоре обычно должно быть не менее 2,5 ат.
3. При сжатии в компрессоре воздух нагревается до 100—
200 °С, и, следовательно, его необходимо охлаждать. Охлажде-
Охлаждение целесообразно проводить до такой максимально возможно
высокой температуры, при которой не снижается продуктив-
продуктивность штамма. Например, продуцент окситетрациклина выдер-
выдерживает температуру входящего воздуха 70 °С. В то же время
продуценты пенициллина и стрептомицина снижают продуктив-
продуктивность при температуре воздуха, поступающего в ферментатор,
выше 40 °С. Высокая температура входящего воздуха предот-
предотвращает конденсацию в воздухе паров воды. Конденсация же
паров воды в фильтре может вызвать развитие микрофлоры и
тем самым способствовать загрязнению воздуха, поступающего
в ферментатор. Воздух охлаждается обычно в теплообменни-
теплообменниках.
4. Охлажденный воздух через брызго- и маслоотделитель,
рассивер поступает в общий фильтр, затем в индивидуальный
фильтр и ферментатор. Общий фильтр представляет собой вер-
вертикальный сосуд с решеткой на дне. Заполняется он, как пра-
правило, активированным углем, а на дне решетки укладывается слой
стеклянной ваты; т^кой же слой ваты находится на угле под
верхней решеткой. Индивидуальные фильтры могут быть раз-
различной конструкции в зависимости от используемого материа-
материала. В зависимости от насадки стерилизация индивидуальных
фильтров осуществляется паром или формалином одновремен-
одновременно со стерилизацией ферментатора. Один из вариантов техноло-
технологической схемы получения стерильного сжатого воздуха пред-
представлен на рис. 42. На протяжении всего процесса биосинтеза
стерильный сжатый воздух непрерывно поступает в фермента-
ферментатор.
Известно, что микроорганизмы в глубинной культуре могут
использовать только растворенный кислород. В культуральнои
жидкости при наличии трехфазной системы (жидкость—твердое
тело — воздух) очень трудно создать и поддерживать необходи-
необходимую для продуцента концентрацию растворенного кислорода.
Трудности усугубляются также тем, что биохимические и фи-
физико-химические показатели среды непрерывно меняются. Ис~
пользование концентрированных питательных сред •приводит к
358
увеличению биомассы, в связи с чем резко возрастает вязкость
культуральной жидкости. Повышенная вязкость ухудшает ус-
условия массопередачи кислорода из воздуха к клетке. В то же
время потребность в кислороде увеличивается пропорциональ-
пропорционально концентрации биомассы.
В настоящее время общепринят барботажный метод аэра-
аэрации культуральных жидкостей при механическом их перемеши-
перемешивании. Растворимость кислорода в водных растворах мала
@,2 мМ/л), поэтому для обеспечения культуральной жидкости
Рис. 42. Технологическая схема очистки воздуха от аэрозоля.
( — масляный фильтр; 2 — компрессор; 3 — теплообменник; 4 — рессивер; 5 — брызгоуло-
витель; 6 — общий фильтр; 7 — индивидуальный фильтр.
достаточным количеством кислорода повышают интенсивность
барботажа воздуха. Увеличение расхода воздуха позволяет
уменьшить интенсивность механического перемешивания. Ин-
Интенсивный барботаж и механическое перемешивание улучшают
массопередачу кислорода в целом. Перемешивание культураль-
культуральной жидкости позволяет также более равномерно распределять
растворенный кислород и питательные компоненты среды по
всей массе, улучшает обмен веществ.
В культуральной жидкости протекают два непрерывных про-
процесса: поступающий кислород абсорбируется жидкостью, и аб-
абсорбированный кислород потребляется клетками продуцента.
Существенное влияние на скорость потребления кислорода
оказывает его концентрация. Концентрация кислорода, при ко-
которой скорость его потребления становится постоянной и не
зависит от дальнейшего повышения концентрации, называется
критической (Скр). Эта концентрация значительно меньше рав-
равновесной (Ср).
В культуральной жидкости в процессе биосинтеза образуют-
образуются колонии продуцента. Следовательно, концентрацию кислорода
в жидкости необходимо поддерживать более высокой, чтобы в
центре колоний концентрация не была меньше критической.
Указанная концентрация кислорода называется кажущейся
критической (Скр.к). Если в какой-либо момент скорость аб-
абсорбции кислорода окажется больше скорости его потребления,
359
то концентрация кислорода в жидкости
возрастет и тем самым понизит скорость
абсорбции. В результате система доволь-
довольно быстро приходит к стационарной кон-
концентрации Сс. Аэрация считается доста-
достаточной, если имеется положительное зна-
значение разности (Сс—Скр.к).
Механическое перемешивание диспер-
диспергирует в газовые пузыри входящий воз-
воздух, создает турбулентные движения,
уменьшая тем самым диффузионное со-
сопротивление, и, следовательно, способст-
способствует массопередаче кислорода. Эффек-
Эффективность механического перемешивания
зависит от конструкции мешалки. Реже
всего используются лопастные мешалки,
чаще всего — турбинные в разных вари-
вариантах. Использование механического пе-
перемешивания в ферментаторах имеет ряд
недостатков, требует для сохранения сте-
стерильности в аппарате специальных уп-
лотнительных устройств. Расход воздуха
велик, полезная работа мешалки мала,
в процессе аэрации образуется много пе-
пены, поэтому исследователи и конструкто-
конструкторы стремятся создать ферментаторы, ра-
работа которых основана на иных принци-
принципах аэрации.
В ряде работ предложена вихревая
система аэрации с засасыванием воздуха
мешалкой через полый вал, трубу-инжек-
трубу-инжектор и т. д. Имеются конструкции фермен-
ферментов, в которых стерильный воздух подает-
подается по крышке аппарата вентилятором.
В посевных аппаратах при малой
концентрации мицелия иногда осуществ-
осуществляют аэрацию только барботажем возду-
воздуха или применением эрлифтного подъем-
подъемника, обеспечивающего циркуляцию жид-
жидкости в аппарате. Немалое влияние на
процесс аэрации оказывают внутренняя
конструкция ферментатора, наличие,
змеевиков, отражательных перегородок.
Глубинное культивирование микроорганизмов — продуцентов
антибиотиков проводится в специальных аппаратах —фермен-
—ферментаторах, представляющих собой закрытые цилиндрические со-
сосуды со сферическим днищем, снабженные мешалкой, барботе-
ром для подачи воздуха, отбойником, рубашкой или змеевиком
У77777/77
Рис. 43. Схема фермента-
ферментатора.
1 — электродвигатель; 2 —
редуктор; 3 — соединитель-
соединительная муфта; 4 — промежуточ-
промежуточный подшипник; 5 — торцо-
торцовый сальник; 6 — отража-
отражательная перегородка; 7 —
стреловидная трехъярусная
мешалка; 8 — растяжка,
центрирующая вал мешалки;
9 —секционная рубашка;
10 — барботер; 11 — подпят-
подпятник; 12 — опора.
360
для нагрева и охлаждения среды. Конструкция ферментатора
должна обеспечивать стерильность культуральной жидкости,
аэрацию,, перемешивание, регулирование температуры и т. д.
Внутреннее устройство ферментатора должно предусматривать
все необходимое для обслуживания его изнутри и ремонта. На
крышке ферментатора имеются смотровые стекла, люк, штуце-
штуцеры для манометра, датчиков дистанционных приборов контроля
и т. д. Неотъемлемой частью ферментатора является барботер.
Конец барботера необходимо устанавливать непосредственно
под мешалкой. В промышленности используются барботеры трех
типов: труба, открытая под мешалкой, квадратный и лучевой
барботеры. Конструкции ферментаторов и его основных узлов
представлены на рис. 43.
§. 15. ПЕНООБРАЗОВАНИЕ В КУЛЬТУРАЛЬНЫХ ЖИДКОСТЯХ
И ПЕНОГАШЕНИЕ
Процесс биосинтеза осуществляется в промышленных усло-
условиях в глубинной культуре в ферментаторах различной емкости
{10—60 м3). За рубежом используются также ферментаторы ем-
емкостью до 100—300 м3. Продолжительность ферментации велика
и часто достигает 10 сут. При этом требуется соблюдать сте-
стерильные условия. Очень важно правильно вести процесс фер-
ферментации и максимально использовать объем ферментатора.
Одна из причин, по которой нарушаются нормальные условия
биосинтеза антибиотиков, — интенсивное пенообразование.
Интенсивное пенообразование обусловлено непрерывным
пропусканием через культуральную жидкость больших коли-
количеств воздуха, диспергируемого барботером и мощной мешал-
мешалкой, наличием в среде растворенных поверхностно-активных ве-
веществ и коллоидных частиц, повышающих прочность оболочек
воздушных пузырьков, а также жизнедеятельностью продуцен-
продуцента. Чрезмерное вспенивание культуральных жидкостей ограни-
ограничивает полезную емкость ферментатора, нередко служит причи-
причиной попадания в аппарат посторонней микрофлоры, приводит
к потерям антибиотика из-за уноса пены с отработанным воз-
воздухом, вынуждает прекращать перемешивание, уменьшать аэ-
аэрацию и т. д.
В настоящее время не существует универсального пеногаси-
тельного средства, пригодного для любых производств, любых
пенящихся жидкостей. Это обусловлено тем, что пены, образу-
образующиеся при различных технологических процессах, сильно
отличаются друг от друга по свойствам. Кроме того, определен-
определенные условия производства и особенности применяемой аппара-
аппаратуры также предъявляют специфические требования к пенога-
сителям. Большое количество исследований, посвященных изу-
изучению пен и пенообразования при биосинтезе антибиотиков, вы-
выполнено в ВНИИА. В этих исследованиях показано, что пены
361
:oo
разных культуральных жидкостей существенно отличаются по
характеристикам. Не существует прямой связи между интенсив-
интенсивностью образования и прочностью пены. Интенсивность пено-
образования, характер образующихся пен неодинаковы не толь-
только у разных продуцентов. Даже у одного и того же продуцента
степень пенообразования на
разных этапах ферментации
различна. О чем ярко сви-
свидетельствуют графики, при-
приведенные на рис. 44.
Существует многа
средств воздействия на пе-
нообразование и методов
борьбы с пенами. Класси-
Классифицируются они следующим
образом: 1) физико-химиче-
физико-химические воздействия; 2) разру-
разрушение пен механическими,
гидро- и аэродинамическими
способами; 3) акустическое
пеногашение; 4) термиче-
термическое пеногашение; 5) элект-
электрическое пеногашение;
6) комбинированные мето*
ды.
Действие пеногаситель-
ных средств неодинаково па
отношению к разным проду-
продуцентам и на разных стадиях
ферментации. Попытка уда-
удалить вспениватели из исход-
исходной питательной среды с по-
помощью адсорбентов (активированный уголь, иониты, коллоид-
коллоидная глина) не дало положительных результатов. Безуспешным
было и применение методов пенного фракционирования. Хоро-
Хорошие результаты в борьбе с пенообразованиями получены цри
использовании метода, основанного на взаимном связывании
вспенивателей. Например, различные соотношения кукурузного
экстракта и соевой муки в питательной среде позволяют взаим-
взаимно ослабить действие вспенивателей, находящихся в них. Для
того чтобы найти состав питательной среды, удовлетворяющей
совокупности технологических требований, необходима большая
экспериментальная работа. Однако это не значит, что нужно ог-
ограничиться только такими методами борьбы с пенообразовани-
ем и отказаться от химических и механических пеногасителей.
Добавление к пенящейся жидкости поверхностно-активных
веществ, уменьшающих прочность пленок пены, является основ-
основным способом пеногашения при производстве антибиотиков.
362
120 744 168 192
Длительность ферментации Гф, ч.
Рис. 44. Динамика пенообразования куль-
культуральных жидкостей различных про-
продуцентов.
Действие химических пеногасителей заключается главным обра-
образом в вытеснении ими молекул вспенивателя с поверхности пле-
пленок пены. В связи с этим в пленке возникают неоднородные, ос-
ослабленные участки и пузырьки разрушаются под действием сил
тяжести, поверхностного натяжения и т. д. Кроме того, некото-
некоторые пеногасители снижают вязкость поверхностных слоев плен-
пленки, вследствие чего увеличивается скорость вытекания жидкости
из нее. Отдельные пеногасители вступают в химическое взаимо-
взаимодействие со стабилизатором пены, вызывая образование поверх-
поверхностно-неактивных комплексов, хрупких пленок и т. п.
Химические пеногасители должны удовлетворять ряду тре-
требований. Важнейшие из них следующие:
1. У пеногасителя поверхностная активность должна быть
выше, чем у вспенивателя. Адсорбция его на поверхности раз-
раздела фаз должна быть значительной. Эта адсорбция для раз-
разбавленных растворов выражается соотношением Гиббса.
г с г
где Г — избыточное количество пеногасителя в поверхностном
слое по сравнению с его количеством в объеме жидкости (на
единицу поверхности); С — количество загруженного пеногаси-
пеногасителя на единицу объема жидкости; R — универсальная газовая
постоянная; Т — абсолютная температура; G — поверхностная
активность пеногасителя, характеризуемая изменением поверх-
поверхностного натяжения жидкости (Да) с повышением концентра-
концентрации (АС):
г Аа
2. Взаимодействие молекул пеногасителя между собой и с
молекулами вспенивателя не должно приводить к образованию
структур, способных укрепить пленку.
3. Растворимость пеногасителя в обрабатываемой жидкости
должна быть минимальной. При этом ДС^С, а уравнение Гиб-
Гиббса приобретает вид:
RT '
4. Пеногаситель должен обладать способностью к образо-
образованию на поверхности жидкости тончайших мономолекулярных
слоев.
Совершенно необходимым требованием к пеногасителям, ис-
используемым при ферментации, является нетоксичность по отно-
отношению к культуре продуцента. Важное значение имеет отсут-
отсутствие отрицательных влияний пеногасителя на биосинтез, на по-
последующее отделение мицелия, а также на выделение и очист-
очистку препарата.
363
В основу химической классификации пеногасителей положе-
положена природа гидрофильных функциональных групп, определяю-
определяющих характер электролитической диссоциации молекул в воде.
Все известные пеногасители в связи с этим делятся на четыре
основных класса: анионогенные вещества, катионогенные ве-
вещества, амфолиты и неионогенные вещества.
В ряде случаев используются кремнийорганические (сили-
(силиконовые) пеногасители. Эти продукты, называемые силиконо-
силиконовыми маслами, по сравнению с обычными жирами требуются
для пеногашения в значительно меньших количествах.
В промышленности антибиотиков в качестве пеногасителей
применяются природные растительные и животные жиры (ка-
шалотовый жир, подсолнечное масло и др.). Однако значение
жиров в биосинтезе антибиотиков не исчерпывается ролью пе-
ногасителя. Жиры являются дополнительным источником угле-
углеродного питания. Во многих случаях они интенсивно потребля-
потребляются продуцентами антибиотиков даже при значительном ко-
количестве углеводов в питательной среде. Добавление жировых
пеногасителей в излишне больших количествах часто угнетает
биосинтез антибиотика; загруженный в конце ферментации жир
остается неиспользованным и затрудняет последующий процесс
отделения мицелия от нативного раствора.
Пеногасители оказывают существенное влияние на газооб-
газообмен между продуцентом и окружающей средой, так как опреде-
определенная часть пеногасители сосредоточивается на поверхности
воздушных пузырьков. Через эту поверхность осуществляется
диффузия кислорода из воздуха, подаваемого в культуральную
жидкость, а также отвод из нее углекислого газа, выделяемого
микроорганизмами. Многие пеногасители способны повышать
интенсивность газообмена между воздушными пузырьками и
клетками продуцента благодаря снижению вязкости поверх-
поверхностных слоев пленок.
Эффективность действия химических пеногасителей тесно
связана со способом их применения. Вопрос этот изучен недо-
недостаточно. Жировые пеногасители, как правило, подаются из
специальных мерников небольшими порциями (от долей литра
до нескольких литров). Пеногашение осуществляется автомати-
автоматически по мере в-спенивания культуральной жидкости. Предва-
Предварительно пеногаситель (жировой) стерилизуется в течение не-
нескольких часов три температуре до 130 °С. Действие пеногаси-
теля можно усилить диспергированием его, например подачей
его через распыляющие форсунки.
Химические пеногасители (жиры животного происхождения—
говяжий, свиной, кашалотовый; растительные масла — подсол-
подсолнечное, льняное, кокосовое, пальмовое и др.; кремнийорганиче-
кремнийорганические пеногасители — полисилоксаны, высшие жирные спирты и
их производные октадеканол, лаурилсульфонат и другие суль-
сульфированные продукты; минеральные масла и др.) рекоменду-
364
ется применять в виде растворов (спиртовые, масляные) или
эмульсий (жировые, иногда водные). Для предотвращения рас-
расслоения эмульсий в них вводятся специальные стабилизирую-
стабилизирующие до>бав'ки.
§ 16. КОЛЛОИДНО-ХИМИЧЕСКИЕ ОСНОВЫ КОАГУЛЯЦИИ
КУЛЬТУРАЛЬНЫХ ЖИДКОСТЕЙ
Важным этапом в производстве антибиотиков является выде-
выделение их из культуральных жидкостей. .Антибиотические жид-
жидкости представляют собой многокомпонентные, полидисперсные
системы переменного состава. Состав культуральных жидкостей
значительно изменяется от фер-
ментации к ферментации и в каж-
дом случае зависит от условий
биосинтеза. В культуральных
жидкостях накапливаются неже-
нежелательные примеси, от которых
необходимо освободиться, с внед-
внедрением концентрированных сред
количество примесей еще больше
увеличивается.
Выделение антибиотика начи-
начинается с разделения твердой и
жидкой фаз. Большинство про-
продуцентов при биосинтезе выде-
выделяют биологически активное на- Рис. 45. Схема строения мицеллы.
чало в водную фазу, поэтому для ^SSS5K°SS Т
Й б
^SSS5KSSh; i-аТрГаГп
ХИМИЧеСКОЙ ОЧИСТКИ аНТИбиОТИКОВ ядро мицеллы; III - собственно кол-
игпптт^итптга мртптттл аи-ртпяк-тттли лоидная частица; IV - диффузный слой
ИСПОЛЬЗуЮТСЯ МеТОДЫ ЭКСТраКЦИИ, мицеллы; V —мицелла.
ионообменной сорбции, осажде-
осаждения. Успешное разделение фаз,
а также последующее выделение антибиотика, особенно при раз-
разработке сокращенных технологических схем, требует получения
нативных растворов повышенной степени чистоты.
Культуральные.жидкости наряду с мицелиальной массой про-
продуцента содержат значительное количество коллоидных при-
примесей, затрудняющих процесс фильтрации и выделения антибио-
антибиотика. Первым этапом технологии выделения является предвари-
предварительная обработка культуральных жидкостей. Она предусматри-
предусматривает максимальный перевод антибиотика в ту фазу, из которой
предполагается его выделение, коагуляцию коллоидных приме-
примесей и, следовательно, улучшение процесса фильтрации, а также
удаление тех примесей, которые затрудняют последующий про-
процесс химической очистки антибиотика.
Для более полного понимания процесса коагуляции коллоид-
коллоидных структур необходимо знать их строение. Коллоидные части-
частицы представляют собой агрегат твердых кристаллов, состоящих
365
'из атомов, молекул или ионов, образующих дисперсную фазу. На
поверхности кристаллов агрегата находятся достраивающие его
ионы, которые и придают ему электрический заряд. Эти ионы
называются шотенциалоопределяющими (рис. 45).
Агрегат твердых кристаллов вместе с адсорбированными на
его поверхности потенциалоопределяющими ионами носит наз-
название ядра мицеллы. В непосредственной близости от него, в
так называемом адсорбционном слое находится часть противо-
ионов, очень прочно связанных с ядром. Ядро вместе с частью
прочно связанных с ним противоионов называется собственно
коллоидной частицей. Вокруг коллоидной частицы имеется диф-
диффузный слой, образуемый противоионами. Они относительно
свободны и, если поместить исследуемый раствор в электриче-
электрическое поле, двигаются к противоположному электроду по отно-
отношению к движению самой коллоидной частицы. Мицелла же в
•отличие от коллоидной частицы электронейтральна. Таким об-
образом, вокруг кристаллов твердой фазы возникает как бы двой-
двойной электрический слой с определенной величиной ^-потенциала,
который и характеризует стабильность (коллоидной структуры.
Большинство коллоидных структур, возникающих природным
путем, имеет отрицательный заряд. Вероятно, это обусловлено
большей деформируемостью анионов, поскольку радиусы анио-
анионов вообще значительно больше радиусов катионов. Поляризуе-
Поляризуемость анионов также выше, чем у катионов . (как правило,
коллоидная структура с большей величиной ^-потенциала бо-
более устойчива).
Поскольку ^-потенциалы в некоторых случаях являются
величиной, характеризующей устойчивость коллоидной систе-
системы, представляется весьма важным рассмотреть влияние на не-
него таких факторов, как введение в систему электролитов, из-
изменение рН и концентрации окружающего частицы раствора,
изменение температуры. При введении электролитов в раствор
следует рассматривать несколько случаев: 1) когда в систему
вводится электролит, один из ионов которого одинаков с про-
противоионами; 2) когда в систему вводится электролит, не имею-
имеющий общих ионов с электролитом-стабилизатором; 3) когда
в систему вводится электролит, один из ионов которого спосо-
способен достраивать кристаллическую решетку дисперсной фазы.
В первом случае введение электролита сжимает двойной
электрический слой. В результате ^-потенциал снижается, пока
не станет равным нулю, что соответствует так называемому изо-
электрическому состоянию системы.
Второй случай отличается от первого лишь тем, что внача-
вначале имеет место явление обмена противоиона коллоидной сис-
системы на эквивалентное количество соответствующего по знаку
иона вводимого электролита.
В третьем случае при введении в систему электролита, один
из ионов которого способен достраивать кристаллическую ре-
366
шетку дисперсной фазы, можно наблюдать различные эффекты
в зависимости от концентрации электролита. При малых кон-
концентрациях происходит достраивание кристаллической решет-
решетки и £-етотенциал увеличивается. При больших концентрациях
электролита, когда достройка кристаллической решетки завер-
завершена, потенциал уменьшается. В связи с этим при введении в
систему все возрастающих количеств такого электролита ^-по-
^-потенциал сначала повышается, а затем снижается.
Коллоидные частицы культуральных жидкостей антибиоти-
антибиотиков природного происхождения имеют отрицательный заряд,
поэтому при.коагуляции электролитами, особенно с поливалент-
поливалентными катионами, может происходить их перезарядка. Многова-
Многовалентные ионы, несущие за1ряд, противоположный заряду по-
поверхности твердой фазы, сильно втягиваются и больше адсор-
адсорбируются на поверхности дисперсной фазы. Ионы металлов мо-
могут адсорбироваться в таком количестве, что будут не только
нейтрализовать заряд твердой поверхности колоидной частицы,,
но и перезаряжать ее. Это явление необходимо учитывать в
практике при коагуляции электролитами с поливалентными по-
нами.
Существенное влияние на состояние коллоидной системы
оказывает рН среды. Водородные и гидроксильные ионы обла-
обладают высокой способностью адсорбироваться, первые — благо-
благодаря тому, что из-за своего малого радиуса близко подходят к
поверхности твердой фазы, а вторые — из-за большого диполь-
ного момента, которым они обладают. Отрицательный заряд
коллоидных частиц культуральных жидкостей и большая под-
подвижность ионов водорода позволяют использовать для их коагу-
коагуляции различные кислоты.
Для улучшения процесса фильтрации культуральные жид-
жидкости часто разбавляют. Этот процесс не безразличен для кол-
коллоидных систем. Исходя из самых общих соображений можно
думать, что при разбавлении всякой коллоидной системы ^-по-
^-потенциал должен возрастать вследствие расширения двойного
электрического слоя из-за уменьшения концентрации противо-
ионов в растворе. С другой стороны, при разбавлении могут
наблюдаться десорбции потенциалообразующего иона с поверх-
поверхности дисперсной фазы и, следовательно, уменьшение ^-потен-
^-потенциала коллоидной частицы. При концентрировании коллоидной
системы положение будет прямо противоположным.
С повышением температуры ^-потенциал должен возрас-
возрастать, вследствие большего теплового движения противоионов и
увеличения толщины двойного электрического слоя. Однако од-
одновременно может усиливаться и десорбция потенциалопреде-
ляющих ионов, а следовательно, ^-потенциал системы может
падать.
От указанного положения зависит возможность использова-
использования тепловой коагуляции для культуральных жидкостей анти-
367
биотиков. В промышленности процесс коагуляции улучшается
"за счет использования комбинированных методов, таких, как
кислотная и тепловая коагуляция, коагуляция электролитами и
тепловая и т. д. Указанные типы коагуляции являются доволь-
довольно «жесткими». Повышение концентрации антибиотиков в пере-
перерабатываемых жидкостях приводит к росту абсолютных потерь
препарата при «жестких» условиях очистки, поэтому в послед-
последнее время много внимания уделяют разработке более «мягких»
методов коагуляции. Эффективным «мягким» методом коагуля:
ции дисперсных систем является обработка их высокомолекуляр-
высокомолекулярными полиэлектролитами — флоккулянтами. Применение флок-
кулянтов значительно улучшает технологические показатели
обрабатываемых суспензий — улучшает седиментационные и
фильтрационные свойства взвесей, способствует осветлению
жидкой фазы, увеличению производительности оборудования.
Особенно интересным и перспективным для обработки нативных
растворов антибиотиков оказался флоккулянт ВА-2 с молеку-
молекулярной массой 30 000 — поли- D-ви'нил-Ы-'бенз(Илтриметиламмо-
ний хлорид). При обработке нативных растворов полимерными
электролитами в отличие от обычной коагуляции образуются
флоккулы или осадки с рыхлой структурой. Это и определяет
значительное улучшение процесса фильтрации. Обработка вы-
высокомолекулярными полиэлектролитами позволяет удалить ряд
пигментных примесей, а также понизить эмульгируемость на-
нативных растворов при экстракционных методах выделения ан-
антибиотиков. Флоккуляция примесей органическими полиэлектро-
полиэлектролитами более эффективна, чем коагуляция неорганическими
электролитами из-за возможности образования с коллоидными
частицами водородных связей.
Флоккулянт ВА-2 и подобные ему являются линейными по-
полиэлектролитами. Возможности коагуляции, или, вернее, флок-
куляции, культуральных жидкостей антибиотиков значительно
расширяются с использованием разветвленных растворимых по-
полиэлектролитов. Если флоккулянт ВА-2 проявляет свои флокку-
ляционные свойства только в нативных растворах, то развет-
разветвленные полиэлектролиты благодаря своей структуре весьма
эффективны в культуральных жидкостях. Это дает возмож-
возможность избавиться от второй фильтрации нативных растворов.
Обработка культуральных жидкостей антибиотиков растворимы-
растворимыми частично разветвленными полиэлектролитами позволяет про-
проводить процесс в «мягких» условиях с высоким выходом анти-
антибиотика со значительным увеличением скорости фильтрации и
депигментацией получаемых растворов.
Из большого количества существующих конструкций как пе-
периодических, так и непрерывно действующих фильтров для от-
отделения мицелия нашли применение лишь несколько типов.
Это, во-первых, барабанные вакуум-фильтры, которые применя-
применяются в производстве пенициллина для отделения мицелия гриб-
368
кового происхождения. В последние годы вакуум-бараба-нные
фильтры с намывным слоем (инфузорная земля, перлит, дре-
древесная мука и т. д.) успешно используются для отделения ми-
целиальной массы актиномицетного происхождения.
Производительность вакуум-барабанных фильтров с намыв-
намывным слоем зависит от качества материала, используемого для
образования намывных слоев, тщательности его нанесения на
поверхность барабана, но особенно от установки ножа и снятия
осадка. Вакуум-барабанные фильтры характеризуются непре-
непрерывностью действия с автоматическим чередованием операций
фильтрации, промывки, сушки и снятия осадка с фильтроваль-
фильтровального полотна. Перфорированная поверхность барабана, медлен-
медленно вращаясь, погружается в емкость, в которую подается сус-
суспензия. Создание зон фильтрации, сушки, промывки и отдувки
осадка осуществляется с помощью неподвижной распредели-
распределительной головки, разделенной на камеры, которые соответствен-
соответственно соединены с вакуум-приемником фильтрата, вакуум-прием-
вакуум-приемником промывных вод и источником сжатого воздуха.
В промышленностц антибиотиков для отделения мицелия ак-
актиномицетного происхождения используются рамные фильтр-
прессы периодического действия. Автоматические непрерывного
действия фильтр-прессы типа ФПАК и ФПАК-М с непрерывной
регенерацией фильтрующей ткани не нашли широкого примене-
применения. Рамные фильтр-прессы состоят из комплекта рам и плит,
между которыми зажата фильтрующая ткань. С помощью гид-
гидравлического или электромеханического зажима плиты и рамы
с фильтрующей тканью между ними плотно прижимаются друг
к другу. Через специальные каналы суспензия подается в ра-
рамы, осадок остается в раме, а фильтрат по плитам спускается
в каналы для отвода фильтрата. Осадок из рам и с фильтрую-
фильтрующей ткани удаляется после снятия давления и раздвигания рам
и плит. Фильтр-прессы довольно просты по конструкции, ком-
компактны, позволяют получать прозрачные фильтраты. Однако об-
обслуживание их — трудоемкий процесс, требующий больших уси-
усилий от работников, занятых сборкой фильтра и промывкой
фильтрующей ткани. Фильтр-прессы в промышленности антиби-
антибиотиков используются для отделения мицелия продуцента от на-
тивных растворов.
Для изготовления вакуум-барабанных фильтров и фильтр-
прессов используется, как правило, углеродистая сталь. В тех
же случаях, когда фильтруются агрессивные жидкости, сталь
покрывают кислотоустойчивым лаком или применяют фильтры
из кислотоустойчивой стали.
Использование для отделения мицелия сепарирующих цент-
центробежных машин не дало положительных результатов. Обычно
они применяются при производстве дрожжей.
24—926
Глава 19
ТЕХНОЛОГИЯ ХИМИЧЕСКОЙ ОЧИСТКИ
И ВЫДЕЛЕНИЯ
§ 1. ЭКСТРАКЦИЯ
Экстракция представляет собой процесс разделения смеся
твердых или жидких веществ с помощью избирательных (селек-
(селективных) растворителей (экстрагентов).
Физическая сущность экстракции состоит в переходе извле-
извлекаемого компонента из одной фазы (жидкой или твердой) в
фазу жидкого экстрагена при их взаимном соприкосновении.
Экстрагируемые компоненты переходят из исходного раствора
в растворитель вследствие разности концентраций, поэтому дан-
данный процесс относится к числу диффузионных. Процесс экстрак-
экстракции проводится обычно в двухфазных системах: твердое тело—
жидкость или жидкость — жидкость.
Экстрагирование как каждый диффузионный процесс в боль-
большой степени зависит от температуры и с повышением ее идет
скорее. Однако из-за свойств обрабатываемых систем экстра-
экстрагирование производится при умеренной температуре (часто она
даже ниже, чем температура окружающей среды). Это позволя-
позволяет в процессе экстрагирования обеспечить разделение исходного
раствора с полным исключением термического распада моле-
молекул.
Процесс экстрагирования обладает большими экономически-
экономическими преимуществами, так как затраты на него меньше, чем на
другие процессы. Низкая стоимость эксплуатации особенно за-
заметно проявляется при малых концентрациях экстрагируемого
компонента.
Процесс экстрации в большинстве случаев проводится при
относительном движении обеих фаз. Широко применяется про-
тивоточное движение фаз, что обеспечивает экономически более
выгодное решение задачи. Экстракция используется для выде-
выделения и очистки многих химических соединений, в том числе
антибиотиков. Для выделения и очистки антибиотиков экстрак-
экстракция успешно производится в тех случаях, когда антибиотик не
ионизирован в водной фазе. Несмотря на существенный недо-
недостаток экстракционных процессов, а именно использование
вредных, взрывоопасных органических растворителей, все же
эти процессы находят широкое применение в промышленности.
370
Во-первых, экстракционные процессы протекают значительно
быстрее, чем ионообменные, коэффициенты распределения для
некоторых систем очень велики, и это позволяет резко сокра-
сокращать объемы перерабатываемых растворов. Аппаратурно этот
процесс сравнительно легко осуществить в непрерывном испол-
исполнении. Особенно интересна экстракция с переносчиком. Часто
этот вид экстракции называют жидкостным ионным обменом.
Основным законом экстракции является закон распределения
Нернста.
где К — коэффициент распределения; Со —концентрация ком-
компонента в водной фазе; Со —концентрация компонента в орга-
органической фазе.
Вещество переходит в органический растворитель в неиони-
зированном состоянии, поэтому для улучшения процесса экст-
равдии изменяют »,рН.раствора с целью уменьшения степени ио-
ионизации экстрагируемого компонента. При экстракции веществ,
способных к ионизации, пользуются несколько видоизмененным
уравнением Нернста:
-§- = A-а)/С,
где К — коэффициент распределения; Со и Со— концентра-
концентрация компонента в одной и в другой фазе в неонизированном со-
состоянии; а — степень ионизации. В процессе экстракции, осо-
особенно в органической фазе, наблюдается ассоциация молекул
экстрагируемого вещества с образованием ассоциатов. В таких
случаях необходимо учитывать степень диссоциации ассоциатов.
Тогда уравнение распределения Нереста будет выглядеть сле-
следующим образом:
Л
где (Х2 — степень диссоциации ассоциатов.
Уравнение справедливо в том случае, если в водной фазе
имеются только мономеры, а в органической — только димеры.
Эта закономерность имеет место при выделении и очистке мно-
многих антибиотиков экстракцией.
Если при экстракции происходят ионизация экстрагируемо-
экстрагируемого компонента в водной фазе и образование ассоциатов в органи-
органической фазе, то коэффициент распределения следует определить
по формуле (обозначения см. выше):
К= A-а)С; •
24* 371
В таких системах при изменении рН раствора ионизация экстра-
экстрагируемого вещества изменяется и, следовательно, изменяется
коэффициент распределения. Для вещества кислотного харак-
характера это изменение можно выразить следующей зависимостью:
где /Са часто можно определить потенциометрическим титрова-
титрованием; К — 'коэффициент распределения — также бьгаает изве-
известен. В данном случае справа мы имеем уравнение Лэнгмюра,
которое показывает, что с повышением концентрации ионов во-
водорода в растворе насыщение органической фазы веществом
кислотного характера увеличивается до определенного значе-
значения и затем не изменяется.
Представителем антибиотиков, для выделения и очистки ко-
которых до сих пор наиболее успешно применяется экстракцион-
экстракционный метод, является пенициллин. После отделения пеницилли-
альной массы продуцента и предварительной обработки натив-
ного раствора его направляют на экстракцию. Экстракцию пе-
пенициллина осуществляют при рН 2,0 бутилацетатом. В этих ус-
условиях коэффициент распределения для водных растворов мо-
может достигать 30. рН раствора обычно доводят до 2,0 серной
кислотой, которая и позволяет подавить кислотную ионизацию
пенициллина в водной фазе. Неионизированная пенициллиновая
кислота легко переходит в органическую фазу. Реэкстракцию
пенициллина из бутилацетата осуществляют слабыми раствора-
растворами щелочей или буферными растворами с рН 6,8—7,0, и пени-
пенициллин в виде соли снова переходит в водную фазу. Для полу-
получения пенициллина более высокой степени чистоты и для
концентрирования раствора бутилацетатную экстракцию повто-
повторяют и проводят вторую щелочную или буферную экстракцию.
В последние годы в промышленности антибиотиков удалось по-
повысить активность пенициллина в культуральных жидкостях
(обогащение сред и получение высокоактивных штаммов), по-
поэтому в настоящее время разрабатываются сокращенные техноло-
гические схемы выделения и очистки пенициллина.
Экстракция с переносчиком. Переход экстрагируемого ком-
компонента из водной фазы в органическую в этом случае экстрак-
экстракции происходит с образованием комплексного соединения с гид-
гидрофобным переносчиком. Переход экстрагируемого компонента
из водной фазы в органическую осуществляется за счет не толь-
только комплексообразования, но и гидрофобизации молекул анти-
антибиотика переносчиком.
В качестве переносчиков, как правило, используются вещест-
вещества кислотного или основного характера с длинной углеводород-
углеводородной цепью (Сю—Сзо), фенолы, сульфокислоты, олеиновая и ун-
дециленовая кислоты, из оснований цетазол (цеталпиридиний
372
бромид). Олеиновая кислота, например, взаимодействует со
стрептомицином, образуя соль с большей энергией сольватации
в органической фазе. В связи с этим экстрагируемый компо-
компонент (стрептомицин) в большей степени переходит в органиче-
органическую фазу.
Переносчик может находится как в водной, так и в органи-
органической фазе. В случае, когда переносчик (В) находится в вод-
водной фазе, экстракцию вещества А можно представить следую-
следующим образом:
(АВ) *=+ (АВ) A>
Водная фаза Органи-
Органическая
фаза
Если переносчик (В~О) находится в органической фазе,
экстракцию вещества А можно представить следующим обра-
образом:
А+ + В"С+ <—>- А+В~ + С+ B>
Водная Органичес- Органичес- Водная
фаза кая фаза кая фаза фаза
В этом случае мы имеем уравнение ионного обмена, и к не-
нему можно применить все законы ионного обмена (об этом
см. ниже). Таким образом, если перенос осуществляется с по-
помощью неионизированного переносчика, то используется урав-
уравнение B), а если переносчик ионизирован, то уравнение A).
Представителем антибиотиков, которые выделяются и очи-
очищаются методом экстракции с переносчиком, являются антибио-
антибиотики тетрациклиновой группы (тетрациклин). Наличие в моле-
молекуле этих антибиотиков одной основной [N(CH3h] и двух кис-
кислотных (фенольный и енольный гидроксил) групп позволяет
использовать в качестве переносчиков этих соединений вещест-
вещества основного и кислотного характера.
Основным достоинством экстракции с переносчиком являет-
является высокая избирательность (переносчик может быть высоко-
высокоизбирательным), и процесс протекает практически мгновенно.
Исследования по экстракции тетрациклина показали возмож-
возможность его экстракции на 95—97% 5% раствором цетазола в бу-
тилацетате или изоактиловом спирте, взятом в количестве 15%
от объема нативного раствора, за 1 мин. Такая высокая ско-
скорость протекания процесса экстракции тетрациклина позволяет
проводить процесс при довольно высоком значении рН раство-
раствора (9,5—10,2) без потерь антибиотика. Не менее важным мо-
моментом является последующая реэкстракция тетрациклина из
органической фазы. Использование для этой цели соляной, фос-
фор'ной, лимонной кислот хотя и позволяет шолучать растворы
с концентрацией 40000—50000 ЕД/мл, но интенсивной темной
окраски. Реэкстракция тетрациклина 4—5% раствором щаве-
373
•левой кислоты позволяет получить растворы с концентрацией
антибиотика 35 000—45 000 ЕД/мл со светлой окраской реэкст-
ракта.
Большинство антибиотиков представляют собой полярные мо-
молекулы, и для их экстракции используются достаточно поляр-
полярные растворители (эфиры, спирты, галоидопроизводные и др.).
В неполярных растворителях (генсане, гентане, октане и др.) по-
полярные молекулы антибиотиков не растворяются. Для успеш-
успешного проведения процесса экстракции органические растворите-
растворители должны обладать целым рядом свойств; остановимся на не-
некоторых из них.
Селективность экстрагента. Основным свойством, которым
должен обладать экстрагент, является селективность. Указан-
Указанное свойство характеризует способность экстрагента препочти-
тельно извлекать один из двух компонентов раствора. С этой
точки зрения наиболее подходит тот экстрагент, который раст-
растворяет максимальное количество одного компонента и мини-
минимальное количество другого. Селективность (C) экстрагента
В по отношению к компоненту С можно определить отноше-
отношением:
где Хсв — весовая доля С в фазе компонента В; ХСА — весовая
доля С в фазе компонента А; ХАв— весовая доля А в фазе ком-
компонента В; ХАА— весовая доля А в фазе компонента А. Для
практических целей необходимо, чтобы величина C была воз-
возможно больше единицы. При значениях |3, мало отличающихся
от единицы, увеличиваются габаритные размеры оборудования,
необходимое число ступеней экстракции, а также в общем
случае возрастают капитальные затраты и эксплуатацион-
эксплуатационные расходы. При |3 = 1 разделение смеси экстракций
невозможно. Высокая селективность экстрагента к экстраги-
экстрагируемому компоненту, доля которого в исходном растворе мала,
обусловливает более экономичное проведение процесса за счет
снижения количества циркулирующего экстрагента. Чем боль-
больше коэффициент распределения, тем выше селективность. Если
какой-нибудь экстрагент обладает малым коэффициентом рас-
распределения, но по другим причинам применение этого экстра-
экстрагента желательно, то коэффициент распределения можно иног-
иногда увеличить изменением рН раствора, а также введением в
систему неэкстрагируемой соли, т. е. применить «высалива-
«высаливание». В таком случае коэффициент распределения в новых ус-
условиях (Кх) можно вычислить по правилу Сеченова:
374
где Кх — коэффициент распределения в присутствии соли;
К—коэффициент распределения в отсутствии соли; А — кон-
константа, отражающая влияние концентрации соли; Хс — концент-
концентрация соли.
Емкость экстрагента. Помимо селективности — избиратель-
избирательности экстрагентов, очень важной характеристикой экстраген-
экстрагента является их емкость. Если экстрагент обладает большой ем-
емкостью, т. е. способностью растворять относительно большое
количество избирательно извлекаемого компонента, то, несмотря
на его высокую селективность, применение экстрагента может
оказаться неэкономичным из-за необходимости иметь в экстрак-
экстракционной системе большое количество циркулирующего экстра-
экстрагента. В связи с этим экстрагент должен иметь не только высо-
высокий коэффициент распределения, но и возможно большую рас-
растворимость по экстрагируемому компоненту.
Немаловажное значение для экстрагента имеет взаимная
растворимость растворителей. Высокая взаимная растворимость
растворителей приводит к большим потерям экстрагента и тре-
требует регенерации растворителя в обеих фазах. В регенерации
экстрагента большую роль играют летучесть и теплота парооб-
парообразования. Высокая летучесть экстрагента будет вызывать
большие потери его. Низкая теплота парообразования облегча-
облегчает организацию процесса регенерации растворителя и умень-
уменьшает затраты тепла на регенерацию. Все эти показатели опре-
определяют экономику многократного использования экстрагента.
Плотность экстрагента. При выборе экстрагента существен-
существенную роль играет также разность плотностей фаз, которая долж-
должна быть возможно большей. Разность плотностей фаз опреде-
определяет скорость расслаивания несмешивающихся жидкостей и тем
самым влияет на производительность экстракционного обору-
оборудования. Для выбора экстрагента недостаточно сравнить плот-
плотности исходного раствора и чистого растворителя, так как вза-
взаимная растворимость этих жидкостей при смешении приводит
к изменению плотностей фаз.
В непрерывных экстракторах важно также по всей рабочей
высоте (длине) аппарата поддерживать определенную разность
плотностей контактирующих фаз.
Межфазовое натяжение. Для ускорения расслаивания несме-
несмешивающихся жидкостей при их разделении (отстаивании) не-
необходимо, чтобы межфазовое натяжение было достаточно вы-
высоким. Чрезмерно большое межфазовое натяжение приводит к
увеличению энергии, затрачиваемой на создание дисперсии.
Жидкости же с малым межфазовым натяжением образуют ста-
стабильные эмульсии, возникновение которых очень трудно предот-
предотвратить.
Поверхностно-активные вещества (ПАВ). Поверхностно-ак-
Поверхностно-активные вещества — вещества, способные адсорбироваться на
поверхностях раздела фаз и понижать вследствие этого их
375
поверхностную энергию (поверхностное натяжение). В водных
растворах этой способностью в той или иной степени обладают
большинство органических соединений, молекулы которых име-
имеют «дифильное» строение, т. е. наряду с полярными гидрофиль-
гидрофильными группами также содержат углеводородные радикалы. Од-
Однако в более узком, обычно принятом смысле слова к поверх-
поверхностно-активным веществам относят те из органических ве-
веществ, которые имеют особенно резко выраженную способность
к адсорбции из той или иной среды (обычно из водных раство-
растворов) на границах жидкость — жидкость, жидкость — воздух,
жидкость—твердое тело.
Экстракция пенициллина из нативных растворов практиче-
практически без ПАВ невозможна, так как нативные растворы содер-
содержат значительное количество примесей, способствующих обра-
образованию стойких эмульсий. Для предотвращения стабильных
эмульсий при экстракции добавляют ряд поверхностно-актив-
поверхностно-активных соединений: авироль — сульфобутилат натрия, контакт Пет-
Петрова— смесь сульфокислот (отход нефтеперерабатывающей про-
промышленности) и т. д. За рубежом для предотвращения образо-
образования стабильных эмульсий используются катексол, ультравет
и т. д., состав этих антиэмульгаторов, как правило, не раскры-
раскрывается.
В то же время некоторые ПАВ даже в незначительных коли-
количествах могут блокировать поверхность раздела фаз, уменьшая
поверхностное натяжение. Под влиянием ПАВ может наблю-
наблюдаться уменьшение подвижности поверхности. Подвижность же
раздела фаз в системе жидкость—жидкость оказывает влия-
влияние на величину массопередачи. Некоторые ПАВ, адсорбируясь
на границе раздела фаз, могут увеличивать ее жесткость и тем
самым препятствовать нормальному перемещению поверх-
поверхности. ПАВ влияют на форму капель, возникающих при диспер-
диспергировании экстрагента и раствора, изменяют скорость паде-
падения и подъема капель и тем самым воздействуют на величи-
величину массопереноса экстрагируемого компленента, изменяя гидро-
гидродинамические условия процесса.
Межфазовая турбулентность. Межфазовая турбулентность
представляет собой самопроизвольную активность поверхности,
возникающую тогда, когда в соприкосновение приводятся две
жидкие неравновесные фазы. Межфазовая турбулентность вы-
вызывается резким различием в свойствах двух соприкасающихся
жидкостей (резкое отличие по вязкости, межфазовому натяже-
натяжению, концентрации и т. д.) Проявление межфазовой турбулент-
турбулентности усиливает интенсивность внутренней циркуляции, увеличи-
увеличивает поверхность массопередачи (в некоторых случаях скорость
массопередачи возрастает в 10 раз). Межфазовая турбулент-
турбулентность облегчает создание турбулентности за счет перемешивания
системы и в целом влияет на производительность экстракционно-
экстракционного оборудования.
376
Аппаратура экстракции. В химико-фармацевтической про-
промышленности используется много принципов экстракции и ти-
типов аппаратов, пригодных для экстракции лекарственных ве-
веществ.
В промышленности антибиотиков для экстракции из жидко-
жидкостей в основном применяются центробежные экстракторы, в ко-
которых смешение и разделение жидкостей происходит в поле
центробежных сил. Центробежные экстракторы обеспечивают
весьма малое время контакта фаз, что очень важно для таких
лабильных соединений, как антибиотики. Обычно такие экстрак-
экстракторы применяются при очень малой разности плотностей фаз,
при образовании стойких эмульсий, что характерно для произ-
производства антибиотиков.
Величина рН нативных растворов колеблется от 2,0 до 10,0.
Необходимость снижения или повышения рН при экстракции
определяется ионизацией антибиотиков в водной фазе. Регу-
Регулирование рН растворов позволяет увеличивать коэффициент
распределения для пенициллина; этот коэффициент достигает
30 при рН 2,5. Как показали исследования, рН экстракции пе-
пенициллина можно повысить до 3,0 вместо 2,0, которое исполь-
используется в промышленности. Практика показывает, что при рН
2,0 основную долю потерь антибиотика составляет не остаточ-
остаточное содержание пенициллина в отработанном растворе, а по-
потери антибиотика за счет инактивации и в результате уноса бу-
тилацетатного экстракта с водной фазой (из-за повышенной
эмульгируемости), а также потери с осадком балластных ве-
веществ. Извлечение пенициллина при рН 3,0 с использованием
противоточных дифференциально-контактных экстрактов (типа
«Подбельняк» и пр.), позволяющих достигнуть 5ч-8 ступеней
экстракции, повышает степень концентрирования экстракта
с 2 до 5—7. В этом случае значительно уменьшаются
потери от уноса бутилацетатного экстракта с отработанным на-
тивным раствором и осадком примесей, так как в этих условиях
количество осаждающихся примесей резко снижается и эмуль-
эмульсия разрушается сравнительно легко. Скорость инактивации
пенициллина при рН 3,0 в 6 раз ниже, чем при рН 2,0. Кроме
того, с увеличением рН экстракции повышается селективность
экстрагента: в бутилацетатный экстракт переходит меньше при-
примесей. Кислотные примеси, сопутствующие пенициллину, как
правило, более сильные кислоты, чем пенициллин (с большей
величиной константы диссоциации), и для них коэффициент
распределения с ростом рН уменьшается значительнее, чем для
пенициллина. Из этих же исследований следует, что повышение
эффективности процесса экстракции пенициллина может быть
достигнуто проведением многоступенчатого противоточного из-
извлечения антибиотика при рН 3,0.
Перед выбором экстрактора рассчитывают число требуемых
ступеней изменения концентрации (п) на основе данных о рав-
377
новесной концентрации антибиотика при контакте двух жидко-
жидкостей.
Коэффициент экстракции рассчитывают по уравнению:
где /( — коэффициент распределения; тэ — масса экстрагента;
тр — масса нативного раствора.
По уравнению противоточной экстракции выход антибиоти-
антибиотика будет составлять:
-100%,
Для осуществления двухступенчатой экстракции (п=2)
создается обычно технологическая схема из двух прямоточных
Экстрагент
Раствор ♦ __\.
Экстракт
Р.ис. 46. Схема двухступенчатой жидкостной экстракции антибиотиков с при-
применением трубчатых смесителей A) и сепараторов САЖ-ЗМ B), снабженных
сдвоенными насосами (<?).
смесителей и двух сепараторов; экстрагент и раствор движут-
движутся между ступенями противотоком (рис. 46).
В струйном экстракторе — сепараторе «Россия» осуществля-
осуществляется двукратное прямоточное смешение с помощью кинетиче-
кинетической энергии струи тяжелой (на первой ступени) и легкой (на
второй ступени) жидкостей и разделение на тарелках сепара-
сепараторной части вертикального ротора. Легкая и тяжелая жид-
жидкости между ступенями в прямоточных экстрактах движутся
противоточно, как показано на рис. 46.
378
§ 2. СОРБЦИОННЫЕ ПРОЦЕССЫ
Сорбционные методы выделения и очистки антибиотиков на-
находят широкое распространение в промышленности. Они обла-
обладают рядом преимуществ по сравнению с другими методами п
поэтому являются исключительно перспективными.
Первые сорбционные методы выделения и очистки антибио-
антибиотиков были основаны на применении молекулярных сорбентоа
(активированный уголь, окись алюминия и т. д.). Молекулярные-
сорбенты такие, как активированный уголь, обладают универ-
универсальной сорбционной способностью, т. е. одинаково хорошо сор-
сорбируют выделяемое вещество и ряд примесей. Исключительна
большие возможности синтеза ионообменных сорбентов, кото-
которые обладают различной избирательностью и, что особенно-
важно, специфической избирательностью по отношению к от-
отдельным антибиотикам, быстро выдвинули ионообменные сор-
сорбенты на первый план. Молекулярные же сорбенты в настоя-
настоящее время находят применение на последних стадиях — до очи-
очистки и удаления пигментных примесей.
Ионообменные сорбенты принадлежат к классу высокомо-
высокомолекулярных соединений, макромолекулы которых имеют сет-
сетчатую и пространственную структуру и в большинстве случаев
представляют собой аморфные вещества. Отдельные атомы
этих гигантских молекул соединены друг с другом ковалентны-
ми связями. Такая структура ионообменных сорбентов и их не-
нерастворимость нарушают в известной степени общепринятое-
представление об электролитах и ионообменных процессах меж»
ду ними. Диссоциация растворимых в воде кислот или основа-
оснований вызывает изменение концентрации водородных ионов.
Ионообменные сорбенты также содержат кислотные и основ-
основные группы, но их погружение в воду не вызывает изменения в
ней концентрации водородных ионов. Иониты — в самом общем
представлении органические или неорганические вещества,,
практически нерастворимые в воде и обычных растворителях,
содержат активные (ионогенные) группы с подвижными иона-
ионами и способные обменивать эти ионы на ионы электролитов при-
контакте с их растворами.
В настоящее время известны следующие иониты: органиче-
органические иониты природного происхождения — целлюлоза, лигнинь
шерсть, казеин, кератин, желатина, крахмал, гумусовые вещест-
вещества почвы, древесина и т. д. и синтетические ионообменные смо-
смолы. Синтетические ионообменные смолы находят широкое при-
применение в промышленности, и все последующее изложение бу-
будет связано именно с ними. Хотя имеется ряд неорганических
природных и синтетических ионитов, они играют меньшую прак-
практическую роль.
Иониты представляют собой особый класс кислот или осно-
оснований, которые характеризуются многовалентностью и, обладая
громоздкой структурой, практически лишены подвижности. Про-
379»
тивоион, входящий в состав ионита, подвижен и только сила
электростатического притяжения препятствует ему отделяться
в растворитель, поэтому вокруг адсорбента создается ионная
«атмосфера». Противоионы в ионной «атмосфере» нера(В*шценны
по силе их электростатического сцепления с ионом адсорбента
(матрицы смолы), в связи с чем процесс ионного обмена пред-
представляет собой многоступенчатую реакцию.
В зависимости от наличия ионогенных групп иониты можно
разделить на два основных класса: ионообменные сорбенты, со-
содержащие кислотные группы — катиониты (нерастворимые
кислоты), и ионообменные сорбенты, содержащие основные
группы — аниониты (нерастворимые основания). Катиониты в
свою очередь делятся на сильнокислотные и слабокислотные,
аниониты — на сильноосновные и слабоосновные. К сильнокис-
сильнокислотным катионитам относятся фосфорнокислотные и сульфо-
кислотные, к слабокислотным — карбоксильные катиониты. К
сильноосновным ионитам относятся иониты, в структуре кото-
которых имеются группы четвертичных оснований, а слабоосновны-
слабоосновными аниояитами являются аниоеиты, в которые входят группы
первичных, вторичных и третичных аминогрупп.
Разграничение ионитов на кислоты и основания не исклю-
исключает возможности существования ионитов-амфонитов, ионоген-
ные группы которых могут вести себя как кислоты или как ос-
основания в зависимости от рН среды. Существуют также иониты,
содержащие одновременно кислотные и основные группы (бипо-
(биполярные иониты).
Все применяемые для сорбции антибиотиков иониты отно-
относятся к молекулярным сорбентам, минеральным ионитам или
ионообменным смолам. Среди последних наибольшее значение
имеют карбоксильные смолы и сульфокатиониты, а так&е ани-
аниониты различной степени основности.
Полимерные смолы — сорбенты могут быть синтезированы
на основе процессов полимеризации или поликонденсации. Смо-
Смолы, полученные первым из этих методов, обычно обладают
большей механической прочностью. Тем не менее ряд поли-1
мерных сорбентов, полученных путем поликонденсации, также
применяется в лабораторной практике и промышленности.
Полимеризационные карбоксильные катиониты получаются
путем совместной полимеризации акриловой и метакриловой
кислот с непредельными соединениями, содержащими по мень-
меньшей мере две двойные связи, с целью получения трехмерного не-
нерастворимого полимера. Типичным представителем карбоксиль-
карбоксильных смол этого типа являются продукты сополимеризации ме-*
такриловой кислоты с дивинилбензолом (смолы КБ-4, КБ-4П-2,
КМД).
Одной из важнейших характеристик ионообменных смол яв-
является их обменная емкость, т. е. количество миллиграмм-экви-
миллиграмм-эквивалентов, поглощенных ионов 1 г сухого сорбента, или количест-
380
во ионообменных групп, приходящихся на 1 г сухого ионита.
Элементарное звено ионита типа КБ-4:
СН3 СН3
—С—СН2—СН—СН2—С—СН2—
соон ^\ соон
—С—СН2—СН—СН2—-С—CH2—
соон соон
м-экв
Обменная емкость ионита составляет 10—11 —~. Путем
сополимеризации с другими «мостикообразующими» агентами
получены карбоксильные кантиониты КМТ, КМ, КМГ и СГ.
Другой тип карбоксильных катионитов получается путем
совместной конденсации по механизму, сходному с образова-
образованием фенолформальдегидных смол. Типичным представителем
этих смол является- (катионит КФУ — продукт ко/нденсации
феноксиуйсуоной кислоты и формальдегида:
ОСН2СООН ОСН2СООН
В последние годы для выделения биологически активных ве-
веществ (особенно ферментов) используется карбоксильный ка-
катионит КМТ — сополимер метакриловой кислоты гексагидро-1,
3, 5, 3 акрилтриазина: элементарное звено этого сорбента:
О СН3
II I I
N—C—СН—СН2—СН2—С—
44 соон
?■ I ?"■ -
—С-СН2—СН2—СН-С—N N--C—СН—СН2—СН2—С—
I II \/ II I I
СООН О О СООН
м-экв
Обменная емкость ионита достигает 10—И ~— . В струк-
структуре этого сорбента сшивающий агент имеет три двойные свя-
связи. Это способствует дополнительному разветвлению сетки
ионита и возникновению неоднородных по плотности участков
смолы. Такие особенности структуры сорбента позволяют до-
381
биться лучшего совпадения «формы» матрицы сорбента и сор-
сорбируемого иона.
Карбоксильные катиониты проявляют свои ионообменные
свойства в нейтральных или щелочных растворах и относятся к
слабым катионитам.
Из сульфокатионитов в производстве антибиотиков наи-
наибольшее применение находят сульфокатиониты, получаемые на
основе сульфостирола. Среди них центральное место занимают
сорбенты, полученные совместной полимеризацией сульфости-
сульфостирола и дивинилбензола (смолы СДВ-3, КУ-5). Структура этого
типа катионитов может быть представлена следующим обра-
образом:
_СН СН2 СН СН9 СН—СНо—
SO3"H+ ,
~СН—СН2-
В результате сополимеризации стирола и дивинила при по-
последующем сульфинировании получены иониты типа СБС-1,
СБС-2, СБС-3.
SO3H+
- СН-СН2-СН2-СН-С
Сульфокатиониты проявляют ионообменные свойства в ши-
широком диапазоне рН (от 2,0 до 8,0) и относятся к сильным ка-
тионитам. Обменная емкость сульфокатионитав достигает
мг-экв
3—5——■
Среди анионитов различают два существенно различных
класса сорбентов — слабоосновные и сильноосновные аниони-
ты. Слабоосновные аниониты содержат в своей структуре пер-
первичные, вторичные или третичные аминогруппы.
Сильноосновные аниониты представляют собой полимеры,
содержащие четвертичные аммониевые основания. Слабооснов-
Слабоосновные аниониты вступают в реакцию ионного обмена лишь в силь-
сильнокислой среде. Сильноосновные аниониты вступают в реакцию
ионного обмена в нейтральной и даже в слабощелочной среде.
382
Из анионитов промышленное значение имеют сорбенты ЭДЭ-10,
АН-2Ф. ЭДЭ-10 используется для нейтрализации и депигмента-
депигментации растворов антибиотиков, а АН-2Ф используется для нейтра-
нейтрализации растворов антибиотиков. Обменная емкость указанных
1 л 11 МГЭКВ
ионитов достигает 10—11 —-—.
Анионит ЭДЭ-10 представляет собой продукт конденсации
этилендиамина и полиэтиленаминов с эпихлоргидрином:
2—сн-сн2—/
ОН
Слабоосновной анионит АН-2Ф получают поликонденсацией
метилольных производных полиэтиленаминов и фенола в кис-
кислой среде.
t-N-(CH2-CH2-NH-)n~CH2-CH2-N-
НО СН2 Н2С ОН
-N-(CH2-CH2-NH-)n-CH2-CH2-N-
Основные требования, предъявляемые к ионитам. Обычно
качество ионитов отражают условной оценкой сорбционных
свойств ионита (полная обменная емкость сорбента, емкость
сорбента при различных рН среды, скорость установления сорб-
ционного равновесия, объем десорбирующего раствора и полно-
полнота десорбции). Важная характеристика ионита — его физические
свойства в определенных условиях (набухаемость и прочность
зерен, теплостойкость и химическая стойкость и т. д.).
Ионит, применяемый для извлечения ценных ионов из рас-
раствора или для очистки того или иного вещества, должен обла-
обладать возможно большей емкостью. Для выполнения этого ус-
условия необходимо, чтобы в синтезируемом сорбенте на едини-
единицу веса сорбента содержалось возможно большее число ионо-
генных групп, полностью ионизированных в конкретных усло-
условиях сорбции. Следует помнить, что ионизация фенольных ок-
сигрупп становится заметной при рН выше 9,0, ионизация кар-
карбоксильных групп — при рН выше 5,0. Сульфогруппы полностью
ионизированы даже в кислой среде, амино- и иминогруппы
вступают в реакцию ионного обмена в кислой среде, четвертич-
четвертичные аммониевые основания проявляют свойства ионообменного
сорбента в нейтральных и слабощелочных средах. Иониты, при-
применяемые для хроматографического анализа, должны содер-
содержать однотипные кислотные или основные группы. В этом слу-
случае легко достигнуть четкого разделения смеси.
383
При выборе ионитов необходимо такж^учитывать относи-
относительную прочность связи подвижных ионов с ионитом, чтобы
десорбция их осуществлялась столь же легко, как и сорбция.
Чем больше энергия ионной связи между подвижным и непод-
неподвижными ионами сорбента, тем труднее сместить равновесие в
период десорбции.
Для выделения крупных органических ионов, в том числе
антибиотиков, используются, как правило, набухающие иониты,
так как на сильносшитых ионообменных сорбентах функцио-
функциональные группы оказываются недоступными для ионов антибио-
антибиотиков. В структуре ионообменных сорбентов органического про-
происхождения имеются исходные мономеры, обладающие значи-
значительной растворимостью в водных растворах. При синтезе ионо-
ионообменного сорбента хорошо растворимые в воде и водных рас-
растворах мономеры сшиваются в полимерную цепь, в целом теряю-
теряющую способность растворяться в воде, но способность погло-
поглощать воду и вследствие этого набухать остается. Способность
ионообменных сорбентов набухать в водных растворах выра-
выражается коэффициентом набухания (/Снаб). Коэффициент набуха-
набухания для ионитов определяется как отношение объема набухше-
набухшего в воде ионита (V\) к объему сухого ионита {У2):
к У*
Анаб. =~"j7~~~«
У производственных сорбентов Кшб. обычно равен 3—4.
Для набухающих ионитов большое значение приобретает
степень набухания его в сорбируемых растворах. С увеличени-
увеличением набухаемости возрастает доступность ионогенных групп, что
в свою очередь увеличивает емкость сорбента и скорость уста-
установления сорбционного равновесия. С величиной набухаемости
тесно связана прочность зерен сорбента; с повышением набухае-
набухаемости сорбента она уменьшается. При резком изменении сте-
степени набухания в зернах ионита возникают внутренние напря-
напряжения, вызывающие их разрушение. Механическая прочность
ионитов имеет огромное значение в технологическом процессе
выделения антибиотиков. Малая механическая прочность сор-
сорбента приводит к быстрому истиранию его в ионообменной ко-
колонне и потере с уходящим раствором.
Равновесная обменная емкость ионитов зависит от многих
факторов, определяющих состояние равновесия в системе рас-
раствор—ионит, и является переменной 'величиной. При 'ионообмен-
'ионообменной сорбции органических ионов-антибиотиков, кроме структуры
сорбента и сорбируемого иона, большое значение имеет величи-
величина рН внешнего раствора. В общем случае по мере увеличения
рН раствора обменная емкость катионита, содержащего одно-
одноименные по заряду активные группы, прямолинейно возрастает,
а обменная емкость анионита прямолинейно падает.
Изменением рН среды можно регулировать степень диссо-
384
циации сорбируемого или десорбируемого вещества, а также са-
самого сорбента. Изменение степени диссоциации веществ явля-
является одним из важнейших факторов, позволяющих создавать
условия, благоприятные для сорбции или вытеснения ионов.
Ярким примером в этом отношении является процесс сорб-
сорбции и десорбции антибиотиков тетрациклиновой группы. Сорб-
Сорбция антибиотиков этой группы сульфокатионитами проводится
при рН 2,0, т. е. в таких условиях, когда они в растворе явля-
являются однозарядными катионами. Десорбция антибиотиков тет-
тетрациклиновой группы проводится щелочными растворами с рН
10,0—11,0, когда антибиотики этой группы превращаются в
двузарядный анион и отталкиваются от сорбента.
Существенное значение при ионообменном процессе имеют
свойства растворителя. Обмен ионов в гетерогенных системах
может протекать не только в водных растворах, но и при ис-
использовании других полярных растворителей, в которых наблю-
наблюдается диссоциация электролитов.
Применение неводных растворов при обмене ионов метал-
металлов, ионов водорода и аммония обычно представляет ограничен-
ограниченный интерес. Совершенно особые возможности открываются при
использовании неводных растворов для обмена ионов органиче-
органических соединений. Во всех изученных системах относительная
сорбируемость ионов органических веществ по сравнению с сор-
бируемостью неорганических ионов заметно падает. Это явле-
явление может быть объяснено двумя причинами. С одной стороны,
в неводных растворах существенно уменьшается сольватация
ионов, причем особенно заметно это должно сказываться на
сильносольватированных в водных растворах небольших ионах.
С другой стороны, ухудшение сорбируемости органических ве-
веществ может быть объяснено уменьшением их степени иониза-
ионизации, что в первую очередь проявляется именно у органических
соединений. Подобное смещение равновесия в сторону вытесне-
вытеснения ионов органических веществ из ионитов при переходе к не-
неводным растворам имеет большое значение как один из спо-
способов десорбции трудно десорбируемых веществ.
Экспериментальные исследования показали, что ионы мно-
многих органических соединений сорбируются ионообменными смо-
смолами с большой избирательностью из растворов, содержащих
одновременно ионы металлов, ионы аммония или водорода. Кон-
Константы ионного обмена для большинства изученных систем с ор-
органическими ионами составляют несколько сотен, а иногда пре-
превышают даже 1000.
Большие величины констант обмена, полученные для ионов
органических соединений, указывают на большую прочность
связи этих ионов с ионитами, которая не может быть объяснена
лишь электростатическими силами. По-видимому, при сорбции
ионов органических соединений проявляется дополнительное
взаимодействие.
25-926 385
Рассмотрение влияния различных факторов на процесс ио-
нообмена позволяет сделать вывод о существовании весьма
сложной зависимости между свойствами среды и твердой фазы
при ионообмене.
Из существующих теоретических уравнений изотерм ионооб-
мена наибольшего внимания заслуживает уравнение Никольско-
Никольского, выведенное им на основе термодинамики и статистической
механики для обмена двух катионов любой валентности. Это
уравнение имеет вид:
1 1
= А
- *2 п 22
а а2
где К — константа ионного обмена; а,\, а2— активности ионов в
растворе; aif а2— активности ионов в сорбенте; zu z2— заря-
заряды ионов.
Рассматривая особенности взаимодействия сорбированных
ионов с сорбентом и окружающей средой на основе статистиче-
статистических представлений, Никольский высказал мысль, что коэффи-
коэффициентами активности ионов в ионите можно пренебречь. В этом
случае уравнение упростится:
г 2 ' 22
т2 а2
где ти т2— концентрация сорбированных ионов в мг-экв на
1 г сорбента.
Для разбавленных растворов, когда активности ионов ,в
растворе примерно равны их концентрациям, уравнение еще бо-
более упрощается:
г»Zl rZl
m 22 r 22
В ряде исследований показана справедливость уравнения для
многих систем.
Если рассмотреть теорию сорбции ионов на набухающих
ионитах, особенно для ионов больших размеров, к которым от-
относятся антибиотики, то можно видеть, что при расчете обмен-
обменной емкости ионитов учитываются только те ионообменные
центры сорбента, которые доступны для органического иона.
Ионообменные группы сорбента, недоступные для иона анти-
антибиотика, занимаются ионами малых размеров, и их количество
определяется по разности (т—т\). В соответствии с этим урав-
386
нение изотермы сорбции для органических ионов принимает
вид:
1 1
, «1
(т —
где т\ — обменная емкость ионита по органическому иону в
присутствии неорганического иона с концентрацией С2; m — об-
обменная емкость ионита по органическому иону при отсутствии
в растворе неорганического иона (С2=О); С\ — концентрация
органического иона.
Последнее уравнение оказалось справедливым для обмена
ионов ряда антибиотиков с неорганическими ионами.
Самое первое и прочное место ионообменный метод завое-
завоевал при выделении и очистке стрептомицина. Стрептомицин в
большинстве стран мира в настоящее время выделяется и очи-
очищается с помощью карбоксильных катионитов в солевой (чаще
всего натриевой) форме. Сорбция, как правило, проводится из
нейтральных растворов карбоксильными катионитами КБ-4П-2,
КБ-4 или КБ-2. Сорбция осуществляется в колоннах открытого
типа (в кипящем слое) при подаче раствора снизу вверх, это
позволяет несколько увеличить емкость ионита, а также избе-
избежать в производстве неприятного явления — слеживания смолы
в колоннах с верхней подачей раствора. \
Процесс сорбции стремятся проводить до полного насыще-
насыщения сорбента стрептомицином или на 75—80% от возможной
емкости сорбции. Перед началом процесса сорбции культураль-
ную жидкость стрептомицина подвергают предварительной об-
обработке для коагуляции белков и удаления поливалентных ме-
металлов. С этой целью ее обрабатывают щавелевой кислотой
(рН около 3,0), и она освобождается от мицелия и осадка поли-
поливалентных металлов (оксалат кальция). Затем для более пол-
полного удаления белковых примесей культуральную жидкость под-
подвергают тепловой коагуляции. В полученный после такой обра-
обработки нативный раствор добавляют определенное количество
триполифосфата натрия, образующего.с ионами магния раство-
растворимый комплекс. При бесфильтрационном методе сорбции
стрептомицина культуральную жидкость подвергают автолизу
при нагревании до 40—50 °С с целью уменьшения вязкости рас-
раствора до 2 сп. Поливалентные металлы, как в предыдущем слу-
случае, переводят в осадок щавелевой кислотой или триполифос*
фатом в неионизированный растворимый комплекс и культу-
культуральную жидкость без фильтрации направляют на сорбцию.
Сорбцию стрептомицина проводят на карбоксильном катионите
КБ-4, КБ-2 в солевой (натриевой) форме при нейтральном зна-
значении рН раствора. Для практически полного удаления из кати-
онита сорбированных ионов магния после окончания процесса
25* -387
сорбции смолу промывают раствором, содержащим триполи-
фосфат натрия и стрептомицин. Избыток этого раствора отмы-
отмывают обессоленной водой и проводят процесс десорбции стреп-
стрептомицина 4—5% раствором серной кислоты.
Процесс сорбции и десорбции можно представить в виде
следующей схемы:
3KB"Na+ + Str+++ > (KB"KStr+++ + 3Na+
Десорбция:
2(KB!3Str+++ + 3H2SO4 >■ (Str+++J(SO4K + 6КБ~Н+
В процессе десорбции получают высокоактивные и низкоак-
низкоактивные фракции. Низкоактивные фракции используют для де-
десорбции на последующей колонне или присоединяются к натив-
ному раствору. Высокоактивные фракции, если они нейтраль-
нейтральные, направляют на деминерализацию и затем на нейтрализа-
нейтрализацию. Кислые активные фракции подвергают сначала нейтрали-
нейтрализации на анионите ЭДЭ-10 в ОН-форме, а затем также направ-
направляют на деминерализацию и нейтрализацию.
Деминерализация и нейтрализация элюатов
стрептомицина наионитах. В основу процесса деми-
деминерализации и нейтрализации положен метод «молекулярных
сит», заключающийся в следующем. Раствор стрептомицина,
проходя через катионит с большим количеством сшивающего
агента, ввиду большого размера молекулы антибиотика и ма-
малой набухаемости сильносшитого катионита, не проникает к
ионообменным центрам сорбента. Ионы металлов натрия, маг-
магния и др., обладая малыми размерами, легко проникают к ионо-
ионообменным центрам сильносшитого катионита и остаются на
нем, обмениваясь с водородом ионита (катионит для деминера-
деминерализации берется в Н-форме), В производстве стрептомицина
для деминерализации элюатов нашли применение сильносши-
тые сульфокатиониты типа СБС и КУ-2-20.
Процесс деминерализации можно записать в виде следую-
следующей схемы:
RiSO3H+ + Z+A" > RiSOsZ* + Н+ + А",
где Z — катион, А — анион.
Как видно из схемы, в раствор выделяются ионы водорода,
х раствор подкисляется, поэтому следующей стадией является
процесс нейтрализации элюатов на анионитах в ОН-форме. Ней-
Нейтрализацию растворов стрептомицина можно проводить на раз-
различного типа анионитах, так как стрептомицин практически не
сорбируется на них.
Процесс нейтрализации на сильноосновном анионите можно
представить в виде следующей схемы:
R^OH" + Н+ + А" >- RjA" + Н2О,
где R2 ОН~ — анионит в основной форме; R2 А- — анионит в
солевой форме.
388
В случае применения слабого анионита нейтрализация про-
протекает по уравнению:
RNH2 + Н+ + А" >- RNHjA",
где RNH2 — анионит в основной форме; RNHjA" — анионит в
солевой форме.
Нейтрализация растворов кислот протекает на многих анио-
нитах различно в статических (в аппарате с'мешалкой) и ди-
динамических условиях (в иоонобменной колонне). В статичес-
статических условиях сильнооснавные группы (четвертичные аммоние-
аммониевые основания) первыми принимают участие в нейтрализации
раствора, после чего оставшиеся неиспользованными слабоос-
слабоосновные группы (первичные, вторичные и третичные аминогруп-
аминогруппы) не могут нейтрализовать раствор, рН которого повысился
до 2,5—3,0. В динамических условиях процесс такой частичной
нейтрализации происходит только во фронтальной части посту-
поступающего в колонну раствора. За фронтом раствора в колонку
поступает исходный раствор высокой кислотности (почти 1 н.
раствор H2SO4). В этих условиях слабоосновные центры также
принимают участие в нейтрализации слабокислотного раствора.
Деминерализация растворов может быть осуществлена как
рассмотренным методом при двухстадийном процессе катионно-
го и анионного обмена, так и при одностадийном процессе с
применением катионита и анионита в одной колонне (деминера-
(деминерализация раствора в смешанном слое ионитов).
Деминерализованные элюаты, если требуется, обрабатыва-
обрабатывают кислым активированным углем для удаления пигментных
веществ и направляют в отделение сушки. Этот процесс прово-
проводят обычно на двухступенчатой распылительной сушилке. Рас-
Раствор стрептомицина, предварительно простерилизованный филь-
фильтрацией через фильтры Зейтца, на первой ступени упаривают
до определенной концентрации и затем при непрерывном поступ-
поступлении его на вторую ступень высушивают до остаточной влаж-
влажности 0,5%. Высушенный продукт расфасовывают в стерильных
условиях.
Стрептомицин является эффективным средством лечения ту-
туберкулеза. Он рассматривается как основной противотуберку-
противотуберкулезный препарат, применяемый при всех формах туберкулеза
легких.
Стрептомицин используют при лечении туляремии, чумы,
бруцеллеза, менингитов различной этиологии, бактериального
эндокардита, перитонитов, инфекций мочевых путей, кишечных
инфекций и других заболеваний, вызываемых чувствительными
к стрептомицину возбудителями. Стрептомицин-сульфат приме-
применяют внутримышечно, местно, в виде аэрозолей, внутритрахе-
ально, внутрикостно и т. д. Стрептомицин-сульфат выпускают
стерильно во флаконах по 250, 500 мг и 1 г B50 000, 500 000 и
1000 000 ЕД).
389
Выделение и очистка антибиотиков тетра-
циклиновой группы ионообменным методом.
Процесс химической очистки начинают с предварительной обра-
обработкой культуральной жидкости. Для удаления ионов кальция
к культуральной жидкости добавляют щавелевую кислоту до
рН кальция 3,0—3,4. При этом тетрациклин, находившийся в
виде нерастворимого кальциевого комплекса, переходит в ща-
щавелевокислый тетрациклин, а ионы Са++ осаждаются в виде
оксалата кальция, выпадающего в осадок при рН 3,0. Затем к
культуральной жидкости добавляют соляную кислоту до рН
1,9 и желтую кровяную соль для удаления ионов Fe++. После
перемешивания и выдержки нативныи раствор отделяют фильт-
фильтрацией от осадка и направляют на сорбцию. Процесс сорбции
тетрациклина ионитом состоит в обмене ионов между раство-
раствором и сорбентом. Каждому миллиграмм-эквиваленту тетрацик-
тетрациклина, сорбированного из раствора, соответствует миллиграмм-
эквивалент десорбированных и перешедших в раствор ионов
водорода в соответствии с уравнением реакции ионного обмена:
RSO JH+ + ТЦ+ >• RSOs ТЦ+ + Н+.
Для сорбции могут использоваться сульфокатиониты типа
СБС-3 (сополимер стирола и бутадиена, обработанный серной
кислотой) или сульфокатионит СДВ-3 (сополимер стирола и ди-
винилбензола). Сорбцию проводят последовательно на несколь-
нескольких колоннах. После полного насыщения смолы в первой колон-
колонне обедненный нативныи раствор подают на сорбцию в следу-
следующую колонну. После окончания сорбции нативныи раствор
вытесняют из колонны обессоленной водой. Затем ионит промы-
промывают от остатков нативного раствора и неорганических ионов:
1) обессоленной водой снизу вверх до появления на выходе из
колонны прозрачной жидкости; 2) 2,5 н. раствором NaCl;
3) обессоленной водой для вытеснения избытка раствора.
Десорбцию тетрациклина с ионита проводят 2 н. раствором
аммиака (рН 10,3—10,5). Элюаты с активностью выше
3000 ЕД/мл собирают и направляют на стадию осаждения тех-
технического основания, а слабоактивные элюаты, подкисленные
соляной кислотой, вновь направляют на сорбцию. В процессе
десорбции тетрациклина ионами аммония происходит перенос
катионов тетрациклина из сорбированного состояния на смоле
в щелочной раствор, в котором антибиотик может находиться
только в виде анионов:
ТЦ+ > Н+ + ТЦ
I-*. н+ + ТЦ-
'-*■ Н+ + ТЦ —
Для осуществления процесса десорбции применяют избыток
аммиака. В противном случае согласно первой ступени схемы
390
тетрациклин переходит в нейтральное состояние и выпадает в
осадок в колонне.
Высокоактивные фракции после десорбции подкисляют со-
соляной кислотой до рН 4,5—5,0. При этом в осадок выпадает
техническое основание. Его отделяют от маточника и направ-
направляют на стадию осветления. Для этого техническое основание
растворяют в 2,5% растворе щавелевой кислоты и обрабатыва-
обрабатывают активированным углем. После отделения угля рН фильтрата
доводят раствором аммиака до 4,0; при этом в осадок снова
выпадает основание тетрациклина. Основание тетрациклина вы-
высушивают и расфасовывают. После десорбции тетрациклина
смолу регенерируют: для удаления органических примесей и пе-
перевода смолы в Н+- или №+-форму регенерацию проводят сна-
сначала 1 н. раствором едкого натра и затем 2 н. раствором тех-
технической соляной кислоты.
После каждой обработки смолы щелочью или кислотой ее
отмывают обессоленной водой.
Аппаратура ионообменной сорбции антибиотиков. Аппара-
Аппаратура для сорбционных ионообменных процессов значительно бо-
более про-ста по устройству по сравнению с экстракционной и до-
доступнее по стоимости. Конструкция ионообменного фильтра,
называемого также колонной, для сорбции в динамических ус-
условиях должна обеспечивать: 1) постоянное нахождение смо-
смолы под слоем раствора; 2) минимальный унос мелких зерен
смолы с уходящим фильтратом; 3) минимальную «слеживае-
мость» слоя смолы; 4) отсутствие «мертвых пространств» в
сорбенте; 5) незначительное разбавление водой обрабатываемых
растворов.
Ионообменные колонны изготавливаются из углеродистой
стали, защищенной от коррозии гуммированием, а также из ви-
винипласта, стекла, плексиглаза. Высота ионообменных колонн
обычно значительно больше, чем диаметр. В производстве ис-
используются два типа ионообменных колонн — закрытые и от-
открытые.
Закрытые ионнобменные колонны представляют собой гер-
герметичную конструкцию, работающую под давлением до 3 ат.
Одна из конструкций таких колонн представлена на рис. 47.
В верхней и нижней частях колонны имеется ложное виниплас-
товое дно с ввинчивающимися колпачками. Колпачки также из-
изготовляются из винипласта и имеют прорези, которые пропускают
раствор и задерживают зерна смолы. В верхней части колонны
над ложным дном имеется распределяющее устройство для
равномерной подачи раствора.
Ионообменный фильтр открытого типа (рис. j48) имеет в
нижней части колонны аналогичное устройство, 5но колпачки
дополнительно покрываются измельченным стеклом, которое
улучшает фильтрацию и предохраняет колпачки от засорения
частицами смолы и механическими примесями.
391
Рис. 47. Ионитовый фильтр закрытого
типа.
/ — нижний патрубок для входа и выхода
жидкости; 2 —днище; 3 —диск нижнего
распределительного устройства; 4 — слой
ионообменной смолы; 5 —корпус; 5 —опор-
—опорная лапа; 7 — слой гуммировки; 8 — верх-
верхнее распределительное устройство; 9 —■
крышка; 10 — загрузочный люк; // — шту-
штуцер для воздушника; 12 — верхний патру-
патрубок для входа и1 выхода жидкости; /3 —
смотровое окно в крышке люка; 14 — щеле-
щелевой колпачок.
Рис. 48. Ионитовый фильтр от-
открытого типа.
/ — днище; 2 —нижний распредели-
распределитель жидкости; 3 — нижний патру-
патрубок для входа и выхода жидкости;
4 — дренажный настил; 5 — царга
корпуса; 6 — слой ионообменной»
смолы; 7 — расширитель-отстойник;
В — переливной патрубок; Р —коль-
—кольцевой карман; 10 — патрубок для
выхода жидкости; 11 — крышка;
12 — патрубок для входа жидкости;
13 — верхний распределитель жид-
жидкости.
Верхнее распределительное устройство для подачи жидкости
представляет собой диспергатор типа душевого рожка. В верх-
верхней части колонны находится расширение, предназначенное для
осаждения под действием силы тяжести взвешенных в выходя-
выходящей жидкости мелких частиц смолы. Частицы смолы оседают
в кольцевом кармане, из которого через патрубок снова возвра-
возвращаются в колонну. Ионообменный 'процесс сорбции обычно осу-
осуществляется в батарее колонн C—5 шт.). Нативный раствор
пропускают через первую колонну до появления в проходящем
растворе активности и затем подключают следующую колонну.
Неактивный отработанный нативный раствор направляют в ка-
канализацию. После насыщения ионита антибиотиком колонну
отключают, промывают обессоленной водой и ионит подверга-
подвергают десорбции. Перед промывкой обессоленной водой сорбент
для дезинфицирования промывают формалином, а после десорб-
десорбции производят регенерацию ионита и снова проводят сорбцию.
Нативный раствор в колонну открытого типа подается сни-
снизу вверх с линейной скоростью несколько больше критической,
при которой сопротивление слоя становится равным погружен-
погруженному весу слоя, и зерна переходят во взвешенное состояние.
Сорбция в таком псевдоожиженном слое протекает быстрее,
увеличивается сорбционная емкость смолы и чистота элюатов.
Количество ионообменных колонн, необходимое для обеспе-
обеспечения заданной мощности производства, может быть рассчитано
несколькими способами. Один из них осуществляется по фор-
формуле:
^с т.'Тцикл.
где GCyT. — количество смолы, необходимое для сорбции суточ-
суточного количества антибиотика; GKOn. — количество смолы в одной
колонне; тЦИкл. — время сорбционного цикла в часах.
§ 3. ОСАЖДЕНИЕ И КРИСТАЛЛИЗАЦИЯ
Наряду с экстракционными и ионообменными методами в
промышленности антибиотиков применяют методы выделения
антибиотиков из нативных растворов осаждением. В основе ме-
методов осаждения лежит способность антибиотиков образовы-
образовывать с неорганическими, а также органическими ионами и це-
целыми молекулами сложные, сравнительно легко распадающие-
распадающиеся химические соединения — комплексы. Подобные нераствори-
нерастворимые комплексные соединения возникают, например, при взаи-
взаимодействии тетрациклиновых антибиотиков с ионами кальция,
магния, цетазола в щелочной среде. Выпавший комплекс отде-
отделяют от отработанного нативного раствора и подвергают разло-
разложению. При этом антибиотик извлекают из смеси экстрак-
экстракцией или перекристаллизацией.
393
Так, в производстве ХТЦ антибиотик осаждают из нативно-
нативного раствора при щелочных значениях рН G,2—7,4) в виде ком-
комплексного соединения с ионами кальция, магния и другими ио-
ионами, находящимися в растворе. Отфильтрованное соединение
высушивают и обрабатывают раствором хлорида кальция в ме-
метаноле. При этом происходит распад кальциевого комплекса, и
хлортетрациклин в виде нового комплексного соединения (с
хлористым кальцием) переходит в метанол, причем благодаря
избирательности растворителя и хорошей растворимости в нем
хлоркальциевого комплекса достигаются очистка и концентри-
концентрирование антибиотика.
Выделение и очистка окситетрациклина методом осаждения
предусматривают использование в качестве комплексообразо-
вания цетазола. Цетазоловый комплекс разлагается разбавлен-
разбавленной кислотой, а из водного раствора осаждают окситетрацик-
лин в виде основания.
В последние годы на концентрированных (обогащенных)
питательных средах удается получить значительные активности
окситетрациклина, поэтому в производстве оказалось возмож-
возможным использовать «прямой метод» осаждения окситетрациклина-
основания непосредственно из нативного раствора. Для реализа-
реализации этого метода требуются нативные растворы повышенной
степени чистоты, что достигается более глубокой очисткой их
при коагуляции и предварительной обработке.
Выделение и химическая очистка методом осаждения свя-
связаны с переводом антибиотиков в твердую фазу. Экстрагирова-
Экстрагирование антибиотика из твердого комплексного соединения, как и
экстракция из раствора, является диффузионным процессом, в
основе которого лежит переход молекул извлекаемого вещества
в растворитель. Однако вследствие твердого состояния исход-
исходной фазы, наличия у нее стабильной поверхности раздела рас-
рассматриваемый вид экстракции имеет существенные отличия от
жидкостной. Поскольку при экстракции антибиотика из твердо-
твердого комплекса взаимодействие с жидкостью имеет место главным
образом на поверхности раздела фаз, очевидно, следует стре-
стремиться к возможному ее увеличению.
Кристаллизация антибиотиков, как и других веществ, осно-
основана на резком уменьшении их растворимости в результате из-
изменения температуры раствора (обычно понижения, но иногда,
например в случае эритромицина, — повышения) или пере-
перевода антибиотика в другую (плохо растворимую) химическую
форму.
Последнее достигается изменением рН раствора или добав-
добавлением соответствующего реагента, часто с одновременным сни-
снижением температуры (например, окситетрациклин-основание
осаждается при подщелачивании кислого водного раствора до
рН 4,0 с одновременным охлаждением до 5—7°С). Кристалли-
Кристаллизация является не только способом получения антибиотика в
394
твердом виде, но и очень эффективным средством его очистки
от сопутствующих примесей.
В твердом состоянии вещества могут быть кристаллически-
кристаллическими и аморфными. Кристаллическое состояние отличается от
аморфного расположением молекул, атомов или ионов в опре-
определенном и строгом порядке. Рентгеновский анализ показал,
что многие аморфные вещества имеют правильное расположение
молекул, но термин «кристаллический» применяется для обоз-
обозначения высокой степени внутренней упорядочности, приводя-
приводящей к образованию определенных наружных граней кристалла.
Кристаллы, образующиеся у этой поверхности, в конце кон-
концов, попадают в раствор и затравляют его, прежде чем в ос-
основной массе раствора достигаются условия, представленные
точкой С1.
На практике чаще всего применяют сочетание охлаждения
и испарения. Такой процесс на рис. 49 представлен линией
АВ"С".
Кристаллизация и растворение. Обычно процессы кристал-
кристаллизации и растворейия считают равновеликими и взаимосвязан-
взаимосвязанными. Если бы они были по природе чисто диффузионными, то
скорость кристаллизации должна равняться скорости растворе-
растворения при данной температуре. В результате при одинаковых дви-
движущих силах, т. е. при одинаковых отклонениях от равновес-
равновесных насыщенных состояний, все грани кристалла должны расти
и растворяться с одинаковой скоростью. Такие условия редко
достигаются на практике. Кристаллы обычно растворяются
быстрее, чем растут, и различные грани чаще всего растут и
растворяются с неодинаковыми скоростями. При росте кристал-
кристалла грани его гладкие, а когда он растворяется, грани его обычно
испещрены ямками, что приводит к увеличению площади кон-
контакта между твердой и жидкой фазой.
Растворимость кристаллов зависит от их размеров (рис. 50).
Растворимость весьма малых кристаллов значительно повыша-
повышается. В результате при слишком малых размерах кристаллы
исчезают из системы, а крупные растут. Это явление несколько
затрудняет начальную фазу кристаллизации. Первые кристал-
кристаллы («зародыши») не могут образовываться из-за весьма малых
размеров.
Величина выпадающих кристаллов зависит от поверхностно-
поверхностного натяжения раствора, которое может изменяться в зависи-
зависимости от содержания примесей. Чем больше поверхностное на-
натяжение, тем сильнее его действие в направлении уменьшения
общей, межфазной поверхности на границе твердое вещество —
жидкость. Это ведет к образованию кристаллов больших разме-
размеров, так как их удельная поверхность (поверхность на единицу
массы) меньше удельной поверхности мелких кристаллов.
Следует отметить, что крупные кристаллы не могут обладать
большой чистотой. Вследствие агломерации они содержат вклю-
395
чения маточного раствора, в то время как мелкие кристаллы
отличаются однородностью (чистотой), но большая поверхность
затрудняет их промывку. Величина кристаллов и их форма су-
существенно влияют не только на степень чистоты продукта, но
и на последующие методы перевода их в соответствующие ле-
лекарственные формы (таблетки и т. д.).
Рост кристаллов. Как только в пересыщенной или переох-
переохлажденной системе образовались устойчивые зародыши, т. е.
I
/
m
Рис. 50. Зависимость рас-
растворимости от величины
кристаллов.
с _ концентрация; d —величина
кристалла.
Температура
Рлс. 49. Пути юоздания условий кри-
кристаллизации.
/ _ лабильная (пересыщенная) область, в
которой спонтанная кристаллизация ве-
вероятна, но не является неизбежной; II —
метастабильная (пересыщенная) область
между кривыми растворимости и пересы-
• шения, где спонтанная кристаллизация не-
невероятна; /// — устойчивая (ненасыщенная)
область, в которой кристаллизация невоз-
невозможна. Остальные обозначения в тексте.
частицы больше критического размера, они начинают расти,
превращаясь в кристаллы видимого размера. Рост кристаллов
зависит от очень многих причин. Остановимся на некоторых из
них
Типичная кривая образования зародышей кристаллизации
показана на рис. 51. Для всех изученных систем установлено,
что оптимальная температура зарождения центров кристалли-
кристаллизации была гораздо ниже той, которая требовалась для макси-
максимального роста кристаллов (рис. 51). При температуре ниже
оптимальной вязкость увеличивалась до такой величины, кото-
которая препятствовала кристаллизации, в то время как при темпе-
температуре выше оптимальной зарождению центров кристаллиза-
кристаллизации препятствует интенсивное молекулярное движение жид-
жидкости.
Влияние перемешивания. Скорость роста кристаллов при
данной темйературе в постоянных условиях пересыщения может
значительно изменяться при перемешивании жидкости или вра-
вращении кристалла относительно жидкости. Скорость роста зна-
396
чительно увеличивается на первых стадиях, но вскоре достига-
достигаются такие условия, когда дальнейшее ускорение перемешива-
перемешивания не оказывает никакого влияния (рис. 52). Из подобных ис-
исследований были сделан вывод, что диффузия не является един-
единственным и наиболее важным фактором, который влияет на
процесс кристаллизации. Кроме того, в кристаллизаторе, в ко-
котором кристаллы поддерживаются во взвешенном состоянии пу-
50" /00°
Рис. 51. Зависимость чис-
числа центров кристаллиза-
кристаллизации (ЧЦК) от степени
переохлаждения.
/О 20 30 40 50 60
Спорость Вращения, о(Цнин
Рис. 52. Влияние скорости (вращения
кристалла на скорость роста кристал-
кристалла тиосульфата натрия при 45 °С и
различных пересыщениях:
/ — 1,5—2; 2 — 1,03; 5 — 0,5 г тиосульфата
натрия на 100 г воды.
тем перемешивания, большие кристаллы будут расти быстрее,
чем маленькие, так как для первых окажутся благоприятными
более высокие относительные скорости реакции между твердым
веществом и раствором.
Форма кристаллов зависит от очень многих факторов:
свойств растворителя, рН раствора, наличия примесей, степени
пересыщения или переохлаждения, скорости охлаждения, темпе-
температуры кристаллизации, интенсивности перемешивания и т. д.
Быстрое охлаждение раствора часто вызывает предпочтитель-
предпочтительный рост кристалла в одном направлении, что приводит к обра-
образованию иголок.
Наиболее частым случаем, вероятно, следует считать изме-
изменение формы кристалла в присутствии примесей в кристалли-
кристаллизующемся растворе. Изменение формы под влиянием примесей
представляет собой в сущности поверхностное явление: молеку-
молекулы или ионы примесей притягиваются к различным граням
кристалла и связываются с поверхностью в результате физиче-
физического или химического процесса. Это явление уменьшает пло-
площадь, пригодную для зарождения центров кристаллизации на
поверхности для осаждения растворенного вещества, и рост на
той грани замедляется.
397
Расчет выхода кристаллов. Расчет выхода кристаллов, обра-
образующихся при кристаллизации в результате охлаждения, очень
прост, если известны первоначальная концентрация и раство-
растворимость вещества при низкой температуре. Расчет может ус-
усложниться, если в процессе охлаждения некоторая часть раст-
растворителя удаляется (или теряется) или вещество само связы-
связывает некоторую часть растворителя, например путем захвата
кристаллизационной воды. Примем обозначения: С\ — первона-
первоначальная концентрация раствора (в граммах безводной соли на
I г растворителя); Сг — конечная концентрация раствора (в
граммах безводной соли на 1 г растворителя); W — первона-
первоначальная масса растворителя в граммах; V— потеря растворите-
растворителя (например, при испарении) в процентах от исходного коли-
количества растворителя; R — отношение молекулярных масс кри-
кристаллогидрата и безводной соли; у — выход кристаллов в грам-
граммах.
Если растворенное вещество остается неизменным при кри-
кристаллизации (например, кристаллизуется в виде безводной со-
соли), то выход кристаллов при полном удалении растворителя
составит:
без потери растворителя —
и при частичной потере растворителя —
Если растворенное вещество кристаллизуется в виде сольвата,
то выход кристаллов при полном удалении свободного раство-
растворителя составит:
без потери растворителя —
Ш?(СХ--С2)
7= 1 — с,(/? — 1)
и при частичной потере растворителя —
у- 1 — с, (/? — 1)
Последнюю формулу можно рассматривать как общую для
всех рассмотренных случаев.
Применение кристаллизации. В производстве антибиотиков
процессы кристаллизации находят широкое применение при вы-
выделении и очистке антибиотиков тетрациклиновой группы, пе-
пенициллина, эритромицина, олеандомицина и др. Рассмотрим не-
некоторые процессы кристаллизации антибиотиков.
Кристаллизация тетрацлклинов. Для получения
кристаллического хлоргидрата окситетрациклина необходимо
398
применять органические растворители (метиловый, изопропи-
ловый спирт и др.), поскольку высокая гидрофильность хлор-
гидрата препятствует выделению его из водных растворов да-
даже при больших концентрациях.
Отмечено также, что растворимость основания окситетра-
циклина в метиловом спирте сильно возрастает с увеличением
концентрации хлорида кальция. С другой стороны, раствори-
растворимость хлоргидрата окситетрациклина в кислом метаноле умень-
уменьшается с повышением концентрации хлорида кальция. Сопо-
Сопоставление этих данных позволило сделать вывод, что оптималь-
оптимальным условием для получения кристаллического хлоргидрата ок-
окситетрациклина является концентрация основания 100 000—
120 000 ЕД/мл в 7—9% растворе хлорида кальция в метаноле-
При подкислении такого раствора соляной кислотой происхо-
происходит быстрая кристаллизация солянокислой соли, растворимость
которой в метанольном растворе данного состава минимальна.
Процесс кристаллизации осуществляют следующим образом.
Техническое основание окситетрациклина растворяют в 8% ме-
метанольном растворе хлорида кальция, раствор обрабатывают
активированным углем, фильтруют и при 3—5°С прибавляют
концентрированную соляную кислоту. Смесь перемешивают в
течение 30 мин при этой же температуре, после чего выпавший
кристаллический осадок солянокислой соли окситетрациклина
отфильтровывают и на фильтре промывают охлажденным ме-
метанолом. Осадок сушат в вакууме при 35/38 °С и получают со-
солянокислую смесь окситетрациклина в виде кристаллов лимон-
но-желтого цвета с активностью 900 ЕД/мг. Аналогичным обра-
образом проводят процесс кристаллизации других представителей
антибиотиков тетрациклиновой группы (тетрациклин, хлортет-
рациклин) с некоторыми изменениями в каждом случае.
Рассмотренный случай осаждения солянокислой соли окси-
окситетрациклина представляет собой не что иное, как осаждение
электролита (хлоргидрат антибиотика) в присутствии другого
сильного электролита с общим ионом (СаСЬ).
Рассмотрев этот процесс с химической точки зрения, можно
показать, что концентрация антибиотика в маточнике (L) пос-
после осаждения будет равна:
где X — концентрация добавленного электролита с общим ио-
ионом (СаСЬ); Lo — исходная концентрация окситетрациклина
(рис. 53).
Таким образом, зная исходную концентрацию антибиотика
и количество добавленного электролита, можно предсказать ос-
остаточную концентрацию хлоргидрата антибиотика в маточнике.
Следует указать также, что процесс кристаллизации как и в
случае получения основания, так и в случае получения соляно-
399
кислой соли окситетрациклина является процессом кристалли-
кристаллизации с химической реакцией. Тетрациклины применяют при
лечении заболеваний, вызываемых чувствительными к ним воз-
возбудителями: ангины, бронхита, гнойного плеврита, пневмоний
различной этиологии, подострого септического эндокардита,
г? 2ог а
i
i
I
12
4 -
2 4 6 8 ЮрН
в ЮрН
4 Рис. 53. Равновесие в системе,
а — окситетрациклин — вода B0 °С); б — тетрациклин — вода B0 °С).
холецистита, бактериальной и амебной дизентерии, коклюша,
гонореи, сыпного и возвратного тифов, трахомы, пситтакоза и
т. д.
Тетрациклины для приема внутрь выпускают в виде табле-
таблеток (драже), капсул и суспензии. Таблетки и капсулы содержат
от 100.до 250 мг антибиотика A00 000 — 250000 ЕД).
Для внутримышечного введения используют гидрохлорид
тетрациклина или окситетрациклина, выпускаемый в стериль-
стерильных флаконах по 100 мг A00 000 ЕД). Препарат для внутримы-
внутримышечного применения растворяют в 3—5 мл стерильного 1—2%
раствора новокаина. Используют тетрациклины также в виде
глазных мазей, зубных конусов, свечей.
Кристаллизация пенициллина. Исключительно
высокая гидрофильность калиевой и натриевой солей пеницил-
пенициллина не позволяет проводить кристаллизацию их из водных ра-
растворов. Кристаллизация калиевой или натриевой соли пеницил-
пенициллина осуществляется из бутилацетатного экстракта добавлени-
добавлением насыщенного водного раствора ацетата калия и натрия. При
этом пенициллин, находясь в бутилацетате в виде пенициллино-
вой кислоты, реагирует с ацетатом калия, в результате хими-
химической реакции образуется калиевая соль, которая вследствие
400
L
S
Иильтуральная
Грибов
Распылительная
сушилка fi==n Выпари
ной
аппа-
аппарат
бактерии Сепаратор
жд ось
актин омицетов
Аппарат для
обработки
Теплообменник
Использование 6 животноводстве
Экстракция т8.
тел жидкостью
Ректиср ,, с г-£ * г мицелий a
колонна. Жидкостная экстракция \ Ионообменная сорбция Осаждение
бодн р-р
D~fl i яЩА/ЧастиЯно {очищенные анти\рш
Перегонный / Им/ZIZ Г
Центрифуга
Сушилка
Чистый антибиотик
'OTUKU
.J
Распылит,
сушилка
Чистый антибиотик
Рис. 54. Основная техно-
технологическая схема выде-
выделения н очистки анти-
антибиотиков.
малой растворимости в бутилацетате выпадает в осадок. Вы-
Выпавшие кристаллы промывают бутанолом для удаления ацета-
ацетата калия и 'высушивают. Натриевая или калиевая соль пени-
пенициллина кристаллизуются также методом азеотропной отгонки
растворителя. В вакуум-выпарной аппарат к водному раство-
раствору пенициллина добавляют бутанол, который с водой при ки-
кипении образует азеотропную смесь, состоящую из 68 частей бу-
танола и 32 частей воды. Отгонку азеотропной смеси бутанола
с водой производят при температуре 20°С и остаточном давле-
давлении 10 мм рт. ст. При полном удалении воды из кубового остат-
остатка после охлаждения в избытке бутанола кристаллизуется на-
натриевая соль пенициллина, которую затем также промывают
бутанолом и высушивают. В последние годы разрабатывают не-
непрерывные методы кристаллизации антибиотиков.
Лекарственные формы бензилпенициллина применяют при
лечении заболеваний, вызываемых чувствительными к этому
антибиотику возбудителями ангины, скарлатины, крупозной,
очаговой и хронической пневмонии, сепсиса, большинства ра-
раневых инфекций, подострого септического эндокардита, гной-
гнойных инфекций и т. д.
Натриевая и кальциевая соли бензилпенциллина предназна-
предназначены в основном для внутримышечных инъекций. Их растворы
можно вводить также внутривенно и внутриартериально. Для
инъекций натриевую и калиевую соли бензилпенициллина раст-
растворяют в стерильном изотоническом растворе хлорида натрия,
в стерильной дистиллированной воде или 0,5—1% растворе но-
новокаина.
Феноксиметилпенициллины не разрушаются в кислой среде
желудка, поэтому могут применяться в виде таблеток.
Препарат выпускают в виде свободной кислоты. Таблетки
содержат 100—250 мг в перечете на химически чистую фенок-
симетилпенициллиновую кислоту. Феноксиметилпенициллин
применяют для лечения тех же заболеваний, что и бензилпени-
циллин.
Принципиальная схема выделения антибиотиков приведена
на рис. 54.
§ 4. ОСНОВЫ ТЕХНОЛОГИИ СУБЛИМАЦИОННОЙ
И РАСПЫЛИТЕЛЬНОЙ СУШКИ АНТИБИОТИКОВ
Большинство антибиотиков представляют собой термола-
термолабильные соединения, и следовательно, не выдерживают длитель-
длительные процессы сушки с воздействием высоких температур. Пер-
Первыми методами, используемыми для сушки растворов антиби-
антибиотиков, были методы сублимации льда в вакууме из предйари-
тельно замороженных растворов — так называемая лиофильная
сушка. Антибиотики, высушенные тем или иным способом, впо-
впоследствии используют как инъекционные препараты в стериль-
стерильном виде, поэтому растворы перед сушкой подвергают сте-
402
рильной фильтрации через соответствующие фильтры (в на-
настоящее время на производстве антибиотиков широко использу-
используют стерилизующие фильтры «Миллипор»). Стерильные растворы
антибиотиков, или, как их называют на производстве, концент-
концентраты, различают в стерильных условиях в стерильные флаконы.
Стерильный раствор антибиотика во флаконах замораживают
в холодильных камерах при температуре от —45 до —60 °С в те-
течение 2—4 ч в зависимости от дозировки. Затем флаконы с за-
замороженным раствором антибиотика помещают в вакуум-су-
вакуум-сушильные шкафы, в которых создают и поддерживают остаточ-
остаточное давление 10—100 мкм рт. ст.
Температуру сушки регулируют подачей горячей воды в пол-
полки шкафа. Испаряющуюся воду удаляют из сушильного шкафа
с помощью специальных масляно-диффузионных насосов. При
этом она конденсируется в конденсаторах с рассольным охлаж-
охлаждением или удаляется с помощью пароструйных эжекторных
насосов.
Условия предварительного замораживания влияют на ин-
интенсивность испарения и структуру препарата. Замораживание
растворов антибиотиков производят до температуры «иже (крио-
гидратиой, при этом образуются кристаллы льда и антибио-
антибиотика. В процессе испарения кристаллов льда остаются кристал-
кристаллы высушиваемого вещества, которые образуют пористую мас-
массу. Замораживание растворов стремятся проводить быстро, так
как медленное замораживание вызывает образование крупных
кристаллов, что уменьшает поверхность испарения. Быстрое
замораживание позволяет получать мелкокристаллическую
структуру и, следовательно, большую пористость продукта.
Медленное замораживание может вызвать неравномерное про-
промораживание раствора, в результате чего в центре флакона мо-
может оказаться участок с высокой вязкостью, что затрудняет
удаление паров из раствора. Для достижения максимальной
скорости замораживания в воздушных холодильных камерах
поддерживают температуру — 50 °С с интенсивной циркуляцией
воздуха. Каждый процесс сушки сопровождается удалением
свободной и связанной влаги. Удаление связанной влаги зани-
занимает обычно 7з общего времени сушки и происходит с уменьша-
уменьшающейся скоростью. Скорость сушки в целом зависит от интен-
интенсивности испарения влаги, которая в свою очередь обусловли-
обусловливается интенсивностью подвода тепла и удаления испаряющей-
испаряющейся влаги. В случае сублимационной сушки эти факторы зависят
от разности температур в сушильной камере и стенок поверх-
поверхности конденсатора, от остаточного давления и т. д.
В процессе сублимации нередко наблюдается вспенивание
содержимого флакона. Это обусловлено недостаточно низкой
температурой замораживания. Более концентрированный вяз-
вязкий раствор продукта при температуре от — 4 до —6°С начи-
начинает плавиться. Плавление продукта нарушает пористость
26* 403
структуры, приводит к образованию смолообразных пленок, с
трудом проницаемых водяными парами. Существенное влияние
на процесс сушки сублимацией оказывает дозировка продукта
во флаконе. При больших количествах раствора процесс диф-
диффузии влаги в процессе сублимации затрудняется. Эксперимен-
Экспериментально установлено, что отношение высоты заливаемого во
флакон раствора к диаметру флакона должно быть в пределах
0,5—0,6. В этих условиях удаление влаги происходит равно-
равномерно как с верхней, так и с боковой поверхности. Начальный
период сушки растворов антибиотиков сублимацией следует
проводить при температуре материала от — 8 до —12°С. Основ-
Основным условием, обеспечивающим качество высушенного препара-
препарата, является сохранение интенсивности испарения несколько
меньшей, чем интенсивность конденсации. Если в камере имеет
место накопление значителньых количеств паро-воздушной сме-
смеси и она не успевает быстро удаляться, то циркуляция паро-
паровоздушной смеси вызывает нагрев продукта и его оплавление.
Интенсивность испарения влаги зависит также от пористости
продукта. Сушильные шкафы, используемые при сушке субли-
сублимацией, должны обладать повышенной герметичностью. В них
необходимо соблюдать стерильные условия. Коммуникации, с
помощью которых осуществляется отвод паро-воздушной смеси
из сублиматора, следует специально рассчитывать. Они не
должны препятствовать интенсивному отводу испаряемой влаги.
Конденсаторы для удаления (влаги разнообразны. Часто ис-
используются конденсаторы с поверхностью, охлаждаемой хла-
доагентом, и непрерывным удалением скребком образующегося
льда. Очищенная поверхность конденсатора улучшает интен-
интенсивность конденсации влаги. Недостатком скребковых конденса-
конденсаторов является трудность создания герметичности. Вакуум в си-
системе обеспечивается механическими и масляно-диффузионными
насосами, а также с помощью пароструйных эжекторных насо-
насосов, хотя это связано с применением пара высокого давления
A2—15 ат.). С помощью эжекторных насосов можно удалить
газ и влагу без конденсации и замораживания водных паров.
Продукты, высушенные методом сублимации, имеют хоро-
хорошее качество, сохраняют первоначальные активности. Однако
установки для лиофильной сушки громоздки и чаще всего ис-
используются при малых производительностях. Принципиальная
технологическая схема сушки растворов антибиотиков методом
сублимации представлена на рис. 55.
Изучение процессов сублимации водяного пара в разряжен-
разряженной среде рассматривается на основе сил межмолекулярного
взаимодействия. Межмолекулярное взаимодействие в опреде-
определенных условиях приводит к объединению молекул пара и газа
в группы, которые, как правило, называются ассоциатами. Про-
Процесс ассоциации молекул происходит за счет действия сил Ван-
дер-Ваальса и за счет водородных связей. Скорость сублима-
404
Рис. 55. Принципиальная технологическая схема сушки растворов антибиотиков методом сублимации льда.
/ — бутыль с раствором; 2 — пипетировочная машина; 3 — кассета с флаконами; 4 — камера для замораживания; 5 - вакуум-сушиль-
вакуум-сушильные шкафы; 5 —конденсатор; 7 — скребок; 8 — льдоприемник; 9 - механический вакуум-насос; 10 — диффузионный масляный насос;
// — насос для создания среднего вакуума; 12 — бойлер; 13 — насос; / — вакуум; // - горячая вода; /// — материальная линия.
ции льда для реальных условий (Sv) может быть рассчитана по
уравнению:
где Р\ — давление в объеме сублиматора; Р2 — давление у по-
поверхности испарения; /' — коэффициент, учитывающий распад
комплексных частиц в объеме и число столкновений молекул
между собой, в результате которых определенная часть моле-
молекул возвращается на поверхность испарения (O^f ^1).
Для рассчета общего времени сублимационной сушки из
слоя предложена следующая формула:
где Wu—WK—начальное и конечное влагосодержание материа-
материала; рс — плотность сухого остатка; h — толщина слоя; №кр —
критическое влагосодержание; рм— давление насыщения, со-
соответствующее температуре материала; рк — давление насыще-
насыщения, соответствующее температуре конденсации; р— коэффици-
коэффициент массоотдачи для данного периода сушки; рс — давление в
сублиматоре; /Кон — конечная температура материала; К\ — ко-
коэффициент скорости сушки при досушивании; тс — время субли-
сублимации; тд — время досушивания.
Количество испаренной влаги (при переменной скорости суб-
сублимации) для каждого периода определяется из выражения
где р — средний коэффициент массоотдачи в данном периоде;
Fc — действительная поверхность сублимации; т — продолжи-
продолжительность периода.
Большой объем производства некоторых антибиотиков не
позволяет использовать малогабаритные установки для сушки
растворов антибиотиков методом сублимации, поэтому в про-
промышленности стали широко применять сушку распыле-
распылением.
Растворы антибиотиков, подаваемые в сушильную камеру,
распыляются в токе нагретого воздуха до мелких капель, ве-
величина которых иногда достигает нескольких микрометров. По-
Поверхность соприкосновения мелких кашель раствора с нагретым
воздухом значительна, и сушка протекает в течение нескольких
секунд. Температуря распыленных частиц раствора обычно
равна температуре мокрого термометра и зависит от темпера-
температуры воздуха и его влажности. Например, при температуре
воздуха 150°С и относительной влажности 3% температура
материала составит 45°С. Такие сравнительно невысокие тем-
406
пературы высушиваемого продукта позволяют использовать этот
метод для сушки тремолабильных препаратов^
Большое значение при распылительной сушке имеет процесс
распыления подаваемого раствора антибиотика. Распыление
растворов можно осуществлять различными способами, но наи-
наибольшее распространение получило пневматическое распыление
под действием сжатого воздуха с давлением 1—1,5 ат с исполь-
использованием специальных форсунок, а также под действием цент-
центробежной силы. Частицы жидкости выходят из форсунки с боль-
большой скоростью, которая под влиянием сопротивления воздуха
уменьшается и капли раствора приобретают так называемую
скорость витания. Скорость витания зависит от свойств высу-
высушиваемого материала, а от нее в свою очередь зависят удары
частиц между собой и их слипание, а следовательно, режим суш-
сушки. Практически установлено, что успешно сушка может проис-
происходить при скорости воздуха в камере 0,2—0,5 м/с.
Вначале в промышленности антибиотиков широко использо-
использовались сушилки одноступенчатые с пневматическим распыле-
распылением подаваемого раствора, при этом методе требовалось пред-
предварительное концентрирование раствора. В процессе упарива-
упаривания раствора в пленочных вакуум-выпарных аппаратах темпе-
температура препарата в пленке повышалась от 40 до 100 °С, что
вызывало его разложение. В связи с этим промышленное полу-
получение препаратов методом распыления пошло по пути исполь-
использования двухступенчатых испарительно-сушильных агрегатов. В
испарительно-сушилъных аргегатах концентрирование раствора
осуществляется в потоке нагретого воздуха, а это позволяет из-
избежать перегрева раствора. Распыление поступающего раство-
раствора обычно осуществляется с помощью центробежного распыли-
распылителя. Раствор поступает на диск, вращающийся со скоростью
18 000 об/мин. Диаметр сушильной камеры должен быть доста-
достаточно большим, чтобы капли не попадали на стенки аппарата.
Высота камеры практически бывает меньше, ч^м у сушилок с
пневматическим распылением. Раствор на диск должен пода-
подаваться при постоянном напоре и строгом регулировании ско-
скорости. При несоблюдении этого условия размер диспергирован-
диспергированных капель окажется неодинаковым и качество высушиваемо-
высушиваемого продукта ухудшается. В сушилках данной конструкции теп-
теплоноситель подается противотоком по отношению к подаваемо-
подаваемому раствору. В ряде случаев это приводит к образованию завих-
завихрений и попаданию порошка на потолок сушилки. Нарушение
факела распыления, даже незначительное, вызывает попадание
капель раствора на стенки камеры, что в итоге уменьшает вы-
выход и качество конечного продукта. Подача растворов методом
распыления под действием центробежной силы иногда вызыва-
вызывает попадание в камеру смазочного масла, отчего растворы вы-
высушенного порошка могут опалесцировать. Для более полного
улавливаания высушенного порошка в технологическом процес-
407
Воздух
Воздух
Рис. 56. Принципиальная технологическая схема двухступенча-
двухступенчатого иопарительно-сушильного агрегата.
/— фильтр для предварительной очистки воздуха; 2 — вентилятор вы-
высокого давления; 3 —фильтр для тонкой очистки воздуха; 4 — паровой
калорифер; 5, 6 — электрические калориферы; 7, 10 — распределители
воздуха; 8 — испарительная 'камера; 9 — циклон для отделения концен-
концентрированного раствора; // — дисковый распылитель; 12 — сушильная
камера; 13 — циклон; 14 — специальное устройство для выгрузки гото-
готового продукта.
I Холодный воздух
11
Сухой порошок на просеб
Влажная паста после центрифуг
В /сонденса топровод
Рис. 57. Технологическая схема установки для
сушки во взвешенном слое. ^
/ — фильтр для воздуха; 2 — электрокалорифер; 3 —
фильтрующая сетка; 4 —система подачи продукта в су-
сушилку; 5 — труба-сушилка; 6 — циклоны; 7 — бункер к
циклону; 8 — приемники готового продукта; 9 — улитка;
10 — матерчатый фильтр; // — вентилятор; 12 — противо-
взрывная мембрана; 13 — автоматическая заслонка; 14 —
вибратор,
се сушки растворов распылением предусматривается установка
циклонов. Выгрузка готового продукта производится в стериль-
стерильных условиях с использованием специальных камер типа бок-
боксов. Принципиальная технологическая схема двухступенчатого
испарительно-сушильного агрегата представлена на рис. 56.
Процесс сушки в двухступенчатом сушильном агрегате осу-
осуществляется воздухом, подаваемым вентилятором высокого дав-
давления. Воздух, поступающий в сушильную камеру, проходит
систему очистки — фильтр грубой очистки, фильтр тонкой очи-
очистки, из которого воздух поступает в паро- и электрокалори-
электрокалориферы. Нагретый воздух A60—180°С) через воздухораспреде-
воздухораспределитель поступает в испарительную камеру, в которую одновре-
одновременно поступает отработанный теплоноситель из сушильной ка-
камеры. Использование теплоносителя (воздух) из сушильной
камеры позволяет повысить коэффициент полезного действия
испарительно-сушильного агрегата по сравнению с одноступен-
одноступенчатым почти в 3 раза (до 75—80%). Высушиваемый раствор,
распыленный на дисковом распылителе до мелких капель, кон-
концентрируется в испарительной камере и поступает в сушильную
камеру на диск центробежного распылителя. Температура воз-
воздуха на выходе из первой ступени 45—50 °С (температура ис-
испарительной камеры), на выходе из второй ступени 95—
100 °С.
При производстве многих антибиотиков технологический
процесс заканчивается получением кристаллического продукта.
Сушка пастообразного или кристаллического влажного мате-
материала обычно осуществляется в вакуум-сушильных шкафах пе-
периодического действия. Этот метод сушки имеет ряд недостат-
недостатков, поэтому в последние годы в производство внедрена сушка
пастообразных материалов во взвешенном слое. Сушильные
установки подобного типа могут быть выполнены в непрерыв-
непрерывном периодическом и полунепрерывном исполнении. Наиболее
широкое распространение в этом плане получили сушилки не-
непрерывного действия. Технологическая схема сушки во взве-
взвешенном слое представлена на рис. 57. По такой схеме в про-
промышленности антибиотиков успешно осуществляется сушка тет-
тетрациклина. Высушиваемый материал с влажностью 25—35%
непрерывно подается в трубу-сушилку шнековым питателем.
Воздух, нагретый до 90—100 °С в электрокалорифере, направ-
направляется в трубу-сушилку. Смесь горячего воздуха со взвешенным
в нем материалом проходит через циклон, в котором отделяет-
отделяется готовый продукт.
Глава 20
ПОЛУСИНТЕТИЧЕСКИЕ АНТИБИОТИКИ
§ 1. ПОЛУСИНТЕТИЧЕСКИЕ ПЕНИЦИЛЛИНЫ
Бензилпенициллин, получаемый биосинтетическим путем, и
многочисленные препараты, созданные на его основе, продол-
продолжают оставаться наиболее эффективными и широко применяе-
применяемыми антибиотиками. Бактерицидный тип механизма действия,
высокая активность в организме больного в сочетании с низким
уровнем токсичности позволяют рассматривать бензилпеницил-
бензилпенициллин как один из идеальных химиотерапевтических препаратов.
Однако р-лактамное кольцо бензилпенициллина легко разру-
разрушается по действием пенициллиназы (р-лактамаза), которая
образуется многими микроорганизмами. Распространение
устойчивых пенициллину микроорганизмов вызвало необходи-
необходимость создания новых типов пенициллина.
В 1957 г. благодаря выделению «ядра» пенициллина —
6-аминопенициллиновой кислоты F-АПК) появилась возмож-
возможность создания полусинтетических пенициллинов. Сохраняя ос-
основные преимущества бензилпенициллина (бактерицидность,
низкий уровень токсичности), они приобретают наряду с этим
новые свойства.
Условно основные группы полусинтетических пенициллинов
можно представить следующим образом:
I. Кислотоустойчивые препараты, инактивируемые пеницил-
линазой (фенестициллин, пропициллин, фенбенициллин).
II. Пенициллиноустойчивые препараты: 1) некислотоустойчи-
некислотоустойчивые (метициллин); 2) кислотоустойчивые (препараты изоксазо-
лиловой группы — оксациллин, клоксациллин, диклоксациллин
и др.).
III. Препараты широкого спектра действия (ампициллин,
карбенициллин).
В настоящее время полусинтитические пенициллины широко
выпускаются промышленностью. 6-АПК получается энзимати-
ческим путем из бензилпенициллина и других биосинтетических
пенициллинов с помощью пенициллинацилазы. Фермент пени-
цилинацилаза образуется главным образом бактериями. Име-
Имеются также химические методы расщепления пенициллинов, по-
позволяющие получить 6-АПК.
411
/ \ /снз
RCONHCH—CH Сч
I XCH3
0=С N СНСООН
(Н20)
HoN—CH
0=С-
/ \
—СН
СНСООН
Известны попытки прямого биосинтетического получения
6-АПК. В настоящее время в нашей промышленности принят
энзиматический метод расщепления пенициллина с помощью
пенициллинацилазы Е. coli. Для выделения 6-АПК из водного
гидролизата используются как экстракционные, так и ионооб-
ионообменные методы-
Из 6-АПК синтезировано много ацильных производных, об-
обладающих антибактериальным действием, но применение в ме-
медицине нашли только некоторые из этих соединений благодаря
их устойчивости к пенициллиназе и особой эффективности в
отношении болезнетворных микроорганизмов, в том числе в от-
отношении стафилококков, устойчивых к пенициллину. В качестве
ацилирующих агентов чаще всего используют хлорангидриды
карбоновых кислот (RC0C1).
Из полусинтетических пенициллинов практическое примене-
применение в медицине нашли метициллин, оксациллин, ампициллин.
Метициллин — препарат, получаемый путем воздействия
хлорангидрида 2,6-диметоксибензойной кислоты с триэтилами-
новой солью 6-АПК в безводном ацетоне с последующей экст-
экстракцией метициллина-кислоты бутилацетатом, получением нат-
натриевой соли и кристаллизацией из водного ацетона в виде мо-
моногидрата:
\ /снз
H2N-CH—СН С
ОСН
N СНСООН. N(C2H5K
-(C2H5KN.HCl
—CONHCH-
ОСНо
0=
/ \ /СНз
-СН Сч
|\сн,
-N СНСООН
NaHCO3
СН
l-cONH— CH— СН
оснч
/СН3
снч
0=С N CHCOONa. Н20
412
Метициллин белый кристаллический порошок с температурой
плавления 197°С (с разл.). Удельное вращение+225° (с = 1, в
воде). Метициллин легко разрушается в кислой среде и может
применяться только парентерально в виде стерильного раство-
раствора. Соль выдерживает стерилизацию при 120 °С в течение 1 ч
без заметного разрушения.
Оксациллин получают путем взаимодействия хлорангидри-
да 5-метил-3-фенилизоксазол-4-карбоновой кислоты с натрие-
натриевой солью 6-АПК в водном ацетоне. Из реакционной смеси ок-
оксациллин извлекают бутилацетатом и выделяют моногидрат
натриевой соли путем реакции обмена с ацетатом натрия:
С6Н5С С—Сч H2NCH—СН Сч
I! ;| ЧС1+ \СН,
N С—СН3 О=С N СНСООН
' 1) -НС1
2) CH3COONa
/ \
С6НбС С CONHCH—CH, C
N С-СНз О=С N CHCOONa.H2O
v
Оксациллин представляет собой белый кристаллический поро-
порошок с температурой плавления 188°С (с разл.). Удельное вра-
вращение [а] о =+201° (с=1, в воде).
Ампициллин получают взаимодействием смешанного ангид-
ангидрида производного D-( — )-а-фенилглицина и эфира хлор-
угольной кислоты с 6-АПК в водном ацетоне. Продукт ацили-
рования после омыления защитной группы кристаллизуется из
воды в виде тригидрата (см. стр. 414).
Амшициллин представляет собой белый кристаллический поро-
порошок с температурой плавления 202°С (с разл.). Удельное вра-
вращение [а]Ь° =+231°.
Метициллин применяют главным образом внутримышечно
при инфекциях, вызванных пенициллиназообразующими, а так-
также устойчивыми к другим антибиотикам штаммами стафило-
стафилококков. Выпускают метициллин стерильно во флаконах, содер-
содержащих 1 г стерильного препарата.
Оксациллин применяют при инфекциях, вызываемых устой-
устойчивыми к бензилпенициллину пенициллинообразующими стафи-
стафилококками. Выпускают оксациллин в виде таблеток и капсул по
250—500 мг и во флаконах, содержащих 250—500 мг стериль-
стерильного препарата.
413
еден— соон.
NHO
с6н.снсоок
кон
сс
нсч
h chococi
II
сн ос.
6АПК
СН.— CHCONHCH-
о о
сн,
I J
CHCOO~HN (С2Н5Ь
|
S
n, , , \ у
-^- C6H.CHCONH_CH-CH C~
CH о
6
L2
CH
-b сн
D - (—) а- Аминобензилпенициллин (ампициллин)
нсоон-зн2о
Ампициллин применяют при лечении инфекций дыхательных
путей, желудочно-кишечного тракта, мочевыводящих путей и
т. д., вызываемых чувствительными к действию антибиотика
микроорганизмами. Ампициллин применяют внутрь в виде таб-
таблеток и капсул, содержащих 250 мг антибиотика, натриевую
соль — внутримышечно и внутривенно. Как инъекционный пре-
препарат натриевую соль выпускают по 250—500 мг в стерильных
флаконах.
§ 2. ЦЕФАЛОСПОРИНЫ
В настоящее время за рубежом по аналогии с полусинте-
полусинтетическими пенициллинами получен ряд полусинтетических пре-
препаратов на основе 7-аминоцефалоспорановой кислоты G-АЦК),
среди которых наибольшее практическое значение приобрели
цефалотин, цефалоридин, цефалексин и цефалоглицин.
2~- ОСОСН3
Цефалоридин — 7- [ B-тиенил) -ацетамидо] -3- A -пиридил-
метил)-3-цефал-4-карбоновая кислота, представляет собой бе-
белый кристаллический порошок, хорошо растворимый в воде
B20—250 мг/мл).
414
Nr
Цефалотин — 7- (тиофен-2-ацетамидо) цефалоспорановая кис-
кислота— белый кристаллический порошок (натриевая соль), хо-
хорошо растворимый в воде.
О
Л-СН.—С— NH— "^ °
О-
ХО—Na
В результате исследований, проведенных в области химиче-
химической трансформации 7-аминоцефалоспорановой кислоты, полу-
получены ее производные; хорошо всасывающиеся при приеме
внутрь в отличие от предыдущих препаратов. К ним относятся
цефалоглицин — 7-(D-a-аминофенилацетамидо)-цефалоспорано-
7-(D-a-аминофенилацетамидо)-цефалоспорановая кислота
Н о
NH2
и цефалексин — 7-(О-а-амино-а-фенилацетамидо)-3-метил-3-це-
фам-4-карбоновая кислота.
Н О
II S
>-С--Ш /
NH2 J—
Основными особенностями антибиотиков этой группы явля-
являются: широкий спектр антибактериального действия, устойчи-
устойчивость к действию стафилококковой пенициллиназы и высокая
активность в отношении устойчивых к бензилпенициллину пени-
циллиназообразующих стафилококков, отсутствие перекрестной
аллергии с пенициллинами. Полусинтетические цефалоспорины
(цефалоридин, ценалотин, цефалоглицин и цефалоксин) исполь-
используются при лечении инфекций, вызываемых чувствительными к
ним микроорганизмами. Они высокоэффективны при заболева-
415
ниях дыхательного тракта (пневмония, абсцесс легкого, эмпие-
эмпиема плевры и др.)» инфекциях мочевыводящих путей, системных
инфекциях (септицемия, (менингит, перитонит и др.). Препараты
применяются в -основном внутримышечно.
§ 3. ПОЛУСИНТЕТИЧЕСКИЕ ТЕТРАЦИКЛИНЫ
Антибиотики тетрациклиновой группы используются в меди-
медицинской практике в виде оснований и гидрохлоридов. Основа-
Основания и гидрохлориды тетрациклинов ограниченно растворимы в
воде и вызывают раздражение кожи при местном применении,
поэтому их назначают в основном внутрь. В то же время при
некоторых острых септических заболеваниях требуется быст-
быстро создать в организме, высокие концентрации антибиотика.
В нашей стране и за рубежом велись исследования по созда-
созданию нейтральных водорастворимых препаратов тетрациклина.
В результате этих исследований были синтезированы аминоме-
тильные (карбоксамидные) производные тетрациклинов, хоро-
хорошо растворимые в воде и оказывающие менее выраженное мест-
местное действие на сосуды.
В нашей стране из многочисленных аминометильных про-
производных тетрациклина нашли применение морфоциклин, син-
синтезированный в ЛНИИА, и гликоциклин, синтезированный во
ВНИИА. В последние годы в ЛНИИА синтезировано еще одно
производное — оксиглюкоциклин (производное N-метилглюко-
зоамина с окситетрациклином).
Реакцию вторичного амина, формальдегида с тетрациклина-
ми (реакция Манниха) можно представить следующим обра-
образом:
CONHCHN'
ОН
R = R =Н — юфациклин
R ===: Н ' Ry=OlI — окситетрациклин
При N
\г
При R = H } R==
При R = 0ll, R== И,
-морфоциклин
—оксиглюкоииклин
416
На основании ультрафиолетовой и инфракрасной спектро-
спектроскопии и реакций гидролиза аминометильным производным
тетрациклинов приписано строение II.
Морфоцикглин имеет меньшую острую токсичность при внут-
внутривенном введении, меньше повреждает вены, лучше проникает
внутрь клеток и в макрофазы, больше растворяется в воде.
Вследствие низкой токсичности и высокой растворимости он не
требует добавки в лекарственную форму.
Морфоциклин применяют только внутривенно в виде 1,25—
2,5% раствора на 5% растворе стерильной глюкозы. Морфо-
Морфоциклин для внутривенного введения выпускают в стерильных
флаконах по 100 и 150 мг A00 000 и 150 000 ЕД), лиофильно-
высушенный.
Гликоциклин назначают внутривенно и внутримышечно.
Препарат малотоксичен, местное раздражающее действие вы-
выражено слабо, не обладает кумулятивными свойствами. Выпу-
Выпускают гликоциклин в стерильных флаконах по 100 и 250 мг
A00 000 и 250 000 ЕД), лиофильно-высушенный.
Указанные антибиотики применяют при лечении инфекций,
вызываемых чувствительными к тетрациклинам микроорганиз-
микроорганизмами (пневмонии, абсцессы легких, перитониты, пиелонефриты
и т. д.).
В последние годы большое значение в медицинской практи-
практике приобрели 6-дезокситетрациклины, в частности метациклин —
б-дезокси-б-деметил-б-метилен-5-окситетрациклин. Это полусин-
полусинтетическое производное окситетрациклина является его струк-
структурным гомологом. Применяют метациклин при острых и хро-
хронических бронхитах, бронхоэктатической болезни, эмпиеме лег-
легких, пневмонии и бронхопневмонии и т. д. Выпускают метацик-
метациклин в виде капсул с содержанием 150—300 мг антибиотика.
он о
ОН Q
27—926
Глава 21
СИНТЕТИЧЕСКИЕ АНТИБИОТИКИ
§ 1. СИНТОМИЦИН И ЛЕВОМИЦЕТИН.
СТЕРЕОИЗОМЕРИЯ И БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Антибиотические вещества нарушают основные жизненные
процессы различных видов микробов. К числу синтетических
антибиотиков, т. е. лекарственных веществ, получаемых путем
химического синтеза и обладающих антибиотическими свойст-
свойствами, следует отнести прежде всего левомицетин. Левомицетин
был открыт в 1947 г. и первоначально назван хлоромицетином.
Уже в 1949 г. было подробно изучено строение его молекулы и
осуществлен полный синтез его. В Советском Союзе большая
работа по синтезу аналогов левомицетина (хлоромицетина) и
изучению связи между их строением и антибиологическими
свойствами проведена под руководством академика М. М. Ше-
Шемякина. Несмотря на то что левомицетин продуцируется при-
природными микроорганизмами, в промышленности его получают
путем химического синтеза. Отчасти это объясняется тем, что
молекула левомицетина сравнительно проста. Трудности в син-
синтезе этого антибиотика объясняются стереохимией его моле-
молекулы.
В молекуле левомицетина имеется два асимметрических
атома углерода, что обусловливает существование четырех сте-
реоизомеров: и D(—)- и Ь( + )-эритроизомеров.
Н NH— СОСНС12 ОН NHCOCHCU
С—С—СН2ОН O2N—/ У- С--С-СН2ОН
I I V_/ i i
он н н н
D(—)-трев-нз«мер (левомицетин) D(—)-эритро-извмер
ОН Н Н Н
O2N—^ )>—С—С—СН2ОН O2N—/~V- С—С-СН2ОН
Н NHCOCHC12 ОН NHCOCHC12
Щ-)-трео-изомер Ь(+)-эритро-изомер
Природный хлоромицетин (левомицетин) соответствует
D ( — ) -трео-изомеру. Смесь D ( — ) - -и L ( + ) -трео-изомеров
(т. е. рацемат, имеющий трео-конфигурацию) названа син-
418
томицином. Синтомицин по терапевтическому действию анало-
аналогичен левомицетину, но обладает лишь 50% его активности.
Оба изомера эритроформы очень токсичны, поэтому не нашли
применения в медицине.
Синтомицин и левомицетин применяются при лечении брюш-
брюшного тифа и паратифа, дизентерии, бруцеллеза, коклюша, ту-
туляремии, гонореи, сыпного тифа и других заболеваний, вызы-
вызываемых бактериями и вирусами. Левомицетин и синтомицин вы-
выпускаются в виде порошков и таблеток.
Оба препарата — белые порошки горького вкуса. Плохо ра-
растворяются в хлороформе, дихлорэтане, воде, нерастворимы в
бензоле, толуоле, четыреххлористом углероде, хорошо раствори-
растворимы в спиртах, ацетоне, этилацетате. Нейтральные и слабокис-
слабокислые водные растворы довольно устойчивы. В щелочной среде
левомицетин быстро гидролизуется и теряет биологическую ак-
активность.
§ 2. ПРИНЦИПИАЛЬНАЯ СХЕМА ПОЛУЧЕНИЯ
СИНТОМИЦИНА И ЛЕВОМИЦЕТИНА
Как уже указывалось, основная трудность химического по-
получения левомицетина заключается в синтезе стереоспецифиче-
ской боковой цепи левомицетииа (D-(—)-трео-1-п-нитро-фенил-
2-дихлорацетиламинопропандиола-1,3)
Н NHCOCHC12
Преимущественное образование трео-изомера при восстановле-
восстановлении п-нитро-а-ацетиламино-р-оксипропиофенона изопропилатом
алюминия сделало синтезы, включающие эту химическую ста-
стадию, более предпочтительными. В связи с этим имеющиеся
промышленные методы получения синтомицина и левомицетина
отличаются способом синтеза промежуточного продукта, пред-
предшествующего п-нитро-а-ацетиламино-р-оксипропиофенону, — п-
нитро-а-ацетиламиноацетофенона
O2N—( ^—С—CH2-NHCOCH3
О
Разберем сначала ту часть синтеза синтомицина и левомици-
тина, которая является общей для различных промышленных
методов получения этого препарата.
Полученный тем или иным способом п-нитро-а-ацетилами-
ноацетофенон (ацетильное производное, как его иногда назы-
называют) оксиметилируют формалином в присутствии гидрокарбо-
гидрокарбоната или ацетата натрия в среде изопропилового спирта при
27* 419
температуре 30—36 °С. Образующийся при этом п-нитро-а-аце-
тиламино-р-оксипропиофенон восстанавливают изопропилатом
алюминия в среде изопропилового спирта при 80—82 °С. В ре-
результате образуется смесь трео- и эритро-изомеров с преобла-
преобладанием в 10 раз трео-формы:
NHCOCHg NHCOCHg
сн2он
СН2О, NaHCO3 /^Х I
> on( Усо--сн-с
Н
Н NHCOCHg ОН NHCOCHg
D.L-трео-форма D.L-эритро-форма
Вещества, обладающие трео- и эритро-формой, имеют раз-
различную растворимость в хлороформе, этилацетате и других рас-
растворителях. Обычно трео-форму выделяют, пользуясь ее мень-
меньшей растворимостью в холодном этилацетате. Выделенную трео-
форму омыляют разбавленной соляной кислотой, вг результате
чего получают рацемат основания синтомицина («трео-амин»)
ОН Н ОН Н
| I HC1, Н2О /=\ | I
—С—С-СН2ОН >■ O2N-{ }—С -С
02N—<( h—С—С—СН2ОН >■ 02N—<C j>— С -С—СН2ОН
Н NHCOCHg H NH2-HC1
NaOH
Н NH2
Трео-амин (рацемат)
Свободную аминогруппу трео-амина ацилируют метиловым эфи-
эфиром дихлоруксусной кислоты и получают синтомицин (раце-
(рацемат):
ОН Н
С12СНСООСН3
:—СН2ОН
ОН н
| I
С—С
Н NH2 H NHCOCHC12
Синтомицин (рацемат)
Левомицетин является левовращающим изомером трео-конфи-
гурации. Промышленное получение его сводится к разделению
рацемата трео-амина «а оптически активные формы и ацетили-
рованию левовращающего изомера метиловым эфиром дихлор-
уксусной кислоты. Разделение рацемата на оптически деятель-
420
ные антиподы проводят, пользуясь различной растворимостью
их солей с d-винной кислотой в воде или метаноле.
СООН
ОН Н
O9N
винная кислота
сосг
он н I
| | Н-С-
е— С-СН2ОН
| | ^
Н NH3+ |
С
ОН
Н-С-ОН
ООН
Левовращающий изомер виннокислой соли в отличие от право-
правовращающего хорошо растворим в метаноле и полностью им
извлекается из смеси. Полученную виннокислую соль левовра-
щающего D(—)-трео-изомера разлагают водным раствором ам-
аммиака и ацетилируют метиловым эфиром дихлоруксусной кис-
кислоты.
Н NH3 OOC—(НСОНJ—СООН
O2N-/V-C—С—СН2ОН +2NH3 >■
ОН Н
Н NH2 COONH4
I I I
—С-~СН2ОН + (НСОНJ
I I
I I I
ОН Н COONH4
D(—)-трео-изомер
H NHCOCHC12
—С—СНоОН
он н
Левомицетин
В настоящее время применяется новый способ разделения
рацемата трео-амина на оптически активные антиподы. Спо-
Способ основан на свойстве трео-амина образовывать пересы-
пересыщенные растворы в горячей воде. При внесении в пересыщен-
421
ный водный раствор хлоргидрата трео-амина затравки соответ-
соответствующего антипода начинается кристаллизация одного из оп-
оптически активных веществ. При нескольких последовательных
кристаллизациях из пересыщенных растворов удается выделить
нужный левовращающий изомер и таким образом избежать
разделения рацемата с помощью солей d-винной кислоты.
§ 3. ПРОМЫШЛЕННЫЕ СПОСОБЫ СИНТЕЗА
п-НИТРО-а-АЦЕТИЛАМИНО-АЦЕТОФЕНОНА
В 1949 г. предложен метод синтеза левомицетина из ацето-
фенона. По этому методу ацетофенон бромировали, а затем че-
через стадию образования уротропинового комплекса превраща-
превращали в аминоацетофенон, который ацетилировали уксусным ан-
ангидридом:
Вг2 /=\ (CH2NN4
COCHg >- { >-СОСН2Вг ►■
(СН3С0JО
1+ НС1 /=\
ВГ >• £ )>-СОСН2Ш.
<v ,?—CO-CH2-NHCOCH3
Далее синтез шел аналогично изложенному § 2, но нитро-
группу в ароматическое ядро вводили на предпоследней стадии,
что вызывало снижение выхода целевого продукта. В связи с
этим такой метод синтеза в СССР не применяется. Промышлен-
Промышленное значение получил разработанный почти одновременно
cvv первым синтез левомицетина из п-нитробензойной кислоты
через стадию образования п-нитроацетофенона:
, SOCl2 /=\ C2H5OMgCH(COOC2H6J
^-СООН >■ O2N-4 >-СОС1 *•
OH2SO4 // ^ Вг2
—СОСН(СООС2Н5J >• O2N-<^ \-COCHg >
O(CH2NN4
—СОСН2Вг >■
]+ НС1
ВГ >
(СН3СОJО
^-COCH2NH2 ►■
DCH2NHCOCH3 >-
>- далее в соответствии с изложенным в § 2
422
Этот способ детально разработан и усовершенствован во
ВНИХФИ. При этом технологический процесс разработан та-
таким образом, что пять стадий, начиная с бромирования п-нитро-
ацетофенона до получения п-нитро-а-ацетиламино-|3-оксипро-
пиофенона проводились без выделения продуктов из реакцион-
реакционной массы, что значительно облегчало организацию крупнотон-
крупнотоннажного производства.
Впоследствии был разработан способ синтеза левомицетина
из дешевого и доступного сырья — стирола:
Са(ОС1J, HNO3f CH3COOH /=
38-40^44,5
HNO3 конц.
ОН С1
Фенил(хлорметил)карбинол
(хлоргидрин стирола)
ch-ch2ci
28-30% HNO3
105OQ
0N02
л-Нитрофенил(хлорметил)карбинол
нитрат («нитроэфир»)
25-30% NaOH
ОН
Хлоргидрин
п-нитростирола
NH3, CH3OH
ch-ch2ci
O2N-Q-
O2N—
CH-CH2
OH NHCOCHg
—С—CH2NHCOCH3
II
О
Na2Cr2O7, H2SO4
5—о С
Левомицетин
Непосредственное нитрование стиролхлоргидрина, помимо
стадии защиты оксигруппы ацетилированием, приводит к сокра-
сокращению числа операций и повышению выхода нужного п-изоме-
ра. Пара-, орто- и мета-изомеры образуются в соотношении 60:
:30: 10, тогда как при предварительной защите оксигруппы они
образуются в соотношении 37:56:7 (т. е. примерно таком же,
как при нитровании толуола). Разделение изомеров в этом слу-
случае также облегчено, так как п-изомер является твердым веще-
веществом, а маслообразные орто- и мета-изомеры уходят с «маточ-
«маточниками при центрифугировании окиси п-нитростирола. Окисле-
Окисление п-нитрофенилацетиламиноэтанола хромовой смесью является
очень чувствительным процессом. Высокий выход «кетона» может
быть достигнут только при тщательном выполнении всех требо-
423
ваний регламента. Особенно важны поддержание низкой E—
8°С) температуры реакционной массы и обеспечение интенсив-
интенсивного перемешивания.
Разработан также другой метод синтеза левомицетина из
стирола:
HNO3
O2N
OCH3
NaOHf CH3OH
■—СН—СН2С1 >■ O2N
ОСИ,
н2о
v > O2N-^ >-СОСНя
н2о
СН2Вг >■ O2N
>—COCHoBr
Раствор стирола в метаноле подвергают хлорированию га-
газообразным хлором. Реакционную массу нейтрализуют кальци-
кальцинированной содой, отгоняют метанол, обрабатывают остаток
водой для растворения хлорида натрия и отделяют технический
метиловый эфир фенилхлорметилкарбинола. Полученный техни-
технический эфир нитруют смесью азотной и серной кислот, нитро-
массу разбавляют водой, а выделившуюся смесь нитропродук-
тов обрабатывают водно-метанольным раствором едкого натра.
Образовавшийся п-нитро-а-метоксистирол отфильтровывают,
промывают метанолом и водой для удаления изомерных нитро-
продуктов и хлорида натрия, а затем подвергают омылению 1%
раствором серной кислоты в 60% изопропиловом спирте. Вы-
Выход п-нитроацетофенона, считая на стирол, составляет 25% от
теоретического.
При этом методе несколько меньше выход п-изомера при
нитровании, но возможность получения п-нитроацетофенона
или п-нитро-а-бромацетофенона позволило быстро внедрить этот
метод на тех заводах, где синтез левомицетина проводился че-
через эти продукты. Кроме того, в этом методе отсутствует тех-
технологически сложная и дорогая стадия окисления хромпиком.
Синтомицин и левомицетин в Советском Союзе производят-
производятся только из стирола в соответствии с двумя изложенными вы-
выше химическими схемами.
В настоящее время ведутся работы по переводу производст-
производства синтомицина и левомицетина на непрерывный метод, который
обеспечит высокую производительность труда при комплексной
механизации и автоматизации производства.
424
§ 4. СТРОЕНИЕ И СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОСЕРИНА
Циклосерин является эффективным антибиотиком противо-
противотуберкулезного действия. По активности циклосерин уступает
стрептомицину, тубазиду и фтивазиду, но действует на мико-
бактерии туберкулеза, устойчивые к этим препаратам и ПАСК.
Циклосерин рассматривается как «резервный» противотуберку-
противотуберкулезный препарат, т. е. препарат, применяемый при лечении
больных хроническими формами туберкулеза, на которых ра-
ранее применявшиеся основные препараты перестали эффектив-
эффективно действовать.
Циклосерин (D-4-амино-З-изоксазолидон) относится к анти-
антибиотикам гетероциклического строения:
Н
H2N~C С=О
Н2С NH
Циклосерин является производным изоксазола.
НС СН Н2С СН2 Н2С С=О
II II II II
НС N Н2С NH H2C NH
V V V
Изоксазол Изоксазолидин З-Изоксазолидон
С другой стороны, циклосерин можно рассматривать как
производное важной аминокислоты — серина.
H2n-CH—с/
I ХОН
сн2
он
Серии
Благодаря относительно простому строению существует мно-
много способов синтеза циклосерина. Мы приведем лишь один спо-
способ получения этого антибиотика. Гидрохлорид метилового эфи-
эфира D,L-cepHHa действием пятихлористого фосфора превращают
в гидрохлорид метилового эфира р-хлор-а-аланина. Последний
при обработке щелочным раствором гвдроксиламина превраща-
превращается в циклосерин:
PC13 H2NOH, NaOH
HCLH2N-CH С=О >■ HCLH2N-CH-C=O >"
СН2ОН ОСН3 СН2 ОСН3
Cl
425
H2N—CH— C=0
* H2C NH
Циклосерин представляет собой белый кристаллический по-
порошок горьковатого вкуса. Хорошо растворим в воде. Кристал-
Кристаллизуется из ацетона и спирта, температура плавления 154—
155 °С. Циклосерин оптически активен. Нейтральные и кислые
растворы циклосерина неустойчивы. Выпускается циклосерин в
виде таблеток и капсул 0,25 г.
СОДЕРЖАНИЕ
Предисловие 3
Введение .'.'.'.'! 5
Часть I
ТЕХНОЛОГИЧЕСКИЕ МЕТОДЫ ПРОИЗВОДСТВА ПРОМЕЖУТОЧНЫХ
ПРОДУКТОВ
Глава 1. Сырье для химико-фармацевтической промышленности . 15
§ 1. Источники сырья для химико-фармацевтической промышлен-
промышленности 15
§ 2. Характеристика некоторых видов основного и вспомогательно-
вспомогательного сырья . 17
§ 3. Стандарты и технические условия 21
Глава 2. Основные химические реакции, лежащие в основе синтеза
лекарственных веществ 23
§ 1. Общая характеристика химической технологии лекарственных
веществ 23
§ 2. Общие сведения о строении промежуточных продуктов. Поня-
Понятие об ароматичности 25
§ 3. Три основных типа реакций в технологии промежуточных про-
продуктов 28
§ 4. Нуклеофильное, электрофильное и радикальное замещение . . 29
§ 5. Электронное влияние заместителей на химические свойства
и реакции 33
Глава 3. Сульфирование и сульфохлорирование 38
§ 1. Общие сведения 38
§ 2. Механизм реакции сульфирования 39
§ 3. Условия проведения процессов сульфирования 41
§ 4. Основные способы сульфирования 42
§ 5. Основные способы выделения сульфокислот 49
§ 6. Сульфирование важнейших ароматических соединений ... 52
§ 7. Примеры сульфирования в промышленности. Производство
натриевой соли бензосульфокислоты 54
§ 8. Контроль сульфирования и характеристика продуктов ... 58
§ 9. Сульфохлорирование. Механизм реакции 58
§ 10. Условия и способы проведения сульфохлорирования ... 59
§11. Техника безопасности при проведении процессов сульфирова-
сульфирования и сульфохлорирования
Глава 4. Нитрование
§ 1. Общие сведения ^3
§ 2. Механизм реакции ^
§ 3. Условия проведения процессов нитрования *>£
§ 4. Основные способы нитромания °й
§ 5. Основные методы выделения нитропродуктов п
427
§ 6. Примеры нитрования ароматических соединений .... 77
§ 7. Производство нитробензола непрерывным методом .... 79
§ 8. Контроль нитрования и характеристика продуктов .... 81
§ 9. Техника безопасности 82
Глава 5. Галогенирование 84
i § 1. Общие сведения 84
§ 2. Механизм реакции '.'.'. 85
§ 3. Условия проведения процессов галогенирования .... 86
§ 4. Основные стадии процесса хлорирования 87
§ 5. Практически важные примеры галогенирования 94
§ 6. Промышленное производство хлорбензола непрерывным мето-
методом 97
§ 7. Технологические особенности процессов бромирования ... 98
§ 8. Контроль процессов 99
§ 9. Техника безопасности 99
Глава 6. Оксилирование и аминирование 101
§ 1. Механизм реакции 101
§ 2. Нуклеофильное замещение сульфогруппы 102
§ 3. Промышленные методы проведения процесса щелочного плав-
плавления ^ 106
§ 4. Обмен хлора на окси- и аминогруппу 109
§ 5. Взаимные превращения амино- и оксисоединений . . . . 121
§ 6. Техника безопасности при ведении реакций оксилирования и
аминирования 124
Глава 7. Алкилирование и ацилирование аминов и оксисоединений . 127
§ 1. Алкилирование аминов 128
§ 2. Алкилирование ароматических оксисоединений 131
§ 3. Ацилирование ароматических аминов 133
§ 4. Ацилирование оксигруппы 138
§ 5. Производство фенацетина 139
§ 6. Техника безопасности при проведении процессов алкилирова-
ния и ацилирования 142
Глава 8. Алкилирование и ацилирование углеводородов . . . . 145
§ 1. Механизм реакции 145
§ 2. Примеры реакций алкилирования и ацилирования углеводо-
углеводородов 148
§ 3. Производство салициловой кислоты 154
§ 4. Техника безопасности 155
Глава 9. Диазотирование и нитрозирование. Превращение диазосоеди-
нений 156
§ 1. Механизм реакции 156
§ 2. Условия проведения реакции 158
§ 3. Превращения диазосоединений. Замена диазогруппы . . . 159
§ 4. Производство гваякола 162
Глава 10. Восстановление и окисление 166
§ 1. Восстановление металлами 166
§ 2. Восстановление сульфидами щелочных металлов . . . . 175
§ 3. Каталитическое восстановление водородом 177
§ 4. Электролитическое восстановление }8у
§ 5. Техника безопасности при проведении процессов восстановления 181
§ 6. Окисление I82
§ 7. Техника безопасности при проведении процессов окисления . 188
428
Глава 11. Образование гетероциклов 189
§ 1. Образование пятичленных гетероциклов 191
§ 2. Образование шестичленных гетероциклов 194
Часть II
ТЕХНОЛОГИЯ СИНТЕТИЧЕСКИХ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Глава 12. Неорганические лекарственные вещества 198
§ 1. Общие сведения о неорганических лекарственных веществах
и их значении в медицине 198
§ 2. Получение лекарственных препаратов брома и йода\ ! . . 200
§ 3. Получение перманганата калия 202
Глава 13. Лекарственные органические соединения алифатического
ряда .204
§ 1. Галогенпроизводные углерводородов алифатического ряда . . 204
§ 2. Спирты и простые эфиры 207
§ 3. Альдегиды, альдегидокислоты и их производные . . . . 210
§ 4. Карбоновые кислоты и их производные 219
§ 5. Алифатические амины, аминокислоты и их производные . . 228
Глава 14. Лекарственные соединения алициклического ряда . . . 233
§ 1. Терпеноиды 233
§ 2. Витамин А 235
§ 3. Заменители плазмы крови 239
§ 4. Витамин К и вещества, влияющие на свертываемость крови . 241
§ 5. Витамин Р, биофлаваноиды 243
§ 6. Такоферолы ((витамины прушш Е) 244
§ 7. Стероидные соединения, гормоны . ' 246
§ 8. Витамин D 249
Глава 15. Лекарственные соединения ароматического ряда . . . 251
§ 1. Фенолы и их производные 251
§ 2. Ароматические карбоновые кислоты и их производные . . . 252
§ 3. Производные ароматических сульфокислот. Сульфаниламидные
препараты 256
Глава 16. Элементорганические лекарственные вещества .... 270
§ 1. Органические соединения мышьяка 270
§ 2. Органические соединения сурьмы и висмута 272
§ 3. Органические соединения ртути 273
§ 4. Фосфорорганические соединения 274
§ 5. Рентгеноконтрастные средства 277
Глава 17. Лекарственные соединения гетероциклического ряда . . 280
§ 1. Производные пятичлеиных гетероциклов 280
§ 2. Производные шестичленных гетероциклов с одним гетероатомом 291
§ 3. Производные шестичленных гетероциклов с двумя гетероатомами 301
Часть III
ТЕХНОЛОГИЯ АНТИБИОТИКОВ
Глава 18. Технология биосинтеза антибиотиков и получения натив-
ного раствора 316
§ 1. Пути использования минеральных и органических азотсодержа-
азотсодержащих веществ микроорганизмами 319
429
§ 2. Основные пути использования азота микроорганизмами . . 322
§ 3. Основные пути метаболизма углеводов у продуцентов антибио-
антибиотиков 324
§ 4. Характеристика источников углерода, применяемых в производ-
производстве антибиотиков, и влияние их на биосинтез 328
§ 5. Участие минеральных веществ в метаболизме продуцентов ан-
антибиотиков 331
§ 6. Значение отдельных элементов минерального питания в жизне-
жизнедеятельности микроорганизмов 333
§ 7. Значение кислотности и других характеристик среды для жиз-
жизнедеятельности продуцентов 335
§ 8. Биосинтез пенициллина . 337
§ 9. Биосинтез тетрациклиновых антибиотиков 340
§ 10. Биосинтез тетрациклина 343
§ 11. Биосинтез окситетрациклина 344
§ 12. Биосинтез стрептомицина 346
§ 13. Приготовление и стерилизация питательной среды . . . 349
§ 14. Очистка воздуха от микроорганизмов, аэрация культуральных
жидкостей. Конструкция ферментаторов 356
§ '15. Ценообразование в культуральных жидкостях и пеногашение 361
§ 16. Коллоидно-химические основы коагуляции культуральных
жидкостей 365
Глава 19. Технология химической очистки и выделения .... 370
§ 1. Экстракция 370
§ 2. Сорбционные процессы 379
§ 3. Осуждение и кристаллизация 393
§ 4. Основы технологии сублимационной и распылительной сутки
антибиотиков 402
Глава 20. Полусинтетические антибиотики 411
§ 1. Полусинтетические пенициллины 411
§ 2. Цафалоспорины 414
§ 3. Полусинтетические тетрациклины 41 6
Глава 21. Синтетические антибиотики 418
§ 1. Синтомицин и левомицетин. Стероизомерия и биологическая ак-
активность 418
§ 2. Принципиальная схема получения синтомицина и левомицетина 419
§ 3. Промышленные способы синтеза п-иитро-а-ацетиламино-ацето-
фенона 422
§ 4. Строение и способ получения циклосерина .425
ИБ № 162
ПАССЕТ БОРИС ВИКТОРОВИЧ,
ВОРОБЬЕВА ВЕРА ЯКОВЛЕВНА
Технология химико-фармацевтических
препаратов и антибиотиков
Редактор Л. Е. Холодов
Художественный редактор Л. Л. Лозовская
Корректор 3. П. Бабуева
Техн. редактор Н. И. Людковская
Переплет художника Т. Ф. Санкиной
Сдано в набор 23/VIII 1976 г. Подписано к печати
26/XI 1976 г. Формат бумаги 60X90/16 Печ. л.
27,0+0,125 печ. л. вкл. (условных 27,125 л.)
27,60 уч.-изд. л. Бум. тип. № 2 Тираж 33 000 экз.
Т-19536. МУ-49. Цена 79 коп.
Издательство «Медицина». Москва,
Петроверигский пер., 6/8
Заказ 926. Московская типография № 11
Союзполиграфпрома при Государственном
комитете Совета Министров СССР по делам
издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская, 1.