/





































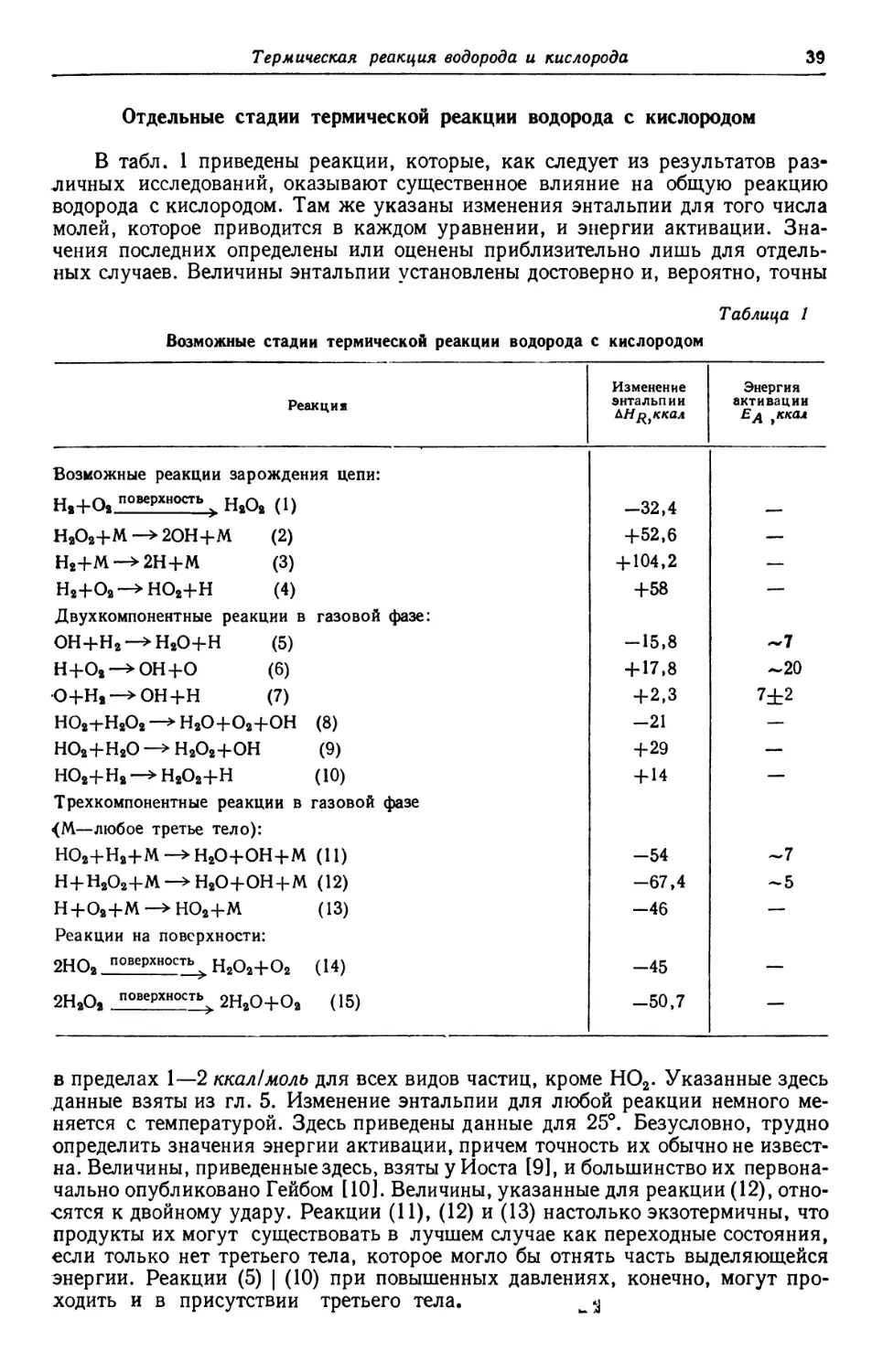

































































































































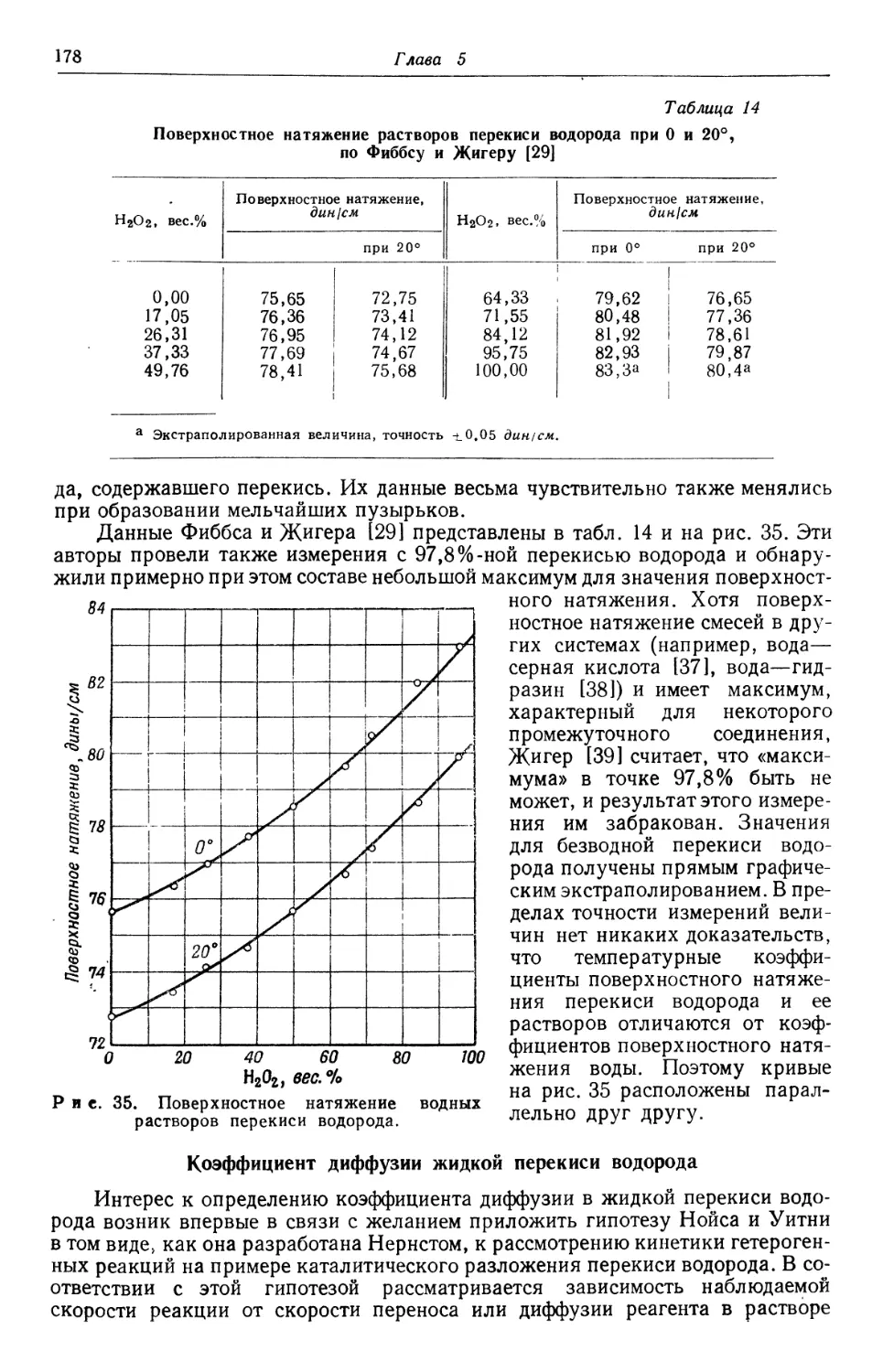



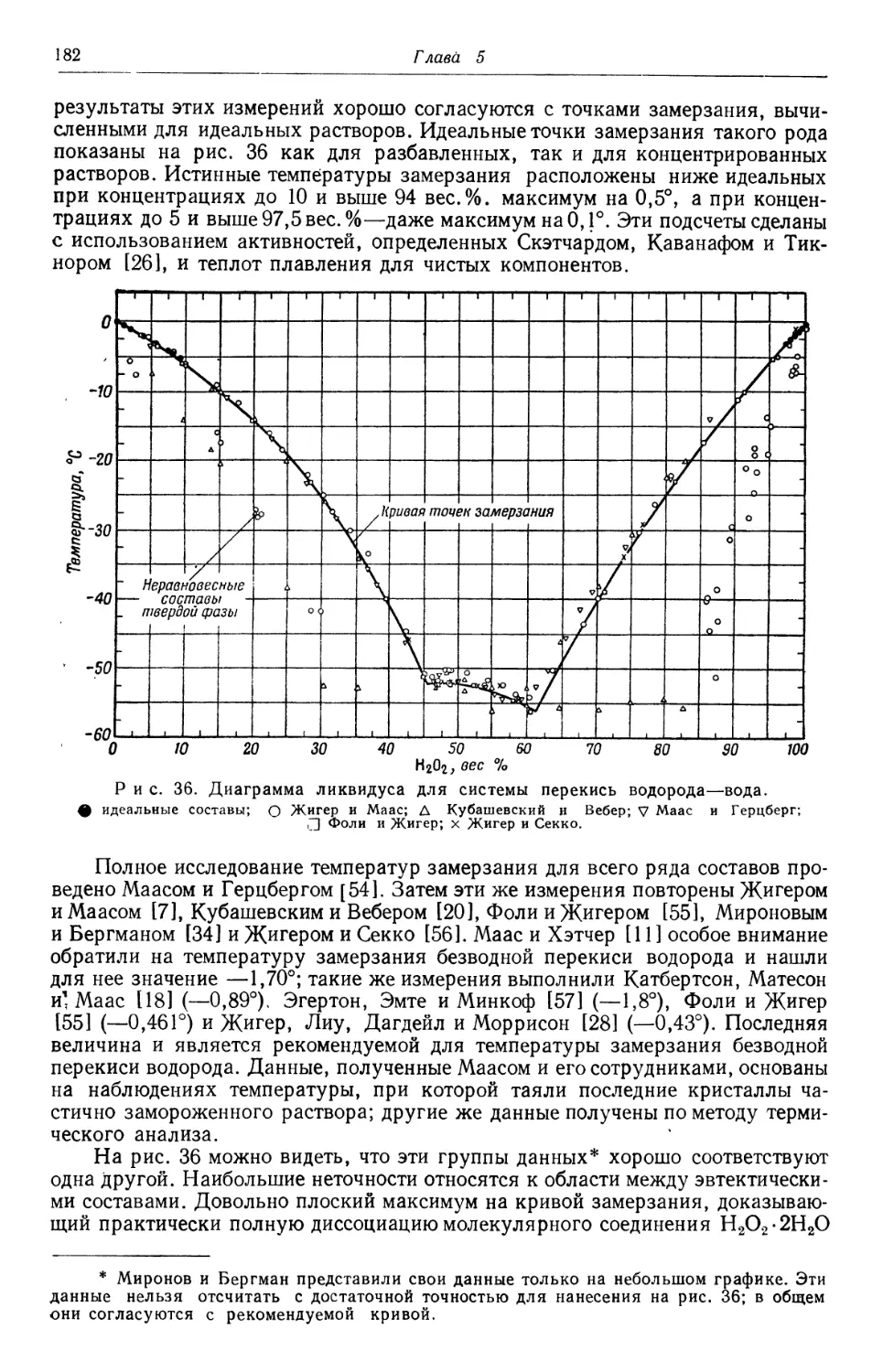








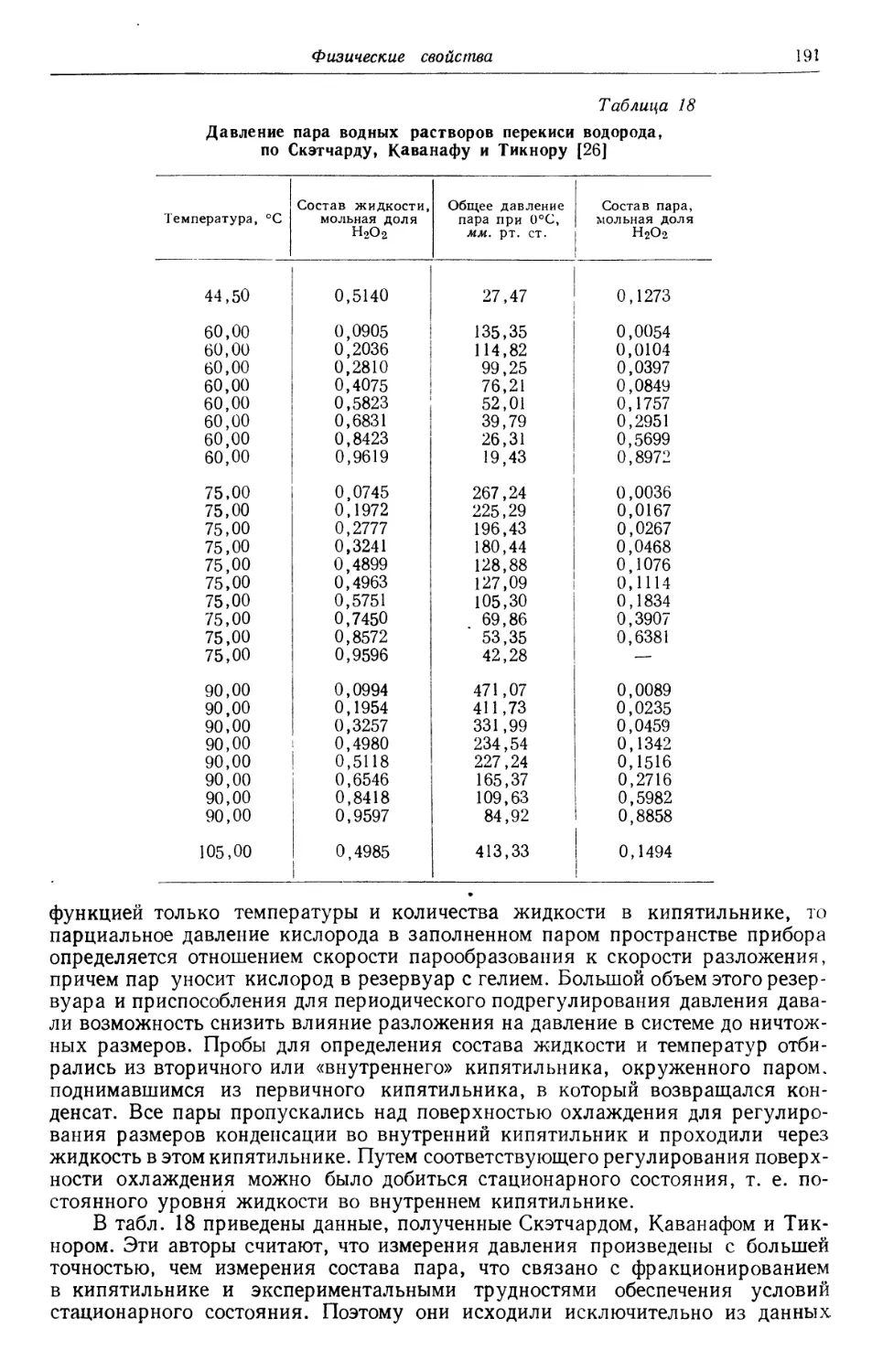

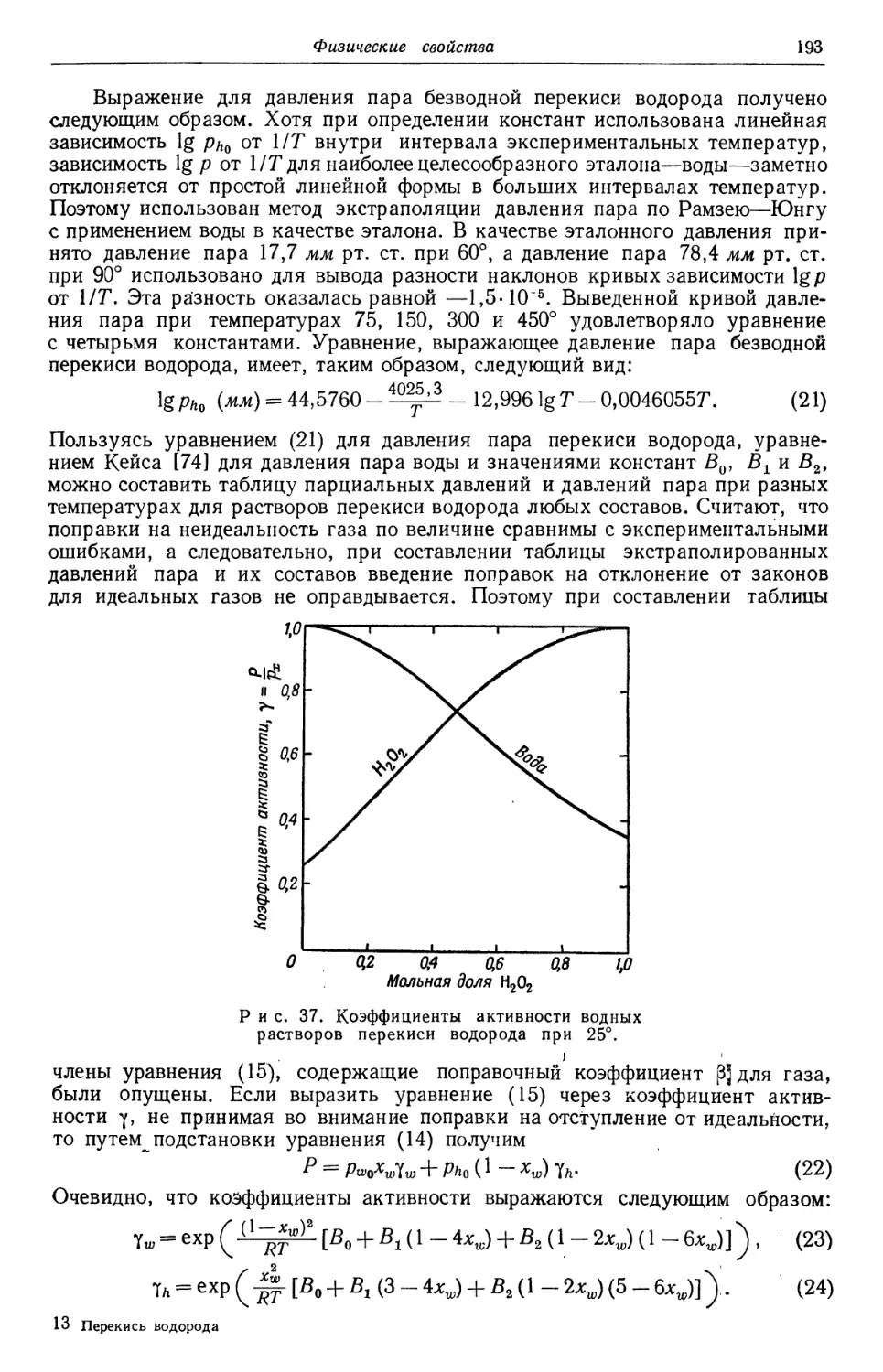





































































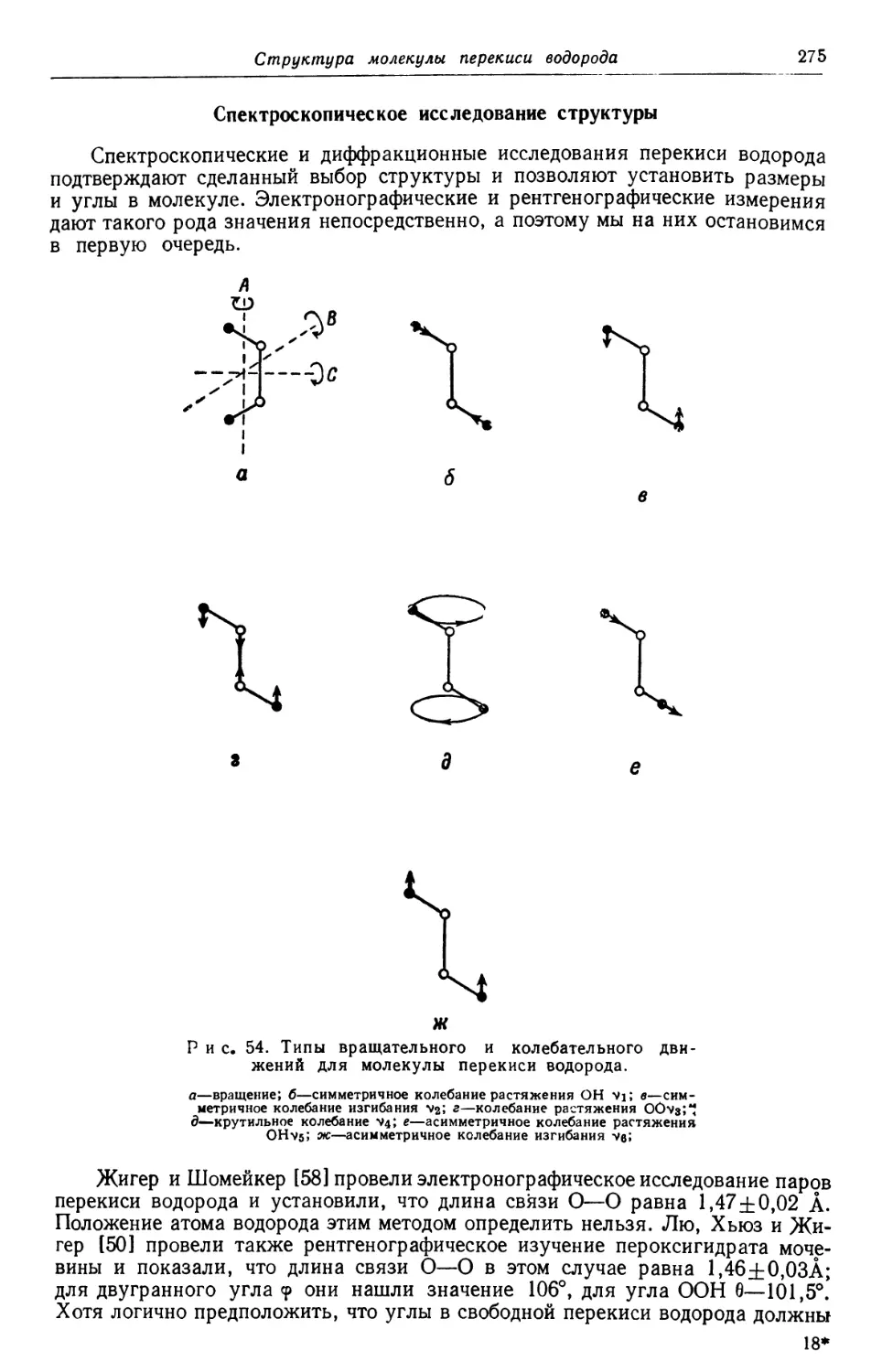
























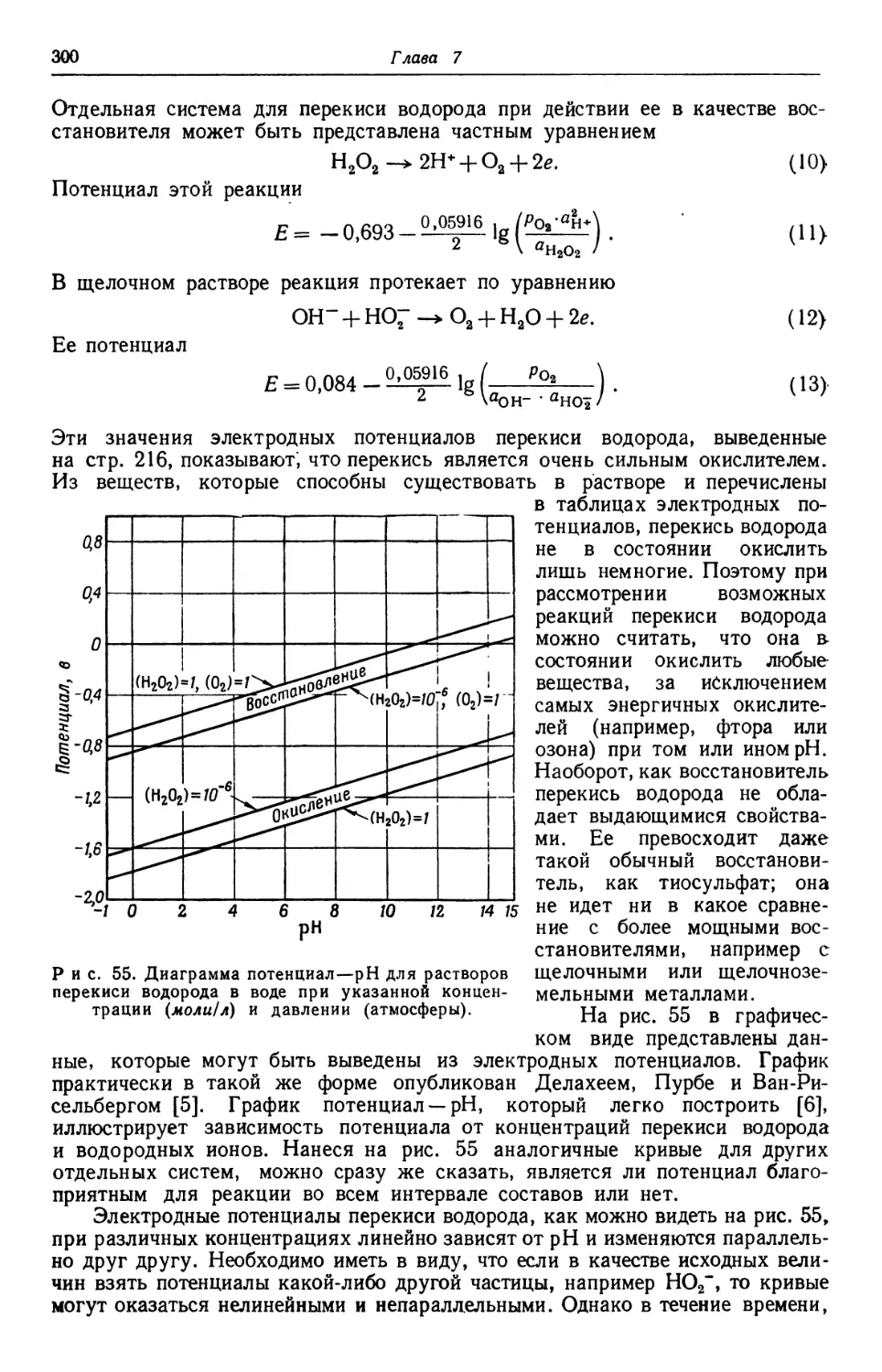

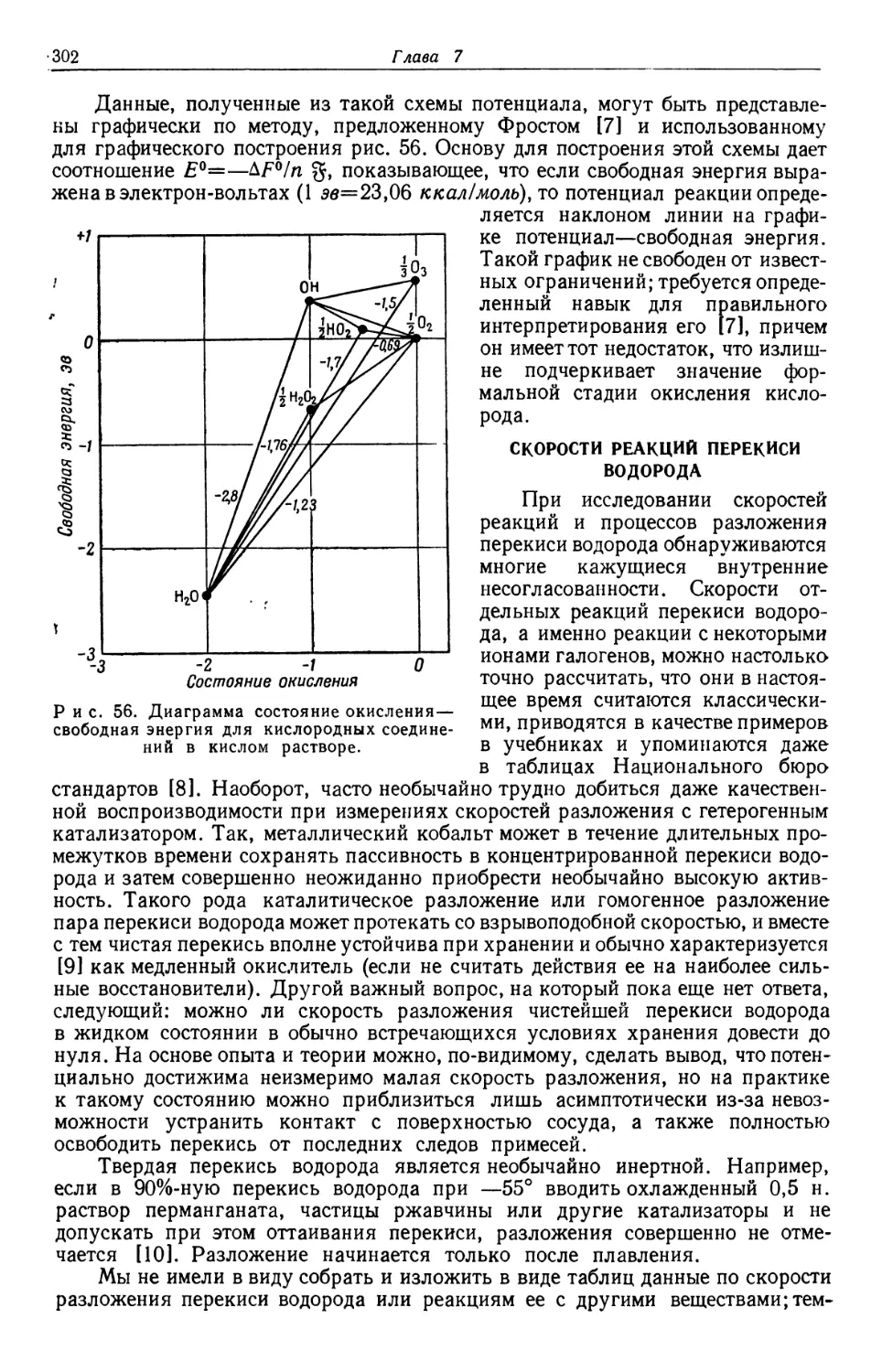

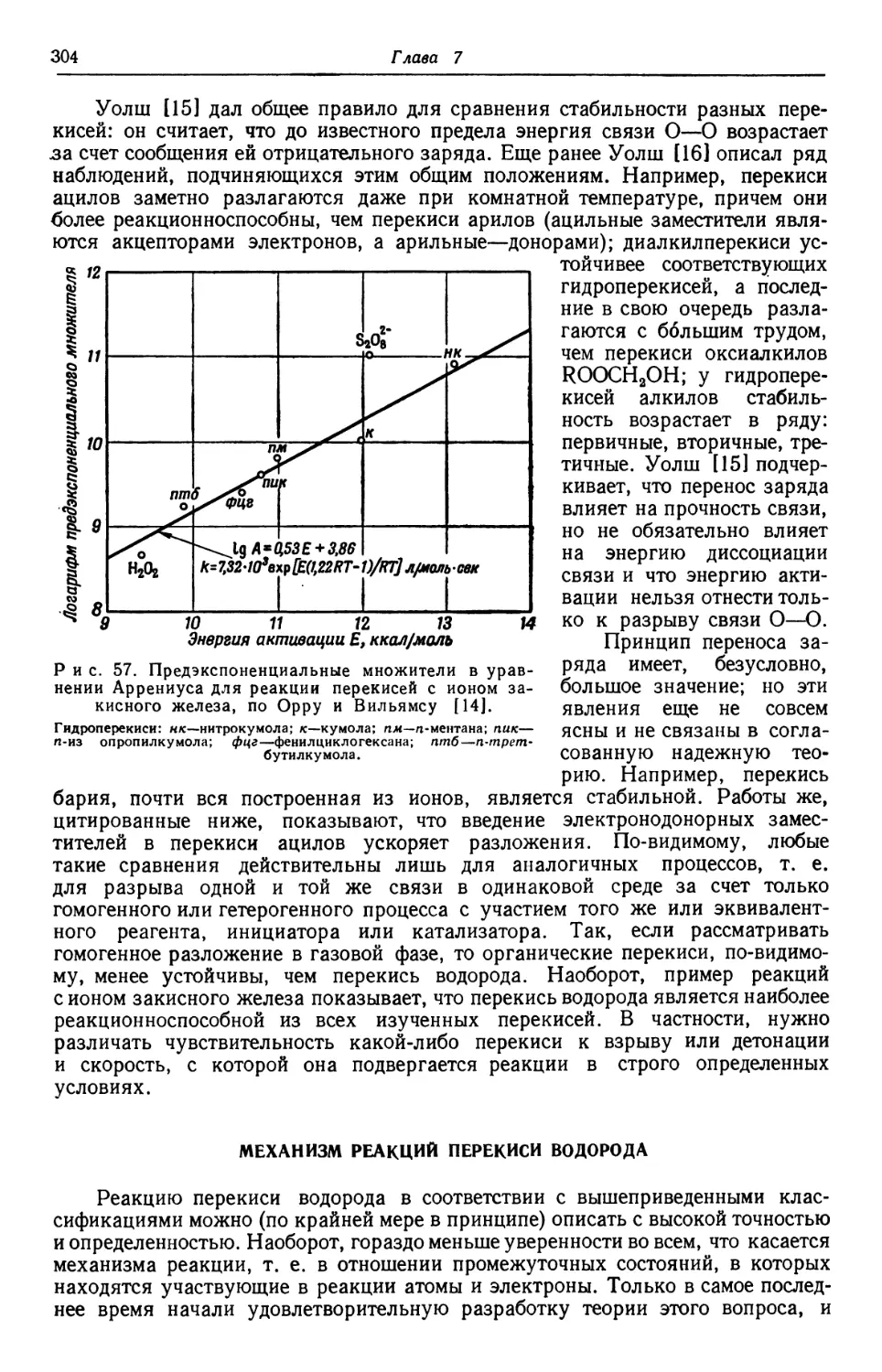































































































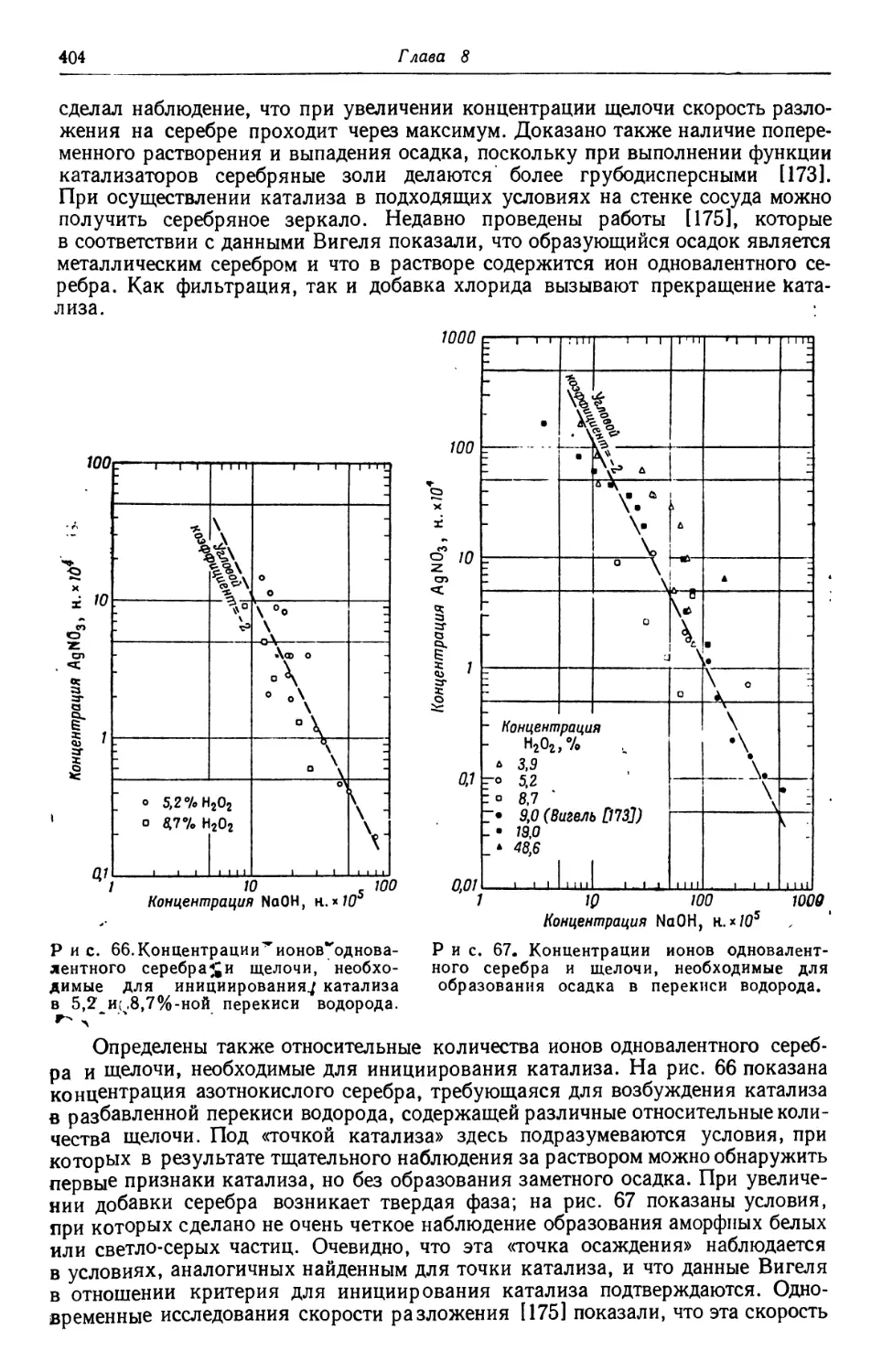












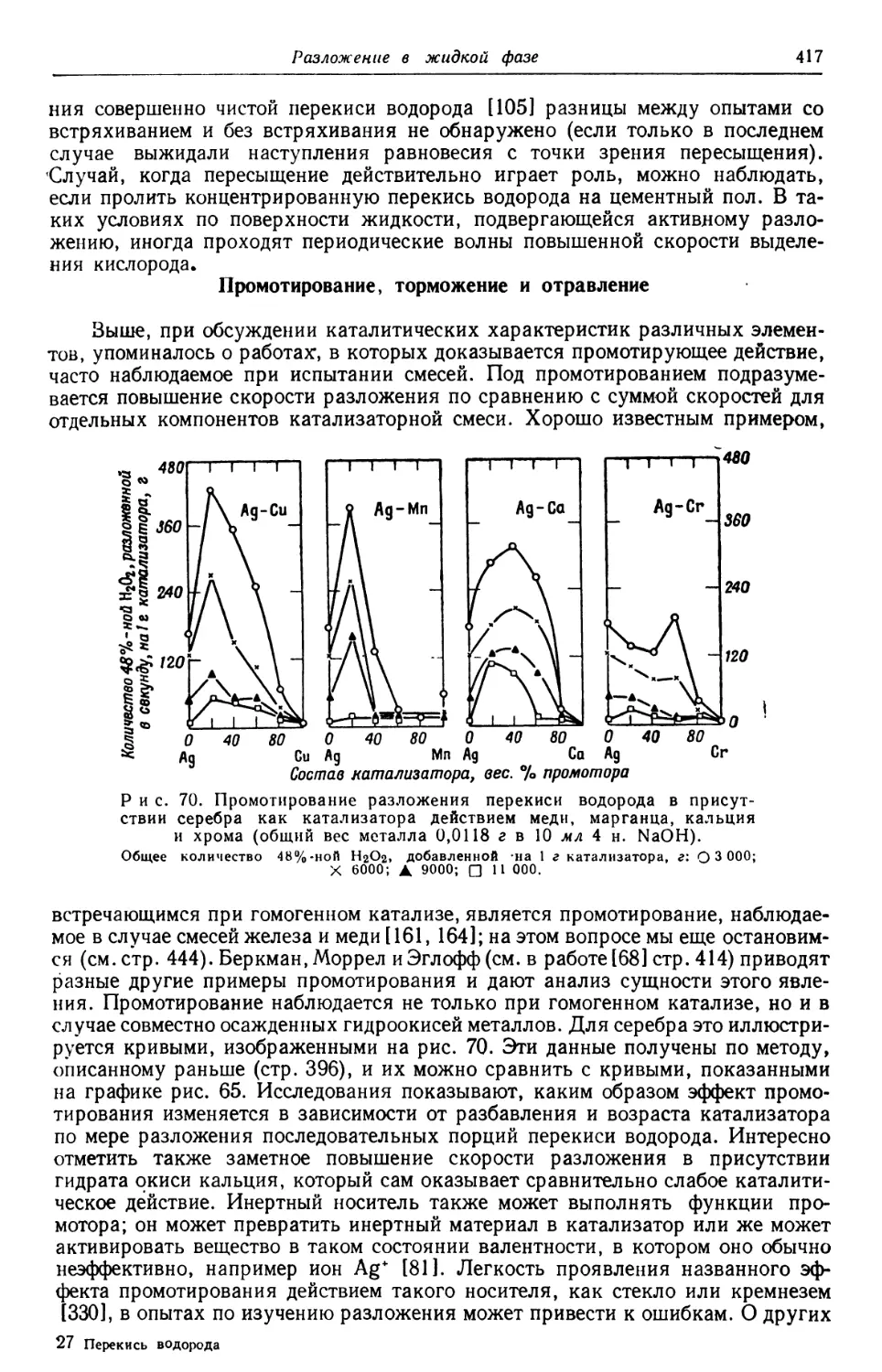





















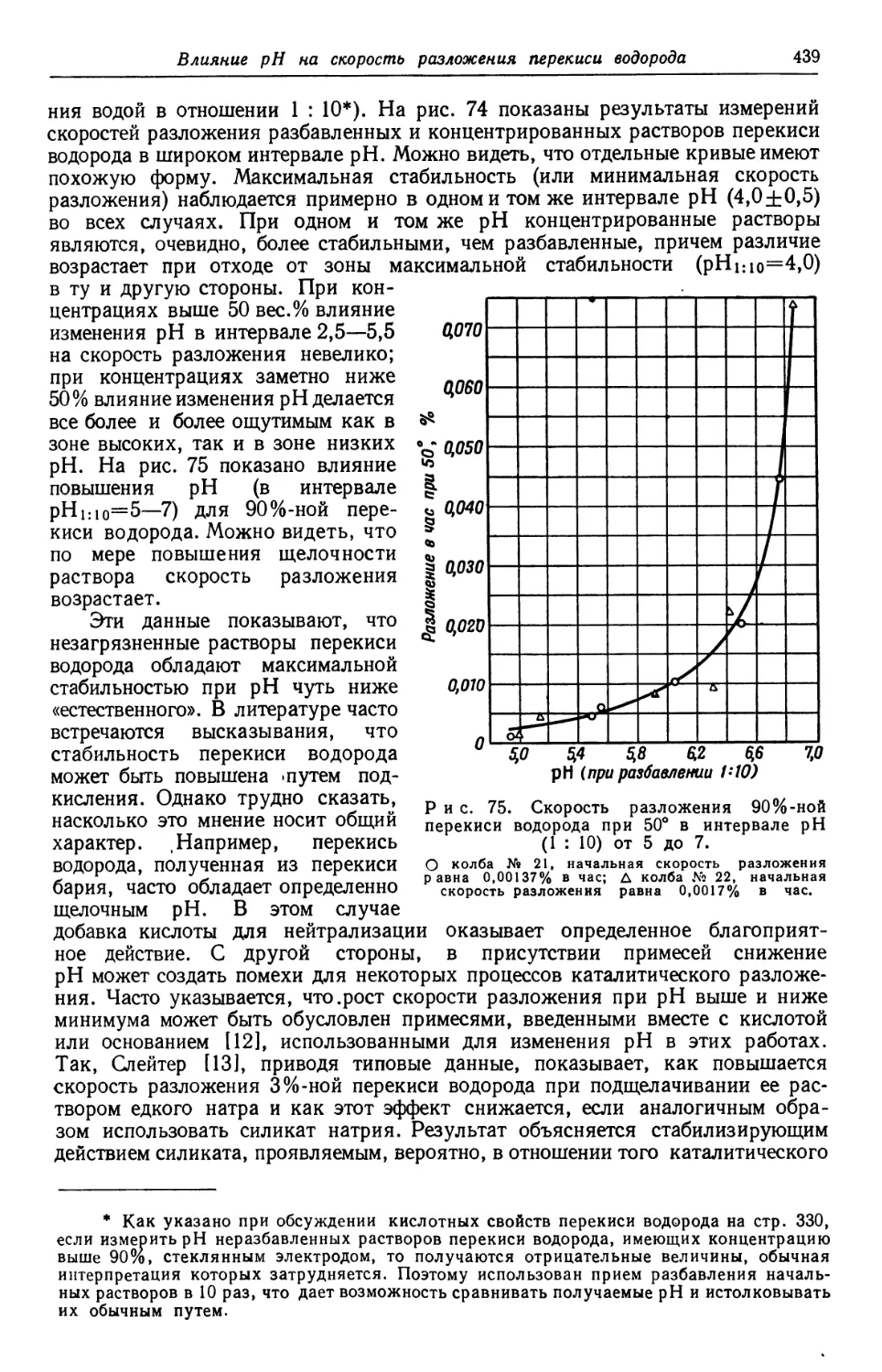

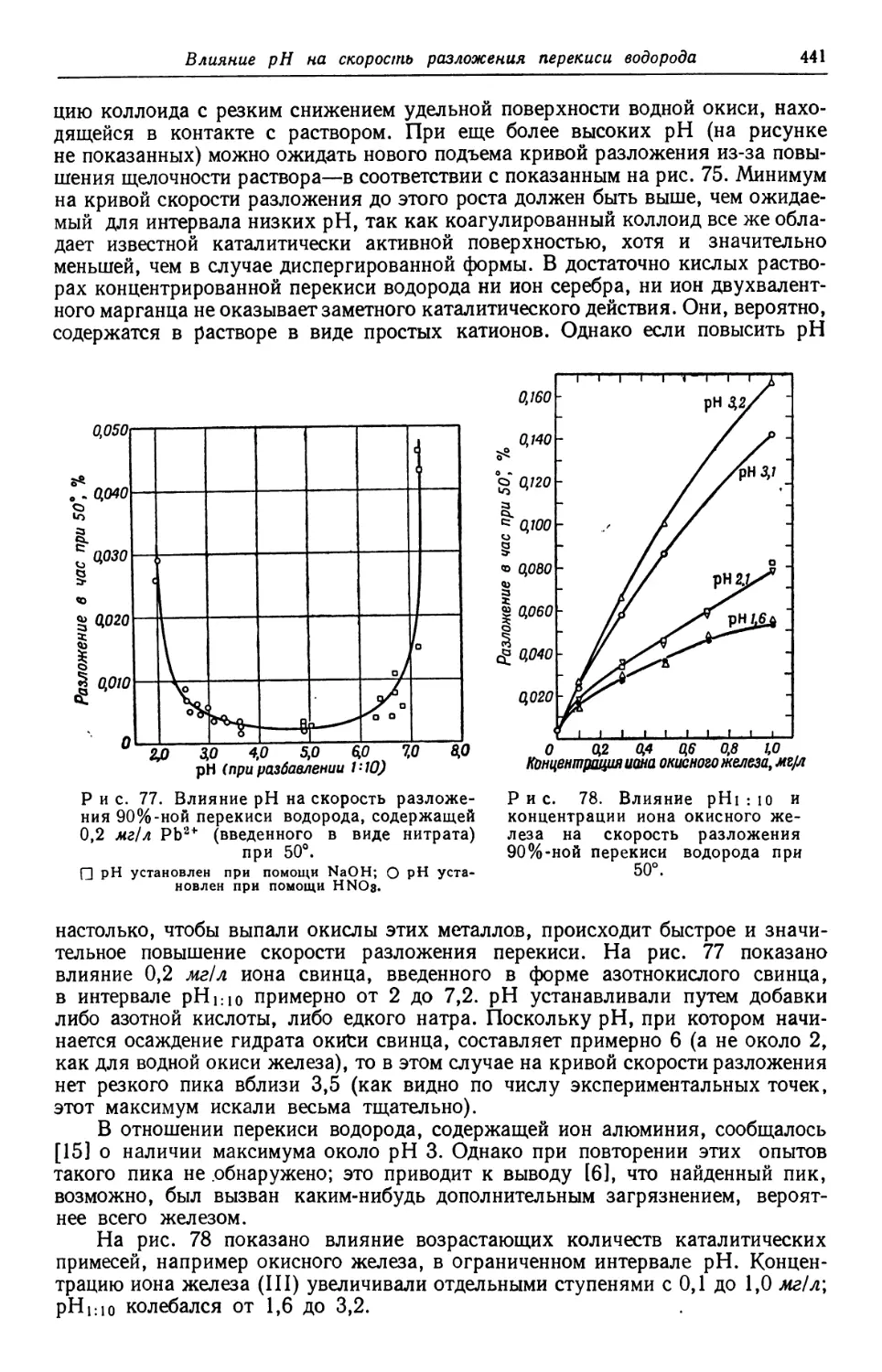










































































































































Text
и * л
Издательство
иностранной
литературы
HYDROGEN PEROXIDE
BY
WALTER С SCHUMB
Professor of Inorganic Chemistry
CHARLES N. SATTERFIELD
Associate Professor of Chemical Engineering
RALPH L. WENTWORTH
Industrial Liaison Officer*
Massachusetts Institute of Technology
American Chemical Society Monograph Series
REINHOLD PUBLISHING CORPORATION
NEW YORK. N.Y.
CHAPMAN «* HALL, LTD., LONDON
У. ШАМБ, Ч. СЕТТЕРФИЛД, Р. ВЕНТВОРС
ПЕРЕКИСЬ ВОДОРОДА
Перевод с английского
Г. Д. ВИГДОРОВИЧА
Под редакцией
доктора техн., наук
А. И. ГОРБАНЕВА
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, 1958
АННОТАЦИЯ
В книге дано описание историй открытия перекиси водо-
водорода, развития способов ее получения и очистки, исследования
физических, химических и термодинамических свойств, условий
разложения и стабилизации, а также методов качественного
и количественного анализа; кроме того, проведено сопоставление
перекиси водорода с другими кислородо-водородными соедине-
соединениями и описаны свойства и методы получения других неорга-
неорганических перекисных соединений. Уделено внимание изложению
результатов исследования реакций перекиси при помощи изо-
изотопного обмена.
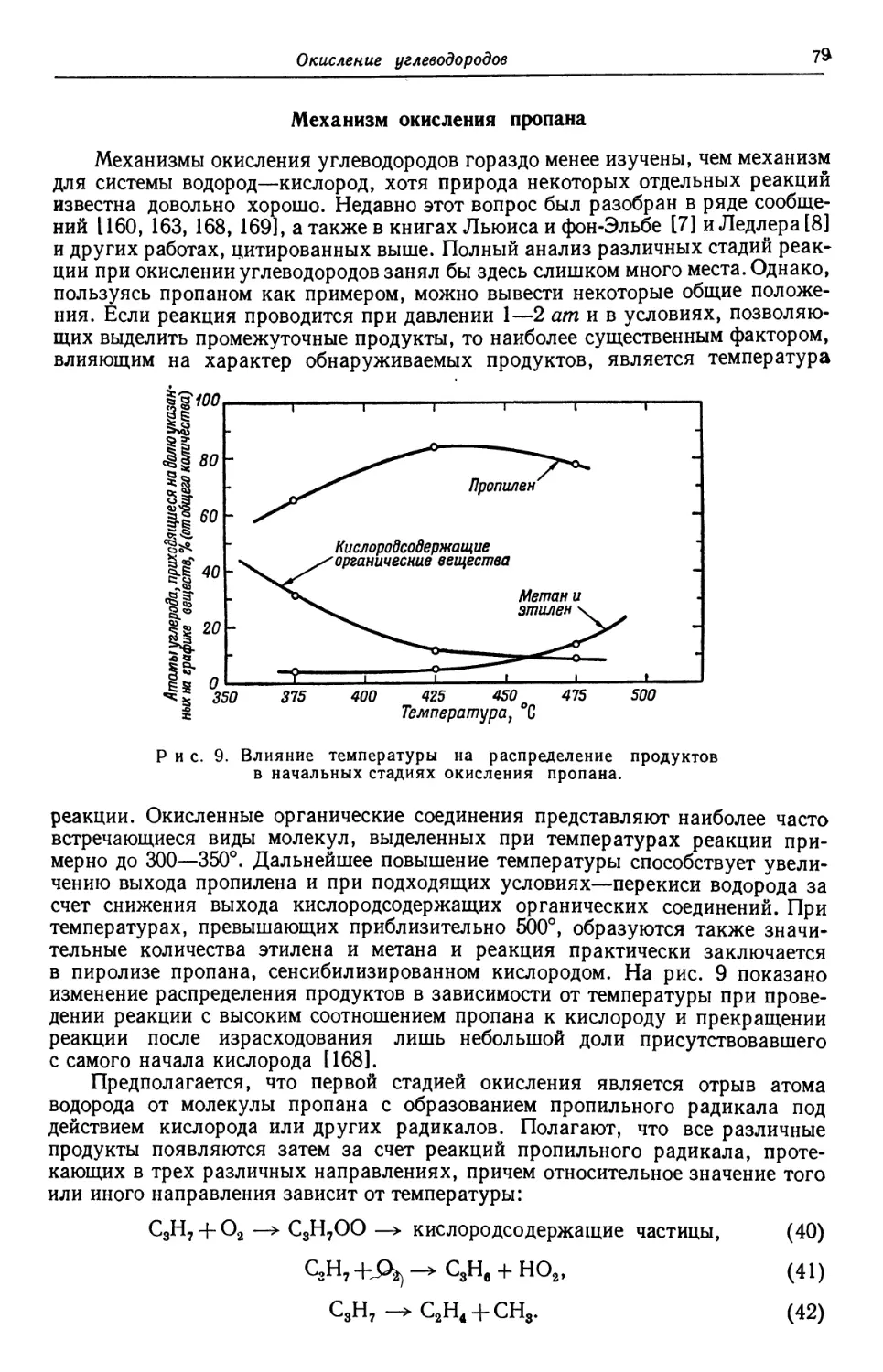
Рассмотрены также примеры широкого применения перекиси
водорода в качестве отбеливающего средства, химического реа-
реагента в текстильной, бумажной и других областях промышлен-
промышленности, значения ее в биологии и медицине, использования в ра-
ракетной технике и т. п.
Предназначена для широкого круга научных работников
и технологов, занимающихся вопросами исследования и приме-
применения перекиси водорода и перекисных соединений.
Редакция литературы по химическим наукам.
Заведующий редакцией профессор О. А. РЕУТОВ.
ПРЕДИСЛОВИЕ
Перевод книги У. Шамба, Ч. Сеттерфилда, Р. Вентворса «Перекись водо-
водорода» в некоторой степени восполняет пробел, существующий в нашей науч-
научной литературе по химии перекисных соединений. В настоящее время эта от-
отрасль химии быстро развивается; в течение последних 10—15 лет количество
печатных работ, посвященных изучению перекиси водорода, значительно воз-
возросло как в СССР, так и за рубежом.
Среди ранее опубликованных трудов, посвященных перекисным соеди-
соединениям, следует указать на оригинальную монографию Л. В. Писаржевского
«Перекиси и надкислоты», в которой освещается состояние этого вопроса
к периоду, предшествовавшему 1902 г. Вышедшая в свет в 1915 г. переводная
монография Гирзевальда «Неорганические перекисные соединения» теперь
устарела и не отвечает современным требованиям. Перевод первого издания
A936 г.) книги Маху «Перекись водорода и перекисные соединения», выпущен-
выпущенный в 1951 г., до настоящего времени был наиболее полным руководством по
химии перекисных соединений на русском языке. Однако монография Маху
по содержанию является компилятивным трудом, в котором расположение мате-
материала носит случайный характер.
Настоящая монография является итогом многолетней работы авторов и по-
посвящена главным образом перекиси водорода. В ней достаточно глубоко и полно
обобщена мировая литература в этой области (приведено свыше 2500 литератур-
литературных источников). Книга содержит описание истории открытия и технического
применения перекиси водорода, способов образования и, как следствие, путей
промышленного ее получения и способов очистки, а также правил обращения
с этим веществом. Из рассмотрения физических свойств перекиси водорода
и сопоставления их со свойствами других кислородо-водородных соединений
вытекает строение молекулы перекиси водорода, которое подтверждается при
последующем рассмотрении химических свойств перекиси водорода и способов
ее, стабилизации. Достаточно полно представлено описание качественного
и количественного анализа, а также способов применения перекиси водорода
для целей отбеливания и окисления, как источника энергии в военном деле,
источника радикалов, а также как агента, влияющего на биологические про-
процессы.
В конце книги сделана попытка вкратце рассмотреть склонность химиче-
химических элементов к образованию перекисных соединений в зависимости от их
места в периодической системе Д. И. Менделеева, а также способов получения
неорганических перекисей, надперекисей, пероксокислот и их производных.
Знание предмета и критическая оценка имеющегося материала позволили
авторам выбрать из массы данных наиболее достоверные числовые характери-
характеристики ряда физических свойств перекиси водорода, которые и рекомендуются
для применения в практике. Подробные таблицы и графики для ряда свойств
системы Н2О—Н2О2, например по удельным весам, показателям преломления
и т. п., окажут большую помощь работникам, работающим как в лаборатории,
так и на производстве. Определенную ценность представляют также указания
Предисловие
авторов на нерешенные вопросы и направление путей дальнейшего изучения
способов получения, свойств и реакций перекиси водорода.
Наряду с этими достоинствами необходимо также отметить оригинальные
иллюстрации, а также описание новой аппаратуры и результатов неопубли-
неопубликованных работ.
Безусловно, оправдано введение авторами единой номенклатуры перекис-
ных соединений, благодаря чему устраняется разнобой, существующий в ми-
мировой литературе. В переводе номенклатура приведена в соответствие с послед-
последними рекомендациями Комиссии по номенклатуре неорганических соединений
Международного союза чистой и прикладной химии A953 г.), что подробнее
оговорено в соответствующем примечании.
Особо следует отметить, что наряду со сравнительно объективной оценкой
приведенных работ, выполненных в последнее время советскими учеными в об-
области изучения перекиси водорода, в книге отсутствуют указания на принци-
принципиальные, основополагающие работы по химии перекисных соединений ряда
выдающихся русских ученых. Например, классифицируя перекисные соеди-
соединения в зависимости от их принадлежности к группам периодической системы,
авторы не упоминают ни о трудах Д. И. Менделеева, ни о классических рабо-
работах П. Г. Меликова и Л. В. Писаржевского (за которые эти ученые в числе дру-
других были названы Д. И. Менделеевым «укрепителями периодического закона»).
При изложении теории самоокисления и роли перекиси водорода в процессах
дыхания ничего не сказано о капитальных исследованиях А. Н. Баха и его
сотрудников. При описании термической реакции водорода с кислородом
совершенно опущены работы школы академика Н. Н. Семенова, хотя упоми-
упоминаются работы Хиншельвуда, Льюиса и фон-Эльбе. В связи с этим в соответ-
соответствующих местах текста русского перевода введены подстрочные примечания
редактора.
В редактировании перевода большая помощь оказана кандидатом химиче-
химических наук К. Е.Мироновым, сличившим текст с оригиналом и составившим
примечания.
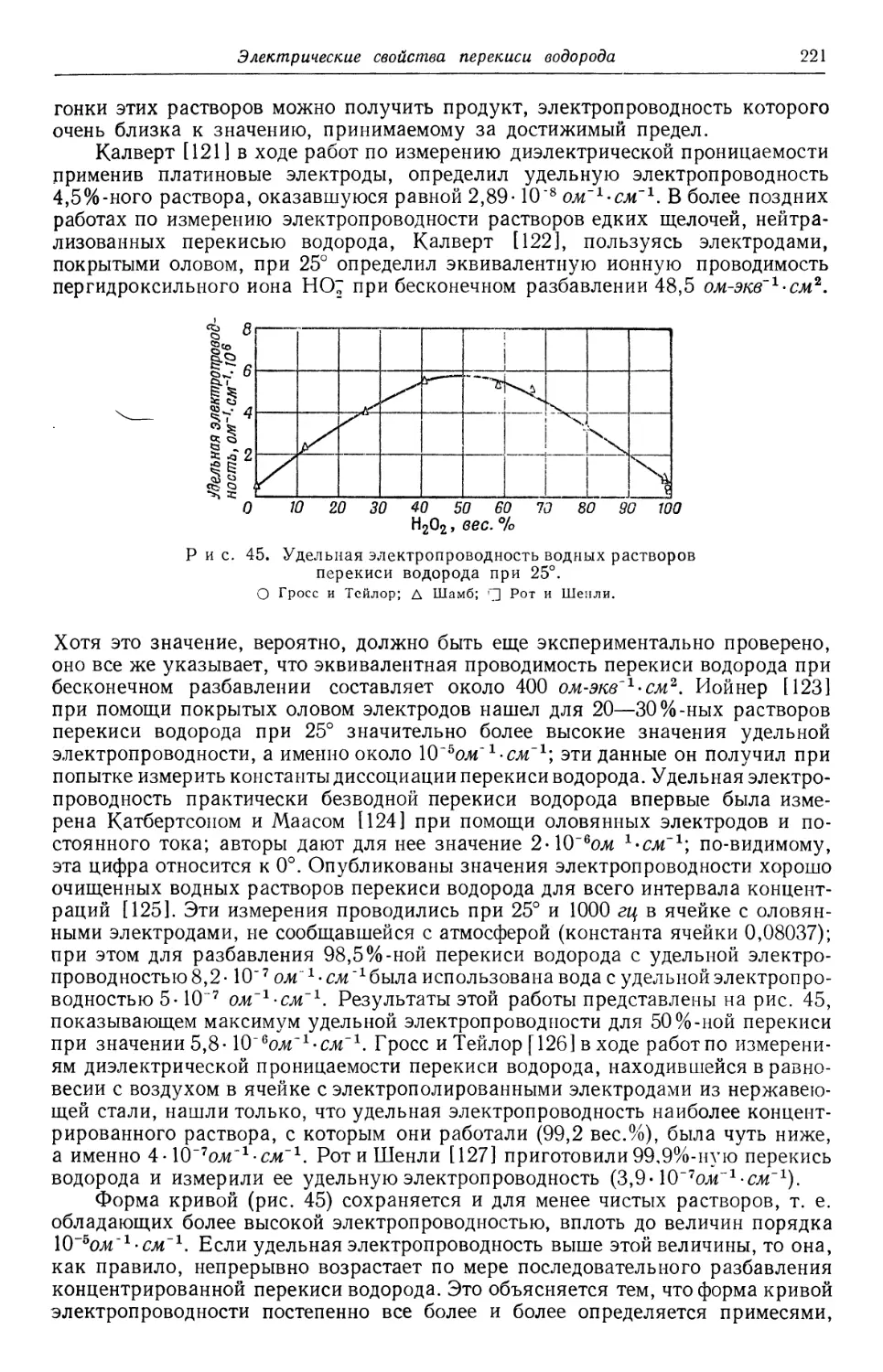
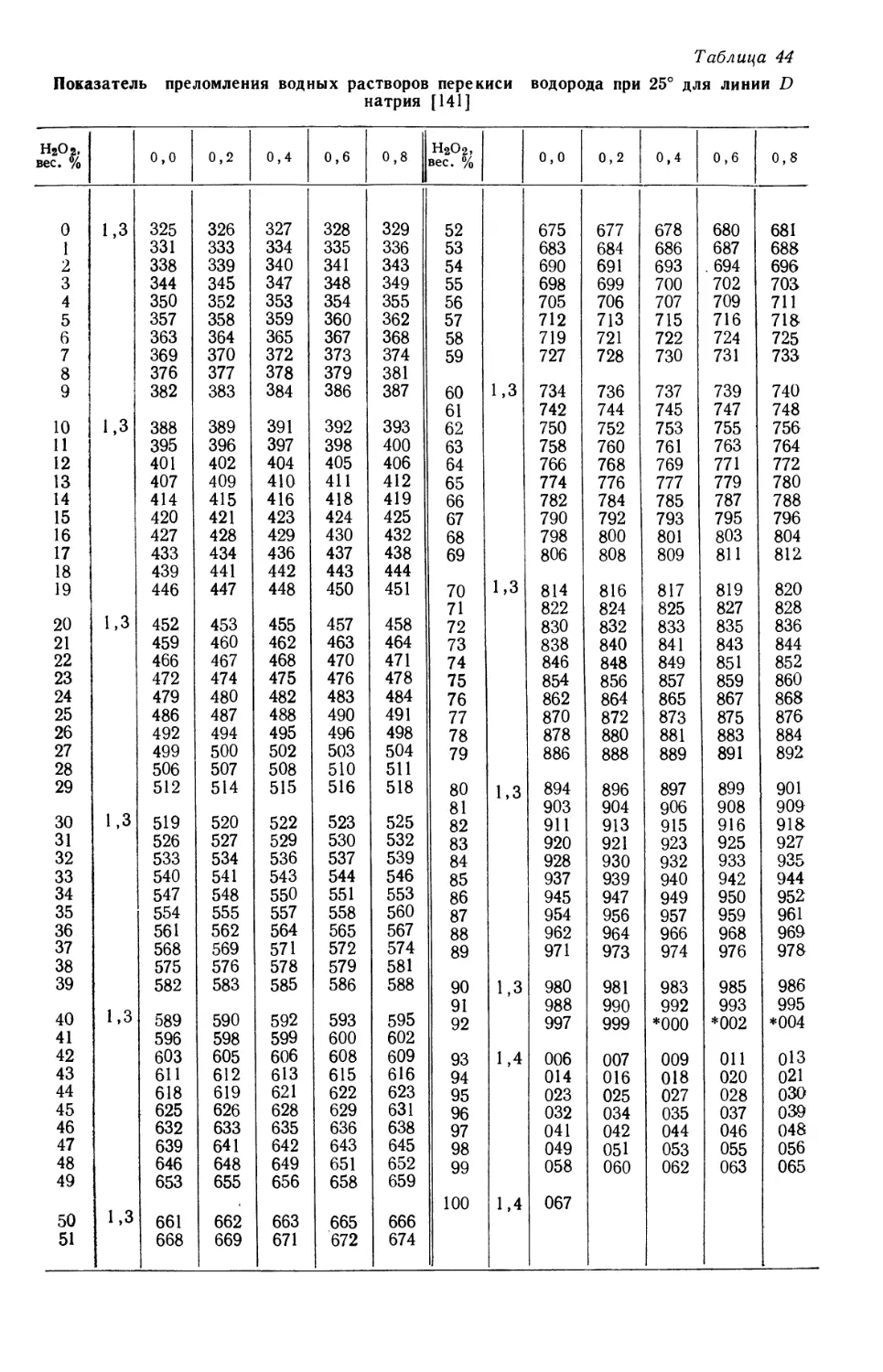
А. Горбачев.
ПРЕДИСЛОВИЕ АВТОРОВ
Начиная с 1945 г. мы занимаемся научно-исследовательской работой по
перекиси водорода в Массачусетсом технологическом институте. В течение
всего этого времени вместе с нами в работе над изучением разнообразнейших
проблем, связанных с перекисью водорода, принимает участие свыше 100 сту-
студентов и научных сотрудников. С самого начала отрицательно сказывалось
отсутствие на английском языке полного источника информации о перекиси
водорода, содержащего критический анализ имеющихся данных. Главная цель,
которая преследовалась при подготовке книги к изданию, состояла в воспол-
восполнении этого пробела с передачей в общее пользование полученных данных и при-
приобретенного опыта. Приводятся также основные сведения по перекиси водо-
водорода, чтобы шире показать ее взаимосвязь с другими веществами. Так, напри-
например, структура перекиси водорода сопоставлена со структурой других кисло-
родо-водородных соединений и проведено сравнение ее реакционной способ-
способности с реакционной способностью других перекисных соединений. При обсу-
обсуждении вопроса о применении перекиси водорода в промышленности сделана
попытка оценить перекись с точки зрения возможной конкуренции ее с дру-
другими аналогичными веществами. Описан также ход исследований с момента
открытия перекиси водорода A818 г.), который постепенно приближал ученых
к пониманию истинной природы этого вещества.
В отличие от технологии многих других неорганических веществ техноло-
технология перекиси водорода за последние годы быстро развивалась. Получавшийся
в начале XX столетия разбавленный нестабильный продукт находил лишь
ограниченное применение, главным образом в качестве дезинфицирующего сред-
средства и отбеливающего агента (для таких веществ, как волосы, перья и шерсть,
которые разрушаются при обработке их другими отбеливающими средствами).
Электрохимический процесс производства перекиси водорода, введенный
в 1908 г., дал возможность получать сравнительно чистый и стабильный про-
продукт, который постепенно находил все большее применение. Широкое исполь-
использование в Германии во время второй мировой войны концентрированной пере-
перекиси водорода в качестве компонента топлива для военных целей показало,
что перекись можно безопасно хранить и перерабатывать в больших количествах.
Это возбудило интерес многих потенциальных потребителей, ранее считавших,
что перекись водорода, даже гораздо менее концентрированная, отличается
нестабильностью и чувствительностью к внешним воздействиям. К тому же
промышленное освоение за последние годы новых производственных процес-
процессов открыло перспективы снижения цен на перекись водорода. Рост интереса
к этому продукту доказан пятикратным увеличением производства и потребле-
потребления его в США в последнее десятилетие и быстро возрастающим числом публи-
публикуемых сообщений по перекиси водорода. И то, и другое показывает, насколько
необходима критическая сводка всех имеющихся данных по перекиси водорода.
Помимо промышленного значения, перекись водорода уже давно интере-
интересует химиков вследствие необычных свойств перекисной связи. Образование
перекиси водорода в самых различных реакциях и роль, которую она играет
8 Предисловие авторов
в обмене живых клеток, объясняется специфическим положением ее в качестве
промежуточного продукта окисления—восстановления между кислородом
и водой. Интерес, проявляемый к перекиси водорода научными работниками
и технологами, работающими в самых различных областях, обусловлен ее
специфическими свойствами и широкой распространенностью. Вероятно, не
все знают, что по разнообразию областей применения перекись водорода не
уступает большинству других широко распространенных химических веществ.
Литература по перекиси водорода настолько обширна и разбросанна, что
практически нет никакой возможности охватить ее полностью и процитировать
все сообщения, в которых упоминается это вещество. Ссылки даны только
на те работы, которые привлекли наше внимание до середины 1954 г. и кото-
которые, по-видимому, представляют определенный интерес и в настоящее время.
Более поздние работы, за отдельными исключениями, привести не представля-
представлялось возможным. В некоторых случаях мы ограничивались ссылками на обзор-
обзорные статьи, не приводя ссылок на упоминаемую в них литературу. Патенты
охвачены таким же образом, как и журнальная литература, а поэтому исчер-
исчерпывающего перечня их тоже не приводится. В отдельных сообщениях прави-
правительственных учреждений США и Великобритании о результатах германской
практики содержатся более полные данные, чем в других источниках, а поэто-
поэтому в некоторых случаях (особенно в главах, посвященных производству)
приведены ссылки именно на эти сообщения. Так называемые доклады
(Р. В. Reports), упоминаемые в литературных источниках, можно приобрести
в Информационном отделе Министерства торговли США. Аналогичные отчеты
опубликованы Объединенной подкомиссией технической разведки США (C.I .O.S.)
и Британской подкомиссией технической разведки (B.I.O.S.). Многие такие
отчеты опубликованы и в виде докладов (Р. В. Reports). Хотя возможно, что
некоторая часть важной литературы в книгу и не попала, мы все же считаем,
что практически все данные и соображения, имеющие определенное значение
в настоящее время, доведены до сведения читателя. К оценке имеющегося ма-
материала мы старались подходить критически, насколько это было возможно,
принимая во внимание доступность оригинальной литературы и уровень наших
знаний.
Вследствие ограниченного объема книги ряда специальных тем пришлось
коснуться только вкратце. В самом начале было решено не касаться (кроме
отдельных случаев) важной родственной области органических перекисей
и ограничиться лишь введением в органическую и биологическую химию пере-
перекиси водорода. В данной книге нет возможности уделить внимание и чисто прак-
практическим вопросам, например связанным с упаковкой и перевозкой перекиси
водорода.
Основными источниками литературных ссылок по перекиси водорода были
реферативные журналы «Chemical Abstracts» и «Chemisches Zentralblatt», две
ранее опубликованные монографии по перекиси водорода (Маху и Кауша),
указатели старых журналов, картотеки ряда библиотек и бюллетени по пере-
перекиси водорода, выпускаемые различными производственными фирмами. Зна-
Значительная часть данных взята из неопубликованных сообщений и диссерта-
диссертаций по перекиси водорода, написанных в Массачусетском технологическом
институте. При ссылке на многие, сравнительно мало распространенные источ-
источники в скобках приведена также ссылка на реферат в «Chemical Abstracts».
Аналогично описан и ряд других рефератов, заметок, переводов или параллель-
параллельных патентов.
Мы приветствовали бы перепечатку любых старых и новых сообщений,
касающихся перекиси водорода.
Уолтер К. Шамб,
Массачусетса технологический Чарльз Н. Сеттерфилд,
институт, апрель 1955 г. Ральф Л. Вентворс*
Глава 1
ВВЕДЕНИЕ
Перекись водорода получила признание как индивидуальное химическое
соединение с 1818 г. В июле этого года Луи-Жак Тенар сообщил Парижской ака-
академии наук о методе получения веществ, которые он сначала считал «окислен-
«окисленными кислотами». Но еще до конца года Тенар сделал ряд новых сообщений,
подводящих итоги изучению этих «кислот», и пришел к заключению, что в дей-
действительности он открыл совершенно новое соединение—окисленную воду.
Тенар тщательно изучил технику получения и химические свойства нового со-
соединения и установил, что оно является довольно неустойчивой новой формой
химического соединения и может подвергаться (это, возможно, самое интерес-
интересное) интенсивному разложению под действием некоторых веществ, которые
при этом сами заметно не изменяются.
Эти три исключительно важных наблюдения обусловили постоянный инте-
интерес к перекиси водорода со времени ее открытия и послужили основой для
дальнейших исследований этого соединения. С годами, по мере накопления
знаний о свойствах перекиси водорода, стало очевидно, что она не является
веществом, неустойчивость которого заложена в самой его природе, хотя оно
и обладает высокой реакционной способностью. Вследствие характерного типа
химической связи перекись водорода занимает особое положение в химической
науке и привлекает к себе усиленное внимание при исследованиях катализа.
Кроме того, необходимо отметить, что наряду с чисто научным интересом
к перекиси водорода развивались и представления о возможности ее техниче-
технического использования. Параллельно с усовершенствованием методов и техники
производства постоянно расширялись области применения перекиси водорода
и увеличивалось ее производство.
Для понимания современного состояния научных и технических знаний,
касающихся этого вещества, а также для оценки старой литературы по этому
вопросу читателю будет полезен краткий исторический обзор, в котором пере-
перечисляются основные этапы исследования и применения перекиси водорода.
ОТКРЫТИЕ ПЕРЕКИСИ ВОДОРОДА
Честь открытия перекиси водорода, несомненно, принадлежит Тенару.
Хотя Дэви [1] и Гей-Люссаки Тенар [2], безусловно, получали перекись водо-
водорода в ходе исследований щелочных металлов, совершенно очевидно, что Тенар
первым обнаружил ее образование. Быстрота, с которой он сообщил о широ-
широких исследованиях в области техники получения и свойств перекиси водорода,
не оставляет никаких сомнений в том, что именно он выяснил природу этого
нового соединения. Правда, Тенар чисто случайно поставил те опыты, которые
привлекли его внимание к перекиси водорода, однако вполне точно можно
проследить ход событий, которые привели Тенара к этому открытию [3].
В 1806 г. Гемфри Дэви на заседании английского Королевского общества
прочел доклад «О некоторых химических воздействиях электричества» и полу-
получил за него премию 3000 франков, учрежденную Наполеоном за крупные иссле-
10
Глава 1
дования в области электричества. Премия, назначенная французским комите-
комитетом, была выдана Дэви, хотя Англия и Франция находились тогда в состоя-
состоянии войны и Наполеон был раздражен тем, что ни один французский ученый
не мог быть удостоен этой премии. Когда Наполеону доложили, что во Фран-
Франции нет вольтова столба, необходимого для постановки таких опытов, которые
были осуществлены Дэви, он приказал построить два таких столба и подарил
их Политехнической школе, причем работа с вольтовым столбом была пору-
поручена Гей-Люссаку и Тенару. Наи-
Наиболее интересным из открытий, сде-
сделанных Дэви при помощи вольтова
столба, было получение металли-
металлических натрия и калия. Пока Гей-
Люссак и Тенар ожидали оконча-
окончания изготовления больших фран-
французских батарей, они обратили свое
внимание на получение щелочных
металлов химическими способами
и добились особенных успехов в
методе восстановления расплавлен-
расплавленных щелочей раскаленным докрас-
докрасна железом. Этот процесс оказал-
оказался лучше электрического метода в
отношении легкости получения и
производства в крупном масштабе.
Предоставленные для их работы
вольтовы столбы, как оказалось,
имели плохую конструкцию, но
работа, начатая в результате заде-
задетого самолюбия Наполеона, приве-
привела Гей-Люссака и Тенара к поста-
постановке широких исследований, имев-
Рис. 1. Луи-Жак Тенар [4] A/77—1857 гг.). ших большое значение для разви-
развития химии [2].
Интересно, что в работу исследователей входило получение окислов ще-
щелочных и щелочноземельных металлов разными способами. В результате Тенар
получил эти вещества и хорошо ознакомился с ними. Именно тогда было сде-
сделано открытие, что многие металлы способны образовать больше одного соеди-
соединения с кислородом и что эти различные соединения отличаются по соотноше-
соотношению количеств металла и кислорода. В согласии с тогдашними представлени-
представлениями, металл, который соединялся более чем с «соответствующим» количеством
кислорода, обычно отдавал этот избыток при обработке кислотой.
Рассказывая об этом утверждении своим студентам и заметив, что описан-
описанное явление экспериментально не доказано, Тенар решил поставить опыт с пе-
перекисью бария, считавшейся в соответствии с господствовавшей в то время
гипотезой соединением с «избытком кислорода». Он растворил перекись бария
в чашке с азотной кислотой, находившейся в ледяной бане, и, к своему удивле-
удивлению, не обнаружил выделения газа. Тенар оставил смесь на ночь и утром
по возвращении в лабораторию обнаружил, что из жидкости выделяются пу-
пузырьки газа. Тенар налил небольшое количество жидкости в пробирку, нагрел
ее и простым погружением тлеющей лучинки в пробирку установил, что выде-
выделяющийся газ представляет собой кислород.
Первым выводом Тенара было предположение, что он окислил кислоту.
Поскольку кислород был тогда в центре интересов химии, желание лучше
изучить его реакции и отношения, в которых он вступает в соединения с дру-
другими веществами, побудило Тенара к постановке обширных исследований новой
Размах работы Тенара 11
«окисленной кислоты». После небольшого периода экспериментирования Тенар
обнаружил, что из активного раствора можно удалить всю кислоту и бариевые
соли, причем остается жидкость, которая при выпаривании не дает твердого
остатка. Отсюда он сделал вывод, что в действительности он получил окислен-
окисленную воду, представляющую собой соединение определенного состава.
РАЗМАХ РАБОТЫ ТЕНАРА
Если проанализировать сообщения Тенара, то можно составить очень точ-
точное представление о характере перекиси водорода, о проблемах, связанных
с ее получением из перекиси бария, и о ее химической природе.
Интересно дать краткий обзор работы Тенара, так как она представляет
собой наиболее полное и квалифицированное исследование перекиси водорода,
проведенное в XIX столетии, и так как существенные части этой работы, по-ви-
по-видимому, не были в достаточной мере учтены в течение нескольких десятилетий
после смерти этого автора. В год, последовавший за открытием, Тенар опуб-
опубликовал 9 сообщений [5] и выводы из своей работы [6]. Полный отчет о работе
был включен в третье и последующие издания «Traite de Chimie» [7]. В даль-
дальнейшем были опубликованы два дополнительных сообщения, посвященных
способу получения [8] и каталитического разложения [9] перекиси водорода
(соответственно в 1832 и 1856 гг.). За этими работами с интересом следили ино-
иностранные ученые, и вскоре появился ряд комментариев и кратких переводов
ПО].
Значительная часть работы Тенара посвящена усовершенствованию прак-
практических способов получения устойчивой концентрированной перекиси водо-
водорода. В отдельных литературных источниках [3] высказываются различные
мнения относительно кислоты, которой Тенар пользовался для первого полу-
получения перекиси водорода, однако, как видно из его первого сообщения [5],
он брал для этой цели азотную кислоту. В дальнейшем, доказав, что многие
другие кислоты также способны реагировать с перекисью бария с образованием
раствора, богатого кислородом, Тенар остановился на соляной кислоте, как
наиболее эффективной. Между этой кислотой и перекисью' бария происхо-
происходила быстрая реакция, течению которой не мешало образование нераствори-
нерастворимых бариевых солей вокруг реагирующих частиц; вместе с тем на следующей
стадии можно было удалить избыток кислоты и образовавшегося хлористого
бария химическими методами. Тенар выяснил также возможность получения
перекиси водорода действием кислоты на перекиси калия, натрия, стронция
и кальция, однако ни одну из них он не использовал на практике для полу-
получения перекиси водорода, поскольку осаждением наиболее удобно отделять
соединения бария.
Тенар рекомендовал получать перекись бария из азотнокислого бария,
предварительно перекристаллизованного для возможно более полной очистки
^го от железа и марганца. Нитрат разлагали нагреванием на воздухе в фар-
фарфоровой реторте и образующуюся окись бария затем переокисляли нагреванием
докрасна в трубке, через которую пропускали быстрый ток кислорода. Затем
перекись бария увлажняли, измельчали и растворяли в соляной кислоте.
К смеси добавляли небольшой избыток серной кислоты для осаждения серно-
сернокислого бария и регенерации соляной кислоты. Тенар доказал, что образова-
образование хорошо фильтрующегося осадка зависит от относительного количества
добавленной серной кислоты; при слишком большом или слишком малом коли-
количестве кислоты отделение сернокислого бария ухудшается. Операцию раство-
растворения перекиси бария и осаждения серной кислотой повторяли несколько раз,
что давало возможность обогатить раствор до накопления в нем способного
к выделению кислорода в количестве 125 объемов на каждый объем раствора,
или 33 вес. % Н2О2. Полученный таким образом концентрированный раствор
\2 Глава 1
все еще содержал соляную кислоту и растворенные примеси от перекиси бария
и кислот. Для устранения этих примесей жидкость сначала охлаждали во
льду и пересыщали ее окисью бария, которая осаждала железо, марганец, крем-
кремнезем, глинозем. Впоследствии Тенар предложил [8] на этой стадии добавлять
фосфорную кислоту для осаждения железа и марганца в виде фосфатов. Фильт-
Фильтрование этого раствора производили возможно быстрее для предотвращения раз-
разложения перекиси водорода на собранном осадке. К фильтрату после этой опе-
операции добавляли сернокислое серебро, лишенное свободной окиси серебра.
Выпадение хлористого серебра служило также для осветления раствора, кото-
который до этого оставался мутным. Далее необходимо было удалить из раствора
серную кислоту осаждением ее в виде сернокислого бария. Эту стадию осу-
осуществляли добавлением окиси бария к холодному раствору.
Для лучшего сохранения раствор перекиси водорода, свободный от серно-
сернокислого бария, оставляли слабо подкисленным. Дальнейшее концентрирова-
концентрирование раствора можно было производить выпариванием воды в вакууме. Этим
путем Тенару удалось получить практически безводную перекись водорода.
Газометрический анализ растворов перекиси водорода на содержание кис-
кислорода проводили без труда, причем кислород выделяли из раствора или путем
нагревания, или введением катализатора, например окиси серебра или дву-
двуокиси марганца. В «Traite de Chimie» Тенар сообщил подробности анализа
пробы плотностью 1,452 г/см3, из которой было выделено 476 объемов кисло-
кислорода на объем жидкости. Температура, при которой была определена плот-
плотность, не указывается, но если принять, что она была близка к 15°, то получен-
полученные значения очень близки к тем, которые принимаются в настоящее время
для чистой безводной перекиси водорода. Кроме того, вес собранного в этом
опыте кислорода всего в пределах 1 % отличался от веса кислорода, вычислен-
вычисленного для навески безводной перекиси водорода, соответствующей весу образца.
Содержание кислорода в перекиси водорода плотностью 1,452 г/см3 нельзя
было увеличить путем длительного выдерживания в вакууме. Это привело
Тенара к заключению, что «наиболее сильно окисленная вода представляет
перекись водорода, которая в сущности содержит вдвое больше кислорода, чем
чистая вода, и что в тех случаях, когда перекись содержит меньше этого коли-
количества кислорода, ее можно рассматривать как смесь чистой воды и перекиси
водорода».
Имея в виду принятое позже утверждение, что перекись водорода является
неустойчивым веществом, нужно считать весьма замечательным факт получения
Тенаром практически безводной чистой перекиси водорода. По-видимому, ни
один химик в течение долгих лет после этого не брал на себя труд получить
перекись водорода, которая по чистоте и устойчивости была бы близка к полу-
полученной Тенаром. Стабильность перекиси Тенара при хранении, вероятно,
нельзя было сравнить с современными стандартами (сам Тенар предлагал хра-
хранить перекись в стеклянном сосуде, помещенном в лед в холодном подвале), но
признание Тенаром необходимости исключения примесей тяжелых металлов
и стабилизирующего действия кислоты, безусловно, явилось самой существен-
существенной подготовкой для изучения природы перекиси водорода.
Для изучения свойств перекиси водорода Тенар использовал как концент-
концентрированные, так и разбавленные растворы. Он обнаружил, что заморозить
чистую перекись невозможно (в настоящее время известно, что концентриро-
концентрированные растворы перекиси водорода очень склонны к переохлаждению),
а непропорциональное распределение ее между твердой и жидкой фазами,
при частичном замораживании 10%-ного (по весу) раствора, было недостаточ-
недостаточно полным, чтобы практически концентрировать перекись водорода кристал-
кристаллизацией. В свете более поздних сообщений, в которых утверждалась противо-
противоположная точка зрения, интересно отметить, что Тенар обнаружил возможность
выпарить перекись водорода при уменьшенном давлении досуха, причем замет-
Размах работы Тенара 13
ного разложения ее не наблюдалось. По-видимому, Тенару не удалось опреде-
определить температуру кипения ни одного из своих растворов; разложение всегда
усиливалось при нагревании до температуры значительно выше комнатной.
Тенар обратил внимание на то, что при выливании перекиси водорода на
раскаленную докрасна металлическую пластинку детонации не происходит.
Тенар испытывал перекись водорода и на вкус; она напоминала ему по
вкусу металлический раствор и была несколько тошнотворной. Он отметил
также повеление кожи при соприкосновении с перекисью водорода. По Тенару,
перекись водорода практически не имеет запаха.
Наиболее интересны обширные исследования Тенара по реакционной спо-
способности перекиси водорода. В общем он приводит данные по реакциям для
130 с лишком веществ, куда входят металлы, окислы, соли, кислоты и основа-
основания, включающие соединения 40 элементов и различные органические веще-
вещества. Самая первая проба, показавшая, что кислая перекись водорода не дей-
действует на золото, еще немного напоминает старую алхимию. Большая часть
веществ, которые Тенар испытывал по их действию на перекись водорода,
вызывала ее разложение. При этом разложении некоторые вещества химически
изменялись; так, мышьяк, молибден, вольфрам и хром окислялись, соединяясь
с частью кислорода перекиси. Установлено, что некоторые металлы, такие,
как олово, сурьма и теллур, не оказывают никакого действия даже на концент-
концентрированную перекись водорода. Разложение перекиси водорода всегда сопро-
сопровождалось выделением значительного количества тепла. Тенар затруднялся
объяснить это явление для реакции, происходящей с выделением кислорода,
в свете существовавшей тогда теории.
Еще в тот период, когда перекись рассматривали как окисленную кислоту,
были сделаны наблюдения по эффективному разложению ее такими вещества-
веществами, как окись серебра, свинец, двуокись марганца и платина. Для некоторых
из этих веществ Тенар дает полное описание химического действия, сопрово-
сопровождающего каталитическое разложение перекиси водорода. Так, например, он
описывает восстановление окиси серебра до металлического серебра при вве-
введений ее в перекись водорода, полное растворение окиси серебра, наблюдаемое
при приливании кислоты, и выпадение металлического серебра и продолжение
разложения перекиси водорода, вызываемое последующей нейтрализацией.
После некоторых таких наблюдений реакций разложения Тенар сделал вывод,
что «в этих разложениях, по-видимому, отсутствует химическое воздействие;
поэтому необходимо приписать эти воздействия физической причине; однако
они не связаны ни с нагреванием, ни со светом, а поэтому они, вероятно, элект-
электрического происхождения». Были исследованы и другие каталитические реак-
реакции, и при суммировании реакций перекиси водорода Тенар точно отметил
различие между явлениями разложения, протекающими с химическим изме-
изменением агента разложения и без его изменения.
Эта работа представляет собой одно из самых первых исследований по ка-
катализу. Механизм этого разложения оставался неясным для Тенара. На осно-
основании наблюдения, что животные продукты, как например фибрин крови
и ткани органов, могут эффективно разлагать ряд порций перекиси водорода
одну за другой, он высказал предположение, что это действие обусловлено той
же силой, которая проявляется металлическими или неорганическими катали-
катализаторами. Это замечание и в дальнейшем нашло отражение в литературе при
классификации таких катализаторов, как платина, под названием «неорганиче-
«неорганических ферментов». Однако Тенар не мог «понять, каким образом вещество, не
подвергающееся абсорбции или изменению, может непрерывно действовать на
жидкость, превращая ее в новые продукты». Только Берцелиусу [11], который
собрал разрозненные наблюдения каталитических эффектов, причем из них
значительная часть была описана Тенаром, принадлежит честь признания этой
важной области химии; им же было предложено и ее название.
14 Глава 1
Хотя Тенар и сделал несколько ценных открытий в области прикладной
химии, он нашел только две области применения для перекиси водорода. Он
предложил использовать ее в медицине в качестве вещества для наружного
раздражения и в искусстве при реставрации старых картин. Безобразные
черные пятна, возникавшие под действием сероводорода из воздуха на свинцо-
свинцовые белила картин, быстро снимались при нанесении кистью разбавленной
перекиси водорода. Сам Тенар успешно использовал этот метод для реставра-
реставрации одной из картин Рафаэля.
Работы Тенара способствовали тому, что перекись водорода заняла вполне
определенное положение в схеме химии. Безусловно, современная химия позво-
позволяет более последовательно интерпретировать работы Тенара, однако исследо-
исследование в целом представляет собой превосходную работу. Благодаря открытиям,
сделанным его другом Гей-Люссаком в более фундаментальных областях, ра-
работы Тенара остаются несколько в тени, хотя в действительности они заслу-
заслуживают значительно большего признания.
НАЧАЛЬНЫЕ РАБОТЫ ПО ПЕРЕКИСИ ВОДОРОДА
Химики, познакомившиеся после Тенара с перекисью водорода, старались
объяснить ее строение и найти ей место в рамках химического сродства. Это был
период становления химии как науки, и проблема выяснения природы пере-
перекиси водорода привлекала к себе значительное внимание. Еще при жизни
Тенара (до 1857 г.) появилось около тридцати-сорока сообщений по перекиси
водорода, ее реакциям, получению и строению, и упоминание о перекиси водо-
водорода нашло отражение в тогдашних курсах химии. В то время возникла анто-
зоновая теория Шенбейна. Это было самое энергичное (хотя и не полезное)-
обсуждение вопросов, связанных с перекисью водорода, со времени начальных
работ Тенара, и исследования Шенбейна представляют основное ядро литера-
литературы по перекиси водорода вплоть до 1870—1880 гг. В эти годы теория
Шенбейна была опровергнута, и материалы, касающиеся природы перекиси
водорода, вошли в химию, содержавшую основы современных воззрений. Семи-
Семидесятые годы, таким образом, являются промежуточным периодом между лите-
литературой, имеющей только исторический интерес, и началом накопления знаний
по перекиси водорода, представляющих значительный шаг вперед по сравнению
с фактами, известными Тенару.
В течение десятилетия после открытия перекиси водорода не появилось
никаких новых исследований; учебники, например Силлимана, Тернера и Гме-
лина [12], содержат только точное изложение работ Тенара. Первые новые
материалы по перекиси водорода, появившиеся в литературе, представляли
собой попытки объяснить механизм ее разложения, и они характеризуются
отсутствием экспериментального подтверждения. Так, по теории, выдвинутой
Фаустом 113] в 1830 г., считается, что разложение перекиси водорода пред-
представляет своего рода электролиз, причем агенты разложения выполняют функ-
функции электродов в электролизе воды. Либих [14], исходя из мнения Тенара [15],
что сероводород аналогичен по характеру перекиси водорода, принимал,
что сродство кислорода к другому веществу в окиси должно быть больше, чем
сила, необходимая для сжатия кислорода от его нормального состояния в атмо-
атмосфере до того небольшого объема, который он занимает в окиси. Поэтому любое
вмешательство могло способствовать стремлению кислорода к обратному пере-
переходу в газообразное состояние, и для разложения окисла достаточно было
превысить сродство. Митчерлих [16] указывает, что скорость разложения пере-
перекиси водорода на твердых телах пропорциональна величине поверхности этих
тел, и отсюда делает вывод, что именно притяжение со стороны атомов твер-
твердых тел может вызвать реакцию и разложение. В 1848 г. Плейфер [17] дал
обзор теории каталитического разложения и показал, что теории Либиха и Мит-
Начальные работы по перекиси водорода 15
черлиха не в состоянии объяснить все примеры наблюдаемых каталитических
разложений. По Плейферу, силы притяжения воды и какого-либо другого
вещества к кислороду могут быть уравновешены таким образом, что из пере-
перекиси начнет выделяться кислород. Однако сам Плейфер указывает, что для
полного понимания этих явлений необходимо более тщательное изучение про-
проблем катализа.
Вышеуказанные обобщения мало способствовали пониманию истинной
природы перекиси водорода. Однако вскоре появилось более подробное объяс-
объяснение, которое заслужило широкое признание и в то же время вызвало ожив-
оживленную критику. Это была озоно-антозоновая теория Шенбейна. Шенбейн
открыл озон в 1840 г., и после этого вся его жизнь [18] была почти полностью
посвящена изучению кислородных соединений.
Развитие озоно-антозоновой теории Шенбейна можно проследить со всеми
подробностями в очень небольшом числе сообщений [19], главным образом
в письмах к Фарадею, хотя Шенбейн опубликовал очень много работ. Действи-
Действительно, в каталоге Королевского общества перечислено примерно 364 сообще-
сообщения Шенбейна, из которых последнее касается вопроса о нахождении пере-
перекиси водорода в атмосфере. Исходной точкой теории Шенбейна было его откры-
открытие, что некоторые вещества, например эфир, скипидар и фосфор, медленно окис-
окисляются на воздухе и при этом делаются эффективными в качестве отбеливаю-
отбеливающих веществ. Помня об аналогичном отбеливающем действии озона, Шенбейн
сделал вывод, что это действие обусловлено образованием озона или, как он
позже выразился, превращением в озон обычного кислорода, который в форме
озона соединяется с веществом, подвергающимся окислению. Теория эта сразу
объясняла, каким образом отбелка, требующая определенного энергетического
эффекта, может осуществляться посредством кислорода, и в то же время
находилась в полном согласии с концепцией Шенбейна о природе озона. Сна-
Сначала он считал, что озон представляет соединение кислорода и водорода, но
впоследствии выяснил, что озон является аллотропной активной формой кисло-
кислорода. По-видимому, он не считал, что кислород и озон различаются по струк-
структуре, а признавал только их неодинаковую активность или полярность.
Сначала Шенбейн сделал вывод, что активный кислород в перекиси водо-
водорода присутствует в виде озонированного кислорода. Зная, что озон превра-
превращается в обычный кислород при нагревании или в контакте с некоторыми веще-
веществами, он объяснял аналогичное поведение перекиси водорода как обусловлен-
обусловленное озонированным кислородом, который в соединении с водой сохраняет все
свойства, присущие ему в свободном виде. Поскольку было известно, что неко-
некоторые окислы металлов восстанавливаются в процессе разложения перекиси
водорода, Шенбейн считал, что кислород в этих окислах также должен нахо-
находиться в озонированной форме и что озонированный кислород должен обладать
способностью связываться сам с собой с образованием обыкновенного кисло-
кислорода.
Вскоре после этого Шенбейн несколько изменил начальную точку зрения
и облек ее в форму, которая, как ему казалось, лучше всего объясняла реак-
реакции кислорода. Он постулировал наличие трех модификаций кислорода: двух
активных с противоположной полярностью и обыкновенного кислорода. Эти
активные модификации относятся друг к другу как плюс и минус, причем
отрицательная представляет собой озон, а положительная—форму кислорода,
которой Шенбейн присвоил название антозон. Знаки были установлены на
основании «электродвижущих свойств».
Эта мысль дала возможность следующим образом систематизировать реак-
реакции кислорода. Обыкновенный кислород химически не активен и образуется
путем взаимной нейтрализации озона и антозона. Озонированный кислород
возникает под действием электрической искры или медленного окисления
фосфора, являясь идентичным с частью кислорода, встречающегося в груп-
16 Глава 1
пе окислов, которым должно быть присвоено название озонидов, напри-
например МпО2, РЬО2.
Другая группа веществ называется антозонидами; в нее входят перекись
водорода, перекиси щелочных металлов и некоторых органических соединений,
например эфира, скипидара и эфирных масел. Соприкасаясь друг с другом,
озонид и антозонид дают реакцию взаимного катализа с выделением обык-
обыкновенного кислорода, причем исходные озонидное и антозонидное соединения
сохраняют только ту часть кислорода, которая ранее не была поляризована.
Примером этого действия является каталитическое разложение перекиси водо-
водорода двуокисью свинца, причем получаются вода, кислород и окись свинца.
Шенбейн обозначает этот процесс следующим образом:
(НО + О) + (РЬО + 6) -> НО + РЬО + 20.
Это был основной тезис Шенбейна, который был высказан им почти полностью
еще в 1859 г., но до самой смерти, последовавшей в 1869 г., Шенбейн неутомимо
ставил новые опыты для получения и обнаружения присутствия озона и анто-
зонов. Он нашел, что амальгамы некоторых металлов образуют перекись водо-
водорода (а следовательно, антозон) при встряхивании с водой в присутствии воз-
воздуха. Разновидность плавикового шпата, обладающего неприятным галогено-
подобным запахом, как обнаружил Шенбейн, дает перекись водорода при расти-
растирании в порошок с водой. Поэтому Шенбейн пришел к выводу, что запах пла-
плавикового шпата обусловлен антозоном. При дальнейшей разработке своей тео-
теории Шенбейну пришлось внести некоторые коррективы в общепринятую хи-
химию. Поскольку озониды, например двуокись марганца, выделяют хлор при
реакции с соляной кислотой, а антозониды, например перекись бария, обра-
образуют перекись водорода, то эти наблюдения привели Шенбейна к выводу, что
по аналогии с водой, которая может химически связываться лишь с антозоном,
соляная кислота может соединяться лишь с озоном, выделяя хлор. В сущно-
сущности, это был возврат к старой кислородной теории кислот, опровергнутой
уже Дэви.
Эти исследования и теории Шенбейна часто оказываются взаимно противо-
противоречивыми. Так, однажды он сообщил, что при обработке перекиси бария сер-
серной кислотой ощущается характерный запах антозона. Впоследствии при до-
добавлении перекиси бария к серной кислоте, содержащей озонид—марганцево-
кислый калий,—он обнаружил запах озона. Несмотря на неувязку в его тео-
теории, у Шенбейна был ряд последователей. В 1869 г. Шер [20] опубликовал под-
подробное изложение теории Шенбейна. Одним из наиболее рьяных последователей
Шенбейна был Мейснер [21]. Он указал на новое явление, связанное с мнимым
антозоном; впоследствии этот опыт получил совершенно другое толкование
и привел к отказу от озоно-антозоновой теории.
При пропускании через воду или во влажный воздух кислорода, подверг-
подвергнутого действию электрического разряда, Мейснер обнаружил образование лег-
легкого тумана. Отсюда он сделал вывод, что электризованный воздух содержит
озон и еще какое-то вещество, которое он назвал атмизоном (от греческого слова,
означающего «дымить»). Туман был приписан присутствию атмизона. Позже
Мейснер сделал заключение, что атмизон идентичен с антозоном Шенбейна,
и затратил много усилий, чтобы отделить это вещество от озона и изучить его
свойства. Мейснеру казалось, что это образование тумана может объяснить
возникновение дыма при курении табака и сжигании топлива или пороха
и что «обильные дожди, которые возникают после больших сражений», могут
быть вызваны действием антозона на атмосферу.
Другие исследователи еще больше приукрашали антозоновую теорию.
Ленсен [22] высказал мнение, что кислород положительно поляризован в под-
подкисленном растворе перекиси водорода и отрицательно поляризован в щелоч-
Начальные работы по перекиси водорода 17
ном растворе. Клаузиус [23] выдвинул теорию, что озон и антозон сохраняют
свои противоположные заряды при образовании кислорода и эти заряды свя-
связывают их вместе в двухатомную молекулу; Озанн [24] предположил, что туман*
вызывается образованием азотнокислого аммония; Фипсон [25] считал, что
перекись водорода является одним из наиболее электроотрицательных веществ.
Эти и другие подробности озоно-антозоновой теории проанализированы Фок-
Фоксом [26] и Биркенбахом [27].
В течение длительного времени возражениям против озоно-антозоновой
теории уделяли мало внимания. В 1854 г. Броди [28] опубликовал результаты
опытов по каталитическому разложению перекиси водорода и тщательно их
проанализировал. Он доказал, что существует два различных типа реакций
разложения перекиси: 1) реакция с восстановлением агента разложения и
2) реакция с дальнейшим каталитическим действием восстановленной формы.
Впоследствии Броди [29] опровергал воззрения Шенбейна, указывая, что нет
никакой необходимости отступать от общепринятой химии и постулировать
наличие специальных модификаций кислорода. Броди допускал, что кислород
может существовать в соединениях в различных полярных состояниях, но
принимал, что способность элементов к образованию соединений зависит от
физических условий и от тех химических веществ, с которыми они реагируют.
Что касается перекиси водорода, то Броди продолжал защищать свою точку
зрения относительно разделения ее реакций на каталитические и стехиометри-
ческие, указывая, что кажущуюся общую реакцию можно хорошо представить
в виде суммы отдельных реакций.
Вельтцин [30] соглашался с Броди в том отношении, что для объяснения
озона и антозона достаточно данных общепринятой химии. Он особенно под-
поддержал мнение Броди о том, что до сих пор при изучении кислородных со-
соединений слишком большое внимание уделялось роли кислорода и совершенно
упускалось значение'природы других элементов, что обнаруживается, напри-
например, в различии между такими окислами, как РЬО2 и ВаО2. Самым существен-
существенным было высказывание Вельтцина о том, что нет никаких причин, чтобы пере-
перекись водорода сама не испарялась в известной мере, например в процессе
образования ее из перекиси бария (т. е. таким образом она в состоянии давать
реакции антозона).
Пожалуй, одни работы Броди и Вельтцина достаточно поколебали теорию
Шенбейна, но все же требовались дополнительные доказательства. Вскоре рабо-
работами Эндрюса и Тета, Соре и других, занимавшихся изучением озона, было
показано, что плотность паров озона в 1,5 раза больше, чем плотность паров
кислорода, что потребовало существенного изменения представления о струк-
структуре озона. Окончательное поражение антозону нанесли работы фон-Бабо
[31] и Энглера и Нассе [32]. В хорошо обоснованном сообщении, подкреплен-
подкрепленном точными опытами, фон-Бабо показал, что ему ни разу не удалось получить
туман, о котором говорил Мейснер, если только отсутствовал азот или другое
вещество, способное к окислению. При испытании методов, будто бы позволяю-
позволяющих отличить озон от антозона, фон-Бабо показал, что проба на антозон может
оказаться положительной в присутствии озона, если только имеются легко
окисляемые вещества, способные образовать перекись водорода.
Энглер и Нассе в 1870 г. доказали возможность образования перекиси водо-
водорода при взаимодействии озона с водой. Эти авторы пропускали кислород через
трубку с электрическим разрядом и улавливали озон до пропускания газа через
воду. При этом не наблюдалось образования туманного облака. Если по ходу
газа до контакта с водой помещали хлористый кальций (который, по предполо-
предположениям, должен был задерживать антозон), чтобы через воду мог проходить
* В английском тексте ошибка: должно быть «фосфорный туман вызывается обра-
образованием азотистокислого аммония».—Прим. перев.
2 Перекись водорода
18 Глава 1
только озон, то туман возникал. Это доказывало, что мнимый антозон являлся
просто перекисью водорода, возникавшей при реакции озона с водой. Кроме
того, Энглер и Нассе привели в своей статье доказательства возникновения
химических реакций, которые противоречили схеме Шенбейна об отношениях
между классами озонидов и антозонидов.
Все эти исследования помогли решить проблему озона и антозона и снова
установить индивидуальность перекиси водорода. Фейрли [33J и Лидс 1341
в конце семидесятых годов опубликовали обзоры, содержащие доказательства
по этому вопросу и критику ошибочных взглядов предыдущего поколения
химиков. Однако оставалась еще задача определения структурной формулы
перекиси водорода, которая могла бы объяснить наличие слабо связанного
атома кислорода и восстановительную способность перекиси. Биркенбах [271
дал критический обзор разных мнений по этому вопросу, в котором были рас-
рассмотрены, по-видимому, все возможные плоскостные и пространственные кон-
конфигурации для символов двух атомов водорода и двух атомов кислорода,
а также некоторые формы, в которых отношение кислорода к водороду превы-
превышало единицу. Структурные теории были построены почти исключительно на
геометрических соображениях; не было создано реальной основы для понима-
понимания свойств перекиси водорода. По-видимому, большинство химиков были склон-
склонны принять, как это сделал Хейс [35], что один атом кислорода в молекуле
перекиси водорода четырехвалентен, а другой не связан ни с одним из атомов
водорода, как это показывает формула t в отличие от формулы II
Н-О-Н н-О-О-Н
II
О
i п
Формула I объясняет образование воды и легкую потерю одного атома кис-
кислорода, который затем может участвовать в окислении. Восстанавливающие
свойства перекиси можно было объяснить одновременной потерей обоих атомов
кислорода с образованием молекулярного кислорода и соединением остающих-
остающихся атомов водорода с кислородом из восстанавливаемого соединения с обра-
образованием воды. Современное представление о структуре перекиси водорода,
отличающееся от вышеизложенного, представлено в гл. 6.
Выше дан обзор начальных работ по перекиси водорода от момента ее
открытия и приблизительно до 1880 г. В течение следующих трех-четырех деся-
десятилетий научный интерес к этому веществу сводился в основном к изучению
явлений каталитического разложения и реакций. Очень мало работ было по-
посвящено количественному исследованию физических свойств перекиси водо-
водорода, и только Отто Маас [36] и его ученики начиная с 1920 г. провели ряд
работ и установили с высокой точностью много важных свойств перекиси.
РАЗВИТИЕ ПРОИЗВОДСТВА И ПОТРЕБЛЕНИЯ ПЕРЕКИСИ ВОДОРОДА
До освоения в XX в. электрохимических способов производства практи-
практическим методом получения перекиси водорода оставался открытый Тенаром
способ действия кислоты на перекись щелочного или щелочноземельного
металла. Первым, кто предложил усовершенствование метода Тенара, был
Пелуз [37], который в 1832 г. рекомендовал использовать кремнефтористово-
дородную кислоту для взаимодействия с ВаО2. Эта реакция приводит к обра-
образованию осадка кремнефтористого бария и ускоряет процесс получения пере-
перекиси водорода. Описанный метод был распространен в течение долгих лет,
вероятно, потому, что получить легко отделяемый осадок при применении крем-
нефтористоводородной кислоты оказалось проще, чем при работе с серной
кислотой.
Развитие производства и потребления перекиси водорода 19
Ряд химиков того периода предложил различные методики получения
перекиси водорода, совершенно непригодные в практическом отношении. Так,
например, существовавшая в то время неопределенность относительно раз-
различий между природой перекисей щелочных металлов и так называемых
«перекисей» типа МпО2 и РЬО2 вызвала появление предложений о получении
перекиси водорода путем действия кислоты на МпО2 или РЬСХ. Такие пред-
предложения и даже уверения в возможности получения этими способами мы нахо-
находим, например, у Кастнера [38], Лампадиуса [39] и Бодримона [40]. Не все
эти предложения были приняты; так, сообщение Кирхнера [41] о возможности
получения перекиси водорода действием хлора на воду, находившуюся в пузы-
пузыре, было вскоре же опровергнуто [42].
Дальнейшие изменения, касающиеся использования перекисей щелочных
металлов для получения перекиси водорода, не нашли практического осуще-
осуществления до тех пор, пока не был серьезно поставлен вопрос о начале ее про-
промышленного производства. Дюпри [43] показал, что для образования перекиси
водорода из шлама перекиси бария можно использовать в качестве кислоты
двуокись углерода. Значительно позднее Бейл и Симе [44] запатентовали цик-
циклический процесс, основанный на реконверсии углекислого бария в перекись.
Впоследствии он был изучен Фоуситом [45]. Однако эти и другие системы,
предложенные в то время, не были приспособлены к существовавшей тогда
технологии. В 1879 г., когда промышленное производство перекиси водорода
уже было налажено, Дэвис [46] писал, что почти вся эта перекись получалась
действием фтористоводородной или кремнефтористоводородной кислоты на
перекись бария. Другие системы, например предложенная Гофманом [47],
по которой кремнефтористоводородную кислоту нужно было вводить в реак-
реакцию с расплавленным калием при продувке воздухом, не нашли практического
применения. Обстоятельством, способствовавшим производству перекиси водо-
водорода из перекиси бария, а возможно даже обусловливавшим практичность
этого процесса, явилось внедрение в промышленность в 1879 г. процесса
Брина 148] для производства кислорода [49], благодаря которому на рынке
в больших количествах появилась перекись бария. Процесс Брииа был осно-
основан ка попеременном окислении окиси бария на воздухе с последующим тер-
термическим разложением полученной перекиси при высоких температурах; он
обеспечил развитие крупного производства перекиси бария, которое составляло
в Европе в 1901 г. до 10 000 т/год [50]. Скуиб [51] подробно описал процесс
производства перекиси водорода на заводе того времени.
На рис. 2 показано типичное производство перекиси водорода того времени.
Рисунок изображает завод перекиси водорода Бернарда Лапорта в Шиплее
(Йоркшир) непосредственно после ввода в эксплуатацию в 1888 г. Операции,
показанные на рисунке, Слейтер [52] описывает следующим образом:
«На рисунке слева показаны два чана для производства перекиси водо-
водорода. Они заполнены слабыми, содержащими незначительное количество пере-
перекиси водорода промывными водами, оставшимися от предыдущей операции,
или же просто водой. В эти чаны добавляли небольшое количество соляной,
фосфорной или фтористоводородной кислоты для получения среды, применяв-
применявшейся для растворения перекиси бария. Перекись бария добавляли в чан одно-
одновременно с серной кислотой, а поэтому непосредственно после растворения
перекиси бария в среде кислот последние почти сразу же переходили в сво-
свободное состояние в результате осаждения сернокислого бария. Весь этот про-
процесс представлял скорее* искусство, чем научно обоснованное производство,
так как рабочие должны были подгонять подачу перекиси бария и серной кис-
кислоты таким образом, чтобы ни на один момент не было избытка ни того, ни
другого реагента: избыток перекиси бария вызвал бы разложение перекиси
водорода, избыток же серной кислоты привел бы к покрытию частиц перекиси
барця слоем сернокислого бария и таким образом исключил бы возможность
2*
20
Глава 1
дальнейшей реакции ВаО2. Рабочий выполнял операции при помощи стек-
стеклянной палочки и лакмусовой бумажки. На рисунке показано, как рабочие
выливают в чан кувшин растворяющей кислоты.
«Когда процесс заканчивался, шлам перекиси водорода и сернокислого
бария перекачивали через фильтрпресс, показанный в правой части рисунка,
в бак для хранения, показанный на заднем плане. Затем сернокислый барий
промывали и промывные воды использовали в следующей операции.
Рис. 2. Старый завод по производству перекиси водорода (картина
Ф. Олдхема, принадлежащая фирме «Laporte Chemicals, Ltd.»).
«Следует обратить внимание на то, что здание, построенное в то время,
когда электрического освещения еще не существовало, имеет газовое освеще-
освещение. Раствор перекиси водорода подвергали конечной обработке для стабили-
стабилизации и отрегулирования кислотности, а затем перекачивали через осветли-
тельный фильтрпресс в бак хранения готового продукта, показанный в крайней
правой части рисунка. Уже из этого бака раствор разливали по стеклянным
бутылям, поступавшим в продажу».
В период, когда было организовано промышленное производство на ос-
основе перекиси бария, был открыт и исследован современный процесс электро-
электролитического производства, а также ряд других процессов, не нашедших прак-
практического применения, но продолжающих привлекать внимание своими воз-
возможностями. В 1853 г. Мейдингер [53] обнаружил образование перекиси водо-
водорода в процессе электролиза серной кислоты. Еще со времен работы Фарадея
было известно, что при электролизе ощущается запах озона и что объем кис-
кислорода, выделяющегося в течение опыта, иногда меньше половины объема
водорода. Мейдингер указал, что недостающий кислород, очевидно^ со-
сохраняется в растворе, вероятно в виде растворенного озона или перекиси водо-
водорода, и пришел к заключению, что это действительно перекись водорода. На
основании своих работ Мейдингер установил, что «перекись водорода» обра-
образуется в наибольших количествах: 1) при удельном весе серной кислоты 1,4,
Развитие производства и потребления перекиси водорода 21
2) при возможно более низкой температуре и 3) при силе* электрического
тока, достаточно большой вследствие применения небольшого электрода.
Другие авторы, Бунзен, Рундшпаден и Леблан [54], подтвердили полу-
получение перекисных соединений при электролизе серной кислоты, но только
в 1878 г. Вертело [55] выяснил сущность этого процесса. Он показал, что в дей-
действительности в начале электролиза образуется надсерная кислота—соеди-
кислота—соединение, которое было им открыто незадолго до этого. Эта кислота медленно под-
подвергается гидролизу с образованием перекиси водорода и серной кислоты.
Дальнейшие исследования, проведенные Эльбсом и Шенгером и другими [56],
обеспечили возможность производства с 19Э8 г. перекиси водорода методом
электролиза. В свою очередь это послужило основой тщательного исследова-
исследования процесса на современном уровне, выполненного в 1912 г. Рейхелем [57],
который описал также историю развития этого процесса.
Другим способом, который давно получил признание, является образова-
образование перекиси водорода в ходе сжигания водорода. Этот способ был изучен
Струве, Беттгером, Шуллером и Траубе [58] в период 1869—1885 гг. и даже
запатентован, но никогда не применялся в промышленном масштабе. Резуль-
Результаты исследований этих и других способов получения и производства перекиси
водорода, так же как и большинство ранних работ по реакциям перекиси водо-
водорода, на которых мы здесь не останавливаемся, не имеют большого значения
для создания правильной перспективы исторического развития перекиси водо-
водорода. Хотя в течение этого периода было исследовано большое число методов
получения, большинство реакций привлекало внимание работников промышлен-
промышленности лишь на очень короткое время. Более подробно на этом вопросе мы оста-
остановимся в гл. 2 и 3.
Примерно к тому времени, когда началось производство перекиси водорода,
возник интерес к вопросам ее стабильности и возможности работы с концен-
концентрированными растворами. По обоим этим вопросам существовала значительная
неразбериха. Типичным для последней явились результаты работы Харкурта
[59], который утверждал, что стекло, нарочно загрязненное маслом или лаки-
лакированное, предотвращает разложение разбавленной перекиси водорода значи-
значительно лучше, чем чистое стекло. Шене [60] в 1878 г. указал, что противоречи-
противоречивые высказывания относительно свойств перекиси водорода, возможно, обус-
обусловливаются присутствием в ней примесей. Он сделал обзор методов получения
с точки зрения чистоты конечного продукта, и сам получил перекись водорода,
имеющую концентрацию 99,1 вес.%. Дэвис [46], пожалуй, впервые дал чис-
численную характеристику для разложения перекиси водорода при хранении.
Проба перекиси водорода, способная к выделению 9 объемов кислорода из
одного объема раствора, стоявшая в лаборатории Дэвиса, за 11 недель разло-
разложилась на 20%; после добавления нескольких капель эфира дальнейшего раз-
разложения не наблюдалось в течение 8 недель. Вертело [61], изучавший скоро-
скорости разложения в течение месяца и более, пришел к выводу, что стабильность
зависит от отсутствия щелочи в растворе.
Большая часть этих выводов была повторным открытием явлений или
свойств, уже известных Тенару, но остававшихся в течение долгого времени
без внимания. Ряд предложений, например сделанных Манном [62] и касаю-
касающихся очистки продажной перекиси водорода путем повторного осаждения
перекиси бария с последующей обработкой кислотой, представляет, в сущ-
сущности, только описание того трудоемкого процесса, которым пользовался Те-
нар. Однако мысль, что различные добавки могут стабилизовать перекись
водорода, была новой и стала общепринятой и практически необходимой. Кинг-
цет 163] провел обширное исследование свойств многих солей и органических
соединений с целью предотвращать разложение 5%-ной перекиси водорода.
1 • Очевидно, имеется в виду плотность тока.—Прим. персе.
22 Глава 1
Наилучшими стабилизаторами оказались спирт, эфир, камфора, ментол и ти-
тимол, причем Кингцет предпочитает спирт другим соединениям. Работы, прове-
проведенные Талботом и Муди [64] в 1892 г. по исследованию продажных раство-
растворов перекиси водорода, показали, что ни один такой раствор не подходит для
аналитических целей вследствие содержания различных примесей и остатков
кислот.
По-видимому, наибольшее значение для опровержения мнения о том, что
неустойчивость перекиси водорода заложена в ней самой, имели работы Анрио
[65] и Вольфенштейна [66]. Анрио в 1885 г. показал возможность выпаривания
перекиси водорода путем перегонки при уменьшенном давлении с улавлива-
улавливанием значительной части продукта в виде конденсата; этим путем он получил
64%-ную перекись водорода. Вольфенштейн доказал, что перекись водорода,
свободная от щелочи, следов соединений тяжелых металлов и твердых веществ,
является весьма стабильной и что 3%-ный раствор перекиси водорода можно
сконцентрировать до 50%-ного выпариванием при пониженном давлении, под-
поддерживая температуру ниже 75°, причем потерь за счет разложения при этом
не наблюдается. Вольфенштейн сам получил 99,1%-ный раствор перекиси
водорода, и на этот результат его исследований очень часто ссылаются. Труд-
Трудности достижения в промышленных условиях стандарта, установленного Воль-
фенштейном, хорошо иллюстрируются Фоуситом [45]. Значительная работа,
проведенная Кеблером, Уорреном и Раддимэиом [67] в отношении испытания
устойчивости продажных 3%-ных растворов, дает возможность судить о фак-
фактической стабильности таких растворов в то время A912 г.), когда техника
производства была вполне разработана. Большинство проб, исследованных
Кеблером с соавторами, было стабилизировано ацетанилидом. Наличие этой
добавки, по-видимому, приводило к большей стабильности проб, но этот эффект
сильно колебался, и продукт одной фирмы без содержания ацетанилида был
не хуже или даже лучше, чем стабилизированные пробы. По-видимому, это было
прямым указанием на различие в тщательности производства и, следовательно,
в чистоте конечной продукции. Пределы разложения в течение годичного
срока испытания колебались от 1 до 90%, однако обычная цифра составляла
примерно 10%, и эта величина считалась вполне допустимой.
Трудно сказать точно, когда впервые было поставлено промышленное
производство перекиси водорода для сбыта. Из данных фон-Шреттера [68]
и Камерона [69] следует, что в 1867 или 1869 г. в продаже появилась 3%-ная
перекись водорода. Это был продукт, который сбывался Тиллаем и, по данным
Гофмана [70], вырабатывался фирмой «Hopkin and Williams» в Лондоне.
Согласно фон-Шреттеру, в Берлине в 1873 г. Шерингом был построен завод
перекиси водорода. Бургуньон [71] сообщает о попытках получения перекиси
водорода в США для продажи в 1873 г. и о производстве ее в промышленном
масштабе в 1878 г. По Дэвису [46], перекись водорода достаточной чистоты
для химических работ имелась в продаже в 1879 г. Ланге [72] указывал, что
заводское производство перекиси водорода началось в 1881 г. Безусловно,
в этом году фирма «Oakland Chemical Co.» (Бруклин, Нью-Йорк) уже зани-
занималась промышленным производством перекиси водорода [73]. Бернард
Лапорт [49] пустил в эксплуатацию завод перекиси водорода в Шиплее
(Йоркшир) в 1888 г. Все эти материалы позволяют заключить, что по меньшей
мере с 1880 г. уже было налажено производство перекиси водорода в большом
масштабе.
Рыночная цена некоторых из первых партий перекиси водорода была очень
высокой; как указывает фон-Шреттер [68], она составляла в 1874 г. около 5 дол-
долларов за фунт. Эта цена была очень высокой, так как Гофман приводит для
1876 г. оптовую цену 1 доллар за фунт или меньше [76]. Лидс [34] сообщает,
что в 1879 г. импортированная перекись водорода продавалась в Нью-Йорке
примерно по 2 доллара за фунт, а 0,8%-ная перекись годом позже стоила
Развитие производства и потребления перекиси водорода
23
щщо
1
45,0 :
IL22'5
1 доллар. Такие же высокие цены существуют и сейчас для небольших партий
перекиси водорода при розничном сбыте, однако промышленное применение
перекиси было бы невозможным, если бы такие цены сохранялись на оптовые
партии. Бургуньон [71] дал анализ производственной себестоимости для 1878 г.,
указав цену около 6 центов за фунт. Расчетная калькуляция, опубликованная
Фоуситом [45] для завода, работавшего в 19Э2 г., дает стоимость около 4 центов
за фунт. К 1909 г. по цензу США [74]
стоимость перекиси водорода, произ-
производимой в США, составляла около
9 центов за фунт. Все эти данные от-
относятся к 10-объемной, или 3%-ной
(по весу), перекиси водорода, кото-
которая обычно продавалась на рынке до
появления процесса электролиза, по-
позволившего без труда получать более
концентрированные растворы.
В дальнейшем цена перекиси во-
водорода франко-завод непрерывно па-
падала, с одной стороны, в результате
конкуренции, с другой—вследствие
роста сбыта. В 1924 г. до появления
электролитического производства пе-
перекиси водорода в США цена на
27,5%-ную перекись, обычно имев-
имевшуюся в продаже, составляла около
36 центов за фунт раствора. После
начала электролитического производ-
производства цена упала примерно до 30 цен-
9,0
I
4,5:
2,25
0,9
1
г
J
А
j
t
А
им
-
-
19Ю 1920 1930
1940
Годы
1950 I960 1970
Рис. 3.
Производство перекиси
рода в США.
во до-
дотов за фунт в 1927 г. и до 24 цен-
центов—в 1929 г. К 1940 г. цена дошла
до 20 центов за фунт и после дальнейшего падения в военные годы до
15 центов за фунт возросла вновь до уровня 20 центов в 1950 г. D40 долла-
долларов за 1 т).
Технологические усовершенствования электролитических процессов,
которые способствовали снижению производственной себестоимости, по-види-
по-видимому, дошли до такой степени, когда дальнейшие затраты уже не оправды-
оправдываются и значительное снижение себестоимости в будущем может быть достиг-
достигнуто только путем разработки новых производственных процессов. Нужно
отметить, что принятая цена на единицу Н2О2 в растворе является мини-
минимальной для растворов промежуточной концентрации. Этот минимум обус-
обусловлен оптимальным балансом между стоимостью перевозки, упаковки и
т. п. для водяного балласта в разбавленных растворах и расходами по кон-
концентрированию.
Производство перекиси водорода непрерывно возрастало, если не считать
небольших сокращений его в отдельные годы. После окончания мировой войны
1914 г. в течение некоторого времени размер производства в США был более
или менее устойчивым, но начиная с 1925 г. рост применения перекиси водо-
водорода, главным образом в текстильной промышленности, вызвал увеличение
ее производства, которое продолжалось после этого непрерывно. Ход произ-
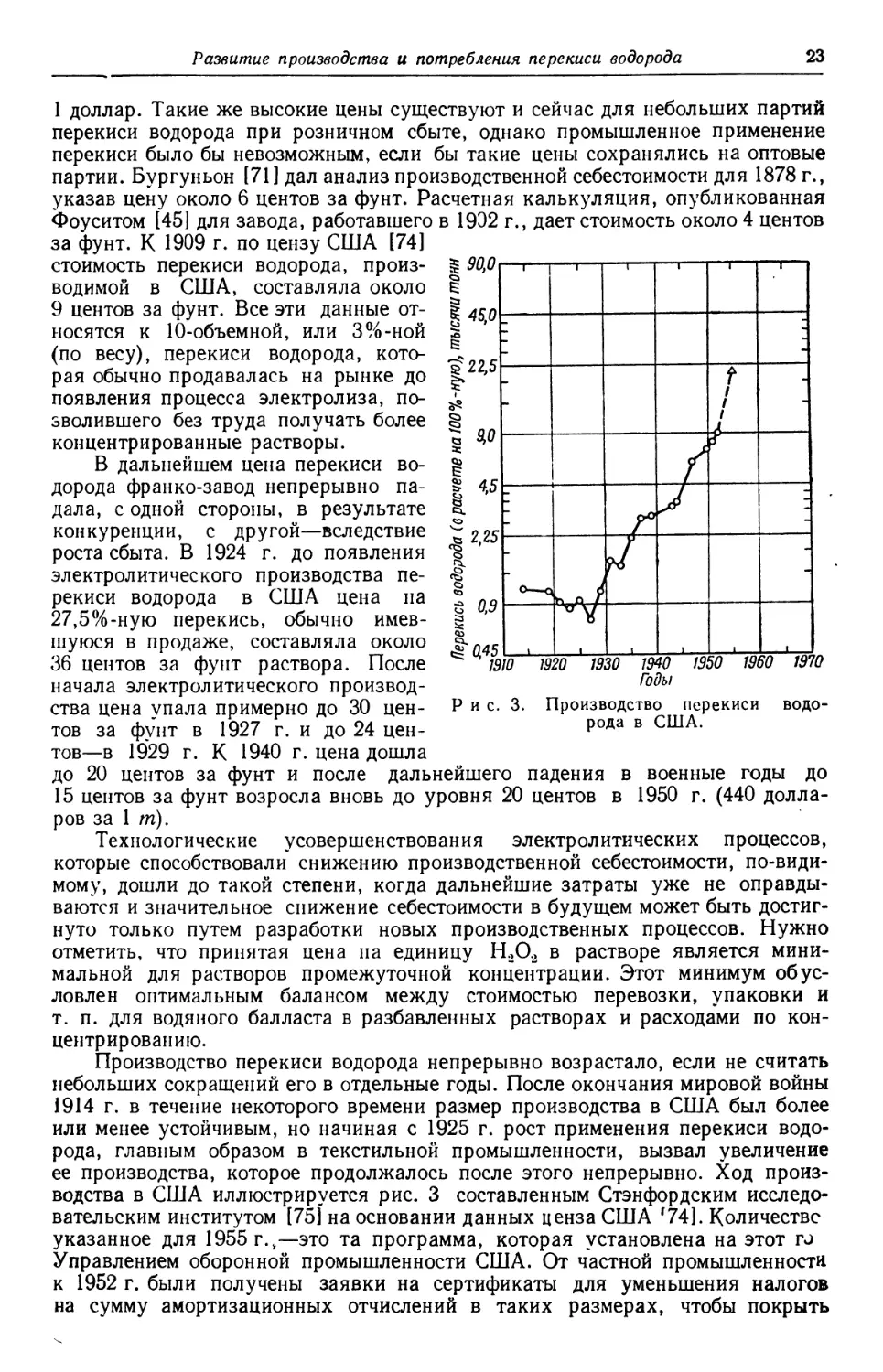
производства в США иллюстрируется рис. 3 составленным Стэнфордским исследо-
исследовательским институтом [75] на основании данных ценза США Г74]. Количестве
указанное для 1955 г.,— это та программа, которая установлена на этот tj
Управлением оборонной промышленности США. От частной промышленности
к 1952 г. были получены заявки на сертификаты для уменьшения налогов
на сумму амортизационных отчислений в таких размерах, чтобы покрыть
24 Глава 1
все намеченное расширение производства [76]. Мировая производственная
мощность в 1939 г. в расчете на 27,5%-ную перекись водорода составила,
45 тысяч тонн; из этого количества около 25% приходилось на США.
Надежных данных относительно размеров послевоенного производства в раз-
разных странах, кроме США, опубликовано очень мало. В настоящее время про-
производство перекиси водорода существует в большинстве промышленно разви-
развитых стран мира. До войны 1939 г. заводы перекиси водорода имелись в Анг-
Англии, Франции, Германии, Австрии и Италии, в большинстве других европей-
европейских стран, а также в Австралии, Корее, Японии и США. Только около 10%
всего произодства приходилось на долю процесса с перекисью бария; в связи
с этим выбор местоположения большинства заводов, пользующихся электро-
электролитическим методом, должен быть сделан с учетом близости к рынкам сбыта
и источникам дешевой электрической энергии.
Наиболее крупной фирмой в Англии, производящей перекись водорода,
является «Laporte Industries, Ltd.», которой принадлежит новый завод в Уор-
Уоррингтоне (Ланкашир), а также старый завод в Лутоне (Бедфордшир). Произ-
Производство перекиси на заводах Германии и Австрии описано в гл. 3.
В Канаде перекись водорода выпускается фирмой «Canadian Industries,
Ltd.» в Шоиниган-Фолсе (Квебек).
Производство перекиси водорода в США было начато в 1881 г. [73].
К 1912 г., как подсчитал Брюер [77], всего имелось свыше 100 производящих
фирм, причем все они зависели от поставок перекиси бария из Европы. В пе-
период мировой войны 1914 г. этот источник был потерян, но потеря в значитель-
значительной мере была возмещена производством фирмы «Rollin Chemical Company»
в1 Чарльстоне (Западная Виргиния), учрежденной в 1912 г. После окончания
первой мировой войны в Калифорнии были построены еще и другие заводы по
производству перекиси бария, которым пришлось столкнуться с острой кон-
конкуренцией с возобновившимся импортом. Один из новых заводов фирмы «Barium
Products, Ltd.» в Модесто (Калифорния) был куплен в 1929 г. «Westvaco Chemi-
Chemical Company». Эта компания (с 1948 г. отделение «Food Machinery and Chemi-
Chemical Corporation») продолжает до настоящего времени выпускать перекись водо-
водорода из перекиси бария; сначала ее завод находился в Калифорнии, а с 1941 г.—
в Картерете (Нью-Джерси).
Впервые производство перекиси водорода электролитическим методом
Гыло поставлено в США фирмой «Roessler and Hasslacher Chemical Company»
в 1926 г. в Ниагара-Фоле (Нью-Йорк). Эта компания начала вырабатывать
перекись водорода в 1909 г. и в 1930 г. была приобретена фирмой «Е. I. duPont
de Nemours and Company». Почти в то же время электролитическое производ-
производство было начато фирмой «Buffalo Electro-Chemical Company», возникшей
в 1925 г. и ставшей в 1951 г. дочерней фирмой компании «Food Machinery and
Chemical Corporation». В настоящее время фирме «duPont Co.» принадлежат
электролитические заводы в Ниагара-Фолсе и в Дрездене (Нью-Йорк). «Buffalo
Electro-Chemical Co.» владеет заводами в Ниагара-Фолсе и в Ванкувере (Ва-
(Вашингтон). Оба эти концерна, по имеющимся данным, выпускают примерно 90%
всей продукции США по перекиси водорода [78].
В течение известного времени фирма «Pennsylvania Salt Manufacturing
Company» также вырабатывала перекись водорода электролитическим мето-
методом. Другие химические заводы 179] вырабатывали небольшие количества
перекиси водорода из перекиси бария, главным образом для фармацевтической
или тонкой химической промышленности. В дальнейшем, в связи с развитием
применения перекиси] водорода, и особенно с разработкой новых способов
производства, можно ожидать, что и многие другие фирмы заинтересуются
этой отраслью промышленности. В 1953 г. фирма «duPont» начала сбывать
перекись водорода, получаемую методом самоокисления на новом заводе вблизи
Мемфиса (Теннесси). В 1955 г. фирмы «Allied Chemical and Dye Corporation»
Литература по перекиси водорода 25
и «Shell Chemical Corporation»"также начали строить заводы по производству
перекиси водорода неэлектролитическим путем. Подробнее история производ-
производства перекиси водорода в США изложена у Хейнеса [80].
Развитие применения перекиси водорода в достаточно значительных мас-
масштабах, которое обеспечило бы возможность организации крупного производ-
производства, сначала происходило медленно. Уже давно появились отдельные пред-
предложения относительно возможности ее применения, например Де-Сондало [81]
в 1843 г. предложил использовать перекись водорода для освежения воздуха
в общественных местах и в шахтах. Однако эти предложения были либо слиш-
слишком непрактичны, либо слишком опережали свое время. Некоторое применение
перекись водорода нашла в медицине, например в виде антисептика, раздра-
раздражающего вещества, для пульверизации по Листеру и т. д.; одно время пред-
предполагалось, что она может найти специфическое применение для лечения диф-
дифтерии. Ричардсон [82] был сторонником использования перекиси водорода
в медицине еще в 1856 г. Применение ее, главным образом в качестве антисеп-
антисептика, продолжалось довольно долго, причем медики периодически то отказы-
отказывались от нее, то вновь признавали.
Применение перекиси водорода для отбелки, имеющее наибольшее зна-
значение, было предложено в 1862 г. Шеврелем [83] и практически использовано
для этой цели в 1867 г. Тесье-дю-Мотеем [84]. Применение перекиси водорода
в косметике как средства для обесцвечивания волос открыло рынок сбыта для
перекиси водорода, впервые выпущенной в промышленном масштабе. И Каме-
Камерон [69] и фон-Шреттер [68], работы которых способствовали началу промыш-
промышленного производства, поставили своей задачей разработать состав жидкости
для обесцвечивания волос; препарат фон-Шреттера носил коммерческое назва-
название «Eau fontaine de jouvence golden». Гофман [70] указывает, что, очевидно,
«именно этот состав придает волосам тот отвратительный белокурый оттенок,
который занимает промежуточное положение между пепельно-серым и ярко-
желтым и привлекает внимание и любопытство наблюдателей в результате
своей пикантной неестественности».
Вскоре после этого перекись водорода начали применять для отбелки
слоновой кости, кости, шелка, щетины и перьев; во всех этих областях она
оказалась вне конкуренции, причем сравнительно большим расходом прида-
придавали второстепенное значение. Преимущества перекиси водорода постепенно
находили признание и в отношении отбелки других материалов—соломы, кож,
шерсти и дерева; поэтому примерно к 1890 г. промышленники стали меньше
зависеть от моды и возник устойчивый рынок. Размеры начального применения
перекиси водорода освещены в сообщениях Эбелла, Геринга и Дельмарта [85].
ЛИТЕРАТУРА ПО ПЕРЕКИСИ ВОДОРОДА
По скромным подсчетам, число источников в технической и коммерческой
литературе, касающихся перекиси водорода, составляет три-четыре тысячи
названий. Для тех, кто специально интересуется этой проблемой, полезно обри-
обрисовать тематику этой литературы и указать на некоторые имеющиеся обзор-
обзорные работы.
Представленный нами исторический обзор ни в коем случае не является
полным; ряд тем, которые подробно изучались задолго до начала XX столетия,
а именно: химические реакции перекиси водорода, содержание ее в атмосфере
и применение в медицине, затронут лишь очень кратко. Известная доля этих
работ упоминается ниже в ссылках, но большая часть их уже устарела и не
представляет интереса.
По перекиси водорода опубликовано 3 книги, все на немецком языке.
Книга Биркенбаха [27] вышла в 1906 г.; основная часть ее посвящена анализу
перекиси водорода, кроме того, имеются и большие разделы по истории, произ-
26 Глава 1
водству и применению. Позднее вышли книги Маху [86] и Кауша [87]; каж-
каждая из них содержит около 2000 ссылок на литературу. В обеих книгах име-
имеется обзор основной литературы вплоть до момента их опубликования по таким
вопросам, как физические и химические свойства, производство и хорошо
известные виды применения. Книга Биркенбаха сохраняет свое значение как
руководство по анализу и может быть использована для исторических справок.
Маху, выпустивший первое издание своей книги в 1936 г., а второе в 1951 г.,
посвящает, помимо перекиси водорода, около 1/5 книги другим перекисным
соединениям. Он дает анализ литературы по перекиси водорода, и особенно оста-
останавливается на ее производстве. Ценной особенностью этой монографии являет-
является аннотированный список патентов. Книга Кауша A938 г.) дает практически
полйую компиляцию литературных ссылок в такой форме, которая, в сущности,
представляет собой аннотированную библиографию. Кроме перекиси водорода,
Кауш не интересуется никакими другими перекисными соединениями, но он
больше, чем Маху, уделяет место вопросу применения перекиси водорода.
Представляют интерес некоторые старые сообщения общего характера.
Мы уже упоминали о сообщениях Дэвиса [46], Лидса [34] и Фоусита [45].
В сообщении Лидса рассматривается озоно-антозоновая теория и приводится
библиография по перекиси водорода до 1879 г. Мезон [88] в 1881 г. написал
полный обзор по всем вопросам, касающимся перекиси водорода. Коллер [89]
в 1892 г. опубликовал полный материал по применению ее для отбелки. Подроб-
Подробный обзор Неймарка [9Э], датированный 1913 г. (т. е. появившийся в конце
периода, когда были разработаны основные области применения перекиси
водорода и достигнуто взаимопонимание), представляет ценность как итого-
итоговый материал.
Из современных обзоров по перекиси водорода можно рекомендовать
работы Рейхерта [91], Слейтера [92], Шенли и Гринспэна [93], Шенли [94],
Миласа 195] и MhttoJ96]. Очень полный общий обзор сделан Вудом в моногра-
монографии № 2 за 1954 г.,'опубликованной Королевским институтом химии. Весьма
ценный итог по химическим свойствам перекиси водорода с особыми ссылками
на аналогичные соединения (и сравнением с ними) опубликовали Иост и Рассел
[97]. Исторические обзоры по производству перекиси водорода принадлежат
Бречгеру и Шенли и Левенштейну [98]. Первые серии Алфавитного каталога
Управления главного военного хирурга [99] дают очень ценный материал по
различным видам использования перекиси водорода в медицине. Некоторые
менее известные области применения рассматриваются Ланге [72], причем
значительное внимание уделяется патентной литературе.
НОМЕНКЛАТУРА ПЕРЕКИСНЫХ СОЕДИНЕНИЙ
Давая названия перекисным соединениям, необходимо указывать на при-
присутствие перекисной структуры и различать перекиси и другие кислородсодер-
кислородсодержащие соединения. Предложены или применяются различные способы обозна-
обозначения перекисных соединений. Наиболее удовлетворительную систему можно
разработать путем рассмотрения разных типов кислородных соединений.
Атом кислорода соединяется с атомами кислорода и других элементов
с образованием химических связей, которые в основном являются ковалент-
ными связями или связями с поделенными электронами. Каждый кислород-
кислородный атом может приобрести путем участия в поделенных электронах до двух
электронов. Молекулы, образующиеся только из атомов кислорода,—это О2,О3
и О4. По-видимому, не наблюдается связывания атомов кислорода в группу,
состоящую более чем из четырех атомов. В соединениях кислорода с другими
элементами постулировано связывание до 4 или даже больше атомов кислорода.
Если ограничиться только такими соединениями кислорода с другими эле-
элементами, в которых кислородо-кислородная связь охватывает не более двух
Номенклатура перекисных соединений 27
атомов кислорода, то соединения кислорода с элементом М, каждый атом кото-
которого способен отдавать по одному электрону для образования ковалентной
связи, могут быть следующими: МО2, МО, М2О2 и М2О. Здесь не рассматри-
рассматриваются соединения кислорода с более электроотрицательным элементом, как
OF2. Отношение числа атомов элемента М и кислорода или доля атома,
присоединяющегося к атому кислорода, в этом ряду составляет 1/а, 1 и 2.
Целесообразно ставить около этих чисел знак минус, чтобы выразить электро-
электроотрицательный или электроно-акцепторный характер кислорода. Если в этот
ряд включить еще молекулярный кислород О2 с отношением, равным нулю, то
номинальные валентности или окислительные коэффициенты для кислорода
будут 0,—V2> — 1 и - 2. Обозначая их таким образом, мы вовсе не намерены
сказать, что связи, образуемые кислородом в этих состояниях, носят по своей
природе преимущественно электростатический или ионный характер; един-
единственное, чего мы добиваемся,—это облегчения идентификации и составления
баланса реакций.
Соединения, которые содержат кислород в виде О^, или с окислитель-
окислительным числом—1/2, в настоящее время называются надперекисями, например
надперекись калия КО2. Этот класс соединений можно рассматривать как
производные кислоты НО2. Молекула или радикал НО2, который нужно логи-
логически называть надперекисью водорода, обычно носит название пергидроксила
[100] (см. ниже). Однако нужно отметить, что термин «надперекись» одно время
употреблялся для обозначения «перекиси» и что терминами Wasserstoffsuper-
oxyd и Wasserstoffperoxyd до сих пор по-немецки обозначают перекись водо-
водорода.
Соединения, содержащие группу —О—О—, или О\", или кислород с окис-
окислительным числом —1, называются перекисями в собственном смысле этого
слова или пероксисоединениями. Были предложены и другие обозначения с при-
применением приставок «супер-», «гипер-», «гол-» [86] или терминов «оксат» [101]
или «перводная кислота» [102], причем имеются некоторые колебания в раз-
разных языках при обозначении родоначальника класса—перекиси водорода,
однако на основании обычной практики и соглашения Международного
химического союза [103] сейчас предпочитают обозначения «перекись» и
«перокси»*.
Наиболее распространенные кислородные соединения, содержащие О2"
или кислород с окислительным числом —2, обычно называют просто окисями
с добавкой приставки типа «моно-», «ди-» (по-русски—«дву-») и т. д. для обоз-
обозначения числа кислородных атомов, например двуокись марганца МпО2.
Если в соединении с окисным кислородом находится еще третий элемент,
то наименование прямо не указывает на присутствие такого кислорода, однако
суффиксы «ат» или «ит» в названии соединения обычно (хотя и не всегда) обо-
обозначают наличие окисного кислорода, поскольку значительная часть радикалов
с этими суффиксами действительно содержит кислород**.
Некоторые окислы типа МО2, например РЬО2 и МпО2, иногда называют
* В английском языке обычно применяют термины hydrogen dioxide и peroxide
of hydrogen (двуокись водорода и перекись водорода). Во французском—сохранено
первое обозначение, данное Тенаром, eau oxygenee (окисленная вода); такой же терми-
терминологии придерживаются и в итальянском и испанском языках (acqua ossigenata, agua
oxigenada). В немецком языке чаще всего пишут Wasserstoffsuperoxyd. Однако этими
названиями как коренными словами для обозначения других перекисных соединений не
пользуются.
** Суффикс «ат» обозначает примерно «тот, который был подвергнут действию»,
например ацетат свинца—«свинец, на который подействовали «ацетатной» (уксусной)
кислотой». Названия многих неорганических радикалов возникли из названий солей,
образованных взаимодействием кислородных кислот со щелочами, например сульфат
от солей серной кислоты. Аналогично применяется суффикс «ит», но для обозначения низ-
низшей валентности.
28 Глава 1
перекисями, но это неправильно. Их следует называть двуокисями, что ука-(
зывает настоящую структуру и исключает возможность смешения с известными
перекисями.
Необходимо подчеркнуть, что приставка «пер-» не является свойственной
только перекисным соединениям [104]. Эта приставка, взятая из латинского
и означающая «полностью» или «совершенно», была впервые введена в химиче-
химическую номенклатуру Томсоном [105] в 1804 г. в виде термина «пероксид» (пере-
(перекись) для обозначения соединения, в котором металл соединен с максимально
возможным количеством кислорода. Позже так обозначали соединения с мак-
максимальным количеством атомов других элементов, например перхлориды;
однако это название сейчас устарело. При такой системе термин «пероксид
водорода» для различия между Н2О2 и Н2О был совершенно правильным. При-
Приставка «пер-» стала общепринятой, но с развитием представлений о природе
химической связи были сделаны два значительных изменения: 1) этимологиче-
этимологическое значение было заменено обратным, а именно приставка «пер-» в названии
соединения подразумевает высшее валентное состояние элемента в соединении
с максимально возможным количеством кислорода или другого элемента 1106];
так, например, в слове «перманганат» достигнуто первоначальное намерение
Томсона указать, что марганец соединялся с максимально возможным коли-
количеством кислорода, но при современном употреблении этого слова основное
внимание обращается на высшее состояние окисления марганца, которое яв-
является следствием малой величины отношения количества атомов марганца
к количеству атомов кислорода; 2) было выяснено, что кислород соединяется
в различных относительных количествах или имеет более чем одно состояние
окисления. Это обстоятельство нельзя отразить одним лишь применением при-
приставки «пер-»; так, если РЬО2, двуокись свинца, называть перекисью свинца,
то исчезает коренное различие в структуре между этим соединением и истин-
истинной перекисью, например перекисью бария ВаО2.
Приставка «пер-» в современной химии используется для следующих со-
соединений [107]: 1) какого-либо соединения или радикала, содержащего эле-
элемент в высшем состоянии окисления, например перхлораты для обозначения
содержания семивалентного хлора; 2) соединения, содержащего перекисную
группу—О—О—, например перекись (по-английски peroxide) натрия Na2O2;
сюда нужно добавить еще третье значение, хотя несколько менее известное,
а именно 3) соединения, подвергнутые полному замещению или присоединению,
например пергидронафталин С10Н18*.
Поскольку в истинных перекисных соединениях состояние окисления,
которое должно быть указано приставкой «пер-», является состоянием кисло-
кислорода, который к тому же формально не находится в состоянии максимального
окисления, то применение приставки «пер-» должно быть оставлено только
для 1-го и 3-го случаев, а для соединений 2-й группы следует применять только
термин «перекись» или приставки «перокси-», «пероксо-»**, если их строение
¦ Этот термин утвержден недавно [108], и можно ожидать, что он войдет в обиход,
так как полностью галогенированные углеводороды приобретают все большее значе-
значение.
** В русской литературе для обозначения присутствия в кислоте перекисной груп-
группы—О—О—Д. И. Менделеев ввел термин «надкислота», который сохранился и до на-
настоящего времени. Этот термин применяется в значении, соответствующем рекомендо-
рекомендованному для международного употребления 17-й конференцией Международного союза
чистой и прикладной химии (Extrait des Comptes Rendus de la Dix-Septieme Conference
Stockholm, 29 Juillet—4 Aout 1953, p. 101 —108) термину «пероксокислота». В слове
«пероксокислота» окончание приставки «пероксо-» отражает координацию двух кислородных
атомов перекисной группы О\~ в сфере центрального атома кислотообразующего эле-
элемента на месте оксогруппы, т. е. О2". На основании высказанных выше соображений
следует отдать предпочтение термину «пероксокислота», в связи с чем соединение HaSO6,
например, называется пероксосерная кислота или пероксомоносерная кислота. Принятое
Номенклатура перекисных соединений 29
должно быть указано точно [109]*. Примером является название перхлорат
натрия для натриевого производного хлорной кислоты НСЮ4, пероксомоно-
сульфат для солей пероксосерной кислоты H2SO5. Поэтому же соединение
№252О8 необходимо называть пероксодисульфат натрия.
Допустимы два исключения из практики применения приставки «перокси-».
Кислотный анион перекиси водорода О2Н~ называют ионом пергидроксила
вместо неудобного названия пероксигидроксил. Радикал НО2, который, соб-
собственно, и является надперекисью водорода, обычно носит название «радикал
пергидроксила», что обусловлено аналогией формул и часто постулируемым об-
образованием из него перекиси водорода при реакциях в газовой фазе. Для О*""
сохранено название перекисного иона.
Аддитивные соединения перекиси водорода, т. е. соединения, содержащие
кристаллизационную перекись водорода, например 2Na2COa-3H2O2, обычно
называются пергидратами по аналогии с термином «гидрат». Однако название
«пергидрат» можно истолковать как обозначение вещества, гидратированного
до максимально возможного содержания воды. Вместе с тем сама комбинация
«пергидро» не является характерной для перекиси водорода. Более желатель-
желательным было бы такое название, которое не допускало бы неправильного толко-
толкования смысла приставки «пер-» и четко обозначало бы, что соединение содер-
содержит перекись водорода, а не воду, как это можно предполагать на основании
присутствия слога «гидро». Вместо термина «пергидрат» были предложены
названия «пероксат», «гидропероксат» или «гидропероксидат»**. Перекисные
соединения или пероксигидраты могут также содержать еще кристаллиза-
кристаллизационную воду, как например октогидрат перекиси натрия Na2O2-8H2O
и дигидратпероксигидрат сульфата натрия Na2SO4-2H2O-H2O2. Строение
некоторых перекисных соединений еще до сих пор четко не выяснено; не решен
вопрос, существует ли в них кислород в виде перекисного или пероксигидрат-
ного. В этом случае такого роде соединения проще всего называть перекисями.
Замещенные производные перекиси водорода могут быть разделены на ряд
категорий: к неорганическим соединениям принадлежат металлические соли
перекиси водорода, гидроперекиси и перекиси металлов, а также пероксокис-
лоты (надкислоты); к органическим соединениям относятся алкильные произ-
производные перекиси водорода, гидроперекиси и перекиси алкилов, гидроперекиси
ацилов, или пероксокислоты (надкислоты), перекиси ацилов и перекиси ацил-
алкилов, или эфиры пероксокислот (надкислот).
Для обозначения замещенных производных перекиси водорода или соеди-
соединений, которые можно рассматривать как производные этой перекиси, при-
применяется следующий практический прием. Однозамещенные органические про-
производные и соли слабой кислоты Н2О2, которые могут быть изображены в виде
ROOH (например, NaOOH или СН3ООН), называют R-гидроперекисями, если
элемент или радикал R не представляет кислотный или ацильный радикал
(—COR). Двузамещенные соединения типа ROOR, ROOR' или RO2 (например,
в русском языке обозначение солей пероксокислот с помощью приставки «пер-», напри-
например Ka^Og—персульфат калия, следует изменить в соответствии с рекомендованным на
пероксодисульфат калия. Приставка «перокси-» сохраняется для обозначения соедине-
соединений, в состав которых входит целая молекула перекиси водорода, например в виде кри-
кристаллизационной.—Прим. ред.
* В немецком языке для устранения путаницы между приставками «пер-» и «пер-
«перокси-» применяют приставку «iiber-» для обозначения состояния максимального окисле-
окисления в веществах, не являющихся перекисями.
** В соответствии с рекомендацией Международного союза чистой и прикладной
химии (см. примечание редактора на стр. 28) в русском переводе принято наименование
«пероксигидрат», которое всюду применяется в дальнейшем (в подлиннике в этом случае
принят термин «гидропероксидат», предложенный проф. О. М. Петерсоном и обозначающий
в соответствии с термином «гидрат» «то, что присоединилось к перекиси водорода или на
что перекись водорода оказала воздействие»).— Прим. ред.
30 Глава 1
NaOONa, СН3ООС2Н5, ВаО2), где R не представляет кислотный радикал, назы-.
ваютИ- или ди-И-перекисями, или RR'-перекисями. Если обе группы R, или
R и R', являются ацильными, то эти соединения также называются R- или
RR'-перекисями.
Соединения ROOH или ROOR, где R представляет кислотный или ациль-
ный радикал, за исключением вышеуказанных, например СН3СО—О—О—Н или
HSO3—О—О—SO3H, называют пероксокислотами (или надкислотами). Эве-
рети Минкоф [110] предпочитают применять термин «гидроперекись ацила» для
соединения типа RCO—О—О—Н вместо термина «пероксокислота», чтобы не
вызвать неверного представления, что эти соединения являются сильными кис-
кислотами. Путем замещения кислот с двумя гидроксильными группами можно
получать 3 типа соединений, например: для угольной кислоты производными
являются НОСО—О—О—Н пероксомоноугольная (мононадугольная) кис-
кислота; НОСО—О—О—СООН пероксодиугольная (наддиугольная) и Н—О—О—
—СО—О—О—Н дипероксоугольная (динадугольная). Этими названиями мы
лишь поясняем, каким образом надо изображать кислотные свойства пере-
перекисей, и вовсе не хотим сказать, что такие кислоты или аналогичные им дей-
действительно существуют. Соли этих кислот обозначаются обычным образом,
например С6Н5СО—О—ONa пероксобеизоат натрия (надбензойнокислый
натрий). Соединения ROOR\ где только одна из групп R является ацильным
радикалом, носят название сложных пероксоэфиров, например СвН5СО—О—О—
—С(СН3K mpem-бутиловый эфирпероксобензойной кислоты или, чаще, трет-
бутилпероксобензоат. Основным различием в свойствах между этими соеди-
соединениями и обычными сложными эфирами является то, что при гидролизе
пероксоэфиры образуют кислоту и гидроперекись, а не пероксокислоту
и спирт. Милас [111] и Тобольский и Месробян [112] тщательно обсуждают
номенклатуру органических перекисей, приводя многочисленные примеры
и разбирая самые сложные случаи.
Среди перекисных соединений, которых известно примерно тысяча или
около этого, встречается много гораздо более сложных перекисей. Если назы-
называть их перекисями или использовать приставки «пероксо-» и «перокси-» для
указания наличия одной или нескольких перекисных групп, не должно возник-
возникнуть никакого недоразумения. Строение группы соединений, образующихся
при реакции непредельных соединений с озоном и известных как озониды [1131
или оксозониды, определенно еще не установлено. Эти соединения, возможно,
действительно содержат перекисный кислород, но они не обозначаются как
перекиси.
ЛИТЕРАТУРА
1. Davy J., ed., The Collected Works of Sir Humphry Davy, Vol. 5, London, Smith,
Elder and Co., 1840.
2. G a y-L u s s а с J. L., Thenard L. J., Recherches Physico-Chimiques, Vol. I,
Deterville, Paris, 1811.
3. P a r i s J. A., The Life of Sir Humphry Davy, Colburn and Bentley, London, 1831;
Flour ens M., in Michaud, Biographie Universelle, Vol. 41, Delagrave et Cit\
Paris, 1843—1865, p. 246; A 1 e x a n d e г С. A., Smithsonian Institution Annual
Report for 1862, Washington, Government Printing Office, 1863, p. 373; С г u m
Brown A., Encyclopedia Britannica, 9th ed., Vol. 23, Charles Scribner's Sons, New
York, 1888, p. 251; Ferguson J., Encyclopedia Britannica, 9th ed., Vol. 10,
Charles Scribner's Sons, New York, 1888, p. 121; Webb K- R., Chemistry and
Industry, 1945, 2; Блох М. in Bugge, Das Buch der grossen Chemiker, Vol. I,
Verlag Chemie, Berlin, 1929; Bouchard G., Une grand Francais, le chimiste
Thenard, Imprimerie Jobard, Dijon (Cote d'Or), France, 1950; [Compt. rend., 232,
665 A951)]; Proc. Roy. Soc, 9, 60 A857); NicklesJ., Am. J. Sci. [2|, 24, 408
A857); 25, 430 A858); P 1 а у f a i r L., J. Chem. Soc, 11, 182 A859); Thenard entry,
The British Museum Catalog of Printed Books, Vol. 53, J. W. Edwards, Ann Arbor,
iMichigan, 1946.
Литература 31
4. Biographic Nouvelle des Contemporains, Vol. 19, Librairie Historique, Paris 1825,
p. 424.
5. ThenardL.J., Ann. chim. et phys., 8, 306 A818), [Ann. Phil., 13, 1 A819); Phil.
Mag., 53, 21 A819)], Ann. chim. et phys., 9, 51 A918), [Ann. Phil., 13, 5 A819); Phil.
Mag., 53, 109 A819)); Ann. chim. et phys., 9, 94 A818); [Ann. Phil., 13, 9 A819);
Phil. Mag., 53, 147 A819I; Ann. chim. et phys., 9, 314, 441 A818); 10, 114, 335
A819); [Ann. Phil., 14, 209 A819); Phil. Mag., 54, 70 A819); Q.M. Sci., 7, 379 A819)];
Ann. chim. et phys., 11, 85 A819); [Ann. Phil., 14, 274 A819); Phil. Mag., 54, 312
A819); Q. J. Sci., 8, 154 A820)]; Ann. chim. et phys., 11, 208 A819); [Q. J. Sci.,
8, 114 A820)].
6. Thenard L. J., Mem Acad. Sci., Paris, 3, 345 A818).
7. T h e n а г d L. J., Traite de Chimie,1 ed. 3, Vol. I, Chez Crochard, Paris, 1821,
pp. 562—601 [Ann. Phil., 3, n. s., 41 A822)]; Les Classiques de la Science, Vol. 3,
Libraire Armand Colin, Paris, 1913.
8. Thenard L. J., Ann. chim. et phys., 50,.80 A832) [Phil. Mag. 2, 403 A833I;
Am J. Sci., 23, 382 A833).
9. T h e n а г d L. J., T h e n а г d P., Compt. rend., 41, 341 A855), Ann. chim. et phys.,
47, 173 A856).
10. Royal Society of London, Catalogue of Scientific Papers A800—1863), Vol. 5, Eyre
and Spottiswoode, London, 1871, p. 946; Ann. Phil., 13, 380 A819); 15, 378 A820);
Phil. Mag., 53, 462 A819); Am. J. Sci., 3, 369 A821); 23, 382 A833); Children
J. G., An Essay on Chemical Analysis, W. Philips, London, 1819.
11. Berzelius J. J., Jahresber. Chem., 15, 237 A836).
12. S i 1 1 i m a n В., Elements of Chemistry, Hezekiah Howe, New Haven, 1830, p. 215;
TurnerE., Elements of Chemistry, 4th ed., John Taylor, London, 1833, p. 225;
Gmelin L, Handbook of Chemistry, tr. by Watts H., Vol. 2, Cavendish Society,
London, 1849, p. 73.
13. Faust E. D., Am. J. Sci., 17, 34 A830).
14. Liebig J., Ann. Chem. Pharm., 2, 19 A832).
15. T h e n а г d L. J., Ann. chim. et phys., 48, 79 A831), [Am. J. Sci., 22, 351 A832)].
16. Mitscherl ich M. E., Pogg. Ann., 55, 209 1842), [Taylor Scientific Mem., 4,
1 A846)].
17. P 1 а у f a i r L., Mem. Proc. Chem. Soc, 3, 348 A848).
18. Oesper R., J. Chem. Ed., 6, 432, 677 A929); Hagenbach J., Smithsonian
Institution, Annual Report for 1868, Government Printing Office, Washington,
1872, p. 185.
19. SchonbeinC. F., Proc. Roy. Soc, 5, 543 A845); J. Chem. Soc, Memoirs, 3,
358 A851); Phil. Mag., [41, 2, 22 A851); 13, 248 440 A857); 15, 24 A858); 16, 178
A858); 18, 510 A859); 21, 88 A861); 23, 466 A862); Am. J. Sci., 27, 19 A859); J.
prakt. Chem., 77, 138 A859); 78, 63 A859); 93, 24 A864) [Chem. News, 11, 25 A864I;
J. prakt. Chem., 98, 65, 257 A866).
20. Schaer E., Vierteljahresschr. pr. Pharm., 18, 1, 371, 497 A869).
21. Meissner G., Untersuchungen iiber den Sauerstoff, Hahn, Hannover, 1863; Neue
Untersuchungen iiber den elektrischen Sauerstoff Dieterich, Gotttingen, 1869.
22. Lenssen E., J. prakt. Chem., 81, 276 A860).
23. Clausius R., Ann. Physik, [21, 103, 644 A858); 121, 250, 330 A864).
24. Osann G. W., J. prakt. Chem., 95, 55¦ A865).
25. Phipson T. L., Smithsonian Institution Annual Report for 1862, Government
Printing Office, Washington, 1863, p. 395.
26. Fox С. В., Ozone and Antozone, J. and A. Churchill, London, 1873; Chem. News,
27, 82 A873).
27. BirckenbachL., Untersuchungsmethoden des Wasserstoffperoxyds, Margosches,
Die chemische Analyse, Vol, 7, Ferdinand Enke, Stuttgart, 1909.
28. В г о d i e В. С, J. Chem. Soc, 7, 304 A854).
29. BrodieB.C, Proc Roy. Soc, 11, 442 A861); J. Chem. Soc, 16, 316 A863).
30. Wei tzien C, Ann. Chem. Pharm., 115, 121 A860); 138, 129, 149 A866); Chem.
News, 14, 1. 15, 39 A866).
31. v о n В a b о, С. Н. L., Ann. Chem. Pharm., Suppl. Vol. 2, 265 A863).
32. EnglerC, Nasse O., Ann. Chem. Pharm., 154, 215 A870).
33. F a i г 1 e у Т., J. Chem. Soc, 31, 1, 125 A877).
34. Leeds A. R., Ann. N. Y. Acad. Sci., I, 363 A880); 3, 153 A884).
35. H e у e s J. F., Phil. Mag., [51, 25, 221 A888).
36. Giguere P. A., Chemistry in Canada, 4, 57 A952).
37. Pelouze T. J., Pogg. Ann., 25, 508 A832).
38. Ka-stner K. W. G., Archiv fur die gesammte Naturlehre, 12, 497 A827)
39. L a m p a d i u s W. A., J. prakt. Chem., 17, 34 A839).
40. Baudrimont A., Compt. rend., 62, 829 A866).
41. Kirchner J. W., Repert f. Pharm., 53, 327 A835).
42. Ann. Chem. Pharm., 17, 40 A836).
32 Глава 1
43. D u р г е у F., Compt. rend., 55, 736 A862) [Phill. Mag., [41, 24, 526 A862)U
44. Bale R.Siras Т. С, англ. пат. 3628 A890); J. Soc Chem. Ind., 10, 482 A891).
45. F a w s i t t С A., J. Soc Chem. Ind., 21, 229 A902).
46. D a v i s G. E., Chem. News, 39, 221 A879).
47. H о f m a n n C, Ann. Chem. Pharm., 136, 188 A865) [Phil. Mag. [4], 31, 143 A865)].
48. Smjth W., J. Soc. Chem. Ind., 4, 568 A885).
49. Laporte Chemicals Ltd., сообщение № 83, «Hydrogen Peroxide; Advances in Pro-
Production and Uses»; Chemistry and Industry, 1951, 401.
50. Heinz R., Chem. Ztg., 25, 199 A901) [Chem. News, 85, 206 A902I.
51. S q u i b b E. R., The Ephemeris of Materia Medica, 4, 1545 A894).
52. S 1 a t e r V. W., частное сообщение.
53. MeidingerH., Ann. Chem. Pharm., 88, 57 A853) [J. Chem. Soc, 7, 251 A855I.
54. В u n s e n R. W., Pogg. Ann., 91, 619 A854); Rundspaden A., Ann. Chem.
Pharm., 151, 306 A869); L e В 1 a n с F., Compt. rend., 75, 537 A872) [J. Chem.
Soc, 25, 242 A873I.
55. Bert helot M., Compt. rend., 86, 71 A878); Bull. Soc. chim. France, [2], 33,
242, 246 A880); GlasstoneS., Hickling A., Electrolytic Oxidation and
Reduction, D. Van Nostrand Co., Inc., New York, 1936, p. 258.
56. E 1 b s K., S с h б n h e г г О., Z. Elektrochem., 1, 417, 468 A895); MullerE.,
F r i e d b e r g e r O., Z. Elektrochem., 8, 230 A902); Marshall H., J. Soc
Chem. Ind., 16, 396 A897).
57. Re i с h e 1 W., Ober die elektrolytische Darstellung der Oberschwefelsaure und deren
Oberfuhrung in Wasserstoffsuperoxyd, Fuller J., Munchen, 1912.
58. В 6 t t g e г R., J. Chem. Soc, 36, 103 A879); StruveH.J. Chem. Soc, 25, 35
A872); Sch ul 1 er A., Ann. Phys. Chem., 251, 289 A882); [J. Chem. Soc, 42,
691 A882I; Traube M., герм. пат. 27163 A883); J. Soc Chem. Ind., 3, 496
A884); Ber., 18, 1894 A885), J. Soc Chem. Ind., 4, 668 A885).
59. H а г с о u г t A. V., J. Chem. Soc, 22, 145 A869).
60. S с h 6 n e E., Ann. Chem. Pharm., 192, 257 A878) [J. Chem. Soc, 34, 931 A878)].
61. В e r t h el о t M., Bull, soc chim. France, [2], 34, 78 A880), [J. Chem. Soc, 40,
16 A881)].
62. Man n, Chem. Ztg., 12, 857 A888), [J. Soc Chem. Ind., 7, 640 A888)].
63. К i n gze t t С. Т., J. Soc Chem. Ind., 9, 3 A890).
64. Moody H. R., S. B. Thesis in Chemistry, Massachusets Institute of Technology,
A892); T a 1 b о t H. P., M о о d у H. R., Technology Quart., 5, 123 A892); J. Soc.
Chem. Ind., 12, 780 A893).
65. H a n г i о t A. A. M., Compt. rend., 100, 57, 172 A885).
66. W о 1 f f e n s t e i n R., Ber., 27, 3307 A894) [J. Soc Ch.em. Ind., 14, 505 A895I.
67. К e b 1 e r L. F., Warren L E., Ruddiman E. A., Bureau of Chemistry,
U. S. Department of Agriculture Bulletin 150, Technical Drug Studies, Government
Printing Office, Washington, 1912, pp. 5—23.
68. von Schrotter A., Ber., 7, 980 A874).
69. С a m e г о n С. A., Chem. News, 33, 16 A876).
70. Hof m an n A. W., Chem. News, 32, 286 A875); 33, 3 A876); J. Franklin Inst.,
101, 212, 262 A876).
71. В о u r g о u g n о n A., J. Am Chem. Soc, 12, 64 A890).
72. Lange O., Chemisch-Technische Vorshriften, Otto Spamer, Leipzig, 1923—1924.
73. U. S. Department of Commerce, Bureau of the Census, Twelfth Census of the U. S.,
Vol. 10, Government Printing Office, Washington, 1902, p. 570.
74. U. S. Department of Commerce, Bureau of the Census, Thirteenth Census of the D. S.,
Vol. 10, pp. 537, 539, 544 A913); Fourteenth Census of the U. S., Vol. 10, p. 629
A923); Fifteenth Census of the U. S., Vol. 2, pp. 654, 655 A929); Sixteenth Census
of the U. S., Vol. 2, p. 830 A939); Census of Manufactures, Vol. 2, Government Prin-
Printing Office, Washington, 1947, p. 388.
75. Stanford Research Institute, Handbook of Chemical Economics, Stanford, 1950; re-
revised Dec, 1953.
76. Chem. and Eng. News, 30, 1215 A952).
77. Brewer J. S., J. Am. Pharm. Assoc, I, 1002 A912).
78. Chem. and Eng. News, 29, 1996 A951).
79. Chemical Week, 71, № 18, Part 2, p. 373, Nov. 1, 1952.
80. H а у n e s N. W., American Chemical Industry, D. Van Nostrand Co., Inc , New
York, 1945.
81. De Sondal o, Compt. rend., 15, 647 A842); 17, 820 A843).
82. R i с h а г d s о n B. W., Trans. Med. Soc. Londoji, 2, 51 A862); The Asclepiad [21,
Vol. 8, Longmans, Green & Co., London, 1891, pp. 1, 101, 201, 301; Lancet, I, 707,
760 A891).
83. ChevreulM. E., Compt. rend., 55» 737 A862) [Phil. Mag., [4], 24, 526 A862)].
84. T e s s i ё d u M о t а у С, Bull. Soc d'Encouragement, [2], 14, 472 A867); Zeit.
ges. Naturwiss., 31, 149 A868); Dingl. polyt. J., 186, 230 A867); Tessie du
Литература 33
М о t а у С, М а г е с h a I R., Les Mondes, 14, 95 A867), Dingl. polyt., J., 184,
524 A867).
в5. E b e 1 1 P., J. Chem. Soc, 42, 1245 A882); Chem. News, 45, 71 A882); [J. Soc. Chem.
Ind. 1, 65 A882)]; 2, 361 A883); 7, 120, 207 A888); GohringC. F., J. Soc. Chem.
Ind., 8, 612, A889); 9, 84 A890); D e 1 m а г t A., J. Soc. Chem. Ind. 7, 120 A888).
вб. Machu W., Das Wasserstoffperoxyd und die Perverbindungen, 2 Aufl., Springer
Verlag, Wien, 1951 [есть перевод первого издания с дополнениями: см. Маху В.,
Перекись водорода и перекисные соединения, Госхимиздат, М., 1951.
87. KauschO., Das Wasserstoffsuperoxyd, Wilhelm Knapp, Halle, 1938; Lithoprinted
by Edwards Brothers, Inc., Ann. Arbor, Michigan, 1943.
88. M a s о n A. H., J. Chem. Soc, 40, 474 A881).
89. Koller Т., Das Wasserstoffsuperoxyd, Sammlung gemeinverstandlicher wissen-
schaftlicher Vortrage, 7, n. s., № 162 A892).
90. Neumark A. S., Sci. Am. Suppl., 75, 182, 200, 221 A913).
91. Rei chert J. S., Chem. Eng. News, 21, 480 A943).
92. SI at er V. W., Chemistry and Industry, 1945, 42.
93. Шенли Э., Гринспэн Ф., «Высококонцентрированная перекись водорода»
в сб. «Физика и химия реактивного движения», № 2, Госиноиздат, М., 1949, стр. 161.
94. Shanley E. S., J. Chem. Ed., 28, 260 A951).
95. М i 1 a s N. A., in К i г k-0 t h m e r «Encyclopedia of Chemical Technology^
Vol. 7, Interscience Encyclopedia, New York, 1951, p. 727.
96. M i t t e a u R., Chimie et industrie, 63, 138 A950).
97. Y о s t D. M., Russell H., Systematic Inorganic Chemistry, Prentice-Hall, Inc.,
New York, 1944.
98. BretschgerM. E., Shanley E. S., J. Electrochem. Soc, 99, 311c A952);
Lowenstein L., Chimia, 5, 79 A951).
99. Index Catalog of the Library of the Surgeon General's Office, U. S. Army, Government
Printing Office, Washington. 1st series, 6, 564 A885); 2nd series, 7, 519 A902);
3rd series, 6, 875 A926).
100. L a t i m e r W. M., The Oxidation States of the Elements and Their Potentials in
Aqueous Solutions, Prentice-Hall, Inc., New York, 1938, p. 41 [есть перевод вто-
второго издания: см. Латимер В. М., Окислительные состояния элементов и их
потенциалы в водных растворах, Издатинлит, М., 19541.
101. Von Antropoff A., Chem. Ztg., 35, 670 A911) (СА 5, 3662).
102. Sperber J., Schweiz. Apoth. Ztg., 53, 717 A915) [CA 10, 1308].
103. J о r i s s e n W. P., В a s s e t t H., D a m i e n s A., F i с h t e г F., RerayH.,
J. Am. Chem. Soc, 63, 889 A941).
104. Murray J. A. H., ed., A New English Dictionary, Vol. 7, Clarendon Press, Oxford,
1905, pp. 667, 714.
105. Thomson Т., A System of Chemistry, Bell and Bradfute, Edinburgh, 1804.
106. Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,1
Vol. 1, Longmans Green & Co., London, 1922, p. 118.
107. Chemical Abstracts, 39, 5954 A945).
108. Crane E. J., Chem. Eng., News, 30, 4514 A952).
109. Yost D. M., J. Am. Chem. Soc, 48, 152 A926).
110. E v e г e t t A. J., M i n k о f f G. J., Trans. Faraday Soc, 49, 410 A953).
111. Mil as N. A., in Kirk-Othmer, «Encyclopedia of Chemical Technology», Vol. 10,
Interscience Encyclopedia, Inc., New York, 1953, p. 58.
112. T о b о 1 s k у A. V., M e s г о b i a n R. В., Organic Peroxides, Interscience Pub-
Publishers, Inc., New York, 1954.
113. Long L., Chem. Revs., 27, 437 A940).
Перекись водорода
Глава 2
ОБРАЗОВАНИЕ ПЕРЕКИСИ ВОДОРОДА И СПОСОБЫ ЕЕ ПРОИЗВОДСТВА
/. ПРЯМОЕ ПОЛУЧЕНИЕ ИЗ ВОДЫ ИЛИ КИСЛОРОДА
Примерно в течение 60 лет после открытия перекиси водорода она остава-
оставалась лабораторной редкостью. Ее получали в небольших количествах по реак-
реакции перекиси бария с кислотой. Введение в Европе в 1879 г. процесса произ-
производства кислорода из перекиси бария по Брину способствовало зарождению
производства последней в промышленных масштабах; к концу последующего
десятилетия в Европе, Великобритании и США уже выпускали небольшие
количества перекиси водорода в концентрации 3 вес.% по реакции перекиси
бария с фосфорной, фтористоводородной, кремнефтористоводородной или
другими кислотами. Эта перекись поступала в продажу в качестве средства для
отбелки шелка, волос, перьев, шерсти и т. д., а также как дезинфицирующее
средство для обработки ран.
В 1878 г. Вертело сделал открытие, что электролизом растворов серной
кислоты можно получать пероксодисерную кислоту, которая легко подвер-
подвергается гидролизу в растворе с образованием перекиси водорода и серной кис-
кислоты. В 1885 г. Анрио показал, что перекись водорода можно выпарить из
гидролизованного раствора, если поддерживать достаточно низкую темпера-
температуру при работе под уменьшенным давлением. Эти открытия привели к тому,
что в 1909 г. было начато промышленное производство перекиси водорода элек-
электрохимическими методами [1], которые позволяли получать сравнительно
чистую и, следовательно, весьма устойчивую перекись водорода значительно
более высокой концентрации, чем раньше. Эти методы почти полностью вытес-
вытеснили способ производства из перекиси бария, который в настоящее время при-
применяется в сравнительно небольшом масштабе лишь там, где имеется на рынке
возможность сбыта получающегося в качестве побочного продукта сернокис-
сернокислого бария. В настоящее время перекись водорода получают главным образом
электрохимическими методами через пероксодисульфат, однако в США недав-
недавно начато промышленное производство перекиси водорода, основанное на само-
самоокислении органических веществ, а некоторые другие методы изучены с точки
зрения потенциальной возможности промышленного применения и доведены
по меньшей мере до стадии опытной установки.
Перекись водорода, возможно, является единственным продуктом, к об-
образованию которого ведет такое большое количество разнообразных реакций.
Это можно приписать тому, что она занимает промежуточное положение между
состояниями окисления кислорода в воде и в молекулярном кислороде, а также
широкому распространению окислительно-восстановительных реакций с уча-
участием кислорода. Значительное число таких реакций подвергалось в разное
время серьезному изучению с точки зрения возможного их использования для
промышленного получения перекиси водорода. Поскольку для понимания
производственных процессов необходимо знакомство с химическими реакция-
реакциями, положенными в основу этих процессов, мы рассмотрим технологию произ-
производства и основной химизм процесса одновременно, причем весь материал рас-
расположим в соответствии с характером затронутых химических реакций.
Образование перекиси водорода из воды термическим путем 35
Реакции, по которым образуется перекись водорода, удобно разделить на
две группы. В данной главе рассматривается первая группа, а именно те
реакции, в которых перекись водорода образуется непосредственно из воды
или кислорода за счет термических, фотохимических и электрохимических про-
процессов, явлений в электрическом разряде и т. п . Вторая группа реакций, кото-
которой посвящена гл. 3, охватывает те реакции, в которых перекись водорода
образуется из других перекисных соединений, например неорганических пере-
перекисей и пероксодисульфатов.
ОБРАЗОВАНИЕ ПЕРЕКИСИ ВОДОРОДА ИЗ ВОДЫ ТЕРМИЧЕСКИМ ПУТЕМ
Небольшие количества перекиси водорода были получены в результате
нагревания воды и кислорода до высоких температур с последующим резким
охлаждением, например путем пропускания смесей водяного пара и кисло-
кислорода с высокой скоростью через сильно нагретые капилляры, изготовленные на
окиси магния, или сжиганием лент магния и направлением пламени на лед [2].
Образование паров перекиси водорода из водяного пара и кислорода яв-
является сильно эндотермической реакцией; по наиболее точным современным
термодинамическим данным, образованию перекиси водорода соответствуют
^298=25,26 ккал/г-моль и ^^98 = 29,38 ккал/г-моль. Хотя образованию пере-
перекиси водорода по этой реакции благоприятствуют высокие температуры и
давления, все же, например даже при 1500° К и 1000 am, смесь, содержащая
сначала 1 моль воды на 0,5 моля кислорода, образует лишь 2,1 • 10"*4 моля пере-
перекиси водорода в равновесных условиях. Таким образом, очевидно, что пере-
перекись водорода, полученная из воды, в опытах, подобных описанному, возни-
возникает за счет ряда реакций, протекающих в условиях, далеких от равновесия.
Начальной стадией, безусловно, является диссоциация молекулы воды, за
которой следуют реакции с участием свободных радикалов, аналогичные неко-
некоторым реакциям, встречающимся при взаимодействии водорода и кислорода.
В недавно выданном французском патенте [3] описывается процесс обра-
образования перекиси водорода из водяного пара и кислорода при давлениях в не-
несколько тысяч атмосфер и температуре 3500—4000°. Предлагается создавать
такие условия быстрым адиабатическим сжатием газов с последующим очень
быстрым охлаждением путем расширения для предотвращения разложения
продукта. Однако в патенте не приводится никаких экспериментальных данных,
и эта идея, по-видимому, не получила практического осуществления. В ука-
указанных условиях возможно равновесное существование значительных коли-
количеств перекиси водорода, но процесс этот связан с очень большими техниче-
техническими трудностями.
ТЕРМИЧЕСКАЯ РЕАКЦИЯ ВОДОРОДА И КИСЛОРОДА
Беттгер [4] в 1878 г. обнаружил образование перекиси водорода при сжи-
сжигании водорода в кислороде. В 1884 г. Траубе [5] получил патент на производ-
производство перекиси водорода путем введения воды в пламя, образующееся при сжи-
сжигании окиси углерода, водорода, водяного газа, светильного газа и т. п. В по-
последующие два десятилетия несколько других исследователей показали, что
для получения перекиси водорода в ощутимых количествах необходимо быстро
охладить газообразные продукты горения, например путем направления пла-
пламени в воду или в лед.
Нельзя было дать полного объяснения полученных результатов до тех
пор, пока значительно позже не были получены точные представления о соот-
соответствующих химических равновесиях, связанных с этим процессом. Выше
показано, что при установлении полного равновесия между кислородом, водо-
водородом, водой и перекисью водорода при любых сочетаниях температуры и дав-
36
Глава 2
ления, технически возможных в настоящее время, количество присутствующей
перекиси водорода исчезающе мало. Однако реакция
С>2 газ — *
A)
экзотермическая, и если бы удалось подавить реакции, ведущие к образованию
воды, то в равновесии с водородом и кислородом могла бы существовать перекись
водорода высокой концентрации. Степень такого превращения показана на
рис. 4 (для построения кривых использованы термодинамические данные гл. 5).
На этом графике представлен процент превращения исходной смеси эквимо-
эквимолекулярных количеств водорода и кислорода в перекись водорода (в условиях
равновесия в зависимости от темпе-
температуры для трех различных общих
давлений). Очевидно, что если бы
удалось предотвратить возможность
образования воды, то практически
все количество водорода и кислоро-
кислорода перешло бы в состоянии равно-
равновесия в перекись водорода пример-
примерно при температурах ниже 900° К;
если же присутствует вода, то в рав-
равновесии с ней могут существовать
лишь ничтожные количества пере-
перекиси водорода.
При реакции водорода и кисло-
кислорода образуется перекись водорода,
очевидно в форме сравнительно не-
неустойчивого промежуточного про-
продукта, как обычная переходная ста-
стадия для целого ряда реакций горе-
горения. Однако, когда достигается со-
состояние полного равновесия с дру-
другими видами присутствующих моле-
молекул, перекись водорода в заметной концентрации существовать не может.
Термическая реакция водорода и кислорода была изучена весьма подробно,
в частности, Хиншелвудом с соавторами [6] в Англии и Льюисом и фон-Эльбе
17] в США*. Общая реакция очень сложна, так как она связана с образованием
и исчезновением такого рода химических частиц, как Н, О, ОН, Н0.2 и Н2О2,
за счет большого числа совместных и консекутивных реакций; относительное
значение каждой из них сильно зависит от температуры, давления, соотноше-
соотношения количеств водорода и кислорода, конфигурации реактора при проведении
опыта и физической и химической природы тех поверхностей, которые сопри-
соприкасаются с реагирующей смесью. Механизм реакции при очень низких давле-
давлениях, по-видимому, установлен достаточно хорошо. Однако для высоких давле-
900 W00 1100 1200
Температура, °К
1300
Рис. 4. Вычисленная конверсия эквимоле-
эквимолекулярной смеси водорода и кислорода в па-
пары перекиси водорода в равновесных усло-
условиях (при отсутствии образования воды).
* Крупные исследования по изучению термической реакции водорода и кислорода
выполнены в Советском Союзе академиком Н. Н. Семеновым и его учениками (С е м е-
н о в Н. Н., Цепные реакции, Л., Госхимтехиздат, 1934; Семенов Н. Нм О некоторых
проблемах химической кинетики и реакционной способности, М., 1954). Академику В. Н.
Кондратьеву принадлежит экспериментальное доказательство образования радикалов
ОН как промежуточного продукта в реакциях окисления (Кондратьев В. Н.,
Свободный гидроксил, М., ГОНТИ, Ред. хим. лит., 1939). Обобщение материалов по воп-
вопросу окисления водорода произведено в монографии «Механизм окисления и горения
водорода», М.—Л., Изд. АН. СССР, 1949, АН СССР, где авторы Налбандян А. Б. и
Воеводский В. В. подробно описывают работы школы Семенова и Хиншелвуда.—
Прим. ред.
Термическая реакция водорода и кислорода 37
ний относительное значение чередования различных возможных реакций
выяснено еще не полностью.
Предложенные механизмы реакции основаны на многочисленных иссле-
исследованиях, в которых различные реакции инициировались фотохимически
или путем электрического разряда, на истолковании спектров испуска-
испускания и поглощения отдельных химических частиц и реакционных смесей, а
также объяснении в различных исследованиях пределов взрыва водорода и кис-
кислорода и измерений скорости реакции вблизи этих пределов—весь этот мате-
материал рассматривался в свете энергетических соотношений и энергий актива-
активации отдельных ступеней реакции. Сначала рассмотрим характеристику преде-
пределов взрыва совместно с совокупностью отдельных ступеней реакций, которым
придается существенное значение. Затем перейдем к частным исследованиям
термической реакции, в которых основное внимание уделялось образованию
перекиси водорода. Ниже рассмотрено образование перекиси водорода из
воды или элементов при возбуждении термическими, электрическими, фото-
фотохимическими или радиохимическими средствами. Более подробный анализ
общей реакции водорода с кислородом и пределов взрыва можно найти у Лед-
лера [8], а также в вышеприведенных ссылках.
Пределы взрыва смесей водорода с кислородом
Смеси водорода с кислородом в широком интервале концентраций само-
самопроизвольно взрываются при температурах газа несколько выше 600° и давле-
давлении не ниже 1 мм рт. ст. При температурах ниже 400° скорость реакции очень
мала и взрыва не происходит, если не возбудить его действием внешнего ис-
источника, например электрической искры или раскаленной проволоки. Напри-
Например, в стехиометрической смеси из 2 молей водорода и 1 моля кислорода при
температуре 550° и давлении 400 мм рт. ст. будет проходить гомогенная реак-
реакция с постоянной измеримой скоростью; если же давление повысить или пони-
понизить, то произойдет взрыв. Так, если происходит постепенное изотермическое
понижение давления, то скорость реакции в течение некоторого времени будет
падать, но при достижении критического давления около 100 мм рт. ст. взрыв
смеси произойдет самопроизвольно. Давление, при котором это происходит,
носит название второго, или верхнего, предела взрыва. Если ту же начальную
смесь при температуре 550° постепенно подвергать изотермическому сжатию',
то скорость реакции будет медленно возрастать и опять-таки произойдет самот
произвольный взрыв при давлении около 1000 мм рт. ст.; этот предел известен
под названием третьего предела взрыва. Аналогично та же начальная смесь
при 550°, но очень низком давлении будет медленно реагировать до тех пор,
пока давление не повысится примерно до 1 мм рт. ст.; при этом давлении дости-
достигается первый предел взрыва. Эти пределы самовоспламенения могут несколько
колебаться в зависимости от соотношения компонентов в смеси и природы сосу-
сосуда, в котором происходит реакция. Общее изменение пределов с температурой
и давлением показано на рис. 5. Очевидно, что существование второго предела
взрыва* нельзя объяснить термическим взрывом, так как в случае термического-
* Термин «предел взрыва» применяется в научной и технической литературе в двух
различных смыслах. В одном смысле (принятом и здесь) он представляет собой те предель-
предельные сочетания давления и температуры, при которых газ данного состава самопроизволь-
самопроизвольно еще не взрывается. В другом смысле он выражает пределы состава, вне которого не
может распространяться взрыв, инициированный внешним источником, например искрой
или пламенем. Последняя концепция иногда носит название «пределы взрывчатых соста-
составов» или «пределы воспламенения», и в дальнейшем мы пользуемся именно этими терминами
во избежание путаницы. Пределы воспламенения несколько изменяются с температурой
и давлением; повышение температур и давлений обычно (но не всегда) расширяет область
взрывчатых составов. Пределы воспламенения для смесей кислорода и водорода составля-
составляют 9,2 и 91,6% водорода при комнатной температуре и атмосферном давлении [9].
38
Глава 2
взрыва минимальная температура самовоспламенения—это та температура,
при которой скорость образования тепла за счет гомогенной реакции только
незначительно превышает скорость потери тепла. В связи с этим увеличение
давления, которое не может заметно влиять на скорость переноса тепла, но
резко повышает скорость реакции, должно способствовать падению темпера-
температуры самовоспламенения.
Для объяснения этих пределов взрыва, а также других наблюдений было
постулировано, что реакция водорода с кислородом происходит с образова-
образованием свободных атомов и радикалов (известных под названием активных цент-
центров цепи) в качестве промежуточных продуктов; эти продукты образуются и рас-
расходуются в простых реакционных стадиях, в которых участвуют одновременно
600
i
I500
1
400
\
\
\
Первый -"*\
(нижний) \
предел \
взрыва >
i
Область
взрыва
/ Третий ^у.
/ предел \
/ взрыва \
/ \
/~— Второй (верхний)
/ предел взрыва
Область медленной реакции
Ю 100
Давление, мм рт. ст.
W00
Рис. 5. Пределы взрыва в системе водород—кислород.
один или два вида частиц, а иногда—при более высоких давлениях—и три.
Если происходит значительное развитие реакций с образованием более
чем одного атома или радикала на каждый израсходованный атом или ра-
радикал (разветвление цепей), то это приводит к самоускоряющейся реакции и
взрыву.
Количественный анализ реакций, происходящих при взаимодействии водо-
водорода и кислорода, может быть сильно упрощен, если ограничиться условиями
для стационарного состояния реакции и исходить из предположения, что в этих
условиях активные центры цепи присутствуют в постоянных, но небольших
концентрациях. Скорости отдельных частных реакций, включая диффузию
к стенкам и последующую реакцию на них, могут быть выражены в виде про-
простых уравнений в соответствии с законом действия масс. Затем эти уравнения
решают совместно, приравнивая результирующую скорость образования
и исчезновения промежуточных продуктов нулю и исключая из уравнений
концентрацию этих продуктов. При этом получается выражение для скорости
образования продукта в виде функции от констант скорости частных реакций
и концентраций исходных реагирующих компонентов.
При тех переменных рабочих параметрах, при которых скорость образова-
образования продукта стремится к бесконечности, должен произойти взрыв. Путем пред-
предположения различных реакционных схем и сравнения предварительно вычис-
вычисленных пределов взрыва с экспериментально найденными можно исключить
из рассмотрения отдельные схемы и прийти к одной или нескольким возмож-
возможным ступеням реакции для каждой группы с общностью экспериментальных
условий. Дальнейшие сведения можно получить в результате анализа харак-
характеристики реакций вблизи предела взрыва, а также других типов иссле-
исследований, о которых говорилось выше.
Термическая реакция водорода и кислорода
Отдельные стадии термической реакции водорода с кислородом
В табл. 1 приведены реакции, которые, как следует из результатов раз-
различных исследований, оказывают существенное влияние на общую реакцию
водорода с кислородом. Там же указаны изменения энтальпии для того числа
молей, которое приводится в каждом уравнении, и энергии активации. Зна-
Значения последних определены или оценены приблизительно лишь для отдель-
отдельных случаев. Величины энтальпии установлены достоверно и, вероятно, точны
Таблица 1
Возможные стадии термической реакции водорода с кислородом
Реакция
Изменение
энтальпии
Энергия
активации
ЕА 9к
Возможные реакции зарождения цепи:
B)
Н2О2+М —>20Н+М
Н2+М-->2Н+М C)
Н2+О2->НО2+Н D)
Двух компонентные реакции в газовой фазе:
ОН+Н2—>Н2О+Н E)
Н+О2 —>ОН+О F)
О+Н2 —>ОН+Н G)
HO2i-H2O2 —» Н2О+О2+ОН (8)
НО2+Н2О —» Н2О2+ОН (9)
НО2+Н2 —>Н2О2+Н A0)
Трехкомпонентные реакции в газовой фазе
<М—любое третье тело):
НО2+Н2+М-*Н2О+ОН+М (Ц)
Н+Н2О2+М —»Н2О+ОН+М A2)
Н+О2+М —»НО2+М A3)
Реакции на поверхности:
2НО* поверхность^ Н2О2+О2 A4)
2Н2О2
2Н2О+О2 A5)
-32,4
+52,6
+ 104,2
+58
-15,8
+ 17,8
+2,3
-21
+29
+ 14
-54
-67,4
-46
-45
-50,7
~,7
-20
7±2
~7
-5
в пределах 1—2 ккал/моль для всех видов частиц, кроме НО2. Указанные здесь
данные взяты из гл. 5. Изменение энтальпии для любой реакции немного ме-
меняется с температурой. Здесь приведены данные для 25°. Безусловно, трудно
определить значения энергии активации, причем точность их обычно не извест-
известна. Величины, приведенные здесь, взяты у Иоста [9], и большинство их первона-
первоначально опубликовано Гейбом [10]. Величины, указанные для реакции A2), отно-
относятся к двойному удару. Реакции A1), A2) и A3) настолько экзотермичны, что
продукты их могут существовать в лучшем случае как переходные состояния,
если только нет третьего тела, которое могло бы отнять часть выделяющейся
энергии. Реакции E) | A0) при повышенных давлениях, конечно, могут про-
проходить и в присутствии третьего тела. ^Я
40 Глава 2
Обычно принимается, что при давлениях ниже первого предела взрыва
(около 0,1—5 мм рт. ст.) горение происходит за счет реакций E), F) и G),
сопровождающихся диффузией радикалов О, Н и ОН к поверхности сосуда,
где они инактивируются. Поскольку диффузия к стенкам является причиной
обрыва цепей реакции, давления первого предела взрыва обратно пропорцио-
пропорциональны диаметру сосуда, а инертные газы, предотвращающие диффузик>
активных центров цепи к стенкам, увеличивают взрывчатость смесей. Нет ника-
никаких доказательств, что при этих очень низких давлениях гомогенно образуется
пергидроксильный радикал или перекись водорода, хотя, как показано ниже,
такое образование возможно при введении в систему значительных количеств
атомарного кислорода. Перекись водорода может образоваться гетерогенно даже
при этих низких давлениях за счет конденсации радикалов ОН на стенках,
если стенки охлаждены ниже —180° (см. ниже).
При более высоких давлениях (выше второго предела взрыва—примерно
от Юдо 100 мм рт. ст.) увеличивается вероятность тройных столкновений и при-
приобретает значение реакция A3), которая, по-видимому, требует участия третье-
третьего тела для отвода части выделенной энергии. В дальнейшем, как видно, воз-
возможны различные пути исчезновения образовавшегося таким образом радикала
НО2 точно т;ак же, как и различные пути возникновения и разложения пере-
перекиси водорода. При некотором давлении, лежащим между вторым и третьим*
пределами, рост температуры благоприятствует увеличению роли реакций F)
и G), способствующих разветвлению цепей, по сравнению с нормальными цеп-
цепными реакциями, которые рассмотрены выше. Имеющихся в настоящее время
экспериментальных данных пока недостаточно для того, чтобы для любого
ряда условий точйо указать относительное значение различных комбинаций
этих реакций. Однако очевидно, что при более высоких давлениях (прибли-
(приближающихся к третьему пределу взрыва—приблизительно выше 400 мм рт. ст.>
продолжает возрастать значение реакции A3), а роль реакций, для которых
имеет значение диффузия к поверхности сосуда, падает по сравнению с реак-
реакциями, включающими прямые столкновения. Способ инициирования термиче-
термической реакции продолжает оставаться предметом умозрительных рассуждений.
Хиншелвуд считает, что инициирование происходит за счет небольшого коли-
количества атомарного водорода, образовавшегося путем столкновений молекул,
тогда как Льюис и фон-Эльбе предполагают, что это инициирование обусло-
обусловлено разложением перекиси водорода, образовавшейся каким-то не известным
еще путем. С другой стороны, изменение энтальпии в реакции D) позволяет
считать ее возможной ступенью инициирования.
Образование перекиси водорода при термической реакции водорода
и кислорода
Подытожим сначала экспериментальные результаты, опубликованные
до настоящего времени, а затем проанализируем эти данные в свете возмож-
возможных механизмов реакции. Одно из первых количественных исследований обра-
образования перекиси водорода по реакции водорода с кислородом принадлежит
Пизу [11], который непрерывно пропускал смесь этих газов при атмосферном
давлении через трубку, нагретую до 550°, и затем в сосуд, охлажденный до
—79°; при этом были получены продукты, содержащие несколько процентов
перекиси водорода. В дальнейших опытах этот автор в течение короткого перио-
периода времени нагревал сферические сосуды диаметром 4,3 см, содержащие 95%
водорода и 5% кислорода, после чего быстро охлаждал их до —79°. Продукты
содержали до 25 вес.% перекиси водорода. При давлениях ниже атмосферного,
но лежащих в интервале пределов взрыва^образования перекиси водорода от-
отмечено не было.
Термическая реакция водорода и кислорода 41
Позже Поляков с соавторами [12] опубликовал ряд сообщений по образо-
образованию перекиси водорода при взрывах водорода с кислородом в неболь-
небольшом стеклянном реакционном сосуде, погруженном в жидкий воздух. Анало-
Аналогичные исследования были проведены и Э^ертоном и Минкофом [13]. Как ока-
оказалось в этих обеих работах, выход перекиси водорода, выраженный в виде
процента перекиси водорода в конденсате, зависел от большого числа перемен-
переменных, в том числе от природы ютенки сосуда, температуры стенки, состава газа,
давления, формы сосуда, метода смешения газов и количества продукта, вымо-
вымороженного на стенке. В работах Полякова на выход перекиси водорода лишь
слабо влияли температура платиновой проволоки, использовавшейся для вос-
воспламенения смеси (несмотря на значительное варьирование этой температуры),
а также продолжительность индукционного периода, предшествовавшего взры-
взрыву, хотя этот период заметно колебался с изменением температуры проволо-
проволоки. Для инициирования взрывов в сосудах небольшого диаметра и при боль-
большом содержании водорода в газовых смесях требовалась более высокая темпе-
температура проволочки, применявшейся для воспламенения. В некоторых опытах
проволочку нагревали до сравнительно низкой температуры; в данных услох
виях смесь газов реагировала медленно, без взрыва. В этих случаях колебач
ния выхода перекиси водорода в зависимости от изменения условий опыта были
аналогичны колебаниям, наблюдавшимся при взрывах, но самые выходы были
раз в 10 меньше. Таннер [14] также нашел, что выход перекиси водорода в ана-)
логичной установке не зависит от того, производится ли инициирование реак-
реакции искрой или нагретой поверхностью катализатора. Таннер указал также,;
что в присутствии паров тетраэтилсвинца значительно повышается выход пере^
киси водорода и снижается интенсивность взрыва. . \ ,
Поляков проводил опыты в сферических стеклянных сосудах диаметром;
25, 35 и 45 мм при общих давлениях от 60 до 120 мм рт. ст. и молярных отно-
отношениях водорода к кислороду от 1 до 4—5, причем более высокое отношение
представляет тот максимум, при котором еще возможен взрыв. До введения
в реакционный сосуд оба газа пропускали над хлористым кальцием и смеши-
смешивали. В каждом сосуде выход перекиси водорода возрастал с падением давле-
давления; он достигал максимума при отношении водорода к кислороду примерно
2,5—4,0. В общем при более низких давлениях максимальные выходы наблю-
наблюдались для меньших отношений водорода к кислороду. Выходы возрастали
с уменьшением диаметра сосуда, причем при диаметре сосуда 25 мм был отме*
чен максимум, составлявший около 3% перекиси водорода. При низких отно-
отношениях водорода к кислороду в конденсате обнаружен озон, хотя авторы не
указывают метода, использованного ими для анализа. Образованием озона
можно объяснить низкий выход перекиси водорода, отмеченный для этих газо-
газовых смесей, так как перекись водорода и озон могут быстро реагировать друг
с другом. Показано также, что если окружить зажигательную проволоку пла-
платиновой сеткой, то на охлажденных стенках сосуда совсем не образуется пере-
перекиси водорода.
В другой серии опытов инициировались последовательные взрывы в ци-
цилиндрических трубках из эмалированного металла диаметром 10—35 мм и
длиной 80—300 мм. В этом случае для инициирования реакции предпочитали
прибегать не к раскаленной проволоке, а к искре. В результате^пропускания
детонационной волны через узкую трубку диаметром 2—3 мм перекись не полу-
получалась. Однако в более широких трубках отмечены существенные выходы пере-
перекиси. Обнаружен значительный разброс данных, но все же можно отметить
следующую тенденцию: так же как и в сферических сосудах, выходы перекиси
водорода возрастают с понижением давления от 150 до 50 мм для любой из взя-
взятых трубок и падают с увеличением диаметра сосуда при постоянной длине.
Эгертон и Минкоф исследовали реакцию водорода с кислородом самыми;
различными методами, в^том числе с применением однократных и многократ-
42 Глава 2
ных взрывов и динамических опытов с непрерывным искровым воспламе-
воспламенением. В опытах с однократным взрывом применяли трубку из стекла пирекс
длиной 130—140 мм и диаметром 14 мм. Методика была аналогичной методике
Полякова, за исключением того, что сосуд, который в момент взрыва находился
в среде жидкого воздуха, нагревали до комнатной температуры после каждого
отдельного взрыва и количество образовавшейся перекиси водорода опреде-
определяли колориметрически путем сравнения с эталонами, применяя в качестве
реактива треххлористый титан. Выходы перекиси водорода были в общем не-
несколько выше, чем указанные Поляковым, но и для них обнаружены та же
склонность к максимальному выходу при отношении водорода к кислороду
около 2,5 и увеличение выхода с падением давления примерно до 40—30 мм
рт. ст. Однако дальнейшее снижение давления до 20 мм вызывало падение
выхода; аналогичный результат отмечен и Поляковым, который сообщает о
максимальном выходе при давлении около 45 мм рт. ст. в одной серии опытов,
проведенных в цилиндрическом сосуде 100x10 мм.
Добавка 5% аргона или азота к водородо-кислородной смеси с отношением
1,7 вызывала наибольшее снижение выхода при изученных давлениях B0—
•60 мм рт. ст.). Ряд опытов с последовательными взрывами, т. е. в соответствии
с методикой Полякова, показал, что выход падает с увеличением числа взры-
взрывов; это было приписано изолирующему влиянию конденсированной пленки
воды и перекиси водорода. Тот же изолирующий эффект был обнаружен и при
динамических исследованиях, в которых Эгертон и Минкоф пропускали
предварительно приготовленную смесь водорода и кислорода над непрерывной
искрой в цилиндрической трубке, погруженной в жидкий воздух. В оптималь-
оптимальных условиях начальные выходы 30—35%-ной перекиси водорода быстро
падали во времени до стационарной величины 10—15%, которая почти не зави-
зависела от толщины ледяной пленки.
В различных типах опытов покрытие стекла целлофаном, алюминием или
•обработка поверхности борной кислотой не изменяли выхода по сравнению
<: наблюдаемым на чистой поверхности стекла; покрытие стекла хлористым
калием снижало выход. Эти эффекты аналогичны хорошо известной активности
указанных поверхностей в отношении разложения перекиси водорода. Зави-
Зависимость выхода от изменения экспериментальных условий для динамических
опытов была в общем такой же, как и в исследованиях с единичным взрывом
по данным Полякова или Эгертона и Минкофа. Выход возрастал с уменьшением
диаметра трубки с 24 до 14 мм\ максимальные значения выхода установлены
при общих давлениях 40—60 мм рт. ст.; при 30 и 80 мм выходы были гораздо
ниже. Повышение температуры стенки с—180 до —79° вызвало падение выхода
-с 15 до 6%.
Эгертон и Минкоф [13] и Холт и Олденберг [15] попытались определить
присутствие перекиси водорода при реакции водорода с кислородом путем
ультрафиолетовой спектрофотометрии, но результаты были недоказательными.
Холт и Олденберг [15] исследовали непрерывную термическую реакцию при
температуре около 540° и давлении 1 am. Величина поглощения при 2537 А,
приписываемого перекиси водорода, как оказалось, сильно зависит от состоя-
состояния поверхности реакционной трубки из стекла пирекс; это состояние зависит
от возраста реактора и предыдущей обработки. В старой трубке, поверхность
которой стала шероховатой в результате большого числа проведенных опытов,
практически не наблюдалось никакого поглощения в условиях, которые в дру-
других случаях были для него благоприятными. При трубке с удовлетворительной
поверхностью поглощение возрастало с повышением отношения водорода
к кислороду.
Эгертон и Минкоф [13] сообщили о значительном ультрафиолетовом погло-
поглощении, наблюдавшемся при взрывах водорода с кислородом в условиях давления
ниже атмосферного. Это явление было приписано в то время образованию пере-
Термическая реакция водорода и кислорода 45
киси водорода. Однако в более поздних работах [16] проведено измерение погло-
поглощения при взрывах смесей метана или окиси углерода с водородом, а также
смесей водорода с кислородом. В каждой системе наблюдалось сильное погло-
поглощение при длине волны ниже 2800 А, причем поглощение возрастало с падением
длины волны. Это показывает, что какие-то другие виды химических частиц,
помимо перекиси водорода, также могут сильно поглощать в указанной обла-
области. Было высказано предположение, что поглощение может быть приписано
«горячему» кислороду.
Мак-Лейн [17] исследовал реакцию между перекисью водорода и водоро-
водородом при 470—500° и давлении 1 am и показал, что в присутствии водорода пере-
перекись водорода исчезает быстрее, чем в присутствии инертного газа, но зави-
зависимость скорости реакции от концентрации была сложной и не позволяла соста-
составить ясное представление о механизме реакции. Скорость реакции водородо-
кислородных смесей заметно увеличивалась от добавки небольших количеств
перекиси водорода в экспериментальных условиях, в которых обычно реак-
реакция имеет очень малую скорость.
В двух патентах, принадлежащих Куку [18], описывается образование
перекиси водорода путем пропускания смеси водорода с кислородом, в которой
содержание О2 меньше 10%, при 5—35am и 400—650° через трубку с наплавлен-
наплавленным покрытием из борной кислоты или боратов для замедления разложения
перекиси водорода. Экспериментальные условия в каждом случае подбирают
таким образом, чтобы температура была на несколько градусов ниже точки,
при которой происходит быстрое сгорание с образованием воды. Приведенные
примеры показывают, что в случае конденсации продукта при комнатной тем-
температуре в перекись водорода превращается 3—5% введенного в реакцию кис-
кислорода или 10—20% прореагировавшего. При добавке водяного пара в количе-
количестве от 1 до 40% выход перекиси водорода повышается; азот в количестве
до 25% оказывает ничтожное влияние; газообразные углеводороды и хлор
в небольших количествах заметно не влияют на основную реакцию, но всту-
вступают в побочные. Двуокись азота или другие окислы азота и окись углерода
мешают проведению реакции.
Механизм образования перекиси водорода при термической реакции]
одорода с кислородом^
Из анализа различных наблюдений становится очевидным, что при давле-
давлении ниже атмосферного и температуре стенки, соответствующей температуре
жидкого воздуха, часть перекиси водорода должна образоваться на стенках
реакционного сосуда, вероятно за счет вымораживания гидроксильных ради-
радикалов
он + он1^^-0™ н2о2- A6)
Имеется хорошее доказательство [19] того, что при пропускании электриче-
электрического разряда через водяной пар и конденсации продуктов разряда в холодной
ловушке перекись водорода возникает за счет реакции A6). Установлено также,
что этот процесс становится малоэффективным с повышением температуры
стенки и размеры его ничтожно малы, если температура стенки значительно
превышает—180°. Возможно также, что в реакциях водорода с кислородом
в известных условиях играет существенную роль реакция]
ил J иП повеРХНОСТ10 тт П Гг? /1А\
НО2 -f НО2 > Н2О2 + O2.j A4)
Однако образованием перекиси водорода из гидроксильных радикалов на стен-
стенке нельзя объяснить присутствие перекиси водорода в газовой фазе, а также
то, что выход перекиси проходит через максимум при давлении около
44 Глава 2
40 мм рт. ст. Эгертон и Минкоф [13], опубликовавшие превосходный анализ
вероятного механизма образования перекиси водорода, предлагают следующее
объяснение этого влияния давления на выход перекиси. Постулируется,
что НО3 образуется в активированном состоянии (в формуле обозначено
звездочкой) за счет прямой ассоциации Н и О2 при двойных столкновениях
-> НО*. A3а)
Хотя активированный радикал HOJ должен быть недолговечным, поскольку
он находится в возбужденном состоянии, все же не исключено, что в известных
условиях значительная доля этих радикалов может подвергнуться столкнове-
столкновениям и прореагировать до своего распада. При низких давлениях, вероятно,
большая часть этих радикалов распадется на ОН и О до столкновения с третьим
телом. Однако при более высоких давлениях и при наличии избытка водорода
излишек энергии в активированном радикале HOJ должен благоприятствовать
следующей реакции:
HO2* + H2->HaOt + H. A0a)
Перекись водорода может образоваться в активной форме, и поэтому, если
только энергия образования не отнимается холодными стенками, она распа-
распадается, вероятнее всего на два гидроксила. Постулируется возможность
образования более значительной доли стабилизированных радикалов НОа
За счет столкновений с другими молекулами, если происходит дальнейшее
увеличение давления. Тогда будет происходить рост доли НО2 вследствие
большого числа тройных соударений, приводящих к образованию стабилизи-
стабилизированного радикала НО2 по реакции
* НО2 + М. A3)
Поскольку реакция A0) эндотермическая, постулируется, что только не-
небольшая доля неактивированных радикалов НО2 будет исчезать по этой реак-
реакции, основная же масса их расходуется за счет таких экзотермических процес-
процессов, как
НОа + Н2-> Н2О + ОН A1)
или
НО2 + Н2О2 -* Н2О + О2 + ОН, (8)
что приводит к снижению концентрации перекиси водорода.
Не следует предполагать, что механизм с участием возбужденного HOJ
будет иметь существенное значение при давлениях, превышающих второй
взрывной предел, где приобретают большую вероятность тройные столкнове-
столкновения. Приведенной выше гипотезой объясняется большая часть наблюдений,
сделанных при низких давлениях; увеличение диаметра трубки вызывает уве-
увеличение времени, необходимого для того, чтобы молекулы перекиси водорода
достигли стенки, и, таким образом, снижает вероятность стабилизации моле-
молекул перекиси водорода; вместе с тем снижается и вероятность рекомбинации
гидроксилов на стенке. Остается неясным, почему тетраэтилсвинец повышает
количество образующейся перекиси водорода, как это установлено Таннером.
Возможно, что тетраэтилсвинец селективно забирает атомарный водород,
обладающий относительно высокой скоростью движения и, как это показал
Гейб [20], особенно быстро разлагающий перекись водорода.
При атмосферном или более высоком давлении взаимодействие, по-види-
по-видимому, идет через НО2 по реакции A3) с последующим образованием перекиси
водорода по реакции A0). В данном случае присутствует неактивированный
радикал НО2 ^необходимая энергия может быть получена за счет тройныхшстолк~
Термическая реакция водорода и кислорода 45
новений. Рост выхода перекиси водорода в присутствии водяного пара может
быть обусловлен эффективностью последнего в качестве третьего тела в реак-
реакции A3) или же тем, что пар способен образовать перекись водорода по реакции
(9), хотя эта реакция довольно эндотермическая. Не исключено также, что
влияние водяного пара обусловлено адсорбцией воды на поверхности трубки
(см., например, у Бэдина [21]), что может способствовать относительному
преобладанию реакции A4) над реакцией A5).
То, что НО2 обычно постулируется в качестве промежуточного продукта
при горении водорода и различных углеводородов, привело к постановке опы-
опытов по экспериментальному доказательству присутствия этого радикала. Пер-
Первое непосредственное доказательство было представлено Элтентоном [22],
который присоединил реакционную камеру к масс-спектрометру типа Демпсте-
ра и обнаружил определенный рост потока положительного иона с массой 33
в пламени смеси пропана и кислорода. Позже Фонер и Хадсон [23] привели
•безупречное доказательство образования пергидроксила в процессе смешения
•кислорода с инертным газом (при давлении в несколько сантиметров ртутного
столба) с атомами водорода, полученными в газовом разряде, и последующего
направления смеси в масс-спектрометр. При включении разрядной трубки
мгновенно возрастала интенсивность пика для массы 33, тогда как для масс 32
и 34 она оставалась практически неизмененной. Смесь находилась в зоне реак-
реакции приблизительно 0,01 сек., причем было установлено, что около 1% всех
атомов водорода переходило в НО2 в течение времени, которое нужно было для
того, чтобы газ попал в пробоотборное отверстие масс-спектрометра. Предпо-
Предполагается, что реакция образования происходит по уравнению A3), выражаю-
выражающему результат тройного столкновения. Обнаружены также заметные количе-
количества гидроксила и воды, что говорит в пользу протекания и реакции A1).
В более ранних работах Фонер и Хадсон [24], несмотря на применение той же
аппаратуры, не обнаружили НО2 в водородо-кислородном пламени, хотя они
проводили работы при давлениях до 0,5 am и с различными отношениями водо-
водорода к кислороду. В аналогичных исследованиях Робертсон [25] обнаружил,
что и НО2 и Н2О2 образуются в начальной стадии реакции атомов водорода
и молекулярного кислорода при давлении около 0,5 мм рт. ст. На этой стадии
реакции он не обнаружил образования Н2О, Н2О4, О3, ОН и О. Нортон [26]
также кратко сообщил об увеличении пиков, обусловленных массами 33 и 34,
при добавлении избытка водорода к кислороду. В 1933 г. Бэйтс [27] подсчи-
подсчитал, что средний период жизни пергидроксильного радикала составляет вели-
величину порядка 10"9 сек. (на основании исследования механизма реакции йоди-
йодистого водорода и кислорода, в которой в качестве промежуточного продукта
принимается без доказательств НО2).
Получение перекиси водорода путем термической реакции]
водорода и кислорода
Возможность промышленного производства перекиси водорода прямой
термической реакцией из элементов интересовала различных исследователей
в течение многих десятилетий, так как такого рода реакция представляет собой
простой прямой процесс, в результате которого можно получить чистый
высокоустойчивый продукт из сравнительно дешевых исходных материалов.
Был выдан ряд патентов на получение перекиси водорода путем сжигания
водорода и кислорода [28], причем для синтеза рекомендуется и охлаждение
до низких температур и применение катализаторов, например палладия. Од-
Однако, по-видимому, ни один из этих патентов не приобрел промышленного зна-
значения и реакция никогда не была осуществлена в промышленном масштабе;
объясняется это тем, что в экономически приемлемых условиях получаются
слишком малые выходы.
46 Глава 2
Факторами, которые особенно влияют на выход, являются характер по-
поверхности сосуда, общее давление, отношение водорода к кислороду и темпе-
температура, до которой охлаждаются продукты. Выше показано, что при очень
низких давлениях перекись водорода путем гомогенной термической реакции,
по-видимому, не образуется; выход ее равен нулю даже при максимально быст-
быстром охлаждении продуктов реакции до комнатной температуры. При повышении
давления в заметной степени возрастают размеры гомогенного образования
и возникает возможность получения перекиси водорода с небольшим выходом
путем охлаждения до комнатной температуры, причем выход возрастает при
работе в условиях давлений выше атмосферного.
При любых давлениях выход существенно возрастает при охлаждении
продуктов до температур значительно ниже комнатной, но этот повышенный
выход в промышленном масштабе неэкономичен из-за высокой стоимости низко-
низкотемпературного охлаждения. Перекись водорода может быть получена прак-
практически из любых смесей водорода и кислорода, но самые большие выходы,
по-видимому, получаются при применении большого избытка водорода. При
атмосферном или более высоком давлении желательно применять смесь водо-
водорода и кислорода, лежащую вне пределов взрывчатых составов (от 9,2 до 91,6%
водорода при 1 am), с целью уменьшения взрывоопасности. При давлении 1 am
и закалке водородо-воздушного пламени в воде выход перекиси водорода
составляет 0,5% количества взятого водорода. Замена воздуха кислородом
повышает выход до 2,5% [29].
В течение тридцатых годов электрохимический отдел фирмы «duPont»
исследовал непрерывную термическую реакцию при атмосферном давлении
и температурах 500—625°, для чего пропускали смеси, содержавшие 1—3%
кислорода в водороде или 1—3% водорода в кислороде или воздухе, через трубку
из стекла пирекс или через кварцевую трубку и промывали продукты водой
при комнатной температуре [30]. Как показано в других работах, ход реакции
весьма чувствителен к поверхностным эффектам. Избирательная способность
реагирующего кислорода к образованию перекиси водорода была максимальной
при наименьшем времени реакции, однако и при этом условии концентрация
перекиси водорода в газовой фазе была необычайно низка. Максимальная легко
воспроизводимая концентрация составляла 0,04—0,06 мол.% перекиси водо-
водорода, полученной из смеси на основе водорода с содержанием 3% кислорода, что
соответствует превращению от 20 до 30% всего прореагировавшего кислорода.
При применении воздуха, содержащего 3% водорода, получалась приблизи-
приблизительно та же концентрация перекиси водорода в газовой фазе, но выход соста-
составлял лишь 10—15%. Однако, как было показано, для ведения технического
процесса необходимо было создать в газообразных продуктах реакции как
минимум концентрацию 0,25—0,50 мол.% перекиси водорода, иначе стоимость
нагрева и циркуляция газовой смеси были бы чрезмерными.
В недавно опубликованном сообщении П. М. Стадник [31] описал иссле-
исследование по горению водородо-кислородного пламени в трубке, по стенкам
которой циркулировала холодная вода. По окончании опыта воду подкисляли
и титровали марганцевокислым калием для определения количества образовав-
образовавшейся перекиси водорода. Затем пробу выдерживали в темноте в течение 24 час.
в присутствии йодистого калия, и за это время происходило выделение свобод-
свободного йода. Стадник приписывает выделение йода наличию другого окисли-
окислителя X, который не титруется марганцовокислым калием. Он предположительно
отождествляет его с Н2О4 и считает, что при некоторых обстоятельствах коли-
количество этого соединения может быть в 3 раза выше, чем содержание перекиси
водорода. Все его выводы кажутся весьма проблематичными, и имеются сом-
сомнения в том, проверял ли автор правильность аналитических методов с точки
зрения возможных источников различных ошибок. Результаты его работы,
например, могут быть приписаны окислению йодистого калия воздухом. До сих
Процессы в электрическом разряде 47
пор еще не получено точных доказательств в пользу существования Н2О4.
Если даже это соединение и образуется в какой-либо момент, структура его
свидетельствует о том, что оно должно быть весьма неустойчивым (см. гл. 6).
ПРОЦЕССЫ В ЭЛЕКТРИЧЕСКОМ РАЗРЯДЕ
Разряд в водяном паре
Изучение реакции продуктов, полученных при действии тлеющего электри-
электрического разряда на водяной пар, было предметом работ многочисленных иссле-
исследователей [32], и особенно Олденберга, Родбаша и их сотрудников [19, 33—37].
Превосходное резюме вместе с интерпретацией различных экспериментальных
данных, полученных до настоящего времени, опубликовали Родбаш, Кейзер,
Мак-Ки и Куалиано [37]. Основные результаты экспериментальных работ
сводятся к следующему. Максимальный выход перекиси водорода наблюдается
при давлении водяного пара в разрядной трубке 0,1—0,2 мм рт. ст. при воз-
возможно более высокой скорости пропускания пара и столь же быстрой конден-
конденсации продуктов в ловушке, охлаждаемой жидким воздухом. Если заменить
жидкий воздух твердой двуокисью углерода, то перекиси водорода не образует-
образуется и часто даже вода конденсируется в очень незначительном количестве. Если
ловушку охлаждать жидким воздухом, но удалить ее от разрядной трубки для
увеличения продолжительности реакции, то количество образующейся пере-
перекиси водорода падает до нуля и в ловушке практически не происходит конден-
конденсации воды. Если ловушку с жидким воздухом поместить в непосредственной
близости к электрическому разряду, то практически весь кислород, поступаю-
поступающий в виде воды, конденсируется в ловушке как вода или перекись водорода.
Хотя в разряде происходят сложные процессы ионизации, спектроскопиче-
спектроскопические определения показывают, что продукты диссоциации на выходе из раз-
разрядной трубки состоят почти целиком из атомарного водорода и гидроксиль-
ного радикала, не несущих заряда; заметного количества атомарного кислорода
не образуется, за исключением тех случаев, когда применяется очень интен-
интенсивный разряд при низких давлениях водяного пара. При разряде умеренной
интенсивности (например, 1000 в при -60 периодах в трубке диаметром 30 мм
и длиной 2 м) происходит полная диссоциация воды, так как вода практически
не конденсируется в ловушке, расположенной достаточно далеко от разрядной
трубки. Предполагается, что первичной стадией образования перекиси водо-
водорода в этсм случае является конденсация гидроксильных радикалов на стен-
стенках ловушки, охлаждаемой жидким воздухом, а не реакция в газовой фазе.
На это указывают хорошие выходы перекиси водорода F0%), а также резуль-
результаты изучения спектров поглощения, проведенного Фростом и Олденбергом [33],
которые не обнаружили следов перекиси водорода в газовой фазе после про-
пропускания электрического разряда через водяной пар, хотя в их приборе можно
было обнаружить перекись водорода уже при парциальных давлениях 0,01 мм
рт. ст. После первоначального образования Н и ОН в разряде соотношение
трех конкурирующих реакций:
ОН + ОН =!!!™ НА. A6)
Н,О, A7)
Н2, A8)
протекающих на холодных стенках ловушки, определяет, по-видимому, состав
продуктов. Перекись водорода при температуре ловушки —78° не образуется.
48 Глава 2
что объясняется низкой температурой конденсации гидроксильных ради-
радикалов.
Многочисленные исследования подтверждают быстрое исчезновение обра-
образующихся в разряде радикалов ОН, что говорит в пользу гомогенного меха-
механизма. Исчезновение может происходить тремя различными путями:
ОН-ЬОН-> НаО2. 06)
ОН + ОН]-* Н2О + О, A9)
ОН + ОН -> Н2 + О2. B0)
Родбаш и его сотрудники пришли к заключению, что при таких низких давле-
давлениях протекает только реакция B0), так как по мере увеличения расстояния
от разрядной трубки до ловушки выходы воды и перекиси водорода падают до
нуля, а обнаружить атомарный кислород в этих условиях невозможно. При
более высоких общих давлениях или других условиях может приобрести зна-
значение реакция A7):
Н + ОН 4-М -> Н2О + М. A7)
Скорость исчезновения радикалов ОН была определена по изучению спек-
спектра поглощения через различные промежутки времени после их образования
в электрическом разряде [33, 34]. Эти радикалы могли существовать в течение
0,4 сек. в том случае, когда разряд пропускался через водяной пар, но исче-
исчезали менее чем через 0,01 сек., если они получались путем разряда в перекиси
водорода, что доказывает удаление их за счет реакции с перекисью.
Реакции кислорода и атомарного водорода
Пропусканием электрического разряда через водород, обычно при давле-
давлениях около 1 мм рт. ст. или меньше, можно получить концентрацию атомов
водорода 20—50%; образовавшийся таким образом атомарный водород спо-
способен к длительному существованию до рекомбинирования. Бонхеффер и Бем
[38] и Гейб и Гартек [39] изучили реакцию кислорода с атомарным водородом
при давлениях 0,1 и 0,5 мм рт. ст. и температурах стенки 30—200° К. Про-
Продукт представлял собой практически 100%-ную перекись водорода при 30° К,
но с повышением температуры стенки выход падал и становился ничтожно
малым при 200° К. Эти авторы указывали, что их продукт энергично выделял
кислород при нагревании приблизительно до —115°, и выдвинули предполо-
предположение, что получаемая на первой стадии перекись водорода обладает структу-
структурой, отличающейся от нормальной, что и обусловливает ее разложение при
нагревании до —115°.
Аналогичное явление было отмечено также Джонсом и Винклером [40] и
Жигером, Секко и Итоном [41] для продуктов конденсации из водяного пара,
подвергшегося диссоциации под действием электрического разряда. Продукты,
сконденсированные при температурах ловушки ниже —150° и имеющие вид
стекла, выделяли кислород при нагревании выше —120°, причем в ловушке
оставались вода и перекись водорода. В другом исследовании [42] пар пере-
перекиси водорода был подвергнут полной диссоциации под действием электриче-
электрического разряда практически на такой же установке, и оказалось, что перекись
водорода образуется только при охлаждении ловушки ниже —120° и что про-
продукты, вымороженные при температуре ниже —150°, выделяют кислород при
нагревании до комнатной температуры. Природа этого продукта рассматри-
рассматривается в гл. 6.
Гейб и Гартек [39] предполагали, что в их опытах перекись водорода обра-
образовывалась по уравнению
+ М A3)
Процессы в электричесском разряде 49*
с последующей реакцией
НО2 + Н + М-> Н2О2 + М B1)
или
НО2 + НОа + М -^ Н2О2 + О2 + М, A4)
где М представляет третью молекулу или, что более вероятно, стенку.
Высказано предположение, что эти реакции протекают преимущественно по
сравнению с реакциями E), F) и G), так как последние требуют значительной
энергии активации. Постулировано, что при повышении температуры стенки
радикалы НО2 или молекулы Н2О2 не могут отдать стенке количества энергии,
достаточного для их возможной стабилизации, а поэтому вместо них образуют-
образуются Н2О, О или ОН. Однако возможно также образование ОН за счет гомогенных
реакций F) и G) с последующей диффузией этих радикалов в стенке, где они
должны реагировать с образованием перекиси водорода, если только темпера-
температура стенки достаточно низка.
Бэдин [21] сообщил о результатах аналогичных исследований реакции
между молекулярным кислородом и атомарным водородом, взятым в большом
избытке, при давлении 0,27 мм рт. ст. Он нашел, что при постоянном состоя-
состоянии поверхности скорость образования перекиси водорода пропорциональна
концентрации кислорода в пределах широкого интервала и что на результаты
не влияет пропускание кислорода, а также и водорода через электрический
разряд. Кинетические данные согласуются с механизмом возникновения НО2
по реакции A3) и последующим образованием перекиси по реакции B1).
При температуре конденсации —196° весь поступающий кислород при-
присутствует в конечных продуктах в виде воды и перекиси водорода, образую-
образующихся примерно в эквимолекулярных количествах. При более высоких тем-
температурах конденсации выход воды возрастает, а выход перекиси водорода
падает; наконец, при —79° образование перекиси вообще прекращается.
Увеличение расстояния между точкой смешения и конденсационной ловушкой
снижает выход перекиси водорода настолько, что остаются лишь ее следы;
количество же образовавшейся воды при этом изменяется лишь незначительно.
Эти данные аналогичны данным, полученным при пропускании электрического
разряда через водяной пар (см. выше).
Чтобы объяснить одновременное образование воды и перекиси водорода,
Бэдин без доказательства принимает, что образуется активированная пере-
перекись водорода по реакции, конкурирующей с реакцией B1) в отношении
потребления радикалов НО2:
НО2 + Н-> H.OJ- B1а)
Активированная молекула может быть затем стабилизована на холодной по-
поверхности или может распасться на два радикала ОН, образующих затем воду
по уравнению
Н + ОН + М -> Н2О + М. A7)
Предположено, что энергия активации, необходимая для реакции A1), умень-
уменьшает вероятность ее осуществления по сравнению с реакцией A7). Однако
поскольку количество образующейся воды не возрастает в зависимости от вре-
времени, очевидно, что часть ОН должна исчезать также за счет реакции
ОН + ОН -> Н2 + О2. B0)
Характер поверхности в зоне реакции оказывает заметное влияние на количе-
количество и природу конденсируемых продуктов, как этого и следовало ожидать
при очень низких давлениях. Количество продуктов, которые могут конден-
4 Перекись водорода
5flh Глава %
сироваться в ловушке с жидким воздухом, расположенной за зоной реакции,
уменьшается с повышением температуры стенки в зоне реакции. При темпера-
температурах поверхности от 0 до 250° поверхность из хлористого калия практически
исключает образование перекиси водорода, поверхность из стекла пирекс
Способствует образованию; промежуточного количества перекиси, уменьшаю-
уменьшающегося с повышением температуры; наконец, поверхность, покрытая фосфор-
фосфорной кислотой, позволяет получить максимальное количество перекиси водо-
водорода, которое, по существу, не зависит от температуры и раз в 10 превышает
выход при применении хлористого калия. Бэдин предполагает, что поверхност-
поверхностные эффекты каким-то образом связаны с количеством воды, присутствующим
на поверхности, причем поверхность, покрытая хлористым калием, является
«сухой», а покрытая фосфорной кислотой—«мокрой». Уменьшение коли-
количества конденсированных продуктов с повышением температуры стенки дол-
должно быть обусловлено вторичным образованием водорода и кислорода из
ОН и НО2.
Выше мы упоминали об исследовании реакции кислорода с атомарным
водородом при помощи масс-спектрометра. Этот метод является весьма много-
многообещающим с точки зрения выяснения некоторых существующих неясных воп-
вопросов, касающихся механизма этой и других реакций.
Если получают в разрядной трубке атомарный кислород и смешивают его
с молекулярным водородом, то при этом не происходит никакой реакции или
она протекает в очень малой степени даже тогда, когда реакционные газы
охлаждают в ловушке при температуре жидкого воздуха. Если и происходит
реакция в каких-либо размерах, то продуктом ее является вода, а не перекись
водорода. Очевидно, что рекомбинация атомов кислорода происходит с гораз-
гораздо большей скоростью, чем реакция их с водородом. На основании этого, а
также вследствие более прочной связи атомов в молекуле кислорода по срав-
сравнению с молекулой водорода следует ожидать, что при пропускании электри-
электрического разряда через смесь избытка водорода с кислородом результаты должны
быть примерно такими же, как при пропускании разряда через один водород
с последующим смешением его с молекулярным кислородом.
Пропускание электрического разряда через воздух или кислород с содер-
содержанием водяного пара также приводит к образованию перекиси водорода [2J,
но выходы ее значительно меньше, чем при применении смеси водорода с кис-
кислородом, содержащей большой избыток водорода (см. ниже). Это приписы-
приписывают быстрому разложению перекиси водорода одновременно образующимся
озоном. Применение избытка кислорода в водородо-кислородной смеси приво-
приводит к низким выходам перекиси водорода, возможно, по той же причине,
Получение перекиси водорода в электрическом разряде
Выше показано, что перекись водорода нельзя получить путем пропуска-
пропускания электрического разряда через водородо-кислородную смесь при низких
давлениях (около 1 мм рт. ст.), за исключением образования ее на стенках,
происходящего при температурах поверхности значительно ниже —79°.
Поэтому промышленность интересуют такие процессы, в которых перекись
водорода образуется по реакциям, протекающим примерно при атмосферном
давлении, по-видимому за счет гомогенных взаимодействий, и ее можно отде-
отделить при охлаждении до комнатной температуры. При этом желательно при-
применение газовых смесей, содержащих менее 8—10 мол.% кислорода, чтобы
исключить работу в пределах взрывчатых смесей. Хотя при давлении 1 am
выход перекиси водорода возрастает при охлаждении до температуры ниже атмо-
атмосферной [43], как и при термической реакции водорода с кислородом, этого
недостаточно для компенсации стоимости процесса в связи с применением низко-
низкотемпературного охлаждения. Во всех случаях желательно иметь коронный,
Процессы в электрическом разряде
или кистевой, тип электрического разряда, а не дуговой. Соответствующие про*
цессы запатентованы Доуси [44] и изучены концерном «L G. FarbenIndustrie*
в Леверкузене (Германия) [29], но максимальные успехи с точки зрения тех-
технического оформления, пожалуй, достигнуты Крущемна заводе «Elektroche-*
mische Werke» в Мюнхене (Германия) в течение 1931—1945 гг. [45].
Экспериментальные исследования проводили в небольшом лабораторном
аппарате мощностью от 4 до 20 вту использованном главным образом для
изучения влияния изменений состава газа, температуры, напряжения, часто-
частоты и т. д. Некоторые опыты были проведены в аппарате на 10 кет. Было почти
. ]истиллированная
вода
Газ в этом месте содер-
содержит 0,1 мол. %Н202
—+> 10% -ный раствор
Рис. 6. Схема электроразрядного процесса получения
перекиси водорода по Крущу.
i—газодувка; 2—теплообменник; 5— ионизационная камера;
4—алюминиевый конденсатор; 5—ректификационная колонна.
завершено строительство опытной установки на 100 кет, но в связи с воен-
военными действиями она не была пущена в эксплуатацию. Описываемый ниже
процесс был изложен Крущем для завода на 2000 кет, рассчитанного на выра-
выработку перекиси водорода в количестве 100 кг/час с концентрацией 1Q вес.%.
Проект и предложенный технологический режим основывались на результатах
исследований в аппаратах на 4—20 ет и 10/сет, пересчитанных на соответствую-
соответствующие масштабы. При работе газодувки, поддерживающей давление несколько
выше атмосферного, в аппарате циркулирует газовая смесь, содержащая 95%
водорода, 5% кислорода и некоторое количество водяного пара (рис. 6). Газы;
нагреваются в теплообменнике и затем попадают в ионизационную камеру,
которая представляет, в сущности, высокоемкостный конденсатор, состоящий иэ
большого числа кварцевых пластин толщиной 5 мм и размерами 50x50 см.
С одной стороны эти пластины покрыты слоем алюминия высокой чистоты,
проводящим электричество, с другой стороны они протравлены плавиковой
кислотой. Стороны, покрытые алюминием, обращены одна к другой, и газ
протекает через 5-миллиметровые промежутки между непроводящими поверх-;
ностями. Пластины вместе с распорками монтируются одна над другой по две
на распорку. Пластины соединены параллельно и присоединены к источнику
переменного тока напряжением 12 000 в и частотой 9500 гц. Мощность электри-
электрического разряда составляет 0,96 кет на каждую пару пластин. Газы, выходя-
выходящие из ионизационной камеры, охлаждают в теплообменнике и затем подают
в ректификационную керамическую колонну с насадкой из глазурованных,
фарфоровых колец. В верхнюю часть колонны подается дистиллированная вода;
для охлаждения служит конденсатор из чистого алюминия. Скорость цирку-^
ляции охлаждающей воды через конденсатор определяет температуру, а еле-.
4*
52 Глава 2
довательно, и содержание водяного пара в газах, подвергающихся рецирку-
рециркуляции. К этим газам добавляют свежий водород и кислород, и газы, освобож-
освобожденные от перекиси водорода, вновь пропускают через систему при помощи
газодувки. Место подачи газов не имеет существенного значения, так как
изменение состава за цикл очень мало.
Исследования, проведенные в 20-ваттном лабораторном аппарате, пока-
показали, что максимальный выход перекиси водорода на единицу израсходован-
израсходованной энергии наблюдается при средней температуре 160° в ионизационной камере
и содержании водяного пара в количестве, соответствующем упругости насы-
насыщенного пара при 60°. Предполагалось, что температура газа возрастает с 60
до 120° в теплообменнике и затем на выходе из ионизационной камеры подни-
поднимается до 200°. Хотя выход по энергии в лабораторном аппарате был макси-
максимальным при частоте 1000 гцу минимальной из изученных частот, все же для
крупного завода была рекомендована частота 9500 гц> очевидно для уменьше-
уменьшения количества дорогих кварцевых пластин, требующихся для обеспечения
определенной производительности.
Сообщалось, что выход по энергии возрастает с понижением средней кон-
концентрации перекиси водорода в газах, что, по-видимому, происходит при
уменьшении времени пребывания в ионизационной камере, а это в свою очередь
имеет следствием меньшую степень реакции. При рекомендованных условиях
работы получалось 70 молей перекиси водорода на 30 молей воды, но конверсия
за время цикла была столь низкой, что концентрация перекиси водорода в газах
на входе в ректификационную колонну составляла лишь 0,1 %. При оптималь-
оптимальных условиях расход энергии в ионизаторе был примерно 40 квт-ч на 1 кг
полученной перекиси водорода (в расчете на 100%-ную Н2О2).
Концентрация перекиси водорода на выходе из колонны определяется
количеством воды, введенной ниже конденсатора. Можно легко получить пере-
перекись водорода концентрации 10 вес.%, однако при попытках получения более
высоких концентраций в колонне существенно увеличивается степень разло-
разложения перекиси водорода.
Теплообменник является узким местом в работе завода. Хотя он повышает
использование тепла в процессе, значительная часть образовавшейся перекиси
водорода может разложиться в теплообменнике до поступления в ректифика-
ректификационную колонну. Разложение, как это было показано, обусловливалось влия-
влиянием поверхности; повышение размеров поверхности раз в 8 при постоянном
объеме уменьшало конечный выход перекиси водорода приблизительно на 20%;
увеличение же времени выдерживания в теплообменнике с 0,35 до 1,1 сек.
при постоянной небольшой поверхности теплообмена не влияло на конечный
выход. Чтобы свести разложение до минимума, пришлось применять алюминий
исключительно высокой чистоты (99,9%). Степень разложения на влажных по-
поверхностях обычно значительно меньше, чем на сухих, вероятно в результате
защитного действия пленки жидкости. Прямая закалка газов на выходе из
ионизатора, возможно, устранила бы эти трудности, но в этом случае вторич-
вторичный подогрев газов потребовал бы дополнительной затраты большого количе-
количества энергии из-за необходимости возвращения в цикл большого количества
газа на единицу полученной перекиси водорода.
Выход по энергии и селективность превращения водорода в перекись водо-
водорода сильно зависели от содержания воды в водородо-кислородной смеси и от
температуры ионизационной камеры. Однако концентрация водяного пара,
которой соответствовал максимальный выход по энергии, не обязательно сов-
совпадала с концентрацией, соответствовавшей максимальному превращению
всего прореагировавшего водорода в перекись.
Изучено большое число диэлектрических материалов с точки зрения влия-
влияния их на выход перекиси водорода по энергии; наиболее удовлетворительными
оказались кварцевые пластины. Пластины из «лувикана» (N-поливинилкар-
Процессы в электрическом разряде 53
базола) давали, правда, более высокие выходы по энергии, чем кварц, но их
нельзя было нагревать выше 150°. Показано, что при применении хороших изо-
изолирующих материалов свойства поверхности диэлектрика оказывали гораздо
большее влияние на выход по энергии, чем внутренние качества, и что дей-
действие поверхности диэлектрика, по-видимому, обусловлено главным образом
его влиянием на электрический разряд, а не на разложение пара перекиси
водорода. Изучено большое число различных поверхностных покрытий на квар-
кварце, однако большая часть их была органического происхождения, и они разла*
гались в условиях эксплуатации.
Работа при давлении 1,5 am давала худшие результаты, чем при 1 ат\
применение смеси, содержащей 8,8% аргона в расчете на сухой газ, несколько
увеличивало выход.
Капитальные затраты на процесс Круща примерно такие же, как и на элек-
электролитический процесс, однако по этому методу издержки на рабочую силу
меньше и оборудование амортизуется медленнее; отпадает необходимый при
электролитическом процессе большой расход охлаждающей воды, и производ-
производственная себестоимость не очень сильно изменяется в зависимости от размеров
производства; поэтому процесс электрического разряда мог бы представлять
интерес для небольших заводов. Перекись водорода, получаемая по этому
методу, отличается высокой чистотой и высокой стабильностью, поскольку
в процессе производства применяются только водород, кислород и вода. Однако
на современной стадии развития этот метод ввиду большого расхода энергии
может заинтересовать промышленность только при наличии очень дешевой
электроэнергии. Тем не менее возможно, что общая стоимость процесса может
быть значительно снижена путем соответствующего видоизменения его, напри-
например при работе ионизатора на меньшей частоте и в случае применения пластин
из более дешевого материала, обеспечивающего большие выходы по сравнению
с кварцем, например пластин из высокополимеров, устойчивых при сравни-
сравнительно высоких температурах. Крущ указывает, что работа при давлении ниже
атмосферного могла бы быть более эффективной. Не исключено также, что
добавка к газу других веществ вместо аргона может увеличить эффективность
процесса. Выход по энергии в процессе «I. G. Farbenindustrie» [29] был значи-
значительно ниже, чем в процессе Круща, причем, по-видимому, первый из этих
процессов вообще менее удовлетворителен. Недавно в США Путнэм [46] изу-
изучил процесс с применением электрического разряда в лабораторном масштабе,
причем выходы по энергии были близки к полученным Крущем.
Перекись водорода или пероксосоединения могут быть получены также
при пропускании искры или тлеющего разряда между электродом и водным
раствором электролита или при установке обоих электродов в газовой фазе
вблизи поверхности жидкости [47]. Выход перекиси водорода зачастую зна-
значительно превышает вычисленный из закона Фарадея, но падение потенциала
в этом случае на один или несколько порядков превышает падение, встречаю8-
щееся в процессах электролиза. Выход зависит от природы электролита, рН
раствора, природы газа, в котором происходит электрический разряд, давле-
давления газа, а также и от направления электрического тока. Клеменц [48] сооб-
сообщил, что из серной кислоты в присутствии кислорода могут быть получены пере-
перекись водорода, пероксосерная и пероксодисерная кислоты с выходом, примерно
в три раза превышающим выход по закону Фарадея; при добавлении станната
натрия, играющего роль ингибитора разложения перекиси водорода, выход
был примерно в 12 раз больше, чем по закону Фарадея. Трудно интерпретиро-
интерпретировать полученные результаты с точки зрения общепринятого механизма, так
как перекись водорода может образоваться в паровой фазе, а также за счет
рекомбинации ионов противоположного знака в газовой фазе или электролити-
электролитических процессов и может исчезать путем разложения в газовой или в жидкой
фазе, причем скорость разложения в жидкой фазе зависит от таких переменных,
С54 Глава 2
как рН, температура, природа присутствующих растворенных веществ и ката*
-литическая природа электрода, погруженного в раствор. На обоих электродах
^наблюдается большое падение потенциала; в аппарате Клеменца оно составляет
около 400 в на катоде и 70 в на аноде. В соответствии с этим расход мощно-
мощности значительно выше, чем в электролитическом процессе или даже в процессе
Круща в газовом разряде. В аналогичных исследованиях, проведенных Мута [49],
выход перекиси водорода составил 9 г/квт-ч. Клеменц 150] опубликовал обзор
работ последних лет по электролизу в тлеющем разряде.
Электролиз в тлеющем разряде был недавно исследован Дэвисом и Хик-
лингом [51], которые работали в достаточно простых условиях, что позволило
получать воспроизводимые результаты и в значительной мере интерпрети-
интерпретировать полученные данные. Они пропускали электрический разряд при умень-
уменьшенном давлении между поверхностью разбавленного раствора инертного
электролита и анодом, находившимся вне раствора. Количество образовав-
образовавшейся сначала перекиси водорода было прямо пропорционально количеству
пропущенного электричества и практически не зависело от плотности тока,
объема электролита и многих других факторов, влияющих на природу разряда,
хотя начальный выход и изменялся с концентрацией электролита и изменением
рН. Постепенно в течение каждого опыта выход падал вследствие разложения
образовавшейся перекиси водорода, а при работе с сильнощелочным раствором
перекись вообще нельзя было обнаружить. Результаты работы согласовыва-
согласовывались с предположением о том, что разряд происходит главным образом через
водяной пар и ионы, образовавшиеся в газовой фазе, вызывают появление гид-
роксильных радикалов в жидкой воде преимущественно за счет электролитиче-
электролитического эффекта. Далее предполагается, что гидроксилы димеризуются с обра-
образованием перекиси водорода, которая в свою очередь разлагается за счет даль-
дальнейшей реакции с гидроксильными радикалами.
ПРОЦЕССЫ ОБЛУЧЕНИЯ
Процессы, возникающие под действием излучения, могут быть разделе-
разделены на две группы, обычно называемые фотохимической и радиохимиче-
радиохимической.
Фотохимические процессы—это процессы, в которых поглощение излу-
излучений ведет к образованию возбужденных молекул, инициирующих затем
в свою очередь вторичные реакции с образованием атомов, свободных радика-
радикалов или молекул. Размер и характер начальных реакций обычно весьма спе-
специфичны в отношении длины волны, а следовательно, и квантовой энергии
излучения. Для протекания фотохимических процессов необходимо, чтобы
излучение лежало в ультрафиолетовой области спектра. Радиохимические про-
процессы возникают в результате поглощения излучений с высокой энергией,
например рентгеновских, у- или катодных лучей, а также быстрых частиц,
например протонов, а- и р-лучей, обычно носящих название «ионизирующих
излучений». В этом случае известная доля поглощенной энергии образует воз-
возбужденные молекулы или радикалы, остальная же часть приводит к образо-
образованию пар ионов, которые затем генерируют новые количества свободных ради-
радикалов и атомов. Радиохимические процессы характеризуются возникновением
ионизации и результатами, которые сравнительно не зависят от энергии отдель-
отдельной частицы, или кванта, но зависят от общего количества поглощенной энер-
энергии. Начальные процессы, протекающие при действии ионизирующих излу*
чений, приводят к одновременному образованию заряженных и незаряженных
частиц. Эти процессы являются гораздо более сложными, чем протекающие при
фотохимическом возбуждении. Однако реакции, следующие за процессами
возбуждения, обычно близки по характеру, причем они выражены тем более
резко, чем выше энергия излучения.
Процессы облучения <35
1 Фотохимическая реакция водорода и кислорода
Сначала рассмотрим образование перекиси водорода при ультрафиолетс*-
вом облучении смесей водорода с кислородом. Коэн и Гроте [52] обнаружили
образование небольшого количества перекиси водорода при быстром пропу-
пропускании смеси водорода с кислородом через кварцевый сосуд, подвергнутый
облучению; при медленном пропускании или в статическом опыте образования
перекиси водорода не наблюдалось. Образование очень небольших концентра-
концентраций перекиси водорода, вероятно, является следствием того, что перекись погло-
поглощает излучение сильнее, чем исходные газы—кислород и водород, что приводит
к разложению возбужденной молекулы. В связи с этим, а также с тем, что эф-
эффективным является только такое излучение, которое поглощается, большин-
большинство исследований проводили с добавкой фотосенсибилизирующего агента»
обладавшего значительно большим поглощением, чем одна смесь водорода
с кислородом. В качестве сенсибилизатора широко применялись ртутные пары
вследствие их большей способности к поглощению при длине волны 2537А,
которую можно легко получить в разрядной трубке с парами ртути. Даже при
низком давлении паров ртути, соответствующем комнатной температуре (напри-
(например, 0,0028 мм рт. ст. при 30°), интенсивность падающего излучения снижается
вдвое при прохождении через слой паров ртути всего в несколько миллиметров.
Возбужденные атомы ртути в свою очередь отдают свою энергию при столкно-
столкновениях с другими видами присутствующих частиц.
Хотя и опубликован ряд исследований по реакции водорода с кислородом,
сенсибилизированной ртутью [53—57], однако результаты даже одного и того
же экспериментатора могут колебаться от опыта к опыту, результаты же раз-
различных авторов часто вообще трудно согласовать.
В самых последних работах смеси водорода с кислородом сначала высу-
высушивали, насыщали парами ртути при комнатной температуре, а затем непрерыв-
непрерывно пропускали через кварцевую трубку такого диаметра, который был доста-
достаточен для практически полного поглощения всего падающего излучения с дли-
длиной волны 2537А смесью водорода, кислорода и паров ртути. Эта трубка облу-
облучалась ртутной лампой. Из выходивших газов вымораживали воду и перекись
водорода в ловушке, охлаждавшейся жидким воздухом или твердой двуокисью
^углерода, или извлекали их, пропуская путем барботирования через.воду.
Количество падающего света (принимали, что излучение поглощалось пол-
полностью), а следовательно, и квантовый выход реакции, т. е. число молекул,
образовавшихся на каждый квант поглощенной энергии, определяли по како-
какому-либо эталону. Так, в качестве актинометра часто применяют оксалат урани-
ла. Реакционный сосуд заполняют раствором оксалата уранила в щавелевой
кислоте и затем по известной реакционной характеристике этой системы
вычисляют количество излучения, поступающего за определенный период.
В общем максимальные выходы перекиси водорода наблюдались при
отношениях водорода к кислороду, равных 5—10. В некоторых опытах свыше
90% всего прореагировавшего кислорода превращалось в перекись водорода,
однако этот процент конверсии сильно колебался для разных опытов и в раз-
различных исследованиях (может быть, в связи с различной степенью разложения
НО2 или перекиси водорода под действием окиси ртути или ртути, которые
могли осаждаться на стенках реакционной трубки). Сначала квантовый выход
для общей реакции водорода с кислородом, по Маршаллу [53], принимался при-
примерно за 6,6, но впоследствии Маршалл пересмотрел это значение и понизил его
до 2,5. По Франкенбургеру и Клинкгардту [561, квантовый.выход составляет
около единицы, но эти авторы измеряли количество света, поглощенного в их
реакционном сосуде, с применением монохлоруксусной кислоты как актино-
актинометра. Другое исследование [58] по свойствам монохлоруксусной кислоты
прказало, что фактически квантовый выход у этих авторов был даже значительно
56 Глава 2
ниже единицы. В более поздней работе Волмэна [57] квантовые выходы для
общей реакции составили всего 0,3—0,5. Эти результаты говорят о том, что—
по меньшей мере при комнатной температуре—реакция водорода с кислородом
при давлении 1 am может осуществляться лишь короткими цепями или вообще
не иметь цепного характера, хотя с увеличением температуры и приближением
к взрывным пределам длина цепей, безусловно, возрастает. За исключением
фотохимического возбуждения реакции, можно считать, что характер отдель-
отдельных стадий реакции должен быть таким же, как и для термической реакции
в тех же самых условиях.
Исследуя смеси, содержащие 1—3% кислорода, Франкенбургер и Клинк-
гардт [56] получили выходы перекиси водорода до 20 г на 1 квт-ч электриче-
электрической энергии, израсходованной на питание ртутной лампы. Лампа излучала
до 5% всей поступавшей электрической энергии в виде волн длиной 2537А.
Выход перекиси водорода не зависел от изменения температуры в пределах
50—100° и давления 1—11 am. Однако нельзя поверить, чтобы давление и тем-
температура не имели существенного значения при более высоких концентра-
концентрациях кислорода. Работы Волмэна при атмосферном давлении и температуре
40° показали, что общее число молей воды и перекиси водорода, образовавшихся
при реакции, незначительно меняется в зависимости от отношения водорода
к кислороду в пределах 0,5—10. Выход перекиси оказывается минимальным
при низких отношениях водорода к кислороду, однако он и не возрастал за-
заметно, если это отношение было примерно больше двух.
В качестве первой стадии фотохимической реакции, инициированной излу-
излучением при 2537 А, предполагается образование атомов водорода из молекулы
водорода при столкновении с атомом ртути, возбужденным поглощением кванта
энергии. Этот постулат основан на том, что энергия кванта A12 ккал/моль)
достаточна для разрыва связи в молекуле водорода A04 ккал/моль), но не до-
достаточна для кислорода A16 ккал/моль). Однако из первичного возбужденного
атома ртути могут образоваться и такие частицы, как HgH, HgO, HgOH или
озон [59], а поэтому характер начальной реакции далеко еще нельзя считать
выясненным.
Тем не менее низкий квантовый выход говорит именно о том, что образо-
образование перекиси водорода, вероятно, протекает в основном за счет реакций:
НО2 + НО2 + М -> Н2О2 + Оа + М A4)
или
Н + НО2 + М->Н2О2 + М, B1)
где М—стенка или третье тело, а не за счет реакций, приводящих к образо-
образованию в качестве одного из продуктов свободного радикала или атома. По-
Поскольку в некоторых условиях достигались высокие выходы перекиси водоро-
водорода, вода, вероятно, образуется главным образом за счет разложения НО3
или перекиси водорода, что может быть обусловлено либо реакциями со ртутью
или ее соединениями на стенках, либо реакциями в газовой фазе, возможно,
с участием озона. Хорошо известно, что при ультрафиолетовом облучении кис-
кислорода образуется озон, и Волмэн [57, 60] сделал вывод, что озон и перекись
взаимодействуют друг с другом в газовой фазе*.
* Проведен ряд исследований по взаимодействию озона с перекисью водорода в вод-
водном растворе; наиболее полно этот вопрос исследовали Таубе и Брей [61], предложившие
следующий механизм:
HaOa-f О8 -> ОН + НО2+О2, B2)
НОа+О8 -> ОН+2О2, B3)
ОН+О8 -* НОа+Оа, B4)
ОН+НаОв - НОа+НаО. B5)
Процессы облучения 57
Предположение, выдвинутое исследователями в прежних работах, об обра-
образовании молекулы перекиси водорода по реакции
20Н + М-* Н2О2 + М A6)
в настоящее время опровергается [19].
Другими возможными объяснениями низкого квантового выхода явля-
являются предположения об инактивировании возбужденных атомов ртути
без образования атомов водорода, рекомбинации водородных атомов на стенке
в молекулярный водород или обратном образовании водорода и кислорода
за счет реакций в газовой фазе типа
ОН->Н2 + О2, B0)
причем необходимые гидроксилы образуются за счет разложения перекиси
водорода или ее взаимодействия с озоном. Исчезновение перекиси водорода
и НО2 может происходить и путем реакции на стенках с участием окиси ртути
или HgOH в качестве промежуточных продуктов.
Несенсибилизированная реакция водорода с кислородом изучена в самое
последнее время Смитом и Кистяковским [62] и Смитом и Направником [63]
в динамическом аппарате с применением ультрафиолетового света в областях
спектра 1719—1725 и 1850—1862А при общих давлениях от 1140 до 95 мм рт.
ст. со всевозможными смесями водорода и кислорода. В качестве продуктов
получены озон, перекись водорода и вода. Понижение концентрации кисло-
кислорода увеличивает квантовые выходы перекиси водорода и воды; повышение
концентрации кислорода вызывает рост квантового выхода озона. Снижение
давления от 1140 мм рт. ст. при постоянном составе газа увеличивает выходы
воды и перекиси водорода и снижает выход озона, причем этот эффект особенно
заметен в области самых низких из изученных давлений. При постоянном дав-
давлении 190 мм рт. ст. повышение температуры с 25 до 280° снижает квантовый
выход озона (до полного его отсутствия при отношении водорода к кислороду,
равном 9, и температуре 150°) и повышает квантовый выход воды и перекиси
водорода, причем температурный коэффициент квантового выхода очень быстро
возрастает при температурах 230—280°.
При давлении 190 мм рт. ст. максимальный квантовый выход перекиси
водорода наблюдался при наименьшей из изученных концентраций кисло-
кислорода A0%), но отношение числа молей воды и перекиси водорода в полученных
продуктах оставалось примерно в пределах 5—10 при изменениях температуры
и давления.
Резкое увеличение квантового выхода с температурой, обнаруженное
Смитом и Направником, аналогично установленному Тейлором и Сэлли [64]
для реакции, сенсибилизованной ртутью, при 1 am и примерно 500° (а также
данным других авторов, изучавших термические реакции). Изменение кванто-
квантового выхода с температурой доказывает падение роли нецепных реакций типа
A4) и B1) и рост значения цепных реакций типов
НО2 + Н2->Н2О2 + Н A0)
и
НО2+Н2->Н2О + ОН (И)
и разветвленных цепных реакций с двумя активными центрами
Н + О2 -> ОН + О F)
G)
Я8 Глава
за когорыми следует
Н2-> Н2О + Н. E)
Вероятно, этим путем можно объяснить и повышение квантового выхода
с понижением давления при постоянной температуре.
Механизм реакции в данном случае отличается от механизма реакции,
сенсибилизованной ртутью, в том отношении, что первичной стадией
при поглощений излучений 1850—1862А является, вероятно, образование
возбужденных молекул кислорода, а излучение 1719—1725 А способствует
диссоциации молекулярного кислорода непосредственно на 1 нормальный
и 1 возбужденный атом. Вслед за этим, по-видимому, происходит взаимодей-
взаимодействие кислородных атомов с молекулами кислорода и водорода за счет реакций
такого типа, которые выше предлагались для объяснения термической реакции.
Озон, вероятно, образуется по реакции
О + О2 + М->О3 + М B6)
или за счет столкновения возбужденной молекулы кислорода с нормальной
молекулой этого газа. Исчезновение озона, происходящее с увеличением тем-
[пературы еще до быстрого возрастания температурного коэффициента, ясно
показывает, что это исчезновение не связано с появлением длинных цепей при
повышении температуры.
В качестве сенсибилизаторов для фотохимической реакции водорода с кис-
кислородом можно предложить и другие вещества, помимо ртути, если только они
обладают достаточно высокой поглощающей способностью в этой спектральной
области.
По этому вопросу проведено очень мало работ, но все же можно принять,
что такие вещества, как галогены, галогеноводороды, сероводород, аммиак
и двуокись азота, должны быть эффективными, поскольку они должны диссо-
диссоциировать при ультрафиолетовом облучении с образованием Н, О или других
атомов [59, 65], которые затем могут уже возбудить дальнейшие реакции водо-
водорода и кислорода» Механизм в этом случае, конечно, будет отличаться от
наблюдаемого для паров ртути, поскольку сенсибилизатор сам разлагается
в процессе генерирования свободных радикалов и атомов, хотя галогены или
галогеноводороды могут затем возникнуть снова за счет других реакций, про-
протекающих после диссоциации.
Фотохимически активированные атомы инертного газа ксенона также могут
обладать эффективностью, поскольку они аналогично ртути [59] вызывают
диссоциацию молекулярного водорода на атомы:
Хе + Ь->Хе*, B7)
Хе*+Н2-> Хе + 2Н. C)
Кернбаум [66] обнаружил следы перекиси водорода при 10-часовом облу-
облучении воды ультрафиолетовыми лучами. Тиан [67] сообщает о появлении по-
постоянной концентрации перекиси водорода при облучении воды светом с дли-
длиной волны 1900А; эта концентрация выше в том случае, когда вода содержит
растворенный кислород. Это же влияние кислорода обнаруживается и при
ионизирующих облучениях.
Ряд исследователей указывает на обнаружение небольших количеств
перекиси водорода @,04—1 мг/л) в атмосферных осадках в виде дождя, снега,
тумана, росы и т. д. Так, Шене [68] в Москве в семидесятых годах прошлого
столетия сообщил, что дожди, принесенные полярными ветрами, содержат
Меньше «перекиси», чем дожди от южных ветров, что летние дожди богаче
перекисью, чем осенние, что содержание перекиси в водяных парах, сконден-
сконденсированных из воздуха, возрастает по мере подъема солнца над горизонтом.
Этот автор сделал вывод, что,обнаруженное явление обязано происхождением
Процессы облучения 59
действию солнечных лучей. Матцуи [69] недавно сообщил об аналогичных
результатах на основании метеорологических работ, проведенных в Японии:
дождевая вода содержала 0,35—0,86 мг/л перекиси водорода, а вода
из снега 0,08—0,15 мг/л> причем это содержание зависело от интенсивности
ультрафиолетового излучения в момент отбора пробы. Метод анализа, исполь-
использованный в этих исследованиях, состоял в окислении йодистой соли до сво-
свободного йода, но эту реакцию могут дать также и другие окислители. Поэтому,
хотя весьма вероятно, что сделанные наблюдения действительно обусловлены
присутствием перекиси водорода, фактический характер окисляющих частиц
нельзя еще считать доказанным. Так, в атмосфере присутствуют озон и различ-
различные окислы азота, а в воздухе, содержащем промышленные загрязнения,
могут содержаться еще и органические перекиси, образовавшиеся в результате
фотохимического окисления углеводородов [70].
Бейтс и Николе [71] провели теоретический анализ стационарной кон-
концентрации различных видов частиц в верхних слоях атмосферы, основываясь
на происходящих там фотохимических и других реакциях. Из их работы выте-
вытекает, что на этих очень больших высотах могут находиться небольшие количе-
количества радикала пергидроксила, но количества перекиси водорода признаны ими
столь незначительными, что в общем анализе этой проблемы их не учитывали.
Имеются данные об образовании перекиси водорода при фотохимическом
окислении различных водных растворов и суспензий, например оксалатов
в присутствии иона двухвалентного марганца [72] и паральдегида [73], при
ультрафиолетовом облучении водных растворов йодистого калия и бромнова-
токислого калия [74] и растворов или суспензий биологических сенсибилиза-
сенсибилизаторов (например, сухого дрожжевого экстракта и хлорофилла [75]).
По имеющимся данным, разные белые пигменты могут функционировать
в качестве сенсибилизаторов различных фотохимических реакций; так, образо-
образование перекиси водорода из воды происходит в присутствии следующих сен-
сенсибилизаторов: ZnO, ZnCO3, а также CdO, SnO2 и ThO2. Был сделан ряд попы-
попыток связать фотохимическую активность таких пигментов со степенью меления
или «выветривания» красок, в состав которых они входят, но до сих пор эти
попытки увенчались лишь очень небольшим успехом. Однако при опрыски-
опрыскивании цитрусовых деревьев окисью цинка увеличенное опадание листвы на-
наблюдалось только при применении окиси цинка, обладающей «перекисноводо-
родной активностью» [76]. ;
Не раз были исследованы водные суспензии окиси цинка в присутствии
кислорода, и оказалось, что в этом случае фотохимическое действие охватывает
всю область от ультрафиолетовой до видимой части спектра в пределах 4000—
4700 А. Последние работы выполнили Чари и Куреши [77], Рубин, Калверт,
Рэнкин, Мак-Невин и Тейрер [78, 79], Веселовский и Шуб [80] и Маркхем
и Ледлер [81]. В сообщениях этих авторов имеется ссылки и на прежние ра-
работы. Пока с достоверностью установлено лишь небольшое количество правил
относительно характера этих реакций. Скорость образования перекиси водо-
водорода возрастает с концентрацией кислорода, однако пока еще не известно,
может ли вообще возникнуть перекись при облучении суспензии в отсутствие
кислорода. В присутствии кислорода концентрация перекиси водорода дости-
достигает стационарной величины около 10'б моль/л. При добавлении к системе
какого-либо соединения из широкой гаммы органических производных обра-
образование перекиси водорода возрастает. В качестве примера можно привести
следующие соединения: глицерин, глюкозу, глицин, бензидин, этиламин,
фенол,, ацетон, спирт, уксусную кислоту, бензойную кислоту, флороглюцин,
резорцин, толуол, ацетанилид, анилин, щавелевокислый натрий, хинон и гид-
гидрохинон. В отдельных сообщениях встречаются различные мнения об их отно-
относительной эффективности в отношении образования перекиси. Обычно прини-
принимается, что эти органические соединения предотвращают цепное разложение
'60 Глава 2
перекиси водорода за счет реакции со свободными радикалами, возникшими при
фотолизе. Однако возможно также, что известное количество перекиси водо-
водорода образуется за счет фотохимического самоокисления органических моле-
молекул. Маркхем и Ледлер [81] приводят некоторые данные по продуктам окисле-
окисления, получающимся о из органических веществ. При применении излучений
с длиной волны 3650А они обнаружили начальный квантовый выход для обра-
образования перекиси водорода 0,25 в отсутствие органических веществ и 0,4 и
0,5 соответственно в присутствии небольших количеств ацетанилида и фенола.
Отношение расхода кислорода к образованию перекиси в отсутствие органи-
органических веществ составляло 1:1; в присутствии ацетанилида и фенола—соот-
фенола—соответственно 2 : 1 и 1 : 1. Имея в виду известные свойства окиси цинка как полу-
полупроводника, они предполагают, что в этих случаях фотохимический механизм
включает перенос электрона от окиси цинка к адсорбированной воде, что спо-
способствует диссоциации последней на атом водорода и гидроксильный ион.
Наиболее вероятной следующей стадией является реакция водородного атома
с молекулой кислорода с образованием НО2, после чего два пергидроксила
взаимодействуют с образованием перекиси водорода и кислорода. Однако
в опытах с водой и с кислородом, обогащенным изотопом О18 в присутствии и в
отсутствие органических добавок, Калверт и сотрудники [79 ] показали,
что кислород в образовавшейся перекиси водорода целиком происходит из газо-
газообразного кислорода, а не из кислорода воды, окиси цинка или органической
добавки. Отсюда эти авторы пришли к заключению, что образование перекиси
водорода происходит путем восстановления фотовозбужденным электроном
молекулярного кислорода, адсорбированного на окиси цинка.
Влияние ионизирующих излучений
При действии ионизирующих излучений на воду в жидкой или паровой
фазе или на водные растворы могут происходить самые различные процессы,
в связи с чем трудно интерпретировать те различные наблюдения, которые
были сделаны относительно образования перекиси водорода при такого рода
бомбардировке. В последние годы отмечен большой рост научно-исследователь-
научно-исследовательских работ в этой области, стимулировавшийся развитием ядерной энергии.
Наши знания в этой области быстро развиваются. Недавно появились сообще-
сообщения о многочисленных экспериментальных работах, но решающие факторы
пока еще недостаточно изучены, и твердо установленным можно считать лишь
некоторое число фактов.
Значительная часть ранее полученных знаний в области этих процессов
связана с работами Фрике и его сотрудников [82]. Современное состояние
этого вопроса можно уяснить из ряда ежегодных обзоров [83]. В недавней
дискуссии Фарадеевского общества [84] по радиационной химии также охва-
охвачен ряд сообщений о роли перекиси водорода. В выступлениях на дискуссии
содержится много ценного материала. Другие работы последнего времени,
посвященные образованию перекиси водорода при бомбардировке воды, при-
принадлежат Аллену и его сотрудникам [85], Дейнтону [86], Кренцу [87] и груп-
группе французских авторов [88—93]. Твердо установлено, что бомбардировка
воды ионизирующими излучениями любого типа приводит к образованию
газообразных водорода и кислорода, а также перекиси водорода. Обычно
принимается, что газообразный водород и перекись являются первыми из
образующихся молекулярных продуктов, газообразный же кислород полу-
получается уже за счет вторичных реакций свободных радикалов с перекисью водо-
водорода. Так, Аллен [85] показал, что при применении быстрых электронов или
излучений атомного реактора (быстрые нейтроны вместе с у-лучами) и очень
малых экспозициях образуется не кислород, а перекись водорода; кислород
появляется только при более длительном облучении. Однако в одном из сообще-
Процессы облучения 61
ний [88] констатировано, что при бомбардировке воды а-лучами выходы пере-
перекиси водорода и кислорода были постоянными в течение начального периодэ
реакции, что позволяет предполагать образование этих двух продуктов в на-
начальной стадии разложения воды.
Природа и количества различных образующихся молекул, скорости их
образования, количества их на единицу поглощенной энергии и другие явле-
явления зависят от большого числа разных факторов, к которым относятся
тип излучения (например, производится ли бомбардировка электронами или
тяжелыми частицами), энергия отдельных частиц, интенсивность и длитель-
длительность бомбардировки, распределение поглощения энергии в жидкости, отно-
отношение объемов жидкой и газовой фаз в реакционном сосуде и наличие или
отсутствие следов растворенных веществ, например кислорода. В настоящее
время отсутствует способ измерения числа ионных пар (положительный ион
плюс электрон), образующихся на единицу количества ионизирующего излуче-
излучения, поглощенного водой. Обычно предполагается, что около половины погло-
поглощенной энергии расходуется на образование молекул воды с возбужденными
электронами, другая же половина энергии идет на образование ионных пар.
Это соображение основано на предпосылке, что для образования одной ионной
пары в жидкой воде требуется такое же количество энергии (т. е. 30—35 эв),
как и в воздухе. Поскольку примерно половина этого количества энергии тре-
требуется на ионизацию одной молекулы воды, приходится принять, что другая
половина расходуется на образование активированных молекул воды. Часть
этих активированных молекул инактивируется затем за счет столкновений,
другие могут образовать радикалы Н и ОН. Однако весьма вероятно, что,
поскольку радикалы, возникшие за счет диссоциации активированной моле-
молекулы воды, находятся близко друг от друга, будет немедленно происходить
их рекомбинация с образованием воды. Степень участия их в других реакциях
неизвестна, но принимается, что она невелика.
Реакции, в которых принимают участие ионизированная молекула воды
и выбитый электрон, имеют большее значение, хотя предложенные механизмы
все еще носят умозрительный характер. Обычно принимают, что первой реак-
реакцией ионизированной молекулы воды Н2О+ является следующая:
Н2О+ -^Н* + ОН B8)
или
Н2О+ 4- Н2О -> Н3О+ + ОН. B9)
Предполагают также, что электрон захватывается по реакции
Н2О + е-^ Н + ОН-, C0)
но Гайсинский и Мага [84, 89] считают, что в действительности протекают
реакции:
. C1)
C2)
Полагают, что при последующих реакциях происходит нейтрализация заря-
заряженных частиц и реакции между ними и незаряженными частицами или моле-
молекулами с дальнейшим образованием радикалов' Н и ОН, которые могут подвер-
подвергаться рекомбинации с образованием либо воды, либо водорода или перекиси
водорода. Затем может появиться кислород за счет реакций с участием пере-
перекиси водорода.
Показано, что тяжелые частицы радиоактивных источников обычно более
эффективны, чем рентгеновские или у-лучи, с точки зрения химического измене-
изменения воды в расчете на количество поглощенной энергии [85, 94, 95]. Так,
62 Глава 2
рентгеновские излучения вызывают очень небольшое разложение чистой воды
(лишенной кислорода); для излучений же с более высокой энергией Аллен 1851
приводит следующие значения для числа молекул воды, разложившихся при
поглощении 100 эв энергии (на основании количества образовавшегося газо-
газообразного водорода): 0,1—0,4 для C-излучения трития (около 5 кэв), 0,2-~0,5
для электронов с энергией 1 мэв, 0,54 для дейтонов с энергией 8 мэв, около 1
для излучений атомного реактора (смешанные у-лучи и быстрые нейтроны)
и 2,0 для а-лучей.
Заметное влияние типа излучении на выход разложения воды по энергии,
вероятно, зависит от степени разделения возникших Н- и ОН-радикалов, обра-
образовавшихся в треке ионизирующего луча, или от аномального распределения
Н- и ОН-радикалов [86, 94]. Например, предполагается, что положительные
ионы, возникшие по каждому следу а-частицы, протона или дейтона, быстро
диссоциируют на Н+ и радикал ОН, тогда как электрон, появившийся от пер-
первичного процесса, захватывается только на некотором расстоянии от этого
пути. Вследствие этого создается избыток ОН-радикалов вдоль центра пути
и избыток Н-радикалов в зоне, окружающей этот центр. Это увеличивает ве-
вероятность рекомбинации двух гидроксильных радикалов с образованием пере-
перекиси водорода и двух Н-атомов в молекулу водорода. При облучении рентге-
рентгеновскими, у- или р-лучами логично предполагать, что ОН- и Н-радикалы
образуются в значительно меньшей концентрации и распределены более равно-
равномерно, что увеличивает вероятность их рекомбинации с образованием исход-
исходной воды. Аллен [96] показал, что потеря энергии быстрыми электронами, про-
проходящими через воду, происходит внезапными толчками, что приводит к обра-
образованию скоплений пар ионов вдоль пути этих электронов, что также должно
влиять на распределение ОН- и Н-радикалов. У нас очень мало сведений об
относительных выходах по энергии в водяном паре по сравнению с выходами
в жидкой воде. Однако близость между молекулами воды и наличие водород-
водородных связей в жидком состоянии, как можно предполагать, обусловливают
значительные различия в механизмах реакций в обеих фазах.
При бомбардировке чистой жидкой воды (не содержащей растворенного
газа или других растворенных веществ) электронами с энергией 1 мэв или
рентгеновскими лучами Ван-де-Граафа возникает стационарная концентрация
перекиси водорода порядка нескольких микромолей в литре [85] с соответ-
соответствующим стационарным парциальным давлением газообразного водорода
над водой, достигающим нескольких миллиметров ртутного столба; концентра-
концентрация перекиси водорода настолько мала, что ряд прежних исследователей
вообще не обнаружил ее присутствия. Факт установления стационарного
состояния говорит о том, что перекись водорода и водород, растворенные в воде,
продолжают реагировать с радикалами Н и ОН с вторичным образованием
воды по реакциям:
E)
Н + НаО2 -> НаО + ОН. A2)
Это—не термодинамическое равновесие, а скорее условие стационарного состоя-
состояния, которое достигается тогда, когда в процессе бомбардировки противодей-
противодействующие друг другу реакции «эдинаковы по величине. Стационарные концен-
концентрации перекиси водорода и водорода возрастают с ростом энергии излучения.
Так, стационарное давление водорода изменяется от 1—2 см рт. ст. для электро-
электронов с энергией 1 Мэв до давления свыше 1 am для а-лучей. Выход по энергии
падает тем сильнее, чем больше размер обратных реакций.
Дополнительные доказательства образования Н- и ОН-радикалов при
облучении чистой, лишенной воздуха воды рентгеновскими или у-лучами полу-
получены при исследовании полимеризации, например акрилонитрила, в разбавлен*
Процесс»1' облучения
ных водных растворах в условиях такого рода бомбардировки [97].
доказано, что свободные радикалы, например ОН, могут инициировать подоб*
ные реакции полимеризации [59, 98] и что группы ОН в полученном полимере*
можно обнаружить анализом в инфракрасном спектре.
Растворение кислорода или воздуха в воде заметно повышает как началь-
начальную скорость образования перекиси водорода, так и ее стационарную концен-
концентрацию при облучении рентгеновскими лучами; вместе с тем присутствие кис-
кислорода не оказывает заметного влияния на результаты бомбардировки а-луча-
ми [92]. Джонсон, Шолс и Вейс [99] сообщили, что в присутствии кислорода
число молей перекиси водорода, образующейся в начальном периоде при
рентгеновском облучении в расчете на единицу поглощенной энергии,
не зависит от рН в интервале 1—12 и что скорость образования перекиси
резко падает при потреблении всего кислорода. С другой стороны, Луазлёр
1100] сообщил, что снижение рН увеличивает образование перекиси водорода
под действием рентгеновских лучей. В последних работах [92] сообщалось,
что при рентгеновском облучении льда, приготовленного из насыщенной кисло-
кислородом воды, количество перекиси водорода, образующейся на единицу погло-
поглощенной энергии, падает с понижением температуры в интервале от 0 до
—116° и при температуре ниже—116° перекись вообще не обнаружена. Вода,
насыщенная кислородом при 0°, образует значительно больше перекиси водо*
рода, чем лед при этой же температуре, и выход перекиси возрастает с повыше-
повышением температуры до 20°, т. е. до максимальной из изученных температур. Для
а-излучения этого влияния температуры не обнаружено, но все же было показа-*
но заметное различие в выходе перекиси водорода при переходе воды в лед.
Выше мы уже останавливались на начальных реакциях, которые были приняты
для ионизированных молекул и электронов. Если присутствует также раство-
растворенный кислород, то за начальную реакцию принимается следующая:
> НО*. A3а)
На основании этого были предложены различные реакционные схемыi
касающиеся образования перекиси водорода из радикала пергидроксила и раз-
разложения перекиси и НО2 в различных реакциях с участием свободных радика-
радикалов. В число этих реакций включены и такие, в которых участвуют ионные
частицы и которые должны объяснить влияние рН на результаты опытов.
Разложение перекиси водорода при действии облучений подробнее рассматри-
рассматривается в гл. 8.
Проведен ряд исследований по влиянию излучений на различные водные
растворы. Кинетика этих процессов очень сложна; результаты исследований во
многих случаях являются противоречивыми, а поэтому можно сделать лишь
небольшое число обобщений. Протекающие процессы обычно согласуются
с постулированным начальным образованием Н- и ОН-радикалов из воды или
(в случае присутствия газообразного кислорода) образованием пергидроксила;
в дальнейшем протекают реакции этих радикалов с растворенным веществом,
хотя Лефор и Гайсинский сообщают о случае, когда арсенит в водном растворе^
по-видимому, перешел в элементарный мышьяк под прямым действием излу-
излучений [90]. В ряде случаев скорость образования перекиси водорода оказы-
оказывается более высокой, чем при облучении чистой воды; так, например, ионы
галогенидов в растворе повышают количество образующейся перекиси водо-
водорода, причем йодид более эффективен, чем бромид, который в свою очередь
эффективнее хлорида. В недавно проведенной дискуссии на заседании Фара-
деевского общества [84] были сообщены результаты ряда новейших исследова-
исследований по влиянию растворенных веществ. В этих сообщениях содержатся также
ценные ссылки на предыдущие работы. Из других новых работ нужно указать
на облучение рентгеновскими лучами водных растворов йодноватокислого
калия [101], йодистого калия [102], дезоксирибонуклеиновой кислоты [103}
64 Глава 2
и молочной кислоты [99], облучение борной кислоты и других электролитов
1104], облучение а- и рентгеновскими лучами водного раствора фермента кар-
боксипептидазы 1105] и облучение растворов бора излучениями от атомного
реактора [106]. Особенно подробно исследовано поведение растворов серно-
сернокислого закисного железа при действии излучений [107], поскольку эта система
обычно используется в качестве эталона для химической дозиметрии ионизи-
ионизирующих излучений.
Образование перекиси водорода при облучении воды, особенно в при-
присутствии кислорода, привело к умозрительному выводу, правда подкреплен-
подкрепленному некоторыми доказательствами, что повреждение живых клеток при
интенсивном облучении отчасти может быть обусловлено химическим механиз-
механизмом с участием перекиси водорода или других перекисей [108, 109]. Конечно,
протекающие при этом процессы очень сложны и связаны, без сомнения, с раз-
различными другими реакциями с участием свободных радикалов, но, поскольку
основное содержимое большинства клеток (около 70%) представляет собой
воду, образование следов перекиси водорода должно быть весьма вероятным.
Угнетающее действие перекиси водорода на биологические процессы рассматри-
рассматривается в другом разделе. Особенный интерес представляют два доказательства:
1) отмечено, что добавление перекиси водорода в среды с бактериями может
вызвать появление мутантных штаммов, аналогичных наблюдаемым при облу-
облучении бактерий, 2) найдено, что различные биологические структуры при
облучении в атмосфере с недостаточным содержанием кислорода разрушаются
меньше, чем при достаточной концентрации кислорода. Так, при более низком
парциальном давлении кислорода, чем в воздухе, крысы в состоянии перенести
значительно большие дозы излучений, а рентгеновское облучение вредителей
растений и фруктов вызывает меньшее количество изменений хромосом и мута-
мутаций. При бомбардировке рентгеновскими лучами это согласуется с установлен-
установленной четкой взаимосвязью между количествами образовавшейся перекиси
водорода и присутствующего кислорода. Дальнейшие данные по этому вопросу
можно получить из недавно проведенного семинара по химии биологических
последействий ультрафиолетовых и ионизирующих излучений [ПО].
Звуковые и ультразвуковые колебания
Доказано, что звуковые волны возбуждают или ускоряют самые различ-
различные химические реакции. Особенно подробно изучены частоты, превышающие
предел слышимости человеческого уха (примерно свыше 15—20 кгц). Обнару-
Обнаружено образование небольших количеств перекиси водорода при действии на
воду ультразвуковых волн частотой 400, 500, 540 и 985 кгц [111—113] и звуко-
звуковых волн частотой 9 кгц [114]. Этот эффект наблюдается только в том случае,
когда вода содержит растворенный кислород, воздух или какой-либо другой
газ. В присутствии воздуха констатировано также образование азотистой и
азотной кислот. Эффект образования перекиси водорода невелик—многочасо-г
вое воздействие ультразвуковых волн дает Н2О2 в количестве всего около
20 мг/л; это увеличивает трудность обнаружения указанных фактов, и меха-
механизм, по которому используется ультразвуковая энергия, до сих пор еще
не выяснен.
Поскольку превращение воды и кислорода в перекись водорода или же
воды в перекись водорода и водород является весьма эндотермическим, дей-
действие ультразвуковых колебаний, очевидно, нельзя объяснить исключительно
тем, что они дают необходимую энергию активации; это может быть их основ-
основной ролью лишь при некоторых экзотермических реакциях. Количество обра-
образующейся перекиси водорода достигает максимума в том случае, когда экспе-
экспериментальные условия способствуют максимальной кавитации—появлению
«полостей» или пузырьков в жидкости. Эти пузырьки, вероятно, образуются
Процессы облучения 66
в течение отрицательной фазы звуковой волны, когда вода подвергается растя-
растяжению, а не сжатию. Кавитация обычно не происходит в обезгаженной жидко-
жидкости; ее появлению мешает также высокое давление. Близкая зависимость между
образованием перекиси водорода и кавитацией доказывается опытами Полоц-
Полоцкого [115], который получал неультразвуковые кавитации пропусканием пара,
имеющего температуру 150 или 250°, из капилляра диаметром 0,5 мм в воду
с температурой 20 или 50°. В том случае когда температура воды была 20°,
л пара 250°, обнаружено 0,255 мг Н2О2 на 50 мл воды через 20 мин. и 0,635 мг
через 60 мин. Количества образовавшейся перекиси водорода в этих опытах
при температуре воды 50° в 2—2,5 раза ниже, чем при 20°; уменьшение темпе-
температуры пара также несколько снижает образование перекиси водорода. Пере-
Перекись водорода не обнаружена при действии ультразвука на воду при давлениях
ниже 100 мм рт. ст., но выше этого давления количество ее возрастает с повы-
повышением давления вплоть до достижения максимума примерно при 1520 мм,
после чего вновь падает с ростом давления и доходит до нуля приблизительно
при 4180 мм рт. ст.
Много авторов выдвинули теории для объяснения зависимости между
кавитацией и химической реакцией. Все предложенные теории можно разбить
на две большие группы. Одна базируется на том, что при кавитации наблю-
наблюдаются различные явления сцинтилляции и люминесценции, и приписывает
химические реакции электрическим явлениям, протекающим на поверхности
раздела между газом и жидкостью в пузырьках. Эти явления способствуют
образованию заряженных частиц и свободных радикалов или только свободных
радикалов, причем как те, так и другие могут реагировать с образованием
обнаруженных продуктов. Другой возможностью является образование фото-
фотонов, способных возбудить химические реакции. По другой группе теорий пред-
предполагается, что мгновенные местные высокие давления и температуры, возни-
возникающие при разрушении пузырьков, возбуждают реакции за счет прямого
образования атомов и свободных радикалов, как например Н и ОН.
Образование перекиси водорода при ультразвуковой бомбардировке
воды было недавно предметом подробной работы Шика [113]. Этот автор дает
обзор и предыдущих исследований химических реакций при ультразвуковой
бомбардировке с исчерпывающим списком литературы. Шик показал, что
в отсутствие растворенного газа перекиси' водорода совсем не образуется.
Наличие кислорода способствовало образованию перекиси, но без выделения
заметных количеств водорода. Этим подтверждаются предыдущие работы.
В присутствии гелия (вместо кислорода) образуются водород и перекись водо-
водорода вместе с некоторым количеством газообразного кислорода (последний мог
возникнуть за счет разложения образовавшейся перекиси водорода). При
изменении рН максимум образования перекиси водорода наблюдается при рН 4;
весьма вероятно, это обусловлено тем, что перекись водорода обладает макси-
максимальной устойчивостью вблизи рН 4 (см. подробно в гл. 9). В интервале темпе-
температур 10—85° максимальная скорость образования отмечена при 45°. Предло-
Предложены различные возможные интерпретации этого обстоятельства, включающие
влияние температуры на: 1) свойства воды, 2) скорость образования центров
реакции, 3) скорость роста пузырьков, 4) механизм реакции в пузырьках и
5) скорость разложения перекиси водорода.
Особенно интересным является наблюдение, что в замкнутой системе,
содержащей только воду и гелий, достигается стационарное давление газа
1—2 см рт. с г. независимо от интенсивности ультразвука (в пределах изучен-
изученных интенсивностей). Газ состоит главным образом из водорода с некоторым
количеством кислорода, а жидкость содержит перекись водорода. Стационар-
* ные условия могут быть достигнуты и в том случае, если исходить из водорода
и перекиси водорода; в таких условиях потребляется водород. Результаты
работы аналогичны наблюдаемым при описанной выше радиохимической бом-
5 Перекись водорода
66 Глава 2
бардировке, причем Шик считает, что в этом случае происходят те же реакции
с участием свободных радикалов. В оптимальных условиях D5° и интенсив-
интенсивность поглощенных ультразвуковых колебаний около 21 вт на 75 мл) выход
перекиси водорода составлял около 0,01 молекулы на 100 эву тогда как по Алле-
ну [85] число молекул воды, разложенных при бомбардировке различными
ионизирующими излучениями, составляет от 0,2 до 2,0. Шик показал также,
что ультразвуковое облучение водных растворов 4,4',4"-гексаметилтриами-
нотрифенилметана или четыреххлористого углерода вызывает окисление их,
которое не может быть приписано перекиси водорода. Вывод Шика совпадает
с данными других исследований явлений окисления при ультразвуковом облу-
облучении [116]. Возможно, что в этих случаях активными частицами являются
гидроксильные радикалы.
Проведены многочисленные исследования других химических реакций
в водной среде при ультразвуковой бомбардировке, причем во многих случаях
в качестве промежуточного продукта образуется перекись водорода. Интере-
Интересующиеся этим вопросом могут найти подробный материал в книге
Бергмана [111].
САМООКИСЛЕНИЕ
Термин «самоокисление» применяется к реакциям между молекулярным
кислородом и различными веществами при температуре, близкой к комнатной.
Обычно эти реакции протекают медленно, но для очень большого числа органи-
органических и неорганических веществ они происходят с измеримыми скоростями.
Вместе с тем такие реакции играют большую роль в важных для жизни биоло-
биологических процессах. Самоокисление наблюдается в таких различных процес-
процессах, как ржавление железа, полимеризация высыхающих масел, выветрива-
выветривание угля, разрушение каучука и резины, прогоркание жиров и масел, обмен
веществ у бактерий и биологические процессы окисления, с которыми связано
усвоение пищи.
Большинство процессов органического самоокисления протекает по меха-
механизму с участием свободных радикалов. Это подтверждается, например, тем,
что реакции часто предшествует индукционный период и скорость ее может
быть сильно увеличена или уменьшена при наличии следов других веществ.
Первичным продуктом самоокисления органической молекулы является пер-
оксосоединение*. Предельные и непредельные углеводороды дают гидропере-
гидроперекись, альдегид образует пероксокислоту. Первой стадией, по-видимому, явля-
является образование свободного радикала R, который затем реагирует с кисло-
кислородом по цепной реакции, состоящей из следующих стадий
R-f <V-^ROO, C3)
ROO + RH -¦> ROOH + R C4)
и приводящей к образованию пероксосоединения. В некоторых случаях послед-
последнее является относительно устойчивым и может быть выделено из реакционной
смеси с высоким выходом (например, гидроперекиси, образующиеся из изопро-
пилбензола или тетралина). Чаще, однако, это пероксосоединение неустойчиво
и быстро разлагается, а поэтому во многих случаях можно обнаружить самое
большее лишь следы пероксосоединений. В газовой фазе или в неионизирую-
щих средах это разложение обычно происходит за счет разрыва связи О—О:
ROOH -» RO + OH. C5)
* На это первым в 1897 г. указал русский ученый академик А. Н. Бах (Б а х А. Н.,
Собрание трудов по химии и биохимии, изд. АН СССР, 1950) и почти одновременно с ним .
немецкий ученый Энглер. Дальнейшее развитие теории самоокисления, сопровождавшее-
сопровождавшееся идентификацией выделяющихся промежуточных продуктов, блестяще подтвердило
теорию Баха—Энглера.—Прим. ред.
Самоокисление 67
Различные свободные радикалы, которые образуются в реакциях C3)—C5),
могут быть причиной самых различных реакций, часто приводящих к изомери-
изомеризации, полимеризации и т. п. В ионных средах реакции могут катализировать-
катализироваться кислотами или щелочами и протекать частично или полностью за счет ионных
механизмов.
Если органическая молекула может легко отдавать атомы водорода, на-
например молекула гидрохинона, то вместо гидроперекиси образуется перекись
водорода (см. ниже). В большинстве случаев перекись водорода быстро реаги-
реагирует с дегидрированным соединением с образованием других соединений. Это
заставляет думать, что так же, как и гидроперекиси, перекись водорода, ве-
вероятно, образуется в качестве нестойкого промежуточного продукта во многих
реакциях, где ее еще практически не обнаружили.
Характер начальных реакций окисления сильно зависит от температур-
температурного уровня. Так, при окислении предельного углеводорода при комнатной
или умеренно повышенной температуре может образоваться гидроперекись,
а при более высоких температурах вместо нее возникает перекись водорода
(см. ниже).
Биологические системы
Роль перекиси водорода в биологических процессах будет рассмотрена
здесь лишь вкратце. Все животные и большое число бактерий получают необхо-
необходимую им энергию путем окисления органических веществ до двуокиси угле-
углерода и воды. Этот процесс представляет цепь реакций, многие из которых не-
неизвестны или еще слабо изучены. Однако в каком-то месте этой цепи должна
происходить реакция с.участием молекулярного кислорода, что благоприят-
благоприятствует образованию перекиси водорода в такого рода процессах. На эти реакции
сильно влияют «биологические катализаторы»—ферменты.
Образование перекиси водорода обнаружено в ряде ферментных и биологи-
биологических систем, но лишь в том случае, когда система не содержит тяжелых метал-
металлов или ферментов каталазы и пероксидазы, которые сбе разлагают перекись
водорода. Как показано на стр. 347, эти ферменты способствуют либо прямому
разложению перекиси, либо ее удалению путем участия в реакциях. Перекись
водорода обнаружена в целом ряде реакций, катализированных разными окис-
окисляющими ферментами (оксидазами) и пуриновой дегидрогеназой молока.
Перекись водорода не обнаружена в клетках, которые требуют кислорода для
своего обмена (аэробных), та^ как такие клетки всегда содержат катал азу,
но она была найдена при действии кислорода на некоторые бактерии, лишенные
каталазы, например на пневмококки и стрептококки с анаэробным существо-
существованием. Перекись водорода угнетает рост анаэробных организмов типов, ука-
указанных выше. Это доказывает, что разрушение таких организмов, наблюдаю-
наблюдающееся при действии воздуха, может протекать за счет образования перекиси
водорода, являющейся ядом для процесса их сбмена. Имеются доказательства,
что антибактериальная активность человеческой слюны обусловлена присут-
присутствием стрептококков. Так, эфирные экстракты стрептококков подавляют рост
дифтерийных бактерий и стафилококков, и этот эффект приписывается содер-
содержанию перекиси водорода [117]. Этот и другие антисептические эффекты
перекиси водорода рассматриваются ниже при анализе применения перекиси
водорода в медицине (см. стр. 512 и ел.).
С точки зрения биологического сбразевания перекиси водорода одной из
наиболее интересных и лучше всего изученных систем является антибиотик,
носящий название глюкозооксидазы, нотатина или пенициллина В. В настоящее
время имеются доказательства, что антибактериальная активность пеницилли-
пенициллина может быть обусловлена образованием из него перекиси водорода под дей-
действием ферментов в присутствии глюкозы и кислорода [118]. В одном ряду
опытов рост Staphylococcus aureus тормозился при содержании всего лиш*>
5*
68 Глава 2
-0,0003% перекиси водорода; концентрации же 0,0027% и выше были бактери-
бактерицидными. В других исследованиях количества перекиси водорода, требовав-
требовавшиеся для предотвращения роста разных грам-положительных и грам-отрица-
тельных организмов, были того же порядка, что и образующиеся в среде с ан-
антибактериальным пенициллином В. Оптимальная антибактериальная актив-
активность пенициллина В наблюдается в условиях, благоприятных для образова-
образования перекиси водорода, а именно: 1) при наличии кислорода, 2) при наличии
глюкозы или подобных ей Сахаров и 3) в отсутствие заметных количеств ката-
лазы. Так, пенициллин В не обладал антибактериальной активностью в ана-
анаэробных опытах. Свежая лошадиная сыворотка или какая-либо другая
сыворотка, содержащая каталазу, является антагонистом для пенициллина В.
При строгом исключении катализаторов разложения этот фермент в состоянии
образовать значительные концентрации перекиси водорода. Франке и Дефф-
нер [119] кратко сообщают, что очищенная глюкоксидаза (пенициллин В) обра-
образовала в одном случае 4%, а в другом 7% перекиси водорода. Представляют
интерес дальнейшие исследования по характеру и предельным возможностям
этого действия.
Более подробные данные о самоокислении и биологическом окислении
читатель может найти в гл. 7, а также в книгах, обзорах и сводных сообщениях,
перечисленных в ссылке [120].
Самоокисление металлов
При действии кислорода и влаги на многие металлы образуются неболь-
небольшие количества перекиси водорода, которую определяли качественно колори-
колориметрическим методом, например с титановой солью, или путем эффекта Рассела.
Этот эффект основан на том, что фотопластинки весьма чувствительны к очень
небольшим количествам перекиси водорода. Так, Рассел показал, что ряд ве-
веществ, в том числе различные металлы, особенно после свежей шлифовки поверх-
поверхности, дают фотографические изображения при выдерживании их вблизи фото-
фотопластинки в темноте. Доказано, что это обусловлено выделением перекиси
водорода. Перекись водорода по одному из указанных методов обнаружена
при окислении следующих металлов: цинка, свинца, олова, серебра, ртути,
меди, алюминия, кадмия, магния и железа [121, 122]. Вполне вероятно, что она
образуется также при окислении многих других металлов. Очень трудно от-
открыть ее на таких металлах, которые являются активными катализаторами раз-
разложения перекиси водорода, например на железе, меди и свинце. По-видимому,
концентрация перекиси водорода, возникающей при самоокислении металлов,
определяется относительными скоростями реакций образования и разложения;
открытие перекиси водорода тем или иным автором зависит от чувствитель-
чувствительности применяемой им методики, а также от условий опыта. Более высокие
концентрации перекиси водорода обнаруживаются на поверхностях свежешли-
фованного] металла, а также (по крайней мере в случае алюминия) в слабо-
или умереннокислых или слабощелочных водных растворах. В процессе окис-
окисления металл приобретает отрицательный потенциал. Анодная поляризация
металла подавляет образование перекиси водорода, катодная поляризация
способствует этому образованию. Сказать точно, требуется ли обязательно
наличие и воды и кислорода для образования перекиси водорода, не представ-
представляется возможным, однако весьма вероятно, что требуется. В одном опыте
образец алюминия в сухом азоте дал слабое фотографическое изображение, но,
вероятно, он адсорбировал кислород и воду (или только воду) из воздуха до
помещения в инертную атмосферу.
Грюнберг [123] приписывает образование перекиси водорода электронам,
отрывающимся от поверхности металла и способным возбуждать, как описано
в предыдущих разделах, реакции с участием свободных радикалов. Испускание
Самоокисление 69
электронов свежешлифованными поверхностями металла при сравнительно
низких температурах недавно доказано Крамером [124] при помощи модифи-
модифицированного счетчика Гейгера—Мюллера, причем Крамер показал, что интен-
интенсивность испускания электронов падает во времени от начала экспозиции.
Грюнберг провел опыты с резанием различных металлов под водой и показал,
что в аналогичных условиях цинк, алюминий, магний и никель образуют при-
примерно одинаковые количества перекиси водорода, причем одновременно возни-
возникает также гидроокись соответствующего металла. При резании меди обнару-
обнаружено ничтожное количество перекиси водорода.
Вероятно, что в определенных условиях кислородная коррозия многих
металлов, например ржавление железа, проходит через образование перекиси
водорода в качестве промежуточного продукта при катодном восстановлении
кислорода. Механизмы таких процессов рассматриваются в этой главе ниже.
Образование перекиси водорода при коррозии было постулировано Данстеном,
Джауэтом и Гулдингом [125] в 1905 г., а также другими авторами, но эти на-
начальные перекисные теории коррозии только затормозили развитие правиль-
правильных представлений о причинах, лежащих в основе образования перекиси при
коррозии.
Перекись водорода легко образуется при действии кислорода на ртуть или
амальгамированные металлы в кислых растворах или в среде, состоящей
в основном из какого-либо спирта. Так, Фармэн и Мюррей [126] показали, что
при совместном встряхивании ртути, соляной кислоты и кислорода образуется
перекись водорода и однохлористая ртуть, причем в начальных стадиях реак-
реакции наблюдается образование некоторого количества соли двухвалентной ртути.
При встряхивании ртути с чистой водой и кислородом образуется ртутное сое-
соединение, но не перекись. Эти данные могут быть обусловлены тем, что катали-
каталитическая активность ртути является минимальной в кислом растворе. Мюллер
и Борхман [127] получили растворы, содержащие до 3,77% перекиси водорода,
путем введения 3%-ной кадмиевой амальгамы в соприкосновение с кислородом
при 0° в среде, состоявшей из этилового спирта и 50%-ной серной кислоты.
Одновременно происходило образование и сернокислого кадмия. Были выданы
патенты на образование перекиси водорода путем приведения в контакт амаль-
амальгамы, содержащей около 0,0003%—0,001% щелочного металла, с кислородом
в присутствии воды или жидкости типа метилового спирта, содержащей неко-*
торое количество воды^{128]; одновременно образуется и соответствующая
щелочь. Амальгама может быть получена электролизом соединений щелочных
металлов на ртутном катоде или растворением щелочного металла в ртути.
Обнаружено образование небольших количеств перекиси водорода и при
растворении золота в водных растворах цианистого калия при перемешивании
воздухом [129] и при окислении щавелевой кислоты ионом трехвалентного
марганца в ограниченном интервале кислотности [130].
Образование перекиси водорода при самоокислении металлов сильно за-
затрудняет проведение микроанализов, если они проводятся при доступе возду-
воздуха, и приводит к искажению их результатов. Так, при микроаналитическом
определении железа этот элемент восстанавливают серебром. Анализ должен
проводиться без доступа воздуха во избежание образования небольших коли-
количеств перекиси водорода, которые могут оказать нежелательное влияние на
результаты [131].
Образование перекиси водорода на металлах путем восстановления кисло-
кислорода на катоде рассматривается ниже (стр. 82).
Самоокисление органических соединений
Уже указывалось, что по современным воззрениям самоокисление органи-
органических веществ связано с начальным образованием органической перекиси
70 Глава 2
или перекиси водорода. Если органическое соединение содержит легко отщеп-
отщепляемые атомы водорода, то происходит дегидрирование с образованием пере-
перекиси водорода. Начиная с работ Шенбейна в середине прошлого столетия,
многие авторы сообщали об обнаружении ими следов перекиси водорода в про-
продуктах различных реакций самоокисления. Однако существенных количеств
перекиси при этом выделить не удавалось вплоть до работы Уолтона и Фил-
сона[132] в Висконсинском университете в 1932 г. Эти авторы показали, что
гидразобензол в спиртовом или бензольном растворе может быть окислен воз-
воздухом или кислородом с образованием перекиси водорода и азобензола при
протекании побочных реакций в ничтожных размерах. Аналогичные результа-
результаты констатированы для разных других арилгидразосоединений:
R-NH-NH-R' + O2 -» R-N = N-R' + H2O2. C6)
В 1936 г. Уолтон и Филсон получили основной патент на окисление само-
самоокисляющихся соединений в качестве метода получения концентрированной
перекиси водорода [133]. В этом патенте перечислено большое количество
пригодных для этого соединений, в том числе антрагидрохинон, а также раз-
различные гидразопроизводные. В течение нескольких лет после 1932 г. выдано
большое число патентов разным лицам в США и других странах; в этих патен-
патентах защищались различные технологические усовершенствования и указыва-
указывались дополнительно пригодные органические соединения [134]. Хотя многие
органические вещества могут образовать перекись водорода при самоокисле-
самоокислении, но в отсутствие катализатора такого рода реакция рбычно протекает срав-
сравнительно медленно. Отдельные исключения, в том числе имеющие интерес для
промышленности, представляют реакции диоксималеиновой кислоты и некото-
некоторых ароматических соединений, к которым относятся такие многоатомные
фенолы, как гидрохинон, гидразосоединения и гидропроизводные (лейкоформы)
некоторых красителей, превращающихся при окислении в хиноны, например
метиленовый голубой и антрахиноновые кубовые красители. Однако во многих
из этих случаев перекись водорода исчезает при последующих реакциях с та-
такой же скоростью, с какой образуется. Сложность этих реакций и зависимость
их от строения самоокисляемого соединения иллюстрируется данными по само-
самоокислению гидрохинона и его гомологов в слабощелочном растворе, получен-
полученными в работе Джеймса, Снел л а и Вайсбергера [135]. Первой стадией являет-
является, вероятно, образование хинона и перекиси водорода:
О
ОН
ш —' 1* w ~ v «I I I Iv w ' * '
П'.
OH
Однако хинон быстро реагирует с перекисью водорода с переходом в оксихи-
нон:
о О
НаО, C8)
в результате чего в системе не могла быть обнаружена перекись водорода до
тех пор, пока указанным авторам не удалось «связать» хинон непосредственно
после образования путем добавки к реакционной смеси 2-метилбензотиазолме-
Самоокисление 71
то-/г-толуолсульфоната. Обычно оксихинон образуется и полимеризуется
в гуминовые кислоты (СвН4О3)х. Если реакционноспособные атомы водорода,
связанные с бензольным кольцом, заменить неактивными атомами или груп-
группами, то самоокисление не сопровождается никакими другими реакциями,
что обусловливает возможность простого выделения образовавшихся хинона
и перекиси водорода. Так, самоокисление дурогщфохинона или антрагидро-
хинона дает количественное образование перекиси водорода и соответствующей
структуры хинонного типа.
ОН *
I Н
н3с- '
Иен 3
Н 1 Н
ж
Дурогидрохинон Антрагидрохинон
В интервале рН от 7,2 до 8,2 скорость окисления различных гидрохинонов
пропорциональна квадрату концентрации гидроксильных ионов, откуда цити-
цитируемые авторы сделали вывод, что первой стадией реакции является образова-
образование аниона хинона с двумя зарядами, который затем уже реагирует с кисло-
кислородом:
О- О
А- &
Реакция сильно ускоряется небольшими количествами хинона, и не всегда
скорость ее пропорциональна концентрации кислорода, что приводит к пред-
предположению, что реакция C9) может происходить различными путями и может
быть связана с промежуточным образованием сложной структуры—возможно,
заряженного радикала.
Образование перекиси водорода в процессе окисления восстановленных
(лейко) антрахиноновых и антрахиноидных кубовых красителей изучено Этер-
тэном и Тарнером [136], которые указывают, что перекись водорода может
образоваться в качестве промежуточной стадии при окислении целлюлозы.
Это окисление инициируется или ускоряется при изменениях системы кра-
красителей.
Производство процессами самоокисления
Использование процесса самоокисления для производства перекиси водо-
водорода подробно изучалось многими компаниями в США и других странах, в том
числе «I. G. Farbenindustrie» в Германии и фирмами «duPont Co.», «Buffalo
Electro-Chemical Co.» и «Mathieson Alkali Works» в США. Впервые этот процесс
был осуществлен в техническом ^^сштабе в Людвигсгафене (Германия) кон-
концерном «I. G. Farbenindustrie», который во время второй мировой войны по-
построил и эксплуатировал небольшой завод, где в качестве самоокисляемого
вещества использовался 2-этилантрахинон. Производительность этого завода
составляла около 5 т 20%-ной перекиси водорода в сутки. Затем предполага-
предполагалось построить два завода промышленного значения в Гейдебреке (Силезия)
и в Вальденберге [137]. Сначала было принято, что эти заводы будут иметь
мощность 2000 т перекиси водорода в месяц, в расчете на ь100%-ный продукт,
72
Глава 2
но в 1944 г. намеченную производительность было решено уменьшить в 3 раза.
Заводы были построены лишь частично. По процессу «I. G. Farbenindustrie»
имеется значительный материал, основанный на послевоенных отчетах бригад,
инспектировавших немецкие заводы [137].
По германскому процессу, часто носящему название «Ридль-Пфлейдерер»,
2-этилантрахинон* растворяют в растворителе, называемом «паральк» и вы-
выбранном на основании тщательных исследований. Этот растворитель представ-
представляет собой смесь равных частей бензола и вторичных спиртов с длиной цепи
С7—Сш получаемых путем кетонизации жирных кислот С4—Св, приготовлен-
приготовленных в процессе окисления парафина, с последующим гидрированием кетонов
до вторичных спиртов. Выбор пал на 2-этилантрахинон, а не на сам антрахинон,
Скелетныр
никелевый __
катализатор f
г Азот I
Охлаждающая
вода —
* Рабочая
жидкость
Кислород
Таким образам
мотет быть отведено
неболыиое количество
" teu жидкости
Вода
Рабочая
жидкость
20-25-ная
Н202
33%-ный раствор
карбоната
1 калия
\ кал(
3\
В концентратор
с возвратом
в виде 50%-ного
раствора
Рис. 7. Поточная схема 2-этилантрахинонового процесса по-
получения перекиси водорода по Ридлю-Пфлейдереру.
/—гидрогенизатор; 2—фильтр; \3—холодильник; 4—окислительный реак-
реактор; 5—экстракционная колонна; 6—слой никельсеребряного катализа-
катализатора; 7—слой глины; 8—сушильная колонна.
по-видимому, вследствие лучшей растворимости этого соединения и большей
скорости его реакции. Гидрирование этого антрахинона в присутствии скелет-
скелетного никелевого катализатора прекращали на промежуточной стадии образо-
образования хингидрона ввиду меньшей растворимости полностью восстановленного
2-этилантрагидрохинона.
Восстановление производится непрерывно при 30—36° со взвешенным
в бензоле скелетным никелевым катализатором, который добавляют непрерыв-
непрерывно со скоростью около 3—5 г катализатора на каждый килограмм готовой
перекиси водорода (см. рис. 7). С целью обеспечения достаточной разно-
разности давлений для последующей фильтрации применяется давление водорода
около 2 am. Бензол должен быть свободным от тиофена, водород должен быть
тщательно очищен во избежание отравления катализатора. В лабораторных
операциях использование водорода доходило до 90%, на опытной установке
оно составляло около 80%, длительность реакции составляла полчаса. Раствор
необходимо тщательно отфильтровать от тонкодиспергированного катализатора
перед началом самоокисления, иначе образующаяся на следующей стадии
перекись водорода будет разлагаться. Поэтому раствор сначала выпускают
через фильтрующую перегородку, а затем еще пропускают через отдельный
* 2-Этилантрахинон получали~конденсацией этилбензола и фталевого ангидрида
в присутствии хлористого алюминия с последующим нагреванием с дымящей серной
кислотой для отщепления воды и замыкания кольца.
Самоокисление
фильтр. После этого раствор охлаждают и переводят в окислительный реактор.
На этой стадии кислород разбавляют азотом во избежание образования взрыв-
взрывчатых паровых смесей. Чтобы избежать потери растворителя, выходящий газ,,
главным образом азот, доукрепляют дополнительным количеством кислорода,
и вновь пропускают через систему. На этой стадии вместо кислорода можно
применять и воздух, заменив паральк другим подходящим и в достаточной ме-
мере нелетучим растворителем и несколько увеличив длительность окисления.
В реакционную смесь до окончания стадии окисления воду не вводят;
только после образования перекиси водорода ее извлекают из раствора путем
йротивоточной экстракции водой в насадочной колонне. Немцы применяли
раствор с концентрацией 100 г этилантрахинона на литр раствора; в этом слу-
случае теоретическая максимальная концентрация перекиси водорода в рабочем
растворе составляет 7,2 г Н2О2 в литре. Фактически концентрация Н2О2 после
получасового окисления достигала примерно 5,5 г/л, а после экстракции водой
получалась 20—25%-ная (по весу) перекись водорода.
После экстракции антрахиноновый раствор содержит около 0,1—0,3% во-
воды, небольшие количества перекиси водорода (в качестве типичной указана
концентрация 0,17 г/л), а также различные окисленные органические вещества,
например органические кислоты, альдегиды, кетоны и т. д. Эти соединения
могут отравить никелевый гидрогенизационный катализатор, а поэтому они
должны быть удалены до повторной циркуляции. По германскому процессу
рабочий раствор подвергают сушке водным раствором углекислого калия с кон-
концентрацией 33% (по весу); этот раствор извлекает также часть перекиси водо-
водорода. Органические вещества и следы воды удаляют путем адсорбции на слое
глины. Остаточную перекись водорода подвергают разложению на слое никель-
серебряного катализатора, причем иногда к возвратной жидкости до подачи
ее на носитель с катализатором для лучшего удаления перекиси водорода
и растворенного кислорода добавляют небольшое количество (около 10%) вос-
восстановленного раствора из гидрогенизатора. При этом образуется небольшое
количество воды, которое остается в рабочем растворе.
Для успеха процесса вследствие необходимости осуществлять рециркуля-
рециркуляцию больших количеств растворителя на единицу продукции решающее зна-
значение имеет выбор растворителя. Растворитель должен обладать возможно
большей растворяющей способностью в отношении самоокисляемого вещества
как в форме хинона, так и гидрохинона; для снижения потерь растворителя,
а также чтобы избежать опасности пожара и взрыва, он должен быть сравни-
сравнительно нелетучим; чтобы можно было извлекать перекись водорода в виде чи-
чистого водного раствора, он не должен растворяться в воде; наконец, он должен
быть устойчив к окислению. Существенное значение имеет и высокий коэффи-
коэффициент распределения перекиси водорода между водой и растворителем, иначе
нельзя получить перекись приемлемой концентрации и с достаточным выходом.
Наряду с паральком, использованным в Германии, предложены и различные
другие растворители; как и паральк, они состоят обычно из смеси двух'раство-
рителей, из которых один хорошо растворяет хинон, а другой—гидрохинон.
В новейших патентах предлагается [138] смесь углеводорода (например, бен-
бензола) с органическим фосфонатом, обладающим следующим строением:
/OR'
R—Р=О
таким, как диоктилстиролфосфонат, или же, смесь бензола с органическим пол-
полным эфиром фосфорной кислоты (например, трибутилфосфатом). В качестве
растворителя, который можно использовать без смешивания с другими, пред-
предложено также применять эфир двухосновной кислоты (например, дибутилсеба-
цитат или дигексилсукцинат). Перекись водорода практически не растворима
74 Глава 2
в некоторых из таких эфиров, что дает возможность выделения высококонцен-
высококонцентрированной перекиси водорода путем прямой декантации. Аналогичное пред-
предложение касается смеси растворителей с использованием какого-либо слож-
сложного эфира циклогексанола или алкилированного циклогексанола [139].
Образующийся раствор перекиси водорода оказывается насыщенным
в отношении органического (рабочею) раствора, и все продукты разложения
и окисления постепенно переходят в полученную перекись водорода. Природа
неорганических и органических примесей в вырабатываемой перекиси водо-
водорода поэтому зависит не только от природы рабочего раствора, но также и от
степени очистки его при повторной циркуляции и от свойств используемых
конструкционных материалов. 20—25%-ный раствор перекиси'водорода, кото-
который вырабатывался на германском опытном заводе, содержал железа около
0,4 мг/л (от гидрогенизационного аппарата, клапанов и т. д.), никеля—около
0,8 мг/л (из скелетного никелевого катализатора) и другие неорганические
примеси, например алюминий. Содержание органических веществ в сырой пе-
перекиси водорода колебалось в расчете на углерод вплоть до 1500 мг/л. Примеси
состояли главным образом из бензола и продуктов окисления (жирных кислот),
образовавшихся из спиртового компонента растворителя (вода, насыщенная
бензолом при 25°, содержит 620 мг/л углерода). Этилантрахинон или его про-
продукты окисления давали, по-видимому, лишь очень небольшое количество
органических примесей в растворе; сами спирты совсем не растворимы. При
б-месячном хранении содержание углерода в сырой перекиси падало примерно
№ 800 мг/л [140].
Процесс самоокисления, подобный описанному, кажется экономически
более выгодным, чем применяемые в настоящее время процессы электролиза,
так как в случае его применения отпадает большой расход электроэнергии
и, по-видимому, несколько снижаются и капитальные затраты, особенно на уста-
установке крупного масштаба. Более точная оценка должна базироваться на ряде
важных факторов, в отношении которых отсутствуют или почти отсутствуют
какие-либо данные. К этим факторам относится скорость выхода из процесса
применяемых растворителей и алкилантрахинона, стоимость строительства
завода и эксплуатации, возможность требования дополнительных стадий очи-
очистки для освобождения перекиси водорода от органических примесей и, нако-
наконец, стабильность конечной перекиси водорода, особенно в процессе дальней-
дальнейшего ее концентрирования, если оно окажется необходимым. Потенциальной
опасностью при этом процессе является возможность образования органиче-
органических перекисей в рабочей жидкости или органических веществ, переходя-
переходящих в готовую перекись водорода. На стадии восстановления часть алкил-
алкилантрахинона восстанавливается до тетрагидроантрахинона, который труднее
окислять на следующей стадии—окисления. По имеющимся данным, рабочая
жидкость в германском процессе содержала 30—40% алкилантрахинона в фор-
форме тетрагидросоединения.
В 1953 г. фирма «duPont» начала эксплуатировать завод перекиси водо-
водорода вблизи Мемфиса (Теннесси), на котором используется процесс само-
самоокисления антрахинона, аналогичный примененному на заводе «I. G. Farben-
industrie». В качестве растворяющей смеси запатентована [141] смесь первич-
первичного или вторичного нонилового спирта с метилнафталином или диметилнафта-
лином, но фактически применяемый растворитель засекречен. В патентной
литературе этой фирмы [141] указано, что 2-трет-бутилантрахинон является
наиболее подходящим хиноновым производным вследствие его сравнительно
высокой растворимости в соответствующих растворителях, однако приводятся
примеры работы и с другими производными. Подобно 2-этилантрагидрохи-
нону, 2-трет-бутильное производное при многократной рециркуляции медлен-
медленно превращается в менее растворимую тетрагидроформу, а поэтому возможно
выпадение нежелательных осадков. По-видимому, превращение в тетрагидро-
Самоокисление 75
производное обратимо, а поэтому стационарную концентрацию этого соедине-
соединения можно поддерживать ниже некоторого предельного максимума путем
соответствующего регулирования процесса гидрирования. Для этой цели не-
необходимо, чтобы парциальное давление газообразного водорода было ниже
0,9 am и размер гидрирования был ограничен 40—65% теоретического путем
регулирования времени выдерживания рабочего раствора в гидрогенизаторе.
Минимально допустимое парциальное давление водорода составляет около
0,3 am, причем лимитирующим фактором является снижение скорости образо-
образования гидрохинона. Для поддержания общего давления газа на уровне около
2 am к водороду добавляют инертный газ, например азот. В качестве катали-
катализатора восстановления алкилантрахинона палладий на активированном гли-
глиноземе, по-видимому, лучше скелетного никелевого катализатора. Указывает-
Указывается, что этот катализатор не отравляется перекисью водорода или кислородом
в рабочем растворе, что позволяет устранить стадию очистки, которая в про-
противном случае необходима. Сообщается также, что такой катализатор обладает
долговечностью, легко регенерируется и, по сравнению с никелем, в меньшей
степени способствует превращению промежуточных продуктов антрахинона
•в вещества, непригодные для ведения процесса.
Фирма «Mathieson Chemical Co.» [142] тщательно исследовала производство
перекиси водорода методом самоокисления с использованием гидразобензола
и родственных ему соединений. Эта фирма в течение 18 месяцев во время второй
мировой войны эксплуатировала опытный завод по этому методу. Гидразо-
соединение растворяют в нереакционноспособном растворителе, не смешиваю-
смешивающемся с водой, например в бензоле. Окисление может производиться воздухом
или кислородом, причем побочные реакции имеют ничтожные размеры. Пере-
Перекись водорода можно затем экстрагировать водой. Однако восстановление
азосоединения до гидразоформы, требующееся для технического циклического
процесса, оказывается значительно более затруднительным, чем соответствую-
соответствующая стадия для алкилантрахинона. Каталитическое восстановление водоро-
водородом вызывает частично разрыв связи N = N. Таким образом, азобензол образует
заметные количества анилина. Предложен метод электролитического восста-
восстановления, но лучшим методом является применение металла в водном растворе
щелочи. Для этой цели предложены олово, цинк, железо и натрий; железо,
однако, вызывает заметное образование анилина; из других восстановителей
самым дешевым является металлический натрий. Применять его, как это
защищается рядом патентов, целесообразно в виде натриевой амальгамы, на-
например с 0,2% натрия. Такую амальгаму можно получить в обычном ртутном
электролизере, и ртуть от стадии восстановления азосоединения вновь пода-
подавать в ванны. При этом в качестве побочного продукта получается едкий натр.
Окисление гидразобензола протекает сравнительно медленно; применение
аминозамещенных соединений с водой повышает скорость, но эти соединения
образуют эмульсии, с трудом поддающиеся разрушению. Некоторые алкил-
замещенные производные, например л-гидразотолуол, оказываются наиболее
подходящими, поскольку они легче окисляются, чем бензольные производные,
и вместе с тем не образуют эмульсии. Этот процесс также изучался в Германии
во время второй мировой войны, но был оставлен, так как давал худшие
результаты, чем антрахиноновый процесс. Фирма «Mathieson Co.» также отка-
отказалась от дальнейшей эксплуатации этого процесса.
Самоокисление гидразин а
При исследовании самоокисления разбавленных водных растворов гидра-
гидразина @,25—Ш) Джилберт [143] и Одрит и Мор [144] обнаружили, что в каче-
качестве промежуточного продукта образуется некоторое количество перекиси
водорода, хотя конечными продуктами являются вода, азот и следы аммиака.
76 Глава 2
В работах с 0,635 М раствором гидразина Одрит и Мор нашли, что добавка
8-оксихинолина, ингибитора разложения перекиси водорода, приводит к зна-
значительно более высоким концентрациям перекиси водорода, чем в других слу-
случаях, и заметно снижает скорость самоокисления гидразина. Однако в кон-
концентрированном гидразине добавка 8-оксихинолина или соединений, катали-
катализирующих разложение перекиси водорода, например каталазы или йодистого
калия, не влияет на основную скорость окисления.
ОКИСЛЕНИЕ УГЛЕВОДОРОДОВ
Реакция углеводородов с кислородом может приводить к образованию
перекисей вместе с другими самыми различными веществами, в том числе
с окисью и двуокисью углерода, водородом, водой, непредельными соедине-
соединениями (из насыщенных углеводородов), органическими кислотами, кетонами,
альдегидами и спиртами. Понятно, что глубокие знания и теория этих весьма
сложных реакций развиты гораздо меньше, чем менее сложных, но все же
достаточно неясных и еще плохо изученных реакций водорода с кислородом.
Если реакция проводится при достаточно высоких температурах, то перекиси
удается выделить только в том случае, когда газовую смесь подвергают резкому
охлаждению через небольшой промежуток времени от начала реакции; однако-
перекиси, вероятно, образуются на начальных стадиях процессов окисления
практически любых углеводородов. Реакции сгорания углеводородов отличают-
отличаются двумя особенностями: 1) после приведения реакционной смеси к температуре
реакции проходит известный промежуток времени различной продолжитель-
продолжительности, называемый индукционным периодом, после которого только наблю-
наблюдается заметное взаимодействие; небольшие количества различных добавок
оказывают заметное влияние на длительность индукционного периода и на
образующиеся продукты; 2) характеристика реакции может быть значительно'
изменена в зависимости от конфигурации реакционного сосуда и природы era
поверхности. Оба эти явления характерны для цепных реакций с участием
свободных радикалов.
Часто трудно определить, представляют ли собой перекиси, выделенные
из реакционной смеси, перекись водорода или же они являются органическими
перекисями; до самого последнего времени было предпринято лишь немного
попыток определить строение этих перекисей. Выводы относительно характера
перекисей могут быть сделаны на основании следующих доказательств: 1) со-
состава газа и жидкости, образующихся при разложении перекиси (например,
перекись водорода дает при этом кислород и воду; гидроперекись оксиалкила
при щелочном разложении дает водород и кислоту; гидроперекись метила при
разложении на платиновой черни [145] дает двуокись углерода); 2) разных
цветных реакций, например реакции с применением титановой соли, которую
считают весьма специфичной для перекиси водорода (см. гл. 10); 3) характери-
характеристики реакции с кислым раствором йодистого калия (гидроперекись метила,
например, реагирует лишь в присутствии сернокислого закисного железа как
катализатора, но не реагирует в присутствии молибдата аммония [146); кроме
того, скорость окисления йодида до йода заметно зависит от характера пере-
перекиси [147, 148]); 4) образования нерастворимых неорганических перекисей,
например перекиси кальция или пероксобората натрия, при введении соответ-
соответствующих добавок к продукту, что доказывает наличие перекиси водорода или
гидроперекисей оксиалкилов; 5) сравнения спектров поглощения с этими
спектрами для известных перекисей [149, 150]; 6) определения коэффициентов
распределения с эфиром [151]; 7) методов хроматографического разделения
[146, 152]; 8) определения скорости термического разложения различных пере-
перекисей при температуре реакционной зоны и 9) методов полярографии [152—154}
(см. гл. 10).
Окисление углеводородов 77
Задача анализа осложняется также последующими реакциями, протекаю-
протекающими между веществами, содержащимися в конденсированной фазе, получаемой
путем охлаждения продуктов окисления углеводородов. Например, перекись
водорода и альдегиды реагируют с образованием перекисей оксиалкилов типа
RCH(OH)OOH и RCH(OH)OOCH(OH)R [155, 156]. Недавно исследована
проблема анализа водных растворов, содержащих смеси перекиси водорода,
ацетальдегида и формальдегида, на отдельные составные части [157].
Доказано, что перекись водорода образуется при окислении метана [158],
этана [159], пропана и высших гомологов [151, 159—165] и при работе двига-
двигателя (без искрения) на «-гептане, изооктане или других углеводородных топ-
ливах [166]. Уровень температуры, требуемой для получения достаточно
значительной скорости реакции, падает в следующем ряду: метан, этан, пропан.
Это явление объясняется сравнительными прочностями связей С—Н в этих
разных видах молекул. Если реакция полностью протекает при температурах
порядка 350° или выше, то по меньшей мере в случае метана, этана и пропана
перекись в конденсированной фазе представляет либо перекись водорода, либо
продукты ее конденсации с присутствующими кислородсодержащими молеку-
молекулами, например с альдегидами. Наоборот, при самоокислении или низкотем-
низкотемпературном окислении различных углеводородов, например при 100—150°,
можно выделить гидроперекиси алкилов ROOH.
Образование перекиси водорода при окислении пропана изучено значи-
значительно подробнее, чем при окислении других углеводородов. Поэтому в качестве
примера мы рассмотрим реакции, наблюдаемые при окислении пропана. Про-
Пропан представляет собой самую маленькую молекулу углеводорода, которая
содержит и вторичный и первичные атомы углерода и, кроме того, представляет
технический интерес, так как пропан получается в значительном количестве
при переработке нефти. Как в статических, так и в динамических опытах ско-
скорость окисления смесей, богатых пропаном или высшими гомологами, для из-
известного интервала температур обладает отрицательным температурным коэф-
коэффициентом. Так, Пиз [151] обнаружил максимум скорости при температуре
около 330° с последующим падением до минимума приблизительно при 380°;
после этого скорость с повышением температуры вновь возрастала. Эти данные
относятся к эквимолекулярной смеси пропана и кислорода при общем давле-
давлении 200 мм рт. ст. Точно так же в новейших работах [160—163], проведенных
со смесью из 5,5 молей пропана на 1 моль кислорода в динамических условиях
при атмосферном давлении, обнаружен максимум скорости реакции при 375° и
снижение до минимума при 425°; при дальнейшем повышении температуры
скорость вновь возрастала. Аналогичные данные сообщены Гаррисом [161],
Черняком и Штерном [162] и Коойманом [159].
В типичном динамическом опыте газы вводят в реакцию на несколько
секунд и затем подвергают их охлаждению до комнатной температуры в поверх-
поверхностном конденсаторе. При этом получается жидкая фаза, содержащая воду,
метиловый спирт, перекись водорода и альдегиды и их продукты присоединения
в сочетании с газовой фазой с непредельными углеводородами (пропиленом
и этиленом), метаном, окисью углерода, непрореагировавшим пропаном и не-
небольшими количествами несконденсировавшихся компонентов жидкой фазы.
В начальных стадиях реакции наблюдается образование небольших или
ничтожных количеств водорода, двуокиси углерода и органических кислот;
в этот период возникают максимальные количества перекиси водорода [159,
160, 163].
Максимальная концентрация перекиси водорода в газообразных продук-
продуктах обнаружена при молярном отношении пропана к кислороду, равном 3 : 10,
температуре реакции около 425—485° и длительности контакта от 3 до 4 сек.
[159, 160, 163]. Максимальный вь^(ход перекиси водорода в расчете на прореа-
прореагировавший кислород наблюдается примерно при тех же условиях, но при
78
Глава!, 2
несколько меньшем времени контакта. При более низких температурах возни-
возникают большие количества частично окисленных продуктов, например альдеги-
альдегидов и спиртов. Типичный ряд экспериментальных данных для реакции при
температуре около 475° показан на рис. 8 [160]. На этом графике дано изме-
изменение состава продукта по мере протекания реакции, причем степень реакции
выражена как отношение проре-
прореагировавшего кислорода к введен-
введенному (в процентах). Можно ви-
видеть, что концентрация перекиси
водорода проходит через макси-
максимум. Падение концентрации пе-
перекиси водорода при увеличении
времени реакции может быть вы-
вызвано гетерогенным разложением
ее на стенках реактора, гомоген-
гомогенным разложением или реакциями
в газовой фазе с другими при-
присутствующими химическими ча-
частицами. Какой именно вид раз-
разложения является наиболее су-
существенным, еще с достоверно-
достоверностью не определено, но при изу-
изученных температурах в приме-
примененном лабораторном оборудо-
оборудовании наиболее вероятным яв-
является, по-видимому, гетероген-
гетерогенное разложение [160]. Количе-
Количества выделенной перекиси водо-
водорода сильно колеблются в зависи-
зависимости от характера поверхности
реактора. Если стенки реактора
покрыты хлористым калием, пе-
перекись водорода вообще не обра-
образуется. Максимальные выходы
наблюдаются в том случае, ког-
когда имеется минимальное отноше-
отношение величины поверхности реак-
гора к его объему или же когда
эта поверхность обладает особой
инертностью, например поверх-
поверхность боросиликатного стекла,
особенно после промывки его
плавиковой или азотной кислотой. В работе такой реактор может «ста-
«стареть», что сопровождается удлинением индукционного периода реакции и
уменьшением1 количества перекиси водорода, которое может быть выде-
выделено [163]. Очистка такого реактора азотной 'кислотой способствует изме-
изменению характеристики реакции в сторону характеристик, соответствующих
новому реактору.
Если к реагирующему пропану добавить до 15% пропилена, то индукцион-
индукционный период реакции сокращается и количества выделенной перекиси водорода
немного снижаются [163]. Перекись водорода обнаружена также при реакциях
этилена с гидроксильными радикалами, образовавшимися при пропускании
тлеющего разряда через водяной пар [1671.
0 20 40 60 80 100
Прореагировавший кислород, % (от поступившего)
Рис. 8. Продукты окисления пропана (тем-
(температура на входе 475°, молярное отношение
пропана к кислороду 5,6:1).
Окисление углеводородов
Механизм окисления пропана
Механизмы окисления углеводородов гораздо менее изучены, чем механизм
для системы водород—кислород, хотя природа некоторых отдельных реакций
известна довольно хорошо. Недавно этот вопрос был разобран в ряде сообще-
сообщений 1160, 163, 168, 169], а также в книгах Льюиса и фон-Эльбе [7] и Ледлера[8]
и других работах, цитированных выше. Полный анализ различных стадий реак-
реакции при окислении углеводородов занял бы здесь слишком много места. Однако,
пользуясь пропаном как примером, можно вывести некоторые общие положе-
положения. Если реакция проводится при давлении 1—2 am и в условиях, позволяю-
позволяющих выделить промежуточные продукты, то наиболее существенным фактором,
влияющим на характер обнаруживаемых продуктов, является температура
80
Пропилен'
Кислородсодержащие
'органические вещества
375
400 425 450 475
Температура, °С
Рис. 9. Влияние температуры на распределение продуктов
в начальных стадиях окисления пропана.
реакции. Окисленные органические соединения представляют наиболее часто
встречающиеся виды молекул, выделенных при температурах реакции при-
примерно до 300—350°. Дальнейшее повышение температуры способствует увели-
увеличению выхода пропилена и при подходящих условиях—перекиси водорода за
счет снижения выхода кислородсодержащих органических соединений. При
температурах, превышающих приблизительно 500°, образуются также значи-
значительные количества этилена и метана и реакция практически заключается
в пиролизе пропана, сенсибилизированном кислородом. На рис. 9 показано
изменение распределения продуктов в зависимости от температуры при прове-
проведении реакции с высоким соотношением пропана к кислороду и прекращении
реакции после израсходования лишь небольшой доли присутствовавшего
с самого начала кислорода [168].
Предполагается, что первой стадией окисления является отрыв атома
водорода от молекулы пропана с образованием пропильного радикала под
действием кислорода или других радикалов. Полагают, что все различные
продукты появляются затем за счет реакций пропильного радикала, проте-
протекающих в трех различных направлениях, причем относительное значение того
или иного направления зависит от температуры:
С3Н7 + О2 -^ С3Н7ОО —> кислородсодержащие частицы,
С3Н7->С2Н4
D0)
D1)
D2)
"80 Глава 2
Перекись водорода, вероятно, образуется за счет реакции радикала пергидро-
ксила с пропаном
НО2 + С3Н8 -> С3Н7 + Н2О2. D3)
Образование и разложение НО2 и перекиси водорода предполагается также
и в реакции между окисью углерода и кислородом, катализируемой водой
{7, 170].
Получение перекиси водорода путем окисления органических веществ
Некоторые авторы [159, 160, 164, 165] изучали возможность промышлен-
промышленного производства перекиси водорода путем частичного окисления этана, про-
пропана и их высших гомологов. Окисление этих парафинов можно инициировать
при значительно более низких температурах, чем те, которые требуются для
окисления метана. Оптимальный рабочий режим окисления парафинов трудно
точно установшь, так как максимальное превращение кислорода в перекись
водорода может происходить при несколько иных условиях, чем те, которым
соответствует наибольший выход перекиси водорода на каждый объем пропу-
пропущенного через реактор газа. Оптимальные температуры лежат внутри интер-
интервала 400—500°*.
Этот процесс производства может представлять интерес в том случае,
когда цена на газообразные углеводороды определяется лишь стоимостью их
как топлива. Преимущества этого процесса заключаются в низкой себестоимо-
себестоимости при работе с газами и жидкостями, в простоте оборудования по сравнению
с необходимым для электролиза, в малом расходе электроэнергии и в образова-
образовании значительных количеств олефинов в качестве побочных продуктов; недо-
недостатками являются небольшая доля газа, превращаемая в перекись водорода
и олефин за один проход, и особенно трудность и высокая стоимость выделения
перекиси водорода из конденсата, в котором она образует перекиси оксиалки-
лов с присутствующими альдегидами. Однако можно было бы технически ис-
использовать содержащий перекись конденсат непосредственно, например в ка-
качестве отбеливающего вещества, без попытки дальнейшего выделения перекиси.
Изучены различные методы отделения перекиси водорода. Один [159, 172]
основан на установлении равновесия в жидкой фазе между перекисью водо-
водорода, альдегидом и перекисями оксиалкилов:
Н2О2 + RCHO ^± RCH (ОН) ООН, D5)
RCH (ОН) ООН + RCHO ^ RCH (ОН) ООСН (ОН) R. D5)
Добавка гидрата окиси кальция или гидроокисей других щелочноземельных
металлов к смеси вызывает выпадение перекиси кальция, которую затем мож-
можно превратить в перекись водорода путем обработки кислотой или двуокисью
углерода. В последнем случае образующийся углекислый кальций можно под-
подвергнуть обжигу до окиси кальция, чтобы последнюю затем вновь использо-
использовать в процессе. Кук [172] сообщает, что при быстром проведении операции
при температурах около 0° 96,8% всей перекиси, содержащейся в водной смеси
в виде эквимолекулярных количеств формальдегида и перекиси водорода,
можно превратить в перекись кальция. Последующая обработка углекислым
газом дает возможность выделить перекись водорода в количестве 89,8% от
начального содержания перекиси в продуктах реакции. В контакте с извест-
известковым шламом смеси формальдегида и перекиси водорода более нестойки, чем
* Оптимальные температуры образования органических гидроперекисей жидко-
фазным окислением углеводородов с более высоким молекулярным весом лежат в ин-
*ервале около 100° [171].
Другие процессы окисления 81
одна перекись водорода или эквивалентные смеси с ацетальдегидом, а поэтому
для достаточно высокого извлечения перекиси необходима тщательная и быст-
быстрая работа при температурах около 0° [173]. Процент выделяющейся перекиси
падает по мере увеличения отношения формальдегида к перекиси водорода.
Если содержание перекиси водорода значительно выше, чем содержание
альдегида, органические перекиси предложено превращать путем нагревания
в неперекисные кислоты и отгонять образовавшиеся таким образом органиче-
органические кислоты в вакууме с получением перекиси водорода в остатке [174].
При этом водный раствор перекиси должен быть достаточно разбавленным,
чтобы не могли образоваться взрывчатые смеси и не происходило потерь зна-
значительной доли образовавшейся в процессе перекиси.
По другому способу конденсат можно нагревать с разбавленной серной
кислотой и метиловым спиртом, в результате чего образуются ацетали, более
летучие, чем альдегиды; их можно отогнать в вакууме, что приводит к смещению
равновесия в реакциях D4) и D5) налево [175]. Недавно проведенное исследо-
исследование этого предположения [173] показало, что добавка метилового спирта
действительно способствует разделению, но этот эффект обусловлен, по-види-
по-видимому, снижением температуры кипения смеси, уменьшающим скорость разло-
разложения перекиси, а не просто образованием ацеталей. Однако заметное разло-
разложение все же наблюдается, и процесс может оказаться взрывоопасным. Сооб-
Сообщается о сильном взрыве, происшедшем при попытках использования этого
процесса [173].
Для разделения предложена также дробная конденсация [161], поскольку
можно было ожидать, что формальдегид, ацетальдегид и метанол обладают
значительно большими летучестями, чем вода и перекись водорода. Однако
большая часть формальдегида оказывается в конденсате вместе с водой и пере-
перекисью водорода [173], т. е. подтверждается предположение, что при конденса-
конденсации имеет место образование продуктов присоединения или другие реакции,
протекающие с большой скоростью.
Запатентовано образование перекиси водорода при газофазном окислении
гидроароматических соединений [165]. Этот процесс, возможно, представляет
определенный интерес в промышленном отношении, поскольку получение
такого соединения, как 1,4-дигидронафталин, можно осуществлять цикличе-
циклическим способом путем восстановления нафталина металлическим натрием в эти-
этиловом спирте и регенерации нафталина окислением. Отношение количеств пере-
перекиси водорода и карбонильных соединений в продуктах реакции может быть
весьма значительным. Указано, что в оптимальных условиях молярные отно-
отношения этих двух продуктов составляют от 10 до 100 при соответствующих
выходах перекиси водорода в расчете на кислород от 60 до 83%. В принципе
этот процесс напоминает циклические процессы самоокисления алкилантраги-
дрохинона или гидразобензола, если не считать, что в данном случае окисление
осуществляется в газовой фазе при повышенных температурах. Однако значи-
значительное превращение «рабочего вещества» при окислении в соединения, непри-
непригодные для работы, заставляют считать этот процесс малопривлекательным.
ДРУГИЕ ПРОЦЕССЫ ОКИСЛЕНИЯ
Окисление спиртов
Некаталитическое частичное окисление первичных или вторичных спиртов
в газовой или жидкой фазе кислородом или воздухом также может дать высо-
высокие выходы перекиси водорода и альдегида или кетона [176]. Например,
при барботировании через ледяную воду эквимолекулярной смеси изопропи-
лового спирта и воздуха, реагировавшей при температуре около 485° в течение
времени, достаточного для превращения 46% поступившего кислорода, был
получен водный раствор перекиси водорода и ацетона при выходах еоответ-
6 Перекись водорода
82 Глава 2
ственно 81 и 96% в расчете на прореагировавший кислород согласно уравнению
(СН3J СНОН + О2 -> (СН3J СО + Н2О2. D6)
Оптимальные температуры реакции в жидкой фазе лежат в пределах 70—160°.
Окисление первичного спирта ведет к образованию соответствующего альде-
альдегида. Вследствие высокой селективности в производственном процессе целесо-
целесообразнее применять не первичный, а вторичный спирт, например изопропило-
вый или emop-бутиловый.
Окисление аммиака
Стефенс и Пиз [177] сообщают, что в определенных условиях газообразные
продукты реакции аммиака и кислорода, проведенной без катализатора, мед-
медленно окрашивают йодокрахмальную бумажку и обесцвечивают подкисленный
перманганат, что позволяет предполагать содержание следов перекиси. Про-
Продукты в основном представляют воду и азот. Лафт и Коэн [178] проанализиро-
проанализировали механизм горения аммиака и постулировали образование радикалов ОН
и НО2.
Реакции с участием фтора
Фтор является чрезвычайно энергичным окислителем и может превратить
водные растворы сульфатов, карбонатов, боратов и фосфатов в растворы соот-
соответствующих пероксосоединений, причем сам фтор восстанавливается до фто-
фтористого водорода. При взаимодействии фтора с одной водой образуется неболь-
небольшое количество перекиси водорода и фтористого водорода, а также различные
другие окислители, например кислород, F2O и, возможно,'озон. Природа этой
реакции очень мало известна ввиду недостаточной надежности методов анализа
продуктов. В одном опыте Фихтер и Бладергрен [179] пропускали фтор в
50 мл воды в платиновой чашке с наружным ледяным охлаждением. Макси-
Максимальная концентрация перекиси водорода 0,2% была обнаружена через
20 мин., после чего концентрация падала во времени. Эти авторы приписали
исчезновение перекиси реакции ее с озоном, поскольку после первых 30 мин.
ощущался «запах озона» и, кроме того, в результате пропускания фтора в вод-
водный раствор едкого кали при —20° получался продукт, свойства которого были
аналогичны свойствам озоната калия. Однако не менее вероятно, что перекись
водорода разлагалась за счет реакции с другими присутствующими в растворе
веществами. Хюккель [180] сообщил, что при реакции фтора с водой или льдом
образуется F2O, перекись водорода, фтористый водород и кислород, но, как
максимум, лишь следы озона.
В недавно проведенных работах Дэвис, Розен, Вингет и Висвол [181]
вводили в реакцию газообразный фтор и водяной пар в динамическом аппара-
аппарате при комнатной температуре и атмосферном давлении; они брали избыток
фтора и азот в качестве разбавителя. Продукты реакции они конденсировали
при температуре жидкого кислорода и фракционировали нагреванием до тем-
температур твердой двуокиси углерода и комнатной, а затем анализировали мето-
методами, описанными в гл. 10. При этом обнаружены фтористый водород, кисло-
кислород, перекись водорода, озон и, вероятно, некоторые окислы фтора.
КАТОДНОЕ ВОССТАНОВЛЕНИЕ КИСЛОРОДА
В 1882 г. Траубе [182] сообщил, что при электрохимическом процессе
наличие кислорода у катода способствует образованию перекиси водорода.
В этой работе, а также в дальнейших работах других авторов показано, что
выход перекиси водорода по току сильно зависит от физического и химического
характера электрода, концентрации растворенного кислорода и перекиси водо-
водорода в электролите, температуры, плотности тока, рН и состава электролита.
Катодное восстановление кислорода 83
При применении электролитической ванны с различными металлическими ка-
катодами, низких плотностей тока (например, 0,02 а/дм2) и разбавленного кислот-
кислотного электролита достигнуты почти теоретические выходы перекиси водорода.
Выход быстро падал по мере возрастания концентрации перекиси водорода или
плотности тока [182, 183]. Однако максимальные концентрации перекиси не
превышали доли процента. Более высокие концентрации перекиси в сочетании
с хорошими выходами по току наблюдались при работе с электролизером под
повышенным давлением кислорода с целью увеличения концентрации раство-
растворенного кислорода. Так, Фишер и Прис [183] тщательно исследовали влияние
давления и доказали возможность получения 2,7%-ной перекиси водорода
с выходом по току 83% и 1,3%-ной—с выходом по току 9Э% в условиях давле-
давления кислорода 100 am (самого высокого из изученных давлений) при приме-
применении катода из золотой пластинки, платинового анода, 1 %-ной серной кислоты
в качестве электролита, плотности тока 2,3-10~2 а/см2 и напряжении на ванне
около 2 в. Попытки получения более высоких концентраций перекиси водорода
неизменно приводили к резкому ухудшению выходов, то же самое наблюдалось
при более низких давлениях кислорода. Низкие концентрации перекиси
водорода, вероятно, получаются вследствие восстановления образовавшейся
перекиси до воды. Мюллер и Мельгорн [184] описали электролизер с ртутным
катодом и специальной мешалкой, при помощи которой они рассчитывали на
быстрый перевод образовавшейся на катоде перекиси водорода в раствор, что
снизило бы возможность последующего восстановления перекиси. Выдан ряд
патентов на методы осуществления катодного восстановления, в частности
с осуществлением операции под давлением и с применением различных спе-
специальных металлических электродов [185], однако техническое значение всех
этих методов в настоящее время ничтожно. Патрик и Вагнер [186] сообщили
недавно о проведенных ими работах по восстановлению кислорода на железном
катоде при очень низких плотностях тока (около 10"в а/см2). В слабокислых
средах (рН 4,25) перекиси не обнаружено, но при рН, равном 6,5—11,1, выходы
составляли от 5 до 36%. Максимальный выход получен при рН 11,1 и наиболее
низкой плотности тока из изученных. Авторы предполагают, что это обуслов-
обусловлено существованием перекиси водорода в щелочной среде главным образом
в виде пергидроксильного иона с отрицательным зарядом, что способствует
отталкиванию его от катода, тогда как в нейтральной или кислой среде моле-
молекулы самой перекиси водорода остаются вблизи электрода и поэтому могут
продолжать восстанавливаться. Другим возможным объяснением является
наличие максимума каталитической активности у соединений железа в слабо-
слабокислой среде, что может привести к исчезновению всей образовавшейся пере-
перекиси водорода (см. гл. 9). Проведен ряд исследований по восстановлению кис-
кислорода на капельном ртутном электроде [187—190], поскольку такая реакция'
иногда встречается при полярографических анализах. Изучены также реакции
на электродах из амальгамированных металлов [191]. Восстановление в поля-
рографе протекает четко в виде двух отдельных стадий. При увеличении напря-
напряжения возникает сначала первая волна, интерпретированная как восстанов-
восстановление кислорода до перекиси водорода, что обычно выражают следующим
образом:
О2 + 2Н^-(-2е->Н2О2 D7)
или
О2 + 2Н2О + 2е -> Н2О2 + 2ОН". D8).
При дальнейшем росте напряжения появляется вторая волна, соответствующая
восстановлению перекиси водорода до ОН" или воды:
-> 2ОН- D9)
6*
84 Глава 2
ИЛИ
_> 2Н2О. E0)
Истинные механизмы этих реакций еще недостаточно изучены, но вопрос об
обратимости их может быть в принципе решен при сравнении изменения потен-
потенциала с рН для каждой точки волны с рассчитанным из независимых
термодинамических данных. Разные авторы высказывают различные мнения
относительно обратимости реакции D7) или D8) в полярографическом аппа-
аппарате, но реакция D9) или E0) признается необратимой. Иофа, Шимшелевич
и Андреева [188] подробно изучили ртутный электрод и пришли к заключе-
заключению, что восстановление кислорода до перекиси водорода на этом электроде
является реакцией первого порядка по кислороду как в кислом растворе, так
и в щелочном. Постулировано, что реакция протекает через О~, НО2 и НО",
причем общая скорость определяется стадией
O2 + e->O", E1)
Восстановление перекиси водорода также оказалось реакцией первого порядка.
Делахей[192] также исследовал восстановление кислорода на поверхно-
поверхностях различных металлов и определял количество образовавшейся перекиси
водорода полярографическим методом. Образованию перекиси благоприятст-
благоприятствовали алюминий, цинк и магний, тогда как на никеле, железе, серебре, меди,
олове и свинце выходы ее были невелики. Эти результаты могут быть объясне-
объяснены, если исходить из относительных скоростей трех одновременно протекаю-
протекающих реакций: образования перекиси водорода на поверхности данного металла,
разложения перекиси за счет каталитического действия этого металла и воз-
возможных других реакций, связанных с расходом металлического электрода.
Представляет интерес также теоретический анализ процессов электровосста-
электровосстановления на металлах, данный Фрумкиным [193].
Берль [194] измерил потенциал, возникающий при пропускании кисло-
кислорода над графитовым электродом, и показал, что изменение потенциала этого
полуэлемента в присутствии разных количеств едкого кали и перекиси водо-
водорода в электролите соответствует вычисленному из независимых термодинамиче-
термодинамических данных для реакции на полуэлементе (с учетом ионизации перекиси водо-
водорода и образования КООН):
ОН- + НО" ^± О2 + Н2О + 2е. E2)
Вычисленный стандартный электродный потенциал этой реакции равен 0,0745 б.
Берль пришел к выводу, что эта реакция полуэлемента обратима и механизм
ее следующий:
01 + 2e^0J", E3)
О;- + НаО ^± НО" + ОН", E4)
Этот механизм кажется более правдоподобным, чем ранее предложенные, по
которым считают, что кислородная молекула сначала распадается на незаря-
незаряженные атомы кислорода, и не учитывают образования пергидроксила или
перекиси водорода.
Фишер и Крёниг [195] обнаружили образование небольших количеств
перекиси водорода при выходах по току до 86% в том случае, когда кислород-
кислородный электрод (на гладкой платине) представлял собой часть гальванического
элемента с водородным анодом и когда элемент замыкали накоротко; однако
при этом происходила значительная поляризация и максимальный отбирав-
отбиравшийся ток составлял только 3 ма. При применении более активных форм пла-
платины в качестве электродов получались лишь следы перекиси водорода. Берль
[194] сообщает также, что при подходящем режиме работы электролитической
Катодное восстановление кислорода 85
ячейки с кислородным электродом электрическую или химическую энергию
можно полностью израсходовать на образование перекиси водорода незави-
независимо от того, как эксплуатируется ячейка: с применением ли внешнего потен-
потенциала (как электролизер) или путем соединения кислородного электрода с под-
подходящим анодом с образованием гальванического элемента, генерирующего
электрический ток и одновременно перекись водорода. Замена графита или
угля металлическим электродом с газообразным кислородом, как правило,
дает низкие и необратимые потенциалы, неправильно изменяющиеся во време-
времени, что обычно приписывают образованию окислов, способных создавать мест-
местные самополяризующие токи между металлом и окислом. Берль не обнаружил
различия в потенциалах при применении графита и активированного угля,
хотя в его электролизерах выход перекиси водорода заметно зависел от харак-
характера электрода.
Производство перекиси водорода методом катодного восстановления
кислорода
Берль [196] изучил возможность применения катода из активированного
угля для технического производства перекиси водорода путем восстановления
кислорода в электролизере. Решающим фактором с точки зрения выхода пере-
перекиси является природа электрода—необходимо, чтобы он был изготовлен из
высокоактивного активированного угля с высокой электропроводностью. Гра-
фитированный ретортный уголь, например, не подходит для этой цели, и наи-
наилучшие результаты получаются при применении угля, приготовленного из
кислого гудрона нефтеперегонных заводов, нейтрализованного калиевыми
соединениями. Такой уголь более активен, чем полученный с применением
натриевых производных. Электрод формуется с полостью в середине и делается
настолько пористым, чтобы кислород мог проходить через него в электроли-
электролизер. Наиболее эффективными угольными электродами являются такие, кото-
которые обладают значительной удельной поверхностью пор; такие электроды яв-
являются и наиболее активными с точки зрения разложения раствора перекиси
водорода, но скорость этого разложения падает при наложении на угольный
электрод внешнего напряжения.
При применении электрода из активного угля и щелочного калиевого элек-
электролита выход по току превышает 9Э% в случае образования 2—5%-ной пере-
перекиси водорода и 65% в случае 25%-ной. Эти выходы по току несколько лучше,
чем достигнутые при применении металлических электродов [185]. Напряжение
на электролизере зависит от плотности тока: при плотности тока «несколько»
ампер на 1 дм2 напряжение составляет 1,4 в, при 20—35 а/дм2 оно равно
2,8—3,3 в. При последней плотности тока можно получить 12—15%-ный рас-
раствор перекиси водорода с выходом по току 70—80% и расходом энергии около
7,5 квт-ч на 1 кг перекиси водорода. Этот расход примерно в 2 раза меньше,
чем тот, который требуется для электролитических процессов с образованием
пероксодисульфата. Однако при данном процессе приходится обязательно при-
применять щелочной электролит (калиевые производные предпочтительнее натрие-
натриевых), а поэтому для извлечения перекиси из электролита его необходимо сна-
сначала нейтрализовать, а затем подвергнуть перегонке. Можно также растворен-
растворенную перекись превратить в нерастворимую и из последней уже регенерировать
перекись водорода с применением такой же аппаратуры, какая предложена
для извлечения перекиси водорода из конденсатов от окисления углеводородов.
Выходы в процессе катодного восстановления несколько снижаются при замене
кислорода воздухом. Этот процесс теоретически открывает возможность одно-
одновременного получения кислорода, хлора или перекисных соединений на аноде
и перекиси водорода на катоде, но технически такой процесс осуществить
затруднительно.
86 Глава 2
Приготовление электрода, пригодного для этого процесса, до-видимому,
является скорее искусством, чем научно обоснованным методом, причем эффек-
эффективность электрода изменяется во времени. На практике придется, вероятно,
довольно часто заменять угольные катоды в электролизерах свежими и подвер-
подвергать отработанные катоды регенерации для восстановления их активности.
В 1930-х годах фирма «Mathieson Alkali Works» провела ряд работ по осуще-
осуществлению процесса Берля; этот процесс изучался также другими фирмами
в США и Германии [197]. Однако практически он никогда не применялся.
Процесс восстановления кислорода на электроде из активированного
угля недавно был изучен Мизуно [198], который получил в общем те же ре-
результаты, что и Берль. Выход по току при электролизе в случае применения
едкого натра в общем низок, хотя и указано, что 8%-ный раствор едкого натра
дает такой же выход по току, как 20%-ный раствор едкого кали. Выход по току
возрастает при равномерном пропускании потока кислорода вокруг катода
и при поддержании температуры раствора ниже 20°. С наилучшим из изучен-
изученных электродов удалось получить 4%-ный раствор перекиси водорода с выходом
по току 80% путем электролиза 150 мл 20%-ного раствора едкого кали (+0,1 %
фтористого натрия) в течение 3 час. при катодной плотности тока 3,51 а/дм2
и концентрации тока 26,7 а/л. Использованные при этом исследовании электро-
электроды из активированного угля не давали обратимого потенциала в полуэлементе,
содержащем, как это делал Берль, 20%-ный раствор едкого кали; оказалось,
что при таком опыте потенциал электрода зависит от активности послед-
последнего и от результатов, которые достигаются при его применении. Сообщается
также о двух исследованиях по образованию перекиси водорода при адсорбции
кислот и кислорода на активированном угле [199].
По-видимому, во всех электролизерах с кислородным электродом кислород
сначала восстанавливается практически количественно до перекиси водорода
или HOg по реакции D7) или D8) или по реакции, предложенной Берлем.
Однако по мере увеличения концентрации перекиси равновесный потенциал
для реакции; разложения D9), E0) или E2) приближается к потенциалу
реакции образования, а поэтому перекись водорода или НОз разлагается.
Кроме того, можно наблюдать каталитическое разложение в электролите,
особенно если он сильнощелочной, а в некоторых случаях и на электроде.
Деполяризация первичных элементов
Восстановление кислорода представляет интерес также и как метод депо-
деполяризации катода первичного элемента, что допускает возникновение более
высоких потенциалов и большего тока. Так, в элементе Эдисона из угольного
катода и цинкового анода в электролите из едкого натра корпус батареи
обеспечивает доступ воздуха к катоду, вследствие чего вместо газообразного
водорода образуется перекись водорода. Однако в данном случае жела-
желательно, чтобы возникающая перекись разлагалась возможно быстрее и можно
было получать максимально возможный потенциал.
Вейс и Джэйф [200] при исследовании потенциалов на угольном кисло-
кислородном катоде подтвердили результаты работы Берля для щелочных растворов
и показали, что в кислых растворах также происходит восстановление кисло-
кислорода, но необратимо. Оказалось, что в щелочных растворах элемента с воздуш-
воздушной деполяризацией образование перекиси водорода подчиняется закону Фара-
дея и что эта перекись не восстанавливается до воды на электроде, а исчезает
из элемента лишь в результате каталитического разложения. Потенциал эле-
элемента при разрядке при любых плотностях тока в случае применения щелочного
электролита соответствовал рассчитанному для термодинамического равнове-
равновесия между кислородом и перекисью водорода после введения поправки на вну-
внутреннее сопротивление. Другое исследование первичных щелочных угольных
Литература 87
элементов с воздушной деполяризацией [201] подтвердило вышеуказанные
наблюдения в том отношении, что поглощенный из воздуха кислород превра-
превращается в перекись водорода. Показано также, что в конкретных изученных
элементах часть перекиси переносится к цинковому аноду, вызывает его депо-
деполяризацию и способствует протеканию коррозии, на 40—70% превышающей
растворение цинка под действием тока. Э.д.с. элементов на 10—20% выше при
введении катализатора разложения перекиси в электрод или при осаждении
его на поверхности. При первом методе оказались наиболее эффективными
азотнокислое серебро и марганцовокислый калий, но лучшие результаты были
получены при осаждении хлористого палладия на поверхности электрода.
Применение электродов, содержащих катализатор, увеличивало также емкость
элемента на 30—50% вследствие снижения коррозии цинка.
ЛИТЕРАТУРА
1. Leeds A. R., Annals N. Y. Acad. Sciences, 1, 405 A880), М а с h u W., Das
Wasserstoffperoxyd und die Perverbindungen, 2 Aufl., Springer Verlag, Wien,
1951 [есть перевод первого издания с дополнениями: см. Маху В., Перекись
водорода и перекисные соединения, Госхимиздат, М., 1951]; Laporte Chemicals
Ltd., Advances in Production and Uses of Hydrogen Peroxide, Communication № 83.
2. Fischer F., Ringe O., Ber., 41, 945 A908).
3. Kouzmine E., франц. пат. 940026 A948).
4. В 6 t t g e г, J. Chem. Soc. (Abstracts) 1879, p. 103.
6. Traube M., герм. пат. 27163 A884).
6. H i nshel woo d С. N., Proc. Roy. Soc. (London), A188, 1 A946); W il 1 bo ur n
A. H., Hi nshel wood C. N., ibid., A185, 353, 369, 376 A946);
Cull is С F., Hinshelwood С N., ibid., A186, 194, 462, 469 A946).
7. L e w i s В., von Elbe G., Combustion, Flames and Explosions of Gases, Acade-
Academic Press, New York, 1951 [есть перевод с раннего издания США: см. Льюис Б.,
Эльбе Г., Горение, пламя и взрывы в газах, Госиноиздат, М., 1948].
8. Lai die г J. К., Chemical Kinetics, McCraw-Hill, New York, 1950.
9. И о с т В., Взрывы и горение в газах, Издатинлит, М., 1952.
10. Geib К. Н., Ergeb. exakt. Naturw., 15, 44 A936), Reaktionen der Wasserstoff-
und Sauerstoffatome, Leipzig, Habilschr., 1937.
11. P e a s e R. N., J. Am. Chem. Soc, 52, 5106 A930).
12. Поляков X. В., С т а д н и к П. М., Труды VI Менделеевского съезда теорети-
теоретической и прикладной химии, 2, 202 A935); Поляков М. В. и др., ДАН СССР,
4, 454 A934); Поляков М. В. и др., Acta Physicochimica U.R.S.S., 1, 551,
817, 821 A934) (на немецком языке); Поляков М. В. и др., там же, 2, 397
A935) (на немецком языке); Поляков М. В., Bicri Ihct. физ. хем. АН УРСР,
5, 179 A936); Поляков М. В., Н е й м а р к И. Е., М а к с и м у к Ф. Г.,
там же, 7, 21 A937); Поляков М. В., Acta Physicochimica U.R.S.S., 9, 517
A938) (на английском языке); Поляков М. В., Л и ф а н о в Е. В., Но-
Носенко Д. С, ЖФХ, 12, 326 A938); см. также Acta Physicochimica U.R.S.S., 10,
441 A939) (на английском языке); ПоляковМ. В., Хим. реферат, журнал, 4,
17 A941).
13. Egerton А. С, М i n k о f f G. J., Proc. Roy. Soc. (London), A191, 145 A947).
14. T a n n e r H. F., J. Am. Chem. Soc, 56, 2250 A934).
15. H о 1 t R. В., Oldenberg O., J. Chem. Phys., 17, 1091 A949).
16. В г о i d a H. P., Everett A. J., M i n к о f f G. J., Fuel, 33, 251 A954).
17. M с L a n e C. K., J. Chem. Phys., 17, 379 A949).
18. С о о к G. А., пат. США 2368640 F. II 1945), 2368806 F. II 1945).
19. Rodebush W. H., J. Am. Chem. Soc, 59, 1924 A937).
20. Gei b К. Н., Z. physik. Chem. A., 169, 161 A934).
21. Б а д и н Э., в сб. «Вопросы горения», № 2, Издатинлит, 1953, стр. 147; В a d i n
Е. J., J. Am. Chem. Soc, 70, 3651 A948).
22. E 1 t e n t о n G. C, J. Chem. Phys., 15, 455 A947).
23. Foner S. N., Hudson R. L., J. Chem. Phys., 21, 1608 A953).
24. Foner S. N.. Hudson R. L., ibid., 21, 1374A953).
25. Robertson A.J.B., частное сообщение, 1954; Applied Mass Spectrometry,
Institute of Petroleum, London, 1954.
26. N о г t о n F. J., Natl. Bur. Standards (U.S.) Circ. № 522, 201 A953).
27. Bates J. R., Z. physik. Chem., .B22, 469 A933).
28. Kahlbaum C.A.F., герм. пат. 197023 A908); Teichner G., герм. пат.
205262 B3.XII 1908); L'Air Liquide, швейц. пат. 140403 [франц. пат. 667349
88 Глава 2
A5.Х 1929)], Ch. Fabrik Coswig-Anhalt G. M. b. H. from E. von Drathen, герм. пат.
558431 G.IX 1932); Henkel & Cie, W. Weber, герм. пат. 296357 C0.1 1917), Rene
M о г i t z, англ. пат. 120045 (8.X 1919).
29. B.I.O.S. Final Report No. 1414, Item № 22, Trip № 1080, N о а с к Е., N i t z -
s с h к е О., пат. США 1890793 A3.XII 1932), швейц. пат. 137302, I. G. Farbenin-
dustrie et al., герм. пат. 442412 B9.111 1927), 560004 B7.IX 1932), 630905 (8.VI 1936).
30. Report P. B. 74329 (frames 1235—1242).
31. С т а д н и к П. М., ДАН СССР, 87, 445 A952); М с К i n 1 е у J. D., Jr., J. Chem.
Phys., 22, 1258 A954).
32. В о n h о е f f ег К. F., Pearson Т. G., Z. physik. Chem. В., 14, 1 A931);
Jackson W. F., J. Am. Chem. Soc, 57, 82 A935); G e i b К. Н., J. Chem.
Phys., 4, 391 A936); О h a r a E., J. Chem. Soc. Japan, 61, 257, 265 A940).
33. Frost A. A., 01 d en b erg O., J. Chem. Phys., 4, 642, 781 A936).
34. О 1 d e n b e r g O., J. Chem. Phys., 3, 266 A935).
35. Smith W. V., J. Chem. Phys., 11, 110 A943).
36. R о d e b u s h W. H., W a h 1 M. H., J. Chem. Phys., 1, 696 A933); Camp-
Campbell R. W., R о deb us h W. H., J. Chem. Phys., 4, 293 A936); Rode-
bush W. H., J. Phys. Chem.., 41, 283 A937); Rodebush W. H., Wen-
de C. W., J., Camp bell R. W., J. Am. Chem. Soc, 59, 1924 A937).
37. Rodebush W. H., Keizer C.R, McKee F. S., Quagliano J. V.,
J. Am. Chem. Soc, 69, 538 A947); О 1 d e n b e r g O., J. Chem. Phys., 17, 1059
A949).
38. В о n hoef f er K. F., В oe h m E., Z. physik. Chem., 119, 385 A926).
39. G e i b K. H., H a r t e с к, Вег., 65, 1551 A932); Z. physik. Chem., 170, 1 A934).
40. J о n e s R. A., W i n к 1 e г С. A., Can. J. Chem., 29, 1010 A951).
41. Gi guere P. A., S e с с о Е. A., Eaton R. S., Discussions Faraday Soc,
1953, № 14, 104.
42. В a t z о 1 d J. S., LunerC, W i n к 1 e г С A., Can. J. Chem., 31, 262 A953).
43. Fischer F., Wolf M., Ber., 44, 2956 A911).
44. D a w s e у L. H., пат. США 2022650 C.XII 1935), 2162996 B0.VI 1939) [канадск.
пат. 344104 B1.VIII 1934)|.
45. С. I. О. S. Report, Item № 22, File № XXXIII-44, Pietzsch А. пат. США 2015040
A7.IX 1935) [Elektrochemische Werke Munchen, A. G., франц. пат. 790916 B9.XI
1935), англ. пат. 453458 A1.IX 1936)].
46. Р u t n a m G. L., D о b о Е., The Trend in Engineering (pub. by Engr. Exp. Sta-
Station, U. of Wash.) 1 B), 28 (IV 1949), D о b о Е. J., диссертация 1948; Sulli-
Sullivan J. F., диссертация 1948; Rundall W. J., Cal dwell N. А., диссер-
диссертация 1950, all at University of Washington.
47. Kl emenc A., Z. physik. Chem., A182, 91 A938); A183, 297 A939); Monatsh.,
75, 42 A944); 76, 38 A946); W i r t h H., К 1 e m e n с A., Monatsh., 82, 868 A951);
Kohl W, Klemenc A, ibid., 84, 365 A953); Павлов В. И., Залес-
с к и й Н. А., Еремеев М. А., Ш и ф р и н X. В., Изв. АН СССР, отд.
хим. наук, 309 A944); см. также Павлов В. И., ДАН СССР, 43, 405 A944);
de В ее о P., Compt. rend., 207, 623 A938); Bull. soc. Chim., 12, 779 A945);
M u t a A., J. Electrochem. Soc. Japan, 17, 265, 298 A949) [C.A., 44, 5233g and
6304b]; Eyraud C, Gill у Р., Compt. rend., 234, 1614A952).
48. Chem. Engr. News, 29, 3971 A951).
49. M u t a A., J. Electrochem. Soc. Japan, 18, 82 A950) [C.A., 45, 7448d|.
50. К 1 e m e n с A., Chimia (Switz.), 6, 177 A952).
51. D a v i e s R. A., H i с k 1 i n g A., J. Chem. Soc, 1952, 3595; H i с k 1 i n g A.,
L i n а с г e J. К., J. Chem. Soc, 1954, 711.
52. Coehn A., Grote G., Nernst Festschrift, Wilhelm Knapp, Halle, 1912, p.
136.
53. M a r s h a 1 1 A. L., J. Phys. Chem., 30, 1078 A926); J. Am. Chem. Soc, 54, 4460
A932).
54. Bates J. R., Salley D. J., J. Am. Chem. Soc, 55, 110 A933).
55. Bates J. R., J. Chem. Phys., 1,457A933).
56. Frankenburger W., KlinkhardtH., Trans. Faraday Soc, 27, 431
A931); [Z. physik. Chem., B15, 421 A932)].
57. Vol man D. H., J. Chem. Phys., 14, 707 A946).
58. S m i t h R. N., L e i g h t о n P. A., L e i g h t о n W. G., J. Am. Chem. Soc,
61, 2299 A939).
59. R о 1 1 e f s о n G. K., Burton M., Photochemistry and the Mechanism of Chemi-
Chemical Reactions, Prentice-Hall, Inc., New York, 1939.
60. Vol man D. H., J. Am. Chem. Soc, 73, 1018 A951).
61. T a u b e H., Bray W. C, J. Am. Chem. Soc, 62, 3357 A940).
62. S m i t h H. A., Kistiakowsky G. В., J. Am. Chem. Soc, 57, 835 A935).
63. S m i t h H. A., N a p r a v n i k A., J. Am. Chem. Soc, 62, 385 A940).
64. T а у 1 о г H. S., Salley D. J., J. Am. Chem. Soc, 55, 96 A933).
Литература 89
65. W a t е г s W. A., The Chemistry of Free Radicals, Second Ed., Oxford, 1948 [есть
перевод первого издания: см. У о т е р с У., Химия свободных радикалов, Ино-
издат, М., 1948].
66. К е г n b a u гп, Compt. rend., 149, 273 A909).
67. Tian A., Ann. Phys., 5, 248 A926).
68. S с h 6 n e E., Ber., 7, 1693 A874); 11, 483, 561, 874, 1028 A878).
69. M a t s u i H., Science (Japan), 19, 564 A949) [C. A., 45, 5469i].
70. В 1 а с e t F. E., Ind. Eng. Chem., 44, 1339 A952); Haagen-Smit, A. J., ibid.,
44, 1342 A952); Haagen-Smit A. J., Bradley С E., Fox M. M.,
ibid., 45, 2086 A953).
71. Bates D. R., Nicolet M., J. Geophys. Research, 55, 301 A950).
72. Д а и н Б. Я-, Куцая Л. Ф., Изв. научн.-иссл. инст. физич. химии имени
акад. Писаржевского, 12, 75 A940).
73. Cantiene R., Z. wiss. Phot., 36, 119 A937).
74. Clark H. L., Сое W. S., J. Phys. Chem., 5, 97 A937).
75. Y a m a h u z i K-, N i s i о e d a M., Biochem. Z., 296, 348 A938); 298, 293 A938);
300, 414 A939); 301, 404 A939); 303, 260 A939); 304, 160 A940); 305, 354 A940).
76. T г u m b 1 e H.P.C., J. Agr. S. Australia, 55, 214 A951).
77. С h а г i С N., Qureshi M., J. Indian Chem. Soc, 21, 97, 297 A944).
78. R u b i n T. R., С a 1 v e г t J. G., R a n k i n G. Т., М а с N e v i n W. N.,
J. Am. Chem. Soc, 75, 2850 A953).
79. С al vert J. G., Theurer K., Ran kin G. Т., MacNevin W. N.,
J. Am. Chem. Soc, 76, 2575 A954).
80. Весел о вс к и й В. И., Шуб Д. М., ЖФХ, 26,509A952).
81. Markham М. С, L a i d 1 е г К. J., J. Phys. Chem., 57, 363 A953).
82. F г i с k e H., Hart E. J., Smith H. P., J. Chem. Phys., 6, 229 A938); F г i-
c k e H., J. Chem. Phys., 3, 364 A934); см. также старые работы.
83. Burton M., Annual Review of Phys. Chem., 1, 113, A950); D a i n t о n F. S.,
С о 1 1 i n s о n E., ibid., 2, 99 A951); Allen A. O., ibid., 3, 57 A952); см. так-
также дальнейшее тома этой серии.
84. Discussions of the Faraday Society, «Radiation Chemistry», № 12, 1952.
85. Allen A. O., J. Phys. and Colloidal Chem., 52, 479 A948); Allen А. О., Но-
с h a n a d e 1 C. J., G h о г m 1 e у J. A., Davis T. W., J. Phys. Chem.,
56,575A952); Hochanadel С J., ibid., 56, 587 A952).
86. D a i n t о n F. S., J. Phys. and Colloidal Chem., 52, 490 A948).
87. Krenz F. H., Proc Conf. Nuclear Chem., Chem. Inst. Can. (Ottawa), 192 A947),
88. Lef ort M., J. chim. phys., 48, 339 A951).
89. Haissinsky M., MagatM., Compt. rend., 233, 954 A951).
90. Lefort M., HaissinskyM., Compt. rend., 230, 534 A950).
91. Ant a A., Haissinsky M., Compt. rend., 235, 170 A952).
92. Bonet-Maury P. et al., Compt., rend., 218, 400 A944); 226, 1363, 1445 A948);
Bull, soc chim. France, 333 A951); Brit. J. Radiology, 24, 422 A951).
93. В о n e t • M a u г у P., Lefort M., Nature, 166, 981 A950), J. chim. phys.,
47, 179 A950).
94. В о n e t • M a u г у Р., Discussions Faraday Soc, № 12, 72 A952).
95. Ebert M., В о a g J. W., Discussions Faraday Soc, № 12, 189 A952).
96. All en A. O., Discussions Faraday Soc, № 12, 79 A952).
97. С о 1 1 i n s о n E., D a i n t о n F. S., Discussions Faraday Soc, № 12, 212 A952).
98. В a x e n d a 1 e J. H., Evans M. G., Park G. S., Trans. Faraday Soc 42, 155
A946).
99. J о h n s о n G. R. A., S с h о 1 e s G., Weiss J., J. Chem. Soc, 1953, 3091.
100. Loiseleur J., Compt. rend., 214, 76 A942); Compt. rend., 230, 1901 A950).
101. Todd N., Whitcher S. L., J. Chem. Phys., 20, 1172 A952).
102. Johnson E. R., J. Chem. Phys., 21, 1417 A953).
103. Daniels M., Sc holes G., Weiss J., Nature, 171, 1153 A953).
104. P u с h e a u 1 t J., Lefort X., Haissinsky X., J. chim. phys., 49, 286
A952), Haissinsky X., Pucheault J., ibid., 49, 294 A952).
105. Dale W. M., D a v i e s J. V., Gilbert C.W., Keene J. P., Gray L. H.,
Biochem. J., 51, 268 A951).
106. Bonet-Maury P., Deysine A., Compt. rend., 232, 1101 A951).
107. Krenz F. H., D e w h u r s t H. A., J. Chem. Phys., 17, 1337 A949); Miller N..
ibid., 18, 79 A950); Hart E. J., J. Am. Chem. Soc, 73, 1891 A951),74, 4174
A952); Rigg Т., Stein G., Weiss J., Proc. Roy. Soc (London), A211,
375 A952); D a i n t о n F. S., S u t t о n H. C, Trans. Faraday Soc, 49, 1011
A953); Dew hurst H. A., ibid., 49,1174A953).
108. S p о е г 1 E., Scientific American, p. 22, XII 1951.
Г09. Atper Т., Discussions Faraday Soc, № 12, 234 A952).
ilO. The Chemistry of Biological After-Effects of Ultraviolet and Ionizing Radiations,
a Symposium, British Journal of Radiology, 27, 36, 117 A954).
90 Глава 2
111. Bergmann L., Der Ultraschall und Seine Anwendung in Wissenschaft und Tech-
nik, 5 Aufl., Hirzel S., Zurich, 1949.
112. Schultes H., Gohr H., Angew. Chem., 49, 420 A936); F 1 о s d о г f E. W.,
Chambers L. A., M a 1 i s о f f W. M., J. Am. Chem. Soc, 58, 1069 A936)-4
Kl i n g A., Kl i n g R., Compt. rend., 223, 33 A946). ^
113. Schick H. L., Ph. D. Thesis. University of Wisconsin, 1951.
114. Fl osdorf E. W., Chambers L. A., Mai i sof f W. M., J. Am. Chem.
Soc, 68, 1069 A936).
115. У р а з о в с к и й С. С, Полоцкий И. Г., Acta Physicochimica U.R.S.S., 13,
443 A940) (на английском языке); Полоцкий И. Г., ЖОХ, 17, 649, 1048
A947). Оуата Н., К u sumo to G., J. Inst. Elec. Engrs. Japan, 62, 549
A942) [C. A., 42, 16c]; A k i у a S., О k и i S., J. Pharm. Soc. Japan, 67, 232
A947) [C.A., 45, 6447Ц; Beuthe H., Furbach E., Sorenson C, Akus.
Zeits, 4, 209 A939).
116. Liu S., Wu H., J. Am. Chem. Soc, 56, 1005 A933).
117. В e t h ge J., Soehr i ng K., T sc hesc he R., Z. Naturforsch., № 2b, 12
A947).
118. Keilin D., Hartree E. F., Biochem. J., 42, 221 A948).
119. Franke W., Deffner M., Liebigs Ann. Chem., 541, 117 A939).
120. Milas N., Chem. Reviews, 10, 295 A932); W i e 1 a n d H., On the Mechanism of
Oxidation, Yale University Press, 1932; Oppenheimer C, Stern G. K.,
Biological Oxidation, Nordemann Publ. Co., New York, 1939; Dufraisse Ch.,
С h о v i n P., Chapter in «Handbuch der Katalyse,» Vol. 2, J. Springer, Vien, 1940
(на французском языке); «Oxidation: A General Discussion», reprinted from Trans.
Faraday Soc, 42, 99 A946) by Gurney and Jackson, London; «Le Mechanisme de
L'Oxydation,» Proc 8th Conf. Inst. Int. Chim. Solvay, Stoops R., Brussels, 1950;
В a w n C.E.H., Journal Oil Colour Chem. Assoc, 36, № 398, 443 A953).
121. Churchill J. R., Trans. Electrochem. Soc, 76, 341 A939).
122. W i e 1 a n d H., Franke W., Ann., 469, 257 A929); 473, 289 A929).
123. Grunberg L., Proc Phys. Soc, B, 66, 153 A953).
124. Kramer J., Z. Phys., 125, 739 A949); 128, 538 A950); 129, 34 A951).
125. Duns tan W. R., J о w e t t H.A.D., Goulding E., J. Chem. Soc, 87,
1548 A905).
126. Fur man N. H., Murray W. M., Jr., J. Am. Chem. Soc, 58, 429, 1843A936).
127. Mull er E., Barchmann H., Z. Elektrochem., 40, 188 A934).
128. К о n i n k 1 i j k e N. V., Ned Zoutindustrie, голл. пат. 58703 A6.XII 1946), англ.
пат. 606745 A9.VIII 1948), герм. пат. 805278 A951); Chem. Engr. News, 24, 1426
A946).
129. Kameda M., Science Repts. Research Inst. Tohoku Univ., Ser. A., I, 223, 435
A949) [C.A., 45, 46151 and 5583d].
130. Та и be H., J. Am. Chem. Soc, 70, 1216A948).
131. Fryling С R, Tool e у F. V., J. Am. Chem. Soc, 58, 826 A936); Miller
С. С, Chalmers R. A., Analyst, 77,2A952).
132. Walton J. H., F i 1 s о n G. W., J. Am. Chem. Soc, 54, 3228 A932).
133. Filson G. W., Walton J. H., пат. США 2059569 C.XI 1936).
134. Pfleiderer G., герм. пат. 649234 A934); I. G. Farbenindustrie, англ. пат.
449360 A934); R e i d 1 H. J., Pfleiderer G., пат. США 2158525 A6.V
1939), [англ. пат. 465070, франц. пат. 812404]; R e i d I H. J., Pfleide-
Pfleiderer G., пат. США 2215883 B4.IX 1940) [герм. пат. 671318], пат. США 2369912
[англ. пат. 508081].
135. James Т. Н., S n е 1 1 J. M., Weissberger A., J. Am. Chem. Soc, 60,
98, 2084 A938); см. также балее старые работы Вайсбергера и его сотрудников.
136. A t h е г t о n W., Т и г п е г Н. A., J. Soc Dyers Colourists, 62, 108 A946).
137. Slater V. W., Clapp D. В., Master man S., C.I.O.S., File Number
XXXI-15; P. B. report 4336; U. S. Naval Technical Mission in Europe, C.I.O.S.
File Number XXV-44, Item № 22; К e r n J. G., Murray R. L., Sudhoff
R. W., C.I.O.S. File Number XXVII-84, P. B. report 385; Brode W. R., P. B.
report 1737; Go г ml e у W. G., P. B. report 395; P. B. report 107717.
138. D a w s e у L. H., U m h о e f e г R. R., Muehlhausser С. К., пат. США
2455238 C0.XI 1948); 2537516 (9.1 1951) [англ. пат. 669274]; 2537655 (9.1 1951)
[англ. пат. 671524].
139. С. W. LeFeuvre, англ. пат. 695779 A9.VIII 1953).
140. Technical Oil Mission Reel 32, Translated by Charles A. Meyer & Co., Inc., PC-S-III,
Vol. II-G A949).
141. E. I. duPont deNemours & Co., англ. пат. 686567, 686572 B8.1 1953); Sprauer
J. W., пат. США 2657980 C.XI 1953) [англ. пат. 6865741, пат. США 2673140 B3.Ш
1954); Harris С. R., Sprauer J. W., пат. США 2668753 (9.11 1954).
142. Soul e E. С, пат. США 2035101 B4.111 1936), Reissued as Re 20769 A938)
[L. Mellersh-Jackson англ. пат. 461589 A9.11 1937)]; Michalek J.C., Soule
Литература 91
Е. С, пат. США 2144341 A I 1939) [англ. пат. 501385 B7.11 1939), 533633 A8.11
1941)]; пат. США 2178640 G.XI 1940); Soule E. С. or Mathieson Alkali Works
англ. пат. 489978 (8.VIII 1938), 489979 (8.VIII 1938), [франц. пат. 823334 A81;
1938), герм. пат. 681960 A939I; М i с h а 1 е к J. С, Soule E. С, or Mathieson
Alkali Works, англ. пат. 479994 A5.11 1938); франц. пат. 801754 A7.VIII 1936);
герм. пат. 683953 A939); 710961 B1.VIII1941).
143. Gilbert Е. С, J. Am. Chem. Soc, 51, 2744 A929).
144. A u d г i eth L. F., Mohr P. H., Ind. Eng. Chem., 43, 1774 A951).
145. R i eche A., Alkylperoxyde und Ozonide, T. Steinkopff, Dresden, 1931.
146. Эверетт А. Дж., Минков Г. Дж., в сб. «Вопросы горения», № 2, Издатин-
лит, М., 1953, стр. 155.
147. С a die R. D., Huff H., J. Phys. Coll, Chem., 54, 1191 A950).
148. E g e г t о n A. C, Everett A. J., M i n k о f f G. J., Nature, 173, 399 A954).
149. Egerton A., Harris E. J., Young G.H.S., Trans. Faraday Soc, 44, 746,
750, 755, 764 A948).
150. Everett A. J., M i n k о f f G. J., Trans. Faraday Soc, 49, 410 A953).
151. Pease R., J. Am. Chem. Soc, 60, 2244 A938).
152. В r usch we i 1 er H., Minkoff G. J., Salooja К. С, Nature, 172,
909 A953).
153. Bernard M. Y., Compt. rend., 236, 2412 A953).
154. Штерн В., Поляк С, Acta Physicochimica U.R.S.S., 11, 797 A939).
155. D u n l с z B. L., P e r r i n D. C, Style D.W.G., Trans. Faraday Soc, 47,
1210 A951).
156. S a t t e г f i e 1 d С N., Case L C, Ind. Eng. Chem., 46, 998 A954).
157. S a t t e г f i e 1 d C. N., Wilson R. E., L e С 1 a i r R. M., Reid R. C,
Analytical Chemistry, 26, 1792 A954).
158. Minkoff G. J., Salooja К. С, Fuel, 32, 516 A953).
159. К о о i j m a n P. L., Rec trav. chim., 66, 5, 205, 491 (with W. L. Ghijsen) 217,
A947) (все на английском языке); Technical Oil Mission reports translated by Chas.
A. Meyer & Co., Translations PC-11 and PC-99.
160. S a t t e г f i e 1 d C. N.. Wilson R. E., Ind. Eng. Chem., 46, 1001 A954).
161. Harris C. R., пат. США 2533581 A2.XII 1950).
162. Черняк Н. Я., Штерн В. Я., ДАН СССР, 78, 91 A951).
163. S a t t е г f i e I d С. N., Reid R. С, in «Fifth International Symposium on Combu-
Combustion», Reinhold, New York, 1955.
164. Lacomble A. E., пат. США 2376527 A5.V 1945) [канад. пат. 415510 B8.IX
1943)]; Cook G. А., пат. США 2416156 A8.11 1947); N. V. de Bataafsche Petro-
Petroleum Mij., голл. пат. 52521, 52522 A5.V 1942) и 59737 A5.VIII 1947) [англ. пат.
540534].
165. Harris С. R., пат. США 2443503 A5.VI 1948).
166. Pahnke A. J., Cohen P. M., S t u r g i s В. М., Ind. Eng. Chem., 46, 1024
A954).
167. M i 1 a s N. A., S t a h 1 L. E., Dayton В. В., J. Am. Chem. Soc, 71, 1448
A949).
168. Sat t erf i el d С N., Reid R. C, J. Phys. Chem., 59, 283 A955).
169. Walsh A. D., Trans. Faraday Soc, 42, 269 A946); 43, 297, 305 A947).
170. Jackson W. F., J. Am. Chem. Soc, 57, 82 A935).
171. Farkas A., Stribley A. F., пат. США 2430864, 2430865 A8.XI 1947);
Brewer P. D., пат. США 2447794 B4.VIII 1948).
172. Cook G. А., пат. США 2614907 B1.X 1952).
173. S a t t e г f i e 1 d C. N., Wilson R. E., Stein T. W., Cooper D. O.,
Ind. Eng. Chem., 46, 1007 A954).
174. К oo ij man P. L., пат. США 2461988 A5.11 1949) [голл. пат. 55114 A6.VIII
1943)].
175. О v е г h о f f J., голл. пат. 61465 A6.Х 1948); Stein Т. W., S. M. Thesis in Che-
Chemical Engineering, M.I.Т., 1952.
176. Harris С R., пат. США 2479111 A6.VIII 1949); N. V. de Bataafsche Petroleum
Maatschappij, англ. пат. 708339 E.V 1954).
177. Stephens E. R., Pease R N.. J. Am. Chem. Soc, 72, 1188 A950).
178. L и f t N. W., С о h e n L., J. Chem. Phys., 22, 348 A954).
179. Fichter F., Bladergroen W., Helv. Chim. Acta., 10, 549 A927).
180. Huckel W., Nachr. Akad. Wiss. Gottingen, Math.-physik. Klasse, 1946, 36 [C. A.,
43, 6793]; ibid., 1946 № 1, 55 [C. A., 44, 4359d].
181. Davis W., Jr., Rosen F. D., W i n g e t R. H., Jr., W i s w a 1 1 R. H.,
Jr., United States Atomic Energy Commission Report AECD-2827, December 27,
1949).
182. T г а и b e M., Ber., 15, 2434 A882); В о r n e m a n n K-, Z. anorg., allgem. Chem.,
34, 1 A903).
183. Fischer P.Priess O., Ber., 46, 698 A913).
92 Глава 2
184. Mull ег Е., Me hi horn К., Z. anorg. allgem. Chem., 223, 199 A935).
185. Henkel & Cie, герм. пат. 266516 B8. X 1913); 273269 B3. IV 1914); 276540 A4.VI 1914);
279073 F.X 1914); 283957 (I.V 1915); 302735 A9.XII 1917); I. G. Farbenindustrie
A. G., герм. пат. 463794 B.VIII 1918); 485053 B4.X 1929); 485714 E.XI 1929);
514172 A0.XII 1930); I. G. Farbenindustrie A. G., швейц. пат. 126402 B1.Ill 1927);
127517B6.1111927); Amrein Werner, швейц. пат. 268529 A6.VIII 1950);
I. G. Farbenindustrie A. G., франц. пат. 636330 F.V 1927).
186. Patrick W. A., Wagner H. В., Corrosion, 6, 34 A950).
187. S i 1 v e s t г о n i P., Ricerca. Sci., 20, 1291 A950) [C.A., 45, 3735b]; К a 1 о u-
s e к М., Collection Czechoslov. Chem. Communs., 13, 105 A948) (на английском
языке) [С. А., 42, 8674с].
188. И о ф а 3. А., Ш и м ш е л е в и ч Я. В., Андреева Е. П., ЖФХ, 23, 828
A949).
189. К о 1 t h о f f I. M., Miller С. S., J. Am. Chem. Soc, 63, 1013 A941).
190. P e 1 1 e q u e г H., J. chim. phys., 47, 590, 927 A950).
191. Сивер Ю. Г., Кабанов Б. Н., ЖФХ, 22, 53 A948); 23, 428 A949).
192. D el a h а у P., J. Electrochem. Soc, 97, 198, 205 A950).
193. Ф р у м к и н А. Н., Вестник МГУ, 7, № 9, 37 A952).
194. Berl W. G., Trans. Electrochem. Soc, 83, 253 A943).
195. Fischer F., Kronig W., Z. anorg. allgem. Chem., 135, 169A924).
196. Berl E., Trans. Electrochem. Soc, 76, 359 A939), Berl E., пат. США 2091129,
2091130 B4.VIII 1937); 2093989 B8.VIII 1937) [герм. пат. 648964 A3.VIII 1937)],
пат. США 2000815 G.V 1935).
197. Р. В. report 74413 (frames 2555-2558).
198. М i z u n о S., et. al., J. Electrochem. Soc. Japan., 17, 262, 288 A949); 18, 48, 80,
116, 167 A950); Bull. Tokyo Inst. Technol., 13, 102, 108 A948) [C. A., 44, 5233e,
6305d, 10549f; 45, 3259a, 7451a].
199. Бурштейн Р., Фрумкин А., ДАН СССР 32, 327 A941) (на английском
языке); Бурштейн Р., М и л л е р Н. В., ЖФХ, 23, 43 A949).
200. Weisz R. S., J a f f e S. S., Trans. Electrochem. Soc, 93, 128 A948).
201. Иофа З. А., Моисеева Н.В., Мирлина С. Я-, Крымакова Е.Е.,.
ЖПХ, 21, 329 A947).
Глава 3
ОБРАЗОВАНИЕ ПЕРЕКИСИ ВОДОРОДА И СПОСОБЫ ЕЕ ПРОИЗВОДСТВА
//. ПОЛУЧЕНИЕ ИЗ ПЕРЕКИСНЫХ СОЕДИНЕНИЙ
В данной главе рассматриваются методы получения и способы промышлен-
промышленного производства перекисных соединений (кроме производства самой перекиси
водорода) из неперекисных соединений, а также реакции этих перекисных сое-
соединений, приводящие к образованию перекиси водорода. Две группы реакций
относятся к этой категории: 1) образование неорганических перекисей и после-
последующие их реакции с кислотами с целью выделения перекиси водорода и 2) об-
образование пероксодисерной кислоты и пероксодисульфатов путем электролиза
и последующий их гидролиз с целью получения перекиси водорода.
Первая группа реакций служила основой старых методов производства.
Хотя в экономике процессы, основанные на этих реакциях, в настоящее время
не играют большой роли, их обсуждение полезно как с научной точки зрения,
так и с целью выяснения возможных путей улучшения и модернизации, которые
в будущем могут привести к их вторичному выдвижению для конкуренции с со-
современными процессами. Вторая группа реакций является основой современ-
современных методов производства перекиси водорода,
НЕОРГАНИЧЕСКИЕ ПЕРЕКИСИ
Перекись бария
Окись бария реагирует с кислородом, образуя перекись бария в соответ-
соответствии с уравнением:
ВаО + уО2 -> ВаО2 Д//= - 17 100 кал. A)
Реакция будет протекать слева направо, если при заданной температуре пар-
парциальное давление кислорода выше того, которое существует в равновесии со
смесью окиси и перекиси бария. Эта система моновариантная, и равновесное
парциальное давление изменяется с температурой следующим образом [1J.
Парциальное Парциальное
Температура, °С давление Температура, °С давление
кислорода, am кислорода, am
620 0,015 835 0,945
700 0,0861 853 1,220
787 0,1855 868 1,534
Из приведенных данных видно, что перекись бария можно получать из
окиси бария путем выдерживания последней на воздухе под атмосферным давле-
давлением при температурах около 790° или ниже, хотя на практике с целью обеспе-
обеспечения достаточной скорости реакции в благоприятных условиях равновесия
применяются температуры около 500°. Процесс образования перекиси обычно
осуществляется путем продувания воздуха над окисью бария, находящейся
в муфельной печи с внешним обогревом. Если окись бария сухая, тонкодисперс-
тонкодисперсная и высокопористая, а воздух освобожден от двуокиси углерода и влаги,
94 Глава 3
то реакция протекает довольно быстро. В этих условиях можно легко получить
продукт, содержащий 88—90% перекиси бария. Замена воздуха кислородом
значительно увеличивает скорость образования перекиси, но экономически
такая замена обычно не оправдывается. До настоящего времени во всех про-
промышленных процессах применяется только воздух.
Для получения высокого выхода основным требованием является приме-
применение достаточно пористой окиси бария; с производственной точки зрения это
более значительная проблема, чем превращение окиси в перекись, так как раз-
разложение бариевой соли при высокой температуре может сопровождаться спе-
спеканием и образованием твердой и нереакционноспособной окиси. В течение
XIX столетия окись бария получали почти исключительно из азотнокислого
бария, который довольно быстро разлагается при 600—800°, однако процесс
был весьма затруднительным и дорогостоящим, поскольку в результате его
всегда образуется расплав нитрата, нитрита и окиси. Это приводило к необхо-
необходимости вести процесс небольшими периодическими операциями в закрытых
тиглях с внешним обогревом, причем возникали трудности, вызванные необ-
необходимостью отвода и регенерации образующейся окиси азота. Поэтому уже
в самом начале XX столетия описанный процесс был вытеснен другим—полу-
другим—получением окиси бария из углекислого бария.
Окись бария из углекислого бария
При нагревании углекислого бария наблюдаются аллотропные превраще-
превращения при 806 и 968°, за которыми следует частичное разложение с образованием
эвтектики (с окисью бария) с температурой плавления 1030°:
ЗВаСО3 —» 2ВаСО3- ВаО + СО2. B)
Для полного обжига при атмосферном давлении требуется температура около
1450°:
2ВаСО3-ВаО -- ЗВаО + 2СО2. C)
Однако при добавке углеродистых веществ к углекислому барию выделяющаяся
двуокись углерода почти полностью превращается в окись углерода. Такой
метод снижения парциального давления двуокиси углерода в системе позволяет
осуществлять полное разложение при значительно более низких температурах,
чем это было возможно другими путями, поэтому он обычно и применяется
при техническом обжиге. Кроме того, добавка углеродистых веществ способ-
способствует получению пористой окиси бария с высокой реакционной способно-
способностью. Необходимо применять вещества с минимальной зольностью, поскольку
глинозем и силикатные соединения', содержащиеся в золе, могут образовать
плотную расплавленную массу с окисью бария. Часто применяют ламповую
сажу, нефтяной кокс или деготь. В некоторых случаях к углекислому барию
до обжига вместо углеродистых веществ добавляют такие соединения, как
азотнокислый барий или перекись бария, которые разлагаются при нагревании
без загрязнения исходного сырья.
Хотя углекислый барий встречается в природе, например в виде минерала
витерита, такой продукт слишком загрязнен, чтобы его можно было непосред-
непосредственно использовать для переработки на перекись бария. Поэтому обычно в ка-
качестве исходного сырья пользуются наиболее распространенным бариевым
минералом баритом (BaSO4), который сначала восстанавливают углем до серни-
сернистого бария. Затем последний отделяют от нерастворимых примесей выщела-
выщелачиванием горячей водой и из раствора осаждают углекислый барий путем обра-
обработки газообразной двуокисью углерода или же углекислым натрием:
BaS + Н2О + СО2 — ВаСО3 + H2S. D>
Неорганические перекиси 95
Обжиг углекислого бария сопровождается значительно большими трудностями,
чем аналогичный и хорошо известный процесс обжига углекислого кальция,
в связи с необходимостью применять вышеуказанные меры для получения
пористой и реакционноспособной окиси бария высокой чистоты и избегать
работы при высоких температурах. Как и при обжиге известняка, при обжиге
углекислого бария требуются большие количества тепла и высокая темпера-
температура, причем окись бария отличается сильными шлакообразующими свойства-
свойствами. В принципе необходимую для обжига теплоту можно получить непосред-
непосредственно в самой шихте сжиганием дополнительного количества угля, например,
путем вдувания во вращающуюся печь угольной пыли вместе с воздухом, либо
смешением с карбонатной шихтой, либо применением обоих этих способов.
Однако окисление угля до окиси углерода дает лишь немногим больше1^ того
количества тепла, которое выделяется при образовании двуокиси углерода.
Таким образом, расход топлива будет очень значительным, и если применять
угольную пыль, то, вероятно, содержание примесей в продукте оказалось бы
недопустимо высоким. На практике обжигательные печи конструируют таким
образом, чтобы не'допускать непосредственного контакта дымовых газов с ших-
шихтой в отличие от обычной эксплуатации известковообжигательных печей.
В связи с этим теплота должна передаваться через стенки реторты, как напри-
например в коксовых печах с улавливанием побочных продуктов. Такая конструк-
конструкция печи значительно удорожает строительство и повышает производственные
расходы по сравнению с обжигом известняка. Применение избытка углерода,
например в количестве 25—30 вес.% от начальной смеси с карбонатом, позво-
позволяет получить более пористый продукт, но избыток должен быть в конце
удален (чтобы вновь не образовался карбонат) окислением при достаточно
высокой температуре. Указывается, что эффективным является нагревание на
воздухе в течение получаса при 1350° или выше.
Углерод может быть удален также путем реакции с паром:
Н2О->СО + Н2, E)
однако это требует температур примерно выше 988° при атмосферном давле-
давлении, чтобы предотвратить образование гидрата окиси бария*. Смесь углекисло-
углекислого бария и углерода обычно подвергают периодическому обжигу в тиглях (при
работе в небольшом масштабе), или в шахтных, или вращающихся печах/
В некоторых случаях в процессах последнего типа для снижения парциального
давления двуокиси углерода, а следовательно, и температуры разложения [2]
применяется вакуум или продувка инертным газом. Избыточный углерод
удаляют затем обработкой сильно нагретым воздухом. Нагрев производят при
помощи электрических приборов сопротивления или косвенным путем—ды-
путем—дымовыми газами. По имеющимся данным, при применении электродуговой печи
образуется твердая непористая, а следовательно, непригодная для перера-
переработки окись бария. При температурах ниже температуры обжига, во избежание
вторичного образования карбоната, полученная окись бария должна быть
защищена от контакта с газами, содержащими двуокись углерода. Аналогично
газы с содержанием водяного пара могут привести к образованию гидрата
окиси бария с температурой плавления около 400°.
Болло и Каденаччо [3] запатентовали процесс с добавкой металлического
катализатора, позволяющего получать перекись бария непосредственно из
карбоната. Ввиду вышеуказанных равновесных соотношений такая возмож-
возможность является весьма сомнительной; вероятно, перекись бария, полученная
* Парциальное давление водяного пара в равновесии с окисью и гидроокисью ба-
бария составляет, например, 1 am при 988° и 0,125 am при 786°. Поэтому можно ожидать,,
что газы, содержащие водяной пар, например дымовые, при температурах ниже пример-
примерно 800—1000 будут превращать окись бария в гидроокись.
96 Глава 3
указанными авторами, образовалась после обжига при охлаждении шихты на
воздухе,
Разработаны процессы превращения углекислого бария в гидроокись
путем обработки паром при высокой температуре или путем обжига в присут-
присутствии силикатов для снижения температуры реакции с последующим выщела-
выщелачиванием водой. Однако попытки дегидратации расплавленной гидроокиси
бария до окиси при тех весьма высоких температурах, которые требуются для
осуществления реакции, сопряжены со значительными техническими трудно-
трудностями. Предложено смешивать гидрат окиси бария с древесным углем и рабо-
работать при температуре около 1200°. Древесный уголь увеличивает пористость
продукта и снижает необходимую температуру за счет реакции с паром по урав-
уравнению E). Однако технически, по-видимому, более выгоден прямой обжиг до
окиси.
Реакция перекиси бария с кислотой
Реакция перекиси бария с избытком кислоты приводит к полному превра-
превращению этой перекиси в перекись водорода в равновесных условиях*. В произ-
производственной практике выбор кислоты зависит от ряда факторов, куда входят:
а) влияние на чистоту и устойчивость продукта и на максимальную концентра-
концентрацию перекиси водорода, достигаемую без существенного разложения; б) ско-
скорость и степень превращения перекиси бария, которые могут быть осуществле-
осуществлены без особых трудностей; в) фильтруемость бариевой соли, если она нераство-
нерастворима; г) намеченное использование образующейся бариевой соли и д) желатель-
желательность получения нерастворимой бариевой соли с целью удаления ее из раствора
продукта. Обычно перекись бария вводится в реакцию либо с фосфорной кис-
кислотой, либо с серной кислотой с небольшими добавками соляной или фосфорной
кислоты. Если применять не серную кислоту, а другую, то образующуюся ба-
бариевую соль превращают затем действием серной кислоты в сернокислый ба-
барий, т. е. в бариевую соль, которая пользуется наибольшим спросом, а поэтому
исходная кислота регенерируется. В принципе бариевую соль можно разло-
разложить до окиси бария и последнюю вновь направить в процесс для получения
перекиси, но, к сожалению, кислоты, наиболее удовлетворительные с точки
зрения реакции с перекисью бария (серная и фосфорная), образуют бариевые
соли, отличающиеся термической стойкостью и с трудом разлагающиеся до окиси
бария; применение других кислот встречает трудности в связи с высокой стои-
стоимостью, малой концентрацией водородных ионов или сложностью превращения
их солей в окись бария. С появлением методов электрохимического производ-
производства, которые могут дать непосредственно более концентрированный и устой-
устойчивый продукт, бариевые процессы были, как правило, вытеснены и в настоя-
настоящее время сохраняются только в тех местах, где имеется подходящий рынок
сбыта для побочного сернокислого бария, известного также под названием
бланфикса. Бариевым методом в США и во всем мире сейчас получают лишь
небольшую часть перекиси водорода. Вследствие простоты оборудования,
необходимого для бариевого процесса, и сравнительной легкости эксплуатации
этот процесс представляет интерес в случае, когда намечено производство в не-
небольшом масштабе, особенно при наличии рынка сбыта для низкоконцентри-
низкоконцентрированной перекиси водорода, расположенного сравнительно близко от завода-
изготовителя. В настоящее время бариевый процесс с повторной переработкой
бариевой соли на окись бария для возвращения ее в цикл нигде не применяется,
тем не менее новейшие усовершенствования в]технике работы с твердыми веще-
* Льюис и Рэндол [4] измерили константу равновесия для реакции:
BaO2TU-f2H2O;?Ba2++H2O2+2OH-
и приводят величину около 15-10~12 для К=(Ва2+) (H2OJ (ОН'J, где концентрации
выражены в молях на литр.
Неорганические перекиси 97
ствами при проведении операций в условиях высоких температур могут
привести к тому, что циклический процесс в производстве перекиси водорода
из перекиси бария окажется конкурентоспособным.
Максимальная концентрация перекиси водорода, которая может быть
получена в результате прямой реакции перекиси бария с кислотой, обычно
ограничивается по экономическим причинам 6—8% Н2О2, так как при более
высоких концентрациях приходится работать с густыми шламами и увеличи-
увеличивается потеря активного кислорода. Можно получать несколько более высокие
концентрации путем рециркуляции разбавленной перекиси с направлением
ее в свежую смесь перекиси бария с кислотой; при этом, однако, возникает
опасность усиления общего разложения, и обычно такой способ является не-
неэкономичным.
Перекись водорода обладает максимальной стабильностью в слабокис-
слабокислых растворах, но она гораздо менее устойчива в щелочной среде, чем в кислых
растворах умеренной концентрации (см. стр. 349). Стабильность снижается при
добавке очень малых количеств ионов тяжелых металлов, вроде ионов железа
или меди, и твердых частиц, или растворении больших количеств почти любых
веществ. Хотя чистая перекись бария сравнительно нерастворима в воде, окись
бария, всегда присутствующая в техническом продукте, растворяется с образо-
образованием основной среды, и поэтому желательно применять достаточно концентри-
концентрированную кислоту и вводить ее в контакт с твердой фазой таким образом,
чтобы раствор все время был кислым. Целесообразно брать кислоту, которая
образует нерастворимую бариевую соль и таким образом позволяет удалить
барий из раствора (например, серную кислоту, двуокись углерода или фосфор-
фосфорную кислоту); вместе с тем при применении этой кислоты необходимо избегать
такого образования осадка, при котором происходит обволакивание частиц
перекиси бария, что исключает возможность дальнейшей их реакции. Так,
практически перекись водорода не обнаружена при введении в реакцию кон-
концентрированной серной кислоты с негидратированной перекисью бария при
20° или 10—50%-ной серной кислоты при температурах ниже 0° [5]. Получав-
Получавшийся твердый остаток содержал большую часть исходной перекиси бария,
вероятно, частицы были покрыты нерастворимым сернокислым барием. Если
перекись бария до обработки кислотой встряхивать с водой для превращения
ее в гидрат ВаО2-8Н2О, то образование перекиси водорода улучшается,
вероятно в связи с большим удалением частиц перекиси бария друг от друга
и лучшей их растворимостью, однако значительная доля исходного активного
кислорода может теряться из-за высокой щелочности на стадии гидратации.
Поэтому для гидратации пользуются разбавленной соляной или фосфорной
кислотой.
Добавка нескольких процентов соляной или фосфорной кислоты к серной
существенно повышает скорость реакции и обеспечивает практически полное
превращение. Механизм этого явления и реакции вообще можно объяснить на
основании пленочной теории, по которой зона, непосредственно окружающая
каждую частицу перекиси бария, представляет собой относительно неподвиж-
неподвижную область, через которую массообмен происходит главным образом за счет
молекулярной диффузии ионов и молекул. Хотя степень турбулентности фак-
фактически возрастает непрерывно с увеличением расстояния от поверхности части-
частицы, все же можно себе представить, что существует совершенно неподвижная
пленка, как это показано на рис. 10. В этой пленке происходит реакция и совер-
совершается массообмен исключительно за счет ионной и молекулярной диффузии.
Ввиду ограниченной растворимости перекиси бария, ионы водорода, диффун-
диффундирующие внутрь, вероятно, реагируют вблизи поверхности смеси твердых
фаз—перекиси бария и окиси бария—с образованием перекиси водорода и ионов
б^зйя, которые затем диффундируют наружу. Если кислотный анион образует
нерастворимую бариевую соль, то анионы также диффундируют внутрь, что
7 Перекись водорода
98
Глава 3
способствует осаждению соли на поверхности частиц, где концентрация барие-
бариевых ионов максимальная. Это приводит к обволакиванию частиц нерастворимым
осадком. Однако если ввести небольшое количество другой кислоты, образую-
образующей растворимую бариевую соль, например соляной кислоты, то в начальной
стадии реакции твердая частица будет окружена пленкой хлор-ионов за счет
селективного осаждения сульфат-ионов из смеси хлор- и сульфат-ионов, окру-
окружающих частицу. В результате этого бариевые ионы успеют продиффундиро-
вать дальше от поверхности частицы, возможно даже дальше основной части
«неподвижной пленки», раньше чем они встретятся с концентрацией сульфат-
ионов, достаточной для образования осадка. Поэтому полученный осадок дол-
должен сравнительно слабо прилипать к исходным частицам.
Неподвижная
пленка
ВаО2
Н202
Ва
•Анион (например, SQj", Cf)
Неподвижная
пленка
Рис. 10. Предположительная'диффузия
различных видов частиц при реакции
кислоты с перекисью бария.
Рис. 11. Предположитель-
Предположительное изменение рН с расстоя-
расстоянием от поверхности части-
частицы перекиси бария во время
реакции.
Величина рН достигнет максимального значения, вероятно соответствую-
соответствующего сильно щелочной среде, вблизи поверхности частицы и будет падать па
мере удаления от частицы в соответствии с рис. 11. Поскольку перекись водо-
водорода образуется в щелочной и поэтому неустойчивой среде, необходимо умень-
уменьшить время, в течение которого перекись водорода остается в неустойчивой
зоне, с целью снижения размеров разложения ее до минимума. Для этого
нужно повысить скорость диффузии перекиси водорода наружу или уменьшить
толщину щелочной зоны. Скорость диффузии можно увеличить повышением тем-
температуры или градиента концентрации. Температуру заметно изменить нельзя
из-за быстрого роста скорости разложения с температурой, но градиент концен-
концентрации можно повысить путем поддерживания концентрации перекиси водоро-
водорода в основной массе жидкости на низком уровне (от 3 до 8%).
Для снижения толщины щелочной зоны можно: а) либо пользоваться кон-
концентрированной кислотой для поддержания низкого рН в основной массе
жидкости, и следовательно, крутого градиента рН; б) либо применять интенсив-
интенсивное перемешивание для создания максимальной скорости перемещения твердых
частиц в жидкости, чтобы таким образом уменьшить до минимума толщину
пленки. Интенсивное перемешивание требуется также для дезагрегации агло-
агломератов частиц и обеспечения хорошего смешения, поскольку в реакторе недо-
недопустимо существование щелочных зон в большом объеме.
Из сказанного видно, что образование нерастворимой бариевой соли вокруг
реагирующих частиц не только снижает скорость реакции, но и уменьшает ско-
скорость диффузии перекиси водорода из щелочной зоны и тем самым уменьшает
выход этой перекиси.
Проведенный анализ показывает, что оптимальные рабочие условия и
последующая обработка сильно зависят от природы примененной кислоты.
Неорганические перекиси 99
Это можно иллюстрировать путем рассмотрения характеристики реакции
с различными кислотами.
Реакция перекиси бария с фосфорной кислотой
Фосфорная кислота образует с барием три соли: в щелочной среде фосфат
присутствует главным образом в форме очень мало растворимого соединения
Ва3(РО4J, в нейтральной и слабощелочной—как слаборастворимый ВаНРО4
и в слабой или концентрированной кислоте—как растворимый Ва(Н2РО4J.
Таким образом, быстрого и полного превращения перекиси бария можно достиг-
достигнуть путем осуществления реакции в кислой среде и регулирования рН
в конечной стадии, например путем добавки гидроокиси бария для осаждения
фосфатов главным образом в виде ВаНРО4. Фосфаты железа, меди и других
примесей выпадают вместе с фосфатом бария, а поэтому раствор перекиси
водорода остается сравнительно устойчивым. Чтобы удалить примеси из
фосфатного осадка, переводят фосфат бария в растворимую форму путем
добавки фосфорной кислоты:
ВаНРО4 + Н8РО4 -+ Ва (Н2РО4J, F)
Примеси, которые остаются нерастворенными, отделяют фильтрацией
и затем путем обработки раствора серной кислотой регенерируют фосфорную
кислоту, что позволяет использовать последнюю повторно:
Ва (Н2РО4J + H2SO4 -> BaSO4 j + 2Н3РО4. G)
Предложены некоторые другие методики, незначительно отличающиеся
от указанной [6]. Так, для регулирования рН можно использовать перекись
бария или углекислый барий; вместо ортсфосфорной кислоты можно брать
и метафосфорную; наконец, фосфорную кислоту или фосфаты можно очистить
перед повторным их использованием и другими способами. Предложено также
обрабатывать фосфорные кислоты избытком концентрированной азотной,
соляной, плавиковой, кремнефтористоводородной или фтороборной кислоты
с последующим осаждением соответствующей бариевой соли и нагреванием
фосфорнокислотного фильтрата для отгонки летучих кислот перед вторичным
использованием. Еще по одному методу фосфорнокислый барий восстанавли-
восстанавливают углеродом до элементарного фосфора и карбида бария. Фосфор можно
затем превратить в фосфорную кислоту, а карбид бария ввести в реакцию
с водой с образованием ацетилена и раствора гидрата окиси бария; последний
затем превращают в углекислый барий действием карбоната щелочного металла.
Однако большое число стадий обработки и технические трудности, с которыми
сопряжено оформление процесса в соответствии с этими предложениями, сви-
свидетельствуют о вероятной их неэкономичности.
Высокая термическая устойчивость фосфатов исключает возможность их эко-
экономического использования путем превращения непосредственно в окись
бария. Поэтому конечным побочным продуктом, получаемым при фосфорно-
кислотном процессе, всегда является то же вещество, что и при пр>ямсм приме-
применении серной кислоты, т. е. сернокислый барий. Дополнительные расходы при.
применении фосфорной кислоты по сравнению с производством на основе серной
кислоты, по-видимому, перекрываются экономией, достигаемой за счет непо-
непосредственного получения несколько более концентрированной перекиси водо-
водорода при исключении излишнего разложения. Раствор перекиси водорода,
получаемый при применении фосфорной кислоты, по имеющимся данным,
является более чистым и устойчивым, чем получаемый при применении серной
кислоты; это объясняется тем, что обычные примеси, например железо и медь,
образуют сульфаты, более растворимые, чем фосфаты, и фосфорная кислота
оказывает общее стабилизирующее действие. i
7*
100 Глава 3
Реакция перекиси бария с другими минеральными кислотами
Реакция соляной или азотной кислоты с перекисью бария никогда не при-
применялась в техническом масштабе из-за трудностей, возникающих в связи
с необходимостью освобождения получающегося раствора перекиси водорода
от растворимых бариевых солей, хотя в ряде патентов и описываются такие
реакции [7]. Малая летучесть перекиси водорода по сравнению с летучестью
воды способствует тому, что при последующей перегонке перекись остается
в перегонном кубе, т. е. в растворе, насыщенном бариевыми солями, и в сопри-
соприкосновении с осадком солей; все эти условия благоприятствуют каталитическому
разложению. Однако при применении ионообменных смол для разделения
стадии образования перекиси водорода и стадии солеобразования (см. ниже,
стр. 105) целесообразнее пользоваться неорганической кислотой, дающей
растворимую соль.
В XIX столетии применение в небольшом масштабе находили и фтористо-
фтористоводородная и кремнефтористоводородная кислоты; получавшиеся нераствори-
нерастворимые бариевые соли обрабатывали серной кислотой для получения сернокислого
бария и регенерации кислоты.
Реакция перекиси бария с двуокисью углерода
Реакция перекиси бария с двуокисью углерода была объектом многочислен-
многочисленных исследований; привлекала возможность путем обжига образующегося
углекислого бария получать окись бария для повторного использования в про-
процессе и, следовательно, избежать получения бариевой соли в качестве побоч-
побочного продукта, требующего рынка сбыта. Однако низкая растворимость дву-
двуокиси углерода и слабая ионизация угольной кислоты повышают щелочную
зону нестабильности вокруг реагирующих частиц и способствуют более значи-
значительному разложению перекиси водорода по сравнению с наблюдаемым при
применении более сильных кислот. Растворимость двуокиси углерода можно
увеличить применением давления, но степень диссоциации при этом не воз-
возрастает; даже при давлении двуокиси углерода, равном 25 am, выход перекиси
водорода резко снижается при попытках увеличить концентрацию ее примерно
выше 7%. В растворе образуются небольшие количества двууглекислого бария
B г/л при давлении двуокиси углерода, равном 1 am), но в твердой фазе его нет;
единственным компонентом твердой фазы является нерастворимый углекислый
барий. Остающийся к концу операции в растворе бикарбонат можно превратить
в нерастворимый карбонат путем добавки основания, например гидрата окиси
бария, или путем продувания воздухом для вытеснения двуокиси углерода.
Как и при образовании нерастворимого сернокислого бария из перекиси бария
и серной кислоты, скорость реакции и выход перекиси водорода увеличиваются
при добавке небольших количеств кислот, дающих растворимые бариевые соли.
Предложено применять муравьиную, уксусную, пропионовую, азотную и дру-
другие кислоты. При сравнении уксусной и соляной кислот оказывается, что по-
последняя несколько более эффективна [5], вероятно вследствие значительно
более высокой степени ионизации. Рекомендуется также добавлять аммониевые
соли [8] или Na2HPO4 [9], который способствовал бы также дезактивации железа
или других примесей, содержащихся в перекиси бария. Согласно недавно
выданному патенту [10], предлагается добавка небольшого количества фосфор-
фосфорной кислоты как наиболее эффективной и приводится пример образования
в этом случае 7%-ного раствора перекиси водорода с выходом 94%.
Наиболее тщательное исследование реакции двуокиси углерода с пере-
перекисью бария проведено Аскенази и Розе [5], которые суммируют также работы
предыдущих авторов и приводят полную библиографию по этому вопросу.
В патенте Мак-Федьена и Джэффа [10] также приводится обширный материал
Неорганические перекиси 101
по оптимальным значениям рабочих параметров. Скорость реакции и выход
перекиси водорода возрастают с увеличением давления по меньшей мере
до 10—20 am. Чем выше концентрация шлама, тем выше и давление двуокиси
углерода, необходимое для получения высокого выхода. Аскенази и Розе сооб-
сообщают, что при 25 am выход перекиси водорода составляет 80—90% при конеч-
конечной концентрации ее 1—2вес.% и 76—80% при концентрации 4 вес.%; попытки
повышения концентрации приводили к значительному снижению выходов.
Можно получить более концентрированную перекись водорода путем введения
в реакцию шлама перекиси бария в разбавленном растворе перекиси водорода,
но при этом происходит дополнительное разложение, а поэтому суммарный
выход оказывается ниже. Увеличение времени выдерживания в реакторе
обычно вызывает снижение выхода; вместе с тем при слишком малом времени
выдерживания шлам после спуска давления оказывается щелочным, что в свою
очередь может вызвать заметное разложение перекиси. В опытах Аскенази
и Розе, которые пропускали ток двуокиси углерода через партию шлама пере-
перекиси бария в автоклаве, оптимальное время реакции составляло около 10 мин.
В работе Мак-Федьена и Джэффа водный раствор фосфорной кислоты поддер-
поддерживали при температуре примерно ниже 20° путем пропускания рассола через
рубашку реактора. Затем нагнетали двуокись углерода до создания нужного
давления и небольшими порциями добавляли шлам перекиси бария с таким
расчетом, чтобы температура все время была ниже 20°, а раствор оставался
кислым. В этом случае оптимальное время для введения всей перекиси бария
составляло от 0,5 до 1,5 часа.
Выход в опытах Аскенази и Розе возрастал при добавке кислоты, образую-
образующей растворимую бариевую соль; так, добавка соляной кислоты в количестве,
соответствующем образованию однопроцентного водного раствора, давала воз-
возможность получать продукт с концентрацией 5,5% и выходом 90%, однако
при этом возникали трудности при освобождении раствора от образовавшейся
бариевой соли. Добавка соляной кислоты способствовала получению грубо-
дисперсного, почти песчанистого осадка, отличающегося легкой фильтруе-
мостью, вместо тонкозернистого и студенистого углекислого бария, который
получался при применении одной двуокиси углерода.
Чтобы избежать образования или выдерживания перекиси водорода
в щелочной среде, реакцию необходимо проводить очень тщательно; это имеет
особенно большое значение в том случае, когда желательно получить более
концентрированный продукт. Так, если исходить из загрузки шлама перекиси
бария, то для скорейшего создания кислой среды необходимо возможно быст-
быстрее создать давление двуокиси углерода, равное 10 am или выше. По методу
Дернера [11] или Мак-Федьена и Джэффа [10] вместо этого перекись бария
вводят в раствор угольной кислоты, находящийся под давлением, со скоростью,
достаточно низкой, чтобы шлам оставался кислым. Спуск давления после окон-
окончания реакции способствует сильному снижению кислотности шлама, а поэтому
если этот шлам оставить после спуска давления, то небольшие количества непро-
реагировавшей перекиси или окиси бария могут сообщить ему щелочную
реакцию, что, как следствие, приведет к значительному разложению перекиси
водорода. При некоторых условиях разложение перекиси водорода может
протекать столь быстро, что наблюдаемое при этом повышение температуры
будет вызывать самоускоряющееся разложение, способное в течение короткого
времени уничтожить всю перекись. Поэтому важно, чтобы после окончания
реакции образовавшийся осадок был выгружен немедленно или же были при-
приняты другие меры для обеспечения слабой кислотности шлама. Значительные
колебания результатов работ и низкие выходы, отмеченные некоторыми автора-
авторами паннего периода, по меньшей мере частично, можно приписать незнаком-
ст^у этих авторов с влиянием указанных факторов, а возможно также приме-
применению менее чистой перекиси бария. Так же, как и при реакции перекиси бария
102 Глава 3
с другими кислотами, и в этом случае необходимо отнимать экзотермическую
теплоту реакции и поддерживать шлам при комнатной или более низкой тем-
температуре, поскольку уже умеренно высокие температуры приводят к заметному
снижению выхода. Проблема удаления тепла приобретает особенно большое
значение при повышенных концентрациях шлама.
Вольфенштейн и Пельтнер [12] работали при атмосферном давлении и мед-
медленно вводили двуокись углерода в шлам перекиси бария, а поэтому раствор
оставался щелочным; перекись бария исчезала без образования перекиси
водорода, но последняя сразу возникала, как только шлам делался кислым
в результате продолжающейся подачи двуокиси углерода. Авторы приписы-
приписывали это явление образованию пероксокарбоната бария—нерастворимого соеди-
соединения, сравнительно устойчивого в щелочной среде, но разлагающегося в кис-
кислой среде с образованием перекиси водорода и углекислого бария. Они полу-
получили это соединение, медленно пропуская двуокись углерода над гидратом
перекиси бария при 0—5°. Образование пероксокарбоната бария в щелочной
среде свидетельствует о возможности технического получения перекиси водо-
водорода умеренных концентраций при атмосферном давлении путем использова-
использования разбавленной двуокиси углерода, например с концентрацией, имеющейся
в дымовых газах. После превращения перекиси бария можно удалить осадок
пероксокарбоната бария и обработать его небольшим количеством кислоты
для получения углекислого бария и перекиси водорода. В одном опыте, про-
продолжавшемся 3 часа, Вольфенштейну и Пельтнеру удалось получить 1,95%-ный
раствор перекиси бария с выходом 87%. Мицуно, Ямада и Ясукава [13] в опы-
опытах по электролитическому восстановлению кислорода выделяли образовав-
образовавшуюся перекись водорода путем добавки гидрата окиси бария к электролиту,
что вызывало осаждение перекиси бария. Для превращения последней в пере-
перекись водорода они вводили ее в реакцию с газообразной двуокисью углерода
в слабощелочной жидкости. В оптимальных условиях они получили
1,65%-ный раствор перекиси водорода с выходом 73,67%. Аскенази и Розе
в своих опытах не получили никакого пероксокарбоната бария, как и следо-
следовало ожидать, поскольку они анализировали только конечный продукт,
а конечные растворы были кислыми.
Ряд авторов сообщает, что перекись водорода, получаемая при реакции
с двуокисью углерода, более чиста и устойчива, чем получаемая при приме-
применении серной кислоты; вероятно, это обусловлено очень малой растворимостью
углекислых солей примесей. Лунге [14] сообщает, что в 189Э г. на одном фран-
французском заводе использовали реакцию перекиси бария с двуокисью углерода
под давлением, однако никаких данных о более позднем применении этого
процесса в техническом масштабе не имеется.
Производство перекиси водорода из перекиси бария
В США по методу с применением перекиси бария работает только один
завод, а именно отделение фирмы «Food Machinery and Chemical Co.» в Карте-
рете (Нью-Джерси). Перекись бария вырабатывается фирмой «Barium Products,
Ltd.» в Модесто (Калифорния) и отгружается в Нью-Джерси для переработки
на перекись водорода [15]. Единственный процесс производства перекиси водо-
водорода из перекиси бария, по которому имеются подробные опубликованные
данные,—процесс на заводе «Kalichemie» в Гённингене (Германия) [16].
Этот завод довольно старый, и в современных условиях технологический
процесс, возможно, значительно видоизменен. Однако ввиду отсутствия
другой информации, приведем здесь краткое его описание. Годовая мощ-
мощность завода составляет 2—3 тысячи тонн перекиси водорода в расчете на
30%-ный раствор и 3—4 тысячи тонн перекиси бария.
Неорганические перекиси
103
В 1945 г. процесс проводили следующим образом: измельченный барит,
содержащий 94% сернокислого бария, смешивали с грубоизмельченным углем
Угольная пыль
и воздух
Дымовые
газы
Смесь угольной
пыли и барита
Отходящий
газ •*—
Клинкер
BaS Вода
) 1
на извлечение
серы
2-5% Н3Р04
2-5% НС1
NaOH для нейтрализации
после реанции
Шла At co/
6% -ныи раствор
Н202
6aS04
Рис. 12. Поточная схема производства перекиси водорода с применением бариевого
процесса на заводе фирмы «Kalichemie».
/—вращающаяся печь; 2—холодильник; 3—чаны для выщелачивания; 4—сосуды для осаждения;
5—барабанная сушилка; 6—стержневая мельница; 7—бегунковая мельница для смешивания;
8—батарея вертикальных реторт (наружный обогрев генераторным газом и газами, выходящими
из реторты); 9—вращающаяся печь (наружный обогрев генераторным газом); 10—мешочный фильтр;
11—смеситель; 12—змеевик для охлаждающей воды.
и восстанавливали до сернистого бария во вращающихся печах, отапливаю-
отапливающихся угольной пылью (см. поточную схему на рис. 12). Две трети необходи-
необходимого угля смешивали с баритом, а остальную треть вдували во вращающуюся
печь вместе с воздухом в противотоке к твердой шихте. При этом происходила
следующая реакция:
BaSO4 + 4С -> BaS + 4CO. (8)
В присутствии твердого угля двуокись углерода образуется в очень незна-
незначительном количестве. Максимальная температура, достигавшаяся в печи,
была 1000°; горячий клинкер по выходе из печи охлаждали в другом вращаю-
вращающемся цилиндре путем продувки через него части воздуха, необходимого
для сжигания топлива. 65—80% всего твердого продукта составлял водорас-
водорастворимый сернистый барий; более высокое содержание относится к случаю при-
применения малозольных углей. Для получения 100 т 100%-ного сернистого бария
требовалось 170 т барита и всего 40 т угля.
Клинкер сернистого бария, выходивший из холодильников с температурой
100—150°, подавался на выщелачивание, которое проводилось в противотоке
в серии баков из углеродистой стали. Каждая серия состояла из 4 баков, рас-
рассчитанных на 1,5 m клинкера каждый. Свежий клинкер вступал в контакт
с наиболее насыщенным раствором, а сравнительно истощенный обрабаты-
обрабатывался свежей водой. На каждой стадии выщелачивания раствор нагревали до 60°
острым паром и через 4—5 час. после отстаивания твердого остатка жидкость
104 Глава 3
откачивали. Остаток, содержавший только 5—6% сернистого бария (в расчете
на сухое вещество), шел в отвал.
Полученный щелок, содержавший 180 г/л сернистого бария, обрабатывали
в противотоке двуокисью углерода в серии цилиндрических чугунных сосудов
объемом по 12 м3 каждый. При этом выпадал углекислый барий в соответствии
со следующей реакцией:
BaS + СО2 + НаО -* ВаСО3 + H2S. D)
И здесь каждая группа состояла из четырех баков, причем первые три содер-
содержали щелок, четвертый же служил лишь для задерживания уноса. Наиболее
старый раствор обрабатывали свежей двуокисью углерода, которая затем
поступала в менее старый раствор и, наконец, в самый свежий. Каждый раствор
находился в контакте с двуокисью углерода в течение 6—9 час, т. е. на каждую
стадию этой операции расходовалось 2—3 часа. Этого времени было достаточно
для вытеснения всех сернистых соединений из раствора. Выходящий газ (при-
(примерный состав 90% H2S и 10% СО2) затем частично окисляли воздухом для реге-
регенерирования серы. Шлам углекислого бария отфильтровывали на вращаю-
вращающихся вакуумных фильтрах из чугуна или углеродистой стали, причем фильтрат
возвращали в баки для выщелачивания, а мокрый отфильтрованный осадок
высушивали в барабанных сушилках из углеродистой стали, обогревавшихся
генераторным газом. Работу проводили в условиях параллельного потока, так
как противоточная операция давала неудовлетворительные результаты. Сухой
продукт содержал 98—99% углекислого бария и 0,8—1,2% общей серы в рас-
расчете на сернокислый барий.
Для превращения в окись бария карбонат смешивали на бегунках с лам-
ламповой сажей (зольность 0,03—0,05%) из расчета 1 часть сажи на 10 частей кар-
карбоната. Восстановление проводили в ретортных батареях, обогревавшихся
снаружи генераторным газом в смеси с газами, отходившими из реторт. Газ
из реторт содержал 18—20% двуокиси углерода (остальное до 100%—прак-
100%—практически чистая окись углерода). Каждая реторта представляла вертикальную
трубу внутренним диаметром 20 см и длиной 4 м, составлявшуюся из отдель-
отдельных секций, изготовленных из кислотоупорного камня. Начальный вес шихты
в такой реторте был 70 кг, а после 12-часового нагрева до 1200°—50 /сг. Шихту
после этого без охлаждения выгружали в стальные тележки. По имеющимся
сообщениям, разгрузка труб не сопровождалась никакими затруднениями;
не было также случаев сплавления труб с окисью бария. Продукт
состоял на 93—94% из окиси бария (остальное—углекислый барий и зола),
причем размер частиц был около 7 мм. На каждую тонну выработанной окиси
бария расходовалось около 1 т угля (на получение генераторного газа для
реторт и на производство перекиси бария, как описано ниже).
Перекись получали периодическим методом в горизонтальных чугунных
вращающихся барабанах, обогреваемых снаружи генераторным газом. Воздух
предварительно подогревали до 150—200° в теплообменнике, установленном
на борове обжигательных печей, при помощи отходящих газов. Воздух проходил
сначала через загрузку окиси бария весом 3 т со скоростью 1000 мг/час, а затем
через мешочные фильтры. По имеющимся данным, температура загрузки соста-
составляла около 300°, хотя эта величина, по-видимому, несколько занижена.
В начале реакции кислород воздуха полностью поглощался. Реакция
заканчивалась спустя 14—16 час. и давала в среднем продукт следующего
состава (%):
ВаО2 87—88 SiO2 0,2—0,3
ВаСО8 .... 3—5 А1аО3 0,1—0,2
BaSO4 .... 1—1,5 Fe ...*... 0,02
Содержание окиси бария не указано, но на ее долю, видимо, падало остальное
количество до 100%.
Неорганические перекиси 10S
Перекись бария получалась главным образом в виде частиц размером ниже
1 мм, но попадались также и крупные куски до 1 см. После охлаждения ее
измельчали на стержневой мельнице до прохода через сито со 100 отверстиями
на линейный сантиметр и затем перемешивали в смесителе для получения равно-
равномерного продукта. Для превращения перекиси бария в перекись водорода в чан,
снабженный охлаждающим змеевиком и мешалкой, вводили сначала 1,4 л*3
воды, а затем 2 т перекиси бария. После этого в течение 2,5—3 час. заливали
в чан 70%-ную серную кислоту, содержащую 5% фосфорной и 5% соляной
кислоты (в некоторых источниках указано только по 2% соляной и фосфорной
кислот), вместе с таким количеством воды, чтобы получалась перекись водорода
приблизительно 6%-ной концентрации. Путем пропускания холодной воды под-
поддерживали температуру массы на уровне 30—35°. По окончании реакции, чтобы
довести конечную величину рН примерно до 4, к смеси добавляли едкий натр.
Сообщается, что выход перекиси водорода из перекиси бария составлял 90%.
Перекись водорода из перекиси натрия
Перекись водорода можно получить по реакции кислоты с перекисью натрия
таким же образом, как это описано для перекиси бария. Однако большая часть
натриевых солей растворима в воде, что, как описано выше, сильно увеличи-
увеличивает затруднения, связанные с выделением перекиси водорода из солевого
раствора без заметного ее разложения.
Перекись натрия получается окислением металлического натрия, в свою
очередь получаемого электролизом хлористого натрия, растворенного во фто-
фторидах щелочных металлов и хлористом кальции. В 1954 г. в США перекись
водорода, полученная электролитическим процессом, стоила по рыночным
ценам примерно на 30% дороже перекиси натрия в расчете на моль активного
кислорода. Если эти два продукта в какой-либо местности в достаточной мере
различаются по отпускным ценам, то перекись водорода можно получать из
перекиси натрия; обычно это оправдывается только в местах непосредствен-
непосредственного потребления перекиси водорода и притом лишь тогда, когда остающаяся
в растворе натриевая соль не мешает применению продукта. В настоящее время
перекись водорода получается этим путем только в том случае, когда конечный
раствор должен немедленно быть использован в процессах щелочной отбелки,
например в производстве древесной целлюлозы или других целлюлозных мате-
материалов. Вопрос о соотношении цен между перекисью водорода и перекисью
натрия рассматривается также в гл. 11.
Реакция с перекисью натрия может быть использована для получения
перекиси водорода еще в том случае, когда последняя является побочным про-
продуктом в процессе, где основное внимание уделено получению подходящей нат-
натриевой соли за счет реакции соответствующей кислоты с основным натриевым
соединением. Так, из фтористоводородной кислоты и перекиси натрия можно
получить умеренно растворимый фтористый натрий, а затем из раствора оса-
осадить искусственный криолит Na3(AlF6) путем добавки фтористого алюминия
[17]. Имеются данные, что этот процесс в течение некоторого времени осуще-
осуществлялся на практике во Франции.
В одном из патентов [18] предлагается использование ионообменных смол
для отделения растворимых солей, образующихся при реакции перекиси натрия
с кислотой. Водный раствор перекиси натрия превращают в раствор перекиси
водорода путем пропускания его через катионитную смолу, которая в самом
начале находится в водородной форме. Для этой цели применяют органические
смолы, полученные из сульфированного продукта конденсации формальдегида
и многоатомного фенола, поскольку неорганические ионообменные вещества
вызывают очень значительное разложение. Последующей обработкой ионита
106 Глава 3
кислотой его вновь переводят в водородную форму и вытесняют адсорбирован-
адсорбированные натриевые ионы в виде натриевой соли. На каждой стадии регенерации
смолу необходимо обрабатывать кислотой, взятой в количестве, превышающем
в 2—5 раз количество кислоты, необходимое для полного удаления только
натриевых ионов; для этой цели применяют 2—20%-ную сильную кислоту,
например соляную, серную или азотную. Эта обработка избыточной кислотой
способствует вытеснению ионов железа и других каталитически действующих
многовалентных тяжелых металлов, которые в противном случае вызвали бы
заметное разложение перекиси. Для снижения разложения перекиси водорода
рекомендуются также следующие меры: 1) предварительное охлаждение рас-
раствора перекиси натрия до 5—10°, поскольку реакция является экзотермиче-
экзотермической; 2) добавка небольших количеств стабилизаторов к раствору, например
силиката натрия, растворимой магниевой соли или растворимого пирофосфата,
и 3) поддерживание щелочности раствора, поступающего на ионит, на приемле-
приемлемом низком уровне путем применения раствора, содержащего лучше всего
0,5—2,5% перекиси натрия. В патенте указывается, что в этих условиях выход
перекиси водорода составляет 9Э—99%. Концентрация перекиси водорода при
желании может быть доведена до 20—30% путем повторного пропускания рас-
раствора перекиси водорода через ионит с предварительной добавкой перекиси
натрия к разбавленному раствору, хотя, вероятно, при этом увеличивается
потеря перекисного кислорода. Согласно патенту, этот процесс может быть
использован и для переработки других перекисей, в том числе и перекиси бария
(хотя никаких экспериментальных данных не приводится). Однако работать
с нерастворимой перекисью труднее, особенно если желательно получить
нерастворимую бариевую соль непосредственно из ионита.
Получение перекисных соединений из окислов других щелочноземельных
металлов
Все прочие неорганические перекиси, за исключением перекисей бария
и натрия, обычно получаются либо из перекиси натрия, либо путем смешения
перекиси водорода с соответствующими солями или гидроокисями. Перекись
водорода может быть регенерирована из этих соединений, как описано выше,
обработкой кислотой.
При данной температуре равновесное парциальное давление кислорода
над перекисью щелочноземельного металла возрастает в ряду: перекись бария,
перекись стронция, перекись кальция. Поэтому значительно труднее получить
из окиси прямой реакцией с воздухом или кислородом перекись стронция или
кальция. Так же, как и при получении перекиси бария, равновесное давление
кислорода быстро возрастает с температурой, а поэтому температура, необхо-
необходимая для обеспечения благоприятных условий равновесия при образовании
перекиси кальция или стронция, настолько низка, даже при давлениях выше
атмосферного, что скорость реакции необычайно мала. Проведено очень мало
работ по образованию перекисей из этих двух окислов.
Равновесное парциальное давление кислорода над смесью перекисни окиси
стронция составляет 1 am при 215°. Следы перекиси стронция получены при
нагревании окиси стронция на воздухе, причем Фишер и Плетце [19] получили
перекись стронция с выходом до 16% путем нагревания окиси строн-
стронция при температуре около 410° под давлением кислорода 100 am. По патенту
Пирса [20] можно получить 85% перекиси стронция при 400—500° и
105—126 кг/см2 A02—122 am). Поскольку спрос на стронциевые соли невелик,
промышленное производство перекиси водорода из перекиси стронция воз-
возможно лишь при оформлении процесса как циклического, например через угле-
.кислый стронций. Однако трудности обжига аналогичны тем, которые наблю-
Получение путем электролитических процессов 107
даются при углекислом барии, так как углекислый стронций разлагается при
температуре около 1340°; кроме того, стадия получения перекиси в этом случае
еще значительно сложнее.
Равновесие в системе перекись кальция—окись кальция—кислород недо-
недостаточно точно изучено ввиду низких скоростей реакции при требующихся
низких температурах, а также трудностей, с которыми приходится сталкиваться
при попытках приближения к равновесию с той и другой стороны. Недавно этот
вопрос был подвергнут анализу [21]. По-видимому, давление кислорода над
твердой фазой в состоянии равновесия должно составлять около 107 кг1смг
при температуре около 111°. Однако из кислорода и окиси кальция были полу-
получены лишь следы перекиси кальция даже при проведении реакции в течение
нескольких дней [21, 22], и эта реакция, по-видимому, не может найти практи-
практического применения для получения перекиси кальция, а следовательно, и пере-
перекиси водорода.
ПОЛУЧЕНИЕ ПУТЕМ ЭЛЕКТРОЛИТИЧЕСКИХ ПРОЦЕССОВ
Перекись водорода и другие перекисные соединения могут образоваться
при электролитическом окислении на аноде. Перекись водорода можно также
получать непосредственно на катоде за счет восстановления кислорода, раство-
растворенного в электролите или барботируемого над катодом. Эти катодные процессы
уже рассмотрены в гл. 2 (стр. 82 и ел.).
При анодном окислении можно обнаружить лишь небольшие1 количества
перекиси водорода и то только в сильнощелочной среде. Так, Ризенфельд и
и Рейнгольд [23] сообщили, что при электролизе насыщенного раствора едкого
кали при —40° обнаружены небольшие количества перекиси водорода, но при
электролизе раствора едкого натра в тех же условиях образования перекиси
водорода не наблюдалось. В случае применения едкого кали при более высоких
температурах выход снижался. Риус [24] в исследованиях, проведенных при
2—4°, применяя 6,3 н. раствор едкого кали вместе с фтористым калием, получил
максимальный выход по току 5,35%, причем этот выход падал во времени. В дру-
других работах, проведенных при комнатной температуре, перекиси водорода
совсем не получено или же получены только следы ее. Большая часть этих
работ поставлена с электродами из платины, которая является катализатором
разложения перекиси водорода, а поэтому трудность обнаружения перекиси
водорода во многих случаях, возможно, вызвана быстрым каталитическим раз-
разложением ее на аноде. Бюргин [25] пользовался гораздо менее активными ката-
катализаторами—цинковыми анодами с защитным слоем из гидроокиси цинка или
углекислого цинка и получил выход по току около 60% при температуре элект-
электролита 0° (электролит содержал 5 молей едкого натра и 1 моль борной кислоты
в литре). Однако этот высокий выход получался только при низких концентра-
концентрациях перекисного кислорода в электролите.
В 1853 г. Мейдингер констатировал образование перекиси водорода при
электролизе воды, сильно подкисленной серной кислотой, что подтверждено
Бунзеном в 1854 г. и Гофманом в 1867 г., а также другими авторами. Однако
Вертело [26] в 1878 г. показал, что вещество, образовавшееся на аноде, является
в действительности пероксодисерной кислотой, которая образует перекись
водорода при гидролизе. В настоящее время хорошо известно, что пероксоди-
сульфаты можно получать с высокими выходами по току из растворов серной
кислоты, а также из кислых или нейтральных растворов сульфатов, причем
эти реакции представляют в настоящее время основу наиболее широко приме-
применяемых производственных процессов получения перекиси водорода.
На аноде можно получать также и другие перекисные соединения, напри-
например, пероксобораты и пероксокарбонаты, но они имеют меньшее промышленное
значение и в данной монографии подробно не рассматриваются.
108 Глава 3
Теория образования на аноде
Наблюдения, касающиеся природы и количества перекисных соединений,
образующихся на аноде, могут быть объяснены, если исходить из стандартных
окислительно-восстановительных потенциалов [27] для реакций, которые могут-
протекать на аноде в воде или в различных растворах (см. также гл. 5):
2Н2О —> Н2О2 + 2Н+ + 2е, (9)
Е° = — 1,76 в A н. кислота),
2Н2О -> О2 + 4Н+ + 4е, A0)
?°= — 1,229 в A н. кислота),
?°= —0,82 в (нейтральный раствор),
4ОН- -> О2 + 2Н2О + 4е, A1)
Е°= +0,401 в A н. щелочь),
Н2О2 _»О2 + 2Н+ + 2е, A2)
?°= -0,693 в A н. кислота),
2HSO4--»S2O°;- + 2Н+ + 2е, A3)
?°= -2,18*,
2S0J- -» S;O28- + 2e, A4)
Е°= -2,06 в.
Эти потенциалы относятся к активностям, равным единице (практически
к 1 н. растворам), различных видов частиц при температуре 25°. В термодина-
термодинамически обратимых условиях по мере повышения наложенного потенциала
будет протекать сначала тот процесс, который имеет минимальный отрицатель-
отрицательный (максимально положительный) потенциал. В таких равновесных условиях
на аноде не может возникать ни перекись водорода, ни пероксодисульфат,
а может образовываться только кислород. Соответствующий теоретический
потенциал ванны составляет 1,229 в в 1 н. кислоте или 0,82 в в нейтральном
растворе. Однако при осуществляемых электролитических процессах можно
добиться протекания реакции, требующей более высокого потенциала, преиму-
преимущественно перед реакцией с более низким потенциалом, но протекающей с выде-
выделением газа, путем увеличения разности потенциалов на ванне до значений,
превышающих больший потенциал, например путем применения высоких
плотностей тока и использования такого материала для электродов, который
требует высокого перенапряжения для выделения газа. Ничтожное образова-
образование перекиси водорода при эксплуатации электролизера с применением потен-
потенциала, достаточно высокого для возможости частичного протекания реакции
(9), можно объяснить тем, что реакция A0) протекает с большей скоростью,
чем реакция (9), или же тем, что уже образовавшаяся перекись водорода, как
только возникает некоторая невысокая ее концентрация, исчезает за счет реак-
реакции A2). Исчезновение перекиси водорода возможно также за счет неэлектро-
неэлектролитического разложения ее в среде с высоким рН, поскольку перекись водо-
водорода очень неустойчива в щелочной среде. Поверхности анодов также могут
быть причиной значительного разложения*.
* Наложение переменного тока на постоянный, протекающий через ванну, может
вызвать последовательное протекание реакций восстановления и окисления на одном
и том же электроде. Описаны некоторые работы по образованию в таких условиях пере-
перекиси водорода на цинковом аноде [28]. Постулируется, что перекись водорода образуется
под действием постоянного тока и затем подвергается восстановлению отрицательной
фазой переменного тока, однако трактовка экспериментальных данных весьма затруд-
затруднительна.
Получение путем электролитических процессов 109
В серной кислоте раствор содержит главным образом бисульфат-ионы,
и образование пероксодисульфата протекает, вероятно, преимущественно за
•счет реакции A3). Нейтральные растворы сульфатов содержат преимуществен-
преимущественно сульфатные ионы, и соответствующая анодная реакция представлена, вероят-
вероятно, уравнением A4).
Большинство материалов обладает слишком высокой каталитической ак-
активностью или недостаточной устойчивостью для использования их в качестве
электродов для этих процессов. По сравнению с другими металлами предпо-
предпочтение отдается платине вследствие ее стабильности и высокого электродного
потенциала, что обеспечивает возможность протекания таких реакций, как
реакции A3) и A4). Однако платина является активным катализатором разло-
разложения перекиси водорода и способствует таким образом ее разложению за счет
других процессов.
Образование пероксодисульфатов с высокой концентрацией и высоким
выходом по току путем электролиза возможно, так как в отличие от перекиси
водорода они не обладают способностью легко разлагаться на платиновом
аноде в условиях эксплуатации ванны. Для получения высокого анодного по-
потенциала, требующегося для их образования, применяют полированные пла-
платиновые аноды. Работа проводится при высокой анодной плотности тока, что
способствует высокому перенапряжению и снижает долю тока, потребляемого
в нежелательной реакции образования кислорода. Некоторые небольшие
добавки к электролиту, например фтористых или роданистых соединений или
мочевины, несколько повышают анодный потенциал и усиливают указанный
эффект, вероятно, вследствие адсорбции их на активных центрах платинового
анода, где они функционируют как ингибиторы образования кислорода.
Относительно механизма образования перекиси водорода и пероксодисуль-
пероксодисульфатов на аноде мнения сильно расходятся [29—33], хотя наиболее вероятным
является предположение о непосредственном соединении ионов, например
по уравнениям реакций A3) и A4) для сульфатных растворов в кислой и ней-
нейтральной среде, или о соединении двух гидроксильных ионов с образованием
перекиси водорода в щелочной среде (при этом возможно промежуточное воз-
возникновение гидроксильных радикалов). Имеются указания на невозможность
образования перекиси водорода на аноде в качестве промежуточного продукта,
реагирующего на следующей стадии с сульфатом с образованием пероксоди-
пероксодисульфата. Максимальное образование пероксодисерной кислоты наблюдается
в растворах серной кислоты, не содержащих сернокислых солей, при умеренно
высоких ее концентрациях, тогда как в слабокислых растворах указанная
кислота образуется в очень малых количествах. Это явление часто приводят
как «доказательство», что пероксодисерная кислота образуется не из сульфат-
сульфатных, а из бисульфатных ионов. Возможно, что это совершенно верно (по-
(поскольку в сравнительно концентрированной кислой среде анионы представ-
представляют в основном бисульфат), однако более низкий выход по току в разбавлен-
разбавленной кислоте, вероятно, обусловлен скорее более низкой общей концентрацией
анионов, а не преимущественным существованием сульфатных ионов вместо
бисульфатных, так как пероксодисерная кислота образуется при соединении
любых двух анионов. То, что в нейтральных растворах сульфатов для
обеспечения высокого выхода по току требуются высокие концентрации солей,
•свидетельствует о правильности изложенной точки зрения.
Имеются данные, что для осуществления реакции образования пероксо-
пероксодисерной кислоты требуется наличие кислорода in statu nascendi:
2HSO- + О + Н2О -» H2S2O8 + 2OH-. A5)
Однако доказательств в пользу этого механизма не приведено, и гораздо более
вероятно, что выделение кислорода на аноде необходимо лишь по той причине,
что его поляризующее действие способствует возникновению того высокого
ПО Глава 3
потенциала, который требуется для образования пероксодисульфата. Поскольку
выделение кислорода протекает при более низком потенциале, термодинами-
термодинамически всегда возможны условия, благоприятствующие его образованию,
если потенциал достаточно высок для образования пероксодисульфата.
Вероятно, полностью исключить образование кислорода невозможно, но на
практике оно может быть сокращено до такой степени, чтобы на него расходо-
расходовалась лишь небольшая доля тока. В сообщении Вуда [34] подытожены раз-
различные другие гипотезы, предложенные для механизма образования пероксо-
дисульфатов. Механизм анодного окисления вообще также является предме-
предметом спора. Так, Глэстон и Хиклинг [29] предполагают, что окисление проте-
протекает через образование перекиси водорода на аноде в качестве промежуточ-
промежуточного продукта. В доказательство они приводят существование параллелизма
между направлением реакций анодного окисления в разных эксперименталь-
экспериментальных условиях и направлением реакций анодного окисления, которого можно
ожидать, если перекись водорода действительно является промежуточным
продуктом, а также влияние различных катализаторов разложения перекиси
водорода и различных материалов анода на обнаруженные реакции. Наоборот,
Гайсинский и Коттэн [35] указывают, что перекись водорода, вероятно, не
является промежуточным продуктом в электролитическом образовании пер-
оксокарбонатов и пероксоборатов.
Пероксодисерная кислота из серной кислоты
При комнатной температуре пероксодисерная кислота разлагается с обра-
образованием кислорода и серной кислоты лишь с очень небольшой скоростью, но
она легко гидролизуется с образованием пероксомоносерной кислоты (кислоты
Каро) HZSO5:
H2S2O8 + H2O -> H2SO5 + HaSO4. A6)
Пероксомоносерная кислота может затем разложиться на аноде:
soj-+o->so;-+olf (i7>
либо образовать перекись водорода:
H2SO5 + Н2О -> H2SO4 + Н2О2, A8)
либо подвергнуться разложению действием перекиси водорода:
H2SO6 + Н2О2 -> H2SO4 + О2 + Н2О. A9>
Реакция гидролиза A6) и реакции A8) и A9) не электролитические и могут
протекать в любой точке раствора. Для обеспечения высокого выхода
пероксодисерной кислоты из серной нужно, по возможности устранить
образование пероксомоносерной кислоты не только потому, что она легко
разлагается в указанных реакциях, но и из-за вызываемой ею деполяризации
анода, снижающей напряжение на зажимах электролизера и тем самым резко
уменьшающей выход пероксодисерной кислоты по току.
Проведено много работ по определению зависимости образования пер-
пероксодисерной кислоты от ряда факторов, в том числе от конструкции ванны и ее
эксплуатации. Эти данные подытожены в книге Маху [36] и у Вуда [34].
Максимальный выход пероксодисерной кислоты по току наблюдается при ми-
минимальном общем времени нахождения электролита в ванне, так как нараста-
нарастание концентрации пероксомоносерной кислоты во времени приводит к потере
активного кислорода. Концентрация же пероксодисерной кислоты в электро-
электролите, наоборот, по мере увеличения времени нахождения в ванне продолжает
возрастать. Поэтому в производственных процессах оптимальное время опре-
определяется путем составления соответствующего экономического баланса. По
Получение путем электролитических процессов 111
мере повышения концентрации пероксодисульфата расход энергии и капитало-
капиталовложения в ванны на единицу производства перекиси водорода возрастают,
расходы же по получению перекиси водорода гидролизом пероксодисульфата
падают. Выше уже указывалось на необходимость применения высокой анод-
анодной плотности тока (около 1 а/см2). Максимальный предел плотности тока
определяется возрастанием скорости гидролиза пероксодисерной кислоты
в пероксомоносерную и, как следствие, потерей активного кислорода при плот-
плотностях тока, значительно превышающих 1,5 а/см2. Отчасти это обусловлено
повышением местной температуры анода. Для снижения влияния этого роста
скорости гидролиза пероксодисерной кислоты с температурой необходимо ра-
работать с возможно более холодным электролитом. Кроме того, рост температуры
снижает напряжение на ванне и тем самым понижает выход по току. Этот выход
заметно падает уже при небольшом повышении температуры, и поэтому заводы,
работающие по пероксодисернокислотному процессу, необходимо строить
по соседству с крупными источниками снабжения холодной водой. Для дости-
достижения достаточной эффективности температура электролита должна быть
около 20° или ниже.
Выше показано, что наиболее значительная доля потери активного кисло-
кислорода обусловлена реакциями, протекающими не на аноде, а в самом электро-
электролите. Поэтому можно ожидать, что максимальный выход по току будет при ми-
минимальном отношении объема электролита к поверхности анода, что должно
повысить величину электродных реакций по сравнению с реакциями, проте-
протекающими в электролите. В связи с этим электролизеры для получения пер-
пероксодисерной кислоты конструируют таким образом, чтобы объем электролита
в анодном пространстве (анолита) был небольшим; работают же на них
с «концентрацией тока» 500 а/л раствора.
Пероксодисульфат аммония
Замена части серной кислоты сернокислым аммонием увеличивает выход
по току по сравнению с получаемым при применении одной серной кислоты,
причем максимум наблюдается при нейтральном растворе сернокислого аммо-
аммония. Это обусловлено тем, что в нейтральных растворах побочные реакции,
особенно образование SO*", происходят лишь в небольших размерах. В этом
случае пероксодисульфатный ион, вероятно, образуется за счет прямого соеди-
соединения двух сульфатных ионов:
2S0J- _>S2O*- + 2e. A4)
Для обеспечения на электролизере высокого напряжения, без которого невоз-
невозможен значительный выход по току, и в этом случае требуется высокая плот-
плотность анодного тока. Однако если применять нейтральный раствор, то катод
становится щелочным и на нем выделяется газообразный аммиак за счет общей
реакции:
2 (NH4J SO4 -+ (NH4J S2O8 + 2NH3 + H2. B0)
В связи с этим в производственных условиях применяют раствор, который
в самом начале обладает несколько более высокой молярной концентрацией
серной кислоты, чем концентрация сернокислого аммония, а поэтому раствор
в целом сохраняет кислотность в процессе электролиза, что обусловливает
ничтожные потери аммиака в ваннах. Присутствующая избыточная кислота
облегчает гидролиз при нагревании, представляющий вторую стадию процесса.
Поэтому начальный электролит можно рассматривать как кислый раствор
бисульфата аммония.
Сульфаты натрия и калия в кислом растворе также могут быть превращены
в пероксодисульфаты путем электролиза и притом с высоким выходом по току
112 Глава 3
[37], однако эти реакции обычно в промышленности не используются ввиду
малой растворимости соответствующих пероксодисульфатов, которые оседают
в ваннах и на электродах и с трудом могут быть удалены. Если желательно
получить эти соли в качестве конечныхдфодуктов, то их готовят путем соответ-
соответствующих реакций из пероксодисерной кислоты или пероксодисульфата аммо-
аммония.
В процессе электролиза бисульфата аммония гидролиз образовавшегося
иона S2Og" протекает значительно медленнее, чем при электролизе пероксоди-
пероксодисерной кислоты ввиду более высокого рН. В связи с этим можно работать со
значительно более низкими концентрациями тока в амперах на литр (но не
q более низкими плотностями тока) и электролизеры могут содержать сравни-
сравнительно большой объем электролита. Точно так же можно применять и более
высокие температуры электролита (до 30—40°) без существенного гидролиза
продукта, причем они действительно нужны для предотвращения выпадения
соли, образующейся как продукт в конце процесса, из раствора.
Независимо от того, применяется ли серная кислота или бисульфат аммо-
аммония, необходимо, чтобы и оборудование и химикаты были очень чистыми во
избежание излишнего разложения, которое может происходить особенно в про-
процессе последующего гидролиза при повышенных температурах. Для конструк-
конструкционных целей большая часть тяжелых металлов, если они должны находиться
в контакте с электролитом, непригодна. Часть циркулирующего электролита
непрерывно подвергается обработке для понижения до минимума концентра-
концентрации каталитически активных ионов тяжелых металлов, например железа
и платины.
Смешанные растворы сульфатов
Согласно одному английскому патенту [38], добавка сульфата натрия или
других сульфатов, например лития, магния или цинка, к водному раствору
сульфата аммония и серной кислоты способствует образованию системы, в кото-
которой общая концентрация пероксодисульфатов, насыщающая раствор, на 50—
100% выше, чем концентрация, достигаемая при применении одного кислого
пероксодисульфата аммония. Увеличение концентрации тока и плотности тока
позволяет почти удвоить производительность установки. В патенте указывается
выход по току 82—85%, т. е. такой же, какой получается при применении
в качестве электролита одного только бисульфата аммония, однако не указы-
указывается напряжение на зажимах ванны и расход энергии; в другом месте сооб-
сообщается, что общий выход по электроэнергии несколько ниже. На основании
приведенных экспериментальных материалов эффективность гидролиза со-
составляет около 80%, т. е. столько же, сколько получается при применении кон-
контрольного раствора бисульфата аммония, но несколько меньше, чем на заводах,
работающих по процессу с бисульфатом аммония без участия твердой фазы.
Конструкция электролизера
В качестве анодов предложено много различных материалов, но только
одна платина удовлетворяет двум основным требованиям, а именно инерт-
инертности к электролиту и высокому перенапряжению на электроде. Поскольку
платина очень дорога, анод конструируют возможно меньших размеров. Обыч-
Обычно ток поступает по неблагородному металлу, в достаточной мере защищенному
против коррозии в кислотной жидкости. В некоторых случаях подвергающаяся
воздействию часть анода состоит из тантала и платины, причем ток подводится
по танталу. В качестве центральной опоры можно применять алюминий, покры-
покрытый эбонитом или каким-либо другим защитным материалом. Благоприятным
фактором является то, что в связи с требуемыми очень высокими анодными
плотностями тока поверхность анода может быть доведена до минимума. Един-
Получение путем электролитических процессов 113
ственное требование, предъявляемое к катоду, состоит в том, чтобы он был инерт-
инертным к электролиту. В связи с этим можно применять катод из графита или свин-
свинца или изготовлять его в виде свинцового змеевика, через который пропускается
ток холодной воды. Свинец, по-видимому, не является активным катализатором
разложения пероксодисульфатов или пероксодисерной кислоты, но активно
разлагает перекись водорода, которая может образоваться при электролизе.
На поверхности электрода образуется сернокислый свинец, сообщающий ей
инертность. При любых электролитических процессах необходимо не допускать
диффузии пероксодисульфатных ионов к катоду, на котором они разлагаются.
Поэтому обычно для отделения анолита от католита применяют пористую диа-
диафрагму из неглазурованного фарфора. В процессе Питча—Адольфа с пероксоди-
сульфатом аммония катод обертывают асбестовым шнуром, служащим диафраг-
диафрагмой и устраняющим необходимость в применении католита. Однако, вообще
говоря, выгода, связанная с применением одного жидкостного потока, исчезает
в результате разложения пероксодисульфата на катоде, что приводит к пониже-
понижению выхода по энергии по сравнению с выходом в ваннах двухкамерных кон-
конструкций, которые поэтому являются более предпочтительными. В любых
ваннах во избежание перегрева скорость потока анолита желательно поддер-
поддерживать выше некоторого минимума. Необходимой концентрации пероксоди-
пероксодисульфата можно достичь путем повторного пропускания жидкости через ванну,
но на крупных промышленных установках целесообразнее пропускать электро-
электролит через серию последовательно включенных ванн, которые часто монтируют
в виде отдельных ступеней, дающих возможность жидкости перемещаться
самотеком. Ванны обычно помещают в керамические сосуды (специального
типа с высоким электрическим сопротивлением), которые снабжают приспособ-
приспособлениями для снижения до минимума всползания жидкости и последующей
кристаллизации на внутренних стенках. Если в ванне данной конструкции
имеются отдельно анолит и католит, то после стадии гидролиза жидкость про-
пропускается сначала через все католитные камеры серии ванн, а затем через ано-
литные. При прохождении электролита через катодные пространства выделяется
водород и происходит удаление остающегося от стадии гидролиза активного
кислорода. Других реакций окисления или восстановления не происходит,
но состав электролита может заметно изменяться в результате диффузии ионов
через диафрагмы или испарения воды. В следующих разделах описаны наиболее
важные промышленные типы электролизеров.
Гидролиз пероксодисерной кислоты и пероксодисульфатов
Соединения, образующиеся в электролизере, подвергаются затем гидро-
гидролизу с целью выделения перекиси водорода. Пероксодисерная кислота подвер-
подвергается гидролизу непосредственно (вейссенштейнский процесс), кислый раствор
пероксодисульфата аммония либо гидролизуется также непосредственно (ле-
венштейнский и лапортский процессы), либо превращается в сравнительно
нерастворимый пероксодисульфат калия, который затем удаляют, подкисляют
и гидролизуют (процесс Питча—Адольфа).
В том или другом случае для гидролиза требуется кислая среда, причем
фактически гидролизу подвергается пероксодисерная кислота. Эта реакция,
как указано выше, протекает в две стадии:
H2S2O8 + Н2О -+ H2SO6 + H2SO4, A6)
H2SO5 + Н2О -+ H2SO4 + Н2О2. A8)
Реакции обратимы, первая из них протекает значительно быстрее второй.
Так, Пальме [39] показал, что при применении смеси пероксодисульфата калия
и серной кислоты при 50° константа скорости для реакции A6) в 40 раз больше,
чем для реакции A8). С повышением концентрации кислоты возрастают обе
8 Перекись водорода
114 Глава 3
скорости, так что при 10 н. кислоте скорости обеих реакций в 8,5 раза больше,
чем при 5 н.
Для обеспечения высокого выхода перекиси водорода при гидролизе необ-
необходимо уменьшить до минимума разложение перекиси водорода и реакцию ее
с пероксомоносерной кислотой:
2Н2О2 -> 2Н2О + О2, B1)
H2SO6 + Н2О2 -> HaSO4 + Н2О + О2. A9)
Реакция A9) протекает в жидкой фазе, а реакция B1) либо в жидкой, либо
в паровой. В связи с этим перекись водорода необходимо отгонять из раствора
практически сразу же по мере образования, для чего операция проводится
при повышенной температуре и пониженном давлении. Для осуществления
гидролиза с низкими потерями активного кислорода [36, 40] предложено
и применяется много различных методов, включая разные способы обеспечения
оптимальных концентраций и температур; наиболее существенные из них из-
изложены в следующем разделе.
Прямой гидролиз раствора пероксодисульфата аммония с большим выхо-
выходом перекиси водорода осуществить труднее, чем гидролиз пероксодисерной
кислоты, поскольку кислый раствор пероксодисульфата аммония необходимо
концентрировать до достижения достаточно большой скорости гидролиза,
а в этих условиях на различных частях аппаратуры может выкристаллизоваться
твердая фаза, в значительной степени способствующая разложению образо-
образовавшейся перекиси. Эта трудность преодолена путем применения испарителей
с всползающей пленкой, в которых концентрирование и гидролиз проводятся
одновременно таким образом, что отложение твердых частиц на стенках ока-
оказывается невозможным (левенштейнский процесс), а позже путем применения
двухступенчатого процесса, при котором раствор сначала концентрируют
в вакууме с частичным гидролизом и затем подают жидкость в перегонный куб,
где гидролиз завершается в присутствии пара (лапортский процесс). В том
и другом случае необходимо применять очень чистый электролит, иначе невоз-
невозможно добиться гидролиза без существенного разложения перекиси.
Превращение в калиевую соль до гидролиза имеет следующие преиму-
преимущества: 1) большая часть каталитически активных примесей, например железо
или платина, остается в растворе, благодаря чему для гидролиза получается
более чистый пероксодисульфати 2) сравнительно низкая растворимость перок-
пероксодисульфата калря снижает скорость гидролиза, а поэтому обеспечивает бо-
более легкую отгонку перекиси водорода из раствора до разложения или реакции
с пероксомоносерной кислотой. Недостатками этого процесса нужно считать
необходимость в дополнительных производственных стадиях, и особенно рас-
расходы, связанные с использованием в процессе твердой фазы. При любом про-
процессе гидролиза дробная конденсация образующихся паров дает раствор с со-
содержанием до 35 вес.% перекиси водорода.
ПРОМЫШЛЕННЫЕ ПРОЦЕССЫ ЭЛЕКТРОЛИЗА
В настоящее время свыше 80% всего мирового производства перекиси водо-
водорода получается электрохимически из пероксодисерной кислоты или пероксоди-
пероксодисульфата аммония путем последующего гидролиза до перекиси водорода и сер-
серной кислоты или бисульфата аммония, из которых два последних вещества
вновь направляются на электролиз.
Эти процессы обладают тем преимуществом перед процессами с перекисью
бария, что дают возможность прямого получения более концентрированного
раствора перекиси водорода (до 35 вес.% вместо 6—8% из перекиси бария),
к тому же более высокой чистоты и стабильности. Основными их недостатками
являются большие капиталовложения, большой расход электроэнергии и не-
Промышленные процессы электролиза
115
обходимость тщательной и непрерывной очистки химикатов и электролита.
Эти процессы впервые разработаны в Германии и Австрии и применялись
в этих странах до второй мировой войны и во время ее в значительно больших
размерах, чем в любой другой стране. Получавшаяся перекись водорода затем
подвергалась концентрированию для применения в военном деле. К концу
войны из отчетов различных исследовательских бригад США и Великобрита-
Великобритании достоянием гласности стало много подробностей, касающихся этих заводов
перекиси водорода. Упомянутые отчеты и служат основой следующих описаний
процессов. Особенно подробным и содержащим очень ценную информацию яв-
является отчет, написанный Бречгером, Крюсоном и Кашингом [41]—крупными
специалистами в этой области. Электролитические процессы, применяемые
в других местах, по-видимому, практически не отличаются от описываемых
ниже.
Установленные мощности для различных процессов в Германии и Австрии
выражались следующим образом (в тоннах 100%-ной перекиси водорода, хотя
фактически вырабатывалась обычно 35%-ная перекись).
' Процесс
Из пероксодисульфата калия ....
Из пероксодисерной кислоты ....
Из пероксодисульфата аммония . . .
Из перекиси бария
Всего
Довоенная
мощность
2600
1700
700
700
5700
Максимальная
мощность
во время
войны A944)
15 800
2 700
800
700
20 000
Кроме того, к концу войны строились дополнительные мощности на
22 000 т/год (из пероксодисульфата калия) и на 52 000 т/год (из этилантрахи-
нона)*. Для сравнения укажем, что до 1939 г. производственная мощность
заводов перекиси водорода во всем мире (в расчете на 100%-ную), включая
и Германию и Австрию, составляла только 12 000 ml год.
Максимальная мощность дистилляционных установок в Германии и Австрии,
на которых перекись водорода концентрировалась до 80—85%-ной, достигла
в 1944 г. 17 000 т/год; дополнительно строились еще мощности на 66 000 т/год
(все данные в расчете на 100%-ную перекись). Значительная часть этих мощ-
мощностей в Германии была затем разрушена или демонтирована.
Данные по мощности японских заводов перекиси водорода во время войны
более скудны и противоречивы [42]. Довоенная мощность (применялся исклю-
исключительно процесс из пероксодисульфата аммония) составляла, по-видимому,
около 700 ml год в расчете на 100%-ную перекись водорода (хотя вырабатыва-
вырабатывалась 30%-ная). В 1944 г. было принято решение построить дополнительные
заводы для производства и концентрирования перекиси водорода на 28 800 т
80%-ной перекиси в год (в расчете на 100%-ную); однако действительно была
построена только небольшая часть этой огромной мощности и значительная
часть ее была разрушена. Результаты подсчетов для максимального, факти-
фактически достигнутого производства 80%-ной перекиси водорода колеблются от
100 до 600 ml месяц, причем меньшая мощность, по-видимому, более вероятна.
Основной фирмой, производящей перекись водорода в Англии, является «La-
porte Industries, Ltd.», имеющая заводы в Лутоне и Уоррингтоне, причем
строительство последнего завода завершено только в 1953 г. После окончания
* На стр. 71 указана мощность 2000 т/месяц.—Прим. перев.
8*
116 й Глава 3
мировой войны опубликовано незначительное количество данных относительно
производства или производственных мощностей по перекиси водорода, несмотря
йа то, что, как указано на стр. 24, заводы перекиси водорода имеются в большин-
большинстве европейских стран и в других промышленно развитых странах мира.
На стр. 22—25 приведены цифры по росту производства перекиси водорода в
США по годам, причем кратко изложена также история развития производствен-
производственных процессов. В 1952 г. Управление оборонного производства при правительст-
правительстве США установило план, по которому производственная мощность к 1 января
1955 г. должна была превысить на 10 100 т мощность, достигнутую в 1951 г.
и составлявшую 10 500 т. Заявки на сертификаты на аммортизационное сни-
снижение налогов в 1952 г. уже покрыли всю намеченную программу расширения.
Все эти данные относятся к 100%-ной перекиси [43]. В Северной Америке
в 1954 г. вырабатывали перекись водорода следующие фирмы: «duPont
Company»—2 электролитических завода в Ниагара-Фоле и Дрездене (штат
Нью-Йорк) и 1 завод по процессу самоокисления в Мемфисе (Теннесси); «Buf-
«Buffalo Electro-Chemical Co.», отделение «Food Machinery and Chemical Co.»—2
электролитических завода в Тонаванде (Нью-Йорк) и Ванкувере (Вашинг-
(Вашингтон); «Westvaco Co.», также отделение «Food Machinery and Chemical Co.»—завод
перекиси водорода из перекиси бария в Картерете (Нью-Джерси); «Canadian
Industries, Ltd.»—завод электролитической перекиси водорода в Шавинигэн
Фоле (провинция Квебеке Канаде) и, наконец, «Pennsylvania Salt Co.»—электро-
Co.»—электролитический завод в Уайандотте (Мичиган).
Основные характеристики каждого из электролитических процессов из-
изложены ниже.
Процесс получения из пероксодисерной кислоты (вейссенштейнский процесс)
Этот процесс является наиболее старым электролитическим процессом
и впервые начал эксплуатироваться в 1908 г. фирмой «Oesterreichische Chemi-
sche Werke» в Вейссенштейне (Австрия). Мощность этого завода в 1945 г. была
около 75 ml месяц в расчете на 100%-ную перекись водорода. Фактически вы-
выпускалась 30%-ная перекись. Этот процесс иепользуется также фирмой
«Deutsche Gold undSilber Scheideanstalt (Degussa)» в Рейнфельдене (Германия),
завод которой в 1944 г. имел мощность 160 т в расчете на 100%-ную перекись.
Кроме того, дополнительные мощности находились в стадии строительства.
Указанный процесс применяется и на многих других заводах в Европе,
а также фирмой «duPont Co.» в Ниагара-Фоле. Поточная схема этого процесса
показана на рис. 13.
В Вейссенштейне и Рейнфельдене в 1944 г. [41, 44] работало 24—26 электро-
электролизеров, включенных последовательно как в отношении раствора, так и электри-
электрического тока; они размещались каскадообразно таким образом, что раствор
перетекал из одной ванны в другую самотеком. На рис. 14 показан цех электро-
электролиза на заводе фирмы «Degussa». Каждая ванна, применяющаяся в этом про-
процессе, представляет керамический сосуд длиной около 100 см> шириной 15 см
и глубиной 50 см. В этом сосуде помещается свинцовый змеевик, через который
пропускается охлаждающая вода и который одновременно служит и катодом
и охлаждающим змеевиком. Внутри свинцового змеевика размещены на рав-
равных расстояниях 10 вертикальных цилиндрических фарфоровых диафрагм.
Внутри каждой диафрагмы установлен стеклянный сосуд, служащий для огра-
ограничения объема анодного пространства. Через стеклянный сосуд проходит
охлаждающая вода. Каждый анод состоит из 11 вертикальных платиновых
полосок, расположенных на одинаковом расстоянии друг от друга в тонком
кольцевом промежутке между стеклянным сосудом и диафрагмой, причем
каждая полоска припаяна к свинцовому кольцу, находящемуся на верхней
части диафрагмы. Все 10 анодных комплектов внутри одного сосуда соеди-
Промышленные процессы электролиза
117
няются параллельно. В соответствии с зарекомендовавшей себя конструкцией
анода нижняя часть каждой полоски изготовлена из платины, а верхняя—из
тантала, причем оба металла соединены точечной сваркой. Этот биметалличе-
биметаллический анод обладает большей долговечностью; несколько меньшее электрическое
сопротивление в такой комбинации немного снижает напряжение ванны (ком-
(комбинация из циркония и платины также была бы удовлетворительной, но воз-
возможно, что изготовить ее сложнее; тантал, цирконий и их соли не вызывают
заметного разложения перекиси водорода [45]).
Раствор H2SO4, возвращаемый
— Водяной пар
Н вакуум-
-насосу-*—
Вода
«резервуару
оля горячей
воды
2-5%-ная
Н202
¦35%-ная
Остаточная кислота
¦— Водяной пар
——, Здесь отбирается '
I часть электролита
для очистки и
г» ' возврата о систему
Рис. 13. Поточная схема электролитического процесса с пероксодисерной кислотой.
7—серия электролизеров с водяным охлаждением; 2—диафрагмы; 3—гидролизер с паровой рубашкой;
4-— сепаратор жидкости и брызгоуловитель; 5—насадочная колонна (второй гидролизер); 6—насадочные
абсорбционные колонны; 7—барометрический конденсатор.
Через катодные пространства каскада последовательно включенных эле-
электролизеров протекает раствор серной кислоты (концентрация 500 г/л); замет-
заметного изменения концентрации или химического состава при этом не происхо-
происходит. По выходе из катодного пространства тот же раствор перекачивается
обратно для подачи в последовательно включенные анодные камеры. В цехе
электролиза происходит выделение водорода, а поэтому требуется усиленная
вентиляция. До поступления раствора в первое анолитное отделение к нему
добавляется раствор роданистого аммония и соляной кислоты в количествах,
соответствующих содержанию 0,15 г HCNS и 0,037 г НС1 на 1 л анолита. Тем-
Температура католита обычно должна быть на 5° ниже температуры анолита.
Через катодные свинцовые змеевики и вставленные в аноды стеклянные сосуды
пропускают охлаждающую воду, чтобы температура анолита была не выше
20—30°. Электролит на выходе из последней ванны содержит 25—30 г пероксо-
пероксодисерной кислоты на 100 см3 раствора. Среднее падение напряжения на зажи-
зажимах электролизера составляет 5,5—5,7 в для анода из чистой платины и 5,2—
5,3 в для комбинации платины с танталом. Работа производится при плотности
тока около 0,86 а/см2; при плотностях тока, превышающих 1 а/см2, наблюдается
значительная потеря платины. Нормальная сила электрического тока состав-
составляет 950 а на каскад. Выход по току заметно колеблется в зависимости от темпе-
температуры. При 950 а на каскад и температуре охлаждающей воды 2° этот выход,
118 Глава 3
по имеющимся данным, равен 80%. С повышением температуры охлаждающей
воды на каждый градус сверх 15°, как это иногда бывает летом, во избежание
сильного уменьшения выхода по току силу тока обычно снижают на 50 а
на каскад, но даже в этом случае при температуре воды 23° и нагрузке 550 а
на каскад выход по току составляет около 68%. В среднем этот выход за год
равняется примерно 72%.
Электролит подвергается гидролизу путем непрерывного пропускания
его через длинный свинцовый змеевик с паровой рубашкой, в котором происхо-
происходит упаривание жидкости приблизительно до половины ее объема; в этих усло-
условиях 80—90% всей пероксодисерной кислоты подвергается гидролизу и полу-
получающаяся перекись водорода испаряется. Почти у самого входа в змеевик
Рис. 14. Электролизерная на заводе фирмы «Degussa», ра-
работающем с пероксодисерной кислотой.
происходит гидролиз пероксодисерной кислоты до пероксомоносерной. Испа-
Испарение воды по всей длине змеевика увеличивает концентрацию кислоты и этим
ускоряет гидролиз пероксомоносерной кислоты до перекиси водорода. Образо-
Образование перекиси водорода, по-видимому, происходит главным образом у выхода,
где находится кислота высокой концентрации. После этого производится разде-
разделение жидкости и пара. Жидкость подается на верхнюю часть насадочной фар-
фарфоровой колонны, а в кубовую часть ее поступает пар в противотоке к жидкости.
Этот пар дает теплоту, необходимую для гидролиза остаточных пероксосер-
ных кислот, одновременно предотвращая образование в жидкости кислоты
слишком высокой концентрации. Большая часть остающегося в жидкости актив-
активного кислорода при этом превращается в перекись водорода и уходит в виде
пара. Жидкость, выходящая из колонны, содержит некоторое количество
перекиси водорода и обе пероксосерные кислоты в количестве, эквивалентном
2,6 г пероксодисерной кислоты на 100 см3 раствора; при протекании этой жид-
жидкости через катодные пространства ванн происходит разложение этих соеди-
соединений за счет электролиза.
Общие реакции могут быть представлены следующими уравнениями:
в электролизере
2H2SO4 -> H2S2O8 + H2, B2)
в гидролизере
H2S2O8 + 2Н2О -* 2H2SO4 + Н2О2. B3)
Промышленные процессы электролиза
119
Пары из гидролизеров поступают в две группы последовательно включен-
включенных насадочных абсорбционных колонн (работающих в вакууме), из которых
выходит водный раствор, содержащий 30—35% перекиси водорода. По имею-
имеющимся данным, этот раствор содержит 4 г! л серной кислоты, хотя, вероятно,
содержание ее можно было бы понизить путем применения более эффективного
брызгоуловителя. Кислоту частично нейтрализуют, чтобы конечный продукт
обладал лишь слабой кислотностью, и перед отгрузкой добавляют к продукту
стабилизаторы.
Общая эффективность использования перекисного кислорода при гидро-
гидролизе и перегонке составляет около 85%. Чтобы довести до минимума концен-
концентрацию примесей, непрерывно отбирают около 5% циркулирующей кислоты,
очищают ее путем перегонки в кварцевой аппаратуре и возвращают в систему.
Ниже приводятся некоторые рабочие параметры, отнесенные к 1 кг продукта
в виде 100%-ной перекиси водорода: расход энергии на ванны составляет 14—
16,2 квт-ч; общий расход энергии в процессе, включая 10%-ные потери при пре»
образовании переменного тока в постоянный, 21,5 квт-ч; расход пара на гидро-
гидролиз и перегонку 27,5 кг; расход серной кислоты 0,5—0,9 кг; рабочая сила около
0,1 человеко-часа; потеря платины 0,0025—0,003 г; при производительности
1000 кг/месяц требуется 815 г платины или (в случае применения биметалли-
биметаллических анодов) 334 г платины и 3340 г тантала. Процесс с пероксодисерной кис-
кислотой имеет преимущество перед описываемым ниже процессом с пероксоди-
сульфатом калия, заключающееся в меньших расходах на рабочую силу,
однако выход по току и эффективность гидролиза в нем ниже.
Процесс с пероксодисульфатом калия (процесс Питча—Адольфа, или E.W.M.)
Этот процесс впервые применен на заводе в Холлригельскрейте в 1910 г.
фирмой «Elektrochemische Werke» в Мюнхене (E.W. М.), а сейчас применяется
/
атмосферу
1
—
Пероксодисульфат
аммония\
KHS04
NH4HSO4+H2SO4
I 4
I I
I
Здесь отбирается часть
электролита для очистки
и возврата в систему
Возвратная жидкость
S02
NH4CNS
К конденсатору Вода
водяного пара ¦ i 1
и вакуум-насосу I '
10
Твердая
фаза
H2SO4
д
Шлам
—Вода
— S0*
и Ферооиианид
^^калия
10-50%-ная
Н202
Рис. 15. Поточная схема
Осадок, перерабатываемый
для извлечения платины
процесса с пероксодисульфатом калия.
/—серия электролизеров с водяным охлаждением; 2—фильтр; 3—вакуумный холодильник; 4—бак
периодического действия для проведения обменной реакции; 5 —центрифуга периодического дей-
действия; 6— гидролизер периодического действия; 7—реакционный сосуд; Я—холодильник; 9—бак
для осаждения; 10—брызгоуловитель; 11—ректификационная колонна.
на заводе «Henkel Co.» в Дюссельдорфе и в других местах. Большая часть заво-
заводов, построенных в Германии во время второй мировой войны, работала именно
по этому методу, причем были построены очень крупные заводы в Бад-Лаутер-
120
Глава 3
берге (мощностью 1100 т/месяц в расчете на 100%-ную перекись ) и в Рум-
шпринге (фактически построенная мощность составляет 700 т/месяц 100%-ной
перекиси). Поточная схема этого процесса показана на рис. 15, а подробности
сообщаются ниже [41, 46].
Сначала кислый раствор' бисульфата аммония подвергают электролизу
с получением пероксодисульфата аммония в растворе:
2NH4HSO4 -> (NH4JS2O8 + H2. B4)
Затем добавляют бисульфат калия для осаждения пероксодисульфата калия,
а бисульфат аммония подвергают очистке и возвращают обратно в ванны:
(NH4JS2O8 + 2KHSO4 -> K2S2O8l+2NH4HSO4. B5)
К шламу пероксодисульфата калия добавляют серную кислоту; смесь перего-
перегоняют с водяным паром, в результате чего получают пар перекиси водорода,
который подвергают дробной конденсации:
K2S2O8
2KHSO4 + H2O2.
B6)
Из шлама отделяют центрифугированием бисульфат калия, который возвра-
возвращают обратно на стадию, представленную уравнением B5). Остаточная серная
кислота после очистки используется вновь для гидролиза.
Р if с. 16. Детали пероксодисульфатного электролизера завода
фирмы «Elektrochemische Werke» в Мюнхене.
На рис. 16 показаны отдельные детали верхней части электролизера типа
Е. W. М., а на рис. 17—цех электролиза на заводе в Бад-Лаутерберге, где
установлены электролитические ванны указанного типа. Каждый сосуд вме-
вмещает несколько электродных комплектов, а каждый комплект в свою очередь
состоит из вертикального ряда анодов A4 в каждом ряду), параллельно им
с каждой стороны расположены ряды катодов A5 в ряду), а параллельно этим
катодам с обеих сторон размещены ряды охлаждающих трубок A6 в ряду).
Все это закреплено на графитовом блоке. Другими словами, каждый электрод-
электродный комплект состоит из пяти параллельных рядов, расположенных в следую-
следующем порядке: охлаждающие трубки, катоды, аноды, катоды, охлаждающие
трубки. В каждой такой ванне имеется 5 электродных комплектов,установлен-
Промышленные процессы электролиза
121
ных в керамическом сосуде размером 71Х80 см и глубиной 86 см (без перегоро-
перегородок). Таким образом, в каждом сосуде находится всего 70 анодов, 150 катодов
и 160 охлаждающих трубок, причем отдельно все аноды и отдельно все катоды
присоединены в электрической цепи параллельно. В Румшпринге в одном
керамическом сосуде помещалось 6 электродных комплектов, а в Бад-Лаутер-
берге 7. Максимальные размеры ванны, очевидно, определяются степенью
трудности ее изготовления. Ванна окружена графитовыми блоками, поддержи-
поддерживающими электродные комплекты; эти блоки лежат на верхней части электро-
электролизера и перекрывают его. Чтобы полностью перекрыть ванну, на концах
каждого керамического сосуда имеется еще два дополнительных блока. Всего,
таким образом, на 1 ванну E.W.M. приходится 7 блоков. На рис. 16 можно
видеть несколько таких блоков ниже токоподводящих шин.
Рис. 17. Цех электролиза на заводе в Бад-Лаутерберге
(ванны типа фирмы «Elektrochemische Werke»).
Конструкция электролизера очень сложна и здесь подробно не рассматри-^
вается. Каждый анод состоит из вертикального алюминиевого стержня, покры-
покрытого эбонитом и несущего ряд вертикальных платиновых проволок, которые
образуют собственно анод. Каждый катод представляет собой вертикальный
графитовый стержень, покрытый парафином для снижения коррозии и плотно
обмотанный по всей длине асбестовым шнуром диаметром 3 мм; эта обмотка
служит диафрагмой ванны. Таким образом, катодное пространство предста-
представлено небольшим объемом между асбестовым шнуром и стержнем. На каждый
блок подвешивается два ряда катодов. Между ними в каждом блоке есть ряд
отверстий для ввода анодов в ванну, а снаружи катодов—два других ряда
отверстий для охлаждающих трубок. Электрический контакт с катодами осу-
осуществляется при помощи алюминиевой плиты, лежащей на блоках и присоеди-
присоединенной с обоих концов к распределительным шинам. В этой плите имеются
отверстия, соответствующие отверстиям в блоках, для установки анодных
комплектов и холодильников. Анодные комплекты в тех местах, где они про-
проходят через отверстия в блоках и алюминиевых плитах, изолированы; они сви-
свисают с алюминиевых брусков, расположенных выше блоков, причем сами бруски
присоединены с обеих сторон к токоподводящим шинам. Катодная плотность
тока составляет 0,03—0,05 а/см2. Более высокие плотности катодного тока
ведут к поляризации и к образованию свободного газообразного аммиака на
катоде. Концентрация тока составляет около 18 а/л. Каждый холодильник со-
122
Глава 3
стоит из двух вертикальных концентрических стеклянных трубок, так что
вода стекает вниз через середину и затем поднимается по кольцевому проме-
промежутку. Эти трубки присоединены к эбонитовым двойным гребенкам, подвешен-
подвешенным над угольными блоками. Для изготовления трубок с целью устранения
возможности коррозии применяется стекло. На рис. 16 показаны эбонитовые
коллекторы и верхняя часть наружного ряда холодильников. Стеклянные
трубки над коллекторами позволяют визуально контролировать поток охлаж-
охлаждающей воды. После монтажа электролизера щели между блоками или между
блоками и самой ванной заплавляют воском. 7—11 сосудов соединяют последо-
последовательно при помощи стеклянных трубок для пропускания электролита, при-
причем все ванны находятся на одном уровне. На заводе в Мюнхене все 28 электро-
электролизеров включены в электрическую цепь последовательно. Вытяжным венти-
вентилятором водород засасывается с одного конца ванны и подается в отводящий
трубопровод, причем для поддержания его концентрации на очень низком уров-
уровне с другого конца ванны открывается доступ воздуху. Затем смесь этих газов
выпускается в атмосферу.
На каждом электродном комплекте номинальная сила тока равна 900 а
при плотности тока 1—2 а/см2. Таким образом, хотя ванны типа Е. W. М.
и рассчитаны на 4500 а, вполне возможно изменять ток на ванны в пределах
нескольких сот ампер. Потеря платины с анодов на каждый ампер-час является
минимальной при плотности тока 2—4 а/см2; в этих условиях аноды могут рабо-
работать от 3 до 5 лет, пока не окажется необходимой их замена. Потеря платины
несколько снижается в результате присадки к ней 0,1% иридия. Каждая ванна
содержит около 340 л раствора, и нормальная скорость пропускания электро-
электролита составляет 1500 л/час через каждую серию последовательно включенных
ванн. Для шин часто используется алюминий, поскольку даже следы меди
в электролите могут быть причиной существенного каталитического разложения.
В процессе работы на катоде медленно возрастает концентрация иона аммония
и в соответствии с этим возрастает рН:
2NHJ + 2е -» 2NH8 + Ht. B7)
Когда раствор у катода становится щелочным, газообразный аммиак вы-
выпускают в атмосферу; при этом происходит выпадение сернокислого калия.
Работу начинают с напряжения на ванне 5,2 в, но в соответствии с практикой
германских заводов это напряжение медленно увеличивают до достижения
к концу шестидневной работы в среднем 6,2 в, так что среднее напряжение за
неделю составляет 5,9 в. После этого, чтобы выпавший сульфат калия вновь
растворился, серию ванн выключают на 1 сутки. На отдельных электролизе-
электролизерах напряжение иногда растет быстрее, а поэтому их выключают из цепи при
достижении напряжения 6,5 в. В связи с этим в среднем работает около 90%
всех ванн, и цех электролиза полностью выключается из работы в течение
одного дня в неделю.
Раствор электролита имеет следующий типовой состав:
Компонент
Пероксодисульфат аммония ....
Сульфат аммония
Серная кислота
Сульфат калия
Роданид аммония, г/л
Нитрат калия (максимум), г /л . . .
Концентрация (нормальность)
на входе
в ванны
0,6
4,0
1,4
0,4
0,1
2,5
на выходе
из ванн
1.2
3,4
0,8
0,4
0
2,5
Промышленные процессы электролиза
123
Допустимо еще содержание железа примерно 40—ПО мг/л. Увеличение
содержания иона нитрата, образующегося за счет окисления аммониевого
иона, приводило бы к снижению выхода по току.
Концентрации пероксодисульфата аммония и сульфата калия в жидкости
на входе определяются соотношениями равновесия в предшествующем осажде-
осаждении пероксодисульфата калия. Поэтому концентрации изменяются с темпера-
температурой и количеством бисульфата калия, вводимого на этой стадии. Для увели-
увеличения степени превращения пероксодисульфата в калиевую соль (с целью сни-
снижения содержания пероксодисульфата аммония на входе в ванны) необходимо
также увеличить содержание сульфата калия. Однако при этом увеличивается
скорость выпадения осадка в ваннах.
Рис. 18. Гидролизеры пероксодисуль-
пероксодисульфата калия на заводе в Бад-Лаутер-
берге.
Рис. 19. Ректификационные колон-
колонны для 35%-ной перекиси водорода
на заводе в Бад-Лаутерберге.
Оптимальная рабочая температура для ванн равна примерно 38°; при
более высоких температурах происходит более сильное выделение перекисного
кислорода, при более низких—слишком большое выпадение соли.
Электролит на выходе из серии электролизеров (см. рис. 15) подается
в вакуумный холодильник, где испарение воды снижает температуру его до 28°,
а затем в баки из нержавеющей стали 18-8 A8% Сг, 8% Ni), снабженные охлаж-
охлаждающими приспособлениями и мешалками. В этих баках к электролиту добав-
добавляют бисульфат калия в форме влажного брикета в таком количестве, чтобы
осадить половину пероксодисульфата в виде калиевой соли. Конечная темпера-
температура составляет 14—16°; выпавший осадок еще содержит 10—20% пероксоди-
пероксодисульфата аммония, окклюдированного калиевой солью. Пероксодисульфат
калия периодически центрифугируют, промывают, а затем подают на гидролиз.
Маточный раствор обрабатывают двуокисью серы с целью разложения перок-
сомоносерной кислоты восстановлением ее до серной, затем добавляют рода-
роданистый аммоний для повышения потенциала анода, и жидкость отводят обратно
124 Глава 3
в ванны. Разложение пероксомоносерной кислоты снижает потерю платины
с анодов.
Обычно гидролиз осуществляют периодически в керамических ретортах,
загружая в каждую 1000 кг пероксодисульфата калия с добавкой 250—300 л
18 н. серной кислоты. Гидролизеры завода в Бад-Лаутерберге показаны на
рис. 18. Через дырчатую трубу в течение 4,5 часа пропускают водяной пар,
и пары отгоняют в вакууме. Реторта работает при температуре 75—85° иЪстаточ-
ном давлении 45—55 мм рт. ст. Пары поступают в насадочную колонну, где
задерживается унесенная жидкость, а затем в ректификационную, колонну,
в которую подается необходимая флегма в виде либо потока воды, либо конден-
конденсата из алюминиевого холодильника. На рис. 19 показаны ректификационные
колонны на заводе в Бад-Лаутерберге. Пары, поступающие в колонну из ретор-
реторты, имеют концентрацию перекиси водорода от 10 вес.% в начале гидролиза
до величины менее 1% к концу его. Жидкость из куба ректификационной колон-
колонны имеет начальную концентрацию перекиси водорода около 50%, которая
под конец падает до 3% и ниже. Средняя концентрация перекиси водорода со-
составляет 35% при общем выходе 85—91%; большая часть потерь обусловлена
разложением, остальное теряется в кубовой жидкости гидролизеров или в парах,
выходящих из ректификационной колонны. Однако обычно пар содержит ни-
ничтожное количество перекиси водорода; его конденсируют, а неконденсируемые
газы откачивают вакуумным насосом. Содержание примесей в перекиси водо-
водорода может быть различным; в отдельных источниках приводятся количества
серной кислоты от 2 до 0,25 г/л и меньше.
После окончания перегонки реторту промывают 15 н. серной кислотой,
шлам охлаждают и центрифугируют. Бисульфат используется вновь для
осаждения пероксодисульфата калия в осадительных баках, а вся отфильтро-
отфильтрованная серная кислота подвергается очистке до возвращения ее в процесс.
В Германии применяется следующий №етод очистки кислоты. Сначала раз-
разлагают перекисные соединения путем нагревания с паром до 105° с добавкой
сернокислого железа (III). Кислоту после выдерживания в ряде керамических
баков в течение 1,5—3 час. охлаждают до 60—80° в танталовом холодильнике
и разбавляют до 10 н. концентрации для осаждения железа. Для удаления сле-
следов оставшегося активного кислорода вводят двуокись серы, после чего кислоту
обрабатывают периодически железистосинеродистым калием при перемешивании
воздухом с целью осаждения остаточного железа в виде берлинской лазури.
Для этой цели применяют гуммированные стальные баки с обкладкой из кера-
керамических плиток. Осадок фильтруют без применения давления через керами-
керамические фильтрующие трубки и подвергают обработке для извлечения примерно
30% платины, потерянной с анодов. Кислотный фильтрат концентрируют до
18 н. концентрации в керамической реторте, нагреваемой змеевиком из золо-
золотого сплава (с 30% серебра) при остаточном давлении 10 мм рт. ст. (серебро
добавляется к золоту для повышения его твердости). После концентрирования
фильтрат содержит железо в количестве 5—10 мг/л.
Железо и платину можно осадить также путем слабого подщелачива-
ния раствора аммиаком, но если это осуществить непосредственно, то коли-
количество необходимого аммиака увеличило бы концентрацию сульфата аммония
в системе сверх желательного. Поэтому если применяется указанная техника
очистки, то отбирают 1—5% электролита после гидролиза и с целью снижения
концентрации серной, кислоты до 0,2 н. медленно пропускают его через ряд
ванн, при этом концентрация пероксодисульфата повышается до 1,8 н. После
этого к жидкости в баке из пластмассы хавег добавляют 0,3 эквивалента аммиа-
аммиака для осаждения железа и глинозема, т. е. вводят в систему ионы аммония
в количестве, достаточном лишь для восполнения потерь из системы. Вместо
этого можно добавить также аммиак для осаждения примесей и избыточный
сульфат аммония отделить путем направления части общего потоку электро-
Промышленные процессы электролиза 125
лита в керамические испарители. При этой операции удаляются также неболь-
небольшие кусочки графита, асбестовых волокон и т. п.
Некоторые технологические параметры приведены ниже. Все они отно-
относятся к 1 кг продукта в расчете на 100%-ную перекись водорода (при выработке
35%-ной перекиси) при производительности завода около 150 ml месяц или
больше: расход энергии на электролиз 12,9—13,2 квт-ч непосредственно на
ванны, 10—15% как потери при преобразовании переменного тока в постоян-
постоянный и 2—2,5 квт-ч на различные производственные нужды; расход технологи-
технологического пара на перегонку 13 кг при общем расходе 25—28 кг\ расход воды
1—2,5 м3 (этот расход сильно колеблется в зависимости от температуры воды);
расход серной кислоты 0,08—0,15 кг; гидрата окиси аммония 0,03—0,1 кг;
сульфата калия 0,02—0,03 кг; роданида аммония 0,025—0,075 кг; ферроциа-
нида калия 0,002—0,01 кг; двуокиси серы 0,005—0,075 кг; сульфата железа
(III) 0,01—0,08 кг; чистая потеря платины 0,0014—0,0027 г; количество при-
применяемой платины 330 г на ЮООлсг месячной производительности; рабочая сила
составляет около 0,28 человеко-часа. Амортизационные отчисления A5%
от стоимости оборудования и 5% от стоимости строений в год) и расходы по
эксплуатации составляли в Германии вместе 35—40% от общей производствен-
производственной себестоимости. Общая себестоимость производства 80%-ной перекиси
водорода по этому процессу, включая накладные расходы и амортизационные
отчисления, равнялась 2,25 герм, марки за килограмм содержавшейся 100%-ной
перекиси; это соответствует 41 центу за фунт при курсе марки 40 центов
(900 долларов за тонну).
Этот процесс имеет то преимущество перед процессом, основанным на
применении одной серной кислоты, что дает более чистый продукт, более высо-
высокий выход по току (около 82—84% против 72% при применении пероксодисер-
ной кислоты и около 84% при применении процесса с пероксодисульфатом аммо-
аммония без участия твердой фазы) и несколько более высокую эффективность гид-
гидролиза (85—91% против 85—88% при работе по пероксодисернокислотному
методу и 85% при применении процесса с пероксодисульфатом аммония по ме-
методу Elchemie). Кроме того, при этом процессе по сравнению с другими мето-
методами можно применять более крупные ванны. Однако осуществление процесса
является несколько более сложным и расходы на рабочую силу значительно
выше. В частности, применение катода с обмоткой из асбестовой диафрагмы
в электролизере способствует выпадению соли на катоде с постепенным ростом
напряжения на зажимах ванны. В связи с этим электролизер необходимо время
от времени выключать. Кроме того, более высокое среднее напряжение на
электролизере сводит к нулю преимущества высокого выхода по току. Для
обеспечения эффективной операции необходим тщательный уход за асбестовой
диафрагмой, а именно: это покрытие должно быть весьма равномерным и его
приходится время от времени заменять свежим. Внутризаводской транспорт
твердых веществ и операция гидролиза, как периодический процесс, требуют
значительной затраты рабочего времени.
По-видимому, из всех трех процессов наиболее эффективным является
процесс с пероксодисульфатом аммония без участия твердой фазы (если только
стадию гидролиза можно осуществлять с приемлемым высоким выходом),
причем этот процесс представляет особый интерес для крупных заводов и для
тех стран, где стоимость рабочей силы высока, например для США. Так,
в 1945 г. подсчитано, что если расходы на рабочую силу в Германии во время
войны составляли при применении процесса с пероксодисульфатом калия
30—35% всей производственной себестоимости, то в США эта цифра составила бы
70%. Процесс с пероксодисульфатом калия при периодическом проведении гид-
гидролиза может оказаться более конкурентоспособным на малых установках.
Решение об использовании этого процесса в Германии во время войны, по-
видимому, было основано не только на чисто технических соображениях, но
отчасти и на политических. Ванну типа E.W.M., как это будет описано ниже,
126 Глава 3
можно с успехом применять в процессе с пероксодисульфатом аммония без
участия твердой фазы.
Процесс с пероксодисульфатом аммония без участия твердой фазы
(левенштейнский или лапортский процесс)
Промышленная эксплуатация этого процесса началась не раньше 1927 г.
из-за трудностей, как указано выше, связанных с реакцией гидролиза. Процесс
этого типа применяется фирмами «ElchemieCo.»B Куфштейне в Австрии (мощ-
(мощность около 65 т/месяц в расчете на 100%-ную перекись), «Laporte Chemicals,
Ltd.» в Лутоне (Англия) и «Buffalo Electro-Chemical Co.» в Буффало (штат
Нью-Йорк). Он изучался также фирмой «Elektrochemische Werke» в Мюнхене
в 1940—1945 гг. в предположении о возможной замене им процесса с пероксо-
пероксодисульфатом калия. Процесс с пероксодисульфатом аммония, по-видимому,
является наиболее экономичным из всех трех электролитических процессов,
если только стадия гидролиза обеспечивает достаточно высокий выход, по-
поскольку выход по току в ванне выше, чем при применении пероксодисерной
кислоты, а стоимость рабочей силы меньше, чем при работе с пероксодисуль-
пероксодисульфатом калия.
Рис. 20. Детали пероксодисульфатного электролизера на за-
заводе фирмы «Elchemie».
Поточная схема напоминает показанную на рис. 13 для процесса с перок-
пероксодисерной кислотой, за исключением того, что для гидролиза применяется
несколько иное оборудование.
В процессе, используемом фирмой «ElchemieCo.» [41,47], носящем назва-
название левенштейнского процесса или процесса Ридель-де-Хаена или Ридель-
Шеринга, электролитические ванны, как обычно, состоят из платиновых ано-
анодов и свинцовых катодов в керамическом сосуде. Однако физическое размеще-
размещение отличается от принятого в ранее описанных электролизерах. Детали одного
такого сосуда показаны на рис. 20, а самый цех электролиза на заводе Elchemie—
на рис. 21. Каждый прямоугольный сосуд разделен на 6 симметрично располо-
расположенных отделений двумя поперечными и одной продольной керамическими
перегородками. Каждое отделение представляет собой в сущности отдельную
ванну, разделенную двумя пористыми керамическими диафрагмами на цен-
Промышленные процессы электролиза 127
тральное анодное пространство и два боковых катодных. Для охлаждения
анодного пространства в центре установлен двойной ряд вертикальных стек-
стеклянных холодильников, параллельно каждой стороне которого размещено
по одному ряду из 4 вертикальных анодов (всего 8). На наружных сторонах
этих рядов анодов и параллельно им находятся пластины диафрагмы (на снимке
не видны), а затем идут свинцовые пластины, представляющие катоды. Охлаж-
Охлаждение катодного пространства осуществляется змеевиками из свинцовых
трубок. На заводе «Elchemie» каждый каскад состоял из 11 таких сосудов,
включенных последовательно, причем уровень каждого сосуда находился
на расстоянии примерно 15 см от уровня соседнего с ним, чтобы обеспечить
движение электролита самотеком. Таким образом, каждый каскад состоял
из двух параллельных линий, по 33 ванны в каждой. Раствор бисульфата аммо-
аммония проходит сначала через все катодные пространства, а затем через все анод-
анодные. Все электролизеры покрыты слабо пригнанными колпаками, в которые
может проходить воздух, разбавляющий выделяющийся водород. Образую-
Образующаяся газовая смесь отсасывается вентилятором. Выход по току составляет
Рис. 21. Цех электролиза с пероксодисульфатными ваннами
на заводе фирмы «Elchemie».
82—84% при анодной плотности тока 0,4—1,1 а/см2 и соответствующих напря-
напряжениях на зажимах ванн E,0—5,2 в). Нагрузка составляет около 220 а
на ванну, или 440 а на каскад. Скорость протекания анолита и католита состав-
составляет в каждой серии ванн примерно по ПО л/час, т. е. 220 л/час продукта
на каскад. Концентрация анодного тока равна примерно 17 а!л\ ванны работают
при температуре от 28 до 33°.
Ниже приводятся типичные концентрации электролита, в граммах
на литр:
Католит Лполит
(на входе) (на выходе)
Серная кислота 260 160
Сульфат аммония 210 76
Пероксодисульфат аммония 0 232
В этих ваннах концентрация католита заметно изменяется при прохож-
прохождении через электролизеры, причем ионы аммония мигрируют через диафрагмы
в сторону католита, а ионы сульфата—в сторону анолита.
128 Глава 3
На заводе «Elchemie» гидролиз осуществляется путем пропускания жид-
жидкости из ванн через два ряда свинцовых испарителей (типа всползающей пленки)
с паровыми рубашками, включенных последовательно. Первый ряд. состоит
из 28 параллельных трубок, расположенных в две линии, причем жидкость
в каждую трубку подают с регулируемой скоростью. На рис. 22 показан ниж-
нижний конец такого ряда. Над рядом испарителей находится водоотделитель,
л. -¦ ¦ ¦
I
ррНИ
11
¦
щ
$ ш
0>я№> mm
Рис. 22.. Трубки (нижняя часть) для гидролиза пероксоди-
сульфата на заводе фирмы «Elcnemie».
который можно видеть на рис. 23 (цех гидролиза и дистилляции). К жидкости до
подачи ее во второй ряд испарителей, после выхода из первого ряда, добавляют
воду, вероятно во избежание выпадения аммониевых солей. Пары подвергаются
Рис. 23. Цех гидролиза и дистилляции на заводе фирмы
«Elchemie».
ректификации в вакууме в серии из трех колонн с получением раствора
с 30—35 вес.% перекиси водорода. Эффективность дистилляции—гидролиза
Промышленные процессы электролиза 129
из расчета выхода перекисного кислорода составляет около 85%; около 10%
теряется в результате разложения. Для изготовления трубок испарителей
пригодно очень небольшое число материалов; свинцовые трубки быстро корро-
корродируют и требуют замены через каждые 30—45 суток. Правда, присадка
к свинцу сурьмы в количестве 0,2—2,0% заметно увеличивает долговечность
трубок.
Более эффективный метод гидролиза применяет фирма «Laporte Chemicals,
Ltd»; аналогичный же метод разработан и фирмой «Elektrachemische Werke» в
Мюнхене (E.W.M.). Поэтому методу операцию проводят в две стадии: на первой
раствор концентрируют и частично гидролизуют (но с небольшим образова-
образованием перекиси водорода), после чего жидкость подвергают перегонке с водя-
водяным паром в вакууме для завершения гидролиза и отгонки перекиси водорода.
Упаривание приблизительно до половины исходного объема проводят в кера-
керамическом сосуде, снабженном паровым змеевиком из нержавеющей стали
V4A Круппа (хромоникелевая сталь 18-8 с 2,2% молибдена). После выдержи-
выдерживания в течение примерно 5 мин. жидкость пропускают в колонну (насадоч-
ную или с ситчатыми тарелками), где она нагревается танталовыми нагрева-
нагревателями в противотоке к острому пару. Общая эффективность процесса гидро-
гидролиза—дистилляции, разработанного E.W.M., 96%.
При использовании ванн типа Питча—Адольфа в процессе без участия
твердой фазы обязательно применение более концентрированной серной кислоты
для гидролиза пероксодисульфата, чем при работе с пероксодисульфатом калия.
Типичная концентрация электролита на входе в ванну составляет 4,0 н. по
сульфату аммония и 5,0 н. по серной кислоте. Повышенное содержание кислоты
вызывает снижение выхода по току приблизительно до 75% и заставляет обя-
обязательно применять более высокую концентрацию тока и более низкую темпе-
температуру ванны (около 27°); однако, поскольку в состав электролита не входит
калий, выпадение соли не происходит и напряжение на электролизере вместо
5,9 в составляет только 4,6 в.
Ниже приведены рабочие показатели для установки «Elchemie», также
в расчете на 1 кг 100%-ной перекиси водорода при выпуске 30%-ного раствора.
Расход электроэнергии на ванну 13,4 квт-ч, а всего вместе с потерями при
преобразовании тока и другими затратами энергии 17,0 квт-ч\ расход пара
на дистилляцию по отчетным цифрам 75 кг, а по расчетам на основании тепло-
теплового баланса 50 кг; вложение платины 522 г на 1000 кг месячной производи-
производительности в расчете на 100%-ную перекись; потери платины 0,006 г; рабочая
сила 0,26 человеко-часа*.
Как указано выше, процесс с пероксодисульфатом аммония без участия
твердой фазы является, вообще говоря, наиболее экономичным, из электроли-
электролитических процессов, если только гидролиз можно провести с достаточно прием-
приемлемым высоким выходом.
Запатентованы или применялись различные небольшие видоизменения
указанного процесса по сравнению с вышеописанным. Эти изменения касаются
конструкции электролизеров и концентрации химикалиев, а также выбора
оборудования для гидролиза. Так, фирма «Henkel Co.» в Дюссельдорфе, кото-
которая в основном вырабатывала перекись водорода по процессу Питча—Адольфа,
эксплуатировала в небольшом масштабе также так называемый процесс Шмидта
в течение 1938—1944 гг. В том виде, в каком этот процесс применяла фирма
«Henkel Co.» [48], работу проводили в больших электролизерах с анодными
* Данные по расходу рабочей силы для всех трех процессов нельзя считать надеж-
надежными, так как они должны будут значительно колебаться в зависимости от величины
установки. Однако указанная здесь величина, по-видимому, несколько завышена, если
сравнить ее со значениями 0,1 человеко-часа для пероксодисернокислотного процесса и
0,28 человеко-часа для процесса Питча—Адольфа, поскольку последний определенно тре-
требует большей затраты рабочей силы, чем оба процесса без участия твердой фазы
9 Перекись водорода
130 Глава 3
и катодными пространствами с электролитом, содержавшим больше кислоты
по сравнению с электролитом, применявшимся в других процессах с пероксо-
дисульфатом аммония. Типичные концентрации (в граммах на литр) следующие:
Католит Анолит
(на входе) (ма входе)
Серная кислота 325 285
Сульфат аммония 195 235
Активный кислород в расчете
на Ы2Оа 4 0
Напряжения на зажимах ванн колебались от 5,0 до 5,2 в, и выход по току
составлял в среднем около 88%. Для гидролиза и дистилляции жидкости да-
давали стекать вниз по внутренним стенкам вертикальных фарфоровых труб
с паровыми рубашками. Пары проходили через брызгоуловитель и ректифи-
ректификационную колонну, аналогичную ранее описанной. Максимальная эффектив-
эффективность дистилляции составляла около 80—85%. Однако установка по Шмидту,
по меньшей мере в том виде, в каком она была смонтирована и эксплуати-
эксплуатировалась на заводе «Henkel Co.», считалась менее удовлетворительной, чем
установка в процессе Питча—Адольфа.
ЛИТЕРАТУРА
1. Н i I d e b г a n d J. Н., J. Am. Chem. Soc, 34, 246 A912).
2. Ul 1 m a n F., Enzyklopadie der technischen Chemie, Vol. 2, p. 112; Vol. 10, p. 420.
Urban & Schwarzenberg, Berlin, 1928.
3. В о 1 1 о V., С a d e n а с с i о Е., англ. пат. 24817 B5.Х 1910), герм. пат. 250417
4. E.IX 1912).
Lewis G. N.. Randall M., J. Am. Chem. Soc, 36, 1969 A914).
5. A s k e n a s у P., Rose R., Z. anorg. allgem. Chem., 189, 1, 10 A930); A s k e n a-
sy P., Hornung G., Rose R., герм. пат. 452266 (8.XI 1927), 460030 A9. V
1928); Askenasy P., Hornung G., герм. пат. 460029 A9.V 1929).
6. Rusberg F., пат. США 2031180 A8.11 1939), [Steatit-Magnesia A. G., герм.
пат. 587888 A1.XI 1933); Kali-Chemie A. G., англ. пат. 398399 A4.IX 1933); франц.
пат. 750592 A2.VIII 1933); Kali-Chemie A. G., герм. пат. 582925 B5.VIII 1933).
585518 D.Х 1933); Budowski I., пат. США 2153658 A1.IV 1939); Von
Draht enE., герм. пат. 620170 A5.Х 1935), 657291 B.III 1938); Semi-
d i e А. С, франц. пат. 833288 A938), Rentschler M. J., пат. США 1978551 (ЗО.Х
1934);Веуег А., герм. пат. 723673 (8.VIII 1942).
7. Martin F., герм. пат. 403253 B5.IX 1924); Jacquelet R., пат. США 1364558
D.1 1921).
8. Rhenania Verein Ch. Fab. A. G., герм. пат. 378609 B4. VII 1923).
9. О s h i m a M. et al., японск. пат. 173428 A6. VIII 1946) [С. А., 46, 2763c]; Oshi-
m a M., японск. пат. 174395 (9.1 1947) [С. A., 44, 1238b].
10. M а с F a d у e n K. W., Jaffa N. E., пат. США 2661268 (l.XII 1953).
11. Doer n er H. А., пат. США 1235664 G. VIII 1917); Chem. and Met. Eng., 26, 1111
A922).
12. Wol f f ens t ei n R., Peltner E., Ber., 41, 275 A908); E. Merck Co., герм.
пат. 179771 A4.XII 1906), 179826 A7.XII 1906).
13. M i z u n о S., Y a m a d a D., Y a s u k a w a S., J. Electrochem. Soc. Japan, 18,
80, 116, 167 A950) [C.A., 45, 7451a].
14. Lunge G., Z. angew. Chemie, 1890, 3.
15. Сагг К. L., Am. Dyestuff Reptr., 24, 441 A935).
16. Lawrence J., Peacock R. В., LeFeuvre C. W., B.I.O.S. Final
Report 1432, Item N° 22 A946).
17. H ul i n P. L., герм. пат. 132090 D. VI 1902); Oesterreichischer Verein fur Ch. und
Metal. Production, герм. пат. 253284 G.XI 1912).
18. Campbell D. J., Lefson E. F., пат. США 2497810 A4.11 1950).
19. Fischer F., Plotze H., Zeit. anorg. Chem., 75, 1, 10 A912).
20. Pierce J. В., англ. пат. 130840 A919).
21. S a t t e r f i e 1 d C. N.. S t e i n T. W., Ind. Eng. Chem., 46, 1734 A954).
22. В 1 u m e n t h a 1 M., Roczniki Chem., 12, 232 A932).
23. R iesenf el d E. H., Rein hold В., Вег., 42, 2977 A909).
24. R i u s A., Helv. chim. Acta, 3, 347 A920).
Литература
25. В й г g i n E., Dissertation, Berlin A911).
26. В е г t h e 1 о t M., Compt. rend., 86, 71 A878).
27. Л а т и м е р В. М., Окислительные состояния элементов и их потенциалы в водных
растворах, Издатинлит, М., 1954.
28. Klemenc A., Z. Elektrochem., 49, 141 A943); Klemenc A., HeinrichG.
ibid., 49, 493 A943).
29. G 1 a s s t о n e S., H i с k 1 i n g A., Chem. Revs., 25, 407 A939).
30. Bancroft W. D., Trans. Electrochem. Soc, 71, 195 A937).
31. Haissinsky M., Discussions Faraday Soc, I, 254 A947).
32. H e i n г i с h G., Klemenc A., Z. physik. Chem., 184A, 347 A939).
33. S i h v о n e n V., Suomen Kemistilehti, 10 B. 27 A937) (на немецком языке) |С. А. 32,
4317].
34. Wood W. S., Chemistry and Industry, 1953, 2.
35. Haissinsky M., Cottin M., Compt. rend., 224, 392 A947).
36. M а с h u W., Das Wasserstoffperoxyd und die Perverbindungen, 2 Aufl., Springer
Verlag, Wien, 1951. [есть перевод первого издания с дополнениями: см. Маху В.,
Перекись водорода и перекисные соединения, Госхимиздат, М., 1951].
37. S k i г г о w F. W., Stein E. R., Trans. Am. Electrochem. Soc, 38, 209 A920);
Solanki D. N., Kamath I.S.K., Proc Indian Acad. Sci., 24A, 305 A946)
[C.A., 41, 33781].
38. Al cock H. E., англ. пат. 633782 C0. XII 1949) [инд. пат. 42617].
39. Palme H., Z. anorg. allgem. Chem., 112, 97 A920); К о 1 t h о f f I. M., M i 1-
1 e г I. K., J. Am. Chem. Soc, 73, 3055 A951), R i u s A., Z u 1 u e t a C, Ana-
les. real soc. espan. fis. у quim. (Madrid), 46B, 79 A950) [C. A., 44, 10468e].
40. Slater V. W., Chemistry and Industry, 1945, 42.
41. В г e t s с h g e г M. E., CrewsonC.G., С u s h i n g R. E., P. B. report 17331
A946); Chem. Eng., p. 102, August, 1948.
42. Cool e у R. A., Chemical Industries, 957 (VII 1946); Cool e у R. A., P. B.
report 48373).
43. Anon., Chem. Eng., News, 30, 1215 A952).
44. Vigers B.E.A., Wood W. S., Hickson G., B.I.O.S. Report 683 (издан
так же как Р. В. report 45252); Р. В. report 95248.
45. P. В. report 73716 (frames 288—294).
46. Me A ul ay J., Clapp D. В., Slater V. W., Cooper K. A., C.I.O.S.
File № XXXIII-42 (ca. 1946) (издан так же как Р. В. report 93482 и как Р. В. report
33489); Hull and H. L, Wood W. S., MacGregor J., F a i г 1 i e,
B.I.O.S. Report 1381 (ca. 1946); Vigers B.E.A., Wood W. S., B.I.O.S.
Trip № 1080 A945) (издан как Р. В. report 80350).
47. M с A u 1 а у J., Clapp D. В., Slater V. W., Cooper K. A., C.I.O.S.,
File № XXXIII-45 ?nd C.I.O.S. File № XXXIII-43.
48 Vigers B.E.A., Wood W. S., Hickson G., P. B. report 60893, B.I.O.S.
Trip № 1080.
Глава 4
КОНЦЕНТРИРОВАНИЕ, ОЧИСТКА, КОНСТРУКЦИОННЫЕ МАТЕРИАЛЫ,
ХРАНЕНИЕ ПЕРЕКИСИ ВОДОРОДА И ОБРАЩЕНИЕ С НЕЮ
Растворы перекиси водорода в том виде, в каком они получаются непо-
непосредственно на производстве, часто могут быть использованы потребителем
без дальнейшей обработки. Однако чаще до отгрузки продукта необходима или
желательна определенная его обработка: концентрирование, очистка, вве-
введение добавок. Высокая реакционная способность перекиси водорода и ее
склонность к разложению заставляют тщательно выбирать конструкционные
материалы, вступающие с нею в контакт, применять специальную технику
и соблюдать необходимые меры предосторожности при хранении и обращении
с нею. Эти вопросы рассматриваются в данной главе.
КОНЦЕНТРИРОВАНИЕ
В промышленных условиях перекись водорода обычно концентрируют
путем перегонки и ректификации. Другие методы концентрирования, такие, как
дробная кристаллизация, не имеют экономического значения, но часто приме-
применяются в небольшом масштабе, например в лаборатории, для приготовления поч-
почти безводной перекиси водорода или концентрированной перекиси водорода,
полученной каким-нибудь иным путем. Под термином концентрирование здесь
всегда понимается снижение содержания воды в растворе. Любые другие со-
составные части раствора перекиси водорода (за исключением очень небольших
количеств стабилизаторов или ингибиторов, добавляемых с целевым назначе-
назначением) считаются загрязнениями, или примесями. Под термином очистка под-
подразумевается процесс удаления таких составных частей; очистка может со-
сопровождаться изменением концентрации или происходить без него.
Во избежание перехода за максимально допустимое предельное содер-
содержание примесей с точки зрения приемлемой стабильности процесс промы-
промышленного концентрирования, как правило, должен включать и очистку.
Для применения перекиси водорода в большинстве областей, не связан-
связанных с военным делом, например для отбелки, требуются только разбав-
разбавленные растворы. Так, продукт производства, полученный прямо по методу
с применением перекиси бария и содержащий до 8 вес.% перекиси водорода,
часто отгружается непосредственно потребителю без концентрирования; однако
высокое содержание воды увеличивает стоимость упаковки, транспортировки
и хранения на единицу веса Н2О2, а следовательно, кладет определенные огра-
ограничения в отношении зоны, в пределах которой такой продукт можно сбывать
по экономически приемлемой цене. Поэтому обычно желательно получать
лерекись водорода значительно более высокой концентрации.
Концентрирование путем перегонки
Как можно видеть из кривой равновесия жидкость—пар, приведенной на
стр. 196, высокая летучесть воды по сравнению с летучестью перекиси в прин-
принципе дает возможность концентрировать разбавленные растворы перекиси
Концентрирование 133
водорода простой отгонкой водьц Однако при этом приходится сталкиваться
с тремя трудностями: 1) нелетучие примеси концентрируются вместе с пере-
перекисью водорода, а поэтому получающийся концентрированный раствор ока-
оказывается менее стабильным, чем исходный. Если содержание примесей доста-
достаточно велико, то даже добавка заметных количеств стабилизаторов не всегда
приводит к получению продукта приемлемой стабильности. Для обеспечения
высокой стабильности требуется высокая степень чистоты; 2) скорость разло-
разложения жидкой перекиси водорода возрастает приблизительно в 2,3 раза при
повышении температуры на каждые 10°; 3) пары перекиси водорода, если ее
концентрация выше известного предела, легко взрываются при контакте с ката-
каталитически активным материалом даже в условиях комнатной температуры,
а с некаталитическими материалами—при несколько повышенных температу-
температурах. Для устранения последних двух трудностей применяют наиболее инерт-
инертные конструкционные материалы, а перегонку проводят в вакууме, чтобы сни-
снизить температуру процесса и избежать зоны взрывчатых составов. Для некото-
некоторых целей допустимо присутствие в перекиси заметного количества приме-
примесей и стабилизаторов и приемлема пониженная стабильность, особенно если
необходимо получить умеренно концентрированный раствор перекиси водо-
водорода, например содержащий ее в количестве 35 вес. % . В этом случае доста-
достаточно простого концентрирования с отгонкой воды в вакууме. При применении
электролитического процесса получается смесь паров воды и перекиси водо-
водорода путем гидролиза пероксодисульфата, и эти пары без труда можно под-
подвергнуть дробной конденсации (ректификации), например в насадочной ко-
колонне, и получить продукт, содержащий 28—35 вес.% перекиси водорода.
Можно получать непосредственно и несколько более высокие концентрации,
но лишь ценой несколько пониженного выхода перекиси водорода.
Электролитический продукт может содержать очень небольшие количества
примесей, попадающих в него в результате механического уноса. В большин-
большинстве случаев такие количества примесей не оказывают вредного влияния, так
как продукт все еще обладает приемлемой стабильностью; поэтому такой про-
продукт непосредственно отгружают заказчику без дальнейшей обработки, если
не считать одной или нескольких из следующих операций: 1) отрегулирования
рН до величины, обеспечивающей максимальную стабильность; 2) добавки не-
небольшого количества стабилизующих соединений для подавления каталити-
каталитического разложения и 3) добавки ингибиторов коррозии для снижения действия
примесей на алюминиевую тару, обычно применяемую для транспортировки.
Однако для некоторых видов применения необходимо, чтобы раствор перекиси
водорода содержал меньше примесей; для этого требуется более тщательный
процесс ректификации или же отдельная дополнительная очистка. Проблема
приобретает особенную остроту в том случае, когда желательно получить
высококонцентрированную перекись водорода, например 80%-ную или выше.
Такие растворы требуют весьма высокой степени чистоты как для обеспечения
высокой стабильности, так и для применения в намечаемой области. Ниже
описываются процессы комбинированного концентрирования и очистки мето-
методом перегонки для получения концентрированного раствора.
Маас и Хэтчер [1] тщательно исследовали методы получения чистой высоко-
высококонцентрированной перекиси водорода в лабораторных условиях и опублико-
опубликовали общий обзор по этому вопросу. Для лабораторных работ такого рода мож-
можно использовать описанную этими авторами аппаратуру и руководствоваться
их методикой. В настоящее время в продаже для лабораторий имеется значи-
значительно более чистый и концентрированный исходный материал, часто не требую-
требующий дальнейшей обработки. Вопрос концентрирования 35%-ной продажной
перекиси водорода до 80%-ной исследован в лаборатории и на небольшой опыт-
опытной установке [2]. Продукт, полученный перегонкой под вакуумом, может
все же содержать следы механически унесенных примесей. Если требуется
134
Глава 4
максимальная чистота и максимальная концентрация, то для устранения уноса
можно применить методику Гросса и Тейлора [3]. Нагретый раствор поддер-
поддерживают при температуре ниже точки кипения с тем, чтобы не происходило
образования пузырьков. Такой процесс отгонки протекает очень медленно,
но дает перекись водорода максимальной чистоты.
Техника получения высококонцентрированных растворов перекиси водо-
водорода в промышленном масштабе впервые разработана фирмой «Elektrochemi-
«Elektrochemische Werke» в Мюнхене (эта работа была начата еще в 1934 г.) и нашла практи-
практическое использование во время второй мировой войны в Германии для военных
Первая
стадия
D аппарата)
Вторая
стадия
A аппарат)
Охлаждающая
вода
30-35%-ная
НгОг
К вакууму
Охлаждающая
вода
Продукт
Р и с. 24. Поточная схема концентрирования перекиси водорода
на заводе фирмы «Elektrochemische Werke» в Мюнхене.
/ — парциальные конденсаторы; 2—ректификационные колонны; 3—брызгоуло-
витель; 4—реторты; 5—паровые змеевики.
целей при приготовлении растворов, содержащих 80—85 вес.% перекиси
водорода. Аналогичные процессы разработаны во время войны в Англии и США,
а также в Японии, располагавшей данными о подробностях производства
по германскому способу.
Необходимая высокая чистота по любому из этих процессов достигается
за счет двух- или трехступенчатой перегонки, при которой раствор перекиси
водорода подвергается полному выпариванию, а нелетучие примеси получаются
в остатке, после чего пары перекиси концентрируют путем ректификации.
В Германии цехи концентрирования работали на заводах в Бад-Лаутерберге
и на заводе «Elektrochemische Werke» в Мюнхене (E.W.M.), а к концу войны
было начато строительство цехов на заводах в Румшпринге, Гейдебреке и Валь-
денбурге, причем последние два цеха должны были концентрировать 20—
30%-ные растворы перекиси водорода, полученные по этилантрахиноновому
способу. В двух фактически работавших цехах применяли 30—35%-ную пере-
перекись водорода, полученную электролитическим путем, причем различие в ме-
методах производства заключалось только во второстепенных деталях. Поточная
схема [4] показана на рис. 24.
Концентрирование 135
В первую реторту непрерывно подавали раствор 30—35%-ной (по весу)
перекиси водорода, к которому были добавлены стабилизаторы*. Если разло-
разложение протекает в ничтожных размерах, то пары на выходе из реторты будут
иметь ту же концентрацию, что и жидкость на входе, но жидкость в реторте
будет находиться в равновесии с паром и таким образом содержать 70—75 вес.%
перекиси водорода. Пары пропускали через брызгоуловитель с насадкой, на-
например с кольцами Рашига или битым кварцем [5], в насадочную ректифика-
ректификационную колонну. Оборудование в основном было керамическим, за исключе-
исключением поверхностей теплообмена.
Рис. 25. Общий вид реторт для концентрирования на заводе
фирмы «Elektrochemische Werke» в Мюнхене.
Флегма получалась на алюминиевом парциальном конденсаторе, как по-
показано на схеме, или путем непосредственного добавления воды. Отходившие
пары, практически 100%-ный водяной пар, поступали в конденсатор смеше-
смешения с барометрической трубой, который был соединен с вакуумным насосом
или паровым эжектором. Очищенные кубовые остатки из первой колонны по-
подавали во вторую реторту, из которой пары пропускали через другой брызгоуло-
брызгоуловитель и другую ректификационную колонну**. Конденсат из второй колонны
возвращали во вторую реторту, откуда непрерывно отбирали концентрирован-
концентрированный продукт—80—85%-ную перекись водорода, которую пропускали через
холодильник. В первой реторте постепенно возрастала концентрация примесей
и стабилизаторов, что сопровождалось повышением скорости разложения
и увеличением пенообразования, а поэтому после 120-часовой непрерывной
работы приходилось прерывать перегонку и опорожнять первую реторту.
Загрязненные периодически получаемые остатки для выделения из них пере-
* В Германии к 30—35%-ной (по весу) перекиси водорода, полученной электроли-
электролитически по способу Питча—Адольфа, добавляли примерно 200 мг/л пирофосфата натрия
и 100 мг/л 8-оксихинолина, а при необходимости получения особенно высококонцентри-
высококонцентрированного продукта—еще немного фосфорной кислоты. К продукту, полученному по
антрахиноновому способу, добавляли предпочтительно станнат натрия. При более чистом
начальном продукте, по-видимому, можно применять меньшие концентрации стабилиза-
стабилизатора.
*¦ На заводе в Бад-Лаутерберге в качестве предохранительной меры конденсат из
первой колонны вместо непосредственного направления во вторую реторту охлаждали
и подавали в хранилище.
136 Глава 4
киси водорода можно было подвергать отгонке с паром в отдельном аппарате»
При возобновлении операции первую реторту заполняли питательным раство-
раствором и конденсат из первой колонны возвращали в эту реторту до достижения
70—75%-ной концентрации, после чего его переключали на вторую реторту.
На каждый аппарат второй ступени требовалось четыре аппарата первой. По
германским способам во вторую реторту также добавляли небольшое коли-
количество стабилизатора, а поэтому последний сохранялся и в конечном продукте.
Если перекись водорода на этой стадии была сравнительно чистой, в нее можно
было не добавлять стабилизатор или же брать его в очень небольшом коли-
количестве. Доказательством может служить то, что в США в настоящее время
в производственном масштабе вырабатывается 90%-ная перекись водорода,
содержащая максимально лишь несколько миллиграммов стабилизатора на
литр. Общий вид реторт для концентрирования на заводе «Electrochemische
Werke» показан на рис. 25.
Решающее значение в конструкции аппарата для концентрирования пере-
перекиси водорода имеет материал, из которого изготовлен паровой змеевик. Он
должен быть устойчивым против коррозии под действием горячего раствора
перекиси водорода или возможных присутствующих примесей и вызывать ми-
минимальное разложение перекиси водорода. В германском процессе применялся
полированный змеевик из нержавеющей стали VI4А Круппа A7,596 Сг, 12,5% Ni,
4,7% Mo и максимально 0,07% С). Такую легированную сталь применяли,
по-видимому, из-за присутствия в питательной жидкости свободной серной
кислоты. В некоторых случаях применяли и тантал. Работу проводили при
остаточном давлении 28—32 мм рт. ст. в верхней части ректификационных
колонн и 50—60 мм рт. ст. в ретортах; разница в основном обусловливалась
перепадом давления при протекании через насадку. Соответствующие темпера-
температуры были 62—66° в первой реторте и 69—72° во второй. В Германии стандарт-
стандартная максимальная концентрация получавшейся перекиси водорода составляла
85 вес.%; в США в 1954 г. в промышленном масштабе вырабатывался продукт
с максимальной концентрацией 90%. Посколькупри всех составах пар богаче
водой, чем находящаяся с ним в равновесии жидкость, путем ректификации
можно в принципе получать практически 100%-ную перекись. Однако в этом
случае приходится работать при очень низких остаточных давлениях, чтобы
избежать образования паров взрывчатого состава (см. стр. 157). Максимальная
концентрация, получаемая в производственных условиях путем перегонки
и ректификации, ограничена взрывными пределами паров при минимальном
давлении, легко достигаемом в последней реторте. Это минимальное давление
в свою очередь в значительной мере определяется минимальным перепадом
давления в ректификационной колонне.
В указанных условиях, при работе по германскому способу, выход пере-
перекиси водорода без учета остатка составлял 95%, а при учете его—98%. При
концентрировании продукта, получавшегося по 2-этилантрахиноновому ме-
методу, достигали эффективности 81—85%. Процесс концентрирования в этом
случае был основан на том же принципе, но существовали некоторые различия
в отношении конструкции аппаратуры. Около 10% потерь падало на кубовые
остатки, которые в данном случае шли в отходы, поскольку содержащиеся
в них органические вещества могли образовывать взрывчатые перекисные смеси;
около 4—8% терялось за счет разложения. Ниже приводятся данные по энерго-
энергозатратам и расходу рабочей силы на концентрирование перекиси водорода
с 35 до 82% в расчете на 1 кг содержащейся И2О2: энергия 0,10 квт-ч; пар
5,8 кг; вода с температурой 11° 0,4 м3; рабочая сила 0,020 человеко-часа; на
1000 кг продукции в месяц необходимо откачивать воздух до остаточного дав-
давления 40 мм рт. ст. со скоростью 8 л*3/час. Калькуляция операций по герман-
германскому методу показывает, что стоимость концентрирования составляла около
V5 стоимости производства 35%-ного продукта по электролитическому методу.
Концентрирование 137
Описанный процесс является потенциально опасным, так как большие
количества (более тонны) высококонцентрированной перекиси водорода выдер-
выдерживают непрерывно при высоких температурах. Правда, ни на одном из гер-
германских заводов не отмечено случаев взрыва при работе в течение нескольких
лет. По одному из английских процессов [6] эта опасность может быть понижена
путем замены реторты испарителем, работающим по принципу всползания
пленки раствора, вследствие чего в любой данный момент в аппаратуре нахо-
находится лишь небольшое количество горячей концентрированной жидкости. Эта
техника безусловно имеет преимущества по сравнению с применяемой по про-
процессу E.W.M. При применении английского метода, в соответствии с патент-
патентными данными, потеря перекиси водорода при концентрировании с 28 до 90 вес.%
составляет лишь 1 %. Аналогичный процесс с применением испарителей с вспол-
всползающей пленкой был разработан во время войны фирмой «Elchemie» в Куф-
штейне (Австрия) и доведен до опытно-заводской стадии независимо от про-
процесса E.W.M., который фактически применялся только в Германии [7]. По
аналогичным процессам выдан ряд патентов [8]. По другому методу [9] общее
количество перекиси водорода в аппаратуре снижают до минимума путем распы-
распыления питательного раствора в вакууме в цилиндрическом испарителе с нагре-
нагретыми стенками, причем жидкость испаряется почти мгновенно. Выходящие
пары можно затем концентрировать описанным выше способом.
При концентрировании путем перегонки и ректификации перекиси водоро-
водорода, полученной через органические соединения, необходимо снижать до миниму-
минимума содержание органических веществ в высококонцентрированном продукте
и вместе с тем не допускать накопления органических веществ до опасных кон-
концентраций в испарительной секции перегонного аппарата. Подробности техно-
технологического процесса зависят от природы и количеств примесей при данном
методе производства. Примеси, отличающиеся относительной летучестью, могут
быть легко удалены, однако трудно рассчитать равновесные соотношения между
жидкостью и паром в любом данном случае, поскольку перекись водорода легко
реагирует с различными органическими веществами, особенно кислородсодер-
кислородсодержащими (например, альдегидами и спиртами), с образованием сложных и не-
неустойчивых соединений, обладающих меньшей летучестью, чем летучесть лю-
любого из реагентов.
20%-ный раствор перекиси водорода, получавшийся по германскому этил-
антрахиноновому процессу, содержал растворенные органические вещества
в количестве, эквивалентном 1300—1600 мг углерода на литр раствора. В ос-
основном эти органические вещества состояли из бензола (входившего в состав
растворителя для этилантрахинона) и жирных кислот, а также других кисло-
кислородсодержащих соединений, образовавшихся за счет окисления вторичных
спиртов, составлявших остальную часть растворителя (см. стр. 72). При концен-
концентрировании этого продукта в опытно-заводской аппаратуре, близкой по кон-
конструкции к применявшейся на германских заводах, выяснено, что содержание
органических веществ в перекиси водорода в первой кубовой реторте (в ра-
расчете на углерод) стремится к некоторой асимптотической величине, характер-
характерной для данного неочищенного раствора. Это показывает, что по мере увели-
увеличения содержания органических веществ в кубовых остатках скорость разло-
разложения их за счет окисления и испарения также возрастает. Отношение орга-
органических углеродистых веществ в кубовых остатках к их количеству в исход-
исходном растворе колебалось от 5 до 30%; это доказывает, что большая их часть
улетучивалась или уносилась механически*. По имеющимся данным, даже
при содержании углерода в кубовых остатках в количестве 20—25 г/л, не
* Перегонка сопровождалась значительными трудностями из-за пенообразования,
вызывавшегося мылами, которые образуются за счет реакции жирных кислот с калиевы-
калиевыми солями, применявшимися при сушке рабочего раствора.
138 Глава 4
отмечено случаев внезапного разложения или взрыва [10]. Однако, как пока-
показано ниже, эта концентрация приближается к пределам взрывчатости рас-
растворов органических веществ в водной перекиси водорода, а поэтому более
высокие концентрации органических веществ, несомненно, могут оказаться
весьма опасными. В одном патенте, полученном Мак-Муллином [11], описы-
описывается метод перегонки, устраняющий эти трудности при концентрировании
и очистке продукта, полученного я-азотолуольным методом.
Другие методы концентрирования
Метод концентрирования путем дробной кристаллизации является неэф-
неэффективным при применении к системе из перекиси водорода и воды. Как пока-
показано на стр. 184, при частичном вымораживании водных растворов перекиси во-
водорода любых концентраций образуется чистая твердая фаза; однако она очень
прочно удерживает маточный раствор. Эта особенность сильно ограничивает
степень разделения на каждой стадии. Тем не менее этот метод можно исполь-
использовать для получения безводного продукта из весьма концентрированных ра-
растворов; правда, он является весьма трудоемким. Процесс этот может быть до-
доведен до такой степени, когда анализ перекиси водорода на содержание оста-
остаточной воды оказывается трудной задачей [12].
Предложен ряд методов концентрирования перекиси водорода путем селек-
селективного экстрагирования растворителями. Экстрагирование перекиси водорода
органической жидкостью может оказаться в высшей степени опасной операцией.
Так, одно время применяли процесс, основанный на использовании этилового
эфира в качестве экстрагирующего растворителя с последующей отгонкой
эфира от перекиси водорода; однако в такой системе возможно образование
неустойчивых органических перекисей. Такая методика необычайно опасна,
и ее ни в коем случае нельзя рекомендовать. Сообщается о сильном взрыве,
происшедшем при попытке использования этого метода [13]. Практически все
органические растворители образуют детонирующие смеси с водными раство-
растворами перекиси водорода в определенных интервалах концентраций, если только
оба вещества обладают значительной взаимной растворимостью. Поэтому лю-
любой процесс экстрагирования с участием перекиси водорода необходимо тща-
тщательно рассчитать таким образом, чтобы состав любой жидкой фазы все время
находился за пределами области детонации (см. ниже). Те же предосторож-
предосторожности необходимо соблюдать и при использовании селективного экстрагирова-
экстрагирования в качестве метода очистки; предложено, например, использовать селектив-
селективное экстрагирование для удаления из перекиси водорода, полученной частич-
частичным окислением углеводородов, органических примесей [14].
Высококонцентрированную или практически безводную перекись водорода
можно получить и путем реакции перекисного соединения с кислотой в невод-
неводной среде, причем выбирают такую среду, которую можно отделить вакуумной
перегонкой от образовавшейся перекиси [15]. В качестве примера приводится
реакция пероксокарбоната натрия с серной кислотой в присутствии абсолют-
абсолютного метилового спирта. При этом выпадает сульфат натрия; спирт же может
быть отогнан в вакууме. Однако низшие спирты, например метиловый, и многие
другие органические соединения, в которых перекись водорода хорошо раство-
растворяется, образуют внутри широкого интервала составов чувствительные и весьма
взрывчатые смеси с перекисью водорода и водой (см. ниже). Поэтому приме-
применение метилового спирта в этом процессе исключительно опасно. Другой ме-
метод состоит в освобождении водного раствора перекиси водорода от воды обра-
обработкой борнокислым эфиром. При этом получают нерастворимую борную кисло-
кислоту и перекись водорода в спиртовом растворе; спирт образуется за счет гидро-
гидролиза борного эфира [16]. В этом патенте указывается, что безводные растворы
перекиси водорода в спиртах, например амиловом, бутиловом и бензиловом,
Очистка 139
устойчивы при комнатной температуре и не обладают взрывчатостью. Если
это так, то отсутствие взрывчатости, вероятно, обусловлено ограниченной ра-
растворимостью перекиси водорода в указанных жидкостях.
ОЧИСТКА
Степень чистоты, которая требуется от технического продукта, обыч-
обычно определяется либо минимальной приемлемой стабильностью при хране-
хранении, либо областью применения, для которой предназначена перекись
водорода.
Так, для медицинских целей, для обработки пищевых продуктов и для
некоторых видов военной продукции необходима весьма чистая перекись водо-
водорода независимо от того, требуется ли для нее высокая стабильность. На ряде
заводов, работающих по электролитическим методам, ректификация паров
от стадии гидролиза пероксодисульфата дает продукт, не требующий дальней-
дальнейшей обработки, если не считать добавки стабилизаторов, описанных в разделе
по концентрированию. Если требуется дополнительная очистка, то ее можно
осуществить вышеописанными методами перегонки, особенно если желательно
получить также более концентрированный продукт. Описаны способы [17)
полной перегонки без концентрирования в целях очистки.
Применяются или предложены также разные другие способы очистки.
По одному из них в растворе перекиси водорода образуют коллоидную суспен-
суспензию гидроокиси олова путем добавки соответствующего соединения олова
и тщательного отрегулирования рН с целью получения осадка гидроокиси олова,
адсорбирующего примеси ионов тяжелых металлов. Затем осадок удаляют пу-
путем отстаивания или фильтрования [18]. Остающееся в растворе олово является
весьма эффективным стабилизатором. По методу очистки, который одно время
применяли на заводе «duPont Company», продукт гидролиза пероксодисерной
кислоты, который может содержать и небольшое количество серной кислоты,
сначала обрабатывали гидратом окиси бария для осаждения сульфата и других
анионов, а затем гидратом окиси олова осаждали каталитически действующие
катионы, например ионы железа [19]. Этот процесс описан подробно в литера-
литературе [20].
Другой интересный способ заключается в удалении ионных примесей
путем наложения электрического напряжения. В перекиси водорода, которая
•ведет себя как неэлектролит (аналогично воде), неорганические соли диссоции-
диссоциируют на ионы. В связи с этим анионы и катионы будут соответственно пере-
переноситься к противоположно заряженным электродам. Выдан ряд патентов по
методам очистки, основанным на этом принципе [21], однако, насколько авторам
известно, ни один из них не нашел промышленного применения для очистки
растворов перекиси водорода. Понятно, что таким способом нельзя удалить
«з перекиси водорода незаряженные примеси, встречающиеся, например,
в продукте производства, полученном по методу с применением органических
веществ.
Новейшим методом, предложенным Юнгом (фирма «duPont Company»
[22]), является использование органических ионообменных смол поливинил-
арильного типа для удаления примесей ионов тяжелых металлов. В таких
адучаях для уменьшения разложения перекиси водорода желательно работать
при температурах ниже комнатной. Ионообменную смолу предварительно об-
обрабатывают 10%-ной серной кислотой для полного освобождения от железа,
а затем промывают водой. Когда связанное количество ионов тяжелых метал-
металлов становится эквивалентным или несколько меньшим 1% общей емкости
смолы в отношении связывания ионов железа (III), смолу подвергают регене-
регенерации или выбрасывают. Ринасевич [23] описал освобождение разбавленных
растворов перекиси водорода от хромата с использованием ионообменной смолы.
140 Глава 4
Сообщается также, что сама перекись водорода обратимо адсорбируется из этих
разбавленных растворов; возможно, что это свойство может найти известное
практическое применение.
Если перекись водорода получается по какому-либо процессу с исполь-
использованием органических веществ (например, путем частичного окисления угле-
углеводородов), при котором одновременно образуются некоторые количества
органических, примесей, может оказаться целесообразной очистка сырого
продукта путем введения подходящего реагента для осаждения перекисного
соединения; последнее затем вновь можно превратить в перекись водорода.
Так, путем добавки гидрата окиси кальция получают нерастворимую перекись
кальция, которую удаляют, промывают и вновь превращают в перекись водо-
водорода действием кислоты (см. стр. 80).
В одном частном случае, когда присутствие нитрата в 30%-ном (по весу)
растворе перекиси водорода оказалось вредным, он был удален из нее в основ-
основном путем адсорбции на активированном угле со сравнительно незначительным
разложением перекиси [24]. В качестве лабораторного метода предложена
также [25] очищать перекись водорода путем быстрого добавления при переме-
перемешивании сначала раствора хлорного железа, а затем углекислого кальция
и быстрого фильтрования смеси через тигель Гуча. Последующим приливанием
концентрированной серной кислоты удаляют остаточную желтую окраску
и осаждают кальций. Первые два добавляемых вещества, вероятно, образуют
осадок водной гидроокиси железа (III), которая, обладая высокой адсорбцион-
адсорбционной способностью, может захватить небольшие количества примесей. Однако
соединения железа являются мощными катализаторами разложения, и даже
небольшие количества, остающиеся после указанной обработки, могут быть
причиной значительного разложения. Трудно себе представить, чтобы такого
рода методика, сопряженная с введением недопустимого загрязнения, обладала
какими-либо преимуществами перед способом осаждения гидратом окиси олова.
В лучшем случае может произойти заметное разложение перекиси; в худшем
случае этот процесс сопряжен с опасностью, связанной с добавкой к перекиси
каталитически действующих веществ, особенно если они введены в заметной
концентрации. Поэтому описанный способ ни в коем случае не может быть ре-
рекомендован.
КОНСТРУКЦИОННЫЕ МАТЕРИАЛЫ
При рассмотрении вопроса о материалах, пригодных для изготовления
того или иного аппарата, приходящего в контакт с перекисью водорода, необ-
необходимо учитывать два следующих фактора: 1) влияние материала на скорость
разложения перекиси и 2) влияние, оказываемое перекисью водорода на этот
материал. Кроме того, при концентрациях перекиси водорода 45 вес.% или
выше существует возможность образования детонирующих смесей за счет
загрязнения органическими веществами. Обычно при выборе подходящего
материала наиболее существенное значение имеет влияние последнего на
устойчивость перекиси водорода. Очень немного материалов пригодно для
манипуляций с перекисью водорода различных концентраций при всех усло-
условиях хранения, производства и применения.
Скорость разложения перекиси водорода зависит от большого числа раз-
различных факторов, а именно от: 1) концентрации, 2) рН, 3) температуры, 4) при-
природы и количества примесей и стабилизаторов, содержащихся в перекиси водо-
водорода, 5) времени контакта (например, раствор перекиси водорода может мед-
медленно выщелачивать примеси из стенки сосуда, и, таким образом, скорость
разложения возрастает во времени, и наоборот, тщательно очищенная поверх-
поверхность инертного металла, практически свободная от вредных примесей, обыч-
обычно медленно пассивируется в контакте с перекисью, что ведет к снижению ско-
скорости разложения во времени) и 6) физической и химической природы поверх-
Конструкционные материалы 141
ности (как правило, чем ровнее поверхность, тем ниже скорость разложения).
Взаимоотношения между влияниями этих переменных факторов часто
весьма любопытны и сложны. Так, в одном случае добавка небольшого коли-
количества фосфатного стабилизатора к перекиси, хранившейся в алюминиевой
таре, заметно увеличила скорость разложения. Впоследствии оказалось, что
сосуд для перекиси был изготовлен из двух различных сортов алюминия;
добавка фосфата увеличила электропроводность перекиси водорода, а тем самым
и скорость электролитической коррозии. В свою очередь это вызвало выделе-
выделение примесей в раствор перекиси, что существенно повысило скорость разло-
разложения. Такой эффект всегда возможен в случае использования разнородных
металлов при монтаже аппаратуры для хранения и транспортировки перекиси.
Например, при применении н?ржав*кчц?й ^тапи и алюминия на последнем
в тбЧКЗх контакта образуется белый порошкообразный -огядпу Дэвис и Киф
[267 предлагают изолировать разнородные металлы один от другого подходя-
подходящими пластиками или по меньшей мере для изготовления большей поверхности
брать не катодный металл, а анодный; эти авторы показали также, что склон-
склонность к электролитической коррозии при более концентрированных растворах
уменьшается.
Поскольку при работе с высококонцентрированными растворами перекиси
требуется соблюдение более строгих требований, чем при работе с разбавлен-
разбавленными растворами, сначала следует рассмотреть материалы, которые могут
найти применение для 90%-ного раствора, причем для некоторых из них будет
дана более подробная характеристика. В прошлом допускалось применение
материалов, оказывавших слабое разлагающее действие на перекись водо-
водорода, особенно когда по экономическим соображениям выбор можно было
сделать лишь из ограниченного их числа. Поэтому в литературе можно найти
высказывания, противоречащие приводимым ниже, особенно в отношении
работ с разбавленными растворами. Кроме того, как и следовало ожидать,
опыты по определению скорости разложения в контакте с большинством по-
поверхностей трудно воспроизводимы даже при тщательном соблюдении условий
испытания, и весьма часто наблюдается, что скорости разложения, тщательно
измеренные в различных лабораториях в совершенно одинаковых условиях,
отличаются одна от другой в несколько раз. Поэтому, чтобы получить общее
представление об условиях, в которых разные материалы могут применяться
для работы с концентрированной перекисью водорода, мы здесь будем поль-
пользоваться качественным методом, используемым фирмой «Buffalo Electro-Chemi-
Electro-Chemical Co.» [27], в соответствии с сообщением Дэвиса и Кифа [26]. Материалы де-
делятся на 4 общих класса.
«Класс 1. Материалы, которые отличаются высокой совместимостью
и могут быть использованы для длительного контакта с перекисью водорода.
Независимо от времени контакта эти материалы не вызывают загрязнения
перекиси. Типичным видом применения материалов этого класса является из-
изготовление емкостей для долговременного хранения, например бочек, вагонов-
цистерн или баков».
«Класс 2. Материалы, которые подходят для неоднократного кратковре-
кратковременного контакта с высококонцентрированной перекисью водорода. Эти мате-
материалы могут быть использованы для кратковременного контакта с раствором
до передачи перекиси в хранилище или до последующего немедленного ее упо-
употребления. Это ограниченное время контакта не должно превышать 4 час.
при 71° или 1 недели при 21°. Типичные виды применения материалов этого
класса—изготовление клапанов или насосов для перекачивания перекиси
по трубам или хранилищ высокого давления».
«Класс 3. Материалы, которые разрешается применять лишь для кратко-
кратковременного контакта с концентрированной перекисью водорода непосредствен-
непосредственно перед ее употреблением. Эти материалы могут быть использованы в уело-
Таблица 2
Классификация материалов в соответствии с рекомендуемым их применением для
работы с 90%-ной перекисью водородаа
Материал
Способ
обработ-
обработки*5
Состав, %
А1
Мп
Си
Сг
Zn
Класс
Металлы
Алюминий 99,6%-ной
чистоты
Сплав 2 S
3
3
3
J
> 43
> 3 S
> 52 S
> 56 S
> 61 S
> 63 S
> 150 S
> В 214
> 355
» 356
> А 360
> 24 S
> 13
i 40 Е
» 53 S
• 214
» 75 S
Медь
П
П
Л
П
П
П
П
П
Л
Л
Л
п
л
п
>>99
#
J
5
0
0
1
5
7
,0
,6
,4
,8
,0
1,2
2,5
0,1
0,6
0,2
5
1
0
3
0
0
1
3
2
,2
,0
,7
,8
,5
,3
,5
,8
,5
0
1
4
1
,25
,3
,5
,6
0,
0,
0,
0
25
1
25
,3
5,6
1
1
2
2
2
2
2
2
2
2
2
2
2
3
4
4
4
4
4
4
Материал
Дуримет 20 •
Дуримет Т
Хаско-О-7
Хастеллой В
» С
Инконель
Чугун или углеродистая
сталь
Свинец
Магниевые сплавы . . .
Малин-уилстабрайт . .
Марганецсодержащие
сплавы
Монель-металл ....
Мультимет N-155 ...
Рефракталлой 26 ...
» 27 ...
» 70 ...
Нержавеющая сталь 303
304
309
310
Fe
Оста-
Остальное
6
6
7,5
1,4
Оста-
Остальное
Si
1
1
0,25
0,1
1
1
1
2
Мп
1
1
0,25
1,0
2
2
2
2
Сг
20
17
15,5
17—19
18-20
22—24
24—26
Состав,
N1
29
60
51
76
67
8—10
8—П
12-15
19—22
%
Мо
2
32
19
0,60
Си
3,5
0,2
30
С
7
0,08
0,15
<0,15
<о,ос
<0,'25
другие
элемен-
элементы
5W
Класс
2
4
2
3
3
4
4
4
4
2
4
4
3
3
3
3
2
2
2
2
Продолжение табл.
Материал
Нержавеющая сталь 316
317
318
321
ооо
347
экстра
304
экстра
446
440
W
Стеллит Мб
Олово х. ч.или99%-ное
Юниверсал сайклопс
19-9 DL
Вортит
Состав, %
Fe
Оста-
Остальное
Si
1
1
1
1
1
1
1
3,25
Мп
2
2
2
2
2
1
1
0,6
Сг
16—18
18—20
17—19
17—19
18—20
23—27
16-18
33
9
20
Ni
10—14
11—14
8—11
9—12
8—11
<0,60
0,60—
1,20
19
24
Мо
2—3
3—4
<0,75
3
Си
1,75
с
<0,08
<0,10
<0,08
<о,ю
<0,03
<0,35
0,60—
1,20
<0,07
другие
элемен-
элементы
Ti>5C
Nb>10C
55% Со,
6%W
Класс
2
2
2
2
о
J,
2
2
2
А
4
4
2
4
1
з
3
Боросиликатное стекло
Бутадиенстирольный кау-
каучук
Бутадиенакрилонитриль-
ный каучук
Джеон 8372
Хайкарв
Коросил 116, 117, 700
(формованный или
шприцованный, не пиг-
пигментированный) . . .
Неопрен
Найлон
Плайовик
Полихлорвинил . . . .
Вискин, полишен, три-
тен, джесколайт 231
Тефлон
Кел-F
Полистирол
Саран
Силиконовая резина . .
Стеклоткань, пропитан-
пропитанная тефлоном ....
Тиокол
Тайгон 2807
Велоформ
Винилит
Дерево, пробка, углево-
углеводородные смазочные
масла и другие окис-
окисляемые вещества . . •
Синтетический сапфир
Полимеры
Полихлорвинил
Полихлорвинил или пластифицированный
Полихлоропреновый каучук
Полимер из адипогексаметнлендиамида или себацино-
гексаметилендиамида
Полиэтилен
Политетрафторэтилен
Политрифторхлорэтилен
Сополимер хлористых винилидена и винила
Вулканизованный полисилоксан
Полисульфидный каучук
Фурановая смола
Сополимер хлористого и уксуснокислого винила
144
Глава 4
Продолжение табл. 2
Материал
Ароклоры
Флуоролюбы
Галоидоуглеводородные
масла
Полимеры кел-фло . . .
Парафин
Перфлуоролюбы ....
Гидравлическая жид-
жидкость RPM
Силиконы
Гидравлическая жидкость
скайдрол
Юкон хайдролюб U-4
Состав, %
Смазочные масла
Хлорированные дифенилы и полифенилы
Трифторхлорвиниловые полимеры
Фторированные углеводороды
Перфторированные углеводороды
Полисил океаны
Класс
а Извлечение из таблицы, составленной «Buffalo Electro-Chemical Co.», Буффало,
Нью-Йорк [26]. Схема классификации описана в тексте.
ОП—прокатка; Л—литье.
в Ввиду отсутствия номера при общем термине не известно, идет ли речь о бута-
диенстирольном, бутадиеннитрильном или акриловом (лактопреновом) СК.—Прим. перев.
виях многократного контакта с перекисью, но длительность каждого периода
не должна превышать непосредственно перед употреблением 1 мин. при 71°
или 1 часа при 21°. Эти материалы могут вызвать заметное загрязнение раство-
раствора, в результате чего он становится непригодным для хранения».
«Класс 4. Материалы, которые вообще не рекомендуются для применения
в контакте с вьгеококонцентрированной перекисью водорода. Они вызывают
мгновенное бурное разложение перекиси водорода, сами быстро разрушаются
под ее действием или же образуют взрывчатые смеси с концентрированной
перекисью водорода».
Эта классификация очень удобна для оценки различных материалов.
В соответствии с ней материал 1-го класса при желании можно использовать
для видов применения, соответствующих 2-му или 3-му классу, но мате-
материал 3-го класса можно применять лишь в тех случаях, для которых он
показан.
В табл. 2 указаны различные материалы и класс, к которому их можно от-
отнести. Необходимо иметь в виду, что эта классификация относится лишь к хо-
хорошо очищенным 9Э%-ным растворам перекиси водорода. Небольшие коли-
количества примесей, которые сами по себе даже не обладают каталитическим дей-
действием, например, минеральных кислот, могут привести к коррозии материала
и, как следствие, существенно увеличить скорость разложения. При концен-
концентрациях перекиси ниже 9Э вес.% иногда оказывается возможным применять
материалы более низкого класса, чем это указано в классификации. Матери-
Материалы, которые, по сообщению «Buffalo Electro-Chemical Co», дали удовлетво-
удовлетворительные результаты при длительном применении, указаны в табл. 2 кур-
курсивом.
Имеются и другие источники такого рода информации. В отчетах из гер-
германской практики приводятся скорости разложения концентрированной пе-
перекиси водорода при контакте с различными материалами [28]. Беллинджер,
Фридман, Бауэр, Истс и Балл [29] сообщают о большом числе количествен-
количественных испытаний стабильности концентрированной перекиси водорода в контакте
с различными металлами и другими материалами, причем эти авторы приводят
Конструкционные материалы 145
также данные по влиянию поверхности (в зависимости от различных способов
ее обработки) на разложение перекиси. Эти вопросы рассматриваются и в гл. 9.
Рейхерт и Пете [30] рассматривают различные конструкционные материалы,
используемые в разных производственных процессах и для разных видов при-
применения. Этот вопрос обсуждается и в гл. 2, 3 и И. Дэвис и Киф [26] описы-
описывают различные типы оборудования, оправдавшие себя в работе с концен-
концентрированной перекисью водорода, например клапаны, насосы и трубопро-
трубопроводы.
Ниже мы остановимся несколько более подробно на условиях примени-
применимости отдельных материалов.
Алюминий
Алюминий с практической точки зрения является наилучшим материалом
для изготовления емкостей под нейтральные растворы перекиси водорода
в тех случаях, когда желательно снизить до минимума размер разложения
(например, для изготовления хранилищ). Скорость коррозии алюминия возра-
возрастает и в щелочной и в кислой средах, а поэтому алюминий нельзя рекомендо-
рекомендовать для работы с разбавленными отбеливающими щелочными растворами*.
Наряду с общей коррозией под действием кислоты или щелочи в присутствии
такого рода загрязнений, как хлориды, возможна и точечная коррозия, кото-
которую можно предотвратить добавкой небольшого количества нитратов, напри-
например 0,05 вес.% нитрата аммония или меньше [32]. Емкости, используемые для
хранения перекиси, должны быть сделаны из очень чистого алюминия (99,6%
А1 или выше) с минимальным содержанием меди. Они требуют тщательной
очистки [33]. Обычная обработка состоит в следующем: поверхность быстро
обрабатывают разбавленным раствором едкого натра, а затем в течение не-
нескольких часов протравливают в серной, фосфорной или азотной кислоте
(концентрации 35—50 вес.%), причем чаще всего применяется азотная кислота
[34]. После этого сосуд промывают водой, причем для последней промывки
обычно берут дистиллированную или деминерализованную воду. Важно,
чтобы на поверхности не было инородных веществ, например остающихся
в результате операций сварки или формовки, а также чтобы поверхности были
возможно более гладкими. Обычно алюминиевая поверхность при длитель-
длительном контакте с перекисью водорода становится постепенно более инертной,
что обусловливается образованием инертной окисной пленки. Поэтому емкости,
предназначенные для концентрированной перекиси водорода, часто «пассиви-
«пассивируют» после очистки, наполняя их разбавленной перекисью водорода и выдер-
выдерживая наполненными в течение некоторого времени; эта операция представ-
представляет также хороший метод испытания на соблюдение правил изготовления
и очистки сосуда.
Путем анодного окисления на поверхности алюминия можно получить
инертную окисную дленку [35]; такого рода покрытие сообщает алюминиевым
сплавам 2-го класса большую инертность, чем та, которая достигается путем
химической обработки. Обработка поверхности алюминия водным раствором
мышьяковой кислоты и гидразингидрата [36] или гидросульфита натрия [37]
в соответствии с данными некоторых патентов придает алюминию «устойчи-
«устойчивость» против концентрированной перекиси водорода, однако, насколько авто-
авторам известно, эти методы на практике не применяются.
Запатентован также способ покрытия поверхности алюминиевых емкостей
* В одном из патентов [31] приводятся результаты кратковременных опытов по
определению устойчивости разбавленных и щелочных растворов перекиси водорода
в контакте с магнием; на основании полученных данных указывается, что магний является
подходящим материалом для изготовления емкостей под эти растворы.
10 Перекись водорода
146 Глава 4
такими веществами, как смесь парафина и битума [38]. Действительно, пара-
парафин образует сравнительно инертную поверхность в отношении перекиси водо-
водорода, и такого рода парафиновые покрытия на стекле когда-то были общеупо-
общеупотребительны, парафиновые же покрытия на алюминиевых резервуарах и сей-
сейчас еще находят применение в Европе. Однако получить равномерный слой
и обеспечить сохранность сплошного покрытия поверхности трудно, а по-
поэтому в США обычно ограничиваются применением весьма чистого алюминия
с тщательно очищенной поверхностью, на которую никаких покрытий не на-
наносят.
Нержавеющая сталь
Многие нержавеющие стали устойчивы к коррозии под действием пере-
перекиси водорода в широком интервале рН, но даже при тщательной очистке
и полировке скорости разложения^nepeKHCHj^gappo^a-ua поверхностях из не-
нержавеющей стали несколько выше, чем на соответствующих алюминиевых по-
поверхностях. Нержавеющая сталь дает превосходные результаты при приме-
применении в качестве обкладки или материала для изготовления баков, чанов
и другого оборудования для хранения разбавленных отбеливающих растворов
перекиси [30]. Коррозионная устойчивость и пассивность этой стали обычно
приписываются образованию на поверхности пленки из окиси хрома или хеми-
сорбированной пленки кислорода, выполняющей функцию механического
барьера между перекисью водорода и металлом. Так же как и для алюминия,
очень важно, чтобы поверхности были тщательно очищены, например азотной»
кислотойди, были всшюжно_бод??ЛУ1адкими. Так, для нержавеющих сталей
типа 300 (хромоникелевых) сравнительно удовлетворительны поверхности,
подвергнутые проковке или механической обработке, тогда как шероховатые
поверхности, образовавшиеся при литье, непригодны. Шероховатая поверх-
поверхность может способствовать выщелачиванию каталитически активного хрома
перекисью водорода, что снизит ее стабильность. Литая поверхность может
содержать включения материала изложниц, который может обладать катали-
каталитической активностью. Если поверхность нержавеющей стали нельзя очистить
простой обработкой азотной кислотой из-за шероховатости, наличия окалины,
включений, брызг сварочного металла и т. д., можно пассивировать ее путем
протравливания (после 'предварительного обезжиривания) выдерживанием
в растворе с 3% плавиковой и 10% азотной кислоты в течение 30 мин.
при 38° или 2—3 час. при 18—21°. Затем поверхность тщательно про-
промывают водой и там, где это возможно, очищают жесткой щеткой. После этого
поверхность необходимо еще раз обработать азотной кислотой. Если некото-
некоторую часть изделия нельзя протравить, например детали, подвергнутые меха-
механической обработке, можно нанести пасту из кислотной смеси с графитом
только на те места, которые должны быть обработаны [26]. Для получения
гладкой и пассивной поверхности нержавеющей стали можно использовать
и метод электрополировки, например описанный Улигом [39]. Как и в случае
с другими поверхностями, электрополированную поверхность можно сделать
более стойкой по отношению к перекиси водорода путем предварительной
обработки, состоящей в выдерживании ее в перекиси водорода той концентра-
концентрации, которая намечается для употребления.
Олово
Олово является пассивным в нейтральной и слабокислой перекиси водо-
водорода концентрацией до 50%, а возможно и выше, но оно корродирует в щелоч-
щелочных растворах, если только не подавить коррозию путем добавки силиката
натрия [30]. Чистое олово является удовлетворительным материалом для
изготовления насосов, трубопроводов и т. п.; однако луженые поверхности
Конструкционные материалы 147
часто оказываются неудовлетворительными, особенно в контакте с концентри-
концентрированной перекисью. Небольшие дефекты поверхности, например отверстия
величиной с булавочный прокол, способствуют контакту перекиси с основным
металлом, на котором она разлагается с выделением газообразного кислорода
и образованием пузырей, отслаивающих полуду. Чистое олово, обработанное
соляной кислотой и промытое чистой водой, годится в качестве прокладочного
материала, применимого в контакте с жидкой перекисью или горячим паром;
оловянная лента находит применение в качестве уплотнения для насосов.
В пределах указанных ограничений массивное олово является, пожалуй,
наиболее инертным изобычны^^ материалов для работы
с ПерекИСьЮ воДбродатТолько высокая стоимость олова и малая его прочность
огрйштчйвЛТШЗго11?именение,в качестве конструкционного материала. Олово
нельзя также применять при слишком высоких или низких температурах из-за
существования нежелательных аллотропных модификаций при температурах
ниже 13 и выше 203°.
Тантал
Тантал весьма инертен и устойчив к коррозии в самых различных усло-
условиях. Ввиду его высокой стоимости применение его ограничивается исполь-
использованием лишь в некоторых весьма жестких условиях, когда более дешевые
материалы не годятся, например для поверхностей нагрева в перегонных уста-
установках для концентрирования перекиси водорода, содержащей небольшое коли-
количество минеральной кислоты.
Свинец
Свинцовые соединения являются активными катализаторами разложения.
Свинцовое оборудование можно успешно применять лишь в условиях, когда
присутствуют сульфаты, вызывающие образование покрытия из инертного
сульфата свинца. Свинец применяется в некоторых случаях на заводах, выра-
вырабатывающих перекись водорода по электролитическому пероксодисульфат-
ному процессу, и при отбелке сырой шерсти в присутствии сульфатов. Однако
применение свинца в контакте с любыми растворами перекиси, за исключением
весьма разбавленных, может быть опасным, и поэтому его следует избегать.
Железо
Обыкновенный чугун или углеродистую сталь нельзя применять в контакте
с перекисью водорода из-за, образования ржавчины, являющейся активным
каталрШТором'разложения. Единственными сталями, устойчивыми к ржавле-
ржавлению в присутствии концентрированной перекиси, являются стали, содержащие
свыше 13% хрома, или же хромоникелевые нержавеющие стали 18-8, при-
причем последние значительно устойчивее первых. Чугунное оборудование
находит применение лишь для весьма разбавленных щелочных раство-
растворов перекиси, содержащих растворимое стекло в качестве ингибитора
коррозии, но даже в этом случае другие материалы более предпочтительны.
Высококремнистые чугуны, например дурайрон*, находят применение при
изготовлении насосов, используемых для перекачивания разбавленных щелоч-
щелочных отбеливающих растворов. В известных условиях поверхность же-
железа приобретает пассивность в растворе перекиси водорода, т. е. железо
* Кислотоупорный чугун, содержащий 82% Fe, 15,5% Si, 0,66% Mn, 0,83% С
и 0,57% Р.—Прим. перев.
10*
148 Глава 4
кажется инертным веществом; однако такое поведение зависит от слишком
большого числа факторов, и, следовательно, на практике нельзя на него
полагаться.
Медь
Медь является катализатором разложения перекиси водорода, а поэтому
оборудование из меди или сплавов', содержащих большее количество меди,
•чем следы, не подходит для работы с перекисью водорода.
Полимеры
Полихлорвинил, полихлорвинилиден и их сополимеры, полиэтилен и гало-
генированные полиэтилены, как правило, пригодны для применения в качестве
труб, рукавов и прокладок для перекиси водорода любой концентрации.
Полностью галогенированные полиэтилены, например тефлон (политетрафтор-
(политетрафторэтилен) и кел-F (политрифторхлорэтилен), по имеющимся данным, устойчивы
к концентрированной перекиси при довольно высоких температурах [26].
Эти вещества, а также полиэтилен являются наиболее удовлетворительными
органическими веществами для прокладок и набивок. Диспергированный по-
политетрафторэтилен, загущенный поливиниловым спиртом [40], представляет
собой уплотняющий материал подходящей консистенции, при помощи кото-
которого можно осуществлять непроницаемые соединения труб, работающих при
повышенных давлениях (с минимальным заеданием резьбы); этот метод может
найти применение и в работе с перекисью водорода. В качестве уплотнения или
замазки для резьбы труб можно с успехом использовать и низкомолекулярный
полиэтилен; в литературе имеется краткое описание такого способа [26].
Концентрированная перекись водорода медленно экстрагирует пластификатор
из полихлорвиниловых полимеров, а поэтому при эксплуатации эти полимеры
постепен но_затвер девают.
" Трифторхлорвиниловые полимеры (флуоролюбы), имеющиеся в продаже
в виде масел и консистентных смазок, в общем инертны по отношению к пере-
перекиси водорода и являются безопасными и каталитически инертными смазками
для шлифованных стеклянных соединений; их можно также применять и в ка-
качестве манометрических жидкостей для работы в контакте с перекисью водо-
водорода. В прошлом рекомендовалась пропитка уплотнения насоса, выполненно-
выполненного из алюминиевого литья, консистентной трифторхлорвиниловой смазкой.
Однако в последнее время обнаружено [26], что алюминий и фторированные
соединения могут образовать детонирующую смесь. В качестве материалов для
уплотнения насосов рекомендуются шнур или лента из галогенированного
полиэтилена (тефлон или кел-F), лента из алюминия, чистое олово или несма-
несмазанные шнуры из стекловолокна.
В условиях работы с концентрированной перекисью применение полиме-
полимеров в качестве материалов для покрытий не рекомендуется, и пока еще не соз-
созданы такие краски, которые могли бы выдержать действие перекиси. Как и в слу-
случае с оловянными покрытиями, перекись водорода проникает под слой краски
и разлагается на основном металле, выделяя кислород, что приводит к отслаи-
отслаиванию покрытия. Большая часть резин в контакте с перекисью водорода быстро
разрушается.
Дерево
В контакте с высококонцентрированной перекисью водорода дерево может
самовоспламениться. Для очень разбавленных растворов перекиси водорода
Можно применять "чаны и трубы из кипарисового или соснового дерева, но срок
их службы исчисляется всего 4—6 годами [30]; там, где возможно, желательно
применение хромоникелевой стали 316 (сталь 18-8 с 2—3% молибдена).
Конструкционные материалы 149
Трансит* и бетон
Баки и трубы из этих материалов дают удовлетворительные результаты
при работе с разбавленными слабощелочными растворами перекиси, приме-
применяемыми при отбелке, особенно в присутствии силиката натрия. Для высоко-
высококонцентрированной перекиси их рекомендовать нельзя; в этом случае лучше
подходит нержавеющая сталь типа 316. Резервуары из кислотоупорных пли-
плиток, а также бетонные резервуары, выложенные плитками, оправдали себя
в качестве сосудов для отбеливающих растворов.
Никель
Никель и инконель оказывают слабый каталитический эффект на перекись
водорода любой концентрации и коррозионно стойки в нейтральной и щелоч-
щелочной средах. Инконель в отличие от никеля устойчив к некоторым растворам
кислот. Монель-металл, содержащий 30% меди, можно применять лишь для
очень слабощелочных отбеливающих растворов. Для таких растворов при-
применяют и никель и инконель.
Стекло и керамика
Стекло весьма инертно и с успехом применяется для хранения и транспор-
транспортировки перекиси водорода различной концентрации. Основным недостатком
его является хрупкость. Стекло с высоким содержанием силиката, например
боросиликатное (стекло пирекс), вызывает меньшее разложение перекиси,
чем более щелочное натриевое стекло; сосуды из стекла щелочного типа не
подходят для хранения высококонцентрированной перекиси. Эмалированные
сосуды находят применение для разбавленной перекиси; можно думать, что они
ведут себя аналогично натриевому стеклу. Скорость разложения перекиси уве-
увеличивается при действии солнечного света; для снижения влияния этого факто-
фактора часто применяют .шетвую_стекля1шукх-1^рул_Недавно проведено исследова-
исследование влияния алюминийфосфатного стекла (флуорекса) на 90%-ную перекись
водорода [41]. В сосуде из этого стекла наблюдалось медленное падение кон-
концентрации перекиси (в пределах нескольких процентов) в течение месяца;
при дальнейшем трехмесячном выдерживании изменения концентрации больше
не наблюдалось. Это свидетельствует о том, что медленное выщелачивание фос-
фосфата оказывает стабилизующее действие. Кварц.по инертности незначительно
превосходит стекло пирекс. Керамика_л„кшшшый товар с низким содержа-
содержанием железа сравнительно ийертиы"и с успехом'применяются при изготовле-
изготовлении резервуаров для хранения перекиси, однако в настоящее время в этом слу-
случае предпочитают пользоваться алюминием.
При решении проблемы о создании поверхностей, максимально инертных
при контакте с перекисью водорода, стеклу было уделено особое внимание.
В гл. 9 дан подробный анализ влияния природы поверхности на устойчивость
перекиси водорода, здесь же мы остановимся лишь на некоторых способах
обработки поверхности стекла, изученных или рекомендованных. Для очистки
стекла пирекс с целью применения при наиболее точных лабораторных изме-
измерениях с него удаляют путем промывки поверхностные загрязнения, затем
обрабатывают раствором кислоты и, наконец, раствором перекиси водорода.
Необходимо избегать любого процесса, который мог бы способствовать щелоч-
щелочному состоянию поверхности. Хукаба и Кейс [42] описали следующую мето-
методику: подходящее стекло пирекс 1) очищают горячей дымящей серной кисло-
* Материал, изготовляемый из асбеста и портланд-цемента и содержащий 15—
35% серы.—Прим. перев.
150 Глава 4
той; 2) промывают дистиллированной водой и дают ей стечь, затем высушивают
стекло досуха в свободном от пыли ящике; 3) если необходимо, то на этой ста-
стадии производят стеклодувные операции, причем при выдувании воздух пред-
предварительно пропускают через фильтр; 4) вновь обрабатывают серной кислотой
и промывают водой для кондуктометрических измерений; 5) испытывают поверх-
поверхность, наблюдая за действием ее на перекись водорода; 6) если необходимо,
обрабатывают зоны с устойчивой каталитической активностью плавиковой
кислотой; 7) вновь промывают и испытывают действие поверхности на перекись
водорода; 8) если поверхность все еще недостаточно пассивна, сосуд бракуют.
Описана другая аналогичная методика [43], основанная на применении азотной
кислоты, выдерживании изделия в кипящей воде для кондуктометрических
измерений и последующем выдерживании в перекиси водорода. Позже на
основании сравнительных испытаний [44] по эффективности отдельных стадий
выяснилось, что наиболее эффективная обработка состоит в выдерживании
стекла в перекиси, лучше всего в горячей. Для серийных лабораторных работ
достаточно очистки с получением чистых «стекаемых» поверхностей стекла,
для чего стекло промывают моющим веществом и затем выдерживают в 15%-
ном растворе азотной кислоты (не следует допускать выделения едких паров
кислоты в атмосферу лаборатории). После этого выдерживают стекло еще
в перекиси водорода, что дает инертную поверхность для большинства видов
применения. Обычная обработка хромовой кислотой недопустима; нельзя
также обрабатывать стекло крепкой щелочью, например для освобождения
от жировых загрязнений.
Описаны различные методы создания покрытия на поверхности стекла.
Арнтзен и Роули [45] указывают, что силиконовое покрытие, пигментирован-
пигментированное двуокисью титана, весьма устойчиво против перекиси водорода. Тамрес
и Фрост [46], наоборот, сообщают, что силиконовое покрытие ускоряет разло-
разложение паров перекиси. Оказалось, что в тех же условиях покрытие из
микрокристаллического парафина лишь слабо снижает скорость разложения,
наблюдаемую на поверхности, очищенной хромовой кислотой. Кук [47] описал
процесс нанесения слоя плавленой борной кислоты, окиси бора или бората
на поверхность из стекла или керамики. При этом поверхность приобретает
такие же свойства, какими обладает сначала боросиликатное стекло; такого
рода покрытия, в частности, рекомендуются как средство для замедления раз-
разложения паров перекиси водорода в некоторых производственных операциях.
ХРАНЕНИЕ И ПЕРЕВОЗКА ПЕРЕКИСИ ВОДОРОДА
Емкости для хранения и транспортировки концентрированной перекиси
водорода обычно изготовляются из алюминия высокой чистоты, предварительно
подвергнутого вышеописанной обработке. Иногда для очень небольших коли-
количеств (примерно меньше 0,5 кг) концентрированной перекиси водорода или не-
несколько больших количеств перекиси с концентрацией ниже приблизительно
35 вес.% применяется стекло, хотя это и рискованно в связи с его хрупкостью.
В отдельных случаях используются эластичные сосуды из подходящего поли-
полимера; так, сосуды из полиэтилена почти вытеснили небольшие стеклянные или
запарафиненные внутри склянки для перекиси водорода марки «Для реакти-
реактивов». Как указано выше, оборудование, покрытое пластмассами, не при-
пригодно для концентрированных растворов. Нормальные скорости разложения,
которые можно ожидать при хранении, рассматриваются дальше, в гл. 9.
Описан и применяется ряд способов покрытия стенок емкостей для перекиси
различными веществами для придания им повышенной инертности, например
покрытие их тонким слоем парафина, обладающего малой непредельностью.
Однако при эксплуатации парафиновый слой постепенно отслаивается. В на-
настоящее время считают, что наиболее удовлетворительный метод обеспечения
Хранение и перевозка перекиси водорода
151
максимальной устойчивости перекиси водорода при хранении ее в металличе-
металлической таре состоит в применении алюминия высокой чистоты, подвергнутого
вышеописанной обработке и затем пассивированного путем длительного кон-
контакта с перекисью водорода.
В 1954 г. в США фирмы выпускали в продажу перекись водорода следую-
следующих концентраций: 27,5; 35; 50 и 90 вес.%. 27,5%- и 35%-ная перекись иногда
транспортируется в стеклянных буты-
бутылях, вмещающих до 54 кг раствора.
Продукт всех этих четырех концентра-
концентраций отпускается также в алюминиевых
бочках емкостью 113 л, а также в желез-
железнодорожных или автомобильных цистер-
цистернах емкостью 15—30 ж3. Емкости такого
рода снабжены соответствующими отду-
отдушниками, обычно устроенными по прин-
принципу клапана Бунзена. На рис. 26 и 27
показаны типичные бочки и вагоны-ци-
вагоны-цистерны.
Бочка, показанная на рис. 26, слева,
применяется для- растворов перекиси во-
водорода умеренной концентрации или для
высококонцентрированных растворов, но
только в том случае, если бочки транспортируются в вагонах партиями.
Бочка, показанная справа, используемая для 90%-ной перекиси при отгрузке
отдельными бочками, имеет «ложную головку», содержащую верхнюю камеру
Рис. 26. Алюминиевые бочки для
хранения высококонцентрированной
перекиси водорода.
Рис. 27. Алюминиевые железнодорожные вагоны-цистерны
для транспортирования перекиси водорода.
непосредственно над самой бочкой. Затворы и отдушники устроены таким обра-
образом, что, если бочка случайно окажется лежащей на боку, любое количество
вытекающей через отдушник жидкости будет уловлено в верхней камере, что
исключает возможность утечки высококонцентрированной перекиси водорода.
Обе эти бочки имеют в верхних ободах сливные отверстия, чтобы дождевая вода
или другие жидкости не скоплялись вокруг отверстия отдушника. Обычно в ка-
качестве стабилизаторов добавляются "небольшие количества станнатных или
фосфатных соединений или тех и других вместе.
Информацию по технике хранения и перевозки и по специальному
оборудованию, например стеллажам для хранения, желобам для спуска
152 Глава 4
и сифонам, можно получить у различных фирм, производящих перекись водо-
водорода, и здесь мы на этих вопросах не останавливаемся. Наиболее существен-
существенные меры предосторожности заключаются в следующем: 1) необходимо избегать
контакта перекиси с активными катализаторами, например материалами, со-
содержащими железо, медь, марганец и большинство других металлов, а также
с пылью и щелочными соединениями, которые могут вызвать быстрое разложе-
разложение; 2) недопустим контакт с органическими веществами, которые могут воспла-
воспламениться или образовать взрывчатые смеси с концентрированной перекисью
водорода; 3) следует всегда обеспечивать надлежащую вентиляцию оборудова-
оборудования, в котором может храниться или временно находиться перекись водорода*;
4) нужно избегать слишком высоких температур. Физиологическое действие
перекиси водорода описано на стр. 153. Перекись, имеющая концентрацию око-
около 50 вес.% или меньше, обычно не вызывает немедленного воспламенения слу-
случайно облитого способного гореть материала, например одежды, но, если дать
ей высохнуть, то, поскольку вода испаряется легче, концентрация перекиси,
увеличивается, что иногда приводит к самовоспламенению. Загрязненные
материалы, содержащие каталитические примеси, или другие горючие вещества,
например дерево или предметы одежды, особенно шерстяной, часто самовозго-
самовозгораются при попадании на них концентрированной перекиси водорода. Во всех
случаях пролитую перекись следует смывать большим количеством воды.
Все емкости для хранения должны иметь отдушники для выделения
кислорода, образовавшегося при разложении, причем все отверстия должны
быть сделаны таким образом, чтобы уменьшить до минимума возможность
загрязнения содержимого. Теплота, выделяющаяся при разложении, проис-
происходящем в условиях хранения с нормальной скоростью, легко рассеивается
в результате теплообмена с окружающей средой. Однако иногда при сильном
загрязнении концентрированной перекиси разложение ее происходит с такой
большой скоростью, что выделяющуюся теплоту отвести невозможно. В этом
случае происходит самоускорение разложения, сопровождающееся повыше-
повышением температуры и ростом давления, которые могут вызвать взрыв даже
вентилируемой емкости, аналогичный взрыву сосуда, работающего под давле-
давлением. Для предотвращения этого крупные емкости для хранения пере-
перекиси водорода обычно снабжаются указателями температуры и приспособле-
приспособлениями для быстрого выливания и разбавления содержимого (или только
разбавления) в случае аномально высокой скорости разложения. Для этой
цели нельзя применять ртутные термометры, поскольку в случае их поломки
возникает опасность загрязнения перекиси водорода каталитически активной
ртутью.
Шэнли [48] составил тепловые балансы для различных комбинаций раз-
размеров емкостей для хранения, концентраций перекиси водорода и скоростей
разложения; на основании этих балансов можно подсчитать критические усло-
условия, при которых происходит самоускоряющееся разложение. Представлены
также расчетные данные по изменениям температуры во времени для неко-
некоторых случаев самоускоряющегося разложения и обсуждаются средства
определения возможной опасности при хранении. Описаны также возмож-
возможные контрольные меры. Чинкель [49] рассматривает противоположную про-
проблему, а именно, проблему умышленного повышения температуры раствора
перекиси водорода при низких окружающих температурах; он описывает
* В трубопроводах, наполненных концентрированной перекисью водорода, опасно
применять краны. Газ, выделяющийся при разложении перекиси водорода, задержав-
задержавшейся в проходе закрытого крана, может создать давление, достаточно высокое, чтобы
разорвать кран, особенно стеклянный. Дэвис и Киф [26] приводят описание конструкции
оборудования, в котором отсутствуют такие ловушки и мертвые пространства, и обсу-
обсуждают желательные способы, обеспечивающие стекание жидкости при выключении этого
оборудования.
Физиологическое действие 153
автоматическое приспособление для каталитического разложения части пере-
перекиси водорода с целью нагревания остальной части.
Перевозка перекиси водорода в США подчиняется правилам Междуштатной
торговой комиссии, которая относит перекись водорода к разряду «едких жид-
жидкостей», требующих наличия опознавательной белой этикетки. Комиссия издала
подробные технические условия относительно типа и конструкции транспорт-
транспортной тары в зависимости от концентрации перевозимой перекиси. Для перекиси
водорода, имеющей концентрацию выше 10 вес.%, допустимо применение лишь
стеклянных или керамических сосудов или бутылей, помещенных в соответству-
соответствующие ящики, снабженные амортизирующими приспособлениями, или же подхо-
подходящих алюминиевых сосудов. Для перекиси водорода, имеющей концентрацию
37—52 вес.%, емкость стеклянной тары не должна превышать 3,8 л; для кон-
концентрации свыше 52 вес.% максимальная емкость стеклотары—не больше
0,95 л. Технические условия, касающиеся упаковки стеклотары внутри другой
защитной тары, тем строже, чем выше концентрация перекиси. В связи с этим
более или менее значительные количества перекиси водорода, имеющей кон-
концентрацию выше 35 вес.%, перевозятся исключительно в алюминиевой таре.
Дополнительные данные можно получить непосредственно от Междуштатной
торговой комиссии или же почерпнуть в справочнике Сэкса [50]. Организация
«Manufacturing Chemists' Association» издала подробный кодекс по методике
разгрузки, хранения, перевозки перекиси водорода и т. п. [51].
В некоторых промышленных центрах приходится сталкиваться с трудно-
трудностью ликвидации сточных вод, содержащих перекись водорода, путем сброса
их в водоемы. Так, концентрации перекиси водорода, превышающие 40 мг1л,
оказывают токсическое влияние на молодь форели, более низкие концентрации
совершенно безвредны в течение 48-часового периода [52]. Наилучший метод
освобождения воды от остаточной перекиси водорода зависит от природы дру-
других содержащихся в воде отходов; так, при наличии восстановителей (гидра-
(гидразина или метилового спирта), например в сточной воде от ракетоиспытатель-
ной станции, желательно вызвать сначала взаимодействие между перекисью
и этими веществами. Поскольку перекись водорода легко разлагается в щелоч-
щелочной среде, а также под действием различных металлических катализаторов,
то по одному из методов [52] обработки остаточной перекиси предложено
к воде добавлять известь для доведения рН до 11, после чего вводить раствори-
растворимую марганцовую соль, например хлорид, чтобы концентрация марганца со-
составила около 4 мг/л. При этом рН марганец, по-видимому, превращается
в тонкодисперсный осадок гидрата окиси марганца, являющегося очень
эффективным катализатором. Смесь следует перемешать до полного разложения
перекиси и, после того как осадок отстоится, сточные воды сбросить в водоем.
Осевший шлам, вероятно, можно использовать вторично.
ФИЗИОЛОГИЧЕСКОЕ ДЕЙСТВИЕ
Перекись водорода не является ядовитой в обычном смысле этого_слова,
но концентрированные растворы (например, содержащие 27 вес.% или выше)
являются сильными первичными раздражителями кожи и дыхательных путей.
Растворы перекиси водорода при соприкосновении с кожей вызывают ее побе-
ление, сопровождаемое зудом или чувством жжения, как от ожога крапивой.
Это неприятное ощущение обычно исчезает при длительном промывании водой,
и кожа, как правило, вновь приобретает через некоторое время нормальную
окраску без каких-либо видимых следов от действия перекиси. При длительном
соприкосновении с перекисью или при более высоких ее концентрациях воз-
возможно образование пузырей на коже. Пары nepeKHCjjjtoao?Ofla_Bbi3biBaioT_cjie-
зотечение и раздражение слизистых оболочек носа и горла. Вдыхание паров,
насыщенных при комнатной температуре 90%-ной перекисью, в течение срав-
154 Глава 4
нительно небольших промежутков времени, например при проливании боль-
большого количества перекиси, не оказывает, по-видимому, значительного вредного
действия на дыхательные пути [53]. Жидкая перекись водорода сильно раз-
раздражает глаза и может вызвать их поражение. Если в глаза попала перекись,
необходимо промыть их водой и немедленнообратиться к медицинской помощи.
Прием внутрь растворов перекиси водорода, если только они не очень разбав-
разбавлены, недопустим, так как вследствие сильного раздражения может произойти
внутреннее кровотечение. Опыты над животными показывают также, что 90%-
ная перекись водорода может всасываться при нанесении на кожу [53]
(см. стр. 357).
При работе с большими количествами концентрированной перекиси реко-
рекомендуется носить спецодежду, спецобувь и перчатки из полихлорвинила, поли-
полиэтилена, акрилового волокна «орлон» или, что лучше, полиэфирного волокна
«дакрон» и принимать все меры предосторожности, чтобы перекись не попадала
на какую-либо часть тела. Работать с растворами, содержащими свыше 3—
5 вес.% перекиси, следует обязательно в очках-консервах или защитных
масках. Соответствующую информацию о спецодежде можно получить от фирм,
производящих перекись водорода.
ВЗРЫВЧАТЫЕ СВОЙСТВА
Данный раздел посвящен практической информации, которая может дать
потребителю понятие о потенциальной взрывоопасности, с которой связано
применение перекиси водорода. Химия разложения обсуждается в гл. 7 и 8.
Проведено много работ, касающихся возможности вызвать взрывное разложе-
разложение чистых водных растворов перекиси водорода. Обычная методика состоит
в том, что пробу подвергают механическому удару или чаще испытывают на
детонацию от капсюля-детонатора или определенного количества взрывчатого
вещества, например тетранитропентаэритрита, и сравнивают разрушения
в этом случае и в аналогичном опыте с применением воды вместо перекиси.
Для водных растворов перекиси водорода без органических примесей наблю-
наблюдается постепенное усиление взрывных эффектов с увеличением концентра-
концентрации перекиси или изменением других условий опыта. Максимальный взрыв-
взрывной эффект инициирующего вещества в присутствии раствора перекиси водо-
водорода вместо воды увеличивается при росте какого-либо из следующих факто-
факторов: температуры, концентрации перекиси водорода, диаметра сосуда, проч-
прочности оболочки или величины инициирующего взрыва. Наибольшее значение
имеет концентрация перекиси.
При комнатной температуре трудно добиться распространения детонации
даже в 100%-ной перекиси. В одной серии опытов [10] с применением сосудов
диаметром 25—30 мм оказалось необходимым в качестве сосуда применять
прочные трубки из нержавеющей стали и 30 г тетранитропентаэритрита в ка-
качестве инициатора, обеспечивающего полную детонацию в каждом опыте. При
менее жестких условиях перекись водорода, находящаяся в непосредственном
соседстве с инициатором, по-видимому, детонирует, но детонационная волна
затухает после пробега определенного расстояния, зависящего от условий.
Так, могут оказаться нетронутыми отрезки трубы различной длины, содержащие
еще более или менее значительные количества неразложенной перекиси. Ниже
приведены некоторые примеры полученных в разных условиях результатов
с целью характеристики взрывоопасных областей. Все описанные в дальнейшем
опыты проведены с перекисью водорода при комнатной температуре. 100%-
ная перекись не детонирует при механическом ударе, например от падающего
молота или при простреле. Она не детонирует даже в наиболее жестких усло-
условиях инициирования с применением взрыва капсюля-детонатора № 8, если
находится в алюминиевой трубке диаметром 21 мм, зарытой в рыхлый влажный
Взрывчатые свойства 155
песок. Однако, если использовать для инициирования взрыва 10 г тетранитро-
пентаэритрита, то в описанных жестких условиях наблюдается полная детона-
детонация (она происходит также и тогда, когда алюминиевая трубка не зарыта).
Капсюль-детонатор № 8 вызывает частичную детонацию, если 100%-ная пере-
перекись находится в армированном сосуде. Частичная детонация обнаружена
также при 90%-ной концентрации перекиси в случае применения 10 г тетрани-
тропентаэритрита в качестве первичного заряда и значительной прочности
оболочки. Если повысить температуру выше комнатной, то при прочих равных
условиях детонация оказывается более полной; частичная же детонация может
происходить при еще более низких концентрациях перекиси (например, до
80—85%).
В одном опыте, проведенном в Германии, отмечена частичная детонация
85%-ного раствора при комнатной температуре в том случае, когда большое
количество перекиси B00 кг) было подорвано взрывом 25 кг смеси равных
частей тринитротолуола и циклонита (симметричного триметилентринитрамина).
Ряд опытов показал, что механический удар в различной форме, например
падающий груз, винтовочный или пулеметный огонь [29], не вызывает детона-
детонации 90%-ной перекиси водорода. Имеется ряд доказательств, что при комнат-
комнатной температуре даже при очень мощном инициаторе и прочной оболочке рас-
растворы, содержащие до 95—96% перекиси водорода, не способны к распростра-
распространению детонационной волны. При испытании в свинцовом блоке Трауцля,
в котором испытуемое вещество в прочной оболочке подвергается удару под
действием взрыва капсюля-детонатора, в перекиси водорода всего 60%-ной
концентрации наблюдается усиление взрывного эффекта. Последний непрерыв-
непрерывно возрастал по мере увеличения концентрации перекиси водорода, причем
изменения были очень быстрыми в интервале нескольких последних процентов
при приближении к 100%-ной концентрации.
Сообщается, что взрывной эффект достигается легче при добавке к перекиси
щелочей, хотя добавка к ней стабилизаторов или катализаторов не оказывает
никакого влияния. Растворы перекиси водорода в твердом состоянии значи-
значительно менее склонны к детонации, чем в жидком.
Скорость детонации водных растворов, содержащих 96—100% перекиси
водорода, в соответствии с определениями, проведенными по Дотришу и по
оптическому методу [10], оказалась равной примерно 6500 м/сек. Хотя при
применении известной смеси перекиси водорода с органическим веществом были
обнаружены три различные скорости детонации (см. стр. 157), для чистой
водной перекиси всегда наблюдалась только одна определенная скорость.
Некоторые органические вещества, особенно если они загрязнены тяже-
тяжелыми металлами или щелочными веществами, могут бурно реагировать с кон-
концентрированными растворами перекиси водорода. В большинстве же случаев
при смешении органических веществ с перекисью немедленных видимых при-
признаков реакции не наблюдается, но образуются способные к детонации смеси
(если только взаимная растворимость этих веществ и перекиси водорода доста-
достаточно высока). Если даже органическое вещество нерастворимо, но образует
эмульсию или хотя бы временную механическую дисперсную смесь с концен-
концентрированной перекисью водорода, смесь может быть способной к детонации.
Чувствительность органических растворов к удару и взрывной эффект их не-
несколько колеблются в зависимости от природы органического вещества, но
еще большее значение имеют концентрация перекиси водорода и отношение
количеств перекиси и органического вещества. Шенли и Гринспэн [54] приводят
данные о границах областей, детонирующих состав для растворов различных
органических веществ в водной перекиси водорода. На рис. 28 показаны данные,
полученные этими авторами для смесей ацетона, перекиси водорода и воды, при-
причем эти данные типичны для различных изученных ими растворов. Минималь-^
пая концентрация растворимого органического вещества, дающая детонирую-
156
Глава 4
100%
ацетона
Стехио-
метрические
соотношения
100%
Нг02
100%
воды
щую смесь с 90%-ной перекисью водорода, обычно составляет 2—5 _вес.%^_
Однако в том случае, когда содержание 90%-ной йере!01Ся15ыло меньше 30 об.%
конечной смеси, детонации не обнаружено.
Наиболее заметные эффекты наблюдаются в том случае, когда отношение
количеств органического вещества и перекиси соответствует полному окисле-
окислению органического вещества(стехиометрическое соотношение). Испытано боль-
большое количество разных органических веществ в концентрированных растворах
перекиси водорода на способность к детонации. Во всех случаях, если только
органическое вещество достаточно
растворимо, отмечалось сущест-
существование обласги детонирующих
составов. В качестве примеров
приводятся [54—58] растворы ме-
метилового, этилового, пропилового
и бутилового спиртов; муравьи-
муравьиной, уксусной и пропионовой ки-
кислот; аллилового и бензилового
спиртов; ацетонитрила; мно'со-
атомных спиртов, например гли-
глицерина, диоксана, хинолина и
этилацетата. Стехиометрические
растворы непредельных спиртов
более чувствительны к детонации,
чем растворы предельных; в свою
очередь последние более чувстви-
чувствительны, чем растворы органиче-
органических кислот. В качестве критерия
при этом исходили из количества
первичного заряда, которое требо-
требовалось для возникновения харак-
характерной высокоскоростной детона-
детонационной волны вместо второй волны, распространяющейся с небольшой
скоростью (см. ниже). Некоторые растворы органических веществ в кон-
концентрированной перекиси водорода весьма чувствительны к детонации от
механического удара. Так, растворы метилового спирта в концентрирован-
концентрированной перекиси, находившиеся в стеклянном сосуде, детонировали при сбра-
сбрасывании их с высоты около метра на бетонный пол. Нужно отметить
также, что многие такие растворы органических веществ в концентриро-
концентрированной перекиси водорода, в которых вначале не наблюдается видимой
реакции, в действительности медленно окисляются, образуя смеси с новы-
новыми взрывчатыми свойствами, иногда с повышенной чувствительностью.
Проведено подробное исследование взрывчатых свойств метилового и эти-
этилового спиртов и глицерина в концентрированной перекиси водорода [10].
Такого рода смеси могут загораться от пламени. Небольшие количества горят
со свистящим звуком, большие быстро детонируют после воспламенения. На-
Наличие примесей может привести к спонтанному разложению, ведущему к сго-
сгоранию и детонации. Детонация или вспышка (в зависимости от условий и ко-
количеств) наблюдается при нагревании примерно до 130—140°.
Методами Дотриша и оптическими методами [10] измерены скорости дето-
детонации стехиометрических смесей этих трех соединений, растворенных в кон-
концентрированной перекиси водорода (80%-ной или выше), так же как и многих
других органических веществ. В подходящих условиях скорость детонации
составляет около 7000 м/сек, причем эта скорость в зависимости от природы орга-
органического вещества существенно не изменяется. Однако по мере снижения кон-
концентрации перекиси или импульса инициирующего заряда или же при посте-
Рис. 28. Интервал взрывчатых составов для
смесей ацетона, перекиси водорода и воды, по
данным Шенли и Гринспэна [54].
X взрыв; О взрыва нет.
Взрывчатые свойства
157
пенном изменении других переменных, например прочности оболочки, диа-
диаметра трубки и т. д., скорость детонации может сразу упасть примерно до
2300 м/сек. При еще более мягких условиях наблюдается третья скорость дето-
детонации—около 750 м/сек. Бризантность, измеренная по степени обжатия свинцо-
свинцового или медного блока, также сразу делается значительно ниже. Другими сло-
словами, путем непрерывного изменения
инициирующего заряда или других
параметров можно получить две со-
совершенно различные и дискретные
степени взрывных эффектов.
В условиях, способствующих до-
достижению максимальной скорости де-
детонации, смеси этих органических ве-
веществ с перекисью водорода облада-
обладают бризантностью, сравнимой с бри-
зантностью пикриновой кислоты или
даже превосходящей ее, но уступа-
уступают в этом отношении нитроглицери-
нитроглицерину. Величина расширения свинцово-
свинцового блока выше, чем при применении
нитроглицерина, вероятно из-за боль-
шого объе^- газа, выделяющегося
при взрыве перекиси*
160
800
100
600
500
400
150
140
1/20
ПО
ь
/ж
'Равное
НОИНИИ
щ
есная -N
тптшЯ'
, паров выше/
/^26 мол. %У/
''Опасная /^
авновеа
h
чая конь
mm 26
{внтрац
мол. %
ия паро
-
-
-
3
i
f 300
200
100
;
: :
¦
¦
1
С
-
-
л...
у
\
о о oodVC
о оо <
° о»
о о
1 *nuf t I Jl I 1 1
2
/
X
\
\
\
О ООО Л>
lilt
-
-
-
-
-
;
-
,"¦¦¦ *^
1111
100
90 80 10 60 50
НгОг в водном растворе, вес. %
Р]и^с. 29. Комбинации температуры
и состава жидкости, приводящие к обра-
образованию равновесных паров взрывчато-
взрывчатого состава. Общее давление 1 am.
40
20 30 40 50
Н2О2 в парах, мол. %
60
рис. 30. Влияние общего давления на пре-
предел воспламенения при искровом иницииро-
инициировании (точки относятся к сосудам без на-
насадки).
О взрыва нет; ¦ взрыв. /—предел для сосуда без
насадки; 2—предел для сосуда с насадкой.
При атмосферном давлении пары, содержащие 26 мол.% (или больше)
перекиси водорода (остальное—инертный газ, например кислород, азот или
водяной пар), могут быть взорваны от искры, за счет контакта с каталитически
активными веществами первоначально при комнатной температуре, или с нека-
некаталитическими материалами, например алюминием, но нагретыми до темпера-
температуры около 150° или выше [59]. Ряд доказательств свидетельствует о том, что
взрывы, которые, по имеющимся данным, происходили при нагревании чистых
водных растворов перекиси водорода до температур, близких к точке кипения,
в действительности могли происходить в паровой фазе, а не в жидкой; этим
158 Глава 4
объясняются те сообщения, в которых температура 150 ° или около этого при-
приводится как «температура взрыва» перекиси водорода. Минимальные темпера-
температуры, при которых возможно существование взрывоопасной смеси паров над
водными растворами перекиси водорода при атмосферном давлении, были вы-
вычислены из известных пределов взрывчатости и по данным равновесия системы
пар—жидкость. На рис. 29 эти температуры показаны как функция концен-
концентрации жидкости. Ввиду высокой относительной летучести воды по сравнению
с перекисью водорода опасность взрыва перекиси в паровой фазе при хранении
ге в жидком виде может встретиться лишь в случае высококонцентрированных
растворов при повышенных температурах, которые, например, могут воз-
возникнуть в случае пожара или при сильном загрязнении перекиси. Однако
возможно, что в таких условиях ускоряющаяся реакция разложения при этих
температурах будет способствовать взрыву закрытого или сообщающегося
с атмосферой сосуда до того момента, когда образуется пар взрывчатого со-
состава. Скорость рассеивания теплоты при нормальном разложении перекиси
в обычной бочке за счет конвекции достаточно велика для того, чтобы темпера-
температура содержимого поднялась самое большее на несколько градусов выше тем-
температуры окружающего воздуха даже в том случае, если последняя равна 65°.
Как показано на рис. 30, при падении давления возрастает минималь-
минимальная концентрация перекиси водорода в парах, которая необходима для
того, чтобы пары стали взрывчатыми. Одна кривая на этом рисунке показывает
взрывной предел, определенный в незаполненной полулитровой-^руглодонной
колбе. Другая кривая относится к взрывному пределу для случая, когда колба
заполнена отполированными на пламени стеклянными кольцами Рашига внеш-
внешним диаметром и длиной по 14 мм. По мере увеличения поверхности в сосуде
возрастает та минимальная концентрация, при которой возможно распростра-
распространение взрыва. На рис. 30 видны те опасные области, которых нужно избегать
при концентрировании перекиси водорода путем перегонки под вакуумом.
Предел взрывчатых составов не зависит от соотношения количеств водяного
пара и кислорода в инертном газе. Взрывчатые свойства парообразной пере-
перекиси водорода рассматриваются также в гл. 8.
ЛАБОРАТОРНЫЕ МЕТОДЫ ГЕНЕРИРОВАНИЯ ПАРОВ ПЕРЕКИСИ ВОДОРОДА
При попытках генерирования паров в лабораторных условиях с целью
изучения их физических и химических свойств приходится сталкиваться с труд-
трудностями, обусловленными чувствительностью водного раствора перекиси
к разложению и другими его свойствами. Мы поэтому дадим краткое описание
этого процесса или по меньшей мере упомянем ряд способов, пригодных для
этой цели; некоторые из них раньше в литературе не описаны.
Если для статического опыта желательно иметь пары с содержанием пере-
перекиси водорода, соответствующим порядку парциального давления в несколько
миллиметров ртутного столба, то обычно некоторое количество концентриро-
концентрированного раствора перекиси водорода нагревают до кипения в сосуде и пропу-
пропускают пары в предварительно эвакуированную колбу, после чего эту колбу
отделяют для исследования. Прибор в экспериментальном отношении отли-
отличается простотой, и различными авторами опубликованы хорошие схемы такого
рода установок [60]. Этот прибор может быть использован и для динамической
системы, но тогда нужно создать приспособления для конденсации паров после
протекания их через секцию для наблюдений и для откачивания кислорода,
образовавшегося при разложении [61]. В одном простом случае осуществления
этого метода испытывавшийся материал просто подвешивали в колбе, в которой
происходило кипение перекиси при пониженном давлении [62]. При помощи
этого прибора в сочетании с системой обратной конденсации для возвращения
конденсата в кипятильник получены весьма надежные результаты при измере-
Лабораторные методы генерирования паров перекиси водорода 159
нии упругости паров над водными растворами перекиси водорода [63]. Фрост
и Олденберг [64] для изучения последовательного статического накопления
пара пользовались атим прибором, периодически прерывая поток паров из
колбы с жидкостью, кипевшей при пониженном давлении. Для этой цели авторы
применяли стеклянный клапан с магнитным управлением (с запаянным в стекло
куском мягкого железа), который позволял работать без разложения перекиси
водорода.
Все эти приборы применяли при давлении ниже атмосферного. Для устра-
устранения необходимости в применении вакуума иногда используют продувание
подходящим инертным газом. В двух работах [65] приводится описание ме-
метода насыщения некоторого постороннего газа перекисью водорода.
Вода отличается более высокой летучестью по сравнению с перекисью водо-
водорода, а поэтому если необходимо получить хотя бы сравнительно низкое пар-
парциальное давление парообразной перекиси, приходится пользоваться высоко-
высококонцентрированным раствором. В обычном случае, когда пары перекиси водо-
водорода не подвергаются конденсации с обратным отводом их в испаритель, со-
состав паров, конечно, изменяется во времени (если только не применяется прак-
практически 100%-ная перекись водорода). В некоторых случаях желательно полу-
получить пары с гораздо более высоким парциальным давлением перекиси водорода,
чем это возможно обычно, и притом таким образом, чтобы концентрация паров
не изменялась во времени. Этого можно достигнуть путем непрерывного введе-
введения водной перекиси водорода в кипятильную колбу со скоростью, соответст-
соответствующей отбору пара, и с концентрацией, соответствующей концентрации этого
пара. Опыт показал, что путем применения подходящей аппаратуры можно
получить концентрированные пары с парциальным давлением перекиси водо-
водорода, превышающим предел взрывчатости, причем, если принять необходимые
меры предосторожности, можно не опасаться заметного разложения. Это зна-
значительно повышает интервалы концентрации, давления и скорости, которые
могут быть достигнуты в указанных условиях. Согласно одной из старых работ
[66], такая непрерывная операция кипячения может быть основана на прин-
принципе «мгновенного испарения». Жидкую перекись водорода распыляют на
нагретую поверхность нержавеющей стали, причем подачу перекиси и тепла
регулируют таким образом, чтобы жидкость мгновенно испарялась при сопри-
соприкосновении с горячим металлом. Для отвода паров применяется стеклянный
колпак, уплотненный на металлической поверхности прокладкой. Такое при-
приспособление частично оправдало себя, но регулирование его сопровождается
затруднениями. Опыт показал, что при недостаточно тонкой пленке жидкости
на поверхности горячего металла происходит значительное разложение и,
если концентрация паров превысит предел взрывчатости, на голом металле
может произойти взрыв в газовой фазе. Кроме того, при высокой скорости кипе-
кипения возможны механический унос и образование тумана. Сама эта техника
не является порочной, однако имеются и другие более простые методы.
Разработана сйециальная методика [67], по которой производят адиабати-
адиабатическое разложение некоторой части потока жидкой перекиси водорода с доста-
достаточно высокой начальной концентрацией (выше 65 вес. %), чтобы вызвать при
помощи выделяющейся теплоты испарение всей жидкости. Однако этим путем
можно получить пары с концентрацией перекиси не выше примерно 10 мол.%,
причем весьма вероятен унос жидкости. Для определения взрывчатых свойств
паров перекиси водорода использован кипятильник непрерывного действия,
представляющий модификацию известного аппарата с всползающей пленкой
и напоминающий по виду «рибойлер», применяемый при дистилляции. В лите-
литературе имеется подробное описание и схема этого прибора [59], иллюстрирую-
иллюстрирующая метод отделения паров от жидкости, систему электронагрева, простую
склянку Мариотта, приспособленную для питания кипятильника, и способ
работы при давлении ниже атмосферного.
160
Глава 4
Разработаны различные модификации этого аппарата, еще не описанные
в литературе, которые позволяют проводить операцию более плавно с интенсив-
интенсивным генерированием пара при давлениях несколько выше атмосферного.
По одному из методов [68] увеличивают поверхность для притока тепла путем
увеличения диаметра кипятильника. Вместе с тем очень важно свести до мини-
минимума среднее время нахождения жидкости в кипятильнике, чтобы по возмож-
возможности уменьшить разложение. Этого достигают при помощи нагревательного
прибора, имеющего вид кипятильника с кольцевым промежутком (рис. 31, б).
Оказалось желательным также для устранения местного перегрева разместить
нагревательную рубашку на расстоянии около 13 мм (ширина кольцевого про-
промежутка) от поверхности стеклянного кипятильника; это целесообразнее, чем,
К обратному
холодильнику 0
+ ВыХОд А Г
пара ^ ^—<Ь^ Выход
пара
К обратному
олодильнику
3
шиокости
К ловушке
пара
К кипятиль-
кипятильнику
Рис. 31. Аппаратура для испарения растворов перекиси во-
водорода.
о—кипятильник с паровым нагревом; б—кипятильник с кольцевым про-
промежутком; в—уравнительный сосуд; 1—конический шлиф; 2—трубка
диаметром 100 мм из стекла пирекс; 3—алюминиевая поверхность на-
нагрева; 4—стандартный фланец для трубки из стекла пирекс; 5—сое-
5—соединение, осуществленное электродуговой сваркой; 6—трубка диаметром
20 мм из стекла пирекс; 7—алюминиевая пластинка; 8—электронагре-
8—электронагреватель; 9—трубка диаметром 10 мм из стекла пирекс; /0—трубка диа-
диаметром 100 мм из стекла пирекс; И—трубка диаметром 75 мм из стекла
пирекс; 12—сферический шлиф; 13—питательный резервуар; 14—трубка
для присоединения к системе буферного газа.
как это предполагалось раньше [59], непосредственно обматывать кипятиль-
кипятильник нагревательной проволокой. Меньший приток тепла в таком аппарате сни-
снижает интенсивность кипения и устраняет необходимость в трубке для возврата
жидкости при наличии достаточного свободного объема над поверхностью
жидкости для разделения и отстаивания тумана. Если необходимо, можно
присоединить наружный туманоотделитель и пароперегреватель. Скорость ки-
кипения в этом случае может быть ограничена имеющейся электрической мощно-
мощностью; в таких случаях можно пользоваться кипятильником с паровым обогре-
обогревом [69], показанным на рис. 31, а. Алюминиевая поверхность теплообмена, если
она чистая и все время погружена в кипящую жидкость, вызывает лишь ничтож-
ничтожное разложение перекиси водорода. При применении водяного пара под давле-
давлением 4,2 am и величине поверхности теплообмена около 200 см2 легко удается
выпаривать 30—50 мл жидкости в минуту с получением пара с концентрацией
перекиси до}35 вес.%. При применении дистиллированной высококачественной
воды для разбавления питающей перекиси водорода разложение в кипятиль-
Лабораторные методы генерирования паров перекиси водорода 161
нике составляет максимум несколько процентов. Необходимо отметить, что,
поскольку конденсация пара и кипение раствора, протекающие соответственно
по обе стороны поверхности теплообмена, являются процессами с высоким
притоком и уносом тепла, разность температур, необходимая для теплопере-
теплопередачи через металл, может быть сравнительно высокой. Поэтому желательно,
чтобы сечение металла, насколько это допустимо с точки зрения давления
в приборе, было возможно меньше; в противном случае уровень температуры
водяного пара, поступающего из имеющегося источника, может быть недоста-
недостаточным для кипячения концентрированной перекиси водорода.
При работе этих кипятильников поступающая жидкость имеет ту же или
почти ту же концентрацию перекиси водорода, что и выходящий пар, хотя
концентрация перекиси в жидкости в самом кипятильнике значительно выше.
Например, при подаче в кипятильник холодной 20%-ной (по весу) перекиси
водорода, смешивающейся в нем с жидкостью, содержащей около 60 вес.%
перекиси, выходящие пары содержат 20 вес.% перекиси или ниже в зависимо-
зависимости от степени разложения. Необходимо устранить охлаждение и разбавление,
обусловленные переполнением капятильника питающей жидкостью, или излиш-
излишнее обратное конденсирование и концентрирование за счет подавления потока
пара. Для обеспечения стационарной работы аппарат, подающий жидкость, дол
жен реагировать на потребность в жидкости и обеспечивать постоянный уро-
уровень ее в кипятильнике в узких пределах. Для достижения этого целесообраз-
целесообразно заменить обычную склянку Мариотта модифицированным уравнительным
сосудом (рис. 31, в), описанным Холмсом [70]. В то же время установка обрат-
обратного холодильника в верхней части кипятильника и создание буферной систе-
системы газа исключают влияние умеренных колебаний общего давления или паде-
падения его, вызванных работой внешнего прибора, в который поступают пары пе-
перекиси. В таком аппарате [68, 69] газ от источника с регулируемым давлением
проходит через буферную емкость в гребенку, откуда ответвляются линии, веду-
ведущие: 1) к верхней части обратного холодильника, 2) к уравнительному сосуду
и 3) к трубке, погруженной на соответствующую глубину в воду для мед-
медленного непрерывного выпуска газа в атмосферу. Общее давление регулирует-
регулируется высотой столба воды над выходом из этой трубки. В обратном холодильнике
между конденсирующимся паром и буферным газом существует поверхность
раздела (обычно видимая); уровень этой поверхности изменяется в зависимости
от перепада давления вне аппарата и расхода пара; этим путем обеспечивается
постоянная скорость кипения за счет саморегулируемого изменения (внутри
известных пределов) отношения выпускаемого и конденсируемого пара. В ка-
качестве буферного газа выбран гелий, который и оправдал себя на практике,
однако, если допустимо смешение паров перекиси с посторонним газом, приме-
применение гелия не обязательно.
Вышеописанные кипятильники, приспособленные для работы при атмо-
атмосферном давлении, используются в сочетании с пароперегревателем для полу-
получения паров с температурой примерно до 250° (если только пары перекиси во-
водорода имеют концентрацию ниже предела взрывчатости). В течение длитель-
длительного промежутка времени, не опасаясь взрыва, можно получать насыщенный
или слабо перегретый (на 20°) пар с концентрацией перекиси до 70 мол.%.
При более высоких концентрациях в течение первого часа работы обычно про-
происходили случайные взрывы. Количественно границы безопасной работы не
установлены. Приведенные данные могут служить только в качестве общего
руководства и справедливы лишь для аппаратов, где пары перекиси приходят
в соприкосновение со скрупулёзно очищенными поверхностями из стекла
пирекс. Нужно иметь в виду, что взрыв паров перекиси при такого рода опытах
может быть очень интенсивным, особенно при высоких концентрациях. Персо-
Персонал, работающий с аппаратурой такого типа, должен работать за защитным
ограждением.
11 Перекись водорода
162 . Литература
ЛИТЕРАТУРА
1. Maass О., Matcher W. Ы., J. Am. Chem. Soc, 42, 2548 A920).
2 К е у e s F. G., Schumb W. С, В г о u g h t о n D. В., Р. В. report 27292
A945), Regnault H., d e Carl an R. L., Congr. chim. ind., Compt.
rend. 18-me Congr., Nancy, Sept.—Oct., 1938, p. 766 [CA., 33, 6534]; Беллинд-
ж ер Ф., Фридман Г., Бауэр В., Истс Д ж., Бул В., веб.
«Физика и химия реактивного движения», № 1, Госиноиздат, М., 1948, стр. 152.
3. Gross P.M., Jr., Taylor R. С, J. Am. Chem. Soc, 72, 2075 A950).
4. В г о d e W. С, С 1 a p p D. В., G о r m 1 e у W. G., P. B. report 13266; M с A u -
lay J., Cl a p p D. В., Slater V. W., Cooper'K. A., C.I.O.S., File
№ XXXIII-42; Chem. Eng., p. 102, August, 1948; В г e t s с h g e r M. E., Crew-
son C.G., Cushing R. E., P.B. report 17331; Slater V. W., Cl a p p
D. В., Master man S., C.I.O.S. File № XXXI-15.
5. Tucker C. W., пат. США 2049979 D. VIII 1936).
6. W о о d W. S., HolmesW.R., W h i t t a k e г Н., пат. США 2520870 B9.VIII
1950), [англ. пат. 590439 A7.VII 1947)].
7. М с A u 1 а у J., С 1 а р р D. В., Slater V. W., Cooper К. A., C.I.O.S.
File № XXXIII-45.
8. Hess L., пат. США 2088239 A9.VII 1938); Kali-Chemie A. G., белы. пат. 443883
C1. I 1942); Schmidt H., герм. пат. 687292 B8.XII 1939); Societe des produits
peroxides, франц. пат. 970103 B9.XII 1950), [англ. пат. 6688741.
9. Hawkinson А. Т., G г а 1 о w В. В., М а у И. Е., пат. США 2491732 B0. XII
1949).
10. Technical Oil Mission*Reel 32, translated and published by Meyer Charles A. and Co.,
PC-S-III, Vols. I and II A948, 1949).
11. M а с M u 1 1 i n R. В., пат. США 2298064 F.X 1942).
12. G i g u ё г e P. A., L i u I. D., D u g d a 1 e J. S., M о г г i s о n J. A., Can J.
Chem., 32, 117 A954).
13. S p r i n g W., Z. anorg. allgem. Chem., 8, 424 A895).
14. F e r n a n d e z J. V., S. M. Thesis in Chemical Engineering, M.I.Т., 1954.
15. Etat Francais (M. le Ministre de L'Armement), франц. пат. 940547 A5.XII 1948).
16. Levitan N. I., Kuoch R., пат. США 2386484 (9.X 1945); Hawkinson
А. Т., пат. США 2017440 A5.X 1935) [англ. нат. 438886A4.XI 1935)].
17. R i с h а г d son С. N.. пат. США 2438252 B3. III 1948). [канад. пат. 434211 A6.IV
1946I.
18. R e i с h е г t J. S., пат. США 2027838, 2027839 A4.1 1936); The Roessler& Hasslacher
Chem. Co., англ. пат. 409361 C0. IV 1934); E. I. duPont de Nemours 8 Co., англ.
пат. 432915 F. VIII 1935), франц. пат. 793423 B4.1 1936).
19. R e i с h e r t J. S., пат. США 2021384 A9.XI 1936), канад. пат. 367172 F.VII 1937).
20. Nees П., Р. В. report 74329 (frames 1177—1185).
21. Р a u 1 Schmidt, герм. пат. 534283 A6. VI 1929); М и 1 1 е г J., пат. США
1966102, 1966103 A0. VII 1934); Osterreische Chem. Werke G. m. b. H., герм,
пат. 588 267 A5.XI 1933).
22. Y о и n g J. H., пат. США 2676923 B7. IV 1954); duPont Co., англ. пат. 695325
E. VIII 1953); Elchemie G. m. b. H., австр. пат. 167414 A0.1 1951) [белы. пат.
503789, C0.VI 1951I.
23. R у n a s i e w i с z J., Anal. Chem., 26, 355 A954).
24. С a n t i n о Е. С, Ind. Eng. Chem., Anal. Ed., 16, 181 A944).
25. D i с k m a n S. R., Bray R. H., Ind. Eng. Chem., Anal. Ed., 12. 279 A940);
Шконде Е. И., Почвоведение, 1952, 374.
26. Davis N. S., Jr., Keefe J. H., Jr., J. Am. Rocket Society, p. 63, March-April
1952, Keefe J. H., Jr., неопубликованные сообщения.
27. Buffalo Electrochemical Co., Engineering Materials for Use with 90% Hydrogen Pero-
Peroxide, Part I, Dec. 1, 1950.
28. С h a p m a n R. M., P. B. report 34739, Nees H., P. B. reports 74329 (frames
1211—1216), 73716 (frames 116—142; 9654—9662), см. также I. G. Farbenindustrie
A. G., Evaluation of Rocket Fuel, Vols. I and II-G, T.O.M. Reel 32, Translation
PC-S-11I, перевод издан фирмой Meyer С. А. & Co., Inc., New York.
29. Беллинджер Ф., Фридман Г., Б а у э р В., Истс Дж., Бул В..в сб.
«Физика и химия реактивного движения», № 1, Госиноиздат, М., 1948, стр. 152.
30. R e i с h е г t J. S., Pete R. H., Chem. Eng., 58, № 10, 263 (X 1951).
31. R e i с h e г t J. S., M с N e i g h t S. А., пат. США 2316487 A3. IV 1943).
32. Reichert J. S., пат. США 2008726 B3. VII 1936), [герм. пат. 662536 A5. VII
1938), франц. пат. 792106 B3. XII 1935).
33. Harris J. С, Metal Cleaning Bibliographical Abstracts, ASTM Special Technical
Publication № 90, 1949.
Литература 163
34. В г е t s с h g е г М. Е., Kauflmann Ы. О., Gilbert F. А., пат. США
2219293, 2219294 B9.Х 1940).
35. Reichert J. SM HinegardnerW. S., пат. США 2224835 A0. XII 1940).
36. Etudes et applications financieres, франц. пат. 952579 B1.XI 1949).
37. Riggs W. S., Lehmann R., пат. США 2346609 A1. IV 1944).
38. Bretschger M. E., пат. США 2056894 F.X 1936).
39. U hi i g H. H., Trans. Electrochem. Soc, 78, 265 A940).
40. T h о m p s о n J. В., Nielsen G. C, Chem. Eng., 60, № 11, 196 (XI 1953).
41. Doyle J. R., неопубликованная работа M.I.Т. Hydrogen Peroxide Project, 1950'.
42. Huckaba С E., Reyes F. G., J. Am. Chem. Soc, 70, 2578 A948).
43. Schumb W. C, Ind. Eng. Chem., 41, 992 A949).
44. Lincoln E. С, неопубликованная работа M.I.Т. Hydrogen Peroxide Project-
1953.
45. Arntzen С E., Rowley R. D., Materials and Methods, 35, Л° 1, 82 A952).
46. Tarn res M., Frost A. A., J. Am. Chem. Soc, 72, 5340 A950).
47. С о о k G. А., пат. США 2368806, 2368640 F. II 1945), 2416156 A8.11 1947).
48. S h a n 1 e у E. S., Ind. Eng. Chem., 45; 1520 A953).
49. Tschinkel J. G., пат. США 2627454 C. II 1953).
50. Sax N. I., Handbook of Dangerous Materials, Reinhold Publishing Corp., New York,
1951.
51. Mfg. Chemists* Assoc, Chem. Safety Data Sheet, Manual Sheet SD-53 A953).
52. E d en G. E., Freke A.M., Melbourne K. V., Chem. and Ind. 1951, 1104.
53. К г а с k о w E. H., Health Hazards of Military Chemicals, Report № 4, Medical
Division, Army Chemical Center, 1950; С о m s t о с k С. С, Hackley E. В.,
Oberst F. W., Lawson L., Greene E., Medical Laboratories Research
Report № 243, Army Chemical Center, 1954.
54. Ш е н л и Э., Гринспэн Ф., в сб. «Физика и химия реактивного движения»,
№ 2, Издатинлит, М., 1949, стр. 161.
55. Bretschger M. E., Shanley E. S., Trans. Electrochem. Soc, 92, 67 A947).
56. М е д а р Д., в сб. «Физика и химия реактивного движения», № 1, Госиноиздат, М.,
1948, стр. 197; М е d а г d L., Mem. Poudres, 31, 273 A949).
57. К у рб а н га л и на Р. X., ЖФХ, 22, 49 A948).
58. Haeuscler E., Explosive Stoffe, 1953, 64.
59. Саттерфилд Ч., Каванаг Дж., Резник Г., в сб. «Вопросы ракет-
ракетной техники», № 4 A6), 90 A953); Satterfield С. N.. С е с с о t t i P. J.,
Feldbrugge A. H. R., Ind. Eng. Chem., 47, 1040 A955).
60. E 1 d e r L. W., R i d e a 1 E. K., Trans. Faraday Soc, 23, 545 A927); Baker
В. Е., О u e 1 1 e t C, Can. J. Res., 23B, 167 A945); Mackenzie R. C,
Ritchie M., Proc Roy. Soc, A185, 207 A946); G i g u e r e P. A., Can. J.
Res., 25B, 135 A947).
61. В a t z о 1 d J. S., L u n e г С, W i n k 1 e г С. A., Can. J. Chem., 31, 262 A953);
Jones R. A., W i n k 1 e г С A., ibid., 29, 1010 A951).
62. Ройтср В. А., Г а у x м а н С. С, ЖФХ, 4, 465 A933).
63. Scatchard G., К a v a n a g h G. M., T i с k n о г L. В., J. Am. Chem. Soc,
74, 3715 A952).
64. Frost А. А., О 1 d e n b e г g O., J. Chem. Phys. 4, 781 A936).
05. Volman D. II., J. Am. Chem. Soc, 73, 1018 A951); Stone F. S., Taylor
11. S., J. Chem. Phys., 20, 1339 A952).
66. К a v a n a g h G. M., Buchorn К. С, неопубликованная работа, The M.I. Т.
Hydrogen Peroxide Project, 1947.
67. W e n t w о г t h R. L., S. M. Thesis in Chemical Engineering, МЛ.Т., 1948.
68. M e e k e n S. R., S. B. Thesis in Chemical Engineering, M I. T., 1950.
(i9. Resnick П., Sc D. Thesis in Chemical Engineering, M.I.Т., 1952; Satter-
Satterfield С N., Resnick H., Wentworth R. L., Chem. Eng. Progress,
50, 460 A954).
70 Holmes F. E., Ind. Eng. Chem., Anal. Ed., 12, 483 A940).
II*
Глава 5
ФИЗИЧЕСКИЕ СВОЙСТВА
В настоящее время измерены или могут быть оценены все существенные
свойства перекиси водорода и ее водных растворов. За последнее время многие
из физических свойств вновь подвергнуты проверке, что в связи с применением
усовершенствованной техники и возможностью сравнения с ранее опубликован-
опубликованными величинами повысило их надежность. В этой главе приводятся рекомен-
рекомендуемые значения свойств перекиси водорода и ее водных растворов; весь мате-
материал разбит по разделам, относящимся к физическим, термодинамическим,
электрическим и радиационным свойствам. При рассмотрении каждого свой-
свойства описаны использованные экспериментальные методы, достигнутая точ-
точность и выбрано надлежащее значение из результатов работ различных экспе-
экспериментаторов. Там, где возможно, приведены данные оригинала в виде таб-
таблиц. При описании наиболее важных свойств или в тех случаях, когда опубли-
опубликованные данные вызывают некоторые сомнения, мы старались возможно
подробнее их проанализировать, чтобы читатель мог сам вынести окончательное
суждение. Ввиду довольно ограниченного количества литературных источников
по физическим и термодинамическим свойствам процитированы почти все
опубликованные сообщения. Таким образом, поскольку знакомство с каким-
либо источником может представлять известный интерес, здесь кратко цитирует-
цитируется также ряд устаревших значений физических свойств. Для иллюстрации раз-
различных зависимостей даны рисунки и кривые, но, за редкими исключениями,
эти рисунки не настолько точны, чтобы при практическом применении они
могли заменить табличные данные. Для этой цели можно составить необходи-
необходимые графики на основании табличных данных или же использовать компиля-
компиляции типа опубликованных фирмой «Buffalo Electro-Chemical Co.» [1].
В повседневной практике, как правило, необходимо располагать значения-
значениями свойств водных растворов перекиси водорода. Мы не останавливаемся от-
отдельно на свойствах безводной перекиси водорода, но и для нее в целях срав-
сравнения со свойствами перекиси дейтерия в виде таблиц приведены данные,
касающиеся ограниченного числа свойств. Если отдельного обсуждения не
требуется, то свойства безводной перекиси водорода представлены повсюду
как предельные значения свойств ряда водных растворов перекиси. В,качестве
других предельных значений использованы значения свойств чистой воды.
Соответствующие данные иногда определялись при исследовании растворов
перекиси водорода; если эти значения кажутся ненадежными, можно пользо-
пользоваться величинами, опубликованными такими авторитетами, как Дорси [2]
или Кинен и Кейс [3].
Проведены также некоторые измерения свойств систем, содержащих, поми-
помимо перекиси водорода и воды, еще и другие компоненты. Результаты этих из-
измерений кратко рассматриваются в одном из разделов этой главы (стр. 248).
Отдельно представлены также физические свойства перекиси водорода,
отличающейся по изотопному составу от обыкновенной; в настоящее время та-
такие сведения имеются лишь по перекиси водорода, обогащенной перекисью
дейтерия. Для возможности сравнения и быстрого отыскания соответствующих
Физические свойства
165
значений на стр. 256 дана краткая сводная табл. 66, в которой суммируются
рекомендуемые величины по избранным свойствам воды, тяжелой воды, без-
безводной перекиси водорода и безводной перекиси дейтерия.
Рассмотрение физических и термодинамических свойств ограничено в ос-
основном экспериментально измеренными величинами. Молекулярная структура
и физическая природа перекиси водорода в различных фазах и в растворе обсу-
обсуждаются в гл. 6, где представлены также дополнительные сведения, выведен-
выведенные из результатов измерений физических свойств и первичных эксперименталь-
экспериментальных данных, полученных с основной целью выяснить структуру.
Обозначения концентрации
В таблицах свойств концентрация перекиси водорода выражена на основе
соотношений весовых или мольных. В литературе по перекиси водорода часто
применяется неудачное выражение «объемная крепость» (volume strength) для
обозначения концентрации. Под этим понимается измеренный при 0° и 1 am
объем газообразного кислорода, выделяющегося при полном разложении од-
одного объема раствора перекиси водорода, также измеренного в стандартных
условиях. Таким образом, объемная крепость раствора перекиси водорода
3%-ной концентрации равна 10, объемная же крепость безводной перекиси
составляет 484,62. Пользование этой системой затрудняется еще и тем, что в ста-
старой американской литературе при обозначении объемной крепости исходили
из объема кислорода, выделяющегося не только в результате разложения са-
самой перекиси водорода, но и за счет реакции перекиси с перманганатом калия
в кислом растворе. Выражение «объемная крепость» сейчас употребляется ре-
реже, чем раньше, и от него лучше отказаться. То же можно сказать и о выра-
выражении «объемные проценты» (объемы безводной перекиси водорода на 1 объем
раствора или иногда на 1 объем чистой воды). Приведенные ниже равенства*
Эквивалентные значения для
различных выражений концентрации
перекиси водорода а
Таблица 3
водных растворов
•
W,
вес. %
0
10
20
30
40
50
60
70
80
90
100
xh
молярная
доля Н2О2
0,0000
0,0556
0,1169
0,1850
0,2610
0,3462
0,4427
0,5527
0,6793
0,8266
1,0000
Молярность
с^ при 25°,
моль! л
0,000
' 3,034
6,286
9,770
13,505
17,511
21,809
26,421
31,373
36,692
42,404
Моляльность
mh,
моль! кг
воды
0,000
3,266
7,349
12,599
19,599
29,398
44,097
68,595
117,59
264,58
оо
V
объемная
крепость
0,00
34,03
71,19
110,96
153,68
199,49
248,66
301,46
358,17
419,16
484,62
м
молекуляр-
молекулярный вес
18,02
18,90
19,88
20,98
22,19
23,55
25,10
26,86
28,88
31,24
34,02
а В табл. 72 (стр. 328) приводятся активности перекиси водорода в водном растворе.
Расшифровку буквенных обозначений см. в приложении на стр. 557.
166 Глава 5
дают взаимную зависимость между различными концентрациями (выраженны-
(выраженными как функции молекулярных весов компонентов) и плотностями растворов:
mh
1000-\- mhlAw 1000p - chMh + chM
!Ah (\00 —W)-\-WMw [A00р0МЛ)B2414/2)] —VMft+VMhMu," ^
Путем перегруппировки уравнения A) получаются следующие удобные
соотношения:
w __\00N-0,0\7 _ \00lAhxh (С?.
W ~~ р \
1000^
m
В табл. 3 приведены эквивалентные значения для различных единиц кон-
концентрации, причем для тех свойств, которые зависят от температуры, указаны
величины, измеренные при 25°. Нормальность N, определяемая как число экви-
эквивалентов перекиси водорода, содержащихся в 1 л раствора, может быть выведе-
выведена из табл. 3 путем умножения молярности на 2. В таблице приведены также
молекулярные веса растворов.
Если в этой книге концентрация перекиси водорода указана в процентах,
то при отсутствии других указаний подразумеваются весовые проценты.
Формульный и молекулярный вес перекиси водорода
Можно считать, что эмпирическая формула перекиси водорода, как в свое
время установил Тенар, является формулой соединения, в котором отношение
кислорода к водороду в два раза больше, чем в воде. Это на основе фор-
формулы Н2О для воды привело для перекиси к формуле НО.
Тамман [4], Каррара [5] и Орндорф и Уайт [6] провели опыты по опреде-
определению молекулярного веса перекиси водорода путем измерения температур за-
замерзания ее водных растворов. Растворы перекиси водорода в воде приближен-
приближенно соответствуют идеальным растворам только в пределах ограниченного интер-
интервала составов; кроме того, на точность определения точки замерзания серьезно
влияет разложение перекиси. Тем не менее эти опыты вполне достоверно
доказали, что значение молекулярного веса перекиси водорода приблизи-
приблизительно равно 34. Сравнение вычисленных и экспериментально измеренных
температур замерзания разбавленных растворов перекиси водорода, проведен-
проведенное Жигером и Маасом [7] и Фоли и Жигером [8], подтверждает эти выводы
авторов старых работ.
Матесон и Маас [9] измерили плотность пара безводной перекиси водо-
водорода при 92° и давлении, равном нескольким сантиметрам ртутного столба, по
методу Виктора Мейера. Результаты измерений также приводят к молекуляр-
молекулярному весу 34 и показывают, что пар йерекиси не ассоциирован. Если исхо-
исходить из принятых химических атомных весов [10] 1,0080 для водорода и
16,0000 для кислорода, то молекулярный вес перекиси водорода составит
34,0160.
При рассмотрении природы жидкой перекиси водорода и ее водных раство-
растворов становится очевидным существование сил притяжения между молекулами
одной перекиси водорода и между молекулами воды, с одной стороны, и пере-
перекиси водорода—с другой; эти силы притяжения приписываются существова-
существованию водородных связей. О существовании таких связей можно предполагать
в связи с ассоциацией отдельных молекул Н2О2 и Н2О; действительно, можно
считать, что раствор перекиси водорода в воде содержит полимеры и сополи-
сополимеры (Н2О2)д;, (Н,О)у и (Н2О)у-(Н2О2)х, причем на долю отдельных таких поли-
Физические свойства 167
меров падает значительная часть взятой пробы. Водородные связи, соединяю-
соединяющие эти мономеры в полимерах, являются сравнительно прочными для межмо-
межмолекулярных связей, но гораздо более слабыми, чем химические связи, соединя-
соединяющие между собою водород и кислород в мономерах; кроме того, эти связи но-
носят переходящий характер; они непрерывно образуются, разрываются за счет
тепловых движений и вновь возникают. Наличие таких полимеров или боль-
больших молекул не находит отражения при определениях температур замерзания;
эффективной единицей для перекиси водорода является Н2О2, а ассоциация за
счет водородных связей отражает лишь межмолекулярные силы, характерные
для жидкости.
Плотность твердой перекиси водорода
Маас и Хэтчер [11] определили в дилатомере плотность твердой безводной
перекиси водорода A,6434 г/см3 при —4,45° и 1,6437 г/'см3 при —7,45°). Срав-
Сравнительно недавно Абрагэмс, Коллин и Липскомб [12] рентгенографическим
методом определили кристаллическую структуру безводной перекиси водорода
при —20°. Объем тетрагональной кристаллографической элементарной ячейки,
содержащей 4 молекулы, оказался равным 131,9А3. Из этих данных ими выве-
выведена плотность для перекиси 1,70 г/см3. Авторы настоящей книги вновь вычис-
вычислили эти значения, использовав уравнение Брегга [13] (приняв для числа
Авогадро [14] значение 6,023-1023). С учетом возможных ошибок измерения для
плотности при —20°* получено значение 1,71^q'q3 г/см3. Рекомендуется при-
применять это значение.
Данные для термического коэффициента объемного расширения твердой
безводной перекиси водорода отсутствуют. Коэффициент объемного расши-
расширения льда, полученного из воды', по Дорси [2], равен 1,53-10 град г. Если
принять, что коэффициент объемного расширения твердой перекиси водорода
имеет величину того же порядка, что и соответствующий коэффициент льда,
полученного из воды, то его можно использовать для определения темпера-
температурного интервала, допускаемого возможной погрешностью 0,08 г/см3 для плот-
плотности твердой перекиси водорода. На этом основании найдено, что для измене-
изменения плотности на величину, соответствующую возможной погрешности в ука-
указанном значении, необходима разность температур, равная интервалу от точки
замерзания до температуры, лежащей заметно ниже эвтектических температур
для системы вода—перекись водорода. До опубликования результатов новых
измерений необходимо для всех температур применять рекомендуемую
плотность.
Жигер и Жоффрион [16] измерили изменения объема, наблюдаемые при
замораживании водных растворов перекиси водорода в дилатометре. В резуль-
результатах измерений была некоторая несогласованность, но все же они совершенно
отчетливо показали, что при замерзании растворы, содержащие меньше
45 вес.% перекиси водорода, расширяются, а растворы, содержащие больше
65 вес.%, сжимаются. Таким образом, растворы ведут себя, как тот компонент,
который содержится в них в большем относительном количестве. По относи-
относительной плотности твердого соединения Н2О2-2Н2О D8,6 вес.% перекиси водо-
водорода) нельзя сделать никаких выводов.
При обсуждении фазовых соотношений в этой системе (см. стр. 181 и ел.)
цитируются работы, доказывающие отсутствие образования истинных твер-
* Таким образом, плотность безводной перекиси водорода возрастает при замерза-
замерзании, изменяясь от 1,47 г/см3 для жидкости при температуре ее замерзания до 1,71 г/см3
для твердого тела. В этом отношении перекись водорода ближе к большинству других
веществ и отличается от воды, которая становится менее плотной при замерзании (плот-
(плотность изменяется от 0,9999 г/мл для жидкости до 0,9167 г/мл для твердой фазы при 0°
1151).
168 Глава 5
дых ратворов. Тем не менее точно установлено, что твердые фазы, образующие-
образующиеся при замерзании водных растворов перекиси водорода, обычно содержат оба
компонента системы. Напрашивается вывод, что твердая фаза окклюдирует ма-
маточный раствор. Объем, занимаемый частично замороженным раствором пере-
перекиси водорода, должен поэтому представлять сумму объемов однокомпонентной
твердой фазы и раствора, в котором эта фаза содержится. Объемы, вычислен-
вычисленные на этой основе по методу конод (по которому количества твердой и жидкой
фаз, присутствующие при данной температуре, выводятся из материального
баланса для воды и перекиси водорода по фазовой диаграмме), хорошо согла-
согласуются с измеренными объемами в работе Жигера и Жоффриона. Объемы, вычис-
вычисленные для растворов, богатых перекисью водорода, могут быть уточнены путем
использования приведенной выше исправленной плотности твердой безводной
перекиси. Остается все же некоторая неточность, поскольку вычисленный
объем не изменяется в заметной степени с составом или плотностью твердой
фазы. Однако с практической точки зрения вычисленные по этому методу объемы
не очень значительно отличаются от истинных.
Плотность жидкой перекиси водорода
В литературе имеются данные ряда определений плотности жидкой пере-
перекиси водорода и ее водных растворов [17]. Однако они либо охватывают недо-
недостаточный интервал составов и температур, либо точность их неизвестна.
Первыми, достаточно удовлетворительными данными, полученными при измере-
измерениях плотности, нужно считать данные Мааса и Хэтчера [11], опубликованные
в 1920 г. и впоследствии исправленные для концентрированных растворов Кат-
бертсоном, Матесоном и Маасом [18]. После 1948 г. опубликованы результаты
новых измерений плотности жидкой перекиси водорода Хукаба и Кейсом [19]
при 0 и 20° для всех составов, Кубашевским и Вебером [20] при 18° и для со-
составов, содержащих до 82 вес.% перекиси, Жигером и Жоффрионом [16] при
температурах от —50 до 0° для всех составов и Истоном, Митчелом и Уинн-
Джонсом [21] для 0, 10, 25, 50 и 96° для любых составов.
Ниже вкратце рассмотрены экспериментальные методы, часть исходных
данных, соотношения между результатами и вероятная точность для измерений
плотности, проведенных Хукаба и Кейсом, Жигером и Жоффрионом и Истоном,
Митчелом и Уинн-Джонсом. Величины плотности, которые рекомендуются для
любых требований, за исключением наиболее точных, можно взять либо из
рис. 32 (стр. 173), либо путем интерполирования данных табл. 12 (стр. 174)
с использованием коэффициентов объемного расширения, приведенных в этой
таблице для уравнения D), с целью введения поправки на температуру. Для
более точных или специальных целей можно рекомендовать аналитическое
выражение, а именно уравнение F), которое соответствует всем приведенным
выше данным. Таким образом, мы располагаем точными значениями плотности
водных растворов перекиси водорода для полного ряда составов при темпера-
температурах от —50 до 96°. По сравнению с другими значениями данные этих трех
групп авторов, принимавших при измерениях более тщательные меры к устра-
устранению разложения, являются более точными. Более высокие величины, полу-
полученные этими авторами, доказывают, что при измерениях Мааса и Хэтчера
и Кубашевского и Вебера происходило значительное разложение перекиси.
Хукаба и Кейс затратили много усилий для разработки технических деталей,
исключающих возможность разложения перекиси в их приборе; "кроме того,
они специально изучили [22] точность использованного ими аналитического
метода, а поэтому их данные и другие согласующиеся с ними значения сле-
следует предпочесть.
Жигер и Жоффрион измерили плотность различных растворов при 0°,
а также вблизи температуры замерзания. Из этих измерений выведены сред-
Физические свойства
169
ние коэффициенты объемного расширения для интервалов температур от 0°
и ниже до области температур замерзания растворов. Результаты их дилатоме-
дилатометрических измерений, согласующиеся в пределах 0,1% с данными Хукаба
и Кейса при 0°, приведены в табл. 4. Коэффициент объемного расширения опре-
определяется по уравнению
Плотность растворов перекиси водорода при 0°
расширения при температурах ниже 0°,
-',)]• D)
Таблица 4
и средние коэффициенты объемного
по Жигеру и Жоффриону [16]
н2о2,
вес.%
9,32
10,21
12,21
18,32
19,27
29,54
41,23
46,72
Плот-
Плотность р
при 0°,
г [мл
1,0377
1,0410
1,0493
1,0748
1,0783
1,1209
1,1722
Средний коэф-
коэффициент объ-
объемного расши-
расширения р,
град.-1-104
1,3±0,5
—
3,24-0,3
4,6+0,2
5,5440,09
5,8140,09
Изученный тем-
температурный
интервал
А/,°С ниже 0°
8,1
14,9
20,0
50,0
50,0
н2о2,
вес.%
49,87
68,37
69,05
78,61
79,63
89,64
100,00"
Плот-
Плотность п
при 0°,
г 1мл
1,2119
1,3006
1,3037
1,3535
1,3590
1,4136 __
Т,4630а
Средний коэф-
коэффициент объ-
объемного расши-
расширения {$,
град.~1 • 1 О4
6,034-0,11
6,7
.
7,1+0,2
—
_ 7,54-0,4
7,9а
Изученный тем-
температурный
интервал
М,СС ниже 0°
50,0
35,0
25,0
—
10,0
Около точки
замерзания
1 Экстраполированная величина.
Уравнение D) может быть использовано для вычисления удельного объе-
объема vtz при температуре t2 из известных значений удельного объема Vtx при
какой-либо основной температуре с использованием данного ря^а коэффициен-
коэффициентов расширения. При помощи этих коэффициентов нельзя выполнить обрат-
обратную задачу, т. е. найти vh из vi2.
Таблица 5
Плотности растворов перекиси водорода
при °С, по Хукаба и Кейсу [19]
н2о2,
вес.%
[0,000
9,657
19,979
40,009
59,171
71,354
80,023
Измеренная
плотность
0,9998
1,0379
1,0803
1,1660
1,2539
1,3139
1,3590
89,379
95,987
96,228
96,282
99,479
99,605
100,0
Измеренная
плотность
о, г\см*
1,4100 '
1,4483
1 ,4493
1,4499
1,4681
1,4685
1,4709аЧ
аЭкстраполированная величина.
В табл. 5 приведены значения плотности водных растворов перекиси водо-
водорода при 0°, определенные в специальных пикнометрах Хукаба и Кейсом [19].
Воспроизводимость измерения плотности равна 0,01%, анализов 0,02%.
170
Глава 5
Данные табл. 5 могут быть выражены через удельные объемы с максимальным
отклонением около 0,25% уравнением
\gv = 0,00009 - 0, \676w. E)
Хукаба и Кейс приводят также таблицы плотностей и удельных объемов (при
0° через интервалы, равные 1 вес.%), вычисленных по уравнению E) в сочета-
сочетании с диаграммой отклонений, что позволяет сохранить максимальную точ-
точность для всех данных.
Таблица 6
Удельные объемы растворов перекиси
водорода при 20° и средние коэффициен-
коэффициенты объемного расширения в пределах
от 0 до 20°, по Хукаба и Кейсу [19]
22,
вес.%
0,00
21,525
39,159
59,083
78,529
96,877
100,000
Эксперимен-
Экспериментальный
удельный
объем
v, см*\г
1,00184а
0,92818
0,87037
0,80836
0,75103
0,69888
Средний коэффи-
коэффициент объемного
расширения,
?•10*, град.-1
0,83
4,326
5,846
6,656
7,344
7,759
7,85
а По Дорси [2].
Хукаба и Кейс определили также плотности растворов перекиси водорода
при 20° дилатометрическим методом и на основании этих измерений вывели
средние коэффициенты объемного расширения в пределах от 0 до 20°. В табл. 6
представлены удельные объемы, измеренные при 20°, и средние коэффициенты
объемного расширения в пределах от 0 до 20°. Коэффициенты объемного рас-
расширения, определенные в соответствии с уравнением D), вычислены из обще-
общего уравнения для всего интервала температур от 0 до 20°; Хукаба и Кейс
дают выравненные значения для этого коэффициента через интервалы, равные
1 вес.% перекиси водорода.
Измерения Истона, Митчела и Уинн-Джонса [21], проведенные при тем-
температурах 0, 10, 25, 50 и 96°, дают возможность иметь величины плотности жид-
жидкости и при более высоких температурах. Эти авторы, пользуясь для измере-
измерений усовершенствованным прибором типа весов Вестфаля, определяли вытес-
вытеснение жидкости стеклянным поплавком. Особое внимание было уделено очист-
очистке стеклянной посуды с целью устранения основного источника ошибок, с ко-
которым связано применение данного метода, а именно: образования пузырьков
на поплавке при разложении перекиси. В процессе измерений при 96° к раство-
растворам перекиси в качестве стабилизатора добавляли сульфат цинка в количестве
81 мг/л.
В табл. 7 приведены экспериментальные результаты, полученные Истоном,
Митчелом и Уинн-Джонсом при 0°. Эти данные прекрасно согласуются с дан-
данными, полученными Хукаба и Кейсом при 0 и 20—25°. Истон, Митчел и Уинн-
Джонс считают, что данные, полученные ими при 25°, имеют точность около
0,02%, а при 96°—0,2%. Данные Истона, Митчела и Уинн-Джонса могут
быть выражены при помощи уравнения
F)
Физические свойства
171
Таблица 7
Плотности растворов перекиси
водорода при 0°, по Истону,
Митчелу и Уинн-Джонсу [21]
Таблица 8
Коэффициенты уравнения F), выражающего
зависимость плотности от весового процента
перекиси водорода, по Истону, Митчелу
и Уинн-Джонсу [21]
НоОо,
вес.%
0,0
7,36
17,14
24,36
33,23
44,62
Плот-
Плотность р,
0,9999
1,0291
1,0690
1,0987
1,1370
1,1860
НоОо,
вес.%
55,69
65,07
74,96
83,27
91,55
96,65
Плот-
Плотность о,
1,2373
1,2822
1,3321
1,3759
1,4221
1,4517
Темпера-
Температура. °С
0
10
25
50
96
0
0
0
0
0
а
,9998
,9997
,9970
,9880
,9612
ь
0,39939
0,36790
0,34672
0,31382
0,27652
с
0,01758
0,06208
0,06995
0,09402
0,11956
0
0
0
0
d
,05470
,02954
,02885
,01910
—
причем согласованность экспериментальных данных, вычисленных по этому
уравнению, составляла около 0,02% для всех температур (кроме 96°, где совпа-
совпадение было лишь 0,07%). В табл. 8 приведены коэффициенты для уравнения F).
Уинн-Джонс [23] провел еще более точные исследования для разбавлен-
разбавленных растворов перекиси водорода (от 0 до 5 вес.%). Пользуясь дилатометром,
он определил плотности разбавленных растворов при 0, 10 и 25° с точностью
порядка 0,01%. Результаты этих измерений приведены в табл. 9 вместе с соот-
соответствующими эмпирическими уравнениями, которыми, однако, можно поль-
пользоваться лишь при концентрациях не выше 5 вес.%.
Таблица 9
Плотность разбавленных растворов
при 0, 10 и 25°
Концентрация Н2Оо
вес.%
0
2,242
3,332
4,971
молярная
доля
0
0,012001
0,017928
0,026958
перекиси водорода
[23]
Плотностьа, г'смЗ
0°
0,999849
1,008725
1,013097
1,019626
10°
0,999680
1,008002
1,012109
1,018265
2 5°
0,997044
1,004835
1,008639
1,014289
а Уравнения, удовлетворяющие этим данным, имеют следующий вид:
Р0о=0,999841+0,3975 w,
pi0o=0,99t9700+0,3725 w,
Р25°=О,997044+0,34725 w.
Для вывода общего уравнения для плотности растворов перекиси водорода
при всех температурах от 0 до 96° и любой концентрации коэффициенты уравне-
уравнения F) можно выразить как функцию температуры. В табл. 10 приведены
коэффициенты уравнений:
G)
(8)
*, (9)
где b, с и d коэффициенты уравнения F).
172
Глава 5
Таблица 10
Коэффициенты уравнений G)—(9) для вывода коэффициентов
уравнения F) в
Коэффициент
уравнения F)
«а
b
с
d
J
0,39763
0,02206
0,05187
зависимости
К • 1 03
-2,8732
3,5357
-1,9414
от температуры
L- 105
3,2488
-6,0947
3,9061
М- 107
-1,6363
3,6165
-2,5500
а а—плотность воды при температуре/.
Коэффициент а в уравнении F) представляет просто плотность воды при
соответствующей температуре, и эту величину проще всего взять по соответству-
соответствующей таблице [2]. Уравнение F) в сочетании с коэффициентами, вычисленными
из уравнений G)—(9), очень близко соответствует точности, указанной Истоном,
Митчелом и Уинн-Джонсом для уравнения F), если применить коэффициенты
для температур, при которых эти авторы проводили измерения. Отклонения
можно видеть, рассматривая данные в табл. 11. Эти отклонения являются так-
Таблица 11
Отклонение уравнения F) от экспериментальных данных при применении общих
коэффициентов табл. 10
Темпера-
Температура, СС
0
0
18
25
96
Среднее отклонение
абс.
0,00034
0,0015
0,0023
0,0003
0,0008
0,026
0,117
0,195
0,022
0,067
Максимальное отклонение
абс.
0,0006
0,0025
0,0056
0,0007
0,0016
%
0,058
0,184
0,478
0,056
0,144
Литература
Хукаба и Кейс [19]
Жигер и Жоффрион [16]
Кубашевский и Вебер [20]
Истон, Митчел и Уинн-
Джонс [21]
То же
же мерой согласованности различных данных. Применять уравнение F) в со-
сочетании с общими коэффициентами можно рекомендовать в тех случаях, когда
желательно знать точные значения плотности при температурах и концентра-
концентрациях, заметно отходящих от экспериментальных условий, или при введении
поправок, основанных на оригинальных данных.
Там, где не требуется большой точности, для определения плотности
водных растворов перекиси водорода при 25° и коэффициентов объемного рас-
расширения [для подстановки в уравнение D)] в интервале от 0 до 96° можно поль-
пользоваться данными, взятыми из табл. 12. Табл. 4 (стр. 169) дает коэффициенты
объемного расширения для температур ниже 0°. Для нахождения плотностей
графическим методом можно также использовать рис. 32. Линейная интерполя-
интерполяция между значениями, показанными в табл. 12 или отсчитанными из рис. 32,
точна в пределах 0,1%. Серийные объемные или денситометрические анализы
обычно не требуют большей точности.
Рассмотрение данных измерений плотности водных растворов перекиси
водорода показывает, что кривые, выражающие зависимость плотности от
100-
о
15 20 25
Температура, °С
Рис. 32. Плотность водных растворов перекиси водорода.
0,2
О
-0,2
^ '0,6
Со
I?? -0,8
/
/
'—-^
f
—^
г—-
Вода
^ —
'О 07 02 05 04 Q5 Q5 Q7 0» о5 ^Ь
Мольная доля Н2О2
Рис. 33. Парциальные моляльные объемы воды и пере-
перекиси водорода при 25°.
174
Глава 5
Таблица 12
Плотность и средние коэффициенты ^объемного [расширения растворов перекиси водорода
при 0 и 25°
НоОо,
вес.%
0
5
10
15
20
25
30
35
40
45
50
i ПЛОТНОСТЬ'?, ZlCM*
при 0°
[19]
0,9998
1,0193
1,0393
1,0598
1,0804
1,1014
1,1226
1,1441
1,1661
1,1883
[,2110
при25°
[21]
0,9971
1,0145
1,0324
1,0507
1,0694
1,0885
1,1081
1,1282
1,1487
1,1698
1,1914
^Сре д н и й ~_ крэфф и ц и -^
ента объемного рас-
расширения C,
град.-
[Т0—2 5°
[19]
0,83
. 1,97
2,92
3,61
4,21
4,70
5,14
5,50
5,83
6,11
6,36
-1-104 ",.
25—96°
[21] j
5,25
5,57
5,91
6,26
6,56
6,82
7,05
7,26
7,46
7,64
7,80
Н2О2,
вес.%
55
60
65
70
75
80
85
90
95
100
Плотность о, г/см$
при 0°
[19]
1,2342
,2579
,2822
,3071
,3326
,3589
,3858
,4136
,4421
,4709
при25°
[21]
1,2137
1,2364
1,2598
1,2839
1,3086
1,3339
,3600
1,3867
,4142
1,4425
Средний коэффици-
ента объемного рас-
расширения C,
град.-1.1 0*
0—25°
[19]
6,57
6,77
6,95
7,11
7,26
7,40
7,53
7,65
7,75
7,85
25—96°
[21]
7,93
8,04
8,15
8,24
8,34
8,44
8,50
8,53
8,56
8,58
а Для подстановки в уравнение D); для температур ниже 0° используются данные табл. 4.
концентрации, во всех случаях обращены вогнутостью вверх. Эта кривизна де-
делается все более отчетливой по мере возрастания температуры. Кривые зави-
зависимости удельного объема от концентрации перекиси водорода также обраще-
обращены вогнутостью вверх, но в данном случае кривизна уменьшается по мере
увеличения температуры. Отсюда видно, что сумма объемов несмешанных
исходных компонентов больше объема смеси. Так, при 0° при смешении одина-
одинаковых весовых количеств перекиси водорода и воды происходит сжатие на
1,7%; при 96° это сжатие равно лишь 0,5%. Неидеальность этих растворов до-
доказывается также отрицательным отклонением парциальных моляльных объе-
объемов воды и перекиси водорода в растворе по сравнению с моляльными объема-
объемами чистых компонентов. На рис. 33 дано значение (v—vo)> т. е. разность
между парциальным моляльным объемом каждого компонента в растворе и мо-
ляльным объемом чистого компонента, вычисленным по уравнению F), при 25°.
Опубликованы аналогичные кривые и для других температур [24].
Коэффициенты объемного расширения растворов перекиси водорода и чи-
чистой перекиси больше соответствующих коэффициентов воды. Таким образом,
рост объема с повышением температуры тем больше, чем больше концентрация
раствора. Эффект, обусловленный наличием максимума плотности чистой
воды при 4°, и влияние его на плотность растворов перекиси водорода изучены
Митчелом и Уинн-Джонсом [24]. Показано, что максимум плотности воды
сохраняется и в разбавленных растворах перекиси водорода. По мере роста
концентрации перекиси водорода снижается температура, при которой наблю-
наблюдается максимум. Это понижение является почти линейной функцией концен-
концентрации. Максимум обнаруживается при 0° для 2,5%-ной концентрации переки-
перекиси водорода, причем методом экстраполяции можно показать, что при —2°
и 3,5%-ной перекиси водорода он должен находиться на кривой замерзания.
Плотность пара перекиси водорода
Измерения плотности пара безводной перекиси водорода,., проведенные
Матесоном и Маасом [9], показали только то, что в условиях опыта, а именно
Физические свойства
175
при 92° и 3,5 см рт. ст., закон идеальных газов соблюдается с очень большим
приближением. Впоследствии Элдер и Ридл [25] подтвердили этот вывод.
В литературе нет никаких данных о других опытах, проведенных с целью
прямого обнаружения отклонений от закона идеальных газов, что по понят-
понятным причинам связано с трудностями проведения точных измерений над
парами, содержащими значительную концентрацию перекиси водорода, в
условиях, в которых могли бы наблюдаться отклонения от этого закона. По
аналогии с водой можно рекомендовать применение закона идеальных газов
при обычных температурах и давлениях с использованием средних молекуляр-
молекулярных весов для смесей перекиси водорода и воды. Если необходимо приближенно
подсчитать отклонения от идеальности для паров, можно использовать диаграм-
диаграмму коэффициентов сжимаемости или, что лучше, вычислить отклонения для воды
по Кинану и Кейсу [3] и приложить их к перекиси водорода и смесям ее с ве-
ведой с использованием теории соответственных состояний. Для этой цели можно
исходить из следующих критических величин для безводного пара перекиси
водорода: Гс=457° С G30° К) и Рс=214 am (см. ниже). Скэтчард, Каванаф
и Тикнор [26], пользуясь этим методом и уравнениями Кейса, Смита и Джер-
Джерри [27], подсчитали, что отклонения от закона идеальных газов могли доходить
до 1 % общего давления пара при некоторых концентрациях паров при их изме-
измерениях давления паров водных растворов перекиси водорода. Для определения
отклонений перекиси водорода от закона идеальных газов использовано также
уравнение состояния Вертело с эмпирически определенными константами [28].
Вязкость жидкой перекиси водорода
Маас и Хэтчер [ШиФиббс и Жигер [29] измеряли вязкость растворов
перекиси водорода при помощи вискозиметра Оствальда, калиброванного по во-
воде. Оба ряда данных хорошо согласуются; данные Фиббса и Жигера, приведен-
приведенные в табл. 13, можно рекомендо-
рекомендовать для практического употреб- zft
ления, так как указанные авторы
пользовались новыми, более точ-
точными значениями вязкости воды и, v цц
возможно, применяли также более й '
чистую перекись водорода. Неко- 8
торые отрывочные данные [30], g^'
полученные при 25° по неизвест- Б
ному методу, не сходятся с реко- §
мендуемыми цифрами. sj4
На рис. 34 можно видеть, | '
что вязкость жидких растворов §
перекиси является примерно линей- «§ ;„
ной функцией состава, выражен-
выраженного в весовых процентах, в пре-
пределах значительного интервала '
концентраций. Однако сложная ' о • 20 4ov 60 80 wo
зависимость между вязкостью и Н262, вес.%
концентрацией для полного интер-
интервала составов свидетельствует
д
¦ —О
А
0°
д
***
18°
д
qf
д 1
•
А
*^ о
Z
Oft-.
^—¦
J
в—
S
Рис. 34. Вязкость жидких водных раство-
растворов перекиси^водорода.
О Фиббс и Жигер; А Маас и Хэтчер.
о том, что обратная вязкость, или
текучесть, не является аддитивным
свойством этой системы. Расчет изменений вязкости растворов в зависи-
зависимости от температуры не является надежным. По определениям Мааса и
Хэтчера, если принять вязкость воды при 0° за 1,778 санпгипуаза, вязкость без-
безводной перекиси водорода составляет 1,828; 1,456; 1,447 и 1,272 сантипуаза
176
Глава 5
при температурах 0,04; 11,90; 12,20 и 19,60° соответственно. Температурные
изменения вязкости безводной перекиси водорода можно определить прибли-
приближенно, пользуясь данными известных измерений по диаграмме типа Дюринга,
с водой в качестве эталонной жидкости [32]. Аналогичным образом раствор
любой концентрации на такой диаграмме можно считать отдельной жидкостью.
Таблица 13
Вязкость жидких растворов перекиси водорода при 0 и 20°,
по Фиббсу и Жигеру [29]
Н2О2, вес.%
0,00
8,68
19,06
25,11
37,33
42,72
49,08
51,93
Вязкость, сантипуазы
0°
1,792
1,746
1,746
1,783
—
1,844
—
1,884
20°
1,005а
1,011
1,045
—
1,120
1,140
1,170
—
Н2О2, вес.%
62,37
69,78
71,55
81,21
91,76
95,75
' 100,00
Вязкость,
0°
1,917
1,933
1,919
—
1,854
1,819
"антипуазы
20°
1,219
1,234
1,266
1,262
1,259
1,249
а Недавно Бюро стандартов США установило новую величину 1,002 [31]. Посколь-
Поскольку в указанной работе для калибрования вискозиметра применяли чистую воду, все ука-
указанные значения следует уменьшить на 0,3%.
Можно легко использовать эмпирическое уравнение i*>L=Aexp(a/RT3),
предложенное для ассоциированных жидкостей Литовичем [33]; для безвод-
безводной перекиси водорода значение константы а, выбранное таким образом, чтобы
удовлетворить данным рис. 34, составляет 8,0-107 кал-град К/моль. Значение
этой константы не является линейной функцией состава, как это найдено для
других смесей ассоциированных жидкостей. Возможно, что такая линейность
наблюдается при несколько более высоких температурах, например выше 25°
где изменения^степени и величины ассоциации в зависимости от состава имеют
меньшее значение-.
Трудно согласовать часто встречающиеся в литературе указания, что
безводная или концентрированная перекись водорода представляет собой сиро-
сиропообразную жидкость, с фактическими данными измерений вязкости. Действи-
Действительно, при комнатной температуре вязкость перекиси водорода не намного
больше вязкости воды. Эта ошибка, вероятно, вызвана наблюдениями при рабо-
работе с загрязненным остатком от выпаривания водной перекиси или неправиль-
неправильным подбором выражений, использованных Тенаром для описания внешнего
вида перекиси водорода при смешении с водой. Однако вязкость растворов пере-
перекиси водорода действительно настолько значительно возрастает при охлажде-
охлаждении ниже 0°, что консистенция оправдывает название «сиропообразной». При
низких температурах измерения не проводились, но вязкость переохлажденных
растворов настолько значительна, что вызывает заметное снижение скорости
кристаллизации при введении зародышей. Миронов и Бергман [34] указывают,
что вязкость 50—80%-ных растворов перекиси водорода резко возрастает
вблизи кривой температур замерзания.
Вязкость паров перекиси водорода
В капиллярном вискозиметре при общем давлении 1 am и температуре
170° определена вязкость парообразных смесей перекиси водорода и воды, со-
содержащих до 65 мол.% перекиси [35]. Опубликованы также результаты ряда
Физические свойства 177
измерений при 200 и 240° с целью определения влияния температуры на вяз-
вязкость. Вискозиметр калибровали по водяному пару; оказалось, что присут-
присутствие кислорода в смеси паров в количестве менее 1 мол.% не влияет на точ-
точность измерений.
Описанные опыты показали, что вязкость парообразных смесей перекиси
водорода и воды имеет линейную зависимость от состава паров. Поэтому для
определения вязкости безводного пара перекиси проведено прямое линейное
экстраполирование. Получены некоторые данные, свидетельствующие о том,
что изменение вязкости парообразной перекиси водорода с температурой мень-
меньше, чем такое же изменение для водяного пара, но точность результатов изме-
измерений была не настолько высокой, чтобы установить различие между темпера-
температурными коэффициентами вязкостей пара перекиси водорода и водяного пара.
Для последнего изменение вязкости при указанных температурах составляет
0,35 микропуаза на Г. Эти данные можно выразить следующим уравнением:
^= 134 + 0,35 (/- 100)-14^. A0)
Это уравнение рекомендуется для вычисления вязкости смесей паров перекиси
водорода и воды внутри интервала температур примерно от 100 до 300° и, как
полагают, с точностью в пределах ,4:2%.
Абсолютная точность этих вискозиметрических измерений, конечно, зави-
зависит от точности значения вязкости для водяного пара. Хотя данные вязкости
для водяного пара, использованные при калибровании, вероятно, можно счи-
считать наиболее точными из имеющихся для больших интервалов давлений и тем-
температур, другие подсчеты свидетельствуют о том, что возможная погрешность
для использованной величины вязкости водяного пара при 1 am и температуре
около 200° не меньше ± 1 %.
Линейное изменение вязкости смеси пара воды и пара перекиси водорода
с температурой подчеркивает аналогию между обоими типами молекул с точки
зрения возможности их молекулярного взаимодействия. Интересно отметить,
что вязкости пара воды и пара перекиси при точке кипения каждого из этих
веществ практически одинаковы.
Поверхностное натяжение жидкой перекиси водорода
Поверхностное натяжение водных растворов перекиси водорода измерено
Шпрингом [36], Маасом и Хэтчером [И] и Фиббсом и Жигером [29] методом
капиллярного поднятия. Судя по сравнению с данными, полученными Шприн-
Шпрингом для воды, его данные для растворов перекиси, по-видимому, отличаются
значительной погрешностью. Маас и Хэтчер установили наличие линейной за-
зависимости между поверхностным натяжением и составом, выраженным в весо-
весовых процентах, и таким образом получили для безводной перекиси водорода
значение 78,76 дин/см при 0° и 75,99 дин/см при 18°. Результаты измерений
Фиббса и Жигера не согласуются с данными Мааса и Хэтчера. Они получали
более высокие значения и определенное отклонение от линейной зависимости.
Частично это расхождение может быть объяснено использованием различных
величин плотности перекиси водорода, но в основном, по-видимому, оно вы-
вызвано загрязнением растворов Мааса и Хэтчера поверхностноактивными приме-
примесями. Это предположение подтверждается тем, что Маас и Хэтчер исходили из
загрязненного 3%-ного раствора, который они подвергали очистке и концен-
концентрированию. Фиббс и Жигер сообщают, что им трудно было получить согласо-
согласованные данные даже при применении весьма чистой 90%-ной перекиси водо-
водорода, полученной промышленным электролитическим методом; особенные за-
затруднения вызывало присутствие поверхностноактивного вещества. Это веще-
вещество оказалось пластификатором, перешедшим в раствор перекиси водорода
в результате выщелачивания им пластмассы, использованной для пробки сосу-
' 2 Перекись водорода
178
Глава 5
Таблица 14
Поверхностное натяжение растворов перекиси водорода при 0 и 20°,
по Фиббсу и Жигеру [29]
Н2О2, вес.%
0,00
17,05
26,31
37,33
49,76
Поверхностное натяжение,
75,65
76,36
76,95
77,69
78,41
дин \см
при
72
73
74
74
75
20°
,75
,41
,12
,67
,68
Н2О2,
64
71
84
95
100
вес.%
,33
,55
,12
,75
,00
Поверхностное натяжение,
при 0°
79,62
80,48
81,92
82,93
83, За
ouhIcm
при 20°
76,65
77,36
78,61
79,87
80,4^
а Экстраполированная величина, точность i0t05 дин/см.
да, содержавшего перекись. Их данные весьма чувствительно также менялись
при образовании мельчайших пузырьков.
Данные Фиббса и Жигера [29] представлены в табл. 14 и на рис. 35. Эти
авторы провели также измерения с 97,8%-ной перекисью водорода и обнару-
обнаружили примерно при этом составе небольшой максимум для значения поверхност-
поверхностного натяжения. Хотя поверх-
поверхностное натяжение смесей в дру-
других системах (например, вода—
серная кислота [37], вода—гид-
вода—гидразин [38]) и имеет максимум,
характерный для некоторого
промежуточного соединения,
Жигер [39] считает, что «макси-
«максимума» в точке 97,8% быть не
может, и результат этого измере-
измерения им забракован. Значения
для безводной перекиси водо-
водорода получены прямым графиче-
графическим экстраполированием. В пре-
пределах точности измерений вели-
величин нет никаких доказательств,
что температурные коэффи-
коэффициенты поверхностного натяже-
натяжения перекиси водорода и ее
растворов отличаются от коэф-
коэффициентов поверхностного натя-
натяжения воды. Поэтому кривые
на рис. 35 расположены парал-
параллельно друг другу.
84
82
S
1 18
|.
14
12
г
0°
-У
20°
А
/
/
/
/
—О-Л
/
с/
/
-/
20
40 60
Н202, вес.%
Рис. 35. Поверхностное натяжение
растворов перекиси водорода.
80
100
водных
Коэффициент диффузии жидкой перекиси водорода
Интерес к определению коэффициента диффузии в жидкой перекиси водо-
водорода возник впервые в связи с желанием приложить гипотезу Нойса и Уитни
в том виде> как она разработана Нернстом, к рассмотрению кинетики гетероген-
гетерогенных реакций на примере каталитического разложения перекиси водорода. В со-
соответствии с этой гипотезой рассматривается зависимость наблюдаемой
скорости реакции от скорости переноса или диффузии реагента в растворе
Физические свойства 179
к поверхности твердого тела, где происходит реакция. В том случае, когда ско-
скорость диффузии по сравнению с возможной скоростью реакции на поверхности
мала, скорость диффузии лимитирует общую скорость и соответствует общей
скорости реакции.
Этим путем Генри [40] проанализировал каталитическое разложение пере-
перекиси водорода коллоидной платиной, причем в качестве приближенной вели-
величины для коэффициента диффузии он использовал величину 0,86 см2/сутки.
Бредиг [41] с большим успехом применил ту же величину для результатов
измерений при разложении на листовой платине. Вейгерт [42] при сравнении
вычисленной и измеренной скорости разложения перекиси водорода базиро-
базировался на измеренном им коэффициенте диффузии. Пользуясь теорией Нернста,
Вейгерт определил кажущуюся толщину пленки жидкости, через которую
перманганат калия, действовавший как деполяризатор, должен был диффунди-
диффундировать к поверхности вращающегося платинового электрода. Зная коэффици-
коэффициент диффузии перманганата калия и измеренную силу тока, он мог определить
путь диффузии как функцию скорости вращения. Эти пути диффузии были за-
затем использованы для нахождения коэффициентов диффузии из резуль-
результатов опытов, в которых в качестве деполяризатора для того же электрода
применялась перекись водорода. В случае с 0,014 н. перекисью водорода Вей-
Вейгерт получил для коэффициента диффузии D значения от 1,2 до 1,37 см2/сутки
при 18°. Для 0,049 н. перекиси водорода он получил D=l,2 см2/сутки. Эти
данные удовлетворяли результатам измерения скорости разложения перекиси
водорода на вращающемся платиновом электроде в отсутствие тока.
Хейман [43] провел опыты, аналогичные опытам Вейгерта, но использовал
для калибрования электрода йодный раствор. Полученные таким образом
пути диффузии использованы при вычислении коэффициентов диффузии
перекиси водорода на основании измерения скоростей разложения.
При 0,1—0,3 н. концентрациях значения коэффициента диффузии при 18°
составляли от 1,17 до 1,20 см2Icyтки. Для проверки этих величин Хейман про-
производил непосредственное измерение коэффициента диффузии перекиси водо-
водорода, для чего заставлял диффундировать слой раствора перекиси водорода
в столб воды. Для 0,1 н. перекиси водорода Хейман получил коэффициент диф-
диффузии 0,97 смУсутки при 11° и принял, что путем экстраполяции допустимо
вывести для 18° значение коэффициента 1,16 см2/сутки.
Сомневаться в правильности гипотезы диффузии Нернста и в ее примене-
применении к разложению перекиси водорода на платине нет никаких оснований. Од-
Однако вряд ли при помощи таких измерений можно просто и прямым способом
получить надежные коэффициенты диффузии. Кислород, выделяющийся при
реакции, может, например, вызвать дополнительное перемешивание, экрани-
экранировать часть поверхности или же способствовать диффузии в паровой фазе.
Технику прямых измерений Хеймана также нельзя признать наилучшей. Бо-
Более приемлемым являются данные Штерна [44], полученные мембранным ме-
методом [45], по которому диффузия осуществляется в стационарных условиях
из резервуара с раствором через пористую диафрагму (в данном случае из иен-
ского стекла G-4) в резервуар с растворителем. Штерн калибровал свою ячей-
ячейку при 20° с применением 0,1 н. раствора йодистого калия (принимая D=
= 1,448 см2/сутки), причем для подавления разложения перекиси водорода на
мембране он использовал добавки мочевины или ацетанилида, из которых ни
одна не влияла на результаты. В табл. 15 представлены отдельные дифферен-
дифференциальные коэффициенты диффузии, вычисленные из результатов опытов Штер-
Штерна в 0,011—4,78 н. водных растворах перекиси водорода, содержащих не более
0,1% ацетанилида. Значения, полученные Штерном для диффузии 0,1 н. пере-
перекиси водорода в водном растворе, содержащем другие добавки, отличаются
не больше чем на 10% от величин, приведенных в табл. 15. Из этих опытов
нельзя вывести заключение о том, какое значение имеет изменение концентра-
И*
180
Глава 5
ции перекиси; зависимость коэффициента диффузии 0,1 н. перекиси водорода от
температуры в пределах точности данных является, по-видимому, линейной,
причем температурный коэффициент составляет около 0,03 см2/сутки на Г.
Штерн считает реальным кажущееся изменение коэффициента диффузии с тем-
температурой ниже 5°, но его заключение вряд ли можно оправдать.
Таблица 15
Коэффициент диффузии перекиси водорода в водных растворах, по Штерну [44]
Концентра-
Концентрация Н2О2,
н.
Темпера-
Температура, °С
Коэффици-
Коэффициент диф-
диффузии ?>,
см^/сутки
Концентра-
Концентрация Н2О2,
н.
Температу-
Температура, °С
Коэффици-
Коэффициент диф-
диффузии D,
с м^/сутки
Концентра-
Концентрации Н2О2,
н.
Темпера-
Температура, °С
Коэффици-
Коэффициент диф-
диффузии D,
с м? {сутки
0,102
0,104
.0,103
4,78
40
30
25
20
1,48
1,18
1,03
1,12
0,85
0,107
0,011
0,098
20
20
20
15
1,18
0,84
0,76
0,78
0,102
0,101
0,102
10
5
0
0,46
0,39
0,53
Интересно сравнить данные для перекиси водорода с данными для само-
самодиффузии воды по Граупнеру и Винтеру [46]. Эти авторы пользовались раство-
растворами окиси дейтерия и воды, обогащенной О18 и содержащей менее 1 ат.%
тяжелого изотопа, и измеряли самодиффузию воды внутри интервала темпера-
температур 15—45°. При 25°, например, они определили, что коэффициент диффузии
воды в воде равен 1,80 см2/сутки. Отношение коэффициентов диф-
диффузии воды и перекиси водорода в концентрациях, изученных Штерном,
внутри исследованного интервала температур, приблизительно равно отноше-
отношению молекулярных весов. Граупнер и Винтер не обнаружили различий в коэф-
коэффициенте диффузии, когда применяли окись дейтерия или воду, обогащенную
О18; это показывает, что диффузия воды осуществляется целыми молекулами.
Штерн провел также измерения коэффициентов диффузии перекиси
водорода в метиловый спирт, этиловый спирт, ацетон и этиловый эфир [44].
Эти коэффициенты заметно отличались от найденных при диффузии в воду.
Для различных растворителей не обнаружено постоянства произведения коэф-
коэффициента диффузии на вязкость растворителя. Попытки вычисления коэффи-
коэффициента диффузии перекиси водорода обобщенным методом, таким, как метод,
предложенный Вилке [47], дали неудовлетворительное совпадение с экспери-
экспериментальными значениями.
Колландер [48] изучал проницаемость коллодиевой мембраны для пере-
перекиси водорода. Использовав молекулярную рефракцию в качестве мерила
молекулярного объема, Колландер выразил скорость переноса через мембрану
для большого числа разных веществ как функцию молекулярной рефракции,
причем получил удовлетворительную зависимость. Найденная им проницае-
проницаемость гораздо ниже коэффициентов диффузии изученных веществ, а проницае-
проницаемость для перекиси водорода по сравнению с ее молекулярной рефракцией ма-
мала; эти данные, вероятно, можно использовать лишь в качестве относительного
мерила диффузионных свойств.
Коэффициент диффузии парообразной перекиси водорода
Мак-Мартри и Кейс [49] измерили скорость диффузии безводного пара пе-
перекиси водорода через воздушный столб по методу Стефана. Жидкий 99%-ный
раствор перекиси водорода испаряли при 60° в трубку сечением 6,32 см%
и длиной 17,62 см таким образом, чтобы пары диффундировали вверх по труб-
трубке. В верхнем конце трубки концентрацию перекиси поддерживали на нуле
Физические свойства 181
путем пропускания через этот конец равномерного тока воздуха. Для вычисле-
вычисления коэффициента диффузии использована скорость уменьшения объема жидко-
жидкости после введения поправки на разложение; при этом применялось уравнение
для стационарной диффузии одного газа через второй, неподвижный газ в од-
одном направлении в условиях постоянного градиента концентрации. Поправки
на параллельную диффузию небольшого количества кислорода, образовавшего-
образовавшегося в результате разложения перекиси, и на небольшое изменение упругости
паров (за счет разбавления водой от разложения) были ничтожно малыми. При-
Пригодность прибора и методики проверена путем измерения скорости диффузии
водяного пара через водород при различных температурах. Мак-Мартри
и Кейс в качестве средней величины для трех определений коэффициента диф-
диффузии паров перекиси водорода через воздух при 60° и 1 am нашли величину
0,188+0,004 см21сек.
Экспериментально определенный коэффициент диффузии хорошо согла-
согласуется с уравнением Джиллиленда [50], в которое входят эмпирические атом-
атомные объемы. По этому уравнению получается значение 0,189 см2/сек. Коэф-
Коэффициент, вычисляемый по обобщенному уравнению Андруссова [51], хуже со-
согласуется с экспериментальным значением. Для коэффициента диффузии
водяного пара в воздухе при 60° и 1 am получается значение 0,320 см2/сек [52].
Соотношения между твердой и паровой фазами для перекиси водорода
В литературе нет никаких данных о прямых измерениях давления пара
твердой перекиси водорода. По данным Скэтчарда, Каванафа и Тикнора [26],
для давления пара в тройной точке (—0,42°) можно принять величину 0,26 мм
рт. ст. Теплота возгонки при температуре замерзания, получаемая путем сум-
суммирования тепл от испарения иплавления, равна457,8/сал/гили 15,58ккал/моль.
По уравнению Клаузиуса—Клапейрона наклон dp/dT кривой равновесия
твердой и паровой фаз в тройной точке равен 3,6-10~5 ami град К.
Соотношения между жидкой и твердой фазами для перекиси водорода
Диаграмма ликвидуса для системы перекись водорода—вода имеет
довольно простой вид и показывает понижение температуры замерзания каж-
каждого компонента при добавке другого, а также наличие двух эвтектических точек
для смесей лед—Н2О2-2Н2О и Н2О2-2Н2О—твердая перекись водорода с кон-
конгруэнтной температурой плавления для соединения Н2О2-2Н2О, лежащего меж-
между ними. Истинные твердые растворы не образуются, но твердые фазы очень
упорно удерживают маточный раствор. На рис. 36 показаны все имеющиеся
данные, за исключением относящихся к разбавленным растворам, богатым
водой, и к составам твердых фаз между эвтектиками; на этом же рисунке пока-
показана и рекомендуемая кривая. Мы не приводим многочисленных определений
температур замерзания очень разбавленных растворов, так как они могут
быть использованы в этом интервале только для проверки идеальных точек за-
замерзания (показанных на рис. 36). В области между эвтектиками точки жидких
и твердых фаз перемешаны друге другом; во избежание путаницы показаны точ-
точки только для жидкостей. Сначала рассмотрим кривую точек замерзания.
Затем будет дано определение составов твердых фаз и приведены наблюдения
по замерзанию и разложению при плавлении. В конце этого раздела изло-
изложены наблюдения и измерения, касающиеся взаимного превращения твердых
фаз при температурах ниже эвтектических. В следующих разделах приведен
материал по переохлаждению и теплоте плавления.
Тамман [4], Каррара [5], Орндорф и Уайт [6] и Менцель [53] провели ряд
измерений по понижению температуры замерзания разбавленных растворов
перекиси водорода (содержащих ее в количестве меньше 5 вес.%). В общем
182
Глава 5
результаты этих измерений хорошо согласуются с точками замерзания, вычи-
вычисленными для идеальных растворов. Идеальные точки замерзания такого рода
показаны на рис. 36 как для разбавленных, так и для концентрированных
растворов. Истинные температуры замерзания расположены ниже идеальных
при концентрациях до 10 и выше 94 вес.%. максимум на 0,5°, а при концен-
концентрациях до 5 и выше 97,5 вес. %—даже максимум на 0,1°. Эти подсчеты сделаны
с использованием активностей, определенных Скэтчардом, Каванафом и Тик-
нором [26], и теп лот плавления для чистых компонентов.
-20
-30
-40
-50
-60
N
о
- о
-
-
-
-
-
¦ н
N
с
&
/
\
/
/
еравновесные
wepdou фазы
!
N
•
\
о с
\.
Кривая точен замерзания
К
S
\
\
1
i/
У
/
V
/
У
1
•/
с
о
о
о
о
о
/
г
8с
°о
о
Л
*
-
-
-
-
ю
20
30
40
10
80
90
100
50 60
я, вес %
Рис. 36. Диаграмма ликвидуса для системы перекись водорода—вода.
ф идеальные составы; О Жигер и Маас; Л Кубашевский и Вебер; V Маас и Герцберг;
Г\ Фол и и Жигер; х Жигер и Секко.
Полное исследование температур замерзания для всего ряда составов про-
проведено Маасом и Герцбергом [54]. Затем эти же измерения повторены Жигером
и Маасом [7], Кубашевским и Вебером [20], Фол и и Жигером [55], Мироновым
и Бергманом [34] и Жигером и Секко [56]. Маас и Хэтчер [11] особое внимание
обратили на температуру замерзания безводной перекиси водорода и нашли
для нее значение —1,70°; такие же измерения выполнили Кдтбертсон, Матесон
а! Маас [18] (—0,89°), Эгертон, Эмте и Минкоф [57] (—1,8°), Фоли и Жигер
[55] (—0,461°) и Жигер, Лиу, Дагдейл и Моррисон [28] (—0,43°). Последняя
величина и является рекомендуемой для температуры замерзания безводной
перекиси водорода. Данные, полученные Маасом и его сотрудниками, основаны
на наблюдениях температуры, при которой таяли последние кристаллы ча-
частично замороженного раствора; другие же данные получены по методу терми-
термического анализа.
На рис. 36 можно видеть, что эти группы данных* хорошо соответствуют
одна другой. Наибольшие неточности относятся к области между эвтектически-
эвтектическими составами. Довольно плоский максимум на кривой замерзания, доказываю-
доказывающий практически полную диссоциацию молекулярного соединения Н2О2-2Н2О
* Миронов и Бергман представили свои данные только на небольшом графике. Эти
данные нельзя отсчитать с достаточной точностью для нанесения на рис. 36; в общем
они согласуются с рекомендуемой кривой.
Физические свойства
183
при переходе в жидкое состояние, затрудняет получение четко определенных
точек в этой области.
Таблица 16
Диаграмма ликвидуса
температура
Первая эвтектика (богатая
водой)
Н2О2, вес.%
45
46
45,8
45,2
46,1
45,2
45,2
температура,
°С
-53
-51,5
-52,5
—52,4
-52,5
-52,2
-52,2
для системы
перекись водорода—вода;
для соединения и эвтектик, по Фоли и
Соединение
Н2О2, вес.%
48,6
48,6
48,6
48,6
48,6
48,6
48,6
Н2О2-2Н2О
температура,
°С
-51
-50,3
-50,3
-52,1
-50,2
— 52,0
-52,0
координаты
\ Жигеру [55]
Вторая эвтектика
Н2О2, вес.%
59
60
59,0
61,2
60,2
61,2
61,2
температура,
°С
-56
-56,5
-55,5
-56,5
-55,7
-56,1
-56,1
состав—
Литература
54
7
20
55
34
56
Рекомен-
Рекомендуется
Таблица 17
Рекомендуемые координаты для диаграммы ликвидуса системы перекись водорода—вода
Концен-
Концентрация
н2о2,
вес.%
0
5
10
15
20
мпера-
ра за-
рзания,
Н н 2о
0
-2,9
-6,4
-10,3
-14,6
25
30
35
40
45
Is 1
Sao,
-19,5
-25,7
-33,0
-41,4
1,7
l
45,2
48,6
50
55
60
мпера-
ра за-
рзания,
-52,2а
-52,06
-52,2
-53,3
-55,5
he
61,2
65
70
75
80
мпера-
ра за-
рзания,
-56,1а
—49,0
-40,3
-32,3
-24,8
и =гО о
85
95
95
100
мпера-
ра за-
рзания,
Н Н So
-17,9
-11,5
-5,6
-0,43
а Эвтектика.
б Соединение.
В табл. 16 приведены координаты, выбранные различными исследователя-
исследователями для указанного соединения и эвтектик; рекомендуемые значения основаны
на наблюдениях Жигера и Секко [56]. Координаты рекомендуемой кривой тем-
температур замерзания даны в табл. 17.
На кривой точек замерзания (рис. 36) не показан излом при —29° и
76,5% перекиси водорода, обнаруженный Мироновым и Бергманом [34].
Эти авторы нашли, что точки замерзания выше эвтектики на стороне перекиси
водорода могут быть соединены кривой, состоящей из двух ветвей, пересекаю-
пересекающихся в точке с вышеуказанными координатами. Этот излом нечеткий. Такого
рода излом, или безвариантная точка, для этой системы свидетельствует о суще-
существовании двух твердых фаз, так же как и в эвтектических точках. Этот излом
может представлять либо перитектическую точку, показывающую равновесие
жидкости с твердым соединением д:Н2О2-Н2О (где х >1), нестабильным при тем-
температуре выше —29°, либо точку превращения, в которой твердая перекись
водорода переходит в другую аллотропную модификацию. Поскольку других
указаний на такую точку не получено, а проведенный позже термический ана-
анализ [28] не показал остановки при —29°, существование этой точки излома
•является весьма сомнительным.
184 Глава 5
Вопрос о составе твердых фаз, получаемых при замерзании раствора пере-
перекиси водорода, долгое время оставался неясным, но благодаря проведенным
в последнее время работам состав теперь изучен гораздо лучше. На рис. 36
показаны экспериментальные данные для состава твердых фаз, которые,
как предполагают, находятся в равновесии с жидкостью. На рисунке
отсутствуют точки для составов твердых фаз в области между эвтектиками.
Такого рода точки лежат в пределах колебаний составов жидких фаз при
разных определениях и во избежание путаницы не помещены на рисунке. Эти
данные по составу твердой фазы основаны на исследовании состава фаз после
разделения, выполненном Жигером и Маасом [7], и на термическом анализе,
проведенном Кубашевским и Вебером [20]. При отсутствии других данных мож-
можно эту кривую температур плавления, или линию солидуса, считать показате-
показателем образования твердых растворов. Действительно, если не считать наиболее
разбавленных и наиболее концентрированных растворов, то твердое вещество,
получаемое при частичном замораживании растворов перекиси водорода,
содержит и воду и перекись и притом даже в условиях, способствующих дости-
достижению равновесия. Однако в настоящее время вполне убедительно доказано,
что эти твердые фазы смешанного состава не являются истинными твердыми
растворами. В связи с тем, что в литературе по этому вопросу имеются зна-
значительные расхождения, мы приведем здесь основные доказательства в пользу
этого заключения и против него.
Сначала рассмотрим основания в пользу образования твердых растворов.
1. Еще со времен Тенара известно, что дробная кристаллизация не являет-
является достаточно эффективным методом для получения концентрированной или
чистой перекиси водорода. Жигер и Маас [7] сообщают, что эффективное повы-
повышение концентрации, достигаемое за одну операцию, обычно составляет около
2% и никогда не превышает 4%, в связи с чем невыгодно пытаться получать
безводную перекись водорода путем вымораживания растворов любой концен-
концентрации ниже 90 вес.%.
2. Кристаллические габитусы твердой воды и твердой перекиси водорода,
гексагональный и тетрагональный, не изоморфны, но имеют близкие размеры.
Известно, что неизоморфные системы даже с меньшей близостью параметров
могут образовать твердые растворы.
3. Оба метода определения составов твердой фазы в точке плавления (путем
разделения фаз с прямым анализом [7, 11] и путем термического анализа [20])
показывают образование твердых растворов.
4. Твердая фаза, получаемая частичным замораживанием раствора пере-
перекиси водорода, может быть настолько полно отделена от маточного раствора,
что она кажется совершенно сухой. Результаты анализа такой твердой фазы
показали, что если она состоит из однокомпонентного твердого вещества с при-
прилипшим или окклюдированным маточным раствором, то это твердое вещество
должно удерживать жидкость в количестве, равном своему весу или даже
большем.
Против образования твердых растворов говорит следующее:
1. Натта и Ригамонти [58] провели рентгенографический анализ раство-
растворов перекиси'водорода при концентрации 82 и 90 вес.% и показали, что пере-
перекись водорода^не образует твердого раствора с водой или с Н2О2 • 2Н2О; кон-
константа решетки твердой перекиси водорода не изменяется в замороженных
растворах указанных составов.
2. Общий |Ъбъем частично вымороженных растворов можно подсчитать
довольно точно на основании предположения об образовании однокомпонентных
твердых фаз.
3. Системы, образующие фазы твердых растворов, в которых одним из
компонентов является лед из воды, весьма редки, если они вообще сущест-
существуют.
Физические свойства 185
4. Жигер и Жоффрион [16] показали, что можно вызвать кристаллизацию
41,8%-ного раствора и затем переохладить систему (до —60°) по отношению
к первой эвтектике. Жидкость, отделенная от полученного твердого вещества,
оказалась 48,4%-ной перекисью водорода; эту жидкость нельзя заставить за-
закристаллизоваться путем введения затравки льда из воды, следы же кристал-
кристалла полностью замороженного 45%-го раствора перекиси водорода вызывают
немедленное затвердевание раствора. На основании этого опыта сделано
заключение, что выпавшая сначала фаза представляет собой лед, и пос-
последний не в состоянии вызвать кристаллизацию эвтектики с соединением
н2о2-2Н2о.
5. Отклонения температур замерзания, вычисленных для идеальных рас-
растворов, от температур замерзания, определенных экспериментально для кон-
концентрированных растворов и приписывавшиеся раньше необходимости введения
поправки на образование твердых растворов, в действительности, как показано
теперь, обусловлены применением заниженного значения теплоты плавления
безводной перекиси водорода.
6. Фоли и Жигер [55] изучили состав фаз, получаемых методом частичного
вымораживания растворов перекиси водорода, с применением метода остатков
Шрейнемакерса и использованием хлористого калия в качестве третьего ком-
компонента. Эти результаты убедительно доказывают отсутствие образования твер-
твердых фаз.
7. Чтобы исключить возможность изменения в этих опытах фазовых равно-
равновесий хлористым калием, проведены аналогичные опыты с измерением пропор-
пропорциональной растворимости следов радиоактивного однозамещенного фосфата
калия в обеих фазах. Эти результаты вновь доказали отсутствие образования
твердых растворов.
8. При термическом анализе Миронов и Бергман не нашли доказательств
образования твердых растворов.
9. Связь между теплоемкостью твердой фазы и температурой в присут-
присутствии небольшого количества воды, по данным точных калориметрических
измерений, говорит против предположения об образовании твердых раство-
растворов [28].
Взвешивая те и другие доводы, можно убедиться, что наблюдения в пользу
доказательства образования твердых растворов не вполне безупречны; более
многочисленные противоположные выводы включают выводы двух работ [по
методу мокрых остатков (пункты 6 и 7) и калориметрические наблюдения
(пункт 9)], основанных на исключительно точных исследованиях и позво-
позволяющих провести выбор на абсолютной основе. Эти работы полностью опровер-
опровергают мнение об образовании истинных твердых растворов.
Расхождения во мнениях относительно состава твердых фаз. безусловно,
вызваны трудностью полного разделения жидкой и твердой фаз при замора-
замораживании растворов перекиси водорода, и на основании таких опытов трудно
судить о том, какими должны быть кажущиеся составы твердой фазы, которых
можно ожидать на практике*. В случае отсутствия других данных по определе-
* Жидкость, окклюдированная твердой фазой, имеет тот же состав, что и свобод-
свободная жидкость, окружающая эту фазу. Хотя такая система остается моновариантной, по-
поведение водных растворов перекиси водорода доказывает возможность охлаждения без
дальнейшего образования твердой фазы после достижения точки явного затвердевания.
Это поведение, характерное для дивариантной системы, и сухой внешний вид твердой
фазы свидетельствуют об устойчивом внедрении жидкости в массу твердых кристаллов.
В этом сочетании твердая и жидкая фазы могут оставаться стабильными с точки зрения
разделения фаз до достижения температур ниже эвтектической, когда окклюдированная
жидкость становится нестабильной в отношении твердого соединения Н2О2-2Н2О. Это
предположение в известной мере подтверждается наблюдениями Жигера и Жоффриона
(в работе [16] по превращениям при образовании эвтектики), изложенными ниже, при
описании разложения при оттаивании.
186 Глава 5
нию составов, которые должны получиться после некоторого замораживания,
однако не слишком длительного, следует исходить из кажущихся составов твер-
твердой фазы по Маасу и Хэтчеру, Жигеру и Маасу и в меньшей степени из дан-
данных Кубашевского и Вебера (рис. 36). Некоторые практические наблюдения по
составам и относительному содержанию веществ в твердых фазах, полученных
при вымораживании растворов перекиси водорода, сделаны Штейндлем [59].
Жигер сообщает [39], что на практике можно добиться изменения состава твер-
твердой фазы против указанного на рис. 36; например, в частично вымороженном
10%-ном растворе, выдержанном в течение длительного времени вне помеще-
помещения в морозную погоду, масса микрокристаллов медленно превращается в боль-
большую прозрачную массу почти чистого льда. Оказалось возможным также выра-
вырастить большой монокристалл чистой перекиси водорода непосредственно
в лаборатории [28].
Жигер и Жоффрион [16] описали внешний вид кристаллов, получаемых
при замораживании растворов перекиси водорода. Твердое вещество, получае-
получаемое из разбавленных растворов (содержащих менее 45 вес.% перекиси), имеет
вид тонких белых хлопьев, появляющихся на стенке сосуда. Образование
этих призм происходит самопроизвольно или его можно легко вызвать; кри-
кристаллы быстро растут, разветвляясь, и по виду несколько напоминают иней.
Твердое вещество, получаемое в концентрированных растворах (выше 65 вес. %
перекиси), имеет вид длинных прозрачных игл, вырастающих в виде пучка из
находящегося у дна слоя жидкости. Концентрированные растворы отличаются
заметной способностью к переохлаждению; даже тогда, когда кристаллизация
начинается, она протекает медленно, несмотря на заметное переохлаждение
растворов, что обусловлено высокой вязкостью раствора при этих низких тем-
температурах. Скорость кристаллизации настолько низка по сравнению со ско-
скоростью теплоотдачи от сосуда, что температура такого переохлажденного
раствора во время затвердевания может подняться очень немного или даже
совсем не измениться.
Практический и теоретический интерес представляет явление разложения
растворов перекиси водорода при плавлении. Это обычно наблюдаемое явление
исключает возможность использования попеременного замораживания и от-
оттаивания для очистки раствора перекиси водорода или для удаления из него
растворенного газа. Жигер и Жоффрион [16] показали, что это разложение
наблюдается лишь в эвтектических смесях. Их наблюдения можно суммировать
следующим образом: замороженные растворы перекиси водорода (обычно для
замораживания применяется двуокись углерода с температурой —78°) начинают
выделять газ при оттаивании; при прекращении плавления прекращается
и газообразование; наличие или отсутствие растворенного газа не оказывает
никакого влияния (например, при замораживании раствора в вакууме или при
плавлении кристаллов полностью замороженного 52%-ного раствора опуска-
опусканием их в свежеперегнанную воду происходит выделение тока пузырьков газа).
Такого рода разложение отсутствует при плавлении первых кристаллов,
полученных при медленном замораживании. Однако, как пишут Жигер и Жоф-
Жоффрион: «...если эти первые кристаллы охладить до достаточно низкой темпера-
температуры (примерно до —70°) и выдержать при этой температуре в течение неко-
некоторого времени, происходит постепенное превращение, обнаруживаемое по
резкому потрескиванию и постепенной усадке массы, которая становится похо-
похожей на тонкопористое сплавленное стекло или спрессованный снег и отрывает-
отрывается от стенок сосуда. При частичном плавлении эта снеговидная масса оказы-
оказывается образованной из мелких игл, короче 1 мм, с неупорядоченной ориен-
ориентацией. В жидкости на концах этих микрокристаллов можно обнаружить
при рассматривании через увеличительное стекло образование газовых
пузырьков».
Таким образом, Жигер и Жоффрион объясняют эти особенности разложе-
Физические свойства 187
нием перекиси водорода в аддитивном соединении Н2О2»2Н2О и считают, что
молекула перекиси водорода подвергается значительному искажению при
внедрении ее в кристаллическую структуру этого соединения, а также что
разрушение кристалла при плавлении способствует не снятию напряжений,
а диссоциации перекиси водорода. Авторы, однако, не исключают и предполо-
предположения, что в данном случае происходит просто каталитическое разложение на
поверхностях кристаллов. Возможно также, что указанный процесс обуслов-
обусловлен разложением перекиси водорода в кристаллическом состоянии с удержи-
удерживанием кислорода, появившегося в результате этого разложения, внутри твер-
твердой фазы.
Из приведенного анализа видно, что твердые фазы, находящиеся в равно-
равновесии с жидкостью, являются льдом из воды, соединением Н2О2-2Н2Оили твер-
твердой перекисью водорода, в зависимости от состава исходного раствора. При
температурах ниже эвтектических полностью затвердевшие растворы состоят
из смесей льда и соединения Н2О2-2Н2Оили твердой перекиси водорода и этого
соединения, в зависимости от содержания в исходном растворе перекиси водо-
водорода (меньше 48,6 вес.% или больше этого количества). Нет никакой необходи-
необходимости считать, что при температурах ниже эвтектических происходят какие-
либо другие превращения в твердом состоянии, как это предполагали Кубашев-
ский и Вебер и как это обязательно происходило бы, если бы вода с перекисью
водорода образовывала твердые растворы. Высказано также предположение,
что при очень низких температурах (около —110°) происходят другие фазовые
превращения. Гейб и Гартек [60] и Джонс и Винклер [61] показали, что пере-
перекись водорода, образовавшаяся в ловушке, охлажденной жидким воздухом,
за счет соединения атомарного водорода и молекулярного кислорода или из
продуктов диссоциации водяного пара, частично разлагается при нагревании
до температур выше —120°, причем внешний ее вид при этом изменяется.
Высказано предположение, что при более низких температурах существуют две
стабильные таутомерные формы перекиси водорода, из которых одна разлагает-
разлагается при температуре выше —120°, а другая представляет собой «нормальную»
перекись.
Природа этого соединения еще не установлена, но показано, что сделанные
наблюдения нельзя объяснить на основании предположения о равновесном
фазовом превращении. При калориметрических измерениях [28, 34], проведен-
проведенных с перекисью водорода при температурах до 12° К, фазового превращения
не обнаружено. Нейдинг и Казарновский [62] также не обнаружили превраще-
превращения при этих температурах, проводя измерения магнитной восприимчивости
с перекисью водорода, которую они охлаждали от комнатной температуры.
Боун и Хогг [63] произвели качественные калориметрические измерения этого
эффекта при —115° и обнаружили тепловой эффект величиной несколько сот
калорий. Они предполагают, что величина этого теплового эффекта доказы-
доказывает существование фазового изменения. Однако более правдоподобно предпо-
предположение, что действительной причиной этого теплового эффекта является
десорбция газа или химическая реакция.
Переохлаждение растворов перекиси водорода
Растворы перекиси водорода легко переохлаждаются. Это прекрасно дока-
доказывается тем, что в северной части США запасы продажной 9Э%-ной перекиси
водорода фирмы-изготовители обычно хранят вне помещений. Зимние темпера-
температуры в этой области часто бывают значительно ниже —11° (температура замер-
замерзания 90%-ных растворов), однако не отмечено ни одного случая замерзания
перекиси водорода в незащищенных складских цистернах. Эта способность
к переохлаждению вместе со значительным понижением температуры замерза-
замерзания обеспечивает возможность безбоязненного наружного хранения любых
188 Глава 5
растворов (за исключением самых разбавленных). Как общее правило, мож-
можно считать, что растворы перекиси водорода любой концентрации способны
к переохлаждению, но особенно заметно это при концентрациях перекиси выше
50%, когда нередки случаи переохлаждения на 10—50° ниже точки замер-
замерзания [1].
Бабинский, Дирдорф и Киф [64] провели количественное исследование пере-
переохлаждения 93%-ной перекиси водорода. Они обнаружили, что в тщательно
очищенных пробирках из стекла пирекс при перемешивании палочками из
этого же стекла неизменно наблюдается переохлаждение на 1—55°. В боль-
большинстве опытов растворы переохлаждались на 40° ниже температуры замерза-
замерзания, причем на переохлаждение не влияют ни перемешивание, ни потирание
стенок пробирки, ни скорость охлаждения. Аналогичные опыты, проведенные
при пониженном давлении с целью удаления растворенного кислорода, дали
такие же результаты. Внесение уже замерзшей перекиси водорода легко вызы-
вызывает кристаллизацию переохлажденной 93%-ной перекиси; показано также,
что даже лед, образовавшийся из воды, может играть роль затравки при тем-
температурах ниже —27°. Присутствие других веществ, например полиэтилена,
нержавеющей стали, алюминия или оттавского песка, приводит к большому
колебанию результатов, хотя, как правило, всегда происходит переохлаждение
по меньшей мере на 10° ниже температуры замерзания; некоторые пробы даже
в присутствии указанных веществ удавалось охладить без замораживания до
очень низких температур.
Склонность к переохлаждению становится еще более заметной у перекиси
дейтерия [56]. Концентрированная D2O2 может быть подвергнута переохлажде-
переохлаждению не только до температуры возгонки двуокиси углерода, но даже до темпе-
температуры жидкого воздуха, когда вязкость раствора настолько велика»
что последний имеет вид прозрачного стекла. Выдерживание стекла при
температуре жидкого воздуха приводит вскоре к спонтанной кристалли-
кристаллизации.
Большую ясность в проблему переохлаждения и причины его возникнове-
возникновения как для воды, так и для перекиси водорода и ее растворов внесли работы,
проведенные Дорси [65] с водой. Систематическими опытами Дорси показал,
что почти любую пробу воды можно переохладить на несколько граду-
градусов без всякой специальной обработки. Очищенные пробы воды можно
легко охладить примерно до —15°, причем одну пробу необработанной водо-
водопроводной воды удалось охладить даже до —22°. Наиболее низкая температура
переохлаждения, найденная Дорси в литературе, составляет —40°. В отли-
отличие от обычных предположений Дорси показал, что температура, названная
им «температурой спонтанного замерзания», до которой проба может быть
переохлаждена, является воспроизводимой и не зависит от длительности выдер-
выдерживания на холоду. Температура спонтанного замерзания любой пробы мед-
медленно изменяется с возрастом в общем в сторону понижения и, как правило,
дискретными ступенями, что создает впечатление о наличии избирательных
температурных уровней. Эффективным методом снижения температуры спон-
спонтанного замерзания является кипячение проб в запаянных сосудах, как содер-
содержащих воздух, так и не содержащих его. Эффективными были и дистилляция
и фильтрация. Внешние удары, выливание и встряхивание (за исключением
самого энергичного) не вызывают кристаллизации. Только потирание стенок
сосуда или двух каких-нибудь поверхностей внутри жидкости способствует
замерзанию; температура, при которой это потирание оказывается эффектив-
эффективным, также является довольно воспроизводимой; она превышает на несколько
градусов температуру спонтанного замерзания.
Дорси пришел к выводу, что эти явления можно объяснить только иници-
инициированием кристаллизации чужеродными веществами, т. е. что образование за-
зародышей вызывается лишь пылинками определенных размеров и с определенной
Физические свойства 189
природой поверхности, причем именно эти размеры и поверхность и определяют
температуру спонтанного замерзания. Данная «гетерогенная теория», по-
видимому, вполне приложима к перекиси водорода и ее растворам. В частности,
можно сделать заключение, что перекись водорода, обладающая высокой
реакционной и растворяющей способностью, сама способна переводить в раствор
или уменьшать размеры пылинок, способных вызвать замерзание. Здесь можно
провести аналогию с кипячением воды. Этим присущим самой перекиси водо-
водорода очищающим действием можно объяснить большую способность ее к пере-
переохлаждению, и притом к очень заметному. Такому переохлаждению способ-
способствует и то, что производство перекиси водорода, по меньшей мере высококон-
высококонцентрированной, осуществляется с особой тщательностью; чистоту продажных
растворов, как правило, можно сравнить с чистотой тщательно перегнанной
воды. Предположение, что присутствие пузырьков в жидкости препятствует
возможности переохлаждения, по-видимому, не обосновано, как это, с одной
стороны, вытекает из опытов Дорси, а с другой—обусловлено природой поверх-
поверхности раздела газ—вода, которая, как указывает Вейл [66], не может при иска-
искажении под действием поляризации принять структуру твердого тела. Вероятно
также, что зародыши в растворах перекиси водорода окружаются пленкой
кислорода, образующегося при разложении. Эта пленка должна радикально
изменить эффективность зародышей.
Таким образом, переохлаждение является, по-видимому, незакономерным
явлением, зависящим от чистоты пробы в отношении определенного типа
загрязнений. В таком случае трудно заранее предсказать поведение данной
пробы, и положение [20] о том, что перемешивание предотвращает возможность
переохлаждения, не может служить общим правилом. Только введением за-
затравки или выдерживанием в условиях интенсивного охлаждения можно дей-
действительно предотвратить переохлаждение.
Теплота плавления перекиси водорода
Форкран [67] определил теплоту плавления безводной перекиси водорода
B700 кал/моль) путем умножения значения теплоты плавления воды на отно-
отношение молекулярных весов перекиси водорода и воды. Экспериментально эта
величина впервые определена Маасом и Хэтчером [11] G4 кал1г, или
2516 кал/моль). Фоли и Жигер [8] определили в ледяном калориметре теплоту
плавления (85,83 кал/г, или 2920 кал/моль) перекиси водорода, полученной из
99,6%-ного раствора путем многократной фракционированной кристаллизации.
По-видимому, оба эти результата страдают неточностью, обусловленной при-
присутствием неизвестного количества воды в перекиси водорода. Более точные
измерения Жигера, Лиу, Дагдейла'и Моррисона [28], осуществленные в ади-
адиабатическом калориметре с перекисью водорода, содержавшей только 0,03 мол. %
воды, дали для перекиси водорода теплоту плавления 87,84 кал/г, или
2987±3 кал/моль. Эта величина и рекомендуется. Ниже мы остановимся
на теплоемкостях твердой и жидкой фаз, которыми пользовались в этой
работе. Принятая величина теплоты плавления воды 79,72 кал/г, или 1436,3
кал/моль.
Пользуясь этой величиной и плотностями твердой и жидкой перекиси
водорода, можно вычислить по уравнению Клапейрона наклон dpIdT, линии
равновесия твердая фаза—жидкость, в тройной точке для перекиси водорода
(—0,42°С, или 272,74° К). Он составляет 148 am/град К. Таким образом, вли-
влияние давления составляет 0,007° К на 1 am, а температура замерзания при
атмосферном давлении равна —0,43° С. Для воды значение dp/dT составляет
—134 am/град К. Величина криоскопической константы для перекиси водо-
водорода, рассчитанная таким же образом, составляет 1,68° на моль; для воды
она равна 1,86° на моль.
190 Глава 5
Соотношения между жидкой и паровой фазами
для перекиси водорода
В системе вода—перекись водорода жидкие компоненты смешиваются во
всех отношениях, причем вода значительно более летуча, чем перекись. В этой
системе обнаружены отрицательные отклонения от закона Рауля; парциаль-
парциальные давления компонентов в паровой фазе над жидкостью меньше их значений,
вычисленных для идеальных растворов. Поскольку давления пара чистых
компонентов сильно отличаются друг от друга, это отклонение не настолько
значительно, чтобы привести к образованию азеотропов; не обнаружено
минимума давления пара или максимума температур кипения.
Измерение давления пара перекиси водорода и ее растворов осложняет-
осложняется разложением, неизбежно сопровождающим нагревание или концентрирова-
концентрирование, если только раствор перекиси водорода не отличается максимальной
чистотой. В то же время большие различия в летучести обоих компонентов соз-
создают возможность легкого концентрирования разбавленных растворов до обыч-
обычных продажных концентраций C0 вес. % или больше) в простом оборудовании.
Эти обстоятельства, безусловно, тормозили постановку точных определе-
определений равновесия между жидкой и паровой фазами для перекиси водорода;
в старой литературе встречаются только отрывочные данные относительно
точки кипения, определенные случайно при изучении техники концентриро-
концентрирования [68, 69]. Более свежие и полные данные по равновесию жидкость—пар
приведены Сидерским [70], а также Учида, Огава и Ямагучи [71] для ограни-
ограниченного интервала условий; Маас и Хиберт [72] и Эгертон, Эмте и Минкоф
[57] провели такие определения для практически безводной перекиси водорода,
а Жигер и Маас [73J и Скэтчард, Каванаф и Тикнор [26]—для широкого
интервала температур и составов. Соответствие результатов всех этих работ
вполне удовлетворительное, особенно при низких температурах, но можно
считать, что максимальная точность и наилучшая экспериментальная методика
достигнуты в работе Скэтчарда, Каванафа и Тикнора, а поэтому здесь приняты
их данные. Поскольку измерения равновесий жидкость—пар имеют большое
значение для установления многих свойств смесей воды с перекисью водорода,
мы остановимся довольно подробно на этих данных и их обработке. Представ-
Представленные здесь некоторые аспекты этого вопроса и вычисления отсутствуют в ори-
оригинальной работе.
Основная проблема, с которой приходится сталкиваться при измерении
давления паров над растворами перекиси водорода, заключается в устранении
или учете возможности изменения состава или давления в результате раз-
разложения. В большинстве цитированных работ измерения проводились в ста-
статической системе с использованием того или иного критерия для отклонения
неправильных результатов; в других случаях применялся метод экстраполиро-
экстраполирования кривой давление—время для учета влияния небольшого, но все же замет-
заметного разложения, происходившего в тщательно очищенных растворах перекиси
водорода, взятых для исследования. Другая техника, снижающая погреш-
погрешности, обусловленные разложением, состоит в проведении измерений в дина-
динамической системе в условиях непрерывного кипения под регулируемым давле-
давлением. Опыты Скэтчарда, Каванафа и Тикнора, а также японских ученых [71]
проведены в таком приборе. В этих опытах происходило непрерывное образо-
образование пара, который затем конденсировали и конденсат отводили обратно
в кипятильник. Внутри конденсатора создавали поверхности раздела между
паром перекиси водорода и гелием, который в свою очередь находился в кон-
контакте с ртутью в манометре. Желательной температуры достигали путем регу-
регулирования давления гелия, подававшегося в систему, скорость же парообразо-
парообразования регулировали путем контроля поступления тепла в кипятильник. Если
принять, что скорость образования кислорода в процессе разложения является
Физические свойства
192
Давление
по <
Температура, °С
44,50
60,00
60,00
60,00
60,00
60,00
60,00
60,00
60,00
75,00
75,00
75,00
75,00
75,00
75,00
75,00
75,00
75,00
75,00
90,00
90,00
90,00
90,00
90,00
90,00
90,00
90,00
105,00
пара водных растворов перекиси
^кэтчарду, Каван а фу и Тикнору
Состав жидкости,
мольная доля
Н2О2
0,5140
0,0905
0,2036
0,2810
0,4075
0,5823
0,6831
0,8423
0,9619
0,0745
0,1972
0,2777
0,3241
0,4899
0,4963
0,5751
0,7450
0,8572
0,9596
0,0994
0,1954
0,3257
0,4980
0,5118
0,6546
0,8418
0,9597
0,4985
Общее давление
пара при 0°С,
мм. рт. ст.
27,47
135,35
114,82
99,25
76,21
52,01
39,79
26,31
19,43
267,24
225,29
196,43
180,44
128,88
127,09
105,30
69,86
53,35
42,28
471,07
411,73
331,99
234,54
227,24
165,37
109,63
84,92
413,33
Таблица 18
водорода,
[26]
Состав пара,
мольная доля
Н2О2
0,1273
0,0054
0,0104
0,0397
0,0849
0,1757
0,2951
0,5699
0,8972
0,0036
0,0167
0,0267
0,0468
0,1076
0,1114
0,1834
0,3907
0,6381
0,0089
0,0235
0,0459
0,1342
0,1516
0,2716
0,5982
0,8858
0,1494
функцией только температуры и количества жидкости в кипятильнике, то
парциальное давление кислорода в заполненном паром пространстве прибора
определяется отношением скорости парообразования к скорости разложения,
причем пар уносит кислород в резервуар с гелием. Большой объем этого резер-
резервуара и приспособления для периодического подрегулирования давления дава-
давали возможность снизить влияние разложения на давление в системе до ничтож-
ничтожных размеров. Пробы для определения состава жидкости и температур отби-
отбирались из вторичного или «внутреннего» кипятильника, окруженного паром,
поднимавшимся из первичного кипятильника, в который возвращался кон-
конденсат. Все пары пропускались над поверхностью охлаждения для регулиро-
регулирования размеров конденсации во внутренний кипятильник и проходили через
жидкость в этом кипятильнике. Путем соответствующего регулирования поверх-
поверхности охлаждения можно было добиться стационарного состояния, т. е. по-
постоянного уровня жидкости во внутреннем кипятильнике.
В табл. 18 приведены данные, полученные Скэтчардом, Каванафом и Тик-
нором. Эти авторы считают, что измерения давления произведены с большей
точностью, чем измерения состава пара, что связано с фракционированием
в кипятильнике и экспериментальными трудностями обеспечения условий
стационарного состояния. Поэтому они исходили исключительно из данных.
192 Глава 5
давления пара для вычисления их состава, экстраполирования данных и вы-
вывода термодинамических свойств растворов.
Метод, примененный Скэтчардом, Каванафом и Тикнором для выравнива-
выравнивания и экстраполирования данных по давлению пара, состоит в следующем.
Принято, что избыточная свободная энергия смешения на моль раствора может
быть представлена уравнением, имеющим форму:
Yl A1)
Избыточная свободная энергия смешения определяется как избыток или раз-
разность между измеренной свободной энергией и свободной энергией для идеаль-
идеального раствора. Свободная энергия в свою очередь связана с химическим
потенциалом или парциальной моляльной свободной энергией и коэффициен-
коэффициентом активности уравнениями:
A3)
A4)
Тогда общее давление пара раствора определяется уравнением:
ехр (-jp- [ji. - (?„ - Vw) (Р - pwo)]) -Ь
A5)
Первый член в правой части уравнения представляет, таким образом, парциаль-
парциальное давление воды, а второй—парциальное давление перекиси водорода.
После умножения уравнения A1) на член (nw-hnh) для выражения FEX
через общее число молей раствора и дифференцирования в соответствии с урав-
уравнением A2) получают следующие выражения для избыточных химических
потенциалов компонентов:
l-2xw)E-<5xw)]. A7)
Уравнения A6) и A7) можно подставить в уравнение A5) и подогнать послед-
последнее к измеренным давлениям пара. При этой подгонке Скэтчард, Каванаф
и Тикнор пользовались следующей методикой:
1. Давление пара воды вычисляли из уравнения Кейса [74].
2. Газовые поправки для воды определяли по методу Кейса, Смита и Джер-
Джерри [27]. После определения критических констант находили поправки для
перекиси водорода из уравнения, рекомендованного для воды.
3. Давление пара безводной перекиси водорода сначала выводили путем
графического экстраполирования данных для растворов, затем из аналитиче-
аналитического экстраполирования последовательных приближений к уравнению, удо-
удовлетворяющему экспериментальным давлениям пара.
4. На основании трех предыдущих операций находили константы, удо-
удовлетворяющие эспериментальным данным при каждой температуре, путем
последовательного приближения по методу наименьших квадратов. Получен-
Полученные константы затем выравнивали как функции температуры, причем были
получены следующие значения:
Яо= -752 + 0,97^= -1017 + 0,97Т, A8)
В1 = 85, A9)
В2=13. B0)
Физические свойства
193
Выражение для давления пара безводной перекиси водорода получено
следующим образом. Хотя при определении констант использована линейная
зависимость lg ph0 от 1/Т внутри интервала экспериментальных температур,
зависимость lg р от 1/Т для наиболее целесообразного эталона—воды—заметно
отклоняется от простой линейной формы в больших интервалах температур.
Поэтому использован метод экстраполяции давления пара по Рамзею—Юнгу
с применением воды в качестве эталона. В качестве эталонного давления при-
принято давление пара 17,7 мм рт. ст. при 60°, а давление пара 78,4 мм рт. ст.
при 90° использовано для вывода разности наклонов кривых зависимости lgp
от 1/Т, Эта разность оказалась равной —1,5-ЮЛ Выведенной кривой давле-
давления пара при температурах 75, 150, 300 и 450° удовлетворяло уравнение
с четырьмя константами. Уравнение, выражающее давление пара безводной
перекиси водорода, имеет, таким образом, следующий вид:
\gPho (мм) = 44,5760-
^?
12,996 lg T -0,00460557.
B1)
Пользуясь уравнением B1) для давления пара перекиси водорода, уравне-
уравнением Кейса [74] для давления пара воды и значениями констант Во, Вх и B2t
можно составить таблицу парциальных давлений и давлений пара при разных
температурах для растворов перекиси водорода любых составов. Считают, что
поправки на неидеальность газа по величине сравнимы с экспериментальными
ошибками, а следовательно, при составлении таблицы экстраполированных
давлений пара и их составов введение поправок на отклонение от законов
для идеальных газов не оправдывается. Поэтому при составлении таблицы
0,2 0,4 Ofi Oft
Мольная доля Н202
Рис. 37. Коэффициенты активности водных
растворов перекиси водорода при 25°.
j •
члены уравнения A5), содержащие поправочный коэффициент р^для газа,
были опущены. Если выразить уравнение A5) через коэффициент актив-
активности у, не принимая во внимание поправки на отступление от идеальности,
то путем^ подстановки уравнения A4) получим
р __ р д- у 4- пи A х ) y B2)
Очевидно, что коэффициенты активности выражаются следующим образом:
B3)
B4)
13 Перекись водорода
Таблица 19
Общее давление пара водных растворов перекиси водорода, по Скэтчарду,
Каванафу и Тикнору [26], мм. рт. ст.
Температура,
°С
0
10
20
25
30
40
50
60
70
80
90
100
ПО
120
130
140
150
0
4,58
9,20
17,5
23,7
31,8
55,3
92,6
149
234
355
526
760
1074
1489
2025
2709
3568
0,1
4,06
8,17
15,6
21,1
28,3
49,3
82,5
133
209
318
471
682
965
1339
1824
2443
3222
Мольная доля перекиси водорода в ;
0,2
3,45
6,96
13,3
18,1
24,3
42,4
71,1
115
181
216
410
595
845
1175
1604
2153
2847
0,3
2,81
5,70
10,9
14,9
20,1
35,2
59,3
96,6
152
233
348
507
722
1008
1381
1860
2467
0,4
2,20
4,49
8,69
11,9
16,0
28,3
48,1
78,7
125
192
289
422
605
848
1168
1580
2105
0,5
1,66
3,42
6,68
9,17
12,4
22,2
37,9
62,6
100
155
235
346
499
704
974
1326
1776
0,6
1,21
2,53
5,00
6,90
9,41
17,0
29,3
49,0
79,0
124
189
280
407
578
807
1105
1489
кидкости
0,7
0,856
1,83
3,66
5,09
6,99
12,8
22,4
37,8
61,8
97,8
150
226
331
474
666
919
1247
У
0,8
0,593
1,30
2,64
3,71
5,14
9,55
17,0
29, W
48,2
77,2
120
182
269
389
552
767
1048
0,9
0,404
0,915
1,89
2,69
3,77
7,14
12,9
22,5
37,8
61,3
96,5
148
221
322
460
645
887
1,0
0,272
0,642
1,36
1,95
2,77
5,36
9,90
17,5
29,8
49,1
78,2
121
182
269
387
546
755
Таблица 20
Состав пара над водными растворами перекиси водорода, по Скэтчарду,
К^ванафу и Тикнору [26], мольная доля Н2О2
Температура,
°С
0
10
20
25
30
40
50
60
70
80
90
100
ПО
120
130
140
150
0, 1
0,002
0,003
0,003
0,003
0,003
0,004
0,005
0,005
0,006
0,007
0,007
0,008
0,009
0,010
0,011
0,012
0,013
0,2
0,006
0,008
0,009
0,010
0,010
0,012
0,014
0,015
0,017
0,019
0,021
0,023
0,025
0,027
0,029
0,031
0,033
Мольная доля перекиси водорода в
0,3
0,015
0,018
0,020
0,022
0,023
0,026
0,030
0,033
0,036
0,040
0,043
0,047
0,051
0,054
0,058
0,061
0,065
0,4
0,031
0,037
0,041
0,044
0,046
0,052
0,057
0,063
0,068
0,074
0,080
0,085
0,091
0,097
0,102
0,108
0,113
0,5
0,060
0,070
0,077
0,081
0,085
0,094
0,103
0,111
0,120
0,128
0,136
0,144
0,152
0,160
0,168
0,175
0,182
0
0
0
0
о,
о,
о,
о,
о,
о,
0,
о,
о,
0,
о,
о,
о,
о,
,6
112
128
138
144
151
163
175
187
199
210
221
231
241
251
260
269
278
жидкости
0,7
0,202
0,224
0,238
0,247
0,255
0,272
0,287
0,302
0,316
0,323
0,342
0,354
0,365
0,376
0,386
0,396
0,405
0,8
0,352
0,381
0,397
0,407
0,417
0,435
0,452
0,468
0,482
0,495
0,508
0,519
0,530
0,540
0,549
0,558
0,566
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
,9
,600
,626
,640
,648
,656
,671
,684
,696
,707
,716
,725
,733
,740
,747
,753
,758
,763
Физические свойства
195
На рис. 37 показаны значения коэффициентов активности при 25°.
Составы пара могут быть вычислены из уравнения
B5)
В табл. 19 и 20 представлены общие давления пара и состав его, вычисленные
при помощи уравнений B2) и B5) для растворов перекиси водорода любых
wo
90
ёв0
со
5 70
| SO
50
1
Г\
1
1
\
\
\
ч-
\
^—
50°
Давление
' пара
\
\
Состав I
s^ пара \
— C0CmmAnnnff\
1 Т г 1 1дг
\
'—¦ —
-—¦—.
** —
==¦
а:
1
S 20
I 20
10
0 Ц/ 0,2 а,3 0,4 0,5 0,6 0,l Ц8 0,9 1ft
Состав шдкости, мольная доля Н202
Рис. 38. Состав и давление пара над смесями
перекиси водорода и воды при 25 и 50° (жидкость
состава х находится под давлением пара у в равно-
равновесии с паром состава г).
составов внутри интервала температур 0—150°*. Частично эти данные представ-
представлены также на рис. 38, 39 и 40 в целях сравнения равновесия жидкость—пар
для растворов при некоторых избранных температурах и давлениях.
Общие давления пара, вычисленные Скэтчардом, Каванафом и Тикно-
ром при помощи уравнения B2), отклоняются в среднем на 0,44% (максимум на
1,38%) от экспериментальных величин, приведенных в табл. 18. При темпе-
температурах до 75° уравнение B1) для давления пара безводной перекиси водорода
согласуется в пределах десятых долей миллиметра с величинами, выведенными
из уравнения, рекомендуемого Маасом и Хибертом:
lgpho {мм) = 8,853 - 2534,7/Г.
Аналогичная согласованность наблюдается и для уравнения
lgp*o (MM) = 8,63-2469/7,
выведенного для безводной перекиси водорода Эгертоном, Эмте и Минкофом;
При температурах выше 75° отклонения возрастают; Скэтчард, Каванаф и
Тикнор дали анализ причин этого отклонения и предложили в целях повыше-
повышения точности использовать более полное уравнение B1) для определения
давления пара безводной перекиси водорода. Мы рекомендуем пользоваться
исключительно этим уравнением.
* Для получения дополнительных данных, отсутствующих в таблице, можно ис-
использовать эти же уравнения или другой метод [75], основанный на графике Дюринга^
13*
196
Глава 5
Из указанных выше данных становится очевидно, что относительная
летучесть воды по сравнению с летучестью перекиси водорода достаточно
велика, особенно для богатых водой растворов перекиси водорода и при низ-
низких температурах.
О 0,1 0,2 0,3 0,4 0,5 0,6
Состав жидкости, мольная доля М20г
РГи с. 39. Температуры кипения и состав пара
над смесями перекиси водорода и воды при общем
давлении 30 и 760 мм рт. ст. (жидкость состава х ки-
кипит при у °С с образованием пара состава z).
Из этих данных можно также подсчитать гигроскопичность растворов пере-
перекиси водорода. Если в качестве эталона для сравнения взять относительную
ИМИ
160т
/
/у
ipm.a
/
у
/
7. / а
/го»
/
1
f
шрт.
j
/
/
ст.
0,1 42 43 Ofl Q5 0,6 Q7 Ц8 Q9 tfl
Состав жидкости, мольная доля НО
Р^и с. 40. Кривые равновесия пар—жидкость для
смесей перекиси водорода и воды при общем дав-
давлении 30 и 760 мм рт. ст.
влажность 50% при окружающей температуре 25°, то растворы с концентра-
концентрацией выше 60 вес.% перекиси водорода будут поглощать водяной пар из
Физические свойства
197
воздуха. Обычно этот эффект не заметен, за исключением только очень концен-
концентрированных растворов или же растворов и кристаллов при низких темпера-
температурах. При хранении концентрированных растворов медленное выделение
кислорода изолирует раствор от атмосферы и способствует сохранению равно-
равновесного состава пара в сосуде над жидкостью.
Таблица 21
Температуры кипения растворов перекиси водорода при атмосферном давлении
Концентрация перекиси
водорода
мольная
доля
0,0
0,1
0,2
0,3
0,4
0,5
вес. %
0
17,34
32,07
44,73
55,73
65,37
Температура,
100,0
103,0
106,9
111,5
116,7
122,3
Концентрация перекиси
водорода
мольная
доля
0,6
0,7
0,8
0,9
1,0
ее. %
73,90
81,50
88,31
94,44
100,0
Температура,
°С
128,2
134,0
139,7
145,1
150,2
Путем интерполирования кривых давление пара—состав для различных
температур выведены температуры кипения растворов перекиси водорода,
приведенные в табл. 21. Если не считать величин для максимальной концент-
концентрации, то значения точек кипения выше значений, найденных Жигером и Маа-
Маасом [73] путем приложения упрощенного правила Рамзея—Юнга к их дан-
данным, а также выше значений, выведенных из этих же данных с применением
диаграммы Кокса [1]. Повышение температуры кипения, вызываемое ростом
концентрации перекиси водорода, составляет около 1° на 0,02 мольной доли.
Точность данных для температур кипения не настолько велика, чтобы можно
было пользоваться ими для определения концентрации, если требуется точ-
точность, превышающая 0,1—0,3 вес.%. Кроме того, при любых концентрациях
вследствие разложения всегда происходит большее или меньшее выделение
кислорода, причем температура кипения снижается на величину, соответствую-
соответствующую влиянию парциального давления кислорода над растворами.
Степень разложения растворов перекиси водорода при кипячении (без-
(безразлично, под атмосферным или другим давлением) зависит от чистоты раствора
и чистоты сосуда и от степени возможного разложения выделяющихся паров.
Авторы старых работ считали невозможным концентрировать растворы пере-
перекиси путем кипячения при любых давлениях, кроме самых минимальных; од-
однако позже было показано, вчто можно спокойно кипятить растворы перекиси
водорода, имеющие концентрацию по меньшей мере до 90% или даже при-
приближающуюся к 100%, с минимальным разложением в том случае, когда
поверхности всей аппаратуры весьма тщательно очищены и изготовлены из
материалов, не оказывающих каталитического действия. В частности, необходи-
необходимо отметить, что описанные случаи взрывного разложения, имевшие место при
кипячении концентрированной или безводной перекиси водорода, как показано
на стр. 157, обусловлены взрывным разложением самих паров. При соблюдении
надлежащих условий можно избегнуть такого взрывного разложения или
добиться, чтобы разложение протекало плавно., как это описано ниже (стр. 374
и ел.) при анализе разложения паровой фазы. Разложение, происходящее в са-
самой жидкости, способствует плавному кипению растворов перекиси водорода,
причем пузырьки кислорода, образующиеся при разложении, предотвращают
толчкообразное кипение, наблюдающееся при кипячении воды.
198
Глава 5
Критические константы безводной перекиси водорода
Скэтчард, Каванаф и Тикнор, основываясь на предположении, что отно-
отношения критической температуры к температуре кипения для перекиси водо-
водорода и воды одинаковы, вычислили критическую температуру для перекиси,
которая оказалась равной 457° С, или 730° К (для воды 374,2°). По этому
же методу Маас и Хиберт [72] получили величину 459° С. При помощи урав-
уравнения B1) вычислено критическое давление; оно равно 163 000 мм рт. ст.,
или 214 am (для воды 218,2 am). Точность этих подсчетов неизвестна; экспери-
экспериментально критического состояния вряд ли можно достигнуть.
Теплота парообразования жидкой перекиси водорода
В табл. 22 приведены значения для теплоты парообразования безводной
перекиси водорода. Значения, полученные Льюисом и Рэндолом [76], Маасом
и Хибертом [72] и Эгертоном, Эмте и Минкофом [57], не зависят от температуры,
так как они выведены путем приложения уравнения Клаузиуса—Клапейрона
Таблица 22
Теплота парообразования для безводной перекиси водорода
Темпера-
Температура, °С
—
—
18
150
25
155
25
0
25
26,9
0
25
150
Теплота парообразования
кал 1 моль
12 300
11610
11300
11950
11084
10 266
10 200
13010
12 593
12 334
12314
12 930
12 588
11260
кал \г
362
341,5
332
351
326
302
300
382
370,2
362,6
362,0
380,3
370,1
331,2
Литература
Льюис и Рэндол J76]
Маас и Хиберт [72]
Эгертон, Эмте и Минкоф [57]
Жигер и Маас [73]
То же
Келли [77]
Ш
Национальное бюро стандартов [78]
Фоли и Жигер [8]
Мориссет и Жигер [79]
То же
Скэтчард, Каванаф и Тикнор [26]
То же
» »
к дифференцированной форме линейного уравнения зависимости lg p от ЦТ
для давления пара. Данные, использованные Лььрисом и Рэндолом, основаны
на отрывочных наблюдениях Вольфенштейна [68] и Брюля [69]. Жигер и Маас
[73] использовали уравнение упрощенной формы Рамзея и Юнга; по этому
уравнению отношение температур, при которых перекись водорода и вода
имеют одинаковое давление пара, одно и то же при всех условиях. На основе
этого приемлемого предположения можно показать, используя уравнение
Клаузиуса—Клапейрона, что отношение теплот парообразования прямо про-
пропорционально отношению температур, при которых давление пара одно
и то же для обоих веществ. При такой методике вводится зависимость теплоты
парообразования от температуры.
На основе данных Мааса и Хиберта и Жигера и Мааса фирма «Buffalo
Electro-Chemical Co.» построила для давления пара диаграмму [1] типа Кокса,
из которой по методу Отмера [80] выведены теплоты парообразования. Един-
Единственными непосредственными измерениями теплоты парообразования являются
Физические свойства
199
данные Фоли и Жигера [8] и Мориссета и Жигера [791. Эти величины выведены
путем измерения тепловых потерь при выпаривании безводной перекиси водо-
водорода в ледяном калориметре и в калориметре с дифениловым эфиром (т. пл. 26,9°);
они, вероятно, самые надежные из имеющихся значений, так как точность
данных, получаемых при прямом измерении, больше той, которая достигается
при вычислении на основании определения наклона кривой даже по самым
точным данным для давления пара. Теплоты парообразования, приведенные
Скэтчардом, Каванафом и Тикнором [26], выведены из уравнения
АЯу = 0,021066Г2-25,817Г+18412, B6)
полученного путем дифференцирования уравнения B1) с приложением его
к уравнению Клаузиуса—Клапейрона.
Значения теплоты парообразования безводной перекиси водорода, вычис-
вычисленные по уравнению B6), превосходят примерно на 300 кал/моль эксперимен-
экспериментально определенные величины [8, 79]. Расхождение уменьшается по мере
повышения температуры; это свидетельствует о том, что данные, на которых
основано уравнение B6), обладают максимальной точностью при температурах,
не намного превышающих 25°. Для определения теплоты парообразования при
температурах, отличающихся от температур, при которых проводились экспе-
экспериментальные измерения @ и 26,9°), но все же лежащих в этом интервале,
величину указанного выше порядка можно рекомендовать вычесть из значений,
получаемых по уравнению B6); несколько более точные данные можно полу-
получить, комбинируя экспериментальные теплоты парообразования с данными для
теплоемкости. При более высоких температурах рекомендуется пользоваться
одним уравнением B6).
Жигер и Маас [73] (а также фирма «Buffalo Electro-Chemical Co.» [1]),
пользуясь теми же методами, которыми они определяли теплоты парообразо-
парообразования безводной перекиси водорода, вычислили и общую теплоту парообразо-
парообразования водных растворов перекиси. При такого рода расчетах не учитывается
теплота смешения или отклонение растворов от идеальности. Пренебрежение
теплотой смешения при вычислении теплот парообразования может привести
к ошибкам, хотя последние часто не имеют существенного значения. Если
нужно вычислить точные теплоты парообразования растворов, то к теплотам
парообразования воды и безводной перекиси водорода следует добавить те-
теплоты смешения, о которых говорится ниже. В табл. 23 приведены точные
теплоты парообразования для температуры 26,9°. Можно также вычислить
дифференциальные или парциальные теплоты парообразования с учетом соот-
соответствующей теплоты разбавления.
Таблица 23
Общая теплота парообразования водных растворов перекиси водорода
при 26,9°, по Мориссету и Жигеру [79]
Концентра-
Концентрация Н2О2,
вес.%
0
20
40
60
80
100
Теплота парообразования
чистых компонентов, кал/г
1
н2о2 1 н2о
0
72,4
144,8
217,2
289,6
362,0
582,1
465,7
349,3
232,8
116,4
0
Теплота смешения,
кал\г раствора
0
4,7
8,4
9,7
7,4
0
Общая теплота паро-
парообразования, кал (г
раствора
582,1
542,8
502,5
459,7
413,4
362,0
По уравнению Клаузиуса—Клапейрона вычислен наклон кривой равно-
равновесия между жидкостью и паром в тройной точке (—0,42°), который для без
200 Глава 5
водной перекиси водорода оказался равным 2,9-10~5 ami град. Соответствующая
величина для воды составляет 4,4-10~4 ami град.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ПЕРЕКИСИ ВОДОРОДА
Необратимость реакций образования и разложения перекиси водорода
в лабораторных условиях и погрешности, вызываемые разложением, исклю-
исключают возможность точного экспериментального определения некоторых ее
термодинамических свойств. Тем не менее проведено достаточное число прямых
измерений термодинамических свойств, а также измерений других свойств,
позволяющих вычислить разные термодинамические величины. На основании
этих измерений оказалось возможным составить большое число ценных и до-
достаточно полных таблиц по термодинамическим свойствам перекиси водорода
во всех фазах. В порядке изложения сперва рассматриваются свойства, кото-
которые в наименьшей степени зависят от свойств, изложенных в следующих
разделах. Энтальпии фазовых превращений уже рассмотрены в предыдущих
разделах.
Термодинамические функции пара безводной перекиси водорода
Путем исследования инфракрасных спектров и спектров комбинационного
рассеяния пара перекиси водорода можно вычислить энергию, требующуюся
для совершения вибрационных и колебательных движений атомов, составляю-
составляющих перекись водорода. Доля этих вибраций, а также доли трансляционного
и вращательного движений в энергии всей молекулы при различных температу-
температурах можно суммировать при помощи уравнений, основанных на квантовой тео-
теории. Эта методика, позволяющая определить соотношение энергетических
состояний молекулы и числа молекул в каждом таком состоянии с общей энер-
энергией системы, приводит к так называемым функциям распределения, подробные
данные по которым можно найти в стандартных руководствах [81].
Если известна энергия системы, термодинамические свойства можно вычис-
вычислить непосредственно. Как правило, результаты таких вычислений выражают
в виде термодинамических функций, а именно величин: S0, С°р, —(F0—ЩIТ
и (Я0—Щ)/Т, где Ср—теплоемкость при постоянном давлении, F0—свободная
энергия, Н°о—энтальпия при абсолютном нуле, Т—абсолютная температура,
Н°—энтальпия исследуемого вещества в виде идеального газа при давлении
1 am. За основу такой схемы принимают, что энергия элементов в стандартном
состоянии (идеальный газ при давлении 1 am) равна нулю при 0°К. Энтальпия
для гипотетического стандартного состояния при абсолютном нуле Н°о эквива-
эквивалентна общей энергии при абсолютном нуле, поскольку E=H+PV=H+RT
для идеального газа. Таким образом, энтальпия при абсолютном нуле равна
теплоте или свободной энергии образования также при абсолютном нуле.
Значения энтропии 5° представляют собой абсолютные значения для идеаль-
идеального пара при давлении 1 am. Значения, приведенные в табл. 25, имеют неболь-
небольшую погрешность (см. сноску на стр. 203).
Для молекулы перекиси водорода можно вывести следующие зависимости
между этими термодинамическими функциями и экспериментально определен-
определенными свойствами; эти зависимости основаны на функциях распределения и под-
подробно объяснены Астоном [82] и Герцбергом [83]:
S) B7>
fa , ff V гй \
\ )
Термодинамические свойства перекиси водорода 201
B9)
В этих суммарных выражениях представлены члены для долей энергии,
падающих на каждый вид движения молекулы перекиси водорода; значения
для вибрации представлены членами со знаком Б, для крутильных колебаний—
указанными функциями параметров -^ и -^Ц-. (плюс дополнительные члены
Ai * red*
для функций свободной энергии); остальные члены представляют собой доли
энергии, падающие на трансляционное и вращательное движения, например,
для теплоемкости 57?/2 обусловлено трансляционным движением, а 3/?/2—
вращательным. Эти уравнения не содержат членов, соответствующих доле
энергии молекулы за счет различия электронных состояний или вращения
ядер; не рассматривается также и эффект присутствия изотопов. Эти эффекты
ничтожно малы или взаимно уничтожаются; доказательство этого можно
найти у Россини [84].
При выводе этих уравнений сделаны три допущения: 1) при вращении моле-
молекула в целом остается жесткой, 2) вибрации могут быть приняты за гармони-
гармонические колебания и 3) потенциальный барьер, ограничивающий внутреннее
вращение групп ОН вокруг оси О—О, может быть представлен функцией
^=4^0A+со5лф), C0)
где V—потенциал, противодействующий вращению группы ОН через угол ф,
Vo—максимальное значение этого потенциала, а п—число положений на каж-
каждое вращение, при которых потенциал имеет минимальное значение. Соотно-
Соотношение между волновым числом а, характеризующим внутреннее вращение,
и потенциалом следующее:
4,186-1010 n2V0 = 8N/red (теаJ. C1)
Хотя эти допущения могут и не точно отражать истинное положение, они
вполне оправдываются при вычислении термодинамических функций перекиси
водорода. Более подробно этот вопрос рассматривается в гл. 6.
Термодинамические функции для перекиси водорода вычислены по только
что описанным методам разными авторами, а именно Цейзе [85], Микли [86],
Жигером [87], Рибо [88] и Жигером, Лиу, Дагдейлом и Моррисоном [28].
Здесь приведены вычисления последних авторов, поскольку они основаны на
самых новых молекулярных данных, измерениях энтропии по третьему закону
термодинамики и выборе правильной величины ni9 т. е. числа неразличимых
положений, которые принимает молекула при внутреннем вращении. Моле-
Молекулярные данные, использованные в этих вычислениях, приведены в табл. 24
(анализ основы для их выбора дан в гл. 6), а результаты вычислений—в табл. 25.
Доля вибрации в общей энергии взята из стандартных таблиц [81], доля же
крутильных колебаний—из таблиц Питцера и Гвина [89]. Мы следуем обычной
практике и даем результаты вычислений с большей точностью, чем та, которая
оправдывается сделанными допущениями. Поскольку крутильные колебания
составляют значительную долю от общей энергии молекулы и точный размер
этой доли, а также способ учета ее недостаточно точны, данные табл. 25 могут
потребовать исправления.
В таблицах Бюро стандартов [78] (серия III) приводятся данные, анало-
аналогичные указанным в табл. 25, для водорода, кислорода и водяного пара, необ-
необходимые для вычисления некоторых других термодинамических свойств пере+
киси водорода.
232
Глава 5
Таблица 24
Молекулярные данные для перекиси водорода, использованные
при составлении табл. 25 [28]
Фундаментальное колебание
vx (а) растяжение связи О—Н
v2 (а) симметричное изгибание
v3 (а) растяжение связи О О
v4 (а) крутильное колебание
v5 (b) растяжение связи О—Н
ve (b) асимметричное изгибание
Волновое число, см-*
3610
1295
890
520
3610
1266
Моменты инерции, г-см2
/л=2,78в.10-*°
/^=34,0.10-40
/С=33,8ЛО-40
/red =/л/4-0,696.1 -
Барьер, отграничивающий внутреннее вращение Vo=3,5 ккал/моль.
Экспериментально определенная абсолютная энтропия S°298jle:=
=55,66 кал/моль-град К-
Таблица 25
Термодинамические функции для пара перекиси водорода при 1 а/я, по Жигеру, Лиу,
Дагдейлу и Моррисону [28]
Температура,
°К
298,16
300
350
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
/ сО rjO \
1 ** —"О |
1 т )'
кал
моль-град
46,33
46,39
47,87
49,17
51,44
53,38
55,09
56,61
57,99
59,26
60,43
61,56
62,52
63,50
64,41
н°~Н°о
Т '
кал
моль-град
9,42
9,43
9,69
9,94
10,42
10,85
11,21
11,58
11,89
12,15
12,41
12,65
12,87
13,09
13,29
5°,
к-.-...
моль -град
55,76
55,82
57,54
59,11
61,84
64,23
66,31
68,17
69,86
71,40
72,83
74,17
75,41
76,58
77,69
СО
р '
кал
моль -град
11,00
11,02
11,47
11,93
12,68
13,28
13,76
14,15
14,51
14,84
15,15
15,45
15,72
15,97
16,20
кал 1 моль
2 810
2 830
3 391
3 976
5210
6511
7 848
9 263
10 702
12 153
13 656
15 186
16 737
18 322
19 935
Теплоемкость безводной перекиси водорода
Маас и Хэтчер [11] определили теплоемкости жидкой и твердой безводной
перекиси водорода с применением адиабатического калориметра Ричардса
с точностью (по их оценке) 2—4%. При этом они получили значения
0,58 кал/г-град для жидкости в температурном интервале от 0 до 18,5° и
0,47 кал/г-град для твердой фазы в интервале температур от —40 до 0°. Так же,
как и значение теплоты плавления, выведенное из этих же опытов, эти данные
Термодинамические свойства перекиси водорода
203
не точны из-за предварительного плавления, вызванного присутствием неко-
некоторого количества воды [8].
Фоли и Жигер [8] провели измерения теплоемкости в ледяном калори-
калориметре. Для твердой фазы они определили изменения теплосодержания в интер-
интервале температур от —25,1 до —9,6° и получили для теплоемкости значения
0,41 ±0,02 кал/г-град. При помощи того же прибора они получили для теплоем-
теплоемкости жидкости (99,8%-ной перекиси водорода) в пределах температур 0—25°
значение 0,632 ±0,003 кал/г -град. Обнаружив небольшую ошибку в калибро-
калибровании термометра, применявшегося в последней работе, Мориссет и Жигер
[79] ввели поправку, экстраполировали данные этих авторов для растворов
и получили для теплоемкости жидкой безводной перекиси водорода в интервале
температур от 0 до 27° значение 0,628 кал/г-град.
Жигер, Лиу, Дагдейл и Моррисон [28] измерили теплоемкость твердой без-
безводной перекиси водорода в интервале температур от 12 до 273°К; эта работа,
проведенная с адиабатическим калориметром, дала очень точное значение для
теплоемкости твердой фазы и позволила вычислить абсолютную энтропию
перекиси водорода. При температурах приблизительно выше 220°К теплоем-
теплоемкость твердой фазы можно вычислить из уравнения
Ср тв = 2,31 + 0,03967Г кал/моль-град.
C2)
Вычисленная из этих данных [28] абсолютная энтропия пара пере-
перекиси водорода как идеального газа при 298,16°К и давлении 1 am равна
55,66 ±0,12 кал/моль-град*. Зная эту величину, можно определить влияние
внутреннего вращения на термодинамические функции перекиси водорода,
для чего достаточно вычесть из экспериментальной величины энтропию, вычи-
вычисленную из трансляционного, вращательного и других вибрационных движе-
движений. Таким способом найдено, что лучше всего удовлетворяет синусоидной
функции потенциала высота барьера 3,5 ккал/моль, вычисленная по уравне-
уравнению C0).
По теплоемкости паров перекиси водорода прямых измерений не произво-
производилось. Льюис и Рэндол [76] рекомендуют использовать для перекиси водорода
выражение Ср=7,5+0,00427\ выведенное путем измерений для аммиака; в то
время это была единственная экспериментально определенная величина тепло-
теплоемкости для четырехатомной молекулы.
Теплоемкость парообразной перекиси водорода вычислена из молекуляр-
молекулярных данных, указанных в последнем разделе. Эти моляльные теплоемкости [28]
представлены в табл. 25. Они могут быть выражены уравнением
Таблица 26
Коэффициенты уравнения C3) для моляльной теплоемкости пара перекиси водорода,
водяного пара и кислорода
н2о2
Н2О
о2 .
Пар или газ
8
7
6
а
,514
,256
,148
102 ь
0,948
0,2298
0,3102
105 с
-0,300
0,0283
-0,0923
Отклонение, %
среднее
0,73
0,45
0,24
максимальное
1,34
0,74
0,65
* В сообщении [28] указана величина 55,76 кал/моль-град. Однако фактическая
поправка для идеального газа составляет лишь 0,003, а не 0,10 кал/моль-град, как это ука-
указано в работе Жигера с сотрудниками. В табл. 25 приведены опубликованные значения.
204
Глава 5
Для этого уравнения в табл. 26 приведены коэффициенты, пригодные в тем-
температурном интервале 298—1500°К. В этой же таблице представлены коэффи-
коэффициенты [90] к аналогичным уравнениям для водяного пара и кислорода.
Теплоемкость растворов перекиси водорода
Впервые прямые измерения теплоемкости растворов перекиси водорода
проведены Пайком и Грином [91], которые, приблизительно при 20° применив
простую экспериментальную методику, нашли для растворов, имеющих кон-
концентрации 28,30; 30,08; 58,87 и 86,33 вес.%, значения 0,874, 0,870, 0,708 и
0,775 кал/г-град соответственно.' Более точные измерения теплоемкости раство-
растворов проведены Мориссетом и Жигером [79] в изотермическом калориметре,
в котором плавящимся веществом служил дифениловый эфир. Результаты этих
измерений даны в табл. 27. В табл. 28 представлены выравненные значения
теплоемкости растворов.
Таблица 27
Теплоемкость водных растворов перекиси водорода
в температурных пределах 0—26,9°, по Мориссету и Жигеру [79]
Концентрация,
вес.%
0
21,2
41,0
Теплоемкость,
калJa»град
1,0023
0,8930
0,8175
Концентрация,
вес.%
60,1
79,1
98,6
Теплоемкость,
кал id'град
0,757!
0,6953
0,6322
Таблица 28
Теплоемкости водных растворов перекиси водорода в температурных пределах 0—27° [79]
Концентрация,
вес. %
0
10
20
30
40
50
60
70
80
90
100
Мол. вес, г 1 моль
раствора
18,02
18,91
19,89
20,97
22,19
23,56
25,Ю
26,86
28,88
31,24
34,02
Теплоемкость раствора
кал /г* град
1,002
0,945
0,899
0,860
0,823
0,790
0,757
0,725
0,692
0,660
0,628
кал/моль -град
18,06
17,87
17,88
18,04
18,27
18,61
19,00
19,48
19,99
20,62
21,36
АСрМ
8,9
6,72
4,84
3,41
2,50
1,71
1 ;is
0,73
0,46
0,20
0,00
а АСрМ =
Н2О2
-MCV — (
н2о
p
раствора
(см. рис. 42)
Эти данные показывают, что теплоемкость водных растворов перекиси
водорода заметно отклоняется от идеальной среднемолярной теплоемкости.
Отклонение от идеальности отрицательное, т. е. теплоемкость растворов пере-
перекиси водорода меньше вычисленной из теплоемкости чистых компонентов.
В связи с этим отклонением теплота растворения перекиси водорода должна
изменяться с температурой. В следующем разделе, где рассматривается термо-
Термодинамические свойства перекиси водорода
205
динамика смешения перекиси водорода с водой, эти данные для теплоемкости
используются с целью истолкования и вычисления теплот растворения. При
вычислениях необходимо пользоваться разностью &СрМ между теплоемкостями
компонентов в растворе и теплоемкостью самого раствора в расчете на 1 моль
содержащейся в растворе перекиси. В табл. 28 даны также значения ЬСрм-
Фазовые превращения безводной перекиси водорода
Значения энтальпии фазовых превращений безводной перекиси водорода,
приведенные в табл. 29, выбраны из данных, относящихся к этому вопросу
и помещенных в предыдущих разделах. Точность этих данных не настолько
велика, чтобы можно было учесть влияние температур, отличающихся одна от
другой лишь на величину, соответствующую разнице температур замерзания
перекиси водорода и воды.
Таблица 29
Энтальпии фазовых превращений безводной перекиси водорода
Равновесие
Твердое тело—пар
Твердое тело—жидкость
Жидкость—пар
» »
» »
» »
» »
Температура, °С
-0,43
-0,43
-0,43
25
50
100
150
Д#у, кал/моль
15510
2 920
12 593
12 334
12 270
11710
11260
Термодинамика смешения перекиси водорода и воды
Смешение перекиси водорода с водой или разделение их смеси, так же как
и изменение состава водных растворов перекиси водорода путем разбавления
или концентрирования, сопровождается заметными тепловыми эффектами. Сме-
Смешение или разбавление является экзотермическим процессом при всех концент-
концентрациях для температур выше 21°. При более низкой температуре некоторые про-
процессы разбавления являются эндотермическими. Величины этих тепловых
эффектов определены прямыми измерениями, а также на основании измерений
теплот разложения при разных концентрациях или измерений давления пара
растворов. Определены также изменения свободной энергии и энтропии для
процесса смешения.
При рассмотрении этих процессов иногда пользуются недостаточно четкой
терминологией, в связи с чем небесполезно дать сначала некоторые точные
определения. Термины «смешение» и «растворение» применяются лишь для
процессов образования раствора путем смешения безводной перекиси водорода
изводы. Снижение концентрации раствора перекиси водорода введением воды
называется разбавлением. Тепловые эффекты* для этих процессов всегда даются
в"расчете на моли, т. е. на 1 моль перекиси водорода, подвергаемый этим про-
процессам, независимо от количества взятой воды. При обсуждении перекись водо-
водорода везде рассматривается как растворенное вещество, а вода как растворитель.
Все значения для приводимых здесь тепловых эффектов представляют собой
* «Теплота растворения» (или теплота другого процесса) имеет то же численное зна-
значение и тот же знак, что и изменение энтальпии процесса. Как теплота растворения, так
и энтальпия будут иметь отрицательное численное значение, если в течение описываемого
процесса происходит перенос тепла из системы в окружающую среду.
206
Глава 5
интегральные значения для конечных изменений концентрации растворенного-
вещества—перекиси водорода. В таблицах рекомендуемых величин приво-
приводятся: а) интегральная теплота растворения для процесса смешения безводной
перекиси водорода с водой с образованием данного раствора и б) теплота раз-
разведения с получением бесконечного разбавления, представляющая тепловой
эффект разбавления раствора бесконечно большим количеством воды.
Эти концепции могут быть представлены следующими символами. Теплота
растворения для случая образования бесконечно разбавленного раствора
из безводной перекиси водорода равна Д#х и
Н2О2
ооН2О
Н2О2ооН2О
C4)
Разность этих уравнений приводит к величине
А^з = АЯ1-ДЯ2
для интегральной теплоты растворения всего процесса
Н2О2 + МН2О -> H2O
Значение Д# может быть положительным (поглощение тепла) или отрицатель-
отрицательным (выделение тепла) в зависимости от того и другого процесса. При беско-
бесконечном разведении раствора, содержащего М молей воды на 1 моль перекиси
водорода, теплота разбавления до бесконечности равна Д#2 и
Н2О2.МН2О-ЬооН2О -» Н202.„(М+оо)"Н20 + ДЯ2. C5)
C6)
C7)
До опубликования Мориссетом и Жигером [79] экспериментальных опре-
определений теплот разбавления и теплоемкостей растворов перекиси водорода
трудно было выбрать правильную величину из многочисленных встречавшихся
в литературе значений теплот разбавления или смешения. В настоящее время
эти неясности устранены и большую часть ранее рекомендованных величин
можно согласовать в свете измерений Мориссета и Жигера. Эти авторы измеряли
тепловой эффект разбавления перекиси водорода концентрацией около 99 вес. %
в изотермическом калориметре с применением в качестве расплавляемого ве-
вещества льда или дифенилового эфира. Они пользовались обычной методикой:
стеклянную ампулу, содержащую один из компонентов, вносили в пробирку
с другим компонентом, выжидали до наступления термического равновесия
и, разбив ампулу, смешивали оба компонента. Применялись пробы перекиси
Таблица 30
Теплоты разбавления растворов перекиси водорода водой при 0 и 26,9°,
по Мориссету и Жигеру [79]
Темпера-
Температура, °С
26,9
26,9
26,9
26,9
26,9
26,9
26,9
26,9
26,9
26,9
26,9
Концентрация
Н2О2, вес. %
начальная
98,70
98,70
98,70
98,70
98,70
98,70
98,70
98,70
98,70
98,70
98,70
конечная
89,70
88,89
79,38
71,00
62,02
54,88
50,25
49,61
42,77
30,10
13,97
Теплота
разбавления,
кал \ моль
Н2О2
— 146,9
-156,7
—295,6
—406,1
—502,0
—575,4
—618,3
—627,4
—668,3
—736,1
—773,0
Темпера-
Температура, °С
26,9
0
0
0
0
0
0
0
0
0
0
Концентрация
Н2О2, вес. %
начальная
98,70
98,70
99,40
99,40
98,70
99,40
99,40
99,40
99,40
99,40
24,5
конечная
5,26
85,97
70,22
60,50
46,27
45,92
38,07
37,24
19,69
19,05
9,34
Теплота
разбавления,
кал 1 моль
Н2О2
—803,0
— 195,0
—417,3
—515,1
—593,6
—597,3
—669,4
—669,9
—671,9
—671,6
+ 42,2
Термодинамические свойства перекиси водорода
207
I-
200
I -400
водорода весом от 0,6 до 5 г, причем измеренные тепловые эффекты составляли
от 8 до 34 кал. Предполагается, что разложение, вызванное наличием битого
стекла, настолько незначительно, что ничтожно малым тепловым эффектом
можно пренебречь. Экспериментальные результаты Мориссета и Жигера при-
приведены в табл. 30. Путем графического экстраполирования найдено, что инте-
интегральная теплота растворения безводной перекиси водорода при 0° с образова-
образованием 99,40%-ного раствора равна —10 кал/моль, с образованием 98,7%-ного
раствора —21 кал/моль; при 26,9° интегральная теплота растворения с образова-
образованием 98,70 %-ного раствора
равна —22 кал/моль. eg о
Эти данные показывают, g
что теплота разбавления рас-
растворов перекиси водорода,
как это и следует из данных
для теплоемкости растворов,
в заметной степени зависит
от температуры. Величина
этой температурной зависи-
зависимости дана на рис. 41, где
показана интегральная тепло-
теплота растворения или смешения
безводной перекиси водоро-
водорода с образованием раствора
определенной концентрации
в виде функции этой концен-
концентрации. На рис. 41 вся кри-
кривая для 26,9° проведена через
экспериментальные точки.
Кривая для 0° вычислена из
данных для 26,9° и теплоемко-
стей растворов, приведенных
в последнем разделе. На этом
рисунке дана температурная зависимость теплоты растворения, которая пока-
показывает, что при более низких температурах разбавление некоторых растворов
перекиси водорода может оказаться эндотермическим процессом. Эксперимен-
Экспериментальное подтверждение этого вывода можно найти в табл. 30, данные которой
показывают, что разбавление 24,5%-ной перекиси водорода до концентрации
9,3% при 0° требует поглощения системой 42,2 кал/моль перекиси водорода для
обеспечения постоянной температуры.
-600
I
5 -800
СО
I -woo
20
40 60
Н202, вес. %
80
100
Рис. 41. Интегральная теплота растворения пе-
перекиси водорода, по Мориссету и Жигеру.
Смешение
AH*-
Т2:н2о2-мн2о
<i+M)cPpflcmAAT
ДНМТ,
Разложение
мн2о 2-*- A+м)н2о •
|(Ср„202+МСрН20)АТ
н2о2+ш2о-
Т,: Н202-МН20
Рис. 42. Процессы смешения и разложения для одного моля пере-
перекиси водорода.
Для определения наилучших значений тепловых эффектов, имеющих
место при смешении перекиси водорода и воды, рекомендуется исходить из
теплот разбавления при 26,9° (найденных Мориссетом и Жигером) в сочетании
208
Глава 5
Таблица 31
Интегральная теплота растворения и теплота разведения
до бесконечного разбавления для перекиси водорода и воды
Концентрация
Н2О2, вес. %
0
10
20
30
40
50
60
70
80
90
100
Интегральная теплота растворе-
растворения, кал(моль Н2О2
26,9°
—830
—818
—795
-759
—707
—640
—548
—440
—310
— 161
0
25°
—813
—805
—785
—752
—703
—636
—545
—438
—310
— 160
0
Теплота разведения
при 25° до беско-
бесконечного разбавления,
кал 1 моль Н2О2
0
— 8
— 27
— 61
— ПО
— 177
—268
—375
—503
—653
—813
с данными теплоемкости. Ниже дан краткий анализ метода такого сочетания
и выведены результаты для стандартной температуры 25°.
На рис. 42 представлен процесс разложения перекиси водорода и смеше-
смешения ее с водой. Если рассматривать только процесс смешения, то окажется,
что разность тепловых эффектов
разделения перекиси водорода и
воды при двух температурах рав-
равна произведению разности тепло-
емкостей несмешанных компонен-
компонентов и раствора на разность темпе-
температур, или с1(&Нм)/сН = АСрМ.
В табл. 28 приведены значе-
значения АСрм- Таким образом, если
известна интегральная теплота
растворения перекиси водорода
при температуре Г2, можно опре-
определить интегральную теплоту
100
О
чоо
?-200
^ -300
* -500
5
-600
-100
-800
-900
-1000
-1100
-1200
\
s.
jw
20\
\56°
\
растворения при
ния
Тг из уравне-
о ю 20
30 40 50 60
н2о2, вес. %
70 80 90 100
C8)
В табл. 31 приведены реко-
рекомендуемые интегральные теп-
теплоты растворения при 26,9°,
взятые из работы Мориссета
и Жигера, интегральные теплоты
растворения при 25°, вычислен-
вычисленные из первых по уравнению C8),
и теплоты разведения до беско-
бесконечного разбавления по урав-
уравнению C6).
Имея надежный метод для
расчета влияния температуры на
теплоту разбавления, можно сравнить полученные результаты с другими
источниками соответствующих данных. На рис. 43 показаны значения теплоты
Рис. 43. Теплота разведения указанных раство-
растворов до бесконечного разбавления.
СгДе-Форкран A2—16°); Л Рот, Грау и Мейхснер B0°);
О Кубашевский и Вебер B0°); Q Пайк и Грин B0°);
V Скэтчард, Каванаф и Тикнор (вычислено);—Морис-
" сет и Жигер A4, 20 и 56°).
Термодинамические свойства перекиси водорода
209
разведения до бесконечного разбавления, выведенные из рабог Форкрана [67],
Рота, Грау и Мейхснера [92], Пайка и Грина [91], Кубашевского и Вебера
[20] и Скэтчарда, Каванафа и Тикнора [26]. Все эги измерения проведены при
20°* только Форкран работал при 12—16°, а Скэтчард, Каванаф и Тикнор не
указали температуры. На рис. 43 не приведены данные Эванса и Юри [93],
не указавших точно условий работы, а также величин, которые могут быть
выведены из измерений теплот разложения. Значения теплот разложения
с этой точки зрения менее точны, и при отсутствии достаточных причин целе-
целесообразно эти данные не учитывать, поскольку без них получается более
четкая диаграмма. Тем не менее рис. 44 дает возможность оценить теплоты
разбавления, выводимые из величин теплот разложения.
Поскольку большая часть
указанных выше данных по теп- '
лотам разбавления не пересчи-
пересчитана на бесконечное разбавле-
разбавление, мы повсюду добавляли
соответствующие теплоты для
разведения до бесконечности,
взятые из работы Мориссета и
Жигера, с целью приведения
всех данных к общей основе.
Если не считать значение Форк-
Форкрана, то все данные хорошо сог-
согласуются. Тем не менее для вы-
вывода теплот растворения необхо-
необходимо базироваться исключитель-
исключительно на данных Мориссета и
Жигера, так как другие авторы
применяли метод адиабатической
калориметрии и пользовались
| -22,8
% -22,9
g4 -23,0
1 -23,1
I-
1-
23,2
-23,3
23,4
-23,5
-23А
-
-
д
«
и
L
1^*
\
\
\
\
N
о
«
40 50 60 70
Н202, вес. %
Рис. 44. Теплота разложения растворов пере-
перекиси водорода при 25° и 1 am по реакции
Н2О2.МНАк->A+М)Н2Ол,+у О2газ.
V Матесон и Маас; Д Рот, Грау и Мейхснер; <*> Фон-
Фонтана; (_, Фоли и Жигер; Д Мор несет и Жигер.
О 10 20 30 40 50 G0 70 80 90 100
недостаточно точными значения-
значениями теп л оем костей, что исключает
возможность выяснения точного
влияния температуры на измере-
измерения. Ранее нельзя было уста-
установить рекомендуемые величины
теплот разбавления в связи
с кажущимися большими рас-
расхождениями результатов работ, основанных на измерениях теплот разложения и
на измерениях давления пара, из-за относительно небольших теплот разбавле-
разбавления для концентраций перекиси водорода ниже 30%, а также вследствие отсут-
отсутствия одного какого-либо ряда данных, охватывающего весь интервал составов.
В свое время Быховский и Россини [94] рекомендовали значения:
460 кал/моль для теплоты разбавления и 910 кал/моль для теплоты разведения
до бесконечного разбавления; Микли [86] рекомендует величину 840 кал/моль,
Национальное бюро стандартов [78] 810кал/моль. Теперь эти значения должны
быть заменены величинами, выведенными из табл. 31 и содержащими соответ-
соответствующую температурную поправку.
Остановимся на методе, использованном Скэтчардом, Каванафом и Тик-
нором [26] для расчета энтальпии, энтропии и свободной энергии смешения.
При рассмотрении давления пара перекиси водорода представлено уравне-
уравнение A1), которое отражает принятую функцию избыточной свободной энергии
смешения ff перекиси водорода и воды. По уравнению Sf = — dFx/dT после
подстановки констант получается следующее уравнение:
-*„) 0,97.
C9)
14 Перекись водорода
210
Глава 5
Аналогично из соотношения H^ — Fx+TSx получается уравнение для избы-
избыточной теплоты смешения на 1 моль раствора:
1017 + 85A ~2xw)+ 13A -2*J«].
D0)
Поскольку тепловой эффект смешения для идеального раствора равен
нулю, избыточная теплота смешения Н1? представляет общий тепловой эффект
для смешения п молей безводной перекиси водорода и A —п) молей воды
с образованием 1 моля раствора, мольная доля воды в котором равна х.
Интегральная теплота растворения для получения раствора с мольной долей
xw по воде будет &H = Hx4/(l—xw).
В табл. 32 приведены термодинамические величины для процесса смеше-
смешения, вычисленные Скэтчардом, Каванафом и Тикнором. Отклонение этих
величин свободной энергии смешения от значений, вычисленных на основа^
нии результатов измерений при отдельных температурах, не превышает
6 кал, причем это справедливо для растворов высокой концентрации, обладаю-
обладающих низким давлением пара. Уравнение A1) (выравненная функция для
свободной энергии) давало значения, которые находились в пределах раз-
разброса индивидуальных значений, вычисленных из результатов измерения
упругости пара, выполненных Жигером и Маасом [73]. Ввиду недостаточно
ясной зависимости функции свободной энергии от температуры температурные
коэффициенты в выражения для энтропии и энтальпии смешения не введены.
Сравнения, показанные на рис. 43, свидетельствуют о том, что теплоты разбав-
разбавления, основанные на данных измерения упругости пара, хорошо со-
согласуются с величинами, полученными путем экстраполяции к 56° зна-
значений Мориссета и Жигера. Это доказывает, что аналогично теплотам паро,
образования теплоты разбавления, основанные на измерениях упругости паров-
лучше всего удовлетворяют диапазону температур, вблизи которого проводи-
проводились измерения F0—93°).
Таблица 32
Термодинамические данные для процесса смешения перекиси водорода и воды,
по Скэтчарду, Каванафу и Тикнору [26]
Концентрация перекиси
водорода
мольная
доля
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
вес, %
0
17,34
32,07
44,73
55,73
65,37
73,90
81,50
88,31
94,44
100,0
Избыточная
свободная
энергия при 75°
F**, кал 1 моль
0
— 66,5
— 116,1
— 149,3
— 166,9
— 169,8
— 158,8
— 135,1
—99,7
-54,2
0
Избыточная
энтропия
кал 1 моль'град.
0
—0,087
0,155
0,204
0,233
0,243
0,233
0,204
0,155
0,087
0
Избыточная
энтальпия
Н^> кал (моль
0
—97
— 170
—220
—248
—254
—240
—206
— 154
—85
0
Теплота раз-
разведения до
бесконечного
разбавления,
кал 1 моль
0
— 119
—239
—356
—469
—581
—689
—795
—897
—995
— 1089
Жигер и Маас [73] на основании измерений упругости пара определили
также парциальные моляльные теплоты смешения. Кубашевский и Вебер [20]
Термодинамические свойства перекиси водорода
211
использовали активности, найденные Жигером и Маасом, для вычисления пар-
парциальных моляльных свободных энергий смешения, которые в сочетании с изме-
измеренными теплотами растворения позволили вывести величины для энтропии
смешения. Мы этих значений не приводим, так как считаем, что при определе-
определении термодинамических величин для процесса смешения надежнее применять
данные Скэтчарда, Каванафа и Тикнора. Хотя эти данные и рекомендованы,
все же было бы желательно проверить их и улучшить путем сочетания со зна-
значениями теплот смешения по Мориссету и Жигеру. При помощи уравнения
d(hF/T)/dT=—АЯ/Г2 можно проверить надежность зависимости свободной
энергии смешения от температуры, по Скэтчарду, Каванафу и Тикнору, путем
сопоставления с данными, вытекающими из исследования Мориссета и Жигера.
Термодинамические величины для разложения перекиси водорода
Разложение перекиси водорода по уравнению Н2О2—>Н20+%02 сопро-
сопровождается выделением теплоты, для определения которой был произведен ряд
экспериментов. Равновесие данной реакции в обычных лабораторных условиях
весьма благоприятствует разложению. Вычислены свободная энергия и кон-
константа равновесия этой реакции с использованием измеренной теплоты разло-
разложения и ранее приведенных термодинамических функций.
Здесь не приводятся значения теплот разложения, определенные в старых
работах [95], так как между ними наблюдаются некоторые расхождения; хотя
одно или два из них находятся в пределах результатов современных определе-
определений, точность этих старых данных трудно все же оценить, и она, вероятно,
не соответствует современным стандартам калориметрической точности. Вычис-
Вычисленное Медаром [96] (из измерений, проведенных в реакторе с постоянным
объемом) значение—24,3 ккал/моль дль теплоты разложения 100%-ной перекиси
водорода при 1 am и 17° точно так же не согласуется с результатами других"
известных измерений, а поэтому и не принимается здесь во внимание.
Остальные экспериментальные значения теплоты разложения перекиси
водорода при давлении 1 am приведены к общей основе 25° путем использования
значений теплоемкости [79] (см. рис. 44). При рассмотрении рис. 42 легко убе-
убедиться, что разность теплот разложения раствора при двух температурах
равна разности feMoeMKOcrefl продуктов разложения и исходного раствора,
умноженной на разность температур, т. е. d (AHo)/dT = &Сро- В табл. 33 приве-
приведены значения &CpD.
Таблица 33
Теплота разложения растворов перекиси водорода при 25° и 1 am, по Мориссету
и Жигеру [79]
1
Реакция: Н2О2-МН2ОШ-
Концен-
Концентрация
н2о2,
вес. %
0
10
20
30
40
50
Моли
воды на
1 моль
Н2О2, М
ОО
16,98
7,55
4,40
2,83
1,89
Теплота
разложения,
ккал1моль
Н2О2
-22,62
-22,63
-22,65
-22,68
-22,73
-22,80
АС а
pD
9,1
6,90
5,02
3,59
2,68
1,89
Концен-
Концентрация
н2о2,
вес. %
60
70
80
90
100
Моли
воды на
1 моль
Н2О2, М
1,26
0,809
0,472
0,210
0
Теплота
разложения,
ккал\моль
Н2О2
-22,89
-23,00
-23,13
-23,28
^23,44^
кг а
pD
1,36
0,91
0,64
0,38
0,18
(см. рис. 42).
212 Глава 5
На кривой (рис. 44) и в табл. 33 даны рекомендуемые значения теплоты
разложения растворов перекиси водорода, основанные на рекомендуемой
теплоте растворения (см. предыдущий раздел) в сочетании со значением
—23,44±0,03 ккал/моль для теплоты разложения безводной перекиси водорода
при 25° по реакции
Н2О2ж -» Н2Ож + уО2газ. D1)
Указанное значение для теплоты разложения основано на работе Мориссета
и Жигера [79]. Доказательство правильности этого выбора приводится ниже
при рассмотрении экспериментальных данных. Изменение стандартной сво-
свободной энергии при 25° для реакции D1) AF°=—27,92 ккал/моль.
Матесон и Маас [9] определяли теплоту разложения 10-граммовых проб
растворов перекиси водорода в адиабатическом калориметре. В качестве ката-
катализатора разложения применялась двуокись марганца. По сообщению этих
авторов, разложение заканчивалось внезапно, причем авторы не вводили
поправки на остаточную перекись водорода. Сделана поправка на водяной пар
и вычислена некоторая часть водяного эквивалента калориметра. Путем линей-
линейной экстраполяции теплоты разбавления, основанной на средней величине
из четырех определений (два определения с 38,05%-ной перекисью и два
с 97,15%-ной), вычислена теплота разложения безводной перекиси водорода
(—23,45 ккал/моль).
Рот, Грау и Мейхснер [92] пользовались той же методикой, что и Матесон
и Маас, но калибровали свой калориметр путем электрического нагревания
остатка от разложения. Эти авторы объединили результаты своей работы
с данными Матесона и Мааса и вычислили теплоту разложения, которая ока-
оказалась равной для безводной перекиси водорода —23,48 ккал/моль, а для бес-
бесконечно разбавленной перекиси водорода —22,65 ккал/моль.
Фонтана [97] измерил теплоту разложения разбавленного раствора пере-
перекиси водорода (Н2О2-8880 Н2О). Калориметр был прокалиброван электрическим
способом, причем в качестве катализатора служила двуокись марганца B г);
через раствор в калориметре^протускали азот со скоростью 125 мл/мин для
устранения неточностей, связанных с присутствием в калориметре кислорода
от разложения (наличие заметных расхождений при пробных измерениях теп-
теплоты растворения твердого магния в соляной кислоте доказало необходимость
пропускания через раствор тока инертного газа). Фонтана ввел поправку на
разложение (около 10 кал), произошедшее за время между анализом раствора
и калориметрическим определением. В качестве среднего значения из трех опре-
определений он получил значение —22,59±0,02 ккал/моль и считал, что эта цифра
подтверждает данные Рота, Грау и Мейхснера.
Фоли и Жигер [8] проводили измерения с пробами весом около 1 г в ледя-
ледяном калориметре в интервале концентраций перекиси водорода 12—100%.
Для катализирования разложения в течение периодов от 3 до 10 час. применяли
отрезок платинированной платиновой проволоки. Скорость реакции (большую
в начале опыта) регулировали путем последовательного опускания проволоки
в раствор и извлечения ее оттуда до тех пор, пока реакция не становилась
менее энергичной в связи с уменьшением концентрации раствора. К концу раз-
разложения закономерного изменения скорости не происходило, а поэтому наблю-
наблюдались нерегулярные периоды активности. В этих опытах разложение до конца
не доводили; неразложенная перекись в некоторых опытах составляла до 10%
начального количества. Выбор метода для введения поправки зависел от состоя-
состояния, в котором находилась остаточная перекись водорода, т. е. от того, пред-
представляла ли она собой капли сравнительно концентрированной перекиси,
распыленные по стенке калориметра, или же разбавленный раствор. Фоли
и Жигер считают, что общий измеренный тепловой эффект равняется сумме
Термодинамические свойства перекиси водорода
213
теплоты разложения прореагировавшей части и теплоты разбавления непро-
реагировавшей части от начальной концентрации до конечной; эта поправка
колебалась от 0 до 96 кал/моль.
Для определения теплоты разложения растворов перекиси (от 24,6%-ной
концентрации до 98,6%-ной при 26,9°) Мориссет и Жигер 179] пользовались
изотермическим калориметром с дифениловым эфиром. Катализатором слу-
служила коллоидная суспензия платины; время от времени небольшие количества
этой суспензии приливали в калориметр для обеспечения устойчивой, но малой
скорости разложения. Разложение проб весом 0,7—1,8 г доходило до конца
примерно за время от 2 до 4 час; в табл. 34 представлены результаты отдель-
отдельных опытов.
Таблица 34
Теплота разложения растворов перекиси водорода. Экспериментальные
значения при 26,9°, по Мориссету и Жигеру [79]
Концентрация,
Н2О2, вес. %
24,64
24,72
25,10
49,58
74,23
98,52
98,62
Теплота разложения,
ккал\моль Н2О2
-22,67
—22,65
-22,67
-22,80
-23,01
-23,45
-23,42
Интегральная тепло-
теплота растворения,
кал 1 моль
-780
-780
-779
— 644
-388
-25
-24
Теплота разложения
100%-ной Н2Оо,
ккал 1моль
-23,45
-23,43
-23,45
-23,44
-23,40
-23,48
-23,44
Из анализа приведенных данных видно, что работа Мориссета и Жигера
проведена с соблюдением ряда важных особенностей, которые частично недо-
недоучитывались в прежних работах. Применение изотермического калориметра
устраняет погрешности в отношении определения точной температуры реакции
и избавляет от необходимости знать теплоемкость. Разбавленный коллоидный
Таблица 35
Теплота, свободная энергия и константа равновесия
для разложения паров перекиси водорода
Реакция: Н202Газ
Температура,
0
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
ЛЯ, ккал}моль
-25,85
-25,26
-25,26
-25,24
-25,26
-25,34
-25,40
-25,50
-25,58
-25,65
—25,73
-25,81
-25,89
-25,97
-26,05
AF, ккал\м.оль
-25,85
-29,39
-29,42
-30,81
-32,20
-33,57
-34,93
-36,28
-37,62
-38,95
-40,27
-41,53
-42,92
-44,20
-45,50
lg К
21,55
21,43
16,84
14,07
12,23
Ю,91
9,91
9,13
8,51
8,00
7,56
7,21
6,90
6,29
214
Глава 5
катализатор обеспечивает медленное, но полное разложение, не оставляя сом-
сомнений в отношении конечной концентрации. Целесообразно также применять
сравнительно концентрированную перекись водорода, пЪскольку при исполь-
использовании разбавленных растворов возможные ошибки в определении концентра-
концентрации оказывают на результаты значительное влияние.
После выбора подходящей величины теплоты разложения перекиси водо-
водорода можно получить значения теплоты реакции и свободной энергии реакции
Н2
^ С>
2газ
D2)
как функции температуры. Для этого нужно сочетать величину теплоты раз-
разложения: 1) с термодинамическими функциями для перекиси водорода, приве-
приведенными в табл. 25 (стр. 202); 2) с термодинамическими функциями для водя-
водяного пара и кислорода, содержащимися в таблицах Национального бюро стан-
стандартов (серия III) [78]; 3) с величиной теплоты парообразования безводной
перекиси водорода при .25° A2,334 ккал/моль) и 4) со значением теплоты паро-
парообразования водыпри 25° A0 ^БЩккал/моль). Эти значения приведены в табл. 35.
Они несколько отличаются от опубликованных Жигером [87], так как для соста-
составления табл. 35 использованы значения теплот разложения и парообразования,
полученные в последнее время.
Термодинамические величины для образования перекиси водорода
Теплоту, свободную энергию и константу равновесия для реакции обра-
образования перекиси водорода из элементов
Нггаз + Оггаз ^-> НгОггаз D3)
можно вычислить по способу, аналогичному описанному в последнем разделе,
Эти значения приведены в табл. 36 и основаны на следующих данных:
Таблица 36
Теплота, свободная энергия и константа равновесия
для образования парообразной перекиси водорода
Температура,
0
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
Реакция: Н2газ + (
АН, ккал\м.оль
-31 ,26
-32,52
-32,54
-32,80
-33,01
-33,15
-33,30
-33,38
-33,48
-33,56
-33,63
—33,69
-33,73
-33,77
-33,79
^2газ —> НгОггаз
AF, ккал1моль
-31,26
-25,24
-25,20 "
-22,71
-20,16
-17,58
-14,98
-12,36
-9,73
-7,08
-4,43
-1,83
+0,92
+3,56
+6,24
Ig К
18,51
18,35
12,41
8,8U
6,40
4,68
3,38
2,36
1,55
0,88
+ 0,33
-0,15
-0,56
-0,91
1) на теплоте разложения жидкой безводной перекиси водорода при 25°,
равной 23,44 ккал/моль; 2) на теплоте парообразования безводной перекиси
Термодинамические свойства перекиси водорода
215
водорода при 25°, которая составляет 12,334 ккал/моль; 3) на теплоте обра-
образования водяного пара при 25° [78], равной—57,798 ккал/моль; 4) на термо-
термодинамических функциях перекиси водорода, приведенных в табл. 25 (стр. 202);
5) на термодинамических функциях водорода и кислорода, взятых из таблиц
Национального бюро стандартов (серия III) [78].
Эти значения свободной энергии образования парообразной перекиси
водорода можно использовать и для вычисления свободной энергии образова-
образования перекиси водорода в жидкой фазе (как безводной, так и в растворе
с моляльной активностью, равной единице при 25°). Стандартная свободная
энергия конденсации пара перекиси водорода при 25° оказывается равной
— 3530 кал/моль при расчете с использованием в уравнении Д/г =
= — RT In (ph2/760) для давления пара при 25° значения 1,95 мм рт. ст.
При сочетании этой величины со свободной энергией образования парообраз-
парообразной перекиси водорода при 25° из табл. 36 для реакции
= Н2О2Ж»
D4)
получается значение AF°= —28,78 ккал/моль.
Кроме того, путем использования данных по давлению пара можно
вычислить моляльную активность безводной перекиси водорода в качестве
растворенного вещества; она равна 214,1 (см. табл. 72 на стр. 328). Свобод-
Свободная энергия-обратимого переноса одного моля безводной жидкой перекиси
водорода в бесконечное количество раствора перекиси водорода с моляль-
моляльной активностью, равной единице, при 25° будет &F = RT\n{2\4,\/\) =
= 3,180 ккал/моль Н2О2.
Этот результат в сочетании с данными, полученными для реакции D4),
позволяет найти значение Д/г0= —31,95 ккал/моль при 25° для реакции
+ Оггаз — H2C>2aq. D5)
Только что вычисленные величины основаны на значительно более точных
данных, но все же можно считать, что они только подтверждают ^величины,
приведенные в книге Латимера [98] и выведенные Льюисом и Рэндолом [76]
из отрывочных данных. Так, Латимер дает для реакции D4) А/70 =
= —28,23 ккал/моль, а для реакции D5) А/70= —31,47 ккал/моль.
Таблица 37
Свободная энергия образования перекиси водорода
* в растворе при 25°
Реакция:
Концентрация
Н2О2, вес. %
0
10
20
30
40
50
Свободная энер-
энергия образования
AF, ккал/моль
Н2О2
-31,19
-30,65
-30,25
—29,98
-29,71
Концентрация
Н2О2, вес. %
60
70
80
90
100
Свободная энер-
энергия образования
AF, ккал\моль
Н2О2
. -29,48
—29,26
—29,07
-28,91
-28,78
Аналогичным образом составлена табл. 37, в которой приведены значе-
значения свободной энергии для процесса образования и смешения безводной
жидкой перекиси водорода с бесконечным количеством раствора указанной
концентрации.
216 Глава 5
Стандартные электродные потенциалы перекиси водорода
Свободные энергии образования, вычисленные в предыдущем разделе,
можно использовать для непосредственного вычисления электродных потен-
потенциалов перекиси водорода. Для системы перекись водорода — кислород *
Н2О2 = О2 + 2Н+ + 2е, Е° = - 0,693в; D6)
при подстановке свободной энергии образования перекиси водорода
( — 31,95 ккал/моль) для моляльности, равной единице, в уравнение Д/7 =
= — п%Е стандартный электродный потенциал Е° оказывается равным
-0,693в.
Путем суммирования уравнения D6) с уравнением для системы вода —
кислород [98]
2 Н2О = О2 + 4Н+ + 4е, Е° = - 1,229в, D7)
для системы вода — перекись водорода получаем
2Н2О = Н2О2 + 2Н+ + 2е, ?°=—1,76в. D8)
Аналогично сказанному в отношении свободной энергии образования, из кото-
которой они выведены, эти электродные потенциалы также подтверждают уже
давно примен&ощиеся потенциалы Льюиса и Рэндола [76].
Приведенные выше электродные потенциалы относятся к стандартному
условию, что раствор кислоты имеет моляльность, равную единице. В следую-
следующем разделе, где рассматривается диссоциация, будет показано, что свободная
энергия образования пергидроксильного иона HO~AF°= — 15,230 ккал/моль.
При помощи этой величины можно вычислить электродные потенциалы в рас-
растворе щелочи с моляльностью, равной единице:
ОН" + НО" = О2 + Н2О + 2е, Е% = 0,084*, D9)
3 ОН" = НО" -f Н2О + 2в, Е% = - 0,87в. E0)
* Следует напомнить, что перекись водорода содержит кислород в степени оки-
окисления, промежуточной между этими степенями для молекулярного кислорода и воды,
как показано на следующей схеме:
Восстановление >
О2 ^± НО2 ^t Н2О2 ~± ОН ^± Н2О
@) С"т) (-1) ("° ()
< Окисление
Таким образом, реакцию D6), происходящую в системе перекись водорода—кислород,
можно рассматривать как окисление перекиси водорода, если читать уравнение D6)
слева направо, или как восстановление кислорода, если читать его справа налево. Эта
двойственность еще больше увеличивается, если рассматривать реакцию D6) при проте-
протекании слева направо как представляющую восстановительный потенциал перекиси водо-
водорода, так как в этом случае должно иметь место одновременное восстановление какого-то
другого реагента.
Точно так же реакцию D8) в системе вода—перекись водорода можно рассматривать
как окисление воды при чтении уравнения слева направо, как восстановление перекиси
водорода при чтении справа налево и как «окислительный потенциал» перекиси водорода
при чтении справа налево с учетом действия перекиси на какой-то другой реагент.
В литературе встречаются все такого рода обозначения в зависимости от привычки
того или иного автора или желания его подчеркнуть известное явление. Пожалуй, на-
наименьшую путаницу дают такие обозначения, как «система перекись водорода—кислород»
или «система вода—перекись водорода», но можно также использовать и любое другое
обозначение для указания направления реакции. Направление, в котором пишутся реак-
реакции на электродах, и знаки, присваиваемые потенциалам, являются условными; мы при-
придерживаемся системы, принятой Латимером [98]. В книге этого автора имеются и другие
необходимые определения, например определение термина «стандартный», рассматривают-
рассматриваются также и теоретические аспекты.
Термодинамические свойства перекиси водорода 217
Делахей, Пурбе и Ван-Рисельберг [99] опубликовали ценную диаграм-
диаграмму электродного потенциала перекиси водорода в виде функции концентра-
концентрации и рН. Аналогичная диаграмма приведена на стр. 300.
Был поставлен ряд опытов с целью прямого измерения электродных
потенциалов перекиси водорода и выяснения точных реакций, определяю-
определяющих эти потенциалы [100]. В значительной мере эти исследования были
посвящены изучению влияния природы электрода и обработки его поверх-
поверхности на потенциал, который он принимает в растворе перекиси водорода.
Изучалось также влияние изменения концентрации перекиси водорода
и водородных ионов, а также присутствия добавок. Пожалуй, наиболее
ценная работа в этой области принадлежит Борнеману [101]. Этот автор
исходил из гипотезы, что наиболее положительный потенциал по отношению
к кислородному электроду [т. е. реакции D7)], который может быть измерен
в перекиси водорода и который подчиняется надлежащей зависимости от
концентрации, ближе всего подходит к значению потенциала системы
перекись водорода—кислород [т. е. реакции D6)]. Наиболее подходящим
электродом оказалась платина, причем был разработан способ химической
и электролитической обработки, которая за счет изменения каталитической
активйюсти поверхности сообщала ей наиболее положительный и воспроиз-
воспроизводимый статический потенциал в разбавленных растворах перекиси водорода
(однонормальных по кислоте). Результаты этой работы при экстраполирова-
экстраполировании к одномолярной перекиси водорода дают потенциал —0,69 в. Борнеман
вывел из этой величины и значения —0,63 в, определенного раньше для потен-
потенциала образования перекиси водорода на электроде, насыщенном кислоро-
кислородом, среднее значение Е° = —0,66 ± 0,03 в для потенциала системы перекись
водорода —кислород. Суммирование с реакцией D7) дает ?° — — 1,80 б для потен-
потенциала системы вода — перекись водорода. Учитывая экспериментальные трудно-
трудности, получение такого результата можно считать значительным достижением.
Берль [102] измерил потенциал системы перекись водорода — кислород
в щелочном растворе. Пористый угольный электрод, через который осуще-
осуществляли барботаж кислорода, приобретал стандартный потенциал Е% = 0,0416 в
в соответствии с реакцией D9), для которой выше вычислена величина
Е% = 0,084 в. Это, по-видимому, является достаточным подтверждением
проведенного измерения. Берль считал, что реагирующей частицей в дей-
действительности является ион пергидроксила НОз и что элемент является
полностью обратимым. Результаты этой работы подтверждены Хиклингом
и Вилсоном [103] в процессе изучения анодного разложения перекиси водо-
водорода на электродах из различных материалов. Они предпочитают писать эту
реакцию следующим образом:
E1)
Хиклинг и Вилсон [103] и Хиклинг [104] дают обзор отдельных стадий,
выражающихся суммарной реакцией E1), и указывают на значение этой
реакции для явления перенапряжения кислорода.
Калузек [105], Хакобян [106], Керн [107] и Корита [108] нашли, что
система перекись водорода — кислород является обратимой на ртутном
электроде в кислой, основной или нейтральной среде, тогда как Кольтгоф
и Лингейн [109] отрицают эту обратимость на указанном электроде. Новак
и Гейеровский [ПО] показали, что потенциал восстановления кислорода до
перекиси водорода не изменяется в растворе тяжелой воды, потенциал же
восстановления перекиси водорода до воды изменяется в тяжелой воде
в такой же мере, в которой изменяется перенапряжение водорода. Нет
никаких доказательств, что восстановление перекиси водорода до воды, т. е.
реакция D8), протекает в какой-либо мере обратимо. Электродные процессы
с участием перекиси водорода рассмотрены также в гл. 2.
218
Глава 5
Термодинамические величины для диссоциации перекиси водорода
Под общим названием «диссоциация» рассматриваются различные моно-
мономолекулярные реакции перекиси водорода. Сюда не относится диссоциация
на молекулярный водород и кислород, которая обсуждалась выше, в разделе
об образовании перекиси водорода, а также разложение, под которым
понимается исключительно реакция перекиси водорода с образованием воды
и молекулярного кислорода.
Таблица 38
Теплота, свободная энергия и константа равновесия
для диссоциации перекиси водорода на гидроксилы
Реакция: Н202Газ —* 2 ОНгаз
Температура,
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
ЛЯ, ккал\моль
51,26
52,66
52,67
52,84
53,13
53,22
53,31
53,31
53,32
53,31
53,29
53,27
53,23
53,19
53,14
AF, ккал/моль
51,26
43,11
43,06
39,80
36,49
33,16
29,81
26,46
23,11
19,75
16,39
13,09
9,65
6,33
2,97
1«Г /С
—31,61
-30,36
-21,75
-15,95
-12,08
—9,31
-7,23
-5,61
-4,32
—3,26
-2,38
-1,62
-0,99
-0,43
В табл. 38 приведены теплота, свободная энергия и константа равновесия
для реакции
E2)
Значения, приведенные в табл. 38, несколько отличаются от величин, опубли-
опубликованных Жигером [87], в связи с использованием вновь найденных значений
для теплот разложения и парообразования перекиси водорода.
Можно написать еще четыре другие неионные реакции диссоциации,
для которых термодинамические величины могут быть вычислены со значи-
значительной точностью [78]. Эти реакции, а также их теплоты и свободные
энергии при 25° приведены ниже:
н2
н2
н2
н.
лЛ газ —
i^2 газ =
!^-'а газ ==
2^2 газ =
Н2ОГаз+Огаз
^Пгаз -г ^2 газ
П2 газ ~т" ^^газ
2Нгаз + 2Огаз
+
+
~г
дя°
33,90
136,72
150,86
255,04
Ар°(ккал/моль)
+ 25,60,
+ 122,41,
+135,23,
+ 232,39.
E3)
E4)
E5)
E6)
Для полноты списка возможных неионных реакций диссоциации нужно
добавить еще следующую:
Н2О2 Газ = Нг
2 газ.
E7)
Для теплоты этой реакции можно дать только приближенное значение,
так как для точного определения необходимо знать термодинамические
Термодинамические свойства перекиси водорода
219
свойства радикала пергидроксила. Сумма теплот реакции E7) и реакции
НО2 газ — Нг
2 газ
О2
E8)
в соответствии со значением, полученным для реакции E4), должна соста-
составлять 137 ккал/моль. В качестве первого приближения можно в соответствии
с данными Юрея, Доуси и Раиса [111] принять, что тепловой эффект при
отрыве водородного атома от перекиси водорода такой же, как и при отрыве
атома водорода от пергидроксильного радикала, что приводит для теплот
Значения,
Таблица 39
приведенные разными авторами для теплот диссо-
диссоциации Н2О2 и НО2
Теплота диссоциации,
ккал/моль
Н2О2, реак-
реакция E7)
68
66—76
86—96
90
102
НО2, реак-
реакция E8)
68
70—60
50—40
46
36
Литература
Юрей, Доуси, Райе [111]
Уолш [112]
Разные авторы [113]
Робертсон [114]
Эванс, Хаш и Юри [115]
реакций E7) и E8) к величине 68 ккал/моль. Однако, так же как и в случае
с водой, вернее считать, что отрыв первого атома водорода от перекиси
водорода должен потребовать затраты большей энергии, чем отрыв второго
атома. В табл. 39 приведены значения, предложенные для указанных дис-
диссоциаций. Наименее вероятными нужно считать значения, определенные
Уолшем [112] на основании чисто теоретических рассуждений. Другие
крайние значения для реакции E7) даны Эвансом, Хашом и Юри [115],
которые для вывода пользовались интересной методикой; к сожалению, мы
не можем здесь на ней остановиться. Ни одно из промежуточных значений
[113], по-видимому, не имеет под собой более надежной основы (цифры,
приводимые Хейтлером [113], были как будто получены опытным путем,
но никаких подробностей об этих опытах не сообщается). Робертсон [114]
методом электронной бомбардировки определил верхний предел теплоты
реакции E7); он равен 90 ккал/моль. Таким образом, окончательная величина
для теплоты реакции E7) не известна; мы примем для этой теплоты значе-
значение 90 ккал/моль, следовательно, значение для реакции E8) оказывается
равным 46 ккал/моль. Было бы весьма желательно поставить дополнитель-
дополнительные опыты для более точного определения необходимых значений.
Из различных возможных реакций ионной диссоциации перекиси водо-
водорода наибольшее значение имеет кислотная диссоциация
H2O2aq = H+ + HO2. E9)
На стр. 329, где обсуждается*~эта реакция, указаны значения Эванса и Юри
[116], использованные для нахождения константы равновесия и теплоты
реакции. При 25° К = (Н+) (HCQ/(H2O2) = 2,24-102, АЯ°= +8,2 ккал/моль
и АР= + 15,89 ккал/моль. Из этих данных вычисляется свободная энергия
образования иона пергидроксила НО2 в растворе при 25° и моляльности,
равной единице; она составляет— 15,23 ккал/моль.
Робертсон [114] изучал диссоциацию паров перекиси водорода при
электронной бомбардировке с образованием ионов Н2О*, Н2О+, НО*, OH+,
220 Глава 5
О* и О+. Показано, что ионы Н2О\ О* и О+ появляются в эксперименталь-
экспериментальных условиях лишь за счет ионизации присутствующих воды и кислорода,
образующихся в качестве продуктов разложения перекиси водорода. Отно-
Относительные интенсивности ионов Н2Оз, НО* и ОН+ равны 100, 10 и 20;
потенциалы возникновения их составляют соответственно 12,1; 16,1 и 16,0 эв.
Из этих видов частиц особого интереса заслуживает положительный гидрок-
сильный ион ОН", поскольку он аналогично пергидроксильному иону может
принимать участие в промежуточных стадиях реакции перекиси водорода
в растворе.
Для ряда реакционных схем, в которых постулируется участие перекиси
водорода, необходимо знать термодинамические значения для радикалов,
образующихся при диссоциации перекиси водорода. Расчеты и оценка термо-
термодинамических свойств таких радикалов и продуктов их диссоциации опу-
опубликованы Ла-Ьшером [98], Эвансом, Хашом и Юри [115] и Юри [117]. По
Латимеру, свободные энергии образования частиц ОН, НО2 и O.J в водном
растворе равны соответственно +8,53, +3,0 и +13,0 ккал/моль. Притчард
[118] дал обзор методов определения и подсчета электронного сродства
таких частиц, как О, О", О2, О~ и НО2.
Термодинамические величины для различных реакций перекиси водорода
Помимо возможности вычисления термодинамических величин для
реакций с участием перекиси водорода на основании изложенных данных
в сочетании со стандартными термодинамическими величинами [78,98],
имеется еще ряд непосредственно измеренных величин и таблиц для измерен-
измеренных или вычисленных величин. Теплота реакции
2Fe2+ + Н2О2 + 2Н+ = 2Fe3+ + 2НаО F0)
измерена Эвансом, Баксендейлом и Юри [119] и оказалась равной
— 65,5 + 2/шх/г, по Фонтана [97], она составляет — 71,78 + 0,12 ккал. Фонтана
[97] измерил также теплоту реакции
U4+ + Н2О2 = UO'f + 2H+ F1)
и нашел величину— 58,00 + 0,02 ккал. Траутц и Пакшвер [120] измерили
теплоту реакции
CaSO3 • 2Н2О + Н2О2ач -> CaSO4 • 2Н2О + Н2О F2)
и нашли значение—81,3 ккал. Эванс, ХашиЮри [115] и Юри [117] опублико-
опубликовали таблицы с вычисленными термодинамическими величинами для разных
реакций.
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ПЕРЕКИСИ ВОДОРОДА
Электропроводность
Поскольку перекись водорода является очень слабым электролитом,
электропроводность ее водных растворов приближается к электропровод-
электропроводности чистой воды, причем измерения электропроводности осложняются
необходимостью удаления следов примесей. Перекись водорода, как и вода,
является превосходным ионизирующим растворителем. Таким образом,
диссоциация электролитов может происходить в любом интервале составов.
Высокая реакционная способность перекиси водорода ограничивает число
металлов, пригодных для измерения электропроводности; наиболее практич-
практичным является выбор для этой цели литого олова. Несмотря на эти трудности,
в продаже имеются растворы перекиси водорода, обладающие удельной электро-
электропроводностью, сравнимой с электропроводностью обыкновенной дистилли-
дистиллированной воды (т. е. 10~5 ом'1-см'1 или меньше), причем путем тщательной пере-
Электрические свойства перекиси водорода
221
гонки этих растворов можно получить продукт, электропроводность которого
очень близка к значению, принимаемому за достижимый предел.
Калверт [121] в ходе работ по измерению диэлектрической проницаемости
применив платиновые электроды, определил удельную электропроводность
4,5%-ного раствора, оказавшуюся равной 2,89- 10~8 ом'1-см'1. В более поздних
работах по измерению электропроводности растворов едких щелочей, нейтра-
нейтрализованных перекисью водорода, Калверт [122], пользуясь электродами,
покрытыми оловом, при 25° определил эквивалентную ионную проводимость
пергидроксильного иона НО" при бесконечном разбавлении 48,5 ом-экв~1-см2.
i
if2
о
V
10 20 30 40 50 60 70
Н202, вес. %
80 90 100
Рис. 45. Удельная электропроводность водных растворов
перекиси водорода при 25°.
О Гросс и Тейлор; д Шамб; [J Рот и Шенли.
Хотя это значение, вероятно, должно быть еще экспериментально проверено,
оно все же указывает, что эквивалентная проводимость перекиси водорода при
бесконечном разбавлении составляет около 400 ом-экв'1-см2. Иойнер [123]
при помощи покрытых оловом электродов нашел для 20—30%-ных растворов
перекиси водорода при 25° значительно более высокие значения удельной
электропроводности, а именно около \^'ъом'1-см~1\ эти данные он получил при
попытке измерить константы диссоциации перекиси водорода. Удельная электро-
электропроводность практически безводной перекиси водорода впервые была изме-
измерена Катбертсоном и Маасом [124] при помощи оловянных электродов и по-
постоянного тока; авторы дают для нее значение 2-10~6сш 1-см~1; по-видимому,
эта цифра относится к 0°. Опубликованы значения электропроводности хорошо
очищенных водных растворов перекиси водорода для всего интервала концент-
концентраций [125]. Эти измерения проводились при 25° и 1000 гц в ячейке с оловян-
оловянными электродами, не сообщавшейся с атмосферой (константа ячейки 0,08037);
при этом для разбавления 98,5%-ной перекиси водорода с удельной электро-
электропроводностью 8,2 • 10 ом~х • см ~г была использована вода с удельной электропро-
электропроводностью 5-10 ом'1-см'1. Результаты этой работы представлены на рис. 45,
показывающем максимум удельной электропроводности для 50%-ной перекиси
при значении 5,8-10'^м'1-см'1. Гросс и Тейлор [126] входе работ по измерени-
измерениям диэлектрической проницаемости перекиси водорода, находившейся в равно-
равновесии с воздухом в ячейке с электрополированными электродами из нержавею-
нержавеющей стали, нашли только, что удельная электропроводность наиболее концент-
концентрированного раствора, с которым они работали (99,2 вес.%), была чуть ниже,
а именно 4- 10~7ом-см~1. Роти Шенли [127] приготовили99,9%-ную перекись
водорода и измерили ее удельную электропроводность C,9- \0~7ом~1>см~1).
Форма кривой (рис. 45) сохраняется и для менее чистых растворов, т. е.
обладающих более высокой электропроводностью, вплоть до величин порядка
Ю^ом^-см'1. Если удельная электропроводность выше этой величины, то она,
как правило, непрерывно возрастает по мере последовательного разбавления
концентрированной перекиси водорода. Это объясняется тем, что форма кривой
электропроводности постепенно все более и более определяется примесями,
222 Глава 5
вероятно (хотя и не обязательно), вводимыми в перекись по мере ее разбавле-
разбавления во все больших количествах. Что касается чистого продукта с низкой
электропроводностью, то в этом случае форма кривой зависит от изменения кон-
концентрации и состояния продуктов диссоциации воды и перекиси водорода.
О влиянии различных добавок на электропроводность растворов перекиси водо-
водорода упоминается еще в последнем разделе, где рассматриваются свойства
многокомпонентных систем, содержащих перекись водорода (стр. 249).
Хотя электропроводность растворов перекиси водорода или воды, приме-
применяемой для их разбавления, часто принимается за показатель чистоты, необ-
необходимо подчеркнуть, что скорость разложения перекиси водорода с малой
электропроводностью может быть иногда значительной, особенно при повышен-
повышенных температурах, например при ускоренных испытаниях на стабильность
или при генерировании пара. Предполагается, что эта значительная скорость
разложения вызвана присутствием веществ в коллоидном состоянии, не влияю-
влияющих на электропроводность. Обычно причиной возникновения таких трудностей
является недостаточное внимание к качеству дистиллированной воды.
Юнг [128] показал, что при применении оловянных электродов для изме-
измерения электропроводности растворов перекиси водорода при 0° получаются
недопустимо заниженные величины. Показано также, что оловянные электроды
дают значения, превышающие приблизительно на 7% величины, получаемые
при применении электрополированных электродов из нержавеющей стали.
Эти эффекты не исследованы. Добавка щелочи к 90%-ной перекиси перед ди-
дистилляцией (до концентрации 0,0015 ы. по едкому натру) является эффективным
способом снижения удельной электропроводности до величины, показанной
на рис. 45. В работах Гросса и Тейлора [126] и Рота и Шенли [127] перегонку
щелочной перекиси водорода (с целью концентрирования изучавшихся раство-
растворов) для устранения возможности загрязнения из-за уноса проводили методом
спокойного выпаривания (без кипячения). Еще большее увеличение степени
очистки перекиси водорода, вероятно, не вызовет заметного снижения ее
удельной проводимости по сравнению с показанной на рис. 45.
Диэлектрическая проницаемость
Из ранних измерений диэлектрической проницаемости растворов перекиси
водорода можно указать лишь на работы Дьюара и Флеминга [129],изучавших
замороженные растворы 6%-ной перекиси водорода и 5%-ной перекиси на-
натрия при температуре жидкого воздуха, и Калверта [121], который нашел для
диэлектрической проницаемости 37%-ной перекиси водорода при 18° значение
84,7; путем довольно сомнительной экстраполяции он вывел для безводной
перекиси величину 92,8. Катбертсон и Маас [124] измерили диэлектрическую
проницаемость при 0° резонансным методом для всего интервала составов
растворов и доказали наличие максимума в 120 единиц для 35%-ной перекиси
водорода, тогда как для воды и безводной перекиси водорода получены соот-
соответственно значения 84,4 и 89,2. В связи с возражениями, которые встретила
эта работа, Линтон и Маас [130] проверили использованную ими технику,
улучшили ее и на основании новых опытов порекомендовали изменить значение
диэлектрической проницаемости безводной перекиси водорода на величину
93,5. Более поздние измерения диэлектрической проницаемости смесей перекиси
водорода с эфиром, проведенные Линтоном и Маасом [131], привели к значе-
значениям 93,7 и 91 для диэлектрической проницаемости 100%-ной перекиси при 0°,
что подтверждает результаты прежних работ.
Гросс и Тейлор [126] проверили измерения диэлектрической проницае-
проницаемости растворов перекиси водорода всех составов внутри интервала от —65 до
30°. Эти авторы считают, что результаты Катбертсона и Мааса ошибочны в свя-
связи с тем, что последние не учитывали влияния электропроводности на резуль-
Электрические свойства перекиси водорода
223
таты измерений. Их мнение подтверждается тем, что Катбертсону и Маасу
не удалось получить для воды значения, соответствующего принятым по
Уаймэну [132] величинам. Гросс и Тейлор применили мостиковую схему нового
типа. Они пользовались ячейкой из электрополированной нержавеющей стали,
кварца и тефлона (политетрафторэтилена), сконструированной таким образом,
что измеренная емкость не зависела от содержащегося в ячейке объема жид-
жидкости. Результаты измерений этих авторов для равновесной проводимости
воды совпадали с данными Уаймэна (погрешность была меньше 0,2%) и не
зависели от электропроводности даже при величине ее 2,5-10~4 ом'1-см.
Измерения проводились примерно при 200 кгц, хотя колебания частоты в пре-
пределах 0,1—1,5 мггц в случае работы с 97%-ной перекисью водорода при —9°
не оказывали заметного влияния на равновесие мостика; доказано, что диспер-
дисперсионных эффектов в интервале частот, использованных для измерений, не
должно быть. Тщательность приготовления проб перекиси водорода и кон-
конструкция ячейки позволили проводить измерения при температурах до 30—35°,
причем признаки разложения появляться не успевали; способность перекиси
водорода к переохлаждению дала возможность провести исследования при тем-
температурах на 20—40° ниже температур замерзания растворов.
Таблица 40
Изотермы диэлектрической проницаемости в зависимости от состава для водных раство-
растворов перекиси водорода а , по Гроссу и Тейлору [126]
Концентра
мольная
доля
0,0000
0,0406
0,0638
0,1582
0,1602
0,2241
0,3151
0,3626
0,3785
0,4947
0,6187
0,7529
0,9377
0,9813
0,9850
ция Н2О2
вес. %
0,0
7,4
11,4
26,2
26,5
35,3
46,5
51,8
53,5
64,9
75,4
85,2
96,6
99,0
99,2
Температура, °С
30
76,7
76,3
G6,9)
G7,6)
G775)
G6,8)
G7,7)
G8,6)
G5,9)
G3,6)
F9,9)
F9,5)
F8,6)
20
80,4
80,7
81,3
81,9
(82,4)
83,1
82,6
83,3
83,9
—
81,3
78,8
74,7
73,2
73,6
10
84,1
85,0
85,7
87,3
87,7
88,8
88,6
88,9
89,8
(89,5)
87,3
84,3
80,1
78,7
79,0
0
88,0
89,4
90,4
92,8
93,1
94,6
94,7
95,3
95,9
(95,0)
93,5
90,4
85,9
84,7
84,9
— 10
(93,7)
(95,3)
98,2
98,4
100,6
101,2
102,3
102,6
101,7
100,1
97,16
92,4
91,3
(91,4)
—20
—
—
103,7
106,7
108,0
109,9
109,6
109,0
107,7
104,56
—
98,7
—30
—
—
113,3
115,4
117,0
116,9
117,1
115,9
112,06
106,3
-40
124,7
125,0
A25,0)
126,5
125,3
122,06
—
а Скобки указывают на экстраполированные значения.
6 Эти значения исправлены на основании опубликованной рукописи.
Величины, полученные Гроссом и Тейлором, представлены в изотермах
диэлектрическая проницаемость—состав, показанных на рис. 46 и в табл. 40;
эти значения взяты из выравненных кривых по графикам зависимости диэлектри-
диэлектрической проницаемости от температуры. Значения для диэлектрической прони-
проницаемости безводной перекиси водорода выведены путем небольшой экстрапо-
экстраполяции и выражаются следующим уравнением:
г = 84,2 - 0,62^ + 0,0032^. F3)
Это уравнение представляет данные со стандартным отклонением на 0,13 и мак-
максимальным наО,2. Талат-Эрбен [ 133] использовал эти данные для доказательства
224
Глава 5
предложенной им формы зависимости диэлектрической проницаемости от тем-
температуры и вывел уравнение
lg (S7) = 4,78792 - 0,0015587\ F4)
Это уравнение хорошо соответствует значениям, полученным по уравнению F3).
Очевидно, что рекомендуемые величины, определенные Гроссом и Тейлором,
ниже значений, приведенных Катбертсоном и Маасом, и что диэлектрическая
проницаемость безводной перекиси водорода ниже диэлектрической проницае-
проницаемости воды при любых температурах. Наличие максимума подтверждается,
и2и2, вес %
О Ю 20 30 40 50 60 70 80 90 ___ 100
130
0,2 0,3
0,4 0,5 0,6
Мольная доля Н2О2
0,7 0,8 0.9
10
Рис. 46. Изотермы диэлектрической проницаемости для водных
растворов перекиси водорода.
О интерполированные данные; • экстраполированные данные.
но последний наблюдается при 0° и концентрации перекиси водорода около
55 вес. %, причем величина диэлектрической проницаемости в максимуме лишь
на 8—9% выше, чем для воды. При снижении температуры максимум стано-
становится более резким и смещается в сторону более высоких концентраций пере-
перекиси водорода. Максимальная величина, а именно 157, найдена для диэлектри-
диэлектрической проницаемости 75,4%-ной перекиси водорода при —67,1°.
Магнитная восприимчивость
При введении в магнитное поле какого-либо вещества напряжение поля
которое проходит через вещество, может быть больше или меньше, чем напря,
жение в пространстве вокруг вещества. Для выражения способности магнит-
магнитного поля проходить через вещество по сравнению со способностью его проходить
через вакуум пользуются величиной, называемой проницаемостью. Такие
вещества, как кислород, которые более проницаемы, чем вакуум, и в которых
магнитное поле концентрируется, называются парамагнитными. Такие веще-
вещества, как вода и перекись водорода, которые обладают меньшей проницае-
проницаемостью, чем вакуум, и в которых магнитное поле ослабляется, называются
диамагнитными. Парамагнитные вещества обладают постоянным магнитным
моментом и притягиваются более интенсивной зоной неоднородного магнитного
поля, тогда как диамагнитные вещества приобретают индуцированный магнит-
магнитный момент при помещении их в магнитное поле и стремятся выйти из более
интенсивных зон неоднородного поля.
Электрические свойства перекиси водорода
225
-70
со
I
L
«5
'3
§-5
-4
Проницаемость какого-либо вещества отличается от проницаемости вакуума
(для которого проницаемость равна единице) на величину 4тс&; таким образом,
Р=1+4тс&. Величина k называется объемной магнитной восприимчивостью
и имеет для парамагнитных веществ положительный знак, для диамагнитных—
отрицательный. Магнитную восприимчивость можно выражать так же, как
удеЛЬНуЮ ВОСПРИИМЧИВОСТЬ (xg = k/p) И МОЛЯрнуЮ ВОСПРИИМЧИВОСТЬ (Km = Mv.g).
Имеется четыре источника данных по магнитной восприимчивости пере-
перекиси водорода и ее водных растворов. Маас и Хэтчер [134] измеряли воспри-
восприимчивость безводной перекиси водорода по методу Квинке при 10° и получили
для объемной восприимчивости значе-
значение— 8,8-10 ед. CGSM на 1 см3. Грей
и Фаркхарсон [135] вывели для без-
безводной жидкости значение хт =—
—A6,73+0,20). 10-6eA.CGSMHa 1моль
на основании измерений над раствора-
растворами, содержащими до 99,78 вес.% пере-
перекиси водорода, при 18° с применением
весов Кюри—Шенево; данных по от-
отдельным измерениям эти авторы не
опубликовали. Савитри и Рао [136]
измеряли восприимчивость 6—32%-
ных растворов перекиси водорода по
методу Гюи без указания темпера-
температуры. Наиболее полное исследование
принадлежит Нейдингу и Казарнов-
Казарновскому [62], которые также применяли
метод Гюи для всего интервала соста-
составов при 10—12°, а также для 96—
98%-ной перекиси водорода в жид-
жидком, переохлажденном и твердом
состоянии для интервала температур
от +18 до—183°.
Данные всех этих авторов пред-
представлены на рис. 47 в виде удельной
восприимчивости; для пересчета цифр
Мааса и Хэтчера была использована
недавно установленная величина плотности, равная 1,459 г/см3. Если судить по
рис. 47, то имеется два согласованных между собой ряда данных, а именно Са-
Савитри и Рао и Мааса и Хэтчера, с одной стороны, и Нейдинга и Казарновского
и Грея и Фаркхарсона—с другой. Мы считаем, что более достоверны значения
второй группы исследователей. Об этом свидетельствуют следующие факты.
Савитри и Рао измеряли плотности своих растворов, но эти плотности не согла-
согласуются с общепринятыми величинами; они (если исходить из комнатной тем-
температуры 10—25°) завышены при низких концентрациях и занижены при высо-
высоких. При пересчете этих данных на основе обычно принятых плотностей получа-
получаются значения, близко сходящиеся сданными Нейдинга и Казарновского. Отсут-
Отсутствует видимая причина отклонения единственной точки, данной Маасом и
Хэтчером, от остальных величин; однако совпадающие результаты работ трех
групп авторов имеют большее значение, чем этот единичный результат. Данным
Грея и Фаркхарсона можно придавать меньшее значение, посколькуJfэти
авторы не опубликовали оригинальных данных. Нейдинг и Казарновский
пользовались цифрами плотности для 18° и таким образом ввели известную
погрешность в расчеты, основанные на данных, полученных при 10—12°*
эта^ ошибка составляет около 0,005% и лежит в пределах точности их измере-
измерений. Ни в одной работе не указано на необходимость устранения возможности
15 Перекись водорода
20
40
60
80
100
Рис. 47. Магнитная восприимчивость
водных растворов перекиси водорода.
О Нейдинг и Казарновский; V Грей и Фарк-
Фаркхарсон; Q Маас и Хэтчер; Л Савитри и Рао;
те же авторы (с учетом предпочтитель-
предпочтительных плотностей).
226
Глава 5
образования пузырьков в трубке с пробой; кроме того, при работах с перекисью
водорода всегда существует возможность пересыщения кислородом. Оба эти
явления [137] должны приводить к получению более положительных значений
магнитной восприимчивости.
В настоящее время спорным представляется вопрос, обязательно ли
магнитная восприимчивость является линейной функцией состава растворов.
Из данных Нейдинга и Казарновского по методу наименьших квадратов полу-
получено уравнение
= - 0,724 +
F5)
Никаких указаний об отступлениях от линейности для растворов перекиси
водорода не получено.
Сравнение этого уравнения с принятой величиной для воды при комнатной
температуре—0,720- 10~6ед. CGSM на 1 г показывает, что к величинам, вычис-
вычисляемым по уравнению F5), для согласования их с величиной, принятой для
воды, необходимо добавлять по 0,004 ед.СОБМ на 1 г. Указанная величина
близка к приведенной в самой статье точности расчетов. На этом основании
можно рекомендовать следующие значения магнитной восприимчивости для
безводной перекиси водорода: k A0°) = —0,73-10 ед. CGSM на 1 см3;
*g= — 0,50- 1СГ6ед. CGSM на 1 г и хт=—17- Ю ед. CGSM на 1 моль. Магнит-
Магнитная проницаемость безводной перекиси водорода равна, таким образом,
A—9,2)-10~6ед. CGSM на 1 см8 или очень близка к магнитной проницаемости
для воды, причем оба вещества в одинаковой степени диамагнитны.
Удельная восприимчивость диамагнитного вещества в соответствии с тео-
теорией не зависит от температуры. Для воды обнаружен лишь небольшой тем-
температурный эффект, для перекиси водорода можно считать, что в пределах
точности данных этот эффект ничтожно мал. Измерения магнитной восприим-
восприимчивости концентрированной перекиси водорода как функции температуры,
принадлежащие Нейдингу и Казарновскому, приведены в табл. 41. Данные не
указывают на наличие действительного изменения даже в области переохла-
переохлаждения. При кристаллизации восприимчивость увеличивается на 2,4% в
сторону положительных значений. Для сравнения укажем, что для воды вос-
восприимчивость возрастает в сторону положительных значений на 2,2%. Мы
не приводим значений плотности, по которым были вычислены величины
табл. 41.
Таблица 41
Магнитная восприимчивость перекиси водорода при разных
температурах, по Нейдингу и Казарновскому [62]
Температура,
°С
18,2
17,0
9,8
- 5,0
- 6,0
- 12,2
- 12,2
- 28,7
- 78,5
-183,0
Агрегатное состояние
Жидкое
»
»
»
Твердое
Переохлажденная жид
кость
Твердое
—xg. 106 ед.
98,15%-ная
Н2О2
0,520
0,523
—
0,523
0,505
0,508
.
0,503
CGSM на 1 г
96,66%-ная
Н2О2
0,526
0,523
0,511
• _
0,508
0,510
Влияние перекиси водорода на различные виды излучений
227
Измерений магнитной восприимчивости парообразной перекиси водорода
не производилось. Можно считать, что, как и для других диамагнитных
веществ, удельная восприимчивость не изменяется при испарении.
ВЛИЯНИЕ ПЕРЕКИСИ ВОДОРОДА НА РАЗЛИЧНЫЕ ВИДЫ ИЗЛУЧЕНИЙ
В этом разделе рассматриваются экспериментальные результаты измерений
преломления видимого света, магнитооптическое вращение, поглощение коле-
колебаний с длиной волны от микроволновой до ультрафиолетовой области, рас-
рассеяние в видимом свете и дифракция рентгеновых лучей и электронов пере-
перекисью водорода и ее растворами. По указанным вопросам имеется значительное
количество литературы и проведено много превосходных работ, однако можно
надеяться на еще большие успехи в будущем, особенно в области абсорбцион-
абсорбционной спектроскопии, так как совершенствование техники позволяет улучшить
разрешающую способность спектрографов. Материал, касающийся струк-
структуры, по возможности рассматривается в гл. 6. Экспериментальные методы,
использованные при некоторых измерениях, нельзя описать кратко и четко,
поэтому для ознакомления с такими подробностями, как описание источников
излучения, типа пленки и измерительных приборов и т. д., необходимо обра-
обратиться к оригинальным работам. Обычная техника работы в этой области
вполне удовлетворительно описана в монографии под редакцией Вайсбергера
[138].
Показатель преломления
Поскольку измерения показателя преломления можно использовать как
точный и легкий метод определения концентрации растворов перекиси водорода,
тщательности этих измерений придается особенно большое значение. Экспери-
Экспериментальная техника отличается достаточной простотой, однако при работе
с перекисью водорода приходится принимать особые меры предосторожности,
Таблица 42
Сводка измерений показателя преломления перекиси водорода
Интервал
концен-
концентраций,
вес. %
8—25
100
96—100
14-89
0—30
0—99
0—99
100
Метод
Минимального
отклонения
—
Минимального
отклонения
Тот же
Фотометр Пул ь-
фриха
Тот же
Точный рефрак-
рефрактометр Аббе
Рефрактометр
Хилджера-
Аббе
.. Длина волны
ИС, F, G; NaO
и'<нс,f, о;
NaD. Ti
NaD
НС, F, С
Na0
Не. f, о- Naz>
NaD
NaD
Темпе-
Температура,
°C
23—25
14—20
22—24
24,5
20
16—28
20—25
15—20
Данные
для безводной
перекиси водорода
п
1,4062
1,4139а
1,3910
—
1,4087
1,4067
1,4058
а Позже [1J2] эта величина была исправлена на 1,4063.
20,4
22
24,5
—
20
25
20
X
NaD
NaD
оо
—
NaD
NaD
NaD
Литература
Kappapa [5]
Брюль [69]
Маас и Хэтчер
[И]
Катбертеон и
Маас [124]
Шурл [139]
Жигер [140J
Жигер и Жоф^
фрион [141]
Эгертон, Эмте и
Минкоф [571
15*
228
Глава 5
чтобы избежать изменения концентрации тех небольших проб, которые обычно
применяются. Испарение раствора недопустимо. Держатель пробы или призма
не должны оказывать каталитического действия. Измерениям мешают также
пузырьки, образующиеся при разложении.
В табл. 42 дана сводка опубликованных данных по показателям прелом-
преломления водных растворов перекиси водорода. Измерения в старых работах,
вероятно, были достаточно точными, но точность определения концентрации
проб осталась неизвестной. Тщательно проведенные Жигером и Жоффрионом
[141] одновременные измерения концентрации и показателя преломления под-
подтверждают это заключение. Анализ экспериментальной техники и сравнение
с принятыми данными Жигера и Жоффриона показывают, что в процессе
измерений Катбертсона и Мааса [124] и Жигера [140] концентрация проб сни-
снижалась. Если принять за стандарт показатель преломления безводной перекиси
водорода, то Брюль [69], Маас и Хэтчер [11], Эгертон, Эмте и Минкоф [57],
опубликовавшие результаты измерений по безводной перекиси водорода, также
работали с перекисью, содержавшей некоторое, хотя и очень малое, количество
воды.
Таблица 43
Экспериментальные значения показателя преломления водных растворов перекиси
водорода, по Жигеру и Жоффриону [141]
н2о2,
вес. %
о,оо
Ю,10
19,98
30,1!
40,03
50,10
25
nD
1,3325х
l,3388i
1,3452!
1,35203
1,35885
1,3661!
20
nD
1,33299
1,33946
1,34603
1,35296
1,35986
1,36724
н2о2,
вес. %
60,6в
70,15
79,8б
92, Зб
96,2б
99,30
25
nD
1,37389
l,3815i
1,38927
1,39998
1,40333
1,40607
20
nD
1,37508
1,38284
1,3907а
1,40157
1,40496
1,40774
Экспериментальные данные Жигера и Жоффриона для линии D натрия,
приведенные в табл. 43, получены с прецизионным рефрактометром Аббе, т. е.
с прибором, который, по общему мнению [142], обеспечивает точность до 5-10~5
(или еще лучшую). При проведении этих измерений открытые места замазки
призмы покрывали церезином, температуру регулировали с точностью в пре-
пределах 0,02°, а для проверки отсутствия разложения в пробах применяли вспо-
вспомогательную линзу. Пробы приготовляли путем разбавления в достаточной
мере устойчивого концентрированного раствора, полученного путем перегонки
из 90%-ного продукта; они были нестабильными при низких концентрациях
перекиси водорода и поэтому к ним добавляли следы станната. Проведено под-
подробное изучение перманганатного метода анализа, использованного для опре-
определения концентрации проб.
В табл. 44 приведены координаты усредненной плавной кривой, проведен-
проведенной через точки экспериментальных значений, взятых из табл. 43. Мы видим,
что показатель преломления перекиси водорода и ее раствора выше, чем пока-
показатель преломления воды, и кривая, связывающая показатель преломления
с составом (выраженным в весовых процентах), обращена вверх вогнутостью.
Температурный коэффициент показателя преломления перекиси водорода и ее
растворов больше, чем для воды. Уравнение
6,5w F6)
представляет температурный коэффициент показателя преломления ап для
интервала 25—20°, дающий возможность вычислить значения показателя пре-
Таблица 44
Показатель преломления водных растворов перекиси водорода при 25° для линии D
натрия [141]
н2о2,
вес. %
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
1,3
1,3
1,3
1,3
1,3
1,3
0,0
325
331
338
344
350
357
363
369
376
382
388
395
401
407
414
420
427
433
439
446
452
459
466
472
479
486
492
499
506
512
519
526
533
540
547
554
561
568
575
582
589
596
603
611
618
625
632
639
646
653
661
668
0,2
326
333
339
345
352
358
364
370
377
383
389
396
402
409
415
421
428
434
441
447
453
460
467
474
480
487
494
500
507
514
520
527
534
541
548
555
562
569
576
583
590
598
605
612
619
626
633
641
648
655
662
669
0,4
327
334
340
347
353
359
365
372
378
384
391
397
404
410
416
423
429
436
442
448
455
462
468
475
482
488
495
502
508
515
522
529
536
543
550
557
564
571
578
585
592
599
606
613
621
628
635
642
649
656
663
671
0,6
328
335
341
348
354
360
367
373
379
386
392
398
405
411
418
424
430
437
443
450
457
463
470
476
483
490
496
503
510
516
523
530
537
544
551
558
565
572
579
586
593
600
608
615
622
629
636
643
651
658
665
672
.,«1
329
336
343
349
355
362
368
374
381
387
393
400
406
412
419
425
432
438
444
451
458
464
471
478
484
491
498
504
511
518
525
532
539
546
553
560
567
574
581
588
595
602
609
616
623
631
638
645
652
659
666
674
Н2О2,
вес. %
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
1,3
1,3
1,3
1,3
1,4
1,4
0,0
675
683
690
698
705
712
719
727
734
742
750
758
766
774
782
790
798
806
814
822
830
838
846
854
862
870
878
886
894
903
911
920
928
937
945
954
962
971
980
988
997
006
014
023
032
041
049
058
067
0,2
677
684
691
699
706
713
721
728
736
744
752
760
768
776
784
792
800
808
816
824
832
840
848
856
864
872
880
888
896
904
913
921
930
939
947
956
964
973
981
990
999
007
016
025
034
042
051
060
0,4
678
686
693
700
707
715
722
730
737
745
753
761
769
777
785
793
801
809
817
825
833
841
849
857
865
873
881
889
897
906
915
923
932
940
949
957
966
974
983
992
*000
009
018
027
035
044
053
062
0,6
680
687
.694
702
709
716
724
731
739
747
755
763
771
779
787
795
803
811
819
827
835
843
851
859
867
875
883
891
899
908
916
925
933
942
950
959
968
976
985
993
*002
011
020
028
037
046
055
063
0,8
681
688
696
703
711
718
725
733
740
748
756
764
772
780
788
796
804
812
820
828
836
844
852
860
868
876
884
892
901
909
91&
927
935
944
952
961
969
978
986
995
*004
013
021
030
039
048
056
065
230
Глава
ломления с точностью ±Ы0~4 (или большей). На основании измерений, про-
проведенных с тщательно очищенной безводной перекисью водорода при помощи
иммерсионного рефрактометра, Жигер и Жоффрион получили значение
34-10~5 град. для среднего температурного коэффициента показателя преломле-
преломления в пределах 15—25°. Если пользоваться табл. 44 для определения состава
по рефрактометрическим измерениям, целесообразно иметь еще таблицу тем-
температурных поправок, чтобы можно было применить указанные значения пока-
показателя преломления для определения состава при других температурах.
В табл. 45 приведены эти поправки. В качестве примера применения этой таб-
таблицы допустим, что мы имеем пробу перекиси водорода с концентрацией около
30 вес.%, показатель преломления, измеренный при 21°, оказался равным
1,3536. По табл. 44 концентрация перекиси водорода должна составить 32,4 вес.%.
Поправка, указанная в табл. 45, равна 1,0, откуда следует, что истинная кон-
концентрация перекиси водорода составляет 31,4 вес.%.
Таблица 45
Температурные поправки, которые следует вычесть из процентного содержания перекиси
водорода, определенного по табл. 44.
Темпера-
Температура, °С
20
21
22
23
24
Приблизительная концентрация, вес. %
10
0,9
0,8
0,6
0,4
0,2
20
1,2
0,9
0,7
0,4
0,2
30
1,3
1,0
0,8
0,5
0,3
40
1,4
1,1
0,9
0,6
0,3
50
1,4
1,1
0,9
0,6
0,3
60
1,5
1,2
1,0
0,6
0,3
70
1,6
1,2
1,0
0,6
0,3
80
1,7
1,3
1,0
0,7
0,4
90
1,8
1,4
1,1
0,7
0,4
100
1.9
1,5
1,1
0,8
0,4
Таблица 46
Константы, выведенные из данных показателя преломления [140, 141]
Константа
Удельная рефракция при 25° rD, см3/г
Молярная рефракция при 25° [R]D, см3/моль
Поляризуемость при 25° а-1024, см3 на 1 молекулу
Молярная дисперсия при 20° [R]g—[R]c, см3/моль
Константа дисперсии при 20° а-10~30, сек.
Характеристическая частота при 20° vo-10~15, сек.1 . . . .
Перекись
водорода
0,1705
5,801
2,30
1,3576
8,479
2,979
Вода
0,2060
3,712
1,47
0,9285
6,532
2,945
В табл. 46 приведены различные константы рефракции и дисперсии, вычис-
вычисленные из измерений показателя преломления. Удельная рефракция для
линии D натрия вычисляется из уравнения
а молярная рефракция—из уравнения [/?Ь=Мгд. Молярная рефракция рас-
растворов обычно эквивалентна средней молярной рефракции несмешанных компо-
компонентов в расчете на молярные доли каждого, т. е.
Влияние перекиси водорода на различные виды излучений
231
Это уравнение справедливо для водных растворов перекиси водорода с точ-
точностью до 0,001. Предполагается, что молярная рефракция пара перекиси
водорода та же, что и молярная рефракция жидкости, или превышает ее самое
большое на несколько процентов. В табл. 46 указаны также поляризуемость
а=3[/?Ь/4те#, молярная дисперсия [Rh—[Rh, константа дисперсии а
и характеристическая частота v0, вычисленная из упрощенной формулы Зел-
мейера [143] п2—\=а/(^1—v2), где v—частота, при которой измеряется
показатель преломления. Последние две константы определены по показате-
показателям преломления при 20°, измеренным Жигером для красной С и синей F линий
водорода. Обе эти константы и вычисленная на основании данных той же
работы молярная дисперсия могут быть несколько неточны, поскольку, как
выше указано, точность измерений показателя преломления в работе Жигера
была низка из-за недостаточной точности определения состава. Дисперсия или
разность показателей преломления перекиси водорода и ее растворов при раз-
разных частотах нормальная, т. е. показатель преломления правильно возрастает
с понижением длин волн в области видимого спектра. Так, показатели пре-
преломления при 20°, определенные Жигером [1401 для трех водородных линий С
F562,8А), F D861,33А) и G D340,46А), оказались следующими: пс= 1,4066,
пг= 1,4136 и по—1,4175 для безводной перекиси водорода.
Магнитооптическое вращение для перекиси водорода
Перекись водорода и ее водные растворы не обладают оптической актив-
активностью, т. е. не вызывают вращения плоскости поляризации пропущенного
через них света. Однако если их внести в магнитное поле, то происходит вра-
вращение плоскости поляризации; это явление называется эффектом Фарадея, или
магнитооптическим вращением. Этот эффект количественно выражается урав-
уравнением
a = VIH cos 60. F7)
Андерсен и Асмуссен [144] определили значения константы Верде при 0° для
<:вета с длиной волны 5460А. Они оказались равными 0,0154; 0,0154 и
0,0152 мин/гаусс-см для растворов перекиси водорода при концентрациях 10,52;
15,67 и 30,42 вес.% соответственно.
Таблица 47
Константы Верде для водных растворов перекиси водорода при 10е, по Жигеру
и Финн [145]
Концентрация Н2О2
вес. %
0
18,1
38,1
50,9
62,0
78,5
96,0
100,0
мольная
доля
0
0,105
0,246
0,354
0,463
0,659
0,927
1,00
Показатель
преломления
п
1,3330
1,3447
,3585
1,3680
1,3766
,3899
1,4052
,4112
Константа Верде V
5893 А
13,09
12,91
12,69
12,53
12,30
11,98
11,60
11,48
5780 А
13,64
13,38
13,15
12,98
12,80
12,45
12,03
11.90а
103» мин /гаусс
5461 А
15,40
15,13
14,86
14,60
14,43
14,07
13,64
13,32
-см
4358 А
25,21
25,00
24,47
24,22
24,11
23,45
22,70
22,65а
а Экстраполированные значения.
232
Глава 5
Жигер и Финн [145] провели исчерпывающие измерения константы Верде
для перекиси водорода. Результаты их измерений при температуре 10±2°
в магнитололяриметре, калиброванном по воде, представлены в табл. 47, где
приводятся также значения показателей преломления растворов, использо-
использованных для определения концентрации. В пределах точности этих данных
(около 1 вес.%), константа Верде для водных растворов перекиси водорода
является линейной функцией молярного состава при каждой длине волны.
В значения, приведенные в табл. 47, можно было бы ввести некоторые поправки
на основе уточненных величин показателя преломления перекиси водорода и
новейших измерений [146] константы Верде для воды, но вводить их, вероятно,
не имеет смысла. Жигер и Финн показали, что дисперсия для перекиси водорода
в зависимости от изменения константы Верде с длиной волны параллельна
дисперсии для воды, и опубликовали значения, вычисленные для констант
дисперсии. Вычисленные величины молекулярного магнитного вращения пере-
перекиси водорода на основе атомных эквивалентов Перкинса оказались ниже
экспериментально найденных значений.
Галле и Вольф [147] измерили магнитооптическое вращение растворов
перекиси водорода при концентрациях 20—80 вес.% при 20°, использовав свет
с длиной волны 5780, 5460 и 4360 А. Эти данные практически совпадают со зна-
значениями Жигера и Финн в отношении величины вращения и зависимости его
от состава.
Точность этих трех рядов данных не настолько велика, чтобы можно было
вычислить температурный коэффициент константы Верде для длины волны
5460 А, исследованной обеими группами авторов. Указывается, что константа
Верде падает с температурой, но этот эффект, как и для воды [146], очень мал.
Микроволновой спектр поглощения
Масси и Бьянко [148] исследовали поглощение излучений в микроволновой
области (длина волны от 1 м до 1 мм) парами перекиси водорода. Проанализи-
Проанализирована область частот от 9000 до 40 000 мггц\ обнаружено 9 линий абсорбции
для Н2О2 и около 100 линий для D2O2 и HDO2. Данные для перекиси водорода
представлены в табл.48, где/—квантовое число углового момента, a J*—боль-
Таблица 48
Микроволновой спектр перекиси водорода, по Масси и Бьянко [148]
Ли-
нияа
Частота, мггц
14 829,5+0,2
37517,6+0,2
22 054,5+0,2
27 639,6+0,2
11 072,4+0,5
35 916+2
39 033+2
39 495+2
37 960+2
Примечания
Квадратичный эффект Штарка
Вероятно, высокое J
» » J
» » J
J*=-A
J*=2
J*muh. = 7, I А
У*мин. = 7, | A J = 1
Вероятно, высокое J
Высокое /, | Д J | —
a Все линии средней или большой интенсивности, кроме сравнительно слабых линий 5, б и 7.
шее из двух участвующих в превращении квантовых чисел. Точность этих
результатов по сравнению с точностью, которая обычно наблюдается для микро-
микроволновой области, не высока. Это вызывается экспериментальными трудно-
Влияние перекиси водорода на различные виды излучений 233
стями работы с парами перекиси водорода. Масси и Бьянко сочли необходимым
разработать динамическую систему, но и в этом случае быстрое разложение
в волноводе не давало возможности работать при давлениях, достаточно низ-
низких, чтобы обеспечить высокую точность. Изучено также квадратичное штар-
ковское расщепление этих линий; следующие уравнения связывают экспери-
экспериментально обнаруженное разделение линий и силу электрического поля Е для
четырех из этих линий. В уравнениях Av'—частотное отклонение т-то ком-
компонента от частоты начальной линии, m—магнитное квантовое число.
Линия 1 Av' = 29,09 ?2.10. F8)
Линия 2 Av' = F,88- 4,25 т2)?2.1(Г6. F9)
Линия 3 Av' = E,65- 0,0874 m2)?2-1(Г6(</*> 8); G0)
Av' = F,07 - 0,0727m2) ?2.10"e (J* > 9); G1)
Av' = F,47 - 0,0623 m2) ?2.10 {J* > 10). G2)
Линия 4 Av' = D,18-0,0537m2)?2.10-6(J*>8); G3)
Av' = D,49 - 0,0459 m2) ?2-10 (J* > 9); G4)
Av' = E,10 - 0,0356 m2) ?2-10~6 {J* > 10). G5)
Для линий З и 4 оказалось невозможным точно определить значение У*,
а поэтому для каждой из них дано несколько уравнений с указанием соответ-
соответствующих минимально допустимых значений У*.
Инфракрасный спектр поглощения
Еще в 1895 г. Фридель [149] показал, что перекись водорода обладает
в инфракрасной области значительным поглощением такой величины, как
и поглощение, наблюдаемое в случае с водой. Этот автор измерил только общее
количество пропущенного излучения, причем успешное доказательство нали-
наличия поглощения обусловлено тем, что примененный им источник излучений
(металлический брусок, нагретый до 393°) испускал больше всего лучей с дли-
длиной волны 4,4 р, вблизи наиболее интенсивных полос инфракрасного поглоще-
поглощения воды и перекиси водорода. Исследование инфракрасного спектра поглоще-
поглощения перекиси водорода начато только в 1935 г., когда Майоне [150] сравнил
интенсивность поглощения излучений с длиной волны 1,0—6,0 р. жидкой
36%-ной перекисью водорода и водой. Майоне нашел, что интенсивность полос
поглощения воды 1,5 и 2,0 [* в растворе перекиси водорода возрастала, полоса
поглощения воды при 3,0 ^ становилась более интенсивной и смещалась в сто-
сторону более длинных волн в перекиси водорода, полоса же поглощения воды
при 4,6 р заменялась в перекиси водорода двумя четкими полосами с максиму-
максимумами при 4,4 и 4,85 {х. Ганц [151] опубликовал результат небольшого числа
измерений влияния температуры на инфракрасное поглощение жидкой 30%-ной
перекиси водорода для излучения с длиной волны от 0,7 до 0,9 [*. Обнаружено
поглощение и в воде и в перекиси водорода близ 0,77 р. Повышение температуры
перекиси с 12 до 55° заметно увеличивает интенсивность поглощения.
Подробное исследование инфракрасного поглощения перекиси водорода
впервые выполнено Бейли и Гордоном [152], которые измеряли поглощение
жидкой 95%-ной перекиси в интервале 2—12 р. и паров перекиси при давлении
около 18 мм рт. ст. в области 2,5—13 [*. Жидкость в области 2,0—8,8 р иссле-
исследовали в виде пленки между слюдяными пластинками; для области выше 8,8 р.
жидкость помещали между пластинками из каменной соли. Во избежание хими-
химических реакций. необходимо было быстро производить измерения. В табл. 49
представлены значения частот для центров полос поглощения, обнаруженных
234
Глава 5
Бейли и Гордоном. Эти авторы указывают, что в отличие от Майоне они не
наблюдали поглощения при 4,4 и 4,85 р..
Таблица 49
Инфракрасный спектр поглощения, по Бейли и Гордону [152]
Положение центров полос
жидкость
длина
волны, ix
11,40
7,50
3,48
2,93
волновое
число, см-1
877
1341
2869
3418
пар
волновое
число, см-\
870
1370
2870
3418
Относительные интенсив-
интенсивности3
жидкость
33
87
55
78
пар
17,8
22,2
15
59
а На произвольной основе для сравнения.
Зумволт и Жигер [153] исследовали поглощение парообразной перекисью
водорода излучений инфракрасной области при длинах волн около 1 р.; иссле-
исследование поглощения этих излучений жидкой перекисью водорода произвел
Жигер [154]. Зумволт и Жигер исследовали область 0,95—1,05 р. путем про-
пропускания паров перекиси водорода при давлении 9Э мм и температуре 100°
через 6-метровую трубку, освещенную угольной дугой, при помощи 6,4-метро-
6,4-метрового решетчатого вогнутого спектрографа с фотографической регистрацией
спектра. Обнаружена пара полос поглощения при 0,972 р; идентифицированы
отдельные частоты линий поглощения и проведен анализ полосатой струк-
структуры. Жигер [154] нашел, что 99,6%-ная жидкая перекись водорода дает в этой
области спектра широкую полосу с центром 1,01 р.
Жигер [155] вновь проверил поглощение перекиси водорода в области,
изученной Бейли и Гордоном, и расширил их исследование. Жидкую 99,5%-ную
перекись водорода изучали в области 2—21 р., а пары (при 90° и давлении 5, 10
и 15 мм)—в области 1,4—15 р.. Жидкость исследовали в виде пленки, зажатой
между пластинками из хлористого серебра, причем пришлось столкнуться
с трудностями, обусловленными разложением*. В абсорбционной трубке для
паров окна также были сделаны из хлористого серебра. Для работ при длинах
волн за пределами 15 р. применяли спектрометр с оптикой из бромистого калия,
причем для устранения помех от поглощения инфракрасного излучения водя-
водяным паром корпус спектрометра подвергали специальному обезвоживанию.
В табл. 50 приведены значения частот в центрах полос поглощения и коэффи-
коэффициенты поглощения, определенные Жигером [155]. Для жидкости полоса
поглощения при 18,3 р. была мало четкой, а полоса при 2,1 р.—очень слабой;
существование последней признано не вполне доказанным, поскольку она обна-
обнаружена не на всех фотометрических кривых. Разрешающая способность спект-
спектрофотометра была достаточно высокой и позволила для пара четко обнаружить
два максимума на полосе при 8 р., причем на некоторых фотометрических кри-
кривых обнаружено еще присутствие Q-ветви. Изучена также полоса 1,4 р.. Для
пара в условиях высокой разрешающей способности идентифицированы отдель-
отдельные частоты обеих составляющих полос и проведен анализ полосатой струк-
структуры.
* Возможно, что применение абсорбционных кювет, изготовленных из политрн-
-фторхлорэтилена [156], при изучении спектра при длинах волн короче 4,5—7,5 \i могло
<бы уменьшить затруднения, обусловленные разложением перекиси водорода.
Влияние перекиси водорода на различные виды излучений
235
Таблица 50
Инфракрасные спектры поглощения, по Жигеру [155]
Положение
жидкость
длина волны, ц
18,3
11,4
7,4
3,6
2,1
1,4
1,4
волновое число.
550
880
1350
2780
4720
—
центров полос
пар
волновое число,
см-1
877
1255
2630
7036,6
7041,8
коэффициент
погашения а
0,025
0,70
0,05
—
0,085
—
Тейлор 1157] изучил инфракрасное поглощение переохлажденной (—30)
99,2%-ной жидкой перекиси водорода и кристаллической перекиси при —30
и —78° в области длин волн 2—25 р. Проведены также измерения поглощения
при —30° жидкой 82,5%-ной перекиси дейтерия в тяжелой воде, содержащей
8—10% водорода. Пробы находились в кюветах из хлористого серебра, причем
в этом случае вследствие низкой температуры почти не было трудностей, свя-
связанных с разложением. Применялись призмы из флюорита, каменной соли или
бромистого калия; снята фотометрическая кривая полос поглощения твердой
перекиси водорода при —78°. Сводка данных Тейлора для перекиси водорода
представлена в табл. 51.
Таблица 51
Инфракрасный спектр поглощения перекиси водорода, по Тейлору [157]
жидкость, —30°
длина волны, \i
21,2
15,8
12,6
11,4
7,4
7,1
3,6
3,1
2,1
Положение
волновое число,
см-1
635 (ср.
878@4.
1353 (ср.
2796 (ср.
4715(оч.
, ш.)
ел.)
с.)
ел., ш.)
ел.)
центров полоса
твердая перекись,
при —30°
—
—
—
1365 (с.)
1418 (ел.)
2743 (ср.)
—
4640 (оч. ел.)
волновое число, см~*
при —78е
у 472'(оч. ел.)
| 660 (с.)
792 (ел.)
878(оч. ел.)
, 1378 (с.)
| 1430 (ел.)
2733 (ср.)
3218@4. с.)
4595@4. ел.)
а Сокращения: оч. с—очень сильная; с—сильная; ср.—средняя; ел. —слабая; оч. ел.—
очень слабая; ш.—широкая.
Новейшие достижения в области инфракрасной спектроскопии дали воз-
возможность расщепить некоторые основные полосы поглощения. Бен и Жигер
U58) получили данные, представленные в табл. 52, и разрешили полосу погло-
236
Глава 5
щения вблизи 21—18 р.. Эта полоса описана как состоящая из двух перекры-
перекрывающих друг друга перпендикулярных полос с центрами около 465 и 575 см'1.
Таблица 52
Инфракрасный спектр поглощения перекиси водорода,
по Бену и Жигеру [158]
Длина волны,
ц.
20—15
7,9
7,8
2,7
2,3
2,2
2,0
2,0
1,6
1,5
1,2
Положение центров полос а
пар
волновое число, слг-1
D90—660) (ср. с.)
1255 (ел.)
1266 (оч. с.)
A280) (сл.)
3610 (с.)
4465 (оч. ел.)
4835 (оч. ел.)
4955 (оч. ел.)
жидкость B 0°)
волновое число, см-*
4290 (оч. сл.)
6100 (оч. сл.)
6805 (оч. сл.)
8040 (оч. сл.)
а Сокращения: с—сильная; оч. с—очень сильная; ср.—средняя;
с л.—слабая; оч. сл.—очень слабая.
Жигер и Бен [159] недавно вкратце опубликовали результаты исследова-
исследования инфракрасного спектра поглощения паров перекиси дейтерия и щелочных
растворов перекиси водорода при 2—25 р. Результаты спектроскопических
измерений перекиси дейтерия приводятся на стр. 255.
В табл. 53 дана сводка экспериментально обнаруженных полос поглощения
перекиси водорода. Указаны также все описанные вторичные максимумы,
показаны изменения с температурой, причем повторены, вероятно, идентичные
полосы поглощения, занимающие, однако, различные положения, по данным
разных авторов. Наиболее вероятные значения и объяснения происхождения
частот главнейших полос приведены в следующей главе, где рассматри-
рассматривается структура перекиси водорода. Необходимо иметь в виду, что инфра-
инфракрасное поглощение перекиси водорода является сравнительно слабым, что
существование некоторых из полос поглощения, упомянутых в старых лите-
литературных источниках, впоследствии не подтвердилось и что точность указан-
указанного местоположения некоторых центров полос не высока. Как упомянуто
выше, отдельные полосы изучены в условиях высокой разрешающей способно-
способности, тогда как другие, безусловно, можно было бы еще разложить. Для срав-
сравнения можно перечислить главные полосы инфракрасного поглощения для'воды
[21: 0,85; 0,98; 1,18; 1,46; 1,98; 2,97 и 6,1 (л. Краткий обзор литературы по
инфракрасному спектру перекиси водорода опубликован Учида и Фукуда [160].
Спектр комбинационного рассеяния
Если освещать вещество монохроматическим излучением, то иногда ока-
оказывается, что рассеиваемое веществом излучение содержит не только возбуж-
возбуждающую длину волны, но и еще одну или несколько других. Это другое излуче-
излучение отличается по частоте от возбуждающего излучения на величину, которая
в первом приближении не зависит от длины волны возбуждающего излучения.
Спектр, состоящий из этих рассеянных частот, носит название спектра комби-
комбинационного рассеяния, или раман-спектра. Хотя этот спектр, обусловленный
Таблица 53
Инфракрасный спектр поглощения перекиси водорода (сводная таблица полос
поглощения и интенсивностей)
Длина волны,
Волновое
число, см-
Интенсивность3
Температура,
оСб
Литература
21,2
15,2
12,6
11,4
7,3
7,3
7,1
7,0
3,6
3,7
3,1
2,2
2,2
18,2
15,8
11,4
7,4
4,85
4,4
3,6
3,6
3,48
2,93
2,3
2,1
1,6
1,5
1,5
1,2
1,01
0,77
.20—15
11,4
8,0
7,9
7,8 *
7,3
3,8
3,5
2,9
2,7
2,2
2,0
2,0
1,4
1,4
0,972
0,972
472
660
792
878
1365
1378
1418
1430
2 743
2733
3218
4 595
4 640
550
635
880
1350
2 000
2 300
2 780
2 796
2 864
3418
4 290
4 720
6 100
6 700
6 805
8 040
9 900
13 000
490-660
877
1255
1266
1280
1370
2 630
2 870
3418
3610
4 465
4 835
4 955
7 037
7 042
10 283,68
10 291,08
Твердое тело
(оч. ел.)
(с.)
(ел.)
(оч. ел.)
(с)
(с.)
(ел.)
(ел.)
(ср.)
(ср.)
(оч. с.)
(оч. ел.)
(оч. ел.)
Жидкость
(оч. ел.)
(ср.)
(ел.)
(ср.)
(оч. ел.)
(оч. ел.)
(ср.)
(ср.)
(ср.)
(с.)
(оч. ел.)
(оч. ел.)
(оч. ел.)
(ел.)
(оч. ел.)
(оч. ел.)
(ел.)
(ел.)
Пар
(ср. с.)
(оч. ел.)
(оч. с.)
(оч. с.)
(ел.)
(ел.)
(ел.)
(оч. ел.)
(ср.)
(с.)
(оч. ел.)
(оч. ел.)
(оч. ел.)
(ел.)
(ел.)
(ел.)
(ел.)
-78
-78
-78
-78
-30
-78
-30
-78
-30
-78
-78
-78
-30
—
-30
—
.—.
—
20
.
20
—
20
20
—
12,55
—
90
90
—
—
90
—
.—.
—
—
—
90
90
100
100
157
157
157
157
157
157
157
157
157
157
157
157
157
155
157
152, 155, 157
152, 155, 157
150
150
155
157
152, 157
152
158
152, 155, 157
158
150
158
158
154
151
158
152, 155
155, 158
158
158
152
155
152
152
158
158
158
158
155
155
153
153
а Сокращения ?83]: оч.с.—очень сильная; с—сильная; ср.—средняя; ел.-слабая; оч. ел.—очень
слабая. Определение интенсивности имеет сравнительную силу только для полос в одной и той же обла-
области спектра.
" Отсутствие численного значения обозначает комнатную температуру.
238 Глава 5
испусканием света, является противоположным спектру поглощения, излуче-
излучение часто обусловлено теми же внутримолекулярными движениями, которые
определяют инфракрасный спектр поглощения. Так, разность длины волны
возбуждающей линии и некоторой линии комбинационной частоты пред-
представляет собой мерило энергии Av, отнятой от возбуждающего излучения
и приложенной к внутримолекулярному движению, причем испускается менее
богатая энергией более низкая остаточная частота, которую и обнаруживают как
комбинационную линию. Возможно поэтому точнее соответствие между волно-
волновым числом а, характеризующим полосу инфракрасного поглощения, и вели-
величиной разности волновых чисел Аа комбинационной линии и линии, возбуж-
возбуждающей ее появление (комбинационная разность). Некоторые внутримолеку-
внутримолекулярные движения могут вызвать появление комбинационных линий (спутников)
без инфракрасного поглощения, и наоборот; это наряду с некоторыми вопро-
вопросами удобства эксперимента и точности диктует желательность определения
спектров комбинационного рассеяния.
Венкатесваран [161 ] первый опубликовал данные по комбинационному рас-
рассеянию перекиси водорода и указал на существование комбинационных линий
30%-ной жидкой перекиси водорода, соответствующих волновым числам
875 и 9ЭЗ см. Линия 875 см'1 оказалась интенсивной, а линия 9ЭЗ см'1 слабой,
причем отмечены также и другие полосы с еще меньшей интенсивностью. Дама-
шун [162] кратко подтвердил существование комбинационной линии 873 см'1,
но не нашел линии 933 см'1. В более детальном сообщении Венкатесваран [163]
обнаружил для 30%-ной жидкой перекиси водорода комбинационные линии
400, 875 и 9ЭЗ см'1, а также полосы для воды, вероятно, представляющие ком-
комбинационную линию около 3400 см'1, наблюдаемую также в спектре комбина-
комбинационного рассеяния воды. Степень деполяризации спутника с комбинационной
частотой 875 см'1 по измерениям Венкатесварана оказалась равной 0,15.
Симон и Фехер [164] проводили измерения в отношении концентрирован-
концентрированной (99,5 вес.%) жидкой перекиси водорода и обнаружили спутников, имею-
имеющих максимумы на расстоянии 877, 1421 и 3395 еж от возбуждающей линии
с длиной волны 4358А. Эти авторы считают, что наличие линии 903 см1, ука-
указанной Венкатесвараном, обусловлено стеклом сосуда. Симон и Фехер опубли-
опубликовали фотометрический снимок спектра комбинационного рассеяния, полу-
полученного ими и указывающего следующие пределы полос и интенсивностей:
877 см'1, четкая полоса, сравнительная интенсивность 15; 1421 см'1, пределы
полосы 1462—1345, сравнительная интенсивность 1; 3395 еж, пределы полосы
3410—3200, сравнительная интенсивность 3. Комбинационная полоса 1421 см'1,
по-видимому, имеет два максимума с расстоянием 27 см'1, она ослабевает с раз-
разбавлением и снижается при меньших концентрациях, а именно эта полоса
является линией 1399 см'1 в 95%-ной перекиси водорода и линией 1392 см'1
в 80%-ной. Линия 3395 см'1 при разбавлении изменяется меньше. В 30%-ной
перекиси эта полоса расширяется и захватывает область 690 см'1, причем
образовавшаяся широкая полоса также расщепляется с образованием двух
максимумов с отдельными линиями около 3130 и 3430 см'1. Край этой полосы
в 3%-ной перекиси перемещается к 3630 еж, тогда как в 99,5%-ной он соот-
соответствует 3410 см. При этом разбавлении появляется комбинационная линия
1630 см'1, приписываемая воде. Симон и Фехер показали также, что все комби-
комбинационные линии, за исключением 3395 см'1, исчезают при нейтрализации пере-
перекиси водорода едким натром.
Кольрауш [165] считает, что комбинационная линия 3395 еж, по Симону
и Фехеру, ошибочна и должна быть заменена линией 3305 см'1. Однако недавно
Бейли и Гордон [152] опубликовали результаты изучения спектра комбина-
комбинационного рассеяния 95%-ной жидкой перекиси водорода, снятого Леки, также
обнаружившего комбинационную линию 3418 см'1. Разность между 3395 и 3418
признана ничтожно малой, что подтверждают данные Симона и Фехера.
Влияние перекиси водорода на различные виды излучений
239
Тейлор [166] недавно тщательно исследовал комбинационные спектры
жидкой перекиси водорода при концентрации 99,3 вес.% и выше, водных ее
растворов, а также кристаллической перекиси водорода. Для возбуждения
использованы различные ртутные линии, причем для возможно большего
W00 2000 3000
Комбинационная линия, см'2
а
1000 2000 3000
Комбинационная линия, см'1
Рис. 48. Спектр комбинационного рассеяния жидкой
99%-ной перекиси водорода при —40°.
а—при интенсивной экспозиции для обнаружения слабых полос при
525 и 2800 сж-1; б—нормальная экспозиция.
подавления всех линий, кроме желательной, применяли подходящие свето-
светофильтры. Результаты экспозиции в течение 12—48 час. записывали при помощи
спектрографа с разрешающей способностью 15А на 1 мм в синем свете. Перекись
водорода отгоняли непосредственно в кювету и анализировали ее до каждого
облучения и после него. При 30° пришлось столкнуться с некоторыми труд-
трудностями из-за разложения вследствие использованного интенсивного облуче-
облучения. Для снижения до минимума этого разложения необходимо было перед
исследованием особенно тщательно обрабатывать кювету, изготовленную из
стекла пирекс.
На рис. 48 и в табл. 54 приведены данные о спектрах, полученных Тейлором,
для 99,3%-ной жидкой перекиси водорода, причем для возбуждения он приме-
применил излучение с длиной волны 4358А. Кривые (рис. 48) отчетливо доказывают
наличие комбинационных линий при 525 и 2815 смг1, а в соответствии сданными
предыдущих исследований также при 880, 1400 и 3400 см'1.
Несимметричное распределение интенсивностей около возбуждающей
линии, наблюдающееся при нормальной экспозиции, также можно рассматри-
рассматривать как комбинационное рассеяние. Однако рассеяние возбуждающей линии
исключает возможность дать характеристику величины смещения для такого
плеча или крыла.
240
Глава 5
Таблица 54
Комбинационные линии, отмеченные в жидкой перекиси
Возбуждаю-
Возбуждающее излуче-
излучение с длиной
волны, А
4358
4358
4358
4046
4046
4046
4358
•
Темпера-
Температура, °С
+30
+30
+30
+30
+30
+30
-40
99,С
Комбина-
Комбинационная
линия
Асу, сж-1
880
1400
3411
879
1397
3417
525
1 вес. %, по
Ширина
полосы
в основа-
основании, с/и~~1а
# (Р)
260
470
(Р)
250
500
•—
водорода
Тейлору [166]
Возбуждаю-
Возбуждающее излуче-
излучение с длиной
волны, А
4358
4358
4358
4358
4046
4046
4046
Темпера-
Температура, °С
-40
-40
-40
-40
-40
-40
-40
при концентрации
Комбина-
Комбинационная
линия
Дет, см-1
882
1402
2815
3364
879
1415
3382
Ширина
полосы
в основа-
основании, сл!-1а
(Р)
300
—
530
(Р)
190
470
а (р)—резкая полоса.
Ртутная линия 4358А, использованная для возбуждения спектра, показанно-
показанного на рис. 48, в действительности представляет собою тесно расположенный трип-
триплет. Об этом свидетельствует формао полосы 880 см'1. Если для возбуждения
применяется указанная линия 4358А, наблюдается еще и другое осложнение:
имеется ряд слабых ртутных линий, отстоящих от линий 4358А на волновые
числа 2800—2900 см'1. При длительной экспозиции релеевское рассеяние*
таких линий может проявиться даже при применении эффективных светофильт-
светофильтров. Поэтому могут возникнуть некоторые сомнения в отношении комбина-
комбинационной линии 2815 см'1 (рис. 48,а); результат комбинационной спектроскопии
нижеописанных растворов заставляет считать, что эта полоса действительно
существует.
Таблица 55
Комбинационные линии, обнаруженные в водных растворах перекиси водорода,
по Тейлору [166]
Мольное
отноше-
отношение
Н2О2 : Н2О
8: 1
9:2
4:1
2: 1
3:2
1: 1
1 : 16
0,85:1
1:2
1:3
1:4
2: 15
1 :9
, 1 : 28
Темпера-
Температура, °С
30
-40
30
-40
30
-40
-40
30
-40
30
-25
30
- 8
30
800
800
880
881
881
878
879
877
878
879
878
878
878
878
878
Комбинационная линияа в разных областях Да, см~1
1400
1396
1411
1406
1422
1416
1431
1427 (ш.)
1430 (ш.)
1465 (ш.)
1445 (ш.)
1455 (ш.)
1450 (ш.)
1445 (ш.)
1440 (ш.)
1650в
1625
1650 (ш.)
1630
1617 (ш.)
1638
1670 (ш.)
1640
1610 (ш.)
1630 (ш.)
1665 (ш.)
2800
2800 (ш.)
2850 (ш.)
.—
2840 (ш.)
2815 (ш.)
2825 (ш.)
2835
2815 (ш.)
2845 (ш.)
—
2805 (ш.)
—
2810 (ш.)
—
3400
—
—.
3207 (ш.)
3260 (ш.)
—
3240 (ш.)
3270
3345 (ш.)
3290 (ш.)
3400
3403
3373
3406
3377
3418
3395
3405
3430
3412
3430
3417
3430
3417
3429
л ш.—широкая размытая полоса, снижающая точность определения местоположения.
6 В этом случае для возбуждения применяли линию 4046А, во всех других случаях-линию 4358А.
в Полоса 1650 c.w-1 обусловлена водой.
* Под релеевским рассеянием подразумевается рассеяние падающего излучения
веществами молекулярных размеров, происходящее без изменения длины волны.
Влияние перекиси водорода на различные виды излучений
241
В табл. 55 приведены данные, по Тейлору, о спектре комбинационного рас-
рассеяния растворов перекиси водорода в воде при комнатной температуре и вблизи
точки замерзания, главным образом при +30 и —40°. На рис. 49 нанесены фото-
фотометрические кривые для спектра растворов при температуре —40° в области
3000 см'1. Чтобы возможно полнее устранить кажущиеся изменения в положе-
положениях максимумов полос, обусловленные различиями в денситометрических свой-
свойствах фотопластинок, данные для построения кривых (рис. 49) выбирались из
спектров, в которых интенсивности комбинационной полосы 3400 см'1 были
примерно одинаковыми. Ввиду того что растворы с мольным отношением пере-
перекиси водорода к воде 1 : 9 и 1 : 4 нельзя переохладить до —40°, отсчеты необхо-
необходимо было проводить при —8 и —25°.
Данные табл. 55 и рис. 49 свидетельствуют о
двух явлениях, имеющих особый интерес: о наличии
комбинационной линии около 2800 см'1 и о расщепле-
расщеплении полосы 3400 см'1 (при увеличении разбавления)
на две составляющих. В отношении существования
комбинационной линии 2800 см'1 возможны некоторые
сомнения в связи с тем, что при применении для
возбуждения линии 4358 А наблюдаются ртутные ли-
линии, находящиеся на расстоянии 2784 и 2866 см'1
от возбуждающей линии. Однако Тейлор устранил
возможность такого возражения, исследовав спектр
комбинационного рассеяния, возбужденного в раство-
растворе с равным молярным содержанием воды и перекиси
водорода, линией 4046А. В этом случае область спектра,
находящаяся на расстоянии 2800—2900 см'1 от воз-
возбуждающей линии, совершенно свободна от релеев-
ского рассеяния, и таким образом наличие полосы
2800 еж в растворе полностью подтверждено.
Исследование комбинационного рассеяния твер-
твердой, почти безводной перекисью водорода, проведен-
проведенное Тейлором, также доказало наличие комбинацион-
комбинационной линии при 2800 см'1 и четкое разделение полосы
3400 см'1 на два максимума. Один опыт проведен с мас-
массой довольно больших кристаллов, ориентированных
преимущественно таким образом, что спектр наблю-
наблюдали вдоль наиболее длинной их оси. Наличие боль-
большого числа кристаллических граней вызывало заметное отражение в
спектрограф, в результате чего уничтожалось любое комбинационное рассея-
рассеяние, которое можно было бы наблюдать в области, отстоявшей на расстоянии
до 2400 см'1 от возбуждающей линии. За этой областью спектр был вполне
четким, если не считать повышенной интенсивности некоторых ртутных
линий. Комбинационная линия 3400 еж была четкой, причем соответствую-
соответствующая полоса расщеплялась на две линии с резко отделенными максимумами.
Сравнение нефильтрованного ртутного спектра и комбинационного спектра
твердой перекиси показало, что это расщепление действительно существует,
а не вызвано появлением в этой точке ртутной линии повышенной интенсив-
интенсивности. Комбинационные линии для массы кристаллов, полученные Тейлором
при применении для возбуждения излучения 4358А, можно суммировать
следующим образом: при —10° максимумы наблюдались у линий 3200 и
3334 ш~\ при —40°—у линий ЗЦ98 и 3328 см'1.
Промежуточные комбинационные линии отмечены Тейлором в спектре
несколько несовершенного монокристалла при —20°. С целью предотвратить
появление спутников, находившихся на расстоянии менее 600 см'1 от возбуж-
возбуждающей линии, и затемнить подробности в области между 600 и 2000 см'1
16 Перекись водорода
2500 3000 3500
Комбинационная
линия, см
Рис. 49. Полоса ОН
в спектре комбинацион-
комбинационного рассеяния водных
растворов перекиси во-
водорода при —40°.
242 Глава 5
опыты проводили в условиях наличия достаточного количества отраженного
света. Совершенно четко обнаружены следующие спутники: 878 + 0,5, 1408±Ю,
2790±20, 3203 + 3 и 3331+4 см'1.
Для комбинационного спектра перекиси водорода рекомендуется приме-
применять данные Тейлора [166]. Выше мы рассматривали влияние на комбинацион-
комбинационное рассеяние температуры, фазового состояния, концентрации и длины волны
возбуждающей линии; для безводной жидкости можно принять следующие
комбинационные линии: 880, 1400, 2810 и 3410 см'1. Как показано ниже
(стр. 279), где речь идет о структуре, последняя полоса должна состоять из
двух основных полос, перекрывающих одна другую или совпадающих друг с
другом. По-видимому, эта полоса сильнее всего зависит от изменения концен-
концентрации или от перехода из жидкого агрегатного состояния в твердое. Наиболее
четкие комбинационные линии для воды соответствуют Аа = 1650 и 3400 см'1.
Дорси [2] дает для воды в виде таблиц также много других комбинационных
линий.
Симон и Маршан [167] изучили влияние на комбинационное рассеяние
добавки щелочи к растворам перекиси водорода. Показано, что наличие щелочи
(гидратов окисей лития, натрия или калия, но не аммония) вызывает появле-
появление комбинационной полосы при 844 см вдобавок к полосе, наблюдаемой при
880 см'1. При полной нейтрализации последняя полоса исчезает, а полоса при
844 см'1 остается. Бен и Жигер [158] не обнаружили такого изменения в инфра-
инфракрасном спектре жидких растворов щелочной перекиси водорода, но в твердой
гидроперекиси натрия наблюдалась частота растяжения связи О—О при 814 см~*.
Симон и Улиг [168] провели сравнение спектра комбинационного рассеяния
перекиси водорода в аммиаке (жидком или водном растворе) и в растворе четы-
рехзамещенного аммониевого основания в воде. Аммиак не влиял на вибрацию
О—О; четвертичная же гидроокись функционировала как сильное основание,
способствуя диссоциации перекиси водорода, и вызывала смещение полосы,
отмеченное выше в случаях с другими сильными основаниями.
Видимый спектр
Обычно указывается, что перекись водорода является бесцветной; действи-
действительно, визуально определить, что содержится в колбе: безводная перекись
водорода, раствор ее в воде или чистая вода, не представляется возможным
(если только не происходит разложения или реакции). В одной из старых работ
по поглощению видимого света различными жидкостями Рассел и Лапрек
[169] утверждали, что 6%-ный раствор перекиси водорода обладает известным
поглощением в оранжевой части спектра. Если это поглощение действительно
наблюдалось, то оно, несомненно, обусловлено наличием примесей, а следова-
следовательно, можно считать, что с практической точки зрения перекись водорода
и ее водные растворы, как и вода, прозрачны для излучений, чувствительных
для глаза, т. е. для области длин волн между 4000 и 8000А. Пределы зоны инфра-
инфракрасной части спектра, где перекись водорода не поглощает, примерно такие же,
как для воды, т. е. лежат близко к 1 р; в ультрафиолетовой области поглощение
делается заметным очень близко от края видимого спектра (рис. 50).
Шпринг [36] изучил релеевское рассеяние видимых излучений концентри-
концентрированной перекисью водорода. Этот автор наблюдал цвет отраженного днев-
дневного света сквозь столб жидкой перекиси водорода и сравнивал его с окраской
различных растворов медного купороса. При этом он определил, что почти без-
безводная перекись водорода за счет рассеяния приобретает окраску, которую
можно описать как синюю с зеленым оттенком; эта окраска очень близка
к окраске воды, но более интенсивна. Шпринг обнаружил, что при образовании
пузырьков внутри перекиси водорода наблюдается окраска от желтой до
зеленой.
Влияние перекиси водорода на различные виды излучений
324
Высокие столбы жидкой перекиси водорода в больших емкостях, в которых
можно было бы наблюдать довольно слабое явление рассеяния, встречаются
редко, тем не менее отдельные наблюдения подтверждают выводы Шпринга,
Если вглядеться в жидкую концентрированную перекись водорода, налитую
1000
100
Ю
1,0
0,1
0,01
0,0001
Предел
видимого
спектра-*
i <
i
¦
со'
0
}
9
d
Л
9
I
р
¦
ё
о
>
о
Р
t
А
4400^ 4000 3600 3200 2800
Длина волны, А
2400
2000
Рис. 50. Ультрафиолетовый спектр поглощения водных раст-
растворов перекиси водорода.
ф Фиббс и Жигер (ж.» 6,5%); ф Фиббс и Жигер (ж., 54,9%); ф Фиббс
и Жигер (ж., 99,3%); О Тейлор и Гросс (ж., 50%); О Тейлор и [Гросс
(ж., 91%); х Холт, Мак-Лейн и Олденберг (ж.); А Холт, Мак-Лейн и
Олденберг (п.); П Олменд и Стайл; (ж.); Д Ледерле и]Рихе (ж.); V Бредиг,
Леман и Кун (ж.); -ф Генри и Вюрмсе (ж.); + Корнфельд (ж.);
¦ Юрей, Доуси и Райе (ж., п.); Ў Фергуссон, Слотин и Стайл (п.); (ж.) —
жидкость, (п.)—пар.
в бочку, то создается определенное впечатление слабой окраски с оттенком от
зеленого до синего. Несомненно, в некоторой мере это зависит от свойств алю-
алюминиевой стенки сосуда, от наличия пузырьков в жидкости и от возможного
присутствия взвешенных частиц, обусловленных наличием стабилизаторов.
Однако вполне логично предполагать, что перекись водорода должна обладать
такими же свойствами рассеяния света, как и вода. Дорси [2] довольно под-
подробно разбирает этот вопрос и указывает на часто совершаемые ошибки.
16*
24;4
Глава 5
Ультрафиолетовый спектр поглощения
В ходе работ по спектроскопии (в настоящее время считающихся класси-
классическими) Хартли I170J показал, что излучение в ультрафиолетовой области
с длиной волны меньше 2925 А полностью поглощается раствором перекиси водо-
водорода при концентрации ниже 1 вес.%. Желая проверить применимость закона
Эйнштейна о фотохимической эквивалентности, Тиан [171] и Генри и Вурмсер
[172] провели первые количественные измерения поглощения ультрафиоле-
ультрафиолетовых излучений с различной длиной волны перекисью водорода. Розанов [173]
в работах по изучению образования перекиси водорода действием радона
на воду точно так же определил, что область поглощения для 3%-ной перекиси
водорода лежит между 3120 и 2480А,. В настоящее время ультрафиолетовый
спектр поглощения перекиси водорода в жидком и парообразном состоянии
исследован для всей области длин волн между краемовидимого спектра, вблизи
4000А, и краем шумановской области, вблизи 2000А. В табл. 56 дана сводка
результатов этих исследований. Опубликован фотоснимок [187], показываю-
показывающий пропускание лучей дугового спектра между 5000 и 2000А и 0,2%-ным рас-
раствором перекиси водорода.
Таблица 56
Сводка исследований по ультрафиолетовому спектру поглощения перекиси водорода
Фаза6
Ж
Ж
Ж
Ж
п
ж
ж
ж
п
п
ж
п
ж
п
п
ж
п
ж
Концентрация перекиси
водорода
0,06%
0,04—0,8%
—
0,02—0,06%
1,5 мм рт. ст. при 20°
0,02—35%
3, Ю, 30%
0,2—5%
0,01 мм рт. ст.
10~6 моля на 1 л
—
—
0,0007—1%
5—20 мм рт. ст.
2 мм рт. ст.
50,90%
2 мм рт. ст.
6, 55, 99%
Длина волны,
А
2980—2144
3600,3110
3500—2000
3750—2150
2750—2150
3650—2540
—
3300—2400
—
2900—2200
2537
2537—1850
3100—2100
2260—2150
2536
3800—2700
2100—2000
4300—3000
Метод
анализа^
Т
—
Ф, В
Ф, В
т
—¦
—
ФЭ
Ф, В
ФЭ
Ф, ФМ,
Ф9
ФЭ
Ф, ФМ
—¦
ФЭ
Литература
Генри и Вурмсер [172]
Ксфнфельд [174]
Ледерле и Рихе [175]а
Юрей, Доуси и Райе [111]
Там же
Олменд и Стайл [176]
Шарма [177]
Бредиг, Леман и Кун [178]а
Фрост и Олденберг [179]
Фергуссон, Слотин и Стайл
[180]*
Холт, Мак-Лейн и Олден-
Олденберг [181]
Там жеа
Эгертон, Гаррис и Юнг
[182]
Там же
Мак-Лейн [183]
Тейлор и Гросс [184]
Идее [185]
Фиббс и Жигер [186]
а Результаты представлены только в графической форме.
6 Сокращения: П—пар; Ж—жидкость; Т—термостолбик; Ф—фотографический метод; В-визуальная
интерпретация; ФЭ—фотоэлемент; ФМ—фотометрическая интерпретация.
Концентрации жидкости, если нет других указаний, даны в весовых процентах.
Поглощение ультрафиолетовых излучений способствует диссоциации моле-
молекулы перекиси водорода. Эта диссоциация рассматривается в гл. 8; здесь же
можно указать только, что первичный фотохимический процесс состоит в обра-
образовании двух радикалов ОН, хотя возможны и другие реакции, протекающие
в небольшой степени. Как видно на рис. 50, спектр поглощения перекиси водо-
водорода является сплошным. На этом рисунке в виде зависимости молекулярного
Влияние перекиси водорода на различные виды излучений 245
коэффициента погашения от длины волны приведены данные исследователей,
перечисленных в табл. 56.
На рис. 50 можно видеть, что различные данные согласуются между собою
в пределах около 10%. Основными исключениями являются лишь результаты
работ Юрея, Доуси и Раиса [ 111 ] и Фергуссона, Слотина и Стайла [ 180].В обоих
этих случаях определялось поглощение пара перекиси водорода. В одном из них
[111] были проведены также измерения поглощения лучей жидкостью, причем
результаты поглощения для обеих фаз совпадали, что указывает на отсутствие
различий между ними. В другом случае [180] измерения проводились только
для пара и было принято, что результаты их указывают на различие в погло-
поглощающей способности парообразной и жидкой фаз. Трудно объяснить отклоне-
отклонение результатов этих измерений, особенно результатов работы Юрея, Доуси
и Раиса, полученных при сравнительно длинных волнах, от других данных;
в обоих случаях интерпретация фотографической записи производилась
визуально. Необходимо отметить, что при работах с паром возникают допол-
дополнительные трудности в отношении точного установления концентрации. Кроме
того, при более коротких волнах из-за высоких коэффициентов поглощения
приходится работать с низкими концентрациями или же пользоваться корот-
короткими кюветами. Особых трудностей из-за фотохимического разложения не
отмечено. Только в одном исследовании [181] сделана небольшая поправка на
поглощение ультрафиолетовых лучей водой.
Можно сделать вывод, что коэффициенты ультрафиолетового поглощения
в пределах точности имеющихся результатов для жидкой и паровой фаз одина-
одинаковы. Однако не исключено, что все же существует небольшое различие, так
как новейшие данные показывают, что растворы перекиси водорода не подчи-
подчиняются закону Бера; это приписывается [186] изменению состояния молеку-
молекулярной ассоциации жидкости с концентрацией. Для коэффициента погашения
пара перекиси водорода мы предлагаем принять результаты работ Холта,
Мак-Лейна и Олденберга [181]. Если не считать вышеупомянутых данных
[111, 180], это единственные имеющиеся количественные данные по погло-
поглощению пара. Другие авторы приводят только качественные результаты. Так,
Фрост и Олденберг [179] указывают, что, пользуясь данными Юрея, Доуси
и Раиса, пар перекиси водорода можно обнаружить в абсорбционной кювете
длиной 150 см при длине волны 2537 А при давлении всего 0,01 мм рт. ст.;
Эгертон, Гаррис и Юнг [182] измеряли длину волны, при которой происходит
полное поглощение для перекиси водорода различных концентраций (в паровой
и жидкой фазах), причем они не обнаружили различий между обеими фазами
и указали, что их результаты согласуются с данными других авторов [172,
175, 111]. Мак-Лейн [183] нашел, что коэффициент поглощения пара при
2536А согласуется в пределах 1,5% с величиной, указанной Холтом, Мак-
Лейном и Олденбергом [181]; Идее [185] тоже указывает, что его опыты под-
подтвердили данные, полученные Холтом, Мак-Лейном и Олденбергом в области
длин волн от 2100 до 2000А.
На рис. 50 можно видеть, что кривая, показывающая зависимость коэф-
коэффициента погашения от длины волны, имеет параболическую форму. Однако
внутри значительного интервала длин волн вблизи видимого спектра кривая
может быть представлена прямой на полулогарифмическом графике. Холт,
Мак-Лейн и Олденберг считают, что при длинах волн, значительно меньших,
чем исследованные, на кривой поглощения возможно наличие максимума.
На кривой рис. 50 можно наметить две точки перегиба, расположенные соот-
соответственно у обоих концов исследованного интервала длин волн. Однако,
прежде чем утверждать, что эти точки перегиба действительно существуют,
потребуются дополнительные исследования. Признаки перегиба вблизи 1920А,
намечающиеся по данным Холта, Мак-Лейна и Олденберга [181], по мнению
Идее [185], подтверждают положение Шарма [177] о существовании «четкого
246
Глава 5
разреза» в спектре поглощения при 2055А. Шарма сделал этот вывод на основа-
основании исследования поглощения 3—30%-ной перекисью водорода в жидкой фазе.
Однако он не дает точного определения, что подразумевается под термином
«четкий разрез», и не приводит количественных измерений коэффициентов
поглощения. Нет никаких теоретических оснований для существования пре-
прерывности или резкого изменения наклона кривой поглощения (если это под-
подразумевается под «четким разрезом»); имеющихся данных для доказательства
существования такой прерывности или точки перегиба на кривой поглощения
также недостаточно.
Работы Тейлора и Гросса [184], а также Фиббса и Жигера [186] обогатили
наши знания в отношении коэффициентов поглощения для ряда концентраций
вплоть до безводной перекиси водорода. Результаты этих работ показывают,
что растворы перекиси водорода не подчиняются строго закону Бера. По мере
увеличения концентрации перекиси водорода растворы обладают все большей
и большей поглощающей способностью, чем следует по указанному закону. Это
видно на рис. 50 по падению молекулярного коэффициента погашения с ростом
концентрации перекиси водорода; указанное падение, по-видимому, прямо
пропорционально концентрации перекиси водорода. Отклонение от закона Бера
не может быть доказано для длин волн меньше 3400А, так как, за исключением
Тейлора и Гросса, другие авторы работали в коротковолновой области лишь
с растворами, имеющими концентрации не более 5%, ввиду сравнительно
высокого коэффициента поглощения перекиси водорода в этой области. Коэф-
Коэффициенты поглощения, найденные Тейлором для 50%-ной перекиси водорода
в области от 3400 до 2700А, в пределах точности данных не отличаются от резуль-
результатов, полученных другими авторами для более разбавленных растворов.
Отсюда можно сделать вывод, что отклонение от закона Бера меньше, чем
возможная погрешность в точности определения коэффициентов поглощения
при растворах, имеющих концентрацию ниже примерно 50 вес.%. Это иллю-
иллюстрируется также данными Олменда и Стайла [176], проводивших измерения
при 3650 А и не обнаруживших заметных изменений коэффициента поглощения
при изменении концентрации растворов вплоть до 35 вес.%. В табл. 57 представ-
представлены средние данные для молекулярного коэффициента погашения, взятые
из рис. 50; они могут служить для общего руководства. Пожалуй, наиболее
точной установленной величиной нужно считать единственное значение при
2537А, определенное Холтом, Мак-Лейном и Олденбергом [181] при помощи
Таблица 57
Молекулярные коэффициенты погашения для ультрафиолетовых
лучей в жидких и парообразных смесях перекиси водорода и воды
Длина волны,
А
4000
3800
3600
3400
3200
3000
Молекулярный
коэффициент
погашен ияа
е, л 1 моль-см
0,00066
0,0022
0,010
0,047
0,22
1,0
Длина волны,
А
2800
2600
2537
2400
2200
2000
Молекулярный
коэффициент
погашенияа
е, л 1 моль • см
4,2
13
19,6-4-0,3
35
76
140
8 1 7о
е=——- lg-у2-. За исключением единственной величины [181] для
2 53 7 А, все остальные представляют собою средние значения, полученные
ин1ерполяцией из рис. 50, а не результаты работы какого-либо одного иссле-
исследователя.
Влияние перекиси водорода на различные виды излучений 247
фотоэлемента и подтвержденное Мак-Лейном [183]; поглощение при этой длине
волны без труда может быть использовано для анализа разбавленных растворов
или паров.
Ледерле и Рихе [175] и Бредиг, Леман и Кун [178] изучили влияние
добавки щелочи на поглощение ультрафиолетовых лучей перекисью водорода.
Показано, что в присутствии щелочи кривая поглощения смещается в направ-
направлении видимого света, т. е. коэффициент поглощения возрастает. Бредиг,
Леман и Кун изменяли молекулярное отношение едкого натра к перекиси
водорода от 0 до 11,2 при использовании растворов перекиси водорода, имею-
имеющих концентрацию от 0,2 до 5 вес. %, и нашли, что смещение коэффициента
поглощения почти прямо пропорционально указанному молекулярному отно-
отношению вплоть до значения, равного 1. Это смещение можно объяснить как
следствие нейтрализации перекиси водорода и приписать его тому, что погло-
поглощение пергидроксильного иона НОг более интенсивно, чем поглощение недис-
социированной перекиси.
На первой ступени нейтрализации, т. е. при отношении (NaOH)/(H2O2)= 1,
один и тот же молекулярный коэффициент погашения получается при-
примерно при длине волны на 500 А большей, чем та длина волны, при которой этот
коэффициент наблюдается для растворов перекиси водорода, не содержащих
щелочи. Из результатов опытов Бредига, Лемана и Куна можно как будто
вывести, что поглощение продолжает несколько смещаться при дальнейшем
увеличении молекулярного отношения щелочи к перекиси водорода. Это непре-
непрерывное смещение, которое можно приписать нейтрализации второго водород-
водородного атома перекиси водорода, значительно слабее, и его нельзя считать вполне
достоверным. В изученных условиях одни растворы едкого натра не поглощают
ультрафиолетового света.
Спектры испускания в парах перекиси водорода
Юрей, Доуси и Райе [111] облучали пар почти безводной перекиси водо-
водорода при давлении 1 мм рт. ст. фильтрованным излучением цинковой искры
и обнаружили в спектре флуоресценции полосу 3064А для воды (происхо-
(происходящую от радикала ОН)*. Это было принято за доказательство одновре-
одновременной диссоциации перекиси водорода и возбуждения гидроксильного радика-
радикала. Олденберг [188] указывает, что такая интерпретация описанного опыта
не является единственно возможной и что не исключено образование еще дру-
других частиц, помимо гидроксильного радикала.
Юрей, Доуси и Райе изучали также влияние электрического разряда на
перекись водорода. При создании неконденсированного разряда в охлажденной
трубке, через которую быстро пропускали ток почти безводного пара перекиси
водорода, отмечена слабая голубая окраска. Спектроскопическое исследование
показало наличие сплошного спектра, причем атомные и молекулярные спектры
водорода отсутствовали, но в спектре наблюдались очень интенсивные полосы
воды. Фрост и Олденберг [189] нашли, что интенсивность испускания радикалов
ОН в паре перекиси водорода, подвергнутом действию разряда, значительно
больше интенсивности испускания тех же радикалов в водяном паре. Согласно
определению по методу поглощения, скорость исчезновения радикалов ОН в пе-
перекиси водорода значительно выше, чем в воде. Реакции, наблюдаемые в парах
перекиси водорода при действии электрического разряда, изучались Бат-
цольдом, Люнером и Винклером [61].
* Олменд и Стайл[176] обнаружили сине-зеленую флуоресценцию жидкой перекиси
водорода. Она была вызвана примесями от парафинового покрытия стенок сосуда. Эта
флуоресценция уменьшалась и затем после длительного освещения полностью исчезала,
причем это явление сопровождалось ростом скорости разложения.
248 Глава 5
Рентгеновская и электронная диффракция перекиси водорода
Фехер и Клетцер [190, 191], Натта и Ригамонти [58], Лю, Хьюз иЖигер
[192] и Абрагэмс, Коллин и Липскомб [12] исследовали диффракцию рентге-
рентгеновских лучей кристаллической перекисью водорода с целью определения ее
структурных параметров. Фехер и Клетцер и Абрагэмс, Коллин и Липскомб
проводили свои работы с безводной перекисью. Натта и Ригамонти исследовали
перекись водорода, имеющую концентрацию 82 и 90 вес.%, а также соединение
Н2О2-2Н2О. В работе Лю, Хьюза и Жигера применялись кристаллы аддитив-
аддитивного соединения пероксигидрата мочевины CO(NH2J-H2O2. Во всех этих
сообщениях приводятся фотоснимки, таблицы с координатами, размерами и
структурными факторами или диаграммы электронной плотности, вычислен-
вычисленные на основании найденных рентгенограмм.
Рэндол [193] исследовал рассеяние рентгеновских лучей жидкой без-
безводной перекисью водорода. Жигер и Шомейкер [194] исследовали электрон-
цую диффракцию в парах перекиси водорода.
Оригинальные данные, получаемые при исследовании диффракции, мало-
малоинтересны. Ценным результатом является определение размеров и простран-
пространственного расположения атомов в молекуле перекиси водорода; эти свойства
обсуждаются в следующей главе, касающейся структуры.
ФИЗИЧЕСКИЕ СВОЙСТВА МНОГОКОМПОНЕНТНЫХ СИСТЕМ, СОДЕРЖАЩИХ
ПЕРЕКИСЬ ВОДОРОДА
В литературе приводятся измерения некоторых физических свойств ряда
систем, содержащих перекись водорода и другие вещества, помимо воды или
совместно с водой. Эти системы можно рассматривать как такие, в которых пере-
перекись водорода или ее водные растворы играют роль растворителя.
Необходимо подчеркнуть, что добавка к. перекиси водорода других
веществ, кроме воды, создает потенциальную опасность взрывного ее разло-
разложения или детонации. Неорганические добавки могут реагировать с перекисью
водорода или катализировать ее разложение со скоростью взрыва, иногда после
вводящего в заблуждение индукционного периода. Органические добавки
могут образовать с перекисью водорода внутри широкого интервала составов
смеси, способные детонировать с энергией, сравнимой по величине с энергией
детонации нитроглицерина. Например, способ концентрирования перекиси
водорода путем экстракции эфиром, широко применявшийся прежде, при-
привел к ряду серьезных несчастных случаев [36]. Поэтому при работе с такими
смесями необходимо соблюдать надлежащие меры предосторожности.
В табл. 58 приведены ссылки на литературу, где сообщаются данные по
физическим свойствам таких смесей. Сами данные не приводятся из-за недо-
недостатка места. Часть из них имеет сомнительную точность, причем нужно иметь
в виду, что указываются те вещества, которые были смешаны с перекисью
водорода; однако во многих случаях сразу же или через некоторое время после
смешения между перекисью и указанными веществами происходила реакция,
а поэтому измеренное значение в действительности могло принадлежать совсем
не тому веществу, которое указано в списке как вводимая добавка.
Основная цель большинства этих измерений заключалась в определении
соотношений растворимости разных веществ и перекиси водорода. Перекись
водорода и ее водные растворы в общем обладают такой же растворяющей спо-
способностью и растворимостью, как и одна вода. Правда, существуют и серьезные
различия, но пока еще нет общей схемы, на основании которой можно было бы
их предсказать. Наиболее полное исследование, проведенное с целью выясне-
выяснения природы этих различий, принадлежит Флойду и Гроссу [19 1, которые
подтвердили прежние работы и расширили их [ 9, 134, 196, 201 „ 2071. Резуль-
Таблица 58
Литературные ссылки на физические свойства многокомпонентных систем, содержащих
перекись водорода
Вещество
Растворимость или
точка замерзания
Электропро-
Электропроводность3
Коэффициент рас-
распределения^
Хлорид натрия
Хлорид калия
Хлораты калия и натрия
Нитрат натрия
Нитрат калия
Фторид натрия
Сульфат »
Сульфат калия
Сульфат аммония
Пероксосульфат калия
Соли ванадия
Сульфат лития
Нитрат лития.
Нитрат свинца
Фосфаты натрия и калия
Пирофосфат бария
Карбонат натрия
Аммиак3
Гидроокиси лития, натрия, калия,
рубидия и цезияв>г
Сернистая кислота (двуокись серы)
Азотная кислотад
Соляная »
Хлорная »
Фосфорная »
Борная »
Муравьиная »
Уксусная, »
Трихлоруксусная кислота
Пропионовая »
Гликолевая »
Щавелевая »
Янтарная »
Виннокаменная »
Лимонная »
Метиловый спирте •
Этиловый спирт6
Изобутиловый спирт
Амиловый »
Гексанол
Циклогексанол
Ацетоне
Хлороформ
Этиловый эфире» ж,3, и, к
Диоксан8»и
Бензол
Нитробензол
Петролейный эфир (пентан, гексан)
Фенол
Анилин
134, 195, 196
9, 195, 196, 199,
200, 201
196
134, 195, 196,
200, 202
195, 196, 199,
200, 201
9, 195, 196
134, 195, 203
9, 195, 196, 201,
204
199
204
195
195
196
203, 206, 207
203
208
134
9, 199, 204, 209
209
199
69
197
124
122
197
122
201
205
122, 123
197
197, 210
197
197
197
211
212, 213
124, 197,
212, 213
197
212, 213
212
213
197
197
214, 215
122, 123, 206,
211, 215-218
215
215
214
134, 214, 216,
219, 220, 221
134, 214, 222
214
214
214
250
Глава 5
Табл. 58 (продолжение)
Вещество
Растворимость или
точка замерзания
Электропро-
Электропроводность3
Коэффициент рас-
распределения^
Диметиланилин
Пиперидин
Диэтиламин
Моно-я-бутиламин
Моно-трет-бутиламин ....
Диизобутиламин
Трипропиламин
Сахар
Изопрен
Хинолин
Ацетофенон
Этилацетат
Пропилацетат
Бутилацетат
Амилацетат
Циклогексилацетат
Пропилформиат
Изоамилпропионат
Пропилбутират
Этилизовалерат
Изобутилбутират
Метилметакрилат
Диэтилфталат и диметилфталат
9
9
9
9
9
9
9
134
224
224
222
214
214, 218
215, 223
215
215
214, 215, 223
215
214
214
214
214
214
а Бобтельский и Симхен [198] приводят результаты кондуктометрического титрования 21-й неорга-
неорганической соли в децинормальном растворе перекиси водорода.
6 Распределение перекиси водорода измерялось между указанными растворителями, с одной сторо-
стороны, и либо водой, либо растворами разных солей или кислот в воде—с другой.
в Определено влияние этих веществ и четырехзамещенного аммониевого основания на инфракрасный
спектр и спектр комбинационного рассеяния [158, 167, 168].
г Отмечено влияние щелочи на ультрафиолетовый спектр поглощения [175, 178].
д Отмечено влияние азотной кислоты на вязкость [210].
6 Штерном [44] определен коэффициент диффузии перекиси водорода в этих веществах.
ж Линтон и Маас [130, 131] измерили плотность смесей с перекисью водорода.
8 Линтон и Маас [130, 131[ измерили диэлектрическую проницаемость смесей с перекисью водо-
водорода.
и Симон и Фехер [225] определили влияние перекиси водорода на спектр комбинационного рас-
рассеяния.
к Рихе и Заутгоф [226J измерили ультрафиолетовый спектр поглощения смесей с перекисью водо-
водорода.
таты этого исследования показывают, что фторид натрия, нитрат калия, раз-
различные фосфаты калия или натрия, хлорид калия и сульфат натрия или калия
лучше растворимы в перекиси водорода, чем в воде. Нитрат натрия, хлорид
натрия, нитраты серебра, свинца и лития и сульфат лития хуже растворимы
в перекиси водорода, чем в воде. Флойд и Гросс [195] изучили также зависи-
зависимость растворимости солей в перекиси водорода от температуры. Найдено, что
растворимость хлорида натрия при данной концентрации перекиси водорода
почти не зависит от температуры в интервале 0—25°. Растворимости нитратов
натрия, калия и лития обладают почти постоянным температурным коэффи-
коэффициентом для всего интервала концентраций перекиси водорода. Растворимость
хлорида калия обладает очень низким температурным коэффициентом в
концентрированной перекиси водорода в отличие от высокого температурного
коэффициента растворимости в воде. Для фторида натрия обнаружена обрат-
обратная зависимость. Найдено, что растворимость фтористого натрия падает
с ростом температуры.
Физические свойства перекиси водорода с изотопным составом 251
Для нитратов лития и калия получены кривые растворимости в зависи-
зависимости от концентрации перекиси водорода с четкими разрывностями. Измере-
Измерения электропроводности показывают, что эти соли диссоциируют в перекиси
водорода так же, как и в воде, но электропроводность кислот в перекиси водо-
водорода ниже, чем в воде, что, вероятно, нужно объяснить меньшей подвижностью
водородного иона в перекиси водорода. Измерения электропроводности исполь-
использованы и для обнаружения реакции кислот или солей с перекисью водорода
с образованием перекисных соединений. Доказано существование ряда адди-
аддитивных соединений с перекисью водорода. Некоторые ссылки, приведенные
в табл. 58, относятся к этим пероксигидратам; дальнейшее обсуждение этого
вопроса и ссылки на эту и другую литературу приведены в гл. 7 и 12. Лишь
очень небольшое число таких систем изучено достаточно подробно для пред-
представления полных фазовых диаграмм.
Измерения интервалов смешиваемости перекиси водорода и органических
веществ показывают, что эфир и бензол практически не растворимы в безводной
перекиси водорода. Последняя обладает несколько большей относительной
растворимостью в эфире, амиловом спирте и хинолине, чем вода. Метиловый
спирт снижает температуру замерзания перекиси водорода меньше, чем тем-
температуру замерзания воды.
Наибольшее внимание привлекало изучение свойств смесей перекиси
водорода с этиловым эфиром. Перекись водорода и эфир образуют неидеальные
растворы с полной смешиваемостью лишь при температуре выше 80° (выше кри-
критической температуры растворения). Линтон и Маас [130, 131] измеряли плот-
плотность смесей перекиси водорода с эфиром или с водой и эфиром при 0 и 10°.
При смешении перекиси водорода и эфира отмечено уменьшение объема и за-
заметное выделение тепла. В образованных растворах диэлектрические прони-
проницаемости перекиси водорода и эфира оказались аддитивными и их можно
выразить следующей формулой:
е = 4,6 +0,608 слМл. G6)
Штерн [44] показал, что скорость диффузии перекиси водорода в эфире больше,
чем в воде, причем коэффициент диффузии для 0,1 н. перекиси водорода в эфире
при 20° составляет 2,407 см2/сутки. Симон и Фехер [225] показали, что наличие
перекиси водорода изменяет спектр комбинационного рассеяния метилового
эфира. Рихе и Заутгоф [226] нашли, что ультрафиолетовый спектр поглощения
перекиси водорода в эфирном растворе несколько смещен в сторону видимого
спектра по сравнению с этим же спектром в водном растворе.
ФИЗИЧЕСКИЕ СВОЙСТВА ПЕРЕКИСИ ВОДОРОДА С ИЗОТОПНЫМ СОСТАВОМ,
ОТЛИЧАЮЩИМСЯ ОТ НОРМАЛЬНОГО
В табл. 59 представлены массы и распространенность изотопов водорода
и кислорода. Поскольку и водород и кислород являются относительно легкими
атомами, изменение изотопного состава молекулы перекиси водорода сравни-
сравнительно сильно изменяет ее молекулярный вес. Однако в связи с тем, что тяже-
тяжелые изотопы присутствуют в природе лишь в очень малых относительных коли-
количествах, необходимо во много раз обогатить пробу естественной перекиси водо-
водорода тяжелыми изотопами, чтобы средний молекулярный вес смеси заметно
повысился. Возможность такого обогащения в процессе производства (напри-
(например, при электролизе или перегонке), по-видимому, ничтожно мала.
Получены препараты перекиси водорода, отличающиеся по изотопному
составу от естественной перекиси как по водороду, так и по кислороду. Так,
приготовлена перекись водорода, обогащенная тяжелым кислородом (О18, так
как О17 менее распространен) в размере нескольких процентов. Эта перекись
использована для изучения механизма реакций, но физические свойства ее
252
Глава 5
Таблица 59
Масса и распространенность изотопов водорода и кислорода
Изотоп
Н1
Н2
Н3
О14
О15
О16
О17
О18
О19
Атомный
номер
1
1
1
8
8
8
8
8
8
Физическаяа
атомная
масса [227]
1,00812
2,01471
3,01402
14,013
15,0078
16,0000
17,0045
18,0049
19,0139
Распростра-
Распространенность^
[228, 229),
атомные доли
0,99985
0,00015
Ю-18
—
0,99759
0,00037
0,00204
Период
полураспада
[230]
Устойчив
»
10—12 лет
76 сек.
2 мин.
Устойчив
»
»
30 сек.
а Отношение физической шкалы к химической равно 1,000275 [10].
б Относительная распространенность может зависеть от источника по-
получения [23], хотя различия лежат в пределах указанной здесь точности.
Значение для НЗ представляет собой равновесие между непрерывным обра-
образованием и распадом.
не измерены. Не отмечено обмена изотопами кислорода между перекисью
водорода и водой [232]. Водород же, наоборот, быстро и полностью обмени-
обменивается между перекисью водорода и водой [233], что дает возможность без труда
получать обогащенную перекись водорода путем растворения перекиси водорода
в тяжелой воде. Описанная методика использована для исследования влияния
дейтерия на реакции перекиси водорода, но, очевидно, мало подходит для изу-
изучения физических свойств, поскольку в результате образуется смесь различ-
различных молекул: Н2О, HDO, D2O, H2O2, HDO2 и D2O2. Поэтому для исследования
физических свойств перекись дейтерия необходимо получать более трудоем-
трудоемкими методами; эти методы описаны Фехером [234], который получал это соеди-
соединение через пероксодисульфат, и Жигером, Секко и Итоном [235], получав-
получавшими его при действии электрического разряда на пар тяжелой воды. При
наличии безводной D2O2 можно легко получить смесь HDO2 с перекисями дей-
дейтерия и водорода за счет использования обменной реакции Н2О2+D2O2 = 2HDO2.
Фехер [236] сделал вывод, что константа равновесия для этой реакции равна
примерно 4; другими словами, из эквимолекулярной смеси перекисей водорода
и дейтерия получаются перекиси водорода, дейтерия и водорода—дейтерия
в относительных концентрациях 1:1:2. Приводимые ниже физические свой-
свойства, за одним исключением, относятся к перекиси дейтерия D2O2, либо без-
безводной, либо растворенной в тяжелой воде D2O. i
Фехер определил плотность растворов перекиси дейтерия при 18° [234],
причем он исходил из предположения, что D2O2 имеет тот же самый моляльный
объем, что и перекись водорода. На основе вычисленных плотностей Фехер
при помощи микроареометра определял концентрации. Фиббс и Жигер [29]
измеряли плотность перекиси дейтерия непосредственно пикнометрическим
методом при 0 и 20°. Результаты этих измерений, полученные для растворов,
содержащих менее 0,25% обыкновенного водорода, приведены в табл. 60.
Фиббс и Жигер определяли для тех же проб перекиси дейтерия показатель
преломления (при помощи прецизионного рефрактометра Аббе), вязкость
(вискозиметром Оствальда) и поверхностное натяжение (по методу капилляр-
капиллярного поднятия). Эти данные также представлены в табл. 60.
Жигер и Секко [56] построили диаграмму ликвидуса для системы перекись
дейтерия—тяжелая вода. Полученная ими кривая точек замерзания (основанная
Физические свойства перекиси водорода с изотопным составом
253
Таблица 60
Плотность, показатель преломления, вязкость и поверхностное натяжение растворов
перекиси дейтерия, по Фиббсу и Жигеру [29]
Концентрация
D2O2
вес.%
0,00
9,05
20,02
33, б8
43,25
56,68
73,78
91,65
мольная
доля
0,0
0,0524
0,1222
0,2203 ,
0,2979
0,4213
0,6102
0,8593
Плотность о, ',
г\мл
0°
1,Ю4в
1,1414
1,1857
1,2440
1,2860
1,3447
1,4236
,5130
25°
1,Ю56
1,1370
1,1780
1,2320
1,270!
1,3284
1,4033
1,4908
25
nD
1
,32806
,3340
,3414
,3510
,3579
,3678
,3809
,3955
Вязкость ц,
саптипуазы
0°
2,350
2,230
2,184
2,18.
2,193
2,192
2,149
2,047
20°
1,244
1,255
1,268
1,300
1,325
1,372
1,397
1,379
Поверхностное на-
натяжение, дины/см
0°
76,1
76,5
77,0
77,9
78,7
79,8
81,4
82,7
20°
73,3
73,5
74,1
75,2
75,8
77,0
78,7
80,0
на термическом анализе проб, содержащих менее 0,3% обыкновенного водорода)
представлена значениями, приведенными в табл. 61. Кривая ликвидуса для
системы с дейтерием параллельна кривой ликвидуса для системы с обыкновен-
обыкновенным водородом, но смещена вверх по вертикали в соответствии примерно со сле-
следующими данными: на стороне, богатой водой, выше на 4°, между эвтектиками
на 0,6° и на стороне, богатой перекисью водорода, на 2°. Экстраполированное
значение для температуры замерзания безводной перекиси дейтерия равно
Таблица 61
Кривая температур ликвидуса для системы перекись дейтерия—тяжелая
вода, по Жигеру и Секко [56]
Концентрация
D2O2 в D2O,
вес.%
0,00
11,00
20,62
30,46
38,5Х
43,5!
44,68
45,84
46,5t
46,9б
Температура
замерзания,
d
3,80
- з,з9
-11,62
-22,34
-35,4!
-45,00
-47,00
-50,10
—51,52
-51,62
Эвтектика
-51,37
—51,32
Концентрация
D2O2 в D2O,
вес.%
48,84
50, Зб
56,03
61,17
66,56
77,54
80,85
95,0
95,85
100,0
Температура
замерзания,
°С
-51,53
-51,64
-53,28
—
-43,76
-25,8х
-20,9б
- 4,34
- з,о7
1,5
Эвтектика
-55,10
-55,0в
1,5° (эвтектики соответствуют: 46,2 вес. % D2O2 и —51,5° и 60,5% D2O2 и —55,1°;
аддитивное соединение D2O2-2D2O, содержащее 47,3 вес. % D2O2, плавится при
—51,5°). Растворы перекиси дейтерия переохлаждаются еще заметнее, чем
растворы обычной перекиси водорода. Растворы, богатые перекисью дейтерия,
можно охладить до температур жидкого воздуха с получением прозрачного
стекла. Примерно через 10 мин. после образования стекло превращается
в кристаллическую форму, причем кристаллизация сопровождается расшире-
расширением.
Мориссет и Жигер [79] измерили некоторые термодинамические свойства
перекиси дейтерия. В табл. 62 приведены значения теплоемкости растворов
перекиси дейтерия в тяжелой воде между 0 и 26,9°. Эти измерения проводили
•в изотермическом калориметре с дифениловым эфиром и перекисью дейтерия,
254
Глава 5
Таблица 62
Теплоемкость растворов перекиси дейтерия в тяжелой воде
между 0 и 26,9°, по Мориссету и Жигеру [79]
Концентрация D2O2 в D2O
вес. %
0
30,46
40,7
74,2
100,0
мольная
доля
0
0,1958
0,2762
0,6152
1,0
Молеку-
Молекулярный
вес рас-
раствора
20,03
23,16
24,45
29,88
36,03
Теплоемкость
кал
моль• град
20,35
20,03
20,17
21,33
22,75»
кал
г • град
1,016
0,865
0,825
0,715
0,632а
Экстраполированная величина.
приготовленной из тяжелой воды с 99,7% D2O. Общая теплота парообразова-
парообразования раствора перекиси дейтерия, имеющего концентрацию 46,96 вес.% при
26,9°, оказалась равной 454,96 кал/г; для безводной D2O2 при 25° найдена [237]
величина 347,8 кал1гу или 12,53 ккал1моль.
Таблица 63
Термодинамические функции для пара перекиси дейтери;
Температура,
°К
298,16
300
350
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1
\ т У
кал
град' мьли
47,33
47,39
47,85
50,22
52,55
54,57
56,35
57,95
59,38
60,75
61,98
63,13
64,22
65,24
66,20
am, по Лиу и Жигеру [233]
Яо-Яо
Т '
кал
град • моль
9,54
9,54
9,87
10,20
10,79
11,31
11,82
12,17
12,52
12,84
13,14
13,39
13,64
13,87
14,09
S0,
кал
град¦ моль
56,86
56,93
58,76
60,42
63,34
65,89
68,12
70,11
71,93
73,58
75,12
76,54
77,87
79,12
80,30
кал
град' моль
11,49
11,51
12,16
12,72
13,58
14,21
14,74
15,18
15,58
15,93
16,25
16,53
16,77
16,98
17,17
[ при
н°-я0,
кал 1 моль
2 843
2 861
3 456
4 078
5 396
6 787
8 275
9 735
И 265
12 838
14 455
16 074
17 733
19 422
21 135
Термодинамические функции для перекиси дейтерия, приведенные
в табл. 63, вычислены Лиу и Жигером [238] из спектроскопических данных, взя-
взятых из табл. 64.
Масси и Бьянко [148] исследовали микроволновой спектр D2O2 и HDO2.
В области между 9000 и 40 000 мггц при применении пробы перекиси, содер-
содержавшей примерно 3 молекулы D2O2 на молекулу HDO2r обнаружено свыше
100 линий поглощения. Выяснение происхождения отдельных линий с учетом
изотопного состава оказалось невозможным. Качественное исследование эффекта
Штарка показало, что такого рода данные можно получить примерно для 25
Физические свойства перекиси водорода с изотопным составом
255
Таблица 64
Молекулярные данные для перекиси дейтерия,
по Лиу и Жигеру [158]
Фундамен-
Фундаментальные
колебания
vi (а)
v2 (a)
^з (а)
Волновое
число, сл-1
2690
965
885
Фундамен-
Фундаментальные
колебания
v4 (a)
* (Ь)
чв (*)
Волновое
число, cjh-1
380
2680
947
Моменты инерции, г-см2
/д = 5,14.Ю-**
/в = 38,3.
с
/red = /A/4=l,29.10-
Барьер, ограничивающий внутреннее вращение Vo =
ккал/моль.
из этих линий, причем происходят превращения как типа IAJ I =0, так и типа
|ДУ| =1.
Тейлор [157] и Жигер и Бен [158, 159] измерили инфракрасный спектр
поглощения перекиси дейтерия. Данные о нем приведены в табл. 65. Тейлор
изучал при —30° спектр жидкой 82,5%-ной D2O2 в пробе, содержащей 8—10%
обыкновенного водорода, в интервале длин волн 3—19 (х. Жигер и Бен иссле-
исследовали спектр пара D2O2 и твердой D2O2 при —70° в интервале 2—25 р.. Жигер
и Бен [158] расшифровали ротационную структуру полосы при 2680 см'1
в парах D2O2; они обнаружили 18 подчиненных полос.
Таблица 5
Инфракрасный спектр поглощения перекиси дейтерияа
Длина
волны, \х.
20,8
18,6
11,4
10,6
9,6
7,2
5,1
4,8
4,0
3,7
3,0
2,9
2,0
1,9
1,6
1,4
Волновое число, см~1
твердое тело
480 (ср.N [159]
—
880 (ел.) [158]
—
• 1000 (с.) [158]
—
—
2090 (ср. ел.) [158]
2640 (оч. с.) [158]
—
жидкость
538 (ср., ш.) [157]
878 (оч. ел.) [157]
—
1004 (с.) [157]
A386) (ел.) [157]
—
2087 (ср. ел.) [157]
2482 (оч. с.) [157]
—
3368 (ср. ел.) [157]
3430 [158]
4980 (оч. ел.) [158]
6110 (оч. ел.) [158]
7215 (оч. ел.) [158]
пар
—
—
923 (?) [158]
947 (оч. с.) [159]
955 [158]
—
—
1985 (ел.) [159]
—
—
2680 (с.) [159]
5236 (оч. ел.) [143]
а Источники данных приведены при каждой полосе.
б При -70°.
Сокращения: с—сильная; оч. с—очень сильная; ср.—средняя; ел.—слабая; оч. ел.—очень слабая;
[.—широкая.
256
Глава 5
Фехер [239] исследовал спектр комбинационного рассеяния перекиси
дейтерия и обнаружил комбинационные линии 877,1009 и 2510 см'1 в D2O2. Была
также приготовлена перекись водорода—дейтерия HDO2 путем составления
эквимолекулярной смеси перекисей дейтерия и водорода. Для этого соедине-
соединения обнаружены комбинационные линии 877, 1009, 1406, 2510 и 3407 см'1.
Фехер убедительно доказывает, что эти линии обусловлены самой молекулой
HDO2, а не являются аддитивными свойствами взятых в отдельности перекисей
дейтерия и водорода. Тейлор [166] обнаружил в 90%-ном растворе перекиси
дейтерия в тяжелой воде комбинационную линию 1990 см'1.
Фиббс и Жигер [186] исследовали ультрафиолетовый спектр поглощения
жидкостей, содержащих 6,5 и 30,7 вес.% перекиси дейтерия и практически
свободных от обыкновенного водорода в интервале 3900—3050А. Перекись
дейтерия в ультрафиолетовой области спектра поглощала меньше, чем перекись
водорода. Таким образом, кривая коэффициента погашения для перекиси дей-
дейтерия смещена в сторону коротких волн по сравнению с кривой для перекиси
Таблица 66
Рекомендуемые значения некоторых физических свойств перекисей водорода и дейтерия
и обыкновенной и тяжелой воды
(все значения, если нет особых указаний, даны для 25° и 1 am)
Свойство
Молекулярный вес
Плотность твердого тела при темпе-
температуре замерзания, г/см3 .....
Плотность жидкости при 20°, г/смъ
Вязкость жидкости при 20°, санти-
пуазы ...
Вязкость пара при температуре кипе-
кипения, микропуазы
Поверхностное натяжение при 20°,
дины 1см
Коэффициент диффузии в воздухе при
60°, см2/сек . . . . . .
Теплота возгонки при температуре
замерзания, ккал/моль
Температура плавления, °С . ' . . .
Теплота плавления при температуре
плавления, кал /моль
Температура кипения, °С . . . . .
Теплота парообразования,, ккал/моль
Критическая температура, °С . . . .
Критическое давление, am
Теплоемкость твердого тела при тем-
температуре замерзания, кал/г-град
Теплоемкость жидкости, кал/г-град .
Теплоемкость пара, кал /моль -град
Теплота образования в парообразном
состоянии, ккал/моль
Свободная энергия образования в па-
парообразном состоянии, ккал/моль
Диэлектрическая проницаемость при
20°
Магнитная восприимчивость, 106 ед.
CGSM на 1 г
Показатель преломления .....
н2о2
34,016
1,71
1,450
1,245
137
80,4
0,188
15,58
-0,43
2987
150,2
12,33
457
214
0,39
0,628
10,22
- 32,52
- 25,24
73,1
- 0,50
1,4067
D2O2
36,028
—
1,534
1,354
81,2
—
1,5
-3030
—
12,53
—
—
—
0,632
11,01
-34,46
-26,48
1,4026
н2о
18,016
0,917
0,998
1,002
134
72,75
0,320
12,17
0
1436
100,0
10,51
374,2
218,2
0,50
0,998
8,025
-57,80
-54,64
80,4
—0,720
1,3325
D2G
20,028
1,017
1,106
1,25
143
72,73
12,60
3,82
1515
101,4
10,85
371,5
218,6
0,53
1,016
—
-59,56
-56,06
79,8
— 0,647
1,3281
Литература 257
водорода. Фиббс и Жигер подсчитали, что размер этого смещения около 390 см х
соответствует по величине вычисленному из различий вибрационной энергии
перекисей дейтерия и водорода в нулевой точке. Для растворов перекиси дей-
дейтерия обнаружено отклонение от закона Бера в таких же размерах, как и для
перекиси водорода.
В табл. 66 для сравнения дана сводка ряда физических свойств перекисей
водорода и дейтерия и обыкновенной и тяжелой воды. Значения для обыкно-
обыкновенной воды взяты у Дорси [2], для тяжелой—у Киршенбаума [231].
ЛИТЕРАТУРА
1. Dierdorff L. H., Jr., Beccio Chemical Division, Food Machinery and Chemical
Corp., Hydrogen Peroxide Physical Properties Data Book, Buffalo, 1955.
2. D о r s e у N. E., Properties of Ordinary Water Substance, Reinhold Publishing Corp.,
New York, 1940 (Reprinted, 1954).
3. К e e n a n J. H., К е у e s F. G., Thermodynamic Properties of Steam, John Wiley
& Sons, Inc., New York, 1936.
4. T a m m a n n G., Z. physik. Chem., 4, 441 A889); 12, 431 A893).
5. Carrara G., Gazz. Chim. Ital., 22, 341 A892).
6. О r n d о r f W. R., White J., Am. Chem. J., 15, 347 A893).
7. Giguere P. A., M a a s s O., Can. J. Res., 18B, 66 A940).
8. F о 1 e у W. Т., G i g u ё r e P. A., Can. J. Chem., 29, 895 A951).
9. Ma t h e s о n G. L., M a a s s O., J. Am. Chem. Soc, 51, 674 A929).
10. W i с h e r s E., J. Am. Chem. Soc, 76, 2033 A954).
11. M a a s s O., H a t с h e r W. H., J. Am. Chem. Soc, 42, 2548 A920).
12. Abrahams S. C, Coll in R. L., Lipscomb W. N.. ActaCryst., 4, 15A951).
13. Bragg W. L., J. Sci. Inst., 24, 27 A947).
14. DuMond J. W. M., Cohen E. R., Revs. Mod. Phys., 21, 651 A949); Am.
Scientist, 40, 447 A952).
15. G i n n i n g s D. С, С о г г и с с i n i R. J., J. Res. Nat. Bur. Stand., 38, 583 A947).
16. G i g u ё r e P. A., G e о f f r i о n P., Can. J. Res., 28B, 599 A950).
17. Spring W., Z. anorg. Chem., 8, 424 A895); Bruhl J. W., Ber., 28, 2855 A894);
С a 1 v e r t H. Т., Ann. physik., D), 1, 483 A900); S с h о о r 1 N., Pharm. Week-
blad, 67, 390 A930).
18. С u t h b e r t s о n A. C, Matheson G. L., M a a s s O., J. Am. Chem. Soc,
50, 1120 A928).
19. Huckaba С E., Key e s F. G., J. Am. Chem. Soc, 70, 2578 A948); 72, 5324A950).
20. K. u b a s с h e w s k i *O., Weber W., Z. Elektrochem., 54, 200 A950); U. S.
Dept. Commerce, PB report 91739, Washington, 1948.
21. E as ton M. F., Mitchell A. G.( Wynne-Jones W. F. K., Trans.
Faraday Soc, 48, 796 A952).
22. H u с k a b а С. Е., К е у e s F. G., J. Am. Chem. Soc, 70, 1640 A948).
23. Wynne-Jones W. F. К., частное сообщение, 1953.
24. M i t с h e 1 1 A. G., Wynne-Jones W. F. K-, Discussions Faraday Soc,
№ 15, Solutions of Non-Electrolytes, 1953, p. 161.
25. E 1 d e r L. W., Rideal E. K., Trans. Faraday Soc, 23, 545 A927).
26. Scatchard G., К a v a n a g h G. M., T i с k n о r L. В., J. Am. Chem. Soc,
74, 3715 A952).
27. Keyes F. G.,Smith L. В., G e г г у Н. Т., Proc Am. Acad. Arts Sci., 70,
319 A936).
28. G i g u ё r e P. A., L i u I. D., Dugdale J. S., Morrison J. A., Can.
J. Chem., 32, 117 A954).
29. P h i b b s M. K., Giguere P. A., Can. J. Chem., 29, 173 A951).
30. Беллинджер Ф., Фридман Г., Бауэр В.,Истс Дж., Э д м о н д с С,
в сб. «Физика и химия реактивного движения», № 1, Госиноиздат, М., 1948, стр. 175.
31. S w i n d е 1 1 s J. F., С о е J. R., G о d f г е у Т. В., J. Research Nat. Bur. Stan-
Standards, 48, 1 A952).
32. Ре г г у J. H., S m i t h E. R., Ind. Eng. Chem., 25, 195 A933); Porter A. W.,
Phil. Mag., 23, 458 A912).
33. L i t о v i t z T. A., J. Chem. Phys., 20, 1088 A952).
34. Миронов К. Е., Б е р г м а н А. Г., ДАН СССР, 81, 1081 A951).
35. S a t t e r f i el d C. NM Wentworth R. L, Demetriades S. Т., J.
Am. Chem. Soc, 76, 2633 A954).
36. S p г i n g W., Z. anorg. Chem., 8, 424 A895).
37. С а б и н и н а Л., Т е р п у г о в Л., Z. physik. Chem., 173A, 237 A935).
38. Одрит Л., Огг Б., Химия гидразина, Издатинлит, М., 1954.
17 Перекись водорода
258 Глава 5
39. G i g u ё г е P. А., частное сообщение, 1952.
40. Hen ri V., Z. Elektrochem., 11, 790 A905).
41. Bred ig G., Z. Elektrochem., 12, 581 A906).
42. W e i g e r t F., Z. physik. Chem., 60, 513 A907).
43. H e у m a n n H., Z. physik. Chem., 81, 204 A913).
44. Stern K. G., Ber., 66, 547 A933).
45. G о r d о n A. R., Ann. N. Y. Acad. Sci., 46, 285 A945).
46. G r a u p n e г К., W i n t e r E. R. S., J. Chem. Soc, 1952, 1145; Wang J. H.r
Robinson С . V., E d e 1 m a n I. S., J. Am. Chem. Soc, 75, 468 A953).
47. W i 1 к е С R., Chem. Eng. Progress, 45, 218 A949).
48. С о 1 1 a n d e r R., Finska Vetenskaps Societeten, Helsingfors, Comm. Biol., 2, № 6
A926) [C. A., 21, 1739).
49. M с M u r t r i e R. L., К е у e s F. G., J. Am. Chem .Soc, 70, 3755 A948).
50. G i 1 1 i 1 a n d E. R., Ind. Eng. Chem., 26, 681 A934).
51. Andrussow L., Z. Elektrochem., 54, 566 A950).
52. Montgomery R. В., J. Meteorology, 4, 193 A947).
53. M e n z el H., Z. anorg. Chem., 164, 10 A927); Z. Elektrochem., 33, 63 A927).
54. M a a s s O., H e r z b e r g O. W., J. Am. Chem. Soc, 42, 2569 A920).
55. Foley W. Т., G i.g u ё r e P. A., Can. J. Chem., 29, 123 A951).
56. G i g u ё r e P. A., S e с с о Е. A., Can. J. Chem., 32, 550 A954).
57. E g e r t о n A. C, E m t e W., Minkoff G. J., Discussions Faraday Soc, № 10,
278 A951).
58. N a t t a G., Rigaraonti R., Gazz. Chim. Ital., 66, 762 A936).
59. S t e i n d 1 , Behavior of Normal H2O2, Special H2O2, and Sodium Permanganate at
Low Temperatures, Peenemunde 1942, P. B. report 98493.
60. G e i b K. H., H a r t e с к Р., Ber., 65, 1551 A932).
61. J о n e s R. A., W i n к 1 e г С A., Can. J. Chem., 29, 1010 A951); Bat z о 1 d
J. S., L, u n e r C, W i n к 1 e г С A., Can. J. Chem., 31, 262 A953); Giguere
P. A., S e с с о E. A., E a t о n R. S., Discussions Faraday Soc, Toronto, № 14,
p. 104 A953).
62. Нейдинг А. Б.Д азарновский И. А., ДАН СССР, 74, 735 A950); ЖФХ,
26, 1167 A952).
63. В awn С. Е. Н., Hogg M. A. P., Discussions Faraday Soc, Toronto, № 14,
p. 141 A953).
64. В a b i n s k у Т. С, D i e r d о r f f L. H., KeefeJ. H., частное сообщение, 1953.
65. D о r s e у N. E., Trans. Am. Phil. Soc, 38, part 3 A948).
66. We у 1 W. A., J. Colloid. ScL, 6, 389 A951).
67. d e Foreran d R., Compt. rend., 130, 1620 A900).
68. Wol f f en s t ei n R., Ber,, 27, 3307 A894).
69. Bruhl J. W., Ber., 28, 2847 A895).
70. S i d e r s k у Н., Dissertation, Berlin, 1934, reported by Machu; J. S. African Chem.
Inst., 18, 44 A935).
71. Uchida S., Ogawa S., Yamaguchi M., Japan Sci. Rev., Eng. Sci.,
1, № 2, 41 A950) (на английском языке) [С. А., 46, 2271а].
72. М a a s s О., Н i e b e r t P. G., J. Am. Chem. Soc, 46, 2693 A924).
73. G i g и ё г е P. A., M a a s s О., Can. J. Res., 18B, 181 A940).
74. К е у e s F. G., J. Chem. Phys., 15, 602 A947).
75. W il 1 i a m s G. C, S a t t e r f i e 1 d C. N.. I-sbin H. S., J. Am. Rocket
Soc, III—IV, 1952, 70.
76. Льюис, Рендалл, Химическая термодинамика, ОНТИ, Химтеорет., Л., 1936;
J. Am. Chem. Soc, 36, 1969 A914).
77. К е 1 1 е у К. К., U.S. Dept. Interior, Bureau of Mines Bull. 383, Government Prin-
Printing Office, Washington, 1935.
78. Rossini E. D., W a g m a n D. D., E v a n s W. H., L e v i n e S., J a f f e I.,
Selected Values of Chemical Thermodynamic Properties, Natl. Bureau Standards Cir-
Circular 500, Government Printing Office, Washington, 1952, p. 10, 539, 825, 1027; см.
также серию III этого же издания в виде отдельных листков, выпускаемых нерегу-
нерегулярно с 1948 г.
79. G i g u ё г е Р. А., М о г i s s e t t e В. G., частное сообщение [Can. J. Chem.,
33, 804 A955)].
80. О t h m e r D. F., Ind. Eng. Chem., 32, 841 A940).
81. T а у 1 о r H. S., Glasstone S., eds., A Treatise on Physical Chemistry, 3 rd
ed., Vol. 1, D. Van Nostrand Co., Inc., New York, 1942; [есть перевод раннего изда-
издания: см. Т е й л о р X. С, Физическая химия, Химтеорет, Л., 1935, 19361; Н о и-
gen О. A., Watson К. М., Chemical Process Principles, Vol. 2, John Wiley
and Sons, Inc., New York, 1947.
82. Aston J. G., Ind. Eng. Chem., 34, 514 A942).
83. Герцберг Г., Колебательные и вращательные спектры многоатомных молекул,
Госиноиздат, М., [1949.
Литература 259
84. R о s s i n i F. D., Chemical Thermodynamics, John Willey and Sons, Inc., New
York, 1950,
85. Z e i s e H., Z. Elektrochem., 48. 693 A942).
86. M i с к 1 e у H. S., M. I. T. Hydrogen Peroxide Project, неопубликованная работа,
1946.
87. G i g u ё r e P. A., Can. J. Res., 28B, 485 A950).
88. R i b a u d G., Publ. sci. et tech. ministere air (France), 1952, № 266, p. 129.
89. P i t z e r K. S.,Gwinn W. D., J. Chem. Phys., 10, 428 A942).
90. Spencer H. M., J. Am. Chem. Soc, 67, 1859 A945).
91. Pike H. H. M., Green H., неопубликованная работа, 1945.
92. R о t h W. A., G r a u R.,Meichsner A., Z. anorg. Chem., 193, 161 A930).
93. Evans M. G., Uri N., Trans. Faraday Soc, 45, 224 A949).
94. В i с h о w s k у F. R., Rossini F. D., The Thermochemistry of the Chemical
Substances, Reinhold Publishing Corp., New York, 1936.
95. F a v r e P. A., S i 1 b e r m a n n J. Т., Ann. chim. et phys., C), 36, 5 A852);
J. Chem. Soc, 6, 234 A854); Thomsen J., Ann. Physik., 227, 194 A874); Thermo-
dynamische Untersuchungen, Vol. 2, Barth J. B. Leipzig, 1882, p. 58; В е г t h e-
1 о t M. P. E., Ann. chim. et phys., E), 6, 209 A875); 21, 194 A880); Compt rend.,
90, 331 A880); de Forcrarud H. R., Compt. rend., 130, 1250, 1620A900);
Ann. chim. et phys., (8), 15, 433 A908); Kiispert F., Natur und Schule, 2, 171,
A903); Henri V., Wurmser R., Compt. rend., 157, 126 A913).
96. M e д а р Л., Взрывные свойства перекиси водорода, в сб. «Физика и химия реак-
реактивного движения», № 1, Госиноиздат, М., 1948, стр. 197.
97. Fontana B.J., Atomic Energy Commission Documents MDDC-542 and CC-3482
A946).
98. Л а т и м е р В. М., Окислительные состояния элементов и их потенциалы в вод-
водных растворах, Издатинлит, М., 1954.
99. Del ah ay P., Pourbaix M., Van Rysselberghe P., Comite in-
intern, thermodyn. et cinet. electrochim., Compt. rend, reunion, 1950, 42.
100. S m al e F. J., Z. physik. Chem., 14, 590 A894); I hi e R., Z. physik. Chem., 22,
114 A897); G 1 a s e r L., Z. Elektrochem., 4, 373 A898); Richarz F., Lon-
n e s C, Z. physik. Chem., 20, 145 A898); H a b e r F., G r i n b e r g S., Z. anorg,
Chem., 18, 37 A898); В о s e E., Z. physik. Chem., 34, 701 A900); H a b e r F.,
Z. elektrochem., 7, 441, 1043 A901); Fredenhagen C, Z. anorg. Chem., 29,
451 A902); В о r n e m a n K., Z. anorg. Chem., 34, 1 A903); 78, 33 A912); Z. Elektro-
Elektrochem., 15, 673 A909); Mazzuchelli A., Barbero C, Atti R. Accad. Lin-
cei, E), 15, 35, 109 A906); H a b e r F., Patterson W. H., Z. anorg. Chem.,
51, 356 A906); H a k о m о r i S., Tech. Repts. Tohoku Imp. Univ., 9, 567, 583,
607 A931) (на английском языке); Wolff R., Compt. rend., 196, 1113 A933);
Bancroft W. D., Murphy N. F., J. Phys. Chem., 39, 377 A935); R о d a-
mov K., Onizuka Т., J. Biochem. (Japan), 25, 573 A937) [CA, 31, 8394];
Д о б р и н с к а я А. А., Н е й м а н М. В., Acta Physicochim. U.R.S.S., 10, 297
A939) (на английском языке); Kordesch K.,MartinolaF., Monatsh., 84,
39 A953); Я б л о к о в а Н. Е., Б а г о ц к и й В. С, ДАН СССР, 85, 599 A952).
101. Bornemann К., Nernst Festschrift, Knapp W., Halle, 1912, p. 118.
102. В erl W. G., Trans. Electrochem. Soc, 83, 253 A943).
103. H i с k 1 i n g A., W i 1 s о n W. H., J. Electrochem. Soc, 98, 425 A951).
104. H i с k 1 i n g A., Quart. Revs. (London), 3, 95 A949).
105. К a 1 о u s e k M., Collection Czechoslov. Chem. Communs., 13, 105 A948).
106. Hacobian S., Australian J. Chem., 6, 211 A953).
107. Kern D. M. H., J. Am. Chem. Soc, 76, 1455 A954).
108. К о г у t a J., Chem. Listy, 46, 593 A952); Collection Czechoslov. Chem. Communs.,
18, 21 A953).
109. К о 1 t h о f.f I. M., L i n g a n e J. J., Polarography, 2 nd. ed., Interscience Publi-
Publishers, New York, 1952, p. 552; [есть перевод первого издания: см. Кольт-
г о ф И. М., Л и н г е й н Дж. Дж., Полярография, Госхимиздат, М.—Л., 1948].
110. Novak J., Heyro vsky J., Collection Czechoslov. Chem. Communs., 9, 203,
273 A937).
111. U г е у Н. С, D a w s e у L. H., Rice F. O., J. Am. Chem. Soc, 51, 1371 A929).
112. Walsh A. D., J. Chem. Soc, 1948, 331.
113. Bodenstein M., S с h e n k P. W., Z. phys. Chem., 20B, 420 A933); Bray
W. C, J. Am. Chem. Soc, 60, 82 A938); H e i 11 e r W., Elementary Wave Mechanics,
Oxford, Clarendon Press, 1945; Haber F., W e i s s J., Proc Roy. Soc, 147A,
350 A933).
114. Robertson A. J. В., Trans. Faraday Soc, 48, 228 A952).
115. E v a n s M. G., Hush N. S., Uri N., Quart. Revs. (London), 6, 186 A952).
116. Evans M. G., U r i N., Trans. Faraday Soc, 45, 224 A949).
117. Uri N.. Chem. Revs., 50, 375 A952).
118. P г i t с h a r d H. O., Chem. Revs., 52, 529 A953).
17*
260 Глава 5
П9. Evans M. G., В а х е n d a 1 е J. H., U r i N., Trans. Faraday Soc, 45, 236 A949).
120. T r a u t z M., P а к s с h w e r S., J. prakt. Chem. N. F., 122, 147 A929).
121. Cal vert H. Т., Ann. Physik, 1, 483 A900).
122. Cal vert H. Т., Z. physik. Chem., 38, 513 A901).
123. J о у n e r R. A., Z. anorg. allgem. Chem., 77, 103 A912).
124. С u t h b e r t s о n A. C, M a a s s O., J. Am. Chem. Soc, 52, 489 A930).
125. Schumb W. C, Ind. Eng. Chem., 41, 992 A949).
126. Gross P. M., Та у 1 о r R. С, J. Am. Chem. Soc, 72, 2075 A950).
127. Roth E. M., Jr., S h a nl e у Е. S., Ind. Eng. Chem., 45, 2343 A953).
128. Y о u n g R. С, неопубликованная работа, М. I. Т. Hydrogen Peroxide Project, 1946.
129. D e w a r J., Fleming J. A., Proc Roy. Soc (London), 62, 250 A897).
130. L i n t о n E. P., M a a s s O., J. Am. Chem. Soc, 53, 957 A931).
131. L i n t о n E. P., M a a s s O., Can. J. Res., 4, 322 A931); 7, 81 A932).
132. Wyman J., Phys. Rev., 38, 623 A930).
133. Talat-Erben, Bull. Soc Chim. France, 1952, 176.
134. M a a s s O., Hatcher W. H., J. Am. Chem. Soc, 44, 2472 A922).
135. Gray F. W., Farquharson J., J. Sci. Insts., 9, 1 A932).
136. Savithri K., Rao S. R., Proc Indian Acad. Sci., 16A, 221 A942).
137. E g gl es t о n B. C, E v a n s D. F., R i с ha r d s R. E., J. Chem. Soc, 1954, 941.
138. Физические методы органической химии, под ред. А. Вайсбергера, т. IV, Издатин-
лит, 1954.
139. Schoorl N., Pharm. Weekblad, 67, 390 A930).
140. Giguere P. A., Can. J. Res., 21B, 156 A943).
141. Giguere P. A., G e о f f r i о n P., Can. J. Res., 27B, 168, A949).
142. Bauer N.. Fajans K-, in «Techniques of Organic Chemictry», Vol. I, Part 2,
I Interscience Publishers, Inc., New York, 1949, p. 1141.
143. Kurtz S. S., W a r d A. L., J. Franklin Inst., 224, 583, 697 A937).
144. Andersen E. В., A s m u s s e n R. W., J. Phys. Chem., 36, 2827A932).
145. Giguere P.A.,Feeny H., Can. J. Res., 21A, 69 A943).
146. Waring C. E., Custer R L., J. Am. Chem. Soc, 74, 2506 A952).
147. Gall ais F., Wolf R., Compt. rend., 231, 1458 A950).
148. Massey J. Т., В i a n с о D. R., Physical Rev., 85, 717 A952); J. Chem. Phys.,
22, 442 A954).
149. Friedel C, Ann. Physik, 55, 453 A895).
150. M a i о n e A., Nuovo cimento, 12, 358 A935).
151. Ganz E., Z. physik. Chem., 33B, 163 A936).
152. Bailey C. R., G о r d о n R. R., Trans. Faraday Soc, 34, 1133 A938).
153. Z u m w a 1 t L R., Giguere P. A., J. Chem. Phys., 9, 485 A941).
154. Giguere P. A., Trans. Roy. Soc Canada, C), 35, III, 1 A941).
155. Giguere P. A., J. Chem. Phys., 18, 88 A950).
156. К i г b y-S m i t h J. S., J о n e s E. A., J. Opt. Soc Am., 39, 780 A949); К a t z J. J.,
Ну man H. H., Rev. Sci., Insts., 24, 1066 A953).
157. Taylor R. C, J. Chem. Phys., 18, 898 A950).
158. Bain O., Giguere P. A., Can. J. Chem., 33, 527 A955).
159. Giguere P. А., В a i n O., J. Phys. Chem., 56, 340 A952).
160. U с h i d a Y., FukudaK., Bull. Inst. Chem. Research, Kyoto Univ., 27, 1 A951)
[CA; 48, 6807f] (Выводы на английском языке).
161. Venkateswaran S., Nature, 127, 406 A931).
162. Damaschun I., Z. physik. Chem., 16B, 81 A932).
163. Venkateswaran S., Phil. Mag., G), 15, 263 A933).
164. Simon A., Feher F., Z. Elektrochem., 41, 290 A935).
165. Kohl r a use h K. W. F., Monatsh., 68, 349 A936).
166. Taylor R. С, частное сообщение, 1953.
167. Simon A., M a r с h a n d M., Z. anorg. Chem., 262, 192 A950).
168. Simon A., U h 1 i g U., Chem. Ber., 85, 977 A952).
169. Russell W. J., L a p r a i k W., Nature, 22, 368 A880).
170. Hartley W. N., J. Chem. Soc, 39, 111 A881).
171. Tian A., Compt. rend., 151, 1040 A910), 156, 1879 A913).
172. Henri V., W u r m s e r R., Compt. rend., 156, 1012 A913); 157, 126 A913).
173. Розанов Н. А., ЖРФХО, 44, 1146 A912).
J74. К о r n f e 1 d G., Z. wiss. Phot., 21, 66 A921).
175. L e d e r 1 e E., Rieche A., Ber., 62, 2573 A929).
176. Al 1 m a n d A. J., S t у 1 e D. W. G., J. Chem. Soc, 1930, 596.
177. S h a r m a R. S., Proc Acad. Sci. United Provinces Agra Oudh, India, 4, 51 A934).
178. В r e d i g G., L e h m a n H. L., К u h n W., Z. anorg. allgem. Chem., 218, Г6
A934).
179. Frost А. А., О 1 d e n b e r g O., J. Chem. Phys., 4, 642 A936).
180. F e r g u s s о n W. C, S 1 о t i n L., S t у 1 e D. W. G., Trans. Faraday Soc, 32,
956 ,A936). ;
Литература 261
181. Holt R. В., М с L a n е С. К., О 1 d e n b e r g О., J. Chem. Phys., 16, 225 A948).
182. Е g е г t о п А. С, Н а г г i s -E. J., Young G. H. S., Trans. Faraday Soc, 44,
745 A948).
183. M с L a n e С. К., J. Chem. Phys., 17, 379 A949).
184. Taylor R. C, G г о s s P. C, J. Am. Chem. Soc, 71, 2266 A949).
185. E d se R., J. Chem. Phys., 18, 244 A950).
186. P h i b b s M. K., Giguere P. A., Can. J. Chem., 29, 490 A951).
187. D h a r N. R., В h a t t а с h a r у а А. К., J. Indian Chem. Soc, 11, 311 A934).
188. О 1 d e n b e r g O., J. Chem. Phys., 3, 266 A935).
189. Frost А. А., О 1 d e n b e r g O., J. Chem. Phys., 4, 781 A936).
190. Feher F., Klotzer F., Z. Elektrochem., 41, 850 A935).
191. F e h ё г F., Klotzer F., Z. Elektrochem., 43, 822 A937).
192. L u C.S.,HughesE.W.,Giguere P. A., J. Am. Chem. Soc, 63, 1507A941).
193. Randall J. Т., Proc Roy. Soc. (London), 159A, 83 A937).
194. Giguere P. A., S с h о m а к e r V., J. Am. Chem. Soc, 65, 2025 A943).
195. Floyd J. P., G г о s s P. M., Jr. J. Am. Chem. Soc, 77, 1435 A955).
196. Akerlof G., Turck H. E.,J. Am. Chem. Soc, 57, 1746 A935).
197. В о b t e 1 s к у M., S i m с h e n A. E., J. Am. Chem. Soc, 64, 454 A942).
198. В о b t e 1 s к у M., Simchen A. E., Bull. soc. chim. France, 1950, 870.
199. Jones H. С, С а г г о 1 1 С G., Am. Chem. J., 28, 284 A902).
200. Jones H.C.BarnesJ. H у d e E. P., Am. Chem. J., 27, 22 A902).
201. Казанецкий П. В., ЖРФХО, 46, 1110 A914).
202. J a h г К. F., В г е с h 1 i n A., Bl anke M., Kubens R., Z. anorg. allgem.
Chem., 270, 240 A952) [C.A., 47, 6740a]; 272, 45 A953) [C.A., 47, 5838a].
203. M u n z b e r g F., Lotos, 76, 351 A928) [C. A., 26, 2132].
204. Price T. S., Proc. Chem. Soc (London), 23, 75 A907); J. Chem. Soc, 91, 531 A907).
205. Meyer J., P a w 1 e t t a A., Z. physik. Chem., 125, 49 A927).
206. H u s a i n S., Z. anorg. allgem. Chem., 177, 215 A928).
207. Menzel H., Gable r C, Z. anorg. allgem. Chem., 177, 187 A928),
208. Макарове. 3.,ЧамоваВ. Н., Изв. АН СССР, отд. хим. наук, № 3, 225A951);
Bull. Acad. Sci. USSR, Div. Chem., Sci., 1952, 589 (на английском языке).
209. Jones H. С, M u г г а у G., Am. Chem. J., 30, 205 A903).
210. H a t с h e r W. H., M а с L a u с h 1 a n D. W., Can. J. Res., 16B, 253 A938).
211. Menzel H., Z. physik. Chem., 105, 402 A923).
212. Hatcher W. H., Sturrock M. G., Can. J. Res., 4, 35 A931).
213. Hatcher W.H., Powell E. C, Can. J. Res., 7, 270 A932).
214. Walton J.H., Lewis H. A., JonesD.O., Brann A., J. Am. Chem. Soc,
38, 633, 1956 A916).
215. A b e g g P. В., report L-73679 (frames 8575-8577).
216. П е р ш к е В., Ч у ф а р о в Г., Z. anorg. Chem., 121, 121 A926).
217. Livingston R., J. Am. Chem. Soc, 50, 3204 A928).
218. Gorin M. H., J. Am. Chem. Soc, 57, 1975 A935).
219. Осипов И., П о п о в С, ЖРФХО, 35, 637 A903).
220. Колосовский Н. A., Bull, soc chim. Beige, 28, 257 A914), Bull, soc chim.,
37, 372 A925) [C. A., 19, 3404].
221. С r i s m e r L., Bull, soc chim., C), 6, 24 A893).
222. Ю р ж е н к о Т. И., Г р о м о в а Г. Н., Хайцер В. Б., ЖОХ, 16, 1505 A946).
223. Колосовский Н. А., Куликов Ф. С, Укра1н. хем. журнал., 9, научно-
техническая часть, 37—40 A934).
224. Bretschger M. E., Shanley E. S., Trans. Electrochem. Soc 92, 67 A947).
225. Simon A., F e h e r F., Z. Elektrochem., 42, 688 A936).
226. Rieche A., Alkylperoxyde und Ozonide, Steinkopf Т., Dresden, 1931.
227. Бете Г., Лекции по теории ядра. Госиноиздат, М., 1949.
228. В a i n b r i d g е К. Т., N i e r А. О., Relative Isotopic Abundances of the Elements.,
Prelim. Rpt. № 9, National Research Council, Washington, 1950.
229. Dole M., Chem. Revs., 51, 263 A952).
230. U. S. Dept. of Commerce, National Bureau of Standards, Circular 499 and^ Supplement,
Government Printing Office, Washington, 1950.
231. Киршенбаум И., Тяжелая вода. Физические свойства и методы анализа,
Издатинлит, М., 1953.
232. Ben tl ey R., Cold Spring Harbor Symp. quart. Biol., 13, 11 A948).
233. Erlenmeyer H., Gartner H., Helv. Chim. Acta, 17, 970 A934).
234. Feher F., Ber., 72B, 1789 A939).
235. Giguere P. A., S e с с о Е. A., EatonR. S., Discussions Faraday Soc, № 14,
Toronto, 1953, p. 104.
236. Feher F., Z. Elektrochem., 43, 663 A937), Angew. Chem., 50, 909 A937).
237. Giguere P. A., Bull. Soc Chim. France, 1954, 720.
238. Liu I. D., Giguere P. А., частное сообщение, 1953.
239. Feher F., Ber., 72B, 1778 A939).
Глава 6
СТРУКТУРА ПЕРЕКИСИ ВОДОРОДА
н—о —о—н
'N.-C
\
н
Структуру перекиси водорода целесообразно рассматривать по частям,
отдельно обсуждая расположение атомов в молекуле и природу ее ассоциации
в растворе и в самой перекиси водорода в различных фазовых состояниях.
Установление структуры основано главным образом на результатах измерений
физических свойств, собранных в гл. 5. Данные по некоторым химическим
свойствам с целью использования их для определения структуры взяты
из гл. 7; вопросы же влияния структуры на химические свойства излагаются
в следующих главах. На стр. 283 приведена табл. 69, в которой подытожены
наиболее вероятные данные различных структурных параметров.
СТРУКТУРА МОЛЕКУЛЫ ПЕРЕКИСИ ВОДОРОДА
Определение молекулярного веса и стехиометрия реакций перекиси водо-
водорода отчетливо доказывают, что формула этого соединения соответствует Н2О2.
В обычном смысле слова молекулы полимеров (Н2О2)Х не существуют. Точно
,,м так же молекулу, в которой отно-
'V шение кислорода к водороду превы-
;л?,'шает единицу, нельзя называть
перекисью водорода. В этом классе
соединений обнаружена только мо-
молекула, имеющая формулу НО2,
но она встречается только в неко-
торых]реакциях и возникает лишь
на очень короткий срок (см. стр. 44).
Сравнительно недавно [1] вторично
высказано предположение о суще-
существовании больших молекул этого
типа, например Н2О4 11], которые
будто*бы существуют отдельно или
в*равновесной смеси сН2О2. Однако
никаких убедительных доказа-
доказательств, подтверждающих это пред-
предположение, не приведено, и возмож-
возможность существования таких молекул
в'обычных условиях маловероятна.
Кловер [2] показал, что непра-
неправильная методика анализа может
привести к таким ошибочным вы-
выводам. Для обсуждения за основу принимаются некоторые предложенные
в разное время приемлемые конфигурации молекулы Н2О2, показанные
на рис. 51. Эти модели представлены только для иллюстрации относительных
положений атомных ядер; тип связи в них не рассматривается. Все модели,
Н
Nn —
IV
;о—о
51. Возможные конфигурации
кулы перекиси водорода.
/—линейная; //—транс-плоскостная; ///—цис-
плоскостная; IV—двухплоскостная; V—плоскостная
тригональная; VI—пирамидальная тригональная.
Р и с.
моле-
Структура молекулы перекиси водорода 263
изображенные на рис. 51, можно разделить на две основные структурные
формы: в одной каждый атом водорода соединен с отдельным атомом кислорода
(модели I—IV); в другой оба атома водорода присоединены к одному и тому же
атому кислорода (модели V и VI). Во избежание'путаницы при анализе этих
моделей необходим осторожный подход. Особенностью моделей I—IV является
цепное расположение атомов. Модели V и VI обладают спицеобразным распо-
расположением обоих атомов водорода и одного атома кислорода вокруг централь-
центрального кислородного атома, что оправдывает название «тригональные формы».
Внутри этих групп, цепных и тригональных, можно различать отдельные
модели, называемые линейными, плоскостными, двухплоскостными или пира-
пирамидальными*.
Первая стадия при анализе структуры перекиси водорода заключается
в выборе одной из этих двух общих форм. После решения этого вопроса можно
остановиться на подробностях, касающихся расстояний и углов между атомами.
Ниже показано, что правильность модели IV в настоящее время можно считать
доказанной.
Химические доказательства
Хотя структура, вероятно, должна проявляться в химических свойствах,
теория этого вопроса еще не настолько разработана, чтобы по этим свойствам
можно было более или менее полно выяснить структуру перекиси водорода.
С одной стороны, разложение перекиси водорода приводит к образованию
воды и кислорода, и легко представить, что механизм такого разложения
состоит в разрыве кислород-кислородной связи в моделях V или VI с образова-
образованием воды и кислородного атома, способного соединиться с другим атомом
кислорода, образующимся аналогичным образом из другой молекулы. С другой
стороны, перекись водорода может образовать какую-нибудь диалкилперекись,
при восстановлении которой получается не эфир, а спирт. Отсюда можно сде-
сделать заключение [3], что перекись водорода обладает одной из цепных конфи-
конфигураций, представленных на рис. 51 моделями I—IV. Эта дилемма оставалась
неразрешенной в течение долгого времени, хотя, по мнению большинства,
наиболее вероятной является тригональная форма (см. стр. 18). Еще в 1928 г.
Райков [4] привел чисто геометрический аргумент в пользу плоскостной три-
гональной формы. Но хотя доказательство, основанное на образовании орга-
органических производных, является, по-видимому, вполне основательным, нет
никаких причин постулировать наличие тригональной структуры только
потому, что она показывает наиболее прямой и очевидный путь к образованию
кислорода. Уже не говоря о весьма большой вероятности очень сложных сту-
ступенчатых механизмов реакции, предположение, что кислород в цепной струк-
структуре экранирован от реакций, по-видимому, не оправдывается. Можно себе
представить конфигурацию смежных молекул с плоскостной цепной структурой
(на плоскости или в пространстве), которая, вероятно, обладает такой же
направленностью реакции в геометрическом смысле, как и тригональная форма.
Имеется еще доказательство, которое истолковывалось в пользу триго-
тригональной формы. Гейб и Гартек [5] в 1932 г. показали, что из продуктов реакции
атомарного водорода и молекулярного кислорода можно при температуре жид-
жидкого воздуха конденсировать стекловидный продукт. Последний частично раз-
разлагается при нагревании до —115°; при дальнейшем нагревании происходит
плавление с образованием перекиси водорода и воды. Качественное исследова-
исследование ультрафиолетового спектра поглощения стекловидного вещества и мате-
материальный баланс доказывают, что в системе присутствуют лишь перекись
водорода и вода. Для объяснения этого явления и свойств вещества Гейб и Гар-
* Двухплоскостную форму можно представить и как пирамидальную; однако этот
термин логичнее сохранить для правильной формы типа модели V.
264 Глава 6
тек постулировали, что только что образовавшееся стекловидное вещества
представляет собой смесь нормальной перекиси водорода и аномальной пере-
перекиси водорода тригональной формы. Разложение при —115° может быть при-
приписано аномальной форме, которая, по-видимому, не стабильна при температу-
температурах выше указанной. Однако необходимо отметить, что предположение о суще-
существовании тригональной формы высказано Гейбом и Гартеком только в виде
гипотезы.
Эти наблюдения еще не нашли объяснения, однако результаты экспери-
экспериментальной работы этих авторов были впоследствии подтверждены и расширены
[6]. Кроме того, Жигер и Секко [7] дали предварительное описание результа-
результатов исследования инфракрасного спектра поглощения этого стекловидного
осадка. У продуктов, полученных при разряде в парах тяжелой и обыкновен-
обыкновенной воды, обнаружена среди других полос поглощения одна полоса, соответ-
соответствующая 1300 см'1 и не изменяющаяся при замене легкого изотопа водорода
тяжелым; таким образом, она не может быть обусловлена вибрацией ОН, а
должна быть приписана вибрации О—О. Как показано при анализе спектра
перекиси водорода (стр. 277), с этой вибрацией молекулы обычно связывается
полоса поглощения при 877 см'1. Сделано заключение, что полоса 1300 см'1
не может возникнуть за счет координационной кислород-кислородной связи;
остается возможность, что в указанных условиях образуется пергидроксильный
радикал НО2. Раньше чем можно будет дать удовлетворительное объяснение
результатов этих опытов, потребуются дополнительные работы. Безусловно,
нужно проанализировать и другие возможности, например адсорбцию газа
и наличие радикалов, таких, как ОН. Пока нет никаких доказательств, что
при обыкновенных температурах существует в измеримых концентрациях
тригональный изомер или что обыкновенная перекись водорода может быть
превращена в другую форму путем резкого снижения температуры.
Признание ионной диссоциации перекиси водорода в водных растворах
привело к выводу, что активной частицей в действительности является ион
НОд, однако этот вывод можно согласовать с любой из предложенных моделей.
Недавно поставленные опыты с применением меченых атомов для выяснения
природы газообразного кислорода, выделяющегося при каталитическом разло-
разложении или окислении перекиси водорода, также нельзя интерпретировать
совершенно однозначно. Приходится сделать заключение, что химические
доказательства не позволяют точно установить структуру перекиси водорода.
Аддитивные и конститутивные свойства
Обычные аддитивные или конститутивные свойства молекулярной струк-
структуры мало что могут дать для установления структуры перекиси водорода.
Под этим заголовком можно рассматривать парахор, молекулярную рефрак-
рефракцию, магнитооптическое вращение, магнитную восприимчивость и молярный
объем.
Катбертсон и Маас [8] на основании измерений вычислили парахор пере-
перекиси водорода F9,6 г1/*-см3-сек~1/*-моАЬ~1У, они считают, что эта величина под-
подтверждает плоскостную тригональную структуру. На основании атомных
и структурных инкрементов Сегдена эмпирически вычислены парахоры для
трех моделей перекиси водорода, оказавшиеся равными 72,5; 74,1 и 97,7. Фиббс
и Жигер [9] путем новых измерений поверхностного натяжения и плотности
определили для парахора значение 70,0. Эмпирическая природа парахора
и зависимость его от молекулярной ассоциации обусловливает сомнительную
ценность его для выяснения молекулярной структуры перекиси водорода.
Экспериментальное значение молекулярной рефракции 5,801 см3/моль,
обладает «экзальтацией» по сравнению с величиной, вычисленной из аддитив-
аддитивных атомных эквивалентов [10]. Брюль [11] и Штрекер и Шпиталер [12] счи-
Структура молекулы перекиси водорода 265-
тают, что это свидетельствует о наличии многократных связей между кислород-
кислородными атомами в перекиси водорода. Линтон и Маас [131 расценивают это как
доказательство правильности плоскостной тригональной формы. Жигер [141
указывает, что экзальтация скорее всего вызывается наличием двух неделенных
пар электронов у каждого кислородного атома в группе О—О. Таким образом,
молекулярная рефракция не дает возможности установить структуру на осно-
основании эквивалентов рефракции, найденных в результате наблюдений, произ-
произведенных в отношении других веществ. Можно считать, что в лучшем случае
она дает величину инкремента рефракции, обусловленную группой О—О.
Милас, Сардженор и Перри [15] таким именно образом использовали данные
по рефракции и обнаружили хорошую согласованность вычисленных и экспе-
экспериментальных значений для рефракции ряда органических перекисей. Молеку-
Молекулярная рефракция дает возможность подсчитать радиус молекулы перекиси
водорода. На стр. 230 указано, что поляризация перекиси водорода составляет
2,30- Ю4 см3 на молекулу, откуда в соответствии с простой электростатической
георией радиус молекулы равен 1,32А. Дисперсионная постоянная а формулы
Зеллмейера дает возможность вычислить [16] число электронов на молекулу
перекиси водорода, обладающих периодом колебаний, соответствующим
характеристической частоте v0, которая лежит около 1000 А (в дальнем ультра-
ультрафиолете). Число электронов на молекулу перекиси водорода оказывается рав-
равным 4; для воды это число равно 2. Вероятно, это число указывает на неделен-
ную пару р-электронов при каждом атоме кислорода. Другое близкое оптичес-
оптическое свойство перекиси водорода, молекулярное магнитное вращение, не со-
согласуется с величиной, вычисленной из принятых атомных эквивалентов [171.
Грей с сотрудниками [18] разработали теоретическую интерпретацию
структурных констант магнитной восприимчивости. Они использовали эту
теорию и для анализа структуры перекиси водорода, но очевидно безуспешно,
так как в одном случае более предпочтительной оказалась плоскостная цепная
молекула (форма II или III), в другом—плоскостная тригональная. Простые
аддитивные магнитные факторы Паскаля также дают для магнитной восприим-
восприимчивости значение, не согласующееся с рекомендуемой величиной. Основное
значение определения магнитной восприимчивости с точки зрения анализа
структуры состоит в доказательстве, что перекись водорода не является моле-
молекулой «с неспаренным электроном» (см. ниже).
Молекулярный объем перекиси водорода при температуре кипения
(около 26,6 см*/моль) точно согласуется с вычисленным из атомных объемов
Коппа [19]. Однако при отсутствии рациональной основы для введения кон-
конститутивного фактора это определение ничего не дает для выяснения структуры.
Дипольный момент перекиси водорода
В 1932 г. Линтон и Маас [13] определили дипольный момент перекиси водо-
водорода методом разбавления, использовав в качестве растворителей диоксан
и этиловый эфир. Найденные значения, по их мнению, подтверждают пра-
правильность плоскостной тригональной модели. Они получили для дипольного
момента перекиси водорода в диоксане значение 2,13-10~18CGSE, а в эфире
2,06-108CGSE. При рассмотрении вопроса о диэлектрической проницаемости
(стр. 222) против метода Линтона и Мааса приведены некоторые возражения,
однако эти возражения не могут поколебать в значительной мере полученные
значения. Эти значения дипольного момента экспериментально подтвердили
Масси и Бьянко [20], показавшие, что их наблюдения эффекта Штарка в
микроволновом спектре перекиси водорода согласуются с дипольным моментом
2,26-Ю"*18 CGSE. Эти значения дипольного момента, безусловно, исключают
возможность линейной цепной и транс-плоскостной цепной моделей. .
266 Глава 6
Смит [21] еще раньше высказывал мысль, что высокая диэлектрическая
проницаемость перекиси водорода, вероятно, доказывает наличие значитель-
значительного дипольного момента, и истолковал это соображение в пользу плоскостной
тригональной модели. Против такого истолкования высказываются Хантер
и Партингтон [22], подчеркивающие, что свободное вращение гидроксильных
групп в перекиси водорода около оси О—О (что допускается для всех форм
плоскостных и двухплоскостной цепных моделей II—IV) также должно при-
привести к большому дипольному моменту молекулы. После опубликования
результатов экспериментальных измерений дипольных моментов Тильакер
[23] сделал такой расчет для молекулы, обладающей свободным вращением ОН.
При этом он исходил из предпосылки, что угол ООН в перекиси водорода
такой же, как и угол НОН в воде (последний тогда принимался за 110°), и что
момент группы ОН в обеих молекулах один и тот же; на этом основании он вычис-
вычислил дипольный момент перекиси водорода (около 2,l-10~18CGSE), который
хорошо совпадает с экспериментальным значением. С другой стороны, имеются
указания [20], что быстрое свободное вращение должно было бы привести
к малому дипольному моменту; однако основа для такого суждения не ясна.
Роджерс и Кемпбел [24] вычислили, что связь ОН в перекиси водорода
имеет момент 1,71 • 10~18 CGSE; при этом они использовали экспериментальный
дипольный момент и значения 105° для угла ООН и 100° для азимутального
угла. Они сравнили это значение с величиной 1,51-108 CGSE для момента
связи ОН в воде. Вектор постоянного дипольного момента молекулы перекиси
водорода лежит вдоль ее оси С2 (т. е. оси С, стр. 275, рис. 54, а). Высшие
моменты [25], квадрупольный, октупольный и т. д., не измерялись и были
вычислены лишь приближенно. На этом вопросе мы кратко остановимся в одном
из следующих разделов.
Проведенное выше обсуждение не дает возможности вывести надежное
заключение; для удовлетворительного выяснения структуры необходимо при-
прибегнуть к данным спектроскопических измерений и квантово-механических
вычислений. Этот вопрос излагается в следующих разделах; обзор его дан
Жигером [26]. Хотя этим путем тригональная модель полностью исключается
и доказывается правильность двухплоскостной цепной формы (модель IV),
все еще встречаются высказывания, что перекись водорода состоит из таутомер-
ной смеси плоскостной тригональной формы (модель V) и предпочтительной
двухплоскостной цепной формы (модель IV). Эта точка зрения, защищаемая
например Сиджвиком [27], в основном базируется на гипотезе Гейба и Гартека.
Квартароли [28], например, истолковал результаты изучения реакции пере-
перекиси водорода с гидратом окиси меди как доказательство существования тауто-
мерной смеси. Физические доказательства, приведенные ниже, устанавливают
с несомненностью, что вероятность существования тригональной формы исче-
зающе мала и что истинная конфигурация молекулы перекиси водорода соот-
соответствует модели IV. Это положение необходимо принять, правда, с небольшой
оговоркой до тех пор, пока не будет выяснена природа вещества, образующегося
при низкой температуре. Однако, поскольку дело касается обыкновенных тем-
температур, приведенный вывод оказывается единственно возможным.
Электронная структура
Изучение расположения электронов в перекиси водорода открывает больше
возможностей для правильного выяснения ее структуры. Кислородный атом
состоит из ядра и восьми электронов, расположенных в двух оболочках,
из которых первая, или /С-оболочка, содержит два электрона, а вторая, или
L-оболочка (валентная оболочка),—шесть электронов. Водородный атом также
состоит из ядра и одного-единственного электрона в /(-оболочке. Это описание
водорода и кислорода и развитие октетной теории позволили Льюису [29]
Структура молекулы перекиси водорода
267
представить перекись водорода в виде молекулы с электронной конфигурацией
Н:6:6:*Н. Эта формула показывает, что электронный октег каждого[атома
кислорода завершается за счет деления валентных электронов атомов, свя-
связанных с атомом кислорода. Хотя такая структура считается стабильной, она
не требует определенной ориентации атомов водорода в отношении кислород-
кислородного атома. Льюис указывает, что «подвижные» атомы водорода могут занимать
любое положение вокруг пары атомов кислорода. Таким образом, любая из
Рис. 52. Изображение электронной структуры
атомарного кислорода и перекиси водорода.
а—кислород; б—перекись водорода»
конфигураций, показанных на рис. 51, оказывается возможной, поскольку
в ней сохраняется структура завершенного октета. Сообщение Лоури [30]
доказывает трудность выяснения структуры перекиси водорода на основе
одной лишь октетной теории. Только развитие квантово-механического описа-
описания электронной структуры дало те необходимые дополнительные подроб-
подробности, которые позволяют выбрать определенную ориентацию водородных
атомов в отношении атомов кислорода.
Квантово-механическое представление электронной структуры основано
на признании индивидуальности пар электронов, которые могут занимать
последовательные и единственные уровни энергии или орбиты внутри атома.
Эти орбиты не представляют собой зафиксированные траектории; лучше всего
их описать как области пространства вокруг ядра, в которых вероятность
нахождения электрона является высокой. Каждая орбита может содержать два
268 Глава 6
электрона, но не больше. Некоторые из таких орбит должны обладать опре-
определенной ориентацией по отношению к другим орбитам. Для понимания струк-
структуры перекиси водорода необходимо знакомство с этой характеристикой напра-
направленности. Поэтому здесь дается элементарное описание этого вопроса. Более
подробное изложение материала можно найти в сжатом описании Ноллера
[31], в книгах Брауна [32] и Палмера [33], представляющих собой введение
в эту область, а также в книгах Паулинга 134] и Кулсона [35].
Электронная структура атомарного кислорода представлена на рис. 52,а.
Точки на рисунке обозначают не положения электронов, а только число их
на определенной орбите. Вышеупомянутая /(-оболочка содержит два электрона
в так называемой ls-орбите. Эта орбита (так же как и все s-орбиты) сферически
симметрична в отношении ядра. Показанная L-оболочка содержит четыре
орбиты 2s, 2рХУ 2ру и 2 р2, из которых каждая может содержать два электрона.
2s-op6nTa является сферически симметричной, тогда как р-орбиты ориентиро-
ориентированы вдоль координат*, у и г. Уровень энергии 2$-орбиты заметно ниже уровня
равноэнергетических р-орбит. Поэтому уровень 2s заполнен двумя электро-
электронами, тогда как остальные четыре могут быть распределены между 2р-орбитами.
Эти четыре электрона распределены таким образом: два находятся в одной р-ор-
бите и по одному в каждой из остальных р-орбит. Изложенное выше является
упрощенным описанием подробной электронной структуры нейтрального атома
кислорода в его основном или наинизшем энергетическом состоянии. Возмож-
Возможность вхождения электрона в каждую из двух не полностью занятых 2р-орбит,
например путем деления электрона или перекрывания орбиты другого атома,
создает возможность образования химической связи. Нейтральный атом водо-
водорода описывается просто как ядро, окруженное ls-орбитой, содержащей один
электрон.
Для частного случая соединения атомов кислорода друг с другом или
с атомами водорода можно получить достаточно удовлетворительное предста-
представление о структуре образовавшейся молекулы путем рассмотрения 2р-орбит
кислорода, ls-орбита кислорода может быть совершенно оставлена без внима-
внимания, а влияние 2s-op6nTbi на химическую связь за счет образования связей
первичной валентности в первом приближении ничтожно мало. В частности,
можно при качественном описании характеристики валентности кислорода
оставить без внимания явление гибридизации, при которой уровень энергии
25-электронов повышается так, что они могут участвовать вместе с 2р-электро-
нами в направленных орбитах. Для других элементов, например азота и угле-
углерода, гибридизацией уже нельзя пренебречь.
Рассмотрим сейчас структуру перекиси водорода в сравнении со структу-
структурой двух рядов кислородных соединений—основанных на атомарном и молеку-
молекулярном кислороде. Виды частиц, которые содержат эти соединения и еще один
ряд кислородных соединений, могут быть представлены следующим образом:
о4
о
он
н2о
о2
но2
на
о.
но3
НА
Мы здесь не рассматриваем ряд, основанный на высокомолекулярных кислород-
кислородных молекулах, например ряды с О3 или О4. Электронная структура озона
более сложна и еще окончательно не выяснена. Имеются сообщения о суще-
существовании простых озонидных соединений, аналогичных НО3 [36] и Н2О3 [37],
но они очень реакционноспособны и о них известно очень мало. Существование
частицы О4 эфемерно, и, во всяком случае [34, 38], атомы в ней не связаны
исключительно силами первичной валентности, как атомы в молекулах О2 и О3.
Необходимо рассмотреть различие между стабильной и реакционноспособ-
ной молекулами. К стабильным молекулам относятся такие, которые обра-
Структура молекулы перекиси водорода 269
зуются из атомов с уменьшением энергии. Во время поступательных движений
два атома, или атом и молекула могут приблизиться друг к другу и занять
относительные положения на расстоянии, сравнимом с расстоянием химической
-связи. Если условия неблагоприятны для связывания, то атомы продолжают
перемещаться по своим отдельным траекториям, сейчас же удаляясь после
этого максимального сближения, или «столкновения». В некоторых определен-
определенных условиях две сближающиеся частицы могут находиться одна около другой
на расстоянии химической связи в течение более длительного промежутка
времени, чем тот интервал, когда обе частицы только коснулись одна
другой и вновь разошлись. Такого рода соединение является устойчивым
даже в том случае, если энергия связывания настолько мала, что образовав-
образовавшаяся молекула может вновь легко расщепиться и поэтому обладает лишь
очень малым вероятным периодом жизни. Таким образом, молекула, которую
•считают стабильной, может оказаться реакционноспособной, если в сложив-
сложившихся условиях перегруппировка или реакция с другой молекулой оказы-
оказывается энергетически более выгодной. Все упомянутые выше молекулы, по
крайней мере теоретически, стабильны, но некоторые из них в обычных усло-
условиях наблюдения из-за своей реакционноспособности обладают лишь очень
непродолжительным периодом жизни. В качестве молекул, не являющихся ста-
стабильными в соответствии с вышеприведенным определением, можно назвать
частицы Н3О и Н3О2. Эти молекулы могут составить продолжение вышеуказан-
вышеуказанных двух рядов, выбранных нами для анализа, однако Н3О и Н3О2 не могут обра-
образоваться из кислорода в его основном состоянии; после образования Н2О и Н2О2
орбиты связи оказываются заполненными.
Рассмотрим теперь, каким образом эти два ряда кислородных соединений
образуются за счет участия электронов. Для этого будем исходить из кислород-
кислородного атома, представленного на рис. 52, а. Когда ls-орбита водородного атома
•с единственным электроном перекрывает одну из 2р-орбит некоторого кисло-
кислородного атома, содержащую только один электрон, например 2ру на рис. 52, а,
и спаривается с нею, образуется стабильная молекула гидроксила, или ОН.
В другой незавершенной 2р-орбите кислорода, например 2/?2, остается единич-
единичный или «неспаренный» электрон. Наличие неспаренного электрона должно
•сообщить гидроксилу парамагнитные свойства и открыть возможность для даль-
дальнейшего образования химической связи. Среднее распределение электронов
в связывающей молекулярной 2ру—ls-орбите (так называемой сигма-орбите)
может быть таким, что электронная плотность становится более высокой около
кислородного атома. В таком случае молекула должна обладать электрическим
моментом. При таком же присоединении другого атома водорода со спариванием
2/?г-орбиты образуется вода. Поскольку 2ру- и 2р2-орбиты, спаренные с ато-
атомами водорода, ориентированы под прямым углом, молекула воды должна
быть V-образной. Это доказывается экспериментально; фактический угол между
ОН-связями составляет 104°ЗГ [39]. Разность между 104 и 90° приписывается
электростатическому отталкиванию между атомами водорода и другим эффек-
эффектам, которые не приняты во внимание в рассматриваемой нами простой модели,
например влиянию гибридизации. Асимметрическая структура должна спо-
способствовать образованию у молекулы воды электрического момента; последний
оказался равным 1,85-10~18 CGSE.
Второй ряд соединений может быть построен путем связывания сначала
двух атомов кислорода с образованием молекулярного кислорода О2. Как пер-
первую ступень можно принять перекрывание двух орбит, содержащих единичные
электроны, с образованием связи вдоль осей ру каждого атома кислорода. Таким
образом, каждый из связанных атомов имеет две р-орбиты под прямым углом
к связывающей орбите, причем из этих двух орбит одна содержит пару элект-
электронов, а другая—только один. Энергетически более благоприятным является
положение, когда орбиты, содержащие по паре электронов, не параллельны друг
270 Глава 6
другу. Конечной ориентацией будет такая, в которой орбиты с парами электро-
электронов параллельны орбитам с единичными электронами. В этом случае возможно
дальнейшее связывание между кислородными атомами. Три электрона в каждой
из двух параллельных орбит могут быть поделены между кислородными ато-
атомами с образованием трехэлектронной связи по Паулингу [34]. Такую связь
нужно рассматривать просто как положение, в котором пара электронов и
неспаренный электрон непрерывно переходят от одного атома кислорода к дру-
другому. Каждая такая трехэлектронная связь обладает половинной прочностью
двухэлектронной а-связи, образовавшейся сначала. Остается возможным раз-
разрыв одной или обеих трехэлектронных связей кислородной молекулы, а поэтому
один или оба неспаренных электрона могут быть поделены с другими атомами.
Если один неспаренный электрон поделен с атомом водорода, то образуется
пергидроксил (или надперекись водорода) НО2. Эта стабильная молекула
содержит о-связь и трехэлектронную связь между кислородными атомами
и о-связь между атомами кислорода и водорода. Ось ОН должна быть ориен-
ориентирована под прямым углом к оси 00, что приводит к V-образной форме моле-
молекулы, так же как это наблюдается в случае воды. Молекула должна обладать
заметным электрическим моментом, и наличие остающегося неспаренного элект-
электрона должно способствовать парамагнитным свойствам пергидроксила. Моле-
Молекула пергидроксил а может теперь связать еще второй атом водорода, трехэлект-
трехэлектронная связь при этом разрывается и оставшийся неспаренный электрон ста-
становится поделенным с атомом водорода, что ведет к образованию Н2О2. По-
Поскольку в кислородной молекуле 02 орбиты с неспареиными электронами
ориентированы под прямым углом, можно ожидать, что, после того как элект-
электроны атомов водорода будут поделены с этими орбитами, плоскости, в которых
расположены оси ОН и которые проходят через ось 00 в перекиси водорода,
также будут ориентированы под прямым углом. Молекула может обладать
электрическим моментом, но, поскольку отсутствует неспаренный электрон, она
будет диамагнитной. Это описание и аргументация, наиболее подробно раз-
развитые Уолшем [40, 41], дают логическую основу для выбора структуры двух-
плоскостной цепи (рис. 51, модель IV), электронное изображение которой пока-
показано на рис. 52, б.
Выше указано, что в некоторых молекулах распределение электронов
может отличаться от распределения их в составляющих атомах, а именно
у какого-либо одного из атомов молекулы электронная плотность может быть
больше, чем у других. Тем не менее подразумевается, что распределение элект-
электронов остается в достаточной степени равномерным. Конечно, возможно и более
крайнее положение. Например, если ядро одного из атомов водорода молекулы
воды оттягивается таким образом, что.его электрон остается в орбитах кислоро-
кислорода, то образуется хорошо известный гидроксильный ион ОН". Аналогичным об-
образом из вышерассмотренных молекул могут быть образованы ионы О", О2", Ог,
HOg и О!;"; последние три частицы называются соответственно надперекисным,
пергидроксильным и перекисным ионами. В нейтральной молекуле перекиси
водорода пергидроксильный и перекисный ионы собственно не существуют.
Это означает, что атомы водорода в какой-либо значительной мере группой 02
за счет электростатических сил не притягиваются; электроны связи поделены
довольно равномерно. Иначе обстоит дело в перекисях, образованных элемен-
элементами, обладающими значительно меньшей электроотрицательностью, чем
водород, например барием. Можно сказать, что перекись бария представляет
электровалентное соединение, в котором силы связи между барием и кислоро-
кислородом являются преимущественно ионными.
Высказана мысль, что ионная связь может способствовать возникновению
тригональной структуры перекиси водорода. Так, Палмер (см. [33], стр. 82)
высказывает предположение, что в водном растворе ионизация перекиси водо-
водорода до пергидроксильного иона может способствовать возникновению тауто-
Структура молекулы перекиси водорода 271
мерного равновесия между тригональной и цепной структурами. По-видимому,,
он исходит из положения, что кислородный атом, с которым связан атом
водорода в ионе НОг, может координировать протон связью преимущественно
ионного характера. Гельман [42] приводит также в пример окись углерода*
где часть связи является ионной в результате перехода электрона из р-орбиты
кислорода в гибридную sp-орбиту углерода. Можно себе представить, что один
из несвязывающих электронов кислородного атома в молекуле воды может
перейти к свободному атому кислорода с образованием ионов Н2О* и О", что
создает положение, аналогичное наблюдаемому для случая с окисью углерода..
Это выражает образование ионной структуры, предложенной Палмером, но
иными словами; электростатические силы связывают эти ионы между собой,
и вместе с тем в связи с перегруппировкой электронов у каждого атома кисло-
кислорода остается неспаренный электрон, деление которого может привести к воз-
возникновению о-связи между кислородными атомами. Этим путем может воз-
возникнуть плоскостная тригональная структура (рис. 51) или, более вероятно,
как указывает Уолш [43], пирамидальная тригональная структура (рис. 51,
модель VI), что обусловлено ориентационной характеристикой кислородных
орбит. Такая структура в соответствии с вышеприведенным определением
является стабильной, но эта стабильность гораздо ниже устойчивости двух-
плоскостной цепи, а поэтому при обыкновенной температуре пирамидальная
структура, требующая для своего возникновения значительных ионных сил,
не может существовать в течение длительного периода. Не исключено, что при
очень низкой температуре такая форма способна к сравнительно длительному
существованию, но это требует еще экспериментального подтверждения.
Вышеприведенное качественное описание электронной структуры перекиси
водорода получило количественное оформление в работе Пенни и Сазерленда
[44] в 1934 г. Поскольку до проведения квантово-механических вычислений
нельзя было сделать совершенно однозначный выбор структуры, эти авторы
предпочли использовать для расчета метод валентной связи или электронной
пары. Этот метод менее точен, чем метод молекулярных орбит, но, поскольку
для решения проблемы при выборе между двумя общими формами достаточно
было знать относительные величины энергии для разных конфигураций, решено
было остановиться на менее трудоемком методе электронной пары. Рассматри-
Рассматривались только четыре 2р-электрона каждого из атомов кислорода, так как
остальные электроны в определении энергии связи играют ничтожную роль.
Пенни и Сазерленд показали, что плоскостная тригональная структура обла-
обладала бы только слабыми ионными, кулоновскими и вандерваальсовыми силами
притяжения, а поэтому существование ее является невероятным. Этот вывод
согласуется с заключением Казарновского [45], который рассчитал, что свя-
связывание атома кислорода с кислородным атомом молекулы воды за счет сил
поляризации может дать связь с энергией всего 0,58 ккал/моль, т. е. значи-
значительно меньшей, чем требуется.
После исключения тригональной формы (пирамидальная тригональная
форма отдельно не обсуждалась) Пенни и Сазерленд подробно исследовали,
какая из всех остальных возможных конфигураций, т. е. из моделей I—IV
на рис. 51, отличается наибольшей относительной устойчивостью. В самом
начале на основе качественных данных типа вышеизложенных, логический
выбор остановился на двухплоскостной структуре (модель IV). После этого
было изучено влияние изменения угла НОО б и двугранного, или азимуталь-
азимутального, угла 9 межДУ плоскостями, в которых расположены гидроксильные
группы, на относительную стабильность молекулы. Предварительно была
сделано заключение, что азимутальный угол 9 равен 90°, но при этом не учи-
учитывалось взаимодействие между атомами, не находящимися в непосредственной
связи, а также наличие вандерваальсовых и ионных сил. Для исследования
этих эффектов были сделаны следующие предположения: 1) влияние одной гид-
72
Глава 6
4J
роксильной группы на второй атом водорода такое же, как влияние, наблюдае-
наблюдаемое при взаимодействии обоих атомов водорода в молекуле воды; 2) угол НОО
0= Ю0°, т. е. такой же, как и угол НОН для воды; 3) длина связи О—О равна
1,4 А; 4) длина связи О—Н равна 1,0А. На основании этих предпосылок вычис-
вычислена энергия для значений 9 между 0 и 180°. Результаты этих вычислений пока-
показаны кривой на рис. 53, которая подтверждает, что выбранная величина <р=90°
почти правильна и требует только небольшого исправления, примерно до 100°.
Хотя априори можно предполагать, что взаимодействие водородных атомов или
гидроксильных связей должно благоприятствовать траяс-плоскостной цепной
структуре, эти вычисления доказы-
доказывают, что преобладающим фактором,
определяющим величину двугранного
угла <р> является отталкивание непо-
деленных пар электронов каждого
атома кислорода, достигающее на-
наибольшей величины в том случае,
когда орбиты с неподеленными парами
параллельны.
После выбора величины для дву-
двугранного угла Пенни и Сазерленд про-
провели расчеты для подтверждения,
что принятая величина для угла НОО
6 100° согласуется с величиной 100°,
определенной для двугранного угла.
Показано, что в случае идеального
спаривания электронов угол НОО
должен составить 90°. Однако оттал-
отталкивание со стороны атомов одной
гидроксильной группы заставляет
сместиться другой атом водорода с оси,
связывающей его в орбите ОН, на угол
ХР1. Связь .ОО не является бесконечно
прочной, поэтому происходит дополни-
дополнительное смещение связывающей орбиты
ОН на угол Ф2, чтобы таким образом
обеспечить максимальное перекрыва-
перекрывание орбит водорода и кислорода.
Поскольку необходимо, чтобы кислородные орбиты оставались перпендику-
перпендикулярными, связывающие орбиты ОО также должны сместиться на угол Ф2 от
линии центров ОО, которая раньше предполагалась совпадающей с осями свя-
связывающих орбит ОО. Как энергии связи ОО и ОН, так и пространственное
взаимное отталкивание атомов водорода и групп ОН и взаимодействие ОН-дипо-
лей зависят от углов Ч\ и W2. Пенни и Сазерленд вычислили, что минимальная
энергия будет наблюдаться при Ч\= ^2 = 5°. Отсюда 6 = 90°+Чг1+*Г2 = 100°.
Это краткое изложение показывает, на каком основании был сделан вывод,
что молекула с двухплоскостной цепью (модель IV) с углами в 6=9=100°
представляет собой истинную структуру перекиси водорода. Пенни и Сазер-
Сазерленд рассчитали, что такая модель должна обладать дипольным моментом
2-10~18CGSE, что хорошо согласуется с экспериментальными результатами.
Такое же совпадение было отмечено при вычислениях дипольного момента
Рэидолом [46] и Джектаром 147].
Энергетические^ барьеры, показанные на рис. 53, свидетельствуют о том,
что вращение одной гидроксильной группы относительно другой ограничено;
движение их в основном должно представлять собой крутильное колебание.
Такое ограничение должно способствовать возможности существования двух
9
Азимутальный угол ср, град.
Рис. 53. Потенциальная энергия как функ-
функция угла кручения для перекиси.
Пенни и Сазерленд; приближен-
приближенная синусоидальная функция.
Структура молекулы перекиси водорода 273
зеркальных или энантиоморфных форм перекиси водорода. Однако в паровой
фазе такое ограничение вращения сохраняется лишь в течение промежутков
времени, которые достаточно велики по сравнению с периодом колебания.
Барьер вращения не настолько высок, чтобы в результате надлежащих моле-
молекулярных столкновений молекула перекиси водорода не могла приобрести
достаточное количество энергии для его преодоления. Кроме того, можно рас-
рассчитать (см. [48], стр. 222), что группа ОН может перейти от одного минимума
потенциала к другому примерно 1 раз за каждые сорок (или даже меньше) коле-
колебаний за счет туннельного эффекта. Наличие этих изомеров не должно сооб-
сообщать оптической активности перекиси водорода, поскольку противолежащие
группы идентичны [49]. В конденсированных фазах, где наличие межмолекуляр-
ной ассоциации накладывает дополнительные ограничения на вращение, воз-
возможно обнаружить наличие изомеров в виде обеих стереохимически различных
форм [50].
Необходимо указать, что кривые, показанные на рис. 53, не являются
в действительности столь точными, как это может показаться. Кривая Пенни
и Сазерленда, вероятно, дает правильное качественное представление о форме,
т. е. позволяет обнаружить наличие двух минимумов, разделенных максиму-
максимумами неодинаковой высоты, причем разность высот является довольно заметной.
Однако представленные абсолютные значения могут потребовать перерасчета*.
Выражение потенциального барьера приближенным синусоидальным уравне-
уравнением
V=4-V0(l+cos29) A)
(на рис. 53для Vo показано значение 5 ккал/моль [51]; величина 3,5 ктл1моль
[52] в настоящее время кажется более правдоподобной) представляет чисто
математический прием. При использовании этого уравнения с правильным
подбором значения для потенциала Vo можно практически рассчитать термо-
термодинамические функции (как это описано на стр. 201).
Лассетр и Дин [53] исследовали перекись водорода в качестве примера при
разработке теории потенциальных барьеров, тормозящих вращение вокруг
однократных связей; при этом они нашли дополнительное подтверждение
структуры, принятой Пенни и Сазерлендом. В трактовке этих авторов учтены
все внутримолекулярные взаимодействия, рассмотренные Пенни и Сазер-
Сазерлендом. Лассетр и Дин выразили эти взаимодействия в форме электростатиче-
электростатических сил между различными распределениями электронов в молекуле; для
перекиси водорода эти силы следующие: 1) между двумя ОН-связями; 2) между
связями ОН и неподеленпой парой у противоположных атомов кислорода и
3) между неподеленными парами для каждого кислородного атома. Выска-
Высказано предположение, что неподеленные пары не обладают дипольным моментом,
так что роль их сводится лишь к влиянию их квадрупольных моментов**.
После расчета этих квадрупольных моментов Лассетр и Дин вычислили дву-
двугранный, или азимутальный, угол, соответствующий минимуму энергии для
молекулы. Эти расчеты проведены для довольно значительного интервала
квадрупольных моментов ввиду недостаточной точности их определения, причем
получены значения для ср от 94 до 113°, хотя значения для высоты барьера
* Пенни и Сазерленд [44] в двух сообщениях приводят разные величины для
кривой потенциала, т. е. для высшего максимума, а именно: 10 ккал/моль в одном слу-
случае и 1 эв B3 ккал/моль)— в другом; по-видимому, первая величина является более пра-
правильной.
** Потенциал на расстоянии от определенного местоположения заряда выражается
как сумма ряда, члены которого являются степенными функциями этого расстояния
[25]. Потенциал, обусловленный дипольным моментом, обратно пропорционален квадра-
квадрату расстояния, а вызванный квадрупольным моментом обратно пропорционален кубу
расстояния.
18 Перекись водорода
274
Глава 6
внутреннего вращения 12 ккал1моль, вычисленные на основе некоторых сделан-
сделанных предпосылок, по-видимому, завышены. Эта величина сильно отличается
от значения 0,2 ккал1мольу выведенного путем исследования микроволнового
спектра [20].
Мюлликен [54] приводит следующее сокращенное выражение для орбит
перекиси водорода с указанием приблизительного расположения орбит в отно-
отношении соответствующих атомов, а также потенциала ионизации электрона
в каждой орбите:
Обозначение электрона .
Местоположение
Потенциал ионизации, эв
BsJ
О
32
BsJ
О
32
[У1]2
00
18
[z1]2
ОН
17,5
{г1}2
ОН
17,5
Bа:J
0
13
Bх
0
13
Работа Робертсона [55] позволяет проверить совпадение этих потенциалов
ионизации с экспериментальными данными и совершенно убедительно опровер-
опровергает любую возможность существования тригональной формы перекиси водо-
водорода при обыкновенных температурах. Робертсон подвергал пар перекиси
водорода действию электронного пучка с известным регулируемым количест-
количеством энергии. При помощи масс-спектрографа определены потенциалы, при
которых возникали различные положительные ионы, и относительные интен-
интенсивности ионов при разных потенциалах. Результаты этих определений пока-
показаны в табл. 67.
Таблица 67
Относительные интенсивности и потенциалы возникновения
некоторых ионов, образующихся в парах перекиси водорода,
по Робертсону [55]
Ион
н2о;
но*
он+
о+
Относительная интенсивность а при
20 эв
100
3,5
200
2
го'эв
100
6
200
2
40 эв
to о о о
Потенциал
возникнове-
возникновения, эв
12,1±0,3
16,1±0,4
16,0±0,3
18,9±0,4
а В качестве произвольного стандарта принята интенсивность для H2O*t равная 100.
Робертсон показал, что обнаруженные ионы Н2О*, OJ и О+ обязаны происхо-
происхождением водяному пару, присутствующему в аппарате вследствие гетероген-
гетерогенного разложения части перекиси водорода. Особенное значение имеет отмечен-
отмеченный потенциал появления для О+18,9 эв.
Это тот же потенциал, что и наблюдаемый при появлении О+ в чистом
водяном паре; вместе с тем Робертсон делает вывод, что потенциал образования
(У из тригональной формы перекиси водорода не должен превышать 15,6 эв.
Робертсон считает, что эти измерения доказывают невозможность присутствия
тригональной формы в количестве, превышающем 1 : 1400. Найденный потен-
потенциал 12,1 эв для образования Н2С>2, очевидно, соответствует отрыву электрона
кислородного атома перекиси водорода; этот электрон не участвует в образо-
образовании связи. Эта величина меньше указанного выше и подсчитанного Мюлли-
кеном значения 13 эв и близка к наименьшему потенциалу ионизации молеку-
молекулярного кислорода A2,25 эв).
В последнее время проведены измерения полупериода существования
позитронов [56] и протонного резонанса [57] в перекиси водорода. Результаты
этих измерений, по-видимому, имеют значение для выяснения структуры моле-
молекулы, но никаких попыток их истолковать пока не предпринималось.
Структура молекулы перекиси водорода
275
Спектроскопическое исследование структуры
Спектроскопические и диффракционные исследования перекиси водорода
подтверждают сделанный выбор структуры и позволяют установить размеры
и углы в молекуле. Электронографические и рентгенографические измерения
дают такого рода значения непосредственно, а поэтому мы на них остановимся
в первую очередь.
Рис. 54. Типы вращательного и колебательного дви-
движений для молекулы перекиси водорода.
а—вращение; б—симметричное колебание растяжения ОН vu в—сим-
в—симметричное колебание изгибания V2; г—колебание растяжения OOvs;*J
д—крутильное колебание V4; e—асимметричное колебание растяжения
OHV5; otc—асимметричное колебание изгибания ve;
Жигер и Шомейкер [58] провели электронографическое исследование паров
перекиси водорода и установили, что длина связи О—О равна 1,47±0,02 А.
Положение атома водорода этим методом определить нельзя. Лю, Хьюз и Жи-
Жигер [50] провели также рентгенографическое изучение пероксигидрата моче-
мочевины и показали, что длина связи О—О в этом случае равна 1,46±0,03А;
для двугранного угла <р они нашли значение 106°, для угла ООН G—101,5°!
Хотя логично предположить, что углы в свободной перекиси водорода должны
18*
276 Глава 6
быть такие же, как в пероксигидрате мочевины, не исключено, что в твердом
производном мочевины эти углы несколько деформированы.
Абрагэмс, Коллин и Липскомб [59] определили длину связи О—О A,49А),
двугранный угол у (93°5Г) и угол ООН 0 (96°52'). В этом исследовании они
пользовались методом рентгенографии кристаллической безводной перекиси
водорода. Проведены измерения длины связи О—О и в других пероксосоеди-
нениях (см. гл. 12), как органических, так и неорганических, методами диффрак-
ции, причем полностью доказано, что эта величина очень мало отличается от
найденной для перекиси водорода.
Основным источником данных для определения размеров молекулы пере-
перекиси водорода остаются результаты исследования инфракрасного спектра
поглощения и спектра комбинационного рассеяния. В связи с эксперименталь-
экспериментальными трудностями, сложностью спектра и слабостью поглощения в некоторых
областях ряд вопросов остался нерешенным, тем не менее основная характе-
характеристика спектра может быть истолкована достаточно безупречно.
Наиболее предпочтительная структура для перекиси водорода (рис. 51,
модель IV) имеет 12 возможных видов движения в паровой фазе, а с некоторыми
ограничениями также и в конденсированных состояниях. Три таких движения
(простые поступательные в пространственных координатах) не имеют значения
для спектроскопии. Остальные движения, из которых три представляют собой
вращения молекулы как целого около осей и шесть—внутримолекулярные
колебания, показаны и соответственно обозначены на рис. 54. Эта модель имеет
одну ось симметрии (ось С на рис. 54, а), а именно линию, пересекающую линию
центров атомов кислорода и лежащую в плоскости, которая делит пополам дву-
двугранный угол 9- Поворот молекулы на 180° около этой оси дает конфигурацию,
не отличающуюся от исходной. Ни при какой другой операции вращения или
отражения исходная конфигурация не воспроизводится. Такая конфигурация
относится по классификации к точечной группе С2, причем считается, что она
обладает двойной осью симметрии.
После выяснения симметрии молекулы можно сделать ряд выводов относи-
относительно влияния на нее излучения или, что еще более важно, определить струк-
структуру ее из спектра. Сложные подробности этой теории можно найти у Герц^
берга [48]. Здесь же мы ограничимся лишь некоторыми элементарными пред-
предсказаниями, касающимися спектра перекиси водорода: 1) поскольку перекись
водорода имеет постоянный дипольный момент, она должна обладать спект-
спектром поглощения, обусловленным вращательным движением; 2) поскольку
каждое колебательное движение (рис. 54) связывается с изменением дипольного
момента, каждому такому движению соответствует полоса инфракрасного
поглощения; 3) точно так же, поскольку каждое колебательное движение вызы-
вызывает изменение молекулярной рефракции, оно дает полосу и в спектре комби-
комбинационного рассеяния. Помимо фундаментальных частот, отмеченных для коле-
колебательных движений, возможны различные обертоны и составные частоты.
Обертоны обусловлены ростом энергии колебания более чем на один квантовый
уровень за один раз, а составные частоты вызваны одновременным повышением
энергии двух колебаний.
Использование этих положений о спектре, или правил отбора, дает воз-
возможность сделать предварительный выбор конфигурации перекиси водорода
из различных моделей, представленных на рис. 51. Так, модель I с линейной
цепью и модель II с mpawc-плоскостной цепью уже раньше были исключены,
поскольку такие модели не должны обладать дипольным моментом. Правила
отбора говорят, что цепная чис-плоскостная модель III должна отличаться
от модели IV тем, что она может обладать лишь пятью возможными инфракрас-
инфракрасными фундаментальными колебаниями, тогда как для модели IV их шесть.
Пять инфракрасных фундаментальных колебаний было обнаружено с досто-
достоверностью, шестое полностью не доказано. Таким образом, это как будто говорит
Структура молекулы перекиси водорода 277
в пользу модели IV, но окончательное решение требует более детального иссле-
исследования.
Вращательный спектр перекиси водорода до настоящего времени изучен
сравнительно слабо. Исследован спектр, обусловленный только чистым вра-
вращением в микроволновой области, и проведены два исследования ротационной
тонкой структуры для двух колебательных обертонов или составных полос.
В настоящее время все фундаментальные частоты колебаний наблюдаются
с ограничениями из-за следующих осложнений: 1) инфракрасное поглощение
обычно бывает слабым, что снижает точность определения местоположения
центров полос; 2) две фундаментальные частоты vt и v3 и спектр комбинацион-
комбинационного рассеяния изучены лишь для конденсированных фаз, так что необходимо
учитывать известное влияние молекулярной ассоциации на наблюдаемые
частоты; 3) два фундаментальных колебания, а именно vt и v5, имеют почти
одну и ту же частоту*, в конденсированных фазах они разрешены, но пол-
полностью их характеристика еще не установлена. С этими оговорками можно
идентифицировать частоты, т. е. приписать определенное происхождение всем
частотам, и выводы использовать для расчета длин связей, углов, моментов
инерции и силовых постоянных перекиси водорода.
Идентификация отмеченных полос поглощения с колебательными движе-
движениями или определение характера частот у перекиси водорода проводилось
Бейли и Гордоном [60], Симоном и Фехером [61] и Жигером [51, 62]. Иденти-
Идентификация, осуществленная первоначально Бейли и Гордоном, а также Симоном
и Фехером, страдала из-за недостаточного исследования спектра (были най-
найдены с достоверностью лишь три комбинационные линии), а также, по-видимому,
и от недостаточной точности в определении интенсивности поглощения и место-
местоположения центров полос в инфракрасном спектре. Так, Бейли и Гордон не
обнаружили заметных различий между инфракрасными спектрами паров
и жидкости, тогда как опытом доказано, что для гидроксильного соединения,
склонного к ассоциации в жидкой фазе, должны наблюдаться существенные
различия.
В табл. 68 дается сводка колебательных спектров для перекиси водорода
в соответствии с идентификацией частот, данной Жигером [62]. Порядок пере-
перечисления частот vlf v2 и т. д. находится в согласии с частотой и симметрией,
соответствующей колебанию (см. [48], стр. 271). Если не учитывать некоторых
различий в местоположении, то идентификация, приведенная в табл. 68, отли-
отличается от идентификации, данной Бейли и Гордоном, Симоном и Фехером,
главным образом интерпретацией комбинационной полосы 1400 см'1. Симон
и Фехер обнаружили, что она состоит из двух максимумов, разделенных рас-
расстоянием около 27 см'1. Они приписали это случайному вырождению, а Бейли
и Гордон идентифицировали эти максимумы как v2 и v4. Величина 1400 см'1
для крутильного колебания была признана Симоном и Фехером недопустимо
высокой, и поэтому они приписали эти два максимума 1400 см'1 колебаниям
v2 и v6. Результаты более поздних работ не подтвердили расщепления полосы
1400 см'1, обнаруженного Симоном и Фехером, а поэтому эта полоса сейчас
приписывается единичной частоте v.2.
Ниже кратко излагается материал, на основе которого произведена иден-
идентификация частот в табл. 68 (подобно тому, как это делал Жигер [62]).
Частота v3, колебание растяжения ОО. Поглощение при 877 см'1
остается одним и тем же в перекиси водорода и перекиси дейтерия, а поэтому
становится совершенно ясным, что частота v3 обусловлена колебанием растя-
растяжения ОО, не зависящим от массы атомов водорода. Правильность такого
* Это явление иногда называют «случайным вырождением», хотя, строго говоря,
вырождение требует наличия одинаковой симметрии для обеих полос. Здесь правильнее
говорить о перекрывании или совпадении.
Таблица 68
Идентифи-
Идентификация
частот
v8
2v4
ve
v2
v2-bve
v&
3v5 или
2v1+v6
nap
465
575
877 @4.
—
1266 @4.
1280
2630 (ел.)
3590 (с.)
3590 (с.)
—
7036,6@4.
7041,8@4
10283,7@4
10291,1@4
Сводка колебательных спектров перекисей водорода и
Перекись водорода
инфракрасный спектр, см-1
СЛ.)
С.)
сл.)
сл.)
сл.)
сл.)
жидкость
635
878
1353
2796
3360
3360
4715
9900
-30°)
(ср.)
@4. СЛ.)
—
(ср. с.)
—
(ср. сл.)
@4. С.)
@4. С.)
@4. СЛ.)
—
@4. СЛ.H
твердое
тело (—78°)
660 (с.)
878 @4. сл.)
1300
1378 (с.)
1430 (сл.)
2733 (ср.)
3218 @4. с.)
3218 @4. с.)
4595 @4. сл.)
—
—
комбинационные полосы
жидкость
(-40°)
525
882
—
—
1402
2815
3364
3364
—
—
—
•
твердое
тело (—20°)
878
—
—
1408
2790
3203
3331
—
—
—
947
1944
2680
2680
5236
дейтерия
а
Перекись дейтерия
инфракрасный спектр, сл<-1
пар
—
—
@4. С.)
—
(СЛ.)
(с.)
(с.)
—
@4. СЛ.)
—
жидкость
(-30°)
538 (ср.)
878@4. сл.)
—
1004 (с.)
—
2087 (ср. сл.)
—
2482 @4. с.)
—
—
—
твердое
тело (—70°)
480 (ср.)
—
—
—
—
—
—
—
—
—
—
комбинаци-
комбинационные
полосы
жидкость
(комнатная
температура)
877
—
—
1009
1990
2510
—
—
—
—
Сокращения: с.—-сильная; оч. с—очень сильная; сл.—слабая; оч. сл.—очень слабая; ср.—средняя.
При комнатной температуре
Структура молекулы перекиси водорода 279
толкования подтверждается также небольшим влиянием изменения агрегатного
состояния на эту частоту и слабостью инфракрасного спектра по сравнению
с комбинационным, как и следовало ожидать для слабого изменения диполь-
ного момента, ассоциируемого с этой вибрацией. Жигер [63] указывает, что для
паров перекиси водорода эту полосу никогда еще не наблюдали с полной оче-
очевидностью. Низкая степень деполяризации, обнаруженная для комбинацион-
комбинационной линии с этой частотой, также показывает, что она обусловлена симметрич-
симметричной вибрацией, т. е. такой, при которой симметрия молекулы сохраняется.
Эта частота обнаружена и в других перекисях [641. В соответствии с правилом
Бэджера [65], выражающим эмпирическую зависимость между длиной связи
и силовой постоянной, ассоциированной с данной очастотой, из этой частоты
можно вывести, что длина связи О—О равна 1,48 А, что хорошо согласуется
с величиной, выведенной электронографическим методом.
Частоты vx и v6, симметричное и асимметричное колебания растяже-
растяжения ОН. Эти колебания идентифицируются с поглощением вблизи 3600 см'1
в парах, 3400 см'1 в жидкости и 3200 см'1 в твердом теле. Аналогичная частота,
обусловленная растяжением ОН, наблюдается и для воды. Частота этой полосы
изменяется примерно на 1/1/2 при переходе от перекиси водорода к перекиси
дейтерия, а это подтверждает, что она обусловлена движением атома водорода.
Для молекулы воды в паровой фазе частоты симметричной и антисимметричной
вибраций ОН разделены примерно на 100 волновых чисел, что обусловлено
жестким сочетанием гидроксильных групп. Близкое совпадение, или вырожде-
вырождение, этих колебаний в перекиси водорода приписывается слабому сцеплению
между гидроксильными группами. Такое предположение, как обнаружил Тей-
Тейлор, подтверждается тем, что комбинационная полоса 3200 см'1 в твердой фазе
может быть расщеплена на два максимума, разделенных примерно на 130 смГ1,
и что приблизительно такое же расстояние наблюдается и в растворе. Правда,
вполне возможно, что при переходе из парообразного состояния в конденси-
конденсированную фазу единичная частота ОН расщепляется на ряд составляющих ком-
компонент в связи с эффектами молекулярной ассоциации, но в этих условиях
вряд ли возможно такое большое расстояние между составляющими линиями.
Поскольку в паровой фазе такого разделения не наблюдается, трудно точно
определить правильную величину для невозмущенной молекулы. Оно не
должно быть столь велико, как в конденсированных фазах, и невозможность
обнаружения его в парах показывает, что расщепление лежит в пределах обыч-
обычной точности для инфракрасной спектроскопии. Колебания vx и v6 правильнее
всего отождествить с колебанием при 3610 см'1 [66]. Приложение правила
Бэджера дает величину 0,98 А для длины связи О—Н.
Частота ve, асимметричное колебание изгибания. Инфракрасная полоса
поглощения, наблюдаемая при 1266 еж в паровой фазе, 1353 см'1—в жидкой
и 1378 см *—в твердой, приписывается асимметричному изгибанию ОН-групп.
Частота ve изменяется при переходе от перекиси водорода к перекиси дейтерия,
что подтверждает связь этого движения с атомом водорода. Эта частота в пере-
перекиси водорода отличается от частоты 1400 см'1 (наблюдаемой лишь в конденси-
конденсированных фазах) большей интенсивностью (как и следовало ожидать для асим-
асимметричного колебания в инфракрасной области), а также наличием Q-ветви,
как это и должно быть для преимущественно параллельного колебания диполя.
Полоса при 1400 см'1 также слаба в инфракрасном спектре, но интенсивна
в спектре комбинационного рассеяния.
Частота v2, симметричное колебание изгибания. У одного из двух остаю-
остающихся фундаментальных колебаний (которое наблюдается при 1400 см'1
в спектре комбинационного рассеяния и для твердого тела при —78° в инфра-
инфракрасном спектре) соответствующая частота изотопически изменяется, что поз-
позволило идентифицировать его с симметричным колебанием изгибания. По-
Поскольку эта полоса, которая должна обладать низкой интенсивностью в инфра-
280 Глава 6
красном спектре, сначала не была обнаружена в спектре паров, пришлось
сделать несколько произвольную поправку на влияние ассоциации. Эта по-
поправка была принята за 100 см, и Жигер и Бен [66] дают для v2 значение
1315 см'1. Позже Бен [67] идентифицировал симметричное колебание изгиба-
изгибания, наложенное на полосу v6 в спектре для паров, и принял величину 1280 см
в качестве центра полосы для v2.
Частота v4, крутильное колебание. Экспериментальное доказательство
существования полосы поглощения, обусловленной крутильным колебанием
гидроксильных групп, получено позже всего, чем и объясняется недостаточная
надежность начальных попыток идентификации частот. Полоса, найденная
вблизи 600 см'1 Тейлором [68] и Жигером [62], обладает надлежащим место-
местоположением для энергии, с которой должно быть связано это движение. Так же
как и при других колебаниях ОН, точно не известна поправка, которую следует
ввести на ассоциацию в конденсированных фазах. Жигер [62] считал, что
в паровой фазе v4 занимает положение около 500 см'1; позднее Жигер и Бен
[66] изменили это положение до 660 см'1. Бен [67] недавно обнаружил широкую
интенсивную полосу в парах, занимающую положение от 660 см'1 до предела
оптики бромистого калия, около 440 смг1. По-видимому, эта полоса состоит
из перекрывающих друг друга полос с центрами около 465 и 575 см'1. Отмечен-
Отмеченная разность значений для этой частоты в инфракрасном спектре и спектре
комбинационного рассеяния жидкости (см. табл. 68) остается пока не выяснен-
выясненной. Дальнейшие трудности внесли результаты исследования микроволно-
микроволнового спектра поглощения перекиси водорода. Масси и Бьянко [20] сделали
вывод, что наиболее вероятная величина для высоты потенциального барьера
составляет 113 см'1. По уравнению A) для синусоидального потенциального
барьера эта величина согласуется с константами вращения, выведенными из
результатов измерений этих авторов. Раньше чем можно будет с точностью уста-
установить энергию этого колебания, потребуется дальнейшая экспериментальная
работа в сочетании с теоретическими исследованиями [52].
Обертоны и составные полосы. Несколько полос в инфракрасном спектре
и одна полоса в спектре комбинационного рассеяния идентифицированы как
обертоны или составные частоты. Эта идентификация кажется довольно бес-
бесспорной, хотя не исключена возможность и другой идентификации. Тейлор [681
предполагает, что крыло при 1300 см'1 на стороне полосы 1400 см'1 с низкой
частотой в инфракрасном спектре кристаллической перекиси водорода предста-
представляет собой первый обертон 2v4 для частоты крутильного колебания. Полоса,
наблюдаемая приблизительно при 2800 см'1 в конденсированных фазах и около
2600 см'1—в паровой, принималась сначала за тройную сумму v2-H3+v4 [62].
Однако величина изменения при переходе к перекиси дейтерия доказывает, что
эта поолса обусловлена движением водородного атома, причем в настоящее
время считается, что ее лучше всего представить суммой v2+ve [66], хотя
имеются предположения, что она в действительности является неразрешенным
обертоном только v2 или только v6 [68]. Обертоны 2v2 и 2v6, особенно послед-
последний, должны быть очень слабыми, и на этом основании можно принять, что
полоса 2600 см'1 не может быть приписана главным образом этим обертонам.
Инфракрасная полоса при 4715 см'1 в жидкости, по-видимому, совершенно бес-
бесспорно может быть приписана v5+>e. На основании изучения тонкой струк-
структуры полосы при 7040 см'1 она идентифицирована как составная vx+v5 [62].
Для полосы 10 290 см'1 можно принять две различные идентификации: либо 3v5,
либо 2v1+v5 [62]. Наблюдение, показывающее, что это полоса гибридного
типа, исключает возможность ^ис-плоскостной структуры перекиси водорода
[69]. Безусловно, существуют еще и другие обертоны и составные частоты,
возможно даже в тех областях спектра, которые уже были подвергнуты иссле-
исследованию. Слабость поглощения перекиси водорода затрудняет открытие и раз-
разрешение такого рода полос.
Структура молекулы перекиси водорода 281
Моменты инерции
Моменты инерции перекиси водорода по трем ее осям (см. рис. 54, а)
определены вычислениями [62, 66], основанными на размерах и углах, измерен-
измеренных для молекулы, изучением тонкой структуры спектра, обусловленного
молекулярным вращением [62, 69], а также исследованием чисто вращатель-
вращательного спектра [20]. Зумволт и Жигер [69] определили константы вращения пере-
перекиси водорода из вращательной тонкой структуры для инфракрасной полосы
10 290 см 1. Сделано предположение, что вращательная энергия перекиси
водорода может быть представлена энергией для симметричного волчка. Это
предположение приемлемо ввиду сравнительно большой массы атомов кисло-
кислорода. Таким образом, момент около оси А на рис. 54, а является единичным,
а моменты, перпендикулярные ему, т. е. 1в и /#, почти одинаковы. Зумволт
и Жигер сделали вывод, что форма молекулы с азимутальным углом 9 90° обла-
обладает ничтожной асимметрией и что некоторые особенности спектра, обусловлен-
обусловленные асимметрией, нельзя было бы объяснить, считая, что <р лежит между 85
и 95°. Зумволт и Жигер для перекиси водорода в основном состоянии вывели
значение меньшего момента инерции /л B,78в- 10~ло г-см?) и значение гармониче-
гармонической средней из двух больших моментов инерции у-~Г- 33,9- КГ40 г-см2.
Найденные Жигером [62] константы вращения на основании изучения
тонкой структуры полосы 7040 см'1 совпали с ранее проведенными исследова-
исследованиями в условиях высокой дисперсии. Жигер [62] вычислил моменты инерции,
соответствующие значениям азимутального угла между 0 и 180°, причем поль-
пользовался величинами 1,48 А (расстояние О—О), 0,98 А (расстояние О—Н) и 102°
(угол ООН) (уравнения, связывающие моменты инерции с атомными массами
и размером молекул, установлены Вилсоном и Бэджером [70]). При варьиро-
варьировании азимутального угла от 0 до 180° получены следующие крайние значения
моментов инерции: 1А 2,89—2,76; 1В 32,0—35,1 и 1С 35,0—32,4-100 г-см2.
Эти моменты совместимы с величинами постоянных вращения, определенными
на основании исследования полос 10 290 и 7040 см'1. Можно видеть, что гармо-
гармоническая средняя больших моментов почти не зависит от величины, принятой
для азимутального угла. Меньший момент инерции при переходе от цис-кон-
фигурации к /прайс-конфигурации изменяется примерно на 5%. Поскольку
этот момент в большей степени зависит от недостаточно точно определенного
расстояния О—Н, чем от азимутального угла, определение точной величины
последнего не имеет существенного значения. Жигер рассчитал, что наилучшее
согласование меньшего момента инерции с найденной постоянной вращения
окажется в том случае, если расстояние О—Н в перекиси водорода будет чуть
больше, чем в воде (где оно равно 0,957А).
Жигер и Бен [66] произвели спектроскопическое исследование перекиси
дейтерия для решения проблемы о выборе молекулярных размеров, совмести-
совместимых с моментами инерции. Они использовали правило произведения Теллера—
Редлиха [48], представляющее собой безразмерное отношение между частным
от деления фундаментальных частот двух изотопических молекул и произведе-
произведением отношений их масс и моментов инерции и коэффициента симметрии. На
основании этого они определили, что отношение /с//с~ 1,12 и отношение
/2/я//и/в=2,113±0,02, где индекс D обозначает момент инерции перекиси
дейтерия. Первое из этих отношений IqIIc не может считаться точным,
поскольку оно вычислено из отношения частот для симметричных колебаний,
часть которых наблюдалась лишь для жидкой фазы и требует введения поправки
(величина последней еще не вполне известна). Значение дроби IaIb/IaIb
известно более точно, поскольку оно вычислено из частот, обнаруженных для
паровой фазы, и содержит лишь небольшую поправку на использование фак-
282 Глава 6
тических величин вместо величин нулевого порядка. Определен интервал
значений азимутального угла 9, согласующегося со значением IaIb/IaIb для
различных размеров молекулы. Длину связи О—О принимали равной 1,49А,
длину связи О—Н варьировали от 0,957 до 1,01 А, а угол ООН—от 96 до 15,5°.
Результаты этих расчетов приведены [66] в форме кривых, показывающих гео-
геометрические места согласующихся между собой значений разных параметров,
причем сделан вывод, что азимутальный угол должен лежать внутри
интервала 82±20°. Жигер и Бен [63] считают, что расстояние О—Нв перекиси
водорода имеет значение 0,?65±0,01А.
Масси и Бьянко [20] вычислили величину меньшего момента инерции 1а
B,959 • 10~*°г-см2) на основании исследования микроволнового спектра поглоще-
поглощения. При расстоянии О—О 1,49 А и О—Н 0,98 А этот момент инерции лучше
всего согласуется с углом ООН 6, близким к 90°.
Силовые постоянные
Идентификация колебательных частот позволяет вычислить силовые
постоянные, т. е. силы восстановления положений двух атомов после единич-
единичного смещения их вдоль оси связи в результате колебания. Бейли и Гордон
[60] вычислили эти постоянные для перекиси водорода, но, поскольку их иден-
идентификации частот были опровергнуты, мы не приводим вычисленных ими зна-
значений. Жигер и Бен [66] определили силовые постоянные для перекисей водо-
водорода и дейтерия после исследования размеров молекул и идентификации частот.
Результаты этих вычислений, выполненных с применением подходящей квад-
ратической потенциальной функции, приведены в табл. 69. Эти силовые постоян-
постоянные не вполне точны, так как положенные в их основу величины фундаменталь-
фундаментальных частот и размеров молекулы могут потребовать исправления.
Конечный выбор структуры
Приведенные в табл. 69 размеры молекулы можно считать наиболее надеж-
надежными для структуры перекиси водорода или перекиси дейтерия в паровой
фазе. Нужно иметь в виду, что эти параметры взаимно связаны в различной
степени.
Значение для длины связи ОО, безусловно, установлено вполне точно.
Пределы, предложенные Жигером и Беном [66] (от 0,957 до 1,01 А), весьма
вероятно, полностью охватывают возможный интервал значений для расстоя-
расстояний ОН в перекиси водорода. Выбранное нами значение 0,97 А несколько выше,
чем величина для воды [39] @,957А) и метанола [71] @,937А), и лишь немногим
меньше, чем величина для гидроксильного радикала [48] @,971 А).
Выбор значения 100° для угла ООН 0 является своего рода компромиссом.
Вряд ли этот угол такой же, как угол НОН для воды, тем не менее он, безусловно,
несколько отклоняется от невозмущенного орбитального угла 90°. По измеренным
постоянным вращения низший предел для него 96°, тогда как по данным микро-
микроволнового поглощения получается величина 90°. Значение 96° близко к значе-
значению, найденному путем рентгенографического исследования кристалла, при
изучении же пероксигидрата мочевины получается величина, близкая к 102°.
Обе эти величины, безусловно, относятся к молекуле в конденсированной фазе,
когда на величину угла оказывает определенное влияние межмолекулярное
притяжение.
Выбор равновесного значения для азимутального угла <р также нельзя
считать полностью доказанным. Значение 82°, основанное на исследовании
моментов инерции, обладает точностью в пределах ±20°. Трудно сделать
дыбор между этим острым углом для азимута молекулы в паровой фазе,
Структура молекулы перекиси водорода
283
Таблица 69
Вероятные значения молекулярных параметров для перекиси водорода
(литературу см. в тексте)
Параметры
Фундаментальные частоты, см
v
V4
V5
ve
Силовые постоянные, дин!смЛ0~ъ
ko—н
?'o—н
?OOH
0 ООН
Тно—он
Длины связей, А
ОН
00
Углы связей, градусы
ср
0
Моменты инерции, г -см2 -10*°
/а
/в
/с
Н2О2
3610
1315
877
660
3610
1266
7,28
<0,01
3,84
0,89
-0,04
0,11
0,30
0,97±0,005
1,49±0,01
95±10
100
2,83
33,6
33,7
D2O2
2670
1000
877
480
2680
947
7,53
0,03
4,04
0,95
-0,09
0,11
0,508
0,97±0,005
1,49±0,01
95±Ю
100
5,14
38,3
37,9
а Константа, добавляемая к потенциальной функции [66].
установленным на основании исследования спектра вращения, и тупым углом
со значениями между 90 и 113°, выводимым из квантово-механических расчетов
и измеренным путем рентгеновской диффракции перекиси водорода в конден-
конденсированных фазах. Имеются как будто доказательства в пользу угла, немного
большего 90°, но все методы определения не чувствительны к небольшим коле-
баниям.
Энергии связи в перекиси водорода
Энергия, ассоциирующаяся со связью между двумя атомами в молекуле,
представляет собой величину, которая может быть использована для сравне-
сравнения и вычисления структуры. Представление о том, что известная часть энер-
энергии молекулы локализуется в одной связи, носит, безусловно, несколько фор-
формальный характер, но эта концепция практически удобна и в известных пре-
пределах точности оправдывается. В соответствии со сказанным необходимо дать
точное определение энергии связи. По этому вопросу можно рекомендовать
обзоры, опубликованные Уолшем [40,72] и Шварцем [73], а также обзоры Глок-
лера [74] и замечания Богана [75]. Энергия связи представляет собой ту часть
общей энергии молекулы, которая относится к связыванию двух ее атомов
точно в тех электронных состояниях, в которых они существуют в молекуле.
Сумма всех энергий связи в молекуле равна теплоте превращения молекулы
в атомы, сохраняющие те же валентные состояния, которыми они обладали
в молекуле. Энергия связи не эквивалентна энергии диссоциации молекулы
на две части, разделенные между собой по месту данной связи, хотя, если
пренебречь сравнительно небольшими количествами энергии вращения, коле*
284 Глава 6
бания и колебания в нулевой точке, то энергия диссоциации двух-
двухатомной молекулы может быть приравнена энергии связи. Для мно-
многоатомной молекулы возможны сопровождающиеся выделением энергии
перегруппировки частей, образовавшихся при диссоциации по месту ка-
какой-либо связи, и поэтому энергия диссоциации, вообще говоря, не эквива-
эквивалентна энергии связи. В то же время трудно или даже невозможно провести
измерения, которые убедительно доказали бы локализацию известной части
энергии молекулы в одной связи. Это заставляет прибегать к тем или иным прие-
приемам, и часто для определения энергии связи используются значения различных
теплот диссоциации. Так, в молекуле XYn энергия связи XY вполне логично
принимается равной \1п от общей теплоты превращения XYn в атомы. Для
асимметричных молекул или в тех случаях, когда возникает вопрос об экви-
эквивалентности данной связи в различных окружениях для двух несравнимых
молекул, эта методика оказывается более затрудненной. Ниже мы остановимся
на проблемах, возникающих при определении энергии связи в перекиси
водорода.
Для вывода энергии связей ОН и 00 в перекиси водорода необходимо рас-
рассмотреть энергии ряда реакций. Нижеследующие значения, выраженные
в килокалориях на моль, справедливы для реакций при 25°, и вычисленные
из них энергии связи несколько завышены, поскольку в реакции учитываются
и изменения давления—объема. Однако для той точности, с которой могут быть
вычислены энергии связи, этим можно пренебречь. Все указанные ниже значе-
значения являются вполне доказанными [76], за исключением двух предпоследних;
основа для выбора этих двух недостаточно надежных значений рассмотрена
на стр. 218 при анализе термодинамических свойств. Причина включения пос-
последней реакции, касающейся фтора, станет ясной из дальнейшего изложения.
B)
C)
D)
E)
F)
G)
(8>
(9)
A0)
A1)
Предполагается, что обе связи ОН в перекиси водорода эквивалентны,
поэтому, если известна энергия связи ОН, можно вывести энергию 00 при
помощи уравнения F), и наоборот. Если известна величина одной из этих энер-
энергий связи в другом соединении, можно в качестве первого приближения при-
принять ту же величину для энергии связи в перекиси водорода. Именно этим
путем Паулинг [34] первый определил энергии связи для перекиси водорода.
Значение 118 ккал/моль из уравнения B) не может быть принято за энергию
связи 00 в перекиси водорода, так как указанное приближение в этом случае
не оправдывается; в молекуле 02 существуют две трехэлектронные связи
наряду с простой единичной связью, имеющейся между кислородными атомами
в перекиси водорода. Наоборот, связи ОН в воде должны быть весьма близкими
к этим связям в перекиси водорода. Если принять половину величины энергии
для уравнения C), а именно 110,5 ккал/моль, за величину для энергии связи
ОН в перекиси водорода, то, применив уравнение F), можно вычислить энергию
о..
н2о
и2о
он
н8о2
НА
НА
на
но3
—>20
-¦2Н + О
—>Н + ОН
—>0 + Н
-^2Н + 2О
— 20Н
--2Н + 0,
->н + н6,
->Н-|-02
-*2F
118,3
221,1
119,9
101,2
254,9
52,6
136,6
¦ 90
46
36,6
Структура молекулы перекиси водорода ' 285
связи 00 в перекиси, которая должна быть равной 33,8 ккал/моль. Пользуясь
несколько иными, более старыми термохимическими данными, Паулинг этим
путем получил значение 34,9 ккал/моль.
Метод выбора значения энергии связи ОН для вышеприведенного рас-
расчета иллюстрирует различие между энергией связи и энергией диссоциации.
Так, в то время как энергия связи ОН по уравнению C) принята равной
110 ккалу энергия диссоциации для отрыва первого атома водорода от воды по
уравнению D) равна 120 ккал, а для отрыва второго атома по реакции E)—
101 ккал. Хотя сумма энергий этих реакций равна сумме энергий связи ОН,
первый атом водорода оторвать труднее, чем второй. Несколько иное положе-
положение наблюдается для перекиси водорода. Термохимические данные для дис-
диссоциации перекиси водорода с отрывом первого и второго атомов водорода
[реакции (9) и A0I точно неизвестны, но, по-видимому, энергия, необходимая
для отрыва первого атома водорода от перекиси водорода, в 2 раза (или не-
немного больше или меньше) превосходит ту энергию, которая требуется для
отрыва второго атома. Однако в обоих случаях выбранная выше энергия
связи ОН больше, чем энергия диссоциации. Очевидно, что при перегруппи-
перегруппировке частиц, получающихся после диссоциации (т. е. НО2 в одном случае
и О2 в другом), выделяется некоторая энергия, из-за которой энергия диссо-
диссоциации оказывается ниже, чем энергия связи. Вероятно, это обусловлено
образованием трехэлектронных связей в НО2 и О2.
Возможно, конечно, что связи ОН в перекиси водорода существенно
отличаются от этих связей в воде, тогда сделанное предположение теряет силу.
Такого рода возражения высказаны Скиннером 1771 и Уолшем [40]. Скиннер
указывает, что перекись водорода аналогично гидроксильному радикалу
не обладает определенной энергией резонанса ионных форм, характерных
для воды. Он поэтому считает, что энергия связи ОН в гидроксиле [реакция
E)] больше подходит для перекиси водорода. На основании принимавшейся
в то время величины энергии диссоциации гидроксила 102 ккал/моль Скиннер
вычислил, что энергия связи ОО равна примерно 52 ккал/моль. Глоклер и Мэт-
лак [78] высказались против основы этого заключения и показали, что пере-
перекись водорода обладает почти той же ионной энергией резонанса, что и вода,
и что в гидроксиле эта энергия отсутствует. В качестве дополнительного дока-
доказательства правильности рассчитанной величины 34 ккал для энергии связи
ОО Глоклэр и Мэтлак сообщают, что эта величина попадает на кривую, выра-
выражающую энергии диссоциации озона и молекулярного кислорода О2 в раз-
различных электронных состояниях в виде функции межатомных расстояний.
Однако последнее соотношение между энергией связи и энергией диссоциации
не поддается четкому и бесспорному истолкованию.
Проблема поисков основы для выбора энергии связи ОН проанализиро-
проанализирована с различных точек зрения Уолшем [40]. Этот автор приводит ряд сообра-
соображений, подтверждающих, что связь ОН в перекиси водорода более полярна,
чем в гидроксиле, и поэтому считает, что она должна быть слабее. Подтверж-
Подтверждение заключается, по его мнению, в том, что длина связи ОН в перекиси
водорода, определенная Жигером, согласно предварительному сообщению [79],
равна 1,01 Л, тогда как в гидроксиле она имеет величину 0,97 Л; в пользу этого
говорит и величина силовой постоянной для растяжения ОН в перекиси водо-
водорода по Бейли и Гордону [601. На этом основании Уолш считает, что энергия
связи ОН в перекиси водорода меньше 100 ккал и дает для энергии связии ОО
низший предел 56 ккал. Им выбраны значения: 96 ккал для связи ОН и 65 ккал
для связи ОО. Это более высокое значение энергии связи ОО является, по
мнению Уолша, более удовлетворительным по ряду причин: при этом полу-
получается более высокое значение энергии связи ОО, чем в случае SS E1 ккал/моль)
или SeSe D1 ккал/моль), что находится в согласии с положением о более проч-
прочной связи для меньших атомов; эта величина сравнима с указываемым в лите-
286 Глава 6
ратуре значением 64 ккал/моль для связи FF, и она позволяет лучше объяснить
характер электронной структуры кислородных связей.
К сожалению, как указывает Жигер [80], данные, на которых основаны
рассуждения Уолша, не выдерживают критики. Как показывает приведенное
выше обсуждение структуры перекиси водорода, значения для длины связи ОН
и силовой постоянной, принятые Уолшем, были ошибочными. Правильная вели-
величина для теплоты диссоциации фтора по реакции A1) [76, 81] сравнима со
значением 34 ккал/моль для энергии связи 00. Меньшую энергию связи 00,
а также связей FF и NN по сравнению с энергиями для более тяжелых ато-
атомов можно легко объяснить. Вкратце это объяснение состоит в том, что у более
тяжелых атомов отталкивание внутренних оболочек снижает перекрывание
орбит и энергию связи; для более легких атомов возможно большее сближение
и образование более прочных связей, за исключением азота, кислорода и фто-
фтора, у которых отталкивание сильно сближенных заполненных орбит ограни-
ограничивает прочность связи.
Таким образом, наилучший расчет энергий связи в перекиси водорода
принадлежит Паулингу; выше были приведены несколько исправленные его
значения. Тем не менее указанные значения все же являются только оценкой,
поскольку другими путями нельзя получить достаточно точных величин для
сопоставления. Хотя Уолш в своих расчетах вывел неприемлемые крайние
значения, все же его обстоятельный анализ зависимостей между энергией
связи, другими свойствами связи и электронной структурой достоин изучения
в связи со многими поднятыми им вопросами. Так, Хаггинс [82] показал, что
свойства перекиси водорода согласуются с предложенным соотношением между
энергией и длиной связи. После того как такого рода соотношения приобретут
более количественный характер и будут возможны более точные измерения,
может быть, окажется необходимой небольшая ревизия значения энергии свя-
связи 00 в перекиси водорода в сторону ее повышения.
Сравнительная структура
Интересно сравнить структуры перекиси водорода и других соединений.
Такие сравнения можно классифицировать следующим образом: 1) с замещен-
замещенной перекисью водорода, 2) с «надсоединениями» гомологов кислорода и 3) с со-
соединениями элементов, смежных с кислородом, а именно с соединениями
углерода, азота и фтора. Остановимся кратко на каждом таком сравнении.
1. Гайсинский [83] предложил рациональную основу для классификации
элементов по способности к образованию перекисей. Он считает, что давать
перекиси могут элементы с такой же электроотрицательностью, как у водорода,
или меньшей. При образовании таких перекисей элемент, по-видимому,
всегда входит в перекись в состоянии максимальной валентности. Это значит,
что при образовании перекиси атом стремится отдавать электроны, а такая
возможность, конечно, наиболее вероятна для элементов с низкой электро-
электроотрицательностью. Уолш [84] объяснил правильность этой классификации,
исходя из того, что для элементов с меньшей электротрицательностью больше
возможность перекрывания атомных орбит элемента и перекисного кислорода.
Таким образом, преимущественно ионная природа перекиси натрия и безус-
безуспешные поиски перекисей галогенов находят общее объяснение в том, что для
надлежащей стабильности перекисной группе должен быть сообщен значитель-
значительный заряд. Эта тема обсуждается еще в гл. 7 и 12.
Указанные соображения должны быть распространены и на область орга-
органических перекисей. Указывается [85], что повышенную реакционную спо-
способность ацилзамещенных перекисей по сравнению с арилзамещенными можно
объяснить большей электроотрицательностью ацильных групп. Пока проведе-
проведено слишком мало исследований соответствующих физических свойств органи-
Структура молекулы перекиси водорода 287
ческих перекисей, чтобы можно было сделать общие выводы. При изложении
материалов по спектроскопии упомянуто, что колебание 00, по-видимому,
одно и то же во многих органических перекисях. Энергии диссоциации по
связи 00, обычно встречающиеся у этих перекисей, как правило, ниже, чем
у перекиси водорода [73]. Относительно ориентации замещающих органических
групп в производных перекиси водорода известно очень мало. Так, логическое
предположение [40], что ди-трет-бутилперекись обладает траяс-конфигура-
цией, как будто противоречит результатам новейших измерений дипольного
момента [24]. Что касается стабильности или реакционной способности пере-
кисной группы, то, по-видимому, неорганические перекиси, как правило, более
стабильны, чем перекись водорода. Для органических перекисей таких обоб-
обобщений сделать нельзя; обычно они считаются менее стабильными, чем перекись
водорода, и возможно, что это правильно, но наличие таких стабильных пере-
перекисей, как перекись бензоила и ди-трет-бутилперекись, и возможность синте-
синтеза в целях идентификации таких производных, как перекиси ацетилена [86]г
не позволяют в настоящее время предсказывать стабильность той или иной
перекиси. Эти вопросы рассматриваются при анализе механизмов реакций
в гл. 7 (стр. 320).
2. Структура молекулы двусернистого водорода H2S2 была подвергнута
тщательному изучению [70, 87]. Эта молекула очень близка к перекиси водо-
водорода, обладает длиной связи SS 2,05 А и азимутальным углом, очень близким
к 90°. Аналогия распространяется на химические свойства, так же как и на
структуру [88]. Некоторые различия в поведении этих двух соединений объ-
объясняются неодинаковыми ассоциативными свойствами [89]. Интересно сравнить
и сопоставить кислородные и сернистые соединения вообще, как это сделано в раз-
разделе по реакциям с производными серы на стр. 335. Наибольший интерес имеет
различие, состоящее в меньшей электроотрицательности серы. Например, сера
способна к полимеризации с образованием молекул S6 и S8; может существовать
также трехсернистый водород H2S3 или сульфаны [90] H2Sn. Кислород не может
образовать таких соединений (за исключением, может быть, типа H2S3). В то
же время невозможно заменить водород галогенами ни в перекиси водорода г
ни в двусернистом водороде. Правда, указывается 191] на существование сое-
соединения O2F2 и сообщается, что вязкость полимерной серы может быть сни-
снижена путем введения хлора с образованием ClSnCl [92], однако структура
соединения S2C12, по-видимому [93], тетраэдрическая, подобная структуре хло-
хлористого тионила SOC12, а доказательство существования O2F2 нельзя считать
бесспорным. К сожалению, нельзя распространить эти сравнения на произ-
производные селена и теллура. Имеется предположение о существовании двуселени-
стого водорода, но доказательств не приводится.
3. В качестве основы для сравнения можно использовать «изоэлектрон-
ные» соединения, т. е. простые структуры, содержащие одно и то же число
электронов во внешней или валентной оболочке. Так, соединения, содержащие
14 электронов в незамкнутых оболочках, изоэлектронны с перекисью водо-
водорода; в эту категорию входят такие молекулы, как F^O*", НО2, H.ZS2 и CIO".
Приложение изоэлектронного принципа особенно подходит для элементов,,
находящихся рядом в периодической системе Менделеева. В углеродных соеди-
соединениях, которые могут быть сравнены с перекисью водорода, значение гибрид-
гибридных орбит в структуре является причиной значительных различий в энергии
связи; частичный s-характер углеродных связей значительно повышает их
прочность. Это заметно для этана, изоэлектронного углеродного производного;
для ацетилена, по формуле аналогичного перекиси водорода, сравнение ока-
оказывается более затруднительным, поскольку в ацетилене имеется т;-связь
(а в перекиси водорода—однократная связь). Такое же возражение можно
выдвинуть и при попытках поисков параллели с дифтордиазином N2F2 [94]
или его водородным аналогом H2N2 (пока еще не обнаруженным). В литературе
288 Глава 6
[41] рассматривается вопрос, каким образом эти молекулы не только содержат
-связь, но и могут существовать в цис- или транс-конфигурации. Более близ-
близкое сравнение можно провести с азотом в форме гидразина N2H4. Этот вопрос
тщательно изучен [95, 96], причем доказано, что гидразин относится таким же
образом к аммиаку, как перекись водорода к воде. Структура гидразина очень
близка у. структуре перекиси водорода [44]. Для гидразина можно представить
себе наличие плоскости, делящей пополам угол HNH и проходящей через оба
атома азота; имеются две такие плоскости, по одной для каждой группы
HNH, причем эти плоскости взаимно перпендикулярны. Расчеты показы-
показывают, что эта структура несколько более стабильна, чем траяс-структура,
ввиду слабого гибридного характера азотных орбит. Гидроксиламин сочетает
свойства и перекиси водорода и гидразина. Анджели [97] произведено
сравнение всех этих веществ. Молекула гидроксиламина, по-видимому, имеет
/(//с-структуру [98], тем не менее полностью исключить возможность существо-
существования транс-структуры или смеси цис- и транс-структур также нельзя. Лафт
1991 провел сравнение перекиси водорода, гидразина и гидроксиламина
с точки зрения возможности внутреннего вращения в каждой из этих
молекул.
Если рассмотреть ряд изоэлектронных соединений N2Hj, NH2OH, H2O2,
FOH и F2, то окажется, что каждый центральный атом имеет пару s-электро-
нов, не участвующих в образовании связи; кроме того, при центральных ато-
атомах имеется последовательно от 0 до 4 рядов р-электронных пар не участвую-
участвующих в образовании связи. Интересно было бы выявить взаимозависимость
между свойствами этого ряда молекул и их структурами. В связи с этим жела-
желательно было бы рассмотреть и малоизвестную молекулу фторамина FNH2 [100],
которую можно считать [43] изоэлектронным аналогом пирамидальной триго-
нальной молекулы перекиси водорода, рассмотренной выше, с аналогичными
валентными связями. Атом фтора можно рассматривать как «составной» атом
группы ОН, т. е. как изоэлектронный аналог гидроксила. Часто отмечается,
что такого рода атом аналогичен по свойствам с группой, которую он представ-
лязт. Это, по-видимому, в достаточной мере оправдывается при сравнении фто-
фтора и гидроксила. Часто перекись водорода изображается как псевдогалоген,
особенно с точки зрения еефотолитической диссоциации [1011.
Можно привести и другие сравнения для перекиси водорода. Можно, на-
например, рассматривать ее как член ряда гидридов 195!. Можно также шире
развить сравнение кислородных соединений, представленное при обсуждении
электронной структуры. Такого рода сравнения должны оказаться весьма цен-
ценными и дать интересный материал по мере совершенствования количественной
теории химической связи.
МОЛЕКУЛЯРНАЯ АССОЦИАЦИЯ ПЕРЕКИСИ ВОДОРОДА
Характер многих обычно наблюдаемых физических свойств определяется
наличием межмолекулярных связей между молекулами перекиси водорода или
между перекисью и другими молекулами, находящимися в смеси с нею. Нали-
Наличие структуры, способной к образованию прочных водородных связей,
имеет очень большое значение с точки зрения определения ассоциативных
свойств перекиси водорода. В дополнение к такой структуре ассоциации замет-
заметно способствует и большой постоянный дипольный момент. Очевидная анало-
аналогия между перекисью водорода и водой в отношении этой характеристики обу-
обусловливает близкое соответствие значений таких свойств, как температура ки-
кипения, поверхностное натяжение и диэлектрическая проницаемость. Водород-
Водородная связь обладает определенной направленной ориентацией; ось ее представ-
представляет продолжение молекулярной связи ОН, в состав которой входит водород,
принимающий участие в водородной связи. Каждый атом кислорода может
Молекулярная ассоциация перекиси водорода 289
притягивать два атома водорода, размеры же атома водорода исключают воз-
возможность участия его более чем в одной связи. Таким образом, два атома водо-
водорода воды образуют водородные связи, кислород же воды может участвовать
в двух других связях. Перекись водорода также может дать два атома водорода,
но два ее кислородных атома могут участвовать в четырех водородных связях.
Водородную связь можно рассматривать просто как силу притяжения, суще-
существующую между остаточными положительными и отрицательными зарядами,
находящимися соответственно у атомов водорода и кислорода в различных
молекулах. Электронная конфигурация, обусловливающая такое притяжение,
является, конечно, в деталях значительно более сложной. В частности, имеет
значение гибридизация 2s- и 2р-орбит, которую можно было не рассматривать
в вышеизложенном качественном описании структур. Хотя такого рода гибри-
гибридизация не в состоянии увеличить число первичных валентностей кислородного
атома, как это происходит при гибридизации бериллия, бора и углерода, она
все же имеет существенное значение для прочности, ориентации и симметрии
связей первичной валентности, а также для характера или существования
других связей, например водородных или координационных. Никаких пред-
предположений о степени гибридизации перекиси водорода нет, однако по анало-
аналогии с водой можно считать, что она должна быть значительной. Для воды это
подтверждается доказанным существованием [102] структуры связанных
тетраэдров у кристаллов льда, а также тем, что угол при вершине A04°) близок
к углу тетраэдра A09°). Это находится в разительном противоречии с ограни-
ограниченной ассоциацией сероводорода, в молекуле которого угол при вершине
равен примерно 90°. Теоретически рассматривался вопрос о том [103], почему
необходима асимметрия электронных пар, не участвующих в образовании
связи, как для создания водородной связи, так и для обеспечения максималь-
максимальной прочности валентных связей ОН в воде. Высказывается и противополож-
противоположное мнение [104], а именно, что орбиты свободного атома кислорода лишь сла-
слабо изменяются при образовании воды. Для перекиси водорода имеет значение
и то, что участие в координационной связи, по-видимому, способствует ее
разложению.
Перейдем сейчас к рассмотрению ассоциации перекиси водорода в различ-
различных фазах.
Ассоциация в твердой фазе
Абрагэмс, Коллин и Липскомб [59] методом рентгеновской диффракции
определили кристаллическую структуру перекиси водорода. Эти авторы
приводят следующие данные: тетрагональный кристалл размерами а=Ь=
=4,06±0,02 А, с=8,00±0,02 А, пространственная группа D^—PA^ (энан-
тиоморфная с DJ—P432J, 4 молекулы в элементарной ячейке, объем элементар-
элементарной ячейки 131,9 А3. Эти результаты подтверждают более ранние данные, полу-
полученные Фехером и Клетцером [1051 и Натта и Ригамонти [106]. Кристалл
является довольно плотным, причем водородные связи образуют основные связи
между молекулами. Расстояние между кислородными атомами через водородную
связь О—Н—О оказалось равным 2,78 А, т. е. примерно таким же, как для
обычного льда. Каждая молекула связана с любой другой сеткой водородных
связей, в кристалле вдоль винтовых осей четвертого порядка вьются бесконеч-
бесконечные спирали. Сделан вывод, что при абсолютном нуле кристалл не должен
обладать остаточной энтропией. Уэллс [107] указывает, что идеальная струк-
структура кристаллической перекиси водорода представляет собой одну из двух
простейших трехсвязных сеток в трехмерном пространстве. Такая сетка пред-
представляет систему связанных десятиугольников.
Абрагэмс, Коллин и Липскомб показывают, как кристалл может быть
модифицирован за счет введения в состав его молекул воды; таким образом
они объясняют образование твердых растворов. Но на стр. 184 показано, что
19 Перекись водорода
290 Глава 6
перекись водорода не образует истинных твердых растворов, а потому эти
рассуждения отпадают. Единственной стабильной ассоциацией перекиси водо-
водорода с водой в твердой фазе является гидрат Н2О2-2Н2О. При рассмотре-
рассмотрении фазовой диаграммы в гл. 5 указано, что в процессе образования этого-
соединения с водородными связями должна происходить известная деформация
молекулы перекиси водорода. Единственным другим аддитивным соединением
перекиси водорода, которое подвергнуто тщательному изучению под углом
зрения структуры, является соединение с мочевиной СО (NH2JH2O2 150].
Конечно, имеются и многие другие аддитивные соединения; их природа ча-
частично рассматривается в гл. 7 (стр. 309).
Ассоциация в безводной жидкой перекиси водорода
При рассмотрении молекулярной структуры указывалось на невозможность
использования для выявления структуры некоторых конституционных дан-
данных, например парахора; это доказывает, что перекись водорода ассоцииро-
ассоциирована. Кроме того, имеется еще другое доказательство в пользу ассоциации.
По мнению Мааса и Хэтчера [108], низкое значение константы Рамзея—Этве-
са—Шилдса (зависящей от поверхностного натяжения и плотности) показывает,
что перекись водорода ассоциирована примерно в таких же размерах, как
и вода. Правило Трутона позволяет сделать несколько более количественные
выводы. В соответствии с этим энтропия парообразования при нормальной
температуре кипения для большинства веществ равна 21 ккал/мол-град. К.
Высокие значения для перекиси водорода B6,6) и воды B6,1) доказывают,
что эти вещества обладают в жидком состоянии некоторыми дополнительными
силами притяжения между молекулами, которых нет у других веществ.
Как перекись водорода, так и вода в парах не ассоциированы, а поэтому
их состояния в парах такие же (если не считать различий в объеме), как
и состояния тех веществ, для которых определено нормальное значение кон-
константы Трутона. Эта дополнительная упорядоченность или ограничение
совершенно неупорядоченного движения в жидкой воде и жидкой перекиси
водорода обусловлено связыванием групп молекул за счет водородных связей.
Таким образом, силы, связывающие молекулы друг с другом в жидком состоя-
состоянии, в среднем имеют большую величину, что приводит к увеличению плот-
плотности, поверхностного натяжения и энергии, необходимой для парообразова-
парообразования. Это иллюстрируется также высокой диэлектрической проницаемостью
перекиси водорода. Диэлектрическая проницаемость пропорциональна ква-
квадрату дипольного момента; следовательно, если молекулы связываются вместе
таким образом, что дипольные моменты агрегатов оказываются увеличенными,
то эффект связывания должен увеличить диэлектрическую проницаемость,
несмотря даже на снижение общего числа диполей. Расчеты Паулинга
[34] показывают, что диэлектрическая проницаемость перекиси водорода
при комнатной температуре в отсутствие ассоциации должна была бы ле-
лежать в интервале от 10 до 20, фактическая же ее величина находится
около 80.
Имеется очень мало предположений, касающихся точной ориентации ас-
ассоциированных молекул перекиси водорода. Рэндол [46] в результате рентге-
рентгеновского исследования пришел к выводу, что безводная жидкость может быть
представлена как скопление гидроксильных групп с плотнейшей упаковкой
гранецентрированного кубического типа, в которой каждая молекула переки-
перекиси водорода имеет от 6 до 12 ближайших соседей. Харт и Матесон [109] счи-
считают, что некоторые процессы разложения можно объяснить образованием
комплекса перекиси водорода с одним или двумя пергидроксильными радика-
радикалами за счет водородных связей (формулы I и II). Лафт [ПО] расширил это
представление, указав на возможность существования циклических димеров
Молекулярная ассоциация перекиси водорода 291
(формула III) перекиси водорода.
н-о/ )о о/ >о-нч н-о/ >о-н
хн-с/ хн-ск >о чн-ск
/
III
Какая бы ни была ориентация молекул перекиси водорода, необходимо иметь
в виду, что тепловые движения молекул в жидкости в состоянии разорвать
и вновь образовать водородные связи; агрегат, образованный за счет водород-
водородных связей, не сохраняется неизменным в течение длительного времени.
Сравнение перекиси водорода с водой
Сравнимые величины константы Трутона для воды и перекиси водорода
свидетельствуют о близкой степени ассоциации в обеих жидкостях при их
температурах кипения. Более значительное различие, по-видимому, существует
между обеими жидкостями при температурах их замерзания, почти совпа-
совпадающих друг с другом. Теплота плавления перекиси водорода B,9 ккал/моль)
примерно в два раза больше, чем для воды A,4 ккал/моль). Поэтому если общие
межмолекулярные энергии ассоциации примерно одинаковы в обоих случаях,
то при плавлении перекиси водорода теряется большая часть ассоциации, чем
при плавлении льда. Паулинг [34] подсчитал, что 15% водородных связей
льда разрывается при плавлении; по оценке Тафта и Сислера 1111], эта вели-
величина составляет 11%. Если применить последний метод, то оказывается, что
при плавлении перекиси водорода разрывается 30% водородных связей,
и эта величина качественно довольно хорошо согласуется с подсчетами [95,
112], показывающими, что в перекиси водорода разрывается около 20% свя-
связей, или в два раза больше, чем в воде.
Изменения инфракрасного спектра поглощения при переходе от пара
к жидкости для перекиси водорода и воды примерно одинаковы. Как указывает
Родбаш [113], фундаментальные частоты растяжения при этом снижаются,
тогда как фундаментальные частоты изгибания увеличиваются; интенсивность
и ширина полос возрастает в том и другом случае. Жигер [95] пришел к заклю-
заключению, что больший сдвиг частоты, наблюдаемый для перекиси водорода,
доказывает, что водородные связи в перекиси водорода немного более прочны,
чем в воде. Тейлор приводит следующие дополнительные доказательства того,
что при плавлении в перекиси водорода водородных связей разрывается боль-
больше, чем при плавлении льда: в переохлажденной перекиси водорода в полосе
для ОН при 3400 см'1 можно обнаружить лишь очень мало деталей, най-
найденных для этой полосы в кристалле. Наоборот, для воды характер этой полосы
в жидкости почти такой же, как и в твердой фазе.
Относительные значения диэлектрической проницаемости также показы-
показывают, что степень ассоциации в жидкой перекиси водорода меньше, чем в воде.
Несмотря на то, что перекись обладает большим дипольным моментом, чем
вода, диэлектрическая проницаемость воды больше, чем перекиси водорода.
Поскольку диэлектрическая проницаемость является функцией как числа
диполей в единице объема, так и величины их, то трудно заранее рассчитать
итоговое влияние одновременного изменения момента мономера, степени агре-
агрегации и специфической ориентации агрегатов. Гросс и Тейлор [115] считают
возможным, что больший момент перекиси водорода компенсируется снижением
степени ассоциации, и приводят лежащие в основе такого вывода теоретиче-
теоретические рассуждения [116], касающиеся взаимозависимости между водородными
связями и диэлектрической проницаемостью перекиси водорода.
Существенное различие между перекисью водорода и водой состоит
в характере изменения объема при плавлении. Молярные плотности обоих
19*
292 Глава 6
веществ в твердой фазе примерно одинаковы, однако плотность воды возрастает
при плавлении, а плотность перекиси водорода снижается. Части кристаллов,
переходящие в жидкость при плавлении без нарушений, по-видимому, про-
продолжают оказывать одинаковое влияние на плотность. Остальные молекулы,
освобожденные от кристаллической ассоциации, оказываются связанными
друг с другом менее прочно, но зато увеличивается число их ближайших
соседей на более значительных расстояниях. Итоговый эффект для воды состоит
в увеличении притяжения; для перекиси водорода возросшее число контак-
контактов не компенсирует снижение прочности каждого из них. По мнению Тейлора
[114], легкость переохлаждения перекиси водорода доказывает, что молекула
может войти в кристалл, только претерпев известную деформацию. Это пра-
правильно, если решающим фактором не является образование зародышей.
Жигер [95] опубликовал сравнение ассоциативных свойств перекиси водорода
и других гидридов.
Ассоциация в растворе
Растворы перекиси водорода в воде не являются идеальными, что обнару-
обнаруживается при исследовании любым из трех обычно применяемых методов:
объем раствора меньше, чем сумма объемов составляющих компонентов, сме-
смешение происходит с заметным тепловым эффектом и величины давления пара
растворов не подчиняются закону Рауля. Дальнейшими доказательствами
являются неправильные зависимости между концентрацией раствора и такими
свойствами, как вязкость, поверхностное натяжение и диэлектрическая про-
проницаемость. Характер отклонения от идеальности в каждом отдельном случае
говорит об увеличении либо числа молекул, либо сил притяжения между моле-
молекулами при образовании растворов, что выражается в уменьшении общего
объема и давления пара и выделении тепла при смешении. Аналогия между
водой и перекисью водорода в отношении природы и размеров межмолекуляр-
межмолекулярных сил приводит к логическому выводу, что это поведение обусловлено обра-
образованием дополнительных водородных связей; иначе говоря, можно предпола-
предполагать, что водородные связи между молекулами воды и перекиси водорода более
стабильны, чем связи между молекулами каждого из этих веществ в отдель-
отдельности. Это подтверждают и измерения Уинн-Джонса [117] по изменению основ-
основности водных растворов перекиси водорода с концентрацией.
В связи с присутствием большого числа водородных связей растворы пере-
перекиси водорода в воде, так же как и сами компоненты, представляют собой
смеси сложных полимерных частиц. Хотя отдельные связи, создающие эту
систему, очень недолговечны, распределение полимерных частиц по размерам
сохраняется. Процесс растворения перекиси водорода в воде можно рассма-
рассматривать как смешение уже полимеризованных воды и перекиси водорода с обра-
образованием полимерного раствора с одновременным изменением ассоциации до
степени, которая соответствует равновесному состоянию. Скэтчард, Каванаф
и Тикнор [1181 указывают, что для смешения этих растворов справедлива теория
Флоури и Хаггинса; чтобы применить ее на практике, требуется лишь знать,
как распределены полимерные частицы. В такой равновесной системе эти виды
частиц должны находиться в соотношениях, выводимых из формул, разрабо-
разработанных Штокмейером и Флоури для реакций полимеризации. В этих уравнениях
распределение видов частиц по размерам представляет функцию доли возмож-
возможных связей, уже образовавшихся в разных растворах. Сделано очень простое
допущение [118], касающееся изменения степени ассоциации с составом: шан-
шансы связывания водорода пропорциональны концентрации несвязанных акцеп-
акцепторов. Поскольку степень ассоциации компонентов при температурах изме-
измерений не была хорошо известна, необходимо было сделать несколько произ-
произвольный выбор этой величины таким образом, чтобы она удовлетворяла экспе-
экспериментальной свободной энергии смешения эквимолекулярных растворов..
Молекулярная ассоциация перекиси водорода 293
Полученные при этом доли значения ассоциации для компонентов [118] оказа-
оказались приемлемыми, хотя несколько завышенными, и совпадение с эксперимен-
экспериментальными данными для других концентраций, кроме эквимолекулярной, было
удовлетворительным. Доли связанных водородных атомов составляли 0,720
для воды и 0,886 для перекиси водорода. Эти значения, однако, не согласуются
с тем положением, что при низких температурах перекись водорода менее полно
ассоциирована, чем вода; в самой предложенной функции для зависимости
степени ассоциации от концентрации подразумевается противоположное
соотношение.
Мориссет и Жигер [119] объясняют следующим образом эндотермический
эффект (анализ его см. на стр. 207), наблюдаемый при разбавлении перекиси
водорода некоторых определенных концентраций при температурах ниже 21°.
Допустим, что добавление известного количества перекиси водорода к воде
при 0° вызывает разрыв большего числа водородных связей между молекула-
молекулами воды, чем то, которое вновь образуется между водой и перекисью; резуль-
результирующее снижение числа водородных связей способствует эндотермическому
тепловому эффекту. При более высоких температурах, т. е. выше 21°, число
водородных связей между молекулами воды меньше. Поэтому при добавлении
перекиси водорода разрывается меньшее число связей и сравнительно большее
число их образуется между водой и перекисью. Таким образом, нагревание
раствора от 0°до температуры выше 21° вызывает разрыв большего числа
водородных связей между молекулами воды, чем между молекулами воды
и молекулами перекиси водорода.
Для объяснения неидеальных свойств водных растворов перекиси водо-
водорода часто приводилось существование аддитивного соединения Н2О2-2Н2О.
Так, Вознесенская и Заславский [120] указывают, что максимальное отклоне-
отклонение от аддитивности в расчете на атомные концентрации (числа атомов в еди-
единице объема) наблюдается для соединения Н2О2-2Н2О. Гросс и Тейлор [115]
указывают на возможное влияние соединения Н2О2-2Н2О на образование
максимума в изотермах диэлектрическая проницаемость—состав. Верно и то,
что максимальное отклонение для многих свойств растворов наблюдается при
составе, близком к Н2О2-2Н2О, что позволяет говорить о специфической
роли этого соединения; однако соотношение неопределенное и неустойчивое.
Большая пологость максимума на диаграмме температур замерзания (который
доказывает существование аддитивного соединения Н2О2-2Н2О) подтверждает,
что это соединение должно быть в жидкости почти полностью диссоциирован-
диссоциированным. Более логично предполагать, что из всех возможных агрегатов между
перекисью водорода и водой только агрегаты состава Н2О2-2Н2О могут
сохранять свою идентичность при переходе в твердую фазу.
Митчел и Уинн-Джонс [121] сравнили водные растворы перекиси водо-
водорода с водными растворами спирта, доказывая на основании избыточных тер-
термодинамических свойств, что перекись водорода легко может внедряться в
структуру воды и стягивать ее, тогда как спирт в структуру воды внедриться
не может. Аналогично молекула перекиси водорода, находясь в окружении
молекул воды, теряет значительную часть кислотности и не может функцио-
функционировать как кислота в такой же степени, как в свободном состоянии.
Другие типы межмолекулярных сил, например притяжения между ионами
и диполями или наведенными диполями, в растворах перекиси водорода изу-
изучались очень мало. Горин [122] показал, что перекись водорода «всаливается»
в ряд растворов электролитов. Сделано заключение, что молекулы перекиси
водорода имеют склонность к вытеснению молекул воды из окружения ионов,
что объясняется более высоким дипольным моментом перекиси водорода. В этом
отношении водородный ион отличается от других ионов, что объясняется пре-
преимущественным образованием иона Н3О+ по сравнению с ионом HgOJ. Все эти
предположения, безусловно, важны с точки зрения определения характера
294 Глава 6
среды, в которой происходят реакции перекиси водорода. В гл. 7 этому вопро-
вопросу посвящен отдельный раздел (стр, 330).
Энергия водородной связи
Паулинг [34] считает, что около х/4 теплоты возгонки льда A2,2 ккал/моль)
должно быть приписано вандерваальсовым силам, а остальная часть—водород-
часть—водородным связям. Поскольку каждая молекула имеет две водородные связи, то
для энергии водородной связи в воде получается значение 4,5 ккал/моль.
Паулинг принимал, что теплота возгонки перекиси водорода равна
14,1 ккал/моль; после вычитания 5 ккал/моль, подсчитанных для вандерваальсо-
выхсил, и деления остатка на две водородных связи на молекулу он получил
ту же величину для водородной связи в перекиси водорода, что и для воды. Если
подсчитанная энергия вандерваальсовых сил верна, то на основании исправ-
исправленной величины для теплоты возгонки перекиси водорода, 15,5 ккал/моль,
получается, что водородная связь в перекиси водорода несколько прочнее,
чем в воде; это согласуется и с выводами, основанными на других фактах.
Скэтчард, Каванаф и Тикнор [118] показали, каким образом на основании до-
долей ассоциации компонентов и раствора в сочетании с теплотой смешения можно
подсчитать энтальпию образования водородной связи. Они рассчитали, что
энергия образования водородной связи при смешении равных частей перекиси
водорода и воды составляет 3,4 ккал/моль. При использовании теплот смешения,
экстраполированных из величин, измеренных [119] при 26,9°, получается
значение 3,8 ккал/моль [123]. Эта величина близка к рассчитанной Паулингом.
Другие подсчеты дают для энергии водородной связи в воде значения 4,25 [111]
и 6,4 ккал/моль [124]. Дейтериевая связь, по-видимому, несколько прочнее,
чем водородная [112].
ЛИТЕРАТУРА
1. К р у г л я к о в а К. Е., Э м а н у э л ь Н. М., ДАН СССР, 83, 593 A952); При-
Природа, № 6, 103, VI A952).
2. С 1 о v е г А. А\., Am. Chem. J., 29, 463 A903).
3. В а е у е г А., V i I I i g e r V., Вег., 33, 3387 A900); Willstatter R.,Ha-
uenstein E., Вег., 42, 1842 A909).
4. Raikow P. N., Z. anorg. Chem., 168, 297 A928).
5. G e i b К. H., H a r t e с к Р., Вег., 65, 1551 A932).
6. J о n e s R. A., W i n k 1 e г С. A., Can. J. Chem., 29, 1010 A951); GiguereP.A.,
S e с с о E. A., E a t о n R. S., Discussions Faraday Soc, Toronto, № 14, 1953, p. 104.
7. Gi guere P. A., S e с с о Е. А., частное сообщение, 1953.
8. С u t h b e г t s о n A. C, M a a s s O., J. Am. Chem. Soc, 52, 489 A930).
9. Phibbs M. K., Giguere P. A., Can. J. Chem., 29, 173 A951).
10. S с h о о г 1 N.. Pharm. Weekblad, 67, 390 A930).
11. Bruhl J. W., Вег., 28, 2847 A895).
12. S t г е с k e г W., Spitaler R., Ber., 59, 1754 A926).
13. L i n t о n E. P., M a a s s O., Can. J. Res., 7, 81 A932).
14. G i g u ё г e P. A., Can. J. Res., 21B, 156 A943).
15. M i 1 a s N. A., S u г g e n о r D. M.,Perry L. H., J. Am. Chem. Soc, 68, 1617
A946).
16. R i с h t m у е г F. К., Introduction to Modern Physics, McGraw-Hill Book Co., Inc.,
New York, 1928, p. 120; Kurtz S.S., Ward A. L., J. Franklin Inst., 224, 583,
697 A937).
17. Giguere P. A., F e e n у H., Can. J. Res., 21A, 69 A943).
18. G г а у F. W., F а г q u h а г s о n J., Phil Mag., G), 10, 191 A930); Gray F. W.,
С r u i k s h a n k J. H., Trans. Faraday Soc, 31, 1491 A935).
19. P a r t i n g t о n J. R., An Advanced Treatise on Physical Chemistry, Vol. 2, Long-
Longmans, Green & Co., London, 1951, p. 17.
20. M a s s e у J. Т., В i a n с о D. R., J. Chem. Phys., 22, 422 A954).
21. S m у t h С. Р., Dielectric Constant and Molecular Structure, Chemical Catalog Co.,
Inc. (Reinhold Publishing Corp.), New York, 1931.
22. H u n t e r E. C. E., P a r t i n g t о n J. R., J. Chem. Soc, 1932, 2817.
23. T h i e 1 а с k e г W., Z. phys. Chem., B20, 142 A933).
Литература 295
24. Rogers M. Т., С a m p b e 1 1 Т. W., J. Am. Chem. Soc, 74, 4742 A952).
25. I n gol d С. К., Structure and Mechanism in Organic Chemistry, Cornell Univ. Press,
Ithaca, 1953, p. 33.
26. Giguere P. A., Bull. Soc. Chim. France, 1954, 720.
27. S i d g w i с к N. V., The Chemical Elements and Their Compounds, Vol. 2, Clarendon
Press, Oxford, 1950, p. 869.
28. Q u а г t а г о 1 i A., Gazz. chim. Ital., 64, 243 A934) [C. A., 28, 4998]; IX Congr.
intern, quim. pura applicada, Madrid, 3, 223 A934) [C.A., 30, 4368].
29. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog
Co., Inc., (Reinhold Publishing Corp.), New York, 1923.
30. L о wry T. M., Trans. Faraday Soc, 18, 285 A923).
31. Noll ег С R., Chemistry of Organic Compounds, W. B. SaundersCo., Philadelphia,
1951, p. 5—12, 43—45, 73—76; J. Chem. Education, 27, 504 A950).
32. Brown G. I., A Simple Guide to Modern Valency Theory, Longmans, Green & Co.,
London, 1953.
33. Palmer W. G., Valency: Classical and Modern, Cambridge Univ. Press, Cambridge,
1944.
34. П а у л и н г Л., Природа химической связи, Госхимиздат, М.—Л., 1947.
35. С о u I so n С. A., Valence, Clarendon Press, Oxford, 1952.
36. W h a 1 e у Т. Р., К 1 e i n b e г g J., J. Am. Chem. Soc, 73, 79 A951); Казар-
Казарновский И. А., Н и к о л ь с к и й Г. П., А б л е ц о в а Т. А., ДАН СССР,
64, 69 A949).
37. Kraus С. A., Why te E. F., J. Am. Chem. Soc, 48, 1788 A926).
38. G г u n d 1 a n d I., Compt. rend., 236, 476 A953).
39. Darling В. Т., D e n n i s о n D. M., Phys. Rev., 57, 128 A940).
40. Wai sh A. D., J. Chem. Soc. 1948, 331, 398.
41. W a 1 s h A. D., J. Chem. Soc, 1953, 2288, 2325.
42. Гельман Г., Квантовая химия, М.—Л., ОНТИ, 1937.
43. Walsh A. D., Discussions Faraday Soc, Toronto, № 14, A953), p. 140.
44. Penney W. G., Su t herl and G.B.B.M., Trans. Faraday Soc, 30, 898 A934);
J. Chem. Phys., 2, 492 A934).
45. Казарновский И. А., ЖФХ, 1, 93 A930). Труды Химического института
им. Л. Карпова, 9, 93 A930).
46. R a n d al I J. Т., Ргос Roy. Soc. (London), 159A, 83 A937).
47. J a k t а г S. К. К., Nature, 153, 316 A944).
48. Г е р ц б е р г Г., Колебательные и вращательные спектры многоатомных молекул,
Издатинлит, М., 1949.
49. D a v i s A. G. (и возражения G i g u ё г е P. A.), Discussions Faraday Soc, To
ronto, № 14 A953), p. 140.
50. L u С. S., H u g h e s E. W., G i g u ё r e P. A., J. Am. Chem. Soc, 63, 1507 A941).
51. G i g u ё r e P. A., Can. J. Res., 28B, 485 A950).
52. G i g u ё г e P. A., L i u I. D., D u g d a 1 e J. S., Morrison J. A., Can. J.
Chem., 32, 117 A954).
53. L a s s e t t г e E. N.. D e a n L. В., Jr., J. Chem. Phys., 17, 317 A949).
54. M u 1 1 i k e n R. S., J. Chem. Phys., 3, 5U6 A935).
55. R о b e r t s о n A. J. В., Trans. Faraday Soc, 48, 228 A952).
56. D e В e n e d e t t i S., Richings H. J., Phys. Rev., 85, 377 A952).
57. G a b i 1 1 а г d R., Compt. rend., 232, 324 A951).
58. G i g u ё г e P. A., S с h о m a k e г V., J. Am. Chem. Soc, 65, 2025 A943).
59. A b г a h a m s S. С, С о 1 1 i n R. L, Lipscomb W. N., Acta Cryst., 4, 15
A951).
60. Bailey C.R.,Gordon R. R., Trans. Faraday Soc, 34, 1133 A938).
61. Simon A., Feher F., Z. Elektrochem., 41, 290 A935); Simon A., Z. angew.
Chem., 51, 794 A938); Feher F., Вег., 72В, 1778 A939).
62. G i g u ё г e P. A., J. Chem. Phys., 18, 88 A950).
63. G i g u ё г е Р. А., частное сообщение, 1953.
€4. L e a d b e a t e r R., Compt. rend., 230, 829 A950); К а р я к и н А. В., Ники-
Никитин В. А., Изв. АН СССР, сер. физ., 17, 636 A953); М i n k о f f G. J., Ргос.
Roy. Soc, A224, 176 A954).
65. В a dger R. M., J. Chem. Phys., 2, 128 A934); 3, 710 A935).
66. G i g u ё г е Р. А., В a i n O., J. Phys. Chem., 56, 340 A952).
67. Bain O., Ph. D. Thesis, Laval University, 1953.
68. T а у 1 о г R. С, J. Chem. Phys., 18, 898 A950).
69. Z u m w a 1 t L R., Giguere P. A., J. Chem. Phys., 9, 458 A941).
70. W i 1 s о n M. K., Badger R. M., J. Chem. Phys., 17, 1232 A949).
71. I v a s h E. V., D e n n i s о n D. M., J. Chem. Phys., 21, 1804 A953).
72. W a 1 s h A. D., Trans. Faraday Soc, 43, 60 A947).
73. Sz ware M., Chem. Revs., 47, 75 A950).
74. G 1 о с к 1 e г G., Annual Review of Physical Chemistry, 3, 151 A952).
296 Глава 6
75. В a ugh an E. С. , Quarterly Revs., 7, 103 A953).
76. R о s s i n i F. D., W a g m a n D. D., E у a n s W. H., Levine S., J a f f e I.
Selected Values of Chemical Thermodynamic Properties, Nat'l. Bureau of Standards
Circular 500, Government Printing Office, Washington, 1952; см. также серию III
по вышеуказанному вопросу, 1948 и ел.
77. S k i п п е г Н. A., Trans. Faraday Soc, 41, 645 A945).
78. G 1 о с к 1 е г G., M a 11 а с к G., J. Chem. Phys., 14, 504 A946).
79. G i g u ё г е P. A., Ann. ACFAS, 9, 88 A943).
80. Giguere P. A., Can. J. Research, 28B, 17 A950).
81. G i 1 1 e s P. W., M a r g г a v e J. L., J. Chem. Phys., 21, 381 A953).
82. Huggins M. L., J. Am. Chem. Soc, 75, 4123, 4126 A953).
83. Haissinsky M., J. Chem. Phys., 15, 152 A947).
84. Walsh A. D., J. Chem. Phys., 15, 688 A947).
85. W a 1 s h A. D., Trans. Faraday Soc, 42, 264 A946).
86. M i 1 a s N. A., M a g e 1 i O. L., J. Am. Chem. Soc, 74, 1471 A952).
87. S t e v e n s о n D. P., В е а с h J. Y., J. Am. Chem. Soc, 60, 2872 A938).
88. Yost D. M., Russell H., Systematic Inorganic Chemistry, Prentice Hall, Inc.,.
New York, 1944.
89. Butler K.H.,Maass O., J Am. Chem. Soc, 52, 2184 A930).
90. Feher F., Schliep E., Weber H., Z. Elektrochem., 57, 916 A953).
91. F r i s с h P., S с h u m а с h e r H. J., Z. anorg. allgem. Chem., 229, 423 A936).
92. Fan ell i R., J. Am. Chem. Soc, 70, 1965 A948).
93. S a m u el R., J. Chem. Phys., 12, 380 A944).
94. Bauer S. H., J. Am. Chem. Soc, 69, 3104 A947).
95. Giguere P. A., Trans. Roy. Soc Canada, C), III, 35, 1 A941).
96. Одрит Л., Or г Б., Химия гидразина, Издатинлит, М., 1954; S isle г Н.,
A u d r i e t h L. F., Trans, 111. State Acad. Sci., 31, 144 A938); Au dri eth L F.,
M о h r P. H., Chem. & Eng. News, 26, 3746 A948).
97. Angel i A. Atti R. Accad. Lincei, E), 19, II, 29, 94 A910); 20, I, 625 A911);
26, I, 480 A917); [C. A. 12, 365]; Gazz. chim. ital., 47, I, 220 A917).
98. G i g u ё г e P. A., L i u I. D., Can. J. Chem., 30, 948 A952).
99. L u f t N. W., J. Chem. Phys., 21, 179 A953).
100. Ruff O., S t a u b L., Z. anorg. allgem. Chem., 198, 32 A931).
101. U г е у Н. С, D a w s e у L H., Rice F. O., J. Am. Chem. Soc, 51, 137 A929).
102. В е г n a 1 J. D., F о w 1 e r R. H., J. Chem. Phys., 1, 515 A933).
103. Fyfe W. S., J. Chem. Phys., 21, 2 A953).
104. F i s с h e r-H j a 1 m а г s I., Arkiv Fysik, 7, 165 A953) (на английском языке).
105. Feher F., Klotzer F., Z. Elektrochem., 41, 850 A935); 43, 822 A937).
106. N a t t a G., Rigamonti R., Gazz. chim. ital., 66, 762 A936).
107. Wells A. F., in Gomer-Smith, Structure and Properties of Solid Surfaces, Univ.
of. Chicago Press, Chicago, 1953, p. 261.
108. M a a s s O., H a t с h e г W. H., J. Am. Chem., Soc, 42, 2548 A920).
109. Hart E. J.,Mathesoh M. S., Discussions Faraday Soc, № 12, Radiation Che-
Chemistry, 1952, p. 180.
110. L u f t N. W., Discussions Faraday Soc, № 12, Radiation Chemistry, 1952, p. 226.
111. T aft R. W., Jr., Si si er H. H., J. Chem. Ed., 24, 175, A947).
112. G i gu ё r e P. A., Sec с о Е. A., Can. J. Chem., 32, 550 A954).
113. R о d e b u s h W. H., Advances in Nuclear Chemistry and Theoretical Organic Che-
Chemistry, Interscience Publishers, Inc., New York, 1945, p. 132.
114. Taylor R. С, неопубликованная работа, 1947.
115. G г о s s P.M., Taylor, R. C, J. Am. Chem. Soc, 72, 2075 A950).
116. OsterG., К i r k woo d J. G., J. Chem. Phys., II, 175 A943).
117. W у n n e -J о n e s W. F. K., J. chim. phys., 46, 337 A949).
118. S с a t с h a r d G., К a v a n a g h G. M., Ticknor L. В., J. Am. Chem. Soc,
74, 3715 A952).
119. G i g u ё г е Р. А., М о г i s s e t t e B. G., частное сообщение, 1952.
120. Вознесенская О. М., Заславский И. И., ЖОХ, 16, 1189 A946).
121. Mitchell A.G., Wynne-Jones W. F. К., Discussions Faraday Soc, ЛЬ \ЪУ
Solutions of Non-Electrolytes, 1953, p. 161.
122. Gorin M. H., J. Am. Chem. Soc, 57, 1975 A935).
123. Kavanagh G. M., частное сообщение, 1954.
124. S e а г с у A. W., J. Chem. Phys., 17, 210 A949).
Глава 7
ХИМИЧЕСКИЕ СВОЙСТВА
Обсуждению химических свойств перекиси водорода посвящены три главы.
В данной главе описывается общее химическое поведение и стехиометрические
реакции перекиси водорода. Следующая глава содержит описание процессов
разложения, иногда сопровождающихся химическими реакциями других
веществ в количествах, которые, однако, не соответствуют стехиометрическим
соотношениям разложившейся перекиси водорода. Наконец, в гл. 9 рассматри-
рассматриваются вопросы стабильности перекиси водорода. Это подразделение является
произвольным, но полезным с точки зрения концентрирования внимания на
двух темах, имеющих существенное значение при обсуждении химических
свойств перекиси водорода. Такое подразделение неизбежно сопровождается
частичным повторением материала и перекрыванием отдельных тем, а поэтому
читателю, желающему ознакомиться с каким-либо специальным вопросом,
рекомендуется прочитать все три главы, так как один и тот же вопрос может
быть затронут в каждой из них с разных точек зрения. Перекисные соединения,
образуемые различными элементами, описываются в гл. 12.
Сначала даются некоторые общие классификации химических свойств
перекиси водорода. После этого рассматривается характер перекиси водорода
в порядке возрастающей сложности с точки зрения термодинамики, кинетики
и механизма реакции. Далее анализируются свойства перекиси водорода как
кислоты и реакционной среды, реакции в неводных средах и опыты с изотоп-
изотопными мечеными атомами с участием перекиси водорода. Затем даются ссылки
на литературу, и отдельные реакции (сгруппированные в соответствии с перио-
периодической таблицей элементов) рассматриваются настолько полно, насколько
это допускает объем книги. Вслед за рассмотрением неорганических химиче-
химических реакций даются разделы по органическим реакциям и биологическому
действию.
КЛАССИФИКАЦИЯ РЕАКЦИЙ ПЕРЕКИСИ ВОДОРОДА
Реакции перекиси водорода можно разделить на четыре категории. При
этом учитывается только итоговый или общий результат реакции.
Процессы разложения
Почти одновременно с открытием перекиси водорода стала известной
и реакция ее разложения
2Н2О2->2Н2О + Оа. A)
Эта реакция интерпретировалась как саморазложение перекиси водорода.
Однако дальнейшие исследования показали, что, если не считать высоких
температур в паровой фазе, реакция A) происходит лишь под действием како-
какого-либо другого вещества. Последнее в большинстве случаев химическому
изменению не подвергается или же это изменение не носит стехиометрического
298 Глава 7
характера; другими словами, число молей взаимодействующего вещества, под-
подвергшихся изменению, не находится в простом или постоянном отношении
к числу молей перекиси водорода, превращенных в воду и кислород. Эти
процессы разложения описываются в следующей главе.
Перекись водорода может реагировать и более упорядоченным путем,
чем здесь указано; эти стехиометрические реакции представляют основную тему
данной главы. Они классифицированы, например Гайсинским [1], следую-
следующим образом: 1) реакции окисления или восстановления, 2) перенос перекис-
ной группы и 3) образование аддитивных соединений.
Реакции окисления или восстановления
При реакциях окисления и восстановления второй реагент и кислород
молекулы перекиси водорода изменяют свои валентности. Примером чистой
реакции окисления под действием перекиси водорода является следующая
реакция:
Н2О2 + 2Fe2+ + 2Н+ ~-> 2Fe3+ + 2H?>. B)
Можно также привести и реакцию восстановления перекисью водорода:
2КМпО4 + 5Н2О2 + 3H2SO4 ~-> 2MnSO4 + K2SO4 + 8H,0 + 5О2. C)
Штейнбах [2] указывает, что для реакций восстановления перекисью водорода
можно выбрать бесконечное число рядов совместимых коэффициентов; однако
это обусловливается лишь возможностью сложения с кратными значениями
коэффициентов реакции A), но без изменения баланса реакции восстановления.
Необходимо подчеркнуть двойственный характер перекиси водорода
{т. е. способность ее действовать в качестве и окислителя и восстановителя),
который в значительной мере увеличивает разнообразие и сложность химии
перекиси водорода, что не всегда учитывается. В широком] смысле слова
эту двойственность можно считать отражением промежуточного положения,
которое перекись водорода занимает в ряду окисления—восстановления
между водой и молекулярным кислородом. Нет никаких оснований считать
это свойство «парадоксальным» [3]; молекулярный кислород является, без-
безусловно, общепризнанным продуктом окисления перекиси водорода.
Перенос перекисной группы
При переносе перекисной группы к другой молекуле изменения валент-
валентности кислорода не происходит. Примером такой реакции двойного обмена
может служить реакция
Н2О2 + Ва(ОНJ -» ВаО2 + 2Н2О. D)
Нужно иметь в виду, что перенос перекисной группы без изменения от одной
молекулы к другой в значительной мере сходен с образованием молекулярного
кислорода из перекиси водорода, хотя в одном случае изменение валентности
происходит, а в другом не происходит.
Образование аддитивных соединений
Молекула перекиси водорода может присоединяться как целое к другой
молекуле с образованием аддитивных соединений или пероксигидратов, ана-
аналогичных гидратам. Например
ЗН2О2 + 2Na2CO3 —> 2Na2CO3- 3H2O2. E)
Термодинамика реакций перекиси водорода
299
Из этих трех классов реакций основное внимание в этой главе уделено
первому классу, или реакциям окисления—восстановления. Реакции обрат-
обратного типа, т. е. образования перекиси водорода, уже рассмотрены в гл. 2 и 3.
Дополнительный материал по реакциям последних двух классов дан в гл. 12.
ТЕРМОДИНАМИКА РЕАКЦИЙ ПЕРЕКИСИ ВОДОРОДА
Для лучшего понимания химических свойств перекиси водорода необходи-
необходимо обратиться к термодинамическим данным, которые позволяют вычислить
химические равновесия. Необходимые для этой цели термодинамические вели-
величины выведены в гл. 5 и подытожены ниже в табл. 70 наряду с аналогичными
Таблица 70
Свободные энергии образования перекиси водорода
и аналогичных частиц при 25° и 1 am
Вещество
Н202газ
Н2О2)И
H202aq
HO-aq
&F°
ккал/моль
-25,24
-28,77
-31,95
-15,23
Вещество
Н2Огаз
Н2О}К
OH-aq
AF°
ккал1моль
-54,635
-56,690
-37,595
данными для некоторых других веществ [4]. Значения свободной энергии можно
комбинировать с аналогичными данными для других реагентов и продуктов
с целью получения общего изменения свободной энергии в той реакции, которая
нас интересует. Зная свободную энергию, можно вычислить константу равно-
равновесия при помощи соотношения A F°=—RT\nK и таким образом определить,
насколько полно может протекать та или иная реакция с участием перекиси
водорода.
Электродные потенциалы отдельных систем
Более удобным методом по сравнению с использованием данных сво-
свободной энергии для выражения способности к реакции является применение
электродных потенциалов отдельных систем (полуэлементов)*.
Отдельная система для перекиси водорода при действии ее в качестве
окислителя может быть написана следующим образом:
2Н2О -> Н2О2 + 2Н+ + 2е. F)
Потенциал этой реакции
?= —1,76 —
0,05916
G)
Для реакции в щелочном растворе, где образуется пергидроксильный ион,
отдельная система выражается уравнением
ЗОН" -» НО7 + Н2О -{- 2е. (8)
Потенциал этой реакции выражается через
?=_0,87-^р-61
* В приложении к этой главе (стр. 359) дан краткий обзор использования электрод-
электродных потенциалов в химии перекиси водорода.
300
Глава 7
Отдельная система для перекиси водорода при действии ее в качестве вос-
восстановителя может быть представлена частным уравнением
Н2О2 -* 2Н+ + О2 + 2е. A0}
Потенциал этой реакции
0,05916 \
Ц.-MM-L., ,VOh>Os
В щелочном растворе реакция протекает по уравнению
ОН" + НО7 -* О2 + Н2О + 2е.
Ее потенциал
? = 0,084-
0,05916
Ро2
\аон- • аио-
;)¦
A2)
A3)
0,8
Эти значения электродных потенциалов перекиси водорода, выведенные
на стр. 216, показывают, что перекись является очень сильным окислителем.
Из веществ, которые способны существовать в растворе и перечислены
в таблицах электродных по-
потенциалов, перекись водорода
не в состоянии окислить
лишь немногие. Поэтому при
рассмотрении возможных
реакций перекиси водорода
можно считать, что она а
состоянии окислить любые
вещества, за исключением
самых энергичных окислите-
окислителей (например, фтора или
озона) при том или иномрН.
Наоборот, как восстановитель
перекись водорода не обла-
обладает выдающимися свойства-
свойствами. Ее превосходит даже
такой обычный восстанови-
восстановитель, как тиосульфат; она
~J4lh не иАет ни в какое сравне-
сравнение с более мощными вос-
восстановителями, например с
Рис. 55. Диаграмма потенциал—рН для растворов щелочными или щелочнозе-
перекиси водорода в воде при указанной концен- мельными металлами.
трации (моли/л) и давлении (атмосферы). На риСж 55 в графичес-
графическом виде представлены дан-
данные, которые могут быть выведены из электродных потенциалов. График
практически в такой же форме опубликован Делахеем, Пурбе и Ван-Ри-
сельбергом [5]. График потенциал —рН, который легко построить [6],
иллюстрирует зависимость потенциала от концентраций перекиси водорода
и водородных ионов. Нанеся на рис. 55 аналогичные кривые для других
отдельных систем, можно сразу же сказать, является ли потенциал благо-
благоприятным для реакции во всем интервале составов или нет.
Электродные потенциалы перекиси водорода, как можно видеть на рис. 55,
при различных концентрациях линейно зависят от рН и изменяются параллель-
параллельно друг другу. Необходимо иметь в виду, что если в качестве исходных вели-
величин взять потенциалы какой-либо другой частицы, например НО2", то кривые
могут оказаться нелинейными и непараллельными. Однако в течение времени,
Термодинамика реакций перекиси водорода 301
лока существует быстроустанавливающееся равновесие между перекисью водо-
водорода и другими видами частиц, практически нет никакой разницы в употребле-
употреблении того или иного графика. Кроме того, если не доказано, что истинным
реагентом является какая-либо другая частица, а не перекись водорода,
выражать изменение потенциала этой частицы (концентрацию которой можно
вывести лишь с большим трудом) не имеет никакого смысла.
После сравнения потенциалов перекиси водорода и того или иного реаген-
реагента может оказаться желательным подытожить полученный материал, как это
показано ниже для системы из серебра и перекиси водорода в интервале кон-
концентраций кислоты от 10~7 до \М в присутствии \М перекиси водорода.
Окисление: Ag —> Ag+ Ag2+ AgO*
* 1
Восстановление: Ag <— Ag* <— Ag2" ч— AgO*
t* I
I
В этой схеме наличие стрелки указывает, что равновесие является таким,
при котором связанные стрелкой вещества могут сосуществовать или при
котором реакция может практически полно протекать в указанном направле-
направлении. Такая же схема может быть выведена и для других пределов концентра-
концентраций.
Относительный термодинамический потенциал
По другому типичному способу выражения склонности перекиси водорода
к реакции не учитывается влияние концентрации, а вместо этого показывается
взаимоотношение реакций перекиси водорода и реакций кислорода в других
валентных состояниях. В качестве примера такого способа приведем следую-
следующую схему окислительного потенциала, взятую у Латимера [4] и показываю-
показывающую значение Е° для группы родственных равновесий:
Кислый раствор
-1,23
I -2,82 —0,72 —1,5 +0,13
2НаО ОН + Н2О Н2О2 НО2 02
! ! 1 1
-1,76 -0,693
-2,8 -1,34
н2о+о2 но+о2 о8
—2,07
-0,89 +0,13
О8 + Н2О НО2 + О2 2О2
I !
-0,38
Щелочной раствор
—2,0 +0,24 —0,41 +0,56
2ОН' ОН + ОН- НОг 07 О2
! i! ; I
—0,87 . 0,08
•302
Глава 7
Данные, полученные из такой схемы потенциала, могут быть представле-
представлены графически по методу, предложенному Фростом [7] и использованному
для графического построения рис. 56. Основу для построения этой схемы дает
соотношение E°=—&F°/n gf, показывающее, что если свободная энергия выра-
выражена в электрон-вольтах A 56=23,06 ккал/моль), то потенциал реакции опреде-
определяется наклоном линии на графи-
графике потенциал—свободная энергия.
Такой график не свободен от извест-
известных ограничений; требуется опреде-
определенный навык для правильного
интерпретирования его [7], причем
он имеет тот недостаток, что излиш-
излишне подчеркивает значение фор-
формальной стадии окисления кисло-
кислорода.
СКОРОСТИ РЕАКЦИЙ ПЕРЕКИСИ
ВОДОРОДА
При исследовании скоростей
реакций и процессов разложения
перекиси водорода обнаруживаются
многие кажущиеся внутренние
несогласованности. Скорости от-
отдельных реакций перекиси водоро-
водорода, а именно реакции с некоторыми
ионами галогенов, можно настолько
точно рассчитать, что они в настоя-
настоящее время считаются классически-
классическими, приводятся в качестве примеров
в учебниках и упоминаются даже
в таблицах Национального бюро
-3
-2 -/
Состояние окисления
Рис. 56. Диаграмма состояние окисления—
свободная энергия для кислородных соедине-
соединений в кислом растворе.
стандартов [8]. Наоборот, часто необычайно трудно добиться даже качествен-
качественной воспроизводимости при измерениях скоростей разложения с гетерогенным
катализатором. Так, металлический кобальт может в течение длительных про-
промежутков времени сохранять пассивность в концентрированной перекиси водо-
водорода и затем совершенно неожиданно приобрести необычайно высокую актив-
активность. Такого рода каталитическое разложение или гомогенное разложение
пара перекиси водорода может протекать со взрывоподобной скоростью, и вместе
с тем чистая перекись вполне устойчива при хранении и обычно характеризуется
[9] как медленный окислитель (если не считать действия ее на наиболее силь-
сильные восстановители). Другой важный вопрос, на который пока еще нет ответа,
следующий: можно ли скорость разложения чистейшей перекиси водорода
в жидком состоянии в обычно встречающихся условиях хранения довести до
нуля. На основе опыта и теории можно, по-видимому, сделать вывод, что потен-
потенциально достижима неизмеримо малая скорость разложения, но на практике
к такому состоянию можно приблизиться лишь асимптотически из-за невоз-
невозможности устранить контакт с поверхностью сосуда, а также полностью
освободить перекись от последних следов примесей.
Твердая перекись водорода является необычайно инертной. Например,
если в 90%-ную перекись водорода при —55° вводить охлажденный 0,5 н.
раствор перманганата, частицы ржавчины или другие катализаторы и не
допускать при этом оттаивания перекиси, разложения совершенно не отме-
отмечается [10]. Разложение начинается только после плавления.
Мы не имели в виду собрать и изложить в виде таблиц данные по скорости
разложения перекиси водорода или реакциям ее с другими веществами;тем-
Скорости реакций перекиси водорода 303
не менее интересно кратко описать интервалы встречающихся скоростей и тем-
температурные коэффициенты, выражаемые в виде энергий активации по Аррениу-
су. Исследования газофазного разложения перекиси водорода при 467—542°,
выполненные Мак-Лейном [11], показывают, что по меньшей мере в некоторой
части разложение является гомогенным. Результаты работ этого автора приво-
приводят к энергии активации от 40 до 50 ккал/моль. Логично предполагать, что
только при таких высоких температурах может иметь место термически акти-
активированное гомогенное разложение перекиси водорода с измеримой скоростью.
Это подчеркивает также Харт [12], который приходит к следующему заклю-
заключению. Рассмотрение термодинамических данных, приведенных на стр. 218,
показывает, что наиболее вероятной начальной стадией гомогенного разложения
перекиси водорода является диссоциация на два гидроксильных радикала.
Если принять изменение энергии для этой реакции в качестве мерила энергии
активации и исходить из предэкспоненциального множителя в уравнении Ар-
рениуса 1014, можно рассчитать, что при комнатной температуре за 1 сек.
такой диссоциации подвергнется только одна молекула перекиси водорода
на 1027 молекул. При температуре около 300° этим путем диссоциирует одна
молекула перекиси на 10е молекул в 1 сек., что может уже обусловить умерен-
умеренную скорость разложения, если за каждой стадией инициирования последует
цепная реакция заметной длины. При 600° за долю секунды даже в отсутствие
цепных реакций должна разложиться вся перекись водорода. Если ввести
даже поправку на возможные погрешности, то все же такой подсчет совершенно
отчетливо доказывает, что в описанных в гл. 8 исследованиях разложения
в паровой фазе при более низких температурах B5—420°) фактически изучалось
только гетерогенное разложение. Соответствующие энергии активации лежат
в интервале 4—20 ккал/моль.
Этот вывод можно приложить и к разложению в жидкой фазе. Как показа-
показано на стр. 376, температурный коэффициент скорости разложения тщательно
очищенной пробы перекиси водорода в сосуде из стекла пирекс соответствует
энергии активации около 15 ккал/моль, а поэтому процесс должен быть в ос-
основном гетерогенным. Гомогенные реакции перекиси водорода в жидкой фазе
с другими веществами также имеют энергии активации, лежащие внутри ука-
указанного интервала, например реакция перекиси водорода с ионами различных
галогенов характеризуется энергиями активации 10—27 ккал/моль [13].
Эти величины можно сравнить с полученными при исследованиях реакций
различных замещенных перекисей. Так, гомогенное разложение ряда переки-
перекисей, например диэтилперекиси, дибензоилперекиси и ди-тре/гс-бутилперекиси,
гидроперекиси кумола и пероксосульфата, протекающее с гомолитическим
разрывом связи О—О в газовой или жидкой фазе, обладает энергиями актива-
активации около 30—40 ккал/моль. Энергии активации при реакциях с другими
веществами лежат ниже этого интервала и составляют всего лишь 10 ккал/моль.
Более детальное сравнение различных перекисей в одном и том же процессе,
а именно при реакции с ионом закисного железа, приведено на рис. 57, взятом
у Орра и Вильямса [14]. Как показывают приведенные на рисунке данные,
для этой частной реакции между предэкспоненциальным множителем и энер-
энергией активации имеется прямая пропорциональность. При заданной температу-
температуре это соотношение показывает, что все приведенные на рисунке заместители
в перекиси водорода снижают скорость реакции. В данном случае положение
как раз обратно наблюдаемому при некаталитическом гомогенном разложении
органических перекисей, где другие перекиси разлагаются быстрее, чем пере-
перекись водорода. Орр и Вильяме [ 14] интерпретируют результаты реакции с ионом
закисного железа (рис. 57) таким образом, что отталкивание избыточного элек-
электрона в связь О—О снижает энергию активации за счет увеличения числа элек-
электронов, способных к координации с закисным железом в активированном ком-
комплексе.
304
Глава 7
Уолш [15] дал общее правило для сравнения стабильности разных пере-
перекисей: он считает, что до известного предела энергия связи О—О возрастает
за счет сообщения ей отрицательного заряда. Еще ранее Уолш [16] описал ряд
наблюдений, подчиняющихся этим общим положениям. Например, перекиси
ацилов заметно разлагаются даже при комнатной температуре, причем они
более реакционноспособны, чем перекиси арилов (ацильные заместители явля-
являются акцепторами электронов, а арильные—донорами); диал кил перекиси ус-
12 тойчивее соответствующих
гидроперекисей, а послед-
последние в свою очередь разла-
разлагаются с ббльшим трудом,
чем перекиси оксиалкилов
ROOCH2OH; у гидропере-
гидроперекисей алкилов стабиль-
стабильность возрастает в ряду:
первичные, вторичные, тре-
третичные. Уолш [15] подчер-
подчеркивает, что перенос заряда
влияет на прочность связи,
но не обязательно влияет
на энергию диссоциации
связи и что энергию акти-
активации нельзя отнести толь-
только к разрыву связи О—О.
Принцип переноса за-
Р и с. 57. Предзкспоненциальные множители в урав- РяДа имеет, безусловно,
нении Аррениуса для реакции перекисей с ионом за-
кисного железа, по Орру и Вильямсу [14].
10 11 12 13
Энергия активации Е, ккал/маль
14
Гидроперекиси: нк—нитрокумола; к—кумола; пм—n-ментана; пик—
n-из опропилкумола; фцг—фенилциклогексана; птб—п-трет-
бутилкумола.
большое значение; но эти
явления еще не совсем
ясны и не связаны в согла-
согласованную надежную тео-
теорию. Например, перекись
бария, почти вся построенная из ионов, является стабильной. Работы же,
цитированные ниже, показывают, что введение электронодонорных замес-
заместителей в перекиси ацилов ускоряет разложения. По-видимому, любые
такие сравнения действительны лишь для аналогичных процессов, т. е.
для разрыва одной и той же связи в одинаковой среде за счет только
гомогенного или гетерогенного процесса с участием того же или эквивалент-
эквивалентного реагента, инициатора или катализатора. Так, если рассматривать
гомогенное разложение в газовой фазе, то органические перекиси, по-видимо-
по-видимому, менее устойчивы, чем перекись водорода. Наоборот, пример реакций
с ионом закисного железа показывает, что перекись водорода является наиболее
реакционноспособной из всех изученных перекисей. В частности, нужно
различать чувствительность какой-либо перекиси к взрыву или детонации
и скорость, с которой она подвергается реакции в строго определенных
условиях.
МЕХАНИЗМ РЕАКЦИЙ ПЕРЕКИСИ ВОДОРОДА
Реакцию перекиси водорода в соответствии с вышеприведенными клас-
классификациями можно (по крайней мере в принципе) описать с высокой точностью
и определенностью. Наоборот, гораздо меньше уверенности во всем, что касается
механизма реакции, т. е. в отношении промежуточных состояний, в которых
находятся участвующие в реакции атомы и электроны. Только в самое послед-
последнее время начали удовлетворительную разработку теории этого вопроса, и
Механизм реакций перекиси водорода 305
можно ожидать, что в дальнейшем результаты работ будут значительно улуч-
улучшены и уточнены. Ниже приводится ряд предположений, касающихся меха-
механизмов реакции. Пожалуй, наиболее важным выводом является тот, что ни оби-
обилие экспериментальных фактов, ни наилучшим образом разработанные теории
не позволяют пока сделать никаких общих выводов, по которым можно было
бы заранее предсказать механизм реакций перекиси водорода или хотя бы одно-
однозначно их классифицировать.
Итоговые уравнения реакции как описание механизма
В химии перекиси водорода имеются прекрасные примеры, иллюстрирую-
иллюстрирующие, насколько итоговые реакции мало дают в отношении описания действитель-
действительного механизма. Если рассмотреть реакцию, изображаемую уравнением C),
т. е. реакцию окисления перекиси водорода перманганатом калия, то немедленно
встает вопрос, действительно ли при этом происходит взаимодействие десяти
молекул. Реакцию C) можно написать также несколько иным путем, а именно
с добавлением одной молекулы серной кислоты, и представить конечный про-
продукт—калиевую соль—в виде бисульфата KHSO4. Эта двусмысленность устра-
устраняется, если учесть, что данная реакция обычно проводится в водном растворе,
где некоторые реагенты и продукты ионизированы. Таким образом, можно
вообще не упоминать о ионах калия, сульфата и бисульфата, поскольку они
не участвуют в реакции, и представить реакцию.
2МпО4- + 5Н2О2 + 6Н+ ^> 2Мп2+ + 8Н2О + 5О2. A4)
Однако такой метод в одном отношении еще больше осложняет дело, а именно
место прежних десяти молекул теперь заняли тринадцать. Кроме того, не-
неизвестно, в какой точке нужно остановиться в смысле применения ионизации.
Известно, что перекись водорода может слабо ионизировать в водном растворе
с образованием ионов водорода и пергидроксила. Перманганат калия можно
с полным правом представить как полностью ионизирующееся вещество, а пере-
перекись водорода слабой кислотой, и если реакция проводится не в щелочном
растворе, то в последнем должно присутствовать очень малое количество пер-
гидроксильных ионов. Можно, правда, утверждать, что пергидроксильный ион
является реакционноспособной формой даже в случае присутствия его
в небольших количествах. Не вдаваясь в обсуждение правильности такого
высказывания, следует лишь указать, что если оно справедливо, то это должно
привести к дальнейшему увеличению числа реагирующих молекул в итоговом
уравнении.
Ступенчатые реакции
Данный выше элементарный обзор показал, насколько итоговое уравне-
уравнение мало подходит для описания механизма реакции. Объяснение несоответ-
несоответствия между этими уравнениями и механизмом заключается в том, что итоговые
реакции являются только суммой ряда отдельных реакций. Указывается, что
в случае применения перекиси водорода в качестве восстановителя, например
при обсуждавшемся выше восстановлении перманганата, перекись водорода
сначала окисляет субстрат, а затем происходит его восстановление действием
воды. Это выражается уравнениями
-» 2М2+ + 2Н2О, A5)
2М2+ + 2Н2О -> 4Н+ + 2М + О2. A6)
Против этого предположения можно выдвинуть сразу два возражения. Часто
кажется невероятным, чтобы окисленное вещество при восстановлении пере-
перекисью водорода с самого начала подвергалось бы еще большему окислению.
20 Перекись водорода
306 Глава 7
Если даже в связи с весьма неблагоприятными энергетическими соотношениями
необходимо принять возможность образования в минимальном количестве
более высокой степени окисления, остается в силе другое возражение, а имен-
именно, что обе реакции, A5) и A6), в действительности включают уже известный
механизм, описанный реакциями F) и A0), выражающими потенциалы
отдельных систем. Между тем форма этих реакций также не имеет ничего обще-
общего с механизмом, поскольку они представляют только итоговые результаты
в отношении энергии, а также природы реагентов и образующихся продуктов.
Приведенные ниже результаты опытов с изотопом кислорода как индикатором
также с совершенной очевидностью доказывают несостоятельность механиз-
механизма реакций A5) и A6).
Можно привести ряд других частных реакций, причем в отсутствие ка-
каких-либо дополнительных оснований для выбора комбинации таких реакций,
а возможно и других, безусловно, заслуживают внимания.
A7)
ОН + ОН"" -> Н2О2 + е, A8)
+ 2ОН--»НОГ + Н8О + е, A9)
НО2~+ ОН" -» О2" + Н2О + е, B0)
B1)
Если бы даже не существовало прямых экспериментальных доказательств
существования некоторых из этих промежуточных продуктов, все же имеет
смысл учесть возможность их существования для упрощения реакций с точки
зрения снижения числа участвующих молекул.
При вышеприведенном анализе механизма вскользь упомянута еще одна
мысль. Часто высказывается предположение, что в реакциях с перекисью
водорода активную роль могут играть необычайно малые количества высоко-
окисленного субстрата, обычно не встречающегося. Эта гипотеза, как показано
в следующей главе, используется, пожалуй, в еще большем размере при объ-
объяснении процессов разложения. Она является приемлемой в связи с довольно
высоким окислительным потенциалом перекиси водорода. Поскольку отсут-
отсутствуют какие бы то ни было прямые доказательства невозможности такого
окисления, единственным ограничением, которое нужно учесть при анализе
этой проблемы, является скорость реакции, возможная при небольших кон-
концентрациях образующегося окисленного субстрата. В принципе можно рассчи-
рассчитать, какой должна быть та небольшая концентрация, которая была бы в со-
согласии с наблюдаемой скоростью; однако надежный расчет, вероятно, в свою
очередь потребует подробного знания механизма реакции, а поэтому указанной
цели достичь нельзя. Часто выдвигалась еще и другая мысль, несколько похо-
похожая на высказанную выше. Возможно, что перекись водорода реагирует с мо-
молекулами субстрата с образованием перекисей или гидроперекисей, являю-
являющихся более сильными окислителями, чем перекись водорода. В тех случаях,
когда выдвигалась такая гипотеза, редко можно было показать фактическое
существование такого рода перекиси или определить ее термодинамический
потенциал.
При рассмотрении этих идей целесообразно использовать новейшие ревизии
концепций окисления и восстановления [17, 181, Согласно Мюлликену [19],
термины «окисление» и «восстановление» лучше всего применять лишь для
описания итоговых процессов и формального приписывания тех или иных заря-
зарядов атомам. Для исследования фактического или предположительного меха-
механизма реакции целесообразнее ввести концепцию склонности к притяжению
или отталкиванию электронов. Некоторые аспекты этого вопроса имеютзна-
Механизм реакций перекиси водорода 307
чение при толковании реакций перекиси водорода, и на них мы остановимся
ниже.
Положение атомов и распределение электронов. В старых теоретических
работах часто указывалось на мнимую геометрическую недоступность кисло-
кислорода в перекиси водорода для реакции, что привело к защите мнения о суще-
существовании тригональной структуры перекиси. Развитие наших знаний тормо-
тормозилось несовершенными наблюдениями. Так, Банкрофт [20] писал: «Химия
перекиси водорода является совершенно безнадежной темой для феноменологи-
феноменологического экспериментатора, или экспериментатора в духе Бэкона, так как
повсюду налицо неправильно ориентирующие опыты». Это интересное поло-
положение многократно цитировалось и даже переведено на французский язык Тис-
се [21]. Оно, по-видимому, означает, что путем теоретических рассуждений
истины можно добиться скорее, чем путем постановки опытов. Нет никакого
сомнения в том, что в огромной литературе по перекиси водорода имеется
большое число сообщений с неудовлетворительным изложением опытов, про-
проведенных к тому же недостаточно точно и охватывавших только очень узкий
интервал условий. Тем не менее можно сделать точные наблюдения, а послед-
последние правильно истолковать. Основой такого толкования должна быть электрон-
электронная теория, использованная на стр. 266 для определения структуры. При таком
подходе, помимо относительного положения атомов, необходимо учитывать также
распределение электронов вокруг этих атомов. Разработка этой теории про-
проводилась в большей степени химиками-органиками [22, 23], чем физико-хими-
ками или химиками-неорганиками, но развитие ее и приложение к реакциям
перекиси водорода вполне возможно.
При изучении и описании положения атомов и распределения электронов
в течение какой-либо реакции целесообразно ее рассматривать как состоящую
из ряда отдельных стадий, хотя экспериментально эти стадии нельзя отделить
друг от друга. Так, Кулсон [24] делит реакцию на следующие стадии: 1) на-
начальную, т. е. сближение реагентов до начала реакции; 2) стадию поляризации,
при которой реагенты еще различимы, но находятся в возмущенном состоянии;
3) переходную, в течение которой энергия проходит через максимум, реагенты
же перегруппировываются и сливаются с образованием переходного комплек-
комплекса, и 4) конечную, при которой выделяются продукты. Таким образом, для
окончательного химического изменения перекиси водорода требуется наличи^
последовательных или параллельных стадий образования активированного
комплекса и разрыв связей в продуктах начальной реакции или в этих
продуктах с присоединившейся перекисью водорода. В] некоторых случаях
вообще не требуется разрыва связей перекиси водорода, в других должна
быть разорвана одна, две или даже все связи.
Большое значение имеет величина энергии, необходимой для образования
активированного комплекса [25], так как она определяет скорость реакции
или даже вообще возможность ее протекания. Энергия, которая может быть
использована для этой цели, не является неограниченной, причем при терми-
термических реакциях она получается за счет кинетической энергии реагирующих
молекул. Обстоятельства, позволяющие рассматривать возможность механизма,
основанного на разрыве прочных связей, обусловлены тем, что разрыв связи
(или любая из других реакционных стадий) не протекает в виде дискретного
акта. Нет необходимости подавать всю эту энергию, требующуюся для полного
разрыва связи, поскольку разрыв и образование связи протекают одновременно.
Этот процесс не ограничен разрывом и образованием ковалентных связей, цо>>
как указывает Ингольд) в работе [22] на стр. 44), может быть обусловлен вме-
вмешательством растворителя (как, например, при разрыве ковалентной связи
и одновременной сольватации ионов). \
Характерной, имеющей очень большое значение чертой, которую следует
учесть при любых анализах механизма, является распределение зарядов в моле-
20*
308 Глава 7
кулах, вступающих в реакцию. Для перекиси водорода наиболее существенным
фактором является концентрация отрицательного заряда в кислороде. Она
может быть выражена различным образом в зависимости от точки зрения того
или иного автора или предлагаемого механизма реакции. Например, можно
в основном исходить из полярной природы ОН-связи. Можно также базировать-
базироваться на присутствии неподеленных пар электронов у атомов кислорода. Обычно
это выражается ссылкой на электроноакцепторную природу кислорода, т. е.
на его электроотрицательный характер. Необходимо отметить, что электро-
электроотрицательность—относительное понятие, пока еще не поддающееся точному
определению. Например, изолированный атом . (или группа) рас-
рассматривается как частица, обладающая известной электроотрицательностью.
Однако в том случае, когда происходит какая-либо реакция или молекулярное
взаимодействие, относительная величина этой электроотрицательности изме-
изменяется. Как подчеркивает Сэндерсон [26], логично предполагать, что при обра-
образовании химической связи средняя электронная плотность взаимодействующих
атомов изменяется в молекуле таким образом, чтобы придать им одинаковую
электроотрицательность. Такой процесс ограничивается возможной степенью
переноса электронов; при комбинировании группы, имеющей высокую электро-
электроотрицательность, с другой группой, обладающей низкой электроотрицательно-
электроотрицательностью, может оказаться, что даже полный перенос электронов не в состоянии
обеспечить равенство; например, перекисный ион О*~ сохраняет электроотри-
электроотрицательность в самых различных комбинациях.
При анализе вопроса о распределении зарядов необходимо различать
кинетический порядок реакции и ее молекулярность. Кинетический порядок
касается лишь того порядка концентрации, которому должна быть пропорцио-
пропорциональна наблюдаемая скорость реакции. Молекулярность реакции обозначает
число молекул, принимающих участие в той или иной стадии реакции. Термин
«принимать участие» интерпретируется по-разному, особенно с точки зрения
участия растворителя. Ингольд (работа [22], стр. 315, 356) в стремлении дать
более строгое определение ограничивает понятие молекулярность «числом
молекул, обязательно подвергающихся изменению ковалентности». Другие
авторы, например Свен [27], подчеркивают значение взаимодействий какого-
либо реагента с растворителем или с другими растворенными веществами,
вследствие чего он становится готовым к реакции. Взаимодействие со смеж-
смежной молекулой, которую обычно считают инертной, способно, по всей вероят-
вероятности, вызвать такое перераспределение заряда в молекуле перекиси водорода,
что один конец молекулы становится положительнее другого; это облегчает
возможность приближения другой молекулы или иона и его сольватацион-
ной оболочки, а следовательно, и реакцию. В данном случае реакцию можно
рассматривать как связанную с направленным отталкиванием и притяжением
под действием растворителя и реагента и считать ее тримолекулярной.
Ингольд [22] придерживается мнения, что такого рода взаимодействия
являются слишком расплывчатыми и общими, чтобы их можно было исполь-
использовать при классификации типа реакции. Эти положения и противоречия
проанализированы в обзорном сообщении Ремика [28]. Другие растворенные
вещества и растворители также могут играть роль в механизме реакции; так,
перенос электронов от Сг2+ к Fe3+ (окисление) катализируется присутствием
ионов хлорида [29]. Вся эта группа эффектов, обусловленная электроста-
электростатическими или другими силами, удачно названа Мюлликеном [19] термином
«содействие среды».
Высказанные мысли в значительной мере являются еще новыми и не сво-
свободными от противоречий. В большой доле они еще носят интуитивный харак-
характер, но главные предпосылки их правильны и дают подходящую основу для
анализа механизма. Мюлликен [19] рассматривает терминологию в этой обла-
области, а также обоснованность различных точек зрения на механизм. В этом
Механизм реакций перекиси водорода 309
сообщении показано также, как далеко можно пойти с систематизацией. Прав-
Правда, многих может не удовлетворить сложность принятой номенклатуры.
Характеристика перекиси водорода в ее реакциях
Образование пероксигидратов. Простейшей реакцией перекиси водорода,
по крайней мере на первый взгляд, является присоединение ее молекулы как
целого к другой молекуле, т. е. образование пероксигидратов. При аналогич-
аналогичном образовании гидратов обычно считаются возможными различные способы
связывания, и гидратационная или кристаллизационная вода поэтому класси-
классифицируется как координационная вода, анионная вода, вода кристаллической
решетки или' цеолитная вода. Имеющегося экспериментального материала
слишком мало, чтобы установить такие же категории для аналогичных соеди-
соединений с перекисью водорода, особенно для менее четко определенных случаев
связывания в кристаллической решетке или цеолитного связывания. Беглое
наблюдение Вильштетера [30], касающееся образования квасцами перокси-
гидрата, возможно, представляет пример введения перекиси водорода в ре-
решетку, но исследование, проведенное Мюнцбергом [31], заставляет усомниться
в правильности этого положения. Недавно сделано наблюдение [32], что амин-
ный пермутит в состоянии абсорбировать перекись водорода. Неясно также,
образуется ли стабильная координационная связь с молекулой перекиси водо-
водорода в целом. Усовершенствование представлений [33] об электронной природе
координационной связи, вероятно, сумеет пролить свет и на этот важный во-
вопрос. Указывается, что нельзя осуществить координационное связывание пере-
перекиси водорода без существенного снижения стабильности молекулы. По-види-
По-видимому, смещение заряда от атомов кислорода, требующееся для образования
координационной связи, должно или вызвать разрыв молекулы, или способ-
способствовать увеличению ее реакционной способности. Такого рода функция, ве-
вероятно, выполняется ферментами пероксидазой и каталазой; в этом отношении
интересно отметить, что почти все тяжелые металлы являются превосходными
катализаторами разложения. Если образование координационной связи спо-
способствует разрыву валентной связи, то весьма логично предположить, что
разрыв произойдет по связи водорода с кислородом, в результате чего получит-
получится пергидроксильный ион.
Несомненно, существующие пероксигидраты правильнее всего представ-
представлять как соединения с водородной связью, аналогичные вышеупомянутому
классу соединений с анионной водой. Механистически процесс образования водо-
водородных связей прост. Природа водородных связей в перекиси водорода кратко
проанализирована при рассмотрении ассоциативных свойств перекиси на стр. 290
и ел. Показано, каким образом электроотрицательный характер кислорода,
полярность связи ОН и малые размеры атома водорода способствуют образова-
образованию таких связей. Указывается также, что эти связи слишком слабы для дли-
длительного сохранения в газовой или жидкой фазе; они сохраняются только
в твердых телах. Из этих свойств вытекает, что пероксигидраты, так же как
гидраты, могут быть образованы соединениями, содержащими высокоэлектро-
высокоэлектроотрицательные атомы, например азот, кислород и фтор. Ссылки на литературу
по этим пероксигидратам см. на стр. 534—535. Большая часть опубликован-
опубликованного материала касается только существования этих соединений, и мало что
известно о механизме их образования, который принимается почти одно-
однозначным надлежаще ориентированным столкновениям и прилипанию. Во мно-
многих случаях данные о существовании пероксигидратов требуют еще подтвер-
подтверждения. Мюнцберг [31] путем микроскопического исследования показал, что
многие пероксигидраты, упоминаемые в литературе, в действительности пред-
представляют лишь включения маточного раствора в кристаллическую массу,
и сообщил об отрицательных результатах, полученных при испытании большого
310 Глава 7
числа солей на образование пероксигидратов. Интересно отметить, что, по
наблюдениям Гузена [34], касающимся природы образующихся связей, спо-
способность фосфатов связывать перекись водорода возрастает в следующем
ряду: первичные фосфаты/ вторичные фосфаты, пирофосфаты. При любом типе
соли калиевая соль прочнее связывает перекись водорода, чем натриевая.
Мюнцберг [31] вывел правило, что связывать перекись водорода способны
только двуосновные и многоосновные кислоты. В результате исследования [35]
образования пероксигидратов шерстью показано, что перекись водорода свя-
связывается аминогруппами прочнее, чем карбоксильными или гидроксильными
группами. Сделано также заключение [36], что поглощение перекиси водорода
ионообменными смолами обусловлено аминогруппами. По наблюдениям Леви
и Баттальино [37], гидратированные пероксигидраты удерживают перекись
водорода прочнее, чем воду.
Процессы разрыва валентной связи. В других реакциях перекиси водорода
разрываются одна или несколько валентных связей молекулы, причем наряду
с этим могут происходить и некоторые процессы переноса электронов. Можно
различать пять возможных процессов, приводящих к разным результатам. Их
можно выразить следующими уравнениями, в которых точки обозначают элек-
электроны ковалентной связи, а пунктирные линии—место разрыва:
НО Х'ОИ -— 2ОН B2)
НО : ;ОН — OH" + ОН+ B3)
НОО >;'Н — НО2 + Н B4)
НОО s |н — НО; + Н+ B5)
hOOJ : Н — НО+ +• н- B6)
Оба случая, в которых электроны оказываются поделенными между продук-
продуктами [реакции B2) и B4)], представляют собой примеры гомолитического раз-
разрыва валентной связи. Поскольку каждая частица обладает в этом случае не-
спаренным электроном, такого рода механизмы носят название свободноради-
кальных. В трех других случаях, когда оба электрона разорванной связи
остаются при одной из получающихся частиц [реакции B3), B5) и B6)], про-
происходит образование заряженных частиц. Эти процессы называет ионными
механизмами; они протекают за счет гетеролитического разрыва связи. Можно
написать и другие процессы ионизации, связанные с отрывом электрона от
молекулы как целого, однако энергия, необходимая для такой ионизации *
(см. стр. 274), во много раз выше, чем для представленных реакций, и поэтому
ни для одного известного вида химических реакций нельзя предположить
механизм такого рода. С развитием исследований процессов разложения,
вызываемых ионизирующим излучением, может оказаться, что такого рода
ионы действительно играют определенную роль [38]. Ион Н2О7, упоминаемый
Вейсом [38], трудно себе представить, хотя возможна комбинация электрон-
электронного удара, расщепления перекиси водорода и присоединения электрона к одной
из образовавшихся частиц. Реакция B6), связанная с образованием гидридного
иона, для перекиси водорода маловероятна и здесь не рассматривается.
Часто для того или иного комплекса реакционных условий не сразу
удается выбрать наиболее вероятную из реакций B2)—B5), хотя и существуют
определенные принципы для такого выбора. В процессах разложения, про-
протекающих в паровой фазе (безразлично, при термическом или фотохимическом
разложении), наиболее вероятной является реакция B2), следующее место
за ней по вероятности занимает реакция B4). Весьма возможно, что реакции
с другими веществами в паровой фазе также протекают исключительно за счет
Механизм реакций перекиси водорода 311
свободных радикалов. В жидкой фазе факторы содействия среды сообщают
большую вероятность реакциям B3) и B5). Это обусловлено снижением не-
необходимости прямого поступления энергии для осуществления этих ионных
реакций; как указано выше, температурный уровень при реакциях в жидкой
фазе слишком низок для возможности значительного термического активиро-
активирования разрыва на свободные радикалы в отсутствие какого-либо вспомогатель-
вспомогательного фактора. Однако если подавать в жидкость энергию в виде фотохимиче-
фотохимического или ионизирующего излучения, то свободнорадикальные реакции ока-
оказываются также возможными.
Поскольку электронные оболочки перекиси водорода заполнены, то, чтобы
сделать ее способной к захвату новых электронов, необходимо предваритель-
предварительное расщепление молекулы. Точно так же, чтобы создалась возможность отдачи
электрона или деления его, вероятно, требуется разрыв молекулы. После этого
основное внимание концентрируется на более своеобразном или единственно
реакционноспособном осколке. В случае реакции B3)—это ион ОН+, электро-
фильная или электроноакцепторная частица, аналогичная, например, иону
СН*. В реакции B5)—это ион НО", нуклеофильная или электронодонорная
частица. Гидроксильный свободный радикал, вероятно, нужно считать элек-
троноакцепторной частицей, тогда как пергидроксильный свободный радикал
может играть роль и акцептора и донора электронов. Другие особенности
процессов с участием свободных радикалов описаны Уотерсом [39]. Тобольский
и Месробян (см. работу [40], стр. 57—59) сравнили между собою свободно-
радикальный и ионный механизмы для органических перекисей.
Перенос электронов. Выше указано, что в течение реакции обмен электро-
электронами может сопровождаться разрывом валентных связей. Это открывает новый
путь типизации реакций перекиси водорода, предложенный Брауном [41],
который указывает, что в том случае, когда перекись водорода функционирует
в качестве окислителя, она захватывает два электрона, тогда как некоторые
другие окислители принимают только один. Браун использовал это явление
как основу для классификации и предложил для обозначения его термины:
«монодэлектроиатор» и «дидэлектронатор». Такое различие действительно
имеет значение, но желательно применение менее громоздких терминов,
а именно «одноэлектронный обмен» и «двуэлектронный обмен». Требуется также
некоторое видоизменение и уточнение определений Брауна. При любых ито-
итоговых реакциях окисления—восстановления с участием перекиси водорода
происходит обмен двумя электронами, но процессы можно представить проте-
протекающими либо в две стадии, либо в одну. Таубе [42] дал весьма четкое опре-
определение. В качестве примера он приводит некоторое итоговое изменение, кото-
которое формально можно представить следующим уравнением:
А2+ + В = А + В2+. B7)
Эта реакция, по-видимому, протекает через активированный комплекс АВ2+.
Различие между одноэлектронным и двуэлектронным обменами не связано
с перемещением электронов в комплексе, а лишь с вопросом о том, можно ли
доказать существование промежуточных частиц А+ и В+. Если такого рода ча-
частицы действительно существуют и могут быть обнаружены, то реакция проте-
протекает при помощи одноэлектронных обменов. Если такое доказательство не
может быть приведено, то предполагается, что мы имеем дело с двуэлектрон-
двуэлектронным обменом.Таубе указал, что нечувствительность методов испытания*
на такого рода промежуточные продукты, как А+ и В\ может привести к оши-
ошибочным выводам. В связи с этим имеются некоторые расхождения в мнениях
* С другой стороны, методы определения наличия свободных радикалов как про-
промежуточных продуктов весьма чувствительны; Тобольский и Месробян [40], например,
приводят 11 методов доказательства их присутствия.
312 Глава 7
относительно того, может ли перекись водорода реагировать как за счет одно-
электронных, так и за счет двуэлектронных обменов. Юри [43] подтверждает
мнение Габера и Вейса, что перекись водорода никогда не реагирует с субстра-
субстратом, требующим двувалентного обмена, или одновременно с двумя субстратами,
требующими моновалентных обменов, с прямым образованием кислорода или
воды. С другой стороны, и Таубе [42, 44] и Христиансен [45] приводят дока-
доказательства, что перекись водорода действительно вступает в двуэлектронные
обмены. Кахил и Таубе [46] дали очень подробный обзор проблем, связанных
с этим вопросом. Эти авторы попытались определить при помощи опытов с изо-
изотопом кислорода в качестве меченого атома, происходит ли окисление иона
закисного железа перекисью водорода как одноэлектронный процесс с образо-
образованием окисного железа или как двуэлектронный с получением четырехвалент-
четырехвалентного железа, т. е. протекает ли реакция B8) или B9) в одну стадию с образо-
образованием лишь одного активированного комплекса:
Fe2+ + Н2О2 = Fe3+ + ОН + ОН", B8)
Fe2+ + Н2Оа = Fe4+ + 2OH". B9)
Начальная стадия разрыва связи в молекуле перекиси водорода должна быть
одной и той же в обоих случаях.
Совершенно ясно, что если бы четырехвалентное железо было конечным
продуктом реакции, то этот итоговый результат можно было бы получить
также за счет реакции B8), как первичной, с последующим переносом электрона
от иона окисного железа к гидроксильному радикалу. Однако если последнее
взаимодействие не количественное, то какая-нибудь другая реакция гид-
роксильного радикала, весьма вероятно, изменяет кинетику процесса или
его итог. Кахил и Таубе считают, что окисление действительно протекает
по обоим механизмам, т. е. с переносом как одного, так и двух электронов.
Этот вопрос с различных точек зрения изучен также Хиггинсоном, Сат-
тоном и Райтом [47], причем на основании опыта они пришли к интересным
выводам, что скорость окисления различных ионов многовалентных металлов
перекисью водорода лучше согласуется с электродными потенциалами метал-
металлов для одноэлектронных стадий, чем для двуэлектронных. Хотя они и ука-
указывают, что найденная зависимость может оказаться несправедливой, но
все же предпочитают механизм одноэлектронных обменов. При этом надо учи-
учитывать ограничения, присущие описаниям, базирующимся на электронном
порядке. В отношении концепции окисления и восстановления, как сообща-
сообщалось выше, часто трудно однозначно выяснить ход фактического переноса элек-
электронов. Эта концепция, по мнению указанных авторов [47], иногда эквива-
эквивалентна лишь утверждению, что протекает итоговая реакция окисления или
восстановления, хотя применяемые термины как будто бы свидетельствуют
о знании механизма этого процесса во всех подробностях.
Этот анализ терминологии и ее недостатков вновь иллюстрирует трудности
сведения в одну систему представлений о механизме, различающихся по
уровню знания отдельных подробностей. Например, реакцию окисления
закисного железа в окисное с образованием гидроксильного иона и гидро-
ксильного радикала из перекиси водорода можно классифицировать по ион-
ионному механизму или по механизму с участием свободных радикалов в зави-
зависимости от того, как происходит взаимодействие перекиси водорода с ионом
Fe2+: по реакции B2) или B3). Наиболее вероятен ионный механизм, хотя
в данном случае и нельзя подробно проследить пути обмена электронами. В то
же время эта реакция имеет важное значение для образования свободных ра-
радикалов, причем последующие реакции после начальной диссоциации перекиси
на свободные радикалы могут быть такими же, как и при ионном механизме.
Кроме того, возможно, что ионные продукты ассоциированы между собою.
Таким образом, при описании ионной реакции, связанной]с переносом электро-
Механизм реакций перекиси водорода 313
на с образованием какого-то свободного радикала и комплексного иона, воз-
возможна значительная путаница. Частично эта неопределенность зависит от
точки зрения; в данном примере интерес концентрируется на гидроксильном
свободном радикале как продукте. Пожалуй, нелишне отметить, что свобод-
свободные радикалы вовсе не заслуживают специального статута как необыкновенные
молекулы. Безусловно, они отличаются очень высокой реакционной способ-
способностью, однако их структура вполне объясняется теорией валентности; сей-
сейчас уже прошло то удивление, с которым они были встречены при первом их
обнаружении [39]. Кроме того, некоторые ионы, например ион закисного же-
железа или Hg\ также можно называть свободными радикалами или радикаль-
радикальными ионами.
Другой особенностью перекиси водорода, увеличивающей трудности оп-
определения механизма реакции ионного разрыва связи О—О, является иден-
идентичность обеих половин молекулы. У других перекисей, типа ROOR' или
ROOH, имеется преимущественное направление переноса заряда. И в элемен-
элементарном, часто приводимом примере с гидроксиламином всегда происходит дис-
диссоциация на' NH* и ОН", а не на NHj и ОН+ ввиду большего электронного
сродства ОН [48]. Для перекиси же водорода, наоборот, тенденция к переносу
заряда в одном направлении отсутствует, если только это не индуцируется
сближением с другими молекулами.
Классификация в соответствии с процессом разрыва связи. На основании
вышеизложенного обсуждения возникает мысль, что первой стадией реакции
перекиси водорода является образование одной из частиц ОН\ HOg, ОН или
НО2 в виде более или менее независимого промежуточного продукта. В лите-
литературе (см. в работе [40] стр. 107—112) обсуждается вопрос о степени незави-
независимости такого рода частиц для аналогичных реакций органических переки-
перекисей. Почти одновременно может происходить электронный обмен. Эта класси-
классификация реакций, выраженных уравнениями B2)—B5) (за исключением НО2),
была предложена Дербишайром и Уотерсом [49], которые придерживаются
мнения, что каждый из этих видов частиц обладает определенной характеристи-
характеристикой, обусловливающей возможность его распознавания. Для классификации
подробного механизма реакций перекиси водорода мы будем придерживаться
следующей практики: реакции, связанные с начальным образованием двух
радикалов ОН, мы называем механизмами с радикалом гидроксила, реакции
же с образованием ОН+ и ОН" обозначаются как механизмы с ионом гидрокси-
гидроксила. Точно так же, если принимается, что первым продуктом является НО2,
то механизм обозначается как механизм с радикалом пергидроксила; наконец,
диссоциации на НО" и Н+ присвоено название механизма с ионом пергидро-
пергидроксила.
Можно заметить, что оба ионных механизма связаны с концепциями кис-
кислотно-основного катализа. Ион Н0'2 представляет собой анион, образующийся
за счет диссоциации перекиси водорода как кислоты. Труднее доказать, что
ион ОН+ существует самостоятельно. Высказано предположение [50] о проте-
протекании реакции
Н+ + НООН = HOOHJ C0)
с последующей диссоциацией этого пероксониевого иона с образованием иона
ОН+:
НООН* = Н2О + ОН\ C1
Однако отсутствие обмена изотопом кислорода между перекисью 'водорода
и водой доказывает, что ион ОН+ как независимый ион не существует. Таким
образом, значительным различием между обоими ионными механизмами являет-
является то, что в одном случае (ОН+) оба кислородных атома молекулы перекиси
водорода оказываются разделенными в самом начале. Если реакция протекает
314 Глава 7
через ион пергидроксила, то кислородные атомы, по крайней мере в начальной
стадии, остаются вместе, а поэтому создается возможность для них сохранять
это связанное состояние при окислении до молекулярного кислорода. Однако
если не применять кислородного изотопа в качестве индикатора, то это различие
может остаться незамеченным. Так, например, и гипохлоритный ион (СЮ")
и молекулярный хлор (С12) реагируют с перекисью водорода с образованием
кислорода и иона хлорида. В первом случае можно представить, что реакция
происходит между С1О~ иОН+ с образованием С1ООН. Во втором случае взаи-
взаимодействие С1+ и HOg также может дать С1ООН. Эта гидроперекись хлора не
выделена, и действительно, как это следует из замечаний, сделанных на стр. 287,
она должна обладать высокой реакционной способностью. Наличие электро-
электроотрицательного хлора должно способствовать и ионизации водорода и отделе-
отделению молекулярного кислорода. Оба ряда предложенных реакций в действи-
действительности могут протекать не совсем точно согласно вышеизложенному, но
в качестве способа представления стадий разрыва связи и ступеней переноса
электронов эта методика вполне подходит; она иллюстрирует, каким образом
может образоваться получающийся молекулярный кислород: исключительно
за счет перекиси водорода или же частично за счет субстрата. Фактически
доказано, что как в случае гипохлорита, так и в случае хлора кислород полу-
получается исключительно из молекулы перекиси водорода [46]; это свидетельству-
свидетельствует о том, что вышепредложенный механизм может быть правильным для моле-
молекулярного хлора, но не для гипохлорита, где в действительности в реакции
должен участвовать НО^. Дальнейшая реакция ионов ОН+ и HOj, безусловно,
не всегда обязательна. Они могут непосредственно привести к гидроксилиро-
ванию или образованию гидроперекиси.
Указаны и другие характеристики реакций перекиси водорода. Часто
наблюдается присоединение кислорода к органическим и неорганическим сое-
соединениям с неподеленными электронами (см. в работе [39] стр. 245). В каче-
качестве примеров можно привести следующие реакции:
As(OHK + Н2О2 = H3As04 + Н2О, C2)
R2S + Н2О2 = R2SO + Н2О. C3)
Работа Галперина и Таубе [51] показывает, что в одном случае такого при-
присоединения атома кислорода, а именно при окислении сульфита до сульфата,
перекись водорода отдает в начальной стадии сульфиту два атома кислорода,
хотя итоговый процесс требует присоединения только одного.
Уотерс (см. в работе [18] стр. 86) указывает, что органические свободные
радикалы, взаимодействующие с перекисью водорода, обладают структурой, спо-
способствующей созданию высокой электронной плотности в области неспарен-
ного электрона. Такие молекулы вызывают развитие цепной реакции; молекулы
же с более равномерным распределением заряда не способствуют такому раз-
разрыву перекиси водорода, сопровождающемуся в дальнейшем образованием
радикалов. Наиболее наглядной реакцией перекиси водорода, протекающей
с образованием гидроксильных свободных радикалов, является окисление
иона металла с повышением его валентности на единицу. Эванс, Баксендейл
и Парк [52] путем инициирования полимеризации показали, что ионы хрома
(II), ртути (I), меди (I), титана (III), марганца (II) и железа (II) способны гене-
генерировать гидроксильные радикалы. Существенное значение для реакции пере-
передачи или развития цепи под действием гидроксильных свободных радикалов
в водном растворе имеет то, что гидроксил может вступать в обменную
реакцию с водой по уравнению
О*Н + НаО = Н2О* + ОН. C4)
Такая возможность [53]доказана]экспериментально [54, 55]. В результате этой
реакции радикал гидроксил может иметь кажущуюся значительную долговеч-
Механизм реакций перекиси водорода
315
ность. Обмен с другими видами частиц может вызвать появление менее актив-
активного свободного радикала, что снижает эффективность процесса.
С другой стороны, радикал пергидроксил может образоваться лишь под
действием окислителя. Возможной роли этого радикала уделялось меньше
внимания, чем роли гидроксила. Основу для суждений о радикале пергидрок-
силе дает работа Бера и Штейна [56], которые исследовали восстановление иона
церия (IV) перекисью водорода и предложили для этого следующие реакции:
Се4+ + Н2О2 = НО2 + Н+ + Се3+, C5)
Се4+ + НО2 = О2 + Н+ + Се3+. C6)
Эти механизмы в корне отличаются от тех, с которыми связана реакция в си-
системе железо—радикал гидроксил. Эти реакции протекают быстро (по сравнению
с медленной реакцией иона закисного железа), и повторное окисление иона
церия (III) совершенно невоз- н
можно. Эта система исследо- •!• н ••• «и •»•
вана при высоких концентра- qC-± q— 0^-J Оп-
Опциях кислоты, причем не /\ */\ /\ */
получено никаких доказа- ^0
тельств наличия реакции
между перекисью водорода
и предполагаемым пергидрок-
силом. Это согласуется с гипо-
тезой, по которой перекись
водорода реагирует только
с надперекисным ионом О",
представляющим продукт кис-
кислотной диссоциации пергид-
роксила. Однако нельзя ска-
зать, что это исследование вое-
становления иона церия (IV)
является совершенно неопро-
вержимым доказательством
существования механизма
с пергидроксильными ради-
калами. Возможен и двуэлек-
тронный механизм с участием
комплексных димеров церия,
предложенный Хейдтом и
Смитом [57].
Распределение электронов
0^-J О
/\ */\
\у # Г
"/9 • /9
i—H
О—
Рис. 58. Схема распределения электронов в р-орби-
тах некоторых водородо-кислородных соединений
и ионов.
В ВОДОрОДО-КИСЛОрОДНЫХ ВИ-
дах частиц. Для подытожи-
вания и обобщения идей, ка-
сающихся общей характерис-
характеристики реакций перекиси водорода, необходимо вновь обратиться к ранее
высказанным положениям относительно электроотрицательного или электро-
ноакцепторного характера кислорода. Весьма целесообразно все время учиты-
учитывать, что это красной нитью проходит через всю химию перекиси водорода.
Рис. 58 дает возможность составить представление об электронном окру-
окружении кислорода в некоторых простейших радикалах и ионах. На этом
рисунке схематически показано вероятное распределение электронов
в трех перпендикулярных 2/?-орбитах кислорода для водородо-кислород-
ных соединений и для ионов, содержащих 1 или 2 атома кислорода. Эффекты
гибридизации при этом не учитываются.
316 Глава 7
Мы видим, что в молекуле воды один кислородный атом приобретает заряд,
сообщаемый ему двумя атомами водорода; в перекиси водорода каждому атому
кислорода сообщается значительно меньший заряд. Эта тенденция в молекуле
кислорода должна была бы продолжаться и дальше, но здесь вмешиваются дру-
другие факторы, способствующие стабилизации этой молекулы за счет образова-
образования трехэлектронных связей. Нужно упомянуть о двух совершаемых иногда
ошибках, обусловленных такого рода связями. Поскольку молекула кислорода
имеет два неспаренных электрона, ее иногда рассматривают как дирадикал,
чем подчеркивают структурную аналогию со свободными радикалами. Однако,
как указывает Уотерс (см. в работе [18] стр. 73), наличие двух трехэлектрон-
трехэлектронных связей в значительной мере сводит на нет такое сравнение; против него
говорит также малая реакционная способность кислорода по сравнению
с реакционной способностью свободных радикалов. Необходимо приложить
значительную энергию, чтобы оторвать эти неспаренные электроны от их
трехэлектронных связей. Другой ошибкой, отмеченной Уолшем [15], является
рассматривание молекулярного кислорода как «двукратно связанного».
В соответствии с этой точкой зрения «размыкание двойной связи» должно спо-
способствовать проявлению полной реакционной способности молекулы кислорода,
причем такая реакционная способность должна наблюдаться и у любого проме-
промежуточного продукта, например у НО2. Однако если рассматривать обе трех-
электронные связи, то легко заметить, что в некоторый момент должен про-
произойти разрыв только одной из них, причем остающаяся связь этого типа должна
придавать значительную остаточную стабильность. На рисунке показано так-
также, почему, согласно указанному предложению, ион надперекиси О\ должен
быть более реакционноспособным, чем радикал пергидроксил НО2. Схема
электронной структуры Оа и НО2 показывает, что, хотя каждый из этих ионов
в состоянии образовать трехэлектронную связь, в ионе надперекиси происхо-
происходит еще дополнительное отталкивание двух параллельных орбит, заполненных
неподеленными парами. Таким образом, нет необходимости предполагать
(см. в работе [18] стр. 74), что при образовании надперекиси RO2 из молекуляр-
молекулярного кислорода проявляется полная реакционная способность кислорода
аналогично тому, как это бывает в насыщенных радикалах в случаях с други-
другими элементами.
Рис. 58 помогает также понять допущение Уолша [15], что хотя перенос
заряда к перекисной группе и увеличивает прочность связи между атомами
кислорода, но это верно лишь с ограничением; дальнейший перенос заряда
вызывает ослабление связи. Имеется ряд аспектов этого вопроса, причем труд-
трудно судить, насколько далеко можно идти в таких рассуждениях. Например,
свободный ион перекиси ОГ имеет структуру, изоэлектронную со структурой
молекулы фтора F2, и можно предполагать, что отталкивание двух рядов
двух параллельных заполненных орбит должно увеличивать реакционную
способность. Такое предположение как будто опровергается сравнительно
малой реакционной способностью перекиси бария. Однако трудно судить, на-
насколько далеко в последнем случае проходит процесс переноса заряда. Оче-
Очевидно, что у иона 01" отсутствует дополнительная стабилизация, вызываемая
наличием добавочных связей в молекуле кислорода, и именно на этом основа-
основании выдвигается положение [46], что отнятие электронов от этого иона упроч-
упрочняет связи между атомами кислорода.
Примеры механизмов реакции перекиси водорода
Механизмы с радикалом гидроксила. Для реакций и процессов разложения
перекиси водорода, протекающих в паровой фазе с термическим или фотохими-
фотохимическим инициированием, или инициированием действием электрического
разряда, предложены механизмы, основанные на образовании радикала гид-
Механизм реакций перекиси водорода 317
роксила. Что касается реакций в жидкой фазе, то этот механизм предложен
лишь для случая фотохимического инициирования. Фрост и Олденберг [58]
доказали фактическое присутствие гидроксильных радикалов при исследовании
разложения паров перекиси водорода в электрическом разряде. Волмэн [59],
исследовавший фотохимическое разложение перекиси водорода в паровой фазе,
и Хант и Таубе [60], изучавшие этот процесс в жидкой фазе, доказали, что
в обоих случаях реакция протекает через образование радикала гидроксила.
Предполагается, что гомогенное термическое разложение перекиси водорода
также протекает в соответствии с этим процессом, однако доказательства явля-
являются либо косвенными, либо результаты не поддаются объяснению в связи
-с одновременным протеканием гетерогенного каталитического разложения,
происходящего по тому же или по какому-либо другому механизму. Мак-Лейн
[11] сообщает, что при температуре около 500° наряду с гетерогенным разло-
разложением происходит еще и чисто гомогенное термическое разложение; отмечено
также взрывное разложение пара, которое, безусловно, развивается по гомо-
гомогенному механизму [61]. Как показано в разделе о кинетике реакции, гомоген-
гомогенное термическое разложение обладает совершенно определенными энергети-
энергетическими ограничениями. Таким образом, термическое разложение, наблюдав-
наблюдавшееся в опытах Стоуна и Тейлора [62] по фотохимическому образованию гидро-
гидроксильных радикалов из перекиси водорода, было, несомненно, гетерогенным.
Отсутствие возможности термического гомогенного разложения пара перекиси
водорода при низких концентрациях и температурах иллюстрируется также
опытами Стоуна и Тейлора, показавших, что окись углерода не реагирует
с паром перекиси водорода при температурах около 100°, несмотря на одновре-
одновременное гетерогенное разложение перекиси. Однако окись углерода интенсивно
реагировала в том случае, когда систему облучали актиническим светом. Ре-
Результаты этой работы доказывают, что не происходит никакого заметного раз-
развития радикалов, возможно генерируемых при гетерогенном разложении,
если последнее протекает при низких концентрациях, и что термическая ак-
активация механизма с радикалом гидроксила невозможна при температуре
100°. Подробности процессов фотохимического и термического разложения
(как гомогенных, так и гетерогенных) описываются в следующей главе. Для
большинства таких процессов считается общепринятым, что основную роль
в них играют механизмы с радикалом гидроксила, причем в вышеприведенном
материале заключаются экспериментальные и теоретические доказатель-
доказательства, подтверждающие такое предположение.
Механизмы с ионом гидроксила. Лишь сравнительно недавно сделано
предположение о том, что реакции перекиси водорода могут протекать по меха-
механизму с ионом гидроксила. Первым, по-видимому, упомянуло такой возможно-
возможности Росс [50]. Он исследовал окисление тиодигликоля и триэтиламина пере-
перекисью водорода в присутствии кислотного катализатора и высказал мысль,
что эффективность перекиси водорода в этом случае обусловлена легкостью
отдачи группы ОН+. Предполагается, что взаимодействие за счет промежуточ-
промежуточного образования пероксониевого иона НООЬЦ протекает быстро, поскольку
при этом не получается иона ОН", что требовало бы разделения заряда на
критической стадии реакции. Эта мысль в дальнейшем была высказана также
Овербергером и Камминсом [63], которые, таким образом, с успехом объяснили
результаты аналогичного исследования образования сульфоокиси из п,п'-
дихлорбензилсульфида при действии перекиси водорода. Те же рассуждения
использованы Дербишайром и Уотерсом [49] для объяснения наблюдений по
гидроксилированию мезитилена перекисью водорода. Эти авторы высказали
предположение, что данный механизм может способствовать также пониманию
реакций, в которых перекись водорода присоединяет атом кислорода за счет
координации с молекулами, обладающими атомами с неподеленной парой
электронов. Эти же соображения были использованы Эдвардсом [13, 64]
318 Глава 7
и по отношению к неорганическим реакциям перекиси водорода в исследовании
по механизму реакций электронодонорных молекул. В общем анализе этого
вопроса Эдварде [64] рассматривает перекись водорода как источник оксиани-
она (HOg), образующего за счет соединения с протонами электронный акцеп-
акцептор, например HOOHJ, безводная форма которого представляет ОН\ В спе-
специальном исследовании реакций перекиси водорода с различными ионами гало-
галогенов Эдварде [13] обобщил большое количество кинетических измерений и
показал, что они согласуются с теориями общего кислотного катализа. Он
показал также [13, 65], что катализ в значительной мере обусловлен действием
растворителя, причем во многих случаях наблюдавшаяся скорость более чем
на 50% была следствием катализа под действием растворителя при рН выше 3.
Автор при этом не защищает никакого определенного механизма, но указывает,
что наблюдавшиеся явления согласуются с тем, что мы здесь называем механиз-
механизмом с ионом гидроксила. В этой работе [65] показано, что кинетический закон
для механизма с ионом гидроксила в то же время согласуется и с таким меха-
механизмом, при котором отсутствует разрыв связи О—О. В этот класс входят
различные примеры окисления перекиси водорода; например, опыты с изото-
изотопом кислорода в качестве меченого атома, проведенные Кахилом и Таубе
[46], показывают, что кислород, выделяющийся при реакции с гипохлоритом
(в одном из примеров Эдвардса), получается исключительно из молекулы пе-
перекиси водорода, а поэтому возможность разрыва связи О—О исключается.
Бантон и Левеллин [66] считают, что механизм радикального иона гидро-
гидроксила для случая разложения перекиси водорода в растворе хлорной кислоты
невероятен, однако они не сообщают о подробностях своей работы с мечеными
атомами и о предложенных итоговых реакциях. По мнению Магдэна и Юнга
[67], некоторые реакции гидроксилирования перекиси водорода, которые,
по имеющимся данным, катализируются определенными металлами, проте-
протекают по механизму с ионом гидроксила, но через образование пероксокислот.
Механизмы с радикалом пергидроксила. По-видимому, нет ни одного вполне
доказанного примера реакции разложения перекиси водорода, возбужденной
термическим или фотохимическим путем, которая протекала бы через образо-
образование радикала пергидроксила. Это вполне понятно, если учесть, что такого
рода механизм является гораздо более неблагоприятным с точки зрения затра-
затраты необходимой энергии, чем другой возможный механизм с радикалом гидро-
гидроксила. Хант и Таубе [60] при исследовании фотохимического разложения жид-
жидкой перекиси водорода не нашли никаких причин, чтобы принять в качестве
инициирующего механизма образование пергидроксила. Правда, интерес к
вопросу открытия радикала пергидроксила проявлен только в самое последнее
время [68]; с другой стороны, в случае перекиси водорода возникновение пер-
пергидроксила за счет саморазложения приводит к одновременному образованию
хорошо известной и кинетически специфической частицы—атомарного водорода.
Возможно, что механизм с радикалом пергидроксила имеет силу в часто посту-
постулированной реакции
ОН + Н2О2 = Н2О + НО2. C7)
Механизмы с ионом пергидроксила. По сравнению с только что описанным
механизмом большее внимание привлек к себе механизм разложения и реакции
перекиси водорода за счет образования иона пергидроксила. Поскольку извест-
известно, что такая диссоциация легко протекает за счет отдачи протона воде или
другим растворенным веществам, и сделано наблюдение, что перекись водорода
разлагается с повышенной скоростью при высоком рН, логично предположить,
что особенно реакционноспособным должен быть ион пергидроксила. Несмотря
на это, механизм, лежащий в основе повышенной скорости разложения в ще-
щелочных растворах, еще окончательно не выяснен. Достаточно прямым доказа-
доказательством механизма с ионом пергидроксила можно считать факт легкого
Механизм реакций перекиси водорода 319
окисления сульфита до сульфата перекисью водорода [51]. На начальной ста-
стадии реакции к сульфиту присоединяются два атома кислорода от молекулы
перекиси с образованием промежуточной монопероксосернистой кислоты.
Поскольку при последующей перегруппировке образуется сульфат, несомнен-
несомненно, что в реакции принимает участие ион пергидроксила. Аналогичное иссле-
исследование с применением методики меченых атомов 146] доказывает, что поэтому
механизму происходит реакция гипохлорита и перекиси водорода. Эдварде
[65, 69] рассматривает такие превращения как реакции замещения в окси-
анионах и, в частности, показывает, какое большое значение имеет скорость
установления равновесия при таких замещениях, чтобы определись, можно ли
доказать присутствие некоторых перекисей, например пероксоборной кислоты
и аналогичных ей спорных соединений. Таким образом, хотя существование
определенной перекиси в растворе и возможно, однако быстрое наступление
равновесия за счет замещения (гидролиза) перекисью водорода может привести
к такому положению, что в действительности удастся обнаружить присутствие
только последней. Указывается [65], что это имеет также значение и для анали-
анализа смеси перекисей, который рассматривается в гл. 10 (стр. 468). Многие за-
замещенные перекиси восстановительной способностью не обладают, что дает
возможность определять перекись водорода в их присутствии. Однако если
равновесие замещения или гидролиза устанавливается быстро, то использовать
такое различие не представляется возможным.
Такие процессы легко представить как процессы, приводящие к обра-
образованию промежуточных гидроперекисей, что воскрешает старое представле-
представление о нестабильных промежуточных перекисях; в связи с этим интересна
работа Дэвиса, Фостера и Уайта [70]. Эти авторы вводили в реакцию с 90%-
ной перекисью водорода спирты, эфиры карбоновых кислот, 2-бутилсульфат
натрия или олефины, что приводило к образованию гидроперекисей алкилов;
эти данные, по мнению указанных авторов, подтверждают, что реакции
проходят по механизму с ионом пергидроксила и участием карбониевых ионов.
В частности, показано, каким образом высокополярная природа кон-
концентрированной перекиси водорода должна благоприятствовать реакциям,
при которых возникают заряды. Бантон, Льюис и Левеллин [71 ] показали, что
образование пероксокислот из меченных изотопом кислорода карбоновых кис-
кислот, по крайней мере в случае пероксоуксусной кислоты, протекает за счет
прямого замещения гидроксильной группы на пергидроксильную. Еще раньше
Бантон [72] доказал, что окисление а-дикетонов перекисью водорода проте-
протекает через образование иона пергидроксила. Пример прямого присоединения
иона пергидроксила дает, по-видимому, реакция перекиси водорода с бен-
золбороновой кислотой [73]. В качестве последнего примера механизма с ионом
пергидроксила можно назвать превращение бензонитрила в бензамид дей-
действием перекиси водорода; итоговый процесс здесь представляет перенос
одного атома кислорода к субстрату путем, требующим действия двух молекул
перекиси водорода. Этот вопрос послужил темой тщательно запланирован-
запланированного исследования, проведенного Вибергом [74], по мнению которого сначала
образуется гидроперекись, разлагающаяся затем под действием второй моле-
молекулы перекиси водорода. Виберг указывает, что первая реакция должна
протекать за счет уже образованного иона пергидроксила, вторая же стадия
вызывается индуцированной диссоциацией второй молекулы перекиси водо-
водорода.
Механизмы реакций других перекисей
Хотя в данной монографии и не описываются с большей или меньшей пол-
полнотой другие перекиси, кроме перекиси водорода, все же исследование других
перекисей может способствовать лучшему выяснению природы родоначаль-
родоначальника этой группы. Имеющиеся данные относятся в основном к органическим
320 Глава 7
перекисям; по неорганическим перекисям и пероксокислотам (возможно,
за исключением пероксодисерной кислоты), к сожалению, не проведено прак-
практически никаких работ, которые имели бы непосредственную ценность для
выяснения механизма реакции. Характеристика механизмов реакций орга-
органических перекисей послужила недавно предметом превосходного обзора 140],
а поэтому здесь нет необходимости на ней останавливаться. Приводимые ниже
замечания направлены лишь к тому, чтобы проиллюстрировать, каким образом
связь между атомами кислорода в перекисной группе зависит от замещения,
и показать, что и для других перекисей имеют силу механизмы реакции,
аналогичные предложенным для перекиси водорода.
Значение обеих этих тем доказывается классификацией, предложенной
Тобольским и Месробяном (см. в работе [40] стр. 107). Эти авторы приводят
следующие основные характеристики для реакции некоторых типов органиче-
органических перекисей: гидроперекиси алкилов, диалкилперекиси и диацилперекиси
разлагаются за счет гомолитической диссоциации, гидроперекиси ацилов
(пероксокислоты) разлагаются гетеролитически, а алкилацилперекиси (пер-
оксоэфиры) могут разлагаться и по ионному и по радикальному механизму.
Эта классификация, по-видимому, хороша, но в новых и малоизученных слу-
случаях ею нужно пользоваться с осторожностью. Леффлер [75] и Бартлет [761
показали, каким образом такие факторы, как растворитель, концентрация
и температура, влияют на механизм данной реакции. Леффлер [77] приводит
пример, показывающий, каким образом эти факторы могут изменять механизм
разложения; разложение перекиси n-метокси-п'-нитробензоила может изме-
измениться от свободнорадикального к ионному в зависимости от условий среды;
это изменение происходит несмотря на то, что симметричное исходное соеди-
соединение—перекись бензоила—разлагается через свободные радикалы в широком
интервале условий, даже таких, которые благоприятствуют ионной диссоциа-
диссоциации.
Важнейшее различие между реакциями перекиси водорода и органиче-
органических перекисей заключается в значительно меньшей величине энергии, необ-
необходимой для диссоциации органических перекисей на свободные радикалы
за счет разрыва связи О—О. Выше неоднократно указывалось, каким образом
необходимая для этого энергия резко снижает возможность разложения пере-
перекиси водорода в результате гомогенного термически активированного свободно-
радикального механизма, а поэтому, чтобы создать возможность разложения
с участием свободных радикалов, требуется наличие катализаторов или вме-
вмешательство электрической или фотохимической энергии. Для многих изучен-
изученных до настоящего времени органических перекисей энергия, необходимая
для разрыва связи О—О, приблизительно на 20 ккал/моль меньше, чем в случае
с перекисью водорода, в связи с чем наиболее предпочтительными оказываются
термически инициируемые свободнорадикальные механизмы. Предполагается,
что в противном случае во многих из использованных неполярных раствори-
растворителей разложения не должно было бы происходить. Это различие необычайно
ярко иллюстрируется наблюдением Брауна [78]. Гомогенное свободнорадикаль-
ное разложение перекиси бензоила протекает с одинаковой скоростью неза-
независимо от увеличения размеров поверхности платины, находящейся в кон-
контакте с системой, тогда как на скорость разложения перекиси водорода платина
не оказывает заметного влияния.
Среди процессов разложения органических перекисей, протекающих
по свободнорадикальному механизму за счет разрыва связи О—О, больше
всего исследованы два: процессы разложения ди-/лрет-бутилперекиси
и перекиси бензоила. Ди-/лрвт-бутилперекись разлагается совершенно
гомогенно, причем природа продуктов зависит от концентрации и среды; эти
результаты подытожены в недавно опубликованных обзорах [40]. Аналогично
и поведение перекиси бензоила; особый интерес представляют исследования
Механизм реакций перекиси водорода 321
по влиянию замещения на скорость ее разложения. Свен, Штокмейери Кларк
[79] показали, что симметрическое замещение перекиси бензоила электроно-
донорными группами увеличивает скорость разложения, тогда как электроно-
акцепторные группы снижают эту скорость; эта зависимость может быть пред-
представлена в виде графика о-функции Гаммета [80]. Для объяснения такого
поведения указывается, что в перекиси бензоила обе оксибензоатные группы
можно представить себе как диполи, соединенные таким образом, что происхо-
происходит взаимное отталкивание (отрицательные концы находятся рядом у перекис-
ной связи); введение электронодонорных групп усиливает это явление в моле-
молекуле, которая сама уже нестабильна, что обусловливает рост скорости разло-
разложения. Эти данные подтверждены другими авторами [81—83], изучавшими
также влияние таких факторов, как природа растворителя [81 ], справедливость
функции Гаммета для этого случая [82, 83] и возможные влияния положения
замещающей группы [82, 83]. Интересен вывод, что влияние замещения на-
находится в противоречии с вышеприведенным предположением, т. е. что перенос
отрицательного заряда к перекисной группе способствует ее стабилизации.
Возможно, что в этом случае максимум благоприятного влияния такого перено-
переноса заряда уже превзойден, но справедливость этого положения пока еще нель-
нельзя доказать. Интересным является мнение Купера [83], который считает, что
в диацилперекиси такого рода возможна эквивалентность атомов кислорода,
как это доказано для аналогичного производного серы, тетраметилтиурам-
дисульфида [84]. Эта эквивалентность, как можно предполагать, способна изме-
изменить характер стабильности перекисной группы. Для доказательства сущест-
существования такого эффекта потребуются дальнейшие работы, особенно в связи
с сообщением Бломквиста и Берштейиа [85] о том, что аналогичное влияние
замещающих групп наблюдается и у mp^/n-бутилпероксобензоата, асимметрич-
асимметричного моноацильного соединения, обладающего тенденцией к разрыву с обра-
образованием радикалов, свойственной обоим его родоначальникам.
Ионные механизмы реакции для органических перекисей, аналогичные
механизму с ионом гидроксила для перекиси водорода, являются, по-видимо-
по-видимому, вполне доказанными. Караш и Барт [86], например, считают, что в общем
уравнении для так называемого кислотного расщепления асимметричных
диалкилперекисей карбинол всегда образуется из радикала R, обладающего
большей электроноакцепторной способностью:
ROOR'^ ROH + R'O+. C8)
Сделан также интересный вывод, что чувствительность гидроперекисей
к такого рода кислотному расщеплению изменяется параллельно легкости
образования таких гидроперекисей путем самоокисления соответствующих
углеводородов. Из гидроперекисей, способных к кислотному расщеплению
(т. е. к ионной диссоциации по связи О—О), к исследованным в достаточной
мере относятся: гидроперекись кумола, пероксоуксусная кислота и гидро-
гидроперекиси декалина и тетралина. Исследование гидроперекисей декалина и
тетралина [87] показывает, каким образом механизм разложения этих пере-
перекисей может измениться от свободнорадикального до протекающего как в виде
гомолитического, так и в виде гетеролитического процесса, для чего достаточно
изменить главным образом полярность растворителя. Аналогичное соотноше-
соотношение наблюдается и для гидроперекиси кумола. При 100° в неполярном раство-
растворе происходит диссоциация на свободные радикалы [88]. При комнатной тем-
температуре в водном растворе легко происходит ионная реакция с закисным же-
железом [89—91]. При изучении этой реакции выявлен ряд интересных фактов.
При переходе от радикального механизма к ионному с участием иона за-
кисного железа предэкспоненциальный множитель уравнения Аррениуса
21 Перекись водорода
322 Глава 7
падает примерно с 1012 до 109, а энергия активации—с 30 до 11 ккал/моль.
Эти значения можно сравнить с аналогичными величинами для перекиси
водорода: энергии активации для радикального разложения перекиси водоро-
водорода, как указано выше, можно практически достичь лишь при температурах в
несколько сотен градусов, тогда какпредэкспоненциальный множитель и энер-
энергия активации для реакции перекиси водорода с ионом закисного железа
примерно те же, что и для гидроперекиси кумола (стр. 304, рис. 57). Таким
образом, при разложении по свободнорадикальному механизму скорости в том
и другом случае представляют величины совершенно разного порядка; при
более легко осуществимом ионном механизме перекись водорода разлагается
раза в 4 быстрее, чем гидроперекись кумола. На основе этого можно сделать
вывод, что замещение водорода группой кумила снижает стабильность связи
О—О в отношении свободнорадикального механизма, но повышает ее в отно-
отношении ионного. Исследование влияния дальнейшего замещения кумильной
группы [14, 92] показывает, что введение групп с возрастающей электронодо-
норной способностью увеличивает скорость реакции гидроперекиси кумола.
Эти факты как будто противоречат вышеприведенному постулату о влиянии
переноса заряда к группе О—О на ее стабильность, однако трудно выделить
относительную долю влияния различных процессов, к которым относятся пере-
перенос энергии, активация и ориентация. Несколько более простой механизм
с ионом гидроксила имеет место, по-видимому, при реакции пероксокислот
с олефинами, в результате которой образуются эпокиси. Суэрн [93] показал,
каким образом в случае пероксоуксусной кислоты замещение олефинов
электронодонорными группами увеличивает скорость реакции с электроно-
акцепторной пероксокислотой, а Уотерс [94] дал превосходный анализ вероят-
вероятного хода этой реакции. Наряду с этим Бартлет [76] предложил другой ме-
механизм, труднее укладывающийся в классификацию по механизму иона гид-
гидроксила или пергидроксила.
Эверёт и Минкоф [95] исследовали кислотную диссоциацию некоторых
гидроперекисей алкилов и ацилов; этот процесс, безусловно, аналогичен
разложению перекиси водорода по механизму с ионом пергидроксила. Ре-
Результаты этого исследования позволяют количественно измерить силу пер-
пероксокислот; они, несомненно, слабее соответствующих карбоновых кислот,
однако значительно сильнее соответствующих неацильных гидроперекисей.
Интересно отметить сравнимое изменение силы кислоты при переходе от
спирта к карбоновой кислоте. Эти соотношения объяснены тем, что влияние
замещения на легкость разрыва ОН-связи должно в гидроперекисях и пероксо-
кислотах передаваться через связь О—О. Для гидроперекисей алкилов, в ко-
которых замена одного атома водорода перекиси водорода на алкильную группу
вызывает смещение заряда в направлении кислорода, группа О—О, обладаю-
обладающая высокой электроотрицательностью, удерживает этот перенесенный заряд,
причем оставшийся водород мало изменяется. В аналогичной системе вода—
спирт этот перенос заряда, безусловно, обнаруживается по увеличению проч-
прочности связи иона водорода в спирте. В гидроперекисях ацилов или пероксо-
кислотах введение замещающей ацильной группы должно оттягивать заряд
от группы О—О, что снижает прочность связи иона водорода, но этот эффект
менее отчетлив, чем при переходе от воды к карбоновой кислоте, которая теряет
свой водородный ион значительно легче. Результаты этого исследования нахо-
находятся в согласии с постулированным упрочением перекисной связи за счет
переноса к ней заряда. Аналогичное положение, как указано Тобольским и
Месробяном [40], наблюдается при разложении третичных гидроперекисей
вТприсутствии основных катализаторов. Эти вещества, как предполагается,
при высоких и низких концентрациях едкого натра образуют соль. При проме-
промежуточной оптимальной концентрации возможна ионизация и последующее
быстрое разложение индуцированным действием гидроперекиси.
Обмен изотопами в реакциях перекиси водорода 32
Этот краткий обзор механизмов реакций других перекисей целесообразно
закончить некоторыми ссылками на поведение пероксодисерной кислоты.
По-видимому, аналогично органическим перекисям это, соединение может
разлагаться как гомолитически, так и гетеролитически. Морган [96] доказал
инициирование полимеризации пероксодисульфатом. Кольтгоф и Миллер [97]
высказали предположение, что некаталитическое разложение связано с обра-
образованием двух сульфатных радикалов. Указывается также на возможность
разложения при каталитическом действии кислоты через промежуточное
образование четырехокиси серы. Показано, что последний механизм не при-
приводит к установлению равновесия между ионами сульфата и пероксосульфата
[98]. Родственная и технически важная реакция—гидролиз пероксомононад-
серной кислоты, по-видимому, протекает за счет переноса иона пергидроксила
166].
ОБМЕН ИЗОТОПАМИ В РЕАКЦИЯХ ПЕРЕКИСИ ВОДОРОДА
Исследование обмена изотопами кислорода для выяснения поведения
перекисного кислорода в реакциях с участием перекиси водорода имеет столь
существенное значение, что оправдывает специальное рассмотрение этого
вопроса независимо от числа посвященных этому вопросу исследований.
Были поставлены опыты с мечеными атомами и для определения хода пре-
превращения атомов молекул субстрата при реакциях с участием перекиси водо-
водорода, однако результаты этих исследований носят менее общий характер и они
будут рассмотрены при обсуждении реакции отдельных элементов. Интерес дол-
должны представлять также опыты с меченым водородом перекиси водорода,
но, как и следовало ожидать, водород перекиси спонтанно и быстро обме-
обменивается с водородом воды. Это установлено Эрленмейером и Гертнером [99],
которые измеряли содержание дейтерия в трех фракциях от перегонки смеси
2%-ной D2O, содержавшей около 10% Н2О2. Не обнаружено никакого разли-
различия в содержании дейтерия в трех фракциях после введения поправки на
влияние самой перегонки с точки зрения обогащения проб дейтерием. Можно
пользоваться меченым водородом для исследования реакций с некоторыми
веществами, которые не обмениваются с водой или перекисью водорода. Такой
опыт проведен Вирцем и Бонгоффером [100]; оказалось, что молекулярный
водород, получающийся при реакции формальдегида с перекисью водорода
в щелочном растворе, образуется исключительно из формальдегида. Ниже
рассматривается вопрос о применении тяжелой воды для исследования эффек-
эффектов растворителя.
В табл. 71 сведены результаты исследований реакций перекиси водо-
водорода, проведенных с применением изотопов кислорода. Эта сводка позволяет
сразу же сделать некоторые выводы. Три опыта с реакциями образования
[103, 108, 111] показывают, что молекулярный кислород сохраняется как
целое при восстановлении до перекиси водорода; в образующуюся перекись не
внедряется кислород от воды или катализатора. Сводка опытов также доказы-
доказывает, что кислород воды, применяемой в качестве растворителя, не участвует
в разложении или реакции перекиси водорода; не отмечено обмена между во-
водой и образовавшимся молекулярным кислородом или окисленными продукта-
продуктами. Интересно и наблюдение за фракционированием изотопов, т. е. за измене-
изменением отношения обоих изотопов при переходе от реагента к продукту. Различ-
Различные авторы формулируют это фракционирование математически немного по-раз-
по-разному. Имеются два пути такого фракционирования для перекиси водорода.
Один путь представляет чисто кинетический процесс, в котором отношение
изотопов в продукте изменяется в зависимости от степени полноты протекания
реакции. Так, Бантон!и Левеллин [66] показали, что О16 в первом проценте
газа, выделяющемся при разложении перекиси, находится в большем коли-
количестве, чем в остальной части, хотя общего изменения отношения изотопов
21*
Таблица 71
Исследование по обмену изотопами
Реакция
Каталитическое раз-
разложение 30%-ной
Н2О2
Каталитическое раз-
разложение Н2О2
Та же
Глюкоза+О2 —> глю-
коновая кислота+
+н2о2
OJ8+H2O —> Н2О18+
-j-Oa
OJ8-f-H2O—>Н2О18+О2
Каталитическое раз-
разложение 10%-ной
Н2О2
15% H2O2-f Na2SO3 ->
-*H2O-{-Na2SO4
5Н2О2@,13-0,4л<олб/уг)
-4-2МпО^ -f-
4-6Н+ -> 2Мп2 + +
+8Н2О+5О2
Каталитическое раз-
разложение Н2О2
Каталитическое раз-
разложение 1%-ной
Н2О2 (полученной из
перекиси натрия)
0*4-Н Оу -+ 0^4-
2+н2о*
Реагент, растворитель,
катализатор и прочие
вещества
Нормальная Н2О, Pt
Каталаза, FeSO4 в
1,6%-ной Н2О18
Н2О18, Ь
При помощи фермента
нотатина в Н2О18
или с OV
Окислы и гидроокиси
металлов или орга-
органические вещества
Н2О2 или катализато-
катализаторы ее разложения:
Pt или каталаза
MnO2, Fe2O8, Pd и Pt
на асбесте в Н2О18,
нагрев
Н2О18
0,4% KMnOJ* в кис-
кислоте, 0,4% Н2О18
при 0—45°
Неуказанный катали-
катализатор в НоО18 при
разных рН
MnO2, Pt,Au,Fe(OHK,
каталаза в нормаль-
нормальной воде или в во-
воде, обогащенной О18
Длительный контакт
с каталазой или
гемоглобином
Результат
Фракционирование,
более легкое выде-
выделение О18
В газообразном О2
О18 не содержится
Результаты доказы-
доказывают, что OH-f
+Н2О2-Н2О+НО2
протекает быстрее,
чем ОН+Н2О ->
-> н2о+он
Образование Н2О|8 из
О|8, но не из Н2О18
Отсутствует обмен
между газообразным
кислородом и водой
Доказан катализ об-
обмена
Н2О18 не теряется в
виде О2
Переноса О18 от Н2О18
к сульфату не про-
происходит
Увеличения отноше-
отношения О18/О16
(=0,21%) в выде-
выделившемся О2 не про-
происходит
Отношение О18/О10 в
выделившемся О2 не
зависит от этого
отношения в воде;
отмечено фракцио-
фракционирование кислоро-
кислорода
Содержание О18 в ки-
кислороде не зависит
от содержания О18
в воде; неорганиче-
неорганические катализаторы
способствуют фрак-
фракционированию кис-
кислорода
Обмена не происхо-
происходит
Литература
Тейлор и Гулд
A934) [101]
Бентли A948)[102]
Коллинсон и Дей-
Дейтон A948) [55]
Бентли и Нейбер-
гер A949) [103]
Маккензи и Мил-
нер A951) [104]
Там же
Винтер и Бриско
A951) [105]
Там же
Берчи A951) [106]
Там же
Дол, Радд, Мачоу
и Конт A952)
[107]
Там же
Продолжение
Реакция
Каталитическое раз-
разложение Н2О2
Та же
H2O*4-2H2SO4 -
-> H2S2Og-f-2H2O
Мочевая кислота+
+OJ8 -> Н2О2
7% O8-f Н2О18 -> О]8+
4- Н2О
7%O3-fH2O18->OJ8-f
4-Н2О
H2O24-H2SO8-H2O-f
4-H2SO4 (образова-
(образование 1% дитионата)
н о1^1/ s о2-Мо°3
2 f / qfB-^38/ h"tTj_
.1/н*4 2
/2
О284-Н2О-,О24Н2О18
О?84-Н2О»О24-Н2О18
OJ84-H2O4-/iv^ O24-
4-Н2О18
Н О _1_ Y Y i
П2^/2Т"Л —> ЛВОССТ."Т*
+О2; концентрации
не указаны
Реагент, растворитель,
катализатор и прочие
вещества
12%-ная Н2О18 с Pt,
Ag, Fe2+, Fe3+, Ce4+,
НВг, Вг2 или же
MnO2, MnOj ,
Сг2О?-, H6JOe и
НОС1, меченные О18
12%-ная Н2О18, содер-
содержащая 60—70%
НСЮ4; катализатор
не указан
Нормальная Н2О
Подробности отсут-
отсутствуют
Нормальная Н2О, ури-
каза
0,04 М НС1О4 в
1,4%-ной Н2О18
Как и выше, с добав-
добавкой 9-Ю-4 М Н2О2
В соляной кислоте
или в ацетатном бу-
буфере с рН 5; либо
Н2О2, либо SOfr-f
4-Н2О с содержа-
содержанием О18
В ацетатном буфере
с рН 5
При рН 11,8
То же с 0,18 М Н2О2
рН от 2 до 12
В нормальной Н2О и
в 1,2%-ной Н2О18
с Се (IV), МпО4,
С12+С1-, НС1О и
Сг2О7
Результат
О18 в газообразном О2
отсутствует
О18 в газообразном О2
не содержится
Наблюдается фрак-
фракционирование, за-
заметное при Fe2+,
Fe3+, Pd и менее
заметное при КМпО4
Перенос перекисных
атомов кислорода
происходит без раз-
разделения
Кислород в Н2О2 про-
происходит целиком от
Оо
Обмена в кислой сре-
среде не происходит;
значительный обмен
происходит в ще-
щелочной среде
Происходит некото-
некоторый обмен
На каждую частицу
SO2,"", окисленную
в SO2,', обнаружены
2 атома кислорода
из Н2О2
На каждую частицу
SO2', окисленную
в SOJ"", обнаружен
1 атом кислорода
из Н2О2
Обмена не происходит
Обмен происходит
Обмен наблюдается
при ^-излучении;
при рН12 Н2О2
тормозит обмен
Отношение О18/О16 не
зависит от содержа-
содержания Н2О18
Литература
Бантон и Левеллин
A952) [66]
Там же
Бснтли и Нейбер-
гер A952) [108]
Форхеймер и 1Тау-
бе A952, 1954)
[54]
Там же
Галперин и Таубе
A952) [51]
Там же
Харт, Гордон и
Хатчисон A952)
[109]
Там же
» »
Кахил и Таубе
A952) [46]
Продолжение
Реакция
Реагент, растворитель,
катализатор и прочие
вещества
Результат
Литература
Каталитическое раз-
разложение Н2О2; кон-
концентрации не ука-
указаны
Реакция или катали-
каталитическое разложе-
30%-ная H202+/iv->02
0,37 М Н2О2 с содер-
содержанием 1,6% О18
Н2О2 -> О2 (концен-
(концентрация не указана)
CeH5CN+2H2O2 »
->C6H5CONH2+O2-f
RCOO18H-bH2O2 -
-> RCOOOH-f-H2O18
ZnO-fO2-fH2O-j-/*v
- H2O2
0,1 M K2S2O8 -> 02.
Термическое разло-
разложение
В нормальной Н2О и
в 1,2%-ной Н2О18
с Fe3+, J-, J2, Br-,
Br2, MnO2 или Pt
В нормальной Н2О
с Fe2+, Fe3+, Sn2+,
Ti3+, Cr2+, Cu+,
Ce3+, Cl2, HOC1,J-,
Br~, Mo (V) с раз-
разными добавками при
разных рН
В 1,6%-ной Н2О18
В нормальной Н2О,
через которую бар-
ботировали нор-
нормальный кислород
В Н2О18
В Н2О18
Уксусная и муравьи-
муравьиные кислоты в нор-
нормальных водных
растворах Н2О2+
Оа8 в Н2О
В Н2О18 при разных
рН
Отношение О18/О1в не
зависит от содержа-
содержания Н2О18
Отмечено фракциони-
фракционирование кислорода
Отношение О18/О16 в
кислороде такое же,
как в Н2О2; фрак-
фракционирование
Выделившийся кисло-
кислород обогащен О18
по сравнению с
Н2О2, что доказы-
доказывает фракциониро-
фракционирование
О2 не содержит О18
Вода, образовавшаяся
избензамида, содер-
содержала на 0,03% боль-
больше О18, чем нор-
нормальная; если бы
кислород получался
из воды, использо-
использованной в качестве
растворителя, то из-
избыток был бы 0,6—
0,7%
Кислород гидроксила
карбоновой кислоты
превращается в во-
воду. Обмена О18 меж-
между Н2О2, RCOOOH,
водой и кислородом
карбоксила не про-
происходит
Кислород в Н2О2 по-
получается исключи-
исключительно из О2
О28 получается из во-
воды в различных со-
соотношениях в зави-
зависимости от рН (с па-
падением рН О18 воз-
возрастает)
Кахил и Таубе
A952) [46]
Там же
Хант и Таубе A952)
[60]
Там же
Берчи A952) [ПО]
(по Христиансе-
ну)
Виберг A953) [74]
Бантон, Льюис и
Левеллин A954)
[71]
Калверт, Тейрер,
Рэнкин и Мак-
Невин A954)
[111]
Кольтгоф и Мил-
Миллер A951) [97]
Обмен изотопами в реакциях перекиси водорода
327
Продолжение
Реакция
Раствор KaSgOj
Раствор KjSS6O«
s,o»-+s*os- -
- SJOJ-+SOS-
s,o*-+s*o?- -
- ssor+soj-
Реагент, растворитель,
катализатор и прочие
вещества
В Н2О18 при 50 и 90°
В K2S2O8 в Н2О
<NH4)S2O8 в водной
H2S36O4
В воде и концентри-
концентрированной СН8СООН
Результат
В щелочном растворе
обмена О18 не про-
происходит; в 0,5 М
H2SO4 обмен равен
2% при 90° и 0,4%
при 50°
Обмена S36 с немечен-
ным Кг^Ов не про-
происходит
Обмена S36 не проис-
происходит
Обмена S35 не проис-
происходит
Литература
Кольтгоф и Мил-
Миллер A951) [97]
Рисбос и Атен
A952) [112]
Элькелес и Броссе
A953) [ИЗ]
Иджер и Мак-Кол-
лам A954) [98]
при переходе от реагента к продукту и не обнаружено. Поскольку связь О—О
при образовании молекулярного кислорода, очевидно, не разрывается, то это
истолковывается как доказательство, что процесс отрыва двух атомов водо-
водорода от О1в происходит легче, чем от О18. Второй тип фракционирования оказы-
оказывается возможным в случае образования более чем одного продукта (напри-
(например, молекулярного кислорода и воды). Дол, Радд, Мачоу и Конт [107] по-
показали, что газообразный кислород, получаемый при полном разложении под
действием некоторых неорганических катализаторов, обогащается О18 (пример-
(примерно 2%), тогда как каталаза такого обогащения не вызывает. Аналогичные
исследования с рядом восстановителей проведены Кахилом и Таубе [46]. Эти
работы позволяют сделать вывод, что при окислении до молекулярного кисло-
кислорода, как указано выше, связь О—О не разрывается и фракционирования не
происходит. Однако если перекись водорода восстанавливается, то связь
разрывается и возможно фракционирование. Различные работы, перечисленные
в табл. 71 (графа «Литература»), дают обширный материал по значению этих
опытов для выяснения механизма; отчасти этот материал уже затронут в выше-
вышеприведенном анализе. Сделаны также попытки объяснения фракционирования
в свете теории абсолютных скоростей реакции. Эти вопросы, а также связь
опытов с мечеными атомами для перекиси водорода с опытами для других
кислородных соединений рассматриваются в обзоре Дола [114].
Интересно сравнить и сопоставить эти данные с аналогичными данными
для пероксосульфата и гидразина. Как показывают последние несколько строк
табл. 71, пероксосульфат или молекулярный кислород, образующийся при
его разложении, могут частично обмениваться меченым кислородом с растворя-
растворяющей водой. В то же время ряд исследований показывает, что сульфат не обме-
обменивается кислородом с пероксосульфатом; отсюда делается вывод, что пере-
кисный кислород в пероксосульфате не обменивается с водой, так же как он
не обменивается и в перекиси водорода. Однако при термическом разложении
в растворе образующийся молекулярный кислород содержит и кислород, полу-
получающийся из воды; в этом отношении положение иное, чем с перекисью водо-
водорода. Опыты с гидразином [115] показывают, что, как и в случае перекиси
водорода, для его реакции существуют различные пути в зависимости от того,
происходит или не происходит разрыв связи N—N; если образуется молеку-
молекулярный азот, то можно считать, что разрыва этой связи не происходит. Инте-
Интересно было бы распространить сравнение и на персульфиды, для которых
предложены аналогичные механизмы [116].
328
Глава 7
РАСТВОРЫ ПЕРЕКИСИ ВОДОРОДА КАК РЕАКЦИОННАЯ СРЕДА
Рассмотрение механизмов реакции показало значение взаимодействия
перекиси водорода не только с реагентами, но и с растворителем и другими
растворенными веществами. В этом разделе рассматриваются некоторые
свойства растворов перекиси водорода, которые влияют на протекающие в них
реакции. Физическая природа растворов перекиси водорода освещена в гл. 6.
Упомянутые в ней сообщения указывают на высокую диэлектрическую прони-
проницаемость перекиси водорода и на близость ее к диэлектрической проницаемости
воды. Правда, существуют определенные различия, но для большей их части
пока еще нет надлежащего объяснения. Так, Бамбергер и Нуссбаум [117]
указали, что вода и перекись водорода очень легко растворяют такие органиче-
органические вещества, которые содержат большое число гидроксильных групп, но
при росте молекулярного веса растворяемого вещества только сравнительно
концентрированная перекись водорода сохраняет растворяющие свойства.
Среди неорганических веществ имеются такие, которые лучше растворимы или
хуже растворимы в перекиси водорода, чем в воде. Электропроводности солей
в растворах перекиси водорода, наоборот, очень близки к электропроводностям
Таблица 72
Активность перекиси водорода как растворенного
вещества в водных растворах
Концентрации
мольные доли
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,80
0,90
0,91
0,92
0,93
0,94
0,95
0,96
0,97
0,98
0,99
1,00
Н2О2
вес. %
0
1,871
3,710
5,517
7,293
9,039
10,755
12,443
14,103
15,735
17,34
32,07
44,73
55,73
65,37
73,90
81,50
88,31
94,44
95,02
95,60
96,17
96,73
97,29
97,84
98,39
98,93
99,47
100,0
Активность Н2О2
мольные доли
0
0,0103
0,0213
0,0329
0,0451
0,0581
0,0718
0,0861
0,1013
0,1171
0,1338
0,3445
0,6406
1,020
1,470
1,967
2,481
2,981
3,445
3,488
3,531
3,574
3,616
3,658
3,699
3,739
3,779
3,819
3,857
молярные еди-
единицы
0
0,5724
1,180
1,824
2,506
3,225
3,980
4,782
5,621
6,502
7,424
19,12
35,56
56,64
81,62
109,2
137,7
165,5
191,2
193,6
196,0
198,4
200,7
203,0
205,3
207,5
209,8
211,9
214,1
Растворы перекиси водорода как реакционная среда 329
их в воде; кислоты в перекиси водорода [118] обладают худшей электро-
электропроводностью, что доказывает пониженную подвижность в ней водородного
иона. В гл. 5 приведены ссылки на литературу по такого рода физическим
свойствам, как растворимость и электропроводность. Этим вопросам при
исследовании реакций перекиси водорода уделялось обычно очень небольшое
внимание. Например, хотя и имеются сообщения [119] о влиянии солей на гете-
гетерогенное каталитическое разложение, но, по-видимому, только в одном случае
[120] изучен первичный солевой эффект для кислотно-основного катализа.
Редко рассматривался также вопрос об активности перекиси водорода в ее
растворах. Данные, приведенные в гл. 5, дают возможность рассчитать актив-
активность перекиси водорода как вещества, растворенного в водных растворах.
Эта активность в мольных долях определяется [121] из уравнения
a=ph(xh/ ph)*y где величина со звездочкой представляет собой предельную
величину при бесконечно большом разбавлении. Вычисленные таким образом
значения (в мольных долях и молярных единицах) приведены в табл. 72. Ли-
вингстон [122] измерил активность 1,5М перекиси водорода в растворах солей.
Кислотные свойства перекиси водорода
Перекись водорода представляет собой слабую кислоту, т. е. термиче-
термически активированный перенос протона от молекулы перекиси водорода к моле-
молекуле воды происходит лишь в небольшой степени
НаО2 + Н2О = Н3О+ + НО7. C9)
Уже давно исследователи, работавшие с перекисью водорода, знали, что она
обладает слабой кислотностью [123], но константа диссоциации ее измерена
впервые лишь в 1912 г. Иойнером [124]. Этот автор пользовался классически-
классическими методами, определяя степень гидролиза гидроперекиси натрия по омылению
эфиров, коэффициенту распределения и электропроводности. Пользуясь этими
методами, он получил для константы кислотной диссоциации перекиси водо-
водорода приО° среднюю величину 7-10~13. Прямыми термохимическими измерения-
измерениями для теплоты диссоциации получено значение 8,6 ккал/моль, откуда вы-
вычислен /С=2,4-10~12 при 25°. Эта величина практически согласуется со зна-
значением, полученным В. А. Каргиным [125] при измерениях, проведенных
с 0,1 и 0,5 М дистиллированной перекисью водорода и приведших к значению
/С=1,55-10'12 при 20°.
Недавно Эванс и Юри [126] повторно определили константы диссоциации
перекиси водорода, причем надлежащим образом учли влияние ионной силы.
В результате измерений при помощи стеклянного электрода с растворами
перекиси водорода концентрацией до 2 М они определили, что константа дис-
диссоциации при 20° и нулевой ионной силе равна 1,78-10~12, что даетрК= 11,75.
Измерения проведены также и при других температурах, причем получены
следующие результаты: при 15° /С= 1,39-102; при 20° 1,78-10'12; при 25°
2,24-10~12; при 30° 3,55-10~12. Рассчитанная из этих данных теплота дис-
диссоциации равна 8,2 ккал/моль. Эти рекомендуемые значения доказываются
также измерениями Эверета и Минкофа [95], которые сравнили константу
диссоциации перекиси водорода с соответствующими константами различных
гидроперекисей арилов и ацилов.
Сделано также заключение, что стеклянный электрод вполне подходит
для измерений рН растворов перекиси водорода. Так, Рейхерт и Хал [1271
сравнили стеклянный электрод и ряд колориметрических индикаторов в каче-
качестве средства для определения рН растворов перекиси водорода при концент-
концентрациях до 30 вес.% и обнаружили сравнительно хорошее совпадение. В ци-
цитированном сообщении, а также и в других [128, 129] приведены типичные
кривые рН—концентрация для титрования перекиси водорода, содержащей
330
Глава 7
кислоту или щелочь. Уинн-Джонс [130] также доказал пригодность стеклян-
стеклянного электрода для измерения рН перекиси водорода, притом даже еще* более
концентрированной, чем указанная, и исследовал возможность применения
метанилового желтого в качестве индикатора. На рис. 59 приведены фактиче-
фактические значения рН для всего интервала концентраций и показано также влия-
влияние двуокиси углерода на рН растворов перекиси водорода; это влияние,
например, способствует тому, что при экстраполяции к нулевой концентрации
кривая не проходит через рН = 7. Поэтому наличие двуокиси углерода может
вызвать расхождение между фактическими и расчетными данными для кривой
титрования рН, как например между результатами измерений Рейхерта
и Хала [127] и расчетами по методам, изложенным Берлем [129].Сдругой сто-
стороны, редко возникает желание вычислить рН на основании данных сложных
¦маня
-— —
—¦ ¦—
lljmjc
одам
зииц\
ПО
10 20 30
40 50 60 ТО
Н?О2,вес.%
80 30 100
Р и с. 59. Измеренная величина рН растворов пе-
перекиси водорода в электропроводной воде.
Д раствор, насыщенный СОг; О—после выдержки на воз-
воздухе.
равновесий в растворах кислотных смесей; даже если не учитывать то, что
наличие двуокиси углерода в обычных атмосферных условиях может вызвать
изменение рН раствора перекиси водорода на несколько десятых долей еди-
единицы рН, воспроизводимость обычных измерений редко бывает лучше ±0,1 еди-
единицы. В экспериментальных работах с перекисью водорода иной раз при-
применяются буферные системы. Преимущества таких систем, безусловно, заслу-
заслуживают рассмотрения. Может возникнуть вопрос о совместимости этих буфе-
буферов со столь реакционноспособной системой и о возможности их распада,
однако систематических исследований, на основании которых можно было бы
рекомендовать эти буферы или отвергнуть их, до настощего времени еще
не проводилось.
Кривые рис. 59 иллюстрируют также сверхкислотные свойства перекиси
водорода при высоких концентрациях. Очевидно, что активность иона водо-
водорода становится очень высокой по мере приближения раствора к безводному
состоянию. Эванс и Юри исследовали этот вопрос и высказали предположение,
что равновесие реакции
должно быть сильно смещено направо; по их оценке, константа равновесия
составляет примерно 103. Митчел, Бек и Уинн-Джонс [131] также показали,
что перекись водорода обладает значительно меньшей основностью, чем вода.
Реакция в необычных растворителях
Понимание природы эффектов сольватации и других взаимодействий
в растворах может иметь большое значение для определения механизмов реак-
реакций. В литературе имеется много сообщений о влиянии изменения природы
Растворы перекиси водорода как реакционная среда 36i
растворителя, но лишь небольшое количество новейших работ получило
удовлетворительное истолкование.
Пожалуй, наименее резко изменяется характер растворителя при заме-
замещении части или всей воды, применяемой в качестве растворителя, тяжелой
водой; правда, этот метод имеет то неудобство, что при его применении одно-
одновременно изменяются и растворитель и растворенное вещество, так как пере-
перекись водорода быстро обменивается водородом с растворителем. Такого рода
исследования показали, что гетерогенное разложение под действием платины
[132, 133], стекла и золота [134] в присутствии тяжелой воды замедляется.
В двух сообщениях [135, 136] утверждается, что в этих условиях гомогенное
разложение йодидом также замедляется, однако в третьей работе [133] влия-
влияние тяжелой воды на реакцию не обнаружено.
Восстановление перманганата перекисью водорода в растворе в тяжелой
воде [136, 137] протекает значительно медленнее, однако катализ реакции
ионом двухвалентного марганца проходит в этих условиях более интен-
интенсивно.
В ряде работ изучалось влияние метилового и этилового спиртов на реак-
реакции перекиси водорода [138]. Было показано, например, что эти вещества
тормозят каталитическое разложение под действием меди или марганца
1139, 140]. Подавляется также и реакция с ионом закисного железа [141];
ацетон и уксусная кислота оказывают значительно меньшее влияние. Это
влияние может заметно зависеть и от концентрации. В случае йодидного ката-
катализа повышение концентрации метилового или этилового спирта или пиридина
в растворителе непрерывно снижает скорость разложения; с другой стороны,
при я-пропиловом спирте скорость проходит через минимум в зависимости от
концентрации [142]. Наоборот, изобутиловый и амиловый спирты, глицерин
и этиленгликоль увеличивают скорость реакции в присутствии йодида, хотя
Уолтон и Джонс [143] указывают, что такие растворители все же качественно
отводы не отличаются. Овербергер и Камминс [63] исследовали окисление суль-
сульфида в таких растворителях, как этиловый и изопропиловый спирты, диизо-
пропиловый эфир, ацетонитрил и пропионитрил, и пришли к заключению,
что окисление протекает быстрее в растворителях, способных к образованию
водородных связей с перекисью водорода, а также, что вода и спирт, возможно,
обладают одинаковой эффективностью в отношении сольватации перекиси водо-
водорода.
К другим исследованным растворителям относится ацетон, который, как
было доказано, тормозит каталитическое разложение платиной [144] и по-
подавляет термическое разложение при 40° в большей мере, чем этанол, диок-
сан или тетрагидрофуран [74]. Стонер и Догерти [145] показали, что окисле-
окисление дитиодикарбоновых кислот перекисью водорода катализируется кислотами
и вследствие этого ускоряется при замене воды диоксаном, который, вероятно,
устраняет тормозящие эффекты сольватации. Этиловый эфир снижает скорость
разложения на платине [146]. Изучена реакция между четыреххлористым ти-
титаном и перекисью водорода в безводном этилацетате [147]. Недавно опубли-
опубликовано сообщение [148], что28%-ная перекись водорода в безводном фтори-
фтористом водороде является эффективной реакционной средой для специальных
целей.
Можно было бы привести данные еще и "многих других опытов, однако
несколько сомнительна возможность отнести их к реакциям в необычных
растворителях—либо потому, что исходные относительные количества этих
растворителей были очень малы, либо потому, что они более или менее энер-
энергично реагировали непосредственно с перекисью водорода. Этот вопрос имеет
особое значение с точки зрения стабилизации перекиси водорода; в гл. 9 будут
приведены другие исследования, которые можно истолковать с точки зрения
влияния растворителя.
332 Глава 7
РЕАКЦИИ С НЕОРГАНИЧЕСКИМИ ВЕЩЕСТВАМИ
В этом разделе дается краткое изложение природы и характера реакций
перекиси водорода с неорганическими веществами, причем материал размещен
по положению элементов в периодической таблице в соответствии с системой^
использованной Латимером 14]. Тесно примыкающие к этому вопросу про-
процессы разложения описываются в гл. 8, а органические и биологические реак-
реакции рассматриваются в следующих разделах. Приводимый материал в зна-
значительной мере взят из очень старых или разнообразных источников, причем
попыток дать исчерпывающий обзор старой литературы по этим вопросам,
которую можно найти у Меллора [149] и Фрейда и Твиса [150], не предприни-
предпринималось. Несколько менее полные, но зато охватывающие и более позднюю ли-
литературу обзоры неорганических реакций опубликованы Банкрофтом и Мэрфи
[20] и Стоуном [151]. Реакции, перекиси водорода с образованием неорганиче-
неорганических перекисей рассматриваются в гл. 12. Хотя приводимый материал представ-
представляет собой обзор довольно большого количества опубликованной литературы,
достаточно авторитетных и обстоятельных исследований таких реакций про-
проведено немного.
При обсуждении этого вопроса нужно иметь в виду, что перекись водо-
водорода может реагировать либо с анионной частью молекулы, либо с катионной,
либо с той и другой. Так, в литературе встречаются часто отрывочные описания
реакции окисления некоторых анионов, например сульфида или сульфита,
где внимание в основном обращено на какую-то другую часть молекулы.
Поэтому здесь приведены не все примеры таких реакций. Перекись водорода
очень часто функционирует как растворитель металлов за счет своего окисля-
окисляющего действия; так, кислый раствор перекиси водорода может конкурировать
в этом отношении с царской водкой. Путем правильного подбора кислоты
можно добиться растворения почти всех металлов; по этому вопросу опубли-
опубликованы многочисленные работы [152]. Окисляющее действие перекиси водо-
водорода имеет значение также и в отношении коррозии, поскольку перекись
(стр. 68) может образоваться как промежуточный продукт при реакции кисло-
кислорода с различными металлами. Представляет интерес влияние перекиси водо-
водорода на форму окисла, образующегося при коррозии [153], и на ход коррозии:
например, сообщается [154], что цинк может корродировать с образованием
особенно гладкой поверхности в присутствии соляной кислоты и Н2О2.
Некоторые исследования, имеющие общий интерес, касаются влияния света и
магнитного поля на реакции перекиси водорода. Дхар и Бхаттачариа [1551
показали, что поглощение света некоторыми реакционными смесями выше,
чем отдельными составными частями. Коллинс и Брайс [156] сообщают,
что, как и следовало ожидать, магнитное поле 12000 гаусс не оказывает
влияния на скорость термического разложения 1—3%-ной перекиси водорода
при 80°.
Все рассматриваемые реакции относятся к перекиси водорода в жидкой
фазе. Исследование реакции пара перекиси водорода ограничивается лишь
данными по взаимодействию с окисью углерода [62, 157], озоном [158], метал-
металлическим магнием [159] и амальгамой натрия-калия [160]. Возможные реак-
реакции в твердой фазе вообще не исследовались.
Кислород, водород
В подкисленном водном растворе избыток перекиси водорода реагирует
с озоном по следующей итоговой реакции:
Н2О + 2О2. D1)
Реакции с неорганическими веществами 333
Если относительное количество перекиси водорода меньше, то озон исчезает
быстрее, чем перекись. Ротмунд и Бургшталлер [161] дали основные характери-
характеристики этой реакции, Таубе и Брей [162] внутри широкого диапазона перемен-
переменных изучили ее кинетику, влияние конкурирующих веществ и ингибиторов и
другие явления, связанные с механизмом. Реакция протекает через образование
свободных радикалов в качестве промежуточных продуктов и некоторыми
авторами рассматривается практически как каталитическое разложение озона
под действием перекиси водорода.
Важный вопрос, происходит ли реакция между перекисью водорода
и надперекисью водорода (пергидроксилом, НО2) или ионом от кислотной дис-
диссоциации НО2, т. е. Од, пока не решен. Джордж [163] на основании результатов
исследования надперекиси калия, диспергированной в растворе перекиси водо-
водорода, пришел к выводу, что реакция не происходит ни по тому, ни по дру-
другому механизму. Однако, поскольку указанный опыт проводился в гетерогенной
системе, справедливость вывода Джорджа оспаривается [164], а поэтому
и сейчас еще предлагаются механизмы, основанные на реакции перекиси водо-
водорода и пергидроксила (т. е. надперекиси водорода). Широко распространено
мнение, что пергидроксил является довольно сильной кислотой и что реакция
в действительности происходит с ионом О~. Описана несколько необычная
реакция—диспропорционирование дипероксигидрата перекиси калия на над-
перекись калия и воду [165]. Гейб [166] рассматривает ход реакции перекиси
водорода с атомарным водородом или с атомарным кислородом.
Галогены
Реакции галогенов и их соединений с перекисью водорода относятся к тем,
изучение которых производилось особенно интенсивно. Вопрос о скорости и
механизме ряда таких реакций с хлором, бромом и йодом неоднократно прив-
привлекал к себе внимание разных авторов, что привело к надежному истолкованию
этих процессов. Опубликованы два обзора, в которых подытоживаются
эти р аботы: один, написанный Бреем [167] в 1932 г., освещает старую литерату-
литературу по этому вопросу, другой, более поздний, принадлежащий Баксендейлу
[168], охватывает литературу до последнего времени. Имеющиеся кинетические
данные сведены вместе в таблицах Национального бюро стандартов [8]. Особен-
Особенное значение для анализа механизмов этих реакций имеют два ранее цитиро-
цитированных сообщения Эдвардса [13, 64].
Реакции с соединениями фтора отличаются от реакций с соединениями
других галогенов. Окисление фтор-иона термодинамически неблагоприятно;
элементарный фтор вызывает разложение перекиси водорода и образование
ряда продуктов, пока еще полностью не идентифицированных. Уже в одной
из старых работ [169] отмечено влияние перекиси водорода на различные сме-
смешанные фториды. Маас и Хэтчер [170] показали, что растворимость элемен-
элементарных хлора и йода в безводной перекиси водорода не очень велика, и нашли,
что бромид и йодид в отношении перекиси водорода обладают значительно
большей реакционной способностью, чем хлорид. Изучено также влияние сме-
смесей из хлорида и бромида [171].
Как элементарный хлор, так и продукт его гидролиза, гипохлорит, вос-
восстанавливается перекисью водорода до хлор-иона [172]. Хлораты не реагируют
в щелочных или нейтральных растворах даже при температуре кипения,
но в кислом растворе перекиси водорода образуется хлор и двуокись хлора
[173]. В свою очередь гидролиз двуокиси хлора не зависит от присутствия
перекиси водорода [174]. Перхлораты также не реагируют с перекисью, в связи
с чем для регулирования рН растворов перекиси водорода особенно подходит
хлорная кислота.
334
Глава 7
Окислительно-восстановительные взаимоотношения брома с перекисью
водорода такие же, как и у хлора, только бромат восстанавливается перекисью
до бромида и элементарного брома. Наиболее обстоятельные исследования по
галогенам проведены с йодистыми соединениями. Обзор старой литературы
дан Уолтоном [175]; нужно также отметить значительные работы в этой обла-
области, принадлежащие Абелю [176]. Недавно по этим реакциям опубликован
обзор Моргана [177]. Сводку реакций, приведенную в табл. 73, составили Брей
и Либхафский [178]; черточки означают, что реакций практически не происхо-
происходит. Необходимо упомянуть о ряде исследований некоторых особенностей реак-
реакций йода с перекисью водорода. Румпф [179] опубликовал спектрографическое
исследование реакции с йодидом; имеется также сообщение об изменениях рН„
происходящих в системе, содержащей йодид и тиосульфат [180]. Окисление
йодида лежит в основе «часовой» реакции* [181]. Эта реакция катализирует-
катализируется железом; по этой так называемой «реакции Шенбейна» опубликован ряд
работ [182].
Таблица 73
рн
13
5
1
J-
J-
Реакции
j-
-j,
йода
h
J,-
J2 "•"
С
J-
J-
перекисью
Виды частиц
|
j
Ja+
J2-
J,-
водородг
> J-
JO;
1
JOa"
JOj->
+J-
Сера, селен, теллур и полоний
Элементарная сера не реагирует с перекисью водорода. Сера в других
валентных состояниях подвергается ступенчатому окислению, которое в под-
подходящих условиях продолжается до шестивалентного состояния с образованием
сульфата или трехокиси серы. Сероводород в кислых водных растворах окис-
окисляется, но только до элементарной серы. Недавно проведенное исследование
[183] показало, что скорость этой реакции обратно пропорциональна концент-
концентрации водородных ионов в степени 0,4 или 0,5 и первого порядка по кон-
концентрации перекиси водорода. В щелочных растворах образуется сульфат
[184]. Эта реакция катализируется железом в такой мере, что существенное
значение имеет даже чистота воды, применяемой в качестве растворителя [185].
Изучены также родственные реакции металлических сульфидов [184]. Суль-
Сульфит легко окисляется перекисью водорода до сульфата, причем выяснению
природы этой реакции в значительной мере помогли два уже описанных выше
исследования, проведенных с мечеными атомами [51, 105]. Интересной особен-
особенностью окисления сульфита является одновременно протекающая (в известной
степени) димеризация его до дитионата Sfii~ [186]. Глэсстон и Хиклинг [1871.
* Эта реакция состоит в следующем. Если к раствору йодистого калия, серной
кислоты и крахмала прилить избыток перекиси водорода, раствор синеет; при добавле-
добавлении небольшого количества тиосульфата раствор обесцвечивается, поскольку тиосульфат
быстрее реагирует с йодом, чем перекись водорода с йодидом. После израсходования всего
введенного тиосульфата в результате продолжающегося взаимодействия перекиси с йоди-
йодидом раствор опять синеет. При добавке новых малых порций тиосульфата обесцвечивание
и появление окраски может повториться несколько раз, причем путем введения строго
определенных количеств раствора тиосульфата можно добиться появления окраски ч.ерез.
определенный промежуток времени.—Прим. перев.
Реакции с неорганическими веществами 335
противопоставили эту димеризацию стехиометрическому образованию дитио-
ната при электролизе сульфита. Они считали также, что при электролитиче-
электролитической реакции в связи с особенно благоприятными условиями высокой местной
концентрации на поверхности анода происходит промежуточное образование
перекиси водорода. Аналогичные усложнения наблюдаются в реакции, при
которой перекись водорода вызывает замещение «сульфидного» атома серы
в тиосульфате S2Ol~ на атом кислорода с образованием сульфата [51]. В этом
случае могут образоваться также, по-видимому, тритионат Sfil' и тетратионат
S4Ojf [188]. Кинетически, по имеющимся данным [189], реакция бимолекуляр-
на; исследовано влияние на нее различных катализаторов, в частности йода
и молибдата [190].
Сравнить и сопоставить между собой соединения серы и кислорода осо-
особенно интересно потому, что это может пролить свет на механизм обсуждав-
обсуждавшихся реакций. Сера и кислород аналогичны друг другу по одинаковой внеш-
внешней $2/?4-электронной структуре, в связи с чем они близки и по валентности.
В то же время они сильно отличаются по электроотрицательности и склон-
склонности к гибридизации. Другое различие, которое может иметь решающее зна-
значение, состоит в том, что для серы возможно использование d-орбит в гибрид-
гибридных электронных структурах, для кислорода же такое положение энергетиче-
энергетически невероятно. В свете изложенного можно понять ряд явлений: очень малый
срок существования гидроксильного радикала ОН при сопоставлении с отно-
относительной нереакционноспособностью сульфгидрильной группы SH, диспропор-
ционирование в известных условиях [191] моноокиси серы SO с образованием
двуокиси серы и элементарной серы (аналогично образованию озона из молеку-
молекулярного кислорода) и, очевидно, полное отсутствие sp-гибридизации в молекуле
сероводорода с углом связи 90°. Особенное значение имеет вопрос об электрон-
электронной структуре серы в шестивалентном состоянии, т. е. о том, содержит ли
эта структура шесть $р3сB-гибридных орбит, что должно привести к наличию
тс-связей в такой структуре, как сульфат. Этот вопрос обсуждается в различ-
различных сообщениях [192] и имеет значение для нашей темы, поскольку возможно,
что легкость окисления производных серы перекисью водорода связана с лег-
легкостью образования этих гибридных структур. Говард и Левит [193] рассмотре-
рассмотрели с этой точки зрения тот факт, что наличие электроноакцепторных групп
в сульфоксиде или сульфиде замедляет соответственно окисление этих веществ
пероксосульфатом и перекисью водорода. В другом предположении указано
[65], что легкость окисления может зависеть от поляризуемости серы и его
следует рассматривать как механизм вытеснения.
Реакции перекиси водорода с остальными членами семейства халькогенов
привлекли к себе меньшее внимание. Селениты легко окисляются перекисью
водорода до селенатов [194], хотя реакция с селенистой кислотой H2Se03, по
имеющимся данным, протекает медленно. Элементарный селен медленно окис-
окисляется до двуокиси селена [195], но может превращаться также в селеновую
кислоту H2Se04. Селенистый водород быстро реагирует с перекисью. Эле-
Элементарный теллур растворяется в достаточно концентрированной перекиси
водорода с образованием теллуровой кислоты Н6Те06 [196]. Эта реакция на-
наблюдается также при пропускании электрического тока через теллуровые эле-
электроды в растворе перекиси водорода, причем можно доказать, что промежу-
промежуточным продуктом является теллуристый водород [196]. В общем при действии
перекиси водорода получаются теллураты, хотя в подходящих условиях воз-
возможно образование и теллуровой кислоты [197]. Сообщается, что двуокись
теллура не растворима в перекиси водорода [67], откуда можно вывести за-
заключение, что окисление до теллуровой кислоты, соответствующей ТеО3, не
протекает через ТеО2, так как последняя в связи с нерастворимостью, вероят-
вероятно, не могла бы реагировать дальше. Сообщается 1198], что элементарный
полоний растворяется в подкисленной перекиси водорода.
336 Глава 7
Азот, фосфор, мышьяк, сурьма, висмут
В разных источниках [199J описан ход и кинетика реакции перекиси водо-
водорода с нитритом. Конечным продуктом является нитрат, причем часто упо-
упоминается об образовании в качестве промежуточного продукта пероксоазотистой
кислоты. Нитрат способен к реакции с перекисью водорода только в растворах,
обладающих высокой кислотностью, в нейтральных растворах он практиче-
практически не реакционноспособен. Массой [200] показал, что при реакции цианида
с перекисью водорода образуются цианат и карбонат; пока существует избыток
цианида, кислород в продуктах реакции отсутствует. Шустер [201] указывает,
что при реакции роданида с перекисью водорода образуется сначала аммиак,
затем нитрат. Аммиак может реагировать с перекисью водорода взрывоподобно
[62]. По наблюдениям Вурстера [202], гидроксиламин, заполняющий струк-
структурный пробел между перекисью водорода и гидразином, окисляется пере-
перекисью водорода до нитрата.
Браун [204] в старой работе сообщает, что перекись водорода реагирует
в кислом растворе с гидразином N2H4 [203] с образованием азотистоводородной
кислоты HN3. Позже Гордон [205] нашел, что при этой реакции в водном раство-
растворе образуются лишь молекулярный азот и вода. Максимальная скорость реак-
реакции наблюдается при рН 10, причем в продуктах реакции нельзя обнаружить
азотистоводородной кислоты или аммиака. Более поздние работы [47, 206, 207]
показали, что в зависимости от условий ход этой реакции может быть различ-
различным. Особенно большое значение имеет необычайно высокая чувствительность
этой реакции к каталитическим эффектам; сделано предположение, что реакция
между перекисью водорода и гидразином в разбавленных водных растворах мо-
может протекать только в присутствии катализаторов. Сделано также наблюдение
[208], что в течение известного времени эти вещества могут даже сосуществовать
в паровой фазе при 100° и давлении в несколько миллиметров ртутного столба.
Однако смешение концентрированной перекиси водорода и гидразина, получен-
полученного из обычных источников, как правило, приводит к взрыву, хотя при этом
возможен индукционный период различной длительности, зависящей от кон-
концентрации, температуры, метода контактирования и наличия других видов
молекул. С точки зрения каталитического влияния на эту реакцию изучены
в различных формах следующие вещества: кобальт [205], титан [47], йодид
[206], медь [139, 206, 209, 210] и железо [139, 209]. Сообщается, что молибдат
и ванадат не являются эффективными как катализаторы [209]; оказалось воз-
возможным [206] подавить катализ добавкой комплексообразователя. Реакция
между гидразином и перекисью водорода привлекла к себе интерес еще и вслед-
вследствие применения этих веществ в ракетных топливах.
Вейл [211] описал реакцию между элементарным фосфором и перекисью
водорода и указал/что при этом образуются фосфористый водород РН3 и фос-
фосфорная кислота. Фосфаты совершенно инертны к действию перекиси водорода
и могут быть использованы даже как ее стабилизаторы (см. гл. 9). В других
валентных состояниях, например в виде фосфористой кислоты, фосфор превра-
превращается в фосфат [212]. Сообщается, что элементарный мышьяк окисляется пере-
перекисью водорода в мышьяковую кислоту H3As04. Лучше изучена реакция окисле-
окисления арсенита до арсената, протекающая количественно [213]. Металлическая
сурьма и ее окислы довольно инертны к перекиси водорода, хотя и сообщается
[214], что сульфид сурьмы растворяется в аммиачном растворе перекиси
водорода с образованием антимоната. Описаны некоторые сложные влияния
перекиси водорода на кристаллографические свойства трехокиси сурьмы
Sb2O3 [215]. Мозер [216] обнаружил, что трехокись висмута Bi2O3 окисляется
в четырехокись Bi2O4, которая в свою очередь может быть восстановлена пере-
перекисью водорода. Показано [217], что азотнокислый висмут Bi(NO3K в щелоч-
щелочном растворе превращается в желто-коричневую гидроокись висмута ВКОН).,
Реакции с неорганическими веществами 337
с одновременным образованием еще одного соединения, недостаточно оха-
охарактеризованного.
Углерод, кремний, германий, олово, свинец
Элементарный углерод не вступает в стехиометрическую реакцию с пере-
перекисью водорода, хотя протекающее при этом разложение вызывает в известной
степени изменение поверхности углерода. Руп и Шлее [218] сообщили, что
перекись водорода окисляет карбонат до муравьиной кислоты и формальдегида,
но позже [219] они выяснили, что это действие обусловлено присутствием при-
примесей. Нет никаких сообщений о реакции перекиси водорода с производными
кремния, если не считать данных об абсорбции [220] и образовании перекисей
1221]. Металлический германий протравливается перекисью водорода [222].
Вопрос об инертности металлического олова уже обсуждался при рассмотрении
техники обращения с перекисью водорода (стр. 146). В растворе двухвалентное
олово превращается перекисью водорода в четырехвалентное [223], причем вод-
водная двуокись олова совершенно инертна, а поэтому применяется даже в каче-
качестве стабилизатора. Сравнительная инертность, наблюдающаяся у этих элемен-
элементов, отсутствует у последнего члена группы, свинца, который является весьма
активным катализатором разложения. Металлический свинец растворяется
в подкисленной перекиси водорода; при повышении рН образуются окислы,
причем в щелочных растворах продуктом реакции, безусловно, является
двуокись свинца [224].
Галлий, индий, таллий
В этой группе изучены, по-видимому, лишь реакции таллия; сообщается, что
металлический таллий превращается в гидрат закиси таллия Т1ОН, а закись тал-
таллия Т12О окисляется до окиси Т12О3.
Цинк, кадмий, ртуть
Металлический цинк может быть превращен в окись цинка действием
перекиси водорода. Сообщается также [225], что перекись водорода вызывает
растворение цинка в щелочных спиртовых растворах. Стоун [151] открыл еще
одно необычное явление, что добавка перекиси водорода к серной кислоте,
из которой амальгамированный цинк выделяет водород, вызывает прекра-
прекращение выделения газа; позже выделение водорода начинается вновь. Двух-
Двухвалентный цинк легко образует перекись, причем по одному из старых сооб-
сообщений эта реакция с карбонатом цинка происходит взрывоподобно. Метал-
Металлическая ртуть растворяется в подкисленной перекиси водорода, при повы-
повышении рН образуются окислы ртути. О реакции кадмия с перекисью водорода,
по-видимому, нет никаких сообщений.
Медь, серебро, золото
Металлическая медь способна растворяться в подкисленной перекиси
водорода. Эта реакция изучена Гляунером и Глёкером [226] с точки зрения кор-
коррозии. Возможно также окисление меди до двух-, трех- и четырехвалентного
состояния в зависимости от условий [227]. Сообщается, что перекись водорода
в соляной кислоте может растворять металлическое золото, окислы золота
восстанавливаются щелочной перекисью водорода [228]. Изучено также влия-
влияние легирования золота на скорость его реакции со смесями перекиси водорода
и хлористого натрия [229]. Элементарное серебро также растворяется в под*
22 Перекись водорода
338 Глава 7
кисленной перекиси водорода, причем оно окисляется до одновалентного со-
состояния. В щелочном растворе также происходит восстановление до металла
[230], хотя часто указывается на образование окиси [231].
Действие перекиси водорода на соединения серебра вызывает эффекты,
имеющие значение в фотографии. В щелочном растворе перекись водорода
способствует проявлению скрытого фотографического изображения [232].
При анализе этого вопроса Мис [233], как и исследователи в старых работах,
указал на структурную аналогию между перекисью водорода и таким вещест-
веществом, как гидрохинон, сточки зрения фотопроявляющего действия. Сделан также
[233] логический вывод, что активной частицей, обусловливающей проявляю-
проявляющее действие перекиси водорода, является ион пергидроксила; однако интересно
было бы доказать это положение путем применения экспериментальной техники,
разработанной Джеймсом [234]. Перекись водорода непосредственно может
вызвать также почернение фотоэмульсии; это явление отмечено еще в 1842 г.,
и, поскольку оно достаточно чувствительно даже в отношении минимальных
концентраций перекиси водорода, его используют в качестве метода для откры-
открытия следов перекиси водорода (см. стр. 68). Особое значение для понимания
этого «эффекта Рассела» имело опровержение защищавшегося еще сравнительно
недавно мнения, что отмеченное действие перекиси водорода обусловлено
испусканием ею каких-то таинственных лучей или радиоактивности.
Никель, палладий, платина
Перекись водорода, подкисленная соляной или серной кислотой (но не
уксусной), способна растворять металлический никель. На подкисленный
сульфат никеля [235] перекись водорода не действует; на гидроокиси никеля,
полученной в щелочном растворе, происходит разложение. Металлическая пла-
платина очень устойчива против атаки подкисленной перекиси водорода [236].
Описаны такие эффекты, оказываемые перекисью водорода на платину, как
пассивирование [237], а также действие ее на окислы платины [238]. Двух-
Двухвалентная платина в комплексных соединениях, например (NH4JPtCl4, окис-
окисляется [239] до четырехвалентного состояния (NH4JPtCl4(OHJ.
Кобальт, родий, иридий
Сообщается [240], что скорость растворения металлического кобальта
в перекиси водорода с образованием иона двухвалентного кобальта соответ-
соответствует реакции первого порядка по разности между мгновенной концентрацией
кобальта в растворе и концентрацией его в состоянии насыщения и зависит
от рН раствора и концентрации перекиси водорода. При более высоких рН
образуется гидрат окиси трехвалентного кобальта. Наряду с этим исследование
реакции кобальта с перекисью водорода [241] показало также, что растворен-
растворенный ион трехвалентного кобальта количественно восстанавливается пере-
перекисью водорода. Скорость реакции соответствует первому порядку по концен-
концентрации как иона трехвалентного кобальта, так и перекиси водорода и обратно
пропорциональна концентрации водородных ионов. Для объяснения этого
предложены две стадии с переносом одного электрона и с участием радикала
пергидроксила; в реакции, возможно, как уже предложено выше для реакции
с ионом четырехвалентного церия, участвуют димерные гидратированные
ионы трехвалентного кобальта. В аммиачном растворе перекись водорода
образует комплекс [(NH3MCoO2Co(NH4M]5+, содержащий как трехвалентный,
так и четырехвалентный положительный кобальт (см. гл. 12). Химия родия
и иридия в реакциях с перекисью водорода не исследована.
Реакции с неорганическими веществами • 339
Железо, рутений, осмий
Окисление и восстановление системы закисное железо—окисное железо
в присутствии перекиси водорода подвергалось интенсивному исследованию
в связи со связанным с этой системой процессом разложения. Обычно в кислом
растворе преобладает окисление двухвалентного железа до трехвалентного,
а при более высоких рН происходит выпадение коллоидной гидроокиси.
Рассматривался вопрос [242] и о возможности окисления до других состояний
окисления, причем Эванс, Джордж и Юри [243] изучили образование комплек-
комплекса между ионами пергидроксила и трехвалентного железа. В подходящих
условиях обработка стали перекисью водорода может дать полированную
и коррозионноустойчивую поверхность [244]. Изучено взаимодействие перекиси
водорода с рядом комплексных производных железа [245]. В подкисленной
перекиси водорода ферроцианид окисляется до феррицианида, в щелочных
растворах феррицианид восстанавливается. Аналогично ведет себя и рутено-
цианид. Некоторые из этих реакций активируются светом.
Марганец, технеций, рений
Из реакций перекиси водорода с производными марганца основное вни-
внимание уделено восстановлению перманганата, которое протекает до образова-
образования иона двухвалентного марганца в кислом растворе и двуокиси марганца
в щелочном. Получающаяся при этом двуокись марганца никогда
по своему составу не соответствует точно формуле МпО2; этот вопрос многократ-
многократно подвергался исследованиям [246]. Реакция перекиси водорода с пермангана-
том протекает исключительно быстро, особенно в присутствии иона двухва-
двухвалентного марганца как катализатора, тем не менее проведены кинетические
измерения этой реакции [247]. Механизм ее рассмотрен Мерцом, Стаффордом
и Уотерсом [248]. По реакциям рения с перекисью водорода [249], по-видимому,
имеется всего лишь одно исследование. В нем сообщается о растворении
семиокиси рения Re2O7 в перекиси. :
Хром, молибден, вольфрам
С этого подраздела начинается рассмотрение тех элементов, которые
легко образуют хорошо известные пероксокислоты. Более полно эти соеди-
соединения описаны в гл. 12.
Металлический хром в подкисленной или щелочной перекиси водорода
сравнительно инертен, он лишь медленно растворяется в ней. Кроме изучения
образования пероксохроматов, много внимания уделялось также восстановле-
восстановлению хромового ангидрата СгО3 перекисью водорода до трехвалентного хрома
[250]. Молибден также в любых валентных состояниях превращается в пере-
перекись [251], причем в присутствии перекиси водорода нельзя осадить молибден
в виде фосфоромолибдата [252]. Сернистый молибден реагирует с перекисью*
водорода с образованием сульфата, что исключает возможность применения
этого сульфида в качестве смазочного вещества в контакте с перекисью. Воль-
фрам может растворяться в перекиси водорода с образованием вольфрамовой
кислоты H2WO4, и последняя может быть превращена далее в пероксоволь-
фраматы.
Ванадий, ниобий, тантал
Реакция метаванадата VOJ или пятиокиси ванадия V2O5 с перекисью водо-
водорода с образованием пероксоформы протекает, как предполагают, через ряд-
стадий [253]. Ниобий и тантал влияют на растворимость друг друга в подки-)
сленной перекиси водорода; по-видимому, они образуют коллоидальные рас-;
22*
340 * Глава 7
творы [254]. Фактические растворимости их в перекиси водорода весьма раз-
различны: танталовая кислота растворима лишь частично, а ниобиевая хорошо.
Различаются между собой и металлы [255]: в подкисленной перекиси водорода
тантал практически инертен.
Титан, цирконий, гафний
Желтое комплексное соединение, образующееся при смешении титановой
соли с подкисленной перекисью водорода, имеет большое значение, так как
эта реакция лежит в основе весьма специфического метода анализа на пере-
перекись водорода (см. стр. 455, 466, 546). Сообщается [47], что при реакции пере-
перекиси водорода с ионом трехвалентного титана образуются свободные гидроксиль-
ные радикалы, причем наблюдается легкий переход в пеюекисное состояние
TiO3. Описан также оксисульфат [256]. Сообщается [257], что гидрат окиси
титана образует в перекиси водорода коллоидальный раствор, или гель. Пере-
Перекись водорода способна катализировать образование титанофтрридного ком-
комплексного иона [258]. Цирконий, безразлично, как металл или как сульфат,
по-видимому, совершенно инертен в перекиси водорода [259].
Бор, алюминий, скандий, иттрий
Коллоидальный элементарный бор превращается перекисью водорода
в борную кислоту Н3ВО3 [260]; дальнейшей реакции не наблюдается. Метал-
Металлический алюминий реагирует с перекисью водорода весьма слабо с обра-
образованием гидрата окиси алюминия. Истинной причиной исключительной
инертности алюминия к перекиси водорода при применении его в качестве
материала для емкостей, безусловно, является образование слоя гидроокиси,
хорошо прилипающей к поверхности. Тем не менее в присутствии] других
веществ, а именно хлоридов, возможна коррозия алюминия; по этому вопросу
проведено много работ [261]. Роль хлорида, как предполагается, состоит
в том, что он способствует разрыхлению пленки гидроокиси алюминия и уве-
увеличению электропроводности раствора. Наличие электролитической коррозии
доказано Акимовым и Олешко [262] путем применения колориметрических
индикаторов. Нет никаких данных о реакциях перекиси водорода со скандием
или иттрием.
Редкие земли и актиниды
Очень немногие из этих элементов изучены с точки зрения реакции с пе-
перекисью водорода. Указывается, что характерным продуктом реакции с ред-
редкими землями является окись R4O9; по-видимому, такая окись обнаружена
в случае лантана и самария. Ион четырехвалентного церия количественно реа-
реагирует с перекисью водорода с переходом в трехвалентное состояние; в гл. 10
эта реакция рассматривается как основа одного из аналитических методов
определения перекиси водорода. Из актинидов торий образует окисел Th2O7
[263], а уран, помимо образования пероксоурановой кислоты, окисляется также
от четырехвалентного состояния до иона уранила UOj+ [264].
Щелочноземельные металлы
Металлический магний медленно растворяется в перекиси водорода с обра-
образованием гидроокиси; другие щелочноземельные элементы под этим углом зре-
зрения, по-видимому, не изучались, но можно думать, что они должны вести себя
Таким же образом. Положение этой системы в растворе определяется равнове-
равновесием между перекисью и гидроокисью металла и перекисью водорода, изучен-
изученным, например, Абегом [265] в отношении гидроокиси бария (см. также гл. 3).
Реакции перекиси водорода в органической химии 341
Описаны реакции галогенидов радия с перекисью водорода, однако при про:
верке оказалось, что реакции в действительности вызываются только излуч
чениями этих соединений.
Щелочные металлы
Щелочные металлы энергично реагируют с перекисью водорода, так же
как и с водой, с образованием газообразного водорода. Бэкер и Паркер [266]
показали, что неодинаковая реакционная способность амальгамы натрия с раз-
различными пробами воды вызывается присутствием перекиси водорода. Пере-
Перекиси этих элементов описаны в гл. 12 (стр. 538 и ел.).
РЕАКЦИИ ПЕРЕКИСИ ВОДОРОДА В ОРГАНИЧЕСКОЙ ХИМИИ
Наибольшее значение перекись водорода имеет для области реакций с орга-
органическими соединениями. Для большей части этих реакций, например
протекающих при отбелке, основные механизмы изучены мало, однако при-
применению перекиси водорода способствует сочетание в ней высокого окисли-
окислительного потенциала с эффективностью и специфичностью действия, а также
безвредность продуктов реакции. В последнее время возросло применение
перекиси водорода для хорошо известных реакций органического синтеза*
например для эпоксилирования, гидроксилирования, образования хинонов,
размыкания кольца, полимеризации и пероксидации. Такого рода реакции
находят применение в производстве восков, смол, полимеров, пластификаторов,
фармацевтических и медицинских препаратов, инсектицидов и многих орга-
органических полупродуктов. Можно считать, что эти виды применения и ис-
исследования соответствующих реакций в дальнейшем, вероятно, сильно
разовьются.
Во многих случаях перекись водорода образует в реакционной смеси
органическую перекись или пероксокислоту, органическую или неорганиче-
неорганическую, причем активным агентом является именно этот вид молекул, а не пе-
перекись водорода. Такого рода образование может быть естественным след-
следствием выбора неводного растворителя, иногда же нарочно подбирают условия
таким образом, чтобы с целью повышения скорости реакции или ее специфич-
специфичности образовалась определенная перекись. Причины неодинаковых свойств
различных перекисей рассмотрены выше при общем обсуждении механизма.
В связи с этими различиями при истолковании результатов, полученных при
применении «перекиси водорода» в качестве реагента, важно выяснить,
какая именно перекисная структура является истинным реагентом. Часто
не имеет значения, вводится ли готовая перекись или же она образуется щ
situ из перекиси водорода, а в некоторых случаях одинаковые результаты
получаются и при разных перекисях. Во многих случаях, когда перекись
водорода только формально числится реагентом, а в действительности им
не является, может оказаться, что фактически действующей перекисью являет-
является какая-либо пероксокислота (пероксомуравьиная или пероксоуксусная,
реже один из высших гомологов их или пероксобензойная кислота). Иногда
in situ образуются пероксокислоты того или иного тяжелого металла, например
осмия или молибдена. Нередко процесс описывается как реакция, протекающая
с участием перекиси водорода, в то время как фактически введенным реаген-
реагентом нужно считать органическую перекись; например, гидроперекись трет-
бутила и перекись бензоила являются обычными наиболее употребляемыми
в таких случаях перекисями: неацильной и двузамещенной ацильной. Если
используются другие перекиси, помимо только что указанных, то гораздо мень-
меньше вероятности, чтобы реакцию можно было приписать перекиси водорода.
К этому списку нужно добавить еще вещества, образующие свободные радика-
342 Глава 7
лы при реакции с перекисью водорода. Особенно большое значение имеет
система перекись водорода—закисное железо, известная как реактив Фентона
[267]. При низких отношениях концентраций перекиси водорода и иона
закисного железа эта система является исключительно активной окислительной
системой; Мерц и Уотерс [268] дали обзор типов органических субстратов,
способных к реакции с реактивом Фентона.
При изучении органических реакций перекиси водорода эти факторы
обусловливают интерес к реакциям пероксокислот и других пероксосоеди-
нений. Превосходным источником такого рода информации является обзор
по пероксокислотам, сделанный Суэрном [269] и содержащий 700 ссылок
на литературу. Другие обзоры органических реакций перекиси водорода
и пероксокислот опубликовали Тейлор [270], Джонсон [271], Фрис [272],
Худлички [273] и Теста [274]. Типичные реакции Шенли и Гринспэн [275]
свели в ценную таблицу. Нижеизложенный материал представляет собой
краткое введение в литературу, где описываются типичные органические
реакции перекиси водорода.
Алканы
Предельные парафины не реагируют даже в случае контакта с концент-
концентрированной перекисью водорода шц с перекисью водорода в присутствии
металлических катализаторов [275], но эмульсии или механические дисперсии
их в концентрированной перекиси водорода могут детонировать, что верно
и в отношении многих других органических веществ. Алкильные реактивы
Гриньяра реагируют с образованием спиртов [276].
Этиленовые соединения—эпоксилирование и гидроксилирование
Соединения RCH=CHR', содержащие непредельную углерод-углерод-
углерод-углеродную связь, могут быть. под действием различных перекисей превращены
в эпоксильную форму RCH=CHR' и далее в многоатомный спирт, или
О
гликоль RCH(OH)CH(OH)R\ Суэрн в двух сообщениях [269,277] дал обзор
литературы по этим реакциям, где обсуждается механизм и эксперимен-
экспериментальная методика. Там же приведены подробные таблицы веществ, всту-
вступающих в эту реакцию. Опубликованы также менее полные обзоры, касающиеся
главным образом практических аспектов этой реакции [278], иногда называе-
называемой реакцией Прилежаева и осуществленной с алифатическими, ароматиче-
ароматическими, алициклическими и гетероциклическими олефинами, непредельными
жирными кислотами и эфирами, жирами и маслами и непредельными спиртами.
Обыкновенная перекись водорода сама по себе неэффективна; так, пентен-1
не реагирует с 90%-ной перекисью водорода даже в присутствии железа или
ванадия [275]. Чтобы реакция протекала, обычно требуется присутствие пер-
оксокислоты с большей электронной асимметрией перекисной группы по
сравнению со структурой перекиси водорода. Остановится ли реакция на
стадии эпокиси или будет продолжаться до образования гликоля, зависит
от условий: для получения эпокиси с высоким выходом требуются более мягкие
условия реакции. Пероксокислоты не способны к эпоксилированию кетонов
и кислот, содержащих двойную связь в положениях a,t3 к карбонильной груп-
группе. Однако в этом случае можно применять щелочную перекись водорода
[270]. При применении пероксокислот эти непредельные кетоны окисляются
в сложные эфиры, а а- и р- непредельные кислоты реагируют в жестких усло-
условиях с образованием соответствующих оксикислот [279]. Разработаны неор-
неорганические катализаторы гидроксилирования, функционирующие в форме
Реакции перекиси водорода в органической химии 343
пероксокислот [67, 280]; катализатор на основе осмия в гидроперекиси трет-
бутила носит название реактива Миласа. Изучено фотохимическое присоеди-
присоединение перекиси водорода к двойной связи [281 ]. В частности, имеется ряд работ
по. алициклическим соединениям. Циклогексан сам по себе не реагирует [275].
Изучено гидроксилирование циклогексанона и превращение его в адипиновую
кислоту [282], хотя это окисление, вероятно, протекает через промежуточное
образование перекиси кетона, в связи с чем оно не должно рассматриваться
в этом разделе. Проведены также работы с терпенами, кадаленом [283] и бра-
зилеином [284], с терпеном цинеолом [285], с терпеновым спиртом сабинолом
1286] и с одним производным индена [287].
Спирты
Этанол на холоду в заметной степени не реагирует с концентрированной
перекисью водорода [275], но в смесях внутри определенного интервала кон-
концентраций может быть вызвана детонация исключительной силы. Однако
в присутствии иона трехвалентного железа реакция с этанолом идет до обра-
образования уксусной кислоты [288] или даже двуокиси углерода [275]. Трет-
алкиловые спирты можно превратить перекисью в гидроперекиси алкилов
[70]; получен ряд продуктов из третичных ароматических спиртов [289].
Многоатомный спирт глицерин дает с перекисью водорода непосредственно
муравьиную кислоту, причем промежуточными продуктами являются глице-
глицериновая и гликолевая кислоты [290]. В присутствии иона окисного железа
образуется глицериновый альдегид, а карбонат кальция способствует обра-
образованию формальдегида и кислоты. Механизм этих реакций со спиртами изучен
Мерцем и Уотерсом [291].
Карбоновые кислоты и кетокислоты, альдегиды и кетоны
Реакция перекиси водорода с простейшими карбоновыми кислотами
часто применяется в качестве метода препаративного получения пероксокислот
[292]. Возможно и дальнейшее окисление, причем кислоты с более длинной
цепью менее чувствительны к такому окислению. При окислении двуосновой
кислоты (щавелевой) кислая среда способствует протеканию окисления до
двуокиси углерода, а основная среда тормозит это окисление [293]. Аналогич-
Аналогичное поведение наблюдается у мезоксалевой кислоты [294]. Наличие гидро-
ксильных групп повышает скорость окисления, например в ряду кислот: янтар-
янтарная, яблочная и винная [295]. Исследованы также непредельные фумаровая
и малеиновая кислоты [296]. Окисление малеинового ангидрида перекисью
водорода предложено в качестве метода производства винной кислоты [297].
Изучены реакции перекиси водорода с кетокислотами, например с глиокси-
ловой и ацетоуксусной [298]; рассмотрены механизмы, которые в состоянии
объяснить наблюдаемый основной катализ при этой реакции [299]. Иссле-
Исследованы реакции перекиси водорода с дикарбонильным соединением—глиок-
салем [300], с родственной ему гликолевой кислотой [301], стрикетонами [302]
и другими кетонами [303].
Альдегиды реагируют с перекисью водорода непосредственно с образованием
перекисей оксиалкилов RCH(OH)OOH и RCH(OH)OOCH(OH)R, а последние
в свою очередь реагируют и дальше с образованием кислот и других продук-
продуктов [304]. Эта реакция катализируется кислотой [305]; изучена кинетика таких
реакций с муравьиным, уксусным и пропионовым альдегидами [306].'Окисле-
[306].'Окисление формальдегида дает муравьиную кислоту [307], но следует отметить, что
в щелочных условиях одним из продуктов является и молекулярный водород
1308].
344 Глава 7
Органические азотистые соединения
Интересно отметить окисление ароматических аминов, например анилина,
до нитробензола и азоксибензола [309]. Первичные амины RNH2 бурно разла-
разлагаются перекисью водорода; вторичные амины R2NH энергично реагируют
с образованием гидроксиламинов R2NOH, а третичные R3N окисляются до
окисей аминов R3NO [275]. Аналогичным образом нитробензол в присутствии
аскорбиновой кислоты и железа превращается в нитрофенилгидроксил-
амин [310]. Нитрилы RCN переходят при действии перекиси водорода в амид^
RCONH2; исследованы реакции бензонитрила [74] и нитрила адипиновой кис-
кислоты [311]. Сообщается также об омылении нитрилов [312]. Аминокислоты
реагируют с перекисью водорода в присутствии иона окисного железа с пере-
переходом в кетокислоты [313]. Азобензол легко превращается в азоксибензол
[314], причем сообщается о сочетании его с образованием трисазоксипроизвод-
ных. Проведены исследования и по обесцвечиванию трифенилметанового кра-
красителя малахитового зеленого [315]. Очень наглядными являются люмине-
люминесцентные реакции перекиси водорода g некоторыми [азотистыми соединениями.
Лучше других известна реакция с люминолом, или гидразидом 3-аминофталевой
кислоты 1316]; при наблюдении этой реакции в темноте видно свечение, напо^
минающее свет от светлячка. Хантрес, Стенли и Паркер [317] подробно опи-
описали условия проведения этой реакции наиболее эффективным и наглядным
образом. Описаны также и другие люминесцентные реакции с масляным
желтым [318], пирогаллолом [319] и рядом других красителей [320]. Имеется
сообщение и о люминесцентных реакциях с неорганическими соединениями[321 ].
Органические сернистые соединения
Сообщается, что перекись водорода может образовать меркаптаны из смеси
сернистого натрия и хлористого бензила [322]. Такого рода соединения с сульф-
гидрильными группами (меркаптаны) могут в свою очередь быть окислены пе-
перекисью до дисульфидов; так, этилмеркаптан QHgSH превращается «в диэтил-
дисульфид QH^SSQHg, хотя обычно эта реакция требует катализаторов.
Примером, имеющим очень большое значение, является превращение цистеина
в цистин [323]; подобным же образом изменяется трипептид (глутатион)
[324]. Эти дисульфиды, которые можно называть также персульфидами,
могут быть в свою очередь превращены в такие продукты, как RSOSOR [3251
(см. на стр. 355 о реакции с шерстью). Цистин реагирует с перекисью
водорода с образованием таурина [326]. Сульфиды окисляются в сульфоокиси;
так, дибензилсульфид (CeH6CH2JS дает дибензилсульфоксид (CeH6CH2JSO;
имеется сообщение об аналогичных превращениях алкилсульфидов и хлорал-
кил сульфидов [327], арилтритилсульфидов [328] и одного аминосульфида
[329]. Дальнейшее окисление может привести к сульфонам R2SO2,
Углеводы
В отсутствие катализатора углеводы сравнительно нереакционноспособны,
хотя и могут образовать смеси, способные к детонации. В присутствии железа
как катализатора может произойти хорошо известная реакция получения
низшей' альдозы из альдоновой кислоты; так, d-арабоновая кислота
СН2ОН (СНОН)зСООН превращается в d-эритрозу СН2ОН(СНОНJСНО.
Моррель и Крофтс [330] провели подробные исследования по окислению
углеводов; изучены также фруктоза [331] и диацетат /-рамнозы 1332].
Органические перекиси 345
Ароматические соединения
Бензол и толуол не реагируют с перекисью водорода в отсутствие ката-
катализатора. Если в слой перекиси ввести соединение железа и смесь перемешать,
происходит окисление до фенола. Если проводить процесс так, чтобы окисле-
окисление протекало дальше, происходит глубокое окисление с образованием темно-
окрашенных и коллоидных веществ [333]. Боттомли и Блэкмэн [334] указы-
указывают, что эта реакция может находиться в определенной связи с образованием
торфа и угля. В присутствии уксусной кислоты наблюдается окисление дру-
других замещенных бензолов; например, анилин окисляется в нитробензол и азо-
ксибензол, а бензальдегид дает бензойную кислоту. Полициклические углево-
углеводороды образуют хиноны; при продолжении окисления происходит размыка-
размыкание кольца; например, при окислении фенантрена образуется дифеновая
кислота. Подобным же образом нафтол превращается в нафтохинон [335], при-
причем происходит и разрыв кольца, например с образованием о-карбоксикорич-
ной кислоты. Перекись водорода не активирует сульфирование нафтола
[336]. С другой стороны, перекись водорода способствует галогенированию ряда
соединений [337], например образованию хлоранила из п-бензокинона.
Гетероциклические .соединения
Проведено много работ по реакциям перекиси водорода с гетероцикличе-
гетероциклическими соединениями. В качестве примеров реагентов можно привести фурфу-
фурфурол [338], тиофен [339], никотиновую кислоту [340],тимин [341], производные
пиразина [342], пиперазин [343],феназиныихиноксалины [344] и мочевую кис-
кислоту [345]. Многие такие соединения представляют биологический интерес;
ниже по ним приведем дополнительный материал. 8-Оксихинолин имеет значе-
значение для стабилизации перекиси водорода. Изучены реакции большого числа
алкалоидов с перекисью водорода, особенно Шером [346] и Фернандесом и Пиз-
зарозо 1347]. К этим исследованиям нужно добавить также работы по никотину
[348], наркотину [349] и колхицеину [350].
Проведены многочисленные исследования по аморфным и полимерным
органическим веществам и их взаимодействию с перекисью водорода. Значи-
Значительная часть этой работы представляет интерес с точки зрения биологиче-
биологического значения перекиси водорода; она рассматривается в соответствующем
разделе. Сюда нужно добавить еще работы по влиянию перекиси на натураль-
натуральный и синтетический каучуки [351], уголь 1352] и торфяной мох [353].
ОРГАНИЧЕСКИЕ ПЕРЕКИСИ
Область органических перекисей настолько значительна, что требует отдель-
отдельного обзора, и охватить ее в этой монографии попыток не предпринималось. Пред-
Представлено достаточно материала по выяснению взаимоотношений между пере-
перекисью водорода и органическими перекисями. Эти вопросы освещены, в частно-
частности, в разделах по номенклатуре, образованию, технике безопасности, обраще-
обращению и хранению, структуре, механизму реакций и применению. Отметим ряд
книг и обзоров, содержащих вполне авторитетное введение к этой теме. Выше
мы уже ссылались на обзор Суэрна [269] и книгу Тобольского и Месробяна [40 ].
Ценное введение в эту область дает также статья Миласа, Маргулиса и Шенли
1354].Прекрасная трактовка препаративных методов, имеющих значение также
для выяснения возможности применения перекиси водорода в процессе получе-
получения органических перекисей и указания на некоторые опасности, сопряжен-
сопряженные с этими веществами, опубликована Криджи [355]. В последнее время по-
появились некоторые другие обзоры [356], которые могут оказаться очень полез-
полезными вследствие различного их содержания и различного подхода к этой теме.
346 Глава 7
ПЕРЕКИСЬ ВОДОРОДА В БИОЛОГИЧЕСКИХ ПРОЦЕССАХ
Выше уже указывалось, что для химии перекиси водорода имеет суще-
существенное значение положение последней *в качестве окислительно-восстано-
окислительно-восстановительного промежуточного продукта между молекулярным кислородом
и водой. Поскольку живые организмы используют атмосферный кислород для
окисления различных питательных веществ до двуокиси углерода и воды в ка-
качестве средства получения необходимой энергии, это соотношение особенно
интересно в жизненно важных процессах биологического окисления. Ясно,
что перекись водорода представляет продукт обмена, который в нормальных
условиях образуется и разлагается самыми различными растениями и живот-
животными, однако до настоящего времени в выяснении механизма биологического
окисления, его ограничений и значения этого аспекта сделаны только первые
шаги. Перекись водорода участвует не только в процессах, обеспечивающих
организм энергией, но и в других процессах, например синтеза и детоксикации.
Все это нормальные и, возможно, обязательно необходимые виды деятельности
живой ткани, в которой перекись водорода участвует в исключительно малых
концентрация^. Интерес представляют также эффекты, которые перекись водо-
водорода оказывает на жизненные процессы при возникновении ее в аномально
высоких концентрациях, за счет ли неправильной функции организма или пред-
преднамеренной или случайной экспозиции. Поскольку перекись водорода в нор-
нормальных условиях встречается в природе лишь в минимальных количествах,
экспозиция возможна лишь в процессах производства и применения. В этом
разделе рассматриваются все эти аспекты химии перекиси водорода, и только
механизм каталитического разложения под действием ферментов обсуждается
на стр. 418. По этим вопросам имеется большая и несколько разбросанная
литература; этот недостаток в известной степени смягчается наличием ряда
превосходных обзоров. По этим причинам приводимый ниже материал
нельзя считать исчерпывающим в отношении полноты охвата литературы.
Биологическое окисление*
В упрощенной форме процесс получения энергии, требующийся для жиз-
жизненно важных процессов, рассматривается как регулируемый процесс окисления,
где субстрат под действием кислорода превращается в воду и двуокись углерода.
Это происходит в виде отдельных стадий; в каждой из них протекает реакция не-
нескольких атомов субстрата в водных растворах, близких к нейтральным, при
температурах, которые химиком рассматриваются как низкие, причем катализ
на этих стадиях осуществляется биологическими катализаторами, или фер-
• ментами. С точки зрения значения перекиси водорода нужно различать три
типа таких реакций. Это—те стадии, в которых перекись водорода образуется,
функционирует в качестве окислителя и каталитически разлагается. Ниже
дается краткая схема, иллюстрирующая, каким образом по современным
воззрениям эти стадии протекают.
Первой стадией окисления субстрата является обычно дегидрирование.
В этой реакции фермент, носящий название дегидразы** и обычно активный
* Проблемы биологических окислительных процессов неразрывно связаны с име-
именем выдающегося советского ученого академика А. Н. Баха, который в течение всей
своей жизни занимался изучением химизма дыхания. О роли перекисных соединений
в процессах биологического окисления см. А. Н. Б а х, Собрание трудов по химии и
биохимии, Изд. АН СССР, М., 1950.— Прим. ред.
** Терминология ферментов находится еще в стадии развития, и пока применение
ее еще не установилось; например, некоторые из этих дегидраз называются иногда ок-
сидазами. Недавно Гофман-Остенгоф изучил проблему номенклатуры в области фер-
ферментов [357]. Б соответствии с данной им схемой, все ферменты, представляющие для нас
интерес, являются оксидоредуктазами.
Перекись водорода в биологических процессах 347
только в отношении одного какого-либо определенного вещества или класса
веществ, активирует два водородных атома субстрата таким образом, что
они легко переносятся к молекуле акцептора. Такое дегидрирование эквива-
эквивалентно окислению субстрата; так, молочная кислота СН3СНОНСООН может
превратиться в пировиноградную кислоту СН3СОСООН. В качестве акцепторов
водорода, отнятого от субстрата, могут служить различные вещества; часто
эти акцепторы называют переносчиками, поскольку они занимают определен-
определенное положение в цепи из отдельных стадий, в которых происходит перенос
водорода до конечной его реакции с кислородом, заканчивающейся образо-
образованием воды. Существенное значение имеет следующее различие: является ли
та молекула, которая отнимает атом водорода от субстрата, молекулярным
кислородом или каким-либо другим веществом. Если это молекулярный кисло-
кислород, то результат переноса водорода состоит в образовании перекиси водорода,
и фермент, катализирующий такой перенос, называется аэробной дегидразой.
Дегидразы, которые катализируют перенос водорода к другим акцепторам или
переносчикам (не к молекулярному кислороду), называются анаэробными
дегидразами.
Дальнейшими стадиями окисления субстрата могут являться декарбокси-
лирование, гидратация и добавочное дегидрирование. При декарбоксилирова-
нии субстрат теряет карбоксильную группу с образованием двуокиси углерода,
причем этот процесс осуществляется карбоксилазой. Стадия гидратации также
может потребовать вмешательства фермента—гидратазы. Другие стадии, имею-
имеющие значение для этой цепи реакций разложения субстрата, регулируются
ферментами, называемыми оксидазами. ~)ти ферменты активируют молекуляр-
молекулярный кислород, делая его способным и к окислению определенных субстратов*
не вызывая в то же время действия со стороны дегидразы или переносчиков
водорода. Результатом действия оксидазы является выделение воды; образо-
образования перекиси водорода в этих случаях не отмечено. Аналогия с действием
оксидаз имеется в классе ферментов, носящих название пероксидаз. Эти фер-
ферменты обнаружены в растениях, молоке и крови, причем они активируют пере-
перекись водорода, сообщая ей способность принять участие в процессах окисления.
По-видимому, субстраты, вступающие в такие реакции, являются некоторыми
фенолами и ароматическими аминами. Однако в этих классах имеется ряд био-
биологически важных соединений, которые не изменяются под действием пер-
оксидазы; вообще же очень мало известно о значении этого действия перекиси
водорода в общей схеме окисления. Продуктами действия пероксидазы являют-
являются окисленный субстрат и вода; молекулярного кислорода совершенно не обра-
образуется. Поэтому любую стехиометрическую реакцию перекиси водорода, про-
протекающую таким образом и, следовательно, не приводящую к образованию
кислорода, можно считать «пероксидатической»; этот термин иной раз приме-
применяется в небиологических системах. Так, при обработке текстиля желательно
и экономически выгодно, чтобы действие перекиси водорода было пероксида-
тическим, т. е. чтобы оно выражалось в эффективной отбелке без потерь пере-
перекиси за счет каталитического разложения.
Задача • каталитического разложения перекиси водорода, и таким образом
устранения ее, осуществляется в биологических системах ферментом, называе-
называемым катал азой. Этот фермент, по-видимому, встречается во всех животных
и растениях (за исключением некоторых микроорганизмов), и одной из целей,
которым он служит, является предотвращение возможности накопления пере-
перекиси водорода в токсических концентрациях. Каталаза исключительно эффек-
эффективна в этом процессе; она активна при очень низких концентрациях перекиси
водорода и способна осуществлять разложение со скоростью, значительно
превышающей скорость для большинства других известных катализаторов.
Разложение под действием каталазы иногда называют «каталатическим»
с целью отличить его от пероксидатического действия и сделать возможным
348 Глава 7
противопоставление. Не исключено, что в дальнейшем это может привести
к известной путанице, поскольку в настоящее время доказано, что каталаза
в состоянии также активировать перекись водорода, в результате чего послед-
последняя приобретает способность к окислению некоторых спиртов до альдегидов.
Это носит название сопряженного окисления, а также пероксидатического дей-
действия. Значение этой функции каталазы еще не выяснено, но некоторые исследо-
исследователи придерживаются того мнения, что это действие может иметь не мень-
меньшее или даже большее значение, чем каталитический эффект фермента, при-
приводящий только к потере перекиси водорода.
Процессы, как описанные выше, так и другие, связанные с ними, рас-
рассматриваются подробно в общепринятых руководствах, например в учебнике
Веста и Тода [358] или в сообщениях по дыхательным ферментам, выпущен-
выпущенным под редакцией Ларди [359]. Сточки зрения изучения роли перекиси водо-
водорода нужно различать два аспекта, которые ограничивают общее количество
имеющегося материала по данному вопросу и интерес к нему. Перекись водо-
водорода представляет простую структуру, повсеместно встречающуюся в биоло-
биологических системах; это лишает ее интереса в качестве специфического продукта
обмена, играющего специальную роль в химии клетки. В то же время воз-
возникает вопрос, действительно ли перекись водорода играет жизненно важную
роль, т. е. является ли она звеном в общей последовательности стадий биоло-
биологического окисления. Сейчас, по-видимому, нельзя еще дать окончательного
ответа на этот вопрос, но проведенная дискуссия [360] показала, что основная
цепь процессов может быть окружена защитной системой торможений и рав-
равновесий, которая обусловливает возможность осуществления жизненно важных
стадий без перерыва; перекись водорода может играть роль и в главных
и в вспомогательных функциях. Необходимо также отметить, что вопросы,
касающиеся образования и реакции перекиси водорода в биологическом окисле-
окислении, привлекали к себе серьезное внимание и гораздо ранее работавших
исследователей и привели к разработке теорий, предшествовавших установ-
установлению вышеприведенной схемы. Особенное значение в работах раннего периода
имели теории Варбурга и Виланда, которые противоречили друг другу. Вар-
бург развил концепцию активации кислорода и рассматривал реакцию моле-
молекулярного кислорода и железа, связанного с ферментом, как первичный акт
при дыхании. Эта точка зрения, особенно ярко выраженная в книге Варбурга
[361], исключает возможность образования перекиси водорода за счет соеди-
соединения водорода с кислородом. Виланд разработал противоположную теорию,
в которой выдвигается на первый план активирование водорода и как след-
следствие образование перекиси водорода за счет действия кислорода в качестве
акцептора. Эта теория, в которой подчеркивается роль дегидрирования, защи-
защищается также в книге, написанной Виландом [362]. В настоящее время
принимается наличие обоих типов действия при биологическом окислении.
Указанные теории окисления, а также более ранние рассматриваются. Оппен-
геймером и Стерном [363]. Интересно отметить, что представления о биологи-
биологическом окислении в значительной части разработаны на основании ряда
«модельных реакций», т. е. систем in vitro, обладающих одним или несколькими
свойствами, аналогичными свойствам, которыми, по-видимому, обладает
и живая система, т. е. содержанием железа и способностью катализировать реак-
реакции типа происходящих в организме. Такого рода сравнения имеют значения
для планирования и проведения биологических исследований, но не являются
вполне надежными. Так, в 1911 г. Дакин [364] указал, что из всех окислителей
только одна перекись водорода в состоянии вызывать реакции тех же типов,
которые осуществляются,ферментами, что дает право приписать ей участие
в биологических системах. С другой стороны, Дакин возражает против той
мысли, что каталаза также может участвовать в окислительной схеме. Как
упомянуто выше, эта мысль только недавно получила дополнительное эксперт
Перекись водорода в биологических процессах 349
ментальное подтверждение, а потому заслуживает внимания при дальнейших
исследованиях.
Природа ферментов-гемопротеинов
Строение ферментов, имеющих значение для биохимических реакций или
разложения перекиси водорода, сравнительно хорошо изучено, что дает возмож-
возможность разобраться в механизме их действия. Ценные введения к природе
всех ферментов опубликованы в книгах Самнера и Сомерса [365] и Ледлера
[366]. Однако, чтобы понять взаимоотношения между перекисью водорода
и ферментами, достаточно ознакомиться с некоторыми основами, касающимися
группы ферментов и родственных им соединений, известных под названием ге-
мопротеинов, а также с соответствующей номенклатурой. Из ферментов наи-
наибольшее значение имеют каталаза и пероксидаза. Эти ферменты состоят из бел-
белка, содержащего активную или простетическую группу, известную как прото-
порфирин железа. Эти ферменты очень близки к переносчику кислорода крас-
красных кровяных шариков, гемоглобину и родственному ему компоненту мышц
Н00ССН2СН2 ^? СН2СН2С00Н
Рис* 60. Протопорфирин железа (гем).
миоглобину, функция которого также состоит в связывании и отдаче кислорода.
Эти вещества менее походят на некоторые другие ферменты, например на
протеолитические ферменты пищеварения, которые, хотя и обладают сложной
белковой структурой, не содержат порфирина тяжелого металла в качестве
простетической группы и часто находятся в организме в форме предшествен-
предшественников, которые требуют активации для приобретения активности в качестве
ферментов.
Хотя в общих чертах все эти четыре гемопротеина, имеющие значение
для биохимии перекиси водорода,—каталаза, пероксидаза, гемоглобин и мио-
глобин—схожи друг с другом, все же между ними существуют и значительные
различия, что объясняет неодинаковое их отношение к перекиси водорода
как в самом организме, так и в экспериментальных условиях. Эти различия
выражаются в неодинаковых молекулярных весах, относительных количествах
составляющих аминокислот, входящих в состав белковой части молекулы,
и в числе и доступности групп протопорфирина железа, связанных с белком.
Эти различия подробно проанализированы Джорджем [367]. Особое значение
имеет то, что каталаза и гемоглобин имеют по четыре протопорфириновых груп-
группы на молекулу, тогда как пероксидаза и миоглобин несут только по одной
группе.
Структурная формула протопорфирина железа, носящего также название
гема, представлена на рис. 60.
Эта структура родственна структуре хлорофилла [368], где комплексо-
образующим металлом является магний, а также структуре фталоцианинов.
350 Глава 7
Как указано, центральный атом железа лежит в плоскости 16-членного пор-
фиринового кольца, причем он связан с атомами азота пиррольных колец
четырьмя из шести имеющихся валентностей, а поэтому для завершения
октаэдрического комплекса остаются две валентности; эти валентности нахо-
находятся выше и ниже плоскости чертежа показанной модели. Кроме того, нужно
учитывать также степень окисления железа; когда железо находится в закисном
состоянии, как показано на рисунке, то протопорфирин носит название гема
или, более четко, феррогема. Когда железо находится в трехвалентном состоя-
состоянии, то порфирин называется ферригемом или более специфически гемином
или гематином (если желательно указать на наличие координационно связан-
связанного аниона). Если гем находится в свободном состоянии в растворе и не связан
с белком, то предполагается, что две молекулы воды находятся в координацион-
координационной связи с пятым и шестым положениями валентности железа. Джордж
[367] обозначает этот феррогем следующим образом: H2OFepH2O. При окис-
окислении он переходит в H2OFep-H2O с одним положительным зарядом. Этот
ферригем может координировать хлор-ион с переходом в H2OFep-Cl и в этом
случае называется гемином; при координированном гидроксильном ионе
ферригем обозначается как гематин H2O-Fep-OH, хотя эти термины иногда
употребляются и менее специфически, лишь для обозначения наличия окисного
железа. Возможна координация и других молекул, причем в первую очередь
играет роль координация с перекисью водорода. Феррогем легко координи-
координирует азотистые основания, например пиридин, образуя гемохромогены
B-Fep-B. Такое же соединение с ферригемом B-Fep-B называется парагема-
тином.
Когда такой протопорфирин железа присоединяется к определенному
белку, образуется собственно фермент. Связывание происходит, по-видимому,
через одну из валентностей железа, а дополнительно и за счет взаимодействия
белка с двумя группами пропионовой кислоты протопорфирина. В случае
каталазы четыре группы ферригема, или гемина, присоединяются к одной моле-
молекуле белка такой величины, что общее содержание железа составляет около
0,1 вес.% [369]. Катал аза из различных источников или разных видов (на-
(например, бактериальная, печеночная или эритроцитная) может обладать разной
активностью. Происхождение этих различий еще не вполне ясно, но оно,
возможно, связано с различиями в доступности геминовых групп или же зави-
зависит от замещения одной или нескольких групп гемина биливердином, произ-
производным гема, но с разомкнутым кольцом [370]. Ферригемы каталазы не легко
восстанавливаются до феррогема; действительно, только в последнее время
выяснена возможность такого восстановления без разрушения фермента.
Фермент пероксидаза также образуется подобным образом путем присоедине-
присоединения ферригема к белку. Весьма отчетливое различие заключается в том, что
в пероксидазе имеется только одна группа ферригема на молекулу. Молекула
белка также меньше и обладает способностью к соединению с протопорфири-
ном марганца без потери пероксидатической активности. Пероксидаза отли-
отличается еще тем, что она труднее инактивируется при нагревании, чем каталаза.
Гемопротеиновые ферменты каталазу и пероксидазу часто сравнивают
с гемоглобином и миоглобином сточки зрения отношения к перекиси водорода.
Хотя такого рода подход и может быть поучительным, все же нет никаких
доказательств, что взаимодействие гемоглобина или миоглобина с перекисью
водорода в организме имеет в обычных условиях существенное значение. Оба
эти вещества образуются за счет связывания белка, называемого глобином,
с восстановленным протопорфирином железа, или феррогемом [370]. Гемогло-
Гемоглобин содержит четыре группы феррогема на молекулу, а миоглобин—только
одну. Основная физиологическая функция этих веществ состоит в координа-
координационном связывании молекулярного кислорода. В таком виде гемоглобин
функционирует в качестве переносчика кислорода из легких к тканям, что
Перекись водорода в биологических процессах 351
может быть представлено следующим образом: 10
Gb. Fep • Н2О + О2 = Gb • Fep ¦ О2 + Н2О. D2)
Гемоглобин Оксигемоглобин
Феррогем как гемоглобина, так и миоглобина может быть окислен до ферри-
гема, например, перекисями с образованием метгемоглобина и метмиоглобина
соответственно. Оба эти вещества (но не их восстановленные формы) обладают
в небольшой мере способностью разлагать перекись водорода и направлять
ее пероксидатическую активность.
В таком кратком введении нельзя дать исчерпывающего списка гемо-
протеинов. По-видимому, аналогичные или близкие структуры имеют и другие
ферменты, например некоторые оксидазы и так называемые цитохромы, однако
они еще не так хорошо охарактеризованы и по взаимоотношению их с пере-
перекисью водорода проведено лишь небольшое число исследований. При описании
структуры гемопротеинов нужно подчеркнуть возможность занятия перекисью
водорода одного из координационных положений при центральном атоме груп-
группы протопорфирина железа. Как указывает Роулинсон [371], атом железа
в этих гемопротеинах является центром активности; протеиновая же и протопор-
фириновая части молекулы выполняют функцию приспосабливания железа
к этой роли, часто весьма специфическим образом, в среде, где более простые
производные железа были бы совершенно инертны или не специфичны. Имен-
Именно это связывание перекиси водорода с"железом способствует активации пере-
перекиси, делая ее чувствительной к разложению или к реакции с другими моле-
молекулами. Проблема изучения механизма реакции перекиси водорода с фермен-
ферментами сводится, таким образом, в значительной мере к выяснению природы
и судьбы этих комплексов. Эти процессы можно сравнить с теми, которые
происходят с другими веществами, присоединяющимися к ферментам, напри-
например с окисью углерода, и особенно с другими кислородными соединениями,
родственными перекиси водорода,—молекулярным кислородом, гидроксиль-
ным ионом и водой.
Реакции с гемопротеинами
В живых системах источником перекиси водорода является восстановление
молекулярного кислорода аэробными дегидразами. Это установлено уже в ста-
старой общепризнанной работе Тарлоу [372], а более поздние работы Кейлина
и Хартри [373] и Кентена и Манна [374] расширили наши знания по биоло-
биологическому образованию перекиси водорода. Особого интереса заслуживает
исследование глюкозооксидазы (в сущности дегидразы) при помощи меченых
атомов, проведенное Бентли и Нейбергером [103]. Этот фермент привлек к себе
большое внимание с точки зрения образования перекиси водорода (см. гл. 2),
а работа с изотопами доказывает, что кислород в образующейся перекиси водо-
водорода получается целиком из молекулярного кислорода и не обменивается с ки-
кислородом воды, функционирующей как растворитель. При образовании в жи-
живой системе перекись водорода либо вступает в реакцию, либо разлагается;
Чане [375] подсчитал, что стационарная концентрация перекиси водорода,
устанавливающаяся в результате равновесия между образованием и разло-
разложением в печени, составляет несколько микромолей на 1 л. Если учесть, что
перекись водорода в такой небольшой концентрации находится в тканевых
жидкостях с рН, близким к нейтральному, то можно сделать вывод, что
перекись водорода фактически не диссоциирована и должна реагировать сначала
именно как перекись.
Один подход к проблеме выяснения природы взаимодействия между пере-
перекисью водорода и гемопротеинами заключается в исследовании свободной
группы протопорфирина железа в том или другом состоянии окисления или
в комплексах. Таким именно путем подошли, например, Горовиц [376] и Ки-
352 Глава 7
кучи [377]; полученный ими материал проанализирован Горовицем [378]
и Джорджем [367]. Показано, что свободный гематин H2O-FepOH образует
комплекс, который может быть представлен как H2O2FepOH, тогда как гем
Н2О- Fep • Н2О не образует такого комплекса. Подобным же образом реагируют
с перекисью водорода парагематины в отличие от гемохромогенов. Например,
пиридиновый парагематин (Py-Fep-Py)+Cr можно заместить последовательно
одной и затем двумя молекулами перекиси водорода; в гемохромогенах при
этом происходит разложение порфирина.
Комплексы, образуемые перекисью водорода с гемопротеинами, изучены
более подробно, сначала методом визуальной спектроскопии, а в более поздних
работах путем применения специальной техники быстрой спектрофотометрии.
Все эти комплексы йастолько неустойчивы, что их не удалось выделить. По-
Показано, что и пероксидаза и каталаза образуют'по три комплекса, тогда как
метгемоглобин и метмиоглобин—только по одному. Эти комплексы различают-
различаются по цвету; и Чане [375] и Джордж [367] в составленных ими обзорах описали
эти различия. Чане характеризует эти комплексы как первичные, вторичные
и т. д. в соответствии с характером спектров. Некоторые из этих комплексов
принимают участие в ферментных реакциях. Проведено много работ для
выяснения их относительных ролей. Чане [375] указывает, что первичные ком-
комплексы наблюдаются лишь для гемопротеинов, активных как ферменты, тогда
как каталитически неактивные гемоглобин и миоглобин их не образуют.
Имеются также различия в константах равновесия при образовании и дис-
диссоциации обоих этих типов комплексов. С механизмом катализа при действии
этих ферментов связано также то, что в отсутствие избытка перекиси водорода
первичные комплексы, относительно говоря, устойчивы. Это дало возможность
титрования гемопротеинов перекисью водорода с применением специальной
техники; такого рода исследования показали, что на каждый атом железа
связывается одна молекула перекиси водорода. Ход этих реакций и форма
образующихся комплексов еще не вполне выяснены. Чане [375] и Джордж
[367] описывают влияние рН и чужеродных ионов на процесс и рассматривают
некоторые возможности. Например, в каждом отдельном случае не ясно,
содержится ли перекись водорода в комплексе в виде недиссоциированной
молекулы или пергидроксильного иона НОа. Таким образом, можно предста-
представить три способа образования комплекса с гемопротеином:
Fep. Н2О + Н2О2 = Fep. Н2О2 + Н2О, D3)
Fep.OH + H2O2 = Fep.OOH + H2O, D4)
Fep. Н2О + Н2О2 = Fep • ООН" + Н+ + Н2О. D5)
Чане указывает, что в виде комплекса, активного как фермент, может высту-
выступать комплекс, содержащий пергидроксильный ион.
Независимо от формы активного комплекса можно считать, что реакция
его с субстратом протекает в соответствии со следующими стадиями:
Фермент + Н2О2 = Фермент • Н2О2, D6)
\ Фермент • Н2О2 + Субстрат = Фермент + Продукты. D7)
При такой упрощенной точке зрения все ферментные реакции являются пер-
оксидатическими; каталатическое действие представляет собой специальный
случай, при котором субстратом является вторая молекула перекиси водорода.
Пероксидатическое действие ферментов
Впервые действие пероксидазы наблюдалось в 1863 г. Шенбейном [379],
название же ферменту дал Линосье [380] в 1898 г. Этот фермент широко рас-
распространен в растительных тканях и находится также в молоке и крови, но
Перекись водорода в биологических процессах 353
наличие его в большинстве животных тканей еще не доказано, хотя некоторый
материал в пользу этого и представлен. Пероксидаза активирует не только
перекись водорода, но может функционировать и в сочетании с некоторыми
другими перекисями*. Роль пероксидазы в биологических процессах еще
с достоверностью не известна, но уже проведено много работ для выяснения
природы ее действия, и здесь можно отметить размах этой работы. Эллиот [381]
исследовал в 1932 г. реакции перекиси водорода, катализируемые пероксида-
зой, и указал, что в реакции вступают триптофан, тирозин, ряд фенолов (но
не резорцин), некоторые диамины, нитрит и йодид. Некоторые из этих соеди-
соединений, например йодид [382], изучались и раньше, но полученные результаты
были противоречивыми. Позже исследовано действие пероксидазы на тиолы
[383] (тиомочевину и дитиоурацил), аскорбиновую кислоту [384], гетероаук-
сии [385], токсины дифтерии и столбняка [386], ароматические амины [387],
производные марганца [388] и аминокислоты и белки [389]. Большая часть ра-
работ, приведенная в этих источниках, направлена к выяснению биологического
значения пероксидазы. Так, Рэндол [383] рассматривает предположения, что
пероксидаза принимает участие в синтезе тироксина, а Кентен и Манн [388]
приводят доказательства роли пероксидазы, в сообщении марганцу функции
незаменимого питательного микроэлемента растений. В работе Сайзера [389]
к списку аминокислот, реагирующих с пероксидазой, добавлены еще цистин
и цистеин; этот автор доказал высокую избирательность действия пероксидазы
наркологическую активность таких белков, как гормоны, токсины, анти-
антитела, ферменты, фибриноген, казеин и глобулин. Сообщается, что пероксидаза
может оказывать регулирующее действие на такие белки. Боле и Хейл [390]
показали, что пероксидатическое действие вызывает нежелательное возникнове-
возникновение коричневого пигмента на свежесрезанной поверхности яблока, и рассмот-
рассмотрели методы борьбы с этим явлением.
Фермент каталаза также оказывает пероксидатическое действие, хотя
исключительная его способность к действию в качестве катализатора разложе-
разложения в течение длительного времени маскировала указанное свойство и отвлека-
отвлекала от него внимание. Это пероксидатическое действие легко понять в свете
реакций D6) и D7), однако имеются значительные различия в отношении
пероксидатического действия между. каталазой и пероксидазой. При иссле-
исследовании этого действия каталазы [373] сделано наблюдение, что оно требует
непрерывного поступления перекиси водорода, например получаемой под
действием дегидразы. Имеются также различия в отношении специфичности
субстратов. Каталаза действует пероксидатически на первичные и вторич-
вторичные спирты и на нитрит. Скорость такой реакции зависит [391] от длины цепи
спирта. С другой стороны, пероксидаза, как указано выше, реагирует с самыми
различными веществами. Таким образом, по-видимому, нет никакой группы,
реакция с которой была бы специфичной для пероксидазы, тогда как каталаза
во всех реакциях вступает во взаимодействие только с гидроксильной группой,
например в RCHOH или Н2О2; замещение водорода в этой группе исключает
возможность реакции с каталазой. Так, комплекс из каталазы и перекиси
водорода может реагировать с перекисью водорода или гидроперекисью
алкила, но не с двузамещенной перекисью водорода [375]. Имеются также
различия в кинетическом порядке пероксидатического действия каталазы
и пероксидазы. Чане [375] считает, что небольшие концентрации перекиси
водорода, как правило, способствуют проявлению пероксидатического действия
каталазы, тогда как при более значительных концентрациях возможно про-
проявление каталатического действия. Показано также [392], что при одновремен-
* Обобщение работ школы академика А. Н. Баха по вопросу о взаимодействии прп
оксидаз с перекисными соединениями см. Михлин Д. М., Пепоксияш и Поппгл .
дазы, Изд. АН СССР, М.-Л., 1947.-Прим. персе. Н ДЫ " ПеР0КС»"
23 Перекись водорода
354 Глава 7
ном присутствии каталазы и пероксидазы в растительном экстракте каталати-
ческое действие подавляется до завершения окисления субстрата. Сообщается
также, что пероксидаза является главным образом растительным ферментом,
а каталаза—животным и первичные роли этих двух ферментов в обоих этих
царствах—растительном и животном—могут быть аналогичны. По литератур-
литературным данным, пероксидазоподобной активностью обладают и другие вещества,
например некоторые альдегиды [393], каротин [394] и аскорбиновая кислота
[395].
Физиологическое действие перекиси водорода
Проведена значительная работа с целью выяснения влияния перекиси
водорода на вещества или процессы, имеющие биологическое значение. Эти
исследования сильно отличаются друг от друга по глубине и методу. В не-
некоторых случаях пользовались заведомо модельными системами, что давало
возможность четко определить условия, однако результаты в отношении жи-
живых систем иногда имели лишь сомнительную ценность. Другие работы про-
проведены с органическими веществами или in vivo, а поэтому они ближе к есте-
естественным условиям, но результаты с точки зрения влияния побочных реакций
и ферментов получились менее определенными. Часто для таких исследований
выбирают бактериальные колонии, а в этой системе возможны неопределен-
неопределенности из-за колебаний концентрации перекиси водорода на различных ста-
стадиях роста, что может быть обусловлено различиями в парциальном давлении
присутствующего кислорода [396] или действием фактора роста [397].
Пейн и Фостер [398] исследовали действие перекиси водорода и осмия на
ряд типичных углеводов. Продуктами реакции оказались кислород, водород,
двуокись углерода, окись углерода, муравьиная и другие кислоты и альдегиды.
Характерным продуктом является водород, происходящий от формальдегида.
Перекись водорода вызывает разложение пектиновых веществ, других поли-
полисахаридов и глюкъзидов [399]. На эти процессы влияет наличие щелочи [400],
железа и меди [401] и аскорбиновой кислоты [402]. Изучение [403] различных
Сахаров в качестве питательных веществ для бактерий показывает, что они
неодинаково влияют на образование перекиси водорода. Родственные вещест-
вещества, рибофлавин [404] и стрептомицин [405], также влияют на образование
перекиси водорода или сами подвергаются ее действию.
Действие перекиси водорода на жиры с точки зрения биологических
процессов изучалось очень мало. Жиры и масла относительно устойчивы,
и большая часть работ с ними [406] проводилась с применением концентриро-
концентрированной перекиси водорода или в присутствии катализаторов—тяжелых метал-
металлов.
Наиболее широкие исследования по действию перекиси водорода на ве-
вещества, представляющие биологический интерес, проведены с белками.
Природа таких реакций может сильно колебаться. Более или менее энергич-
энергичная обработка перекисью водорода может вызвать агрегацию [407] или жела-
желатинирование [408] альбумина, желатины или тканевых экстрактов или даже
разложение их до аммиака, кетонов и альдегидов [409]. В менее жестких усло-
условиях влияние перекиси водорода слабее, что позволяет говорить о фактиче-
фактическом физиологическом действии. Такого рода исследования были проведены
с фибрином и фибриногеном [410] (белками, участвующими в свертывании кро-
крови), глобулином [411] (имеющим значение для иммунитета) и миозином [412]
(белком мышцы, обусловливающим ее способность к сокращению); для этой
темы имеют интерес и работы Сайзера [389] по действию пероксидазы на белки.
Этот уровень реакции обладает известной специфичностью; так, сделано
наблюдение [413], что обработка казеина перекисью водорода лишает питатель-
питательной ценности только содержащиеся в нем метионин и триптофан. Эти бел ко-
Перекись водорода в биологических процессах 355
вые реакции имеют особое значение для ферментов, многие из которых подвер-
подвергаются действию перекиси водорода. Проведены работы по выживанию спермы
[414], уменьшению ядовитости змеиного яда [415], изменению промежуточных
продуктов в процессах фиксации азота [416] и инактивации бактериальной
и дрожжевой дегидразы [417, 418], папаина [418, 419], фосфатазы [420] и фи-
фиговой протеазы [421] под действием перекиси водорода. В большинстве слу-
случаев (если не считать уреазы и гексокиназы [418]) перекись водорода угнетает
активность ферментов, и в этом отношении интересно отметить сообщение
[422] о стимулировании образования каталазы в плесневых грибах при обработ-
обработке перекисью водорода. К другим белковым веществам, которые разрушаются
под действием перекиси водорода, относятся гормоны гипофиза [423] и эри-
эритроциты. Гемолиз электроцитов уже известен давно, однако только недавно
сделано наблюдение [424], что на этот процесс заметно влияет а-токоферол,
или витамин Е.
Особого интереса с точки зрения взаимодействия между перекисью водо-
водорода и белками заслуживает влияние перекиси на волосы, шерсть и кожу.
Все эти вещества состоят из белковых соединений, которые аналогичны друг
другу, хотя и различаются по аминокислотному составу. В общем нужно счит
тать, что все эти вещества сравнительно устойчивы против перекиси водорода,
что дает возможность применять последнюю в строго определенных условиях,
например для отбелки шерсти и волос или для окисления ранее нанесенных
красителей. Тем не менее эти вещества все же подвергаются атаке со стороны
перекиси водорода, и интенсивность этой атаки зависит от времени контакта,
температуры, концентрации, рН и присутствия ионов тяжелых металлов..
Очевидно, что частью молекулы белка, наиболее чувствительной к реакции
с перекисью водорода, является дисульфидная связь RSSR, содержащаяся
в аминокислоте (цистине). Это имеет особое значение, поскольку структура ци-
стина обусловливает его специфическую роль в сшивании длинноцепочечных
аминокислотных полимеров, составляющих молекулу белка. По-видимому,
происходит атака и на другие связи, например между углеродом и азотом в пеп-
пептидном звене RCONHR, и на солевые или сложноэфирные связи, но эта атака
менее энергична и изучена в меньшей степени. В основном работы проводились
по исследованию шерсти; в литературе описаны химические и физические
результаты обработки этого вещества перекисью водорода в различных условиях
[425, 426]. Практические аспекты этой обработки обсуждаются на стр. 486.
Широкое применение перекиси водорода в косметике заставляет уделить
внимание ее влиянию на человеческие волосы. И в этом случае опять-таки
основной атаке подвергается цистин, притом даже интенсивнее, чем в других
случаях, так как человеческие волосы особенно богаты этой аминокислотой.
Стовс [427] провел экспериментальную работу и дал обзор литературы по
этому вопросу, указав, что при повышении рН интенсивность действия переки-
перекиси водорода возрастает во много раз и что это влияет на способность волос
принимать завивку. Стовс считает, что взаимодействие с дисульфидной груп-
группой приводит к образованию групп RSH и RSOH; Гоффарт [428] указывает,
что перекись водорода вступает в обратимую реакцию соединения с сульф-
гидрильными группами, образованными в коже; Смит и Гаррис [426J еще
раньше высказали мысль о ступенчатом присоединении кислорода к дисуль-
фидным группам с образованием RSOSR, RSOSOR, RSO2SOR и т. д. Действие
на волосы катализируется некоторыми металлами, например остающимися от
предыдущей обработки, в частности от крашения. Вельварт [429] сообщает о
возможности обугливания волос и ожога кожи головы при применении пере*
киси водорода в присутствии меди. С другой стороны, если перекись водорода
используется для окисления красителей, примененных для окраски волос,
то сами волосы страдают очень мало, так как реакция с красителями происхо-
происходит избирательно [427].
23*
356 Глава 7
Применение перекиси водорода для обесцвечивания волос связано с обес-
обесцвечиванием пигмента меланина. Меланин—это высокомолекулярное произ-
производное тирозина, и высокая эффективность перекиси водорода с точки зрения
обесцвечивания меланина может быть обусловлена тем, что тирозин представ-
представляет одну из небольшого числа аминокислот, которые легко реагируют с пе-
перекисью водорода. Обесцвечивания меланина в коже не происходит потому, что
перекись водорода не проникает в глубоко расположенный слой кожи, содер-
содержащий этот пигмент. Наряду с обесцвечивающим действием перекись водорода
в известной мере, как указано выше, вызывает также ослабление и разруше-
разрушение волоса; другие факторы, которые, возможно, имеют значение при этой
реакции, например действие перекиси водорода на естественные масла или
пластифицирующие фракции волос, по-видимому, не изучены.
Экспериментатору редко приходится встречаться с такими ощущениями,
как вкус и запах перекиси водорода. Как и вода, перекись водорода лишена
вкуса, однако она дает ощущение вяжущего привкуса, иногда обозначаемого
хак «металлический» привкус. При разложении перекиси водорода наблюдает-
наблюдается такое же шипение, как у содовой воды, что создает впечатление пощипы-
пощипывания во рту. При высоких концентрациях все эти эффекты в полости рта на-
настолько усиливаются, что они становятся болезненными, не говоря уже об
•опасности ожогов; поэтому такой контакт, возможный, например, при всасы-
всасывании перекиси водорода пипеткой, недопустим. При длительном пользовании
перекисью водорода для медицинских целей в небольших концентрациях
поверхность языка может прийти в состояние, которое можно охарактеризовать
как «шерстистое»; к счастью, это состояние обратимо и быстро исчезает.
Трудно охарактеризовать запах перекиси водорода. Еще не выяснено, влияет
ли она на обонятельные клетки или только раздражает общие нервы слизистой
оболочки носа. Концентрированная перекись водорода обладает очень слабым
запахом, если только нарочно не вдыхать пары ее вблизи поверхности жидкости
в сосуде или не пролить значительное количество этой жидкости. Ощущение
в этом случае напоминает запах озона или галогенов. Последний факт не лишен
интереса с точки зрения химической характеристики перекиси водорода как
псевдогалогена. Если по каким-либо причинам в воздухе диспергировано
большое количество перекиси водорода, например в виде тумана, то через
небольшой промежуток времени наблюдается значительное раздражение сли-
слизистой оболочки носа. При долговременном дыхании в такой атмосфере появ-
появляется удушье, как то, которое наблюдается в случае аммиака или дву-
двуокиси серы. Это ощущение сопровождается резким чувством жжения в носовых
ходах при вдыхании и выдыхании.
Исследование токсичности
К счастью, такое реакционноспособное соединение, как перекись водорода,
обладает минимальной токсичностью; как указано на стр. 153, основными источ-
источниками опасности при работе с перекисью водорода, которых следует избегать,
являются последствия проливания ее на одежду с возможным затем воспла-
воспламенением и потенциальная разрушительная способность концентрированной
перекиси при разложении ее в неконтролируемых условиях. Что касается
прямых химических эффектов, то организм прекрасно приспособлен против
действия перекиси водорода, кожа к ней сравнительно инертна, а жидкости
тканей организма обладают эффективными средствами для разложения пере-
перекиси водорода, попадающей внутрь. Отсутствует или почти отсутствует дена-
денатурирование, растворение или обугливание, наблюдаемые при других первич-
первичных раздражителях. Однако это нельзя понимать в том смысле, что при работе
с перекисью водорода не нужно соблюдать никаких мер предосторожности;
наоборот, указанные на стр. 154 основы безопасной работы с перекисью должны
Перекись водорода в биологических процессах 357*
неукоснительно выполняться. В этом разделе приводятся результаты некоторых
работ, направленных к выяснению природы и границ токсических эффек-
эффектов перекиси водорода. Поскольку такие эффекты редко встречаются на прак-
практике, эти исследования базируются главным образом на данных, полученных
с лабораторными подопытными животными. Случаи астмы и экземы, отмечен-
отмеченные среди рабочих одного итальянского завода по производству перекиси водо-
водорода, как оказалось, были вызваны не действием перекиси, а чувствительностью
к пероксосульфату аммония [430].
Токсичность перекиси водорода для низших организмов хорошо известна
и подвергалась многочисленным исследованиям; ссылки на эти работы приведе-
приведены при обсуждении применения перекиси в медицине^стр. 512 и ел.). Интересной
особенностью является существование пороговой концентрации, ниже которой
перекись не оказывает никакого действия. Проведенные опыты [431] с дрож-
дрожжами при рН 5,5 с применением пероксоуксусной, пероксофталевой и пер-
оксомалеиновой кислот и перекиси водорода показали, что каждое из этих сое-
соединений имеет свой характеристический порог, причем выше всего он для
перекиси водорода; кроме того, при концентрациях, выше пороговой, перекись
дает максимальную долю выживших организмов. Аналогичный эффект был
обнаружен и для молоди форели [432]. Для высших форм организмов также
наблюдается такой результат, но значительно большая дифференциация функ-
функций у этих организмов требует более точного указания характера действия
перекиси. Опыты над животными можно разбить на группы в зависимости
от того, проводились ли они на поверхностях тела, как например на коже или
роговой оболочке глаза, были ли они предназначены для выяснения действия
вдыхания паров или аэрозоля или изучалось действие перекиси при при-
приеме внутрь. В опытах с кроликами показано [433], что нанесение 90%-ной
перекиси водорода на выбритую кожу способствует всасыванию этого
вещества со смертельным исходом из-за газовой эмболии. Однако отдельные
виды животных сильно отличались в этом отношении один от другого. Кошки,
морские свинки, крысы, свиньи и собаки значительно менее чувствительны
к такому воздействию; у них наблюдается соответственно значительно боль-
большая реакция кожи в месте нанесения. Другими факторами, определяющими
разницу между кроликами и другими животными, являются большая чув-
чувствительность кролика к эмболии и различные содержания каталазы в коже.
При исследовании влияния 90%-ной перекиси водорода на человеческую кожу
[434] выявлена разница между действием ее ка ладони и кончики пальцев,
с одной стороны, и на иные участки кожи—с другой. На ладонях и кончиках
пальцев имеется толстый слой кератина, и эти места богаты нервными окон-
окончаниями. В этом случае даже тонкий, мазок концентрированной перекиси водо-
водорода вызывает сильное покалывание и образование непрозрачных белых пятен.
Особенно болезненные ощущения вызывает перекись водородаспод ногтями.
Повеление, очевидно, вызвано преломлением в газе, образующемся при раз-
разложении под первым слоем кожи и удерживаемым в толще этого слоя. Более
интенсивная обработка может вызвать покраснение кожи, появление папул,
иногда с последующим уплотнением. На других участках кожи с более тонким
слоем,'кератина также наблюдается раздражение, но с меньшим зудом, причем
побеление распространяется лишь на отдельные участки у основания волос.
Проникания перекиси водорода глубже первого слоя кожи, рогового (stratum
сотеит), не отмечено, и все эти эффекты исчезают бесследно. Более интенсив-
интенсивное действие оказывает перекись водорода на роговую оболочку глаза кролика
[433, 435]. Количества около 1 мл 90%-ной перекиси водорода вызывают
обратимые явления, аналогичные явлениям на коже [433]. Большие количе-
количества, около 3 мл, вызывают стойкое помутнение роговой оболочки. Отсюда
ясно, что в случае попадания перекиси водорода в глаз его необходимо быстро
промыть водой. Опыты по токсичности аэрозолей перекиси водорода при вдыха-
358 Глава 7
нии проведены 1433, 436, 437] на мышах, крысах, собаках и кроликах,
причем опубликованы подробные анализы крови и мочи собак после такой
экспозиции [437]. При такой экспозиции обнаружены ожоги носа и лап, по-
повреждение роговицы глаза и гиперемия легких. Повреждение роговицы у крыс
носит затяжной характер и медленно развивается после экспозиции у живот-
животных,выживших после вдыхания доз, смертельных для отдельных экземпляров.
У собак при вдыхании перекиси водорода в концентрации 0,0007% в течение
6 месяцев наблюдалось хроническое раздражение; предполагается [437], что
люди без вреда для себя могут переносить в течение длительного времени
0,0004%-ную концентрацию перекиси водорода в воздухе. Трудно определить
действительную концентрацию диспергированной перекиси в этих опытах,
так как последняя легко конденсируется и разлагается. У различных видов
животных изучено также влияние внутривенной инъекции [433, 438]. В суб-
сублетальных дозах перекись влияет на уровень каталазы в печени и метгемогло-
бина в крови. При количестве 0,015 мл 90%-ной перекиси водорода на 1 кг
веса тела 50% кроликов погибало. Сообщается [439], что, по наблюдениям, эта
доза падает со снижением концентрации и составляет, например, 0,003 мл/кг
для 4%-ной перекиси. Причина большей токсичности более разбавленного
вещества состоит, по-видимому, в том, что разбавленная перекись глубже
проникает в систему до разложения и блокирования кровотока. Концентри-
Концентрированная перекись наиболее полно разлагается в месте самой инъекции.
Описывается смерть человека, вызванная непреднамеренной инъекцией пере-
перекиси водорода [440]. Другим значительным явлением при попадании перекиси
водорода в организм является некроз пораженных мышц [441]. Прием раз-
разбавленной перекиси водорода 'через рот не оказывает заметного влияния;
более концентрированные растворы вызывают раздражение и кровоизлияние,
а также опасность разрыва внутренних оболочек от интенсивного выделения
газа.
Интересен вопрос и о физиологическом действии перекиси водорода на
молекулярном уровне. Показано, что перекись водорода может вызвать му-
мутации, и в ряде литературных источников [442] описываются условия и приро-
природа этого эффекта. Последний иногда считают радиомиметическим эффектом,
причем он представляет интерес с точки зрения образования перекиси водо-
водорода в живых организмах при действии ионизирующих излучений (см. стр. 60).
Механизм этого мутагенного действия точно еще не известен, а поэтому заслу-
заслуживают внимания различные высказанные мнения и точки зрения. Процессы
мутации находятся в близком родстве с карциногенезом, и, как указывает
Дженсен (см. в работе [443] стр. 159), необходимо различать возникновение опу-
опухоли и ее развитие; факторы, имеющие значения для одного из этих явлений,
могут не оказывать влияния на другое. Мутагенное действие перекиси водо-
водорода изменяется также в зависимости от легкости доступа ее к клеточным ядрам
(см. в работе [443] стр. 116). Процесс может зависеть и от возможного изме-
изменения содержания каталазы в разных частях клетки. Шнейдер (см. в работе
[359] стр. 273) считает, что катал аза в клеточном ядре почти отсутствует
и находится в растворимой форме в цитоплазме; однако мнения по этому пред-
предположению расходятся [443]. Тем не менее установлено [444], что каталаза
устойчива против рентгеновского облучения. Логическим выводом из того, что
рентгеновские лучи и подавляют опухоли и вызывают образование перекиси
водорода, была мысль, что перекись водорода может оказывать благоприятное
влияние на лечение рака. Такого рода опыты проводились (см. в работе [443]
стр. 149; [445]) и проводятся сейчас, но пока еще положительных результатов
не получено. Возможно, что перекись, образующаяся при действии излуче-
излучения, представляет органическую перекись или перекись водорода в форме
аддитивного соединения, причем высказана мысль (см. в работе [443] стр. 149),
что эти соединения не разлагаются каталазой. Большинство авторов в на-
Приложение 359
стоящее время склоняются к мнению, что действие излучения следует при-
приписать образующимся свободным радикалам, а не самой перекиси водорода,
хотя она и может образоваться за счет взаимодействия этих свободных
радикалов [418]. Это не изменяет того положения, что сама перекись водо-
водорода может в известных условиях вызвать мутацию; однако, как подчеркивает
Грей [446], несмотря на аналогий) между этим процессом и процессом, вызы-
вызываемым излучением, доказано, что рентгеновские лучи вызывают разрыв хро-
хромосом по всей их длине, тогда как химические агенты способствуют разрыву
только одной связи. Длина цепи в процессах, происходящих с участием свобод-
свободных радикалов (см. в работе [443] стр. 159), также может иметь значение. Другие
специфические эффекты перекиси водорода на молекулярном уровне указаны
или исследованы лишь Ь очень небольшой степени. Сообщается о том, что
перекись водорода играет роль в образовании вирусов [447] и что она может
способствовать возникновению анемии [448]. Мозговая ткань легко отравляет-
отравляется перекисью водорода [449], однако инъекция перекиси водорода в белок
куриного яйца не влияет на развитие зародыша [450].
ПРИЛОЖЕНИЕ. ПРИМЕНЕНИЕ И ЗНАЧЕНИЕ ЭЛЕКТРОДНЫХ ПОТЕНЦИАЛОВ
Для тех, кто не знаком с электродными потенциалами, прилагаемое крат-
краткое объяснение их применения и значения (с особым учетом перекиси водо-
водорода) может быть не лишенным интереса. Условно [4] все уравнения отдельных
электродных реакций для отдельных систем пишут всегда таким образом, чтобы
электроны были в правой части, а восстановленная форма вещества—в левой,
потенциалы же считают от водорода вверх или вниз; потенциалы имеют поло-
жительнй знак для тех реакций, где восстановленная форма является луч-
лучшим восстановителем, чем молекулярный водород. Значения электродных
потенциалов ?° относятся к тому потенциалу, который наблюдался бы, если
бы растворы, содержащие реагенты и продукты, обладали единичной актив-
активностью. Концентрации газообразных соединений измеряются в атмосферах,
а растворенных веществ—в моляльных единицах. Таким образом, значения
Е° для реакций F) и A0) относятся к реакции перекиси водорода приблизи-
приблизительно при одномоляльной концентрации и при рН=0; значения ?° для реак-
реакции (8) и A2) относятся к рН=14.
Если имеется список таких отдельных реакций и их потенциалов, можно
с первого взгляда определить, будет ли та или иная реакция протекать в замет-
заметной мере. Восстановленная форма вещества способна к окислению, если потен-
потенциал ее отдельной реакции более положительный, чем потенциал такой же
реакции для окислителя. Другими словами, обращение какой-либо отдельной
реакции (т. е. протекание ее справа налево), если выражать ее согласно выше-
вышеприведенному правилу, будет способствовать протеканию реакции с более
положительным потенциалом в соответствии с написанием, т. е. слева направо.
В качестве примера окислительной способности перекиси водорода
можно показать, что потенциал реакции
2Н2О = Н2О2 + 2Н+ +'2е, Е° = - 1,76*
является вполне достаточным для возможности окисления
2Н2О f Pb2+ = PbO2 + 4Н+ + 2е, Е° = - 1,46*.
Другими словами, реакция
Н2О2 + РЬ2+ = РЬО2 + 2Н+, Е° = -!- 0,30*
может протекать слева направо таким образом, как показано, в связи со
значительным потенциалом, который может быть достаточным для превра-
превращения иона двухвалентного свинца в двуокись свинца. В химии обычно сама
360 Глава 7
двуокись свинца считается довольно сильным окислителем, и то, что перекись
водорода способна образовать такую двуокись из восстановленной формы
свинца, иллюстрирует высокую окислительную способность перекиси водо-
водорода. Действительно, по окислительному потенциалу лишь немного окисли-
окислителей превосходят перекись водорода.
Для иллюстрации восстановительного действия перекиси водорода возь-
возьмем следующий пример. Потенциал реакции
СГ= уС12 + е, ?°= -1,34*
в достаточной мере менее положителен, чем потенциал реакции
Н2О2 = 2Н+ + О2 + 2е, Е° = - 0,67*,
а поэтому может протекать реакция
Н2Оа + С12 = 2Н+ + 2СГ + О2.
С другой стороны, потенциал реакции системы
F2 + 2e, ?°= -2,65*
настолько отрицателен, что окисление фтор-иона по реакции
Н2О2 + 2F" + 2Н+ = 2Н2О + F2
не может происходить в заметной степени. Точно так же положительное-
значение потенциала системы
Fe = Fe2+ -j- 2е, Е°= + 0,44*
доказывает невозможность протекания реакции
Н2О2 + Fe2+ = Fe + 2Н+ + О2.
Это означает, что перекись водорода не является настолько сильным вос-
восстановителем, чтобы превратить заметное количество закисного иона же-
железа в металлическое железо.
Расхождение между принятым способом обозначения, по которому
каждую отдельную реакцию пишут в совершенно определенном порядке,
и обычным ходом мыслей, по которому перекись водорода считается реаген-
реагентом, т. е. пишется всегда с левой стороны уравнения, может сначала при-
привести к путанице. Можно считать искусственным приемом писать так, как
указано выше:
2Н2О = Н2О2 + 2Н+ + 2е, Е° = - 1,76*,
вместо
Н2О2 + 2Н+ + 2е = 2Н2О, Е° = + 1,76*.
Однако если всегда следовать принятой системе, то можно [не опасаться
недоразумений, связанных для какой-нибудь реакции со знаком и величи-
величиной электродного потенциала. При таком методе в отношении [направле-
[направления какой-либо отдельной реакции всегда будет один и тот же подход.
Действительно, если в вышеприведенных реакциях считать воду восстано-
восстановителем или кислород окислителем вместо перекиси водорода, то изменяется
лишь внешняя форма, а не суть самих реакций. Эти обратные способы
представления реакций относятся к категории образования перекиси водо-
водорода или ее роли в качестве промежуточного продукта при превращении
кислорода в воду или наоборот. Обе эти темы более подробно рассмотрены
в гл. 2 и 3.
Литература 361
Приведенные выше примеры показывают возможность суммирования
отдельных реакций для того, чтобы получить выражение полной реакции.
При такой методике можно суммировать также отдельные потенциалы для
определения потенциала полной реакции, т. е. того потенциала, который
получился бы, если бы был создан электрический элемент, в котором про-
происходит данная реакция. Необходимо подчеркнуть, что этот потенциал имеет
силу лишь для обратимого элемента, функционирующего так, как это выра-
выражено уравнением. Если отдельные стадии реакции необратимы, то потен-
потенциал будет другим. Иногда предполагаемая реакция оказывается невоз-
невозможной из-за того, что какая-либо промежуточная стадия ее не может
быть осуществлена.
Можно также суммировать отдельные электродные реакции для вывода
третьей отдельной реакции. Это иллюстрируется способом вывода данных
для системы перекись водорода — вода из известных величин для систем
перекись водорода — кислород и вода — кислород, а именно
2Н2О = 4Н+ + О2 + 4е, Е° = - 1,23*,
Н2О2 = 2Н+ + О2 + 2е, Е° = - 0,69в,
2Н2О = Н2О2 + 2Н+ + 2е, ?° = - 1,76*.
Способ вычисления ?° = [4.(- 1,23)-2-(-0,69)]/2 = - \J6e показывает,
что при такого рода суммировании отдельных потенциалов нужно всегда
складывать вольт-эквиваленты.
Электродные потенциалы можно, конечно, пересчитать и на значения
свободной энергии путем использования уравнения &F°= —n%E°9 где g
имеет значение 23060 кал/в-экв. В свою очередь из величины свободной
энергии можно вычислить константу равновесия для реакции: Д/70 = — RT In К
или при 25° А/70 = — 1364,3 lg/С. Так, например, потенциал окисления иона
двухвалентного свинца перекисью водорода был выше найден равным 0,30 в.
Изменение свободной энергии для этой реакции
А/70= -2.23060-0,30 = —13840 кал.
Этот результат можно получить и непосредственно из таблиц для свобод-
свободной энергии образования. Константу равновесия можно вычислить по урав-
уравнению /C = antilg (— 13840/— 1364,3)= 1,4-1010. Таким образом, если пре-
пренебречь различием между концентрациями и активностями, то концентра-
концентрации раствора перекиси водорода, находящегося в контакте с двуокисью
свинца, будут подчиняться следующему соотношению:
jg^-tf- 1,4.10».
При рН равном нулю в одномоляльном растворе перекиси водорода (около*
3 вес. %) двуокись свинца может появиться после того, когда концентра-
концентрация иона двухвалентного свинца достигнет всего лишь 7-10~иМ (если
только реакция вообще будет протекать). При более высоких рН та концен-
концентрация иона двухвалентного свинца, которая способна дать заметное коли-
количество двуокиси свинца, будет еще меньше.
Более подробное обсуждение применения и значения электродных потен-
потенциалов вообще, а также численные значения их можно найти у Латимера [4].
ЛИТЕРАТУРА
1. Haissinsky М., Discussions Faraday Soc, № 1, 254 A947); J. chira. phys., 44,.
181 A947).
2. S t e i n b а с h O. F., J. Chem. Education, 21, 66 A944).
362 Глава 7
3. Feigl F., J. Chem. Education, 21, 347 A944).
4. Л а т и м е р В. М., Окислительные состояния элементов и их потенциалы в водных
растворах, Издатинлит, М., 1954.
5. Delahay P., Pour bai x M., Van RysselbergheP., Comite intern.
thermodynam. et cinet. electrochim. Compt. rend, reunion 1950, 42 (Pub. 1951).
6. Delahay P., Pourbaix M., Van Rysselberghe P., J. Chem.
Education, 27, 683 A950); Pourbaix M., Corrosion, 5, 121 A949).
7. Frost A. A., J. Am. Chem. Soc, 73, 2680 A951).
8. Tables of Chemical Kinetics, Homogeneous Reactions, U. S. Dept. of Commerce, Bureau
of Standards, Circular 510, Government Printing Office, Washington, 1951.
9. L a t i m e г W. H., H i| debrand J.H., Reference Book of Inorganic Chemistry,
MacMillan Co., New York, 1940, p. 32.
10. Davis N. S., К e e f e J. H., частное сообщение, 1954.
11. M с L a n e С. К., J. Chem. Phys., 17, 379 A949); S t e i n T. W., Sc. D. Thesis in Che-
Chemical Engineering, M. I. T., 1955.
12. H a r t А. В., частное сообщение, 1954.
13. Edwards J. О., J. Phys. Chem., 56, 279 A952); J. Am. Chem. Soc, 76, 1540 A954).
14. О г r R.J., Williams H. L., J. Phys. Chem., 67, 925 A953).
15. Wai sh A. D., J. Chem. Soc, 1948, 331, 398.
16. Walsh A. D., Trans. Faraday Soc, 42, 264 A946).
17. VanderwerfC. A., J. Chem. Education, 25, 547 A948).
18. Waters W. A., in «Le Mecanisme de L'Oxydation», Proc. 8th Conf. Inst. Int. Chem.
Solvay, Stoops R., Bruxelles, 1950, p. 63.
19. M u 1 1 i k e n R. S., J. Phys. Chem., 56, 801 A952).
20. Bancroft W. D., Murphy N. F., J. Phys. Chem., 39, 377 A935).
21. Thiesse X., Bull. Soc. Chim., 7, 495 A940).
22. I n g о 1 d С. К., Structure and Mechanism in Organic Chemistry, Cornell University
Press, Ithaca, 1953.
23. Hammett L. P., Physical Organic Chemistry, McGraw-Hill Book Co., New York,
1940; R e m i с k A. E., Electronic Interpretations of Organic Chemistry, 2nd ed.,
John Wiley and Sons, Inc., New York, 1949; [есть также перевод первого издания:
см. Р е м и к А., Электронные представления в органической химии, Издатинлит,
М., 1950]; Waters W. A., Physical Aspects of Organic Chemistry, 4th ed., D. Van
Nostrand Co., Inc., New York, 1950.
24. С о u 1 s о n С A., Research, 4, 307 A951).
25. Глесстон С, Лейдлер К., Эйринг Г., Теория абсолютных скоростей
реакции, Госиноиздат, М., 1948; Frost A. A., Pearson R. G., Kinetics and
Mechanism, John Wiley and Sons, Inc., New York, 1953.
26. S a n d e r s о n R. Т., J. Chem. Education, 31, 2 A954).
27. S w a i n С G., Record Chem. Progr. (Kresge-Hooker Sci. Lib.), 12, 21 A951).
28. R e m i с k A. ?., Record Chem. Progr. (Kresge-Hooker Sci. Lib.), 11, 52 A950\
29. T a u b e H., M у e r s H., R i с h R. L., J. Am. Chem. Soc, 76, 4118 A953).
30. W i 1 1 s t a t t e г R., Ber., 36, 1828 A903).
31. Munzberg F., Lotos, 76, 351 A928) [C.A., 26, 2132].
32. В e r m e j о Martinez F., Anales real soc. espafi. ffs. у quim., 45B, 533 A949).
33. N у h ol m R. S., Revs. Pure Applied Chem., 4, 15 A954).
34. H u s a i n S., Z. anorg. allgem. Chem., 177, 215 A928).
35. A 1 e x a n d e г Р., С a r t e г D., Earland C, Biochem. J., 47, 251 A950).
36. R у n a s i e w i с z J., Anal. Chem., 26, 355 A955).
37. L e v i G. R., В a t t a g 1 i n о F., Gazz. Chim. ital., 67, 659 A937).
38. Weiss J.," Advances in Catalysis, 4, 343 A952).
39. Уотерс У., Химия свободных радикалов, Госиноиздат, М., 1951.
40. Т о b о 1 s k у А. V., Mesrobian R. В., Organic Peroxides, Interscience Publi-
Publishers, Inc., New York, 1954.
41. Kirk R. E., В г о w n e A. W., J. Am. Chem. Soc, 50, 337 A928).
42. T a u b e H., Annual Review of Physical Chemistry, 4, 290 A952).
43. Uri N., Chem. Revs., 50, 375 A952).
44. T a u b e H., J. Am. Chem. Soc, 64, 161 A942).
45. С h г i s t i a n s e n J. A., Acta Chem. Scand., 6, 1056 A952).
46. С a h i 1 1 A. E., Та u b e H., J. Am. Chem. Soc, 74, 2312 A952).
47. H i g g i n s о n W.C. E.f Sutton DM Wright P., J. Chem. Soc, 1963, 1380.
48. Evans M. G., in «Le Mecanisme de L'Oxydation», Proc. 8th Conf. Inst. Int. Chim.
Solvay, R. Stoops, Bruxelles, 1950.
49. D e г b у s h i r e D.H., Waters W. A., Nature, 165, 401 A950).
50. Ross S. D., J. Am. Chem. Soc, 68, 1484 A946).
51. H a 1 p e r i n J., T a u b e H., J. Am. Chem. Soc, 74, 380 A952).
52. Evans M. G., В a x e n d a 1 e J. Т., Р а г к G. S , Trans. Faraday Soc, 42, 155
A946).
.53. Waters W. A., Discussions Faraday Soc, № 2, The Labile Molecule, 1947, p. 176.
Литература 363
54. F о г с h e i m е г О. L., T a u b e H., J. Am. Chem. Soc, 74, 3705 A952); 76, 2099
A954).
^55. D a i n t о n F. S., Ann. Reports, 45, 27 A948); неопубликованная работа Col-
Collins о п E., Dainton F. S., на которую ссылается U г i N., Chem. Revs.,
50, 409 A952).
56. В а е г S., S t e i n G., J. Chem. Soc, 1953, 3176.
57. H e i d t L. J., S m i t h M. E., J. Am. Chem. Soc, 70, 2476 A948); H e i d t L. J.,
McMillan A. F., J. Am. Chem. Soc, 76, 2135 A954).
58. F г о s t А. А., О 1 d e n b e г g O., J. Chem. Phys., 4, 781 A936).
59. Volman D. H., J. Chem. Phys., 17, 947 A949).
60. Hunt J. P., T a u b e H., J. Am. Chem. Soc, 74, 5999 A952).
61. S a t t e r f i e 1 d C. N., К a v a n a g h G. M., Resnick H., Ind. Eng. Chem.,
43, 2507 A951).
62. S t о n e F. S., T а у 1 о г H. S., J. Chem. Phys., 20, 1339 A952).
63. О v e г b e г g e г С. G., С u m m i n s R. W., J. Am. Chem. Soc, 75, 4783 A953).
64. Edwards J. O., Chem. Revs., 50, 455 A952).
65. Edwards J. О., частное сообщение, 1954.
66. В u n t о n C. A., LI e wel 1 у n D. R., Research, 5, 142 A952).
67. M u g d a n M., Y о u n g D. P., J. Chem. Soc, 1949, 2988.
68. F о n e г S. N., H u d s о n R. L., J. Chem. Phys., 21, 1374 A953).
69. Edwards J. O., J. Chem. Education, 31, 270 A954).
70. D a v i e s A. G., F о s t e г R. V., W h i t e A. M., J. Chem. Soc, 1953, 1541.
71. В u n t о n С A., L e w i s T. A., L 1 e w e 1 1 у n D. R., Chemistry and Industry,
1954, 191.
72. В u n t о n С A., Nature, 163, 444 A949).
73. К u i v i 1 a H. G., J. Am. Chem. Soc, 76, 870 A954).
74. Wi berg К. В., J. Am. Chem. Soc, 75, 3961 A953).
75. Lef f 1 er J. E., Chem. Revs., 45, 385 A949).
76. В а г tl e t t P. D., Record Chem. Progr. (Kresge-Hooker Sci. Lib.), II, 47 A950).
77. L e f f 1 e г J. E., J. Am. Chem. Soc, 72, 67 A950).
78. Brown D. J., J. Am. Chem. Soc, 62, 2657 A940).
79. S w a i n С G., S t о с k m а у е г W. H., С 1 а г k e J. Т., J. Am. Chem. Soc, 72,
5426 A950).
SO. J af f e H. H., Chem. Revs., 53, 191 A953).
81. В a w n С E. H., M e 1 1 i s h S. F., Trans. Faraday Soc, 47, 1216 A951).
82. В 1 о m q u i s t А. Т., В u s e 1 1 i A. J., J. Am. Chem. Soc, 73, 3883 A951).
83. Cooper W., J. Chem. Soc, 1951, 3106.
34. С r a i g D., D a v i d s о n W. L., J u v e A. E., G e i b I. G., J. Polymer Sci.,
6, 1 A951).
85. В 1 о m q u i s t А. Т., В e r s t e i n I. A., J. Am. Chem. Soc, 73, 5546 A951).
86. К h а г a s с h M. S., Burt J. G., J. Org. Chem., 16, 150 A951).
87. Tipper С F. H., J. Chem. Soc, 1953, 1675; H а у J. E., Johnstone N. M.,
Tipper С F. H., W i 1 1 i a m s R. K., J. Chem. Soc, 1954, 629.
88. FordhamJ.W.L, Williams H. L., Can. J. Res. 27B, 943 A949).
89. Kol t hof f I. M., Medal i a A. I., J. Am. Chem. Soc, 71, 3789 A949).
90. F о r d h a m J. W. L., Williams H. L., J. Am. Chem. Soc, 72, 4465 A950).
91. Boardman H., J. Am. Chem. Soc, 75, 4268 A953); Boardman H., Hulse
G. E., J. Am. Chem. Soc, 75, 4272 A953).
92. О г r R. J., W i 1 1 i a m s H. L., Can. J. Chem., 30, 985 A952).
93. S w e г n D., J. Am. Chem. Soc, 69, 1692 A947); Chem. Revs., 45, 1 A949).
94. W a t e г s W. A., in Gilman «Organic Chemistry, An Advanced Treatise», Vol. 4,
John Wiley and Sons, Inc., New York, 1953, p. 1165.
95. E v e г e t t A. J., M i n k о f f G. J., Trans. Faraday Soc, 49, 410 A953).
96. Morgan L. В., Trans. Faraday Soc, 42, 169 A946).
97. К о 1 t h о f f I. M., M i 1 1 e г I. K., J. Am. Chem. Soc, 73, 3055 A951); Levitt
L. S., Can. J. Chem., 31, 915 A953).
98. E a g e r R. L, McCall um K. J., Can. J. Chem., 32, 692 A954).
99. E г 1 e n m e у е г H., G a r t n e г Н., Helv. Chim. Acta, 17, 970 A934).
100. Wirtz К., В on hoef f er K. F., Z. physik. Chem., B32, 108 A936).
101. T а у lor H. S., Gould A. J., J. Am. Chem. Soc, 56, 1823 A934).
102. В e n 11 e у R., Cold Spr. Har. Symp. quart. Biol., 13, 11 A948).
103. Bentley R., Neuberger A., Biochem. J., 45, 584 A949).
104. Mackenzie H.A. E., Mil пег A. M., J. S. African Chem. Inst., [N. S.I, 4,
79 A951) [C.A., 47, 5742f].
105. Winter E. R. S., В г i s с о e H. V. A., J. Am. Chem. Soc, 73, 496 A951).
106. В а е г t s с h i P., Experientia, 7, 215 A951).
107. Dole M., R u d d D. P., M u с h о w G. R., С о m t e C, J. Chem. Phys., 20,
961 A952).
108. Bentley R., N e u b e г g e г A., Biochem. J., 52, 694 A952).
364 Глава 7
109. Hart E.G., Gordon S., Hutchison D. A., J. Am. Chem. Soc, 74, 554$
A952).
ПО. В a e r t s с h i P., неопубликованная работа, которую цитирует Christian-
s e n J. A., Acta Chem. Scand., 6, 1056 A952).
111. Cal ver t J. G., T h eurer K., R a nk i n G. Т., М а с N e v i n W. N., J. Am.
Chem. Soc, 76, 2575 A954).
112. R i e s e b о s P.C.Aten A. H. W., Jr., J. Am. Chem. Soc, 74, 2440 A952).
113. E 1 k el e s H., В г о s s e t C, Svensk. Kern. Tidskr., 65, 26j A953) (на английском
языке).
114. Dole M., Chem. Revs., 51, 263 A952).
115. H i g g i n s о n W. С E., S u t t о n D., J. Chem. Soc, 1953, 1402.
116. С о о p e J. A. R., В г у с е W. A., Can. J. Chem., 32, 768 A954).
117. Bamberger M., Nussbaum J., Monatsh., 40, 411 A919).
118. Шэнли Э., Гринспэн Ф., в сб. «Физика и химия реактивного движения»,.
№ 2, Издатинлит, М., 1949, стр. 161.
119. Heath M. A., W а 1 t о n J. H., J. Phys. Chem., 37, 977 A932); К о л а р о в Н.,
Известия на Химическия институт Българска Академия на науките, 1, 185 A951).
120. Bell F., Gill R., Н о 1 d e n D., Wynne-Jones W. F. К., J. Phys. and
Colloid Chem., 55, 874 A951) [C. A., 45, 9342 i|; W a 1 t о n J. H., J о n e s D. O.,
J. Am. Chem. Soc, 38, 1956 A916); Bray W.C., Livingston R. S., J. Am»
Chem. Soc, 45, 1251, 2048 A923); Livingston R. S., ibid. 48, 53 A926);.
H e n d e г W. С, R о b i n s о n R. A., Trans. Faraday Soc, 29, 1300 A933),
121. Льюис Г.,Рендалл М., Химическая термодинамика, ОНТИ, Химтеорет, Л.,.
1936.
122. Livingston R., J. Am. Chem. Soc, 50, 3204 A928).
123. Carrara G., Bringhenti A., Gazz. chim. ital., 33, 362 A903) [C. Z. 1904, I.
246]; Meyer J., J. Prakt. Chem., 72, 278 A905); M u m m O., Z. physik, Chem.^
59, 459, 492, 497 A907).
124. Joy пег R. A., Z. anorg. Chem., 77, 103 A912).
125. К а р г и н В. A., Z. anorg. allgem. Chem., 183, 77 A929).
126. Evans M. G., U r i N., Trans. Faraday Soc, 45, 224 A949).
127. Reichert J.S.,Hull H. G., Ind. Eng. Chem., Anal. Ed., 11,311 A939).
128. С r a b t r e e J. I., E a t о n G. Т., M u e h 1 e r L. .E, J. Soc. Motion Picture Engrs.r
35, 484 A940).
129. Berl W. G., Trans. Electrochem. Soc, 83, 253 A943).
130. Wynne-Jones W. F. K., J. chim. phys., 46, 337 A949).
131. Beck W. H., W у n n e - J о n e s W. F. K., J. chim. phys., 49, C97 A952); M i t-
c h e 1 1 A. G., W у n n e -J о n e s W. F. K., Discussions Faraday Soc, 1953>
№ 15, A953), p. 161.
132. Oliveri-Mandala E., Indovina R., Gazz. chim. ital., 67, 53 A937);
D e 1 e о E., Indovina R., P a r 1 a t о A., Gazz. chim. ital., 79, 451 A949).
133. D e 1 e о Е., Indovina R., Parlato A., Ann. chim. (Roma), 40, 251 A950).
134. Giguere P. A., M a a s s O., Can. J. Res., 18B, 84 A940).
135. Abel E., R e d 1 i с h В., S t r i с k s W., Naturwissenschaften, 22, 525
A934).
136. Chang T.L.Wei Y.C, J. Chinese Chem. Soc, 7, 138 A940)[C. A., 35, 5022];
Science Record, L, 132 A942); Science, 100, 29 A944).
137. Chang T. L., Science, 100, 29 A944) [C. A., 38, 5142).
138. В о b t e 1 s k у M., J. Chem. Soc, 1950, 3615; F 1 о г e s с о N., Bull, faculte Stiinte
Cernauti, 2, 184 A928) [C. A., 26, 4233].
139. Graham D. P., J. Am. Chem. Soc, 52, 3035 A930).
140. Eden G. E., Freke A.M.,Mel bourne K. V., Chem. and Ind., 1951, 1104.
141. Kolthoff I. M., Medal i a A. I., J. Am. Chem. Soc, 71, 3777, 3784, 3789-
A949).
142. В о h n s о n V. L., J. Phys. Chem., 24, 677 A920); H e n d e r W. C, Robin-
Robinson R. A., Trans. Faraday Soc, 29, 1300 A933).
143. Walton J. H., J о n e s D. O., J. Am. Chem. Soc, 38, 1956 A916).
144. Гликман Т. С, Изв. АН СССР, отд. математ. и естеств. наук, 1934, 1593.
145. Stoner G. G., D о u g h e r t у G., J. Am. Chem. Soc, 63, 1291 A941).
146. Писаржевский Л. В., Гликман Т. С, Изв. АН СССР, отд. математ.
и естеств. наук, 1934, 281.
147. Nicholson D. G., R e i t е г М. A., J. Am. Chem. Soc, 59, 151 A937).
148. H u d 1 i с k у M., Chem. Listy, 45, 380 A952); Collection Czechoslov. Chem. Communs^
16,283, 611 A951) (на английском языке).
149. M el 1 о r J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans, Green and Co., London, 1922, Vol. 1, Ch. 14, p. 877—956.
150. Friend J. N., Twiss D. F., A Textbook of Inorganic Chemistry, Charles Griffin
and Co., Ltd., London, 1924, Vol. 7, Part I, Oxygen, p. 324—349.
151. Stone С. Н., J. Chem. Education, 21, 300 A944).
Литература 365
152. Н о d g к i n s о n W. R. E., С о о t e A. H., Chem. News, 92, 38 A905); Salkow-
ski E., Chem. Ztg., 40, 448A916); J. Soc. Chem. Ind., 35, 741 A916) [C. A., 10, 2559];
HaissinskyM., Cottin M., Compt. rend., 224, 467 A947) [C. A., 41, 4044ij;
Ka warn ura K., Japan Analyst, 2, 417 A953) [C. A., 48, 6319i].
153. AbeR.,J. Phys. Soc. Japan, 6, 345 A951) (на английском языке) [С. А.,47, 952g].
154. Д е р я г и н а О. Г., А к и м о в Г. В., ДАН СССР, 85, 1305 A952).
155. Dhar N. R., В h a t t а с h а г у а А. К., J. Indian Chem. Soc, 11, 311 A934).
156. Collins S., В г у с e W. A., J. Chem. Phys., 18, 1297 A950).
157. von Elbe G., J. Am. Chem. Soc, 54, 821 A932); 55, 62 A932).
158. V ol m a n D. H., J. Am. Chem. Soc, 73, 1018 A951).
159. К u b a s с h e w s k i O., E b e г t H., Z. Metallk., 38, 232 A947).
160. D e n i s о f f A. K., R i с h а г d s о n O. W., Proc Roy. Soc. (London), A150, 495
A935).
161. Rothmund V., 8th Intern. Congr. Appl. Chem., Orig. Comm., 26, 611 A912);
RothmundV., BurgstallerA., Monatsh., 38, 295 A917); J. Chem. Soc,
114, II, 16 A918).
162. Bray W. C, J. Am. Chem. Soc, 60, 82 A938); T a u b e H., В г а у W., J. Am.
Chem. Soc, 62, 3357 A940); T a u b e H., J. Am. Chem. Soc, 64, 2468 A942).
163. George P., Discussions Faraday Soc, № 2, 196 A947).
164. Discussions Faraday Soc, JNfe 2, p. 217—221 A947).
165. Казарновский И. А., Нейдинг А. Б., ДАН СССР, 86, 717 A952).
166. G e i b К. H., Z. physik. Chem., A169, 161 A934) [С. А., 28, 6614]; GeibK. H.,
Z. Elektrochem., 47, 761 A941).
167. Bray W. C, Chem. Revs., 10, 161 A932).
168. В a x e n d a 1 e J. H., Advances in Catalysis, 4, 35 A952).
169. P i с с i n i A., Z. anorg. allgem. Chem., 10, 438 A895).
170. M a a s s O., H a t с h e г W. H., J. Am. Chem. Soc, 44, 2472 A922).
171. Bobtelsky M., Radovensk y-C h о 1 a t n i k о v C, Z. anorg. allgem.
Chem., 206, 161 A932).
172. С о n n i с k R. E., J. Am. Chem. Soc, 69, 1509 A947).
173. M а с к E. S., J. Phys. Chem., 21, 238 A917).
174. Brown R. W., Tappi, 35, 75 A952).
175. Walton J. H.,Jr., Z. phys. Chem., 47, 185A904); Bredig G., ibid., 48, 368
A904).
176. Abel E., Z. physik Chem., 96, 1 A920) [C. A., 15, 199].
177. Morgan K. J., Quarterly Revs., 8, 123 A954).
178. Bray W. C, L i e b h a f s k у Н. A., J. Am. Chem. Soc, 53, 38 A931).
179. R u m p f P., Compt. rend., 198, 256 A934).
180. SorumC. H.,Charlton F. S., N e p t u n e J. A., E d w a r d s J. O., J. Am.
Chem., Soc, 74, 219 A952).
181. Me Alpine R. K-, J. Chem. Education, 22, 387 A945).
182. Памфилов А. В., Петин Н. Н., Изв. Иваново-вознесенского политехниче-
политехнического института, 6, 221 A922); Goard А. К., R i d е а 1 Е. К., Proc Roy. Soc.
(London), 105A, 148 A924); Кг a use A., E r n s t Z., Вег., 70В, 443 A937).
183. S a t t e r f i e 1 d С. N., Reid R. С, В г i g g s D. R., J. Am. Chem. Soc, 76,
3922 A954).
184. Classen А., В a u e г Е., Ber., 16, 1062 A883).
185. Wassermann А., Вег., 65В, 704 A932); Ann. Chem., 503, 249 A933).
186. A 1 b u H. W., v о n S с h w e i n i t z H. D., Ber., 65, 729 A935).
187. Glasstone S., Hickling A., Electrolytic Oxidation and Reduction, D. Van
Nostrand Co., Inc., New York, 1936, p. 257.
188. Willstatter R., Ber., 36, 1831 A903); T а г u g i N., V i t a 1 i G., Gazz.
chim. ital., 39, 1, 418 A909) [C. A., 5, 437].
189. Sand ved K., Holte J. В., Kgl. Norske Videnskab. Selskabs, Forh., 11, 189
A938) (Pub. 1939) [C. A., 33, 4856].
190. Abel E., Monatsh., 28, 1239 A907); 34, 821 A913); Abel E., Z. Electrochem., 18,
705 A912); A b el E., Monatsh., 81, 798 A950); Uzumasa Y., Ok ига Т.,
J. Chem. Soc. Japan, Pure Chem. Sect., 71, 48 A950) [C. A., 45, 4540d].
191. С о г d e s H., Schenk P. W., Z. anorg. allgem. Chem., 214, 33 A933); К a s s el
L. S., M о n t g о m e г у С. W., J. Chem. Phys., 2, 417 A934).
192. Phillips G.M.,Hunter J. S., Sutton L. E., J. Chem. Soc, 1945, 146;
M о f f i t t \\\, Proc. Roy. Soc, A200, 409 A950); Gillc'spie R. J., J. Chem.
Soc., 1952, 1002; M ul 1 i k e n R. S., J. Phys. Chem., 56, 816 A952); Dewar
M. J. S., J. Chem. Soc, 1953, 2885; Craig D. P., M а с с о 1 1 A., N у h о 1 m
R.S.,OrgelL.E.,Sutton L. E., J. Chem. Soc, 1954, 332; Nyholm R. S.,
Revs. Pure Appl. Chem., 4, 15 A954); Craig D. P., Revs. Pure Appl. Chem.,
4, 4 A954); J a f f e H. H., J. Phys. Chem., 58, 185 A954); Bird G. R., T о w-
nes С. Н., Phys. Rev., 94, 1203 A954).
366 Глава 7
193. Howard E., Jr., Levitt L. S., J. Am. Chem. Soc, 75, 6170 A953).
194. G i 1 b e r t s о n L I., King G. В., Inorganic Syntheses, Vol. Ill, McGraw-Hill
Book Co., New York, 1930, p. 137; J. Am. Chem. Soc, 58, 180 A936).
195. Roseman R., Neptune R. W., A 1 1 e n B. W., пат. США 2616791 D. XL
1952).
196. S с hi иск G., Monatsh., 37, 489 A916).
197. Gutbier A., Wagenknecht W., Z. anorg. Chem., 40, 260 A904); Gut-
bier A., R e s e n s с h e с к F., Z. anorg. Chem., 42, 174 A904).
198. Joliot F., J. chim. physique, 27, 119 A930).
199. W e i t h W., Weber A., Ber., 7, 1745 A874); Трифонов И., Z. anorg. all-
gem. Chem., 124, 123, A922); G 1 e u K., Hubold R., Z. anorg. allgem. Chem.,
223, 305 A935) [C. A., 29, 7849]; Шилов Е, А., С т е п а н о в а 3. С, ЖФХ, 24,
820 A950); Halfpenney E., Robinson P. L.,J. Chem. Soc, 1952, 928.
200. Masson O., Proc. Chem. Soc, 23, 117A907) [C. A., 1, 20894; J. Chem. Soc, 91,
1449 A907) [C. A., 2, 35].
201. Schuster F., Z. anorg. allgem. Chem., 186, 253 A930).
202. Wurster C, Ber., 20, 2631 A887).
203. Одрит Л., О г г Ю., Химии гидразина, Издатинлит, М., 1954; Clark С. С,
Hydrazine, Mathieson Chemical Corp., Baltimore, 1953, p. 8.
204. Browne A. W., J. Am. Chem. Soc, 27, 551 A905); Kirk R. E., Browne
A. W., J. Am. Chem. Soc, 50, 337 A928).
205. Гордон Э., в сб. «Вопросы горения», № 2, Издатинлит, М., 1953, стр. 167.
206. Davis P., H i g g i n s о n W. С. Е., J. Chem. Soc, 1953, 1902.
207. С a h n J. W., P о w el 1 R. E., J. Am. Chem. Soc, 76, 2568 A954).
208. В о w e n E. J., В i r 1 e у A. W., Trans. Faraday Soc, 47, 580 A951).
209. H о d g к i n s о n W. R., J. Soc Chem. Ind., 33, 815 A914).
210. Nelson D. H., S. B. Thesis in Chemistry, Massachusetts Institute of Technology,
1947; С a n t w e 1 1 Т., М а с h i e W. H., S. B. Thesis in Chemical Engineering,
Massachusetts Institute of Technology, 1948.
211. Weyl Т., Ber., 39, 1307 A906).
212. N e г z K., W a g n e г С, Ber., 70B, 446 A937).
213. J a m i e s о n G. S., Am. J. Sci., 44, 150 A917); W e d e к i n d E., Chem. Ztg.,
48, 185 A924) [C. A., 18, 2449].
214. R a s с h i g F., Ber., 18, 2743 A885).
215. С a g 1 i о t i V., M i 1 a z z о G., Ricerca Sci., 9, 11, 358 A938) [C. A., 33, 9100].
216. M о s e г L., Z. anorg. Chem., 50, 33 A906).
217. Hasebrock K., Ber., 20, 213 A887); H a n u s J., К a 1 1 a u n e r O., Z. anorg.
Chem., 70, 232 A911).
218. R u p p E., S с h 1 e e H., Biochem., Z., 172, 373 A926).
219. R u p p E., S с h 1 e e H., Biochem., Z., 181, 250 A927).
220. К о m а г о v s к i i A., Chem. Ztg., 38, 121 A914) [C. A., 8, 1715]; Fells H. A.,
Firth J. В., Proc. Roy. Soc. (London), П4А, 517 A927).
221. Serdaroglu N., Bull. Tech. Univ. Istanbul, 2, № 2, 87 A944) (на немецком
языке).
222. Ellis R. С, Jr., Wol sky I. P., J. Appl. Phys., 24, 1411 A953).
223. T h о m s e n, Ann. Phys., B), 151, 194 A874); Hasselskog S., Svensk. Farm.
Tids., 25, 149 A921) [C. A., 15, 2047]; End6 H., Y о k о у a m a G., Science
Repts. Research Insts., Tohoku Univ., Ser. A, 2, 449 A950) [C. A., 46, 3937d].
224. Zofier V., Bull, soc Chim., D), 13, 61 A913); 21, 241 A917); Бах А., М о н о с-
c о н М., Ber., 57B, 735 A924).
225. К л я ч к о Ю. А., Б л е щ у н о в а Е. И., ДАН СССР, 56, 823 A947).
226. Glauner R., G 1 о с к е г R., Z. Metallkunde, 20, 144 A928) [С. А., 22, 4445].
227. Al d r i d ge J., A p pi eb ey M. P., J. Chem. Soc, 121, 238 A922); Sp e r-
b e r J., Schweiz. Apoth. Ztg., 53, 717 A915) [C. A., 10, 1308]; Бунтин, Вла-
Власов, Труды Воронежского гос. университета, 8, №4, 6 A935).
228. Q u а г t а г о 1 i A., Gazz. chim. ital., 55, 619 A925).
229. R a u b E., Engel A., Z. Metallkunde, 44, 298 A953).
230. Kl unstuck M., Ber., 51, 108 A918); J. Chem. Soc, 114,11,107A918).
231. S a 1 k о w s k i E., J. prakt. Chem., 102, 194 A921).
232. L e R о у G. A., Compt. rend., 119, 557 A894); A n d г e s e n M., Brit. J. Phot., 46,
262 A899); Andresen M., Phot. Korr., 36, 260 A899); S h e p p а г d S. E.,
M e e s С. Е. К-, Investigations on the Theory of the Photographic Process, London,
Longmans, Green and Co., 1907; Siradjian J., Chem. and Eng. News, 31, 4198
A953).
233. M e e s С. Е. К., The Theory of the Photographic Process, Revised Ed., The MacMillan
Co., New York, 1954.
234. James Т. Н., J. Franklin Inst., 240, 15, 83, 229, 327 A945).
235. Watson G., Chem. News, 46, 9 A882); P e 1 1 i n i G., M e n e g h i n i D., Z.
anorg. Chem., 60, 178 A908).
Литература 367
236. Marie С, J. Chim. phys., 6, 596 A908).
237. Ruis A., Llopis J.,Tordesill as I. M., Anales real soc. espan. fis. у quim.,
48B, 23 A952) [C. A., 47, 2063i].
238. В о r n e m a n n K., Z. Elektrochem., 15, 673 A909); S i e v e г t s А., В г fi-
fining H., Z. anorg. allgem. Chem., 204, 291 A932).
239. Ч у г а е в Л., X л о п и н В., Z. anorg. allgem. Chem., 151, 253 A926); Бабаева
А. В., ДАН СССР, 23, 653 A939); Бабаева А. В., Compt. rend. URSS, 24, 145
A939) (на английском языке).
240. BroughtonD. В., Went wort h R. L., FarnsworthM. E., неопублико-
неопубликованная работа, M I. T. Hydrogen Peroxide Project, 1947.
241. McConnel 1 A. H., H an es E. S., J. Chem. Soc, 71, 584 A897); D u r r a n t R. G.,
ibid., 87, 1781 A905); P a a 1 C.Boeters H., Вег., 58В, 1539 A925); В e r n а г d .
R., J о b P., Compt. rend., 190, 186 A930); G 1 e u K-, R e h m R., Z. anorg. allgem.
Chem., 237, 79 A938); Barb W. G., В a x e n d a 1 e J. H., G e о г g e P., H a r-
gra ve K. R., Trans. Faraday Soc, 47, 591 A951).
242. G о а г d A. K-, R i d e a 1 E. K., Proc Roy. Soc (London), 105A, 148 A924); M a n-
c h о t W., L e h m a n n G., Ann., 460, 179 A928); Hale D. R., J. Phys. Chem.,
33, 1633 A929); Manchot W., Pflaum W., Z. anorg. allgem. Chem., 211,
1 A933).
243. Evans M. G., George P., U г i N., Trans. Faraday Soc, 45, 230 A949).
244. Marshall W. A., Metal Finishing, 50, 78 (XI. 1952); 67 (XII. 1952).
245. К i s t i a k о w s k у W., Z. physik. Chem., 35, 431 A900); L u e с k E., Apoth.
Ztg., 34, 87 A919) [C. A., 13, 2823]; К о h n M., Monatsh., 66, 393 A935); В a u-
dischO., Science, 108, 443 A948); L a 1 В. В., Proc. Indian Acad. Sci., 36A, 244
A952) [C. A., 47, 5803d); DeFord D. D., Davidson A. W., J. Am. Chem.
, Soc, 73, 1469 A951).
246. T а н а н а е в H. A.,Z. anorg. allgem. Chem., 143, 118 A925); О рловский СТ.,
ЖРФХО, 61, 1185 A929); D u В о i s P., Compt. rend., 196, 1401 A933); 199, 1310
A934); P ug h W., J. Chem. Soc, 1934, 1150; Porter L. E., С a d e G. N.. Jr.,
J. Am. Chem. Soc, 57, 1604 A934).
247. Z a wi ds k i J., Roczniki Chemji,!, 135 A921) [C. A., 16, 521]; L imanowskiW.,
Roczniki Chemji, 12, 519,638 A932) [C. A., 27, 5621]; Bailey К. С, Т a y-
1 о г G. Т., J. Chem. Soc, 1937, 994; С h a n с е В., J. Franklin Inst., 229, 737 A940);
M с К i n n e у С. D., Jr., К i 1 p a t г i с k M., Rev. Sci. Instruments, 22, 590
A951).
248. M e г z J. H., S t a f f о г d G., W a t e г s W. A., J. Chem. Soc, 1951, 638.
249. H a g e n H., S i e v e г t s A., Z. anorg. allgem. Chem., 208, 367 A932).
250. Шпитальский E., Z. anorg. Chem., 69, 179 A910); P i e t s с h E., К о t о w-
s k i А., В e г e n d F. G., Z. physik. Chem., 5B, 1 A929); N i с k e г s о n D. G.,
J. Am. Chem. Soc, 58, 2525 A936); M u 1 1 e г F., Pietsch R., Korrosion u.
.Metallschutz, 15, 122A939) [C. A., 33, 7205]; H икол аев Л. А.ДобозевН.И.,
ЖФХ, 19, 562 A945).
251. Martinez J. В., Anales real soc. espan. fis. у quim. (Madrid), 46B, 665 A949)
[C. A., 44, 3828П; M e л и к о в П., ЖРФХО, 44, 605 A912).
252. Macri V., Boll. chim. farm., 56, 417 A917) [С. А., 12, 795].
253. Cain J. R., H о s t e t t e r J. C, J. Am. Chem. Soc, 34, 274 A912); R i v e n q F.,
Bull. Soc. Chim., 1946, 677; T г u j i 1 1 e R., M а г t i n e z J. В., Anales real soc.
espan. fis. у quim., 47B, 705 A951) [C. A., 46, 8589a].
254. H a h n O., G i 1 1 e H., Z. anorg. allgem. Chem., 112, 283 A920).
255. Nees H., P. B. Report 74717 (frame 203).
256. M e h t a S. M., P о n с h a R. P., J. Am. Chem. Soc, 74, 3469 A952) [C. A., 46,
HOOlh).
257. К a t z о f f S., R,o s e m a n R., J. Am. Chem. Soc, 57, 1384 A935).
258. К л е й н е р К. Е., ЖОХ, 22, 17 A952).
259. Nees H., P. B. Report 74717 (frames 199—205).
260. A g e n о F., Barzetti E., Atti R. Acad. dei Lincei di Roma C), 191, I, 381.
261. Ma ass E., Wi ederhol t W., Z. Metallkunde, 17, 115A925) [C. A., 19, 3244];
R а с k w i t z E., S с h m i d t E. К. О., Korrosion u. Metallschutz, 3, 5 A927)
[C. A., 21, 19591; Wache X., С h a n d г о n G., Rev. Metal., 26, 209 A929>
[C. A., 23, 4661]; Rohrig H., S с h б n h e г г К-, Korrosion u. Metallschutz, 11,
136 A935) [C. A., 29, 6196]; W i e d e r h ol t W., Korrosion u. Metallschutz, 8,
4 A932) [C.A., 26, 2683]; M i a 1 k i W., Aluminum, 20, 315 A938) [C. A., 32, 6218];
Do b b el m a n n T. A. H. M., Verlag. Natl. Luchtvaartlab, 12, M1-M4 A943)
(C. A., 42, 6732al; LoganH.L, H e s s i n g H., J. Research Natl. Bur. Stan-
Standards, 41, 69 A948) (Research Paper 1905); Томашов Н. Д., Матвеева Т. В. t
ЖФХ, 24, 1281 A950); Павлов С. Е., Труды Института физической химии АН
СССР, 2, Исследования по коррозии металлов, № 1, 75 A951); Томашов Н. Д.,
Матвеева Т. В., там же, 146 A951); Barnett F. R., Australian Engr.,
Feb., 1953, 58, [С. А., 48, 5399d].
368 Глава 7
262. Акимов Г. В., О л е ш к о А. С, Korrosion u. Metallschutz, 11, 125 A935).
263. de BoisbaudranL, Compt. rend., 100, 605 A885); С 1 e v e P. Т., Bull. Soc
Chim., B), 43, 53 A885); Танатар С, Z. anal. Chem., 28, 255 A889).
264. Font an a B. J., MDDC-542, AEC, 1946.
265. A b e g g A., P. B. Report L-73679, frames 8525—8530.
266. Baker H. В., P a r k e r L. H., J. Chem. Soc, 103, 2060 A913).
267. F e n t о n H. J. H., J. Chem. Soc, 65, 899 A894).
268. M e r z J. H., W a t e г s W. A., J. Chem. Soc, 1949, S15.
269. Swern D., Chem. Revs., 45, 1 A949).
270. Taylor H. V., The Industrial Chemist, 29, 9 A953).
271. Johnson A. W., Science Progress, 39, 96 A951).
272. F г i e s s S. L., in Weissbcrger, Investigation of Rates and Mechanisms of Reactions,
Vol. 8, Technique of Organic Chemistry, Interscience Publishers, Inc., New York,
1953, p. 412.
273. Hudlicky M., Chem. Listy, 46, 567 A952) [C. A., 46, 11104A].
274. Testa E., Oxydationen durch Wasserstoffsuperoxyd und Persauren, die zur Spal-
tung von С—С Bindungen Fiihren, Juris—Verlag, Zurich, 1950 [C. A., 46, 5614d].
275. HI э н л и Э., Г р и н с п э н Ф., в сб. «физика и химия реактивного движения»,
№ 2, Издатинлит, М., 1949, стр. 161.
276. О d d о В., В i n a g h i R., Gazz. chim. Hal., 51, II, 343 A921);, Od d о В., В i-
n a g h i R., Atti. R. Acad. dei Lincei di Roma E), 32, 349 A923).
277. С в е р н Д., в сб. «Органические реакции», № 7, Издатинлит, М., 1956, стр. 476.
278. Greenspan F. P., G а 1 1 R. J., Ind. Eng. Chem., 45, 2722 A953); Green-
spanF. P., Modern Plastics, 31, № 7, 123, 202, 204 A953); Epoxidation and Hydro-
xylation, A Literature Review, E.I. duPont de Nemours and Co., 1953; Epoxidation
and Hydroxylation, Bulletin 16, Buffalo Electro-Chemical Co., 1950.
279. English J., G г e g о г у J. D., J. Am. Chem. Soc, 69, 2120 A947); Boese-
ken J., J а с о b s J., Rec. trav. chim., 55, 786, 804 A936).
280. M i 1 a s N. А., пат. США 2395638 B6.11. 1946), 2414385 A4. I. 1947).
281. A n s 1 о w W. P., S. B. Thesis in Chemistry, Massachusetts Institute of Technology,
1936.
282. Vene J., Bull. Soc. Sci. Bretagne, 20, 11 A945) [C. A., 41, 41 lid]; Bull. soc. sci.
Bretagne, 23, 123 A948) [C. A., 44, 6395d]; Institute of Synthetic Organic Chemical
Research, япон. пат. 155800 A.IV. 1943) [С. А., 44, 3009g].
283. I b u k i E., J. Chem. Soc. Japan, 70, 284 A949) [C. A., 45, 4701h].
284. P e г k i n W. H., Jr., Proc Chem. Soc Lond., 23, 166 A907).
285. Kawamoto Т., О g u r a I., J. Chem. Soc. Japan, 72, 526 A951) [C. A., 46, 6617gl
286. Henderson G.G., Robertson A., J. Chem. Soc, 1926, 2761.
287. T a i p a 1 e V., Ober die Einwirkung von Wasserstoffperoxyd auf die Natriumyerbin-
dung des 2-Phenyl-Indandions-l,3 in wassriger Losung, Vaitoskirja, Helsinki, 1952
[C. A., 47, 4356g].
288. Walton J. H., Christensen С J., J. Am. Chem. Soc, 48, 2083 A926),
Hobaica J. C, S.B. Thesis in Chemical Engineering, Massachusetts Institute of
Technology, 1947.
289. К h а г a s с h M. S., F о n о A., Nudenberg W., P о s h k u s A. C, J. Org. Chem.,
15, 775 A950).
290. Miner C. S., D a 1 t о n N.N., Glycerol, Reinhold, Publishing Corp., New York, 1953.
291. M e г z J. H., W a t e r s W. A., Discussions Faraday Soc, № 2, 179 A947).
292. d'Ans J., F г е у W., Ber., 45, 1845 A912); G г e e n s p a n F. P., J. Am. Chem.
Soc, 68, 907 A946); Hatcher W. H., H о 1 d e n G. W., Trans. Roy. Soc. Can.
C), 18, Sect. Ill, 231. A924); WielandH., LovenskioldH., Ann. Chem.,
436,241 A924); W i t z e m a n n E. J., J. Am. Chem. Soc, 48, 202, 208, 211 A926);
Hatcher W. H., H i 1 1 A . C, Trans. Roy. Soc Can., 23, III, 213 A929).
293. Hatcher W. H., Trans. Roy. Soc Can., 17, III, 119 A923).
294. Hatcher W.H.,Hill A. C, Trans. R. Soc Canada C), 22, III, 211 A928);
23, 213 A929).
295. Walton J. H., G г a h a m D. P., J. Am. Chem. Soc, 50, 1641 A928).
296. Наг vey J. P., S. B. Thesis in Chemistry, Massachusetts Institute of Technology, 1923.
297. Church J. M., Bl umberg R., Chem. Eng., 58, № 1, 261 A951).
298. Hatcher W.H.,Holden G. W., Trans. Roy. Soc. Can., 19, III, 11 A925);
NathM.C, Islam A. H. M. H., Indian J. Med. Research, 36, 61 A948) [C. A.,
47, 5527g].
299. В u n t о n C. A., Nature, 163, 444 A949); К а г г е г Р., Н a a b F., Helv. Chim.
А с ta, 32, 950 A949) (на немецком языке).
300. F r i e d m a n n Т. Е., J. Biol. Chem., 73, 331 A927).
301. Hatcher W. H., H о 1 d e n G. W., T о о 1 e F. J., Trans. Roy. Soc. Can.,
20, III, 407 A926); Goldschmidt S., Askenasy P., Pierros S.,
Ber., 61, 223 A928); Fry H. S., M il s tea d K. L, J. Am. Chem. Soc, 57, 2269
A935).
Литература 369
302. Н a s s a 1 1 С. Н., J. Chem. Soc, 1948, 50.
303. Hudl icky M., Chem. Listy, 45, 380 A952); Collection Czechoslov. Chem. Commun.,
16, 283, 611 A951) (на английском языке) [С. А., 47, 8012i].
304. Hatcher W. Н., Н о 1 d е n G. W., Т о о 1 е F. J., Trans. Roy. Soc. Can., C),
III, 20, 395, 415 A926).
305. Reiner L., Z. anorg. allgem. Chem., 127, 187 A923); 141, 363, A924).
306. J a i 1 1 e t J. В., О u e 1 1 e t C, Can. J. chem., 29, 1046 A951); D u n i с z B. L.,
P e г г i n D. D., S t у 1 e D. W. G., Trans. Faraday Soc, 47, 1210 A951); Satter-
field С N., С a s e L. C, Ind. Eng. Chem., 46, 998 A954).
307. Fry H.S., Payne J. M., J. Am. Chem. Soc, 53, 1973, 1980 A931).
308. В u r k i F., Schaaf F., Helv. Chim. Acta, 4, 418 A921); W i г t z K-, В о n-
hoeffer K. F., Z. physik. Chem., B32, 108 A936).
309. Greenspan F. P., Ind. Eng. Chem., 39, 847 A947); Eramons W.D..J. Am.
Chem. Soc, 76, 3470 A954).
310. Г у р е в и ч А. А., ДАН СССР, 91, 543 A953).
311. W i 1 e у R. H., M о r g a n H. S., Jr., J. Org. Chem., 15, 800 A950).
312. Dubsky J. V., Chem. Weekblad, 17,220A920) [C.A., 14,3644].
313. Johnson G. R. A., S с h о 1 e s G., Weiss J., Science, 114, 412 A951).
314. Costa G., Ann. triestini univ. Trieste, sez. 2, 2223, 151 A953) [C.A., 48, 4332i|;
К a uf f man n H. J., J. Am. Chem. Soc, 73, 4311 A951).
315. J о s t W., Fahlbusch K. G., Z. anorg. Chem., 262, 79, A950); Sparrow
D. В., S.B. Thesis in Chemistry, Massachusetts Institute of Technology, 1949; Barb
W. G., В a x e n d a 1 e J. H., G e о г g e P., H a r g г a v e K. R., Trans. Fara-
Faraday Soc, 47, 462 A951).
316. RytzW., Mitt, naturforsch. Ges. Bern., Sitzber, 1942[C.A., 41, 5920a]; Weber K.,
Arkiv. Kern., 23, 173 A951) [C.A., 47, 11993g].
317. Huntress E. H., S t a n 1 e у L.N., Parker A. S., J. Chem. Education, 11,
142 A934).
318. Anderson W., Nature, 160, 892 A947); 163, 44 A949).
319. Гринберг А. А., ЖРФХО, 52, 151 A920).
320. Biswas N. N.. Dhar N. R., Z. anorg. allgem. Chem., 186, 154 A930).
321. Jablcznski K., Cholewicki A., Roczniki Chem., 17, 387 (на немецком
языке, 391) A937) [С. А., 31, 25171); Groh Р., К i г г m a n n A., Compt. rend.,
215, 275 A942).
322. de Seixas Palma J., Ber., 39, 3317 A906).
323. Schoberl A., Hamm R., Chem. Ber., 81, 210 A948) [C.A., 43, 1014h].
324. von S z e n t-G у 6 г g у A., Biochem. Z., 178, 75 A926).
325. К a m b а г a S., 0 k i t a K., Tajima M., Chem. High Polymers, 5, 199 A948)
[C.A., 46, 1795].
326. Schoberl A., Hoppe-Seylers Z. physiol. Chem., 216, 193 A933).
327. I s h i d a S., J. Chem. Soc Japan, 64, 165 A943) [C.A., 41, 3752a].
328. Gregg D.C., Blood С A., Jr., W e i m a n D. E., J. Am. Chem. Soc, 75, 4344
A953).
329. К a z u m i F., J. Pharm. Soc. Japan, 65, № 5/6A, 4 A945) [C.A., 45, 5652c].
330. M о г г e 1 1 R. S., Crofts J. M., J. Chem. Soc, 75, 786 A899); 77, 1219 A900);
81, 666A902); 83, 1284 A903); Мог г ell R.S., Bellars A. E., Proc Chem.
Soc, 21, 78 A905).
331. Kiichin А. Т., Biochem. Z., 261, 411 A933).
332. LemaireH., S.B. Thesis in Chemistry, Massachusetts Institute of Technology, 1942.
333. M e r z J.H., Waters W. A., J. Chem. Soc, 1949, 2427; Anderson V. S.,
Acta Chem. Scand., 4, 207 A950) (на английском языке); Baxendale J. H.,
Ma gee J., Discussions Faraday Soc, № 14, Toronto, 1953.
334. В о t t о m 1 e у G. А., В 1 а с k m a n J. В., Nature, 171, 620 A953).
335. Greenspan F. P., Ind. Eng. Chem., 39, 847 A947); R а а с k e -F e 1 s I. D.,
W a n g С H., Robins R. K., Chr istensen B. E., J. Org. Chem., 15,
627 A950); В a d e г A. R., J. Am. Chem. Soc, 73, 3731 A951).
336. Богданов СВ., М и г а ч е в а И. В., ЖОХ, 20, 124 A950).
337. L e u I i е г М. A., Bull. Soc Chim. (France), D), 35, 1325 A924); M а г s h J. E.,
J. Chem. Soc, 1927, 3164; Johnson В., J. Am. Chem. Soc, 65, 1218 A943);
V i t e x S. А., франц. пат. 971654 A9.1.1951); J u г d L., Australian J. Sci. Re-
Research, 2A, 595 A949).
338. Rice R. G., К е г e s z Z. I., S t о t z E. H., J. Am. Chem. Soc, 69, 1798 A947).
339. D a v i e s W., G a m b 1 e N. W., James F. C.Savige W. E., Chemistry
and Industry, 1952, 804.
340. Brown E. В., T h о m a s J. M., В i n a A. F., J. Biol. Chem., 162, 221 A946).
341. В a u d i s с h O., BassL. W., J. Am. Chem. Soc, 46, 184 A924).
342. Baxter R. A., N e w b о 1 d G. Т., S p г i n g F. S., J. Chem. Soc, 1948, 1859.
343. Bennett G. M., Glynn E., J. Chem. Soc, 1950,211.
344. Maffei S., Gazz. chim. ital., 76, 239 A946).
24 Перекись водорода
370 Глава 7
345. V е n a b 1 е С. S., Moore F. J., J. Am. Chem. Soc, 39, 1750 A917); V e n a b 1 e
C. S., J. Am. Chem. Soc, 40, 1099 A918); Chrometzka F., Z. Physiol. Chem.,
162, 219 A926—1927); G a t e w о о d E. S., S.B. Thesis in Chemistry, Massachu-
Massachusetts Institute of Technology, 1922.
346. Schaer E., Arch. Pharm., 248, 458 A910).
347. Fernandez O., Pizzaroso A., Annales Soc. espafiola fis у quim., 20,
589 A922).
348. Pinner A., W о 1 f f e n s t e i n R., Ber., 24, 63 A891).
349. D r u m m о n d A. M., McMill an A., J. Chem. Soc, 1926, 2702.
350. С w ech J., S a n t a v у F., Collection Czechoslov. Chem. Communs., 14, 532 A949)
(на английском языке) [С.А., 44, 5887h].
351. Kambara S., Muramatsu Y., J. Soc Rubber Ind. Japan, 16, 676 A943)
[C.A., 44, 3283c]; J. Soc. Rubber Ind. Japan, 16, 761 A943) [C.A., 44, 5137c]; Kam-
Kambara S., 0 k i t а К., О z a w a N.. Chem. High Polymers, 5, 380 A948) [C.A., 46,
1796 fgl; Kambara S., О о k i t a K., I m a d a M,. Chem. High Polymers, 3,
81 A946) [C.A., 44, 11151d]; T о m о n а г i Т., J. Electrochem. Assoc. Japan, 15,
32A947) [C.A., 44, 9824a], Hand а Т., Chem. High Polymers, 6,382A949) [C.A.,
46, 1794c].
352. Веселовский В. С, Орлеанская Г. Л., Изв. АН СССР, отделение
техн. наук, 1951, 1041.
353. Кондратьев Е. В., ЖПХ, 22, 882 A949).
354. М i I a s N. А., М а г g u I i e s P. H., Shanley E. S., in Kirk-Othmer, Encyclo-
Encyclopedia of Chemical Technology, Vol. 10, Interscience Encyclopedia, Inc., New York,
1953, p. 58.
355. С r i e g e e R., in Houben-Weyl, Methoden der Organischen Chemie, 4th ed., Vol. 3
(Sauerstoffverbindungen), G. Thieme Verlag, Stuttgart, 1952.
356. Rieche A., Die Bedeutung der Organischen Peroxyde fur die Chemische Wissen-
schaft u. Technik, Sammlung, 34, 1436; Swern D.,Scanlon J. Т., Knight
H. В., J. Am., Oil Chemists Soc, 25, 193 A948); Leffler J. E., Chem.
Revs., 45, 385 A949); С r i e g e e R., Fortschs. Chem. Forsch., 1, 508 A950); Haw-
Hawkins E. G. E., Quart. Revs., 4, 251 A950); Frank С. Е., Chem. Revs., 46, 155
A950); Eggersgluss W., Organische Peroxyde, Verlag-Chem., Weinheim,
1951 [C.A., 45, 3860b).
357. И о f f m a n n-0 s t e n h о f O., Advances in Enzymology, 14, 219 A953).
358. West E. S., T о d d W. R., Textbook of Biochemistry, Mac-Millan, New York, 1951.
359. L a r d у Н. A., ed., Respiratory Enzymes, Burgess Publishing Co., Minneapolis, 1949.
360. Trans. 4th Conf. Biological Antioxidants, Josiah Macy, Jr. Foundation, New York, 1950,
p. 96—99.
361. Warburg O., Heavy Metal Prosthetic Groups and Enzyme Action, Clarendon Press,
Oxford, 1949.
362. W i e 1 a n d H., On the Mechanism of Oxidation, Yale Univ. Press., New Haven, 1932.
363. О p p e n h e i m e г С, S t e г n K. G., Biological Oxidation, Nordemann Publi-
Publishing Co., Inc., New York, 1939.
364. D a k i n H. D., Oxidations and Reductions in the Animal Body, 2nd ed., Longmans,
Green and Co., London, 1922.
365. С а м н е р Дж. Б., С о м е р с Г. Ф., Химия ферментов и методы их исследования,
Госиноиздат, М., 1948.
366. L a i d 1 е г К. J., Introduction to the Chemistry of Enzymes, McGraw-Hill Book Co.,
Inc., New York, 1954.
367. George P., Advances in Catalysis, 4, 367 A952).
368. G r a n i с k S., Record Chem. Progr. (Kresge-Hooker Lib.), 15, 27 A954).
369. Herbert D.,Pinsent J., Biochem. J., 43, 193 A948).
370. G r a n i с k S., Ann. N. Y. Acad. Sci., 48, 658 A947).
371. R a w 1 i n s о n W. A., Revs. Pure Appl. Chem., 4, 15, A954).
372. T h u r 1 о w S., Biochem. J., 19, 175 A925).
373. К e i 1 i n D., H a r t г e e E. F., Biochem. J., 39, 293 A945).
374. К e n t e n R. H., M a n n P. J. G., Biochem. J. 52, 130 A952).
375. Chance В., Advances in Enzymology, 12, 153 A951).
376. Haurowitz F., Brdieka R., Kraus F., Enzymologia, 2, 9 A937);
H a u г о w i t z F., ibid., 4, 139 A937).
377. К i k u с h i G., J. Japan. Biochem. Soc, 22, 213, 235 A950) [C.A., 45, 9091 ij; 23, 38
A951) [C.A., 46, 8165c]; 23, 137 A951) [C.A., 47, 2794b]; К a j i г о К-, К i k u -
с h i G., J. Biochem. (Japan), 38, 213 A951) [C.A., 46, 548b|; 39, 63 A952) [C.A., 46,
7597b]; 39, 193 A952) [C.A., 46, 9129b]; 39, 357 A952) [C.A., 47, 17491].
378. Haurowitz F., Progress in Biochemistry, Interscience Publishers, New York,
1950.
379. S с h 6 n b e i n С F., Verhandl. Naturforsch. Ges. Basel, 3, 339 A863).
380. L i n о s s i e г M. G., Compt. rend. soc. biol., 50, 373 A898).
381. Elliot К. А. С, Bicchem. J., 26, 10, 1281 A932).
Литература 371
382. Бах А. II., Вег.. 37, 3785 A904); Abel К., Z. Elcktrochcm., 28, 489 A922).
383. Randall L. О., J. Biol. Chem., 164, 521 A946).
384. Fischer M., Biochem. Z., 292, 271 A937); Околов Ф., Биохимия, 16, 108
A951).
385. G о 1 d а с г e P. L., Australian J. Sci. Research, B4, 293 A951).
386. Agner K., Nature, 159, 271 A947); J. Exptl. Med., 92, 337 A950).
387. Zamboni P., Delia-Bella D., Atti soc. lombarda sci. med. biol., 3, 322
A948) [C.A., 45, 3442dl; Daniels D. G. H., N а у 1 о r F. Т., SaundersB.C,
J. Chem. Soc, 1951, 3433.
388. К e n t e n R. H., Mann P. J. G., Biochem. J., 45, 255 A949); 46, 67 A950); 50,
29 A951); 52, 125 A952).
389. S i z e г I. W., В г i n d 1 e у С. О., W a g 1 e у P. F., Intern. Congr. Biochem.,
Abstrs. of Communs. 1st Congr., Cambridge, Engl., 1949, 361; Sizer I. W.,
Advances in Enzymology, 14, 129 A953).
390. Balls A. K., Hale W. S., Ind. Eng. Chem., 27, 335 A935).
391. Chance В., J. Biol. Chem., 182, 643, 649 A950).
392. Stern R., В i г d L. H., Biochem. J., 49, 335 A951).
393. I s h i d a t e M., Okano S., T a d a K., J. Pharm. Soc. Japan, 63, 228, 231
A943) [C. A., 44, 6898cI.
394. Балаховский С. Д., Троицкая Н. А., ДАН СССР, 82, 285 A952).
395. L е г о и х Н., Bull. soc. chim. biol., 33, 705 A951).
396. An near D. I., Dor man D. C, Australian J. Exptl. Biol. Med. Sci., 30, 192
A952).
397. К о d i t s с h e k L. K., II e n d 1 i n D., W о о d r u f f H. В., J. Biol. Chem., 179,
1093 A949).
398. Payne J. H., Foster L., J. Am. Chem. Soc, 67, 1654 A945).
399. К е г t e s z Z. I., Plant Physiol., 18, 308 A943); Griffin J. H., К e r t e s z Z. I.,
Botan. Gaz., 108, 279 A946); SondheimerE., Kertesz Z. I., Food Research,
17, 288 A952).
400. Gust us E. L., Ph. D. Thesis, Northwestern Univ., 1926.
401. Brown \V. R., J. Biol. Chem., 113, 417 A936); G a s t i n e a u С F., Shep-
p а г d F., E v e r e t t M. R., Proc. Oklahoma Acad. Sci., 23, 53 A943) [C.A., 38,
19411; Yamafuji K., U r a k a m i M., J. Faculty Agr. (Kyushu Univ.), 9,
333 A950) [C.A., 46, 11117h|.
402. Robertson W. V. В., Ropes M.W., Bauer W., Biochem. J., 35, 903
A941).
403. Jordan R. M. M., Brit. J. Exptl. Path., 33, 36 A952).
404. S e e 1 e у H. W., d e 1 R i o-Est r ad aC, J. Bact., 62, 649 A951); L e v i t о n A.,
J. Am. Chem. Soc, 68, 835 A946).
405. V a n d о 1 a h R. W., Chr i stenson G. L., Arch. Biochem., 12, 7, A947).
406. I s h i i Y., J. Soc. Chem. Ind. Japan, 44, Suppl. binding 219 A941) [C.A., 44,3163i];
Dhar N. R., Raghavan B. V. S., Proc. Natl. Acad. Sci. India, I6A, 14
A947).
407. R о n d о n i P., В a s s i M., Enzymologia, 15, 70 A951).
408. Fernandez O., Garcia de Mirasierra M., Rev. real acad. cienc.
exact, fis. у nat. (Madrid), 44, 63 A950) [C.A., 46, 543d].
409. N e u b e г g C, Miura S., Biochem. Z., 36, 37 A911).
410. Jacques L. В., В e 1 1 H. J., Can. J. Research, 24E, 79 A946).
411. D e u t s с h H. F., J. Am. Chem. Soc, 68, 2625 A946).
412. Dickens F., Glock G. E., Biochim. et Biophys. Acta, 7, 578 A951).
413. Bennett M. A., T о e n n i e s G., J. Biol. Chem., 145, 671 A942).
414. Tosic J., Walton A., Nature, 158, 485A946); Biochem. J., 47, 199 A950);
T о s i с J., Nature, 159, 544 A947); Biochem. Soc. Symposia, № 7, 22 A951) [C.A.,
46, 7606Ы.
415. В о q u e t P.,Compt. rend., 208, 770 A939); Compt. rend. soc. biol., 131, 7 A939);
132, 418 A939); 135, 781 A941); Ann. Inst. Pasteur, 66, 379 A941); С h a u d h u г i
D. K., Ann. Biochem. and Exptl. Med. (India), 10, 71 A950); D e S. S., Indian J.
Med. Research, 27, 793 A940) [C. A., 34, 7438].
416. Virtanen A. I., Jorma J., Linkola H., Linnasalmi A., Acta
Chem. Scand., 1, 90 A947); Федоров М. В., ДАН СССР, 67, 557 A949).
417. G r u n b с r g-M a n a g о M., S z u 1 m a j s t e г J., Del a vier C, Ann. Inst.
Pasteur, 83, 102 A952).
418. Guzman Barron E. S., SekiL., Johnson P., Arch. Biochem., Biophys.,
41, 188 A952).
419. Feigenbaum J., Exptl. Med. Surg., 8, 102 A950).
420. Ross M. II., Ely J. O., J. Cellular Сотр. Physiol., 34, 71 A949).
421. Krishnamurti C. R., Subrahmanyan V., Indian J. Dairy Sci., 2, 19
A949).
422. Кита в и и Г. С, Биохимия, 17, 336 A952).
24*
372 Глава 7
423. Binder E., Rev. suisse zool., 64, 236 A947).
424. Gordon H. H., D e M e t г у J. P., Proc. Soc. Exptl. Biol. Med., 79, 446 A952);
RoseC.S., G у 6 г g у P., Am J. Physiol., 168, 414 A952); G у 6 г g у P., С о-
g a n G., R о s e C. S., Proc. Soc. Exptl. Biol. Med., 81, 536 A952).
425. von Bergen W., Proc. ASTM, 35, Part 2, 705 A935); E 1 6 d E., N о w о t n у Н.,
Z a h n H., Melliand Textilber., 23, 313 A942) [C.A., 38, 3136]; Blackburn S.,
L о w t h e r A. G., Biochem. J., 49, 554 A951).
426. Smith A. L, Harris M., J. Research Nat'l. Bur. Standards, 16, 301, 309 A936);
Textile Mfr.,62, 278 A936); Am. Dyestuff Reptr., 25, P180.P183 A936); Ruther-
f о r d H. A., H a r r i s M., J. Research Nat'l. Bur. Standards, 20, 559 A938).
427. Stoves J. L., Trans. Faraday Soc, 38, 501 A942); Mfg. Chemist, 22,387 A951).
428. Gof f art M., Arch, intern, pharmacodynamie, 74, 9 A947).
429. Wei war t, Seifensieder-Ztg., 62, 1013 A935) [C. A., 30, 2323].
430. Barsotti M., Parmeggiani L, Sassi C, Med. Lavoro, 42, 49 A951)
[C. A., 45, 6321c]; 43, 160 A952) [C.A., 48, 11739a].
431. К i n s e у V. E., G г a n t W. M., Robinson P., P e n t z H., P. B. Report
41021 A944).
432. Eden G. E., F г е к e A.M.,Mel bourne K. V., Chem. and. Ind., 1951, 1104.
433. К г а с к о w E. H., Health Hazards of Military Chemicals, Rpt. № 4, Army Chemical
Center, Edgewood, Md., 1950.
434. Buffalo Electro-Chemical Co., Properties of Hydrogen Peroxide, 1946, в которой цити-
цитируется Toxicological Study at Huntsville Arsenal, 1945.
435. Hughes W. F., Jr., Bull. Johns Hopkins Hosp., 82, 338 A948).
436. P u n t e С L, Saunders L Z., Krackow E. H., Chem. Corps Med. Labs.
Research Rpt. 189, Army Chemical Center, Edgewood. Md., 1953.
437. С о m s t о с k С. С, Н а с к 1 е у Е. В., О b е г s t F. W., L a w s о n L., Gre-
Greene E., Chem. Corps Med. Labs. Research Rpt. 243, Army Chemical Center, Edge-
wood, Md., 1954.
438. Lorincz A. L., Jacob у J. J., Livingstone H. M., Anesthesiology,
9, 162 A948); F e i n s t e i n R. N.,Butler С L, Hendley D. D., Science,
111, 149 A950); Петров В. П., Фармакология и токсикология, 3, № 5, 6106
A940).
439. Hrubetz М. С, Conn L. W., G i t t e s H. R., M а с N a m e e J. K., W i 1-
1 i a m s J. H., Chem. Corps Med. Labs. Research Rpt. 75, Army Chemical Center,
Edgewood, Md., 1951.
440. L i с u г z i A., Rev. assoc. med. argentina, 52, 970 A938) [C.A., 33, 2207].
441. Baker R. D., Arch. Surg., 48, 300 A944).
442. W у s s O., S t о n e W. S., С 1 a r k J. В., J. Bact., 54, 767 A947); A 1 p e г Т.,
Nature, 162, 615 A948); D u s t i n P., Jr., G о m p e 1 C, Compt. rend. soc. biol.,
143, 874 A949); Sherman F. G., Biochim. et Biophys. Acta, 5, 569 A950); D u-
b о u 1 о z P., D u m a s J., V i g n e J., Compt. rend. soc. biol., 144, 1080 A950);
Demerec M., Bertani G., Flint J., Am. Naturalist, 85, 119 A951);
Dean A.C. RM Hinshel wood C, J. Chem. Soc, 1951, 1157, 1159; Jen-
s e n K. A., Kirk I., К о 1 m a r k G., W e s t e г g а а г d M , Cold Spring
Harbor Symposia Quant. Biol., 16, 245 A951); Alexander P., F о х М., Nature,
170, 1022 A952); Kimball R. F., G a i t h e r N., Proc. Soc. Exptl. Biol. Med.,
80, 525 A952); D i P а о 1 о J. A., Am. Naturalist, 86, 49 A952); К 6 1 m а г k G.,
W e s t e r g а а г d M., Hereditas, 39, 209 A953).
443. Trans. 5thConf. Biological Antioxidants, Josiah Macy, Jr., Foundation, New York, 1951.
444. Guzman Barron E.S., Dickman S. R., S i n g e г Т. P., M u n t z J. A.,
in Zirkle, Biological Effects of External x-and Gamma Radiation, Part I., McGraw-
Hill Book Co., New York, 1954, p. 388.
445. T a k e d a K., Japan J. Obstet. Gynecol., 24, № 2, 20, A941) [C. A., 36, 551].
446. Gray L. H., Chem. and Eng. News, 31, 2774 A953).
447. Y a m a f u j i K-, T a k e о k a S., F u j i k i Т., J. Agr. Chem. Soc. Japan, 19,
225 A943) [C.A., 45, 8057h]; Yamahuzi K., Yosihara H., Biochem. Z.,
317, 81,87 A944) [C.A., 39, 41031; Yaraafuji К., С h о Т., Biochem. Z., 318, 95
A947) [C. A. 42, 3085Ы; Y a m a f u j i K., F u j i k i Т., Biochem. Z., 318, 101
A947) [C.A., 42, 3030g]; Y a m a f u j i K., Y u k i Т., Biochem. Z., 318, 107
A947) [C.A., 42, 3085d];Osawa M., Igaku to Seibutsugaku (med. and Biol.),
21, 154 A951) [C.A., 46, 10467c]; I s h i m о r i N.. О s a w a M., ibid., 22, 81,
172 A952) [C.A., 46, 10467e].
448. H e u b n e г W., Klin. W о с h s с h r ., 20, 137 A931); Keith С. К., В г о а с h
W. J., Warren D., DayP.L, Totter J. R., J. Biol. Chem., 176, 1095A948).
449. Mann P. J. G., Q uastel J. H., Biochem. J., 40, 139 A946); Dickens F.,
ibid., 40, 145 A946); Givre A., R e x e d В., Acta Psychiat. et Neurol., 23, 247
A948); Excerpta Med., Sect. VIII, 2, 840 A949).
450. Kaufman L., Ann. univ. Marie Curie Sklodowska Lublin-Polonia, Sect, E, 2. 19
A947) [C.A., 42, 8981b).
Глава 8
ПРОЦЕССЫ РАЗЛОЖЕНИЯ
Легкость разложения перекиси водорода на воду и молекулярный кисло-
кислород является характерным свойством этого соединения, известным уже с мо-
момента открытия перекиси. Это свойство является в одних случаях весьма
полезным, в других—нежелательным, причем разложение может быть осущест-
осуществлено различными способами. Оно изучается уже давно и весьма интенсивно,
и тем не менее даже сейчас понимание процесса разложения еще во многих
отношениях ограниченно. В гл. 7 рассматривались характеристики перекиси
водорода, определяющие ее термодинамические свойства, а также механизмы
реакций. Процессы, обсуждаемые в этой главе, подчиняются тем же основам,
но в понимании их большей частью достигнуты меньшие успехи. Имеется ряд
факторов, объясняющих такое положение. Все процессы разложения похожи
один на другой в том отношении, что они отображают итоговую реакцию
2Н2О2-»2Н2О + О2, A)
включающую как окисление, так и восстановление перекиси водорода. Однако
многие пути, по которым протекает этот процесс, по существующим теориям
представляют цепные реакции; в случае гетерогенного катализа способ, по ко-
которому проявляется действие катализатора, известен лишь качественно.
В настоящей главе дается общий обзор, который может служить также введе-
введением в литературу. Анализ основных реакционных механизмов дан только
в тех областях, где имеется достаточная экспериментальная основа для опре-
определенных выводов. Проблема стабильности перекиси водорода заключается
в основном в снижении до минимума процессов разложения; этот вопрос рас-
рассматривается отдельно в гл. 9 ввиду его большого практического значения,
а также в связи с тем, что скорости разложения, которые интересны с точки
зрения стабильности, представляют величины совершенно другого порядка,
чем рассматривающиеся в этой главе. В гл. 11 приведены данные по примене-
применению процессов разложения для генерирования энергии или получения кисло-
кислорода. Тема разложения перекиси водорода в целом до сих пор еще не охвачена
исчерпывающим обзором. По отдельным вопросам имеются превосходные
обзоры (в которых, однако, излагается лишь ограниченный материал); ссылки
на них даются в соответствующих разделах.
РАЗЛОЖЕНИЕ В ПАРОВОЙ ФАЗЕ
Только сравнительно недавно в известной степени выяснены явления,
имеющие отношение к разложению перекиси водорода в паровой фазе, особен-
особенно взрывного характера. В этом разделе рассматриваются экспериментальные
и теоретические данные, касающиеся разложения пара перекиси водорода.
Экспериментальные методы получения пара перекиси водорода обсуждены
на стр. 158. Разложение пара перекиси под действием излучения кратко рас-
рассмотрено на стр. 382.
374 Глава 8
Экспериментальный материал
Разложение перекиси водорода в паровой фазе впервые исследовано
в 1923 г. Хаузером [1], который наблюдал это разложение на ряде поверх-
поверхностей различного типа, а также Хиншелвудом и Причардом [2], сообщив-
сообщившими на основании краткого исследования, что разложение в стеклянных
сосудах при 76° представляет собой поверхностную реакцию первого порядка.
В 1927 г. Элдер и Ридл [3] сообщили, что в кварцевом сосуде реакция прекра-
прекращается после разложения примерно 20% всех паров перекиси водорода; если
же этот сосуд нагревать примерно до 300—400°, происходит полное разложе-
разложение. Однако Кистяковский и Розенберг [4] в 1937 г. констатировали полное
разложение пара перекиси водорода на кварце, после чего не появлялось
никаких сообщений о наблюдениях, подтверждающих внезапное прекраще-
прекращение реакции до полного ее окончания. Жигер [5] высказал правдоподобное
объяснение, что Элдер и Ридл в действительности измеряли разложение пара
в соединительных трубках, ведущих к сосуду, тогда как фактическая скорость
реакции в самом кварцевом сосуде была ничтожно малой.
Указанные выше работы представляют в основном исторический интерес.
Более поздние исследования [5—8] доказали, что при температурах примерно
до 400° и парциальных давлениях перекиси водорода в несколько миллимет-
миллиметров ртутного столба реакция является полностью гетерогенной даже в сосудах
с наиболее инертными из изученных поверхностей, например изготовлен-
изготовленных из стекла пирекс. Для температур в области 470—540° и парциальных
давлений 1—2 мм рт. ст. (общее давление 1 am) доказано, что разложение,
по меньшей мере частично, является гомогенным, если оно проводится в стек-
стеклянных сосудах, обработанных с целью снижения активности поверхности
борной кислотой [9]. В работах, проведенных совсем недавно динамическим
методом при парциальных давлениях в несколько миллиметров ртутного
столба [10] (общее давление 1 am) и температурах 120—490° в приборе из
стекла пирекс, обнаружено при 425° резкое изменение характера разложения—
с гетерогенного на гомогенное. При атмосферном давлении пары, содержащие
26 мол.% перекиси водорода или больше (парциальное давление перекиси
198 мм рт. ст.), в остальном же состоящие из инертного газа, могут быть взор-
взорваны при контакте с каталитически действующей или сильно нагретой поверх-
поверхностью [11]. Это доказывает, что в случае достаточно высоких концентраций
возможно развитие вполне гомогенной реакции при значительно^более низкой
начальной температуре пара. Парциальное давление перекиси водорода для
предельного взрывчатого состава падает при снижении общего давления
и составляет, например, 22 мм рт. ст. при общем давлении 40 мм, т. е. при
минимальном из изученных (взрывчатые свойства паров перекиси водорода
рассмотрены на стр. 154 и ел.).
Скорость гетерогенного разложения в паровой фазе характеризуется
необычайной чувствительностью к небольшим изменениям физической при-
природы поверхности реакционного сосуда, а также ее химического состава.
В связи с этим неудивительно, что различные авторы получили сильно рас-
расходящиеся данные относительно скорости реакции, энергии активации или
влияния водяного пара, а также других газов на реакцию. Ниже в сжатой
форме приведены результаты некоторых существенных наблюдений, сделан-
сделанных в последнее время.
Кондратьева и Кондратьев [7] пропускали влажный воздух с содержа-
содержанием перекиси водорода, соответствовавшим парциальному давлению 0,01 —
0,4 мм рт. ст., через трубку из «молибденового стекла» при температурах
от комнатной до 175°. Они сообщили, что в этих условиях протекает реакция
второго порядка, имеющая энергию активации 8,5 ккал/моль. При промывке
реакционного сосуда раствором хлористого калия или азотнокислой меди
Разложение в паровой фазе 375
поверхность его активируется настолько, что уже при комнатной темпера-
температуре наблюдается значительное разложение перекиси водорода.
Маккензи и Ритчи [8] исследовали разложение манометрическим методом
в кварцевых сосудах при 80°, варьируя парциальное давление в пределах 1 :3
при начальном общем давлении от 0,6 до1,2 мм рт. ст. Методика опытов состояла
в эвакуировании реакционного сосуда с последующим введением перекиси
водорода путем испарения из известного количества высококонцентрирован-
высококонцентрированной жидкости. Скорость реакции определяли по росту давления. Эти авторы
сообщают, что водяной пар оказывает определенное замедляющее действие.
Наличие кислорода или азота с высоким отношением его парциального давле-
давления к парциальному давлению перекиси водорода также несколько замедляет
реакцию; это, возможно, вызывается тем, что указанные газы снижают ско-
скорость диффузии перекиси водорода к поверхности сосуда. На основании этих
исследований Маккензи и Ритчи пришли к выводу, что разложение является
реакцией второго порядка по перекиси водорода, они также ввели в уравнение
скорости еще один член для выражения замедляющего действия водяного пара.
В приборе аналогичного типа Бэкер и Уэллет [6] провели работу с при-
применением паров с начальным содержанием около 70—75% перекиси водорода
(остальное—водяной пар) при давлениях 10—20 мм рт. ст. и температурах 70—
200°. Они показали, что реакция сохраняет первый порядок по меньшей мере
до 80%-ного разложения перекиси при температурах приблизительно до 140^.
При более высоких температурах порядок реакции в их аппарате колебался
и температурный коэффициент был очень мал. Исследования, проведенные
в интервале температур 80—130°, показали, что скорость разложения в сосу-
сосудах, изготовленных из оплавленного стекла пирекс, ниже, чем в изготовлен-
изготовленных из обыкновенного стекла пирекс, а в сосудах из известково-содового
стекла реакция протекает быстрее, чем в случае применения стекла пирекс
любого вида. Скорость возрастает с ростом отношения поверхности к объему,
но невыясненные различия между сосудами как будто бы с одной и той же
поверхностью вызывают изменение скорости в несколько раз. Кажущиеся
энергии активации для различных поверхностей из стекла пирекс колебались
от 13,4 до 19,0 кшл/моль. Добавка небольших количеств воздуха, кислорода,
двуокиси углерода или водяного пара не изменяет скорости реакции.
Жигер [5J продолжил вышеописанную работу в аналогичном приборе,
причем очень тщательно и подробно исследовал влияние обработки поверх-
поверхности на скорость реакции при общих давлениях около 5 мм рт. ст.; пары
получались путем испарения 95—99%-ной перекиси водорода с известным
фракционированием. В каждом из рассмотренных ниже случаев реакция
изучена в однолитровом сферическом реакционном сосуде и уравнение для
константы скорости первого порядка выражено в виде функции парциального
давления перекиси водорода. В табл. 74 показаны различия в активности
некоторых поверхностей. С целью создания общей основы для сравнения
в каждом случае приводится приближенная температура, при которой кон-
константа скорости первого порядка (сек.) равна 103 (что найдено интерполя-
интерполяцией по данным Жигера), а также кажущаяся энергия активации, по Жигеру.
Мы видим, что оплавление поверхности стекла пирекс резко снижает
его активность, но обработка горячей хромовой кислотой эту инертность сни-
снимает. Возможно, что на поверхности стекла остаются очень небольшие коли-
количества каталитически активных окислов хрома, хотя известно, что обработка
хромовой кислотой существенно повышает также величину удельной поверх-
поверхности стекла, в связи с чем может также увеличиться скорость разложения
на единицу кажущейся поверхности (в одной из работ найдено, что обработка
хромовой кислотой способствует образованию более активной поверхности, чем
та, которую можно получить, применяя иные методы; активность сохраняется
даже после многократных промывок). Повторное оплавление вновь снижает
376
Глава 8
Таблица 74
Активность некоторых поверхностей в отношении влияния на разложение пара
перекиси водорода, по Жигеру [5]
№
п/п
Поверхность
Приблизительная
температура,
при которой
k (сек.-1)=103,
°С
Энергия
активации,
ккал\моль
1
2
3
4
5
6
7
8
9
10
11
12
13
14
Сосуд из стекла пирекс (III)
Сосуд III с оплавленной поверхностью
Сосуд III, очищенный горячей хромовой кисло-
кислотой
Сосуд III после повторного оплавления . . . .
Сосуд III, промытый горячей водой
Сосуд III, покрытый тринатрийфосфатом . . . .
Сосуд III, протравленный плавиковой кислотой
Сосуд из содово-известкового стекла (V) ....
Сосуд из содово-известкового стекла (VI) с по-
поверхностью, идентичной поверхности сосуда V
Сосуд из прозрачного кварца (витреосила) (VII)
Сосуд VII после оплавления
Сосуд из прозрачного кварца (амерсила) (VIII)
Сосуд из стекла пирекс с поверхностью, покры-
покрытой оловом (IX)
Сосуд из стекла пирекс с поверхностью, покры-
покрытой алюминием (X)
140
240
125
210
157
65
170
100
60
125
150
70
70
90
12,6
18,7
7,8
15,8
16,1
21,6
8,4
П,4
10,5
12,2
11,6
10,3
13
12
активность поверхности до прежнего низкого уровня, причем эти поперемен-
попеременные изменения активности можно повторить по меньшей мере несколько раз.
Промывка горячей водой или травление плавиковой кислотой повышает актив-
активность; покрытие поверхности стекла солью делает ее значительно более актив-
активной, чем любая другая обработка. Путем экстраполирования температуры
показано, что поверхность сосуда из стекла пирекс, покрытая солью, способ-
способствует протеканию реакции разложения со скоростью, приблизительно
в 10б раз большей, чем в сосуде из этого же стекла с оплавленной поверхностью.
Два одинаковых сосуда из содово-известкового стекла, подвергнутые одина-
одинаковой обработке, дают скорости реакции, которые отличаются одна от другой
в несколько раз. Сосуды из прозрачного кварца двух разных партий сильно
различаются по своей активности. Оплавленное стекло при старении до дости-
достижения им своего рода равновесного состояния становится более активным.
Активирующее действие промывки горячей водой или кислотой обычно при-
приписывается избирательному растворению более растворимых компонентов стек-
стекла, в результате чего остается губчатая структура. После покрытия сосудов
чистым оловом или алюминием, осажденным методом испарения до образования
зеркальной поверхности, получались скорости, сравнимые со скоростями для
сосудов из кварца и мягкого стекла. Однако Жигер указывает, что такие метал-
металлические поверхности могут обладать активностями, сильно отличающимися
от активностей других поверхностей, образованных каким-либо иным спосо-
способом на массивном металле. Как видно, наиболее инертная поверхность наблю-
наблюдается в случае оплавленного стекла пирекс. Более высокая активность по-
поверхности кварца по сравнению с активностью поверхности стекла пирекс
обусловлена, вероятно, более высокой пористостью кварцевой поверхности.
В двухлитровом сосуде из стекла пирекс с низкой активностью температур-
температурный коэффициент соответствует кажущейся энергии активации от 7,5 до
8,0 ккал/моль при любых температурах до 420°, т. е. максимальной из изучен-
изученных; это с очевидностью доказывает, что в этих условиях гомогенной реакции
не происходит.
Разложение в паровой фазе 377
Устойчивой взаимозависимости между кажущейся энергией активации
и скоростью реакции не отмечено. Жигер приписывает это отсутствие взаимо-
взаимозависимости изменению числа адсорбированных молекул, что связано с пред-
экспоненциальным множителем А в видоизмененном уравнении Аррениуса
k=Ae-(E-ViRT, где Е—истинная энергия активации, а X—энергия адсорбции
реагента. Жигер [5], Бэкер и Уэллет [6], Маккензи и Ритчи [8] и Тамрес
и Фрост [12] обнаружили в известных обстоятельствах адсорбцию пара при
введении его в реакционный сосуд, что доказано начальным падением давления
или наличием индукционного периода перед последующим ростом давления,
которого вследствие разложения и следовало ожидать. Другие различные
опыты подтвердили, что по меньшей мере часть адсорбированного пара пред-
представляет собой перекись водорода. В исследованиях, проведенных Жигером
с кварцем, это явление наблюдалось при температурах вплоть до 175°. Адсорб-
Адсорбция, вероятно, может происходить и при более высоких температурах, хотя
в приборе, применявшемся для исследования, этого нельзя было обнаружить.
Как и следовало ожидать, это явление выражено тем более резко, чем ближе
парциальное давление перекиси водорода к давлению пара, которое должно
наблюдаться над конденсированной фазой. Так, Тамрес и Фрост [12], про-
проводившие опыты при комнатной температуре и начальном давлении пара 0,5—
0,7 мм рт. ст. и работавшие с высококонцентрированным паром перекиси водо-
водорода, обнаружили, что количество адсорбированной перикиси водорода раза
в 3 выше, чем содержание ее в паровой фазе; эти опыты проведены в сосуде
с насадкой из мягкой стеклянной ваты. Величина адсорбции заметно изменяет-
изменяется также с характером поверхности. Так, Жигер отметил, что обнаружен-
обнаруженный им «индукционный период» в кварцевом сосуде отсутствует, если кварц
оплавлен. По-видимому, при оплавлении закрывается пористая структура
кварца, в результате чего сильно снижается величина поверхности, способной
к адсорбции; одновременно, возможно, изменяется и характер самой поверх-
поверхности.
В двух патентах Кука [13] сообщается, что покрытие поверхности реак-
реакционных сосудов борной кислотой или различными комбинациями этой кисло-
кислоты, боратов и некоторых окислов снижает скорость разложения, особенно
при температуре 300° или выше. Принимается, что этот эффект в основном
обусловлен наличием борной кислоты или окиси бора, а также образованием
гладкой поверхности при оплавлении.
Мак-Лейн [9] сообщает, что покрытие сосуда из стекла пирекс борной
кислотой с последующим оплавлением покрытия в печи при 500—520° в тече-
течение ночи вызывает снижение скорости разложения по сравнению с наблю-
наблюдаемой в сосуде из непокрытого стекла пирекс; этот автор указывает также,
что при очистке сосуда и повторном покрытии его по этому методу удава-
удавалось получать устойчивые результаты в различных сосудах (или в одном
и том же сосуде). Мак-Лейн исследовал разложение пара перекиси водорода
в таких сосудах при парциальных давлениях 1—2 мм рт. ст. в присутствии
кислорода или азота под давлением 1 am; опыты проводились в динамической
системе при температурах 470—540°. Применялось два рода сосудов: в одном
из них отношение поверхности к объему составляло около 7 см'1, в другом—
около 3 см'1. Скорость реакции подчинялась первому порядку, и константы
скорости в сосудах с более высоким отношением поверхности к объему, как
правило, были на 0—50% выше, чем в сосудах с меньшим отношением. Скорости
реакции приблизительно на 30% выше в азоте, чем в кислороде. Кажущиеся
энергии активации составляли 40 ккал/моль в сосудах с отношением поверх-
поверхности к объему 7 см как в присутствии азота, так и кислорода и 50 ккал/моль
в сосудах с отношением 3 см'1 в присутствии азота. При опытах в последних
сосудах с кислородом получено слишком мало данных, а поэтому вычислить
энергию активации не удалось. На основании влияния отношения поверхно-
378
Глава 8
500
100,01—
I I i
сти к объему и величин энергии активации сделан вывод, что, вероятно, частич-
частично реакция происходит гомогенно в газовой фазе.
Результаты небольшого исследования, проведенного Гаррисом [141,
согласуются с материалом, полученным другими авторами. Гаррис барботи-
ровал ток азота при давлении 1 am через примерно 65%-ный водный раствор
Т мпе m а °С перекиси водорода с темпе-
450 400 " збо1^0' зоо 250 ратурой 52°, а затем про-
пропускал газ череа нагретую
кварцевую реакционную
трубку. Доля перекиси,
которая претерпевала раз-
разложение, в разных опытах
сильно колебалась и возра-
возрастала при покрытии
трубки хлористым калием,
добавке ртути или запол-
заполнении трубки насадкой.
В чистом эксперименталь-
экспериментальном сосуде без насадки
время полураспада пере-
перекиси водорода составляло
0,5 сек. при 505—525°;
эта величина очень близка
к периодам полураспада
0,7 и 0,9 сек., найденным
Мак-Лейном для перекиси
водорода соответствен но
в присутствии азота или
кислорода при применении
сосудов, покрытых борной
кислотой, при температуре
52Г. Если учесть сильные
колебания, обнаруженные
для активности разных
поверхностей, то необы-
Рис. 61. Влияние температуры на разложение пара пере- чайно хорошее совпадение
киси водорода (парциальное давление Н2О2=0,02 am).
G трубка № 100, кО трубка № 103; Д трубка № 104.
ДЕ& 10 ккал/моль
«5
16 1,7 (*
Ю00/Т, (°КГ
и
скоростей реакции, опре-
определенных независимо друг
от друга Мак-Лейном и
Гаррисом при указанном уровне температур, приводит к заключению,
что в обоих случаях реакция в значительной части была гомогенной.
Работы, совсем недавно проведенные Штейном [10], дают ведьма четкое
доказательство перехода гетерогенной реакции в гомогенную при повышении
температуры. Этот автор пропускал смесь воды и перекиси водорода в паро-
паровой фазе под давлением 1 am через тщательно очищенную трубку из стекла
пирекс в динамическом приборе; парциальное давление перекиси водорода
составляло несколько миллиметров ртутного столба; скорость разложе-
разложения определяли путем анализа газов на входе и выходе в стационарных
условиях. Скорость реакции была первого порядка в гетерогенной области
и приблизительно второго порядка в гомогенной. На рис. 61 даны как
функция температуры скорости разложения, найденные для каждой из
трех трубок из стекла пирекс идентичных размеров, при парциальном
давлении перекиси водорода 0,02 am и постоянной скорости потока пара.
Отчетливо виден резкий переход от гетерогенной реакции к гомогенной при
температуре около 425°. Такое изменение механизма доказывается также
HaOt
-..#¦ y»v^vt- Увеличенная
/\/\/\/X частьРисУнка
Инерт- 25 SO 75 HZO
76% H2O2
0% инертного газа
257.Нг0
25%Н202 35
50% инертного газа
25% Н20
45 55
Н20, мол. %
25% Н20г
0% инертного газа
75% Н20
Рис. 62. Предел воспламенения смесей перекиси водорода, водя-
водяного пара и инертного газа при общем давлении 200 мм рт. ст.
о2
о
¦]
д
N2
6
Не
°т
V
Число взрывов на 5
пропускании
5
0
от 1 да
попыток при
искры
4
Увеличенная
2о часть рисунка
20%Н2О2
50% С0-г
30% И?0
40
50 60
НоО, мол. %
10
20%Н2О2
0%С02
80% Нг0
Р и с. 63. Предел воспламенения смесей перекиси водорода,
водяного пара и двуокиси углерода при общем давлении
200 мм рт. ст.
Число взрывов при пяти попытках: О 5, J 0, Л от 1 до 4.
380 Глава 8
и тем, что две разные реакционные трубки заметно различаются по активности
в области низких температур, тогда как в области высоких температур ско-
скорости разложения в обоих случаях совпадают.
Мицуватари и Нагаи [15] и Суито [16] провели дополнительные исследо-
исследования по разложению пара перекиси водорода, но результаты их работ труд-
трудно интерпретировать. Очень небольшое число работ, посвященных фото-
фотохимическому разложению паров, рассматривается на стр. 385.
Если нагревать концентрированный водный раствор перекиси водорода
до температуры, при которой образующиеся пары лежат внутри интервала
взрывчатых составов (см. стр. 154 и ел.), можно зажечь пары и получить пламя,
способное к непрерывному распространению. Так, при нагревании сосуда,
содержащего концентрированную перекись водорода, до температуры, близкой
к точке кипения, можно воспламенить пары действием искры накаленной
проволоки или каталитически активной поверхности; пары могут воспла-
воспламениться даже непосредственно от стенки сосуда. Если только пары воспла-
воспламенились, то теплота, переходящая сверху вниз от пламени, вызывает непре-
непрерывное испарение жидкости. В результате пламя «горит» весьма равномерно
без притока внешнего тепла, пока не истощится запас концентрированной
жидкой перекиси водорода. Пламя такого типа наблюдалось Хартом [17]
при общем давлении 20 мм рт. ст. или выше; оно наблюдалось также и в усло-
условиях атмосферного давления при исследованиях, проведенных в других лабо-
лабораториях. Это явление очень эффектное. Так, если поставить стакан с 90%-ной
перекисью водорода на электроплитку, то жидкость начинает пениться, а после
начала кипения выделяет туман и пар. Если происходит воспламенение паров,
то туман мгновенно исчезает, поверхность жидкости успокаивается, пено-
образование прекращается и объем жидкости очень быстро уменьшается.
На стр. 157 приведен предел воспламенения смесей паров перекиси водо-
водорода и воды при атмосферном и уменьшенном давлении. На рис. 62 и 63 пока-
показано влияние изменения природы и концентрации присутствующего инертного
газа на предел воспламенения при общем давлении 200 мм рт. ст. [18]. Замена
части водяного пара гелием, азотом или кислородом не изменяет предела
воспламенения; двуокись углерода оказывает известный тормозящий эффект.
Истолкование этих данных затруднительно, так как роль инертного газа может
быть обусловлена его теплоемкостью, отражающейся на температуре адиаба-
адиабатической реакции, теплопроводностью, влияющей на скорость отвода тепла
из реакционной зоны, действием его на скорость, с которой образовавшиеся
в реакции свободные радикалы могут уходить путем молекулярной диффу-
диффузии, или эффективностью этого газа в отношении переноса энергии при трой-
тройных соударениях. Вероятно, наиболее существенное значение имеет тепло-
теплоемкость. Адиабатическая температура реакции предельного воспламеняю-
воспламеняющегося состава для системы перекись водорода—вода составляет, например,
780° при общем давлении 1 am и 880° при 200 мм рт. ст.; эти значения
гораздо ниже встречающихся в большинстве систем из топлива и окислителя.
Механизм
Во многих случаях оказывалось, что относительные активности различ-
различных поверхностей с точки зрения влияния на разложение в паровой фазе
примерно такие же, как и наблюдаемые в жидкой фазе. Так, для обеих фаз
стекло пирекс более инертно, чем натриевое, а кислые поверхности более
инертны, чем основные. При той и другой фазе обработка стекла пирекс хро-
хромовой или плавиковой кислотой повышает поверхностную активность. Ройтер
и Гаукман [19] пришли к тому же заключению для каталитически активных
поверхностей. Они сообщают, что скорость разложения возрастает в следую-
следующем порядке: двуокись свинца, двуокись марганца, платина; измеренные
Разложение в паровой фазе 381
ими относительные активности этих трех поверхностей примерно одинаковы
в жидкой и паровой фазах. Эти различные наблюдения позволяют заключить,
что механизм гетерогенного разложения в обеих фазах при некоторых экспе-
экспериментальных условиях один и тот же. Размеры адсорбции, которая, по лите-
литературным данным, происходит при разложении в паровой фазе, в ряде слу-
случаев соответствуют адсорбированному слою толщиной в несколько молекул.
Такой слой во многих отношениях ведет себя подобно жидкости. Однако эту
аналогию можно принять лишь с оговорками. Количество адсорбированной
перекиси водорода должно снижаться с повышением температуры; вероятно,
при этом происходит и изменение механизма. Кроме того, разложение в жид-
жидкой фазе часто связано с растворением материала сосуда в массе раствора,
что может способствовать искажению результатов наблюдений из-за неожидан-
неожиданной реакции.
Механизм гомогенного разложения в газовой фазе может быть выведен
на основании энергетических расчетов. Начальная ступень, разрыв молекулы
перекиси водорода, может происходить только по одному из двух путей: путем
разрыва связи О—О с образованием двух ОН-радикалов или путем разрыва
связи О—Н с образованием радикала НО2 и Н-атома. В первом случае тре-
I буется 52 ккал/моль вместо 90 ккал/моль для расщепления связи О—Н, так
что первый путь значительно более вероятен. Правильность предположения,
что разрыв связи О—О действительно является инициирующей стадией,
подкрепляется данными Юрея, Доуси и Раиса [20], которые изучали спектр
испускания перекиси водорода при быстром пропускании ее через холодную
разрядную трубку. Они обнаружили полосы воды, обусловленные ОН-ради-
калами, и отсутствие молекулярного или атомарного водорода.
Изменения энергии для всех возможных ступеней цепной реакции с уча-
участием частиц, которые могут присутствовать в системе, нетрудно рассчитать из
значений, приведенных на стр. 218, где указаны также литературные источники.
Все возможные механизмы разветвления сопровождаются неблагоприятными,
т. е. эндотермическими изменениями энергии, и поэтому гомогенная реакция,
вероятно, распространяется через газ при помощи неразветвленных цепей.
Единственный механизм прямых цепей с экзотермическими реакциями, ини-
инициированными активированными ОН-радикалами, возникшими в начальной
стадии, следующий:
ОН + Н2О2 -» НО2 + Н2О, B)
НО2 + Н2О2 -> ОН + Н2О + О2. C)
Эти реакции, в которых участвуют свободные радикалы, вероятно, не тре-
требуют энергии активации, или же она очень мала.
Активные центры уничтожаются одной или несколькими из следующих
реакций:
+ М->Н2О2 + М, D)
2НО2 + М -» Н2О2 + О2 + М, E)
F)
В качестве третьего тела функционируют стенка, инертный газ или молекула
перекиси водорода. Теплоты, генерируемой общей реакцией в случае газофаз-
газофазного пламени и взрывов, вероятно, достаточно для выделения новых ОН-ра-
ОН-радикалов из молекул Н2О2 и, следовательно, для распространения реакции,
которая, в сущности, по своей природе является термической.
Приведенный механизм согласуется с наблюдениями пределов воспламене-
воспламенения пара перекиси водорода, рассмотренных выше в этой главе и на стр. 154 и ел.
382 Глава 8
Непрерывный рост парциального давления перекиси водорода близ предела
воспламенения с ростом парциального давления присутствующих инертных
газов также свидетельствует о том, что взрыв носит фактически термический
характер, а не является взрывом с разветвлением цепей.
Если пропускать пар перекиси водорода над активной поверхностью
катализатора, то возможная скорость разложения на поверхности может
оказаться значительно выше скорости поступления перекиси водорода к этой
поверхности. Таким образом, создается положение, когда фактическая ско-
скорость разложения лимитируется скоростью диффузии перекиси водорода
к поверхности. В этом случае дальнейшее повышение активности катализатора
не изменяет измеренную скорость реакции. Исследованы скорости разложе-
разложения пара перекиси водорода при пропускании его через цилиндрическую
трубку, стенка которой представляла собою весьма активный катализатор,
а также через слой каталитически действующих шариков [21]. Показано,
что скорость разложения определяется скоростью диффузии, причем фак-
фактически измеренные скорости разложения точно согласуются с рассчитанными
по уравнениям для массообмена, выведенным для нереагирующих систем.
В такой реакции, скорость которой зависит от диффузии, поверхность твер-
твердой фазы нагревается до значительно более высокой температуры, чем темпе- ч
ратура основной массы газа, проходящего над поверхностью, и эта темпера-
температура может быть подсчитана на основании изучения характеристик одновре-
одновременного теплообмена и массообмена между основным потоком газа и твердой
поверхностью.
РАЗЛОЖЕНИЕ ПОД ДЕЙСТВИЕМ ИЗЛУЧЕНИЙ
Реакции, вызываемые излучениями, обычно подразделяют на две группы:
фотохимические процессы, как правило обусловленные ультрафиолетовыми
лучами, и радиохимические процессы, возникающие под действием поглоще-
поглощения излучений с высокой энергией, например рентгеновских или у-лучеи,
а также излучений, состоящих из частиц, например а-и р-изл учений. Послед-
Последняя группа процессов характеризуется возникновением ионизации. Вопрос
об образовании перекиси водорода в процессах излучения обсуждался на
стр. 54 и ел.
Фотохимические процессы
В старой литературе имеется большое число отрывочных и качественных
данных по разложению перекиси водорода под действием света и по влиянию
различных растворенных веществ на разложение. Систематические исследо-
исследования были начаты лишь примерно в 1910 г., но обобщение данных разных
исследователей представляется затруднительным в связи с тем, что авторы
прежних работ не указывали значения некоторых переменных, которые, как
оказалось в дальнейшем, имеют существенное значение для определения
квантовых выходов. Из этих переменных наиболее важными являются интен-
интенсивность и частота излучений, рН, природа чужеродных веществ, например
продажных ингибиторов, содержащихся в растворах, природа стенок сосуда
и поправки на протекающую одновременно темновую реакцию. Особенно
большие трудности вызывает необычайная чувствительность скорости разло-
разложения к следам примеси; воспроизводимые результаты как для термического,
так и для фотохимического разложения можно получить лишь при самой
скрупулезной технике эксперимента.
Недавно проведенный описываемый ниже опыт [22] иллюстрирует ско-
скорость фотохимического разложения, которая может наблюдаться на практике.
10 мл 90%-ной водной перекиси водорода, не содержащей ингибиторов, под-
подвергли облучению при 25° в кварцевом сосуде светом стоваттной ртутной
лампы, которая, по имеющимся данным, обладала мощностью излучения
Разложение под действием излучений 383
2,2 вт в близком ультрафиолете, 3200—3800 А. Скорость разложения перекиси
водорода в этом случае составляет 0,25°о в час, или в 70 раз больше скорости
разложения в отсутствие облучения при прочих равных условиях.
Вопрос о поглощении ультрафиолетового излучения перекисью водорода
рассмотрен на стр. 244. Ультрафиолетовый спектр является сплошным бес-
бесструктурным спектром и охватывает область приблизительно от 3800 до 2000 Л
и ниже. Коэффициент погашения практически не зависит от концентрации
перекиси водорода при содержании ее ниже 50 вес.%, хотя точно закон Бера
и не соблюдается. Аналогично коэффициенты погашения для водных раство-
растворов и смесей пара перекиси водорода с воздухом практически одни и те же,
если сравнивать их при одинаковых произведениях концентрации на длину
пути света.
Экспериментальные результаты. В настоящее время считается обще-
общепризнанным, что в растворах, имеющих концентрацию от низкой до средней
(приблизительно до 0,2 УИ), при умеренной интенсивности поглощенного
излучения (ниже примерно 1017 квант/л-сек) скорость разложения в молях
н& Литр в секунду прямо пропорциональна концентрации перекиси водорода
и квадратному корню из интенсивности [23—26]. Для более высоких концен-
концентраций и умеренных интенсивностей облучения данные разных авторов значи-
значительно расходятся. Так, по Куреши и Раману [27], квантовый выход стано-
становится независимым от концентрации для концентраций от 0,3 до 0,9 М\ в работе
Олменда и Стайла [23] обнаруженное влияние концентрации колебалось
в зависимости от того, как снижали концентрацию: путем ли добавки дистил-
дистиллированной воды или путем выдерживания раствора под действием излуче-
излучения до частичного его разложения. Однако в недавно проведенной, весьма
тщательно выполненной работе Дейнтон и Роуботтом [26] нашли, что при
интенсивностях приблизительно до 1017 квант/л-сек и концентрациях до 20 М
скорость все еще прямо пропорциональна концентрации перекиси водорода
и квадратному корню из интенсивности излучения; расхождения, отмечен-
отмеченные в старых работах, можно с полным правом приписать наличию следов
ингибиторов или примесей в перекиси водорода.
По некоторым старым сообщениям, выход становится независимым от ин-
интенсивности излучения при очень низких интенсивностях и концентрациях
ниже 0,1—0,5 М [23, 24, 28]. При очень высоких интенсивностях излучения
(приблизительно выше 3-1017 кеант/л-сек) Ли [25] обнаружил, что квантовый
выход делается независимым от концентрации, начиная от 0,01 и кончая
по меньшей мере 0,04 М. Хейдт [291, применявший излучения с интенсивно-
интенсивностью от 5-1017 до 12-1017 квант/л-сек, пришел к выводу, что квантовый выход
имеет тенденцию расти с повышением концентрации от 1,8 до 4,2 М. Однако
этот рост едва значимый, если учесть общую воспроизводимость данных, и, во
всяком случае, гораздо меньший, чем линейный.
При экспозиции на свету с длиной волны выше 3800 А перекись водорода
не изменяется. В инфракрасной области спектра перекись обладает полосами
поглощения, но светом с такой частотой не разлагается. Данные по влиянию
длины волны ультрафиолетового света на разложение не позволяют сделать
окончательных выводов. Генри и Вурмсер [281 сообщают, что с повышением
длины волны от 2080 до 2800 А при концентрациях 0,02—0,05 М и интенсив-
интенсивностях излучения около 1011 квант/л сек квантовый выход падает на 25%;
Олменд и Стайл [231 сообщают о росте квантового выхода на 100% с увеличе-
увеличением длины волны от 2750 до 3650 А при концентрациях 0,5—11,5 М и интен-
интенсивностях в той же области, что указана выше.
В табл. 75 приведены температурные коэффициенты для скорости раз-
разложения при излучениях с различной длиной волны. По-видимому, с умень-
уменьшением длины волны наблюдается тенденция к снижению температурного
коэффициента. Эти величины можно сравнить с экспериментально измерен-
384
Глава 8
Таблица 75
Влияние длины волны на температурный коэффициент
скорости фотохимического разложения
Длина волны, А
3650
3650
Полный спектр
ртутной лампы
То же
3130
3070
2750
2600
Температурный
коэффициент, аа
1,42
1,50
1,43
1,38 и 1,41
1,32
1,38
1,28
1,15
Интервал темпе-
температур, °С
2—22
21—45
—
10—30
2—22
2—22
2—22
Литература
1
226
2
39
19
1
1
1
а G2-Ti) Iff a=\0 gB/i)
6 Без поправки на термическое разложение.
ным коэффициентом, несколько превышающим значение 2, при термическом
разложении в отсутствие излучений (см. стр. 437).
Сообщается о влиянии различных неорганических кислот, оснований
и солей на скорость фотохимического разложения [28, 30, 31]. Интервалы
интенсивности излучения не указаны, но почти наверняка можно считать,
что они были ниже 10" квант/л-сек. При этих интенсивностях добавка силь-
сильных неорганических кислот к раствору вызывает заметное торможение, кото-
которое, по-видимому, стремится к пределу при увеличении концентрации кис-
кислоты. Поскольку коэффициент погашения в присутствии кислот заметно
не изменяется [28, 30], этот эффект является истинным торможением и не может
быть объяснен исключительно экранированием от радиации. Корнфельд [24],
приводя данные по этому эффекту для серной кислоты, приходит на основа-
основании теоретических рассуждений к выводу, что скорость в сильной кислоте
при умеренных интенсивностях излучения должна быть близка к 33% скоро-
скорости в нейтральном растворе. По Ли [25], при очень высоких интенсивностях
(примерно выше 3-1017 квант/л -сек) и 0,02—0,05 М концентрациях перекиси
водорода квантовый выход становится независимым от рН в интервале 1—6.
Справедливость этого положения для высоких интенсивностей подтверждается
и данными Хейдта [29J, который обнаружил лишь небольшое влияние сер-
серной кислоты при концентрациях ее до 4,Ш в том случае, когда интенсив-
интенсивность составляла около 5-1017 квант/л^ сек, а концентрация перекиси водо-
водорода—около 4М.
Сильные основания являются значительно более эффективными ингиби-
ингибиторами, чем кислоты. Их присутствие сказывается также в значительно^
увеличении коэффициента погашения [28, 29]. Интересно отметить, что, когда
излучение отсутствует, основания оказывают противоположное влияние, силь-
сильно ускоряя разложение (см. стр. 438 и ел.). Следовательно, измерение раз-
разложения под действием излучений в присутствии оснований затрудняется
в связи с необходимостью введения большой поправки на темновую реакцию.
Щелочные и щелочноземельные соли сильных кислот (за исключением
галогенидов) не оказывают на разложение никакого влияния. Относительно
действия галогенидов имеются противоречивые данные. По Мэтьюсу и Кур-
тису [311, они оказывают лишь слабое тормозящее действие; наоборот, Андер-
Андерсон и Тейлор [30] сообщают о существенном ингибирующем действии, менее
заметном в присутствии солей, имеющих общий катион с галоидной солью.
Разложение под действием излучений 385
Хант и Таубе [32] сообщают, что бромид и хлорид не оказывают влияния
на разложение.
Андерсон и Тейлор [30] приводят данные относительно влияния боль-
большого числа органических соединений о на фотохимическое разложение при
длинах волн 2000, 2650, 2930 и 3050 А. Они сравнили тормозящее влияние
при различных длинах волн с коэффициентом поглощения для каждого соеди-
соединения при каждой длине волны и обнаружили параллелизм для кислот, слож-
сложных эфиров, амидов, кетонов, бензола и алкалоидов: высокое поглощение
соответствует высокой активности торможения. Тормозящее действие спиртов
больше, чем этого можно ожидать на основании их поглощения. Для аминов
тако% зависимости не обнаружено, и возможно, что они действуют по иному
механизму, связанному с их основной природой. Все эти соединения менее
эффективны при применении их в качестве экранирующего светофильтра,
чем при непосредственной добавке к перекиси водорода. На этом основа-
основании Андерсон и Тейлор пришли к заключению, что такие органические инги-
ингибиторы не только действуют как поглотители излучения, но влияют и на меха-
механизм разложения. Мэтьюс и Куртис [31] также исследовали различные орга-
органические добавки при применении смешанного света с длиной волны около
2500 А.
Колеблющиеся результаты, часто отмечаемые разными исследователями,
#и плохое количественное совпадение результатов исследования, взятых
из разных источников, доказывают, что квантовый выход сильно зависит
от возможных случайных примесей. Выше уже было указано на осо-
особую чувствительность к рН. По Генри и Вурмсеру [28], уже 2 мг/л едкого
натра снижают скорость до 60% нормальной ее величины. Таким образом,
может иметь значение влияние щелочи, извлеченной из стеклянной посуды,
следов аммиака в дистиллированной воде или атмосферной двуокиси углерода.
Тиан [33] сообщает, что скорости заметно зависят от чистоты дистиллирован-
дистиллированной воды, взятой для разбавления (эта чистота определялась кондуктометри-
ческими измерениями). По Корнфельду [24], можно добиться согласованных
данных о влиянии определенных переменных при проведении опытов с про-
пробами из одной и той же партии продажной перекиси водорода известной марки,
но для различных партий ее количественные выходы могут заметно колебаться.
Райе и Килпатрик [34] сообщают, что скорость фотохимического разложения
прямо пропорциональна концентрации частиц пыли, измеренной методом
светорассеяния. Очевидно, что, как и при исследовании стабильности пере-
перекиси водорода, необходима особая тщательность в очистке и работе, иначе
невозможно получить согласованные результаты. Дейнтон и Роуботтом [26]
описывают применявшиеся ими меры предосторожности, которые требовали
большой затрать! труда.
По фотохимическому разложению пара перекиси водорода имеется очень
мало работ. Проведены некоторые отрывочные исследования [12, 35]; наибо-
наиболее поздняя и полная работа принадлежит Волмэну [361, который облучал
пар перекиси водорода с начальным парциальным давлением 1,23 мм рт. ст.
(в присутствии водяного пара давлением 0,39 мм) светом с длиной волны 2537 А.
Квантовый выход равен 1,7±0,4 и не зависит от давления перекиси водорода
и от температуры в интервале 25—50°. Продукты разложения не содержат
водорода, добавка кислорода или азота не влияет на скорость разложения.
Механизм. По литературным данным, квантовые выходы колеблются
приблизительно от 1 до нескольких сот. В соответствии с опытом при малой
интенсивности излучения и высоких концентрациях перекиси водорода можно
ожидать образования длинных цепей. При повышении интенсивности излу-
излучения длина цепи снижается, причем ряд экспериментаторов сообщает о «пре-
«предельном квантовом выходе» при максимальной интенсивности, где цепные
реакции, вероятно, сведены к минимуму или даже совсем не протекают.
25 Перекись водорода
386
Глава 8
Таблица 76
Предельный квантовый выход для фотохимического
Длина волны,
А
2537
3130
2537
2537
2537
Концентрация
Н2О2, М
ю-2
1,7—4,5
0,02—0,2
ю-<
0,1
разложения
Предельный кванто-
квантовый выход
1,39±0,11
1,7±0,4
0,98±0,05
(при 25°)
0,76±0,05
(при 0°)
1.7±0,4
1,9±0,1
Литература
Ли [25|
Хейдт |29|
Хант и Таубе [32]
Волмэн [36|
Дейнтон и Роубот-
том [26]
Влияние случайных примесей в этих условиях также доходит до минимума.
В табл. 76 приведены минимальные квантовые выходы, обнаруженные раз-
разными авторами.
В новейших исследованиях принимается, что начальная фотохимическая
стадия разложения может быть представлена следующим уравнением:
В доказательство Юрей, Доуси и Райе [20] приводят данные, показывающие,
что спектр испускания пара перекиси водорода при быстром пропускании
через холодную разрядную трубку состоит преимущественно из полос воды,
которые, согласно известным независимым данным, обусловлены гидроксиль-
ными радикалами. Эти авторы указывают также, что при облучении перекиси
водорода светом с длиной волны 2025—2138 А спектр флуоресценции содер-
содержит полосы воды.
Ли [25] в подтверждение этой начальной реакции указывает, что пере-
перекись водорода является фотосенсибилизатором для полимеризации винило-
виниловых соединений аналогично тому, как реакция переноса электрона
Fe2+ + НаО2 -> Fe3+ + ОН + ОН",
(8)
также являющаяся источником гидроксильных радикалов, может играть
термосенсибилизующую роль. Фон Эльбе [37] сообщает, что если в началь-
начальной реакции в качестве продуктов образуются Н2О+О, то дальнейшей реак-
реакции с водородом или с окисью углерода не должно происходить, так как атомы
кислорода значительно быстрее реагируют с перекисью водорода, чем с водо-
водородом или окисью углерода. На основании того, что при облучении в дейст-
действительности протекает реакция между перекисью водорода и каждым из ука-
указанных двух газов, этот автор делает вывод, что начальная реакция не при-
приводит к образованию одноатомного кислорода, а, вероятно, идет по уравнению
G). Единственное доказательство против этого предположения дают новей-
новейшие работы Ханта и Таубе [32], которые осуществляли фотохимическо^раз-
ложение в условиях довольно значительной интенсивности поглощения с при-
применением перекиси водорода вместе с водой, обогащенной О18. Весь образо-
образовавшийся кислород оказался производным перекиси водорода. Отмеченные
эффекты фракционирования оказались несовместимыми с предположением,
что единственными продуктами начальной диссоциации являются радика-
радикалы ОН. Поэтому эти авторы выражают первичную стадию уравнением
НаОа
Ь -> Н2О + О,
(9)
Разложение под действием излучений 387
за которой следует реакция
O-t-H2O2-»OH + HO2. A0)
Они считают также возможным, что сначала происходит реакция G), но затем
в «клетке» растворителя гидроксильные радикалы дают воду и атомарный
кислород по уравнению
2ОН-^Н2О + О. A1)
Габер и Вильштетер [38] постулировали развитие цепей по следующим реак-
реакциям:
Н2О2-»Н2О+НО2, A2)
A3)
протекающим после образования гидроксильных радикалов по уравнению G).
На основании схем, включающих эти реакции, Вейс [39] установил кинети-
кинетические закономерности системы перекись водорода—озон, и ряд авторов
рассчитал таким же образом кинетику для системы перекись водорода—соли
железа. По мнению Ли [25], квантовые выходы при высоких интенсивностях
излучения согласуются с предположением, что именно реакции A2) и A3)
обусловливают развитие цепей. Корнфельд [24] рассчитал кинетику фото-
фотохимического разложения перекиси водорода в нейтральных и кислых раство-
растворах на основании предположения, что реакция A3) протекает через' посред-
посредство иона О2, возникающего в результате диссоциации, т. е. по уравнениям
A4)
О + Н2О2-»О2
Обычно постулируемыми реакциями обрыва цепей являются следующие
(одна из них или обе):
2НО2-»Н2О2 + О2, A6)
A7)
Корнфельд [24], исходя из предположения, что общая реакция протекает
по уравнениям G), A2), A4), A5) и A7), вывел кинетические уравнения,
из которых следует, что внутри широкого диапазона (где цепи обладают до-
достаточной длиной) квантовый выход должен быть пропорционален концентрации
перекиси водорода в первой степени и обратно пропорционален квадратному
корню интенсивности излучения и первой степени концентрации ионов водо-
водорода. Эти выводы подтверждаются основной массой данных о разложении
в нейтральной и кислой средах. Однако, по схеме Корнфельда, при добавке
основания квантовый выход должен возрастать, тогда как в действительности,
по сообщениям многих авторов, реакция в этом случае сильно тормозится.
Очевидно, что в основной среде могут приобрести значение другие процессы
поглощения, связанные с высоким коэффициентом поглощения растворов
перекисей щелочных металлов.
Кинетические уравнения, выведенные Корнфельдом, дают возможность
вычислить кажущуюся энергию активации фотохимической реакции (измерен-
(измеренную по изменению квантового выхода с температурой) из энергии активации
отдельных реакций. Кондратьев [40] вычислил энергию активации отдель-
отдельных реакций теоретическим путем и рассчитал, что кажущаяся энергия акти-
активации должна составлять 5,5 ккал. Это значение соответствует температур-
температурному коэффициенту около 1,4, который лежит внутри интервала данных,
имеющихся в литературных источниках (см. табл. 74 на стр. 376). Однако
'25*
388 Глава 8
Кондратьев не сообщает ни подробных расчетов, ни величин энергии акти-
активации для отдельных реакций.
Ли [25] указывает, что в соответствии с приведенной схемой реакции
при достаточно высоких интенсивностях излучения и низких концентрациях
перекиси водорода фотолиз дол жен. утратить характеристику цепной реак-
реакции. В этих условиях из кинетических уравнений следует, что квантовый
выход не должен зависеть от интенсивности и концентрации перекиси
водорода. Действительно, Ли показал, что при интенсивностях выше
3-Ю17 квант/л-сек квантовый выход независимо от интенсивности излучения
составляет 1,39 (при концентрации перекиси от 0,010 до 0,034 М и рН в пре-
пределах от 1 до 6).
Отсутствие зависимости квантового выхода от рН при указанных высоких
интенсивностях считается доказательством, что в этом случае реакция A3)
протекает так, как она написана, а не по ионному механизму [уравнения A4)
и A5)], предложенному Корнфельдом. Габер и Вейс [41] также указывают,
что ингибиторное действие кислоты при малых интенсивностях излучения
может быть обусловлено ускорением реакции обрыва водородными ионами,
а не ионным механизмом Корнфельда, представленным уравнениями A4) и A5).
Дальнейшие подробности, касающиеся механизмов разложения и сравнения
механизмов фотолиза и радиолиза растворов перекиси водорода, можно найти
в недавно опубликованных сообщениях Дейнтона и Роуботтома [26] и Вей-
са [42],
Разложение под действием ионизирующих излучений
Явления, наблюдаемые при бомбардировке перекиси водорода ионизи-
ионизирующими излучениями, труднее истолковать, чем те, которые происходят
при ультрафиолетовом облучении. В этом случае наряду с частицами, харак-
характерными для фотолиза, возникают также ионизированные частицы; кроме
того, свободные радикалы, образующиеся по линии движения быстрых частиц,
могут распространяться в растворе неравномерно. В связи с этим приобре-
приобретают большое значение скорости диффузии и другие осложняющие обстоятель-
обстоятельства. Большая часть исследований радиохимического разложения прово-
проводилась с применением рентгеновских или у-лучей. В разбавленных водных
растворах, с которыми проведена основная часть работ, первичной стадией
является разложение воды на Н и ОН, причем некоторые из этих частиц в свою
очередь тут же непосредственно образуют Н2 и Н2О2 (см. гл. 2). Если усло-
условия радиолиза таковы, что передатчики цепи распределены сравнительно
равномерно, как это наблюдается, например, при высоких дозах излучения
и сранительно долговечных радикалах, то реакции, следующие за первич-
первичной стадией, должны быть аналогичны наблюдаемым при фотохимическом
разложении. Единственными добавочными реакциями активных центров
цепи являются превращения атомов водорода:
Н + Н2О2—>ОН + Н2О, A8)
Н + О2-^НО2. A9)
При равномерном распределении радикалов математическая формула
для зависимости скорости радиолиза от концентрации перекиси водорода
и дозы излучения такая же, как и при фотолизе, т. е. в условиях, когда обра-
образуются длинные цепи, число молей перекиси водорода, разложенных в еди-
единицу времени, должно быть пропорционально концентрации перекиси водо-
водорода и квадратному корню из интенсивности излучения (общего количества
поглощенного излучения в единицу времени на единицу объема). Эта зави-
зависимость от квадратного корня из интенсивности излучения экспериментально
подтверждена рядом авторов [26, 43—45], которые применяли рентгенов-
Электролитическое разложение 389
ские лучи и у-лучи Со60. При высоких концентрациях скорость реакции также
приблизительно пропорциональна концентрации перекиси водорода [46];
однако при более низких концентрациях она пропорциональна этой концен-
концентрации лишь в степени 0,5 [44,45] или даже совершенно не зависит от кон-
концентрации [47]. Тем не менее все эти результаты согласуются с предположе-
предположением, что при достаточном снижении концентрации реакционные цепи делают-
делаются более короткими или даже совсем исчезают. Математические зависимо-
зависимости приобретают более сложный характер в том случае, когда реакция про-
протекает с участием коротких цепей. Однако зависимость от концентрации
в степени 0,5 была в последнее время обнаружена Хартом и Матесоном [431
и для столь высоких концентраций, как одномолярная. Для объяснения этих
данных сделано предположение, что происходит цепная реакция, обрываемая
реакцией третьего порядка типа
2НО2 + НА -> 2Н А + О2. B0)
Опубликованы также результаты опытов [45, 47—50] по влиянию раз-
различных растворенных веществ, добавленных к растворам перекиси водорода,
при бомбардировке рентгеновскими или у-лучами; в частности, рассматри-
рассматривается роль водорода, так как последний является одним из первичных про-
продуктов, образующихся из продуктов радиолиза воды. Кайлан [48] исследо-
исследовал процесс разложения водных растворов перекиси водорода при бомбар-
бомбардировке р- и у-лучами радия, а Хопвуд [51]—при действии у-лучей и ней-
нейтронов. Недавно проведенные исследования Эберта и Бога [52] по образова-
образованию и разложению перекиси водорода показали, что имеются заметные раз-
различия между результатами опытов, полученными, с одной стороны, при дей-
действии электронов с энергией 1 мгэв и рентгеновских лучей с энергией 1,2 мгэв
и, с другой стороны, при действии рентгеновских лучей с меньшим содержа-
содержанием энергии B00 кэв). Эта область исследований находится сейчас в стадии
бурного развития; можно ожидать, что в ближайшем будущем удастся раз-
разработать значительно более четкую картину процессов, происходящих при
радиационной бомбардировке. Дейнтон и Роуботтом [26] и Вейс [42, 531
опубликовали обзоры, иллюстрирующие состояние наших знаний в этой
области (см. также обсуждение вопроса образования перекиси водорода под
действием излучений на стр. 60 и ел.).
ЭЛЕКТРОЛИТИЧЕСКОЕ* РАЗЛОЖЕНИЕ
Водные растворы перекиси водорода можно подвергать электролизу
с получением молекулярного водорода и кислорода; при высоких плотно-
плотностях тока общий процесс выражается уравнением
1 НА->Н2 + О2. B1)
При снижении плотности тока выход водорода все более падает по сравнению
с указываемым этой реакцией и под конец процесса становится равным нулю.
Однако во всех случаях количество кислорода точно соответствует 1 молю
на 2 фарадея электричества. Эти факты установлены рядом исследователей E4,
55], начиная с Тенара. Причины изменения относительного образования
водорода на катоде не исследованы. Вейс [56] отметил, что в зависимости
от плотности тока и концентрации перекиси водорода на катоде выделяется
водород, смесь его с кислородом или даже один кислород. Альбареда [571,
как и авторы, работавшие раньше, пришел к выводу, что это необходимо
объяснить протеканием конкурирующего процесса каталитического раз-
разложения перекиси. Танатар [55] предполагает, что между водородом и пере-
перекисью водорода происходит реакция с образованием двух молей воды. Пере-
Перекись водорода влияет [581 на перенапряжение водорода.
390 Глава 8
Лучше изучена реакция на аноде, где происходит выделение кислорода
159—61]. Хиклинг и Вилсон 159) построили кривые сила тока—напряжение
для растворов перекиси водорода, находившихся в контакте с различными
электродными материалами. В 0,01 М растворе перекиси водорода ток начи-
начинает проходить через раствор приблизительно при напряжении 0,1 в. Тогда
на кривой сила тока—напряжение наблюдается горизонтальная часть, высо-
высота которой зависит от концентрации и интенсивности перемешивания. Эш
авторы пришли к выводу, что в щелочном растворе анодный процесс выра-
выражается уравнением
Н(Х = О2 + Н+ + 2г. B2)
В кислом или нейтральном растворе для любых электродов, за исключением
платины, основной процесс, по-видимому, возникает при том потенциале,
когда начинается электролиз фонового электролита. Поэтому можно сделать
вывод, что разложение перекиси водорода в этих условиях зависит от взаимо-
взаимодействия с каким-то промежуточным продуктом, с которым связано обычное
выделение кислорода на аноде (весьма вероятно, с радикалом гидроксила).
Дальнейшие исследования и объяснения механизмов анодного разложения
перекиси водорода принадлежат Риусу и Окон Гарсиа [60J, а также Красиль-
щикову, Волчковой и Антоновой [61]. Последние авторы вывели уравнение,
связывающее перенапряжение с рН и концентрацией перекиси водорода.
Эти рассуждения имеют значение также для полярографии [621 неза-
независимо от того, образуется ли перекись водорода путем восстановления кисло-
кислорода или она присутствует с самого начала [63]. Особого интереса заслужи-
заслуживает разложение перекиси водорода, вызываемое кислородом [64] и протекаю-
протекающее, по-видимому, под действием иона надперекиси. Полярографическое
восстановление перекиси водорода катализируется также ионами некоторых
металлов [65] (см. также стр. 84).
РАЗЛОЖЕНИЕ В ЖИДКОЙ ФАЗЕ ПОД ДЕЙСТВИЕМ НЕОРГАНИЧЕСКИХ
КАТАЛИЗАТОРОВ
Хотя имеется огромное количество литературы по каталитическому раз-
разложению жидкой перекиси водорода, большая часть ее имеет лишь описатель-
описательное значение. Это обусловлено отчасти трудностями самой темы и медлен-
медленностью развития знаний в области катализа вообще. Однако еще в большей
степени успешное изучение разложения перекиси водорода тормозилось недо-
недостаточно квалифицированной постановкой экспериментов и малой надеж-
надежностью результатов. Во многих случаях можно только сказать, что дейст-
действительно происходит катализ, однако в опыте не получено никаких данных
о влиянии концентрации, рН, температуры, наличия примесей или добавок
и состояния катализатора (или же эти данные имеют сомнительную ценность).
Между тем именно такие данные требуются для любого кинетического иссле-
исследования; поэтому для трактовки механизмов реакции можно использовать
только те системы, в которых было обращено тщательнейшее внимание на все
указанные факторы. Особенно большое значение имеет в данном случае влия-
влияние следов чужеродных веществ в перекиси водорода,например случайных
примесей, примесей, обусловленных производственным процессом, или пред-
преднамеренно добавленных стабилизаторов. Много работ проведено с перекисью
водорода сомнительной чистоты, особенно в процессе начальных исследований,
описанных в литературе.
Вероятно, наибольшее внимание привлекло каталитическое разложение
под действием галогенов, платины, железа и ферментов. Некоторые другие
элементы исследованы лишь слабо или вообще не изучены. На рис. 64 схема-
схематически изображены известные нам данные в отношении каталитического
Разложение в жидкой фазе
391
разложения перекиси водорода*. Каталитические свойства, несомненно, при-
присущи многим веществам, в том числе тяжелым металлам и металлам переход-
переходной группы, обладающим высокой каталитической активностью и во многих
других системах. При интерпретации рис. 64 нужно соблюдать известную
осторожность. Данные, на которых основана эта схема, в некоторых слу-
случаях взяты из литературных сообщений сомнительной ценности, а, кроме
Рис. 64. Периодическая таблица, показывающая элементы
или их соединения, способные и не способные катализировать
разложение перекиси водорода (по литературным данным).
. О катализаторы; 0 с сомнительной каталитической активностью; П не
\ обладающие каталитической активностью; Д не исследованные; Р. 3.—
редкоземельные элементы.
того, небольшие размеры рисунка не дают возможности поместить на нем
все, что характеризовало бы их достоверность (как это сделано ниже при
рассмотрении отдельных элементов). Отдельные обозначения на рис. 64 могут
быть неравноценны из-за неодинаковой надежности основных источников,
изменений формы катализатора (гомогенный или гетерогенный, элемент или
соединение), различных значений рН и особенно из-за различий и колебаний
в скорости катализа. Интервал скоростей разложения очень велик. Например,
в одних и тех же условиях металлическая сурьма почти инертна, тогда как
осмий и свинец могут вызвать разложение со скоростями, которые можно
считать взрывоподобными. Наблюдаются градации интенсивности среди род-
родственных элементов, например в ряду цинк, кадмий и ртуть. Наиболее резким
является изменение от стабилизатора к активному катализатору при переходе
от олова к свинцу.
Ниже приводятся некоторые общие объяснения по характеристике гомо-
гомогенного и гетерогенного разложения. Затем следует обзор литературы по
каталитическому разложению, в котором материал размещен в соответствии
с положением элементов в периодической таблице по системе, предложенной
Латимером 166]. После этого кратко рассмотрены вопросы периодического
разложения, промотирования, торможения и отравления. Имеются некоторые
характеристики механизмов катализа, общие для ряда элементов, например
критерий произведения растворимости для инициирования катализа. Мы
* Указание, что тот или иной элемент не является катализатор'ом разложения
перекиси водорода (рис. 64), еще не доказывает возможности использования его в качестве
аппаратостроительного материала для перекиси водорода (см. стр. 140).
392 Глава 8
попытались рассмотреть типичный пример такой характеристики полностью
по меньшей мере для одного случая. Для справок общего характера и в каче-
качестве введения в катализ можно рекомендовать книгу Шваба, Тейлора и Спенса
[67], монографию Беркмана, Моррела и Эглоффа [68] и серию книг под редак-
редакцией Эмметта [69]. Только в отдельных случаях дано сравнение процессов
разложения перекиси водорода и других перекисей, например перекиси ба-
бария [70] и гидроперекиси тетралина [71]. На стр. 319 рассмотрены некоторые
причины, обусловливающие фундаментальные различия между перекисью водо-
водорода и другими перекисями. Экспериментальная техника для исследования раз-
разложения перекиси водорода не отличается большими особенностями, тем не
менее здесь все же описаны некоторые специальные методики [72].
Гомогенный катализ
Гомогенными катализаторами называются те вещества, которые оказы-
оказывают каталитический эффект, присутствуя в виде истинного молекулярного
раствора. Такого рода катализ разложения перекиси водорода послужил не-
недавно предметом превосходного обзора, составленного Баксендейлом [73];
к этому обзору мы и отсылаем читателя за более подробными сведениями, об-
общего характера и описаниями гомогенного катализа в присутствии галогенов,
железа, меди, перманганата, хрома, молибдена и вольфрама. Баксендейл
указывает, что для объяснения механизма гомогенного катализа выдвинуты
две общие теории. По одной из них постулируется возникновение весьма актив-
активных промежуточных продуктов, обычно перекисей, образующихся из перекиси
водорода и катализатора и затем разлагающихся с регенерированием ката-
катализатора. По второй теории предполагается попеременное окисление и вос-
восстановление катализатора в следующей последовательности:
Восстановленный катализатор+Н2Оа-* Окисленный катализатор+НаО, B3)
Окисленный катализатор f H2O2 -> Восстановленный катализатор +О2. B4)
Итоговым результатом такого цикла является разложение перекиси водорода
в соответствии с реакцией A). Эта теория, как отмечает ряд авторов, в частности
Гайсинский [741, особенно подтверждается тем, что многими из самых актив-
активных катализаторов являются элементы, способные существовать в нескольких
валентных состояниях. Отметим, что хотя схема окисления—восстановления,
соответствующая реакциям B3) и B4), согласно расчетам, может быть термо-
термодинамически вполне допустима, тем не менее, как показал Мак-Олпайн [75]
для нескольких случаев взаимодействия перекиси водорода, одна или даже обе
эти реакции в действительности могут и не происходить.
Все гомогенные каталитические процессы (а также многие гетерогенные)
можно качественно объяснить одной из этих двух реакций, хотя в действитель-
действительности механизмы, безусловно, являются гораздо более сложными. Так, Бак-
Баксендейл классифицирует процессы катализа, протекающие по механизмам сво-
свободных радикалов, как циклическое окисление и восстановление, и подчерки-
подчеркивание роли иона пергидроксила [76] также не противоречит такой схеме,
а. является лишь ее развитием. Некоторые другие схемы механизма, в особен-
особенности основанные на таутомерных формах перекиси водорода, в настоящее
время не выдерживают критики. Другой стороной проблемы является так
называемое «термическое разложение», т. е. разложение очищенной перекиси
водорода, особенно при повышенных температурах. Как показывают работы
Раиса и Рейфа [77], Ливингстона [78], а также соображения, изложенные
на стр. 434, это термическое разложение должно быть в значительной мере при-
приписано гетерогенному катализу под действием сосуда, в котором находится
перекись, или взвешенных в ней частиц. Хотя в некоторых условиях возможно
Разложение в жидкой фазе 393
образование длинных цепей с многократным повторением гомогенных процес-
процессов, эти цепи могут зарождаться только на стенках или на следах примесей.
Даже при чистейшей исследованной перекиси водорода и при повышенных
температурах наблюдавшееся разложение в жидкой фазе не является гомоген-
гомогенным процессом саморазложения собственно перекиси водорода.
Взаимоотношения между гомогенным и гетерогенным катализом изу-
изучены лишь слабо главным образом потому, что элементы, способные дать начало
обоим видам катализа, не исследованы по всему интервалу переменных (на-
(например, рН и концентрации), определяющих состояние катализатора. В каче-
качестве катализатора, при котором можно наблюдать переход от гомогенного
механизма к гетерогенному, можно назвать железо. В кислом растворе реак-
реакция чисто гомогенная. Однако если увеличивать рН, начинает появляться
коллоидное, вещество и одновременно происходит изменение скорости (см,
рис. 76 на стр. 440). При еще более высоких рН может наблюдаться образова-
образование макроскопического осадка, а также и другие кинетические изменения. На
скорость катализа могут влиять и изменения физической формы (наличие
носителя для катализатора, спекание катализатора или изменение кристалли-
кристаллической структуры). Хотя еще не вполне точно определен рН, при котором начи-
начинает появляться коллоидное вещество, не подлежит никакому сомнению факт
перехода от гомогенного разложения к гетерогенному при повышении рН.
Однако существуют еще значительные неясности по вопросу природы измене-
изменения механизма. В некоторых случаях оба вида разложения могут быть каче-
качественно объяснены одним и тем же механизмом, например циклическим окис-
окислением и восстановлением. В то же время образование комплекса или осажде-
осаждение катализатора в коллоидном или твердом состоянии может определить ту
долю от общего количества имеющегося катализатора, которая способна
фактически участвовать в реакции и таким образом. влиять на наблю-
наблюдаемую скорость разложения. Такого рода случай комплексообразования
встречается при катализе полимеризации действием перекисей [79]. При чисто
гетерогенном катализе наблюдаемая скорость зависит от степени дисперсности
твердого катализатора, так как эта дисперсность определяет размер поверх-
поверхности, находящейся в контакте со средой. Наоборот, вполне возможно, что при
переходе от гомогенной системы к гетерогенной коренным образом изменяется
и характер реакции, которой подвергается перекись водорода, например ион-
ионный механизм может перейти в радикальный. Возможно, что при изменении
условий имеется сравнительно тонкая градация в переходе от одного меха-
механизма к другому. При выяснении различий гомогенного и гетерогенного ката-
катализа нужно всегда учитывать возможное влияние адсорбции из раствора на
гомогенный катализ. Так, одновалентное серебро, не обладающее каталитиче-
каталитическими свойствами при гомогенном диспергировании, легко адсорбируется
стеклом [80]. В адсорбированном состоянии оно может приобрести каталити-
каталитические свойства в результате либо истинного восстановления до металла,
либо только поляризации [81]. Последующее использование поверхности
стекла в контакте с более щелочным раствором также может активировать
адсорбированное серебро. Это особенно заметно в случае поверхности стеклян-
стеклянного электрода.
Гетерогенный катализ
Успехи в области теоретического или экспериментального определения
механизма разложения перекиси водорода под действием гетерогенных катали-
катализаторов развивались необычайно медленно; эта область изучена гораздо хуже,
чем гомогенный катализ. Такое состояние характерно не для одной перекиси
водорода, но и для большинства реакций гетерогенного катализа. Исследова-
Исследование разложения перекиси водорода заслуживает особого интереса в этой об-
области вэиду наличия большого числа эффективных катализаторов, способных
394 Глава 8
возбуждать именно только этот процесс. Хотя число способов начального раз-
разрыва самой молекулы перекиси водорода и ограниченно, последующие ступени
каталитического механизма могут сильно различаться. Дополнительные ослож-
осложнения обусловлены многообразием форм твердой фазы, которая может нахо*
диться в коллоидном, аморфном или кристаллическом состоянии, а также на
носителе.
Современное состояние знаний по гетерогенному катализу вообще пре-
прекрасно изложено в серии книг под редакцией Эмметта [69]. Основные аспекты
относительно природы твердых веществ описали Зейц [82], Шокли [831
и Гомер и Смит 184]. Доуден [85] рассмотрел основные факторы, которые необ-
необходимо учитывать при выводе зависимости между катализом и электронной
структурой. Несколько менее обширным, но все же ценным понятием является
поляризация атомов на поверхностях твердых фаз, разработанная Вейлом [81,
84]. Все эти обобщения подчеркивают огромную роль, которую должен играть
процесс хемосорбции перекиси водорода на поверхности катализатора. Как
указывает Уилер [84] в очень сжатой форме, эта стадия связана с довольно
значительными электронными перегруппировками, выражающимися в делении
или переносе электронов. В соответствии с концепцией Будара [84] поверх-
поверхность можно рассматривать как резервуар частиц или как активный агент,
способствующий разрыхлению или диссоциации перекиси водорода на осколки,
удерживаемые одним или несколькими участками поверхности. Эти осколки
могут затем реагировать с ударяющимися о них молекулами из окружающей
среды. Одновременно должны протекать процессы (строго говоря, их нужно
рассматривать как гомогенные), распространяющиеся в основной массе среды
или же происходящие на пленке вблизи поверхности. Состояние и концентра-
концентрация веществ в этой пленке могут значительно отличаться от состояния и кон-
концентрации их в основной массе среды или на поверхности твердого тела, что
обусловлено как физическими, так и химическими кинетическими процес-
процессами.
Имеется несколько точек зрения на гетерогенный катализ перекиси водо-
водорода и общие его характеристики, на которых необходимо остановиться.
Уже давно Бредиг и Фон-Бернек [86] опубликовали подробное исследование
многих факторов, влияющих на характеристику катализатора разложения
перекиси водорода. Поскольку это разложение протекает в условиях, весьма
отдаленных от состояния равновесия, можно было ожидать, как отметили эти
авторы, что давление кислорода] не окажет влияния на ход реакции. Для
гомогенного катализа это верно, но при гетерогенных реакциях происходят
другие явления, оказывающие существенное влияние на механизм реакции
и сильно зависящие от давления кислорода. Например, Ройтер [87] указы-
указывает, что скорость разложения на платиновой фольге возрастает с падением
давления, вероятно потому, что при этом поверхность катализатора освобо-
освобождается от адсорбированного газа. Другие авторы исследовали влияние солей
[88], природы растворителя [89], света [90] и магнитного поля [91] на катализ
перекиси водорода. Большое внимание уделено проблемам невоспроизводи-
мости и изменения активности в зависимости от размеров частиц, общей по-
поверхности и удельной активности. Хотя значение этих факторов известно уже
давно, успехи в этой области тормозились недостаточно развитой эксперимен-
экспериментальной техникой определения важнейших факторов. В последнее время раз-
разработка методики определения микроструктуры при помощи электронной
микроскопии и измерений величины поверхности [92] или магнитной воспри-
восприимчивости [93] улучшила это положение, тем не менее и сейчас еще отсутст*
вуют вполне надежные средства для характеристики факторов, определяющих
активность. Так, например, Шваб [94] отметил, что если константу
скорости каталитического процесса выразить через уравнение Аррениуса
к=А -ехр (—E/RT), то член для энергии активации Е является характеристикой
Разложение в жидкой фазе 395
системы, а коэффициент Л определяется факторами, обусловливающими случай-
случайные колебания и невоспроизводимость, в связи с которыми так трудно получить
согласованные данные. В качестве примера для перекиси водорода, который
можно объяснить этим путем, приведем опыты разложения пара перекиси,
проведенные Штейном [10]. Этот автор показал, что при гетерогенном режиме
кривые для стеклянных трубок одинаковой природы (см., например, кривые
на рис. 61), как правило, параллельны, но смещены одна относительно дру-
другой; это является доказательством аналогичных величин энергии активации,
но различных величин Л, что можно объяснить случайными изменениями изу-
изученных поверхностей.
Как показал вышеприведенный анализ гомогенного разложения, при пере-
переходе от гомогенной системы через системы, в которых твердая фаза находится
в различных состояниях дисперсности и активности, часто наблюдается резкое
изменение каталитической активности. Это изменение обусловлено рядом факто-
факторов, причем некоторые системы обладают активностью не как гомогенные
катализаторы, а лишь как гетерогенные. В сравнительно старой работе Мади-
навейтиа и Агирреке [95] изучили влияние добавки коагулянта на скорость
разложения перекиси водорода на золотом золе. При изменении степени дис-
дисперсности скорость разложения проходит через максимум. Истинный рас-
раствор золота лишен каталитического эффекта, макроскопические частицы ме-
металла также сравнительно инертны. Таким образом, чтобы катализатор был
эффективным, требуется присутствие твердой фазы в состоянии высокой дис-
дисперсности. Другие работы показали, что некоторые элементы, не обладающие
каталитическими свойствами в совершенно гомогенной системе, например
серебро, марганец, свинец и кобальт, приобретают способность катализиро-
катализировать разложение перекиси водорода только при достижении определенного
произведения растворимости. При более высоких концентрациях металл или
его гидроокись образует твердую фазу, наличие которой, по-видимому, обяза-
обязательно, несмотря на то, что циклическая схема окисления—восстановления
термодинамически возможна даже в отсутствие твердого вещества. Когда такое
вещество образовалось, скорость разложения на нем может быть прямо про-
пропорциональна размерам возникшей поверхности. Это доказано для двуокиси
марганца Паркером, Коэном и Смитом [96], которые провели точные измерения
удельной поверхности методом адсорбции азота. В связи с необходимостью
располагать таким характеристическим произведением растворимости суще-
существенное значение имеют амфотерные свойства большинства, а возможно и всех,
металлов. Так, например, Райт и Ридл 197] сделали вывод, что твердые катали-
катализаторы должны обладать максимальной активностью в изоэлектрической
точке соответствующей твердой фазы. Эта гипотеза удовлетворительно согла-
согласуется с опытом. Указанные характеристики носят несколько поверхностный
характер, и, как сообщалось выше, прежде чем удастся вполне удовлетворитель-
удовлетворительно описать механизм катализа, нужно будет определить более фундаментально
роль поверхности с точки зрения регулирования концентрации или создания
возможностей реакционных Путей с пониженной энергией активации. Нет
никаких указаний на возможность универсального механизма, действитель-
действительного во всех случаях 198].
Очень важный вопрос об обмене электронами между гетерогенным ката-
катализатором и субстратом для случая с перекисью водорода только начинают
изучать. Первым, кто предложил возможность такого обмена, по-видимому,
был Вейс [56]. Он считает, что стадией, определяющей скорость при разложе-
разложении перекиси водорода металлами, может быть следующая:
Н2О2 + электрон металла —> ОН" + ОН. B5)
Указывается, что для медноникелевых сплавов такой перенос электрона
зависит от состава. Он протекает быстрее в области сплавов с высоким содер-
396
Глава 8
жанием меди и падает до небольшой величины с повышением относительного
количества никеля, что соответствует отдаче электрона с d-yровня. Вейс под-
подчеркивает, что эта характеристика должна иметь существенное значение
лишь при такой кислотности раствора, при которой невозможно коррозионное
действие, и только в том случае, если отсутствует окисная пленка, толщина
которой может предотвратить перенос электронов. Капустинский и Шмелев [991
измерили каталитическую активность ряда сплавов свинца с кадмием, олова
с висмутом и сурьмы с висмутом. Выведена зависимость между эффективностью
этих сплавов в отношении разложения перекиси водорода и характером диа-
диаграммы температур плавления, а следовательно, и составом соответствующей
системы. Активность кон-
концентрировалась на грани-
границах фаз. Доуден и Рей-
Рейнольде [1001 проводили
опыты с фольгами^из мед-
ноникелевого сплава. Эти
фольги сначала обрабаты-
обрабатывали кислотой, чтобы они
были совершенно свободны
to
р
отокисной пленки, а затем
испытывали в разбавлен-
разбавленном растворе нейтральной
перекиси водорода при
20—80°. В исследованных
пределах содержания меди
70—100% скорость разло-
разложения падала с уменьше-
нием доли меди в соответ-
ствии с эанее упомяну-
SI ГИПОТЕЗОЙ Об^аруже-
на линейная зависимость
между предэкспоненциальным множителем А и энергией активации ?, анало-
аналогичная зависимости, показанной для гомогенного процесса переноса электро-
электронов на рис. 57 (стр. 304). Результаты, полученные для сплавов, отличаются от
найденных для гомогенного процесса в том отношении, что здесь меняется
катализатор, а субстрат остается одним и тем же. Доуден и Рейнольде привели
доказательства, что указанная зависимость не может быть объяснена образо-
образованием промежуточной окисной пленки. Однако Шваб [84, 1011 показал,
что эта характеристика может быть обусловлена и первичным окислением
катализатора с последующей быстрой десорбцией кислорода. Это заключение
основано на экспериментальных работах с железными шпинелями и некото-
некоторыми системами сплавов. Дальнейший материал по роли переноса электронов
при гетерогенном катализе можно найти у Лича [102].
Как с практической, так и с теоретической точки зрения интересно срав-
сравнить эффективность различных гетерогенных катализаторов, хотя и трудно
разработать способ иллюстрации влияния широкого диапазона пере-
переменных. Качественное сравнение этого типа провели Кэстл и Кларк [103];
аналогичное сравнение [1041 показано на рис. 65. Эти данные получены путем
измерения времени, требующегося для разложения ряда последовательных
порций 48%-ной перекиси водорода (по 10 мл), введенных в раствор, содержа-
содержащий 0,0118 г соответствующего металла в виде нитрата. Сначала перекись во-
водорода (порциями по 10 мл) смешивали с раствором соли металла и 4 н. раство-
раствором едкого натра для осаждения гидроокиси металла. Разложение проводили
в большой>олбепри температуре 95—100°, причем колба была снабжена обрат-
обратным холодильником, через который могло уходить менее 10% образовав-
О 5 Ю 15 20
Общее количество 48%-Hou)\tPi на 1мг катализатора, г
Р и с. 65. Относительная каталитическая активность
гидроокисей некоторых металлов [образованных из
0,0.18 г металла (в виде нитрата)+10 ммЛ н. NaOH].
Разложение в жидкой фазе 397
шегося водяного пара. Хотя количественных выводов сделать было нельзя,
все же при этом сравнении обнаружены необычайно большие колебания ско-
скорости разложения и доказано влияние разбавления и старения.
Кислота и щелочь
Данные, приведенные в гл. 9 для иллюстрации вопроса о влиянии рН
на стабильность чистой перекиси водорода, доказывают, насколько значитель-
значительными могут быть изменения скорости разложения, вызванные варьированием
концентрации водородных ионов. Совершенно бесспорно, что добавка как
кислоты, так и щелочи к незагрязненным растворам перекиси водорода повы-
повышает скорость разложения. В случае с кислотами наиболее надежной основой
для этого вывода является работа Рота и Шенли [105], которые изучали фос-
фосфорную, хлорную, плавиковую и серную кислоты и показали, что рост ско-
скорости разложения с падением рН не зависит от природы кислотного аниона
(возможно, за исключением фосфатного). Поскольку кислоты обладали экви-
эквивалентным эффектом, а хлорная и плавиковая кислоты не могут образовать
пероксокислоты, сделано заключение, что скорость разложения зависит только
от изменения концентрации водородных ионов. Однако имеются системы,
в которых на разложение влияет и кислотный анион. Очень ярким примером
нужно считать азотную кислоту. Хэтчер и Мак-Лочлэн [106] описали бурное
разложение концентрированной перекиси водорода при добавке азотной кис-
кислоты в количестве 50% или больше. Это разложение катализируется поверх-
поверхностью и характеризуется индукционным периодом, появлением бурых паров
и возникновением запаха, напоминающего запах озона. Сделано предполо-
предположение, что этот механизм разложения включает стадию образования пер-
пероксокислоты, однако это нельзя еще считать доказанным. Форбес [107] указы-
указывает, что при добавке восстановителя к бесцветной азотной кислоте реакции
почти не происходит вплоть до появления желтой двуокиси азота.
Больше внимания уделялось усилению разложения перекиси водорода при
добавке щелочи, хотя механизм этого процесса изучен не лучше, чем механизм
предыдущего. Дойл [108] исследовал разложение 5%-ной перекиси водорода
при 70° в интервале рН от 5,3 до 9,3. По данным Бюрки и Шаафа [109],
скорость реакции соответствовала первому порядку по концентрации
перекиси водорода внутри всего интервала рН. На графике логарифма
константы скорости реакции в виде функции рН обнаружены две прямые
линии, пересекающиеся при рН 8,7. Переход весьма резкий: наклон ветви
прямой ниже рН 8,7 равняется 0,2, при более высоких рН—2,6; таким образом
получаются нецелые обратные порядки по концентрации водородных ионов
0,2 и 2,6. Хотя эти опыты и не охватывают широкой области, все же можно
принять, что при рН 8,7 происходит изменение механизма, определяющего
общую скорость, причем при более низком рН разложение, вероятно, гетеро-
гетерогенное, при более высоком—гомогенное. Можно сделать предварительное за-
заключение, что порядок по концентрации водородных ионов при более высоких
рН равен в действительности—2 (если вычесть ту долю разложения, которая
должна быть приписана первому механизму, и учесть степень точности работы).
Этот переход находится также в согласии с наблюдением Слейтера [110] отно-
относительно изменения скорости разложения при рН между 5 и 6 и между 8 и 9.
В других сообщениях также указывается, что при дальнейшей добавке щелочи
к перекиси водорода скорость разложения переходит через максимум [111]
или минимум [112].
Пока еще нет вполне приемлемого объяснения механизма разложения,
вызываемого щелочью или кислотой, главным образом из-за недостатка надеж-
надежных кинетических данных. Особенно важным требованием является отсутствие
случайных примесей [110, 113]. Часто при рассмотрении разложения, катали-
398 Глава 8
зируемого основаниями, указывается на роль иона пергидроксила; так,
Абель [114] предполагает, что механизм этой реакции может быть следующий:
Н2О2 + НО" -* Н2О + О2 + ОН". B6)
При кислотном катализе важную роль может играть ион перексония H3Og.
Дальнейшие точные опыты должны привести к выяснению этого механизма,
вероятно представляющего пример специфического кислотно-основного ката-
катализа, в котором скорость разложения может быть выражена в виде суммы двух
или трех кинетических уравнений, основанных на действии различных частиц.
Кислород, водород
Радикалы, например гидроксил или пергидроксил, по-видимому, играют
значительную роль в процессах разложения перекиси водорода, но в связи
с образованием их в качестве промежуточных продуктов рассматривать эту
роль целесообразнее при дальнейшем анализе разложения в присутствии от-
отдельных элементов. Лучше всего вопрос о значении этих частиц разработан
при исследовании гомогенного разложения в присутствии железа.
Галогены
Галогены (за исключением фтора) катализируют разложение перекиси
водорода при помощи гомогенного циклического механизма окисления—вос-
окисления—восстановления, который можно представить следующим образом:
Н2О2 + 2Х- + 2Н+ -> Х2 + 2Н2О, B7)
Н2О2 + Х2 -» 2Х- + 2Н+ + О2. B8)
Эта система исследована весьма подробно, и в настоящее время не подлежит
сомнению, что суммарные процессы действительно соответствуют указанным.
Кроме того, исследованы по отдельности индивидуальные реакции, причем
выведена их кинетика с указанием взаимозависимости между концентрацией,
константами скорости реакции, предэкспоненциальными множителями и энер-
энергиями активации [73, 115, 116]; на основании этих данных можно рассчитать
скорость катализа для определенного ряда условий. Чтобы катализатор
в окисленной или восстановленной форме не истощался, необходимо, очевидно,
равенство скоростей обеих реакций. Таким образом, когда начальный состав
отличается от «стационарного состояния», то в зависимости от условий проис-
происходит окисление или восстановление до тех пор, пока не будут достигнуты ста-
стационарные концентрации обоих видов частиц—галогенида и галогена.
По катализу разложения в присутствии галогенов имеются прекрасные
обзоры [73, 117]. Хотя кинетика приведенных выше итоговых реакций легко
поддается математической обработке [118], фактический механизм, безусловно,
гораздо сложнее. Баксендейл [73] рассмотрел подробности кинетики и пред-
предложил механизмы реакций, а Эдварде [116] показал, что эти механизмы имеют
характеристику реакций, которые можно истолковать в соответствии с общими
принципами кислотно-основного катализа. Были сделаны предположения, что
реакция протекает через свободные радикалы [119], но впоследствии эти
предположения опровергнуты [73, 120]. В отношении экспериментальных ра-
работ, касающихся как обеих отдельных реакций, так и общей реакции разло-
разложения и послуживших основой для выводов, можно сослаться на ряд работ
(по хлору [121], по брому [122], по йоду [123]). Лучше всего изучен йод,
в частности исследованы солевые эффекты [124], влияние йода, присутствую-
присутствующего в негомогенной фазе [125], и эффекты растворителя (см. стр. 330).
Разложение в жидкой фазе 399
Несколько слабее эти вопросы изучены для хлора и брома, а также в отноше-
отношении катализа в присутствии йодата [126], который, по-видимому, протекает
по схеме
5Н2О2 + 2 Ю8- + 2Н+ -> J2 + 6Н2О + 5О2, B9)
5Н2О2 + J2 -> 2JO8- + 4H2O + 2Н\ C0)
У этой системы обнаружена интересная периодичность. Исследовано также
влияние света на каталитическое разложение, вызванное бромом и йодом [127].
Сера, селен, теллур, полоний
Сера не катализирует разложение перекиси водорода. Селен вызывает это
разложение [128] путем циклического процесса окисления—восстановления
между селенатом SeOJ" и селенитом SeOg" без участия пероксоселената, о суще-
существовании которого сообщили Ворсли и Бекер [129], что, однако, подвергается
сомнению [128]. Боне-Мори и Лефор [130] показали, что полоний способен
каталитически разлагать перекись водорода при более низкой концентрации,
чем та, при которой на платине наблюдается измеримая скорость разложения.
Азот, фосфор, мышьяк, сурьма, висмут
Если исключить действие азотной кислоты, о котором упомянуто выше,
то нет никаких данных о каталитическом разложении с участием азота или
фосфора. Леви и Гирон [131] в ходе безуспешных попыток получения пероксо-
арсенатов показали, что арсенаты обладают каталитическим действием. Сурьма
отнесена [132] к веществам, обладающим безусловно малым каталитическим
эффектом (если он вообще существует). Более достоверным следует считать
катализ в присутствии висмута или окиси висмута [133, 134], однако по этому
вопросу имеются лишь качественные данные.
Углерод, кремний, германий, олово, свинец
Различные формы углерода, например графит и активные угли из разных
источников, являются гетерогенными катализаторами разложения перекиси
•водорода, отличающимися рядом интересных особенностей. Активность
углерода зависит от его происхождения [135]; кроме того, ее можно изменять
специальной обработкой. Фоулер и Уолтон [136] исследовали влияние добавки
солей или желатины на каталитическую активность активированного угля из
сахара [136]; другие авторы изучали влияние температуры, размеров частиц,
концентрации водородных ионов, излучения [137], концентрации перекиси
водорода и химической природы поверхности угля. По-видимому, из всех
описанных до настоящего времени свойств наиболее существенную роль играет
адсорбционная способность поверхности [138]. Однако эффективность катализа
не является прямо пропорциональной этой адсорбции. Обработка поверхности,
например нагреванием или пропусканием над ней азота [139], заметно изме-
изменяет активность. Чистый активированный уголь из сахара при взбалтывании
с растворами перекиси водорода вызывает лишь слабое выделение кисло-
кислорода, однако действие этого угля можно сильно интенсифицировать, если пред-
предварительно нагреть его в вакууме при 600°. Активированный уголь из целлю-
целлюлозы и рисового крахмала, высушенный при 100°, обладает максимальной ак-
активностью; более слабым действием отличается уголь из декстрина, инулина
и пшеничного крахмала; уголь из декстрозы, лактозы, мальтозы или карто-
картофельного крахмала едва ли обладает какой-либо активностью. Сырой костяной
уголь или кровяной уголь вызывает лишь медленное разложение перекиси
400 Глава 8
водорода, если только эти сорта угля не нагреть предварительно в вакууме
до 600—900°.
Бринкман [140] сделал вывод, что активность угля возрастает с числом
основных поверхностноактивных групп, определяемых по способности адсор-
адсорбировать соляную кислоту, и падает с ростом .числа кислотных поверхностно-
активных групп, определяемых по сорбции едкого натра. В качестве меры ката-
каталитической активности служит время, которое требуется в определенных усло-
условиях для снижения активности до половины начального значения. Этим путем
можно открыть и количественно определить очень небольшие изменения со-
состояния поверхности активного угля. Однако прямой зависимости между
каталитической активностью и адсорбционной способностью активирован-
активированного угля, очевидно, не существует: активные формы углерода с высокой
адсорбционной способностью могут обладать в отношении перекиси водо-
водорода очень малой каталитической активностью и, наоборот, способность к
интенсивному разложению иногда сочетается с малой адсорбционной способ-
способностью.
Что касается механизма разложения перекиси водорода активированным
углем, то Бринкман выдвинул гипотезу, по которой сначала происходит реак-
реакция обмена между ОН-группой на поверхности угля и ООН-ионом в растворе.
Затем ион пергидроксила, активированный таким образом путем присоеди-
присоединения к поверхности, претерпевает саморазложение или же вступает в реакцию
с молекулой перекиси водорода с вторичным образованием гидроксильного
иона. Показано, что активированный уголь, имеющий на поверхности практи-
практически только кислотные группы (в результате окислительной обработки),
несмотря на высокую адсорбционную способность, не обладает измеримой
каталитической активностью в отношении перекиси водорода в нейтральном
растворе, если только все группы находятся в водородной форме (например,
в виде группы СООН). Однако если кислотные группы действием щелочи
превратить в солеобразные группы COONa, то в отношении перекиси водорода
возникает каталитическая активность, правда очень небольшая по сравнению
с величиной ее для активированного угля с присоединенными основными
группами.
При анализе величины каталитической активности активированного угля
в отношении разложения перекиси водорода Бринкман показал, что в быстром
выделении газа с поверхности угля принимает участие только небольшая доля
имеющихся основных групп, а именно только те группы, которые расположены
в тонкой наружной зоне; те же группы, которые покрывают внутреннюю по-
поверхность в порах угля, не взаимодействуют с молекулами или ионами
перекиси водорода вследствие сильного торможения диффузии перекиси
в поры из-за выделения кислорода. Доказательством решающего значения
этого фактора является четкая зависимость скорости разложения от раз-
размеров частиц угля. Чтобы можно было точно сравнивать каталитические
активности путем таких измерений, размеры частиц должны лежать в узком
интервале.
Лемуан [141] сообщал, что кремнезем является хорошим катализатором
разложения, однако более поздние работы [142—144] не подтвердили этого
мнения. По наблюдениям Феллса и Фирта [142], в силикатном растворе разло-
разложения перекиси водорода не происходит до начала образования геля; на осно-
основании этого сделан вывод, что для возбуждения активности требуется появле-
появление коллоидной или твердой фазы. Однако Пеннер [144] пришел к заключению,
что разложение протекает рарномерно как в растворе, так и в геле и вызывается
загрязнениями на поверхности сосуда с перекисью. Сделаны также наблюде-
наблюдения, касающиеся формы пузырьков, возникающих в силикагеле [142], а также
фотохимического разложения в этой системе [144].
По-видимому, не производилось никаких исследований каталитической
Разложение в жидкой фазе 401
активности германия как катализатора разложения перекиси водорода. Олово,
как совершенно отчетливо доказывают данные, приведенные на стр. 146 и 446,
лишено каталитических свойств. Опубликованное .сообщение [134J, что желтый
осадок олова обладает небольшой каталитической активностью, основано на
наблюдениях при работе с загрязненной пробой.
Свинец—один из наиболее активных гетерогенных катализаторов. Опубли-
Опубликованы разные качественные характеристики этого каталитического процесса
A34, 145, 146], а именно: двухвалентный свинец в кислом растворе не оказы-
оказывает никакого действия на перекись водорода; для разложения ее требуется
щелочная среда, в которой образуется двуокись свинца. В результате изучения
I147J механизма этого катализа сделан вывод, что его можно описать как
окислительно-восстановительный цикл между двухвалентным свинцом РЬ(ОНJ
и свинцовым суриком РЬ3О4. Условия высокой каталитической активности
возникают тогда, когда оба эти вещества присутствуют как твердые фазы;
в сильнощелочном растворе образуются высшие окислы. Влияние различных
интервалов рН можно охарактеризовать следующим образом. Азотнокислый
свинец растворяется в перекиси водорода с образованием прозрачных устойчи-
устойчивых растворов. При добавке щелочи выпадает беловато-желтый осадок и воз-
возникает небольшая активность. При дальнейшей добавке щелочи осадок перехо-
переходит в оранжево-красный и начинается бурное разложение перекиси. Как ока-
оказалось, количество щелочи, требующееся для достижения этой точки, обратно
пропорционально количеству растворенного свинца; на это явление наклады-
накладывается еще четко не установленное влияние старения. Количество пирофосфата,
требующееся для прекращения катализа, примерно эквивалентно количеству,
необходимому для образования пирофосфорнокислого свинца РЬ2Р.2О7. Катали-
Каталитическая активность проходит через максимум приблизительно при 0,2 н. кон-
концентрации щелочи; при более высокой концентрации возрастает растворимость
свинца в виде плюмбита и плюмбата и каталитическая активность снижается.
Сделана попытка [147] доказать наличие циклического процесса окисления—
восстановления при помощи радиоактивных индикаторов, однако она закончи-
закончилась неудачей в связи с тем, что даже в отсутствие перекиси водорода происхо-
происходит обмен между ионом двухвалентного свинца и двуокисью свинца в азотной
кислоте (что соответствует литературным данным [148, 149]) и между плюмби-
том и плюмбатом в основном растворе (что противоречит опубликованным
данным [149]).
Влияние указанных факторов на металлический свинец сказывается очень
резко. Если погрузить полированный свинец, лишенный окисной пленки,
в перекись водорода, то активность его оказывается весьма низкой. Постепенно
образуется белый осадок, который после накопления превращается в свинцовый
сурик с последующим бурным проявлением каталитической активности. Если
металлический свинец ненадолго погрузить в раствор перекиси водорода и сей-
сейчас же извлечь, то небольшое количество жидкости, приставшей к металлу,
в течение короткого промежутка времени остается в спокойном состоянии,
а затем после образования на металле пленки свинцового сурика РЬ3О4 резко
отрывается от поверхности под действием бурного разложения. В этом про-
процессе происходит растворение свинца, безусловно связанное с наблюдаемым
уничтожением пассивности свинца при действии перекиси водорода [150],
однако перекись не влияет на рост на нем дендритов [151]. Описано практиче-
практическое применение свинцовых катализаторов для разложения концентрирован-
концентрированной перекиси водорода в системах, применяемых для генерирования энергии
[152].
Галлий, индий, таллий
Элементы галлий, индий и таллий, по-видимому, не исследованы в отноше-
отношении возможного каталитического действия на перекись водорода.
26 Перекись водорода
402 Глава 8
Цинк, кадмий, ртуть
Цинк обладает необычными свойствами; он может функционировать и как
катализатор и как стабилизатор. Как указывается на стр. 451, цинк в растворе
90%-ной перекиси водорода обладает стабилизирующим действием. Сделано
наблюдение [153], что при снижении концентрации перекиси водорода это дей-
действие ослабевает и что в растворах с содержанием ниже 40 вес. % перекиси
водорода цинк действует уже как катализатор разложения. Это каталитическое
действие обнаружено также [154] в смесях с другими катализаторами. Вейс
[56] показал, что металлический цинк разлагает перекись водорода с выделе-
выделением водорода и кислорода. До сих пор не предложено механизма, которым
можно было бы объяснить это двоякое действие цинка. Влияние кадмия изуче-
изучено лишь в слабых растворах, причем ему приписываются либо слабые катали-
каталитические свойства 1134, 154], либо он считается совсем неэффективным [155].
В отличие от кадмия ртуть обладает совершенно определенными каталити-
каталитическими свойствами и вместе с тем отличается интересной особенностью вы-
вызывать периодическое проявление катализа. Это явление, воспроизводимое лишь
при довольно точном соблюдении условий, описано рядом авторов [132, 156],
в первую очередь Бредигом. Скотт и Соренсон [157] описывают четыре области
рН, различающиеся по действию перекиси водорода на металлическую ртуть.
В очень кислом растворе (при рН< 0,5) ртуть сохраняет блестящий вид, причем
катализ фактически отсутствует, но происходит растворение ртути. В интер-
интервале рН между 1 и 4 на поверхности ртути образуется ее окись, а в растворе
появляются ионы закисной и окисной ртути, концентрация которых возрастает
во времени. Концентрация иона двухвалентной ртути значительно выше, чем
концентрация, соответствующая равновесию Hg2++Hg=Hgg+. До рН 2,5обра-
2,5образуется объемистая окись, которая переходит в основную массу раствора; однако
иногда ртуть сохраняет блестящую поверхность вплоть до рН 2,0. Периодиче-
Периодический катализ наблюдается в интервале рН между 4,0 и 6,5, хотя Бредиг и
Штарк [158] обнаружили особую длительность его при рН 7. Этот катализ
характеризуется периодом затухания, когда поверхность ртути покрыта тонкой
золотисто-коричневой пленкой, с последующим периодом активности в усло-
условиях блестящей поверхности ртути. Эта смена циклов, нарушаемая при пере-
перемешивании, может продолжаться в течение короткого времени, тогда как при
более низком рН в растворе образуется осадок. Эрнст [159] обнаружил индук-
индукционный период перед началом этих циклов; в более старых работах изучено
влияние добавок, температуры и поляризации на этот процесс. В интервале рН
выше 8 разложение носит непрерывный и быстрый характер и происходит на
блестящей поверхности ртути, причем количество растворенной ртути состав-
составляет меньше 10'7моля в 1 л. Сделано наблюдение, что в щелочном растворе про-
происходит восстановление ионов окисной и закисной ртути, а также окислов ртути
до металла, в интервале рНот 1 до 4 закись ртути окисляется до окиси, а при
рН ниже 1 оба окисла растворяются.
Механизм ртутного катализа не выяснен, хотя, по предположению Кварта-
роли [134], имеется основание считать, что он связан с циклическим процессом
окисления—восстановления между металлической ртутью и окисью ртути.
Периодичность лучше всего объяснить как проявление равновесия между фи-
физическими и химическими процессами, происходящими во времени, в резуль-
результате местного истощения водородных ионов, вызванного растворением окиси
ртути в пленке, смежной с поверхностью ртути, и последующей диффузии водо-
водородных ионов в эту пленку из массы раствора. Для выяснения этого механизма
желательно было бы поставить опыты по определению природы градиентов
концентрации вблизи поверхности. Интересно также сравнить каталитическое
действие ртути и благородных металлов. В случае с ртутью наблюдается пере-
переход от условий, при которых ртуть, очевидно, подвергается изменениям валент-
Разложение в жидкой фазе 403
ности, обусловливающим катализ или сопутствующим ему, к условиям, при ко-
которых отсутствует заметное изменение ртути; в последнем случае можно про-
провести аналогию, например, с блестящей платиной, вид которой при катализе
не изменяется.
Медь, серебро, золото
Медь функционирует и как гомогенный и как гетерогенный катализатор.
В этих отношениях медь весьма близка к железу, хотя она менее активна и зна-
значительно меньше изучена. По-видимому, можно с полным правом считать, что
катализ под действием обоих этих элементов протекает в основном по одному
и тому же механизму [73]. Так, при гомогенном катализе наблюдается чере-
чередующееся образование одновалентной и двухвалентной меди. В литературе
опубликованы кинетические данные и анализ причин сравнительной медлен-
медленности проявления каталитического действия меди [160, 161]. Сульфонамиды
тормозят этот катализ [162]. Весьма точно и подробно исследован катализ
в присутствии меди в виде комплексов, например с аммиаком, аминами и цитра-
цитратом [163]. Результаты показывают, что наблюдаемые скорости разложения
в этих случаях могут быть выше, чем при коллоидной гидроокиси меди.
Выдвинуто несколько гипотез относительно механизма действия этих комплек-
комплексов, в том числе и предположение о наличии стадии образования перекиси
меди, но ни одно из этих предположений не является достаточно обоснованным.
Медь исследована также в качестве промотора или составной части катализа-
катализатора, главным образом в сочетании с железом [161, 164], а также с вольфрамом
и молибденом [154, 165, 166], марганцем [167] и кобальтом или никелем [168].
В случае гетерогенного катализа в присутствии меди [134, 168—170]
наблюдается тот же интересный максимум скорости при росте рН, как и при
катализе в присутствии железа. Эту характеристику меди весьма четко описали
Радел и Хэринг [146] и Шпитальский, Петин и Коновалова [171]. Так, при
добавке 0,4 мг/л иона двухвалентной меди к 30%-ной перекиси водорода по-
последняя оставалась бесцветной при рН ниже 3,8, но приобретала травянисто-
зеленую окраску при рН выше 4,4. При промежуточных рН возникала желтая
муть из-за образования гидрата окиси меди Си(ОНJ, причем именно в этом
интервале и проявлялась максимальная скорость разложения. Сама металличе-
металлическая медь не обладает особенной активностью, хотя все же эта активность
настолько велика, что исключает возможность применения меди в качестве
аппаратостроительного материала для использования при работах с пере-
перекисью водорода.
В этой группе элементов максимальной активностью отличается серебро.
Растворенное одновалентное серебро не обладает заметной активностью; так,
90%-ную перекись водорода можно насытить азотнокислым серебром без ка-
какого-либо эффекта. Металлическое серебро и окись серебра Ag2O в небольших
количествах также растворимы и при этом не обладают каталитическими свой-
свойствами. Однако при наличии небольшого количества шелочи эта нечувстви-
нечувствительность к серебру заметно снижается, и при добавке иона одновалентного,
серебра, металла или окиси быстро возникает осадок, инициирующий разло-
разложение. Имеется много литературных источников [170, 172], посвященных это-
этому катализу. Выдвинуты различные теории, приписывающие каталитический.4
эффект металлу, окиси или перекиси, но механизм его до настоящего времени
еще не установлен. Вигель [173] выдвинул гипотезу, что непрерывное катали-
каталитическое разложение наблюдается в том случае, когда достигается произведе-
произведение растворимости гидроперекиси серебра AgOOH. Он постулирует наличиет
окислительно-восстановительного цикла между металлом и гидроперекисью.^
Вейс [56] интерпретировал работу Вигеля как доказательство, что при реакции
между ионом Ag+ и ионом пергидроксила на металлическом серебре образуется
радикал пергидроксил, который приводит к цепной реакции. Мак-Интош [1741.
404
Глава 8
сделал наблюдение, что при увеличении концентрации щелочи скорость разло-
разложения на серебре проходит через максимум. Доказано также наличие попере-
попеременного растворения и выпадения осадка, поскольку при выполнении функции
катализаторов серебряные золи делаются более грубодисперсными [173].
При осуществлении катализа в подходящих условиях на стенке сосуда можно
получить серебряное зеркало. Недавно проведены работы [175], которые
в соответствии с данными Вигеля показали, что образующийся осадок является
металлическим серебром и что в растворе содержится ион одновалентного се-
серебра. Как фильтрация, так и добавка хлорида вызывают прекращение ката-
катализа.
woo с
WOc
10
I
\
\
%4sk
-
?%\
\ '
" о 5,2% H2O2
о 47% H
2О2
о
о
\ °
\
о\
1
о
о
0
VaD 0
\
о\
\
о \
о
-
-
-
-
_
;
-
Y
100 -
Ю к
I
/ 10 . 100
Концентрация NaOH, к.х/0
Рис. 66.Концентрации""ионов^однова-
лентного серебра?и щелочи, необхо-
необходимые для инициирования./ катализа
в 5,2 И(,8,7%-ной перекиси водорода.
0,1 =-,
0,01
ш
:
-
-
- \
i
¦
%
х-
\-.а
\
° \
\
¦
- Концентрация
- н2о2,%
* 3.9
:о 5,2
:° 8.7 *
. • 9,0 (В
-• 19.0
_* Щ6
игель
> U /JJJ
1 1 J
••
-*-
& т
Г й
к
N
О
•
\ о
\
\
V
-
:
-
':
-
:
•
Ю ЮО
Концентрация NaOH, н.х/05
1000
Рис. 67. Концентрации ионов одновалент-
одновалентного серебра и щелочи, необходимые для
образования осадка в перекиси водорода.
Определены также относительные количества ионов одновалентного сереб-
серебра и щелочи, необходимые для инициирования катализа. На рис. 66 показана
концентрация азотнокислого серебра, требующаяся для возбуждения катализа
в разбавленной перекиси водорода, содержащей различные относительные коли-
количеств^ щелочи. Под «точкой катализа» здесь подразумеваются условия, при
которых в результате тщательного наблюдения за раствором можно обнаружить
первые признаки катализа, но без образования заметного осадка. При увеличе-
увеличении добавки серебра возникает твердая фаза; на рис. 67 показаны условия,
при которых сделано не очень четкое наблюдение образования аморфных белых
или светло-серых частиц. Очевидно, что эта «точка осаждения» наблюдается
в условиях, аналогичных найденным для точки катализа, и что данные Вигеля
в отношении критерия для инициирования катализа подтверждаются. Одно-
Одновременные исследования скорости разложения [175] показали, что эта скорость
Разложение в жидкой фазе
405
после достижения измеримой величины возрастает, достигает максимума и за-
затем при дальнейшем увеличении концентрации щелочи падает до постоянной
величины. Скорость соответствует первому порядку по концентрации перекиси
водорода в области, где отсутствует видимая твердая фаза, и приобретает нуле-
нулевой порядок после появления осадка. На рис. 68 показано это действие для
90%-ной перекиси водорода. Очевидно, что рис. 66—68 можно объединить
в виде трехмерной фигуры с тремя координатами состава и скорости и выделить
зону, показывающую активную область для серебра.
При поисках механизма, объясняющего эти наблюдения в отношении
серебра, следует учесть, что внутри всего изученного интервала рН условия
благоприятны для реакции окисления—восстановления между металлом и
ионом одновалентного серебра, и все же при выполнении критериев рис. 66 и 67
0,010
3,5 4,0 4,5
рН (приразбавлении V-W)
Рис. 68. Влияние растворенного серебра на скорость
разложения 90%-ной перекиси водорода I(Ag+)=l мг/л].
металл приобретает стабильность. Форма кривых на этих рисунках свидетель-
свидетельствует о том, что критерием является установление произведения растворимо-
растворимости. Это произведение не может относиться к гидроокиси серебра AgOH, так
как произведение (Ag4) (ОН") никогда не превышает 101, т. е. оно всегда зна-
значительно меньше принятой величины [176] 4 • 10"8. С другой стороны, вероятно,
в данном случае имеет значение произведение растворимости гидрата окиси
двухвалентного серебра Ag(OHJ,TaK как произведение (Ag2+) (ОНJ в этих
опытах в среднем равно 10'28, тогда как произведение растворимости для
Ag(OHJ составляет 10~82. Перекись водорода вряд ли может образовать замет-
заметные концентрации двухвалентного серебра (разве только в условиях высокой кис-
кислотности [177]), но реакция с двухвалентным серебром, по имеющимся данным
[178], очень интенсивна. Несмотря на это совпадение и очевидный угловой
коэффициент—2 на рис. 66 и 67, можно показать, что наклонд олжен равнять-
равняться —1 для произведения растворимости гидратов окиси одновалентного (или же
двух- или трехвалентного) серебра или гидроперекиси одновалентного серебра.
Видимое отсутствие влияния иона одновалентного серебра и необходимость
наличия металлического серебра подтверждают предположение об окислитель-
окислительно-восстановительном цикле между металлом и серебром в состоянии высшего
окисления, но как этот, так и другие механизмы носят пока чисто умозрительный
характер.
Золото, как и другие благородные металлы, например платина, палладий
и т. д., является типичным гетерогенным катализатором. Золотой золь, образо-
образованный либо in situ восстановлением хлорного золота [179], либо действием
внешнего фактора, дуговым методом [1.80], является катализатором, актив-
активность которого возрастает с ростом концентрации щелочи. Бредиг и Рейндерс
[180] превосходно описали характер катализа при помощи этого катализатора,
включая влияние солей и ядов. Проведены также исследования влияния на
406 Глава 8
процесс методов получения золей золота и наблюдаемых температурных коэф-
коэффициентов [181], а также степени дисперсности этих золей [95, 182].
Никель, палладий, платина
Никель каталитического действия на кислые растворы перекиси водорода
фактически не оказывает. Коллоидная гидроокись никеля Ni(OHJ, осаж-
осажденная действием щелочи, является лишь слабым катализатором [75, 183].
Исследовано каталитическое действие смесей закиси-окиси никеля Ni3O4 с дру-
другими окислами [168] и сернокислого никеля на носителе [184] в отношении пе-
перекиси водорода. Из ряда изученных носителей никеля только силикагель в из-
известной степени активирует катализ. Это усиливающее действие проходит через
максимум при увеличении относительного содержания никеля на силикагеле.
По каталитическому действию палладий, по-видимому, очень близок
к платине; детали катализа под действием палладия изучены, в частности,
Бредигом и Фортнером [185]. Предполагается [56], что действие обоих катали-
катализаторов основано на отрыве электронов от металла; сообщается [186], что
активность этих металлов с удлинением времени контакта проходит через мак-
максимум. Различие между этими веществами связано с высокой абсорбционной
способностью палладия по газообразному водороду. В состоянии насыщения
водородом палладий приобретает более высокие каталитические свойства [185].
Интересно сопоставить это с указанием [187], что насыщенный водородом пал-
палладий способен ускорять также образование перекиси водорода из кислорода.
Другое различие между палладием и платиной заключается в том [188], что
палладиевая чернь не обладает большей активностью в качестве катализатора
для разложения перекиси водорода, чем массивный металл.
Из числа благородных металлов, являющихся катализаторами разложения
перекиси водорода, больше всего изучали платину. В подробном исследовании
этого катализатора Бредиг и Фон-Бернек [86] охарактеризовали его как
«неорганический фермент»; этот термин часто используется для того, чтобы
подчеркнуть аналогию между действием платины и ферментов. Платину изучали
главным образом в коллоидном состоянии или в виде платиновой черни. В этих
формах она обладает обычно высокой активностью; блестящая же листовая
платина, по-видимому, как катализатор значительно менее активна. Катализ
на платине, как правило [189, 190], соответствует первому порядку по концен-
концентрации перекиси водорода, однако имеются данные и о кинетике второго по-
порядка [191]. Присутствие щелочи увеличивает активность платины, а кислота
оказывает тормозящее действие [86, 190, 192]. Тартар иШэффер [192] пришли
к заключению, что в кислой среде влияние оказывают как водородный ион, так
и кислотный анион, тогда как в щелочном растворе активность обусловлена
лишь гидроксильным ионом. На основании исследований платины, защи-
защищенной желатиной, они сделали также вывод, что как кислота, так и щелочь
влияют и на каталитический процесс и на защитное действие желатины. Актив-
Активность катализатора изменяется и при других видах воздействия, например при
старении [193], нагревании [194, 195], электрической поляризации [196]
или рентгеновском облучении [197]. Хотя термическая обработка снижает
активность платины, отношение скоростей разложения при двух температурах
не зависит от этой обработки [194]. Взаимозависимость между потенциалами,
которые платина принимает в перекиси водорода [198], и механизмом разложе-
разложения не нашла еще достаточного объяснения (см. также раздел о стандартных
электродных потенциалах на стр. 216). По вопросу о влиянии степени дисперсно-
дисперсности на активность мнения расходятся. Сивертс и Брюнинг [190] показали, что
по сравнению с коллоидной платиной и массивным металлом платиновая чернь
обладает промежуточной активностью, другие же авторы [188] пришли к заклю-
заключению, что полированный металл и платиновая чернь в расчете на одинаковую
Разложение в жидкой фазе 407
поверхность имеет одинаковую активность. Легирование платины 10% иридия,
родия, палладия, рутения или осмия [199] дает ряд катализаторов разложе-
разложения перекиси водорода, активность которых убывает в порядке перечисления
присадок. Как указано на стр. 331, замена воды другими растворителями
в присутствии платины также влияет на скорость разложения.
Сделан ряд наблюдений по влиянию на платину различных ингибиторов
и ядов. К изученным соединениям относятся ртуть и свинец [200], окись угле-
углерода [201], азидный ион [202], цианиды [203] (о разногласиях в отношении
действия цианидов см. [204]), защитные коллоиды [205], многие другие соли
1203, 206, 207] и неэлектролиты [203, 207]. Относительно отравления ртутью
Мэкстед и Льюис [208] констатировали, что снижение активности пропорцио-
пропорционально количеству ртути (до известной величины этой добавки). Кубокава
1209] показал, что отношение скорости разложения к количеству адсорбирован-
адсорбированной ртути в логарифмических координатах обеих осей является линейным.
Соли в качестве ингибиторов для платины оказывают различное влияние. Хит
и Уолтон [206] нашли, что ионы алюминия, тория, натрия, нитрата, сульфата
и фторида не оказывают никакого эффекта, а хлорид, нитрит и цианид являются
ядами. Гексаплатинат Pt (ОН)*~ не оказывает каталитического действия на раз-
разложение и не влияет также на действие платины. Нейлсон и Браун [207j на
основании исследования влияния различных натриевых солей и хлоридов при-
пришли к заключению, что катионы тормозят разложение перекиси водорода в при-
присутствии платины, а анионы ускоряют это действие.
Предложены различные механизмы для объяснения каталитического раз-
разложения перекиси водорода платиной: к этим механизмам относятся окис-
окислительно-восстановительный цикл с участием окислов платины, образование
атомарного кислорода, взаимная деполяризация атомарного водорода и гидро-
ксильного радикала [210] и обмен электроном [56]. Недавно этот вопрос про-
проанализирован Рокстро [211], пришедшим к выводу, что активной формой
платины является металл, который сам выполняет функции реакционного
центра, но не входит как компонент в схему окисления—восстановления:
Это положение согласуется со значительно ранее сделанным выводом Мак-
Иннеса [212], который отвергает теорию образования промежуточного окисла
и подчеркивает значение адсорбции. В этом отношении интересно отметить [ 190],
что перекись водорода восстанавливает двуокись платины, но не изменяет
моноокиси. По-видимому, можно считать доказанным, что наблюдаемая ско-
скорость разложения на активной поверхности платины определяется скоростью
диффузии перекиси водорода к поверхности катализатора [211, 213] (о коэффи-
коэффициенте диффузии см. также стр. 178). Указанный вопрос освещается также в ра-
работе Пенникуика [214] о природе коллоидной платины. Этот автор пришел
к заключению, что гидрофобный золь, образующийся в системе, содержащей
кислород, заряжен отрицательно, причем на поверхности частиц находятся
как металл, так и двуокись платины, а в лиосфере, окружающей частицы,
содержится гексаплатинатный ион. Кипячение или старение вызывает переход
гексаплатината в основную массу раствора, что ведет к коагуляции. В атмосфере
водорода или азота отсутствуют окись или платинат, и система стабилизируется
под действием распределения водородных и гидроксильных ионов. Это описа-
описание, очевидно, согласуется с выводом, что активностью обладает именно сам
металл. Поэтому золи, образовавшиеся в атмосфере азота или водорода, в не-
несколько раз более активны, чем золи, полученные в присутствии кислорода
[215], и помещение золя, образовавшегося в атмосфере кислорода, в атмосферу
с низким парциальным давлением кислорода увеличивает его активность [87,
216]* (хотя значительное увеличение давления кислорода не оказывает ника-
* В цитированных советских работах [87, 216] авторы применяли не золь, а глад-
гладкую или платинированную платину.—Прим. перев.
408
Глава 8
кого эффекта [217]). Выше уже отмечалась неэффективность повышенных доз
гексаплатината. С этой точкой зрения согласуется и вывод, что эффективность
таких веществ, как хлорид и цианид, в качестве ядов для катализатора обуслов-
обусловлена их способностью к образованию комплексов с платиной. Вероятно, катали-
каталитическая активность обусловлена тем, что сам металл отдает электрон перекиси
водорода.
Кобальт, родий, иридий
В металлическом состоянии кобальт проявляет специфическую пассив-
пассивность: при погружении его в перекись водорода на металле образуется окисная
пленка и) возникает слабое каталитическое разложение, но вслед за этим
иногда совершенно неожиданно происходит огромный рост активности. Однако
при истощении начального количества перекиси водорода и добавке нового
количества восстанавливается начальная малая скорость разложения. При
12
Интенсивное
разложение
Н202
77«
признаки
7Н11Я
стойкого
темно-
коричневого
осадка
Начальные растворы
125мл 0,36М CoSQ*
I без Н20г
I 10 мл 0,36 М CoSO*
ХгОмл Ю%-ной Hz0i
\ Стойкий
^осадок
Мутный ""
.раствор
Местное образование л
осадка, растворяющею
при перемешивании
20
60 100
Добавленный NaOH,
140
180
Рис. 69. Сравнение результатов титрования сульфата
двухвалентного кобальта щелочью в присутствии перекиси
водорода и в отсутствие ее.
качественном исследовании этого явления [218] не обнаружено никакой зави-
зависимости между внешним видом поверхности металла, электродным потенциалом,
рН, температурой или временем нахождения в перекиси, с одной стороны,
и началом быстрого катализа—с другой. По-видимому, высокая активность
возникает лишь в концентрированной перекиси водорода и связана со значи-
значительными изменениями электродного потенциала, обычно в сторону более
катодных значений.
Ион двухвалентного кобальта не обладает заметным каталитическим дей-
действием на растворы перекиси водорода, но при добавке щелочи выпадает гидро-
гидроокись трехвалентного кобальта и происходит разложение перекиси 1219].
Трайхорн и Джессоп [220] показали, что активность этого катализатора может
сильно колебаться и характер его действия еще не выяснен, хотя и сделаны
описанные ниже наблюдения [218]. На рис. 69 дано сравнение кривых титрова-
титрования раствора, содержащего ионы двухвалентного кобальта, едким натром в при-
присутствии перекиси водорода и в отсутствие ее (относительное смещение вдоль
абсциссы не имеет значения, так как в этих опытах с самого начала раствор
содержал некоторые количества кислоты). Результаты показывают, что в при-
присутствии перекиси водорода происходит непосредственное окисление кобальта
Разложение в жидкой фазе 409
до трехвалентного состояния и осаждение гидрата окиси кобальта до достиже-
достижения условий, в которых наблюдается осаждение гидрата закиси кобальта.
Другие опыты [218] показывают, что концентрация кобальта, требующаяся
для инициирования катализа, обратно пропорциональна концентрации щелочи,
причем критерием катализа является достижение произведения растворимости.
При концентрациях щелочи ниже примерно 6 н. весь осажденный кобальт
находится в трехвалентном состоянии, весь же кобальт в растворе—в двухва-
двухвалентном. В более щелочной среде происходит некоторое растворение трехвалент-
трехвалентного кобальта. Пирофосфат, карбонат, сульфид и арсенат в качестве ингиби-
ингибиторов этого катализа неэффективны, и ультрафиолетовый спектр поглощения
щелочных растворов, содержащих ион двухвалентного кобальта и гидрат
окиси кобальта, не изменяется при добавке перекиси водорода. Исследования
при помощи радиоактивных индикаторов [221] показали отсутствие обмена
между ионом закисного кобальта и гидратом окиси кобальта, безразлично
в присутствии или в отсутствие перекиси водорода. Эти факты, очевидно,
исключают возможность катализа по механизму окислительно-восстановитель-
окислительно-восстановительного цикла. Однако, возможно, что катализ происходит по свободнорадикаль-
ному механизму. Этот механизм предложен, между прочим, для объяснения
каталитического разложения озона [222] и гидроперекиси кумола [223] ко-
кобальтом. Далее, исследование [224] окисления воды до кислорода ионом окис-
ного кобальта показало, что эта реакция в состоянии вызвать полимеризацию
виниловых соединений; постулировано, что при этом образуются гидроксиль-
ные радикалы путем переноса электрона от гидроксильного иона к окисному
иону кобальта, причем последний, возможно, находится в растворе в виде ди-
мерного комплекса с водой. Оказывают каталитическое действие на перекись
водорода [225] и другие комплексы кобальта, например с аммиаком и цитратом.
Кобальт на носителе [184, 226] также обладает каталитическими свойствами.
Сообщается и о промотировании катализаторов разложения перекиси водорода
кобальтом [168, 227].
Слабо изучены с точки зрения каталитического действия на разложение
перекиси водорода родий и иридий. Имеется лишь краткое указание, что ро-
родиевая чернь более активна, чем массивный металл [188]. Более подробные
исследования, проведенные с золями иридия, показали, что разложение пере-
перекиси водорода на этих золях кинетически имеет первый порядок по концен-
концентрации перекиси [217, 228]. Действие иридия по виду весьма напоминает дей-
действие платины, например в отношении торможения этого катализа сульфидом,
ртутью или цианидом; однако, согласно исследованию Бросса [228], иридий
отличается от платины тем, что щелочь на катализ иридием не оказывает ника-
никакого действия, а кислота усиливает каталитическое разложение.
Железо, рутений, осмий
Гомогенный катализ разложения перекиси водорода в присутствии железа
изучен очень подробно и глубоко. В связи с этим имеется довольно подробное
описание этого процесса, хотя на многие вопросы еще не получено ответа. Ниже
рассматриваются следующие аспекты этого катализа: разложение перекиси
водорода с одновременным окислением иона закисного железа в окисное, меха-
механизм гомогенного разложения, вызываемого ионом трехвалентного железа,
торможение и усиление этого катализа, разложение под действием железа
в комплексной форме и гетерогенный катализ на железе. По этому вопросу
(кроме гетерогенного катализа), имеется ряд обзоров [73, 229—232]; из них
особенно можно рекомендовать сообщения Баксендейла [73], Медалиа и Кольт-
гофа [229] и Юри [230].
Начальные исследования в присутствии железа [233—239] касались как:
явления катализа с одновременным окислением иона закисного железа, так.
4 10 Глава 8
и катализа, вызываемого ионом окисного железа; однако первый вопрос при-
привлек к себе особое внимание, поскольку он был связан с окисляющим действием
смеси из ионов закисного железа и перекиси водорода (носящей название реак-
реактива Фентона) [229]. Ион закисного железа окисляется перекисью водорода
по следующей стехиометрической реакции:
2Fe2+ + Н2О2 + 2Н+ -*[2Fe3+ + 2Н2О. C1)
Эта реакция протекает количественно при избытке иона закисного железа,
но при избытке перекиси водорода наряду с окислением закисного железа про-
происходит также разложение перекиси водорода с выделением кислорода. В до-
дополнение к сказанному Маммери [234] показал, что при избытке перекиси
начальное выделение кислорода происходит с очень большой скоростью.
После того как преобладающей формой оказывается трехвалентное железо
и возникает катализ под действием этой формы железа, скорость разложения
сильно падает. Габер и Вейс [240] ввели для этого явления концепцию коэффи-
коэффициента расхода перекиси A(H2O2)/A(Fe2+). Очевидно, что величина этого коэф-
коэффициента должна составлять 0,5 в том случае, если реакция C1) протекает
количественно, если же наряду с окислением иона закисного железа происхо-
происходит каталитическое разложение перекиси водорода, то этот коэффициент воз-
возрастает. Относительно возможной предельной величины этого коэффициента
высказаны разные мнения; но в настоящее время в результате работ Барба,
Баксендейла, Джорджа и Харгрейва [161] можно считать установленным, что
этот коэффициент стремится к пределу, определяемому условиями реакции,
и не возрастает до таких больших величин, о которых сообщали указанные
выше авторы [240]. >
Выдвинуты различные механизмы для объяснения разложения перекиси
водорода, сопровождающегося окислением иона закисного железа. На осно-
основании спектроскопических исследований Бонсон и Робертсон [235] постули-
постулировали промежуточное образование иона феррата FeOJ". Однако наблюдавшая-
наблюдавшаяся ими окраска раствора в настоящее время приписывается наличию примесей.
Предполагалось также образование такого высшего окисла железа, как
Fe2O5 [237]. Существуют мнения и об образовании различных комплексов
железа и перекиси водорода. Брей и Горин [238] предложили механизм
с участием иона феррила FeO2+. Однако принятый в настоящее время меха-
механизм основан на возникновении в качестве промежуточных продуктов
свободных радикалов — гидроксила и пергидроксила. Впервые этот механизм
был предложен Габером и Вилыытеттером [241], а затем Габером и Вейсом
[240]. Выдвинутая теория имеет значение не только для установления данного
механизма катализа, но и для развития знаний в области понимания
реакционных механизмов вообще. Этот механизм связан со следующей
•схемой реакций:
Fe2+ + Н2О2 -^ Fe3+ + ОН + ОН", C2)
C3)
Н2О2 Ч ОН -> НО2 + Н2О, . C4)
Fea+ 4 НО2 -> Fe3+ + НО", C5)
НО2 -» Fe2+ + Н+ + О2, C6)
установленной работой [161] Барба, Баксендейла, Джорджа и Харгрейва,
* которой мы и отсылаем читателя за обширными экспериментальными
.доказательствами. Их схема отличается от схемы, выдвинутой вначале
Разложение в жидкой фазе 411
Габером и Вейсом [240], в том отношении, что место реакций C5) и C6),
по Габеру и Вейсу, занимает реакция
Н2О2 + НО2 -+ ОН + Н2О + О2. C7)
Можно видеть, что кислород образуется по реакции C6), причем степень
протекания ее обусловлена конкуренцией за взаимодействие с гидроксильным
радикалом между ионом закисного железа и перекисью водорода.
Аналогично протекает и каталитическое разложение перекиси водорода
при действии окисного железа [161], однако ряд взаимодействий в этом
случае начинается реакцией
Fe8+ + НО" -> Fe2+ + НО2, C8)
за которой следуют реакции C2), C4), C5) и C6). Эта схема согласуется
с кинетическим выражением
d (Н2О2) __ , (Fe*+) (HaOa)
38 * (Н*)
установленным для высоких отношений (H2O2)/(Fe3+). Объясняется
и отношение
d (Н2О2) __ и, (НаОаK/* (Fe3+)
dO -* [(н+) + /(Г '
наблюдаемое при очень малых относительных концентрациях, а также и
другие встречающиеся кинетические выражения для промежуточных относи-
относительных концентраций перекиси водорода. Этот механизм отличается и от
механизма, выдвинутого в свое время Габером и Вейсом [240]. Дальнейший
экспериментальный материал [242, 243] и обсуждение [244, 245] кинетики
и механизма содержится в работах Андерсена и Христиансена [242], Оната и
Партса [243], Вейса [244] и Абеля [245]. В связи с большим числом работ,
проведенных по инициированию полимеризации системой из железа и перекиси
водорода [246], вряд ли приходится сомневаться в том, что катализ происхо-
происходит с участием свободных радикалов. Тем не менее ряд вопросов еще не получил
ответа. Например, участие иона закисного железа в катализе при дейртвии
окисного железа нельзя считать твердо установленным [73], хотя и имеются
серьезные доводы [247] в пользу этого предположения. Доказано образование
комплекса [Fe(HO2)]2+ из иона окисного железа и иона пергидроксила [248],
но не ясно, в какой именно степени он непосредственно участвует в указанных
-схемах. В новейших обсуждениях механизма авторы склонны отводить замет-
заметную роль иону надперекиси Oj в качестве фактического реагента вместо радика-
радикала пергидроксила, из которого этот ион должен образоваться [249, 250]. Пожа-
Пожалуй, особого интереса заслуживает вновь защищаемая гипотеза о роли соеди-
соединений железа с высшей валентностью (в качестве промежуточных продуктов).
Медалиа и Кольтгоф [229] указывают, что феррильный ион Брея и Горина
обладает рядом свойств гидроксильного радикала и, следовательно, может
играть роль в указанных выше схемах. Кахил и Таубе [251] также считают, что
промежуточным продуктом может быть четырехвалентное железо; они указы-
указывают, что образование его с переносом двух электронов может протекать одно-
одновременно с переносом одного электрона, а поэтому в общем процессе принимают
участие как ион окисного железа, так и ион железа в состоянии более высокой
валентности. Исследования процесса окисления иона закисного железа пере-
перекисью водорода позволили определить значения предэкспоненциального мно-
множителя и энергии активации в уравнении Аррениуса [161, 252], а также теп-
теплоты реакции [253].
* /(—константа гидролиза иона окисного железа.
412 Глава 8
Изучено также влияние различных анионов, например нитрата [254]г
фторида [161, 250, 255], хлорида [161, 256], сульфата [256], цитрата [257],
фосфата [255, 258] и ацетата [255], на каталитическое разложение перекиси
водорода солями железа. Цитрат в различных концентрациях может вызывать
и активирование реакции и торможение ее, а хлорид (в зависимости от кон-
концентрации иона окисного железа) может играть роль ингибитора или быть
инертным. Другие ионы, например фторид, фосфат и ацетат, являются инги-
ингибиторами, так же как и ацетанилид [259]. На стр. 447 и 449 представлены
иллюстративные данные, показывающие влияние фосфата и станната.
Превосходными промоторами для этого катализа служат и другие добавки;
в этом отношении изучено влияние меди [161, 164], молибдена [260], смеси
молибдена и вольфрама [261] и одного вольфрама [262].
Железо в виде различных комплексов также обладает каталитическим дей-
действием, хотя реакции этих комплексов еще не настолько подробно исследова-
исследованы, чтобы можно было установить механизм. Такого рода катализ заслуживает,
однако, особого внимания вследствие связи его с катализом при действии фер-
фермента каталазы, т. е. катализатора, содержащего железо в комплексной форме.
При исследовании механизма катализа железом в виде закисного или окис-
окисного иона одновременно изучено разложение в присутствии комплексов с ди-
пиридилом и фенантролином [237, 239, 263]. При общем обсуждении процесса
катализа действием комплексов железа Баксендейл [73] указал, что во время
образования этих комплексов в присутствии перекиси водорода наблюдается
более быстрое каталитическое разложение, чем разложение, которое происходит
в присутствии одного трехвалентного железа или на следующей стадии, когда
действует только один комплекс. Сделано заключение, что и в этом случае
разложение происходит по свободнорадикальному механизму. Представляет
интерес изучение разложения перекиси водорода цианидными комплексами же-
железа [73,264], поскольку это разложение зависит от действия излучения. Кистя-
ковский [265] сделал наблюдение, что слабый каталитический эффект смеси
ферроцианида с феррицианидом существенно увеличивается под действием
света и что высокая скорость разложения сохраняется и после прекращения
освещения. Срикантан и Рао [266] считают, что в этом катализе принимает
участие комплекс, содержащий молекулу перекиси водорода, а результаты
подробного исследования, проведенного Л ал ом [267], доказали важное значе-
значение, которое имеет в механизме реакции водный комплекс, образующийся при
гидролизе цианида. Эта работа показывает также, что гидрат окиси железа не
может функционировать в этом катализе в качестве активной частицы. Изучена
действие нитропруссида [268] и ферроцианидов различных металлов [269].
Разложение перекиси водорода в присутствии гетерогенных железных ката-
катализаторов еще не настолько изучено, чтобы можно было в достаточной мере
выяснить его механизм. Гидрат окиси железа является активным катализато-
катализатором [236, 270]; на стр. 440 приведен пример действия этого катализатора при
малых соотношениях его и перекиси водорода, а также дано объяснение полу-
полученных результатов как коллоидного поверхностного эффекта. Другие авторы
предполагают, что истинным катализатором в этом случае является ион окис-
окисного железа, адсорбированный на коллоиде [271], или же что имеет место цикл
окисления—восстановления [272]. Проведено сопоставление эффективности
этого катализатора со степенью его кристалличности [273]. Каталитическая
активность окиси железа FeLO3, по-видимому, изучена лишь под углом зрения
влияния кристаллической структуры [236, 274]. Из других гетерогенных
железных катализаторов разложения перекиси водорода изучены доменный
шлак [275], железный колчедан [276] и шпинели [84]. Шваб, Рот, Гринтцос
и Мавракис [84] постулировали, что активность феррита магния обусловлена
присутствием ионов закисного железа в участках тетраэдрической решетки
вследствие несовершенств кристалла; феррит цинка, не содержащий ионов.
Разложение в жидкой фазе 413
закисного железа, лишен активности. Сообщается также о гетерогенном ката-
катализе в присутствии смешанных гидроокисей железа и меди [277] и железа на
носителе [239, 278].
Рутений как катализатор разложения перекиси водорода, по-видимому,
не исследован. Осмий в щелочном растворе является довольно активным ката-
катализатором. Фрицман [279] наблюдал катализ разложения разбавленной пере-
перекиси водорода уже при концентрациях четырехокиси осмия 10~в моля в 1 л.
При концентрации осмия в 1000 раз большей скорость разложения не зависит
от концентрации и соответствует первому порядку по концентрации пере-
перекиси водорода. При увеличении концентрации щелочи скорость проходит через
максимум. Нис, Реймонд и Ювинг [280] исследовали этот катализ при высоких
концентрациях щелочи | * *Q ч =400 в приборе, который допускал исключи-
исключительно быстрое перемешивание, и показали, что скорость разложения в усло-
условиях перемешивания пропорциональна квадрату концентрации четырехокиси
осмия. В присутствии уксусной или пропионовой кислоты каталитическое
разложение и окисление кислоты, ничтожно малые в отсутствие перекиси водо-
водорода, протекают, по-видимому, параллельно и независимо друг от друга [2811..
Марганец, технеций, рений
Катализ разложения перекиси водорода в присутствии марганца отличается
значительной активностью и многократно подвергался изучению. Растворен-
Растворенный марганец в двухвалентном состоянии сам не активен [282], но он может
функционировать как промотор других катализаторов, например меди [283].
Сообщалось также, что стехиометрическое восстановление перманганата
(см. стр. 339) может сопровождаться гомогенным катализом разложения
перекиси водорода. Однако эти работы были проведены при значительно более
высоких значениях рН, чем значения, применяемые при обычной методике
анализа. Фуэна [284] и Абель [285] приводят данные, доказывающие, что
катализ инициируется перманганатом; эти же данные приводятся также
в обзоре Баксендейла [73].
Катализ в присутствии перманганата объясняется под углом зрения обра-
образования промежуточных радикалов; такие механизмы предложены и для дру-
других реакций перманганата [286]. Однако хорошо известно, что при реакции
перманганата с перекисью водорода может образоваться двуокись марганца
[287], и не исключено, что в действительности именно эта двоукись функциони-
функционирует как катализатор. Кроме того, вычисления, проведенные авторами этой
книги с использованием литературных данных [284], показывают, что во всех
опытах Фуэна, в которых происходило полное разложение перекиси водорода,
произведение (Мп2+) (ОН"J превышает 10~14. Этот факт согласуется с рассматри-
рассматриваемым ниже механизмом катализа под действием двуокиси марганца. Некото-
Некоторые авторы [288] описали экспериментальные методы изучения необычайно
быстро протекающего восстановления перманганата с последующим катализом
двуокисью марганца.
Катализ в присутствии двуокиси марганца [96, 289], по-видимому, проис-
происходит при помощи окислительно-восстановительного цикла. Исследование
этого вопроса [282] показывает, что при добавке иона двухвалентного марганца
или перманганата к очень разбавленным растворам перекиси водорода разной
щелочности катализа не происходит до тех пор, пока не достигается произве-
произведение растворимости гидрата закиси марганца, равное примерно 10'14. Логарифм
величины произведения (Мп2+) (ОН J, которое должно быть превышено, преж-
прежде чем начнется катализ, представляет обратную линейную функцию концен-
концентрации перекиси водорода. Пирофосфат предотвращает этот катализ, причем
необходимая для этого концентрация пирофосфата пропорциональна концен-
414 Глава 8
трации иона двухвалентного марганца. Опыты, проведенные с применением
радиоактивных индикаторов, доказывают наличие обмена между ионом двух-
двухвалентного марганца и коллоидной двуокисью марганца в процессе разложе-
разложения, что позволяет постулировать следующий механизм реакции:
МпОг н- Н2О2 Н
Мпг+ + 2Н
Мп (ОН), +
-2Н*
Н2О2
-> Мпг* -t 2HaO +
> Mn(OHJ t 2Н%
-^ MnOa + 2H.0.
о„
C9)
D0),
D1)
Однако доказательства наличия обмена в отсутствие перекиси водорода [2911
спорны, и, поскольку эти опыты с изотопами включали операцию кипячения
растворов, их нельзя считать вполне убедительными [284, 290].
Металл, очевидно, катализирует разложение по тому же механизму.
Измерен потенциал, который принимает металлический марганец с пленкой
из двуокиси марганца в водных растворах с различным рН и различными кон-
концентрациями иона двухвалентного марганца и перекиси водорода [292]. Про-
Проведено сравнение полученных результатов измерения с аналогичными потен-
потенциалами для платины и с теоретическими потенциалами, выведенными на осно-
основании различных предложенных механизмов каталитического разложения
перекиси водорода. Наблюдения согласуются с гипотезой, что потенциал опре-
определяется реакцией восстановления двуокиси марганца до иона двухвалентного
марганца. Шольдер и Кольб [293], однако, предполагают, что в механизме
катализа могут играть роль пероксоманганаты. Прежде чем удастся удовлетво-
удовлетворительно решить вопрос об истинной природе катализатора, потребуется про-
провести дальнейшие работы. В частности, известно, что состав двуокиси марганца
редко в точности соответствует формуле МпО2 [294]; кроме того, сообщается
[294], что окисел, в точности соответствующий этой формуле, обладает лишь
слабым каталитическим действием. Активность различных форм двуокиси мар-
марганца [295], по-видимому, находится в определенном соотношении с их деполя-
деполяризующими свойствами [296]. Термическая обработка также влияет на актив-
активность двуокиси марганца как катализатора разложения перекиси водорода
[297]. Марганцовые катализаторы на носителе [298], по-видимому, представляют
системы, допускающие лучшее изучение, чем в случае с МпО2. Муи и Селвуд
[299] исследовали разложение перекиси водорода на окислах марганца на гли-
глиноземе и рутиле методом магнитной восприимчивости [93] и показали, что ак-
активность находится в определенной зависимости от степени окисления катали-
катализатора. Она становится максимальной, когда эта степень занимает промежуточ-
промежуточное положение между + 3 и + 4. В связи с этим предложен окислительно-вос-
окислительно-восстановительный цикл, в котором участвуют двуокись марганца и окись марган-
марганца (Мп2О3), причем приведены данные, доказывающие, что этот цикл более ве-
вероятен, чем приводимый в реакциях C9)—D1). Разбирался также вопрос о
сравнительной легкости возникновения зародышей и образования каталитиче-
каталитически активной твердой фазы. Это имеет значение для всех тех случаев, при кото-
которых гетерогенный катализатор образуется in situ. Согласно логичному предпо-
предположению, необходимость достижения произведения растворимости до начала
катализа свидетельствует о том, что именно образование твердой фазы пред-
представляет общую основу каталитического действия; например, в данном кон-
конкретном случае активным марганцовым катализатором является двуокись
марганца. Однако, хотя равновесие и благоприятствует образованию этой
твердой фазы, она не может возникнуть непосредственно из раствора; для этого
требуется предварительное осаждение гидрата закиси марганца в качестве
твердой фазы, на основе которой уже в дальнейшем может образоваться ста
бильное соединение марганца с более высокой валентностью. Муи и Селву
Разложение в жидкой фазе 415
указывают также, что подробный механизм катализа может включать и стадию
передачи электронов с образованием гидроксильных радикалов.
О возможном каталитическом действии технеция или рения нет никаких
данных.
Хром, молибден, вольфрам
Эти три элемента особенно интересны, так как гомогенное каталитическое
разложение перекиси водорода в их присутствии дает четкое доказательство
существования механизмов с участием пероксосоединений как промежуточ-
промежуточных продуктов. Данные по этому вопросу имеются в обзоре Баксендейла [73].
Качественная характеристика гомогенного катализа в присутствии хромата
описана многими авторами старых работ; подробные кинетические исследова-
исследования впервые проведены Ризенфельдом [300] и Шпитальским [301], а после
этого еще и рядом других авторов [302—304]. Активным является хром в ше-
шестивалентном состоянии (и как хромат CrOJ , и как бихромат Ст2О27')щ хотя
в кислом растворе перекись водорода вызывает известное восстановление ше-
шестивалентного хрома до трехвалентного Сг3*. В постулированных механизмах
принимается, что хромат реагирует с перекисью водорода с образованием пер-
оксохромата, который затем разлагается с выделением молекулярного кисло-
кислорода. Окончательно не решен вопрос о дальнейшем поведении хрома, т. е. о том,
восстанавливается ли он до иона Сг3+ и затем вновь окисляется до хромата [301 ]
или же хромат регенерируется непосредственно из пероксохромата [303,
304]. Однако можно с уверенностью принять наличие окислительно-восстанови-
окислительно-восстановительного цикла с участием пероксохромата. Описано явление промотирования
хромата кобальтом [227], марганцем [305, 306], молибденом [307] и медью,
железом и никелем [306]; ванадий тормозит катализ [308].
Как гетерогенный катализатор хром не очень активен. Металл оказывает
лишь слабое каталитическое действие, мало зависящее от рН, образования
окисла на поверхности или присутствия растворенного хрома. В концентри-
концентрированной перекиси водорода может происходить некоторое растворение металла;
возможно и избирательное растворение хрома из нержавеющей стали. Изучены
также коллоидная гидроокись хрома [309] и хром на носителе [299, 310,
311].
Гомогенный катализ в присутствии молибдена [312], по-видимому, проте-
протекает через образование и разложение пероксомолибдатов McOJ" и MoOg",
а возможно, также и МоО*~, которые образуются в растворе перекиси водо-
водорода из молибдата MoOf". Другие состояния валентности в реакции, вероятно,
не участвуют. Юри [154], а в особенности Богданов [260, 261, 307, 313, 314]
изучили промотирование молибдатного катализа другими металлами.
Вольфрамат—менее активный катализатор, чем молибдат (в кислоте он
практически становится инертным), но, по-Тзидимому, действует также путем
образования аналогичных пероксосоединений WO^" и WO*". Богдановым [315]
исследована кинетика этого катализа; изучено также промотирование этого
катализа [261, 262, 313, 316]. В одной небольшой работе рассматривается ката-
катализ разложения перекиси водорода вольфрамом в металлическом состоянии
или в виде карбида [317]. В таком состоянии каталитическая активность воль-
вольфрама быстро падает во времени.
Ванадий, ниобий, тантал
Ванадат описан как гомогенный катализатор разложения перекиси водо-
водорода, действующий посредством окислительно-восстановительного цикла с
участием различных пероксованадатов [308, 318, 319]. Ванадий на рутиле или
глиноземе как носителе не активен [299]. Металлический ниобий является
медленно действующим катализатором, тантал же вообще инертен [320].
416 Глава 8
Титан, цирконий и гафний
Нет никаких данных по вопросу о том, насколько активными катализато-
катализаторами разложения перекиси водорода являются титан, цирконий и гафний.
Бор, алюминий, скандий, иттрий
Борат образует с перекисью водорода равновесную смесь, содержащую
устойчивый пероксоборат. Инертность металлического алюминия и стабили-
стабилизирующие свойства его водной окиси рассмотрены на стр. 145 и 436. Опубли-
Опубликованы также и другие доказательства, свидетельствующие о некаталитиче-
некаталитической природе алюминия [141, 321]. Скандий и иттрий в качестве катализа-
катализаторов перекиси водорода, очевидно, не подвергались изучению.
Редкие земли, актиниды
Лемуан [141] указывает, что окислы церия и тория представляют умеренно
активные катализаторы разложения перекиси водорода. Других данных по
этой серии элементов не опубликовано.
Щелочноземельные металлы
Щелочноземельные металлы иногда включаются в список катализаторов
разложения перекиси водорода, однако логично предполагать, что эти элементы
не являются собственно катализаторами, а функционируют за счет щелочности
некоторых их растворов. В определенных условиях ион магния оказывает даже
стабилизирующее действие.
Щелочные металлы
Щелочные металлы в форме нейтральных соединений каталитического
действия на разложение перекиси водорода не оказывают; в виде металлов
они, конечно, бурно реагируют с ней.
Почвы, воды и различные агенты
Отмечено и изучено влияние различных почв [322], фуллеровой земли
C23], металлизированного бентонита [324], минеральных вод [325], морской
воды [326], синтетических моющих веществ [327] и сплавов [99] в качестве
катализаторов разложения перекиси водорода. Наиболее активными компо-
компонентами естественных вод являются, по-видимому, железо и хлорид.
Периодический катализ
У многих катализаторов разложения перекиси водорода обнаружено заме-
замечательное явление периодичности действия. Наиболее известными примерами
таких катализаторов могут служить йодат [126] и ртуть [156]; это же явление
отмечено и в случаях с металлическим железом и медью [328], а в монографии
Хеджеса и Майерса [329] приведены и другие примеры. Несомненно, что перио-
периодичность возникает тогда, когда физические процессы, протекающие во време-
времени, например диффузия, способствуют периодическим изменениям концентра-
концентрации какого-либо химического вещества, участвующего в катализе. Постулиро-
Постулировано, что это явление может быть вызвано попеременными процессами: пере-
пересыщением раствора кислородом и устранением этого пересыщения. Однако в слу-
случае йодатного катализа [126] доказано, что для возникновения периодичности
необходимо предварительно устранить пересыщение. При изучении разложе-
Разложение в жидкой фазе
417
ния совершенно чистой перекиси водорода [105] разницы между опытами со
встряхиванием и без встряхивания не обнаружено (если только в последнем
случае выжидали наступления равновесия с точки зрения пересыщения).
Случай, когда пересыщение действительно играет роль, можно наблюдать,
если пролить концентрированную перекись водорода на цементный пол. В та-
таких условиях по поверхности жидкости, подвергающейся активному разло-
разложению, иногда проходят периодические волны повышенной скорости выделе-
выделения кислорода»
Промотирование, торможение и отравление
Выше, при обсуждении каталитических характеристик различных элемен-
элементов, упоминалось о работах:, в которых доказывается промотирующее действие,
часто наблюдаемое при испытании смесей. Под промотированием подразуме-
подразумевается повышение скорости разложения по сравнению с суммой скоростей для
отдельных компонентов катализаторной смеси. Хорошо известным примером,
Ag-Cr
480
360
240
120
О 40
80 0 40 80 0 40 80 О
Си Ад Мл Ад Са Ад
Состав катализатора, вес. % промотора
40 80
Сг
Рис. 70. Промотирование разложения перекиси водорода в присут-
присутствии серебра как катализатора действием меди, марганца, кальция
и хрома (общий вес металла 0,0118 г в 10 мл 4 н. NaOH).
Общее количество 48%-ной Н2О2, добавленной на 1 г катализатора, г: О 3 000;
X 6000; А 9000; П И 000.
встречающимся при гомогенном катализе, является промотирование, наблюдае-
наблюдаемое в случае смесей железа и меди [161, 164]; на этом вопросе мы еще остановим-
остановимся (см. стр. 444). Беркман, Моррел и Эглофф (см. в работе [68] стр. 414) приводят
разные другие примеры промотирования и дают анализ сущности этого явле-
явления. Промотирование наблюдается не только при гомогенном катализе, но и в
случае совместно осажденных гидроокисей металлов. Для серебра это иллюстри-
иллюстрируется кривыми, изображенными на рис. 70. Эти данные получены по методу,
описанному раньше (стр. 396), и их можно сравнить с кривыми, показанными
на графике рис. 65. Исследования показывают, каким образом эффект промо-
промотирования изменяется в зависимости от разбавления и возраста катализатора
по мере разложения последовательных порций перекиси водорода. Интересно
отметить также заметное повышение скорости разложения в присутствии
гидрата окиси кальция, который сам оказывает сравнительно слабое каталити-
каталитическое действие. Инертный носитель также может выполнять функции про-
промотора; он может превратить инертный материал в катализатор или же может
активировать вещество в таком состоянии валентности, в котором оно обычно
неэффективно, например ион Ag+ [81]. Легкость проявления названного эф-
эффекта промотирования действием такого носителя, как стекло или кремнезем
[330], в опытах по изучению разложения может привести к ошибкам. О других
27 Перекись водорода
418 Глава 8
катализаторах на носителях уже упоминалось выше. Кинетика разложения,
наблюдаемая в случаях с катализаторами на носителях, часто является свое-
своеобразной [331].
В вышеприведенном обсуждении упоминалось о веществах, которые умень-
уменьшают или прерывают разложение перекиси водорода, т. е. об ингибиторах или
ядах. Такого рода вещества находят практическое применение для стабилиза-
стабилизации перекиси водорода (см. гл. 9). Известны самые различные вещества, дей-
действующие как яды; некоторые из них были упомянуты при рассмотрении от-
отдельных катализаторов. Большинство этих веществ является неорганическими
по своей природе, но и многие органические вещества представляют сильные
ингибиторы таких катализаторов, как серебро или платина [332]. Механизм
отравляющего действия часто остается неясным; так, иногда два вещества,
которые по отдельности являются катализаторами, в смеси взаимно тормозят
действие друг друга [333]. В старых работах [86, 334] имеются превосходные
качественные описания явления отравления катализаторов, но только в послед-
последнее время, основываясь на электронной структуре, мы приблизились к пони-
пониманию механизма отравления. Общий обзор по этому вопросу имеется у
Мэкстеда [335].
ОРГАНИЧЕСКИЕ И БИОЛОГИЧЕСКИЕ КАТАЛИЗАТОРЫ
Среди простых низкомолекулярных органических веществ нет истинных
катализаторов разложения перекиси водорода. По-видимому, единственными
органическими катализаторами разложения, которые могут быть легко синте-
синтезированы, являются фталоцианины [336]. Если не считать этих соединений,
то все изученные до настоящего времени органические катализаторы являются
веществами биологического происхождения; примером может служить кол-
коллоидный холестерин [337]. Это верно и для таких искусственных катализаторов
разложения, как смеси железа с альбумином [338] и гемин на активированном
угле как носителе [339], которые действуют наподобие ферментов. Хотя про-
проведено много наблюдений с недостаточно охарактеризованными биологиче-
биологическими катализаторами, например с бактериями [340], дрожжами [341], мор-
морскими водорослями [342] и кровью [343], наибольшее внимание привлек к себе
фермент каталаза.
Структура и биологическая функция каталазы в отношении перекиси водо-
водорода и других гемопротеиновых ферментов описаны на стр. 349 и ел. Название
этому ферменту дал в 1901 г. Лёв [344], который изучал влияние рН, раствори-
растворителей и солей на действие, производимое на перекись водорода сырыми экс-
экстрактами, содержащими каталазу. Последующие работы [345] установили
общую кинетику катализа разложения перекиси водорода и сопровождаю-
сопровождающую его инактивацию фермента. Позже показано [346], каким образом инак-
инактивация может быть устранена путем применения экспериментальных методов,
значительно превосходящих классическую технику. В настоящее время опубли-
опубликовано много данных по кинетике и механизму действия каталазы на перекись
водорода; сюда относятся работы Кейлина и Хартри [347], 'Чанса [348],
Джорджа [349], Сайзера и Бирса [350], Теорела и Эренберга [351] и других
авторов [352]. Описан также ряд ингибиторов этого катализа [353]. Все
явления и объясняющие их механизмы подробно рассмотрены в недавно
опубликованных обзорах Чанса [346, 354] и Джорджа [232]. В настоящее
время преобладают две точки зрения: согласно одной из них, катализ протекает
путем образования и разложения комплексных соединений из каталазы
и перекиси водорода (см. реакции D6) и D7) на стр. 352), в соответствии с другой
точкой зрения в реакции принимают участие также промежуточные свободные
радикалы. Последняя гипотеза была высказана сначала Габером и Вильште-
ттером [241], затем рассматривалась другими авторами [102, 355] и энергично
Литература 419
защищается Джорджем [232]. Интересен вывод [232], что высокая активность
разложения перекиси водорода каталазой эквивалентна той активности,
которая выводится путем экстраполирования действия ионного железа к усло-
условиям, фактически невозможным из-за осаждения его в щелочных растворах.
С другой стороны, предварительные сообщения [232] показывают, что система
катал аза—перекись водорода не вызывает полимеризации, в связи с чем
сейчас пользуется большим признанием механизм, основанный исключительно
на диссоциации комплекса [354].
ЛИТЕРАТУРА1
1. На user M., Вег., 56, 888 A923).
2. Hinshelwood С. N.. Р г i с h а г d С. R., J. Chem. Soc, 123, 2725 A923).
3. Е 1 d е г L. W., Jr., R i d e a 1 Е. К., Trans. Faraday Soc, 23, 545 A927); Elder
L. W., Jr., J. Am. Chem. Soc, 59, 2737 A937).
4. К i s t i a k о w s k у G. В., R о s e n b e г g S. L., J. Am. Chem. Soc, 59, 422
A937).
5. G i g u ё г e P. A., Can. J. Research, B25, 135 A947).
6. Baker В. Е., О u e 1 1 e t C, Can. J. Research, B23, 167 A945).
7. Кондратьева Е., Кондратьев В. Н., ЖФХ, 19, 178, A945).
8. M а с k e n z i e R.C., Ritchie M., Proc Roy. Soc. (London), A185, 207 A946).
9. M с L a n e С. К., J. Chem. Phys., 17, 379 A949).
10. S t e i n T. W., Sc D. Thesis in Chemical Engineering, Massachusetts Institute of
Technology, 1955.
11. Sa t t er f iel d С N.. Kavanagh G. M., ResnickH., Ind. Eng. Chem.,
43, 2507 A951).
12. Tamres-M., Frost A. A., J. Am. Chem. Soc, 72, 5340 A950).
13. С о о k G. А., пат. США 2368640, 2368806 F.11. 1945).
14. Harris E. J., Trans. Faraday Soc, 44, 764 A948).
15. M i z u w a t а г i E., N a g a i S., Repts. Chem. Research Inst., Kyoto Univ., 16,
29 A947) [C. A., 45, 8332d].
16. S u ; t о E., J Electrochem. Soc. Japan, 17, 156 A949) [C. A., 44, 5196h]. "
17. H а г t А. В., Nature, 163, 876 A949).
18. Sa t ter f i el d С N., С е с с о t t i P. J., Fel dbrugge A. H. R., Ind. Eng.
Chem., 47, 1040 A955).
19. P о й т е р В. А., Г а у к м а н С. С, ЖФХ, 4, 465 A933).
20. U г е у Н. С, D a w s e у L. H., Rice F. O., J. Am. Chem. Soc, 51, 1371 A929).
21. S a t t e r f i e 1 d C. N., R e s n i с k H., W e n t w о г t h P. L., Chem. Eng.
Progress, 50, 460, 504 A954).
22. S с h u m b W. С, неопубликованная работа MIT Hydrogen Peroxide Project, 1949.
23. A 1 1 m a n d A. J., S t у 1 e D., J. Chem. Soc (London), 1930, 596.
24. К о г n f e 1 d G., Z. Phys. Chem., B29, 205 A935).
25. Lea D. E., Trans. Faraday Soc, 45, 81 A949).
26. D a i n t о n F. S., R о w b о t t о m J., Trans. Faraday Soc, 49, 1160 A953).
27. Q u г e s h i M., Rahman M. K., J. Phys. Chem., 36, 664 A932).
28. Henri V., W u г m s e г R., Compt. rend., 156, 1012 A913); 157, 126, 284 A913).
29. H e i d t L. J., J. Am. Chem. Soc, 54, 2840 A932).
30. A n d e г s о n W. Т., Т а у 1 о г H. S., J. Am. Chem. Soc, 45, 650 A923).
31. Matthews J. H., С u г t i s H. A., J. Phys. Chem., 18, 166, 521 A914).
32. Hunt J. P., T a u b e H., J. Am. Chem. Soc, 74, 5999 A952).
33. Tian A., Compt. rend., 156, 1879A913).
34. R i с e F О., К i 1 p a t r i с k M. L., J. phys. Chem., 31, 1507 A927).
35. S t о n e F. S., T а у 1 о г H. S., J. Chem. Phys., 20, 1339 A952).
36. V о 1 m a n D. H., J. Chem. Phys., 17, 947 A949).
37. V о n Elbe G., J. Am. Chem. Soc, 54, 821 A932).
38. H a b e г F., Will statter R., Ber., 64, 2844 A931).
39. Weiss J., Trans. Faraday Soc, 31, 668 A935).
40. Кондратьев В. Н., Проблемы кинетики и катализа, Сборник трудов конфе-
конференции, посвященной 40-летию перекисной теории Баха—Энглера, 4, 63 A940);
Хим. реф. ж., 4, № 1, 22 A941).
41. Haber F., Weiss J., Proc. Roy. Soc (London), 147A, 332 A934).
42. Weiss J., Discussions Faraday Soc, № 12, 161 A952).
43. H а г t E. J., M a t h e s о n M. S., Discussions Faraday Soc, № 12, 169 A952).
44. F г i с k e H., J. Chem. Phys., 3, 364 A935); Symposia Quant. Biol., 3, 55 A935).
45. JohnsonE.R., J. Chem. Phys., 19, 1204 A951).
27*
420 Глава 8
46. D a i n t о n F. S.( Rowbottom J., Nature, 168, 370 A952).
47. R isse O., Z. physik. Chem., 140A, 133 A929).
48. Kail an A., Z. phys. Chem., 95, 215 A920); 98, 474 A921); Monatshefte fur Chemie,
32, 1019 A911); 33, 1329 A912); 44, 35 A923).
49. Lefort M., Compt. rend., 233, 1194 A951).
50. A 1 1 e n A. OM Hochanadel C. J., G h о г m 1 e у J. A., D a v i s T. W.,
J. Phys. Chem., 56, 575 A952).
51. H о p w о о d F. L., Brit. J. Radiol., 13, 221 A940).
52. E b e г t M., В о a g J. W., Discussions Faraday Soc, № 12, 189 A952).
53. Weiss J., Annual Review of Physical Chemistry, 4, 143 A953); см. также даль-
дальнейшие выпуски этой серии.
54. В е с q u е г е 1 Е., Ann. Chim. phys. C), 11, 179 A844); Baudrimont A.t
Compt. rend., 62, 829 A866); S с h 6 n e E., Ann. Chem., 197, 137 A879) [J. Chem
Soc., 36, 878, A879I; В e r t h el о t M., Compt. rend., 95, 8 A882) [J. Chera.
Soc, 42, 1157 A882)]; H a n г i о t M., Compt. rend., 100, 172 A885).
55. Та на тар С, Ber., 36, 199 A903) [J. Chem. Soc, 84, 202 A903)].
56. Weiss J., Trans. Faraday Soc, 31, 1547 A935).
57. Albareda уНеггега J. M., Rev. acad. cienc. Madrid, 24, 514 A929) [C. A.,
25, 4801].
58. A z z a m A. M., Bockris J. O'M., Conway B. E., Rosenberg H.,
Trans. Faraday Soc, 46, 918 A950).
59. H i с k 1 i n g A., Wilson W. H., J. Electrochem. Soc, 98, 425 A951).
60. Rius А., О con Garcia J., Anales ffs. у quim. (Madrid), 41, 38 A945) [C. A.,
43, 5677e].
61. Красильщиков А. И., Волчкова Л. М., Антонова Л. Г., ЖФХ,
27, 512 A953).
62. Kolthoff I. M., Lingane J. J., Polarography, Interscience Publishers,
New York, 1952 [есть также перевод: см. Кольтгоф И. М., Л ингейн
Дж. Дж., Полярография, Госхимиздат, М.—Л., 19481, Del a hay P., New
Instrumental Methods in Electrochemistry, Interscience Publishers, Inc., New York,
1954.
63. И о ф а 3. А., Ш и м ш е л е в и ч Я. В., Андреева Е. П., ЖФХ, 23, 828
A949); Б а го ц к и й В. С, Я б л о к о в а И. Е., там же, 27, 1663 A953); Van
Rysselberghe P., Murdock G. A., Chem. Eng. News, 31, 2702 A953);
Green J. H., Australian J. Chem., 7, 197 A954).
64. Kolthoff I. M., Jordan J., J. Am. Chem. Soc, 74, 570, 4801 A952);
Anal. Chem., 24, 1071 A952).
65. Strnad F., Collection Czech. Chem. Commun., II, 391 A939); Kolthoff
I. M., Parry E. P., J. Am. Chem. Soc, 73,3718 A951);, D e 1 a h а у Р.,
S t i e h 1 G. L., J. Am. Chem. Soc, 74, 3500 A952).
66. Л а т и м е р В. М., Окислительные состояния элементов и их потенциалы в вод-
водных растворах, Издатинлит, М., 1954.
67. S с h w a b и. М., Taylor H. S., Spence R., Catalysis from the Standpoint
of Chemical Kinetics, D. Van Nostrand, New York, 1937.
68. Беркман С.Моррелл Д., Э г л о ф ф Г., Катализ в неорганической и ор-
органической химии, Гостоптехиздат, М.—Л., 1949.
69. Eramett P. H., ed., Catalysis, Vol. 1, Fundamental Principles (Part 1), Reinhold
Publishing Corp., New York,. 1954, см. также след. тома.
70. Kendall J. К., F u с h s F. J., J. Am. Chem. Soc, 43, 2017 A921).
71. R о b e r t s о n A., Waters W. A., Trans. Faraday Soc, 42, 201 A946); Ива-
Иванов К. И., С а в и н о в а В. К., Михайлова Е. Г., ДАН СССР, 25, 34,
40 A939); Cook A. H., J. Chem. Soc, 1938, 1774.
72. Santesson С. G., Skand. Arch. Physiol., 42, 129 A922) [C. A., 16, 2871];
Lamp V. A., H a r i n g M. M., J. Chem. Education, 17, 577 A940); M c-
K i n n e у CD., Jr., К i 1 p a t r i с k M., Rev. Sci. Instruments, 22, 590 A951);
For m wal t J. M., пат. США 2658819 A9.XI.1953).
73. Baxendale J. H., Advances in Catalysis, Vol. 4, Academic Press, Inc., New
York, 1952, p. 31.
74. Haissinsky M., J. chim. phys., 44, 181 A947).
75. M с Al p i n e R. K., J. Chem. Education, 23, 301 A946).
76. Abel E., Z. anorg. allgem. Chem., 263, 229 A950).
77. Rice F. O., R e i f f О. М., J. Phys. Chem., 31, 1353 A927).
78. L i v i n g s t о n R., J. Phys. Chem., 47, 260 A943).
79. О г r R. J., Williams H. L., J. Am. Chem. Soc, 77, 3715 A955).
80. H e r s h e n s о n H. M., R о g e r s L. В., Anal. Chem., 24, 219 A952).
81. W e у 1 W. A., Trans. N. Y. Acad. Sci., 12, 245 A950).
82. S e i t z F., The Modern Theory of Solids, McGraw-Hill Book Co., New York, 1940.
83. Shockley W., Electrons and Holes in Semiconductors, D. Van Nostrand Co.,
Inc., New York, 1950; American Scientist, 42, 41 A954).
Литература 421
84. Go mer R., .Smith С. S., eds., Structure and Properties of Solid Surfaces, Univ.
of Chicago Press, Chicago, 1953.
85. Do wd en D. A., Research (London), 1, 239 A948); J. Chem. Soc, 1950, 242.
86. В г e d i g G., Von Berneck M., Z. physik. Chem., 31, 258 A899); В г e-
dig G., Anorganische Fermente, Engelmann W., Leipzig, 1901.
87. P о й t e p B. A., Bicri Ihct. физ. хем. АН УРСР, 2, 42 A929).
88. N е i 1 s о п С. Н., В г о w n О. Н., Am. J. Physiol., 10, 225, 335 A904).
89. П и с а р ж е в с к и й Л. В., Г л и к м а н Ф. С, Acta Physicochim., URSS, 6,
575 A937).
90. С ы р к и н Я- К., Г о д н е в И. Н., ЖФХ, 5, 32 A934); Писаржевский
Л. В., К о р а б е л ь н и к Р. К., Рынская Е. С, Изв. АН СССР, отд. мате-
мат. и естеств. наук, 1934, 931; Acta Physicochimica URSS, 7, 261 A937); W a-
t a na be A., Iwata Inst. Plant Biochem., Pub., 2, 129 A936) [C. A., 30, 6650];
Красновский А. А., Гуревич Т. Н., ДАН СССР, 75, 715 A950).
91. С о 1 1 i n s S., В г у с е W. A., J. Chem. Phys., 18, 1297 A950).
92. М с С а г t п е у J. Т., S е 1 i g m а п В., Hall W. К., An der son R. В.,
J. Phys. and Colloid Chem., 54, 505 A950).
93. S el woo d P. W., Advances in Catalysis, 3, 28 A951).
94. Schwab G. M., Trans. Faraday Soc, 42, 689 A946).
95. Madinaveitia A., Aguirreche F. D., Ann. soc. espafi. fis. у quim. B),
19, 124 A921) JC. A., 16, 7].
96. P а г k e г W. G., Cohen M., Smith P., неопубликованная работа Royal
Aircraft Establishment, 1948.
97. W г i g h t W. M., R i d e a 1 E. K., Trans. Faraday Soc, 24, 530 A928).
98. Кобозев Н. И., Acta Physicochim. URSS, 21, 469 A946).
Ш. Капустинский А. Ф., Шмелев Б. А., Изв. АН СССР, отд. хим. наук,
1940, 617; Шушунов В. А., Ф е д я к о в а К- Г., ЖФХ, 23, 936 A949).
100. Dowden D., Reynolds P., Discussions Faraday Soc, № 8, 184 A950).
101. Schwab G. M., Chimica e industria, 35, 810 A953) [C. A., 48, 4948d].
102. Leach S. J., Advarrces in Enzymology, 15, 1 A954).
103. К a s 11 e J. H., С 1 а г k e M. E., Am. Chem. J., 26, 518 A901).
104. BroughtonD. B.,MaukeD. M., Laing M. E.,'Wentworth R. L.,
неопубликованная работа MIT Hydrogen Peroxide Project, 1946.
105. Roth E. M., S h a n 1 e у Е. S., Ind. Eng. Chem., 45, 2343 A953).
106. Hatcher W. H., MacLauchlan D. W., Can. J. Res., I6B, 253 A938).
107. Forbes G. S., J. Chem. Education, 27, 402 A950).
108. Doyle J. R., неопубликованная работа M.I.T Hydrogen Peroxide Project, 1951,
109. В u г k i F., Schaaf F., Helv. Chim. Acta, 4, 418 A921).
110. SI a ter V. W., Chemistry and Industry, 1945, 42.
111. Pierron P., Compt. rend., 222, 1107 A946); Do г felt C, Monatschr. Textil-
Ind., 50, 37, 67, 90, 117 A935) [C. A., 29, 5278]; E г d e у L., Acta Chim. Acad.
Sci. Hung., 3, 95 A953) [C. A., 47, 10399h].
112. S i m о n A., M а г с h a n d M., Z. anorg. Chem., 262, 192 A950).
113. Pana C, Trans. Faraday Soc, 24, 486 A928); Phragmen G., Medd. Kgl.
Vetenskaps akad. Nobelinst, 22, 1 A919).
114. Abel E., Monatsh., 83, 422 A952).
115. Mohammed A., Liebhafsky H. A., J. Am. Chem. Soc, 56, 1680 A934).
116. Edwards J. O., J. Phys. Chem., 56, 279 A952).
117. Bray W. C, Chem. Revs., 10, 161 A932).
118. T h i e J., J. Phys. and Colloid Chem., 51, 540 A947); S k r a b al A., Monatsh.,
81, 239 A950).
119. Weiss J., Ann. Repts., 44, 60 A947); Abel E., Monatsch., 79, 178 A949).
120. Szwarc M., L. Farkas Memorial Volume, Research Council of Israel, Jerusalem
A952), p. 144.
121. M a a s s O., H i e b e r-1 P. G., J. Am. Chem. Soc, 46, 290 A924); Living-
Livingston R. S., Bray W. C, ibid., 47, 2069 A925); 48, 405 A926); Budge E. A.,
ibid., 54, 1769 A932); M a k о w e г В., Bray W. С, ibid., 55, 4765 A933);
M a k о w e г В., ibid., 56, 1315 A934); С о n n i с k R. E., ibid., 69, 1509 A947);
Кобозев Н. И., Краснопольская В. Н., ЖФХ, 21, 277 A947);
С к о п и н ц е в Б. А., ДАН СССР, 68, 869, 1053 A949).
122. Livingston R. S., J. Am. Chem. Soc, 48, 53 A926); 54, 2393 A932); Bray
W. C, L i v i ngs to n R. S., ibid., 45, 1251,2048 A923); 50, 1654 A928); Bray
W. C, Davis P. R., ibid., 62, 1427 A930); Bray W. C, Young H. A.,
ibid., 54, 4284 A932); Bobtelsky M., Z. anorgan. Chem., 206, 161 A932);
Mohammed A., Liebhafsky H. A.,J. Am- Chem. Soc, 56, 1680 A934);
Griffith R. О., М с К е о w n A., ibid., 58, 2555 A936); Young H. A.,
ibid., 72, 3310 A950).
123. Magnanini G., Gazz. chim. ital., 19, 476 A889); 21, 476 A891); N о у е s A. A.,
Z. physik. Chem., 18, 119 A895); 19, 601 A896); В г о d e J., ibid., 49, 208 A904);
422 Глава 8
Walton J. H., ibid., 47, 185 A904); В r e d i g G., ibid., 48, 368 A904); В г е-
dig G., Walton J. H., Jr., Z. Elektrochem., 9, 114 A903); A b e 1 E., Z.
Elektrochem., 14, 598 A908); Z. physik. Chem., 96, 1 A920); Monatsh., 41,
405 A920); Z. physik. Chem., 136, 161 A928); BohnsonL.,J. Phys. Chem., 24,
677 A920); qh г i s t i a n s e n J. A., Z. physik. Chem., 117, 433 A925); 128, 430
A927); G о о d i n g R.U., Walton J. H., J. Phys. Chem., 35, 3612 A931);
Bray W. C./L i e b h a f s к у H. A., J. Am. Chem. Soc, 53, 38 A931); L i e b-
h a f s к у H. A., ibid., 54, 1792, 3499 A932); 56, 2369 A934); H e n d e г W.C.K.,
Robinson R. A., Trans. Faraday Soc, 29, 1300 A933); О к a b e K., J. Chem.
Soc. Japan., 61, 1235 A940) [C. A., 35, 2060]; G а г g a 1 1 о М. A., Anales asocn.
quim. Argentina, 36, 150^A948) [C. A., 43, 2121П; Morgan K. J., Quarterly
Reviews, 8, 123 A954).
124. H a r n e d H. S., J. Am. Chem. Soc, 40, 1461 A918); L i e b h a f s к у Н. А.,
J. Phys. Chem., 38, 857 A934); Bell F., Gill R., H о 1 d e n D., Wynne-
Jones W. F. K., J. Phys. Chem., 55, 874 A951).
125. Thomas A. W., Co h e n В., J. Am. Chem. Soc, 61, 401 A939); Cohen В.,
ibid., 64, 1340 A942).
126. В г а у J,W. С, С a u 1 к i n s A. L., J. Am. Chem. Soc, 43, 1262 A921); 53, 44 A931);
W i el a n d H., Fischer F.G., Ber., 59, 1171 A926); Bray W. C, Lieb-
hafsky H. A., J. Am. Chem. Soc, 53, 38 A931); L i e b h a f s к у Н. А.,
ibid., 53, 896, 2074 A931); Abel E., Monatsh., 80, 122 A949); Abel E.,
Z i f f e г J., Monatsh., 80, 585 A949); P e a r d M. G., С u 1 1 i s С F., Trans.
Faraday Soc, 47, 616 A951).
127. Livingston R., S с h о e 1 d E. A., J. Am. Chem. Soc, 58, 1244 A936); Grif-
Griffith R. О., М с К е о w n A., J. Am. Chem. Soc, 58, 2555 A936); Callow
A. E., G г i f f i t h R. О., М с К е о w n A., Trans. Faraday Soc, 35, 412 A939);
Bennett J. G., Griffith R. O., ibid., 44, 471 A948).
128. Edwards J. О., частное сообщение, 1954.
129. W о r s 1 e у R. R., В а к е г Н. В., J. Chem. Soc, 123, 2870 A923).
130. Bonet-Maury P., LefortM., Compt. rend., 22Б, 173 A948).
131. Levi G. R., Ghiron D., Atti Acad. Lincei, 21, 454 A935).
132. Fl о r e s с о N., Bull, fac Stiinte Cernauti, 2, 308 A928) [C. A., 26, 5253].
133. Hasebroek K., Ber., 20, 213 A887); Bamberger M., Trautzl K..
Z. anal.^Chem., 64, 9 A924).
134. Q u а г t а г о 1 i A., Gazz. chim. Hal., 54, 713 A924) [C. A., 19, 1524].
135. Lemoine G., Compt. rend., 144, 357 A907) [Am. Sci. Suppl., 82, 235 A916)];
H о n i g P., Kolloid-Beihefte, 22, 345 A926); MezzadroliG., Varenton
E., Ann. chim. appl., 19, 415 A929) [C. A., 24, 922]; S h i v о n e n V., Suomen
Kemistilehti, 10B, 25 A937) [C. A., 32, 417]; К e e g e 1 J. F., S u г u d a W. A.,
S с h w о b C, J. Am. Chem. Soc, 60, 2483 A938); T i s i г о I., Mitt. med. Akad.
Kioto, 27, 808 A939) [C. A., 35, 1809]; А с h a r у a H. K., J. Indian Chem. Soc,
18, N° 1, 15 A941); К г a u s e A., Przemysl Chem., 29, 377 A950) [C. A., 46, 6916e].
136. Fowler D.( Walton J. H., Rec trav. chim., 54, 476 A935); К о л а р о в Н.,
Известия на Химический институт Българска Академия на науките, 1, 185 A951).
137. ПисаржевскийЛ. В., Корабел ьник Р. К., Рынская Е. С,
Изв. АН СССР, отд. математ. и естеств. наук, 7, 931 A934).
138. Skumburdis К., Kolloid-Z., 55, 156 A931); King A., J. Chem. Soc, 1936,
1688; L а г s e n E. C, W a 1 t о n J. H., J. Phys. Chem., 44, 70 A940).
139. Firth J. В., W a t s о n F. S., J. Chem. Soc, 123, 1750 A923); J. Soc Chem. Ind.,
42, 371T A923); Trans. Faraday Soc, 19, 601 A924); 20, 370 A924); BenteP.F.,
Watson J. H., J. Phys. Chem., 47, 329 A943); Watanabe J., Shiramo-
to Т., J. Electrochem. Soc. Japan, 19, 274 A951); 20, 135, 386 A952).
140. В г i n k m a n n G., Kolloid-Z., 123, 116 A951); Angew. Chem., 61, 378 A949).
141. Lemoine G., Compt. rend., 162, 702 A916).
142. Fells H. A., Firth J. В., Proc Roy. Soc, Al 14, 517 A927).
143. Wright W. M., Trans. Faraday Soc, 24, 539 A928).
144. P e n n e r S. S., J. Am. Chem. Soc, 74, 2754 A952).
145. Gawalowski A., Rundschau fur Pharmacie u. Chemie, 4, 79 A894); Z о t i -
er V., Bull, soc chim., 13, 61 A913); 15, 402 A914); 21, 241 A917).
146. R u d e 1 H.W.,Haring M. M., Ind. Eng. Chem., 22, 1234 A930).
147. В г о u g h t о n D. В., W e n t w о г t h R. L., L a i n g M. E., M a u k e D. M.,
неопубликованная работа M.I.Т. Hydrogen Peroxide Project, 1946.
148. Von Hevesy G., Phys. Z., 16, 52 A915).
149. Z i n 11 E., R а а с h A., Ber., 57B, 1739 A924).
150. Rius A., Knecht J. A., Anales Fis. у quim., 43, 367 A947) [C. A., 41,
679a].
151. P i о n t e 1 1 i I. R., Canonica L., M о n t e g g i a G., Gazz. chim. itai., 78,
599 A948).
152. M a s t er so n S., Peers С H., Willows R., англ. пат. 672860 B8. V
Литература 423
1952); Жабр ов а Г. М., Р о г и н с к и й С. 3., Фокина Е. А., ДАН
СССР, 52, 313 A946).
153. Doyle J. R., неопубликованная работа M.I.T. Hydrogen Peroxide Project, 1950.
154. U г i N.. J. Phys. and Colloid Chem., 53, 1070 A949).
155. G о о d i n g R. U., Walton J. H., J. Phys. Chem., 35, 3612 A931).
156. В г e d i g G., W e i n m а у е г J., Z. physik. Chem., 42, 601 A903); F г е d e n-
hagenC.Z. Elektrochem., 11, 859 A905); В г е d i g.G., vonAntropoffA.,
ibid., 12, 585 A906); В r e d i g G., Biochem. Z., 6, 283 A907); von Antro-
p о f f A., Z. physik. Chem., 62, 513 A908); J. Prakt. Chem., B), 77, 273 A908);
В г e d i g G., W i 1 k e E., Biochem. Z., 11, 67 A908); L e m о i n e G., Compt.
rend., 162, 580 A916); Hedges E. S., M у е г s J. E., J. Chem. Soc, 125, 1282
A924); В e t h e A., Z. Naturforsch., 3b, 69 A948) [C. A., 43, 927a].
157. Scott E. A., Sorenson M. O.,S. В., Thesis in Chemical Engineering, M.I.Т., 1948.
158. Bredig G., Stark A., Z. physik. Chem., B2, 282 A929).
159. Ernst K. H., Pflugers Arch. ges. Physiol., 254, 79 A951) [C. A., 47, 10978i].
160. von Kiss A., Lederer E., Rec. trav. chim., 46, 453 A927).
161. Barb W. G., В a x e n d a 1 e J. H., George P., Hargrave K. R.,
Trans. Faraday Soc, 47, 462, 591 A951).
162. Schuler W., Meier R., Helv. Physiol. Pharmacol, 2, 83 A944) [C. A., 39,
2337].
163. В о b t e 1 s k у M., Kirson В., Compt. rend., 199, 573 A934); Kirson В.,
Bull. soc. chim., France, 1952, 957; 1953, 404; 1954, 157, 950; НиколаевЛ.А.,
Кобозев Н. И., ЖФХ, 19, 323 A945); Н и к о л а е в Л. А., Вестник МГУ № 2,
105 A946); № 6, 115 A947); 3, № 10, 159 A948); «Методы изучения катализаторов»
в сб. ориг. и перев. статей «Проблемы кинетики и катализа», № 5, Химтеорет, Л.,
1948, стр. 297; Gl a sner A., J. Chem. Soc, 1951, 904; Erkut H., Rev. f ас.
sci. univ. Istanbul, ISA, 153 A953) [C. A., 48, 31cl.
164. В о h n s о n V. L., R о b e г t s о n A. C, J. Am. Chem. Soc, 45, 2512 A923); 47,
1299 A925); Robertson A. C, ibid., 47, 1300 A925); Wai ton J. H.,Chr i-
s t i a n s e n, ibid., 48, 2083 A926); Шпитальский Е., Коновало-
Коновалова Б. А. ЖРФХО, 62, 1033 A930); Quartaroli A., Del С а г г a t о г е A.,
Ann. chim. applicata, 39, 527 A949) [C. A., 45, 4121 g]; Andersen V. S.,
Acta Chem. Scand., 6, 1090 A952).
165. Abel E., Monatsh., 81, 965 A950).
166. Коновалова Б. А., Монахова 3. Д., Ангарская М. Н., П е т и н
Н. П., ЖФХ, 10, 313 A937); Б о г д а н о в Г. А., П е т и н Н. Н., ЖФХ, 12,
598, 607 A942).
167. В о b t е 1 s k у М., В о b t е 1 s к у-С h a i к i n L., Compt. rend., 201, 604 A935).
168. Зворыкин А. Я., Перельман Ф. М., ЖФХ, 20, 1095 A946).
169. von El isafoff G., Z. Elektrochem., 21, 352 A915); Quartaroli A.,
Gazz. chim. ital., 55, 264 A925); 61, 466 A931); Ann. chim. appl., 24, 70 A934);
Хармандарьян М. О., Алексеева Е. А., ЖРФХО, 62, 1677 A930);
Kohlshutter V., Helv. Chim. Acta, 14, 1215 A931); Bailey К. С, Sci.
Proc Roy. Dublin Soc, 21, 153 A935) [C. A. 29, 2830); Holmes J.W., Tay-
Taylor E. H., J. Am. Chem. Soc, 63, 2906, 2911 A941); Heinz W., Z. anorg. allgem.
Chem., 247, 240 A941); Б у б е н Н. Я-, Ф р а н к-К а м е н е ц к и й Д. А., ЖФХ,
20, 225 A946).
170. v о n Euler H., Jansson В., Monatsh., 53—54, 1014 A929).
171. Шпитальский Е. И., Петин Н. Н., Коновалова Б. А., ЖРФХО,
60, 1237 A928).
172. Bert helot №., Ann. chim. phys., E), 21, 164 A880); Baeyer A., V i 1-
1 i g e г V., Ber., 34, 749 2769 A901); Mulder E., Rec, chim. Pays-Bas, 22,
388 A903); M a nc hot W., Ber., 42, 3948 A909); L e m о i n e G., Compt.
rend., 155, 9, 15 A912); Kleinstuck M., Ber., 51, 108 A918); Salkow-
ski E., J. prakt. Chem. B), 102, 194 A921); Fodor A., Kolloid-Z., 39, 173
A926); Quartaroli A., Gazz. chim. ital., 57, 234 A927); Wright W. M.,
R i d e a 1 E. K., Trans. Faraday Soc, 24, 530 A928); W i 1 k e E., G a n s e r H.,
Kolloid-Z., 70, 132 A935); В a 1 а г е v D., Kolloid-Z., 101, 47 A942), 105, 26A943);
G a 1 e с k i A., Bull. soc. amis sci, Poznan, ser. B, Sci. math et nat., № 10, 87
A949) [C. A., 46, 319Ы; Mills G. F., пат. США 2515609 A8. VII. 1950); К г а-
u s е А., О 1 e g n i k G., Roczniki Chem., 27, 17 A953) [C. A., 48, 5624g].
173. Wiegel E., Z. physik. Chem., A143, 881 A929); Kolloid-Z., 47, 323 A929); 51,
112A930).
174. Mclntosh D., J. Phys. Chem., 6, 15 A902).
175. Wise W. Т., G a d e b u s с h P., T a n g J. C, S. B. Theses in Chemical Engine-
Engineering, M.I.Т., 1948 and 1949; W e n t w о г t h R. L., неопубликованная работа
M.I.Т. Hydrogen Peroxide Project, 1951.
176. J о h nso n H. L.,C u t a F., G а г г e t t А. В., J. Am. Chem. Soc, 55, 2311 A933);
177. N о у e s А. А., К о s s i a k о f f A., J. Am. Chem. Soc, 57, 1238 A935).
424 Глава 8
178. Yost D. M., J. Am. Chem. Soc, 48, 152 A926); О к а с A., Yecera Z., Chem.
Listy, 40, 211 A946) [C. A., 44, 9294h); Barbieri G. A., Malaguti A.,
Atti accad. nazl. Lincei, Rend, classe sci. fis, mat. e nat., 8, 619 A950) [С. А., 45„
55e].
179. Van i no L., Seemann L., Ber., 32, 1968 A899).
180. В г e d i g G., Reinders W., Z. physik. Chem., 37, 323 A901).
181. Galecki A., Bincer K., Bull, intern, acad. polon sci., 1925A, 93 A925)
[C. A., 20, 27721; G'al eckiA., Krzeczkovskal., ibid., Ill; G a I e с к i A.,
Jerke G., Roczniki Chem., 7, 1 A927) [C. A., 22, 1265].
182. Rusznyak S., Z. physik. Chem., 85, 681 A913); T e Ji e т о в И. С, Браун-
штейн Н. А., Труды 1нституту хем. Харк. держ. унив., 3, 7 A938); Т е л е-
то в И. С, Г р и ц а н Д. Н., Записки Харьковского сельскохозяйственного
института, 2, № 1—2, 47 A939); Бюллетень Всесоюзного химического общества
им. Менделеева, 1940, № 8, 22; Труды Института химии Харьковского Гос. уни-
университета, 5, 235 A940).
183. Watson G., Chem. News., 46, 9 A882); V е i 1 SM Compt. rend., 180, 932 A925);
Greene G. U., Trans. Electrochem. Soc, 76, 391 A939).
184. Николаев Л.А.Добозев Н. И., ЖФХ, 20, 145 A946).
185. Bred ig G., Fort пег М., Вег., 37, 798 A904).
186. De G. Rocasolano A., Compt. rend., 171,301A920).
187. Зелинский Н. Д., Б о р и с о в П. П., Вег., 56В, 396 A923).
188. Жуков И. И., Глаголева А. А., С т р у к о в а В. И., ЖОХ, 4, 9 A934).
189. В г е d i g G., T e 1 e t о v J., Z. Elektrochem., 12, 581 A906); S a n о I., Bull.
Chem. Soc. Japan, 13, 118 A938) [C. A., 32, 4865]; HasegavaS., Rev. Phys.
Chem. Japan, Shinkichi Horiba Commen. Vol., 1946, 21 [C. A., 41, 4698П; Bias-
с h k о Н., Biochem Z., 175, 68 A926).
190. S i e v e r t s A., Bruning H., Z. anorg. allgem. Chem., 204, 291 A932).
191. Dyer G., Dale А. В., Proc. Chem. Soc. London, 29, 55 A913); S u i t о Е.„
Rev. Phys. Chem. Japan, 15, 155 A941) [C. A., 45, 937el.
192. Tartar H. V., Schaffer N. K., J. Am. Chem. Soc, 50. 2604 A928).
193. Damianovich H., Nicola O.F.F., Anales asocn. quim. Argentina, 17,
142 A929) [C. A., 24, 1017].
194. Maxted E. В., М о о п С. Н., J. Chem. Soc, 1935, 393.
195. S u i t о E., Rev. Phys. Chem. Japan, 13, 74 A939) [C. A., 33, 91091; 15, 1 A941)
[C. A., 35, 6503]; Николаева М. И., Ш л ы г и н А. И., ЖФХ, 24, 427,
. 534 A950).
196. Lebeden A., Bull. Soc. chim. France, 3, 56 A907); Шпитальский E.^
Каган M., Ber., 59, 2900 A926); Ройте р В. А., Л е п е р с о н М. Г.,
BicTi Ihct. физ. хем. АН УРСР, 4, 41, 49 A934).
197. Писаржевский Л., Чрекашвили С., Савченко Г., Acta Physi-
cochim. URSS, 7, 289 A937); ЖФХ, 10, 534 A937).
198. Grube G., Z. Elekctrochem., 16, 621 A910).
199. L e v i G. R., Atti Accad. Lincei, 8, 409 A928).
200. Maxted E. В., J. Chem. Soc, 121, 1760 A922); 1934, 26; M a x t e d E. В.,
Ball G. Т., J. Chem. Soc, 1953, 1509.
201. S u i t о E., Rev. Phys. Chem. Japan, 18, 96 A944) [C. A., 41, 4698П; 1946, 35 [C. A.,
44, 4319].
202. О 1 i v e г i-M a n d a 1 a E., Gazz. chim. Ital., 59, 699 A929); 60, 878 A930).
203. В г e d i g G., I k e d a K., Z. physik. Chem., 37, 1 A901).
204. В 1 e s a A. A., Rev. acad. cienc. exactas, fis.-quim. у nat. Zaragoza, Ser 2A, 3, № 2y
39 A948) [C. A., 44, 6712]; BA), 4, 61 A949).
205. I г e d a 1 e Т., J. Chem. Soc, 119, 109 A921); de G. Rocasolano A., Nacm\
Ges. Wiss. Gottingen, 1924, 177 [C. A., 20, 1348].
206. Heath M. A., Walton J. H., J. Phys. Chem., 37, 977 A933).
207. N e i 1 s о n С. Н.., Brown О. Н., Am. J. Physiol., 10, 225 A904); 12, 374 A904).
208. Maxted E. В., Lewis G. J., J. Chem. Soc, 1933, 502.
209. Kubokawa M., Rev. Phys. Chem. Japan, 11, 202 A937) [C. A,, 32, 2817).
210. Wolff R., Compt. rend., 196, 1113 A933).
211. R о с k s t г о h R. K., S. M. Thesis in Chemical Engineering, M.I.Т., 1951.
212. M а с 1 n n e s D. A., J. Am. Chem. Soc, 36, 878 A914).
213. Телетов И., ЖРФХО, 39, 1145 A908); Senter G., Z. physik Chem., 52,
737 A905).
214. P e n n у с u i с k S. W., J. Am. Chem. Soc, 52, 4621 A930); 61, 2234 A939).
215. SuitoE., Rev. Phys. Chem. Japan, 16, 1 A942) [C. A., 44, 10474c]; T h о г е n F.,
Svensk Kem. Tids., 42, 134 A930) [C. A., 24, 5585].
216. P о й т е р В. А., Ш а ф р а н И. Г., ЖФХ, 4, 461 A933).
217. Spear E. В., J. Am. Chem. Soc, 30, 195 A908).
218. Broughton D. В., W e n t w о r t h R. L, Farnsworth M. L., неопубли-
неопубликованная работа M.I.Т. Hydrogen Peroxide Project, 1947.
Литература 425
219. В а у 1 е у Т., Phil. Mag., E), 7, 126 A879).
220. Т г у h о г n F. G., J e s s о р G., J. Chem. Soc, 127, 1320 A925).
221. В г о u g h t о n D. В., W e n t w о г t h R. L., F а г n s w о г t h M. E., J. Am.
Chem. Soc, 71, 2346 A949).
222. Hill G. R., J. Am. Chem. Soc, 70, 1306 A948); 71, 2434 A949).
223. К h а г a s с h M. S., Fono A., N u d e n b e г g W., В i s с h о f В., J. Org.
Chem., 15, 763 A950).
224. В a w n С E. H., W h i t e A. G., J. Chem. Soc, 1961, 331.
225. S h i b a t a Y., К a n e к о Н., J. Chem. Soc, Japan, 44, 166 A923) [С. А., 17„
2812]; В о b t e 1 s к у M., R a p p о р о г t M., Compt. rend., 205, 234 A937);
В о b t e 1 s к у M., S i m с h e n A. E., J. Am. Chem. Soc, 64, 2492 A942).
226. К г a u s e A., W о 1 s к i W., Roczniki Chem., 27, 321 A953) [C. A., 48, 6050g].
227. Robertson A. C, J. Am. Chem. Soc, 48, 2072 A926).
228. В г о s s a G. A., Z. physik. Chem., 66, 162 A909).
229. Medalia A. I., К о 1 t h о f f I. M., J. Polymer Sci., 4, 377 A949).
230. Uri N., Chem. Revs., 50, 375 A952).
231. Uri N., L. Farkas Memorial Volume, Research Council of Israel, Jerusalem, 1952,
p. 193; Weiss J., Advances in Catalysis, Vol. 4, Academic Press, New York,
1952, p. 343; L. Farkas Memorial Volume, Research Council of Israel, Jerusalem,
1952, p. 233.
232. George P., Advances in Catalysis, 4, 367 A952).
233. В г о d e J., Z. physik. Chem., 37, 257 A901); M a n с h о t W., W i 1 h e 1 m s O.,
Ann. Chem., 325, 105 A903); Ma nc ho t W., Pflaum W., Z. anorg. Chem.,
211, 1 A933); D u с 1 a u x J., Bull, soc chim. France, 31, 961 A922); von В е r-
t a 1 a n J., Z. physik. Chem., 95, 328 A920); P о i z a t J., Bull. soc. chim. Fran-
France, D), 33, 1606 A923); G о а г d A. K., R i d e a 1 E. K., Proc Roy. Soc, A106,
148 A924); L i m a n о w s к i W., Roczniki Chem., 12, 782 A932) [C. A., 27,
5626).
234. Mummery С S., J. Soc. Chem. Ind., 32, 889 A913).
235. В о h n s о n V. L., J. Phys. Chem., 25, 19 A921); В о h n s о n V. L., R о b e г t-
s о n A. C, J. Am. Chem. Soc, 45, 2493 A923).
236. Ш п и т а л ь с к и й Е. И., П е т и н Н. Н., Z. phys. Chem., ИЗ, 161A924); Шпи-
т а л ь с к и й Е. И., П е т и н Н. Н., Б у р о в а Е. И., ЖРФХО, 60, 1271 A928).
237. Ma nc hot W., Lehmann G., Ann. Chem., 460, 179 A928).
238. Bray W. C, G о г i n M. H., J. Am. Chem. Soc, 54, 2124 A932).
239. К u h n R., Wassermann A., Ann. Chem., 503, 203 A933).
240. Haber F., Weiss J., Naturwissenschaften, 20, 948 A932); Proc Roy. Soc,
A147, 332 A934).
241. Haber F., Wills tatter R., Ber., 64, 2844 A931).
242. Andersen V. S., Acta Chem. Scand., 2, 1 A948); 4, 914 A950); Christian-
Christiansen J. A., A n d e г s e n V. S., ibid., 4, 1538 A950); 5, 1406 A951); 6, 1056 A952).
243. Parts A. G., Nature, 168, 79 A951); О n a t E., Parts A. G., Australian J.
Sci., 15, 30A952); Communs. fac. sci. univ. Ankara, 4B, 17 A952) [C. A., 48, 7410c].
244. Weiss, J., Discussions Faraday Soc, № 2, 212 A947); Experientia, 7, 135 A951).
245. Abel E., Monatsh., 79, 457 A948); 80, 776 A949); 81, 685 A950); 82, 1104 A950);
84, 416, 419 A953).
246. В a x e n d a 1 e J.H., Evans M. G., Park G. S., Trans. Faraday Soc, 42,
155 A946); Evans M. G., J. Chem. Soc, 1947, 266; Evans A. G., Tyr-
ral E., J. Polymer Sci., 2, 387 A947); M a r v e 1 C.S., Dean in R., Cla-
u s С J., W у 1 d M. В., S e i t z R. L., J. Polymer Sci., 3, 350 A948); Abel
E., Monatsh., 80, 186 A949); 83, 841 A952).
247. К о 1 t h о f f I. M., Parry E. P., J. Am. Chem. Soc, 73, 3718 A951).
248. Evans M. G., George P., Uri N., Trans. Faraday Soc, 45, 230 A949).
249. George P., Discussions Faraday Soc, № 2, 196 A947); Barb W. G., Baxen-
dale J. H., George P., Har grave K. R., Nature, 163, 692 A949).
250. Weiss J., HumphreyC. W., Nature, 163, 691 A949).
251. С a h i 1 1 A. E., T a u b e H., J. Am. Chem. Soc, 74, 2312 A952).
252. В а у s al В., Communs. faculte sci. univ. Ankara, 3, 17 A950) [C. A., 46, 3845h];
Taylor W., W e i s s J., J. Chem. Phys., 21, 1419 A953); R i g g Т., Tay-
Taylor W., Weiss J., ibid., 22, 575 A954); Experientia, 10, 202 A954).
253. Evans M. G., В a x e n d a 1 e J. H., U r i N.n Trans. Faraday Soc, 45, 236-
A949).
254. P e t e r s 0 n S., J. Am. Chem. Soc, 73, 3521 A951).
255. Simon A., H a u f e W., R e e t z Т., Р г e i s s 1 e r R., Z. anorg. allgenu
Chem., 230, 129 A936).
256. von Kiss A., Led ere r E., Z. physik. Chem., 129, 186 A927).
257. Bobtelsky M. M., К i г s о n В., Compt. rend., 208, 1577 A939).
258. M а л к о в А. М., Ц в е т к о в а Н., Biochem. Z., 246, 191 A932).
259. Bray W. С, PetersonS., J. Am. Chem. Soc, 72, 1401 A950).
426 Глава 8
260. Богданов Г. А., П е т и н Н. Н., ЖОХ, 12, 369 A942); Б о г д а н о в Г. А.,
ЖФХ, 21, 439 A947).
261. Богданов Г. А., ЖФХ, 21, 51 A947).
262. К о н о в а л о в а Б. A., Z. anorg. allgem. Chem., 222, 82 A935); ЖФХ, 6, 704 A935).
263. Simon A., H a u f e W., Z. anorg. allgem. Chem., 230, 148 A936).
264. Q u i n с к e J., Z. anal. Chem., 31, 1 A892); W e i g e г t F., Ann. Physik, D),
24, 261 A907); Winther C, Danske Videnskabernes Selskab. Math. fys. Med-
delelsar, 2, 1 A920) [C. A., 14, 3377]; К г a u s e A., Z i el i n s к i S., Roczniki
Chem., 27, 327 A953) [C. A., 48, 6050d].
265. К i s t i а к о w s к i W., Z. physik. Chem., 35, 431 A900).
266. S г i к a n t a n B. S., Rao A. R., J. Indian Chem. Soc, 16, 299 A933).
267. L a 1 В. В., J. Indian Chem. Soc, 16, 321 A939); L a 1 В. В., S i n g h a 1 СР.,
Current Sci., 13, 78 A944); L a 1 В. В., Current Sci. (India), 16, 118 A947);
16, 219 A947); 16, 281 A947); Proc. Indian Acad. Sci., 38A, 522 A953) [C. A.,
48, 7441c].
268.YAmann J., Kolloid-Z., 8, 11 A911); Q u e г e s h i M., J. Phys. Chem., 35, 656
A931); Lai В. В., Proc. Indian Acad. Sci., 14A, 652 A941).
269. E г к u t H., Rev. fac. sci. univ. Istanbul, 19C, 1 A954) [C. A., 48, 12529i].
270. D u с 1 a u x J., J. chim phys., 20, 18 A923); Телетов И. С, Алексеева
Е. А., Украш. хем. журнал, 6, научная часть, 75 A931); Rot ini О. Т.,
Snassel F., Magyar Chem. Folyoirat, 39, 65 A933) [С. А., 28, 3975]; Kohl-
schutter H. W./iSiecke H., Z. Elektrochem., 41, 851 A935); Kapi-
t a n с z у k H., Roczniki Chem., 18, 261 A938) [C. A., 33, 3283]; P 1 о е n E. A.,
Mindermann W. A., S. B. Thesis in Chemical Engineering, M.I.T., 1947
and 1948.
271. Ермоленко Н., Новикова E., Z. anorg. allgem. Chemie, 225, 333 A935).
272. Kepfer R. J., Walton J. H., J. Phys. Chem., 35, 557 A931).
273. KrauseA., KaniowskaD., Ber., 69B, 1982 A936).
274. Krause A., Krzyzansky S., Z. anorg. allgem. Chem., 227, 417 A936);
Кг a use A., Krach H., Ber., 69B, 2708 A936); В a u d i s с h O., Ber.,
70B, 218 A937); F r i с k e R., Wiedmann H., Kolloid-Z., 89, 178 A939);
KleraraW, ed., Fiat Review of German Science, 1939—1946, part VI, Dieterich'-
sche Verlag, Wiesbaden, 1949, p. 246; Веселовский В. И., ЖФХ, 23,
1095 A949); К г a u s е А., Ар pelt К., Kotkowski S., Przemysl Chera.,
5 B8), 351 A949) [С. А., 45, 10019а]; Stumpf К. Е., Z. anorg. u. allgem. Chem.,
270, 114 A952).
275. N е 1 1 е n s t е у n F. J., S t e f f e 1 а а г G. M. A., Chem. Weekblad, 36, 438 A939).
276. Schwab G. M., Karatzas A., Kolloid-Z., 106, 128 A944).
277. Krause A., Kwintkiewiczowna L., Grochowska A.,
Trzeciakowski Z., Ber., 72B, 161 A939); Krause S., Kotkowski S.,
Karolewicz S., Przemysl. Chem., 6 B9), 25 A950) [C. A., 45, 10021c|.
278. Блох О., Кобозев Н. И., Acta physicochim. USSR, 5, 417 A936); Кобо-
Кобозев H. И., 3 у б о в и ч И. А., ДАН СССР, 52, 131 A946); Krause A., Rocz-
nikiJChem., 26, 3 A952) [С. А., 46, 11026а]; Krause A., Kotkowski S.,
DzialoszynskaB., Przemysl Chem., 9, 277 A953).
279. Fritzmann E., Z. anorg. allgem. Chem., 172, 213 A931).
280. N e a s С C, Raymond M. W., E w i n g CO., неопубликованная работа
M.I.Т. Hydrogen Peroxide Project, 1946.
281. Foster L. M., Payne J. H., J. Am. Chem. Soc, 63, 223 A941).
282. В г о u g h t о n D. В., W e n t w о r t h R. L., J. Am. Chem. Soc, 69, 741 A947).
283. В о b t e 1 s k у М., В о b t e 1 s k y-C h a j k i n L., Compt. rend., 201, 604
• A935).
284. F о u i n a t F., Compt. rend., 226, 1619 A948); 228, 1593 A949); Fouinat F.,
M a g a t M., J. chim. Phys., 47, 514 A950); Fouinat-Walch F., ibid., 50,
527 A953).
285. Abel E., Monatsh., 80, 455 A949); 81, 681 A950); 82, 642 A951).
286. Duke F. R., J. Am. Chem. Soc, 70, 3975 A948); S у m о n s M. C. R., J. Chem.
Soc, 1953, 3956.
287. Bowman M. I., J. Chem. Education, 26, 103 A949).
288. Bellinger F., Friedman H. В., Bauer W. H., Eastes J. W.,
L a d d J. H., Ross J. E., Ind. Eng. Chem., 38, 160 A946); N e a s С. С, Sc.
D. Thesis in Chemical Engineering, M.I.Т., 1947; McKinney C. D., К i 1 p a t-
rick M., Rev. Sci. Insts., 22, 590 A951); L a p о r t e M., Bull. soc. chim. Fran-
France, 1950, 178; Le Vide, 6, 927 A951) [C. A., 45, 5539f].
289. Bredig G., Marck A., van Bemmelen Festschrift, Polytechnikum, Zurich,
1910, p. 342 [CZ, 1911, II, 1140]; Lottermoser L., Lehmann R., Kol-
Kolloid-Z., 29, 250 A921); АльтшулерО. В., Коновалова Б. А., Пе-
Пети н Н. Н., ЖФХ, 13, 907 A939).
290. Н a i s s i n s k у М., J. chim. Phys., 47, 957 A950).
Литература 427
291. Р о 1 i s s а г М. J., J. Am. Chem. Soc, 58, 1372 A936); L i b b у W. F., J. Am.
Chem. Soc, 62, 1930 A940); H a 'i s s i n s к у M., Pullman В., J. phys.
Radium, 8, 36 A947).
292. BroughtonD.B., Wentworth R. L, LaingJM. E., J. Am. Chem. Soc,
69, 744 A947).
293. S с h о 1 d e r I. R., К о 1 b A., Z. anorg. Chem., 260, 41 A949).
294. В г e n e t J., В r i о t A. M., Compt. rend., 232, 726 A951).
295. W a d s 1 e у A. D., Walkley A., Rev. Pure Applied Chem. (Australia), 1, 203
A951).
296. A m i e 1 J., R о d i e г G., В г e n e t J., Compt. rend., 227, 1356 A948); Bull.
soc. chim. France, 1949, D507.
297. В al a rev D., Kolloid-Z., 99,73A942).
298. von E 1 i s s a f о f f G., Z. Elektrochem., 21, 352 A915); D u v a 1 C, Lecom-
t e J., Compt. rend., 234, 2445 A952); К г a u s e A., Bull, soc amis. sci. et let-
tres Poznan. Ser. В., № 12, 43 A953) [С. А., 47, 7875e].
299. M о о i J., S e 1 w о о d P. W., J. Am. Chem. Soc, 72, 4333 A950); 74, 1750 A952).
-300. R i e s e n f e 1 d E. H., W о h 1 e г s H. E., К u t s с h W. A., Ber., 38, 1886
A905); R i e s e n f e 1 d E. H., Ber., 38, 3578 A905); 41, 2826, 2832 A908); Z. anorg
Chem., 74, 48 A912).
301. Шпитальский Е., Z. anorg. Chem., 53, 184 A907); 56, 72, A907); 69, 179
A910); Ber., 43, 3187 A910); Шпитальский Е., Кобозев Н. И., Z. phys.
Chem., 127, 129 A927).
302. Орлов Е., ЖРФХО, 44, 1576, 1598, 1623 A912); R i u s A., Helv. chim. Acta,
3, 347 A920); Abel E., Monatsh., 81, 955 A950); Rynasiewicz J., Anal.
Chem., 26, 355 A954).
303. Bo b t el s k у M., Glasner А., Bobtelsky-Chajkin L., J. Am.
Chem. Soc, 67, 966 A945).
304. Кобозев Н. И., Г а л ь б р а й к Е. Е., ЖФХ, 14, 1550 A940); Acta Physicochim
URSS, 20, 479 A945).
305. Robertson A. C, J. Am. Chem. Soc, 49, 1630 A927).
306. В о b t el s k у М., В о b t e 1 s k y-C h a j k i n L., Compt. rend., 203, 1158A936).
307. Богданов Г. А., ЖФХ, 26, 766 A952).
308. Robertson А. С, Ргос Nat. Acad. Sci., 13, 192 A927).
309. Do wd L. E., S. B. Thesis in Chemical Engineering, M.I.Т., 1946.
310. Vol tz S. E., Wei 1 er S. W., J. Am. Chem. Soc, 76, 1586, A954).
311. H и ко л а ев Л. А., К о б о з е в Н. И., ЖФХ, 19, 562 A945).
312. Шпитальский Е., Функ A., Z. phys. Chem., 126, 1 A927); Abel E.,
Z. Elektrochem., 18, 545, 705A912); J a h г К. F., Вег/Ges. Freunden tech. Hoch-
schule Berlin, 1939, 91 [C. A., 36, 6934]; Кобозев Н. И., С о к о л о в Н. Н.,
Z. anorg. allgem. Chemie, 214, 321 A933).
313. Богданов Г. А., П е т и н Н. Н., ЖОХ, 12, 381 A942); Богданов Г. А.,
ЖФХ, 25, 323 A951).
314. Богданов Г. А., ЖФХ, 25, 332 A951); Богданов Г. А., П е т и н Н. Н.,
ЖОХ, 12, 598 A942).
315. Богданов Г. А., ЖОХ, 17, 887 A947); ЖФХ, 24, 1450 A950); 25, 49 A951).
316. Bobtelsky M., В о b t e 1 s k y-C h a j k i n L., Compt. rend., 203, 872
A936); Коновалова Б. А., Монахова 3. Д., Ангарская М. Н.,
П е т и н Н. Н., ЖФХ, 10, 313 A937); Богданов Г. А., Беркенгейм
Т. И., ЖФХ, 25, 1313 A951).
317. Van Liempt J. A. M., Kolloid-Z., 32, 118 A923); Lottermoser A.,
Eichler W., Z. Elektrochem., 35, 610 A929); Креймер Г. А., В а х о в-
c к а я М. Р., С а ф о н о в а О. С, Б о г и н о Е. Е., Зав. лаб., 15, 159 A949).
318. Belt r an Martinez J., Trujillo R., Anales real soc espau. tts. у qufm.,
45B, 719 A949) [C. A., 44, 5752d].
319. J a h г К. F., Z. Elektrochem., 47, 810 A941).
320. N e e s H., P. B. report 74717 (frame 203).
321. Lemoine G., Compt. rend., 162, 702 A916); Кг a use A., Roczniki Chem.,
28, 3 A954) [C. A., 48, 9168b].
322. Ермоленко Н. D., Новикова Е. Н., Гутерман В. Я-, Белорусск.
АН, Инст. хим., Сб. прац., I, 113 A934); Gunther W., Z. Pflanzenernahr,
Dungung Bodenk., 34A, 30 A934); Scharrer К., Forschungsdienst, 1, 824
A936); Mi su H., J. Agr. Chem., Soc, Japan, 17, 521 A941) [C. A., 41, 4598f].
-323. R i d eal E. K., Thomas W., J. Chem. Soc, 121, 2119 A922); Fi n dl а у
A., Thomas W., J. Chem. Soc, 125, 1244 A924).
-324. В rough ton G., J. Phys. Chem., 44, 180 A940); JohnsonR. L,S. B.
Thesis in Chemical Engineering, M.I.Т., 1937.
325. Fresenius L., Fichler A., LedererH., Z. anorg. Chem., 160, 273
A927); Mo n gego t A., Au ber tot V., Compt. rend, biol., 98, 905 A928);
Retovsky R., Bui. soc chim biol., 18, 1106 A936); Okabe K., J. Chem.
428 Глава 8
Soc Japan, 62, 300, 843 A941) [С. А., 41, 5655h]; 63, 27, 1025 1030 A942); 4, 351
A943) [С. А., 41, 3555i]; С г о п е i m G., J. Phys. Chem., 46, 328 A941); Frank
M., Hidrol. Kozlony, 30, 416 A950); [C. A., 45, 8680e];J N a g a s a w a S.,
J. Japan Chem., 6, 484 A952) [C. A., 48, 13381h).
326. Matsudaira C.'Tohoku J. Agr. Research., 1, 177 A950) [C. A., 46, 8487g];
3, 135 A952) [C. A., 47, 9739g].
327. U hi O., Fette u. Seifen, 51, 64 A944).
328. Marshall W. A., Metal Finishing, 50, 78 (XI, 1952); 67, (XII, 1952).
329. Hedges E. S., Myers J. E., The Problem of Physico-Chemical Periodicity,
Longmans-Green, Inc., New York, 1926.
330. Wright W. M., Z. Elektrochem., 34, 298 A928); Jacobson H. W., Smith
G. W., J. Am. Chem. Soc, 76, 4494 A954).
331. С ы р к и н Я. К., Васильев В. Г., ДАН СССР, 1, 513 A935); Кобозев
Н. И., Р е ш е т о в с к а я Н. А., ЖФХ, 23, 388 A949).
332. Н a n z I i k P. J., Cutting W. С, Science, 98, 389 A943).
333. К г a u s e A., R о с z n i к i Chem., 27, 9 A953) [С. А., 48, 5624h].
334. Lovenhart A. S., К a s 11 е J. H., Am. Chem. J., 29, 397, 563 A903).
335. M a x t e d E. В., Adwances in Catalysis, 3, 129 A951).
336. Cook A. H., J. Chem. Soc, 1938, 1761; С a h i 1 1 A. E., T a u b e H., J. Am.
Chem. Soc.,^73, 2847 A951).
337. Ремезов И., Сепалова О., Biochem. Z., 266, 330 A933); Ремезов И.,
Вег., 67В., 134 A934).
338. Rohman R., Shmamine Т., Biochem. Z., 42, 235 A912).
339. Кобозев Н. И., Николаев Л. А., Зубович И. С., Гольдфельд
Ю. М., ЖФХ, 19, 48 A945).
340. Fouassier M., Compt. rend., 170, 145 A920); AussenB. S., Kingman
Boltjes T. Y., Wostmann B. S. J., Antonie van Leeuwenhoek, J. Micro-
biol. Serol., 16, 65 A950); P r e v о t A. R., T h о u v e n о t H., Ann. inst. Paste-
Pasteur, 83, 443 A952).
341. von Euler H., Blix R., Z. physiol. Chem., 105, 83 A919);
342. Emerson R., Green L., Plant. Physiol., 12, 537 A937). French C.S,,
J. Gen. Physiol., 18, 209 A934).
343. В 1 e у е г L., Biochem Z., 161, 91 A925).
344. Loew O., U. S., Department of Agriculture, Rpt. № 68, Government Printing.
Office, Washington, 1901.
345. Morgulis S., J. Biol. Chem., 47, 341 A921); Northrop J. H., J. Gen.
Physiol., 7, 373 A925); Williams J., J. Gen. Physiol., 11, 309 A927); H a 1-
d a n e J. B. S., Proc Roy. Soc, B108, 559 A931).
346. Chance В., Trans. 4th Conf. Biological Oxidants, Josiah Macy, Jr. Foundation,
New York, 1950, p. 54, 86.
347. Keilin D., Наг tree E. F., Proc. Roy. Soc. London, B121, 173 A936);
B124, 397 A938); Nature, 144, 787 A939); 152, 626 A943); Biochem. J., 39,
148, 293 A945).
348. Chance В., Acta. Chem. Scand., 1, 236 A947); Nature, 161, 914 A948); J. Biol.
Chem., 179, 1299,1311,1341A949); 180, 865, 947 A949); 182, 643, 649 A950); 194,
471 A952); Biochem. J., 46, 387, 402 A950); Rev. Sci. Instr., 22, 619 A951).
349. George P., Nature, 160, 41 A947); Biochem. J., 43, 287 A948); 44, 197 A949).
350. S i z e г I. W., J. Biol. Chem., 154, 461 A944); Beers R. F., Jr., S i z e г I. W.»
J. Biol. Chem., 195, 133 A952); J. Phys. Chem., 57, 290 A953); Beers R. F.,
J. Phys. Chem., 58, 197 A954).
351. Theorell H., Ehrenberg A., Arkiv Fysik, 3, 299 A951); Arch. Biochem.
Biophys., 41, 442, 462 A952).
352. Кобозев Н. И., Acta Physicochim. URSS, 21,469 A946); LembergR.,
Foulkes E. C, Nature, 161, 131 A948); Sv end sen A., Acta Chem. Scand.,
7, 545 A953); Koutecky J., Brdicka R., Hanus V., Collection Czech.
Chem. Communs., 18, 611 A952).
353. A 1 у e a H. N., Pace J., J. Am. Chem. Soc, 55, 4801 A933); F r a n k e W.,
S i e w e г d t-K i b a t D., Z. physiol Chem., 281, 162 A944); Kreke С W.,
В а г 11 e t t M. D., S u t e г М. A., Nature, 154, 268 A944); Hoffmann-
O s t e n h о f О., В i а с h E., GiererS.,' Experientia, 3, 108 A947); Monatsh.,
76, 319 A947); Gol dacre P. L., Australian J. Sci. Research, B4, 293 A951).
354. Chance В., Advances in Enzymology, 12, 153 A951).
355. M о e 1 w у n-H u g h e s E. A., Ergebnisse Enzymforsch., 6, 23 A937); Weiss J.,
J. Phys. Chem., 41, 1107 A937); Wei ss J., Weil-Mai her be H., Nature,
144, 866 A939); M а с k i n n о n D. J., W a t e r s W. A., J. Chem. Soc, 1953,
323.
Глава 9
СТАБИЛИЗАЦИЯ
Работами различных исследователей с очевидностью установлено, что
чистая перекись водорода любой концентрации, свободная от загрязняющих
катализаторов, в совершенно чистом сосуде из некаталитического материала
представляет собой весьма устойчивое соединение. Так, типовые данные, полу-
полученные для высококачественной нестабилизированной перекиси водорода,
находившейся в сосуде с отношением поверхности к объему 0,7 см'1, показы-
показывают, что скорость разложения 90%-ной перекиси водорода при 50° не превы-
превышает 0,0010% в час, а возможно даже значительно меньшего процента. В при-
присутствии небольшого количества стабилизатора, например станната натрия
или 8-оксихинолинпирофосфата, эту величину можно уменьшить примерно
до 0,0003% в час при 50°. Если принимать особенно тщательные меры [1]
по очистке и концентрированию перекиси водорода, можно снизить и скорость
разложения нестабилизированной перекиси водорода до этого же уровня.
Соответствующее значение при 30° равно примерно 0,00006% в час, или около
0,5% в год. Тщательные измерения, проведенные с применением нестабилизи-
нестабилизированной, менее концентрированной перекиси водорода, полученной путем
разбавления возможно более чистых концентрированных растворов очень
чистой водой, показали, что скорость разложения (в процентах) возрастает
лишь слабо в интервале концентраций от 95 до 40% перекиси водорода. Однако
если вода, использованная для разбавления, не подвергнута особо тщательной
очистке или разбавление не проведено с достаточной тщательностью, чтобы
устранить возможность заметного загрязнения, то скорость разложения разбав-
разбавленной перекиси может оказаться значительно больше скорости разложения
начального концентрированного раствора. Так, Жигер и Жоффрион [2] наблю-
наблюдали, что при разбавлении нестабилизированной перекиси водорода дважды
дистиллированной водой получаются растворы, которые неизменно обладают
заметно повышенной способностью к разложению, а поэтому для обеспечения
стабильности, требующейся при измерении показателя преломления раство-
растворов, необходимо добавлять к ним небольшое количество станната натрия.
Точно доказано, что, хотя скорость разложения наилучшим образом очищен-
очищенных растворов перекиси водорода и очень низка, практически, конечно, невоз-
невозможно достичь таких идеальных условий, как совершенная чистота раствора
и полное отсутствие каталитического эффекта со стороны стенок сосуда или же
растворенных или взвешенных примесей; в связи с этим наилучшие из имею-
имеющихся данных относительно стабильности, присущей самой перекиси водорода,
нужно рассматривать, с одной стороны, лишь как приближение к идеально
достижимым результатам, а с другой—как цель, к которой следует стремиться
в условиях производства, хранения и перевозки. На практике разложение
в период хранения или в процессе применения сводится к минимуму при
соблюдении следующих трех условий: 1) изготовления с самого начала перекиси
водорода в состоянии высокой чистоты; 2) добавки некоторых веществ (назы-
(называемых стабилизаторами), которые противодействуют эффекту каталитических
примесей или поверхности сосуда, и 3) контроля среды, с которой перекись
430 Глава 9
водорода приходит во взаимодействие. Как показано в историческом обзоре,
изложенном в гл. 1, вначале в качестве средства, противодействующего разло-
разложению перекиси водорода, особенно охотно применяли различные добавки.
Постепенное выяснение того, что сама перекись водорода не является веще-
веществом с присущей ей внутренней неустойчивостью, и развитие электролитиче-
электролитических производственных методов, которые открыли возможность получения зна-
значительно более чистой перекиси водорода, изменили этот подход. В настоящее
время считается общепризнанным, что производство чистой перекиси и сохра-
сохранение этой чистоты на дальнейших этапах являются наилучшей гарантией
стабильности перекиси водорода при хранении. Правда, применение стабили-
стабилизаторов все еще не утратило своего значения, например для защиты от загряз-
загрязнений или в тех случаях, когда особо тщательная очистка может оказаться
неэкономичной. Однако в связи с изменением подхода к мерам для повышения
стабильности перекиси водорода относительное количество стабилизаторов,
добавляемых к перекиси водорода, сейчас значительно меньше, чем прак-
практиковалось ранее. Вместе с тем в настоящее время гораздо лучше изучен
механизм действия стабилизаторов и присущие им ограничения.
Для полного понимания проблем стабилизации необходимо знать при-
природу процессов разложения, рассмотренных в двух предыдущих главах.
Отличительной чертой процессов стабилизации является то, что разложение
происходит со сравнительно небольшой скоростью и обычно бывает вызвано
веществами неопределенной природы. В этой главе рассматриваются экспе-
экспериментальные методы определения таких малых скоростей разложения, влия-
влияние поверхности, температуры, рН и излучения на скорость разложения
и дается обзор соединений, используемых или рекомендуемых в качестве
стабилизаторов, и соответствующей методики стабилизации.
ОПРЕДЕЛЕНИЕ СТАБИЛЬНОСТИ РАСТВОРОВ ПЕРЕКИСИ ВОДОРОДА
Прямой анализ пробы перекиси водорода
Простейшим методом определения скорости разложения раствора перекиси
водорода является анализ раствора (например, путем титрования стандарт-
стандартным раствором марганцовокислого калия в присутствии кислоты) в начале
и к концу определенного промежутка времени. Этот метод дает удовлетвори-
удовлетворительные результаты, особенно при длительном хранении при комнатной темпе-
температуре, при которой скорость разложения может быть очень низкой, или
при испытаниях, в процессе которых происходят значительные изменения
концентрации. В любом таком случае описываемый ниже метод выделения
газа может оказаться непрактичным. Для получения точных данных нужно-
учитывать такие источники ошибок, как колебания температуры и потеря
перекиси водорода и в особенности воды из раствора за счет испарения. Так,
проводя испытания при достаточно повышенной температуре, желательно
сосуд с пробой снабдить простым воздушным или водяным обратным холодиль-
холодильником с целью задержки воды и перекиси водорода, которые в противном слу-
случае могут быть унесены в форме паров вместе с выделяющимся кислородом.
Возможно также увеличение содержания воды в пробе вследствие поглощения
ею влаги из окружающей атмосферы, если последняя обладает достаточно .
высокой влажностью, а перекись водорода сильно концентрирована.
При ведении опытов в условиях повышенной температуры, при которой
скорости разложения высоки, перед анализом к концу исследуемого про-
промежутка времени может оказаться желательным быстро охладить испытуемые
пробы. Например, если исходить из пробы перекиси водорода известного
веса и концентрации, выдерживаемой при постоянной температуре, можно
отбирать через заданные промежутки времени определенные порции перекиса
Определение стабильности растворов перекиси водорода 431
и немедленно замораживать их в смеси твердой двуокиси углерода с ацетономf
чтобы таким образом снизить скорость разложения до ничтожно малой вели-
величины. После этого можно нагреть отобранную часть пробы до комнатной
температуры, а затем анализировать ее на содержание перекиси водорода.
Если известно, что раствор содержит практически только перекись водорода
и воду, то концентрацию можно определить путем точного измерения показа-
показателя преломления или плотности раствора. При измерении нужно тщательно
контролировать температуру. Более подробно о методах анализа, применимых
для определения перекиси водорода, см. в гл. 10.
Даже при благоприятных условиях воспроизводимость измерений, про-
проведенных в различных сосудах, иногда оставляет желать лучшего в связи
с тем, что причины появления некоторых встречающихся ошибок остаются
неизвестными. В результате для получения надежных средних данных обычно
приходится исследовать ряд параллельных проб. Исходную пробу можно
разлить в несколько небольших пробирок из стекла пирекс или из кварца
(например, с введением в каждую пробирку точной навески около 1 мл рас-
раствора), снабженных притертыми стеклянными колпачками и капиллярными
трубками для сообщения с атмосферой. Через различные промежутки времени
из термостата вынимают по одной пробирке, охлаждают и определяют содер-
содержание перекиси водорода таким образом, как это описано выше. Зная количе-
количество перекиси водорода в исходной пробе, можно рассчитать скорость разло-
разложения в условиях опыта на основании предположения, что начальная ско-
скорость разложения в каждой из пробирок одна и та же.
|!Иетод выделения газа
Другим методом, который оказался удобным и надежным, особенно при
испытании в лабораторных условиях, является газометрический, или метод
выделения газа, осуществляемый при температуре, обеспечивающей полу-
получение достаточного количества газообразного кислорода за счет реакции
разложения в течение приемлемого промежутка времени. Этот анализ может
производиться в условиях либо постоянного давления, либо постоянного
объема. На рис. 71 показан прибор для анализа при постоянном давлении [3].
Кислород собирают в газовой бюретке над ртутью, и по его объему, приведен-
приведенному к нормальным условиям, можно вычислить количество разложившейся
перекиси водорода в пробе. На ртуть в бюретке наливают около 1 мл воды,
чтобы собирающийся газ был насыщен влагой при температуре измерения.
При низких скоростях разложения необходимо применять микробюретку,
лучше всего с водяной рубашкой, и соединять бюретку при помощи капил-
капиллярной трубки из стекла пирекс с сосудом, содержащим пробу и находящимся
в термостате при желательной температуре. Растворы перекиси водорода могут
пересыщаться кислородом, а поэтому измерения обычно проводят лишь после
выдерживания раствора в течение 4—16 час. при температуре термостата
(иначе нельзя получить согласованные результаты при последовательных
определениях).
Газометрический метод можно оформить таким образом, чтобы измерять
изменение давления кислорода при постоянном объеме газа. В этом случае
вместо газовой бюретки пользуются манометром, а чтобы запирающая жидкость
в манометре всегда находилась на том же уровне, что и в колене, непосред-
непосредственно соединенном с реактором, используется уравнительный сосуд. Опыт
с такого рода приборами показывает, что они мало чем отличаются от обычных,
работающих в условиях постоянного давления; при высоких скоростях разло-
разложения оба метода непрактичны. Описано приспособление манометра Варбурга
для измерения изменения давления в приборах с постоянным объемом 11].
432
Глава 9
Беллинджер, Фридман, Бауер, Истс и Бал [4] использовали очень простой
прибор, в котором запаивают 5-миллилитровые пробы для анализа.
При исследовании проб перекиси водорода средней стабильности целесо-
целесообразно работать при температурах 50—60°. В тех случаях, когда ожидаются
низкие скорости разложения, можно рекомендовать применение термостата
с несколько более высокой температурой, иначе объем кислорода, накопляю-
накопляющегося за приемлемый промежуток времени, будет слишком мал для точного
Рис. 71. Прибор для определения скорости разложения газометрическим
методом.
/—деревянный ящик; 2—стеклянная вата; 3—сосуд из стекла пирекс диаметром 300 мм;
4—микробюретка на 10 мл с водяной рубашкой; 5—капиллярная трубка из стекла пирекс;
6—стеклянные шлифы 10/30; 7—манометр; 8—колба на 250 мл со стеклянным шлифом 14/35;
9—ртутный уравнительный сосуд; 10—стальной штатив для закрепления уравнительного
сосуда; //—резиновая трубка; 12—мотор мешалки; 13—погружной нагреватель; 14—
ртутный регулятор; 15—приспособление для поддерживания постоянного уровня жидко-
жидкости в термостате; 16—трубка для подачи водопроводной воды; 17—трубка для поступле-
поступления воды в термостат; 18—переливная трубка; 19—резиновая пробка с отверстием для сооб-
сообщения с атмосферой; 20—дырчатая медная пластинка для установки колб; 21—термометр
с делениями 0—100°; 22—провода к реле.
измерения. Для технических целей часто применяется ускоренный метод испы-
испытания при 100° в течение 24 час, однако постоянство и воспроизводимость
результатов таких испытаний несколько хуже, чем при более низких темпе-
температурах; возможно также, что результаты, полученные при столь высокой
температуре, окажутся менее точными при пересчете их на низкие температуры.
Эти вопросы рассмотрены в сообщении Рота и Шенли [1].
При скорости разложения раствора перекиси водорода порядка 0,1% в час
или выше газометрический метод может оказаться неподходящим главным
образом потому, что объем измеряемого газа очень быстро возрастает и при
измерении его возникают неудобства. Кроме того, в этом случае нельзя считать,
как это допустимо при малых скоростях разложения, что концентрация рас-
раствора перекиси водорода сохраняется практически неизменной в течение
всего периода измерений.
Газометрический метод анализа можно в известной мере автоматизиро-
автоматизировать; это уменьшает затрату труда на различные манипуляции и визуальные
отсчеты, что особенно важно при необходимости проведения большого числа
Методы выражения стабильности 433
определений. Однако такая автоматизация оправдывается только при значи-
значительном объеме программы серийных анализов, проводимых в течение длитель-
длительного промежутка времени.
Метод потери веса
Следующий метод состоит в том, что пробу наливают в пробирочку, снаб-
снабженную колпачком с капилляром определенной длины. Последний функцио-
функционирует в качестве воздушного конденсатора, задерживающего воду (а возможно,
и перекись водорода), которая в противном случае улетучивалась бы в виде
пара вместе с выделяющимся кислородом. Количество этого кислорода нахо-
находят путем точного взвешивания пробирочки вместе с колпачком до опыта и после
него и из полученных данных вычисляют скорость разложения перекиси
водорода.
Дрю и Уиттейкер [5] разработали метод анализа по потере веса с приме-
применением кварцевых ампул весом 0,3 г, содержащих около 1,5 мл перекиси водо-
водорода, которые подвешивают к спирали из кварцевой нити. Таким образом
можно непрерывно наблюдать за потерей веса при помощи катетометра без
проведения каких-либо манипуляций с самой пробой.
МЕТОДЫ ВЫРАЖЕНИЯ СТАБИЛЬНОСТИ
Физико-химики обычно выражают скорость реакции по изменению моляр-
молярной концентрации за единицу времени (например, в молях на литр в секунду).
Однако, если основное внимание уделяется не кинетике, а лишь скорости
потери ценного ингредиента, может оказаться более наглядным выражение
результатов в процентах интересующего нас вещества, разложенного за еди-
единицу времени. Этот метод больше всего и испольвуется.
Высококачественный 90%-ный раствор перекиси водорода без стабилиза-
стабилизатора может обладать скоростью разложения около 0,0010% в час при 50°; это
означает, что за 1 час разлагается 0,001% того числа молекул Н2О2, которые
присутствовали в самом начале. Если эта скорость достаточно мала, то коли-
количество присутствующей перекиси водорода в течение всего периода измерения
можно рассматривать как практически неизменное. Результаты можно выра-
выражать и в единицах обратного времени; так, например, 1% в час обозначается
как 0,01 час. Не нужно путать изменение концентрации в процентах в час
и разложение в процентах в час. Так, снижение концентрации на 1% в час
в том случае, когда начальное содержание перекиси водорода в растворе
составляет 50 вес.%, должно привести через 1 час к образованию раствора,
содержащего не 49,0% Н2О2, а 49,5%. В строго стандартизированных условиях
испытания* результаты удобно выражать в виде количества газообразного
кислорода в миллилитрах (в нормальных условиях давления и температуры),
выделяемого из 1 г или 1 см3 раствора в течение определенного времени, напри-
например в течение 24 или 48 час. при определенной температуре, обычно 50, 55, 60
или 100°.
Различные выражения для скорости разложения связаны между собой
следующим образом (см. таблицу обозначений на стр. 557; Mh обозначает
* В качестве примера стандартной методики германские авторы часто используют
так называемое Z-число (Zersetzungszahl), представляющее процент исходной перекиси
водорода, разложенной за 24 часа при 96 . Вероятно, эта температура соответствует тем-
температуре нагревания раствора перекиси водорода в лабораторных условиях на паровой
бане. Изменение состава раствора перекиси в этом случае определяется титрованием проб
раствором перманганата в начале и конце испытания.
28 Перекись водорода
434
Глава 9
здесь молекулярный вес перекиси водорода):
разложения
в
dc
"pW
1,
При нанесении на график данных по скорости разложения в виде функции
концентрации перекиси водорода (при применении этих различных методов)
получаются кривые, сильно различающиеся по форме. Это можно видеть
на рис. 72, где один ряд экспериментальных данных представлен тремя раз-
различными способами i6].
Обсуждавшиеся выше данные относятся
к дифференциальной скорости разложения.
Если в течение опыта происходит значи-
значительное изменение концентрации, то вычис-
вычисленная скорость представляет среднюю или
интегральную величину, которую иногда
трудно интерпретировать, если порядок
реакции в данных экспериментальных
условиях не известен. Это действительно
часто наблюдается. Порядок реакции опре-
определяется как показатель степени х в выра-
выражении скорости разложения в виде функ-
функции концентрации посредством уравнения
-?=kcx. Данные рис. 72 соответствуют вы-
выражению для реакции нулевого порядка
по концентрации перекиси водорода; если
учитывать также концентрацию водородных
ионов, порядок реакции может измениться.
Однако такого рода нулевой порядок на-
наблюдается только при исследовании исклю-
исключительно чистой перекиси водорода и при-
принятии особых мер для очистки воды,
используемой для разбавления концентри-
концентрированной перекиси. В других условиях
может наблюдаться совершенно другой
кинетический порядок.
40 60 80
, вес. %
Рис. 72. Различные методы выра-
выражения данных скорости разложе-
разложения. Дважды дистиллированная
90%-ная Н2О2, разбавленная водой
для измерения электропроводности
при отношении s/i/=0,7 см.
+ разложение (проценты в час); Д
разложение (моли на литр в час); О вы-
выделение кислорода (миллилитры на 1 г
раствора в час).
ВЛИЯНИЕ ОТНОШЕНИЯ ПОВЕРХНОСТИ К ОБЪЕМУ И ПРИРОДЫ ПОВЕРХНОСТИ
Обычная перекись водорода, выпускаемая в настоящее время, обладает
столь высокой чистотой, что разложение, наблюдаемое при хранении, часто
обусловлено в основном гетерогенной реакцией на стенках емкости. В некото-
некоторых условиях применения перекись водорода может также содержать тонко-
диспергированные вещества, способствующие гетерогенному разложению.
В последнем случае степень дисперсности твердых частиц и, следовательно,
размер поверхности контакта с перекисью оказывает решающее влияние
' на наблюдаемую скорость разложения. Относительная роль поверхности
сосуда определяется свойственной материалу сосуда инертностью к перекиси
водорода и физическими свойствами поверхности (например, степенью глад-
гладкости стенок).
Отношение поверхности к объему
Опыты, проведенные в сосудах из стекла пирекс в лабораторном масштабе
при 50°, показали, что скорость разложения чистых растворов перекиси водо-
водорода в этих условиях приблизительно пропорциональна отношению величин
Влияние отношения поверхности к объему и природы поверхности
435
поверхности к объему в широком интервале; другими словами, гетерогенная
реакция разложения на стенках сосудов лабораторных размеров обычно идет
значительно быстрее, чем гомогенное разложение.ч Типичные данные пред-
представлены на рис. 73, иллюстрирующем данные исследований [1, 6], проведен-
проведенных над высокоочищенными пробами нестабилизованной перекиси водорода
(90%-ной и безводной).
На рисунке можно видеть, что при высоких отношениях поверхности
к объему кривые приблизительно линейны, а при низких отношениях они
стремятся к линии, параллельной оси абсцисс, и соответствуют скорости
разложения около 0,005—0,001% в час при 50°*. Кривые смещаются вниз в при-
присутствии стабилизаторов; как указано выше (стр. 429), отмеченные минималь-
минимальные скорости разложения при 50° не превышают по крайней мере 0,0003%
в час. Нанесенные на рис. 73 кривые относятся к лабораторным пробам,
0,012
0,010
Цилиндр Шар
диаметром диаметром
k 5см Ъ \5см
2 3 4 5
Отношение s/vt см
Рис. 73. Влияние отношения поверхности к объему
на скорость разложения.
Д Рот и Шенли, 100%-ная Н2О2 (при пересчете на 50°); О Шамб,
90%-ная Н2О2.
которые, несмотря на соблюдение тщательных мер предосторожности, все же
содержали минимальные количества каталитических примесей, захваченных
в процессе лабораторных работ с пробами и, вероятно, вызывавших несколько
завышенные минимальные скорости разложения.
Рот и Шенли [1] показали, что общая скорость разложения может зависеть
в значительной мере и от той доли поверхности сосуда, которая находится
в контакте с паровой фазой. В исследованиях, проведенных с чистой и прак-
практически безводной перекисью водорода в приборах из стекла пирекс, разло-
разложение, вызванное контактом паровой фазы с поверхностью сосуда, составляло
от V4 до V3 от скорости разложения, вызванного контактом поверхности сосуда
с жидкой фазой, в расчете на единицу поверхности.
Экспериментальные результаты, показанные на рис. 73, нужно поэтому
рассматривать как сумму трех различных механизмов: 1) гетерогенного разло-
разложения жидкости на поверхности сосуда, соприкасающейся с жидкостью;
2) гомогенного разложения в жидкой фазе и 3) гетерогенного разложения
паров на поверхностях сосуда и присоединенных к нему частях прибора. Гомо-
Гомогенное разложение в паровой фазе не имеет существенного значения, гетеро-
гетерогенное же разложение, обусловленное тонкодисперсным взвешенным вещест-
веществом, по мнению авторов, было исключено. При подходящих условиях скорость
• На рис. 73 значения абсциссы приблизительно соответствуют отношениям по-
поверхности к объему в некоторых обычных лабораторных сосудах для проведения изме-
измерений стабильности. Для шара радиусом г отношение поверхности к объему равно 3/г,
а для цилиндра отношение боковой поверхности к объему равно 2/г. На рисунке показаны
значения s/v, соответствующие цилиндрам диаметром 1,6 см (пробирки) и 5 см (стаканы)
и шару диаметром 5 см (колбы).
28*
436 Глава 9
разложения, получаемая при экстраполяции кривой к нулевому отношению
поверхности к объему, должна представлять скорость гомогенной реакции.
Очевидно, что гомогенное разложение обладает конечной величиной даже
в исследованных высокоочищенных пробах.
Поверхность сосудов для хранения перекиси водорода
Активность поверхности сосудов, изготовленных из обычного содового
стекла, в основном обусловлена щелочной природой поверхности, действующей
как катализатор гетерогенного разложения. При стоянии, возможно, проис-
происходит растворение щелочи перекисью водорода, что обусловливает нежела-
нежелательный рост рН. Поэтому сосуды из содового стекла совершенно не годятся
для хранения перекиси водорода, не содержащей стабилизаторов. Процесс
гетерогенного разложения усиливается в случае шероховатой поверхности
или наличия тонких царапин на внутренних стенках стеклянного сосуда.
Можно наблюдать, как с какой-либо мельчайшей точки на поверхности такого
сосуда периодически поднимается цепочка пузырьков кислорода, хотя даже
под микроскопом нельзя обнаружить, что это NjecTO на поверхности имеет
какую-то неправильность или отличается по внешнему виду от других ее частей.
Хоркхеймер [7] изучил стабильность разбавленных растворов перекиси
водорода в стеклянных сосудах и показал, что результаты наблюдений в основ-
основном обусловлены природой применяющейся дистиллированной воды и каче-
качеством стеклянного сосуда и что при применении стекла «максимальной чистоты»
добавки стабилизирующих веществ не требуется, особенно при низких темпе-
температурах (около 16—18°). Иванов и Дочикян [8] (наряду с другими авторами)
исследовали стабильность разбавленной перекиси водорода при комнатной
температуре при хранении ее в склянках из обычного стекла, из коричневого
стекла или пропарафиненных изнутри. Эти авторы показали, что концентрация
перекиси за 4 месяца упала с начальных 2,93 до 0,02% в сосудах из бесцветного
стекла, до 0,48%—в сосудах из коричневого стекла и до 2,71%—в пропара-
пропарафиненных сосудах (безразлично, из бесцветного или коричневого стекла).
Это почти полное разложение перекиси водорода в стеклянных сосудах без
внутреннего покрытия, несомненно, обусловлено щелочностью, сообщенной
раствору действием содового стекла в сочетании с возможной шероховатостью
стеклянных поверхностей. Уолтон и Джад [9] еще в 1912 г. сообщили, что
колбы из иенского стекла, а еще лучше из кварцевого стекла, при пропарке
и обработке соляной кислотой вызывают минимальные скорости разложения
перекиси.
Боросиликатное стекло (стекло пирекс) значительно превосходит содовое
по инертности к перекиси водорода, однако более широкому его распростране-
распространению мешает высокая стоимость. Стекло пирекс является превосходным мате-
материалом для лабораторных сосудов. Его инертность можно повысить путем
тщательной очистки концентрированной азотной кислотой с последующей
промывкой горячей водой (применяемой для измерений электропроводности)
и выдерживанием этой воды в сосуде; особенно удовлетворительные результаты
дает предварительная обработка концентрированной перекисью водорода.
Чистый металлический алюминий, олово, магний или некоторые магниево-
алюминиевые сплавы, а также некоторые нержавеющие стали оказывают
минимальное каталитическое действие на процесс разложения и представляют
интерес как материалы при изготовлении емкостей для перекиси водорода.
Что касается металлического алюминия, то влияние примесей в металле можно
обнаружить по увеличению вдвое скорости разложения 70%-ной перекиси
водорода при 30° в случае замены сосуда из чистого алюминия сосудом
из 99,0%-ного алюминия. Примеси в металле в количестве 0,5% или даже
меньше могут вызвать ясно выраженный рост скорости разложения перекиси
Влияние температуры на стабильность 437
водорода. Процесс анодирования алюминия, способствующий образованию
цельной поверхностной пленки из окиси алюминия (с последующим нагрева-
нагреванием с водой для «заплавления пор»), повышает пригодность металла в каче-
качестве материала при изготовлении резервуаров для хранения растворов пере-
перекиси водорода. Однако этот процесс мало подходит для обработки поверхностей
крупных емкостей. Удовлетворительное состояние внутренней поверхности
алюминия в больших резервуарах может быть достигнуто путем заполнения
резервуара концентрированной азотной кислотой A:1; иногда после предва-
предварительного выдерживания в нем раствора едкого натра); при этом происходит
«пассивирование» металла вследствие образования окисной пленки с высокой
адгезией. Изучено и влияние многих других поверхностей на стабильность
перекиси водорода [4] (см. также стр. 140 и ел.).
Некоторые пластмассы, например полиэтилен, хотя и в меньшей степени,
также находят применение для хранения перекиси водорода. При комнатной
температуре сосуды из такого материала обладают удовлетворительными
показателями, однако при более высоких температурах диффузия жидкости
через пластмассу часто не дает возможности применять их для указанной цели.
Стенки металлических сосудов иногда покрывают пленками из различных
стойких веществ, но в этом случае приходится считаться с опасностью, связан-
связанной с наличием пор или трещин в слое покрытия, что приводит к контакту пере-
перекиси водорода с нижележащим металлом. Разложение перекиси водорода
в этом случае вызывает отслаивание покрытия. Даже сплошное пластмассовое
покрытие нельзя считать удовлетворительным, так как перекись водорода,
диффундируя через покрытие, может вызывать отслаивание его по выше-
вышеуказанному механизму. Покрытие поверхности стекла парафином нельзя
считать вполне удовлетворительной мерой защиты против разложения пере-
перекиси, особенно при высоких ее концентрациях [10]. Для хранения перекиси
водорода при обыкновенных температурах сосуды из полиэтиленовой пласт-
пластмассы, которая хорошо подходит для этой цели,'#в значительной мере уже вытес-
вытеснили стеклянные сосуды с парафиновым покрытием.
Значение состояния поверхности сосуда в отношении влияния на ста-
стабильность хранящейся в сосуде перекиси водорода нужно рассматривать
не только с точки зрения того, что в большинстве случаев очень большая доля
разложения перекиси обусловлена гетерогенным процессом; следует иметь
в виду еще, что поверхности при правильной обработке становятся неактив-
неактивными (или пассивными) к растворам перекиси водорода. Концентрированные
растворы перекиси водорода и пассивированные поверхности взаимно влияют
одни на другие; обработанные определенным образом поверхности вызывают
значительно меньшее гетерогенное разложение, чем необработанные, вместе
с тем сама концентрированная перекись может функционировать как конди-
кондиционирующий агент для поверхностей соответствующих сосудов, что приводит
к улучшению характеристики стабильности хранящейся в них перекиси водо-
водорода. Трудно сказать, действует ли перекись водорода в этом случае только
как агент, вызывающий очистку поверхности, или же она образует на поверх-
поверхности каталитических загрязнений на стенках сосуда защитную пленку.
Однако, во всяком случае, можно рекомендовать завершать процесс конди-
кондиционирования сосуда (безразлично, алюминиевого или стеклянного) выдержи-
выдерживанием в нем концентрированной перекиси водорода.
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА СТАБИЛЬНОСТЬ
Измерения скорости разложения высокоочищенных нестабилизированных
растворов перекиси водорода при температурах 50—70° показали, что повы-
повышение температуры на 10° увеличивает скорость разложения в 2,2±0,1 раза.
438
Глава 9
Этот температурный коэффициент а определяется следующим выражением:
где kx и k2—константы скорости реакции при температурах Тг и Т2 соответ-
соответственно. Получаемая величина а хорошо согласуется с вычисленной из резуль-
результатов других измерений [11], проведенных с растворами перекиси водорода
при 96° во всем возможном интервале концентраций. Эти измерения показы-
показывают, что отношение между скоростями разложения при 96 и 50° составляет
приблизительно 40 : 1. По вычислению при помощи вышеприведенного урав-
уравнения это отношение равно примерно 38 : 1. Такая величина температурного
коэффициента в общем согласуется со значениями, найденными Ротом
и Шенли [1] для высокоочищенной безводной перекиси водорода (как одной,
так и в смеси с кислотами или различными стабилизаторами).
В присутствии каталитических загрязнений температурный коэффициент
должен быть ниже 2,2, что в действительности обнаружено при добавке к рас-
раствору следов солей окисного железа. Для таких растворов отмечены значения а,
равные всего 1,6. Наоборот, в присутствии станнатного стабилизатора коэф-
коэффициент а никогда не падал ниже 2,2; иногда он был несколько выше, а именно
составлял 2,3—2,4. Необходимо учесть, что температурный коэффициент,
определяемый по вышеприведенному уравнению, является только приближен-
приближенным к коэффициенту, вычисляемому из уравнения Аррениуса; поэтому он не
пригоден для широкого интервала температур.
Экспоненциальный рост скорости разложения с температурой можно
иллюстрировать некоторыми типичными данными. При комнатной температуре
90%-ные чистые нестабилизированные водные растворы можно хранить в тече-
течение нескольких месяцев со снижением содержания перекиси менее чем на 1 %.
При 100° потеря за счет разложения составляет, как правило, около 2% в сутки.
При температуре кипения (около 140°) происходит быстрое разложение 90 %-ной
перекиси водорода, причем выделяющаяся теплота может способствовать про-
продолжению кипения концентрированного раствора даже без внешнего подвода
тепла.
ВЛИЯНИЕ рН НА СКОРОСТЬ РАЗЛОЖЕНИЯ ПЕРЕКИСИ ВОДОРОДА
Влияние рН на чистую перекись водорода
Если представить графически скорость разложения нестабилизированного
водного раствора перекиси водорода данной концентрации, по возможности
Ifl 2JS
3,0 3,5 4,0 4.5 5,0
рН (при разбавлении V-10)
5,5
Рис. 74. Скорость разложения перекиси водорода раз-
различной концентрации в зависимости от рН при 50° (рН
установлен при помощи серной кислоты).
лишенного загрязняющих примесей, в виде функции рН, регулируя послед-
последний добавкой чистейшей серной кислоты или едкого натра, то получается кривая,
обладающая минимумом при рН около 4,5—5,0 (рН измеряли после разбавле-
Влияние рН на скорость разложения перекиси водорода
439
0,070
0,060
ния водой в отношении 1 : 10*). На рис. 74 показаны результаты измерений
скоростей разложения разбавленных и концентрированных растворов перекиси
водорода в широком интервале рН. Можно видеть, что отдельные кривые имеют
похожую форму. Максимальная стабильность (или минимальная скорость
разложения) наблюдается примерно в одном и том же интервале рН D,0±0,5)
во всех случаях. При одном и том же рН концентрированные растворы
являются, очевидно, более стабильными, чем разбавленные, причем различие
возрастает при отходе от зоны максимальной стабильности (pHi:io=4,O)
в ту и другую стороны. При кон-
концентрациях выше 50 вес.% влияние
изменения рН в интервале 2,5—5,5
на скорость разложения невелико;
при концентрациях заметно ниже
50% влияние изменения рН делается
все более и более ощутимым как в
зоне высоких, так и в зоне низких
рН. На рис. 75 показано влияние
повышения рН (в интервале |.
рНi:iо=5—7) для 90%-ной пере-
перекиси водорода. Можно видеть, что
по мере повышения щелочности
раствора скорость разложения
возрастает.
Эти данные показывают, что
незагрязненные растворы перекиси
водорода обладают максимальной
стабильностью при рН чуть ниже
«естественного». В литературе часто
встречаются высказывания, что
стабильность перекиси водорода
может быть повышена -путем под-
кисления. Однако трудно сказать,
насколько это мнение носит общий
характер. .Например, перекись
0,040
0,030
0,020
0,010
— ¦¦¦
—--
¦
/
/
J
f
w
\
\
\
/
f
5,0 5,4 5,8 6,2 6,6
рН (при разбавлении 1*10)
7,0
Рис. 75. Скорость разложения 90%-ной
перекиси водорода при 50° в интервале рН
A : 10) от 5 до 7.
О колба № 21, начальная скорость разложения
равна 0,00137% в час; Л колба »Nb 22, начальная
скорость разложения равна 0,0017% в час.
водорода, полученная из перекиси
бария, часто обладает определенно
щелочным рН. В этом случае
добавка кислоты для нейтрализации оказывает определенное благоприят-
благоприятное действие. С другой стороны, в присутствии примесей снижение
рН может создать помехи для некоторых процессов каталитического разложе-
разложения. Часто указывается, что.рост скорости разложения при рН выше и ниже
минимума может быть обусловлен примесями, введенными вместе с кислотой
или основанием [12], использованными для изменения рН в этих работах.
Так, Слейтер [13], приводя типовые данные, показывает, как повышается
скорость разложения 3%-ной перекиси водорода при подщелачивании ее рас-
раствором едкого натра и как этот эффект снижается, если аналогичным обра-
образом использовать силикат натрия. Результат объясняется стабилизирующим
действием силиката, проявляемым, вероятно, в отношении того каталитического
* Как указано при обсуждении кислотных свойств перекиси водорода на стр. 330,
если измерить рН неразбавленных растворов перекиси водорода, имеющих концентрацию
выше 90%, стеклянным электродом, то получаются отрицательные величины, обычная
интерпретация которых затрудняется. Поэтому использован прием разбавления началь-
начальных растворов в 10 раз, что дает возможность сравнивать получаемые рН и истолковывать
их обычным путем.
440
Глава 9
агента или поверхности, которые обусловливают обнаруженный рост скорости
разложения.
Опыты [1], проведенные по тщательно продуманному плану с исключи-
исключительно чистой безводной перекисью водорода, показали, что снижение рН
всегда вызывает падение стабильности независимо от содержания стабилизатора
или природы использованной кислоты—плавиковой, серной, хлорной или
фосфорной; последняя, правда, может оказать одновременно и весьма слабое
стабилизирующее действие. Необходимо иметь в виду, что даже перекись водо-
водорода, подвергнутая особо тщательной очистке, способна к каталитическому
разложению согласно механизмам, которым благоприятствуют рН выше или
ниже, чем pHi:i0=4—5. Возможная природа этих механизмов обсуждалась
на стр. 380 и ел.
Влияние рН на загрязненную перекись водорода
В присутствии следов добавленных ионов металлов, например Fe3+, Cu2+ или
Сг3+, на графике скорости разложения растворов перекиси водорода как функ-
функции рН при любом постоянном количестве добавленного каталитически дей-
действующего иона обнаруживается
Q/2|—|—i—|—|—|—\—\—|—| 1 резкий подъем, или «пик», в
интервале рН^ю около 3,5,
отсутствующий у чистых рас-
растворов перекиси водорода. Для
катионов других металлов, на-
например Ag+, максимум может
лежать при более высоком рН.
Этот максимум может иногда на-
наблюдаться лишь в небольшом
интервале рН, например состав-
составляющем всего одну единицу,
а поэтому если для выяснения
влияния рН на стабильность
определить только несколько
точек какой-либо пробы, то этот
максимум можно и не обнару-
обнаружить.
На рис. 76 показана кри-
кривая скорость разложения—рН
в присутствии железа в качестве
загрязняющей примеси. С ростом
рН наблюдается резкое повыше-
повышение скорости разложения с по-
последующим быстрым падением
этой скорости при дальнейшем
росте рН.
Предложено приемлемое объяснение этого явления [3,14]. Резкий рост
скорости разложения при изменении рН объясняется прогрессирующим гидро-
гидролизом растворенного катализатора (например, окисножелезной соли) с образо-
образованием коллоидной водной окиси (или, возможно, слаборастворимых основ-
основных солей), обладающей значительной каталитически активной поверхностью,
находящейся в контакте с раствором перекиси водорода. Известно, что в вод-
водных растворах солей трехвалентного железа рост мицелл достигает максимума
в том же интервале (приблизительно около .3,6); логично поэтому принять,
что каталитическое действие, иона окисного железа должно быть приписано
этому образованию мицелл. Последующее падение скорости разложения при
дальнейшем повышении рН объясняется тем, что рост рН вызывает коагуля-
коагулят 2,6 3,4 4,2
рН (приразбавлении t-IO)
5,0
Рис. 76. Влияние рН на скорость разложения
90%-ной перекиси водорода, содержащей
0,3 мг/л Fe8+ при 30°.
Влияние рН на скорость разложения перекиси водорода
441
цию коллоида с резким снижением удельной поверхности водной окиси, нахо-
находящейся в контакте с раствором. При еще более высоких рН (на рисунке
не показанных) можно ожидать нового подъема кривой разложения из-за повы-
повышения щелочности раствора—в соответствии с показанным на рис. 75. Минимум
на кривой скорости разложения до этого роста должен быть выше, чем ожидае-
ожидаемый для интервала низких рН, так как коагулированный коллоид все же обла-
обладает известной каталитически активной поверхностью, хотя и значительно
меньшей, чем в случае диспергированной формы. В достаточно кислых раство-
растворах концентрированной перекиси водорода ни ион серебра, ни ион двухвалент-
двухвалентного марганца не оказывает заметного каталитического действия. Они, вероятно,
содержатся в растворе в виде простых катионов. Однако если повысить рН
0,050
о. 0,040
8
0,030
0,020
0,010
?0 ДО 4,0 S,0 6,0 7,0 8,0
рН (приразбавлении 1-10)
Рис. 77. Влияние рН на скорость разложе-
разложения 90%-ной перекиси водорода, содержащей
0,2 мг/л РЬ2+ (введенного в виде нитрата)
при 50°.
G рН установлен при помощи NaOH; О рН уста-
установлен при помощи HNOs.
0,020
О 0,2 0,4 0,6 0,8 1,0
Концентрация иона окисного железа, мг/л
Рис. 78. Влияние pHi : ю и
концентрации иона окисного же-
железа на скорость разложения
90%-ной перекиси водорода при
50°.
настолько, чтобы выпали окислы этих металлов, происходит быстрое и значи-
значительное повышение скорости разложения перекиси. На рис. 77 показано
влияние 0,2 мг/л иона свинца, введенного в форме азотнокислого свинца,
в интервале рН1:10 примерно от 2 до 7,2. рН устанавливали путем добавки
либо азотной кислоты, либо едкого натра. Поскольку рН, при котором начи-
начинается осаждение гидрата окиси свинца, составляет примерно 6 (а не около 2,
как для водной окиси железа), то в этом случае на кривой скорости разложения
нет резкого пика вблизи 3,5 (как видно по числу экспериментальных точек,
этот максимум искали весьма тщательно).
В отношении перекиси водорода, содержащей ион алюминия, сообщалось
[15] о наличии максимума около рН 3. Однако при повторении этих опытов
такого пика не .обнаружено; это приводит к выводу [6], что найденный пик,
возможно, был вызван каким-нибудь дополнительным загрязнением, вероят-
вероятнее всего железом.
На рис. 78 показано влияние возрастающих количеств каталитических
примесей, например окисного железа, в ограниченном интервале рН. Концен-
Концентрацию иона железа (III) увеличивали отдельными ступенями с 0,1 до 1,0 мг/л;
pHi:i0 колебался от 1,6 до 3,2.
442
Глава 9
. Для правильного сравнения стабильностей различных проб перекиси
водорода, очевидно, нужно точно указать рН раствора; это требование осо-
особенно важно в том случае, когда раствор загрязнен. Верно также, что стабили-
стабилизатор может обладать большим эффектом в определенном интервале рН.
ВЛИЯНИЕ ИЗЛУЧЕНИЯ НА СТАБИЛЬНОСТЬ
Обсуждение процессов фотохимического разложения (стр. 382 и ел.)
показывает, что на стабильность перекиси водорода оказывает заметное дей-
действие ультрафиолетовый свет. Это явление учтено уже давно, так как растворы
перекиси водорода часто выпускались в продажу в склянках из цветного
стекла или в другой упаковке, преграждавшей доступ свету. Бухи и Курер,
например, [16] рассмотрели защитное действие сосудов из цветного стекла
Видимый
— спектр
Ультрафиолетовый
спектр •
Флуоресцентная
лампа дневного
света
5000
Рис. 79.
4000 ф3000
Длина волны, А
Факторы,, влияющие на стабильность
водорода при освещении.
2000
перекиси
в качестве тары для разбавленной и концентрированной перекиси водорода.
Однако степень разложения, которая может быть вызвана солнечным светом
или другими лучами, в обычных условиях перевозки и применения четко
не выяснена. Очевидно, что выдержка на солнечном свету может способствовать
нагреванию с одновременным повышением скорости разложения наряду
с фотохимическим разложением. В то же время применение цветного стекла
порочно в своей основе, так как в стремлении преодолеть один источник разло-
разложения увеличивают опасность другого из-за низкого качества цветного стекла
и наличия в его составе металла или щелочи. К этому нужно добавить еще, что
стекло (безразлично, твердое или мягкое) обычно считается достаточно непро-
непроницаемым для большей части ультрафиолетового спектра.
Возможное влияние нагревания и относительной активности поверхно-
поверхностей в качестве сопутствующего фактора при предположительном фотохими-
фотохимическом разложении перекиси водорода обычным путем установить нельзя,
однако, как указывает Хейдт [17], явления, касающиеся вероятности фото-
фотохимического разложения, можно определить качественно по рис. 79.
Из анализа этого рисунка следует, что источники искусственного света
вряд ли могут вызвать заметное разложение перекиси; излучение этих источ-
источников света падает почти до нуля при таких длинах волн, при которых погло-
поглощение перекисью водорода только начинается, а квантовые выходы, вероятно,
очень низки. С другой стороны, при действии прямого солнечного света всегда
имеется большой риск значительного разложения, особенно при высоких
концентрациях. Около 4% всего солнечного излучения происходит при длинах
Типовые исследования стабильности 443
волн, лежащих ниже выступа в нижней части кривой интенсивности; в отно-
отношении разложения перекиси водорода эту часть можно считать эффективной.
Далее, опыт показывает, что при прямом облучении солнечным светом проб
90%-ной нестабилизированной перекиси водорода в стеклянных сосудах раз-
разложение их при комнатной температуре может достигнуть примерно 1 % в сутки.
На основании кривых рис. 79 следует также, что сосуды из бесцветного стекла
не в состоянии задержать фотохимическое разложение, вызванное обычными
источниками света; это особенно верно для стекла пирекс, обычно применяемого
в лаборатории. Стекло (кроме кварцевого или специального) задерживает
ультрафиолетовое излучение с длиной волны ниже 2800 А. Этим объясняется,
что перекись водорода в сосудах из стекла пирекс не изменяется при облу-
облучении специальной ультрафиолетовой лампой; такие лампы, называемые
бактерицидными, концентрируют свое излучение в области ртутной линии
2537 А, стекло же пирекс полностью задерживает такое излучение.
Эти выводы, конечно, носят только общий характер; в каждом отдельном
случае необходимо учесть абсолютную интенсивность и спектральное распре-
распределение падающего света, а также квантовый выход, поглощение света в пере-
перекиси соответствующей концентрации и конфигурацию сосуда. Следует отме-
отметить, что по мере изменения длины волны и концентрации скорость разложе-
разложения быстро изменяется. Поскольку диффузное отражение эффективно сни-
снижает и интенсивность солнечного света и относительную долю в нем ультра-
ультрафиолетовых лучей, можно принять, что в обычных лабораторных условиях
рассеянный дневной свет мало влияет на скорость разложения растворов пере-
перекиси водорода, а поэтому этот фактор не вызывает заметной ошибки в резуль-
результатах. Если необходимо провести сравнительные измерения в условиях облу-
облучения с различной интенсивностью и частотой, то фотохимическое разложение
может исказить результаты. Поэтому в некоторых случаях измерения стабиль-
стабильности осуществляются без доступа света. При анализе фотохимического разло-
разложения на стр. 384 был приведен ряд веществ, которые в значительной мере
тормозят эффективность разложения, однако в технике эти вещества не
находят применения.
ТИПОВЫЕ ИССЛЕДОВАНИЯ СТАБИЛЬНОСТИ
Вопрос о влиянии различных факторов (в отдельности или в сочетании)
на стабильность перекиси водорода был предметом исследования в многочис-
многочисленных работах, однако только сравнительно недавно удалось глубже рассмо-
рассмотреть природу этих факторов. Так, результаты, полученные Вильямсом [18]
в 1928 г., иллюстрируют трудность разделения гетерогенной и гомогенной
реакций и определения кинетического порядка реакции. Марканд, Мак-Элрой
и Кетли [19] сообщают о различных результатах, полученных ими при добавке
разных стабилизаторов к 3- и 30%-ной перекиси водорода. Пожалуй, наиболь-
наибольшее значение для выяснения явлений, связанных со стабильностью и стабили-
стабилизацией, имеют некоторые новейшие работы [1,3—5, 12, 20, 21], касающиеся
концентрированной перекиси водорода.
Шенли и Гринспэн [12] сообщают данные о скоростях разложения 90%-ной
нестабилизированной перекиси водорода при температурах от 30 до 140°:
при 30° 1 % в год; при 66° 1 % в неделю; при 100° 2% в сутки; при 140° быстрое
разложение с кипением. Эти авторы сообщают, что практически достижимые
давления не оказывают заметного влияния на скорость разложения. В табл. 77
показано влияние добавки небольших количеств ионов разных металлов
на разложение.
Как и следовало ожидать, скорость разложения возрастает с ростом концен-
концентрации катализатора.
Интересно выяснить влияние смешанных катализаторов на каталитическое
разложение. В случае твердых катализаторов, содержащих определенные
444
Глава 9
Таблица.77
Влияние добавленных ионов на скорость разложения концентрированной
перекиси водорода, по Шенли и Гринспэну [12]
Природа добавлен-
добавленного иона
Без добавки . . .
А13+
Sn4+
Zn2+
Количество
добавки,
мг\л
10
10
10
Потеря
исходного
активного
кислорода
за сутки
при 100°, %
2
2
2
10
Природа добавлен-
добавленного иона
Fe3+
Cu2+
Cu2+
Сг3+
Количество
добавки,
мг\л
1,0
0,01
0,1
0,1
Потеря
исходного
активного
кислорода
за сутки
при 100°, %
15
24
85
96
0,10
0,06
1
смеси совместно осажденных гидроокисей, влияние смеси в некоторых случаях
гораздо больше, чем вычисленное суммированием эффектов для отдельных
окисей. Точно так же если добавить к пробе 90%-ной нестабилизированной
перекиси водорода растворы солей
0,12\ л „2+ 1—|—|—|—|—|—, трехвалентного железа и двухва-
двухвалентной меди с таким расчетом,
чтобы в конечном растворе содер-
содержалось 0,1 мг/л Fe3+n0,l мг/лСи2*,
то скорость разложения перекиси
при постоянной температуре оказы-
оказывается значительно большей, чем
сумма скоростей разложения того
же раствора перекиси при примене-
применении в таких же условиях каждого
из этих катализаторов в отдель-
отдельности. Эти данные иллюстрируются
рис. 80. В случаях с другими
парами каталитических агентов
также можно, хотя и не всегда,
получить аналогичные результаты;
например, при применении смеси
солей, дающих ионы трехвалентного
железа и серебра, скорость разло-
разложения не выше той, которая соот-
соответствует сумме действия обоих
этих ионов в отдельности. При
исследовании каталитического дей-
действия катионов различных простых
металлов на разложение перекиси
водорода Юри [22] отметил, что
наличие хлор-иона вместе с этими металлическими катионами увеличивает
скорость разложения раз в 40 по сравнению с начальной. Поэтому в дан-
данном случае промотирующее действие вызывается добавкой аниона, а не вто-
второго катиона.
В принципе перемешивание раствора перекиси водорода также может
влиять на ее стабильность; однако в пределах широкого интервала скоростей
перемешивания такого влияния не установлено. Взбалтывание раствора пере-
перекиси водорода может вызвать в нем образование пузырьков в связи с выделе-
выделением кислорода, находившегося в пересыщенном состоянии, но фактического
роста скорости разложения при этом не происходит. Это подтверждается дан-
данными Джильберта и Майхлиса [23], которые показали, что 90%-ная перекись
А Си**
о Fe3+
о Cu2++Fe3+
J
2-
/
У
/
У
/
f /
/
/
w-
/
•
•
/
•
- —
/
•
•
}
0,02
0 0,1 42
Общее количество катализатора, мг/л
Рис. 80. Влияние смешанных катализаторов
на скорость разложения 90%-ной перекиси
водорода.
— — — параллельные опыты.
Стабилизаторы 445
водорода в стандартных бочках разлагается только со скоростью около 1 % в год
{за три года, в течение которых бочки перевозились на далекие расстояния).
Недавно появилось сообщение по исследованию безводной перекиси водо-
водорода, в результате которого, вероятно, был почти достигнут конечный возмож-
возможный уровень стабильности. Рот и Шенли [1] настолько хорошо очистили
перекись водорода, что дальнейшее заметное изменение ее электропроводности
уже не оказывало влияния на стабильность; они показали, что на такую пере-
перекись водорода стабилизаторы существенным образом не влияют в широких
диапазонах температур и рН.
СТАБИЛИЗАТОРЫ
Если бы можно было получать перекись водорода и хранить ее в условиях
полного отсутствия каталитически активных веществ, то для обеспечения
возможности ее длительного хранения при обыкновенной температуре без
заметных потерь от разложения стабилизаторы были бы не нужны. Такая
высоко концентрированная перекись водорода выпускается в продажу почти без
добавок, и, несмотря на это, ее можно безопасно транспортировать и хранить
в алюминиевых бочках.
Хотя логически и возможно, что при наличии каталитически активных
загрязнений в количествах более чем минимальных задержать разложение
перекиси водорода нельзя даже при добавке стабилизаторов, все же вопрос
стабилизации перекиси водорода усиленно изучается в целях увеличения
устойчивости технического продукта при хранении.
Основные принципы стабилизации
Если рассматривать процесс стабилизации растворов перекиси водорода
(не считая контроля рН и температуры) в самом широком аспекте, то он сво-
сводится к инактивированию каталитически активных веществ, которые находятся
в растворенном или взвешенном состоянии в растворе или содержатся на стен-
стенках сосуда. Стабилизация—это не предотвращение некоторых процессов само-
саморазложения перекиси водорода. Таким образом, добавка стабилизаторов к кон-
концентрированным растворам перекиси водорода исключительно высокой чистоты,
находящимся в чистом и инертном сосуде, уже заметно не снижает скорости
разложения раствора перекиси. Если бы природа примеси была известна,
то можно было бы выбрать стабилизатор, руководствуясь специфическим его
действием на эту примесь. Однако чаще всего природа загрязнения неизвестна,
и поэтому может оказаться более желательным применение неспецифического
стабилизатора. Дальнейшие ограничения в выборе приемлемых стабилизаторов
обусловлены их разрушением под действием перекиси водорода и тем, в какой
области затем применяется эта перекиси.
Как правило, при обыкновенных температурах органические вещества
медленно окисляются перекисью водорода. Поэтому они могут оказаться
непригодными как стабилизаторы для длительного хранения, особенно в слу-
случаях с высококонцентрированной перекисью. Тем не менее при некоторых
применениях, например в медицине, для разбавленных растворов перекиси
водорода предпочитают пользоваться органическими стабилизаторами вместо
более сильных неорганических, которые могут оказать вредное влияние
(см. раздел по применению в медицине, стр. 512 и ел.). При использовании пере-
перекиси водорода для фармацевтических целей может оказаться существенным
наличие таких соединений, которые действуют на разложение Н2О2 каталазой
[24, 25], хотя о влиянии ингибиторов на разложение в присутствии ферментов
существуют противоречивые мнения [26, 27]. Кемпбел [28] дал анализ ряда
факторов, которые влияют на стабильность, и указал пути выбора стабили-
стабилизатора для отбеливающих ванн из перекиси водорода. При таком применении
446 Глава 9
стабилизирующему действию может способствовать периодическая очистка;
например, предложено [29] адсорбировать примеси на листах из целлюлозных
волокон. Комбинированный эффект смеси двух различных стабилизаторов может
быть больше, чем тот, который соответствует сумме эффектов каждого из них
в отдельности [30]. Это напоминает описанное выше обратное явление, касаю-
касающееся влияния смеси двух катализаторов, в случае которого скорость разло-
разложения перекиси водорода также была больше аддитивной величины. Очевидно,
необходимо, чтобы рН раствора перекиси водорода находился в интервале,
в котором каждый отдельный стабилизатор является эффективным.
Часто к перекиси водорода добавляют вещества, способствующие сохра-
сохранению в пассивном состоянии поверхности сосуда, в частности изготовленного
из алюминия. Имеются данные [15], показывающие, каким образом вели-
величина рН и ионные примеси влияют на точечную коррозию и растворение
алюминия в технической перекиси водорода. Сообщается, что сульфат и хлорид
повышают растворимость алюминия, фосфат на нее не влияет, а пирофосфат
ее снижает. Нитрат тормозит действие сульфата и хлорида, а поэтому его
иногда добавляют к перекиси водорода для предотвращения точечной коррозии
алюминиевой тары [31]. Существует также возможность коррозии алюминия
под действием пара перекиси водорода в части емкости, заполненной паром,
или у поверхности раздела жидкость—пар. Некоторые стороны вопроса
о влиянии алюминия на стабильность при хранении еще мало изучены. Хотя
растворение алюминия нежелательно влияет на стабильность, все же алюми-
алюминиевые соединения предложены в качестве стабилизаторов [32]. Испытание [6]
действия алюмината натрия, фосфата и пирофосфата алюминия показало, что
эти вещества не обладают значительной стабилизирующей способностью, но они
по крайней мере не снижают стабильности перекиси. Часто наблюдается обра-
образование хлопьевидного осадка, содержащего алюминий, вероятно образовав-
образовавшегося из растворенного алюминия. Влияет ли этот осадок положительно или
отрицательно на стабильность, неизвестно. Возможно, что снижение стабиль-
стабильности, наблюдаемое при растворении алюминия, из которого изготовлены
бочки для хранения перекиси, вызвано одновременным переходом в раствор
примесей, имеющихся в алюминии.
Испытано очень большое число различных неорганических и органических
стабилизаторов с разбавленными и концентрированными растворами перекиси
водорода. Наибольший успех достигнут (особенно для высококонцентрирован-
высококонцентрированной перекиси) при применении станната натрия или 8-оксихинолина (оксина),
в том и другом случаях в присутствии растворимого пирофосфата или смеси
фосфата и пирофосфата. Рассмотрим отдельно каждое из этих трех веществ
(станнат, оксин и фосфаты), а затем другие стабилизаторы.
Станнат
Станнат натрия Na2SnO3-3H2O при гидролизе дает коллоидальную вод-
водную двуокись олова SnO2-*H2O, которая эффективно адсорбирует каталити-
каталитически действующие ионы, например окисного железа, и таким образом повы-
повышает стабильность растворов перекиси водорода. При применении избытка
станната натрия гидролиз его приводит к такому росту рН раствора перекиси
водорода, что ее стабильность снижается. Существует также оптимальный
интервал рН, внутри которого станнат оказывает максимальное стабилизи-
стабилизирующее действие. Так, для 90%-ной перекиси водорода станнат натрия осо-
особенно эффективен в том случае, когда рН раствора перекиси (при десяти-
десятикратном разбавлении) находится в пределах 3,5—6. При рН, равном 2 или 7,
скорость разложения того же раствора может быть выше в 20 раз, чем в ука-
указанном интервале. Рейхерт и Хоукинсон [33] рекомендуют добавку буфера,
например какой-либо предельной алифатической кислоты с константой иони-
Стабилизаторы 447
зации 10—10~в (лучше всего адипиновой кислоты) в сочетании со стабилиза-
стабилизацией смесями станната и пирофосфата. Раствор перекиси водорода (по-видимому,
с концентрацией не свыше 30%) при этом приобретает pHi-.ю от 3,5 до 4,0.
В растворе перекиси водорода водная двуокись олова находится в виде
отрицательно заряженного коллоида, который может коагулировать при
нейтрализации положительными ионами. Отрицательные ионы оказывают
пептизирующее действие, причем сообщается [34], что их эффективность изме-
изменяется, как указано, в ряду OH">P2O*">POJ">SOJ"">NOe. Таким образом,
добавка пирофосфата натрия @,15% или ниже) должна улучшить свойства кол-
коллоидной двуокиси олова благодаря замедлению процесса выпадения олова
в осадок. По другому методу в качестве стабилизатора применяется соединение
SnCl2 • 2Н2О, нагревавшееся в течение часа при температуре 300° с 85%-ной
фосфорной кислотой (для превращения последней в пирофосфорную кислоту).
Для получения оптимальных результатов pHj:ю раствора перекиси водорода
должен быть ниже 5. Все эти способы описаны в патентной литературе [35],
касающейся стабилизации станнатом.
Вместо добавки станната натрия в качестве стабилизатора можно добиться
аналогичных результатов путем приготовления водной двуокиси олова в виде
студенистого осадка (который тщательно промывают многократной деканта-
декантацией) и введения этого водного геля в раствор перекиси водорода; этот опыт
подтверждает предположение, что стабилизация в данном случае вызвана
«очистным» действием коллоидной водной двуокиси олова [36]. Возможным
преимуществом этого метода является отсутствие изменения рН при смешении.
Измерения скорости разложения концентрированных растворов перекиси
водорода, стабилизированных станнатом натрия, показали, что стабилизирую-
стабилизирующее действие для достижения максимального эффекта требует известного
времени; обычно скорость разложения медленно падает в течение 3—4 суток,
пока не становится практически постоянной. Жигер иЖоффрион [2] сообщают
о небольшом запаздывании C0—60 мин.) в проявлении стабилизирующего дей-
действия при добавке небольшого количества станната натрия к разбавленным
растворам перекиси водорода. По-видимому, эта задержка обусловлена тем,
что требуется известное время для образования в растворе (в результате
гидролиза станната) достаточного количества коллоидной двуокиси олова,
необходимой для достижения желательного стабилизирующего действия.
На активность влияет также температура и ход ее изменения; например, ста-
стабильность перекиси водорода со станнатным стабилизатором увеличивается
при подогреве раствора до 50° перед проведением измерений при 30°.
При добавке возрастающих количеств стабилизатора к данной пробе пере-
перекиси водорода скорость разложения сначала падает до минимума, а затем мед-
медленно возрастает. Таким образом, существует оптимальное количество стаби-
стабилизатора, являющееся функцией природы и количества присутствующих при-
примесей. В табл. 78 приведены те количества станната [3], которые являются
наиболее эффективными с точки зрения торможения каталитического действия
иона трехвалентного железа при концентрации 0,1—4,0 мг/л в 85%-ной
перекиси водорода. Скорость разложения начальной незагрязненной перекиси
водорода, как это показано в первой графе, несколько колеблется (примерно
от 0,003 до 0,004% в час при 50°). Скорость разложения стабилизированной
перекиси водорода составляет от 0,0003 до 0,004% в час, оптимальное количе-
количество SnO2 (введенной в виде Na2SnO3-3H2O) колеблется приблизительно
от 1 до 60 мг/л\ отношение SnO2/Fe3* в различных опытах изменяется от 13
до 26 при содержании железа свыше 0,3 мг/л. Станнат натрия не обладает
очень высокой эффективностью против каталитического действия примесей
соединений меди. Для подавления каталитического влияния таких малых
количеств иона двухвалентной меди, как 0,1 мг/л, требуется добавка SnO2
(введенной в виде станната) до 65 мг/л.
448
Глава 9
Таблица 78
Оптимальные относительные количества SnO2-jcH2O, необходимые
для стабилизации 85%-ной перекиси водорода» загрязненной ионом
окисного железа [3]
Начальная ско-
скорость разложения
в час при 50°,
0,0029
0,0029
0,0034
0,0020
0,0037
0,0031
0,0046
0,0029
0,0040
0,0029
0,0020
0,0023
0,0040
Fe3+ (в виде
сульфата),
мг\л
0,10
0,27
0,29
0,31
0,35
0,47
0,55
0,85
1,09
1,52
1,80
2,72
3,99
Оптимальное
количество SnO2
(в виде станната),
мг\л
0,83
1,98
1,75
7,00
5,24
7,79
11,1
11,3
17,5
23,3
47,0
63,1
61,5
Конечная ско-
скорость разложения
в час пои 50°,
0,0003
0,007
0,0013
0,0007
0,0013
0,0015
0,0006
0,0013
0,0005
0,0017
0,0020
0,0021
0,0037
Отношение
в опыте
8,3
7,3
6,0
22,6
15,0
16,6
20,2
13,3
16,0
15,3
26,1
23,2
15,4
8-Оксихинолин (оксин)
Из стабилизаторов, которые изучены (особенно в отношении высококонцен-
высококонцентрированной перекиси водорода), чаще всего упоминаются такие органические
вещества, как 8-оксихинолин (оксин), нередко находящий применение в виде
пирофосфатного производного или в сочетании с растворимым фосфатом или
пирофосфатом*. Механизм процесса стабилизации с участием оксина еще
подробно не изучен. В присутствии железа один оксин не оказывает или почти
не оказывает защитного действия, но вместе с фосфатом или, что еще лучше,
с пирофосфатом он оказывается активным стабилизатором против действия
небольших концентраций некоторых каталитических примесей, в том числе
соединений железа. Оксин широко применялся в Германии в качестве стабили-
стабилизатора 85%-ной перекиси водорода, которой пользовались для различных воен-
военных целей во время второй мировой войны; однако, как и другие органические
добавки, при длительном хранении оксин должен постепенно окисляться пере-
перекисью водорода. Такое окисление особенно вероятно при загрязнении раствора
перекиси водорода окисным железом, так как окисление оксина перекисью
водорода заметно катализуется производными трехвалентного железа. Однако,
если стабилизатор содержит (как в вышеописанном примере) также и ион пиро-
фосфата, то ион трехвалентного железа инактивируется за счет реакции с ионом
пирофосфата; в результате окисление оксина сильно замедляется и стабили-
стабилизирующее действие смеси может сохраняться в течение многих месяцев (даже
при небольшом загрязнении ионом окисного железа). Сам оксинат трехвалент-
трехвалентного железа [37] вызывает бурное разложение 90%-ной перекиси водорода [6].
* Раствор оксина, пригодный для стабилизации концентрированного раствора
перекиси водорода, можно приготовить следующим образом. Растворяют 19 г пирофосфор-
ной кислоты в 200 мл воды и к раствору добавляют 31 г оксина; затем раствор нагревают
в течение примерно 20 мин. на водяной бане при энергичном перемешивании до растворе-
растворения всего оксина. Бурый раствор доливают водой до 1 л и отфильтровывают от возможного
осадка в виде темных хлопьев. Для большинства целей добавка 1—2 мл такого раствора
на 1 л раствора перекиси водорода сообщает достаточную стабильность последнему (при
содержании 85—90% Н2О2) и приводит к скорости разложения, не превышающей 0,002%
в час при 50°, если только перекись водорода в исходном растворе была достаточно чистой.
Другой рецепт для стабилизации с помощью оксина следующий: на 1 л раствора вводят
304 мг оксина, 230 мг Na4P2O7-10 H2O и 202 мг Н8РО4.
Стабилизаторы
449
Фосфаты
Один пирофосфат натрия Na4P2O7- 10H2O может предотвратить катали-
каталитическое действие иона железа при концентрации Fe3+ по меньшей мере 10 мг/л>
но не обладает достаточной защитной способностью против ионов меди даже
с концентрацией несколько миллиграммов на литр. Механизм его защитного
действия состоит, по-видимому, в реакции с такого рода растворенными ката-
каталитическими ионами, как Fe3\ которые при этом либо выпадают в осадок, либо
(в присутствии большого избытка пирофосфата) превращаются в сравнительно
стабильные комплексы; в том и другом случаях каталитический эффект в зна-
значительной мере исчезает. Если исключить способность пирофосфата инакти-
вировать ионы металлов, которые в противном случае могли бы в результате
гидролиза превратиться в каталитически активные твердые продукты, то пред-
представляется невероятным, чтобы пирофосфат непосредственно влиял на процесс
гетерогенного разложения. Нужно иметь в виду также, что сам ион пирофос-
пирофосфата при обыкновенных температурах в водных растворах способен подвер-
подвергаться постепенному гидролизу P2O7~ + H2O=2HPOJ\ Конечно, очень важно,
чтобы пирофосфат, применяемый для стабилизации, был химически чистым
и в особенности свободным от примесей железа.
Таблица 79
Оптимальные относительные количества пирофосфата, необходимые для стабилизации
при загрязнении перекиси водорода ионами окисного железа [3]
Fe3+ в виде
сульфата,
мг\л
0,08
0,16
0,24
0,32
Средняя концентра-
концентрация РгО*- (при
введении в виде
мг\л
0,30
0,60
1,10
1,60
Отношение
3,8
3,8
4,6
5,0
Fe8* в виде
сульфата,
мг\л
0,48
0,96
1,08
1,92
Средняя концентра-
концентрация P2QJ- (при
введении в виде
N^P-iO?- IOH2O),
мг\л
2,50
7,40
10,00
17,20
Отношение
P2O*-/Fe3+
5,2
7,7
9,3
9,0
В табл. 79 показано соотношение между ионами пирофосфата и окисного
железа, необходимое для максимальной стабилизации концентрированных
растворов перекиси водорода при 50°.
В рецептах стабилизации одним пирофосфатом Рейхерт [38] рекомендует
величину рН в пределах от 1 до 7. В одном из сообщений [4] указывается, что
фосфаты (как класс соединений) являются наилучшими стабилизаторами и един-
единственными веществами кислотного характера, которые могут быть рекомендо-
рекомендованы в качестве стабилизаторов. Как указано выше (см. также [39]), фосфаты
или пирофосфаты сочетают с другими стабилизаторами. Тетраэтилпирофосфат
в количестве 50 мг на 1 л 90%-ной перекиси водорода не защищает перекись,
если она загрязнена ионами Fe3+ в количестве, большем 0,1 мг/л [61. Гипо-
фосфит натрия NaH2PO2 несколько менее эффективен против иона окисного
железа, чем пирофосфат, но обладает такой же эффективностью против иона
двухвалентной меди [61. Пирофосфат окисного железа обладает определенно
каталитическим действием [6].
Другие стабилизаторы
В начальный период развития концепции стабилизации прежде всего была
произведена оценка действия органических веществ. Были предложены такие
соединения, как эфир, спирт, камфара или нафталин, но их применение в доста-
достаточной мере не обосновано и в настоящее время для этих целей они не исполь-
29 Перекись водорода
450 Глава 9
зуются. В качестве стабилизаторов предложены буквально сотни разных
веществ, перечислять которые здесь вряд ли имеет смысл. Перечень этих
веществ можно найти у Кауша [40] и Маху [41], здесь же мы ограничимся
только некоторыми дополнительными ссылками и указаниями.
Из различных органических веществ, предложенных для стабилизации
(особенно разбавленных растворов перекиси водорода), очень часто применя-
применялись ацетанилид и другие жаропонижающие вещества. Относительно эффек-
эффективности ацетанилида в различных литературных источниках приводятся
противоречивые данные. Сообщается [42], например, что ацетанилид дает
хорошие показатели при одномесячном испытании при 50°, однако известно [ 15],
что перекись водорода превращает ацетанилид в уксусную кислоту и анилин,
а последний окисляется затем до нитробензола. Стабилизированные таким
образом 3%-ные растворы перекиси водорода при хранении на полке устой-
устойчивы лишь около 6 мес; кроме того, ацетанилид, по опубликованным данным
[43], окисляется легче, чем фенацетин, также применяющийся для стабили-
стабилизации [44]. В качестве стабилизирующего вещества, устойчивого против раз-
разрушения, часто упоминается метиловый эфир л-оксибензойной кислоты, так
называемый нипагин-М [45]. Относительно сравнительных достоинств фена-
фенацетина [43] и нипагина [46] мнения расходятся. Сравнение обоих этих веществ
с хининсульфатом [25] показывает, что последний дает лучшие результаты.
Что касается других возможных органических стабилизаторов, то некото-
некоторые из них были подвергнуты кратковременному испытанию в 90% -ной пере-
перекиси водорода при 50° [6], причем получены следующие результаты: акридин
столь же эффективен, как оксин; 4,5-диоксиакридин обладает лишь слабым
стабилизирующим действием; ализарин эффективен в течение нескольких
дней до разрушения его вследствие окисления; купферрон (аммониевая соль
фенилнитрозогидроксиламина) не обладает никаким действием (ни положи-
положительным, ни отрицательным); салицилальдоксим неэффективен против двух-
двухвалентной меди в количестве 0,1 мг/л. В качестве стабилизаторов рекомендуется
применять пиридинкарбоновые кислоты [47]. Попытки обобщения свойств
органических соединений, обусловливающих возможность их применения
как стабилизаторов, были не очень успешны. Либкнехт [48] в своих патентах
указывает на возможность применения веществ, содержащих серную кислоту,
связанную с углеводородом, например бензолсульфоновой кислоты, или соеди-
соединений с карбоксильной группой, связанной с ароматическим радикалом, напри-
например бензойной кислоты. Добавка бензойной кислоты @,1%) оказалась эффек-
эффективной при концентрировании растворов перекиси водорода методом дистил-
дистилляции [49]. Сульфонамиды также оказывают стабилизирующее действие [50].
Этилендиаминтетраацетат (вещество, образующее клешневидные комплексные
соединения), как показали Дэвис и Хиггинсон [51], защищает разбавленную
перекись водорода от каталитического действия следов ионов металлов; однако
изучение действия продажных комплексообразователей в 90%-ной перекиси
водорода при 50° показало, что они обладают слабым каталитическим дей-
действием [6].
Нередко упоминается о применении в качестве стабилизаторов поверх-
ностноактивных веществ, но часто неясно, применяется ли этот термин в наи-
наиболее употребительном значении и речь идет о благоприятном эффекте, обуслов-
обусловленном снижением поверхностного натяжения [52], или же под этим термином
подразумевается вещество, избирательно адсорбируемое поверхностью сосуда
для перекиси или поверхностью катализатора [53]. Часто в эту категорию
включаются силикаты. Добавка силиката натрия или магния к щелочным
растворам, содержащим перекись водорода, оказывает антикаталитическое
действие, которое, по-видимому, следует приписать коллоидам, образующимся
в результате гидролиза силикатов. Применение последних часто практикуется
при отбелке хлопчатобумажных и других текстильных изделий.
Стабилизаторы 451
Фольгнер и Шнейдер [54] исследовали стабилизирующее действие неболь-
небольших добавок алифатических кетонов, сложных эфиров и алкилсульфонатов,
а также аммиака и некоторых неорганических солей (бората, силиката, фос-
фосфата и пирофосфата натрия) к отбеливающей жидкости при температуре 40,
50 или 70°; испытание длилось 96 час. Данные этих авторов, изложенные в виде
таблиц, доказывают, что пирофосфат натрия обладает более высокой эффектив-
эффективностью, чем другие неорганические стабилизаторы, в частности силикат, осо-
особенно при высоких температурах. Оказалось, что некоторые из органических
веществ отличаются хорошими смачивающими свойствами наряду со стабили-
стабилизирующими, что имеет положительное значение-при отбелке текстиля. Мета-
силикат натрия с рН 5,5 в количестве 35 мг/л увеличивает скорость разложения
чистой 90%-ной перекиси водорода при 50° [6]; кремнефтористый натрий
Na2SiFe обладает слабым стабилизирующим действием. При отбелке часто при-
применяется силикат в виде магниевой соли [55] или в сочетании с магнием в той
или иной форме [56]. Сами магниевые соли [57] или окись магния [58] также ока-
оказывают стабилизирующее действие. Д'Анс и Маттнер [59] изучили стабилиза-
стабилизацию щелочных растворов перекиси водорода против каталитического разложе-
разложения под действием гидрата окиси меди путем добавки гидрата окиси магния. Они
указывают, что осажденная Mg(OHJ увлекает в осадок следы тяжелых метал-
металлов (Со2+, Ni2b и Мп2* и особенно медь), что способствует очистке раствора
перекиси водорода. Серебро и свинец при этом остаются в растворе. В концен-
концентрированной перекиси водорода при 50° жженая MgO в количестве 70 мг/л
не оказывает никакого влияния [6], водная же Mg(OHJ немного снижает
скорость разложения, что показывает влияние физической формы стабилиза-
стабилизатора на характер его действия.
Интерес представляет также поведение водной окиси цинка или окиси
кадмия [60]. При добавке надлежащего количества цинковых солей, например
сульфата, к весьма концентрированным растворам перекиси водорода (порядка
90%-ной) они оказывают стабилизирующее действие на последнюю, по крайней
мере в узком интервале рН. Однако в более разбавленных растворах перекиси
(например, содержащих 20—30% Н2О2) этот эффект меняется на обратный,
и скорость разложения перекиси водорода при добавке растворимых цинковых
солей даже немного увеличивается. Кадмий заметным действием не обладает [6].
Действие ряда веществ, находящихся в коллоидном состоянии, послужило
толчком к исследованию [3] ряда естественных продуктов, например агар-агара,
желатины и камеди карая*. Перечисленные вещества оказались неэффек-
неэффективными; это доказывает, что одна физическая форма не является достаточ-
достаточным критерием стабилизирующего действия. Лучшие результаты получены
при попытках воспроизведения действия станната при помощи производных
сурьмы. Добавка пиросурьмянокислого натрия к концентрированным рас-
растворам перекиси водорода вызывает небольшое, но измеримое повышение
стабильности раствора, что, безусловно, вызвано присутствием коллоидной
водной пятиокиси сурьмы. Фтористая сурьма неэффективна [3]. При испы-
испытании ряда неорганических соединений в качестве стабилизаторов 90%-ной
перекиси водорода при 50° получены следующие результаты [6]: молибдат
натрия обладает слабым стабилизирующим действием; фтор-ион, сульфат
одновалентного таллия, пятиокись тантала, бура и пятиокись ниобия не про-
являют никакого действия; сульфаты бериллия и висмута способствуют ката-
каталитическому разложению.
Выбор стабилизатора
Ни один стабилизатор нельзя считать наилучшим главным образом потому,
что стабилизатор следует выбирать с учетом условий последующего примене-
* Вид бассорской акациевой камеди.—Прим. перев.
29*
452 Глава 9
ния перекиси водорода и типа вероятных ее загрязнений, а также таких данных,
как необходимая продолжительность хранения и вероятные температуры раство-
растворов перекиси водорода во время хранения. Как указывают Шенли и Грин-
спэн [12], станнат тормозит разложение под действием меди сильнее, чем
пирофосфат, а последний лучше защищает от каталитического действия хрома.
Если продолжительность хранения сравнительно невелика, может оказаться
удовлетворительным органический стабилизатор; при длительном же хранении
эффективность органического соединения может исчезнуть в связи с его мед-
медленным окислением [61]. С другой стороны, при некоторых видах применения
известные неорганические стабилизаторы могут оказаться непригодными из-за
того, что при разложении значительного количества перекиси водорода в про-
процессе употребления может образоваться недопустимое количество нежелатель-
нежелательного остатка. Несомненным кажется вывод [4], что избыток стабилизатора
хуже, чем полное его отсутствие. В той мере, в которой вообще можно давать
рекомендации, допустимо, по-видимому, считать, что наилучшую защиту
против небольших концентраций загрязнений могут оказывать станиат,
фосфат, оксинфосфат или их комбинации, упомянутые выше.
Механизм стабилизации
Во многих случаях практика стабилизации основана па чисто эмпириче-
эмпирических началах и не подкреплена теоретическим толкованием. До настоящего
времени еще отсутствует вполне удовлетворительное объяснение различных
механизмов, в силу которых разные органические и неорганические вещества,
нашедшие применение до настоящего времени, оказывают стабилизирующее
действие на перекись водорода; правда, в отдельных случаях доказательства
в пользу определенного механизма можно считать вполне убедительными.
Выше мы уже рассматривали влияние добавки небольших количеств некоторых
кислот с точки зрения подавления кислотной диссоциации перекиси водорода.
Однако подавлением ионизации перекиси водорода путем добавки кислот
(например, серной, фосфорной или пирофосфорной, щавелевой или бензойной)
еще нельзя объяснить повышение стабильности растворов перекиси; это под-
подтверждается тем, что ряд других кислот обладает действием, меньшим, чем
то, которого можно было бы ожидать, судя по их силе. Более того, если добавить
достаточное количество кислоты для снижения рН (при десятикратном раз-
разбавлении) до 2 или ниже, то стабильность перекиси водорода даже снижается.
Высказано предположение, что эффективность кислоты в известной мере
зависит от ее способности образовать соединение с перекисью водорода.
Роль различных добавок, способных давать комплексы с некоторыми катали-
каталитически активными ионами, присутствующими в растворе перекиси, хорошо
известна. Показано также, что некоторые стабилизаторы, очевидно, действуют
как защитные коллоиды. Тормозящее действие таких коллоидных веществ,
как желатина, крахмал или клей, на разложение перекиси водорода объясняют
повышением ими вязкости растворов перекиси или изменением поверхностного
натяжения этих растворов. Более вероятно, что их действие связано с образо-
образованием покрытия на каталитически активной поверхности. Некоторые кол-
коллоидные стабилизаторы, например водная двуокись олова, действуют за счет
адсорбции каталитически активных ионов или коагуляции диспергированных
активных твердых частиц. Если принять, что процесс разложения зависит
от образования активированных молекул под действием положительных
катализаторов, то логично предположить, что стабилизаторы предотвращают
образование таких активированных молекул или инактивируют их. Поскольку
роль многих катализаторов, очевидно, связана с возможностью участия в цикле
быстро сменяющих друг друга процессов окисления и восстановления какого-
либо вещества, стабилизация может происходить в результате обрыва или
Литература 453
разрыва такой реакционной цепи. Если каталитически действующее вещество
находится в стенках сосуда, то стабилизатор может служить для такого изме-
изменения характера этой поверхности, при котором она потеряет способность
катализировать разложение.
Окись цинка не стабилизирует разбавленных растворов перекиси водорода
или даже увеличивает скорость их разложения; Пьеррон [58] объясняет такое
поведение окиси цинка на основании гипотезы, что окислы металлов, функцио-
функционирующие в качестве стабилизаторов, превращаются в гидратированные пере-
.ООН
киси по типу МО-»М^ , или МО2-Н2О, а возможно, МО2-Н2О2, или
/Оон он
M<f . По Пьеррону, если отношение количеств окиси металла и перекиси
ХООИ
водорода такое, что образование перекиси металла не доходит до конца,
то наблюдается либо небольшая стабилизация, либо слабое ускорение процесса
разложения. В отношении окислов металлов, несомненно являющихся ката-
катализаторами разложения (например, окислов свинца, меди, марганца, серебра
или ртути), предполагается, что образование перекиси происходит в очень
малых размерах или даже вообще не происходит из-за неустойчивости таких
перекисей.
Логично приписать соотношение между концентрацией перекиси водорода
и стабилизирующим действием такого соединения, как окись или гидроокись
цинка, возможности смещения равновесия в реакции следующего типа:
.ОН .ООН
м ( + ноон ;± м < + н2о.
хон хон
Если в соответствии с мнением Пьеррона гидроокись металла, содержащая
гидроперекисную группу, является активным стабилизатором, то она должна
обладать максимальной эффективностью в присутствии избытка перекиси
водорода; в разбавленном растворе перекиси водорода она будет подвергаться
гидролизу с вторичным образованием гидроокиси металла (или водной или
гидратироваиной окиси); неясно только, почему смешанная гидроперекись—
гидроокись является эффективным стабилизатором, а гидроокись или окись
лишены этой способности.
ЛИТЕРАТУРА
1. Roth E. M., S h a n 1 е у Е. S., Ind. Eng. Chem., 45, 2343 A953).
2. G i g u e г е P. A., G e о f f r i о n P., Can. J. Res., 27B, 168 A949).
3. S с h u m b W. C, Ind. Eng. Chem., 41, 992 A949).
4. Белли ii джер Ф., Фридман Г., Бауер В., Истс Дж., Бал В.,
в сб. «Физика и химия реактивного движения», Госиноиздат, М., 1948, стр. 152.
5. Drew С. М., W h i t t a k e r A. G., U. S. Naval Ordnance Test Station Tech,
Memo. 1602, Inyokern, Calif., 1954.
6. S с h u m b W. С, неопубликованная работа по M.I.Т., Hydrogen Peroxide Project.
7. Horkheimer P., Pharm. Ztg., 80, 507 A935); 79, 582 A934); Anon., Ceram
Ind., 56, № 6, 80 A951).
8. И в а н о в И. В., Д о ч и к я н А., Советская фармакология 6, № 6, 25 A935).
9. W а 1 t о n J. Н., Judd R. С, 8th Intern. Congr. Appl. Chem., Orig. Comm.,
26, 621 A912).
Ю.Лурье С. Н., Колбин Н. И., ЖХП, 14, 757 A937).
И. I. G. Farbenindustrie Aktiengesellschaft, Evaluation of Rocket Fuel, Rept. 597 of
Tech. Test Stand of I. G. Oppau, 1944, T.O.M. Reel 32, Translation PC-S-III, Vol.
I, published by C. A. Meyer & Company, New York.
12. Ill э н л и Э., Г р и н с п э н Ф., в сб. «Физика и химия реактивного движения»,
№ 2, Издатинлит, М., 1949, стр. 161.
13. Slater V. W., Chemistry & Industry, 1945, 42.
454 Глава 9
14. S с h u m b W. C, Doyle J. R., Abstracts of Papers, XHth Int. Congr. of Pure
and Appl. Chem., p. 508, IX, 1951.
15. Nees H., P. B. report 74329 (frames 1211—1216).
16. Buchi J., Kurer V., Pharm. Acta Helv., 15, 59 A940).
17. H e i d t L. J., частное сообщение, 1954.
18. W i 11 i a m s В. Н., Trans. Faraday Soc, 24, 245 A928).
19. M a r q u a n d СВ., McElroy О. Е., К e t h 1 e у Т. W., P. В. report 13673
A942).
20. В r e t s с h g e г M. E., S h a n 1 e у E. S., Trans. Electrochem. Soc, 92, 67 A947).
21. К eyes F. G., Schumb W. С, В rough ton D. В., Р. В. report 27292.
22. U r i N., J. Phys. & Colloidal Chem., 53, 1070 A949).
23. G i 1 b e r t F. A., Michaelis N.. P. B. report 98779 A948).
24. T r i t t о n S. M., Quart. J. Pharm. Pharmacology, 12, 446 A939).
25. V e g t e r J. J. M., Pharm. Weekblad, 81, 363 A946).
26. F г a n с к W., Siewar t-Ki ba t D., Z. physiol. Chem., 281, 162 A944).
27. Richter D., J. Chem. Soc, 1934, 1219.
28. Campbell D. J., Am. Dyestuff Rep., 25, 67 A936).
29. Reichert J. S., пат. США 2091186 B4.VIII. 1937).
30. H e n к H. J., Fette u. Seife, 48, 508 A941) [C. A., 37, 2524].
31. Reichert J. S., пат. США 2008726 B3.VII. 1935).
32. S с h a i d h a u f А., пат. США 1275765 A3. VIII. 1918).
33. R e i с h e r t J. S., И a w к i n s о n A. J., пат. США 2426154 A9. VIII. 1947).
34. Nees H., P. B. report 74329 (frames 1217—1218).
35. S h a n 1 e у E. S., Kauffmann H. О, пат. США 2658818 A0. XI. 1953);
Elston А. А., пат. США 2497814 A4.11.1950); [англ. пат. 645225 B5. X. 1950)];
Gilbert H. N.. Reichert J.S., пат. США 2091178 B4. VIII. 1937);
Reichert J. S., англ. пат. 460208 B2. I. 1937); пат. США 2027839 (I. I. 1936);
Gilbert H. N.. Reichert J. S., англ. пат. 433470 A5.VIII. 1935);
пат. США 2004809 A1. VI. 1935); Deutsche Gold-und Silber-Scheideanstalt (vorm.
Roessler), герм. пат. 610185 G. III. 1935); франц. пат. 755562 B7. XI. 1933);
R e i с h e r t J. S., пат. США 1958204 (8. V. 1934); канадск. пат. 337601 E. XII.
1933); Liebknecht О., Schaidhauf А., пат. США 1213921 C0. I. 1917).
36. I s h i k a w a F., Haga H., Haga Y., J. Chem. Soc Japan, Pure Chem.
Sect., 69, 35 A948); N a g a s a w a S., ibid., 69, 72, 73 A948).
37. С a s t a g n о u R., Gaucher G., Bull. trav. soc. pharm. Bordeaux, 88, 98
A950).
38. Reichert J. S., пат. США 2027838 A4. I. 1936).
39. Weber I. E., Wood W. S., англ. пат. 435401 A7. IX. 1935).
40. Kausch О., Das Wasserstoffsuperoxyd, W. Knapp, Halle, 1938, p. 51—59.
41. M а с h u W., Das Wasserstoffperoxyd und die Perverbindungen, 2 Aufl. Springer-
Verlag, Wien, 1951, pp. 195—208.
42. Mar qua nd С. В., М с Е 1 г о у О. E., Kethley Т. W., М с N a m a-
г а В. P., report 11188 A942).
43. Н 6 1 1 К., Deut. Apoth. Ztg., 50, 252 A935); 54, 946 A939).
44. Sonol J., Rev. faculted cienc. quim. (la Plata Univ.), 9, 15 A934).
45. M a n с i n i M. A., BasiliscoL., Boll, soc ital. biol. sper., 9, 327 A934).
46. L i n d h о 1 m M., Farm Tidende, № 5 A933); Anales farm, bioquim. (Buenos Aires),
Suppl. 4, 70 [C. A., 28, 3524].
47. G г e e n s p a n F. P., пат. США 2624655 F. I. 1953).
48. L ieb k nech t О., пат. США 1002854 A2.IX.1911), 1058070 (8. IV. 1913).
49. P а г с h о m e n k о Р., Pharm. Zentralhalle, 82, 493 A941).
50. С а г г a r a G., M о n z i n i G., Chimica e industria (Italy), 23, 391 A941).
51. Davis P., Higginson W.C.E., J. Chem. Soc 1953, 1902.
52. L i s i e v i с i-D r a g a n e s с u A., Bui. soc. chim. Romania, 6, 42 A924) [C. A.,
18, 2449]; Porzky J., Z. ges. Textil-Ind., 39, 451 A936) [C. A., 30, 6951].
53. H e n k H. J., Seifensieder-Ztg., 67, 75 A940) [C. A., 34, 3028]; Deut. Parfum-Ztg.,
27, 1 A941) [C. A., 38, 2169].
54. F о 1 g n e г R., Schneider G., Melliand Textilber., 14, 452, 502 A933); 15,
24 A934); 18, 619 A937); Folgner R., Monatschr. Textil-Ind., 52, 257 A937).
55. Reichert J. S., Hi neg a rdner W. S., Potter H. L., пат. США 2160391 C0.V.
1939).
56. Schaidhauf А., пат. США 1181409, 1181410 B.V. 1916); канал, пат. 230967 (8.V.
1923).
57. Contant P., франц. пат. 782933 E.VII. 1935).
58. Pierron M. P., Bull. Soc. Chim. France E), 17, 291 A950); ibid. A949), p. 754.
59. D'Ans J., Mattner J., Angew. Chem., 64, 448 A952); 65, 368 A953).
60. Cooper K. A., Hill Т. С (to Minister of Supply), англ. пат. 649019 A7. I. 1951).
61. Isemura J., Nakamura S., Science (Japan), 19,379,A949).
Глава 10
МЕТОДЫ АНАЛИЗА
При анализе растворов, содержащих перекись водорода, может быть два
различных подхода: с одной стороны, очень важно иметь возможность открыть
или количественно определить в таких растворах перекись водорода (незави-
(независимо от того, содержится ли она в небольших количествах или представляет
основной компонент смеси); с другой стороны, приходится определять наличие
примесей или добавок в концентрированных или разбавленных растворах
перекиси водорода, в частности таких веществ, которые, несомненно, влияют
на стабильность раствора. В первом разделе описаны качественные пробы для
открытия и идентификации перекиси водорода без точного выяснения относи-
относительного ее содержания. Эти пробы не только должны быть чувствительны
к небольшим количествам перекиси водорода, но часто желательно также,
чтобы они были избирательными и давали возможность отличить перекись
водорода от других веществ, обычно от окислителей. Такие окислители иногда
фактически могут образовать перекись водорода или же последняя может
образоваться в результате восстановления атмосферного кислорода, например
под действием некоторых металлов. Поэтому результаты испытания на при-
присутствие или отсутствие перекиси водорода необходимо интерпретировать
с соблюдением надлежащей осторожности и с полным учетом всех других при-
присутствующих видов молекул, особенно в случае разбавленных растворов пере-
перекиси водорода. Вопрос о помехах при качественном и количественном анализе
обсуждается при рассмотрении отдельных методов анализа.
В связи с тем, что в настоящее время обычно выпускается исключительно
чистая перекись водорода, определение присутствия в ней других веществ
не имеет такого значения, как раньше. Такие анализы в основном предназна-
предназначены для проверки, не превышает ли содержание примесей в перекиси водорода
предел, установленный техническими условиями.
Методы анализа перекиси водорода достаточно полно рассмотрены в книге
Биркенбаха, опубликованной в 1909 г. [1 ]. Эта книга и до сих пор не утратила
своей ценности, поскольку содержит нужные ссылки на более старую литера-
литературу и оценку почти всех методов, обычно предлагаемых и в настоящее время
для анализа перекиси водорода. Более поздние обзоры, но не столь полные,
опубликованы Рейхертом, Мак-Нейтом и Радел ом [2] и Дювалем [31.
КАЧЕСТВЕННЫЙ АНАЛИЗ
Пробы, чувствительные на перекись водорода
Из различных реакций, которые можно использовать для открытия пере-
перекиси водорода или другого перекисного соединения, способного образовать
перекись водорода, чаще других упоминается реакция, основанная на образо-
образовании желто-оранжевой окраски титановой кислотой в присутствии перекиси
водорода. Эта проба весьма характерна и требует тщательного выполнения [41.
Ниже описан способ приготовления кислых растворов сернокислого титана
для этой цели.
456 Глава JO
Уже при содержании 1 или 2 мг перекиси водорода в литре ее можно
открыть по окраске, возникающей при взаимодействии такого подкисленного
раствора с указанным реактивом. Эта окраска приписывается образованию
иона [TiO2(SO4J]2~ [5]. Бериссо и Акино [6] описывают микрохимическую
форму выполнения этой пробы на присутствие перекиси водорода; для этого
применяется 10%-ный раствор сернокислого титана в б н. серной кислоте
и чистый амиловый спирт. Сливают по 3 мл каждой из этих жидкостей, встря-
встряхивают, выжидают до расслоения, отбирают 1 мл спиртового слоя, переносят
его в другую пробирку и добавляют одну каплю испытуемого раствора пере-
перекиси водорода.
Для качественного открытия широко применяется также реакция образо-
образования синей пероксохромовой кислоты. В пробирку, содержащую около 1 мл
анализируемого раствора (подкисленного несколькими каплями разбавленной
серной кислоты) и несколько миллилитров эфира, приливают несколько
капель разбавленного раствора двухромовокислого калия. Если при встря-
встряхивании эфирный слой приобретает синюю окраску, приписываемую пероксо-
пероксохромовой кислоте НСЮ5, то в испытуемом растворе содержится перекись
водорода; этот метод дает надежные результаты при наличии в пробе около
0,2 мг перекиси водорода с предельной концентрацией около 10 мг/л. Чувстви-
Чувствительность этого метода анализа можно еще более повысить путем применения
дифенилкарбазида, который реагирует с пероксохромовой кислотой с образо-
образованием красновато-фиолетовой окраски эфирного слоя, тогда как синей пе-
пероксохромовой кислоты может быть почти не видно. Этим способом можно
открыть уже 0,005 мг перекиси водорода в 5 или 10 мл жидкости [71.
Одним из очень чувствительных качественных методов открытия перекиси
водорода является обесцвечивание черного сернистого свинца с образованием
белого сернокислого свинца. Эта реакция служит основой микрохимического
испытания, по которому сернистый свинец получают на фотобумаге следующим
образом: после фиксации бумагу обрабатывают сначала разбавленным раство-
раствором уксуснокислого свинца, а затем сероводородной водой. При нанесении
на эту бумагу раствора, содержащего меньше 0,001 мг перекиси водорода,
окраска сернистого свинца светлеет [81. Сообщается о дальнейших усовер-
усовершенствованиях этого метода [9], обеспечивающих возможность применения его
для открытия Ю~5мг перекиси водорода при предельной концентрации 0,02мг/л.
Другой чувствительной цветной реакцией является так называемая реак-
реакция Шерна-Шельгазе [10], основанная на образовании синей окраски при
введении в реакцию с перекисью водорода свежеприготовленного спиртового
разбавленного раствора гваяковой смолы, содержащего диастазу; указывается,
что при помощи этой реакции можно открыть перекись водорода в разбавле-
разбавлениях до 0,02 мг/л. Эта реакция применяется для открытия перекиси водорода
в молоке [11], но она требует присутствия каталазы [12].
Для открытия перекиси водорода используются как ее восстановительные,
так и окислительные свойства. Так, Фейгль [13] описывает несколько капель-
капельных реакций на перекись водорода, в том числе реакцию, основанную на восста-
восстановительном действии перекиси в отношении разбавленной смеси хлорного
железа и железосинеродистого калия, с образованием берлинской лазури.
Этим путем можно открыть уже 0,0001 мг перекиси водорода при концентрации
не намного выше 1 мг/л. Другая проба [14] состоит в восстановлении солей
золота с образованием синих или красных коллоидных растворов золота; она
обладает примерно такой же чувствительностью, как и вышеуказанная. Пере-
Перекись водорода восстанавливает также окись трехвалентного никеля, что при-
приводит к значительному осветлению окраски этого соединения; микрохимиче-
микрохимическую пробу [15], основанную на этом эффекте, по имеющимся данным, можно
использовать для открытия всего только Ю мг перекиси водорода в кон-
концентрации 0,2 мг/л. По желто-красному осадку, образуемому перекисью водо-
Качественный анализ 457
рода с кислыми растворами роданидов щелочных металлов, можно открыть
7-Ю мг перекиси водорода [14]. Весьма чувствительную капельную реак-
реакцию на перекись водорода описали Кульберг и Матвеев [16]: при действии
о-толидина на перекись в присутствии Fe2+ получается синяя окраска уже при
наличии 2,5-10 б мг перекиси водорода при концентрации около 40 мг/л.
Другая чувствительная капельная реакция [17] базируется на применении
раствора трехвалентного сернокислого церия, который под действием избытка
раствора углекислого калия превращается в малорастворимую двойную соль
с промежуточным осаждением углекислого церия (III). Достаточно нанести
одну или две капли такого бесцветного раствора на фарфоровую пластинку,
в углублении которой содержится раствор перекиси водорода, чтобы в резуль-
результате окисления церия до четырехвалентного состояния получилась желтая
или коричневато-красная окраска.
Дениже [18] описал цветную реакцию, которую можно использовать для
обнаружения перекиси водорода при концентрации 0,2 мг/л. Реакция основана
на восстановлении феррицианида перекисью в аммиачном растворе до ферро-
цианида с выделением кислорода; получающийся оранжевый Ag3Fe(CN)g
хорошо растворим в разбавленном аммиачном растворе, тогда как белый
Ag4Fe(CN)e нерастворим. В реакцию с перекисью водорода вводится реактив,
содержащий железосинеродистое серебро, растворенное в аммиачном растворе;
при этом образуется белая муть. Кристаллы железистосинеродистого серебра
имеют характерную крестообразную форму. Если концентрация перекиси
очень мала, то для возникновения мути может потребоваться от 5 до 10 мин.
Аналогичный метод, а именно восстановление железосинеродистой меди или
цинка, предложен Коном [19].
Штам [20] описал цветную реакцию, посредством которой можно открыть
0,1 мг/л свободной перекиси водорода в эфире. Щелочной раствор, содержащий
фенолфталеин, восстанавливают цинковой пылью. К пробе эфира (около 1 мл)
добавляют каплю этого разбавленного обесцвеченного раствора и каплю раз-
разбавленного раствора медного купороса. В случае присутствия перекиси водо-
водорода на поверхности соприкосновения возникает розовая окраска. Лекок [21]
исследовал методику открытия перекиси водорода при помощи фенолфталеина
и чувствительность этой реакции.
Гле и Пфаннштиль [22] предложили интересную качественную реакцию
на перекись водорода, основанную на люминесценции, вызываемой ею в при-
присутствии люминола (гидразида 3-аминофталевой кислоты) с гемином как ката-
катализатором. Впоследствии эту реакцию изучали также Лангенбек и Руге [231
и Штейгман [24]. Штейгман рекомендует применять растворы люминола и мед-
медного купороса и свежеприготовленный раствор пероксодисульфата натрия
Na2S2O8. Co смесью этих трех растворов, взятых в надлежащих количествах,
люминесценцию можно обнаружить уже при наличии 2-Ю мг перекиси
водорода. Другие чувствительные методы открытия перекиси водорода, осно-
основанные на явлениях люминесценции, проанализировал Шалее [25].
Для открытия перекиси водорода предложены также азидная каталаза
[26], тетраметил-п-фенилендиамин [27], солянокислый я-фенилендиамин [28],
солянокислый бензидин [29], 2,7-диаминофлуорен [29] и ванилин [30].
Пробы для идентификации перекиси водорода
Интерес к вопросу о возможности различения перекиси водорода и разных
других окислителей впервые возник в связи с появлением антозоновой теории и
полемики вокруг нее [31 ] (см. стр. 16). Проблема открытия небольших количеств
перекиси водорода или других окислителей в атмосфере стоит уже давно,
причем и в последнее время вопрос о природе этих окислителей в атмосфере
приобрел определенное значение в связи с исследованиями загрязнения воз-
458
Глава 10
духа. Биркенбах [1] опубликовал обзор старой литературы по избирательным
методам открытия перекиси водорода и дал в виде таблиц сводку о специфич-
специфичности различных методов открытия; он считает, что единственно надежным
способом идентификации перекиси водорода является проба с титановым рас-
раствором. Кейзер и Мак-Мастер [32] оценили ряд способов испытаний и пришли
• к выводу, что для открытия перекиси водорода в присутствии озона и нитрита
специфично образование берлинской лазури из железосинеродистого калия
и хлорного железа. Они указывают, что при пропускании смеси озона, перекиси
Таблица 80
Испытания на открытие перекиси водорода в присутствии других окислителей,
по Меллору [33]
Реактив
Йодистый калий и
i/nov \Я Ск П
1\ Uo.AM.cLJi
Раствор индиго
Раствор лакмуса
Чистая серебряная
фольга
Ртутные шарики
СгО3 и эфир
Титановая кислота
Тетраметиловое ос-
нованиеа
Раствор КМпО4
Перекись водорода
Синяя окраска
Обесцвечивание
Совершенно без
изменений
То же
Посинение
Пожелтение
Совершенно без из-
изменений
Обесцвечивание
Озон
Синяя окраска
Обесцвечивание
Почернение
Оставляет след
на стекле
Совершенно без
изменений^
То же
Фиолетовая ок-
окраска
Без изменений
Хлор
Синяя окраска
Обесцвечивание
Белая пленка
Побелен ие
Совершенно без
изменений
То же
Синяя окраска
Обесцвечивание
Двуокись азота
Синяя окраска
Обесцвечивание
Без изменений
» »
Совершенно без
изменений
То же
Окраска от жел-
желтой до корич-
иоплиг
ncbUll
Обесцвечивание
а Тетраметилди-л-диаминодифенилметан. Реактивные бумажки пропитывают спиртовым раствором
этого реактива и применяют во влажном виде.
б По другим источникам озон при этом испытании дает синюю окраску.
водорода, хлора и закиси азота через раствор марганцовокислого калия один
только озон уходит из поглотителя в неизменном виде и может быть обнаружен
обычным путем по реакции с реактивом из йодистого калия и крахмала. Мел-
лор [33] привел ряд специфических проб, касающихся перекиси водорода,
озона, хлора и двуокиси азота. Все эти пробы приведены в табл. 80. Позже
Дэвис, Розен, Винджет и Уисвол [34] сравнили между собою шесть капельных
проб с точки зрения применения их для идентификации перекиси водорода
в присутствии фтора, фтористого кислорода, озона и некоторых комбинаций
этих веществ. Результаты приведены в табл. 81. Сокращение Р. н. обозначает
«реакции нет».
Желтая окраска пероксотитановой кислоты, по-видимому, является чув-
чувствительной и специфической для перекиси водорода. Так, ни озон [35],
ни пероксосульфаты [36] не дают желтой окраски с Ti(SO4J; окраска пробы,
содержащей перекись водорода, блекнет не так быстро, как это характерно
для некоторых других окрашенных соединений с меньшей стабильностью.
Возможно, что в том случае, когда другой окислитель дает эту реакцию, в дей-
действительности происходит предварительное образование перекиси водорода.
Вопрос о специфичности других методов испытания нельзя считать решенным.
Так, не все авторы сходятся во мнениях по вопросу о специфичности испыта-
испытания на образование пероксохромовой кислоты в присутствии озона. Другие
Качественный анализ
459
Таблица 81
Капельные реакции для открытия летучих окислителей, по Дэви су, Розену,
Винджету и Уисволу [34]
Реактив
Тетраметилди-
л-диаминоди-
фенилметано-
вая бумажка
Бензидин
К В г—флуорес-
цеин
Активирован-
Активированное серебро
KJ—крахмал
Кислота—осно-
Кислота—основание
Зелено-синяя
окраска
То же
Красная окрас-
окраска
Черная или жел-
желтая окраска
Пурпурная ок-
окраска
Покраснение,
затем обес-
обесцвечивание
OF2
Фиолетовая ок-
окраска
Желто-коричне-
Желто-коричневая окраска
Красная окрас-
окраска
Р. н.
Пурпурная ок-
окраска
Р. н.
Оз
. Фиолетовая
окраска
Желто-ко-
Желто-коричневая
окраска
Р. н.
Черная
окраска
Пурпурная
окраска
Р. н.
Р. н.
Р. н.
Р. н.
Р. н.
Пурпурная
окраска
Р. н.
HF
Р. Н.
Р. Н.
Р. Н.
Р. Н.
Р. Н.
Красная
окраска
Продолжение табл. 81
Реактив
Тетраметилди-
я-диамииоди-
фснилметано-
вая бумажка
Бензидин
КВг—флуорес-
цеин
Активирован-
Активированное серебро
HF+O8
Фиолетовая ок-
окраска
Сине-зелено-ко-
Сине-зелено-коричневая ок-
окраска
Желтая окрас-
окраска
Р. н.
OF2+O3
Фиолетовая ок-
окраска
Грязно-зелено-
коричневая
окраска
Красная окрас-
окраска
Р. н.
OF2+HF
Фиолетовая
окраска
Желто-ко-
Желто-коричневая
окраска
Желтая
окраска
Р. н.
OF2+O2+HF
Фиолетовая
окраска
Грязно-зе-
лено-ко-
лено-коричневая
окраска
Слабо-крас-
Слабо-красная окраска
Черная
окраска
O84-F2
Фиолетовая
окраска
Сине-зеле-
Сине-зеленая или
черная
окраска
Красная
окраска
Светло-ко-
Светло-коричневая
окраска
окислители, например озон, хлор или азотистая кислота, также дают цветную
реакцию с гваяковым раствором, даже без добавки диастазы. Реакцию с сер-
сернистым свинцом дают также другие окислители. Расходятся мнения и в том,
реагирует ли озон с водой с образованием перекиси водорода. Решение этого
вопроса осложняется тем, что сами перекись водорода и озон также реагируют
между собой. При низкой концентрации в газовой фазе при комнатной темпера-
температуре скорость этой реакции может быть ничтожной. Однако очень большое раз-
различие между летучестями перекиси водорода и озона дает возможность разли-
различить эти два соединения.
Вопрос о том, как отличить перекись водорода от какого-либо другого
неорганического или органического перекисного соединения, иногда очень
трудно решить, особенно в связи с тем, что гидролиз этого перекисного соеди-
соединения может привести к образованию перекиси водорода. Испытание на хеми-
люминесценцию не является полностью специфическим; некоторые пероксо-
соединения, например пероксобензойная кислота и пероксодисульфат аммо-
аммония, также способны давать хемилюминесценцию, хотя требуемая для этого
концентрация не должна быть ниже примерно 100 мг/л.
460 Глава 10
Некоторые органические перекисные соединения, например диоксиэтил-
перекись, способны давать реакцию, аналогичную той, которая наблюдается
при взаимодействии перекиси водорода с фенолфталеином, правда несколько
более медленную. Вопрос о том, как различить перекись водорода и разные
органические перекиси, рассматривался также в гл. 2 при обсуждении про-
проблемы окисления углеводородов (стр. 76).
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Наиболее общепринятые методы количественного определения перекиси
водорода следующие: 1) сочетание весового и объемного анализов, основанное
на титровании навески исследуемого раствора перманганатом или растворами
солей четырехвалентного церия или йодида; 2) объемный анализ, основанный
на титровании известного объема пробы с использованием соответствующей
диаграммы удельных весов; 3) газометрический анализ, т. е. измерение коли-
количества кислорода, выделяющегося при каталитическом разложении известного
количества раствора перекиси водорода; 4) колориметрический анализ, осно-
основанный на интенсивности окраски, появляющейся при реакции с перекисью
водорода, и 5) физические методы, например прямое измерение плотности
или показателя преломления раствора в отсутствие других растворенных
веществ. Кроме того, предложены различные способы оформления этих методов,
на чем кратко остановимся.
Следует отметить, что содержание воды в водных растворах перекиси
водорода предложено также определять путем титрования'реактивом К. Фишера,
состоящим из йода, двуокиси серы, пиридина и метанола, с определением ко-
конечной точки по коричневой окраске от избытка йода [37].
Объемные методы
Титрование перманганатом. Этот метод является одним из наиболее точных
и надежных способов, обычно употребляемых для анализа перекиси водорода.
Он применим как для разбавленных, так и для более концентрированных рас-
растворов и обладает предельной чувствительностью около 0,1 мг/л. При этом
методе определению мешают органические или неорганические восстанови-
восстановители, способные реагировать с перманганатом.
Характер протекающей реакции впервые выяснен Броди [38] и Шенбей-
ном [39] и впоследствии экспериментально подтвержден работами Ашофа
[40]. Эта реакция соответствует уравнению
2КМпО4 + 5Н2О2 + 3H2SO4 -» K2SO4 + 2MnSO4 + 8Н2О + 5О2. A)
Ряд авторов занимался вопросом об источнике кислорода, выделяющегося
при этой реакции, т. е. получается ли он из перекиси водорода и перманганата
или же только из одной перекиси водорода. В настоящее время бесспорно
доказано, что только одна перекись водорода является источником выделяю-
выделяющегося кислорода; кислород же, который сначала частично входит в состав
иона перманганата, находится в продуктах реакции в виде воды. Как показы-
показывает уравнение реакции, для проведения анализа необходимо наличие кислоты,
которая обычно добавляется к титруемому раствору в виде разбавленной
серной кислоты. Применение соляной кислоты, так же как и других источников
хлор-ионов, недопустимо, так как перманганат может окислить хлор-ион
до хлора и это исказит результат анализа. Если нельзя избегнуть наличия
хлорида, особенно в присутствии иона окисного железа, рекомендуется доба-
добавлять соль двухвалентного марганца, например сульфат, в присутствии которой
окисление хлорида перманганатом сильно тормозится. Присутствие даже
Количественный анализ 461
следов иона двухвалентного марганца в титруемом растворе оказывает на про-
процесс окисления—восстановления также каталитическое влияние и способ-
способствует мгновенной реакции между перманганатом и раствором восстановителя.
При полном отсутствии соли двухвалентного марганца розовая окраска,
образующаяся при приливании первых нескольких капель перманганата
к титруемому раствору, иногда исчезает лишь через некоторое время в резуль-
результате взаимодействия перманганата с этим раствором; но как только образо-
образовалось минимальное количество Мп2+, скорость реакции возрастает и розовая
окраска при смешении с раствором мгновенно исчезает. Наличие органических
веществ в растворе перекиси водорода, например салициловой кислоты, гли-
глицерина и т. п., очевидно, может исказить результаты, так как сами эти вещества
также реагируют с перманганатом. Если проба содержит оксалат (например,
в отбеливающих ваннах), то, по Симону и Ритцу [41], определяют сначала
сумму оксалата и перекиси водорода путем восстановления перманганата.
Затем в свежей пробе до определения оксалата каталитически разлагают
перекись водорода. Наличие фтор-иона неблагоприятно отражается на
результатах титрования перманганатом [42]; в присутствии больших
количеств кальциевых солей или титановой кислоты получаются заниженные
результаты.
Титр раствора марганцовокислого калия, применяемого для этого ана-
анализа, устанавливается лучше всего по щавелевокислому натрию в соответствии
с тщательно разработанной методикой Фаулера и Брайта [43]. Такие растворы
сохраняются в течение длительного промежутка времени, если после пригото-
приготовления их нагревают и затем фильтруют через пористую стеклянную пла-
пластинку (до установления титра). Склянку с раствором нужно хранить в тем-
темноте или защищать ее черной тканью или каким-либо другим покрытием,
непроницаемым для света.
Методика [43]. «В стакан емкостью 600 мл вносят 0,3 г щавелевокислого
натрия, высушенного при 105°. К нему приливают 250 мл разбавленной серной
кислоты E : 95), предварительно прокипяченной в течение 10—15 мин. и затем
охлажденной до 27^3°. Смесь перемешивают до растворения оксалата. Затем
приливают при медленном перемешивании 39—40 мл 0,1 н. раствора КМпО4
со скоростью 25—35 мл в минуту. Раствор выдерживают до исчезновения розо-
розовой окраски, на что требуется около 45 сек. Если розовая окраска сохраняется
из-за слишком высокой концентрации перманганата, раствор выливают и опе-
операцию начинают снова, причем раствор перманганата приливают уже в мень-
меньшем (на несколько миллилитров) количестве».
«Смесь нагревают до 55—60° и завершают титрование путем приливания
раствора перманганата до тех пор, пока слабо-розовая окраска не будет сохра-
сохраняться в течение 30 сек. Последние 0,5—1 мл приливают по каплям, пережидая,
пока не обесцветится каждая предыдущая капля».
«Определяют избыток перманганата, необходимый для сообщения рас-
раствору розовой окраски. Для этого готовят раствор путем добавления
перманганата к такому же объему прокипяченной и охлажденной разбав-
разбавленной серной кислоты при 55—60°. Эта поправка обычно составляет 0,04—
0,05 мл».
«При применении потенциометрического титрования поправка ничтожно
мала, если только приближение к конечной точке осуществляется медленно».
Однонормальный раствор КМпО4 содержит 0,2 моля перманганата на литр,
а поэтому 0,1 н. раствор содержит КМпО4 в количестве 3,1606 г/л. Таким обра-
образом, 1 мл децинормального раствора марганцовокислого калия эквивалентен
1,701 мг перекиси водорода. Обычно целесообразно, чтобы проба раствора
перекиси водорода, которая титруется перманганатом, содержала около 0,3 г
перекиси водорода (в расчете на 100%-ную, т. е. около 1 г 30%-ной перекиси
водорода) при титровании 0,5 н. раствором перманганата или около 0,05 г
462 Глава 10
перекиси (в расчете на 100%-ную) при титровании 0,1 н. раствором перманга-
ната. Удельный вес раствора перманганата слабо изменяется с температурой.
Так, нормальность 0,5 н. раствора перманганата снижается приблизительно
на 0,0002 при повышении температуры на Г; при особо точных работах может
оказаться необходимым учет разности рабочей температуры и температуры,
при которой был установлен титр перманганата.
Концентрацию раствора перманганата обычно подбирают таким образом,,
чтобы она соответствовала концентрации испытуемого раствора перекиси
водорода. Децинормальный раствор пригоден для анализа растворов перекиси
водорода в широком интервале концентраций, но часто применяют даже столь
высокие концентрации перманганата, как 0,5 н., причем соответственно увели-
увеличивают навеску анализируемой перекиси. Выбор концентрации перманганата
может зависеть также от того, желательно ли разбавить взятую навеску пере-
перекиси водорода и использовать ее аликвотную часть для титрования или же
непосредственно внести пробу в бюксе в сосуд для титрования. При титрова-
титровании нужно обязательно иметь в виду, что не следует приливать перманганат
к раствору со слишком высоким содержанием перекиси водорода. При 0,1 н.
перманганате концентрация перекиси водорода не должна превышать при-
примерно 0,1—0,2 вес.%.
Анализ раствора перекиси водорода производят следующим образом.
Методика. Во взвешенный чистый сухой бюкс наливают раствор в коли-
количестве, не превышающем 0,7 г перекиси водорода в расчете на 100%-ную.
Для 90%-ного раствора это составляет около 0,75 г, или приблизительно 10 ка-
капель; для 50%-ного раствора—1,4 г; для 30%-ного—2,3 г и для 6%-ного—12 г.
Для определения веса пробы бюкс с раствором вновь взвешивают. Содержи-
Содержимое бюкса при помощи воронки и промывалки переносят в мерную колбу
на 500 мл\ по заполнению почти всего объема колбы ее содержимое осторожно
перемешивают путем вращения колбы; затем доливают колбу до метки и тща-
тщательно перемешивают раствор путем взбалтывания.
Пипеткой отбирают 25 мл раствора из мерной колбы в колбу Эрленмейера
на 250 мл и приливают 1 мл концентрированной серной кислоты. Из бюретки
приливают 0,1 н. раствор КМпО4 со скоростью не выше 0,5 мл в секунду
при постоянном перемешивании. Достижение конечной точки определяют
по появлению светло-розовой окраски, устойчивой в течение одной минуты.
Если слишком быстро приливать перманганат (или недостаточно энергична
производить перемешивание, или брать недостаточное количество кислоты),
может возникнуть бурая окраска, обусловленная образованием двуокиси
марганца. Независимо от причины образования двуокиси эта проба бракуется,
так как результаты такого анализа будут ошибочными. Содержание перекиси
водорода в весовых процентах вычисляется из следующего соотношения:
Т4 п / о/\ inn (мл КМпО4) (н.КМпО4) (мэкв Н2С>2)(коэффициент разбавления)
Н2и2(вес. /о) = 1UU Навеска пробы с Н2Оа (г) '
При вышеуказанной методике анализа, если взять аликвотную часть
в размере г/20 от исходного раствора, эта формула приобретает следующий вид:
НО (п^г цч 34,0(*лКМпО4) (н. КМпО4)
п2и2^вес. /oj- Навеска пробы с НаОа(г) '
Для низкоконцентрированных растворов можно обойтись без разбавления
в мерной колбе и непосредственно титровать пробу после подкисления и добавки
около 100 мл воды. Для 30%-ного раствора берут навеску 0,1 г, для 6%-ного—
около 0,5 г. В этом случае при расчете отпадает необходимость введения коэф-
коэффициента разбавления.
Для серийных анализов, не требующих очень высокой точности, пробу
можно отбирать пипеткой, а вес ее находить путем использования диаграммы
Количественный анализ 463
для плотности*. Если требуются точные результаты, необходимо учитывать
также изменение плотности с температурой.
В специальной литературе, выпускаемой фирмами, производящими пере-
перекись водорода, описаны также другие способы анализа, не отличающиеся
по принципу от вышеизложенного, но приспособленные для определенных
условий концентрации, а также и для требуемой скорости и точности.
Другие варианты перманганатного метода основаны на реакции раствора
перекиси водорода с известными количествами солей закисного железа или
двухлористого олова с оттитрованием избытка этих восстановителей раствором
перманганата. Перманганат может быть также использован для выделения газо-
офазного кислорода, количество которого определяется газометрически. Однако
при отсутствии помех прямое титрование, описанное выше, оказывается вполне
удовлетворительным. Если условия такие, что конечная точка не появляется
резко, то титрование можно выполнить потенциометрическим методом или же
использовать о-фенантролин в качестве индикатора [2].
Перманганатный метод, как доказали Жигер и Жоффрион [44], дает луч-
лучшие результаты, чем тиосульфатный. Хукаба и Кейс [45] исследовали точность
титрования перманганатом путем сравнения полученных результатов сданными
каталитического газометрического разложения, представляющего собой абсо-
абсолютный метод. Результаты этих методов сходились в пределах 0,02%. Конечно,
обычно точность такого кропотливого исследования не может быть достигнута.
При серийных анализах и применении взвешивания пробы точность обычно
не превышает 0,1%; если же для отбора пробы применяется пипетка на 0,1 мл,
можно ожидать точности лишь около 1% [46].
Йодометрические методы. Несколько менее точным, чем перманганатный,
но тем не менее находящим широкое применение, особенно в интервале кон-
концентраций перекиси водорода 1—6%, является метод Кингцета [47], по кото-
которому применяется слабоподкисленный раствор йодистого калия (лучше всего
в присутствии следов молибдата аммония в качестве катализатора) [48].
При этом происходят следующие реакции (в которых ион йодида играет роль
восстановителя):
Н2О2 + 2KJ + H2SO4 -» J2 + K2SO4 + 2Н2О, B)
J2 + 2Na2S2O3—>Na2S4Oe + 2NaJ. C)
Типичная методика состоит в приливании соответствующим образом разбавлен-
разбавленного раствора перекиси водорода, содержащего молибдатный катализатор,
к 50 мл 1%-ного раствора йодистого калия, подкисленного 1 мл раствора
серной кислоты A : 4) и нагретого до 40°. После перемешивания и выдержи-
выдерживания в течение 5 мин. выделившийся йод оттитровывают раствором тиосуль-
тиосульфата в присутствии крахмала как индикатора или без крахмала. Поскольку
при длительном стоянии подкисленного раствора йодистого калия в контакте
с воздухом возможно выделение йода даже в отсутствие каких-либо реагентов,
желательно ввести поправку в результаты анализа путем постановки контроль-
контрольного опыта с применением таким же образом подкисленного раствора йоди-
йодистого калия, который выдерживают на воздухе в течение такого же времени
при той же температуре.
Очевидно, что этот метод неприменим в присутствии других окислителей,
способных, выделять йод из йодида, например хлора, озона, окиси азота или
азотистой кислоты, или же непредельных органических веществ, способных
к реакции с выделившимся йодом. В отсутствие таких соединений йодидный
* Для этой цели рекомендуется составить диаграмму соотношения между нормаль-
нормальностью и количеством перекиси водорода в весовых процентах с использованием отношения.
(Плотность раствора Н2О2)-(Весовой процент Н2О2)=1,7 (нормальность Н2О2).
464 • Глава 10
метод дает хорошие результаты даже в присутствии органических веществ
типа Сахаров или глицерина, нежелательно влияющих на титрование перман-
ганатом. Этот метод широко применяется [49] для определения органических
перекисей, например в растворе в изопропаноле; сделано наблюдение [50],
что он может быть с успехом использован для определения перекиси водорода
в растворе ацетона. Боле и Хейл [51] показали, что добавка молибдатного
катализатора не только ускоряет окисление йодида, но может способствовать
также взаимодействию органических веществ с перекисью водорода. В то же
время при стоянии возможно медленное дополнительное образование йода;
предложено вместо МоО^" применять пирогаллол, который устраняет это явле-
явление и одновременно предотвращает реакцию перекиси водорода с органиче-
органическими веществами. В литература [52] имеется оценка различных других йодо-
метрических методов в приложении к титрованию смесей перекиси водорода
и альдегидов на общее содержание перекиси.
Титрование цериевыми соединениями. Для определения концентрации
растворов перекиси водорода, особенно в присутствии органических веществ,
рекомендуется также объемный метод, основанный на восстановлении иона
четырехвалентного церия. Сообщается [53], что в присутствии эфира, лишен-
лишенного перекисей, этанола или смеси эфира с этанолом титрование ионом четырех-
четырехвалентного церия до конечной точки, определяемой при помощи 1,10-фенан-
тролинового комплекса закисного железа или электрометрически, дает более
удовлетворительные результаты, чем титрование перманганатом. Можно при-
применять растворы сульфата четырехвалентного церия по спосебу, совершенно
аналогичному титрованию перманганатом, но желтая окраска в конечной
точке несколько хуже различается, чем розовая от перманганата. Поэтому
целесообразнее потенциометрическое титрование [54]. Исследован также ряд
индикаторов [55] для этой реакции, а именно родамин 6G и кристаллический
фиолетовый [56]; закисножелезный комплекс нитрофенантролина [57] обес-
обесцвечивается слабее, чем такой же комплекс с 1,10-фенантролином. Хурдис
и Ромейн [57] изучили точность определения перекиси водорода путем титро-
титрования четырехвалентным церием и показали, что точность этого метода
такая же, как и при абсолютном газометрическом методе или при титровании
перманганатом. Эти авторы приводят подробности методики установления
титра раствора церия, рассматривая этот метод также с точки зрения надеж-
надежности и стоимости различных источников церия.
Другие объемные методы. По другому объемному методу [58] приливают
избыток арсенита, который затем обратно оттитровывают раствором йодата ка-
калия в присутствии большого количества соляной кислоты. Выделяющийся йод
окисляется до однохлористого йода в присутствии индикатора; при наличии
следов избыточного йодата индикатор обесцвечивается. В качестве индикаторов
можно применять красители: амарант (прочный красный D), бриллиантовый
пунцовый 5R и нафтоловый сине-черный (British Colour Index № 184, 195
и 246 соответственно) [59]. Эти индикаторы не разлагаются в присутствии кон-
концентрированной соляной кислоты.
Кнехт и Хибберт [60] рекомендуют объемный метод, основанный на при-
применении разбавленного раствора треххлористого титана. Этот раствор при-
приливают к подкисленному раствору перекиси водорода, причем последней
приобретает сначала желтую окраску, затем темно-оранжевую (из-за образова-
образования пероксотитановой кислоты) и наконец обесцвечивается в связи с восстано-
восстановлением пероксокислоты до двуокиси титана. Этот метод применим и в при-
присутствии органических веществ, но титр раствора треххлористого титана необ-
необходимо все время проверять при помощи раствора хлорного железа, в свою
очередь приготовленного из известного количества соли Мора. Поскольку
конечную точку определить трудно [21, Триттон [61] считает, что методе трех-
хлористым титаном уступает перманганатному или йодометрическому. Синг
Количественный анализ . 465
и Малик [62] применяли сернистокислый натрий в качестве восстановителя
в кислом растворе с избытком йодистого калия в процессе потенциометриче-
ского титрования. Швикер [63] для восстановления перекиси водорода приме-
применял избыток KHSO3 с последующим определением свободной серной кислоты
титрованием раствором едкого натра после добавки достаточного количества
3%-ного раствора формальдегида для связывания избытка бисульфита. Рупп
и Вегнер [64] пользовались непосредственно двуокисью серы.
Далее нужно упомянуть о потенциометрическом титровании щелочным
раствором гипохлорита натрия [65], применимом, по литературным данным,
к определению перекиси водорода в присутствии таких пероксосолей, как
K,S2Od, KLSO5 или пероксофосфаты. Фере [66] использовал в качестве основы
для объемного метода реакцию между перекисью водорода и хлоргипохлоритом
кальция СаОС12, при которой получается хлористый кальций и кислород.
Перекись водорода выделяет бром из гипобромита; эта реакция положена
в основу метода анализа, при котором свободный бром оттитровывается арсе-
нитом [67]. Описано также потенциометрическое титрование железосинероди*
стым калием в присутствии едкого кали [65]. Дениже [68] разработал объем-
объемный феррицианидный метод. Белчер и Вест [69] для объемного определения
перекиси водорода использовали азотнокислую закисную ртуть в качестве
восстановителя. Новинкой среди методов, основанных на восстановлении,
можно считать применение аскорбиновой кислоты в качестве титрующего
агента [70]; этот так называемый «аскорбиметрический метод», по данным его
авторов, применим для определения многих окислителей, в том числе и Н2О2.
Рейхерт, Мак-Нейт и Радел [2] сравнили данные потенциометрического
титрования перекиси водорода такими реагентами, как перманганат, соль
Мора, тиосульфат, арсенит, двухлористое олово, треххлористый титан, суль-
сульфит и нитрит. Они считают, что наилучшие результаты при окрашенных рас-
растворах, содержащих органические вещества, дает нитрит, и описывают подроб-
подробную методику применения последнего.
Газометрические методы
Эти методы основаны на измерении объема кислорода, выделяющегося
из известного количества раствора перекиси водорода, либо при каталитиче-
каталитическом разложении
2НаО2 -> 2НаО + О2, D)
либо при взаимодействии с окислителем, например с кислым раствором пер-
манганата, щелочным раствором феррицианида или нейтральным раствором
гипохлорита натрия [71]:
2КМпО4 + 5Н2О2 + 3H2SO4 -» K2SO4 + 2MnSO4 + 8Н2О + 5О2, A)
2K3Fe (CN)e + 2КОН + НаО2 -> 2K4Fe (CN)e + 2H2O + O2, E)
NaOCl + H2Oa -> NaCl + H2O + O2. F)
При каталитическом разложении 1 моль перекиси водорода дает 0,5 моля
кислорода, тогда как при реакции перекиси водорода с окислителями на каждый
моль перекиси водорода выделяется 1 моль кислорода.
При каталитическом способе перекись водорода в пробе можно раз-
разложить при помощи тонкодисперсной платины или серебра, коллоидных
растворов этих металлов или твердой двуокиси марганца; объем выделившегося
кислорода измеряют в соответствующем приборе для газового анализа. В био-
биохимических работах часто используется каталаза [72].
30 Перекись водорода
466 Глава 10
Независимо от того, получается ли кислород непосредственно путем ката-
каталитического разложения перекиси водорода или при взаимодействии послед-
последней с окислителем, в результатах могут быть ошибки двух типов, а именно
из-за растворимости кислорода в воде и возможности пересыщения растворен-
растворенным кислородом применяемого раствора или запирающей жидкости прибора
для измерения газа [73]. При применении бюретки с ртутью на мениск ртути
в бюретке наливают 1—2 мл воды, насыщенной кислородом, чтобы собираю-
собирающийся кислород был насыщен водой при температуре бюретки. Трудности га-
газометрического анализа и методика его рассмотрены на стр. 431. При анализе
(в отличие от измерения скорости) желательно полное разложение; этот фактор
влияет на выбор необходимого прибора. Описаны подробные методики, обес-
обеспечивающие в опытах получение точных результатов [45, 57]. Хаит [74] опи-
описал очень простой и доступный метод, основанный на применении аммиачного
раствора сернокислой меди для разложения перекиси водорода в градуирован-
градуированной ферментационной трубке. Матзура [75] предложил определять газ, выде-
выделяющийся при разложении перекиси водорода, измеряя количества возни-
возникающей пены, стабилизированной сапонином. Меры предосторожности [76],
предложенные при применении метода выделения кислорода в предварительно
эвакуированную камеру, по-видимому, не оправдываются. Эллиот [77] пока-
показал, что при определении перекиси водорода путем разложения двуокисью
марганца при наличии белка получаются завышенные количества кислорода.
Хотя классический газометрический метод определения перекиси водо-
водорода нашел применение раньше других методов и при соблюдении надлежащих
условий дает абсолютные результаты, в обыденной практике в настоящее
время его все же применяют редко, так как он менее удобен, чем некоторые
другие методы.
Колориметрические методы
Перекись водорода может быть определена различными колориметриче-
колориметрическими методами [78]. Так, можно использовать окраску, образующуюся при
введении пробы, содержащей перекись водорода, в реакцию со смесью крах-
крахмала и йодистого калия; эта окраска с известными ограничениями может
считаться показателем содержания перекиси водорода в пробе [79]. Хотя этот
метод, по-видимому, отличается высокой чувствительностью, он все же не вполне
надежен, так как разбавленные растворы перекиси водорода медленно реаги-
реагируют с йодистым калием и при длительном стоянии в контакте с воздухом ион
йодида в известной мере непосредственно окисляется до свободного йода.
Интервал концентраций перекиси водорода, для которых этот метод применим,
колеблется примерно от 0,08 до 0,5 мг/л. Если йода выделяется больше 1 мг/Лу
то интенсивность синей окраски раствора в присутствии крахмала не может
быть количественно измерена.
Реакция перекиси водорода и иона йодида изучена в отсутствие крахмала
спектрофотометрированием получаемого раствора йода при 320 ммк, и в этом
случае оказалось возможным открыть еще 0,3 мг/л перекиси водорода [80].
Эта методика использована в системе для непрерывного измерения [81].
Проба с образованием пероксотитановой кислоты приспособлена для
колориметрического количественного определения небольших количеств пере-
перекиси водорода в водном растворе [82—84]. Боие-Мори [85] применял следую-
следующий раствор реактива: 10 г Ti(SO4J, 50 мл воды и 20 г концентрированной
серной кислоты; этот реактив выдерживали 24 часа и затем центрифугировали
для осветления раствора* (высокая кислотность необходима для предотвраще-
предотвращения выпадения гидроокиси титана). На 1 мл испытуемого раствора берут
* Раствор Ti (SO4J можно приготовить также, по Эйзенбергу [83], путем растворения
Х~г TiOa в 100 мл концентрированной серной кислоты, нагреванием раствора в течение
15 час. при 150° с последующим охлаждением и прибавлением 400 мл воды.
Количественный анализ 467
1 каплю реактива, развивающуюся окраску измеряют в фотоэлектрическом
колориметре и отсчитывают результаты по кривой, построенной по данным
измерения известных количеств перекиси водорода. Сообщается, что этим
путем можно еще открыть 0,1 мг/л перекиси водорода.
Ринасевич [86] показал, что силикат, молибдат или вольфрамат при
содержании их в 10 М перекиси водорода в количестве порядка 100 мг/л
не ^мешают колориметрированию по окраске пероксотитановой кислоты. Хромат
мешает этой реакции, однако для устранения этой помехи разработаны особые
методы. Раствор пятиокиси ванадия в концентрированной серной кислоте, как
показало сравнение его с титановым реактивом, обладает несколько меньшей
чувствительностью, чем реактив Ti(SO4J; он также был использован для колори-
колориметрического определения перекиси водорода [82]. Окраска в этом случае
изменяется от зеленой до коричневой.
Муса, Хигасино и Дой [87] описали применение ферронового реактива,
или 7-йод-8-оксихинолин-5-сульфокислоты, который при микроопределении
перекиси водорода образует темно-зеленый комплекс с ионом окисного железа
(но не закисного). Измеряют количество Fe3+, образовавшегося из Fe2+ при
окислении перекисью водорода, и отсюда вычисляют количество перекиси.
Ионы тяжелых металлов, так же как окисляющие или восстанавливающие
ионы, мешают этому анализу.
Колориметрическое определение [2] красной окраски роданистого железа,
возникающей при реакции раствора .сернокислого закисного железа с пере-
перекисью водорода в присутствии роданистого аммония [88], обладает, по имею-
имеющимся данным, чувствительностью около 10 мг/л перекиси водорода. Для
колориметрического определения перекиси водорода предложены также
молибдат [89], окись меди [90], фенолфталеин [91], флуоресцеин [21, дихлор-
диоксидифениламин [92] и аминопирин [93]. Аллен [94] сравнил между собой
ряд микрохимических колориметрических методов определения перекиси
водорода и озона и пришел к заключению, что для перекиси водорода наилуч-
наилучшим реактивом является перманганат.
Полярографический анализ
Полярографическое микроопределение перекиси водорода [95—97] осно-
основано на том, что при растворении кислорода в воде получаемая полярограмма
имеет два плато, из которых одно обусловлено восстановлением кислорода
до перекисного состояния, а второе—восстановлением перекиси. Этим путем
можно открыть еще 10 мг в пробе 2 мл. Жигер и Жайе [96] изучили
полярографический анализ разбавленных растворов перекиси водорода в
основном с целью выяснения применимости капельного ртутного электрода
с неподвижным платиновым электродом для непрерывного анализа движущих-
движущихся растворов; они пришли к заключению, что твердые микроэлектроды мало
подходят для этой цели. Полярографический метод использован также для
контроля чистоты растворов перекиси водорода путем открытия в них таких
примесей, как соединения железа, свинца и меди [97], и для определения
содержания стабилизаторов, например станната. Реймерс [98] разработал
полярографический метод для открытия перекиси водорода в присутствии
перекиси эфира.
Физические методы анализа
Для серийных определений перекиси водорода в водных растворах,
не содержащих других растворенных компонентов, можно использовать чисто
физические методы анализа, например измерение показателя преломления
или плотности раствора. Показатель преломления [99] может быть измерен
быстро, например при помощи иммерсионного рефрактометра; концентрацию
30*
468 Глава 10
перекиси водорода можно затем найти по соответствующим таблицам (стр. 229).
Результаты вполне сравнимы с получаемыми методами титрования. Очевидные
преимущества этого метода с применением иммерсионного рефрактометра—
удобство и быстрота—снижаются в связи с затратами на приобретение прибора,
а также необходимостью иметь довольно значительную пробу. Если же поль-
пользоваться более удобным рефрактометром Аббе, для которого нужна небольшая
проба, приходится сталкиваться с проблемами тщательного контроля темпера-
температуры и защиты пробы перекиси водорода от каталитического разложения. Если
необходимо поставить большое число серийных анализов, то рефрактометриче-
рефрактометрический метод часто оказывается наиболее быстрым й удобным. Рефрактометр
должен быть снабжен специальными призмами (поставляемыми той же фирмой),
позволяющими снизить размер разложения до минимума.
При анализе чистых водных растворов перекиси водорода в качестве
непосредственного метода измерения концентрации можно использовать и опре-
определение плотности раствора [100]. Для этой цели пригодны данные типа приве-
приведенных на стр. 174. При определении плотности при помощи пикнометра, арео-
ареометра или весов Мора—Вестфаля возможны значительные ошибки из-за обра-
образования пузырьков кислорода вследствие разложения перекиси водорода [101].
Разработано [102] автоматическое приспособление с температурной компенса-
компенсацией для непрерывного измерения и записи плотности растворов Н2О2.
Инфракрасное поглощение перекиси водорода не настолько интенсивно,
чтобы его можно было использовать в качестве удовлетворительной основы
метода анализа; к тому же спектр перекиси весьма близок к спектру воды.
Ультрафиолетовое поглощение перекиси водорода отличается интенсивностью,
и хотя растворы перекиси не вполне строго подчиняются закону Бера, все же
анализ, если эти растворы бесцветны и совершейно прозрачны, можно произ-
производить спектрофотометрическим методом даже при концентрации перекиси,
не превышающей 8 мг/л Н2О2 [103].Жигер [104] описал прибор, применяемый
для этой цели, и методику анализа. Ультрафиолетовая спектрофотометрия
особенно подходит для анализа разбавленных растворов и паров. Однако этот
метод может быть использован [103] и для определения концентрации жидко-
жидкости вплоть до содержания по меньшей мере 50 вес. % перекиси водорода, если
измерять разность поглощения известной и неизвестной проб.
Точным методом определения концентрации перекиси водорода является
измерение температуры замерзания ее чистых водных растворов; так, у рас-
растворов с высоким содержанием перекиси водорода температура замерзания
изменяется приблизительно на Г на каждый весовой процент состава. Однако
этот метод следует рассматривать лишь как специальный; в качестве обычного
экспериментального метода он не подходит. В особых условиях использована
для открытия перекиси водорода и масс-спектрометрия [105]. Другие физиче-
физические свойства растворов перекиси водорода применяются очень редко для
анализа. Хотя диэлектрическая проницаемость как будто и является удобным
показателем концентрации перекиси водорода, но, поскольку кривая диэлек-
диэлектрической проницаемости как функция концентрации обладает максимумом,
для ее использования необходимо предварительно знать приближенный состав
раствора. Кроме того, этот метод пригоден лишь для анализа чистейших проб,
так как уже следы примеси электролитов влияют на электропроводность и таким
образом обусловливают ошибки в измерениях [106]. Для определения перекиси
водорода предложено также применение гальванических элементов [107].
АНАЛИЗ СМЕСЕЙ
Определение перекиси "водорода
При электрохимических методах производства перекиси водорода необ-
кодимо определять относительное содержание ее в смеси, в которой имеются
'также пероксоеерная и пероксодисерная кислоты. По этому вопросу имеет-
Анализ смесей 469
ся обширная литература; частично обзор ее дан Риусом [108] и Берри [109].
Методика определения всех этих трех веществ, оправдавшая себя на прак-
практике [ПО], принадлежит Гле [111]. По этому методу сначала определяют
пероксосерную кислоту с бромидом, затем перекись водорода при помощи
перманганата и, наконец, пероксодисульфат с арсенитом. Описаны [112]
потенциометрические способы оформления этого анализа. Некоторые авто-
авторы считают, что перекись водорода в этой смеси можно определить непо-
непосредственно титрованием перманганатом [108, ИЗ], но это предположение
оспаривается [114], ив качестве другой методики предложено титрование четы-
четырехвалентным церием [109]. Другие описанные способы включают примене-
применение арсенита [115], гипохлорита натрия или феррицианида калия [65], суль-
сульфита [108] и йодида одновалентной меди в качестве катализатора для титро-
титрования йодидом [116].
Вопрос анализа смесей других перекисей изучался очень мало. Гринспэн
и Маккеллар [117] показали, что перекись водорода в смеси с алифатическими
пероксокислотами и диацилперекисями можно определять при помощи четы-
четырехвалентного церия. В свою очередь пероксокислоты можно определить дей-
действием йодида на холоду без помех со стороны диацилперекисей. С другой
стороны, в работе Эгертона, Эверета и Минкофа [118] показано, что титрование
гидроперекисей алкилов йодидом нельзя считать надежным методом их опре-
определения. Все эти методы основаны на различной скорости реакции йодида
с разными перекисями; эти различия иллюстрируются данными Кедла и Хаффа
[119]. Брушвейлер, Минкоф и Салуджа [120] кратко описали хроматографиче-
ский и полярографический методы анализа смесей перекисей. По одной мето-
методике, предложенной этими авторами, перекись водорода удаляют из раствора
путем осаждения ее в виде перекиси лантана. Мак-Невин и Юрон [121 ] описали
аналогичнуюметодику отделения перекиси водорода от органических гидропере-
гидроперекисей путем образования комплекса и осаждения титаном. Описаны количе-
количественные методы определения перекиси водорода в присутствии других веществ,
а именно: оксалата [41], нитрита [122], озона (путем отделения выморажива-
вымораживанием или абсорбцией) [123], альдегидов и других кислородсодержащих орга-
органических веществ [52] и хромата (путем ионного обмена) [86].
Определение добавок или загрязняющих примесей
Часто имеет большое значение качественное или количественное опреде-
определение в растворах перекиси водорода различных небольших добавок, напри-
например стабилизаторов, или примесей, особенно каталитически действующих ионов
металлов. На стр. 467 указано, что при этом можно использовать полярографи-
полярографические методы [97], например для открытия присутствия таких каталитически
действующих ионов, как окисного железа, двухвалентной меди или свинца,
или же для установления присутствия станнатного стабилизатора. Вместо
этого можно поступить еще следующим образом: выпаривают и разлагают
достаточно большую пробу перекиси в таких условиях, чтобы получающийся
остаток был загрязнен лишь минимальным количеством материала стенок
сосуда, и то только известного состава, после чего остаток исследуют спектро-
спектроскопически. Если в процессе выпаривания раствора возможно разложение
пробы перекиси, необходимо принимать меры против уноса в брызгах заметных
количеств растворенных веществ, возникающих при разложении; в противном
случае эти вещества будут потеряны для последующего анализа остатка.
Совершенно очевидно, что, если даже сосуд сделан из столь инертных мате-
материалов, как алюминий высокой чистоты или боросиликатное стекло, все же
небольшое попадание этих веществ в раствор неизбежно. Этот вопрос
обсуждается в литературе [46].
470 Глава 10
Существует ряд технических условий на перекись водорода. В этих техни-
технических условиях обычно устанавливаются следующие показатели: концентра-
концентрация Н2О2, остаток от выпаривания, содержание свободной кислоты, хлорида,
нитрата, фосфата, сульфата, аммиака, тяжелых металлов и железа. В случае
трехпроцентного раствора по Фармакопее США (XIV издание) требуется,
чтобы нелетучий остаток, получаемый при выпаривании 20 мл раствора досуха
на водяной бане с последующим высушиванием при 105° в течение 1 часа,
не превышал 30 мг. Тяжелые металлы не должны присутствовать в количествах
больше 5 мг/л, а содержание мышьяка должно быть меньше 2 мг/л. Содержание
веществ, используемых для стабилизации раствора, не должно превышать
500 мг/л. Кислотность раствора должна быть такой, чтобы на нейтрализацию
25 мл раствора перекиси расходовалось не более 2,5 мл 0,1 н. раствора едкого
натра. Для 30%-ной перекиси водорода как реактива в технических условиях
Американского химического общества [124] даются несколько иные пределы.
Опубликованы результаты анализов некоторых типовых проб 30 и 90%-ной
перекиси водорода различных фирм [46, 125]. Разработаны методики [124]
для испытания перекиси на органический остаток, нитрат, хлорид, фосфат
[126], сульфат, аммоний, тяжелые металлы и железо. Иногда определяют
также фторид и оксалат. В операциях отбелки может иметь значение и опреде-
определение кремнезема в перекиси водорода [127]. Широко распространенная пре-
прежде стабилизация растворов перекиси водорода ацетанилидом привела к раз-
разработке метода анализа на это соединение [128].
В прежние времена вопросы анализа составляли значительную часть
литературы по перекиси водорода. Как технические условия, так и методы
анализа все время подвергались критике, особенно в отношении анализа
на кислотность. Общее содержание этих проблем можно оценить по сообще-
сообщениям Эндрьюса [129] и Вёлера и Фрея [130], где рассматриваются технические
условия Фармакопеи США и методы определения свободной кислоты.
ЛИТЕРАТУРА
1. Birckenbach L., Untersuchungsmethoden des Wasserstoffsuperoxyds, in Mar-
gosches, Die Chemische Analyse, vol. 7, Enke F., Stuttgart, 1909.
2. R e i с h e г t J. S., M с N e i g h t S. A., RudelH. W., Ind. Eng. Chem., Anal.
Ed., 11, 194 A939).
3. D u v a 1 C, Chim. anal., 35, 265 A953).
4. S с h 6 n n L., Z. anal. Chem., 9, 41, 330 A870).
5. Kolthoff I. M., Sandell E. В., Textbook of Quantitative Inorganic Analysis,
Revised Ed., Macmillan Co., New York, 1943 [есть также перевод: см. Кольт-
г о ф И. М., С е н д э л Е. Б., Количественный анализ, изд. 3-е, Госхимиздат,
М.—Л., 1948].
6. В е г i s s о В., Aquino R. M., Pubs. inst. invest, microquim., Univ. nacl.
litoral. (Rosario, Arg.), 14, 119 A950).
7. L a p i n L. N., Z. anal. Chem., 102, 418 A935).
8. К e m p f R., Z. anal. Chem., 89, 88 A932).
9. F г е у t a g H., Z. anal. Chem., 131, 77 A950) [C. A., 44, 9867f].
10. Sc hern K., Schellhase W., Zentr. Biochem. Biophys., 12, 768 A912).
11. Eyrard A., Jouffret R. J., Lait, 23, 141 A943) [Chem. Zentr., 1943,11,
1509].
12. PienJ., DesirantJ., Lafontaine D., Ann. fals. et fraudes, 46, 416A953).
13. Feigl F., «Spot Tests», Vol. 1, Inorganic Applications, transl. by Oester R. E.,
Elsevier Publ. Co., Houston, 1954 [есть русский перевод со второго немецкого
издания; см. Ф а й г л ь Ф., Капельный анализ, ОНТИ, Госхимтехиздат, М.—Л.,
1937].
14. Feigl F., Fran k el E., Mikrochemie, 12,304A933).
15. F e i g 1 F., Klanier K., Weidenfeld L., Z. anal. Chem., SO, 5 A929).
16. Кульберг Л., Матвеев Л., ЖПХ, 9, 755 A936).
17. Plank E., Z. anal. Chem., 99, 105 A934).
18. D«niges G., Compt. rend., 211, 196 A940); Bull. trav. soc. pharm. Bordeaux,
80, 5 A942).
Литература 471
19. Kohn M., Universidad Habana, Nos. 40, 41, 42, 187 A942) [С. А., 37, 845];
Anal. Chim. Acta, 3, 38 A949).
20. Stamm J., Pharmacia (Estonia), 18, 71, 106 A938) (на немецком языке).
21. L e с о q H., Bull. soc. chim. Belg., 54, 186 A945).
22. Gleu K., Pfannstiel K-, J. prakt. Chem., 146, 137 A936).
23. Langen beck W., R u g e V., Вег., 70В, 367 A937).
24. S t e i g m a n n A., J. Soc. Chem. Ind., 61, 36 A942).
25. S с h a 1 e s О., Вег., 71B, 447 A938).
26. Keil i n D., H artree E. F., Biochem., J., 39, 148 A945).
27. К у л ь б е р г Л. М., С о й ф е р П. А., Журн. анал. хим., 1, 263 A946).
28. Н a w k Р. В., Oser В. L., Summer son W. H., Practical Physiosogical
Chemistry, 12th ed., The Blakiston Co., New York, 1947, p. 211.
29. M с С 1 u n g L. S., В i 1 1 m a n J. H., Reid W. В., Arch. Biochem., 9, 57
A946).
30. A r г h e n i u s S., Svensk. Kern. Tid., 64, 260 A952) (на английском языке) [С. А.,
47, 444с].
31. Engler С, Wild W., Вег., 29, 1940 A896); М с L е о d H., Chem. News,
40, 307 A879); В a m b е г g е г М., Т г a u t z 1 К., Z. anal. Chem., 64, 9 A924).
32. К е i s е г Е. Н., McMaster L., Am. Chem. J., 39, 96 A908).
33. M e 1 1 о г J. W., ed. A Comprehensive Treatise on Inorganic and Theoretical Chemi-
Chemistry, Vol. I. Longmans, Green &. Co., London and New York, 1922, p. 951.
34. D a v i s W., Rosen F. D., W i n g e t R. H., W i s w a 1 1 R. H., A.E.C.D.
2827, Tech. Inf. Div., O.R.E., Oak Ridge, Tenn., Dec. 27, 1949.
35. T p e д в е л л Ф. П., Г о л л В. Т., Курс аналитической химии, т. I, Качественный
анализ, 2-е изд., Госхимиздат, М.—Л., 1946.
36. Kl e n k, Klepzig's Textil-Zeit., 42, 549 A939) [С. А., 34, 1179).
37. Mitchell J., Jr., Anal. Chem., 23, 1069 A951); Митчел Д ж., Смит Д.,
Акваметрия, Издатинлит, М., 1952; Mitchell J., Jr., Smith D. N., Ash-
by E.C., Bryant W. M. D., J. Am. Chem. Soc, 63, 2927 A941).
38. В г о d i e B. C, Phil. Trans., 1850, II, 769.
39. S с h 6 n b e i n С F., J. prakt. Chem., 77, 131 A859); 79, 78 A860).
40. A s с h о f f H., J. prakt. Chem., 81, 401 A860).
41. S i m о n A., R e e t z Т., Z. anal. Chem., 104, 249 A936); 105, 321 A936).
42. P u g h W., Trans. Roy. Soc. S. Africa, 22, 71 A934) [C. A., 28, 6084].
43. Fowler R. M., Bright H. A., J. Res. Natl. Bur. Standards, 15, 493 A935).
44. G i g u ё г e P. A., G e о f f г i о n P., Can. J. Res., 27B, 168 A949).
45. HuckabaC.E., К е у e s F. G., J. Am. Chem. Soc, 70, 1640 A948).
46. Беллинджер Ф., Фридман Г., Бауэр В., Истс Дж., Э д м о н д с
С, в сб. «Физика и химия реактивного движения», № 1, Госиноиздат, М.,
1949, стр. 175.
47. К i n g z e t t С. Т., Chem. News, 41, 76 A880); см. также Т г е a d w е 1 1 Р.,
Hall W. Т., Analytical Chemistry, 9th ed., vol. 2, John Wiley & Co., Inc., New
York, 1942, p. 609 [есть также перевод с немецкого издания: см. Тредвелл
Ф. П., Г о л л В. Т., Курс аналитической химии, т. II, Количественный анализ,
ОНТИ, М.—Л., 1935].
48. К о 1 t h о f f I. M., Chem. Weekblad, 17, 197 A920); T h о г s e 1 1 P., Bengts-
so n E., Svensk Farm. Tid., 43, 567 A939).
49. К о k a t n u г V. R., Jelling M., J. Am. Chem. Soc, 63, 1432 A941); К о 1-
t h о f f I.M., Medalia A.I., Anal. Chem., 23, 595 A951); S k e 1 1 о n J. H.,
Wills E. D., Analyst, 73, 78 A948).
50. W i b e г g К. В., J. Am. Chem. Soc, 75, 3961 A953).
51. Balls A.K., Hale W. S., J. Assoc Official Agr. Chem., 16, 395 A933).
52. S a t t e г f i e 1 d С N., W i 1 s on R. E., L e С 1 a i г R. M., R e i d R. C,
Anal. Chem., 26, 1792 A954); Bailey H. С, К n о x J. H., J. Chem. Soc,
1961, 2741; Sa tterfi el d C. N.. Bonnell A. H., Anal. Chem., 27, 1174
A955).
53. H i с k m a n J. W., Proc W. Va. Acad. Sci., 23, 76 A951).
54. A n a s t a s i u J. A., S t e f a n e s с u V., Вег., 61В, 1343 A928); F u г m a n N. H.,
Wallace J. H., Jr., J. Am. Chem. Soc, 51, 1449 A929); G u z m a n J.,
Rancano A., Anales soc. espan. fis. quim., 32, 590 A934).
55. W i 1 1 a r d H. H., Young P., J. Am. Chem. Soc, 55, 3260, A933).
56. Rao B. S. V. R., Current Sci (India), 16, 378 A947) [C. A., 42, 4485i]; M u r t h у
Т. К. S., Rao В. S. V. R., J. Indian Chem. Soc, 27, 37 A950).
57. Hurdis E. C, Romeyn H., Anal. Chem., 26, 320 A954).
58. J a m i e s о n G. S., Am. J. Sci., 44, 150 A917); Volumetric Iodate Methods, Chemi-
Chemical Catalog Co. (Reinhold Publishing Co.), New York, 1926, p. 61.
59. S m i t h G. F., W i 1 с о х С. S., Ind. Eng. Chem. Anal. Ed., 14, 49 A942).
€0. К n e с h t E., H i b b e г t E., Вег., 38, 3324 A905); 36, 1551 A903); M а с h F.,
Lederle P., Landw. Vers. Sta., 90, 191 A917); J. Chem. Soc, 112, 11,580
472 Глава 10
A917) [С. А., 12, 1952]. L е е К. W., J. Chem. Soc Japan, 53, 25 A932) [С. А.,
27, 38].
61. Т г i t t о n S. M., Analyst., 64, 469 A939).
62. S i n g h В., Malik I. I., J. Indian Chem. Soc, 14, 435 A937) [C. A., 32, 880].
63. S с h w i с к е г A., Z. anal. Chem., 108, 89 A937).
64. R u p p E., W eg пег W., Arch. Pharm., 261, 202 A923).
65. Rius Miro A., Beltran Martinez J., Anales fis. quim., 38, 347 A942).
66. Fehre W., Z. anal. Chem., 87, 185A932).
67. M a n с h о t W., О b e r h a u s e r F., Z. anorg. allgem. Chem., 139, 40 A924).
68. Deniges G., Bull. trav. soc. pharm. Bordeaux, 88, 41 A950) [C. A., 46, 1919i].
69. Belcher R., West T. S., Anal. chim. Acta, 5, 360 A951).
70. E г d e у L., Magyar Kern. Folyoirat, 56, 262 A950) [C. A., 46, 849i]; L e h о n g г е
G., Neumann J., L a v о 1 1 а у J., Bull. soc. chim. biol., 32, 1023 A950).
71. Bury A., J. pharm. chim., 15, 189 A917); Malagelada J., Farm, nueva
(Madrid), 13, 580 A948).
72. С 1 а г к J. В., W у s s O., Haas F., Stone W. S., Proc. Soc. Exptl. Biol.
Med., 72, 32 A949); A p p 1 e m a n D., Anal. Chem., 23, 1627 A951) [C. A., 44,
1159c]; Fu j i t а А., Ко da ma Т., Biochem. Z., 232, 15 A931).
73. Bosshard E., Furrer E., Helv. Chim. Acta, 7, 486 A924).
74. H a i g h t M., J. Chem. Education, 6, 2234 A929).
75. M a t s u u г a N., J. Chem. Soc. Japan, Pure Chem. Sect., 71, 375 A950) [C. A.^
45, 6527d].
76. Szabo Z., So os I., Z. anal. Chem., 127, 181 A944).
77. E 1 1 i о t t К. А. С, Biochem. J., 26, 10 A932).
78. S n e 1 1 F. D., S n e 1 1 С. Т., Colorimetric Methods of Analysis, 3rd ed., D. Van.
Nostrand Co., Inc., New York, Vol. 1 1948, Vol. 2, 1949, Vol. 3, 1953.
79. S a v a g e D. J., Analyst, 76, 224 A951).
80. P a t г i с к W. A., Wagner H. В., Anal. Chem., 21, 1279 A949); Ovenston'
Т. С J., R e e s W. Т., Analyst., 75, 204 A950).
81. L i t t m a n F. E., В е п о 1 i e 1 R. W., Anal. Chem., 25, 1480 A953).
82. All sop p С. В., Analyst, 66, 371 A941).
83. E i s e n b e r g G. M., Ind. Eng. Chem., Anal. Ed., 15, 327 A943).
84. M i 1 1 e r N., MacPherson E., Natl. Research Council Can., At. Energy
Proiect Div. Research CRC 352 (N. R. С № 1617) A949) [С. А., 42, 2202i]; H u m-
poletz J. E., Australian J. Sci., 12, 111 A949).
85. Bonet-Maury P., Compt. rend., 218, 117 A944) [C. A., 40, 1425].
86. R у n a s i e w i с z J., Anal. Chem., 26, 355 A954).
87. M u s h a S., H i g a s h i n о H., D о i Т., J. Chem. Soc, Japan, Pure Chem. Sect.,
72, 995 A951) [C. A., 46, 7938e]; 73, 363 A952) [C. A., 47, 2553h].
88. E г d m a n n H., S e e 1 i с h F., Z. anal. Chem., 128, 303 A948).
89. Isaacs M. L., J. Am. Chem. Soc, 44, 1662 A922).
90. Q u a r t а г о 1 i A., Ann. chim. applicata, 15, 32 A926); 24, 70 A934).
91. McCabe L. C, Ind. Eng. Chem., 45, № 9, 111A A953).
92. G 1 a v i n d J., Hartjnann S., Acta Chem. Scand., 5, 975 A951); H а г t m a n
L, White M. D. L., J. Sci. Food Agr., 3, 112 A952).
93. S с h u 1 e r W. A., Pharmazie, 4, 459 A949) [C. A., 44, 689i].
94. Allen N.. Ind. Eng. Chem., Anal. Ed., 2, 55 A930).
95. P e 1 1 e q u e г Н., J. Chim. Phys., 47, 386 A950); Compt. rend., 222, 1220 A946);
Михайлова М. П., Нейман М. Б, Зав. лаб., 9, 166 A940); Д о б р и н-
с к а я А. А., Н е й м а н М. В., Acta physicochim. URSS, 10, 297 A939); Кольт-
гоф И. М., Л ингейн Дж. Д ж., Полярография, Госхимиздат, М.—Л.,
1948; К о г у t a J., Chem. Listy, 46, 593 A952) [С. А., 47, 1530h]; V i t e к V.,
Chimie & Industrie, Special № 215 (VI. 1933) [С. А., 27, 5274].
96. G i g u ё г e P. A., J a i 1 1 e t J. В., Can. J. Research, 26B, 767 A948).
97. P i g n a t a r о N.. Ann. Chim. applicata, 37, 448 A947).
98. R e i m e r s F., Quart. J. Pharm. Pharmacol., 19, 473 A946).
99. Giguere P. A., Can. J. Res., 21B, 156 A943); G i g u ё г е P. A., Geof-
frion P., ibid., 27B, 168A948).
100. H u с к a b a C. E., Keyes F. G., J. Am. Chem. Soc, 70, 2578 A948); 72, 5324
A950).
101. H i d n e r t P., Peffer E. L., Density of Solids and Liquids, Natl. Bureau Stds.
Circular 487, Government Printing Office, Washington, 1950.
102. Дэвис Н., Киф Д ж., в сб. «Вопросы ракетной техники», № 1, 124 A953).
103. S h a n 1 е у Е. S., частное сообщение, 1954.
104. Giguere P. А., пат. США 2617940 A1. XI. 1952).
105. Е 1 t e n t о n G. С, J. Chem. Phys., 10, 403 A942); 15, 455 A947); F о n e r S. N..
Hudson R. L., J. Chem. Phys., 21, 1608, 1374 A953); Robertson A. J. В.,
Trans. Faraday Soc, 48, 228 A952).
106. Watson R. O., S. B. Thesis in Chemical Engineering, M.I.Т., 1949.
Литература 473
107. Todt F., Arch. Metallkunde, 1,469 A947); Kamienski В., Bull, intern.
acad. polon. sci., Classe sci. math, nat., 1949A, 87 (C. A., 44, 7676g].
108. Rt us A., Trans. Am. Electrochem. Soc, 54, 347 A928).
109. Berry A. J., Analyst., 58, 464 A933).
110. R e i с h e г t J. S., частное сообщение, 1954.
111. Gl e u K., Z. anorg. allgem. Chem., 195, 61 A931).
112. Д е н и с о в Е. И., Труды Ленингр. Инд. инст., № 9, отд. физ.-мат., № 2, 40 A936);
Miiller E., Holder G., Z. anal. Chem., 84, 410 A931).
113. N а у а к U. M., J. Indian Chem. Soc, 8, 535 A931); R i и s A., Z и 1 и e t a C,
Anales real soc. espaft. fis. y., quim, 44B, 923 A948); Zulueta de Haz C,
Inform, quim. anal. (Madrid), 4, 45 A950).
114. Van der Meulen J. H., Rec. trav. chim., 58, 553 A939).
115. I si da Т., Yukawa M., J. Soc. Chem. Japan, 43, suppl. 46 A940).
116. Бод и н М. А., Зав. лаб. 7, 124 A938).
117. Green sp a n F. P., Mackell ar D. G., Anal. Chem., 20, 1061 A948).
118. E g e г t о n A. C, Everett A. J., Minkoff G. J., Nature, 173, 399 A954).
119. С a die R. D., Huff H., J. Phys. Colloid Chem., 54, 1191 A950).
120. Bruschweiler H., Minkoff G. J., Salooja К. С, Nature, 172,
909 A953).
121. M а с N e v i n W. M., U г о n e P. F., Anal. Chem., 25, 1760 A953).
122. Q и а г t а г о 1 i A., Gazz. chim. ital., 48, I, 102 A918).
123. В a m b e г g e г M., T г а и t z 1 K., Z. anal. Chem., 64, 9 A924).
124. American Chemical Society, Reagent Chemicals, Am. Chem. Soc, Washington,
1951, p. 176.
125. Bellinger F., Friedman H. В., В а и e r W. H., E a s t e s J. W.,
Bull W. C, Ind. Eng. Chem., 38, 310 A946).
126. D a 1 i e t о s J., Z. anorg. allgem. Chem., 217, 346 A934).
127. Phila. Quartz Co., Textile World, 85, 1884 A935); В a 1 1 о и К. D., R о а г к e J. J.,
G a n t z G. M., Am. Dyestuff Reptr., 40, Proc Am. Assoc Textile Chem. Colo-
rists A951) p. 218—22 [C. A., 45, 5413i].
128. Waller E., J. Ind. Eng. Chem., 1, 262 A909).
129. Andrews L. W., Trans. Am. Inst. Chem. Eng., 2, 238 A909).
130. Woehler L., Frey W., Z. angew. Chem., 23, 2353 A910).
Глава 11
ПРИМЕНЕНИЕ ПЕРЕКИСИ ВОДОРОДА
Впервые перекись водорода нашла техническое применение в качестве
антисептика и отбеливающего вещества (особенно для продуктов животного
происхождения, например волоса и шерсти, которые легко разрушаются при
действии других отбеливающих агентов). Однако до 1910 г. перекись водорода
получали исключительно из перекиси бария; малая стабильность конечного
продукта наряду с большими затратами на хранение и перевозку получавшихся
разбавленных растворов (приблизительно до 5 вес.%) сильно ограничивали
ее применение. Электрохимические процессы (в значительной мере вытеснив-
вытеснившие химические процессы производства из перекиси бария в большинстве про-
промышленных стран в течение 1910—1925 гг.) включают стадию выпаривания
перекиси водорода, образующейся при гидролизе известного пероксосоедине-
ния, с последующей дробной конденсацией продукта. Применение этих про-
процессов позволило получать гораздо более чистый продукт, обладающий повы-
повышенной стабильностью и большей концентрацией, что и способствовало широ-
широкому его внедрению.
Для невоенных целей наибольшее значение имело и имеет потребление
перекиси водорода в качестве отбеливающего вещества. Отбелка веществ живот-
животного происхождения чаще всего осуществляется перекисью водорода. При
отбелке целлюлозных веществ перекись водорода постепенно вытесняет гипо-
хлориты или дополняет их. Так, в 1954 г. основная масса хлопчатобумажных
тканей в США отбелена перекисью водорода. За последние несколько лет
стали широко применять перекись натрия и перекись водорода для отбелки
древесной массы, а частично и химической целлюлозы как дополнение к обра-
обработке хлором и гипохлоритом. Все большее количество перекиси водорода
применяют и в производстве органических химических веществ. Во время вто-
второй мировой войны очень большие количества концентрированной перекиси
водорода были использованы в качестве ракетного топлива (особенно в Гер-
Германии). Эти виды применения, так же как и другие, описаны ниже более
подробно.
На стр. 23, 115, 116 приведены данные о размерах производства перекиси
водорода во всем мире и в США. В США в 1951 г. оно было примерно в 3 раза
выше, чем в 1945 г., и раз в 10 больше, чем в среднем за период 1915—1930 гг.
В табл. 82 приводятся данные о распределении потребления перекиси водорода
в США в 1947 г. для различных целей (на основании отчетов правительства
США [1]).
Можно видеть, что текстильная промышленность является наиболее круп-
крупным гражданским потребителем перекиси водорода. В 1946 г. 20—30% всего
отбеленного хлопчатобумажного товара было отбелено перекисью водорода,
65—75%—гипохлоритом и около 5%—обоими этими веществами [2]. Однако
развитие процессов непрерывной отбелки перекисью водорода, особенно
после 1946 г., способствовало вытеснению большей части применявшегося
гипохлорита. В 1954 г. на всех текстильных фабриках перекисью водорода
Применение перекиси водорода
475
Таблица 82
Сравнительные данные по расходу перекиси водорода в США в 1947 г.
Применение
Для гражданских целей:
на хлопчатобумажные и вискозные товары ....
на производство неорганических химических веществ
на фармацевтические и косметические препараты . .
на обработку мехов
на пищепродукты
на желатину и клей
на шерстяные и камвольные товары
на производство органических химических веществ .
на мыло и родственные продукты
на прочие гражданские цели
Всего для гражданских целей
Для военных целей
35
9
4
2
3
3
3
3
3
5
70
30
отбелено около 85% хлопчатобумажного товара; на это израсходовано около
7300 т перекиси водорода в расчете на 100%-ную. В США расход перекиси
водорода на отбелку шерстяных товаров по сравнению с расходом ее на отбелку
хлопчатобумажных товаров невелик; в Великобритании же наблюдается
обратное соотношение. С 1947 г. количество перекиси водорода, применяемой
в производстве органических химических веществ, сильно возросло. По Кауф-
Кауфману, распределение расхода перекиси водорода в различных отраслях про-
промышленности за последние годы приблизительно соответствует данным, при-
приведенным в табл 83 [3].
Таблица 38
Распределение расхода перекиси водорода
по различным отраслям промышленности и видам применения, %
Потребители
По отраслям промышлен-
промышленности:
текстильная ....
прочие
По видам применения:
на отбелку
на другие цели . . .
1947
70
30
78
22
1949
60
40
80
20
1951
55
45
70
30
Импорт перекиси водорода в США сравнительно невелик (по крайней мере
в течение последних 20 с лишним лет). Так, например, за период 1946—1949 гг.
стоимость импорта перекиси водорода составляла меньшеJ25 тысяч долларов
в год, так как в цензе США за соответствующий период перекись водорода
отдельно не указана.
В табл. 84 приведены оптовые продажные цены на перекись водорода
в США [41.
Средняя продажная цена 27—35%-ной перекиси водорода в Нью-Йорке
в течение периода 1940—1954 гг. колебалась от 1210 до 1540 долларов за тонну
в расчете на 100%-ную перекись. В промышленном цензе США за 1947 г. ука-
476 ' Глава И
Таблица 84
Оптовые продажные цены на перекись водорода в 1954 г.
Концентрация,
вес. %
Цена за тонну водного
раствора, доллары
3
27,5
35
90
81,5—110
385—430
455—520
1480—1650
Цена за тонну в расчете
на 1 00%-ную перекись,
доллары
2750—3650
1400—1550
1310—1480
1640-1840
зано, что цена франко-завод на 27,5%-ный раствор составляет 1133 доллара
за тонну (в расчете на 100%-ную перекись).
Различные виды применения перекиси водорода, основанные на некото-
некоторых важных ее свойствах, следующие:
/. Окислитель. В основном перекись водорода применяется в качестве
отбеливающего вещества. Поэтому этот вид применения рассматривается
отдельно от других, где она также используется в качестве окислителя. Другие
виды применения следующие: а) в качестве окислителя для кубовых красите-
красителей и для удаления остаточного гипосульфита в фотографии; б) для разделе-
разделения металлических ионов путем избирательного окисления; в) как реактив,
в аналитической химии; г) как деполимеризатор при модификации смол, клеев,
растворимого крахмала и т. д. и д) для отбелки бумажной макулатуры.
2. Источник энергии. Концентрированные растворы перекиси водорода
могут служить в качестве весьма компактного источника энергии, которая
может быть выделена при разложении перекиси или при реакции ее с горючим.
3. Образование газа при разложении. Разбавленные растворы могут быть
использованы в качестве пенообразователей в производстве пористых веществ,
например пенистой резины или пористых стройматериалов.
4. Источник свободных радикалов. Перекись водорода и другие перекисные
соединения используются для инициирования процессов полимеризации
и других реакций с участием свободных радикалов.
5. Влияние на биологические процессы.
6. Применение в химическом синтезе. Перекись водорода является наиболее
удобным исходным веществом для получения большей части неорганических
и органических перекисных соединений, а также часто применяется в качестве
реагента в органическом химическом синтезе.
Все эти виды применения ниже рассмотрены подробно в указанном порядке.
Перекись водорода действует, так же как восстановитель в реакциях с некото-
некоторыми высокоокисленными соединениями, например с перманганатами, бихрома-
тами и солями четырехвалентного церия, причем во всех случаях образующийся
газообразный кислород происходит от перекиси водорода. Однако эти реакции
не имеют большого технического значения (они рассмотрены в гл. 7).
Перекись водорода и различные другие перекисные соединения при при-
применении в водных средах в значительной мере взаимозаменяемы. Например,
в тех случаях, когда активной действующей частицей является перекись водо-
водорода или ион НО^, можно вводить готовую перекись водорода и установить
рН на надлежащем уровне или же получить перекись водорода in situ из дру-
другого перекисного соединения путем гидролиза или нейтрализации, например
из перекиси натрия или же из соли, содержащей кристаллизационную пере-
перекись водорода. Наоборот, активным началом может в действительности быть
более сложное перекисное соединение, например пероксоуксусная кислота,
которая также может быть введена в готовом виде или образована in situ
Отбелка 477
из уксусной кислоты и перекиси водорода. Большинство перекисных соедине-
соединений получается из перекиси водорода, как описывается на стр. 522.
Ниже исчерпывающе рассматриваются не все виды применения перекисных
соединений, а лишь те, при которых используется или использовалась ранее
готовая перекись водорода.
ОТБЕЛКА
Перекись водорода в основном применяется в качестве отбеливающего
вещества. От перекиси водорода требуется, чтобы она разлагала или обесцве-
обесцвечивала окрашенные вещества или превращала их в форму, растворимую в воде
или в отбеливающем веществе. Для отбелки можно использовать как восстано-
восстановитель, например двуокись серы, гидросульфит или тиосульфат, так (и притом
чаще) и окислитель, например перекисное соединение, хлор или кислород-
кислородсодержащие хлоропроизводные, например гипохлориты, хлорит натрия и дву-
двуокись хлора. В небольшом количестве для отбелки применяются также бихро-
маты, озон и перманганаты. В последнем случае образующуюся двуокись
марганца удаляют путем последующей обработки перекисью водорода, уксус-
уксусной или щавелевой кислотой. Из других перекисных соединений, которые также
используются в качестве отбеливающих веществ, следует указать на перекись
натрия, которая может заменить перекись водорода при отбелке древесной
целлюлозы и для удаления краски с бумажной макулатуры или же применяться
в сочетании с нею. В прачечных производственного типа, если требуется отбе-
отбеливающее вещество слабого действия, или в тех случаях, когда отбелка произ-
производится случайно или в небольших масштабах, иногда применяют пероксо-
борат натрия. За последние несколько лет быстро возрастает применение
пероксобората в виде сухого отбеливающего вещества в домовых прачечных.
Водный раствор пероксобората в действительности представляет собой раствор
перекиси водорода с буфером, рН которого равен примерно 10.
Выбор отбеливающего вещества для того или иного вида применения
зависит от таких факторов, как стоимость химических веществ и операций
отбелки, эффективность этих веществ, их стабильность, проблемы коррозии,
хранения и перевозки, снижение прочности обрабатываемого материала,
устойчивость отбеливающего действия, легкость окраски или набивки отделен-
отделенного материала, а также и другие менее ощутимые эффекты, как мягкость
на ощупь и внешний вид отбеленного материала, особенно в текстильной про-
промышленности. В некоторых случаях, например в случае хлопчатобумажных
товаров, ткань сначала обрабатывают щелочью или кислотой и вся операция
рассматривается не только как процесс отбелки, но как общий процесс очистки,
предназначенный для удаления таких веществ, как крахмал, жиры и воски,
а также шелуха семян и других остатков, которые могли бы сохраниться
в ткани.
Природа химических реакций, на которых основано разложение красящих
веществ перекисями, очень мало изучена. Недавно Карабинос и Боллан [5]
изучили механизм обесцвечивания продуктов конденсации таллового масла
с окисью этилена действием перекиси водорода; Джонс [6] опубликовал работу
по исследованию процесса отбелки древесной массы перекисью, а Холст [7]
недавно дал общий о,бзор химии и механизма отбелки, рассмотрев, в частности,
изменения энергии, с которыми связаны протекающие химические процессы.
Очевидно, что для раскрытия механизмов реакций, происходящих в процессах
отбелки, требуется значительное расширение фундаментальных исследований.
Сделаны попытки разработать систему для ускорения отбеливающего действия
перекиси водорода. Желательный эффект должен быть аналогичен наблюдае-
наблюдаемому при действии фермента пероксидазы [8]. Эти попытки были безуспешными
главным образом потому, что вещества, обладающие таким действием, одновре-
одновременно способствуют и разложению перекиси без какой-либо пользы для про-
478 Глава 11
цесса отбелки; помимо потери перекиси водорода, выделяющийся кислород
может вызвать порчу отбеливаемого материала.
Скорость, с которой происходит отбелка того или иного вещества пере-
перекисью водорода, возрастает с ростом рН, откуда следует, что активной частицей
в данном случае является ион пергидроксила НО~, который образуется в резуль-
результате ионизации перекиси водорода
Повышение щелочности раствора перекиси снижает концентрацию водо-
водородных ионов и увеличивает концентрацию пергидроксильных. Хорошо
известно, что кислород, выделяющийся при разложении перекиси, как пра-
правило, лишен отбеливающего действия и может даже оказаться вредным 19].
Например, целлюлозные волокнистые вещества в сильнощелочной среде под-
подвергаются, хотя и медленно, действию кислорода, что приводит к снижению
длины цепи целлюлозных молекул и как следствие—к снижению прочности.
Поэтому операции отбелки необходимо проводить таким образом, чтобы снизить
до минимума разложение перекиси с выделением кислорода, например вызы-
вызываемое мельчайшими примесями таких металлов, как железо, медь и марганец»
Различные виды материалов, подвергаемых отбелке, целесообразно раз-
разбить на две группы: на состоящие в основном из целлюлозы и на имеющие
преимущественно белковую структуру.
Отбелка целлюлозных материалов
Текстильные волокна. Практически все целлюлозные волокна, например
волокно хлопка, льна, джута или вискозного шелка, можно отбеливать как
производными хлора, так и перекисью водорода (или обоими этими отбели-
отбеливающими агентами). До тридцатых годов текущего столетия для этой цели
обычно применяли гипохлорит кальция или натрия, что объяснялось сравни-
сравнительно высокой стоимостью перекиси водорода на единицу отбеливающей спо-
способности и малой изученностью методов сохранения ее в достаточно устойчивом
состоянии на складе и при употреблении. В 1954 г. стоимость химических
веществ, расходуемых на обработку одной тонны хлопчатобумажных товаров-
перекисью водорода, составляла примерно 5,5—7,7 доллара против 3,3—
5,5 доллара при обработке гипохлоритом. Тем не менее гипохлорит был в зна-
значительной мере вытеснен перекисью водорода в результате усовершенствова-
усовершенствования техники стабилизации, перевозки и хранения перекиси, а также благо-
благодаря приобретению опыта, который показал следующее:
1. Гипохлорит является более мощным окислителем, чем перекись водо-
водорода. Правда, можно добиться одинаковой степени белизны с минимальной
потерей разрывной прочности волокна при применении как того, так и дру-
другого отбеливающего вещества, однако для этого при использовании гипохло-
рита требуется более тщательное соблюдение производственного режима, чем
при отбелке перекисью водорода. В последнем случае меньше возможность
деструкции волокна и снижения его прочности из-за слишком сильного дей-
действия отбеливающего агента.
2. Продуктами разложения перекиси водорода являются кислород и вода,
а поэтому упрощается необходимая обработка после отбелки.
3. Пригодность перекиси водорода для отбелки различных видов волокна
обусловливает возможность использования одного и того же оборудования
для разных текстильных товаров.
4. Перекись водорода можно применять для отбелки товара из пряжи,
окрашенной кубовыми или другими красителями, который может быть повре-
поврежден при отбелке хлором,
Отбелка 479
5. Хлопок, отбеленный перекисью, хорошо поддается окраске, мягок
на ощупь и обладает более устойчивым белым цветом.
Рост применения перекиси водорода в США обусловлен также разработ-
разработкой непрерывных процессов, в которых перекись водорода используется для
отбелки в крупном масштабе (например, 360 тыс. метров или больше за 120-ча-
120-часовую рабочую неделю). Впервые такой непрерывный процесс использован
фирмой «duPont» в 1939 г., по окончании же второй мировой войны как эта
фирма, так и фирма «Buffalo Electro-Chemical Company» применили этот про-
процесс на многих отбельных фабриках. Хотя капитальные затраты на необхо-
необходимое для непрерывного процесса оборудование высоки, тем не менее он отли-
отличается следующими преимуществами: по сравнению с периодическими про-
процессами отбелки перекисью водорода для него требуется меньшая производ-
производственная площадь, сокращаются расходы на зарплату, а также снижается
потребление пара, воды и химических веществ.
В некоторых операциях перекись водорода используют в качестве «анти-
«антихлора» после отбелки гипохлоритом. Товар, отбеленный гипохлоритом, иногда
со временем медленно желтеет. Это изменение цвета, очевидно, обусловлена
образованием хлораминов из гипохлорита и белков, содержащихся в сыром
хлопковом волокне. Последующая обработка перекисью водорода предотвра-
предотвращает пожелтение в результате разложения хлораминов, причем ослабление
волокна от перекиси водорода меньше, чем в случае применения такого анти-
антихлора, как тиосульфат натрия. В зависимости от условий обработки на долю»
перекиси водорода может падать также существенная часть общего отбели-
отбеливающего эффекта.
Техника отбелки целлюлозных волокон сильно зависит от природы обра-
обрабатываемого материала, причем для отбелки новых волокнистых веществ, как
правило, приходится разрабатывать эту технику более или менее эмпириче-
эмпирическими методами. Опубликованы буквально сотни различных рецептов по от-
отбелке разных веществ; эти рецепты различаются по таким показателям, как
время, температура, концентрация, природа и количество стабилизаторов,
способ предварительной и последующей обработки. Здесь можно привести
лишь несколько типичных методик.
Наиболее важными характеристиками отбеливаемого материала, опреде-
определяющими вид последующей обработки, являются содержание целлюлозы
и количество присутствующего лигнина. Чистая целлюлоза сравнительно
инертна к действию щелочи без доступа кислорода, но лигнин и низкомоле-
низкомолекулярные вещества целлюлозного типа, например гемицеллюлозы, легко
подвергаются атаке щелочью. Таким образом, допустимая интенсивность
обработки волокна зависит от относительного содержания лигнина и других
нецеллюлозных веществ, которые играют роль цементирующих и упрочняю-
упрочняющих веществ для волокна. Так, хлопок, представляющий весьма чистую форму
целлюлозы, можно обрабатывать при высокой температуре едким натром для
освобождения от посторонних примесей, например восков, без снижения
прочности волокна, причем его можно отбеливать перекисью водорода в срав-
сравнительно жестких условиях, например при значениях рН примерно до 11
и при температурах кипения. Слейтер и Ричмонд [10] делят все растительные
волокна, кроме хлопка, на 3 группы в зависимости от содержания целлюлозы
и допустимой интенсивности обработки: 1) волокна, содержащие свыше 85%
целлюлозы и небольшое относительное количество лигнина или пектиновых
цементирующих веществ, например лен; 2) волокна, содержащие меньше 85%
целлюлозы и 6—18% лигнина, например джут, сизаль или Phormium tenax
(новозеландский лен); 3) волокна с исключительно высоким содержанием
лигнина, например кокосовое с 34% лигнина.
Отбелка волокнистых веществ, содержащих заметные количества лигнина
или других нецеллюлозных веществ, обычно производится путем обработки
480 Глава It
сначала гипохлоритом, а затем перекисью водорода. Ниже приводится описа-
описание типичной операции отбелки льняной пряжи как пример обработки во-
волокон 1-й группы [10]. Пряжу сначала бучат в течение 3 час. при 100° в рас-
растворе углекислого натрия (вместо едкого натра, применяемого при бучении
хлопка). Далее следует обработка разбавленным раствором гипохлорита
кальция, содержащего 5,6 г активного хлора в 1 л, кисловка 0,1—0,2%-ной
серной кислотой и выдерживание при 71—77° в течение 4—5 час. в растворе,
содержащем 0,1—0,2% перекиси водорода с добавкой углекислого натрия
и силиката натрия. Если льняную пряжу отбеливать до полной белизны,
то волокно заметно теряет в прочности, а поэтому часто вырабатывают и вы-
выпускают в продажу частично отбеленные ткани.
В джуте, материале, принадлежащем к 2-й группе, целлюлозные волокна
сравнительно коротки B—5 мм) и сцементированы вместе лигнином и геми-
целлюлозой, которые, хотя сами по себе и обладают малой разрывной проч-
прочностью, обусловливают основную часть механической прочности волокна.
Поскольку* они легче подвергаются атаке, чем целлюлоза, беление и другие
операции обработки должны производиться в более мягких условиях, чем
применяемые для хлопка и материалов 1-й группы. Типичная обработка джу-
джутовой пряжи заключается в кратковременном вымачивании ее в растворе гипо-
гипохлорита кальция, содержащем 2,5—3,0 г активного хлора и 0,7 г углекислого
натрия в 1 л, с последующей отбелкой тем же раствором перекиси, который
указан выше, но в течение 1,5—2 час. Таким же образом отбеливают и коко-
кокосовые волокна, но в этом случае требуется еще предварительная обработка
бисульфитом и конечная обработка гидросульфитом натрия. Недавно опубли-
опубликована статья [11] о влиянии обработки перекисью водорбда на химические
составные части и физические свойства джута.
Искусственное волокно из целлюлозы, например вискозный или ацетат-
ацетатный шелк, требует сравнительно умеренного режима отбелки. Особенно под-
подвержен гидролизу при высоких рН ацетатный шелк. Типичная операция
отбелки заключается в выдерживании в течение 30—45 мин. в ванне с 0,1—
0,2%-ной перекисью водорода с добавленными в нее необходимыми стабили-
стабилизаторами; температура ванны должна поддерживаться на уровне 71—82°,
рН—в пределах 8,8—9,5.
Основная часть перекиси водорода в текстильной промышленности рас-
расходуется для отбелки хлопка. Хлопок можно обрабатывать в самых различ-
различных формах, например в виде пряжи, кускового или готового товара в опе-
операциях периодического типа, либо в форме жгута или врасправку—в линии
непрерывной отбелки. Периодические операции обычно производятся в желез-
железных котлах, выложенных цементом (бучильных котлах), либо в чанах или
баках, изготовляемых главным образом из нержавеющей стали. Облицовка
из керамических плиток при высоких температурах отбелки может дать тре-
трещины. Начальная обработка хлопка может быть различной, но во всех
случаях требуется предварительная тщательная очистка. Типичная отбельная
ванна на основе перекиси водорода содержит 0,1—0,4 вес. % последней,
около 1—1,5% силиката натрия й достаточное количество едкого натра для
доведения рН до 10,5—11,5. При периодических операциях ткань обрабаты-
обрабатывают этим раствором в течение 2—8 час. при 80—100°, обычно с использова-
использованием того или иного вида перемешивания. Для отбелки товара, окрашенного
нафтоловыми красителями, применяется ванна, рН которой во избежание
разложения красителя поддерживают в пределах 7,0—9,0.
Большая часть хлопка, отбеливаемого в США, обрабатывается непрерыв-
непрерывным методом. На типичной установке этого типа очищенный хлопчатобумаж-
хлопчатобумажный товар пропитывают 3—4%-ным раствором едкого натра и затем непрерыв-
непрерывно пропускают б верхнюю часть пологого J-образного ящика, в котором ткань
выдерживается в течение 1—1,5 часа и подвергается пропарке при 100°. Ткань
Отбелка 481
непрерывно отбирается из нижней части J-образного ящика, промывается,
пропитывается отбеливающим раствором, содержащим около 0,2—0,5% пере-
перекиси водорода, 1—1,5% силиката натрия и 0,05—0,25% едкого натра для
создания рН около 10,5, и затем пропаривается примерно в течение часа при
99—100° во втором J-образном ящике. Сообщается, что ткань движется со
скоростью 73—275 м/мин.
Роль силиката натрия, который обычно вводится в состав отбеливающего
раствора, многосторонняя. Он действует в качестве стабилизатора, снижая
бесполезные потери перекиси водорода из-за разложения, а так же как буфер,
способствующий поддержанию рН на желательном уровне. Вместе с тем он
обладает определенной моющей и пропитывающей способностью и антикорро-
антикоррозионными свойствами в отношении металлического оборудования. В отбели-
отбеливающий раствор можно добавлять также смачивающие и пропитывающие
вещества. Оптимальный рН устанавливается путем компромисса между двумя
крайними возможностями: при высоких рН (примерно выше 11,5) происходит
быстрая отбелка, но и перекись водорода быстро разлагается, причем требуется
также весьма тщательный контроль для обеспечения надлежащей отбелки без
снижения прочности волокна. При значительно более низких рН скорость
отбелки становится настолько малой, что процесс оказывается неэкономичным.
Для отбелки 1 т хлопка требуется приблизительно 12 кг 27,5%-ного раствора
перекиси водорода и 5 кг силиката натрия [12].
Синтетические волокна, например вискозный шелк, найлон, орлон (по-
лиакрилонитрил) и т. д., уже в процессе производства получаются в общем
удовлетворительной белизны. И если отбелка оказывается все же необходимой,
то обычно лишь для удаления пятен или желтизны, возникающих при обра-
обработке пряжи до вязания или ткачества или при ширении ткани для обеспече-
обеспечения надлежащего размера. Вискозный шелк обычно подвергают отбелке
методами, аналогичными применяемым для хлопка. Для ацетатного волокна
во избежание гидролиза необходимо применять нейтральную или слабо-
слабокислую ванну. Часто применяют ванну, содержащую пероксоуксусную кис-
кислоту, нейтрализованную едким натром, и некоторое количество полифосфата.
Пероксоуксусная кислота с успехом применяется и для отбелки найлона.
Перекись водорода, по-видимому, не оказывает никакого .действия на орлон,
дайнел (сополимер хлористого винила и акрилонитрила) и акрилан (сополи-
(сополимер винилацетата и акрилонитрила).
Недавно комиссией Американской ассоциации химиков-текстильщиков
и колористов [13] опубликована ценная библиография. В ней приводятся
краткие рефераты всех статей по отбелке текстиля, напечатанных за период
1900—1950 гг. В этой библиографии содержится 245 ссылок на работы по
отбелке перекисью, 153—на труды по отбелке гипохлоритом (из них значи-
значительная часть относится к отбелке одновременно обоими этими веществами)
и 321—на работы по применению для отбелки других химических агентов,
по оборудованию и т. д. Дополнительную подробную информацию по технике
отбелки целлюлозного текстиля можно извлечь из этих работ, а также полу-
получить у различных фирм, производящих перекись водорода [14—16], или
прочесть в книгах по целлюлозе и хлопку [17, 18]. В одном из сообщений
Белла и Стэлтера [19] рассматривается влияние различных свойств хлопчато-
хлопчатобумажного кускового товара на процесс непрерывной отбелки перекисью
водорода. Приводятся количественные данные, показывающие, каким обра-
образом при этом изменяются белизна, текучесть, впитывающая способность, со-
содержание масел, жиров, восков и золы, а также рН.
Отбеливающее действие перекиси водорода используют также при выводе
пятен и отбелке белья при домашней стирке. Трехпроцентный раствор пере-
перекиси водорода разбавляют в отношении 20 : 1 тепловатой мыльной или про-
прозрачной водой, содержащей небольшое количество аммиака, и выдерживают
31 Перекись водорода
482 Глава 11
белье в этой воде в течение 30 мин., потом полощут и сушат; таким образом,
и в домашних условиях используют преимущества отбелки перекисью водо-
водорода [20]. Вследствие разницы в цене и необходимости применения двух
жидкостей исключается возможность конкуренции перекиси водорода с гипо-
хлоритом для отбелки при домашней стирке; однако применением порошко-
порошкообразных отбеливающих веществ на основе пероксобората натрия отмеченные
недостатки устраняются, и вещества эти, несмотря на повышенную стоимость,
находят значительное применение. С точки зрения расширения рынка инте-
интересно, что такого рода отбеливающие ванны в Европе уже давно пользуются
популярностью, а в США начали распространяться лишь недавно. Причина,
по-видимому, заключается в том, что для проявления отбеливающего дей-
действия такого пероксигидрата необходима несколько более высокая темпера-
температура, чем обычно; в Европе, насколько известно, широко практикуется стирка
с кипячением, а поэтому уже используется высокая температура, в то время
как в США стирка при требуемых высоких температурах начала распростра-
распространяться только в связи с недавним появлением бытовых автоматических сти-
стиральных машин.
Отбелка древесной массы. Дерево содержит около 45—55% целлюлозы,
остальное же состоит из углеводов (например, пентозанов и гемицеллюлоз)
и неуглеводов (например, смол, жиров, восков, лигнина, дубящих веществ
и золы).
Бумага и бумажные изделия вырабатываются из продуктов переработки
древесины, получаемых двумя способами. Древесная масса получается путем
окорки и последующего механического измельчения древесины и содержит
почти все составные части, присутствовавшие в дереве. Бумага, содержащая
древесную массу, быстро разрушается, особенно при действии солнечного
света и воздуха, поскольку в ней присутствуют лигнин и другие относительно
нестойкие вещества. Из химической целлюлозы можно получать значительно
более устойчивую бумагу, поскольку в этом случае практически все неугле-
неуглеводные компоненты древесины удалены в процессе варки и при последующих
операциях отбелки. В некотором количестве (путем менее полной обработки)
вырабатывают и продукты промежуточного состава, известные под названием
полухимической целлюлозы.
Существует три химических процесса удаления лигнина из древесины:
1. Сульфитный процесс, основанный на применении горячего водного
раствора двуокиси серы в сочетании с бисульфитом кальция или магния.
Обычно такой раствор применяют для варки хвойной древесины, например
ели и тсуги.
2. Натронный процесс, имеющий в настоящее время меньшее значение
и основанный на применении раствора едкого натра; как правило, его при-
применяют для варки лиственной древесины, например тополя.
3. Сульфатный, или крафт-процесс, в котором используется раствор
едкого натра и сернистого натрия. Обычно по этому методу варят сосновую
и аналогичную ей древесину с высоким содержанием смолы, хотя этот метод
пригоден и для древесины любого типа. Щелочная варочная жидкость дей-
действует слабее, чем варочная кислота, применяемая в сульфитном процессе;
поэтому крафт-целлюлоза обладает более высокой прочностью, но содержит
также большее количество нецеллюлозных компонентов древесины. В связи
с этим такая целлюлоза, если желательно достижение высокой белизны, требует
более интенсивной отбелки.
При любом из этих трех процессов химической варки желательно уда-
удалить максимально возможное количество лигниновых и аналогичных веществ
при минимальной деструкции молекул целлюлозы. Лучше всего это дости-
достигается прекращением процесса варки на такой стадии, когда еще удалены
не бее нецеллюлозные вещества, с последующей очисткой и отбелкой полу-
Отбелка 483
ченной целлюлозы. Назначение этой последующей обработки состоит в раз-
разложении окрашенных веществ, сохранившихся в волокне, и удалении нецеллю-
нецеллюлозных веществ. Обычно процесс проводится по одному из двух следующих
методов: 1. Целлюлозу обрабатывают раствором гипохлорита кальция или
гипохлорита натрия (который получается взаимодействием кальциевой соли
с углекислым натрием или едким натром) с рН от 8 до 11 в присутствии целлю-
целлюлозы; в этих условиях получается целлюлоза, окрашенная в кремовый, цвет.
2. При так называемом двухступенчатом или многоступенчатом методе целлю-
целлюлозу обрабатывают водным раствором хлора (хлор реагирует с водой с дис-
пропорционированием до хлорноватистой и соляной кислот и дает рН ниже 2).
После этого нейтрализуют массу гидроокисью кальция (или едким натром),
которая экстрагирует вещества, перешедшие в раствор при обработке хлором.
Затем целлюлозу обрабатывают гипохлоритом или перекисью. Обработка
хлором и промывка щелочью сравнительно мало снижают интенсивность
окраски и обычно рассматриваются как одна из стадий очистки. Но после
такой предварительной очистки последующая отбелка является особенно
эффективной. Если желательно обеспечить надлежащую белизну, то до ста-
стадии отбелки попеременную обработку хлором и щелочью можно повторить
два или три раза. Интенсивность отбелки и тип ее зависят от отличительных
особенностей исходной древесины, процесса варки и конечного применения
целлюлозы.
Многие типы бумаги вырабатываются из смесей древесной массы и хими-
химической целлюлозы, чтобы таким образом при минимальных затратах получить
надлежащую комбинацию белизны, стойкости, способности принимать пе-
печать, непрозрачности, механической прочности, впитывающей способности
и т. п. в соответствии с намечаемым применением. Например, масса, исполь-
используемая для газетной бумаги, где самым основным требованием является низ-
низкая себестоимость, состоит преимущественно из древесной массы; для высоко-
высококачественной бумаги (для книг) применяют исключительно смеси химических
видов целлюлозы. В производстве различных бумаг непродолжительного при-
применения, например для журналов и каталогов, а также в производстве обер-
оберточной, папиросной и фильтровальной бумаги используют смеси химической
целлюлозы и древесной массы. Все такие материалы требуют значительно
более интенсивной очистки и большего количества отбеливающих веществ
на единицу веса обрабатываемого материала, чем текстиль. Поскольку стои-
стоимость химических веществ при применении перекиси водорода или перекиси
натрия больше, чем стоимость отбеливающих растворов на основе гипохло-
гипохлорита, перекиси оказываются экономичными лишь в том случае, когда каче-
качество конечного продукта настолько значительно превышает качество, дости-
достигаемое при гипохлорите, что компенсирует более высокие затраты.
Почти бесконечное число вариантов в отношении видов сырья и жела-
желательных свойств конечного продукта является причиной того, что в разных
условиях применимы самые различные процессы отбелки; это способствует
также тому, что в конкуренции перекиси с хлором, гипохлоритом и другими
агентами перекись оказывается далеко не в таком благоприятном положении,
как например при отбелке хлопка. Очевидно, однако, что особенное значение
перекиси имеют при отбелке древесной массы. Гипохлорит обычно для этой
цели не применяют, так как им приходится пользоваться в такой высокой
концентрации, которая может привести к значительному снижению выхода
конечной массы. Обработка перекисью может повысить степень белизны дре-
древесной массы примерно на 10—12 единиц G. Е.* при ничтожно малом умень-
* В Северной Америке обычно принято выражать степень белизны бумаги в виде
отношения отражающей способности бумаги при свете с длиной волны 458 ммк к отражаю-
отражающей способности углекислого магния, произвольно принятой за единицу (в процентах).
Для этой цели обычно пользуются прибором фирмы «General Electric Company».
484 Глава 11
шении выхода массы. Это дает возможность использовать значительное коли-
количество такой дешевой массы в смеси, предназначенной для выработки печатной
или папиросной бумаги удовлетворительной белизны. Наряду с низкой себе-
себестоимостью древесная масса обладает определенными преимуществами,
например большей непрозрачностью (позволяющей применять более легкую
бумагу) и некоторыми желательными качествами для печати (упругостью).
Однако необходимо иметь в виду, что такого рода отбелка не увеличивает
в заметной степени низкую устойчивость древесной массы к действию солнеч-
солнечного света.
В 1950 г. древесную массу подвергали отбелке перекисью водорода или
перекисью натрия на 24 заводах общей производственной мощностью ИЗО т
в сутки, смеси же древесной массы и сульфитной целлюлозы подвергали
отбелке перекисями на ряде заводов общей производительностью около 360 т
печатной бумаги в сутки [21]. Для этой цели можно применять как пере-
перекись водорода, так и перекись натрия или смесь обоих этих веществ. При
рыночных ценах 1954 г. перекись натрия стоила примерно на 25% дешевле,
чем перекись водорода, в расчете на моль перекисных ионов. Для обеспечения
надлежащего рН отбеливающей ванны перекись натрия должна быть частично
нейтрализована кислотой типа серной; с другой стороны, к перекиси водорода,
как правило, необходимо добавлять едкий натр. Таким образом, при приме-
применении соответствующей смеси обеих перекисей отпадает необходимость в при-
применении каких-либо дополнительных химических веществ, кроме стабилиза-
стабилизаторов.
Наиболее экономично применение перекисей для отбелки древесной массы
в системах с высокой консистенцией, т. е. со сравнительно высоким отноше-
отношением массы к отбеливающему раствору. Желательная щелочность после вве-
введения древесной массы в такую ванну соответствует рН от 10,0 до 10,5; этот
рН падает по мере протекания процесса отбелки. Время отбеливания зависит
от консистенции массы, температуры, щелочности, содержания перекиси
и желательной белизны. Типичный режим основан на применении 12%-ной
консистенции массы, перекиси водорода в количестве около 1% в расчете
на сухой вес массы с добавкой силиката натрия и сульфата магния (или одного
из них) при температуре 38—43° [21]. При двухчасовой обработке степень
белизны возрастает приблизительно на 12 единиц G. Е. Отбелка перекисью
может осуществляться периодическим или непрерывным методом в том же
оборудовании, которое используется для отбелки гипохлоритом.
Другая методика состоит в нанесении раствора перекиси на бумагу или
целлюлозу с выдерживанием при комнатной температуре в течение одного
или нескольких дней. Отбелка может производиться, например, во время
хранения или перевозки. Наряду с обычными мерами предосторожности, при-
принимаемыми для устранения разложения перекиси, в процессах отбелки дре-
древесной массы требуется тщательно устранять возможность развития бактерий
путем применения подходящих ингибиторов, так как многие из этих бакте-
бактерий вырабатывают фермент каталазу, активно разлагающий перекись водо-
водорода. Для отбелки древесной массы применяется также гидросульфит натрия
Na2S2O4 или гидросульфит цинка; часто гидросульфит используют последо-
последовательно с перекисью водорода.
Сульфитная целлюлоза на разных заводах также подвергается отбелке
перекисями в сочетании с гипохлоритами, но в этой области перекись до на-
настоящего времени не нашла еще значительного технического применения.
Чаще всего она употребляется в настоящее время для производства специаль-
специальных сортов бумаги, от которых требуется повышенная степень белизны.
Отбелка перекисью водорода может быть экономичной только как конечная
стадия обработки после предварительных более дешевых процессов очистки
хлором и окисления гипохлоритом (или одного из них). Основными преимуще-
Отбелка 485
ствами этого метода перед гипохлоритным процессом являются значительно
повышенная белизна и устойчивость цвета, достигаемые без заметной деструк-
деструкции, что оправдывает более высокую себестоимость. Обработка перекисью
позволяет также получить более высокие выходы целлюлозы в процессах
отбелки и повышает теплостойкость беленой целлюлозы, что имеет существен*
ное значение при дальнейших операциях бумажного производства. Раствор
перекиси отличается от других отбеливающих веществ в том отношении,
что он обесцвечивает лигнин и другие окрашенные вещества, но не растворяет
их в заметной степени. Так, сульфитную целлюлозу можно отбелить пере-
перекисью водорода за одну операцию с получением очень высокого выхода цел-
целлюлозы, но стоимость химических веществ в этом случае настолько значи-
значительна, что процесс не оправдывается экономически. Для отбелки крафт-
целлюлозы можно применять перекиси вместе с гипохлоритом. В этом случае
на стадии экстракции щелочью можно использовать перекись натрия. При
таком применении перекись натрия обходится дешевле перекиси водорода.
С перекисью водорода при конечной отбелке химической целлюлозы может
конкурировать газообразная двуокись хлора, сообщающая целлюлозе очень
высокую степень белизны при минимальном снижении прочности. Недостат-
Недостатками этого метода являются высокая себестоимость двуокиси хлора, корро-
коррозионные ее свойства, а также ядовитость и взрывчатость при концентрации,
превышающей известный предел. Двуокись хлора, обычно получаемую вне
цеха отбелки из хлората натрия, абсорбируют водой.
Механизм отбелки целлюлозы перекисями изучен очень мало. В недавно
проведенном исследовании процесса отбелки еловой древесной массы пере-
перекисью натрия Джонс [6] показал, что существенного химического изменения
компонентов массы при этом не происходит. На лигнин тратится около 40%
всей израсходованной перекиси, на голоцеллюлозу (целлюлоза+гемицеллю-
лозы)—около 60%. Метилирование экстрагированного лигнина заметно снижает
расход перекиси на реакцию с ним; на основании этого сделан вывод, что пер-
первичная реакция перекиси с лигнином происходит в результате взаимодей-
взаимодействия с карбонильными, а возможно, и с фенольными гидроксильными груп-
группами. Характеристики реакции между перекисью натрия и экстрагированным
лигнином показывают, что высокая эффективность перекисной отбелки может
быть обусловлена высокой специфичностью и реакционной способностью пере-
перекиси в отношении наиболее сильно окрашенных лигниновых фракций. Иссле-
Исследование влияния времени и температуры на отбелку древесной массы пере-
перекисью водорода и последующее желтение этой массы при ультрафиолетовом
облучении изучено Брехтом, Жеймом и Шустером [22], которые сравнили
эти результаты с полученными при действии бисульфита и гидросульфита.
В недавно опубликованной монографии Технической ассоциации целлю-
целлюлозной и бумажной промышленности в главе, принадлежащей Биману и Рей-
херту [21], содержится превосходное описание применения перекисей для
отбелки древесной массы и целлюлозы. Эти авторы дают также отчасти и исто-
исторический обзор этих процессов и полную библиографию до 1950 г. включи-
включительно. В других главах этой книги содержится прекрасно изложенный ма-
материал по различным другим аспектам отбелки целлюлозы.
Дополнительные данные можно получить от фирм, вырабатывающих
перекись водорода [14, 16, 23], и найти в многочисленных патентах. Мак-
Юэн [24] недавно опубликовал сообщение, где по этому вопросу содержится
очень обширная библиография.
Очистка бумаги от печатных красок. Путем комбинирования механиче-
механических и химических процессов удаления типографской краски бумажную маку-
макулатуру можно обработать таким образом, чтобы использовать полученную
массу для нового производства печатной или какой-либо другой светлоокра-
светлоокрашенной бумаги. Обычно для этой цели используют горячие растворы едкого
486 Глава 11
или углекислого натрия или же смеси их обоих. Они вступают в реакцию
с различными связующими веществами в бумаге, особенно со связующим
веществом краски, и этим способствуют отделению волокна от других присут-
присутствующих примесей. Щелочные растворы перекиси эффективнее едких щело-
щелочей с таким же рН, причем хотя стоимость химических веществ в первом слу-
случае и выше, но перекись дает возможность перерабатывать более низкокаче-
низкокачественную бумажную макулатуру, проводить варку при более низких темпе-
температурах и получать более высокие выходы бумажной массы и более чистый
продукт. Помимо того, что перекись оказывает отбеливающее действие, она
функционирует еще как вещество, ускоряющее гидролиз и деполимеризацию
белков, крахмалов и масел, которые представляют компоненты клеев и свя-
связующего вещества печатной краски, в связи с чем пигменты переходят в рас-
растворимое состояние и отрываются от бумаги. Как и при других видах при-
применения, допустимо использование и перекиси водорода, и перекиси натрия,
и смеси их с отрегулированием необходимого рН и добавкой стабилизаторов.
Применение перекисей особенно желательно для удаления краски с бумаги,
содержащей древесную массу, так как такая бумага заметно темнеет при обыч-
обычной щелочной обработке.
Отбелка других целлюлозных материалов. Другие целлюлозные мате-
материалы также можно отбеливать перекисями с применением оборудования,
аналогичного употребляемому для отбелки целлюлозных волокон и древес-
древесной целлюлозы. Отбелка соломы представляет длительную и трудную опера-
операцию; она должна производиться в мягких условиях путем длительного и много-
многократного попеременного выдерживания в ваннах из перекиси и восстановителя,
например двуокиси серы или гидросульфита натрия. Перекись водорода осо-
особенно пригодна для отбелки древесины, применяемой, например, для про-
производства светлой мебели, корпусов радиоприемников и телевизоров и бит
для бейсбола. По одному часто применяемому методу [25] на поверхность
дерева наносят кистью или распылителем 5%-ный раствор едкого натра, со-
содержащего силикат натрия, а после некоторой выдержки покрывают ее 27—
35%-ным раствором перекиси водорода; эти операции можно осуществлять
и в обратном порядке. После достижения намеченной белизны щелочь ней-
нейтрализуют слабой уксусной кислотой и дерево подвергают сушке. Вместо
этого дерево можно отбеливать одноступенчатой операцией с применением
смеси перекиси, силиката натрия и аммиака или едкого натра. В результате
отбеливается только тонкий поверхностный слой, а поэтому последующие
отделочные операции требуют осторожногоfи четкого выполнения.
Отбелка веществ, содержащих белки
Вещества животного происхождения, например шерсть, волос, мех,
перья, шелк и кожа, представляют в основном белки, которые могут быть
повреждены при действии высокоактивных отбеливающих растворов или
при обработке в щелочных средах. Поэтому уже в течение многих лет для
отбелки таких веществ пользуются разбавленным раствором двуокиси серы
или же перекисью водорода, а иногда и обоими этими агентами, причем сначала
производят обработку перекисью.
Отбелка шерсти. В типичной операции отбелки шерстяные одеяла под-
подвергают очистке и промывке, затем пропитывают отбеливающим раствором,
содержащим около 1,3% перекиси водорода, с добавкой силиката натрия для
доведения рН до 9. Часто вместо силиката натрия применяют четырехзамещен-
ный пирофосфат натрия в сочетании с небольшими количествами аммиака.
При этом получается материал более мягкий на ощупь. После выжимания
шерстяные одеяла оставляют еще на 24 часа или больше [10] и затем промывают.
Огбелку шерстяных изделий можно проводить при температурах, достигаю-
Отбелка 487
щих 38—49°, что позволяет снизить продолжительность отбелки до 1—12 час.
при применении 0,4—0,8 вес.% перекиси водорода [14—16]. Значительную
часть шерсти, подвергающейся отбелке, в США обрабатывают методом «внут-
«внутренней сушки», применимым лишь для шерстяных материалов; в этом случае
отмытую шерсть закладывают в отбеливающий раствор, содержащий 0,3—
0,9% перекиси водорода, при рН от 3 до 5.
При повышенных температурах все шерстяные изделия обладают склон-
склонностью к потемнению, причем этот эффект возрастает при наличии остаточной
перекиси. Одной промывки оказывается недостаточно, и поэтому остаточную
перекись водорода (независимо от метода отбелки) приходится удалять из
шерсти путем обработки гидросульфитом натрия или шламом каталазы. Эта
операция исключается лишь в том случае, если перекись достаточно быстро
разлагается сама [26]. Лэксер и Уивелл [27] измеряли влияние обработки
шерсти перекисью водорода на кривые графика напряжение—растяжение,
считая эти характеристики мерилом степени деструкции шерсти.
В прачечных как целлюлозные вещества, так и материалы животного
происхождения отбеливают иногда и пероксоборатом натрия, но последний,
в сущности, действует лишь как щелочной раствор перекиси водорода. Тех-
никаГотбелки не отличается от вышеизложенной.
Видоизменение свойств шерсти. Механическая обработка шерсти во влаж-
влажном и нагретом состоянии способствует сцеплению или «свойлачиванию» во-
волокон. Различные виды шерсти и других животных волокон могут сильно
различаться по способности к свойлачиванию. В производстве фетра для шляп
необходимая способность к свойлачиванию часто достигается путем окисле-
окисления волокна в тщательно контролируемых условиях. В прошлом для этой
цели применяли азотную кислоту, содержащую соли ртути в качестве ката-
катализатора, но в 1936 г. сделано открытие, что ядовитый раствор ртутной соли
может быть заменен раствором перекиси водорода, содержащим минераль-
минеральную кислоту [28]. Окисляющее действие, по-видимому, состоит в присоеди-
присоединении кислорода к дисульфидным группам цистина шерсти [29] с последую-
последующим разрывом этих цистиновых мостиков в структуре белка [30]. Раньше
считали [30], что перекись водорода и аминная или карбиминная группа
волокна образуют фактически аддитивное соединение. Описано влияние такого
рода обработки на кроличий пух [31]. Сообщается также о видоизменении
пуха ангорских кроликов путем последовательной обработки тиогликолятом
натрия, пепсином и окислителем вроде перекиси водорода [32].
Щелочные растворы перекиси водорода снижают способность шерсти
к свойлачиванию и сообщают ей сопротивляемость усадке. Поэтому обработку
перекисью водорода используют иногда в сочетании с хлорированием шерсти
для сообщения последней устойчивости против усадки. Недавно выпущен
технический бюллетень, в котором описано видоизменение свойств шерсти
путем обработки перекисными соединениями; в этом бюллетене содержится
и полная библиография по этому вопросу [33]. Вуд [34] опубликовал недавно
статью, касающуюся вопроса снижения способности смеси волокон чистой
шерсти или смесок к свойлачиванию и обеспечения их формостойкости путем
обработки различными химическими веществами; в статье содержатся также
многочисленные ссылки на литературу.
Различные операции отбелки
Перекись водорода применяется и|для отбелки волос человека, а также
и других волосяных волокон, например конского волоса, козьей и коровьей
шерсти, различных видов мехов, перьев и кожи. В тех случаях, когда белковое
вещество может легко подвергнуться деструкции за счет переокисления,
слишком высокой температуры или высокого рН, например при отбелке ка-
488 Глава 11
шемира, рекомендуется применять смесь перекиси водорода с пероксодисуль-
фатом аммония, которая лучше подходит для этой цели, чем одна перекись
водорода. Небольшие количества перекиси водорода находят применение
и для отбелки таких материалов, как требуха, кости, пуговицы из перламутра
или растительной слоновой кости (оболочки плодов пальм Phytetephas), клей,
могильные плиты, белая оленья кожа, яйца, табак, пробка, губка и оболочки
для колбас. Почти во всех этих случаях процессы отбелки должны быть мяг-
мягкими во избежание повреждения отбеливаемого материала.
Жиры, масла, воски, мыла и соки можно обесцвечивать окисляющим дей-
действием перекиси водорода, но чаще красящие вещества отделяют путем абсорб-
абсорбции на измельченной инфузорной земле или активированном угле. Для жиров
и масел существенное значение имеет концентрация перекиси водорода [35],
и успех отбелки зависит от химической природы жира или масла [36]. Для
каждого такого применения разработана своя методика отбелки [14—161.
ДРУГИЕ ВИДЫ ПРИМЕНЕНИЯ ПЕРЕКИСИ ВОДОРОДА КАК ОКИСЛИТЕЛЯ
Производство вискозного шелка
Молекулы целлюлозы обладают линейной полимерной структурой, кото-
которую можно рассматривать как состоящую из большого числа звеньев глю-
глюкозы, соединенных своими концами при помощи кислородных эфирных мости-
мостиков. Средний молекулярный вес обычно определяют путем измерения вяз-
вязкости пробы, растворенной в водном медноаммиачном или каком-либо дру-
другом аналогичном растворе; молекулярный вес почти пропорционален вяз-
вязкости. Длина цепи, или молекулярный вес, обычно выражается как степень
полимеризации, представляющая собой среднее число звеньев глюкозы в моле-
молекуле целлюлозы. Целлюлоза, используемая для производства вискозного
волокна, обычно представляет химическую древесную целлюлозу специаль-
специальной очистки с начальной степенью полимеризации от 800 до 1000. Степень
полимеризации должна быть понижена примерно до 350, чтобы при после-
последующем растворении целлюлозы в смеси сероуглерода и едкого натра с обра-
образованием ксантогената целлюлозы раствор обладал такой низкой вязкостью,
при которой его можно было бы продавливать через отверстия фильеры,
В США для снижения длины цепи целлюлозу замачивают в растворе едкого
натра и оставляют ее созревать в течение 20—40 час. в строго определенных
условиях. В щелочной среде кислород воздуха вступает во взаимодействие
с цепями целлюлозы и снижает степень полимеризации (если тщательно
защитить целлюлозу от доступа воздуха, то такой деполимеризации не наблю-
наблюдается). Скорость деполимеризации увеличивается при действии небольших
количеств ионов многовалентных металлов, например марганца, железа
и никеля, которые действуют в качестве активаторов. Поэтому во избежание
неконтролируемых колебаний деполимеризации содержание таких примесей
должно быть доведено до минимума. Время, требующееся для деполимери-
деполимеризации, может быть значительно снижено путем добавки к смеси целлюлозы
и щелочи таких окислителей, как гипохлориты или перекись водорода. Дей-
Действительно, перекись водорода используется для этой цели в производстве
вискозного волокна в некоторых европейских странах, но, очевидно, не в США.
Дальнейшие подробности по этому виду применения и по использованию
перекиси для деполимеризации целлюлозы вообще можно найти в сообщении
Маргулиса [37] и в одном техническом бюллетене, где приводится обширная
библиография [38 .
Обработка щелочью в производстве вискозы имеет многостороннее зна-
значение и помимо снижения средней длины цепи целлюлозы до желательных
размеров. Такая обработка применяется для очистки с целью освобожде-
Другие виды применения перекиси водорода как окислителя 48^
ния требуемой а-целлюлозы (наиболее инертного компонента всей целлюлозы)
от других углеводов, например гемицеллюлоз, и она также способствует
процессу набухания целлюлозы, который желательно проводить до превра-
превращения ее в ксантогенат. Добавка окислителей, например перекиси водорода,
для ускорения деполимеризации не влияет на скорость образования алкали-
целлюлозы или на скорость растворения гемицеллюлоз [37, 38]. Поэтому
применение перекиси в производстве вискозы ограничено в связи со сложной
природой различных происходящих при этом процессов и как следствие—
недостаточного знакомства с характером влияния такого рода обработки на
свойства конечного волокна. Возможно, что ускоряющее действие примесей
многовалентных металлов на обычный процесс созревания обусловлен обра-
образованием перекиси водорода под действием самоокисления (как описано
на стр. 68 и ел.). Добавка перекиси водорода к ксантогенату целлюлозы
перед прядением ускоряет также его созревание [39].
Перекись водорода как антихлор
Перекись водорода может быть использована для разложения избытка
хлора или соединений, содержащих последний, которые могут остаться в том
или ином веществе в результате предыдущей обработки. Например, при
отбелке шеллака гипохлоритом применение перекиси водорода для трудной
операции удаления гипохлорита, остающегося к концу отбелки, не только
исключает возможность сохранения остатков хлора, но допускает также
отбелку с применением избытка гипохлорита, ускоряющего процесс. В опе-
операциях отбелки действие перекиси водорода как антихлора может быть только
одной из нескольких функций, выполняемых перекисью. Например, при
обработке шеллака характеристика старения и растворимость шеллака улуч-
улучшаются при применении перекиси водорода; эти виды применения описаны
в разделе «Отбелка». Перекись водорода рекомендуется также в качестве
химического реагента для разложения хлора, остающегося в воде после хло-
хлорирования. Избыточную перекись предложено затем разлагать пропусканием
воды через слой двуокиси марганца [40]. Однако разложение перекиси водо-
водорода (как описано на стр. 414) может способствовать растворению небольших
количеств марганца, в связи с чем возникает вопрос о пригодности такой воды
для питья.
Окисление красителей
При крашении кубовыми красителями (т. е. антрахиноновыми или инди-
гоидными) сначала восстанавливают нерастворимый пигмент до растворимой
бесцветной лейкоформы действием восстановителя, обычно гидросульфитом
натрия в щелочной среде. После адсорбции красителя на волокне последний
окисляют, в результате чего вновь образуется исходный нерастворимый пиг-
пигмент, который фиксируется на ткани. Превосходная прочность красителей
этой группы объясняется очень плохой растворимостью пигмента в окислен-
окисленном состоянии. Раньше окисление проводилось исключительно действием
воздуха; сейчас для этой цели используется химическая ванна. Типичная
окислительная ванна может иметь один из следующих составов: 1) 2 вес. %
бихромата калия +2 вес.% уксусной кислоты;,2) 1 вес.% пероксобората нат-
натрия или 3) 1 вес.% перекиси водорода. Соответственно нужно регулировать
рН и температуру ванны [17, 41].
Процесс окисления, как периодический, так и непрерывный, зависит
от типа красителя и природы ткани. Применение перекисных соединений спо
собствует получению ярких и чистых тонов, высокой всасывающей способ-
490 Глава 11
ности ткани, облегчает последующую промывку и обеспечивает общую чис-
чистоту всей операции.
Сернистые красители, весьма широко применяемые для крашения хлоп-
хлопка-сырца, а также вискозного волокна, сначала восстанавливают (обычно
сернистым натрием), а затем после нанесения окисляют таким же образом,
как и кубовые красители. В некоторых случаях в качестве окислителя при-
применимы и пероксодисульфаты. Перекись водорода может найти применение
и для протравного крашения окисляемыми полупродуктами красителей;
этот процесс изучен с точки зрения использования его в отделке меха [42].
Фотохимическое окисление и деструкция хлопка на влажном воздухе
сенсибилизируются желтыми, оранжевыми и красными антрахиноидными
кубовыми красителями, но не красителями, принадлежащими к синей, фио-
фиолетовой или зеленой группе [43, 44]. Эффект деструкции сопровождается обра-
образованием окислителя (его можно открыть реакцией с йодокрахмальной бумаж-
бумажкой), по-видимому представляющего перекись водорода; по всей вероятности,
в основном деструкция обусловлена атакой со стороны этой перекиси. Напри-
Например, пропитка волокна сенсибилизирующим красителем может способствовать
атаке близрасположенного неокрашенного волокна даже в отсутствие кон-
контакта между обоими волокнами [43]. Это явление объясняют улетучиванием
перекиси водорода, образовавшейся на окрашенном волокне, с последующим
поглощением ее неокрашенным. Если рядом лежат окрашенные и неокрашен-
неокрашенные вискозные или найлоновые волокна, неокрашенные заметно не изме-
изменяются, и, согласно некоторым данным, деструкция волокон из этих материа-
материалов протекает по другому механизму. Хлопок, окрашенный окисью цинка,
двуокисью титана или сернистыми и основными красителями, также под-
подвергается фотохимической деструкции, хотя в волокнах, окрашенных серни-
сернистыми красителями, перекиси водорода не обнаружено. Эти результаты могут
сильно измениться при пропитке хлопка, окрашенного кубовыми красите-
красителями, небольшими количествами солей металлов. При испытаниях на содер-
содержание окислителя уменьшения его образования после пропитки окрашенных
волокон иногда совсем не наблюдалось (никель), иногда снижение было неболь-
небольшим (хром) или довольно значительным (марганец), в случаях же с медью,
железом или кобальтом окислитель вообще отсутствовал [44]. Результаты
испытаний показывают совершенно определенный параллелизм с каталити-
каталитической активностью соответствующих металлов в отношении разложения
перекиси водорода.
Степень фотохимической деструкции, однако, подчиняется более слож-
сложным зависимостям. Так, введение меди в волокно снижает степень фотохи-
фотохимической деструкции для хлопка, окрашенного наиболее чувствительными
красителями, но увеличивает деструкцию неокрашенного хлопка или хлопка,
обработанного кубовыми красителями темного цвета; таким образом, сте-
степень деструкции фактически оказывается независящей от природы взятого
красителя. Железо и другие металлы также влияют на фотохимическую
деструкцию. Возможное объяснение заключается в том, что различные тяже-
тяжелые металлы способствуют не только разложению перекиси водорода, но
и образованию ее в результате самоокисления в атмосфере (см. стр. 68),
а поэтому в некоторых случаях размер деструкции значительно больше зави-
зависит от природы и количества присутствующих тяжелых металлов, чем от при-
природы красителя. В этом отношении интересно было бы изучить влияние
металлов, обладающих сравнительно ничтожными каталитическими свойствами,
.а также неметаллических катализаторов на фотохимическую деструкцию
хлопка. Шеффер [45] обнаружил перекись водорода также при щелочной
обработке одной целлюлозы и привел доказательства, подтверждающие, что
щелочная деструкция целлюлозы 'происходит в результате гидролиза глюко-
пиранозных колец целлюлозы с последующим окислением перекисью*
Другие виды применения перекиси водорода как окислителя 491
Применение в фотографии
О действии перекиси водорода на фотоэмульсию см. на стр. 68 и 338, где
описываются процесс ее образования и химические свойства. Проявляющее
действие щелочной перекиси водорода, на котором мы остановимся ниже, не
находит практического применения в фотографии ввиду его неселективности,
образования вуали и значительно меньшей эффективности и удобства по срав-
сравнению с обычными органическими проявителями. Некоторое применение
перекись водорода нашла лишь как .проявитель для синек.
Подкисленная перекись водорода, содержащая растворимые бромиды,
оказывает восстанавливающее действие (в фотографическом смысле этого
слова) на серебряное изображение проявленной пластинки или отпечатка,
а также размягчает и'растворяет желатину на тех участках, где имеется изобра-
изображение. Этот эффект используется для получения желатинового рельефа,
который может найти применение для механического воспроизведения. Упо-
Упомянутый процесс описан Андресеном [46], Люппо-Крамером [47] и Ланге [48].
Сама перекись водорода большим применением как общий фотовосстанови-
фотовосстановитель не пользуется; для этой цели широко используется пероксодисульфат
аммония.
Перекись водорода применяется в двух процессах фотопечатания [49].
По первому из них обычный негатив опускают в эфирный раствор разбавлен-
разбавленной водной перекиси водорода и выдерживают короткое время в контакте
с бумагой, покрытой желатиной с пигментом. Затем бумагу переносят в рас-
раствор соли закисного железа. Поверхность покрытой желатиной бумаги, по-
поглотившая больше перекиси водорода из участков негатива, свободных от
изображения, способствует окислению впитывающейся соли закисного желе-
железа в размерах, пропорциональных количеству перекиси водорода, находя-
находящейся на разных участках поверхности. Образующаяся соль окисного железа
сообщает желатине нерастворимость на большую или меньшую глубину;
при растворении растворимой желатины в теплой воде получается отпечаток,
где основная часть пигмента находится в тех зонах, которые соответствуют
частям поверхности негатива, лишенным изображения.
. Второй процесс печатания [50], известный под названием кататипии,
аналогичен предыдущему и отличается от него лишь тем, что покрытая
желатиной бумага не содержит пигмента. После контакта с негативом обыч-
обычная желатиновая пленка с абсорбированной перекисью водорода вносится
в раствор соли двухвалентного марганца или щелочной раствор азотнокислого
серебра. Металл поглощается и осаждается в виде двуокиси марганца или
металлического серебра в зонах, содержащих перекись водорода, в резуль-
результате чего получается позитивный отпечаток.
Перекись водорода способна давать скрытое изображение на фотопластин-
фотопластинке без действия света; это свойство использовано для получения прямых
изображений структуры разных поверхностей. Поверхность покрывают рас-
раствором перекиси водорода и прижимают к этой поверхности фотографиче-
фотографическую пленку. Некоторые участки поверхности способствуют более быстрому
разложению перекиси или сильнее поглощают ее, причем после проявления
пленки эти участки оказываются слабее почерневшими или даже совершенно
не чернеют. Кречмер [51] и Абрамсон [52] использовали этот метод для
-снятия структуры кожи. Кречмер [53] и Фрейтаг [54] применили его при
изучении пороков бумаги, для обнаружения подделок на документах, а также
для воспроизведения печатных материалов.
Фотопластинки или фотопленки можно гиперсенсибилизировать (умень-
(уменьшить время экспозиции или повысить чувствительность к определенной
области спектра) путем обработки в ванне с перекисью водорода. Шмишек [55]
показал, что скорость съемки на панхроматических пластинках и чувствитель-
492 Глава 11
ность их к красному свету могут быть значительно повышены, если опустить
их в подкисленный или щелочной раствор, содержащий 0,075 вес.% перекиси
водорода и растворенное серебро, а затем промыть и высушить. Для таких
ванн он рекомендует следующий рецепт: 1 мл 25%-ного раствора аммиака,
1 мл 30%-ного раствора перекиси водорода, 400 мл воды и серебряную соль
в количестве, дающем 0,025 г серебра. Наиболее подходящими серебряными
солями являются молибдат или вольфрамат. Они лучше других предотвра-
предотвращают образование вуали, но с точки зрения сенсибилизирующего действия
многие другие соли серебра обладают такой же эффективностью. Применение
перекиси водорода для гиперсенсибилизации отличается двумя недостат-
недостатками: конечный эффект зависит от типа эмульсии и обработанные пластинки
должны быть использованы вскоре после обработки, иначе становится замет-
заметным образование вуали. В литературе дано сравнение перекиси водорода
с другими неорганическими сенсибилизаторами [56].
Перекись водорода оказывает интенсифицирующее действие (увеличение
плотности или проявляемости скрытого изображения) на пластинку после
экспозиции. Уайтмэн и Кирк [57] показали, что обработка 0,004—0,016%-ной
перекисью водорода с последующим немедленным проявлением дают замет-
заметную интенсификацию; для этого эффекта, особенно заметного при высоко-
высокочувствительных пластинках, требуется наличие растворимого бромида. Бар-
нес, Уайтгорн и Лоуренс [58] изучили влияние разных проявителей на интен-
интенсифицирующее действие перекиси водорода.
Больше всего перекись водорода применяется в фотографии для удале-
удаления гипосульфита (тиосульфата); применение перекиси для этой цели было
впервые предложено еще в 1866 г. Смитом и Спиллером [59]. Перекись водо-
водорода окисляет тиосульфат, остающийся на фотоотпечатках после фиксации,
до сульфата. Тиосульфат (или продукт его частичного окисления—тетратио-
нат) реагирует с серебром медленно в обыкновенных условиях и быстро при
высокой температуре и большой влажности, причем в результате образования
сульфида серебра получается блеклое и мутное изображение бурого цвета.
Удаление последних следов тиосульфата из фотобумаги только промывкой
практически невозможно. Для получения высококачественных отпечатков
требуется химическое удаление тиосульфата.
Грэбтри, Итон и Мюлер [60] составили обзор литературы по этому воп-
вопросу и подробно изучили применение перекиси водорода для удаления избыт-
избытка гипосульфита. Они рекомендуют следующий рецепт (известный под назва-
названием «Кодак НЕ-1»): 500 мл воды, 125 мл 3%-ной перекиси водорода, 100 мл
3%-ного аммиака. Смесь разводят водой до 1 л. Отпечатки промывают в тече-
течение 30 мин. (при бумаге двойного веса —1 час) при температуре 18—21°, выдер-
выдерживают в ванне вышеуказанного состава в течение 6 мин., промывают 10 мин.
и сушат. 4 л этого раствора достаточно для обработки 50 отпечатков размером
20x25 см или эквивалентного количества отпечатков другого размера. Для
массовой фотографии рекомендуется несколько иной рецепт.
Очистка растворов солей металлов
При очистке различных химических соединений, особенно соединений
металлов, приходится сталкиваться с общей проблемой, а именно с пробле-
проблемой освобождения их от железа. Такая задача, например, ставится при про-
процессах получения разных чистых металлов или при очистке гальванических
ванн. Для этой задачи целесообразно использовать следующую методику:
к очищаемому раствору добавляют основания в количестве, достаточном для
того, чтобы повысить рН раствора до значений, при которых гидрат окиси
(или закиси) железа нерастворим, а гидроокиси других металлов остаются
в растворе.
Другие виды применения перекиси водорода как окислителя
493
В каждом отдельном случае надлежащий рН определяется желательной
степенью очистки от железа, концентрацией очищаемого металла, который
должен оставаться в растворе, а также другими факторами, например амфо-
терным характером некоторых металлов. Интервалы рН, представляющие
интерес для работы, приведены в табл. 85. В ней даны максимальные значе-
значения рН, при которых раствор, содержащий 0,001 моля данного металла на
Таблица 85
Значения рН, при которых различные гидраты окисей металлов
образуют миллимолярный насыщенный раствор
Гидрат окиси металла
Fe(OH)8
А1(ОН)8
Сг(ОН)8
Си(ОН)8
Zn(OH)*
Со(ОН)а
рН осаждения
2,5
4,1
4,9
5,9
7,3
7,6
Гидрат окись металла
РЬ(ОН)а
Fe(OH)a
Мп(ОНJ
Ni(OHJ
Mg(OHJ
рН осаждения
7,7
8,1
8,4
8,6
9,9
1 л, может существовать при комнатной температуре; этот рН назван условно
«рН осаждения». Значения рН вычислены из данных по произведению раство-
растворимости Латимера и Гильдебранда [61]. Очевидно, что чем выше концентра-
концентрация любого металла, который желательно сохранить в растворе, тем ниже
должен быть рН.
Из таблицы видно, что легче всего выпадает в осадок гидрат окиси желе-
железа; он почти полностью осаждается даже в довольно кислых растворах. Гид-
Гидрат закиси железа, наоборот, теряет растворимость практически только в ней-
нейтральном растворе, т. е. в таких условиях, в которых многие другие присут-
присутствующие металлы также выпадают в виде гидроокисей. В связи с этим для
полного осаждения железа из большинства растворов металлов необходимо
окислить закисное железо до окисного. Окисление может быть осуществлено
продуванием через раствор воздуха, но скорость окисления воздухом мала,
и поэтому часто вместо воздуха применяют окислитель, например перекись
водорода, перекись натрия или хлор. После окисления гидрат окиси железа
осаждают, добавляя соответствующее основание, и затем вызывают коагуля-
коагуляцию осадка нагреванием с последующим отделением его фильтрованием или
отстаиванием. Чтобы снизить количество перекиси, теряемой за счет разло-
разложения, железо желательно окислять до повышения рН, поскольку такие
металлы, как железо, обладают большей каталитической активностью в форме
коллоидного осадка окиси или гидроокиси, чем в форме растворенных
ионов.
Хотя теоретически металлы, обладающие значениями рН осаждения,
значительно превышающими величину рН осаждения для закисного железа,
могут быть очищены по указанному методу и без предварительного окисле-
окисления закисного железа до окисного, на практике разделение происходит легче
после предварительного окисления. Например, при обработке хлористого
магния до процесса его электролиза (с целью получения металлического
магния) раствор обычно обрабатывают окисью магния для осаждения
примесей; однако отделение железа может быть достигнуто лишь путем
предварительного окисления раствора, например перекисью водо-
водорода [62].
494 Глава 11
Перекись водорода (или перекись натрия) находит применение для очист-
очистки сульфатов двухвалентных марганца, никеля или меди, хлорида цинка,
сульфата цинка или бериллия от железа [15, 16], Во всех тех случаях, когда
наличие натриевой соли в конечном растворе недопустимо, лучше пользо-
пользоваться не перекисью натрия, а перекисью водорода. Процесс обработки,
вообще говоря, заключается в добавке некоторого стехиометрического избыт-
избытка перекиси против содержания присутствующего железа, в отрегулирова-
нии рН путем введения соответствующего основания (часто окиси или гидро-
гидроокиси обрабатываемого металла) и в отделении осадка. В некоторых случаях
выпадает примесь более чем одного металла, например при очистке лантано-
вых солей путем осаждения гидратов окисей церия и железа из соответствую-
соответствующего раствора [63]. Точно так же можно использовать перекись водорода для
отделения марганца от железа и редких земель [64]. Иногда разделение можно
осуществить, превратив ион того или иного металла в нерастворимую форму—
перекись металла. Например, перекисью водорода пользуются для осаждения
перекиси урана из растворов ураниловых солей в одном из процессов про-
производства весьма чистого металлического урана. Опубликованы подробности
этого метода [65]. При некоторых гальванотехнических процессах операции
очистки можно проводить путем окисления перекисью водорода и осажде-
осаждения в самой ванне, что связано с некоторыми дополнительными преимуще-
преимуществами; например, при этом методе исключается возможность точечной кор-
коррозии при высоких плотностях тока. В присутствии перекиси водорода повы-
повышается также «белизна» осадков олова и цинка.
Известны и другие процессы разделения, в которых используется окис-
окисление перекисью водорода и которые базируются на том, что растворимость
разных солей многовалентных металлов изменяется с изменением состояния
валентности. Так, покрытие изделий из железа и стали фосфатом цинка нару-
нарушается в случае присутствия в растворе фосфата закисного железа. Однако
этот фосфат может быть осажден путем окисления его перекисью водорода
до фосфата окисного железа [66]. В операциях переработки цветного лома
переход олова в раствор или осаждение его из смесей солей металлов можно
регулировать путем окисления перекисью водорода [67]. В других случаях
перекись водорода или другие окислители можно использовать для сохране-
сохранения компонентов соли металла в растворе, вместо того чтобы путем окисления
способствовать осаждению определенного вещества; окислитель может слу-
служить и для сохранения раствора в желательном состоянии окисления. Напри-
Например, для защиты нержавеющей стали в процессе механической обработки
часто применяется раствор, содержащий щавелевую кислоту и ион окисного
железа. В этом случае может оказаться желательной добавка перекиси водо-
водорода для предотвращения осаждения оксалата закисного железа, обладаю-
обладающего лишь очень малой растворимостью, а также и для других целей [68].
Окислительная способность перекиси водорода используется также для
регенерации отработанных растворов от демеркаптанизации при нефтеперера-
нефтепереработке. Отработанный раствор плумбита натрия обычно превращают в окись
свинца путем продувки воздухом. В одном сообщении [69] указывается, что
добавка перекиси водорода в стадии регенерации воздухом снижает общую-
стоимость процесса в результате инициирования окисления, чтоЧпособствует
уменьшению затраты времени и экономии пара и энергии. Перекись водорода
или иные перекиси с успехом используются и в других процессах, где необхо-
необходимо окисление воздухом, с целью повышения начальной скорости реакции.
Перекись водорода находит также применение и для очистки от небольших
количеств примесей путем окисления; например, окись азота в серной кислоте
можно окислить до азотной кислоты [70].
Макстед [71] описал способ регенерации катализаторов гидрирования,
отравленных производными серы или фосфора, путем окисления их перекисью
Другие виды применения перекиси водорода как окислителя 495
водорода. Действие перекиси водорода в этом случае заключается в окислении
яда на катализаторе с превращением его в «экранированную», или^нетоксиче-
скую, форму. Лучше всего в этом случае использовать окислительную спо-
способность пероксокислот, например пероксомолибденовой, пероксовольфрамо-
вой, или их солей, которые можно получить и в самой реакционной среде.
Путем применения избытка перекиси водорода в присутствии, например,
соли молибдена или вольфрама можно обеспечить непрерывное регенериро-
регенерирование перекисного соединения, вследствие чего необходимое количество доро-
дорогой соли будет сохраняться все время минимальным.
Обработка металлических поверхностей
Опубликовано большое число методов, особенно в патентной литературе,
по различным видам обработки поверхностей металлов окислителями, напри-
например перекисью водорода или другими перекисными соединениями. Эта обра-
обработка может преследовать самые различные цели, например: 1) осаждение
некоторых соединений или сохранение известных веществ в растворе, как
описано выше; 2) образование окисной пленки на поверхности; 3) освобо-
освобождение поверхности металла от нежелательных или специфических компо-
компонентов; 4) растворение металла с поверхности и удаление его. Опубликован-
Опубликованные методы получения окисных пленок являются обычно весьма специфиче-
специфическими, и выбор их сильно зависит от намеченной цели; как правило, они
заключаются в многократных окунаниях материала в две или большее число
ванн разного состава, одна из которых содержит окислитель. Образующаяся
таким образом окисная пленка может улучшить внешний вид металлического
изделия, способствовать возникновению более устойчивого покрытия, пас-
пассивировать поверхность металла, сообщая ей большую инертность, или давать
какой-либо другой эффект (например, в производстве селеновых или купрокс-
ных выпрямителей).
При очистке металлических поверхностей для травления обычно при-
применяется ванна, состоящая из водного раствора кислоты, причем природа
и концентрация кислоты определяются природой обрабатываемого металла
и желательным эффектом. Добавка окислителя к ванне может служить самым
различным целям; так, окислитель может способствовать образованию на
металле более блестящей или слабее 'окисляемой пленки, возможно за счет
того или иного пассивирующего действия, или же повысить скорость или
эффективность операции травления, например за счет деполяризующего дей-
действия (стр. 496). В различных операциях травления и снятия слоя металла,
применяемых, например, в металлургических лабораториях, можно исполь-
использовать перекись водорода и перекисные соединения [72]. С применением ванн
для травления близко соприкасаются различные процессы «глянцевого тра-
травления» для сообщения повышенной гладкости и увеличения отражательной
способности поверхностей металлов. Такого рода ванны обычно предста-
представляют собой растворы кислот, часто содержащие окислитель, например' пере-
кисное соединение. Выпущены различные технические бюллетени, bJкоторых
приводятся более подробные данные об обработке поверхностей металлов
и даются необходимые ссылки на литературу [73].
Другим видом применения перекиси водорода в обработке поверхности
является использование ее для флотации руд. Проведенные исследования [74]
показали, что перекись водорода может изменить поверхность частиц^рудного
концентрата с улучшением селективности разделения при флотации. Это дей-
действие, конечно, зависит от химической природы руды; например, Маху [75]
описывает случай, когда присутствие перекиси водорода, наоборот, тормо-
тормозило флотацию.
496 Глава И
Перекись водорода как деполяризатор
В ряде реакций твердых тел с жидкостями, когда происходит выделение
газообразного водорода (например, при растворении металлов в кислотах
или при осаждении золота из цианистого раствора цинком), скорость реакции
часто снижается из-за образования защитной пленки водорода на твердой
фазе. В таких случаях скорость может быть значительно повышена путем
введения в раствор окислителя (деполяризатора), окисляющего водород до
воды, в результате чего становится возможным более быстрый контакт реа-
реагентов. При достаточно высокой концентрации окислителя можно полностью
предотвратить выделение газообразного водорода на твердой фазе. Для этой
цели используется как перекись водорода, так и нитраты и другие окисли-
окислители [76—78]. Перекись водорода, очевидно, играет аналогичную роль и в не-
недавно запатентованном способе использования ее в электролитическом про-
производстве хлоритов щелочных металлов [79]. Согласно данным этого патента,
при пропускании через пористый катод раствора двуокиси хлора и перекиси
водорода (в стехиометрическом избытке по отношению к выделяемому водо-
водороду) возрастет выход хлорита в электролизере.
Опубликованы различные предложения и методы использования пере-
перекиси водорода для производства коррозионных испытаний металлов. Однако
если не считать коррозионных исследований легких металлов, например
алюминия, ее нельзя рекомендовать для таких целей. Перекись водорода
легко разлагается уже в присутствии ничтожных количеств металлических
ионов, образующихся в процессе коррозии; поэтому концентрация перекиси
в коррозионном растворе меняется во времени, что затрудняет воспроизведе-
воспроизведение и интерпретацию результатов. При исследовании влияния коррозионных
растворов на алюминий оказалось, что перекись водорода ускоряет точечную
коррозию, вероятно в результате деполяризующего действия. Сравнительная
инертность перекиси водорода по отношению к алюминию как металлу позво-
позволяет рекомендовать ее для осуществления ускоренного испытания алюминия
на коррозию (см. также раздел «Самоокисление металлов», стр. 68).
Модифицирование углеводов, белков и каучука
При обработке высокополимеров, например крахмала, белков, каучуков
и т. п., перекисью водорода или перекисными соединениями происходят
окисление и деполимеризация. Снижение среднего молекулярного веса при
частичной деполимеризации уменьшает вязкость растворов полученных про-
продуктов, что позволяет легче перевести их в раствор и использовать в каче-
качестве клеев, шлихт, связующих веществ и т. д. [15, 80]. Деполимеризация
целлюлозы путем реакции с перекисью рассматривалась уже выше (стр. 488).
Механизм деполимеризации целлюлозы и крахмалов еще недостаточно выяс-
выяснен, хотя известно, что этой деполимеризации всегда предшествует увеличе-
увеличение содержания кислорода в целлюлозе в форме карбонильных или карбо-
карбоксильных групп [18, 81].
Натуральный латекс можно обработать растворами перекиси водорода
(взяв последнюю в количестве, приблизительно равном содержанию сухого
каучука в латексе), причем получаются латекс с клеящими свойствами и пла-
пластифицированный каучук [82]. Обработка перекисью водорода вызывает
такую же деполимеризацию каучука, как и обработка на вальцах (пласти-
(пластикация); продукты, полученные при обеих этих операциях, обладают почти
идентичными химическими и физическими свойствами. В обоих случаях,
возможно, действует один и тот же химический механизм, так как при обра-
Другие виды применения перекиси водорода как окислителя 497
ботке на вальцах каучук поглощает небольшие количества кислорода из воз-
воздуха, причем образуются перекиси, что, очевидно, и приводит к разрыву
углеводородных цепей.
Применение перекиси водорода для удаления волос, вероятно, основано
на ее деполимеризующем действии. По одному из предложенных ранее мето-
методов дубления шкуру или мех перед самым процессом дубления подвергают
окислению перекисью водорода [83].
Завивка волос (перманент)
«Холодная» завивка волос (перманент) основана на восстановлении их
действием раствора тиогликолята аммония, после чего волосы подвергают
завивке и последней сообщают стойкость путем обработки раствором бро-
мата, пероксобората натрия или перекиси водорода. Перекисные соединения
обладают тем недостатком, что при неправильном применении они могут
вызвать нежелательное обесцвечивание волос; броматы действуют удовлет-
удовлетворительно, но являются опасными, так как при приеме внутрь вызывают
поражение почек и даже смерть.
Применение в анализе
Еще в 1883 г. Классен и Бауэр указали на ценность перекиси водорода
в качестве аналитического реактива; в настоящее время она применяется
в самых различных методиках анализа, обычно для одной из следующих трех
целей:
1. Перекись водорода может образовать комплексное перекисное соеди-
соединение с металлическим ионом. Возникающая окраска может быть использо-
использована для идентификации или для колориметрического количественного ана-
анализа. В качестве примеров можно привести образование пероксосоединений
ванадия, титана, молибдена и церия или образование окрашенного соедине-
соединения из хромата в присутствии перекиси водорода и эфира [84].
2. Перекись водорода, обычно вместе с серной кислотой, может быть
использована для окисления и разложения органических веществ до анализа.
Этим путем можно быстро перевести в раствор такие вещества, как ткани,
волокна, мясо и т. д. Например, при определении азота по Кьельдалю добавка
перекиси водорода на предварительной стадии разложения способствует
получению прозрачных, как вода, растворов со значительным снижением за-
затраты времени на анализ в целом [85].
3. Перекись водорода можно использовать для окисления известных
видов молекул до состояния высшей валентности, легче определяемого ана-
аналитически, например для окисления сернистого цинка [86] или двуокиси
серы 187] до шестивалентного состояния серы. Окислительная способность
перекиси водорода может быть использована также для устранения окисле-
окислением каких-нибудь других мешающих примесей или для осаждения опре-
определенных примесей из раствора (см. стр. 492). Добавка перекиси водорода
может также повысить скорость растворения пробы металла при анализе
(см. стр. 496).
В большинстве этих случаев перекись водорода предпочтительнее других
окислителей, так как продукты ее реакции не мешают анализу. Методы при-
применения перекиси водорода в различных анализах описаны у Файгля [84],
Тредвелла и Голла [88] и Фурмана [89]. В табл. 86 приводится ряд специфи-
специфических видов применения перекиси водорода в анализе, причем большинство
этих методов описано сравнительно недавно.
32 Перекись водорода
498
Некоторые
виды
применения
Глава 11
перекиси водорода
Применение
в качестве
Таблица 86
аналитического реактива
Литература
и церия
Колориметрическое определение ванадия
Спектрофотометрическое определение церия в УФ-области
Спектрофотометрический анализ ванадия, ниобия, кобальта
в УФ-области
Определение следов титана в стали
Применение вместе с хромотроповой кислотой для открытия избытка
титанового реактива при качественном анализе на фосфат
Определение фтора в минералах, основанное на обесцвечивании желтой
окраски перекисного раствора титана небольшими количествами фторида
Определение кобальта путем окисления до трехвалентного состояния,
что вызывает выпадение его из раствора . ,
Определение урана окислением до нерастворимой перекиси ,
Определение ртути
Определение ртути в микрограммовых количествах
Определение ниобия
Определение осмия
Определение олова в медных сплавах
Окисление железа при колориметрическом определении железа при по-
помощи роданида
В качестве окислителя при определении молибдена в молибденотитано-
вых сплавах
Опрелеление нитрата в присутствии нитрита, основанное на окислении
азотистой кислоты до азотной • .
Определение йодида в присутствии бромида и хлорида, а также броми-
бромида в присутствии хлорида, основанное на неодинаковой способности
к окислению различных ионов галогенидов в кислом растворе перекиси
водорода
Опрелеление серы в виде сернистого цинка путем окисления до сульфата
Опрелеление двуокиси серы путем окисления до сульфата
Опрелеление чистых многосернистых щелочных металлов путем окисле-
окисления ло сульфатов
Устранение сульфита при определении хлоридов по Мору
Непрерывное определение концентрации двуокиси серы в воздухе при
помощи автометра Томаса (пробу воздуха непрерывно протягивают че-
через разбавленный раствор перекиси водорода; в качестве мерила кон-
концентрации двуокиси серы служит рост электропроводности, обуслов-
обусловленный образованием серной кислоты)
Окисление соединений галогенов, серы и фосфора для анализа . . . .
Определение метионина путем окисления до соответствующего суль-
фоксида
Опрелеление закиси азота в воздухе
Использование при определении составных частей сополиамидов . . . .
Опрелеление ацетальдегида
Качественная проба на платифиллин
Определение каталазы по каталитическому действию ее на перекись во-
водорода
Определение содержания пероксидазы в бланшированной брюссельской
капусте в качестве показателя достаточности бланшировки
Разложение до анализа на азот по Кьельдалю .
Разложение до определения фосфора
Разложение до определения меди в фруктах и овощах
90, 91
92
93
94
95
96
97
98
99
100
101
102
103
104, 105
106
107
108
86
87
109
ПО
111
112
113
114
115
116
117
118
119
85
120, 121
122
ПЕРЕКИСЬ ВОДОРОДА КАК ИСТОЧНИК ЭНЕРГИИ
В большинстве приспособлений, вырабатывающих энергию за счет горе-
горения, используется метод сжигания топлива в воздухе. Однако существуют
два обстоятельства, когда может оказаться желательным или необходимым
применение не воздуха, а иного окислителя: 1) при необходимости генериро-
Перекись водорода как источник энергии 499
вания энергии в таком месте, где снабжение воздухом ограничено, например
под водой или высоко над поверхностью земли; 2) когда желательно получить
в течение короткого времени очень большое количество энергии из компакт-
компактных ее источников, например в_орудийных метательных ВВ, в установках
для взлета самолетов (ускорителях) или" в ракетах. В некоторых таких
случаях*"! принципе можно использовать воздух, предварительно сжатый
и хранящийся в соответствующих сосудах под давлением; однако такой метод
часто является непрактичным, так как вес баллонов (или хранилищ других
видов) составляет около 4 кг на 1 кг воздуха; вес же тары для жидкого или
твердого продукта равен 1 кг/кг или даже меньше.
В том случае, когда применяется небольшое приспособление и основное
внимание уделяется простоте конструкции, например в патронах огнестрель-
огнестрельного оружия или в небольшой ракете, используется твердое топливо, содер-
содержащее тесно смешанные между собой горючее и окислитель. Системы жидкого
топлива сложнее, но обладают двумя определенными преимуществами по
сравнению с системами твердого топлива:
1. Жидкость можно хранить в сосуде из легкого материала и нагнетать
в камеру сгорания, размеры которой должны удовлетворять только требо-
требованию обеспечения желательной скорости сгорания (техника вдувания твер-
твердого вещества в камеру сгорания под высоким давлением, вообще говоря, не-
неудовлетворительна; следовательно, вся загрузка твердого топлива с самого
начала должна находиться в камере сгорания, которая поэтому Должна быть
большой и прочной).
2. Скорость генерирования энергии можно изменять и регулировать
путем соответствующего изменения скорости подачи жидкости. По этой при-
причине комбинации жидких окислителей и горючих находят применение для
различных сравнительно крупных ракетных двигателей, для двигателей под-
подводных лодок, торпед и т. п.
Идеальный жидкий окислитель должен обладать многими желательными
свойствами, но наиболее важны с практической точки зрения следующие три:
1) выделение значительного количества энергии при реакции, 2) сравнитель-
сравнительная устойчивость" к удару и повышенным температурам и 3) низкая производ-
производственная себестоимость. Вместе с тем желательно, чтобы окислитель не обла-
обладал коррозионными или токсическими свойствами, чтобы он быстро реагировал
и обладал надлежащими физическими свойствами, например низкой точкой
замерзания, высокой точкой кипения, большой плотностью, малой вязко-
вязкостью и т. д. При применении в качестве составной части ракетного топлива
особое значение имеет и достигаемая температура пламени и средний моле-
молекулярный вес продуктов сгорания. Очевидно, что ни одно химическое соеди-
соединение не может удовлетворять всем требованиям, предъявляемым к идеаль-
идеальному окислителю. И очень мало веществ, которые вообще хотя бы приблизи-
приблизительно обладают желательной комбинацией свойств, причем только три из
них нашли некоторое применение: жидкий кислород, концентрированная
азотная кислота и концентрированная перекись водорода.
Перекись водорода обладает тем недостатком, что даже при 100%-ной
концентрации она содержит лишь 47 вес.% кислорода, который может быть
использован для сжигания топлива, тогда как в азотной кислоте содержание
активного кислорода составляет 63,5%, а для чистого кислорода возможно
даже 100%-ное использование. Этот недостаток компенсируется значитель-
значительным выделением тепла при разложении перекиси водорода на воду и кисло-
кислород. Фактически мощности этих трех окислителей или силы тяги, развивае-
развиваемые единицей веса их, в любой определенной системе и при любом виде горю-
горючего могут различаться максимум на 10—20%, а поэтому выбор того или иного
окислителя для двухкомпонентной системы обычно определяется другими,
соображениями.
32**
500 Глава* 1J .
Экспериментальное исследование применения перекиси водорода в каче-
качестве источника энергии было впервые поставлено в Германии в 1934 г. при
поисках новых видов энергии (независимых от воздуха) для движения под-
подводных лодок, Это потенциальное военное применение стимулировало про-
промышленное развитие метода фирмы «Electrochernische Werke» в Мюнхене
(Е. W. М.) по концентрированию перекиси водорода с получением водных
растворов высокой крепости, которые можно было бы транспортировать и хра-
хранить с приемлемой низкой скоростью разложения. Сначала для военных нужд
выпускали 60%-ный водный раствор, но впоследствии эту концентрацию по-
повысили и наконец начали получать 85%-ную перекись. Увеличение доступ-
доступности высококонцентрированной перекиси водорода в конце тридцатых годов
текущего столетия привело к применению ее в Германии во время второй
мировой войны в качестве источника энергии для других военных нужд.
Так, перекись водорода впервые была использована в 1937 г. в Германии
в качестве вспомогательного средства в топливе для двигателей самолетов
и ракет.
Высококонцентрированные растворы, содержащие до 90% перекиси во-
водорода, производились также в промышленном масштабе к концу второй
мировой войны фирмами «Buffalo Electro-Chemical Co» в США и «В. Laporte,
Ltd.» в Великобритании. Воплощение идеи процесса генерирования тяговой
мощности из перекиси водорода в более ранний период представлено в схеме
Лишолма [1.231, предложившего методику генерирования энергии путем тер-
термического разложения перекиси водорода с последующим сжиганием горю-
горючего в полученном кислороде. Однако на практике эта схема, по-видимому,
не нашла применения.
Концентрированную перекись водорода можно использовать и в каче-
качестве однокомпонентного топлива (в этом случае она подвергается разложению
под давлением и образует газообразную смесь кислорода и перегретого пара)
и в качестве окислителя для сжигания горючего. Механически однокомпо-
нентная система проще, но она дает меньше энергии на единицу веса топлива.
В двухкомпойедтной системе можно сначала разложить перекись водорода,
а затем сжечь горючее в горячих продуктах рааложения или же ввести в реак-
реакцию обе жидкости непосредственно без предварительного разложения пере-
перекиси водорода. Второй метод проще механически оформить, но при нем может
оказаться затруднительным обеспечение воспламенения, а также равномер-
равномерное и полное сгорание. В любом случае энергия или тяга создается за счет
расширения горячих газов. Различные видь? ракетных двигателей, основан-
основанных .на действии перекиси водорода и использованных в Германии во время
второй мировой войны, весьма подробно описаны Вальтером [124], который
был непосредственно связан с разработкой многих видов военного применения
перекиси водорода в Германии. Опубликованный им материал иллюстрируется
также рядом чертежей и фотоснимков.
В Германии до второй мировой войны и во время ее была проведена зна-
значительная работа, по изысканию горючих, обладающих способностью само-
самовоспламеняться в контакте с концентрированной перекисью водорода или
азотной кислотой («гиперголи»). Обычным критерием пригодности является
необходимый минимум времени, протекающего от начала контакта обеих
жидкостей до появления пламени (запаздывание воспламенения). Поскольку
результаты сильно зависят от техники смешения, различные исследователи
пришли к сильно расходящимся выводам для одной и той же комби-
комбинации горючего и окислителя. Наличие небольших количеств солей некото-
некоторых металлов в горючем может также оказать большое влияние на снижение
запаздывания воспламенения или сообщение смеси способности самовоспла-
самовоспламеняться вообще. В некоторых таких случаях активность соли металла за-
заключается, по-видимому, в первую очередь в том, что она способствует ката-
Перекись водорода как источник энергии
501
800
§ 700
I
литическому разложению перекиси водорода и, как следствие, повышению
температуры системы до точки воспламенения горючего; чаще роль катали-
катализатора более сложна. Так, подходящие катализаторы, вероятно, образуют
комплексы с горючим, но такие комплексы не должны обладать ни слишком
большой, ни слишком малой прочностью. Горючие, обладающие свойствами
гиперголей в сочетании с концентрированной перекисью водорода, принадле-
принадлежат к одной из следующих групп химических структур.
1. Неорганические амины (например, гидразин), особенно в присутствии
растворенных солей меди или железа, например куприцианида калия или
нитропруссида натрия, а также некоторые органические амины (например,
толуидин и различные диамины).
2. Соединения с непредельными свя-
связями (не считая связей в ароматических
кольцах), например с несколькими сопря-
сопряженными двойными связами, с непредель-
непредельной связью в сочетании с кетогруппой или
оксигруппой или с непредельным пяти-
членным кольцом (такие, как фуран, пир-
пиррол и циклопентадиен).
3. Альдегиды (например, кротоновый).
4. Высшие алифатические спирты и
соединения, содержащие гидроксильные
группы (например, пирокатехин).
В каждом из таких случаев действие
может быть заметно интенсифицировано
путем растворения в горючем соли железа,
меди, никеля, ванадия или кобальта.
Имеется значительная подробная ин-
информация по проведенным в Германии
работам [125]. Бротч [126] опубликовал
описание прибора для измерения запаз-
запаздывания воспламенения, где указываются
также результаты, полученные при приме-
I
| 600
I
§500
I
300
|
§200
Г
I wo
/
/
36,2 кг
/см2
1
36.2кг/см2 II
s^r 1 7
/jjfi кг/см2 1
1атм
i
/
/
1
Латм
0,2 0,4 0,6 0,8
Доля разложенной О
Рис. 81. Влияние давления на
температуру адиабатического раз-
разложения водного раствора переки-
нении 80%-ной перекиси водорода в каче- си водорода с начальной концентра-
стве окислителя. В двигателях, работаю- цией 90 вес-%-
щих на невоспламеняющемся горючем,
воспламенение можно обеспечить в течение первых секунд работы путем
введения гиперголя. Если он тяжелее основного горючего и не смешивается
с ним, его можно просто загрузить в бак для основного горючего, где оно
образует нижний слой и первым попадает в зону сгорания. Эта техника
была использована в некоторых двигателях в Германии, а недавно на нее
был выдан патент в США [127].
Теплота разложения водных растворов перекиси водорода в • адиабатиче-
адиабатических условиях обеспечивает испарение всей воды, как присутствующей с са-
самого начала, так и образующейся при разложении, если только начальная
концентрация перекиси водорода превышает приблизительно 68 вес.%.
В условиях, когда испаряется вся жидкость, конечная достигаемая темпера-
температура практически не зависит от давления. Вычислены температуры адиабати-
адиабатического разложения для различных условий [128]. На рис. 81 показаны тем-
температуры, достигаемые при различных степенях разложения концентриро-.
ванных растворов перекиси водорода, начиная с комнатной температуры
при абсолютном давлении 36,2 кг/см2. Развиваемая энергия зависит от при-
применяемой механической системы, начального и конечного давления и кон-
концентрации перекиси. Например, полное разложение 85%-ной перекиси водо-
водорода при абсолютном давлении 36,2 кг/см2 в динамическом стационарном
502 Глава И
процессе с последующим расширением газообразных продуктов через обра-
обратимый (эффективный на 100%) детандер до атмосферного давления дает не-
несколько более 0,22 л. с/час на каждый килограмм исходного раствора. При сжи-
сжигании вместе с горючим, например со спиртом или углеводородом, можно
до расширения получить вдвое больше энергии в расчете на килограмм горю-
горючего вместе с раствором перекиси водорода.
Техника разложения
Генерирование энергии путем разложения концентрированной перекиси
водорода (с последующей реакцией с горючим или без такой реакции) требует
применения техники, обеспечивающей быструю и полную реакцию в мини-
минимальном объеме. В принципе такого рода разложение может быть осуществлено
чисто термическим методом, как это предложено Лишолмом [123], но проблемы
надежного пуска и работы привели к тому, что на практике всегда приме-
применяется катализатор, если не считать тех случаев, когда используется прямая
реакция с самовоспламеняющимся горючим. Ряд соединений может выполнять
роль очень активных катализаторов разложения (особенно окислы различ-
различных тяжелых металлов). По одному из простых методов вместе с раствором
перекиси водорода в камере разложения непрерывно распыляется водный
раствор перманганата. Ион перманганата немедленно восстанавливается
с образованием тонкодисперсных частиц твердых окислов марганца, которые
фактически и представляют катализатор. Путем достаточно тонкого распы-
распыления можно добиться разложения до 1 кг концентрированной перекиси во-
водорода в секунду на 1 л объема реактора [124]. Эта техника использована
в Германии для запуска с катапульты и для генерирования пара с целью при-
привода насосов ракеты V-2. Растворимость марганцовокислых солей в воде воз-
возрастает в следующем порядке: соли калия, натрия, кальция; поэтому приме-
применялись концентрированные растворы перманганата натрия или перманганата
кальция. Перманганат кальция особенно пригоден в зимнее время ввиду низ-
низкой точки замерзания его высококонцентрированных растворов.
Однако недостаток применения растворов перманганата для разложе-
. ния перекиси заключается не только в усложнении конструкции вследствие
необходимости подачи дополнительной жидкости, но и в том, что вместе с па-
ро-кислородной смесью непрерывно выносится поток твердого вещества, ока-
оказывающего очень вредное влияние, если смесь должна быть пропущена через
турбину. В связи с этим в Германии разработаны многочисленные твердые
катализаторы, оформленные в виде слоя, на который непрерывно разбрызги-
разбрызгивается перекись водорода. В литературе приводятся данные по некоторым
отдельным рецептам, которые применялись в последних работах [129]. В дви-
двигателе, работавшем на перекиси водорода, на немецкой подводной лодке при-
применялся катализатор, состоявший из пиролюзита (природной двуокиси мар-
марганца) в смеси с цементом и песком; эту смесь формовали в виде цилиндриков.
Другой катализатор представлял высокопористые фарфоровые камни, кото-
которые пропитывали раствором перманганата кальция, высушивали, вновь про-
пропитывали смесью перманганата кальция и хромата калия и опять высушивали.
Последняя обработка имела целью обеспечить активность поверхности в те-
течение продолжительного времени. В обоих случаях размер зерен составлял
1—1,5 см. Сообщается [124], что в случае работы при 25 am 1 кг катализатора
того или другого типа обладает способностью разлагать перекись водорода
в количестве 150—200 г/сек и что продолжительность его службы соответ-
соответствовала разложению 2000 кг перекиси водорода на 1 кг катализатора.
Наличие небольших примесей в перекиси водорода (попавших в нее в про-
процессе производства или от преднамеренно добавленных стабилизаторов) посте-
постепенно снижает активность катализатора. Снижение активности в большой
Перекись водорода как источник энергии 503
степени зависит от природы катализатора, а также от количества и природы
примесей, содержащихся в перекиси водорода. Поэтому при таком примене-
применении перекиси водорода особенно важно, чтобы она была возможно более чистой
и содержала стабилизаторы в минимально допустимой концентрации.
Очевидно, что процессы, происходящие в камере разложения, исключи
тельно сложны, так как они начинаются реакцией в жидкой фазе на входе
(при комнатной температуре и в концентрированной перекиси водорода),
после чего следует разложение смеси газовой и жидкой фаз и, наконец, на
выходе происходит разложение в паровой фазе (при высокой температуре
и исчезающе малой концентрации перекиси водорода). Лафт [130] дал теоре-
теоретический расчет такой камеры разложения, сделав ряд упрощающих пред-
предпосылок, но практическое конструирование, очевидно, должно в значитель-
значительной мере носить эмпирический характер.
Лафт описал также ряд практических задач, которые встречаются при
конструировании и эксплуатации камеры разложения. Хотя такая камера
работает под значительным давлением, в начале работы давление равняется
атмосферному, а в этих условиях требуется значительно больший слой ката-
катализатора, чем при высоких давлениях. Далее, поскольку слой катализатора
сначала бывает холодным, от него на входе в камеру и при инициировании
реакции требуется особенно высокая активность. По этой причине прихо-
приходится прибегать к таким специальным приемам, как нанесение слоя особо
активного вещества на частицы обычного катализатора; этот слой, возможно,
содержит вещество, которое растворяется в начальном потоке перекиси водо-
водорода и вызывает быструю гомогенную реакцию. Конечно, такой метод можно
рекомендовать на практике только для системы однократного действия. На-
Начальная зона катализатора подвергается высоким термическим и механиче-
механическим напряжениям. Кроме того, в этой зоне активность катализатора падает
особенно легко вследствие реакции с примесями или стабилизаторами пере-
перекиси водорода или в результате покрытия пленкой за счет испарения. Для
предотвращения этой постепенной инактивации катализатор предложено
размещать таким образом, чтобы происходило его медленное механическое
разрушение с непрерывным обнажением свежих поверхностей. Однако это
возможно только тогда, когда в газовом потоке допустимо наличие уносимого
материала.
Из других трудностей, с которыми приходится встречаться, нужно ука-
указать на колебания или пульсации потока. Последние могут происходить в том
случае, если часть слоя содержит катализатор с низкой или умеренной актив-
активностью, например после длительной эксплуатации, когда часть катализатора,
находящаяся выше по течению, уже потеряла в значительной степени свою
активность. Мгновенное падение давления способствует тому, что в систему
поступает увеличенное количество перекиси водорода, причем часть катали-
катализатора «заливается». Эта перекись после очень короткого, но конечного про-
промежутка времени сразу разлагается, в связи с чем резко возрастает давление
и снижается поступление перекиси. Падение давления после выхода образо-
образовавшегося газа из камеры вызывает вновь увеличенное поступление пере-
перекиси водорода, и цикл повторяется. Этого явления не происходит, если весь
катализатор обладает очень высокой активностью; и наоборот, указанная пуль-
пульсация проявляется тем сильнее, чем ниже активность катализатора, так как
в этом случае возможно накопление перекиси водорода до наступления резкого
разложения. В качестве корректирующей меры, помимо применения более
активного катализатора, рекомендуется увеличивать перепад давления через
сопла, по которым перекись водорода поступает в камеру, или же пользо-
пользоваться другими приемами, которые делают поступление перекиси водорода
независимым от давления в камере. Характеристика разложения перекиси
водорода в жидкой и паровой фазах дана в гл. 8 (стр. 373, 390 и ел.).
504
Глава 11
Ниже описаны различные виды военного применения перекиси водорода
в том объеме, в котором они освещены в литературе*.
Применение в двигателях для подводных лодок
До второй мировой войны подводные лодки обычно приводились двига-
двигателями Дизеля при надводном плавании и электрическими моторами, дей-
действовавшими от аккумуляторов,—при подводном. Имеющаяся емкость и ско-
скорость разрядки аккумуляторных батарей резко снижали тяговую мощность
подводных лодок в подводном положении. Для подводной лодки особенно важ-
важно обладать большой скоростью подводного хода, так как это позволяет ей
Перекись
водорода
Дизельное
топливо
Выхлоп
Выхлоп У
Рис. 82. Схема двигателя германской подводной лодки типа 26 (по Мак-Ки
[131]).
/—насос; 2—приводной вал; 3—камера сгорания; 1—водяной холодильник; 5—змеевик
с циркулирующей морской водой; б—винт; 7—паровая турбина; «—конденсатор.
быстро уйти от преследования после атаки, между тем общее количество полу-
получаемой энергии при быстрой разрядке аккумуляторов значительно меньше,
чем при более медленной разрядке. Для обычной подводной лодки периода
начала второй мировой войны можно привести в качестве типичных следую-
следующие показатели (для германской подводной лодки 7С): надводная скорость
17 узлов, скорость подводного хода 7,5 узла [131] (однако 7,5 узла можно
было поддерживать лишь в течение очень небольшого времени). Во время
войны в Германии была построена подводная лодка типа 21, которая могла
развивать надводную скорость 15,5 узла и подводную 16,5 узла в течение
одного часа; это достигалось путем применения аккумуляторов большой
емкости и с очень тонкими решетками.
Применение перекиси водорода для двигателей подводных лодок было
разработано на верфях «Walter Werke» в Киле, где до войны была построена
экспериментальная подводная лодка водоизмещением 80 т, имевшая скорость
подводного хода 25—26 узлов. В дальнейшем во время войны были построены
4 учебные лодки и 5 боевых типа 17В. Эти лодки имели водоизмещение 380 т,
надводную скорость 8,5 узла и подводную 25 узлов. Однако фактически они
не были ни разу использованы ввиду дефицита перекиси водорода. К концу
войны в стадии строительства находилась подводная лодка типа 26 водоизме-
водоизмещением 900 т, которая должна была развивать скорость 11 узлов в надводном
* В ряде описаний работ, проведенных в Германии, в частности в переводах не-
немецких документов и другой литературе, помимо печатаемой в обычных технических жур-
журналах, перекись водорода обозначается под различными немецкими военными шифрован-
шифрованными названиями: тимиоль, Т-штоф (в виде различных марок TN, TSS или TS), реналь,.
инголин, аураль, нейтралин, DiR, D2R или субсидоль.
Перекись водорода как источник энергии 505
положении и 24 в подводном (с цистернами приблизительно на 90 т перекиси
водорода); этого количества должно было быть достаточно для поддержива-
поддерживания максимальной скорости подводного хода в течение примерно 6 час. Дви-
Двигатель, работавший на перекиси водорода, имел мощность 7500 л. с.
Силовая установка схематически показана на рис. 82. Концентрирован-
Концентрированную перекись водорода накачивают в камеру, заполненную катализатором,
где она и разлагается. Катализатор состоит из пористых гранул, пропитан-
пропитанных перманганатом и затем высушенных. Для этой цели применяют предпо-
предпочтительно перманганат натрия или кальция, поскольку они хорошо раство-
растворимы в воде. Для снижения температуры рабочих газов приблизительно до
550° (максимально допустимой температуры на входе в турбину) вместе с горю-
горючим (дизельное топливо или декалин) в камеру сгорания впрыскивается также '
вода. Отработанные газы из турбины охлаждаются, часть конденсата возвра-
возвращается в камеру сгорания, остаток же вместе с неконденсированными газами
откачивается за борт. Перекись водорода хранится в сжимаемых полихлор-
полихлорвиниловых мешках вне прочного корпуса. Недостатком подводной лодки
с двигателем, работающим на перекиси водорода, является большой расход
дорогостоящей перекиси. Кроме того, для достижения высоких показателей
подводной лодки в подводном положении необходимо было пожертвовать
некоторыми другими боевыми качествами (дальностью действия, внутренней
кубатурой и т. д.). Дополнительные подробности по немецким подводным
лодкам с двигателями, работающими на перекиси водорода, опубликованы
Мак-Ки [131].
В справочнике «Jane's Fighting Ships» за 1950—1951 гг. [132] сообщается,
что военно-морской флот США запроектировал строительство подводной лод-
лодки водоизмещением 2200 т (с двигателем, работающим на перекиси водорода)
стоимостью 37 млн. долларов, но никаких дальнейших сведений о том, была ли
действительно построена эта подводная лодка, не опубликовано. В 1954 г.
в Великобритании сообщено о спуске на воду подводной лодки длиной 68 м
под названием «Эксплорер», в которой в качестве источника энергии исполь-
используется перекись водорода. Вторая такая подводная лодка спущена в 1955 г.
Применение как источника энергии для торпед
Торпеда представляет собой в сущности миниатюрную автоматически
действующую подводную лодку. В начале второй мировой войны большин-
большинство боевых торпед, применявшихся различными воюющими державами,
приводилось в действие либо от электрических аккумуляторов и моторов,
либо путем сжигания горючего (например, спирта, керосина или декалина)
в воздухе, поступавшем из резервуаров со сжатым воздухом. Горячие газы
сгорания поступали в поршневой двигатель или турбину, которая приводила
гребные винты. Замена воздуха кислородом или перекисью водорода давала
возможность при неизменных технических показателях снизить вес или по-
повысить скорость, радиус действия или размеры боевого зарядного отделения.
В японской торпеде типа 93 применяли жидкий кислород, но это было связано
с различными трудностями в отношении хранения, ухода и взрывоопасности.
В Германии были сконструированы различные торпеды, в которых в ка-
качестве окислителя применялась 80%-ная перекись водорода и в качестве горю-
горючего—декалин. Горение инициировалось впрыскиванием в камеру сгорания
в течение первых нескольких секунд 80%-ного водного раствора гидразин-
гидрата, содержащего в качестве катализатора растворенный куприцианид
калия или другие соединения меди. В присутствии концентрированной пере-
перекиси водорода это горючее самопроизвольно воспламеняется. Дополнительное
преимущество торпед, приводимых в движение при помощи перекиси водо-
водорода, по сравнению с торпедами, приводимыми воздухом, состоит в том, чт
506 Глава 11
в первом случае газы сгорания легкорастворимы в воде и поэтому торпеда
движется, фактически не оставляя следа. В отчете Колмэна и Килпатрика [129]
приводятся многие подробности, касающиеся двух различных немецких тор-
торпед, приводимых при помощи перекиси. Мэксфилд [133] также опубликовал
данные, касающиеся механической конструкции и показателей двух различ-
различных немецких торпед, в которых применялась перекись водорода, и сравнил
их с данными для других торпед. Сообщается, что во время второй мировой
войны в Германии было произведено около тысячи торпед различных типов
с перекисью водорода как источником энергии.
Перспективы использования перекиси водорода в военном деле были
учтены военно-морским флотом США еще до 1933 г., причем до вступления
США в войну на морской торпедной станции в Ньюпорте производились
строго засекреченные работы по торпедам, в которых в качестве окислителя
должна была применяться перекись водорода. В двигателе 50%-ный раствор
перекиси водорода разлагается под давлением водным раствором перманганата
или другого окислителя, и продукты разложения используются для поддер-
поддержания горения спирта. Продукты реакции затем пропускаются через тур-
турбину. Двигатель был значительно улучшен во время войны, но по различным
причинам торпеды, приводимые при помощи перекиси водорода, до оконча-
окончания военных действий не нашли боевого применения во флоте США.
Применение в качестве компонента ракетных топлив
Сравнительно высокая стоимость концентрированной перекиси водорода
по сравнению с жидким кислородом или азотной кислотой ограничивает при-
применение ее (в качестве окислителя) для ракет, особенно крупных. Наиболь-
Наибольшее применение она нашла в ракетном двигателе самолета-перехватчика Ме163,
сконструированном в Германии во время второй мировой войны; это был пер-
первый управляемый самолет с ракетным двигателем.
На первых стадиях конструирования самолета Mel63 применялся про-
простой двигатель, в котором использовалось разложение одной концентриро-
концентрированной перекиси водорода. Однако удельный импульс такой системы оказался
очень низким, и она была заменена двухкомпонентной установкой с исполь-
использованием горючего, состоящего из смеси 30% гидразингидрата с растворенным
катализатором (куприцианидом калия), 57% метилового спирта и 13% воды.
В присутствии перекиси водорода это горючее самовоспламеняется. Метиловый
спирт вводили в горючую смесь для снижения расхода гидразингидрата (чтобы
запаса гидразингидрата хватило на длительное время), а воду добавляли для
снижения температуры сгорания. Самолет с полным запасом топлива весил
4100 /сг, и ракетные двигатели развивали тягу 1500 /сг, что давало возмож-
возможность самолету подняться на высоту 9 км примерно за 2 мин. Хотя около 49%
всего взлетного веса самолета приходилось на топливо, радиус его действия
был весьма ограничен и он обладал продолжительностью полета максимум
10—20 мин. При максимальном расходе запаса горючего хватало только на
4 мин. Дальнейшие подробности, включая и технические характеристики
самолета Ме163, описываются Вальтером [124] и Цукровом [134], который
сообщает также данные, характеризующие различные системы жидкого топ-
топлива для ракетных двигателей.
Перекись водорода применяли также в сочетании с горючим, содержав-
содержавшим гидразингидрат, в некоторых немецких ускорителях, которые сообщали
дополнительную тягу во время взлета самолета, а затем сбрасывались. В ка-
качестве такого типичного ускорителя можно указать на HWK R1-209, который,
применялся в сочетании с бомбардировщиками Хейнкель 111 и Юнкере 88.
Сам двигатель весил 227 /сг; он брал 221 кг раствора перекиси водорода, 28 кг
горючего и развивал тягу 1500 кг в продолжение 30 сек. Однако применялись
Перекись водорода как источник энергии 507
и ускорители, работа которых была основана на разложении одной перекиси
водорода с использованием водного раствора перманганата натрия или пер-
манганата кальция в качестве катализатора, особенно для небольших самоле-
самолетов, где очень простая конструкция установок вполне перекрывала понижен-
пониженный удельный импульс, который они давали. 1\ак пример можно привести
ускоритель Фокке-Вульф АТО FW-56, развивавший тягу 300 кг в течение
30 сек., и ускорители Хейнкель Не112 и Не12б, развивавшие соответственно
максимальную тягу 1000 и 500 кг. Другие подробности можно найти у Валь-
Вальтера [124].
В 1952 г. фирма «De Havilland Engine Co» в Англии сообщила о выпуске
«холодной», или одно компонентной, ракеты под названием «Спрайт» для общего
применения в качестве ускорителя при взлетах реактивных самолетов, как
военных, так и гражданских. Для взлета требуются две такие установки.
Каждая из них содержит 177 л 80%-ной перекиси водорода, 11,3 л раствора
перманганатного катализатора, весит 420 кг и развивает тягу 2270 кг в тече-
течение 9 сек. Опубликованы полные конструктивные данные по этим ускорите-
ускорителям [135]. В 1953 г. сообщено о выпуске «Супер-спрайта» с применением
перекиси водорода в сочетании с твердым катализатором и керосином или
бензином в качестве горючего. Такая установка весит без топлива 275 кг
и развивает тягу 1820 кг примерно в продолжение 40 сек. Ускорители
с применением порохового заряда проще по конструкции, чем ускорители,
работающие на жидком топливе, и часто применяются при взлетах самолетов.
Ракетное топливо находит применение и для вертолетов. В 1954 г. появи-
появилось сообщение, что для армии США выпущен легкий вертолет на одного
человека с расположенными в роторах ракетными двигателями, работающими
на перекиси водорода.
Коллинс [136] провел детальное исследование работы небольшого ракет-
ракетного двигателя, развивающего тягу около 4,5 кг и работающего на 90%-ной
перекиси водорода с одним из следующих двух видов самовоспламеняющегося
горючего: 1) 85%-ным водным раствором гидразингидрата, содержащим
в качестве катализатора соли железа и меди, и 2) метиловым спиртом, содер-
содержащим тригидрат (З-нафталинсульфоната закисного железа в количестве
200 г/л. Показано, что по характеристике такой небольшой ракетный двига-
двигатель очень близок к большому двигателю при приблизительно одинаковом
времени контакта реагентов. Экспериментальные результаты сравнены с теоре-
теоретическими расчетами, причем при таких сравнениях особое внимание уделено
вопросу учета тепловых потерь. Приводится также много данных по работе
инжекторов различных типов.
Весьма популярная книга, в которой рассматривается развитие ракетных
двигателей, написана Вилли Леем [137]. Более детальный технический анализ
ракетных реактивных двигателей можно найти в книгах Цукрова [138] или
Саттона [139]. Основы ракетной техники, а также конструкция и работа
жидкостных ракетных двигателей даны также Зейфертом, Милсом и Саммер-
филдом [140] и Диплоком, Лофтсом и Гримстоном [141].
Применение в качестве вспомогательного источника энергии
При непрерывном полном разложении перекиси водорода получается
смесь кислорода с перегретым паром, имеющая постоянную температуру, опре-
определяемую концентрацией использованной перекиси водорода, и поэтому пере-
перекись водорода может быть простым, надежным и компактным вспомогатель-
вспомогательным источником энергии. В качестве такого источника перекись водорода
нашла применение в германской ракете V-2 для привода топливных насосов,
которые нагнетали основные компоненты топлива (жидкий кислород и смесь
75% этилового спирта и 25% воды) в камеру сгорания. В этой ракете поток
508- Глава 11
перекиси водорода разлагался в специальной камере путем непрерывного
впрыскивания в нее водного раствора перманганата натрия или перманганата
кальция, хотя механическая система с пропусканием перекиси водорода через
слой активного катализатора была бы проще. Особое преимущество перекиси
водорода при таком применении состоит в том, что температура газов разло-
разложения достаточно низка, а поэтому их не надо разбавлять перед подачей
в турбину.
Применение для запуска при помощи катапульты
Германский самолет-снаряд V-1 (представлявший собою миниатюрный
робот-самолет, который приводился в действие пульсирующим воздушно-
реактивным двигателем) запускался с земли при помощи катапульты поршне-
поршневого типа. Источником энергии для катапульты служило разложение переки-
перекиси водорода раствором перманганата натрия или перманганата кальция. Для
каждого такого запуска требовалось около 100 кг 80%-ного раствора перекиси
водорода и 5 кг раствора перманганата кальция.
Обе жидкости впрыскивались в казенную часть катапульты из нагнета-
нагнетателей при помощи сжатого воздуха из баллонов; разложение перекиси водорода
способствовало развитию внутреннего давления 56—70 am. При этом дости-
достигалась пусковая скорость 400 км/час при длине трубы катапульты 45 лс, что
соответствует среднему ускорению около 15 g. Необходимо было, чтобы реакция
практически заканчивалась в течение пускового периода 0,75 сек. Было скон-
сконструировано остроумное механическое приспособление для соединения само-
самолета-снаряда над трубой с поршнем внутри нее.
Беллинджер, Фридман, Бауэр, Истс, Лэдд и Росс [1421 описали характер-
характерные особенности германской катапульты и сообщили также о результатах ис-
исследования разложения перекиси водорода перманганатами в тяговых двига-
двигателях. Эти опыты были проведены в предварительном исследовании, постав-
поставленном химической службой США, по вопросу о применении жидких топлив
для катапультного запуска американского аналога самолета-снаряда V-1.
При военном применении жидких топлив в камерах разложения могут
возникать весьма высокие давления. Барр и Вилсон [143] опубликовали резуль-
результаты вычислений термодинамических свойств продуктов реакции 100%-ной
перекиси водорода и 100%-ного гидразина при давлениях 700—13 000 am.'
Приводятся конечные температуры равновесия, плотности и составы газов для
реакции при постоянном давлении и при постоянном объеме.
§
Перекись водорода в качестве взрывчатого вещества
Как указано на стр. 155, некоторые смеси или растворы органических
веществ в концентрированной перекиси водорода образуют весьма чувствитель-
чувствительные и мощные взрывчатые вещества. Однако для возможности технического
применения необходимо, чтобы взрывчатое вещество обладало очень низкой
чувствительностью. Некоторые смеси глицерина, перекиси водорода и воды
не чувствительны к механическому импульсу (например, от винтовочного
выстрела), но способны детонировать от взрыва капсюля-детонатора [144].
Каждая из эти* двух жидкостей в отдельности не имеет взрывчатых свойств;
это открывает возможность безопасного транспортирования и хранения, так
как взрывчатую смесь можно готовить только на месте использования и неза-
незадолго до применения. Тем не менее никаких сообщений о фактическом исполь-
использовании таких смесей в технике не имеется. Было предложено использовать
реакцию перекиси водорода с некоторыми веществами (например, с порошко-
порошкообразными алюминием и углем) как основу для изготовления взрывателей
замедленного действия [145], хотя эти реакции и трудно, по-видимому,
регулировать.
Применение газа% образующегося при разложении Н2О2 509
ПРИМЕНЕНИЕ ГАЗА, ОБРАЗУЮЩЕГОСЯ ПРИ РАЗЛОЖЕНИИ ПЕРЕКИСИ ВОДОРОДА
Выделение газа при разложении перекиси водорода или некоторых ее
реакциях является основой использования ее в производстве пористых изде-
изделий. Она находит, например, применение при изготовлении пористых бетонных
строительных блоков, в производстве пенистой резины; предложено также
применять перекись в качестве средства для поднятия теста. Для многих из
этих видов применения перекись водорода обладает тем преимуществом перед
другими химическими веществами, что она может выделять газ без нагревания
и образующийся остаток, а именно вода, совершенно безвреден. Перекись
водорода может служить также удобным источником чистого кислорода. Газ,
образующийся при разложении, можно использовать так же, как флотацион-
флотационный реагент, например для регенерации бумажной массы из отходов фабрик.
Применение в производстве пористого бетона и пористой керамики
Во многих случаях строительства для снижения общего веса здания
или повышения теплоизоляционных свойств желательно применять строитель-
строительные блоки или другие стройматериалы низкого удельного веса. В США в бетон-
бетонную смесь для этой цели часто вводят вспученный перлит или другой легкий
заполнитель. В Германии как до второй мировой войны, так и во время нее
проводилась значительная экспериментальная работа по применению перекиси
водорода для получения пористых изделий из цементных смесей [146]. Техни-
Техника производства была различной в зависимости от природы заполнителя и же-
желательных свойств конечного продукта. Типичная смесь имеет нижеследующий
состав (причем отдельные ингредиенты вводятся в порядке перечисления):
1) 600 кг тонкозернистого песка;
2) 300 кг цемента;
3) вода;
4) 3,5 л 40 % -ной перекиси водорода;
5) гипохлорит кальция в количестве, эквивалентном перекиси водорода
в соответствии с реакцией B).
По имеющимся данным, применение катализаторов для разложения
перекиси водорода в этом случае не приводит к удовлетворительным резуль-
результатам. Гипохлорит кальция действует не как катализатор, а реагирует по
следующему уравнению:
СаОС12 + Н2О2 -» СаС12 + Н2О + О2. B)
Сообщается, что получавшиеся таким образом строительные блоки обла-
обладали удельным весом около 1, прочностью на сжатие примерно 25—30 кг/см*
и изолирующей способностью, превышавшей в 2—3 раза изолирующую способ-
способность обыкновенных строительных блоков. Эти блоки в значительных количе-
количествах использовались германской армией в качестве стройматериала. При удвое-
удвоении количества перекиси водорода и добавке около 0,7% целлюлозной крош-
крошки из древесной целлюлозы (в расчете на сухой вес цемента вместе с песком)
можно получать блоки удельного веса около 0,5 и прочностью на сжатие
20 кг/см2. Вырабатывались также пористые гипсовые блоки, причем в данном
случае применялись еще и катализаторы разложения перекиси водорода;
блоки имели удельный вес около 0,3 и сопротивление сжатию около 4 кг/см*.
В новейших патентах [147] также описываются различные способы полу-
получения аналогичных пористых веществ и пористой керамики. Типичный рецепт
включает следующие составные части: глину, гипс, воду, небольшое количе-
количество смачивающего агента, перекись водорода, едкий натр для повышения
рН до 8—10 и катализатор разложения перекиси водорода, например сульфат
двухвалентного марганца. Пористый бетон, полученный с применением пере-
510 Глава 11
киси водорода, хуже сцепляется со стальной арматурой в железобетоне, чем
обычный бетон, так как перекись водорода разлагается на поверхности металла
с образованием газовой пленки между металлическим стержнем и бетоном.
Применение в производстве пенистой резины
Изделия из пенистой резины готовят из натурального или синтетического-
латекса, смешивая его не только с обычными агентами для вулканизации,
наполнителями, пигментами и т. п., но еще и с химическим веществом, выделя-
выделяющим газ. Обычно в качестве газообразователя используется перекись водо-
водорода; на различные рецепты для этой цели выданы патенты [148]. В некоторых
случаях добавляют еще органический агент разложения (например, каталазу
или такой полиамин, как этилендиамин), в других же случаях сами присут-
присутствующие ингредиенты вызывают достаточно быстрое разложение перекиси
водорода. Вспененному латексу обычно дают расширяться до желательных
размеров в форме, после чего его подвергают желатинизации и вулканизации.
По одному из процессов латексную пену подвергают быстрому замораживанию
в форме для сохранения ее конфигурации и ячеистой структуры, а затем
вулканизуют и промывают. Качество конечного продукта сильно зависит от
последовательности введения отдельных ингредиентов и времени, протекаю-
протекающего между различными добавками; перекись водорода обычно, хотя и не все-
всегда, вводится последней.
Ниже как пример приводится одна из патентных формул [148]. К 1 л
латексной смеси, содержащей 100 частей латекса, 3 части серы, 8 частей окиси
цинка, 1,5 части ускорителя и 1 часть наполнителя, добавляют 10 мл 30%-ной
перекиси водорода. Смесь перемешивают до тех пор, пока она не займет объем
5 л, после чего добавляют 150 мл 15%-ного раствора казеината кальция и 20 мл
20%-ного раствора хлористого аммония. Пена, которая, по патентным данным,
устойчива в течение 3—5 час, подвергается вулканизации при 95°. Конечный
общий объем обычно определяется количеством добавленной перекиси водоро-
водорода, причем для получения желательных размеров пузырьков в латекс можно
вводить мыло или другие поверхностноактивные вещества.
Применение для поднятия теста
Выдан ряд патентов по применению перекиси водорода или перекисных
соединений в качестве веществ, способствующих поднятию теста [149]. В тесто
вводят перекись водорода в количестве, эквивалентном 0,5—2% от веса
сухой муки; перед выпечкой тесто выдерживают до исчезновения большей
части этой перекиси. Остальная перекись разлагается в процессе выпечки при-
присутствующей каталазой. Поскольку каталаза сама разлагается при повышен-
повышенных температурах, операцию выпечки необходимо проводить таким образом,
чтобы разложение всей перекиси водорода заканчивалось до достижения тем-
температуры разложения каталазы. Бейли и Ле-Клерк [150] сравнивали хлеб,
выпеченный на дрожжах и на перекиси водорода; они провели тщательный
анализ хлебной корки и мякиша на содержание золы, жира, редуцирующих
Сахаров, общего сахара, водорастворимых веществ и т. д. Не обнаружено почти
никаких заметных различий, если не считать, что по вкусу перекисный хлеб
оказался хуже хлеба, выпеченного на дрожжах! К числу недостатков относит-
относится то, что хлеб или пироги, выпеченные на перекиси водорода, обладают слиш-
слишком тонкой и правильной структурой; прочность продукта слишком мала, и во
рту создается чувство сухости вследствие огромной адсорбционной способно-
способности, которой продукт обладает из-за тонкой текстуры. В качестве ингредиентов
пекарных порошков предложены также твердые перекисные соединения,,
например перекись кальция и перекись мочевины [151].
Перекись водорода в качестве источника свободных радикалов 511
Применение в качестве источника кислорода
Перекись водорода как специальное средство для генерирования кислоро-
кислорода широким распространением не пользуется, если не считать лабораторного
применения. Перекись водорода предложена для пополнения атмосферы кисло-
кислородом (например, в подводной лодке); она находит также некоторое применение
для увеличения содержания кислорода в воде при перевозке рыбной молоди.
Представляет интерес и может быть экономически оправдано использование
концентрированной перекиси водорода в качестве источника кислорода для
сварки и других целей в отдаленных местностях, поскольку при перевозке
90%-ной перекиси водорода достигается экономия в весе больше чем на 50%
по сравнению с перевозкой эквивалентного количества кислорода в обычных
баллонах; еще большая экономия достигается на возврате опорожненной та-
тары. Однако если требуются большие количества кислорода, то экономически
целесообразнее построить установку для производства жидкого кислорода.
Разбавленные растворы перекиси водорода являются удобным источником
получения небольших количеств кислорода, которые могут потребоваться,
например, в лабораторных работах или для демонстрации на лекциях. Удобным
методом является введение раствора перекиси водорода в сосуд или прибор,
работающий по принципу аппарата Киппа и содержащий твердый катализатор,
например двуокись марганца.
ПЕРЕКИСЬ ВОДОРОДА В КАЧЕСТВЕ ИСТОЧНИКА СВОБОДНЫХ РАДИКАЛОВ
Полимеризацию большей части непредельных соединений (например,
стирола, винилацетата, метилметакрилата, каучукового латекса и т. д.) мож-
можно ускорить путем добавки перекиси водорода или других перекисных соеди-
соединений [152]. Это действие обусловлено свободными радикалами, образующи-
образующимися от разложения перекиси, поскольку эти радикалы, как известно, способ-
способны инициировать полимеризационные цепи. Варьирование концентрации
и природы используемой перекиси, а также экспериментальных условий поз-
позволяет в значительной мере видоизменять средний молекулярный вес и дру-
другие свойства продукта. Скорость образования радикалов из перекиси может
быть значительно повышена путем применения так называемых «редокс-систем»,
обеспечивающих возможность достижения значительных скоростей полимери-
полимеризации при температурах гораздо ниже обычных. Этим путем можно получать
полимеры, обладающие превосходными физическими свойствами (например,
«холодный каучук»). Типичная редокс-система содержит соль многовалентного
металла, например железа, в сочетании с таким восстановителем, как сахар.
Начальные реакции могут быть написаны следующим образом:
Fe2+ + Н2О2 -» Fe3+ + ОН~ + ОН,
R R'
Н Н
Ион окисного железа затем восстанавливается до начальной закисной формы
восстановителем. Вместо соли многовалентного металла (или вместе с нею)
можно использовать различные амины или другие соединения. В качестве
катализатора полимеризации можно выбрать любое из большого числа пере-
кисных соединений. Так, в сороковых годах в США для сополимеризации
бутадиена и стирола применяли пероксодисульфат калия; обычно применяе-
применяемыми органическими перекисями являются перекись бензоила и гидроперекись
512 Глава 11
кумола. В тех случаях, когда желательна растворимость в воде, можно исполь-
использовать перекись водорода. Если более существенное значение имеет раствори-
растворимость в мономере, то применяют какую-нибудь органическую перекись.
Перекиси могут катализировать и другие органические реакции, протекаю-
гЦие по свободнорадикальному механизму. В качестве примеров можно привести
присоединение таких полигалогенированных углеводородов, как бромоформ,
или присоединение треххлористого фосфора к двойной связи олефина [153].
ВЛИЯНИЕ ПЕРЕКИСИ ВОДОРОДА НА БИОЛОГИЧЕСКИЕ ПРОЦЕССЫ
Применение перекиси водорода в медицине
Впервые перекись водорода была использована в медицинской практике
Ричардсоном в 1856 г. В результате энергичной пропаганды Ричардсона раз-
разработаны различные методы лечения с применением перекиси водорода, при-
причем Ричардсон [154] и Валлиан [155] опубликовали обзор этих методов. Все эти
виды лечения различных болезней были в основном симптоматическими,
и лишь очень немногие из них могли бы претендовать на признание в настоящее
время. Введение перекиси водорода в практику в качестве антисептического
или дезинфицирующего средства точно так же произошло еще в то время,
когда не была осознана и научно обоснована необходимость асепсиса в меди-
медицинской и хирургической практике. В результате обширная старая литерату-
литература по применению перекиси водорода в медицине сейчас может представлять
почти только исторический интерес.
В настоящее время можно более четко ограничить область пригодности
перекиси водорода в качестве антисептического и дезинфицирующего сред-
средства*, а так же как лечебного препарата против некоторых болезней. Для этих
видов медицинского применения используется незначительная доля производи-
производимой перекиси водорода, однако применение ее в качестве антисептика в быту
широко распространено. Хотя в настоящее время перекись водорода отчасти
вытеснена другими антисептиками, обладающими еще и красящей способ-
способностью, знакомство с перекисью водорода в быту в значительной мере основа-
основано на применении ее в качестве антисептика и как средства для обесцвечива-
обесцвечивания волос. В медицинскую практику вошли также многие другие перекисные
соединения. Например, подробно изучен и, по-видимому, обладает значитель-
значительной эффективностью пероксигидрат мочевины в глицерине. Рейд и Альтемей-
ер [156] провели сравнение различных органических и неорганических пере-
перекисей в качестве составных частей мазей для антисептического применения;
они нашли, что особенно подходящей является перекись цинка. Неорганиче-
Неорганические перекиси, например цинка и кадмия, в связи с их антисептическим дей-
действием используются, так же как составные части косметических средств и зуб-
зубных паст. Перекись водорода находит применение, тэк же как компонент паст,
таблеток, мазей и т. д.
Проведено много подробных исследований по бактерицидному действию
перекиси водорода и перекисных соединений против различных микроорганиз-
микроорганизмов [157], причем эффективность ее была сравнена [158] с эффективностью
других бактерицидов, например соединений ртути, йода, фенолов, этилового
спирта и др. Однако опубликованные данные по относительной эффективности
сильно зависят от выбранного метода сравнения, типа изученных организмов,
концентрации антисептика и длительности контакта.
* Термины «антисептик», «дезинфицирующее средство», а также аналогичные
термины «бактерицид» «бактериостат», «гермицид», «вирусцид» и «стерилизующий агент»
иногда употребляются как синонимы. Строго говоря, под антисептиком подразумевается
вещество, которое угнетает рост микроорганизмов, но не обязательно уничтожает
их в отличие от дезинфицирующего средства, которое полностью устраняет инфекшю.
путем уничтожения микроорганизмов и инактивации вирусов.
Влияние перекиси водорода на биологические процессы 513
Испытание антисептика или дезинфицирующего средства in vitro аналогич-
аналогично измерению скорости химической реакции, где параметрами являются кон-
концентрация, температура, а в ограниченных размерах также растворитель или
культуральная среда. При попытках экстраполирования этих результатов
к вероятному действию в жидкостях тканей организма приходится сталкивать-
сталкиваться со множеством других факторов, которые трудно или невозможно одновре-
одновременно воспроизвести в опытах in vitro. Кроме того, необходимо учесть, особен-
особенно при обработке ран, поверхности кожи или слизистых оболочек, еще и ско-
скорость переноса антисептика к пораженной поверхности и длительность нахо-
нахождения его в условиях, при которых действуют факторы, способствующие
разложению и разбавлению. Такие физические процессы, протекающие во
времени, могут оказывать решающее влияние на эффективность антисептика.
Эти проблемы рассматриваются в монографии Мак-Каллоча [159], а в отно-
отношении перекиси водорода, в частности, Брауном [160]. Мы здесь не можем при-
привести подробные результаты этих исследований. В общем растворы перекиси
водорода в глицерине более эффективны против грам-положительных, чем
против грам-отрицательных организмов, хотя бактериостатические эффекты
наблюдаются в отношении организмов обоих типов и перекись водорода можно
классифицировать как неспецифический бактерицид. Для уничтожения споро-
образующих бактерий, например В. subtiis, требуются более высокие кон-
концентрации перекиси водорода и большая продолжительность контакта, чем
для бактерий типа кишечной палочки, как Е. coli. Особенное преимущество при-
применения перекиси водорода для дезинфекции заключается в том, что она срав-
сравнительно слабо угнетает фагоцитоз (т. е. активность некоторых кровяных и дру-
других клеток в отношении уничтожения отбросов организма, вредных бактерий
и т. д.). В общем опыты in vitro показали, что перекись водорода в концентра-
концентрациях порядка 0,001—0,1% при комнатной температуре угнетает рост микро-
микроорганизмов, в концентрации же приблизительно 0,1% или выше убивает эти
организмы. Такие концентрации уничтожают также пирогенные вещества
в растворах, предназначенных для инъекции [161]. Этот вопрос рассмотрен
также в гл. 2 (стр. 67).
Сильные и слабые стороны применения перекиси водорода в качестве
антисептика можно представить следующим образом:
Преимущества Недостатки
Отсутствие слишком большой специфичности Медленность действия
Малая токсичность для тканей Сравнительная неустойчивость при хра-
хранении
Отсутствие аллергенного действия Высокое поверхностнее натяжение
Безвредность продуктов разложения Инактивация тканями
Отсутствие красящей способности
Растворимость в жидкостях организма
Наличие очищающего дейстия
Сравнительная дешевизна
Сравнительная безболезненность
Указанные выше недостатки можно в значительной мере преодолеть, если
заменить воду в качестве растворителя безводным глицерином. Такой препарат,
в который перекись водорода вводится в виде пероксигидрата мочевины, содер-
содержащего оксин в качестве стабилизатора, разработан Брауном, Горином и Абрам-
соном [162] и в настоящее время имеется в продаже. Вязкая глицериновая
среда способствует действию перекиси водорода в течение более продолжитель-
продолжительного времени, так как она предотвращает испарение, обеспечивает прилипание
к месту нанесения и создает резервуар перекиси, которая не разлагается быст-
быстро под действием окружающих тканей, а непрерывно диффундирует к обраба-
обрабатываемой поверхности. Гигроскопический глицерин имеет еще и то положитель-
положительное значение, что он притягивает тканевую жидкость к месту нанесения. Бра-
33 Перекись водорода
514 Глава 11
ун [163] показал, что такой препарат хорошо переносят ткани, не обладающие
к нему чувствительностью. Исследования [164] этого препарата, а также
других антисептических препаратов перекиси водорода (в том числе и аэро-
аэрозолей) показывают, что приемлемая стабильность может сохраняться
в течение длительных промежутков времени. Комбс [165] испытал ряд
органических растворителей для перекиси водорода и рекомендует при-
применение раствора ее в трет-бутаноле в качестве фунгицида. Такого рода
препарат имеется в продаже; он, по-видимому, [166] более эффективен не
как бактерицид, а как спорицид.
Основной механизм антисептического или дезинфицирующего действия пе-
перекиси водорода не выяснен. Известно [167], что перекись водорода являет-
является нормальным продуктом обмена веществ при росте бактерий; таким образом,
увеличение концентрации перекиси водорода в культуральной среде, где
происходит рост бактерий, способствует угнетающему действию продуктов
жизнедеятельности, замедляя или приостанавливая рост. Выделение кислорода
также может тормозить рост анаэробов. Некоторую роль может играть и синер-
гетическое действие фагоцитов [168]. С другой стороны, некоторые природ-
природные вещества тормозят действие перекиси водорода, например за счет разложе-
разложения ее каталазой [169] или защитного, действия солей и эфиров пировиноград-
ной кислоты [170]. Показано [171J, что добавка некоторых солей к перекиси
водорода, например железа, меди, хрома (с марганцем или кобальтом в каче-
качестве активаторов) и молибдена, заметно увеличивает ее дезинфицирующую спо-
способность, откуда можно сделать вывод о роли, которую играет образование
свободных радикалов. Здесь можно провести аналогию со способностью
перекиси водорода, одной или в реактиве Фентона, вызывать мутации [172]
в бактериях таким же образом, как это происходит при действии излучений.
Постулировано также образование перекиси водорода в качестве промежу-
промежуточного продукта в механизмах антисептического действия или детоксикации,
представляющих интерес с медицинской точки зрения. Действие сульфанил-
сульфаниламида отчасти приписывается [173] угнетению или нейтрализации активности
каталазы, в связи с чем концентрация перекиси водорода (возникающей
в результате обмена веществ бактерий) может возрасти до размеров, при
которых она уже начинает функционировать как ингибитор. Однако это толь-
только одна из многих теорий действия сульфаниламида; эти теории рассмотрены
в сообщении Норти [174]. Точно так же антибиотик, носящий названия нота-
тина и пенициллина В, представляет фгрмент, действие которого может зави-
зависеть от влияния перекиси водорода, образующейся в ходе катализирования
аэробного окисления глюкозы до глюконовой кислоты [175]. Сообщается, что
слюна обладает известным антисептическим действием, причем оказалось [176].
что оно обусловлено присутствием перекиси водорода. Выяснено [177], что
в присутствии пероксидазы и перекиси водорода действие фенолов на бакте-
бактерии усиливается, однако нельзя быть уверенным, что без преднамеренной
добавки этих веществ действие фенола должно быть приписано наличию перок-
пероксидазы и перекиси водорода. Перекиси водорода приписывается [178] роль
промежуточного вещества в детоксикации циклических соединений аскорби-
аскорбиновой кислотой. Образование перекиси водорода под действием бактерий
и ферментов рассматривается выше (стр. 67 и 351).
Остановимся на недавно опубликованных описаниях применения перекиси
водорода при лечении отдельных заболеваний или поражений. Получены хоро-
хорошие результаты при лечении хронических инфекций уха [179], ряда воспали-
воспалительных инфекций кожи и слизистых оболочек бактериального и грибкового
происхождения [180], отравления цианидами [181], хронической эмпиемы
и инфекций легких [182]; перекись водорода снижает или предотвращает пора-
поражение кожи при трении о шероховатую бумагу [183], она применима в ано-
ректальной хирургии [184], при лечении ожогов люизитом [185] или неболь-
Влияние перекиси водорода на биологические процессы 515
ших ожогов и для удаления бородавок. Эммерсон [186] рассматривает примене-
применение перекиси водорода в ветеринарии; она может найти применение и как гли-
глистогонное, или антигельминтовое средство [ 187], а также полезна для инъекции
в места, ужаленные оводами (для уничтожения яичек) [188]. Результаты, полу-
полученные при лечении газовой гангрены [189], гриппозной пневмонии [1901г
язвы желудка [191] и реакции ядовитого плюща, являются противоречивыми
или безуспешными. Опыты по лечению перекисью водорода фосфорного отрав-
отравления [192], гипоксии, отравления окисью углерода, кровоизлияния и шока,
проведенные над животными, дали отрицательные результаты [193].
Протравливание семян
Опубликовано большое число сообщений по применению перекиси водоро-
водорода или перекисных соединений для протравливания семян. Основная задача
состоит в том, чтобы дезинфицировать семена и улучшить их всхожесть. Сооб-
Сообщается 'также о попытках применения перекиси водорода для размягчения
оболочки семян и сообщения ей повышенной гибкости с целью улучшения об-
обрушивания. Сравнительно легко определить эффективность дезинфицирующей
обработки семян, но далеко не ясно, действительно ли повышение всхожести
семян, наблюдаемое в некоторых случаях протравливания перекисью водорода,
обусловлено иммобилизацией микроорганизмов, которые в противном случае
нарушали бы ход прорастания, или специфическим действием на способность
семян к прорастанию. При слишком интенсивной дезинфекции всхожесть мо-
может значительно снизиться. Стерилизующее действие перекиси водорода изме-
изменяется в зависимости от характера оболочки семян и типа микроорганизма.
Киссер и Портгейм [194] показали, что семена цветов, бобовых и злаков мож-
можно полностью или частично стерилизовать 15—30%-ными растворами переки-
перекиси водорода с очень небольшим снижением всхожести.
Шероховатые семена, покрытые шелухой или слипшиеся в комки, труднее
или даже совсем невозможно стерилизовать. Те виды обработки, которые
благоприятствуют прониканию раствора перекиси водорода в семена, снижают
их всхожесть. Пихлер [195] сообщает, что обработка пшеницы и ячменя раство-
растворами перекиси водорода концентрацией до 3% не снижала развития вонючей
головни пшеницы (Tilletia caries) или каменной головни ячменя (Ustillago
hordei), обработка же овса 4—5%-ным раствором перекиси водорода не обез-
обеззараживала его от пыльной головни овса (Ustillago avenae). Олджией [196]
показал, что для дезинфицирования зерна, зараженного головней, при дли-
длительности контакта 5 мин. требовались растворы перекиси водорода примерно
20%-ной концентрации; всхожесть в этом случае, однако, немного снижалась.
В литературе имеется ряд сообщений, что обработка перекисью водорода
оказывает лишь слабое действие в борьбе с заражением зерна или даже со-
совершенно неэффективна, но во всех этих случаях, по-видимому, применялись
слишком низкие концентрации перекиси водорода (например, 5вес.% или ни-
ниже). Имеются также некоторые указания, что пероксоуксусная кислота может
оказаться эффективной при протравливании зерна.
Выдерживание семян в разбавленных растворах перекиси водорода (на-
(например, при концентрации от 0,01 до 1,5 вес.%) в течение около суток (или
даже до начала прорастания) повышает процент всхожести семян и скорость,
или интенсивность, прорастания. Такое улучшение отмечено для пшеницы
[197], семян цветов и бобовых [198] и помидоров [199]. Проведен ряд работ
по ячменю, доказавших улучшение прорастания его при действии перекиси
водорода, что имеет особенное значение для пивоваренных заводов. Типичные
растворы перекиси водорода в воде для замочки должны иметь 0,01—0,5%-нуф.
концентрацию; оптимальная температура равна 20° [200]. Единственным
остатком при такого рода обработке является вода, что в данном случае име-
33*
516 Глава 11
ет особое значение. Ро [201] опубликовал обзор по основным работам, проведен-
проведенным до 1939 г., по применению перекиси водорода для обработки ячменя в про-
производстве солода. Более поздние исследования, проведенные Боуденом [202],
показали, что небольшие концентрации перекиси водорода в воде для замоч-
замочки оказывают более благоприятное влияние на прорастание, чем растительные
гормоны. Применение перекисных химических веществ для обработки семян
описывается в одном из технических бюллетеней [203], в котором содержится
17 литературных ссылок по этому вопросу. Практически та же самая информа-
информация опубликована и в одном из технических журналов [204].
Сообщается, что введение перекиси водорода или некоторых других оки-
окислителей в почву повышает урожай кукурузы и соевых бобов [205] и рост
сеянцев [206]. Причина этого неясна. Высказано предположение [206], что
окислитель ускоряет разложение органического вещества в почве; не исключено
также, что он, как указано выше, непосредственно действует на семена.
Другая возможность состоит в том, что реакция перекиси водорода с неоргани-
неорганическими компонентами почвы может перевести их в более легко усвояемую
форму. Так, в одном подробном исследовании [207] показано, что обработка
перекисью водорода превращает содержащийся в почве азот в аммиак и увели-
увеличивает также содержание водорастворимых органических и неорганических
веществ. Однако способность минеральной части почвы к обмену катионами
при этом не изменяется. Возможно, что механическое действие, обусловленное
выделением кислорода при разложении, способствует тому, что почва делается
более пористой. По мере повышения концентрации перекиси водорода это
явление интенсифицируется; при проливании 90%-ной перекиси водорода на
землю разложение протекает очень интенсивно и после разложения поверх-
поверхностный слой почвы делается порошкообразным и его кажущийся объем воз-
возрастает. Благодаря выделению тепла перекись водорода способствует высыха-
высыханию даже сырой почвы. Различные почвы и различные компоненты их по-раз-
по-разному реагируют на перекись водорода; в частности, изучено влияние переки-
перекиси на слюдяной компонент почвы [208].
Приготовление и консервирование пищевых продуктов и напитков
Предприняты попытки или сделаны предложения использовать перекись
водорода для приготовления или консервирования ряда пищевых продуктов
и напитков, но лишь в отдельных случаях применение ее для этих целей дей-
действительно получило общее признание. Описаны разные способы консервирова-
консервирования продуктов путем добавки небольших количеств перекиси водорода, а имен-
именно молока и разных молочных продуктов (стр. 518), рыбы [209] (путем введе-
введения перекиси водорода в лед, применяемый для упаковки), консервированного
бульона [210], яиц [211], саке (рисовой водки), уксуса, кетчупа, кофейного
сиропа, соевого творога, вермишели [212], сиропа какао [213] и напитков
{214], желатина [215], лецитина, пикулей и крахмала.
Изучен вопрос очистки воды путем обработки перекисью водорода [216].
По этому вопросу имеется общий обзор Рейхеля [217], который на основании
собственных опытов пришел к заключению, что для эффективной дезинфекции
воды необходимо добавлять к ней 5 вес.% перекиси водорода, иначе нельзя
добиться завершения дезинфекции за практически приемлемый промежуток
времени. При такой концентрации вода приобретает ощутимый вкус остаточ-
остаточной перекиси водорода, а поэтому последняя должна быть разложена путем
добавки безвредного катализатора. Если допустимы более длительные периоды
обработки, то можно дезинфицировать воду с применением меньшего количе-
количества перекиси водорода; например, 0,5% ее достаточно для дезинфекции за
период в 24 часа. Перекись водорода можно использовать и для устранения при-
Влияние перекиси водорода на биологические процессы 517
вкуса от хлора в воде, обработанной обычными методами. Стерилизация емко-
емкостей, труб, фильтроз и т. п., применяемых в пищевой промышленности, часто
производится перекисью водорода [218].
Бактерицидное и фунгицидное действие перекиси водорода находит при-
применение в пищевой промышленности: например, ее предложено употреблять
для улучшения заплесневевшего зерна [219], подобная же обработка применяет-
применяется для предотвращения порчи свежесобранных фруктов и овощей [220].
Широкое применение нашел аналогичный процесс, а именно опрыскивание
помидоров раствором пероксоуксусной кислоты [221] (возможно, что оба про-
процесса имеют одинаковый основной механизм). Путем добавки перекиси водо-
водорода предотвращается при хранении нежелательное брожение виноградного
сусла 1222].
Другие виды применения перекиси водорода в обработке пищевых про-
продуктов, не связанные с ее консервирующим действием, следующие: пекарная
добавка для поднятия теста (см. стр. 510); улучшение вин и коньяка [2231
(ускоряет созревание, вероятно путем превращения сивушного масла в альде-
альдегиды в результате реакции с перекисью водорода; перекись способствует
также устранению побурения белых вин); обесцвечивание красного свекловичного
сока [224]; регенерация культуральных сред [225]; очистка дрожжей от горь-
горьких начал [226]; стабилизация лецитина [227]; улучшение органолептических
свойств пищевых масел [228]; модификация пищевых крахмалов и камедей
[229]; улучшение вкуса кофейных экстрактов [230]; отбелка яиц в скорлупе
[231]; устранение изменения цвета пищевых крабов [232]; бланшировка ово-
овощей [233]; отбелка мононатрийглутамата; отбелка требухи, желатина и яично-
яичного желтка и отбелка орехов и сушеных фруктов для улучшения их внешнего
вида. Новым достижением [234] является процесс, устраняющий один из источ-
источников неприятного развития запаха и вкуса в сушеных пищевых продуктах.
Это—ферментативный процесс, в котором глюкозооксидаза потребляет сбра-
сбраживаемые вещества, например из яичного белка; перекись водорода является
наиболее удобным источником кислорода для этой аэробной реакции. Большие
количества перекиси водорода находят косвенное применение в пищевой про-
промышленности в виде перекиси бензоила, широко применяемой для отбелки
муки. Такая отбелка, обусловленная окислением ксантофилла, происходит
быстро и равномерно, однако она не сопровождается одновременным созре-
созреванием, т. е. улучшением пекарных свойств муки.
В том случае, когда перекись водорода используется для консервирования
пищевых продуктов, она может оказывать двоякого рода действие: разрушать
ферменты, обусловливающие нежелательное изменение состава, и предотвра-
предотвращать или тормозить рост патогенных или вредных организмов. Обычно кон-
концентрацию перекиси водорода рекомендуется подбирать таким образом, чтобы
взаимное разложение добавленной перекиси и того субстрата, который имеет
значение в данном случае, привело к исчезновению обоих этих веществ, так
чтобы не оставалось избыточной перекиси; правда, иногда оказывается возмож-
возможным разложить остаточную перекись водорода действием добавляемого на
следующей стадии катализатора, например каталазы [235]. В связи с тем, что
чувствительность, состав и характер обработки и хранения могут колебаться
в очень широких пределах (притом даже при разных пробах одного и того же
пищевого продукта или напитка), трудно сделать окончательный вывод о воз-
возможной эффективности обработки, об изменении вкуса, питательной ценности
или физиологических последствий, которых можно ожидать в каждом отдель-
отдельном случае в результате обработки перекисью водорода. Ответ на этот вопрос
трудно дать еще и потому, что иногда в качестве причины порчи пищепродук-
тов выдвигается процесс образования перекиси водорода за счет реакций
самоокисления. Очевидно, нельзя считать, что применение перекиси водорода
(или другого аналогичного консервирующего вещества) может заменить соблю-
518 Глава И
дение правил гигиены при обработке, хранении и перевозке пищепродуктов
(например, в случае предложения использовать перекись водорода для улучше-
улучшения испорченного мяса). С другой стороны, однако, не ясно, является ли
применение перекиси водорода в качестве консервирующего вещества жела-
желательным и экономичным процессом для пищевой промышленности. Наиболее
широко дебатировался вопрос о применении перекиси водорода в качестве
консервирующего средства для молока. Ниже приводится довольно подроб-
подробное описание этого потенциально выгодного применения перекиси водорода,
усиленно защищавшегося многими исследователями, в качестве типичного
примера проблем, с которыми приходится сталкиваться при обработке пище-
пищевых продуктов или напитков.
Амберг [236] и Ансельми [237] дали обзор ранней истории исследований
по применению перекиси водорода для консервирования молока; впервые этот
способ был предложен Болди в 1881 г. и нашел применение во Франции и в Гер-
Германии [238] во время первой мировой войны, но потом был оставлен. В настоя-
настоящее время во многих местах запрещено консервировать молоко каким-либо
другим способом, помимо пастеризации и охлаждения, однако применение
перекиси водорода для этой цели вновь получило развитие в Италии во время
второй мировой войны, и большая часть новейших исследований в этой области
принадлежит итальянским авторам.
Очень часто цитировалась работа Чик ' [239] 1901 г., с которой начинает-
начинается ряд работ на эту тему. Чик пришла к выводу, что молоко можно полностью
стерилизовать, добавляя к нему перекись водорода в количестве 0,2 вес.%
(в расчете на 100%-ную). После добавки такого количества перекиси водорода
сначала происходит частичное ее разложение, а затем концентрация сохраняет-
сохраняется неизменной в течение длительного времени. Поскольку Чик сообщает, что
она в состоянии открыть изменение вкуса молока при наличии в нем 0,01%
перекиси водорода, этот метод стерилизации, по ее мнению, не может претен-
претендовать на практическое применение (если обработка достаточна для стерилиза-
стерилизации, то молоко теряет вкусовые качества).
Розам [240] показал, что 0,2% перекиси водорода недостаточно для стери-
стерилизации, и предложил комбинировать добавку перекиси водорода с термиче-
термической обработкой. Затем Будде [241] развил эту идею и разработал в Дании
производственный процесс, состоящий в добавке к молоку 0,9 г!л перекиси
водорода (в расчете на 100 %-ную) с последующим нагреванием в течение не-
нескольких часов до 50° (если в молоке все же сохраняется перекись, ее разлагают
введением дрожжей). Другие авторы впоследствии высказывали различные мне-
мнения о необходимом количестве перекиси водорода и рекомендовали разные до-
добавки—вплоть до 1%. Показано, что эффективность зависит от ряда факторов,
таких, как характер проб, температура обработки и хранения, рН, тип микро-
микроорганизмов, присутствующих в молоке; все эти факторы влияют на количества
перекиси, необходимые для стерилизации молока, и на изменение его вкуса.
Общепризнанным считалось, что створаживание молока в присутствии перекиси
водорода невозможно, однако мнения расходились по вопросу, может ли
присутствие перекиси водорода в количестве, достаточном для предотвращения
скисания молока, быть действенным также против патогенных организмов или
последние при этом не погибают. Опубликованы различные отчеты о длитель-
длительном питании молоком, обработанным перекисью водорода, причем ни в одном
случае не отмечено нежелательных физиологических эффектов.
Более поздние работы способствовали лучшему пониманию влияния добав-
добавки перекиси водорода к молоку. Кроме того, общедоступность электролитиче-
электролитической 30%-ной перекиси водорода привела к тому, что отпали ранее высказы-
высказывавшиеся возражения о нежелательности слишком сильного разбавления моло-
молока и введения в него металлических примесей (попадавших при применении
3%-ной перекиси водорода, получаемой из перекиси бария). Сейчас произ-
Влияние перекиси водорода на биологические процессы 519
водственные фирмы выпускают специальную чистую перекись водорода «для
пищевых продуктов».
В настоящее время бесспорно установлено, что добавка перекиси водорода
в количестве около 0,1 вес.% в расчете на 100%-ную перекись к сырому молоку
непосредственно после выдаивания снижает содержание патогенных организ-
организмов до приемлемо низкого уровня и" устраняет его скисание при хранении при
комнатной температуре в течение нескольких дней. Этот вывод подкрепляется
работами Моранди [242], о которых сообщают также Дален и Кросли [243].
Сатта, Моранди и Моджи [244] показали, что если молоко хранить при более
низкой температуре, то может оказаться приемлемой и меньшая добавка пере-
перекиси водорода. По Романи [245], если добавку перекиси водорода не снижать,
а температуру поддерживать на низком уровне, то молоко будет сохраняться
дольше (например, 32—40 суток при 5°). При комбинировании пастеризации
с добавкой перекиси водорода для предотвращения скисания можно брать
и меньшие дозы перекиси; Сэндерс и Сэджер [246] показали, что 0,1 вес.%
перекиси достаточно для предотвращения створаживания пастеризованного мо-
молока, содержащего 2,5% сырого, в течение 10—21 суток. Такие результаты
получены ими в опытах по использованию перекиси водорода для консерви-
консервирования проб молока до анализа.
Моранди [242] показал, что число бактерий в молоке снижается приблизи-
приблизительно на 95% при добавке 0,1% перекиси водорода. Перагалло (цитируемый
в работе [243]), изучая молоко, искусственно зараженное В. coliy В. typhicus,
Brucella melitensis, Brucella abortus и бациллой туберкулеза рогатого скота,
нашел, что добавка 0,1% перекиси водорода к молоку сразу после выдаивания
в тех случаях, когда измерения проводились в течение промежутков времени
от 12 до 48 час, оказывало бактерицидное действие на все микроорганизмы,
кроме туберкулезных бацилл. Если проходило определенное время от доения
до добавки перекиси водорода, то последняя действовала лишь бактериостати-
чески и в течение указанных промежутков времени происходила заметная реге-
регенерация бактерий. Эти выводы подтверждаются работами Гейнемана [247],
Розати [248], Джентили и Романьоли [249] и Моначи [250]. Арнауди и Трек-
кани [251] показали, что добавка 0,05% перекиси водорода к молоку эффектив-
эффективна против Clostridiutn butyricum, Streptococcus lactis и молочных термобакте-
термобактерий.
В табл. 87 приведены данные по влиянию добавки перекиси водорода на
физические и химические свойства молока и содержание в нем ферментов и
витаминов. Ансельми [237] и Казерио (цитируемый в работе [243]) также изу-
изучали скорость роста кислотности после добавки различных количеств переки-
перекиси водорода и в разных условиях хранения и заражения; они подтвердили
прежний вывод, что молоко заметно не скисает в течение всего того времени,
пока оно остается стабилизированным благодаря присутствию перекиси водо-
водорода. Ансельми [237] привел качественные данные по повышению склонности
молока к образованию пены и пленки агломерированного вещества на стенках
сосудов.
Перекись водорода оказывает заметное, но не длительное действие на
органолептические свойства молока. Почти все авторы, изучавшие этот вопрос,
считают, что и запах и вкус молока заметно ухудшаются при добавке перекиси
водорода в эффективных концентрациях, если только не разложить ее избыток
до потребления молока. По этой причине в различных предложениях, касающих-
касающихся применения перекиси водорода, рекомендуется сочетать эту добавку с тер-
термической обработкой, введением катализатора или длительной выдержкой или
же использовать перекись только для консервирования молока в период меж-
между выдаиванием и пастеризацией. Такого рода комбинация может даже пред-
представлять определенный интерес; в ряде новейших патентов [261] подчеркивает-
подчеркивается, что при добавке перекиси водорода с последующим нагреванием возможна
520
Глава 11
Таблица 87
Влияние добавки перекиси водорода на физические и химические свойства молока
и содержание в нем ферментов и витаминов
Свойство или содержание вещества
Приблизитель-
Приблизительная добавка
Н2О2, вес. %
Действие
Литература
Удельный вес
Точка замерзания
Показатель преломления сыворотки . . .
Содержание жира
Общее содержание азота
Содержание аминокислот
» казеина
» альбумина ...
» лактозы
Число Поленске
» Рейхерт-Мейссля
Показатель преломления жира
Размер и структура глобул жира . . . .
Скорость образования сливок
рН . . . .
рН
Окислительно-восстановительный потенциал
Створаживание сычугом
Содержание каталазы
редуктазы
пероксидазы
амилазы
липазы . .
триптазы .
фосфатазы
витамина А
» В
» В
» С
0,1
0,1
о
о
о
о;з
о,
о,
о,
о,
о,
о,
о,
о,
о,
о,
о,
о,
0,1
0,1
0,1
0,1
о
D
1
0,1
оТТ
0,1
0,3
0,1
Не действует
» »
» »
Снижает
Увеличивает
Не действует
Снижает
Не действует
Изменяет
Тормозит
Снижает или
уничтожает
То же
» »
Не действует
Снижает
Не действует
Снижает или
разлагает
Не действует
243
243
243
243, 252
243, 252, 251
253
252
252
243, 252, 25L
243
243
243
243
243
243
252
254, 255
243
243, 256
243, 256
256
256
256
256
256
246, 257
244, 252
243, 244
258
259, 260
244
полная стерилизация, а при стерилизации путем одной только пастеризации
молоко приходится нагревать до столь высоких температур (или в течение та-
таких длительных промежутков времени), что его вкус и физические свойства
при этом заметно ухудшаются.
Нужно различать два влияния, которые перекись водорода оказывает на
вкус молока. Первое заключается в том, что при достаточной концентрации ее
в молоке появляется характерный вкус перекиси водорода, который описывает-
описывается как «металлический». Второе влияние сложнее: перекись водорода участвует
в реакциях и изменяет их ход, что обычно вызывает изменение вкуса моло-
молока. Необработанное молоко при хранении может постепенно приобрести окис-
окисленный или «сальный» привкус, который, по Круковскому и Гатри [262],
вызывается окислением непредельных жирных кислот, главным образом оле-
олеиновой, в липидной фракции молока. Наибольшей чувствительностью к оки-
окислению обладают фосфолипиды, содержащиеся в мембране глобул жира [263],
хотя Понт [264] и сомневается в том, чтобы фосфолипид был источником при-
привкуса.
Гринбэнк [255] нашел, что с повышением концентрации перекиси водорода
от 0,001 до 0,016 вес.% ее влияние на сальный привкус изменяется от усиливаю-
Влияние перекиси водорода на биологические процессы 521
шего к тормозящему и что торможение вызвано изменением окислительно-вос-
окислительно-восстановительного потенциала. Круковский и Гатри [262] считают, что предел
тормозящей способности лежит несколько ниже 0,001%, и показали, что ес-
если молоко содержит меньше перекиси водорода, то у него через различные про-
промежутки времени появляется сальный привкус. Эти авторы объясняют появ-
появление привкуса окислением аскорбиновой кислоты до дегидроаскорбиновой.
Они считают, что окисление липидов является индуцированной реакцией, зави-
зависящей от известного равновесия между аскорбиновой и дегидроаскорбиновой
кислотами, причем возможно, что она инициируется катализаторами.
Скорость окисления аскорбиновой кислоты перекисью водорода обратно
пропорциональна концентрации перекиси ввиду инактивации фермента,
катализирующего окисление (по Круковскому [265], пероксидазы). Следова-
Следовательно, при низких концентрациях перекиси водорода окисление аскорбино-
аскорбиновой кислоты происходит достаточно быстро для создания такого соотношения
концентраций между этой кислотой и дегидроаскорбиновой, которое благо-
благоприятствует окислению липидов. При более высоких концентрациях перекиси
водорода скорость окисления аскорбиновой кислоты меньше скорости последую-
последующего разложения лабильной и чувствительной дегидроаскорбиновой [266],
а поэтому до полного окисления аскорбиновой кислоты не может установиться
соотношения концентрации ее и дегидроаскорбиновой кислоты, благоприят-
благоприятного для окисления липидов; это и тормозит развитие сального привкуса.
В этом сложном процессе определенное влияние оказывают и другие фак-
факторы; анализ их дан в работах Чилсона, Мартина и Парриша [267] и Гринбэн-
ка [268]. В качестве практического метода для задержания развития сального
привкуса в замороженном молоке Бел и Муча [260] рекомендуют добавку
аскорбиновой кислоты, которая селективно окисляется при повышенной кон-
концентрации ее и этим самым увеличивает период времени до окисления липидов;
добавка перекиси водорода также эффективна, но дает менее удовлетворитель-
удовлетворительные результаты.
При выдерживании на солнечном свету у молока иногда появляется неже-
нежелательный привкус. Этот так называемый «привкус от солнечной активации»,
связанный с белковой частью молока, вызывается [269] фотолизом метионина
и катализируется рибофлавином. Добавка перекиси водорода к молоку предо-
предотвращает развитие этого вкуса [270].
В заключение можно сказать, что добавка перекиси водорода к молоку,
если эта перекись сохраняется в молоке, поступающем к потребителю, не обла-
обладает никакими преимуществами перед пастеризацией, а поэтому этот метод
нельзя рекомендовать для замены пастеризации. С физиологической точки зре-
зрения обработка перекисью водорода, очевидно, безвредна и существенно не влия-
влияет на питательную ценность молока, однако, поскольку эта обработка, по-ви-
по-видимому, не поддается точной стандартизации и является настолько эффектив-
эффективной, что может замаскировать антисанитарную практику при доении или хра-
хранении, ее следует осуществлять обязательно под компетентным техническим
надзором. В качестве крайней меры, например в отсталых районах или на теат-
театре военных действий, можно использовать консервирование молока перекисью
водорода, но и в этих случаях целесообразно комбинировать такую обработку
с той или иной термообработкой.
Эти возражения против применения перекиси водорода отпадают, если пе-
перекись используется только для начальной стерилизации молока, а затем под-
подвергается разложению. При этом имеются даже определенные преимущества,
как указано выше, с точки зрения сохранения вкуса молока. Такой процесс
разработан для производства сухого молока [8]; по этому же методу [261]
в настоящее время в США [271] вырабатывается консервированное цельное
молоко. Указывается, что по ряду причин молоко, обработанное перекисью
водорода, дает сыр [272] и масло [273] повышенного качества.
522 Глава 11
ПРИМЕНЕНИЕ В ХИМИЧЕСКОМ СИНТЕЗЕ
Особенным преимуществом применения перекиси водорода в качестве оки-
окислителя является безвредный характер продуктов ее разложения (кислорода
и воды). В 1954 г. цена перекиси водорода в расчете на эквивалент окислитель-
окислительной способности была довольно высокой и составляла около 44 долларов на
килограмм-атом активного кислорода [для сравнения укажем, что цена
килограмм-атома активного кислорода в трехокиси хрома («хромовой кислоте»)
равна 40 долларам, килограмм-атома хлора в газообразном хлоре—2,2 дол-
доллара]. Поэтому перекись водорода экономично применять в качестве окис-
окислителя только в таких случаях, когда невозможно получить необходимую
структуру образующейся окисленной молекулы или желательную реакцию
с использованием дешевого окислителя. В связи с этим продукты, получаемые
путем окисления перекисью водорода, обычно принадлежат к двум типам:
1) продукты, стоимость единицы веса которых сравнительно высока, и 2) про-
продукты, в которые вводят 1 или 2 атома кислорода в молекулу, обла-
обладающую гораздо большими относительными размерами (т. е. когда количество
потребляемой перекиси невелико по сравнению с количеством образуемого
продукта).
Перекись водорода является исходным материалом для приготовления
большей части перекисных соединений, находящих промышленное применение.
Неорганические перекисные соединения рассматриваются в гл. 12, где указа-
указаны также методы их получения. Единственными неорганическими перекисны-
ми соединениями, обладающими промышленным значением и получаемыми не
через перекись водорода, являются следующие: перекись натрия (сжиганием
натрия в воздухе); перекись и надперекись калия; перекись бария (переокисле-
(переокислением окиси бария на воздухе) и пероксодисульфаты калия и других металлов
(непосредственно из пероксодисульфата аммония и пероксодисерной кислоты,
образующихся в качестве промежуточных продуктов при электролитическом
производстве перекиси водорода). Пероксоборат натрия также может быть полу-
получен электролитически из буры, но чаще его получают из смеси буры с перекисью
водорода и перекисью натрия или только с перекисью натрия.
Органические гидроперекиси можно получать прямо окислением углево-
углеводородов или других органических соединений кислородом или воздухом; этим
путем, например, получают гидроперекись кумола. В том случае, когда при пря-
прямом окислении получается целая гамма продуктов, в промышленной практике
обычно используется синтез через перекись водорода или другое перекисное
соединение.
Различные типы органических реакций с участием перекиси водорода или
какой-либо пероксокислоты описаны на стр. 341 и ел. Наряду с образо-
образованием органических перекисных соединений к этим реакциям относятся
еще следующие: 1) реакция с непредельной связью с образованием эпо-
~ ОН ОН
/°\ I I
ксидного производного (—R—R—) или структуры гликоля —R — R—;
II II
2) различные реакции окисления, например окисление замещенного нафта-
нафталина до соответствующего хинона или производство виннокаменной кислоты
окислением малеинового ангидрида в присутствии окиси вольфрама как ката-
катализатора; 3) реакции размыкания кольца, например превращение фенола в
муконовую кислоту. Многие способы применения перекиси водорода в промыш-
промышленном синтезе еще не разработаны, но такие возможности представляют
большой интерес и заслуживают дальнейшего изучения. Так, было указано
[274], что присутствие перекиси водорода может ускорить дубление кожи пу-
путем превращения таннинов в хиноны. Высококонцентрированная перекись
Литература 523
водорода находит применение [275] в производстве белых мыл, не изменяющих
цвета при хранении (из растительных масел, содержащих у-токоферол). Общее
представление о применении перекиси водорода в качестве окислителя с особым
учетом реакций, которые могут приобрести промышленное значение, дает обзор,
опубликованный Тейлором [276]. Такие реакции описаны и в различных тех-
технических бюллетенях, где приводится также обширная библиография [277].
Техническое применение органических перекисных соединений или орга-
органических процессов, в которых используются какие-либо другие перекисные
соединения, помимо перекиси водорода, здесь не рассматриваются. Некоторые
эпоксидные соединения используются непосредственно, например диэлдрин,
применяемый в качестве инсектицида. Другие эпоксидные соединения служат
для производства смол и поверхностноактивных веществ или как промежуточ-
промежуточные продукты в органическом синтезе. Так, при синтезе кортизона можно
ввести 17-оксигруппу путем образования сначала промежуточного эпоксид-
эпоксидного производного с последующим восстановлением эпоксигруппы до гидро-
ксильной. Эпоксидное кольцо можно получить также путем нагревания соответ-
соответствующего хлоргидрина со щелочью, что экономичнее в том случае, когда процесс
не сопровождается заметными побочными реакциями. Например, эпихлор-
гидрин, один из мономеров, применяемых в быстро развивающемся производ-
производстве эпоксидных смол, получается в промышленном масштабе именно этим
путем. В настоящее время наблюдается повышенный интерес к эпоксидации
дешевых непредельных растительных масел и непищевых животных жиров
с целью получения пластификаторов для виниловых смол и других продуктов.
В качестве окислителя обычно применяются пероксоуксусная или пероксо-
муравьиная кислоты, легко образующиеся in situ или получаемые на отдель-
отдельной стадии из соответствующей кислоты и концентрированной перекиси
водорода.
ЛИТЕРАТУРА
1. U. S. Dept. of Commerce, Bureau of te Census, Facts for Industry—Hydrogen
Peroxide in the War Program, U. S. Government Printing Office, Washington A0.1.
1947); Industry Reports. The Inorganic Chemicals Industry, A Manufacturing Sector
in the 1947 Interindustry Relations Study, BLS Report № 31, U. S. Dept. of Labor,
May 1953.
2. Manufacturing Chemists' Association, Inc., Chemical Facts & Figures, 2nd. ed., Wa-
Washington, 1946.
3. К a u f f m a n n H. О., частное сообщение, 1951.
4. Chemical and Engineering News, разные номера за 1954 г.
5. К а г a b i n о s J. V., В а 1 1 u n А. Т., J. Am. Oil Chemists Soc, 31, 71 A954).
6. Jones G. W., TAPPI, 33, 149 A950).
7. Hoist G., Chem. Revs., 54, 169 A954).
8. NeesH., P. B. report 74329 (frames 1219—1234) A939).
9. Kind W., Melliand Textilber., 29, 57, 96, 133 A948).
10. S 1 a t e г V. W., Richmond K. W., Dyer, 98, 658 A947).
11. D a s D. В., M i t r a M. K., W а г e h a m J. F., J. Textile Inst., 43, T433, T449
A952).
12. Kastens A., Chem. & Engr. News, 31, 3135 A953).
13. Literature Survey of Textile Bleaching, American Dyestuff Reporter, 39, 663, 703, 739,
782, 820 A950).
14. Hydrogen Peroxide, Pennsylvania Salt Manufacturing Co., Philadelphia, Pennsyl-
Pennsylvania.
15. Technical Data Bulletins (разные), Buffalo Electro-Chemical Co., Division of Food
Machinery and Chemical Corp., Buffalo, New York.
16. Technical Data Bulletins (разные), duPont Co., Peroxygen Products Division, Wil-
Wilmington 98, Delaware.
17. Merrill G. R., M а с о r m а с A. R., M a u e г s b e г g e r H. R., American
Cotton Handbook, 2nd ed., Textile Book Publishers, Inc., New York, 1949.
18. О t t E., editor, «Cellulose and Cellulose Derivatives, Interscience Publishers», Inc., New
York, 1943; HeuserE., Chemistry of Cellulose, J. Wiley and Sons, New York,
1944.
19. Bell T. E., Staltcr N. Т., Am. Dyestuff Reporter, 41, 110 A952).
524 Глава И
20. О'В rien R., Hoi brook H. S., U. S. Dept. of Agriculture, Farmers Bulletin,
1947, Government Printing Office, Washington, D. C, 1937.
21. BeemanL. A., Reichert J. S., Peroxides in Pulp Bleaching Processes, Chapter
XII, of The Bleaching of Pulp, TAPPI Monograph Series, №10, Technical Associa-
Association of the Pulp and Paper Industry, New York, 1953.
22. В г е с h t W., J а у m e G., S с h u s t e r G., Das Papier, 4, 174 A950) [C. A., 44,
8648 g].
23. Pulp Bleaching with Hydrogen Peroxide, разные другие технические бюллетени,
Buffalo Electro-Chemical Co., Division of Food Machinery and Chemical Corp., Buffa-
Buffalo, New York, см. также М с E w e n R. L., S h e 1 d о n F. R., NelsonD.H.,
TAPPI, 34, No 5, 193 (Y. 1951).
24. Me E wen R. L., Paper Trade Journal A4. XI. 1952).
25. D о w n s L. E., Bleaching Wood, Report № R1705, U. S. Dept. of Agriculture Forest
Service, Forest Products Lab., Madison, Wise, VII, 1950.
26. S h a n 1 e у E. S., К a u f f m a n n H. О., К i b b e 1 W. H., American Dyestuff
Reporter, 40, 1 A951).
27. L a x e r G., Whewell С S.f J. Soc. Dyers Colourists, 68, 256 A952).
28. F a b i a n C. F., S а с h a n e n A. N., пат. США 2048645 B1.VII. 1936).
29. R u t h e r f о r d H. A., HarrisM., Report of Investigations № 1090, U. S. Na-
National Bureau of Standards, 20, № 4, 559 A938), см. предыдущие отчеты этой серии.
30. Alexander P., Carter D., E a r 1 a n d C, Biochem. J., 47, 251 A950).
31. H u с k e 1 P., Frohlich H. G., Textil-Praxis, 7, 381 A952).
32. Sakaguchi I., О k u M., J. Soc. Textile Cellulose Ind. (Japan), 8, 226 A952)
(выводы на английском языке).
33. Modification of Wool with Peroxygen Compounds, Bulletin №35, Buffalo Electro-
Chemical Co., Div. of Food Machinery and Chemical Corp., Buffalo, New York,
Nov., 1951.
34. Wood F. C, J. Soc. Dyers Colourists, 68, 485 A952).
35. S t i e p e 1 C, Seifensieder Ztg., 53, 291 A926) [C. A., 20, 2422].
36. L a n g e n k a m p P., Z. deut. Oel-Fett-Ind., 45, 621 A925) [C. A., 20, 2758].
37. M a r g u 1 i e s P. H., Rayon and Synthetic Textiles, 32, № 11, 36 A951).
38. Treatment of Cellulose and Cellulose Derivatives with Peroxygen Compounds, Bul-
Bulletin № 43, Buffalo Electro-Chemical Co., Division of Food Machinery and Chemical
Corp., Buffalo, New York, June 1952.
39. Lenzinger Zellwolle und Papierfabrik A. G. & Kunstseide-Ring G. m. b. H.,
белы. пат. 450628 (VI. 1943); Thuringische Zellwolle A. G. & Zellwolle & Kunst-
Kunstseide-Ring G. m. b. H., белы. пат. 451163 (VII. 1943).
40. G 6 m e z P. L., R i v a L. H., Ion, 8, 514 A948).
41. Oxidation of Vat Dyes on Cotton and Synthetics with Hydrogen Peroxide, Bulletin
№ 38, Buffalo Electro-Chemical Co., Buffalo, N. Y., II. 1952.
42. S t a t h e r F., Schubert R., Ges. Abhandl. deut Lederinsts. Freiberg/Sa.,
No 6, 50 A951); StatherF., Konigfeld, ibid., N2 7, 5 A951) [C. A., 46,
5852i].
43. EgertonG.S., J. Textile Inst., 39, T305 A948); Textile Recorder, 18, 659A948).
44. A s h t о n D., С 1 i b b e n s D., P г о b e r t M. E., Soc. Dyers and Colourists, Proceed.
of Symp., IX, 1949, p. 651.
45. S с h a e f f e r A., Textil Rundschau, 6, 489 A951).
46. AndresenM., Phot. Korr., 36, 260 A899); Archiv f. wiss. Phot., 2, 28 A900).
47. Luppo-Cramer H., Phot. Korr., 48, 466 A911).
48. L a n ge O., Chemisch-Technische Vorschriften, Vol. 2, Otto Spamer Verlag, Leipzig,
1923; BelinE., Drouill ard С, герм. пат. 230386 B0.1.1909).
49. Luppo-Cramer H., Phot. Korr., 49, 501 A912); GrosO,, герм. пат. 147131
B5. XI. 1903); Neue Phot. Gesell. A. G., герм. пат. 153769 B5. VII. 1904), 158368
A4. II. 1905).
50. G г о s О., О s t w a 1 d W., J. Soc. Chem. Ind., 22, 379, 380, 963 A903); 23, 1044
A904).
51. Kretschmer P. M., Med. Klin., 41, 381 A946) [C. A., 42, 8836h].
52. Abramson H. A., J. Investigative Dermatology, 15, 19 A950).
53. К r e t s с h m e r P. M., Z. Naturforsch., 3b, 60 A948) [C. A., 43, 1668b]; Woclibl.
Papierfabr., 76, 35 A948) [C. A., 42, 7041e].
54. FreytagH., Z. Naturforsch., 3b, 379 A948) [C. A., 43, 6097h]; 5b, 123 A950)
[C. A., 44, 670П].
55. Schmieschek U., Brit. J. Phot., 77, 276 A930).
56. К i k u с h i S., T о m о d a Y., J. Soc. Sci., Phot. Japan, 10, 55 A945) [C. A., 45,
2343e].
57.W i g h t m a n E. P., Q u i r k R. F., J. Frank Inst., 203, 261 A927); Wight-
man E. P., Brit. J. Phot., 74, 447 A927).
58. В a r n e s С E., W h i t e h о r n e W. R., L a w r a n с e W. A., J. Phys. Chem.,
35, 2637 A931).
Литература 525
59. Smith A., Brit. J. Phot., 13, 226 A866); S p i 1 1 e r J., Brit. J. Phot., 13, 283
A866); Phot. J., II, 58 A866).
60. С г a b t г e e J. I., E a t о n G. Т., M u e h 1 e г L. E., J. Soc. Motion Picture Engrs.,
35, 484 A940); 36, 555 A941).
61. L a t i m e г W. M., HildebrandJ. H., Reference Book of Inorganic Chemistry,
Revised edition, Macmillan, New York, 1940, p. 506.
62. В u t t С. А., пат. США 2398493 A6.IV. 1946).
63. В а 1 1 a r d A. E., Mart i nson L E., пат. США 2364613 A2. XII. 1944).
64. L u i g g i M. F., M a r q u e z M. С, С i s a F. P., Technica у economia (Bahra Blan-
ca, Arg), 11, 69 A952) [C A., 47, 7366a].
65. В г a d у L. J., S u s a n о С. D., L а г s о n С. E., U.S.A.E.C.D. 2366 B1.V.1948);
J а у К- Е. В., Britain's Atomic Factories, Her Majestry's Stationery Office, 1954.
66. R о m i g G. С, пат. США 2326309 A0. VIII. 1943).
67. Stack J. R., пат. США 2286240 A6. VI. 1942).
68. Gibson R. С, пат. США 2617749 (И. XI. 1952).
69. WilsonV. W., Refiner Natiural Gasoline Mfg., 18, 96 A939).
70. Ammoniaque synthetique et derives S. А., белы. пат. 508748 A. V. 1952).
71. M a x t e d E. В., J. Chem. Soc, 1945, 204, 763, 766; 1946, 23; англ. пат. 644239
D. X. 1950); Advances in Catalysis, Vol. 3, Academic Press, New York, 1951, p. 129;
M a x t e d E. В., В a 1 1 G. Т., J. Chem. Soc, 1953, 1509.
72. American Society for Metals, Metals Handbook, 1939 Edition, 1472, pp. 726—735.
73. Buffalo Electro-Chemical Company Inc., Industrial Bulletins № 39 (III. 1952), № 51
(V. 1953).
74. G a u d i n A. M., S p e d d e n H. R., С о г г i v e a u M. P., частное сообщение,
1953.
75. M а с h u W., Wasserstoffperoxyd und die Perverbindungen, 2 Aufl., Springer Verlag,
Wien., 1951, p. 54; [есть перевод первого издания с дополнениями: см. М а х у В.,
Перекись водорода и перекисные соединения, Госхимиздат, М., 1951].
76. П л а к с и н И. А., С у в о р о в с к а я II. А., Изв. АН СССР, отдел, техн. наук,
1949, 407.
77. К i n g С. V., Braverman M. M., J. Am. Chem. Soc, 54, 1744 A932).
78. Kawamura K., Japan Analyst, 2, 347 A953).
79. V a n h а г e n L., Rossier M., Vandeur L, пат. США 2584824 E. И. 1952).
80. H lggins СМ., пат. США 466240B9. XII. 1891); Muller J., пат. США 2173040,
2173041 A2. IX. 1939); N i vl i ng W. А., пат. США 2204615 A8. VI. 1940);
HieronymusR. H., пат. США 2274983 C. III. 1942); К a u I f m a n n H. O.r
пат. США 2276984 A7. III. 1942), 2307684 E. I. 1943); G 1 о b e E. F., пат. США
2513638 D. VII. 1950); The Calico Printers Assn., Ltd., агл. пат. 120183 C1. X.
1918); К e r n о t J. С, англ. пат. 556187 B3. IX. 1943); Societe Anonyme «Trust
Chimique», герм. пат. 134301 D. IX. 1902); Z i 1 1 i с h E., герм. пат. 310388 A1.
I. 1919); К и h 1 H., SoltauG., герм. пат. 522555 A0. IV. 1931).
81. Krajcinovic M., Arkiv Kern., 19, 104 A947); Experientia, 4, 271 A948) [С. А.,
42, 8005 b).
82. Sa i n t-M i e u x С, пат. США 2430481 A1. XI. 1947).
83. Fahrion W., герм. пат. 148796 D. VI. 1902).
84. Feigl F., Spot Tests, 4th ed., translation by Oesper R. E., Elsevier Pub. Co.,
Houston, 1954 [есть перевод: см. Ф а й г л ь Ф., Капельный анализ, Госхимтехиздат,
М.-Л., 1933]; Feigl F., Chemistry of Specific Selective, Sensitive Reactions,
translated by R. E., Oesper, Academic Press, New York, 1949.
85. N a p о n e n G. E., D г i e s M. J., O'B г i e n A. S., Paper Trade J., 125, № 5, 45,
A947); H a d о r n II., J u n g k u n z R., В i e f e r K. W., Mitt. Lebensm. Hyg.,
44, 14 A953); Kleeman В., Z. angew. Chem., 34, Aufsatzteil, 625 A921).
86. Feher F., Heuer E., Angew. Chem., 62, 162 A950).
87. Tschirch E., Chem. Ztg., 65, 193 A941).
88. T p e д в е л л Ф. П., Г о л л В. Т., Курс аналитической химии, Госхимиздат,
М.-Л., 1946.
89. Furman N. Н., editor, Scott's Standard Methods of Chemical Analysis, 5th ed.,
D. Van Nostrand Co., Inc., New York, 1939.
90. W r i g h t E. R., M e 1 1 о n M. G., Ind. Eng. Chem., Anal. Ed., 9, 375 A937).
91. F о s t e r M. D., U. S. Geological Survey Bulletin 950 (Contributions to Geochemi-
Geochemistry 1942—1945).
92. TelepG., BoltzD. F., Anal. Chem., 25, 971 A953).
93. Telep G., Doctoral Dissertation, Wayne Univ., V. 1952.
94. Milner O., Proctor K. L., WeinbergS., Ind. Eng. Chem., Anal. Ed.,
17, 142 A945).
95. NuttenA. J.,StephenW. I., Anal. Chim. Acta, 5, 448 A951) (на английском
языке).
96. Steiger G., J. Am. Chem. Soc, 30, 219 A908).
526 Глава 11
97. В и г d e t t L. W., Anal. Chem., 23, 1268 A951); В а к е г L. C. W., McCut-
cheonT.P., Anal. Chem., 22, 944 A950).
98. U. S. Atomic Energy Comm., Analytical Chem. of the Manhattan Project, Rodden C. J.,.
Ed., McGraw-Hill Book Co., New York, 1950, p. 18; Groenweghe, Industrie chim.
beige, 17, 1060 A952).
99. Васильева А. А. ШвайковаМ. Д., Аптечное дело, № 1, 46 A953).
100. MellesJ. L., deBreeW., Rec. trav. chim., 72, 576 A953) (на английском языке).
101. Chariot G., S a u 1 n i e r J., Chim. anal., 35, 51 A953) [C. A., 47, 4789a].
102. Kolthoff I. M., ParryE. P., Anal. Chem., 25, 188 A953).
103. Rapport R., Foundry, 81, № 8, 115 A953).
104. Houlihan J. E., Farina P. E. L., Analyst, 78, 559 A953).
105. PetersC. A., MacMastersM. M., FrenchC. L, Ind. Eng. Chem., Anal.
Ed., II, 502 A939).
106. N о r w i t z G., С о d e 1 1 M., Anal. Chem., 25, 1428 A953).
107. Busch M., Ber., 39, 1401 A906).
108. Jannasch P., Ber., 39, 3655 A906).
109. FeherF., Ber thol d H. J., Z. anal. Chem., 138, 245 A953).
110. S h e e n К. Т., К a h 1 e r H. L., Ind. Eng. Chem., Anal. Ed., 10, 628 A938).
111. P e r 1 e у G. A., L a n g s d о r f B. F. Т., «Air Pollution», Ed. by L. С McCabe,
McGraw-Hill Book Co., New York, Proc. U. S. Tech. Conf. Air Pollution, 1950
(Pub. 1952).
112. Capitani C, Chimica e industria (Milano), 34, 633 A952).
113. Koib J. J., ToenniesG., Ind. Eng. Chem., Anal. Ed., 12, 723 A940).
114. L u g g G. A., Wright A. S., Australia, Dept. Supply, Defense Research Labs,.
Circ. № 14, 1 A953).
115. E с о с h a r d G., D u v e a u N., Makromol. Chem., 7, 148 A951).
116. К i r j a k k a P., P i h a P., Suomen Kemistilehti, 23B, № 3, 1 A951) (на англий-
английском языке).
117. К р а м а р е н к о В. Ф., Аптечное дело, 2, № 2, 52 A953).
118. ApplemanD., Anal. Chem., 23, 1627 A951); G u t t е г m a n В. М., J. Assoc. Offic.
Agr. Chemists, 35, 181 A952).
119. M a s u r e M. P., D i e t r i с h W. C, Lindquist F. E., BlackwoodL.O,
Food Technol., 7, 363 A953).
120. Vorbrodt W., Bull, intern, acad. polon. sci., Classe sci. math, nat., 1939, BI
JVb 1/5, I (на французском языке) [С. А., 44, 3407 с].
121. HeimannW., Heiman n-G e i e r h a a s A., Z. Anal. Chem., 133, 255 A951).
122. Tucek A., Chemie (Prague), 8, 139 A952).
123. L у s h ol m А., пат. США 2325618, 2325619 C. VIII. 1943).
124. WalterH., Nat'l. Advisory Comm. Aeronautics, Tech. Mem. Jsfe 1170, 1947; Jet
Propulsion, V—VI, 1954, p. 166.
125. Technical Oil Mission, Reel 32, Translated by Charles A. Meyer &. Co., Translation
PC-S-III, New York, Vol. I.
126. Broatch J. D., Fuel, 29, №5, 106 A950).
127. T sc h i n k el J. G., пат. США 2637161 E.V. 1953).
128. Williams G. C, S a t t e r f i e 1 d С N., I s b i n H. S., J. Am. Rocket Soc,
22, 70 (III—IV. 1952).
129. С о 1 e m a n W. F., Kil pa trick M., P. B. report 94005.
130. L u f t N., Heterogeneous Catalytic Decomposition of Flowing Concentrated Hyd-
Hydrogen Peroxide, Fife № B. I. G. S.-175, Joint Intelligence Objectives Agency, Wa-
schington, D. C, 27. XI. 1946; P. B. report 97602.
131. Me К ее L., Mechanical Engineering, 68, 1045 A946).
132. Jane's Fighting Ships, 1950-51, Blackman R. V. В., ed., McGraw-Hill Book Co ,
Inc., New York, 1950.
133. M a x f i e 1 d F. A., J. Am. Rocket Soc, XII. 1949, p. 166.
134. Z u с г о w M. J., Trans. A. S. M. E., 69, 847 A947).
135. Hawthorne R., Aviation. Age, 17, 35 A952).
136. С о 1 1 i n s A. S:, Sc. D. Thesis in Chemical Engineering, M. I. T., 1948.
Iff. Ley W., Rockets, Missiles, and Space Trevel., Viking Press, New York, 1951.
138. Z u с г о w M. J., Principles of Jet Propulsion and Gas Turbines, John Wiley & Sons,.
Inc., New York, 1948.
139. S u t t о n G. P., Rocket Propulsion Elements, John Wiley & Sons, New York, 1949.
140. S e i f e r t H. S., M i 1 1 s M. M., Summerf iel d M., Am. J. Phys. 15, 1, 121
A947).
141. D i p 1 о с k В. R., Lofts D. L., G г i m s t о n R. A., J. Roy. Aeronaut. Soc,
57, № 505, 19 A953) [C. A., 47, 5686a].
142. Беллинджер Ф., Фридман Г., Бауэр В., Истс Дж., в сб. «Физика
и химия реактивного движения», № 1, Госиноиздат, М., 1948, совместно
с Л э д д о м Дж. и Р о с с о м Дж., стр. 132; совместно с Б у л л о м В., стр. 152;
совместно с Эдмондсом С, стр. 175.
Литература 527
143. Barr W. J., Wilson E. J., Jr., J. Chem. Phys., 19, 470 A951).
144. ShanleyE.S., К a u f f m a n n H. О., пат. США 2452074 B6. X. 1948).
145. Hajek V., Hajek J., Research, 4, 186 A951).
146. P. B. report 74413 (frames 2312—2324).
147. К a u f f m a n n H. O., W h a 1 e n J. F., Jr., пат. США 2662825 A5. XII. 1953);
Mflller J., австр. пат. 176493 B6. X. 1953).
148. G u m m i Werke Richterswil A. Q., швейц. пат. 261167 A949); В e t h e E. J., пат.
СЩА 2567988 A8. IX. 1951); К е с к 1 e г N. F., пат. США 2598127 B7. V. 1952);
Eckert С. F., пат. США 2617840 (И. XI. 1952); Baker D. L., пат. США
2540040 C0. I. 1951).
149. Reichert J. S., Sparks W. J., пат. США 1953567 C. IV. 1934) [англ. пат.
433471J, 2051745 A8. VIII. 1936), 2075468 C0. III. 1937); duPont Co., англ. пат.
445768 A4. IV. 1936); Zeelandia H. J., Doeleman N. V., дат. пат.
64925 A949); Kiyohara S., Ashida H., Takagi Т., японск. пат.
174753 (9. VIII. 1947).
150. В a i 1 е у L. H., L е С 1 е г с J. A., Cereal Chem., 13, 119 A936).
151. Kiyohara S., A s h i d a H., T a k a g i Т., японск. пат. 174748, 174749
(9. VIII. 1947).
152. Marks В. М , пат. США 2109595 A. III. 1938); W h i t e F. Т., D a 1 у A. J.,
пат. США 2440318 B7. IV. 1948); В а с о n R. G. R., В u 11 е г Е. Т., I s а а с s E.
and Imp. Chem. Ltd., англ. пат. 586988 (9. IV. 1947); W h i t e F. Т., D a 1 у A. J.,
англ. пат. 578759A0. VII. 1946); I. G. Farbenindustrie A. G., герм. пат. 662121
E. VII. 1938); Ушаков С. Н., Бинкина М. М., Егорова
А. В., Б р о й т м а н И. А., ЖПХ, 13, 1208 A940); Юрженко Т. И., Гро-
Громов а Г. Н., X а й ц е р В. Б., ЖОХ, 16, 1505 A946); W e s t f a h 1 J. С, пат.
США 2588947; 2588948; 2588949 A1. III. 1952); Societe auxiliare de Tinstitut
Fran$ais du caoutchouc, франц. пат. 972455 C0. I. 1951).
153. К h а г a s с h M. S., Jensen E. V., Urry W. H., Science, 102, 128 A945); J.
Am. Chem. Soc, 67, 1864 A945); 68, 154 A946).
154. R i с h а г d s о n B. W., Lancet, 1, 707, 760 A891).
155. W a 1 1 i a n S. S., N. Y. Med. J., 56, 594 A892).
156. R e i d M. R., Altemeier-W. A., Ann. Surg., 118, 741 A943).
157. S 1 a n e t z L. W., В г ow n F. A., J. Dent. Res., 25, 223 A946); Science, 105, 312
A947); J. Dent. Res., 28, 313 A949); В г о w n E. А., К г a b e k W. В., S k i f f i n-
gtonR. E., New England J. Med., 234, 468 A946); Archives Otolaryngology, 48,
327 A948); В г о w n E. A., Tufts Dental Outlook, 20, № 2, 17 A946); Ohio State
Med. J., 42, 600A946); К a v a n a g h F., J. Bact.,54, 761 A947); KunzmannT.,
Fortschr. Med., 52, 357 A934); AspissovS., Zentr. Biochem. Biophys., 16, 280
A913) [C. A., 8, 1947]; KoefoedH., Dansk Tids. Farm., 21, 31 A947) [(C. A., 41,
3175f]; Nambudripad V. K. N., Laxminarayana H., I у а К. К-,
Indian J. Dairy Sci., 2, 65 A949) [C. A., 43, 9159h]; 4, 38 A951) [C. A., 45,
10304h); N о 1 t e W. A., E d w a r d s M. A., G г u b b T. C, J. Am. Pharm.
Assoc, 38, 258 A949); ReinerL, Leonard С S., Proc. Soc. Exper. Biol. Med.,
28, 951 A932); H a a s e L. W., Pharmazie, 5, 436 A950) [C. A., 45, 2148e].
158. T i z z a n о A., Atti accad. Lincei, Classe sci. fis., mat. e nat., (8), 1., 225 A946);
Grubb T. C, E d w а г d s M. A., J. Am. Pharm. Assoc, Sci., Ed. 36, 385
A947); H err его F. J., Arch. farm, у bioquim. Tucuman, 2, 181 A945) [C. A.,
41, 3924e]; В г о w n E. А., К г a b e k W. В., S k i f f i n g t о n R. E., J. Bact.,
53, 793 A947); Am. J. Surg., 77, 630 A949); Journal-Lancet, 67, 405 A947); Brown
E. А., С r u i с k s h a n k G. A., J. Dent. Res., 26, 83 A947); FriedenthalH.,
Biochem. Z., 94, 47 A919) [C. A., 13, 3210]; Bryson V., P. B. report 6298.
159. McCulloch E. C., Disinfection and Sterilization, 2nd. ed., Lea and Febiger,
Philadelphia, 1945.
160. Brown E. A., Bull. New England Medical Center, 7, 274 A945).
161. Campbell D. H., С h e г k i n A., Science, 102, 535 A945); TaubA., Hart F.,
J. Am. Pharm. Assoc, Sci. Ed., 37, 246 A948); Menczel E., J. Am. Pharm.
Assoc, 40, 175 A951).
162. В го wn E. A., Go г i n M. H., A b r a m s о n H. А., пат. США 2430450 A1. XI.
1947).
163. Brown E. A., Ann. Allergy, 4, 33 A946).
164. Brown E. A., Abramson H. A., Gorin M. H., Kauffmann H. O.,
Shanley E. S., J. Am. Pharm. Assoc, Sci. Ed., 35, 304 A946); В г о w n E. A.,
К r a b e k W. В., J. Am. Pharm. Assoc, Sci. Ed., 40, 294 A951); В г о w n E. A.,
К r a b e k W. В., S k i f f i n g t о n R. E., С r u i с k s h a n k G. A., J. Am.
Pharm. Assoc, Sci. Ed., 37, 34 A948); W о о d w а г d W. A., P i с k 1 e s J., Quart.
J. Pharm. Pharmacol., 7, 418 A934); A b г a m s о n H. A., Science, 96, 238 A942);
. В г у s о n V., D e m e г е с M., F г e m о n t-S m i t h F., Reiss A., P. B. report
6298 A946); Steiger K., H e g n a u e r R., Pharm. Acta Helv., 21, 57 A946).
165. CombesF. C, N. Y. State J. Med., 37, 1927 A937).
528 Глава 11
166. Pearl E. N.. S. В. Thesis M. I. T. 1939.
167. Me Leo d J. W., Gordon J., Biochem. J., 16, 499 A922); J. Path.-Bact., 26,
326 A923); BergerU., Z. Hyg. Infektionskrankh., 132, 47 A951) [C. A., 45,
5228e].
168. F u j i о к a J., Nippon Geka Hokan (Arch. Japan. Chir.), 18, 511 A941) [C. A., 42,
4647e].
169. M ul 1 er A., Z. Hyg. Infektionskrankh., 93, 348 A921) [C. A., 16, 2571].
170. Thompson T. L., M e f f e r d R. В., Jr., WyssO., J. Bact., 62, 39 A951).
171. Dittmar H. R.,Baldwin I. L., Miller S. В., J. Bact., 19, 203 A930);
Троицкий М. В., НазаренкоИ В., Журнал микробиологии, эпидемиоло-
эпидемиологии и иммунобиологии, 1946, 68.
172. 'DemerecM., Bertani G., F 1 i n t J., Am. Naturalist, 85, 119 A951); D e-
a n A. C. R., HinshelwoodC, J. Chem. Soc, 1951, 1157.
173. L о с k e A., M a i n E. R., M e 1 1 о n R. R., Science, 88, 620 A938); H a n z 1 i k P.J.,
С u t t i n g W. C, Science, 98, 389 A943).
174. N о r t h e у Е. И., The Sulfonamides and Allied Compouds, Reinhold Publishing
Corp., New York, 1948, p. 506.
175. KeilinD., HartreeE. F, Biochem., J., 42, 221 A948).
176. D о 1 d H., Z. Hyg. Infektionskrankh., 124, 519, 578 A942) [C. A., 37, 6691]; Beth-
ge J.Jschesche R., et al., Z. Naturforsch., 2b, 12A947) [C. A., 41, 7446f];3b,
330 A948) 1С. A., 43, 5079iJ; 6b, 22 A951) [C. A., 45, 6677c]; Thompson R.,
Johnson A., J. Infectious Diseases, 88, 81 A951); Berger U., Z. Hyg.
Infektionskrankh., 133, 371 A952).
177. KojimaS., J. Biochem. (Japan), 14, 95 A931) [C. A., 26, 745].
178. E k m a n В., Acta Physiol. Scand., 8, Suppl. 22 A944); Acta Pharmacol. Toxicol.,
3, 261 A947) 1С. A., 42, 5558a].
179. В г о w n E. A., KelemanG., The Laryngoscope, 56, 556 A946); В г о w n E. A.,
OwenW.E., Arch. Otolaryngology, 43, 605 A946); AagesenW. J., Brown
E. A., Weiss L. R., Eye, Ear, Nose, Throat Monthly, 26, 27 A947).
180. Farrell G. W., Me Nichols W. A., J. Am. Med. Assoc, 108, 630A937);
HaleyP. S., J. Am. Dental Assoc, 26, 612 A939); В г о w n E. A., J. Maine Med.
Assoc, 37, 181 A946); Thurmon F., Brown E. A., J. Investigative Dermato-
Dermatology, 8, 11 A947); Arch. Dermat. Syphil., 55, 801 A947).
181. GettlerA.O., BlaineJ. O., Am. J. Med. Sci., 195, 182 A938); Сидеро-
в а С. Г., Фармакология и токсикология, 7, 39 A944) [С. А., 39, 3843].
182. AbramsonH.A., Bull. New England Med. Center, 8, 97 A946); O'B rienW.B.,
Brown E. A., Pearse H., Am. Review Tuberculosis, 56, 190 A947).
183. В a t e s J. V., Brit. Med. J., II, 154 A945).
184. J e n k i n s J. Т., Am. J. Surg., 74, 428 A947).
185. M a r q u a n d С. В., М с Е 1 г о у О. Е., К е t h 1 е у Т. W., М с N a m a r а В. Р.,
Bowers R. V., Р. В. reports 11188, 13654, 13663 A942).
186. Emmerson J. H., Veterinary Medicine, 43, A948).
187. ButzL. W., Lalande W. A., Jr., J. Am. Pharm. Assoc, 23, 1088 A934);
S с h w a r t z В., Р о r t e г D. A., Trop. Diseases Bull., 34, 876 A937); Oest-
г e i с h e r F., К о k D. J., Acta Brevia Neerland Physiol. Pharmacol. Microbiol.,
10, 22 A940).
188. Spann J., .Forschungsdienst, 5, 472 A938).
189. MilchH., Ann. Surg., 93, 1220 A931); Index Medicus, 25, 558 A939); G w i n n А. В.,
Am. J. Surgery, 65, 430 A944).
190. DuttonW. F., Intravenous Therapy, F. A. Davis Co., Philadelphia, 1924.
191. С u 1 m e г С U., At kinson A. J., I v у А. С, Am. J. Dig. Dis. Nutrition, 4,
219 A937).
192. Rase da Y., Folia Pharmacol., Japan., 15, № 1 A932) [C. A., 27, 136].
193. Lorincz A. L., Jacoby J. J., Livingstone H. M., Anesthesiology, 9,
162 A948).
194. Kisser J., P о r t h e i m L., Phytopath. Z., 7, 409 A934).
195. Pichler F., Phytopath Z., 8, 245 A935).
196. Von Olgyay M.,Z. Pflanzenkrankh. Pfianzenschutz, 46, 1 A936).
197. MullerJ., пат. США 1927988 B6. IX. 1933), 1962996 A2. VI. 1934).
198. S p a r k s W. J., пат. США 2006967 B. VII. 1935).
199. Abad Manrique J. В., Ion, 9, 590 A949).
200. EndersC, Nowak G., Schneebauer F., Pfahlen A., Wochschr.
Brau., 57, 81, 89 A941).
201. Raux J., Brasseur Fran^., 3, 78, 97 A939).
202. BawdenR. F., Am. Soc. Brewing Chem., Proc, II, 10 A946).
203. Seed Treatments with Peroxygen Chemicals, Bulletin № 33, Buffalo Electro-Chemical
Co., Division of Food Machinery and Chemical Corp., Buffalo, New York, VIII.
1951.
204. Anon., Can. Chem. Process, 36, 84 (IX. 1952).
Литература 529
205. Mel s ted S. W., KurtzT., Bray R., Agron. J., 4!, 97 A949).
206. Subrahmanyan V., Harihara Iyer C. R., Rajogopholan R.,
Current Science, 2, 384 A934).
207. Гедройц К. К., Удобрение и урожай, 3, 814, 910 A931).
208. D г о s d о f f M., M i I e s E. F., Soil Sci., 46, 391 A938).
209. D г е е s s e n А., герм. пат. 506597 B6. IV. 1929); W i 11 г a t h H. H., Chem. Ztg., 54,
51 A930); Partmann W., Z. Lebensm. Untersuch. u. Forsch., 94, 246 A952)
[C. A., 46, 7247b]; H jorth-Hansen S., К a r 1 s e n O., Arsberet. Vedkom.
Norg. Fiskerier, 1936, 19 A939) [C. A., 33, 7414]; Martens, Fischwaren-u. Fein-
kost-Ind., 22, 146 A950) [C. A., 47, 8279e]; LuningO.,Z. Nahr. Genussm.,
48, 120 A924) [C. A., 18, 3656].
210. Nobel & Co., герм. пат. 307135 A3. IX. 1919), 309180 A4. XI. 1919).
211. Neumark A. S., Sci. Am. Suppl., 75, 182 A913).
212. M a t u i H., Y a m a d a M., J. Agr. Chem. Soc. Japan, 15, 704, 826 A939) [C. A., 34,
1124].
213. Lowy А., пат. США 1587485 A. VI. 1926).
214. W i 1 s о n J. В., T u г n e г W. R., S a 1 e J. W., Am. Food J., 22, 244, 347 A927).
215. StadlingerH., Kunstdunger u. Leim., 28, 435 A931) [C. A., 26, 1358].
216. RouquetteE., герм. пат. 270239 B2. III. 1912); BonjeanE., Compt. rend.,
140, 50 A905); Courmont J., Nogier Т., Rochaix M., Compt. rend.,
150, 1453 A910).
217. Reichel H., Z. Hygiene, 61, 49 A908) [C. A., 3, 466].
218. W о о 1 d г i d g e H. В., J. Inst. Brewing, 22, 437 A916).
219. Heinrich M., Landwirtsch. versuchsstat., 88, 399 A916).
220. Sannino F. A., Boll. chim. farm., 55, 392 A916) [C. A., 10, 2483].
221. Greensp a n F. P., Mac к el 1 а г D. G., Food Tech., 5, 95 A951).
222. G а г i n o-C a n i n a E., Am. accad. agr. Torino, 87, 335 A943) [C. A., 41, 4887d];
Por t el e K., Wiener Landw. Ztg., 65, 311 A915).
223. Герм. пат. 200395; С h a u v i n А. С, Moniteur Scientifique D), 23, II, 567 A909);
24, 1, 12 A910); Ann. Chim. anal, appl., 17, 258A910); F e г г ё L., Ann. fals., 13, 475
A920) [C. A., 15, 3362]; Venezia M., Ann. sper. agrar (Rome) [N. S.]
5, 859 A951), [C. A., 46, 2233g]; В е г t о n i L. J., Rev. fac. cienc. quim., 23, 37
A948) [C. A., 48, 65981].
224. Кг a use A.; Witkowska A., Bull. soc. amis sci. lettres Poznan, BU, 100
A951) (на английском языке) [С. А., 46, 7414h].
225. U n g e m а с h A. G., Els. Conserven-Fabrik Import Ges., герм. пат. 298133 C0. V.
1917).
226. M. E 1 b, герм. пат. 157626 B. I. 1905).
227. Заявка на герм. пат. D26130, кл. 12 q.
228. V i 1 1 о n A. H., Bull. Soc. Chim. (France), C), 9, 749 A893).
229. Buffalo Electro-Chemical Co., Division of Food Machinery and Chemical Corp., Data
Sheet 27 A950); v a n d e г М е е г W. А., голл. пат. 60862 A5. III. 1948).
230. А г n d t W., H a n s e n S. & Son, Ltd, англ.. пат. 548889 B8. X. 1942).
231. A 1 m q u i s t H. J., пат. США 2655449 A3. X. 1953); M u 1 v a n у Н. А., пат. США
2668772 (9. II. 1954).
232. Idler D. R., MacLeod R. A., Fisheries Research Board Can., Progr. Repts.
Pacific Coast Stas., № 95, 56 A953) [C. A., 48, 2283b).
233. V i г m a n i R. S., T a n d о n G. L., Indian J. Hort., 7, № 2, 1 A950) [C. A.,
48, 1600i].
234. BaldwinR.R., Campbell H. A., Thiessen R., LorantC. J., Food
Tech., 7, 275 A953); ScottD., J. Agr. Food Chem., 1, 727 A953).
235. Baker D. L., пат. США 2635069 A4. IV. 1953).
236. Am berg S., J. Biol. Chem., 1, 219 A906).
237. AnselmiS., Ann. chim. applicata, 36, 321 A946) [C. A., 41, 4862i].
238. Singer G., Archiv. Hygiene, 86, 263 A917); Office Intern. Hyg. Publique, 19,
1395 A927).
239. ChickH., Centr. Bakt., 7, B), 705 A901).
240. Rosam A., Centr. Bakt., 8, B) 739 A902).
241. BuddeCCL, Milch Ztg., 32, 690 A903); 33, 359 A904).
242. Morandi L., Chimica e industria, 25, 45 A943) [C. A., 38, 5988].
243. D a h 1 e n M. А., С г о s s 1 e у М. L., Quartermaster Corps., Hydrogen Peroxide of
High Purity and Its Use for Sterilization of Milk, U. S. Department of Commerce,
P. B. report 31003 A945).
244. S a t t a E., M о г a n d i L., M о g g i D., Medicina e. Biologia, 3, 333, 441 A943).
245. R о m a n i В., Chimica e industria, 29, 143 A947) [C. A., 42, 2687i].
246. Sanders G. P., S a g e г О. S., J. Dairy Sci., 32, 166 A949).
247. Heinemann P. G., J. Am. Med. Assoc, 60, 1603 A913).
248. R os a t i Т., Riv. ital. igiene, 4/5, 455 A945); Chimie & industrie, 58, 73 A947)
[C. A., 42, 34971].
34 Перекись водорода
530 Глава 11
249. G e n t i 1 i G. В., R о m a g n о 1 i A., Boll. soc. ital. biol. sper., 25, 93 A949) [C. A.,
45, 6313h].
250. Monaci V., Boll. ist. sieroterap. milan., 28, 357 A949) [C. A., 44, 4593e].
251. ArnaudiC, TreccaniV., Intern. Dairy Congr., Proc. 13th Congr. (Hague),
2, 403 A953) [C. A., 47, 107561].
252. G i ol i t t i G., Atti soc. ital. sci. vet., 3, 543 A949) [C. A., 46, 4137i].
253. Previtera A., Atti regia accad. fisiocrit. Siena, Sez. med.-fis., 13, 89 A945)
[C. A., 43, 3115c].
254. С u г r a n H. R., E v a n s F. R., L e v i t о n A., J. Bact., 40, 423 A940).
255. Green ban к G. R., J. Dairy Sci., 23, 725 A940).
256. CiminoG., Atti regia accad. fisiocrit. Siena, Sez. med.-fis., 13, 19 A945) [C. A.,
43, 3115a].
257. Fuchs W., Proc. 13th Intern. Dairy Congr. (Hague), 3, 1018 A953) [C. A., 48,
4136 c].
258. NambudripadV. K. N., LaxminarayanaH., Iya К. К., Indian J.
Dairy Sci., 5, 135 A952) [C. A., 48, 2940 f].
259. BisogniG., CalendoliG., Boll. soc. ital. biol. sper., 18, 322 A943) [C. A.,
41, 538i].
260. В el 1 R. W., M u с h a T. J., J. Dairy Sci., 32, 827, 833 A949).
261. W i n g e г L. Т., пат. США 2596753 A3. V. 1952), 2622983 B3. XII. 1952); Rei-
с h e г t J. S., McAllister R. W., пат. США 2125398 B. VIII. 1938); Rei-
ch er t J. S., Me Al 1 i s t er R. W., Hinega r d n er W. S., пат. США 2053740
(8. IX. 1936].
262. К r u к о v s к у V. N., G u t h r i e E. S., J. Dairy Sci., 28, 565 A945); 29, 293 A946).
263. OlsonF.C, BrownW.C, J. Dairy Sci., 25, 1027 A942).
264. PontE. G., J. Dairy Research, 19, 316 A952) [C. A., 47, 4003i].
265. К r u к о v s к у V. N., J. Dairy Sci., 32, 163 A949); 35, 21 A952).
266. Steinman H. G., DawsonC. R., J. Am. Chem. Soc, 64, 1212 A942).
267. С h i 1 s о n W. H., M a r t i n W. H., P a r r i s h D. В., J. Dairy Sci., 32, 306 A949).
268. G г e e n b a n к G. R., J. Dairy Sci., 31, 913 A948).
269. PattonS., J о s e p h s о n D. V., Science, 118, 211 A953).
270. W e i n s t e i n B. R., T г о u t G. M., J. Dairy Sci., 34, 559 A951).
271. Anon., Food Processing, 14, № 3, 58 A953); Dairy World, 32, № 10, 4 A953).
272. M о г r i s A. J., L a r s e n P. В., Johnson J. D., Food Eng., 24, 167 A952);
Can Dairy Ice Cream J., 32, № 3, 72 A953); Armour and Co., Bulletin EC-10 A953);
Arnaudi C, Mondo latte, 1949, 271, 273; T г е с с a n i V., Ann. microbiol., 5, 17
A952).
273. N egret ti F., Mondo latte, 1952, 10, 13, 17, 19, 21.
274. E r n s t R. L., G u t m a n F., J. Soc. Leather Trades' Chemists, 34, 454 A950).
275. LangeW., F о 1 z e n 1 о g e n R. G., пат. США 2473154 A4. VI. 1949].
276. T а у 1 о г Н. V., Industrial Chemist, 29, № 336, 9 A953).
277. Buffalo Electro-Chemical Co., Division of Food Machinery & Chemical Co., Technical
Bulletin on Epoxidation and Hydroxylation A950); Electrochemicals Dept., duPont
Co., Epoxidation and Hydroxylation with Organic Peracids, 1954.
Глава 12
НЕОРГАНИЧЕСКИЕ ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ
Известно большое число неорганических производных перекиси водорода,
в том числе перекисей металлов и неметаллов, пероксокислот и их солей, кото-
которые все содержат кислород в так называемой активной или способной к выде-
выделению форме. Перекиси и пероксокислоты можно рассматривать как производ-
производные от Н2О2, получаемые замещением одного или обоих атомов водорода атома-
атомами металла или кислотными радикалами. Так, при замене первого атома водо-
водорода атомом натрия получается NaOOH (гидроперекись натрия, термически
малостойкое соединение); замещением обоих атомов водорода получают NaOONa
или обыкновенную перекись натрия. Аналогично замещение одного или обоих
атомов водорода сульфокислотным радикалом—SO3H дает пероксомоносерную
кислоту HSO3OOH и пероксодисерную кислоту HSO3OOSO3H.
Неорганические продукты замещения перекиси водорода менее изучены,
чем их родоначальник или соответствующие органические производные. Связь
между неорганическими перекисями и перекисью водорода кратко рассмотрена
в гл. 7, а приводимый ниже обзор, посвященный образованию и химии этих
соединений, дает представление о современном состоянии знаний в этой обла-
области. Мы не пытались дать здесь исчерпывающий материал; более подробно см.
у Прайса [1], Неймарка [2], Маху [3], Йоста и Рассела [4], Слейтера 15],
Яра [6] и Шенли, Джильберта, Реймонда и Клейнберга [7]. Органические пе-
перекиси в данной монографии не рассматриваются [если не считать отдельных
замечаний, сделанных в гл. 7 (стр. 319 и ел.)].
КЛАССИФИКАЦИЯ ПЕРЕКИСНЫХ СОЕДИНЕНИЙ
Взаимосвязь между перекисями и другими кислородными соединениями
Напомним, что истинные перекиси обязательно должны содержать груп-
группу —О—О— и что часто отдельные окислы называют перекисями, хотя в стро-
строгом смысле слова они ими не являются, так как не содержат кислородного мо-
мостика. Так, серебро образует обыкновенную окись Ag2O, а также соединение
черного цвета AgO, которое в старых справочниках иногда называется «пере-
«перекисью серебра», причем часто формулу его удваивают до Ag2O2, чтобы подчерк-
подчеркнуть сходство с перекисью водорода. Однако показано, что в это соединение
входит двухвалентное серебро, причем AgO не может быть получено непосред-
непосредственно из перекиси водорода, а также не образует перекиси водорода при
обработке кислотой. Двуокись свинца и двуокись марганца также иногда не-
неправильно называют перекисями. При действии на них разбавленной соляной
кислотой эти соединения выделяют хлор, а не перекись водорода, как этого
следовало бы ожидать, если бы они представляли собой истинные перекиси*.
Атомы свинца и марганца в этих соединениях четырехвалентные, а не двух-
двухвалентные (как было бы в случае перекисной структуры).
* Эта проба на присутствие группы —О—О— требует в известной мере осторож-
осторожного подхода; например, при действии концентрированной соляной кислоты на перекись
бария получается некоторое количество хлора.
34*
532 Глава 12
Образование каким-либо элементом перекиси с соответственным увеличе-
увеличением относительного количества связанного им кислорода (по сравнению с со-
содержанием кислорода, присущим нормальным окислам этого элемента) не
означает повышения состояния окисления этого элемента сверх допускаемого
по положению его группы в периодической системе Д. И. Менделеева. Так,
в перекиси ТЮ3-2Н2О валентность титана равна +4 (т. е. та же самая, что
и в ТЮ2). Точно так же в случае перекиси хрома СЮ5 наряду с двумя перекис-
ными группами О*~ имеется также обычный атом О2", а поэтому хром сохраняет
валентность Сгв + (т. е. ту же самую, что и в СгО3); правда, аутентичность
этого пероксосоединения нельзя считать вполне доказанной.
Чтобы то или иное соединение можно было бесспорно классифицировать
как перекись металла, оно не только должно содержать группу —О—О—, но
еще и относиться к перекиси водорода так, как соль относится к кислоте,
производной которой эта соль является. Перекиси металлов содержат ион О^",
тогда как такие соединения, как AgO, PbO2 или МпО2, все содержат ион О2"
с двумя отрицательными зарядами. Кроме этих ионов, некоторые соединения
сильно электроположительных металлов содержат и ион надперекиси, напри-
например NaO2, КО2, а возможно, и ВаО4. Кислота—родоначальник этих соединений
НО2 до сих пор не выделена.
В гл. 6 структура этих соединений рассматривается более глубоко с точки
зрения электронной конфигурации. Одним из способов, которым могут быть
охарактеризованы различия между ними, являются межатомные расстояния
у атомов кислорода в соединениях разных классов. Результаты рентгеногра-
рентгенографических и электронографических измерений, описанных на стр. 248, показы-
показывают, что во молекуле Н2О2 межатомное расстояние в группе —О—О— равно
1,49 ± 0,01 А. Изменение характера группы—О—О— при переходе от перекиси
водорода к другим перекисным соединениям состоит в основном в изменении
степени электроотрицательности или ионного характера соединения; сама
связь между атомами кислорода практически при этом не изменяется. Таким
образом, в перекисях металлов (или других перекисных соединениях) расстоя-
расстояние —О—О—, очевидно, заметно не отличается от этого расстояния в переки-
перекиси водорода. К такому заключению приводят измерения межатомных расстоя-
расстояний в таких перекисях, как перекись бария [8, 9], перекиси кальция и строн-
стронция [9], пероксодисульфаты аммония и цезия [10], диметилперекиси [11] и ди-
бензоилперекиси [12]. На основании некоторых ранее проведенных измерений
[13, 14] считали, что длина связи—О—О—в перекиси бария меньше, чем в пе-
перекиси водорода, но результаты последующих измерений межатомного рас-
расстояния в перекиси бария [8, 9], а также приведенные выше данные для дру-
других пероксосоединений, по-видимому, опровергают это меньшее значение
длины связи. Тем не менее кажется несколько странным, что увеличение степени
смещения заряда к перекисной группе при переходе от перекиси водорода
к такому чисто ионному соединению, как перекись бария, не оказывает влия-
влияния на длину связи —О—О—. Возможно, что при дальнейших более точных
измерениях обнаружится существование заметных, хотя и небольших раз-
различий.
У иона надперекиси, имеющеговодну трехэлектронную связь, расстояние
—О—О— становится равным 1,29 А [15];о у кислородной молекулы с двумя
трехэлектронными связями оно равно 1,21 А [16]. У озона оно почти такое же,
как у иона надперекиси, а именно 1,28 к [17]. Эти изменения межатомного
расстояния по сравнению с длиной связи —О—О— в перекисях обусловлены
существенным различием в отношении характера связи между атомами
кислорода.
Имеются некоторые данные, доказывающие, что более тяжелые и крупные
атомы щелочных металлов, особенно рубидий и цезий, способны к образованию
перекисей типа МаОд, которые занимают промежуточное положение между
Классификация переписных соединений
533
типами соединений М2О2 и МО2 [7]. Однако фактически эти соединения пред-
представляют собой, вероятно, лишь сочетания надперекисей и перекисей.
Итоговые данные по известным перекисям
В табл. 88 и 89 перечислены известные перекиси и надперекиси элементов
в порядке положения, занимаемого этими элементами в периодической системе.
Хотя, строго говоря, надперекиси принадлежат к совершенно отдельному клас-
классу соединений, все же целесообразно рассмотреть их вместе с перекисями ввиду
аналогии, существующей между способами образования и реакциями соедине-
соединений обоих типов. Ниже приведены дальнейшие подробности, касающиеся ха-
характеристики надперекисей.
В связи с тем, что соединения щелочных и щелочноземельных металлов
имеют очень большое значение, они выделены в отдельную табл. 88. Табл. 89
с совершенной очевидностью доказывает, насколько еще не полны наши зна-
знания по перекисям многих других элементов. В этой таблице существование
пероксокислот различных элементов групп III—VI или их солей (при отсут-
отсутствии выделенных перекисей) обозначено буквой а со скобкой при символе
элемента, например О>. В эту категорию входят и предположительные пероксо-
кислоты кремния, германия и тория. По группе V,a имеются данные о суще-
существовании перекиси протактиния [19].
Таблица 88
Неорганические перекиси и надперекиси" I и II групп периодической системы
Д. И. Менделеева
Группа I
а б
Н2О2; (НО2)а)
Li2O2; (LiO2)
Na2O2; NaO2
К2О2;(К2Оз);КО2;(КО8)а> (CuO2H2O)
Rb2O2; (Rb2O8); RbO2
Cs2O2; (Cs2O3); CsO2
(NH4JO2
a
(MgO2.*H2O)
CaO2; (CaO4)
SrO2; (SrO4)
BaO2; (BaO4);
K2Ba(O2JO2
Группа II
6
(ZnO2)
(CdO2)
(Hg2O2); (HgO.,)
Соединения в скобках представляют такие вещества, в существовании которых имеются сомнения,
и такие, которые еще не выделены в чистом виде. Наличие значительного числа таких соединений доказыва-
доказывает, насколько еще несовершенны современные знания по химии перекисей. В эту таблицу не включены гидро-
гидроперекиси, например NaOOH. Соединение КОз, которое, по Уоли и Клейнбергу [18], образуется путем
реакции озона с едким кали, предположительно содержит ион О~3и поэтому представляет степень окис-
окисления выше надперекиси. [Впервые в чистом виде соединение КОз выделено И. А. Казарновским с сотруд-
сотрудниками; см. ДАН СССР, 64,69 A949).—Прим. ред.]
Гайсинский [20] указывает, что, за небольшими исключениями, элементы
с электроотрицательностью ниже 2,1 (т. е. ниже величины, присвоенной водо-
водороду), по-видимому, способны к образованию перекисных соединений. Однако
некоторые элементы с большей электроотрицательностью, например углерод,
азот, сера и ртуть, также образуют (или, вероятно, образуют) перекисные соеди-
соединения. Эти наблюдения, возможно, послужат основой для выработки определен-
определенного правила, которое дало бы возможность отличать истинные перекисные
соединения от мнимых.
534
Глава 12
Таблица 89
Неорганические перекиси III—VIII групп периодической системы Д. И. Менделеева
III
а б
<Y4O9/iH2O)
(La4O9nH2ON)
или .
(La2O5/zH2ON)
VI
а б
(SO«)
(СгО5)
Sea>
Моа>
Wa>
UO4-2H2Ob)
IV
а б
Са>
Si»
ТЮ82Н2О
(ZrO8-2H2O)
(SnOa-/iH2O)
НЮ8-2НаО
ThO82H2O
VII
a 6
1(СЮ4J]
(Re2O8) ((Ю4Ц
V
a 6
(NO3);(NO4);(N2O7)
pa)
Va)
Nba)
Taa>.
VIII
(FeO2); (Fe2Oe);
(CoO2 *H2O);
(NiO2*H2O)
а) Эти элементы образуют периоксокислоты, но перекиси их еще не выделены.
б) Из редкоземельных элементов Се дает СеО8«2Н2О, обозначаемый, так же как Се(ОН)з*ООН;
Nd, Pr, Sm дают водные соединения, аналогичные производным лантана.
в> Так же NpO4.4H2O, PuO4*H2O.
Пероксигидраты
Характер связи перекисной группы в ряде перекисных соединений являет-
является сомнительным; другими словами, неизвестно, чистая ли это перекись с ха-
характеристической связью —О—О— (содержащая еще, может быть, молекулы
гидратной воды) или структура, принадлежащая нормальному соединению, со-
содержащему «присоединившуюся перекись водорода» или (в твердом состоянии)
«кристаллизационную перекись водорода». Например, углекислый натрий
образует продукты присоединения с перекисью водорода, на которых мы более
подробно остановимся ниже, причем одно из них по составу приближается
к формуле 2Na2CO3-3H2O2, имеет вид белого порошка и вполне устойчиво
в обычных условиях. Эти соединения не являются солями таких истинных
пероксокислот, как гидратированная пероксоциркониевая или пероксотитано-
ваякислота (H2ZrO4H2O или Н2ТЮ4-Н2О). Положение осложняется еще тем,
что истинные перекиси могут содержать ассоциированную кристаллизацион-
кристаллизационную перекись водорода, а также игидратную воду. Например, в соединении со
структурой Li2O2-H2Oa-3HaO имеется истинная перекись металла, кристал-
кристаллизующаяся с тремя молекулами гидратной воды и одной молекулой кристал-
кристаллизационной перекиси водорода. Аналогичный случай можно привести с так
Получение неорганических перекисей и надперекисей 535
называемым техническим «перборатом натрия», обычно изображаемым форму-
формулой NaBO2H2O2-2H2O, если не считать того, что в этой структуре перекис-
ная группа представлена только кристаллизационной перекисью водорода.
Такого рода аддитивные соединения перекиси водорода, содержащие также
гидратную воду, можно называть гидратами пероксигидратов.
При взаимодействии устойчивых истинных перекисных соединений, напри-
например пероксодисульфата калия K2S2O8, с раствором йодистого калия при рН
от 7,5 до 8,0 выделяется не кислород, а йод. Наоборот, аддитивные соединения,
содержащие кристаллизационную перекись водорода, и неустойчивые перекис-
ные соединения с группами, легко гидролизуемыми до перекиси водорода
и нормальной кислоты (например, —О—ОН+Н2О-^—ОН+Н2О2), в этих
условиях не выделяют йод; вместо этого образовавшаяся перекись водорода
разлагается с выделением кислорода. Эти реакции были использованы в про-
пробе, предложенной, чтобы различить истинные перекисные соединения и соеди-
соединения аддитивного типа. Иногда эта проба называется пробой Ризенфельда—
Либхафского. Партингтон и Фаталлах [21] недавно высказали сомнения в до-
достоверности результатов этой пробы. Вероятно, результат пробы можно
считать надежным, если при анализе обнаруживается выделение йода; однако,
если получается отрицательный результат (т. е. выделяется кислород, а не
йод), остается сомнение в том, имеем ли мы дело с легко гидролизуемым пере-
кисным соединением или соединением, содержащим кристаллизационную
перекись водорода.
Ниже приводятся соединения, в отношении которых имеются данные об
образовании пероксигидратов в безводном или гидратированном состоянии:
Na2SO4 [22-25]; KF [22,23]; NaBCX [26,27]; (NH4J SO4, A12(SO4K, NaOOCCH3,
MA1(SO4J [28]; Na2Si03 [29, 30]; Na3PO4, Na2HPO4, NaH2PO4, Na(NH4)HPO4,
Na4P2O7 и аналогичные калиевые соли [24, 31—33]; Mg(NH4)As04, Na2HAs04,
K4Sb207 [24]; (NH4JCO3, Cs2CO3, MgOMgCO3, BiONO3 [34]; Na2CO3 [7, 35,
36]; NH3 [37]; CO(NH2J [38]; многие другие амины [39, 40]; маннит, пинакон
[40] NaO2H (см. в работе [3] на стр. 227). Дополнительные данные по таким со-
соединениям приводятся выше (на стр. 248) при рассмотрении многокомпонентных
систем, содержащих перекись водорода, и на стр. 309 при классификации реакций
перекиси водорода. Аутентичность некоторых таких соединений является сом-
сомнительной, так как доказательства обычно основаны на результатах анализа
твердых соединений, полученных кристаллизацией из смесей перекиси водоро-
водорода и растворов соответствующих солей; фазовых диаграмм для этих соедине-
соединений определено очень мало [24]. Эти соединения обычно представляют белые
сыпучие твердые вещества, умеренно растворимые в воде; их растворы дают
реакции перекиси водорода. Из всех этих веществ только небольшое число полу-
получается в промышленном масштабе и находит практическое применение, напри-
например боратное производное, пероксигидрат углекислого натрия, пероксигидрат
пирофосфорнокислого натрия и пероксигидрат мочевины. Последнее соединение
часто называют «гиперолом» или «твердой перекисью водорода».
ПОЛУЧЕНИЕ НЕОРГАНИЧЕСКИХ ПЕРЕКИСЕЙ И НАДПЕРЕКИСЕЙ
Общие методы, ведущие к образованию перекисей
Наиболее подходящим методом получения перекиси или надперекиси
определенного металла является один из следующих трех: 1) нагревание метал-
металла или окиси его в кислороде или на воздухе; 2) окисление в жидком аммиаке;
3) реакция соединения металла с перекисью водорода, обычно в присутствии
добавленной щелочи.
По первому из этих методов металл с избытком кислорода или воздуха
нагревают до необходимой температуры. Так, например, при нагревании метал-
металлического натрия в сухом воздухе приблизительно до 300° он превращается
536 Глава 12
в двухступенчатом процессе с 95%-ным выходом в светло-желтую перекись
Na2O2. Если к спиртовому раствору этой перекиси, охлажденному до темпера-
температуры льда, добавить концентрированную минеральную кислоту, можно полу-
получить гидроперекись NaOOH или пероксигидрат ее NaOOH-^ H2O2. Аналогич-
Аналогичное нагревание лития в кислороде дает нормальную окись Li2O —белое твердое
тело, содержащее лишь следы перекиси; калий, рубидий и цезий образуют
в этом случае надперекиси, из которых КО2 имеет оранжево-желтый цвет,
a RbO2 и CsO2 описываются в различных источниках как красновато-желтые
или темно-коричневые вещества.
Из элементов второй группы для бериллия образование перекиси с опреде-
определенностью не доказано; из щелочноземельных металлов только стронций
и барий дают перекиси при нагревании соответствующего металла или окиси
его с кислородом под атмосферным давлением*. Образуются также следы пере-
перекиси кальция, но лишь при взаимодействии окиси с кислородом при высоком
давлении и низкой температуре (см. ниже).
Доказано, что реакция
2SrO-+O2 -» 2SrO2 A)
обратима при давлениях 200—250 am и температурах 350—400° [42]. Обрати-
Обратимость реакции окиси бария с кислородом
2ВаО + О2 ->2ВаО2 B)
при 500—550° была в свое время использована в процессе разделения кисло-
кислорода и азота воздуха (процесс Брина); направление реакции можно регулиро-
регулировать по желанию, изменяя давление газа, находящегося в контакте с твердой
фазой при этой температуре.
Второй общий метод отличается от первого тем, что осуществляется окис-
окисление растворов щелочных или щелочноземельных металлов в жидком аммиаке
при температуре около —33° или ниже. Например, раствор металлического
натрия в жидком аммиаке может быть превращен действием кислорода в пере-
перекись Na2O2; из других щелочных металлов калий, рубидий и цезий аналогич-
аналогично дают перекиси в качестве первой стадии, ведущей к образованию надпере-
надперекиси. Литий, а из щелочноземельных металлов—кальций, магний и бериллий
совершенно не реагируют по этому методу или же претерпевают лишь неполное
превращение. Несомненно, неспособность атомов лития и бериллия к образо-
образованию соответствующих перекисей в этих условиях должна быть приписана
небольшому размеру атомов этих металлов.
Перекиси некоторых металлов, не образующиеся в обычных условиях пу-
путем прямого взаимодействия металла с кислородом (например, перекись
лития), можно получить по третьему методу, т. е. действием перекиси водорода
на концентрированные водные растворы, содержащие ион металла. Так, можно
получить твердую Li2O2-H2O2-3H2O при добавке 30%-ной перекиси водорода
к концентрированному раствору гидроокиси лития с последующим разбавле-
разбавлением смеси спиртом. Из пероксигидрата путем высушивания его над пяти-
* Давление диссоциации SrOa достигает 760 мм рт. ст. при 357°. По имеющимся
данным, твердая окись стронция при 400° и 100 am присоединяет только около 15%
теоретического количества кислорода, необходимого для превращения в перекись [41К
но если применять раствор окиси стронция в расплавленной едкой щелочи и пропускать
через него кислород под давлением, образование перекиси может быть практически пол-
полным (см. работу [3], стр. 249). Выщелачиванием охлажденного плава спиртом растворяют
щелочь, в остатке же получают слаборастворимую перекись стронция. Как указано
ниже, для получения довольно чистой перекиси можно использовать реакцию окиси
стронция с кислородом в условиях повышенных давлений и температур.
** Если применять более концентрированный водный раствор перекиси водорода,
то смесь со спиртом может быть очень чувствительной к взрывчатому разложению (см.
стр. 156).
Получение неорганических перекисей и надперекисей 537
окисью фосфора в вакууме можно получить безводную перекись Li2O2. Однако
перекись бериллия по этому методу получить нельзя; магний дает продукт,
содержащий смесь некоторого количества перекиси с большим количеством
гидроокиси. Из соединений других щелочноземельных металлов можно по-
получить гидратированные соединения (например, СаО2-8Н2О, SrO2-8H2O
или ВаО2-8Н2О, которые при дегидратации превращаются в безводные соеди-
соединения*).
В литературе встречаются противоречивые высказывания относительно
существования окислов промежуточного типа М*О3 или М2 О3 (например,
К2О3, Rb2O3 или ВаО3), которые, по некоторым данным, образуются при нагре-
нагревании надперекиси или при действии кислорода на растворы соответствующих
металлов в жидком аммиаке. В последнем случае предполагается, что окисле-
окисление металла, например калия, проходит через ступени, представленные форму-
формулами К2О2, К2О3 и КО2. Жоанни [43] окислял металлический калий, раство-
растворенный в жидком аммиаке, пропусканием тока кислорода через раствор до
тех пор, пока осадок не принимал определенной кирпично-красной окраски
[но до изменения этой окраски в желтую (КО2)]. Де-Форкран [44] сообщил, что,
нагревая надперекись калия КО2 при 480° и давлении 1 мм рт. ст., он получил
продукт, формула которого соответствует чистому К2О3.Жобер [45] заявил, что
он получил то же соединение путем медленного регулируемого окисления
сплава калия со свинцом, оловом или натрием в условиях медленного нагре-
нагревания при постепенной подаче воздуха. Однако химическую индивидуальность
этих промежуточных окислов нельзя считать полностью доказанной, и очень
вероятно, что они в действительности являются простыми смесями перекисей
и надперекисей; правда, некоторые авторы предпочитают рассматривать их как
соединения определенного стехиометрического состава, выражаемого такой
формулой, как Rb2O2-2RbO2. Сейб и Клейнберг [46] описали смешанный оки-
окисел, состав которого изображается формулой К2Ва(О2JО2. Рентгеновское
исследование смешанных окислов рубидия и цезия привело к установлению
для этих соединений формул Rb4Oe и Се4Ов [47]. Поскольку отсутствуют доказа-
доказательства, что такие соединения содержат ион Of", эту формулу «полуторной
окиси» следует считать сомнительной.
Получение надперекисей
Как указано выше, реакция металлического калия, рубидия или цезия
с кислородом при нагревании в условиях атмосферного давления или в рас-
расплаве с азотнокислым калием протекает бурно и дает непосредственно надпере-
надперекиси, например КО2. Технический метод получения надперекиси калия заклю-
заключается в следующем: расплавленный металл распыляют в камере, содержащей
смесь кислорода с азотом A3—35% О2), в таких условиях, что получаемый
продукт быстро затвердевает в виде легкого порошка. Окисление натрия этим
путем, как правило, останавливается на стадии перекиси Na2O2. Надперекись
калия, рубидия и цезия можно получить также путем быстрого окисления
этих металлов, растворенных в жидком аммиаке [48]. Недавно проведенные
работы [49, 50] показали, что натрий тоже способен образовать надперекись
NaO2, взаимодействуя с кислородом при температуре около —77° в растворе
в жидком аммиаке. При повышении температуры это соединение превращается
в перекись и кислород. Предполагается [49], что в этих условиях при —78°
возможно образование также надперекиси лития, хотя последняя еще и не
выделена [49, 51]. Опубликованы данные о существовании надперекисей каль-
* Недавно сообщалось [9] о получении небольших монокристаллов СаО2-8Н2О путем
диффузии газообразного аммиака в раствор азотнокислого кальция,1 содержащий пере-
перекись водорода.
538 Глава 12
ция и бария [48]. В продуктах реакции 30%-ной перекиси водорода с гидрати-
рованными перекисями бария и кальция установлено наличие 8—9% надпере-
кисей этих металлов, однако совершенно чистые соединения СаО4 и ВаО4,
вероятно, еще не выделены.
Надперекиси отличаются от соответствующих перекисей легкостью выделе-
выделения кислорода при введении в воду с температурой льда. В аналогичных
условиях в отсутствие катализатора перекиси не разлагаются. Нагревание
сухой надперекиси калия не сопровождается заметной потерей кислорода
вплоть до достижения температуры плавления, лежащей приблизительно при
400°. Надперекись натрия, по-видимому, устойчива лишь при низких темпера-
температурах.
Подобно перекисям металлов, которые можно рассматривать как соли
кислоты—родоначальника Н2О2, надперекиси можно считать солями неустой-
неустойчивой кислоты НО2. В свободном виде эта кислота спонтанно разлагается на
перекись водорода и кислород:
2НО2-> Н2О2 + О2. C)
Вероятно также, что реакция озона с твердым едким кали, при которой обра-
образуется желтовато-коричневая твердая пленка, обусловлена образованием над-
надперекиси калия
О3 + 2КОН -> 2КО2 + Н2О. D)
Джильберт [52] описал несколько необычную бурную реакцию между над-
перекисью калия и металлическим калием. Корка из окислов калия толщиной
6—12 мм, образовавшаяся при постепенном действии атмосферного кислорода
на бруски этого металла в процессе их хранения, вызывала сильный взрыв
в случае принудительного введения в тесный контакт с нижележащим метал-
металлом, вероятно находившимся под слоем моноокиси, которая в нормальных
условиях функционировала как изолятор. Этот взрыв был приписан взаимо-
взаимодействию металла с надперекисью, содержавшейся в наружном слое. В метал-
металлическом натрии такого явления не происходит, вероятно в связи с тем, что
он не образует надперекись при медленном окислении в условиях постепенного
притока воздуха.
Перекиси металлов могут обладать разной термической стойкостью, начи-
начиная от сравнительно очень устойчивых производных щелочных металлов, ме-
менее устойчивых перекисей щелочноземельных металлов, а также перекисей
цинка и кадмия и кончая относительно неустойчивыми перекисями меди и рту-
ртути; такое расположение отражает порядок возрастания электроотрицатель-
электроотрицательности соответствующих металлов. Ниже даются сведения об отдельных соедине-
соединениях, размещенных в соответствии с расположением элементов в периодической
системе.
ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ I ГРУППЫ
Из производных щелочных металлов практическое значение имеют лишь
перекись натрия и надперекись калия. Для получения LiOOHH2O
(его можно так же представить и как Li2O2H2O2-2H2O) применяются
реакции между этилатом лития LiOC2H5 и перекисью водорода в этиловом
спирте [53]. При этом получаются ромбические кристаллы, тогда как Li2O2
кристаллизуется в тетрагональной системе.
Перекись натрия
Перекись натрия (наиболее известная и чаще всего встречающаяся из
перекисей металлов) обычно получается действием сухого воздуха, лишенного
двуокиси углерода, или кислорода на металлический натрий, лучше всего при
Переписные соединения I группы 539
300—400°. В производственном масштабе она выпускается в виде желтовато-
белого порошка, содержащего 96% (или больше) Na2O2. Окисление можно про-
проводить либо непрерывным методом, для чего металлический натрий нагревают
и одновременно вводят в атмосферу, все более богатую кислородом, либо по
двухступенчатому процессу, по которому сначала получают окись натрия
в условиях ограниченного доступа кислорода и низкой температуры A20—
200°), а затем превращают эту окись в перекись в печи с температурой 200—
390°, где порошкообразную окись поддерживают в состоянии непрерывной
циркуляции в атмосфере чистого кислорода или сухого воздуха, обогащенного
кислородом. Путем применения вращающихся печей (вместо обычных непод-
неподвижных) достигается значительная экономия в производственной площади
и затрате труда. Нантц [54] рекомендует способ, по которому окись натрия
подвергают действию циклически изменяющихся давлений сухого воздуха при
температурах 350—490°.
Некоторые авторы считают, что желтоватый цвет перекиси натрия обу-
обусловлен содержанием в ней примесей (например, железа). Однако при нагрева-
нагревании окраска чистой перекиси натрия углубляется до коричневой, при охлажде-
охлаждении же начальный цвет ее восстанавливается. Перекись натрия ведет себя
подобно окислам цинка, олова, индия и т. д., причем аналогичные изменения на-
наблюдаются и для надперекиси натрия; в последнем случае изменение окраски оп-
определенно не связано с изменением кристаллической структуры [55]. При прев-
превращении поверхностного слоя перекиси натрия в контакте с воздухом в карбонат
(как это наблюдается, если держать твердую перекись в течение некоторого
времени в неплотно закрытом сосуде) желтая окраска переходит в белую. При
температуре выше точки плавления D60°) становится заметным разложение
перекиси натрия с потерей кислорода, а при температуре красного каления
оно протекает быстро [56].
Перекись натрия легко поглощает атмосферную влагу и двуокись углерода
и образует соответственно гидроокись и карбонат. Твердая перекись натрия при
обыкновенных температурах бурно реагирует с капельножидкой водой с выде-
выделением кислорода и сильным разогреванием; если, однако, применять ледяную
воду, а твердую перекись натрия вводить постепенно, можно существенно умень-
уменьшить разложение. Разбавленные растворы, содержащие около 2% перекиси
натрия, можно получать без потери активного кислорода, даже не пользуясь
водой, охлажденной до температуры льда, если только вводить перекись в воду
медленно, при интенсивном перемешивании. В получающемся щелочном раство-
растворе при комнатной температуре можно обнаружить присутствие перекиси водо-
водорода. При более высоких температурах наличие свободной щелочи способствует
быстрому разложению перекиси водорода.
В различных видах применения перекись натрия при растворении функ-
функционирует в качестве удобного источника перекиси водорода. Если перекись
натрия ввести при комнатной температуре в соприкосновение с воздухом, на-
насыщенным водяным паром и лишенным двуокиси углерода, или же растворить
ее примерно в четырехкратном по весу количестве холодной ледяной воды,
можно получить октагидрат Na2O2-8H2O. Последний при выпаривании раство-
раствора получается в виде бесцветных прозрачных пластинчатых кристаллов. При
30° гидрат растворяется в собственной кристаллизационной воде; при темпера-
температурах выше 40° щелочной раствор начинает разлагаться. Известны также
моногидрат и дигидрат. Бюссе [57] приводит данные для давления водяного
иара над гидратами перекиси натрия. Одно из основных применений пере-
перекиси натрия состоит в использовании ее для отбелки (например, древесной
целлюлозы). В этом отношении она конкурирует с перекисью водорода, на
что уже указывалось на стр. 484. Разложение водных растворов перекиси
натрия может быть использовано также в качестве удобного метода гене-
генерирования небольших количеств кислорода.
540 Глава 12
В качестве окислителя перекись натрия находит разнообразное примене-
применение как в промышленности, так и в аналитической лаборатории. При исполь-
использовании для анализа плавленая перекись натрия (одна или в смеси с соответ-
соответствующим количеством активированного угля из сахара) при сплавлении
с различными веществами представляет очень мощный окислитель, действую-
действующий также на многие огнеупорные материалы [59, 60]. Такое сплавление обыч-
обычно проводят в никелевых или серебряных тиглях, поскольку платина при
этом быстро разрушается в самых обычных условиях*. Железо несколько
устойчивее платины; алюминий и медь быстро взаимодействуют с расплавлен-
расплавленной перекисью.
Смеси плавленого едкого натра с небольшими добавками перекиси натрия
используются для очистки нержавеющих сталей от окалины [58]. Рекомендует-
Рекомендуется применять перекись натрия для повышения извлечения металлов из руд;
для этого тонкоизмельченный материал спекают с перекисью натрия и затем
проводят дополнительную обработку, например выщелачивание [62].
Перекись натрия является довольно устойчивым веществом при темпера-
температуре ниже точки плавления и не подвержена взрывному разложению при ударе
или нагревании в пламени. Тем не менее смеси перекиси с самыми различными
легкоокисляемыми веществами органического и неорганического происхо-
происхождения могут давать взрывные реакции. Если смесь перекиси натрия с желез-
железными опилками, порошкообразным алюминием, карбидом кальция или тонко-
измельченной серой увлажнить водой или концентрированной серной кислотой
или сильно нагревать такую смесь, то может произойти взрыв; в аналогичных
условиях и многие органические вещества, например сахар, глицерин, ледя-
ледяная уксусная кислота и эфир, также могут привести к сильным взрывам или
к раскаливанию смеси. Дерево, бумага или ткань при соприкосновении с пере-
перекисью натрия могут воспламениться.
Перекись натрия легко окисляет соединения низшей валентности, например
соли трехвалентного хрома до хромата и соли двухвалентного марганца до
двуокиси марганца; железный порошок при этом превращается непосредствен-
непосредственно в феррат натрия Na2Fe04. Окись или закись азота реагируют при температуре
около 150° с перекисью натрия с образованием нитрита натрия, причем из заки-
закиси азота выделяется свободный азот.
Взаимодействием перекиси натрия с ангидридами или хлорангидридами
органических кислот в подходящих условиях можно получать органические
перекиси и пероксокислоты.
Перекись натрия образует не только гидраты, но, как указано выше, при
обработке на холоду спиртовым раствором концентрированной серной, соляной
или азотной кислоты она образует «кислую соль» NaOOH (гидроперекись нат-
натрия), представляющую собой довольно неустойчивое кристаллическое белое
вещество. Гидроперекись в свою очередь может давать пероксигидрат, например
2NaOOHH2O2, и его гидратное производное 2NaOOH-H2O2-4H2O. Эти
соединения настолько неустойчивы, что нельзя даже рассчитывать на их при-
применение в технике. Известны также аналогичные производные калия.
Перекись и надперекись калия
Калий, подобно натрию, может образовать перекись К2О2 (гигроскопич-
(гигроскопичный порошок желтого цвета, напоминающий серу), например путем окисления
металлического калия, растворенного в жидком аммиаке, пропусканием кис-
кислорода через этот раствор при —50° (до обесцвечивания раствора). При нагре-
* Если предварительный нагрев производить осторожно, чтобы температура не
возрастала выше 20Q* до наступления собственно реакции, то в отсутствие легко окисляе-
окисляемых веществ (например, сульфидов) максимально достигаемая температура, как правило,
не превышает 540°; в этих условиях разъедания платины не происходит F1).
Перекисные соединения I группы 541
вании перекиси в воздухе или кислороде она переходит в надперекись КО2.
Поэтому продуктом окисления металлического калия при нагревании в кисло-
кислороде или на воздухе до 60—80° является надперекись КО2, которая по цвету
напоминает желтый крон. Надперекись может быть получена также при низко-
низкотемпературном окислении калия в жидком аммиаке с применением избытка
кислорода по сравнению с количеством, необходимым для образования переки-
перекиси. Надперекись калия весьма устойчива при нагревании до температуры плав-
плавления 380°. При более высоких температурах она диссоциирует на перекись
калия и кислород. Под действием атмосферной влаги надперекись быстро раз-
разлагается с образованием едкого кали и выделением кислорода, с капельножид-
капельножидкой водой она интенсивно реагирует таким же образом. Она реагирует как
с двуокисью, так и с окисью углерода с образованием карбоната и кислорода.
Рубидий и цезий образуют аналогичные надперекиси. Все они имеют темно-
желтый цвет, при нагревании переходящий в коричневый, а при плавлении
превращающийся в черный.
Авторы прежних работ [43] считали, что надперекись калия является ди-
мерной, т.е. должна выражаться формулой К2О4, однако рентгенографическое
исследование твердых соединений не подтверждает этого вывода. Совершенно
бесспорно доказано, что кристалл надперекиси калия состоит из правильно
чередующихся ионов К+ и О2 в гранецентрированной решетке типа карбида
кальция. Кристаллическая структура КО2, так же как и СаС2, может быть
выведена из структуры каменной соли путем замещения иона СГ соответствен-
соответственно ионом О2 или С2~. Для возможности замены иона СГ на О2 необходимо сни-
снижение симметрии кристалла до тетрагональной системы. Рассматриваемое
соединение парамагнитное; магнитный момент равен 2 магнетонам Бора, что
соответствует структуре с одним неспаренным электроном, как и следует из
электронной формулы соединения. Это также указывает на неправильность
димерной формулы К2О4.
Интенсивная реакция надперекиси калия с водой, в результате которой
выделяется кислород, используется в противогазах для пожарных и спасатель-
спасательных работ; влага и двуокись углерода, выдыхаемые носителем противогаза,
вступают в реакцию с надперекисью, содержащейся в коробке противогаза,
и выделяют кислород в количестве, достаточном для нужд носителя противо-
противогаза, с одновременным превращением двуокиси углерода в углекислый калий.
В этих условиях противогаз функционирует как изолированная система, совер-
совершенно не зависящая от окружающей атмосферы.
Перекиси группы 1,6
По современным воззрениям так называемая перекись серебра в действи-
действительности представляет не истинную перекись, а только окись двухвалентного
серебра AgO. Однако не исключена возможность существования и перекиси
одновалентного серебра. Имеющиеся данные не позволяют дать на этот вопрос
однозначный ответ.
Известна гидратированная красная перекись меди СиО2Н2О, а также
и основное соединение, представляющее собой, по литературным данным,
CuO2-Cu(OHJ [63]. Предположение о существовании таких соединений вы-
выдвинуто для объяснения заметного каталитического действия соединений двух-
двухвалентной меди на разложение перекиси водорода [64]. Происходящий при этом
цикл изменений может быть выражен уравнениями:
СиО + Н2О2 -> СиО2 + Н2О, E)
СиО2 + Н2О2 -» СиО + Н2О + О2. F)
542 . Глава 12
ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ II ГРУППЫ
Щелочноземельные металлы—кальций, стронций и барий (но, вероятно, не
бериллий) образуют перекиси, которые в случае бария и стронция могут быть
получены либо поглощением кислорода окисью щелочноземельного металла,
либо по реакции с перекисью водорода. Так, реакция поглощения кислорода
с образованием перекиси
2SrO + O2 -> 2SrO2 G)
яри 600—700° и 150 am дает перекись стронция 90—95%-ной чистоты. Окись
бария может быть превращена практически полностью в перекись при атмосфер-
атмосферном давлении. В процессе переокисления получается более плотный продукт,
чем при осаждении путем добавки перекиси водорода к раствору соли (на этом
мы остановимся ниже). По-видимому, перекись магния нельзя получить непо-
непосредственно из окиси. Струве [65], а также авторы более поздних работ [66, 67]
сообщают об образовании следов перекиси кальция из окиси кальция и кисло-
кислорода. ЭтимпутемРизенфельдуи Ноттебому [68] не удалось синтезировать пере-
перекись кальция.
Проведены исследования по давлению диссоциации перекиси кальция при
низких температурах [66,67]. На основании исследований скорости разложе-
разложения при разных температурах Блюменталь [66] постулирует существование
двух модификаций перекиси кальция: а-формы, устойчивой при низких темпе-
температурах и медленно диссоциирующей, и р-формы, устойчивой при высоких тем-
температурах и легко подвергающейся диссоциации. Однако это предположение
не подкреплено достаточными доказательствами, а потому описанная гипотеза
носит только умозрительный характер и является весьма неправдоподобной.
Вопрос об образовании перекисей щелочноземельных металлов рассматривал-
рассматривался и на стр. 106.
Второй метод получения перекисей этих металлов основан на введении
в реакцию шлама гидроокиси металла [69] с концентрированным раствором пе-
перекиси водорода; эта реакция в сущности является обратной процессу по-
получения перекиси водорода путем гидролиза перекиси металла. Пример та-
такой реакции:
Ва (ОНJ + Н2О2 -> ВаО2 + 2Н2О. (8)
Можно также обработать перекись водорода и раствором нитрата или дру-
другой соли металла в присутствии щелочи:
М (NO3J + Н2О2 + 2NaOH -> МО2 + 2NaNO8 + 2H2O (9)
или проще
М2+ + Н2О2 + 2ОН- -> МО2 + 2Н2О. A0)
Перекиси металлов способны присоединять до 8 молекул воды с образованием
гидратов. При охлаждении раствора может выпасть в виде твердой фазы окто-
гидрат МО2 • 8Н2О, при высоких же температурах возможна кристаллизация без-
безводной перекиси. Гидратированные кристаллы легче фильтровать и промывать,
чем слизистые осадки безводной перекиси. Гидратная вода связана в них
довольно слабо и может быть удалена осторожным нагреванием без разложе-
разложения перекиси.
При действии на перекись металла холодной концентрированной перекиси
водорода получаются монопероксигидратные соединения, например ВаО2 • Н3О2;
при нагревании такие соединения теряют кристаллизационную перекись водо-
водорода. Соединение магния, хотя и способно к существованию, но в чистом виде
не выделено.
В сухом виде перекиси щелочноземельных металлов достаточно устойчивы
и нереакционноспособны, а поэтому их можно хранить годами при обыкновен-
Перекисные соединения II группы 543
ных температурах без заметного разложения. Как и другие перекиси, они
способны, приходя в соприкосновение с горючими веществами, вызывать их
воспламенение, особенно при совместном измельчении. Альтаба [70] сообщает,
что растворимость перекисей щелочноземельных металлов в ацетоне ничтожно
мала. Дополнительные данные по отдельным соединениям II группы приведены
ниже.
Перекись бария
В группе II, а перекись бария является наиболее важной и наиболее устой-
устойчивой из всех щелочноземельных перекисей. Вероятно, эта перекись была от-
открыта самой первой (она изучена еще до получения перекиси водорода). До
развития электролитического метода производства перекиси водорода перекись
бария служила основным источником ее получения. Современное применение
перекиси бария и потенциальные промышленные процессы рассматриваются
в гл. 3. Получение перекиси бария по обратимой реакции A1) было положено
в основу процесса производства кислорода по Брину:
2ВаО + О2 -* 2ВаО2 + 34,2 ккал A1)
(в настоящее время этот процесс устарел и вытеснен способом разгонки жид-
жидкого воздуха). Перекись бария ВаО2 легко получается при нагревании окиси
на воздухе при 600—700°. Процесс этот обратим; увеличение давления способ-
способствует образованию перекиси, а рост температуры смещает равновесие влево.
Давление диссоциации достигает 760 мм при 840° [71]. Сообщается, что следы
влаги (приводящей к образованию гидроокиси бария) необходимы для катали-
зирования как разложения перекиси, так и образования ее из окиси и кислоро-
кислорода. По Кендалу и Фуксу [72], каталитическое действие различных количеств
добавленных окислов снижает температуру, при которой становится заметной
скорость диссоциации при атмосферном давлении.
В чистом виде перекись бария представляет белый порошок (уд. вес 4,96);
перекись, выпускаемая промышленным способом, обладает иногда, если она
не вполне свободна от следов железа, слабо-желтоватым оттенком. Технические
перекиси бария и стронция содержат переменные количества окисей и гидро-
гидроокисей. При нагревании в условиях атмосферного давления, но без доступа
влаги перекись бария должна быть устойчивой примерно до 740°; при этой
температуре давление ее диссоциации равно 0,21 am. В связи с непрерывным
ростом давления диссоциации при еще более высоких температурах точка плав-
плавления этой перекиси не определена, но получающаяся масса, представляющая
собой смесь перекиси и окиси, размягчается примерно при 800° и обладает
свободной текучестью при температуре ярко-красного каления.
Как указано выше, перекись бария образует вполне определенный окто-
гидрат ВаО2*8Н2О. Кристаллы этого соединения имеют вид блестящих чешуек,
легко выветриваются и слаборастворимы в воде (около 1 г/л при комнатной
температуре).
Раньше перекись бария находила значительное применение как исход-
исходный материал для получения разбавленных растворов перекиси водорода
и кислорода и в связи с этим также использовалась при отбелке и дезинфекции;
кроме того, вследствие высокого окислительного действия перекись бария
используется и сейчас в пиротехнике (зеленое пламя), а в смеси с магниевым
порошком применяется в воспламеняющих составах, например для иницииро-
инициирования термитной реакции. Перекись бария используется для катализаторов,
применяемых в крекинге жидких углеводородов [73], и как промотор окисно-
серебряного катализатора для синтеза гидразингидрата из аммиака и кисло-
кислорода [74].
544 Глава 12
Перекиси кальция и магния
Перекиси кальция и магния (как правило, в смеси с соответствующими
гидроокисями) применяются для отбелки и дезинфекции (например, на скла-
складах зерна и семян и при перевязке ран); перекись кальция используется также
в зубных пастах и порошках, в косметике и даже как компонент жеватель-
жевательной резины. В хлебопекарнях расходуются значительные количества перекиси
кальция с целью улучшения процесса механической обработки теста. Перекись
кальция, так же как двуокись свинца, двуокись марганца и перекись стронция,
находит применение для регенерации серных мостиков в вулканизованном бу-
тилкаучуке, в котором произошла реверсия под действием высоких температур
[751. Перекиси магния и кальция находят применение также для приема внутрь
с целью нейтрализации избыточной кислотности и как составы против бро-
брожения. Это белые порошки, обычно выпускаемые не в виде чистых соединений,
а с содержанием 50—60% перекиси. Они вполне стойки и в сухом виде могут
сохраняться годами при комнатной температуре; выше 300° они быстро разла-
разлагаются, и для полной потери всего активного кислорода достаточно нагревать
их в течение нескольких часов при 350°. При приливании перекиси водорода
к раствору гидроокиси кальция (или при введении перекиси натрия в раствор
кальциевой соли) выпадает гидрат СаО2-8Н2О. Он теряет кристаллизационную
воду при слабом нагревании.
Перекись цинка
Перекись цинка ZnO2, как и перекиси кальция и магния, обычно выпускает-
выпускается в виде смеси, содержащей 40—50% гидроокиси. Ее можно получить обработ-
обработкой тонкого шлама окиси цинка перекисью водорода [761. Более чистый про-
продукт можно получить взаимодействием цинкэтила или амида цинка с перекисью
водорода в эфирном растворе. В чистом виде перекись цинка представляет
белый порошок, но технический продукт может обладать слабо-желтой окрас-
окраской. Это соединение бурно разлагается с выбросом продукта при нагревании
его до температуры выше 200°. Вода вызывает лишь слабый гидролиз продукта.
Перекись цинка находит применение как индивидуально, так и в сочетании
с другими веществами в качестве антисептика и вяжущего средства, например
в сочетании с сульфамидными препаратами при обработке ран и лечении кож-
кожных заболеваний. Перекись цинка часто используется в составе косметических
средств, дезодораторов, мазей и присыпок. Она находит также применение
в производстве инсектицидов и в вулканизации каучука.
Перекись кадмия
Перекись кадмия СсЮ>, желтый порошок, обычно образуется в нечистой
форме как основное соединение A;CdO2«r/Cd(OHJ по методам, аналогичным
тем, которые применяются для получения перекиси цинка. Как и перекись
цинка, она также полностью разлагается при температурах выше 180°. В на-
настоящее время технического применения не имеет.
Перекись ртути
Сообщается, что ртуть образует очень нестойкую кирпично-красную пере-
перекись HgO2, напоминающую по внешнему виду красный фосфор. Ее можно полу-
получить либо действием 30% -ной перекиси водорода на спиртовый раствор хлор-
хлорной ртути с последующей добавкой щелочи, либо действием 30%-ной перекиси
водорода на красную окись ртути. Эта перекись—одна из наиболее легко-
легкоразлагающихся среди неорганических перекисей, которые можно выделить
Переписные соединения III—VIII групп 545
в твердом состоянии. Она взрывается при быстром нагревании или от удара.
Даже при 0° перекись ртути быстро разлагается на красную окись ртути
и кислород. В воде она энергично выделяет свободный кислород с образованием
желтой окиси ртути, причем одновременно в растворе образуется некоторое
количество перекиси водорода. Сообщается и об образовании перекиси закис-
ной ртути Hg2O2 действием концентрированной перекиси водорода на закись
ртути при низкой температуре, но доказательство существования этой перекиси
нельзя считать вполне убедительным.
ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ HI—VIII ГРУПП
Бор
Пероксоборат натрия [22, 26, 27] можно получить [77] по реакциям
раствора буры с перекисью водорода и едким натром или с перекисью натрия:
Na2B4O7 + 2NaOH -> 4NaBO2 + Н2О, A2)
NaBO2 + Н2О2 + ЗН2О -» NaBO2. H2Oa. ЗН2О A3)
или путем электролиза [78] раствора буры и углекислого натрия:
Na2B4O7 + 2Na2COs + 21Н2О ->
-> 4 (NaBO2. Н2О2. ЗНаО) + 2NaHCO3 + 4Н2. к v
При охлаждении раствора до 10° выпадает гидрат, который можно
отфильтровать, промыть и высушить. Описано также осаждение соответству-
соответствующей бариевой соли [79]. Натриевое соединение может служить источни-
источником перекиси водорода в щелочном растворе (насыщенный раствор имеет
рН около 10) и находит широкое применение не только для отбелки текстиля
и окисления красителей, но также и в качестве составной части зубных
порошков, антисептиков для приема внутрь и т. д. По свойствам это соеди-
соединение соответствует продукту присоединения перекиси водорода, а не соли
какой-то гипотетической пероксоборной кислоты. Поэтому его формулу
лучше изображать так, как это указано выше, т. е. в виде тригидрата
монопероксигидрата метабората натрия, а не в форме тетрагидрата перо-
ксобората NaBO3. Однако возможность существования истинного пероксобо-
рата не исключается, так как синтезы, осуществленные Фаталлахом и
Партингтоном [80], и результаты измерений рН в растворах боратов, про-
произведенных Вибергом [81] и Эдвардсом [82], подтверждают образование
пероксобората*.
Иттрий и редкие земли
В III группе известны гидратированные перекиси для иттрия, лантана,
церия и некоторых других редкоземельных металлов. Сообщается, что пе-
перекись иттрия имеет состав: Y4O9-nH2O. Церий образует соединение красно-
коричневого цвета СеО3-2Н2О, для которого предложена структура
(НОKСеО —ОН. Титан, цирконий, гафний и торий из группы IV,a обра-
образуют аналогичные соединения, причем производное титана обладает харак-
характерным желтым цветом, а остальные —белым. Состав перекисей других
редкоземельных элементов достоверно не установлен. Для соединений
лантана, неодима, празеодима и самария еще не выяснено, обладают ли они
общей формулой R4O9-mH2O или R2O6-nH2O. При нагревании в кислороде
под высоким давлением окись празеодима, по имеющимся данным, превра-
* Как и при других перекисных соединениях, для которых не ясен характер распо-
расположения группы —О —О— в структуре, это соединение называется просто пероксобо-
ратом натрия.
35 Перекись водорода
546 Глава 12
щается в двуокись РгО2, хотя в обычных условиях состав продукта близок
к Рг6Оп.
Углерод
При выпаривании перекиси водорода и углекислого натрия в виде раство-
раствора, содержащего необходимые относительные количества обоих соединений
(или при введении перекиси водорода в концентрированный раствор угле-
углекислого натрия), выпадает пероксигидрат 2Na2CO3-3H2O2 [7,83]. Это соеди-
соединение прочно удерживает перекись водорода при комнатной температуре
в течение значительных промежутков времени. Белое твердое вещество
содержит около 14% активного кислорода.
Сообщается о существовании некоторых других аддитивных соединений
углекислого натрия и перекиси водорода, а также их гидратов, к которым
относятся соединения с соотношением Н2О2:Н2О, равным 1,5:1 и 2:1.
Макаров и Чамова [36] изучили термические свойства гидратированных
пероксигидратов и процесс их обезвоживания, а также обнаружили существо-
существование безводного продукта Na2CO3-2H2O2; они, однако, не подтвердили су-
существования ранее найденных соединений: 2Na2CO3-3H2O2, Na2CO3H2OH2O2
или Na2CO3H2O2,5H2O2.
Указанные соединения, безусловно, не являются солями истинной
пероксоугольной кислоты. Калиевое производное этой кислоты, пероксо-
карбонат калия К2С2Ов, впервые получено [84] в 1896 г. путем электролиза
[85] насыщенного раствора углекислого калия при — 15°. Это соединение,
так же как и аналогичные соединения натрия и рубидия, содержит ион
[С2Ов]2", который может быть представлен как
_о-с-о-о-с-о-п2-
л & ] •
Соль, которая во влажном состоянии окрашена в светло-голубой цвет,
довольно устойчива при хранении в сухой атмосфере, но легко разлагается
влагой воздуха с образованием двуокиси углерода, кислорода и углекислого
калия.
Истинные пероксокарбонаты натрия, калия и рубидия приготовлены
также действием газообразного фтора на соответствующие растворы карбо-
карбонатов при температуре от —13 до —16° с равномерным добавлением
растворов едких щелочей [86]. Устойчивость получающихся пероксокарбо-
натов в значительной мере зависит от чистоты исходных веществ. Очень
важно отсутствие даже минимальных количеств каталитически действующих
ионов, а именно ионов, способных снижать устойчивость собственно пере-
перекиси водорода. Образование пероксоугольных кислот в растворах солей,
содержащих кислород и двуокись углерода, установлено полярографически
[87]. Партингтон и Фаталлах [21] опубликовали обзор по пероксокарбона-
там и изложили результаты своих исследований по этому вопросу.
Титан, цирконий, гафний и торий
Наиболее четко определенным типом перекисей металлов группы IV,a
является гидратированная трехокись МО3-2Н2О [88], характерная для всех
членов подгруппы а. Интенсивная окраска (от желтой до оранжево-красной),
наблюдаемая при приливании перекиси водорода к растворам титана в
присутствии разбавленной серной кислоты, является хорошо известной
качественной пробой на перекись водорода. Шварц и Гизе [89] считают,
что эти перекиси имеют формулу, аналогичную формуле цериевого произ-
производного, т. е. М4+(ОНK (ООН), Титановое производное растворимо в едком
Перекисные соединения III—VIII групп 547
кали в присутствии перекиси водорода с образованием таких продуктов,
как К4ТЮ8-6Н2О (соли ортокислоты H4Ti (O2L). В кислом растворе интен-
интенсивная желтая окраска, возникающая при приливании перекиси водорода
к раствору титана, приписывается [6,90J иону пероксотитанила [ТЮ2-л:Н2О]2+,
который связан с перекисным анионом, существующим в щелочном растворе,
посредством следующего равновесия:
[ТЮ2Г + ЗН2О2 ^ [Ti (О2L]«- + 6Н+. A5)
Получен ряд пероксотитанатов с еще большим содержанием кислорода,
например Na2TiO5-3H2O [91] и (NH4JTiO6-H2O. Натриевое производное
пишут также в виде TiO2-Na2O2-2H2O, хотя такие формулы мало дают
в отношении фактического молекулярного строения соединения. Цирконий дает
аналогичные пероксоцирконаты, например K4ZrO8-6H2O и Na4Zr2On-9H2O,
но соответствующие соли гафния и тория неизвестны. Однако из растворов
гафния и тория приготовлены соответственно гидратированные перекиси
НЮ3-2Н2О [92] и ThO3.2H2O [93].
Фосфор
Из пирофосфата натрия можно получить [94] соединение Na4P2O7 • 2Н2О2
действием концентрированной перекиси водорода на суспензию пирофосфата.
Свойства этого соединения напоминают свойства ранее описанных продук-
продуктов присоединения. Получены различные продукты присоединения перекиси
водорода к ортофосфатам щелочных металлов, аммония и щелочноземель-
щелочноземельных металлов, особенно к двух- и трехзамещенным солям, путем прямой
реакции составляющих соединений [7].
По способу, аналогичному указанному для сульфатов и карбонатов,
удалось также получить истинные пероксофосфаты, в частности электро-
электролитическим методом. Так, при электролизе раствора фосфата К2НРО4 полу-
получается пероксофосфат калия К4Р2О8 на аноде [95]; основная реакция при
этом соответствует выражению
A6)
Пероксофосфаты можно получить также действием фтора на водные рас-
растворы динатрийфосфата или пирофосфата [96]. Твердая соль весьма устой-
устойчива, но растворы ее быстрее подвергаются гидролизу, чем растворы пер-
оксосульфатов, с образованием перекиси водорода и фосфорной кислоты.
Ванадий, ниобий и тантал
В соответствии с правилом, что устойчивость пероксокислот элементов
любой подгруппы периодической системы возрастает с ростом атомного веса
элементов группы, устойчивость в группе V,a возрастает от ванадия к
танталу. Однако сами безводные перекиси, по-видимому, в чистом состоянии
не выделены. Мышьяк, сурьма и висмут, входящие в подгруппу б, вероятно,
не образуют перекисей.
Элементы группы V,a относятся к перекиси водорода аналогично титану
и цирконию. Действием перекиси водорода на растворы ванадата в кислой
или щелочной среде можно получить ряд пероксованадатов с окраской от
желтой до красной. Сама пероксованадиевая кислота не выделена; боль-
большинство перекисных соединений ванадия легко разлагается. При приливании
перекиси водорода к раствору такой ванадиловой соли, как (VOJ (SO4)a
(образующейся при растворении пятиокиси ванадия в умеренно концентри-
концентрированной серной кислоте, например 15 — 20%-ной), раствор окрашивается
в темный красно-коричневый цвет в результате образования пероксована-
35*
548 Глава 12
дилового производного [V->O->O]2(SO4K [97]. Ривенк [98] предполагал
образование V2O7. Эту окраску можно использовать для качественной пробы
на перекись водорода или пятиокись ванадия. Яр [6,90] пришел к выводу,
что при взаимодействии пятивалентного ванадия с перекисью водорода обра-
образуются два перекисных соединения, которые в растворе участвуют в следу-
следующих равновесиях:
[ VO • щГ + Н2О2 ? [ VO2 • aq]8+ + H2O, A7)
Красно-коричневый
A8)
Красно-коричневый Желтый
Желтый ион, производимый от дипероксоортованадиевой кислоты, способен
к существованию в разбавленных слабощелочных или слабокислых пере-
перекисных растворах. Свободная кислота H3[V(O2)O4] весьма напоминает орто-
фосфорную. Описаны также окрашенные в синий цвет соединения, получаемые
взаимодействием ванадатов и перекиси водорода при низкой температуре
[99].
Сообщается, что при взаимодействии V2O6 или ортованадата с щелоч-
щелочными растворами перекиси водорода получаются кристаллические пероксо-
ванадаты довольно точного состава, в том числе Na3VO8 и KH2VOe-H2O.
Найдено [100], что скорость гидролиза перекиси ванадия меньше, чем скорость
гидролиза перекиси молибдена или вольфрама. .
Пероксониобаты несколько более устойчивы, чем пероксованадаты, и
свободная к-ислота H4Nb05-aq (монопероксониобиевая) выделена в виде
светло-желтого твердого вещества. Перекись водорода реагирует с щелоч-
щелочными растворами ниобатов щелочных или щелочноземельных металлов
с образованием ряда таких пероксониобатов, как K3Nb08 или K4Nb2On-3H2O,
часть которых содержит гидратную воду, а другие являются безводными.
Пероксотанталаты, например Na3Ta08- 14H2O и К3Та08, походят на
соответствующие производные ниобия, причем из раствора пероксотанталата
адлия действием серной кислоты можно получить твердую свободную вод-
цую кислоту Н (ТаО4) aq. Ее можно нагревать до 100° без заметного разло-
разложения.
Хром, молибден, вольфрам и уран
В группе VI,а хром дает ряд перекисных соединений, в том числе
синюю перекись СгО5, растворимую в эфире и образующую продукты при-
присоединения с органическими основаниями, например пиридином. Легко
различимы два ряда пероксосолей хрома, а именно синие соли типа МСгОв
(например, КСгОв-Н2О), образующиеся при приливании раствора СЮ5
к*щелочному спиртовому раствору перекиси водорода, и красные соли типа
М^СгО8, изоморфные с пероксованадатами. К3Сг08 образуется из слабоще-
слабощелочного раствора хромата при приливании к нему на холоду перекиси во-
водорода.
Вилсон [14] считает, что в красных солях (например, в К3Сг08) ион
CrOg" имеет тетраэдрическую структуру, причем атом хрома находится
внутри тетраэдра, а центры четырех групп О2 расположены в вершинах
последнего. Пероксониобаты и пероксотанталаты также обладают той же
кристаллической структурой; это позволяет сделать заключение, что степень
окисления хрома в красных солях также равна 5. Синяя калиевая соль
диамагнитна, и, чтобы представить ее структуру без участия нечетного числа
Перекисные соединения III—VIII групп 549
электронов, необходимо принять, что ее молекула является димерной (напри-
(например, М2 [ОСг (О2J — О — О — Сг (О2J — О] с шестивалентным хромом). При
действии щелочей перекись СгО5 не образует солей, а разлагается. Предпола-
*О2
гается, что в ее структуре содержатся две группы О2 в виде О<—Сг<"
No2
(с шестивалентным хромом) [89]. Перекисная группа СгО2, по Сиджвику
[101], имеет структуру Сг—>О—>О или Сг = О—>О, что предпочтительнее
замкнутого кольца G/ | . Бельтран Мартинес и Портер [102] для красного
пероксохромата кальция предложили формулу Са3Сг2015- 10Н2О.
Известен еще один тип перекисных соединений коричневой окраски,
содержащих группы СгО4 (например, CrO4-3NH3), образующийся при прили-
вании перекиси водорода к аммиачным растворам хромата при 0° и после-
последующем нагревании до 50°. Предполагается, что это соединение имеет
следующую структуру [101]:
Сг( -(NH3K.
NOO
При введении цианистого калия в водный раствор этого соединения обра-
образуется CrO4-3KCN. Точно не известно, является ли хром в этих соедине-
соединениях шестивалентным или четырехвалентным. Вопрос об образовании и
строении пероксохроматов изучен Глазнером [103].
В отличие от хрома молибден при взаимодействии МоО3 и RO2 образует
пероксокислоту Н2МоОб- 1,5Н2О; этот продукт представляет желтое твердое
тело, нерастворимое в зфире [1041. Однако для молибдена, как и для хрсма,
по-видимому, существуют также несколько рядов пероксосолей. При введении
молибдата натрия при 0° в реакцию с 30%-ной перекисью водорода образуется
раствор, содержащий очень нестойкую красную соль Na.McO8, которая при
нагревании до комнатной температуры превращается в более устойчивое желтое
соединение Na.^MoOe [105]. Положение осложняется тем, что в сильнокислых
растворах молибдатов образуются полимолибдаты, которые в свою очередь
способны к образованию перекисных соединений [106J.
Пероксокислоты вольфрама и их соли более устойчивы, чем ссотБетств) ю-
щие соединения хрома и молибдена, и подвергаются лишь весьма постепенному
разложению при обыкновенных температурах, являясь одними из наиболее
устойчивых известных пероксокислот [107]. Известны как бесцветные соеди-
соединения, содержащие ион (Н\Ю6)~, так и желтые с ионом (UO8J~. На основании
интенсивности возникающей желтой окраски Румф-Нордман [108] пришел
к выводу, что максимальное образование пероксосолей наблюдается при моляр-
молярном отношении перекиси водорода к вольфрамату натрия 2,0. Известны также
полипероксовольфраматы, аналогичные полипероксомолибдатам. В литературе
нет данных об их строении. Яр и Бланке [109] предполагают существование
иона IW2O3(O -OLJ-aq.
Уран образует желтую гидратированную перекись UO4-2H2O, устойчи-
устойчивую при температурах ниже 100° и иногда неправильно называемую «надурако-
вой кислотой». Показано, что это соединение не ионизовано, а поэтому его
нельзя рассматривать как кислоту—родоначальника пероксоуранатов. Пере-
Перекись урана легко образуется при одновременном введении перекиси водорода
и основания в раствор уранилсульфата (с регулированием рН) [110]. Сооб-
Сообщается о существовании соединений, содержащих одновалентные металлы
типа M4UO4, M2UOe и МвЦ,О18. Трансурановые элементы нептуний и плу-
550 Глава 12
тоний [93] также способны к образованию водных перекисей NpO4H2O
и РиО4хН2О. Конник и Мак-Вей [111] сообщают о существовании двух
пероксокомплексов четырехвалентного плутония—коричневого и красного.
Для них предложены разные возможные структуры и показано, что эти соеди-
соединения содержат в молекуле более одного атома плутония.
Другие металлы
Нет недостатка и в сообщениях о перекисях других, еще не рассмотрен-
рассмотренных металлов, но в большинстве случаев опубликованные данные не являются
в достаточной мере доказательными. Так, Ноддак И. и Ноддак В., описывая
соединение Re2O8(T. e. O3Re—О—О—ReO3, или перекисную форму семиокиси
рения) [112], сообщают, что оно имеет белый цвет и неустойчиво в присутст-
присутствии воды; однако существование этого соединения нельзя считать вполне уста-
установленным. Хотя железо и не дает чистую перекись, все же возможно существо-
существование такого соединения в качестве промежуточного продукта, образующегося
при реакциях окисления, в которых участвует перекись водорода в присут-
присутствии иона окисного железа. Сообщается, что при действии перекиси водорода
при низких температурах на взвесь водной окиси железа в спирте образуется
красное соединение, принимаемое за FeO2; различные исследователи сообща-
сообщают и о более богатых кислородом соединениях FeO3 и даже FeO4.
Сообщается, что при взаимодействии осажденного гидрата закиси
никеля с перекисью водорода образуется зеленое никелевое производное
NiO2JcH2O; но возможно, что это соединение является фактически лишь
продуктом присоединения перекиси водорода к гидрату закиси никеля. По
свойствам оно отличается от безводной черной NiO2, в которой никель четырех-
четырехвалентен. Гайсинский и Квесни [115] описали процесс получения на аноде
водной перекиси никеля неопределенного состава. При действии избыт-
избытка гипохлорита натрия на разбавленный раствор сульфата двухвалентно-
двухвалентного кобальта в присутствии сульфата натрия образуется водная двуокись
кобальта СоО2л:Н2О. Судя по методу получения, это соединение, вероятно,
не является перекисью. Недавно Томсон и Вильмарт [113] подтвердили суще-
существование ряда зеленых координационных соединений кобальта, представляю-
представляющих двуядерные перекисные соединения трех- и четырехвалентного кобальта,
типа (А5Со3+—О—О—Со4+А5), где А—одна или несколько обычных ком-
плексообразующих групп (впервые эти соединения получены Вернером). Эти
соединения были подвергнуты [114] спектроскопическому исследованию.
. Перекиси неметалле г
Для неметаллов известно только небольшое число неустойчивых и плохо
изученных перекисей. Сюда относится NO3 (а возможно, N2O7), существование
которой доказано только спектроскопически, и NO4, для которой имеются
и кинетические данные [116] (см. ниже данные по пероксоазотной кислоте).
Перекиси аммония, сообщения по которым имеются в литературе, по-видимо-
по-видимому, правильнее считать пероксигидратами [37]. Указывается, что при действии
фтора на серную кислоту или сульфаты щелочных металлов при низких темпе-
температурах получается четырехокись серы SO4, причем предполагается, что это
соединение функционирует как промежуточный продукт в реакциях пероксо-
сульфатов. Сообщается о существовании производных хлора и йода, например
СЮ4 (или С12О8), получаемых при взаимодействии перхлората серебра и йода
в эфирном или бензольном растворе, но лишь в разбавленном. Доказательства,
что эти производные серы и галогенов являются перекисями, не убедительны.
Пероксокислоты и их соли 551
пероксокислоты и их соли
Пероксодисерная кислота
Пероксодисерная кислота H2S2O8, иногда называемая также надсерной
кислотой или кислотой Маршалла, представляет собою белое кристаллическое
вещество, плавящееся с разложением при температуре около 65°. Она образует-
образуется в качестве промежуточного продукта в производстве перекиси водорода по
одному из электролитических методов. В данном процессе эта кислота исполь-
используется в растворенном виде, и в чистом состоянии ее не выделяют. Как указано
на стр. 109 и ел., серную кислоту (примерно 10 н.) подвергают электролизу на
платиновых анодах в условиях высокой плотности тока, для чего эту кислоту
пропускают через ряд ванн и в конце концов получают раствор пероксодисер-
ной кислоты примерно 2,5 н. концентрации.
При гидролизе теплого раствора кислоты получается перекись водорода:
?. ?. нон <? ?.
НО — S — О-О — S — ОН > НО—S—OOH + HO-S—ОН
о *б о о
JHOH
о
HO-S-OH + H — О-О-Н A9)
о
с промежуточным образованием пероксомоносерной кислоты (кислоты Каро)
H2SO6.
Структура иона S20jJ~ установлена [10] рентгенографическим анализом
пероксодисульфатов цезия и аммония; она представляет два тетраэдра SOf~,
соединенных между собою в вершинах за счет связи О—О.
Хотя растворы пероксодисерной кислоты довольно устойчивы на холо-
холоду, они быстро подвергаются гидролизу при кипячении в присутствии избытка
серной, кислоты. В связи с этой тенденцией к гидролизу, а также из-за наличия
сильных окислительных свойств у пероксодисерной кислоты ее трудно полу-
получить в очень чистом состоянии; обычно ее и не выделяют из растворов. Эта кис-
кислота действует на различные органические вещества: вызывает обугливание
парафина, а смеси ее с некоторыми органическими растворителями могут
даже взрываться.
В свежеприготовленном виде растворы пероксодисерной кислоты при ком-
комнатной температуре не дают характерных реакций перекиси водорода с титано-
титановым раствором или перманганатом; из раствора йодида они лишь медленно
выделяют йод. Однако сильный окислительный характер пероксодисульфата
проявляется при превращении иона двухвалентного марганца в перманга-
нат (в присутствии следов иона серебра как катализатора). Основное
практическое значение пероксодисерной кислоты заключается в ее применении,
как указано выше, для производства перекиси водорода электролитическим
методом.
Соли пероксодисерной кислоты
Аммониевая и калиевая соли (NH4JS2O8 и K2S2O8, представляющие
кристаллические белые твердые вещества, имеют наибольшее значение из
всех солей. Особенно устойчива в воздухе калиевая соль; аммониевое
производное также вполне устойчиво в сухой атмосфере, но, обладая некоторой
гигроскопичностью, слеживается во влажном воздухе. Пероксодисульфат аммо-
аммония значительно более растворим, чем калиевая соль (соответствующие раство-
растворимости при 0° равны 58 и 1,8 г в 100 г воды), а поэтому калиевая соль
552 Глава 12
может быть осаждена при приливании раствора бисульфата калия к рас-
раствору пероксодисульфата аммония. Обе соли в водных растворах медленно
гидролизуются при комнатной температуре с образованием перекиси водорода
и других продуктов, например
(NH4JS2O8 + 2Н2О -* 2NH4HSO4 + Н2Оа. B0)
Эти пероксосульфаты, так же как и другие, образуются при электролити-
электролитическом окислении сульфатов [117]. Так, можно получить раствор пероксоди-
пероксодисульфата аммония на аноде (в ванне с платиновыми анодами) электролизом
раствора бисульфата аммония, причем пероксодисульфат кристаллизуют
непосредственно из раствора, промывают и сушат. Работа с пероксодисульфа-
тами аммония и калия и перевозка их не связаны с опасностью пожара или
взрыва, причем в качестве тары для них обычно применяют деревянные
или фибровые ящики [71. Пероксодисульфат бария BaS2O8 резко отличается
от сульфата бария по своей растворимости в воде. Сообщается и о получении
пероксодисульфата серебра [119].
В присутствии каталитически действующих следов серебряных солей пер-
пероксодисульфат аммония способен к различным реакциям окисления, например
к окислению аммиака до азота:
3 (NH4JS2O8 + 8NH3 -> 6 (NH4JSO4 + N.. B1)
Тиосульфаты окисляются им до тритионатов или тетратионатов, а металлы типа
меди растворяются с образованием сульфатов.
Пероксодисульфат калия (обычно называемый персульфатом калия)
K2S2O8 во многих отношениях аналогичен соответствующей аммониевой соли.
Он, однако, гораздо менее растворим (насыщенный раствор при 0° содержит
около 2% твердого K2S2O8, при 20°—около 5%), а также более устойчив в кон-
контакте с воздухом, чем пероксодисульфат аммония. В производственном масшта-
масштабе эта соль получается приливанием раствора бисульфата калия к раствору
пероксодисульфата аммония, получаемому электролизом раствора бисульфата
аммония; осадок пероксодисульфата калия центрифугируют для отделения
его от маточного раствора бисульфата аммония.
Пероксодисульфаты широко применяются как инициаторы полимеризации.
Они находят также применение для отбеливания жиров и мыл и окисления кра-
красителей [7]*.
Пероксомоносерная кислота
Как упомянуто в предыдущем разделе, пероксомоносерная кислота, или
кислота Каро (HaSO5 или НО—SO2—О—ОН),- образуется в качестве про-
промежуточного продукта при гидролизе пероксодисерной кислоты. Как и пер-
оксодисерную, вряд ли ее когда-либо получают в чистом виде из-за неустойчи-
неустойчивости и высокой реакционной способности в отношении окисляющихся ве-
веществ. В том виде, в котором она получается при кислотном гидролизе перок-
содисульфатов или при реакции концентрированной перекиси водорода
с серной кислотой, она представляет собою белое твердое вещество (т. пл.
около 45°). Ее можно отличить от перекиси водорода, пользуясь тем, что при
введении ее в раствор йодида сейчас же происходит выделение йода, тогда
как другие типы перекисных соединений реагируют довольно медленно. Подоб-
Подобно пероксодисерной кислоте, но в отличие от перекиси водорода кислота Каро
не реагирует с марганцовокислым калием. Растворы кислоты Каро применя-
применяются главным образом в различных процессах окисления органических
веществ.
* Фирма «Buffalo"Electro-Chemical Co.» выпустила ценную библиографию по раз-
различным видам применения пероксодисульфатов [118]; см. также монографию Прайса [1].
Пероксокислоты и их соли 553
Другие пероксокислоты
Другие пероксокислоты и их соли можно получать действием перекиси
водорода на соответствующий анион или ангидрид кислоты или путем анодного
окисления. Так, пероксомонофосфорная кислота Н3РОб (аналогичная кислоте
Каро) образуется при приливании холодной концентрированной перекиси
водорода к Р.2Об или НРО3 [120]:
2НаО2 + Р2Об + Н2О -* 2Н3РОб B2)
или при анодном окислении растворов ортофосфорной кислоты. Известны
также производные типа М4Р2О8, аналоги пероксодисульфатов. Калиевую
соль пероксодифосфорной кислоты можно получить в твердом виде.
По методу, аналогичному тому, который применяется для пероксодисуль-
пероксодисульфатов, можно получить светло-синие пероксокарбонаты К2С2Ов или Rb2C2O6,
подвергая электролизу концентрированные растворы карбоната калия или
рубидия при — 10°; они, вероятно, имеют структуру МООС — О — О — СООМ.
Эти же пероксосоединения можно получить и действием элементарного
фтора на раствор карбоната щелочного металла при низкой температуре [86].
Сообщается о существовании и других типов пероксокарбонатов, напри-
например Na2CO4[21j, который образуется при взаимодействии фосгена и Na2O2
с выделением кислорода. При действии двуокиси углерода на перекись
натрия или на спиртовый раствор гидроперекиси натрия получается другой
пероксодикарбонат Na2C2Oe, изомерный с продуктом электролитического
процесса получения, а также, возможно, и пероксобикарбонат NaHCO4,
вероятно имеющий следующее строение:
NaO4
>с = о.
НО -(К
Окончательно структура этих веществ не установлена, но приведенные
выше формулы являются вполне приемлемыми.
К пероксокислотам других элементов IV группы периодической системы
относятся соединения титана, германия и олова. Пероксотитановую кислоту,
отличающуюся глубокой желтой окраской, обычно пишут таким образом:
(HOKTi —ООН или чаще в виде гидратированной перекиси ТЮ3-2Н2О.
Ее получают при взаимодействии перекиси водорода с раствором титанового
соединения, и образование ее представляет хорошо известную качественную
пробу на перекись водорода.
Осажденная водная двуокись олова образует с концентрированной
перекисью водорода продукт, который принимают за пероксодиоловянную
кислоту H2Sn207; сообщается также о получении производного пероксоди-
германиевой кислоты K2Ge2O7-4H2O по реакции Н2О2 с раствором метагер-
маната. Элементы группы V,a, включая ванадий, ниобий и тантал, не обра-
образуют перекисей, но в растворе дают при действии перекиси водорода анионы
пероксокислот. В присутствии перекиси водорода наблюдается темно-желтая
окраска подкисленного раствора ванадата; эта реакция, так же как и соот-
соответствующая реакция с титановыми производными, используется для откры-
открытия перекиси водорода. Пероксониобат и пероксотанталат калия K3Nb08
и К3Та08 напоминают красный пероксохромат К3Сг08.
Одна из наиболее широко применяемых качественных проб на перекись
водорода основана на образовании темно-синей пероксохромовой кислоты
при добавке перекиси водорода к подкисленному раствору хромата. Обычно
формула этой кислоты пишется как НСгО6. Менее известны соответствующие
перекисные соединения других металлов группы VI,а, молибдена и воль-
554 Глава 12
фрама. Пероксоанионы этих элементов вступают в реакции, аналогичные
известным для ранее упомянутых элементов.
Д'Анс еще в 1911 г. [121] сообщил об образовании пероксоазотной
кислоты HNO4 при взаимодействии пятиокиси азота с безводной перекисью
водорода при низкой температуре; однако, поскольку это вещество неустой-
неустойчиво даже при низких температурах, в чистом виде оно не получено;
из него не получены также и устойчивые соли. Сообщается и о других
методах синтеза пероксонитратов, например действием озона на азиды
щелочных металлов [122] или фтора на разбавленные растворы нитрита
натрия [123], но применимость этих методов вызывает сомнения.
ЛИТЕРАТУРА
1. Price Т. S., Peracids and Their Salts, Longmans Green and Co., London, 1912;
Гризевальд, Неорганические перекиси и перекисные соли, М., 1915.
2. N е u m a r k A. S., Sci. Am. Suppl., 75, 200, 221 A913).
3. Machu W., Wasserstoffperoxyd und die Perverbindungen, 2 Aufl., Springer Ver-
lag, Wien, 1951 [есть перевод первого издания с дополнениями: см. Маху В.,
Перекись водорода и перекисные соединения, Госхимиздат, М., 1951].
4. Yost D. M., Russell H., Jr., Systematic Inorganic Chemistry, Prentice-Hall,
Inc., New York, 1944.
5. SlaterV. W., Chemistry and Industry, 1945, 42.
6. J a h г К. J., FIAT Rev. Ger. Sci., Part III, 1948, p. 170.
7. Shanley E. S., Gilbert H. N., R а у m о n d D. N., Kleinberg J., in
Kirk-Othmer, «Encyclopedia of Chemical Technology», Vol. Interscience Encyclo-
Encyclopedia, Inc., New York, 1953, p. 38.
8. Б у т у з о в В. П., ДАН СССР, 58, 1411 A947); A b г a h a m s S. С, .частное со-
сообщение, 1954.
9. Н а г г Т. Е., Ph. D. Thesis in Chemistry, Syracuse University, 1952; S h i n e m a n
R. S., К i n g A. J., Acta Cryst., 4, 67 A951).
10. Z а с h а г i a s e n W. H., Mooney R. С L., Z. Krist., 88, 63 A934).
11. Shand W., Ph. D. Thesis, California Inst. Tech., 1946.
12. К а с а т о ч к и н В., П е р л и н а С, А б л е с о в а К., ДАН СССР, 47, 37 A945).
13. BernalJ. D.,Дятлова Е., Казарновский И. А.,Рейхштейн С,
W a r dA. G.,Z. Krist.,92, 344A936); К а с а т о ч к и н В., ДАН СССР, 47, 199
A945); Miller W. S., Ph. D. Thesis in Chemistry, Syracuse University, 1936.
14. Wilson I. A., Arkiv Kemi., Min. Geol., 15B, JSfe 5 A941) [C. A., 36, 308].
15. К а с а т о ч к и н В., К о т о в В., J. Chem. Phys., 4, 458 A936), ЖТФ, 7, 1468
A937); Ж д а н о в Г. С, 3 в о н к о в а 3. В., ДАН СССР, 82, 743 A952); Теш-
р 1 е t о п D. H., DaubenC. H., J. Am. Chem. Soc, 72, 225 A950).
16. GlocklerG., M a 11 а с k G., J. Chem. Phys., 14, 504, 505 531 A946); H u m e-
Rothery W., Proc. Roy. Soc. (London), A197, 17 A949).
17. T r a m b a r u 1 о R., G h о s h S. N., В u г г u s C. A., G о г d у W., J. Chem. Phys.,
21, 851 A953).
18. W h a 1 e у Т. Р., К 1 e i n b e r g J., J. Am. Chem. Soc, 73, 79 A951).
19. Haissinsky M., Discussions Faraday Soc, № 1, 255 A947).
20. H a i s s i n s k у M., J. Chem. Phys., 15, 152 A947), W a 1 s h A. D., J. Chem. Phys.,
15, 688 A947).
21. P a r t i n g t о n J. R., F a t h a 1 1 a h A. H., J. Chem. Soc, 1950, 1934.
22. Танатар С, Z. anogr. allgem. Chem., 28, 255 A901).
23. Танатар С, Вег., 32, 1013 A899).
24. Munzberg F., Lotos, 76, 351 A928) [C. A., 26, 2132].
25. L e v i G. R., В a t t a g 1 i n о F., Gazz. chim. ital., 67, 659 A937).
26. Меликов П., Писаржевский Л., Вег., 31, 678, 955 A898).
27. Foerster F., Z. angew. Chem., 34, 354 A921).
28. Willstatter R., Вег., 36, 1828 A903).
29. KraussF., OettnerC, Z. anorg. Chem., 222, 345 A935); KraussR, ibid.,
204, 318 A932); С a n о n n e J. J., франц. пат. 877384 D. XII. 1942); белы. пат.
450403 (V. 1943).
30. Spiegler J. J., пат. США 2367971 B3. I. 1945).
31. Menzel H., Ga bl er С, Z. anorg. allgem. Chem., 177, 187 A928).
32. HusainS., Z. anorg. allgem. Chem., 177, 215 A928).
33. H u s a i n S., P a r t i n g t о n J. R., Trans. Faraday Soc, 24, 235 A928).
34. К а з а н е ц к и й П. В., ЖРФХО, 46, ПО A914).
Литература 555
35. Thorpe's Dictionary of Applied Chemistry, Vol. 6, Longmans Green and Co., London,
1943, 345.
36. Макарове. 3., Ч а м о в а В. Н., Изв. АН СССР, отд. хим. наук, 1961, 255;
1952, 632.
37. М a a s s О., Hatcher W. H., J. Am. Chem. Soc, 44, 2472 A922).
38. М а г g u 1 i e s P. H., Shanl ey E.S., in Kirk-Othmer, «Encyclopedia of Chemical
Technology», Vol. 10, Interscience Encyclopedia, Inc., New York, 1953, p. 83.
39. M a t h e s о n G. L, MaassO., J. Am. Chem. Soc, 51, 674 A929); RobozZ.,
венгерск. пат. 113335 A5. XI. 1935) [С. А., 30, 2716]; Krepelka J. H.,
BuksaR., Chem. Listy, 31, 447 A937) [C. A., 32, 4522].
40. Танатар С, ЖРФХО, 40, 376 A908).
41. Fischer F., P 1 о e t z e H., Z. anorg. Chem., 75, 10 A912).
42. H о 1 t e r m a n n C, L a f f i t t e P., Compt. rend., 208, 517 A939).
43. Joannis A., Compt. rend., 116, 1370 A893).
44. d e F о г с г a n d R., Compt. rend., 158, 991 A914).
45. J a u b e r t G. F., франц. пат. 738418; герм. пат. 82982, см. также Joannis А.,
ссылка [43].
46. S е у b E., Jr., К 1 е i n b е г g J., J. Am. Chem. Soc, 73, 2308 A951).
47. H el m s А., К 1 e m m W., Z. anorg. Chem., 241, 97 A939); 242, 201 A939).
48. К 1 e i n b e г g J., Chem. and Eng. News, 31, 4012 A953).
49. Da vi dson A. W., Kl e i n b er g J., J. Phys. Chem., 57, 571 A953).
50. S t e p h a n о u S. E., S с h echte'r W. H., А г g e г s i n g e г W. J., Jr., Klein-
ber gJ., J. Am. Chem. Soc, 71, 1819 A949); S с h e с h t e г W. H., S i s 1 e г Н. Н.,
KleinbergJ., ibid., 70, 267 A948); S с h e с h t e г W. H., T h о m p s о n J. К.,
Kl e i n b e r g J., ibid., 71, 1816, A949); Schech ter W. Н.Д 1 einbergJ.,
J. Chem. Ed., 24, 302 A947).
51. T h о m p s о n J. K., KleinbergJ., I Am. Chem. Soc, 73, 1243 A951).
52. Gilber't H. N., Chem, and Eng. News, 26, 2604 A948); см. также J о a n n i s A.,
ссылка [43].
53. Cohen A. J., J. Am. Chem. Soc, 74, 3762 A952).
54. Nantz D. S., пат. США 2633406 C1. V. 1953).
55. С a r t e г G. F., T e m p 1 e t о n D. H., J. Am. Chem. Soc, 75, 5247 A953).
56. В u n z e 1 E. G., К о h 1 m e у е г E. J., Z. anorg. Chem., 254, 1 A947).
57. Busse, P. B. report 73716 (frames 9541—9554).
58. Francis С. В., пат. США 2569158 B5. IX. 1951).
59. MarvinG. G., Schumb W. C, J. Am. Chem. Soc, 52, 574 A930).
60. Hosking K. F. G., Mining Mag., 89, 137 A953).
61. Rafter T. A., Analyst, 75, 485 A950).
62. S e e 1 у e F. Т., R a f t e г Т. А., англ. пат. 661527 B1. XI. 1951).
63. Wieland H., Liebig's Ann. Chem., 434, 185 A923).
64. Glasner A., J. Chem. Soc, 195], 904.
65. StruveH., Z. anal. Chem., 11, 22 A872).
66. Blumenthal M., Roczniki Chemji, 12, 119, 232 A932); 13, 5 A933).
67. S a t t e r f i e 1 d C. N., S t e i n T. W., Ind. Eng. Chem., 46, 1734 A954).
68. R i e s e n f e 1 d E. H., N о t t e b о h m W., A. anorg. Chem., 9, 371 A915).
69. Young J. H., пат. США 2533660 A2. XII. 1950).
70. Al taba M. F., Rev. real acad. dene, exact., ffs у nat. Madrid, 44, 271 A951)
[C. A., 46, 6467].
71. Hildebrand J. H., J. Am. Chem. Soc, 34, 256 A912).
72. Kendall J. K., FuchsF. J., J. Am. Chem. Soc, 43, 2017 A921).
73. Olson L. E., пат. США 2573481 C0. X. 1951).
74. Marshall W. H., Jr., пат. США 2583584 B9. I. 1953).
75. ZappR. L., FordF. P, J. Polymer Sci., 9, 97 A952).
76. Wood W. S.,ClennettG., пат. США 2563442 G. VIII. 1951).
77. Borax Consolidated Ltd., англ. пат. 679877 B4. IX. 1952) [С. А., 47, 4564 b].
78. Дрозин Н. Н., ЖПХ, 24, 86 A951).
79. Y о u n g J. H., пат. США 2556953 A2. VI. 1951).
80. Fat hall ah A. H., Parti ngton J. R., Nature, 164, 952 A949).
81. Wiberg К. В., J. Am. Chem. Soc, 75, 3961 A953).
82. Edwards J. O., J. Am. Chem. Soc, 75, 6154 A953).
83. SI a t er V. W., Woo d W. S., англ. пат. 568754 A9. IV. 1945) [С. А., 41, 428H];
пат. США 2448058 C1. VIII. 1948); Young J. H., пат. США 2541733
A3. II. 1951).
84. Constam Е. J., von Н a n s е п А., герм. пат. 91612 A896); Z. Elektrochem.,
3, 137 A896); L у m n A. H., J. Soc Chem. Ind., 16, 406 A897).
85. Н a i s's i n s k у М., С о t t i n M., Compt. rend., 224, 392 A947).
86. F i с h t e г F., В 1 a d e r g г о e n W., Helv. chim. Acta, 10, 566 A927).
87. Van RysselbergheP., Delahay P., Gropp A. H., McGeeJ. M.,
WilliamsR. D., J. Phys. & Colloid Chem., 54, 754 A950).
5&6 Глава 12
88. Billy M., Compt. rend., 172, 1411 A921); 186, 760 A928); В i 1 1 у М., S a n-G a 1-
1 i I., ibid., 194, 1126 A932).
89. S с h w a r z R., G i e s e H., Ber., 65, 871 A932).
90. J a h г К. J., Z. Elektrochem., 47, 810 A941); FIAT Rev. Ger. Sci., Part III, 1948,
p. 170.
91. Wai ton J. H., Jr., J. Am. Chem. Soc, 29, 481 A907).
92. D u к e F. R., В г e m e г R. F., Iowa State Coll. J. Sci., 25, 493 A951) [C. A., 45,
6947d].
93. К о s h 1 a n d D. E., Jr., Kroner J. C, Spector L, Natl. Nuclear Energy
Ser., Div. IV, 14B, Transuranium Elements, part I, 731 A949) [C. A., 44, 7699b].
94. Kali-Chemie A.-G., англ. пат. 679908 B4. IX. 1952) [С. А., 47, 4567b].
95. FichterF., MullerJ., Helv. chim. Acta, 1, 297 A919).
96. F i с h t e r F.,Gu tz will er E., Helv. chim. Acta, 11, 323 A928); 10, 551 A927).
97. Mayer J., Z. anorg. allgem. Chem., 161, 321 A927).
98. R i ven q F., Bull. soc. chim., 12, 283, 289 A945).
99. Beltran Martinez J., RodriguezRiosB., Anales real soc. espafi. fis.
y. quim. (Madrid), 48B, 388 A952).
100. R i u s A., M a r t i n M., Anales fis. y. quim. (Madrid), 40, 1034, 1057 A944).
101. S i d g w i с к N. V., The Chemical Elements and Their Compounds, Vol. 2, The Oxford
Press, Oxford, 1950, p. 1006.
102. Beltran Martinez J., Porter C, Anales real soc. espSn. fis. y. quim.
(Madrid), 48B, 879 A952) [C. A., 47, 5837 a].
103. Glasner A., J. Chem. Soc, 1950, 2795.
104. M u t h m a n W.,[Nagel W., Ber., 31, 1836 A898).
105. R о s e n h e i m A*, H а к к i M., KrauseO., Z. anorg. allgem. Chem., 209, 175
A932); G 1 e u K., ibid., 204, 67 A932); КобозевН. И., С о к о л о в Н. Н.>
ibid., 214, 321 A934).
106. К о 1 t h о f f I. M., P a r г у Е. P., J. Am. Chem. Soc, 73, 5315 A951).
107. J a h r K. F., L о t h e г Е., Ber., 71, 894, 903, 1127 A938).
108. R u m p f-Nor dma nn M. E., Compt. rend., 212, 485 A941).
109. J a h r K. F., В 1 a n k e M., Z. anorg. allgem. Chem., 272, 45 A953).
ПО. М о h г Р., пат. США 2551543 (I. V. 1951).
111. Co n n i с k R. E., Me Vey W. H., J. Am. Chem. Soc, 71, 1534 A949);*NatL
Nuclear Energy Ser., Div. IV, 14B, Transuranium Elements, part I, 445 A949).
112. N о d d а с k I., N о d d а с k W., Z. anorg. Chem., 181, 1 A929).
113. T h о m p s о n L. R., W i 1 m a r t h W. K., J. Phys. Chem., 56, 5 A952).
114. YamadaS., ShimuraY., T s u с h i d a R., Bull. Chem. Soc. Japan, 26, 72
A952) [C. A., 48, 445e].
115. Ha'issinskyM., QuesnyM., J. chim. phys., 49, 302 A952).
116. Ogg R. A., Jr., J. Chem., Phys., 21, 2079 A953).
117. Изгарышев Н. А., Петрова А. А., ЖФХ, 24, 88A950).
118. Buffalo Electro-Chemical Co., Buffalo, N. Y., Research and Development Dept., Bull.
№ 34, Uses of Persulfates—A Bibliography, 1951.
119. Tettamanzi A., Atti accad. sci. Torino, Classe sci. fis mat. e nat., 86, 265
A951 — 1952) [C. A., 47, 3746a].
120. Sc h m i dl i n J., Ma ss i n i P., Ber., 43, 1162 A910); d'A n s J., Friede-
r i ck W., ibid., p. 1880.
121. d'Ans J., Z. Elektrochem., 17, 850 A911).
122. G 1 e u K., R о e 1 1 E., Z. anorg. Chem., 179, 233 A929); Gleu К., Н ubol d R.,
ibid., 223, 305 A935).
123. Fichter F., В runner E., Helv. Chim. Acta, 12, 305 A929)
ОБОЗНАЧЕНИЯ11
Буквенные обозначения
Л —коэффициент в уравнении вязкости Литовича
а —показатель степени в уравнении вязкости Литовича
а —температурный коэффициент скорости реакции
а— активность (безразмерная)
а —константа дисперсии в формуле Зеллмейера, сек.
а —коэффициент в уравнении F), г/см3
а —коэффициент в уравнении C3), кал/моль-г рад
Ви В2 — коэффициенты избыточной свободной энергии в уравнении A1), кал/моль
6 —коэффициент в уравнении F), г/см3
6 —коэффициент в уравнении C3), кал/'моль- град2
Ср — теплоемкость при постоянном давлении, кал/моль-град
с—скорость света в вакууме, 2,998-1010 см/сек
с — состав в молярных единицах, моли растворенного вещества на 1 л раствора
с —коэффициент в уравнении @), г/см3
с —коэффициент в уравнении C3), кал/моль-град3
D — коэффициент диффузии в жидкости, см2/сутки
d — толщина слоя, поглощающего излучение, см
d — коэффициент в уравнении F), г/см3
d — коэффициент в уравнении C3), кал/моль-град1
d — знак дифференциала
?—электродвижущая сила или потенциал, в; Eq относится к стандартному со-
состоянию основного раствора с активностью, равной единице
? —внутренняя энергия, кал/моль
Е — напряженность электрического поля в уравнениях F8) — G5), в/см
? —энергия активации, кал/моль
е — основание натуральных логарифмов =-2,718 ...
У7 —свободная энергия, кал/моль; /^—избыточная свободная энергия раствора
с мольной долей х
3 —константа Фарадея, 23 060 кал/в-экв
Я—энтальпия, кал/моль
Я —напряженность магнитного поля, гауссы
[Л —константа Планка, 6,624-10~27 эрг-се к/молекула
/ — момент инерции, г-см2\ /^, /д, Iq относятся к осям Л, В, С; fre^ =
/ — интенсивность излучения (/о — падающего, /Л — выходящего)
У —коэффициент в уравнениях*G), (8) и (9), г/см3
J — квантовое число углового момента
К — коэффициент в уравнениях G), (8) и (9), г/см3-град
К— константа равновесия
К-
коэффициент погашения, (ат-см)'1; (K = -Tjlgr )
ч Р « 1Х/
* Применяемые обозначения соответствуют рекомендациям Американской ассо-
ассоциации стандартов, изложенным в их циркулярах Z 10.1 от 1 октября 1941 г., Z 10.6
ют 6 октября 1948 г. и Z 10.12 от 12 октября 1946 г. В том случае, когда указаны номера
уравнений, они относятся к гл. 5.
558 Обозначения
k — константа Больцмана, 1, 380 эрг /град.» молекула
k— объемная магнитная восприимчивость, ед. CGSM/сж3
k—константа скорости реакции
k — силовая константа, дины/см
L—коэффициент в уравнениях G), (8) и (9), г/см3-град2.
I—длина, см
М — химический молекулярный вес, г /моль
М — число молей воды на 1 моль перекиси водорода в смеси
М—коэффициент в уравнениях G), (8) и (9), г/см3-град3
т~— моляльный состав, моли растворенного вещества на 1 кг растворителя
т—магнитное квантовое число, уравнения F9) —G5)
N — число Авогадро, 6,023* 1028 молекул на моль
л — число молей
л — показатель преломления, безразмерная величина
л — число потенциальных минимумов при внутримолекулярном вращении, для
перекиси водорода равное 2 [уравнения C0) и C1)]
П{ — число неразличимых положений вращающихся групп при внутримолекуляр-
внутримолекулярном вращении, для перекиси водорода равное 1 [уравнения B7) —B9)]
л—число эквивалентов
Р — общее давление, мм рт. ст.
Р—проницаемость (безразмерная величина)
р — парциальное давление, мм рт. ст.
р' — парциальное давление, am
R—газовая постоянная, 1,987 кал /г рад.» моль
[/?] —молярная рефракция, см3/моль
г—удельная рефракция, см3/г
S — энтропия, кал/моль-град.
Т—температура, °К
t—температура, °С
V — моляльный объем, см3/моль [уравнение A5)]
V — потенциальный барьер, тормозящий внутримолекулярное вращение, кал/моль;
Vo — максимальное значение [уравнения B7) —C1)]
V — константа Верде, мин/гаусс*см
V — объемная крепость (стр. 165), число объемов кислорода, выделяющихся
при стандартной температуре и давлении, на 1 объем раствора перекиси
водорода при этих же условиях
V— удельный объем, см3/г=\/р
iT— парциальный молярный объем ( = dv/dn), см3/моль
W—состав, вес.% перекиси водорода (безразмерная величина)
w—состав, весовая доля перекиси водорода (безразмерная величина)
х — мольная доля в жидкости (безразмерная величина)
X—ЧИСЛО
х— группа hcQ/kT (безразмерная величина) [уравнения B7) —B9)]
# —мольная доля в паре (безразмерная величина)
ап—температурный коэффициент показателя преломления, град.'1
а —поляризуемость, см3/молекула
а — угол оптического вращения, минуты дуги
р—объемный коэффициент расширения, град.'1
Э—поправочный коэффициент , для газов, см3/моль
7 — коэффициент активности (безразмерная величина)
7 —силовая константа, дины/см
А — конечное приращение
Ь—силовая константа, дины/см
s — молекулярный коэффициент погашения, л/моль-см; е=— lg ~
Си I х
Обозначения 559
е —диэлектрическая проницаемость (безразмерная величина)
60 — угол между магнитным полем и направлением света
6 —угол ООН в молекуле перекиси водорода, градусы
6 — время
X—длина волны A (\ = c/v)
jjt — вязкость, микропуазы
{л — химический потенциал (=dF/dn), кал/моль
V —частота (v = oc), гц
V— частота, мггц
те — 3,1416 ...
р —плотность, г/см3 при нормальных температуре и давлении
а—волновое число (a = v/c= 1/X), см
о' —число симметрии в уравнении B9) для вращения молекулы как целого; для
перекиси водорода оно равно 2
ср —азимутальный угол между плоскостями, в которых расположены группы ОН
в молекуле перекиси водорода, градусы
Хо~-массовая магнитная восприимчивость, ед. CGSM/г
Хт — молярная магнитная восприимчивость, ед. CGSM/лсоль
Верхние индексы
Е — обозначение избыточного количества
М— обозначение величины, относящейся к смешению
0 —относится к величине или процессу для каждого вещества в стандартном
состоянии
Нижние индексы
А — относится к активации
а —обозначение свойства вещества а вообще
Ь — относится к коэффициенту Ь в уравнении F)
С—относится к свету с длиной волны 6562,8 А в воздухе
с —относится к коэффициенту с в уравнении F)
с — относится к свойству в критической точке
D — относится к процессу разложения
D— относится к свету с длиной волны 5893 А в воздухе
d — относится к коэффициенту d в уравнении F)
F — относится к свету с длиной волны 4861,33 А в воздухе
G — относится к свету с длиной волны 4340,46 А в воздухе
Л —обозначение свойства перекиси водорода
1 — обозначение свойства вещества i
у —обозначение свойства вещества /
L — относится к свойству жидкости
М — относится к процессу смешения
т—обозначение свойства смеси
/? — относится к реакции
Г —обозначение свойства или процесса при температуре Т
t — обозначение свойства или процесса при температуре t
V —относится к свойству пара или к процессу испарения
w—обозначение свойства воды
я —обозначение свойства при мольной доле х
х—число
(/ — ЧИСЛО
1, 2, 3 относится к последовательным состояниям или к разным процессам
0 —обозначение свойства или процесса при 0°К
0 —обозначение свойства чистого компонента
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агар-агар, стабилизация П. В.* 451
Аддитивные соединения с П. В., образова-
образование 298
Адипиновая кислота, из циклогексанона
действием П. В. 343
Азимутальный угол П. В. 271 и ел., 276 и ел.
Азобензол
восстановление в процессе получения
П. В. 70
окисление его П. В. 344
Азот
влияние на пределы воспламенения
П. В. 379
— на реакцию водорода и кислорода
42
определение по Кьельдалю с П. В. 498
Азота двуокись, помеха при анализе П. В.
458
Азота закись, определение при помощи
П. В. 498
Азота окись, помеха при анализе П. В. 463
Азота перекиси 550
Азотистая кислота, помеха при анализе
П. В. 463, 469
Азотистые соединения, реакции с П. В.
344
Акридины, стабилизаторы П. В. 450
Акриловое волокно, спецодежда для П. В.
154
Активированные комплексы 307
Активность П. В. 328
Актиниды
катализ разложения П. В. 416
реакции с П. В. 340
Актинометр 55
Ализарин, стабилизатор П. В. 450
Алканы
окисление с образованием П. В. 77
реакции с П. В. 342
Алкилгидроперекиси
кислотная диссоциация 322
от окисления алканов 77 и ел.
типы реакций 320
Алкилсульфонаты, стабилизация П. В. 451
Альдегиды
окисление П. В. 343
пероксидазоподобная активность 354
получение из альдоновых кислот дей-
действием П. В. 344
• П. В.—перекись водорода (везде).— Прим.
рев.
помеха при определении П. В. 469
равновесие с П. В. 80, 81
реакции с П. В. 343
Алюминиевые квасцы, пероксигидрат 535
Алюминий
анодное окисление 145
пассивирование 145
покрытие поверхности 145
Алюминий и его соединения
катализ разложения П. В. 416, 441,
444
реакции с П. В. 340
Алюминий и его сплавы, конструкционный
материал для работы с П. В. 52, 142,
145, 151, 153, 376, 436
Амальгама, реакция с П. В. 332
Аминокислоты, окисление их П. В. 344
Аминопирин, колориметрическое опреде-
определение П. В. 467
Амины, пероксидатические реакции с П. В.
353
Аммиак
окисление с образованием П. В. 82
реакции с П. В. 336
система с П. В. 249
Аммиака пероксигидраты 535, 550
Аммония карбонат пероксигидрат 535
Аммония пероксодисульфат 551 и ел.
гидролиз 114, 127
межатомные расстояния 532
причина астмы и экземы 357
электролитическое получение 126 и
ел.
Аммония сульфат, система с П. В. 249
Анализ
газометрический П. В. 12, 431 и ел.,
465 и ел.
качественный П. В. 455
количественный П. В. 460
колориметрический П. В. 466 и ел.
объемный П. В. 460 и ел.
полярографический П. В. 83, 467, 469
потенциометрический, количественное
определение П. В. 465
применение П. В. 497
хроматографический при определении
П. В. 469
Анаэробные организмы, подавление их 67
Анемия от П. В. 359
Анилин
окисление его П. В. 344, 345
система с П. В. 249
Анодное окисление, получение П. В. 107
Предметный указатель
561
Аноректальная хирургия, применение П. В.
514
Антигельминтные средства, применение
П. В. 515
Антисептическое действие П. В. 25, 67,
512 и ел.
Антитела, пероксидатические реакции с
П. В. 353
Антихлор 479, 489
Антозон 15 и ел., 457
Антрагидрохинон, образование П. В. при
окислении 71
Аппараты для отбелки действием П. В. 480
Аргон, влияние на реакции водорода с
кислородом 42
Ароматические углеводороды, реакции с
П. В. 345
Арсенит, количественное определение П. В.
464, 465, 469
Аскорбиновая кислота
количественное определение П. В. 465
окисление ее П. В. 521
пероксидазоподобна я активность 354
пероксидатическая реакция с П. В.
353
Ассоциация П. В. 166, 288 и ел., 292
Атмизон 16
Атмосфера
открытие П. В. 457
содержание П. В. 59
Атмосферные осадки, содержание П. В. 58
Ацетали, использование образования для
очистки П. В. 81
Ацетальдегид, определение с П. В. 498
Ацетанилид, стабилизатор П. В. 22, 450
Ацетатный шелк, отбелка 480
Ацетилена перекись 287
Ацетон
взрывчатые составы с П. В. 155
система с П. В. 249
торможение разложения П. В. 331
Ацетонитрил
влияние на реакции с П. В. 331
детонирующие составы с П. В. 156
Ацетоуксусная кислота, окисление ее П. В.
343
Ацетофенон, система с П. В. 250
Ацилгидроперекиси, кислотная диссоциа-
диссоциация 322
Бактерии
влияние на образование П. В. 354
действие на них П. В. 513
Бактерицидное действие П. В. 512 и ел.
Бария карбонат
образование при. реакции ВаОа с СО2
100
получение и отжиг 94, 104
прямое превращение в перекись 95
Бария окись
переокисление до перекиси 104
получение из карбоната 94
Бария перекиси октогидрат 97, 537, 543
Бария перекись
«антозонид» 16
межатомные расстояния 532
образование неподвижной пленки 98
опыты Тенара 10 и ел.
36 Перекись водорода
получение и свойства 93, 95, 542 и ел,
производственное получение 105
реакция с СО2 100
— с кислотами 96, 97 и ел.
способы превращения с П. В. 18 и ел.,
105
Бария пероксокарбонат, образование при
реакции ВаО2 с СОа 102
Бария пирофосфат, система с П. В. 249
Бария сульфат, восстановление до BaS
103
Бария сульфид, получение и превращение
в карбонат 95, 103 и ел.
Бария фосфаты 99
Безопасность работы с П. В. 161
Белки
деполимеризация П. В. 496
пероксидатическая реакция с П. В.
353 и ел.
Бензальдегид, окисление его П. В. 345
Бензамид, окисление его П. В. в Н2О18 326
Бензидин, идентификация П. В. 459
Бензидинхлоргидрат, реактив на П. В.
457
Бензойная кислота, стабилизатор П. В.
450
Бензол, система с П. В. 249
Бензолсульфокислота, стабилизатор 450
Бера закон для П. В. 245 и ел., 468
Бериллий и его соединения, катализ разло-
разложения П. В. 451
Берлинская лазурь,образование как реак-
реакция на П. В. 456
Бетон
конструкционный материал для работы
с П. В. 149
пористый, получение при помощи П. В.
509
Биологические катализаторы разложения
П. В. 418
Биологические процессы, роль П. В. 346
Биологические фотосенсибилизаторы обра-
образования П. В. 59
Бор и его соединения
катализ разложения П. В. 416, 451
реакции с П. В. 340
Борная кислота, покрытие поверхности
стекла 44, 377 и ел.
Борнокислый эфир для обезвоживания П.
В. 138
Боросиликатное стекло
влияние на разложение П. В. 375 и ел.
для реакторов для окисления про-
пропана 78
— реакции Н2 и О2 46
как .поглотитель излучения 443
— конструкционный материал для ра-
работы с П. В. 143, 149, 436
пассивирование поверхности 149 и ел.
Бочки для хранения П. В. 151
Бризантность П. В. 157
Бром
катализ разложения П. В. 398
реакции с П. В. 333
Бумага, очистка макулатуры от краски
действием П. В. 485
Бутиламины, системы с П. В. 250
2- mpem-Бутилантрахинон, производство
П. В. путем окисления его 74 и ел.
562
Предметный указатель
Бутилкаучук, регенерация при помощи
СаО2, SrO2, PbO2 и МпО2 544
В
Валентная связь, процесс разрыва в П. В.
310 и ел.
Ванадий и его соединения
катализ разложения П. В. 415
реакции с П. В. 399
Ванадия соединения, определение при
помощи П. В. 498
Ванадия соли, системы с П. В. 249
Ванилин, реактив на П. В. 457
Вдыхание П. В. 358
Велоформ, конструкционный материал для
работы с П. В. 143
Верде константа для П. В. 231
Ветеринария, применение П. В. 515
Взрыва пределы водородо-кислородных си-
систем 37 и ел.
Взрывчатость П. В. 154
Взрывчатые вещества, содержащие П. В.
508
Вина, улучшение их при помощи П. В. 517
Винилит, конструкционный материал для
работы с П. В. 143
Винная кислота, окисление ее П. В. 343
Виноградное сусло, предотвращение бро-
брожения его при помощи П. В. 517
Вирусы, роль П. В. в образовании 359
Вискозный шелк
отбелка 480, 481
ускорение созревания при помощи
П. В. 488
Висмут и соединения его
катализ разложения 399, 451
реакции с П. В. 336
Висмута основного нитрата пероксигидрат
535
Витамины, действие П. В. 355, 520
Вкус П. В. 13, 356
Вода обыкновенная
свойства 256
очистка ее П. В. 516
Вода тяжелая, свойства 256
Водород
атомарный, образование при фотохими-
фотохимическом облучении Н2 56
— реакции с Оа 48 и ел.
пределы взрыва смесей с кислородом
37 й ел.
термическая реакция с кислородом 39
и ел.
фотохимическая реакция с кислородом
55 и ел.
электронная структура 266 и ел.
Водорода дисульфид, структура 287
Водородные связи
П. В. 167, 288 и ел., 292 и ел.
пероксигидратов 309
энергия 294
Водородных ионов концентрация, влияние
на скорость разложения П. В. 438 и
ел., 440 и ел.
Воды, катализ разложения П. В. 416
Воздух, освежение действием П. В. 25
Волос, удаление его П. В. 496
Волосы
завивка, использование П. В. 497
обесцвечивание П. В. 25, 487 и ел.
реакция с П. В. 355
Вольфрам и его соединения
катализ разложения П. В. 415
реакции с П. В. 339
Вольфраматы, помехи при анализе П. В.
466
Вортит, конструкционный материал для
работы с П. В. 143
Воспламенение П. В., пределы 379 и ел.
Восстановление П. В. 298
Всаливание у П. В. 293
Всасывание П. В. 154
Выводка пятен при помощи П. В. 481
Выпаривание П. В., различные приборы
159 и ел.
Вязкость
жидкой П. В. 175 и ел., 256
— перекиси дейтерия 256
пара П. В. 176 и ел., 256
Газы инертные, влияние на реакцию На
и О2 42, 43
Галлий и его соединения
катализ разложения П. В. 401
реакции с П. В. 337
Галогены
воспроизводимость реакции с П. В.
302
катализ разложения П. В. 398
применение П. В. при анализе 498
реакции с П. В. 333
Гальванотехника, применение П. В. для
очистки 494
Гаммета о-функция 321
Гафний и его соединения
катализ разложения П. В. 416
реакции с П. В. 340
Гафния перекись 545, 547
Гваяковая смола, реактив на П. В. 456,
459
Гелий, влияние на пределы воспламенения
П. В. 379
Гем 349
Гемицеллюлозы, влияние на отбеллу 479
и ел.
Гемоглобин, строение 349
Гемопротеины
реакции 351 и ел., 418
строение и роль 349
Германий и его соединения
катализ разложения П. В. 400
реакции с П. В. 337
Гетероциклические соединения,» реакции с
П. В. 345
Гибридные структуры 268, 271
Гигроскопичность растворов П. В. 196
Гидравлические жидкости, материал для
П. В. 144
Гидразин
гиперголь 501
обмен меченого кислорода 327
реакция с П. В. 336
самоокисление 75
структура 288
Предметный указатель
563
Гидразингидрат в сочетании с П. В. в уско-
ускорителях 506
Гидразобензол, производство П. В. путем
окисления его 75
Гидразосоединения, образование П. В. при
окислении 70
Гидратация в биологии 347
Гидроксиламин
реакция с П. В. 336
структура 288
Гидроксильные ионы, механизм реакций
П. В. 317 и ел.
Гидроксильные радикалы 39 и ел., 43 и ел.,
47 и ел., 56 и ел., 61 и ел., 78, 82, 268 и ел.,
316 и ел., 335, 386 и ел., 398
Гидроперекиси органические, определение
в присутствии П. В. 469
Гидроперекиси, терминология 29
Гидропероксидаты, терминология 29
Гидросульфиты для отбелки 477
Гидрохиноны, образование П. В. при
окислении 70
Гиперголи 500
Гиперол 535
Гипобромиты, количественное определе-
определение П. В. 465
Гипосульфит, удаление его с фотоотпечат-
фотоотпечатков при помощи П. В. 492
Гипохлориты
для отбелки древесной массы и целлю-
целлюлозы 483
текстиля 477
сравнение с П. В. в отбелке 478 и ел.
(см. также Натрия гипохлорит, Каль-
Кальция хлоргипохлорит).
Глаза
действие П. В, 357, 358
защита от П. В. 154
Гликолевая кислота, окисление ее П. В.
343
Глиоксаль, окисление его П. В. 343
Глиоксиловая кислота, окисление ее П. В.
343
Глицерин
детонирующие составы с П. В. 156
реакция с П. В. 343
сочетание с П. В. в антисептических
средствах 513
Глюкозооксидаза 67, 351, 517
Головня, уничтожение ее П. В. 515
Горение П. В. 380
Гормоны, реакции с П. В. 353, 355
Д
Давление пара П. В. 181 и ел., 190 и ел.
Двуплоскостная форма П. В. 262 и ел.,
272, 277
Двуэлектронные обмены при П. В. 311
Дегидразы и их действие 346 и ел.
Дегидрирование в биологии 347
Дегидрогеназа пуриновая, катализатор об-
образования П. В. 67
Дезинфекция, применение СаО2 и MgO2
544
Дейтерий, влияние на свойства П. В. 251
и ел.
Дейтерия перекись
колебания 277 и ел.
переохлаждение 188
свойства 252 и ел.
Декалина гидроперекись, кислотное рас-
расщепление 321
Декарбоксилирование в биологии 347
Деполяризатор, П. В. 496
Деполяризация восстановлением кислорода
86 и ел.
Дерево, конструкционный материал для
работы с П. В. 143, 148
Детонация
П. В. 154 и ел.
скорости ее 155 и ел.
Джут, отбелка 480 и ел.
Диалкилперекиси
терминология 30
типы реакций 320
Диаминофлуорен, реактив на П. В. 457
Диацилперекиси, терминология 30
Дибензоил перекись
межатомные расстояния 532
механизм разложения 320 и ел.
отбелка муки 517
структура 287
энергия активации реакций 303
Ди-трет-бутилперекись
механизм разложения 320
структура 287
энергия активации реакций 303
1,4-Дигидронафталин, образование П. В.
при окислении 81
Диизобутиламин, система с П. В. 250
Диизопропиловый эфир, влияние на реак-
реакции П. В. 331
Диметиланилин, система с П. В. 250
Диметилперекись, межатомные расстоя-
расстояния 532
Диоксан
детонирующие составы с П. В. 156
система с П. В. 249
торможение разложения П. В. 331
Дипероксоортованадиевая кислота 548
Дипероксоугольная кислота, терминоло-
терминология 30
Дипольный момент П. В. 265 и ел., 272, 276
Дисперсии П. В. константа 230, 265
Дисперсия молярная П. В. 230
Диссоциация П. В.
кислотная 219
—постоянная 329
при электронной бомбардировке 219
и ел.
различные типы 219 и ел.
Дифтордиазин, структура 287
Диффузия
жидкой П. В. и ее растворов 178 и ел .
пара П. В. 180, 256
при реакции ВаОа с кислотой 98
Дихлордиоксидифениламин, колориметри-
колориметрическое определение П. В. 467
Диэлдрин 523
Диэлектрическая проницаемость
в анализе П. В. 468
П. В. и ее растворов 221 и ел., 251,
256, 291
Диэтиламин, система с П. В. 250
Диэтилперекись
типы реакций 320
энергия активации реакций 303
564
Предметный указатель
Длины связей
в П. В. 272, 275, 282 и сЛ.
— перекисях 532
Древесная масса
белизна 483 и ел.
отбелка при помощи П. В. и др. 482
. и ел.
Дрожжи, очистка их П. В. 517
Дубление кожи, роль П. В. 522
Дуримет, конструкционный материал: для
работы с П. В. 142
Дурогидрохинон, образование П. В% при
окислении 71
Емкости для П. В. 150
Ж
Жевательная резинка, .применение СаО2 544
Желатина, стабилизация П. В. 451, 452
Железа перекись 550
Железа соли в анализе П. В. 456, 463
Железа (III) сульфат в очистке HaSO4 124
Железо и его соединения
катализ разложения П. В. 409 и ел.,
416 и ел., 440 и ел., 444, 447 и ел.
определение при помощи П. В. 498
реакции с П. В. 339, 349 и ел.
Жиры и масла
действие П. В. 354
отбелка при помощи П. В. 488
Запах П. В. 13, 356
Зарядов распределение в молекуле 307
Звуковые колебания, образование П. В. 64
Зерно, улучшение его П. В. 517
Золото и его соединения
катализ разложения П. В. 405
реакции о П. В. 337
Золото коллоидное, образование как реак-
реакция на П. В. 456
Зубные пасты и порошки, применение СаОа
544
И
Изопрен, система с П. В. 250
Изотопный обмен при реакциях П. В.
323 и ел.
Изотопы водорода и кислорода, масса и
распространенность 252
Изоэлектронные структуры 287, 316
Ингибиторы коррозии, добавка к П. В.
133
Индиго, идентификация П. В. 458
Индий и его соединения
катализ разложения П. В. 401
реакции с П. В. 337
Индикаторы для арсенитометрического ана-.
лиза П. В. 464
Индукционный период
образования П. В. 78
разложения П. В. 377
Инконель, конструкционный материал для
работы с П. В. 142, 149
Инсектициды, применение ZnO2 544
Ионизирующие излучения
образование П. В. 60 и ел., 358
разложение П. В. 388 и ел.
Иониты
использование в очистке П. В. 139
— в получении П. В. из Na2O2 105
Ионные механизмы реакций П. В. 313
Иридий и его соединения
катализ разложения П. В. 409
реакции с П. В. 338
Испарители для концентрирования П. В.
137
История открытия и развития П. В. 9 и ел.
Иттрий и его соединения
катализ разложения П. В. 416
реакции с П. В. 340
Иттрия перекись 545
Йод и его соединения
катализ разложения П. В. 398, 416
реакции с П. В. 334
Йодиды, пероксидатические реакции 353
Кавитация, влияние на химические реак
ции 65
Кадмий и его соединения
катализ разложения П. В. 402
реакции с П. В. 337
Кадмия перекись 544
Кадмия соединения, стабилизация П. В.
451
Калия бихромат, реактив на П. В. 456
Калия бромат, фотосенсибилизатор образо-
образования П. В. 59
Калия йодид
для количественного определения П. В.
463, 466
идентификация П. В. 4Б8, 459
фотосенсибилизатор образования П. В.
59
Калия надперекись 536, 541
Калия нитрит, система с П. В. 249
Калия перекись, получение и свойства
540 и ел.
Калия перманганат
идентификация П. В. 458, 469
реактив для количественного опреде-
определения П. В. 460 и ел.
Калия пероксодисульфат 551 и ел.
гидролиз 124
получение и переработка на П. В.
119 и ел.
разложение в Н2О18 326 и ел.
система с П. В. 249
Калия пероксокарбонат 546
Калия пироантимоната пероксигидрат 535
Калия сульфат
обмен S*5 с пероксодисульфатом 327
система с П. В. 249
Калия ферроцианид
очистка им серной кислоты 124
реактив на П. В. 456
Калия фосфат, система с П. В. 249
Калия фторид пероксигидрат 535
Калия хлорат, система с П. В. 249
Калия хлорид
покрытие поверхности 50, 78
система с П. В. 249
Предметный указатель
565
Кальция окись
очистка П. В. 140
условия образования 107
Кальция перекиси октогидрат 537, 544
Кальция перекись 107, 536, 542, 544
для выделения П. В. 80
межатомные расстояния 532
Кальция перманганат для разложения П. В
502
Кальция хлоргипохлорит, количественное
определение П. В. 465
Камедь, стабилизация П. В. 451
Камеры разложения П. В. 502 и ел.
Карбонаты, реакции с П. В. 337
Каротин, пероксидазоподобная активность
354
Карциногенез, связь с П. В. 358
Каталаза
азидная, реактив на П. В. 457
определение с П. В. 498
пероксидатическое действие 353
разложение ею П. В. 347 и ел., 418
строение 349
устойчивость 358
Катализ
гетерогенный, механизм 393 и ел.
гомогенный, механизм 392 и ел.
Катализаторы
галогенирования П. В. 345
разложения П. В. 502
— места их в периодической
системе 391
— — — определение 469
регенерация отравленных катализато-
катализаторов при помощи П. В. 494
Каталитическое восстановление при про-
производстве П. В. методом самоокисления
75
Кататипия с П. В. 491
Катодное окисление при получении П. В.
107
Каучук
вулканизация с ZnO2 544
деполимеризация его П. В. 496
конструкционный материал для рабо-
работы с П. В. 143
реакции с П. В. 345
Квадрупольный момент 273
Квантовый выход
при фотохимическом образовании П. В.
55 и ел.
разложении П. В. 383 и ел.
Кварц
материал для пластин в процессе
Круща 52
работы с П. В. 149
прозрачный', разложение П. В. на
нем 376 и ел.
Керамика
в концентраторах П. В. 135
— перевозке П. В. 153
— разном оборудовании для П. В.
149
пористая, получение действием П. В.
509
Кетокислоты, реакция с П. В. 343
Кетоны алифатические, стабилизация П. В.
451
Кетоны, реакции с П. В. 343
37 Перекись водорода
Кислород
атомарный, отсутствие реакции с Н2 50
влияние на пределы воспламенения
П. В. 379
катодное восстановление 82 и ел.
межатомные расстояния в молекуле
532
меченый, отсутствие обмена между
П. В. и водой 323 и ел.
получение из П. В. 511
пределы взрыва смесей с водородом 37
и ел.
распределение электронов в молеку-
молекулах и ионах 315
реакция с атомарным водородом 48 и ел
термическая реакция с водородом 39
и ел.
электронная структура 266 и ел.
Кислорода фторид, помеха при анализе
П. В. 459
Кислотные свойства П. В. 329
Кислоты
влияние на разложение и стабиль-
стабильность П. В. 397 и ел., 438 и ел.
действие на П. В. 452
неорганические, влияние на фотохи-
фотохимическое разложение П. В. 384
— системы с П. В. 249
органические, детонирующие составы
с П. В. 156
— реакции с П. В. 343
— (с О18), восстановление П. В. 326
Кислоты — основания, идентификация
П. В. 459
Классы конструкционных материалов для
работы с П. В. 141 и ел.
Клей, стабилизация П. В. 452
Кобальт и его соединения
катализ разложения П. В. 408 и ел.,
451
применение П. В. в анализе их 498
реакции с П. В. 338
Кобальта перекись 550
Кожа
дубление при помощи П. В. 522
отбелка П. В. 487
реакция с П. В. 355
чувствительность к П. В. 357 н ел.
Колебания
изгибания П. В. 279
крутильные П. В. 280 ,
растяжения П. В. 277, 279
Колхицин, реакция с П. В. 345
Консервирование пищепродуктов с П, В.
516
Конструкционные материалы для П. В.
140 и ел.
Концентрация П. В.
влияние на выбор тары 151
— — комбинационное рассеяние 238
значение в производстве 132
различные зависимости 165
различные обозначения 165
Концентрирование П. В. 132 и ел.
разные методы 138
экономика 136
Коррозия
испытание с применением П. В. 496
образование П. В. 69
566
Предметный указатель
Кортизон 523
Косметика, применение СаОа и ZnO> 544
Кость, отбелка П. В. 25, 488
Кофейные экстракты, улучшение вкуса их
при помощи П. В. 517
Крабы, сохранение цвета действием П. В.
517
Красителей лейкоформы, образование П. В.
при окислении 70 и ел.
Красители
окисление действием П. В. 489
— пероксодисульфатами 552
реакция с П. В. 344
Крахмал
деполимеризация действием П. В. 496
пищевой, модификация его П. В. 517
стабилизация П. В. 452
Кремнезем, катализ разложения П. В. 400,
417
Кремнефтористоводородная кислота в ре-
реакции с ВаО3 18, 100
Кремний и его соединения, реакции с П. В.
337
Криоскопическая константа П. В. 189
Кристаллизация
гетерогенная, теория 188
дробная для концентрирования П. В.
138
Кристаллы П. В., внешний вид 186
Критическая температура П. В. 256
Критическое давление П. В. 256
Кротоновый альдегид, гиперголь 501
Круща процесс, описание 51 и ел.
Ксенон, фотосенсибилизатор 58
Культуральные среды, регенерация их
П. В. 517
Кумола гидроперекись
кислотное расщепление 321
механизм разложения 321
энергия активации реакции 303
Купферрон, стабилизатор П. В. 450
Лакмус, идентификация П. В. 458
Лантана перекись 545
Латекс
деполимеризация П. В. 496
переработка на пенистую резину при
помощи П. В. 510
Летучесть относительная воды и П. В.
196
Лецитин, стабилизация его П. В. 517
Лягнин, влияние на отбелку 479 и ел., 485
Ликвидуса диаграмма
для системы П. В.— вода 181 и ел.
— — перекись дейтерия — тяжелая
вода 252 и ел.
Линейная структура П. В. 18, 262 и ел.,
276
Литература по П. В. 25 и ел., 345, 481
Лития надперекись 537
Лития нитрат, система с П. В. 249
Лития перекиси пероксигидрат 536
Лития перекись 537
Лития сульфат, система с П. В. 249
а-Лучи, влияние на образование П. В.
61 и ел.
Э-Лучи, влияние на образование П. В.
62 и ел.
f-Лучи
влияние на образование П. В. 60 и ел.
влияние на разложение П. В. 388 и ел.
Люизит, лечение ожогов при помощи П. В.
514
Люминесцентные реакции
для качественного открытия П. В.
457, 459
с П. В. 344
Л юминол
реактив на П. В. 457
реакция с П. В. 344
М
Магний и его составы, конструкционный
материал для работы с П. В. 142, 436
Магнитная восприимчивость П. В 224
и ел., 256, 265
Магнитное вращение П. В. 265
Магнитное поле, влияние на реакции П. В.
332
Магнитооптическое вращение П. В. 231
Магния-аммония арсената пероксигидрат
535
Магния основного карбоната пероксигидрат
535
Магния перекись 544
Магния силикат, стабилизатор П. В. 450,
451
Магния соединения, стабилизация П. В.451
Малахитовый зеленый, обесцвечивание его
П. В. 344
Малеиновая кислота, окисление ее П. В.
343
Малин-уилстабрайт, конструкционный ма-
материал для работы с П. В. 142
Маннит пероксигидрат 535
Марганец и его соединения
катализ разложения П. В. 413 и ел.,
451, 453
реакции с П. В. 339
Марганца двуокись
как «озонид» 16
отсутствие перекисного характера 531
Марганца соединения, пероксидатические
реакции с П. В. 353
Марганца сплавы, конструкционный мате-
материал для работы с П. В. 142
Масла для П. В. 143, 144
Масляный желтый, реакция с П. В. 344
Масс-спектрометрия в анализе П. В. 468
Меди окись, колориметрическое определе-
определение П. В. 467
Меди перекись гидратированная 541
Меди перекись основная 541
Медицина, применение П. В. 14, 25, 512 и
ел.
Медь
значение образуемых перекисей для
разложения П. В. 541
и ее соединения, катализ разложения
П. В. 148, 403, 416 и ел., 440, 444,
451 и ел.
реакции с П. В. 337
конструкционный материал для рабо-
работы с П. В. 142
определение при помощи П. В. 498
Предметный указатель
567
Мезоксалевая кислота, окисление ее П. В.
343
Меланин, обесцвечивание П. В. 356
Меркаптаны, образование действием П. В.
344
Металлы
очистка П. В. 492 и ел., 495
разнородные, в конструкционных мате-
материалах для работы с П. В. 141
самоокисление 68
травление поверхности с применением
П. В. 495
Метанол
детонирующие составы с П. В. 156
использование в очистке П. В. 81
— — получении концентрированной
П. В. 138
Метил-л-оксибензоат, стабилизатор П. В.
450
Метионин, определение с П. В. 498
Меха, отбелка П. В. 487
Механизм
образования П. В. из водорода и кис-
кислорода 43 и ел.
окисления пропана 79 и ел.
разложения П. В. 380 и ел.
реакций П. В. 304, 313
— — — с ионом гидроксила 317 и ел.
__ _ _ пергидроксила 318
_ — радикалом гидроксила 316
— — — — — пергидроксила 318
— разных перекисей 319 и ел.
стабилизации 452
фотохимического разложения 385
и ел.
Миласа реактив 342
Миоглобин, строение 349
Многокомпонентные системы с П. В. 248
и ел.
Молекулярный вес
П. В. 166, 256
перекиси дейтерия 256
Молекулярный объем П. В. 265
Молибдаты
колориметрическое определение П. В.
467
помехи при анализе П. В. 466
Молибден и его соединения
катализ разложения П. В. 415
определение при помощи П. В. 498
реакции с П. В. 339
Молоко
влияние добавки П. В. на свойства 520
замороженное, устранение сального
привкуса 521
консервированное П. В. 518 и ел.
устранение привкуса от солнечной
активации 521
Моляльные объемы П. В. 173
Моменты инерции П. В. 232, 281, 283
Монель-металл, конструкционный мате-
материал для работы с П. В. 142, 149
Мох торфяной, реакции с П. В. 345
Мочевины пероксигидрат 290, 535
антисептик 513
Мультимет, конструкционный материал для
работы с П. В. 142
Мутации от П. В. 358
Мыла белые, цветостойкие 523
Мышьяк и его соединения
катализ разложения П. В. 399
реакции с П. В. 336
Н
Надперекиси ион 270, 315 и ел. л
Надперекиси
межатомные расстояния 532
неорганические, получение 537 и ел.
определение 97
Надперекись Н2О4, вопрос существовании-
46
Наркотин, реакция с П. В. 345
Натрия арсената (втор) пероксигидрат 535
Натрия борат, стабилизация П. В. 451
Натрия гидроперекиси и пероксигидраты
536, 540
Натрия гидроперекись 536, 540
Натрия гипофосфит, стабилизатор П. В.
449
Натрия гипохлорит, количественное опре-
определение П. В. 465, 469
Натрия глутамат, отбелка 517
Натрия карбоната пероксигидрат 534 и ел.,
546
Натрия карбонат, система с П. В. 249
Натрия кремнефторид, стабилизация П. В.
451
Натрия метабората пероксигидрат 535
Натрия молибдат, стабилизация П. В. 45!
Натрия нитрат, система с П. В. 249
Натрия перекиси октогидрат 539
Натрия перекись 536, 538 и ел.
как источник П. В. 105, 539
— окислитель 540
применение для отбелки 477, 539
— — очистки 493, 540
производство из металлического натрия
105
реакция с лигнином 485
Натрия перманганат для разложения П. В.
502
Натрия пероксоборат 545
в завивке волос 497
для отбелки в быту 482
Натрия пероксокарбонат 546
как источник П. В. 138
Натрия пирофосфат, стабилизация П. В.
135, 449, 451 и ел.
Натрия пирофосфата пероксигидрат 535,
547
Натрия силикат
роль в отбелке 481, 486
стабилизация П. В. 439, 450, 451
Натрия силиката пероксигидрат 535
Натрия станнат, стабилизатор П. В. 135,
446 и ел., 452
Натрия сульфат, система4 с П. В. 249
Натрия сульфата пероксигидрат 535
Натрия сульфит
количественное определение П. В.. 464,
469
реакция с П. В. в Н2О18 324
Натрия тетраэтилпирофосфат в стабилиза-
стабилизации П. В. 449
Натрия фосфат
влияние на поверхность сосудов 376
и ел.
37*
388
Предметный указатель
система с П. В. 249
стабилизация П. В. 451
Натрия фосфата пероксигидраты 535
Натрия фторид, система с П. В. 249
Натрия хлорат, система с П. В. 249
Натрия хлорид, система с П. В. 249
Натрия хлорит для отбелки 477
Нафталины, окисление их П. В. 522
Нейтроны
влияние на образование П. В. 60 и ел.
— — разложение П. В. 389
Неодима перекись 545
Неорганические вещества, жидкофазные
реакции с П. В. 332
Нептуния перекись 549
Нефтепереработка, регенерация отработан-
отработанных растворов при помощи П. В. 494
Никель и его соединения
катализ разложения П. В. 149, 406, 451
реакции с П. В. 338
Никеля (III) окись, реактив на П. В. 456
-Никеля перекись 550
Никотин, реакции с П. В. 345
Никотиновая кислота, реакция с П. В. 345
Ниобий и его соединения
катализ разложения П. В. 415, 451
применение П. В. в анализе их 498
реакции с П. В. 339
Нитрилы, окисление их П. В. до амидов
344
Нитриты
окисление их П. В. при анализе 498
пероксидатические реакции с П. В. 353
Нитробензол
окисление его П. В. 344
система с П. В. 249
Номенклатура перекисных соединений 26
и ел.
Обертоны в инфракрасном спектре П. В. 280
Обмен электронами при гомогенном ката-
катализе 395
Образование П. В.
биологическое 64, 67 и ел., 348
в Н2О18 324 и ел.
из Н2 и О2 35, 40 и ел.
из воды 35
при действии ионизирующих излуче-
излучений 60 и ел.
— — кислорода на ртуть 69
=— катодном восстановлении О2 82 и
ел., 86
— коррозии 69
— растворении золота 69
— реакциях с участием фтора 82
— самоокислении аммиака 82
— — гидразина 75
— — гидроароматики 81
— — металлов 68
— — органических веществ 69 и ел.
— — пропана 77 и ел.
— — спиртов 81
— — щавелевой кислоты 69
—ультразвуковых и звуковых коле-
колебаниях 64 и ел.
— фотохимическом окислении и дес-
деструкции хлопка 490
при электрическом разряде47 и ел., 53
фотохимическое 55 и ел.
Объемная крепость П. В. 23, 165
Объемного расширения растворов П. В.
коэффициент 174
Овощи, П. В. в бланшировке 517
Одноэлектронные обмены при П. В. 311
Ожоги, лечение П. В. 515
Озон
межатомные расстояния 532
молекулярный вес 17
образование им надперекиси калия 538
П. В. 17
— при реакции фтора с водой 82
— — фотохимической реакции О2 и Н3
57
помеха при анализе П. В. 458 и ел.,
463, 469
реакции с П. В. 332
структура 268
Озоно-антозоновая теория 15 и ел., 457
Окисление П. В. 298
Окисление сопряженное 348
Окислитель жидкий
П. В. как окислитель в двухкомпонент-
ных ракетных топливах 499, 506
и ел.
сравнение П. В., хлора и СгО3 533
Оксианион 318
Оксидазы, катализаторы образования П. В.
67
8-Оксихинолин, стабилизатор П. В. 135,
345, 448
Олова гидроперекись для очистки П. В. 139
Олова двуокись коллоидная, стабилизатор
П. В. 446, 452
Олова дихлорид для количественного опре-
определения П. В. 463
Олово
определение при помощи П. В; 498
прокладочный материал для работы
с П. В. 143, 147, 436
Олово и его соединения
катализ разложения П. В. 400, 444
реакции с П. В. 337
Орбиты молекулярные 289
Органические вещества
влияние на взрывобезопасность П. В.
137
— — разложение П. В. 155
— — фотохимическое разложение
П. В. 385
реакции с П. В. 341 и ел.
самоокисление 69 и ел.
Осмий и его соединения
катализ разложения П. В. 413
определение при помощи П. В. 498
реакции с П. В. 339
Отбелка
гидросульфитами 477, 484
гипохлоритами 477 и ;сл., 483
двуокисью хлора 485
катализаторы отбелки 477
пероксодисульфатами 552
применение П. В. 25, 477 и ел., 484
— СаО2 и MgO2 544
— стабилизаторов 451
Отбора правило 276
Отравление П. В:. 154
Предметный указатель
569
Очистка
П. В. 11 и ел., 80, 139 и ел.
солей металлов 492
П
Палладий и его соединения
катализ разложения П. В. 406
реакции с П. В. 338
Паральдегид, фотосенсибилизатор образова-
образования П. В. 59
Парафин
материал для работы с П. В. 144
пригодность в контакте с П. В. 437
Парахор П. В. 264
Пар П. В.
вдыхание 153
взрывчатые свойства 159
лабораторное генерирование 158
разные свойства (см. Плотность, Вяз-
Вязкость и т. д.)
Пассивирование поверхности 145, 149
437, 446
Пенициллин В, антибактериальная актив-
активность, связанная с П. В. 67
«Перводная» кислота 27
Пергидроксил, терминология 29
Пергидроксила ион 270, 318
Пергидроксильные радикалы 39 и ел., 43
и ел., 48 и ел., 56 и ел., 61 и ел., 268
и ел., 315, 333, 387 и ел., 398.
Перевозка П. В. 150, 153
Перегонка П. В. 132 и ел., 139
Перекиси
взаимозаменяемость с П. В. 476
ион 270
неорганические, идентификация П. В.
в их присутствии 459
— получение 535 и ел.
— таблица 533 и ел.
органические, библиография 345
— идентификация П. В. в их присут-
присутствии 459
— из ангидридов кислот и Na2O2 540
— определение в присутствии П. В.
469 '
— от самоокисления органических ве-
веществ 69 и ел.
— структура 287
характер перекисей, образующихся
при самоокислении углеводородов
76 и ел.
Перекисная группа, перенос в П. В. 298
Перенапряжение, значение для электро-
электролитического получения П. В. 109
Переохлаждение
основы его 188
П. В. 187 и ел.
Перитектическая точка П. В. 183
Перманганат, окисление П. В. в Н2О18 324
Перманганатный метод анализа (см. Калия
перманганат)
Пероксигидраты 534
образование 309
терминология 29
Пероксидаза
биологическое значение 353
определение при помощи П. В. 498
строение 349
Пероксидатические реакции 348, 352 и ел.
Пероксоазотная кислота 554
Пероксобораты, анодное получение 107
Пероксованадиевая кислота 548, 553
Пероксовольфрамовая кислота 549
Пероксодигерманиевая кислота 553
Пероксодиоловянная кислота 553
Пероксодисерная кислота
гидролиз 113, 118 и ел.
получение 53, 107, 551
помеха при анализе П. В. 468
энергия активации реакций 303
Пероксодисульфаты, обмен меченого кисло-
кислорода 327
Пероксодиугольная кислота, терминология
30
Пероксокарбонаты, анодное получение 107
Пероксокислоты
из кислот и Na2O2 540
определение в присутствии П. В. 469
регенерация ими отравленных катали-
катализаторов 495
терминология 28 и ел.
типы реакций 320
токсичность 357
Пероксомолибденовая кислота 549
Пероксомоносерная кислота
получение 53, 552
помехи при анализе П. В. 468
Пероксомонофосфорная кислота 553
Пероксониобиевая кислота 548, 553
Пероксосоединения 27
образование при самоокислении 66
и ел.
Пероксотанталовая кислота 548, 553
в анализе П. В. 458, 466, 534, 547, 553
Пероксоугольная кислота 546, 553
терминология 30
Пероксоуксусная кислота
как активное начало при применении
П. В. 476
кислотное расщепление 321
опрыскивание помидоров 517
Пероксофосфорная кислота 547
Пероксохроматы, образование 339
Пероксохромовая кислота 548, 553
образование в качественной реакции
на П. В. 456
специфичность как реактив на П. В. 458
Пероксоциркониевая кислота 534, 547
Пероксоэфиры
терминология 30
типы реакций 320
Перья, отбелка П. В. 25, 487
Петролейный эфир, система с П. В. 249
Пинакона пероксигидрат 535
Пиперазин, реакция с П. В. 345
Пиперидин, система с П. В. 250
Пиразины, реакция с П. В. 345
Пирамидальная тригональная структура
П. В. 262 и ел., 272 и ел.
Пирекс (см. Боросиликатное стекло)
Пиридинкарбоновые кислоты, стабилизато-
стабилизаторы П. В. 450
Пирогаллол, реакция с П. В. 344
Пирокатехин, гиперголь 501
Пиррол, гиперголь 501
Плавиковая кислота, влияние на поверх-
поверхность сосудов 376
570
Предметный указатель
Платина и ее соединения
катализ разложения П. В. 406 и ел., 418
реакции с П. В. 338
Платифиллин, определение при помощи
П. В. 498
Плоскостная тригональная структура П. В.
262 и ел., 272
Плотность
в анализе П. В. 468
водных растворов П. В. 168 и ел.
жидкой П. В. 168 и ел., 256
жидкой перекиси дейтерия 256
пара П. В. 174 и ел.
твердой П. В. 167 и ел., 256
смесей П. В. с эфиром 251
Плутония перекись 549
Поверхностноактивные вещества
влияние на свойства П. В. 177
катализ разложения П. В. 416
стабилизаторы П. В. 450
Поверхностное натяжение
жидкой П. В. 177 и ел., 256
перекиси дейтерия 456
Поверхность
влияние на разложение П. В. 375 и ел.
стабильность П. В. 375 и ел.,
436 и ел.
пассивирование 145, 149, 437, 446
Погашения коэффициент П. В. 243, 245 и ел.
Подводные лодки, П. В. как источник
энергии 500, 504 и ел.
Позитронов полупериод существования в
П. В. 274
Показатели преломления
в анализе П. В. 467
П. В. и ее растворов 227 и ел., 256
перекиси дейтерия 256
Полиакрилонитрил (орлон), отбелка 481
Полиамидные волокна, отбелка 481
Полиамиды, конструкционный материал
для работы с П. В. 143
N-поливинилкарбазол, материал для пла-
пластин в процессе Круща 52
Полимеризация
с пероксодисульфатами как инициато-
инициаторами 552
с П. В. 511
фотосенсибилизация действием П. В.
386 и ел.
Политетрафторэтилен, конструкционный ма-
материал для работы с П. В. 143, 148
Политрифторхлорэтилен, конструкционный
материал для работы с П. В. 143, 148
Полихлорвинил, применение для работы
с П. В. 143, 148, 154
Полиэтилен . о
галогенироваыный, применение для ра-
работы с П. В. 143, 148
конструкционный материал для рабо-
работы с П. В. 143, 148, 154, 437
Полиэфирное волокно, спецодежда для
П. В. 154
Полоний
катализ разложения П. В. 399
растворение в П. В. 335
Поляризация П. В. 230, 265
Потенциалы
возникновения радикалов и ионов из
П. В. 220, 274
ионизации 274
окисления—восстановления П. В. 108,
216
электродные П. В. 216, 299 и ел., 359
и ел.
Потенциальный барьер П. В. 272 и ел.
Почвы и минералы, катализ разложения
П. В. 416
Празеодима перекись 545
Применение П. В. 23 и ел., 475 и ел.
анализ 498
ВВ 508 и ел.
газообразователь 509 и ел.
деполяризатор 496 и ел.
завивка волос 497
источник кислорода 511
источник свободных радикалов511 и ел.
— энергии 498 и ел.
консервирование пищепродуктов 516
и ел.
медицина 25, 512 и ел.
металлообработка 495 и ел.
модификатор 496
окислитель 488 и ел.
освежение воздуха 25
отбелка 25, 477 и ел.
очистка 492 и ел.
реставрация картин 14
сельское хозяйство 515 и ел.
статистика 474 и ел.
фотография 491 и ел.
химсинтез 522 и ел.
Примеси
влияние на безопасность П. В. 137
— — сохранность П. В. 133
Пробка, конструкционный материал для
работы с П. В. 143
Производство П. В.
восстановление кислорода 85 и ел.
из перекиси бария 19, 24, 102 и ел.
— — натрия 105
постановка производства 22
самоокисление 71 и ел., 80 и ел.
статистика 115 и ел., 474
термическая реакция Н2 и О2 45 и ел.
цены 22, 475 и ел.
электрический разряд 50 и ел.
электролитические методы 24, 114 и
ел., 119 и ел., 126 и ел.
Проницаемость коллодия для П. В.
180
Пропан, исследование окисления 77
Пропилен, влияние на окисление пропана
78
Пропионитрил, влияние на реакции П. В.
331
Пространственная структура П. В. 289
Протонный резонанс П. В. 274
Равновесие
диссоциации П. В., константа 218 и ел.
образования пара П. В. 214
разложения П. В., константа 213
реакции Н2 и О2 36
Разбавления П. В. теплота 206 и ел.
Разложение П. В. 13 и ел., 297
адиабатическое 159
Предметный указатель
571
влияние температуры при наличии ка-
катализаторов 438
возможность снижения до нуля 302
гетерогенное 303, 378 и ел., 393 и ел.
гомогенное 303, 378, 381, 392 и ел.
действием ионизирующих излучений
388 и ел.
изотопный обмен 324 и ел.
каталитическое жидкофазное 390 и ел.
парофазное 373 и ел.
периодический катализ 416
при кипячении 197
при плавлении 186
промотирование катализаторов 417
скорость 443 и ел.
— в присутствии ^стабилизаторов 447
— и порядок реакции 434
техника разложения П. В. для гене-
генерирования энергии 502 и ел.
торможение катализа 418
факторы его 140
фотохимическое 382 и ел., 442
— в Н2О18 326
электролитическое 389 и ел.
Ракета V-2, П. В. как источник энергии 507
Ракетное топливо, одно- и двухкомпонент-
ное 500, 506 и ел.
Рамзая — Этвеса ~~ Шилдса константа для
П. В. 290
/-Рамноза, реакция с П. В. 344
Распределение разных веществ между П. В.
и водой или растворами солей 249 и ел.
Рассела эффект 68, 338
Растворители
для производства П. В. но методу са-
самоокисления 72 и ел.
необычные для реакций П. В. 330 и ел.
Растения, улучшение развития действием
П. В. 516
Рауля закон, отклонения для П. В. 190
Реакции П. В. 13
скорости 302
с отдельными веществами 332 и ел.
типы 17, 306 и ел.
Реакционная среда с П. В. 328
Редкие земли
катализ разложения П. В. 416
реакции с П. В. 340
Редокс-системы с П. В. 511
Резина пенистая, получение при помощи
П. В. 510
Релеевское рассеяние П. В. 242 и ел.
Рений и его соединения
катализ разложения П. В. 415
реакции с П. В. 339
Рения перекись 550
Рентгеновская диффракция П. В. 248
Рентгеновские лучи 359
влияние на образование П. В. 61 и ел.
в разложении П. В. 388 и ел.
Рентгеновское исследование П. В. 275
Реставрация картин, применение П. В. 14
Реторты для концентрирования П. В. 135
Рсфракталлой, конструкционный материал
для работы с П. В. 142
Рефракции
молекулярная П. В. 230, 264, 276
удельная П. В. 230 (см. также Показа-
Показатели преломления)
Рибофлавин, влияние на образование П. В.
354
Родаииды
колориметрическое определение П. В.
467
реактив на П. В. 457
реакция с П. В. 336
Родий и его соединения
катализ разложения П. В. 409
реакции с П. В. 338
Ртути (I) нитрат, количественное опреде-
определение П. В. 465
Ртути перекись 544 и ел.
Ртуть, сенсибилизатор фотохимической ре-
реакции О2 и Н2 55
Ртуть и ее соединения
катализ разложения П. В. 402, 416, 453
реакции с П. В. 387
Рубидия надперекись 536
Рубидия пероксокарбонат 546
Рубидия «полуторная окись» 537
Рутений и его соединения
катализ разложения П. В. 413
реакции с П. В. 339
Салицилальдоксим, стабилизатор П. В.
450
Самария перекись 545
Самолет-снаряд V-1, П. В. как источник
энергии 508
Самоокисление 66 и ел.
Сапфир синтетический, конструкционный
материал для работы с П. В. 143
Сахар, система с П. В. 250
Свет солнечный, влияние на стабильность
П. В. 442
Свинец, детали в производстве и примене-
применении П. В. 142, 147
Свинец и его соединения
катализ разложения П. В. 401, 441, 453
реакции с П. В. 337
Свинца двуокись, отсутствие перекисного
характера 531
Свинца нитрат, система с П. В. 249
Свинца сульфид
его окисление П. В. 14
реактив на П. В. 456
Свободная энергия
диссоциации П. В. 218 и ел.
образования пара П. В. 214, 256
— — перекиси дейтерия 256
— П. В. в растворе 215
— радикалов из П. В. 220
смешения П. В. 209
разложения П. В. 213 и ел.
Свободнорадикальные механизмы реакций
П. В. 313
Свободные радикалы, П. В. как источник
511
Свойлачивание, влияние П. В. 487
Селен и его соединения
катализ разложения П. В. 399
реакции с П. В. 335
Семена
протравливание при помощи П. В. 515
рост всхожести и прорастания в ре-
результате действия П. В. 515
572
Предметный указатель
Сера и ее соединения
катализ разложения П. В. 399
применение П. В. при анализе 498
Серебра (II) окись, отсутствие перекисного
характера 531
Серебра феррицианид, реактив на П. В. 457
Серебро активированное, идентификация
П. В. 459
Серебро и его соединения
катализ разложения П. В. 403 и ел.,
417 и ел., 440 и ел., 453
реакция с П. В. 337
Серебряная фольга, идентификация П. В.
458
Серная кислота
очистка 124
реакция с П. В. в Н2О18 325
Сернистая кислота, окисление П. В. в
Н2О1Й 325
Серы двуокись для отбелки 477
Серы соединения
реакции с П. В. 334, 344
сравнение структуры их и кислород-
кислородных соединений 335
Силикаты, помехи при анализе П. В. 466
Силиконы
защитный слой для стекла 150
материал для П. В. 143
Силовые постоянные П. В. 282 и ел.
Силовые установки, работающие на П. В.,
для подводных лодок 505
Симметрия молекулы П. В. 276
Скайдрол, материал для П. В. 144
Скандий и его соединения
катализ разложения П. В. 416
реакции с П. В. 340
Слоновая кость, отбелка П. В. 25, 488
Смешение П. В.
с водой 205 и ел., 292 и ел.
— органическими веществами 251
Содействие среды 308
Соединение D2O2-2D2O 253
НО2 538
Н2О2.2Н3О 181 и ел., 293
К2О3 537
Соли, влияние на фотохимическое разложе-
разложение П. В. 384 и ел.
сравнительная растворимость в воде
и П. В. 250
Солома, отбелка 25, 486
Сополиамиды, определение при помощи
П. В. 498
Составные полосы в инфракрасном спектре
П. В. 280
Спектр видимый
П. В. 242 и ел.
влияние на разложение П. В. 442
Спектр инфракрасного поглощения
использование в анализе П. В. 468
П. В. 233 и ел., 275 и ел.
перекиси дейтерия 255
Спектр испускания в парах П. В. 247
Спектр комбинационного рассеяния
метилового эфира, влияние П. В. 251
П. В. 236 и ел., 277 и ел.
перекиси дейтерия 256
Спектр микроволновой
П. В. 232
перекиси дейтерия 254
Спектр ультрафиолетового поглощения
П. В. 243 и ел.
в анализе П. В. 468
влияние на разложение П. В. 442
в эфире 251
перекиси дейтерия 256
Спецодежда для работы с П. В. 154
Спирты
влияние на реакции П. В. 331
детонирующие составы с П. В. 138, 156
окисление с образованием П. В. 81
реакции с П. В. 343, 348
системы с П. В. 249
Сплавы
катализ разложения П. В. 416
каталитическая активность в зависи-
зависимости от состава 395 и ел.
Стабилизаторы П. В. 21 и ел., 132 и ел.
выбор, 451 и ел.
определение в П. В. 469
принцип действия 445 и ел.
Стабильность молекул 268
Стабильность П. В. 12
влияние излучений 442
— рН 438 и ел., 440 и ел.
— природы поверхности 436 и ел.
— температуры 437 и ел.
— удельной поверхности сосудов 434
и ел.
выражения для скорости разложения
433 и ел.
газометрический анализ 431 и ел.
определение по потере веса 433
прямой анализ 430 и ел.
Сталь, определение титана при помощи
П. В. 498
Сталь нержавеющая
конструкционный материал для рабо-
работы с П. В. 136, 142 и ел. 147, 436
коррозионная устойчивость и пригод-
пригодность для работы с П. В. 146
Сталь углеродистая, применение в кон-
контакте с П. В. 147
Сталь хромистая, применение с П. В. 147
Стекло
влияние обработки поверхности на
разложение П. В. 375 и ел.
-, — — _ реакцию О2 и Н2 42
конструкционный материал для рабо-
работы с П. В. 21, 149, 153
содово-известковое, разложение П. В.
на нем 376, 436
цветное для работы с П. В. 442
Стекловидная форма П. В. 48, 263 и ел.
Стеклянный электрод для измерения рН
в П. В. 329
Стеллит, конструкционный материал для
П. В. 143
Стирка белья с отбелкой П. В. 481
Сточные воды от П. В., ликвидация 153
Стрептококки, содержание П. В. 67
Стрептомицин, влияние на образрваиие
П. В. 354
Стронция карбоната пероксигидрат 535
Стронция окись, условия образования 106
Стронция перекиси октогидрат 537
Стронция перекись 536, 542
межатомные расстояния 532
условия образования 106
Предметный указатель
573
Структуры молекулы П. В. 262
Ступенчатые реакции, возможность для
П. В. 305
Сульфаниламиды, значение П. В. для ме-
механизма их действия 514
Сульфаты металлов, очистка П. В. и Na2O2
493
Сульфиды, окисление их П. В. 344
Сульфоксиды, получение окислением суль-
сульфидов 344
Сульфонамиды, стабилизаторы П. В. 450
Сульфоны, получение из сульфоксидов 344
Сурьма и ее соединения
катализ разложения П. В. 399
реакции с П. В. 336
стабилизация П. В. 451
Таллий и его соединения
катализ разложения П. В. 401, 451
реакции с П. В. 337
Тантал, конструкционный материал для
работы с П. В. 147
Тантал и его соединения
катализ разложения П. В. 415, 451
реакции с П. В. 339
Твердые растворы, образование из П. В.
и воды 167, 184 и ел., 289
Текстильная промышленность, потреби-
потребитель П. В. 474 и ел.
Текстильные волокна, отбелка 478
Теллура производные, реакции с П. В. 335
Температура
взрыва П. В. 158
влияние на комбинационное рассеяние
П. В. 237 и ел.
замерзания в анализе П. В. 468
кипения водных растворов П. В. 196
и ел., 256
— перекиси дейтерия 256
плавления П. В. 182, 256
Температурный коэффициент реакций П. В.
303
Теплоемкость
П. В. 202 и ел., 256
перекиси дейтерия 254, 256
Теплота
возгонки П. В. 256
диссоциации П. В. 218 и ел.
парообразования П. В. 198, 214, 256
— перекиси дейтерия 256
плавления П. В. 189, 198, 256
разложения П. В. 211 и ел.
растворения и разведения П. В. 206
и ел.
смешения П. В. парциальная 210 и ел.
Терапия, П. В. как лечебное средство 514
Термодинамика
диссоциации П. В. 218
образования П. В. 214
перекиси дейтерия 253
разложения П. В. 211
реакций П. В. 220, 299
смешения П. В. и воды 205 и ел.
Термодинамические свойства П. В., вычи-
вычисление их 200 и ел.
Термодинамические функции П. В. 200
Термодинамический потенциал П. В. 301
Терпены, реакции с П. В. 343
Тетрагидрофуран, торможение разложе-
разложения П. В. 331
Тетралина гидроперекись, кислотное рас-
расщепление 321
Тетраметилди-л-диаминодифенилметан,
идентификация П. В. 458 и ел.
Тетраметил-л-фенилендиамин, реактив на
П. В. 457
Технеций и его соединения
катализ разложения П. В. 415
реакции с П. В. 339
Технические условия на П. В. 469
Тимин, реакция с П. В 345
Тиолы, пероксидатические реакции с
П. В. 353
Тионилхлорид, структура 287
Тиосульфат
восстановление П. В. в Н2О18 325
отбелка им 477
Тиофен, реакция с П. В. 345
Титана перекись 532, 545 и ел.
Титана сульфат, реактив на П. В. 455 и ел.
Титана трихлорид, количественное опре-
определение П. В. 464
Титан и его соединения
катализ разложения П. В. 416
реакции с П. В. 340
Титан, определение при помощи П. В.
498
Токсины, пероксидатические реакции с
П. В. 353
Токсичность П. В. 356 и ел.
о-Толидин, реактив на П. В. 457
Тория перекись 545, 547
Торпеды, П. В. как источник энергии 505
Трансит, конструкционный материал для
работы с П. В. 149
Транс- плоскости а я структура П. В. 262,
272, 276
Трехэлектронная связь 270 и ел., 316, 532
Тригональная форма П. В. 262 и ел., 272,
276
Трипропиламин, система с П. В. 250
Трубопроводы для П. В., применение кра-
кранов 152
Тяжелая вода, свойства 256
Углеводороды
окисление их П. В. 345
самоокисление 76 и ел.
Углеводы, действие П. В. 344, 354
Углерода двуокись
влияние на пределы воспламенения
П. В. 379
реакция с ВаО2 100 и ел.
Углерода окись
реакция с П. В. 332
структура 271
Углерод и его соединения, катализ раз-
разложения П. В. 399 и ел.
Угловой момент П. В., квантовые числа 232
Углы связей П. В. 271 и ел., 283
Уголь активный
катализ разложения П. В. 400
катоды для восстановления О2 85 и ел
реакция с П. В. 345
574
Предметный указатель
Ультразвуковые колебания, образование
П. В. 64 и ел.
Уравнения реакций, несоответствие с ме-
механизмом 305
Уран, определение при помощи П. В. 498
Урана перекись 549
Ускорители (взлетные установки), П. В.
как источник энергии 506
Фазовые превращения П. В. при низких
температурах 187
Фармацевтические препараты из СаОо и
MgO2 544
Феназины, реакция с П. В. 345
Фенантрен, окисление его П. В. 345
Фенацетин, стабилизатор П. В. 450
л-Фенилендиаминхлоргидрат, реактив на
П. В. 457
Фенол
размыкание кольца действием П. В. 522
система с П. В. 249
Фенолфталеин
колориметрическое определение П. В.
467
реактив на П. В. 457
Фенолы, пероксидатические реакции с П. В.
353
Ферменты 346, 418
действие на них П. В. 355, 520
пероксидатические реакции с П. В.
353 и ел.
Феррицианиды, количественное определе-
определение П. В. 465, 469
Ферроновый реактив в колориметрическом
определении П. В. 467
Фетр, применение П. В. в производстве 487
Физиологические действия П. В. 153, 354
Фишера реактив для определения воды 460
Флотация, влияние П. В. 495
Флуоресцеин
идентификация П. В. 459
колориметрическое определение П. В.
467
Формульный вес П. В. 166
Фосфатизация с П. В. 494
Фосфаты
влияние на разложение П. В. 141
применение П. В. в анализе их 498
Фосфор и его соединения
применение П. В. в анализе их 498
реакции с П. В. 336
Фосфорная кислота
в очистке П. В. 12
реакция с ВаО2 99
Фотография
гиперсенсибилизация действием П. В.
491
интенсификация изображения П. В.
492
П. В. как проявитель 338, 491
почернение фотоотпечатков от П. В.
68, 338
удаление гипосульфита при помощи
П. В. 492
Фотопечатание с П. В. 491
Фотосенсибилизаторы образования П. В.
55 и ел.
Фотохимические реакции (см. Образование
П. В., Разложение П. В.)
Фруктоза, реакция с П. В. 344
Фрукты и овощи, предотвращение порчи
их при помощи П. В. 517
Фталоцианины, катализаторы разложения
П. В. 418
Фтор
образование П. В. при реакции с водой
82
определение в минералах при помощи
П. В. 498
помеха при анализе П. В. 459
Фтороводород
помеха при анализах П. В. 459
реакционная среда для П. В. 331
реакция с ВаО2 100 (см. также Плави-
Плавиковая кислота)
Фторуглероды, материал для работы с П. В.
144
Фумаровая кислота, окисление ее П. В.
343
Фуран, гиперголь 501
Фурановая смола, конструкционный мате-
материал для работы П. В. 143
Фурфурол, реакция с П. В. 345
Хаско, конструкционный материал для
работы с П. В. 142
Хастеллой, конструкционный материал для
работы с П. В. 142
Химический синтез, применение П. В. 522
Хиноксалины, реакции с П. В. 345
Хинолин
^j детонирующие составы с П. В. 156
система с П. В. 250
Хлебопечение
применение П. В. 510
— СаО2 544
Хлопок
отбелка 479 и ел.
фотохимические окисление и деструк-
деструкция 490
Хлор
катализ разложения П. В. 398
помеха при анализе П. В. 458, 463
применение для отбелки 477
реакция с П. В. 314, 333
Хлора двуокись, применение для отбелки
477, 485
Хлорированные дифенилы и полифенилы,
материал для П. В. 144
Хлориты, использование П. В. в электро-
электролитическом производстве 496
Хлороформ, система с П. В. 249
Хранение П. В. 150
Хром и его соединения
катализ разложения П. В. 415, 440,
444, 453
реакции с П. В. 339
Хрома перекись 548
Хрома трехокись, идентификация П. В. 458
Хромовая кислота
влияние на поверхность сосудов 375
и ел.
помеха при определении П. В. 469
Хронические инфекции, использование
П. В. 514
Предметный указатель
575
Ц
Цвет П. В. 242
Цветной лом, применение П. В. в перера-
переработке 494
Цезия надперекись 536
Цезия пероксодисульфат, межатомные рас-
расстояния 532
Цезия «полуторная окись» 537
Целлюлоза
деполимеризация ее П. В. 488, 496
отбелка 483 и ел.
химическая, методы получения 482
Цены на П. В. 22 и ел., 475 и ел.
Цепные реакции 38, 303, 381
Церия перекись 545
Церия (IV) соли
количественное определение П. В.
464, 469
применение П. В. в анализе их 498
реакция с П. В. в Н2О18 325
Церия (III) сульфат, реактив на П. В. 457
Цианиды, реакция с П. В. 336
Циклогексан, инертность к П. В. 343
Циклогексанон, окисление его П. В. 343
Циклопентадиен, гиперголь 501
Цинка перекись, получение и свойства 544
Цинка соединения
стабилизация П. В. 451, 453
фотосенсибилизатор образования П. В.
59
Цинк и его соединения
катализ разложения П. В. 402, 444
реакции с П. В. 337
Цинк, снижение коррозии в элементах 87
Цирконий и его соединения
катализ разложения П. В. 416
реакции с П. В. 340
Циркония перекись 545
Дне-плоскости а я структура П. В. 262
и ел., 272, 276
Цистеин, окисление его П. В. 344
Цистерны для П. В. 151
Цистин, получение из цистеина и окисление
до таурина 344
Часовая реакция йодида калия с П. В. 334
Частота характеристическая П. В. 230
Частоты фундаментальные П. В. 277, 278,
283
Чистота, влияние на стабильность П. В. 429
Чугун, конструкционный материал для
работы с П. В. 142, 147
Ш
Шелк, отбелка П. В. 25
Шерсть
отбелка 486
реакция с П. В. 355
Штарка момент 265
Штарка эффект квадратичный П. В. 232
Щ
Щавелевая кислота
окисление П. В. 343
помехи при определении П. В. 469
фотосенсибилизатор образования П. В.
59
Щелочи
влияние на комбинационное рассеяние
П. В. 242
— — разложение и стабильность П. В.
397 и ел., 438 и ел., 440 и ел.
— — ультрафиолетовое поглощение
П. В. 247
— — фотохимическое разложение
П. В. 384 и ел.
системы с П. В. 249
Щелочноземельных металлов соединения
катализ разложения П. В. 416
реакции с П. В. 340
Щелочных металлов соединения
катализ разложения П. В. 416
— — реакции с П. В. 341
Щетина, отбелка П. В. 25
Эвтектики в системе П. В.— вода 181 и ел.
Экстракция селективная дли концентри-
концентрирования П. В. 138
Электрический метод очистки П. В. 139
Электрический разряд
в водяном паре 47
влияние на П. В. 247
Электролиз
как источник П. В. 20 и ел., 107 и ел.
растворов П. В. 389 и ел.
Электролизеры, конструкция 118, 120 и ел.,
126 и ел.
Электролитическое восстановление при про-
производстве П. В. методом самоокисления
75
Электронная диффракция П. В. 248
Электронная структура 266
квантово-механическое объяснение 267
и ел.
Электронные оболочки 266 и ел.
Электронные орбиты 268 и ел., 315
Электронов перенос при реакциях П. В.
310, 311
Электронов распределение 307, 313 и ел.
Электронографическое исследование П. В.
275
Электроны
влияние на образование П. В. 60 и ел.,
68
неспаренные 269 и ел.
Электроотрицательность, влияние на обра-
образование перекисей 533
Электропроводность
П. В. 220
растворов неорганических веществ в
П. В. 249 и ел.
Элементарная ячейка П. В. 289
Элементы, деполяризация 86 и ел.
Эмалированные сосуды, конструкционный
материал для работы с П. В. 149
Эмболия от П. В. 357
Энергии источник, П. В. 499
Энергия активации
разложения П. В. 394
реакций водорода с кислородом 39
— органических и неорганических пе-
перекисей 303
— П. В. 303
576
Предметный указатель
Энергия
водородной связи 294
диссоциации П. В. 218, 283
связи П. В. 283
Энтальпия (см. Теплота, Термодинамика)
Энтропия (см. Теплота, Термодинамика)
Эпихлоргидрин, получение при помощи
П. В. 523
Эпокиси, образование действием П. В. 522
Эпоксидные смолы 523
d-Эритроза, реакции с П. В. 344
Эритроциты, реакции с П. В. 355
Этанол
детонирующие составы с П. В. 156
реакция с П. В. 343
торможение разложения П. В. 331
2-Этилантрахинон, производство П. В. его
окислением 72 и ел.
Этилацетат
действие на реакцию П. В. с TiCl4 331
детонирующие составы с П. В. 156
Этилен, реакция с ОН-радикалами 78
Этилендиаминтетраацетаты, стабилизаторы
П. В. 450
Этиленовые соединения, гидроксилирова-
ние и эпоксилирование П. В. 342
Этиловый эфир
система с П. В. 249
торможение разложения П. В. 331
Эфиры сложные
системы с П. В. 250
стабилизация П. В. 451
Ю
Юкон хайдролюб, материал для работы
с П. В. 144
Юниверсал сайклопс, конструкционный
териал для работы с П. В. 143
ма-
Я
Яблочная кислота, окисление ее П. В. 343
Яйца, отбелка их П. В. 517
Янтарная кислота, окисление ее П. В. 343
ОГЛАВЛЕНИЕ
Предисловие 5
Предисловие авторов 7
Глава 1. Введение 9
Открытие перекиси водорода 9
Размах работы Тенара • 11
Начальные работы по перекиси водорода . . . • М
Развитие производства и потребления перекиси водорода 18
Литература по перекиси водорода 25
Номенклатура перекисных соединений • 20
Литература 30
Глава 2. Образование перекиси водорода и способы ее производства 34
Образование перекиси водорода из воды термическим путем 35
Термическая реакция водорода и кислорода 35
Процессы в электрическом разряде 47
Процессы облучения «54
Самоокисление 66
Окисление углеводородов 76
Другие процессы окисления 81
Катодное восстановление кислорода 82
Литература 87
Глава 3. Образование перекиси водорода и способы ее производства 93
Неорганические перекиси 93
Получение путем электролитических процессов 107
Промышленные процессы электролиза 114
Литература 130
Глава 4. Концентрирование, очистка, конструкционные материалы, хранение
перекиси водорода и обращение с нею 132
Концентрирование 132
Очистка 139
Конструкционные материалы 140
Хранение и перевозка перекиси водорода 150
Физиологическое действие 153
Взрывчатые свойства 154
Лабораторные методы генерирования паров перекиси водорода 158
Литература 162
Глава 5. Физические свойства 164
Термодинамические свойства перекиси водорода 200
Электрические свойства перекиси водорода 220
Влияние перекиси водорода на различные виды излучений 227
Физические свойства многокомпонентных систем, содержащих перекись водорода 248
Физические свойства перекиси водорода с изотопным составом, отличающим-
отличающимся от нормального 251
Литература 257
Глава 6. Структура перекиси водорода 262
Структура молекулы перекиси водорода 262
Молекулярная ассоциация перекиси водорода 288
Литература 294
578 Оглавление
Глава 7. Химические свойства 297
Классификация реакций перекиси водорода 297
Термодинамика реакций перекиси водорода 299
Скорости реакций перекиси водорода 302
Механизм реакций перекиси водорода 304
Обмен изотопами в реакциях перекиси водорода 323
Растворы перекиси водорода как реакционная среда 328
Реакции с неорганическими веществами 332
Реакции перекиси водорода в органической химии 341
Органические перекиси 345
Перекись водорода в биологических процессах 346
Приложение. Применение и значение электродных потенциалов 359
Литература 361
Глава 8. Процессы разложения 373
Разложение в паровой фазе 373
Разложение под действием излучений 382
Электролитическое разложение 389
Разложение в жидкой фазе под действием несрганических катализаторов . . 390
Органические и биологические катализаторы * 418
Литература 419
Г л а ё а 9. Стабилизация 429
Определение стабильности растворов перекиси водорода 430
Методы выражения стабильности 433
Влияние отношения поверхности к объему и природы поверхности 434
Влияние температуры на стабильность * 437
Влияние рН на скорость разложения перекиси водорода 438
Влияние излучения на стабильность 442
Типовые исследования стабильности 443
Стабилизаторы 445
Литература 453
Глава 10. Методы анализа 455
Качественный анализ 455
Количественный анализ 460
Анализ смесей 468
Литература 470
Глава 11. Применение перекиси водорода ¦ . . 474
Отбелка 477
Другие виды применения перекиси водорода как окислителя 488
Перекись водорода как источник энергии 498
Применение газа, образующегося при разложении перекиси водорода .... 509
Перекись водорода в качестве источника свободных радикалов 511
Влияние перекиси водорода на биологические процессы 512
Применение в химическом синтезе 522
Литература 523
Глава 12. Неорганические перекисные соединения 531
Классификация перекисных соединений 531
Получение неорганических перекисей и надперекисей 535
Перекисные соединения I группы 538
Перекисные соединения II группы 542
Перекисные соединения III—VIII групп 545
Пероксокислоты и их соли 551
Литература • 554
Обозначения • 557
Предметный указатель 560
Шамб
ПЕРЕКИСЬ ВОДОРОДА
Редактор В. В. АРНОЛЬДОВ
Художник Л. Г. Ларскип
Технический редактор А. Д. Хомяков
Сдано в производство 14/ХН 1957 г.
Подписано к печати 6/III 1958 г.
Бумага 70ХЮ81/1в=18,1 бум. л.
49,7 печ. л. Уч.-изд. л. 53,7.
Изд. № 3/3785
Цена 39 р. 60 к. Зак. 1604
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, Ново-Алексеевская, 52
16-я типография Московского город-
городского Совнархоза. Москва, Трехпруд-
Трехпрудный пер., 9