


























































































































































































































































































































































































































































































































































































Author: Бочков Н.П.
Tags: общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез внутренние болезни медицина биология генетика медицинская генетика популяции
ISBN: 978-5-9704-1683-9
Year: 2011




Н.П.Бочков
В.П. Пузырев
С.А. СмирнихинаУЧЕБНИКЧЕТВЕРТОЕ ИЗДАНИЕПод редакцией
академика РАМН
Н.П. БочковаЩІ-ИЗДАТЕЛЬСКАЯ ГРУППА«ГЭОТАР-Медиа»
Н.П. Бочков,В.П. Пузырев, С.А. СмирнихинаКЛИНИЧЕСКАЯ
ГЕНЕТИКАПод редакцией академика РАМН Н.П. БочковаУЧЕБНИКЧЕТВЕРТОЕ ИЗДАНИЕ,
ДОПОЛНЕННОЕ И ПЕРЕРАБОТАННОЕМинистерство образования и науки РФРекомендовано ГОУ ВПО «Московская медицинская академия
имени и.м. Сеченова» в качестве учебника для студентов
учреждений высшего профессионального образования,
обучающихся по специальностям 060101.65 «Лечебное дело»,
060103.65 «Педиатрия», 060104.65 «Медико-профилактическое
дело» по дисциплине «Медицинская генетика|^ ^Регистрационный номер рецензии 228 от 02 июля 201*0 года
ФГУ «Федеральный институт развития образования»МоскваИЗДАТЕЛЬСКАЯ ГРУППА«ГЭОТАР-Медиа»>2011
УДК 1б16;575](075.8)ББК54.ІЯ73-1Б86Рецензенты:д-р мед, наук, проф. кя({)едры медицинской генетики РМАПО Минздравсоцраз-
вития РФ с, и. Козлова,зав. кафедрой медицинской генетики МГМСУ Минэдравсоцразвития РФ,
д-р мед. наук, проф. Л.В. Акуленко.Бочков Николай ПавловичБ86 Клиническая генетика: учебник/ Н. П. Бочков, В. П. Пузырен,
С. А. Смирнихина; под ред. Н. П. Бочкова, — 4-е изд., доп. и пере-
раб. - М. : ГЭОТАР-Медиа, 2011. - 592 с, : ил.ISBN 978-5-9704-1683-9Все главы переработаны и дополнены в спязи с развитием медицинс¬
кой науки и практики. Существенно дополнены главы по многофактор¬
ным заболеваниям, профилактике, лечению наследственных болезней,
экологической генетике и фармакогенетике. Весь теоретический матери¬
ал проиллюстрирован схемами и рисунками.В учебнике представлены новые, выявленные в последние годы зако¬
номерности направлений генетики (эпигенетика, малые РНК, однороди-
телыгкие дисомии, генетический полиморфизм и др.).В приложении на компакт-диске размещены дополнительные статьио лечении наследственных болезней, мутагенезе, евгенике.Предназначен студентам медицинских вузов, обучающимся по спе¬
циальностям «лечебное дело», «педиатрия», «медико-профилактическое
дело» по дисциплине «медицинская генетика».УДК[616;575](075.8)ББК54.ІЯ73-1Права на данное издание принадлежат ООО Издательская группа ^ГЭОТАР-Медиа».
Воспроизведение и распространение в каком бы то ии было виде части или целого изда¬
ния не могут быть осуществлены без пись.»енного разреаіения ООО Издательская группа
^ГЭОТАР-Медиа».© Бочков Н.П., Пузырев В.П., СмирнихинаС.А., 2009
© ООО Издательская фуппа «ГЭОТАР-Медиа», 2010
© ООО Издательская группа «ГЭОТАР-Медиа», оформ-
ISBN 978-5-9704-1683-9 ление,2010
ОГЛАВЛЕНИЕСписок сокращений и условные обозначения ЮПредисловие 12Глава 1. Введение в клиническую генетику 14Основные понятия 14Краткая история медицинской генсі ики 16Домен дел евский период 16Открытие законов Менделя 1820-е годы XX века 1930-40-е годы XX века 2050-е годы — конец XX века 20Аксиомы медицинской генетики 22Геномика и клиническая медицина 24Характеристика генома человека 28ДНК-уровень 28Повторы 29Внсхромосомныс и кольцевые молекулы ДНК 31Полиморфизм 31Митохондриальный геном 34Генный уровень 35Функции генов 39Генетические карты хромосом 44Значение генетики для медицины 47Заключение 50Ключевые слова и понятия 52Рекомендуемая литература 53Глава 2. Наследственность и патология 54Изменчивость наследственных признаков как основа патологии.. .54Роль наследственности и среды в развитии патологии 60Мутации как этиологический фактор наследственных болезней .. .63Наследственность и патогенез наследственных болезней 64Наследственность и клиническая картина болезней 66Наследственность и исходы заболеваний 67Классификация наследственной патологии 69Генетическая классификация наследственных болезней .... 70
Клиническая классификация наследственных болезней .... 71Генетические основы гомеостаза 72Ключевые слова и понятия 76Рекомендуемая литература 76
4 ОглавлениеГлава 3. Семиотика и клиническая диагностика наследственных
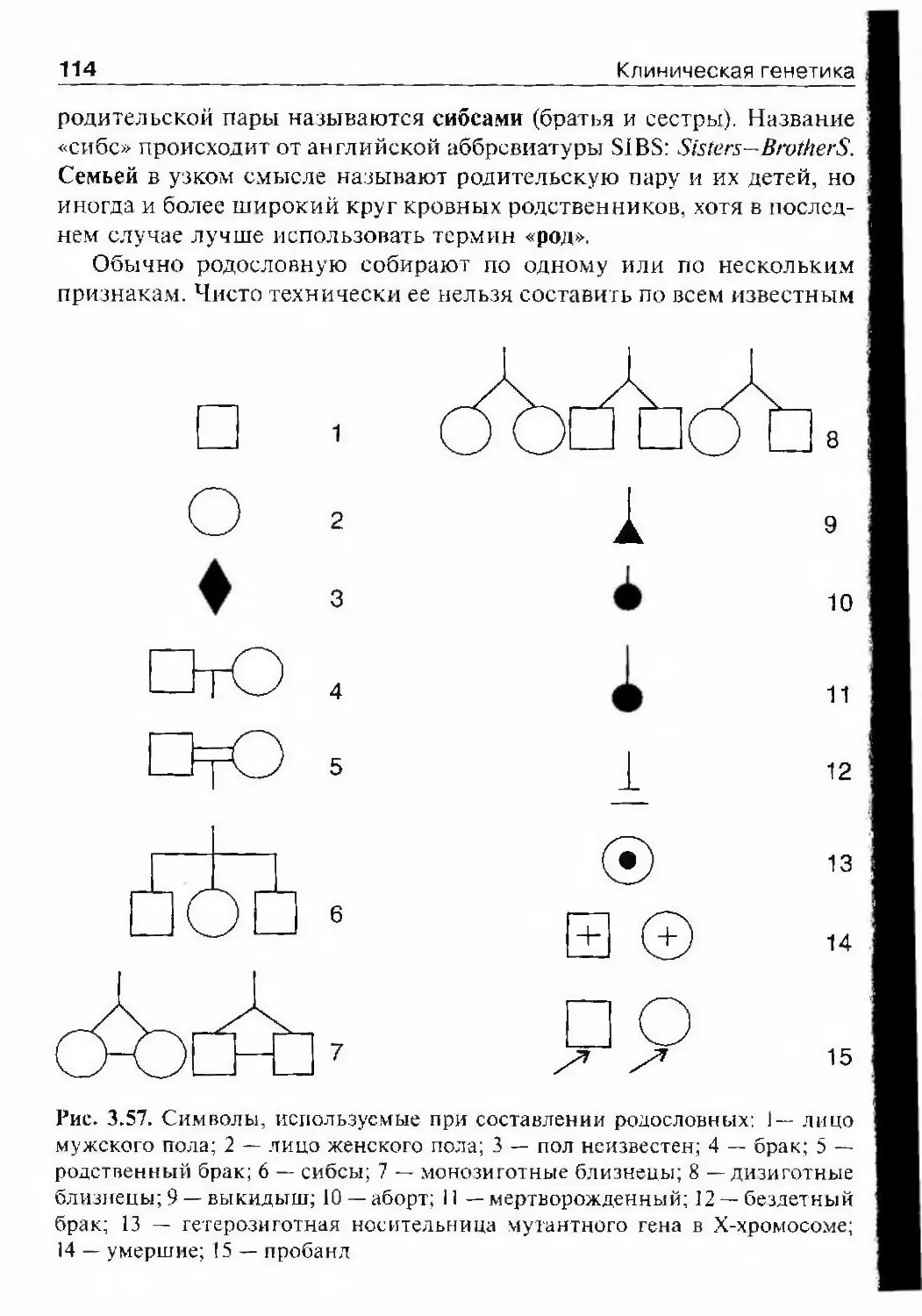
заболеваний 78Общие замечания 78Особенности клинических проявлений наследственнойпатологии 79Семейный характер заболевания 79Хроническое прогредиентное рецидивирующее течение .... 79Специфические симптомы наследственных болезней 80Множественные патологические изменения органов и систем ... 82Врожденный характер заболевания 84Резистентность к наиболее распространенным методамтерапии 84Обшие принципы клинической диагностики наследственныхболезней 85Осмотр и обследование пациентов и их родственников 87Врожденные пороки развития. Генетические механизмыэмбрионального развития 87Классификация и этиология врожденных пороков 90Антропометрия 94Признаки дисморфогенеза в диагностике наследственнойи врожденной патологии 94Признаки дисморфогенеза 95Течение беременности 112Клинико-генеалогический метод 112Составление родословной ИЗГенеалогический анализ 120Болезни с аутосомно-доминантным типом наследования— 121
Болезни с аутосомно-рецессивным типом наследования ... 122
Болезни с X-сцепленным доминантным типомнаследования 125Болезни с Х-сцепленным рецессивным типомнаследования 126Y-сцепленный тип наследования 128Митохондриальная наследственность 128Синдромологический подход к диагностике наследственныхболезней 129Параклинические исследования в клинической генетике 131Компьютерные программы диагностики наследственных
болезней 132
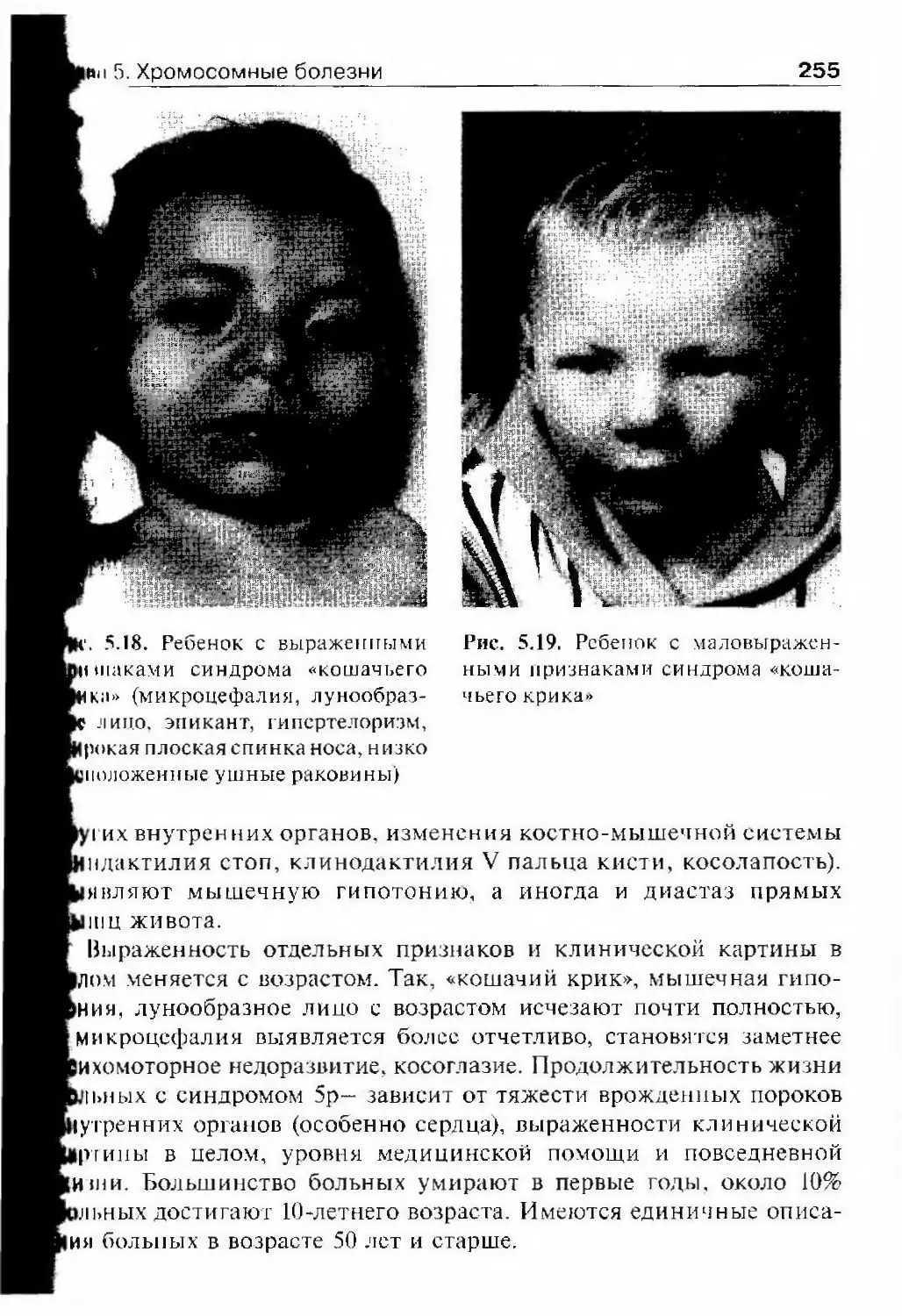
Оглавление 5Ключевые слова и понятия 133Рекомендуемая литература 134Глава 4. Генные болезни 136Этиология 136Классификация 144Общие закономерности пачогенеза 145Патогенез болезни на молекулярном уровне 146Клеточный уровень патогенеза генных болезней 150Органный уровень патогенеза 152Организменный уровень 153Главные черты клинической картины 153Особенности клинической картины 153Клинический полиморфизм и его причины 157Генетическая гетерогенность 165Клиника и генетика некоторых генных болезней 168Нейрофиброматоз (болезнь Реклингхаузена) 168Миотоническая дистрофия 172Семейная гинерхолестеринемия 175Синдром Марфана 178Синдром Элерса—Данло 182Фенилкетонурия 188Муковисцидоз 190Адреногенигальный синдром 196Миодистрофия Дюшенна—Беккера 199Синдром умственной отсталости с ломкой Х-хромосомой.... 203Эпидеми0.|]0гия 206Ключевые слова и понятия 216Рекомендуемая литература 217Глава 5. Хромосомные болезни 219Общие вопросы 219Этиология и классификация 221Эффск гы хромосомных аномалий в онтогенезе 225Летальность 225Врожденные пороки развития 227Эффекты хромосомных аномалий в соматических клетках 228Патогенез 228Клинико-цитогенетические характеристики наиболеераспространенных хромосомных болезней 234Синдром Дауна 234
6 ОглавлениеСиндром Патау (трисомия 13) 240Синдром Эдвардса (трисомия 18) 242Трисомия 8 244Полисомии по половым хромосомам 246Синдром трипло-Х (47,XXX) 247Синдром Клайнфелтера 248Синдром дисомии по Y-хромосоме (47,XYY) 249Синдром Шерешевского-Тернера (45,X) 250Синдромы частичных анеуплоидий 252Синдром «кошачьего крика» 254Синдром Вольфа—Хиршхорна (частичнаямоносомия 4р—) * 256Синдром частичной трисомии по короткому плечухромосомы 9 (9р+) 257Синдромы, обусловленные микроструктурнымиаберрациями хромосом 258Факторы повышенного риска рождения детей с хромосомнымиболезнями 265Ключевые слова и понятия 268Рекомендуемая литература 268Глава 6. Болезни с наследственной предрасположенностью 270Общая характеристика 270Подходы к изучению наследственной предрасположенностик болезням человека 275Клинико-генеалогические доказательства наследственнойпредрасположенности 275Близнецовые исследования 278Популяционные исследования 280Генетические ассоциации 280Гены подверженности некоторым многофакторнымзаболеваниям 287Сердечно-сосудистые заболевания 287Иммунозависимые болезни 290Инфекционные болезни 294Злокачественные новообразования 297Значение наследственной предрасположенности в общейпатологии человека и клинической практике 308Ключевые слова и понятия 310Рекомендуемая литература 311
Оглавление 7Глава 7. Экологическая генетика 312Общие вопросы 312Индуцированный мутационный процесс 313Патологические проявления экспрессии генов 316Генстичсскис основы биотрансформации чужеродныхвеществ (ксенобиотиков) 319Наследственно обусловленные патологические реакциина действие внешних факторов 320Профессиональные вредности 323Пищевые вещества и пищевые добавки 324Физические факторы 330Чувствительность к биологическим агентам 330Изменение генофонда популяций как результат нарушениягенетического равновесия 331Заключение 332Ключевые слова и понятия 334Рекомендуемая литература 334Глава 8. Фармакогенетика 335Общие вопросы 335Фармакогенетические закономерности Т фазыбиотрансформации 337Фармакогенетические закономерности П фазыбиотрансформации 341Фармакогенетические закономерности транспорталекарственных средств (ІП фаза биотрансформации) 345Фармакодинамика и генетический полиморфизм 349Заключение 350Ключевые слова и понятия 352Рекомендуемая литература 352Глава 9. Лабораторные методы диагностики 353Общие вопросы 353Цитогенетические методы 355Получение препаратов митотических хромосом 356Окраска препаратов 358Молекулярно-цитогенетические методы 362Показания для проведения цитогенет№іеских исследований—365Биохимические методы 366Молекулярно-генетические методы 373Общие процедуры 373
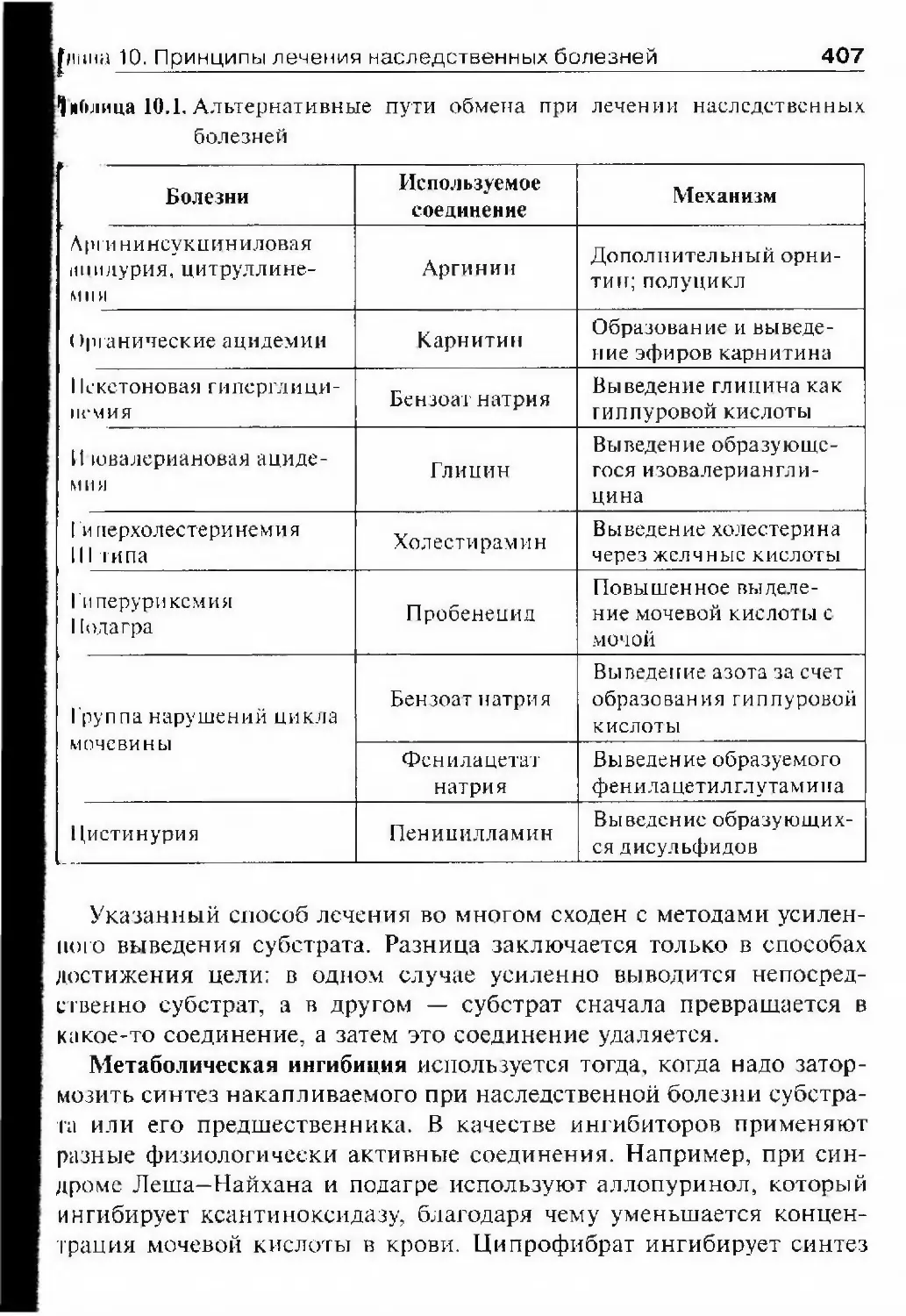
8 ОглавлениеМетоды ДНК-диаі ностики наследственных болезней 378Прямые методы диагностики мутаций 378Косвенное выявление мутаций 390Ключевые слова и понятия 394Рекомендуемая литература 395Глава 10. Принципы лечения наследственных болезней 397Общие вопросы 397Симптоматическое лечение 399Патогенетическое лечение 402Коррекция обмена на уровне субстрата 404Коррекция обмена на уровне продукта гена 408Коррекция обмена на уровне ферментов 4ПМодификация ферментативной активности 412Возмещение фермента 414Хирургическое лечение 417Этиогропное лечение: клеточная и генная терапия 419Введение 419Клеточная терапия 421Генная терапия 424Лечение трансгенными клетками 426Изменение экспрессии генов как метод лечения 429Риски клеточной и генной терапии 430Заключение 431Ключевые слова и понятия 432Рекомендуемая литература 432Глава 11. Профилактика наследственной патологии 434Груз наследственной патологии в медицинском и социальномаспектах 434Генетические основы профилактики наследственной патологии .. .437Общие положения 437Первичная профилактика 437Вторичная профилактика 437Третичная профилактика 438Управление экспрессией генов 439Элиминация эмбрионов и плодов с наследственнойпатологией 443Генная инженерия на уровне зародышевых клеток 443Планирование семьи 445Охрана окружающей среды 447
Оглавление 9Медико-генетическое консультирование 448Общие положения 448Функции врача-гснетика 449Диагностика 449Прогноз для потомства 451Заключение медико-генетического консультированияи советы родителям 451Организационные вопросы 453Анализ обращаемости в медико-генетическуюконсультацию 455Эффективность медико-генетических консультаций 456Пренатальная диагностика 458Общие вопросы 458Скрииин!' беременных на основе определениябиохимических маркеров (просеивающие методы) 460Инвазивные методы 465Заключение 469Предьшплантационная диагностика 471Доклиническая диагностика, просеивающие программыи профилактическое лечение 474Фенилкетонурия 477Врожденный гипотиреоз 478Врожденная гиперплазия надпочечников 479Галактоземия 479Муковисцидоз 481Ключевые слова и понятия 482Рекомендуемая литература 483Глава 12. Этические вопросы медицинской генетики 484Рекомендуемая литература 493Контрольные вопросы 494Приложение 539Генетические термины 539Признаки дисморфогенеза 559Предметный указатель 564СОДЕРЖАНИЕ ДИСКАДополнительные статьи о диагностике, лечений и профилактике
наследственных болезней
список СОКРАЩЕНИЙ
И УСЛОВНЫЕ ОБОЗНАЧЕНИЯА — аденин
ADA — аденози н дезами наза
ADH — алкогольдегидрогеназа
ALDH — альдегиддегидрогеназа
С — цитозинCGH — сравнительная геномная гибридизация {Comparative
Genome Hybridization)FISH — метод флюоресцентной гибридизации in situ
G — гуанинGST — глутатион-8-ЗН-трансферазаHLA — человеческий лейкоцитарный антиген {Human Leucocyte
Antigen)Ig — иммуноглобулин (используется при указании класса
иммуноглобулинов)IQ — коэффициент умственного развития, интеллекта {Intel¬
ligence Quotient)NAT — N-ацетилтрансферазаОМТМ — ои/те-версия каталога В. Маккьюсика «Менделевская
наследственность человека» (Online Mendelian Inheritance
in Man)OR — отношение шансовPAPP-A —• ассоциированный с беременностью плазменный бе¬
лок А {Pregnancy Associated Plasma Protein A)PON — параоксоназа
SULT — сульфотрансферазаT — ТИМИНU — урацилUGT — УДФ-глюкуронилтрансферазаVNTR — полиморфизм no количеству копий {Variable Number
of Tandem Repeates)АКТГ — адренокортикотропный гормонАФП — а-фетопротеинВИЧ — вирус иммунодефицита человекаВОЗ — Всемирная организация здравоохраненияГ6ФДГ — глюкозо-6-фосфатдегидрогеназа
Список сокращений и условные обозначения11ДНК — дезоксирибонуклеиновая кислотаДПДГ — дигидропиримидиндегидрогеназаЖКТ — желудочно-кишечный трактИЛ — интерлейкинИФН — интерферонкДНК — комплементарная ДНККТ — компьютерная томографияЛ ПИП — липопротсиды низкой плотностимРИК — матричная (информационная) РНКМРТ — магнитно-резонансная томография
мтДНК — митохондриальная ДНКОНП — однонуклеотидные полиморфизмыПЦР — полимеразная цепная реакцияРНК — рибонуклеиновая кислотарРНК — рибосомная РНКСПИД — синдром приобретенного иммунодефицитаТПМТ — тиопуринметилтрансферазатРИК — транспортная РНКУДФ — уридиндифосфоглюкуроновая кислотаУЗИ — ультразвуковое исследованиеФНО — фактор некроза опухолейХГЧ — хорионический гонадотропин человекаЦНС — центральная нервная системап.н. — пар нуклеотидовп.о. — пар оснований* — торговое название лекарственного средствар — лекарственное средство в России не зарегистрировано® — лекарственное средство в России аннулировано, т.е.исключено из официального Регистра лекарственных
средств
ПРЕДИСЛОВИЕСреди научных дисциплин, изучающих наследственность человека
медицинская генетика и ее важный раздел — клиническая генетика
занимают особенное место. Основываясь на результатах фундаменталь¬
ных исследований в области генетики человека, медицинской генетике
сегодня под силу решение трех кардинальных вопросов.Где в геноме локализованы гены болезни?Какова функциональная вариабельность последовательности ДНК
в этих генах?Как реализовать полученные данные в клинической практике (диа¬
гноз, прогноз, лечение).Медицинская генетика выросла из небольшой специальности, зани¬
мавшейся редкими наследственными болезнями, в значимую медицин¬
скую специальность, концепции и подходы которой стали важными
компонентами диагностики и лечения многих болезней, и редких и
частых. Она стала частью более широких областей — молекулярной
и геномной медицины, которые применяет широкий анализ генома
человека, включая контроль экспрессии генов, изменчивость генов
человека, взаимодействие генов и среды, ради улучшения медицинской
помощи пациенту и его семье. В последние годы открыты новые гене¬
тические закономерности: экспансии нуклеотидов, однородительские
дисомии, эпигенетические регуляции, роль малых интерферирующих
РНК. Обнаружены многие сотни генов, ассоциирующих с заболевания¬
ми. Фактически можно говорить о начале революции, интегрирующей
знания генетики и геномики в здравоохранение и практику медицины.
Генетика быстро становится организующим принципом медицинской
практики, закладывая основы персонализированной медицины.Будущий врач должєіі быть подготовлеіі к восприятию всего ново¬
го, что несет с собой генетика, ибо никакие ее достижения не могут
быть реализованы в практике здравоохранения без грамотных вра¬
чей. Студенту медицинского вуза важно постоянно развивать в себе
потребность в освоении новых генетических знаний.С момента выхода предыдущего издания, в медицинской генетике
накоплены новые факты, появились новые взгляды на те или иные
болезни, на закономерности возникновения и развития давно извест¬
ных заболеваний, новые подходы в лечении и профилактике не толь¬
ко редких моногснных болезней. Все это нашло отражение в новом
издании учебника.
предисловие 13Методический прорыв в постгеномной эре о поисках ассоциа¬
ций генетических полиморфизмов с многофакторными болезнями
отражен в главе «Болезни с наследственным предрасположением», в
которой описаны результаты поисков патогенетических молекулярных
механизмов развития широко распространенных болезней.В последние годы отмечен прогресс в области изучения генов
биотрансформации ксенобиотиков (генов метаболизма), поэто¬
му в настоящем издании учебника появились новые главы
«'•Экологическая генетика человека» и «Фармакогенетика». Эти
сведения закладывают врачу фундамент знаний, во-первых, для
<1)армакогенетического понимания индивидуального подхода к
лекарственному лечению, и, во-вторых, для обеспечения более
безопасной среды обитания человека: профессиональная деятель¬
ность, особенно на химических производствах, индивидуальная
диета (токсикогенетика, нутригенетика).В приложении на компакт-диске представлены дополнительные
с і атьи о лечении наследственных болезней, мутагенезе, евгенике.В учебнике обобщен опыт преподавания кафедр медицинской
генетики Московской медицинской академии им. И.М. Сеченова и
Сибирского государственного медицинского университета (г. Томск).
Нам представляется важным подчеркнуть положение о том, что истин-
пое образование — это, прежде всего, понимание основных прин¬
ципов, а «не запоминание вороха мелочей и тонкостей». Будущим
врачам важно войти в парадигмы современной генетической науки и
научиться методам приложения усвоенных принципов во врачевании
или клиническом мышлении.Задача учебника научить студентов языку науки, показать логику
рождения новых генетических знаний о формах патологии (мендели-
рующей, хромосомной, многофакторной), обозначить законченные
(|)рагменты понятого в патологии и очертить области еще неизвест¬
ного на примере лекций и практических занятий на кафедре меди¬
цинской генетики.Авторы выражают искреннюю благодарность студентам, ордина¬
торам и врачам за проявленный интерес к учебнику, который стиму¬
лировал обдумывание сложных генетических процессов и изложение
их в понятной для врача форме.Особую признательность выражают коллегам по кафедре А. Ю. Асанову,Н.А. Жученко, Т.И. Субботиной, М.В. Тихопой, М.Г. Филипповой,
Т.В. Филипповой, в совместных дискуссиях с которыми формировалась
программа преподавания клинической генетики и учебные планы.
Глава 1ВВЕДЕНИЕ В КЛИНИЧЕСКУЮ ГЕНЕТИКУ*
ОСНОВНЫЕ понятияГенетика наряду с морфологией, физиологией и биохимией служит
теоретическим фундаментом современной медицины. Наследственность
лежит в основе всех жизненных проявлений. Без наследственности
и изменчивости была бы невозможна эволюция жизни на Земле.
Поскольку человек — продукт длительной эволюции живой природы,
все общебиологические закономерности отражены в его формирова¬
нии как биологического вида Человек разумный {Homo sapiens).Генетика человека изучает явления наследственности и измеі£чи-
вости на всех уровнях его организации и существования: молеку-
лярном> клеточном, организменном, популяционном, биохорологи-
ческом, биогеохимическом. с периода зарождения (начало XX в.) и
особенно в период интенсивного подъема (50-е годы XX в.) генетика
человека развивалась не только как теоретическая, но и как клиниче¬
ская дисциплина, постоянно подпитываясь как обтцебиологическими
концепциями (эволюционное учение, онтогенез), так и генетически¬
ми открытиями [законы наследования признаков, хромосомная тео¬
рия наследственности, информационная роль ДНК (дезоксирибону¬
клеиновой кислоты)]. В то же время на процесс становления генетики
человека как науки постоянно и существенно влияли достижения
теоретической и клинической медицины. Человек как биологический
объект изучен детальнее, чем любой другой высокоорганизован¬
ный организм (дрозофила, мьтпіь и др.). Изучение патологических
вариаций (предмет врачебной профессии) стало основой для позна¬
ния наследственности человека. В свою очередь, развитие генетики
человека ускоряло развитие теоретических дисциплин (например,
молекулярной биологии) и клинической медицины (например, новой
области в медицине — учения о хромосомных болезнях).Медицинская генетика изучает роль наследственности в патоло¬
гии человека, закономерности передачи от поколения к поколению* Исправлено и дополнено при участии д~ра биол. наук И.Н. Лебедева.
Глава 1. Введение в клиническую генетику 15наследственных болезней, разрабатывает методы диагностики, лече¬
ния и профилактики наследственной патологии, включая болезни с
наследственной предрасположенностью. Результатом исследований в
JTOM. направлении становятся медицинские и генетические открытия
и достижения, направленные на борьбу с болезнями и улучшение
здоровья людей.Медицинская генетика, составляя важнейшую часть теоретической
медицины, рассматривает в связи с патологией следующие вопросы:• какие наследственные механизмы поддерживают гомеостаз орга¬
низма и определяют здоровье индивида;• каково значение наследственных факторов (мутации или сочета¬
ние определенных аллелей) в этиологии болезней;• каково соотношение наследственных и средовых факторов в пато¬
генезе болезней;• какова роль наследственных факторов в определении клиниче¬
ской картины болезней (и наследственных, и ненаследственных);• влияет ли (и если влияет, то как) наследственная конституция на
процесс выздоровления человека и исход болезни;• как наследственность определяет специфику фармакологическо¬
го и других видов лечения.Как теоретическая и клиническая дисциплина медицинская гене¬
тика продолжает интенсивно расширяться в разных направлениях:
геномика, цитогенетика, молекулярная и биохимическая генети¬
ка, иммуногенетика, генетика развития, популяционная генетика,
клиническая генетика, фармакогенетика, экологическая генетика,
нутригенетика, токсикогенетика.Образование по медицинской генетике включает изучение основ
общей генетики (менделизм, учение о хромосомах, химические основы
наследственности), основных положений генетики человека (человек
как объект генетического исследования) и клинической генетики.Клиническая генетика прикладной раздел медицинской генети¬
ки. Ее достижения применяются для решения клинических проблем
пациентов или их семей. Она дает ответ на вопросы: какая болезнь
у пациента (диагноз), как ему помочь (лечение), как предупредить
рождение больного потомства (прогноз и профилактика), как диагно¬
стировать и уменьшить вероятность развития болезни с наследствен¬
ным предрасположением. В настоящее время в клинической генетике
используются, с одной стороны, генетические методы (генетический
анализ, молекулярно-биологические, цитогенетические, биохими¬
16Клиническая генетикаческие, иммуногенетические) и, с другой стороны, все современные
методы клинического обследования [ультразвуковое исследование
(УЗИ), магнитно-резонансная томография (МРТ), компьютерная
томография (КТ), позитронно-эмнссионная томография (ПЭТ)].КРАТКАЯ ИСТОРИЯ МЕДИЦИНСКОЙ ГЕНЕТИКИДоменделевский периодУчение о наследственности человека зарождалось в медицине из
эмпирических наблюдений семейных и врожденных болезней. Уже
в трудах Гиппократа отмечалась роль наследственности в происхожде¬
нии болезней: «...эпилепсия, как и другие болезни, развиваются на
почве наследственности; и действительно, если от флегматика проис¬
ходит флегматик, от желчного —
желчный, от чахоточного —
чахоточный, от страдающего
болезнью селезенки — страдаю¬
щий болезнью селезенки, то что
может помешать, чтобы болезнь,
которою страдают отец и мать,
поразила бы также одного из их
детей». Однако в дальнейшем
интерес к роли наследственно¬
сти в происхождении болезней
был утрачен, и на первое место в
теориях медицины выдвигались
внешние этиологические факто¬
ры. Лишь в XVIII—XIX вв. появи¬
лись отдельные работы о значении
наследственности в происхожде¬
нии болезней (полидактилия,
гемофилия, альбинизм).Понятие о патологической
наследственности у человека
утвердилось во второй половинеXIX в. и было принято многими
врачебными школами. С пони¬
манием патологической наслед-Рис. 1.1. Б.М. Флоритюкий (1834-
1899). Акушер-гинеколог и педиатр.
Автор книги «УсовершенстБОБание
и вырождение человеческого рода»
(1865). Основатель первого в Сибири
учебного заведения — Сибирского
университета в Томске (1880-1888)
Глава 1. Введение в клиническую генетику17сі'венности зародилась концеп¬
ция о вырождении человеческого
рода и необходимости его улуч¬
шения, причем одновременно
(1865 г.) и независимо друг от друга
сс высказывали В. М. Флори некий
и России и Ф. Гальтон в Англии
(рис. 1.1, 1.2).Предпосылки развития уче¬
ния о наследственности человека
н XIX в. вытекали из биологи¬
ческих открытий, революциони-
жровавших развитие медицины:
клеточной теории (Т. Шванн) и
доказательства клеточной пре¬
емственности (Р. Вирхов); пони¬
мания идеи онто- и филогенеза;
объяснения эволюции на основе
естественного отбора и борьбы за
существование (Ч, Дарвин),Не меньшее влияние, чем
биологические открытия, на раз¬
витие учения о наследственных
болезнях оказали общемеди¬
цинские предпосылки, в XIX в.
изучение причин заболеванийРис, 1.2. Френсис Гальтои (1822—
1911). Один из основоположни¬
ков генетики человека и евгеники.
Основные труды в этой области:
«Наследственный талант и харак¬
тер» (1865); «Наследственный гений:
исследование его закоттов и следствий»
(1869); «Очерки по евгенике» (1909)стало главным направлением вмедицине. Начался период выделения отдельных болезней как
гюзологических единиц, в том числе наследственных. Например,
были описаны болезнь Дауна, нейрофиброматоз, врожденная дис-
млазия соединительной ткани и др. Изучение патологических сим¬
птомов сменилось изучением нозологических форм болезненных
процессов, которые можно было прослеживать в родословных как
дискретные формы.Несмотря на то, что в XIX в. учение о наследственных болезнях и
закономерностях наследственности человека существенно продвину¬
лось, в целом еще было много противоречий, в больщинстве работ
этого периода были перемешаны факты и ошибочные представления.
Критериев правильной интерпретации наследования болезней еще не
18 Клиническая генетикасуществовало. Генетика человека находилась на «донаучной» стадии
развития. Этот период можно назвать доменделевским.Открытие законов МенделяТолько с переоткрытием законов Менделя в 1900 г. появились
уникальные возможности «инвентаризации» наследственных болез¬
ней. На примере то одной, то другой болезни законы Менделя под¬
тверждались либо врачами, либо биологами. Наследственность как
этиологическая категория прочно ношла в медицину. Природа мно¬
гих болезней стала понятной.Так, в самом начале XX в. английский врач А. Гаррод объяснил
наследственный характер необычного метаболического состояния —
аутосомно-рецессивной алкаптонурии — в соответствии с законо¬
мерностями наследования признаков, открытых Менделем, Более
того, он объяснил своей идеей и другие биохимические аномалии,
опубликовав в 1909 г. книгу «Врожденные ошибки метаболизма»,
благодаря которой он был признан отцом биохимической генетики.В первых двух десятилетиях XX в. в результате эйфории от
менделевской интерпретации многих болезней была существенно
преувеличена роль наследственности в формировании поведения
человека и наследственной отягощенности населения. Концепция
обреченности и вырождения семей с наследственной патологией
стала ведущей для объяснения отягошенности общества потом¬
ством таких больных. Диагноз наследственной болезни считал¬
ся приговором больному и даже его семье. На этом фоне стала
набирать силу евгеника — ранее сфор.мулированное Ф. Гальтоном
направление (или даже наука) об улучшении породы (или природы)
человека (позитивная евгеника ~ преимущественное размножение
лиц с положительными качествами).Под негативной евгеникой понимали ту ее часть, которая ста¬
вила своей целью освобождение человечества от индивидуумов с
наследственной патологией. Евгеника в конечном счете «обосно¬
вывала» насильственное ограничение репродуктивной свободы.
Евгенику правильнее считать не наукой, а социальным или обще¬
ственным движением. Подробная информация о евгенике пред¬
ставлена на компакт-диске (см. «Евгенику»).Евгеника — один из примеров широкого необоснованного внедре¬
ния непроверенных результатов в практику (головокружение от успе¬
хов). В целом она сыграла отрицательную роль в развитии и генетики,
Глава 1. Введение в клиническую генетику19и медико-биологической науки. Подробно и объективно история
евгеники в России изложена в книгах ЕЛ, Пчелова и В.В. Бабкова
(см. рекомендуемую литературу).20-е годы XX векаГенетика человека продолжает развиваться. На основе использо¬
вания менделизма и хромосомной теории наследственности (фор¬
мальная генетика) приходило понимание общих закономерностей
наследственной патологии, при¬
чин клинического полиморфиз¬
ма, генетической гетерогенности,
признание роли внешней среды в
развитии болезней с наследствен¬
ной предрасположенностью.В нашей стране медицин¬
ская генетика в 20—30-х годах
успешно развивалась. В пер¬
вую очередь следует упомянуть
основоположника клинической
генетики С.Н. Давиденкова,
одновременно и генетика, и невропа¬
толога (рис. 1.3), который наряду
с огромным вкладом в изучение
генетики нервных болезней на
несколько десятилетий опреде¬
лил разработку общегенетиче¬
ских проблем. Он первым в мире
поставил вопрос о составлении
каталога генов человека, сформу¬
лировал понятие о генетической
гетерогенности наследственных
болезней, организовал медико¬
генетическую консультацию.Поддержал развитие меди¬
цинской генетики И.П. Павлов.В 20-х годах XX в. он приступил к
изучению генетики высшей нерв¬
ной деятельности, о его роли в
становлении медицинской гене¬Рис, 1.3. С.Н. Давиденков (1880-
1961). Генетик, невропатолог.
Основатель клинической генетики
в СССР. Впервые поставил вопрос
о создании каталога генов (1925).
Организовал первую в мире медико¬
генетическую консультацию (1929).
По генетике наследственных болез¬
ней нервной системы опубликовал
несколько книг: «Наследственные
болезни нервной системы» (1-е изд.
в 1925 г., 2-е изд. в 1932 г.); «Проблема
полиморфизма наследственных
болезней нервной системы» (1934);
«Эволюционно-генетические проб¬
лемы в невропатологии*» (1947)
20Клиническая генетикаРис. 1.4. С.Г. Левит (1894-1937).Директор Медико-биологического
института, преобразованного в і 935 г.
в Мсдико-генетический институт.Руководил работами в разных обла¬
стях генетики человека (цитогетте-
тика, близнецовые исследования,
клиническая генетика, формальная
генетика)нять их. Наши врачи должны каказбуку знать законы наследственности. Воплощение в жизнь научной
истины о законах наследственности поможет избавить человечество
от многих скорбей и горя».тики см. в статье Н.П, Бочкова
на компакт-диске (см. «Статью
об И.П.Павлове».)30-40-е годы XX векаС 1930 по 1937 г, медицин¬
ская генетика развивалась в
Медико-биологическом инсти¬
туте, переименованном в 1935 г.
в Медико-генетический. Это
был передовой институт, в кото¬
ром проводились первоклассные
близнецовые и цитогенетические
исследования. Институт, к сожа¬
лению, был закрыт, а его дирек¬
тор проф. С.Г. Левит репрессиро¬
ван (рис, 1.4).В 30-х годах XX в. генети¬
ка твердо и широко вошла в
медицинскую науку и практи¬
ку. Наиболее точно значение
генетики для медицины того
периода выразил И.П. Павлов
(1935): «Жизнь требует всемер¬
ного использования открытых
Менделем законов наследствен¬
ности. Генетические истины
достаточно изучены для того,
чтобы интенсивно начать приме-50-е годы — конец XX векаНаиболее эффективный период развития генетики человека
начался с 50-х годов XX в, В 1959 г, была открыта хромосомная
природа болезней, и цитогенетика на несколько лет стала веду-
Глава 1. Введение в клиническую генетику ^тим направлением в медицине. В этот период сформировалась
клиническая генетика как результат слияния трех ветвей генетики
человека — цитогенетики, формальной (менделевской) генетики и
биохимической генетики. Человек был главным объектом обшеге-
пстических исследований. Взаимовлияние генетики и медицины
дало колоссальный рывок в исследовании наследственности чело-
иска и реализации достижений в практике.Именно в 1960-е годы в нашей стране возобновилось раз-
питие медицинской генетики. Старшее поколение генетиков и
ученых смежных дисциплин (В.Д. Тимаков, С.Н. Давиденков,B.П. Эфроимсон, А.Д. Прокофьева-Бельговская, Е.Ф. Давиденкова,C.А. Нейфах, А.А. Малиновский, Е.Е. Погосянц, Н.Н, Медведев,
Ю.Я. Керкис) приняло активное участие в ее возрождении.
В 1969 г. в Москве был создан Институт медицинской генетики
АМН СССР, преобразованный в 1989 г. во Всесоюзный научный
центр медицинской генетики АМН СССР (ныне Учреждение
Российской академии медицинских наук Медико-генетический
научный центр РАМН). Информация об ученых-генетиках стар-
іиего поколения представлена на компакт-диске.На рубеже XX и XXI вв. медицинская генетика заняла лидирую¬
щее место в медико-биологической науке, аккумулировав передо¬
вые методы и концепции разных медицинских и биологических
дисциплин.Интенсивному развитию медицинской генетики во второй поло¬
ни не XX в. способствовало несколько обстоятельств. Благодаря
снижению уровня инфекционных и алиментарных заболеваний
после Второй мировой войны больше внимания стали уделять
болезням эндогенной природы, в том числе наследственным,
прогресс лабораторной и инструментальной медицины, широкий
обмен информацией обеспечили более точную нозологизацию
синдромов и болезней, прогресс общей генетики и биологии
принципиально изменил методологию генетического изучения
человека (молекулярная биология, цитогенетика, генетика сома¬
тических клеток).Главным итогом медицинской генетики к концу XX в. стало
создание генетических технологий, которые позволяют уско¬
ренно решать трудные вопросы в медицине и здравоохранении
(табл. 1.1).
22Клиническая генетикаТаблица 1.1. Генетические технологии в медицине и здравоохраненииОбласть медициныРешаемые вопросыТеоретическаяУглубление «инвентаризации» болезней по нозо¬
логическому принципу. Расшифровка патогенеза
болезней, причины клинического полиморфиз¬
ма. причины хронического течения болезней.
Фармакогенети каКлиническаяДиагностика наследственных и инфекционных
болезней. Патогенетическое лечение наследствен¬
ных болезней, производство лекарств на осно¬
ве генной инженерии. Все виды профилактики
наследственных болезнейПрофилактическаяГенетико-гигиеническое нормирование факторов
окружающей среды. Предупреждение мутагенных,
тератогенных и канцерогенттых эффектов. Создание
новых вакцинСовременные успехи генетики человека заставляют врача по-новому
их осваивать. «Как наша современная медицинская практика опира¬
ется на уточненные знания в области анатомии человека, физиологии
и биохимии, так в будущем изучение генетических болезней потребует
детального понимания молекулярной патологии, физиологии и био¬
химии генома человека. Нам потребуются врачи, настолько осведом¬
ленные в молекулярной анатомии и физиологии хромосом и генов,
насколько кардиохирург знает работу сердца и структуру сосудистого
дерева». Это высказывание лауреата Нобелевской премии П. Берга,
относящееся к 1981 г., особенно актуально в настоящее время, когда в
результате международной программы секвенирован и во многом рас¬
шифрован геном человека, а молекулярная медицина стала основой
клинической и профилактической медицины.АКСИОМЫ МЕДИЦИНСКОЙ ГЕНЕТИКИ- Наследственные болезни — часть общей наследственной измен¬
чивости человека. Нет резкой границы между наследственной
изменчивостью, ведущей к вариациям нормальных признаков,
и изменчивостью, результатом которой являются наследствен¬
ные болезни, в одних и тех же генах могут возникать и нейт¬
ральные, и положительные, и патологические мутации.
Глава 1. Введение в клиническую генетику ^- В развитии наследственных признаков или болезней принима¬
ют участие наследственная конституция (генотип) и внешняя
среда. Во всех жизненных проявлениях действие любых генов
осуществляется в тесном взаимодействии с факторами среды.
Хотя в развитии одних признаков или болезней определяющую
роль играет наследственность (генотип), а в развитии других
существенное значение имеет внешняя среда, нет таких при¬
знаков, которые бы зависели только от наследственности или
только от среды, при различных условиях среды экспрессия
гена может изменяться и, следовательно, возможна разная
выраженность фенотипа.- Человечество отягощено огромным грузом разнообразных мута¬
ций, которые накапливались в процессе длительной эволюции.
Постоянный мутационный процесс поставляет новые мутации
в генофонд человечества, а естесп ненный отбор либо сохраняет
и умножает их число, либо приводит к исчезновению.~ Наследственная отягощенность современного человечества
состоит из двух компонентов. Один — это накопленные в про¬
цессе эволюции и истории человечества патологические мута¬
ции, другой — вновь возникающие наследственные изменения
в половых клетках. Количество вновь возникающих мута¬
ций может увеличиваться под влиянием мутагенных факторов
среды (ионизирующей радиации, химических веществ и других
факторов).- Среда обитания человека в широком смысле слова, границы
браков, планирование семьи принципиально изменились и
продолжают изменяться. Человек постоянно сталкивается с
новыми факторами среды, ранее никогда не встречавщимися
на протяжении всей его эволюции, а также испытывает боль¬
шие социальные и экологические нагрузки. Это приводит к
появлению новых видов наследственной патологии ~ экогене-
тическим болезням. Расширился крут потенциальных брачных
партнеров, больших масштабов достигла миграция населения,
увеличивается мутагенная нагрузка; все это меняет генетиче¬
скую структуру популяций человека. В то же время популяци¬
онные генетические процессы обладают большой инерцией,
поэтому не следует ожидать, что всеобъемлющее расширение
границ браков на всей планете, мутационный процесс и эко-
генетические реакции могут в короткий срок (1—2 поколения)
24 Клиническая генетикавызвать опасный взрыв наследственности человека или резкий
подъем частоты наследственных болезней.— Прогресс медицины и общества приводит к увеличению про¬
должительности жизни больных с наследственными болезня¬
ми, восстановлению у них репродуктивной функции и, следо¬
вательно, к увеличению их числа в популяциях. Больной или
носитель патологического гена является полноправным членом
общества и имеет равные права со здоровыми людьми. Такие
концепции, как евгеника, вырождение семей с наследственной
патологией, неизлечимость наследственных болезней, запре¬
щение браков или стерилизация по генетическим показаниям,
ушли в прошлое.ГЕНОМИКА И КЛИНИЧЕСКАЯ МЕДИЦИНАГеномом называют полный состав ДНК клетки, т.е. совокупность
всех генов и межгенных участков.Общие принципы построения геномов и их структурно¬
функциональную организацию изучает геномика. Методы геномики
направлены на расщифровку новых закономерностей биологических
систем и процессов и включают секвенирование, картирование и
идентификацию функций генов и внегенных элементов. Геномика
человека — основа молекулярной медицины и имеет важнейшее зна¬
чение для разработки методов диагностики, лечения и профилактики
наследственных и ненаследственных болезней. Для медицины также
большое значение имеют исследования в области геномики патоген¬
ных микроорганизмов, поскольку они проливают свет на природу
инфекционного процесса и создание лекарств, направленных на
специфические мишени бактерий.Геномика подразделяется на несколько почти самостоятельных
направлений; структурную, функциональную, сравнительную, эво¬
люционную и медицинскую геномику.Структурная геномика изучает последовательность нуклеотидов в
геномах, определяет локализацию (картирование) и строение генов,
распределение генетических элементов в пространстве всего генома.
Этот раздел в геномике медицины иногда называют «анатомией
генома человека», подчеркивая, что расположение генов на наших
хромосомах является важнейшей особенностью нашей анатомии, как
Глава 1. Введение в клиническую генетику ^псе, 4ТО было описано в XVI в. А. Везалием в трактате «О строении
человеческого тела в семи книгах». Неовезалианская метафора часто
исиользуется генетиками в характеристике генома человека.Функциональная геномика направлена на идентификацию функ¬
ций каждого гена и любых элементов генома, изучение их взаимодей-
С1 ВИЯ и регуляции на всех уровнях организации — от молекулярного,
клеточного, органного и тканевого до организменного. В связи с этим
(|)ункциональная геномика оперирует следующими понятиями.— Транскриптом — полный набор транскриптов, производимых
клеткой. Основные компоненты транскриптов — первичный
РНК-транскрипт (РНК — рибонуклеиновая кислота) и молеку¬
лы матричной РІІК (мРНК).— Протеом — полный комплект белков, вырабатываемых данной
клеткой; это белковый комплемент генома клетки.— Метаболом — совокупность продуктов метаболизма клетки.
Различают метаболомику и метабономику. Первая изучает про¬
дукты метаболизма клеток. Вторая — изменения метаболизма
биологических систем под действием различных факторов.— Интерактом — интегративная система всех межбелковых взаи¬
модействий как основа любых биологических процессов.— Феном, по аналогии с понятием «геном!^, определяется как пол¬
ный набор фенотипических характеристик клетки.Сравнительная геномика изучает сходства и различия в организа¬
ции геномов разных организмов с целью выяснения общих законо¬
мерностей их строения и функционирования.Эволюционная геномика объясняет пути эволюции геномов, при¬
чины происхождения генетического полиморфизма и биоразнообра¬
зия, роль горизонтального переноса генов. Эволюционный подход к
изучению генома человека позволяет проследить за длительностью
формирования комплексов генов, отдельных хромосом, стабильно-
сгью его частей, недавно обнаруженными элементами непостоян¬
ства генома, процессом расообразования, эволюцией наследственной
иатологии.Медицинская геномика — самостоятельное направление, решающее
прикладные вопросы клинической и профилактической медицины
на основе знаний генома человека и геномов возбудителей инфекций.
В этом направлении есть место анатомической метафоре, и неко-
горые специальные разделы называют «патологической анатомией
26 Клиническая генетикагенома человека». В этом случае генетики в концентрированном виде
представляют клиницистам информацию о тех мутациях или поли¬
морфизмах генов, которые являются причиной болезней или вовле¬
чены в их патогенез. Пример такой информационной базы в отно¬
шении наследственных болезней человека — каталог В. Маккьюсика
«Менделевская наследственность человека» {Mendelian Inheritance in
Man — МІМ) и его злектроніїая ол//яе-версия — ОМШ.Все шаги эволюции живой природы, несомненно, должны были
закрепляться в информационной системе ДНК (для некоторых
существ — в РНК), а также в ее организации в клетке для выпол¬
нения функции сохранения наследственности и противоположной
функции — поддержания изменчивости. Это наиболее обоснован¬
ное представление о формировании генома каждого вида, поэтому
применительно к геному человека можно сказать, что эволюция
человека — это эволюция его генома. Такое положение подтверждает¬
ся многочисленными молекулярно-генетическими исследованиями,
поскольку стало возможным сопоставление геномов разных видов
млекопитающих, в том числе человекообразных обезьян, а в преде¬
лах вида Homo sapiens — геномов разных рас, этносов, популяций и
отдельных индивидов.Организация генома каждого эукариотического вида представляет
собой последовательную иерархию элементов: нуклеотидов, кодонов,
доменов, генов с меж генным и участками, сложных генов, хромосом,
гаплоидного набора вместе с внехромосомной и внеядерной ДНК.
В эволюционном преобразовании генома каждый из этих иерархи¬
ческих уровней мог вести себя совершенно различно (изменяясь,
комбинируясь с другими и т.д.).Учение о геноме человека — обширная область генетики чело¬
века, включающая следующие разделы: «инвентаризация» генов,
группы сцепления, картирование генов (локализация), секвениро-
вание всей ДНК (генов, их мутаций и хромосом в целом), мейоти-
ческие преобразования, функционирование отдельных генов и их
взаимодействие, интеграция структуры и функции генома в целом.
На решении всех этих вопросов была сосредоточена обширная
многолетняя Международная программа «Геном человека» (с 1990
по 2003 г.). Главным направлением работ было последовательное
секвенирование участков генома. Успешные разработки в этой
области сразу придали программе и клинико-генетическую значи¬
мость (табл. 1.2).
Глава 1. Введение в клиническую генетику27Таблица 1.2. Клинические приложения сведений о геноме человекаЭтап изучения наследственной
болезниКлиническое приложениеРегистрация болезни как наслед¬
ственной формыМед и ко-генетическое консультиро¬
ваниеЛокализация гена в хромосомеДифференциальная диагностика на
основе анализа сцепления геновН(.1деление генаГенотерапияОпределение дефекта генаДиагностика (ДНК-спеиифичсская)Обнаружение первичного про¬
дукта генаДиагностика (биохимическая),
Улучтпение лечения на основе пони¬
мания патогеггезаСистематическое изучение генома человека фактически нача¬
лось с применения менделевского анали:за наследственных призна¬
ков человека (начало XX в.). Генеалогический метод вошел тогда в
широкую практику, и шаг за шагом стал накапливаться материал по
«инвентаризации» дискретных наследственных признаков человека.
{)днако этот процесс постепенно замедлялся (за 50 лет было откры¬
то не более 400 менделируюших признаков и 4 группы сцепления).
ІІОЗМОЖНОСТИ клинико-генеалогического метода в чистом виде для
>| их целей были исчерпаны.Бурный прогресс цитогенетики человека, биохимической гене-
гики и особенно генетики соматических клеток в 60-х гг. XX в.
ІІ комплексе с генеалогическим подходом поставил изучение генома
человека на новые теоретические основы и высокий методический
уровень. Обнаружение новых менделируюших признаков человека,
особенно биохимических и иммунологических, стало быстро про¬
двигаться, появились возможности изучения сцепления и локализа¬
ции генов.Особый импульс изучению генома человека дали молекулярно-
генетические методы, или технология генной инженерии (70-е годыXX в.). Процесс познания генома углубился до выделения гена в
чистом виде и его секвенирования.в отличие от классической генетики, в современной генетике
изменился подход к анализу генов. В классической генетике последо¬
вательность изучения наследственности была следующей: идентифи¬
кация менделируюш,сго признака локализация гена в хромосоме
28Клиническая генетика(или группе сцепления) -> первичный продукт гена -> ген. В совре¬
менной генетике стал возможным и обратный подход: выделение гена
секвенирование первичный продукт.ХАРАКТЕРИСТИКА ГЕНОМА ЧЕЛОВЕКАДНК-уровеньОбшее количество ДНК в соматической клетке составляет
6,2x10^ пар оснований, следовательно, гаплоидный набор состоит из
3,1x10'^ пар нуклеотидов. Основное количество ДНК локализовано в
хромосомах (99,5%). Внехромосохмная часть генома человека — это
ДНК митохондрий (0,5%). Совсем небольшое количество состав¬
ляют отдельные кольцевые молекулы ДНК в ядре и цитоплазме.
Структурные классы ДНК человека представлены на рис. 1.5.В ядерной или хромосомной ДНК только 25-35% составляют
гены и их регуляторные участки (это уникальные последователь¬
ности), Лишь 10% относящейся к генам ДНК является кодирующей.Кодирующиепоследовательности(уникальные)10%Некодирующиепоследовательности(уникальные)90%V УПовторяющиеся
последовательности
(от уникальных
до умеренных)
60%^ Повторяющиеся
последовательности
{от умеренных
до частых)1 40% JРис. Ь5. Структурные классы ДНК человека
Глава 1. Введение в клиническую генетику 29Следовательно, 2,5-3,5% всей ядерной ДНК имеют отношение к
синтезу белков. Что делает остальная часть генома, пока неизвестно.
Однако вряд ли она не имеет функций.ПовторыВ составе геномной ДНК выделяют несколько классов повторяю¬
щихся последовательностей. Участки ДНК различаются по длине
каждого повтора и числу повторяющихся единиц (их называют тан¬
демными). Различают умеренно повторяющиеся последовательности
{до 1000 повторов в одном локусе) и высокоповторяющиеся (свыше
1000 повторов). Они могут быть локализованы в одном локусе или
IU) многих локусах одной или разных хромосом. Одна и та же после¬
довательность может повторяться в разных локусах разнос число
1^аз. Такие повторы называют гипервариабельными тандемными
повторами.Если повтор состоит из 2—6 пар нуклеотидов, то такие повторы
называют микросателлитами. Число повторяющихся копий микро¬
сателлитов варьирует от 5 до 50, а суммарная протяженность может
достигать несколько сотен нуклеотидов, другая группа повторов —
мини-сателлиты, представлена повторяющимися элементами разме¬
ром от 10 до too пар нуклеотидов. Этот умеренно повторяющийся
класс повторов формирует тракты протяженностью 102—10^ нуклео-
гидов. Значительная часть мини-сателлитов равномерно рассеяна
по геному. Некоторые гипервариабельные повторы этого класса
кластеризуются в субтеломерных областях хромосом.Мини- и микросателлитные тандемные повторы разбросаны по
иссму геному и представляют собой уникальную для каждого чело¬
века комбинацию по числу тандемных повторов в разных локусах и
по числу таких локусов. Их выявление характеризует генетический
полиморфизм каждого человека, оценка которого используется в
медико-генетических и судебно-медицинских целях (см. гл. 9).Высокоповторяюшиеся последовательности размером от 100 пар
нуклеотидов и более, формирующие тракты протяженностью до 10^—
10^ нуклеотидов, составляют фракцию сателлитной ДНК. Данный
класс повторов локализован преимущественно в областях конститу¬
тивного гетерохроматина и в прицентромерных регионах хромосом.Особый класс низкокопийных повторов составляют крупные блоки
рассеянных по геному дупликаций. Многие из них имеют достаточно
протяженные размеры (до 100 тыс. пар нуклеотидов) и обнаруживают
высокую степень идентичности нуклеотидных последовательностей
зоКлиническая генетика(>95%). Существует две категории сегментных дупликаций — вну-
трихромосомные и межхромосомные. Часто они кластеризуются в
прицентромерных и субтеломерных районах хромосом, В геноме
человека блоки сегментных дупликаций локализованы в хромосомах
7, 15, 17, 22, X (рис. 1.6). Высокая степень гомологии нуклеотидных1.5МЬ(8%)2МЬ ■(2%) JСиндром Шарко-Мари-Тус, тип 1А
Наследственная нейропатия с
параличами от сдавления1.5МЬЗМЬ(85-90%)^ Синдром ДиДжорджи
Велокардиофациальный
Ц] синдром1.5МЬНейрофиброматоз I типа5МЬСиндром
"•7 Смита-МагенисаРис. 1.6. Сегментные дупликации п геноме человека
Глава 1. Введение в клиническую генетику 31последовательностей в пределах сегментных дупликаций определяет
возможность прохождения между ними неравного кроссинговера,
ч го приводит к возникновению микроделеций и микродупликаций
и хромосомных сегментах. Многие из этих нарушений проявляются
хромосомными или генными заболеваниями (в зависимости от раз¬
мера затрагиваемого участка).Наконец, недавно в геноме человека было идентифицировано
155 областей с крупными, размером от 100 тыс. пар нуклеотидов
,40 2,2 млн пар нуклеотидов, блоками повторов (CNV — Сору Number
Variation). В настоящее время ведутся исследования, направленные
на установление структуры и функции данного класса повторов.11олучсны первые данные, указывающие на ассоциацию числа круп¬
ных блоков повторов с системной красной волчанкой, псориазом,
риском инфицирования вирусом иммунодефицита человека (ВИЧ)I типа.Внехромосомные и кольцевые молекулы ДНКОбнаруживают в цитоплазме и ядре. У человека они изучены
fiue недостаточно. В строгом смысле они являются не составны¬
ми элементами генома, а его продуктом. Их размер колеблется от
150 пар нуклеотидов до 20 тыс. пар нуклеотидов. Являются ли эти
молекулы продуктом фрагментации хромосомной ДНК в клетке или
они образуются в результате других генетических процессов (гомо¬
логичной рекомбинации, обратной транскрипции), пока не ясно.
Исследованные к настоящему времени у млекопитающих большие
кольцевые ДНК размером от 150 до 900 тыс. пар нуклеотидов, локали-
joiuiHHbie только в ядрах, представляют собой амплифицированные
участки онкогенов или генов устойчивости к ядам и антиметаболи-
г;ш. С этими молекулами предположительно связывают устойчи-
иос'1 ь клеток к лекарствам и способность клеток к неограниченному
|)осту. Их происхождение объясняют делениями соответствующих
областей хромосом.1 ІолиморфизмИзменения в структуре ДНК (в хромосомах или митохондриях)
иедут к генетическому полиморфизму. Под полиморфизмом понима-
к) I’ такие варианты последовательностей ДНК, которые распростране-
мы в общей популяции с частотой не менее 1%. Эти изменения могут
быть качественными, когда они обусловлены заменой или потерей
нуклеотидов, или количественными, когда в определенном локусе
32Клиническая генетикаварьирует число нуклеотидных повторов различной протяженности,
И те, и другие нарианты генетического полиморфизма встречаются
как в смысловых (внутриэкзонных), так и в несмысловых (внсгенных
или интронных) последовательностях молекулы ДНК.Существует несколько типов полиморфизма ДНК;• полиморфизм по числу и распределению мобильных генетиче¬
ских элементов;' полиморфизмпочислу копий тандемных повторов ДНК (VNTR —
variable number of tandem repeats);• однонуклеотидные замены в последовательности ДНК (однону¬
клеотидные полиморфизмы — ОНП).онп ~ одна из наиболее частых форм генетического полимор¬
физма. Под этим термином понимают варианты последовательностейИндивидПоследовательность1AGАGTTС2AGGGTTАTG CTCG
TG CG CG1CGTTСGGGА АТСС2CGTTАGGАА АТОТ1ТСТТтG АСG АСТС2тсттАG АGG АСТСРис, 1.7. Примеры однонуклсотидного полиморфизма (ОНП) удвухиндиви-
доп (объяснение в тексте)
I пава 1. Введение в клиническую генетику ^ДНК у разных людей с вовлечением одной пары оснований (рис. 1.7).
Ihi рисунке представлены 3 фрагмента последовате.'[ьносіей оі двух
ІПІДИВИДОВ. В прямоугольниках выделены однонуклеотидные разли¬
чия в г еномных последовательностях. ОНП — наиболее характерный
ИСІ очник вариаций между людьми. Эти вариации встречаются на
протяжении всей ДНК (в экзонах, нитронах, межгенных промежут¬
ках, повторах) и отражают ранее возникшие мутации.Секвенированием геномов или их частей у разных людей установ-
испо, что од1юиуклеотидные различия обнаруживаются в среднем с
частотой 1 замена на 600—1200 нуклеотидов. Число картированных
н рамках проекта НарМар ОНП составляет более 4,5 млн. Расчеты
іижазьівают, что 2 человека на 99,9% идентичны по нуклеотид-
мым последовательностям, т.е. только 0,1% различий по одному
нуклеотиду создает огромные индивидуальные фенотипические
нариации.Предполагают, что различия по одному основанию между опреде¬
ленными отрезками геномов лежат в основе не только генных болез¬
ней, но и чувствительности к возбудителям или защиты от них, в
основе приспособительных реакций и наследственной предрасполо¬
женности к многофакторным болезням.Число ОНП на один ген у человека колеблется от нуля до
мескольких десятков, причем в кодирующих последовательностях
ієна содержится в среднем по 4 полиморфных сайта. В генах
человека около 50% некодирующих ОНП, 25% синонимичных
кодирующих (не изменяющих аминокислоту в кодируемом белке)
и 25% несинонимичных кодирующих ОНП. Типичный индиви¬
дуум гетерозиготен примерно по 24 000—40 ООО несинонимичных
замен. Хотя информация об ОНП еще неполная (основные сведе¬
ния получены в последние несколько лет), уже известно, что все
1СНЫ содержат ОНП.с помощью карт ОНП выясняют вклад индивидуальных генов
и болезни комплексной (многофакторной) и поли генной природы.
Сравнение частот определенных типов ОНП у пациентов и в кон¬
трольных группах позволяет идентифицировать ОНП, с которы¬
ми ассоциируется заболевание. Несмотря на большие перспективы,
которые открываются для объяснения заболеваний человека на осно¬
ве понимания природы и размаха полиморфизма ОНП, необходимо
остерегаться «геномомании». Гены и геномы действуют не в вакууме.
34 Клиническая генетикаСреда не менее важна для биологии человека, чем гены. При пра¬
вильном подходе с помощью карт ОНП можно лучше понять роль
природы (генотипа) и среды в развитии человека в целом и патологии
в частности.Выше представлены характеристики основной хромосомной части
генома человека, но во всех клетках активно функционирует еще
и другая его часть, локализованная в митохондриях. Организация
генома митохондрий по сравнению с хромосомным имеет некоторые
отличия.Митохондриальный геномМитохондрии содержаткольиевую двухцепочечную ДНК, которую
иногда называют 25-й хромосомой человека (мтДНК — митохондри¬
альная ДНК). В каждой соматической клетке в среднем содержится
около 1000 митохондрий. Суммарно ДНК митохондрий составляет не
более 0,5% общего количества ДНК в организме. ДНК митохондрий
реплицируется полуавтономно от ядерной ДНК.Геном митохондрий человека был полностью секвенирован еще в
1981 г. Он содержит 16 569 пар оснований и кодирует 2 рибосомные
РНК (рРНК) [12S и 16S], 22 транспортные РНК (тРНК) и 13 поли¬
пептидов. Полипептиды являются субъединицами ферментативных
комплексов окислительного фосфорилирования. Другие 66 субъеди¬
ниц дыхательной цепи кодируются в ядре.Митохондриальный геном как целое отличается от ядерного гено¬
ма несколькими признаками;• мтДНК наследуется по материнскому типу. В зиготе содержится
от 1 до 4 отцовских митохондрий, а материнских — 25 ООО. К тому
же не исключается, что после оплодотворения репликация отцов¬
ских митохондрий вообще блокируется.• Комбинативная изменчивость мтДНК (мейоз) отсутствует.
Нуклеотидная последовательность меняется в поколениях только
в результате мутаций.• Митохондриальный геном непрерывен, т.е. не содержит интронов.
в нем имеется всего лишь несколько межгенных пар оснований
(или их вообще нет). Известно только одно исключение — около
1000 пар нуклеотидов является интроном в области промоторов
(Д-петля). в мтДНК нет защитных гистонов и системы репарации
ДНК. Такая организация определяет примерно в 10 раз большую
скорость мутирования по сравнению с ядерной ДНК.
lll.lHil 1, Введение в клиническую генетику ^* Ьольшинство генов мтДНК чередуются с генами тРНК, которые
служат разделяющими си талами для дальнейшего процессинга
первичных транскриптов.* Ипутри одной клетки могут функционировать митохондрии с
разными типами мтДНК. Это состояние называют гетероплазми-
ей. Присутствие в клетках митохондрий с одним типом мтДНК —
юмоплазмия.* И мтДНК транскрибируются или транслируются обе цепи. Код
мтДНК лишь частично отличается or универсального (UGA
кодирует триптофан, AUA кодирует метионин, АСА и AGG явля-
К) гея стоп-кодонам и).Мутации генов мтДНК лежат в основе митохондриальных болез-
ік’іі, отличающихся от моногенных болезней не только особенностя¬
ми передачи из поколения в поколение по материнской линии, но и
I мособразными чертами клинической картины.Патологические мутации міДНК открыты в каждом типе мито¬
хондриальных генов.Генный уровеньОсновное внимание в генетике всегда уделялось гену. Благодаря
комплексному подходу к изучению генов (от фенотипа на уров¬
не организма до расипіфровки нуклеотидной последовательности)
накопилась обширная информация о строении и функции генов.
Ген — последовательность нуклеотидов в ДНК, колирующих опре¬
деленную мРНК и соответствующий белок, либо РНК, несущие
I груктурные или регуляторные функции. Больщинство генов явля-
ц)1ся участками ДНК, когорые несут информацию о последователь¬
ности аминокислотных остатков в белке, однако некоторые гены
колируют только РНК. Со всеми генами связаны регуляторные
последовательности ДНК, т.е. участки, к которым присоединяются
Ослки, определяющие, будет ли ген экслрсссирован в данное время
и в данном месте.На основе данных по секвенированию определено, что в геноме
чс.'ювека около 30 ООО генов, а не 70 000—100 ООО, как считали ранее.
По уточненным данным Национального центра биотехнологической
информации США на март 2008 г. в геноме человека насчитывается
}\ 809 генов, включая исевдогены, гены, кодирующие микроРНК. В
базу ОМІМ (Gene Мар) включены только гены, влияющие на различ-
36Клиническая генетикаТьк.30-25-20 Н15-100^1873 1975 1877 1979 1981 1983 1985 1987 19ВЗ 1931 1993 1995 1ЭЭ7 2000 2002 2004 2007 2009Рис. 1.8. Динамика картирования генов человеканые заболевания. На май 2009 г. зарегистрировано 10 752 таких генов.
Динамика картирования генов представлена на рис. 1.8. Сотни генов,
вероятно, получены человеком в результате горизонтальной передачи,
начиная от бактерий. Более 6500 генов человека (примерно Ve часть
генома) охарактеризованы экспериментально (по функции продукта,
наличию мутаций, тканеспецифичности, размеру транскрипта).Гены человека более сложные, чем у других изученных организ¬
мов (например, у дрозофилы). Благодаря альтернативному сплай¬
сингу, число синтезируемых белковых продуктов, очевидно, в
1,5—2 раза больше, чем число генов. Явление альтернативного сплай¬
синга заключается н следующем. Из одного и того же первичного
РНК-транскрипта в процессинге РНК в разных тканях образу¬
ется не один, а несколько разных по длине мРНК-транскриптов.
Соответственно синтезированные полипептиды также будут раз¬
личными. Таким образом, одна и та же ДНК-последовательность
может кодировать не один, а несколько разных белковых продуктов.
Предполагается, что 40-60% генов человека подвергается альтерна¬
тивному сплайсингу. Это существенным образом увеличивает раз¬
нообразие кодируемых геном продуктов.Размер генов человека, число экзонов и интронов в них варьируют
в широких пределах (табл. 1.3).
I лава 1. Введение в клиническую генетику37Таблица 1.3. Классификация генов человека по размеру (примеры)Категория, название первичного
продуктаГеномный
размер, тыс.
п.н.кДНК
(мРНК), тыс.
п.н.ЧислонитроновМалыек-Глобин0,80,52П-Глобин1,50,62Инсулин1J0,42Л иол и поп роте и и Е3,61,231 Іаратиреоидньїй гормон4,21,02Средние1)Слок с11,01,047Коллаген 1 про-а-118,05,050Коллаген 1 про-а-238,05,050Альбумин25,02,114ADA32,01511Фактор IX спертыпания крови34,02,87Каталаза34,01,612Рецептор ЛПНП45,05,517БольшиеФемилаланингидроксилаза90,02,412ГигантскиеФактор Vin свертывания
крови186,09,026Тиреоглобулин>300,08,7>36Трансмембранный регулятор
транспорта ионов хлора-2306,527СупергигантскиеДистрофии>2000,0-16,0>60Примечание. ADA — аденозиндезаминаза. ЛПНП — липопротеиды низкой
плотности, п.н. — пар нуклеотидов.Большинство генов имеет размеры до 50 ООО пар нуклеотидов
( табл. L4). Средняя длина участка хромосомы, приходящегося на ген,
составляет 27 ООО пар нуклеотидов. Однако есть гены, размер кото¬
рых в 100 раз меньше или в 100 раз больше этой средней величины.
38Клиническая генетикаТаблица 1.4. Распределение генов человека по раз.меруРазмер генов, тыс. п.н.% общего числа<1023,310-2535,626-5020,251-10013,0101-5006,7>5001,2Примечание, п.н, — пар нуклеотидов.Как известно из мендслевской генетики, различные аллели могут
проявляться в доминантном, рецессивном и кодоминантном вариан¬
тах. В геноме человека это правило в отдельных случаях нарушается.В таблице 1.5 приведены примеры доминантного или рецессивно¬
го проявления одних и тех же фенотипов, обусловленных различны¬
ми мутациями в одном и том же гене.Таблица 1.5. Доминантные и рецессивные формы одних и тех же патологи¬
ческих состояний, обусловлен и ие различными мутация.ми в
одном и том же генеБолезньБелковый продукт (символ гепа)Тромбофилия вследствие недоста¬
точности антитромбина IIIАнтитромбин 111 {ЛТЗ)Генерализованная резистентность к
тиреоидному гормонуРецептор-1 к тиреоидному гормону
{THR})Дистрофический буллезный эпидер-
молизКоллаген, тин VII (C0L7A1)Комбинированная недостаточность
гормонов гипофизаГипофизспеиифический фактор
транскрипции [PIT]), POU{F)Пигментный ретинитРодопсин {RF)Врожденная миотонияХлоридный канал-1 скелетных
мышц (CLCN1)Р-Талассемияр-Глобин (НЬЬ)Болезнь ВиллсбрандаФактор ВиллебрандаИзолированная недостаточность
соматогропного гормонаСоматотропный гормон-І {GH1)Инсулинорезистентный сахарный
диабет с пигментно-сосочковой дис¬
трофией кожи (acanlosis nigricans)Инсулиновый рецептор {1NSR)
I лава 1. Введение в клиническую генетику ^Эти данные необходимо принимать во внимание при медико-
їсиетическом консультировании, когда родословная может не укла-
лілнаться в рамки привычных типов наследования.Функции геновНакопленные сведения о генах человека позволяют выделить их
|[)уипы по функциям первичного продукта: ферменты; модуляторы
Гіслковой функции; рецепторы; транскрипционные факторы; белки
имутриклеточного матрикса; белки внеклеточного матрикса; транс-
мсмбранные переносчики; структуры ионных каналов; молекулы
клеточных сигналов; гормоны; экстраклеточные переносчики; имму¬
ноглобулины.Кроме того, необходимо отметить наличие генов, продуктом кото¬
рых являются различные классы РНК (тРНК, рРНК, малые ядерные
РНК). В настоящее время идет активное изучение новых видов РНК.
К моменту написания данного учебника открыто более 20 видов
1*НК, которые участвуют в синтезе белка, посттранскрипционной
регуляции, репликации ДНК. Например, микроРНК в эукариоти¬
ческих клетках и малые интерферирующие РНК могут блокировать
і рансляцию мРНК или усиливать се распад, что приводит к пода-
илению экспрессии гена. Этот феномен назван РИК-интерферениией
и по эффекту напоминает эпигенетическую регуляцию экспрессии
ІСИОВ путем их метилирования. Малая ядерная РНК участвует в
сплайсинге путем удаления интронов из мРНК. Очевидно, что основ¬
ин я роль большинства видов РНК — регуляция экспрессии генов и
их продуктов. Полагают, что 30-50% регуляции транскрипционіюй
лктивности выполняется упомянутыми выше видами РНК.Безусловно, есть еще и гены с неизвестным пока действием.Наибольшую функциональную категорию (31,2% общего числа
идентифицированных генов) составляют гены, кодирующие фер¬
менты. В 2 раза меньше генов-модуляторов белковой структуры и
функции (13,6%). Они стабилизируют, свертывают полипептидные
цепи или влияют на функции белка. Каждая из остальных категорий
генов составляет менее 10% общего числа.Сроки развития наследственных болезней во многом зависят от
функции вовлеченного в патологию гена. Болезни, ассоциированные
с генами, кодирующими белки во всех функциональных катего¬
риях, могут проявляться в любом периоде жизни. Гены, кодирую¬
щие транскрипционные факторы, чаще представлены среди генов,
вызывающих болезни с началом во внутриутробном периоде. Это
40Клиническая генетикаговорит о том, что транскрипционные факторы, вероятно, играют
важную роль в «оркестровке» развития на ранних стадиях онтогенеза.
Неудивительно, что гены, кодиру]ощие транскрипционные факторы,
составляют более 30% генов, ассоциируемых с фенотипами врожден¬
ных пороков развития.Особенно высока доля болезней с началом на 1-м году жизни,
вызванных мутаыиями в генах, кодирующих ферменты (47%).
Развивающийся плод имеет доступ к материнской метаболической
системе гомеостаза через плаценту. Таким образом, дети с врож¬
денными нарушениями, вызванными недостаточностью ферментов,
обычно нормальны при рождении, но симптомы нарушения гомео¬
стаза развиваются после рождении, когда у ребенка включается соб¬
ственная дефектная система метаболизма.Болезни, вызванные дефектами генов, кодирующих ферменты,
наследуются по аутосомно-рецессивному типу, а связанные с генами,
кодирующими модуляторы белковой функции или рецепторы, — по
аутосомно-рсцессивному или аутосомно-доминантному. Болезни,
вызванные генами транскрипционных факторов, обычно относятся
к группе аутосомно-доминантных.Таким образом, временные закономерности формирования наслед¬
ственных болезней строго соответствуют роли и месту первичных
продуктов в онтогенезе. Болезни транскрипционных факторов раз¬
виваются внутриутробно, патология ферментов — в течение 1-го года
жизни, рецепторов — в возрасіе от I года до пубертатного периода,
модуляторов белковой функции — у взрослых до 50 лет.Клетка живет и работает благодаря строго скоординированным
действиям генов. Количественное распределение функций генов,
участвующих в основных процессах типичной клетки человека, сле¬
дующее: синтез РНК и белков — 22%; клеточное деление — 12%: кле¬
точные сигналы — 12%; защита клетки — 12%; обмен (метаболизм) —
17%; клеточные структуры — 8%; неизвестная функция — 17%.Немаловажная роль в регуляции активности генов отводится
эпигенетическим механизмам, которые обеспечивают наследуемые, но
потенциально обратимые изменения экспрессии генов, не связанные
с нарушениями их нуклеотидной последовательности. Молекулярную
основу эпигенетической регуляции составляют ковалентные моди¬
фикации ДНК (метилирование цитозиііа) и гистоновых белков (аце-
тилирование, метилирование, фосфорилирование и ряд других) в
составе хроматина, обеспечивающие формирование уникальной для
I пава 1. Введение в клиническую генетику ^к:іжлой клетки функциональной организации ее генома, так назы-
ii;ievioro эпигенотипа. Совершенно очевидно, что клетки организ-
м;и обладагощие одинаковым генотипом, могут иметь бесконечное
множество эпигенотипов, при этом реализация генотипа в фенотип
<к уіцествляетсн сквозь призму эпигенотипа.С ледует признать вполне закономерным, что с прогрессом в обла¬
ет эпигенетики начали формироваться представления и о новом
1ч (і:іссе патологии человека — эпигенетических болезнях, связанных
IIMCIIно с нарушениями эпигенотипа — стабильными и наследуемы¬
ми изменениями генной экспрессии, приводящими к возникновению
і;іГн>лсвания при отсутствии структурных мутаций в кодирующих
и'мах. Примерами таких заболеваний являются хроматиновые болез¬
ни, болезни геномного импринтинга. Весом вклад эпи1енетических
нарушений в развитие опухолевых процессов. Обсуждается роль эпи¬
генетических модификаций генома и в этиологии многофакторных
шГиыеваний человека.Одним из основных правил наследования признаков традиционно
сипалось правило эквивалентности реципрокных скрещиваний, т.е.
р;п»1означной функции аллеля, полученного от отца или от матери.
О-чнако, как показали подробные исследования, это правило может
мс соблюдаться. Функции генов взаимосвязаны и могут изменяться
иилоть до дифференциального выключения одного из аллелей на
протяжении всего онтогенеза. Случаи наследования с выклеочснисм
одиоі'о из аллелей (материнского или отцовского) объясняют генети¬
ческим импринтингом,Генетический импринтинг — это эпигенетический процесс, диф-
(|)сренциально помечающий локусы хромосом одного из родителей,
4 10 приводит к выключению экспрессии генов, в них расположеіі-
ных. Следовательно, в участках генома, подверженных имнринтингу,
обнаруживается моноаллельная (а не биаллельная) экспрессия генов,I.e. если имприитирован материнский аллель, то экспрессируется
и)лько отцовский, и наоборот. Неэквивалентный вклад родителей в
I сиом потомства обусловливает отклонение от строгих менделевских
законов, согласно которым вклад каждого из родителей в наслед-
сшснность потомков равнозначен. Таким образом, фенотипические
проявления конкретного 1сна могут меняться из-за трех причин: не
только из-заегоделеции или мутации в нем, но и за счет эпигенетиче¬
ского выключения экспрессии. Речь идет о стойких функциональных
различиях экспрессии гомологичных генов у потомства. Механизмом
42Клиническая генетикаимпринтинга в большинстве случаев является дифференциальное
полоспецифическое метилирование цитозиновых оснований ДНК,
устанавливаемое при созревании половых клеток, которое и выклю¬
чает в конечном итоге транскрипцию гена у потомства.Известные импринтируемые регионы всего генома человека пред¬
ставлены на рис. 1.9,1 м
І02 34 513 14 15■1JmoHdi17 2018 П П 21 22М — материнскаяО — отцовскаяРис. 1.9. Карта им при нтированных генов генома человека. Хромосомные реги¬
оны, содержащие один ген или более, экспрессируемые только с материнской
копии, отмечены белым; регионы, содержащие один или более генов, экс¬
прессируемых только с отцовской копии, отмечены серым. Некоторые регио¬
ны содержат кластеры импринтиропанных генов: материнский импринтинг
(т.е. экспрессируется только отцовский аллель) и отцовский импринтинг (т.е.
экспрессируется только материнский аллель)
(ліиш 1. Введение в клиническую генетику43Ii настоящее время в геноме человека известно около 70 генов, подвер-
жі'нкьіх импринтингу. Большинство них сгруппировано в кластеры.
1;жіія организация импринтированных генов возникла и поддерживает-
ш н ходе эволюции геномов млекопитающих, обеспечивая тесную коор-
'ППІІІИИЮустановления и поддержания моноаллельной экспрессии генов
и пределах кластеров. В геноме человека кластеры импринтированных
ii-ium локализованы в регионах 7q32. 11р15, 15qll—13. Микроделсции или
литкродупликации в этих регионах, мутации в самих импринтированных
I I'iKix или нарушения характера их дифференциального метилирования,
,1 глкже ошибочное наследование одной пары гомологичных хромосом
1И одного из родителей нри отсутствии гомолога другого родительского
ігроисхождения (однородитсльская дисомия хромосом) приводят к изме¬
нению дозы импринтированных локусов. Эти изменения обусловливают
«К'иомен «потери импринтинга» — появление биаллельной экспрессии
пмириитированного гена, либо, напротив, полное исчезновение продук-
i:i гена в клетке. Нарупісния функций импринтированных генов имеют
прямое отношение к наследственной патологии человека, обусловли-
и;ія ({)ормированис особого класса заболеваний — болезней геномно-
11» имприптинга (синдромы Рассела—Сильвера, Видемана-Беквита,I Ірадера—Вилли, Ангельмана, транзиторный неонатальный сахарный
;тлбет и ряд других).Генетический импринтинг может проявляться не только на уровнеI сна или кластера генов. Он может затрагивать целую хромосому (одно¬
родительские дисомии) и даже геномы. Эффекты геномного имприн-
іинга у человека изучены на примере пузьіріїого заноса (табл. 1.6).И:і представленных в табл. 1.6 данных можно сделать вывод, что раз-
тггие плаценты в большей степени обеспечивается геномом отца, а ран¬
нее развитие эмбриональных структур обеспечивается геномом матери.Таблица 1.6. Последствия ра:іньіх париантов импринтинга целого генома у
человекаГенетическая композицияПоследствияХромосомный [іабор2п.Я й цеклстка без ядра.Два сперматозоида с
Х-хромосомами либо опло¬
дотворение яйцеклетки одним
ли плои д и ы м сперматозоидом.
ДиандрияАндрогенез. Рагтний эмбриогенез нор¬
мальный. Далее ткани собственно эмбри¬
она не формируются. Бурно разрастается
трофобласт с образованием полного
пузырного заноса. Имеется высокий риск
малиі'низации
44Клиническая генетикаОкончание таблицы 1.6Генетическая композицияПоследствияХромосомный набор
2п. Яйцеклетка с двой¬
ным набором хромосом.
Сперматозоиды не участвуют
в оплодотворении. Д и ген и яГипогенез. Образуется тератома, пклю-
чаюшая все 3 эмбриональных слоя.
Плацентарная ткань отсутствуетХромосомный набор Зп (2
отцовских + I материнский).
Диа!тдрическая триплоидияАндроид. Большая голова плода.
Маленькое веретенообразное тело.
Отставание в росте и развитии. Большая
кистозная плацента. Частичітьій пузыр¬
ный заносХромосомный набор Зп (2 мате¬
ринских + 1 отцовский набор).
Дигеническая триплоидияГиноид. Плацента недоразвита. Эмбрион
и плод не развиваются (недифференциро¬
ванная клеточная масса)Хромосомный набор 2п (мате¬
ринский и отцовский), однако
па материнских хромосомах
отсутствует метилирование
импринтированных геновБи родительский полный пузырный занос.
Фенотипическая картина полностью
соответствует классическому варианту
полного пузырного заноса лиандриче-
ского происхождения. Обусловлен гло¬
бальным нарушением записи геномного
импринтинга в женском мейозе. Типичен
для семейных и повторяющихся случа¬
ев пузырного заноса. Прослеживается
аутосомно-рецессивный тип наследо¬
вания. Показано сцепление с регионом
19ql3.42, У женщин идентифицированы
мутации в гене NALP7, продуктом кото¬
рого является белок, негативный регу¬
лятор ИЛ-1р-плейотропного цитокина,
которЕ,1й в большом количестве синте¬
зируется в матке в период имплантации,
обеспечивая инвазию трофобластаПримечание. ИЛ — интерлейкин.Генетические карты хромосомГеном человека наряду с секвенированием нуклеотидной после¬
довательности охарактеризован к настоящему времени и по генети¬
ческим картам хромосом. Карты — это схемы, отражающие порядок
расположения генов и других генетических элементов на хромосоме с
указанием расстояния между ними. Генетическое расстояние измеря-
1. Введение в клиническую генетику45I'K H МО частоте рекомбинаций между гомологичными хромосомами
(I иыражается в сантиморганидах (сМ, названа в честь Т.Х. Моргана).
0)1ма сМ соответствует частоте рекомбинаций, равной 1%. Длина
ПИК) генома человека равна примерно 3000—3500 сМ.И іучение групп сцепления и составление карт хромосом первона-
•Kiji.MO основывались на анализе расщепления фенотипов в потомстве
фи|)мально-генетическими методами. Результат генетического карти-
ропания зависит от трех основных условий: точности клинического
пи;м моза; обширности родословных; использования информативных
и-нстических маркеров. Соблюдение этих условий и международный
и1)мсн информацией обеспечили фундамент для составления карт.Применение молекулярно-генетических методов значительно
ускорило картирование генов, а секвенирование генома позволяет
составить полные генетические карты для всех хромосом, как это
гдслано впервые для хромосом 21 и 22.На рис. 1.10 представлена в качестве примера карта хромосомы 3
1ю генам, патологические мутации в которых ведут к наследственнымСиндром фон Хиппсля-Линдау
Резистентность к тирепнлному гормону
Мелкоклеточкая кіфцинома легких
Пссвдосиндром Цел.тьвегераМ1 -ганглиозвдоз
ИНД[Х)М Моркіїо, ТИЛ В
П>-эырчатый дистрофичсскнй -:»пщсрмсшиэ
Карцинома клеток п«чениНедостаточность беяка S
1'емолитичсскіія анемии в результате
)іЕДО(гіаточности тл^татион пероксид и ы
Болезнь RJi-нольОротик8Ш1дурииПропионацидемия,рссВ-тнп
АтраисферринемияI Гипоиерулоплалминсмия. наследственная |
Пигментный рстинит-5
Постаиестеанчсское апноа
HcfiuptHOCHMOCTb caxupoiw— Блефарофнмоз, обратный эпикант и птачТромппфияия в результате избытка HRG
Недостаточ ность тиро iponин-рилн^инг-
гормонаРис. 1.10. Патологическая анатомия хромосомы 3
46Клиническая генетикаболезням. Такие карты называют патологической анатомией генома
человека. Это далеко не полная карта, которая постоянно пополня¬
ется, как и по другим хромосомам.Примером полной генетической карты может быть геном мито¬
хондрий (рис. 1.11), наиболее точно расшифрованный и секвениро-
ванный. На рисунке видно, что каждый ген митохондрий занимает
свое положение. Известны все особенности генома митохондрий
(см. выше).12s р,ND4LАТРавв бATpBSa ВРис. 1.11. Структура митохондриального генома и примеры митохондриаль¬
ных болезней. ADPD — Болезнь Альцгеймера/болезнь Паркинсона; DEAF —
нейросенсорная потеря слуха; LHON — наследственная нсйроофтальмопа-
тия Лебера; LDYT — LHON и дистония MELAS (митохондриальная миопа-
тия, эннефалопатия, \толочнокислый ацидоз и приступы судорог); MERR F —
миоклопальиая эпилепсия в сочетании с необычно красными мышечными
волокнами; NARP — нейропатия, атаксия и пигментный ретинит; РЕМ —
летальная прогрессирующая энцефаломиопатия
I лава 1. Введение в клиническую генетику47Знание генетических карт необходимо в разных разделах меди¬
цинской генетики — для диагностики болезней методом сцепления,
опенки патологических эффектов хромосомных транслокаций, реше¬
ния вопросов эволюционной и популяционной генетики.ЗНАЧЕНИЕ ГЕНЕТИКИ ДЛЯ МЕДИЦИНЫПрогресс в развитии медицины и общества приводит к относи-
юлыюму возрастанию доли генетически обусловленной патологииІІ заболеваемости, смертности, социальной дезадаптации (инвали-
дизации).Половина спонтанных абортов обусловлена генетическими при¬
чинами.Известно уже около 5000 наследственных болезней. Примерно 5-5,5%
де гей рождаются с наследственными или врожденными болезнями.Не менее 30% перинатальной и неонатальной смертности обуслов¬
лено врожденными пороками развития и наследственными болезня¬
ми с другими проявлениями. Анализ причин детской смертности в
целом (табл. 1.7) также показывает существенное значение генетиче¬
ских факторов.'1'аблица 1.7. Вклад наеледстпенных и врожденных болезней в младенческую и
детскую смертность в развитых странах по сравнеігию с другими
причинамиГлавные причины смер¬
ти в возрасте
до 1 годаДоля среди
умерших, %Главные причины
смерти в возрасте
от 1 года до 4 летДоля среди
умерших,%Перинатальные фак¬
торы28Несчастные случаи31Врождетгные и наслед¬
ственные болезни25Врожденные и наслед¬
ственные болезни23Синдром внезапной
смерти ребенка22Опухоли16Инфекции9Инфекции11Другие16другие19С возрастом меняется профиль наследственной патологии, но груз
патологии не уменьшается. Хотя частота тяжелых форм наследствен¬
ных болезней снижается в результате достаточно высокой летально-
48Клиническая генетикасти в детском возрасте, в пубертатном возрасте и позже проявляются
новые болезни. После 20—30 лет начинают проявляться болезни с
наследствен ной предрасположенностью.Не менее 25% всех больничных коек занято пациентами, страдаю¬
щими болезнями с наследственной предрасположенностью.Как известію, большая доля социальных расходов в развитых
странах идет на обеспечение инвалидов с детского возраста. Огромна
роль генетических факторов в этиологии и патогенезе ипвалидизи-
рующих состояний в детском возрасте (табл. 1.8).Таблица 1.8. Вклад генетического компонента в хронические инвалидизиру-
ющис врожденные состояния в распитых страігах (по материалам
Всемирной организации здравоохранения)Тип нарушенийЧастота на 1000
рожденийГенетическийкомпонентУмственная отсталость:
слабая (IQ 50-70)
умеренная (1Q 35-50)
тяжелая (IQ 20-35)
глубокая (1Q <20)10-30
8,5-25,5
1-3
0,3-і,2
0,1-0,3Для большинства
форм — 30—60%Детский церебральный паралич2.5Очень малыйСлепота0,6Глухота (тяжелая)*1,0>50%Врожденные пороки развития>50«50%Примечание. IQ — коэффициент умственного развития, интеллекта
(Intelligence Quotient).Доказана существенная роль наследственной предрасположенно¬
сти в возникновении широко распространенных болезней (ишеми¬
ческой болезни сердца, эссенциальной гипертонии, язвенной болез¬
ни желудка и двенадцатиперстной кишки, псориаза, бронхиальной
астмы и др.). Следовательно, для лечения и профилактики этой
группы болезней, встречающихся в практике врачей всех специаль¬
ностей, необходимо знать механизмы взаимодействия средовых и
наследственных факторов.Медицинская генетика помогает понять взаимодействие биологи¬
ческих и средовых факторов (включая специфические) в патологии
человека.
hiilua I. Введение в клиническую генетику 49И развитых странах улучшается медицинское обслуживание,
iKiiu.iijJuercH уровень жизни, что меняет направленность и интснсив-
мпсіь отбора. Новая среда, большие социальные и экологические
ширузки (избыток информации, стрессы, загрязнение атмосферы и
ip.) могут усилить мутационный процесс или изменить проявляе-
\юс\ъ генов; и то и другое приведет к дополнительному появлениюII.(следственной патологии.Недавно была предложена концепция ^feдицины 4-х «П» (Hood,
,’(M)S), в которой отражены направления приложений геномных и
юнстических знаний к практической медицине.- Предсказательная (предикативная) медицина рекомендует и раз¬
вивает проекты по диагностике вероятных (возможных) 60jTC3-
ней на основе анализа ДНК и оценок профилей экспрессии
патологических генов у конкретного человека. Термину «пред¬
сказательная медицина» ближе всего понятие, предложенное
ранее А. Боде (1998), — «геномная медицина», определяемая как
стандартное генотипирование ради улучшения качества меди¬
цинской ПОМОШ.И.- Профилактическая (превентивная) медицина организует лечебно-
профилактические мероприятия для лиц с высокой вероятно¬
стью развития той или иной болезни с наследственным пред¬
расположением.— Персонализированная медицина проводит лечебно-профилак¬
тические мероприятия в отношении конкретных пациентов
на основе их индивидуальных геномных особенностей (фар¬
макогенетика, профессиональные вредности, образ жизни,
диета и т.п.).— Партнерская медицина означает информированное и активное
участие самого пациента в сотрудничестве с врачами при выбо¬
ре вариантов лечения и образа жизни.Знание основ медицинской генетики позволяет врачу понимать
механизмы индивидуального течения болезни и выбирать соответ-
сіїіующие методы лечения. Медико-генетические знания лежат в
основе диагностики наследственных болезней. Они помогают напра¬
вить пациента и членов его семьи на медико-генетическое консульти¬
рование для первичной и вторичной профилактики наследственной
патологии.Медико-генетические знания способствуют формированию у
врача четких ориентиров в восприятии новых медико-биологических
50 Клиническая генетикаоткрытий, поскольку прогресс науки быстро и глубоко изменяет
клиническую практику.Наследственные болезни долго не поддавались лечению, а един¬
ственным методом профилактики была рекомендация воздержаться
от деторождения. Эти времена прошли.Современная медицинская генетика вооружила клиницистов
метода\ти ранней досимптомной (доклинической) и даже пренаталь¬
ной диагностики наследственных болезней. Интенсивно развиваются
и в некоторых центрах уже применяются методы предымплантацион-
цой (до имплантации зародыша) диагностики. Понимание молеку¬
лярных механизмов патогенеза наследственных болезней и высокие
медицинские технологии обеспечили успешное лечение многих форм
патологии.Сюжилась стройная система помоши больным с наследственны¬
ми болезнями: медико-генетичсское консультирование, пренатальная
диагностика, массовая диагностика у новорожденных наследствен¬
ных болезней обмена, поддающихся диетической и лекарственной
коррекции, диспансеризация больных с наследственными заболе¬
ваниями и членов их семей, Внедрение этой системы в практику
здравоохранения обеспечивает снижение частоты рождения детей с
врожденнымн пороками развития и наследственными болезнями на
60-70%. Врачи и организаторы здравоохранения обязаны активно
участвовать во внедрении достижений медицинской генетики в прак¬
тику ради счастливой семьи со здоровым потомством.ЗАКЛЮЧЕНИЕИнтенсивное развитие геномики человека обеспечили новый этап в
развитии медицины и переход ее на молекулярный уровень. Геномика
человека является основой молекулярной медицины. Понятие «молеку¬
лярная медицина» и «молекулярные болезни» введено л. Полингом
в 50-х годах XX в. Это понятие шире, чем «гено.мная медицина».
По мнению Л. Полинга, основная роль в биологических системах и
проявлениях жизнедеятельности должна быть отведена разнообра¬
зию типов связей, обусловливающих межмолекулярные воздействия.
Молекулярная медицина выявляет связи между свойствами и функ¬
циями молекул в общих проявлениях жизнедеятельности.Резкое увеличение геномной информации стало началом пере¬
осмысления процессов развития человека и ею болезней. Развитие
I пава 1. Введение в клиническую генетику ^иііголоі'ии прослеживается на молекулярном уровне от первичного
продукта гена до исхода :іаболевания.Полные данные по нуклеотидной последовательности генома
V4'коря ют генетический анализ человека. В связи с этим изменяются
імііравления биомедицинских исследований.Впредыдущиегодыосновноевниманиевизучении наследствен¬
ности человека было сосредоточено на структурной геномике
(секвенировании генома). Теперь исследования направлены на
функциональную геномику (межгенные сети, протеомику).— С середины 80-х годов XX в. обнаружение генов (их идентифи¬
кация вплоть до нуклеотидной последовательности) осущест¬
влялось главным образом через картирование генов (метод
позиционного клонирования). Сведения по геному человека
позволяют идентифицировать новые гены на уровне нуклео¬
тидных последовательностей быстрее и точнее.— До последнего времени акцент в изучении наследственной патоло¬
гии был на моногенных болезнях и на анализе одного гена. Теперь
он сдвиїается в сторону многофакторных болезней, анализа мно-
жсс г венных генов и мониторинга предрасположенности.— Изучение действия гена (первичных продуктов) всегда счита¬
лось «высшим пилотажем» в генетике, но теперь исследования
должны больше концентрироваться на механизмах регуляции
действия гена.— С точки зрения обшей патологии достижения геномики направ¬
ляют мысль от изучения этиологии наследственных болезней
(специфических мутаций) к их патогенезу (механизмам форми¬
рования патологического фенотипа).При обсуждении значимости секвенирования генома человека для
медицины и здравоохранения нередко звучат необоснованные обе-
ищния. В науке (например, в онкологии) не раз вполне объективно
прогнозируемые результаты разработок не сбывались, потому что
проблема (явление, болезнь) оказывалась сложнее, и прямое плани-
ронание успехов прогресса не оправдывалось. Знание генома чело¬
века, несомненно, приведет к прогрессу во многих (если не во всех)
разделах медицины, но маловероятно, что это будет единственным
направлением развития медицины.Исходя из уже применяемых в практическом здравоохранении
достижений генетики, можнр предіюлагать"следуЮ1 цие^ п
использования результатоів г(їномі^нх исследований:
52Клиническая генетика• широкое применение генодиагностики наследственных болез¬
ней, в том числе пренатальной;• техническую доступность предымплантационной диагностики в
основных мсдико-генетических центрах;• генетическое тестирование на болезни с наследственной предрас¬
положенностью и принятие профилактических мер;• разработку новых подходов и методов лечения, в том числе генной
терапии отдельных заболеваний;• создание новых типон лекарств на основе геномной информации
(фармакоген о м ике).Накопление генетической информации будет аккумулироваться
медициной в широком плане и использоваться здравоохранением
для разных контингентов населения. Уже есть предпосылки для
выявления детей с высоким риском раннего атеросклероза, чтобы
начать их лечение на ранних этапах и предупредить изменения в
сосудах во взрослом состоянии. Супруги \югут получить сведенияо своем генетическом статусе в отношении наследственной болез¬
ни у ребенка до планирования деторождения. Население среднего
и более старшего возраста может быть обследовано по риску мно¬
гих болезней, которые удастся предупредить (или облегчить) путем
диетических рекомендаций или лекарственного лечения. Проверку
индивидуальной чувствительности к лекарствам можно проводить
молекулярно-генетическими методами, и это должно стать стандарт¬
ной процедурой перед началом лечения.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯАксиомы медицинской генетики
Генетика человекаГенетические технологии в медицине
Генетический импринтинг
Геномика
Евгеника
Карты хромосом
Клиническая генетика
Медицинская генетика
Менделизм в генетике человека
Молекулярная медицина
Митохондриальный геном
\ пава 1. Введение в клиническую генетику WОднонуклеотидный полиморфизм
Причины детской смертности
ПротеомикаЧастота наследственной патолоі ии
On и гене ти каРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАВабков В.В. Заря генетики человека. Русское евгеническое движе-
мис и начало генетики человека. — М.: Прогресс-Традиция, 2008. —
S(M) с.Генетика: учебник / под ред. акад. РАМН В,И. Иванова. — М.:
Академкнига, 2006. — 638 с.Генетический паспорт — основа индивидуальной предиктивной
медицины / под ред, B.C. Баранова, — СПб.: Изд-во Н-Л, 2009. —
527 с.Геномика — медицине: науч. изд. / под ред. В.И. Иванова,
Л.Л, Киселева. — М.: Академкнига, 2005. — 392 с.Гинтер Е.К. Медицинская генетика; учебник. — М.: Медицина,
;003. - 448 с.Гиэд Д. Будущая эволюция человека / Евгеника двадцать первого
иска. — М.: Захаров, 2005. — 176 с.МакКонки Э. Геном человека / Пер, с англ. — М.; Техносфера,
2008. - 288 с.Марахоное А.В., Баранова А.В., Скоблов М.Ю. РНК-интерференция:
фундаментальные и прикладные аспекты // Медицинская генети¬
ка. - 2008. - № 10. - С. 44-55.Пчелов Е.Л. Родословная гениальности: из истории отечественной
иауки 1920-х годов — М.: Старая Басманная, 2008. — 350 с.Рогаев E.ff., Бори некая С. А., ИсламгуловД.В., ГригоренкоА.П. МикроРНК
человека в норме и патологии. Молекулярная биология, — 2008, —
'Г. 42. - № 5. - С. 751-764.Nakamura К DNA variations in human and medical genetics: 25 years of
iny experience // Journal of Human Genetics. — 2009. — V. 54. — N 1. —•
P. 1-8.Свердлов Е.Д. Взгляд на жизнь через окно генома. Т. 1. Очерки
сгруктурной молекулярной генетики. — М.: Наука, 2009. — 525 с.
Глава 2НАСЛЕДСТВЕННОСТЬ
И ПАТОЛОГИЯ*ИЗМЕНЧИВОСТЬ НАСЛЕДСТВЕННЫХ ПРИЗНАКОВ
КАК ОСНОВА ПАТОЛОГИИСтабильность генетического аппарата и обусловливаемый этим
аппаратом консерватизм наследственности — лишь одна сторона
биологических закономерностей. Другая ее сторона, столь же неотъ¬
емлемая, как и первая, — изменчивость. Наследствен кость и измен¬
чивость в совокупности обеспечили и сохранение жизни на Земле,
и непрекрашающуюся биологическую эволюцию. Наследственная
изменчивость организма обеспечивает его приспособляемость к усло¬
виям существования как н пределах жизни одного индивида, так и в
рамках существования биологического вида в целом.Наследственное многообразие человека — результат длительной
эволюции живой материи. Эволюция человека как биологического и
социального существа имеет свои особенности. У человека как соци¬
ального существа естественный отбор со временем принимал все более
специфические формы, что, безусловно, расширяло наследственное
разнообразие популяций. Сохранялось то, что могло «отметаться» у
животных, или, наоборот, терялось то, что животным необходимо.
Например, более полноценное обеспечение пищей и возможность
удовлетворять потребность в витамине с привели к тому, что чело¬
век в процессе эволюции утерял ген L-гулонолактоноксидазы, ката¬
лизирующей у животных синтез аскорбиновой кислоты. Этот ген у
животных предохраняет их от цинги, а человек из-за такой «всеоб¬
щей врожденной ошибки метаболизма» подвержен авитаминозу С.
В процессе эволюции человек приобретал и другие нежелательные
признаки, имеющие прямое отношение к патологии. Большинство
видов животных невосприимчивы к дифтерийному токсину и виру-* Исправлено и дополнено при участии д-ра биол. наук И.Н. Лебедева.
I iiiinoi 2. Наследственность и патология ^1'У полиомиелита, потому что у животных отсутствуют компоненты
мембраны клеток, обеспечивающие восприятие ряда патогенных
фикюров, у человека эти компоненты есть. Гены, их детермини¬
рующие, уже идентифицированы. Например, ген для восприятия
ни(|и ерийного токсина локализован в хромосоме 5, для вируса полио-
миелита — в хромосоме 19.Ьольшинство мутаций увеличивают полиморфизм человеческих
ікніуляций (группу крови, цвет волос, рост, разрез глаз и др.), но ино-
IIUI мутации затрагивают жизненно важные функции, а это приводит к
Полсзни. Таким образом, наследственная патология — это часть наслед-
С1 иен ной изменчивости, накопившейся за время эволюции человека.
Чі'ловек, став биологическим видом Homo sapiens, как бы заплатил за
•саииентацию» своего вида накоплением патологических мутаций. На
основе этих положений формулируется одна из главных концепций
медицинской генетики об эволюционном накоплении патологических
мутаций в человеческих популяциях. Подтверждением этой концеп¬
ции служат патологические мутации у животных, сходные по проявле¬
ниям с наследственными болезнями у человека (ахондроплазии, гемо-
(|)илии, мышечные дистрофии и др.). Наглядным примером служат
И1КСЫ и кошки-манчкины. Необычные зверьки появились в результате
сиомтанной мутации. При этом мутация коротких лапок «навязыва-
jiaci)» человеку природой достаточно давно. Многочисленные известияо забавных кошках-таксах будоражили Европу (Англию, Германию)
CIIIC в 30-х годах XX в. Немцы дали название коротколапым кошкам
•кошка-кенгуру» за то, что они любили садиться столбиком, внима-
1'сльно осматривая свою охотничью территорию (рис. 2.1).Наследственные болезни встре¬
чал ись у людей, живших несколь¬
ко тысячелетий назад, об этом
можно судить по находкам скеле-
'IOB с признаками патологических
процессов в раскопках и по про¬
изведениям искусства.На рис. 2.2 представлена
скульптура супружеской пары
с двумя детьми периода 2563-
2423 гг. до н.э. («музей египет¬
ского искусства» в Каире). Дети и рис. 2.1. Такса и кошка-манчкин как
женщина нормальные, у мужчи- пример ахондроплазии у животных
56Клиническая генетикаРис. 2.2. Больной с іипохоидро-
плазией, живший 4500 лет назад, в
скульптурном изображенииРис. 2.3. Больной с ахоїшроилазией
(с картины Р. Веласкеса)иы укороченные колечности, умсньшенлые кисти и стопы. Диагноз:
одна из форм хондродистрофий, наиболее вероятно гилохондропла-
зия. На некоторых картинах знаменитого испанского художника
Р. Веласкеса (1599—1660) изображены люди низкого роста. На одном
из его лучших полотен, «Sebastian, de Morra«> (1628), у мужчины
большая голова, запавшая переносица, ризомелически укороченные
конечности. Диагноз: ахондроплазия (рис. 2.3).Эволюция любого вида, в том числе и человека, в конечном счете
сводится к эволюции генотипа. В биологической эволюции человека
болезнь как фактор естественного отбора могла играть существенную
роль, а эволюция генотипа, в свою очередь, меняла нозолоі ию пато¬
логических процессов. Зависимость эволюции болезни от эволюции
генотипа вряд ли может вызывать сомнение. Выше были приведены
конкретные формы этой зависимости (цинга, дифтерия, полиомие¬
лит), Факторы эволюции долго влияли не только на формирование
биохимических, иммунных, физиологических или морфологических
I пипа 2. Наследственность и патология ^снойств органи:зма, но и на его патологические реакции, обусловли-
и:ін іначи'гельно большее многообразие нозологических форм болез-
игіі у человека, чем у животных, поскольку человек как социальное
t VIHCCTBO помогал выживать слабым больным членам общества.Основным источником многообразия наследственных призна-
кпи и их непрекращающейся эволюции служит мутационная измен¬
чивость. Способность ДНК мутировать сложилась в эволюции и
i;iкренилась отбором, по-видимому, так же, как и способность про-
тностоять мутационным изменениям, т.е. репарировать их. В орга-
пичации ДНК заложена возможіюсть оигибок ее репликации наряду
(, ио іможностью изменения первичной структуры, Вероятность сбоя
I» 104 ноет и репликации молекулы ДНК невелика и составляет 1:10-5—
II) К Однако, принимая во внимание исключительно большое число
муклеотидов в геноме (3,2 X 10^ на гаплоидный набор), следует при¬
щать. что в сумме на геном клетки на одно се поколение приходится
несколько мутаций в структурных генах. По мнению разных авторов,
каждый индивид наследует 2—3 новые мутации, которые могут давать
метальный эффект или способствовать усиленному размножению,
уиеличивая генетическое разнообразие человеческих популяций.Изменение нуклеотидной последовательности молекулы ДНК
может отразиться на первичной (аминокислотной) структуре белка
или на регуляции его синтеза. Так, большой опыт изучения молеку-
)| ирной природы гемоглобинозов показывает, что значительная часть
мутаций не изменяет функций гемоглобина, Некоторые мутации
нейтральны и не подвергаются отбору. Другие мутации приводят к
функциональным отклонениям в молекуле белка. Эти отклонения
могут оказаться полезными в некоторых условиях жизни организ-
МІІ, т.е. иметь адаптивное значение, поэтому сохранятся, а иногда
и умножатся в последуюш,их поколениях. Таким путем возникали
п сохранялись в популяциях разнообразные варианты структур¬
ных, транспортных и ферментных белков организма. Свойственный
организму человека широкий белковый полиморфизм, благодаря
которому каждый индивид биохимически неповторим, исходно
обусловлен мутационной изменчивостью и отбором адаптивных
белковых вариантов.Однако если структурные отклонения несовместимы с выпол¬
нением белком его функции, а она жизненно важна для клетки
(организма), мутация становится патологической и в дальнейшем
либо исключается из популяции вместе с нежизнеспособной клет¬
58Клиническая генетикакой (организмом), либо сохраняется, обусловливая наследственную
болезнь. В отдельных случаях гетерозиготные носители патологиче¬
ской мутации подвергаются положительному отбору. Примером этого
служит серповидно-клеточная анемия, которая широко распростра¬
нилась в популяциях, обитающих в эндемичных по малярии райо¬
нах, вследствие большей устойчивости гетерозиготных носителей
аномального гена (мутантного аллеля) к малярийному плазмодию,
чем индивидов с нормальными генотипами.Различные гены, а следовательно, и признаки организма
по-разному устойчивы к мутационным изменениям, что связано,
по-видимому, с их значением в системе организма и с их эволюци¬
онным «возрастом». Например, гистоновые белки, входящие в состав
хромосом, или сократительные белки актин и тубулин, или фер¬
ментные белки репликации и транскрипции весьма консервативны
и одинаковы не только у разных представителей человечества, но
и у биологических видов значительной филогенетической отдален¬
ности. По-видимому, мутации в соответствующих генах летальны.
Большинство белков организма, особенно ферментных, существует
в нескольких изоформах и подвержено мутационным изменениям,
ведущим к патологии.Мутации имеют различные способности сохраняться и распро¬
страняться в популяциях. Одни из них, позволяющие их носителю
сохранять плодовитость и не вызывающие серьезных неблагопри¬
ятных сдвигов в фенотипе, могут долго передаваться из поколения
в поколение. Признаки, обусловленные такими мутациями, сегре¬
гируют в поколениях согласно законам Менделя, и обусловленный
ими генетический груз в популяциях может долго сохраняться.
Некоторые комбинации условно-патологических рецессивных алле¬
лей могут давать селективное преимущество индивидам (выжи¬
ваемость, плодовитость). Частота таких аллелей в популяции будет
повышаться до определенного уровня в ряду поколений, пока не
наступит равновесие между интенсивностью мутационного процес¬
са и отбора. Частота разных мутантных аллелей этого рода может
быть неодинаковой в различных популяциях, что определяется
популяционными закономерностями (эффектом родоначальника,
частотой кровнородственных браков, миграцией и экологическими
условиями).Если вновь возникшая мутаиия имеет доминантное патологиче¬
ское проявление и ведет к легальному генетическому исходу (индивид
I iiniifi 2. Наследственность и патология Ммг ііс іянляет потомства), то такой мутационный груз не передается* 'п чующему поколению. Это обычно доминантные формы тяжелых
Понс ІНЄЙ, а также большая часть хромосомных болезней.Н целом, эффекты генетического груза у человека выражены в
июиоционно-генетических явлениях балансированного полимор-
«|и1 летальности и сниженной фертильности.ІІЛ основе постоянного изменения наследственности (мутаций)II отбора генотипов в процессе длительной эволюции человека в
іииіуляциих сформировался балансированный полиморфизм. Под
ним названием понимают следующее. В популяции представлены(|)1>рмы аллелей одного гена или более, причем частота редкого
іі'ілсля составляет не менее 1%. Поскольку возникновение мутаций —
pL'iiKoe событие (1x10^), частоту мутантного аллеля в популяции
пп'їсе 1% можно объяснить только каким-то селективным преимуще-I I И1)м этого аллеля для организма и постепенным накоплением в ряду
поколений после его появления, примерами балансированного поли-
м1>1м1)изма являются группы крови ABQ, резус-фактор, гены муко-
ннсцидоза, фенилкетонурии, первичного гемохроматоза. Генетическое
миоі'ообразие человека основано на балансированном полиморфиз¬
ме. (|юрмировавшемся в течение десятков и сотен тысячелетий. Такое
М1И)гообразие — основа развития человека как биологического вида
и социального существа. Вероятность возникновения и фиксации в
популяциях какой-либо мутации с положительным эффектом в эво-
люционно отшлифованном человеческом организме существует и в
иис гоящее время, но она крайне мала. Новые мутации практически
ікеї да дают отрицательный эффект.К эффектам мутационного груза относится летальность. Она про-
иіиіяется іибелью гамет, зигот, эмбрионов, плодов, детей. Наиболее
интенсивно летальные эффекты в человеческих популяциях выраже¬
ны на уровне зигот. Примерно 60% зигот погибают до имплантации,
i.e. до клинической регистрации беременности. Исходы всех клини¬
чески зарегистрированных беременностей следующие; спонтанные
пОорты — 15%, мсртворождения — !%, живорождения — 84%, Из
1000 живорожденных не менее 5 умирают в возрасте до 1 года по
причине наследственной патологии, несовместимой с жизнью. Таков
об кем летального груза мутационной изменчивости в популяциях
человека с медицинской точки зрения.Большинство наследственных болезней сопровождается снижен¬
ной фертильностью из-за нарушения репродуктивной функции. Это
60 Клиническая генетикаведет к уменьшенному воспроизводству потомства (и больного, и
здорового) в семьях с наследственной патологией.другие медицинские и социальные последствия мутационного
процесса — социа^іьная дезадаптация больных (инвалидность), повы¬
шенная потребность в медицинской помощи и сниженная продолжи¬
тельность жизни — изложены в главе 11.РОЛЬ НАСЛЕДСТВЕННОСТИ И СРЕДЫ
В РАЗВИТИИ ПАТОЛОГИИЛюбые проявления жизнедеятельности организма являются
результатом взаимодействия наследственных и средовых факторов.
Болезнь также развивается на основе тесного взаимодействия внеш¬
них повреждающих и внутренних факторов. Если внутренние факто¬
ры наследственно изменены, то возникает патологический процесс.
Факторы внутренней среды — это в конечном счете результат взаимо¬
действия генетических и средовых факторов в онтогенезе, потому что
уровень гормонов в организме, особенности обмена веществ, иммун¬
ные реакции исходно определяются функционированием соответ¬
ствующих генов, другими словами, генетической конституцией.Наследственные факторы, определяющие основу внутренней
среды организма в широком смысле слова, принимают самое непо¬
средственное участие в формировании патологических процессов,
либо выступая в роли этиологического фактора, либо участвуя в
патогенезе заболевания. Процессы выздоровления и исход болезни
при прочих равных условиях во многом определяются генетиче¬
ской конституцией организма. Более того, генетические факторы
существенно определяют даже смертность в возрасте от 20 до 60 лет.
Об этом можно судить на основании близнецовых исследований.
Конкордантность близнецов по смертности в возрасте 20—60 лет от
всех болезней составила у монозитотных пар 30,1%, а у дизиготных —
17,4%. Даже по смертности от травм конкордантность у монозиготных
близнецов выше (6,9%), чем у дизиготных (3,9%).С генетической точки зрения все болезни в зависимости от относи¬
тельной значимости наследственных и средовых факторов в их развитии
можно разделить на наследственные болезни, болезни с наследствен¬
ной предрасположенностью и ненаследственные болезни (рис. 2.4).
|м«1им 2. Наследственность и патология
100%It ||Ч1)|П|И1161Порогпенетрантности{проявлениязаболевания)0% Спорадическое
заболеваниеНаследственноемногофакторноезаболеваниеНаследственное
полигенное
заболеваниеНаследственноемоногенноезаболевание-Типы заболеваний-2.4. Спектр (гаследственпых заболепаний: влияние на фенотипНаследственными болезнями называют болезни, вызванные
мутниями. Проявление патологического действия мутации как
миологического фактора практически не зависит от среды. Среда
может только менять выраженность симптомов болезни и тяжесть
1Ч‘ течения, к заболеваниям этой группы относятся хромосом¬
ные и генные наследственные болезни с полным проявлением
(Гюлезнь Дауна, нейрофиброматоз, гемофилия, фенилкетонурия,
мукоиисцидоз, ахондроплазия и др.). Болезнь может проявляться
МО обязательно в детском возрасте, она диагностируется у человека
любого возраста в соответствии с временными закономерностя¬
ми іенной экспрессии (например, средний возраст начала хореи
Гсм і ингтона равен 38—40 годам, болезнь Альцгеймера начинается
tiocjie 50 лет).О болезнях с наследственной предрасположенностью говорят тогда,
когда болезнь развивается у лиц с определенной генетической харак¬
теристикой под влиянием факторов окружающей среды. Эти болезни
называют также многофакторными. Наследственность служит и
этиологическим, и патогенетическим фактором. Для пенетрантности
мутантных генов необходим соответствующий фактор окружающей
среды. К таким заболеваниям относятся, например, некоторые формы
подагры, диабета, фармако- и экогенетические болезни. Подобные
заболевания развиваются после контактов с проявляющим болезнь
нисшним воздействием, специфичным для каждого мутантного гена.иологическими факторами могут быть и средовые влияния, но
62Клиническая генетикачастота возникновения и тяжесть течения болезней существенно
зависят от наследственной предрасположенности (как в индивиду¬
альном, так и в групповом варианте). К таким болезням относятся
атеросклероз, гипертоническая болезнь, туберкулез, экзема, псориаз,
язвенная болезнь и лр. Они возникаю! под действием внешних фак¬
торов (иногда не одного, а сочетания многих), гораздо чаще у лиц с
наследственной предрасположенностью.В происхождении ненаследственных болезней определяющую роль
играет среда. Сюда относится большинство травм, инфекционных
болезней, ожогов и т.д. Генетические факторы могут влиять только
на течение патологических процессов (выздоровление, восстанови¬
тельные процессы, компенсация нарушенных функций).Приведенное выше объяснение происхождения многофакторных
заболеваний (см. рис. 2.4) в некоторой степени условно, но оно помо¬
гает оценить относительное значение наследственности и среды в
развитии болезней человека. При диалектическом понимании про¬
цессов развития необходимо признать значение и наследственных, и
средовых факторов в этиологии и патогенезе многофакторных болез¬
ней. Анализ патологических процессов возможен только с учетом
взаимодействия наследственных и средовых факторов.Генетическая программа индивида может участвовать в разви¬
тии патологии прямо или опосредованно. Передаваясь из поколе¬
ния в поколение, она обеспечивает воспроизведение типологических
характеристик человека как биологического вида и создает каждый
раз (на основе генетических закономерностей) индивида, уникаль¬
ного по генотипу, в том числе по патологическим вариациям. При
образовании гамет и затем зигот возможна любая перекомбинация
аллелей от отца и матери, пополняемая новыми мутациями.Факты, накопленные медицинской генетикой, показывают много¬
образие относительной роли наследственности и среды в развитии
любых видов патологии, кроме двух крайних ситуаций, т.е. полной
независимости от генетических или средовых факторов. Вклад каж¬
дого компонента может быть различным при разных видах пато¬
логии. Так, мутации этиологически обусловливают возникновение
наследственных болезней. Факторы среды будут влиять в этом случае
только на клиническую картину. Известно, что даже при жесткой
генетической детерминации патологии условия внешней срсды и
весь генотип в целом могут существенно модифицировать характер
и степень проявления эффектов патологического гена.
tiimi.i 2. Наследственность и патология W1’ия пнешнесредовых причин, несомненно, обусловливает заболе-
итііиі при любом генотипе (ожоги, травмы). Однако и в этом случае
цтч і;іновление, интенсивность и разнообразие клинических прояв-
Ліміпй, спектр возможных осложнений и исходов могут определяться
ІИ' только собственно повреждением, но и генетической конституци-прііпшзма,Мутации как этиологический фактор наследственных
болезней' )гиологическими факторами наследственных болезней являют¬
ся геномные, хромосомные и генные мутации. Заболевания, обу-
г11о|1111‘нные геномными (изменением числа хромосом) и хромосом-
имми (изменением структуры хромосом) мутациями, называются
хромосомными болезнями. Как правило, при хромосомных болезнях
мпрутаются сбалансированность набора генов и строгая детсрми-
ИИроианность нормального развития организма. Это приводит к
ЙИУ I риутробной гибели эмбрионов и плодов, появлению врожденных
ио|Х)ков развития и других симптомов клинической картины хромо-
С1)МНЫХ болезней.Ьольшинство форм наследственных заболеваний обусловлено
Геимыми мутациями, т.е. молекулярными изменениями на уровне
ЛИК (м уковисцидоз, гемофилия, фенилкетонурия, нейрофиброма-
И* І, миопатия Дюшенна и др.). Это генные болезни.Фенотипически генные мутации могут проявляться на молеку¬
лярном, клеточном, тканевом и органном уровнях.Множественность метаболических путей, функций белков в орга¬
ни »мс, ограниченность наших представлений о нормальном метабо-
ЛИ1МС затрудняют разработку обоснованной этиологической клас¬
сификации генных болезней. Даже число генных болезней можно
определить только ориентировочно (4500-5000), потому что нет сгро-
ГИх критериев для нозологических форм ни с клинической, ни с
1’спегической точек зрения. Например, с клинической точки зрения
миопатии Дюшенна и Беккера являются разными формами, а с
гене гической точки зрения - это результат мутации в одном и том
же локусе. Более определенно можно говорить о тех генах, в которых
ИЛСМ'ї’ифипированьї обусловливающие болезнь мутации. В настоящее
крем я известно около 1500 таких генов. Однако в ближайшее время на
осноие знаний генома человека обнаружение генов и мутаций в них,
Нсронтно, будет ускорено. В связи с тем, что различные мутации в
64 Клиническая генетикаодном и том же гене часто приводят к различным нарушениям, общее
число болезней с установленной мутационной природой можно счи¬
тать равным 2000.Унаследование патологического гена (а в случае рецессивных
мутаций — двух аллелей) не всегда сопровождается развернутой
клинической картиной. Выше уже говорилось о возможном влия¬
нии факторов внешней среды на проявление генов. Однако и другие
гены, формирующие генотип особи, т.е. генетическую конституцию
индивида, могут влиять на проявление патологического гена. В слу¬
чаях отклонения от выраженной картины болезни говорят о непол¬
ной пенетрантности и варьирующей экспрессивности гена. Поскольку
генетическая среда для патологического гена всегда индивидуальна,
возіїикают широкие возможности для разного проявления этого гена
у различных индивидов.Многие генные мутации обусловливают возникновение таких
молекулярных форм белков, патологическое действие которых выяв¬
ляется не в обычных условиях, а только при взаимодействии со
специфическими факторами внешней среды. Это так называемые
экогенетические варианты. Например, у лиц с мутациями в локусе
глюкозо-6-фосфатдегидрогеназы (Г6ФДГ) при лечении сульфанила¬
мидами возникает гемолиз эритроцитов, у лиц с аномальной холи-
нэстеразой введение дитилиііа* приводит к длительной остановке
дыхания (см. главу 8).Наследственность и патогенез наследственных болезнейМногие специфические стороны патогенеза наследственных болез¬
ней определяюгся характером повреждения генетических структур,
но формируются на уровне целостного организма, что и обусловли¬
вает индивидуальные особенности патологических процессов.При хромосомных болезнях отклонения от нормального раз¬
вития коррелируют, как правило, с выраженностью хромосомно¬
го дисбаланса. Чем больше хромосомного материала вовлечено в
мутацию, тем раньше заболевание проявится в онтогенезе и тем
значительнее будут нарушения в физическом и психическом раз¬
витии индивида.Сопоставление фсно- и кариотипа при хромосомных болезнях пока¬
зывает, что специфические проявления синдрома зависят от небольигих
сегментов хромосом. Дисбаланс по большому объе.му генетического
материала приводит к более неспсцифической картине поражения.
tiiiin.i 2. Наследственность и патология WКііких-либо специфических черт патогенеза хромосомных болез-
МГІІ ме обнаружено ни на молекулярном, ни на клеточном уровне.
l)i ионная черта хромосомного дисбаланса — множественность поро-
ьии ра 1ВИТИЯ разных органов и систем (черепно-лицевые дисморфии,
11нр<1ки развития скелета, сердечно-сосудистой, нервной и мочеполо-
»1)11 систем).Механизмь[ патогенеза моногенных заболеваний весьма разноо-
Гмьины. Специфичность этих механизмов во многом определяется
Гіипчимическими нарушениями, обусловленными данной мутацией.Некоторые общие закономерности патогенеза менделируюшей
ішіилогии можно рассмотреть на примере наследственных болезней
оимсна веществ, для которых установлена связь между мутантным
и чшм и биохимической реакцией.Патологические проявления развиваются как следствие слож-
ІИИЧ) іізаи.модействия биохимических сдвигов и физиологических
If іменсний в организме. Даже при сходстве вида нарушения, напри-
Mi'p накопления субстрата, патогенетические механизмы развития
|>1пличных заболеваний будут различны. В одном случае накапли-
иіпощийся субстрат может откладываться в клетках, приводя их к
{иИсли, в других он легко покидает клетки и его концентрация в
Оиологических жидкостях может многократно превысить нормаль-
11 III й уровень, в результате возникают условия для существенного
иімскения кислотно-основного равновесия крови, конкуренция с
физиологическим аналоюм при транспорте через гематоэнцефали-
чсский барьер, накопление вещества в разных тканях.Роль генетических факторов в патогенезе болезней неинфекци-
4)Мной природы выявляют через обнаружение ассоциаций таких
Полезней с мендслирующими признаками (их называют генетиче¬
скими маркерами). Для некоторых заболеваний уже выяснена пато-
ігнсіичсская роль маркеров [туберкулез, синдром приобретенного
иммунодефицита (СПИД)|.Специфичность патогенеза многих наследственных и нена-
слслсгвенных болезней во многом может определяться состояниелі
иммунной и эндокринной систем, функции которых генетически
детерминированы. Неблагоприятный наследственный фон может
сшть провоцирующим моментом в развитии любой патологии.
Иинример, как правило, бессимптомная гетерозиготность по гену
(і-тіїлассемии во время беременности приводит к развитию выра¬
женной; анемии, требующей терапевтического вмешательства. При
66 Клиническая генетикамутациях в генетических системах репарации ДНК мутагенные
и канцерогенные факторы ускоряют развитие злокачественных
новообразований.Наследственность и клиническая картина болезнейМногоплановость клинических и лабораторных проявлений любо¬
го заболевания охватывается понятием клинического полиморфизма.О причинах клинического полиморфизма в общей форме можно
сказать, что он обусловлен взаимодействием генетических и средовых
факторов. В связи с этим объяснение клинического разнообразия
болезней тесно связано с такими фундаментальными понятиями
генетики, как генетическая гетерогенность, пеыетрантность, экс¬
прессивность, плейотропия.Генетические причины полиморфизма наследственных болезней
обусловлены генетической уникальностью каждого индивида, а кон¬
кретные механизмы обусловлены либо генетической гетерогенностью
(мутации в разных локусах или множественные аллели), либо моди¬
фицирующим влиянием всего генотипа особи, т.е. генотипической
конституцией индивида.Истинный клинический полиморфизм наследственных болезней
может быть обусловлен модифицирующим влиянием генотипа на
проявление патологического гена, т.е. взаимодействием генов. На
эту сторону клинического полиморфизма впервые обратил внимание
СН. Давиденков еще в 1930-х годах, изучая наследование отдель¬
ных симптомов нервных болезней среди родственников больного.
Согласно его гипотезе, клинические проявления заболевания стано¬
вятся наибольшими, если в одном генотипе объединяются «патоло¬
гический задаток» и другие наследственные факторы сходно направ¬
ленного действия. Усилительный тропизм, по С-Н. Давиденкову,
специфичен для каждого гена.Несмотря на общепринятые представления о значении наслед¬
ственности, в реализации патологических процессов, до недавне¬
го времени роль наследственности представляли в виде некоего
недифференцированного фона. Однако многочисленные примеры
связей некоторых генетически детерминированных полиморфных
систем с особенностями патологических процессов убедительно
свидетельствуют о значимой роли наследственности в индивиду¬
альной патологии. Таким образом, наследственная конституция
организма — это база, которая может во многом определять инди-
Іиіиі.і 2. Наследственность и патология ^ітиуіі.'п.иую специфику клинической картины наследственных и
т*||;и.лсдстве1111ых болезней. Хорошая иллюстрация сказанного —
0<|||М11ие индивидуальные различия иммунного ответа.1к‘ меньшую роль, чем генотипическая среда, в происхождении
МПІИИЧССКОГО полиморфизма наследственных болезней могут играть
фикюры внешней среды, взаимодействующие с наследственными
«||цк юрами на любом этапе внутриутробной или постнатальной
«мит. Например, богатая фенилаланином пища беременной уси-
iHiniiri развитие фенилкетонурии у будущего гомозиготного ребенка.
Ьоягс юго, у генетически нормальных потомков женщин с фенилке-
гииурисй наблюдаются внутриутробная задержка роста, отставание
и умпиенном развитии, микроцефалия. Эти нарушения связаны с
но niL-iicгнием на плод высоких концентраций фенилаланина и его
МІ'ІІІООЛИТОВ в сыворотке крови беременной.Ікиїсзни с наследственной предрасположенностью имеют еще
Оон.тий клинический полиморфизм по сравнению с моногенными
иИю ісианиями, поэтому при многих многофакторных болезнях речь
И/К'1 о клиническом континууме с многообразием форм — отсубкли-
мичсских до тяжелых.Наследственность и исходы заболеванийI Іа гологическое действие мутации (или мутаций) может приводить
к леїальному исходу на разных стадиях онтогенеза. Существенный
вклад летальных и полулегальных мутаций во внутриутробную
ГИОсль и в раннюю постнатальную смертность не вызывает сомне¬
ний. хотя и не всегда можно определить, прямое это действие
('ІТИ0Л01 ическос) или опосредованное через патогенез. Летальный
яф(|)скт мутаций может проявиться сразу после оплодотворения.
Но 'Видимому, 60% всех зачатий не реализуется в беременность и в
Оильпіинстве случаев — в результате наследственных нарушений.
Около 50% всех спонтанных абортов связано с генетическими фак¬
торам и, В первой половине беременности происходит наибольшая
йлимииация эмбрионов и плодов. При этом чем раньше прерывает-беременность, тем вероятнее, что причиной аборта были хромо¬
сомные аномалии. Хотя механизмы гибели различны, в целом они
оиизаны с нарушениями генетического контроля различных этапов
!»мСриогенеза — от невозможности имплантации бласгоцисты до
неспособности кариотипически аномальных клеток формировать
ткни и вые структуры.
68Клиническая генетикаНе только хромосомные аномалии, но и генные мутации обуслов¬
ливают внутриутробную гибель. Извесіно более 150 таких нозологи¬
ческих форм. Генетические факторы имеют существенное значение
и в перинатальной смертности. Почти у каждого 3-го перинатально
умершего обнаруживается наследственная и врожденная патология.Значение генетических факторов в летальных исходах не отрицает
и не умаляет значения внешних факторов в структуре смертности,
а лишь подчеркивает, что повреждающие воздействия (гипоксия,
родовая травма, интоксикация, гипотрофия, инфекции) у детей с
аномальным генотипом скорее приведут к гибели, чем у нормальных
детей. Таким образом, наследственные болезни могут приводить к
смерти больных с наследственной патологией в качестве первопри¬
чины либо служат неблагоприятным фоном, усложняющим течение
ненаследственных болезней.Среди причин детской смертности существенное значение имеют
хромосомные болезни и такие наследственные генные болезни, как
муковисцидоз, болезни обмена веществ.Патологические мутации как этиологический фактор могут быть
причиной хронических болезней. Наследственные болезни практиче¬
ски всегда относятся к хроническим процессам, если только мутация
не приводит к гибели на эмбриональной стадии либо к смерти в
раннем детстве. Хроническое течение свойственно как генным, так
и хромосомным наследственным болезням. Большинство наслед¬
ственных болезней (в том числе болезни обмена веществ) имеют, как
правило, прогредиентное течение. Генные мутации могут не только
специфически проявляться, но и обусловливать неспедифическое
снижение сопротивляемости организма сопутствующим заболевани¬
ям, обусловливая их хронизацию.Наследственная конституция может существенно изменять эффек¬
тивность лечебных мероприятий. Во-первых, это широко известные
наследственно обусловленные патологические реакции на различные
лекарственные вещества; во-вторых, это полиморфизм по скорости
выведения или окисления некоторых лекарственных веществ либо
метаболитов, модифицирующих фармакокинетику ряда лекарствен¬
ных препаратов (см. главу 8).Значение генетических факторов в летальном эффекте или хро¬
ническом течении известно не только для наследственных, но и
для ненаследственных болезней. Хотя их роль в выздоровлении при
ненаследственных болезнях изучена недостаточно, но в общей форме
fimitii 2. Наследственность и патология Мчто отдельные мутации или их сочетания приводят к понижен-
it(t(l способности организма выдерживать повреждающее влияние
(гены иммунитета, ферментопатий и т.д.). Следовательно, у
HiKii.x лиц выздоровление будет затягиваться, что обусловит пере¬
пой! маюлогического процесса в хронический. Действие конкрет-
ИІ.ІН ісЕюв в хронизации ненаследственных болезней осуществляется
•и'рс» измененную направленность биохимических реакций, нару-
UU'imo гормонального статуса, снижение иммунного ответа и т.д.
Иіиг|)имер, при отсутствии каталазы в крови (наследственная аката-
ЛЙ1ИЯ) наблюдаются хронические воспаления слизистых оболочек,
при наследственных иммунодефицитных состояниях — хронические
іиОолеішния верхних дыхательных путей, носоглотки.КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ
ПАТОЛОГИИИ связи со сложной природой наследственной патологии суще-
I'Miycr несколько вариантов ее классификации (и с генетической, и
V клинической точки зрения). Прямое отношение к классификации
Мнслсдственной патологии имеет терминология, употребляемая в
Мі'лпцинекой литературе.1'ермин «наследственные болезни» не равен понятию «врожденные
0»;іе:иіи». Под врожденными болезнями понимают такие состояния,
которые существуют уже при рождении ребенка, врожденные болез¬
ни могут быть обусловлены наследственными и ненаследственными
фпкгорами. Последние вызывают врожденные пороки, возникшие в
результате тератогенного действия внещних факторов, врожденные
ии(|)скиии (сифилис, цитомегаловирус, краснуха и др.). В то же время
мс нее наследственные болезни являются врожденными (очевидно,
около 50%). Некоторые заболевания проявляются в детском (миопатия
Дкмпенна, муковисцидоз), другие в зрелом (миотоническая дистрофия,
Корея Гентингтона) и даже в пожилом (болезнь Альцгеймера) возрасте.Термин «семейные болезни*» не является синонимом термина
•ипследственные болезни». Семейные болезни могут быть наслед-
стнснными и не наследственными. Этот термин говорит только о том,
Ч'1Ч> заболевание встречается у членов одной семьи, да и само понятие
«семья» включает родственников от двух до нескольких поколений.
Ьолезнь может быть обусловлена влиянием одного и того же вредно¬
70Клиническая генетикаго фактора в семье: неправильного питания, плохой освещенности,
сырой квартиры, одной и той же профессии (шахтеры, ткачи и др.).
Иногда заболевания разделяют на семейные и спорадические. При
этом для семейною заболевания подразумевается его наличие у
родственников, а для спорадического отсутствие в семье. Таким
образом, при подобной классификации большинство рецессивных
заболеваний будет относиться к спорадическим, поскольку в родос¬
ловных (особенно если родословная не очень большая) часто не
наблюдается других случаев этого заболевания. Термин «споради¬
ческий» можно с известной долей условности применять в случаях
доминантных и хромосомных болезней. Спорадические случаи про¬
тивопоставляю іся унаследованным от больного родителя, т.е. термин
«спорадичность» подчеркивает первичное возникновение мутации.Генетическая классификация наследственных болезнейВ основу генетической классификации наследственных болезней
положен этиологический принцип, а именно тип мутаций, в том
числе эпимутации, тип клеток и характер взаимодействия со средой.
Всю наследственную патологию можно разделить на 3 гругшы.— Болезни, обусловленные мутациями в половых клетках:
•хромосомные (например, синдромы Дауна, Клайнфелтера,«кошачьего крика»);•генные (например, гемофилия, ахондроплазия, фенилкето-
нурия);* многофакторные или болезни с наследственным предраспо¬
ложением (как правило, полигенньте; например, шизофрения,
эссенциальная гипертензия, псориаз);•эпигенетические (например, синдромы Ретта, Коффина-
Лоури, Прадера—Вилли),— Болезни, обусловленные мутациями в соматических клетках:
•хромосомные (например, мозаичные формы хромосомныхболезней, лейкозы);• генные (например, опухоли на фоне мутаций онкогенов);
•многофакторные (например, вторичные иммунодефициты,врожденные пороки развития, опухоли);‘ эг[и генетические (например, опухоли пищевода, молочной
железы).— Болезни, обусловленные мутациями в половых и соматических
клетках:
IhfiitH ?. Наследственность и патология71•хромосомные (например, лейкозы у больных с синдромом
Дауна);• генные (например, ретинобластома, опухоль Вильмса);• многофакторные (например, аутоиммунные заболевания);• эпигенетические (например, семейный неполипозный коло¬
ректальный рак).Генные болезни — это болезни, вызываемые генными мутациями.
Хромосомные болезни определяются хромосомными и геномными
муі-’чіиями. Деление наследственных болезней на эти две группы
т*(|іормальное. Генные мутации передаются из поколения в поколе-
ИИ1* и соответствии с законами Менделя, а большинство хромосом¬
ных болезней, обусловленных анеуплоидиями, вообще не наследу-
«I1VH (летальный эффект с генетической точки зрения), структурные
нгрссгройки (инверсии, транслокации) передаются с дополнитель-
HI.IMH перекомбинациями, возникаюшими в мейозе у носителя пере-
(гіройки.Ьолеэни с наследственной предрасположенностью, или многофак¬
торные. Для их реализации недостаточно соответствующей генетиче¬
ской конституции индивида, нужен еще фактор или комплекс фак-
topot} среды, запускающих формирование мутантного фенотипа (или
Пплсзни). С помощью средового фактора реализуется наследственная
It рея расположенность.Группа многофакторных болезней крайне сложная, и генетиче¬
ские механизмы развития большинства нозологических форм далеки
от расшифровки. Особенно это касается синтропных (встречающих-
ия имеете) и дистропных («взаимно отталкивающихся») болезней,
когорые будут подробнее разобраны в главе 6.Эпигенетические болезни обусловлены нарушениями регуляции
функций генов без изменения первичной структуры ДНК (эпиму-
гнции).Клиническая классификация наследственных болезнейКлиническая классификация наследственных болезней по орган¬
ному, системному принципу или по типу обмена веществ очень услов¬
но. Наследственные болезни едины по этиологическому принципу
(мутации), и основу их классификации составляет, прежде всего,
системный и органный принцип: нервные, нервно-мышечные, пси¬
хические, болезни опорно-двигательного аппарата, кожи, зубочелюст¬
ной системы, крови и др. Естественно, такой подход неоднозначен.
72Клиническая генетикаНапример, больные с нейрофиброматозом (аутосом и о-доминантное
заболевание) встречается и в нейрохирургических клиниках (у боль¬
ных развиваются опухоли мозга), и в дерматологических клиниках,
поскольку у этих больных первоначально появляются обширные
светло-коричневые пятна и нейрофиброматозные узелки на коже, и
в клиниках нервных болезней в связи с глубокими нейрофибромами.
Больные с хореей Гентингтона являются наииентами и невропато¬
лога, и психиатра, больные с гепатолентикулярной дегенерацией —
терапевта и невропатолога. При очень немногих наследственных
болезнях избирательно поражается одна система. Даже моногенно
детерминируемые болезни вследствие илейотропного действия гена
и вторичных патогенетических звеньев затрагивают разные органы
и системы. Большинство генных мутаций, а тем более хромосомные
и геномные, вызывают генерализованное повреждение какой-либо
ткани (например, болезни соединительной ткани) или захватывают
несколько органов. В связи с этим многие наследственные болезни
проявляются в виде синдромов или комплекса патологических при¬
знаков, на первый взгляд не связанных между собой.Классификация наследственных болезней, выражающихся в нару¬
шении обмена веществ, проведена по типу повреждения первично¬
го звена обмена. Такая биохимическая классификация объединяет
генетический и физиологический (клинический) подходы. По такому
принципу различают наследственные болезни обмена углеводов,
липидов, аминокислот, витаминов, пуринов и пиримидинов, био¬
синтеза гормонов и т.д.ГЕНЕТИЧЕСКИЕ ОСНОВЫ ГОМЕОСТАЗАБолезнь — одно из проявлений приспособительных реакций на
действие повреждающих факторов окружающей среды. Поскольку
каждый человек с іенетической точки зрения строго индивидуален
и неповторим, то и реакции каждого человека специфичны. То, что
является благоприятным фактором для одного, может быть резко
патогенным для другого.По мере развития медицины и проникновения биологической
науки в глубинные механизмы «построения» и жизнедеятельности
человека будут полнее распознаваться и условия, при которых проис¬
ходит гармоничное развитие человека, сохраняется и укрепляется его
здоровье или, наоборот, возникают нарушения функций и болезнь.
fimnii'?. Наследственность и патология73Здоровье — г омвооталБолезнь — ДисгомеостазI) >мс()стаз как способность организма сохранять равновесие своей
Эиу||)^‘|и1сй среды в условиях постоянно изменяющейся окружающей
ypiMM исегда основан на норме реакции, под которой понимают размах
♦іоікчіііиий реакций организма на внешние воздействия без патологи-
♦іічкпч отклонений. Эти реакции
<»Пус11<шлсны врожденными харак-
и'Рік-гиками организма, которые
1>гкч 111,‘мивают ему возможность
иирпниий любых признаков или
мири MC I ров (физиологических,Mop<l>l>-fI<>rH4eCKHX, биохимиче-
1'кич, иммунологических) в допу-I (1IMI.IX пределах без неблагопри-
HIIIUIX последствий. Не вызывает
і’оміїспий изначальная наслед-
гик'миая обусловленность и индш-
ии лулльных, и видовых признаков
оріанизма.Наследственная информация,
річімизующаяся в индивидуаль¬
ном развитии организма через
биосинтез РНК и белков, обе-
1‘псчивает формирование при-
iiiuKOB и свойств организма не
клк стабильных, инвариантных
ltd отношению к изменяющейся
среде, а как способных к опреде¬
ленной вариабельности. Размах
пой вариабельности, ее ниж¬
ние и верхние границы строго
индивидуальны. Таким обра-
юм, норма реакции генетиче¬
ски обусловлена и формируется
и процессе онтогенеза как один
и-t элементов фенотипа в целом.В эволюционном формировании
как самой нормы реакции, так и
се генотипической обусловленно¬
сти естественный отбор закрепилБолезнь — ДисгомеостазРис. 2.5. Здоровье как гомеостаз (по
Ч. Скрайверу, мoдифициpoвa^fo):
А — здоровье поддерживается на
основе генетически детерминиро¬
ванной нормы реакции, обеспечи¬
вающей гомеостаз при умеренном
возлтействии факторов среды (гомео¬
стаз); Б болезнь, обусловленная
усиленным действием факторов
среды, которые выходят за преде¬
лы возможной нормы реакции (дис¬
гомеостаз); В — болезнь, обуслов¬
ленная генетически уменьшенной
нормой реакции, при которой дис¬
гомеостаз возникает при умеренных
воздействиях факторов среды
74Клиническая генетикагибкие и варьирующие реакции организма на внешние воздействия
или, другими словами, закрепил норму реакции. Следовательно, с
позиции генетики гомеостаз — генетически обусловленный компонент
фенотипа.Взаимодействие факторов среды и генетически детерминирован¬
ных норм реакций в обеспечении здоровья или развития болезни
представлено на рис. 2.5.Здоровье поддерживается при условии сохранения гомеостаза всех
компонентов внутренней среды организма на основе нормы реакции.
Нарушение гомеостаза (дисгомеостаз) проявляется в виде болез¬
ни. Причинами дисгомеостаза могут быть либо усиленные воздей¬
ствия факторов среды, либо ограниченные возможности врожденной
нормы реакции организма.Основу надежности генотипа составляют дублированность его
структурных элементов, матричный принцип биосинтеза, способ¬
ность к репарации, регуляция генной активности.В эволюционном развитии живого появление диплоидных орга¬
низмов обеспечило пересортировку и рекомбинацию генетического
материала при образовании каждой новой половой клетки и при
оплодотворении, что увеличивает размах генетической вариабельно¬
сти. Существование всех генетических локусов в двойном количестве
повышает надежность генетической детерминации признаков.Стабильность генотипа обеспечивается не только дублирован-
ностью генетических элементов. Постоянство генотипа заложено в
матричном принципе биосинтеза ДНК (репликация) и РНК (транс¬
крипция). Этот принцип обеспечивается двумя замечательными осо¬
бенностями молекулы ДНК: двуспиральностью молекулярной струк¬
туры и способностью каждой из полину к леотидных нитей-спиралей
служить матрицей для синтеза новой нуклеотидной нити, которая
комплементарна исходной нити и поэтому полностью соответствует
ей. в процессе репликации самой ДНК обеспечивается точное вос¬
произведение генетической информации в ряду последовательных
этапов синтеза ДНК и последующих клеточных делений. В процессе
транскрипции матричный синтез гарантирует точную, неискаженную
трансформацию закодированной в ДНК генетической информации
через нуклеотидные последовательности РНК в первичную амино¬
кислотную последовательность молекул специфических белков.Эволюция снабдила клетки разносторонними механизмами устра¬
нения (или репарации) повреждений генетических структур (ДНК и
I iimta 2. Наследственность и патология 75•ipoMt)C()M)- в организме не может быть ничего абсолютно стабилъно-
и>, \\ ЮМ числе не может быть абсолютно устойчивым генетический
иіііі;і|>ат клеток. Первичная структура ДНК может изменяться при
реп пикании ДНК, хотя и с малой частотой. Эти события известны
Кик о шибки репликации». ДНК повреждается гораздо больше от
іиімсйствия мутагенов окружающей среды или мутагенов, возни-
♦^.ікчцих в организме,К настоящему времени открыто несколько механизмов, с помо-
1111.10 которых устраняются те или иные повреждения ДНК. В их
1к iitmc лежат ферментативные процессы, познакомиться с которыми
можно на компакт-диске в разделе «Современный взгляд на мутани-
мииый процесс у человека».Гомеостаз внутренней среды организма должен обеспечивать-
1И. помимо только что изложенных фундаментальных механизмов,
ик'кжностью генетического контроля генной активности. Механизмы
ІІІКОІО контроля на молекулярном и надмолекулярном уровнях пока
мг раскрыты. Однако кажется несомненным, что такой контроль
VWIивительно помехоустойчив.Хотя еще не полностью выяснено, какими генетическими механиз-
мими обеспечивается постоянство внутренней среды организма (фено-
іитімеский уровень), можно предположить, что эго молскулярно-
и’мсгические и биохимические цепочки событий протягиваются от
1гпа до признака, В ряде примеров можно расчленить физиологиче¬
ские механизмы гомеостатической реакции на составляющие.ІІ общей форме можно говорить о двух видах генетической детерми-
нации гомеостаза. Один из них — контроль элементарных проявлений
юмсостаза (выделение гормона, синтез фермента и т.д.), другой —
L-истемные проявления. Разумеется, границы между элементарными и
сис гемными проявлениями гомеостаза условны. Чем больше цепочек
расшифровывается генетической детерминации элементарных про-
и плен ИЙ гомеостаза, чем глубже познаются звенья каждой из них,
ГСМ полнее и предметнее становятся наши представления о генетике
и физиологии гомеостаза в целом. В качестве примера генетической
обусловленности элементарной гомеостатической реакции можно при-
иссги генетический контроль свертываемости крови. (См. статью
О.В. Сироткиной и др. «Генетика тромбофильных состояний» на
Kt)M пакт-диске).Генетический анализ системных проявлений гомеостаза представ¬
ляет грудную задачу. Эти проявления интегральны, их невозможно
76Клиническая генетикасвести к сумме элементарных реакций, за которыми стоят конкрет¬
ные цепочки: ген —> его первичный продукт метаболические пре¬
вращения продукта. На более высоком системно-органном уровне
вступают в действие физиологические механизмы регуляции функ¬
ций. Однако и в этом случае глубинную основу таких регуляций
составляют унаследованные нормы реакций.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯБолезни с наследственной предрасположенностьюВрожденные болезниГенетическая классификация болезнейГенетическая несовместимость матери и плодаГенетическая обусловленность гомеостазаГенетические болезни соматических клетокГенетические маркерыГенетические основы хронических болезнейКлиническая классификация наследственных болезнейМенделируюшие болезни и признакиМоногенные болезниНаследственность и выздоровлениеНаследственность и исходы болезнейНаследственные болезниНорма реакцииПричины клинического полиморфизма болезнейРоль наследственности в патогенезеСемейные болезниСпорадический случайУнаследованные болезниХромосомные болезниЭволюция генотипа человекаЭлементы стабильности генотипаЭтиология наследственных болезнейРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАГенетика: учебник / под рсд, акад.
Академкнига, 2006. — 638 с.РАМН В.И, Иванова.М.
I літа 2. Наследственность и патология 77іолуСювский М.Д. Век генетики: эволюция идей и понятий. — СПб.:
|мц)сй Арт, 2000. — 262 с.іиіішер Е.К. Медицинская генетика: учебник. — М.; Медицина,
ЛМИ. - 448 с.Кпаг У., Каммингс М. Основы генетики. / Пер. с англ. — М.:
Іі'чііосфера, 2007. — 896 с.Примроуз С, Тваймен Р. Геномика. Роль в медицине. — М.: БИНОМ.
ІІіііюратория знаний, 2008. — 277 с.Иузырев В.П., Степанов В.А. Патологическая анатомия геномачело-
іи-кії. — Новосибирск.: Наука, Сибирское предприятие РАН, 1997. —с.Пусебаум Р., Мак~Иннес P.P., Виллард Х.Ф. Медицинская генетика:
V'K’f). пос. / Пер. с англ. А.Ш. Латыпова; под ред. Н.П. Бочкова, —
М.; ГЭОТАР-Медиа, 2010. - 624 с.: ил.
Глава ЗСЕМИОТИКА и КЛИНИЧЕСКАЯ
ДИАГНОСТИКА НАСЛЕДСТВЕННЫХЗАБОЛЕВАНИЙОБЩИЕ ЗАМЕЧАНИЯОбшая врачебная подготовка предполагает знание клинико-
генеалогического метода, синдромологического подхода к диагности¬
ке наследственных болезней, оценки результатов параклинических
исследований, основных признаков и особенностей клинических
проявлений наследственной патологии, общих принципов клини¬
ческой диагіюстики, особенностей осмотра и физикального обсле¬
дования пациентов и их родственников. Решающее слово и диа¬
гностике наследственных болезней имеют лабораторные анализы:
иитотенетические, молекулярно-генетические, биохимические и др.
Информация о них будет изложена в отдельной главе.Термин «синдром» в клинической генетике употребляется не толь¬
ко для обозначения совокупности симптомов, объединенных еди¬
ным патоіенезом, но и для болезней, составляющих самостояіель-
ные нозологические единицы. Нозологически идентифицированные
наследственные болезни называют синдромами. Это обусловлено
тем, что такие нозологические формы были первоначально описа¬
ны как симптомокомплексы без понимания их этиологии. Хотя в
дальнейшем расшифровывалась наследственная природа (этиология)
данного симптомокомплекса или синдрома вплоть до его полной
генетической характеристики (хромосомные болезни, генные болез¬
ни, митохондриальные болезни), за наследственными болезнями,
сначала описанными как синдромы, сохранился термин «синдром».Например, после расшифровки этиологии синдрома Клайнфелтера
была попытка называть его болезнью Клайнфелтера, но она оказа¬
лась безуспеигной.Термины «болезнь» и «синдром» для наследственной патологии
равнозначны. Для обозначения некоторых нозологических форм упо¬
требляются оба термина (например, болезнь Дауна, синдром Дауна).
IМііиа 3. Семиотика и клиническая диагностика ... 79Олііііко люди, имеющие синдром Дауна, и их опекуны, по понятным
мричимям, чувствительны относительно используемых для описания
jHiM) хромосомного состояния терминов, в связи с этим после идсн-
1и<1)11кации хромосомной основы синдрома Дауна в 1959 г. постепен¬
но I ГІІЛИ применять термин «трисомия 21».ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ
НАСЛЕДСТВЕННОЙ ПАТОЛОГИИЛюбой вид патологии (инфекции, ожоги, травмы) имеет свои
нікомомерности клинического проявления, в основе которого лежит
и »1П1модействие повреждающего фактора с организмом. Знание этих
шкомомерностей помогает врачу в диагностике заболеваний и лече¬
нии больных. Наследственная патология, несмотря на огромное
ко и)логическое многообразие, имеет специфические черты, кото¬
рые необходимо знать врачу в качестве ориентиров в диагностиче¬
ских поисках.\\ основе клинических проявлений наследственной патологии
лі'Жіїт генетические закономерности действия и взаимодействия генов.
Ниже изложены общие признаки наследственных болезней, позволя-
кнаие врачу заподозрить роль наследственных факторов в этиологии
и патогенезе заболевания.Семейный характер заболеванияЕсли врач при обследовании больного получает сведения о сход¬
ных случаях заболевания в семье, то это прямо указывает на их
1ю шожную наследственную природу. При семейных случаях заболе-
намия необходим второй этап обследования больного, направленный
1111 дифференциальную диагностику наследственной болезни. В то же
иремя заболевание только у одного члена родословной не исключает
наследственного характера болезни, поскольку заболевание может
бьггь результатом новой доминантной мутации у одного из родите¬
лей или гетерозиготности обоих родителей по рецессивной болезни
(сеі регация мутантного фенотипа).Хроническое прогредиентное рецидивирующее течениеНаследственные болезни, начинающиеся в любом возрасте, имеют
хроническое течение с прогредиентной клинической картиной.
80Клиническая генетикаПриведем несколько примеров, хроническая пневмония с
бронхоэктазами формируется у детей с легочной формой муко-
висцидоза. Длительные расстройства пищеварения возникают
при целиакии (синоним: глютеновая энтеропатия), кишечной
форме муковисдидоза, дисахаридазной недостаточности. Дети
с миодистрофисй Дюшенна постепенно теряют двигательную
активность из-за атрофии мыши. В связи с прогредиентным
течением эту болезнь называют прогрессирующей мышечной
дистрофией.Многие новые формы наследственных болезней были открыты
при обследовании людей с хронической патологией.Хронический процесс при наследственных болезнях раз¬
вивается в результате постоянного действия мутантного гена.
Хронизация и прогредиентность одного и того же заболевания
по-разному выражены у разных больных, что объясняется взаи¬
модействием генов (генотип каждого человека индивидуален).
Рецидивирующее течение наследственных болезней обусловле¬
но и генетическими, и средовыми факторами. К генетическим
причинам относятся особенности функционирования генов у
больного, т.е. регуляция их активности в установленных геноти¬
пом пределах. Средовые факторы — это и осложнения основного
патологического процесса (активация микробного фактора, нару¬
шение питания), и дополнительные повреждающие воздействия
(охлаждение, инфекции, стрессы).Специфические симптомы
наследственных болезнейРедко встречающиеся специ¬
фические симптомы или их
сочетания дают основание
думать о наследственной при¬
роде заболевания. Например,
вывих или подвывих хрусталика
глаза характерен для синдромов
Марфана, Вейля—Марчезани и
гомоцистинурии. Голубые скле¬
ры бывают при несовершенном
остеогенезе и некоторых других
болезнях соединительной ткани.Рис. 3.1, грубые черты лица у маль¬
чика с мукополисахаридозом (син¬
дром Хантера)
? hirtiM 3. Семиотика и клиническая диагностика811‘ис. 3.2. Деформиротнанная грудная
К.;к-1ка (килсвидная или «куриная»
I руліО при синдроме Марфа наРис. 3.3. Женшина с ахондропла-
зией11ри алкаптонурии моча на пеленках темнеет. От больных фснилкето-
иурией исходит мышиный запах. При кровоточивости можно думатьо болезни фон Виллебранда или о гемофилии. Грубые черты лица
имеют больные с мукополисахаридозами (рис. 3.1), Лстеническое
іслосложение с деформированной і рудной клеткой встречается при
синдроме Марфана (рис. 3.2). Непропорциональные конечности и
туловище, низкий рост, своеобразный лицевой череп говорят об
ахондроплазии (рис. 3.3). Право-, левосторонняя асимметрия раз¬
меров лица и конечностей позволяет предполагать наследственную
іемигипертрофию (рис. 3.4).
82Клиническая генетикаРнс. 3.4. Гемигиаертрофия лица (а, б) и нижних конечностей (в) (нозологиче¬
ская форма — гемигиисртрофия)Множественные патологические изменения органов
и системПервичное вовлечение в патологический процесс многих органов
или даже систем позволяет думать о наследственной причине заболе¬
вания. Большинство мутантных генов, вызывающих наследственные
болезни, дают плейотроггный эффект, в результате чего в процесс
вовлекаются многие органы.Плейотропное действие гена (плейотропия — влияние одного гена
на формирование нескольких признаков) — универсальПая гене¬
тическая закономерность, имеющая прямое отношение к клиниче¬
ским проявлениям наследственной патологии. Хорошо известно, что
любая моногенно детерминируемая наследственная болезнь всегда
проявляется НС отдельным симптомом, а специфическим сочетанием
или комплексом нарушений разных органов и систем. С клинико¬
генетической точки зрения необходимо различать первичную и вто¬
ричную плейотропию.Важность концепции плейотропии для медицинской генетики
не раз пересматривалась. Первоначальное мнение, что все аспекты
фенотипа и, следовательно, все проявления менделируюшего син¬
дрома зависят от одной функции (или дисфункции) мутантного
аллеля, постоянно подкреплялось доказательствами. Однако вос¬
приятие важности плейотропии постепенно уменьшалось, особенно
в 1940-х годах, когда была сформулирована гипотеза «один ген — один
fiwiin :і. Семиотика и клиническая диагностика ■■■ М|)н'|»М1.‘|| IИсследования млекопитающих и наблюдение за больными
мшлслсгвенными нарушениями соединительной ткани поддер-
UtlllMlor мнение, что генной плейотропии, вероятно, не существует.
()/И1.1ко отказ от понятия плейотропии был бы преждевременным.
( мпіико-генетической точки зрения конпеппия о существовании
птмкиропии правомерна и в некоторых случаях помогает уяснить
111ММ1)связь клинических симптомов болезней.Иерничная плейотропия обусловлена биохимическими механизма¬
ми ж’йствия мутантного белка или фермента — первичных продуктов
MViiiMTHbLX аллелей. Для иллюстрации этого положения приведем
М1Ч'к().1и>ко примеров.Мутантные аллели нескольких генов, контролирующих синтез
М>'| iiai сна и фибриллина, приводят к нарушению свойств волокнистой
14K-/imнательной ткани. Поскольку соединительная ткань — осно-
\и\ IK.CX органов и тканей, становятся понятными множественные
йлиииия этих мутаций на клиническую картину (фенотип) при таких
ИП1 к’лственных болезнях соединительной ткани, как, нанример, син¬
дромы чЭлерса—Данло, Марфана: нарушения строения сосудистой
УІСМКИ (особенно аорты), подвывих хрусталика, пролапс митрального
Нлинаиа, гиперрастяжимость кожи, гиперподвижность суставов и т.д.11ри фенилкетонурии нарушается обмен фенилаланина, в резуль-
цгге чего в организме не синтезируется тирозин. Вследствие этого
умот.шается или прекращается образование меланина, что ведет
к мпюпигментации кожи, волос и радужки. С другой стороны,
шггологические метаболиты (фенилпировиноградная кислота и др.)
іиіруїііают процессы развития и функционирования нервной системы
(щяялшенная возбудимость, тремор, судорожные припадки, умствен-
ІІІІЯ отсталость). В основе всех этих очень разнородных симптомов
лежит первичный эффект недостаточности (или отсутствия) актив¬
ности фенилаланингидроксилазы.Вторичная плейотропия обусловлена осложнениями первичных
Iапологических процессов. Например, при талассемии утолщение
костей черепа и гспатолиенальный синдром — результат вторичных
процессов, возникающих в связи с усиленным кроветворением и
Г'с моей дер озом паренхиматозных органов.Муковисцидоз обусловлен ошибкой в синтезе трансмембранного
белка, обеспечивающего ионный транспорт в клетках. Нарушение
ионного транспорта натрия и хлора ведет к образованию густой слизи в
бронхах и в экзокринной части поджелудочной железы. За этим следу¬
84 Клиническая генетикают вторичные легочные инфекции и нарушения переваривания пищи.
И то и другое относится к вторичным плейотрогшым эффектам.Таким образом, плейотропное действие генов обусловливает одну из
особенностей клинического проявления наследственных болезней —
вовлеченность в патологический процесс многих органов и систем.
Этот важный обобщенный диагностический признак наследствен¬
ной патологии должен служить ориентиром для врача.Врожденный характер заболеванияИ нормальные, и патологические аллели включаются в работу
в разные периоды онтогенеза — от эмбрионального до старческого.
Как подчеркивалось выше, врожденность патологических признаков
не всегда свидетельствует о наследственной природе заболевания.
Однако не менее 25% всех форм генных наследственных болезней
и почти все хромосомные болезни начинают формироваться вну¬
триутробно. Если ребенок рождается с комплексом патологических
признаков, то болезнь считают врожденной. Примеры врожденных
наследственных болезней — хромосомные синдромы, ахондропла-
зия, ихтиоз, Х-сцепленная гидроцефалия, аутосомно-рецессивная
микроцефалия и др. Примеры врожденных, но ненаследственных
болезней — краснушный, талидомидный, сифилитический, алко¬
гольный, гидантоиновый и некоторые другие синдромы, этиология
которых устанавливается при целенаправленном сборе анамнеза,
относящегося к первым неделям беременности.Врожденными нередко бывают наследственные болезни обмена
веществ. Показания для биохимической и молекулярно-генетической
диагностики таких болезней у младенцев — рвота, отказ от пиши,
судороги, гипервентиляция, летаргия, кома, желтуха, гипертермия,
измененный тонус мышц.Резистентность к наиболее распространенным
методам терапииОдна из особенностей наследственных болезней — неэффектив¬
ность лечения, хотя и не абсолютная. Это вполне понятно, потому
что «исправить» первичные звенья, даже если известен первичный
продукт мутантного гена, далеко не всегда удается (мукополисаха-
ридозы, миодистрофия Дюшенна, нейрофиброматоз).Естественно, что толерантность к лечению свойственна не всем
болезням. Если расшифрованы ключевые звенья патогенеза, то раз-
I niiuii 3. Семиотика и клиническая диагностика ... М|»ііилгмваются и успешные методы лечения. Некоторые заболевания из
t румим устойчивых к терапии переходят в группу поддающихся лече¬
нию <ГСПатолентикулярная дегенерация, иелиакия, муковисцидоз).ОБЩИЕ ПРИНЦИПЫ КЛИНИЧЕСКОЙ
ДИАГНОСТИКИ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙКак подчеркивалось выше, по мере развития медицины и здра-
hooxранения наследственные болезни занимают все большую долю
♦Кмцсй патологии человека. Большинство наследственных болезней
имссг хроническое течение, вследствие чего высока повторная обра¬
ні не мость таких больных. Особенно много больных с наследственны¬
ми формами заболеваний поступает в специализированные клини¬
ческие и диагностические отделения. В то же время наследственные
||к)рмы диагностируются не всегда даже в клинических условиях.ІІ определенной степени это понятно, поскольку диагностика наслед-
І’І ИСННОЙ патологии — сложный и трудоемкий процесс.Т рудности диагностики обусловлены прежде всего тем, что нозо¬
логический спектр наследственных болезней, каждая из которых
имеет очень разнообразную клиническую картину, весьма широк
(около 5000 форм). Так, известно более 500 наследственных форм
периных болезней; в дерматологии и офтальмологии таких форм
Htwiee 250. Некоторые формы встречаются крайне редко, и врач может
их никогда не видеть. В связи с таким разнообразием и сходством
некоторых наследственных форм с ненаследственными болезнями
((|)Єиокопиями), а также в связи с редкостью наследственных болез¬
ней (1:200 ООО и реже) врач не может активно владеть веем запасом
ІНПНИЙ, необходимых для диагностики не только всех наследствен¬
ных болезней, но даже редких форм по его специальности. Знание
псновных принципов клинической генетики поможет заподозрить
нечасто встречающиеся наследственные болезни, а после дополни-
гельных консультаций с врачом-гснетиком, проведения паракли-
мических и лабораторно-генетических обследований — установить
точный диагноз.Клиническая диагностика наследственных болезней основывает¬
ся на данных клинического, генеалогического и параклинического
обследования. Чтобы не пропусгить наследственную болезнь, врач
должен помнить о том, что наследственные болезни могут про¬
86Клиническая генетикатекать под маской ненаследственных. В ряде случаев наследствен¬
ная патология может сопутствовать основному нснаследственно-
му заболеванию, по поводу которого больной обратился к врачу.
Диагностика должна быть двухэтапной: общее клиническое обследо¬
вание больного в соответствии с современными требованиями, опи¬
санными в соответствующих руководствах, и специализированное
дифференциально-диагностическое обследование при подозрении на
конкретную наследственную болезнь.При обшем клиническом обследовании любого больного диа¬
гностика должна завершиться четким диагнозом ненаследственного
заболевания; четким диагнозом наследственной болезни; подозре¬
нием на наследственную эгиологию основной или сопутствующей
болезни. Первые две группы заключений составляют большинство,
третья группа, как правило, требует применения специальных допол¬
нительных методов обследования (параклинических, лабораторно¬
генетических).Для установления диагноза ненаследствснного заболевания
достаточно общего клинического и лабораторного обследования.
Диагностика, например, конъюнктивита, острой пневмонии, дизен¬
терии не требует генетического обследования.Общие клинические методы также часто бывают основными в
диагностике наиболее известных и распространенных наследствен¬
ных болезней. Клиническая картина таких болезней была хоро¬
шо известна еще до установления их наследственной природы.
Например, синдром Дауна (трисомия 21) с большой вероятностью
можно диагностировать на основании данных клинического обсле¬
дования больного. Однако известны случаи ошибочной диагностики
синдрома Дауна, особенно на 1-м году жизни. Так, этот диапюз (без
анализа кариотипа) на основании особенностей черт липа без учета
других признаков синдрома Дауна иногда ставят больным с врож¬
денным гипотиреозом.Полного клинического обследования, включая использование
параклинических методов, обычно достаточно для диагностики
таких наследственных заболеваний, как ахондроплазия, нейрофи-
броматоз, хорея Гентингтона, рстинобластома, буллезный эпидермо-
лиз и т.д. Классические случаи, как правило, затруднений у врача не
вызывают, хотя возможны диагностические ошибки, особенно при
неполном проявлении того или иного синдрома или при сходных по
клиническим признакам других наследственных болезнях.
Мійим 3. Семиотика и клиническая диагностика ... ^K:i ііїлось бы, вычленение наследственных форм заболевания — не
1йК(н’ уж трудное дело, но это не так. Кажущиеся на первый взгляд
и» ||;к'лслственными заболевания могут быть осложнением или про-
иіикчіисм скрытого наследственного патологического процесса. Вот
МП колько примеров.Острая пневмония часто возникает у больных с хромосомны¬
ми іпболеваниями, с генерализованной патологией соединительной
ІКІ1ІІИ, с наследственными болезнями обмена веществ, и она чаще,
»н*м у «здоровых», принимает затяжное или хроническое течение.
Итмюнефрит чаще возникает, а затем рецидивирует у больных с
йрчж донным и аномалиями мочевой системы. Нарушение ритма серд-
liit может быть проявлением синдрома Элерса—Данло, наследственно-
И1 удлиненного интервала Q—T.ОСМОТР и ОБСЛЕДОВАНИЕ ПАЦИЕНТОВ
И ИХ РОДСТВЕННИКОВВыше изложены особенности клинических проявлений наслед-
сшснной патологии. На их основе можно построить схему обследо-
Нппия больного, чтобы диагностировать наследственную болезнь или
■ишодозрить ее.врожденные пороки развития. Генетические механизмы
эмбрионального развитияСначала рассмотрим наиболее очевидные признаки наследствен¬
ной патологии — врожденные пороки развития.Полный осмотр больного дает возможность выявить врожден¬
ный порок развития, который может быть наследственной болез¬
нью, составной частью наследственной болезни либо следствием
тератогенеза.Под термином «врожденный порок развития» понимают морфо¬
логический дефект органа, части органа или большой области тела,
исдущий к нарушению его/ее функции. Врожденные пороки раз¬
лития — результат нарушенного органогенеза. В литературе можно
истретить еще более общий термин — «врожденные аномалии» (или
дефекты). Под этим термином подразумевается любая функцио¬
нальная или структурная аномалия, которая есть у новорожден¬
ного или появляется позже, аномалия либо унаследованная, либо
88Клиническая генетикавызванная средовым событием, предшествующим рождению. Это
понятие охватывает не только пороки развития, но и наследствен¬
ные болезни обмена.Морфогенез — это реализация генетической программы в трех¬
мерном пространстве и во времени, осуществляемая под влиянием
многих факторов среды. В строго определенный период онтогенеза,
в строго определенном месте начинается активация или репрессия
строго определенных генов, ведущая к дифференцировке клеток и
органогенезу. Вариации морфогенеза в конечном счете ведут к необо¬
зримому индивидуальному разнообразию людей. Выполнение мор¬
фогенетической программы начинается с оплодотворения, интен¬
сивно продолжается во внутриутробном периоде, а загем в детстве и
даже во взрослом состоянии.Пренатальный период можно разделить на преэмбриональную,
эмбриональную и плодную (фетальную) стадии. Временные характе¬
ристики основных событий эмбрио- и фетогенеза, которые помогают
врачу в правильной интерпретации внутриутробного нарушения раз¬
вития представлены в таблице 3.1.Таблица 3.1. Основные события в пренатальном развитии человекаСтадия, событияВремя
от оплодотворенияДлина эмбриона/
[ нлодаПреэмбриональнаяПервое клеточное деление30 ч-Зигота попадает в полость матки4 дня-Имплантация5—6 дней-Образование двухслойного диска12 дней0,2 ммЛайонизация хромосомы X16 днейI ммОбразолание трехслойного диска
и первичной полоски19 днейЭмбриональнаяОрганогенез4-8 нед4-40 ммФормирование головного и
спи иного мозга; первые при¬
знаки сердца и закладки конеч¬
ностей4 нед4 ммБыстро развиваются мозг, глаза,
сердце и конечности6 нед17 мм
\\ (Семиотика и клиническая диагностика ...89Окончание таблицы 3.1С гадия, событияВремя
от оплодотворенияДлина эмбриона/
плодаfbitiiuiwiorcM пальцы; разпивают-
viim. почки, печень и мышцы8 нед4 см1нкр|.1м;10тся нёбо, образуются10 нед6 смЦ|> икшя лифферснцировка
іііканчивается12 нед9 смfl ііо;иііія (фета.іьная)())iiviti;icrcH движение плода16-18 нед20 см0|к|1ы гы пеки, при специальном
укм;к‘ іиіод жизнеспособен24-26 нед35 смІІІ.К 1 р;ія прибавка массы тела
И( iiML-i nne роста и аккумуляции28-38 нед40-50 смМорфогенез на эмбриональной стадии проявляется в установ-
краииокаудальной и дорсовентральной осей. Клеточная
Іігрічаиия и дифференциация ведут к образованию тканей и
ppiaiioFj, На плодной стадии развития происходят быстрый рост
К ріииитис органов.Хотя сведения о генетических факторах, определяющих эмбрио¬
генез, быстро пополняются, все же они недостаточны. Гены, ответ-
0тисч1ные за «оркестр» морфогенеза, особенно органогенеза, много-
Я1ы: транскрипционные факторы ДНК, ростовые факторы и
СИ шальные молекулы, лиганды, гены сигнальных путей трансдук-
ІІИМ, белки внеклеточного матрикса, энзимы.Миогие вопросы генетического контроля морфогенеза пер-
Нопамально были изучены на экспериментальных животных.
Идентифицированы многие гены и генные семейства, играющие
йпжпую роль в раннем развитии, Гены эмбрионального разви¬
тии человека гомологичны по нуклеотидным последовательностям
Гсиам дрозофилы и других животных. Большинство этих генов
отне гственны за выработку белков, называемых транскрипционными
фнкторами. Они контролируют транскрипцию РНК с ДНК путем
снизывания специфических регуляторных последовательностей
дик, образующих комплексы, которые начинают транскрипцию с
помощью PH К-полимеразы.
90Клиническая генетикаТранскрипционные факторы могут активировать или подавлять
экспрессию генов. Важнейшие транскрипционные факторы кон¬
тролируют многие гены в координации последовательного каска¬
да, включающего такие фундаментальные эмбриологические про¬
цессы, как сегментация, индукция, миграция и дифференциация
клеток, апоптоз (программированная гибель клеток). Очевидно,
эти процессы опосредуются факторами роста, клеточными рецеп¬
торами и др.За 20 лет изучения генов эмбрионального развития установлена
их большая роль в нормальном развитии и болезнях. Можно пола¬
гать, что дальнейший прогресс в этой области позволит определить
генетически обоснованные подходы к лечению и профилактике
многих болезней человека. Более подробная информация изложена
в статье «Эмбриональные гены и транскрипционные факторы в
эмбриогенезе человека» на компакт-диске.Классификация и этиология врожденных пороковКлассификация врожденных пороков развития затруднена из-за
многообразия их форм и сочетаний, исчисляющихся тысячами.
Наиболее объективные критерии классификации — локализация и
этиология пороков.Врожденные пороки развития подразделяют на изолированные (в
одном органе, например, стеноз привратника), системные (в пределах
одной системы органов, например, хондродисплазии), множественные
(в органах двух систем и более).Синдромом множественных врожденных пороков развития называют
такие сочетания пороков, при которых очевидна их этиологическая
и патогенетическая связь, а также клинически очерчена морфоло¬
гическая картина. Множественные аномалии, которые являются
каскадом одного первичного нарушения, называют последователь¬
ностью (не причинная, а патогенетическая связь). Если множествен¬
ные аномалии в определенных сочетаниях появились неслучайно у
нескольких больных, то говорят об ассоциациях.Этиология врожденных пороков развития может быть наслед¬
ственной, экзогенной и многофакторной.Наследственно обусловленные врожденные пороки развития воз¬
никают либо при генных мутациях, эффект которых проявляется
в виде эмбрионального дисморфогенеза, либо при хромосомных и
геномных мутациях (хромосомные болезни). Мутации в определен-
Ijijitm 3. Семиотика и клиническая диагностика91II 1.1 \ лпкусах могут нарушать процесс морфогенеза в эмбриональном
и IK1CIэмбриональном периодах. Этому есть многочисленные дока-
♦іиг.'іі.ства, полученные в экспериментальной генетике и клинико-
it’iu'1 11 ческой практике. Нарушения морфогенеза (дисморфогснез)
Мін VI быть выражены а разной степени и различаться по спсцифич-
ІПН ІИ. Мутации в определенных локусах ведут к наследственным
ічііідромам врожденных пороков развития.ІІ результате интенсивного развития генетических технологий и
применения их для изучения некоторых этапов эмбриогенеза чело-
йі'кл установлена молекулярно-генетическая природа многих изоли-
(и)илмных и синдромальных форм врожденных пороков, в табл. 3.2
приисденьг примеры таких генов.ІііГмнка 3.2. Фенотипы (синдромгл), обусловленные генными мутациямиГенhrr _
smj_НІГ1ИХ5Локализация
в хромосомеЗр12Бо.іеінь, синдромСиидром Ваарденбурга, тип 24ql2Альбинизм неполный7q36Голопрозэнцефалия9q22Синдром ГорлинаlOqrБолезнь Гиршпрунга12q24Синдром Холта—ОрамаHSASIXq28ГидроцефалияКак видно из табл. 3.2, генетические нарушения морфогенеза
Могут затрагивать любые системы. Большинство этих мутаций явля-
кися вновь возникшими.Классификации генов, нарушающих морфогенез, еше нет.'Окзогенно обусловленные пороки развития становятся следствием
действия тератогенных факторов в эмбриональном периоде, когда
осуществляется органогенез. Механизм их действия во многом нея-
CL'ii. Тератогены могут оказывать нитоповрсждающее действие, вызы-
иигь нарушение дифференцировки клеток в зачатках органов или
мутации (генетические соматические повреждения). Хорошо доказа¬
но тератогенное действие ионизируюш^ей радиации, лекарственных
lieiiiecTB (тал и дом ид стрептомицин, гидантоин^% варфарин, валь-11 роевая кислота, аминоптерин^'^, стероидные гормоны и др.), нико¬
тина и алкоголя, недостаточного питания (дефицита витаминов и
92Клиническая генетикамикроэлементов), биологических факторов (краснухи, цитомегалии).
Синдромы, вызываемые тератогенами, имеют специфические наборы
признаков дисморфогеиеза, поэтому их выделяют в самостоятельные
нозологические формы (гидантоиновый, краснушный синдромы,
алкогольная эмбриофетопатия).Чувствительность зародыlua человека наибольшая в конце 1-й —
начале 2-й недели гестации и между 3-й и 6-й неделями. Эти два
срока называют критическими периодами развития.Промежуток времени, в течение которого повреждающий фактор
может вызвать в органе развитие порока, называют тератогенным
терминационным периодом. Чувствительность закладок разных орга¬
нов к действию экзогенных факторов в разные сроки пренатального
онтогенеза сильно различается (рис. 3.5). На видно, что раньше
других органов и систем нарушается развитие центральной нервной
системы (ЦНС) и сердца. Выраженные врожденные пороки разви¬
тия всех органов формируются в первые 7—8 недель пренатального
развития.В экспериментах на животных под влиянием тератогенов полу-
ЧСЕ1Ы пороки развития, сходные с наследственными. Их называют
фенокопиями. Однако в клинической практике достоверных случаевРис. 3.5. Тератогенные терминационные периоды для разных органов
ійм 'Л Семиотика и клиническая диагностика ...93ІфоНОІ'ОПИЙ врожденных пороков развития генной или хромосомной
їІІ'Иіиіоі ИИ не описано,ІУІііоіифактормьіми врожденными пороками ралзития называют
fHKMr мороки, которые вызваны совместным действием наследствен-
КМн и жюгенных факторов, причем ни один из них сам по себе не
ІИииі- іся причиной порока.Омюсительный вклад разных этиологических факторов в воз¬
ник иомен ие врожденных пороков развития можно оценить лишьI иГмцсй форме. Согласно данным разных авторов, генетически
tftvi'iK)пленные формы (генные и хромосомные) составляют при-
Mrpiu) 20-30% всех врожденных пороков развития, многофактор-
HliK' 30—40%, экзогенные (тератогенные) — 2—5%, случаи неясной
ііиомогии — 25-50%.И зависимости от стадии онтогенеза, на которой действовал пато-
^Фииый фактор, врожденные пороки развития бывают следствием
Щмстопатий, бластопатий, эмбриопатий и фетопатий.Гакис состояния, когда в гаметах есть мутации, нарушающие
нормальное развитие организма, называются гаметопатиями. Этим
Термином обозначают также и аномалии гамет ненаследственной
Природы, которые приводят к нарушению оплодотворения или гибе¬
ли )июты. Все врожденные наследственные пороки развития — след-
втиис гаметопатий.Мороки, возникающие в результате поражения бластоцисты,
Мн іьінают бластопатиями. Следствием бластопатий являются такие
Ш)роки развития, как циклопия, сирсномелия, а также мозаичные
формы хромосомных и реже генных болезней.'’^мбрнопатии — нарушение развития зародыша (эмбриона). В стро¬
гом смысле этого слова все врожденные пороки развития незави¬
симо от этиологии являются эмбриопатиями, поскольку именно в
эмбриональном периоде происходит формирование органов. Однако
К )мбриопатиям целесообразно относить лишь пороки тератогенной
природы, т.е. те пороки, которые возникают в результате действия
повреждающего фактора в период от 15-го дня после оплодотворения
до конца 8-й недели внутриутробного развития.Пороки или аномалии, возникающие на плодной (фетальной) ста¬
дии развития, называются фетопатиями. Они возникают в результате
но’шействия повреждающих (тератогенных) факторов в период от 9-й
недели внутриутробного развития до родов, к фетопатиям относят¬
ся нарушения развития плода, вызванные интоксикацией у матери
94 Клиническая генетика(диабетическая, алкогольная, инфекционная). В плодном периоде
действие тератогенных факторов формирует в основном функцио¬
нальные нарушения.АнтропометрияВажным методом при обследовании больного с клинико-
генетической точки 'зрения является антропометрия. Нарушения
роста скелета (замедление или ускорение, избыточность или недо¬
развитие в целом), диспропорциональность отдельных частей скелета
создают специфические антропометрические и визуальные характе¬
ристики наследственных болезней. Приведем несколько примеров.
Высокий рост (более 180 см), определяемый в основном длиной ниж¬
них конечностей, длинные руки, длинные пальцы (арахнодактилия),
долихостеномелия указывают на синдром Марфана. Укороченные
конечности по сравнению с длиной туловища, запавшая переносица
указывают на ахондроплазию. Уменьшение черепа (микроцефалия) —
симптом многих наследственных болезней.Для диагностики наследственных болезней полезны показатели
роста, массы тела, телосложения, длины конечностей (иногда их
отдельных частей), окружности груди и черепа, а также соотношение
сагиттального и латерального размеров черепа. Все эти данные срав¬
нивают с популяционными нормами. Антропометрические показате¬
ли у лиц с наследственной болезнью, имеющих нарушения роста и
развития, выходят за пределы допустимых вариаций (перцентилей).Признаки дисморфогенеза в диагностике наследственной
и врожденной патологииМногочисленные признаки дисморфогенеза или пороки развития
являются составной частью многих наследственных и врожденных
болезней. Они встречаются практически во всех системах и имеют
весьма разнообразные проявления. Некоторые представления об
их видах и числе можно найти в словаре признаков дисморфо¬
генеза (см. «Приложение:»). Большинство признаков дисморфоге¬
неза нарушают функцию того органа, к которому они относят¬
ся (кожа, глаза, нёбо, конечность и т.д.), хотя несколько десятков
признаков функцию не нарушают. Это микроаномаяии развития,
или врожденные морфогенетические варианты, они выходят за пре¬
делы нормальных вариаций, но не нарушают функцию органа (в
отличие от врожденного порока развития). Они являются неспе-
ішщч :г Семиотика и клиническая диагностика ...954фи'11скими признаками эмбрионального дисморфогенеза н отража-
(||Ин) небольшие отклонения в гомеостазе развития, либо наслсд-
ГИічпіуіо патологию, либо отклонения, вызванные тератогенными
1КМ)|>:«ми. Врожденные морфогенетические варианты встречаются
ідороїшх людей, но наличие нескольких признаков требует более
ІИмптсльного обследования больного на предмет врожденной или
III')11* ПС г lie н ной патологии.Поскольку любое нарушение морфогенеза имеет диагностическую
Іймимость, необходимо внимательно осмотреть больного для выяв¬
им и н признаков дисморфогенеза.Ниже приводится перечень наиболее распространенных призна¬
ки 11рС“ и постнатального дисморфогенеза, оценка которых необ-
Цілимії для дифференциальной диагностики наследственных син-і Лромоіі и болезней. Часть из них представлена на рис. 3.6-3.56. На
ІІЖЛ.ПМ рисунке, как правило, можно видеть не один, а несколько
|ри «маков дисморфогенеза.рииіаки дисморфогенезаКожа: ангиомы, телеанги-
зктазии, венозная сеть, пиг¬
ментные пятна, депигмен¬
тация, темно-коричневые
веснушки (более 20), гипер¬
трихоз, гирсутизм, липо¬
мы, фибромы, келоидные
рубцы, повышенная рас¬
тяжимость, складчатость,
»ялость (рис. 3.6), нару¬
шение потоотделения, ги¬
перкератоз (рис. 3.7).- Подкожная жировая клет¬
чатка: избыточное отложе¬
ние, уменьшенное количе¬
ство.- Мышцы: гипертрофия,
гипотрофия, аплазия.- Волосы: сухие, редкие, шер¬
стистые (рис. 3.8); алопеция
(тотальная, гнездная), седаяРис. 3.6. Складчатая вялая кожа
(девочка 6 лет) {cutis laxa — кожа
вялая)
96Клиническая генетикаРис. 3.7. Гиперкератоз (ихі иозоформпая эритроясрмия)Рис. 3.8, Шерстистые волосы (син¬
дром скрученных иолос и глухоты)Рис. 3.9. Седая прядь полос (синдром
Ваарденбурга)прядь над лбом (рис, 3.9), «мыс вдовы», низкий рост волос па лбу
или на шее.- Череп: гидроцефалия (рис. 3.10), микроцефалия, макроцефа¬
лия, брахицефалия, долихоцефалия, тригоноцефалия, акро¬
цефалия (рис. 3.11), выступающий лоб, выступающий затылок
(рис. 3.12), плоский затылок.— Ушные раковины: апотия, макротия (рис. 3.13), микро-
тия (рис. 3.14), деформированные, низко расположен-
^ИС. .VIO. Гидроцефалия (синдром
піі'іілснной гидроцефалии)Рис. 3.12. Выступающий затылок,
низко посаженные и отклоненные
назад ушные ракопины (трисо-
мия 18)Рис. 3.11. Акроцефалия, широкая
Пв|км10сииа, низко расположенные
уши, 6о.'[ьшие щеки, короткие глаз¬
ные тели, короткая шея (акропефа-
Линолисиндактилия, или синдром
Кяр'к^нтера)
98Клиническая генетикё ;Рис. ЗЛЗ. Макротия и другие аномалии (синдром Коэна)
tNM ;i С:пмиотика и клиническая диагностика ...99Ї.І4. Микротия, микроснатия, Рис. 3.15. Упрошенная форма завит-
|Щик|нк гомия (врожденная аномалия ка и утолщенный противозапитокІІОІІ Л'ИОЛОГИИ)(дистрофическая дисплазия)..Я. 16. Прсдушнаяфистула,отсут- Рис. 3.17. Предушные папилломы«типе мочки уха, гипертел о ризм (разные формі,і прожденных нару-ІСМ1 пиром «кошачьего глаза» — пато- шсний слуха)
дш ИИ хромосомы 22)
Рис. 3.20. Монголоидный разрез глаз Рис. 3.21. Телекаит, эпикант, пло-(синдром Грей га)ская переносица; открытые вперед
ноідри (синдром Элерса—Данло)
It^it .1 с<іМиотика и клиническая диагностика ...1013.22. Гинертелоризм, птоз,
ИИ1КП 1И)саженные деформирован-
Иіііі* утпыс раковины (сиїїлром
Ай|н коїа)ные (рис. 3.11), оттопырен¬
ные, отклоненные назад
(рис. 3,12), завитки со сгла¬
женным упрощенным рисун¬
ком (рис. 3.15), предушные
фистулы (рис. 3-16), преду-
шныс папилломы (рис. 3.17).- Лицо: плоское, круглое, треу¬
гольное, вытянутое, грубые
черты (рис. 3.18).— Область глаз и глаза: антимон¬
голоидный (рис. 3,19) и мон¬
голоидный (рис. 3.20) разрез
глаз, эпикант (рис. 3.21), теле-
кант (рис. 3.21), гипертелоризм
(рис. 3.22), гипотелоризм, птоз
(рис, 3.23), блефарофимоз, косо¬
глазие (рис. 3.24), микрофтальм,
экзофтальм, короткая глазнаяРис. 3.23. Птоз, косоглазие, огюпыренные, низко посаженные ушные рако-
нимы с упрошенным рисунком, маленький рот (синдром Халлермана—
111 грайфа)
102Клиническая генетика'!Г--І- п:ь';Рис. 3.25. Телсангиэктазии склеры
(а гаксия-телеангиэктазим)Рис. 3.24. КосоглазиеРис. 3.26. Синофриз (синдром Кор¬
нелии дс Ланге)Рис. 3.27. Седловидная переносица,
ант и монголоидный разрез глаз (син¬
дром Пфайфера)
тйшіСемиотика и клиническая диагностика ...103двойной или тройной
|ні;і рсспиц, колобома радужки,
и'к'рохромия радужек, голу-
(ii.if склеры, телеангиэктазии
(|М1с. 3.25), миопия, гиперме-
іроііия, синофриз (рис. 3.26).
Ппс; короткий, клювовид-
мі.ііі. седловидная переносица
<рпс. 3.27), широкая плоская
111-|)сиосица, плоские крылья
носа, открытые вперед ноздри
(рис. 3.28).'1>нльтр; длинный (рис. 3.29),
короткий, плоский, глубокий.
Мслюсти: прогения, perpo¬
li-и ия, макрогения (рис. 3.30) иРис. 3.29, Длинный фильтр, корот¬
кий нос с недоразвитыми крыльями,
микрогения, низко посаженные уши
(диабетическая эмбриопатия)Рис. 3.28. Открытые вперед ноздри.
Короткий нос, антимоиголоилиый
р1Г)рез глаз, гипертелоризм, широкая
ипкиосица (синдром Аарскога)Рис. 3.30. Макрогения (синдром
Х-сцепленной умственной отстало¬
сти)
104Клиническая генетикаРис- З.ЗТ. Микрогения, крупные
ушкые раковины (синдром Смита-
Лсмли-Опитца)микрогения (рис. 3.31), микро-
гнатия и макрогнатия.Губы и полость рта: макро-
стомия и ми крестом ия; губы
гонкие, толстые; нёбо пло¬
ское, высокое, арковидное,
готическое, расщелина нёба
(рис. 3.32); раздвоение языч¬
ка; макроглоссия (рис. 3.33)
и микроглоссия, короткая
уздечка языка, множественные
уздечки 1 уб.Зубы; неправильное располо¬
жение, неправильная форма,
врожденный избыток или
врожденное отсутствие одного
или нескольких зубов, гипо¬
плазия эмали, диастема (верх¬
няя, нижняя) (рис, 3.34), тремы
(рис. 3.35).Рис. 3.32. Расшелина нёба (врожден¬
ный порок как следствие протипосу-
дорожной терапии беременной)Рис. 3.33. Макроглоссия (синдром
Бе к ви та— В и л ем а и а)
ІрПйи.і 3. Семиотика и клиническая диагностика105ы. J.34. Верхняя и нижняя диасте-
Ми (|і;ісііространсна среди нубийцевh ИИ 111)3,35. Тремы — широкие промс-
Шуїки между зубами (изо^тирован-
надклапанный стеїгоз аорты)> Шея: короткая, длинная, кри-
иотся, крыловидные складки,11 и'жая линия роста волос.
Грудная клетка и туловище:
долихостеномелия, вор он ко-
оГфазная, килеьидная, доба-
ночные соски (полителия)
(рис. 3.36), гипертелориз\т
сосков, сколиоз, лордоз, ки-
(|)оз, пилонидальная ямка
(рис. 3,37).Конечности: укороченные, уд¬
линенные, вальгусная дефор-Рис. 3.36. Полителия (добавочный
сосок) (встречается при частичной
трисомии 12)Рис. 3.37. Пилонидальная ямка
(пстречастся ири разных хромосом-
ттых и генных синдромах, а также
при диабете у маїсри)
106Клиническая генетикаРис. 3,38. Варусная деформация
нижних коисчностей; гинертело-
ризм сосков (гипофосфатемия или
витамин D-резистентный рахит)мация(Х-образные)иливарус-,
пая деформация (О-образные)
(рис. 3.38),тюлидактилия(пре-
аксиальпая и постаксиальная)
(рис. 3.39, рис. 3.40), олигодак-
тилия (рис. 3.4І), брахидак-
тилия (рис. 3.42), укорочение
отдельных пальцев (рис. 3.43,3.44), арахкодактилия, син¬
дактилия (рис. 3.45, рис. 3.46),
клинодактилия (рис. 3.47),
камптодактилия (рис. 3.48),
широкий 1 палеп, гииопла-
зия I пальца, трехфаланго-
вый 1 палец кисти (рис. 3.49),
конусовидная форма пальцев
(рис. 3.50), поперечная ладон¬
ная складка (рис. 3,51), сидне-
евская складка, одна складка
на V пальце кисти, глубокая
складка на стопе (рис. 3.52),
сандалевидная шель на стопе,
полая стопа, конская стопа,
косолапость, плоскостопие,Рис. 3.39. Полидактилия преаксиальная (обозначено стрелкой)
ик :i Семиотика и клиническая диагностика ...^Hv. '-40. Полидактилия кисти и стопы постаксиальная (синдром Смита-
Опитца)107. .4..; ' - • • ■ ' f ^^И1‘. 3.41. Олигодактилия кистей и стоп, гипоплазия огдельных пальцев и
ІНОІ 1<.'й (nocraKCHajjbHbiii акрофациалміий дизостоз)1*ис. 3.42. Брахидактилия (тип А1)Рис. 3.43. Укороченные і пальцы
стоп, гипоплазия ногтей (синдром
Вольфа—Хиршхорна — делспия хро¬
мосомы 4р)
108Клиническая генетика!Рис. 3.44, Короткие пальцы стопы. Рис.3.45. Синдактилия кожная (син-
короткие ногти, широкий I палеи, дром Аарскога)
саіідалепидная шслі> (перифериче¬
ский дизостоз)Рис. 3,46. Синдактилия 11—1П пальцев стоп разной выраженности (аминон-
териновый синдром)
3. Семиотика и клиническая диагностика109|»ис. 3.47. Клинодактилия (синдром трисомии 9)рис. 3.48. Камптодактилия 11 палг,-
Цн правой кисти и III пальца левой
ЦИСГИ (вальпроевый синдром)Рис. 3.49. Трехфалаиговый I палец
кисти (анемия и трехфаланговые
1 пальцы)
110Клиническая генетика]Рис. 3.51. Четьірсхпа.іьцевам попереч¬
ная («обе-^ьянья») ладоріная складка
(часто всірсчается при хромосомных
Рис. 3.52, Глубокая складка на стопе и генпых болезнях, а также у 2-4%(трисомия 8, мозаичная форма)здоропых людей)Рис. 3.53. Подколенная складка (синдром подколенного птсригиума)
: :i Семиотика и клиническая диагностика111иорсразгибание суставов, ісмиї игтертрофия, подколенная
(.•кJ[aдкa (рис. 3.53).Ногти: широкие, короткие (рис. 3.54), ізогнутьш, аплазия, гипо-и.;іа 5ия (рис. 3.55), дистрофия, «часовые стекла».Мочеполовая система; крипторхизм, гипоспадия, талевидная
мошонка (рис. 3.56), увеличенный клитор.Ииг. 3.54* Гипоплазия концет>іх
фіі>і;іім (брахияактилия, тип Д)Рис. 3,56. ІІІалепидігая мошонка
(іти[Лром Аарскога)Рис, 3,55. Гипоплазии концевых
фаланг и ногтей (гилангоиновый
синдром)
112Клиническая генетикаТечение беременностиНа наследственную или врожденную патологию тератогенной
природы может указывать нарушение течения беременности и пре¬
натального развития плода.Угроза прерывания беременности, мало- и многоводие, малая
подвижность плода могут быть признаками врожденных и наслед¬
ственных болезней плода.Например, ограничение движений плода в матке бывает при
артрогрипозах гетерогенной генетической этиологии.При задержке внутриутробного развития или пренатальной гипо¬
плазии размеры и масса плода или новорожденного не соответствуют
гестационному сроку. Это состояние обобщенно отражает неблагопо¬
лучие пренатального периода развития и требует дифференциальной
диагностики наследственной патологии.Возможные причины задержки внутриутробного развития:
хромосомные аномалии, генные мутации (например, семейная
дизавтономия, синдромы Корнели де Ланге, Дубовица и др.),
хронические инфекционные заболевания плода (иитомегалия,
врожденная краснуха, сифилис), радиационное поражение, мно-
гоплодная беременность, аплазия поджелудочной железы у плода.
Развитие плода может задерживаться также под влиянием некото¬
рых факторов материнского организма (токсикоз, курение, гемо¬
глобинопатия и др.).Внутриутробную задержку развития необходимо отличать от гене¬
тически обусловленных малых размеров плода (врожденная гипо¬
функция щитовидной железы, различные формы наследственной
карликовости).При некоторых наследственных болезнях происходит избыточное
развитие в пренатальном периоде (внутриутробная макросомия).
Дети с синдромами Сотоса, Беквита-Видемана, диабетической фето-
патией рождаются с повышенной массой тела.КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОДГенеалогия в широком смысле слова — это родословная.
Генеалогический метод — метод родословных, т.е. прослеживание
болезни (или признака) в семье или роду с указанием типа родствен¬
ных связей между членами родословной, В медицинской генетике
этот метод называется клинико-генеалогическим, поскольку речь идет
DiiMit 'Л. Семиотика и клиническая диагностика ...113т иіИніюдснии патологических признаков с помощью клинического
ІІЙІ 'к-лоиания.Ггіісіь'іогический метод относится к наиболее универсальным
^Цріоііам п медицинской генетике. Его широко применяют в целях
іК^лико-іенетического консультирования для установления наслед-
lltWi'HMoi o характера признака, при определении типа наследования
К тчк'грантности гена, анализе сцепления генов и картировании
Циімосом, изучении интенсивности мутационного процесса, рас-
glMiltpoijKe механизмов взаимодействия генов,' )м11ирические наблюдения родословных с наследованием патоло-
ГИ'кч к их признаков известны давно. Например, в Талмуде отражено
Понимание сцепленного с Х-хромосомой наследования гемофилии.
14Ч'1К‘лине XVIII в. П. Мопертюи описал наследование доминантного
Й1»и ииіка полидактилии и правильно проанализировал расщепление
При така в потомстве. В начале XIX в. Дж. Адамс на основе эмпи-
||И‘К“ског о анализа родословных описал доминантный и рецессивный
Itmii-i наследования. Несколько врачей подробно разобрались в наслс-
[|ЙЛ1Л11ии гемофилии и цветовой слепоты. Эти и некоторые другие
їоііі.ігки анализа родословных можно рассматривать как предпо-
>І»иіки формирования генеалогического метода, которое закончилось
|| Міічале XX в., вскоре после рождения генетики как науки. С этого
Ірсмсни генеалогический метод широко использовали в генетике
Цвлоиска и медицинской генетике. Его дальнейшее усовершенство-
ійтіе шло по линии как составления родословных, так и (особенно)
||>й іработки методов статистического анализа данных. Метод находил
Оолсе широкое применение в клинической генетике и в генетике
Человека (изучение мутационного процесса, сцепления генов и др.).Суть генеалогического метода сводится к выявлению родослов¬
ных связей и прослеживанию признака или болезни среди близких
И ллльних прямых и непрямых родственников. Технически он
вкладывается из двух этапов; составления родословной и генеало¬
гического анализа.Составление родословнойСбор сведений о семье начинается с консультирующегося или
6 пробанда. Консультирующимся называется лицо, обратившееся к
прачу или первым попавшее в поле зрения исследователя. Пробанд —
1ТО больной или носитель изучаемого признака. Во многих случаях
консультирующийся и пробанд — один и тот же человек. Дети одной
114Клиническая генетикародительской пары называются сибсами (братья и сестры). Название
«сибс» происходит от английской аббревиатуры S1BS: Sisters—BrotherS.
Семьей в узком смысле называют родительскую пару и их детей, но
иногда и более широкий круг кровных родственников, хотя в послед¬
нем случае лучше использовать термин «род».Обычно родословную собирают по одному или по нескольким
признакам. Чисто технически ее нельзя составить по всем известнымО♦-н©о9101112131415Рис. 3.57. Символы, используемые при составлении родословных: 1— лицо
мужского пола; 2 — лицо женского пола; 3 — пол неизвестен; 4 — брак; 5 —
родственный брак; 6 — сибсы; 7 — монозиготные близнецы; 8 — дизиготные
близнецы; 9 — выкидыш; 10 — аборт; 11 — мертворожденный; 12 — бездетный
брак; 13 — гетерозиготная носительргица мутантного гена в Х-хромосоме;
14 — умершие; 15 — пробанл
fUann 3. Семиотика и клиническая диагностика ...115М|имн;1кам, да в этом и нет надобности. Врач всегда интересуется
цнк им го коїікретньїм заболеванием или признаком либо нескольки¬
ми -ишолнительными признаками, сопутствующими основному,It і;іі5исимости от цели исследования родословная может быть пол¬
ной или ограниченной. Желательно, конечно, стремиться к наиболее
Циііііому составлению родословных по восходящему, нисходящему
И ьпконым направлениям. Эта задача не такая легкая, как может
ІМж.пліься на первый взгляд. Чем больше поколений вовлекается
И рилословную, тем она обширнее. Это влечет за собой неточность
{Изучаемых сведений и, следовательно, неточность родословной вllrilOM.< осгавление родословной сопровождают краткой записью о каж-
Лпм со члене с точной характеристикой его родства с пробандом
<||1Ч1‘1гда родословной). В дальнейшем для наглядности (или при
Иуїмикации) родословную изображают графически. Для этого обыч¬
но пользуются стандартными символами (рис. 3.57). Перечислить все1)0(1 шачения невозможно. Если рассматриваемых признаков в родос-
^Мииюй много, то можно прибегать к буквенным или штриховым
РЦ І.ІИЧ ИЯМ внутри символов. Изображение родословной обязательно
іміііроіюждается описанием обозначений. Пример составления родос-
;имп1ых приведен на рис. 3.58.! вИис. 3.58, Пример родословной. Обозначения стандартные (см, рис. 3.57);
й Гюлыпле диабетом; б — больные нейрофиброматозом; в — лично обсле-
лоианныс. 1, I), III, IV— поколения
116Клиническая генетикаПоколения обозначают римскими цифрами сверху вниз, обычно
слева от родословной. Арабскими цифрами нумеруют потомство
одного поколения (весь ряд) последовательно слева направо. Братья
и сестры располагаются в родословной в порядке рождений. Таким
образом, каждый член родословной имеет свой шифр, нанример 11-2,
1П-8. Супругов членов родословной можно обозначать тем же номе-»8?,® 10 II 12Рис. 3.59. Большая родословная с концентрическим расположением поколе¬
ний; а — больные гемоглобинозом Е; б — больные талассемией; в — больные
гемоглобинозом Е и талассемией
Іййи 'Л. Семиотика и клиническая диагностика ...117МО со строчной буквой вслед за цифрой, если супруги кровно
1»>ианы с членами родословной. Если супруг не обследован на
♦Ійоичис рассматриваемого признака и его родословная не приводит-
жімаїельно его вообще не изображать. Внесение такого значка в
|И»1ин'||()иную не дает никакой информации, но затрудняет восприятие
#1Ч1и(ик)й части родословной. Все индивиды должны располагаться
^1|И)и> по поколениям в один горизонтальный ряд. «Подвешивание»
вимполов между рядами поколений — довольно грубая отоибка.1ч')|и родословная очень обширная, то разные поколения распола-
|*йии ис горизонтальными рядами, а концентрически (рис. 3.59).И иастояш,ее время все шире используются вспомогательные
рімір'гдуктивньїе технологии, в связи с этим предложены новые
VtiMiiojiN в записях родословных для таких семей (рис. 3.60).11|>и применении генеалогического метода в родословной важно
Шмочать обследованных на наличие признака (можно использоватьI 1 Донорство
спермыО [д6илио2. Донорство
яйцеклеток■Оу®О3. Суррогатное
материнство и
донорство
яйцеклеток^ИС.З .60, Символы вспомогательных репродуктивных технологий в РОДОС-
д«|»1ых: Д — доноры спермы и яйцеклеток; С — суррогатная мать (вынаши-
ййюіцая беременность)
118Клиническая генетикаІІсведения из объективного источника, например из ис тории болсзни)|
и необследованных, сведения о которых почерпнуты из ответов про-*;
банда или родственников, а также из анкет. Грубая ошибка — искус-1
ственное укорочение звеньев родословной из-за трудностей обследо^
вания лиц II и ІІІ степеней родства, особенно если не указывается,
у кого из членов родословной действительно не было родственников,
а у кого сведения не собраны.Получить сведения о родственниках непросто. Во-первых, не все
пациенты знают о болезнях родственников, во-вторых, они неред¬
ко скрывают семейные случаи из-за ложного стыда или, наоборот,
«открывают» их у родственников супруга, стараясь свалить на них
вину за болезнь ребенка.Для получения семейных сведений можно применять анкетирова¬
ние. При правильном перечне вопросов и доступности формулировок
для понимания членами семьи, не имеющими медицинского образо¬
вания, анкетирование дает достагочно полную информацию. Очень
важно провести личный осмотр и дополнительное обследование
родственников больного, если 'JTO необходимо. При сборе семейного
анамнеза желательно использовать и другие источники медицинской
и генеалогической информации (выписки из истории болезни, ломо¬
вые книги, церковные записи и т.д.).Подробное клинико-генеалогическое исследование проводится в
случае подозрения на наследственную болезнь при первичном кли-
ни’іеском осмотре. Обследование членов семьи должно быть подроб¬
ным, в отличие от первичных элементов семейного анализа, когорые
применяются при любом первичном осмотре больного.Одна из распространенных ошибок в применении генеалогиче¬
ского метода — ограничение анализа только опросом родственников
(или о родственниках). Даже подробного опроса, как правило, недо¬
статочно. Некоторые члены родословной часто нуждаются в пол¬
ном клиническом, параклиническом или лабораторно-генетическом
обследовании (цитогене гическом, биохимическом и т.п.), что требует
дополнительных расходов. План такого обследования необходимо
тщательно рассмотреть с генетической точки зрения в соответствии
с принципом «меньше нельзя, а больше не нужно».Помощь клинико-генеалогического метода в диагностике наслед¬
ственной патологии очевидна. Так, если в родословной обнаружена
наследственная болезнь и анализ показывает возможность ее переда¬
чи пробанду, то даже при стертой клинической симптоматике у про-
i(tM ‘Л Семиотика и клиническая диагностика ...119IMiiti (’lit) и сіало причиной подробного генеалогического обследо-
ИНИІ) можно установить диагноз данной наследственной болезни.11)М1 тематическом целенаправленном применении клинико-
|ИИ(Мо| ичсского метода данные лучше регистрировать в таблице
1Пп Из таблипы 3.3 видно, что у пробанда имеется отягощен-
if к. МО злокачественным новообразованиям мочевого пузыря.ІЛИІІ1І 3,3. Табличный способ регистрации генеалогических сведенийГенеалогическая карта № фамилия, имя, отчество пробандаЧленыродословнойпткшдІІМІ. материOicii матери‘Мин* отцаПол,возрастЖ., 4465Болезни*Возраст и причина
смерти63, рак мочевого пузыря73, инсульт28, погиб на фронте25, смерть в родахОігцотца73, рак мочевого пузыря4.'и()сыМІ1ІЧН1Ж., 68М.18, менингитМ.64, рак мочевого пузыряЖ.60, заболевание легкихлпкиой родословной отмечены такие болезни как: сахарный диабет (1), ише-
Мичсекая болезнь сердца (2), злокачественные новообразования (3), гипертони-
ИК’кии болезнь (4).
120Клиническая генетика]Генеалогический анализПервая задача при анализе родословной — установление наслед¬
ственного характера признака. Если в родословной встречается один]
и тот же признак (или болезнь) несколько раз, то можно думать о его
наследственной природе, но, прежде всего, надо исключить возмож-»]
ность фенокопии. Например, если патогенный фактор действовал на)
женщину во время всех беременностей, то могут родиться несколько]
детей с врожденными пороками. Другой пример; одни и те же про-
фессиональные вредности или внешние факторы могут вызывать]
сходные заболевания у членов одной семьи.Если исключается действие сходных внешних факторов (для раз- -
ных поколений оно исключается с большей вероятностью), то говорятІо наследственном характере болезни. С помощью генеалогического^
метода были открыты многие наследственные болезни.Вторая задача — установление типа наследования, если будет!
обнаружен наследственный характер признака (болезни). Для этого і
используют принципы генетического анализа и различные статисти¬
ческие методы обработки данных не из одной, а из многих родослов¬
ных, что является уже исследовательской задачей.Нетрудно понять, что в большинстве случаев простое отношение;
числа больных детей к числу здоровых даст неправильное представ-;
ление о типе наследования, потому что, например, при рецессивном,
заболевании в поле зрения врача не попадают сЄіМЬи-носитєли, в і
которых родились только здоровые дети.Теоретически можно представить полное выявление супружеских
пар, гетерозигот по патологическому гену, в том числе имеющих здо¬
ровых детей. Практически регистрация всегда начинается от больного
потомка. В таком случае невыявленные семьи составляют, напри¬
мер, при одном ребенке и доминантном типе наследования V2^ ^ при
рецессивном — Долю невыявленных гетерозиготных семей можно
определить для любого числа детей при различных типах наследова¬
ния. Следовательно, в расчеты отношения числа больных и здоровых
детей нужно вводить поправки па долю невыявленных семей.Освоение методов количественной оценки сегрегации (расщепле¬
ния) болезней или дискретных менделирующих признаков в потом¬
стве требует специальной подготовки. Здесь они излагаться не будут.Определение типа наследования в конкретной родословной —
всегда серьезная генетическая задача, хотя на первый взгляд она
может показаться довольно легкой. Для решения генетических задач
IKii л. Семиотика и клиническая диагностика ...121«мани 5V родословных врач должен иметь специальную подготов-
|у I I 'III необходим углубленный клинико-генеалогический анализ,
н|ц|ч обшей практики направляет семью в медико-генетическую
IMU У'и.тацию к врачу-генетику. Вместе с тем врачу общей практики
ІЙПО ma i ь основные критерии разных типов наследования, которые
|р111И>ЛЯТСЯ ниже.ІЛІ* III и с аутосомно-доминантным типом наследования
1|М1 гаком типе наследования для развития болезни достаточно
^|И1г(к‘ловать мутантный а;ілель от одного из родителей. Большинство
1»|цч11сй этого типа вызывают такие патологические состояния,
|01и()|,|с не нанося'1 серьезного ущерба з^|,оровью и в большинсі не слу-
Ійі'и НС влияют на способность иметь потомство. Родословные таких
|Ип. <»собенно описанные в прошлом, когда в браках было много
ЧГІІ. дали возможность установить типичные признаки аутосомно-
|Шинамтных форм наследственной патологии (рис. 3.61).Ііолезнь встречается в каждом поколении родословной, что
называют передачей болезни по вертикали.Соотношение больных и здоровых приближается к 1:1.У нормальных детей больных родителей рождаются нормаль¬
ные дети.Число больных мальчиков и девочек равное.Вольные мужчины и женщины с равной вероятностью переда-
Ю'1 болезнь своим дочерям и сыновьям.£D16Д6І ah iotb ifoi с5оЬ1 2 3^4 5 6 7 8 9 10 11 12 13 14 15 16f НС. 3.61. Родословная с аутосомно-доминантным типом наследования болез¬
ни (синдром Марфана)
122Клиническая гєнетин— Чем больше болезнь отражается на репродукции, тем больше
доля спорадических случаев (новые мутации).— Гомозиготы могут рождаться от двух больных родителей.!
Болезнь у них протекает обычно тяжелее, чем у гетерозиготі
(так называемое полудоминантное наследование). Доминаитно]
наследуемые состояния имеют полиморфные клинические!
проявления не только в разных семьях, но и у членов одной!
семьи. Например, при ыейрофиброматозе у одних больных в|
семье могут быть множественные нейрофибромьт, а у других —1
лишь единичные кожные проявления. Особенность ряда доми¬
нантных болезней — высокая вариабельность сроков начала)
болезни даже в пределах одной семьи. Наглядным примером}
служит хорея Гентингтона. Распределение больных по возра-|
сту начала болезни описывается нормальным распределением]
со средним значением 38—40 лет. Объясняется это различным]
числом унаследованных триплетов (см, ниже).При тяжелых заболеваниях, когда у больных снижена возмож-1
ность иметь потомство (сниженная фертильность), родословные не]
типичны. Если мутация возникает впервые в зародышевых клетках,]
то родословная показывает спорадический случай,Наиболее часто встречаются следующие генные болезни с|
аутосомно-доминантным типом наследования; нейрофиброматоз!I типа (болезнь Реклингхаузена), синдромы Марфана, Элерса—Данло,[
ахондроплазия, несовершенный остеогенез, миотоническая дистро-1
фия, хорея Гентингтона.Болезни с аутосомно-рецессивным типом наследованияЗаболевания с данным типом наследования проявляются толь-]
ко у гомозигот. Гетерозиготы фенотипически (клинически) не|
отличаются от здоровых людей с двумя нормальными аллелями^
(рис. 3.62).При редких аутосомно-рецессивных заболеваниях обычно отме¬
чают следуюш;ее.— Родители обычно клинически здоровы.~ Чем больше детей в семье, тем больше вероятность иметь более іодного больного ребенка.— Чем реже встречается мутантный ген в популяции, тем чаще роди¬
тели больного ребенка являются кровными родственниками,— Если больны оба супруга, то все дети будут больными.
|Ц(1 :i Семиотика и клиническая диагностика123[>'-Л2, Родословкан с аутосомно-рецессивным типом наследования болез-
|Н (< ип дром Тея—Сакса — СМ2-ганглиозидоз)В браке больного со здоровым рождаются нормальные дети
(ссли адороізьій не гетерозиготен).ІІ браке больного с носителем мутантного аллеля рождается
50% больных детей, что имитирует доминантный тип наследо-
иа 11 и я (псевдодом и н и рова н ие).Оба пола поражаются одинаково.Ьрпки, в которых оба родителя гетерозиготны, встречаются наи-
'U4' часто. Сегрегация потомства соотвстсгвусг мснделевскому соот-
[ИШсмию 1 (здоровый): 2 (гетерозиготы): 1 (больной). Риск появления11.11 ого ребенка в таком браке составляет 25%. Малодетпость совре-
Івііііі.іхссмсй затрудняет установление рецессивного типа наследования
Ж ЛІИ. Однако если родители ребенка являются родственниками, то
Met' І СЯ высокая вероятность аутосомно-рсцсссивної о заболевания.Mil рис. 3.63 показаны примеры кровнородственных браков.
Пропорция общих генов и коэффициент инбридинга — очень
ІСОКИС. при оценке гснстичсской ситуации в таких семьях надо
римимать во внимание высокую вероятность встречи редких алле-
й и, следовательно, гомозиготность, т.е. рецессивное заболевание.
124Клиническая генетикїГ дПримеры кровнородственных браковПоказателиА.ДвоюродныеБ.ДваждыдвоюродныеВ.Полу-двоюродныеГ.Двоюродныедядя-племянницаД-троюродныеСтепеньродства32445Пропорция
общих генов1/81/41/161/161/32Коэффициент
инбридинга
ребенка(F)1/161/81/321/321/64Рис. 3.63. Примеры кровнородственных бракопБраки двух гомозиготных людей очень редки. Естественно, что все
дети в этих семьях будут гомозиготами, а потому больными. В тех
семьях, в которых у больных родителей (например, альбиносов) рож-']
дались здоровые дети, такое несоответствие объясняется мутациями
в разных генах. Такие дети являются двойными гетерозиготами.Браки гетерозигот (здоровых) с гомозиготами (больными) в основ¬
ном кровнородственные. Соотношение числа больных и здоровых
равно 1:1.Наиболее типичными болезнями с аутосомно-рецессивным
типом наследования являются муковисцидоз, фенилкетону-
рия, галактоземия, гепатолентикулярная дегенерация (болезнь
Вильсона-Коновалова), адреногенитальный синдром, мукополи-
сахаридозы.
їй .'1 Оомиотика и клиническая диагностика ...125^11* мій с Х-сцепленным доминантным типом наследованияOiоік'иііости наследования этих болезней обусловлены тем, что у
інтмії ц\чс Х-хромосомы, а у мужчин одна. Унаследовав от одного из
^іми-'к-й патологический аллель, женщина является гетерозиготой,
іужчимії ~ гемизиготой. Основные характеристики родословных
ІИ »и)М ІИПЄ наследования следующие (рис. 3.64).Поражаются и мужчины, и женщины, но больных женщин в
ра ш больше, чем мужчин,
вольные женщины передают патологический аллель в среднем
М)% сыновей и 50% дочерей.Ьольной мужчина передает патологический аллель всем доче¬
рям и не передает сыновьям, поскольку они получают от отца
Y-хромосому.И среднем женщины (если они гетерозиготны) болеют менее
іяжело, чем мужчины (если они гемизиготны). Болезнь более
нприабельна по клиническим проявлениям у гетерозиготных
женщин.Ии Х-сцепленному доминантному типу наследуется витамин
«|1с IMLтентный рахит (наследственная гипофосфатемия). Если
ІІМИІ. тяжелая и летальна у гемизигот (синдром недержания пиг-
ІПІ, ротолицепальцевой синдром, синдром Гольтца—Горлина), то
Міїльчики погибают. Больными бывают только девочки./10 11 12 139. .1.64. Родословная с Х-сцепленным доминантным типом наследования
1C )ми (витамин D-резистентный рахит)
126Клиническая генетик!Болезни с Х-сцепленным рецессивным типом наследованияПри редко встречающихся болезнях с этим типом наследования!
женщины практически всегда гетерозиготны, т.е, они фенотипи-^1
чески нормальны (здоровы) и являются носителями. Больным!
бывают только мужчины. Характеристики болезней этого типа разч
личаются в зависимости от рспродукгивноіо статуса. Если ренро*
дуктивная функция у больных нарушена (мышечная дистрофия)
Дюшенна—Беккера), то родословные имеют следующие характери-j
стики (рис. 3.65).— Больные — только мальчики.— Около 2/з случаев передается от матерей-носительниц, '/3 возни¬
кает в результате новых мутаций в Х-хромосоме матери.— В унаследованных случаях у больных мальчиков могут быть]
больные братья и дяди по матери.— Новые мутации являются спорадическими случаями.— Сестры больных братьев при унаследованных случаях имеют|
50% всроя1 ность быть носителями иаго.'югического аллеля.ОРис. 3.65. Родословная с Х-сцепленным рецессивным типом наследования
болезни (патология нарушает репродуктивную функцию — миодистрофия
Дюшенна)
Ійіі Семиотика и клиническая диагностика ...12722 3 4 5 6 79 10 11 12 13 14 15 16 17Ii', ,^.66. Родословная с Х-сцепленньтм рецессивным типом наследования
}(И' ІИИ (репродукция не нарушена — гемофилия)Сестры-носительницы передают ген 50% сыновей (они больны)
и 50% дочерей (они носительницы).Здоровые мужчины не передают болезни.1'сли репродукция при конкретной болезни не нарушена (гемо¬
филии, недостаточность Г6ФДГ), то наследование будет следующим
т. 3.66).Доля унаследованных случаев — более ^/3.Больные мужчины передают патологический ал лел ь всем своим
дочерям и никому из сыновей.- Все фенотипически нормальные дочери больных мужчин явля¬
ются носительницами.в браке женщины-носительницы с больным мужчиной среди
дочерей 50% — больные, 50% — носительницы, среди сыновей
50% — больные, 50% — здоровые.Иногда гетерозиготные женщины могут быть больными в связи
со случайной гетерохроматинизацией хромосомы с нормаль¬
ным аллелем во всех или почти во всех клетках.К Х-сцепленным рецессивным болезням относятся гемофилия,
Шііісмная дистрофия Дюшенна-Беккера, синдром Хантера (муко-
лисахаридоз ІЇ типа), синдром Леша—Найхана.
128Клиническая генетик■тООтШто3hOlhOiiiQ1 2 З 4^5 6« □ боо12 зРис. 3.67. Родословная с Y-сцепленным типом наследования признака (ово¬
лосение ушной раковины)Y-сцепленный тип наследованияДолго полагали, что Y-хромосома содержит только іетерохроматинс
вые участки (без генов). Новейшие исследования позволили обнаружит
и локализовать в Y-хромосоме ряд генов: детерминирующий развитие
семенников, отвечающий за сперматогенез (фактор азооспермии), кон*'
тролируюший интенсивность роста тела, конечностей и зубов, определяй
юощй оволосение ушной раковины. На примере этого признака можнс
видеть характерные черты У-сцепленного типа передачи (рис. 3.67).Признак передается всем мальчикам. Естественно, что патологиче¬
ские мутации, затрагиваюш[ие формирование яичек или сперматоге-|
нез, наследоваться не могут, потому что такие индивиды стерильны.]Митохондриальная наследственностьМитохондрии передаются с цитоплазмой яйцеклеток. Спермии;|
не имеют митохондрий, поскольку цитоплазма элиминируется прі
созревании мужских половых клеток, в каждой яйцеклетке содер-|
жится около 25 ООО митохондрий. Каждая митохондрия имеет коль-|
цевую хромосому. Описаны мутации различных генов митохондрий,}
Генные мутации в мтДНК обнаружены при атрофии зрительного]
ittM Семиотика и клиническая диагностика ...129Оті1 2□т£~5 оДtp піі3 4 5 6і і • і7 86оЬ 6 і і 6 бо і*10 11 12 13 142 З 4 5 6 7 8 9 10 11 12іг. Ї.68. Родословная, иллюстрирующая передачу нейтрального признака
|И?|ІОМ июхондрии (фрагмент ДНК)Ifipiui Лебера, митохондриальных миопатиях, доброкачественной
Иіу.чоли (оикоцитоме), прогрессирующих офтальмоплегиях.Митохондриальная наследственность имеет следующие признаки3.68).Болезнь передается только от матери.Гюльны и девочки, и мальчики.Больные отцы не передают болезнь ни дочерям, ни сыновьям.СИНДРОМОЛОГИЧЕСКИЙ подход
к ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙИ наследственной патологии не существует патогномон и чных
іри иіяков. Один и тот же признак встречается при нескольких или
ІДИЖс при многих формах. Например, деформация грудной клетки в
||ИДс воронки или киля бывает не менее чем при 30 наследственных
Шолсзнях, искривление позвоночника — более чем при 50 наслед-
^Ітисмньїх синдромах. Аномалии почек известны при 30 синдромах.
Іриііоплазия или дисплазия ногтей может наблюдаться при 25 раз-
Іііичньїх наследственных болезнях. Люди с этими синдромами часто
бынают пациентами отделений сердечно-сосудистой хирургии.Гели внимательно осматривать больного, то можно выявить при-
>Лнки, существенно облегчающие дифференциальную диагностику.
ї<нііример, у больного с врожденным пороком сердца нужно тща¬
тельно осмотреть руки: укорочение I пальца кисти или 3 фаланги
130Клиническая генети)іна нем вместо 2 сразу наводит на мысль о доминантно наследуем
мом синдроме Холта—Орама (или, как его еше называют, синдроіі
«рука-сердце»).Важное место в диагностике наследственных синдромов илі
болезней занимает анализ строения лицевой области. Так, резкс
выступающие надбровные дуги могут быть признаком синдрои
фронтометафизарной дисплазии (X-сцепленная форма остеодиспла¬
зии Мелника—Нидлза), а запавшая переносица — мукополисахари-
доза или ахондроплазии. Гипертелоризм позволяет заподозрить одщ
из 50-60 наследственных синдромов.Искривление нижних конечностей — результат не только рахитаJ
как полагали ранее. Оно может быть следствием нарушенного обмен?
в костях при 25 различных наследственных болезнях.Большое диагностическое значение имеет осмотр зубов, особеннс
у .молодых людей. Более чем при 20 синдромах наблюдаются измене¬
ния зубов: неправильная форма, раннее выпадение, множественный
кариес, сверхкомплектность или срастание, врожденное отсутствие
резца или клыка. Изменения зубов нередко отмечаются при муко-|
полисахаридозах, синдроме Элерса—Данло, пахионихии и многих]
других наследственных болезнях.Умственная отсталость сопровождает многочисленные моноген-]
ные болезни (более 700 форм) и большинство хромосомных.Можно перечислить около 200 внешних симптомов и признаков^
которые выявляют при наследственных болезнях без применения]
специальных дополнительных методов обследования. Однако чтобц]
выявить эти признаки, их нужно прицельно искать.Большинство ішследственньїх болезней встречаются редко;
(1:100 ООО и реже). Однако среди больных какого-либо профиля веро-^
ятность обнаружения конкретного вида наследственной патология^
существенно увеличивается. Так, больных с синдромом Марфана'
и гомоцистинурией можно встретить в глазных (высокая миопия)
и хирургических (деформация грудной клетки) клиниках. Больные
низкого роста чаще наблюдаются у эндокринолога; к ортопедам
обращаются пациенты с наследственными деформациями костей и
суставов и т.д.Наследственные формы часто встречаются в практике офтальмо¬
логов. Атрофия зрительных нервов наблюдается, по крайней мере,
при 15, катаракты и помутнения хрусталика — более чем при 30
наследственных болезнях. Птоз, косоглазие, нистагм, помутнение
Мй 1 Г.(^миотика и клиническая диагностика131N•1111IIм. отслойка сетчатки и г.л. — симптомы многих наслсдствен-
)щ (и)1кчией, распознавание которых улучшается, если врач знает
|МЛ|1пмилогию наследственных и врожденных болезней.|f Хиш наследственных болезней очень много, их представительство
||111|н‘)к“лснных клиниках ограничено небольшим числом форм, как
ІИІІМП, с более легким течением. Освоение этих форм не прслістав-II ірулііостей для врача любой специальности; нетрудно изучить и
|М111ом1>1, на основании которых можно заподозрить синдром.ПАРАКЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
В КЛИНИЧЕСКОЙ ГЕНЕТИКЕ![иптления наследственных болезней весьма разнообразны по
риилсниости и глубине изменений многих органов и систем, что
lyi iiniL'ieHO большим числом и тяжестью нозологических форм.1Н с этим параклинические исследования занимают существен-
»г моею в диагностике наследственных болезней.Уже в самом начале XX в., когда генетика человека еще только
лучила основы для своего развития, английский врач А. Гаррод
>им^'11ил биохимический анализ мочи для диагностики наслед-
♦гимой болезни обмена вешеств — алкаптонурии. В 30-х годахи. норвежский врач И.А. Феллинг открыл метод диагностики
Иіилкстонурии на основе реакции мочи с хлоридом железа (если в
'U' есть фенилпировиноградная кислота, появляется сине-зеленая
фиска). Однако широкое (трименение параклинических методов
|и /тлгностики наследственных болезней началось с периода инген-
іикого развития клинической генетики (50 годы XX в.).И настоящее время для диагностики наследсгвенных болезней и
^цсики состояния больного используют клинико-биохимические,
>М1пч)Логические, иммунологические, эндокринологические, элек-
іофизиологические, рентгенорадиологические методы. Более
^І'Лубленньїе варианты составили отдельную группу лабораторно-
Яістических методов (см, главу 9).'Значение параклинических методов для клинической гене-
ІКИ трудно переоценить, но подробно описать их невозможно.
Іриисдем лишь отдельные примеры их использования в диагно-
Іітических целях. Клинико-биохимические исследования ПРОВО¬
ЖАЙ! при муковисцидозе, семейной гипсрхолестеринемии, фенил-
132Клиническая генети»кетонурии, болезни Вильсона-Коновалова. Гематологическї
исследования проводят для подтверждения гемоглобинопатиі
наследственного гемохроматоза. Эндокринологические исследовї
ния назначают при врожденном гипотиреозе, адреногенитальної
синдроме, нанизме. Иммунологические исследования необходим^
при первичных иммунодефицитах, атаксии-телеангиэктазии (сиі
дром Луи-Бар). Электрофизиологичсские исследования проводят пр|
нервно-мышечных заболеваниях, многих наследственных болезні
нервной системы. УЗИ обязательно назначают при врождснні
пороках внутренних органов, аномалиях половой дифференцировкі
Рентгенорадиологичсские исследования необходимы при диагностик
ке хондродистрофии, нейрофиброматоза.КОМПЬЮТЕРНЫЕ ПРОГРАММЫ ДИАГНОСТИКИ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ«Охота» за генами и практическая потребность в диагності
ке многочисленных форм наследственных болезней и врожденн!
пороков развития невозможны без сравнительного анализа данны!
литературы. В связи с этим на протяжении последних 20 лет сталі
создаваться компьютерные информационные базы данных и диагнс
стические программы. Их назначение — ускорить и объективизирс
вать выбор из множества генетически разнородных, но клиничесі
сходных синдромов и болезней.Генетическая консультация и помощь при наследственных болезі
нях возможны при точном диагнозе. Поскольку тенетические нар5
шения редки, большинство врачей н даже медицинских генетикої
имеют собственный опыт только по нескольким случаям данноі
болезни. Следовательно, крайне важно знакомство с описанными
литературе случаями. Из-за генетической гетерогенности надо тонне
определить нозологическую форму, чтобы родители знали величинуЦ
риска при повторных деторождениях. Базы данных особенно полез¬
ны для дифференциальной диагностики.Каталоги необходимы для практической работы клинических]
генетиков. Они обеспечивают информацию или помогают ориенти*-!
роваться в информации по диагностике, лечению и генетическому)
консультированию. Поиски по отдельным словам или комбинациям!
слов дают перечень форм, в которых появляется слово или комбина-1
:і Семиотика и клиническая диагностика ...133|М ГІІОИ. Это хорошее начало в поисках диагноза неясной болезни
Ш М|И1 пильного лечения и консультирования конкретной семьи.Ии рисунке 3.69 представлен принцип диагностики с использова-
компьютерных программ. Симптомы, выявленные врачом, вво-
и компьютер. На их основе осуществляется компьютерный поиск
|ИПо111.‘о вероятных диагнозов. После этого врач может обратиться за
ірнпкой в базу данных по выбранным диагнозам и получить опи-
ІИГ синдрома (или болезни) и даже фотографии больных. Таким
ш »ом, врач принимает решение о диагнозе и выбирает способ его
іифіїкииии, если в этом есть необходимость, в случае, представ-
Миюм на рис. 3.69, была нужна цитогенетическая верификация.
ІШ'і' подробная информация по интерактивны.м базам данных
1г;к1 пилена на компакт-диске в разделе «Компьютерные базы дан-
)1 1И постановки диагноза наследственного заболевания».ПациентСимптомы||іуі мая голова
Иижий затылок
KW1H) лицо
tfiyi мое лицо
||Н)КИЙ лобпереносица
кшан переносица
Н(1ипоидный разрез глазИКВНТ1)тепоризм
[Кореикий нос
[^корнутые ноздри
[^нный фильтр
I Г^кий фильтрКомпьюгерный поискПерчень наиболее вероятных
синдромов при выбранных
симптомах:9рТрисомия по хр. 21 5Стинклера синдром 45р 4Апера синдром 4Триметадионовый синдром 3Сетре-Чотс0на синдром 3Верификацияmono 9 (p21-pter)
46, XX, del 9 (р21)I!. 3.69. Компьютерная диагностика наследственных оолезнеиКЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯИрожденные морфогенетические варианты
Гамето-, бласто-, эмбрио- и фетопатии
Генеалогический анализ
Генетические основы морфогенеза
134Клиническая генетикЗначение антропометрии в диагностике
Классификация врожденных пороков развития
Клиническая диагностика наследственных болезней
Критические периоды развития
Области применения генеалогического \тетода
Особенности клиники наследственных болезней
Параклинические методы в диагностике
Плейотропия (первичная и вторичная)
Последовательности и ассоциации
Пренатальная гипоплазия
Пробанд, сибсы, семья, род
Семиотика наследственных болезней
Синдромологический подход
Тератогенез, тератогены
Тератогенный терминациоиный период
Термин «синдром»Типы наследования (критерии)ФенокопииХарактеристика генеалогического метода
Эмбриональный дисморфогенез
Этиология врожденных пороков развитияРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРААкуленко Л.В., Богомазов Е.А., Захарова О.М. и др. Медицинская И;
клиническая генетика для стоматологов. — М.: ГЭОТАР-Медиа^
2008. - 398 с.Баранов B.C., Кузнецова ТВ, Цитогенетика эмбрионального разви¬
тия человека: научно-практические аспекты. — СПб.: Изд-во Н-Л,
2007. - 640 с.; ил.Беляков Ю.Л. Наследственные болезни и синдромы в стоматологи¬
ческой практике. — М.: Мединина, 2008. — 238 с,Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК-
диагностика и медико-генетическое консультирование в невроло¬
гии. - М.: МИА, 2002. - 591 с.Кадуршш Т.И., Горбунова В.Н. Дисплазия соединительной ткани.
Руководство для врачей. — СПб.: Элби-СПб, 2009. — 704 с.Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-
генетическое консультирование: атлас-справочник. — 3-є изд., доп.
)|1нп 3. Семиотика и клиническая диагностика ...135Ш'рораб. — М.: Т-во научных изданий КМК; Авторская академия,
«)П7 448 с.JhrooK Г.И. Тератология человека. — М.: Медицина, 1979. — 440 с.
Мо(Уі)овцев В.Н., Мордовцева В.В., Мордовцева В.В. Наследственные
О'имии и пороки развития кожи. Клиника. Морфология. Лечение:
И'ПК-. — М.: Наука, 2004. — 174 с.Наследственные болезни нервной системы: руководство / под ред.
t) I Ікльтишева, ПЛ. Темина, — М.: Медицина, 1998. — 496 с.
ГлаваГЕННЫЕ БОЛЕЗНІэтиологияГенные болезни — разнородная по клиническим проявлениям груп*]
па заболеваний, обусловленных генными мутациями. Основой для обт
единения их в одну іруппу служат этиологическая генетическая чарак*
теристика и соответственно закономерности наследования в семьях
популяциях. Коль скоро мутации в индивидуальных генах являютс
этиологическим фактором генных болезней, то закономерности И)
наследования соответствуют менделевским правилам расщепления
потомстве, т.е. формальная генетика генных наследственных болезнеі
ничем не отличается от «поведения» в семьях любых менделирующи}
признаков. «Поведение*» некоторых патологических генов может откло-|
няться от Менделе веко-моргановских правил в связи с фенотипиче¬
скими эффектами (летальность, стерильность). Необходимо, однако,]
сразу сделать пояснения в отношении содержания понятий «генньм
мутации» и «менделирующая наследственность» у человека.Во-первых, согласно многочисленным исследованиям разных!
наследственных болезней и генома человека в целом, можно гово-|
рить о многообразии видов мутаций в одном и том же гене, которс
является причиной наследственных болезней, у человека описаны]
все типы генных мутаций, обусловливающие наследственные болез-і
ни: миссснс, нонсенс, сдвиг рамки считывания, дслеции, вставки!
(инсерции), нарушения сплайсинга, увеличение числа (экспансия)!
тринуклеотидных повторов. Любой из этих видов мутаций может*]
вести к наследственным болезням. Даже одна и та же генная болезнь!
может быть обусловлена разными мутациями одного и того же гена.!
Например, в гене муковисцидоза описано около 300 вызывающих]
болезнь мутаций (всего их более 1500) следующих типов: делении,!
миссенс, нонсенс, сдвиг рамки считывания, нарушения сплайсинга.
Для гена фенилкетонурии известно более 30 патолої ических мутаций
(миссенс, нонсенс, делеции, нарушения сплайсинга).Во-вторых, современная генетика, принимая в полной мере мен¬
делизм, делает поправки в определенных случаях. К ним относятся
іип ^ Tt^HHbie болезни137иишосіь понятий о доминантности и рецессивности, неодно-
11, проявления аллеля, унаследованного от отца или матери
<инг), сложное взаимодействие генов, гонадный мозаииизм
'1 (I Ьолее того, установлено, что мутации в разных частях одного
кМо исдут к различным болезням. Например, мутации в разных
'MIS КІ’.Т-онкогена ведут к 4 клинически разным наследственным
ijjr тим; двум формам полиэндокрин ного аденоматоза (ZA) и (ZB),
ЦИ1*()1к»й медуллярной тиреоидной карциноме, семейной болезни
)|1111ирукга.Мм апии, вызывающие наследственные болезни, могут затрагивать
іурньге, транспортные и эмбриональные белки, ферменты.
Ьгмковые классы, ассоциированные с моногенными болезнями,
«і'нися фактически во всех составных элементах клетки (табл. 4.1).ІЙЛИЦИ 4.1, Примеры классов белков, ассоциированных с моногенными
болезнямиЧасти клеток,
функцииПродуцируемый белокПримеры болезнейІЛІ»» f’pm U'к р и 1 шион н ы й
ік іор развитияРАХ6АниридияBRCAI, BRCA2Рак молочной железы[вииміїая интеграцияБелок репарации
неправильно спарен¬
ных нитей ДНКНаследственный нсно-
липозпый рак толстой
кишки«уляция трансля-
1И РНКFMRP (подавляет
трансляцию путем свя¬
зывания РНК)Синдром ломкой
Х-хромосомыцшмагин-ассоии-
лшанные белкиМсСР2 (репрессия
транскриииии)Синдром Ретта[СУ11р‘.:ссоры опухолейRb-белокРетинобластомаІМКОІ еныBCR-Abl онкогенХроническая миелоид-
ная лейкемияиоилазма^Мс гаПолические фер-
,МсптыФенилаланингидро-ксилазаADAФенил кетонурияТяжелый комбиниро¬
ванный иммунодефицит
138Клиническая генетикПродолжение таблицы 4.Части клеток,
функцииПродуцируемый белокПримеры болезней IЦитоскелетДистрофииМиодистрофия J
Дюшенна 1Органеллы jМитохондрии 1Окислительное фос-
форилированиеNDl-белок транспорт¬
ной цепи электроновНаследственная зритель-!
ная нейропатия ЛебераТрансляция мито¬
хондриальных белковtRNAleuМ итохондриал ьная
энцефалопатия с лакто¬
ацидозом и инфарктопо¬
добными эпизодами12S RNAСенсо-нс врал ьная глу¬
хота 1Лизосомы ]Лизосомальные фер¬
ментыГексозами нидаза АБолезнь Тея-Сакса JНедостаточ ность
tt-L-идуронидазыСиндром Гурлер jКлеточная оболочкаРецепторы гормоновРецептор андрогеновНечувствительность к
андрогенамРецепторы факторов
ростаFGFR3-peuenT0pАхондроплазияМетаболическиерецепторыLDL-рецепторГ иперхолестеринемияТранспорт ионовCFTRМуковисцидозПрезентация анти¬
геновНЬАлокус DQPiСахарный диабет
1-го типаВнеклеточные белкиТранспортр-АдреноглобинСерповидно-клеточнаяанемияР-ТалассемияМорфогенезSonic hedgehogГолопрозэнцефалияИнгибирование про¬
теазагАнтитрипсинЭмфизема, заболевания
печениГемостазФактор VIIIГемофилия А
ift л Генные болезни139Чисги клеток,
функцииНик 'К' точныйpIlKt'Окончание таблицы 4.1|(к іілік'иис, 01’вет на
(ИфскциюПродуцируемый белокИнсулинКоллаген 1-го типаФактор комплемента НПримеры болезнейРедкие формы сахарного
диабета 2-го типаНесопершенный остео¬
генезВозрастная макулярная
дегенерацияімі'пниие. CFTR — кистофиброзный трансмембранный регулятор. HLA —
цот’ческий лейкоцитарный антиген (Human Leucocyte Antigen). RNA —
іПі III уклеи новая кислота.Существует несколько уровней регуляции синтеза белков: пре-
цк'крипционный, транскрипционный, трансляционный. Можно
пол ожить, что на всех этих уровнях, обусловленных соот-
Нтиующими ферментативными реакциями, могут проявляться
Іулслсгвенньїе аномалии. Если принять, что у человека примерноООО генов и каждый ген может мутировать и контролировать
ІИК’ч белка с другим строением, а многим генам свойственно
АЄ явление альтернативного сплайсинга, то, казалось бы, должноIII. ие меньшее число наследственных болезней. Более того, по
«ременным данным, в каждом гене может возникать до несколь-
tx сотен вариантов мутаций (разные типы в различных участках
ІНП). 11а самом деле более чем для 50% белков изменения генети-
Ккой природы (первичная структура) приводят к гибели клетки,
муіиция не реализуется в наследственную болезнь. Такие белки
цыиаются мономорфными. Они обеспечивают основные функции
1С1КИ, консервативно сохраняя стабильность видовой организации
рпй клетки.Так или иначе, число генных болезней действительно велико.
Івііделируюших болезней, согласно справочнику ОМ1М, на 2008 г.
ірсі истрировано около 4000, из них 84% (более 3300) обусловле-
мутациями в 1990 генах. Оставшиеся 16% болезней (0М1М)
Гчстливо менделирующие, но мутантные гены их еще неизвестны,
личество болезней с известными генетическими причинами и
іичество генов, мутации в которых могут вызывать болезни, не
І^ниадают. Обусловлено это тем, что разные мутации в одном и
140Клиническая генетиктом же гене могут вызывать различные болезни, а мутации в рЕ
ных генах могут вызывать неразличимые болезни. Непосредствен!
с развитием генетических болезней связано около 8% генов. Этс
безусловно, пока существенная недооценка. Впереди еще больші
работа по «инвентаризации» наследственных болезней, если их рао|
сматривать не только с клинической (фенотипической), но и с геш
тической точек зрения.При рассмотрении генных болезней как менделируюших призш
ков организма речь идет о так называемых полных формах, т.е. фоі
мах, обусловленных га мет и чески ми (в зародышевых клетках) мута
циями. Это могут быть новые или унаследованные от предыдущий
поколений мутации. Следовательно, в этих случаях патологическі
гены присутствуют во всех клетках организма. Однако теоретическі
можно представить возможность появления и мозаичных, а не толы
полных форм, подобно хромосомным болезням. Любые мутации,
том числе и генные, могут возникать на ранних стадиях дробленні
зиготы в одной из клеток, и тогда индивид будет мозаичен по данном^
гену. В одних клетках у него будет функционировать нормальны!
аллель, в других — мутантный или паїологический. Если эта мутаї
ция доминантная, то она проявится в соответствующих клетках
очевидно, приведет к развитию менее тяжелой формы болезни. Есл1
возникшая мутация в одной из клеток на ранних стадиях развитш
зародыша рецессивная, то ее эффект проявится только у гетерози-*}
готы. Вероятность появления двух рецессивных мутаций в одном Ш
том же локусе гомологичных хромосом в одной соматической клетке}
чрезвычайно мала.Проблема мозаичных форм генных болезней и в генетическом, и i|
клиническом плане исследована недостаточно. Частота возникнове«|
ния мозаичных форм не может быть высокой, поэтому выявлять HJ
трудно. Современные молекулярно-генетические методы П03В0ЛЯІ
диагностировать мозаицизм на клеточном или тканевом уровне.!
В одной и той же ткани обнаруживают клетки, несущие разные*
генотипы по изучаемой патологической мутации. Соматические]
мутации, появляющиеся на ранних стадиях развития организма,
дают больший эффект, чем мутации на поздних. В последние годы,
соматический мозаицизм был доказан при 30 генных болезнях,
среди которых нейрофиброматоз I типа, миотоническая дистрофия,
миодистрофия Дюшенна, гемофилия А и В, синдром Альпорта, син¬
дром Марфана, синдром андрогенной нечувствительности, тубероз-
la A Генные болезни141ІИ I киероз и лр. Соматический мозаицизм был обнаружен также
|И і'юкамсственньїх новообразованиях (колоректальный рак и рак
ли мюльной железы).с' мочаичными формами генных болезней не следует путать моза-
1И юиад. Мозаицизм гонад — частный случай органного мозаи-
возникающего на более поздних стадиях эмбрионального
Мг»111ия в процессе органогенеза. Мозаицизм гонад у клинически
(1|»1|»ич1 индивида может обусловить несколько случаев рождения
'(\ I- полной формой доминантной наследственной болезни.ІІІІ рис. 4.1 приведена родословная здоровых родителей (француз-
іч'мья), у которых трое из четверых детей больны ахондропла-ІГІІЛчомдроплазия — аутосомно-доминантное заболевание с пол-
И1 іісіістрантностью гена. Клиническая и рентгенологическая диа-
101'I и ка этой болезни (в частности, в упомянутой выше семье) не
11.1 наст сомнений. Объяснить семейные случаи можно гонадным
ІЯІИЦИЗМОМ у отца, поскольку больные дети родились в двух его
Ійках. Возможно еще одно объяснение подобных случаев: болезнь
пмикла в результате премутации в одном из аллелей этого гена уII1C. 4.1. Родословная семьи с 3-мя случаями ахондроплазии в одном ноко-
|1(ци 01' двух браков: I: 1 — рост 163 см; 2 ~ рост 166 см; 3 — рост 164 см;
ІИИИЧССКИХ и радиологических признаков ахондроплазии не имели; II: I —
дилась от {теродствениого брака (мать 17 лет, отец 30 лсч). Ахоидроплазия
Июдозрена при рождении, позже подтверждена; 2 — здоровый мальчик;больная ахондроплазией. Диагноз установлен после рождения; 4 — во
Юром браке отиа диагноз ахондроплазии у его ребенка был установлен пну-
|fJ>My rpo6H0 на 7-м месянс [сначала с помощью ультразвукового исследования
НУ ІИ), затем рентгенографически!. При рождении диагноз подтвержден
142Клиническая генети»родителя, которая реализуется в мутацию при прохождении чсре
мейоз. Однако в гене ахондроплазии премутантных состояний пої
не обнаружено.Современные молекулярно-генетические исследования показал!
что родительский \гозаицизм (в том числе гонадный) ответствен
менее чем за 5—15% случаев доминантных и Х~ецепленных рецессив^
ных болезней. Мозаицизм гонад у здоровых родителей убедительщ
доказан в случаях рождения детей (по соответствующим генам)
несовершенным остеогенезом, синдромом Элерса-Данло, гемофи^і
лией (факторы VIII и IX).В связи с многообразием мутаций в одном и том же гене возни^І
кает вопрос об этиологической зависимости клинической картині
болезни от характера мутаций. Ответ на этот вопрос неоднозлачеї
и пока не всегда ясен. Определенно можно сказать, что в болы
шинстве случаев такой зависимости нет, хотя в некоторых случаяз
она присутствует. Объяснение этому можно найти в первичныз
механизмах развития генных болезней, т.е. в первичных эффекта?
мутантных аллелей. С клинико-генетической точки зрения эт1
аллели называют патологическими для отличия от друї их мутант*]
ных состояний этого же гена, которые ведут к биологическим!
межиндивидуальным вариациям без патологических проявлений]
признака.Первичные эффекты мутантных аллелей могут проявляться в 4-х І
вариантах: отсутствии синтеза полииептидной цени (белка); синтезві
аномальной по первичной структуре нолипсптидной цепи (белка);!
количественно недостаточном синтезе полипепгидной цепи (белка);]
количественно избыточном синтезе полипепгидной цепи (белка).Независимо от характера изменений первичного продукта гена]
эффект мутаций может выражаться в разных вариантах нарушения!
функций:— Потеря функции белка в результате либо ингибирования про¬
цессов транскрипции/трансляции, либо изменения его струк¬
туры и функциональных свойств.— Появление новой функции, при мутациях такого типа у мутант¬
ного белка наряду с нормальной функцией появляются новые
цитотоксические свойства, приводящие к гибели клеток.— Доминантный негативный эффект проявляется тогда, когда
первичный продукт мутантного аллеля ингибирует функцию
нормальііьіх белков.
wIfltlll 4. Генные болезни143Изменение дозы гена (делеции или дупликации) может приш¬
ли гь к нарушению пространственной структуры молекулярно¬
го продукта,1а ()снове первичного эффекта мутантного аллеля развертывается
IjKtvii сложнейший патогенез генной болезни, проявляюш^ийся в раз-
[ЦноИризных фенотипических эффектах или клинической картине.IV іультатом действия патологической мутации (фенотипический
[||)м|к‘кт) может быть, прежде всего, летальность на ранних стадиях
Ьйііпігия зародыша, до имплантации. Механизмы такой леталь-
Шт I и еще НС выяснены, но ее сушествование у человека не вызы-
сомнений. Это проявляется в виде несостоявшсгося зачатия
КИМ плантации) у фертильных женщин при нормальной половой
ІІКИ ШИ. У молодых женщин зачатие наступает в среднем через 3 мес
[|>Г1 улярной половой жизни (без контрацеппии). Примерно 50% несо-
ІОЯІЯІ1ИХСЯ зачатий обусловлены гибелью зиготы по генетическим
||м1чимам (генные, хромосомные и геномные мутации). Если раз¬
ім шс эмбриона с патологической генной мутацией не остановилось
1>ам них стадиях, то фенотипические эффекты в зависимости
)1 повлеченного гена и характера мутации формируются в виде
пириантов; дисморфогенеза (врожденных пороков развития), нару-
Цгкного обмена веществ, смешанных эффектов (дисморфогенеза и
Аномального обмена веществ).Илияние патологических мутаций начинает реализовываться в
ІИІ.ІС периоды онтогенеза ~ от внутриутробного до пожилого воз-
^йСіа. Большая часть патологических мутаций проявляется внутриу-
jfpofjtio (до 25% всей наследственной патологии) и в допубертатном
<)ірасте (45%). Еще 20% проявляется в пубертатном и юношеском
Otpiit^Te, и лишь 10% моногенных болезней развивается в возрасте
Цтдрше 20 лет.Для понимания природы генных болезней очень важно иметь
Шрслставление о том, что клиническая картина заболевания может
Сформироваться вследствие включения разных патогенетических
•вей ьев, которые могут быть обусловлены фенотипическими эффек-
Тйми мутаций разных генов. Следовательно, в одну группу будут
тп ючсньт разные с генетической точки зрения заболевания (мутации
[| разных локусах). Такие случаи называются генокопиями. Наряду
)1им, хотя и редко, могут встречаться фенокопии генных болез-
йей. Это те случаи, при которых повреждающие внешние факторы,
Действующие, как правило, внутриутробно, вызывают болезнь, по
144Клиническая генетикклинической картине в общих чертах сходную с наследственно!
Противоположное состояние, когда при мутантном генотипе инді
вида в результате средовых воздействий (лекарства, питание и т.п^
болезнь не развивается, называют нормокопированием.Понятия о гено- и фенокопиях помогают установить правильный
диагноз, а также более точно определить прогноз здоровья пациент
или вероятность рождения больного ребенка. Понимание принципе
нормокопирования дает врачу возможность предупредить развил
болезни у ребенка, унаследовавшего патологический ген.КЛАССИФИКАЦИЯКак и для любой группы заболеваний, классификация генны|
болезней условна и многокомпонентна. В основу классификациі
генных болезней можно положить генетический, клинический ил^
патогенетический принпип.в соответствии с генетическим принципом классификации генні
болезни можно подразделить на группы согласно типам наследования^
аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленны^
доминантные, Х-сцепленные рецессивные, Y-сцепленные (голан;
дрические) и митохондриальные. Отнесение болезни к той илй
иной группе помогает врачу сориентироваться относительно ситуа-^
ции в семье и определить вид медико-генетической помощи. ВыШ(!
(см. главу 3) были рассмотрены характеристики наследования каждоі
из этих групп.Клинический принцип классификации генных болезней учиты-;]
вает систему или орган, наиболее вовлеченный в патологическиі
процесс. Так, различают наследственные болезни нервные, нервно-]
мышечные, кожные, глазные, опорно-двигательного аппарата, эндо-1
кринные, крови, сердечно-сосудистой системы, психические, моче-|
половой системы, желудочно-кишечного тракта (ЖКТ), легких. Для!
некоторых групп болезней установились даже специальные термины:]
нейрогенетика, дерматогенетика, офтальмогенетика. Условность кли-^
нического принципа классификации очевидна. Некоторые болезни у !
одних больных больше проявляются в одной системе, у других — в і
другой. Например, муковисцидоз может преимущественно поражать!
или ЖКТ, или легкие. Нейрофиброматоз I типа может выражаться
либо кожными изменениями (пигментные пятна, нейрофибромы),
шА Генные болезни145омухолями нервных стволов и мозга. Несмотря на некоторую
iMiiHDCTb, клиническая классификация помогает врачу соответ-
ІУІОІІІСГО профиля концентрировать внимание на наследственных
тих, встречающихся в его практике.Никиенетическая классификация наследственных болезней под-
|(|Л1"1ясг их на 3 группы в зависимости от того, в чем проявляется
ІИоинос патогенетическое звено. Патогенез болезни может привести
|Ии|>УИ11^||ному обмену веществ, аномалиям морфогенеза или комби-
ІИИИ того и другого. В соответствии с этим различают наследствен-
ІС (и)лсзни обмена веществ, врожденные пороки развития (моно-
itiiHi природы) и комбинированные состояния. Наследственные
ігиііі обмена веществ, в свою очередь, подразделяют по типам
ІМімііі (углеводный, аминокислотный, обмен витаминов, липидов,
И»и1.)1ои и др.).ОБЩИЕ ЗАКОНОМЕРНОСТИ ПАТОГЕНЕЗАМлчало патогенеза любой генной болезни и его ключевая точка
ІИїлііьі с первичным эффектом мутантного аллеля, поэтому прин¬
ці иальные звенья патогенеза генных болезней можно представить
їедующим образом: мутантный аллель -> патологический первич-
|й продукт (качественно или количественно) -> цепь последующих
((Химических процессов клетки органы -> организм. Это
Ійіииіи и общая закономерность развития генных болезней при всем
многообразии.Мутации могут вызывать болезни через множество различных
ІХИНИЗМ0В. Они могут затрагивать посттрансляционный процес-
IIII, (|)ормирование клеточных компартментов, функцию белка и
Щимодействие первичных продуктов. Молекулярные основы пато-
|Нс la большинства болезней еще не полностью понятны. Предстоит
проследить последствия мутации от эффектов на молекулярном
juiiiic до физиологии и клиники болезни, что является задачей
)Лекулярной медицины,
функция больщинства генов определяется трехмерной структурой
Ослковых продуктов. Хотя многие патологические мутации в генах
Жализованы в некодирующих областях, большинство охарактери-
>ттных мутаций поражает структуру и функцию белков. В целом,
)Ч1> при этом идет о болезнях, ассоциированных с одним геном с
146Клиническая генети»высокой пснетрантностью. Большие делеции, вставки или инверсиі
в протеинкодируюших областях гена почти неизбежно нарушают]
функцию белка. Наследуемые болезни обусловлены патологически*!
ми мутациями, не ведущими к смерти гетерозиготного носителя дО|
пострепродуктивного возраста.'MПатогенез болезни на молекулярном уровнеВ зависимости от контролируемого конкретным геном продукта й і
от характера его нарушения при мутации соответствующим образом]
развертывается патогенез болезни на молекулярном уровне.Если в результате мутации будет вырабатываться избыточно^!
количество продукта, то патогенез болезни в целом будет обуслов-іг|
лен усиленной генной активностью. Существование таких вари-^|
антов можно предполагать, но обнаружен он лишь в единичных!
формах наследственных болезней. Пример такой генной мутации —.
мутация в гене FII, приводяидая к усиленному синтезу протром-і
бина.При другом варианте патологического эффекта мутантного гена!
синтезируется аномальный белок. За этим следуют нарушения той|
системы (клетки, органа), функции которой обеспечиваются нор¬
мальным белком. Эти нарушения первоначально развертываются і
на молекулярном уровне. Примером такого варианта патогенез^^
болезни может быть серповидно-клеточная анемия. В результате^
замены урацила на аденин в кодоне GUA синтезируется цепь моле¬
кулы глобина с глютамином, заменившим валин. Замены одной
аминокислоты оказывается достаточно, чтобы изменить функ¬
циональные свойства гемоглобина (пониженную растворимость,
повышенную полимеризацию). Такой гемоглобин уже не может
выполнять кислородакцепторную функцию и кристаллизуется при
недостатке кислорода, а эритроциты приобретают серповидную
форму (отсюда и название болезни), склеиваются, тромбируют
капилляры и т.д. (рис. 4.2).Третий вариант патологического эффекта мутантного алле-
ля — отсутствие выработки первичного продукта. Этот вариант,
очевидно, встречается наиболее часто. Естественно, что в этих
случаях нарушается тот или другой процесс нормального биохи¬
мического гомеостаза. Это выражается в накоплении токсичных
продуктов-предшественников (рис. 4.3). На схеме представлены
hum. I 4. Генные болезни147рис. 4,2. Мазок крови больного серповидно-клеточной анемией (б) по срав-
jH'iiHio с нормой (а). Патология: серповидные эритроциты, пойкилоцитоз,
ЙКИ 50ЦНТОЗ, склеенные эритроциты. Пояснения в текстеТироксин4ФанилаланинТирозин Uc> ДофаФен илп ировиноградная
кислота {выделяется
с мочой при
фенилкетонурии)г омогентизиновая
кислота {выделяется
с мочой при
алкаптонурии)Пигмент меланинfАцетоуксусная кислотааСОг+Нр1*ис. 4.3. Биохимические «блоки» при наследственных нарушениях обмена
Ямпмокислот: 1 — феиилкетонурия; 2 — альбинизм; 3 — алкаптонурия; 4 —
Црожлснная недостаточность тироксина (дисгормоногенез)
148Клиническая генетиіірезультаты наследственных нарушений а\тинокислот фенилала-^
нина и тирозина. При фенилкетонурии в крови накапливаются
фенилаланин и продукты его патологического метаболизма (I),
поскольку он из-за отсутствия фенилаланингидроксилазы Щ
превращается в тирозин. Нарушение обмена тирозина приво-j
дит к патологии образования меланина (2) или тироксина (4>*
Недостаточность синтеза оксидазы гомогентизиновой кислоты;
(сущность мутации в этом гене) ведет к накоплению гомоген^
тизиновой кислоты в крови (3), которая из-за высокой концен*
траиии откладывается в хрящах и клапанах сердца. В конечном
счете с возрастом это приводит к артритам и порокам сердца;
Возможны и другие (обходные) пути обмена, часто также с пато¬
логическим исходом. В результате отсутствия первичного про¬
дукта гена может задерживаться какой-либо важный процесс,
постоянно осуществляющийся R организме. Так, мутации генов^
детерминирующих синтез ферментов репарации ДНК, приво¬
дят к невозможности восстановления постоянно возникающих
нарушений в структуре ДНК, что обусловливает развитие злока^
чественных новообразований (пигментная ксеродерма, атаксия-
тел еангиэктазия).Известен и 4-й вариант первичного патологического эффек¬
та мутантного аллеля — выработка уменьшенного количества
нормального первичного продукта (Р-талассемия, акаталазия).^
Патогенез таких заболеваний различен, поскольку наряду с нор¬
мальным путем обмена веществ будут протекать и патологиче¬
ские процессы,Выще были описаны общие закономерности патогенеза ген¬
ных болезней на молекулярном уровне на примерах нарушения
обмена веществ. Тот же самый принцип патогенеза (мутантный
аллель -> патологический первичный продукт) действует и для
генов морфогенетического контроля, мутации в которых приво¬
дят к врожденным порокам развития (полидактилия, синдромы
Холта—Орама, Крузона, Нунан, Лоренса—Муна, Меккеля, Робертс,
Эллиса-Ван-Кревельда, Гольтца; рис. 4.4, 4.5). Начальное звено
врожденного порока развития связано с нарушением дифферен-
цировки клеток. Запрограммированные в геноме диффсренци-
ровка клеток, а затем и органогенез осуществляются путем смены
процессов активации и выключения определенных генов в строго
4. Генные болезни149Рис. 4.4. Синдром Крузона, Мать и сын
150Клиническая генети»ограниченных временных (по
отношению к онтогенезу) про¬
межутках (транскрипционные
факторы). Если первичный про¬
дукт морфогенетического гена
аномальный, то необходимая
для дальнейшего правильно¬
го развития органа дифферен-
цировка клеток не последует.
Естественно, что морфогенети¬
ческих генов много, они дей¬
ствуют в разные периоды онто¬
генеза. Соответственно мута¬
ции в них будут приводить к
специфическим врожденным
порокам развития.Рис. 4.5. Синдром Меккеля (энцв|
фалоцеле)Клеточный уровень патогенеза генных болезнейПатогенез генных болезней не заканчивается на молекулярном уров-[
не даже в первичных звеньях. Для многих болезней главное звено пато-;|
генеза — клетка. Во всех генетических процессах клетка — дискретная
самостоятельно регулируемая единица, и в ней осуществляются все]
процессы реализации генетической информации (транскрипция, TpaHcJ
ляция, синтез белка). Это — обшебиологическая аксиома. Клеточный]
уровень патогенеза генных болезней означает, что в определенных типаа{1
клеток разыгрываются основные патологические процессы, присущие
конкретной нозологической форме. Клетка как бы не выпускает из себя і
патологические явления, а принимает на себя удар первичного патогенг
ного эффекта гена. Точкой приложения первичного действия мутантно-^
го гена являются отдельные структуры клетки, разные при различных
болезнях (лизосомы, пероксисомы, мембраны, митохондрии).Патогенетические процессы на клеточном уровне развертываются при
болезнях накопления (или лизосомных) в связи с нарушением активностн
лизосомных ферментов. Так, накопление в клетках, а затем и в основт
ном межклеточном веществе, гликозаминогликанов (мукополисахаридов)
приводит к развитию тяжелых заболеваний — мукополисахаридозов.
Причина избыточного содержания полимеров — гликозаминогликанов —
заключена в отсутствии их деградации в лизосомах. Нарушение деграда¬
ции гликозаминогликанов связано с дефектами в группе специфических
iHit A. Генные болезни151Ц1МСИТ0В, катализирующих весь цикл деградации. Более подробную
1фн|>м;щию о клинической картине, диагностике и лечении мукополи-
INnpiuiojOB см. в статье С,В. Михайловой с соавт. «Мукололисахарозы:
іф)|а'ренциальня диагностика и лечение» на компакт-диске.Лі’угим примером болезней накопления могут служить гликогено-ІІ клетках печени и мышц накапливаются полимеры гликогена,
nupidc не подвергаются деградации даже тогда, когда организму
И>ь\(>лима глюкоза в крови. Патогенез гликогенозов принципиаль-
(I І11К0Й же, как и мукополисахаридозов. В клетках печени и мышц
h’V t c (вует определенный фермент (их уже известно много), который
^Миі гиует в цикле расщепления гликогена до глюкозы.другие внутриклеточные структуры — пероксисомы — также
IMVT быть точкой приложения первичного действия мутантного
рма. в этих случаях развиваются так называемые пероксисомные
нчни. Описано уже 18 нозологических форм. Основное патоло-
IML'CKoe звено при всех пероксисомных болезнях локализовано в
Ііроксисомах в виде биохимических нарушений, обусловленных ген-
1ЫМИ мутациями. Биохимическая сущность многих пероксисомных
ож* ІПЄЙ уже раскрыта на уровне мутантных ферментов. Клинически
ле И1И проявляются в виде множественных врожденных поро-
ра звити я, в целом сходных при разных нозологических формах
тожественных черепно-лицевых дисморфиях, катаракте, кожных
ишдках на шее, почечных кистах и др.). Пероксисомные болезни —
Іример наследственных болезней обмена, при которых множествен-
Ibic пороки развития объясняются молекулярным дефектом.Различают 3 группы пероксисомных болезней: генерализованные
и шененным числом пероксисом (пример — цереброгепатореналь-
ІІІІЙ синдром, или синдром Целлвегера); с неизмененным числом
іероксисом и нарушением нескольких биохимических функций
іример ~ целлвегерподобный синдром); с неизмененным числом
[Йсроксисом и нарушением единственной биохимической функции
[(пример — болезнь Рефсума).Мембраны, так же, как и структуры клеток, могут быть ключевыми
[•лементами патогенеза генных болезней. Так, отсутствие специфи-
[ївских белковых молекул-рецепторов на клеточной поверхности,
^вйизывающих ЛПНП, приводит к семейной гиперхолестеринемии.С'индром полной нечувствительности к андрогенам (синоним:
синдром тестикулярной феминизации) вызывается мутациями в
Х-сцепленном гене, который кодирует синтез внутриклеточного
152Клиническая генетикрецептора андрогенов. Отсутствие чувствительности клеток к андр(
генам приводит к развитию женского фенотипа при хромосомної
наборе XY. У таких больных, несмотря на женское строение нару}
ных половых органов, имеются семенники в брюшной полости
нормальный уровень андрогенов в крови.Клиника ви тамин-D-резистентного рахита (аутосом но-доминанні
пюе заболевание) обусловлена дефектом рецепторов 1,25-диіі
гидроксихолекальциферола.При муковисцидозе нарушается регуляция транспорта хлоридо<
через мембраны эпителиальных клеток. Такая регуляция в норме о(
ществляется белком-продуктом гена, названным кистофиброзны!
трансмембранным регулятором (CFTR). Одни мутации в гене CfTi
веду г к снижению синтеза данного белка из-за незавершенности пр(
цессинга РНК, другие — к качественным изменениям мембранні
хлорных каналов. Одна первичная биохимическая аномалия (нарушен!
транспорта хлоридов) обусловливает возникновение мультиорганног
патологического процесса (прогрессирующее поражение дьіхательнні
путей, хронические синуситы, недостаточность экзокринной секрето{
ной функции поджелудочной железы, стерильность у мужчин).Клеточный уровень патогенеза генных болезней может прояві
ляться не только в конкретных органеллах, но и в виде нарушения
скоординированности функций клетки. Так, мутации, затрагивающі
области онкогенов, ведут к снятию контроля размножения клетої
(репрессия антионкогенов) и соответственно к злокачественном!
росту (наследственный рак толстой кишки, ретинобластома).Клетка может быть главным звеном при реализации патогене;
на молекулярном уровне. Так, прекрап^ение синтеза мышечного
белка дистрофина при мутациях в соответствующем гене приводш
к постепенной деградации мышечных клеток. Это спусковой крючої
патогенеза тяжелой наследственной болезни — миопатии Дюшснна.ІОрганный уровень патогенезаОрганный уровень патогенеза наследственных болезней, безуслої
но, производный от молекулярного и клеточного, в результате пер**^
вичных или вторичных процессов при разных болезнях мишены
патологического процесса служаг различные органы. Например, отло^
жение меди в печени и экстрапирамидной системе мозга при гепа-|
толентикулярпой дегенерации (болезнь Ви-'[ьсона-Коновалова)
первичный процесс, а гемосидероз паренхиматозных органов при nep^j
ІЯНІІ 4. Генные болезни15314 пом I емохроматозе или талассемии развивается вторично вследствие
ifM'u-iiiioro распада эритроцитов. При алкаптонурии отложение гомо-
ИІІІНІ1ІІ0В0Й кислоты в хрящах суставных поверхностей и клапанах— вторичный процесс, обусловленный высокой концентрацией
Шіиі'іп изшювой кислоты в крови (она не превращается в малеилаце-
іукі усную кислоту в результате мутационно обусловленного отсутствия
Ш’ииа 5Ы гомогентизиновой кислоты). Это ведет к медленному развитию
орик»)н сердца и ч'уі оподвижности суставов (примерно к 40 годам).Ірганизменньїй уровеньИ целом организме взаимосвязь патогенетических процессов
Ірочиляется сочетанно на молекулярном, клеточном и органном
ІОІИІЯХ. Патологический процесс, запущенный первичным эффек-
му тантного аллеля, приобретает пелостность с закономерными
индивидуальными вариациями. Тяжесть и скорость развития
іііі'иіи при прочих равных условиях (пол ребенка, одинаковый
Ірії к 1 ер мутации) зависят от генотипа организма (соматический
Цинииизм, гены-модификаторы) и условий среды.Патогенез любой наследственной болезни у разных индивидов
ihi н сходен по первичным механизмам и этапам, но формируется
Грого индивидуально.ГЛАВНЫЕ ЧЕРТЫ КЛИНИЧЕСКОЙ КАРТИНЫt)r>iij,we характеристики клинической картины обусловлены генети-
ІУКОЙ природой болезней этой группы, т.е. принципами экспрессии,
Шрессии и взаимодействия генов. В то же время очевидно, что в пол-
объеме все общие черты клинической картины при одном заболе-
ІНИИ наблюдать трудно. Знание общих черт генных болезней поможет
Июлозрить наследственную болезнь даже в спорадическом случае.Ниже приведены 3 главные характеристики генных болезней и их
^логические основы: особенности клинической картины, клиии-
ІСКИЙ полиморфизм, генетическая гетерогенность.юбенности клинической картинык таким особенностям относятся многообразие проявлений, раз¬
уй возраст начала болезни, прогредиентность клинической кар-
1ИЫ и хроническое течение, тяжесть течения, обусловливающая
іниллидность с детства и меньшую продолжительность жизни.
154Клиническая генеткСимптоматика каждой генной болезни очень многообразна. К|
правило, патологическим процессом затрагивается не одна систеї
или орган, а несколько органов уже на первичных этапах формиї
вания болезни. Это касается болезней, проявляюшихся в нарушеш
процессов эмбрионального развития (врожденные пороки разві
тия), наследственных болезней обмена веществ и комбинированнї
болезней. Биологической основой многообразия проявлений генні
болезней служит генный контроль первичных механизмов обмеї
или морфогенетических процессов.Для некоторых групп болезней вовлечение в патологический пр(
иесс многих органов и тканей обусловлено тем, что первичный дефеї
локализован в клеточных или межклеточных структурах мної
органов. Например, при наследственных болезнях соединительж
ткани нарушен синтез специфического для каждой болезни белка
или иной волокнистой структуры. Поскольку соединительная ткаї
есть во всех органах и тканях, то и многообразие клинической си*
птоматики при этих болезнях — следствие аномалии соединительж
ткани. Так, при синдроме Марфана в патологический процесс вовл<
чены скелетно-мышечная система, глаза, сердечно-сосудистая сист
ма, наружные покровы, легкие, ЦНС; при синдроме Элерса—Данло
кожа, суставы, глаза, сердце, сосуды, грудная клетка, мозг, зубы.Наряду с понятными механизмами многообразных проявлю
ний генных болезней имеются примеры необычайно широкої
клинического полиморфизма с пока неизвестными механизмам!
Нейрофиброматоз I типа проявляется пигментными пятнами, ко^
НЫМИ, подкожными и плексиформными нейрофибромами, K0CTH1
ми изменениями, опухолями нервных стволов и головного М031
снижением способности к обучению. Эти многообразные проявл<
ния пока не удается связать в единый патогенетический комплеї
хотя уже известны структура гена и его первичный продукт,
исключается, что в этом и других подобных случаях речь идет
первичной плейотропии, т.е. множественных эффектах гена в раз
ных органах.Другая черта клинической картины генных болезней, поми^
мо многообразия проявлений, — разный возраст начала болезне!
В целом, для наследственной патологии возраст начала практическі
не ограничен: от ранних стадий эмбрионального развития (вроз
денные пороки развития) до пожилого возраста (хорея Гентингтош
болезнь Альцгеймера). Из всех генных болезней 25% развиваютсіі
IMri 4. Генные болезни155ly I l»iiy I робно, T.c. как врожденная патология. За первые 3 года
ІІІИ проявляется еще почти 50% генных болезней (в сумме с
Іуі|М!уіробньім формированием — 70% всех болезней). На конец
|^Оі'|)г;ігного периода приходится 99%.ІЬпраст дебюта различен при многих генных заболеваниях,
три мер, хорея Гентингтона (ау госомно-доминантное заболевание)
іик’г начинаться в любом возрасте: от детского (описаны случаи
заболевания в 6-летнем возрасчс) и до 60-леінего, средний
ирис I начала заболевания 38 лет. Клиническая картина миотониче-
1Ик аистрофии (аутосомно-доминантное заболевание) может возни-
И1> пиутриутробно — врожденная форма, в юношеском возрасте —
им цільная, у взрослых — классическая. Возможна мягкая форма с
или им началом.Ио фаст начала заболевания различен и при рецессивных болез-Н. Муковисцидоз может развиваться внутриутробно (мекониаль-
Itt илеус), в грудном возрасте или после 3—7 лет жизни.
Ьиологическая основа разного возраста начала в целом для группы
1МИЫХ болезней заключается в строгих временных закономерностях
(тогснетической регуляции экспрессии генов, функционирование
Іждоі о гена в ііорме начинается и заканчивается в строго опреде-
пюе в отношении онтогенеза время и в строго определенных клет-
і%. ‘Ого правило относится и к мутантному гену.I І1)ичинами разного возраста начала одной и той же болезни могут
|ть индивидуальные характеристики генома больного. Действие
мих генов на эффект мутантного гена (взаимодействие генов)
ЇЖЄ1 менять время развития болезни. Какие-то комбинации генов
(^ду’1 способствовать более раннему проявлению действия патологи-
ІСКИХ генов, какие-то — тормозить его. Небезразличны для време-
мроявления патологических генов и условия среды в онтогенезе
♦дивида, особенно во внутриутробном периоде.lie с это больше гипотетические предположения о причинах раз-
ЇГ» возраста начала конкретной генной болезни. Вместе с тем
)лскулярно-биол01 ические исследования позволяют конкретизи-
іингь биологические основы клинических проявлений отдельных
)рм генных болезней в разном возрасте. Так, установлено, что сроки
ІЗІ1ИГИЯ хореи Гентингтона могут быть связаны с импринтингом
ітнетствуюшего гена у отца (унаследовавшие ген с увеличением
«ела повторов от отцов заболевают раньше), а при миотонической
(строфии — с числом тринуклеотидных повторов, определяемых
156Клиническая генетиів мейозе у женщин (чем больше повторов, тем раньше развивает
болезнь и тем тяжелее она протекает).Генным болезням свойственны прогредиентность клинической
тины, а также затяжное течение с рецидивами. ^1При многих болезнях клиническая картина и тяжесть течеш
усиливаются по мере развития патологического процесса. Приведе
несколько примеров.Нейрофиброматоз I типа начинается с возникновения безобидні
пигментных пятен цвета кофе с молоком, веснушек в подмышечн!
и паховых областях. Затем появляются единичные нейрофибр<
мы, опухоли или косгные изменения и т.д. При фенилкетонурі
прогрессируют умственная отсталость, гипомеланоз кожи и воле
Наруїиение свертываемости крови при гемофилии с возрастом
ослабевает, а усиливается. При СМ2-ганглиозидозе с 6-меся4но|
возраста начинает развиваться демиелинизаиия нервных волоке
что продолжается вплоть до летального исхода в возрасте 2—4 л<
Затяжное или хроническое течение имеют многие генные болез!
(муковисцидоз, болезнь Рандю—Ослера—Вебера, гепатолентикуля|
ная дегенерация и др.).Как видно из приведенных примеров, прогредиентность КЛИНІ
ческой картины и хроническое течение наблюдаются при генні
болезнях с разными типами наследования. Первичная биологичесі
основа этой характеристики — непрерывность функционировані
патологического гена (либо отсутствие его продукта). К этому щ
соединяются вторичные процессы (воспаление, дистрофия, наї
шенный обмен веществ, гиперплазия и т.д.), которые усиливаї
первично запущенный патологический процесс.Естественно, что прогредиентность присуща не всем болезні
При развитии некоторых болезней к определенному возрасту дості
гается конечный фенотип. Например, при ахондроплазии болезі
полностью формируется по мере роста костей (нарушен хондрогеж
пропорционально возрасту. Развитие болезни как бы запрограмми]
вапо без прогредиентности.Течение большинства генных болезней тяжелое, что приводит
инвалидизации в детском возрасте и сокращению продолжител!
ности жизни. Тяжесть течения болезни не всегда связана с вр01
денным характером заболевания. Такие тяжелые формы, как xoj
Гентингтона, гепатолентикулярная дегенерация, миотоническая ди<
трофия, наследственные кардиомиопатии, развиваются у взрослі
ійіі л Генные болезни 157мііжпее место, которое занимает моногенно детерминируемый
imccc в обеспечении жизнедеятельности, ЮМ клинически тяжелее
ПШІ.1ЯЄТСЯ мутация в соответствующем локусе.1ИНИЧЄСКИЙ полиморфизм и его причинык циническая генетика всегда опиралась в своих принципах на
IfcoHOMCpHOCTH, устаиовле1П1ые жспериментальной генетикой. Это
1ИЧ1МОЙ мере относится к анализу клинического полиморфизма.
1‘М4 г. Н.В. Тимофеев-Ресовский в статье «Связь между геном
ИМС1ПНИМ признаком (феноменология ироявлеиия генов)» писал
„jHitiJb первый шаг к генетической физиологии развития, а имен-
И к 1'ак называемой феііоменологии проявления генов. Этим я
Иимлмаю расчленение и классификацию всеобщих явлений в
|/Д1)иищно многогранной и изменчивой области проявления самых
іличньїх наследственных признаков». Он обратил внимание и
їопллюстрировал на конкретных экспериментальных материалах
їГтіис феномены проявления генов», среди них такие, как гете-
Иснпые гены, полифенные (плейотропные) гены и константно и
Ірмабельно повторяющиеся гены. Он проявил интерес к конпеп-
ІІйііі.ности и практическому использованию таких знаний «прежде
Ml) Ii области наследственной патологии человека». Спустя иочти1 лет В. Маккьюсик эти феномены проявления генов у человека, не
linui сути в их интерпретации, назвал «принципами клинической
ІНСІИКИ»: клинический полиморфизм, генетическая гетероген¬
ні. и плейотропизм. Еще раньше в России, как уже отмечалось,
,Н. Давиденков, практикуя в клинике нервных боігезней, в 1930-е
И>1 ими широко пользовался.Олпа из ocjfOBHHX и наиболее старых аксиом клинической меди-
|||Ы сводится к тому, что болезнь любой этиологии (инфекционной,
іиматической, алиментарной, гормональной и др.) проявляется
одинаково у разных индивидов, поэтому нужно лечить не болезнь,
^вольного, в ряде случаев клиническая картина одного и того же
Іолсвания варьирует от стертых форм до тяжелейших клиниче-
(их проявлений. Формирование клинической картины связывают с
)Г)Синостями действия этиологических факторов (например, виру-
пи ость возбудителя), исходного состояния организма (иммунный
|тус, обмен веществ), сопутствующих условий (стресс, температу-
Кроме того, признается роль врожденных характеристик орга-
іама rt патогенезе и клинической картине болезней.
158Клиническая генеттКазалось бы, можно ожидать более или менее унифицированна
клинической картины какой-либо нозологической формы генш
болезней, поскольку этиологический фактор для всех больных с Э1
формой одинаков (мутация в соответствующем геле), а патогенез р£
вертывается на фоне жестко детерминированного контроля генн<
активности. Такой вывод подсказывал общегенетический взгляд
моногенно детерминируемые события. Однако клиническая практ!
ка показала, что симптоматика наследственных болезней различі
При накоплении наблюдений одних и тех же нозолоіических фоі
оказалось, что клинический полиморфизм генных болезней выражу
не меньше, чем ненаследственной патологии.При многих заболеваниях, достаточно хорошо изученных на кл1
ническом, генетическом и молекулярном уровнях, нет строгой корі
ляции между генотипом и фенотипом. Неясно, почему заболеван!
вызванные строго доказанными одними и теми же мутациями
молекулярном уровне, имеют разные клинические проявления ИШ
гда даже у идентичных близнецов. Нужны дальнейшие исследов!
ния с анализом генных сетей, активности факторов транскрипции
транспортных белков и других модификаторов экспрессии генов.Клинический полиморфизм генных болезней проявляется в раз
ных сроках начала заболевания, полноте и тяжести симптоматиі
(глубина патологического процесса), продолжительности болезн^|
степени инвалидности, толерантности к терапии, в сокращеї
продолжительности жизни. Вместе с тем следует подчеркнуть,
генные болезни не имеют плавных переходов от нормы к патологиіі
Даже самая легкая форма болезни обязательно имеет минимальні
диагностические критерии. Генетическое правило гласит: нормалі
ный генотип детерминирует нормальный фенотип, а мутантні
генотип детерминирует мутантный фенотип (болезнь).Генетической причиной полиморфизма может бьтгь явление взаі
модействия главного гена и генов модификаторов (эпистаз, особеї
ности инактивации и дозовая компенсация Х-хромосомы, цитоплаз
магический геном), с другой стороны — это могут быть и факторі
внешней среды, в которых осуществляется развитие индивида.На рис. 4.6 изображено влияние названных двух групп факторе
(генетических и внешнесредовых) на фенотипы моногенных болез*
ней. Так, фенилкетонурия в пространстве двух обозначенных коор
динат занимает срединное положение, отражая заметное влияни<
средовых и случайных факторов, а также эффектов других гено!
А I пимые болезни159ІАиіримсские проявления болезней, в то же время для болезни
“І икса влияние этих факторов менее выражено, а в клинических
Щж-ииях недостаточности Г6ФДГ преобладающим модифици-
filtut (|)LiKTopoM является внешняя среда.»uii гоя щему времени накопился огромный фактический мате-
ш> (|)Сноменологии клинического полиморфизма отдельных11 <|)акторам, его определяющим. В первую очередь следует
імімриішть значение характера мутации в конкретном локусе
Мрияиления болезни или формирования фенотипа (мутантно-
Мі’рі'ично возникшие и унаследованные от предыдущих поко-
у гации имеют достаточно сходное фенотипическое прояв-
|Иг. I.e. длительность унаследования мутации не отражается на
ІМИ'іеском полиморфизме генных болезней. Как подчеркивалось
110. лссятки и даже сотни разных мутаций (и даже разных типов)
1Ш)М и том же локусе ведут к одной и той же болезни, в боль-
li’inc случаев характер мутации не определяет клиническуюЕ ^ ■ Недостаточность Г6ФДГ■ ГемохроматозI■ Острая перемежающаяся порфирия■ ССгЭнтитрипсин ZZш Аро E-2/F-2 гиперлипопротеинемия
■ Синдром Марфана^ Болезнь
Хартнапа■ Фенилкетонурия■ Семейная гиперхолестеринемия
■ Серповидно-клеточная анеми?■ Муковисцидоз■ Хорея Гентинггона■ Дистрофия ДюшеннаБолезнь Тэя—СаксаРоль других локусов генотипаf. 4.6. Потенциальное влияние генетических (G) и негенетических (Е)
«торов на фенотип некоторых моногенных болезней (по Ч. Скрайверу иI, Уотерс)
160Клиническая генеїкартину болезни. Фенотип определяет первичный эффект гена (і
продукта или мало продукта).Однако накапливается все больше данных о зависимости фене
па (клинической картины болезни) от генотипа (разных мутациі
одном и том же локусе). Такие формы болезней, при которых М]
иии не полностью блокируют выработку первичного продукта,
известны. Расшифровка корреляций между гено- и фенотипом ста
возможной благодаря молекулярно-биологическим исследовані
структуры генов, мутаций и их первичных продуктов.Мутации в одном и том же локусе, ответственные за синтез ді
трофина, приводят к двум клиническим формам: миопатии Дюшен!
(тяжелой) и миопатии Беккера (легкой). Установлено, что миопат
Дюшенна развивается при полной блокаде, а Беккера — при част*
ной блокаде синтеза РНК для дистрофина (при миопатии Бекі
делеции гена меньшего размера).Сплайсинговые мутации, как правило, не полностью блокируї
образование мРНК, поэтому соответствующие формы болезни бы1
ют мягкими по клиііической картине и течению.Четкая корреляция между генотипом (характером мутации) и феї
типом (клинической картины болезни) отмечена покалишь при одне
виде мутаций — экспансии тринуклеотидных повторов в гене.Чем больше повторов в мутантном аллеле, тем тяжелее протека
болезнь. Особенно четко это проявляется при синдроме МартиШ
Белл и миотонической дистрофии. Поскольку экспансия повтор
формируется в мейозе у одного из родителей (у мужчины или ЖЄНЩИІ
при различных болезнях), это обусловливает явление антиципации’
более тяжелое течение наследственной болезни в последующих П0І
лениях. До открытия этого типа мутаций (экспансии триплетов)!
молекулярно-генетического прослеживания числа повторов в пої
лениях явление антиципации рассматривалось как артефакт наблі
дений. Теперь стало ясно, что феномен антиципации существует,
него уже обнаружена биологическая основа при небольшой групі
болезней.В последнее время появились данные, свидетельствующие о TOJ
что развитие некоторых наследственных заболеваний может бі
обусловлено резким изменением числа копий не только трину клее
тидных повторов, но и тандемных повторов ДНК большей прот
женности (тетра- и пентануклеотидов, мини- и мегасателлитов), пі
їй 4 Генные болезни161їм іипможно как увеличение, так и, напротив, уменьшение числа
повторяющегося элемента. Примеры наследственных забо-
Ійіііііі, связанных с так называемыми динамическими мутациями,
1ЙП1СИЫ в табл. 4.2.ІНИН 4.2. Заболевания человека, обуслопленныс динамическими мута¬
циямиПатологияГен(локализа¬ция)Тип повтораЧисло повторовнормаболезнь'иікіром ломкой«Л|1П\П)СОМЫ^ИЛХЛ)FMR!(Xq27.3)(CGG)n6-60>200ЇИИиром тремора
Ійіііксии, ассоции-
ІИшіііьій с ломкой>>1|>ОМОСОМОЙ»XIAS)FMR1(Xq27.3)(CGG)n6-6060-200jiom-
ІЙ X хромосомы
fHAXE)FMR2(Xq27.3)(GCC)n4-39200-900Илксия ФридрейхаfA^/V(9ql3)(GAA)n6-32200-1700ilin ііомозжсчковая
rHKL'IIH, тип 1ATXNI (6p23)(GAG)n6-3940-82Ли ііомо;іжечковая
(НКСИИ, тип 2ATX N2
(12q24)(GAG)n15-2432-200Ї M И H о мозжеч кова я
Гіксия, тип З
ІНПДром Мачадо—
пцоі0(|)а)ATXN3(14q24,3-q31)(GAG)n13-3661-84^Іінііомозжечковая
Ґйксия, тип 6CACNAIA(19р13)(GAG)n4-2020-29ЇПтіобульбарная
ІМіїїсчная атрофия
ІОлс иіь Кеннеди)AR(Xqll-ql2)(GAG)n9-3638-62вея ГентингтонаЯЛ (4р 16.3)(GAG)n11-3440-121ірся Гентингтона,
2JPH3(16q24,3)(CTG)n7-2866-78
162Клиническая генеткОкончание таблицы 4j|ПатологияГен(локализа<ция)Тил повтораЧисло повторовнормаболезіОкулофарингеальиая
мышечная дистрофияPABPN1(14qll.2-ql3)(GCG)n8-13Миотоническая дис¬
трофия, тип 1DMPK(19ql3.3)(GTG)n5-3750-10(Миотоническая дис¬
трофия, тип 2ZNF9(3ql3.3-q24)(CCTG)n10-2675-
И 000IСпиномозжечковая
атаксия, тип 10ATXN10(22ql3)(АТТСТ) п10-29280-4500Синдром Ушера, тип
1CPDZ73(1ІРІ5.1)45-нуклеотид-
нын повтор
(VNTR)b5-m
интроне-<6Лицевая и плечелопа¬
точная дистрофия(4q35)мегасател¬
лит D4Z4
(3,3 тыс, п.н.)11-150<10Примечание. FMR — fragile mental retardation; VNTR — полнморфи:ім по кол]!
честву копий (variable number of tandem repeats); п,н, — nap нуклеотидов.Большинство болезней, обусловленных нестабильными нуклес
тидными повторами, фенотипически (клинически) проявляютс
главным образом неврологической симптоматикой (атаксии, когні
тивные нарушения, деменции, нистагм, паркинсонизм), хотя иногл
в болезнь включаются и другие органы (семенники — макроорхі
дизм, дисплазия соединительной ткани, нарушение сердечной пр<
води мости и др.).Патогенетические механизмы болезней экспансии нестабильш
повторов разнообразны. Их можно разделить на три класса.— Класс 1: экспансии нскодирующих повторов, вызывающи^
нарушение функции белков и транскрипции патологи чес ког
гена преРНК (синдром Мартина-Белл, атаксия Фридрейха).— Класс 2: экспансии некодирующих повторов, вызывающи^
появление новых свойств РНК (миотоническая дистрофия \2, синдром тремор/атаксии с ломкой Х-хромосомой).
Ій'1 I fJHHbie болезни163к luicc 3; экспансии кодирующих повторов, приводящие к новымI иойствам мутантного белка (хорея Гентингтона, спиномозжсч-
коиая атаксия).іі;істояіцему времени обнаружено несколько генов-
liicjiitкаюров при моногенных заболеваниях человека. Приведем
(ІЦ1 It.KO примеров. Хорошо изучено улучщение состояния у ГОМОЗН¬
ІМ) мутации, приводящей к р-талассемии, унаследовавших также
<ї-галассемии. Последний выступает как ген-модификатор.
^'OtiHiiHC синтеза цепей глобина, обусловленный р-талассемией,
f'Uiiiio iCH за счет снижения синтеза а-цепей, вызванного мутаци-
И|1и (ї-галассемии. Еще один установленный факт — пациенты с
ІІІиііпісцидозом, гомозиготные по наиболее частой мутации, имеют
М1Ь ііариабельную паїологию легких. Показано, что это связано с
ІИ'іисм по меньшей мере одного гена-модификатора.Кипиическая картина болезни может зависеть от «дозы» генов
аллелей). Так, гомозиготность (два аллеля) при аутосомно-
тиимтных болезнях определяет более тяжелую клиническую
ииму, а иногда даже внутриутробную гибель плода (ахондро-
И1Я. синдром Элерса^-Данло). Аутосомно-рецессивные болезни
}М11.||мются Б полной мере при условии гомозиготного состояния
муіантному аллелю. Однако некоторые признаки заболевания
"уг проявляться и у гетерозигот (один аллель, легкая форма), они
ІЛИікіются до клинических проявлений при действии провоии-
итих факторов. Например, симптомы кислородной недостаточ-
Л и (ири подъемах на большую высоту) проявляются у гетерозигот
серповидно-клеточной болезни; беременность у гетерозигот по
їлассемии приводит к развитию анемии.Іспетические причины клинического полиморфизма могут быть
;лоилены не только патс^югическим геном, но и генотипом в целом,
генотипической средой в виде генов-модификаторов. Геном в делом
^Нкпионирует как хорошо скоординированная система. Вместе с
ологическим геном индивид наследует от родителей комбинации
М их генов, которые могут усиливать или ослаблять действие патоло-
гского гена. В правильности этого положения не приходится сомне-
fUCH, хотя реально гены-модификаторы только начинают идентифи-
Іроиагь. Сравнение выраженности клинической картины у членов
семьи и разных семей показывает, что межсемейные различия
1Ы11С, чем внутрисемейные, но внутрисемейные тоже существуют.
164Клиническая ген<Примером гема-молификагора может служить ген, локализо!
ный 8 длинном плече хромосомы 1, кодирующий «повсеместщ
транскрипционный фактор {upstream stimulatory factor /, USFl),
ствуюший li метаболизме углеводов и липидов у больных с семей!
комбинированной i иперлипидемией. Показано, что разные алл<
ные варианты этого гена определяют разный уровень его Tpt
крипции и, как следствие, USF-факторы активируют или подавлі
экспрессию многих генов, вовлеченных в липидный {АРОЕ, АРІ
АР0Л5) и углеводный (глюкокиназа, рецептор глюкагона) обмеї
а также участвующих в регуляции уровня артериального давлеї
(ренин, ангиотензиноген). Важно, что аллельные варианты
вносят заметный вклад в риск сердечно-сосудистых заболеваний^
популяционном уровне. Таким образом, модифицирующая функі
гена USF} в отношении генов, вовлеченных в регуляцию липидної
углеводного обменов, а также уровня артериального давления, дел|
этот ген отвеїственньїм за признаки, составляющие метаболичесі
синдром.Одним из факторов вариабельности фенотипа или разной э|
прессивности МОЖЄ1 быть соматический мозаииизм.На клиническом уровне явление индивидуальной модификаї
действия патологического гена изучал С.Н. Давилен ков на примеї
наследственных болезней нервной системы. Многие «мелкие», на(
дуемые независимо от основного патологического гена, признг
усиливают клинические нроявлеиия болезни. Это явный призі
взаимодействия генов. С.Н. Давиленков (1947) высказал гипотї
«условного тропизма патологических нервных задатков»: «Помиї
своего прямого влияния на развитие нервной системы, патологи*
ский задаток обладает еще способностью усиливать эффект от ді
гих наследственных факторов, обладающих сходно направленні
тропизмом».В развитии генной болезни, как и любого наследственного щ
знака человека, имеет значение не только генотип, но и внешні
среда, Выше рассмотрена роль таких генетических факторов,
характер мутаций патологического аллеля и генотипическая С]
(взаимодействия генов) в формировании клинического полиморф»
ма. В то же время не вызывает сомнений влияние окружающей срел
в широком смысле слова на развитие болезни. Этому положені
есть много доказательств из клинической практики и специальш
исследований. Приведем несколько примеров.
А Іпиньїе болезни 165Цими і’оматика фенилкетонурии у ребенка более тяжелая, если во
<4 0 инутриутробного развития в рационе матери было много
іукмш, богатых фенилаланином. Обострение наследственных
Ии1|нх1)ий наблюдается после стрессов, охлаждений, переутом-
|н Клиническая картина гемофилии у ребенка усиливается с
ІИ'ігііисм у него кровоизлияний от падений и травм. У женщин,
^Нмх мейрофибромагозом I типа, резко усиливается рост нейро-
ІПМ при беременности.этическая гетерогенностьЬиип ие генетической гетерогенности означает, что клиническая
(It ЮМ ной болезни может быть обусловлена мутациями в разных
или разными мутациями в одном локусе (множественные
Ми>. Фактически это разные нозологические формы с этиоло-
К‘К1)Й точки зрения, объединенные в связи с клиническим сход¬
ім (1>смотипа.fuit’I и ческую гетерогенность наследственных болезней впер-
полметил (и ввел этот термин) С.Н. Давиденков в 1930-х годах.
И4>рые высказывания о генетической гетерогенности болез-
п М)-х годах XX в. принадлежат ряду авторов. В 1960-х годах
Іиккьюсик сформулировал принцип изучения болезней с точки
ІИН их генетической гетерогенности. Этот принцип оказался
щычайно полезным для изучения нозологии генных болезней и
>ои()1 ипических различий.ІИЛЄИИЄ генетической гетерогенности носит общий характер,
уже можно назвать правилом, поскольку оно распространяет-
1Н все белки организма, не только на патологические, но и fia
тльные варианты. С молекулярно-генетической и биохимико-
ІТИческой точек зрения вполне объяснимо, что различные пато-
ІЧССКИЄ гены могут иметь примерно одинаковый фенотип при
(Нимеской оценке. Конечный эффект поломки какого-либо про-
}й на клиническом уровне может быть обусловлен наследствен-
нарушением синтеза разных белков или разных вариантов
И’о И того же белка.^выяснение степени генетической гетерогенности при любой
ісдственной болезни проходит через все этапы: описание про¬
ший на клиническом уровне, изучение типа наследования и
іилизации гена, выяснение первичного биохимического дефекта,
иювление молекулярной сущности мутации на уровне ДНК.
Клиническая генетик^Генетическая гетерогенность, обусловленная мутациями в разні
Локусах, — межлокусная гетерогенность отчетливо видна на приме<
^НнДрома Элерса-Данло (6 форм), нейрофиброматоза (по меньше!6 форм), гликогенозов (более 10 форм), гипертрофической кар*
^^ио^иопатии (ІІ форм), врожденной катаракты (29 генов), витамин^
^'Резистентного рахита и т.д. Гетерогенность в упомянутых формг
‘Прослеживается даже при применении клинико-генеалогическої
в этих группах имеются и аутосомно-доминантные,
‘‘Утосомно-рсиессивные, и сцепленные с Х-хромосомой вариант
^)олез}^ей, т.е. мутации в локусах, расположенных в разных хромо!^:ом^х.Источником генетической гетерогенности в том же локусе — вву*'Р**^1окусной гетерогенности — могут быть множественный аллелиз!''енетические компаунды. Разные мутантные аллели могут прояв*<
■^'^'ться фенотипически неодинаково (например, разные р-талассемиї
[ * * Кгіторьіе м у копол исахари дозы).^eнeтичecкиe компаунды, иногда неправильно называемые двой-
гетерозиготами, — это сочетание двух разных патологическі
одного локуса у индивида. Фенотипы генетических компас
отличаются от фенотипов гомозиготных форм по обоим унаіі
і^ііоуанньїм аллелям (например, фенотип при гемоглобинопатиі
отличается от фенотипов обеих мутантных гомозигот — HbSJНьсС).li^ля некоторых групп болезней генетическая гетерогенность про<
Ніется и на межлокусном, и на внутрилокусном уровнях (мукопо-і
бцхаридозы, гликогенозы).||>асшифровка гетерогенности генных болезней интенсивно про^
сается одновременно в клиническом и гснстическом напраї
1ях. Общая задача сводится к выявлению корреляции мєі
гипом и фенотипом.Анализ фенотипа (клинической картины болезни) — первый этап і
іифровке генетической гетерогенности. Чем точнее изучен фено«
тем больше возможностей в открытии новых форм болезней,
Ьлении изучаемой формы на несколько нозологических единиц*?
[: помощью классических клинических методов открыто несколь-
Іформ нервно-мышечных дистрофий, наследственных фор!
Ііиковости. Клинико-биохимическими методами разделен]
Іедственньїе несфероцитарные анемии, гемоглобинопатии, гли^
(нозы. Иммунологическими методами дифференцированы пер«
fjiiiiia 4. Генные болезни167ИИ'їньїс иммунодефииитные состояния. По результатам клинико-
ф|| тологичсских исследований описана гетерогенность гемофилии,
(Ііи-Ц)ІЮЙ слепоты.Исс перечисленные методы с некоторыми усовершенствованиями
N іі.іраклиническом плане и сейчас применяются для расшифровки
природы наследственных болезней.Л нал из фенотипа не должен ограничиваться организменным уров¬
нем. Перспективное направление — изучение клеточного уровня, т.е,
111ч лсдование клеток в культуре ткани (клеточная гибридизация,
Мім;іГ)Олическое кооперирование, физиологическая комплемента-
имя). Генетическая гетерогенность нескольких групп болезней была
И1К[>ыта с помощью методов культуры клеток (мукополисахаридозы,
Оіиісчни репарации ДНК).Генетические методы включают весь арсенал генетического ака¬
ли i:i болезни от применения клинико-генеалогического метода до
И'киенирования гена. Накопление родословных по какому-либо забо-
йеніпшю и их генетический анализ позволяют разделять ранее опи-
вйиную одну болезнь на реально существующие формы, если в этой
fpyiine встречаются мутации с доминантным и рецессивным типами
Ни следования. Так были разделены синдром Марфана (доминант-
:Цо1‘ наследование) и гомопистинурия (рецессивное наследование),
[Кмі'ющис сходную клиническую картину (высокий рост, подвывих
хрусталика, деформация грудной клетки). Разные типы наследова-
[Иии обнаружены во многих гетерогенных группах болезней (син-
[дром Элерса—Данло, мукополисахаридозы, витамин-О-резистентный
ІХИТ, амиотрофия Шарко—Мари).Изучение аллелизма рецессивных мутапий дало возможность
ijfCTaiiOBHTb гетерогенность наследственной глухоты (у глухих роди-
IftJieii рождаются нормальные дети). Возможности этого подхода в
Нйі^тоящее время уже ограничены.Наиболее полную информацию о гетерогенности клинической
[формы 5оле,зни дает применение современных методов анализа генов
[человека. Отнесение гена к одной или разным группам сцепления,
Локализация гена, его структура, сущность мутаций позволяют одно-
[ІНично идентифицировать нозологические формы.Концепция генетической гетерогенности генных болезней откры-
ІІЙСГ много возможностей в понимании сущности отдельных форм
и иричин их клинического полиморфизма, что крайне важно для
Прикгической медицины (правильная диагностика, выбор методов
168Клиническая генетиілечения, медико-генетическое консультирование). Если врач не пр1
нимает во внимание генетическую гетерогенность наследственш
болезней, то он может дать пациенту неправильные или неоправдаї
ные советы и необоснованные прогнозы.КЛИНИКА И ГЕНЕТИКА НЕКОТОРЫХ
ГЕННЫХ БОЛЕЗНЕЙНейрофиброматоз (болезнь Реклингхаузена)Это тяжелая полисистемная болезнь с аутосомно-доминантны!
типом наследования. Наиболее тяжело поражается нервная системі
поэтому болезнь считают неврологической. Термин «нейрофибрс
матоз» охватывает по меньшей мерс две болезни: нейрофибромя
тоз I типа и нейрофиброматозII типа, сначала считавшиеся
двумя формами одного и того же
заболевания (периферический
нейрофиброматоз и центральный
нейрофиброматоз). Это яркий
пример генетической гетероген¬
ности наследственных болезней.Ниже будет описан только ней¬
рофиброматоз I типа.Симптоматика нейрофибро-
матоза I типа разнообразна, в
патологический процесс вовле¬
кается несколько систем, что
подтверждает плейотропн ы й
эффект гена. Диагноз нейрофи-
броматоза Ї типа можно устано¬
вить при наличии не менсс двух
из перечисленных ниже призна¬
ков, но при условии, что они не
являются симптомами какой-либо другой болезни. Рис, 4.7. Типичные свстло-коричн<— Светло-коричневые пиг- вые пятна у молодого мужчины
ментные пятна (рис. 4.7). иейрофибромагозом I типа
|ци ^ Генные болезни169у детей их должно быть НС менее 5, а диаметр пятен не менее
S мм. У взрослых число пятен должно быть не менее 6, а диа¬
метр пятен не менее 15 мм. Для выявления пятен необходимо
хорошее освещение. Пигментные пятна появляются обычно к
^ годам жизни, их число увеличивается с возрастом. Они встре¬
чаются у 95% больных.Решающий признак — две нейрофибромы любого типа и более
или одна плексиформная нейрофиброма (по данным анамнеза
пли клинического обследования) (рис. 4.8). Нейрофибромы
могут возникать в любом участке тела, захватывая кожные
нервы, часто располагаются по ходу нервных стволов, иногда
}лхватывают крупные нервы и нервные сплетения (плекси-
()юрмные нейрофибромы), в месте локализации нейрофибром
больные часто ощущают зуд, жжение и боль. У одного больного
могут быть тысячи нейрофибром, а их масса может достигать15 кг и более, если своевременно ис сделано их иссечение.У детей таких нейрофибром
мило. Их число увеличива¬
ется с возрастом, особенно
у женщин при беременно-
с ги. к 30-летнему возрасту
пейрофибромы отмечают¬
ся у 95% больных.Множественные, похожие
па веснушки нигментные
пятнавподмышечнойямке
(рис. 4.9), паховой области,
на других участках тела со
складками. Они обычно
возникают в детстве, их
число трудно определить.Пигментные пятна обна¬
руживают у 80% больных.Костные изменения (дис¬
плазия крыла клиновид¬
ной кости, врожденное
искривление или утонче- Рис. 4.8. Многочисленные нейрофи-
ние длинных трубчатых бромы разных размеров у мужчины с
костей, ложный сустав). нейрофибромато^ом 1 типа
170Клиническая генетіРис. 4.9. Мелкие пигментігьіе пятна и
одно крупное пятно в подмышечной
области у женшины с неврофибро¬
матозом I типа. Видны также нейро¬
фибромы на шее и грудной клеткеРис, 4.10. Плексиформная орбитал!
ная нейрофиброма у ребенкаДисплазия глазницы сочетается с плексиформной нейрофі
бромой глазницы (рис. 4.10).— Глиома зрительного нерва. Эта опухоль характерна для не!
рофиброматоза I типа. С помощью КТ и МРТ (магнитж
резонансной томографии) обнаруживается утолщение зрі
тельного нерва. Глиома зрительного нерва протекает обыч!
бессимптомно, но иногда может вызывать ухудшение зренШ
косоглазие, зрачковые аномалии, проптоз и гипоталамическ}
дисфункцию.— Узелки Лиша (два и более) на радужной оболочке. Узелки пре;
ставляют собой гамартомные новообразования и не влияют
зрение. После полового созревания узелки Лиша наблюдаютс
практически у всех больных. Для обнаружения узелков гла;
осматривают с помош,ью щелевой лампы.— Нейрофиброматоз I типа по приведенным выше критерия*
у родственника I степени родства (родитель, сибс, потомок)
Нейрофиброматоз I типа относится к полностью пенетраіітині
la л 1<жные болезни171нуюсомно-доминантпым бoлeзия^f, поэтому нейрофиброматоз
I гина у родственника можно использовать как диагностиче¬
ский критерий.|||^ (и>.лі,іиинства больных диагноз очевиден уже к 3 годам.^Тг'И'иис заболевания прогрессирующее, с очень большим раз-
1Ш клинической картины. Наряду с описанными выше симпто¬
мі (они либо врожденные, либо зависят от возраста) у 20—30%
наблюдаются когнитивные нарушения (трудности в обучении).
(I0i>iicc опасными проявлениями становятся опухоли — иногда
июкачественности, иногда из-за места расположения (черепные
Uti. малый таз, ЖКТ). Неблагоприятное течение нейрофиброма-
і ипа отмечается у 30% больных.Ипряду с основными симптомами у больных нейрофиброматозом
1'П» исгречаются осложнения: низкий рост у 25-35% пациентов,
||il‘H(l>OpMHbie нейрофибромы у 20%, сколиоз у 10%, головные боли
|(№, мейрофиброматоз 1-ассоциированные злокачественные фут-
М11>1с опухоли периферических нервов у 7—12%, глиома зрительного
)Нп у 7%. К более редким осложнениям нейрофиброматоза 1 типа
«ісягся: эпилепсия у 1—2% пациентов, гидроцефалия у 2%, псев-
іріроч у 3%, стеноз почечной артерии у 1—2%, ксантогранулемы —
1%, (}>еохромоцитома — менее чем у 1%.Патогенез заболевания пока недостаточно изучен, посколь-
трудно объединить разнообразнейшие проявления: пигмент-
|в нятна, костные изменения, когнитивные нарушения и т.д.
нения могут возникать в любой системе организма, в состав
)рой входят ткани, происходящие из эктодермы, мезодермы и
>Ниой трубки. Фенотипические проявления нейрофиброматоза
|па значительно различаются даже у членов одной семьи. Однако
ICCTHO, что ключевую роль играют шванновские клетки, кото-
н результате соматической мутации теряют гетерозиготность.
как этот ген является геном-супрессором опухолей, то после
Гсри функции обоих алле.тей развиваются опухоли и заболевание
1ИИ(1)сстирует.Ьольные нейрофиброматозом Т типа встречаются в практике
ІЧСЙ многих специальностей. Пациенты обращаются в первую
>рсдь к дерматологам с жалобами на косметический дефект в
пигментных пятен и нейрофибром. Наиболее часто больные
Ірофиброматозом 1 типа являются пациентами невропатологов
172Клиническая генетти нейрохирургов. Определенная часть таких больных лечитсЯ)
хирурюв и ортопедов.Генетика нейрофиброматоза I типа на генеалогическом уро!
была ясна уже в начале прошлого столетия. В последние год
она изучена до уровня гена. Локус нейрофиброматоза I типа pi
положен в коротком плече хромосомы 17qll.2. Величина гена:?
350 ООО пар оснований, в нем 59 экзонов. Ген полностью секі
нирован. Он относится к группе генов-супрессоров опухолв(
чем и объясняются его опухолевые эффекты. Обнаружено бо;
500 мутаций (транслокации, делеции, вставки, точковые замен!
Однако корреляция клинической картины с типом мутаций еще
установлена. Также обнаружены гены-модификаторы, в настояі
время идет их изучение. Первичный продукт гена назван нейр<
фибромином.Распространенность нейрофиброматоза I типа равна примері
1:3500—5000 новорожденных, одинакова у обоих полов, у всех
и этнических групп. Аутосом но-доминантный тип наследован*
не нарушается ни в одной популяции. Доля спорадических случї
составляет 50—70%. Частота мутаций по этому гену высокая: 1 мут|
ция на 10 ООО гамсі' на поколение.Лечение нейрофиброматоза 1 типа в основном хирургическое
симптоматическое. В настояш;ее время идет разработка патогене!
ческой терапии (антифиброзные средства).Миотоническая дистрофияСинонимы: болезнь Штайнерта, дистрофическая миотош
Миотоническая дистрофия — аутосомно-доминантное многосистеї
ное заболевание с сильно вариабельной экспрессией гена, обусловлі
вающей клинический полиморфизм по началу заболевания и тяже(
течения. Главные клинические проявления: миотония, мышечш
слабость, катаракты, аритмии сердца, облысение со лба, нарушенш
толерантность к глюкозе, умственная отсталость. Мышечные суде
роги особенно выражены в руках, челюстях, языке (в виде фибри;
ляции). Одновременно постепенно усиливается мышечная слабое!
в связи с дегенерацией отечных мышечных клеток и атрофив
волокон. Миотония и мышечная слабость у пациентов сочетаются
нарушением речи и глотания. Начальные признаки миотоническс
дистрофии различны, Миотония сначала выявляется только п|
специальном тестировании. Мышечные подергивания и слабості
la <1 I (інньїе болезни173ІЧІІП асимметричны, в пер-
пчі-редь в патологический
ііч г вовлекаются лицевые
|Иіі»|ІІІ>]Є мышцы (миотони-
Itkur лицо), затем шейные,
fMrilMO. бедренные мышцы
■I II, 4.12).И п.кюяшсе время известно
^Иіііі миотонической дистро-
|К Миотопичсская дистрофия
иіікі составляет около 98%
ІІІПМИЧССК0Й дистрофии и
|ик 1г|п1зуется началом мышсч-
||.|Г>ости от дистальных
Him к проксимальным. При
іииіической дистрофии 2-го
ІЙ м 1.1 темная слабость разви-
Ш1. наоборот, от проксималь-
к чистильным отделам.Рис. 4.11. Врожденная форма миотони-
чсской дистрофии (мышечная гипото-
н и я, ам и м и ч н ое л и цо, рот треу гол І.ТІОЙ
формы — миотоническое лицо)взрослая пацисн гка с миотонической листрофисй (птоз, слабость
мышц, атрофия желательных мыши)
174Клиническая генетикНаряду с нервно-мышечными си\тптомами при миотоническс
дистрофии отмечаются катаракта (очень ранний симптом), гипого^
надизм (атрофия семенников), аменорея, дисменорея, кисты яичні
ка, облысение со лба, изменения проводимости сердтщ с аритмиеі
абдоминальные симптомы (на почве холелитиаза), прогрессирующал
умственная отсталость.Тяжесть клинических проявлений очень сильно различается даз
в пределах одной семьи.Начало миотонической дистрофии возможно от пренатальної
периода до возраста 50-60 лет. Выделяют 4 формы заболевания
зависимости от возраста манифестации): врожденную, юнопіескуі
классическую (20—30 лет) и минимальную (50—60 лет). Это обт
ясняется различиями в числе три нуклеотидных повторов в локус
миотонической дистрофии (см. ниже).Смерть при миотонической дистрофии наступает в возраст
50—60 леї (при классической форме) вследствие пневмонии, сердеч«(
ных осложнений или других интеркуррснтных заболеваний.Частота болезни может различаться в этносах и популяция)
Эффект родоначальника описан у канадцев французского проис*
хождения. В Израиле распространенность болезни в среднем ранні1 : 6369 с различиями между общинами: у евреев-ашкенази 1 ; 17 54<
у евреев-сефардов 1 : 5000, у евреев-йеменитов 1 : 2114. Обобщені
распространенность миотонической дистрофии можно оценить кг1 : 7500-10 ООО.Генетика миотонической дистрофии хорошо изучена на геш
логическом, формально-генетическом и молекулярно-генетической
уровнях, при миотонической дистрофии 1-го типа у пациенте
всех стран обнаружена одна и та же мутация в гене протеинкина!
зы мышечной дистрофии (символ гена DMPK), локализованном }
хромосоме 19ql3.2-19ql3.3. Суть мутации — экспансия (увеличен!
числа) нестабильных CTG-повторов в З'-нетранслируемой облает
гена. В норме число CTG-повторов колеблется от 5 до 30. При мш
тонической дистрофии этот показатель значительно увеличиваете
и составляет 50—2000 и более. Обнаружена корреляция между тяжб-j
стью и числом тринуклеотидных повторов. Чем больше повторої
тем раньше начинается заболевание и тяжелее протекает болезні
Клиническая картина у гомозигот более тяжелая. При миотоиичЫ
ской дистрофии 2-го типа найдена другая мутация — в гене цинке
Iflhil 4 Генные болезни175ыч іііільцев (ZNFP), локализованном в хромосоме 3q21.3. Мутация
Ijirmшпляет собой нестабильную экспансию CCTG-тетраплета с
^іігшірлми от 75 до 11 ООО.ІІІ) многих семьях с миотонической дистрофией в нескольких
іикшкчшях отмечается антиципация, т.е. более тяжелая мани-
иция болезни и начинающаяся в более молодом возрасте в
iHA.'iDM последующем поколении. Этот признак описан для мио-
hm и ческой дистрофии давно и рассматривался в 1940-х годах
ІИК ». іатистический артефакт. Однако сведения о молекулярном
Ігфгк'і'с указывают на возможность увеличения числа триплетов
поколениях.()||исаны семьи с более чем тремя поколениями с миотонической
1»к ірофией: в 1-м поколении — только катаракты, во 2-м поколе-
|Ии - умеренная слабость мышц, в 3-м поколении — врожденная
{^ормл.Мри миотонической дистрофии выражен импринтинг. Пациенты,
цжлспные от больных матерей, имеют более тяжелую форму болез-
|И I.- более ранни.м началом, чем пациенты, рожденные от больных
titoii. Врожденная форма миотонической дистрофии наблюдается
iJihKo у детей от больных матерей. Механизм импринтинга выяснен:
амлисия триплетов происходит в мейозе у женщин, а при сперма-
0U4IC3C она отсутствует.Уменьшение длины мутантного повтора (почти до нормы) у потом-
)it с легкой клинической картиной или бессимптомным заболева-
ІИСМ наблюдается при передаче гена от отца. Одним из объяснений
Ц'пго может быть селекция против длинных аллелей в мужском
їмсіогенезе.Івмейная гиперхолестеринемия'Оіа генетически гетерогенная аутосомно-доминантная болезнь
1И и и чески выражается в чрезвычайно высокой гиперхолестерине-
кИи. Следовательно, клинически это единая нозологическая форма.
іОолевание связано с наследованием мутантных генов, кодирующих
juciiTop ЛПНП, Ген локализован в хромосоме 19р13.2.Н настоящее время идентифицировано 6 классов мутаций рецеп-
»ри ЛПНП. В результате этих мутаций происходит иарущенис одной
следующих функций:• синтез рецепторов;
176Клиническая генетк• транспорт рецепторов из эндоплазматического ретикулума в кс
плекс Гольджи;• связывание ЛПНП рецептором;• кластеризация рецепторов в окаймленных ямках;• рециклизация рецептора (неспособность освобождать ЛПНП'
эндосомах);• доставка рецептора в базолатеральную мембрану (табл. 4.3).
Таблица 4.3. Генетическая гетерогенность семейной гиперхолестеринемииМутантныйK.iaccНарушенные функцииКлеточные структурыСинтез рецептораЭндоплазматический рети-
кулумТранспорт рецептораЭндоплазматический рети-
кулум, комплекс ГольджиСвязывание рецептора с
ЛПНПКлеточная стенкаКластеризация рецеп¬
торов в окаймленных
ямкахОкаймленная ямкаОсвобождение ЛПНП п
эндосомахЭндосомаДоставка рецептора в
базолатеральную мем¬
брануВторичная везикула, кле¬
точная стенкаМутации в гене АРОВ, кодирующем ароВ-100, могут выз1
вать гетерозиготный фенотип семейной гиперхолестеринемии (|
часто). Кроме того, обнаружен ген ARH, вызывающий гомозі
готный фенотип семейной гиперхолестеринемии, и ген PCSKi
определяющий гетерозиготный фенотип семейной гиперхолест<
ринемии. Однако эти ЛПНП-рецепторные гены редко бывают прі
чиной болезни, в справочнике ОМІМ указано еше много гено!
влияющих на обмен липидов.Таким образом, семейная гиперхолестеринемия — наглядны!
пример генетической гетерогенности наследственной патологш
Более того, по каждому классу мутаций, которые фактически явл*
ЮТСЯ отдельными нозологическими формами с генетической ТОЧК1
А I пнные болезни177ІИИ (разная этиология), обнаружены уже десятки мутантных
H'jt с патологическим действием.[липичсская картина полной (гомозиготной) формы семейной
^рколсстеринемии включает необычно высокую ги перхол естери-
ІНІ и появление уже в детском возрасте ксантом на коже, на сухо-
ІИИЧ. Ііозникает липоидная дуга на роговице. В период полового
ЇРИИНИЯ формируются атероматозное поражение устья аорты, а
||цс сісноз венечных артерий сердца, что проявляется систоличе-
|М шумом на аорте и ангиографически определяемым сужениемIII ;к)рты и стенозом коронарных артерий. Клинически развива-
ишимная картина ишемической болезни сердца. До применения
пчн'ппых методов лечения пациенты с семейной гиперхолестс-
<»?мисй внезапно умирали в возрасте до 30 лет в связи с острой
пиарной недостаточностью. Патологоанатомически обыаружива-
массивный атероматоз аортального клапана и восходящей части
порты. В крупных артериях наблюдаются сходные, но менее
їижоїіньїе изменения.Гижссть клинической картины и возраст, в котором развивается
ЯІН., определяются состоянием рецепторов ЛПНП. По этому био-
іичсскому признаку всех больных можно разделить на рецептор-II и иных и рецептордефицитных. Наиболее ранняя форма ишеми-
Ікой болезни сердца отмечается у рецепторнегативньтх гомозигот,
ipiicre до І0 лет у 65% из них развивается ишемическая болезнь
lita, 20% больных умирают в возрасте до 25 лет. У рецептордефи-
[•ных гомозигот ишемическая болезнь сердца развивается после
|Лс 1, а к 25 годам умирают 4% больных.Поражаемость атеросклерозом и ишемической болезнью сердца
обоего пола при гомозиготной семейной гиперхолестеринемии
інакова.Гетерозиготная форма семейной гиперхолестеринемии часто
Гмеюя невыявленной до взрослого состояния, пока не разви-
ІТСЯ сердечно-сосудистая недостаточность. У таких больных
іечаются гиперхолестеринемии, липоидная дуга на роговице,
Щтслазмы или холестериновые отложения в коже (рис. 4.13,
К ксантомы сухожилий (тыльная поверхность кисти, локте-
сустав, пяточные и коленные сухожилия), к 50 годам у 50%
|(Ж'1ин-гетерозигот по семейной гиперхолестеринемии развива¬
ем ишемическая болезнь сердца (на 20 лет раньше, чем в попу¬
178Клиническая генетіРис. 4.13. Липоилная дуга на рого-
пиие и ксантелалмы у 47-лстнего
мужчины с гетерозиготной формой
г и п е рхол е С1 ер и н с м и иРис. 4.14. Множественные ксаі
лазмы на ногах у мужчины с гомо!
гот ной формой семейной гиперхс
стеринемииляции). у жсншин-гетерозигот по семейной гиперхолестеринемі
первые признаки ишемической болезни сердца появляются
9-10 лег раньше, чем у мужчин.Лечение гомозиготных больных семейной 1’иперхолестеринемией<
необычайно трудная задача. Диета и лекарственные препараты неї
фективны. Эффективной мерой может быть плазмаферез с двухнеде
ным интервалом. Более радикальная мера — пересадка печени.При гетерозиготных формах семейной гиперхолестсрииемии пр^
меняют лекарственную терапию, направленную на снижение урові
холестерина, исключение факторов риска ишемической болезі
сердца (особенно курения), позже выполняют шунтирование.Распространенность обеих форм семейной гиперхолестеринемі
составляет в большинстве популяций 0,2%. В основном наблюдаеі
гетерозиготная форма. Частота мутантных аллелей по всем 6 класс
нарушения рецепторов ЛПНП равна 1 : 500, а гомозиготная форі
встречается с частотой 1 : 250 ООО или даже реже. Подробные сведен!
по диагностике семейной гиперхолестеринемии в России можно най1
на компакт-диске и в статье М.Ю. Мандельштама др. «Диагностиі
семейной гиперхолстеринемии в России; достижения и проблемы».Синдром МарфанаСиндром Марфана — наследственная аутосомно-доминантш
болезнь соединительной ткани. Синдром клинически идентифищ
ровал В, Марфан в 1886 г. Причиной синдрома Марфана являют(
А 11‘нные болезни179^йииіі н гене фибриллияа (локализация в хромосоме 15q21), Уже
ІМ'ігмп несколько типов мутаций (в основном миссенс), веду-
Ih к I трушен ИЮ синтеза фибрилл и на. Обнаружение связи гена
Іриіімина с синдромом Марфана дает возможность проводить
ІРКупярпо-генетическую диагностику, в том числе цренаталь-
И < иммтоматика синдрома Марфана многосистсмная и разноо-
І4ІІІІИ: от легких форм, трудноотличимых от нормы, до инвали-
ІИрУІОІЦСГО течения.||іиіія)лее специфичны для синдрома Марфана нарушения ске-
ІЧ.ИІИХ хрусталика, сердечно-сосудистые изменения, эктазия
|(Я1ПП мозговой оболочки.Мышечно-скелетная система; арахнодактилия, долихостено-
мс.пия, высокий рост, длинные конечности, деформация позво¬
ночника (сколиоз, груднойII орд 03, гиперкифоз), дефор¬
мация передней стенки
I рудной клетки (BflaBjrcH-
мая грудь, «куриная» грудь
или оба варианта), ненор¬
мальная подвижность сус¬
тавов (гиперподвижность,
врожденные контрактуры
пли оба варианта), плоская
сгона, высокое арковидное
исбо, недоразвитие верт¬
лужной впадины, мышеч¬
ная гипотония (рис. 4.15-
4.17).“ Глаза: вывих хрусталика,
миопия, отслоение сет¬
чатки, большая роговица,
удлиненная ось глазного
яблока, уплощение рого¬
вицы.- Сердечно-сосудистая сис-
гсма: аортальная регур-I итация, аневризма вос¬
ходя шей части аорты,
расслоениеаорты,митраль-Рис. 4.15. Два брата: слева — нор¬
мальный мальчик 10 лет; справа —
8-летний мальчик с синдромом
Марфана (подвывих хрусталика, в
связи с чем ребенок п очках и у ftero
наблюдается высокий рост, отсут¬
ствие подкожной клеічатки, ско¬
лиоз, деформация грудной клетки)
180Клиническая генетипая регургитация, заст<
ные сердечные наруї
ни я, пролапс митрально!
клапана, кальцификац^
митрального отверсти<
аритмия.— Наружные покровы; пах<
вые грыжи, атрофическ!
стрии.— Легочная система; спої
таниый пневмоторакс.— Нервная система; эктаз!
твердой мозговой обол(
ки, включая поясничнс
крестцовое менингоцеж
аномалии развития нері
ной системы.Система диагностических прі
знаков синдрома Марфана пр»
недена в табл. 4.4. Для иостаної
ки диагноза синдрома Марфаі
необхоііимо по меньшей мерс по одному главному критерию в ДІ
системах и одного малого — в третьей системе органов.Рис. 4.16. Кисти и стопы в норме
(слева) и при синдроме Марфана
(справа)Рис. 4.17. Диагностические кртерии арахнодактилии при синдром
Марфана
ІЦй 4 Генные болезни181INiiu 4,4. Диагностические признаки синдрома Марфана('ис'1'ема0|)1'НН0ВtliriK* 1'маяГлавные критерииМалые критерииКилевидная деформа¬
ция грудной клетки.
Воронкообразная деформа¬
ция грудной клетки, требую¬
щая хирургического лечения.
Отношение длины верхне¬
го сегмента тела к нижнему
<0,86 или размаха рук >1,05
Сколиоз (>20°)Ограничение разгибания
в локтевом суставе (<170°)
Плоскостопие
Протрузия вертлужной впа¬
диныВоронкообразная дефор¬
мация грудной клетки
Г иперподвижность
суставовВысокое нёбо и деформа¬
ция прикуса
Характерное лицоІИісльнаяДвусторонний подвывих
хрусталиков (экто1[ия хруста¬
ликов)Уплощение роговицы
Увеличение аксиального
размера глазного яблока
(миопия)Гипоплазия радужки или
цилиарной мышцыірдсчіго-^удистаяДилатация основания аорты
Расслоение восходящей аортыПролапс митрального
клапанаДилатация легочной
аортыКальцификация
митрального клапана
Дилатация или расслое¬
ние иных участков аортыиагельнаяНетСпонтанный пневмото¬
раксЖровная)Ж1|)НетАтрофические стриифдая моз-
III я обо-
|каПротрузия в областипояснично-крестцовогоотделаНет
182Клиническая генеттОкончание таблицыСистемаоргановГлавные критерииМалые критерииГенетическиепризнакиНаличие заболевания у роди¬
телей, детей или сибсов
Наличие мутаций в гене
фибриллина-1 (FBNI)
Наличие маркерного гапло-
типа ДНК, cцeплeFrFroгo с
сиилромом Марфана п семьеНетДиагностические критерии синдрома Марфана должны стр(
учитываться, поскольку за синдром Марфана могут быть принят
некоторые другие врожденные дисплазии соединительной ткан^
(наследственной или еше невыясненной природы),при несомненном синдроме Марфана у родственника I стеШ
ни родства этот диагноз можно установить у пробанда, имеющеі
проявления болезни в двух системах (органах) и более. Наибол*
специфичными проявлениями для диагностики считаются выв1
хрусталика, расширение аорты, расслоение аорты, эктазия тверде
мозговой оболочки.При синдроме Марфана ростовые скачки и закрытие зон рос
скелета наблюдаются на 2,4 года раньше у лиц мужского пола и
2,2 года раньше у лиц женского пола. Рост взрослых мужчин равеї
среднем І91 см, женшин — 175 см.Частота синдрома Марфана в популяции равна 1 : 10 000—15 0(
Популяционных и этнических различий в частоте и клиничесі
картине болезни не отмечено.Синдром Марфана — типичная аутосомно-доминантная болезі
хорошо изученная в клинико-генетическом плане. Клиническі
полиморфизм выражен очень ярко, но его причины неясны. Разниі
в клинической картине случаев, унаследованных от больных рОДНІ
телей, и спорадических случаев нет. С увеличением возраста оті
(особенно после 35 лет) повышается вероятность рождения ребен!
с синдромом Марфана.Синдром Элерса-ДанлоСиндром Элерса-Данло — гетерогенная группа наследственні
болезней соединительной ткани с разными типами наследовани)
Ійп л Генные болезни183(Ницими клиническими признаками: гипермобильность суставов,
М.І[ценная растяжимость кожи, скелетные изменения, повышен-
lit р;1ИИМ0СТЬ кожи, проявления со стороны внутренних органов,
яичное клиническое описание этого синдрома впервые было
^лтю СІЦЄ в 1657 г. голландским хирургом Д. ван Мекреном с зари-
ІКПІІ ІШЄШНСГ0 вида больного. Первая документальная фотография
ЛІ.КОЙ позировал, сильно растянув кожу на груди) относится к
I, (рис. 4.18). В России синдром подробно описал А.Н. Черногубов
1^1). назвав его генерализованным нарушением соединительной
iMit. Однако он не продолжил этих наблюдений, а за границей
ипубликован только реферат его работы. Позже этот синдром
описан Э. Элерсом (1901) и Х.А, Данло (1908) и назвал по имени
h ученых.С индром Элерса-Данло проявляется врожденной гиперрастяжи-
ІІ’іьіо соединительной ткани в связи с нарушениями синтеза кол-
М1ІІ, обусловленными мута¬
ции в разных генах коллагена
1|>угих белков экстраклеточ-
матрикса. Клинически,>кимически, молекулярно-
ІГП1ЧССКИ идентифицировано
ІІІОІІ синдрома Элерса—Данло,
fopi.ic с клинико-генетической
ІКІІ фения должны считаться
|()С I оятельными нозологиче-
|ми форма.ми. Их симптомы
їкрьіваются, поэтому целесо-
>щпо познакомиться со всем
1ПЛСКС0М признаков, встре-
)|цихся при различных типах
ІИ0Г0 заболевания.Кожа: сверхрастяжимость
(щеки, под наружными
концами ключиц, локти,
колени), бархатистость,
хрупкость, кровоточи-иость, темно-коричневые Рис. 4.18. Фотография больного с
веснушки (более 20), рубцы синдромом Элерса-Данло с вы ста п-
(множественные, типа ки(1885)
Рис. 4.19. Повышенная растяжи¬
мость кожи липа, рубцы на лбуРис. 4.20. Попг,ииенная растяз
мость кожи под ключицейпапиросной бумаги, келоидные), стрии в области пояснш
просвечивающие вены, расхождение послеоперационных щ|
(рис. 4.19, 4.20).— Суставы: пассивное разгибание мизинца на 90“ и более, щ
ведение большого пальца кисти к предплечью, псреразгибаї
локтевого сустава на 10° и более, псреразгибание колсннс
сустава на 10° и более, свободное касание ладонями пола
несогнутых коленях, переразгибаиие межфаланговых, запя(
ных, голеностопных и других суставоБ, привычный вый
суставов, плоскостопие (рис. 4.21, 4,22).— Глаза: птоз, избыточное развитие псриорбитальной клстчаті
отслойка сетчатки, остатки эпиканта, разрыв глазного яблокї~ Уши: сверхрастяжимость.— Зубы: частичная адентия, сверхкомплектные зубы, опалесі
руюпшя ^)маль, пародонтоз, множественный кариес,— Грудная клегка: сколиоз, кифоз, лордоз, плоская снина, вд2
лсние грудины.
Щ'і i (їННЬїе болезни185ЖIIнот; грыжи (пупочная,
1к‘лой линии, паховая, диа-
(ІФагральная), спонтанная
1к-р(1юрацил кишечника.
К(И1счности; варикозные
т-пы, подкожные подвиж-
мыс узелки на голенях,
плоскостопие.( срдие: пролапс митраль¬
ного клапана, аритмии,
иогсюсосудистая дисто¬
пия.внутренние органы; птоз
желудка, почек и матки.
Мозг; аневризмы сосудов
мозга, су б арах НОИ дальнее
кровоизлияние.('трем и тельные роды.
кИК видно из перечня сим-
при синдроме Элерса-
ию имеются нарушения (пср-
ІИІІІС или вторичные) во всех
1'Гсмах организма. Наиболее
ИІІ1ІС диагностические при-
|Ки: гиперэластичность кожи,
ІКожньїе узелки (сферулы),
(С прощупываемые на перед-
ноперхности голени; пере-
ІГиОание суставов; повышен-
! pin I и мость тканей; сим птом ы
»ррагического диатеза; про-
І* митрального клапана.
Клиническая картина разных
щи cиндpo^fa Элерса—Данло
;крывастся почти по всем
щтомам. Для дифференци-
Н<)й диагностики учитывают
ггания выраженности сим-Рис. 4.21. Псреразгибание V пальцаРис. 4.22. Псреразгибание коленного
сустава. Рубцы в области коленных
суставов
186Клиническая генеїПРОМОВ и осложнений. Для этого нужен опыт лечения бОЛЬН!
врожденными нарушениями соединительной ткани.с клинико-генетической точки зрения наиболее приемЖ
последняя классификация синдрома (1997), основанная в цві
на этиологическом принципе (табл. 4.5). Следует подчерки)
что клиническая диагностика синдрома Элерса—Данло ocj
няется из-за перекрывания клинических признаков разли'
типов данной патологии и других соединительнотканных 3J
леваний.Таблица 4.5. Классификация синдрома Элерса—ДанлоТип синдромаМолекулярный дефектТип наследовіІНКлассическийC0L5A1, COL5A2,
C0L1AIAy TOCOMHO- И
доминантный ЩГ ипермобильный?(C0L3AI,C0L1A1)Аутосомно- 1
доминантный 'jСосудистый (васкуляр¬
ный)COL3A1Аутосомно- 1
доминантныйЛртрохалазияподтип АC0L1A1Аутосомно- ^
доминантный ЧИОДТИП вC0L1A2Кифосколиотическийподтип АPLODIАутосомно- 1
рецессивныйподтип вДерматоспараксисADAMTS2Аутосомно-
рецессивный 1Основные клинические характеристики разных типов синдрс
Элерса-Данло представлены ниже.Классический тип. Основные диагностические критерии: гип<
растяжимость кожи, атрофичные («папиросные») рубцы, гипер*
бильность суставов. Кожа гладкая, бархатистая, тонкая. Атрофичі
рубцы развиваются на местах, наиболее подвергающихся механик
ским воздействиям (колени, локти, лоб). Патологическая подві
ность суставов проявляется повторными вывихами и подвывиха
А Iонные болезни187ми Ml (особенно часто плечевых, височно-челюстных и надколен¬
ім) Характерны сколиоз, плоскостопие, грыжи, опущение оиу-IIIм\ органов (гениталий у женщин и выпадение прямой кишки у
I) У беременных с классическим типом синдрома Злерса—Данло
ікіуіііегся преждевременный разрыв плодных оболочек, преждев-
РИІИ.ІС роды.і обильный тип. Этот тип более доброкачественный, чем
рИі'. и нем доминирует выраженная гипермобильность суставов.
flcMu.ic изменения отсутствуют. Кожные проявления относитель-
<рптчительны (растяжимость вариабельна), Атрофичные рубцы
Ірак гсрны. Наиболее характерны боли в суставах и мышцах. Они
іиіио іся в подростковом возрасте и носят хронический и тяжелый|КП‘Р.^’т'удистый (васкулярный) тип. Наиболее опасный тип синдрома
Н'11 Ланло в связи с разрывами стенок сосудов среднего и круп-
калибра, а также стенок полых органов (кишечника, матки,
Ми>ю пузыря). Чаше всего разрываются артерии среднего кали-
И случае внезапной смерти необходимо исключить синдром
Ipni Данло, особенно в семейных случаях. Отмечаются следую-
ломолнительные признаки этого типа патологии: артериове-
1ЫС каротидно-кавернозные фистулы, пневмоторакс, атрофия
лссен.фгрохалазия. При этой форме отмечается сильно выраженная
ірмобильность суставов с повторными вывихами или подвы-
ІЙМИ. кифосколиоз, врожденный вывих бедра, низкий рост,
»рмй является следствием кифосколиоза. Подтипы выде-
|Ы па основании молекулярно-генетических исследований
гибл. 4.5).ііКифосколиотический тип. Для этого типа характерна мышечная
i'1'оі-іия, задержка моторного развития, прогрессирующий сколиоз
ідсния. В последующем развивается кифосколиоз, из-за которо-
tocjie 20—30 лет больной теряет способность к самостоятельному
ІДІ1ИЖЄНИЮ. Характерным признаком является глазная патология:
>ыны глазного яблока, хрупкость склер, миопия, микрокорнеа.
1ТИПЫ клинически не различаются,
врматоспараксис («рвущаяся кожа^). Основные критерии диагно-
;и следующие: хрупкая, отслаивающаяся, «избыточная» кожа,
|Ылие грыжи (пупочные и паховые). Избыток кожи на лице напо¬
рист cutis laxa, но отличается тем, что для сиШ laxa не характер¬
188Клиническая генеткРис. 4,23. Структура пучка колла-
геновых волокон при синдроме
Элерса-Данло и в норме (правый
нижний угол). У больных волокна
неправильной формы, разных разме¬
ров и расположены неупорядоченноны синяки и хрупкость коз
Дерматоспараксис описан то;
у шести пациентов.Синдром Элерса—Данло
типичный пример разнолокз
ной гетерогенности. Все Л01
сы, мутации в которых ВЫЗЫ!
ют синдром, имеют отношен^
к синтезу белков волокнисті
элементов соединительной ТКІ
ни (главным образом кoлJ
гена). Коллагеновые волокі
имеют неправильную форму^
расположены неупорядочені
(рис. 4.23).Наличие синдрома Элерс*
Данло мало отражается
репродуктивной функции, хс
у больных снижено коли*
ство потомков. Имеются из
ляты с выраженным эффекте
родоначальника на протя:
НИИ нескольких поколений,
которых больные с СИНДРОМА
Элерса—Данло составляют К
всего населения.ФенилкетонурияКлассическая фенилкетонурия — аутосомно-рецессивная болезі
аминокислотного обмена. Фенилкетонурия клинически выделена^
самостоятельную форму А. Фелингом в 1934 г. Патологические щ
явления связаны с недостаточностью печеночного фермента фені
лаланингидроксилазы. Мягкая форма фенилкетонурии и доброї
чественная гиперфенилаланинемия обусловлены мутациями другі
генов, также затрагивающих обмен фенилаланина. С генетическс
точки зрения это самостоятельные формы.Недостаточность фермента ведет к нарушению процесса гидр<
силирования фенилаланина в тирозин. Вследствие этого пром
ходят накопление фенилаланина в крови (фенилаланинемия), обі
ihii Л. Генные болезни189іиіиіис избыточного количества
Mill іііировиноградной кисло-
когорая выделяется с мочой,(рушение формирования мие-
Иктой оболочки кокруг аксо-
}И м ЦІ 1C.Ді' I и с фенилкетонурией рож-
ІИііі'я адороиыми, но в первые
в связи с поступлением
Миі іаланина с молоком матери
І'ииінаются клинические про-
ілгіїмя: повышенная возбуди-
|Пі'11>. гиперрефлексия, повы-
МІИМЙ тонус мышц, тремор,|/ди|южные эпилептиформные
Ишалки, характерный «мыпги-
ІІІ» ;іапах. Позже отмечают
И'I псиную отсталость, микро-
(фалию. Поскольку наруше-
(С ибмена фенилаланина ведет
ІЧІИЖЄНИЮ уровня тирозииа,ІИО и;} фенотипических прояв-
Иіиіі фенилкетонурии — сни-
Мтс уровня или нрекраілснисїііювания меланина, поэтому уменьшена пигментация кожных
№|>о1юв, волос, радужной оболочки глаз (рис. 4.24). Гсчснис болезни
)<)! редиентное. Без лечения умственная отсталость может достигать
ІЖслой степени. Диагноз устанавливают на основании клинической
Іріииьі и результатов биохимического исследования мочи (фенил-
іровиноградная кислота) или крови (фенилаланинемия).Рииняя диагностика фенилкетонурии и профилактическое лече-
(искусственное вскармливание) иредунрежлаюг развитие клини-
ІСКІЖ картины болезни (см. главу П).Генетика фенилкетонурии хорошо изучена. Уже через юд после
ІИИИЧССКОГО описания болезни Л. Пенроуз доказал аутосомно-
Щсссивный характер наследования,Локус фенилкетонурии (фснилаланингидроксилазы) располо-
|Н и длинном плече хромосомы 12 (12q22—24). Ген секвениро-
И1. Для большинства семей сушествует возможность выполненияРис, 4.24. Больной феггилкетону-
рией. Слабая пигментация кожи,
полос, радужной оболочки глаз; уме¬
ренная степень олигофрении
190Клиническая генетиімолекулярно-гснсгичсской ііренаїальной диагностики и выявлещ
гетерозигот.Популяционная генетика фен ил кетой у рии, как и болыиинст!
аугосомно-редессиБных болезней, сложная. Частота фенилкетонурі
в болыпинстве европейских стран в среднем составляет, ію-видимоїI : 10 ООО новорожденных, в России 1 : 8000. Однако по этому показ
телю имеются значительные различия между популяциями: 1 : 26
в Турции, 1 : 4500 в Ирландии, 1 : 6000 в Белоруссии, I : ]6 ООО'
Китае, 1 : 30 ООО в Швеции, 1 : 119 ООО в Японии. Частота гетерозиг<
в большинстве европейских популяций составляет 1 : 100. Механизї
накосіления гетерозигот в популяциях неизвестны. Вероя'гно, эч
обусловлено селективным ііреимуіцеством гетерозигот, потому 41
гомозиготы, т.е. больные, без лечения ранее не оставляли потомства,а
доказательств повышенного мутационного процесса в соответствуї
щем локусе нет.МуковисцидозСиноним: кистозный фиброз. Это аутосомно-рецессивна|
болезнь, в основе патогенеза которой лежит нарушение транспорт
ионов хлора и натрия через клеточные мембраны. Ген муковисцидоз
детермиііирусі син'іез белка, называемого муковисцидозным транс
мембранным регулятором проводимости.Патогенез болезни обусловлен тем, что при отсутствии синтез
первичного продукта гена (трансмембранного регулятора) нарушает
транспорт хлоридов в эпителиальных клетках. Э10 приводит к избі
точному выведению хлоридов. Следствием становится гиперсекрещ
густой слизи в клетках эндокринной части поджелудочной железі
эпителии бронхов, слизистой оболочке ж КТ. Выводные протокі
поджелудочной железы закупориваются, слизь не выводится, обрг
зуются кисты (отсюда второе название муковисцидоза — кистозны!
фиброз) (рис. 4.25). Ферменты поджелудочной железы не поступаю!
в просвег кишечника. Гиперпродукция слизи в бронхиальном дсреї
ведет к закупорке мелких бронхов и последующему присоединениі
инфекции (рис. 4.26). Подобные процессы развиваются в придаточ¬
ных пазухах, в канальцах семенников. В потовой жидкости повышена
концентрация ионов натрия и хлора, это основной диагностический
лабораторный признак.Клинически болезнь проявляется в 4 формах (иногда в одной и]
той же родословной но несколько форм) с большим клиническим]
!1И(1 Л. Генные болезни191Рис. 4.26. Патогистологическая кар¬
тина легкого при муковисиидозе
(бронхоэктазы со слизистым воспа¬
лительным экссудатом в просвете)>л и морфизмом — от врожденных состояний до легких форм у
іросльїх.- Мекониевый и;[еус новорожденных — врожденная форма болез-
1и с избыточным заполнением кишечника густым меконием к
моменту рождения, в первые дни внеутробной жизни болезнь
проявляется признаками полной кишечной непроходимости,
которая трудно разрешается без оперативного вмешательства.
Врожденная форма встречается редко — не более 1% всех слу¬
чаев.- Кишечная форма начинается в раннем детском возрасте, часто
после перевода ребенка на искусственное вскармливание из-за
недостаточности панкреатических ферментов. Нарушение
пищеварения ведет к сниженному питанию, отставанию в
развитии, обильному зловонному стулу, светлому, с большим
количеством жира. Животу ребенка всегда вздут. Со временем в
192Клиническая генеткпатологический процесс вовлекается печень (жировая инфш
траиия, холестатический гепатит, цирроз). Частота кишечні^
формы составляет 5—10% всех больных муковисцидозом.— Бронхолегочная форма обусловлена гиперпродукцией вязке
секрета в бронхолегочной системе. Первые клинические щ
знаки появляются на фоне острой респираторной инфекщ
Вязкий секрет приводит к обструктивному синдрому, прис(
динению вторич ной инфекции. Рецидивирующий хрони ческі
инфекционно-воспалительный процесс осложняется ГНОЙ1
обструктивным бронхитом, тяжелыми пневмониями, В03Ї
кающими несколько раз в год. К вторичным изменениям от
сятся бронхоэктазы, эмфизема, пневмосклероз, легочное сер/
(рис. 4.27). в бронхиальном содержимом в основном выявляют
синегнойная палочка, золотистый стафилококк и гемофильн|
палочка, нередко в ассоциации. Ф;гора часто устойчива к ані
биотикам. Дети умирают от тяжелой дыхательной и сердечнунедостаточности. Брон?
легочная форма встречас
ся у 15-20% всех больн!
муковисцидозом.— Смешанная (легочно-і
шечная) форма — наибо;
распространенная (65—7f
всех больных муковисщ
дозом). При этом варш
те клинической кари
ны от.мечается сочетані
кишечных и бронхолегс
ных симптомов с разнс
выраженностью то одни|
то других.Тяжесть клинической ка|
тины зависит от типа мутаці
Также отмечен тот факт, что оді
и те же мутации могут приводі
к разной клинической картш
заболевания. В настоящее вре>
это объясняется мутация!
генон, модулирующих степеїРис. 4,27. Мукописцидоз. Легкое на
аутопсии (бронхоэктазы и пневмо¬
склероз)
|Э<1 4. Генные болезни193і|кіжсния легких (например, гены HLA класса II, фактора некроза
Іучімісй а (ФНОа), глутатион-5-трансферазьт Ml, трансформирую-
ми фактора роста Р| и др.).Иг нее мутации в гене муковисцидоза приводят к соответствую-
кіЕинической картине. Очевидно, что многие мутации нейтраль-
кик и по ряду других генов (глобины, рецептор ЛПНП и др.),
♦14 \х с тем установлено, что более 10 мутаций, не приводящих к
ІИІІИЧЄСК0Й картине муковисцидоза, способствуют развитию дис-
^мимированных бронхоэктазов неизвестной природы, цирротиче-
1ИЧ процессов 13 печени. Возможна также связь с эмфиземой легких.
Иі'розиготность по патологическим мутациям встречается в 2 раза
IliU' у больных хроническим панкреатитом. У 4% больных муковис-
ідіп диагностируется во взрослом состоявши.Прогноз при муковисцидозе всегда серьезный. Требуются при-
|лыюе внимание со стороны врача и большое терпение со стороны111 и СИ та (или родственников). Почти 50 лет назад, когда был опи-
|Н муковисцидоз, больные умирали в первые годы жизни. Сейчас
(»л»)лжительность жизни существенно увеличилась, в настоящее
;мя основными направлениями лечении больных с муковисци-
h(>M являются: назначение микросферических панкреатических
Ірмсиїов с рН-чувствительной оболочкой, антибиотикотерапия
тисе начало, длительное лечение, применение с профилактиче-
ЇЙ целью), прием урсодезоксихолевой кислотьт.<^\ Также в настоя-
|е 1$ремя активно ведутся разработки в области генной терапии
>{>левания и поиск веществ, способных стимулировать синтез,
ИісіЕорт и функции неполноценного кистофиброзного трансмем-
ІМІІОГО регулятора (CFTR) (например, фенилбутират стимулирует
кМФ-зависимый хлоридный поток (цАМФ — циклический аде-
пиимонофосфат)]. Благодаря такому подходу в развитых странах
'Последние годы отмечается рост числа больных муковисцидозом
фосткового, юношеского возраста и взрослых, что свидетельству-0 постепенной его трансформации из фатального заболевания
гского возраста в хроническую патологию взрослых. Средняя про¬
їж ительность жизни больных муковисцидозом в развитых странах
♦«чале XXI столетия составила 32 года, в России — 25 лет.
Диагносіика муковисцидоза основана на клинической картине,
Иулыатах биохимического определения ионов натрия и хлора в
ГС (натрий более 70 ммоль/л, хлор более 60 ммоль/л). По непонят-
ІМ причинам у 1—2% пациентов с муковисцидозом концентрация
194Клиническая генетіхлоридов в поте бывает нормальной. В затруднительных случаях
диагностики используют молекулярно-генетическую технолог
Для просеивающей преклинической диагностики муковисцидс
лучший метод измерение уровня иммунореактивного трипсі
в каплях высушенной на фильтровальной бумаге крови, коте
позволяет судить об активности трипсиногена. Разработаны спеї
альные наборы для такой диагностики. Однако по поводу массої
обследований на муковисцидоз есть две точки зрения: проводит1-й месяц жизни и не проводить совсем. Вторая точка зрения арі
ментируется тем, что раннее выявление муковиспидоза ничего і|
дает для больных. Лечение начинается по сушеству только при о(
стрении болезни, которое легко распознается клинически.Во многих западных странах проводится скрининг на мукоі
цидоз, и расходы на него считаются оправданными. В России CKJ
НИН г на муковисцидоз введен с 2006 г. в рамках национальнс
проекта «Здоровье». На 4-й день жизни ребенка в роддоме забира|
кровь на тест-бланк и передают (или пересылают) в лабораторі
Ответ о результатах первичного обследования поступает в лечебі
профилактическое учреждение к 9-10 дню. Подробности неонатг
ного скрининга на муковисцидоз см. также в гл. И.Генетика муковисцидоза (формальная, клиническая, молеї
лярная, популяционная) всесторонне изучена. Ген муковисцидс
локализован в хромосоме 7 (7q31—32), его размер составляет 250
пар оснований, ген включает 27 экзонов. Зрелая мРНК состоит,
6500 оснований, кодирует полипептидную цепь длиной 1480 а*
нокислотных остатков. Физиологическая роль и строение перві
ного белка (трансмембранного регулятора проводимости) хорої
изучены, что и позволило расшифровать многие стороны патогене
муковисцидоза. Экспрессия гена ограничена главным образом
телиальными клетками, она проявляется в наибольшей степени;
экзокринных железах (слюнных, поджелудочной и потовых), семе
никах и кишечнике. В эпителии легких ген функционирует, но слаС
и поэтому дефект хлоридного транспорта там четко выражен.В гене муковисцидоза обнаружено более 1500 мутаций, из ж
около 300 дают патологический эффект (миссенс, делеции, Н01
сенс, сдвиг рамки считывания, нарушения сплайсинга). Наибо;
частая мутация (до 70% всех случаев) — делеция трех пар основані
ведущая к отсутствию аминокислотного остатка в 508-м поло5
НИИ (отсюда ііазвание этой мутации: FSOSdet) полипептидной цепі
lUlt A. Генные болезни195i;if работ показано, что наиболее тяжелая и ранняя манифестация
пи ииния наблюдается у гомозигот именно по этой мутации.II OI рафи4сские и этнические различия в частоте муковисцидоза
цірікіптах мутаций гена муковисцидоза очень значительны.
Діїїіиьіе по частоте муковисцидоза в Европе представлены в
■1.6.'iHitii 4.6, Частота муковисцидоза по данным неонатального скрининга в
Европе (2007)РегИОТ!|йиіі;іііая ЧехияЧастота встречаемости, 1 на9100'иикобритания2700-2850ІІІИІІІИ2500-52004000-10 5004700lit іьіііа5000М К) касается частоты муковисцидоза в России, то величины сильно
ЛіМілются — от 1 : 4000-5000 (Приморский край, Томская область)I : 17 ООО (Северо-Запад). Это можно объяснить популяционными
цличиями или малыми выборками, с одной стороны, или, несовер-
IHL гвом скрининговых программ — с другой (на муковисцидоз они
Елсмы в России только в 2006 г.). Наиболее строгие критерии диа-
«ct ики и достаточные величины в выборках выполнены в МосквеII 500) и Северо-Западном регионе (1 ; 17 ООО). В любом случае
ІЖНО говорить о существенно меньшей частоте муковисцидоза в
ссии по сравнению с европейскими странами.Муковисцидоз редко встречается в восточных популяциях и у
>риканского чернокожего населения (1 : 100 ООО). Причина таких
Шуляционных различий неясна. Частота гетерозигот в Европе
ІСііь высока (до 5% населения), что можно объяснить селективным
їимуществом гетерозигот, в чем заключается это преимущество,
1C ПС выяснено. Не исключено, что гетерозиготы ио муковисцидозу
П'ойчивы к туберкулезу, в ряде популяций описан эффект родона-
ільника как причина высокой концентрации мутантных аллелей.Молекулярно-генетическая диагностика муковисцидоза и носи-
Л1>с[ ва соответствующего гена возможна для больщинсгва мутаций
Юсиове полимеразной цепной реакции (ПНР). Пренатальная диагно-
196Клиническая гежетика муковисцидоза вошла в широкую практику. Диагности4«
панель праймеров для 15-86 мутаций (из 200-300) позволяет
познавать 60-80% носителей мутаций среди населения евролеоі
расы. В России такие лаборатории есть в Москве, Санкт-Петер(
Томске, Ростове-на-Дону и Уфе.Адреногенитальный синдромСинонимы: врожденная вирилизируюшая гиперплазия коры
почечников; врожденная надпочечниковая гиперплазия; врождеї
дисфункция коры надпочечников. Адреногенитальный синдром
сится к группе наследственных нарушений биосинтеза стерої
гормонов. Как известно, процесс образования стероидов многосту^
чатый. Каждая ступень катализируется соответствуюш,им фермент
Известны, по меньшей мере, 5 разновидностей наследственных дефї
тов ферментов, обеспечивающих си нтез стероидов (21-гидроксилаза, ^
гидроксистероиддегидрогеназа, 11-р-гидроксилаза, 17-а-гидроксі20-а-холестеролгидроксилаза), а также транспортного St AR-проте
осуществляющего перенос холестерина на внутреннюю мембрану М1
хондрий, где начинается ферментативное превращение холестері
стероиды. Все варианты этих наследственных нарушений наследуі
по аутосомно-рецессивному типу.Наиболее распространена форма адреногенитального сиш
ма (90-95% всех случаев), обусловленная дефицитом ферї21-гидроксилазы (цитохром CYP21), катализирующего превращу
прогестерона в дезоксикортикостерон и 17-гидроксипрогестер(11-дезоксикортизол. Это заболевание очень разнообразно по клі
ческим проявлениям (более подробно об этом можно прочитать в!
тье Н,С. Осиновской «Врожденная гиперплазия коры надпочечні
(ВГКН) как дефицит аргидроксилазы: современные представлю
и генетическая диагностика» на компакт-диске. Известны два
сических клинических варианта этой болезни — сольтеряющі
простая вирильная формы.Сольтеряющая форма характеризуется полным дефицитом
мента и проявляется в нарушении солевого обмена (дефицит мі
ралокортикоидов). В патологический процесс вовлечена реї
альдостероновая система. Клиническая картина возникает в пері
дни после рождения. У новорожденного от.мечаются срыгива!
рвота, симптомы недостаточности периферического кровоо(
щения, сонливость, потеря массы тела. Обезвоживание вызы]
їй л. Генные болезни197Рис. 4.28. Вирилизация наружных
половых органов у девочки с адрсно-
генитальным синдромом1ы111С!1ную жажду, что про-
ІМІЧСЯ активным сосанием,
химическое исследование
iH'iuf i гипсркалиемию, гипо-
мню, ацидоз.Простая вирильная форма
іроиождастся проірессирую-
ііирилизацией, ускоренным
іимгісским развитием, повн¬
іш иі экскрецией гормонов
мадночечников. У новорож-
ІІМ'їх девочек при кариотипе
iXN отмечается маскулиниза-
1>;| 5ЛИЧНОЙ выраженности —
умеренной гипертрофии клитора (рис. 4.28) до полного срастания
Ми мошоночных складок с формированием мошонки и пениса,
чрсиние половые органы сформированы правильно по женскому
(у, У мальчиков вирильная форма адреногенитального синдрома
рождении обычно НС распознается (гениталии нормальные).
Иіюз устанавливают лишь на 5—7-м году жизни при появлении
Инііх признаков преждевременного полового развития.
^Исклассическая форма дефицита 21-гидроксилазы, или поздняя
»мл, вызвана снижением активности фермента на 40-80% по
Ііісішю с нормальны.ми значениями и клинически проявляется
кко в подростковом возрасте. У девочек наблюдаются умеренное
ІИЧСНИЄ клитора, раннее развитие молочных желез, более ран-
іакрьітие зон роста скелета, нарушение менструального цикла,
гу'гизм, аспе, у женщин ~ нарушение менструального цикла и
їлодие. Симптомом избытка андрогенов у мальчиков может быть
>рс!1ное закрытие зон роста костей, олигоспермия с последующим
<ением фертильности у мужчин, у детей обоего пола наиболее
|ТЫм симптомом неклассической формы заболевания является
|нее оволосение на лобке и под мышками (адренархе).Іиіснтная форма не имеет клинических проявлений, но в сыво-
сс крови умеренно повышен уровень предшественников корти-I, Некоторые авторы не выделяют такие состояния в отдельную
ЇМ у болезни.Средняя частота дефицита 21-гидроксилазы составляет 1 : 13 500
Юрожденных (в Швеции — 1 : 9800, в США — 1:15 981, в Европе —
198Клиническая генеї1 : 14 970, в Японии — 1 : 18 ООО), а в некоторых изолировані
популяциях она в 5—10 раз выше. В России частота адреногенитї
ного синдрома составляет 1 : 8000—1 : 10 ООО. Адреиогениталы
синдром относят к группе заболеваний, подлежащих просеиі
щей диагностике у новорожденных. Чем раньше установлен диап
адреногенитального синдрома, тем более аффективной оказывав!
терапия глюкокортикоидами и миі{ералокортикоидами, хотя не
проявления заболевания поддаются коррекции. Лучгие всего лечЩ
неклассическая форма. Болес целесообразно проведение прената
ной диагностики и пренатального лечения дефицита 2і-гидроксилі
в семьях, где оба родителя гетерозиготны и, как правило, уже име
больного ребенка. Это позволяет родителям решить вопрос о сої
нении беременности. Если беременность решено сохранить,
проводят внутриутробную терапию глюкокортикоидами (дево«
избавляется от хирургической коррекции іениталий). У мальчиї
своевременно диагностируется сольтеряющая форма, диагност!
которой в постнатальном периоде часто задерживается.Ген стероид-21-гидроксилазы {CYP21A2) локализован в короті
плече хромосомы 6 (6р21.3), это часть гена цитохрома Р450.
мутантные формы вызывают недостаточность 21-гидроксилазы.
разных клинических формах адреногенитального синдрома,
правило, имеются свои спектры мутаций. Однако причины ши^
кой вариабельности клинических симптомов адреиогениталы
синдрома полностью не ясны. Строгой корреляции между ген<
пом и фенотипом (сольтеряющая или простая вирильная фо|
нет. Одни и те же мутации, идентифицированные на молекуля|
генетическом уровне, имеют разные клинические проявления,
было проведено ДНК-обследование более 200 больных с адреної
нитальным синдромом с описанием степени вирилизации женсі
наружных половых органов; секреции андрогенов в ответ на стш
ляпию адренокортикотропным гормоном (ЛКТГ); реакции на б<
солевую диету путем оценки дефицита альдостерона и потери со;
Больные были разделены на 26 групп с полностью идентичны!
мутациями. Оказалось, что в половине групп фенотипические щ
явления и генотип не коррелируют.Однако неклассическая форма дефицита 21-гидроксилазы в 90%
чаев обусловлена двумя миссенс-мутация ми в гене цитохрома CYP1Среди причин клинического полиморфизма наряду с особен!
стями мутаций можно назвать также и модифицирующее влияі
ін 4 Генные болезни199пич гспов. Так, тяжелая сольтеряюшая форма адреногениталь-
шплрома ассоциируется с антигенами системы HLA: АЗ,
I/. DR7.Юдистрофия Дюшенна-Беккераho одна из частых форм многочисленных наследственных нервно-
М1И"И1ых заболеваний. Мышечные дистрофии характеризуются
щюссирующими дегенеративными изменениями в поперечно-
(Пніїїой мускулатуре без первичной патологии периферического
1'пікмірона.М иодистрофия Дюшенна-Беккера вызвана мутацией в гене, ответ-
ІГММОМ за синтез белка дистрофика. Этот белок находится в боль-
количестве в области сарколеммы, поддерживая, по-видимому,
lotшость мембраны (рис. 4.29). Структурные изменения в сарко-
iMt’ приводят к дегенерации цитоплазматических компонентов,
Ілсчіііому входу внутрь волокон, что вызывает гибель мио-
іПрилл.Ісіістически единая форма миодистрофии Дюшенна-Беккера
[Ниически разделяется на миодистрофию Дюшенна и миодистро-
ІИІ 1>окксра.4.29. Гистохимическая картина мышц в норме (а) и при миопатии
Ліа-ііна (б)
200Клиническая генетиіМиодистрофия Дюшенна встречается с частотой 1 : 5000 жив(
рожденных мальчиков. Генетически она относится к Х-сцепленні
рецессивным летальным нарушениям. Болезнь проявляется раї
Первые симптомы появляются в Bo:jpacTe до 2 лет: дети позднее начі
нают ходить, не умеют бегать и прыгать. Симптомы становятся вьц
женными в 2—3-летнем возрасте. Это изменения походки («утинаї
походка), лсевдогипертрофия икроножных мышц (рис. 4.30). Процеї
атрофии мышц постепенно приобретает восходящее направлені
мышцы бедра тазовый пояс -> плечевой пояс -> руки. Наблюдает
псевдогипертрофия не только икроножных мыпгц, но и ягодичный
дельтовидных, мышц живота, языка. У детей развиваются поясни*
ный лордоз, крыловидиость лопаток (рис. 4.31). Из наклонение
положения больные с трудом распрямляются, опираясь на колеї
(рис. 4.32).Атрофический процесс развивается и в сердце (кардиомиоі
тия). Острая сердечная недостаточность — причина летальніРис. 4.30. Псевдогииертрофия икро¬
ножных мышцРис. 4.31. Лордоз, псевдогииертрС
фия икроножных мышц, атрофі
бедренных мышц
ti«n;i 4. Генные болезни201|ічп,юв. Нарушается моторика
,К1. Обнаруживаются вторич-
|ыо изменения в костной систе¬
мі' Интеллект у больных детей
И) и жен. Корреляции между
Мжі'С'гью мышечного дефекта и
1‘іпісмью снижения интеллекта
|{>| 11а самой последней стадии
1|)1>(|)ия (слабость) захватывает11.|иты лица, глотки и дыхатель-
мышцы. Больные умирают
М 2 3-м десятилетии жизни.Из биохимических показате-
И» (1ЛЯ миодистрофии Дюшенна
|ци<>олее характерен резко повы-
ісмньїй (в 10—100 раз) уровень
инфосфокиназы в сыво-
ІНКС крови. Активность этого
фермента повышена в первые
|Ии ішеутробной жизни и, воз-
Южно, даже во внутриутробном
рриоде.при клинической картине
ІИодистрофии Дюшенна у дево-
|К следует исключить моносо-
|ИК) по Х-хромосоме (синдром
іриера). Редкая возможность
ІИолистрофии Дюшенна у
Мючек с кариотипом 46,XX не
Жлючается из-за инактива-
IHV1 Х-хромосомы с нормальным
ілелем во всех (или почти всех)
ЇС'і'ках на ранних стадиях раз-
Пин (16—32-клеточная бласто-
1ста).Гетерозиготные носитель-
ШЫ миодистрофии Дюшенна
»|'ут иметь субклинические
імиї'омьі; увеличенные икро-Рис. 4,32. Трудности при распрямле-
иии [юсле наклона
202Клиническая генвіножные мышцы, повышенную утомляемость при физичес!
нагрузке, изменения электромиограммы, увеличение уровня KJ
тинфосфокиназы в крови. Болес или менее выраженные симі
мы миодистрофии Дюшенна отмечаются у 70% гетерозиготі
носителей.Миодистрофия Беккера — доброкачественная форма нері
мышечных болезней. Частота миодистрофии Беккера у нової
денных мальчиков составляет 1:20 ООО. По клиническим симптої
миодистрофия Беккера напоминает миодистрофию Дюигенна,
в менее выраженной форме. Начало болезни не ранее 10—15течение мягкое, больные сохраняют работоспособность в B03fте 20-30 лет. Фертильность не снижена. Нарушения интеллект
кардиомиопатии не отмечаются. Активность креатинфосфокин!
заметно повышена, но не в такой степени, как при миодистрс
Дюшенна.Мягкая (или доброкачественная) форма миодистрофии Бекі
объясняется тем, что, в отличие от миодистрофии Дюшенна, КС
полностью прекращается синтез дистрофика, при миодистро(
Беккера синтез дистрофина детерминируется, но либо вырабат!
ется мало белка, либо продуцируется аномальный дистрофии.В настоящее время разрабатывается терапия миодистро(
Дюшенна—Беккера: генная (заместительная), клеточная (миоблг
стволовые клетки), аминогликозидная терапия, применение инп
торов протеосом и др.Генетика миодистрофии Дюшенна и миодистрофии Беккера хс
шо изучена (еще с 1930-х годов). Это типичный пример Х-сиеплені
рецессивного наследования. Ген дистрофина локализован в короті
плече Х-хромосомы, он уже клонирован и секвенирован. Это сам^
длинный ген из всех изученных. Б нем более 2x10^ пар основаї
(более 60 интронов); длина мРИК 16 ООО пар оснований. В связ
большой длиной в гене часто наблюдаются перестройки, ведущі
мутациям. Доля свежих мутаций среди всех случаев миодистро(
Дюшенна-Беккера составляет 30%. Поскольку ген секвенирої
то молекулярно-генетическая диагностика геми- и гетерозиготі
состояний возможна почти во всех семьях.Миодистрофия Дюшенна-Беккера в 60—70% случаев вызы!
ется большими делециями гена дистрофина (по меньшей М(
одного экзона). Дупликация больших сегментов обнаруживаеН
у 10% пациентов. Остальные 30% случаев обусловлены точкові
А I t;nHbie болезни203Ииіими (нуклеотидные замены и делении одного нуклеотида),
іт.н іся также о случаях малых вставок и делеций (2—5 пар
Іґп(іідок). Делеции в экзонах размером более 5 нуклеотидов
?Ч11И)1ся крайне редко.Ьмііііиіо был идентифицирован ген-модификатор миодистрофии
ген миогенного фактора-6 (MYF6), локализованный в хро¬
пи- 12q21. Мутации в этом гене совместно с легкими изменениями
ііін'ірофина ведут к тяжелой клинической картине миодистро-
(и клиническом полиморфизме мышечной дистрофии Дюшенна
1'Нк.жс п статье О.В. Напалковой и др. на компакт-диске).фом умственной отсталости с ломкой Х-хромосомой'и МОН им: синдром Мартина-Белл. Ранее эту болезнь называ-
jMiiiii>0M0M олигофрении с маркерной Х-хромосомой. Синдром
Иинл-Ьелл — одна из наиболее часто встречающихся (после
mm Дауна) форм умственной отсталости. Популяционная часто-
цГииісвания составляет 1:2000-5000 живорожденных. Больных
[И и ков в 2—3 раза больше, чем девочек, мальчики болеют тяжелее,
fniii носительства от 1:165 до 1:1540.Інсіиность больных неспецифична, но определенное диагности-
«П‘ значение имеют удлиненное лицо, высокий выступающий
MiiKpo- и долихоцефалия, гипоплазированная средняя часть
{особенно в сравнении с
Гуинющими и часто увсли-
1ЫМИ щеками), выступаю-
іюдбородок (рис. 4.33). Нёбо
ІИО дугообразное, губы тол-I, нижняя губа часто вывер-I, Оімечаются также увсли-
1ЫС оттопыренные ушные
)Мипы, большие кисти и
1М. Одно из типичных про¬
міни синдрома — макроор-
(»м. При рождении у детей
ІКИ нормальных размеров, но
|«с гуплеиисм пубертатного
<()да они существенно уве-
іипются в результате избы-
ІИОГО роста соединительнойРис. 4.33. Лиио подростка с синдро¬
мом Мартина—Белл
204Клиническая генетіткани и накопления жидкости. Однако половая активность таї
больных минимальна, к половой жизни способны единицы.Возможны повышенная растяжимость кожных покровов, слаб(
связочного аппарата суставов (что приводит к самопроизвольні
вывихам, обычно пальцев кисти), плоскостопие. У многих больн!
формируется пролапс митрального клапана. Все эти симптомы
словлены врожденной дисплазией соединительной ткани.Умственная отсталость, типичная для синдрома Мартина—Беї
как правило, умеренная или выраженная, хотя у Ю—15% больн!
обнаруживаются глубокая олигофрения и еще у такого же процеї
больных — мягкая умственная отсталость. Большинство пациент
социально адаптированы, выполняют несложную физическую рав
ту. Психологи оценивают их как контактных и доброжелательні
Лишь в тяжелых случаях таких больных приходится помешать в сг
циализированные интернаты. Неврологические симптомы включг
мышечную гипотонию и в 01 дельных случаях судороги.Пороки развития и нарушения других органов сравнител!
редки; это расщелины нёба, нистагм, страбизм, птоз, катаракт
кривошея в сочетании со сколиозом, кифоз, дефект межпредсерднв
перегородки.Помимо определенного комплекса клинических аномал!
синдром Мартина—Белл сопровождается характерной цитогене
ческой картиной: ломкостью в дистальной части длинного
Х-хромосомы (в зоне Xq), что внешне напоминает «спутник*- длі
ного плеча (рис. 4.34). Эта ломкость выявляется лишь при кулыliMfРис. 4.34. Ломкая Х-хромосома при синдроме Мартина-Белл; слева — наб
Х-хромосом женшины (одна хромосома ломкая); справа — мужской наС
(ломкая Х-хромосома)
іи 4 Генные болезни205»ІИ1.ІІІИИ лимфоцитов в условиях дефицита фолиевой кислоты,
Мому для обнаружения ломкости нужно либо использовать куль-
)Н'11.11Ые среды, лишенные фолиевой кислоты, либо вводить вІІ.І уральную среду антагонисты фолиевой кислоты. Однако даже
|Н1 л их условиях ломкость Х-хромосомы выявляется не во всех
и К.ІХ (до 60%).Диш о считали, что синдром наследуется по Х-сцепленному рецес-
|ит>му типу. Однако в родословных отмечались случаи тяжелойIII и у женшин и легкой у мужчин, у женш,ин-гетерозигот отме-
1и некоторое снижение интеллекта, чаше оцениваемое как погра-
|Ми;н1 умственная отсталость. У некоторых из них в небольшом
>jmi‘ine клеток обнаруживали ломкую Х-хромосому. Таким обра-I, ниследование синдрома Мартина-Белл не укладывалось в стро-
рпмки Х-сцепленного рецессивного наследования. Более того, в
*1К' 'К)вных отмечалась антиципация. Этиология этого заболевания
ііі.іяснсна с помощью методов молекулярно-генетического ана-
|1и Обнаружена экспансия нестабильных тринуклеотидных повто-
{( GG) в 5’-нетранслируемой области гена FMR1 (fragile mental
rnUuion). В норме в этом гене число повторов варьирует от 6 до
Чистота экспансии премутационных триплетных повторов в гене
ІНІ и полную мутацию в оогенезе является функцией длины пре¬
йти онного аллеля, который имеется у гетерозиготной женщины
4.35). Хромосомы, в которых имеется 50-200 повторов, считают
)м утаи ней. Для этого состояния характерны незначительные про¬
їси и и; задержка развития, признаки аутизма, атаксия. В следую-
моколении число повторов может увеличиться (экспансия) до
)11 и более, что и обусловит выраженную клиническую картину,
іисяні^ую от числа повторов. Огромное влияние оказывает также
(Т(>>иновое метилирование повторов. Такое состояние называют
лмой мутацией. Соответственно если женщина унаследовала боль-
число повторов, то она будет больной. Обнаружены еще два
імдрома (FXTAS (синдром тремора и атаксии, ассоциированный с
ІМКОЙ Х-хромосомой) и FRAXE (синдром ломкой Х-хромосомы)],
условленные динамическими мутациями в локусе Xq27.3, но эти
и»1 {FMR J и FMR2) являются причиной болезни намного реже. При
ом клиническая картина сходна с синдромом Мартина—Белл, но
идентична.Диагноз ставят на основании клинической картины и по резуль-
Гим клинико-генеалогического и цитогенетического исследований.
206Клиническая ген<56-59 еО-69 70-79 60-89 90-99 >100
Число повторов в премутационном аллеле гена FMR1Рис. 4.35. Зависимость экспансии триплетных повторов в гене FMR/отдлі
прсмутационного аллеля у гетерозиготной женщиныНаиболее точный метод — молекулярно-генетическая диаі ностш
том числе с использованием методов оценки метилирования, ?В настоящее время возможна пренатальная диагностика дані
синдрома. Этиотропной терапии пока не существует.ЭПИДЕМИОЛОГИЯКлинико-генеалогические наблюдения свидетельствуют о С1
дартном или своеобразном поведении генов в семьях, что и
сматривалось в предыдущих главах. Эти, безусловно, важные сі
ния должны быть дополнены знанием судьбы генов в популят
реальных закономерностей их распространения среди населеї
Это особенно важно в настоящее время, потому что происх<
интенсивные социальные процессы, с которыми человек ранее п(
не сталкивался: массовая миграция населения, ломка этничес!
национальных, религиозных, классовых, географических грг
браков, увеличение численности населения планеты (неравноме|
в разных популяциях), планирование деторождения (сокращв
числа детей), улучшение медицинской помощи. Все эги факт
4 I пмные болезни207II іипіяіь на распространенность генных болезней в ближайшем
#fl)U4iiioM будущем.Ииьіі-миология генных болезней включает сведения о распро-
(П1 мости этих болезней, частотах гетерозиготного носительства
их обусловливающих. Первичная осіюва возникновения
ІРІІІ I исмньтх болезней — мутационный процесс, а распространен-
fvdiic жей (или частота больных) в популяции определяется уже
І^лніїїіпннмми закономерностями: интенсивностью мутационно-
Ipnuccciu давлением отбора (скоростью элиминации), который
плодовитость мутантов и гетерозигот в конкретных усло-I рі'дьі, миграцией населения, изоляцией, дрейфом генов.
Ійіііюлее объективная оценка распространенности наследствен¬
ен ис ж ей в разных популяциях — определение их частоты среди
р<»ждснных, включая мертворожденных, В последующем частотаI' наследственными болезнями меняется в связи с повышенной
гипс гью в раннем возрасте. Смертность детей от генных болезнейII различаться в разных странах и в разные годы, поскольку этот
11111ель зависит от многих факторов (уровня медицинской помо-
социалыю-экономических условий, культурных традиций и т.п.).
і к II частоты определенных форм наследственных болезней средиIX контингентов (в детских больницах, в специализированных
їйх-интернатах для инвалидов, на амбулаторном приеме и т.д.)
||4Ич> отношения к эпидемиологии генных болезней не имеет.
fH) оценку можно использовать только для косвенных расчетов
Ipoc'i раненности болезни. Социальное перераспределение больных
М1ЫС группы населения, безусловно, зависит от экономического
|нн, религии, государственного устройства и других факторов.
L4»H3H с большим числом нозологических форм генных болез-
их редкой встречаемостью, неполной клинической и патолого-
ft)M и ческой диагностикой наследственной патологии данные по
фос граненности наследственных болезней в целом еше отрывоч-
Однако по формам, по которым проводятся массовые диагно-
(секис или профилактические программы, собран убедительный
іриал для суждения об эпидемиологии генных болезней.^1иая частота новорожденных с генными болезнями в популяциях
ЮМ составляет примерно 1%, из них с аутосомно-доминантным
>м наследования — 0,5%, с аутосомно-рецессивным — 0,25%,
;иемленным — 0,25%; Y-сцепленные и митохондриальные болезни
їчаются крайне редко. Распространенность отдельных форм болез-
208Клиническая генеті*ней колеблется от 1:500 (первичный гемохроматоз) до 1 : 100 ООО и низ
(гепатолснтикуляркая дегенерация, атаксия-телеангиэктазия и др.).Распространенность генной болезни условно можно считать выс<
кой, если 1 больной встречается на 10 ООО новорожденных и чг
средней — 1 : 10 000—40 ООО, низкой — очень редкие случаи. В груї
пу распространенных входит не более 15 генных болезней, но 01
обусловливают почти 50% общей частоты больных с наследственнс
патологией. Данные по распространенности некоторых наиб<
изученных генных болезней представлены в табл. 4.7.Таблица 4.7. Распространенность генных болезнейБолезньРаспространенностьТип наследования іПервичный гемохроматоз1:500А-Р 1Неполипозный рак тол¬
стой кишки (синдром
Линча)1:200-1:2000А-Д ^Умственная недо¬
статочность с ломкой
Х-хромосомой (синдром
Мартина—Белл)1:1250 (мальчики) 1:2500
(девочки)Х-сцепленный SМуковисцидоз1:3000-1:10 000А-Р "1Нейрофиброматоз1:4000-1:5000А-ДСпинальная мышечная
атрофия1:6000А-РМиотоническая дистро¬
фия1:8000-1:10 000А-Д jМиопатия Дюшснна—
Беккера1:3000-1:5000 (маль¬
чики)Р-Х-сцепленный ^Синдром Элерса-Данло
(все формы)1:5000-1:10 ОООА-Д, А-Р, Р-Х- 1
сцепленный JСиндром Марфана1:10 000-1:15 000А-ДФенилкетонурия1:10000А-РАхондроплазия1:100 ОООА-Д fПримечание. А — аутосомный, Д — доминантный, Р ~ рецессивный.Как видно из табл. 4.7, распространенность разных форм генні
болезней сильно различается независимо от типа наследованиі
Причины таких различий во многом неясны. Распространенное!
1Й<1 4. Генные болезни209IMIII.IX болезней определяется частотой вновь возникающих мутаций
M1KJ10M детей у больных родителей и гетерозигот. Биологические
^11ои1>1 распространенности доминантных и репессивных болезней
рил.1) паковые.Рлсиространенность многих доминантных болезней определяет-
п основном новыми мутациями. Практически все доминантные
(иг 111 и ведут к снижению фертильности, а некоторые — даже к сте¬
пи.нос ги. Репродуктивная функция у больных в целом снижена по
4ІГІ10І ическим или социальным причинам. Так, только 10% случаев
Ипияроплазии относятся к семейным. Семейные формы нейрофи-
іома ї'оза I типа составляют 50-70%. Снижена репродуктивная спо-
(Юмость при синдроме Марфана. Поздно начинающиеся аутосомно-
«мииантные болезни (хорея Гентингтона, болезнь Альцгеймера) не
Нрпжаются на репродуктивной способности (число детей). К началу
ними и (после 35—40 лет) деторождение уже заканчивается.Распространенность рецессивных болезней определяется частотой
11с|н>зигот в популяциях. Популяционные закономерности распре-
їлсиия генов и генотипов таковы, что частота гетерозигот во много
I выше частоты гомозигот по мутантному аллелю.Накопление гетерозигот в популяциях обусловлено их репро-
|ук1ивным преимуществом (оставление потомства) по сравнению
гомозиготами по нормальному и патологическому аллелям. Если
Іріпімущсства никакого нет и не было в истории популяции, то
|*си)та гетерозигот, казалось бы, должна приближаться к частоте
іугаиионньїх событий (равное число появляющихся и элимини-
^кицихся мутаций). На самом деле по всем рецессивным мутациям
)храняется повышенная частота гетерозигот, как бы подхватывае¬
те отбором для размножения. Популяции не только человека, но
нссх живых существ отягощены грузом рецессивных мутаций. Эта
ііцсбиологическая закономерность была открыта русским генети-
*)м С.С, Четвериковым в 1926 г.Распространенность генных болезней в общем плане определяется
Юнуляционнымй закономерностями поведения генов (см. ниже).|t Мутационный процесс — одна из биологических характеристик
Мк>6ого вида. Он постоянно протекает у человека в зародыщевых
№ соматических клетках и является основой возникновения и под-
(дсржаиия генетического разнообразия человека. В то же время это
[ Черничный источник наследственных болезней. По разным оценкам,
і Чистота возникновения мутаций (спонтанный уровень) у человека
210Клиническая ген(ориентировочно составляет 1x10“^’—1x10“^ гена на поколение,
мутационные события в каждом гене достаточно редки. Лиш1
нескольких генах мутации возникают с повышенной частотой (І!
10^ гамет). Эти гены отличаются от других необычайно больші
размерами (360 ООО пар оснований в гене нейрофиброматоза и 2х1(
в гене миопатии Дюшенна—Беккера).Таким образом, текущий мутационный процесс на генном урої
в одном поколении не может обеспечивать наблюдаемую высок!
частоту патологических аллелей в популяциях. По приблизителы
косвенным оценкам (а точные прямые пока невозможны) обі
вклад мутационного процесса в распространенность наследствен!
болезней составляет около 20%.Отбор в любых популяциях обусловлен дифференциальной см<
тностью и плодовитостью особей с разными генотипами, чт<
приводит через какое-то число поколений к различной концент
ции аллелей в популяциях. Поскольку отбор (его направленності
интенсивность) тесно связан с условиями окружающей среды, на;
основе возникают разные концентрации аллелей в различных пс
ляциях. Элиминация или преимущественное размножение зави<
от дифференциальной приспособленности гетерозигот, нормалы
или мутантных гомозигот к условиям окружающей среды.В качестве примера этой закономерности можно привести
торы распространения аутосомно-рецессивных гемоглобинопат)!
(серповидно-клеточная анемия, талассемия, аномальные гемоп
бины — гемоглобин с, и и Е) в регионах с высокой заболе
емостью малярией. Малярия у гомозигот по гену нормальне
гемоглобина сокращает жизнь и репродукцию у здоровых лю^
Гомозиготы по мутантным аллелям (больные гемоглобинопат!
ми) умирают от наследственной болезни, не оставляя потомст
Гетерозигогы выживают, потому что малярийный плазмодий
поражает людей с аномальным гемоглобином, а сама гетерозиг<
ность не вызывает патологического процесса в нормальных ус
виях. Преимущественное размножение гетерозигот приводило^
высокой частоте носительства гена среди населения в прошлс
<«малярийных» регионов (Юго-Восточная Азия, Африка, Италі
Греция, Кипр, Азербайджан, Узбекистан). Такая частота сохран^
ется и до настоящего времени.Отбор, безусловно, продолжает действовать и сейчас в поі
ляциях человека не только во внутриутробном, но и Б пості
INn 4. Генные болезни211|Л1<1м)м периоде онтогенеза, несмотря на социальную и меди-
Инкую помощь больным, в детском возрасте умирают больные
I аііглиозидозом, миопатией Дюшеіша, с моногенными врож-
ІИМММИ пороками развития. Репродукция понижена у больных
ик|)илией, ахондроплазией, нейрофиброматозом 1 типа и т.д. В
жг }фсмя необходимо обращать внимание на снижение давле-
|н оібора в современных условиях (разное для разных болезней),
Ипрос в популяциях человека идет двумя путями. Во-первых,
(учшсние медицинской и социальной помощи больным (особенно
ІЧгііис наследственных болезней) приводит к тому, что гомозиготы
тример, больные фенилкетонурией, муковисцидозом), ранее не
итавшие до репродуктивного периода, теперь не только живут
М) лет и более, но и вступают в брак, имеют детей. Следовательно,
JIIV ІЯЦИИ пополняются гетерозиготами по патологическим генам,
'июрых, планирование семьи (произвольное сокращение рож-
ІМОСТИ, чаще всего 1—2 или 3 ребенка) изменяет действие отбора
}ии ж С репродуктивной компенсацией.Суіь этого явления в современных популяциях заключается в
рлуюшсм. Наследственно отягощенные супружеские пары, у кото-
|к повышена смертность детей из-за наследственных болезней, в
Цу;п>тате большего числа беременностей по сравнению с таковыми
іислсдственно неотягощенньтх пар «достигают» того же числа детей,
m положение иллюстрирует табл. 4.8, составленная по даннымII, Ьольшаковой (1986).Популяционные последствия репродуктивной компенсации оче-
1ЛМ1>|, хотя развиваться они будут медленно (десятки, а для нско-
5ЫХ генов сотни поколений). Патологические аллели в этих слу-
|йх будут иметь большую вероятность сохранения и увеличения
гтоты, чем при естественной репродукции индивидов с разными
ІНОІ ипами.}ли11.а 4.8. Репродуктивная компенсация п разных популяцияхЧисло беременностейЧисло детей в семьяхПопуляциянеотяго-отягощен¬неотяго-отягощен¬шенныхныхщенныхныхрусские:'продские жители2,13,72,12,2Іїльские жители3,66,03,63,4
212Клиническая ген(Окончание таблицы 4Число беременностейЧисло детей в семьях^Популяциянеотяго-отягощен¬неотяго-отягощеі1 шенныхныхщенныхныхСреднеазиатские:Изолированныепопуляции6,29,75,86,1 ■Неизолированныепопуляции5,89,55,35,4Дрейф генов — это случайное повышение частоты какого-то aj
ля в результате нескольких совпадающих событий, имеющих стол
стический характер (соответствующий брак, большая семья, yi
следование детьми патологических генов, снова «подходящие^» брг
этих детей, хорошее материальное положение и т.п.). Это явлсі
наблюдается для редко встречающихся признаков (или болезне
которые не отметаются отбором. Благодаря дрейфу генов патологи*
ские гены могут долго сохраняться в роду или в небольшой популії
ции, особенно в изоляте (популяция из 500—1500 человек, в КОТОІ
практически отсутствует миграция).Неравномерное распределение патологических генов в популящ
ях, а точнее, их высокие частоты, могут быть обусловлены так наз|
ваемым эффектом родоначальника. Это явление по популяционі
генетическому смыслу близко к дрейфу генов — накоплен!
какой-либо генной болезни (или многих), унаследованной от одн<й|
или нескольких индивидов, переехавших в другое место. Xopol
документированных историческими материалами примеров зффеї
родоначальника в генетике человека уже много.В XVII в. иммигранты из Европы (Голландии, Дании, Гсрмани!
прибыли в Южную Африку (современная ЮАР). Среди них бы^
носители генов порфирии (мягко текупісе аутосомно-доминантиС
заболевание), хореи Гантингтона (аутосомно-доминантная болезнь
поздним началом), семейного полипоза толстой кишки (аутосомнс
доминантная болезнь с поздним началом), липопротеиноЗ
(аутосомно-рецессивная болезнь). Семьи иммигрантов были болі
шими (более 10 детей), поэтому людей с этими болезнями в Ю>
теперь во много раз больше, чем в Голландии и Дании. Родословна1|
лиц с аутосомно-доминантными болезнями прослеживается до одш
го брака иммигрантов, а ген липопротеиноза — до брата и сестрі
|я^ I онные болезни213КМ.ііиііих D 1652 г. в теперешнюю ЮАР. Они, их дети и виуки имели
семьи, что и способствовало увеличению частоты этого
|Р111ІМІ10Г0 гена.'Кні;иіский иммигрант, прибывший в Южную Африку, имел
|ги 0(1 страдал аутосомно-доминантным заболеванием ~ дис-
міі'іі костей и зубов, вызывающей полную потерю зубов к
и передал этот ген 70 из 356 прослеженных потомков в1И»КО..'1СГ!ИЯХ.М (11 |;ме Пенсильвания (США) живут изолированно амиши (пере-
ІРІІІІМ из Европы), переехавшие туда в XVI11 в. В 60-х годах XX в.
И ікїсслении обнаружены 82 человека с аутосомно-рецессивной
инло (карликовость и 6 пальцев), все эти люди — потомки одной
іружеской пары.ч к'с гйенно, имеется тенденция к элиминации патологических
он 1м популяций путем естественного отбора, поэтому эффект
|оміі'ки]ьника сам по себе не может объяснить долгое существова-
нлтологического гена в популяции.ІІПІ радия населения по регионам или странам с оседлостью
(оном месте — теперь уже неизбежный спутник многих соци-ІІ.ІХ процессов (беженцы, переезд в поисках работы и т.д.).
р;п1ия может отражаться на эпидемиологии генных болезней.
ІН уменьшает или увеличивает частоту носителей патологи-
}ких генов в «донорских» или «реципиентных» популяциях
Um> до эффекта родоначальника или уравнивания частот в
^Пуляниях.Кровнородственные браки имеют особенно большое значение в рас-
)СГ]Х)ненности рецессивных генных болезней. Такие браки встре-
)тся R западных странах с частотой около 1% на уровне двоюродных
li.eit и сестер. Однако в ряде этнических групп частота кровнород-
1СИИЫХ браков на уровне двоюродных родственников составляет
и даже до 30%, если включается троюродный уровень.Н кровнородственных браках существенно повышается вероятность
<дспия потомства, гомозиготного по патологическому рецессивному
|у, по сравнению с таковой в потомстве от неродственного брака.
4()гис редкие рецессивные болезни встречаются в основном у детей от
)1тородственных браков (табл. 4.9). Более того, пос гнатальная смерт-
iJTi) существенно выше у детей, рожденных в кровнородсгвенных
ІК1ІХ (рис. 4.36). Особую опасность представляют инцестные браки, т.е.
<ки между родителями и детьми или \тежду братом и сестрой.
214Клиническая ген<0 12 3 4 5 6
Возраст, годыРис. 4,36. Постнатальная смертность у двоюродных братьев и сестер в
ственных и неродстпенных бракахТаблица 4.9. Частота редких аутосомно-рсцессивных болезней в ЯпонииЧастота бо.іьньїх среди потомства отБолезньнеродственныхбраковбраков между двоюродны1|
родственниками ,Альбинизм (общий)1:40 0001:3000Микроцефалия1:77 ООО1:4200 іГеиатолентикул яркая
дегенерация1:87 ООО1:4500Акаталазия1:360 ООО1:9600 1Врожденный ихтиоз1:1 ООО ООО1:16 000Вариации распространенности генных болезней (этнические, ге
графические, популяционные) подтверждены во многих иссле;
ваниях. Выше были рассмотрены факторы, влияющие на pacnj
страненность генных болезней. Каждый из них может действої
отдельно в конкретной популяции в отношении конкретной НОЇ
логической формы или действует в комбинации с другими фак1
рами. Накопилось много доказательств того, что распростране!
ность множества генных болезней различна в разных популяциі
Высокая частота редких генных болезней в отдельных этничес!
группах объясняется эффектом родоначальника и дрейфом генов]
изолированных популяциях. Отбор в таких случаях имеет мены
значение.
А I f'HHbie болезни215ііииіицс 4.10. приведены примеры высоких частот наследствен-
Пії.кчисй в связи с длительной генетической изоляцией.ІІІН -<.(0. Гснстичсские изоляты с пысокой частотой аутосомных болезнейі Іоііуляция1|ии>ирцдцы-амиши
МП иимшмия)(( III) Блас) индей-
Іаііііма)1111 иидсйиы
loil.l)IIл, а<|)|-)оамериканиыII и IIМлБолезньХондроэктодермалытая дисплазия; синдром
Эллиса-Ван-Кревельда; гииоилазия хрящей
и волосАльбинизмАльбинизмСерповидно-клеточная анемия (генотип S/S)Недостаточность арантитрипсина (генотиіі
ZZ)Муковисиидоз (все мутантные аллели)Иіика, регионыМиотоігическая дистрофия, семейная гиперхо-
лестеринемияшля Африка (буры)Семейная гиперхолестеринемия^Им.1 индейцы (Юго-
ІІІІЛ (. ША)Инсулино независимый сахарный диабетВрожденная хлорилная диарея; аспартилглико-
заминурия; врожденный нефротический син¬
дром; MULIBREY-нанизмII и к эскимосыВрожденная гиперплазия надпочечников^(н1каниы (Южная
«гри ка)Порфирия песірая; семейная гиперхоле-
стеринемия; липоидный протеиноз: хорея
Гентингтона; склероостеоз|рі;и-іиикеназиБолезнь Тея-Сакса; болезнь Гоше; дизавтоно-
мия; болезнь Канавана||рси-караимыБолезнь Верди-Хофманаиитовые острова
ІПоиия)Спинальная мышечная атрофия|иир, Сардиния{J-Талассемияімочание. MULiBREY-нанизм — MUscle-LIvcr-BRain-Eye-нанизм.
216Клиническая генетиРедкие наследственные болезни относительно часто (в 10—100 р|
чаще, чем в других популяциях) обнаруживаются в ряде полуляци!
У армян независимо от места их проживания отмечается высок!
встречаемость семейной средиземноморской лихорадки (период!
ческая болезнь). В Финляндии часто встречается несколько редкі
болезней: врожденный нефроз, лизинурическая нелереносимосі
белка, аспартилгликозаминурия, липофусдиноз (детский тип) и
У евреев-ашкенази, в нескольких поколениях постоянно ііроживаї
щих в США, Европе и Израиле, отмечается высокая частота болезш
накопления (СМ2-ганглиозидоз, болезнь Нимана—Пика, болез!
Гоше), синдрома Блума, семейной дизавтономии, абеталипощ
теинемии, брахидакгилии и др. В некоторых областях Японии час
встречаются акаталазия, болезнь Огаши, наследственный дисхрои
тоз. В Азербайджане в отдельных районах отмечается высокая част
та синдрома Элерса-Данло (классический тип), хореи Гентингтої
гемофилии. Среди канадцев французского происхождения мне
больных тирозинемией, СМ2-ганглиозидозом.Как видно из приведенных примеров (подобной информац!
много и по другим популяциям), во всех случаях существует дост
точно выраженная брачная изоляция, иногда даже вне географи*
ских ограничений (евреи-ашкенази, армяне).Различия в распространенности касаются не только редко,
и часто встречающихся форм генных болезней. В последнем ел]
чае возможно как уменьшение, так и увеличение частоты болезі
Объяснения колебаний распространенности часто встречающи?
генных болезней надо искать, скорее всего, в условиях отбора,
уже упоминалось для гемоглобинопатий. Например, две широко ра
пространенные аутосомно-рецессивные болезни — фенилкетонурі
и муковисцидоз — встречаются с неравной частотой в разных геогі
фических зонах или больших популяциях. Муковисцидоз встречает]!
в среднем у 1 из 2000—3000 новорожденных в Европе и США, а среі
чернокожих африканцев и японского населения его частота составляі|
1:100 ООО. Частота фенилкетонурии в большинстве популяций раві
1:10 ООО, а в Финляндии, Израиле (для евреев-ашкенази), Япощ
болезнь встречается крайне редко (1:100 000—1:200 ООО). В Белоруссі
и Ирландии ее частота составляет 1:6000, в Шотландии 1:5300.В заключение следует подчеркнуть, что понимание зпидемиІІ
логи и генных болезней необходимо врачу любой специальност|
поскольку он может столкнуться с «пучковостью» редкой насле
Ірм.і 4. Генные болезни 217Іііи ііііой болезни в пределах обслуживаемого им района или кон-
пиопта. Знание закономерностей и механизмов распространения
ИНМ.1Х болезней поможет своевременно разработать меры профи-
ІЙКІІІКИ (обследование на выявление гетерогенности, генетическое
шк ультирование и др.).КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯЛиіиципация
Ьолезни накопленияЬолезни с экспансией триплетных повторов
Ннды генных мутаций у человека
Имутрилокусная гетерогенность
Mo jpacT начала болезни
Гс-нетическая гетерогенность
І ометические компаундыI смокопии
дрейф геновИмпринтинг на генном уровне
Классификация генных болезней
Корреляция генотип-фенотип
Кровпородственные браки
Легальный эффект мутаций
Межлокусная гетерогенность
Мутационный процесс и болезни
МормокопииОпределение генных болезнейОсобенности клинической картины генных болезнейПервичные эффекты мутантных аллелейПолные и мозаичные формы болезнейПримеры клеточного уровня патогенезаПричины клинического полиморфизмаПрогредиентность клинической картиныпроявления клинического полиморфизмаРепродуктивная компенсацияС'оматический и гонадный мозаицизмУровни патогенеза генных болезнейФенокопииФенотипические эффекты мутаций
218Клиническая генеткЭпидемиология генных болезней
Эффект родоначальникаРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАВалиев P.P., Хусайнова Р.И., Хуснутдинова Э.К. Синдром Марфі
клинические и молекулярно-генетические аспекты. Медицине!
генетика. - 2003. - Т. 2. - № 11. - С. 450-458.Васильев В.Б. Генетические основы митохондриальных болезней^,
СПб.: Изд-во «Нестор-История», 2006. — 146 с.Вахарловский В.Г., Романенко О. П., Горбунова В. Н. Генетика в практ
педиатра: руководство для врачей. — СПб.; Феникс, 2009. — 288 с.Генетический паспорт — основа индивидуальной предиктивной ме
цины / под ред. B.C. Баранова. — СПб.: Изд-во Н-Л, 2009. ~ 527 с.Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткаї
руководство для врачей. — СПб.: Элби-СПб, 2009, ~ 704 с.: ил.КапрановН.И., Каширская Н.Ю., Петрова И.В. Муковисцидоз: досі
жения и проблемы на современном этапе // Медицинская генет
ка. - 2004. - Т. 3. - № 9. - С. 398-412.Козлова С.И., Демикова И.С. Наследственные синдромы и медиі
генетическое консультирование: атлас-справочник. — 3-є изд., дс
и перераб, — М.: Т-во научных изданий КМК; Авторская академі
2007. - 448 с.: 236 ил.Краснопольская К.Д. Наследственные болезни обмена вещесі
Справ, пос. — М.: РОО «Центр социальной адаптации и реабилі
ции детей «Фохат», 2005. — 364 с.; ил.Курникова М.Л., Блинникова Е.О., Мутовин ГР. и др. Совремеш
представления о синдроме Элерса-Данлоса // Медицинская гене
ка. - 2004. — Т. 3. - №1. — С. 10-17.Наследственные болезни в популяциях человека / под
Е.К. Гинтера. — М.: Медицина, 2002. — 304 с.Ferner R.E. Neurofibromatosis I. // European Journal of Hui
Genetics. - 2007. - N 15. - P. 131-138.Forest M.G, Recent advances in the diagnosis and management
congenital adrenal hyperplasia due to 21-hydroxylase deficiency. // Hui
Reproduction Update. - 2004. - Vol. 10, - N 6. - R 469-485.Sibley C., Stone N.J. Familial hypercholesterolemia: A challenge of dit
nosis and therapy. I I Cleveland Clinic Journal Of Medicine. — 2006,
Vol. 73. - N. 1. - P. 57-64.
Глава 5ХРОМОСОМНЫЕ БОЛЕЗНИ"ОБЩИЕ ВОПРОСЫхромосомные болезни — большая группа наследственных болезней
множественными врожденными пороками развития. В их основе
ІЖігі хромосомные или геномные мутации. Эти два разных типа мута-
|И лля краткости объединяют термином «хромосомные аномзігии».Нозологическое выделение по меньшей мере трех хромосомных
їлічией как клинических синдромов врожденных нарушений раз-
Itmi сделано до установления их хромосомной природы.I Іаиболее часто встречающаяся болезнь, трисомия 21, клинически
ІЛІІ описана в 1866 г. английским педиатром Л. Дауном и получила
шаиие «синдром Дауна». В дальнейшем причина синдрома не разйлисргалась генетическому анализу. Высказывались предположения
ломинантной мутации, о врожденной инфекции, о хромосомной
)ироде.Первое клиническое описание синдрома моносомии по
ккромосоме как отдельной формы болезни было сделано русским
ІИИИЦИСТОМ Н.А. Шерешевским в 1925 г., а в 1938 г, Г. Тернер
ІКЖЄ описал ЭТОТ синдром. По фамилии этих ученых моносо-110 по Х“Хромосоме называют синдромом Шерешевского—Тернера.
шрубежной литературе в основном используют название «синдром
ірисра», хотя никто не оспаривает заслугу Н.А. Шерешевского.Лпомалии в системе половых хромосом у мужчин (трисомия XXY)
1К клинический синдром впервые описал г. Клайнфелтер в 1942 г.
Перечисленные заболевания стали объектом первых клинико-
ітогснетических исследований, проведенных в 1959 г. Расшифровка
ГИологии синдромов Дауна, Шерешевского—Тернера и Клайнфелтера
ГК рыла новую главу в медицине — хромосомные болезни.II 60-х годах XX в. благодаря широкому развертыванию цито-
їнстических исследований в клинике полностью оформилась как
іспиальность клиническая цитогенетика. Была показана роль хро-Исправлено и дополнено при участии д-ра биол. наук И.Н. Лебедева.
220Клиническая генеттмосомных и геномных мутаций в патологии человека, расшифроваї
хромосомная этиология многих синдромов врожденных пороке
развития, определена частота хромосомных болезней среди неї
рожденных и при спонтанных абортах.Наряду с изучением хромосомных болезней как врожденні
состояний начались интенсивные цитогенетические исследования^
онкологии, особенно при лейкозах. Роль хромосомных изменени!
опухолевом росте оказалась очень значимой.По мере совершенствования цитогенетических методов, особен!
таких, как дифференциальная окраска и молекулярная цитогеї
тика, открывались новые возможности для обнаружения ранее
описанных хромосомных синдромов и установления связи МЄ5
кариотипом и фенотипом при небольших изменениях хромосом.в результате интенсивного изучения хромосом человека и xpoj
сомных болезней на протяжении 45—50 лет сложилось учение о xi
мосомной патологии, которая имеет большое значение в современш
медицине. Данное направление в медицине включает не толі
хромосомные болезни, но и патологию внутриутробного перж
(спонтанные аборты, выкидыши), а также соматическую пато;
ГИЮ (лейкозы, лучевую болезнь). Число описанных типов хром^
сомных аномалий приближается к 1000, из них несколько сот
имеют клинически очерченную картину и называются синдромаї
Диагностика хромосомных аномалий необходима в практике вра^
разных специальностей (генетик, акушер-гинеколог, педиатр, нев{
патолог, эндокринолог и др.). Во всех многопрофильных современш
больницах (более 1000 коек) в развитых странах имеются цитогеї
тические лаборатории.О клинической важности хромосомной патологии можно суді
по частоте аномалий, представленных в табл. 5.І и 5.2.Таблица 5.1. Примерная частота новорожденных с хромосомными аномалия*Тип аномалииЧастот]Аттеуплоидия по половым хромосомам у новорожденных маль¬
чиков1:3601Анеуплоидия по половым хромосомам у новорожденных депочек1:58^1Анеуплоидия по аутосомам у живорожденных1:700]Структурные аномалии у живорожденных1:375 ]Аномалии по всем хромосомам у живорожденных1:1541
ijHtiii 5. Хромосомные болезни221іПіініт 5.2. Исходы родов иа 10 ООО беременностейИсходы111 <4 0||ч|)мальные хромо-I ПМІ.ІЧислоберемен¬ностей10 000Спонтанные абортычисло1500%15Числоживорожденных850092007508450\|)|1М()сомные ано-
Мпмии8007509450Клк видно из таблиц, цитогенетические синдромы составля-
1)1 йольшую долю в репродуктивных потерях (50% среди спонтан-
[ІІІ.ІЧ абортов первого триместра), врожденных пороках развития и
умещенного недоразвития. В делом хромосомные аномалии встреча-
пси у 0,7—0,8% живорожденных детей, а у женщин, рожающих после
15 'IC1, вероятность рождения ребенка с хромосомной патологией
(Пристает до 2%.ЭТИОЛОГИЯ и КЛАССИФИКАЦИЯ'■)'гиологическими факторами хромосомной патологии являются
14' ииды хромосомных мутаций и некоторые геномные мутации.геномные мутации в животном и растительном мире много-
)0|>:1 зны, у человека обнаружены только 3 типа геномных мутаций:
)1 [)аплоидия, триплоидия и анеуплоидия. Из всех вариантов анеу-
1Л()идий встречаются только трисомии по аутосомам, полисомии по
toJioubiM хромосомам (три-, тетра- и пентасомии), а из моносомий
стречается только моносомия X.Что касается хромосомных мутаций, то у человека обнаруже-
|ы нсс их типы (делеиии, дупликации, инверсии, транслокации).
у к.'іинико-цитогенетической точки зрения делеция в одной из гомо-
ІОІ ичмых хромосом означает нехватку участка или частичную моно-
^в»>мию по этому участку, а дупликация — избыток или частичную
Грисомию. Современные методы молекулярной цитогенетики позво-
ІМЮТ выявлять мелкие делец и и на уровне гена.Реципрокная (взаимная) транслокация без потери участков вовле-
Ысмиых в нее хромосом называется сбалансированной. Как и инвер-
кии, она не ведет к патологическим проявлениям у носителя. Однако
222Клиническая генеттв результате сложных механизмов кроссинговера и редукции числ
хромосом при образоваїши гамет у носителей сбалансированні
транслокаций и инверсий могут образовываться несбалансировані
гаметы, т.е. гаметы с частичной дисомией или с частичной нуллис<
мией (в норме каждая гамета моносомна).Транслокация между двумя акроцентрическими хромосомами
потерей их коротких плеч приводит к образованию одной мета- ю
субметацентрической хромосомы вместо двух акроцентрическиї
Такие транслокации называются робертсоновскими. Формально
носители имеют моносомию по коротким плечам двух акроце*
трических хромосом. Однако такие носители здоровы, потому Ч1
потеря коротких плеч двух акроцентрических хромосом КОМПЄІ
сируется работой таких же генов в остальных 8 акроцентрическі
хромосомах. У носителей робертсоновских транслокаций мо2
образовываться 6 типов гамет (рис, 5.1), но нуллисомные гаметі
должны приводить к моносомии по аутосомам в зиготе, а так^
зиготы не развиваются.Рис. 5.1. Типы гамет у носителей робертсоиопской транслокапии 21/14; 1
моносомия 14 и 21 (норма); 2 — моносомия 14 и 21 с робертсоновской траК!
локацией; 3 — дисомия 14 и моносомия 21; 4 — дисомия 21, моносомия
5 — нуллисомия 21; 6 — нуллисомия 14
ПйМіІ 5. Хромосомные болезни223q§Xеi{Xq)i(Xp)Клиническая картина про-
Гыч и транслокаиионных форм
»тч)мии 110 акроцснтрическим
^омосомам одинаковая.It случае концевых делеций
оооих плечах хромосомы воз¬
ій к ист кольцевая хромосома.имливида, унаследовавшего
1)Л1.цевую хромосому от одного
|1 родителей, будет частичная
І1ИОС0МИЯ по двум концевым
ІНІ 1кам хромосомы.Иногда разрыв хромосомы
)(1п\ОДИТ через центромеру.Ійжлое плечо, разъединенное
ІНЛС репликации, имеет две
fcipM некие хроматиды, сое-
|И11с11Ные оставшейся частью
Миромеры. Сестринские хро-
Щгилы одного и того же плеча
Глиовятся плечами одной хро-
1К’омы (рис. 5.2). Со с.тедуюшего митоза эта хромосома начинает
Иілицироваться и передаваться из клетки в клетку как само-
1»и гельиая единица наряду с остальным набором хромосом. Такие
омосомы называют изохромосомамн. У них одинаковые по набору
ІМОН плечи. Каков бы ни был механизм образования изохромосом
сте полностью не выяснен), их наличие вызывает хромосомную
Ггологию, потому что это одновременно и частичная моносомия
оотсутствующему плечу), и частичная трисомия (по присутствую-
!му плечу).ІІ основе классификации хромосомной патологии лежат 3 прин¬
та, позволяющие точно охарактеризовать форму хромосомной
ітологии и ее варианты у обследуемого.Первый принцип — характеристика хромосомной или геномной
гмиии (триплоидия, простая трисомия по хромосоме 21, частичная
жосомия и т.д.) с учетом конкретной хромосомы. Этот принцип
}Ж1ю назвать этиологическим.Клиническая картина хромосомной патологии определяется
ІІІОМ геномной или хромосомной мутации, с одной стороны, иРис. 5.2. Изохромосомы X подлинно¬
му li(Xq)J и короткому li(Xp)J плечу
224Клиническая генвіиндивидуальной хромосомой — с другой. Нозологическое ПОД|
деление хромосомной патологии основывается, таким образом*
этиологическом и патогенетическом принципе: для каждой фо(
хромосомной патологии устанавливается, какая структура ъов}
на в патологический процесс (хромосома, сегмент) и к чем сос
генетическое нарушение (недостаток или избыток хромосомі
материала). Дифференциация хромосомной патологии на основа!
клинической картины не имеет существенного значения, поскол!
разным хромосомным аномалиям свойственна большая общие
нарушений развития.Второй принцип — определение типа клеток, в которых В03НИІ
мутация (в гаметах или зиготе). Гамеїические мутации ведут к
ным формам хромосомных болезней. У таких индивидов все кл«
несут унаследованную с гаметой хромосомную аномалию.Если хромосомная аномалия возникает в зиготе или на рані
стадиях дробления (такие мутации называют соматическими, в отл
чиє от гаметических), то развивается организм с клетками разі
хромосомной конституции (два типа и более). Такие формы хрої
сомных болезней называют мозаичными.Для возникновения мозаичных форм, по клинической карт)
совпадающих с полными формами, нужно не менее 10% клет
аномальным набором.Третий принцип — выявление поколения, в котором возникла
ция: возникла она заново в гаметах здоровых родителей (спораді
ские случаи) или родители уже имели такую аномалию (наследуем!
или семейные, формы).О наследуемых хромосомных болезнях говорят тогда, когда м]
ция имеется в клетках родителя, в том числе и в гонадах. Это м(
быть и случаи трисомии. Например, у индивидов с синдромаї
Дауна и трипло-Х образуются нормальные и дисомные гамет
Такое происхождение дисомных гамет — следствие вторичш
нерасхождения, т.е, нерасхождения хромосом у индивида с три<
мией. Большая часть наследуемых случаев хромосомных болезі
связана с робертсоновскими транслокациями, сбалансированны*
реципрокньтми транслокациями между двумя (реже более) хрої
сомами и инверсиями у здоровых родителей. Клинически значи!
хромосомные аномалии в этих случаях возникли в связи со сл<
ными перестройками хромосом в процессе мейоза (конъюгат
кроссинговер).
їм f). хромосомные болезни225ГІНКИМ образом, для точной диагностики хромосомной болезни
Очолимо определить:I* 11111 мутации;І» иоилеченную в процесс хромосому;I» форму (полную или мозаичную);• ІИ I речаемость в родословной — спорадический или наследуемый
с.'іумай,Ііікаи диагностика возможна только при цитогенетическом обсле-
Кіиии пациента, а иногда и его родителей и сибсов.ЭФФЕКТЫ ХРОМОСОМНЫХ АНОМАЛИЙ
В ОНТОГЕНЕЗЕХромосомные аномалии вызывают нарушение общего генетическо-
Оиланса, той скоординированности в работе генов и системности
[•упииии, которые сложились в процессе эволюции каждого вида,
^ііпіштельно, что патологические эффекты хромосомных и геномных
ІІіпций проявляются на всех стадиях онтогенеза и, возможно, даже на
JHMC гамет, влияя на их формирование (особенно у мужчин).
jlnM человека характерна высокая частота репродуктивных потерь
ранних стадиях постимплантационного развития по причине
11Мс)сомных и геномных мутаций. Подробные сведения о цито-
<С1Ике эмбрионального развития человека можно найти в книге
Ь ара нова и Т. В, Кузнецовой (см. рекомендуемую литературу)
|И и статье И.Н. Лебедева «Цитогенетика эмбрионального разви-
|И человека: исторические аспекты и современная концепция» на
иіакт-диске.И лучение первичных эффектов хромосомных аномалий началось
«мчале 1960-х годах вскоре после открытия хромосомных болезней и
і)должается до сих пор. Главные эффекты хромосомных аномалий
онвляются в двух связанных между собой вариантах: летальности
йИрожденных пороках развития.стельностьИмеются убедительные свидетельства того, что патологические
фскты хромосомных аномалий начинают ігроявлягься уже со
Гйдии зиготы, будучи одним из главных факторов внутриутробной
iftcJiM, достаточно высокой у человека.
226Клиническая ген(Выявить количественный вклад хромосомных аномал!
гибель :1игот и бластоцист (первые 2 нед после оплодотвореї
в полной мере трудно, поскольку в этот период беременност!
клинически, ни лабораторно еще не диагностируется. Оді
некоторая информация о разнообразии хромосомных наруї
ний на самых ранних этапах развития зародыша может 61
получена из результатов предымплантационной генетичес!
диагностики хромосомных заболеваний, проводимой в раї
процедур искусственного оплодотворения, с использован!
молекулярно-цитогенетических методов анализа показано,
частота числовых нарушений хромосом у предымпланташ
ных зародышей варьирует в пределах 60—85% в зависимостї
обследуемых групп пациентов, их возраста, показаний для
ведения диагностики, а также числа анализируемых хромо<
при проведении флюоресцентной гибридизации in situ (FISH)
интерфазных ядрах отдельных бластомеров. До 60% зароды!
на стадии 8-клеточной морулы имеют мозаичную хромосомі
конституцию, а от 8 до 17% эмбрионов, по данным сравнителы
геномной гибридизации (CGH), обладают хаотичным кари<
пом; разные бластомеры в составе таких зародышей несут
личные варианты числовых хромосомных нарушений. Ср<|
хромосомных аномалий у предымплантационных эмбрио!
выявлены трисомии, моносомии и даже нуллисомии аутосі
все возможные варианты нарушений числа половых хромосої
также случаи три- и тетраплоидии.Столь высокий уровень аномалий кариотипа и их разнообрая
безусловно, негативно влияют на успешность протекания преді
плантационных этапов онтогенеза, нарушая ключевые морфоі
тические процессы. Около 65% эмбрионов с хромосомными анс
ЛИЯМИ останавливают свое развитие уже на этапе компактизаі
морулы.Такие случаи ранней остановки развития можно объяснить
что нарушение геномного баланса вследствие развития како<
определенной формы хромосомной аномалии приводит к дискооі
нации включения и выключения генов на соответствующей ст
развития (временной фактор) или в соответствующем месте блас
цисты (пространственный фактор). Это вполне понятно: поскс
ку в процессах развития на ранних стадиях участвуют приме
1000 генов, локализованных во всех хромосомах, хромосомная
() хромосомные болезни227|Ии нарушает взаимодействие генов и инактивирует какие-то кон-
liii.li' процессы развития (межклеточные взаимодействия, диффе-
||И1[)1шка клеток и др.).Ітиочисленные цитогенетические исследования материаласпон-
M.IS ііГюртов, выкидышей и мертворожденных позволяют объек-
40 еудить об эффектах разных типов хромосомных аномалий во
І'ірпуї робном периоде индивидуального развития. Летальный или
ІМі^рфоіенетический эффект хромосомных аномалий обнаружи-
(1’н ма всех стадиях внутриутробного онтогенеза (имплантации,
>И()ги*неза, органогенеза, росте и развитии плода). Суммарный
i;i хромосомных аномалий во внутриутробную гибель (после
1м пшии) у человека составляет 45%. При этом чем раньше пре-
1н»‘1ся беременность, тем вероятнее, что это обусловлено анома-
|ми развития эмбриона, вызванными хромосомным дисбалансом.
4 недельных абортусов (эмбрион и его оболочки) хромосомные
ііиіии обнаруживают в 60—70% случаев. В I триместре гестации
ІМосомньїе аномалии встречаются у 50% абортусов. У плодов-
іидишей II три мес гра такие аномалии находят в 25—30% случаев,
М.'и)дов, погибших после 20-й недели гестации,— в 7% случаев,
к (*1*ди перинатально погибших плодов частота хромосомных ано-
ІНІІ составляет 6%.^Н«иболее тяжелые формы по дисбалансу хромосомного набора
)счаются у ранних абортусов. Это полиплоидии (25%), полные
і:омии по аутосомам (50%). Трисомии по некоторым аутосомам
^3; 6; И; 19) встречаются крайне редко даже у элиминированных
JpiioHOB и плодов, что свидетельствует о большой морфогене-
ісікой значимости генов в этих аутосомах. Данные аномалии
Ірьіііают развитие в доим плантационном периоде или нарушают
істоіснез.= Высокая морфогенетическая значимость аутосом еше более отчет-111 мри полных аутосомных моносомиях. Последние редко обна-
іиваются даже в материале ранних спонтанных абортов из-за
їйльного эффекта такого дисбаланса.їжденньїе пороки развитияtcjiM хромосомная аномалия не дает летального эффекта на ран-
сгадиях развития, то ее последствия проявляются в виде врож-
1МЫХ пороков развития. Практически все хромосомные анома-
|И (кроме сбалансированных) приводят к врожденным порокам
228 Клиническая генвіразвития, сочетания которых известны как нозологические фоі
хромосомных болезней и синдромов (синдромы Дауна, Воль(
Хиршхорна, кошачьего крика и т.д.).С эффектами, вызываемыми однородительскими дисомами,
робнее можно ознакомится на компакт-диске в статье С.А. Назаре
«Наследственные болезни, детерминированное однородительскі
дисомами, и их молекулярная диагностика».Эффекты хромосомных аномалий в соматических клетка]Роль хромосомных и геномных мутаций не ограничивается их
янием на развитие патологических процессов в ранних периодах oj
генеза (незачатие, спонтанный аборт, мертворождение, хромосомі
болезнь). Их эффекты прослеживаются в течение всей жизни.Хромосомные аномалии, возникающие в соматических клеи
в постнатальном периоде, могут вызывать различные последстї
остаться нейтральными для клетки, обусловить гибель клеті
активировать деление клетки, изменить функцию. Хромосомі
аномалии возникают в соматических клетках постоянно с неї
сокой частотой (около 2%). В норме такие клетки злиминируї
иммунной системой, если они проявляют себя чужеродно. Оді
в некоторых случаях (активация онкогенов при транслокащ
делениях) хромосомные аномалии становятся причиной злокйіі
ственного роста. Например, транслокация между хромосомам!
и 22 вызывает миелолейкоз. Облучение и химические мутаи
индуцируют хромосомные аберрации. Такие клетки гибнут,
наряду с действием других факторов способствует развитию лу!
вой болезни, аплазии костного мозга. Имеются :>ксперименталы
доказательства накопления клеток с хромосомными аберрацияі
процессе старения.ПАТОГЕНЕЗНесмотря на хорошую изученность клиники и цитогенет!
хромосомных болезней, их патогенез даже в общих чертах еще
сен. Не разработана общая схема развития сложных патологичес!
процессов, обусловленных хромосомными аномалиями и прив(
щих к появлению сложнейших фенотипов хромосомных болез!
Ключевое звено в развитии хромосомной болезни ни при 0Д1
Idii 5. Хромосомные болезни229ірмс не выявлено, Некоторые авторы предполагают, что это звено —
Ііиіиіансированность генотипа или нарушение общего генного
Ілііііса. Однако такое определение ничего конструктивного не дает.
»(Гі;і;іансированность генотипа — условие, а не звено патогенеза,
in ц(Х1жна реализовываться через какие-то специфические био-
Імичсские или клеточные механизмы в фенотип (клиническую
Ірпіну) болезни.(Истематизация данных о механизмах нарушений при хромосом-
IX Полезнях показывает, что при любых трисомиях и частичных
>||1)сомиях можно выделить 3 типа генетических эффектов; специ-
'к*ские, полуспецифическис и неснецифические.Специфические эффекты должны быть связаны с изменениемII 1Л структурных генов, кодирующих синтез белка (при три-
ЇМИИ их число увеличивается, при моносомии уменьщается).
||циочислснные попытки найти специфические биохимические
!|фскты подтвердили это положение лишь для немногих генов илипродуктов. Часто при числовых хромосомных нарушениях не
>о11сходит строго пропорционального изменения уровня экспрес-III ІЄІЮБ, что объясняется разбалансировкой сложных регулятор-
IX процессов в клетке. Так, исследования больных с синдромом
іуііа позволили идентифицировать 3 группы генов, локализо-на хромосоме 21, в зависимости от изменения уровня их
ІТИПН0СТИ при трисомии. в первую группу вошли гены, уровень
іирсссии которых значительно превышает уровень активности в
1СОМНЫХ клетках. Предполагается, что именно эти гены опреде-
|И)| формирование основных клинических признаков синдрома
lynii, регистрируемых практически у всех пациентов. Вторую
/пну составили гены, уровень экспрессии которых частично пере¬
сыпается с уровнем экспрессии при нормальном кариотипе. Как
їліііают, эти гены определяют формирование вариабельных при-
ІЙКОВ синдрома, отмечаемых не у всех пациентов, Наконец, в тре-
группу вошли гены, уровень экспрессии которых в дисомных и
СИсомных клетках практически не различался. По всей видимости,
fM гены наименее вероятно вовлечены в формирование клини-
ItKnx признаков синдрома Дауна. Следует отметить, что только
)% генов, локализованных на хромосоме 21 и экспрессирующихся
лимфоцитах и 69% генов, экспрессирующихся в фибробластах,
^ииадлежали к первым двум группам. Некоторые примеры таких
|Иоп приведены в табл. 5.3.
230Клиническая генїТаблица 5.3. До:зозависимые гены, определяющие формирование клині
ских признаков синдрома Дауна при трисомии 21ГенЛокали¬зацияФункцияКлиничесіпризнакRCANI{DSCR1)21q22.I2Ген критического региона син¬
дрома Дауна. Продукт rcita
является регулятором кальии-
неврина. Гиперэкспрессируется
в головном мозге плодов с
трисомией 21 и взаимодей¬
ствует с кальциневрином
А. Сверхэкспрессия RCAN1
ингибирует кальциневрино-
зависимую транскрипцию генов
вследствие нарушения транс¬
порта в ядро транскрипционно¬
го фактора NFAT, регулирую¬
щего развитие позвоночных. Ген
/?Cy4/V/экспрессируется также в
сердечной и скелетггой муску¬
латуреУмственная
отсталость,
пороки сердца'SUM032lq22.3Малый убиквитинподоб-
ный модификатор 3-го типа.
Обеспечивает посттрансляци-
онные модификации белков,
включая р53. В отличие от
убиквитиновых белков, контро¬
лирующих деградацию протеи¬
нов, SlJMO-белки принимают
участие в ядерном транспорте,
транскрипционной регуляции,
апоптозе, поддержании стабиль¬
ности протеиновНарушения пр<1
цессов репара- _
ции ДНК и апс
тоза. Старение^PRMT221q22.3АргининоваяN-метилтрансфераза 2, осущест¬
вляющая посттрансляиионное
метилирование аргининовых
остатков. Является ядерным
фактором, ингибирующим
NF-kB сигнальный путь и сти¬
мулирующим апоптоз
Nil Г). Хромосомные болезни231Окончание таблицы 5.3ГенЛокали¬зацияФункцияКлиническийпризнакU\AIU''\AR22lq22.121q22.1Продукты генов вовлечены в
ILlORB-сигнальный путь\rsj21q22.1-q22.2Ослабление
клеточного и
гуморального
иммунитета21q22.3Онкоген HTS2, вовлеченный
в закладку эмбриональной
передне-задней оси и форми¬
рование скелета. Участвует в
регуляции плгорипотентности
стволовых клеток, клеточного
старения и гибели, контролирует
активность теломеразы и длину
теломер. Вовлечен в канцероге¬
нез. Активирует промотор гена
Р-амилоидного протеина через
связывание с Ets-сайтами в про-
моторной области гена АРРСклонность к
злокачествен¬
ным новооб¬
разованиям,
умственная
отсталость|»иохимическое изучение фенотипа хромосомных болезней пока
привело к пониманию путей патогенеза возникающих вслед-
IfiMc хромосомных аномалий врожденных нарушений морфогенезаI и роком смысле слова. Обнаруженные биохимические отклонения
^Кіі грудно связать с фенотипическими характеристиками болезней
органном и системном уровнях. Изменение числа аллелей гена
исегда вызывает пропорциональное изменение продукции соот-
jlTc! ($угощего белка. При хромосомной болезни всегда существенно
Иіястся активность других ферментов или количество белков, гены
вторых локализованы на не вовлеченных в дисбаланс хромосомах.
|Н I» одном случае не обнаружено белка-маркера при хромосомных1С1ПЯХ.[Іолуспецифические эффекты при хромосомных болезнях могут
||'Г1> обусловлены изменением числа генов, в нор.ме представленных
[Циде многочисленных копий, к таким генам относятся гены рРНК
тРНК, гистоновых и рибосомных белков, сократительных белков
ІТИ на и тубулина. Эти белки в норме контролируют ключевые
fMlibi метаболизма клетки, процессов ее деления, межклеточных
інимодействий. Каковы фенотипические эффекты дисбаланса этой
232Клиническая генїгруппы існов, как компенсируется их недостаток или избыток,
неизвестно.Неслецифические эффекты хромосомных аномалий связьіваї
изменением гегерохроматина в клетке. Важная роль гетерохром*
на в клеточных делениях, клеточном росте и других бйологичесі
функциях не вызывает сомнений. Таким образом, неспеті,ифичес|
и частично полуспсиифические эффекты приближают нас к
точным механизмам патогенеза, безусловно, играющим важнейщ|
роль при врожденных пороках развития,Большой фактический материал позволяет провести сопостаї
НИС клинического фенотипа болезни с цитогенетическими ИЗМЄІ
ниями (фенокариотипичсские коррелянии).Общее для всех форм хромосомных болезней — множественное
поражения. Это черепно-лицевые дисморфии, врожденные пор(
развития внутренних и наружных органов, замедленные внутрі
тробные и постнатальные рост и развитие, отставание психическ(
развития, нарушения функций нервной, эндокринной и иммун]
систем, при каждой форме хромосомных болезней наблюдается 30-
различных отклонений, частично перекрывающихся (совпадающ!
при разных синдромах. Лишь небольшое число хромосомных бо;
ней проявляется строго определенным сочетанием отклонений Б
витии, что и используют в клинической и иатолого-анатомичесї
диагностике.Патогенез хромосомных болезней развертывается в раннем bhj
утробном и продолжается в постнатальном периодах. Множествені
врожденные пороки развития как главное фенотипическое проявлеї
хромосомных болезней формируются в раннем эмбриогенезе, поэт
к периоду ностнатального онтогенеза все основные пороки развит
уже налицо (кроме пороков развития половых органов). Раннее и М1
жественное поражение систем организма объясняет некоторую
ность клинической картины разных хромосомных болезней.Фенотипическое проявление хромосомных аномалий, т.е. фс
мирование клинической картины, зависит от следующих главн!
факторов;‘ индивидуальности вовлеченной в аномалию хромосомы или
участка (специфический набор генов);• типа аномалии (трисомия, моносомия; полная, частичная);• размера недостающего (при делении) или избыточного (nj
частичной трисомии) материала;
|Й Хромосомные болезни233III'иен и мозаичности организма по аберрантным клеткам;И ц-(м)гииа организма;I?» VI іоїшй срсды (внутриутробной или постнатальной).( u'liciib отклонений в развитии организма зависит от качсствен-II количественной характеристики унаследованной хромосом-
ииомалии. При исследовании клинических данных у человека
1И<к1|,ю подтверждается доказанная у других видов относитель-
Іігіи.ісокая биологическая ценность гетерохроматиновых районов
liMncoM. Полные три сом и и у живорожденных наблюдаются только
«уюсомам, богатым гетсрохроматином (8; 9; 13; 18; 21). Так же
»И1'мястся полисомия (до пентасомии) по половым хромосомам, в
Ippoii Y-хромосома имеет мало генов, а добавочные Х-хромосомы
IhiiKM гетерохроматинизированы.к іиііическое сопоставление полных и мозаичных форм болез-
тжазываст, что мозаичные формы протекают в среднем легче,
ііидимому, это объясняется присутствием нормалы1ых клеток,
ItiiMiio компенсирующих генетический дисбаланс. В индивиду-
||іН<>м прогнозе прямой связи тяжести течения заболевания и соот-
Лі'мия аномальных и нормальных клонов не обнаруживается.ІІО мере изучения фено- и кариотипичсских корреляций при
1ИЫХ протяженностях хромосомной мутации выясняется, что наи-
ЇСС специфичные для того или иного синдрома проявления обу-
)илены отклонениями в содержании сравнительно небольших сег-
И гов хромосом. Дисбаланс по значительному объему хромосомного
Гсриала делает клиническую картину более неспецифичной. Так,
Иіифические клинические симптомы синдрома Дауна проявля-
гя мри трисомии по сегменту длинного плеча хромосомы 21q22,L
|И развития синдрома «кошачьего крика» при делениях короткою
j'la аутосомы 5 наиболее важна средняя часть сегмента (5р15).
IpiiK iepiibie черты синдрома Эдвардса связаны с трисомией ссгмен-
Хромосомы ISqll.Кііждой хромосомной болезни свойствен клинический поли-
>р<()изм, обусловленный генотипом организма и условиями среды.
Іриации в проявлениях патологии могут быть очень широкими;легального эффекта до незначительных отклонений в развитии,
|к, 60-70% случаев трисомии 21 заканчиваются гибелью во вну-
Іиу гробном периоде, в 30% случаев рождаются дети с синдромом
іуна, имеющим различные клинические проявления. Моносомия
Х-хромосоме среди новорожденных (синдром Шерешевского—
234 Клиническая ген<Тернера) — это 10% всех моносомных по Х-хромосоме зароды!
(остальные погибают), а если учитывать еще доимплантационі
гибель зигот ХО, то живорожденные с синдромом ШерешевскоіІ
Тернера составляют только 1%.Несмотря на недостаточное понимание закономерностей пат
неза хромосомных болезней в целом, некоторые звенья общей
событий в развитии отдельных форм уже известны и их число П(
янно увеличивается.КЛИНИКО-ЦИТОГЕНЕТИЧЕСКИЕ
ХАРАКТЕРИСТИКИ НАИБОЛЕЕ
РАСПРОСТРАНЕННЫХ ХРОМОСОМНЫХ
БОЛЕЗНЕЙСиндром ДаунаСиндром Дауна, трисомия 21, — наиболее изученная хромос<
ная болезнь. Частота синдрома Дауна среди новорожденных
1:700—1:800, не имеет какой-либо временной, этнической или
графической разницы при одинаковом возрасте родителей. Час^
рождения детей с синдромом Дауна зависит от возраста матери fi
меньшей мере от возраста отца (рис. 5.3).С возрастом существенно увеличивается вероятность рождеї
детей с синдромом Дауна. Так, у женщин в возрасте 45 лет
составляет около 3%. Высокая частота детей с синдромом
(около 2%) наблюдается у рано рожающих женщин (до 18
Следовательно, для популяционных сравнений частоты рождеї
детей с синдромом Дауна надо принимать во внимание распредв
ние рожающих женщин по возрасту (доля женщин, рожающих п(
30-35 лет, в общем числе рожающих). Это распределение инс
меняется в течение 2—3 лет для одного и того же населения (наї
мер, при резком изменении экономической ситуации в стране),
частоты синдрома Дауна с увеличением материнского возраста из
стен, но большинство детей с синдромом Дауна все-таки рожде
матерями моложе 30 лет. Это связано с большим числом беремі(
ностей в этой возрастной группе по сравнению с женщинами бс
старшего возраста.
t) Хромосомные болезни235ЧастотаВозраст матери, годыА. 'Зависимость частоты рождения детей с синдромом Дауна от возраста!Н иигературе описана «пучковость» рождения детей с сии-
ЇМ(»м Дауна в определенные промежутки времени в некоторых
ІЙІІИХ (городах, провинциях). Эти случаи можно объяснить
>рсс стохастическими колебаниями спонтанного уровня нерас-
ІДСНИЯ хромосом, чем воздействием предполагаемых этиологи-
|Ких факторов (вирусной инфекцией, низкими дозами радиа-
I, хлорофосом).1итогенетические варианты синдрома Дауна разнообразны.
ІЙКО основную долю (до 95%) составляют случаи полной трисо-
21 вследствие нерасхожден ИЯ хромосом в мейозе. Вклад мате-
<ског о нерасхождения в эти гамстические формы болезни состав-
Х5-90%, а отцовского — только 10-15%. При этом примерно
мирушений возникает в первом делении мейоза у матери и
|Ько 25% — во втором. Около 2% детей с синдромом Дауна имеют
Ійичньїе формы трисомии 21 (47,+21/46). Примерно 3—4% больных
(К) і транслокационную форму трисомии по типу робертсоновских
ІНслокаций между акроцентриками (D/21 и G/21). Около V4TpaHC-
Сиционных форм наследуются от родителей-носителей, тогда как
)анслокаций возникают de novo. Основные типы хромосомных
іуіііений, обнаруживаемых при синдроме Дауна, представлены в
I. 5.4.
236Клиническая генвіТаблица 5.4. Основные типы хромосомных аномалий при синдроме ДауіТип хромосомного нарушенияЧастота
среди больні
с синдромом,1.1,1.1.1.1.1.2.1.2.1.2.1.1.2.2.Трисомия 21 мейотического происхожденияНерасхождение в мейозе у матери (85—90%
мейотических мутаций)Ошибки мейоза I(75% нарушений в материнском мейозе)Ошибки мейоза II(25% нарушений в материнском мейозе)Нерасхождение в мейозе у отца
(10—15% мейотических мутаций)Ошибки мейоза I(25% нарушений в отцовском мейозе)Ошибки мейоза II(75% нарушений в отцовском мейозе)2.Трисомия 21 митотического происхождения
(хромосомный мозаицизм 47,+21/4б)Л-23.Робертсоновские транслокации (D/21, G/21)3.1.Семейные формы (25% транслокаций)3.2.Перестройки de novo (75% транслокаций)Соотношение мальчиков и девочек с синдромом Дауна сос]
ляет 1:1.Клиническая симптоматика синдрома Дауна разнообразна; Э1
врожденные пороки развития, и нарушения постнатального разві
нервной системы, и вторичный и.ммунодефицит и т.п. Дети с
дромом Дауна рождаются в срок, но с умеренно выраженной пр<
тальной гипоплазией (на 8—10% ниже средних величин). Многие
птомы синдрома Дауна заметны уже при рождении и в последую!
проявляются более четко. Квалифицированный педиатр устана]|
вает правильный диагноз синдрома Дауна в родильном доме не мі;
чем в 90% случаев. Из черепно-лицевых дисморфий отмечаются
голоидный разрез глаз (по этой причине синдром Дауна долго Hf
вали монголоидизмом), брахицефалия, круглое уплощенное л|
плоская спинка носа, эпикант, крупный (обычно высунутый) яі
деформированные ушные раковины (рис. 5.4), Мышечная гип<
Ніі Ь. хромосомные болезни237\\ ?і,4. Дети разного возраста с характерными чертами синдрома Дауна
||||чицсфалия, круглое лицо, макроглоссия и открытый рот, эпикаит,
ЦИ'ртслоризм, широкая переносица, «карний рот», косоглазие)И1 сочетается с разболтанно-
.1о суставов (рис. 5.5). Часто
}1)К'чаются врожденный порокIII, клииодактилия, типич-
W изменения дерматоглифики
|ггі.ірехпальцевая, или «обезья-
”, складка на ладони (рис, 5.6),
кожные складки вместо трех
МИІИІШЄ, высокое положение
Ырадиуса и др.). Пороки ЖКТ
іСліодаются редко.Диагноз синдрома Дауна уста-
іилиііают на основании сочс-
ИИ1Я нескольких симптомов.
К’луюшие 10 признаков наи-
дсс важны для установления
гиоза, наличие 4—5 из них
)111>верно указывает на сип¬
ким Дауна;•уплощение профиля лица
(90%);• отсутствие сосательного ре-
(|)лекса (85%);Рис. 5.5, Резкая гипотония у пациен¬
та с синдромом ДаунаРис. 5.6. Ладони взрослого мужчины
с синдромом Дауна (усиленная мор¬
щинистость, на левой руке четырех¬
пальцевая, или ^обезьянья», складка)
238Клиническая генеї• мышечная гипотония (80%);• монголоидный разре:і глазных щелей (80%);• избыток кожи на шее (80%);• разболтанность суставов (80%);• диспластичный таз (70%);• диспластичные (деформированные) ушные раковины (60%);• клииодактилия мизинца (60%);• четырсхлальцевая сгибательная складка (поперечная лині
ладони (45%).Большое значение для диагностики имеет динамика физическої
умственного развития ребенка — при синдроме Дауна otio задержиі
ся. Росі взрослых больных на 20 см ниже среднего. Задержка умсті
ного разви ГИЯ може і достигать уровня имбецильности без специальї
методов обучения. Дети с синдромом Дауна ласковые, вн имательнІ
послушные, терпеливые при обучении. Коэффициент умственного
вития {IQ) у разных детей может составлять от 25 до 75.Реакция детей с синдромом Дауна на воздействия окружаюі
среды часто патологическая в связи со слабым клеточным и гуї
ральным иммунитетом, снижением репарации ДНК, недостаточна
выработкой пищеварительных ферментов, ограниченными комп(
саторными возможностями всех систем. По этой причине детй]
синдромом Дауна часто болеют пневмониями, тяжели перенос
детские инфекции, у них от.мечается недостаток массы тела, выраз
гиповитаминоз.Врожденные пороки внутренних органов, сниженная приспос
бляемость детей с синдромом Дауна часто приводят к смерти в п<
вые 5 лет. Следствием измененного иммунитета и недостаточш
репарационных систем (для поврежденной ДНК) являются лейкоз
часто возникающие у больных с синдромом Дауна.Дифференциальная диагностика проводится с врожде
ным гипотиреозом, другими формами хромосомных аномалі
Цитогенетическое обследование детей показано не только при по^Е
зрении на синдром Дауна, но и при клинически установлений
диагнозе, поскольку цитогенетическая характеристика пациеі
необходима для прогноза здоровья будущих детей у родителей и
родственников.Этические проблемы при си ндроме Дауна многоплановы. Несмот
на повышение риска рождения ребенка с синдромом Дауна и другиі
хромосомными синдромами, врач должен избегать прямых рекоме!
itm [}. Хромосомные болезни239іиііі 110 ограничению деторождения у женщин старшей возрастной
Mtm.i, так как риск по возрасту остается достаточно низким, осо-
Ии<1 с учетом возможностей пренатальной диагностики.
Неудовлетворенность у родителей часто вызывает форма сообще-
И1 ирачом о диагнозе синдрома Дауна у ребенка. Диагностировать
Дауна по фенотипическим признакам обычно можно сразу
JU' родоразрешения. Врач, пытающийся отказаться от установле-
|н диагноза до исследования кариотипа, может потерять уважениеI НС и ников ребенка. Важно сообщить родителям как .можно ско-
I# после рождения ребенка, по крайней мере, о ваших подозрениях,ис следует полностью информировать родителей ребенка о диа-
|uu'. Нужно дать достаточно сведений, отвечая на непосредствен-
иопросы, и поддерживать контакт с родителями до того дня,II с ганет возможным более детальное обсуждение. Немедленная
(формация должна включать объяснение этиологии синдрома для
ІКЛІОЧЄНИЯ взаимных обвинений супругов и описание исследова-
|И и процедур, необходимых для того, чтобы полностью оценить
|)роиье ребенка.I Іолпое обсуждение диагноза нужно провести, как только родиль-
Иш более или менее оправится от стресса родоразрешения, обычно
сутки после родов, к это.му времени у матерей возникает множе-10 вопросов, на которые необходимо отвечать точно и определенно.
ІЖІК) приложить все усилия, чтобы на этой встрече присутствовали
родителя. Ребенок становится предметом непосредственного
суждения, в этот период еще рано нагружать родителей всей
(((юрмацией о заболевании, так как новые и сложные понятия тре¬
тяя ііремени для осмысления.Не пытайтесь давать прогнозы. Бесполезно пытаться точно пред-
ідсть будущее любого ребенка. Древние мифы вроде: «По крайней
рре, он будет всегда любить и наслаждаться музыкой» — непро-
|1И1сльны. Нужно представить картину, написанную широкими
І'іками, и отметить, что способности каждого ребенка развиваются
(ди видуально,детей с синдромом Дауна, рожденных в России (в Москве —
)%), родители оставляют на попечение государства. Родители (а
1)10 и педиатры) не знают, что при правильном обучении такие дети
їіуі стать полноценными членами семьи.Лечебная помощь детям с синдромом Дауна многопланова и
ісііецифична. Врожденные пороки сердца устраняются оперативно.
240Клиническая ген(Постоянно проводится общеукрепляющее лечение. Питание ДОЛ)
быть полноценным. Необходимы внимательный уход за больн(
ребенком, защита от действия вредных факторов окружающей cj
(простуда, инфекции). Большие успехи в сохранении жизни детві
синдромом Дауна и их развитии обеспечивают специальные м<
ды обучения, укрепления физического здоровья с раннего детс!
некоторые формы лекарственной терапии, направленные на улучі
ние функций ЦНС. Многие больные с трисомией 21 теперь спосоС
вести самостоятельную жизнь, овладевают несложными професс!
ми, создают семьи. Средняя продолжительность жизни таких б<
ных в промыщленно развитых странах составляет 50-60 лет.Синдром Патау (трисомия 13)Синдром Патау выделен в самостоятельную нозологичесі
форму в I960 г. в результате цитогенетического обследования д€
с врожденными пороками развития. Частота синдрома Патау с\
новорожденных равна 1 : 5000—7000. Существуют цитогенетичес
варианты этого синдрома. Простая полная трисомия 13 как следст!
нерасхождения хромосом в мейозе у одного из родителей (главн!
образом у матери) встречается у 80—85% больных. Остальные слу^
обусловлены в основном передачей дополнительной хромосомы (тс
нее, ее длинного плеча) в робертсоновских транслокациях типа D/13
G/13. Обнаружены и другие цитогенетические варианты (мозаициз
изохромосома, неробертсоновские транслокации), но они встречаї
крайне редко. Клиническая и патолого-анатомическая картина щ
стых трисомных форм и транслокационных форм не различается,Соотношение полов при синдроме Патау близко к J : 1. Дети с сі
дромом Патау рождаются с истинной пренатальной гипоплазией (і
25—30% ниже средних величин), которую нельзя объяснить небел
шой недоношенностью (средний срок гестации 38,3 нед). Характері
осложнение беременности при вынашивании плода с синдроме
Патау — многоводие: оно встречается почти в 50% случаев. Синдр<
Патау сопровождается множественными врожденными норокав
развития головного мозга и лица (рис. 5.7). Это патогенетически ед|
ная группа ранних (и, следовательно, тяжелых) нарушений формі
рования головного мозга, глазных яблок, костей мозговой и лицев(
частей черепа. Окружность черепа обычно уменьшена, встречает
и тригоноцефалия. Лоб скошенный, низкий; глазные щели узки<
переносье запавшее, ушные раковины низко расположенные и дефо|
1Н<1 !v Хромосомные болезни241И'. 5.7. Новорожденные с синдромо\т Патау (триюноцефалия (б); двусто-
ІІІІІИИ расщелина верхней губы и нёба (б); узкие глазные щели (б); низко
положенные (б) и деформированные (а) ушные раковины; микрогения (а);
IfCKCopHoe положение кистей)1|><и$аиные. Типичный признак синдрома Патау — расщелины
Ірчней губы и нёба (обычно двусторонние). Всегда обнаруживают-иороки нескольких внутренних органов в разных комбинациях:
И|к'кты перегородок сердца, незавершенный поворот кишечника,
<С1Ы почек, аномалии внутренних половых органов, дефекты под¬
ол удочиой железы. Как правило, наблюдаются полидактилия (чаще
іусюронняя и на руках) и флексорное положение кистей. Частота
і'ігіїлх симптомов у детей с синдромом Патау по системам следую-
|я: лицо и мозговая часть черепа — 96,5%, опорно-двигательный
Iпарат — 92,6%, ЦНС — 83,3%, глазное яблоко — 77,1%, сердечно-
:удистая система — 79,4%, органы пищеварения — 50,6%, мочевая
мсма — 60,6%, половые органы — 73,2%.Клиническая диагностика синдрома Патау основывается на соче-
ІМИИ характерных пороков развития. При подозрении на синдром
may показано УЗИ всех внутренних органов.
242Клиническая ген(В связи с тяжелыми врожденными пороками развития больШ!
ство детей с синдромом Патау умирают в первые недели или мес
жизни (95% умирают до 1 года). Однако некоторые больные жиІ
несколько лет. Более того, в развитых странах отмечается тендені
увеличения продолжительности жизни больных с синдромом Пі
до 5 лет (около 15% больных) и даже до 10 лет (2—3% больных).Другие синдромы врожденных пороков развития (синдр<
Меккеля и Мора, тригоноцефалия Опитца) по отдельным приз
кам совпадают с синдромом Патау. Решающий фактор в диаі
стике — исследование хромосом. Цитогенетическое исследоваї
показано во всех случаях, в том числе у умерших детей. Точі
цитогенетический диагноз необходим для прогноза здоровья бз
щих детей в семье.Лечебная помощь детям с синдромом Патау неспецифичес!
операции по поводу врожденных пороков развития (по жизнен^
показаниям), обіцеукрепляющее лечение, тщательный уход, пр(
лактика простудных и инфекционных болезней. Дети с синдроі
Патау практически всегда глубокие идиоты.Синдром Эдвардса (трисомия 18)Почти во всех случаях синдром Эдвардса обусловлен прос
трисомной формой (гаметическая мутация у одного из родителе
Встречаются и мозаичные формы (нерасхождение на ранних стаі
дробления). Транслокапионные формы крайне редки, и, как праві
это частичные, а не полные трисомии. Клинических различий мез
цитогенетически различающимися формами трисомии нет.Частота синдрома Эдвардса среди новорожденных состаї
1:5000-1:7000. Соотношение мальчиков и девочек 1 : 3. Причины
обладания девочек среди больных пока неясны.При синдроме Эдвардса отмечается выраженная задержка npei
тального развития при нормальной продолжительности беремен!
сти (роды в срок). На рис. 5.8-5. П показаны пороки при синдр(
Эдвардса. Это множественные врожденные пороки развития лицеї
части черепа, сердца, костной системы, половых органов. Череп дс
хоцефал и ческой формы; нижняя челюсть и отверстие рта маленькі
глазные щели узкие и короткие; ушные раковины деформировані
и низко расположенные. Из других внешних признаков отмечаю!
флексорное положение кистей, аномальная стопа (пятка выстуш
свод провисает), I і шлеп стоп короче П пальца. Спинно-мозгої
1^11 Хромосомные болезни243II'. 5.8. Новорожденный с синдро-
')двардса (выступающий заты-
(, микрогения, флексорное поло¬
умно кисти)Рис. 5.9. Характерное для синдрома
Эдвардса положение пальцев (воз¬
раст ребенка 2 мес)ІС. 5.10. Стопа-качалка (пятка
(ступает, свод провисает)Рис. 5.11. Гипогенитализм у мальчи¬
ка (крипторхизм. гипоспадия)
244Клиническая генеткгрыжа и расщелина губы встречаются редко (5% случаев синдро!
Эдвардса).Многообразная симптоматика синдрома Эдвардса у каждого б<
ного проявляется лишьчастично: лицо и мозговая часть черепа — 10(
опорно-двигательный аппарат — 98,1%, ЦНС — 20,4%, глаза — 13,(
сердечно-сосудистая система — 90,8%, органы пищеварения
54,9%, мочевая система — 56,9%, половые органы — 43,5%.Как видно из представленных данных, наиболее значимьі^І
диагностике синдрома Эдвардса изменения мозгового черепа*
лица, опорно-двигательного аппарата, пороки развития сердечі
сосудистой системы.Дети с синдромом Эдвардса умирают в раннем возрасте (S
до 1 года) от осложнений, обусловленных врожденными поро!
ми развития (асфиксии, пневмонии, кишечной непроходиї
сти, сердечно-сосудистой недостаточности). Клиническая и Д£
патолого-анатомическая дифференциальная диагностика синдр(
Эдвардса сложна, поэтому во всех случаях показано цитогенетик
ское исследование. Показания для него те же, что и при трисомии 1
(см. выше).Трисомия 8Клиническая картина синдрома трисомии 8 впервые описана
ными авторами в 1962 и 1963 гг. у детей с отставанием в умствені
развитии, отсутствием надколенника и другими врожденными noj
ками развития. Цитогенетически был констатирован мозаицизм
хромосоме из группы С или D, поскольку индивидуальной идет
фикации хромосом в тот период еще не было. Полная трисомия 8,
правило, летальна. Ее часто обнаруживают у пренатально погибі
эмбрионов и плодов. Среди новорожденных трисомия 8 встречаете
частотой не более чем 1 : 5000, преобладают мальчики (соотношеї
мальчиков и девочек 5 : 2). Большинство описанных случаев (ок<3
90%) относится к мозаичным формам. Заключение о полной три^
мии у 10% больных основывалось на исследовании одной ткани,
в строгом смысле недостаточно для исключения мозаицизма.Трисомия 8 — результат вновь возникшей мутации (нерасхоз
ние хромосом) на ранних стадиях бластулы, за исключением реді
случаев новой мутации в гаметогенезе.Различий в клинической картине полных и мозаичных ф(
не выявлено. Тяжесть клинической картины широко варьирз
рис. 5.13.10-летний мальчик с трисо-
<исй 8 (умственная недостаточность,
ІОЛ ьшие оттопыренные ушные раке*
ІІИНЬІ с упрощенным рисунком)Рис. 5.14. Контрактуры межфаланго-
вых суставов при трисомии 8Причины таких вариаций неиз¬
вестны. Корреляций между тя¬
жестью заболевания и долей
трисомных клеток не обнару¬
жено.Дети с трисомией 8 рождают¬
ся доношенными. Возраст роди¬
телей из общей выборки не выде¬
ляется.
246Клиническая генетиіДля болезни наиболее характерны отклонения в строєні
лица, пороки опорно-двигательного аппарата и мочевой системі
(рис. 5.12—5.14). Это выступающий лоб (у 72%), косоглазие, эпикан1|
глубоко посаженные глаза, гипертелоризм глаз и сосков, высо!
нёбо (иногда расщелина), толстые губы, вывернутая нижняя губа
80,4%), большие ушные раковины с толстой мочкой, контрактур^
суставов (у 74%), камптодактилия, аплазия надколенника (у 60,7Я6|
глубокие борозды между межпальцевыми подушечками (у 85,55
четырехпальцевая складка, аномалии ануса. При УЗИ выявляют
аномалии позвоночника (добавочные позвонки, неполное закрыт!
позвоночного канала), аномалии формы и положения ребер HAlj
добавочные ребра.Число симптомов у новорожденных составляет от 5 до 15 и болеПри трисомии 8 прогноз физического, психического развития
жизни неблагоприятный, хотя описаны пациенты в возрасте 17 л<
Со временем у больных проявляются умственная отсталость, гиді
цефалия, паховая грыжа, новые контрактуры, аплазия мозолист
тела, кифоз, сколиоз, аномалии тазобедренного сустава, узкий та(
узкие плечи.Методов специфического лечения нет. Оперативные вмешателї
ства проводят по жизненным показаниям.Полисомии по половым хромосомамЭто большая группа хромосомных болезней, представленная
личными комбинациями дополнительных X- или Y-XpOMOCOM, ai
случаях мозаицизма — комбинациями разных клонов. Общая частої
полисомии но X- или Y-хромосомам среди новорожденных cocTasJijj
ст 1,5 : 1000-2 : 1000. В основном это полисомии XXX, XXY и Х’’
Мозаичные формы составляют примерно 25%. В таблице 5.5 пре|
ставлены типы полисомий по половым хромосомам.Таблица 5.5. Типы полисомий по половым хромосомам у человекаТипы полисомийхромосомные наборы jХ-полисомии при отсутствии
Y-хромосомы47, XXX; 48, ХХХХ; 49, ХХХХЩХ-полисомии с одной Y-хромосомой47, XXY; 48, XXXY; 49, XXXXY^У-]юлисомии с одной Х-хромосомой-47, XYY; 48, XYYY; 49, XYYYYПолисомии по X- и Y-хромосомам48, XXYY; 49, XXXYY
)шііа 5. Хромосомные болезни247Обобщенные данные по частоте детей с аномалиями по половым
Іріїмосомам представлены в табл. 5.6.іПлица 5.6. Примерная частота детей с аномалиями по половым хромосомамЗаболеваниеКариотипРаспространенность47,XXY( ипдром
к ішйнфелтера48,XXXYДругие (48,XXYY;
49,XXXYY; мозаицизм)1:10001:25 0001:10 00047.XYY47,XYY1:1000Другие аномалии X
IIII и Y-xpoMOCOM1:1500Мужчины с XX46.ХХ1:20 ОООІксго; 1:400 мужчинСиндромШсрсшевского—Тернера45,Х46,X,i(Xq)Другие (делеции,
мозаицизм)1:50001:50 ООО1:15000Трисомия X47ЛХХ1:1000Другие аномалии
Х-хромосомы1:3000Женщины с XY46,XY1:20 ОООСиндром тестикуляр¬
ной феминизации46ДУ1:20 ОООПсего: 1:650 женщинСиндром трип л о-X (47,ХХХ)Среди новорожденных девочек частота синдрома составляет
[} : 1000. Женщины с кариотипом XXX в полном или мозаичном
ірианте имеют в основном нормальное физическое и психическое
Гінитие, обычно выявляются случайно при обследовании. Это
Объясняется тем, что в клетках две Х-хромосомы гстерохромати-
зированы (два тельца полового хроматина), а функционирует
іиіиь одна, как и у нормальной женщины. Как правило, у жснщи-
ІІ.І с кариотипом XXX нет отклонений в половом развитии, она
імеет нормальную плодовитость, хотя риск хромосомных наруше¬
нии й у потомства и возникновения спонтанных абортов повышен.
248Клиническая генеїИнтеллектуальное развитие нормальное или на нижней гран(
це нормы. Лишь у некоторых женщин с трипло-Х есть наруї
ния репродуктивной функции (вторичная аменорея, дисменої
ранняя менопауза и др.). Аномалии развития наружных полові
органов (признаки дизэмбриогенеза) обнаруживаются лишь nj
тщательном обследовании, выражены незначительно и не слу)
поводом для обращения к врачу.Варианты синдрома Х-полисомии без Y-хромосомы с числ^
Х-хромосом более 3 встречаются редко, С увеличением числа доп<
нитсльных Х-хромосом нарастают отклонения от нормы, У
щин с тетра- и лентасомией описаны отклонения в умствен!
развитии, черепно-лицевые дисморфии, аномалии зубов, ске/
та и половых органов. Однако женш;ины даже с тетрасомией
Х-хромосоме имеют потомство. Правда, у таких женщин есть поі
шенный риск родить девочку с трипло-Х или мальчика с синдрої
Клайнфелтера, потому что триплоидные оогонии образуют мої
сомные и дисомныс клетки.Синдром КлайнфелтераВключает случаи полисомии по половым хромосомам, nj
которых имеется не менее двух Х-хромосом и не менее одн^
Y-xpoMOCOMbi. Наиболее часто встрсчаюшийся и типичный
клинической картине синдром — это синдром КлайнфелтерЕ
набором 47,XXY. Этот синдром (в полном и мозаичном вариа|
тах) встречается с частотой 1 : 500—750 новорожденных мальч
ков. Варианты полисомии с большим числом X- и У-хромос|
(см. табл. 5.6) встречаются редко. Клинически они также относят
к синдрому Клайнфелтера,Присутствие У-хромосомы определяет формирование мужскс
пола. До периода полового созревания мальчики развиваются поч^
нормально, лишь с небольшим отставанием в психическом разві|
тии. Генетический дисбаланс в связи с добавочной Х-хромосоМ(!
клинически проявляется в период полового созревания в виде НЄІ
развития яичек и вторичных мужских половых признаков.Больные имеют высокий рост, женский тип телосложенц!
гинекомастию, слабое оволосение лица, подмышечных впаді
и лобка (рис, 5.15). Яички уменьшены, гистологически обн|
руживаются дегенерация герминативного эпителия и гиалиш
семенных канатиков. Больные бесплодны (азооспермия, олиг1|
спермин).
iHJi 5. Хромосомные болезни249illIliiilrїй:ійШІ&ІЦ5.15. Синдром Клайнфелтера.
іісокий рост, гинекомастия, оволо-
ІИС EUI лобке ПО женскому типуСиндром дисомии
по Y-хромосоме (47ДYY)Встречается с частотой 1:1000
новорожденных мальчиков. Боль¬
шинство мужчин с таким набо¬
ром хромосом слегка отличаются
от лиц с нормальным хромосом¬
ным набором по физическому и
умственному развитию. Ростом
они немного выше среднего,
умственно развиты, не дисмор-
фичны. Заметных отклонений
ни в половом развитии, ни в
гормональном статусе, ни в пло¬
довитости у большинства XYY-
индивидов нет. Повышенного
риска иметь хромосомно ано¬
мальных детей у XYY-индивидов
нет. Почти для половины мальчи¬
ков 47, XYY требуется дополни¬
тельная педагогическая помощь
в связи с задержкой речевого
развития, затруднений в чтении
и произношении. Коэффициент
умственного развития (JQ) в
среднем ниже на 10-15 пунктов.
Из поведенческих особенностей
отмечаются дефицит внимания,
гиперактивность и импульсив¬
ность, но без выраженной агрес¬
сии или психопатологического
поведения, в 1960—70 годы ука¬
зывалось, что пропорция XYY
мужчин повышена в тюрьмах
и психиатрических больни¬
цах, особенно среди высоких,
в настоящее время эти предполо¬
жения считаются некорректны¬
ми. Тем не менее невозможность
250Клиническая ге>предсказать исход развития в индивидуальных случаях делает
фикацию XYY-плода одной из наиболее трудных задач при rei
оком консультировании в пренатальной диагностріке.Синдром Шерешевского—Тернера (45,X)Это единственная форма моносомии у живорожденных. Не
90% зачагий с кариотипом 45,X абортируется спонтанно. Монос<
составляет 15-20% среди всех аномальных кариотипов абортЧастота синдрома Шерешевского-Тернера равна 1 : 2000*^
новорожденных девочек. Цитогенетика синдрома многооб|
Наряду с истинной моносомией во всех клетках (45,X) встре^
другие формы хромосомных аномалий по половым хромосома!
делеции короткого или длинного плеча Х-хромосомы [46,Xg
46,X,Xq~], изохромосоімьі 146,X,i(Xq); 46,X,i(Xp)j, кольцевые x\
сомы [46,X,R(X)J, a также различные варианты мозаицизма.
50-60% пациенток с синдромом Шерешевского-Тернера и|
простую полную моносомию (45,X). Единственная Х-хромс
в 80-85% случаев имеет материнское происхождение и лИ1
15—20% — отцовское.В остальных случаях синдром обусловлен разнообразным м<
цизмом (в целом 30—40%) и более редкими вариантами дел<
изохромосом, кольцевых хромосом.Клинически синдром Шерешевского-Тернера проявляете
направлениях:• гипогонадизм, недоразвитие половых органов и вторичных
вых признаков;• врожденные пороки развития;• низкий рост.Со стороны половой системы отмечаются отсутствие гонад (at
зия гонад), гипоплазия матки и маточных труб, первичная аменс
скудное оволосение лобка и подмышечных впадин, недоразі
молочных желез, недостаточность эстрогенов, избыток гипофі
ных гонадотропинов. У детей с синдромом Шерешевского—Tej
часто (до 25% случаев) встречаются разные врожденные noj
сердца и почек.Внешний вид больных достаточно своеобразен (хотя и не
да), У новорожденных и детей грудного возраста короткая Ш(
избытком кожи и крыловидными складками, лимфатический
стоп (рис. 5.16), голеней, кистей рук и предплечий, в школьно!)
особенно в подростковом возрасте выявляется отставание в рост
16. Лимфатический отек стопы
Порожденного с синдромом
Hiii'iiCKoro—Тернера. Маленькие
ногтиРис, 5.17. Девочка с синдромом
Шерешепского—Тернера (шейные
крыловидные складки, широко рас¬
положенные и недоразвитые соски
молочных желез)ІНТМИ вторичных половых признаков (рис. 5.17). У взрослых отме-
Т «кірушсіїия скелета, черепно-лицевые дисморфии, вальгуспую
ІІ1ИИЮ коленных и локтевых суставов, укорочение метакарпаль-11 метатарзальных костей, остеопороз, бочкообразную грудную
ГК у, ЇШЗКИЙ рост волос на шее, антимонголоидный разрез глазных
?Й, птоз, эпикант, ретрогению, низкое расположение ушных
)иии. Рост взрослых больных на 20-30 см ниже среднего. Тяжесть
|Иических (фенотипических) проявлений зависит от многих пока
Ізнссгньїх факторов, в том числе от типа хромосомной патологии
ІНПСОМИЯ, делеция, изохромосома). Мозаичные формы болезни,
правило, имеют более слабые проявления в зависимости от соот-
1СИИЯ клонов 46ХХ:45Х.} таблице 5.7 представлены даніїьіе о частоте основных симптомов
синдроме Шерешевского-Тернсра.
252Клиническая генепТаблица 5.7. Клинические симптомы синдрома Шерешевского—Тернера W
встречаемостьСимптомыВстречаемость,% от общего числа больны^Маленький рост100 dВрожденная лимфедема65Крыловидные складки65Низкий рост волос на шее75Уплощенная грудная клетка55 1Короткая шея50Вальгусное искривление45Изменение ногтей на стопах и кистях75Высокое нёбо70 ^Лечение больных с синдромом Шерешевского—Тернера W
плексное:• реконструктивная хирургия (врожденные пороки внутрені
органов);• пластическая хирургия (удаление крыловидных складок и т.п.)• гормональное лечение (гзстрогены, гормон роста);• психотерапия.Своевременное применение всех методов лечения, включая nj
менение генно-инженерного гормона роста, дает больным возм<
ность достичь приемлемого роста и вести полноценную жизнь.Синдромы частичных анеуплоидийЭта многочисленная группа синдромов обусловлена хромосом!
ми мутациями. Какой бы вид хромосомной мутации ни был ис>
но (инверсия, транс локация, дупликация, делеция), возникновеї
клинического хромосомного синдрома определяется либо избыт!
(частичной трисомией), либо недостатком (частичной моносоми^
генетического материала или одновременно тем и другим эффект
разных измененных участков хромосомного набора. К настояще
времени обнаружено около 1000 разных вариантов хромосомі
мутапий, унаследованных от родителей или возникших в рані
эмбриогенезе. Однако клиническими формами хромосомных С1
дромов считают только те перестройки (их около 100), но которі
itllil 5. Хромосомные болезни253іин'ано несколько пробамдов с совпадением характера цитогенети-
»1'кпх изменений и клинической картины (корреляция кариотипа
(|к'мотипа).Частичные анеуплоидии возникают главным образом в результате
нічного кроссинговера в хромосомах с инверсиями или транс-
(ікациями. Лишь в небольшом числе случаев возможно первичное
()тикновение делеций в гамете или в клетке на ранних стадиях
)иОления.Частичные анеуплоидии, как и полные, вызывают резкие откло¬
ни ил в развитии, поэтому относятся к группе хромосомных болез-
\ti\. Большинство форм частичных трисомий и моносомий не повто-
IIOI клиническую картину полных анеуплоидий. Они являются
1мостоятельными нозологическими формами. Лишь у небольшого
|М1‘ла пациентов клинический фенотип при частичных анеуплоидиях
)іиіадает с таковым при полных формах (синдром Шерешевского—
fpiicpa, синдром Эдвардса, синдром Дауна). В этих случаях речь
|Д1' I о частичной анеуплоидии по так называемым критическим для
піития синдрома районам хромосом.Какой-либо зависимости тяжести клинической картины хромо-
І0ММОГО синдрома от формы частичной анеуплоидии или от инди-
Iдуальной хромосомы нет. Величина вовлеченного в перестройку
•♦UK г ка хромосомы может иметь значение, но случаи подобного рода
<С11ьшая или большая длина) должны рассматриваться как разные
іидромьі. Обш,ие закономерности корреляций клинической карти-
[Ь1 и характера хромосомных мутаций выявить трудно, потому что
|||<)|'ие формы частичных анеуплоидий элиминируются в эмбрио-
ІЛІ.ИОМ периоде.Фенотипические проявления любых аутосомных делеционных син-
омов состоят из двух групп аномалий: неспецифических находок,
JiHHx для многих различных форм частичных аутосомных анеуплои-
1й (задержка пренатального развития, микроцефалия, гипертелоризм,
Іикаит, явно низко расположенные уши, микрогнатия, клинодак-
<лия и т.д.); комбинации находок, типичных для данного синдрома.
Іииболее подходящее объяснение причин неспецифических находок
5()Лі>іиинство из которых не имеют клинического значения) — это
|вс1[сцифические эффекты аутосомного дисбаланса как такового, а не
р чультагы делеций или дупликаций специфических локусов.Хромосомным синдромам, обусловленным частичными анеу-
їлоидиями, присущи общие свойства всех хромосомных болезней:
254Клиническая генеїврожденные нарушения морфогенеза (врожденные пороки развиті
дисморфии), нарушение постнатального онтогенеза, тяжесть клиі
ческой картины, сокращенная продолжительность жизни.Синдром «кошачьего крика»Это частичная моносомия по короткому плечу хромосомы 5 (5р«
Синдром моносомии 5р- был первым описанным синдромом,
словленным хромосомной мутацией (делецией). Это открытие еде
Дж. Лсжен в 1963 г,У детей с такой хромосомной аномалией отмечается необі
ный плач, напоминающий требовательное кошачье мяуканье
крик. По этой причине синдром был назван синдромом «кошачье!
крика». Частота синдрома достаточно высокая для делеционн!
синдромов — 1 : 45 ООО. Описано несколько сотен больных, поэТ|
му цитогенетика и клиническая картина этого синдрома изучеі
хорошо.Цитогенетически в большинстве случаев выявляется делеци!
утратой от V3 ДО V2 длины короткого плеча хромосомы 5. Потв|
всего короткого плеча или, наоборот, незначительного учасТ1|
встречается редко. Для развития клинической картины синдро<
5р — имеет значение не величина утраченного участка, а конкреД
ный фрагмент хромосомы. За развитие полного сиіідрома отв<
ствен лишь незначительный участок в коротком плече хромосомі
(5р15Л-15.2). Помимо простой делеции, при этом синдроме o5id
ружены и другие цитогенетические варианты: кольцевая хромое!
ма 5 (естественно, с делецией соответствующего участка короткої
плеча); мозаицизм по делепии; реципрокная транслокация кор(
кого плеча хромосомы 5 (с потерей критического участка) с друї
хромосомой,Клиническая картина синдрома 5р— довольно сильно разлії
чается у отдельных больных по сочетанию врожденных порої
развития органов. Наиболее характерный признак — «кошачі
крик» — обусловлен изменением гортани (сужение, мягкость XI
щей, уменьшение надгортанника, необычная складчатость слі
зистой оболочки). Практически у всех больных имеются те ш
иные изменения мозговой части черепа и лица: лунообразное лиі
микроцефалия, гипертелоризм, микрогения, эпикант, антимонг^
лоидный разрез глаз, высокое нёбо, плоская спинка носа (рис. 5.11
5.19). Ушные раковины деформированы и расположены низі
Кроме того, встречаются врожденные пороки сердца и некоторі
рйи 5. Хромосомные болезни2551‘. 5.18. Ребенок с выражеп1ТЕ.1МИ
)и таками синдрома «кошачьего
^Ика» (микроцефалия, лунообраз-
ЛИІГО, эпикант, гипсртелоризм,
<рокая плоская спинка носа, низко
іііоложеипьіе ушные раковины)Рис. 5.19. Ребенок с маловыражен-
ными признаками синдрома «коша¬
чьего крика»а их внутренних органов, изменения костно-мышечной системы
Шдактилия стоп, клинодактилия V пальца кисти, косолапость).
ІЯНЛЯЮТ мышечную гипотонию, а иногда и диастаз прямых
Ц[1Ц живота.Иілраженность отдельных признаков и клинической картины в
ЮМ меняется с возрастом. Так, «кошачий крик», мышечная гипо-
|я, лунообразное лицо с возрастом исчезают почти полностью,
микроцефалия выявляется болсс отчетливо, становятся заметнее
їихомоторное недоразвитие, косоглазие. Продолжительность жизни
льиых с синдромом 5р~ зависит от тяжести врожденных пороков
/тренних органов (особенно сердца), выраженности клинической
іргиііьі в целом, уровня медицинской помощи и повседневной
пш. Большинство больных умирают в первые годы, около 10%
зльных достигают 10-летнего возраста. Имеются единичные описа-
ІИЯ больных в возрасте 50 лет и старше.
256Клиническая генет*Во всех случаях больным и их родителям показано цитогенс
ческое обследование, потому что у одного из родителей может
реципрокная сбалансированная транслокация, которая при прохс
дении через стадию мейоза может обусловливать делсцию учас!
5р15.1-15.2.Синдром Вольфа-Хиршхорна (частичная моносомия 4р-)Обусловлен делецией сегмента короткого плеча хромосомы;]
Клинически синдром Вольфа-Хиршхорна проявляется многочі
ленными врожденными пороками с последующей резкой задержі
физического и психомоторного развития. Уже внутриутробно ОТІ
чается гипоплазия плода. Средняя масса тела детей при рождеї
от доношенной беременности составляет около 2000 г, т.е. ]
тальная гипоплазия выражена больше, чем при других частичн(
моносомиях. У детей с синдромом Вольфа-Хиршхорна отмечаї
следующие признаки (симптомы): микроцефалия, клювовиді
нос, гипертелоризм, эпикант, аномальные ушные раковины (ч2
с преаурикулярными складками), расщелины верхней губы и
аномалии глазных яблок, антимонголоидный разрез глаз, малеїРис, 5.20. Дети с синдромом Вольфа-Хиршхорна (микроцефалия, типі
телоризм, эпикант, аномальные уишые раковины, косоглазие, микр
ния, птоз)
Ilia 5. Хромосомные болезни257IИ рот, гипоспадия, крипторхизм, сакральная ямка, деформация
h)ii и др. (рис. 5,20). Наряду с пороками развития наружных орга-
)h более чем у 50% детей имеются пороки внутренних органов
|грдца, почек, ЖКТ).Жизнеспособность детей резко снижена, большинство умирают в
ііріїстс до 1 года. Описан лишь 1 больной в возрасте 25 лет.I Іитогенетика синдрома довольно характерная, как и многих деле-
іоііиьіх синдромов. Примерно в 80% случаев у пробанда выявляется
лоция часги короткого плеча хромосомы 4, а у родителей кариоти-иормальные. Остальные случаи обусловлены транслокационными
їмГ)инациями или кольцевыми хромосомами, но всегда при этом
[■мочастся потеря фрагмента 4р16.Цитогенетическое обследование больного и его родителей пока-
Ик> для уточнения диагноза и прогноза здоровья будуш;их детей,
Н’кольку родители могут иметь сбалансированные транслокации.
Ійі'іота рождения детей с синдромом Вольфа—Хиршхорна невысо-
1И (I : 100 ООО).^Иилром частичной трисомии по короткому плечу хромосомы 9 (9р+)■ )го наиболее частая форма частичных трисомий (опубликовано
М1..Ю 200 сообщений о таких больных).Клиническая картина многообразна и включает внутриутробные
гшстнатальные нарушения развития: задержку роста, умственную
Гсшлость, микробрахицефалию, антимонголоидный разрез глаз,
40(|)'гальм (глубоко посаженные глаза), гипертелоризм, округлый
>п*1ик носа, опушенные углы рта, низко расположенные оттопырен-
JC ушные раковины с уплош;енным рисунком, гипоплазию (иногда
^силазию) ногтей (рис. 5,21). Врожденные пороки сердца обнаруже-
у 25% больных.Реже встречаются другие врожденные аномалии, свойственные
ЇСМ хромосомным болезням: эпикант, косоглазие, микрогнатия,
jcoKoe арковидное нёбо, сакральный синус, синдактилии.Ьольные с синдромом 9р+ рождаются в срок. Пренатальная гипо-
11Г»ия выражена умеренно (средняя масса тела новорожденных
^00-3000 г). Жизненный прогноз сравнительно благоприятный.
)Л1>ные доживают до пожилого и прекіюнного возраста.Цитогенетика синдрома 9р+ многообразна. Большая часть слу-
кси — результат несбалансированных транслокаций (семейных или
Юрадических). Описаны и простые дупликации, изохромосомы 9р.
258Клиническая геніРис. 5.21. Синдром трисомии 9р+ (гипертелоризм, птоз, эпикант, лукові
бразпый нос, короткий фильтр, большие, низко расположенные ушные]
НИНЫ, толстые губы, короткая шея): а — ребенок 3 лег; б — женшина 21Клинические проявления синдрома однотипны при разных цитої
тических вариантах, что вполне объяснимо, поскольку во всех сл]
имеется тройной набор генов части короткого плеча хромосомыСиндромы, обусловленные микроструктурными
аберрациями хромосомВ эту группу входят синдромы, обусловленные незначительн!
размером до 5 млн и.о., делециями или дупликациями строго ощ
ленных участков хромосом. Соответственно их называют мі
делеционными и микродупликационными синдромами. Мноп
этих синдромов первоначально были описаны как доминант
заболевания (точечные мутации), однако позднее с помощью с(
менных высокоразрешающих цитогенетических методов (ос(
молекулярно-цитогенетических) была установлена истинная
логия данных заболеваний. С использованием CGH на микрочі
стало возможным обнаруживать делеции и дупликации хром^
протяженностью до одного гена с примыкающими областями,'
позволило не только существенно расширить список микродел«
онных и микродупликационных синдромов, но и приблизит!
Idii 5. Хромосомные болезни259III манию генофенотипических корреляций у пациентов с микро-
їуктурньїми аберрациями хромосом.И мен но на примере расшифровки механизмов развития данных
можно видеть взаимное проникновение цитогенетиче-
14 методов в генетический анализ, молекулярно-генетических
Шяов к клиническую цитогенетику. Это позволяет расшиф-
м.|иать природу ранее непонятных наследственных болезней,
І'цкже выяснять функциональные зависимости между генами.
ІСИИДНО, что в основе развития микроделеционных и микроду-
(каиионных синдромов лежат изменения дозы генов в участке
)М1)сомы, затронутом перестройкой. Однако пока не установ-
И), что именно составляет основу формирования большин-III таких синдромов — отсутствие конкретного структурного
in или более протяженного участка, содержащего несколько
ІОИ. Болезни, которые возникают вследствие микроделеций
сгка хромосомы, содержащего несколько генных локусов,
слложено называть смежными генными синдромами. Для фор-
|р1>нания клинической картины данной группы заболеваний
іііципиально важно отсутствие продукта нескольких генов,
[■рагиваемых микроделецией. По своей природе смежные генные
Цідромьі находятся на границе между менделируюшими моно-
1НЫМИ заболеваниями и хромосо.мными болезнями (рис. 5.22).<п генетическом
злезни*1змер
зстройкиСмежные генные
синдромыМенделирующиезаболеванияХромосомныеболезни10“10^ 10"10®10' 10®10*Геном человека (п.о.)5.22. Размеры геномных перестроек при различных типах генетических
Яечией. (По Stankiewicz Р., Lupski J.R. Genome architecture, rearrangementsI gciiomic disorders //Trends in Genctics. — 2002. — V. 18 (2). — P. 74—82.)
260Клиническая гєнвіТипичным примером такого заболевания является синд|
Прадера-Вилли, возникающий вследствие микроделеции
мером 4 млн и.о. в регионе qll—ql3 на хромосоме 15 отцово^
происхождения, Микроделеция при синдроме Прадера-Ви|
затрагивает 12 импринтированных генов (SNRPN, N~DN, МАО\
и ряд других), которые в норме экспрессируются только с ОТІ
ской хромосомы.Остается также пока неясным, как влияет на клиниче<
проявление микроделеционных синдромов состояние локу(
гомологичной хромосоме. По-видимому, природа клиничео!
проявлений разных синдромов различна. Патологический
цесс при некоторых из них развертывается через инактиваї
опухолесупрессоров (ретинобластомы, опухоли Вильмса),
ника других синдромов обусловлена не только делециями
таковыми, но и явлениями хромосомного импринтинга и о;
родительских дисомий (синдромы Прадера-Вилли, Ангельм!
Беквита-Видемана). Клинические и цитогенетические харг
ристики микроделеционных синдромов постоянно уточня]
в таблице 5.8 приведены примеры некоторых синдромов,
словленных микроделециями или микродупликациями неб(
ших фрагментов хромосом.Таблица 5.8. Общие сведения о синдромах, обусловленных микроделещ
иди микродупликациями хромосомных регионовНазвание
синдрома
(или болезни)Вовлечен¬ныйучастокхромосомыРазмер наи¬
более частой
делении
или дунлнка-
цин (млн н.о.)Основные проявления 1СиндромСотосаdel(5)(q35)2,2Гигантизм, макроцефалия
лицевые аномалии (высо-Ч
кий широкий лоб, напо- л
минающий перевернутую
грушу, смещение скуловойj
кости, заостренный подбоч
родок, облысение в лобно{|,
височной части), умственн!
отсталость ^
ІНІІ 5. Хромосомные болезни261Продолжение таблицы 5.8Название
синдрома
(или болезни)Вовлечен¬ныйучастокхромосомыРазмер наи¬
более частой
лелеции
или дуплика¬
ции (млн н.о.)Основные проявленияI(ипдром
|Иіілі.ямса—
[|»оИ|-)сна илиjl'HIIJlpOMji.'uma зльфаі»del(7)(qll.23)1,6Лицевые аномалии (эпикант,
короткий нос с открытыми
вперед ноздрями, уплощен¬
ная средняя часть лица,
широкая верхняя челюсть,
микрогнатия, оттопыренные
уши), надклапанный стеноз
аорты или легочной артерии,
гиперкальциемия[(’ипдром
[Трихорино-
[фліішге-
|М;м>иый
(О типа
ЦЛингера-
Гн.'їсона)
пис. 5.23)del(8){q24.Il-q24,13)4.0Дисморфии лицевого черепа,
большие оттопыренные уши,
множественные экзостозы,
низкий рост, клинобрахидак-
тилия, умеренная умственная
отсталостьопухоль
Іильмса либо
Щиридияdel(ll)(pl3)Различный
размер деле¬
ний (до 4,0
млн п.о.)Отсутствие радужной обо¬
лочки, нейробластома,
гонадобластома, умственная
отсталостьм иноблас-
Гі)маdel(13)(ql4.l-ql4.2)Различный
размер деле-
цийОпухоль сетчатки (одно- или
двусторонняя) в детском воз¬
расте.ипдром
Ірпдера-
Іилли
[(рис. 5.24)del(15)
(qll-ql3) (в
хромосоме
от отца)4,0Ожирение туловища и прок¬
симальных отделов конеч¬
ностей, дисморфии лицевого
черепа, гипотония, гипогона-
дизм, умственная отсталость,
ма^ченькие кисти и стопы[Синдром
Пиельмана
f(pHc. 5.25)del(15)
(qll-ql3) (в
хромосоме
от матери)4,0Необычное лицо, атаксия,
гипотония, эпилепсия,
пароксизмы смеха, микроце¬
фалия, отсутствие речи
262Клиническая генвіОкончание таблицыНазвание
синдрома
(или болезни)Вовлечен’НіЛЙучастокхромосомыРазмер наи¬
более частой
делеции
или дуплнка-
цнн (млн л.о.)Основные проявлениякСиндром
М иллера—
Дикераde!(17)(РІЗ.З)1,5Агирия (лиссэнцсфалия),
микроцефалия, пороки серії
ца, пороки почек, дисморфй
лицевого черепа, гипотония
судорожные припадки чСиндромСмита-Магенисаde](17)(рИ.2)5,0Умерен ітая или глубокая
степень умственІЇОЙ отстало
сти, агрессивное поведение,
склонность к наггесению ,
самоповреждений, наруше-
ргия сна IНейро-
фиброматоз
I типаdel(17)(qIl-2)L5Наличие на коже пятен цвв1
кофе с VtOJlOKOM, нейрофи- 'бромы, глиома зрительного
нерва *СиндромДиДжорджииливелокардио-
фациальный
синдром
(рис. 5.26)del (22)
(ql1.2)3,0Судороги (гипокальииеми'^і
ские), аплазия или гипоплй
зия тимуса, лисморфия лиш
вого черепа, пороки сердцаСиндром
Беквита-
Видемана
(рис. 5.27)dup(]l)(pl5)Различный
размер
дупликаций
(до 5,0 млн
и.о.)Грыжа пупочного канатика;
мякроглоссия, гигантизм,
гипогликемия, микроце- і
фал ИЯ, врожденные пороки
внутренних органов, верти-*,
кальныс бороздки на мочка!
ушей ^Большинство микроделеционных/микродупликационных сщ
дромов встречается редко (1 : 50 000—100 ООО новорожденных),
клиническая картина, как правило, отчетливая. Диагноз мож|
поставить по совокупности симптомов. Однако в связи с проп
зом здоровья будущих детей в семье, в том числе у родственнш
Цмм.і 5. Хромосомные болезни263йі‘.5.23.СиндромЛангера-Гидеона. Рис, 5.24. Мальчик с синдромомМЮЖеСТВСННЫе ЭКЗОСТОЗЫПрадера-ВиллиРис. 5.26. Ребенок с синдромом
Д и Джорджи5.25. Девочка с синдромом
ІАніельмана
264Клиническая ген«родителей пробаида, необходи!
провести высокоразрешающ|
цитогенетическое исследован!
у пробанда и его родителей.
Клинические проявлені
синдромов сильно варьируї
в связи с разной протяжені
стью делеции или дупликаш
а также в связи с родительс*
принадлежностью микроне]
стройки — унаследована ли
от отца или от матери. В послі
нем случае речь идет об импрі
тинге на хромосомном уі
не. Это явление было откр!
при цитогенетическом изучеі
двух клинически различают!
ся синдромов (Прадера—Вш
и Ангельмана). В обоих cjfj
чаях микроделеция наблю/
ется в хромосоме 15 (участі!
qll-ql3). Лишь молекуляр!
цитогенетическими метода!
установлена истинная приї
синдромов (см. табл. 5.8), Участ
qll-ql3 в хромосоме 15 да
настолько выраженный эфф«
импринтинга, что синдромы могут быть вызваны однородительски!
дисомиями (рис. 5.28) или мутациями с эффектом импринтинга.Как видно на рис. 5.28, дисомия по материнским хромосомам]
вызывает синдром Прадера-Вилли (потому что отсутствует участ
qll—ql3 отцовской хромосомы). Такой же эффект дает делеция эт
же участка или мутация в отцовской хромосоме при нормалы
(биродительском) кариотипе. Прямо противоположная ситуаі
наблюдается при синдроме Ангельмана.Более подробные сведения об архитектуре генома и наследству
ных болезнях, обусловленных микроструктурными нарушения,]!
хромосом, можно найти в одноименной статье С.А. Назаренко
компакт-диске.Рис. 5.27. Поперечные насечки на
мочке уха — типичный симптом при
синдроме Беквита-Видемана (ука¬
заны стрелкой)
ІВИ.1 5. Хромосомные болезни265м рАктивный
локус СА□Актив¬ныйпокуиСПВспвСАМО мм МО МО 00 МОАII IIII jknДелеция Материнская Имприґ^тині Депсция Отцовская И\"пртітииг-
ОРД мутация ОРД мутация►И1’. 5.28. Три класса мутнций при синдроме Пралера-Вилли (СПВ) и (СА)
ІИІ ічіьмана: М — мать; О — отец; ОРД — одпородительская дисомияФАКТОРЫ ПОВЫШЕННОГО РИСКА РОЖДЕНИЯ
ДЕТЕЙ С ХРОМОСОМНЫМИ БОЛЕЗНЯМИU последние десятилетия многие исследователи обращались к
|ричинам возникновения хромосомных болезней, Не вызывало
ом пений, что образование хромосомных аномалий (и хромосомных,
1СНОМНЫХ мутаций) происходит спонтанно. Экстраполировались
^ультаты экспериментальной генетики и предполагался индуциро-
11111 ый мутагенез у человека (ионизирующая радиация, химические
іутагеньт, вирусы). Однако реально причины возникновения хромо-
)М1(ых и геномных мутаций в зародышевых клетках или на ранних
Шдиих развития зародыша до сих пор не расшифрованы.Проверялись многие і’ипотезьі нсрасхождения хромосом (сезон-
<»С1Ь, расово-этническая принадлежность, возраст матери и отца,
Ідержанное оплодотворение, порядок рождения, семейное нако-
ІЛСІІИЄ, лекарственное лечение матерей, вредные привычки, негор¬
омал ьная и гормональная контрацепция, флюридины, вирусные
)лозни у женщин), в большинстве случаев эти гипотезы не под-
fi^cpдилиcь, но генетическая предрасположенность к болезни не
иключается. Хотя в большинстве случаев нерасхождение хромосом
I человека спорадическое, можно предполагать, что оно в определен-
ІПЙ степени генетически детерминировано. Об этом свидетельствуют
ісдующие факты;
2вбКлиническая генвті• потомство с трисомией появляется у одних и тех же ЖЄН1
повторно с частотой не менее 1%; л' родственники пробанда с трисомисй 21 или другими анеупл<
днями имеют несколько повышенный риск рождения ребенї
анеуплоидией;' кровное родство родителей может повышать риск трисоми!
потомства;• частота зачатий с двойной анеуплоидией может быть выше,
предсказывается в соответствии с частотой отдельных ан|
плоидий.К биологическим факторам повышения риска нерасхожден(
хромосом относится возраст матери, хотя механизмы этого явл
ния неясны (табл. 5.9, рис. 5.29). Как видно из табл. 5.9, риск р(
дения ребенка с хромосомной болезнью, обусловленной анеупл<
дией, с возрастом матери постепенно повышается, но особен|
резко после 35 лет, У женщин старше 45 лет каждая 5-я берем<
ность завершается рождением ребенка с хромосомной болезш
Наиболее четко возрастная зависимость проявляется для три<%
40
3530252015105О>-ґ' “T pfflr' " -yj»:* іТ'.-'ІЗч...,. . . ^^ ii Til-ll I*-1 .. S f . 1.y-b- ? ■■ _r.20 25 30 35 36 38 39 40 41 42 43 44 45 46 47 48 49Материнский возраст в годахРис. 5.29. Зависимость частоты хромосомных аномалий от возраста мат
1 — спонтанные аборты при зарегистрированных беременностях; 2 — обі
частота хромосомных аномалий во И триместре; 3 — синдром Дауна во ТІ'
местре; 4 — синдром Дауна среди живорожденных
(Ita 5. Хромосомные болезни267|Міі 21 (болезнь Дауна). Для анеуплоидий по половым хромосомам
||)лст родителей либо совсем не имеет значения, либо его роль
їгіи> незначительна.|А.||нца 5.9. Зависимость частоты рождения детей с хромосомными болезня¬
ми от возраста материИо фаст матери,
годыЧастота рождения детейс болезнью Даунас любой хромосомной
болезнью201:18001:500251:13001:500301:10001:400351:3001:200401:1001:70451:301:20491:121:8На рис. 5.29 видно, что с возрастом повышается также частота
пинанных абортов, которая к 45 годам увеличивается в 3 раза и
олее. Такое положение .можно объяснить тем, что спонтанные абор-
но многом обусловлены (до 40-45%) хромосомнь[ми аномалиями,
liToia которых имеет возрастную зависимость.И|>1ше были рассмотрены факторы повышенного риска анеуплои-
ІІІ у детей от кариотипичсски нормальных родителей. По существу,
многочисленных предполагаемых факторов только два имеют зна¬
нию для планирования беременности, а точнее, являются строгими
Жіізаниями для пренатальной диагностики. Это рождение ребенка
«ису плои дией по аутосомам и возраст матери старше 35 лет.
Цитогенетическое исследование у супружеских пар позволяет
шшть кариотипические факторы риска: анеуплоидию (в основном
I мозаичной форме), робертсоновские транслокации, сбалансирован-
|Ыс реципрокныс транслокации, кольцевые хромосомы, инверсии,
|«т,иисние риска зависит от типа аномалии (от 1 до 100%); напри-
|ер, если у одного из родителей в робертсоновскую транслокацию
)Н;ючены гомологичные хромосомы (13/13, 14/14, 15/15, 21/21, 22/22),
члорового потомства у носителя таких перестроек быть не может,
временности будут заканчиваться либо спонтанными абортами (во
}ох случаях транслокаций 14/14, 15/15, 22/22 и частично при транс-
268Клиническая ген<локациях 13/13, 21/21), либо рождением детей с синдромом П|
(13/13) или синдромом Дауна (21/21).Для расчета риска рождения ребенка с хромосомной болезі
в случае аномального кариотипа у родителей были составлю
таблицы эмпирического риска. Теперь в них почти нет нео<
димости. Методы пренатальной цитогенетической диагност!
позволили перейти от оценки риска к установлению диагноз
эмбриона или плода.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯИзохромосомыИмпринтинг на хромосомном уровне
ИзодисомииИстория открытия хромосомных болезнейКлассификация хромосомных болезнейКольцевые хромосомыКорреляция фено- и кариотипаМикроделеционные синдромыОбщие клинические черты хромосомных болезнейОднородительские дисомииПатогенез хромосомных болезнейПоказания для цитогенетической диагностикиРобертсоновские транслокацииСбалансированные реципрокныс транслокацииТипы хромосомных и геномных мутацийФакторы риска при хромосомных болезняхХромосомные аномалии и спонтанные абортыЧастичные моносомииЧастичные трисомииЧастота хромосомных болезнейЭффекты хромосомных аномалийРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАБаранов B.C., Кузнецова ТВ. Цитогенетика эмбрионального разви!
человека: научно-практические аспекты. — СПб.: Научная литер*
ра, 2007. - 640 с.
Шіі 5. Хромосомные болезни269luiimep Е.К. Медицинская генетика. — М.: Медицина, 2003. —Кошова С.И., Демикова Н.С. Наследственные синдромы и медико-
Ііігтическое консультирование: атлас-справочник. — 3-є изд., доп.
Іпі’р^'раб. — М.: Т-во научных изданий КМК; Авторская академия,
||0/. - 448 с.: 236 ил.Иашрепко С.А. Изменчивость хромосом и развитие человека. —
І|ім(.к; Изл-во Томского государственного университета, 1993. —
П с.IІрокофьева-Бельговская Л.Л. Основы цитогенетики человека. — М.:
(сппцина, 1969. — 544 с.Пушрёв В.П., Степанов В.А. Патологическая анатомия генома чело-
І^Кії. — Новосибирск: Наука, 1997. — 223 с.Смирнов В.Г Цитогенетика. — М.: Высшая школа, 1991. — 247 с.
ГлаваБОЛЕЗНИ с НАСЛЕДСТВЕННІ
ПРЕДРАСПОЛОЖЕННОСТЬ!ОБЩАЯ ХАРАКТЕРИСТИКАНаряду с болезнями, этиологически строго детерминированньЦ
наследственностью (генные и хромосомные) или факторами сре
(травмы, ожоги), есть большая группа болезней, развитие котор|
определяется взаимодействием определенных наследственных
торов (мутаций или сочетаний нормальных аллелей разных геж
и факторов среды. Их называют болезнями с наследственной п(
расположенностью или многофакторными заболеваниями. Терми!
«болезни с наследственной предрасположенностью» и «многофакт
ные заболевания» означают одно и то же. В русской литературе чаї
пользуются термином многофакторные (или мультифакториальш
заболевания.Многофакторные болезни могут возникать внутриутро(
(врожденные пороки развития) или в любом возрасте пості
тальною развития. При этом чем старше индивид, тем больї
вероятность развития у него многофакторного заболевания,
отличие от моногенных, многофакторные болезни относятся^
распространенным заболеваниям (табл. 6.1). Большинство mj
гофакторных болезней с генетической точки зрения ЯВЛЯ!
ся полигенными, т.е. в их формировании участвуют несколь!
генов. Болезни с моногенной наследственной предрасполож<
ностью (недостаточностью Г6ФДГ, аномальной холинэстераз(
злокачественной гипертермией и др.) обычно рассматривают
как моноген ные болезни.Врожденные пороки развития, такие, как расщелина губы и нё(
анэнцефалия, гидроцефалия, косолапость, вывих бедра и другі
формируются внутриутробно к моменту рождения и, как прави;
диагностируются в самые ранние периоды постнатального от* Исправлено и дополнено при участии д-ра биол. наук ИМ. Лебедеві
Цйіін 6. Болезни с наследственной предрасположенностью271|тма. Их развитие — результат взаимодействия многочисленных
Ик’гических факторов с неблагоприятными материнскими факто-
IMI1 или факторами среды (тератогены) в период развития плода,
bill истречаются в популяциях человека по каждой нозологической
нечасто (см. табл. 6.1), но суммарно — у 3—5% популяции.I Ісихические и £1ервные болезни, а также соматические болезни,
|ііи»сящиеся к группе многофакторных заболеваний, являются
|0'1игенными (генетически гетерогенными), но развиваются во
|’4ітмодействии с факторами внешней среды в постнаталъном
|Р|1И()де онтогенеза у взрослых индивидов. Эта группа относится
гоциально значимым распространенным болезням (в англий-
|кімі і'ранскрипции они обозначаются как «commone diseases»):
?|1лсчно-сосудистые (инфаркт миокарда, артериальная гипертен-
|и. инсульт), бронхолегочные (бронхиальная астма, хронические
)Пируктивные заболевания легких), психические (шизофрения,
пюлярный психоз), злокачественные новообразования, инфск-
1И»)МНЫе болезни и др.іблица 6.1. При.меры болезней с наследственной предрасположенностыоГруппы и нозологические формыРасиространенность
на 1000 человек
(в соответствующей
возрастноп группе){Йрожденные пороки развитияіЛісіцелина губы и нёба1-2(’і)инномозговая грыжаСісиоз привратника0,5-3Ан энцефалия и черепно-мозговая грыжаІІІ.П1ИХ бедра2-5Гидроцефалия0,5(нноспадияКосолапостьПсихические и нервные болезниШизофрения10-20'•)!1ИЛСНСИЯ8-юЬиполярный психоз2-5Риі;і;еянннй склероз0,02-0,7
272Клиническая генвТ|Окончание таблицыХГруппы и нозологические формыРаспространенное!на 1000 человек ]
(в соответствующе
возрастной rpynnijСоматические болезниПсориаз10-20 'Бронхиальная астма2-5Язвенная болезнь желудка и двенадцатиперстной
кишки20-50 1Ишемическая болезнь сердца50-100Гипертоническая болезнь100-200ДиабетЮ-20Для многофакторных болезней можно предложить следуЮ1
схему причин их развития (рис. 6Л).Схема включает несколько положений. Во-первых, многофаі
ные заболевания представляют собой результат сложного взаиі
действия генетических и внешнесрсдовых факторов, причем и те,^
другие многочисленны. Во-вторых, в популяиии эти факторы р|
пределяются и комбинируются случайным образом. В-третьих,
факторы действуют аддитивно, т.е. их суммарный эффект на болез
теоретически равен сумме эффектов каждого из них в отдельно<
хотя в действительности это более сложный процесс взаимодейст!
факторов, до конца не изученный.Передача многофакторных болезней в семьях не соответствз
законам Менделя. Распределение таких заболеваний в популяциїПРИЧИНЫ1- 1 ПСтохастическиеСредовыеГ енетические(случайные)Семейные Популяционные Гены Генетический
предрасположения фонРис. 6.1. Причины болезней с наследственной предрасположенностью
iHii 6. Болезни с наследственной предрасположенностью273і|Ч’і ация (распределение больных и здоровых) в семьях приниипи-
|hiu> отличается от моногенных (мендслируюших) болезней.Риск развития заболевания у ребенка зависит от состояния здо¬
ровья родителей. Так, если один из родителей больного ребенка
гакже страдает бронхиальной астмой, вероятность развития
заболевания у ребенка колеблется от 20 до 30%; если больны оба
родителя — она достигает 75%. В целом считается, что риск воз¬
ії икновения бронхиальной астмы у ребенка, родители которого
имеют признаки атопии, в 2—3 раза выше, чем в тех семьях, в
которых родители не имеют этих признаков. Риск в семье, когда
болеют оба родителя, значительно превышает индивидуальные
риски каждого из родителей и, что очень важно, не является
простой суммой рисков каждого из родителей, при сравнении
потомков здоровых людей и потомков больных бронхиальной
астмой оказалось, что риск для ребенка заболеть бронхиальной
астмой в 2,6 раза выше, если болеет мать, в 2,5 раза выше, если
болен отец, и в 6,7 раза выше, если болеют оба родителя. Эти
результаты могут быть объяснены аддитивным взаимодействи¬
ем многих генов предрасположенности к развитию бронхиаль¬
ной астмы у ребенка.- Если в популяции многофакторные заболевания встречаются
с разной частотой среди лиц разного пола (половой димор¬
физм), то риск заболевания выше в семьях пробандов реже
поражаемого пола (и наоборот). Так, инфаркт миокарда в
30-40 лет случается чаще у мужчин, чем у женщин. Однако
если речь идет о прогнозе, например, инфаркта миокарда для
потомков больной матери инфарктом миокарда в этом же воз¬
расте, то для них риск поражения будет выше, чем для потом¬
ков больного отца.- Если в двух сравниваемых популяциях многофакторные забо¬
левания встречаются с разной частотой, то риск для родствен¬
ников выше, если семья принадлежит к более поражаемой
популяции. Для моногенных болезней, как известно, риск для
родственников не зависит от популяционной частоты. Для
муковисцидоза, например, встречающегося среди европей¬
ского населения чаще, чем среди африканского, вероятность
поражения для детей в браке здоровых родителей-гетерозигот
одна и та же — 25%. Другое дело для многофакторных болез¬
ней, например, артериальной гипертензии. Ее частота среди
274Клиническая гвнвісибирских народностей составляет не более 5%, в то время;
у белого населения Южной Африки соответствующего bo3J
та — 25—30%. В отношении этой патологии риск выше для;
ственников из семей, принадлежащих к южно-африканс
популяции.— Риск заболеть для родственников зависит от степени тяже(
болезни у пробанда. Если заболевание манифестирует в рані
возрасте, протекает более тяжело и за короткий срок призе
к необратимым изменениям, риск для следующих детей cyt
ственно возрастает, а наследственная отягощенность прояі
ется в этих семьях большим процентом лиц с субклиническ!
проявлениями болезни.в целом генетический риск для родственников в отношении мс
генной патологии, как правило, выше, чем в случае многофакторі
Частота многофакторного заболевания среди родственников болі
го 1-й степени родства приблизительно равнагде q — частота болезни в популяции.Это подтверждается расчетами с использованием коэффицив
риска (Я), указываюш[его на степень генетического вклада в болез
Если X >2, то утверждается, что генетический вклад в болезнь
любой исследуемый признак значителен. Некоторые примеры и
мулы для расчета риска болезни приведены в табл. 6,2,Таблица 6.2. Коэффициенты риска (X) для многфакторных и моноген|
заболеванийБолезниX 1С наследственной предрасположенностью ]Болезнь Альцгеймера3-4 1Диабет I типа15 1Диабет 2 типа3-4 jШизофрения8-9 jРассеянный склероз20-30]Аутизм75-150^Моногенные IМуковисцидоз500 jХорея Гентинг тона (доминантная форма)5000 '
|впіі 6. Болезни с наследственной предрасположенностью275ПОДХОДЫ к ИЗУЧЕНИЮ НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТИ К БОЛЕЗНЯМ
ЧЕЛОВЕКА( рсди основных подходов к оценке роли наследственных
ІЦК горев в этиологии и патогенезе широко распространенных
|Ц(иофакторных заболеваний следует назвать три; клинико-
цимлогический, близнецовый и популяционный. Долгое время
|и были главными способами доказательства существованияI ислственной предрасположенности. Этот период исследова-
|Ии Гюлезней с наследственной предрасположенностью называют
Ііірміїльно-генетическим, так как названные три подхода надежно
і\\ іьівали на значение наследственных факторов в возникновении
(ш иштии таких болезней, давали возможность измерить степень
<11С1 ИЯ их в этих процессах, но не позволяли идентифицировать
)икретные гены, составляющие основу наследственной пред-
Ісіюложенности. До недавнего времени возможность описания
рруктуры наследственной компоненты многофакторного заболе-
ИІПМ в терминах задействованных генов практически отсутство-
ІЛіі. Она появилась с осуществлением международного проекта
Геном человека» — 90-е годы XX в. открыли «охоту за генами»
шей, в том числе и многофакторных. У медицинских генети-
И1 появился, по выражению В. Маккьюсика (1997), «свой» объект
Следования — геном.Однако прежние методы изучения наследственной предраспо-
)Жеиности человека к многофакторным заболеваниям (клинико-
^Нсалогический, близнецовый, популяционный) не утратили своего
(«чей и я. Более того, они являются базовыми в идентификации
подверженности болезням при использовании современных
ІИсгических технологий {полногеномные SNP-микрочипы для гено-
щирования, полногеномные экспрессионные микрочипы, высоко-
;>оизводительное секвенирование и др.).(Линико-генеалогические доказательства наследственной
>драсположенности’Эмпирическое наблюдение за семьями с сердечно-сосудистыми,
||лудочно-кищечными, психическими, аллергическими заболе-
ІНИЯМИ давно уже побуждало врачей предполагать роль наслед-
276Клиническая гениственности в возникновении этих болезней. Более строгие Д{
зательства можно получить любым из трех способов анал!
генеалогических данных.~ Всех опраіииваемьіх разделяют на группы в зависимості
наличия больных в родословной. При этом способе анал!
пробандами являются только больные, от которых получ(
генеалогические сведения в отношении изучаемой болеі
у родственников определенной степени родства. Напрш
у лиц с гипертонической болезнью собирают сведения о
чии гипертонии у живых или умерших родителей. Затем
пробандов разделяют на группы: а) имеющих здоровых pdj|
телей (без гипертонической болезни); б) имеющих одного б(
ного родителя; в) имеющих двух больных родителей, в зак)
чение проводится сравнение величин выборок этих трех гр>
при болезнях с более ранним проявлением (например, броні
альная астма) для разделения пробандов на группы за ocHJ
принимают наличие больных братьев и сестер.— Частоту больных среди родственников больных пробаї
сравнивают со специально подобранной контрольной rj
пой, т.е. с группой лиц, не страдающих изучаемой болезны
учетом пола, возраста, этнической принадлежности, быто|
условий, производственных вредностей и т.д.). Всю работ}
сбору клинико-генеалогических данных проводят строго ш
тично для родственников соответствующей степени родсі
Например, из большого числа студентов отбирают две приї
но равные по величине группы: больные язвенной болезні
здоровые. Далее собирают клинико-генеалогичсские данні
наличии язвенной болезни у родителей, братьев, сестер.
сравнивают частоту язвенной болезни у родственников здс^
вых и больных студентов.— Заболеваемость в семьях больных (обобщенно) можно cj
нивать с популяционной частотой этой же болезни. Tai
подход — фактически комбинация клинико-генеалогичесі
и популяционно-статистического методов. Его широко
меняли для изучения наследственной природы тизофренц
аллергии, ревматизма и др.Генетический анализ родословных на основе менделевского,
дискретного разделения потомства по фенотипу для болезне!
іиіі 6. Болезни с наследственной предрасположенностью277Іічнглственной предрасположенностью, можно применять только в
flKi ни ценных пределах и на больших выборках, чтобы в них вошел
т риал по всему многообразию клинического проявления данного
Упования. При этом, по-видимому, следует ограничивать иссле-
Лтмие данными о родственниках ТІ степени родства (в крайнем
іУ'іас, не далее III степени).1ри клинико-генеалогическом изучении болезней с наследствен-
lilt предрасположенностью анализировать родословные следует осо-
ИНІО тщательно. Наряду с получением обычной для моногенных
^ирм информации (наличие болезни) необходимо обращать особое
ІІIIмание на следующие методические вопросы.Поскольку диагностика стертых форм многофакторной пато¬
логии трудна, нужно в полной мере обеспечить точность диа¬
гностики заболевания у всех членов семьи.- При оценке выраженности болезни у разных членов семьи сле¬
дует оценивать сходство фенотипического проявления среди
различных пораженных родственников (течение болезни, воз¬
раст начала, тяжесть, реакция на лекарства и т.д.). Большое
сходство клинической картины обычно указывает на суще¬
ственное значение генетической компоненты и, следовательно,
на повышенный риск развития заболевания у близких кровных
родственников.- Степень кровного родства среди больных членов семьи должна
быть установлена точно. При полигенном характере болезни
это важно знать для оценки риска, так как степень риска у близ¬
ких родственников резко возрастает.- Необходимо собирать подробные и точные сведения о действии
факторов среды в пре- и постнатальном периодах. Это позволя¬
ет существенно уточнить прогноз.Особый способ клинико-генеалогического изучения пред-
»с110Л0женн0сти к болезням ~ изучение заболеваемости среди
|Иологических и приемных родственников больных или метод
Іриемньїх детей, в таких семьях имеется своеобразный кон-
Іроль — генетически неродственные индивиды, имеющие с про-
Іймдом общие семейные средовые влияния. Анализ этих семей
^Кнзался особенно полезным для доказательства генетической
Іредрасположенности к шизофрении и алкоголизму: заболе-
Інсмость в 2—3 раза выше у биологических родственников по
278Клиническая генвісравнению с таковой у членов семьи, не имеющих крові
родства. В частности, частота алкоголизма у приемных дв|
коррелирует с этим показателем у биологических poдитeJ
У приемных детей масса тела и явная полнота коррелируют с
ВЫМИ у биологических родителей, а не у членов семей, в KOTOJ
выросли приемные дети. Такая же закономерность отмечается
сравнении супругов. Например, частота желчнокаменной боле^
среди сибсов больных выше по сравнению с общепопуляционі
а у супругов ниже, чем у пробандов.Близнецовые исследованияПри изучении многофакторных болезней большие надо
возлагались на близнецовый метод как достаточно объективны!
чувствительный. Исследования многофакторной патологии щ
водились путем сравнения конкордантности моно- и дизиготі
близнецов. Второй вариант близнецового метода — сравнеї
конкордантности монозиготных близнецов, выросших вмест
порознь, не применялся из-за трудности подбора соответствз
щих групп, хотя для изучения болезней с наследственной преді
положенностью он был бы более чувствительным. Обобщен!
результаты использования близнецового метода для понимаї
природы разных заболеваний представлены в табл. 6.3.Таблица 6.3, Конкордантность близнецовых пар для разных групп болезнеТип заболеванияКонкордантность близнецов,монозиготныхДИЗИГОТНЬЦБолезни с наследственной предрас¬
положенностью40-604-18 jАутосомно-доминантные болезни10050 1Аутосомно-рецессивные болезни10025 1С помощью близнецового метода проведены многочислен!
исследования природы предрасположенности ко многим заболв|
ниям, в том числе к сердечно-сосудистым болезням, в таблице
представлены некоторые обобщенные результаты изучения генет
ской компоненты в происхождении болезней этой группы. Как ви^
из данных табл. 6.4, во всех случаях конкордантность монозиготн]
близнецов выше, чем дизиготных.
iDii 6. Болезни с наследственной предрасположенностью279діица 6.4. Результаты исследований предрасположеттности к сердечно¬
сосудистым болезням близнецовым методомБолезньКонкордантность близнецов, %монозиготныхдизиготныхл М с 1) гон и я26,210,0Інфаркт миокарда19,615,5ІМІ Л'льт22,410,8ГПМПТИЗМ26,010,5( помощью близнецового метода изучали роль наследственности
ироисхождснии опухолей. Несмотря на низкую конкордантность
рншиготных близнецов, в 30—40-х годах XX в. был сделан вывод об
цюдсленном значении наследственной предрасположенности для
ІИІИКНОВСНИЯ злокачественных опухолей. В целом на основании
іииієцовьіх исследований было сделано заключение, что значе¬
не внешних факторов в возникновении рака намного больше, чем
Цслсдственных. Теперь ЯСІ10, что подобная постановка вопроса в
IIIIЮМ случае неправомерна. Главным итогом близнецовых иссле-
^иаиий природы рака стало привлечение внимания к генетической
||срминации злокачественных новообразований. В дальнейшем для
01 о уже использовали клинико-генеалогический, цитогенетический
[Молекулярно-генетический методы, которые и определили прогресс
Грисшифровке природы злокачественных новообразований.1>лизиецовый метод применяли для изучения наследственной
>ироды такого сложного явления, как аллергия, Было показано, что
Яікордантность монозиготных близнецов по разным проявлениям
ілсргических реакций выше, чем дизиготных.Ьлизнецовым методом также показана наследственная предрас-
)Л()жеыность к некоторым инфекционным болезням (полиомиели-
|f, туберкулезу). Конкордантность монозиготных близнецов по этим
(Оолеваниям в несколько раз больше, чем дизиготных.Выше были приведены доказательства наследственной предраспо-
)Жснности к широко распространеі[ньім заболеваниям, полученные
іимико-генеалогическим и близнецовым методами по отдельности,
жазательства генетической предрасположенности к широко рас-
їостраненньїм болезням, полученные этими методами для одних и
IX же нозологических форм, совпадают.
280Клиническая ген<Популяционные исследованияПопуляционные подходы к исследованию вопросов генетики
гофакторных заболеваний используются в двух аспектах. Во-пер!
они дают дополнительные доказательства существования наспедсті
ной предрасположенности в отношении конкретных заболеваї
Во-вторых, популяционно-статистические методы вместе с КЛИНІ
генеалогическим анализом составляют основу для картирован!
идентификации генов подверженности болезням. Их объедиш
вылилось в самостоятельное научное направление медицинской Г1
тики — генетическая эпидемиология, краткая характеристика кот(
представлена на компакт-диске в разделе «Модели исслсдованиі
наследственность многофакторных болезней. Идентификация гв|
предрасположенность многфакторным заболеваниям».ГЕНЕТИЧЕСКИЕ АССОЦИАЦИИИсследования генетических ассоциаций осуществляют с исп<
зованием двух принципиально различных подходов. Один из
основан на том, что в качестве генетического маркера высту!
аллельный вариант гена, белковый продукт которого участвуе!
патогенезе исследуемого заболевания. То есть выбор гена и его п(Я
морфизмов определяется исходя из конкретной гипотезы разви]
заболевания, и ген этот называют «геном-кандидатом» подверг
ности многофакторному заболеванию. Задача исследования сосі
в получении доказательств участия его первичного продукта в
витии заболевания и измерении (оценке) силы его эффекта. Tajj
«каїідидатньїй» подход был первым вариантом исследования ген<
ческих ассоциаций. С его помощью проверяют гипотезу о naTord
тической основе возможной связи полиморфизма гена и болезни,]Другой подход предполагает сканирование генома с плотк|
набором ОНП (SNP), вплоть до I млн в одном исследовании для пс
ка функциональных вариантов генов. Такое полногеномное accoi
тивное исследование является масштабным по охвату включение
генома человека при отсутствии сведений о функции и расположен
связанных с болезнью генов на хромосоме.Оба подхода оценивают степень риска развития болезни у н<
теля определенных аллелей гена в сравнении с индивидами,
являющимися носителями такового. Существует много показате
и методик расчета эпидемиологических характеристик риска, Oz
Ill Н. Болезни с наследственной предрасположенностью281[НИХ - показатель отношении шансов (OR — odds ratio, отношение
|Miон). Его получают путем сравнения частот двух сравнивасхмых
(И Ііі іелей (М> и М2) у больных (с — «случай») и здоровых (к —
)Н I роль»),OR = (М1хМ^/(М2хМ1).і М ношение шансов принимает значение от О до бесконечности и
i^'it I только при отсутствии влияния одного показателя (маркера)
лруюй. Отношение шансов — надежная мера ассоциации генети-
ІКПГО маркера и болезни, а также хорошая оценка относительного
‘K;i. Термины «отношение шансов» и «относительный риск» (OR)
ІРІОІ сходный смысл и в исследованиях типа «случай—контроль»
Ці» бывают взаимозаменяемыми. Они показывают, во сколько раз
|)М1чивается (или уменьшается) вероятность быть здоровым (или
Н1> больным), ссли индивид является носителем конкретного алле-
кютветствующего гена по сравнению с индивидом-неносителем
1»|(ио генетического маркера.К настоящему времени проведено большое число исследований по
<отипированию, т.е. изучению генетических ассоциаций с много-
горными заболеваниями, и получены доказательства связей кон-
Мпых генетических маркеров как с фенотипами болезней в целом,II с разнообразными их клиническими проявлениями, а также
И нотом пациентов на лекарственные средства (фармакогеномный
ІЛИ1). Результаты таких исследований представлены в табл. 6.5.Ілика 6,5. Число выявленных полиморфизмов генов для отдельных групп
болезней (к 2009 г.)ХарактеристикаЧисло выявленных
полиморфизмовїЛолевания:в|1)1СЧП0-С0СуДИСТЫе26501сйродегенеративные2100Микологические6100Інрушения обмена веществ160011101' на лекарственные препараты (фар-
Ійкоісномика)1500Следует констатировать, что OR, пригодные уже сегодня для
^пользования опенки риска, определены для ограниченного числа
282Клиническая генімногофакторных заболеваний и признаков, В таблице 6.6 предс
лены OR для полиморфизмов (все они являются ОНП и обоэн!
ны в таблице как «rs» — номер в базе данных, reference site) Ті
в отношении болезней, для которых OR достигает 20. Напримі
индивидов, являющихся носителями рискового аллеля G (rs 382!
гена L0XL1, вероятность иметь тяжелую форму глаукомы (эксф(
тивная глаукома) в 20 раз выше, чем у индивидов, имеющих
полиморфизмы этого гена.Таблица 6.6. Показатели относительного генетического риска для неї
рых многофакторных заболеваний по данным полногеної
исследованийНазваниеболезниРегионхромо¬сомыСимволгенаОНП-
рнсковый
аллель (rs)Наименование
продукта генаOR у ГЄ1
зигот (9!
довериї
ныН
интеріЭксфол натив¬
ная глаукома15q24.1lOXLi3845942-GЛизоловая
оксидаза 120Д (Ю;
37,4).^Старческая
дегенера¬
ция желтого
пятна сет¬
чаткиЦ31CFH380390-СФактор И ком¬
племента4,6 (2,0-jАльцгейме¬
ра болезнь
(поздняя
форма) у
носителей
АР0Е*е4GAB22373115-GАдаптерная
молекула, глав¬
ный активатор
одной из киназ,
активирующей
рецептор IgE4,1 (2,81
14,6SМиопатия,статин-индупиро-ванная12р12SLC02E14149056-СБелок-транспортсрорганическиханионов4,5 (2,6-1Воспали¬тельныезаболеваниякишечника(IBD)1р31IL23RII209026-АРецепторил-233,8 (2,3-
iHii 6. Болезни с наследственной предрасположенностью 283Окончание таблицы 6.6ІіиіваниеГкиїезниРегионхромо¬сомыСимволгенаОНП-
рисковый
аллель (rs)Наименование
продукта генаOR у гетеро¬
зигот (95 %
доверитель¬
ный
интервал)[ііммі'ииая1НЬІМікмитиаз)2р24.2ABCG811887534-ССемействотранспортеровАТФ2,2 (1,8-2,6)^имі'чание. IBD — Inflammatory bowel disease; IgE — иммуноглобулин E;- аденозинтрифосфат.It габлице 6.7. приведены примеры OR в отношении некоторых
>и таков, а для одного из признаков — частоты рекомбинации,
)Торая ассоциирует с полиморфизмом rs 3796619-Т гена RNF212.
>01»снъ рекомбинации как у мужчин, так и у женщин сутественно
ірсделяется тем, присутствует ли в их геномах данный аллель гена
|А//'2/2. OR достигает величин 70,7 и 88,2 у мужчин и женщин, соот-
ІТсівенно.Ілица 6.7. Признаки с высоким и умеренным относительным генетическим
риском по данным полногеномных исследованийМйіваниепризнакаРегионхромо¬сомыСимволгенаОНП
рисковый
аллель (rs)Наименование
продукта генаOR у гетеро¬
зигот (95%
доверитель¬
ный
интервал)Григлице-1ИДЫ7qll.23MLXfPL3812316-СТранскрипци¬
онный фактор в
сети генов
{МАХ, MYC),
вовлеченных
в регуляцию
клеточной про¬
лиферации10,5 (5,3-
17,7)
284Клиническая гекіОкончание таблиціНазваниепризнакаРегионхромо¬сомыСимволгенаОНП
рисковый
аллель (rs)Наименование
продукта генаOR у гет
зигот (S
довері
ный'
интеріПигмента¬
ция кожи15q21.1SLC24A5I834640-GБелок-регулятор
трансмембран¬
ною обмена
ионов Na. К. Са12,5(8,3-20j15ql5.1MYEF246258816Миелинэкс-
прессирующий
фактор 254,7(13,1-23llql4.3TYR1042602-СТирозиназа4,4 (2,6*Частотарекомби¬нации4р16.3RNF2123796619-ТДНК-
связываюший
белок {ringfinger),
участвующий
в регуляции
мейотической
рекомбинации70,7
(57,1-8^
у мужчі88,2:^
(63,7-11І
у ЖЄНЩІв целом следует указать на три основные особенности обнар)
ваемых генетических ассоциаций.Во-первых, большая часть генетических ассоциаций вьізі
эффекты небольшой величины (риски 1,1—1,5, т.е. 10—15% увеличв
вероятности развития заболевания). Любой отдельный полимор(
гена обычно объясняет только 1—8 % от общего риска заболевї
в популяции, т.е. добавочный предсказательный эффект отдел!
генетических полиморфизмов небольшой. Однако исследовані
этом направлении только начаты и в специальных исследо!
ях (когортные, проспективные) риски уточняются. Подтвержда|
целесообразность введения генетической рискометрии.Во-вторых, у индивидов-носителей комбинаций (ансам(
аллельных вариантов генов отмечается заметное увеличение рї
болезней и их осложненного течения. Расчеты показывают, что ал
тивный эффект нескольких таких полиморфизмов может соста!
до 20—70% общего риска, обусловленного генетическими фактої
1ft,1 6. Болезни с наследственной предрасположенностью285ГМ. идет о совместном действии генов и их аллельных вариантов
ЛИ!п ивное, эпистатическое), взаимодействии между полиморфиз-
імі\ генов и модифицирующими факторами, такими, как возраст,
||1|, лечение и многие средовые факторы.ІІ-третьих, для человека характерно наличие обших генов, вовле-
Ііміьіх в патогенез разных, но нередко сочетающихся болезней,
ниросы, касающиеся идентифицируемых генов подверженности
ігпространенньїм болезням человека, освещены в одноименной
Гип.е В.П. Пузырева на компакт-диске.Одним из феноменов хронических заболеваний человека является
Илиматия, множественность болезней, у одного пациента сочетает-мссколько болезней. Около 40% лиц в возрас'іе 15—75 лет имеют
щоиременно две болезни, а четыре болезни встречаются почти у
I ж лого пятого терапевтического больного. Такие сочетания болезней
01 у I носить случайный характер — это «соседство болезней». Однако
ц)сдко болезни, сочетающиеся у одного пациента, встречаются и у его
іижайших родственников. Их обозначают как «семейство болезней»,
ut ношении последней категории болезней немецкими патологами
I, ! Іфаундлером и Л. фон Зехтом в 1921 г. предложен термин «синтро-
1Н». В современном определении его подчеркивается, что синтропия —
1о ириродно-видовое явление сочетания двух и более патологических
)С1ояний (нозологий, синдромов) у индивидуума и его ближайших
оде г вен ни ков, неслучайное и имеющее эволюционно-генетическую
Яюиу. Однако есть и такие патологические явления, которые редко
ї'їсіаются у одного человека, «упорно» не ассоциируют, «взаимно
П'илкиваются». Такой феномен авторы назвали дистропией.I Іаиболее известным примером синтропии является метаболиче-
1ИЙ синдром («квартет»): гипертония, гиперхолестеринемия, гипер-
ІНКСМИЯ и ожирение. Они встречаются у одного пациента, а также
родственников в таких же сочетаниях или отдельно. Известны и
)угие синтропии — иикквикский синдром, включающий ожирение
жірколепсию; сахарный диабет I типа, аутоиммунный тиреоидит
цслиакия; бронхиальная астма, атопический дерматит и высокий
)()иснь иммуноглобулина Е. Антагонистические взаимоотношения
іисіропии) известны, например, для туберкулеза легких и брон-
іальной астмы. Пролиферативные процессы на базе лимфоидного
миелоидного типов кроветворения не ассоциируются, что также
)жет быть примером стойкой дистропии. Отмечают, что сахарный
шОст I типа редко сочетается с язвенной болезнью.
286Клиническая генвіНеслучайность сочетания отдельных патологических фо|
объединенных сходством патогенеза у индивидуума и его
жайших родственников, указывает на участие общих генетиче<
факторов. Гены, вовлеченные в развитие синтропий, назві
синтропными генами. Более точно — синтропвые гены есть ш
функционально взаимодействующих корегулируемых генов, л(
лизованных во всем пространстве генома человека, вовлечет
в общий для данной синтропии биохимический или иной П1
в том случае, когда регуляторные связи складываются так, чт<
особенностью являются альтернативные отношения, объясж
шие взаимоисключения на клиническом уровне таких феноти!
(дистропии), такие гены называют дистропными в отношеї
соответствующих фенотипов.в качестве примеров назовем некоторые синтропные гены, кс
рые вовлечены в синтропию «метаболический синдром» и синтрог
«аллергические заболевания» (табл. 6.8).Таблица 6.8. Синтропные гены для двух вариантов синтропийГенПродукт гена1Хромосомная
локализация цСинтропня: метаболический синдром ^АСЕАнгиотензин-1-конвертирующий
фермент17q23АРОЕАполипопротеин Е19ql3.2 ]СЕТРБелок-переносчик эфиров холе¬
стерина16q21 .SERPINE1Ингибитор плазменного
активатора-1i|LPLЛипопротеинлипаза8р22 1Синтропия: аллергический синдром ^HLA~DRB)Антиген гистосовместимости
И класса DRp,6р21.3 «IL4ИЛ-45q31.1TGFB1Трансформирующий ростовой
фактор-Рі19ql3.2 IMS4A2Fc-фрагмент высокоаффинного
рецептора IgEllql3
pnii 6. Болезни с наследственной предрасположенностью287ГЕНЫ ПОДВЕРЖЕННОСТИ НЕКОТОРЫМ
МНОГОФАКТОРНЫМ ЗАБОЛЕВАНИЯМДостижения в геномике человека определили возможность
Ишружения множества генетических вариантов, ассоцииро-
1М11ЫХ с риском предрасположенности к заболеванию. Такой
|Пир генетических маркеров называют «геномным профилем»,
ігнтификация совокупности генов, которые в комбинации с
ІИ'метическими факторами приводят к патологическому фено-
|пу. сталкивается с необъятностью генома и высокой степенью
іяиішдуальной изменчивости. Такого рода вопросы относятся к
Ііік'й патологии.( истсматическое накопление данных о связи полиморфных
Ірпантов генов с многфакторными заболеваниями, анализ их
^произведения в разных популяциях и этнических группах,
И>Г)1цение всей известной информации о патогенетической и
/икииональной значимости конкретных генных вариантов —
Щрііменние задачи клинической генетики. Результаты этих
^следований будут кратко изложены для сердечно-сосудистых,
1муиозависимых, инфекционных болезней и злокачественных
)иообразований.>дечно-сосудистые заболеванияСердечно-сосудистые заболевания продолжают оставаться глав-
IIричиной смертности населения индустриально развитых стран
Іропьт (в том числе России) и США. Ишемическая болезнь сердца
цртериальная гипертония, их осложнения — инфаркт миокар-
инсульт и внезапная смерть составляют основную долго среди
сердечно-сосудистых заболеваний. В патогенезе этих болезней
іьшое значение отводится так называемым факторам риска —
>ушению липидного обмена (дислипидемии), сахарному диабе-
|f, ожирению, курению и др. После почти 20-летних дискуссий и
Івііиальньїх исследований получены убедительные доказательства
>ниеппии сердечно-сосудистого континуума (Dzau V. et al., 2006).гь ее состоит в том, что сердечно-сосудистые заболевания рас-
іитриваются как цепь событий, вызванных многочисленными
ІН’іанньтми и несвязанными между собой факторами риска, про-
>сссирующими через вовлечение многих физиологических, мета-
288Клиническая ген*Повреждение тканей (инфаркт
миокарда, инсульт, почечная
недостаточность, периферическая
артериальная недостаточность)Атеротромбоз и
лрогрессирование ССЗIРанняя дисфункция тканей\Стресс
вт.ч. оксидатианыйПатологическоеремоделі/ірование\Повреждвниі
op гана-мишениIОрганная недостат
(сердечная недостат
конечная стадия забола
почек)Факторь( рискаСмертьРис. 6.2. Сердечно-сосудистый и почечный патофизиологический К0НТ1
ум (Dzau V. ct al., 2006)болических путей и завершающихся развитием конечной стг
заболевания сердца (рис. 6.2).Более того, было показано, что вмешательство в любое звено
событий, ведущих к сердечно-сосудистым заболеваниям, модифі
рует течение этой группы заболеваний. Следует заметить, что боле
сердечно-сосудистого континуума соответствуют представления!
синтропных заболеваниях и сердечно-сосудистый континуум мс
быть также обозначен как синтропия.В специальном исследовании для 7 болезней, относящихсі
сердечно-сосудистому континууму (гипертония, коронарная болеа
дислипилемии, инсульт, ожирение, метаболический синдром и (
ный диабет 2-го типа), среди более 2,5 тыс. исследованных
идентифицирован 21 ген, которые ассоциируют с этими семью б(
нями и, следовательно, являются общими для данной синтрої
Их назвали синтропными генами сердечно-сосудистого континуз
(табл. 6.9).
Ihil 6. Болезни с наследственной предрасположенностью289|лн1^6.9. Общие гены синтропии «сердечно-сосудистый континуум»СимволюнаПродукт генаЛокализация на
хромосомеOMIMШ'Л!АТФ-связывающий кассет¬
ный переносчик I подсемей¬
ства А9q22-q21600046Ангиотензин1-коипертирующий ферментI7q23106180Рг-Адренергический рецептор5q32-q34109690Ангиотензин 1Iq42-q43106150UUIR!Рецептор ангиотензина 2,
тип 13q21-q25106165M'OAlАполипопротеин А1llq23107680Аполипопротеин Е19ql3.2107741\ТГГБелок-переносчик эфиров
холестерина16q2I118470iirc_inG-белок, р-су6ъединица ЗI2pl3139130ИЛ-67p21147620Печеночная липаза15q21~q23151670Л и поп ротеи н л и паза8р22609708'IHFRМетилентетрагидрофолат-редуктазаІрЗб.З607093Эндотелиальная синтаза
оксида азота7q36163729U:i.EГА//'Селектин ЕIq23-q25131210ФНОа6р21.3191160у-Рецептор, активируемый
пролифератором пероксисомЗр25601487\DIPOQАдиноцектин C1Q3q276054411'Л«/Рецептор эстрогена6q25.1.1133430Ша-Лимфотоксин6р21.3153440РАНИнгибитор плазминогенного
активатора 17q21.3-q22173360імечание. АТФ — аденозинтрифосфат.
290Клиническая геніЭти синтропные гены, конечно, не «закрывают» всю наследсі
ную компоненту подверженности этой группе заболеваний, в нее в>
сушественно больше генов. Однако сегодня общность для СИН1
сердечно-сосудистого континуума доказана только для этой вы1
генов из генома человека. Выявленные гены вовлечены в мета(
липидов, функционирование ренин-ангиотензин-альдостероно!
сим патоадреналовой систем, воспаление, эндотелиальную дис(
цию. Именно они составляют основу патогенетики болезней серде
сосудистого континуума. О генетических факторах развития инфв
миокарда у мужчин молодого возраста можно узнать из одноимві
статьи С.Н. Пчелиной с соавт. на компакт-диске.Иммунозависимые болезниК данной группе болезней, условно названной иммунозависиі
относятся неинфекционные заболевания человека, для которых от*
ния в функционировании иммунной системы приобретают важное
генетическое значение. Приобретенные иммунодефициты, аллерг
ские, аутоиммунные и лимфопролиферативные заболевания, среди
атопии (бронхиальная астма, ринит), сахарный диабет 1 типа,
Крона, ревматоидный артрит, хроническая обструктивная болезнь л«
коллагенозы и другие батезни — предмет изучения иммуногенетикіОдна из наиболее активно развиваемых областей в генетике
ней с наследственной предрасположенностью — генетика ai
ческих заболеваний человека, или атопий. Под атопией пониі
индивидуальную или семейную предрасположенность к выраС
IgE-антител в ответ на малые дозы аллергенов и развитию типич|
симптомов астмы, риноконъюнктивита или дерматита/экземы.Успехам в области генетики атопических заболеваний неї
способствует достаточно полное понимание механизмов реализ
IgE-опосредованных иммунных реакций (рис. 6.3).Аллергены, попадая в организм, взаимодействуют с дендритні
клетками- Результат этого — презентация доминирующего эпиЦ
аллергена с молекулами HLA-П на поверхности дендритных кл«
Связывание этого комплекса с рецепторами CD4 Т-лимфоцитов^
мулируетдифференцировку ThO-лимфоцитов в Th2 (Th — Т-хелп<
способных к секреции цитокинов, функция которых тесно СВЯЗЕ
гуморальным иммунным ответом: ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-9, HJ
GM-CSF {granulocyte-macrophage colony stimulating factor — колонне
мулирующий фактор гранулоцитов-макрофагов).
ІМії ti. Болезни с наследственной предрасположенностью291ДендритнаяклеткагАшкіргенI |<(4ктерииLвирусыв клеткаАллергенТучнаяклеткаМедиаторывоспаленияДендритнаяклеткаТЫ клеткаЭозинофилр. (».3. Механизмы развития атопических реакций (Holgate ST., 1999)При действии антигенов \4икробактсрий и некоторых вирусов
И клетки превращаются в Th 1-лимфоциты, секрстирующис
11-2, у-интерферон (у-ИФН) и ФНО. Это приводит к активации
ікрофагов и элиминации с их помощью патогенных микроорга-
ІМОВ. Сдвиг в сторону ТЬ2-ответа активируется ИЛ-4 и ингиби-
иси ИЛ-12, у-ИФН и а-ИФН. Th 1-ответ требует высвобождения
1-12 макрофагами и дендритными клетками и супрессируется
И).Цитокины, высвобождаемые Т1і2-клетками, главным образом ИЛ-4
взаимодействуют со своими рецепторами на В-лимфоиитах,
[Тинируют транскрипцию іенного локуса тяжелой цепи типа е
(муноглобулинов и индуцируют переключение изотопов с |Д на є
синтезируемый активированными В-клетками, связываются с
(сокоаффинным (FceRl) и низкоаффинным (РскКП; CD23) рецеп-
)«ми тучных клеток, инициируя высвобождение медиаторов вос-
Лсиия и хемокинов: гистамина, простагландинов, лейкогриенов,
IKiopa активации тромбоцитов, дегранулированных протеаз и др.
292Клиническая геніМедиаторы воспаления синтезируют также эозинофилы, акти!
ванные ИЛ-3, 5 и GM-CSF.Действуя в совокупное!и, эти факторы приводят к микро*
нениям сосудов стенок «шокового органа», сокращению гл1
мускулатуры, гиперсекреции слизи и другим симптомам, служат
основой клинических проявлений атопических заболеваний. К|
того, цито- и хемокины ответственны за миграпию и активацию^
ток воспаления, главным образом, эозинофилов, и, таким обрі
внося I вклад в сохранение патологического процесса.В отношении частой формы атопии — бронхиальной астмы, типі
го многофакторного заболевания, в исследовании значения наследст
ных факторов предрасположенности был использован весь арсенал'
диционных и новых исследовательских подходов, принятых в гене
многофакторных болезней; близнецовый, клинико-генеалогиче(
картирование (анализ ассоциаций и сиепленик), по;нюгеномные
циативные исследования, экспрессионные и транскритомные ме
а также изучение на модельных животных.К настоящему времени число генов, протестированных в свз
бронхиальной астмой или обнаруженных в нолногеномных исс;
ваниях, составляет около 550. Для 35 из них ассоциации с брс
альной астмой продемонстрированы в независимых исследої
не менее 10 раз (рис. 6.4).Функционально ассоциирующиеся гены объединяются в че
группы; гены врожденного иммунитета и иммунорегуляции (j
TLR4,1Ы0)\ гены, связанные с дифферениировкой и функционирої
ем Т-хелперов 2-го ти па (IL4RA, 1L4R, і ИЗ), гены иммунитета слизиі
оболочек и физиологии эпителия (CCL5, FLG); гены, ассоциироваї
с легочной функцией, ремоделированием дыхательных путей и бї
альной гиперреактивностью (ADRB2, OSTMJ, GSTP1, LTA, M0S1).Как уже отмечалось, для многофакторных ных заболеві
характерен малый эффект отдельного генетического вариаі
отношении фенотипа болезни. Это свойственно и для бронхиал^
астмы. Однако в синергиз.ме с другими аллельными вариант
(межгенные взаимодействия) этот эффект может заметно увелі
ваться. Так, при исследовании более 1000 немецких детей в возі
9—11 лет было показано, что при пошаговом комбинировании
ИЛ {IL4, 1113), гена кодирующего а-цепь рецептора ИЛ-4 {IL*
и гена внутриклеточного активатора транскрипции (STAT6)
повышенного содержания IgE возрастал в 10,8 раза, а бронхиал!
ІНії в. Болезни с наследственной предрасположенностью293АІШ2
rNF
IL 13
(Ю14
1L4
IL4R
fi.sTP/
At іЛМЗЗ
СиНШІ
ICFB1
MS4A2
І iSTT1
ГІЯ4
ІАППВІ
IDO
CCL5
І ГС43
LTA
NOS3
IFNG
IL1RN
\ЛООВ1
ГУ^іТНІ
FLG
ЛіОХЗ
IL1ATLR2TLR9CTLA4418IL1BITGDRCCL11N0S11020ЗО4050607080L‘. 0.4. Гены, для которых продемонстрированы ассоЕіиации с бронхиаль-
.істмой не менсс 10 раз в независимых исследованиях. Горизонтальные
>і)лГ)ики соответствуют количеству опубликованных работ о позитивттых
інжиациях с заболеваниемРТмы — в 16,8 раза по сравнению с суммарным эффектом отдельных
JU1 морфизмов.Наряду с анализом ассоциаций генов-кандидатов для иденти-
ікации генов подверженности бронхиальной астме, использова-
полногеномное ассоциативное исследование, которое, подтвер-III некоторые ранее известные гены подверженности, обнаружило
>иые. Среди них: два близко расположенных гена на хромосоме
l21 — 0RMDL3 и GSDML, а также ген CH13LL кодирующий хити-
|> >аподобный протеин YKL-40.
294Клиническая геНІВажнейший аспект в исследованиях генетики бронхиал!
астмы — фармакогенетичсский, оиениваюіций индивидуалі
ответ на наиболее часто применяемые противоастматические
иараты (бронходилататоры, кортикостероиды и антилейкотри!
Так, показана ассоциация полиморфизмов гена ADRB2, кодиру»
го Р2-яДР^нсргический рецептор, с бронходилататорным ответо!
высокие дозы р-агонистов короткого действия, например, салі
мола (альбутерола*®) при лечении приступов удупіья: сальбутІ
у гомозигот по 01у 16 лучше купирует и предупреждает прї
удушья, чем у гомозигот по Arg 16. Известен генетический марї
вариант гена (3-иммуноглобулинового рецептора тучных к;
{FCER), кодирующего низкоаффинный рецептор IgE. ассоции):
ный с повышенным риском приступа бронхиальной астмы у дв<
принимающих ингаляционные кортикостероиды, несмотря на
тсктивный эффект этих препаратов в лечении астмы.Инфекционные болезниИнфекционные болезни — одна из главных проблем здравое
нения во всем мире; они характеризуются высоким уровнем за?
ваемости и смертности. С генетической точки зрения важно, что
отличаются от неинфекционных многофакторныхных болезней
что имеют специфическую причину ~ возбудителя болезни (пі
геи). Однако развитие болезни, характер течения инфекцион!
процесса, чувствительность организма к возбудителю определяї
сложным взаимодействием факторов окружающей среды, патої
наследственных факторов «хозяина» (рис. 6.5).Основной взгляд на генетику подверженности распространеі
инфекциям (малярии, ВИЧ/СПИДу, микобактериальным и вирус
инфекциям) обозначается как «одна инфекция — множество rej
Однако у лип с первичными иммунодефицитами часто развиваї
самые разнообразные инфекционные заболевания, которые рассмг
ваются как осложненное течение моноген ного иммунодефицита,
случаи обозначаются как «один ген — многочисленные инфекциіВ настояшес время активно осуществляется поиск локусов (!
ных вариантов) чувствительности к инфекционным заболевай!
Основной подход в этих исследованиях — скрининг генов-кандид!
и анализ их ассоциаций с инфекционными болезнями. К решеї
этой задачи и последнее время привлекаются и полногеномные а<
циативные исследования.
їй Г). Болезни с наследственной предрасположенностью295S. 6.5. Триединство факторов патогенетики инфекционного процессаВ отношении туберкулеза, наиболее активно исследуемого с
ІСІ ИЧЄСКИХ позиций заболевания, накоплена достаточно большая
^формация (табл. 6.10.).|блица 6.10. Гены, для которых показана ассоциация с туберкулезом\ ГенБелковый продуктШи-DQ/DRЛейкоцитарные антигены II классаfoRРецептор к витамину DJfNGИФН-у^l.CriAJ(NRAMPJ)NRAMP1, катионный транспортертоИЛ-10ТАРТранспортер, ассоциированный с процессингом
антигеновТШТоП-Ике рецептор 2ФНОа//.<УИЛ-8
296Клиническая гвні|Окончание таблицы |ГенБелковый продукт ]МВІМаннозаспязывающий лектин jIL1RAРецепторный антагонист ИЛ-1 jJLJBИЛ-1р jP2RX7Р2х7 рецептор JILI2RBIPi-субъединииа рецептора к ИЛ-12 jILI2BР40 субъединиц ИЛ-12 ^CYP2E1Цитохром Р450 1HSP70-1Белок теплового шока 70-1 ’LAMA2Ламинин-азPARK2Паркин 2PACRGП арк и н-корегул яторв таблице 6.11 приведены результаты ассоциативных исследс
ний для вирусных гепатитов В и С.Таблица 6Л1. Гены, для которых показана связь с вирусным гепатитом В и]
ассоииировапными клиііическими фенотипамиГен (белковый продукт)Патология или фенотип |TNFA (ФНОа)вгв, вгс Чвгс 1ІЬІВ(ИЛ-Ір)вгс 1ILIRA (рецепторный антагонист
ИЛ-1)прогрессия до цирроза при ВГуIL6{m-6)вгс, персистенция вируса, -4
тяжестыечсния 1П10(ш-т)вгс, ответ на терапию, исход j
заболевания ]MBL (маннозасвязывающий лектин)вгв, псрсистенция вирусаМРО (миелопероксидаза)вгс, фиброз печени ^МХА (ген устойчивости к миксовиру-
сам)Ответ на терапию при ВГВ; исх^
ВГС ,OAS! {1, 5-олигоаденилатсимтетаза)Исход ВГС 1PKR (РНК-записимая протеин киназа)Исход ВГС 1>4/^0£'(аполипоііротеин Е)Исход ВГС
Ійп ь. Болезни с наследственной предрасположенностью297Окончание таблицы 6.11Ген (белковый продукт)Патология или фенотипІН \МР! (катионный транспортер)ВГС, прогрессия фиброза печениIЛ і\ IKS (хе мок и н)Воспаленис псчеіги при вирусном
гепатитеП 'H5(хемокиновый рецептор 5)ВГС, ответ на терапию\ыт (трансформирующий факторВГС, персистенция вируса,
тяжесть фиброза печени[bVO(MOH-Y)ВГВ, ВГСВГС, тяжесть фиброза печени^/'/•(иротеин наел едет пен но го гемох-
«1м птоза)ВГС, тяжесть фиброза печениіиі'чаиие. ВГВ — вирусный гепатит В; ВГС — вирусный гепатит С.Генетика инфекционных заболеваний, еще совсем недавно счи-
|цтаяся мало перспективной в силу небольших коэффициентов
Іі'псдования (h2 — 10—20%) этой патологии, сегодня активно иссле-
jfcicH всем современным арсеналом генетических методов.Юкачественные новообразованияСреди многочисленных и широко распространенных многофак-
^рммхпых болезней особую группу составляют злокачественные
^цообразования,(' позиций генетической классификации заболеваний человека
лыпинство злокачественных новообразований относится к болез-
обусловленным мутациями в соматических клетках. Наряду с этим
^іксствуют и наследственные формы опухолей^ обязательным ком-
)мснтом развития которых является наследование генной мутации
||Х‘ і половые клетки родителей (иногда их называют герм и нативны-
|И). Подробно о наследственной предрасположенности к онкологиче-
ІИМ заболеваниям см. в статье Е,Н. Имянитова на компакт-диске.Следует подчеркнуть, что злокачественные опухоли — болсз-
соматических клеток, способных к делению (стволовые клетки,
ісгки-предігіествснники). Опухоль возникает в результате накопле-
|И» различных типов генетических нарушений, приводящих к поте-
гсномного контроля над процессами деления, роста и естествен¬
ной убыли клеток. ГТрсдполагается, что для индукции солидных
|И11»в рака необходимо накопление не мснсе 5—7 мутаций. Развитие
298Клиническая генізлокачественных опухолей протекает через 3 основные стадии:
циаиию, промоцию и прогрессию, при этом скорость данных пр<
сов определяется частотой возникновения мутаций, численж
клеточной популяции, скоростью пролиферации и преимуще(
в размножении мутантных клеток.Выделяют 3 основные группы факторов риска возникнов<
злокачественных новообразований. Первую из них составі
физические факторы, а именно ионизирующее и ультрафиолет
излучение, повышающее частоту генных мутаций в соматиче<
клетках.Вторую группу составляют химические факторы, к которым
сятся канцерогены — широкий спектр химических соединений
нической и неорганической природы, способных вызывать м)
в соматических клетках (мутагены, или опухолевые инициаі
или индуцировать опухолевый рост клеток, подвергшихся ДЄЙСІ
мутагена (митогены, или опухолевые промоторы). Рак возник!
результате последовательного действия инициатора и пром<
при этом риск злокачественной трансформации клеток зависйі
дозы канцерогенов, продолжительности и периодичности их
действия.Следующей группой факторов риска являются биологичс
Среди них особое место занимают вирусы. ДНК-содержащие bhj
представители семейств паповавирусов (папилломавирус), геш
вирусов (вирус гепатита В) и герпес-вирусов (вирус Эпстайна—1
способны к интеграции в геном клетки-хозяина и к индукции
планированных циклов ее репликации. Многие вирусные онкої
способны при этом блокировать ключевые опухолесупрессо!
механизмы клетки, РНК-содержащие вирусы семейства ретровиі
(вирус Т-лейкоза человека J типа и ВИЧ) имеют геном, предстаї
ный двумя молекулами одноцепочечной РНК. В геноме этих ВИ1
закодирован фермент обратная транскриптаза, необходимый
синтеза копий вирусной ДНК. Встраивание вирусной ДНК в г<
клетки-хозяина — обязательный этап жизненного цикла ретріі
руса. Подобное встраивание может сопровождаться инсерцион!
мутагенезом, ведущим к появлению структурных мутаций в
рующих последовательностях генов.Среди биологических факторов риска отдельное место,
мненно, занимает наследственная предрасположенность к злої
•и Ь. Болезни с наследственной предрасположенностью2991П1ИЫМ [ювообразованиям, молекулярные основы которой будут
^гмотрены ниже.На каждом из этапов формирования опухоли развертываются
ифлинированныс в патологическом иаправлсиии процессы спа-
клл на молекулярном, а затем на клеточном уровне. Рассматривая
ІИік'рогепез как результат нарушения геномного контроля баланса
ІІ'ИЧІНОСТИ клеточной популяции, можно выделить две группы
мутации которых этиологически связаны с развитием рака.
|>и> ІІДСІ о протоонкогснах и опухолесупрсссорах.11(К1тоонкогены (первая группа) — нормальные клеточные гены,
III полирующие рост и размножение клеток. Продукты протоон-
Цічіов осуп^ествляют контроль роста и деления клсюк и представ¬
шим ростовыми факторами и их рецепторами, внутриклеточными
Цп-датчиками сигналов, факторами транскрипции. Измененные
іргіультате мутации протоонкогены или гены, вносимые в (сном
Шкшорыми вирусами, получили название онкогенов. Онкогены ока-
(imior стимулирующие влияние на клеточную пролиферацию. Их
^Ип пие носит доминантный характер, т.е. для развития рака д:оста-
|М1К) мутации в одном из аллелей онкогена.Ныло замечено, что некоторые вирусы приводят к развитию зло-
І'іоственньїх опухолей у животных. Поскольку вирус привносит
^Клетку хозиииа свой генетический материал, то предположили,
]о геномы вирусов содержат онкогены, в конце 70-х годов XX в.
:1’Н К-содержащем вирусе куриной саркомы Рауса был выделен
IticiOK РНК, ответственный за злокачсствешіую трапсформа-
IIU клеток. Так был открыт первый онкоген — v-src. Позднее
la іалось, что в нормальных клетках есть гены, по своей струк-
близкие к онкогенам — протоонкогены. По всей видимости,
ходе эволюции вирусные онкогены возникли из нормальных
ІСІ очных генов в результате рекомбинации между пред ковы м
М'рансформирующим вирусом и ДНК клетки-хозяина. В нор-
ільньїх клетках протоонкогены находятся под контролем дру-
(X клеточных генов. Интеграция вируса в геном клетки-хозяина
яюбождает их от этого контроля, приводит к нерегулируемой
Пивации и превращению в онкоген. К настоящему времени
Ідімітифипировано более 100 протоонкогенов и их число продол-
|й1*т расти. Характеристика некоторых протоонкогенов и онкоге-
)и приведена в табл. 6.12.
300Клиническая гві>Таблица 6.12. Характеристика протоонкогенов и онкогеновПрогоонкоген,локализацияОнкогенФункциябелкаОпухолШPDGFB — ген р-цели
фактора роста
тромбоцитов PDGF
(22ql3.I)v-sis — онкоген
вируса саркомы
обезьянРостовойфакторГлиобластоМменингиомії{IEGFR — ген рецепто¬
ра эпидермального
фактора роста {7р12)V-erb-B — OFIKOreH
пируса эритробла-
стоза птицРецепторростовогофактораГлиобластоіІI/RAS— гомолог
вирусного онкогена
крысиной саркомы
Харвея (11р15.5)v-ras — онкоген
вируса крысиной
саркомы ХарпеяЦитоллаз-
м ати че¬
ски й пере¬
датчик
сигналов в
клеткуРак моченої^
пузыря, моЩ
ной железы, j
легких, киЩІ
ника іSRC — гомолог
вирусного онкогена
куриной саркомы
Рауса (20ql2-ql3)v~src — онкоген
вируса куриной
саркомы РаусаЦитоплаз¬
матиче¬
ский пере-
латчик
сигналов в
клеткуРак кишечви
ка, миелоим
лейкоз 1MYC— гомолог
вирусного онкогена
куриного миелоци¬
том ато за (8q24.12)v-myc — онкоген
вируса куриного
миелоцито-
матозаТраЕгскрип-ционпыйфакторЛимфома 1
Беркитта, ці
робластома,^
легких ]В клетке иротоонкогены могут трансформироваться в он ко:
через следующие механизмы.— Рекомбинация с ретровирусной ДНК. Интеграиия вир;
нуклеиновой кислоты в геном клетки-хозяина имеет два псе,
ствия. Во-первых, это нарушение структуры гена, в кот
произошло встраивание чужеродного генетического мате
(инсерционный или вставочный мутагенез). Во-вторых,
стимулирование незапланированных циклов репликации
и клеточного деления.— Хромосомные транслокации. Перемещение участка хро
мы, содержащего протоонкоген, на другую хромосому Мі
привести либо к изменению структуры гена, либо к наруше
іиіі Ь. Болезни с наследственной предрасположенностью301регуляции его активности, Например, транслокация t(9;22)
k|34;qll), или так называемая «филадельфийская хромосома»,
IIC1 рсчается у 95% больных хроническим миелоидным лейкозом
{рис. 6,6), Она возникает в клетках костного мозга еще до про¬
явления основных симптомов заболевания и имеет важное про-1 иостическое 'значение. При этой перестройке участок длин¬
ного плеча хромосомы 22 транслоцирован на длинное плечо
хромосомы 9, а небольшой фрагмент хромосомы 9, содержащий
ICH ABL, присоединен к участку хромосомы 22, содержащему
ген BCR. В результате транслокации на хромосоме 22 образуется
химерный ген ABL-BCR, продукт которого (тирозиновая проте-
инкиназа) стимулирует непрерывное клеточное деление. Важно
отметить, что расшифровка цитогенетических и молекулярных
механизмов формирования химерного белка позволила соз¬
дать новый лекарственный препарат (иматиниб), являющийся
ингибитором тирозиновых протеинкиназ и демонстрирующий
обнадеживающие результаты в лечении больных хроническим
миелолейкозом. Другим примером, демонстрирующим роль
хромосомных перестроек в активации протоонкогенов, явля¬
ется реципрокная транслокация t(8;14)(q24;q32), выявляемая
при лимфоме Беркитта. Транслокация отделяет протоонко¬
ген C-MYC (8q24,12) от его
нормального промотора. В
новом месте он попадает
под сильный регулятор¬
ный элемент гена иммуно¬
глобулина Н, расположен¬
ного на хромосоме 14. В
результате перестройки в
клетке резко повышается
продукция белкаС-MYC —
транскрипционного фак¬
тора, обладающего онко-
генными свойствами.Амплификации прото-
онкогенов. Этот процесс
существенно увеличиваетчисло копий протоонкоге- Рис. 6.6. транслокация t(9;22) при
на в клетке, что приводит к хроническом миелолейкозе
302Клиническая геніобразованию большого количестїіа соответствующих онк<
теинов. Часто амплкфикацил специфических протоонко!
ЯВЛЯЄ!ся признаком опрелеленных опухолей (например,!
мелкоклеточном раке легкого обнаруживается амплифиК!
C-MYC, N-MYC и і-Л/УСиротоонкоі енов).— Точковые мутации в кодирующих последовательностях
тоонкогеиов приводят к сингезу онкогснных белков. Haj
с гениыми мутациями может наблюдаться его существе»
амплификация в оіікогсне, что усиливает трансформиру»
клетку свойства.Вторая группа генов, мутации в которых приводят к разві
злокачественных новообразований, — опухо-иссупрессорные
Их функция заключается в ингибировании клеточного дело!В отличие от протоонкогенов, действие опухолесупрессоров реї
сивно, т.е. для развития опухоли необходимо наличие муташ
обоих аллелях одного гена. С мутациями опухолесупрсссорных
связаны наследственные формы новообразований, обусловлен^
комбинацией герминативных и соматических мутаций. Некот
опухолесупрессоры обладают свойством так называемых мутатої
1’енов — их инактивация обусловливает резкое возрастание часі
мутаций в других локусах генома. Как правило, продукты таких
вовлечены в регуляцию процессов репликации и репарации Д|
клеточного цикла и аиоптоза.Механизмы действия опухолесупрессорных генов были открі
при исследовании ретинобластомы ~ злокачественной опухоли
чатки глаза у детей. Впервые рстинобластома как самостоятел!
заболевание с аутосомно-доминантным типом наследования
описана еще в 1902 i'. В 1971 г. А. Кнудсен предположил, что
возникновения болезни необходимы мутации в каждой копии
отвечающего за развитие опухоли.Семейные случаи ретинобластомы (примерно 40% всех слу^
заболевания) характеризуются более ранним возрастом начала б(
ни, предрасположенностью к другим типам злокачественных onj
лей и являются в основном двусторонними или мультифокальный
Согласно предположению А. Кнудсена, семейные формы возник!
в результате наследования одной мутации от родителей, а втс
мутация возникает уже в соматических клетках в нормальном алл<
Шанс второй мутации достаточно высок, что и приводит к «до1
нантной предрасположенности» к развитию опухоли.
lUii 6. Болезни с наследственной предрасположенностью303Спорадические случаи ретинобластомы возникают в результа-
инух соматических мутаций. Для них характерен поздний воз¬
ії начала заболевания, а также отсутствие предрасположенности
Л|)уіим типам злокачественных новообразований. Как правило,
іор;ідическая ретинобластома односторонняя (имеет один фокус
Іііілкновения).Дниные рассуждения А. Кнудсена легли в основу его «двухудар-
IIII ипотезы» канцерогенеза, нашедшей хорошее подтверждение для
jrwiiMHCTBa наследственных форм злокачественных новообразова*
111 (рис. 6.7). Индивид наследует от одного из родителей мутацию
^«яіухолесупрессорном гене. Это может быть точковая мутация или
1к[к>делеция. Гетерозиготность индивида по данному локусу стра-
‘I его от возникновения опухоли. Однако в течение жизни в сома-
моских клетках может возникнуть мутация и в нормальном аллеле
іухолесупрессорного гена. Это приведет к потере гетерозиготности и,
|к результат, к полному отсутствию продукта опухолесупрессорного
н клетке. Очевидно, что подобные события (две мутации) могут
|) тикать и у индивидов, не несуших герминативных мутаций, одна-
псроятность их возникновения будет значительно ниже, посколькуНОРМАСПОРАДИЧЕСКИЙ РАКРедкие мутации в
одном из аллелей
іуколесупрессорного
генаНАСЛЕДСТВЕННЫЙ РАКГерминативная
мутация (1 удар)Соматическая мутация
(1 удар)Соматическая
мутация (2 удар)ттСоматическая
мутация {2 удар)ОПУХОЛЬВремяОПУХОЛЬ^нс. 6.7. «Двухударная гипотеза» канцерогенеза Кнудсена
304Клиническая генітребуется последовательное повреждение обоих нормальных алл<
конкретного опухолесупрессорного гена.Ген ретинобластомы {RB1) стал первым открытым опухолесуп)
copfiMM геном у человека. Это открытие было сделано в 1986 г.,
спустя 15 лет после формулирования «двухударной гипотезы»,
RB] локализован в сегменте I3ql4 и кодирует ретинобластом!
белок (pRB) — негативный регулятор клеточного цикла. В гипо(|
форилированном состоянии pRB связывает семейство TpaHCKj
ционных факторов E2F, подавляя таким образом деление клеї
В нормальных условиях запуск клеточного деления осущсствля!
комплексом никлинов и циклинзависимых киназ, которые
рилируют pRB и освобождают тем самым факторы транскриш
Точковые мутации в гене RB1 или аберрантное гиперметилирої
его промоторного региона, а также микроделеции участка хромое
I3ql4 приводят к отсутствию опухолесупрессорного белка. В pe3j
тате клетки, имеющие повреждения и в других локусах генома^
останавливаются в своем размножении, что увеличивает шансы
вития опухолевого процесса.У человека описан ряд злокачественных новообразований,
никающих за счет потери гетерозиготности (табл. 6.13). Для мн<
форм известны не только хромосомная локализация опухолесущ
сорного гена, но и его структура, мутации и первичные продуі
Часто для возникновения одной и той же опухоли необходима п(
гетерозиготности не в одном, а в нескольких локусах. Кроме тс
нужны еще мутации в онкогенах. Многокомпонентность генети^
ских событий канцерогенеза очевидна.Таблица 6.13. Опухоли, возникающие п связи с потерей гетерозиготностиСиндром или опухольХромосомная локалиэаШРетинобластома13qI4 ^Остеосаркома13ql4, 17р13Опухоль ВильмсаПр13Болезнь фон Хиипеля-ЛиндауЗр25-26Рак мочевого пузыря9q, Ир15, 17р13Рак легкихЗр, I3ql4, 17р13Рак молочной железыIq, Зр, 13ql2,17q2lСиндром Бсквита-ВидеманаЗр, ]]р, 13q, 17рРабдомиосаркома11р15
Цйил 6. Болезни с наследственной предрасположенностью 305Окончание таблицы 6.13Синдром или опухольХромосомная локализацияh(K печени11р15|г11;11оиеллюлярный ракПр15|',1к желудка13q{1‘мсйный аденоматозный полипозКо.юрсктальный рак5q,17р, I8qІІсіірофиброматоз I типа17qll.2Іігіїрофибрсматоз II типа22ql2.2' Мсмиигиома22qМиожестветіная эндокринная неоплазия I типаllqИиеулиномаllqМедуллярный рак щитовидной железы1рИчмхромоцитома1р( ммдром Ли-Фраумени17р13{докачественмые новообразования, развивающиеся на основе уна-
рдглованных мутаций, иногда имеют характер семейных синдромов.
Іріинаки таких синдромов:’ нысокая частота опухолей у родственников I и II степени родства;• наличие в родословной несколько близких родственников со
сходными формами опухолей, например, молочной железы и яич¬
ника, кишечника, эндометрия;• наличие двух членов семьи со сходными редкими формами
опухолей;* необычно ранний возраст начала заболевания;• двусторонние опухоли парных органов;* синхронность или непрерывность возникновения опухолей;♦ опухол и органов разных систем у одного индивида.Одним из примеров таких семейных форм злокачественных ново¬
образований является синдром Ли—Фраумени, впервые описанный1988 г. как заболевание с аутосомно-доминантным типом наследо-
ІИИИЯ. Диагноз основывается на нахождении от 2 до 6 типов опухо¬
лей в одной родословной и более. Саркомы начинаются в возрасте
[до 5 лет, остеосаркомы — в юношеском возрасте, а опухоль мозга,
[Іиолочной железы, аденокарцинома желудка или лейкемия проявля-
[Кпся до 30-летнего возраста. В 1990 г. было показано, что больные
306Клиническая геніимеют мутации в гене-супрессоре опухоли ТР53 (17р13.1). В не
продукт данного гена белок р53 практически не обнаруживаете
клетках вследствие чрезвычайно короткого времени полурасш
Функция р53 заключается в контроле целостности ДНК, регуляї
клеточного цикла и апоптоза. Белок определяет, произошла ли {
рация повреждений ДНК в клетке. Если повреждение не может
репарировано, то запускается программа апоптоза. При повр«
НИИ ДНК р53 активируется и стимулирует транскрипцию гена
который кодирует ингибитор циклинзависимой киназы. Белок
присоединяется к комплексу циклина и циклинзависимой киї
и инактивирует его. Б результате клетка останавливается в Gl-(
клеточного цикла. Очевидно, что мутации в гене делают не|
можным прохождение такого каскада реакций и увеличивают і
злокачественной трансформации клеток. Мутации данного гена
дены более чем в 50% разных форм злокачественных новообраз
ний. Скрининг мутаций гена ТР53 имеет важное клиническое
чение для прогноза как в семьях с синдромом Ли—Фраумени,
при спорадических формах опухолей,Около 5—10% случаев рака молочной железы составляют нас;
ственные формы, обусловленные передачей потомству мутанті
вариантов высокопенетрантных генов репарации ДНК и апопт(
Особое место среди них занимают гены BRCA1 (17q2l) и BRCA2{\M
Продукты этих генов активируются в ответ на повреждение ДН|
вместе с белком RAD51, вовлеченным в осуществление гомологичі
рекомбинации, формируют комплекс, участвующий в репараї
двухцепочечных разрывов ДНК. Открытие генов BRCA1 и
имеет большое значение для прогноза возникновения наследст
ных форм рака молочной железы. Женщины, несущие мутации
генов, имеют высокий риск развития опухолей молочной желез(
яичников. Этот риск особенно повышается в семьях, где есть
случая рака молочной железы и более, а также отмечается раї
начало заболевания — в возрасте до 40 лет.Наиболее распространенной из наследственных форм опухс
кишечника является наследственный неполипозный колорект||
ный рак. В его развитии существенную роль играют мутации в
мисс-матч репарации ДНК - MSH2 (2рН), MSH6 (2р22), MLHI (Зр2
MLH3 (14q24.3), PMS1 (2q31) и PMS2 ij^22). Инактивация обоих
лей одного из этих генов приводит к резкому накоплению ОШІ
репликации ДНК (гены с мутаторньш эффектом), что выявляв
[ііпна 6. Болезни с наследственной предрасположенностью307[и<» иысокой изменчивости числа копий микросателлитных повто¬
ри. Подобный феномен микросателлитной нестабильности, помимо
[tiiK'ji едет венного колоректального рака, наблюдается примерно в
20% случаев спорадических опухолей разных типов, свидетель-
[^И'уя о том, что дефекты в генах репарации ДНК — один из общих
||игчапизмов канцерогенеза.Гассмотренные выше события канцерогенеза не исчерпывают всей
[)йн<’гокомпонентности генетической предрасположенности к раку и
|1И)гофакторности причин развития опухолевого процесса. Можно
1н1|)сделить еще целый ряд наследственных характеристик индивида,
їмсюших отношение к канцерогенезу. Так, например, метаболизм
іпіщсрогенов в клетках определяется биохимическими системами,
щогие из которых генетически полиморфны. Полиморфизм активи-
||»\иицих ксенобиотики ферментов (эстеразы, оксигеназы, изоформы
[мм юхрома Р450) и детоксицирующих ферментов (различные транс-
)1'1>лзы) определяе т индивидуальную чувствительность организма к
Ііпіиерогенньїм воздействиям (см. главу 7).Метаанализ данных литературы показывает, что наследственный
Полиморфизм генов, вовлеченных в биотрансформацию ксенобио¬
тиков, демонстрирует наиболее значимые ассоциации с риском раз-
ІИТИЯ онкологических заболеваний. Некоторые примеры ассоциаций
Ірпнеденьї в табл. 6.14.іб.'іица 6.14. Ассоциации генетических полиморфизмов с различными типа-
.ми опухолейГенПолиморфизмТип ракаOR, отношение шансов
(95% доверительный
интервал)USTMIНулевой аллельРак мочевого
пузыря1,50(1,30-1,60)Лейкоз1,20(1,14-1,25)ХПЕК2*1100delCРак молочной
железы2,40 (1,80-3,20)VYP19ТТТА10Рак молочной
железы1,59(1,01-2,48)\XRtClArg399GlnРак молочной
железы1,60(1,10-2,30)Рак легкого1,34 (1,16^-1,54)
308Клиническая геніїОкончание таблицы \ГенПолиморфизмТип ракаOR, отношение шага
(95% доверительйМ
интервал) ^Г'УРІА /Tle462ValРак пищевода2,52 (1,62-3,91) 1J л / /\ іMspIРак легкого2,36(1,16-4,81) 2MTHFRС677ТРак желудка1,52 (1,31-1,77) JТР53Arg72ProРак желудка0,84 (0,72-0,99) ^DCLRE1Brs376l936Глиома0,36 (0,20-0,65) “1BRIP/rs4968451Менингиома1,61 (1,26-2,06) jARGGN16Рак простаты1,31 (1,06-1,61) іXRCC3Thr24lMetРак кожи0 J6 (0,62-0,93) ^Таким образом, медико-генетическое консультирование сем1
наследственной предрасположенностью к злокачественным н<
образованиям — сложнейшая задача. Требуются специальная
готовка врачей-гсиетиков в вопросах генетических основ канцере
неза и хорошая лабораторная база. Только такое сочетание позв(
правильно выявить предрасположенность членов семьи, определ|
необходимость их диспансерною наблюдения и характер нрофиі
тических мероприятий, с молекулярно-биологическими технолог
ми в современной онкологии можно ознакомиться в одноимен!
статье С.П. Коваленко на компакт-диске.ЗНАЧЕНИЕ НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТИ В ОБЩЕЙ ПАТОЛОГИ!
ЧЕЛОВЕКА И КЛИНИЧЕСКОЙ ПРАКТИКЕИдентификация генетических вариантов подверженно^!
широко распространенным заболеваниям, обозначаемая иш
как генетическое тестирование многофакторного заболеваш
является активно развивающейся областью исследований, кс
рая имеет важное теоретическое и практическое значение. Чж
публикаций по генетическим ассоциациям ежегодно в послед!
десятилетие удваивается, и эта информация излагается в 1;
научных журналах на различных языках. Направления, но ко1
Кйіі.і 6. Болезни с наследственной предрасположенностью3094і\ 6.8, Применение результатов исследования генетических ассоциацийH.IV) осуществляется систематизация накапливаемой информации,
^Ірсдставленьї на рис. 6.8.Полногеномные ассоциативные исследования, анализирующие
^иювременно до 1 млн геномных вариантов, раскрывают биологичс-
ркис основы многофакторных заболеваний, открывая новые, до сих
Юр неизвестные метаболические пути формирования патологических
|к*нотипов (болезней), обнаруживая терапевтические мишени, что
їііособствует созданию новых лекарственных средств. Идентификация
іиомаркеров позволяет надеяться, что риск заболевания может быть
[еиижсн путем проведения оптимальных схем лечения. Даже умерен-
[ііі.іс ассоциации «генотип—фенотип» могут быть использованы для
[fiojiee широких возможностей перехода от теории к практике.Однако следует заметить, что большинство идентифицированных
[к настоящему времени ассоциаций генетических полиморфизмов с
|и иогофа кторн ыми болезн я м и объясн я ют небол ьшой процент (2—10%)
■Индивидуальной вариабельности в риске заболевания. В связи с этим,
Прежде чем «генетические профили» станут пригодны к широкому
Использованию в клинической практике, необходимы дополнитель-
Пмс уточняющие исследования и разработки. Они касаются совер-
Йіснствования подходов к расчету рисков заболеваний, создания
Правового обоснования для применения генетических тестов, согла-
310Клиническая генісованныхдействий исследователей, врачей и гтаииентов. Клииичесі
практика должна опираться на доказательную медицину, Результі
генетического тестирования многофакторного заболевания ник^
да не будут единственными ориентирами в принятии решений
лечебно-диагностическим и профилактическим мероприятиям
генетические тесты не вместо, а вместе с фенотипическими марі
рами составляют основу в персонализированном прогнозе, всв1
вероятностном.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯАддитивное действие геновАссоциации болезней с маркерами (обшая концепция)
Ассоциация антигенов HLA с болезнями
Ассоциация антигенов А ВО с болезнями
Болезни с наследственной предрасположенностью (определена
классификация)Генетико-эпидемиологический подход
Генетические основы индивидуальных различий метаболии
канцерогеновГенетические основы предрасположенности
Генетический маркерГетерозиготность как фактор предрасположенности
Главные гены предрасположенностиДоказательства предрасположенности близнецовым методом
Клинико-генетические доказательства предрасположенности
Молекулярно-генетические механизмы канцерогенеза
Наследственная предрасположенность и профилактика мної
факторных заболеванийОнкогены и гены-супрессоры опухолей
Основные группы болезней с наследственной предрасположен!
стьюПорог предрасположенности
Потеря конституциональной гетерозиготиости
Предрасполагающие к раку наследственные синдромы
Причины болезней с наследственной предрасположенностью
Характеристика родословных при болезнях с наследственна
предрасположенностью
ійіі 6. Болезни с наследственной предрасположенностью311РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАЛксепович ти. Статистические методы генетического анализа при-
|йК(И) человека: учеб, пос. — Новосибирск: Новосиб. гос. ун-т,
;м. - 128 с.Ісріс'іический паспорт — основа индивидуальной предиктивной
►липины / под ред. B.C. Баранова, — СПб.: Изд-во Н-Л, 2009. —
\Н с,Ичюмика — медицине. Научное издание / под ред. В,И, Иванова,
IJI, Киселева. — М.: ИКЦ Академкнига, 2005. — 392 с.Имяпитов Е.Н., Хансон К.П, Молекулярная онкология; клинические
Л11-кты. - СПб.: СПб МАПО, 2007. - 212 с.Новик Л.А., Камилова Т.А., Цыган В.Н. Введение в молекулярную
(ологию канцерогенеза; учеб. пос. / под ред. Ю.Л. Шевченко. — М.;
5)()ТАР-Медиа, 2004. — 224 с.Пузырев В.П., Фрейдин М.Б., Кучер А Н. Генетическое разнообразие
іродонаселения и болезни человека. — Томск: Печатная мануфак-
/ри. 2007. - 320 с.
ГлаваЭКОЛОГИЧЕСКАЯ ГЕНЕТИІ
ОБЩИЕ ВОПРОСЫЭкологическая генетика человека изучает влияние фактоц
среды обитания на наследственность. Основы экологической геї
тики человека лежат в общебиологических закономерностях эвол|
ции. Одна из парадигм медицинской генетики состоит в том, что,
всех жизненных проявлениях действие любых генов осуществляет
в тесном взаимодействии с факторами среды.На протяжении сотен тысяч лет окружающая человека ср<
постоянно менялась. К ее изменениям человек приспосаблив!
ся как биологический вид с широкой нормой реакции. Челої
как мыслящее существо активно изменял элементы среды свов|
обитания. Одновременно на групповом и популяционном ур<
нях происходил отбор ГЄІСОТИПОВ. Окружающая среда обеспечи]
ла отбор, выживание, процветание популяций или групп люд^
в зависимости от их наследственных характеристик. Эволюц|
человека шла через эволюцию его генотипа. Формировалась
биологическая природа, Человек приспосабливался к окружающ^
среде от первобытной пещеры до современного коттеджа как сої
альное и биологическое существо.От собирательства и охоты человек перещел к более эффекти|
ной добыче пищи (разведение полезных растений и животны!
Это делало его менее зависимым от борьбы за существование,
следовательно, естественный отбор приобретал характерные ді
человека формы.При воздействии неблагоприятных факторов окружающї
среды на человека могут наблюдаться нежелательные эффект
в виде:• изменения наследственных структур (индуцированный мутащ
онный процесс);• патологических проявлений экспрессии генов в ответ на спещ
фические факторы среды;
ІМії A Экологическая генетика313II «менений генофонда популяций в результате нарушения гене-
IІІЧССКОГО равновесия между основными популяционными про-
псссами (мутацрюнным процессом, отбором, миграции, дрей-
фом генов).‘‘Эффекты 1-го типа — это мутационный процесс, индуцирован-
ІІІ мутагенными факторами окружающей среды (в широком смысле
ЮІПІ), ведущий к повышению темпов наследственной изменчивости
рлипска на индивидуальном и популяционном уровнях.’’Эффекты 2-го типа у человека проявляются на индивидуальном
ЇОІІІІС с виде патологических реакций (болезней), а на популяци-
lihJM уровне — в виде большей или меньшей приспособленности
Цлиигации, акклиматизации).'’)ффекты 3-го типа — изменения генофонда популяций являются
)лго1феменными и реализуются через десятки и даже сотни поколе-
|И(1. Ьиологически стабильному виду свойственно постоянное равно-
основных генетических процессов (мутационного процесса,
>1>ра, миграции, дрейфа генов). Современный период характери-
4CW большей скоростью и объемом изменений среды обитания.
Іні'исдственность человека на популяционном уровне так быстро
Ігіпггься не может. Следствием высоких темпов и большого объема
іменений среды обитания человека (измененные экологические
5ло1>ия) могут стать изменения в генофонде конкретных популяций
|ли человечества в целом.ИНДУЦИРОВАННЫЙ
МУТАЦИОННЫЙ ПРОЦЕССИсточником наследственной изменчивости как основы эволю-
|ии служат мутации. Мутационный процесс — одна из существен-
IIJX характеристик человека как биологического вида. Хорошо
Ііспшов.іено, что определенный, оптимальный для человека уро-
мп> мутационного процесса может изменяться под влиянием
ИІОГИХ физических, химических и биологических факторов.
Универсальность и всеобшность явления индуцированного мута-
меза в современных условиях не вызывают сомнений, и, следо-
Щ'гельно, все его закономерности распространяются на человека
Итпбл. 7.1).
314Клиническая гвНІТаблица 7.1. Последствия индуцированного мутагенезаПоследствия мутацийВ клетках гонадВ клетках эмбриона
и плодаВ соматических клс
постнатально1. Расширение гене¬
тического полимор¬
физма.2. Повышение
частоты наслед¬
ственной патологии1. Снижение нормы
реакции (приспосо¬
бленности).2, Повышение частоты
врожденных пороков
развития (мутагенез
тератогенез)1. Повышение частоті
злокачественных ноі(
разований (мутагене!;
канцерогенез).2. Нарушение иммуНІ
тета.3. Преждевременное 1
рение І1Если мутации возникают в зародышевых клетках, то повыш<
частота наследственной патологии.Мутации в клетках эмбриона и плода ведут к снижению ш
реакции, или приспособленности будущего ребенка, к coxpai
гомеостаза при повышенных нагрузках среды, повышению чг
врожденных пороков развития, гибели эмбриона или плода, вн]
тробной задержке роста.Мутационный процесс в соматических клетках в постнаталі
периоде повышает частоту возникновения злокачественных но|
разований, нарушает иммунитет, обусловливает преждевремві
старение.Характеристика мутационного процесса у человека на coBpejj
ном уровне знаний представлена в статье «Современный взгляд
мутационный процесс у человека» на компакт-диске.В целом биологическую и медицинскую значимость ПОСІ
ствий мутационного процесса у человека на современном э1
надо рассматривать по показателям мутагенеза, канцерогенвІ
тератогенеза.В 50-х годах XX в. в связи с расширенными испытаниями
ного оружия на нашей планете стал повышаться радиационный
Широкое Применение ионизирующих излучений в медицине, т<
генные катастрофы с ядерным материалом (атомные предпрйЯІ
подводные лодки, атомные электростанции) приводят к увеличв!
накапливаемой дозы облучения у каждого индивида и повьішеі
частоты мутаций у сотен тысяч людей, что хорошо известж
последствиям Челябинских и Чернобыльской аварий.
їм /. Экологическая генетика315И S(l -60-х годах XX в. во всех странах стали резко увеличиваться
ни кюдство и применение химических веществ, в том числе активно
^И»и>|цих Исі хромосомы. Химические вещества (до 60 ООО наимено-
iMiti) в среде обитания человека входят в состав пищевых продук-
II'!и являются отходами производства, В мире производятся уже
4'ніки миллиардов тонн химических веществ в год. Многие есте-
sriiHuie и синтезированные химические вещества в воздухе, воде,
ІИІГ, иа рабочем месте, в коммунальной среде, в лекарствах являются
ТгIIанальными мутагенами, канцерогенами, тератогенами.
Принимая во внимание наводнение среды обитания человека хими-
уцими веществами и увеличение контактов человека с ионизирующи-
IIUIучениями, можно сделать вывод о возможности реального повы-
ІПІІЧ частоты мутирования в зародышевых и соматических клетках.И XX в. были выяснены основные закономерности индуциро-
Иніого мутагенеза, свидетельствующие о неотложности мер по
кроплению окружающей среды. Среди главных характеристик
Ілуиированного мутагенеза можно указать на:(и сутствие порога действия;• іанисимость эффекта от дозы;• с габильность мутаций;• іілдитивность эффектов от действия разных мутагенов;' гсроченность действия.I лавный результат фундаментальных генетических исследований
Гіроблемам мутагенеза — разработка методов выявления мутаген-
|И опасности. Их широкое применение позволит экологам сделать
влу обитания человека более безопасной с генетической точки
>виия, поскольку:• тгрязненнос! ь окружающей среды, в том числе мутагенными
(І)акторами, увеличивается в целом; к тому же быстро изменяется
спектр факторов;• комплексное воздействие многочисленных химических и радиа¬
ционных загрязнителей дает суммарный эффект, превышающий
допустимые уровни токсичности;• и среде обитания человека все чаше появляются факторы с дли¬
тельной устойчивостью (диоксины, радионуклиды), постоянно
пополняющие мутации;• современное жилище с его полимерной и пластмассовой «начин¬
кой» может быть источником мутагенных, тератогенных и канце¬
рогенных влияний;
316 Клиническаягеиеі• стресс и широчайшее распространение психоактивных вешв<
(курение, наркотики) усиливают мутационный процесс.ПАТОЛОГИЧЕСКИЕ ПРОЯВЛЕНИЯ
ЭКСПРЕССИИ ГЕНОВНа протяжении эволюции в человеческих популяциях в связ
постоянно текущими мутационными и генетико-автоматическ!
(дрейф генов) процессами, а также под влиянием отбора сфорї
ровался широкий наследственный сбалансированный полим<
физм. Какой-либо ген считается полиморфным, если он npncj
ствует в популяции в виде двух аллелей и более, причем част
редкого аллеля составляет не менее 1%. Распространенность п<
морфизмов генов в современных популяциях человека громадн|
Не менее 25%, т.е. около 8000 генов, детерминирующих антигі
ную, ферментативную, рецепторную системы и другие элеме!
молекулярно-биохимической конституции человека, предстаї
ны в виде полиморфных систем — 2 аллеля и более, следован
но, число индивидуальных вариаций генотипов может быть 2®*
Чтобы представить себе реальную величину такого многообраз
укажем, что вариации всего лишь по 25 полиморфным систеї
(225) дают число индивидуальных генотипов, приближающе!
к численности населения нашей планеты. С другой сторої
около 99% всех полиморфизмов являются молчащими, и тол|
1% проявляется под действием того или иного фактора окружі
щей среды. Кроме того, следует отметить, что значимость
дого полиморфизма определяется занимаемым им положені
в геноме. Генетические полиморфизмы, так же, как и мутаці
могут приводить к синтезу аномального белка, снижению
повышению его количества, что не может не отразиться на
функции.Многочисленные вариации в ферментных системах, трансш
ных белках, антигенах и рецепторах клетки обусловливают инді
дуальные особенности метаболизма химических веществ, реакциЙ|
биологические агенты или физические факторы.Концепция экогенетики человека к настоящему времени уже с(
мировалась. Ее основы начали закладываться в середине 50-х г<
прошлого столетия, когда впервые были обнаружены генетичес
Пінна 7. Экологическая генетика317|1*и‘|>минированные патологические реакции на лекарства, обуслов-
Цоііііі.іс недостаточностью ферментов. Для описания таких состояний
предложен термин «фармакогенетика» (см. гл. 8), который вскоре
ІІІ.ГІ расширен до понятия «экогенетика«>.И связи с большой практической важностью знаний фармакоге-
1ПІ1ЧЄСКИХ реакций для клинипистои фармакогенетика подробно
пожени в следующей главе, хотя, безусловно, она является частью
Ікіиснетики человека, Накопление экспериментальных данных, при-
высокой чувствительности и толерантности к ксенобиотикам
(чужеродным веществам) у отдельных индивидов, а также молеку-
Мірііая расшифровка механизмов наследственных различий био-
Грпмсформации ксенобиотиков поставили вопрос о наследственных
1ЛИЧИИХ реакций на разнообразные внешние факторы химической,
|лпической и биологической природы.Разработка проблем экогенетики человека ускорилась в связи с
1-см, что среда обитания человека пополнялась новыми факторами
(лекарствами, пестицидами, пищевыми добавками, профессиональ-
(».1ми вредностями и др.), на которые появлялись патологические
)гакции. В процессе эволюции человек не соприкасался с такими
ісіцествами (или факторами), поэтому на действие этих веществ не
>ыло никакого отбора. Какой-то аллель мог ранее распространиться
популяции из-за своих селективных преимуществ или дрейфа, но
других условиях окружающей среды он будет проявлять патоло¬
гические эффекты. Речь идет о таких, как бы «молчащих», аллелях,
союрые начинают функционировать в новых условиях среды. ЭтотДействие факторов
окружающей среды
в процессе
эволюцииГенетический
и фенотипический
полиморфизм попу¬
ляций и индивидовНовыефакторысредыРавновесиеИзменениегенетическихэкспрессии генов-процессовэкогенетическиев популяцияхболезни^ис. 7.1. Попу;]яционно-гснетическая основа возникновения экогенетиче-
5КИХ болезней
318Клиническая генетіфеномен называется экогенетическим действием факторов (рис, 7«j
а патологические проявления мутантных аллелей под влияні
факторов окружающей среды — экогенетическими реакциями
болезнями.Понятие о «молчащих» (или нейтральных) генах весьма усЛС
но. Биологический или патологический эффект какого-либо алл<
зависит от воздействия специфического фактора среды.К настоящему времени не только сформулировано понятие і
экогенетике, но и определены основные направления иссл<
ваний в этой области. Оказалось, что наследственные разлш
могут проявляться в реакциях не только на лекарства, но и
физические факюры, на пищу и особенно на пищевые добавки,
загрязнения атмосферы, профессиональные вредности. Соглас
концепции экогенетики, необходимо изучение действия ВНЄШІ
факторов (особенно новых) с целью выявления наследственно
словленных патологических реакций. Это будет основой создаї
адаптивной среды для каждого человека: подбор индивидуалы
диеты и климата, исключение приема лекарств с патологичесі
ми реакциями, обоснование профессионального отбора и
Такой подход приближает нас к персонализированной медиці
(см. гл. 6).Все болезни многофакторной природы можно рассматривать
примеры экогенетики человека, потому что их развитие становиі
результатом взаимодействия генов предрасположенности и фактої
внешней среды.Генетические различия в реакциях на действие факторов вив^
ней среды можно установить с помощью генеалогического (сеі
ного) анализа, близнецового или популяционно-статистическс
метода. Каждый из этих методов имеет свои разрешающие
можности и ограничения в экогенетике. В выявлении новых эко|
нетических вариаций генотипов все методы дополняют друг дру|
кроме того, наряду с применением генетических методов нуж|
проводить биохимические исследования молекулярных механі
мов патологических реакций (вариантов ферментов, рецептору;
транспортных белков, ионных каналов). Одновременно с генет
чески м анализом нужно применять токсикологические и фарї
кологические методы для определения концентрации различи!
веществ в организ.ме и путей их метаболизма.
iria 7. Экологическая генетика319ГЕНЕТИЧЕСКИЕ ОСНОВЫ БИОТРАНСФОРМАЦИИ
ЧУЖЕРОДНЫХ ВЕЩЕСТВ (КСЕНОБИОТИКОВ)Некоторые специфические мутации являются основой высокой
мк'гвительности или толерантности их носителей к определен-
|ым (факторам внешней среды. Потенциально токсические факторы
И>ужающей среды поражают не все население в одинаковой мере.
1»л1.ко часть населения, генетически предрасположенная, т.е. имею-
1(01 определенные мутации, высоко чувствительна, для которой
ітчіциальная токсичность переходит в реальную. Доказано, что
человека существует генетический контроль метаболизма посту-
ИОІЦИХ в организм химических соединений (биотрансформации),
ЇО.ІІ и морфизм по этим генам и создает экогенетические вариации.Современные данные позволяют говорить о трех фазах деток-
ІКИЦИИ или элиминации чужеродных веществ (ксенобиотиков), из
«яорых первые две осуществляются с помощью генетически детер-
|Ц11ИруемЫХ ферментов.На основе многочисленных сведений о природе экогснетических
1рнаций можно сделать вывод, что они обусловлены сбалансиро-
1МИЫМ полиморфизмом в генах ферментов, участвующих в первых
іух фазах детоксикации. Полиморфизм генов детоксикации выра-
5и достаточно сильно. По каждому гену уже известны десятки или
ІЖС сотни аллелей различной природы. «Инвентаризация» их еще
швершена.Первую фазу биотрансформации ксенобиотиков называют фазой
ітивации (функционализации или модификации). В этой фазе осу-
іссівляются биохимические реакции, в процессе которых ксено-
ІИОТИКИ освобождаются от активных функциональных групп (-ОН,
•NHj, -SH) и превращаются из липофильных соединений в более
шрофильные. Первую фазу детоксикации обеспечивают следующие
ферменты: семейство изоферментов цигохрома Р450, параоксона-
(PON), алкогольдегидрогеназа (ADH) и альдегиддегидрогеназа
LDH), бутирилхолинэстераза, эпоксидгидролаза и др. При нали-
|Ии мутаций в перечисленных выше генах теряется ферментатив-
||и активность, активация не будет происходить, и, следовательно,
гсиобиотики будут вызывать повреждающий эффект сначала на
1ЄГ0ЧН0М, а потом и на организменном уровне.
320Клиническая геніВторая фаза — фаза ней ірализации (дезактивации, детоксикаціСуть процессов в этой фазе сводится к синтетическим реакІ
ям. к активированным в первой фазе продуктам присоединяі
ся ацетильные, мстильные, суфгидрильные группы, глутатион,
приводит к образованию гидрофильных конъюгатов. Это водо|
творимые нетоксичные компоненты, которые и выводятся из 0(
низма. Вторую фазу биотрансформации обеспечивают следуюі
ферменты: глутатион-8-трансферазы и N-ацстилтрансферазы (Ni
тиопуринметилтрансфераза (ТПМТ), сульфонтрансферазы, зпої
гидролазы, УДФ-глюкуронилтрансферазы (UGT, УДФ — уриди!
фосфоглюкуроновая кислота). Если вторая фаза биотрансформаї
не состоится по причине мутантной формы фермента, то продуі
первой фазы детоксикации (промежуточные электрофильные м(
болиты), накапливаясь, будут вызывать, как и неактивирован!
ксенобиотики, окислительный стресс, токсические эффекты, MJ
ции, злокачественные новообразования и другие нежелателі
последствия.Третья фаза детоксикации обеспечивается работой физиолог!
ских систем выделения (кожа, почки, кишечник, легкие).Активная секреция метаболитов (а иногда и просто ксенобиотиі
в мочу, желчь, пот осуществляется гликопротеином Р, а также ті
Портерами органических анионов и катионов. За препятствие вс£
ваниию ксенобиотиков в кишечнике отвечает гликопротеин Р. Бс
подробные сведения о третьей фазе детоксикации («транспорте
метаболитов) можно найти в следующей главе и в статье В.С.Бараї
и др. «Научные основы предиктивной медицины» на компакт-дисіНАСЛЕДСТВЕННО ОБУСЛОВЛЕННЫЕ
ПАТОЛОГИЧЕСКИЕ РЕАКЦИИ НА ДЕЙСТВИЕ
ВНЕШНИХ ФАКТОРОВЭкогенетика человека имеет дело с вариациями ответов органі
различных людей на воздействие факторов среды. На основе
фактов генетики пытаются объяснить, почему поражается то;
некоторая часть подвергающегося вредному воздействию населеї
и как индивиды различаются по адаптации к среде,Экогенетические реакции могут быть обусловлены редк!
мутантными аллелями, которые вызывают патологический ответ
Ийііа 7. Экологическая генетика321^іипсинкразию. Однако существуют и полиморфные системы, обу-
юпливающие количественные вариации ответа. Экогенетические
|1К‘Г1,1 могут контролироваться одним или несколькими генами.
(н|>;іктер сегрегации признака в потомстве в этих случаях будет соот-
мп повать моно- или полигенным системам с учетом воздействияо I КС гствуюшего фактора внепгней среды.<|)акторы срсды обитания человека, которые могут влиять на про-
ІИікчіие патологических эффектов мутантных аллелей, необычайно
||И)образны (в атмосфере, пище, среди антропогенных факторов,
|р(1(|>ессиоиальных вредностей и т.д.).Н широком плане изучение токсических наследственно обуслов-
мтых реакций на факторы среды выделено в отдельное направле-
ІИІ', названное токсикогенетикой, а в еше более широком смысле —
аксикогеномикой. Базы данных по геному человека и современные
отиые информационные технологии позволяют прогнозировать
иксические проявления отдельных факторов среды у лиц с опреде-
рмиыми генотипами.кігрязненис атмосферы выхлопными газами автотранспорта,
гиюбразными продуктами многочисленных фабрик и заводов пред-
Ml ИЛ лет серьезную гигиеническую npo6jreMy глобального масштаба,
некоторых городах в атмосферу выбрасывается до 1000 кг плотных
епдков на человека в год. Химические соединения и пылевые части-
|М попадают в организм через легкие, кожу и слизистые оболочки,
чывая патологические реакции. Все это в широком понимании
цпдит в среду обитания человека. С одними факторами человек
)мрикасается постоянно, с другими — изредка. Проявления наслед-
[■ненных вариаций возможны в ответ на воздействие любых факто-
)». Часть таких факторов уже известна генетикам и врачам.Для людей, занятых на некоторых производствах, доза {или кон-
гитрация) этих веществ намного больше допустимых, но они пере-
іосит это без профессиональной болезни. У некоторых индивидов
|же меньшая доза ведст к профессиональному заболеванию (сили-
иу, антракозу, хронической пневмонии).Наиболее изученная мутация, приводящая к недостаточности белка
1'Лнтитрипсина, обус;ювливает патологическую реакцию на загряз-
Я1ИС атмосферы. Этот белок сыворотки крови называют также инги-
і'юром протеин аз. В норме его концентрация повышается при раз-
1ИЧНЫХ состояниях (беременность, воспаление, введение эстрогенов).
5ИСТИЧЄСКИЄ варианты белка обнаружены во многих популяциях. Его
322Клиническая rehформы различаются по антитрипсиновой активности и электрофс
ческой подвижности (аллели М, S, Z). Неактивность белка обуслс
аллелем Z (рецессивный признак). Частота гомозигот ZZ у еврогм
составляет 0,05%, гетерозигот — 4,5%. Лица с наследственной ш
точностью ингибитора протеиназ, если они гомозиготны по дані
признаку (генотип ZZ), чрезвычайно склонны к развитию хрони»
воспалительных заболеваний и эмфиземы легких. Эмфизема леї
таких людей развивается в 30 раз чаще, чем в популяции у лиц
30—40 лет, и протекает очень тяжело. Основа этой предраспаложені
к эмфиземе лежит в том, что антитрипсиновая система играет ■
ную роль в ограничении воспалительного процесса. При любых,
незначительных повреждениях легочной ткани (воспалении, на;
НИИ микроциркуляции) протеолитические ферменты вскоре начИ!
разрушать измененные участки. В норме включается синтез ингиї
протеиназ, который нейтрализует действие протеолитических фв|
тов и приостанавливает разрушение. При недостаточной щ
ингибитора протеиназ (мутантный генотип) протеолитические \{
менты разрушают поврежденные участки, что и приводит к эм<
легких. Курение и запыленность воздуха существенно ускоряют
тие эмфиземы. Некоторые авторы описывают и более тяжелые
проявления недостаточности ингибитора протеиназ у детей —
жение печени. Даже у лиц, гетерозиготных по гену недостаточі
ингибитора протеиназ (генотип MZ), частота которых в отде
популяциях превышает 10%, выражены патологические реакщ
повышенную запыленность и курение, т.е. повышен риск эмф|
легких. Следовательно, необходимо исключить влияние произволе
ной пыли на этих людей, чтобы предотвратить развитие у них
заболевания. Методы определения недостаточности арантитри!
разработаны, их можно применять при профессиональных осмот
отборе для работы на соответствующих производствах.В среде обитания человека содержится много углеводород<
том числе полициклических, которые после гидроксилирові
арилгидрокарбонгидроксилазой образуют в организме активные
сиды. Эта ферментная система у человека хорошо изучена; индуі
синтеза этого фермента очень вариабельна. Существуют гомозиі
с большим количеством фермента, гетерозиготы и гомозиготы<
малым. Необходимость этих сведений для понимания химичес
канцерогенеза очевидна, потому что эпоксиды являются аки
ми канцерогенными формами полициклических углеводороде!
in.I 7. Экологическая генетика323lliiicpoiенная активность зависит от относитслыюй активности
^пксидобразующих ферментов, с одной стороны, и систем, разла-
Uittimx ’зпоксиды, — с другой. Таким образом, эти соединения явля-
Ми потенциальными мутагенами и канцерогенами. Например, до
(юльных раком легких имеют высокий уровень фермента, а в
|ик‘й популяции этот признак встречается очень редко. Люди с
ішжой индукцией ари л гидрокарбон гидроксилазы должны отка-
ІІІ.ІЯ от курения и исключить профессиональный контакт с углс-
рлородами.I развитие легочных заболеваний влияет полиморфизм гена
Гюхондриальной эпогидроксилазы. Датіьій фермент присоединяет
;;iv к эпоксидам, превращая их в трансгидродиолы и далее в конъю-
lihi с глюкуроновой кислотой и 1’лутатионом. Так же как и в случае
^vnix полиморфизмов, данный ген имеет «быстрый» и «медленный»
|1К'ли. Выявлена положительная корреляция «медленного» аллеля
[;іаиолеваниями органов дыхания. В сочетании с курением у таких
|п чаше, чем в среднем в популяции, развиваются респираторные
мыевания, а также эмфизема и обструктивная пневмония.)фессиональные вредностиИ производственных условиях рабочие контактируют с бснзпире-
)м, ароматическими соединениями, лакокрасочными материала-
|И, продуктами изготовления резиновых изделий, солями тяжелых
па л лов и другими многочисленными факторами. В последнее
>смя обнаружена взаимосвязь заболеваемости с производственными
ікіорами улиц с мутациями в группе генов глутатион-5-трансфераз
ісііа NAT2 {NAT2).Глутатиоиопосредованная детоксикация и]раст ключевую роль в
)с ифеживании продукюв псрскиспого окисления липидок и иерок-
ІДОВ ДНК, восстанавливает органические гидроперекиси в спирты
июмеризует некоторые стероиды и простагландины.Центральное место в семействе генов глутатион-8-трансфсраз
іпимают гены GSTTL GSTMlm GSTPI, функционально ослабленные
ЇЛСЛИ которых ассоциированы с развитием злокачественных иово-
іріізований, хронического бронхита, эмфиземы легких. Ген GSTP!
к'спечивает выведение из организма ароматических соединений и
Мізпирена. Полиморфизм IlelOSVal повышает активность фермента
Ароматическим соединениям в 7 раз и снижает его детоксикацион-
свойства по отношению к бензпирену в 3 раза.
324Клиническая гв^Геи NAT2 участвует в детоксикации ксенобиотиков, с одер)
ароматические амины или гидразиновые группы, путем их а|
лирования. Выделяют 4 полиморфных варианта: 3 «медленны»
«быстрый». Доказана связь между «медленным» ацетилятором
вйтием рака мочевого пузыря. Риск особенно повышается npltj
действии соответствующих факторов среды (курение, произі
резиновых изделий, красок).в ряде сообщений описывается различная чувствительн(
солям тяжелых металлов (свинца, ртути, кадмия и др.). Нап|
отравление органическими соединениями ртути вызывает у
людей нервно-психические расстройства различной выражен!
Гетерозиготные носнтелн генов цистиноза и анемии Фанкони
быть предрасположены к токсическому действию металлов или і
гих почечных ядов. Повышенный, хотя и не токсический yj
свинца может быть «спусковым крючком» гиперактивного пов<
у детей с наследственной предрасположенностью.Пищевые вещества и пищевые добавкиРазвитие таких постгеномных направлений, как протеол
метаболомика и новые генетические методики, заложили
изучения генетического контроля в нутрициологии. Нутригеї
изучает влияние основных пищевых ингредиентов на геїюм,
вает влияние пищевых молекул на метаболические пути и коні
гомеостаза. Сегодня уже доказано, что некоторые нутриенты
оказывать влияние на ДНК, эпигенетические (например, меі
рование), транскрипционные (влияние на мРНК) и посттраї
ционные процессы (фосфорилирование, гликозилирование
Нутригенетика оценивает, как индивидуальные особенности
типа определяют ответ на пищу, т.е. как генетические варі
организмов влияют на усвоение пищи.Основные постулаты нутригеномики следующие.— Больтпинство нутриентов прямо или косвенно воздейсі вуї
геном, изменяя экспрессию генов или их структуру.— при определенных условиях у некоторых индивидов nntl
может быть серьезным фактором риска развития многих
леваиий.— Некоторые регулируемые пищевыми факторами гены, возі
но, играют роль в возникновении, течении и тяжести хрої
ских заболеваний.
itt / : Экологическая генетика325( іспень влияния питания на баланс между здоровьем и: заболс-
иниисм может зависеть от индивидуального генотипа.
Коррекция диеты осиовывасгся на знании пиіпсвой нотрсбно-
I I и, состояния питания и генотипа индивида (персонализиро-
ианнос питание),
к '|;н'симескими примерами нутригенетических заболеваний явля¬
ли (|>сиилкетонурия (см. гл. 4), галактоземия, непереносимость
Ции>1. глютсиовая энтеропатия, семейная гиперхояестеринемия.
Гіі.:і:іктоземия — заболевание, связанное с дефицитом галактозо-
(»сфатуридилтрансферазы или галактокиназы, в результате чего
ірііиіи'зме накапливается галактоза и галактозо-1-фосфат, приво-
к кіиаракте, циррозу печени, задержке психического развития и
Чіису. Исключение галактозы из пищевого рациона способствует
|Ми|)1)ианию практически всех симптомов заболевания.I live одним из наглядных примеров нутригенетических состояний
ик'тся непереносимость лактозы, приводящая к дискомфорту в
|ик“||[ике и диспепсическим явлениям после употребления молока.
!-fcmtie4HHK.e не вырабатывается лактаза, в результате чего лактоза
расщепляется и служит хорошим субстратом для размножения
|Ил<?стной микрофлоры. Мутантные аллели гена лактазы широко
Л1ространены у восточных народов (до 95-100%), среди американ-
|их индейцев и афроамериканцев (70-75%). У европейцев ^іастота
шзигот по этим мутациям невелика (5—10%).Один из вариантов синдрома нарушенного всасывания у детей
с непереносимостью глютена (белок пшеницы и других зла-
>И). Это заболевание называется целиакией. Дети тяжело заболе-
iHvr, как только начинают получать прикорм пищей, содержащей
ІИКИ. При исключении злаковых продуктов (хлеба, манной каши)
ІКИО дети развиваются нормально. Близнецовым и генеалогиче-
{им методами показано значение наследственности в этих реакци-I, 1 (редраспо.аоженность к целиакии определяется взаимодействи-
диух генов главного комплекса гистосовместимости II класса1-І и р-1). Интересно, что некоторые сорта пшеницы не вызывают
|Т(5логичсских реакций. Они отличаются от других сортов заме-
)й одной или нескольких аминокислотных остатков в молекуле
іютена.Конские бобы вызывают гемолиз улиц с наследственной недоста-
пюстью Г6ФДГ. Если своевременно не приняты меры, за гемолизом
1C дует поражение \ючек. Носители соответствующего гена часто
326Клиническая гені(10% и выше) встречаются в районах, где была распространена Ml
рия, потому что гетерозиготы НС болеют малярией. Недостаточ!
Г6ФДГ наследуется как сцепленный с Х-хромосомой рецессиі
признак.Гетерозиготные женщины встречаются чаше, чем гемизигої
мужчины. Все эритроциты мутантного происхождения у Г0М(XX и у гемизигот XY чувствительны к провоцирующим факторе!
конским бобам, лекарствам, промышленным окислителям. Ге?
у таких лиц вызывают и многие лекарства. В полном соответсі
с гипотезой М. Лайон у гетерозиготных женш;ин может быть раЗ
соотношение нормальных и мутантных эритроцитов (от 1 : 9І
99 : J). Примерно у 30% гетерозиготных женишн после воздей(
провоцирующих факторов наблюдается гемолиз эритроцитов с В1
женными клиническими симптомами.Структурные мутации гена приводят к синтезу аномальной м(
кулы Г6ФДГ. Нарушения в этой молекуле изменяют ее каталиті
скую активность, кинетические свойства, стабильность и элек!
форстическую подвижность. Известно более 200 вариантов Г6<
но развитие патологических реакций обусловливают лишь немж
из них. При наличии аномальной молекулы Г6ФДГ в эритро!
снижаются связывание кислорода, скорость восстановления
моглобина и устойчивость к воздействию различных потенция
ных окислителей, при аномальном варианте Г6ФДГ нарушав
основная функция фермента — поддержание стабильности мем(
эритроцитов.Катехоламины, содержащиеся в сыре, у некоторых людей м(
вьізьіваїь мигрень. Это связано с пониженной конъюгацией тир*
на. Иногда мигрень провоцирует шоколад, что объясняется ни^
активностью моноаминоксидазы.Известны специфические реакции людей на алкоголь. У больші
ства представителей монголоидных популяций после уиотреблеї
малых количеств алкоголя немедленно краснеет лицо, вознике
тахикардия, жжение в желудке, мышечная слабость и другие при;
ки отравления. Это врожденное свойство сохраняется на всю жи1
и не зависит от привыкания к алкоголю. Такая реакция на алког
объясняется наследственными вариациями в молекулах двух ферв
тов, расщепляющих этанол.Гены ADH печени представлены тремя полиморфными лок]
ми {ADH-1 ADH-2, ADH-31 ALDH - двумя {ALDH-1 и ALDH%
ІЙІ1 /. Экологическая генетика327|й питая выше токсическая реакция на малые количества алкоголя
ОІІІ I пенна людям, у которых отсутствует изоформа ALDH-1.'I и) же касается многофакторных заболеваний, то здесь на первый
ІІИІ И1>1ступают полиморфизмы генов, участвующих в расщеплении,I и нации, детоксикации и выведении нутриентов, попадающих в
^мшизм с пищей. Считается, что пищевые факторы ответственны
химерно за 30% всех злокачественных новообразований. Велика
і же их роль в развитии сахарного диабета 1-го и 2-го типа, ише-
1'к‘ской болезни сердца, ожирения, гипертонической болезни,
|к()|'орых пороков развития и другой не менее часто встречаемой
Ікміогии.Наиболее изученным заболеванием с этой точки зрения является
щнрный диабет (1-го и 2-го типа). Генетическая предрасположен¬
ии» к сахарному диабету 1-го типа, по мнению авторитетных эндо-
»и пологов, хорошо нивелируется «правильной» диетой. Заболевание
ижно отсрочить и даже вылечить с помощью создания «персона-
шрованной диеты» на основании сведений о значимых полимор-
»мах для этой патологии. Так, показано, что частота сахарного
(и»бста 1-го типа находится в прямой зависимости от энергетической
>мности потребляемой пищи и в обратной — от доли растительной
ІИіііи в ежедневном рационе человека. Отмечен также выраженный
(ротективный эффект грудного вскармливания. А неблагоприятные
ілели генов DQAI и DQB1 повышают риск заболевания при употре-
ІСНИИ животных белков (например, мяса).Очевидно, что в этиологии сахарного диабета 2-го типа большую
)ль играет воздействие окружающей среды, в частности питание.
ІИННОЄ заболевание относительно легко корректируется диетой.
)днако не у всех пациентов изменение диеты бывает эффективным.
1а основании этого был сделан вывод об индивидуальных различиях
реакции пациентов на диету. Полиморфизмы затрагивают не толь-
(0 гены, включенные в метаболизм глюкозы, но и обмен инсулина,
ІИІІИДОВ, водно-солевой гомеостаз тканей, артериальное давление,
іммунньїе реакции и др. Сведения обо всех значимых полимор¬
физмах помогут составить персонализированную диету для каждого
інциента, страдающего сахарным диабетом 2-го типа.Целый ряд заболеваний и нарушений связан с дефицитом фолие-
'|ой кислоты, которая участвует в синтезе нуклеотидов и в реакции
превращения гомоцистеина в метионин. Очевидно, что дефицит
фолиевой кислоты может вести к. различным генетическим нару-
328Клиническая ген*шениям (например, врожденным порокам развития) и к наког
и ИЮ гомоцистеина, оказывающего токсическое влияние на п
Гомоцистеинсмия может приводить к развитию ишемической б(
ни сердца, некоторым формам злокачественных новообразован!
патологии беременности, врожденным порокам развития пл<
аутоиммунным реакциям. Основными генами, продукты коте
участвуют в метаболизме фолиевой кислоты, являются гены: м(
яентетрагидрофолатредуктазы {MTHFR), метионинсинтетазредуі
зы {MTRR), метионинредуктазы {MTR) и транскобаламинсинтет
{ТС). Всс «медленные» аллели данных ферментов способствуют
личению концентрации іомоцистеина в крови и тканях и снижеі
фолатов. Наиболее изучен ген MTHFR, для которого доказано, '
полиморфизм С677Т является «медленным» аллелем и носители:
аллеля подвержены риску невынашивания беременности в 14
большему, чем носители «дикого» аллеля. Частота носителей ме
ных аллелей в Европе составляет около 10%. Таким лицам показі
повышенные дозы фолиевой кислоты, особенно женщинам во Bf
беременности.Выше были описаны наиболее изученные процессы, связан]
с индивидуальным ответом организма на нутриенты. Однако
и обратное явление — влияние нутриентов на геном. Жиі
кислоты, холестерин, глюкоза, жирорастворимые витамины
ствуют на геном через факторы транскрипции, чувствительш
ним. Это гак называемые регуляторные рецепторные белки,
зывающие стеролы (SREBs) и углеводы (ChREBP), а также яд(|
ные рецепторы. Дефицит холина, метионина, фолатов, витамШ
Bg и В|2 оказывает влияние на ДНК-метилтрансферазы, котої
участвуют в процессе метилирования/деметилирования Д]
Такое влияние может изменять экспрессию ряда генов, прив(
к увеличению риска врожденных дефектов заращения нерві
трубки, сердечно-сосудистым заболеваниям и злокачественЩ
опухолям.Как видно из этого раздела, полиморфизмы генов, связанні
метаболизмом нутриентов, играют существенную роль в возі
новении, развитии, течении и исходах многих заболеваний. Зі
заранее профиль полиморфизмов пациента, врач может состаї
индивидуальную, персонализированную диету, которая пом(
если и не предотвратить эти заболевания, то, как минимум, от<
чить их манифестацию, облегчить симптомы.
Чті.1 7. Экологическая генетика329Рассмотрение влияния факторов окружающей среды на геном
|М'|<> бы неполной без упоминания эпигенетики. Как известно,
|о.'1 )мигенетикой понимают способ регуляции экспрессии генов
изменения их первичной структуры. Основными механизмами
реіуляции являются метилирование ДНК и модификация
fm ioHOB (ацетилирование, метилирование, фосфорилирование).
)Пп зти механизма тесно связаны и взаимодополняют друг друга,
|Н‘чультатс чего хроматин уплотняется и практически полностью
выключается экспрессия гена. Природная пластичность эпигено-
1(1 позволяет также репрограммировать пищевые, химические и
>И«ические факторы. Импринтированные гены и метастабильные
цмлллели являются двумя классами генов, которые особенно чув-
|'{ ии гсльны к средовым факторам, потому что их регуляция особен-
|41 ICCHO связана с эпигенетическими механизмами. Разъяснение
Иаимодействия среды с эпигеномом способствовало бы развитию
ІОІЮЙ эпигенетически обоснованной стратегии диагностики, про-
Ііилііктики и лечения болезней человека. Потенциально эпигене¬
тической модификации подвергаются CpG-островки транспозонов,
іромоторньїх регионов генов «домашнего хозяйства» и регулятор-
|Ыс элементы импринтированных генов. Во время эмбрионального
||>ития данный механизм регуляции позволяет экспрессировать-
определенным «нужным» генам в конкретные периоды раЗБИ-
И1, определяя тем самым правильное развитие зародыша. Не так
Иию в литературе стали появляться данные о том, что факторы
фужающей среды, такие, как питание, химические и физические
ікторьі, могут влиять на метилирование CpG-островков, изменяя
|м самым экспрессию, в том числе и во время внутриутробного
лштия. Так, богатое фолиевой кислотой или бетаином питание
Інтсри во время беременности приводит к увеличению метилиро-
ІИИЯ ДНК, к такому же эффекту приводит и чрезмерное употре-
ісчше генистеина (компонент сои), и, наоборот, такие компонен-
как бисфепол Л, подавляют метилирование. Данное явление
ожет никак не отражаться на внутриутробном или постнатальном
}риодах жизни. Однако, если метилирование ДНК происходит в
(нчимых или импринтированных генах, это потенциально может
Іриводить к нарушению развития плода и к развитию социально-
|Н11‘1имых многофакторных болезней уже во взрослом состоянии
Всрдечно-сосудистых заболеваний, сахарного диабета 2-го типа,
:ирения и др.).
330Клиническая генет*Физические факторыНесмотря на то что хорошо известна индивидуальная чувсті
тельность к теплу, холоду и солнечному свету, роль наследств<
ных факторов в этой чувствительности стала изучаться ли1
недавно.Четкие расовые различия обнаружены для реакции на хс
довой фактор. Например, представители негроидной расы б<
чувствительны к холоду, чем представители европеоидной ра(
Это объясняется разным уровнем теплопродукции и расширен^
сосудов.Твердо установлены индивидуальные и расовые различия pel
ции на ультрафиолетовое облучение. Крайний пример генетичесі
чувствительности — редкая аутосомно-рецессивная болезнь пигмеї
ная ксеродерма: под воздействием солнечного света возникают ожої
затем язвенные поражения кожи и, наконец, злокачественные не
образования. Молекулярно-генетический механизм этого явлсі
хорошо изучен. При пигментной ксеродерме поражается репариру(
щая система, восстанавливающая нормальное строение ДНК п(
повреждения ультрафиолетовыми лучами. Речь идет о мутация!
нескольких локусах, обусловливающих процессы репарации Д1
(экзо- и эндонуклеазы, полимеразы, лигазы). Эти гены клониров£
и хорошо изучены. Возможна преклиническая и пренатальная ді
гностика указанной болезни.Предполагается, что наследственно обусловленное различі
репарирующих системах может иметь определенное значение в ч]
ствительности к ионизирующим излучениям, но достаточных ДО|
зательств этому еще нет.Чувствительность к биологическим агентамГенетические основы детерминации иммунитета не вызыва
сомнений. Неодинаковая выраженность иммунитета у разных инл
видов ~ общебиологическая закономерность. Хорошо известна
личная чувствительность людей к вакцинам при введении одни!
тех же доз. У некоторых людей появляются клинические призн!
инфекции, у других реакция на иммунизацию совершенно от
ствует. Это является следствием генетического полиморфизма р<
ций на действие внешних биологических факторов.Первичные иммунодефицитные состояния — наследствен]
дефекты клеточного и гуморального иммунитета. Они предраспс
Ifmuia 7. Экологическая генетика331jtniur К бактериальным и грибковым заражениям, причем для ра'-зных
форм характерны соответствующие типы инфекций {вирусные, бак-11 і-риилькме, грибковые).Классическим примером наследственной устойчивости к биоло-
Иичсским агентам служат гемоглобинопатии (серповидно-клеточная
ІІІИ. МИИ, талассемия) и энзимопатии (недостаточность Г6ФДГ), при
UuiDpbix малярийный плазмодий не может размножаться в эри-
Ироцитах мутантных гомозигот и гетерозигот. В связи с тем, что
(лица с гемоглобинопатиями и недостаточностью Г6ФДГ устойчивы к
ІМії їярийному плазмодию, патологические мутации в соответствую-
Ьиих локусах широко распространились в местносгях с высокой
[іжюлеваемостью малярией (Африке, Греции, Италии, Филиппинах,
!А и'рбайджане, Узбекистане).ИЗМЕНЕНИЕ ГЕНОФОНДА ПОПУЛЯЦИЙ
КАК РЕЗУЛЬТАТ НАРУШЕНИЯ ГЕНЕТИЧЕСКОГО
РАВНОВЕСИЯНа основании приведенных выше примеров можно сделать одно-
Щамный на первый взгляд вывод: необходимо углубленное изучение
11>л и морфизмов генов, отвечающих за метаболизм ксенобиотиков,
Ггобы в дальнейшем индивидуально подходить к назначению лскар-
^гиснной терапии, составлению диеты и подбору профессии. Однако
(С следуег забывать, что наличие того или иною полиморфизма не
Определяет на 100% развитие какого-то заболевания или какой-либо
Щюлогической реакции на экзогенное вещество. Речь идет лишь
предрасположенности, о статистически доказанном повышенном
ЇИСКС, но не о 100% предсказании. Ввиду этого сразу встает вопрос
целесообразности ДНК-диагностики огромного количества гене¬
тических полиморфизмов. Помимо экономической составляющей,
(иньтй вопрос затрагивает и этическую сторону: как осуществлять
>ар материала, его анализ, хранение и использование. В любом
лучае сейчас научный мир разделен на 2 лагеря: выступающие за
інслрение диагностики полиморфизмов и противники этого. И у
иждой стороны есть веские аргументы в свою пользу.Ускоренный научно-технический прогресс без учета экологиче-
ІИХ проблем может приблизить человечество к нарушению био¬
логической гармонии со средой. При чрезвычайном загрязнении
332Клиническая геніРис. 7.2. Сохранение наслсдстпенной изменчивости гг популяциях по(
ством равновесия между «поступающими» мутациями («втекающая во,
оі бором («вытекающая вода»)окружающей среды может нарушиться равновесие і’енетических
цсссов в популяциях (рис. 7.2).Среда обитания человека изменилась и продолжает мені
ся. Человек постоянно сталкивается с ноізьіми факторами среді
также испытывает большие социальные и экологические нагруд
Увеличилась мутагенная нагрузка, расширился круг потенциал!
брачных партеров, увеличилась миграция населения, планирова|
семьи сократило естественное воспроизводство. Все это может MCF
генетическую структуру популяций. Однако популяиионнь[Є генетї
скис процессы обладают большой инерцией, поэтому не следует б1
дать, что мутационный процесс и экогспетическис реакции в течЄ|
одного-двух поколений вызовут опасные изменения наследственж
человека или резкое увеличение частоты наследственных болезі
Утверждения об ухудшении генофонда человечества надо относ
скорее к политическому пиару, чем к научно обоснованным выво;ЗАКЛЮЧЕНИЕДанные о потенциальном воздействии с родовых загрязнений і
наследственность человека и их способность повреждать насл(
ственные структуры, репродуктивные функции, внутриутробі
развитие справедливо вызывают озабоченность мировой общесті
іиіі t Экологическая генетика333Ці ні Необходимы глубокие разработки в области экологической
Ilf I ПКИ человека и мероприятия по охране среды его обитаиия.
Ищк менные научные методологии существенно улучшили опенку
‘miuix влияиий факторов окружающей среды на наследственность
|ио1к-ка. Оценка риска должна посіоянно подвергаться коррекции,
н ко м.ку становятся доступными новые генетические технологии, а
происходит развитие клеточной биологии и информационныхРК111))|ОГИЙ.Нередко высказываются предложения приблизить среду обитания
мюиска к естественной экологии. Это невозможно, так как практи-
ІГКИ вся среда обитания современного человека в широком смысле
imva выстроена самим человеком. Необходимо стремиться не к воз-
(>11IV и прошлое, а к оценке тех изменений, в том числе и в наслед¬
ниц пости, которые возникают при создании новых технологий, и
(р;|мее предупреждать их.Илжность проблем, изучаемых экологической генетикой человека,
(I |ц>еменем будет возрастать и относительно, и абсолютно. Во-первых,
Ипоситсльная значимость экогенети ческой патологии будет у велич и-
ІПІ1.СЯ по мере улучшения медицинской помоши и успешной борьбы
(кісирос'і'раненньїми болезнями. Обычные медицинские меры про¬
филактики не снизят частоту экогенетических болезней. Во-вторых,
|(1 кременем можно ожидать увеличения экогенети ческой патологииI ||(>солютном выражении, поскольку вследствие научно-технического
1|)о1 ресса будут появляться все новые факторы, повысится специфич-
10Lть новых производственных условий и т.д.1^ыявление экогенети ческой патологии и идентификация ее форм
[представляют трудную задачу, поскольку надо найти и суть биохи-
шическою полиморфизма в популяциях человека, и конкретные фак-
[І'орьі среды, обусловливающие патологическое действие «молчащего»
гопа, В этом процессе познания трудно переоценить роль врача,
Іааметившего «непонятный случай». Это особенно касается вопро¬
сов профессиональной патологии и лекарственной терапии: именно
чдссь можно чаще обнаружить проявление еще не описанных форм
■жогенетической патологии.В профилактической медицине концепции экологической гене¬
тики человека крайне важны, поскольку они направляют усилия на
создание оптимальной среды (пища, лекарства, работа) для каждого
индивида с целью предупреждения патологического проявления
жогснетического биохимического полиморфизма.
334Клиническая гвШКЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯГенетика чувствительности к алкоголю
Изменение генофонда популяций
Индуїщрованньїй мутационный процесс
Локусы биогрансформации ксенобиотиков
Методы выявления экогенетическиX реакций
«Молчащие» гены
Непереносимость молочного сахара
Нутр и генетика
Н утр и геном и каОбъем наследственного полиморфизма
Примеры реакций на загрязнение атмосферы
ТоксикогенстикаФазы биотрансформаиии ксенобиотиков
ЦелиакииЭволюция генотипа
Зкоіенетика человекаРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАБонков H.1L, Рослова Т.Л., Якушина И. И. Меди ко-генетическое
сультирование по поводу мутагенных и гсратогенных воздействий^
Медицинская генетика. — 2009. — № 1. — С. 3-8.Бонков И. П., Чеботарев Л.Н. Наследственность человека и мутагеї
внеіпней среды. — м.: Медицина, 1989. — 272 с.Генетический паспорт — основа индивидуальной предиктиві
медицины / под ред. B.C. Баранова. — СПб.: Изд-во Н-Л, 2009,
527 с.Спицын В.Л. Экологическая генетика человека: эволюционн|
адаптация. Профессиональная деятельность. Спортивная гено»
ка. Популяционная фармакогене і и ка. Мультифакториальные боле
ни. — М.: Наука, 2008. — 503 с.Dolinoy D.C., Jirtle R. Environmental Epigenomics in Human Health ai
Disease. — Environmental and Molecular Mutagenesis. — 2008. — V. 49.
P. 4-8.
Глава 8ФАРМАКОГЕНЕТИКАОБЩИЕ ВОПРОСЫФармакогенетика изучает индивидуальные различия в ответах на
ркарства, обусловленные аллельными вариациями в генах, опре-
ІСЛЯЮЩИХ метаболизм лекарства, эффективность и токсичность.И) направление как раздел экологической медицинской генетики и
і;інкической фармакологии зародилось в результате практической
Ишребности разобраться в осложнениях лекарственного лечения,
(К іиническая фармакология накапливала наблюдения патологиче-
рк и X реакций на лекарства, а медицинская генетика расшифровывала
[Механизмы их возникновения.Врач сталкивается с повышенной чувствительностью индивида к
[лгкарству, похожей на передозировку, хотя больному назначена доза,
{соответствующая его возрасту и полу; с частичной или полной толе-
рннтностью больного к лекарству, даже несмотря на увеличение дозы;
t парадоксальными реакциями на лекарство, включающими совсем
другие осложнения, чем те, которые могли бы быть обусловлены
механизмами действия лекарства.Основные положения фармакогенетики были сформулированы в
ІЧ30—1970 гг. Термин «фармакогенетика» был введен в 1958 г. немец¬
ким ученым Ф. Фогелем. Развитие фармакогенетики основывалось
Иа регистрации нежелательных лекарственных реакций с их анали-
:к)м сначала клинико-генеалогическим и близнецовым методами, а
I» последующем — молекулярно-генетическим. При этом изучался
ПС только конечный патологический фенотип, но и биохимические
с гупени метаболизма лекарства, что давало возможность понять сущ¬
ность нежелательных лекарственных реакций и их ключевые точки.Генетическое разнообразие человека — основа индивидуальных
различий биотрансформации ксенобиотиков, к которым и относятся
лекарства (см. гл. 7). Следовательно, теоретической базой фарма¬
когенетики является функциональная геномика человека, а именно
сведения о полиморфизме генов, вовлеченных в биотрансформацию
лекарств и в генетический контроль их взаимодействия. Таким обра-
336Клиническая remзом, основная задача фармакогенетики ~ изучение аллельных
антов генов, определяющих индивидуальные особенности фарМ!
кинетических и фармакодинамических характеристик организм!Расшифровка генома человека и прогресс фармакологии выдв!
ли фармакогенетику на одно из первых мест в персонализироваї
медицине (индивидуализированное лечение).Индивидуальные вариации в ответе на лекарства осуществляї
двумя путями. Во-первых, за счет фармакокинетических процес
(всасывания, транспортировки, метаболизма и выведения лека|!
или метаболитов). Во-вторых, за счет фармакодинамики лекар(
Вследствие аллельных вариаций наблюдаются различия в миі
(рецепторах, энзимах) или метаболических путях. Таким образ
говоря обобщенно, фармакогенетика изучает любые генетич!
детерминированные вариации в ответе на лекарства в отношбі
эффективности и токсичности.Для понимания фармакогенетическнх закономерностей необхоіі
усвоить принципы биотрансформации (детоксикации) ксенобиоті
изложенные в главе 7.Все ступени биотрансформации лекарственных средств осущ(
Бляются соответствующими ферментами и белками. Список осі
ных из них представлен в табл. 8.1.Таблица 8.1. Ферменты и белки биотрансформации лекарственных средсі1 фаза11 фазаТранспортеры ^Цитохромы Р450UGTГликопротеин Р JДПДГNATТранспортные сисТІБутирилхолинэстеразатпмтмы олигопептидов,4(псевдохол и НЭС1 ераза)SULTнуклеотидов, орга-PONГлутатиоптрансфсразыничсских анионов, ^ADH hALDHЭпоксидгидролазыорганических катиаи другие ферменты,нов, мпожественноЯотвечающие за микро-лекарстпенной устоїсомальнос окислениечивоети "Примечание. ДПДГ — дигидропиримилиндегидрогеназа. SULT — суді
трансферази.Генетический полиморфизм определяет три главных фенот!
метабол изаторов (лиц, принимающих лекарства): экстенсивные,
ленные и быстрые.
Ійо ti. Фармакогенетика337Экстенсивные метаболнзаторы -- индивиды с нормальной скоростью
ИШюлизма рассматриваемых лекарственных средств. К этой группе
ІИНіідлежит большинство населения. Они являются чаще всего гомо-II о lit ми по «дикому» аллелю соответствующего фермента.
Медленные метаболизаторы (иногда нулевые) характеризуютсяІІ1ЖСІІН0Й скоростью метаболизма рассматриваемого лекарствен-
средства. С генетической точки зрения они являются гомози-
ІІІМИ (при аутосомно-репессивном типе наследования) или геге-
ии готами (при аутосомно-доминантном типе наследования) по
|| им гном у («медленному») аллелю соответствующего фермента,
І1ІКИХ лиц синтез фермента отсутствует или синтезируется неак-
Ишый («дефектный») фермеіїт, в результате чего лекарственное
Nici Bo накапливается в высоких концентрациях, что и приводит к
пилению нежелательных побочных реакций. Отсюда ясно, что для
1Л..1С11НЫХ метаболизаторов доза лекарства должна быть меньшей
(и назначают другое лекарство.Ьмстрые (или сверхактивные) метаболизаторы характеризуются
>111.1 теныой скоростью метаболизма определенных лекарств. В основ-
ш )1'о гомозиготы (при аутосомно-рецессивном типе наследования)III гетерозиготы (при аутосомно-доминантном типе наследова-
|Ин) по «быстрому» аллелю соответствующего фермента. Достаточно
|1чо встречаются индивиды с копиями функциональных аллелей,гакже приводит к повышенно.му метаболизму лекарства. Быстрый
Іі'ііболизм лекарства не позволяет при стандартных дозах достичь
С) герапевтической концентрации в крови, поэтому доза лекарства
1н быстрых метаболизаторов должна быть выше, чем для нормаль-
IX метаболизаторов.ФАРМАКОГЕНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
I ФАЗЫ БИОТРАНСФОРМАЦИИІІаттбояьшее значение в вариациях фармакокинетических реакций
ІІИОСТ цитохром Р450, обеспечивающий Ї фазу метаболизма лекарств,
[итохром Р450 — большое семейство из 56 дифференциально функ-
|иоиальных ферментов, каждый из которых кодируется отдельным
}ном CYP. С фармакогенетической точки зрения особенно важны
jcci b генов - CYPIA1, CYP1A2, CYP2C9, CYP2C19, CYP2D6n CYP3A4.
))1и ответственны за I фазу биотрансформации 90% широко распро¬
338Клиническая гв>страненных лекарств. Например, СFPJL44 вовлечен в метаболизм
40% всех лекарств, используемых в клинической медицине, а СУІ
метаболизирует более 70 различных лекарств. Вполне понятно,
вариации в метаболизме обусловлены аллелями с различной
циональной значимостью. Есть аллели, повышающие метабс
другие понижают его, а третьи вообще не участвуют в биотран(
мации. В таблице 8.2 приведены избранные примеры полимо{
генов цитохрома Р450, участвующих в метаболизме лекарств.Таблица 8.2. Примеры генов цитохрома
лекарствР450, вовлеченных в метаб<ГенНаличие аллелейЛекарства і
(избранные apHMt||повышаю¬щихметаболизмпонижающихметаболизмнулеваяактив¬ностьCYPIA2++-Кофеин (?), пропрІНОЛОЛ ICYP2C9+++Блокаторы рецепте
ра ангиотензина
нестероидные пр<Н
тивовоспалительНІ
срсдства, метронйА
дазол (?), оральны|
гипогликемическш
варфарин jCYP2C19-++АнтиэпилептичесИ
антидепрессанты,!
анксиолитики ]CYP2D6+++Антиаритмическм
ант и д еп рессанты J
антипсихотическм
Р-адрснергическяі
блокаторы, наркот|
ческие анальгети|С|CYP3A4+++Парацетамол, проТ|
вогрибковые, кокай
кодеин, циклоспо- ]
рин А, диазепам, эп
тромицин, статним
паклитаксел, варфя
Ійи в. Фармакогенетика339Ііолсе подробные сведения о семействе цитохрома Р450 и биотранс-
ї|»м;іции лекарств представлены в книге В.Г. Кукеса, Н.П. Бочкова
іпиііическая фармакогенетика», гл. 2.I і'ііетические вариаиии в I фазе биотрансформации отмечены по
ІРиуїсідим ферментам — ДПДГ, PON, псевдохолинэстеразе (бути-
♦лчолинэстеразе), ADH, ALDH.ДМДГ отвечает за восстановление урацила и тимидина, а также
іііюолизирует фторурацил, применяемый в составе комбинирован-
||)И химиотерапии злокачественных новообразований многих орга-
}и Низкая активность ДПДГ — причина осложнений лечения фтор-
^ишыом. Эта особенность наследуется по аутосомно-рсцессивному
|иу. Молекулярно-генетическими исследованиями выявлены мута-III п гене, колирующем синтез ДПДГ, наличие которых приводит
Ч’пижснной активности фермента и, следовательно, к повышенной
^ін ііштельности к фторурацилу. Распространенность мутантных
ІМОІИГОТ определена только в Японии. Она составляет 1:10 ООО
ІІЧМЄНИЯ. Гетерозиготы, по-видимому, также имеют сниженный
їинснь ферментов. Современные представления о генетическом
1Л1І морфизме ДПДГ позволяют рекомендовать внедрение фено- и
Мк’і ипирования этого фермента в генетическую практику.PON — фермент из группы арилэстераз — метабол и зирует фосфор-
їіаііические антихолинэстеразные соединения (параоксон, мети-
<к’факол, дихлорвос, зарин, табун и др.), эфиры уксусной кислоты
їсиилацетат, тиофенилацетат, венилацетат и др.), органофосфорные
глинения (EPN-OKCOH, этилнитрофенилэтилфосфонат), карбаматы
г»ии, N-диметил карбамилфлуорид). Генетический полиморфизм
[)N не вызывает сомнений. Из известных 3 изоформ PON наибо-
К важным с фармакогенетической точки зрения является PON1,
Іушция в этом гене (Glnl92Arg) ведет к повышенной чувствитель-
к’ги к фосфорорганическим соединениям. Распространенность
гой мутации достаточно высокая: среди испанского населения —
|Ь%. североевропейского — 9%, японского — 41,4%. Именно высокой
РИстотой мутации у японского населения объясняется большое число
5р'| в после применения зарина при террористическом акте в токий-
*ом метро в 1995 г.Псевдохолинэстераза (бутирилхолинэстераза) катализирует реак-
|ИК) гидролиза ацетилхолина. В фармакогенетике этот фермент
ІИНІЮ уже известен в связи с его участием в гидролизе деполяри-
/ЮІЦЄГО миорелаксанта суксаметония, широко применяющегося в
340Клиническая геніанесте:зиологии, В гене бутирилхолинэстеразы (аномальной
дохолинэстеразы) обнаружено несколько мутаций, которые
к синтезу фермента со сниженной активностью, а это прив(
к продолжительной остановке лыхания (апноэ) при примет
суксаметония (вместо 2—3 мин — 2 ч и более). Наследуется эта
мальная реакция по аутосомно-рецессивному типу. Повыше»
чувствительность к суксаметонию наблюдается у гомозигот. РаЗ
мутации этого гена ведут к апноэ разной длительности, и гомозиі
встречаются с разной частотой (от 1 : 3000 до 1 : 150 ООО). Часі
гомозигот по всем мутантным аллелям, определяющим снижен!
активность бутирилхолинэстеразы, согласно литературным дані
следующая: у европейцев — 1 : 2500, у чехов и словаков — 1 : 4(
жителей Ирана и Ирака — 1 : 400. Распространенность гетерозі
следующая: у европейцев 2—4 : 100, у чехов и словаков — 7 : И
жителей Ирана и Ирака — 10 ; 100.Профилактика осложнений, вызываемых мутантными фор»
псевдохолинэстеразы, может осуществляться путем фенотипі
ван ИЯ с помощью так называемого дибукаи нового теста или п]
генотипирования, поскольку структура гена и мутаций хорошо
чена. Генетическая и биохимическая расшифровка данного фарм!
генетического варианта позволяет точно выявить лиц с повыше!
чувствительностью к суксаметонию и обеспечить безопасность!
применения.ADH экспрессируется в печени и является ключевым фермеї
в окислении этанола и других спиртов до альдегидов. Ген этого
мента хорошо изучен, особенно его полиморфный вариант GM
Следовательно, возможна и его ПЦР-диагностика. Аллель А обуо^
ливает повытиепную активность фермента, что ведет к накоплеї
альдегидов (весь алкоголь «перерабатывается»), которые облі
выраженным токсическим эффектом. Такие индивиды имеют
повышенную чувствительность к этиловому спирту и поэтому
подвержены алкоголизму. Даже небольшие дозы алкоголя вєі
сильнейшему отравлению.ALDH экспрессируется в печени в двух формах: ALDH-1 (ш
зольная) и ALDH-2 (митохондриальная). С генетической точки
ния лучше изучен ген ALDH-2, мутации в котором ведут к алкогІ
ной интоксикации. Фермент ALDH-2 вовлечен в патогенез различ!
злокачественных новообразований, связанных со злоупотреблеї
алкоголем. Распространенность мутантных форм ALDH-2o4tnh в1
|ца Н. Фармакогенетика341|и среди населения монголоидной расы (до 50%). Молекулярно-
і'ііічсская диагностика гетеро- и гомозигот по патологическим
' mil ИЯМ возможна.ФАРМАКОГЕНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
II ФАЗЫ БИОТРАНСФОРМАЦИИІІО 11 фазе биотрансформации лекарственных средств осуществля-
К'и конъюгация их или их метаболитов с эндогенными веществами
іИірл юванием гидрофильных конъюгатов.Глюкуронирование является наиболее важной реакцией
)|);иы метаболизма лекарств. К лекарственному средству при-
інминяется УДФ за счет катализа с помощью ферментов [УДФ-
цккуронилтрансферазы (UGT)], включающих два семейства и более
О (поферментов. Они катализируют большое число лекарств (мор¬
щим, хлорамфеникол, парацетамол и др.), их метаболитов, гормонов,
рпицидов, канцерогенов. Физиологической функцией UGT явля-
|'и*я глюкуронирование эндогенных соединений (например, били-
^()ина). Глюкуронированию подвергаются лекарственные средства
|1 следующих групп:• ([)енолы (нропофол, парацетамол);• спирты (хлорамфеникол, кодеин, оксазепам);• алифатические амины (ламотриджин, амигриптилин);• карбоновые кислоты (фенилбутазон и др.);• карбоксильные кислоты (напроксен, кетопрофен).
Наследственное нарушение глюкуронирования билирубина наблю-1нс I ся при синдромах Жильбера и Криглера—Найяра. Мутации в гене
/(/77 приводят к синтезу UGT с активностью на 25—30% меньшей
К) сравнению с нормой, поэтому у больных с синдромом Жильбера
иіОлюдается снижение клиренса тол бутам и да, парацетамола, рифам-
іицина. Другие генетические полиморфизмы (мутации) генов, коди¬
рующих разные изоформы UGT, влияют на фармакокинетику и фар-
іакодинамику лоразепама, морфина, карведилола и других лекарств.
|Иі;следованис полиморфизма гена У разрещено в США для кор-;киии терапии иринотеканом (высокоэффективным цитостатиком)
целью профилактики развития гипербилирубинемии.
Ацетилирование. Эта реакция осуществляется двумя NAT (NATI и
INAT2). NATI не обладает генетическим іюлиморфизмом, а для NAT2,
342Клиническая гв>напротив, важная роль в фармакогенстике хорошо доказана, Геи
лизован в хромосоме 8р23, известно более 20 мутантных алі
В зависимости от активности фермента NAT2 все люди разделяї
«быстрых», «промежуточных» и «медленных» ацетиляїхзров. Впервь
макогенстические закономерности М4Г2были установлены в 1960-в |
на примере лечения изониазидом больных туберкулезом. У «медл<
ацетиляторов обнаруживается повышенная чувствительность не
к изониазиду, но и к сульфаниламидам, ариламинам, гидразин!
некоторым антиаритмическим и другим препаратам. Механизм
ческого действия препаратов связан с медленным выведением леї
из-за сниженной скорости ацетилирования, а следовательно, и вы|
ния препарата. Происходит накопление препарата (рис. 8.1).Распространенность «медленных» аиетиляторов составляет 10-1|
монголоидного населения и почти 50% у населения европеоидной
Помимо ассоциации полиморфизма гена NAT2 с неблагоприят
побочными эффектами лекарств, обнаружена также связь с разлі
ми многофакторными заболеваниями. Частота рака мочевого пузі2—3 раза выше у «медленных» ацетиляторов, чем у «быстрых», а
последних почти в 2 раза чаще встречается колоректальный рак.S-метилирование. Реакцию S-метилирования катализирует
мент ТПМТ. Это основной путь метаболизма эффективныхЧасыРис. 8,1. Распределение индивидов по скорости ацетилирования изониа^
по оси абсцисс — время после введения, часы; по оси ординат — число,
М Фармакогенетика343(меркаптопурина, азатиоприна и тиогуанина). Ген ТРМТ
Ото и зучен (локализован в хромосоме 6q22.3). Хотя низкая эффек-
Imhl i I. ТПМТ наследуется по аутосомно-репессивному типу, повы-
ІІІІІМ чувствительность к тиопуринам отмечается не только у
(пшют, но и у гетерозигот. Известно уже 8 различных аллелей,
|И(пчощих фермент с низкой активностью, что ведет к нарушению
РйСшлизма меркаптопурина. При наличии таких аллелей требуется
ІЖІЧІИЄ стандартной дозы цитостатика в 2-4 раза.
Ькмространенность гомозигот по всем аллельным вариантам
ГРМТ среди европейского и афроамериканского населения
Ц|и»ляет 4-5%. Безопасные дозы меркаптопурина для пациентов-10 III гот по мутантным аллелям в 10—15 раз ниже среднетерапев-
<CLKHX, для гетерозигот — в 2-4 раза. Для обеспечения безопас-
'111 химиотерапии меркаптопурином (острый лимфобластный
ІКО лимфомы) необходимо проводить фенотипирование (актив-
Л«> ТРМТ в эритроцитах) или генотипирование на мутантные
)И1И(ты гена ТРМТ. В клиниках Европы и США одна из этих про-
Лур типирования является обязательной перед началом лечения.Сульфатирование. В организме человека сульфатированию под-
)П1К)тся фенолы (экзогенные), гормоны щитовидной железы, кате-
шмины, некоторые стероидные гормоны. Идентифицировано
и юферментов SULT, которые кодируются Ш генами. С фармако-
лической точки зрения наибольший интерес представляют две
)мы изофермента. SULT1A1 метаболизируетпарацетамол, морфин,
^{)лукты распада лидокаина, эстрадиол и другие лекарсгвенные
:параты фенольной структуры. Субстратами SULT1A3 являются
Иишин, серотонин, норэпинефрин и некоторые другие соединения.11 и обнаружен широкий генетический полиморфизм SULT, данных
иссоциации полиморфизмов генов этих ферментов с дозами соот-гствующих лекарственных препаратов пока не выявлено.Водная конъюгация. Эту реакцию, важнейшую в детоксикации боль-
(1Г0 количества ксенобиотиков, ката;і изирует фермент эпоксидгидрок-
(лпза (ЕРАХ). Известны две его изоформы и их гены. Большая часть
;/1ной конъюгации токсических метаболитов лекарственных препаратов
4И11ример, фенитоина) осуществляется с помощью ЕРАХ1. Обнаружен
іиетический полиморфизм ЕРАХ}. Точечная мутация является причи-
*)й снижения активности фермента (меньше 30% от нормы), что ведет
повышенному риску врожденных пороков развития, если женщина во
гмя беременности принимает фенитоин. Медленный аллель mEPHXl
344Клиническая генеттвстречается примерно у 6% европейского населения. У носителей MJ
1ЩЙ нарушен процесс окисления ксенобиотиков. Выявлена ассоциаі
этого аллеля с заболеваниями органов дыхания, особеїіно у курилыци!
(рак, эмфизема, обструктивные пневмонии), а также с нарушениям!
репродуктивной системе (спонтанные аборты, рак яичников).Конъюгация с глутатионом. Среди лекарственных препаратов ко1
югации с глутатионом подвергаются этакриновая кислота и renal
токсический метаболит парацетамола — N-ацетилбензохинонимі
превращающиеся в нетоксические соединения. Конъюгацию с
татионом катализируют ферменты глутатион-8-5Н-трансфер1
(GST). Выделено пять изоферментов GST, ген принимает В1нейшее участие в инактивации канцерогенов. Распространенж
носителей нулевого аллеля, у которых отсутствует экспрес<
GSTM1, составляет 40-45% у европейского населения и
у негроидного.Ассоциации между аллельными вариантами генов и изменениі
фармакологического ответа представлены в табл. 8.3.Таблица 8.3. Ассоциации между аллельными вариантами генов глутатион^
SH-трансфсраз и изменениями фармакологического ответа
Кукесу В.Г. и Бочкову Н.П., 2007)ГенА.1лельныевариантыИзменениеактивностиферментаЛекарственныесредстваИзменение 1
фармаколога
ческого отв<|GSTT1НулевыеаллелиСнижение актив¬
ности глутатион-
трансферазы
GSTT1Пиоглитазон?’Гепато- 1
токсичностУGSTM1НулевыеаллелиСнижение актив¬
ности глутатион-
трансферазы
GSTM1Пиоглитазон<"Гепато- j
токсичності^ІneFTHUHJi^aMHHПовышение \
эффектив- і
иости терапЦ
ревматоидна
го артрита JGSTP1GSTP*A/*ВСнижение актив¬
ности глутатион-
трансферазы
GSTP1ДоцетакселМиело- ^ТОКСИЧНОСЦі
іина 8. Фармакогенетика345Глутатионопосредованная детоксикация имеет важнейшее значе-
u сохранении резистентности клеток к перекисному окислению
Ктидов, алкилированию белков, освобождению от свободных ради-
пю», а также она предотвращает поломки ДНК. Таким образом,
[||\'|атион-5-8Н-трансферазы прежде всего представляют интерес с
ікигоксикологической точки зрения (см. гл. 7). Их значение в фар-
411 к< і генетике требует дальнейшего изучения.ФАРМАКОГЕНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
ТРАНСПОРТА ЛЕКАРСТВЕННЫХ СРЕДСТВ
(III ФАЗА БИОТРАНСФОРМАЦИИ)Ферменты, обеспечивающие фармакокинетические функции
|і:ісьівания, распределения и выведения из организма лекарствен-
<1.1 X средств, называют «транспортерами лекарств». К ним относят-
^11 гликопротеин Р, транспортеры органических анионов и катио¬
нов и др. Характеристика основных транспортеров лекарственных
средств (название, локализация, функция, лекарственные средства
их метаболиты-субстраты, ингибиторы, индукторы) представлена
кииге В.Г. Кукеса и Н.П. Бочкова (с. 218-226).Наибольший И1{терес с фармакогенетической точки зрения пред-
Маиляетполиморфизм гена MDRI, кодирующий гликопротеин Р (локус
?q2l.l). Этот фермент контролирует выброс различных ксенобиоти-
fcou из клетки, препятствует всасыванию лекарственных средств из
Иппечника. Субстратами гликопротеина Р являются сердечные гли-
*0чиды, блокаторы медленных кальциевых каналов, статины, макро-
Шды, цитостатики, противовирусные препараты. Полный перечень
субстратов, индукторов и ингибиторов гликопротеина Р представлен
книге В.Г. Кукеса и Н.П. Бочкова (с. 214-217).Четыре ОНП гена MDRI изучены детально (табл. 8.4).ГмОлица 8.4. Поли.морфные маркеры гена, кодирующего гликопротеин Р1 Іолиморфньїй
маркерЭкзонИзменения
в нуклеотидной
последовательности ДНКРезультатполиморфизма[(J2677T212677GTAla893SerГ(г2677А212677GAAla893Thr
346Клиническая г0>Окончание таблиіПолиморфныймаркерЭкзонИзменения
в нуклеотидной
последовательности ДНКРезультя1|лолнморфіС1236Т121236СТСнижен!экспресс!С3435Т263435СТСнижен»экспресс»^Наиболее значимой мутацией гена MDR1 является С3435Т. 3|
цитозина на тимин в 26-м экзоне ведет к серьезному наруї
функции гликопротеина Р, что может быть причиной тяжелой И1
сикации в случае применения многих лекарств. Частота алл«
генотипов по полиморфному аллелю С3435Т значительно варЫ
в разных этнических группах, в том числе на территории РФ. .Хотя многое из фармакогенетики гликопротеина Р еще ті
клинической проверки, но на основании уже проведенных ис(
ваний обнаружены ассоциации полиморфных маркеров с изм<
ем фармакологического ответа на многие лекарства. Эти резул!
представлены в табл. 8.5 (по В.Г. Кукесу и Н.П. Бочкову).Таблица 8.5. Ассоциации между носительством генотипов по полимої
вариантам гена MDR1, кодирующим гликопротеин Р, и
нием фармакологического отпетаПолиморф¬ныемаркерыИзменениеактивноститранспортераЛекарственныесредстваИзменение
фармакологического отіС3435ТG2677TG2677AС1236ТСнижение
активности
гликопро-
теин Р/
экспрессии
гена MDR1ДигоксинГликозидная интоксикаїЛоперамидМиоз (сужение зрачка) \ЦиклоспоринНефротоксичность, ней)!
токсичность іТакролимусНейротоксичностьФексофенадинСонливостьБлокаторымедленныхкальциевыхканаловг и перплази я десенДоцетакселМиелотоксичность
irt Н. Фармакогенетика347Окончание таблицы 8.5|«;ІИМОрф-ІІМС[|1)1(1КСрЫИзменениеактивноститранспортераЛекарственныесредстваИзменение
фармакологического ответаИнгибиторыпротонногонасосаУсиление антисекреторного
действияАнгиконвуль-сантыПовышение эффективности
лечения эпилепсииГалоперидолПовышение эффективности
лечения ШИ-ЧОфреНИИАнтагонисты
5-ТНЗ рецеп¬
торов (тро¬
пи се трон,
ондансетрон,
гранизетрон)Усиление противорвотного
действияАторвастатинУсиление гиполипидемиче-
ского действияНелфинавирУсиление антиретровирусно¬
го действияll)Rl-h4IDRl-hlOФлувастатинБолее HHTeHCHBffoe снижение
концентрапии холестерина
ЛПНПМенее интенсивное снижение
концентрации триглицеридовОтсюда следуют приведенные ниже практические рекомендации
ірмакологов клиницистам при обнаружении у пациента полиморф-
JK) маркера (Сычев Д.А. и др., 2007);
следует снижать дозу лекарств-субстратов гликопротеина Р
с узкой терапевтической широтой (дигоксин, циклоспорин);* не следует применять лекарства-субстраты гликопротеина Р,
нежелательные лекарственные реакции которых связаны с их
проникновением через гистогематические барьеры (фексофена-
дин, лоперамид);* назіїачать препараты с низкой биодоступностью, так как они
могут оказаться у лип с мутациями наиболее эффективными (ста-
гины, ингибиторы ВИЧ-протеипазы);
348Клиническая гені• назначать препараты, мииіени которых расположены в Ц1
проникновение через гематоэнцефалический барьер затрудн^
так как у этой категории они могут оказаться наиболее эффект
ными (противосудорожные, галоперидол).Исходи из вышеизложенных сведений о генетическом П0ЛИ1
физме гликопротеина Р, необходимость их использования для И]
видуализации лечения не вызывает сомнений.К транспортерам лекарственных средств также относятся tj
мембранные белки — транспортеры органических анионов и катио!
К субстратам этих транспортеров относятся широко применяв^
лекарства (антибиотики, диуретики, противовирусные, против(
холевые средс'1 ва, статины). Ассоциации между носитсльством алл^
ных вариантов гена ОЛТР-С (органический анионтранспортируюі
полипептид С) и неблагоприятным фармакологическим ответом
с'гавлены в табл. 8.6 (по Кукесу В.Г. и Бочкову Н.П., 2007).Таблица 8.6. Ассоциации носительства генотипов гена ОАТР-С с изменвк
фармакологического ответаПолиморфизмыИзменениеактивноститранспортераЛекарственныесредстваИзменение
фармакологическа
ответа0АТР-С*1Ь,ОАТР-С*15,Т521С,G-1H27AСнижениеактивностиОАТР-СПравастатин,аторвастатин,симвастатинОслабление ГИПОЛІ
пидемического дсЙСТВИЯ jT1628GСнижениеактивностиОАТР-СПравастатин,аторвастатинПовышение риска
развития миопати!G-1U87A.. , .... ,СнижениеактивностиОАТР-СРепаглинид1ГипогликемияВ фармакокинетике (всасывание, распределение, вьіведеї
лекарственных средств принимают участие и другие транспорту
(олигопептидов, нуклеозидов, множественной лекарственной уст
чивости), генетический полиморфизм которых в настоящее Б
интенсивно изучается.Более подробную информацию о фармакогенетике препаї
для лечения сердечно-сосудистых заболеваний и антитромботі
ін.і в. Фармакогенетика349ііі\ препаратов можно посмотреть в статьях В.В. Ляхович с соавт.
^Кііпцьі фармакогенетики в клинике сердечно-сосудистых заболе-
imtji» и О-В. Сироткиной с соавт. «Фармакогенетика антитромбо-
't(4 ких препаратов» на компакт-диске.ФАРМАКОДИНАМИКА И ГЕНЕТИЧЕСКИЙ
ПОЛИМОРФИЗММутации в генах, кодирующих белки-мипюни для лекарственных
fpt-'icТВ (рецепторы, ферменты, ионные каналы), ведут к изменениям
їіірмакологического ответа. Эти генетические полиморфизмы актив¬
но изучаются и сведения о них уже применяются в клинической
Ірик іике. Перечислим некоторые из наиболее изученных полимер¬
ен ІМОВ (габл. 8.7).("мГитцавЛ. Примеры аномальных ответов у носителей мутаций в фармако-
линамичсских реакцияхМишеньПатологические проявления или изменение
ответа у носителей мутацийОтсутствис бронхолитического эффекта при
применении короткодействующих агонистов
р2-адрснорецепторой ЛПФИнгибиторы АПФ у больных гипертонией менее
эффективны у лиц с генотипом DD(V'-Брадикининовые1К‘Ие1ГГОрЫОсложнение п виде сухого кашля на фоне лече-
ния гипертонии ингибиторами АПФ И<)5гные каналыУдлинение интервала Q—TКіФДГГемолиз эритроцитов при применении многих
лекарств1’иаиодиновые реиел-
горы 1-го типаЗлокачествеїшая гипертермия при применении
местных анестетиков, средств для ингаляцион¬
ного наркозаИ|)нмечянне. АПФ — ангиотензинпреврашающий фермент.Таким образом, генетический полиморфизм играет существенную
роль в вариациях фармакодинамических процессов.
350Клиническая геміЗАКЛЮЧЕНИЕКак известно, судьба лекарств в организме определяется всасы!
ем, распределением (по органам, клеткам, органеллам), взаимодейст^
с клеточными элементами, метаболизмом и выведением. Все сту(
кинетики лекарства и динамики его действия осуществляются с
щью специфических и неспсцифических ферментов и белков. Учиті
широкий биохимический полиморфизм человеческих популяций, м<
предполагать, чго судьба каждого лекарства на каком-то фармаї
нетическом или фармакодинамическом этапе связана с полимо{
системой фермента, белка, рецептора и других клеточных мишеней,
и обусловливает весьма разнородные реакции индивидов на лекареС фармакологической точки зрения вариации ответов на лН
ства могут быть обусловлены изменением либо метаболизма лекаї
в организме, либо динамики их действия (нарушение клеточі
мишеней лекарств).По поводу аномалий метаболизма лекарств (первая группа) мс
сказать, что генетическая детерминация ферментов, обеспечиї
ших метаболизм или фармакокинетику лекарств, не вызывает cos
ний. Возникновение мутаций в таких генах приводит к отсутстІ
синтеза фермента или потере его ферментативной активности,
правило, эти мутации наследуются по аутосомно-рецессивному т|
поэтому дефект фермента проявляется только у гомозигот, СЛ€
тельно, не очень часто, хотя в некоторых популяциях частота мут|
ного аллеля и соответственно частота лиц с патологической реакі
на лекарства могут быть высокими.Вторая группа неадекватных реакций на лекарства — это фармаі
гические эффекты через взаимодействие с белками-мишенями, такі
как рецепторы, ферменчы, белки сигнальной трансдукции, коч1
ля клеточного цикла и других событий. Молекулярно-генетическІ
исследованиями показано, что многие гены, кодирующие такие л«
ственные мишени, полиморфны, Их мутантные формы приводят
ветственно к нарушению специфических взаимодействий лекарс!
мишени, а отсюда и к аномальной реакции на уровне организма.Во многих работах показано, что судьба большинства лека|
определяется функционированием нескольких взаимодействую!
генов, поэтому кривые распределения индивидов в зависимости
концентрации лекарств в крови при введении стандартной дозы с<
ветствуют кривым полигенного наследования (рис. 8.2).
іим в. Фармакогенетика351Рис. 8.2. Распределение индивидов
по концентрации лекарства в пла;і-
ме крови после введения стандарт¬
ной дозы при полиген ной детерми¬
нации: по оси абсцисс — условная
коїіцентратдия веіцества в плазме; по
оси ординат — условное число лиц;
1 — отсутствие эффекта от лекар¬
ства; 2 — оптимальный эффект; 3 —
токсический эффектИ и их случаях фармакогене-
ІМіч кий подход мало применим
|м индивидуализации лекар-
^цспиой терапии.К:ік было показано в данной
11К‘. число генов, мутации в
11>рмх ведут к фармакогенети-
VKIIM последствиям, достаточно
Л1.П10С, и перечень их постоян-
іюгіолняется. Патологические
МКИИИ на лекарства касаются
цимх функций и систем орга-
1ма при разных заболеваниях,
пмоіінтельно, ознакомление с
|црмакогенетикой необходи-
ирачу любой специальности.ІІИІИЄ фармакогенетических
^иЬснностей обеспечит лучшую^(|)ск'гивность и большую безопасность назначаемой лекарственной
Іраііии. Однако для этих целей необходима разработка недорогих,
іісірьіх, адаптированных к клинической практике методов тестиро-
IIUIH аллельных вариантов соответствующих генов (генотипированис)
1И концентрации лекарств (фенотипирование).Что касается методов генотипирования, то в их основе лежит
|ЦР-реакция, а современные разработки по созданию биочипов
ІЛПЮТ реальным и доступным обследование пациента на фарма-
їіенетические варианты в еще более широком масштабе. В онко-
м ической практике во многих странах используются биочипы для
стирования пациентов с повышенным риском токсичности цито-
Гнтических препаратов на основе меркаптопурина. В США при-
Іеияется несколько фармакогенетических тестов для индивидуали¬
тии выбора лекарственных средств и их доз (антидепрессантов,
ІІсйролептиков, меркаптопурина, варфарина и других лекарств).Безусловно, в некоторых случаях практичнее ориентироваться на
(стоды фенотипирования ферментов и продуктов биотрансформации
!'Юниазидовый тест, антипириновьтй тест, дибукаиновый тест).В заключение следует подчеркнуть, что фармакогенетика решает не
проблемы персонализации лекарственной терапии. Анализ ситуа¬
ции на сегодня показывает, что предсказательным гено- и феноти-
352Клиническая rehпированием может быть обеспечено примерно 15—20% случаев
ви дуального подбора лекарств или их доз, что позволяет из(
нежелательных лекарственных реакций. Для 15—40% случаев аі
генетического полиморфизма имеет меньшее значение из-за
генного влияния на исход лекарственного лечения, а для 50%
ентов фармакогенетичсский подход никак не будет влиять на П(
лекарств, потому что другие физиологические и средовые фаі
влияют сильнее, чем наследственные.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯАцетилирование
Белки-мишени
Водная конъюгацияГенетические основы фармакодинамики
Генетические основы фармакокинетики
Глюкуронирование
Конъюгация с глутатионом
S-мети лирова ниеПатологические реакции на лекарстваПерсонализированная медицинаСубстратСульфатированиеТипы метаболизаторовФазы биотрансформацииФармакогенетикаРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАГенетический паспорт — основа HHflHBHflyajibHoii предиктивной
цины / под ред. B.C. Баранова. — СПб.: Изд-во Н-Л, 2009. — 527 с.Середенин C.R Лекции по фармакогенетике, — М.: МИА, 20(
303 с.Сычев Д.Л., Раменская Г.В., Игнатьев И.В., Кукес В.Г Клинич*
фармакогенетика // под ред. В.Г. Кукеса, Н.П. Бочкова: учеб. п<
М.: ГЭОТАР-Медиа, 2007. - 245 с.Сычев Д.А., Савельева М.И., Кукес В.Г. Проблема внедрения фї
когенетики в реальную клиническую практику: Медицинская
тика. - 2008. - № I. ~ С. 21-27.
Глава 9ЛАБОРАТОРНЫЕ МЕТОДЫ ДИАГНОСТИКИ*
ОБЩИЕ ВОПРОСЫМногие формы наследственной патологии проявляются настоль-
4'исиифическим фенотипом, ЧТО клинический анализ с син-
імологическим ПОДХОДОМ позволяет установить точный диагноз,
щи u)j[некие к методам клинической диагностики применяют гене-
ІИІІІ'іеСКИЙ метод, который еше больше повышает вероятность
Синильного диагноза. Однако широкий клинический полиморфизм
,мк'дет венных болезней, их фенокопии, частичное совпадение сим-
ІІМОИ разных заболеваний (наследственных и ненаследственных),
О(»чодимость выявления гетерозиготных носителей или носителей
шпсированных транслокаций и инверсий требуют применения
||01>раторных методов диагностики, которые при наследственной
1’ЮЛОГИИ всегда более точные, чем клинические методы.Хотя история применения лабораторных методов диагностики
|слсдственных болезней насчитывает почти 100 лет, первая иолови-
jroro пути отмечена лишь единичными примерами диагностики
ііа'льньїх болезней с использованием качественных биохимических
фикций (моча) или патогистологических методов. Применение био-
ІМИЧЄСКИХ методов началось с диагностики алкаптонурии в началеII., что и позволило А. Гарроду открыть наследственные болезни
імена веществ, обусловленные блоком ферментативной реакции.
ІУ30-Х годах была открыта простая реакция мочи с хлоридом железа
вленая окраска) при фенилкетонурии. Морфологическими мето-
ІМИ подтверждались диагнозы нейрофиброматоза, наследственных
)жных болезней (конец XIX в.).Широкое применение лабораторных методов диагностики наслед-
[•иенных болезней началось в 50-х годах XX в. Это было связа-
по-видимому, не только с прогрессом лабораторной диагности-
(клинической биохимии, гематологии, цитологии, цитохимии,
Іммунологии), но и с повышенным интересом в этот период кИсправлено и дополнено при участии д-ра мед. наук Т.В. Филипповой.
354Клиническая Г9>наследственной патологии. Кроме того, усовершенствование
генетических методов в 50-х голах позволило открыть новую Г|
болезней человека — хромосомные болезни.Таким образом, генетика человека и медицинская генетика
на вооружение многочисленные методы лабораторных иссл<
ний (биохимические, иммунологические, цитологические,
тологические, цитогенетические, немного позже и молекулі
биологические). Это и обусловило формирование клинич<
генетики как медицинской дисциплины и ее интенсивное раэ!Лабораторная диагностика наследственных болезней (фен<
генотииирование индивидов) может быть направлена на идені
кацию одной из трех ступеней болезни. Во-первых, і)Т0 ВЫЯ1
причины наследственной патологии, или характеристика геї
па, т.е. определение конкретной мутации (генной, хромосо!
геномной). Эти цели достигаются с помощью цитогенетически!
молекулярно-генетических методов. Во-вторых, лабораторные
химические и иммунологические) методы позволяют регист|
вать первичный продукт гена. В-трстьих, возможна регисті
специфических метаболитов, возникших в процессе патологичен
действия мутации. Такая регистрация возможна на уровне !||
костей (крови, мочи, секрета) или клеток. Следовательно, наї
ступени можно применять биохимические, иммунологичес!ЯПШШіІII IliSll . ■ ■'"■■'"‘“ІЙнІшИаШьРис. 9.1. Препараты мазков крови: а
лисахаридозепри гайглиозидозе; б — при MJ
In ч Лабораторные методы диагностики355fu ioi ические методы, что и нашло подтверждение в клинической
Ікіикс. Например, иммунологические методы широко применяют
лил ностики первичных (наследственных) иммунодефипитов,
fMU'UHOH несовместимости матери и плода, биохимические — для
ігиос і ики наследственных болезней обмена веществ.
Цшологические клинические анализы помогают выявить неко-
И»|1' наследственные болезни обмена веществ. Например, на
‘М. гтредставлсны препараты мазков крови при ганглиозидозеII \1\кополисахаридозе (б). Эти болезни имеют диагностические
hiiioi ические признаки, выявляемые при клинико-лабораторном
ІЛИ крови.ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫМикроскопические методы изучения хромосом человека при-
ІІїнются с конца XIX в. Соединение цитологического наблюдения
t^^u)coм с генетическим анализом сегрегации и сцепления генов
Ihiilmo к рождению цитогенетики. Первоначально цитогенетика
Ииситрировалась на проблемах корреляции генетических и цито-
гимсских (хромосомных) признаков. В последующем цитогенети-
мсголически отделилась от генетики. Под термином «цитогенс-
ui'> понимают область науки, изучающей структуру и функцииЇМ0С0М.Цитогенетические методы предназначены для изучения структу-
хромосомного набора или отдельных хромосом. Наиболее рас-
4K’ граненный метод в цитогенетике человека — световая микро-
IIИЯ, а электронная и конфокальная лазерная микроскопия
ІИменяется только с исследовательскими целями. Во всей медико-
ІКіической практике используется световая микроскопия (глав-
|м образом в проходящем свете), в том числе люминесцентная
ІКроскопия.Объектом цитогенетических наблюдений могут быть соматиче-
ІИС делящиеся, мейотические и интерфазные клетки. Каждый из
fMx объектов имеет свои преимущества и недостатки. Выбор объекта
Ірсяеляется целью исследования.Ьольщинство цитогенетических исследований выполняют на
^магических клетках, поэтому остановимся на описании этих
П'одов.
356Клиническая гіПолучение препаратов митотических хромосомПервое условие питогенетической диагностики — наличі
щихся клеток в материале для цитологического исследования.Костный МОІГ, ткани семенника и хорион имеют достак
митотический индекс для использования в цитогенетике,
как показал опыт, несравненно информативнее исследован<
культурах клеток: клетки освобождены от элементов соедините
ткани и хорошо суспендируются. Митотический индекс в ку/
клеток много выше, чем в тканях организма.Культуры клеток можно получать из кусочков кожи (растут (
бласты), костного мозга, эмбриональных тканей, хориона,
амниотической жидкости. Наиболее удобным объектом для
цинских генетиков оказалась культура лимфоцитов перифериЧІ
крови (рис. 9.2). Для ее получения достаточно взять 1-2 мл вен<1-2 мл венозной кровиДобавлениевередуИнкубация
в течение
2-3 дней
при 37 “СП ІД п !UiИ » п II П II IIКариогипДобавление
колхицина
на 2-3 чРаспределение клеток
на предметном стекле
путем раскапывания ^Центрифу¬гированиеФиксация клетокIЦентрифугированиеIГипотонизациясолевымрастворомРис. 9.2. Приготовление цитогенетических препаратов путем культиві
имя лимфоцитов периферической крови
'I Лабораторные методы диагностики357III и добавить ее в смесь питательной среды с фитогемагглюти-
♦ом (белок бобовых растеиий). Он вызывает иммунную трансфор-
|и»|> и деление лимфоцитов, продолжительность культивирования
Ііііі'іяет 48—72 ч.ІИіорьім методическим условием цитогенетических исследований
(’кя использование колцемида (или колхицина), разрушающего
'ifiK> деления и останавливающего клеточное деление на стадии
|ф»;ы. Колцемид добавляют в культуры kjictok за 2-3 ч до окон-
111 н культивирования; митотический индекс в культуре клеток за
'I повышается в 2—3 раза. Даже без KVJtbTHBnpoBaHHR экспозиция
іїмцсмидом увеличивает число метафаз. Хромосомы в присутствии
itu'M ида укорачиваются в результате продолжающейся конденсации,
Моїкп'сльно, в препарате они легче отделяются одна от другой.ІЧМІН необходим детальный анализ определенного района хро-
сильно конденсированные хромосомы на стадии метафазы
под называется метафазным) непригодны для анализа. Клетку
/ЖІИ) зафиксировать на стадии, предшествующей метафазе, когда
)М1)Сома редуплицировалась, но еще не полностью конденсиро-
сь. Это стадия прометафазы. Хотя хромосомы на стадии про-
шфазы плохо разъединены (они еще очень длинные), и в препа-
if много наложений одной хромосомы на другую, что, безусловно,
Грудняет анализ, все же в отдельных клетках можно найти нужный
исток, пригодный для анализа. Этот метод (или подход), в отли-
от метафазного метола, называют прометафазным, или методом
Ісокоразрешающей цитогенетики. Суть модификации метода состо-
п прекращении процесса спирализации и конденсации хромосом
лрофазе с помощью препаратов, например мегатрексата, которые
Іодит в культуру клеток за несколько часов до фиксации.
Следующее условие для получения хороших метафазных пла-
рипок — гипотоннзация клеток (і ипотонический шок). Обычно для
furo используют гипотонический раствор хлорида калия или цитра-
иатрия. В гипотоническом растворе клетки набухают, ядерная
Волочка разрывается, межхромосомные связи рвутся, и хромосомы
щОодно плавают в цитоплазме.Культивирование клеток, применение колцемида и гипотониза-
стали условиями, на основе которых сформировались современ-
|мс цитогенетические методы.Клеточную суспензию фиксируют смесью метанола и уксусной
1ИСЛ0ТЫ (3:1), затем суспензию центрифугируют и меняют фиксатор.
358Клиническая rt^iСмесь клеток с фиксатором может сохраняться при темпе{
+4 °С в течение нескольких недель, при нанесении такой суспі
на чистое предметное стекло метафазная клетка расправляется
пределах располагаются отдельно лежащие хромосомы. При ві
НИИ фиксатора хромосомы прикрепляются к стеклу.Выше описана методика получения препаратов из культуры
фоцитов, которая используется наиболее часто. Для диатности»
целей МОЖНО готовить препараты из хориона, кос гного мозга, е{
ников, культуры фибробластов, культуры амниоцитов, ПроЩ
для каждого объекта отличаются от описанной выше, но о4
принцип сохраняется: накопление метафаз, гипотонизация,
ция, капанье на предметное стекло, окраска, кариотипированіОкраска препаратовСледующая стадия цитогенетических методов — окраска
паратов. Методы окраски бывают простыми, дифференциалы
флюоресцентными.Наиболее распространен метод окраски по Гимзе, или
стая окраска (в русскоязычной литературе распространен
термин «рутинная окраска»). Краситель Гимзы окрашивае
хромосомы равномерно по всей длине (рис. 9.3) При этом к<
рируются центромера, спутники (иногда со спутничными
ми) и вторичные перетяжки. Механизм связывания красіГимзы хромосомами неїл.■л Лli'■/гРис. 9.3. Метафазная пластинка при
простой окраскеОн не является специфик
для какого-либо азоти(
основания ДНК,При простой окраске
можна только групповая
тификания хромосом, nos
данный метод используется
ориентировочного опредс
числовых аномалий карио!
Структурные хромосомные 1
малии (делеции, транслокі
инверсии), выявляемые при!
стой окраске, должны быть;
тифицированы с помощью
ференциальной окраски.
їй '» Лабораторные методы диагностики359vAMji*'\vі». ‘>.4. Метафазная пластинка с
III II ц ионно-индуцированными
jMDcoMHbiMM аберрациямиішиаріїіпі:ЯІІЇрІІИ■ЩіШі^:;■iUл' І!• ;ГГ!г!!Рис. 9.5. Метафазная пластинка
(неполная) с химически-индуциро-
ванными аберрациямиПростая окраска широко применяется для изучения хромосомного
пи еиеза (учет хромосомных аберраций) при проверке факторов окру-
ІЮІЦЄЙ среды на мутагенность. На рис. 9.4, 9.5 хорошо видны аберра-
III. іюзникшие под влиянием радиации и химических мутагенов.
Метод простой окраски хромосом как единственный метод изуче-
1И кариотипа человека применялся до начала 70-х годов XX в. С его
)М01ДЬЮ за 10 лет были открыты основные хромосомные болезни,
жазана роль хромосомных аномалий в спонтанных абортах, врож-
|миых пороках развития и канцерогенезе, разработаны принципы
ііологической дозиметрии.Морфологическая однородность хромосомы по длине на стан д ар-
U) приготовленных и окрашенных по Гимзе препаратах обманчива.
Ірогресс цитогенетики человека позволил выявить глубокую линей-
1ук> дифференцированность не только функции, но и структуры
омосом. в 70-х годах в практику вошли методы дифференциального
(рашивания.Под дифференциальной окрашиваемостью хромосом понимают их
Цособность к избирательному окрашиванию подлине без прижизнен-
|ой модификации какими-либо воздействиями. Дифференциальное
Окрашивание хромосом обеспечивается сравнительно простыми
Температурно-солевыми воздействиями на фиксированные хромо¬
сомы. При этом выявляется структурная дифференцировка хромосом
К) длине, выражающаяся в чередовании эу- и гетерохроматиновых
360Клиническая генірайонов (темные и светлые полосы). Протяженность этих у»
ков специфична для каждой хромосомы, соответствующего nj|
и района. Как видно на рисунках 9.6, 9,7, при дифференциал!
окраске идентифицируются все хромосомы, плечи и даже onj
ленные районы. Каждая хромосома имеет свой рисунок исчеі
кости. При дифференциальной окраске метафазиых хромос<кариотиле можно оценить о|
200—400 участков (разрешаї
способность метода), на СТ1
прометафазы — до 2000.Первоначально при сп<
альном окрашивании хроме
использовали флюоресці
ное алкилирующее веще*
акрихин-иприт. Этот варі
был назван Q-методом, он
бует быстрой обработки
парата, что не всегда удо<
Для просмотра препарата
пользоваться люминесценті
микроскопом.В дальнейшем была разрі
тана методика дифференци!
ной окраски без фл юоресцентРис. 9.6. Метафазная пластинка
после дифференциальной окраски1ІIi; IIій иг« гга бРис. 9.7. Кариотипы при простой (а) и дифференциальной (б) окраске
Чйна 9. Лабораторные методы диагностики361кителей. Наиболее птироко используется G-окраска (по Гимзе).
хромосомы нужно предварительно обрабатываг ь (инкубация в соле¬
вім растворе либо обработка иротеазой). Предварительная обработка
#4І ИЧН0 нарушает структуру хромосом, в некоторых участках она
кіч ганавливается при окраске, что и придает хромосоме иидиви-
|ул'м>ную исчерченность. Механизм образования сегментов пока
глостаточно ясен. Предполагается, что окрашенные сегменты — это• к'рохроматиновые, поздно реплицирующиеся участки хромосом с
|и1поряющимися послсдоваї ел hi (ОСТЯМИ ДНК, а неокрашенные —
1<| )ухроматиновые участки, в которых расположены кодирующие
|1н лодовательности.Для идентификации хромосом, помимо методов выявления
Іинсйной структурной дифференцированности, можно восполь-
^пиигься одной из важных характеристик хромосом человека —
[Синхронностью их репликации по длине. Этот метод разработал
^нлпитливый отечественный цитогенетик А.Ф. Захаров о его роли
цитогенетике можно прочитать на компакт-диске. «Рисунки»
іК'лсдовательности репликации (рано или поздно реплицирующи-
ІН участки) специфичны для каждой хромосомы. Для выявления
(к'ледовательности репликации применяется аналог тимидина —
(»|)омдсзоксиуридин. Участки хромосомы, включившие этот ана-
|(И. окрашиваются плохо. Используя этот метод, можно идентифи-
[ировать любую хромосому или хромосомную перестройку.‘і-бромлезоксиуридин вводят в культуру на 24 ч и более
ЛИ дифференциальной окраски сестринских хроматид. Если
«Промдезоксиуридин ввести на полный клеточный цикл, то вновь
^разуемая хроматида включитІИіиюг тимидина и будет окрапти-
111,ся слабо. Другая хроматида
[К'іарая) окрашивается, как обыч¬
но, интенсивно (рис. 9.8). Этот
ІСТОЛ позволяет легко выявлять
Умены между сестринскими
)(>матидами, число которых уве-
ІИ'іивается при наследственных
Іолсзнях с хромосомной неста¬
бильностью (анемия Фанкони,
іиі'ментная ксеродерма и др.)
же. 9.9), Число обменов сестрин-аШшмаРис. 9.8. Метафазная пластин¬
ка с дифференциальной окраской
сестриїтских хроматид
362Клиническая гвНІJРис. 9.9. Хроматидные аберрации (а) и сестринские хроматидные обме)
при заболеваниях с хромосомной нестабильностьюских хроматид увеличивается также при мутагенных воздейсті
поэтому метод учета обменов сестринских хроматид широко ис<
зуется при изучении мутационного процесса у человека.Молекулярно-цитогенетические методыБлагодаря успехам в молекулярной генетике человека разра<
принципиально новый метод изучения хромосом — метод
Принцип этого метода показан на рис. 9.10.Для изучаемой хромосомы или ее конкретного участка (в
со специфичностью последовательности оснований ДНК) к
однонитевой участок ДНК, к которому присоединяется биотиі
дигоксигенин. Такой помеченный участок ДНК называется зоїНа микроскопическом препарате ш situ при обработке щел|
хромосомная ДНК денатурируется, т.е. разрываются связи Mf
двумя нитями ДНК.Зондом обрабатывают препарат. Поскольку последователі
оснований ДНК зонда и соответствующий участок хромосомы]
имно комплементарны, зонд присоединяется к хромосоме. В
участке происходит ренатурация ДНК.После этого препарат обрабатывают веществом, которое спое
избирательно присоединиться к биотину или дигоксигенину.
биотина это стрептавидин, для дигоксигенина — антидигоксиі
новое антитело. К этим веществам могут быть присоединены в
Ill 9. Лабораторные методы диагностики363гтпуглтугп-, 1 .i-j—Li-i—l-i J-..L.Lо:зЗонд9.10. Двойная специфическая флюоресцентная гибридизация in situ
riSII): 1 — биотин; 2 — стрептавидин; З — родамин; Г-дигоксигенин;
»йМ1идигоксигениновое антитело; 3', 4'-флюоросцеина изотиоцианат111 ДІШ этапа флюоресцентные красители (родамин — красный цвет
ІИ <|)люоресцеина изотиоцианат — зеленый цвет).( помошью люминесцентного микроскопа окрашенные хромосо-
1^1 можно увидеть на фоне неокраптенных.11л рисунке 9.10 приведена двойная гибридизация, однако современ-
ІС методические возможности позволяют увеличить число цветов.
Метод FISH применяется очень широко — от локализации
ІІІІІ до расшифровки сложных перестроек между несколькими
^омосомами. Следует подчеркнуть, что сочетание молекулярно-
ІИстических и цитологических методов делает почти неограни-
|циыми возможности диагностики хромосомных аномалий, как
;мь сложных, так и очень мелких. Двух- и трехцветная FISH
вменяется для учета симметричных хромосомных аберраций уIII, много лет назад получивших дозу ионизирующего излучения,
(стод требует меньше времени, чем кариотипирование дифферен-
(ильно окрашенных метафаз.В клинической цитогенетике метод FISH занимает все боль-
1С место. В случаях сложных хромосомных перестроек, захва-
ІІШЮШИХ более двух хромосом, дифференциальная G-окраска не
Ссгда позволяет идентифицировать измененные сегменты хромо-
)м. В этих случаях применяют трехцветный вариант метода F1SH.
(«пример, у ребенка с множественными врожденными аномалиями
(|ри G-анализе обнаружены сложные перестройки в 6 хромосомах
4, 7, 8, 9 и 12) с І0 разрывами. Полная идентификация разрывов
ііможна только с помошью FISH-окраски.
364Клиническая ГіІМетод FISH можно применять для диагностики анеуплоі
интерфазных ядрах. Принцип метода в этом варианте такой
и для метафазиых пластинок, описанный выше. Например,
фичный для хромосомы 21 зонд ДНК, соединенный с би(
гибридизируется с денатурированными клетками из амниоти*
жидкости ifa предметном стекле. В норме, т.е. если у пло;
дисомия по хромосоме 21, в ядре будут видны 2 флюоресцирі
соответствующим пветом точки. Если плод трисомный, то
будут видны 3 точки (рис. 9.И). Такой методический прием назі
интерфазной цитогенетикой. Метод прост, экономичен, анализ
мает всего несколько часов.Метод CGH {comparative genome hybridization). Область исП(
вания — онкологическая цитогенетика, назначение — опреде/
районов хромосом, которые делетируются или амплифицируї
определенном типе опухоли. Районы делений, как правило, С(|
жат гены-супрессоры опухолевого роста, а районы амплификаї
онкогены. Таким образом, метод используется в большей ст<
для картирования и клонирования генов, вовлеченных в кані
генез. Иногда достаточно сложно получить хромосомные преп(
хорошего качества из солидной опухоли или у пациентов с *
тологическими онкологическими заболеваниями. В связи с
был разработан оригинальный метод косвенного анализа xjсом в опухоли. Суть метода!
состоит в том, что из оп
выделяют ДНК и метят ее <
деленным флюорохромом,
выделенную из норма
ткани, метят другим фл
ромом. Хромосомные преп
приготавливают стандар
способом из лимфоцитов
ферической крови контр
го индивида. Меченую ДН!
опухоли и неизмененной
гибридизуют с хромосо
Рис. 9.11. Флюopecцeнтr^aя гибри- препарагом. По интенси
дизаиия in siiu (FISH). Вверху ядро с свечения метки опредітрисомисй по хромосоме 6 и дисоми- области делеций и ампл
ей по хро.мосомс 8 каций. Область разрешен
iNii Лабораторные методы диагностики365|i|<) vuiH пар нуклеотидов. Для обработки данных используют про¬
їм мы компьютерного аиализа хромосом.( иок I роскопический анализ хромосом (SKY), При этом методе исполь-
011я і|)л юоресцентные красители, имеющие сродство к определенным
III кам хромосом. При использовании набора специфических зондов
тими красителями каждая пара хромосом имеет свои уникальные
^К1|1алы1ьге характеристики. Особенность метода — использование
|И'|х[)ерометра, аналогичного используемым для измерения спектра
{шмомических объектов. Незначительные вариации в спектраль-
|м составе, не различимые человеческим глазом, учитываются при
Іміїї.іоіерной обработке, и затем программа назначает каждой паре
имосом легко распознаваемые цвета. Результат в виде цветного изо-
рижсиия чаше используется в цифровой форме. Анализ кариотипа
іичи ісльно облегчается, поскольку гомологичные хромосомы имеют
111 и 11 тот же цвет, а аберрации становятся легкоразличимыми. Кроме
И), спектральное кариотипирование используется для выявления
післокаций, не распознаваемых традиционными методами. Область
Люльзования метода — онкоцитогенетика. Благодаря такому подходу
Illi'гея точно описать множественные структурные перестройки хро-
«foM, происходящие в опухолевых клетках.Н клинической цитогенетике удается определять очень незна¬
ні сльные по величине транслокации, инсерции и маленькие
^иркерные хромосомы. Однако использование метода ограничено
Jcoкoй стоимостью оборудования для анализа. Более подробные
имения о цитогенетических методах см. на компакт-диске в статье
І'оіфеменная клиническая цитогенетикаи в статье Н.Б. Рубцова и
Р|И, Карамышевой «Прямая и обратная цитогенетка».жазания для проведения цитогенетических исследованииI Іоказани я для ци і огенети ческого исследования достаточно широ-
|ис, особенно при акушерско-гинекологической и детской патологии.
ІИЖЄ приводится перечень (возможно, неполный) состояний, при
«горых для диагностики надо иметь результаты цитогенетического
Ісследования у пациента (пробанда) и в случае необходимости у его
родственников:— Подозрение на хромосомную болезнь по клинической симпто¬
матике (для подтверждения диагноза).- Наличие у ребенка множественных врожденных пороков раз¬
вития, не относящихся к генному синдрому.
366Клиническая генві— Многократные (более двух) спонтанные аборты, мертвор(
ния или рождения детей с врожденными пороками развит!— Нарушение репродуктивной функции неясного генеза у
тин и мужчин (первичная аменорея, бесплодный брак и ді— Существенная задержка умственного и физического разві
у ребенка.— Пренатальная диагностика (по возрасту, в связи с нали^
транслокации у родителей, при рождении предыдущего
ка с хромосомной болезнью).— Подозрение на синдромы с хромосомной нестабильно<
(учет хромосомных аберраций и сестринских хроматид).— Лейкозы (для дифференциальной диагностики, оценки эф1]
тивности лечения и прогноза).— Оценка мутагенных воздействий (радиационных, хими'
ких).Медицинских ограничений для применения цитогенетичЄ(
методов нет. Однако необходимо помнить, что эти методы
емкие, дорогие, их назначение наугад не оправдано (по прині
«если неясно, то давайте назначим»). Правильнее назначать
генетическое исследование по рекомендации вpaчa-гel^eтикa п<
проведения медико-генетического консультирования.Опыт работы зарубежных медицинских учреждений показал
ходимость создания цитогенетических лабораторий при болі
многопрофильных больницах и медико-генетических консультаі
комплексно обслуживающих какой-либо район или город. В Рс
цитогенетические исследования проводятся в медико-1 снетиче*
кабинетах и медико-генетических консультациях.БИОХИМИЧЕСКИЕ МЕТОДЫБиохимические методы в лабораторной диагностике наследсі
ных болезней применяются с начала XX в. Биохимические показ
ли (первичный белковый продукт гена, накопление патологии*
метаболитов внутри клетки и во внеклеточных жидкостях)
отражают сущность болезни, че.м клинические симптомы, не т<
в диагностическом, но и в генетическом аспекте. Значимость
химических методов повышалась по мере описания наследстве»
болезней и совершенствования этих методов (электрофорез, хр01
графия, спектроскопия и др.). В 80-х годах XX в. был выделен ш
lf<iiina 9. Лабораторные методы диагностики367|р(11ЛСЛ — наследственные болезни обмена веществ, т.е. заболевания
fi' |ш иичными биохимическими нарушениями.Ьиохимичсские методы направлены на выявление биохимического
Іфґштипа организма. Уровни, на которых оценивается фенотип, могут
разными; от первичного продукта гена (полипептилной цепи)
hui конечных метаболитов в крови, моче или поте. Биохимические
шс1Ч)ды чрезвычайно многообразны, и их значение в диагности-
[♦ir наследственных болезней постоянно возрастает. Разработка
іиіміскулярно-гснетических методов диагностики наследственных
[йн.'К'зней частично снизила интерес к биохимическим исследова¬
ниям, но вскоре стало ясно, что в большинстве случаев указанные
щ-юды дополняют друг друга, поскольку молекулярно-генетически
Описывается генотип, а биохимически — фенотип. Болезнь — это
[н конечном счете фенотип. В связи с этим, несмотря на сложность,
миогда и дороговизну биохимических методов, им принадлежит
Іс.чушая роль в диагностике моногенных наследственных болезней,
ипременные высокоточные технологии (высокоэффективная жид-
IliUL' I ная хроматография, хроматомасс-спектрометрия, газовая хрома¬
тография, тандемная спектрометрия) позволяют идентифицировать
Побые метаболиты, специфичные для конкретной наследственной
Іилсзни.Иа первый взгляд может показаться, что самым точным методом
1И:н ностики является определение мутации на уровне ДНК. Однако
1П) не всегда так. Реализация действия гена — сложный процесс,
ho /гому «нормальная» структура гена, а точнее, необнаружение мута-
1ИИ, не всегда бь[вает полной гарантией нормального биохимическо¬
го (І)енотипа.Принципы биохимической диагностики наследственных болез-
u*ii менялись в процессе развития генетики человека и биохимии.
Гак, до 50-х годов XX в. диагностика была направлена на поиски
Яісцифичньїх для каждой болезни метаболитов в моче (алкаптону-
>ия, фен ил кетон ури я). С 50-х до 70-х годов упор в диагностике был
Цдслан на выявление энзимопатий. Разумеется, поиски метаболитов в
юпсчных реакциях при этом не исключались. Наконец, с 70-х годов
ІЬЛімтьім объектом при диагностике стали белки разных групп.
|К настоящему времени все эти объекты являются предметом био-
{имической диагностики.Оценка метаболитов в биологических жидкостях — необходимый
fiTjm диагностики аминоацидопатий, органических ацидурий, муко-
368Клиническая геніполисахаридозов, митохондриальных и пероксисомных болезі
дефектов метаболизма пуринов и пиримидинов и т.д. Для этих ц|
используют методы качественного химического анализа, спектре
гометрические методы количественной оценки соединений, а TJ
различные виды хроматографии.Хроматографические методы анализа играют важнейшую роль в
гностике наследственных болезней обмена. Это обусловлено тем,
современный арсенал хроматографических технологий чрезвычв!
широк и позволяет эффективно и информативно разделять слож!
многокомпонентные смеси, к которым в том числе относится и бис
ческий материал. Для количественного анализа маркеров-метаболI
ііаследственньїх болезней обмена успешно применяются такие xj
тографические методы, как газовая и высокоэффективная жидкое!
хроматография, а также хроматомасс-спектрометрия. Газовая и выс
эффективная жидкостная хроматография являются универсальні
методами разделения сложных смесей соединений, отличаются выс
чувствительностью и воспроизводимостью, в обоих случаях разделе
осуществляется в результате различного взаимодействия компо»
тов смеси с неподвижной и подвижной фазами хроматографичес
колонки. Для газовой хроматографии подвижной фазой является
носитель, для жидкостной хроматографии — жидкость (элюент). Bi
каждого соединения фиксируется деіектором прибора, сигнал коте
преобразуется в пики на хроматограмме. Каждый пик характерна^
временем удерживания и площадью, Следует отметить, что га^
хроматография проводится, как правило, в вьісокотемтіературном р<
мс, поэтому ограничением для ее применения яіиіяется термичв(
неустойчивость соединений. Для высокоэффективной жидкостной:
матографии не существует подобных ограничений, так как в этом СЛ5
анализ проводится в мягких условиях.Масс-спектрометрия — аналитический метод, с помощью котої
можно получать как качественную (структура), так и количествен!
(молекулярная масса или концентрация) информацию от аналі
руемых молекул после их преобразования в ионы. Существен!
отличие масс-спектрометрии от других аналитических физі
химических методов состоит в том, что в масс-спектрометре ощ
ляются непосредственно масса молекул и их фрагментов. Резулы
представляются графически (так называемый масс-спектр). Ин<
невозможно анализировать многокомпонентные, сложные сї
молекул без их предварительного разделения. Разделить молек)
9. Лабораторные методы диагностики369їжмо либо хроматографически (жидкостная или газовая хромато-
«фпя), либо использовать два последовательно соединенных масс-
(|«?к I рометра ~ тандемная масс-спектрометрия.Ііпілемная масс-спектрометрия позволяет охарактеризовать
PjH'K iypy, молекулярную массу и провести количественную оценку
}1И) соединений одновременно. При этом не требуется длитсль-
подготовки проб для проведения анализа {как, например, для
ППІЮЙ хроматографии), а время исследования занимает несколько
>куил. Нозологические формы наследственных болезней обмена,
|)|ч)рые можно диагностировать с использованием тандемной масс-
Пі'к ?рометрии, приведены в табл. 9.І.іП іица 9.1. Заболевания, диагностируемые с помощью тандемной масс-
спектромстрииЛминоацидопатииОрганическиеацидурииДефектымитохондриальногоокисленияЛі'ІІЦИНОЗ (болезньI' i;i пахом мочи «клсново-ІІ» сиропа»);искстотическая гипергли-цііиіімия;ііірозинемия;іомоцистинурия;іііітруллинемия;шпсрорнитинемия;(|)і.'Иилкетонурия;лсдостаточность орни-I и итранскарбам илазьі;недостаточность арі ина-ІІ.Г.синдромІ иперориитинемии-
гнпераммониемии-
гомоцитруллинсмии;
недостаточность карба-
моилфосфатсинтетаз;
недостаточность аргини-
носукииназьі;
недостаточность аргинин-
сукішнатлиазьіНедостаточность
биотинидазы;
недос'іаточность
синтетазы голо-
карбоксилаз;
изовалериановая
ацидурия;
глутаровая ациду-
рия тип 1;
глутаровая аішду-
рия тип 2;
нропионовая аци-
дурия;метилмалонопаяацидурияНедостаточность средне¬
цепочечной аиил-KoA-
дегидрогеназы жирных
кислот;недостаточность корот-
коиспочечной ацил-
КоА-дегидрогеназы
жирных кислот;
недостаточность длин¬
ноцепочечной ацил-
КоА-дегидрогеназы
жирных кислот;
первичная недостаточ¬
ность карнитина;
недостаточ ноет ь
карнитинпальмитоил-
транслоказы I;
недостаточное! ь
карнитинпальмитоил-
транслоказы 2;
недостаточность
Р-оксотиолазы;
недостаточность
HMG-лиазы
370Клиническая гвьТандемная масс-спектрометрия является одним из перепої
ных направлений в развитии программ диагностики наследствві
болезней обмена, поскольку позволяет количественно и в
количествах биологического материала определять множество
болигов. В настоящее время в некоторых странах данный
применяется для массового скрининга новорожденных. Для
подробного ознакомления см. статью Г.В. Байдакой и Е.Ю. Захара
«Тандемная масс-спектрометрия с ионизацией в электроспрее: П|
цип метода и нримененис для диагностики наследственных болвЯ
обмена» на компакт-диске.В связи с многообразием биохимических методов, применяем!
лабораторной диагностике наследстве[шых болезней, в использоі
этих методов должна быть определенная система. У пробанда
члена его семьи нереально исключить все наследственные болвІ
которые могут быть в поле зрения при обследовании. Если примсі
максимально возможное число методов диагностики, то каждое о(
дование станет очень трудоемким и долгим. Исходная схема
вания строится на клинической картине болезни, генеалогичї
сведениях и биохимической сі'ратегии, которые позволяют опредв
ход обследования на основе поэтапного исключения определеї
классов наследственных болезней обмена (просеивающий метод).Необходимо подчеркнуть, что биохимические методы (в отл^
от цитогенетических) многоступенчаты. Для их проведения треб)
аппаратура разных классов. Материалом для биохимической ди£
стики могут быть моча, пот, плазма и сыворотка крови, формеї
элементы крови, культуры клеток (фибробласты, лимфоциты),
птаты мышц. При использовании просеивающего метода в биохі
ческой диагностике можно выделить два уровня: первичный и
няющий. Каждый из этих уровней может быть по-разному шагру)
реакциями в зависимости от возможностей лаборатории.Основная цель первичной диагностики заключается в том, чт
выявить здоровых людей и отобрать пациентов для последую!
уточнения диагноза. В таких программах первичной диагности!
качестве материала используются моча и небольшое количество кі
Программы первичной биохимической диагностики наследствен!
болезней могут быть массовыми и селективными. Массовые
сеиваюшие программы в диагностике фенилкетонурии, врождені
гипотиреоза, адреногенитального синдрома, врожденіїьіх аном£
развития нервной трубки и болезни Дауна описаны в главе U.
ініі 9. Лабораторные методы диагностики371( V.iicKTHBHbie диагностические программы предусматривают про-
IpKv биохимических аномалий обмена (моча, кровь) у пациентов с
личр^^нием на генные наследственные болезни. Фактически такие
РИММЫ должны функционировать в каждой большой больнице.
Іокії іания для их применения достаточно широкие, стоимость каж-
Ло анализа невысокая.И селективных программах могут использоваться простые качествен-
AV реакции (например, тест с хлоридом железа для выявления фенил-
монурии или с динитрофенилгидразином для выявления кетокислот)
|;п1 ()олес точные методы, позволяющие обнаруживать большие группы
I к .'юнений. Газовая хроматография применяется для выявления наслед-
цк'пных болезней обмена органических кислот, ряда аминоацидопатий.
I' |1»)М0ш;ью электрофореза гемоглобинов диагностируют все заболевания
и іруппьі гемоглобинопатий. На рисунке 9.12 представлены результаты
»11 химической диагностики лизинурической непереносимости белка.Ісредко приходится углублять биохимический анализ от количе-
^шсиного определения метаболита до определения активности фер-Рис. 9.12. Количественное содержание диа.минокислот в образце суточ¬
ной мочи больного с лизинурической непереносимостью белка. Выпол¬
нено на аминокислотном анализаторе: по оси абсцисс — время
ілюиропания, мин; по оси ординат — концентрация, мкМ; сплош-
Ніім линия — больной; пунктирная линия — норма, 1 — лизин; 2 — гистидин;
3 - аммиак; 4 — аргинин
372Клиническая генімента (использование нативных тканей или культивированны)
ток), например, с помощью спектрофлуориометрии.В современных условиях очень многие этапы биохимич!
диагностики осуществляются автоматически, в частности ам|
нализаторами.Селективные диагностические программы обеспечивают
предположительное выявление больных с наследственными
ми обмена веществ. Методы подтверждающей диагностики вклі
количественное определение метаболитов, исследование их кин<
энзимодиагностику, ДНК-диагностику (табл. 9.2).Таблица 9,2. Методы подтверждаюіцей диагностикиКласс болезнейМетоды подтверждения диагноза jАминоацидспатииКоличественное определение аминокислЯ
кропи, мочи, спинномозговой жидкости, -І
ДНК-диагностика gОрганические ацидурииКоличественное определение органичесКіІ
кислот мочи, плазмы ]Болезни углеводного
обменаКоличественное определение МОНО- и ДИСЦІ
РИДОВ и их метаболитов в крови, моче, ЭН31|
диагностика, ДНК-диагностика jjМ итохондриал ьи ые
болезниНагрузочные тесты (глюкозная кривая), эи
модиагностика комплекса дыхательной цЦ
ДНК-диагностика JБолезни парушения мито¬
хондриального р-окис-
ления жирных кислотКоличестветпюе определение карнитина, f
эфиров, жирных кислот, экзимодиагности!
ДНК-диагностика 1Пероксисомные болезниКоличественное определение очень длин™
почечных жирных кислот, ДНК-лиагност||Лизосомиые болезниЭнзи модиагностика, ДНК-диаі ностика 1Нарушение обмена пури¬
нов и пиримидиновКоличественное определение пуринов, ЛИ|
мидинов, мочевой кислоты, ДНК-диагнос|Болезни холестеринового
обменаКоличественное определение холестерина!
его производных в кровиБолезни иейротрансмит-
терного обменаКоличественное определение катехоламин!
аминокислот (кровь, моча, спинномозговм
жидкость) ^Как видно из табл. 9.2, методы подтверждения диагноза мноі
разны и специфичны для разных классов болезней.
ІЙІ4ІІ9. Лабораторные методы диагностики373Показаниями для применения биохимических методов диагно-
ІМК1І у новорожденных являются судороги, кома, рвота, гипотония,
?туха, специфический запах мочи и пота, ацидоз, нарушенное
«I )и>| но-основное равновесие, остановка роста. У детей биохимиче-
UU' методы используют во всех случаях подозрения на наследствен-
|Mi' болезни обмена вешеств (задержка физического и умствен ноіо
КПП ИЯ, потеря приобретенных функций, клиническая картина,
|К'|Ц1<1)ичная для какой-либо наследственной болезни).Ьиохимические методы применяют для диагностики наслед-
|К1синых болезней и гетерозиготных состояний у взрослых (гепа-
іик'нтикулярная дегенерация, недостаточность арантитрипсина,
ІРііостаточность Г6ФДГ и т.д.). Для диагностики многих болезней
|ш>химические меюды заменяют молекулярно-генетическими в
с их большей точностью или экономичностью. Более подроб-
(ую информацию по диагностике болезней накопления можно найти
К г:п ье Е.Ю. Захаровой и др. «Лабораторная диагностикализосомных
иле шей накопления» на компакт-диске.МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ МЕТОДЫ)бщие процедурыМолекулярно-генетические методы — большая и разнообразная
Группа методов, в конечном счете предназначенных для выявле-
||ин вариаций в структуре исследуемого участка ДНК (аллеля, гена,
^С1 иона хромосомы) вплоть до расшифровки нсрвичной последова¬
тельности оснований. В основе этих методов лежат манипуляции с
II!К и РНК. В результате бурного развития молекулярной генетики
Чс.'ювека в 70-80-х годах XX в. и последуюиіего успешного изучения
I'dIома человека молекулярно-генетические методы прочно вошли в
йсдико-генетическую практику. Для идентификации и поиска ДНК-
Іцолиморфизмов (мутаций) применяются уже более 100 разных мето-
|Д1)и. Однако широко используется только незначительная их часть.Ниже схематично описаны основные этапы и варианты
[Тиіиіекулярно-тенетических методов. Освоение этих методов, как и
[других методов лабораторной диагностики, требует специальной под-I Гоговки в соответствующих лабораториях.Получение образцов ДНК (или РНК) — первый этап всех методов.
Он включает выделение всей ДНК (тотальной или геномной) из
374Клиническая геміклеток или накопление определенных фрагментов, которые пр|
лагается анализировать с помощью П1ДР.Источником геномной ДНК могут быть любые ядросодерл
клетки. Выделенная из клеток ДНК представляет собой весь
организма, поэтому такие образцы называют геномной ДНК. На і
тике чаще используют периферическую кровь (лейкоциты),
амниотические клетки, культуры фибробластов. Для одного ан|
необходимо иметь (в зависимости от используемого метода) от не01
ких нанограммов до нескольких микрограммов ДНК. Для этого^
буется небольшое количество биологического материала, Hanj
20—40 мг хориона, 1 мл крови, 5-10 мг культуры клеток. При ио|
зований некоторых методов достаточно иметь одну каплю кров>
соскоб эпителия со слизистой оболочки шеки либо несколько
сяных луковиц. Возможность проведения молекулярно-генетичв<
анализа с небольшим количеством легкодоступного биологич<
материала — преимущество методов этой группы. Можно добаї
что выделенная ДНК одинаково пригодна для проведения разлив
вариантов методов и может долго сохраняться в замороженном
В большинстве случаев для успешной диагностики болезнн!
гетерозиготного состояния достаточно исследовать небольшой
мент генома. Необходимо получить достаточное количество
фрагментов, т.е. амплифицировать (умножить) их. Ранее реї
этой задачи было трудоемким: создание рекомбинантной плазІ
введение плазмиды в бактериальную клетку размножение^
териальных клеток выделение заданных фрагментов ДНК,
накопление нужных фрагментов ДНК обеспечивает ПІД Р. Отк1
этой реакции совершило революцию в изучении генома чел^
и молекулярно-генетической диагностике наследственных болі(
ПЦР ~ метод амплификации ДНК in vitro. За несколько
можно размножить определенную последовательность ДНК в
стве, превышающем исходное в миллион раз и более. Следоват<
исходно требуется незначительное количество материала. Для
дения ПЦР нужно знать нуклеотидную последовательность
фицируемого фрагмента.В соответствий с нуклеотидной последовательностью КОНЦ0ІІ
3' исследуемого участка синтезируется два олигонуклеотидных
мера (затравки). Длина праймеров составляет 20-30 нуклеотид^
Процесс амплификации состоит в повторяющихся ци1
Каждый цикл включает 3 стадии: температурную денатуїЕ
Nil f). Лабораторные методы диагностики375Исхоймая ДНКс=) м
ПраймерыСйнтеэйрувмая ДНКДенатурация ДНК,
отжиг праймеров,
полимеризацияt Денатурация ДНК.
X отжиг праймеров,полимеризацияіО и»Денатурация ДНК.
Ni отжиг праймеров,
полимеризация{. 9.13. Полимеразная цепная реакция (ПЦР)1НК (разделение двухцепочечной ДНК на одноиетючечные моле-
^лы) присоединение праймеров к комплементарным последо-
цельностям одноцепочечных молекул (отжнг) синтез поли¬
су к л еотидных цепей на однопепочечных молекулах в границах
Ірисоедйиенньїх праймеров с помощью полимеразы (рис. 9.13).ПЦР можно подробнее прочитать в на компакт-диске в статьеIV. Охапкиной «ПНР — основа современной генодиагностики».Рестрикция ДНК яа фрагменты -- необходимый этап молекулярно-
ЇМСТИЧЄСК0Й диагностики, осуществляется рестриктазами, относя¬
щимися к группе бактериальных эндонуклеаз. В генетике челове-
используют несколько десятков разных рестриктаз (EcoRl, Rsal,
Ipall, Ksp22I и др.). Они способны разрывать двухцепочечную ДНК
пределах строго определенных для каждого фрагмента последова-
сльностей нуклеотидов протяженностью 4-6 пар оснований (редко
зльше). При обработке геномной ДНК определенной рестриктазой
376Клиническая гвн<получается закономерный для данного фермента набор фрагмеї
различной длины.Электрофорез фрагментов ДНК обеспечивает разделение этих
ментов при их распределении на поверхности агарозного или пс
криламидного геля. Фрагменты ДНК движутся в геле, помещеі
в постоянное электрическое поле, от отрицательного полюса к
жительному в зависимости от размеров (чем больше относител!
молекулярная масса фрагмента, тем медленнее он движется в
трическом поле). После окончания электрофореза каждый фраги
ДНК занимает определенное положение в виде дискретной поло(
конкретном месте геля. Длину каждого фрагмента можно опредс
путем сравнения пройденного фрагментом расстояния с расстоян!
пройденным стандартным образцом ДНК с известными размерам
Визуализация и идентификация фрагментов ДНК в геле становятся.
конечным этапом диагностики, либо элементом дальнейшего аналі
Визуализация фрагментов ДНК после ПЦР осуш.ествляется с{
нительио легко. После окончания ПЦР проводят электрофора
агарозном геле, после чего гель обрабатывают этидия бромиі
который связывается с ДНК. При ультрафиолетовом облуче(
поверхности геля выявляется свечение в красной области спект[
Разработаны и другие методы окраски ПЦР-фрагментов. В
торых вариантах методов возможна автоматическая регистрі
результатов.Идентификацию конкретных фрагментов в геле среди генол
ДНК провести труднее. Из-за больших размеров генома челе
после рестрикции образуется так много рестриктных фрагмеї
что агарозный гель после электрофореза и окраски этидия бромі
в ультрафиолетовых лучах выглядит более или менее равноь
окрашенным, поэтому специфические фрагменты ДНК выяв;
путем блот-гибридизации по Саузерну. Эта методика состоит из
дующих этапов (рис. 9.14).— После окончания электрофореза гель помещают в рао1
основания (щелочи), в котором двухцепочечные фрагм<
ДНК теряют связи и становятся одноцепочечными.— Перенос ДНК с геля на нитроцеллюлозный или нейлоне
фильтр производится в буферном растворе. Непосредств!
на поверхность геля кладут фильтр и стопку фильтровал!
бумаги, В результате капиллярного эффекта создается'
буфера, перпендикулярный плоскости геля. Вьтмьіваемаї
9. Лабораторные методы диагностики377Ijln 1>>1|
нппиМеченыйДНК-зондЛигорадиографияДетекция флуоресцентной метки9.14. Блот-гибридизация по Саузернугеля ДНК задерживается фильтром и практически полностью
оказывается на его поверхности. После переноса одноцепочеч¬
ные нити фиксируют на фильтре. Расположение фрагментов на
фильтре точно соответствует их расположению в геле.- Для того чтобы визуально выявить нужные фрагменты (фик¬
сированная на фильтре ДНК не видна), проводят гибридиза¬
цию со специфическим по нуклеотидной последовательности
меченым радионуклидом или флюоресцентной меткой оли-
гонуклеотидным синтетическим зондом (такой зонд состоит
из 16-30 пар оснований) либо клонированным фрагментом
ДНК. Нуклеотидная последовательность зонда должна быть
378Клиническая геніїполностью или частично комплементарна изучаемому учаСІ
геномной ДНК.- При инкубации фильтра с раствором, содержащим
ный зонд, происходит гибридизация комплементарных цв1
ДНК зонда и фрагмента на фильтре. Неспецифически
занные молекулы зонда отмываются с помощью специалы
процедуры. Радиоактивно меченные участки выявляют пу1
экспонирования фильтра с рентгеновской пленкой (аутораї
ография). После проявления на пленке видны полосы ме*
ной зондом ДНК. Нерадиоактивные метки визуализир)
с помощью флюоресценции или опосредованно с П0М0І
антител.Методы ДНК-диагностики наследственных болезнейДНК-диагносгика бывает подтверждающей, пресимптомати<
ской. Пренатальной, а также ДНК-диагностикой носительства.Принципиально различают прямую и косвенную Д1
диагностику моногенных наследственных болезней. Прямые м|
ды возможны лишь при условии, что ген заболевания клониро!
известна его экзон-интронная организация или нуклеотидная П(
довательность полноразмерной комплементарной ДНК. При пряї
диагностике предметом анализа являются мутации гена.Однако для ряда наследственных заболеваний ген не клониро!
или заболевание является генетически гетерогенным, т.е. обусі
лено повреждениями в разных генах, либо молекулярная органі
ция гена не позволяет использовать прямые методы. Эти трудж
можно преодолеть с помощью косвенных методов ДНК-диагност1
основанных на использовании сиспленных с геном полиморфі
маркеров. В этом случае определяется гаплотип хромосомы, несуі
мутантный ген в семьях высокого риска, т.е. у родителей больної
его ближайших родственников. Такой подход возможен практиче<
для всех моногенных заболеваний с известной локализацией геніПрямые методы диагностики мутацийВ ДНК-диагностике в настоящее время используются два
прямых методов, проще всего обнаруживаются мутации, измені
щие длину амплифицированных фрагментов ДНК, которые выя1
ются при электрофоретическом анализе.Методы детекции известных мутаций. Если мутации известны,
можно их выявлять либо с помощью ферментов-рсстриктаз, KOTOJ
Пиша 9. Лабораторные методы диагностики379EcoRIt■ GAATTC-
•СТТАА^-EcoRIEcoRIt’GAGTTC-
■ CTCA/^G-EcoRIПолученные фрагменты
і іG + ААТТС 3'СТТАА G 5'GAGTTC-CTCAAG-Злектрофорез ДНК4EcoRIНорма Мутация
Рис. 9.15. Рестрикционный анализ с фер\тентом EcoRIраспознают строго определенные нуклеотидные последовательности,
либо на основе ДНК-гибридизации.Рестрикционный анализ — наиболее простой метод прямой детек-
иии мутаций (рис. 9.15). Его суть состоит в том, что рестрикцион¬
ные эндонуклеазы (бактериальные ферменты) разрезают двойную
МИТЬ ДНК в определенных последовательностях из 4—8 нуклеоти¬
дов. Разрезанные участки мутантной ДНК, отличающиеся по длине
от нормальных участков, выявляются на электрофореграмме. Если
И состав сайта рестрикции входит полиморфный нуклеотид, эту
мутацию можно выявить абсолютно достоверно. Если полиморфные
380Клиническая rt>нуклеотиды лежат в неузнаваемых рестриктазой участках, то
рестрикционного анализа неприменим.Лллельспецифичная ПЦР используется для выявления ТОЧІ
мутаций, небольших делеций и инсерций в исследуемых генах,
позволяет многократно увеличить уникальную последователі
ДНК, а затем проанализировать ее на предмет мутации. С пом<
специфических олигонуклеотидных праймеров проводят амги
кацию кодирующих участков геномной ДНК.На рисунке 9.16 представлены результаты ДНК-диагностики в
с мальчиком, страдающим муковисцидозом. Молекулярная стру
этого гена хорошо известна, ПЦР-амллификаиия проведена по
(сайту) расположения мутации Р508ъ гене муковисцидоза у детей и
телей. Как видно из электрофоретической картины, отец, матьи дочь|
здоровы) имеют две полосы (98 и 95 пар нуклеотидов), следовательн(
гетерозиготы, у больного мальчика одна полоса (95 пар нуклеотидої
он гомозиготен по мутантному аллелю F508. Обычно параллельно
реакции с праймерами, соответствующими мутантному и нормалі
аллелям. Использование нормального праймера служит иоложитсл!
контролем, свидетельствующим о нормальном ходе амплификации^
В 1988 г. был предложен еще один метод обнаружения мутаЦІІ
мультиплексная ПЦР. Метод позволяет проводить одновреї
несколько амплификаций в одной пробирке. Это осущест(
ся путем добавления в реакцию сразу нескольких пар прайїМультиплексная ПЦР н(
применение во многих
медицины: детекния муті
скрининг, обнаружение полі
физмов, диагностика инфек(
ных заболеваний. За счет одні
мснного проведения нескс
реакций в одной пробирке]
ный метод позволяет экон<
время и деньги. Однако, нес*
на явные преимущества,
типлексная ПЦР требует
тельной оптимизации прої
(подбор температур отжига
меров, концентрации реакт!
параметров амплификации)»-98Ьр-9ЙЬрРис. 9.16. Д Н К-диагностика муковис-
цидоза с помощью технологии поли¬
меразной цепной реакции (ПЦР) в
сайте ДР508. Объяснение в тексте
9. Лабораторные методы диагностики381‘^їитічивает ее широкое применение. Информация о методе муль-
ІІІІИ КСНОЙ ПЦР более подробно изложена на компакт-диске в статье
Полякова с соавт. «Применение мультиплексной полимеразной
цтой реакции для диагностики наследственных заболеваний».I І;іряду с двумя разобранными выше прямыми методами детекции
ЦИЧ1НЫХ мутаций есть еще несколько методов, не менее точных,
(I оолее трудоемких. Это гибридизация с аллельспецифичными
пионуклеотилами, аллельспецифичная лигазная реакция, мини-
Ціпснирование, пиросеквенирование. Характеристики этих методов
ижмо найти в специальной литературе.И начале 90-х годов XX в. был предложен принципиально новый
идчод в диагностике заболеваний с помощью ПЦР изобретена
UU’ » реальном времени. Суть метода заключается в том, что благодаря
ікі.іішению в пробирку специальных флуорофоров можно обнаружи-II |> ПЦР-продукт на протяжении всей амплификации. Флуорофор
;)|>;1ивается в двухцепочечную ДНК и приобретает способность флюо-
[!1 пировать, что детектируется прибором. Чем больше синтезировано
-продуктов, тем более интенсивная наблюдается флюоресценция.
Другой подход заключается в том, что к олигонуклеотиду при-
цкмишют флуорофор и гаситель флюоресценции. Если происходит
Гибридизация, то флуорофор отделяется от гасителя и фиксируется
|)Л1<юресценция.11ЦР в реальном времени позволяет не только обнаруживать ПЦР-
1|){)дукт еще во время амплификации, но также и определять его
Цоличество и исходную концентрацию ДНК. Данный метод в основ-
40М применяют для оценки экспрессии генов. Для этого из ткани
И.1ЛЄЛЯЮТ РНК, проводят реакцию обратной транскрипции (синтез
КДНК.) и далее ПЦР в реальном времени.Новые технологии позволяют проводить синтез кДНК и ПЦР
ідповременно- Из преимуществ можно отметить высокую чувстви-
[тсльность метода, небольшое количество требуемой РНК (или кДНК),
кісутствие необходимости проводить электрофорез после реакции,
[йизможность одновременно проводить несколько сотен реакций на
шлпом приборе. Из недостатков метода следует отметить следующее:
[он существенно дороже обычной ПЦР, требователен к качеству матри¬
цы, позволяет амплифицировать лишь небольшие фрагменты ДНК.В целом же ПЦР в реальном времени постепенно вытесняет обыч¬
ную ПЦР. На сегодняшний день данный метод является одним из
шмых используемых.
382Клиническая генеїВ последнее время эпигенетическим процессам придают болы
значение в этиологии многих болезней, R 1'ОМ числе и наследстмі
ных. Такие заболевания, как синдром Прадера—Вилли, Ангельмаі
Беквита—Видемана, связывают с нарушением метилирован!
Нарушение метилирования является также диагностическим крИ1
рием для многих онкологических заболеваний. В связи с этим 6j
предложена качественная реакция для определения метилирован
метилсцецифическая ПЦР. Суть этой реакции состоит в следз
щем. ДНК обрабатывают бисульфитом натрия, в результате чего
неметилированные остатки цитозина конвертируются в урацилу
метилированные не изменяются. После этого проводят ПЦР с прі
мерами, соответствующими метилированной и неметилировані
последовательности. По тому, с какой парой праймеров происхоі!
амплификация, можно судить о метилировании. Из недостаті
можно отметить сложность в подборе праймеров, неполную KOHI
сию, возможность оценки только единичных CpG-динyклeoти^
Однако метод относительно дешев, специфичен и прост, что ПОЗІ
ляет его широко использовать в диагностике злокачественных но|
образований и болезней импринтинга.Чувствительность и специфичность прямых методов диагност!
высокие. Наиболее распространены три метода: рестрикционн!
анализ, аллельспецифичная ПЦР и ПЦР в реальном времени. Bj
лаборант или исследователь в каждом случае останавливается
каком-то методе, а потом может перейти на другой. Все методы 0Ч1
точные. Они позволяют однозначно определять мутацию ДНК, д|
если разница составляет одно основание.Методы мутационного скрининга. Если характер мутации ив|
вестей, а клиническая картина заболевания позволяет преді
дожить, в каких генах могла произойти мутация, то в лаборат(
ной диагностике применяются следующие методы мутапионн<
скрининга:• анализ перестроек ДНК-блотингом по Саузерну;• анализ полиморфизма конформапии одноцепочечиой ДНК;• электрофорез двух цепочечной ДНК в градиенте денатуранта;• гетеродуплексный анализ;• денатурирующая высокоэффективная жидкостная хроматог|
фия;• химическое обнаружение неспаренных нуклеотидов;
|/iiina 9. Лабораторные методы диагностики383Чувствительность скрининговых методов обнаружения мутаций
1(1' абсолютна. Однако соотношение между их инфорлтативностью и
Іі ниімостью достаточно высокое, поэтому они широко используются
\\ практике. В то же время их отрицательная сила невелика, т.е. если
iti' обнаружено ИЗМЄІІЄНИЙ, это не значит, что их на самом деле нет.Ниже будут приведены характеристики некоторых методов мута-
щюиного скрининга.SSCP {Single Strand Conformation Polymorphism) — метод анализа
коїіформационного полиморфизма однонитевой ДНК — основан на
|)1ч истрации различий в электрофоретической подвижности однони-
1Г11ЫХ ДНК, одинаковых по величине, но различающихся вследствие
нуклеотидных замен по пространственной организации молекул
(рис. 9.17). Конформация небольших однонитевых ДНК зависит
и1 нуклеотидной последовательности, поэтому замена даже одно-Участоккомплементарности. внутри однойДНКі/В результате мутации
образование внутри-
цепочечного дуплекса
нарушаетсяI—Электрофорез ДНК
в неденатурирующем гелеНорма МутацияНорма +
мутацияРис. 9.17. Анализ полиморфизма конформации (SSCP) одноцепочечной
ІДНК.
384Клиническая геніїго нуклеотида приводит к изменению пространственной струї
ры. Метод включает амплификацию фрагментов ДНК размероц
300 пар нуклеотидов, денатурацию продуктов ПНР и высокор*
шаюший электрофорез в полиакриламидном геле.Конформационный метод выявления точковых мутаций
чил широкое распространение вследствие относительной просі
и способности обнаруживать любые типы замен. Ограничен!
является размер исследуемого фрагмента ДНК, так как высс
эффективность детекции мутаций, составляющая 80—90%, покї
при длине фрагментов менее 200 пар нуклеотидов, а для фрап
тов более 400 пар нуклеотидов вероятность обнаружения мутв
уменьшается до 50%.В настоящее время разрешающая способность метода значит!
но повышена. В частности, разработаны подходы для идентифї
ции точковых мутаций методом SSCP-анализа в амплифицирої
ных фрагментах ДНК размером до 800 пар нуклеотидов. Для
используется полиакриламидный гель с низким pH.НА {Heteroduplex Analysis) — гетеродуплексный анализ позволяет В|
лять мутации, находящиеся в гетерозиготном состоянии, а также и1
ции и делеции. Принцип этого метода заключается в следующем.При амплификации фрагментов генов гетерозигот, последуі
денатурации и медленной ренатурации полученных продуктов
в амплификационной смеси наряду с двумя типами гомодуплві
образуются гетеродуплексы между нормальной и мутантной
ми ДНК (рис. 9.18). Такие гетеродуплексные молекулы отличаї
по электрофоретической подвижности от гомодуплексов из-за
формационных особенностей в местах несовпадения нуклеон
поскольку электрофоретическая подвижность гетеродуплексов,
чительно ниже, чем гомодуплексов. Эти различия обнаруживе
при электрофорезе в обычном полиакриламидном геле.Наиболее распространенный способ скрининга мутаций —
бинация анализа гетеродуплексов и метода однонитевого конфс
ционного полиморфизма, позволяющая выявить точковые муті
почти в 100% случаев и не требующая больших затрат времени.DGGE {Denaturating Gradient Gel Electrophoresis) — денатурируї
градиентный гель-электрофорез (рис. 9.19). ДНК-дуплексы поді
ются миграции в геле с градиентом денатурирующих условий (мс
использовать и температурный градиент). Миграция продолж!
до тех пор, пока ДНК-дуплексы не достигают в геле точки плав;
Ийка 9. Лабораторные методы диагностики385Денатурация ДНКнагреваниемITГомодуплексыіОхлаждение,G,Г етеродуплексыфс;IшXсгкSи<йе-5Норма Мутация Норма +
мутацияяс. 9Л8. Гетеродуплексный анализ (НА)
386Клиническая г«£шу;Рис. 9.19. Электрофорез в градиенте денатуранта (DGGE)и не разделяются, после чего миграция фрагментов останавлі
ся. Однонуклеотидные различия в ііормальной и тестируемой
выявляются по различной электрофоретической подвижности в
Высокая чувствительность метода (95%) достигается благодаря*
цифическим праймерам с так называемым GC-зажимом, пі
ленным чередованием гуанина и цитозина в пределах до 20 нуі
тидов. В результате температура плавления продукта амплифиі
сильно увеличивается, что повышает эффективность опреде;
мутации. Однако праймеры с GC-зажимом достаточно дороги, п<
му применение метода ограничено.ССМ {Chemical Cleavage of Mismatch) — метод химического расі
ния несларенных оснований. Метод основан на гибридизации pi
активно меченой ДНК-пробы с тестируемой ДНК. Места ощ|
затем выявляют с помощью серии химических реакций (модис
ция с использованием тетрахлорида осмия), которые происхс
однонитевой ДНК в сайтах неправильного спаривания. Этот
можно применять для тестирования фрагментов ДНК размерої
1000 пар нуклеотидов, он выявляет локализацию ошибки и до|
но чувствителен. Однако метод не нашел широкого распростї
ния вследствие токсичности химических реагентов и методич€
сложности. Подобное расщепление неспаренных нуклеотидов М1
быть и энзиматическим, что позволяет исключить ИСПОЛЬЗО!
ія я. Лабораторные методы диагностики387;||‘тых химикатов. Метод основан на расщеплении неспаренных
ІМИІІІИЙ в гетеродуплексе, образуемом между тестируемой ДНК и
Іміии.іюй последовательностью посредством таких ферментов, как
фага Т4 или эндонуклеаза VTI. Однако этот метод еше более
|)|р|ииый», чем ССМ.іиключительньш этап анализа мутаций — их секвенирование,
ипрсделение нуклеотидной последовательности фрагмента ДНК,
И1 іаишего аномальную электрофоретическую подвижность,
іиїдовательность нуклеотидов этого фрагмента сравнивается с
>М1)й, в результате чего патология приобретает свою окончатель¬
на) характеристику.Меюл секвенирования является «золотым с гандартом» молекуляр-
|И і еметики. Поиск новых, редких и подтверждение известных мута-
UI можно провести с помощью этого метода. Диагностика многих
Л»к‘дственных заболеваний включает проведение секвенирования:
ґлрасположснность к раку яичников и молочной железы {BRCA1 и
U'A2), нейрофиброматоз, фенилкетонурия, муковисцидоз и другие.
М\ отсутствии мажорных мутаций, приводящих к наследственному
'ко.иеванию, часто секвенируют ген на предмет новых мутаций. Для
|коюрых генов, имеющих небольшие размеры, прямое секвенирова-
HV с успехом применяется как основной метод сканирования мута-
1Й. Гак, в частности, особенно удобным оказалось его применение
И) детекции мутаций в сравнительно небольших генах, таких, какII фактора ТХ свертывания крови (гемофилия В).Любые типы мутаций можно обнаружить путем прямого секвени-
)1»ания мутантной ДНК или отдельных экзонов. Первичный поиск
Ірушений в кодирующих областях гена часто осуществляют именно
ІКИМ образом.Разработанные в последние годы модификации методов ПЦР зна-
мельно облегчили секвенирование амплифицированных фрагмен-
)и и повысили эффективность секвенирования. Так, в частности,
Ірсяложен вариант асимметричной ПЦР, когда при амплификации
^)ицен грация одного из олигопраймеров в несколько десятков раз
Ірсвосходит концентрацию другого праймера, в результате чего син-
?іируется преимущественно только одна, нужная для секвенирова-
ІИЯ цепочка ДНК.Один из существенных недостатков технологии секвенирова-
|ия -- его дороговизна. Однако применение новых методик позво-
ІИСТ из года в год снижать стоимость секвенирования.
388Клиническая г<В настоящее время всё большее распространение получаї
венаторы нового поколения, которые обеспечивают паралл!
секвенирование сотеи тысяч последовательностей. В подавлй!
большинстве случаев это дорогостоящее оборудование испол!
в научных целях. Однако стремительное развитие данного Н1
технического направления приводит к неуклонному повьм
производительности и снижению стоимости оборудования и
ведения анализа. В ближайшие годы можно ожидать приме!
секвенаторов в клинических целях — для неинвазивной преж
ной диагностики, быстрого и недорогого определения всех И9
ных моногенных заболеваний, определения сотен иолиморф»
связанных с рисками многофакторных болезней.В табл. 9.3 представлены характеристики разных скрині
вых методов ДНК-диагностики, Врач лаборант-генетик эа|
определяет стратегию поиска в соответствии с оснащенн<
лаборатории.Таблица 9.3. Эффективность основных скрининговых методов ДНІ
гностикиМетодРазмерпродукта,п.н.Эффективность
определения
мутаций,%Токсич¬ныехимикатыОпределениелокализациимутацииПреимИТВМнедостмSSCP180-25080-85НетНетЛегко, щНА200-25080-85НетНетЛегко, Д||DGGE60095ЕстьНетДороЯпрайм|ССМ1700>95ЕстьЕстьСложЯэкспермтальмСеквени¬рование600>99НетЕс'іьДорогиярудоваїреакт|іПримечание. ССМ — метод химического расщепления несларснных ОС
ний (Chemical Cleavage of Mismatch); DGGE — денатурирующий градиеі
гель-электрофорез (Denaturating Gradient Gel Elcctrophoresis); HA —
дуплексиы]1 анализ (Heteroduplex Analysis); SSCP — метод анализа КС
мационного полиморфизма однонитсвой ДНК (Single Strand Conforl
Polytnorphism); п.н. — пар нуклеотидов.
m U. Лабораторные методы диагностики389Многие мутации прерывают синтез белкового продукта. В этих
f*m\x образуются укороченные полипептидные цепи, функпиональ-
ипиачимые. Для диагностики таких мутаций можно применять
«д трансляции белкового продукта. Он проводится in vitro на основе
1 ученной специфической мРНК с добавлением лизата ретикулоцитов
tu*M содержатся все необходимые компоненты для синтеза белка).
|1«П(»й системе синтезируется белковый продукт соответствующего гена,
ил у КТ трансляции анализируют с помощью электрофореза. Изменение
I рофоретической подвижности белка свидетельствует о наличии
пинии (нонсенс-мутация, нарушение сплайсинга РНК, сдвиг рамки
(Ип.пшния), приводящей к обрыву синтеза полипептидной цепочки.и связи с бурно развивающимся направлением по изучению гене-
|»К‘ского полиморфизма и его роли в предрасположенности к много-
1к Iорным заболеваниям разработаны или усовершенствованы новые
гтды детекции разных вариантов генетических полиморфизмов,
ним можно отнести следующие методы, которые предназначены
hi автоматического или полуавтоматического исследования часто
Ирсчающихся полиморфизмов:»денатурирующая жидкостная хроматография высокого раз¬
решения — DHPLC (Denaturation High Performance Liquid
Chromatography);• методы биочипов и др.Метод DHPLC фактически является вариантом метода гетеро-
^плексного анализа ДНК с последующим автоматическим учетом
ічультатов с помощью жидкостного хроматографа. Его чувстви-
лыюсть и специфичность близки к 100%. Этот метод позволяет
іределять ОНП, делении и инсерции длиной до 1500 п.о. в течение
1^3 мин. Его широко применяют для генотипирования ОНП и в
)угих разделах изучения генетического полиморфизма, в том числе
1я картирования генов.Методы биочипов в последние годы заняли существенное место в
інонаправленньїх молекулярно-биологических исследованиях.
Чипы представляют собой миниатюрные гелевые пластинки с
іногочисленньїми правильно расположенными углублениями или
І'іейками на стекле или какой-либо мембране. В углубления (ячейки)
помещаются иммобилизованные зонды: ДНК, РНК, белки, клетки.зависимости от этих зондов различают: ДНК-чипы, РНК-чипы,
JCilKOBbie микрочипы, клеточные микрочипы.
390Клиническая riВ клинической и медико-генетической практике наиболыт
пространение получили ДНК-чипы. Они исполъ:зуются для 41
спектра мутаций или аллельных вариантов разных генов,
чипа заполняются (иммобилизуются) ДНК-зондами применит
к поставленной задаче исследования. Микрочиповая техн(
позволяет проводить реакцию в микрообъемах, а, следоват
требуется незначительное количество материала, и в то же
проводить многосторонний анализ многих чипов одного и
объекта. Выбор зондов для иммобилизации и их расположение
программирует исследователь.Чувствительность метода высокая, сопоставимая с дру|
ДНКовьгми методами диагностики. С помошью ДНК-чипов можнС
лизировать транслокации, дупликации, делеции, как протяжен ні
и короткие. ДНК-чипы могут содержать несколько сотен ДНК-з<
что очень важно либо для массовых популяционных исследований'
го гена, либо для исследований сотен тысяч генов одного индиви;Технология приготовления микрочипов в настоящее время
мерциализирована, и многочисленные фирмы (в том числе в
могут по заказу подготовить любой вариант чипа.Интерес к использованию микрочипов в исследовательской
тике в последнее время снизился, а в клинической практике поі
ется, особенно в связи с поиском ассоциаций генетических полі
физмов с заболеваниями (предсказательная медицинская практ
Создаются биочипы для диагностики конкретных заболеваний^
поиска полиморфных систем генов детоксикации и фармакогеї
чески оправданного лекарственного лечения лейкозов, туберкуі
бронхиальной астмы. Для более подробного ознакомления см.
тью М.В. Голубенко с соавт. «ДНК-чет для исследования стру»
наследственной предрасположенности к сердечно-сосудистым
леваниям» на компакт-диске.Другие методы или варианты вышеописанных методов pacnd
вания мутаций (метод поверхностного плазмонового резонаї
surface plasmon resonance, метод масс-спектрометрии, метод «Ж1
биочипов», секвенирование ДНК с помощью нанопор), хотя и
но трудоемкие, и в клинической практике используются толі
узких целях (см. раздел по биохимической диагностике).Косвенное выявление мутацийКогда нуклеотидная последовательность гена еще неизвест}
вместе с тем имеется информация об относительном положении
Лабораторные методы диагностики391true г и ческой карте, применяют косвенное выявление мутаций,
ііічсски это соответствует диагностике с помощью метода сце-
НІІИІ генов.Кі>і-і$снная ДНК-диагностика, по существу, сводится к анализу
Щморфных генетических маркеров у больных и здоровых членов
І.ІІ. Эти маркеры должны быть расположены в том же хромосом-
регионе, где и ген болезни, т.е. они сцеплены. Такими маркерами
ІУI быть участки ДНК, существующие в популяции в нескольких
И' и.мых вариантах. Различия могут быть по составу нуклеоти-I, 110 числу динуклеотидных повторов. На основе вариабельности
мина маркерных участков ДНК можно дифференцировать мате-
кос или отцовское происхождение конкретного варианта мар-
сцепленного с геном болезни. Сцепление означает, что маркер
ІГИ болезни располагаются близко друг к другу; они передаются
fiiDC іаве одного хромосомного сегмента. Благодаря анализу поли-
||цых генетических маркеров можно проследить в ряду поколе-
1Й наследование каждой из родительских хромосом.Технические приемы в косвенной диагностике те же самые, что
и прямой (получение ДНК, рестрикция, электрофорез и т.д.).
м сственно, к этому добавляется математический анализ сцепления
Иниаков.Использование косвенных подходов оказалось возможным бла-
Ш!1ря существованию в геноме полиморфных участков (локусов)
[НК. Нуклеотидные замены достаточно часто встречаются в неко-
ірующих участках ДНК. Значительное число нуклеотидных замен
Іриіюдит к изменению мест рестрикции. Эти изменения можно
ікнить с помощью блот-гибридизации по Саузерну, поскольку
і:імсняется длина рестриктных фрагментов. Эта разновидность
Х'іиморфизма ДНК получила название полиморфизма по длине
К‘триктных фрагментов.Расположенный вблизи изучаемого гена или внутри него поли-
Юрфный сайт может служить маркером аллельных вариантов этого
5на, в том числе маркером патологических мутаций.Полиморфизм, обусловленный нуклеотидными заменами или
Велециями, как правило, диаллелен, а, значит, его информационная
ценность ограничена. Более информативны кластеры тандемных
Юнторов, которые обусловливают полиморфизм по количеству копий
NVNTR — variable number of tandem repeats), так называемый полимор-
Іфизм мини- и микросателлитных последовательностей.
392Клиническая riСемья А
М Р оСемья В
М Р ОМикросателлиты —
кие тандемные повторы,
двугекса нуклеотидные,
распространенный из
СА-повтор. Показано, ЧТ<
стары СА-повторов ветре*
в среднем 1 на 30 ООО пар И}
тидов. Они локализован!
правило, в некодирующщ
он ах ДНК.. Блоки СА-п<
имеют менделевское наслК
ние в семьях и не обнар)
ют новых мутаций (рис,
Немаловажным положит*
фактором является отноС!
ная простота обнаружения:
повторов в геноме чел<
Кроме СА-повторов, досі
но распространены GA-n(
и другие кластеры тан;
повторов (ТТТА)хп, (ТС1
(ТТТС)хп, также обнаруз
щие вариабельность по
повторов. Широкая pacnj
ненность в геноме (частота различных микросателлитов,
вместе, составляет I на 6000 пар нуклеотидов) и высокий yj
полиморфизма делают микро- и мини-сателлиты идеальными!
морфными маркерами для картирования генов наследственньи
леваний и проведения косвенной ДНК-диагносгики.Полиморфные ДНК-маркеры и интегральная карта их pj
жения позволяют определить и проследить в поколениях хр<
му, несущую патологический ген, а также подробнейшим о(
охарактеризовать определенный хромосомный район, выявит!
микроскоиические перестройки, определить наименьший ра(
перекрывания и локализовать ген-кандидат, ответственный
левание.Основной недостаток косвенных методов диагностики
зательное предварительное изучение генотипа (гаплотипа) хс
одного пораженного родственника. В случае отсутствия пора)Рис. 9.20. Семейный анализ фраг¬
ментов ДНК с помощью блот-
гибридизации по Саузерну: М —
мать, Р — ребенок, О — отец
lit Лабораторные методы диагностики393jriш'мпикои, доступных для обследования, диагностика (за ред-
|м исключением) становится невозможной.|1і;ік, существует достаточно много молекулярно-генетических
диагностики наследственных болезней. Эти методы ока-
ІИі I. настолько универсальными, что нашли применение не
Л».кп в медицинской генетике, но и диагностике инфекцион-
h *;іГ)0леваний. Каждый из представленных в табл. 9.4 методов
много вариантов. Одни и те же болезни можно диагно-
(ИР‘яиіть разными методами. Можно диагностировать болезнь
likL- н трудных случаях (невозможность обследования родителей,
|лос количество биологического материала, отсутствие сведе-
|И І) 1'СНС и т.д.).їлица 9.4. Сравнительные характеристики прямых и непрямых методов
ДНК'ДиагностикиПрямые методыКосвенные методыII (|(.‘имуществанедостаткипреимуществанедостатки||1 1МОЖНЫ при
||1М11ИТеЛЬНОМ
ІНіи позеТребуют знания
генаНе требуют зна¬
ния гена и мута¬
ции в немТребуют абсо¬
лютной уверен¬
ности в диагнозеІіиможньї при(ультилокусном|Оо.і[еванииНе всегда
информативныИнформативныдля всех семейПрименимы
только для
моналокусных
заболеванийlit іможна бес-
!|ю6андпая диа¬
гностикаОбязателен
семейный ана¬
лизИся методология молекулярно-генетической диагностики наслсд-
hicHHHX болезней представлена на рис. 9.21 в виде алгоритма.Поскольку генодиагиостика проводится в специализированных,
)рошо оснащенных лабораториях и сопряжена с немалыми рас-
Д11МИ, для проведения анализа должны быть строгие клинические
жазания. Лечащий врач и врач-гснетик совместно составляют схему
5слсдования больного. Такое взаимодействие, как правило, обеспе-
рииает успех.Автоматизация сушсствующих методов и разработка принципи-
Льмо новых подходов к изучению структуры нуклеиновых кислот
394КлиническаяРис. 9.21. Алгоритм генодиагностики наследственных заболеванийнаряду с ускоренными темпами изучения генома человека и
рования генов, ответственных за развитие моногенной пат(
позволяют прогнозировать появление в недалеком будущем О]
диагностики большинства известных наследственных бо/
человека.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯАсинхронность репликации ДНК
Блот-гибридизация по Саузерну
Высокоразрешаюшие цитогенетические методы
Диагностика путем анализа полиморфизма по длине рестрї
фрагментовДиагностика путем секвенирования гена
Интерфазная цитогенетика
!». Лабораторные методы диагностики395fjliitiopai орная идентификация ступеней болезни
Мікч.о»ыс просеивающие программы• Міисриал для цитогенетических методов
Мі’юдические условия цитогенетических исследованийо IIII го нуклеотидные зонды
ІІОКІГШНИЯ для биохимических исследований
Міжа іания для цитогенетических исследований
Полимеразная цепная реакцияПринципы биохимической диагностики наследственных болезнейП|и)сеивагощий подходПрямая детекция мутаций (варианты)(Ч'лективные диагностические программы
( псцифические зонды ДНК{'>чцность молекулярно-генетической диагностики
Мсчод флюоресцентной гибридизации in situРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАИаіідакова LB,, БукинаА.М., Гончаров В.М, идр. Диагностика наслед-
*синых болезней обмена веществ на основе сочетания методов так¬
ім ной масс-спектрометрии и энзимодиагностики // Медицинская
^сгика. - 2005. - Т. 4. - № 1. - С. 28-32.існетический паспорт — основа индивидуальной предиктивной
1ЛИЦИНЫ / под ред. B.C. Баранова. — СПб.; Изд-во Н-Л, 2009. —
17 с.Захаров Л.Ф., Бенюш В.А,, Кулешов И.П. Хромосомы человека;
Глнс. — М.: Медицина, 1982. — 263 с.Иллариошкин С.Н, ДНК-диагностика и меди ко-генетическое кон-
IfjibTMpoBaHHe. — М.; МИА, 2002, — 591 с.Клиническая биохимия / под ред. В.А. Ткачука. — М.: ГЭОТАР-
Ісдиа, 2008. — 264 с.Коряков Д. Е., Жимулев И.Ф. Хромосомы. Структура и функции. —
Іопосибирск; Изд-во СО РАН, 2009. — 258 с.Краснопольская К.Д, Наследственные болезни обмена веществ.
Справочное пособие для врачей, — М.: РОО Центр социальной адап-
щии и реабилитации детей «Фохат», 2005, — 364 с.; ил.ПЦР в реальном времени / под ред. Д.В. Ребрикова. — 2-е изд., доп.
іісрераб. — М.; Бином. Лаборатория знаний, 2009. — 221 с.
396Клиническая г<Рубцов Н.Б. Методы работы с хромосомами млекопитающихИ
пос- — Новосибирск: Новосиб. гос. ун-т., 2006. — 152 с.Свердлов Е,Д. Взгляд на жизнь через окно генома: Т. 1.
структурной молекулярной генетики. — м.: Наука, 2009. — 5J
Глава 10ПРИНЦИПЫ ЛЕЧЕНИЯ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ*ОБЩИЕ ВОПРОСЫ■)м11ирические попытки лечить больных с наследственной пато-
lifi ii, предпринимавшиеся в течение 200 лет вплоть до 30-х годовII, не дали положительных результатов. Диагноз наследственной
і)ігти оставался приговором больному и его семье: такие семьи
|Иі;иіи вырождаю!гшмися. Эта позитшя в медицине в первые дссяти-
^гии XX в. опиралась, по-видимому, также на генетическую конпсп-
|1о об очень строгой детерминации менделирующих наследственных
^шпаков. В связи с этим в начале XX в. возникла негативная евге-
|Н11, призывавшая насильственно ограничить деторождение у лиц
Иііследстіїенной патологией. К счастью, практическая реализация
>итшной евгеники была недолгой из-за общественного давления.Переломным периодом в отношении лечения наследственных
5л1‘ !ней можно считать 20—30 годы, так, в середине 20-х годов в
Кспериментах на дрозофиле были по.тучены факты, показываю-
|11с разную степень проявления действия генов в зависимости от
Щяпия генотипической или внешней среды. На основе этих фактов
,1ли сформированы понятия о пенетрантности, экспрессивности и
ІК'пифичности действия генов. Стала возможной .тогическая экс-
)1111оляция: если среда влияет на экспрессивность генов, то, следо-
ІЙісльно, можно уменьшить или исключить патологическое действие
5І10В при наследственных болезнях. На основе этих положений
рыпающийся русский биолог Н.К. Кольцов предложил и обосновал
Цоиое направление в медицинской генетике — евфенику — учение о
Ііпроіііем проявлении наследственных задатков. По его мнению, евфе-
1И ка должна изучать все условия срсды, стимулирующие проявления
Положительных и непроявлсния отрицательных (наследственные
іолсзни) наследственных свойств.♦ Исправлено и дополнено при участии д-ра мед. наук, проф. А.Ю, Аса-
398Клиническая гіВпервые в мире невропатолог и генетик С.Н. Давиденков,
ваясь на собственном клиническом опыте и достижениях экспв;
тальной генетики, в начале 1930-х годов указал на ошибочност!
ния о неизлечимости наследственных болезней и вырождении
такими болезнями. Он, как и Н.К. Кольцов, исходил из признанИ)
факторов внешней и внутренней среды в проявлении наел едет*
болезней. С.Н. Давиденков настаивал на принципиальных bo'JM(
стях вмешательства в функционирование патологических аллелей]
много сделал для разработки методов лечения наследственных б(
нервной системы. Такая исходная позиция позволяла разрабат
различные подходы и методы лечения лиц с наследственными
нями на основе достижений генетики, теоретической и клини»
медицины. Однако отсутствие сведений о патогенетических мв>
мах наследственных болезней в тот период ограничивало возмож|
разработки методов. Все подобные попытки, несмотря на прави^
теоретические установки, оставались эмпирическими.Лечение различных наследственных болезней может включа!
традиционные в медицине подходы (лекарственные препараты^
цифические диеты, хирургическую коррекцию и др.), так и
ствия на наследственные структуры, «повиниьте«> в развитии бо;
Уровни, на которые направлено терапевтическое воздействі
многом определяются состоянием знаний о первичном генетиЩ
дефекте, его клинических проявлениях, взаимодействии с фа|
ми среды и пониманием путей, на которых возможно испраї
дефекта. Обобщенная схема точек приложения лечебных воздей^
приведена на рис. 10.1.В настоящее время благодаря успехам генетики в целом и
ственному прогрессу теоретической и клинической медициныГЕНГенный продуктМетаболические Функциональные Структурные
эффекты эффекты эффектыРис, 10.1. Принципиальная схема «мишеней» для лечения наследств^
болезней
іиіі 10, Принципы лечения наследственных болезней3991'Игрждать, что уже многие наследственные болезни успешно лечат-
, Іакая установка должна быть у врача.(Инцие подходы к лечению наследственных болезней сходны с
їіім»дами к лечению болезней любой другой этиологии, при наслед-
fm nitb[x болезнях полностью сохраняется принцип индивидуализи-
[ИІ1ІИОГО лечения, ведь врач и при наследственной патологии лечит
ti(>0CT0 болезнь, а болезнь конкретного человека. Возможно, что
И1 наследственной патологии принцип индивидуализированного
ІЧГМИЯ должен соблюдаться еще строже, потому что гетерогенность
If ісдственньїх болезней далеко не расшифрована, а, следовательно,
1му и ту же клиническую картину могут вызвать разные наследствен-
|Ы<' болезни с различным патогенезом. В зависимости от условий
и постнатального онтогенеза, а также от всего генотипа человека
^гпптипические проявления мутаций у конкретного человека могут
ифицироваться в ту или другую сторону. Следовательно, необходи-
|м разная коррекция наследственной болезни у разных пациентов.Как и при лечении других хорошо изученных болезней (например,
Імфскционньїх), можно выделить 3 подхода к лечению наследственных
іиіи іней и болезней с наследственной предрасположенностью: симпто-
ІИІИ ческий, патогенетический, этиотропный. Применительно к наслед-
Iионным болезням в отдельную группу можно выделить хирургические
моды, поскольку иногда они выполняют функции симптоматической
?рапии, иногда — патогенетической, иногда — и гой, и другой.При симптоматическом и патогенетическом подходах используют
tc виды современного лечения (лекарственное, диетическое, рент¬
ген о радиологическое, физиотерапевтическое, климатическое и т.д.).
Генетический диагноз, клинические данные о состоянии больного и
|си динамика болезни определяют поведение врача на протяжении
ІССГО периода лечения с постоянным и строгим соблюдением гип-
цжратовского принципа «не навреди». При лечении наследственных
Полезней надо быть особенно внимательным в соблюдении этических
[к деонтологических норм: часто такие больные имеют тяжелую хро-
жическую патологию с детского возраста.СИМПТОМАТИЧЕСКОЕ ЛЕЧЕНИЕХотя неспецифическое лечение не является главным, оно фак"
іТимески используется всегда, в том числе при лечении пациентов с
Ниследственными болезнями. Симптоматическое лечение применяют
400Клиническая rwпри всех наследственных болезнях, даже если врач располагает
дами патогенетической терапии. Для многих форм наследсті
патологии симптоматическое лечение остается единственным,
Лекарственная симптоматическая терапия разнообразна и
от формы наследственных болезней. Один из древних примероі^
птоматической терапии, сохранившейся до наших дней, — примі
колхицина при острых приступах подагрического артрита. Таков
ние использовали еше греки в античном периоде. Другими приї
симптоматического лечения могут быть применение анальгетиі
наследственных формах мигрени, специфических транквилизато]:
психических проявлениях наследственных болезней, пpoтивocy^
ных препаратов при судорожных симптомах и т,д. Успехи этого
терапии связаны с прогрессом фармакологии, обеспечивающим всв1
широкий выбор лекарств. Вместе с тем расшифровка патогенеза к|
болезни позволяет понять причину возникновения симптома, а Н1
основе становится возможной более тонкая лекарственная кор|;
симптомов, если первичная патогенетическая терапия еаіе невозі
В качестве примера можно привести общую схему много»
нентного симптоматического лечения муковисцидоза. Перві
звено патогенеза (нарушение транспорта ионов натрия и хлора)
ригировать при этом заболевании еше не удастся.— В связи с тем, что у больных выделяется много хлорида HI
с потом, детям с муковисцидозом в жарком сухом климате ]
мендуется дополнительно добавлять поваренную соль в
В противном случае иногда может наступить коллапс с
вым ударом,— Недостаточность функции поджелудочной железы у б<
(рано или поздно это наступает) восполняется npenaf
сухих экстрактов поджелудочной железы животных ИЛИ(
ментов в капсулах (панкреатин, панзинорм*, фестал*) й'^
чегонных средств. При клинических признаках наруї
функции печени проводится курс соответствующей Тв1
(эссенциале*, метионин, холин и др.).— Наиболее серьезными и трудными для лечения ЯВЛЯІ
нарушения дыхательных путей. Закупорка просветов
бронхов густой слизью обусловливает развитие инфекї
легочной ткани. На закупорку бронхов и инфекцию нап;
на симптоматическая (почти патогенетическая) терапия,]
уменьшения обструкции применяют бронхоспазмолитичІ
ihii 10. Принципы лечения наследственных болезней401и отхаркивающие смеси (изопреналин, эуфиллин*, атропин,
эфедрин и др.), препараты муколитического действия, в основ¬
ном тиолы. Способ введения препарата (в ингаляциях, внутрь,
внутримышечно) зависит от выраженности клинической кар¬
тины. Применяют лекарства, уменьшающие внутриклеточную
продукцию слизи, например мукодин* (карбоцистеин).
Лечение воспалительных осложнений в легких при муковисци-
дозе представляет трудную задачу, поскольку эти осложнения
обусловлены несколькими видами бактерий, а иногда и грибов.
С этой целью проводят интенсивную микробиологически кон¬
тролируемую антибиотикотерапию (цефалоспорины третьего
поколения и др.), а также лечение фторхинолонами для борьбы
с синегнойной инфекцией. Антибиотики выбирают в зависи¬
мости от чувствительности микрофлоры. Наибольший эффект
дает введение антибиотиков в ингаляциях и парентерально.К'сік видно на примере лекарственного лечения муковисцидоза,
|1ииосимптомные болезни требуют применения нескольких фарма-
окинетически совместимых лекарств,( имптоматическое лечение бывает не только лекарственным.
ІНОГИЄ виды физических методов лечения (климатотерапия, бальне-
щс'іение, разные виды электротерапии, теплолечение) применяются
|ри наследственных болезнях нервной системы, наследственных
олсзнях обмена веществ, заболеваниях скелета. После таких курсов
Цсмсиия больные чувствуют себя намного лучше, продолжительность
жизни увеличивается.Практически нет таких наследственных болезней, при которых не
,1ло бы показано физиотерапевтическое лечение. Например, лекар-
Тпснное лечение муковисцидоза постоянно подкрепляется многообраз¬
ными физиотерапевтическими процедурами (ингаляции, массаж и др.).К симптоматическому можно отнести рентгенорадиологическое
ІСЧСНИЄ при наследственно обусловленных опухолях до и после
Іирургического вмешательства.Возможносп'и симптоматического лечения при многих болезнях
ІІЦС далеко не исчерпаны, особенно это касается лекарственной и
мистической терапии.Следует подчеркнуть, что симптоматическое лечение будет
использоваться в большом объеме и в будущем наряду с самым
роі5ерщенньш патогенетическим или даже этиотропным лечением
^следственных болезней.
402Клиническая гвиіПАТОГЕНЕТИЧЕСКОЕ ЛЕЧЕНИЕЛечение любых болезней путем вмешательства в патогенез
эффективнее, чем симптоматическое лечение. При наследствеї
болезнях патогенс'1 ические методы также наиболее обоснованы,
и НС противопоставляются симптоматическому лечению. По
изучения патогенеза каждой болезни появляются различные
можности вмешательства в этот процесс, в течение болезни ИІ
выздоровление. Клиническая медицина развивалась на основе
ретических представлений о патологических процессах. Таки!
путем идет клиническая генетика в разработке методов лечениЛЛДля патогенетического лечения наследственных болезнк
последние годы применяют принципиально новые подходы,
ванные на достижениях молекулярной и биохимической генет|
при описании генных болезней (см. гл. 4) приводились приї
расшифрованных нарушенных звеньев обмена, всех биохимичев
механизмов, по которым развивается наследственно обусловлен]
патологический процесс, — от аномального генного продукта до
нической картины болезни. Естественно, что на этой основе м<
целенаправленно вмешиваться в патогенез болезни, а такое леЧ(1
фактически равнозначно зтиотропному. Хотя первопричина
мутантный ген) и не устраняется, но иепь патологического прої
прерывается, и патологический фенотип (болезнь) не развивается]
происходит нормокоиирование),Патогенетическое лечение должно расширяться по мере прог]|
генетики развития. Пока ее вклад в разработку методов лечения нас
ственной патологии незначителен, хотя успехи последних лет не в1
вают сомнений, В настоящее время лечение основано на корреї
отдельных нарушенных звеньев, но более эффективно было бы вм^
ваться в патологический процесс на уровне системных реакций.При патогенетических подходах к лечению наследственных бс
ней исходят из того, что у больных л ибо образуется аномальный
(фермент), либо нормального белка вырабатывается недостаточж
полного отсутствия). За этими событиями следуют изменения
превращения субстрата или его продукта. Знание этих принці
и конкретных путей реализации действия гена помогает npasHJ
разрабатывать схемы лечения и даже терапевтическую стратеі
Это особенно четко можно проследить на примере наследствен!
болезней обмена веществ.
ІЙИІІ 10. Принципы лечения наследственных болезней403Кофермент—W—Фермент А Измененный фермент вСубстратФермент С■> С->DПродуктКоррекция обменаIДиетическое ограничение
Диетическое добавление
Усиленное выведение субстрата
патологической реакцииАльтернативные пути обмена
Метаболическая ингибицияВозмещение или добавление недостающего продуктаДобавление кофактора
Модификация ферментативной
активностиВозмещение фермента1’ис, 10.2. Возможные подходы к патогенетическому лечению наследственных
ПолезнейВ обобщенном (может быть, немного упрошенном) виде возмож-
иые подходы к лечению наследственных болезней обмена веществ
представлены на рис. 10.2. Видно, что для различных болезней могут
быть использованы разные пути коррекции. Для одной и той же
Оолезни можно использовать вмешательства в разных звеньях и на
различных этапах развития патологического процесса.В целом патогенетические подходы к лечению наследственных
болезней в зависимости от уровня биохимического дефекта можно
представить следующим образом. Лечение схематично сводится к
нозмешению или выведению чего-либо. Если ген не работает, то
необходимо возместить его продукт; если ген производит не то, что
404 Клиническая генетиінужно, и образуются токсичные продукты, то необходимо удален!
таких продуктов и возмещение основной функции; если ген npoHJ
водит слишком много продукта, то ei'o избыток удаляют.Коррекция обмена на уровне субстратаТакое вмешательство — одна из наиболее частых форм лечені
наследственных болезней. Коррекцию можно обеспечить разн!
ми путями, примеры которых приведены ниже. Субстратом в даі
ном случае называется тот компонент пищи, который подвергает
метаболизму с помощью генетически детерминируемого фермені
(например, фенилаланин, галактоза), а при наследственной болез»
он является участником патологической реакции.Ограничение определенных веществ в пише (диетическое ОГІ
ничение) было первой успешной мерой в лечении наследственн!
болезней обмена, при которых отсутствуют соответствующие фе|
менты для нормального превращения субстратов в продукпах пи1
ния. Накопление некоторых токсичных соединений или продуі
тов их обмена приводит к постепенному развитию болезни. Пі
фенилкетонурии назначают диету с низким содержанием фени;
данина. Несмотря на отсутствие фенилаланингидроксилазы печені
тем самым прерывается патогенетическое звено развития болезн!
Ребенок, находившийся несколько лет на искусственной диете, уі
не будет страдать тяжелой формой болезни, Спустя несколько л<
чувствительность нервной системы к фенилаланину и продукті
его превращения резко снижается, и диетическое ограничение moj
быть уменьшено. Ограничоше диеты не обязательно означает состав
ление специального пищевого рациона. Например, новый меті!
ограничения поступления фенилаланина с пишсй при фенилкеп
нурии основан на приеме внутрь желатиновых капсул, содсржащі
растительный фермент, который освобождает пищевые продукт
от фенилаланина. При таком лечении концентрация фенилаланиі
в крови уменьшается на 25%. Этот метод особенно целесообразІ
применять у более взрослых пациентов с фенилкетонурией и беї
менных, не нуждающихся в строгом соблюдении диеты.Диетическое ограничение применяется при лечении МНОГІ
наследственных болезней обмена углеводов и аминокислот (галакт
земии, наследственной непереносимости фруктозы и лактозы, арг|
нинемии, цитруллинемии, цистинурии, гистидинемии, мeтилмaJ
новой ацидемии, тирозинемии, пропионовой ацидемии) и другі
I лава 10. Принципы лечения наследственных болезней405<>олезней с известным первичным дефектом. Применяются диеты,
специфичные для каждого заболевания.Ограничением определенных веществ Б диете можно также лечить
(и)лезни, для которых еще не расшифрован дефект первичного про¬
дукта гена. Эмпирически установлено, например, что при целиакии
(см. гл. 7) постоянные диспепсические явления провоцирует глютен.
Для лечения этой болезни достаточно исключить из пищи продукты,
содержащие клейковину.Хотя селективное ограничение определенных веществ в пище игиро-
ко используется для повышения эффективности лечения некоторых
наследственных болезней обмена веществ, остается еще много нерешен¬
ных вопросов. Например, несмотря на 35-летний опыт лечения фенил-
кетонурии, еще не полностью определены оптимальные границы диеты,
продолжительность курса лечения для детей, необходимость ограни-
'іения при менее тяжелых формах ферментативной недостаточности,
принципы индивидуализации диеты. Диетическое ограничение должно
проводиться под строгим биохимическим контролем обмена веществ.Диетическое добавление применяется реже, чем ограничение, но
)тот прием также эффективен при патогенетическом лечении и
пошел в практику лечения двух болезней обмена.При синдроме Хартнапа в результате дефекта транспортной функции
клеток слизистой оболочки кишечника возникает мальабсорбция трип-
юфана. Биохимическим следствием этого становится отсутствие трип-
гофана в крови, гипераминоацидоз, эндогенный дефицит никотиновой
кислоты. У пациентов наблюдаются дepмaтoJЮгичecкиe, неврологиче¬
ские и психические проявления пеллагры. Симптомы болезни умень¬
шаются или даже исчезают при введении в рацион ребенка продуктов
с высоким содержанием белка (4 г/кг в сутки) и добавлении никотина-
ми да или никотиновой кислоты (по 40—200 мг 4 раза в сутки).Особетю убедительный аргумент в пользу лечения наследствен¬
ных болезней с помошью диетического добавления дает лечение
їликогеноза ПІ типа (амило-1,6-глюкозидазная недостаточность).
Это заболевание сопровождается гепатоспленомегалией, гипоглике¬
мией натощак, прогрессирующей миопатией, мышечной атрофией,
кардиомиопатией в результате нарушения аланиноглюкозного цикла
(низкая концентрация аланина). Это приводит к распаду аминокис¬
лот в мышцах при глюконеогенсзе. У большинства больных детей
наступает улучшение, если белки обеспечивают 20-25% энергетиче¬
ской ценности пиши, а углеводы — не более 40-50%.
406Клиническая геніУсиленное выведение субстрата патологической реакции можетществляться разными методами, которые снижают концентр
токсичного субстрата. Полного освобождения от патологичв<
продуктов обмена добиться трудно. ПримерОхм усиленного ВЫ1
ния субстрата является влияние хелатов при гепатолентикуля!
дегенерации. Например, пеницилламин связывает, мобилизуй
ускоряет выведение внутриклеточно накопленных ионов меди.При гемоглобинопатиях необходимо усиленное выведение ж<
чтобы не развивался гемосидероз паренхиматозных органов.Применяемый для этих целей дефероксамин (десферал*) Н1
пливает ферритины и освобождает организм от излишнего желіМожно эффективно применять и непрямые метаболичео!
пути для выведения субстрата. Например, нормальный ур01
мочевой кислоты в крови можно обеспечить выведением остат
ного азота в форме не только мочевины, но и ее метаболитов. Те
прием применяется для лечения наследственных болезней,
словленных многими энзимопатиями цикла мочевины. Подо(
примеры известны и для других форм наследственных болсз
обмена веществ.Выше были приведены примеры усиленной элиминации субсі
тов с помощью лекарств. Этих же целей можно добиться с noMOL
физико-химических методов освобождения от накопленного в Kf
субстрата (плазмафереза и гемосорбции).С помощью плазмафереза удаляется большой объем плазі
содержащей токсичное вещество. Плазмаферез можно примвн|
для освобождения крови от излишка липидов, жирных кислот, фї
новой кислоты. Этот метод эффективно используется при ЛЄЧЄ!
болезни Рефсума. Сделаны первые успешные попытки лечения ш
маферезом двух лизосомных болезней накопления — болезни Фг
и болезни Гоше.Гемосорбция помогает селективно удалять вещества или клї
веществ путем их связывания с родственными лигандами,
метод уже применяется для лечения семейной гиперхолестеринем!
в качестве лиганда для экстракорпорального связывания ЛП1
используют гепарин-агарозу, что, к сожалению, дает кратковремі
ный эффект. Уровень холестерина возвращается к исходному че|
3—7 сут после лечения.Альтернативные пути обмена при лечении наследственных 6oj
ней приведены в табл. 10.1.
miuna 10. Принципы лечения наследственных болезней 407[•IиГмица ЮЛ. Альтернативные пути обмена при лечении наследственных
болезнейБолезниИспользуемоесоединениеМеханизмЛ|и и нинсукииниловая
іиіипурия, цитруллине-
мииАргининДополнительный орни-
тиіг; полуцикл()|1| акические ацидемииКарнитиЕ!Образование и выведе¬
ние эфиров карнитинаI к'кетоновая гиперглици-
мгчияБензоат натрияВыведение глицина как
гиппуровой кислотыИ ювалериановая ациде-
м и >1ГлицинВыведение образующе¬
гося изовалериангли-
цинаI и перхолестеринеми я
111 чипаХолестираминВыведение холестерина
через желчные кислотыI иперуриксмия
I ІолаграПробенеиидПовышенное выделе¬
ние мочевой кислоты с
мочойГруппа нарушений цикла
мочевиныБензоат натрияВы ведение азота за счет
образования гиппуровой
кислотыФенилацетаїнатрияВыведение образуемого
фенилацетилглутамииаЦистинурияПенииилламинВыведение образующих¬
ся дисульфидовУказанный способ лечения во многом сходен с методами усилен¬
ною выведения субстрата. Разница заключается только в способах
достижения цели: в одном случае усиленно выводится непосред-
игвенно субстрат, а в другом — субстрат сначала превращается в
какое-то соединение, а затем это соединение удаляется.Метаболическая ингибиция используется тогда, когда надо затор¬
мозить синтез накапливаемого при наследственной болезни субстра¬
та или его предшественника. В качестве иніибиторов применяют
разные физиологически активные соединения. Например, при син¬
дроме Леша—Найхана и подагре используют аллопуринол, который
ингибирует ксантиноксидазу, благодаря чему уменьшается концен¬
трация мочевой кислоты в крови. Ципрофибрат ингибирует синтез
408 Клиническая генатіглицеридов и поэтому эффективно снижает концентрацию липидС
пациентов с гиперхолестеринемией (тип ІИ). Стрихнин конкури)
в связывании глицина с рецепторами в ЦНС, что улучшает дьН
тельную и моторную функции, угнетение которых вызвано высок!
содержанием глицина в спинномозговой жидкости при тяжо!
некетоновой гиперглицинемии.Коррекция обмена на уровне продукта генаЭтот подход применяется уже давно, поскольку во многих случ!
в клинической медицине для некоторых болезней была устано!
на патогенетически ключевая роль отсутствия некоторых вещі
(инсулина, гормонов роста, антигемофильного глобулина и др.).Возмещение продукта (или добавление) с целью коррекции обм<
применяется при таких нарушениях, патогенез которых обуслойі
аномальным ферментом, не обеспечивающим выработку продуі
или другим биологически активным соединением.Примеров эффективных подходов к «исправлению» наследст!
ных нарушений обмена путем возмещения продукта уже мн<
введение необходимых стероидов при врожденной гиперплазии
почечников, тироксина нри гипотиреоидизме, гормона роста
гипофизарной карликовости, уридина при оротовой ациду(К сожалению, пока еш;е нет примеров возмещения внутриклеточ!
белков, хотя попытки в этом направлении предпринимались (нап|
мер, при лечении лизосомных болезней).Подобные примеры известны не только для нарушений обмена,
для других наследственных болезней. Так, введение антигемофилы
глобулина предупреждает кровоточивость при гемофилии, у-глобу|
помогает при агаммаглобулинемии, инсулин — при диабете.При энтеропатическом акродерматите развивается недостат
ность цинка из-за дефекта цинксвязываютцего фактора в ки]
нике. В этом случае состояние больных одинаково улучшаї
введение грудного молока, содержащего цинксвязывающий фак1
и прием препаратов цинка внутрь. Как только концентрация цИ|(
в крови достигает нормального уровня, состояние больных q
улучшается.Для лечения по принципу возмещения продукта надо зї
тонкие механизмы патогенеза и вмешиваться в эти механизмы (!
мещать продукт) осторожно и внимательно. Так, прсдварителЫ
попытки лечения болезни Менкеса путем возмещения меди не ПІ
(лава 10. Принципы лечения наследственных болезней409ИСЛИ к успеху, хотя концентрация меди и церулоплазмина в крови
(юл ьных достигала нормального уровня. Оказалось, что дефект при
данной болезни обусловлен нарушением регуляции синтеза медь-
снизывающего белка, обеспечивающего внутриклеточное содержа-
мие меди. По этой причине препараты меди не улучшали состояние
вольных.Необходимость знания тонких механизмов обмена для лечения
можно показать на примере сцепленной с Х-хромосомой гипофос-
<|);ітемии. При этом заболевании первичный почечный дефект вса¬
сывания фосфата ведет к нарушению (снижению) минерализации
костей (рахит) и гипокальциемии. Прием внутрь фосфата и 1,25-
■чигидроксихолекальциферола улучшает минерализацию костей и
уменьшает гипокальциемию, но не изменяет первичного дефекта
потери фосфата с мочой. В связи с этим имеется большая опасность
шиникновения гиперкальциемии, а значит, в процессе лечения надо
контролировать содержание кальция в крови.В целом можно ожидать дальнейших сдвигов в патогенетическом
лечении путем возмещения продуктов (белков, гормонов) в связи с
успехами физико-химической биологии, генной инженерии и био¬
технологии. Генно-инженерными методами уже получают специфи¬
ческие белки и гормоны человека, необходимые для восполнения
нарушенного звена обмена при лечении наследственных болезней
(инсулин, соматотропин, ИФН и др.).Хорошо известны успехи в получении и разведении трансгенных
лабораторных животных. Хотя технически создание трансгенных
сельскохозяйственных животных намного труднее, чем лаборатор¬
ні,IX, это решаемая задача. От крупных животных можно получить
Гюльшое количество белка. Трансгенных животных, чьи клетки
производят нужные белки, можно называть биореакторами. От них
можно получать потомство, т.е. возможно воспроизводство из поко¬
ления в поколение.Создание трансгенных животных начинается со сшивки двух
генов, каждый из которых клонирован отдельно. Один ген кодирует
нужный белок, другой взят из железы или другого органа, который
Оудет производить этот белок. Например, если белок продуцируется
с молоком, то специфическими органными генами будут гены из
М()ЛОЧНОЙ железы.Гибридная ДНК инъеиируется в оплодотворенную яйцеклет¬
ку или в эмбрион. Примерно в 1—5% случаев ДНК встраивается
410Клиническая rej'Рис. 10.3. Трансгенная свинья, кото¬
рая продуцирует гемоглобин чело¬
векаРис. 10.4. Трансгенный бык с
человеческого лактоферрина^
него получены телята с такиї
геномв геном. Все яйцеклетки подсаживают в матку самок, а р<
шихся животных проверяют на присутствие гибридного
От животного-основателя получают потомство и таким обр|
создают стадо.Один из примеров живых биореакторов — свинья, продуцируї
человеческий гемоглобин (рис. 10.3). Она «сконструирована» в И
Около 15% эритроцитов свиньи содержат человеческий гемоглобин^можно отделить от свиного
глобина с помощью препа{
ных методов. Такой гемоглобі
содержит вирусы человека,
отдельных случаях не ис»
ются аллергические реакцииі<
Другим трансгенным Ж1
ным стала корова, которая
изводит человеческий лв]
феррин, выделяемый с мол<
в результате подсадки т|
генной яйцеклетки родилсяРис. 10.5. Трансгенная коза, в молоке <РИ<=- ставший отцомкоторой содержится плазминоген- трансгенных телок, в ПСный активатор (тромболитический дующем производящих леїфермент) феррин с молоком.
Импа 10. Принципы лечения наследственных болезней411Получены и другие трансгенные животные, трансгенная коза
10.5) выделяет с молоком активатор плазминогена, который
гиоряет тромбы, трансгенные кролики — фермент а-глюкозидазу
||Л11 лечения болезни Помпе, трансгенные куры несут яйца с челове¬
ческими антителами.И последние годы отечественные ученые разработали менее
Іингий и недорогой способ трансгеноза органов-мишеней.
Км1бходимый ген вводят не в яйцеклетку, а непосредственно в
<1М10чную железу, трансген у таких животных присутствует только
измени. Получены соматические трансгенные коровы, свиньи и
служащие биореакторами для фармацевтической промыш-ІҐПИОСТИ.[Коррекция обмена на уровне ферментовМногоступенчатый путь превращения субстрата в процессе
|t)f>McHa осуществляется с помощью соответствующих ферментов,
[^‘►лыпая группа наследственных болезней обусловлена мутация-
[Ми в генах, детерминирующих синтез ферментов (энзимопатии).
[Нмсшательство в развитие болезни (коррекция) на уровне фермента
[цпляется примером патогенетического лечения первичных этапов,
[Т.с. приближающегося к этиотропному лечению. Этот вид лече-
1иия применяется для коррекции наследстненных болезней обмена
Ьсществ, при которых известен функционально аномальный фер-
[Мснт. Для такого лечения можно вводить кофактор или индуци-
[роиать (угнетать) синтез фермента с помощью лекарств либо воз-
[Мсщать недостаток фермента.Введение кофактора используется при многих наследственных
[болезнях. Как известно, некоторые врожденные аномалии обмена
[снязаны с нарушением синтеза или транспортировки специфических
I кофакторов, что изменяет нормальную каталитическую активностьI фермента. В этих случаях добавление соответствующего кофактора
поиыщает активность фермента и в значительной мере исправляет
метаболический дефект. Показано, что при витаминозависимых
состояниях повышение остаточной активности мутантных фермент¬
ных комплексов обеспечивает не только биохимическое, но и клини¬
ческое улучшение состояния. Известны многочисленные примеры
лечения наследственных болезней путем добавления кофакторов,
далеко не исчерпывающая классификация которых представлена в
табл. 10.2.
412Клиническая геніТаблица 10.2. Нарушения обмена, при лечении которых добавляют коф|ДефектВигамки или
кофакторБолезнь і]НарушенныйTpaF<cnopTКобаламинМетилмалоновая аиидемия, j
гомоцистинурия JНарушенныйКобаламинМетилмалоновая ацидемия, 1
гомоцистинурия 1процессингБиотинМножественная карбоксилая
недостаточностьНарушенное свя-ЧКІ КЯНМРКобаламинМетилмалоновая аиидемия jПиридоксинГомоцистинурия ^041 Г1 Г1 WТиаминЛейцинозНарушенныйсинтезБиоптеринГиперфенилаланинемияДефект замеще¬
нияМенадион/
аскорбатДефекты дыхательных цепей*1Нарушенныйтранспорт/реабсорбцияВитамин D (боль¬
шие дозы)Группа гипофосфатемий/виІІ
мин D-резистентный рахит *|НеопределяемыйРибофлавинНедостаточность множествви
ной СоА-дегидрогеназы jИз табиіицьі 10.2 видно, что при лечении наследственных
один и тот же кофактор может выполнять разные функции. По-ви;
му, будет перспективным введение кофактора для внутриутро^
лечения плода (как в случае ^-зависимой метилмалоновой ациде»Модификация ферментативной активностиЭто уже сложившийся подход при лечении наследственных
ней обмена. Стратегия такого лечения отражена в табл. 10.3, в кот
приведены отдельные примеры.Таблица 10,3. Лечение наследственных болезней путем модификацкИ.
ментативной активностиТип модификации (болезнь)ФерментыМодифихііПодав.іение 1ГиперурикемияПодаграКсантиноксидазаАллопурИЩСиндром Леша-Найхана
і|йііа 10. Принципы лечения наследственных болезней413Окончание таблицы 10.3Гни модификации (болезнь)ФерментыМодификатор111 к- |)Л и попротеи нем и я
II ІІІІ1І1Ферменты синтеза
липидовЦипрофибрат(I Ік'рл и попротеи [гемия
I I11IUIН MG - СоА-редуктазаЛовастатин|||)111>11ПСНИе индукции{ ммдромы Криглера—
ІІіііічра и ЖильбераГлюкуронилтранс-феразаФенобарбиталАт ионевротический отекИнгибитор эстеразы СДаназолІІГ ,достаточность ингибитора
ИрогеазИнгибитор протеазДаназолПодавление обратной связи)1|1">кденная гиперплазия
мп-иючечниковФерменты синтеза
АКТГКортизонИндукцию синтеза фермента можно использовать для повышения
кіагочной ферментативной активности путем введения лекарств.Ill пример, фенобарбитал и родственные ему препараты стимулируют
пункцию эндоплазматического ретикулума и синтез специфичных
него ферментов, в связи с этим фенобарбитал применяют для
[}і:':сііия синдромов Жильбера и Криглера—Найяра. При этом снижает-
Цм уровень билирубина в плазме крови. Такой подход имеет определен-
(ос значение при болезнях, обусловленных недостаточной продукцией
JicpMCHTOB, вырабатываемых в эндоплазматическом ретикулуме.
Индукция синтеза ферментов с помощью даназола (дериват этинил-
гстостерона) применена для лечения недостаточности аі-антитрипсина
[и ІІН1 ионсвротического отека. При недостаточности аранттриисина
ірименение даназола в течение 30 дней существенно повышает уровень
И<51 о белка в сыворотке. Таким образом, данный метод можно исполь-
[;і«ішть для предупреждения легочных осложнений.Ангионевротический отек сопровождается снижением количества
[функционально активного сывороточного ингибитора эстеразы С на
Применение андрогенов повышает в 3—5 раз уровень ингиби-
[гора эстеразы. Профилактический прием внутрь даназола снижает
.или предупреждает острый ангионевротический отек, оказывает
|Минимальную вирилизацию и связан с наименьшей токсичностью
[для печени.
414Клиническая генет*Подавление синтеза фермента используют для лечения остр!
порфирий, биохимическая основа которых заключается в повыше!
ной выработке аминолевулинатсинтстазы. Гематин подавляет сині
этого фермента и быстро снимает острые приступы порфирии.Возмещение ферментаУспехи современной энзимологии позволяют выделить этот раз
в патогенетическом лечении наследственных болезней. Это вмвш|
тельство на уровне первичного белкового продукта гена. Современн!
методы позволяют получить такое количество активного фермсі
для экспериментальных и клинических целей, которое необхс
МО для его восполнения при определенных наследственных болгі
нях. Выше разбирались случаи возмещающей терапии: ropMOJ
при эндокринопатиях, антигемофилъный глобулин при гемофил!
у-глобулин при агаммаглобулинемии. По такому же принципу
ного соответствия недостающего продукта строится стратегия ф(
ментотерапии.Главный вопрос современных разработок в области ферментот
рапии — это методы доставки фермента в клетки-мишени и субі
точные образования, вовлеченные в патологию обмена.Рабочая гипотеза экзогенного введения фермента основывалась і
том, что лизосомы часто являются местом патологического процес
и в то же время играют основную роль в клеточном метаболии
Возможность доставки ферментов в лизосомы, сохранение их аки
ности в клетке и взаимодействие с субстратом были проверен*
опытах с культурами фибробластов, полученных от лиц с различні
лизосомными болезнями накопления. Ферменты, введенные в ку|
туральную среду, улучшали обмен соответствующего соединен!
Такая коррекция продемонстрирована при различных гликосфиш
липидозах, мукополисахаридозах, гликогенозах и гликопротеиноз
Опыты показали, что возможно возмещение фермента, который П|
ни кает внутрь клетки, достигает лизосом и нормализует превращеі
субстрата. Однако внутримышечное, внутривенное и внутритрахег
ное введение ферментов, полученных из грибов или органов крупне
рогатого скота, ослабленным больным с гликогенозом, мукополисг
ридозами, метахроматической лейкодистрофией и болезнью Фабри,1(
дало серьезных положительных результатов. Следовательно, в стрї
гии ферментотерапии надо было определить основные направлені
которые в суммарном виде представлены ниже.
іава 10. Принципы лечения наследственных болезней415- Возможность получения достаточного количеств! стабильных,
неиммуногенных и стерильных ферментов с высокой специфи¬
ческой активностью.- Защита введенной активности от биотрансформации и иммун¬
ного надзора, а также доставка фермента в ткань-мишень и
субклеточные образования, вовлеченные в патологический
процесс.— Модельная проверка на млекопитающих для опенки и выбора
наилучшей стратегии ферментотерапии.— Соответствующим образом запланированные и разрешенные
биохимические и клинические испытания на больных.В 70-х годах XX в. была показана возможность получения фермен¬
ти из тканей человека и разработаны системы наблюдения за судьбой
«])Срментов в организме млекопитающих. Первые клинические испы-
Гііішя были проведены при различных лизосомных нарушениях. Это
Пмли СМ2-ганглиозидоз (р-гексозаминидаза А из мочи), гликогеноз
И типа (плацентарная а-галактозидаза), болезнь Фабри (плацентар-
ІШЯ а-галактозидаза), болезнь Гоше (плацентарная р-глюкозидаза).
Перед клиническим испытанием было установлено, что высокоочи-
|ци*нные ферменты человека гидролизируют естественный субстрат.
Проверка показала, что ферменты при внутривенном или подкожном
нисдении обнаруживаются в печеночной ткани. При этом концентра¬
ция ферментов в крови уменьшается, а в печени повышается. Однако
[они не проникают в мозг из-за барьерных функций мозговых оболо-
[чск. Отсюда следует вывод о необходимости специфической достав-
!ки ферментов в клетки-мишени при каждой болезни. Их доставка
И разные клеточные структуры может потребовать специфической
очистки или какой-либо химической модификации фермента.В разработке методов лечения наследственных болезней фер¬
ментами в первую очередь надо ориентироваться на патогенетиче¬
ские механизмы болезней; в каких клетках, каким путе.м и в какой
форме откладывается субстрат реакции, с одной стороны, и каким
путем фермент в норме достигает субстрата, каковы промежуточ¬
ные стадии обмена — с другой. Именно вмешательство в патофи¬
зиологический механизм, ответственный за синтез, распределение
и накопление субстрата, можно использовать с терапевтической
целью: в одних случаях надо увеличить время циркуляции фермен¬
та в крови, в других — способствовать доставке фермента в строго
определенные клетки.
416Клиническая генвіИз анализа первичной клеточной патологии при различных ли|
сомных болезнях накопления видно, что даже близкие по сути л
левания отличаются друг от друга.Первичный дефект локализуется в нейронах (сфинголипИІ
зы, гликопротеинозы), в клетках ретикулоэнлотелиальной систої
(болезнь Нимана—Пика, болезнь Гоше), эндотелии, шваннонс!
клетках, поперечно-полосатой мускулатуре.Экспериментальные разработки в области ферментотераї
наследственных болезней позволили объективно оценивать зах|
молекул фермента рецепторами, гепатоцитами, клетками ретику^
эндотелиальной системы, фибробластами, клетками эндотелия с(
дов и т.д. Это увеличило возможности направленных разрабс
лечения наследственных болезней, в первую очередь с использовИ
ем новых методов доставки ферментов к клеткам-лійшеням в СИ1
тических пузырьках-носителях или микрокапсулах-липосомах лиС
естественных элементах — аутологичных эритроцитах. Такие мет(
доставки разрабатываются для лечения не только наследствен!
болезней, но и другой патологии. Направленная доставка лекарст
ных веществ в органы, ткани и клетки — актуальная проблема
медицины в целом.Современные успехи физико-химической биологии П03В0ЛІ
создавать новые формы микроинкапсулированных ферментных
паратов (опосредованная доставка) или обеспечивать более
ный захват циркулирующего в крови фермента рецепторами клс
мишеней (опосредованная рецепция).Липосома представляет собой многослойный пузырек с мере;
щимися водными и липидными слоями, при формировании Л|
сом можно изменять заряд стенки, их величину, число слоев, к
бране липосом можно пришить антитела к клеткам-мишеням,-
обеспечит более точную доставку липосом. Липосомы, нагружеі
ферментами, при различных путях введения хорошо захватыва!
клетками. Их липидная оболочка разрушается эндогенной липі
а освободившийся фермент взаимодействует с субстратом.Наряду с созданием искусственных носителей — липосом —
рабатьтвают методы нагрузки эритроцитов ферментами. При
можно использовать гомологичные или даже аутологичные эр1
циты. Нагрузка ферментами может осуществляться путем гипот
зации, или диализа, или с помощью хлорпромазининдуцироваї
эндоцитоза.
Iпава 10. Принципы лечения наследственных болезней417Перспективы лечения наследственных болезней возмещением
фі-рментов зависят от успехов энзимологии, клеточной инженерии,
фи тко-химической биологии. Новые подходы должны обеспечить
иііілслсние высокоочищенных ферментов из специфических тканей
человека, введение их в активной форме в клетку путем опосредо-
ИНН ной рецепции или опосредованной доставки, предупреждение
Пиоинактивации, исключение иммунных реакций. Уже имеются
1и>дходы к репіению каждой из этих задач, поэтому можно надеяться
1111 еще более успешное развитие ферментотерапии наследственных
П<1лезней.ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕХирургическое лечение наследственных болезней занимает суще-
і'гиснное место в системе медицинской помощи больным. Это свя¬
тно с тем, что, во-первых, многие формы наследственной патоло-
ІИИ сопровождаются морфогенетическими отклонениями, включая
пороки развития. Во-вторых, расширение возможностей хирурги-
•а'ской техники сделало доступными многие трудные операции.
И-третьих, реанимация и интенсивная терапия сохраняют жизнь
Новорожденным с наследственными болезнями, а такие пациенты
нуждаются в последующей хирургической помощи.Хирургическая помощь больным с наследственной патологи-
в общем виде включает удаление, коррекцию, трансплантацию.
[Операции часто направлены на устранение симптомов болезни.I Однако в некоторых случаях хирургическая помощь выходит за рамки
[симптоматического лечения, приближаясь по эффекту к патогенети-
1»1сскому лечению. Например, для изменения пути патологического
превращения субстратов патологических реакций можно использовать
хирургическое шунтирование. При гликогенозах I и III типов делают
йнасгомоз между воротной и нижней полой венами. Это позволяет
части глюкозы после всасывания в кишечнике обходить печень и не
откладываться в ней в виде гликогена. Аналогичный обходной путь
предложен при семейной гиперхолестеринемии (тип Па) — анастомоз
между тощей и подвздошной кишками. Это приводит к снижению
Исасывания холестерина.Примерами общехирургических видов лечения могут быть опе¬
рация по поводу наследственного полипоза толстой кишки (ее уда¬
ление), спленэктомия при гемоглобинопатиях, удаление глаза при
418Клиническая ген*1ретинобластоме, почки при опухоли Вильмса и др. В ряде сл|
ев хирургическое лечение является частью комплексной терв!
Например, при муковисцидозе возможен мекониалъный илві
новорожденных, в процессе развития болезни встречается пнв!
торакс. И то и другое устраняется хирургическим путем.Большое место в лечении наследственных болезней зани!
реконструктивная хирургия: при незаращении верхней губы, врожі
ных пороках сердца, атрезии отделов ЖКТ, гипоспадии, для ко^
иии костно-мышечной системы и т.д. іТрансплантация органов и тканей как метод лечения наследсті
ных болезней все больше входит в практику. АллотрансплантіІ
может рассматриваться как передача нормальной генетичв(
информации пациенту с нарушением обмена веществ. Такой
ход предполагает пересадку клеток, тканей и органов, содержав
нормальную ДНК, для продукции активных ферментов или д{
продуктов гена у реципиента. Он особенно эффективен тогда,
патологический процесс ограничен одним органом или ткаї
которые и пересаживают.Аллотрансплантация уже выполняется при разных наследсті
ных болезнях и позволяет непрерывно восполнять недостаток
мента, гормона, иммунных функций или эффективно предохрі
орган от функциональных нарушений, обусловленных мутаї
структурного гена. В таблице 10.4 перечислены наследствеї
болезни, при которых применяется аллотрансплантация.Таблица 10.4. Применение аллотрансплактации для патогенетического,
и ИЯ наследственных болезнейОрганБолезни 1Вилочкопая железаСиндромы ДиДжорджи и Незелофа (поражение!
Т-лимфоцитоп) 2СелезенкаБолезнь Гоше (III тип), гемофилия А *ПоджелудочсіаяжелезаСахарный диабет іСердцеПервичные кардиомионатии ]ПеченьБолезнь Вильсона—Коновалова, недостаточносТІ
aj-антитрипсина, тяжелый комбинированный і
иммуподефииит (печень плода), болезнь НиманШ
Пика, тирозииемия
I Гііііва 10. Принципы лечения наследственных болезней419Окончание таблицы 10.4ОрганБолезниПочкаБолезнь Фабри, иистиноз (Фан кон и), болезнь Гоше
(IT тип), амилоидоз, подагра, оксалоз, синдром
ногтей-надколенной чашечки, синдром Альпорта,
семейная средиземноморская лихорадка, кистоз
мозгового вешества почки, семейный поликистоз
почек, диабетНадпочечникАдренокорти кальная недостаточностьСовременная трансплантология обладает большими возмож¬
ностями, и ее успехи можно использовать в лечении наследствен-
имх болезней. Имеются многочисленные сообщения об успешных
іаресадках органов (костного мозга, вилочковой железы, пече¬
ни плода, донорской печени, поджелудочной железы, селезенки
и особенно почки) при упомянутых в табл. 10.4 состояниях.
Трансплантация исправляет патологические механизмы наслед-
сгиенных нарушений.Помимо пересадки органов разрабатываются методы пересадки
клеток, функция которых занимает ключевое место в патогенезе
наследственных нарушений обмена. Лечение стволовыми клетками
Судет рассмотрено ниже.В заключение следует обратить внимание на огромные возможно¬
сти хирургического лечения наследственных болезней, используемые
еще не в полной мере. В этом плане весьма перспективны микрохи¬
рургия и эндоскопическая хирургия.ЭТИОТРОПНОЕ ЛЕЧЕНИЕ: КЛЕТОЧНАЯ
и ГЕННАЯ ТЕРАПИЯВведениеЭтиотропное лечение любых болезней оптимально, поскольку оно
устраняет первопричину заболевания и в результате полностью его
излечивает. Несмотря на успехи симптоматической и патогенетиче¬
ской терапии наследственных болезней, вопрос об их этиотропном
лечении не снимается. Чем глубже будут знания в области теорети-
420Клиническая геніческой биологии, тем чаще будет подниматься вопрос о радикалі
лечении наследственных болезней.Однако устранение причины наследственной болезни озН1
такие серьезные манипуляции с генетической информаци(
человека, как доставка нормального гена в клетку, выключ#|
мутантного гена, обратная мутация патологического аллеля,
задачи достаточно трудны даже при вмешательствах у просі
ших организмов. К тому же, чтобы провести этиотропное леч(
какой-либо наследственной болезни, надо изменить струїсі
ДНК и не в одной клетке, а во многих функционирующих клеї
(и только в функционирующих!), прежде всего, для этого нуї
знать, какое изменение произошло в гене в результате мутаї
т.е. наследственная болезнь должна быть описана в химиче<
формулах.Сложности этиотропного лечения наследственных болезней Q
видны, но уже имеются многочисленные возможности для их прв<
ления, создаваемые успешной расшифровкой генома человека и
грессом молекулярной медицины.Несколько принципиальных открытий в генетике и молекул
ной биологии создали предпосылки для разработки и клиниче{
проверки методов этиотропного лечения наследственных болі
(генная и клеточная терапия).В экспериментах с РНК- и ДНК-содержащими вирусами ОП]
лей (начало 1970-х годов) выявлена способность вирусов переж
гены в трансформированные клетки и сформулирована концеП|
использования вирусов как переносчиков генов, другими сл(|
ми, концепция создания векторной системы (рекомбинантная ДІ
Успех, достигнутый в экспериментах с рекомбинантной ДНК, у)
середине 1970-х годов обеспечил почти неограниченные возможнс
в изоляции генов эукариот (в том числе и человека) и манипуля!
с ними. В начале 80-х годов была доказана высокая эффективнс
переноса генов на основе векторных систем в клетки млскопитаюі
in vitro и in vivo.Принципиальные вопросы генной терапии у человека реш«
Во-первых, гены можно изолировать вместе с фланкирующї
(пограничными) областями, содержащими в себе, по меньшей ці
важные регуляторные последовательности. Во-вторых, изоли
ванные гены нетрудно встроить в чужеродные клетки. «Хирург)]
трансплантации генов многообразна.
іГмава 10. Принципы лечения наследственных болезней421Условия генной терапии разрабатывались удивительно быстро.
Первый протокол генной терапии у человека был составлен в 1987 г. и
проверен в 1989 г., а с 1990 г. уже началась генная терапия больных.Этиотроиное лечение наследственных болезней может осушест-
иияться на уровне клеток или генов. Организм больного должен
Получить дополнительную генетическую информацию, способ¬
ную исправлять наследственный дефект, с геномом аллогенной
♦(І10ТКИ или в виде специально созданной генно-инженерной кон-
11|)укции.Под термином «клеточная терапия» понимают способ лечения
ііуіем трансплантации клеток. Пересаженные клетки сохраняют
л потип донора, поэтому пересадку можно рассматривать как форму
ичютерапии, поскольку она приводит к изменению соматического
к'иома. Генная терапия — способ лечения путем введения допол-
імггельной генетической информации в клетки индивида на уровне
[ДНК или РНК (генно-инженерных конструкций) или путем измене¬
ния экспрессии генов.в целом, к настоящему времени определились четыре направле¬
ния этиотропного лечения:* трансплантация аллогенных клеток (клеточная терапия);‘ введение генно-инженерных конструкций в ткани больного (ген¬
ная терапия);• трансп лантация трансгенных клеток с целевой генно-инженерной
конструкцией (комбинированная терапия);' изменение экспрессии генов (генная терапия).Клеточная терапияТрансплантация клеток или клеточная терапия — это в настоя¬
щее Бремя часть бурно развивающейся регенеративной медицины.
Применительно к лечению наследственных болезней речь идет о
трансплантации аллогенных клеток, потому что аутологичная пере¬
садка не изменяет мутантного генома клеток. Наиболее эффектив¬
ных результатов клеточной терапии можно добиться при трансплан¬
тации стволовых клеток. Они обладают способностью размножаться
й недифференцированном состоянии, а другая их часть дифферен¬
цируется в клетки патологически измененного органа, улучшая его
функцию. Что такое стволовые клетки, где они находятся, каковы их
разновидности и функции см. в книге «Биология стволовых клеток и
клеточные технологии в 2 т.» под ред. м.А. Пальцева.
422Клиническая гене'Источники столовых клеток представлены в табл. 10.5.Таблица 10.5. Типы ство.'ювых клеток, применяемых для лечения нас/ственных болезнейИсточники клетокКостный мозгПуповинная кровьТип стволовых клетокМононуклеарная фракция костного
мозга (смесь стволовых и нестволоШ
клеток)Гемопоэтические стволовые клетки
Мультипотентные мезенхимальные
стволовые клеткиГемопоэтические стволовые клетки
Мул ьти потентные мезенхимальные
C'1'ВОЛОВЫе клеткиПеченьГепатоциты, гепатобластыМышцы (поперечно-полосатые)Миобласты, миоцитьт, мезангиоблавПервым по времени применения и объему проведенных клетощ
трансплантаций является костный мозг и полученные при его KJ
тивировании гс.мопоэтичсские стволовые клетки, а также мульи
тентные мезенхимальные стромальныс клетки. В конце 60-х г(
прошлого века впервые для лечения первичных иммунодефиці
применили трансплантацию костного мозга. В последние гох
качестве источника гемопоэтических стволовых и мезенхимал!
стромальных клеток используется и пуповинная кровь.Печень эмбрионов — хороший источник стволовых клеток
ночной и непеченочной (после культивирования) дифференцирс
Клеточная фракция эмбриональной печени после транспланта!
в организм реципиента выполняет функции печени, что особо|
важно в экстренных случаях поражения печени.Поперечно-полосатые мышцы в культуре образуют миобла(
миоциты, мезангиобласты, когорые обладают способностью к С1
воспроизведению и дифференцироБке в обратном направлені
поперечно-полосатые мьешєчііьіє клетки.Трансплантация гемопоэтических стволовых клеток применя!|
как эффективная терапия наследственных болезней обмена, главі
образом лизосомных болезней накопления и пероксисомных. Вс<
мире сделано около 1000 трансплантаций при более чем 20 болез
Лечение трансі 1ла][тацией гемопоэтических стволовых клеток
IdiiBa 10. Принципы лечения наследственных болезней423іі;іс:іедственньіх болезнях обмена основано на выработке недостаю¬
щих в организме ферментов за счет функционирования донорских
меток. Из всех клинических проколов по более чем 20 болезням
іилько по трем формам получены убедительные результаты, позво¬
ляющие рекомендовать трансплантацию таких клеток как метод
печения. Это синдром Гурлер, Х-сцепленная адренолейкодистрофияII болезнь Краббе (глобоидноклеточная лейкодистрофия). Для этих
форм отработаны условия кондиционирования, претрансплантаци-
1)1 тая терапия, строгие показания, возраст детей.Большой раздел в клеточной терапии занимают болезни крови и
к[1оветворных органов, ассоциированных с недостаточностью про¬
дуктов костного мозга. Важнейшим условием является подбор доно-
|Х)п по НLA-антигенам, чтобы снизить реакцию «трансплантат про¬
пив хозяина». Не останавливаясь на технической стороне клеточной
к'рапии, перечислим болезни, которые уже лечат гемопоэтическими
1Т1ЮЛ0ВЫМИ клетками. При этом не исключаются другие виды лече¬
ния. Трансплантацию гемопоэтических стволовых клеток используют
при лечении следующих болезней: анемии Фанкони, первичных нмму-
иодефицитов, гемоглобинопатий. Переливание моноцитарных фракций
kticTHoro мозга дает худшие результаты из-за большей антигенности
ірельїх клеток по сравнению с гемопоэтическими стволовыми.Более 15 лет назад клеточная терапия была применена для лечения
наследственных заболеваний костей — ахондроплаэии и несовершен¬
ного остеогенеза. Трансплантировали мезенхимальные строма л ьные
клетки, полученные из костного мозга. Лечение было направлено
па усиление роста костей. И действительно, применение мезенхи¬
мальных стромальных клеток давало эффект ускоренного удлине¬
ния костей при дистракционном остеогенезе при ахондроплазии
и существенное прибавление в росте у больных с несовершенным
остеогенезом.Для клеточной терапии заболеваний нервной системы имеется
много источников стволовых клеток: из нервной системы, жировой
ткани, костного мозга и др. Мезенхимальные стромальные клетки
костного мозга могут дифференцироваться в нейтральные стволовые
клетки. Хотя и проводятся многочисленные экспериментальные раз¬
работки, обосновываются новые подходы, проверяются новые кли-
иические протоколы лечения больных стволовыми клетками таких
сложных по патогенезу болезней, как болезнь Альцгеймера, хорея
Гентингтона, болезнь Паркинсона, миопатия Дюшенна, пока одно¬
424Клиническая генізначных результатов лечения не получено. Все клинические
токолы клеточной терапии нервной системы являются первичкі
проверка\1И на токсичность и биобезопасность.Эффективностьлечения стволовыми клетками, как правило,
сокая, и терапевтический эффект сохраняется лишь первые 6
поэтому клеточная терапия должна рассматриваться как доп(
тельный, а не основной метод лечения. Важным методом лбЧ<
является сочетание клеточной терапии с лекарственной, осо(
ферментативной, для наследственных болезней обмена. Впереди]
много работы по доведению первых результатов до эффективЖ
безопасных методов лечения. Несмотря на многочисленные кл1
ческие исследования клеточной терапии, утвержденных протої
лечения для конкретных нозологических форм еще нет (тип КЛІ
количество, способ введения клеток, сроки повторного введеннірг CMVагл|BGH poly
fi oriori ColEI 'V Pr SV40SV40 PolvAРис. 10.6. Карта генетической кон¬
струкции (плазмиды pAngl) с геном
ангиогснипа. Обозначения: Ang —
кДНК гена ангиогенина; PrCMV —
немедленно ранний промотор/энхан-
сер цитомега лови руса; PrSV40 — ран¬
ний промотор/ориджин вируса SV40;
BGH polyA — сигнал иолиаденилиро-
вания гена гормона роста быка; SV40
роІуА — лозяний сигнал полиадсни-
лирования вируса SV40; пео'' — ген
устойчивости к нсомицину; amp^ —
ген устойчивости к ампициллину;
ori — ориджин репликации (fl — фага
П; Со1Е] — плазмиды ColEl)Генная терапияГенная терапия путем
дения генно-инженерных
струкций в клетки и ткани
ного (трансген03 in vivo) м(
стимулировать рост тк1
функцию органа. В этом
терапии создаются функі
нально способные генетичеі
конструкции (генетиче<
вектор) в лабораторных ус;
ях. Эти конструкции долі
включать целевой ген (или1
главную часть), вектор, пром<
(рис. 10.6).Генная терапия в предстаї
ном виде испытывалась глаї
образом для лечения серде
сосудистых заболеваний: і
ми ческой болезни сердца и
нической ишемии нижних КОІ
ностей.Хотя ангиогенез осущест
ется целой группой генов (01
Wna Ю. Принципы лечения наследственных болезней425ц|>1браиы два наиболее критичных целевых гена для проверки
ффоктивности генной терапии. При ишемической болезни сердг^а (в
;1И)м и хроническом СОСТОЯНИЯХ) применяли введение гена VEGF
инютелиального фактора роста сосудов).Ііммшй препарат на основе плазмидной конструкции, содер-
ІІНІІИЙ ген VEGF165 человека, вводится на заключительном этапе
>іи'І>аций (аортокоронарного шунтирования, трансмиокардиальной
к'рной реваскуляризации, миниинвазивной реваскуляризации
Иижарда) в зону, нуждающуюся в неоаги иогенезе. У всех больных
|Н|>С1 истрировали клиническое улучшение: отмечен переход в более
иоприятный класс стенокардии напряжения, снизилась доза при-
П'пяемых нитропрепаратов; проба с физической нагрузкой выявляла
Ііирастанис порога толерантности; все больные отмечали улучшение
кпмества жизни. При сцинтиграфии отмечалось уменьшение общей
||| пощади, а также выраженности дефектов накопления радиофарм-
1|»сIIарата по сравнению с дооперационной картиной.Проведено лечение нескольких тысяч больных с ишемической
Йолознью сердца на разных стадиях. Процедура введения генети-
[Чгских конструкций в миокард безопасна. Положительный эффект
[гемотерапии отмечается в большинстве клинических исследований,
он небольшой (8-10%),Терапевтический ангиогенсз в лечении критической ишемии
[Яижних конечностей осуществлялся разными авторами с помощью
Ьнсдения в мышцы голени и бедра нативной ДНК, кодирующей
[белок VEGF, ген FGF (фактора роста фибробластов), рекомбинант-
[Нме конструкции на основе разных аденовирусов с геном ангиоге-
[Нина — ANG.В нашем исследовании пациентам вводили генно-инженерные
■Цомструкции с геном ANG путем прямых внутримышечных инъек-
ІЦИЙ в тибиальную группу мьіш[і, пораженной конечности троекратно
|П равных дозах (3x10^^ бляшкообразующих единиц) с интервалом 3 сут.
Каждая процедура включала 4—5 прямых внутримышечных инъек¬
ций по 0,3—0,5 мл раствора, равномерно распределенных на площади
15-20x5-6 см. Результаты лечения оценивали через 6-24 мес.В клинических наблюдениях во всех случаях отмечался положи-
І тельный эффект: увеличился показатель времени (расстояния) без-
йолевой ходьбы, увеличился показатель плечелодыжечного индекса,
уменьшились или даже зажили трофические язвы, увеличилась пер¬
фузия мыши нижних конечностей.
426Клиническая rttiДанные литературы и наши наблюдения свидетельствуютчто ГЮЗИТИКНЫЙ эффект сохраняется в течение 6—18 МеС, ПОСЛІДвозникает потребность в повторных инъекциях препарата,
образом, генно-инженерные конструкции, содержащие гены.^
и VEGF, способствуют выработке факторов неоангиогенеза и
мулируют рост кровеносных сосудов в ишемизированных ТІО состоянии, проблемах и перспективах генной терапии см. в
именной статье А.В. Киселева с соавт. на компакт-диске.Лечение трансгенными клеткамиЛечение трансгенными к^іетками с целевой ічгнно-инженерной
стру кцией может быть названо комбинирован ной терапией. Для осу
вления этого типа клеточно-генной терапии необходимо осущс
введение целевого гена в клетку. Такая комбинация сочетает свої
клеточного вектора, генной функции и эффект клеточной терапии.!Трансгеноз (перенос генетического материала) in vitro нап|
на соматические клетки-мишени, заранее выделенные из орган!
(например, резецированная печень, культура лимфоцитов, косі
мозг, культура фибробластов, опухолевые клетки). Для введения Д1
клетки млекопитающих уже опробованы многие подходы: химичс
(микропреципитаты фосфата кальция, DEAE-декстран, диметил(
фоксид); слияние клеток (микроклеток, протопластов); физич<
(микроинъекции, электропорация, лазерная микроинъекция); виї
(ретровирусы, аденовирусы, аденоассопиированные вирусы). Mi
невирусные методы малоэффективны (за иск^іючением злектропої
и лазерной микроинъекции). Наиболее эффективными переносчиї
ДНК в клетки являются «природные шприцы» — вирусы.Процедура трансгеноза клеток должна заканчиваться провс|
ее успешности. Трансгеноз можно считать успешным, если не м|
5% всех обработанных клеток будут иметь введенный генетичес!
материал.Конечная процедура генной терапии через трансгеноз сомі
ческих клеток in vitro — это реимплантация трансгенных кл<
мишеней. Она может быть органотропной (печеночные клетки
дят через воротную вену) или эктопической (клетки костного
вводят через периферическую вену).Клеточно-генная терапия была принята в клинической праї
быстрее, чем можно было ожидать. Варианты ее применения м(
показать на примере трех болезней.
fiHna 10. Принципы лечения наследственных болезней427Недостаточность ADA. Девочка 4 лет (США) страдала редкой
Ійі'лслственной болезнью — первичным иммунодефицитом (тяжелая
І^имОинированная форма), обусловленным мутацией в гене ЛОЛ, Все
и»да девочка жила в стерильном боксе. (Пациенты с этим заболе-
Ійііисм не переносят никаких контактов с любой инфекцией из-за
оіального отсутствия иммунитета.)Лимфоциты больной заранее были отделены от остальных эле-
[MfiiTOB крови, Т“Лимфоциты стимулированы к росту. Затем in vitro
hi 11 их был введен ген ADA с помощью ретровирусного вектора.
Приготовленные таким образом «генно-инженерные» лимфоциты
(йыли возвращены в кровоток.Указанное событие произошло 14 сентября 1990 г., и эта дата счи-
[lueгея днем рождения реальной генной терапии. С этого года стал
Иыходить журнал «Генная терапия».Из протокола клинического испытания стало ясно, что, во-первых,
[лимфоциты пациентов с тяжелым иммунодефицитом могут быть изо-
[лированы, выращены в лабораторных условиях, в них можно ввести
ІГН, а затем возвратить в крово¬
ток больного. Во-вторых, лече¬
ние больной было эффективно.Общее количество лимфоцитов
нозросло до нормального уров¬
ни, а количество ADA-белка в
'1-клетках увеличилось до 25%
нормы. В-третьих, в течение 6 мес
перед очередным курсом лече¬
ния число «генно-инженерных»
лимфоцитов и ADA-фермента в
клетках оставалось постоянным.Из стерильного бокса девочку
перевезли домой (рис. 10.7).Выбор болезни для начала
использования генотерапии был
хорошо продуман. Ген ADA к
)тому времени был клонирован,
имел средние размеры, хоро¬
шо встраивался в ретровирус¬
ные векторы. Ранее при транс¬
плантации костного мозга приРис. 10.7. Первые две депочки, лечен¬
ные методом генной терапии по
поводу тяжелого комбинированного
первичного иммунодефицита, обу¬
словленного недостаточностью аде-
нозиндезаминазы (ADA), примерно
через 2,5 года после начала лечения
428Клиническаянедостаточности ADA было показано, что ключевую роль в
ни играют Т-лимфоци'гы. Следовательно, на эти клетки-мИ1
должна быть направлена генотерапия. Важным моментом craj
что функционирование иммунной системы возможно при yj
А DA-бел ка 5—10% контрольного. Наконец, Л/)у4-«генно-инженер|
Т-Лимфоциты имели селективное преимущество перед ИСХОД!
дефектными клетками.Семейная гиперхолестеринемия. Рецепторы ЛПНП, играї
ключевую роль в обмене холестерина, синтезируются в кл<
печени. Соответственно на гепатоциты (клетки-мишени) д(
быть направлена генотерапия. Одна попытка такою лечения сдс
в США у женщины 29 лет с резко выражении м атеросклерозом а#)(
ных артерий. Эффект предыдущего хирургического шунтиро!
уже сошел на нет. Брат больной умер от такой же болезни, не д<
до 30 лет. Генотерапия больной была проведена в несколько эта|
Больной была сделана частичная (около 15%) гепатэкт
Удаленную долю печени промыли раствором коллагеназы длЯ'
деления гепатоцитов. Получили около 6 млн гепатоцитов. ЗатеМ<
клетки выращивали в 800 культуральных чашках на питате;
среде. Во время роста в культуре для включения нормального
ЛПНП в качестве передающего агента использовали ретрови]
вектор. Трансгенные гепатоциты были собраны и введены паци(
через катетер в воротную вену (чтобы клетки достигли печени). Ч|
несколько месяцев при биопсии печени обнаружили, что в некок
клетках функционирует новый ген. Содержание ЛПНП в крови
на 15-30%. Улучшение состояния больной позволило провс
лечение только лекарствами, снижающими уровень холестерина^
Рак. Необычайно быстрый прогресс в изучении генома
века и методов генной инженерии позволяет развивать ген|
терапию не только для моногенно наследуемых болезней, но и-
таких многофакторных болезней, как рак. Генная терапия ЗЛ1
чественных новообразований уже начата, хотя на ее пути м1
трудностей, обусловленных необходимостью обеспечения селек!
ности, специфичности, чувствительности и безопасности переї
генов, в настоящее время применяется следующая стратегия
терапии рака: повышение иммуногенности опухоли путем всті
цитокиновых генов, генов, кодирующих главный комплекс ги<
совместимости, лимфоцитарных лигандов; направленная досі
ка (векторирование) опухолевых цитокинов в клетки, которі
flinita 10. Принципы лечения наследственных болезней429рі'мслііх опухоли локально могут реализовать токсические эффекты
|»ц|мример, в лимфоциты, инфильтрующие опухоли); использование
^шучолеспецифичсских пролекарствснных активаторов, т.е. вставка
(п-рмснтативно пролекарственно-активирующих генов, сливаюших-
[ои с промоториыми системами, которые реализуются через диф-
Іфгрсмциально контролируемую (идеально опухолеспсцифическую)
нрамскригщию; введение маркирующих генов, которые могут обе-
[«Ш'мивать выявление минимально оставленных после операции или
Ьп іристающихся опухолей; искусственная репрессия функций генов
[livie.M вставки генов.Небольшое число попыток генотерапии злокачественных опу-
\%и'\си связано с введением в клетки резецированной опухоли генов
[ИЛ-2 или ФНО. Затем эти клетки вводят подкожно в область бедра.
|Чі-|)сз 3 нед удаляют регионарный лимфатический узел (для места
Щк-дения смеси трансгенных опухолевых клеток). Культивируют
и )1имфоцитът, выделенные из этого узла, кроме того, размножают
[лимфоциты из опухоли (инфильтрирующие опухоль). Пациенту вво-
лм г об[цую массу лимфоцитов, что обеспечивает иммунную реакцию
|ціі опухолевые клетки. Так лечили больных злокачественной мелано¬
мой, раком почки, запушенным раком разных органов.[Изменение экспрессии генов как метод леченияЭто направление генной терапии открылось для научных разрабо¬
ток в связи с прогрессом функциональной геномики как части генома
человека, другими словами, с увеличением знаний об основах нор-
[міїльной и патологической экспрессии генов. Изменения экспрессии
генов можно достичь путем фармакологической модуляции или РНК-
Интерференции. На сегодня можно говорить о трех направлениях
[разработки методов лечения наследственных болезней путем измене-
I НИИ .экспрессии генов: повышение экспрессии в гене, определяющим
ійолезнь; повышение экспрессии в гене, не относящимся к болезни;
уменьшение экспрессии продукта аномального доминантного гена.-При наследственном ангионевротическом отеке (аутосомно-
доминантиая болезнь) у больных непредвиденно развивается
подслизистый и подкожный невротический отек. Обусловлено
это недостаточной выработкой ингибитора эстеразы компонен¬
та комплемента CL Из-за быстрой природы атак отека профи¬
лактически назначают лечение синтетическими андрогенами
(даназолом). Андрогены значительно увеличивают количество
430Клиническая Г0НІмРНК ингибитора Cl (возможно в нормальном и мутані
локусах). Частота серьезных приступов у больных резко уМ1
шается,— Терапия путем фармакологической модуляции экспрессии і
может быть направлена на увеличение экспрессии нормалі
го гена с целью компенсации эффекта мутации в другом
Гипометилирование ДНК увеличивает количество феталь»
гемоглобина у взрослых. Увеличение уровня фетального Г1
глобина (а2у2) вполне адекватно для пациента с серповил
клеточной анемией, поскольку гемоглобин F (фетальный):
ется нормальным переносчиком кислорода и препятст!
полимеризации гемоглобина S. Суть модуля ции в следуюия
метилирование промотора тормозится приемом аналога
дина деиитабином (5-аза-2’-деоксицитидин), который В)
чается вместо цитидина. Блокада метилирования привод!
увеличению экспрессии гена у-глобина и доли гемоглобині
крови. Такая комбинация, очевидно, окажется полезной
лечения р-талассемии.— Уменьшения экспрессии доминантного гена можно досі
путем РНК-интерференции (информацию о малой интер(
рующей РНК см. в гл. I). При многих наследственных болвЗ
патологические изменения вызваны токсическими продуі
(белки при болезнях экспансии нестабильных повторов)
снижением вклада нормального белка (аномальный кoлJ
при несовершенном остеогенезе). Патогенетически ясно,
надо уменьшить объем синтеза мутантного белка без на(
ния синтеза белка с нормального аллеля. Эта цель может
достигнута РНК-интерференцией. Цепи коротких РНК С1
ваются с целевой РНК и вызывают их распад. Ориентируя;
быстрый прогресс в изучении малых РНК (малых интері
рующйх РНК), можно надеяться на большой потенциал
технологии для лечения наследственных болезней, хотя
интерференционная терапия находится еще на раннем
развития.Риски клеточной и генной терапииКак видно из приведенных выше примеров, эра генотерапии
века уже началась. Определены принципы и методические под)
генотерапии, отобраны болезни, потенциально подлежащие
ІІ^і.іна 10. Принципы лечения наследственных болезней431[ЛС'юиию. Работа продолжается одновременно в разных странах и
|);иличных направлениях. Уже очевидно, что генотерапия будет
Применяться для лечения не только наследственных и сердечно-
іііи'удистьіх болезней, но и злокачественных опухолей и хронических
|йм|)усных инфекций.Имеете с тем необходимо отметить, что применять эти методы надо
К|>;ійне осторожно (это относится именно к применению, а не к раз-
р.иютке!). Это особенно важно при лечении наследственных болезней
(ік'обекно расширенном), даже если будут еще более решительные
прорывы R способах доставки генов в клетки-мишени. Необходимо
имимательно наблюдать за отдельными результатами лечения и стро¬
мі соблюдать этические и деонтологические принципы.Гри типа рисков клеточной и генной терапии уже обозначились.— Неблагоприятный ответ на вектор или комбинацию вектор/
бо.'[езнь. По крайней мере, один пациент погиб из-за патологи¬
ческого иммунного ответа на введенный ген с аденовирусным
вектором. Вывод из этого случая уже сделан — при выборе
вектора необходимо учитывать патофизиологические характе¬
ристики наследственного заболевания.— Инсерционный мутагенез, приводящий к злокачественным
новообразованиям. Существует вероятность, что переданные
клетка или ген (неважно — в чистом виде или с трансгенной
клеткой) может активизировать протоонкогены или нарушить
супрессоры опухолевого роста. Непредвиденный ранее меха-
низ.м онкогенеза был обнаружен у некоторых пациентов после
генотерапии Х-сцеп л енного комбинированного иммунодефи¬
цита. Перенос гена у этих больн ых содействовал развитию лим-
фопролиферативного заболевания.— Онкологический риск при клеточной терапии в связи с генети¬
ческой нестабильностью клеточных трансплантатов, в культуре
которых нередко возникают аномальные хромосомные клоны.Все типы рисков могут быть сведены к минимуму при правильной
проверке методов на безопасность.ЗАКЛЮЧЕНИЕИтак, лечение наследственных болезней — необычайно трудная
шдача, не всегда эффективно решаемая. Несмотря на это, оно долж¬
но быть постоянным и настойчивым. Нестойкость, а часто и недо-
432 Клиническая геністаточная выраженность эффектов терапии не означают отказа
постоянного проведения не только с клинической точки зрени#І
и по деонтологическим соображениям, при этом следует принт
во внимание две особенности лечения наследственных болезнейИ
’ необходимость долговременного контроля лечения;• исходная диагностическая точность до назначения лечені
связи с генетической гетерогенностью наследственных боле:)^КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯВиды симптоматического лечения
Генная терапия (общая схема)Генная терапия злокачественных новообразований
Генная терапия моногенных болезней (примеры)
ЕвфеникаКоицепиия вырождающихся семей
Коррекция обмена на уровне продукта
Коррекция обмена на уровне субстрата
Клеточная терапия
Стволовые клетки
Негативная евгеникаПримеры лекарственного симптоматического леченияПринципы патогенетического леченияТрансгенозФерментотерапия наследственных болезней
Хирургические методы леченияРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАБиология стволовых клеток и клеточные технологии: в 2 т. /
ред. м.А. Пальцева. — М.; Медицина, 2009. — 728 с.Долгих М.С. Возможности генной терапии, ее методы, объект!
перспективы II Успехи современной биологии. — т. 124. — № 2.
С. 123-143.Марахонов А,В., Баранова А.В., Скоблов М.Ю. РНК-интерференщ
фундаментальные и прикладные аспекты // Медицинская гене
ка. - 2008. - № 10, - С. 44-55.
I (miMi) 10. Принципы лечения наследственных болезней433Притчард Д.Дж., Корф Б,Р. Наглядная медицинская генетика /
Мі р. с англ.; под ред. Н.П. Бочкова. — М.; ГЭОТАР-Медиа, 2009. —
^00 с.РогаевЕЖ, Боринская С.А., ИсламгуловД.В., ГригоренкоАМ. МикроРНК
)и*.'|()»ека в норме и патологии // Молекулярная биологргя. — 2008. —
I. 42. - № 5. - С. 751-764.
ГлаваПРОФИЛАКТИКА НАСЛЕДСТВЕН!ПАТОЛОГ!ГРУЗ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
В МЕДИЦИНСКОМ И СОЦИАЛЬНОМ АСПЕКТАХ!Каждая семья мечтает иметь здоровых детей. Это становится
бенно актуальным после рождения больного ребенка. Уменьши
числа детей в семьях в развитых странах делает чрезвычайно важ1
оптимальный исход каждой беременности. В этом смысле профиі
тика наследственных болезней должна занимать ведущее место К|
работе врача, так и в системе здравоохранения.Известно, что вся наследственная патология определяется
зом мутаций, вновь возникающих и унаследованных из пред!
тих поколений. Эффекты мутационного процесса для популя(
человека выражаются в эволюционно-генетическом, мсдидинскС
социальном аспектах. Эволюционно-генетические последствия
ц ион кого npoiLecca (балансированный полиморфизм, летал ьн<
рассмотрены в гл. 1,Медицинские последствия мутационного груза — повыш!
потребность в медицинской помощи и сниженная продолжителы
жизни больных.Медицинскую помощь лицам с наследственными болезня)
поликлинических условиях оказывают в 5—6 раз чаще, чем л|
без такой патологии. В детских больницах общего профиля от И
20% пациентов составляют дети с наследственной патологией,
в 5—10 раз выше частоты таких больных в популяции. Более ч{
обращение к врачу людей с наследственной патологией вполне пс
но, так же как и более длительная их госпитализация. Во-пер|
сама болезнь требует большого объема медицинской помощи, а
гда и постоянного лечения. Во-вторых, наследственная болезні
исключает ожог, травму, инфекционные заболевания. Напротив,Исправлено и дополнено при участии канд. мед. ііаук Т. И. Субботш
І)»ііва 11. Профилактика наследственной патологии435Ніиііикают чаще, протекают тяжелее и длительнее в связи с мень¬
шими возможностями поддержания биохимического, иммунного и
гормонального гомеостаза у больных с наследственной патологией.В обобщенной форме медицинские последствия врожденных поро-
K01J развития и наследственных болезней представлены в табл. 11.1.Тпйлица 11.1. Последствия врожденных аномалий различгтых типов в раз¬
витых странах (по материалам Всемирной организации здраво-
охранеиия)АномалииПримерная
частота при
рождении
(на 1000)Последствияранняясмертность,%хроническое
состояние, %успешное
лечение,%Врожденные
пороки развития
^серьезные)30222454ХромосомныеҐюлезни3464Генные болезни1058Зі11Іісего4431,329,239,5Продолжительность жизни больных с наследственной патологией
шкисит не только от самой болезни, но и от уровня медицинской
иомощи. Хотя точные расчеты еще не сделаны, для стран с хорошо
развитой системой здравоохранения можно с большой уверенностью
полагать, что не менее 50% всех пациентов с наследственными болез¬
нями умирают в детском возрасте. В Канаде проведена комплексная
иценка ожидаемой продолжительности жизни для всех больных с
наследственной патологией (с разным возрастом начала болезней и
разной их тяжестью). Она оказалась на 20 лет меньше средней по
С1 ране (50 лет вместо 70).О социальной и медицинской значимости профилактики наслед¬
ственных болезней говорят высокий уровень инвалидности больных и
ікономические затраты на их содержание. В течение многих лет такие
Сольные остаются инвалидами, которые не могут себя обслуживать.
В домах-интернатах для детей-инвалидов средние расходы на 1 ребенка в
месяц равны среднемесячной зарплате по стране. Такие дети в интерна¬
тах живут в среднем до 10 лет. Из 1 млн новорожденных примерно 5000 —
кандидаты на многолетнюю тяжелую инвалидность с детства.
436Клиническая геніНаряду с медицинской и социальной значимостью профилакт^
наследственных болезней не менее важны психологические аспі
в семье с больным ребенком. Тяжесть и прогредиснтность TC4J
болезни создают, как показывают наблюдения, психологичес
напряженность даже в очень дружных семьях. Супруги или родсі
ники выясняют (или подозревают), кто виноват в рождении болы
ребенка. Члены семьи имеют разное мнение о передаче ребені
интернат (отказе от ребенка), особенно если он жил с родител»|
Постоянный уход за больным ребенком требует больших матери!
ных затрат, моральных и физических сил, что так или иначе всі
конфликтам. К тревоге за больною ребенка присоединяется стрнЛ
возможную болезнь у других детей.Хотя наследственные болезни, с обывательской точки зреї
встречаются редко, жизнь конкретной семьи концентрируетсЯіі
больном ребенке.Наконец, необходимость профилактики наследственных
ней диктуется и популяционными закономерностями их распрооі
нения, при улучшении медицинской 1ЮМ0ЩИ больные будз
только дольше жить, что автоматически повышает число больн!
наследственной патологией в популяции, но и передавать муті
следующим поколениям. Например, за последние 100 лет в Ан|
повысилась частота мутантного гена, обусловливающего вро;і
ный стеноз привратника. Операция по рассечению мышцы
вратника превратила эту аномалию из смертного приговора в
на брюшной стенке. Носители мутантного гена (после операцик]
уже не являются больными в строгом смысле) оставляют п(
ство, часть которого также имеет мутантный ген, а в попул>
дополнительно возникают новые случаи заболевания в резул!
мутационного процесса.В связи с планируемым размером семьи (как правило, 1—3 pd
ка) разница в числе детей у здоровых и наследственно отягощен]
супругов во многом нивелируется (репродуктивная компенса’1
Естественный отбор перестает регулировать численность потол
в наследственно отягощенных семьях бывает больше беременнЫ
(понятно, что часть беременностей заканчивается гибелью потоц|
на любой стадии внутриутробного развития), но число живых
такое же, как и в неотягощенных семьях. Часть таких детей rei
зиготны, в результате искусственно поддерживается повышен
уровень репродукции мутантных аллелей.
Іиіша 11. Профилактика наследственной патологии437ГЕНЕТИЧЕСКИЕ ОСНОВЫ ПРОФИЛАКТИКИ
НАСЛЕДСТВЕННОЙ ПАТОЛОГИИОбщие положенияс профилактической точки зрения всю наследственную патоло-III К) целесообразно подразделить на 3 категории:• вновь возникающие мутации (в первую очередь это анеуплоидии
и тяжелые формы доминантных мутаций);• унаследованные от предыдущих поколений (как генные, так и
хромосомные);• болезни с наследственной предрасположенностью.Различают 3 вида профилактики наследственной патологии,I к'рвичная профилактикаПод первичной профилактикой понимают действия, которые
/и>.'1жны предупредить зачатие больного ребенка; это планирование
Лг [ч)рождения и улучшение среды обитания человека.Планирование деторождения включает 3 основные позиции:• оптимальный репродуктивный возраст, который для женщин
составляет 21—35 лет (более ранние или поздние беременности
увеличивают вероятность рождения ребенка с врожденной пато¬
логией и хромосомными болезнями) (см. рис, 5.29);• отказ от деторождения в случаях высокого риска наследствен¬
ной и врожденной патологии (при отсутствии надежных методов
дородовой диагностики, лечения, адаптации и реабилитации
больных);• отказ от деторождения в браках с кровными родственниками и
между двумя гетерозиготными носителями патологического гена.Улучшение среды обитания человека должно быть направлено глав¬
ным образом на предупреждение вновь возникающих мутаций путем
жосткого контроля содержания мутагенов и тератогенов в окружающей
среде. Это особенно важно для профилак і ики всей группы соматиче¬
ских генетических болезней (врожденные пороки развития, злокаче¬
ственные новообразования, иммунодефицитные состояния и т.п.).Вторичная профилактикаВторичная профилактика предполагает прерывание беременно¬
сти при высокой вероятности заболевания плода или пренатально
438Клиническая генвТідиагностированной болезни. Прервать беременность можно ТОЛІ
ко в установленные сроки и с согласия женщины. Основан Ж
для элиминации эмбриона или плода является наследствени(
болезнь.Прерывание беременности — не самое лучшее решение, но П(
это единственный метод для вторичной профилактики большинст
тяжелых и смертельных генетических дефектов.Третичная профилактикаПод третичной профилактикой наследственной патологии пон|
мают коррекцию проявления патологических генотипов. Это можі
назвать и иормокопированием, поскольку при патологическом ген^
типе стремятся получить нормальный фенотип.Третичная профилактика проводится как при наследственні
болезнях, так и (особенно часто) при болезнях с наследствен!
предрасположенностью. С ее помощью можно добиться ПОЛ!
нормализации функций или снижения выраженности патоло(
ческого процесса. Для некоторых форм наследственной патолог|
она может совпадать с лечебными мероприятиями в обшемедиш
ском смысле.Предотвратить развитие наследственного заболевания (норме
пирование) можно внутриутробно или после рождения.Для некоторых наследственных заболеваний возможно внутрі
тройное лечение (например, при резус-несовместимости, НЄК0Т0І
ацидуриях, галактоземии).Развитие заболевания в настоящее время можно предотвраї
путем коррекции (лечения) после рождения больного. Типичн!
примерами болезней, для которых эффективна третичная пр01|
лактика, могут быть галактоземия, фенилкетонурия, гипотщ
(см. ниже) и др. Например, целиакия проявляется с началом
корма ребенка. В основе болезни лежит непереносимость глют
Исключение зтоі'о белка из пищи полностью гарантирует избавлен
от тяжелейшей патологии ЖКТ.Профилактика наследственных болезней и болезней с нас
ственной предрасположенностью должна включать несколі
этапов и проводиться на популяционном уровне. Совремеш
представления о наследственной патологии и методические возї
ности позволяют осуществлять профилактику на разных уро|
онтогенеза. Их характеристики и целевые установки npeACTaeJ
в табл. 11.2.
I лава 11. Профилактика наследственной патологии439Таблица 11,2. Характеристика основных типов популяционно-генетических
профилактических программ1'ил ирофилактической
программыПервичная цельВторичная цельI ІреконцепционнаяСнижение риска зача¬
тия больного ребенкаИнформированное
ретение о деторожденииПренатальнаяВыявление беременных
с риском рожления
больного ребенка в
период возможного пре¬
рывания беременностиДиагноз пораженного
плода, пренатальное
или неонатальное лече¬
ние, прерывание бере¬
менностиНеонатальнаяВыявление больных
для раннего леченияЛечение больныхОбщая популяционнаяВыявление факторов
высокого рискаРанняя диагности¬
ка и лечение широко
распространенных
болезней, оздоровление
среды обитания Как видно из табл. 11.2, мероприятия по профилактике можно
проводить до зачатия и заканчивать общепопуляционным обсле¬
дованием. При этом желательно использовать одновременно два
принципиально разных подхода: семейный и популяционный.
Каждый из этих подходов имеет свои разрешающие способности
и ограничения.Современная основа профилактики наследственной патологии —
теоретические разработки в области молекулярной природы наслед¬
ственных болезней, механизмов и процессов их развития в пре- и
мостнатальном периодах, закономерностей сохранения мутапий (а
иногда и распространения) в семьях и популяциях, а также изучение
процессов возникновения и становления мутаций в зародышевых и
соматических клетках.В генетическом плане можно выделить 5 подходов к профилактике
іиіследственной патологии, которые рассмотрены ниже.Управление экспрессией геновВ середине 20-х годов XX в. в экспериментах были обнаружены
явления пенетрантности и экспрессивности, которые вскоре стали
предметом изучения медицинской генетики. Выше отмечалось, что
440Клиническая генвтИІН.К. Кольцов сформулировал понятие «евфеника», под которым
понимал формирование хороших качеств или исправление болезнеї
ньтх проявлений наследственности у человека путем создания сос
ветствующих условий (лекарства, диета, воспитание и др.). Эти и;
стали реализовываться только в 60-х годах XX в., когда накопили^
сведения о первичных продуктах патологического гена и молекулЯ|
ных механизмах патогенеза наследственных болезней. Зная механщ
мы действия патологических генов, можно разрабатывать методы
фенотипической коррекции, другими слонами, управлять пенетряі
ностью и экспрессивностью.По мере прогресса науки накапливаются сведения о мете
профилактики наследственной патологии на разных стадиях онт
генеза — о лечебных или диетических воздействиях, Клиническі
примером управления экспрессией генов, уже прошедшим ДІ
тельную проверку практикой, является предупреждение после
сгвий фенилкетонурии, галактоземии и врожденного гипотиреоі
Клиническая картина этих болезней формируется в раннем пості
тальном периоде, в связи с чем принцип третичной профилакткІ
сравнительно простой. Болезнь должна быть диагностирован*!
течение нескольких дней после рождения, чтобы сразу иримен!
профилактическое лечение, предупреждающее развитие патологи*
ского фенотипа (клинической картины). Нормокопирование м(
достигаться диетическими (при фенилкетонурии, галактозем|
или лекарственными (при гипотиреозе) методами.Коррекция проявления патологических генов может начинат|
с эмбриональной стадии развития. Закладываются основы так Н{
ваемой преконцепционной и перинатальной профилактики наследст|
ных болезней (в течение нескольких месяцев до зачатия и до ро|
Так, например, гипофснилаланиновая диета для матери во BJ
беременности уменьшает проявления фенилкетонурии в постнац
ном периоде у ребенка. Отмечено, что врожденные аномалии
ной трубки (полигенный характер наследования) реже встречаї
у детей женщин, получающих достаточное количество витам]
Дальнейшая проверка показала, что если провести лечение
щин в течение 3—6 мес до зачатия и на протяжении первых мес*
беременности гипервитаминной (витамины С, Е, фолиевая кис;
диетой, то вероятность развития у ребенка аномалий нервной Тї
существенно уменьшается. Это важно для семей, в которых уже
больные дети, а также для популяций с высокой частотой патоЛ|(
I лава 11. Профилактика наследственной патологии441■ІССКИХ генов (например, по врожденным аномалиям нервной трубки
с()еди населения Ирландии). Подробнее о проблемах преконцепци-
«)ННОй профилактики репродуктивного здоровья см. в статье Л.Ф.
Курило на компакт-диске.В перспективе могут быть разработаны новые методы внутриу-I робкой коррекции патологического проявления генов, что особенно
важно для семей, в которых по религиозным соображениям непри¬
емлемо прерывание беременности.в таблице 11.3 приведены примеры врожденных аномалий, для
которых уже разработаны методы внутриутробного лечения.Габлица 11.3. Примеры внутриутробного лечения врожденных болезнейБолезниЛечениеЛдрсттогенитальный синдром у
девочекПрименение дексаме газонаМстилмалоновая ацидурияПрименение цианкобаламина
(витамина В12*)Множественная недостаточность
карбоксилазыВведение биотина*®Аритмия у плодаКардиотропныс препаратыЛллоиммунная тромбоцитспения и
другие формы болезней кровиОбменное переливание кропи или
тромбоцитарной массы плодуДиафрагмальная грыжа, обструк¬
ция мочевых путей, тератомы,
кисты легкихОперации на открытой или закры¬
той (аспирация иглой) маткеОпыт пренатальной терапии плодов женского пола с дефицитом
21-гидроксилазы может служить отправной точкой для разработки
методов лечения других наследственных болезней. Лечение Прово-
ІІИТСЯ по следующему плану.Беременным, имеющим риск рождения ребенка с врожденной
гиперплазией коры надпочечников, до Ю-й недели беременности
назначают дексаметазон (20 мкг/кг) независимо от состояния и
пола плода. Дексаметазон подавляет секрецию андрогенов эмбрио¬
нальными надпочечниками. Одновременно необходимо провести
пренатальную диагностику пола плода и ДНК-диагностику мутаций
и гене (путем либо биопсии хориона, либо амниоцентеза). Если
обнаруживается, что плод мужского пола или плод женского пола
пе поражен, то пренатальную терапию прекращают, а если у плода
442Клиническая генвтиженского пола находят мутации в гомозиготном состоянии, то лвЧ
ние продолжают до родов.Пренатальное лечение низкими дозами дексаметазона вряд
дает побочные эффекты. При наблюдении за детьми до 10-леТ|
возраста не обнаружено никаких отклонений, У женщин, ПС
чающих дексаметазон, наблюдаются небольшие побочные эффв1
(колебания настроения, прибавка массы тела, повышение артв!
ального давления, обший дискомфорт), но они согласны перенос)
эти неудобства ради здоровья дочерей. Положительные резулы
лечения плодов женского пола с дефицитом 21-гидроксилазы (ai
ногенитальный синдром) существенно перевешивают отрицатї
ные моменты.Третичная профилактика на основе управления экспресс!
генов особенно важна и эффективна для предупреждения бс
ней с наследственной предрасположенностью. Исключение из с]
факторов, способствующих развитию патологического феноті
а иногда и обусловливающих его, — прямой путь к профилакт(
таких болезней.Профилактике поддаются все моноген ные формы наследствеї
предрасположенности путем исключения из среды обитания
являющих факторов, в первую очередь фармакологических сре
у носителей недостаточности Г6ФДГ, аномальной псевдохолин!
разы, мутантной ацетилтрансферазы. В этих случаях речь и;
первичной (врожденной) непереносимости лекарств, а не о П1
ретенной лекарственной болезни (см. гл. 8).Для работы в производственных условиях, провоцирующих б(
ненные состояния у лиц с мутантными аллелями (например, кот
со свинцом, пестицидами, окислителями), необходимо проводиты
рабочих в соответствии с установленными принципами (см. гл. 7)ИХотя профилактика многофакторных состояний более слож!
поскольку они вызываются взаимодействием нескольких факт
среды и полигенных комплексов, все же при правильном ceMeJ
анамнезе и молекул ярко-генетическом анализе полиморфных
керов генов предрасположенности к заболеваниям можно вы і
«слабые» звенья в здоровье индивида и создать благоприятные
вия для замедления или приостановки развития многофакт(
го заболевания (предупредительная медицина). На этом прині
основана профилактика гипертонической болезни, атероскло]
рака легких.
I пава 11. Профилактика наследственной патологии443Элиминация эмбрионов и плодов с наследственной
патологиейМеханизмы элиминации нежизнеспособных эмбрионов и плодов
отрабатывались эволюционно. У человека это спонтанные аборты и
преждевременные роды. Конечно, не все они происходят по причине
неполноценности эмбриона или плода; часть из них связана с усло-
ииями вынашивания, т.е. с состоянием женского организма. Однако
определенно не менее чем в 50% случаев прерванных беременностейV плодов имеются либо врожденные пороки развития, либо наслед-
і гіїєнньіє болезни.Таким образом, элиминация эмбрионов и плодов с наследствен¬
ной патологией заменяет спонтанный аборт как природное явление.
Методы пренатальной диагностики быстро развиваются, поэтому
пот профилактический подход получает все большее значение.
Установление диагноза наследственного заболевания у плода служит
показанием для прерывания беременности.Процедура пренатальной диагностики и особенно прерывание
f)opeMeHHOCTH должны проводиться с согласия женщины. Как ука¬
пывалось выше, в некоторых семьях по религиозным соображениям
Осремснность не может быть прервана.Естественный отбор у человека в течение внутриутробного периода
позволил американскому эмбриологу Дж. Уоркани в 1978 г. сформу¬
лировать концепцию тератаназии. Под термином «тератаназия» пони¬
мается естественный процесс просеивания (или отсеивания) плодов
с врожденной патологией. Тератаназия может осуществляться путем
С{>здания «непереносимых»’ условий для плода с патологией, хотя такие
условия вполне приемлемы для нормального плода. Эти факторы как
Г>ы выявляют патологическое состояние и одновременно вызывают
г ибель плода. Некоторые экспериментальные доказательства в пользу
такой точки зрения уже имеются. Научные разработки могут быть
направлены на поиск методов индуцированной селективной гибели
іиюда с патолоі’ическим генотипом. Методы должны быть физиологич¬
ны ми для матери и абсолютно безопасными для нормального плода.Генная инженерия на уровне зародышевых клетокПрофилактика наследственных болезней может быть наиболее
полной и эффективной, если в зиготу будет встроен ген, по функ¬
ции заменяющий мутантный. Устранение причины наследственной
болезни (а именно это и есть наиболее фундаментальный аспект
444Клиническая геніїпрофилактики) означает достаточно серьезное манипулировії
генетической информацией в зиготе. Это могут быть: введение
мального аллеЯ^ї в геном путем трансфекции, обратная муті
патологического аллеля, включение нормального гена в работу,
он блокирован, выключение мутантного гена. Сложности этих
очевидны, но иї^т'^^сивньїе экспериментальные разработки в облі
генной инженерии свидетельствуют о принципиальной В03М0ЖІ
сти их решений- Генно-инженерная профилактика наследствен!
болезней стала we утопией, а перспективой, хотя и неблизко!Предпосылка^ для коррекции генов человека в зародышевых
ках уже созданы- Их можно обобщить в виде следующих положені— Расшифр<^®ка генома человека завершена, особенно на yj
не секве1Н‘^Р0в^ния нормальных и патологических алл«
Интенси0>^о развивается функциональная геномика, благод(
которой будут известны меж генные взаимодействия.— Любые г^^ны человека нетрудно получать в чистом виде
основе х^^^ического или биологического синтеза. Интеї
что ген глубина человека был одним из первых искусстве!полученных генов. и— Разработаны методы включения генов в геном человека с
ными век^рами или в чистом виде путем трансфекции.— Методы направленного химического мутагенеза позвол!
индуцирс^вать специфические мутации в строго определен!
локусе (получение обратных мутаций — от патологичесі
аллеля к ^нормальному).— В экспериментах на разных животных получены доказателм
трансфекіїии отдельных генов на стадии зигот (дрозофї
мышь, КО^а> свинья и др.). Введенные гены функционируї
организме-реципиенте и передаются по наследству, хотя »'
всегда по законам Менделя. Например, ген гормона роста Kf
введенный^ и геном зигот мышей, функционирует у родивш]
мышей. Т^кие трансгенные мыши значительно больше по
мерам и мзссе тела, чем обычные.Генно-инжеН^рная профилактика наследственных болезней
уровне зигот разработана пока слабо, хотя выбор способов си!
за генов и способов их доставки в клетки уже достаточно шиї
Решение вопросов трансгеноза у человека сегодня упирается не т<
ко в генно-инж^н^рї^ьіе трудности, но и в этические проблемы,
речь идет о комП^>^иции новых геномов, которые создаются не Э|
Іл.'ша 11. Профилактика наследственной патологии445шоцисй, а человеком. Эти геномы вольются в генофонд человечества.
к;1кова будет их судьба с генетической и социальной точек зрения,
(іудут ли они функционировать как нормальные геномы, готово ли
(К)шество принять на себя последствия неудачных исходов? Сегодня
14 ветить на эти вопросы трудно, а без ответа на них нельзя начинать
клинические испытания, поскольку произойдет безвозвратное вме-
ш-ггельство в геном человека. Без объективной оценки эволюцион-
МІ.ІХ последствий генной инженерии нельзя применять эти методы у
'к’ловека (даже с медицинскими целями на стадии зигот). Генетика
человека еще далека от полного понимания всех особенностей функ¬
ционирования генома. Неясно, как геном будет работать после введе¬
ния в него дополнительной генетической информации, как он будет
иссти себя после мейоза, редукции числа хромосом, в сочетании с
ІІОІЮЙ зародышевой клеткой и тлі.Все сказанное выше дало основание специалистам в области биоме-
іиіцинской этики на международном уровне [ВОЗ (Всемирная органи-
іация здравоохранения), ЮНЕСКО (Организация Объединенных Наций
110 вопросам образования, науки и культуры), Совет Европь[] рекомендо-
іі;гіъ временно воздержаться от проведения экспериментов, а тем более от
клинических испытаний с трансгенозом зародышевых клеток.Планирование семьиПри кысоком (более 20%) риске рождения больного ребенка и
отсутствии возможностей пренатальной диагностики рекомендуется
отказ от деторождения. Понятно, что такая рекомендация должна
быть дана после квалифицированной медико-генетической консуль¬
тации, когда нет методов пренатальной диагностики или для семьи по
различным соображениям неприемлемо прерывание беременности.Как известно, кровнородственные браки повышают вероятность
рождения ребенка с наследственной болезнью. Отказ от кровнород¬
ственных браков или ограничение деторождения в них может рассма¬
триваться как метод профилактики наследственной патологии. Об
■)гом говорят следующие факты.Кровнородственные браки на уровне двоюродных сибсов пред¬
почитает не менее 20% населения всего мира. По меньшей мере,
8,4% детей рождаются от родителей-родственников. Этот обычай
распространен в Восточном Средиземноморье и Южной Индии, а
ткже среди многих популяций, ведущих племенной образ жизни на
протяжении тысячелетий.
446Клиническая Г0ИІВ США, Канаде, России, большинстве европейских страї
Австралии, Новой Зеландии частота кровнородственных бр|
менее 1%, в среднеазиатских республиках, Японии, Северной Ині
южноамериканских странах — 1—10%, в странах Северной Афр»
Среднего Востока, Южной Индии — от 10 до 50%.Обычай кровнородственных браков в прошлом поддерживал
щину и семью. Однако эго отражается на частоте рождения дет
рецессивными болезнями. Для родителей-неродственников обі
риск мергворождений, младенческой и детской смертности
серьезных врожденных пороков развития равен примерно 2,5%,
умственного недоразвития составляет еще 3%. Суммарно эти р»
примерно удваиваются для детей супружеских пар — двоюрод!
сибсов. Если младенческая смертность в регионе высокая, то
эффект мало заметен, а если она низкая, то эффект кровного род(
в виде врожденных пороков развития и хронических инвалид!
рующих заболеваний становится явным.В популяциях с высокой частотой какой-либо болезни, в кот<
проводится диагностика носительства, возможен отказ от бр
гетерозиготных носителей.Для женщин после 35 лет существенно повышается вероятнс
рождения ребенка с хромосомными болезнями (см. гл. 5), для
чин — с генными (табл. 11.4).Таб.іица 11.4. Средний возраст отцов к моменту рождения детей с аутосои
доминантными заболеваниями (спорадические случаи)БолезниВозраст отца, годы |пробандконтрольная вьіборІ|Синдром Марфана36,629,8 "1Синдром Аперта34,830,2Нейрофиброматоз 1 типа34,230,7 1Ахондроплазия36,429,9 ^Синдром Варден бурга34,829,9 цРазница в возрасте отцов пробандов и отцов в контрольной вы(
ке составляет в среднем 5 лет. Причины этого явления неясны,!
для профилактики наследственных болезней его надо при ним j
внимание.
I кава 11. Профилактика наследственной патологии 447Таким образом, окончание деторождения до 35 лет и даже ранее
нилиется одним из факторов профилактики наследственных болез-
И1‘й. При планировании рождения 2—3 детей такого периода вполне
;ичтаточно для большинства семей.Охрана окружающей средыНаследственная изменчивость человека постоянно пополняется
1и>ными мутациями. Вновь возникающие спонтанные мутации опрс-
лі'ляют в целом до 20% всей наследственной патологии. Для некото-
t»Mx тяжелых доминантных форм новые мутации являются причиной
*)(!% наследственных болезней и более. Наследственные болезни,
поусловленные вновь возникшими мутациями, фактически нельзя
предсказать. Это случайные события, редкие для каждого гена.Пока нет предпосылок вмешиваться в процесс спонтанного мута-
цчіеза у человека, хотя интенсивные исследования антимутагенеза
н антитератогенеза могут привести к созданию новых методов
||[)офилактики наследственных болезней и врожденных пороков
|і;ізвития.Наряду со спонтанным мутагенезом у человека возможен индуци-
роиапный мутагенез (радиационный, химический, биологический).
Универсальный характер индуцированного мутагенеза на всех уров-
пих организации наследственности для всех живых суіцеств не вызы-
н;)ст сомнений. Естественно, что индуцированный мутагенез может
служить дополнительным источником наследственных болезней.
(’ ючки зрения профилактики наследственных болезней он должен
Оыть полностью исключен.Необходимо подчеркнуть, что индуцированный мутационный
процесс опасен в плане не столько индивидуального прогноза, сколь¬
ко популяционного. Отсюда вытекает, что исключение мутагенных
факторов из среды обитания человека является методом популяци¬
онной профилактики наследственных болезней.Методы проверки внешних факторов на мутагенность раз¬
работаны, их можно ввести в гигиенические регламентации по
охране окружающей среды. Этот вопрос очень важен, потому что
мутагенные эффекты факторов окружающей среды проявляются
не в экспонированной популяции, а в потомстве в нескольких
поколениях.к охране среды обитания человека относится также исключение
из нее факторов^ вызывающих экогенетические патологические реак-
448 Клиническая reHjции. Например, для лиц с пигментной ксеродермой (гомозигот)
исключить контакт с ультрафиолетовыми лучами, для лиц с нвд(
точностью ингибитора протеаз — с пылью, для носителей муті
порфири нового гена — с барбитуратами и т.д.МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИІОбщие положенияМедико-генетическое консультирование — специализирован!
вид медицинской помощи — является наиболее распространен!
методом профилактики наследственных болезней.Его суть заключается в определении прогноза рождения реб(
с наследственной патологией на основе уточненного диагноза,
яснении вероятности этого события консультирующимся и ПОМІ
семье в принятии решения о дальнейшем деторождении.Еше в конце 20-х годах XX в. С.Н. Давиденков впервые в
организовал медико-генетическую консультацию при Инстиі
нервно-психиатрической профилактики. Он четко сформулир<
задачи и методы медико-генетического консультирования. Оді
развитие данной области профилактики и генетики человека в це
затормозилось в 30-х годах практически во всех развитых CTpj
Это было связано с тем, что в нацистской Германии для обе
вания геноцида использовали генетические концепции и В1
насильственную стерилизацию как метод «оздоровления рї
Евгеническая стерилизация широко проводилась в США, Даі
Швеции и других странах. Во многом в связи с евгеникой, а lej
по политическим соображениям в Москве был закрыт Меді
генетический институт (1936).Хотя в США медико-генетические консультации (кабинеты) н1
ли организовываться уже в 40-х годах, действительно интенсиі
развитие такой помощи в разных странах (в том числе в РоссиІ
Германии) началось в 60-70-х годах. К этому времени произо!
большой прорыв в изучении хромосомной патологии и наследств
ных болезней обмена веществ.Термин «медико-генетическая консультация» определяет два п(
тия: врачебное заключение врача-генетика и специализироваї
учреждение здравоохранения (как самостоятельное, так и в сос1
объединения).
(imua 11, Профилактика наследственной патологии449Показания для медико-генетического консультирования:• наличие установленного или предполагаемого наследственного
шболевания в семье;• рождение ребенка с врожденным пороком развития;• іадержка умственного или физического развития ребенка;• повторные спонтанные аборты, выкидыши, мертворождения;• высокий риск патологии плода по результатам биохимического
скрининга маркерных сывороточных белков беременной;• наличие УЗИ-маркеров наследственного заболевания у плода;• возраст беременной 35 лет и старше;• близкородственные браки;• воздействие тератогенов в первые 3 мес беременности.В принципе каждой супружеской паре желательно пройти медико-
кпетическое консультирование до планирования деторождения
(проспективно) и, безусловно, необходимо после рождения больного
1>сГ)снка (ретроспективно).функции врача-генетикаврач-генетик выполняет две основные функции. Во-первых, он
с помощью других «узких» специалистов устанавливает диагноз,
используя при дифференциальной диагностике специальные генети-
чі'ские методы; во-вторых, он определяет прогноз здоровья будущего
(или уже родившегося) потомства. Перед врачом всегда возникают
ирачебные, генетические и деонтологические проблемы; на разных
этапах консультирования преобладают то одни, то другие.Медико-генетическая консультация включает 4 этапа: диагности¬
ку, прогнозирование, заключение, совет. Общение врача-генетика с
семьей больного должно быть доверительным и доброжелательным.ДиагностикаКонсультирование всегда начинается с уточнения диагноза наслед¬
ственной болезни, поскольку точный диагноз остается необходимой
предпосылкой любой консультации. Прежде чем направить паци-
снга н медико-генетическую консультацию, лечащий врач должен с
помощью доступных ему методов максимально уточнить диагноз и
определить цель консультации. Если необходимо дополнительно при¬
менять генеалогический, цитогенетический, биохимические и другие
специальные генетические методы (например, определить сцепление
генов или использовать молекулярно-генетические методы и т.п.),
го пациента направляют на медико-генетическую консультацию,
450Клиническая гвн$іи врач-іенетик помогает лечащему врачу в установлении диап
При этом может возникнуть необходимость направления пацИ!
или его родственников на дополнительное обследование. Со св(
стороны врач-генетик может поставить перед другими специали<
ми (невропатологом, эндокринологом, ортопедом, офтальмол(
и др.) конкретную задачу — распознать симптомы предполагаоі
наследственной болезни у пациента или его родственников,
врач-генетик не может имеїь столь универсальных знаний, Ч1
в полном объеме обеспечить клиническую диагностику несколЫ
тысяч наследственных болезней.На первом этапе консультирования перед врачом-генети!
возникает много сугубо генетических задач (генетическая гетб
генность болезни, унаследованная или вновь возникшая мутаЦ|
средовая или генетическая обусловленность данного врожден!
заболевания и т.д.).Диагноз уточняют в медико-генетической консультации с noMOt
генетического анализа. С этой целью врач-генетик пользуется клЩ
ко-генеалогическим, цитогенетическим и молекулярно-генетич<
ми методами, а также анализом сцепления генов, методами гене
соматических клеток. Из негенетических методов широко исполі
ются биохимические, иммунологические и другие параклиничв(
методы, которые помогают установить точный диагноз.Клинико-генеалогический метод при условии тщательного с(
родословной дает определенную информацию для установлі
диагноза наследственной болезни. Клинико-генеалогический
позволяет описать впервые встретившуюся, новую форму заб<
ния. Если в родословной четко прослеживается тип наследовї
то консультирование возможно даже при неустановленном ди1
зе (особенности использования клинико-генеалогического м<
и его разрешающие возможности рассмотрены выше). В медї
генетической консультации указанный метод применяется во
случаях без исключения.Цитогенетическое исследование, как свидетельствует опыт ра(
многих консультаций, применяется НС менее чем в 10% случаев.'
обусловлено необходимостью прогноза для потомства при уста
ленном диагнозе хромосомной болезни и потребностью в yTOHHf
диагноза в неясных случаях при врожденных пороках разв!С такими проблемами часто встречаются в практике консультиі
ния. Обследуют, как правило, не то.[ько пробандов, но и родит
Іііііва 11. Профилактика наследственной патологии451Биохимические, иммунологические и другие параклинические мето¬
ды не являются специфичными для генетической консультации, но
применяются так же широко, как и при диагностике ненаследствен-II мх болезней. При наследственных болезнях часто применяют одни и
II’ же тесты не только у пациента, но и у других членов семьи (состав¬
ление биохимической или иммунологической «родословной»).в процессе генетического консультирования часто возника¬
ет [ютребность в дополнительном параклиническом обследовании.
М гаких случаях больного или его родственников направляют в соот-
исгствующие специализированные учреждения.В конечном счете в медико-генетической консультации диагноз
уючняется путем генетического анализа всех полученных сведений,
и том числе (если это необходимо) данных о сцеплении генов или
[кчультатон исследования культивированных клеток. Врач-генетик
должен быть высококвалифицированным специалистом в разных
|)Г>ластях медицинской генетики.I Ірогноз для потомстваПосле уточнения диагноза определяют прогноз для потомства.
1$рач-генетик формулирует генетическую задачу, решение которой
исиовывастся либо на теоретических расчетах с использованием
методов генетического анализа и вариационной статистики, либо
ii:t эмпирических данных (таблицы эмпирического риска). Ясно,
'IIо обычная подготовка врача общей практики не позволяет ква¬
лифицированно сделать такой прогноз. Ошибка врача при непра-
иильном прогнозе для семьи может быть роковой: повторно родится
тяжелобольной ребенок либо семья неправомерно откажется от
лсторождения.Если применяется пренатальная диагностика, не требуется реше¬
ние генетической задачи. В таких случаях не прогнозируется рожде¬
ние ребенка с болезнью, а диагностируется заболевание у плода.Заключение медико-генетического консультирования
и советы родителямЗаключение медико-генетического консультирования и советы
родителям можно объединить. Заключение врача-генетика обяза¬
тельно должно быть письменным, потому что члены семьи могут
возвратиться к обдумыванию ситуации. Наряду с этим необходимо
устно в доступной форме объяснить смысл генетического риска и
помочь семье принять решение.
452Клиническая Г0ЬЗаключительные этапы консультирования требуют самого пр»
него внимания. Как бы ни совершенствовались методы расчета
(эмпирического или теоретического), как бы полно ни внедрі
достижения медицинской генетики в работу консультаций, ко»
тирование будет неэффективным, если пациенты неправильно п(
объяснение врача-генетика. Помогает и контакт с семейным Bf
которому супруги доверяют, поэтому очень важна согласованност»
ствий семейного (лечащего) врача и врача-генетика. Например, да)
установленном в пренатальном периоде диагнозе у плода не все жв1
ны принимают решение о прерывании беременности. При тяжелых|
мосомных болезнях (трисомии 13, 18, 21) прерывают беременж
женщин, при пороках нервной трубки — 76%, при синдроме Тер>
70%, при других формах хромосомных аномалий — 30%.Для достижения цели консультирования при беседе с пациені
следует уч итывать уровень их образован ия, социально-экономич<
положение семьи, структуру личности и взаимоотношения суп|
Многие пациенты не подготовлены к восприятию информац!
наследственных болезнях и генетических закономерностях,
склонны чувствовать вину за случившееся несчастье и страда!
комплекса неполноценности, другие вполне серьезно доверяКУГ’і
сказам знакомых, третьи приходят в консультацию с нереалі
запросами или ожиданиями, в связи с тем, что были неправиІ
осведомлены о возможностях генетической консультации (*'
числе иногда лечащими врачами). Необходимо иметь в видy,j
почти все консультирующиеся супруги хотят иметь ребенка (|
бы они не обратились за консультацией). Это значительно
шает профессиональную ответственность и лечащего врача, и
генетика. Каждое неточное слово может быть интерпретиі
в том направлении, в котором настроены супруги. Если cj
сильно опасаются иметь больного ребенка и хоіят родить здор<
то каждая неосторожная фраза врача об опасности усиливает с1
хотя на самом деле риск может быть небольшим. Наоборот, же
иметь ребенка бывает настолько сильным, что даже при болі
риске супруги принимают решение о деторождении, потому ЧТ04
сказал о некоторой вероятности рождения здорового ребенка.Изложение сведений о риске должно быть индивидуально прі
соблено к каждому случаю. В одних случаях следует говорить
вероятности иметь больного ребенка, в других — о 75% вероят!
рождения здорового ребенка. Однако всегда нужно убедить
ijina 11. Профилактика наследственной патологии453I'll гов в случайном распределении наследственных факторов, чтобы
устранить чувство вины за рождение больного ребенка. Иногда это
'(VBCTBO бывает очень сильным.На медико-генетическую консультацию иелесообразно направ-
кягь супругов не раньше чем через 3—6 мес после установления
лиагноза наследственной болезни, так как в этот период происходит
илаптация к ситуации в семье, а раньше какая-либо информация о
Оудущих детях воспринимается плохо.Тактика врача-генетика в помощи пациентам в принятии решения
окончательно не определена. Безусловно, она зависит от конкретной
і'пгуапии. Хотя решение принимают сами пациенты, в принятии
решения семьей роль врача может быть активной или сводиться толь¬
ко к объяснению смысла риска. По нашему мнению, врач-генетик и
лечащий врач (особенно семейный) должны помогать советом в При¬
пяти решения, так как при сушествующем уровне знаний в области
ІСНЄТИКИ у населения консультирующимся трудно самостоятельно
принять адекватное решение.Медицинские задачи консультирования решаются легче, чем
социально-этические проблемы. Например, при одной и той же
Гюлезни, при одной и той же вероятности рождения больного ребенка
разная обстановка в семье (обеспеченность, взаимоотношения супру¬
гов и др.) требует различных подходов к объяснению риска. В любом
случае принятие решения о деторождении остается за семьей.Организационные вопросыПри организации медико-генетических консультаций как струк¬
турных подразделений нужно опираться на сложившуюся в стране
систему здравоохранения и учитывать степень развития медицины в
целом, в том числе уровень знаний генетики у врачей. Консультации
функционируют как звено существующей системы медицинской
помощи населению.В большинстве зарубежных стран с развитым здравоохранением
система консультирования является 3-х ступенчатой: в простых слу¬
чаях прогноз для потомства определяется семейным врачом; более
сложные случаи попадают к врачу-генетику, работающему в крупном
медицинском центре; консультирование в сложных генетических
ситуациях осуществляется в специальных генетических консульта¬
циях. Для реализации этой в целом эффективной системы необхо¬
димо, чтобы каждый семейный или лечащий врач хорошо понимал
454Клиническая геніклиническую генетику, а организация медицинской помощи Н|
ВИЮ должна быть адекватной.Медико-генетические консультации как структурные подраз
ния лечебно-профилактических учреждений могут быть как
профильными, так и специализированными.Пробанды, обращающиеся в консультацию общего профиля по
логическому принципу, имеют самую различную патологию. Поскс
работа по уточнению диагноза в консультации занимает большое
разнообразный профиль заболеваний пробандов заставляет обсл«
и пробандов, и родственников. В связи с этим генетические КОНО]
тации целесообразно создавать на базе крупных многопрофилі
лечебно-профилактических учреждений республиканского или
ного подчинения. Больной и его родственники в этом случае
получить консультацию у специалистов и при необходимости
госпитализированы. Кроме того, консультация должна иметь возі
ность направлять на специализированное (томография, исследоаі
гормонального профиля и др.) обследование в другие учреждения,
больница, на базе которой функционирует консультация, не расг
гает такими возможностями. Тесный контакт с другими отделені
и их правильная соподчинснность — важный принцип работы мед|
генетической консультации общего профиля.Специализированные медико-генетические консультации
быть организованы при крупных специализированных больни!
в которых врач-генетик приобретает опыт консультирования
наследственным болезням одного профиля. В трудных случаях
сультации общего профиля могут направлять больных в специ!
зированную консультацию.Две консультации — общего профиля и специализированн!
могут функционировать параллельно, но независимо.В штат консультации обіцего профиля должны входить ві
генетики, цитогенетики и биохимики-генетики. Врач-генетик,
щий прием населения, должен иметь всестороннюю генетичео!
подготовку, так как ему приходится решать самые разнообраз
генетические задачи. Объектом исследования врача-генетика явл)
семья, а пробанд — лишь отправное лицо в этом исследовании. Лі
консультация требует сбора сведений о родственниках, а иногда
обследования. Заключение врача-гснетика о повторном риске за(
вания предназначается непосредственно семье, обратившейся за п(
щью, поэтому смысл заключения надо разъяснять в доступной
І/ііша 11, Профилактика наследственной патологии455(ш'редко нескольким членам семьи). Все это занимает гораздо больше
ирсмсни, чем прием больного любым другим специалистом. На пер-
|||1чкь[й осмотр пробанда и его родителей, а также на сбор семейногоI ниамнеза требуется от I до 1,5 ч. Повторная консультация (письменное
шключение, объяснение в доступной форме, помощь в принятии реше¬
ния) занимает в среднем 30 мин. Таким образом, один врач-генетик в
и'чсние рабочего дня может принять не более 5 семей.Из всех специальных исследований наибольшая потребность воз¬
никает в цитогенетических анализах (в среднем 1 исследование наI семью). Большая потребность в применении цитогенетического
метода обусловлена направлением на медико-генетическую консуль-
шцию прежде всего пациентов с хромосомной патологией, врожден-
іи.іми пороками развития и акушерской патологией. При этом, как
правило, обследуются не 1 человек, а 2 или 3.Биохимические исследования необходимы примерно 10% паци-
1’нтов, обратившихся в консультацию. Это довольно высокая цифра.
Олиако при большом разнообразии наследственных болезней обмена
HCLUCCTB повторное прііменение одних и тех же биохимических мето¬
дов в консультации бывает очень редко. В крупных городах целесоо-
Оразно создавать специализированные биохимические лаборатории с
широкими методическими возможностями для обследования боль¬
ных с разнообразными нарушениями обмена веществ.Таким образохм, тенетическая консультация как структурное под¬
разделение представляет собой звено поликлинической службы,
состояш;ее из кабинета врача-генетика, процедурной (взятие крови)
и лаборатории для проведения цитогенетических и просеивающих
Пиохимических исследований. Клинические, параклинические,
молекулярно-генетические, биохимические, иммунологические и
другие исследования проводят в специализированных лабораториях
и лечебно-профилактических учреждениях, к которы.м прикреплена
консультация. Такие консультации в больницах не исключают орга¬
низации высокоспепиализированных медико-генетических центров
со всеми необходимыми подразделениями.Анализ обращаемости в медико-генетическую
консультациюДо сих пор только незначительное число семей (вряд ли более
10%), которым требуется совет врача-генетика, обращается за такой
специализированной помощью. При этом более 50% направлен-
456Клиническая мных на консультацию лиц имеют неправильные показания
проведения. Это несоответствие связано с недостаточным ур(
медико-гснетических знаний у врачей и населения и с недостаї
пониманием организаторами здравоохранения значения мі
генетического консультирования как метода профилактики Н1
ственных болезней.Поскольку основным проводником идеи мcдикo-гeнeтичв^
консультирования является врач общей практики, от его знаН!
понимания задач консультаций зависит направление на такую
сультацию. Осведомленность населения по вопросам наследство»
болезней также влияет на обращаемость в медико-генетическую:
сультацию. Однако обоснованность обращений целиком зависі
компетентности врача.Соотношение пациентов, направленных врачами и самостоят
по обратившихся в консультацию, сильно колеблется. В разных
сулыациях доля самостоятельно обратившихся составляет от
80%. Это зависит от того, на кого (на врачей или на население)
направлена пропаганда, которая в значительной мере определі
обоснованность обратцений, т.е. точный диагноз и правильные
зания для проведения консультации.Распределение обратившихся в консультацию по группам за<
ваний должно соответствовать относительной частоте таких б(
ней в популяциях человека. Однако анализ обрашаемости по н<
логическому принципу в консультациях разных стран показы!
отклонения от теоретически ожидаемого распределения.Чаще всего в консультации обращаются семьи, имеющие д|
с хромосомными болезнями, врожденными пороками развитиі
нервно-психическими заболеваниями.Социальная характеристика пациентов в разных консультаці
однотипна. Большинство пациентов имеют высшее образовані
хорошо обеспечены. Мотивами для обращения в консультацию Я1
ются желание иметь здорового ребенка (около 90% опрошенньїї
желание вылечить больного ребенка (около 10% случаев). В 50% св1
отмечаются конфликтные взаимоотношения супругов.Эффективность медико-генетических консультаций мЦелью генетического консультирования в обтцепопуляцион!
смысле является снижение груза патологической наследственное!
а цель отдельной консультации — это помощь семье в принят
Ifinina 11. Профилактика наследственной патологии457Иі|і;іііильного решеиия по вопросам планирования семьи, лечения и
щпи ноза здоровья больного. Следовательно, критерием эффектив-
мсдико-генетического консультирования в широком смысле
инужит изменеиие частоты патологических генов, а результатом
ИжГюты отдельной консультации — изменение поведения супругов,
[імірашающихся в консультацию по вопросам деторождения.При широком внедрении медико-генетического консультирова¬
ния можно достигнуть некоторого уменьшения частоты наследствен¬
ных болезней, а также снижения смертности (особенно детской).
І’ііечетьі показывают, что из каждых 100 проконсультированных
го чей в 3—5 не рождаются больные дети (без консультации они
Г)ы родились), несмотря на то, что 25—30% проконсультированных
но следуют совету врача-генетика. Если бы лечащие (или семей-
ні-іе) врачи помогли супругам следовать таким рекомендациям, то
іффективность медико-генетического консультирования была бы
пце выше.Популяционные эффекты медико-генетического консультирова¬
ния выражаются в изменении частоты патологических аллелей. Этот
показатель изменится мало, потому что основной вклад в частоту
ІСНОВ в популяциях вносят гетерозиготные носители, а их частотаII результате консультирования практически не изменится. Если
консультируемые будут следовать советам врача-генетика, то умень¬
шится только число ГОМОЗИГО'1'НЫХ носителей. Снижение частоты
тяжелых доминантных болезней в популяциях в результате медико¬
генетического консультирования не будет существенным, потому что
ХО-90% из них являются результатом новых мутаций.Кабинеты медико-генетического консультирования должны быть
()рганизованы во всех областных и крупных городских больницах.
Объем меди ко-генетического консультирования, безусловно, зависит
от уровня медицинской помощи в стране.При развитом здравоохранении реальные потребности в мед и ко-
генетическом консультировании достаточно большие. Например,
всем семьям, где родились дети е врожденной и наследственной
патологией (их около 5%), требуется медико-генетическая помощь.
Следовательно, в России при расчетном числе 1 500 ООО родов в год
гак их семей будет 75 ООО. В медико-генетическом консультировании
нуждаются женщины старше 35 лет, решившие родить ребенка. В годІІ России рожает более 70 ООО женшин старше 35 лет. Другие расчеты
консультаций но поводу ранних форм сердечно-сосудистых заболева-
458Клиническая геніїний, рака, нервных, психических и других болезней иоказываюти
каждая 5-10-л семья нуждается в обшем или специализировш
медико-генетическом консультировании.ПРЕНАТАЛЬНАЯ ДИАГНОСТИКАОбщие вопросыПод термином «пренатальная диагностика» понимается с<
купность всех методов обследования состояния эмбриона
плода, направленных на выявление врожденных пороков раз
тия, наследственных болезней и любых других форм (инфекций
ных, травматичсских) заболеваний, развивающихся внутриут|
но. Цель такой диагностики — предупреждение рождения деі
врожденными и наследственными заболеваниями. Пренаталі
диагностика как научно-практическое направление возникла
70-х годах прошлого века и быстро прогрессировало, опираясі
успехи генетики и клинических дисциплин. Число пренаталы
диагностических процедур в настоящее время исчисляется де<
ками миллионов в год.Пренатальная диагностика наследственных болезней — компл!
ная быстро развивающаяся область медицины, использующ*
УЗИ, и оперативную технику (хорионбиопсия, амнио- и кордоценч
биопсия мышц и кожи плода), и лабораторные методы (цитоген!
ческие, биохимические, молекулярно-генетические).Забота семьи о здоровье будущего ребенка (а иногда и
боснованная обеспокоенность) требует не только оценки ге
тических и средовых факторов риска исхода беременн(
(медико-генетическая консультация), но и использования .мен
пренатальной диагностики.При организации и развитии системы пренатальной диагності
должны выполняться следующие условия,— Врачи, определяя показания к исследованиям, должны зн|
вероятность ложноположительных и ложноотрицатсльных ДІ
гнозов, или, другими словами, ограничения метода.— Пренатальная диагностика должна включать два этапа:• первый этап — выявление и отбор женщин (точнее, семвіповышенным риском неблагополучного в генетическом nj
исхода беременности при медико-генетическом коіїсультиі
I пива 11. Профилактика наследственной патологии459вании или первичном обследовании беременных, в том числе
с использованием методов просеивающей диагностики;• второй этап — уточняющая пренатальная диагностика. Любые
методы уточняющей диагностики (инвазивные или неинва¬
зивные, лабораторные, дорогостоящие, трудоемкие) применя¬
ют только у женщин с факторами риска.- Специалисты по пренатальной диагностике (акушер-гинеколог,
врач-генетик, врач лаборант-генетик) должны знать диагности¬
ческие ограничения метода не вообще, а конкретно в их лабора¬
тории (ультразвуковая гехника, возможность взятия образцов
тканей и клеток плода и др.). Нужно учитывать, что соответ¬
ствующая лабораторная диагностика может быть недоступной
или ограниченной.— Специалисты должны строго соблюдать стандарты определе¬
ния показаний и выполнения процедур и лабораторных анали¬
зов, осуществлять текущий контроль качества работы, а также
иметь статистику исходов беременностей и расхождений диа¬
гнозов (контроль после абортов или после рождения).Важность соблюдения всех перечисленных условий связана не
только с медицинскими, но и с деонтологическими соображениями:
исс эти вопросы обостряются в семье в ожидании ребенка.Методы пренатальной диагностики подразделяют на непрямые и
прямые.Непрямые методы “ акущерско-гинекологическое, серологическое
исследование, а также анализ эмбриоспецифических маркеров [РАРР-А
(ассоциированный с беременностью плазменный белок А ~ Pregnancy
Associated Plasma Protein А), АФП (а-фетопротеин), ХГЧ (хорионический
гонадотропин человека), несвязанный эстриол|. Перечисленные мар¬
керы составляют суть так называемых просеивающих лабораторных
методов.прямые методы — не инвазивное или инвазивное исследование
плода. Неинвазивное исследование практически ограничено УЗИ,
хотя в редких случаях применяют рентгенографию и др. К инва¬
зивным методам относятся хорион- и плацснтобиопсия, амнио- и
кордоцентез, биопсия тканей плода.Для каждого метода есть показания и противопоказания, разрешаю¬
щие возможности и осложнения. Выбор метода и вся тактика пренаталь¬
ной диагностики должны быть строго индивидуализированы в соответ¬
ствии с конкретной ситуацией в семье и с состоянием беременной.
460Клиническая ген<Скрининг беременных на основе определения
биохимических маркеров (просеивающие методы)Такие методы позволяют выделить женщин, имеющих [101
шенный риск рождения ребенка с наследственной или врожден!
болезнью. Методы должны быть доступными для широкого прИ1
нения и недорогими.Безусловно, медико-генетическое консультирование семей пр{
ивает их на предмет пренатальной диагностики. Оптимальным ы
антом просеивания с целью профилактики наследственной пат(
ГИИ путем пренатальной диагностики было бы медикО“іенетичс<
консультирование с проведением генеалогического анализа
семей, планирующих деторождение. В этом случае, по-видим(
около 10% женщин нуждались бы в более глубоком обследоваї
При меди ко-генетическом консультировании на пренатальную
гностику направляют женщин по следующим показаниям:• возраст 35 лет и старше (мужчин 45 лет и старше);• наличие в семье или в популяции пренатально выявляо!
наследственной болезни;• неблагоприятный акушерский анамнез (повторные спонтаї
прерывания беременности или рождение ребенка с врожденн!
пороками развития);• сахарный диабет;• эпилепсия;• инфекции у беременной;• лекарственная терапия;• контакты с тератогенными факторами.К просеивающим методам, определяющим необходимость И1зивной пренатальной диагностики, относятся УЗИ плода и onj
ление в сыворотке крови береіменной веществ, получивших наз1
сывороточных маркеров матери;• концентрации АФП; И• уровня ХГЧ;• уровня несвязанного эстриола;• РАРР-А.а-фетопротеин вырабатывает желточный мешок и печень плода,
белок экскретируется с мочой в амниотическую жидкость, откуда
никает в кровь беременной через плодные оболочки и плацент>\і|І
содержание меняется в течение беременности, в каждой лабої
должны быть установлены нормы в медианном выражении содерл
Іііііва 11. Профилактика наследственной патологии461OtMi ка для каждой недели беременности, потому что концентрации АФП
коисблются у предсгаоительниц разных рас и в различных геоірафи-
•114'к их зонах, причем распределение концентраций не подчиняется
шкоиу нормального распределения. Отклонение от среднего (нормаль-
жно) уровня показателя (обозначается в единицах МОМ — multiples of
mviiian) оценивается по отношению величины содержания АФП в крови
конкретной женщины к усредненной величине (медиане) содержания
/111II його белка у многих женщин при том же сроке нормальной беремен¬
ности. Этот метод позволяет заподозрить врожденные дефекты нервнойI рубки и брюшной стенки. При такой патологии концентрация АФП в
і'і.ііюротке крови беременной во П триместре существенно выше, чем
h норме (рис. п.1). Повышение уровня АФП регистрируется также при
ілстрошизисе, омфалоцеле, аномалиях почек.Поскольку в некоторых популяциях аномалии развития нервной
t рубки встречаются в несколько раз чаще, чем в среднем, в таких
популяциях необходимо определять концентрацию АФП у всех бере¬
менных. Показанием к данному исследованию является также отяго¬
щенная родословная, т,е. наличие в ней больного с аномалией нервной
трубки в пределах П1 степени родства по обеим линиям супругов.Концентрация АФП снижена с 15 по 18-ю неделю беременности
h крови женщин, вынашивающих плод с болезнью Дауна (рис. 11.2)
или другими хромосомными болезнями.ЧастотаЧастота1 2Умноженная медианаРис. 11.1 Концентрация (поосиабсцисс)
ш-фетопротеина (АФП) в сыворотке
крови беременной при вынашивании
нормального плода и плода с врож¬
денным дефектом нервной трубки: 1 —
непораженные; 2 — открытая spina
bifida: З — анэнцефалияУмноженная медианаРис. 11.2. Концентрация (по оси
абсцисс) а-фетопротсина (АФП) в
сыворотке крови беременной при
вынашивании плода с синдромом
Дауна: I — синдром Дауна; 2 — непо¬
раженные
462Клиническая геиіМеханизм этой ассоциации неясен, но ее существование не Н1
вает сомнений. Такое обследование беременных позволяет ВЫ1
до 20% случаев болезни Дауна.Медицинских противопоказаний для определения концентр
АФП нет. Женщину с измененным уровнем АФП направляї
дополнительное обследование. Если концентрация белка пові
на, то для уточнения диагноза аномалии нервной трубки про|
УЗИ и определяют концентрацию АФП в амниотической жид»
Если концентрация белка понижена, то назначают цитогенетичв!
исследование клеток (амниоцитов или лимфоцитов) плода.
Повысить эффективность просеивающей диагностики б(
Дауна путем анализа АФП позволяет определение уровня ХГЧ в і
ротке крови будущей матери. В норме содержание ХГЧ уменьш!
до низких значений после I триместра беременности. У 68% жені
вынашивающих плод с хромосомной болезнью, этот показатель
ется повышенным до родов. Медиана концентрации ХГЧ при chhj
ме Дауна повышена в 2 раза и более (рис. 11.3). Ложноположител!
результаты получают редко.Введение в просеивающую программу определения содері
неконъюгированного эстриола в сыворотке крови беременной
более расширяет диагностические возможности метода, правда,*
этом существенно увеличивается относительное число ложног
жительных ответов. Концентрация этого гормона значительноЧастотаЧастота1.0 2.0 5.0 10.0Умноженная медианаРис. 11.3. Концентрация (по оси
абсцисс) хорионического гонадотро¬
пина человека (ХГЧ) в сыворотке
крови беременной при вьпіашипа-
нии плода с синдромом Дауна: I —
непораженные; 2 — синдром Дауна0.10.51.0 1.5Умноженная мв1Рис. 11.4. Концентрация (по,
абсцисс) неконъюгироваН(
эстриола в сыворотке крови
менной при вынашивании
с синдро.мом Дауна: 1 — син|
Дауна; 2 — непораженные
Ііі.іва 11. Профилактика наследственной патологии463Аналитическаяд
концентрация11|)м вынашивании плода с болез¬
нью Дауна (рис. 11.4).Наибольшие диагностические
IU) ІМОЖНОСТИ предоставляет ком-
Ьітация трех описанных указан-
П1.ІХ тестов (рис. 11.5).В последние годы актив¬
на обсуждается возможность
использования и некоторых
иругих сывороточных маркеров
матери (например, РАРР-А),II іменение которых также тесно
коррелирует с трисомиями у
плода уже в 1 триместре.Компьютерные программы
позволяют сопоставлять разуль-
1;ггы и использовать полученные
показатели с достаточной сте-
ifCHbra достоверности. С путя¬
ми повышения эффективно-
иги биохимического скрининга
можно ознакомиться в однои¬
менной статье Т.К. Кащеевой на
компакт-диске.Хотя возможность достоверного неинвазивного пренатального
определения патологии или пола плода по периферической крови
посредством предварительного обогащения клеток или ДНК сомне¬
нию не подвергается, по причине дороговизны применение этих
методов остается в пределах научных исследований см. статью А.В.
Лаврова «Фетальные клетки и свободная плодная ДНК в крови мате¬
ри в неинвазивной перснатальной диагностике» на компакт-диске.К неинвазивным методам относится УЗИ. Радио- или рентге¬
нография применялась 20—30 лет назад (да и то не очень широко)
иа начальных этапах пренатальной диагностики. В последние годы
постепенно становится возможным применение с целью визуализа¬
ции плода МРТ. Несмотря на высокую разрешающую способность,
ценность метода значительно снижается из-за небольшой скорости
формирования изображения (секунды и десятки секунд), что вслед¬
ствие подвижности плода может приводить к неверным результатам.13 14 15 16 17 18 19 20 21 22 23 24НеделиРис. 11.5. Комбинация результатов
просеивающей биохимической диа¬
гностики врождсггных аномалий
нервной трубки и синдрома Дауна:
по оси абсцисс — срок беремен¬
ности; по оси ординат — аналити¬
ческая концентрация; Л — низкий
риск; Б — высокий риск; НЭ —
нсконъюгированиый эстриол
464Клиническая генетикі1УЗИ позволяет выявить как врожденные пороки развития, так И'
функциональное состояние плода, плаценты, пуповины, оболочвкН
Сроки проведения УЗИ в России определены приказом Министерстве}
здравоохранения. Это 10—13-я, 20—22-я и 30-32-я недели беременно»
сти. УЗИ можно также использовать для выявления задержки росі
эмбриона или плода, начиная с 6—8-й недели беременности.УЗИ \шжно применять и как просеиваюший, и как уточняющи!
метод, В некоторых странах УЗИ проводят всем беременным,
позволяет предупредить рождение 2-3 детей с серьезными врождеї
ными пороками развития на 1000 новорожденных, что составляв!
примерно 30% всех детей с такой патологией. Для проведения деталі
ного повторного УЗИ как уточняющей диагностической процедур
можно выделить следующие показания;• выявление отклонений (маркеров патологии) или пороков разв!
тия плода в ходе просеивающего УЗИ;• несоответствие размеров плода сроку беременности;• рождение предьщущего ребенка с врожденными пороками развитии• болезни у женщины (сахарный диабет, эпилепсия, алкоголизм ^
др.), повышающие риск рождения ребенка с врожденными по(
ками развития;• воздействие тератогенного фактора (радиация, химические веі
ства, инфекции) в первые 10 нед беременности;• врожденные пороки развития у кого-либо из супругов (или у р<
ственников 1—III степени родства по линиям обоих супругов).Краткий перечень врожденных пороков развития, диагностир5
мых с помощью УЗИ приблизительно в 80-90% случаев, представлі
в табл. 11.5. Диапазон пороков, распознаваемых с помощью этс
метода, достаточно широк. Этими сведениями должен владеть К1
дый врач. О возможностях пренатальной диагностики врожденні
пороков сердца можію узнать в одноименной статье И.М. Волкої
соавт. на компакт диске.Таблица 11.5. Врожденные пороки развития, диагностируемые с помощі
ультразвукового исслеловаг{няСистема или органыПороки §цнсАнэнцефалия, spina bifida, голопрозэнцефалия, 1
энцефалоцеле ijКонсч ностиРедукционные пороки, тяжелые формы карликово*!
сти с укорочением конечностей, олиго- и полидак-J
тили и. тяжелый несовершенный остеогенез J
Глава 11. Профилактика наследственной патологии 465Окончание таблицы П. 5Система или органыПорокиСердечно¬
сосудистая системаРазнообразные пороки сердца и крупных сосудовМочеполоваясистемаАгенезия почек, поликистоз почек, удвоение почки,
выраженные гидронефрозы, опухоли яичниковЖКТАтрезия двенадцатиперстной кишки, дефекты
передней брюшной стснки, диафрагмальная грыжаЛегкиеКистозно-адсноматозпая мальформация легкихИнвазивные методыПервоначально к инвазивным методам относилась только фето-
скопия. Теперь инвазивными методами получают клетки и ткали
эмбриона, плода и провизорных органов в любом периоде гестаиии.
Разработка методов взятия материала стимулировалась появлением
более совершенных методов лабораторной диагностики наследствен¬
ных болезней. Инвазивные методы совершенствуются в нескольких
направлениях: более раннее получение образцов для исследования,
более широкий спектр образцов, более безопасные для беременной
и плода методы взятия образцов.К настояш,ему времени в мировой практике имеется достаточный
опыт (миллионы обследованных) по применению хорион- и пла-
центобиопсии, получению амниотической жидкости (амниоцентез),
биопсии тканей плода, взятию крови плода (кордоцентез).Хорион- и плацентобиолсия применяются для получения неболь¬
шого количества ворсин хориона или кусочков плаценты в период
с 7-й по 16-ю неделю беременности, процедура осуществляется
трансабдоминально или трансцервикально под контролем УЗИ
(рис. П.6, П.7). Принципиальной разницы между показаниями
для применения этих двух способов биопсии нет. Результативность
процедуры зависит от того, каким методом специалист владе¬
ет лучше. Хотя хорионбиопсия технически проста, необходимы
достаточный опыт и постоянное техническое совершенствование.
Хорошие результаты получают акушеры, делающие не менее 200—
400 хорионбиопсий в год, неудачи составляют 1%, На основе боль¬
шого материала (несколько миллионов случаев) сделаны выводы
об осложнениях после хорионбиопсии. После трансцервикальной
хорионбиопсии примерно у 10—30% женщин возникает небольшое
466Клиническая гвиіУльтразвукРис. 11.6. Трансабдоминальная хорион- или плацентобиопсияВорсинкихорионаМаткаОграничительВлагалище Шприц
Рис, 11,7. Трансцервикальная хорион- или плацентобиопсия
I лава 11. Профилактика наследственной патологии467кровотечение, очень редко ~ маточная инфекпия, после транс-
:іГ)ломинального способа у 2,5% женщин возможна угроза преры-
ИІІНИЯ беременности.Одним из осложнений хорионбиопсии является спонтанный аборт
Ои>1кидыш). Общие потери плода после хорионбиопсии составляют вV педнем 2,5—3%, в эти цифры входит и частота спонтанных выкиды¬
шей. Собственно хорионбиопсия индуцирует, очевидно, не более 2%
случаев прерывания беременности.Каких-либо нарушений плаценты, роста плода, появления врож¬
денных пороков развития и увеличения перинатальной смертности
после хорионбиопсии не наблюдается. В некоторых центрах отмече¬
но, что ранняя хорионбиопсия (в срок до 8 нед беременности) может
индуцировать поперечные врожденные ампутации конечностей, так
называемые редукционные пороки. В связи с этим (с 1992 г.) хорион-
биопсию рекомендуется проводить после 8-й недели беременности, а
после 11-й недели делают плацентобиопсию.Образцы хориона (ворсины) подлежат цитогенетическому,
молекулярно-генетическому, биохимическому исследованию с целью
выявления наследственной патологии. При аспирации ворсин хорио¬
на в материал могут попадать клетки децидуальной оболочки матки,
что может приводить к диагностическим ошибкам. Считается, что в
4% случаев лабораторная диагностика биоптатов хориона дает лож-
ноположительные результаты (например, в 1,5% анализов отмечается
хромосомный мозаицизм, который является мозаицизмом хориона, а
не эмбриона), а иногда (хотя и крайне редко) — ложноотрицательные
результаты. Точность анализов во многом зависит от квалификации
врача лаборанга-генетика.Амниоцентез — пункция плодного пузыря с целью получения около¬
плодной жидкости с находящимися в ней амниоиитами. Используется
для пренатальной диагностики с начала 1970-х годов. Накоплен
оіромньїй опыт проведения этой процедуры. Диагностическая зна¬
чимость метода не вызывает сомнений. Обычно процедура осущест¬
вляется на 15-18-й неделе беременности, ранний амниоцентез прово¬
дят на 12-15-й неделе беременности. Риск осложнений беременности
при амниоцентезе меньше, чем при хорионбиопсии, по данным
некоторых авторов, всего 0,2%. По этой причине во многих центрах
пренатальной диагностики предпочитают делать амниоцентез, а не
хорионбиопсию. В случае неудавшегося анализа хорионбиоптатов
пренатальную диагностику повторяют с помощью амниоцентеза.
468Клиническая генвіРис. 11.8. АмниоиентезАмниоцентез проводят 40J
переднюю брюшную СТЄІ
(трансабдоминально) женщи!
под контролем УЗИ (рис. 11,|
Чрезцервикальный амниоцемі
возможен, но применяется ред|
Из амниотической полосі
извлекают 3-30 мл жидкости.Предлагавшиеся ранее бя<
химическое и вирусологиЧЄСІ
исследования амниотичсс!
жидкости малоинформатии!
для пренатальной диагностиі
Из биохимических показателей жидкости только концентрат
АФП диагностически значима. Уровень АФП существенно ПО!
шается при аномалиях нервной трубки и дефектах передней брі
ной стенки.Основным диагностическим материалом при амниоцентезе Я1
ются клетки. Их обязательно надо культивировать (на это затрачі
ется 2—4 нед) и для цитогенетических, и для биохимических ИСОІ
дований. Только молекулярно-генетические варианты диагностик!
помощью ПЦР не требуют культивирования клеток,Кордоцентез ~ внутриматочная пункция сосудов пуп(
ны для получения крови плода (рис. 11.9). Сроки кордоцентезлч
18-22-я неделя беременности. Образцы крови используют для цитої
нетической (культивируются лимфоциты), молекулярно-генетичес
и биохимической диагностики наследственных болезней. ItКордоцентез применяют для диагностики хромосомных б<
ней, наследственных болезней крови (гемоглобинопатии, KOgлопатии, тромбоцитопенЩ
иммунодефицитов, гемат(
гического статуса при pej
сенсибилизации, внутриутрї
ных инфекций. 1|Поданным мультицентро!
исследования, частота ослож!
ний при кордоцентезе суммаї
по 16 российским центрам п|
Рис. 11.9, Кордоцентез натальной диагностики не Щ
Іііава 11. Профилактика наследственной патологии469тишает 1%. Первая попытка получить материал успешна в 80—97%
случаев. Преимущество кордоцентеза по сравнению с амниоцен-
ІСЮМ заключается в том, что кровь удобнее для исследования,
чем клетки амниотической жидкости. Лимфоциты культивируют¬
ся быстрее (2—3 дня) и надежнее, чем амииоциты. Молекулярные
методы быстрого кариотипирования в пренатальной диагностике
см. на компакт-диске в одноименной статье В.А. Тимошевского и
И.Н. Лебедева,Биопсия тканей плода как диагностическая процедура осуществля-
сгся во II триместре гестации под контролем УЗИ.Для диагностики тяжелых наследственных болезней кожи (ихти¬
оз, эпидермолиз) делают биопсию кожи плода с патоморфологиче¬
ским (а иногда и с электронно-микроскопическим) исследованием
материала. Морфологические критерии наследственных болезней
кожи позволяют установить точный диагноз или уверенно отвер¬
гнуть его.Для диагностики мышечной дистрофии Дюшсина на внутриу¬
тробной стадии разработан иммунофлюоресцентный метод. Для
)того производят биопсию мышц плода. Биоптат обрабатывают моно-
кJюнaльными мечеными антителами к белку дистрофину, который у
больных не синтезируется. Соответствующая флюоресцентная обра¬
ботка высвечивает белок. При унаследовании патологического гена
сиечение отсутствует. Этот прием является примером диагностики
наследственной болезни на уровне первичного продукта гена. В слу¬
чае миопатии Дюшенна такой метод дает более точные результаты,
чем молекулярно-генетическая диагностика.ЗаключениеВрачу общей практики необходимо иметь представление 6 мето¬
лах пренатальной диагностики, их возможностях и ограничени¬
ях, о показаниях для направления на исследования. Конкретные
сроки ее проведения и выбор метода (а иногда методов) опреде¬
ляет группа (команда) пренатальной диагностики (врач-генетик,
акушер-гинеколог и врач лаборант-генетик), основываясь на состоя¬
нии здоровья беременной, течении беременности, психологической
готовности женщины к процедуре. Объем и возможности вторичной
профилактики наследственных болезней путем элиминации эмбрио¬
нов и плодов после пренатальной диагностики представлены в сум¬
мированном виде в табл. И.6—11.8.
470Клиническая генвіТаблица 11.6. Методы пренатальной диагностикиМетоды (группа)Вид методасрок для проведения (недїя
беременности) JМеди ко -генети ческое
коне ул ьт и рова н иеДо и во время беремен HOOTijОпределение в сыворотке крови беременной J
содержания: _j• АФП16-20-я ^Просеивающие• хгчІ6-20-Я ^• нсконъюгиропанного
эстриола16-20-я 1• ацетил холинэстс разы16-20-я '• РАРР-АІ1-12-Я .jjНеинвазивныеСкрининговое (просеи¬
вающее) УЗИШ-20-30-Я іУточняющее УЗИВ течение всей беременное!МРТв течение всей беременной!Хорионбиопсия9-11-я "іПлацентобиопсия11-18-я JАмниоцентез15-17-яИнвазивныеКордоцентез18-22-я JБиопсия кожи14-16-я 1Биопсия мышц18-22-я jФетоскопия18-22-я 1Таблица 11.7. Сравнительная характеристика методов пренатальной диаг
стики с использованием трансабдоминальпой техники
образцов (по материалам Всемирной организации здравое
нения)УспешныеСрокКариотипированиеЧястоЯМетодпроие<беремен¬время донадеж¬ПОТеМдуры, %ности, нед.результатаностьплода, 1Биопсияхориона>99>9Часы-
10 сутОченьвысокая'-»3
I лава 11. Профилактика наследственной патологии471Окончание таблицы И.7Лмниоцентез>991510-20 сутОчспьвысокаяКордоцентез>95183-4 сутОченьвысокая1-21'аГілица 11.8. Показания для применения разных метояоп инвазивной прена¬
тальной диагностикиМетод (материал)ПоказанияЦитогенетическое исслелопание
(клетки хориона, культивиро-
ианпыс амниотические клетки
или лимфоциты плода)Возраст женщины к моменту родов
35 лет и старше; хромосомная мутация у
одного из родителей; рождение преды-
душсго ребенка с хромосомной болез¬
нью; низкий ypOBCFIb АФП в сыворотке
крови беременной; результаты УЗИ,
предполагающие хромосомную болезнь
у ллодаМолекулярно-генетическое,
биохимическое или иммуноло-
1'ическое исследование (хорион,
амниотические kjjctkm, кровь)Высокий риск рождения ребенка с ген¬
ной болезнью по результатам медико¬
генетического консультирования
(ретро- или проспективного) или про¬
сси ваюишх программ выявления гете¬
розиготного носигельсгва; диагностика
инфекции плода, иммунодефицитов,
иммунной несопместимости матери и
плодаПатоморфологическое исследо¬
вание (кожа и мышцы плода)Высокий риск рождения ребенка с
наследственным заболеванием кожи
(ихтиозы, эпидермолизы), с мышечной
дистрофией ДюшсннаПРЕДЫМПЛАНТАЦИОННАЯ ДИАГНОСТИКАБлагодаря развитию методов вспомогательных репродуктивных
гехнологий [экстракорпоральное оплодотворение, интрацитоплаз-
матическая инъекция сперматозоида п ооцит (ICSI)], с одной сто¬
роны, и усовершенствованию методов лабораторной диагностики
наследственных болезней, с другой, в конце 90-х годов прошлого века
зародилась предымплантационная диагностика. Материалом для
472Клиническая геніїпредымплантаиионной диагностики являются полярные тельцн
отдельные бластомерьт, полученные с помощью МИКрОіМанИПуЛЯІ
из бластоцист.Такая диагностика относится к методам первичной профи ли I
к и наследственных болезнеіі. Ее преимущество заключается в
что она помогает избежать повторных абортов после обычной
натальной диагностики в семьях с высоким риском наследстЕісні
патологии,Предымллантационная диагностика успешна при следующ)
условиях:• получение зародыша на предымплантапионной стадии развит
(до 5—7-го дня после оплодотворения);• наличие диагностических (аналитических) микрометодон
уровне одной или нескольких клеток;• микрохирургическая техника (микробиопсия) для взятия МИ1
мального числа клеток без повреждения зародышевого пузырь!•точные медицинские показания со стороны семьи для диаг!
стики.Получение прсдымплантационных эмбрионов возможно нехиі
гическим маточным лаважем и оплодотворением в пробирке.С помощью маточного лаважа можно получить еще не импл!
тировавшийся зародыш в течение 90—130 ч после оплодотворен!
К этому времени зародыш спускается из маточной трубы в матку,
процедура безболезненная и безопасная. Соответствующие присг
собления (улавливатель, проводник и катетер) уже прошлрі испЫ1
нис. Процедура не влияет на последующие овариальные циклы И;
препятствует будущим беременностям.После подсадки зародыша в матку нормальная беременное
наступает в 50% случаев.Экстракорпоральное оплодотворение и интрадитоплазматичес!
инъекция сперматозоида в ооцит (1CS1) хорошо зарекомендовали се{
в акушерской практике. Эти методы применяют для преодолеЩ
различных видов бесплодия.Микрохирургическая процедура выделения клетки для лабої
торной диагностики осуществляется с помощью микроманипулят
ра (рис. П.10). От зародыша на стадии 8—16 клеток можно отдел!
1—2 клетки. Иногда исследование ограничивается вторичным полі
ным тельцем (оно несет геном яйцеклетки). Зародыш сохраня!
і пава 11. Профилактика наследственной патологии47311 условиях глубокой заморозки
шли зародыш продолжает раз¬
ки паї ься в искусственных усло-
тіях), пока проводится анализК'ІСТКИ,Подсадка после заморозки
ію іможна во время любого дру-
ІОГ0 овариального цикла.Диагностика на уровне од¬
ной или нескольких клеток в
кастояшее время реальна при
многих болезнях. Ее проводят
с использованием ПЦР, моно¬
клональных антител, улътра-
микроаналитических методов.Уже появились сообщения об
успешной диагностике на пред-
і.імплантапионной стадии син¬
дрома Марфана, миотоничсской
дистрофии, хореи Геитингтона,
семейного полипозного рака
1'олстой кишки, муковисцидоза,(}М2-ганглиозидоза (болезнь Тея—Сакса), синдрома Леша-Найхана,
талассемии, спинальной мышечной атрофии, мышечной дистрофии
Дюшенна, умственной отсталости с ломкой Х-хромосомой, фенил-
кетонурии.На сегодняшний день предымплантационная диагностика доступ¬
на примерно для 50 нозологических форм моиогенной и хромосомной
природы.Можно надеяться, что в ближайшие годы методические возмож¬
ности предымплантапионной диагностики расширятся как в обла¬
сти получения диагностического материала, так и аналитических
методов (культивирование предымплантационных эмбрионов и их
бластомеров, микроманипуляции, криопрезерваиия).Предымплантационная диагностика — крайне важное направ¬
ление в системе новых репродуктивных технологий, потому что
но непонятным пока причинам частота анеуплоидий у эмбрионов
человека, согласно данным отечественных исследователей, оченьРис. 11.10. С помощью микромани¬
пулятора удаляется одна клетка (с
ядром) из человеческого умбриона
на стадии 12 клеток. Фото с видео¬
записи
474Клиническая генетик!высокая: 30—50% аномальных зародышей при оценке анеуплоидий
по хромосомам 13, 16, 18, 21, 22, X и Y. Подробнее о нредымплон*;
тационной диагностике можно узнать из статьи А.В. Светлакоші «|
соавт. «Задачи и перспективы предимплантационной генетической]
диагностики» на компакт-диске.ДОКЛИНИЧЕСКАЯ ДИАГНОСТИКА,
ПРОСЕИВАЮЩИЕ ПРОГРАММЫ
И ПРОФИЛАКТИЧЕСКОЕ ЛЕЧЕНИЕИдея просеивания (скрининга) родилась в США в начале XX в. (осмої
школьников, профилактические осмотры на выявление туберкулез
регулярные обследования рабочих и др.). Перечисленные приемы увв
ренно вошли в практику мирового здравоохранения. Скрининг nj
полагает массовое и безотборное обследование, профи л акти ческ)
направленность и двухэтааность (по меньшей мере) диагностики.Просеивание (скрининг) можно определить как идентификаииі
нераспознанных болезней с помощью быстро осуществляемых тестс
Это обеспечивает отбор лиц с вероятным заболеванием. Их повторі
обследуют с применением уточняющих диагностических методе
позволяющих либо отвергнуть предполагавшийся на первом эт|
диагноз, либо подтвердить его.Идея массового обследования новорожденных на наследстве!
ную болезнь стала проверяться в 60-х годах XX в. К настоящв!
времени уже окончательно сложились основные положения Ml
совой диагностики наследственных болезней на доклиническс
стадии (критерии отбора наследственных болезней для скриниї
и диагностические методы).Массовое просеивание новорожденных на наследственные болеэ!
проводится, если они:• без своевременного профилактического лечения существе!
снижают жизнеспособность, приводят к инвалидности и к нес
ходимости специальной помощи больному;• поддаются точной биохимической или молекулярно-генетичес!
диагностике на доклинической стадии;• поддаются эффективному профилактическому лечению;• имеют частоту 1 : 10 ООО и выше. Лишь в некоторых странах
наличии исследовательской группы просеивание новорожд<
Глава 11. Профилактика наследственной патологии 475ных осуществляется для болезней, встречающихся с частотой1 : 20 000-1 : 40 ООО.Диагностические методы массового просеивания новорожденных
должны отвечать следующим критериям.— Экономичность. Методы должны быть технически простыми и
дешевыми Б массовых исследованиях.— Диагностическая значимость. Ложноотрицательных результатов
практически не должно быть, а соотношение истинно положи¬
тельных и ложноположительных должно быть не менее 1:5. Это
можно назвать чувствительностью и специфичностью метода.— Надежность или воспроизводимость. Результаты обследования
должны одинаково воспроизводиться в работе разных исследо¬
вателей.— Доступность биологического материала. Метод должен быть
приспособлен к анализу биологического материала^ легко полу¬
чаемого в малом количестве, хорошо сохраняемого (хотя бы
несколько дней) и приемлемого для пересылки Б централизо¬
ванную лабораторию.Основная цель программ массового просеивания новорожден¬
ных на наследственные болезни — раннее выявление заболевания
на доклинической (досимптомной) стадии и организация лечения.
Программа обязательно включает следующие этапы:• взятие биологического материала для исследования у всех новорож¬
денных и доставка материала в диагностическую лабораторию;• лабораторная просеивающая диагностика;• уточняющая диагностика всех случаев с положительными резуль¬
татами при просеивании;• лечение и диспансеризация больных с контролем хода лечения;• медико-генетическое консультирование семьи.Таким образом, программы массового обследования на наследствен¬
ные болезни, поддающиеся профилактическому лечению, могут учреж¬
даться только в рамках федерального или регионального (в том числе
городского) здравоохранения. Для этого нужны организация специ¬
ального звена в структуре здравоохранения и немалые экономические
затраты, которые в общегосударственном масштабе компенсируются
уменьшением числа инвалидов с детства. Многочисленные исследо¬
вания, проведенные в разных странах, показали, что экономическая
эффективность просеивающих программ (сохранение здоровья леченых
индивидов) дает государству 5—Ю-кратную экономическую выгоду.
476Клиническая генвпПервая программа скрининга новорожденных на фенилкетої
рию была организована в США примерно 25 лет назад. За прош(
шее время в разных странах проверялись также программы б0Л1
чем для 10 наследственных болезней обмена веществ. В результі
были отработаны изложенные выше критерии массовой диагносТ!
ки наследственных болезней. В конечном счете страны с развиты!
здравоохранением стали проводить массовое просеивание новоро)
денных лишь для нескольких болезней, характеристики которі
представлены в табл. 11.9. Следует отметить, что эти рекомендац!
справедливы для популяций европеоидной расы. Для других рао,'
иногда популяций частоты указанных болезней могут быть ниже,
тогда не будет показаний для их массовой диагностики.Таблица 11,9. Характеристики болезней, по которым проводится массої
просеивание новорожденныхБолезньЧастота
среди ново¬
рожденных
(в среднем)КлиническиепоказанияВозмож¬
ность профи¬
лактичес¬
кого леченияНалнчи# ^
метода 1
просенваюиМІ
диагностики!Фснилкетонурия1:10 000+++ шврожденныйгипотиреоз1:5000+++ JВрожденная
гиперплазия над-
почечникоп1:5000+++ JГалактоземия1:40 ООО++Муковисцидоз1:3500+-+ щС 2006 г. в России проводится неонатальный скрининг ш
наследственных заболеваний: адреногенитального синдрома, галі
тоземии, врожденного гипертиреоза, муковисцидоза, фенилке
нурии — с целью их раннего выявления, своевременного лечо!
профилактики инвалидности, развития тяжелых клиничеС!
последствий, снижения детской смертности.Для проведения неонатального скрининга забирают обрі
крови из пятки новорожденного на 4-й день жизни (у доношенШ
и на 7-й день у недоношенных через 3 ч после кормления. 3*
образцов крови осуществляется на специальные фильтровал]
тест-бланки, которые выдаются медико-генетической консультаї
Глава 11. Профилактика наследственной патологии477учреждениям здравоохранения, оказывающим медицинскую помощь
женщинам в период родов. С результатами, проблемами и перспекти¬
вами неонатального скрининга можно ознакомиться в одноименной
статье Л.П. Назаренко с соавт. на компакт-диске.Фенилкетонурияв России в последние десятилетия введена федеральная программа
скрининга, основанная на флюорометрическом количественном мето¬
де определения фенилаланина в крови. В разных странах применяются
различные методы. Суть диагностики фенил кетону рии сводится к
количественному определению концентрации фенилаланина в крови.
Опыт показал, что пропущенные случаи фенил кетон у рии являются
не ошибками лабораторных методов, а следствием недобросовестности
или небрежности при взятии крови в родильных домах.в случае положительного результата скрининга у детей проводят
уточняющую биохимическую диагностику. Это уже более сложная,
иногда многоэтапная процедура. Во-первых, необходимо подтвердить
гиперфенилаланинемию, во-вторых, следует разобраться в ее причине.
Она может быть обусловлена типичной фенил кетону рией (недоста¬
точность фенилаланингидроксилазы), вариантными или атипичны¬
ми формами этой болезни, наследственной гиперфен ил ал анинемией
(доброкачественной), другими формами нарушения метаболизма.При подтверждении диагноза фенилкетонурии ребенка переводят
на искусственную бесфенилаланиновую диету.В таблице 11,10 приведены названия питательных смесей для
питания детей с фенилкетонурией.Таблица НЛО. Бесфенилаланиновые питательные смесиНазваниеВозраст,годыФирмаСтрана-производительАфенилак0-1«Нутритек»РоссияАфенилак1-3«Нутритек»РоссияТстрафен 403 и старше«Нутритек»РоссияМД мил ФКУ-00-1«Херу»Россия-ИспанияМД мил ФКУ-11-7«Херу»Россия—ИспанияМД мил ФКУ-37 и старше«Херу»Россия-ИспанияХР аналог0-1«Нутриция»ГолландияПАМ универсальный1 и старше«Нутриция»Голландия
478Клиническая генетиіВитамины и минеральные соли дают в виде фармакологически!
препаратов. Со временем диету расширяют. Дети старше 1 года лег»
переносят пищевой фенилаланин. Лечение диетой проводится ПС
регулярным биохимическим контролем концентрации фенилалаН!
на в крови: 2 раза в неделю в 1-й месяц (обычно это период rocnJ
тализации), еженедельно до 6-месячного возраста, 2 раза в месяц
возрасте 6 мес — 1 год и ежемесячно в дальнейшем. Этот контр(
позволяет определять адекватность терапии.При своевременном начале лечения бесфенилаланиновой ДИ1
той в первые месяцы после рождения у детей, гомозиготных
гену недостаточности фенилаланингидроксилазы, не отмеч!
ется никаких клинических признаков задержки психическог
или физического развития. С 9—11 лет у таких пациентов дис1
можно существенно расширить, но они остаются под наблюдение!
специалиста-генетика. Это особенно актуально для женщин, болы
ных фенилкетонурией, так как во время беременности повышенны!
уровень фенилаланина и его дериватов в сыворотке женщины тої
сичен для генетически здорового плода. Это требует специальн!
профилактических мероприятий.Врожденный гипотиреозПод названием «врожденный гипотиреоз» понимают сумі
наследственной и ненаследственной патологии; агенезию щитовил
ной железы, эктопию щитовидной железы, дисгормоногенез {наслс
ственные болезни), аутоиммунные процессы. Основные клиническ!
проявления: умственная отсталость, резкое отставание в росте, от«
ность кожных покровов, а при дисгормоногсііезе — и развитие 3(
Для всех форм болезни приемлема одна и та же программа массово!
просеивания, поскольку биохимическими маркерами являются сн1
жение в плазме крови содержания тироксина и увеличение содо|
жания тиреотропного гормона (ТТГ). Диагностическая значиме
просеивания в полной мере проявляется при определении обо1
маркеров, но по экономическим соображениям часто останавлиі
ются на определении ТТГ.Применяют радиоиммунный и иммуноферментный (им!
нофлюоресцентный) методы просеивающей диагностики,
чувствительность и специфичность примерно одинако!
Иммуноферментный метод предпочтительнее по техническ!
соображениям. Тироксин и ТТГ определяют в образцах крб^
Глава 11. Профилактика наследственной патологии 479новорожденных, высушенных на специальной фильтровальной
Г>умаге (см. выше).При положительном результате диагноз должен быть подтвержден
>ндокринологом в клинических условиях и результатом лабораторно-
ю анализа сыворотки крови на тироксин, ТТГ и другие гормоны.Заместительную терапию левотироксином натрия (L-тироксин*)
нужно начинать у детей с положительным просеивающим тестом еще
до окончательного подтверждения диагноза. Эффективность терапии
достаточно высока, но лечение, начатое после 2-го месяца жизни,
неэффективно, хотя к этому возрасту заболевание клинически про¬
является только у 4% больных. Это делает особенно важной раннюю
диагностику.Врожденная гиперплазия надпочечниковЭта клиническая форма объединяет 9 наследственных нару¬
шений ферментных процессов в трех взаимосвязанных метаболи¬
ческих путях стероидогенеза. Наиболее часто встречается недо¬
статочность 21-гидроксилазы, на основе чего разработаны методы
просеивающей диагностики у новорожденных. Эти методы выяв¬
ляют биохимический маркер болезни — увеличение содержания
17-а-оксипрогсстерона в крови. Разработаны радиоиммунный и
иммуноферментный методы, позволяющие четко улавливать повы¬
шенный уровень 17-а-оксипрогестерона. Чувствительность обоих
методов достаточно высокая, но по техническим соображениям
предпочтительнее иммуноферментный метод.при установлении диагноза по клинической картине необходимо
лабораторное подтверждение.Лечение — замести гельная гормональная терапия, обычно успешное.ГалактоземияВ России, начиная с 2006 г., проводится скрининг на галактозе-
мию. Это заболевание — следствие мутаций ферментов, участвующих
в метаболизме галактозы. Из-за недостаточности этих ферментов
в организме накапливаются токсические метаболиты (галактоза и
іалактозо-1-фосфат), которые негативно действуют на внутренние
органы (печень, мозг, почки, кишечник). Помимо этого для галакто-
земии характерно угнетение активности лейкоцитов, что чаще всего
приводит к сепсису. Заболевание манифестирует на 1—2-й неделе
жизни. Дети без лечения живут не дольше полугода.
480Клиническая генетикСкрининг новорожденных проводят на 4—5-й день у доношен HI
детей и на 7-й день — у недоношенных. Важно, чтобы ребенок на>
дился на естественном вскармливании или на питании с помощі
галактозосодержаших смесей.Существует несколько подходов выявления галактоземии. В ннш<
стране оценивают уровень метаболитов и галактозы в сыворот!
новорожденіїьіх с помощью тандемной масс-спектрометрии. П|
уровне галактозы >7 мг% в сыворотке новорожденного тест повтор
ют, при уровне >10 мг% — считают положительным. Одновременж
этим проводят и ферментный анализ флюорометрическим методе
Основное преимущество ферментного анализа — возможность ВЫ1
вить недостаточность вне зависимости от характера питания. Однії
эп’от метод позволяет выявить лишь гомозигот по мутации галактоі
1-фосфатуридилтрансферазы (в гене GALT), тогда как гетерозиготі
гомозиготы по мутациям других ферментов (галактокиназы и УД<
галактозо-4-эпимеразы) могут быть пропушены.Основным недостатком биохимического скрининга новорс
денных на галактоземию является достаточно большое количес!
ложноположительных результатов. Связано это с тем, что услої
получения, транспортировки и хранения материала (температур
влажность) могут привести к снижению активности фермента.Подтверждают диагноз молекулярно-генетическими методаї
Найдено уже более 180 различных мутаций в гене GALT, но паиб(
часто встречаются Q188R и K285N, В совокупности они составля!
около 70% случаев заболевания классической формой галактозем!
Описана также мутация N314D в этом же гене, приводящая к raj
тоземии Дуарте. Этот вид галактоземии отличается относителі
мягким течением, уровень фермента снижается незначительно,
приводит к стертой клинике. Галактоземию Дуарте чаще всего мо>
выявить только путем скрининга. .VIДо сих пор внедрение скрининга новорожденных на галактозеМ!
считается спорным вопросом, так как данное заболевание не отвеч
всем критериям ВОЗ для массового скрининга: заболевание явлж
редким, оно может манифестировать еще до получения резулы
скрининга, лечение не всегда полностью купирует все симпк
Поэтому в последнее время все чаще говорят о селективном скрині
ге галактоземии, включающем проведение метаболических ис<
дований совместно с молекулярно-генетическими в группах ри<
Именно таким образом можно исключить ложноположителы
Глава 11. Профилактика наследственной патологии 481результаты скрининга и определить вид галактоземии, что суще¬
ственно повысит эффективное! ь лечения.Лечение галактоземии предполагает исключение из пищевого
рациона галактозы. Это позволяет уменьшить и предупредить раз¬
витие осложнений со стороны внутренних органов. Однако раннее
начало лечения не влияет на возникновение отдаленных последствий.
У больных галактоземией часто отмечается задержка умственного и
речевого развития, развиваются эндокринологические и неврологи¬
ческие нарушения, отклонения со стороны половых органов. Узнать
подробнее о галактоземии можно в статье Е.Ю. Захаровой с соавт.
«Гальктоземия тип I: клинические проявления, диагностика и лече¬
ние» на компакт-диске.МуковисцидозНеонатальный скрининг на муковисцидоз основан на значи¬
тельном повышении концентрации иммунорсактивного трипсина в
крови новорожденных, страдающих этим заболеванием. Протокол
скрининга на муковисцидоз включает 4 этапа.— Первичный тест на иммунореактивный трипсин. Если уровень
иммунорсактивного трипсина больше или равен 70 нг/мл, то
проводят 2-й этап.— Ретест на иммунореактивный трипсин проводят на 21—28-й
день. Если уровень иммунорсактивного трипсина больше или
равен 40 нг/мл, то переходят к 3-му этану.- Потовая проба — определение хлоридов в поте биохимическим
методом. Если содержание хлоридов 60—80 ммоль/л (погранич¬
ный результат), то проводят 4-й этап. Если больше 80 ммоль/л,
то считают скрининг на муковисцидоз положительным.- ДНК-диагностика (молскулярно-гсиетическое обследование
проводится в том случае, если потовая проба имеет сомнитель¬
ные результаты или по желанию родителей).Молекулярно-генетическое подтверждение доступно лишь в неко¬
торых регионах России, поэтому ключевым этапом скрининга явля¬
ется потовая проба, которую обычно делают дважды.Ранние лечебно-реабилитационные мероприятия, включающие
заместительную ферментную терапию, приводят к улучшению нутри-
ТИБНОГО статуса, что ведет к улучшению состояния и замедлению
необратимых процессов в бронхолегочной системе, а следователь¬
но, определяет и более высокую продолжительность жизни. Раннее
482Клиническая генвтйівыявление больных муковисцидозом способствует профилактиі
данного заболевания путем проведения пренатальной диагностикиїїИтак, предотвратить клинические проявления наследственж
патологии можно путем профилактического лечения болезни
предсимптоматической стадии. Прогресс молекулярной и клини>
ской медицины позволяет идти дальше по пути нормокопирован!
патологических генетических состояний. Уже разрабатываются мет(
ды пренатального лечения (см. табл. 11.3), и имеется опыт лечсні
метил малоновой ацидурии на внутриутробной стадии больши!
дозами витамина Bj2- Недостаточность карбоксилазы лечат преИ!
тально введением биотина. Лечение дексаметазоном врожденш
недостаточности 21-гидроксилазы можно начинать с 9-й недели бе(
менности, если проведена пренатальная диагностика. Женщинаї
больным фенилкетонурией и гетерозиготным по гену фенилкетої
рии, во время беременности рекомендуется диета с низким содер!
нием фенилаланина.В последнее время развивается гипотеза преконцепционной Л|
филактики. Период такой профилактики включает несколько ме(
цев до зачатия и ранние сроки развития зародыша. Предполагаете
что подготовка организма женщины (полноиенная витаминизир(
ванная диета, антиоксидантная терапия, коррекция иммунитет
отсутствие стрессов) до зачатия и на ранних стадиях развития зац
дыша (до 10-й недели) способствует уменьшению частоты врожде
ных пороков развития многофакторной природы. Особенно чеі
это показано для аномалий нервной трубки (различные вариаі
спинномозговых грыж) и врожденных пороков сердца. Част<
повторного рождения ребенка с таким пороком в среднем pal
4,6%, а у женщин, которые принимали фолиевую кислоту и ВИТ
мин с, — 0,7%.КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯГенная инженерия и первичная профилактикаГруз наследственной патологии (медицинские последствия)Лабораторная пренатальная диагностикаМедико-генетическое консультированиеМетоды пренатальной диагностикиМетоды просеивающей пренатальной диагностики
Глава 11. Профилактика наследственной патологии 483Первичная, вторичная и третичная профилактика наследствен¬
ных болезнейПреконцепционная профилактика
Показания для пренатальной диагностики
Предьшплантационная диагностика
Пренатальное лечение
Прогноз здоровья потомстваПpoceивaюIJ^иe программы диагностики болезней обмена для
новорожденныхПрофилактическое лечение
ТератаназияУльтразвуковая диагностика врожденных пороков развития
Фенотипическая коррекция
Функции врача-генетикаРЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАЛавров А. в. Фетальные клетки и свободная плодная ДНК в крови
матери в неинвазивной пренатальной диагностике // Медицинская
генетика. — 2009. — Т. 8. — № 7. — С. 3-8.Пренатальная диагностика наследственных и врожденных болез¬
ней / под ред. Э.К. Айламазяна, B.C. Баранова. — М.: МЕДпресс-
информ, 2006. — 416 с.
Глава 1ЭТИЧЕСКИЕ ВОПРОСІ
МЕДИЦИНСКОЙ ГЕНЕТИКІБольные с наследственными заболеваниями и их семьи состаал!
ют большую группу населения, по отношению к которой суш,еств)
много этических вопросов, возникающих при оказании им врачвї
ной номоши. Все элементы врачебной деонтологии и обшечеловеЧ|
ской морали, сформулированные обществом со времен Гиппократ
и сейчас остаются в силе для этой группы больных. Однако свов(
бразный характер течения большинства наследственных болезнв!
а именно: их пожизненность, прогрсдиентность, тяжесть и особв!
но их свойство передаваться от поколения к поколению — ста!
перед врачами и обш^сством специфические этические вопросы
фоне новейших успехов в генетике человека. Суть успехов в том, ЧІ
они обеспечили такие генетические технологии, которые ПОЗВОЛЯІ
вмешиваться в геном человека. Не все в освоении бурного научнс
прогресса поддается сразу законодательной или правовой регуі
ции для зашиты индивида. Многое остается для решения на ур01
моральных позиций общества.Предпосылками для выработки правовых и законодательН!
регуляций любого характера являются моральные нормы общее!
Следовательно, биоэтическое рассмотрение новых научных дости)
ний — первый шаг к предупреждению отрицательных последсті
научных достижений. Данное положение особенно явно просі
тривается на примере бурно развивающихся дисциплин, к которыЦ
несомненно, относится генетика во всем ее многообразии.Необходимость осмысления этических аспектов использовакІІ
новых технологий существовала всегда. Отличие современного пв(
ода состоит в том, что скорость реализации идеи или научной pajj
ботки резко повысилась. Например, от рождения идеи пренатальї
диагностики наследственных болезней до ее широкого применен^
в клинической медицине прошло лишь 3 года.Формирование правовых положений для медико-биологичесі
науки и практической медицины на основе моральных принципов об1
ства не может быть отделено от формирования правосознания в разН1|
социальных группах (ученые, врачи, пациенты, политики и т.д.).
Глава 12. Этические вопросы медицинской генетики 485Главным результатом биоэтических разработок становится сво¬
евременное обсуждение морально-правовых проблем, возникающих
в новых областях науки и практики. На основе обсуждений и науч¬
ных исследований разрабатываются национальные и международные
законы, рекомендации, правила — как для проведения исследований,
так и для практической реализации их результатов,В генетике человека четко прослеживаются непосредственная
связь научных исследований с этическими вопросами, а также
зависимость научных поисков от этического смысла их конеч¬
ных результатов. Генетика шагнула вперед настолько, что человек
рано или поздно сможет определять свою биологическую судьбу.
В связи с этим использование всех потенциальных возможностей
медицинской генетики реально только при строгом соблюдении
этических норм.прогресс медицинской генетики поставил этические вопросы в
связи с:• генной инженерией (генодиагностика и генотерапия);• разработкой методов ранней диагностики наследс і венных болез¬
ней (см. статью В.Л. Ижевской «Этико-правовые аспекты генети¬
ческого тестирования и скрининга» на компакт-диске);• новыми возможностями медико-генетического консультирования
(оценка гетерозиготных состояний, оплодотворение in vitro и др.);■ пренатальной и предымплантационной диагностикой наслед¬
ственных болезней (см. статью «Этические аспекты пренатальной
диагностики» на компакт-диске);• охраной наследственности человека от повреждающего действия
новых факторов окружающей среды.Поскольку медицинская генетика имеет дело с больным чело¬
веком или его семьей, она должна опираться на выработанные и
проверенные веками принципы медицинской деонтологии. Однако
в современных условиях этого недостаточно, потому что в биоэтике
возникают новые вопросы:• внедрение принципиально новых медипинских и генетических
технологий (искусственное оплодотворение, суррогатное мате¬
ринство, пренатальная диагностика, генетическое тестирование
донора, генотерапия) стало массовым в медицинской практике;• медико-генетическая помощь и генетические технологии все
больше коммерциализируются как на Западе, так и у нас в
стране;
486Клиническая генетик! і* появились новые формы взаимоотношений врача и пациент(1|
формируются общества пациентов и их родителей (с болезнью|
Дауна, муковисиидозом, фенилкетонурией и др.); \І• потребовалось этическое и правовое регулирование научньЦ
исследований, их направлений и итогов, поскольку они затр
гивают интересы общества (дополнительное финансирование! і
угроза войны и т.д.).Большинство этических вопросов современной генетики человеКІ
можно решить в рамках 4 принципов (делай благо, не навреди, автОі||
номия личности, справедливость) и 3 правил (правдивость, конфН#]
денциальность, информированное согласие).Принцип «делай благо» изменялся в медицинской генетике КІ
протяжении 100 лет в зависимости от моральных устоев общества
прогресса генетических знаний.Применение этого принципа на практике сталкивается с протиВ0|
речием между благом конкретного человека и благом группы людс
или общества в целом. На этой основе возникли евгенические пр(
граммы насильственной стерилизации пациентов с отклонениям!
в умственном и физическом развитии в США, Дании, Швециі
Германии и других странах. Главным обоснованием таких мероприі
тий был приоритет общего блага нации над индивидуальным,
привело к тому, что в США в результате евгенической программ!
было стерилизовано более 100 ООО человек. В скандинавских стран!
доля стерилизованных в населении была даже выше, чем в СШ^В Германии было стерилизовано более 350 ООО человек.Современные моральные принципы обязывают искать компі
мисс между интересами общества и отдельного человека. В pj
международных документов утверждается норма, согласно котор<
интересы пациента ставятся выше интересов общества.Соблюдая принцип «делай благо», не во всех случаях можно onj
делить, что является благом для пациента, а что — благом для
семьи. Если раньше право решать принадлежало врачу-генетиі
(например, директивное консультирование считалось нормой),
современная мораль общества принципиально изменила ситуаци!
Принимает решение пациент вместе со своей семьей, а недиректиіі
ное консультирование стало нормой работы врача-генетика.Принцип «не навреди» запрещает исследовательские и терапевтии
ские действия, связанные с неоправданным риском неблагоприятна
последствий для пациента. Однако на стадии клинических испы1
Глава 12. Этические вопросы медицинской генетики 487НИИ моральная ответственность врача занимает большее место, чем
правовая. С принципом «не навреди» медики и биологи столкнулись
при проведении клинических испытаний методов генной терапии.
Выход был найден в создании биоэтических комитетов в учреждени¬
ях, где проводятся такие исследования или испытания.Принцип автономии личности — это признание свободы и досто¬
инства пациентов или участников эксперимента. Их следует уважать
как собственников своей жизни и здоровья. Никакие вмешательства
нельзя проводить без их согласия. Ярким примером нарушения
принципа автономии личности являются медицинские опыты в
фашистской Германии на военнопленных. Применительно к меди¬
цинской генетике этот принцип может легко нарушаться врачом или
исследователем при передаче образцов ДНК по запросу, сохранении и
размножении клеток и т.п. В современной генетике принцип автоно¬
мии личности должен распространяться на потомков обследуемого в
той же мере, в какой сохраняется право наследования имущества.Принцип справедливости учитывает равную доступность ресур¬
сов меди ко-генетической помоши через систему государственного
здравоохранения, с одной стороны, и моральную оправданность
неравенства уровня медико-генетической помощи в частном секто¬
ре здравоохранения, обусловленного рыночными отношениями, —
с другой. Реализация этих двух подходов в чистом виде оказалась
невозможной. Сейчас идет поиск оптимального сочетания обеих моде¬
лей применения принципа справедливости. Принцип справедливости
относится к распределению общественных ресурсов между уже живу¬
щими и представителями будущих поколений, с меди ко-генетической
точки зрения общество должно обеспечить заботу о здоровье потомков.
Предполагается, что общество или семья путем ограничения своих
ресурсов вложат их в здоровье внуков и правнуков. Здесь возможен
поколенческий эгоизм, т.е. изъятие ресурсов у потомков. Однако вряд
ли будет принят принцип безусловного приоритета прав и интересов
человека будущего перед правами и интересами уже живущих людей.Наряду с 4 принципами современной биоэтики выделяют еще3 правила.Первое правило — правило правдивости. Моральный долг врача и
ученого обязывает говорить правду пациентам или участникам экс¬
перимента. Без этого они не могут сами принять правильное решение.
В генетическое обследование вовлекается не только один человек,
но и члены его семьи, что и создает этически трудные ситуации для
488Клиническая генвтитирача-генетика. Например, должен ли сообщать правду врач-генети|
при обнаружении несоответствия биологического и паспортного
отцовства. Доверие между врачом и пациентоїм может поддерживат!
ся только при соблюдении обоюдно правдивых отношений меж^
ними. Если пациент скрывает сведения своей родословной, это обі
зательно отразится на заключении врача.Второе правило — правило конфиденциальности. На первый BjrjjK^
его легко соблюдать, но это не всегда гак. Чем глубже обследует
нациент (например, на генном уровне), тем больше затруднений
соблюдении ЭТ010 правила. Например, разглашение информации
генетической характеристике пациента может нанести ему вред (от»
в приеме на работу, отказ от предстоящего брака). Правило конфк
денциальности требует полного согласия пациентов на передав
полученной при генетическом исследовании информации. Наибол!
трудные случаи соблюдения правила конфиденциальности соз
ет изучение родословной. Например, может ли пациент получи!
информацию от врача о генетическом здоровье своих родственнике
если они на это не сог.тасны, могут ли родственники узнать о rei
тическом диагнозе пациента. И в том и в другом случае это мс
затрагивать моральные интересы каждой стороны.Третье правило — правило информированного согласия. Оно
многом уже вошло в правовые и юридические нормы, регламент
рующие проведение медицинских испытаний и вмешательств. Лі
генетическое обследование должно проводиться с согласия пациеі
или его законных представителей на основе достаточной инфор*
ции, выраженной в понятной для пациента форме.Соблюдение 4 принципов и 3 правил биоэтики в современні
условиях нередко затруднено многоплановостью возникающих си1
аций. Например, какое нужно принять решение, если соблюден!
конфиденциальности не совпадает с соблюдением принципа «де;
благо»; как поступить врачу и администрации предприятия, еслі
хорошего работника выявлена генетическая предрасположенное!
профессиональному заболеванию (уволить его в интересах его бу;
щеі'О здоровья или оставить на работе в интересах предприятия).Из приведенных примеров следует вывод, что все принципы!
правила биоэтики не удается соблюдать абсолютно точно и однозні
но. Каждая ситуация требует индивидуальной оценки, Для принят
решения врачу в этически сложных случаях требуется поддержка
заключение этического комитета при учреждении. ц
Глава 12. Этические вопросы медицинской генетики 489В качестве конкретного примера меди ко-генетической практи¬
ки с соблюдением этических норм приводим основные положения
«Руководства по генетическому обследованию» в Японии.— Генетическое консультирование должно проводиться генети-
ком^ имеющим достаточные знания и опыт в медицинской
генетике («не навреди»).— Консультирующие генетики должны стараться давать паци¬
ентам наиболее свежую и точную информацию. Она вклю¬
чает данные о распространенности болезни, ес этиологии и
генетическом прогнозе, а также информацию о генетических
тестах, таких, как определение носительства, пренатальная
диагностика, доклиническая диагностика и диагностика пред¬
расположенности к болезни. Врачи должны помнить о том, что
внутри одной наследственной болезни могут быть различные
генотипы, фенотипы, прогнозы, реакции на терапию и т.д. («не
навреди»),— При объяснении всех процессов консультирующий генетик
должен стараться использовать простые и понятные слова.
Пациент может прийти на прием с одним или несколькими
сопровождающими, если он этого хочет и/или если для него
предпочтительнее присутствие третьего лица. Все объяснения
следует заносить в журнал регистрации и хранить в течение
определенного времени («автономия личности»).— при консультировании перед генетическим тестированием кон¬
сультант должен предоставить пациенту точную информацию
относительно цели, методики, точности и особенно ограниче¬
ний тестирования (сверх требований обычного генетического
консультирования). Должна быть предоставлена письменная
информация о болезни, чтобы гарангировать отсутствие упу¬
щений («правдивость»).— В равной степени должны соблюдаться право пациента и его
семьи знать и право не знать результаты. Следовательно, гене¬
тическое консультирование и генетическое тестирование с
использованием персональных данных пациента должны быть
основаны на независимом решении, сделанном человеком,
которому проводится исследование. Консультант не должен
принуждатьк какому-либо решению. Пациент может отказаться
от тестирования, и ему нужно объяснить, что он не пострадает
при отказе, но это плохо для прогноза. Особенно для доклини-
490Клиническая генатической диагностики генетических болезней, начинающихся и|
взрослом возрасте, рекомендуется неоднократное проведені
консультаций до назначения любых тестов, и окончательнс
решение пациент должен принимать сам («информированн<
согласие», «конфиденциальность», «автономия личности»).— Генетическое тестирование должно проводиться только ПОСЛІ
получения информированного согласия («информированН(
согласие»).— Врач может отказать пациенту в проведении тестирован и I
если это противоречит социальным или этическим нормш
или принципам самого врача. Если сушсствует личное несогл!
сие, то врач может направить пациента в другие медицинскі
учреждения («автономия личности», «делай благо»),— Если пациент не способен самостоятельно принимать решенИІ
и это делает за него его представитель, то решение относ и тел 1
но генетического тестирования должно защищать интерес!
пациента. Следовательно, следует избегать тестирования дет
на генетические заболевания, начинающиеся во взрослом во1
расте, не имеющие эффективного лечения или средств профі
лактики («не навреди»),— Пациенту, проходящему тестирование на предрасположение
к раку или многофакторным болезням, нужно объяснит!
что клинические черты болезни могут различаться у разні
людей и зависят от пенетрантности и что даже при отсутстві
генотипа предрасположенности существует вероятность
никновения болезни. Следует рассказать ему о медицинскі
мероприятиях, которые могут понадобиться после тестиро!
ния («не навреди»).— Генетическое тестирование должно проводиться только с пол
щью общепринятых методик. Лаборатории или организациі
обеспечивающие исследования, должны соответствовать ус1
новленным стандартам и всегда стараться повышать точнооі
диагностики («не навреди»).— Результаты генетического тестирования нужно объяснить
доступной форме. Даже если тестирование неуспешно
результаты сомнительны, ситуацию нужно объяснить паци0|
ту («правдивость»).— Если консультирующему генетику кажется, что лучше со<
щить пациенту результаты теста в присутствии третьего ли1
Глава 12. Этические вопросы медицинской генетики 491которому пациент доверяет, генетик должен предложить это
пациенту. Пациент может остановись проведение анализа в
любое время, а также может отказаться получить результат.
Кроме того, пациент никогда не должен ощущать ущерб при
принятии этого решения («автономия личности»).— Обязательно следует проводить консультирование после тестиро¬
вания; оно должно продолжаться столько, сколько необходимо.
Кроме того, должна быть подготовлена медицинская поддержка,
включающая психологическую и социальную («делай благо»).— Вся личная генетическая информация должна оставаться
конфиденциальной, ее нельзя сообщать другому лицу, если
пациент этого не позволяет. Нужно заботиться, чтобы эта
информация не использовалась как источник дискриминации
(■^<конфиден циа л ьность»).— Если результаты тестирования можно использовать для предот¬
вращения развития болезни или для ее лечения у членов семьи
пациента, ему предлагают рассказать о результатах членам его
семьи, чтобы они тоже прошли тесты (относительно не только
моногенных, но и многофакторных болезней) («конфиден¬
циальность»). Если пациент отказывается передавать инфор¬
мацию своей семье и если эта информация действительно
может предотвратить заболевание семьи, то по просьбе семьи с
этических позиций допустимо раскрытие генетической инфор¬
мации (только для диагностики, профилактики и лечения)
(«делай благо»). Однако решение о том, делиться информациейо результатах теста с членами семьи пациента или нет, должно
приниматься этическим комитетом, а не консультантом.— Образцы для генетического тестирования должны сохраняться,
но использовать их в других исследованиях (кроме того, для
когорого их изначально собирали) недопустимо. Если обра¬
зен может представлять интерес для будущих исследований,
необходимо получить письменное согласие пациента, которому
следует объяснить, что вся идентифицирующая его личность
информация при сохранении образца будет уничтожена («кон¬
фиденциальность», «информированное согласие»),— Инвазивные процедуры пренатального тестирования/диагно¬
стики (амниоцентез, биопсия ворсин хориона) проводятся по
желанию беременной. Ведение пациентки после диагностики
целиком определяется ее желанием; врач генетик-консультант
492Клиническая генетикіне должен принимать участия в принятии решения. Вне зани»
СИМОСТИ от принятого решения пациентке и ее семье ДОЛЖНІ'
быть оказана психологическая и социальная поддержка (§-
настоящий момент срочно требуется создание служб такой под*
держки) («автономия личности»).Правила, принципы, руководства по общим или конкретными
этическим вопросам медицинской генетики утверждены не только^!
Японским обществом генетиков человека, но и генетическими общО'
ствами других стран, а также рассмотрены комитетом экспертов ВОЗ,^
Все документы отражают рекомендации этического плана. Они Htj
имеют законодательной или правовой силы.Более обязательна для выполнения принятая Парламентской^
ассамблеей Совета Европы в 1996 г. «Конвенция о зашите прав И і
достоинства человека в связи с использованием достижений биоло<*|
гии и медицины», коротко называемая Конвенцией о правах человек*!
и биомедицине, В этой Конвенции один раздел посвящен Bonpocai
медицинской генетики. Приводим его полностью.Часть VI. Геном человека.Статья 11 {Запрет дискриминации).Запрещается любая форма дискриминации по признаку генетич<
ского наследия того или иного лица.Статья 12 {Ггнетическое тестирование).Проведение тестов на наличие генетического заболевания hj
на наличие генетической предрасположенности к тому или ином)
заболеванию может осуществляться только в целях охраны здорові
или связанных с ними целях медицинской науки и при условии не
лежащей консультации специалиста-генетика.Статья 13 (Вмешательства в геном человека).Вмешательство в геном человека, направленное на его моді
фикацию, может быть осуществлено только в профилактически?
терапевтических или диагностических целях и только при условш
что подобное вмешательство не направлено на изменение геном|
наследников данного человека.Статья 14 {Запрет выбора пола).Не допускается использование медицинских технологий, напраї
ленных на оказание помощи в продолжении рода, в целях выбо|
пола будущего ребенка, за исключением случаев, когда это делается
тем, чтобы предотвратить наследование этим ребенком заболевані
сцепленного с полом.
Глава 12. Этические вопросы медицинской генетики 493Следует отметить, что современная биоэтика не только решает
рассмотренные выше врачебные вопросы, но иногда и разреша¬
ет научные конфликты, т.е. моральные стороны замысла научных
исследований, их целей, а также плана реализации научных дости¬
жений на благо общества в целом и каждого человека в отдельности.
Сообщество ученых со времен Галилея утверждало и отстаивало
идеалы свободы научного исследования, а также самостоятельности
в принятии принципиальных решений. Однако в последние десяти¬
летия, в основном в связи с резким увеличением финансовых вкла¬
дов в науку, появилась необходимость опенки эффективности этих
вкладов и переосмысления системы контроля научной деятельности
со стороны налогоплательщиков. Однако система контроля, скорее
всего, будет бюрократической, некомпетентной и тормозящей раз¬
витие науки. Научное сообщество стало создавать такие механизмы
взаимодействия с обществом и государством, которые показывают
всему обществу стремление ученых предвидеть и предотвращать
неблагоприятные с точки зрения общества в целом последствия
новых научных открытий и новых технологий, а также согласие
ученых на социальное и этическое регулирование их деятельно¬
сти. Исходя из этого, можно сформулировать принципы этического
регулирования научных исследований: общество не должно ограни¬
чивать свободу науки, а научное сообщество должно обеспечивать
защиту прав и интересов людей.РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРАБиоэтика: принципы, правила, проблемы. — М.: Эдиториал УРСС,
1998. - 472 с.Введение в биоэтику: учеб. пос. — М.; Прогресс-Традиция, 1998. —
384 с.Генетический паспорт — основа индивидуальной предиктивной
медицины / под ред. B.C. Баранова. — СПб.; Изд-во Н-Л, 2009. — 527 с.Зыбкая грань или опасная сделка. — М.: Издательство
Душепопечительского Православного Центра св. прав, Иоанна.
Кропштадского, 2007. — 456 с.Кэмпбелл А., Джиллетт Г., Джонс Г. Медицинская этика: учеб. пос. /
пер. с англ.; под ред. Ю.М. Лопухина, Б. Г. Юдина — М.: ГЭОТАР-
Медиа, 2004. — 400 с.
КОНТРОЛЬНЫЕ ВОПРОСЫк ГЛАВЕ 11. Концепция «наследственность как этиологический фактор» сформИ*
ровалась:а) в конце XIX века;в) начале XX века (менделизм);г) одновременно с хромосомной теорией наследственности;д) одновременно с установлением структуры ДНК.2. Объектом изучения клинической генетики являются:а) больной человек;б) больной и больные родственники;в) больной и все члены его семьи, в том числе и здоровые.3. Генетические технологии в медицине и здравоохранении применяютС
для:а) классификации болезней;б) создания новых вакцин;в) диагностики наследственных и инфекционных болезней.4. Основоположник клинической генетики Б России:а) Н.К. Кольцов;б) С,Г. Левит;в) С.Н. Давиденков;г) В,М. Флоринский.5. Наследственная отягощенность человеческой популяции включ«|
в себя:а) накопленные в процессе эволюции патологические мутаци|б) вновь возникающие мутации в соматических клетках;в) вновь возникающие мутации в половых клетках.6. Частота наследственных и врожденных заболеваний среди ново(
денных составляет:а) 5-5,5%;б) 3-3,5%;в) 9-10%;г) 0,1-1,0%.
Контрольные вопросы 4957. Число известных клинических форм наследственных заболеваний
составляет примерно:а) до 3000;б) 4000-4500;в) 6000-10 ООО;г) 80 000-100 ООО.8. Исключите неправильные утверждения:а) нет таких признаков, которые бы зависели только от наслед¬
ственности или только от среды;б) наследственная изменчивость, ведущая к вариациям нормаль¬
ных признаков и ведущая к наследственным болезням, —
два разных вида изменчивости;в) в ближайшее время ожидается резкий рост частоты наслед¬
ственной патологии вследствие увеличения мутагенной
нагрузки, миграции населения и разрушения брачных
границ;г) новые мутации могут закрепляться в популяции путем есте¬
ственного отбора.9. Доля наследственных и врожденных болезней среди причин смерти
детей на 1-м году жизни составляет:а) 50%;б) 70%;в) 25%;г) 5%.10. Когда чаще всего возникают симптомы наследственного заболева¬
ния, связанного с дефектом фермента:а) во внутриутробном периоде;б) в период с рождения до 1 года;в) в детском возрасте (до 14 лет);г) с 15 до 50 лет;д) позже 50 лет.11. Генетический импринтинг — это:а) «маркировка*» локуса хромосом одного из родителей;б) вид генетической памяти (например, безусловные реф¬
лексы);в) составление карт хромосом.
496Клиническая генетикіОТВЕТЫ1 - а,2 - в;3 — а, б, в;4 — в;5 — а, в;6-а;7-6;8 - б, в;9 - и,10 - б,
П - а.К ГЛАВЕ 21. Врожденные заболевания — это:а) заболевания, обусловленные мутацией генов;б) заболевания, проявляющиеся на 1-м году жизни ребенка;в) заболевания, проявляющиеся при рождении;г) заболевания, не поддающиеся лечению.2. Выберите верное утверждение:а) механизмы естественного отбора у человека и животных Н|
различаются;б) в процессе эволюционного развития человеческой популяци!
происходило накопление патологических мутатшй, что при»|
вело к большему количеству нозологических форм у человеї
по сравнению с животными;в) в процессе эволюции выработались механизмы репарациі
мутационных повреждений ДНК.3. Генетическая гетерогенность клинически схожих заболеваний о(
словлена:а) разными аллелями одного гена;б) мутациями в разных локусах;в) взаимодействием генетической конституции и среды.4. Спорадический случай наследственной болезни — это:а) пациент с наследственной болезнью, впервые обратившийС
за медицинской помощью;б) первый случай аутосомно-домипантной или хромосомне
болезни в родословной;в) единственный случай данной наследственной болезни
родословной;г) пациент с наследственной болезнью, имеющий здоровИ^
родителей.
Контрольные вопросы 4975. При ненаследственных болезнях генетические факторы не влия>
ют на:а) этиологию;б) сроки выздоровления;в) исход заболевания;г) эффективность лечения.6. К генетическим болезням соматических клеток относятся:а) злокачественные новообразования;б) сахарный диабет;в) некоторые спорадические случаи врожденных пороков раз¬
вития;г) психические заболевания.7. Хромосомные болезни обусловлены:а) генными мутациями;б) геномными мутациями;в) изменениями межгенных участков структуры ДНК;г) изменением структуры хромосом.8. Балансированный полиморфизм — это наличие в популяции двух
форм аллелей одного гена или более, при этом частота редкого аллеля
составляет не менее:а) 10%;б) 5%;в) 1%;г) 0,1%.9. Проявления клинического полиморфизма этиологически единой
формы заболевания выражаются:а) различным временем манифестации;б) различной тяжестью течения;в) вариантами ответов на лечение;г) числом больных родственников.10. К эффектам мутационного груза относятся:а) акселерация;б) летальность;в) сниженная фертильность;г) повышение приспособляемости на популяционном уровне;д) снижение продолжительности жизни.
498Клиническая генетикіИ. Выберите верное утверждение:а) фенотипические проявления небольших по протяженно¬
сти мутаций более специфичны, чем проявления крупных
мутаций;б) межгенные взаимодействия не влияют на клинический поли¬
морфизм;в) около 90% всех спонтанных абортов связано с генетическим1|
нарушениями у эмбриона;г) клинический полиморфизм болезней с наследственной пред¬
расположенностью больше, чем моногенных заболеваний.12. Возможный исход изменений нуклеотидной последовательности!
ДНК:а) изменение аминокислотной структуры белка;б) изменение функции белка;в) синтез белка — продукта другого гена;г) изменение регуляции синтеза белка;д) отсутствие изменения функции белка.13. Стабильность генотипа обеспечивается:а) системой репарации ДНК;б) дублированностью структурных элементов генотипа;в) пол у консервативным характером редупликации ДНК;г) матричным принципом биосинтеза;д) адаптацией организма к факторам внешней среды.14. Наследственные болезни человека появились:а) в связи с уменьшением инфекционной заболеваемости;б) в связи с улучшением условий жизни и медицинской ПОМОШіІв) в процессе эволюционного формирования человека как 6І
логического вида;г) в процессе социального формирования человеческого обі
ства.15. Возможными причинами различия клинической картины nacj
ственного заболевания могут быть:а) неполная ненетрантность гена;б) пол больного;в) варьирующая экспрессивность гена;г) воздействие факторов среды;д) возраст больного.
Контрольные вопросы 49916. Выберите правильное утверждение;а) термины «наследственная болезнь» и «врожденная болезнь»
равнозначны;б) термиііьі «семейная болезнь» и «наследственная болезнь»
равнозначны;в) спорадические случаи болезни относятся к наследственной
патологии.ОТВЕТЫ1 — в; 7—6, г; 13 — а, б, г;2 — б, в; 8 — в; 14 — в,3 — а, б; 9 — а, б, в; 15 — а, в, г,4 — 6; 10 — б, в, д; 16 — в.5-а; 11 ~ а, г;6 — а, в; 12 — а, б, г, д;К ГЛАВЕ 31. Укажите наиболее верное определение клинико-генеалогического
метода:а) составление родословной с последующим обследованием про¬
банда;б) составление родословных;в) прослеживание передачи наследственных признаков среди
родственников одного поколения;г) прослеживание передачи наследственных признаков среди
родственников больного в ряду поколений.2. Чем обусловлена прогредиентноеть наследственных болезней;а) ростом и старением организма больного;б) неэффективностью лечения;в) непрерывностью функционирования мутантных аллелей.3. Укажите положения, характеризующие аутосомно-доминантный тип
наследования:а) родители больного ребенка фенотипически здоровы, но ана¬
логичное заболевание встречается у сибсов пробанда;б) сын никогда не наследует заболевание от отца;в) заболевание встречается одинаково часто у мужчин и женщин;
500Клиническая генетиківг) заболевание передается от родителей детям в каждом поко*
лении.4. Плейотропия — это:а) влияние нескольких генов на формирование одного признака;б) взаимодействие генов с факторами среды;в) влияние одного гена на формирование нескольких при-,
знаков.5. Наследственной патологии свойственныеа) ранняя манифестация клинических проявлений;б) острое течение;в) вовлеченность в патологический процесс многих органов f|
систем;г) прогредиентное течение;д) широкая распространенность каждой формы в популяции;е) резистентность к стандартной терапии.6. Пробанд — это:а) больной, обратившийся к врачу;б) здоровый человек, обратившийся в медико-генетическуЛ|
консультацию;в) человек, впервые попавший под наблюдение врача-генетикаіг) индивидуум, с которого начинается сбор родословной.1, Укажите признаки Х-сцепленного доминантного типа наследованим|а) одинаковая частота заболевания у женщин и мужчин;б) сыновья больного отца будут здоровы, а дочери больны;в) заболевание может прослеживаться в каждом поколении;г) если больна мать, то вероятность рождения больного ребені
независимо от пола равна 50%.8. Критические периоды эмбрионального развития:а) конец 1-й ~ начало 2-й недели гестации;б) конец 2-й — начало 3-й недели гестации;в) 3—6-я неделя гестации;г) 7—8-я неделя гестации.9. Врожденный морфогенетический вариант — это морфологичесі
изменение органа:а) не выходящее за пределы нормальных вариаций и не наї
шающее функцию органа;
Контрольные вопросы 501б) выходящее за пределы нормальных вариаций, но не нарушаю¬
щее функцию органа;в) приводящее к нарушению функции органа.10. Сибсы — это:а) все родственники пробанда;б) дети одной родительской пары;в) братья и сестры;г) родственники пробанда, лично обследованные врачом-
генетиком.И. Синдромологический анализ — это:а) анализ генотипа больного с целью установления диагноза;б) обобщенный анализ всех фенотипических (клинических)
проявлений с целью выявления устойчивого сочетания при¬
знаков для установления диагноза;в) анализ результатов параклинических методов исследования;г) диагностика заболевания на основе анамнестических данных.12. Укажите признаки, не характерные для аутосомно<рецессивного
типа наследования:а) заболевание одинаково часто встречается у мужчин и женщин;б) у больных родителей могут быть здоровые дети;в) женщины болеют чаще мужчин;г) родители больного здоровы;д) родители являются кровными родственниками.13. Выберите правильные утверждения:а) гаметопатии приводят к нарушению оплодотворения или
гибели зиготы;б) к бластопатиям относят мозаичные формы хромосомных
болезней;в) эмбриопатии возникают в результате действия повреждающе¬
го фактора в период от 9-й недели внутриутробного развития
до родов;г) фетопатии возникают в результате действия повреждающего
фактора в первые дни после рождения.14. Признаки Х-сцепленного рецессивного типа наследования:а) заболевание наблюдается преимущественно у мужчин;б) все фенотипически нормальные дочери больных мужчин
являются носительницами;
502 Клиническая генетикДв) больные мужчины передают патологический аллель 50Я
сыновей;г) сыновья женшины-носительницьт будут больны с вероятно»
стьго 50%.15. Термин «врожденный порок» относится к морфологическому изм«<;
нению органа или части органа:а) выходящему за пределы нормальных вариаций, но не нару»
шающему функцию органа;б) не выходящему за пределы нормальных вариаций и не пару*
шающему функцию органа; .в) выходящему за пределы нормальных вариаций и нарушающей!
му функцию органа.16. Признаки митохондриального типа наследования:а) болезнь передается только от матери;б) заболевание одинаково часто встречается у мужчин и жсн*
шин;в) больные женщины передают заболевание 50% детей;г) все дети больных отцов здоровы.17. Информапия о происхождении супругов и их родителей из одної
или близко расположенных населенных пунктов имеет значение дд|
диагностики болезней:а) Х-сцепленных рецессивных;б) аутосомно-рецессивных;в) аутосомно-доминантных с неполной пенетрантностью;г) цитоплазматически наследуемых.18. Выберите правильные утверждения:а) оксицефалия — один из вариантов «башенного» черепа;б) камптодактилия — сгибательная контракгура проксимальны^
межфаланговых суставов;в) прогнатия — нижняя челюсть, выступающая вперед по отнС
шению к верхней;г) синофриз — это опущенные веки;д) брахицефалия — это увеличение поперечного размера черег
относительно продольного;е) эпикант — это сросшиеся брови;ж) арахнодактилия — это увеличение длины пальцев;з) микрогнатия — это малые размеры верхней челюсти;
Контрольные вопросы 503и) гипертелоризм — это опущенные наружные углы глаз;
к) фильтр — это кожная крыловидная складка.19. К признакам Y-сцепленного типа нас«іедования относятся:а) передача только по мужской линии всем детям;б) передача только по мужской линии всем мальчикам;в) передача только от матери к сыну;г) отсутствие потомства у больного мужчины.ОТВЕТЫ1 — г; 8 — а, в; 15 — в;2 — в; 9—6; 16 — а, б, г;3 — в, г; 10 — б, в; 17 — б;4 — в; И — б; 18 — а, б, д, ж, з,5 — а, в, г, е; 12 — б, в; 19 — б.6 — в; 13-а, б;7 — б, в, г; 14 — а, б, г,К ГЛАВЕ 41. Действие мутантного гена при моногенной патологии проявляется;а) только клиническими симптомами;б) на клиническом, биохимическом и клеточном уровнях;в) только на определенных этапах обмена веществ;г) только на клеточном уровне.2. Диагноз нейрофиброматоза устанавливают на основании:а) характерной клинической картины и биохимического ана¬
лиза;б) клинической картины;в) клинической картины, результатов исследования гормональ¬
ного профиля, биохимического анализа и патоморфологиче¬
ского исследования,3. Этиологические факторы моногенной наследственной патологии:а) перенос участка одной хромосомы на другую;б) изменение структуры ДНК;в) взаимодействие генетических и средовьтх факторов;г) мутации генов;д) делеция, дупликация, транслокация участков хромосом.
504Клиническая генетщЩІ4. Вероятность повторного рождения больного ребенка у супруго|||
имеющих больную девочку с феннлкетонурией:а) 50%;б) близко к нулю;в) 75%;г) 25%.5. Диагноз синдрома Марфана устанавливают на основании:а) жалоб больною и данных семейного анамнеза;б) характерного сочетания клинических признаков;в) результатов биохимического анализа;г) клинических симптомов, данных биохимического и патомор!
фологического исследований.6. Классификация генных болезней возможна на основе:а) возраста начала заболевания;б) преимущественного поражения определенных систем и орг|
нов;в) типа наследования;г) мутации.7. Диагноз муковисцидоза устанавливают на основании:а) результатов биохимического анализа мочи и крови;б) данных осмотра офтальмологом, кардиологом и параклит
чсских методов исследования;в) клинических симптомов, концентрапии ионов натрия и хл(
в потовой жидкости;г) характерных клинических симптомов, данных электром!
ографии и определения уровня креатининфосфокиназы
сыворотке крови.8. Вероятность рождения в семье больного с адреногенитальным СИ1
дромом нри условии, что сын (от 1-й беременности) имеет
синдром, а девочка (от 2-й беременности) здорова, составляет:а) 50%;б) около 0%;в) 25%;г) 100%.9. К внутрилокусной гетерогенности наследственных болезней ОТІ
сятся:а) болезни, обусловленные мутациями в разных генах;
Контрольные вопросы 505б)клинически разные болезни, обусловленные разными мутант¬
ными аллелями одного гена;в) сочетание двух аллелей одного гена с разными мутациями у
конкретного больного.10. Признаки синдрома Элерса-Данло:а) гиперрастяжимость кожи;б) повышенная ранимость кожи;в) умственная отсталость;г) пролапс митрального клапана.11. Вероятность рождения больного ребенка в семье, в которой мать
больна фенилкетонурией, а отец гомозиготен по нормальному алле-
лю, составляет:а) 50%;б) около нуля;в) 25%;г) 100%.12. Генные болезни обусловлены:а) потерей участка хромосомы;б) дупликацией части хромосомы;в) потерей двух генов и более;г) мутацией одного гена.13. Диагноз мышечной дистрофии Дюшенна устанавливают на основа¬
нии:а) концентрации ионов натрия и хлора в потовой жидкости;б) неврологической симптоматики, времени начала и харак¬
тера течения, уровня креатининфосфокиназы в сыворотке
крови;в) типичного внешнего вида, данных электрофизиологических
исследований и молекулярно-генетических методов;г) результатов гистологического исследования,14. Вероятность рождения ребенка с синдромом Марфана, если 1-й
ребенок имеет этот синдром, а родители здоровы, составляет при¬
мерно:а) 50%;б) около нуля;в) 25%;г) 75%.
506Клиническая гeнвт^15. Клинический полиморфизм генных болезней определяют:а) множественность мутаций гена;б) действие факторов окружающей среды;в) наличие генов-модификаторов;г) эффект дозы генов.16. Диагностические критерии муковисцидоза:а) хронические бронхоэктазы, правостороннее расположен!
сердца, хронические синуситы;б) грубые черты лица, кифосколиоз, деформация грудины, и ИЗ
кий рост, порок клапанов сердца, умственная отсталость;в) рецидивирующие хронические пневмонии, нарушение фун(
ции поджелудочной железы, мальабсорбция, обильный зл(
вонный стул;г) задержка роста, множественный дизостоз, помутнение рог
вицы, повышенная экскреция гликоэаминогликанов (мукоп<
лисахаридов) с мочой.17. Диагностические критерии нейрофиброматоза:а) врожденный порок сердца и порок развития лучевой костй'|
сс производных;б) множественные пигментные пятна на коже, опухоли кожні
подкожные и по ходу нервных волокон, сколиоз, глиомы і
тельного нерва;в) себорейные аденомы на щеках, депигментированные пяц
«кофейные» пятна, судороги, умственная отсталость;г) анемия, гепатоспленомегалия, «башенный» череп, водяї
плода.18. Диагноз семейной гиперхолестеринемии устанавливают на осі
ваиии:а) семейного анамнеза;б) клинической картины;в) результатов биохимического исследования крови;г) данных гормонального исследования.19. Синдром Марфана диагностирован у матери и дочери. Вероятна
повторного рождения ребенка с синдромом Марфана в другом б|
матери:а) 25%;б) около нуля;Ч
Контрольные вопросы507в) 100%;г) 50%.20. Распространенность моногенного заболевания считается высокой,
если его частота составляет:а) 1:100;б) 1:5000;в) 1:10 ООО;г) 1:20 ООО;д) 1:50 ООО.21. Диагностические критерии фенилкетонурии:а) двойственное строение наружных половых органов, рвота,
дегидратация;б) прогрессирующие бледность и гипотрофия, спленомегалия,
выступающие скулы и лобные бугры, «багиенный» череп,
анемия;в) множественные пигментные пятна на коже, опухоли кожные,
подкожные и по ходу нервных волокон;г) отставание в психомоторном развитии, микроцефалия, гипо¬
пигментация волос и кожи.22. Характерные признаки синдрома Элерса—Данло;а) гиперподвижность суставов;б) варикозное расширение вен;в) микроцефалия;г) мышечная слабость;д) грыжи.23. Критерии диагностики миодистрофии Дюшенна:а) прогрессирующая мышечная слабость;б) псевдогипертрофия икроножных мышц;в) умственная отсталость;г) повышение уровня креатининфосфокиназьт в сыворотке
крови.24. Клинический полиморфизм моногенной наследственной патологии
обусловлен:а) генотипической средой;б) полилокусной и/или полиаллельной детерминацией клини¬
ческой картины болезни;в) различной частотой генов в популяции;
508Клиническая генетикіг) близкородственным браком;д) действием тератогенных факторов.25. Диагноз адреногенитального синдрома устанавливают на осної
вании:а) клинической картины и определения уровня гормонов;б) концентрации ионов натрия и хлора в потовой жидкости;в) клинических симптомов, данных цитогенетического анализ,і
параклинических методов исследования;г) результатов молекулярно-генетических методов, биохимичв*
ского анализа.26. Дети с муковисцидозом в Европе рождаются с частотой:а) 1:700;б) 1:2500;в) 1:10 ООО;г) 1:20 ООО.27. Диагноз синдрома умственной отсталости с ломкой Х-хромосом(
окончательно подтверждается на основании;а) результатов биохимических исследований мочи и крови;б) данных электроэнцефалографии;в) молекулярно-генетического анализа;г) результатов психологического тестирования;д) данных семейного анамнеза.28. Диагностические критерии синдрома Марфана:а) отставание в психомоторном развитии, микроцефалия, ги1
пигментация;б) подвывих хрусталика, гиперподвижность суставов, вор<
кообразное вдавление грудины, высокий рост, аномальні
рост зубов;в) умственная отсталость, макроорхидизм, длинное лицо, выо<
кий лоб, массивный подбородок, оттопыренные уши.29. Вероятность рождения больного ребенка в семье, в которой оба
теля являются гомозиготами по гену фенилкетонурии, составляет^а) 25%;б) 100%;в) около нуля;г) 50%.
Контрольные вопросы 50930. Диагностические критерии адреногенитального синдрома:а) гипертелоризм, брахидак гилия, крипторхизм, низкий рост,
паховые грыжи, умеренная умственная отсталость;б) гонады представлены яичками, наружные половые органы
сформированы по женскому типу, вторичные половые при¬
знаки недоразвиты, кариотип 46, XY;в) прогрессирующая вирилизация, ускоренное соматическое
развитие, повышенная экскреция гормонов коры надпочеч¬
ников;г) умственная отсталость, макроорхидизм, оттопыренные уши,
длинное лицо, массивный подбородок.31. Какие заключения неверны:а) генетически летальными считаются только те заболевания,
которые приводят к смерти индивида до достижения пубер¬
татного периода;б) мутантный ген, унаследованный от родителей, присутствует
в организме внутриутробно и при рождении, поэтому все
наследственные болезни являются врожденными;в) если больной не имеет родственников с тем же заболеванием,
маловероятно, что его болезнь наследственная.32. Женщина с муковисцидозом — единственный случай заболевания
в семье. Определите риск носительства мутантного гена для ее —1) отца; 2) матери; 3) дочери; 4) внука; 5) брата; 6) племянников:а) 100%;б) 50%;в) 67%;г) 33%.33. Выберите из списка термин, соответствующий нижеследующей
ситуации:1) 25-летняя дочь с атрофией и слабостью скелетных мышц
65-летнего мужчины с катарактой без симптомов миотониче-
ской дистрофии родила ребенка с тяжелой мышечной слабо¬
стью и задержкой развития.2) При пестрой порфирии (аутосомно-доминантном наруше¬
нии биосинтеза порфирина) возможны фоточувствительность
кожи, боли в животе, периферическая нейропатия и эпизоды
психических нарушений (психозы).
510Клиническая генетик!3) У сестры мужчины с тяжелым сколиозом и множественными
подкожными нейрофибромами имеются плексиформыые ней¬
рофибромы, а у ее 30-летнего сына обнаружены узелки Лиша
и веснушчатость в подмышечных областях.4) Редкая форма аутосомно-рецессивной недостаточности сома¬
тотропного гормона обнаруживается только в некоторых
маленьких деревнях в Швейцарских Альпах.5) Как нонсенс-мутации, так и делеции гена орнитинтранскар-
бамилазы обусловливают развитие летальной неонатальной
гипераммониемии вследствие отсутствия орнитинтранскар-
бамилазы — важного печеночного фермента цикла мочеви*
ны.6) Существуют как аутосомные, так и Х~сцепленные формы
пигментного ретинита,а) аллельная гетерогенность;б) плейотропность;в) вариабельная экспрессивность;г) антиципация;д) кровнородственные браки;е) локусная гетерогенность.34. Выберите неверное утверждение:а) генокопии — разные проявления одной наследственной болез*
ни;б) фенокопии — ненаследственные болезни, схожие по клини¬
ческим признакам с наследственными;в) нормокопирование — отсутствие признаков заболевания пр1
патологическом генотипе.35. Дрейф генов — это:а) изменение клинической картины заболевания через несколі
ко поколений;б) изменение частоты аллеля в популяции в результате стоха*
стических событий;в) увеличение доли рецессивной патологии в результате кровнОї
родственных браков.ОТВЕТЫ1 - б;2-6;3 - б, г;4 — г;
5-а, б;
6 — б, в;7-в;8 — в;9 - б, в;
Контрольные вопросы51110 - а, б, г;11 - б;12 - г;13 — б, в;14 - б;15 — а, б, в, г;16 — в;17 - б;18 — а, б, в;19 - г;20 — а, б, в;21 - г;22 — а, б, д;23 — а, б, г;24 — а, б;25 — а, г;26 - б;27 - в;28 - б;29 - б;30 - в;31 — а, б, в;32: і - а, 2 - а, 3 - а,4 — б, 5 — в, 6 — г;
33: 1 - г, 2 - б, 3 - в,4 — д, 5 —а, 6-е;34 — б, в;35 - б.К ГЛАВЕ 51. Какие виды хромосомных аномалий не встречаются у живорожденных:а) трисомии по аутосомам;б) трисомии по половым хромосомам;в) моносомии по аутосомам;г) моносомия по Х-хромосоме;д) нуллисомия по Х-хромосоме,2. Какие мутации относятся к геномным:а) инверсии, транслокации, дупликации, дслеции;б) полиплоидии, анеуплоидии;в) триплоидии, тетраплоидии;г) внутрихромосомные и межхромосомные перестройки.3. Выберите основные показания для исследования кариотипа:а) в анамнезе умершие дети с множественными пороками раз¬
вития;б) хроническое прогредиентное течение болезни с началом в
детском возрасте;в) неврологические проявления (судороги, снижение или повы¬
шение мышечного тонуса, спастические парезы);г) олигофрения в сочетании с пороками развития,4. Укажите формулы кариотипа при синдроме Шерешевского-Тернера:а) 45,Х/46,XX;б) 47,ХХХ;в) 45,Х;г) 46,XX.
512Клиническая генвтк5. Метод точной диагностики хромосомных болезней:а) клинический;б) дерматоглифический;в) цитогенетический;г) клинико-генеалогический;д) специфическая биохимическая диагностика.6. Риск рождения ребенка с хромосомными аномалиями существеиі
повышается в возрастных интервалах:а) 20-25 лет;б) 25-30 лет;г) 30-35 лет;д) 35-40 ЛЄ']’.7. К хромосомным относятся мутации:а) делеция;б) триплоидия;в) инверсия;г) изохромосома.8. Формула кариотипа при синдроме «кошачьего крика»:а) 45,X;б) 46,ХХ,9р+;в) 46,ХХ,5р-;г) 45,Х/46,XX.9. Показания для проведения цитогенетического анализа:а) гепатоспленомегалия, катаракта, умственная отсталость;б) привычное невынашивание беременности и мертворожден!
в анамнезе;в) непереносимость некоторых пищевых продуктов, ГЄМОЛИТІ
ческие кризы;г) умственная отсталость, микроаномалии развития или ej
денные пороки развития.10. Формулы хромосомного набора у больного с синдромом Клайнфеліа) 45,X;б) 47,XXX;в) 47,XYY;г) 46,XY,5p-;д) 48,XXYY;е) 47,XXY.
Контрольные вопросы 51311. Полиплоидия — это:а) уменьшение числа хромосом в наборе на несколько пар;б) диплоидный набор хромосом в гамете;в) увеличение числа хромосом, кратное гаплоидному набору.12. В основе хромосомных болезней лежат хромосомные и геномные
мутации, возникающие:а) только в половых клетках;б) в соматических и половых клетках;в) только в соматических клетках.13. Укажите формулу кариотипа при синдроме Патау:а) 47,XX,+18;б) 47,XY,+13;в) 46,ХХ,5р~;г) 47,XXY;Д) 45,X.14. Летальные нарушения кариотипа:а) моносомии по Х-хромосоме;б) трисомии по половым хромосомам;в) моносомии по аутосомам;г) трисомии по аутосомам.15. Набор симптомов, включающий умственную отсталость, долихоце¬
фалию, деформированные ущные раковины, флексорное положение
пальцев рук, врожденный порок сердца, указывает на:а) синдром Эдвардса;б) синдром Патау;в) синдром Дауна;г) синдром «кошачьего крика».16. Показания для проведения кариотипирования:а) задержка физического и полового развития, гипогонадизм,
гипогенитализм;б) задержка психомоторного развития в сочетании с диспластич-
ным фенотипом;в) приобретенные деформации позвоночника и грудины, помут¬
нение роговицы, 1'спатоспленомегалия;г) прогредиснтная утрата приобретенных навыков, судорожный
синдром, спастические параличи.
514Клиническая генетикі17. Анеуолоидия — это:а) увеличение хромосомного набора на целый гаплоидный
набор;б) изменение числа хромосом в результате добавления одной илИ
нескольких хромосом;в) изменение числа хромосом в результате утери одной илй|
нескольких хромосом.18. правильная форма кариотипа при синдроме Эдвардса:а) 47,XY,+21;б) 47,XXY;в) 47,ХХ,+13;г) 47,XY,+18;д) 46,ХХ,9р+;е) 45,t(l3;21).19. Наиболее тяжелые последствия вызывают:а) моносомии по половым хромосомам;б) трисомии по половым хромосомам;в) моносомии по аутосомам;г) трисомии по аутосомам.20. Симптомокомплекс, включающий микроцефалию, расщелину губы:
нёба, полидактилию и поликистоз почек, наиболее характерен д;а) синдрома Эдвардса;б) синдрома Дауна;в) синдрома Вольфа—Хиршхорна;г) синдрома Патау,21. Клинически хромосомные болезни проявляются:а) множественными признаками дисморфогенеза;б) врожденными пороками развития;в) отставанием в умственном развитии;г) необычным цветом и запахом мочи.22. Возможные формулы кариотипа при синдроме Дауна:а) 47,XX,+13;б) 47,ХХ,+22;в) 46,XY,-14,t(l4;21);г) 47,XXX;д) 47,XX,+21.
Контрольные вопросы 51523. Более тяжелые клинические проявления имеют хромосомные болез¬
ни, обусловленные:а) недостатком генетического материала;б) избытком генетического материала.24. Признаки синдрома Беквита—Видемана:а) макроглоссия;б) гипогликемия;в) эпилепсия;г) экзостозы;л) большие рост и масса тела новорожденных.25. Причины возникновения трисомий;а) отставание хромосом в анафазе;б) иерасхождение хромосом;в) точечные мутации.26. Возможные формулы кариотипа при симптомокомплексе, включаю¬
щем низкий рост, короткую шею, бочкообразную грудную клетку,
задержку полового развития:а) 47,XXY;б) 45,X;в) 45,Х/46,ХХ;г) 47,XYY.27. Исследование кариотипа показано;а) у женщины с 1 спонтанным абортом в анамнезе;б) у родителей ребенка с простой формой трисомии 21;в) у супружеской пары с мертворождением и 3 спонтанными
абортами в анамнезе.28. Носители робертсоновских транс локаций:а) клинически здоровы;б) имеют кариотип, состоящий из 45 хромосом;в) имеют риск развития опухолей;г) имеют риск рождения ребенка с хромосомной болезнью.19. Выберите термин, соответствующий описанной ситуации:1) У 7-летнего мальчика с умственной отсталостью, низким ростом,
маленькими кистями и стопами, полифагией (синдром Прадера—
Вилли) при молекулярно-генетическом исследовании обнару¬
жили 2 материнские хромосомы 15 и ни одной отцовской.
516Клиническая генетиі2) При цитогенетическом обследовании 6-летней девочки с ТЯЖ#У
лей умственной отсталостью, судорогами, атаксией, прогеиИ«|
ей (синдром Ангельмана) обнаружили интерстициальную]
микроделецию материнской хромосомы 15.3) При ДНК-исследовании гена FMR-1 у 32-летней женщині
имеющей сына с синдромом ломкой Х-хромосомы, обнару«і
жили 1 аллель с 21 CGG-повтором и I аллель с 92 СО О*
повторами.а) премутация;б) геномный импринтинг;в) однородительская дисомия.ОТВЕТЫ1 — в, д;И - в;21 — а, б, в;2 — б, в;12 - б;22 — в, д;3 - а, г;13 - б;23 - а;4 — а, в;14 — в;24 - а, б, д;5 — в;15 - а;25 - б;6 — д;16 — а, б;26 — б, в;7 — а, в, г;17 - б, в;27-в;8 — в;18 — г;28 ~ а, б, г;9 - б, г;19 - в;29: 1 - в, 210 — д, е;20 - г;3 - а.К ГЛАВЕ 61. Многофакторные болезни:а) гемофилия, талассемия, серповидно-клеточная анемия;б) врожденные пороки сердца, почек, диафрагмальная грыжа|в) шизофрения, эпилепсия, маниакально-депрессивный психсг) рак желудка, рак поджелудочной железы,2. Для доказательства многофакторной природы болезни нспользуі
методы:а) близнецовый;б) исследование ассоциации генетических маркеров с болезнів) цитогенетический;г) клинико-генеалогический;д) попул я ц ион но-статистический.
Контрольные вопросы 5173. Многофакторным болезням свойственны:а) высокая частота в популяции;б) низкая частота в популяции.4. Ассоциация многофакторной болезни с полиморфными системами
означает;а) более высокую частоту определенного маркера у больных по
сравнению с таковой у здоровых;б) расположение гена, обусловливающего болезнь, и гена мар¬
керного признака на одной хромосоме;в) наличие рекомбинации между геном болезни и геном поли¬
морфной системы.5. В основе клеточного онкогенеза могут лежать:а) структурные хромосомные перестройки;б) увеличение копий онкогена;в) наличие мутантного аллеля антионкогена;г) изменение последовательности ДНК в протоонкогене.6. Повышенный риск многофакторной болезни оценивают на основании:а) близкого родства супругов;б) данных клин и ко-генеалогического анализа;в) вредных привычек;г) наличия специфического биохимического маркера.7. Многофакторные болезни имеют:а) различия больных по полу и возрасту;б) широкий спектр клинических проявлений;в) менделирующий характер;г) популяционные различия в частоте.8. Этиологические генетические факторы при многофакторной
патологии:а) действие двух аллелей гена одного локуса;б) микроделеции и другие микроперестройки какой-либо хро¬
мосомы;в) эффект единичного гена;г) аддитивный эффект многих генов с различным относитель¬
ным вкладом каждого в патогенез.9. Риск многофакторной болезни повышают:а) аналогичная болезнь у кровных родственников;
518Клиническая генетикаб) гетерозиготность по аутосомно-рецессивной болезни;в) вредные факторы oKpyxaroutcM среды;г) большое число детей в семье.10. Возможные механизмы потери конституциональной гетерозигот-
ности по антионкогену:а) потеря участка хромосомы с нормальным аллелем антионко-
гена;б) амплификация антионкогена;в) мутация нормального аллеля антионкогена;г) митотический кроссинговер между хромосомами с нормаль¬
ным и мутантным аллелем.11. Реализации предрасположенности к ишемической болезни сердца
способствуют:а) жирная пиша;б) физическая активность;в) высококалорийная диета;г) курение.12. Болезни с многофакторно обусловленной предрасположенностью:а) шизофрения;б) ишемическая болезнь сердца;в) язвенная болезнь двенадцатиперстной кишки;г) галактоземия.13. Индивида можно отнести в группу повышенного риска по много*
факторной болезни на основании:а) генеалогических данных;б) иммунологических или биохимических показателей;в) тяжести течения болезни;г) результатов цитогенетического исследования.14. Доказательства генетической обусловленности многофакторных
болезней:а) болезнь передается соответственно менделсвским законам
наследования;б) более высокая конкордантность у монозиготных близнецо
по сравнению с таковой у дизиготных близнецов в сходных’
средовых условиях;в) заболеваемость у биологических родственников выше, чеі
у родственников, не имеющих кровного родства.
Контрольные вопросы 51915. Коэффициент наследуемости отражает:а) тяжесть заболевания;б) вероятность развития заболевания у родственников пробанда;в) вклад генетических факторов в подверженность заболеванию;г) часть вариации количественного показателя, определяемую
наследственными факторами.16. Степень генетической детерминации многофакторно обусловленного
признака отражает:а) коэффициент инбридинга;б) коэффициент наследуемости;в) показатель пенстрантности;г) долю клеток с мутацией хромосом при мозаичном кариотипе.17. О семейной форме рака может свидетельствовать:а) двусторонний рак легких у работника асбестового производства;б) опухоль мозга и молочной железы у женщины;в) рак толстой кишки у 32-летнего мужчины;г) двусторонняя опухоль почек у 37-летней женщины,18. к многофакторным болезням относятся:а) дефекты нервной трубки;б) семейная гиперхолестеринемия;в) муковисцидоз;г) бронхиальная астма, нейродермит, атопический дерматит.19. Повышенный риск развития многофакторной болезни можно
выявить:а) клинико-генеалогическим методом;б) цитогенетическим методом;в) биохимическим методом;г) нагрузочными тестами.20. Реализации наследственной предрасположенности к гипертониче¬
ской болезни препятствуют:а) занятия физкультурой;б) эмоциональные нагрузки;в) правильное чередование труда и отдыха;г) употребление алкоголя.21. Болезни с многофакторно обусловленной предрасположенностью:а) гемохроматоз;
520Клиническая генетикіб) псориаз:в) болезнь Вильсона-Коновалова;г) болезнь Бехтерева.22. При высокой семейной предрасположенности к бронхиальной астме
следует избегать:а) приема антибиотиков;б) инфекций верхних дыхательных путей;в) контакта с аллергенами;г) инсоляции.23. О многофакторной природе заболевания свидетельствуют:а) некоторые заболевания возникают чаще у женщин, некоторые
чаще у мужчин;б) частота болезни в популяции составляет 4%, а среди детей
больных родителей — 50%;в) заболевание возникает чаще у детей больных, чем у их вну¬
ков;г) повторный риск для 2-го ребенка выше, когда больны обі
родителя.24. Подберите правильные ответы для следующих характеристик:1) могут активироваться амплификацией, транслокацией или
мутацией гена;2) потеря функции ведет к развитию опухоли;3) присутствуют в клетках здоровых людей;4) являются аутосомно-доминантными.а) онкогены и протоонкогены;б) гены-супрессоры опухолей.ОТВЕТЫ1 — б, в, г;2 — а, б, г, д;3 - а;4-а;5 — а, б, г;6 — б, в, г;7 — а, б, г;8 - г;9 - а, в;10 — а, в, г;11 — а, в, г;12 — а, б, в;13 - а, б;14 - б, в;15 - в, г;16 - б;17 — б, в, г;18 — а, г;19 — а, в, г;20 — а, в;21 — б, г;22 - б, в;23 — а, в, г;24; 1 - а; 2 - б;3 - а, б; 4 - б.
Контрольные вопросы 521К ГЛАВЕ 71. Верные утверждения о мутагенезе:а) стойкое нарушение структуры или функции организма в ответ
на действие мутагенов во внутриутробном периоде;б) служит частой причиной врожденных пороков развития;в) является частой причиной аутосомно-доминантных заболеваний;г) является частой причиной аугосомно-рецессивных заболеваний;д) может затрагивать как соматические, так и зародышевые клетки,2. Соотнесите фазу биотрансформации ксенобиотиков и активный
фермент:1) 1-я фаза;2) 2-я фаза.а) глутатион-$-трансфераза;б) CYP2C19;в) CYP2D6;г) N-ацетилтрансфераза;д) алкогольдегидрогенагза.3. Недостаточность каких ферментов может привести к развитию эмфи¬
земы легких при действии неблагоприятных факторов:а) митохондриальная эпоксидгидролаза;б) моноаминоксидаза;в) метионинредуктаза;г) арантитрипсин;д) транскобалами нсинтетаза.4. Дефицит фолиевой кислоты, наблюдающийся у людей с недостаточ¬
ностью ферментов фолатного цикла« приводит к накоплению гомоци-
стеина в тканях организма. Это может вести к:а) невынашиванию беременности;б) ИБС;в) бронхиальной астме;г) ВПР плода;д) непереносимости глютена.5. Каковы прогнозы заболеваемости экогенетическими болезнями на
ваш взгляд:а) доля экогенетических заболеваний будет уменьшаться, так как в
скором времени будет внедрена персонализированная медицина;
522Клиническая генетикіб) доля экогенетических болезней увеличится, гак как появятси
новые факторы, вызывающие эти болезни при определенных
генотипах;в) доля экогенетических болезней в целом не изменится.6. Отметьте верные утверждения:а) факторы внешней среды не влияют на генотип;б) факторы внешней среды могут изменять экспрессию генов;в) генотип определяет действие факторов внешней среды;г) для развития экогенетической болезни самым важным явля¬
ется наличие провоцирующего фактора;д) внешнесредовые факторы не влияют на эпигенетические про¬
цессы.7. Известно, что богатое фолиевой кислотой питание беременной может
приводить к метилированию CpG-ocTpOBKOB генома плода. Как это
может отразиться на его здоровье:а) ребенок может родиться с болезнями Ангельмана, Прадера-
Вилли и другими, патогенез которых связан с нарушением
импринтинга;б) во взрослом состоянии у такого ребенка с большей долей
вероятности разовьются такие заболевания, как ожирение,
сахарный диабет и др.;в) такой ребенок будет подвержен большему действию факторов
внешней среды, чем ребенок, чья мать во время беременности
не злоупотребляла фолиевой кислотой;г) никак не отразится.ОТВЕТЫ1 - д,2: 1 — б, в, д, 2 — а, г,3 - а, г,4 — а, б, в,5 б,6 — б, в,К ГЛАВЕ 81. Перед оперативным вмешательством 49-летняя женщина сообщил!
анестезиологу, что ее мать умерла во время операции, а у брата под воз¬
действием анестезии появилась высокая лихорадка. У этой женщины:а) можно предположить аутосомно-доминантное состояние —
злокачественную гипертермию;
Контрольные вопросы 523б) необходимо изучить истории: болезни родственников для
уточнения причин отмечавшихся осложнений;в) хирургические вмешательства должны проводиться только в
экстренных случаях;г) следует применить нсингаляционнътй наркоз.2. Подберите ферментные нарушения, соответствующие следующим
клиническим ситуациям:1) Появление гемоли тической анемии после приема примахи-
на.2) Возникновение приступа резких болей в 1 плюснефаланговом
суставе справа у мужчины после приема асиирина,3) Появление периферической нейропатии у женщины с тубер¬
кулезом легких, принимающей изониазид.4) Длительная остановка дыхания у мальчика во время нарко¬за.5) Возникновение приступа острых болей в животе с мышечной
слабостью и помрачением сознания у подростка после приема
фенобарбитала.6) Отсутствие эффекта от стандартных доз гидралазина у муж¬
чины с артериальной гипертонией.а) повышенная активность апетилтрансферазы;б) аномальная холинэстераза;в) недостаточность порфобилиногендеаминазы;г) недостаточность глюкозо-6-фосфатдегидрогеназы;д) недостаточность фосфорибозилпирофосфатсинтетазы;е) сниженная активность апетилтрансферазы.3. В анамнезе у 40-летнего мужчины приступ затрудненного дыхания
после хирургического вмешательства. У этого пациента:а) необходимо определение активности сукиинилхолинэстсра-
зы;б) отсутствие подобных случаев в семье с большой долей вероят¬
ности исключает наследственную природу приступа;в) после хирургического вмешательства необходимо тщательное
наблюдение;г) родственники пациента должны быть осведомлены о возмож¬
ности возникновения у них таких приступов;д) анамнез нужно учитывать при выборе препарата для анесте¬
зии.
524Клиническая генетика4. Дибуканновый тест является диагностическим для определения
активности:а) параоксоназы;б) псевдохолинэстеразы;в) алкогольдегидрогеназы;г) дигидропиримидин дегидрогеназы.5. При синдроме Жильбера наблюдает нарушение процессов:а) глюкуронирования;б)ацетилированИЯ;в) S-метилирования;г) конъюгации с глутатионом;д) сульфати рован и я.6. Для какого числа людей, вероятно, фармакогенетический подход не
будет иметь никакого влияния:а) будет влиять на всех;б) 10-15%;в) 30%;г) 50%;д) 80%.ОТВЕТЫ1 - а, б, г;2: 1 — г, 2 — д, 3 — е,4 — б, 5 — в, 6 — а;3 — а, в, г, д;4-6;5-а;6 — г.К ГЛАВЕ 91. В основу современной классификации хромосом положены:а) интенсивность окрашивания;б) характер поперечной исчерченности при дифференциальной
окраске;в) размер и расположение пентромсры;г) длина плеч хромосом.2. Массовый биохимический скрининг предполагает:а) обследование детей из учреждений для слабовидящих;б) исследование крови и мочи новорожденных на содержание
гликозаминогликанов (мукололисахаридов);
Контрольные вопросы 525в) обследование новорожденных с целью выявления опреде¬
ленных форм наследственной патологии в доклинической
стадии;г) обследование детей с судорожным синдромом, отставанием в
психомоторном развитии, параплегией.3. Для проведения цитогенетического анализа используются:а) клетки костного мозга;б) клетки печени;в) лимфоциты периферической крови;г) биоптат семенника.4. Показания для проведения биохимического исследования:а) задержка психического развития в сочетании с признаками
мочекислого диатеза;б) легкая олигофрения, задержка полового созревания;в) олигофрения в сочетании с общей диспластичностью;г) мышечная гипертония, гипопигментация, задержка моторно¬
го и речевого развития.5. Молекулярный зонд — это:а) комплементарный участок ДНК;б) протяженный участок ДНК, комплементарный нуклеотидной
последовательности ДНК, содержащей мутантный ген;в) синтетическая олигонуклеотидная меченая (радиоактивно
или флюоресцентно) последовательность, комплементарная
мутантному или нормальному гену.6. Хромосомы с концевым расположением центромеры называются:а) метацентриками;б) акроцентриками;в) субметацентриками;г) дицентриками.7. Показания для проведения специальных биохимических тестов:а) умственная отсталость, врожденные пороки развития различ¬
ных органов и систем;б) привычное невынашивание;в) катаракта, гепатоспленомегалия, отставание в развитии;г) расторможенность, нарушение поведения, имбецильность,
необычный запах мочи.
526Клиническая генетика8. Эухроматиновые участки хромосом содержат:а) множественные повторы последовательностей ДНК;б) гены;в) нетранскрибируемьте локусы;г) регуляторные области.9. Биохимическая диагностика показана при:а) сочетании задержки психомоторного развития с гипопигмеи-
тацией и необычным запахом мочи;б) гипогенитализме, гипогонадизме, бесплодии;в) прогредиентном утрачивании приобретенных навыков.10. Для диагностики болезней, для которых мутантный ген неизвестен
и не локализован, применяется:а) прямая детекция с использованием специфических молеку¬
лярных зондов;б) семейный анализ распределения нормального полиморфизма
длины рестриктных фрагментов;в) метод специфических рестриктаз;г) прямой сиквенс.11. с применением цитогенетических методов диагностируются:а) наследственные дефекты обмена веществ;б) многофакторные болезни;в) болезни, обусловленные изменением числа и структуры хро¬
мосом.12. Показания для проведения биохимического исследования:а) повторные случаи хромосомных перестроек в семье;б) отставание в физическом развитии, гепатоспленомегалия,
непереносимость каких-либо пищевых продуктов;в) множественные врожденные пороки развития;г) повторные спонтанные аборты.13. Для диагностики небольших структурных перестроек применяются
методы окраски:а) простой (рутинный);б) дифференциальный;в) флюоресцентный,14. Массовому биохимическому скринингу подлежат заболевания:а) нейрофиброматоз;
Контрольные вопросы 527б) гсмохроматоз;в) мукополисахаридозы;г) фенилкегонурия;д) адреногенитальный синдром,15. Эндонук^1еазные рестриктазы — это:а) ферменты, разрезающие ДНК в строго специфических
местах;б) ферменты, сшивающие разрывы молекулы ДНК;в) ферменты, обеспечивающие соединения, осуществляющие
репарацию ДНК.16. При повторных спонтанных абортах (более 3) на ранних сроках
беременности и при мертворождениях в анамнезе цитогенетический
анализ назначается:а) обоим супругам;б) одной женщине;в) родителям женщины;г) плоду.17. Проведения специальных биохимических исследований требуют:а) мышечная гипотония, рвота, отставание в психомоторном
развитии, нарушение координации движений, тромбоцито-
пения;б) хронические пневмонии, нарушение всасывания в кишечни¬
ке, гипотрофия;в) шейный птеригиум, лимфатический отек кистей и стоп, низ¬
кий рост;г) снижение зрения, кифосколиоз, гепатоспленомегалия,
умственная отсталость.18. Наиболее часто используются в пренатальной диагностике методы
разделения фрагментов ДНК;а) центрифугирование в градиенте плотности солей цезия;б) методы одномерного электрофореза.19. Для диагностики геномных мутаций применяют:а) метод G-окраски;б) метод С-окраски;в) рутинную окраску;г) метод с использованием флюоресцентных красителей.
528Клиническая генетика20. Одно из условий проведения массового биохимического скрининга
новорожденных:а) низкая частота гена болезни в популяции;б) отсутствие методов патогенетического лечения;в) наличие быстрого, точного, простого в выполнении и недо¬
рогого метода диагностики биохимического дефекта;г) выраженный клинический полиморфизм болезни.21. Явление полиморфизма но длине рестриктных фрагментов обуслов¬
лено:а) химической и функциональной гетерогенностью ДНК;б) наследуемыми, фенотипически не проявляющимися разли¬
чиями в последовательности групп оснований в геноме;в) существованием различных уровней конформационной орга¬
низации ДНК.22. Гетерохроматические участки хромосом содержат:а) множественные повторы последовательностей ДНК;б) гены;в) нетранскрибируемые локусы;г) регуляторные области.23. Подлежат массовому биохимическому скринингу:а) врожденный гипотиреоз;б) маннозидоз;в) синдром Марфана;г) множественная эндокринная неоплазия;д) фен ил кетон ури я.24. Амплификация генов — это:а) идентификация последовательности оснований ДНК;б) многократное повторение какого-либо участка ДНК;в) выделение фрагмента ДНК, содержащего изучаемый ген.25. Цитогенетический метод является решающим для диагностики:а) моногенной патологии с известным первичным биохимиче¬
ским дефектом;б) синдромов с множественными врожденными пороками раз¬
вития;в) хромосомной патологии;г) многофакторных болезней.
Контрольные вопросы 52926. Показания для проведения специальных биохимических исследований:а) комплексы врожденных пороков развития и микроаномалий
развития на фоне лре- и постнатальной задержки физическо¬
го развития;б) рвота, дегидратация, нарушение дыхания, асцит у ребенка
1-го года жизни при исключении пороков развития ЖКТ;в) прогредиентная умственная отсталость и нсвролої ическая
симптоматика после периода нормального развития различ-
]10Й длительности.27. Для диагностики болезней, обусловленных мутантным геном извест¬
ной последовательности, применяют;а) специфичную рестриктазу;б) прямую детекцию с использованием специфических молеку¬
лярных зондов;в) семейный анализ распределения нормального полиморфизма
длины рсстриктных фрагментов.28. Для проведения цитогенетического анализа используют:а) мышечные клетки;б) эритроциты;в) биоптат хориона;г) эмбриональную ткань.29. Проведения биохимических исследований требуют:а) микроцефалия, умственная отсталость, лицевые дизморфии,
пороки развития почек и сердца;б) судороги, повышенная возбудимость, отставание в психомо¬
торном развитии;в) повышенная фоточувствительность кожи, тетраплегия, поли¬
невриты, изменение цвета мочи;г) низкий рост, пороки развития сердца и ЖКТ, брахидактилия,
эпикант, мышечная гипотония.30. Секвенирование ДНК — это:а) идентификация последовательности оснований ДНК;б) многократное повторение какого-либо участка ДНК;в) выделение фрагмента ДНК, содержащего изучаемый ген.31. Современные цитогенетические методики:а) исследование полового хроматина;
530Клиническая генетикаб) интерфазный аналщ хромосом;в) молекулярно-цитогенетический метод;г) метод рутинной окраски.32. Массовому биохимическому скринингу подлежат болезни с:а) тяжелым течением, летальностью в раннем возрасте незави¬
симо от проводимого лечения;б) высокой частотой гена болезни в популяции;в) курабельностью при назначении специфической патогенети¬
ческой терапии.33. Для получения образцов ДНК можно использовать:а) кровь;б) сыворотку;в) ворсины хориона;г) амниотическую жидкость;д) клетки амниотической жидкости;е) биоптаты кожи, мыщц^ печени.34. Микрохромосомные перестройки (микроделеции, микродупликации,
транслокации небольших участков хромосом) выявляются с помощью:а) прометафазного анализа хромосом;б) метода С-окрашивания;в) анализа полового хроматина;г) молекулярно-цитогенетических методов.35. Для проведения блот-гибридизании по Саузерну необходимы:а) нитроцеллюлозный или нейлоновый фильтр;б) ДНК пациента;в) последовательность ДНК используемого зонда;г) специфичная рестриктаза;д) ДНК-зонд.36. Верные утверждения относительно аллельспецифичной гибридиза¬
ции с олигонуклеотидными зондами:а) необходимо знание мутации, обусловливающей данное забо¬
левание;б) перед началом ДНК-д и агностики необходимо знание после¬
довательности всего Гена, включая фланкирующие регулятор¬
ные последовательности;в) может использоваться для диагностики серповидно-клеточной
анемии;
Контрольные вопросы 531г) для диагностики достаточно ДНК нескольких членов семьи;д) этот диагностический метод применим для небольшого числа
генных болезней.ОТВЕТЫ1 — б, в, г; 13 — б, в; 25 — в;2 — в; 14 — г, д; 26 — б, в;3 — а, в, г; 15 — а; 27 — б;4 — а, в, г; 16 — а, г; 28 — в, г;5 — в; 17 — а, б, г; 29 — б, в;6-6; 18-6; 30 - а;7 — в, г; 19 — в; 31 — б, н;8 — 6; 20 — в; 32 — б, в;9 — а, в; 21 — б; 33 — а, в, д, е;10 -б; 22 - а, в, г; 34 - а, г;11 — в; 23 — а, д; 35 — а, б, г, д;12 — 6; 24-6; 36 — а, в, г, д.К ГЛАВЕ 101. В настоящее время наиболее часто применяется терапия наследствен¬
ных болезней:а) симптоматическая;б) патогенетическая;в) этиотропная;г) заместительная;д) клеточная.2. Поддаются коррекции специальными диетами:а) нейрофиброматоз;б) фснилкетонурия;в) муковисцидоз;г) галактоземия;д) умственная отсталость с ломкой Х-хромосомой.3. Метаболическая ингибиция как один из видов коррекции обмена
включает:а) ограничение поступления вещества с пищей;б) выведение из организма субстрата патологической реакции;в) снижение интенсивности синтеза патологического субстрата;
532Клиническая генетикаг) защиту органов от поступления избыточных количеств про¬
дуктов катаболизма.4. Разработка генной терапии наследственного заболевания воз¬
можна при:а) условии, что мутантный ген должен быть идентифицирован
и секвенирован;б) отсутствии другого эффективного лечения;в) аутосомно-рецессивном типе наследования болезни;г) возрасте манифестации болезни не ранее 5 лет;д) знании патогенеза болезни для определения мишени генной
терапии.5. Верно утверждение:а) при трансгенозе соматических клеток происходит замена
аномального гена нормальным, при трансгенозе зародышевых
клеток добавляется нормальный ген;б) трансгеноз соматических клеток, в отличие от трансгеноза заро¬
дышевых, не отражается на генотипе потомства больного;в) трансгеноз соматических клеток осуществляется постнаталь-
но, зародышевых — пренатально;г) после трансгеноза соматических клеток, в отличие от транс¬
геноза зародышевых, требуется пожизненная иммуносупрес-
сивная терапия;д) трансгеноз соматических клеток может применяться только
при моногенных заболеваниях, зародышевых клеток — еще и
при хромосомных и многофакторных болезнях.6. Относительно трансплантации органов и тканей как метода лечения
наследственных заболеваний верны утверждения:а) помимо пересадки органов, можно пересаживать отдельные
клетки, участвующие в патогенезе болезней обмена;б) трансплантацию костного мозга от здорового индивидуума
его сибсу с наследственным заболеванием можно расценивать
как трансгеноз зародышевых клеток;в) при наследственных болезнях отторжение пересаженного
органа происходит редко, так как аномальный генотип пре¬
пятствует реакции трансплантат против хозяина;г) пересаженный донорский орган через некоторое время также
может поражаться наследственным заболеванием.
Контрольные вопросы 5337. К какому подходу в лечении наследственных заболеваний можноотнести примеры:1) назначение соматотропного гормона ребенку с наследствен¬
ной формой карликовости вследствие сниженной функции
гипофиза;2) назначение фенобарбитала для профилактики судорог у
ребенка с гиперам мониемией вследствие недостаточности
орнитинтранскарбамилазы;3) назначение больших доз витаминов ребенку с умственной
отсталостью вследствие хромосомной аномалии;4) назначение D-пеницилламина для связывания внутриклеточ¬
ных ионов меди при синдроме Вильсона-Коновалова;5) пересадка печени больному семейной гиперхолестсрине-
мией;6) назначение карнитина ребенку с органической ацидемией для
образования эфиров карнитина и их выведения;7) назначение диеты без молочных и кисломолочных продуктов
при галактоземии;а) диетическое ограничение;б) альтернативные пути обмена;в) усиленное выведение субстрата;г) возмещение продукта;д) ничего из перечисленного.ОТВЕТЫ1 — а, б, г, д; 4 — а, б, д; 7: 1 — г, 2 — д, 3 — д,2 — б, г; 5 — 6; 4 — в, 5 — д, 6 — б,3 — в; 6-а; 7-а.К ГЛАВЕ 111. Определение концентрации АФП в крови беременной является скри-
нирующим методом дородовой диагностики:а) хромосомной патологии;б) наследственных ферментопатий;в) врожденных пороков развития;г) гетерозиготности по гену Ст2-ганглиозидоза (болезни
Тея—Сакса).
534Клиническая генетика2. С помощью молекулярно-генетических методов пренатально диа¬
гностируют:а) муковиспидоз;б) синдром «кошачьего крика*>;в) талассемию;г) хронический лимфолейкоз.3. Определение концентрации АФП и ХГЧ в крови беременной является
скринирующим методом дородовой диагностики;а) наследственных дефектов обмена аминокислот;б) наследственной патологии крови;в) пороков развития;г) наследственных дефектов обмена углеводов.4. Требования к методам биохимического скрининга:а) диагностическая значимость (небольшой процент ложнополо¬
жительных и отсутствие ложноотрицательных результатов);б) стоимость диагностической программы не больше стоимо¬
сти содержания обществом больных;в) использование легкодоступного биологического материала
в малом количестве;г) при положительном результате отсутствие необходимости в
проведении повторного исследования с целью подтверждения
диагноза.5. Понятие генетического риска включает:а) повышенную вероятность иметь определенное заболевание
в течение жизни;б) вероятность возникновения наследственной болезни или
болезни с наследственной предрасположенностью;в) вероятность внутриутробной гибели плода,6. с помощью УЗИ у плода диагностируют:а) фенилкетонурию;б) анэнцефалию;в) редукционные пороки конечностей;г) синдром Марфаііа.7. С целью диагностики наследственной патологии у плода проводят
амниоцентез в сроки гестации:а) 7-8 нед;б) 11-12 нед;
Контрольные вопросы 535в) 16-18 нед;г) 24-26 нел.8. Высокий генетический риск составляет:а) 100%;б) 5-10%;в) 10-20%;г) 20-25%.9. Массовую преклиническую диагностику проводят, если:а) частота болезни среди населения до 1%;б) эффективны профилактические мероприятия;в) есть методы ДНК-диагностики;г) заболевание имеет тяжелое течение.10. Оптимальные сроки проведения биопсии хориона:а) 10-12 нед;б) 7-9 нед;в) 4-6 нед.11. С помощью биопсии хориона диагностируют:а) наследственные дефекты обмена веществ;б) множественные врожденные пороки развития;в) хромосомные синдромы;г) изолированные врожденные пороки развития.Щ 12. Средний генетический риск составляет:а) 10-20%;б) 50%;в) 6-10%;г) 20-25%.13. С помощью УЗИ диагностируют;а) анэнцефалию;б) галактоземию;в) мукополисахаридоз;г) ахондроплазию.14. Кордоцентез проводят в сроки гестации:а) 5-8 нед;б) 9-11 нед;в) 16—18 нед;г) 20-22 нед.
536Клиническая генетика15. Кордоцентез проводят при повышенном риске по:а) хромосомным синдромам, обусловленным структурными
мутациями;б) наследственным болезням крови;в) порокам развития;г) хромосомным синдромам, обусловленным числовыми мута¬
циями.16. Диагностировать пренатально до 20 недель гестации можно:а) адреногенитальный синдром;б) гемофилию;в) изолированную расщелину нёба;г) синдром Эдвардса.17. Первичная профилактика — это:а) комплекс мероприятий, направленных на предупреждение
рождения или зачатия детей с наследственными болезнями;б) комплекс мероприятий, направленных на предотвращение
развития унаследованного заболевания;в) фенотипическая коррекция дефекта.18. Неинвазивные методы пренатальной диагностики:сО фетоскопия;б) УЗИ;в) хорионбиопсия;г) анализ ХГЧ в сыворотке беременной;д) кордоцентез.19. Пренатальная диагностика — это:а) комплекс мероприятий, направленных на предупреждение
развития заболевания у ребенка;б) предотвращение беременности при высоком риске рожде¬
ния больного ребенка;в) диагностика болезни у эмбриона или плода;г) оценка риска развития заболевания у будущего ребенка;д) диагностика гетерозиготного носительства рецессивных пато¬
логических генов у беременной.20. Третичная профилактика используется при:а) фенилкетонурии;б) врожденном гипотиреозе;в) хорее Гентингтона;
Контрольные вопросы 537г) альбинизме;д) целиакии.21. Женщине 27 лет был проведен амниоцентез на 16-й неделе беремен¬
ности в связи с множественными аномалиями у плода по результатам
УЗИ. При цитогенетическом исследовании у плода выявили трисо-
мню 21. Тактика врача-генетика:а) рекомендовать прерывание беременности;б) предоставить семье полную информацию о вероятном
состоянии здоровья ребенка, возможностях его лечения и
социальной адаптации;в) предоставить право окончательного решения о пролонги¬
ровании или прерывании беременности родителям;г) рекомендовать повторную беременность.22. Для просеивающей диагностики фенилкетонурии у новорожденного
берут кровь:а) в процессе родов (пуповинная кровь);б) на 7-10-й день жизни;в) на 3-5-й день жизни.23. Женщина 31 года на 6-й неделе беременности очень обеспокоена тем,
что ее сестра недавно родила дочь с синдромом Дауна. Пациентка
хотела бы провести амниоцентез. Тактика врача:а) амниоцентез на сроке 15—16 недель;б) детальное УЗИ плода на 18—20-й неделе гестации;в) запросить результаты кариотипирования больного ребенка;г) специфическая пренатальная диагностика в данном случае
не требуется.24. Показания для пренатального цитогенетического исследования:а) возраст маїери 39 лет;б) рождение ребенка с синдромом Дауна в анамнезе;в) кистозная гигрома шеи у плода при УЗИ;г) расщелина позвоночника (spina bifida) у троюродного брата;д) робертсоновская транслокация у отца.25. К каждой ситуации подберите наиболее вероятный вариант этио¬
логии:1) Повторные выкидьшти на ранних сроках беременности.2) Аутосомно-домиііантное заболевание вследствие новой
мутации.
538 Клиническая генетика3) Аутосомно-рецессивиое заболевание.4) Х-сцепленное заболевание.5) Трисомия 13.а) сходная клиническая картина отмечается также у двух
дядей по материнской линии;б) возраст отца 50 лет;в) возраст матери 40 лет;г) сбалансированная транслокация;д) кровнородственный брак.26. Для каждой ситуации подберите наиболее целесообразный метод
пренатальной диагностики:1) 30-летняя женщина, в анамнезе рождение мертвого ребенка
с множественными пороками развития (полидактилия, рас-
шелина нёба, порок сердца) и нормальным кариотипом.2) В семейном анамнезе миодистрофия Дюшенна, беременная
— носительница семейной делеции Б гене дистрофина.3) У плода на 9-й неделе гестации при УЗИ обнаружили уве¬
личение толщины шейной складки, атрезию или стеноі
двенадцатиперстной кишки,4) 25-летняя женщина очень обеспокоена возможностью рож¬
дения ребенка с синдромом Дауна. Индивидуальный и
семейный анамнез без особенностей.5) 36-летняя женщина на 14-й неделе беременности.а) биопсия ворсин хориона;б) определение концентрации АФП в сыворотке матери;в) амниоцентез;г) детальное УЗИ;д) кордоцентез.ОТВЕТЫ1 - а, в;10 - б;19 - в;2 - а, в;11 — а, в;20 — а, б, д;3-в;12 — а, в;21 - б, в;4 - а, в;13 — а, г;22 - в;5-6;14 — г;23 —в; 24 — а, б, в, д;6 — б, в;15 — а, б, г,25: 1 ~ г, 2 - б, 3 - д,7-в;16 —а, б, г;4 —а, 5 — в;8 - а, г;17 - а;26: 1 — г, 2 — а, в, д,9 - б, в, г;18 - б, г;3 ~ а, в, д, 4 — б, г,
ПРИЛОЖЕНИЕГЕНЕТИЧЕСКИЕ ТЕРМИНЫАберрация хромосомная (или хромосомная аномалия) обоб¬
щенное название любого из типов хромосомных мутаций: делений,
транслокаций, инверсий, дупликаций. Иногда обозначают и геном¬
ные мутации (анеуплоидии, трисомии и т.д.)-Аллель — одна из двух или более альтернативных форм гена, каж¬
дая из которых характеризуется уникальной последовательностью
нуклеотидов.Аллель молчащий — мутантный ген, не имеющий обнаруживаемых
фенотипических признаков.Аллельная гетерогенность — наличие в рамках единой нозологи¬
ческой формы различных вариантов заболевания, обусловленных
различными мутантными аллелями одного гена.Аллельные заболевания — фенотипически различные заболевания,
обусловленные разными мутациями в одном и том же локусе (гене).Аллель-специфический олигонуклеотид — олигонуклеотидный
зонд, синтезированный в полном соответствии с конкретной после¬
довательностью ДНК. Дает возможность распознавать аллели, отли¬
чающиеся одним или несколькими основаниями.Альфа-фетопротеин (а-фетопротеин — ЛФП) — эмбриональный
белок, обнаруживаемый в крови плода, новорожденного, беремен¬
ной, а также в амниотической жидкости.Амниоцентез — прокол амниотического мешка с целью получения
амниотической жидкости, содержащей клетки плода.Амплификация — увеличение числа копий определенного фраг¬
мента ДНК.Анализ сегрегации - статистический метод опенки распределения
индивидов по фенотипу для определения наиболее вероятного типа
наследования болезни или признака.Анеуплоидия — измененный набор хромосом, в котором одна
или несколько хромосом из обычного набора или отсутствуют, или
представлены дополнительными копиями.Антикодон — три основания тРНК. комплементарные кодону в
мРНК.
540Клиническая генетикеАнтимутагенез — процесс предотвращения закрепления (станов¬
ления) мутации, т.е. возврат первично поврежденной хромосомы или
гена в исходное состояние.Антиципация — более раннее появление и более тяжелое течение
заболевания в каждом последующем поколении.Ассортативные браки — браки, при которых выбор брачного пар¬
тнера по одному или нескольким признакам неслучаен.Ассоциация — 1) в генетической эпидемиологии: явление, при кото¬
ром частота встречаемости конкретного аллеля значительно больше
или меньше в группе больных по отношению к частоте в популяции,
к которой относится группа больных; 2) в дисморфологии: сочета¬
ние аномалий неизвестной этиологии и патогенеза, встречающихся
совместно чаще, чем при случайном сочетании.j^TOCOMa — любая неполовая хромосома, У человека имеется 22
пары аутосом.Аутосомно-доминантное наследование — тип наследования, при
котором одного мутантного аллеля, локализованного в аутосомс,
достаточно для проявления болезни или признака.Аутосомно-рецессивное наследование — тип наследования призна¬
ка или болезни, при котором мутантный аллель, локализованный Я
аутосоме, должен быть унаследован от обоих родителей.Бивалент - сцепленная пара гомологичных хромосом, наблюдае¬
мая в метафазе первичного мейотического деления.Биоинформатика — вычислительный анализ и хранение биологи¬
ческих и экспериментальных данных. Применяется в геномных ИПрОТеОМНЫХ исследованиях. 'П]Биопсия хориона — процедура, осуществляемая на 7—11-й неделе бере^
менности, с целью получения клеток для пренатальной диагностики.Блот-гибридизация по Саузерну — идентификация участка ДНХ^
содержащего искомую нуклеотидную последовательность, путем
гибридизации разделенных гель-электрофорезом и фиксированных!
на твердом материксе фрагментов ДНК с меченым комплементарныщ
ДНК-зондом. 'їїВектор (генетический) — материал, применяемый для переноса
клетку генетической информации. Наиболее часто применяют плаЗ
МИДЫ и вирусы (ретровирусы, аденовирусы и др.). Возможно ПрИММ;|
нение синтетических конструкций. idВектор для клонирования — любая небольшая плазмида, фаг илї
ДНК-содержащий вирус животных, в которые может быть ветров!
чужеродная ДНК.
■ Генетические термины 541Вестерн-блоттинг — метод, сходный с блот-гибридизацией по
Саузерну, используется для детекции белков, обычно иммунологи¬
ческими методами.Врожденные болезни — болезни, имеющиеся при рождении, необя¬
зательно генетической природы.Вставка — см. инсерция.Гамета — зрелая половая клетка.Гаплоидный — содержащий одинарный набор хромосом.
Гаилонедостаточность - состояние организма, когда вклад нор¬
мального аллеля недостаточен для предупреждения болезни в связи
с мутацией с утратой функции в другом аллеле.Гаплотип — комбинация конкретных аллелей на одной хромосоме.
Гемизиготность — состояние организма, при котором какой-то ген
представлен в одной из гомологичных хромосом, но отсутствует или
инактивирован на другой.Ген — последовательность нуклеотидов в ДНК, кодирующих опре¬
деленную мРНК и соответствующий белок, либо РНК, несущие
структурные или регуляторные функции.Ген гомеозисный — консервативная последовательность из 180 пар
оснований, названная гомеобоксом в связи с тем, что она кодирует
участок, известный как гомеодомен.Ген-модификатор — ген, изменяющий фенотип, вызванный мута-
п,иями в неаллельном гене.Генетическая ассоциация — математическая корреляция между
частотой или характером заболевания и определенным аллелем изу¬
чаемого гена.Генетический маркер — полиморфный участок ДНК строго
определенной локализации, разные аллели которого позволяют диф¬
ференцировать различные по происхождению хромосомы и анализи¬
ровать их сегрегацию в родословной.Генетический риск — вероятность появления определенного
наследственного заболевания.Генная инженерия — совокупность приемов, методов и технологий
получения рекомбинантных РНК и ДНК, выделения генов из орга¬
низма (клеток), осуществления манипуляиий с генами и введения их
в другие организмы.Генная терапия (генотерапия) — способ лечения путем введения
дополнительного генетического материала (ДНК или РНК) в клетки,
функции которых (или функцию организма) изменяет данный ген,
или путем изменения экспрессии генов.
542Клиническая генетикаГенодиагностика (ДНК-диагностика) — совокупность методов но
выявлению мутаций, приводящих к наследственной патологии,Генокопия — клинический синдром, манифестирующий под
маской известного наследственного заболевания с установленной
генетической природой, но обусловленный мутацией в другом гене
(локусе).Геном — полная последовательность ДНК организма; общаи
генетическая информация, содержащаяся в клетках организма, или
генетический состав клетки. Термин «геном» иногда употребляется
для обозначения гаплоидного набора хромосом.Геномика — область исследования структуры и функций генома.Генотип: 1) набор генов индивида или вся генетическая информа¬
ция организма; 2) конкретная пара аллелей, которые индивид имеет
в данном участке генома.Гены служебные («домашнего хозяйства») — экспрессируемые в
большинстве или во всех клетках гены; их продукты обеспечивают
основные функции клетки.Гетерогенность генетическая — формирование одинаковых или
похожих фенотипов при разных генетических причинах.Гетерогенность локусная — идентичные фенотипы вследствие
мутаций в двух или более разных локусах.Гетерозигота — клетка (или организм), содержащая два различных
аллеля в конкретном локусе гомологичных хромосом.Гетерозигота двойная — индивид, гетерозиготный по двум разным
локусам. Не путать с компаундной гетерозиготой.Гетерозигота компаундная (компаунд) - индивид с двумя различ¬
ными мутантными аллелями в одном локусе.Гетерозиготный организм — организм, имеющий две различные
формы данного гена (разные аллели) в гомологичных хромосомах.Гетероплазмия — ііаличие в к^тетках нормальных и мутантных
молекул митохондриальной ДНК.Гетерохроматин — бедная генами область хромосомы (иногда
даже целая хромосома), имеющая плотную компактную структуру
в интерфазе.Гибридизация (ренатурация, отжиг) — взаимодействие компле¬
ментарных цепей ДНК (или ДНК и РНК), приводящее к образованию
двухцепочечной молекулы.Гибридизация геномная сравнительная (CGH — Comparative Genonit
Hybridization) — техника флюоресцентной гибридизации, используе¬
Генетические термины 543мая для сравнения двух образцов ДНК относительно содержимого
конкретного сегмента или сегментов ДНК.Гибридизация in situ — гибридизация между денатурированной
ДНК клеток на предметном стекле и меченной радионуклидами или
флюоресцентными соединениями одноцепочечной РНК или ДНК.Голаидрическое наследование — наследование, сцепленное с
Y-хромосомой.Гомозигота — клетка (или организм), содержащая два одинаковых
аллеля в конкретном локусе гомологичных хромосом.Гомозиготный организм — организм, имеющий две идентичные
копии данного гена в гомологичных хромосомах.Гомологичные хромосомы — хромосомы, одинаковые по набору
составляющих их генов.Гомоплазмия — наличие в клетках одного вида митохондриальной
ДНК (нормальной или мутантной).Груз генетический — итоговая сумма емері ей и болезней, вызван¬
ных мутантными генами.Группа сцепления — все гены, локализованные в одной хромосоме.Дактилоскопия генная — выявление вариаций в числе и длине
тандемных повторов ДНК.Деления — тип хрохмосомной мутации, при которой утрачивается
участок хромосомы; тип генной мутации, при которой выпадает
участок молекулы ДНК.Денатурация — переход молекулы ДНК из двухиепочечной формы
в одноцепочечную вследствие разрыва водородных связей между
комплементарными основаниями.Диплоидный — содержащий двойной набор хромосом.Дискордантность — ситуация, в которой один член пары имеет
определенный качественный признак, а другой нет.Дисомия однородительская — присутствие в кариотипе двух копий
конкретной хромосомы, унаследованных от одного из родителей, без
участия другого родителя.Дицентрик — структурно аномальная хромосома с двумя центро¬
мерами.ДНК-полимераза — фермент, осуществляющий комплементарный
синтез (репликацию) ДНК.Доза гена ~ количество копий конкретного гена в геноме.Домен — участок аминокислотной последовательности белка,
связанный с определенной функцией.
544 Клиническая генетикаДоминантный — признак или соответствующий аллель,
проявляющийся у гетерозигот.Доминантный негативный — патологический аллель или эффект тако¬
го аллеля, нарушающий функцию аллеля дикого типа в той же клетке,Дрейф генов — изменеиие частот генов в ряду поколений, обусловлен¬
ное случайными событиями митоза, оплодотворения и размножении,Дупликация — тип хромосомной лтутации, при которой удвоен
какой-либо участок хромосомы; тип генной мутации, при которой
удвоен какой-либо участок ДНК.Евгеника — научная и практическая деятельность, направлен¬
ная на повышение распространенности желательных признаков по
популяции (положительная евгеника) и снижение частоты патоло¬
гических аллелей (негативная евгеника) благодаря управляемому,
выборочному разведению (стерилизация, насильственные браки).Заболевание генетическое — болезнь целиком или частично вызван¬
ная аномалией генов.Заболевание моногенное - болезнь, обусловленная одним или
парой мутантных аллелей в единственном локусе.Заболевание хромосомное — болезнь, вызванная аномальной хро¬
мосомной или геномной конституцией.Заболевание экогенетическое — болезнь, вызванная взаимодей¬
ствием генетической предрасположенности с конкретным фактором
окружающей среды.Закон Харди-Вайнберга - закон, связывающий частоту аллеля
с часі'отой генотипа; используется в популяционной генетике длл
определения частот аллелей и гетерозигот при известной встречае*
мости заболевания.Зонд генетический — короткий отрезок ДНК или РНК (16—30 п.и.)
с известной последовательностью нуклеотидов, несущий метку (ради¬
оактивный изотоп, флюоресцентное соединение, биотин и др.).Изолят — малая популяция, браки в которой происходят в грани»
цах этой популяции.Изохромосома — аномальная хромосома с удвоением одного плеч»
(формируются два плеча равной длины, с теми же локусами в обраТ"
ной последовательности) и отсутствием второго плеча.Иммунофлюоресаентные зонды — см. зонд генетический. .Имнринтинг геномный (генный или хромосомный) — феномен
различной экспрессии аллелей в зависимости от родительского про¬
исхождения.
Генетические термины 545Инбредные браки — браки между кровными родственниками IJ и
других степеней родства.Инверсия — тип хромосомной мутации, при которой последователь¬
ность генов в участке хромосом изменена на обратную; тип генной
мутации, при которой в определенном участке ДНК последователь-
носгь оснований изменена на обратную.Инсерция (или вставка) — тип генной мутации, при которой име¬
ется вставка отрезка ДНК в структуру ієна.Интрон — сегмент ДНК в гене, не содержащий информации о
структуре белкового продукта гена.Информативная семья — семья с наследственным заболеванием,
в ко'юрой имеется достаточное число больных и здоровых родствен¬
ников из разных поколений (информативные мейозы), что позволяет
оценивать расхождение признаков в изучаемой родословной при ана¬
лизе генетического сцепления. Впервые описана Мэри Лайон.Исследование типа «случай-контроль» - эпидемиологический
.метод, при котором пациентов с болезнью («случаи») сравнивают с
должной группой контроля (без болезни) относительно частоты раз¬
личных предполагаемых факторов риска.Кариотип ~ хромосомный набор клетки или организма.Картирование — определение локализации гена на хромосоме:• генетическое — определение расположения изучаемого гена
по отношению к другим генам в определенной хромосомной
области;• физическое — установление точной последовательности сайтов
и физического расстояния между ними (исчисляемого в парах
оснований) в изучаемой хромосомной области.Кариотипирование спектральное (SKY - Spectral Karyotyping) - про¬
цедура, использующая технику FISH для различного окрашивания
каждой из 24 хромосом человека.Карта сцепления — хромосомная карта, показывающая относи¬
тельные позиции генов и других ДНК-маркеров в хромосомах на
основе анализа сцепления.Килобаза - единица измерения, равная 1000 парам оснований
ДНК или РНК.Клетки половые — линия клеток, из которой происходят гаметы.Клетки стволовые — клетки, способные к самовоспроизводству,
пролиферации и дифференцировке; имеются почти во всех органах.Клон — генетически однородное потомство одной клетки.
546 Клиническая генетикіКлонирование гена — встраивание определенного участка ДНК и
генетическую конструкцию, например, плазмиду, с целью получения
большого числа коиий данного фрагмента путем введения в микро¬
организмы.Кодоминантные аллели — аллели, каждый из которых проявляетси
в іетерозиготе (например, группа крови по системе АВО).Кодон — элементарная единица генетического кода, состоящан
из трех рядом находящихся оснований, сочетание которых кодирует
включение специфического аминокислотного остатка в полипептид-
ную цепь, либо сигнал начала или завершения транскрипции.Коммитирование (детерминация) — приобретение клеткой
структурно-функциональных признаков терминальной стадии диф-
ференцировки.Комнаунд-гетерозиготность — состояние организма, при котором
один и гот же локус на гомологичных хромосомах представлен рав¬
ными мутантными аллелями.Комплементарная ДНК (к ДНК) — последовательность ДНК, получен¬
ная с помощью обратной транскриптазы с информационной РНК.Комплементарность — способность нуклеотидов образовывать |
пары по правилу А-Т, C-G в результате формирования водородных
связей между ними в двухцепочечной молекуле ДНК или в гибридной I
молекуле ДНК/ РНК.Конкордантность - понятие, описывающее пару родственников,
каждый из которых имеет определенный качественный признак илИ
сходные величины количественного признака.Консультирование генетическое — обеспечение информации
помощи больному или членам его семьи о риске заболевания, с
последствиях, вероятности передачи и путях предохранения илПОМОШИ. гмКордоцентез — процедура взятия крови из пупочной вены ПЛОДІІКорреляция — статистический метод оценки связи между парны
измерениями. Когда большему значению показателя в первом из
рении соответствует большее значение во второ.м — это положит
ная корреляция. Отрицательная корреляция — чем больше пери
значение, тем меньше второе. !Коэффициент инбридинга — вероятность того, что у одного ИНД
вида два аллеля в данном локусе происходят от одного предка, к;Коэффициент корреляции (г) - мера корреляции, изменяющаяся
1 для полной положительной связи до -I для полной отрицатель
Генетические термины 547связи. Когда между парами измерений взаимосвязи нет, коэффици¬
ент корреляции равен нулю.Кровнородственный — происходящий от общего предка.Кроссинговер — обмен участками между гомологичными (не
сестринскими!) хроматидами в процессе мейоза.Лайонизация — инактивация одной из двух Х-хромосом в женской
соматической клетке на ранних стадиях развития, благодаря кото¬
рой обеспечивается сбалансированность генетического материала у
индивидов мужского и женского пола. Инактивация отцовской и
материнской хромосом носит случайный характер,Леталь — мутация, вызывающая гибель клетки или особи до
достижения репродуктивного возраста. Генетическая леталь — инди¬
вид, не способный к размножению.Лизосомные болезни — группа наследственных болезней с унаследо¬
ванной недостаточной продукцией лизосомных ферментов.Локус — область локализации определенного генетического эле¬
мента на хромосоме.Локусная гетерогенность — наличие в рамках единой нозологической
формы различных генетических вариантов заболевания, вызываемых
мутациями самостоятельных генов в разных хромосомных локусах.Маркер — аллелъ (или признак), наследование которого прослежива¬
ется в потомстве.Метилирование ДНК — один из способов эпигенетической регу¬
ляции, при котором происходит добавление метильного остатка в
5’-положение пиримидинового кольца цитозина в ДНК, в результате
чего образуется 5’-метилцитозин.Мейоз — два последовательных (1-е и 2-е) делении ядра зароды¬
шевой (половой) клетки при одном цикле репликации, в результате
чего образуются гаплоидные клетки.МикроРНК— класс некодирующей белок РНК, процессирующей
в короткие интерферирующие РНК (siRNA), двуцепочечные РНК
длиной около 22 нуклеотидов, влияющие на устойчивость мРНК
или трансляцию (феномен РНК-интерференции). siRNA участвуют
в регуляции генов в процессе развития и дифференцировки.Миссенс-мутации — см. мутация.Митохондриальное наследование — наследование признаков,
передаваемых через ДНК митохондрий.Множественные аллели — наличие в популяции (или у вида) более
двух аллелей одного и того же локуса.
548 Клиническая генетикаМозаик — индивид, у которого есть клетки с различными
хромосомными наборами.Мозаицшм — наличие у индивида клеток с двумя и более вари¬
антами хромосомных наборов или генных мутаций.Мозаицизм, ограниченный плацентой — мозаицизм в препаратах,
полученных из ворсин хориона или плаценты, отсутствующий у
самого эмбриона или плода.Моносомия — состояние клетки, при котором какая-либо хромо¬
сома представлена в единственном числе, а не парой гомологичных
хромосом.Морфогенез - процесс изменения формы, адгезии, переме¬
щений и числа клеток, ведущий к формированию трехмерной
структуры.Морфогенетические варианты врожденные (микроаномалии раз¬
вития, признаки или стигмы дисэмбриогепеза) — отклонения н
развитии, выходящие за пределы нормальных вариаций, но не нару¬
шающие функции органа.Мультифакториальные (многофакторные) болезни — болезни, кото¬
рые развиваются в результате взаимодействия определенных комбИ"
наций аллелей разных локусов и специфических воздействий факто¬
ров окружающей среды.Мутаген - агент, вызывающий изменения в ДНК или хромосомах
и увеличивающий частоту спонтанных му гаиий.Мутант — организм, несущий мутантный аллель.Мутация — изменение в наследственных структурах (ДНК, ген,
хромосома, геном):• генная — изменение последовательности нуклеотидов в опреде¬
ленном участке молекулы ДНК; '• геномная — изменение числа хромосом (кратное гаплоидному —
полиплоидия, некратное — анеуплоидия);• динамическая — мутация по типу экспансии тандемных тринуь^|
клеотидных повторов;• мажоріная — встречающаяся с высокой частотой в определение
популяции;• миссенс — замена нуклеотида, сопровождающаяся изменением|
аминокислотного шифра кодона и ведущая к замене аминокислс
ты в составе белка; ■ і• нейтральная (молчащая) — не сопровождающаяся изменениеі
фенотипа;
Генетические термины 549• нонсенс — замена нуклеотида, приводящая к замещению, коди¬
рующего аминокислоту, на стоп-кодон и сопровождающаяся
преждевременным обрывом трансляции;• нулевая — приводящая к отсутствию синтеза либо утрате функ¬
ции белка;’ регуляторная — затраіивающая регуляторные последовательно¬
сти гена и нарушающая его экспрессию;• со сдвигом рамки — приводяпіая к нарущению нормального
отсчета кодирующих гриплетов (делении или вставки участков
молекулы ДНК, размеры которых некратны трем основаниям);
обычно приводит к изменению аминокислотной последователь¬
ности белка;• сплайсинговая — затрагивающая сайт сплайсинга и приводящая к
неправильному вырезанию интрона либо к удалению из молекулы
РНК информационно значимой экзонной последовательности;• структурная — приводящая к протяженному (мультинуклеотид-
ному) дефекту гена;•точковая (точечная) — затрагивающая один нуклеотид либо 1-2
соседних нуклеотида;• хромосомная — любое нарушение структуры хромосом (делеция,
дупликация, инверсия, транслокация).Наследование комплексное (многофакторное) — неменделируїощий
тип наследования. Признаки с комплексным наследованием обычно
вызваны аллелями более чем одного локуса, взаимодействующими с
влиянием факторов окружающей среды.Наследование материнское — передача генетической информации
только через мать.Наследственная болезнь — болезнь, для которой этиологическим
фактором является генная, хромосомная или геномная мутация.Наследуемость (h^) — часть общей фенотипической изменчивости
количественного признака, обусловленная генетическими факторами.Неполно доминантный (синоним полудоминантный) - признак,
наследующийся по доминантному типу, но более тяжело проявляю¬
щийся у гомозигот, чем у гетерозигот.Неравновесное сцепление — неслучайное распределение частот
аллелей генетических маркеров в исследуемой группе лиц по срав¬
нению с общей популяцией.Нерасхождение — неправильное разделение двух хромосом
одной пары в ходе мейоза I или двух хроматид хромосомы в тече-
550 Клиническая генетикание мсйоза И или митозе, что приводит к тому, что оба гомолога
передаются в одну дочернюю клетку, тогда как в другую не пере¬
дается ничего.Нозерн-блоттинг — техника, аналогичная блоттингу по Саузерну,
позволяющая обнаружить молекулы PHК-гибридизацией с компле¬
ментарным ДНК-зондом,Нонсенс-мутации — см. Мутация.Норма реакции — границы выраженности фенотипов при одном и
том же генотипе в различных условиях среды.Носитель — индивид, имеющий одну копию гена, который
обусловливает рецессивную болезнь, и одну копию нормальног'о
аллеля.Нуклеотид — молекула, состоящая из азотистого основании,
5-углеродного сахара и фосфатной группы. Нуклеиновая кислота -
полимер множества нуклеотидов.Обмен хроматидный сестринский — обмен сегментами ДНК между
сестринскими хроматидами.Обратная транскриптаза — фермент, осуществляющий перевод
молекулы РНК в молекулу кДНК (обратная транскрипция).Ограниченный полом — признак, который проявляется толь¬
ко у одного пола, хотя ген, определяющий признак, не сцеплен с
Х-хромосомой.Однонуклеотидный полиморфизм (SNP) — варианты последо¬
вательностей ДНК у разных людей, отличающиеся по одной паре
оснований и встречающиеся в популяции с частотой более 1%,
Варианты, встречающиеся менее чем у 1% популяции относят к
мутациям. \Олигонуклеотид — короткая однонитсвая молекула ДНК или РНК
(приблизительно 16—30 нуклеотидов).Онкоген — доминантный ген, повыщение экспрессии которого
играет важную роль в развитии опухолей.Ортологичный — ген другого биологического вида, имеющий ана»|
логичную последовательность ДНК и кодирующий белки с той
функцией. Ортологичные гены происходят от общего предка.Отбор — действие сил, определяющих относительную приспо«
собленность генотипа в популяции, таким образом влияющих
частоту интересующего гена. НОтношение шансов — сравнение шансов того, что индивиды, име«
ющие конкретный фактор (например, генотип, влияние окружаї
Генетические термины 551щей среды или лекарства), будут иметь болезнь или признак, против
таких ж:е шансов для индивидов, у которых нет этого фактора.Панмиксия — случайный подбор лиц, вступающих в брак, в преде-
лах всей популяции.Паралогичный — относится к двум или более генам одного био¬
логического вида, имеющим аналогичные последовательности ДНКи, вероятно, кодирующие белки с аналогичными, возможно перекры¬
вающимися, но не идентичными функциями. Паралогичные гены,
вероятно, происходят от общего гена-предшественника. Например,
гены а- и р-глобина.Пенетрантность — частота проявления фенотипа (признака или
болезни), детерминируемого доминантным аллелем или рецессив¬
ным аллелем в гомозиготном состоянии,Пероксисомные болезни — наследственные болезни обмена веществ,
обусловленные нарушением биогенеза или функции пероксисом.Плазмиды - самостоятельно копирующиеся внехромосомные
циклические молекулы ДНК у бактерий и дрожжей; используются
в молекулярной биологии в качестве векторов для клонирования
сегментов ДНК.Плейотропность — влияние одного гена на развитие двух или
более фенотипических признаков.Плюрипотентный — характеристика эмбриональной клетки, спо¬
собной создавать различные типы тканей или структур в зависимо¬
сти от положения и влияния окружающей среды.Повторы тандемные — две или больше копий одинаковой (или
аналогичной) последовательности ДНК, располагающиеся последо¬
вательно вдоль хромосомы.Полигенные признаки — признаки, обусловленные многими гена¬
ми в различных локусах, каждый из которых оказывает лишь неболь¬
шое влияние на степень экспрессии данного признака.Полимеразная цепная реакция (ПЦР) — метод циклического син¬
теза in vitro огромного числа копий определенного участка ДНК дли¬
ной от десятков до нескольких тысяч пар оснований:• количественіїая — реакция, по результатам которой оценивается
количественный выход продуктов амплификации с помощью
соответствующих сканирующих устройств;• мультиплексная (мультипраймерная) — одновременная
амплификация в одной реакции нескольких участков исследуе¬
мого гена;
552 Клиническая генетика• обратнотранскриптазная — реакция, в которой матрицей служат
молекулы кДНК.Полиморфизм — наличие в популяции двух или более альтернатив¬
ных генотипов с частотой, превыпшющей поддерживаемую только
за счет повторных мутаций. Условно считается, что локус поли¬
морфный, если редкий аллель имеет частоту 0,01, что дает частоту
гетерозигот по крайней мерс 0,02. Любой аллель, встречаемый реже,
считается редким вариантом.Полиморфизм однонуклеотидный — полиморфизм п последователь¬
ности ДНК, состоящий из изменений в одном основании.Полиморфизм сбалансированный — полиморфизм, поддерживае¬
мый в популяции преимуществом гетерозигот, позволяя даже опас¬
ному в гомозиготном состоянии аллелю нерсистировать в популяции
со сравнительно высокой частотой.Полиморфизм длин рестрикционных фрагментов (ПДРФ) — вариабель¬
ность длин фрагментов ДНК, полученных при действии поределенной
рестриктазы на разные образцы ДНК. Отражает отличия в нуклеотид¬
ном составе участков ДНК, узнаваемых данной рестриктазой.Полиморфизм коротких тандемных повторов (STRP - Short Tandem
Repeat Polymorphysm) — полиморфный локус, состоящий из варьирую¬
щего числа последовательно расположенных повторяющихся блоков
динуклеотидов, тринуклеотидов и тетрануклеотидов.Полиплоид — клетка (ткань или организм), имеющая 3 хромосом¬
ных набора или более.Половые хромосомы — хромосомы, определяющие пол индивида
(у человека X- и Y-хромосомы),Последовательность фланкирующая — область, предшествующая
гену или следующему транскрибируемому участку.Потеря гетерозиготности — возникновение мутации или делеции
в соматической клетке в том локусе гомологичной хромосомы, по
которому организм гетерозиготен. Типично для многих случаев рети-
нобластомы, рака молочной железы и других опухолей, вызванных
мутациями в генах-супрессорах опухолевого роста. ^Поток генов — постепенное проникновение генов из одной попу¬
ляции в другую через барьер. Барьер может быть физическим или
культурным и может нарущаться миграцией или смешиванием.Праймер — олигонуклеотид, выполняющий роль «затравки» и
инициирующий синтез полинуклеотидной цепи на ДНК- или РНК-
матрице.
Генетические термины 553Предрасположенность генетическая ~ комбинация av^лeлeй раз¬
ных локусов, предрасполагаюїних к более раннему воз никн овен иго
заболеваний под влиянием факторов окружающей среды и более
тяжелому их течению.Пренатальная диагностика — диагностика наследственных болез¬
ней или других нарушений в период внутриутробного развития.Пробанд — лицо, с которого начинается сбор родословной.Прогностическое (предсказательное) ДНК-тестирование — проведение
ДНК-анализа у клинически здорового человека с целью установить его
генетический статус, который может привести к развитию наследствен¬
ной болезни или болезни с наследственной предрасположенностью.Промотор — регуляторный участок гена, с которым связывается
PH К-полимераза перед началом транскрипции.Просеивание — см. Скрининг.Протеем — полный набор белков, кодируемых геномом.Протоонкоген — нормальный ген, участвующий в отдельных
аспектах деления или пролиферации клеток, способный вследствие
мутаций или других механизмов становиться онкогеном.Процессинг РНК (созревание РНК) — процесс превращения
первичного РНК-транскрипта в молекулу зрелой мРИК путем уда¬
ления интронов и полиаденилирования.Псевдоген — участок ДНК, очень сходный по последовательности
оснований с известным геном, но не выполняющий такую функ¬
цию либо из-за потери сигнализации трансляции (промоторной
последовательности), либо несущий мутацию, которая предотвра¬
щает трансляцию.Пул генов - все аллели, представленные в данном локусе или,
более широко, во всех локусах популяции.Регуляторный участок гена - сегмент ДНК (промотор, энхансер
или регуляторный регион локуса внутри или около гена), регулирую¬
щий экспрессию данного гена.Рекомбинантная ДНК — молекула ДНК, «собранная» в пробирке с
использованием сегментов ДНК из различных организмов.Рекомбинация — образование новых комбинаций аллелей в связи
с кроссинговером между локусами.Ренатурация — см. гибридизация.Репарация — исправление повреждений в молекуле ДНК и
восстановление ее нативной первичной структуры.Рестриктаза (рестрикционная эндонуклеаза) — фермент бакте¬
554 Клиническая генетикариального происхождения, распознающий специфическую нуклео¬
тидную последовательность длиной от 4 до 10 пар оснований и раз¬
резающий двунигевую молекулу ДНК R этом месте.Рестрикционный анализ — метод анализа ДНК с использованием
рестрикционных эндонуклеаз.Ретровирусы — вирусы с геномом, состоящим из РНК, размножаю¬
щиеся преобразованием РНК в ДНК, благодаря ферменту обратной
транскриптазе.Рецессивность — свойство соответствующего аллеля проявляться
в виде признака только в гомозиготном состоянии.Рибосома — цитоплазматическая органслла, состоящая из рибосом-
ной РНК и белка, синтезирующая полипептиды на основе мРНК.Риск — вероятность появления события. Часто вычисляется как число
случаев появления конкретного события, разделенное на число всех воз¬
можных событий. Как и все вероятности, риск изменяется от о до 1.Риск эмпирический ~ вероятность того, что признак будет наблю¬
даться у члена семьи, основанная не на знании причин болезни,
а на наблюдаемых частотах больных и здоровых лиц в семейных
исследованиях.РНК-полимераза — фермент, осуществляющий синтез РНК на
ДНК-матрицс.Родословная — схема, показывающая родство между членами
одной семьи в ряду поколений.Сайт — определенное место (позиция) в молекуле ДНК:рестрикции — специфическая нуклеотидная последовательность
длиной 4—10 п.и., распознаваемая рсстриктазой;сплайсинга — специфическая нуклеотидная последовательность
на границе между экзоном и интроном, играющая сигнальную роль
для правильного протекания сплайсинга.Сайт рестрикции - короткая последовательность ДНК, распозна¬
ваемая и разрезаемая специфической эндонуклеазой (рестриктазой),Сантиморганида (сМ) — единица генетического расстояния между
локусами; 1 сМ соответствует частоте рекомбинационных событий
между локусами в 1% случаев (эквивалентна в среднем примерно 1
млн. пар оснований).Сегрегация — расхождение гомо.'югичных хромосом в мейозе.Секвенирование — определение последовательности нуклеотидов
в молекуле ДНК или последовательности аминокислот в молекуле
белка.
Генетические термины 555Семейные болезни — болезни, наблюдающиеся у нескольких чле¬
нов семьи в одном или нескольких поколениях.Сибство — все сибсы в семье.Сибсы — родные братья и сестры (от англ. Sisters — Brothers).Синдром — характерный набор аномалий, предположительно
вызванный одной причиной.Синдром протяженной делеции — синдром, вызванный микроделе-
цией хромосомной ДНК, захватывающий два или более последова¬
тельно расположенных локуса (син. сегментная анеусомия).Синтения - физическое присутствие в одной хромосоме двух или
более локусов генов, независимо от того, достаточно ли они близки
друг к другу, чтобы иметь сцепление.Скрещивание ассортативное — выбор супруга с предпочтением
конкретного генотипа, то есть неслучайное скрещивание.Скрининг (просеивание) — обследование больших групп людей на
какие-либо состояния (болезни или носительство) с целью активной
профилактики болезней; выявление не диагностированной ранее
болезни с помощью простых методов, дающих быстрый ответ.Сплайсинг — процесс удаления нитронов и объединения экзонов
в зрелую мРНК.Спорадический — случай болезни, не являющийся результатом
наследования патологического аллеля от больного родителя, т.е.
результат новой половой или соматической мутации.Стоп-кодон — нуклеотидный триплет, являющийся сигналом
окончания трансляции.Сцепление генов — совместная передача генов (признаков) с веро¬
ятностью, превыщающей случайную.Тандемные повторы ~ множественные и расположенные друг за
другом копии определенной последовательности ДНК, число кото¬
рых варьирует у разных индивидов (варьирующее число тандемных
повторов — от англ. VNTR):• динуклеотидные — из 2 нуклеотидов (обычно СА); используются
в качестве генетических маркеров;* тринуклеотидные — из 3 нуклеотидов; увеличение их числа (экс¬
пансия) приводит к наследственным болезням нервной системы
(динамические мутации).Теломера — конец плеча хромосомы. У человека теломеры окан¬
чиваются тандемными копиями последовательности (TTAGGG)j^,
необходимой для правильно репликации концов хромосом.
556 Клиническая генетикаТеломераза — обратная гранскриптаза рибонуклеопротеинов,
использует свой собстоенный шаблон РНК, чтобы добавлять в тсломе-
ру видоспецифические гсксамеры (например, TTAGGG у человека).Тельца Барра — половой хроматин. Представляет неактивную
Х-хромосому,Терапия генная — способ лечения путем введения дополнительной
генетической информации в клетки индивида на уровне ДНК или
РНК (генно-инженерных конструкций) или путем изменения экс¬
прессии генов.Тератоген — агент, вызывающий врожденные пороки развития или
увеличивающий их встречаемость.Технология рекомбинантной ДНК — методы создания молекулы
ДНК in vitro из сегментов более чем одной родительской молекулы
ДНК.Трансверсия - мутация, вызванная заменой пуринового основа¬
ния на пиримидиновое или наоборот.Трансген — ген, переносимый в клетку при генной терапии в
составе вектора.Трансгенные организмы — животные, растения, микроорганизмы,
вирусы, генетическая программа которых изменена с помощью мето¬
дов генной инженерии.Трансгеноз — процедура (или процесс) передачи дополнительной
чужеродной генетической информации в организм, клетку.Трансдукция — введение в клетку гена и как следствие изменение
ее генома.Транскрннтаза обратная — фермент, РНК-зависимая ДНК-
полимсраза, катализирует синтез ДНК по шаблону РНК.Транскрипционный фактор — белок, связывающийся с регулятор¬
ной областью гена и регулирующий его экспрессию.Транскрипция — считывание наследственной информации при
экспрессии гена.Транслокация — хромосомная мутация. Перенос сегмента одной
хромосомы на другую.Транслокация робертсоновская - транслокация между двумя акро-
центрическими хромосомами со слиянием их в центромере с утратой
коротких плеч.Трансляция — передача наследственной информации; синтез бел¬
ковой молекулы или перевод последовательности оснований мРНК в
последовательность аминокислот в полипептидной цепи.
Генетические термины 557Транспозон — один из классов мобильных элементов генома; транспо-
зон может перемешаться вну їри і енома («прыгающий участок ДНК»).Триплоидный — клетка с тремя копиями каждой хромосомы или
индивид, состоящий из таких клеток.Трисомия — добавочная хромосома в кариотипе диплоидного
организма; вид полисомии, при котором имеются 3 гомологичные
хромосомы (индивид с трисомией называется трисомиком).Фармакогенетика — раздел медицинской биохимической гене¬
тики, изучающий роль наследственности в реакциях организма на
лекарства.Фармакогеномика — создание новых типов лекарств на основе
геномной информации; предсказание возможных ответов организма
на лекарства на основе анализа генома.Фенокопия “ клинический синдром, сходный по проявлениям с
наследственным заболеванием, но имеющий негенетическую природу.Фенотип — признаки, проявляющиеся в результате действия генов
в определенных условиях среды.Ферментопатия — метаболическое заболевание, вызванное недо¬
статочностью или аномалией конкретного фермента.Фетоскопия — процедура, позволяющая визуально обследовать
плод в матке при помощи волоконно-оптической техники.Фетотерапия (плодная терапия, пренатальная терапия) — лечение
плода до рождения.Химера - индивид, содержащий клетки, производные от двух
генетически разных зигот.Хромосомная мутация (аберрация) — изменение н структуре
хромосомы.Хромосомный набор — совокупность хромосом в ядре гаметы,
зиготы или соматической клетки.Хромосомы бактериальные искусственные (BACs — Bacterial
Artificial Chromosomes) — векторы, способные нести 100-130 кило¬
баз клонированной ДНК человека. Размножаются в бактериях и
используются для картирования генов высокого разрешения и сек-
венирования ДНК.X-сцепленное наследование — тип наследования признаков, гены
которых локализованы в Х-хромосоме.Центромера - первичная перетяжка в хромосоме, место соедине¬
ния сестринских хроматид и формирования кинетохор. Необходима
для нормального расхождения в митозе и мейозе.
558 Клиническая генетикаЦентросомы - два центра, организующих рост микротрубочек
митотического веретена; видны на полюсах делящейся клетки в
конце профазы.Цис-положение - обозначает положение между двумя последо¬
вательностями в одной хромосоме, буквально «с одной стороны».
Противоположно транс-положению.Экзон — отдельный фрагмент прерывистого гена, сохраняющийся
в зрелой РНК после удаления интронных фрагментов.Экспансия тринуклеотидных повторов — патологическое увели¬
чение числа копий внутригенных тандемных последовательностей,
состоящих из 3-х нуклеотидов; этот тип мутаций называют также
динамическими мутациями.Экспрессивность — степень фенотипической выраженности
(проявления) генетически детерминируемого признака.Экспрессируемая короткая последовательность (от англ. expressed
sequence tag — EST) — короткая последовательность (100—500 п.н.)
обычно из одного конца клонированной ДНК; используется для
идентификации генов, альтернативного сплайсинга и распределения
транскриптов в тканях.Экспрессия гена ~ активизация транскрипции гена, в процессе
которой на смысловой нити ДНК синтезируется мРНК.Электропорация — один из методов переноса чужеродной ДНК в
клетки основанный на пермеабилизации клеточной мембраны под
действием электрического поля.Эндонуклеаза рестрикционная (рестриктаза) — фермент, получае¬
мый из бактерий, способный распознавать специфическую после¬
довательность ДНК и расщеплять молекул я ДНК в области сайта
распознавания или рядом с ним.Энхансер — короікий регуляторный сегмент ДНК, который влия¬
ет на уровень экспрессии примыкающих к нему генов, увеличивая
частоту инициации транскрипции.Эпигенетический - термин, обозначающий любой фактор, влияю¬
щий на функцию гена без изменения генотипа. К эпигенетическим
факторам относятся изменения в метилировании ДНК, структуре
хроматина, модификация гистонов.Эписома - элемент ДНК, способный существовать как автоном¬
ная копируемая последовательность в цитоплазме или внедренная в
хромосомную ДНК.Эуплоидия — наличие у индивида полных наборов хромосом.
Генетические термины 559Эухроматин — богатые генами участки хромосом; слабо красится
при G-окраске, конденсируясь и становясь прозрачным в интерфазе.Эффект основателя (родоначальника) — высокая частота идентич¬
ных аллелей как результат наследования от единого предка.Эффект положения (гена) — изменение экспрессии гена при его
переносе из нормального хромосомного окружения в новое (напри¬
мер, в область гетерохроматина).ПРИЗНАКИ ДИСМОРФОГЕНЕЗААгенезия (аплазия) — полное врожденное отсутствие органа или
его части.Акромикрия — непропорционально маленькие кисти и стопы.Акроцефалия («башенный» череп) — череп с высоким лбом,
сглаженными надбровными и височными выступами вследствие
преждевременного закрытия венечного и затылочного швов.Алопеция -- полное или частичное отсутствие волос на голове.Аниридия — отсутствие радужной оболочки.Анодонтия — отсутствие постоянных зубов.Анонихия — врожденное отсутствие любого числа ногтей.Анофтальмия — врожденное отсутствие глазного яблока.Антимонголоидный разрез глаз — наружные углы глаз расмолаїа-
ются ниже внутренних.Арахнодактилия (паукообразные пальцы) — узкая длинная ладонь
с длинными пальцами.Артрогрипоз — множественные врожденные контрактуры суставов.Ателия — отсутствие сосков.Атрезня — полное отсутствие канала или естественного отверстия.Афакия — отсутствие хрусталика.Блефарофимоз — короткая и узкая глазная щель.Брахидактилия — укорочение пальцев рук или ног.Витнлнго очаговая депигментация кожи.Вормиевы косточки — вставочные косточки в черепе.Гетерохромия радужки — неодинаковый цвет различных участков
радужки.Гиперодонтия — наличие сверхкомплектных зубов.Гипертелоризм — широко расставленные глаза (оценивается по
межорбитальному индексу: в числителе — расстояние между орбита¬
560 Клиническая генетиками в сантиметрах, умноженное на 100; и знаменателе — окружность
головы в сантиметрах. При гипертелоризме межорбиталъный индекс
более 6,8).Гипертрихоз — замена пушковых волос грубыми пигментирован¬
ными волосами.Гиподонтия — уменьшенное число зубов по сравнению с возраст¬
ной нормой.Гипоплазия — недоразвитие органа, проявляюш;ееся дефицитом
относительной массы или размера.Гипоспадия — нижняя расщелина уретры со смещением наружно¬
го отверстия мочеиспускательного канала.Гипотелоризм — близко расположенные глаза; межорбиталъный
индекс менее 3,8 (см. Гипертелоризм).Гипотрихоз — редкие волосы вследствие недостаточного их роста.Гирсутизм — чрезмерное оволосение по мужскому типу у женщин.Диастема — промежуток между верхними или нижними централь¬
ными резцами 3 мм и более.Дискория («кошачий глаз») — щелевидный зрачок.Дистихиаз — двойной ряд ресниц.Долихостеномелия — длинные тонкие конечности.Изодактилия ~ примерно равная длина 1J, III, IV и V пальцев,Камптодактилия — сгибательная контрактура в межфаланговых
суставах пальцев кисти.«Карний рот» — рот с опущенными углами рта и дугообразной
верхней губой,Клинодактилия — латеральное или медиальное искривление пальца.Клювовидный нос — нос в виде птичьего клюва.Колобома радужки — тцелевидный дефект радужной оболочки глаза.Конская стопа (pes equinus) — контрактура i оленостопного сустава,
при которой стопа фиксирована в положении чрезмерного подошвен¬
ного сгибания.Короткий мизинец (симптом Дюбуа) — конец мизинца не достигает
складки концевой фаланги IV пальца.Краниостеноз — уменьшение объема черепа с задержкой развития
головного .мозга вследствие преждевременного закрытия швов.Крипторхизм — отсутствие одного или обоих яичек в мошон¬
ке, обусловленное задержкой их внутриутробного перемещения из
забрюшинного пространства. Яички могут располагаться в паховом
канале или в брюшной полости.
Генетические термины 561крыловидные складки (птеригии) — вертикальные кожные склад¬
ки на боковых поверхностях шеи.Лагофтальм — неполное смыкание век,Макроглоссия — увеличенный язык.Макроорхизм — увеличенные яички.Макросомия — чрезмерно увеличенные размеры и масса тела.
Макростомия — чрезмерно широкая ротовая щель.Макротня — увеличенные ушные раковины.Макрофаллос — увеличенный половой член.Макроцефалия — череп, увеличенный более чем на 10% возраст¬
ной нормы.Микрогеиия — уменьшенная нижняя челюсть.Микроглоссия — уменьшенный язык.Микрогнатия — уменьшенная верхняя челюсть.Микромел ИЯ — укороченные конечности, имеющие все сегменты.
Микроорхизм — уменьшенные яички.Микросомия — чрезмерно уменьшенные размеры и масса тела.
Микростомия — уменьшенная ротовая щель.Микротия — уменьшенные размеры ушной раковины.
Микрофаллос — уменьшенный половой член.Микрофтальмия — уменьшенные размеры глазного яблока.
Микроцефалия — череп, уменьшенный более чем на 10% по сравне¬
нию с возрастной нормой.Монголоидный разрез глаз ~ наружные углы глаз располагаются
выше внутренних.«Мыс вдовы» — клиновидный рост волос на лбу.Оксицефалия — асимметричная акроцефалия (см.) или смешение
«башни» черепа в одну сторону.Олитодактилия — уменьшение числа пальцев на кистях или стопах.
Палатосхиз — расщелина нёба.«Папиросные» рубцы — рубцы, напоминающие папиросную бумагу.
Пахионихия — утолщение ногтей.Пилонидальная ямка (сакральный синус) — слепо заканчиваю¬
щийся канал в межъягодичной складке у копчика.Плагиоцефалия — асимметрия правой и левой сторон черепа.
Платицефалия — череп с плоским сводом.Полая стопа (pes excavatus) — чрезмерно высокий свод стопы.
Полидактилия постаксиальная — дополнительные пальцы со сто¬
роны V пальца кисти или стопы.
562 Клиническая генетикаПолидактилия преакснальная — дополнительные пальцы со сторо¬
ны I пальца кисти или стопы.Полителия — избыточное число сосков.Поперечная ладонная складка («обезьянья» складка) — поперечная
борозда, идущая через всю ладонь.Преаурикулярные папилломы — доброкачественные опухоли в виде
сосочков впереди ушной раковины.Преаурикулярные фистулы (ямки) — слепо оканчивающиеся ходы
перед ушной раковиной (рудимент I жаберной дуги).Прогення — выступающая нижняя челюсть.Прогнатия — выдвинутая вперед верхняя челюсть.Птоз — опущение века или внутренних органов.Пяточная стола (pes calcaneus, выступающая пятка) — контрак¬
тура голеностопного сустава с фиксацией стопы в положении раз¬
гибания.Расщелина позвоночника (spina bifida) — неполное закрытие
позвоночного канала.Ретрогнатия — смещение верхней челюсти назад.Сандалевидная щель — большой промежуток между I и П паль¬
цами стопы.Седловидный нос — впадина в средней части спинки носа с
выступающими вперед ноздрями из-за недоразвития костной части
носовой перегородки.Синдактилия кожная или костная — частичное или тотальное
сращение пальцев.Сиыофриз — рост бровей над переносьем, создающий объединен¬
ную линию бровей (слившиеся или сросшиеся брови).Скафоцефалия (ладьеобразный череп) — удлиненный череп с
выступающим гребнем на месте преждевременно закрытого сагит¬
тального шва.Стопа-качалка — стопа с плосковыпуклой подошвенной поверх¬
ностью и выступающей кзади пяткой.Стрии — линейные полоски истонченной кожи.Телекант — латеральное смещение внутренних углов глазных
щелей при нормально расположенных орбитах и глазных яблоках.Тригоноцефалия — расширение черепа в затылочной и сужение в
лобной части.Тристихиаз — тройной ряд ресниц.
Генетические термины 563Уздечка языка короткая укорочение или прикрепление уздечки
в области кончика языка, приводящее к ограничению eio подвиж¬
ности.Фильтр — расстояние от нижней гочки носовой перегородки до
красной каймы верхней губы.Череп в форме трилистника — высокий выбухающий лоб, плоский
затылок, выпячивание височных костей с вдавленисм в местах их
соединения с теменными костями.Шалевидная мошонка - мошонка, окружающая валиком спинку
полового члена.Экзофтальм — смещение глазного яблока вперед с расширением
I ■ глазной щели.f Эктродактилия — клешневидные кисти или стопы из-за отсут¬ствия одного или нескольких пальцев.I ; Энофталъм — смеиіенис піазного яблока назад; более глубокое,чем в норме, расположение глазного яблока в глазнице.Эпикант — вертикальная кожная складка, прикрывающая вну¬
тренний угол глазной щели.Эписпадия — верхняя расщелина уретры.f і
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬАберрации хромосомные, 228
Аборт спонтанный, 228,443,467
Агаммаглобулинемия, 408,414
Агенезия
гонад,250
почек, 465щитовидной железы, 478
Агирия,262Аденокарцинома желудка, 305
Аденоматоз полиэндокринный, 137
Адренархе, 197Адреногенитальный синдром, 124,196
Адренолейкодистрофия
Х'сцспленная,423
глобоид но клеточная, 423
Акаталазия, 69, 148, 212, 215
Акродерматитэнтеропатический, 408
Акроцефалия, 96
Акроцефалополисиндактилия, 97
Алкаптонурия, 81, 131, 147, 153,353,367Алкогольдегидрогеназа, 319, 340
Aллoтpa^[cплaнтaция, 419
Альбинизм, 17, 147,213
неполный, 91
Альдегиддегидрогеназа, 319, 340
Альфа-фетопротеин, 459,460
Амилоидоз, 419Аминоаиидопатии, 367, 369, 371
Амиотрофия Шарко-Мари, 167
Амниоцснтез, 441, 465, 467, 469, 491
Амплификация, 301, 364, 374,380-387, 507Анализбиохимический, 131, 371
генеалогический, 120
гетеродуплексный, 382, 384
клинико-генеалогический, 121
конформационного
полиморфизма, 383
молекулярно-генетичес¬
кий, 374
рестрикционный, 379
родословной,120
спекірометрический,367
спектроскопический, 365
фенотипа, 166
хроматографический, 368
цитогенетический, 354
Анастомоз, 417Ангиогенез терапевтический, 425
Андрогенез, 43
Андроид, 44
Анемиясерповидно-клеточная, 58, 138,
146,210, 214,331
Фанкони, 324, 361, 423
Анеуплоидия, 71, 220
частичная,253
Аниридия, 137, 261
Аномалииврожденные, 87, 257
метаболизма, 350
нервной трубки, 440,462
хромосомные, 224, 227
Анотия, 96Антиципация, 175, 205
Антракоз, 321
Антропометрия, 94
предметный указатель565Анэнцефалия, 270,464
Арахнодактилия,94, 106, 179
Аргининемия, 404
Аритмия, 172, 180
плодная, 441
Артритподагрический, 400
ревматоидный, 290, 344
Артрогрипоз, 112
Артрохалазия,187
Аспартилгликозаминурия, 215
Ассоциация, 90
генетическая, 294
Астма бронхиальная, 272, 276, 285,290Атаксияспинномозжечковая, 161, 163
телеангиэктазия, 102, 132,148,207
Фридрейха, 161, 162
Атеросклероз, 52, 62,177
Атопия,273,290, 292
Атрезия двенадцатиперстной
кишки, 418, 465
Атрофиязрительного нерва Лебера, 128, 130
мышечная спинальная, 208,215
спинно-бульбарная, 161
Аутизм, 205, 274
Ауторадиография, 378
Аутосомно-доминантное
наследование, 40, 121
Аутосомно-рецессивное
наследование, 40, 122
Ахондроплазия, 56,61, 70, 81, 84, 86,
94, 122, 130, 138, 141,423,446
Ацетилирование, 40, 329, 341
Ацидемияизовалериановая, 407
метил малоновая, 404, 412органическая, 407
пропионовая, 404
Ацидурия, 369, 438аргининсукииниловая, 407
органическая, 367, 369, 372
оротовая, 408Беременность, 59, 67, 112, 117
планирование, 437, 445
прерывание, 112, 437
скрининг, 460элиминация эмбриона (плода), 67,
443, 438
Биопсияплода, 458, 459, 465,469
хориона, 441, 458, 465
Биотрансформация 319ксенобиотиков, 307, 317, 319
лекарственных средств, 336,339. 341Биохимические методы, 353, 355, 366,
449, 451
Биочипирование, 351, 389
Биоэтика, 484
прави.'іа, 487
принципы, 486
Бластопатия, 93
Блефарофимоз, 101
Близнецовый метод, 278
Блот-гибридизация,376
Болезнь/и, 78, 121Х-сцепленные доминантные, 125
Х-сцепленные рецессивные, 126
Y-сцепленные, 128
Альцгеймера, 61, 154, 209, 274, 423
аугосомно-доминантные, 121, 141
аутосомно-рецессивные, 122, 190
566Клиническая генетикаВердпича-Гофмана, 215
Виллсбранда, 38. 81
Вильсона—Ко!Ювалова, 124, 152, 418
вн>'триутробное лечение, 441
воспалительные кишечника, 282
врождеггные, 69наелсдстенные, 84
иенаследстпенные, 85
генные, 63, 70, 136
геномного импринтинга, 41
гипертоническая, 272
Гиршпрупга, 91, 137
Гоше, 215, 406,415,418
Дауна, 61, 78
ДНК-диагностика, 376
желчнокаменная, 283
иммунозависимые, 290
инфекциоігньїе, 294
ишемическая сердца, 177, 272, 287
Канавана, 215
Кеннеди,161
Краббе, 423
Крона, 290
лизосомныс, 150, 372
Менкеса, 408митохондриальные, 128, 372
многофакторные, 61, 70, 270
молекулярные, 50
моногенные, 137
мультифакториальные, 270
накопления, 150
наследственнаяпредрасположенность, 60, 70,
270наследственные, 22,61, 71, 219,437
врожденные, 84
симптомы, 80
неинфекционные, 290нейротрансмиттерного обмена, 372
ненаследственныс, 62
врожденные, 85
Нимана-Пика, 416, 418
нутригенстические, 325
Огаши, 215
Паркинсона, 423
патогенетическое лечение, 402
иероксисомные, 151, 372
Помпе, 411
профилактика, 434
Рандю-Ослера-Вебера, 156
Реклингхаузена, 122, 168
Рсфсума, 151, 406
семейные, 69
сердечно-сосудистые, 287
симптоматическое лечение, 399
спорадические, 70
ступени,351
Тея-Сакса, 138,215,473
углеводного обмена, 372
Фабри, 406,414, 419
Фон Хиппеля—Линдау, 304
хирургическое лечение, 417
холестеринового обмена, 372
хромосомные, 63, 70, 219
мозаичные, 224
наследуемые, 224
хроническая обструктивная
легких, 290
Штайнерта, 172
экспансии,162
эпигенетические, 41, 70
этиотропное лечение, 419
язвенная,275,285
Бракимиестный, 213кровнородственный, 124,213, 445
Брахидактилия, 107, 111,215
предметный указатель567Брахицефалия, 96, 236
Бронхит, 192хронический,323
Бронхоэктазия, 80, 192
Бутирилхолинэстераза, 339ВВарианты экогенетические, 64
Векторная система, 420
Вирусиммунодефицита человека, 294
Эпстайна-Барр, 298
Врач-генетик, 449
Вредности профессиональные, 323
Врожденные аномалии, 87
Врожденные пороки развития, 87
изолированные, 90
многофакторные, 93
множественные, 90
наследственные, 90
системные, 90
хромосомные аномалии, 227
экзогенные, 91
Выкидыш, 467Гатактоземия, 124, 325, 438, 479
Дуарте, 480
скрининг, 480
Гаметопатия, 93
Ганглиозидоз, 123, 156, 473
Гастрошизис, 461
Гель-электрофорез градиентный
денатурирующий, 384
Гемигипертрофия, 81, 111
Гемоглобинопатии, 166, 331,406,417,
423,468Гемолиз, 325
Гсмосидероз, 152,406
Гемосорбция, 406Гемофилия, 70, 81, 127, 138, 387,408,414,418
Гемохроматоз, 153
первичный, 152, 207
Ген/ы, 35дистропные, 286
дрейф, 211, 316импринтированные, 42, 44, 329
модификаторы, 163
молчащие, 318
нейтральные, 318
опухолесупрессорные, 302
плейотропия, 83
полиморфизм, 316
синтропные, 286,288
функции, 39
человека, 36
Генеалогический метод, 27, 112,353
Генеалогия, 112
Генетика, 14биохимическая, 21
классическая, 27
клиническая, 15
медицинская, 14, 22, 485
принципы, 486
этические вопросы, 484
менделевская, 21
современная, 27
формальная, 21
цитологическая, 21
человека, 14
экологическая, 312
Генетическая изоляция, 214
Генетическая чувствительность, 330
Генетические ассоциации, 280
Генетические карты хромосом, 44
568Клиническая генетикаГенетические маркеры, 65, 287
Генетический контроль, 75
Генетическое консультирование, 489
Генетическое расстояние, 44
Генетическое тестирование, 308
Генная инженерия, 409
зародышевых клеток, 443
Генные болезни, 136, 206
Генокопия, 143
Геном, 24митохондриальный, 34,46
человека, 26, 30
Геномика, 24медицинская, 25
сравнительная, 25
структурная, 24
функциональная, 25, 335
эволюционная, 25
Геномный профиль, 287
Гепотерапия, 424
Генотип, 23
Генотипирование, 351
Гепаднавирус, 298
Гепатит, 297холестатический, 192
Герпес-вирус, 298
Гетерогенностьвнутрилокусная, 166
генетическая, 166
межлокусная, 166
Гетерозиготности потеря, 303
Гибридизацияблот Саузерна, 376
геномная срапнительная, 364
флюоресцентная, 362
Гидронефроз, 465
Гидроцефалия, 84, 91, 96,270
Гиногенез, 44
Гиноид, 44Гипергликемия, 285
Гиперглицинемия
некетоноваіі, 407
Гиперкератоз, 96
Гиперлипопротеинемия, 413
Гиперметропия, 102
Гиперплазиякоры надпочечников
врожденная, 441,479
вирилизирующая, 196
скрининг, 479
надпочечниковая врожденная, 196,
215,413Гипертслоризм, 99, 101,106, 130, 246,
254сосков, 105, 106
Гипертония артериальная, 285, 287
Гипертрихоз, 95
Гипсрурикемия, 407,412
Гиперфенилаланинемия, 412,477
Гиперхолестсринемия, 131,285, 407
семейная, 151, 175,215,417,428
Гипоспадия, 111, 243, 271
Гипотезадвухударная, 303
канцерогенеза, 303
один ген — один фермент, 82
преконцепционной
профилактики, 481
условного гропизма, 164
Ги потел оризм, 101
Гипотиреоз, 438
врожденный, 478
скрининг, 478
Гипотиреоидизм, 408
Гипотонизаиия клеток, 357
Гипофосфатемия, 106, 125, 409
Х-сцепленная, 409
наследственная, 125
предметный указатель569Гипохондроплазия, 56
Гирсутизм, 95
Гистидинемия, 404
Гчаукома эксфолиативная, 282
Гчикогеноз, 151,414
Гликопротеиноз, 414
Гликосфииголипидоз, 414
Глиобластома, 300
Глиома, 308зрительного нерва, 170
Глутатионтрансфераза, 344
Гіухота, 48наследственная, 167
сенсо-невральная, 138
Глюкуроїшрование, 341
Глюкуроновая кислота, 341
Голопрозэнцефалия, 91, 138, 464
Гомеостаз, 73
Гомоцистеинемия, 328
Гомоцистинурия, 80, 130, 167,412
Гонадотропинхорионический,462
Грыжа, 185диафрагмальная, 441, 464
паховая,180пупочного канатика, 262
спинномозговая, 242,271,482
черепно-мозговая, 271Дегенерациягепатолентикулярная, 124, 152,
213,406
желтого пятна старческая. 282
маЕсулярная возрастная, 139
Дезоксирибонуклеиновая
кислота, 28, 362
і'еномная, 29, 374гибридная, 409
методы диагностики, 376
рекомбинантная, 420
Делеция, 221Деонтология медицинская, 485
Дерматит атопический, 285
Дерма'юспараксис, 187
Детоксикация, 319
Дсфект/ыбрюшной стенки, 461
врожденные, 87
митохондриального
окисления, 369
молекулярный, 151, 175, 186
нервной трубки,461
Дефицитаденозиндезаминазы, 427
галактозофосфатуридил-
трансферазы, 325
га.'Шктокиназы, 325
гидроксилазы,196,197, 441
гидроксистероидаегидро-
геназы, 196
глюкозо-6-фосфатдегидрогеназы, 325
карбоксилазы,441
фолиевой кислоты, 205,327
холестерол гидроксилазы, 196
арантитрипсина, 321,413
Деформация
вальгусная, 105
варусная,106
конечностей, 106
Диабет сахарный, 38, 139, 215, 285,
288, 290, 326,418,460, 464
Диагностикабиохимическая, 366
компьютерная, 132
лабораторная, 353
570Клиническая генетикамолекулярно-генетическая, 372
наследственныхболезней, 85, 378, 449
ненаследственных
болезней, 86
предымплантационная, 471
пренатальная, 458, 470
ультразвуковая, 463
хромосомных болезней, 225
цитогенетическая, 356
Диандрия, 43
Диастема, 104, 105
Дигения, 44Дигидропиримидиндегидро-
геназа, 339
Диета, 404бесфенилаланиновая, 477
Дизавтономия, 215
семейная, 112, 215
Дизостозакрофасциальный, 107
периферический, 108
Диморфизм половой, 273
Дисгомеостаз, 74
Дислипадемия, 287
Дисморфогенез, 90-94
постнатальный, 95
пренатальный, 95
эмбриональный, 94
Дисплазиядистрофическая, 99
соединительной ткани, 17,162, 182,
204фронтом етафизарная, 130
хондроэктодермальная, 214
Дистропия, 285
ДистрофияДюшенна, 469,473
лицевая, 162миотоническая, 122, 160, 162, 172,
214,473
мышечная, 126, 469
окулофарингеальная, 162
плечелопаточная, 162
Дисфункциягипоталамическая, 170
коры надпочечников
врожденная, 196
эндотелиальная, 290
Долихостеномелия, 94, 105, 179
Долихоцефалия, 94, 96, 105,203
Доминантное Х-сцепленное
наследование, 125
Дрейф генов, 211, 316
Дупликация, 221Евгеника, 18
негативная, 397
позитивная, 18
Евфеника, 397,440ЖЖивотные трансгенные, 410Заболевания, см. Болезнь/и
Задержка внутриутробного
развития, 112
Законы Менделя, 18,58, 71, 444
Занос пузырный, 44
Зигота, 34, 224,444Злокачественные новообразования, 297
Зоб, 478
Зонд,377, 362
Предметный указатель571ИИзменчивость, 54
мутационная, 57
Изоляпия генетическая, 214
Изохромосома, 223
Илеус мекониальный, 155, 191,418
Иммунодефицит, 330
Иммунозависимые болезни, 290
Иммунологические методы, 451
Имиринтинг генетический, 41
Ингибиция метаболическая, 407
Индекс митотический, 356
Инженерия геЕтная, 409, 421, 425
зародышевых клеток, 443
Инсерции, 136
Инсулинома, 305
Инсульт, 287
Интерактом, 25
Инфаркт миокарда, 287
Инфекционные болезни, 294
Иниестные браки, 213
Инъекцияинтрацитоплазматическая, 472
Ихтиоз, 84,469,471
врожденный, 213ККамптодактилия, 106, 108
Канцерогенез, 299, 303
Канцерогеїгьі, 298, 323
Кардиомионатия, 200,405
первичная, 418
Кариотилиропание спектральное, 365
Карликовость, 464
Картирование, 24
хромосом, 44
Карцинома тиреоидная, 137Клеточная терапия,421
Клеточные культ\фьт, 356
Кл и н и ко-генеал огически й
метод, 112,450
наследственнаяпредрасположенность, 275
Клинодактилия, 106, 109,237
Коагулопатии, 468
Коллагсноз, 290
Колхицин, 357
Колцемид, 357Компаунды генетические, 166
Компеьгсаииярепродуктивная,211, 436
Компьютерные диагностические
программы. 132
Конкордантность, 279
Консультированиегенетическое, 448, 489
спеииализиропанное, 454
эффективность, 456
Континуум сердечно-сосудистый, 288
Контроль генетический,75
Конъюгация
водная, 343
глутатионовая, 344
Кордоцентез, 458, 466,468
Косоглазие, 101, 102
Коэффициентнаследования, 297
риска многофакторного
заболевания.274
Крипторхизм,111, 243
Кровнородственные браки, 124, 213,
445Ксантоматоз, 177
Ксенобиотики,319
Ксеродерма пигментная, 330, 361
Культивирование клеток, 356
572Клиническая генетикаЛЛабораторная диагностика, 353
Лаваж маточный, 472
Легенда родословной, 11 5
Легочное сердпе, 192
Лейкемия, 305миелоидпая хроническая, 137
ЛейкодистрофияvieiaxpoMaTHMecKafl, 414
Лейкоз, 220, 238, 307
лимфобластный, 343
миелоидный, 228, 301
Лейциноз, 369, 412
Летальность, 59, 225хромосомные аномааии, 226, 227
Лимфома Беркитта, 301
Липопротеиноз, 212
Липосома, 416
Липофусциноз, 215
Лиссэнцефалия, 262
Лихорадка средиземноморская, 419мМакрогения, 103
Макроглоссия, 104, 262
Макрогнатия, 103
Макроорхидизм, 203
Макросом и явнутриутробная,112
Макростомия, 103
Макротия, 96
Макроцефалия, 96, 203
Маловодне, 112Мальформация легких кистозно¬
аденоматозная, 465
Малярия, 210, 294
Маннозидоз, 100Маркеры іенстические, 65, 287, 391
Масс-спектрометрия, 368
тандемная, 369
Медико-генетическое
консультирование, 448
специализированное, 454
эффективность, 456
Медицина
геномная, 49
молекулярная, 50
партнерская, 49
персонализированная, 49
превентивная, 49
предикативная, 49
предсказательная, 49
профилактическая, 49
Мейоз, 34Мекониальный илеус, 155, 191,418
Менделя законы, 18, 58, 71,444
Мегшнгиома, 300, 305, 308
Метаболизаторы, 337
Метаболизм
аномалии,350
ксенобиотиков, 319
лекарственных средств, 336
нутриентов, 324
Метаболом, 25
Метафаза, 357
Метилирование, 342
Метод/ыбиохимические, 353, 366, 451
биочипов, 389
близнецовый, 278
высокоразрешающей
генетики, 357
генеалогический, 27, 212, 353
генетический, 167
геномной гибридизации, 364
детекции мутаций, 378
предметный указатель573дифференциального
окрашивания, 359
ДНК-диагностики, 372, 378
иммунологические, 355, 451
клинико-генеалогический, 112,
276, 450лабораторной диагностики, 353
метафазный, 357
молекулярно-генетическис, 373
молекулярно-цитогенетические, 362
мутационного скрининга, 382
оценки сегрегации, 120
параклинический, 131,451
цлазмонового резонанса, 390
подтверждения диагноза, 372
популяционный, 281
пренатальной диагностики, 459,470
приемных детей, 277
прометафазный, 357
просеивающие, 370,459,474
простой окраски, 358
расшеплсния неспаренных
оснований,386
родословных, 112
рутинной окраски, 358
секвенирования, 387
скрининговые, 460,474
спектрометрический,368
спектроскопический,365
трансляции белкового
продукта, 389
флюоресцентітой
гибридизации,362
хроматографический, 368
цитогенетические, 355,450
цитологические, 355
Механизматопической реакции, 291
импринтинга, 42опухолевой трансформации, 300
репарации,74
элиминации эмбриона, 443
эмбрионального развития, 87
эпигенетический, 40
Миелолейкоз, 228, 301
Микрогения, 103,104, 254
Микроглоссия, 104
Микрогнатия, 99, 103
Микросателлиты, 392
Микроскопия, 335
Микростомия, 103
Микротия, 99
Микрофтатьм, 101
Микроцефалия, 84, 94, 96, 213, 254
МиодистрофияБеккера, 160, 199, 202
Дюшенна, 80, 138, 140, 199, 469
Дюшенна-Беккера, 126, 199, 202
МиопатияБеккера, 63, 160
Дюшенна, 63, 152, 160, 423
Дюшснна-Беккера, 208
стати и индуцированная, 281
Миопия, 102
Миотония, 122
врожденная, 38дистрофическая, 123, 141, 155, 160,
172Митогены, 298Митохондриальное наследование, 128
Митохондрии, 34
Многоводие, 112Многофакторные болезни, 270, 297
Мозаицизм
гонадный, 141
соматический,140
Молекулярно-генетические
методы, 373
574Клиническая генетикаМолекулярно-цитогенетические
метол г,к 362
Монголоияизм, 236
Моносомия, 223
4р-, 256
5р-, 254
X, 250
Морфогенез, 88Муковисиидоз, 80, 82, 124, 130, 136,
І44, 152, 156, 190, 208, 214, 272, 274,
380, 387, 401,418,472,476,481
симптоматическое лечение, 400
скрининг, 481
Мукополисахаридоз, 81, 124, 414
Мультифакториаіьньїе болезни, 270
Мутагенезиіщуцированный, 314, 447
инсерционный, 431
спонтанный, 447
Мутагены, 298, 323
Мутации, 57, 63, 209,313
гаметические, 140, 224
генные, 63, 91, 136
геномные, 63, 219, 225
динамические, 161
зародышевых к.'1сток, 140,314
зиготическис,224
летальные, 67
патологические, 67, 143
плодных клеток, 314
половых клеток, 70
соматических клеток, 70, 140, 228,
314ючковые, 302
хромосомные, 63, 219
эмбриональных клеток, 314
эффекты, 142
Мутационный процесс, 209
индуцированный, 313нНаслелованиеХ-сцепленноедоминантное, 125
Х-сцепленнос рецессивное, 126
Y-сцспленное, 128
аутосом н о-до м инантное ,121
а>тосомно-рецсссивное, 122
митохондриальное, 128
Наследственность, 54, 64
митохондриальная, 128
Недостаточностьадеиозиндезаминазы, 427
адренокортикальная, 419
галактозофосфатуридил-
трансферазы, 325
галакгокиназы, 325
гилроксилазы, 196, 197, 441
1'идроксистероидаегидро-
геназы, 196
глюкозо-6-фосфатдепадрогеназы, 325
карбоксилазы,441
фолиевой кислоты, 205, 327
холестсрол гилроксилазы, 196
араититрипсина, 321, 413
Нейробластома, 300
Нейропатия Лебера, 139
Нейрофиброма. 169, 262
плексиформная, 169
Нейрофиброматоз, 72, 86, 122, 154,
168,208,262,305,446
Неоплазия эндокринная
множественная, 305
Непереносимость
белка, 215, 371, 438
і'лютеїіа, 325, 438
лактозы, 325,404
Предметный указатель575фруктозы, 404
Новообразованиязлокачественные, 297
Норма реакции, 73
Нормокопирование, 144,438, 440
Нормокопия, 144
Нутригенетика, 324
Нутригенетические болезни, 325
Нутригеномика, 324Ожирение, 285
Окраска препаратов, 358
хромосом, 359
Оксалоз, 419Олигодактилия, 104, 106, 464
Олигофрения, 204
Омфалоцеле, 461
Онкогены, 137, 299
Онкоцитома, 129
Онтогенезнесовершенный, 464
хромосомные аномалии, 225
Оплодотворениеэкстракорпоральное, 472
Опухоль/и, 47Вильмса, 70, 261, 304, 418
злокачественные, 297
потеря гетерозиготности, 303
Остеогенез несовершенный, 80, 122,
139, 142,423,464
ОстеодисплазияМелника-Нидлза, 130
Остеосаркома, 304
Отек ангионевротический, 429, 413
Относительный риск, 281
Отношение шансов, 281
Отсталость умственная, 48, 130, 204пПапиллома предушная, 99
Папилломавирус, 298
Параклинические исследования, 131
Паралич церебральный детский, 48
Параоксоназа, 339
Пахионихия, 130
Пенетрантность
неполная, 64
управление, 439
Период/ыкритические развития, 92
пренатальный, 88
терм инационный
тератогенный, 92
Перцентили,94
Пиелонефрит, 87
Питание лечебноебесфенилаланиновое, 447
диетическое возмещение, 408
диетическое добавление, 405,408
диетическое ограничение, 404
Плазмаферез, 406
Планированиебеременности, 437,445
Плацентобиопсия, 465
Плейотропия, 82, 154
вторичная, 83
первичная, 83, 154
Пневмония, 174обструктивная, 323, 344
острая, 86хроническая, 80, 321
Пневмосклероз, 192
Повторы, 29Подагра, 400, 407, 412,419
Полидактилия, 106,148, 464
Поликистоз почек, 419, 464
576Клиническая генетикаПолимеразная цепная реакция, 374
аллельспецифичная, 380
метилспецифическая, 382
мультиплексная, 380
в реальном времени, 381
Полиморфизмбалансированный, 59
белковый, 58генетический, 307, 316,349
дезоксирибонуклеиновой
кислоты, 31
длины рестриктивных
фрагментов, 391
клинический, 66, 157
количества копий, 391
конформационный, 383
однонуклеотилный, 32
Полипоз, 212, 417, 473
аденоматозный, 305
Полисомии половых хромосом, 246
Полител ИЯ, 105
Популяционный метод, 280
Пороки развития врожденные, 87,
См. также Врожденные пороки
развития
Порфирия, 212
острая, 414
пестрая,215
Последовательность, 90
Правила этические, 488
П редрасположен н остьнаследственная 60, 71, 279, 298
близнецовый метод, 278
клинике-генеалогический ме¬
тод, 275
популяционный метод, 280
П редым плантационная
диагностика, 471
Пренатальная диагностика, 458,470инвазивная, 465, 471
Пренатальный период, 88
Прерывание беременности, 437
Принципы этические, 486
Пробаантипириновая, 351
дибукаиновая, 351
динитрофенилгидразиновая, 370
изониазидовая, 351
нагрузочная, 371
потовая, 481
с хюридом железа, 370
трипсиновая, 481
фармакологическая, 351
Пробанд, 113, 114
Прогения, 103
Программы
компьютерныедиагностические, 132
профилактические, 438
Профедиентность клинической
картины, 156
Прометафаза, 357
Протеиноз, 215
Протеом, 25
Протоонкогены, 299
амплификация, 301
Профессиональные вредности, 323
Профилактика наследственных
болезней, 434
вторичная, 437
генетические основы, 437
генно-инженерная, 444
медико-генетическое
консультирование, 448
неонатальная, 439
первичная,437,471
перинатальная, 440
популяционная, 439
Предметный указатель577прекониепционная, 439,440,482
пренатальная, 439
программы, 439
скрининг, 474
іретичная, 438
элиминация плода, 443
Профиль геномный, 287
Псевдохолинэстераза, 339
Психоз биполярный, 271
Псориаз, 70, 272
Птоз, 100, 101
Пузырный занос, 43
полный, 43Рабдомиосаркома, 304
Равновесие генетическое, 331
Радиография, 463Развитие человека пренатальное, 88
Рак, 428генетический полиморфизм, 305
генотерапия, 428
гепатоцеллюлярный, 305
желудка, 305, 308
кожи, 308колоректальный, 305
легкого, 304, 307молочной железы, 137, 304, 306, 307
мочевого пузыря, 304, 307, 324
печени, 305
пищевода, 308
простаты, 308
щитовидной железы, 305
Расстояние генетическое, 44
Расщелина нёба, 104
Рахит, 409витамин D-резистентный, 125,152, 167,412Реакция/иатоп№іеские, 291
полимеразная цепная, 374, 380
экогенетические, 318, 320
Рентгенография, 463
Репарация,74
Репликация, 74
Репродуктивнаякомпенсация, 211, 436
Рестрикция, 375
Ретинит пигментный, 38
Ретинобластома, 70, 86,137, 261, 302,304,418
Ретровирус, 298
Ретрогения, 103
Рецессивное Х-сцепленное
наследование, 126
Рибонуклеиновая кислота, 373
Ринит, 290Риск относительный, 281
Род,114Родословная,112,115
анализ, 113, 120
биохимическая, 451
иммунологическая, 451
символы, 117
составление, 113
Роды преждевременные, 443Сантиморганила, 45
Саркома, 305
Рауса, 299, 300
Харвея, 300
Сахарный диабет, 38, 139, 215,285,
288, 290, 326,418,460,464
Секвенирование, 387
Семейство болезней, 285
578Клиническая генетикаСемья, 114Сердечно-сосудистые
:іаболсвания, 287
Сибсы, 114
Силикоз, 321Симптомы наследственных
болезней, 80
Синдактилия, 106, 108
кожная, 106
Сиидром/ы, 78
45,X, 247
47,XXX, 246
47,XXY, 247
47,XYY, 247Х-сцепленной умственной
отсталости, 103, 203
Аарскога, 101, 103, 111
алреногенитальный, 124, 196,441
а^хгісргический, 286
Альпорта, 140, 419
аминоптериновый, 108, 110
Ангельмана, 43,260, 261,263, 382
Аперта, 446Беквита-Видемана, 104, 112, 260,
262,264, 304,382
Блума, 215Ваарденбурга, 91, 96,446
вальпроевый, 109
Вейля-Марчезани, 80
вело кард иофациальный, 262
Видемана-Беквита, 43
Вильямса-Бойрена, 261
внезапной детской смерти, 47
Вольфа-Хиршхорна, 107, 256
гидантоиновый, 111
Гольтца, 148
Гольтца—Горлина, 125
Горлина, 91
Грейга, 100Гурлер,138, 423Дауна, 70, 86, 219, 224, 230, 234, 461
ДиДжорджи, 262, 263, 418
дисомии Y-хромосомы, 249
Дубовица, 112
Жильбера, 341,413
Карпентера, 97Клайнфелтера, 70, 219, 247, 248
Корнелии де Ланге, 102, 112
Коффина-Лоури, 70
кошачьего глаза, 99
кошачьего крика, 70, 233,254
Коэна, 98Криглера-Найяра, 341, 413
Крузона, 148, 149
Лангера-Гидеона, 263, 261
Леша-Найхана, 127,407, 412,473
Линча, 208
Ли-Фраумени, 305
лица эльфа, 261
ломкой Х-хромосомы, 137, 161
ломкой Х-хромосомы иумственной отсталости, 203
Лоренса—Муна, 148
Луи-Бара, 132Мартина-Белл, 160, 203,208
Марфана, 80, 94, 121, 130, 154,167,
176, 208,446,473
Мачадо-Джозефа, 161
Меккеля, 148, 150, 242
метаболический, 286
Миллера-Дикера, 262
множественных врожденных
пороков развития, 90
Мора, 242недержания пигмента, 125
Незелофа, 418нефротический врожденный, 215
Нунан, 100, 148
Предметный указатель579Патау, 240
пикквикский,285
подколенного птеригиума, 110
Прадера-Вилли, 43, 70, 260, 261
263,382
приобретенногоиммунодефицита, 294
Пфайфера, 102
Рассела-Сильвсра, 43
Ретта, 70, 137
Робертс, 148
ротолицепальцевой, 125
рука—сердце, 130
скрученных волос и глухоты, 96
Смита-Лемли-
Опитиа, 104,107
Смита-Магениса, 262
Сотоса, 112, 260
тератогенные, 91,92
Тернера, 201, 452
тестикулярнойфеминизации, 151, 247
Тея-Сакса, 123
трипло-Х, 247
три сом ИИ 9, 109
трихоринофалангеальный, 261
Ушера, 162фронтометафизарной
дисплазии,130
Халлермана-Штрайфа, 101
Хантера, 80, 127
Хартнапа, 405
Холта-Орама, 91, 130, 148
хромосомные, 84
хромосомных аберраций, 258
Целлвегера, 151
целлвегероподобный, 151
церебро гепаторенальный ,151
частичной трисомии 9+, 257частичных анеуплоидий, 252
Ш ере шевс кого-Тернера, 219, 233, 247,250
Эдвардса, 233, 242
Элерса-Данло, 83, 87, 100, 122,
130, 154, 167, 182, 208
Эллиса-Ван-Крепельда, 148,214
Синофриз, 102
Синтез белка, 139
Синтропия, 285, 288
Сиреномелия, 93
Склероз рассеянный, 271, 274
Склероостеоз, 215
Скрининг, 474
беременных, 460
га;іактоземии, 479
гиперплазии коры надпочечников
(врожденной), 479
гипотиреоза (врожденного), 478
муковисцидоза, 476
мутационный, 382
новорожденных, 474
фенилкетонурии, 477
Слепота, 48Соседство болезней, 285
Спленэктомия, 417
Спорадичность, 70
Стволовые клетки, 421
гемопоэтические, 422
стромальныемeзeнxимaль^^ыe, 422
Стенозаорты,105, 261
венечных артерий, 177
коронарных артерий, 177
легочной артерии, 261
почечной артерии, 170
привратника, 90, 271, 436
Сулы^затирование, 343
580Клиническая генетикаТалассемия, 38, 83, 138, 148,153, 163,
210,215, 331,430,473
Тслекант, 101
Терапиявнутриутробная, 441
генная, 419,421,424
клеточная, 419, 421
патоіенетическая, 400,402
симптоматическая, 400
ферментная, 411
Тсратаназия, 443
Тератогены, 91
Тератома, 441
Тестантипириновый, 351
дибукаиновый, 351
динитрофенилгидразиновый, 371
изониазидовый, 351
нагрузочный, 372
лотовый, 481
с хлоридом железа, 371
трипсиновый, 481
фармакологический, 351
Тестирование генетическое, 308
Тиопуринметилтрансфераза, 343
Тиреоидит аутоиммунный, 285
Тирозинемия, 404,418
Токсикогенетика, 321
Токсикогеномика, 321
Томография, 463
Трансгенные животные, 409
Трансгеноз, 409, 424, 426
Транскриптом, 25
Транскрипционные факторы, 89
Транскрипция, 74
Транслокациянесбалансированная, 222реципрокная, 221
робертсоновская, 222
сбалансированная, 221
хромосомная, 300
Трансляция, 389
Транспла^гтация ,418
аллогенных клеток, 421
трансгенных клеток, 426
Транспортеры, 345
Трансформация
биологическая,319
опухолевая, 297, 298
Тремы, 105
Тригоноцефалия, 96
Опитца, 242
Триплоидия, 223
диандрическая, 44
дигеническая, 44
Трипсин иммунореактивный, 481
Трисомия, 223
13,240, 452
18,97,242,452
21,79, 86, 229, 234, 452
8, 110, 224
9,109
9р+, 257
X, 247
Тромбофилия, 38
Тромбоцитопения,468
аллоиммунная, 441
Туберкулез, 285, 295Узелки Л иша, 170
Ультразвуковое исследование, 463
Умственная отсталость, 48, 130, 203
Уридинд ифосфогл юкуро новаякислота, 341
Предметный указатель581ФФакторы транскрипционные, 89, 300
Фармакогенетика, 317, 335, 348
Фармакодинамика, 349
Фармакокинетика, 349
Фенилаланинемия, 188
Фенилкетонурия, 70, 81,83, 124, 137,
148, 156, 188,216, 367,404,438,
440,473,477
скрининг, 477
Фснокопия, 143
Феном, 25
Феноменантиципации, (61, 175,205
потери импринтинга, 43
репродуктивной компенсации, 211
экогенетического действия, 312
Фенотип биохимический, 367
Феохромоцитома, 170, 305
Ферментывозмещение, 414
модификация активности, 412
Фетопатия, 93диабетическая,112
Фетоскопия, 465
Фиброз кистозный, 190
Флюоресцентная гибридизация, 362жидкостнаявысокоэффективная, 368
жидкостнаяденатурирующая, 389
Хромосомные аномалии, 219
онтогенез, 225
Хромосомные болезни, 219
Хромосомыаберрации,258
генетические карты, 44
дифференциатьное
окрашивание, 359
колы1епые, 223ЦЦелиакия, 80, 285, 325, 438
Циклопия, 93
Цирроз, 192
Цистиноз, 324
Фан кони, 419
Цистинурия, 404, 407
Цитогенетика, 21, 355высокоразрешающая, 357
интерфазная, 364
Цитогенетические методы, 355, 450
Ци'іруллинемия, 404Холелитиаз, 283
Хондродисплазия, 90
Хондродистрофия, 56
Хорея Гентингтона, 61, 72, 86, 122,
І54, 160,209,215,274,423
Хорионбиопсия, 458, 465
Хроматография, 368
газовая, 368, 341Чипы, 351,389
Чу вствител ы юсть
генетическая, 330
иммунная, 330шШизофрения, 70, 271, 274
Шок гипотонический, 357
582Клиническая генетикаЭкзофтальм, 101
Экогенетика, 312, 317
Экогснетические варианты, 64
Экспансия, 136
Экспрессивность
варьирующая, 64
управление,439
Экстракорпоральное
оплодотворение, 472
Электрофорезгсмоглобиноп, 371
дезоксирибонуклеиновой
кислоты,376
Элиминация эмбриона, 443
Эмбриопатия, 103
диабетическая, 103
Эмфизема легких, 192, 322, 323
Эндокринопатии, 414
Энзимопатии, 331, 406, 411
Энтеропатия глютсновая, 80
Ззиіефалопатиямитохондриальная, 46, 138
Эицефалоцеле, 150,464
Эпигенетика, 329
Эпигенетические механизмы, 40
Эпигеиотип, 41
Эпидермолиз, 469,471
буллезный, 38, 86
Эпикант, 101, 184, 236, 246, 251, 254
Эпилепсия, 271,464
Эпимутации, 71
Эпоксидгидроксилаза, 343
Эритродермия ихтиозоформиая, 96
Эстриол неконъюгированный, 462
Этика, 484
правила, 487
принципы, 486Эффект/ымутантного аллеля, 143, 145, 148
мутаций, 67, 140, 142
окружающей среды, 308
родоначальника, 212
хромосомных аномалий, 225, 228
Клиническая генетика.
Геномика и протеомика
наследственной патологии3-є изд., перераб. и доп.Мутовин Г. Р.Отличительные особенностии832с., 2010г.ІІІІІттшІЇІІІІІІІІВ книге рассмотрены основные положения
и понятия клинической генетики с учійтґіш:;і
результатов международной научной
граммы «Геном человека« (1988-2005'чт.).';
Представлены история, положения, прнйтшиі
и достижения клинической генетики,'ИЙЛО-Й
жены особенности обмена веществ в КЛбТКвН:
и организме в целом, пути управлений ;мет^:і::
болизмом, характеристика рецепторсщ^и сйг-^м
нальных молекул и их роль в формировании: ■:
наследственной патологии. Основное
ниє уделено этиологии, патогенезу( подхода^
к диагностике, лечению и профилактике йа-іг
следственной патологии.В пособие вошли болезни НЄТраДИЦИ(МЙЬІЮ:іі
наследования; накопления, геномной памяти;?!■
митохондриальные, пероксисомные^ : прион-і
ные, экспансии нуклеотидных повторор*іДа-іі;
ется характеристика перспективных напрай-:;
лений развития молекулярной м^иЦИНЙі-;;'
генотерапии, клеточной и тканевой терний/
нанобиотехнологий и наномедицины; показа*;
на их медицинская и социальная Значимо(|^к;;:
Учебное пособие предназначено СтудівнтаМ'
медицинских и биологических спеиййльно?:-,
тей вузов, биологам, а также врачам |а&эйых|,
специальностей. , ’';-:іНіІ.';:* іIIilii
Все главы учебника переработа¬
ны и дополнены в связи с развити¬
ем медицинской науки и практики.
Существенно дополнены главы по
многофакторным заболеваниям, про¬
филактике, лечению наследственных
болезней, экологической генетике
и фармакогенетике. Весь теоретичес¬
кий материал проиллюстрирован схе¬
мами и рисунками.В учебнике представлены новые,
выявленные в последние годы зако¬
номерности направлений генетики
(эпигенетика, малые РНК, однороди¬
тельские дисомии, генетический по¬
лиморфизм и др.).В приложении на компакт-диске
размещены дополнительные статьи
о лечении наследственных болезней,
мутагенезе, евгенике.Предназначен студентам меди¬
цинских вузов, обучающихся по
специальностям «лечебное дело»,
«педиатрия», «медико-профилакти¬
ческое дело» по дисциплине «меди¬
цинская генетика».> Введениев клиническую генетику> Наследственность
и патология> Семиотикаи клиническая диагностиканаследственныхзаболеваний> Генные болезни> Хромосомные болезни> Болезни с наследственной
предрасположенностью> Экологическая генетика> Фармакогенетика> Лабораторные методы
диагностики> Принїїипм лечения
нвсяе^1ственных
болезней> Профилактика
наследственной
патологии> Этические вопросы
медицинской генетикиМедицинская генетикаwww.geotar.ru
WWW. medkn igaservis. ruISBN 978-5-9704-1683-99 785970 416839 >